
International Programme on Chemical Safety
Poisons Information Monograph 335
Chemical
Methanol
Aliphatic alcohol
carbinol
colonial spirit
columbian spirit
methanol
metanolo (Italian)
methylated spirit(s)
methyl alcohol
methyl hydroxide
monohydroxymethane
pyroxylic spirit
wood alcohol
wood naphtha
wood spirit.
67-56-1
NIOSH: PC 14OOOOO
Local only.
Not relevant.
The main risks are severe recurrent metabolic acidosis with increased anion gap, caused by accumulation of formic acid and in late stages also lactic acid. The acidosis and the metabolite formic acid cause the central nervous system depression/toxicity, and visual disturbances that may be permanent.
Complete blindness is possible.
Target organs: central nervous system and retina.
Neurological: Early signs are slight inebriation and drowsiness. Delayed signs appear after 8 - 36 h: headache, vertigo, drowsiness, coma, and, occasionally, convulsions. Dilated pupils, with sluggish or absent light reflex, occur in conscious patients.
Visual disturbances: Vision becomes blurred or dimmed, and may be sufficient to impair perception of light or cause complete blindness. There is impaired pupillary response to light, and contraction of visual fields, scotoma, and flashing lights. Visual disturbances can be permanent. Hyperaemia of the optic disc is common in the acute stage.
Other: Abdominal pain is frequent; acute pancreatitis may occur.
Severe recurrent metabolic acidosis with increased anion gap is caused by formate accumulation and is related to the severity of poisoning . The osmolal gap is also typically elevated in the early stages before the methanol is metabolised to formic acid. Serum amylase may be elevated. Hyperkalaemia may occur related to the metabolic acidosis, and red blood cell mean corpuscular volume may be increased.
If determination of methanol cannot be performed, the anion and osmolal gap should always be calculated (Jacobsen & McMartin, 1997). Formate determination in blood and urine by an enzymatic method may also be performed, but is rarely available in the acute phase.
First-aid measures
Acute poisoning occurs principally by ingestion. All cases of ingestion should be sent to hospital.
If exposure occurred by inhalation or contact with skin, remove the patient from exposure, remove contaminated clothes, and wash the skin and eyes carefully.
Management principles:
Consider emptying the stomach by gastric lavage following recent (< 1 hour) ingestions of large amounts.
Correct metabolic acidosis with sodium bicarbonate, adjusting the administration rate according to repeated and frequent measurement of acid/base status.
Administer ethanol or fomepizole as an antidote to inhibit the formation of toxic metabolites.
Haemodialysis removes methanol and its metabolites, and also helps in correcting the metabolic acidosis.
If specific treatment is started early enough it may prevent death and permanent visual damage.
Methanol was originally obtained by the destructive distillation of wood, but is now usually manufactured from hydrogen and carbon monoxide or carbon dioxide, and also by oxidation of hydrocarbons (Windholz, 1983).
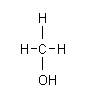
Formula: CH3OH
Molecular weight: 32.04
Colour, odour: clear, colourless liquid, with a slight alcoholic odour.
Normal state at room temperature: liquid.
Boiling point: 65°C
Melting point: 97.8°C
Flash point: 12°C (54°F)
Autoignition temperature: 470°C (878°F)
Relevant density (water = l): d20/4: 0.7915
Relative vapour density (air = l): 1.11
Vapour pressure at 25°C: 125 mmHg (16.2 KPa) at 20°C: 94 mmHg
Explosive limits (vol.% in air): 6.0 to 36.5
Dangers associated with vapour, its dispersion, and possible ignition: the liquid is flammable and, at normal room temperatures, it evolves vapours which form explosive mixtures over a wide range of concentrations. The extent of danger from inhaling vapours depends on the concentration and the duration of the exposure (Parmeggiani, 1983).
Possible chemical reactions: violent reaction with oxidising agents such as CrO3, Pb(ClO4)2, HClO4, P2O3, (Lewis, l996).
Fire hazards: dangerous when exposed to heat, flame, or oxidising agents (Aquilonius et al., 1978).
Spontaneous heating: no (Sax et al., 1989).
Explosion hazards: modest when exposed to flame (Sax et al., 1989). See also "possible chemical reactions", above.
In some countries, a dye is added to methanol to distinguish it from ethanol (e.g., in the United Kingdom, purple dye).
|
Industrial/Commercial Product Intended For Non-Domestic Use |
||
|
|
Remover; Industrial - Other product remover; industrial |
|
|
|
Fuel/Source Of Ignition; Industrial - Fuel; liquid; industrial |
|
|
|
Solvent; Industrial |
|
|
|
Vehicle Maintenance; Industrial |
|
|
|
|
- Antifreeze (vehicle); Industrial |
|
|
|
- Fuel additive; industrial |
|
|
|
- Screenwash; industrial |
|
|
Miscellaneous Industrial Function - Denaturant; industrial |
|
|
|
Chemicals Used In Synthesis; Not Otherwise Specified |
|
|
Household/Leisure Product |
||
|
|
Painting Material; Domestic - Paint stripper; domestic |
|
|
|
Fuel/Source Of Ignition; Domestic - Fuel liquid; domestic |
|
|
|
Vehicle Maintenance; Domestic |
|
|
|
|
- Antifreeze; vehicle; domestic |
|
|
|
- Screenwash; domestic |
|
Food/Beverage |
||
|
|
Food Contaminant - Chemical food contaminant |
|
Industrial solvent.
Raw material for making formaldehyde and methyl esters of organic and inorganic acids.
Antifreeze for radiators and air brakes.
Ingredient of gasoline and diesel oil antifreeze.
Windshield-washing fluids.
Liquid fuel for small engines used in hobbies
Fuel for picnic stoves/soldering torches (Windholz; Merck Index, 1983)
Solvent for inks, dyes, resins, and adhesives
Ingredient of paint and varnish removers (Parmeggiani, l983).
Denaturant for ethanol that is not intended for human consumption, at concentrations up to 5% or more (occasional use).
The majority of poisonings occurs by ingestion. Industrial exposure to methanol vapour is also possible.
Sporadic cases of methanol poisoning are generally due to accidental or suicidal ingestion of the product, which may be available in chemistry laboratories, industries, or sometimes in the home.
Poisoning epidemics pose very severe problems and generally result from the ingestion of adulterated spirits.
Exposure may occur in jobs related to the various uses of the product. Workers should not be exposed to harmful concentrations of vapour. However, there is ample evidence from the photographic film industry that repeated exposure to air levels well in excess of the threshold limit value of 200 ppm does not cause significant discomfort or illness (Finkel et al., 1983). In any case, persons with any disorder of the CNS and/or reduced visual acuity should not be exposed to methanol (Parmeggiani, 1983).
Note: Since methanol poisonings often occur as accidents that involve many people, the clinician is often dealing with "disaster medicine", and must modify the approach to treatment accordingly (Jacobsen & McMartin, 1986).
Poisoning may occur from ingestion, inhalation, or percutaneous absorption (Dutkiewicz et al., l980; Kahn & Blum, l979).
Readily absorbed.
Readily absorbed.
Readily absorbed.
Possible.
Possible.
No data available.
Methanol is readily absorbed from the gastrointestinal and respiratory tracts, and also by the percutaneous route (Dutkiewicz et al., 1980; Kahn & Blum, 1979).
After absorption, methanol is widely distributed in total body water with a volume of distribution of 0.6 to 0.7 L/kg (Jacobsen & McMartin, 1997). There is no protein binding (Jacobsen & McMartin, 1986). Undissociated formic acid readily crosses the blood-brain barrier. Aggressive alkali treatment is therefore important to keep most formic acid dissociated (Jacobsen & McMartin, 1997). It is distributed poorly in fatty tissues.
The elimination of methanol is of zero order with a rate of 8.5 g/L/hour, i.e. about half of that of ethanol (Jacobsen et al., 1988). If methanol metabolism is blocked by ethanol or fomepizole, methanol elimination is very slow (about 50 h) and occurs by pulmonary and renal excretion (Brent et al., 2001, Jacobsen et al., 1983).
The majority of methanol is converted to formaldehyde, principally in the liver, by alcohol dehydrogenase. Formaldehyde is then converted to formate by aldehyde dehydrogenase and other enzymes (Jacobsen & McMartin, 1997). In the monkey and rat, methanol is further metabolised to carbon dioxide by means of a folate-dependent one-carbon pool pathway (Vale & Meredith, 1981; Gosselin et al., 1984; Jacobsen & McMartin, 1986). Studies have shown that the rate of formate oxidation is regulated by the hepatic concentrations of tetrahydrofolate (Eells et al., l982). The tetrahydrofolate concentrations in the livers of monkeys and human beings are much lower than in the liver of rats (Johlin et al., 1986), which probably explains the slower metabolism of formate in primates (Jacobsen & McMartin, 1986).
The metabolism and toxicity of methanol are characterized by important differences among species; in non-primate laboratory animals, methanol itself, not its metabolites, is the major toxic agent (Roe, 1982, Gosselin et al., 1984). (See also Sections 6.4 and 7.2.2.)
Methanol oxidation also occurs in the kidneys (Winchester, l983). The rate of metabolism is independent of the plasma concentration, is slow, and is approximately one-seventh that of ethanol. Complete oxidation and excretion of methanol can require several days. Since ethanol has an affinity for alcohol dehydrogenase that is at least 20 times greater than that of methanol, it preferentially serves as the substrate for this enzyme (Gossel & Bricker, 1984). Administration of ethanol (or fomepizole) reduces the rate of oxidation of methanol and delays its clinical and biochemical effects.
Thirty per cent of the ingested dose is excreted unchanged by the respiratory tract. The kidney excretes less than 5% of unchanged methanol. Formate is detectable in the urine from 4 to 10 days after a single exposure (Baselt, 1982a). Urine formate excretion >70 mg/24 hours confirms the diagnosis of methanol intoxication (Bozza-Marrubini et al., l987). This compares with an average urinary formate concentration of 2 to 30 mg/L in non-exposed subjects (Baselt, 1982a; Shaller & Triebig, 1985).
The average urine-blood concentration ratio for methanol in man is 1.30 (Baselt, 1982a).
Methanol poisoning is characterized by metabolic acidosis and ocular damage. The toxicity of methanol is due to its metabolites rather than to methanol itself. The severity of toxicity correlates with the degree of metabolic acidosis rather than to the concentration of methanol (Jacobsen & McMartin, 1986; 1997; Schwartz et al., 1981). The mechanism of ocular damage is not completely understood (Jacobsen & McMartin, 1997).
Experimental findings suggest that ocular damage is due to formate toxicity, independent of the acidosis. However, acidosis probably causes a more rapid development of ocular damage (Jacobsen & McMartin, 1986; Martin-Amat et al., 1977; Martin-Amat et al., 1978).
Severe metabolic acidosis is characteristic of methanol poisoning. Initially, acidosis is caused by formic acid itself. In a more advanced stage, it is sustained also by lactic acidosis, which is possibly induced by the cytochrome-oxidase inhibition (see Section 7.2.3.3) due to formic acid and tissue hypoxia (Jacobsen & McMartin, 1986). Blood formic acid concentrations are proportional to the increase in the anion gap (Sejersted et al., 1983). For this reason, serum bicarbonate levels and base excess values are reliable early indices of the severity of methanol poisoning (Bozza-Marrubini et al., 1987).
Pathological findings
At autopsy, the liver, kidneys, and heart show parenchymatous degeneration. The lungs show desquamation of epithelium, emphysema, oedema, congestion, and pneumonia. The brain may show oedema, hyperaemia, petechiae, and infarction of the putamen which, in severe cases, may be haemorrhagic (McLean et al., 1980; Aquilonius et al., 1980). The eye shows degenerative changes in the retina and oedema of the optic disc, and there may be atrophy of the optic nerves. The corneal epithelium may show degenerative changes. Pancreatic necrosis has also been observed (Bennet et al., 1952).
Volunteers
After ingestion of 4 mL methanol, urinary formic acid levels reach a maximum of about 56 mg/L within 2 hours and then declined rapidly (Kendal and Ramanathan, 1953).
A peak blood methanol concentration of 117 mg/L (3.6 mmol/L) was achieved 1 hour after an adult male volunteer weighing 78.5 kg ingested 7 mL methanol (Leaf & Zathan, 1952).
Occupational exposure data
In workers exposed to methanol at vapour concentrations of 85 to 134 ppm, urine formic acid concentrations increased from an average of 13 mg/L in the morning to 20 mg/L by late afternoon (Baselt, 1982a).
Clinical case data
Acute ingestion of as little as 4 to 10 mL of methanol may cause permanent blindness (Vale & Meredith, 1981; Bozza-Marrubini et al., 1987; Gossel & Bricker, 1984; Litovitz, 1986). However, individual susceptibility varies widely, possibly because of the frequent concurrent ingestion of ethanol and recovery after the ingestion of 500 to 600 mL has been recorded (Gossel & Bricker, 1984; Litovitz, 1986). The retrospective analysis of data from large scale poisoning by methanol-adulterated wine in Italy, l986, showed that no one with urine formate less than 200 mg/L developed any true symptoms or objective clinical signs.
Other data in human beings are (Sax et al., 1989):
Inhalation human TCLo: 86000 mg/m3:IRR
Eye human: 5 ppm.
Very limited experience with infants suggests that the clinical picture is similar to that in adults in all important respects (Gosselin et al., 1984), but that children could be more sensitive to the toxic effects (Bozza-Marrubini et al., 1987).
Poisoning occurred in a child as young as 10 weeks old when methanol was mistaken for distilled water and mixed in the child's food (Wenzl et al., 1968).
Pediatric poisoning may occur through skin absorption or inhalation (Litovitz, l986). An 8-month-old child died from methanol toxicity after a common cold was treated by rubbing the chest with olive oil, and then applying warm methanol compresses (Kahn & Blum, 1979).
|
oral rat: |
LD50: 5628-l3000 mg/kg |
|
oral dog: |
LDLo: 7500 mg/kg |
|
oral monkey: |
LDLo: 7000 mg/kg |
|
skin rabbit: |
LD50: 20 g/kg |
|
inhalation monkey: |
LCLo: l000 ppm |
|
inhalation rat: |
LC50: 64000 ppm/4H |
(Sax et al. l989)
The metabolism and toxicity of methanol differs markedly between species (Jacobsen & Martin, 1986). In primates and human beings, methanol causes metabolic acidosis and ocular damage. In non-primate laboratory animals, however, it acts as a CNS-depressant (Gosselin et al., 1984; Roe, 1982).
Several enzyme systems involved in methanol metabolism have been reported, including the specific formaldehyde dehydrogenase found in numerous species and tissues (Tephly & McMartin, l984).
The rate of formate oxidation is regulated by the hepatic concentration of tetrahydrofolate (Eells et al., 1982) (see also Section 6.4). This observation could explain some differences in methanol toxicity between species and justifies therapeutic trials with folic acid (Jacobsen & McMartin, 1986).
Formate can inhibit cytochrome C oxidase activity in intact mitochondria, in submitochondrial particles, and in the isolated cytochrome aa3. The inhibition increases with decreasing pH (Nicholls, 1976). This finding could explain the lactate formation that occurs at a late stage in severe methanol poisoning (Jacobsen & McMartin, 1986).
TLV (threshold limit value): 200 ppm (Sax et al., 1989)
TWA (time-weighted average) OSHA:200 ppm 260 mg/m3
NIOSH: 800 ppm/15 minutes short-term exposure limit
The Food Additives and Contaminants Committee recommended that methanol should be used as a solvent in food for extraction purposes only, and recommended a maximum concentration of 8 ppm in food.
No data available.
Methanol is associated with birth defects in rats following both oral (Infurna et al., 1986) and inhalational exposure (Nelson, et al., 1985; Rogers et al., 1993; Bolon et al., 1994).
No data available.
The major metabolic interaction occurs with ethanol, and is described in detail in Sections 6.4 and 10.6.
Blood and urine should be collected for methanol and formic acid determination.
Methanol and formate are stable in biological samples. Methanol, however, is a volatile substance and samples should be stored in tightly capped containers.
During all procedures, precautions should be taken to minimize the loss of alcohols by evaporation.
If prolonged storage before assay is necessary, it is advisable to keep the samples frozen at –20 °C.
Rapid procedures to detect methanol in blood (spot test):
Protein-free filtrate of serum (1 mL) is added to 0.1 mL KMnO4 solution (5 g in 100 mL H2O). The test tube should be gently swirled and, after 5 minutes, sufficient powdered sodium bisulfite should be added to decolourise the permanganate. Freshly prepared chromotropic acid solution (0.2 mL, 0.5 g in 100 mL H2O) and 6 mL concentrated H2SO4 should be added and mixed and heated in a boiling water bath for 5 minutes. A red-violet colour is positive and specific for methanol.
Quantitation can be performed by colorimetry at 570 nm.
This method involves measuring the colour intensity after oxidation of methanol to formaldehyde, and then developing a colour by reacting formaldehyde with chromotropic acid (CTA). The presence of ethanol invalidates a methanol procedure based on oxidation followed by CTA colour development if the calibration curve has been set using pure methanol standards. This method, therefore, may be used for the initial detection of methanol but is unsuitable for monitoring methanol blood levels during antidotal treatment with ethanol (false-positive results) (Tietz, 1986).
To measure methanol in blood, gas-chromatography is the method of choice. Plasma is diluted with:
an equal volume of the internal standard (2 mL of methylethylketone in 1:1 water, procedure A (Baselt, 1982b)
or:
1:10 with internal standard (3 mL of 1-propanol in 1:1 of water), procedure B (Blanke, 1975).
The mixture is injected directly into a gas chromatograph. The use of a pre-column is strongly recommended.
Instruments
Procedure A: Gas chromatograph with replaceable glass injector sleeve and flame-ionization detector: 6' by 1/8" stainless-steel column containing 0.2% Carbowax on 60/80 mesh Carbopack C (Supelco).
Injector 200 °C; column 125 °C; detector 200 °C.
Nitrogen flow-rate, 17 mL/minute.
Procedure: inject 0.5 mL of sample; retention times: methanol 0.6 minutes; (ethanol 0.9 minutes); internal standard 2.3 minutes.
Procedure B: Gas chromatograph with replaceable glass injector, sleeve and flame-ionization detector; 5' x 1/4" glass column containing l0% polyethylene glycol (PEG) 400 on 100 to 120 Anaknom 80.
Injector 110 °C, column 85 °C, detector l25 °C.
Nitrogen flow rate, 70 mL/minute
Procedure: inject 1 mL of sample; retention times: methanol 0.585 minutes, ethanol 0.654 minutes, internal standard 6 minutes.
A quality control specimen containing 1 g/L of each chemical is analyzed daily.
To measure formate in blood and urine several methods are available. The enzymic method of Shaller & Triebig (1985), modified by Zoppi & Motalbetti (1986), is precise (cv at 1 mmol/L 1.5%), simple, economical, and rapid. It should be the method of choice for large-scale screening in the event of an epidemic of methanol poisoning (Zoppi & Montalbetti, 1986), if gas chromatographic determination of methanol cannot be performed. Following poisoning by methanol-adulterated wine in Italy (1986), this method was chosen as a screening test because formate is responsible for the most severe effects of methanol poisoning, and formate concentrations can more accurately predict toxicity than can methanol concentrations (Osterloh, 1986). In addition, the enzymic determination of formate can be performed easily (Zoppi & Montalbetti, 1986).
Other methods for formate determination are high-performance liquid chromatography (HPLC) (Distler, 1978) and gas-chromatography described by Triebig et al. (1974).
Enzymic method
Principle: formic acid is oxidized by NAD+ in the presence of formate dehydrogenase (FDH). The quantity of NADH produced, measured at 340 nm, is equivalent to the quantity of formic acid present in the sample.
HCOOH + NAD+ FDH CO2 + NADH + H+
The optimal pH for FDH is between 7 and 7.5; the reaction proceeds at environmental temperature. Formate can be measured in any biological fluid (urine, plasma, serum, or whole blood) deproteinized with perchloric acid 0.33 mmol/L Apparatus: Hitachi 705 (Boheringer Mannhein; Rotochem CFA 2000 (Kontron s.p.a.). The detailed procedure and its advantages over other enzymic methods are discussed by Zoppi and Montalbetti (1986).
HPLC
Principle: formic acid is separated on a column (250 mm x 4 mm) of nucleosil - 10 C18 having as a mobile phase double-distilled H2O with the pH set at 3.53 by sulfuric acid (flow 1 mL/minute pressure on column 75 bar).
Eluate is monitored at 210 nm. Retention time at pH 3.53 for formic acid is 3.03 min.
Sensitivity threshold is 250 mg (formic acid has a low UV absorbance).
Gas chromatography
Gas chromatography of formic acid can be done by two procedures:
degradation of formic acid with benzoic acid
or:
oxidation of formic acid to CO and catalytic reduction on column to methane; methane is measured by flame-ionization.
In late stages (severe metabolic acidosis), all the ingested methanol may be metabolized and it cannot therefore be detected in serum (Jacobsen, 1984).
Methanol:
To convert conventional units (mg/L) to mmol/L, divide concentrations of methanol by 32.04 (Osterloh et al., 1986).
Normal blood concentrations:
Normal blood methanol concentrations derived from endogenous production and dietary sources are of the order of 15 mg/L (0.05 mmol/L) (range 2 to 30 mg/L) (Baselt, 1982a).
Toxic blood concentrations:
Plasma methanol concentrations >5000 mg/L (157 mmol/L) have been reported (Gosselin et al., 1984; Proudfoot, 1982).
Lethal concentrations:
Blood methanol concentrations are not necessarily a reliable prognostic index (Baselt, 1982a; Jacobsen & McMartin, 1986). The toxicity is clearly correlated to the degree of metabolic acidosis, i.e. formic acid accumulation.
Bennet et al. (1953) reported blood concentrations ranging from 0 to 3900 mg/L (0 to 122 mmol/L) (average 1300 mg/L or 40 mmol/L) in 11 patients who survived, and concentrations of 0 to 4000 mg/L (0 to 125 mmol/L) (average 1600 mg/L or 50 mmol/L) in 7 who died during treatment (Bennet et al., 1953).
Formic acid:
To convert conventional units (mg/L) to milliequivalents per L divide concentrations of formate by 45.02 (Osterloh et al., 1986).
Normal blood concentrations:
From the literature and the experience of the collective poisoning by methanol-adulterated wine in Italy in 1986, blood formic acid concentration in normal subjects can range from 0 up to 18 mg/L.
According to Baselt (1982a), blood formic acid concentrations in normal subjects average about 5 mg/L, and Osterich et al. (1986) report that normal formate concentrations are considered to be less than 12 mg/L.
In occupationally exposed workers, blood formic acid increased from 3.2 mg/L in pre-exposure specimens to 7.9 mg/L by the late afternoon (Baselt, 1982a).
Toxic blood concentrations:
Recently it has been suggested that serum formate concentrations correlate better with the clinical condition than do methanol concentrations, and that formate concentrations are a more direct indicator of toxicity (Osterloh et al., 1986; Bozza-Marribini et al., 1987; Jacobsen et al., 1983a,b; McMartin et al., 1980).
Concentrations of formate in the blood that exceed 200 mg/L may be expected to produce ocular injury or acidosis (Osterloh et al., 1986).
Lethal concentrations: Not reported.
Urine concentrations:
Urine normal formic acid concentrations average from 2 to 30 mg/L if determined by the enzymic method (Shaller & Triebig, 1985), and from 2.1 to 289.2 mg/L if determined by gas-chromatography (Triebig et al., 1974).
According to Baselt (1982a), urine formate concentrations range from 2 to 30 mg/L in unexposed subjects as a result of normal metabolism.
Arterial blood gases, serum electrolytes, osmolality, BUN, creatinine and blood glucose.
The diagnosis and the severity of poisoning by methanol and by ethylene glycol may also be evaluated with reasonable accuracy by indirect methods, i.e. by measuring the degree of metabolic acidosis together with calculating the anion and osmolal gaps (Bozza-Marrubini et al., 1987; Jacobsen & McMartin, 1986; 1997).
Acute systemic poisoning can occur by ingestion, inhalation, skin exposure and parenteral exposure, but ingestion is the major route of exposure. The summary of clinical effects in acute poisoning is given in Section 2.2.
Ingestion is the major route of exposure. Clinical effects after acute poisoning by ingestion consist initially of transient inebriation and drowsiness (similar to ethanol poisoning). After a latency period of 6 to 30 hours, the effects of the toxic metabolites of methanol can cause vomiting, vertigo, abdominal pain, diarrhoea, dyspnoea, acidosis (Kussmaul's respiration), blurred vision, hyperaemia of the optic disc, blindness, dilated pupils (absent light reflex). In very severe cases, restlessness and delirium may occur (Winchester, 1998). Toxic and lethal doses are reported in Section 7.2.3.1.
Acute intoxication by inhalation is rare (Parmeggiani, 1983; Morton Grant, 1986). Indications of intoxication can include irritation of the mucous membranes, headache, tinnitus, vertigo, insomnia, nystagmus, dilated pupils, clouded vision, nausea, vomiting, colic and constipation (Parmeggiani, 1983). For toxic exposure, see Sections 7.2.1 and 7.2.4. However, the described harmful effects were caused by prolonged exposure to concentrations very much in excess of officially recommended limits (Parmeggiani, 1983).
Poisoning by this route has been demonstrated in animals (see Section 7.2.2), and has occurred in humans beings (Kahne & Blum, 1979). However, in one case allegedly involving percutaneous absorption, it is also likely that vapour inhalation occurred (Kahn & Blum, 1979).
External contact of methanol with the eye causes slight local effects (Morton Grant, 1986) (see Section 9.4.9).
No data available.
Whether there is a chronic form of methanol poisoning is open to question; if it exists it is very rare (Morton Grant, 1986). Nevertheless, methanol should be regarded as a cumulative poison (Lewis, 1996; Bozza-Marrubini et al., 1987) because of the slowness with which it is eliminated.
There is ample evidence that repeated exposure to air concentrations well in excess of the threshold limit value of 200 ppm does not cause significant discomfort or illness (Finkel et al., 1983). Nevertheless, people with any disorders of the central nervous system and/or reduced visual acuity should not be exposed to methanol (Parmeggiani, 1983).
Cumulative effects of small but repeated ingested doses can be very severe (see Section 11.2: Milan Poison Centre observations from poisoning by wine adulterated with methanol in Italy, l986).
Daily exposure to methanol vapour may result in the accumulation of toxic concentrations (Lewis, 1996). Chronic poisoning can occur from inhalation during occupational exposure (Parmeggiani, 1983) to air concentrations of 800 - 3000 ppm (Leaf and Zatman, 1952; Baselt, 1982a).
In chronic poisoning due to inhalation, visual impairment may be the first sign of poisoning. It can begin with mild blurring of vision and progresses to contraction of visual fields and, theoretically, to complete blindness.
No data available.
No data available.
No data available.
No data available.
Course
Methanol, when ingested alone, initially causes only a mild and transient inebriation and drowsiness. Characteristically, this early stage is followed by a latent period of 8 - 36 h (Vale and Meredith, 1981), with an average of 12 h (Proudfoot, 1981). In 2 patients who drank ethanol for about 2 days, the latent period lasted 90 h (Jacobsen & McMartin, 1986). This latent period is explained by the slow rate of production of formaldehyde and formic acid. Nausea, vomiting, headache, dizziness, or abdominal pain then supervene.
Visual disturbances, such as blurred vision and diminished visual acuity, occur frequently at this stage. Metabolic acidosis with an increased anion gap and osmolal gap is characteristic, even in the early stages.
Coma, convulsions, and death can occur in the advanced stages of the poisoning.
Prognosis
The prognosis is theoretically good if treatment is started early. Most patients, however, are admitted to hospital at a late stage and often without appropriate facilities for analysing methanol, when delays in diagnosis can lead to death (Jacobsen & McMartin, 1986).
Severe methanol poisoning is fatal in about 25% of cases (Bozza-Marrubini, 1987). During an epidemic in Atlanta, Georgia, USA, 323 people were poisoned and there was an overall mortality rate of 6.2% (Bennet et al., l953).
After poisoning by methanol-adulterated wine in Italy in l986, the Milan Poison Centre reported 34 patients who were admitted to several hospitals. Two patients recovered but had permanent visual damage, and nine patients died.
Bradycardia, hypotension, and severe acidosis indicate a poor prognosis (Proudfoot, 1981).
In 115 patients with serum bicarbonate levels of less than 20 mEq/L, the mortality rate was 19%. An increase in the mortality rate to 50% was noted in 30 patients with serum bicarbonate levels below 10 mEq/L (Bennet et al., 1953).
The most common long-term outcome is visual impairment, usually central scotomata or complete blindness secondary to optic atrophy. Patients who have gross visual symptoms or signs on admission are unlikely to improve (Proudfoot, 1981). Others report that vision is not likely to show much improvement after 6 days (Morton Grant, 1986).
Prevention of ocular damage depends on prompt treatment to eliminate methanol, and particularly its toxic metabolites, from the body.
With haemodialysis and cardiopulmonary support, severely poisoned patients who would other have died are will survive (McLean et al., 1980). Unfortunately, they may be permanently blind with serious damage to the CNS ranging from "moderate polyneuropathy" (Aquilonius et al., 1978), to rigidity, spasticity, and hypokinesis (Guggenheim et al., 1971) as well as a Parkinsonian-like extrapyramidal syndrome with mild dementia (McLean et al., l980).
Causes of death
Death results from complications of metabolic acidosis including coma, convulsions, cardiovascular collapse, cerebral oedema and pulmonary oedema.
Death may be rapid or occur several hours after the onset of coma (Winchester, 1996).
The patient often looks pale and has cold and clammy extremities (Proudfoot, 1981; Winchester, 1983).
Pulse rate and blood pressure are usually well maintained until late in the course of poisoning (Proudfoot, 1981) when cardiovascular failure, bradycardia, hypotension, and myocardial infarction can supervene (observations from the Milan Poison Centre).
Hyperpnoea develops as the metabolic acidosis becomes severe and the patient attempts to compensate.
Respiratory failure can complicate the coma. Pulmonary oedema can supervene (Bozza-Marrubini et al., 1987).
Severe metabolic acidosis is common, but Kussmaul's respiration (slow, deep respirations) is found in only 25% of cases (Proudfoot, 1981).
Early: Inebriation and drowsiness. These signs are much milder than those caused by ethanol (Baselt, 1982a; Bozza-Marrubini et al., 1987; Jacobsen & McMartin, 1986).
Delayed: Headache, vertigo, weakness, motor restlessness. Some patients exhibit a phase of excitement (delirium is possible) before drowsiness and coma supervene.
Convulsions occur occasionally (Proudfoot, 1981; Bozza Marrubini et al., 1987). Dilated pupils with sluggish or absent light reflex are characteristic, and impaired pupillary response to light is accompanied by correspondingly impaired visual acuity (Morton Grant, 1986).
Cerebral oedema and necrosis in the putamen have been documented (McLean et al., 1980). Nuchal rigidity and other meningeal signs have been reported (Litovitz, 1986).
The development of visual symptoms parallels or precedes the development of other symptoms (Jacobsen & McMartin, 1986).
The visual symptoms described range from spots before the eyes and flashing lights, or mistiness of vision, to markedly reduced visual acuity and complete blindness (Morton Grant, 1986). In the acute phase, visual acuity is typically impaired by central or cecocentral scotomata. In the early phase, these are rarely accompanied by constriction of peripheral fields.
Impaired visual acuity is accompanied by correspondingly impaired pupillary response to light; pupils continue to react normally to accommodation (Morton Grant, 1986).
Hyperaemia of the optic disc is the most common abnormality on retinoscopy in the acute stage, and subsides over 2 to 7 days. Peripapillary oedema is also frequent but develops more slowly and persists for longer (up to 8 weeks) than optic disc hyperemia (Proudfoot, 1981). Venous engorgement and retinal haemorrhages are not common (Proudfoot, 1981).
The subsequent course can range from complete recovery, with disappearance of retinal oedema and return of normal vision, to persistent oedema of the retina for as long as 2 months. Optic atrophy can develop in 1 to 2 months and vision may be lost (Morton Grant, 1986).
Between these extremes are many cases of partial but permanent visual impairment.
In patients whose vision does not recover, the scotomata in the central field remain and, as optic atrophy develops, the peripheral fields may constrict (Morton Grant, 1986).
Electroretinography in methanol poisoning has shown minor alterations (reduction of the b-wave), and in many cases was essentially normal although the patients remained blind. This is consistent with the methanol's principal injurious action on retinal ganglion cells and optic nerve fibres (Morton Grant, 1986).
No data available.
No data available.
Impaired pupillary response to light is linked with impaired visual acuity; i.e. it is due to damage of the afferent branch of the light reflex.
Abdominal discomfort and pain are very frequent. Violent attacks of abdominal colic are common and nausea and vomiting occur in about 50% of cases; only a minority develop diarrhoea. Abdominal pain may precede acute
pancreatitis associated with hyperamylasaemia and hyperamylasuria (Gosselin et al., 1984; Winchester, 1983). Back pain can be present (Jacobsen & McMartin, 1986). However, in the only patients in whom serum isoamylase or lipase analysis were studied, the striking rise in the serum total amylase activity was entirely due to salivary-type isoamylase, and the serum lipase activity remained entirely normal. Thus in methanol intoxications, hyperamylasaemia should not be interpreted as firm evidence of pancreatitis (Eckfeldt et al., 1986).
Hepatic effects have not been reported. Liver parenchymatous degeneration has been described at autopsy.
Oliguria and renal failure can sometimes occur in severe methanol poisoning (Closs et al., 1970).
No data available.
Not reported (both in acute and in chronic poisoning) (Advisory Panel, 1985).
Skin injuries can arise from the local irritant and solvent action of methanol (Parmeggiani, 1983).
Visual disturbances occurring in systemic methanol poisoning have been described in detail in Section 9.4.3.2.
External contact of methanol with the eye has been alleged to cause corneal opacities, but this seems to be the exception rather than the rule. Tests on rabbit eyes indicate that the danger is slight (Morton Grant, 1986).
Corneal staining with fluorescein was observed for 1 month in a patient who had had one eye splashed with a mixture of chloroform and methanol (Morton Grant, 1986).
In mucous membranes, the irritant effect of methanol can cause local injury.
In one group of patients, mean corpuscular red cell volumes (MCV) were significantly increased in moderately to severely poisoned patients (Schwarz et al., 1981).
Not reported (either in acute or chronic poisoning).
The underlying disorder in methanol poisoning is a metabolic acidosis with increased anion gap.
Initially acidosis is due to formic acid itself. In a more advanced stage it is also sustained by lactic acidosis (see Section 7.1).
The serum bicarbonate levels and the base excess value are early and reliable indices of the severity of poisoning (Bozza-Marrubini et al., 1987; Jacobsen & McMartin, 1986). In fact, serum bicarbonate levels are inversely correlated with the accumulation of formate (McMartin et al., 1980).
The metabolic acidosis can be severe and recurrent, in spite of appropriate and repeated infusions of alkaline solutions (Bozza-Marrubini et al., 1987).
The increased anion gap is a consequence of formic acid accumulation (in later stages, lactic acid can also accumulate). Methanol-poisoned patients present with an early increased osmolal gap. The osmolal gap is determined by subtracting the calculated from the measured osmolality (Jacobsen & McMartin, 1986; Saxena et al., 1987).
Normal osmolality is 286 mOsm/kg H2O, and the normal osmolal gap is 10 mOsm/kg H2O (Litovitz, 1986; Jacobsen & McMartin, 1986; Saxena et al., 1987).
The calculated osmolality can be derived using the following equation (Saxena et al., 1987):
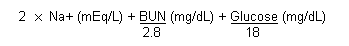
or by the following formula, using SI units (Jacobsen & McMartin, 1986):
(1.86 x Na+ + urea + glucose) / 0.93 (SI units).
Elevated osmolal gap is due to the presence of osmotically active agents, such as methanol, ethanol, mannitol, etc.
The osmolal contribution of a 100 mg/dL concentration of methanol is 34 mOsm/kg H2O (Jacobsen & McMartin, 1986).
Hyperkalaemia may complicate severe metabolic acidosis or rhabdomyolysis.
No data available.
No data available.
No data available.
Pregnant women are not at any specific increased risk from acute methanol toxicity.
Breast feeding: No data available.
Enzyme deficiencies do not confer greater susceptibility to the toxicity of either acute or chronic methanol exposure.
There are no known reports of toxic effects of methanol on any phase of reproduction in avian and mammalian species or on the reproductive cycle of human beings (Advisory Panel, 1985).
Based on the available data, it is not likely that adverse reproductive effects will result if exposure does not exceed the current TLV of 200 ppm (Advisory Panel, 1985).
No data available.
Treatment consists of:
|
· |
emptying the stomach (if indicated); |
|
· |
correction of acidosis; |
|
· |
ethanol or fomepizole administration to inhibit the formation of toxic metabolites; |
|
· |
rapid reduction of the body burden of methanol and formate by haemodialysis; |
|
· |
intensive supportive care for multiple organ/system failures. |
Intensive supportive care for multiple organ/system failure is frequently necessary.
Since severe, recurrent metabolic acidosis is the underlying feature of methanol poisoning, the correction of acidosis is imperative, possibly life-saving, and may prevent ocular damage (Proudfoot, 1981; Bozza-Marrubini et al., 1987). The degree of acidosis has been found to correspond closely to the severity of poisoning (Bozza-Marrubini et al., 1987; Jacobsen et al., 1983; Morton Grant, 1986). Repeated and frequent assessment of the acid/base status is necessary.
Sodium bicarbonate must be used. Avoid sodium lactate, since lactate metabolism may be impaired (Bozza-Marrubini et al., 1987; Gosselin et al., 1984).
Correction of acidosis may require as much as 400 to 600 mmol of bicarbonate during the first few hours (Jacobsen & McMartin, 1986).
Fluids must be given orally or intravenously to maintain adequate urine output (Bozza-Marrubini et al., 1987).
Hyperkalemia is usually corrected by bicarbonate administration (see also treatment guide: hyperkalaemia).
Special treatment for cerebral oedema may be considered (Bozza-Marrubini et al., 1987; Gosselin et al., 1984).
Convulsions should be controlled (see treatment guide: convulsions).
The usual decontamination procedures are required in cases of percutaneous exposure, or exposure to vapours: removal from the exposure, removal of clothes, adequate prolonged washing of skin and eyes.
Consider emptying the stomach by gastric lavage only following recent ingestion (< 1 hour) of a large amount.
Haemodialysis
Extracorporeal haemodialysis removes methanol and formate, and, if started early enough, may prevent permanent visual damage and death (Proudfoot, 1981; Jacobsen & McMartin, 1986; 1997). Methanol is readily dialysable due to its low molecular weight, lack of protein binding, and a low volume of distribution of 0.7 L/kg (Jacobsen & McMartin, 1986; Gonda et al., 1978). The clearance values lie between 150 and 200 mL/minute (Jacobsen & McMartin, 1986), depending on the blood flow and the surface area in the dialyser (Jacobsen & McMartin, 1986).
Formate also has a low molecular weight and a low volume of distribution (0.5 L/kg) (Jacobsen, 1983b) and formate clearance values range from 140 to 150 mL/minute (Jacobsen, 1983b; Jacobsen & McMartin, 1986).
It is difficult to give precise indications for haemodialysis in methanol poisoning. The only absolute indications are pronounced metabolic acidosis and acidosis combined with any degree of visual disturbances. If fomepizole is given in the early stage of poisoning, before significant acidosis develops, the patient may not need haemodialysis regardless of the blood methanol concentration as long as acidosis does not develop. The following may serve as relative indications for haemodialysis:
|
a) |
Blood methanol concentrations greater than 500 to 600 mg/L (16-20 mmol/L) (Gonda et al., 1978; Jacobsen & McMartin, 1986; Gosselin et al., 1984; Proudfoot, 1981; Vale & Meredith, 1981; Winchester, 1983; Gossel & Bricker, 1984). |
|
|
Haemodialysis should be continued until the methanol concentration drops below 200 to 300 mg/L (6 to 10 mmol/L) (Jacobsen & McMartin, 1986; Proudfoot, 1981; Gossel & Bricker, 1984; Winchester, 1983; Gosselin et al., 1984). |
|
b) |
When 20 to 40 mL or more of methanol have been ingested by adults (Bozza-Marrubini et al., 1987; Jacobsen & McMartin, 1986; Vale and Meredith, 1981, Gosselin et al., 1984). |
|
c) |
Severe acidosis; base deficit >15 mmol/L or anion gap >30 mmol/L (Gosselin et al., 1984; Vale & Meredith, 1981; Jacobsen & McMartin, 1986). |
|
|
If blood methanol concentrations are not available, haemodialysis should be continued for at least 8 h and rapidly repeated if acidosis recurs (Jacobsen & McMartin, 1986). |
|
d) |
When symptoms progress rapidly and do not respond to ethanol and sodium bicarbonate. |
|
e) |
Oliguria (Closs et al., 1970). |
|
f) |
Serum formate concentrations may be a better criterion for haemodialysis than methanol concentrations themselves (Osterloh et al., 1986). Concentrations of formate that exceed 200 mg/L may be expected to cause ocular injury or acidosis (Osterloh et al., 1986). |
|
g) |
Some authors have suggested that any degree of visual impairment or fundoscopic abnormalities should be considered as indications for haemodialysis (Gonda et al., 1978; Jacobsen & McMartin, 1986). |
Even during haemodialysis, it is crucial to administer bicarbonate and especially ethanol. Since a profound acidosis is common, it is prudent to use a bicarbonate-based dialysate (Winchester, 1983). In two patients, the mean dialysis ratio of ethanol/methanol was reported as 149/157 and 164/176 mL/minute, respectively (Jacobsen, 1982).
In this study, the removal of ethanol by haemodialysis was 8.9 and 9.8 g/hour, respectively. This means that in adults a total ethanol dose of 16 to 20 g/hour would be needed to replace ethanol lost due to metabolism and dialysis. Similar values were found in other studies (Gonda et al., 1978; Jacobsen & McMartin, 1986). Ethanol loss by dialysis can be replaced during haemodialysis by increasing the ethanol infusion rate by 7 to 10 g/hour (Jacobsen & McMartin, 1986; Vale & Meredith, 1981; Winchester, 1983; Bayer et al., 1984 (103 mg/kg/hour); McCoy et al., 1979).
Alternatively, the addition of ethanol to the dialysate (22 mmol/L) has been recommended (Peterson et al., 1981) but this approach has not been verified (Jacobsen & McMartin, l986).
Rebound rises in methanol levels of as much as 20 mg/dL after haemodialysis have been reported even up to 36 hour after haemodialysis (Winchester, 1983), reflecting the movement of methanol from tissues to the plasma compartment.
Peritoneal dialysis has also been used on occasion for methanol elimination (Wenzl et al., 1968) but is much less efficient than haemodialysis. Its efficiency can be greatly increased by adding the alkalinizing agent THAM (trometanol or tromethamine) to the dialysis liquid (Bozza-Marrubini et al., 1987).
There are two alternative antidotes, both of which act by blocking the alcohol dehydrogenase-mediated metabolism of methanol: ethanol, fomepizole.
(i) Ethanol
Effective because it has a much greater affinity for alcohol dehydrogenase than methanol. A blood ethanol concentration of 100 mg/dL (22 mmol/L) will almost completely block methanol metabolism (Jacobsen & McMartin, 1986). However, ethanol is sometimes technically difficult to administer because of its rapid and unpredicatable rate of metabolism (Jacobsen & McMartin, 1986). A loading dose followed by titrated maintenance therapy is necessary.
Suggested dosing regime:
|
|
Oral |
Intravenous |
|
Loading dose |
1 mL/kg of 95% ethanol, diluted |
10 mL/kg of 10% ethanol in 5% dextrose over 30 minutes |
|
Maintenance dose |
0.1 – 0.2 mL/kg/hour of 95% ethanol, diluted |
1-2 mL/kg of 10% ethanol in 5% dextrose over 30 minutes |
Notes:
In an emergency, an equivalent amount of any alcoholic drink may be administered orally.
The maintenance dosing needs to be adjusted according blood ethanol concentration, ideally measured hourly, to maintain the concentration >100 mg/dL.
Prolonged ethanol administration may cause hypoglycaemia, especially in children, and frequent blood glucose determinations are mandatory (Goldfrank et al., 1986; Bayer et al., 1984). If haemodialysis is started, the ethanol infusion should be increased as detailed in Section 10.5.
(ii) Fomepizole is easily administrated intravenously as a loading dose of 15 mg/kg, followed by bolus doses of 10 mg/kg every 12 hours. After 48 hours, the bolus doses should be increased to 15 mg/kg every 12 hours because of induced metabolism over time. The same dose may be administered orally. No side effects have been reported with this dosage regimen and effectiveness is clearly demonstrated (Brent et al., 2001). If dialysis is performed, the dose of fomepizole must be increased as fomepizole is eliminated at the same rate as urea.
Although there have been fewer reports of ethanol therapy in children, comparable doses may be used. Ethanol is more likely to cause hypoglycaemia in children (Bayer et al., 1984).
Ethanol (or fomepizole) alone may be the only treatment in minor or suspected cases admitted to hospitals at a very early stage. Ethanol should be administered as soon as possible after exposure, ideally before the onset of clinical and laboratory findings that indicate extensive metabolism of methanol. Continuous and accurate control of ethanol levels is rarely achieved even in intensive care units, especially over long time periods (Litovitz, 1986; Osterloh et al., 1986).
In patients who have a history of severe exposure and ingestion of near-lethal quantities, and in patients who have high methanol blood levels, early haemodialysis is mandatory to reduce the toxic burden of the body rapidly.
In advanced stages, or in cases where there have been repeated exposures to low doses or concentrations of methanol, ethanol is of doubtful value. When laboratory findings demonstrate the absence of unchanged methanol in the body and the accumulation of toxic metabolites (very low or absent methanol in the blood, high levels of formate in blood and urine), ethanol is probably useless. However, haemodialysis can still be useful, since both methanol and formate are dialysable substances.
Folic acid or related folates
Ethanol and 4-MP prolong the elimination of methanol, inhibiting its metabolism. A more effective treatment would be to increase the elimination of the toxic metabolites (formic acid) by increasing their metabolism.
Primates are sensitive to methanol poisoning because they do not oxidize formate as readily as do rodents due to a relative deficiency in the function of the folate system (Eells et al., 1982; Jacobsen & McMartin, 1986).
Folic acid treatment in monkeys, even if started as late as 10 h after methanol, prevents formate accumulation (Noker et al., 1980).
In a recent study, four methanol-poisoned patients were treated with intravenous folate and ethanol, combined in two cases with haemodialysis (Osterloh et al., l986). An apparent reduction in formate half-life was obtained. Folate treatment is safe and seems advisable since many alcoholics are folate deficient (Osterloh et al., 1986; Saxena et al., 1987).
Intravenous folate 1 mg/kg (50 to 70 mg) should be given intravenously every 4 hours for the first 24 hours, for a total of 6 doses (Osterloh et al., 1986; Jacobsen & McMartin, 1986).
The two pediatric cases reported by Wenzl (1968) and Kahn and Blum (1979) described in Section 7.2.1.2.
Widespread poisoning by methanol-adulterated wine occurred in Italy in l986 (Bozza-Marrubini, 1987), when a law was passed that abolished the heavy government taxes and fiscal controls on methanol. Methanol then became cheaper than ethanol resulting in its use for the adulteration of wine. In 2 days (12 to 13 March 1986) three cases with striking clinical features in common were referred to the Milan Poison Unit: visual blurring, vomiting, headache, metabolic acidosis, followed in two cases by deep coma with dilated unreactive pupils.
Summary of 415 cases observed (176) or followed through the Poison Unit Information Service (239), 13 March-30 April 1986 (Bozza-Marrubini, 1987)
|
381 cases |
Formate in urine <100 mg/L. Symptoms: none, or very mild. Discharged, no treatment |
|
34 cases |
Formate in urine >100 mg/L (up to 12000 mg/L). Admitted to hospital (11 to the Milano P.U., 23 to other hospitals). |
|
5 cases |
Clinical signs: none, or very mild. Good recovery, no sequelae. |
|
16 cases |
Visual impairment, from single scotoma to total blindness. 14, good recovery; 2, permanent visual damage. |
|
13 cases |
Comatose. 4, good recovery; 9 deaths. |
It was imperative quickly to set up reliable and rapid methods of triage to determine the amount of formate in the urine. Patients who had <100 mg/L of formate in their urine, even if mild symptoms were present, received no treatment and were discharged. Patients who had urine formate levels >100 mg/L even if they were asymptomatic, were admitted to hospital and treated with forced alkaline diuresis, which was very effective in speeding the elimination of formic acid.
Patients with severe clinical signs were admitted to the intensive care units of various hospitals and treated with forced alkaline diuresis alone or with ethanol and/or haemodialysis according to the levels of methanol and formate found in their blood and urine.
Some features of the methanol wine syndrome differed from those of acute pure methanol poisoning and indicated cumulative effects of relatively small but repeated doses:
The latent period was extended over at least 2 to 3 days.
On admission, the methanol concentration in blood was, as a rule, absent or low; in patients with severe symptoms, blood methanol never exceeded 200 mg/L while formate in plasma and in urine was consistently high.
Some patients had a benign and even asymptomatic course in spite of high urine formate levels. They were probably "protected" by the fact that the adulterated wine contained both methanol and ethanol. Therefore the toxic metabolite increased very slowly.
After a prolonged period, when the illness proceeded to the full toxic syndrome (blindness, coma, metabolic acidosis), a very high mortality (70%) was observed in spite of intensive treatment.
The determination of formate in urine was a quick and reliable diagnostic and screening tool. The value adopted a priori as a toxic threshold (100 mg/L) was considerably higher than the accepted "normal" levels (17-30 mg/L) (Baselt, 1982; Schaller and Triebig, 1985).
The retrospective comparison of clinical and laboratory data showed that the actual toxic threshold was even higher, since no patient with urine formate below 200 mg/L had any true symptoms or objective clinical signs.
Store product in appropriately labelled containers, out of reach of children.
Add dyes to methanol to distinguish it from ethanol.
Provide appropriate information on methanol toxicity in the occupational setting and for potential users.
No data available.
Advisory Panel (1985) Effects of toxic chemicals on the reproductive system. Prepared by the Council on Scientific Affairs' Advisory Panel on Reproductive Hazards in the Workplace. American Medical Association, Chicago, Illinois.
Aquilonius SM, Asmark H et al (1978). Computerized tomography in severe methanol intoxication. Br Med J 2: 929-930
Baselt RC (1982a). Disposition of toxic drugs and chemicals in man. 2nd ed. Biomedical Publ. Davis, California: 492.
Baselt RC (1982b). Analytical procedures for therapeutic drugs monitoring and emergency toxicologic. Biomedical Publication: 298-299.
Bayer MJ, Rumack BH, Wanke LA (1984). Toxicologic emergencies. Robert J. Brady, Co., Bowie.
Bennet Jr, IL, Cary FH, Mitchell GL Jr, Cooper MN (1953). Acute methyl alcohol poisoning: a review based on experiences in an outbreak of 323 cases. Medicine 32: 431-463.
Bennet Jr, IL, Nation TC, Olley JF (1952). Pancreatitis in methyl alcohol poisoning. J Lab Clin Med 40: 405-409.
Blanke RV (1975). Methanol. Type C procedure in: Sunshine 1 ed., Methodology for Analytical Toxicology. CRC Press, Cleveland: 239-240.
Bolon B, Welsch F, Mortan K (1994). Methanol-induced neural tube defects in mice: pathogenesis during neurulation. Teratology 49:497-517.
Bozza-Marrubini M (1987). Collective poisoning by methanol-adulterated wine in Italy, 1986. Newsletter of the European Association of Poison Control Centres, April.
Bozza-Marrubini M, Ghezzi Laurenzi R, Ucelli P (1987). Intossicazioni acute: meccanismi, diagnosi e terapia. II ed. OEMF, Milano.
Brent J, McMartin KE, Phillips S, Aaron C, Kulig K (2001). Fomepizole for the treatment of methanol poisoning. N Engl J Med 344: 424-429.
Closs K, Solberg CO (1970). Methanol poisoning. J Am Med Assoc 211: 497-499.
Distler W, Trenung von Milch (1978). Glyoxyl-Ameisen-Essig-und Propionsaeure mit Hilfe der Umkehrphasen Hochdruck-fluessigkeits-Chromatographie. J Chromatogr 152: 250-252.
Dutkiewicz B, Konczakik J, Karwacki W (1980). Skin absorption of administration of methanol in men. Int Arch Occup Environ Health 48: 81-88.
Eckfeldt JH, Kershaw MJ (1986). Hyperamylasemia following methyl alcohol intoxication. Source and significance. Arch Int Med 146: 193-194.
Eells JT, Black KA, Makar AB et al (1982). The regulation of one-carbon oxidation in the rat by nitrous oxide and methionine. Arch Biochem Biophysics 219: 316-326.
Eells JT, McMartin KE, Blanck K et al (1981). Formaldehyde poisoning, rapid metabolism to formic acid. J Am Med Assoc 246: 1237-1239.
Finkel AJ, Hamilton A, Hardy HL (1983). Hamilton and Hardy's Industrial Toxicology. Fourth ed. John Wright PSG Inc. London.
Goldfrank LR, Flomenbaum NE, Lewis NA et al (1986). Goldfrank's Toxicologic Emergencies Third ed. Appleton Century Crafts, Norwalk.
Gonda A, Gault H, Churchill D et al (1978). Hemodialysis for methanol intoxication. Am J Med 64: 749-758.
Gossel TA, Bricker JD (1984). Principles of clinical toxicology. Raven Press, New York.
Gosselin, Smith Hodge (1984). Clinical Toxicology of Commercial Products. Fifth ed.
Guggenheim MA, Couch JR, Weinberg W (1971). Motor dysfunction as a permanent complication of methanol ingestion. Arch Neurol 24: 5540-5554.
Infurna R et al (1986). Neonatal behavioural toxicity in rats following prenatal exposure to methanol. Teratology 33: 259-65.
Jacobsen D, Jansen H, Wilk-Larsen E, Halvorsen S (1983). Toxicokinetics studies during haemodialysis for methanol poisoning. Hum Toxicol 2: 408.
Jacobsen D, Jansen H, Wilk-Larsen E et al (1982). Studies on methanol poisoning. Acta Med Scand 212: 5-10.
Jacobsen D, McMartin KE (1986). Methanol and ethylene glycol poisonings. Mechanism of toxicity, Clinical Course, Diagnosis and treatment. Medical Toxicology 1: 309-334.
Jacobsen D, Ovrebo S, Sejersted OM (1983b). Toxicokinetics of formate during hemodialysis. Acta Med Scand 214: 409-412.
Jacobsen D, Øvrebø S, Arnesen E, Paus PN (1983) Pulmonary clearance of methanol in man. Scand J Clin Lab Invest 43, 377-9.
Jacobsen D, Webb R, Collins TD, McMartin KE (1988) Methanol and formate kinetics in late diagnosed methanol intoxication. Med Toxicol 3, 418-23.
Jacobsen D, McMartin KE. (1997) Antidotes for methanol and ethylene glycol poisoning. J Toxicol Clin Toxicol 35, 127-43.
Johlin Jr, FC, Fortman CS, Nghiem DD, Tephly TR (1986). Studies in human hepatic folate biochemistry. Fed Proceed 45: 277.
Kahn A, Blum D (1979). Methyl alcohol poisoning in an 8 month old boy. An unusual route of intoxication. J Pediatr 94: 841-43.
Kendal LP, Ramanathan AN (1953). Excretion of formate after methanol ingestion in man. Biochem J 54: 424-426.
Leaf G, Zatman LJ (1952). A study of the conditions under which methanol may exert a toxic hazard in industry. Brit J Ind Med 9: 19-31.
Lewis RJ (1996). Sax’s dangerous properties of industrial materials, 9th ed. Van Nostrand Reinhold.
Litovitz T (1986). The alcohols: ethanol, methanol, isopropanol, ethylene glycol. Ped Clin North Am 33: 311-323.
Martin-Amat G, McMartin KE, Hayreh SS et al (1978). Methanol poisoning: ocular toxicity produced by formate. Toxicol App Pharmacol 45: 201-208.
Martin-Amat G, Tephly TR, McMartin KE et al (1977). Methyl alcohol poisoning II. Development of a model for ocular toxicity in methyl alcohol poisoning using the rhesus monkey. Arch Ophthalmol 95: 1847-1850.
McCoy HG, Cipolle RJ, Ehlers SM et al (1979). Severe methanol poisoning. Application of a pharmacokinetic model for ethanol therapy and hemodialysis. Am J Med 67: 804-807.
McLean DR, Jacobs H, Mielke BW (1980). Methanol poisoning: a clinical and pathological study. Ann Neurol 8: 161-167.
McMartin KE, Ambre JJ, Tephly TR (1980). Methanol poisoning in human subjects: role for formic acid accumulation in the metabolic acidosis. Am J Med 63: 414-418.
McMartin KE, Hedstrom KG, Tolf B et al (1980). Studies on the metabolic interactions between 4 MP and ethanol using the monkey as an animal model. Arch Biochem Biophys 199: 616-614.
McMartin KE, Martin-Amat G, Noker PE, Tephly TR (1979). Lack of a role for formaldehyde in methanol poisoning in the monkey. Biochem Pharmacol 28: 645-649.
Morton Grant W (1986). Toxicology of the eye. Third. ed. Charles C. Thomas, Springfield.
Nelson BK et al (1985). Teratological assessment of methanol and ethanol at high inhalation levels in rats. Fundam Appl Toxicol 5: 727-736.
Nicholls P (1976). The effect of formate on cytochrome aa and on electron transport in the intact respiratory chain. Biochemica et Biophysica Acta, 430: 13-29.
Noker PE, Eells JT, Tephly TR (1980). Methanol toxicity: treatment with folic acid and 5-formyl tethrahydrofolic acid. Alcoholism Clin Exp Research 4: 378-383.
Osterloh JD, Pond SM, Grady S, Becker CE (1986). Serum formate concentrations in methanol intoxications as a criterion for hemodialysis. Ann Int Med 104: 200-203.
Parmeggiani L (1983). ILO - Encyclopedia of Occupational Health and Safety. Third revised ed. International Labour Office, Geneva.
Peterson CD, Collins AJ, Keane WF et al (1981). Reply to letter of Freed et al. New Eng J Med 304: 977.
Proudfoot A (1981). Diagnosis and management of acute poisoning. Blackwell Scientific Publications, London.
Roe O (1982). Species differences in methanol poisonings. Critical Reviews in Toxicology 10: 275-286.
Rogers J, Mole M, Chernoff N et al. (1993). The developmental toxicity of inhaled methanol in the CD-1 mouse, with quantitative dose-response modelling for estimation of benchmark doses. Teratology 47:175-188.
Rumack BH et al (1982). Poisindex, Micromedex, Englewood Co.
SAX NI, Lewis RJ Jr (1989) Dangerous Properties of Industrial Materials, 7th ed., 3 volumes, Van Nostrand Reinhold, New York, NY, USA
Saxena K, Lerud K, Cicero JJ (1987). Methanol intoxication in multiple trauma. Am J Emerg Med 5: 60-63.
Shaller KH, Triebig G (1985). Formate: determination with formate dehydrogenase. In: Bergmeyer, H.V. ed. Methods of enzymatic analysis, 3rd ed., vol. VI, Verlag Chemie, Weinheim.
Sejersted OM, Jacobsen D, Øvrebø S, Jansen H. Formate concentrations in plasma from patients poisoned with methanol. Acta Med Scand 1983, 213, 105-10.
Swarz RD, Millman RP, Billi JE et al (1981). Epidemic methanol poisoning: clinical and biochemical analysis of a recent episode. Medicine 60: 373-382.
Tephly TR, McMartin KE (1984). Methanol metabolism and toxicity. In: Stegink and Filer (Eds): Aspartame: physiology and biochemistry, pp. 111-140, Marcel Dekker, New York.
Tietz NW (1986). Textbook of Clinical Chemistry l68l-82. WB Saunders Company, Philadelphia.
Triebig G, Vostrowsky O, Shaller KH (1974). Die quantitative Bestimmung von Ameise sauere in Urin mit hilfe von Zweigaschromatographischen und einem enzymatischen Analyseverfahren. J Clin Chem Clin Biochem 17: 194-195.
Vale and Meredith TJ (1981). Poisoning, Diagnosis and Treatment. Update Books, London.
Wenzl JE, Mills SD, McCall JT (1968). Methanol poisoning in an infant: successful treatment with peritoneal dialysis. Am J Dis Child 116: 445-447.
Winchester J (1998). Methanol, isopropyl alcohol, higher alcohols, ethylene glycol, cellosolves, acetone and oxalate. Chapter 35 in: Haddad LM, Shannon M, Winchester JF. Clinical management of poisoning and drug overdose. WB Saunders Co., Philadelphia.
Windholz M, Budavar S, Blumetti F, Otterbein E (1983). The Merck Index l0th ed. MERCK and Co. Inc. Rahway, NJ, USA.
Zoppi F, Montalbetti N (1986). Large-scale determination of formate as a tool for assessing severity of methanol intoxication. Clin Chem 32: 2002-2003.
|
Authors: |
Bozza-Marrubini M, Brucato A, Locatelli C, Ruggeroni ML |
|
|
Ospedale Niguarda - Cá Granda |
|
|
Centro Antiveleni |
|
|
Piazza Ospedale Maggiore, 3 |
|
|
20162 Milan |
|
|
Italy |
|
|
Tel: 39-2-66101029/64442470 |
|
|
Fax: 39-2-6442768 |
|
Date: |
February l988 |
|
Peer review: |
Adelaide, Australia, April 1991 |
|
Update: |
August 2001 (Professor Dag Jacobsen) |
|
Peer Review: |
L. Lefebvre, M. Mathieu-Nolf, L. Murray, A. Nantel, Edinburgh, Scotland, September 2001. |
|
Format edited: |
May 2002, Joanna Tempowski, IPCS Geneva |
See Also:
Toxicological Abbreviations
Methanol (EHC 196, 1997)
Methanol (HSG 105, 1997)
Methanol (ICSC)
Methanol (FAO Nutrition Meetings Report Series 48a)
METHANOL (JECFA Evaluation)