
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 207
ACETONE
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Mr D J Reisman, US Environmental Protection
Agency, Cincinnati, USA
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1998
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall
objectives of the IPCS are to establish the scientific basis for
assessment of the risk to human health and the environment from
exposure to chemicals, through international peer review processes, as
a prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization, the United Nations
Institute for Training and Research, and the Organisation for Economic
Co-operation and Development (Participating Organizations), following
recommendations made by the 1992 UN Conference on Environment and
Development to strengthen cooperation and increase coordination in the
field of chemical safety. The purpose of the IOMC is to promote
coordination of the policies and activities pursued by the
Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing in Publication Data
Acetone.
(Environmental health criteria; 207)
1. Acetone 2. Environmental exposure
I. International Programme on Chemical Safety II. Series
ISBN 92 4 157207 8 (NLM Classification: QD 305.K2)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1998
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR ACETONE
PREAMBLE
ABBREVIATIONS
1. SUMMARY
1.1. Properties
1.2. Uses and sources of exposure
1.2.1. Production
1.2.2. Uses and emissions into the environment
1.3. Environmental transport, distribution and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism
1.6. Effects on laboratory mammals and in vitro systems
1.7. Effects on humans
1.8. Effects on other organisms in the laboratory and field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Chemical identity
2.2. Physical and chemical properties
2.2.1. Physical properties
2.2.2. Chemical properties
2.3. Conversion factors
2.4. Analytical methods
2.4.1. Biological media
2.4.2. Environmental media
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.2. Uses
3.2.3. Releases
3.2.3.1 Air
3.2.3.2 Water
3.2.3.3 Soil
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution among media
4.1.1. Air
4.1.2. Water
4.1.3. Soil
4.2. Biotransformation
4.2.1. Bioconcentration and biomagnification
4.2.2. Biodegradation
4.2.2.1 Microbial degradation
4.3. Bioavailability from environmental media
4.4. Interaction with other physical, chemical or biological
factors
4.5. Ultimate fate following use
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.1.1 Indoor air
5.1.2. Water
5.1.3. Soil and sediment
5.1.4. Food
5.1.5. Other environmental levels
5.2. General population exposure
5.3. Occupational exposure
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
6.1. Absorption
6.1.1. Inhalation exposure
6.1.1.1 Human studies
6.1.1.2 Experimental animal studies
6.1.2. Oral exposure
6.1.2.1 Human studies
6.1.2.2 Experimental animal studies
6.1.3. Dermal exposure
6.1.3.1 Human studies
6.1.3.2 Experimental animal studies
6.1.4. Absorption summary
6.2. Distribution
6.2.1. Inhalation exposure
6.2.1.1 Human studies
6.2.1.2 Experimental animal studies
6.2.2. Oral exposure
6.2.3. Injection exposure
6.2.4. Distribution summary
6.3. Metabolism
6.3.1. Human studies
6.3.2. Experimental animal studies
6.3.3. Metabolism summary
6.4. Elimination and excretion
6.4.1. Human studies
6.4.1.1 Occupational exposure studies
6.4.2. Experimental animal studies
6.4.3. Elimination/excretion summary
6.4.4. Physiologically based pharmacokinetic model
6.5. Retention and turnover
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Short-term toxicity
7.1.1. Skin and eye irritation
7.2. Longer-term toxicity
7.3. Reproductive toxicity, embryotoxicity and teratogenicity
7.4. Mutagenicity
7.5. Carcinogenicity
7.6. Immunotoxicity
7.7. Special studies
7.8. Factors modifying toxicity; toxicity of metabolites
7.9. Mechanisms of toxicity - mode of action
8. EFFECTS ON HUMANS
8.1. Effects on humans
8.1.1. Non-occupational exposure
8.1.2. Occupational exposure
8.2. Subpopulations at special risk
9. EFFECT ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Aquatic organisms
9.1.1. Acute toxic effects on aquatic fauna
9.1.2. Chronic effects on aquatic fauna
9.1.3. Effects on aquatic plants
9.2. Effects on bacteria and protozoa
9.3. Terrestrial organisms
9.3.1. Effects on fauna
9.3.2. Effects on flora
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health effects
10.2. Evaluation of effects on the environment
11. FURTHER RESEARCH
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
REFERENCES
RÉSUMÉ
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41
22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number
5 U01 ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and in
vitro studies provide support and are used mainly to supply evidence
missing from human studies. It is mandatory that research on human
subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
 All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR ACETONE
Members
Dr D. Anderson, British Industrial Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United Kingdom
Dr Sin-Eng Chia, Department of Community, Occupational and Family
Medicine, National University of Singapore, Faculty of Medicine,
Singapore
Mr J. Fawell, National Centre for Environmental Toxicology, Medmenham,
United Kingdom
Dr L. Fishbein, Fairfax, Virginia, USA ( Chairman)
Dr H. Hansen, Division of Toxicology, Agency for Toxic
Substances and Disease Registry, Atlanta, Georgia, USA
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, United Kingdom ( Co-Rapporteur)
Dr M.V. Park, Edinburgh Centre for Toxicology, Edinburgh, United
Kingdom
Mr D.J. Reisman, National Center for Environmental Assessment, US
Environmental Protection Agency, Cincinnati, Ohio, USA
( Co-Rapporteur)
Dr A. Wibbertman, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany ( Vice-Chairman)
Observers
Dr D. Morgott, Toxicological Sciences Laboratory, Health, Safety and
Environment, Eastman Kodak Company, Rochester, New York, USA
(representing the American Industrial Health Council)
Dr D. Owen, Shell Chemicals Europe Limited, London, United Kingdom
(representing the European Centre for Ecotoxicology and Toxicology of
Chemicals)
Dr P. Montuschi, Department of Pharmacology, Catholic University of
the Sacred Heart, Rome, Italy (representing the International Union of
Pharmacology)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR ACETONE
A WHO Task Group on Environmental Health Criteria for Acetone met
at the British Industrial Biological Research Association (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom, from 1
to 5 December 1997. Dr S. Jaggers opened the meeting and welcomed the
participants on behalf of the host institute. Dr E. Smith, IPCS,
welcomed the participants on behalf of the Director, IPCS, and the
three IPCS cooperating organizations (UNEP/ILO/WHO). The Task Group
reviewed and revised the draft criteria monograph and made an
evaluation of the risks for human health and the environment from
exposure to acetone.
Mr D.J. Reisman, US Environmental Protection Agency, Cincinnati,
USA, prepared the first draft of this monograph. The second draft,
incorporating comments received following the circulation of the first
draft to the IPCS Contact Points for Environmental Health Criteria
monographs, was also prepared by Mr. Reisman.
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the overall scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
* * *
The US Environmental Protection Agency funded the preparation of
this Environmental Health Criteria monograph, financial support for
the Task Group meeting was provided by the United Kingdom Department
of Health, and the meeting was organized by the British Industrial
Biological Research Association (BIBRA).
ABBREVIATIONS
BOD biochemical oxygen demand
CAS Chemical Abstracts Services
DOT/UN/NA/IMCO Department of Transportation/United Nations/North
America/International Maritime Dangerous Goods Code
EINECS European Inventory of Existing Chemical Substances
EPA Environmental Protection Agency
FID flame ionization detector
GC gas chromatography
HPLC high performance liquid chromatography
HRGC high resolution gas chromatography
HSDB Hazardous Substances Data Bank
IC ion chromatography
LOEL lowest-observed-effect level
MS mass spectrometry
NCI National Cancer Institute
NIOSH National Institute for Occupational Safety and Health
NOEL no-observed-effect level
OHM/TADS Oil and Hazardous Materials/Technical Assistance Data
System
ppbv parts per billion (by volume)
RBC red blood cell
RCRA Resource Conservation and Recovery Act
RGD reduction gas detector
RTECS Registry of Toxic Effects of Chemical Substances
TWA time-weighted average
UV ultraviolet
v/v volume per volume
WBC white blood cell
1. SUMMARY
1.1 Properties
Acetone (relative molecular mass = 58.08) is a clear colourless
flammable liquid (flash point -17°C closed cup, -9°C open cup;
flammability limits in air at 25°C = 2.15-13% v/v). The explosive
limits in air are 2.6-12.8% v/v. It has a high evaporation rate
(vapour pressure 181.72 mmHg at 20°C) and a low viscosity (0.303 cP at
25°C). It is miscible with water and organic solvents.
1.2 Uses and sources of exposure
1.2.1 Production
Acetone is manufactured mainly by the cumene peroxidation or
isopropyl alcohol dehydrogenation processes. The cumene peroxidation
process produces trace quantities of benzene as a by-product.
1.2.2 Uses and emissions into the environment
Acetone is used mainly as a solvent and intermediate in chemical
production. Major uses are in the production of methyl methacrylate,
methacrylic acid and higher methacrylates, bisphenol A, methyl
isobutyl ketone, drug and pharmaceutical applications, and as a
solvent for coatings and for cellulose acetate. There are also food
uses as an extraction solvent for fats and oils, and as a
precipitation agent in sugar and starch purification.
Atmospheric emissions occur from consumer products including nail
polish removers, particle board, carpet backing, some paint removers,
and liquid/paste waxes or polishes. Certain detergents/cleansers,
adhesives, and automobile carburetor and choke cleaners also contain
acetone.
Acetone is released into surface water in wastewater effluents
from a wide range of manufacturing processes and industries, such as
paper, plastic, pharmaceuticals, specialty cleaning and polishing
products, paint and allied products, gum and wood chemicals, cyclic
intermediates, industrial organic chemicals, gypsum products, paper
board products, and energy-related industries, such as
coal-gasification and oil shale processing.
Sources of acetone release into soil include disposal of
agricultural and food waste, animal waste, atmospheric wet deposition,
household septic tank effluents and chemical waste disposal sites.
1.3 Environmental transport, distribution and transformation
Acetone released to the atmosphere is degraded by a combination
of photolysis and reaction with hydroxyl radicals. The average
half-life for acetone degradation in the atmosphere is approximately
30 days. Acetone can be physically removed from air by wet deposition.
The dominant degradation process for acetone in soil and water is
biodegradation, and acetone is readily biodegradable. Volatilization
of acetone from the aquatic environment can be a significant transport
process. Acetone is a volatile compound that will evaporate from dry
surfaces. Since acetone is miscible in water, it can leach readily in
most types of soil. Concurrent biodegradation may diminish the general
significance of leaching if biodegradation occurs fast enough.
1.4 Environmental levels and human exposure
Exposure to acetone results from both natural and anthropogenic
sources. Acetone also occurs as a metabolic component in blood, urine
and human breath. It occurs as a biodegradation product of sewage,
solid wastes and alcohols, and as an oxidation product of humic
substances. Acetone has been detected in a variety of plants and foods
including onions, grapes, cauliflower, tomatoes, morning glory, wild
mustard, milk, beans, peas, cheese and chicken breast. Natural
emissions from a variety of tree species contain acetone vapour. Human
sources of emissions to the aquatic environment include waste-water
discharges from many industries and leaching from industrial and
municipal landfills. A major source of human emission to air is
evaporation of acetone solvent from coating products such as paints,
cleaners, varnishes and inks. Acetone is an emission product from the
combustion of wood, refuse and plastics. It is also emitted in exhaust
from automobile, diesel and turbine engines. Concentrations of acetone
monitored in the atmosphere range from 0.5 to 125.4 µg/m3 (0.2-52.9
ppb).
1.5 Kinetics and metabolism
Acetone is one of three ketone bodies that occur naturally
throughout the body. It can be formed endogenously in the mammalian
body from fatty acid oxidation. Fasting, diabetes mellitus and
strenuous exercise increase endogenous generation of acetone. Under
normal conditions, the production of ketone bodies occurs almost
entirely within the liver and to a smaller extent in the lung and
kidney. The process is continuous, and the three products are excreted
into the blood and transported to all tissues and organs of the body
where they can be used as a source of energy. Two of these ketone
bodies, acetoacetate and ß-hydroxybutyrate, are organic acids that can
cause metabolic acidosis when produced in large amounts, as in
diabetes mellitus. Acetone, in contrast, is non-ionic and is derived
endogenously from the spontaneous and enzymatic breakdown of
acetoacetate. Endogenous acetone is eliminated from the body either by
excretion in urine and exhaled air or by enzymatic metabolism. Under
normal circumstances, metabolism is the predominant route of
elimination and handles 70-80% of the total body burden.
Acetone is rapidly absorbed via the respiratory and
gastrointestinal tracts of humans and laboratory animals, as indicated
by the detection of acetone in blood within 30 min of inhalation
exposure and 20 min of oral administration. Studies of rats indicate
that orally administered acetone is extensively absorbed, whereas
during inhalation exposures humans absorb approximately 50% of the
amount of inhaled acetone. However, lower and higher respiratory
absorption values have been reported. The nasal cavities of humans and
laboratory animals appear to have a limited ability to absorb and
excrete acetone vapour, compared with the remainder of the respiratory
tract.
Acetone is uniformly distributed among non-adipose tissues and
does not accumulate in adipose tissues. In mice, maximum acetone
concentrations in adipose tissues were reported to be about one-third
of those in non-adipose tissues following inhalation exposure. Acetone
is rapidly cleared from the body by metabolism and excretion.
Half-times for acetone in human alveolar air and venous and arterial
blood are -4, 6 and 4 h, respectively. Exhalation is the major route of
elimination for acetone and its terminal metabolite (CO2) and the
fraction of administered acetone that is exhaled as unchanged acetone
is dose-related. Urinary excretion of acetone and its metabolites
occurs, but this route of elimination is minor compared with
exhalation of acetone and respiratory CO2.
Exogenously supplied acetone enters into many metabolic reactions
in tissues throughout the body, but the liver appears to be the site
of most extensive metabolism. Carbon from orally administered acetone
has been detected in cholesterol, ammo acids, fatty acids and glycogen
in rat tissues, urea in urine and unchanged acetone and CO2 in
exhaled breath. Metabolically, acetone is degraded to acetate and
formate; this accounts for the entry of carbon from acetone into
cholesterol, fatty acids, urea and amino acids, and formation of
3-carbon gluconeogenic compounds.
Gluconeogenesis from acetone has been proposed to proceed by two
pathways. The first pathway proceeds through the initial catalytic
action of acetone monooxygenase and acetol monooxygenase, which
convert acetone to acetol and acetol to methylglyoxal, respectively.
Both of these enzymatic activities are induced by acetone and have
been identified as an isozyme of ethanol-inducible, hepatic eytochrome
P-450IIE1. The second gluconeogenic pathway involves the formation of
1,2-propanediol from acetone catalysed by acetone monooxygenase and a
non-characterized enzyme capable of converting acetol to
1,2-propanediol.
1.6 Effects on laboratory mammals and in vitro systems
Oral LD50 values in adult rats are in the range of 5800-7138
mg/kg. The 4-h inhalation LC50 value is 76 000 mg/m3 (32 000 ppm).
Acute exposure to acetone has been found to alter performance in
neurobehavioural tests in laboratory animals at concentrations greater
than 7765 mg/m3 (>3270 ppm).
Experimental animal data characterizing the effects of long-term
oral or inhalation exposure to acetone are not available, due probably
to its low toxicity and its endogenous characteristics.
Prolonged acetone inhalation exposure of rats to 45 100 mg/m3
(19 000 ppm), 3 h/day, 5 days/week for 8 weeks, produced a reversible
decrease in absolute brain weight. No consistent changes were noted in
weights of other organs or the whole body, in blood chemical indices,
in liver triglyceride levels or in the histology of the heart, lung,
kidney, brain or liver.
In a 90-day gavage study of rats, increased blood parameters
(increased haemoglobin, haematocrit) were observed at dose levels
>500 mg/kg per day, and a NOAEL of 500 mg/kg per day was identified.
In a 13-week drinking-water study, toxic effects were noted in male
rats exposed to concentrations >20 g/litre (approx. 1700 mg/kg body
weight per day), namely increased relative organ weights, altered
haematological indices and mild nephropathy. In female rats
administered the highest concentration, 50 g/litre (approx. 3400 mg/kg
body weight per day), the effects were increased organ relative
weights and altered haematological indices. In addition, a 13-week
exposure to 50 g/litre caused altered relative testis weight and
altered sperm motility and morphology in male rats. Female mice given
50 g/litre (approx. 11 298 mg/kg body weight per day) in
drinking-water had altered liver and spleen weights and a marginally
increased incidence of centrilobular hepatocellular hypertrophy. No
toxic effects were observed in male mice administered 20 g/litre (4858
mg/kg body weight per day), the highest acetone level administered to
male mice. Thirteen-week exposures to concentrations < 10 g/litre
(900 mg/kg body weight per day) in drinking-water were associated with
no toxic effects in male rats; concentrations < 20 g/litre were
NOELs for female rats (1600 mg/kg body weight per day) and mice (male
4858 mg/kg body weight per day; female 5945 mg/kg body weight per day)
of both sexes.
In a preliminary 14-day drinking-water study of rats and mice,
dose-related centrilobular hepatocellular hypertrophy was noted in
male mice exposed to concentrations of 20-100 g/litre.
Pretreatraent of rodents with acetone enhances the hepatotoxic
effects of a number of compounds, notably halogenated alkanes, It is
hypothesized that the potentiation of the hepatotoxicity is mediated
by acetone-induced elevations of enzymatic activities (hepatic
mixed-function oxidases) that are responsible for the generation of
toxic intermediates from administered halogenated alkanes.
Acetone has tested negatively for genetic toxicity in numerous
non-mammalian systems, as well as in in vitro and in vivo
mammalian systems. Positive results are restricted to a single test
for aneuploidy in a yeast species exposed to high concentrations of
acetone (6.82%) in its growth medium. Acetone is not considered to be
genotoxic or mutagenic.
In a study of pregnant rats and mice exposed to acetone vapour
during days 6-19 of gestation, slight developmental toxicity was
observed following exposures of rats to 26 100 mg/m3 (11 000 ppm) for
6 h/day (increased percentage of litters with at least one fetal
malformation) and following exposures of mice to 15 670 mg/m3 (6600
ppm) for 6 h/day (small decrease in fetal weight and small increase in
percentage incidence of late resorptions). An atmospheric
concentration of 5200 mg/m3 (2200 ppm) was identified as a NOAEL for
developmental toxicity in both mice and rats. In a gavage study,
treatment at 3500 mg/kg per day during organogenesis impaired
reproduction in a screening test in mice. Negative results in vivo
in two different species, using oral and intraperitoneal routes,
indicated that no mutagenic changes were produced in mammals exposed
to acetone.
Reports of other reproductive effects of acetone include
observations of testicular effects and changes in sperm quality in
rats administered drinking-water containing 50 g acetone/litre for 13
weeks. No investigations of the effect of oral doses of acetone on
fetal development (fetotoxicity and teratogenicity) were available.
Acetone has been used extensively as a solvent vehicle in skin
carcinogenicity studies and is not considered carcinogenic when
applied to the skin.
1.7 Effects on humans
Acetone is relatively less toxic than many other industrial
solvents; however, at high concentrations, acetone vapour can cause
CNS depression, cardiorespiratory failure and death. Acute exposures
of humans to atmospheric concentrations as high as approx. 4750 mg/m3
(approx. 2000 ppm) have been reported to produce either no gross toxic
effects or minor transient effects, such as eye irritation. More
severe transient effects (including vomiting and fainting) were
reported for workers exposed to acetone vapour concentrations >25 500
mg/m3 (>12 000 ppm) for approx. 4 h. Acute exposures to acetone have
also been reported to alter performance in neurobehavioural tests in
humans at 595 mg/m3 (250 ppm). Females exposed to atmospheric
concentrations of 2370 mg/m3 (1000 ppm) were reported to suffer
menstrual irregularities.
1.8 Effects on other organisms in the laboratory and field
For most freshwater and saltwater animal species, 48- and 96-h
LC50 and EC50 values are >5540 mg/litre.
Growth of the alga Chlorella pyrendoidosa exposed to acetone at
257.4 mg/litre for 76 h was inhibited. There was inhibition of growth
of Chlamydomonas eugametos exposed to acetone for 48 h at 790
mg/litre. Photosynthesis was increased in Scendesmus quadricauda and
C. pyrenoidosa exposed to 79.0 and 790 mg/litre.
The 7- to 8-day toxicity thresholds for the green alga
S. quadricauda and the cyanobaeterium (blue-green alga)
Microcystis aeruginosa were 7500 and 530 mg/litre, respectively,
indicating that the green alga was more resistant to the toxic action of
acetone. The diatom Nitzschia linearis also seemed very resistant,
with a 5-day EC50 of 11 493 to 11 727 mg/litre. Similarly, the saltwater
diatom Skeletonema costatum was very resistant with 5-day EC50
values of 11 798 and 14 440 mg/litre.
Bacteria appear more resistant to acetone than protozoans.
Photobacterium phosphoreum, Pseudomonas putida and a mixed
microbial culture had EC50 values of 1700 to 35 540 mg/litre, and the
protozoan Entosehon sulcatum had an EC50 of 28 mg/litre. This may
be related to cell wall differences.
Quails and pheasants had oral 5-day LC50 values > 40 g/kg
diet. Fertile mallard eggs were not affected when immersed in 10%
acetone for 30 seconds; however, immersion in 100% acetone resulted in
decreases in survival, embryonic weight and embryonic length, but it
is not clear if this was due to the toxic or the solvent properties of
acetone. White Leghorn chicken eggs injected with 5 µl acetone did not
appear to have any significant changes in mortality or malformations.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Chemical identity
Chemical name acetone
Synonym(s) dimethyl ketone; 2-propanone;
beta-ketopropane
Chemical formula C3H6O
Chemical structure
O
"
H3C - C - CH3
Identification numbers:
CAS registry 67-64-1
NIOSH RTECS AL3150000
EPA Hazardous Waste
(RCRA) U002; F003
OHM/TADS 7216568
DOT/UN/NA/IMCO shipping UN1090
HSDB 41
EINECS 200-662-2
Relative molecular mass 58.08
2.2 Physical and chemical properties
2.2.1 Physical properties
Acetone is a clear and colourless liquid with a strong "fruity"
odour. It is miscible with water and organic solvents such as ether,
methanol, ethanol and esters (Nelson & Webb, 1978). The physical
properties of acetone, such as high evaporation rate, low viscosity
and miscibility, make it suitable for use as a solvent (Krasavage et
al., 1982). The physical properties of acetone are shown in Table 1.
2.2.2 Chemical properties
Acetone shows reactions typical of saturated ketones (SRI, 1996).
These reactions include addition, oxidation-reduction and
condensation, and yield alcohols, ketals, acids and amines (Papa &
Sherman, 1981). The chemical reactivity of acetone is commercially
important for the synthesis of methyl methacrylate, diacetone alcohol,
bisphenol A and other derivatives (SRI, 1996).
Table 1. Physical and chemical properties of acetone
Property Value/descriptiona Reference
Relative molecular mass 58.08 Riddick et al, (1986)
Colour Clear colourless Sax & Lewis (1987)
Physical state Liquid Sax & Lewis (1987)
Melting point -95.35°C Weast (1987}
Freezing point -94.7°C at 1 atm Riddick et al. (1986)
Boiling point 56.2°C at 1 atm Weast (1987)
(760 torr)
Density:
at 20°C 0.78996 g/ml Riddick et al. (1986)
at 26°C 0.78440 g/ml Riddick et al. (1966)
at 30°C 0.78033 g/ml Riddick et al. (1986)
Odour threshold:
Acetone in water 20 mg/litre Amoore & Hautala
(1983)
Air (absolute) 30-48 mg/m3 Amoore & Hautala
(13-26 ppm (v/v)) (1983)
Air (detection) 9.5 mg/m3 (4 ppm) Wysocki et al. (1997)
100% odour recognition 237-332 mg/m3 Hellman & Small
(100-140 ppm) (1974); Leonardos et
al. (1969)
Table 1 (contd).
Property Value/descriptiona Reference
Solubility:
Water at 20°C Completely miscible Windholz (1963)
Organic solvent(s) Soluble in organic
solvents Windholz (1983)
Viscosity at 25°C 0.303 cP Riddick et al. (1986)
Partition coefficients:
Log Kow -0.24 Sangster (1989)
Log Koc 0.73b Lyman (1982)
KB/A 301 ± 22 Dills et al. (1994)
Vapour pressure 181.72 mmHg (at 20°C) Riddick et al. (1986)
231.06 mmHg (at 25°C) Riddick et al. (1986)
Henry's law constant: 4.26 × 10-5 atm-m3/mol Rathbun & Tai (1987)
at 25°C
Flashpoint (closed cup) -17°C Riddick et al. (1986)
(open cup) -9°C Riddick et al. (1986)
Flammability limits Lower, 2.2%; Clayton & Clayton
in air at 25°C upper, 13.0% (1982)
Minimum ignition 465°C Riddick et al. (1986)
temperature
Explosive limits Lower, 2.6% in air (v/v); Sax & Lewis (1987)
upper, 12.8% in air (v/v) Sax & Lewis (1987)
a w/v = weight per volume, v/v = volume per volume.
b Estimated by regression equation 4-13 in Lyman (1982).
2.3 Conversion factors
Conversion factors in air at 25°C:
1 ppm = 2.374 mg/m3
1 mg/m3 = 0.421 ppm
2.4 Analytical methods
A number of analytical methods is available for the detection,
sampling and monitoring of acetone and its metabolites in the various
media. Acetone is a well-studied chemical and is used frequently in
the laboratory. This section is a review of the more established and
standard practices in use today.
2.4.1 Biological media
Methods for determining the presence of acetone in biological
organisms are listed in Table 2. Acetone is found in almost every
tissue and organ in the human body. Acetone and two other chemicals,
beta hydroxybutyrate and acetoacetate, are collectively referred to as
"ketone bodies". In the last 30 years much has been learned of acetone
in biological tissue since the discovery that acetone levels in
diabetes mellitus patients with severe hyperketonaemia may be
significant (Trotter et al., 1971). Higher acetone levels may be found
in the blood levels of individuals or animals after strenuous exercise
or prolonged dieting. Acetone production is also increased in animals
in disease states such as diabetes and anorexia.
The development of biological analytical methods can be done to
measure, but this does not distinguish acetone from either endogenous
and exogenous sources or from acetone in ketone levels in body fluids,
since acetone is produced within the biological system by breaking
down lipids and stored fats. Most of the methods for measuring acetone
in expired air use gas chromatography (GC/FID) and involve the
breakdown of beta-hydoxybutyrate and acetoacetate into acetone, which
is isolated and quantified by any of the techniques listed in Table 2.
The differences between these methods have been mainly concerned with
the nature of the column packing and with the various methods of
sample collection.
The determination of acetone in blood is difficult because it is
a metabolite and the quantity produced depends on storage time, even
when the blood samples are stored at 4°C (Trotter et al., 1971). The
delay between sample collection and analysis could lead to spuriously
elevated acetone concentrations because of the spontaneous
decarboxylation of acetoacetate (Van Stekelenburg & Koorevaar, 1972).
One method for the determination of acetone in the clinical laboratory
involves deproteinizing with acetonitrile and derivitization of the
sample with 2,4-dinitrophenylhydrazine, followed by isolation and
quantification of the hydrazone by high pressure liquid chromatograph
Table 2. Analytical methods for determining acetone in biological media
Sample matrix Preparation method Analytical Sample detection Reference
method limit
Whole blood, urine Centrifuged and deproteinized GC-HPLC 33 µg/ml Gavino et al. (1986)
with acetonitdle and 2,4-DNPH
added
Whole blood Deproteinized with HClO4 and GC-FID 0.4 µmol/litre Mangani & Ninfali
subjected to purge-and-trap (1988)
Whole blood Purge-and-trap GC-MS 0.2 µg/ml Ashley et al. (1992)
Serum Deproteinized with sodium HRGC-FID <58 µg/ml Smith (1984)
tungstate and cupric sulfate (<1 nmol/litre)
Serum Sample centrifuged and clear GC-FID 5.8 mg/ml Cheung & Lin
filtrate injected (0.1 nmol/ml) (1987)
into GC
Urine Diluted sample derivatized with GC-FID 0.2 µg/ml Kobayashi et al.
pentafiuorobenzyloxyl ammonium (3.45 µmol/ml) (1983)
chloride and extracted with
hexane
Liver perfusate, Reduction to isopropanol using GC-HPLC 33 µmol/ml Gavino et al. (1987)
blood, urine sodium borohydrid and
separation by HPLC
Liver Liver perfusion medium reduced GC-FID 3.78 µg/ml in Holm & Lundgren
with NaBH4 and an aliquot of perfusate (1984)
reduced solution injected (65 µmol/litre)
into GC
Liver, kidney, lung Purge-and-trap GC-FID No data Holm & Lundgren
and adipose tissue (1984)
Breath Direct injection into GC GC-FID No data Trotter et al. (1971);
Jansson & Larsson (1969)
(HPLC) (Brega et al., 1991). This method prevents acetoacetate, which
is present in plasma, from being thermally degraded to acetone on the
column when using a GC method (Gavino et al., 1987). The HPLC method
can also be used to measure acetone in urine or liver perfusate. This
method can be used in experiments requiring multiple samples and thus
can be used for diabetic patient monitoring, as well as for
occupational exposure monitoring.
2.4.2. Environmental media
Analytical methods for determining acetone in air, water and soil
are presented in Table 3. The commonly used methods are direct GC/MS
of a sample concentrate or HPLC of the 2,4-dinitrophenyl-hydrazine
derivative. In the United Kingdom, the 2,4-dinitrophenyl-hydrazine
HPLC method is applied to the analysis of acetone in water and there
is a standardized validated method (UK SAC, 1988).
When sampling for acetone, the incorrect use of Tedlar bags and
activated carbon may lead to spurious results.
Table 3 Analytical methods for determining acetone in environmental samples
Sample matrix Preparation method Analytical method Sample detection Reference
limit
Air (occupational) Air passed through charcoal and components GC-FID (NIOSH 7 µg/litre NIOSH (1994)
desorbed with CS2 method 1300)
Air Air passed through a cryogenic trap and the GC-RGD 10 ppt O'Hara & Singh
(ambient) trapped component injected into GC (1988)
Air Air passed through HPLC-UV <3 µg/litre Risner (1995)
(ambient) 2,4-dinitrophenylhydrazine-coated cartridge and eluted reversed-phase
with acetonitrile and tetrahydrofuran column
Air (indoor) Diffusive sampler with silica gel tape impregnated with HPLC-UV 15 µg/litre Brown et al.
2,4-dinitrophenylhydrazine and eluted with acetonitrile (1994)
Air Air passed through a 1% sodium bisulfite solution and Spectrophotometry <0.5 ppm (in Amlathe & Gupta
absorbed acetone reacted with alkaline vanilla solution solution) (1990)
Rural air Air passed through silica gel coated with HPLC-UV No data Shepson et al.
2,4-dinitrophenylhydrazine and eluted with acetonitrile (1991)
Water Sample reacted with alkaline diazotized anthranilic Spectrophotometry 500 µg/litre Rahim & Bashir
acid solution (1981)
Fresh and seawater Sample derivatized with 2,4-dinitrophenylhydrazine HPLC-UV detection 0.5 nmol/litre Kieber & Mopper
passed through a C18 cartridge and absorbed (0.03 µg/litre) (1990)
compound eluted with acetonitrile
Waste water, soil or Sample or sample mixed with reagent water subjected GC-MS 100 µg/litre US EPA (1986b)
sediment to purge-and-trap (EPA method 8240) (water)
100 µg/k9
(sediment and
soil)
Fresh fruit Vacuum distillation followed by solvent extraction HRGC-MS No data Takeoka et al.
of pulp (1988)
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Acetone occurs as a metabolic component in blood, urine and human
breath (Conkle et al., 1975). Because endogenous acetone formation is
so closely linked with the utilization of stored fats as a source of
energy, background levels can fluctuate depending on an individual's
health, nutrition, and level of activity (Morgott, 1993). The acetone
level in the human body at any instant is reflective of acetoacetate
production and ketogenesis. It occurs naturally as a biodegradation
product of sewage, solid wastes and alcohols and as an oxidation
product of humic substances. Acetone has been detected in a variety of
plants and foods, including onions, grapes, cauliflower, tomatoes,
morning glories, wild mustard, milk, beans, peas, cheese and chicken
breast (Day & Anderson, 1965; Grey & Shrimpton, 1967; Palo & Ilkova,
1970; Lovegren et al., 1979). Natural emissions from a variety of tree
species contain acetone vapour (Isidorov et al., 1985) and another
source is direct emission from the ocean (Zhou & Mopper, i990).
3.2 Anthropogenic sources
There are many anthropogenic sources of acetone, with various
levels and concentrations that cover a broad range. Human sources of
emissions to the aquatic environment include wastewater discharges
from many industries (Perry et al., 1978; NLM, 1992) and leaching from
industrial and municipal landfills (Sabel & Clark, 1984; Brown &
Donnelly, 1988). A major source of emission to the air is from
evaporation of acetone solvent from coating products such as paints,
cleaners, varnishes and inks. Acetone is an emission product from the
combustion of wood, refuse and plastics (Lipari et al., 1984; Graedel
et al., 1986), and is emitted in exhaust from automobile, diesel and
turbine engines (Barber & Lodge, 1963; Lloyd, 1978; Jonsson ct al.,
1985; Graedel et al., 1986; Westerholm et al., 1988; Zweidinger et
al., 1988).
Other important anthropogenic sources of acetone in the air are
chemical manufacture (Graedel et al., 1986), tobacco smoke (Manning et
al., 1983), wood burning and pulping (Lipari et al., 1984; Graedel et
al., 1986; Kleindienst et al., 1986), polyethylene burning (Hodgkin et
al., 1982), refuse combustion (NAS, 1976), petroleum production
(Graedel, 1978), and certain landfill sites (LaRegina et al., 1986;
Militana & Mauch, 1989; Hodgson et al., 1992). Acetone is formed in
the atmosphere from the photochemical oxidation of propane (Singh &
Hanst, 1981; Arnold et al., 1986) and possibly from propylene oxide
and epichlorohydrin (Spicer et al., 1985).
In a US EPA-sponsored survey of household products analysed by
purge-and-trap GC/MS for volatile organic compounds, acetone was found
in 314 of 1005 products (31.2%). Of the eight product categories, the
highest categories were paint-related (51.5% contained acetone),
adhesive-related (24.3%) and automotive (22.7%) products (Sack et al.,
1992).
3.2.1 Production levels and processes
In 1994, world acetone capacity amounted to almost 3.83 million
tonnes (SRI, 1996). Since approximately 80% of acetone is produced as
a co-product of phenol, demand for phenol largely determines acetone
production levels. World production in 1994 was estimated to be 3.22
million tomes, and demand for acetone was expected to grow at an
average annual rate of 3.3% annually from 1994 to 1999 (SRI, 1996).
The USA is the largest producer of acetone. Table 4 depicts the
capacity of the largest manufacturers in the USA in 1995, while Table
5 shows the capacity of other countries. The annual capacity in the
European Union in 1992-1994 was 1.1-1.2 million tonnes.
Most acetone is manufactured by one of two processes, cumene
peroxidation (94% yield) or isopropyl alcohol dehydrogenation (IPA)
(95% yield) (SRI, 1996). In the peroxidation process, cumene is
oxidized to hydroperoxide, which is cleaved to yield acetone and
phenol. In the dehydrogenation process, isopropyl alcohol is
catalytically dehydrogenated to yield acetone and hydrogen (Nelson &
Webb, 1978). The cumene peroxidation process accounts for 96%; IPA
accounts for the other 4%. Production grade acetone is 99.5% acetone,
0.5% water. Fermentation of corn starch and molasses to produce
acetone, using Clostridium acetobutylicium, is utilized in several
countries, including Russia, Egypt, Brazil and India (Sifniades,
1985). Although acetone is more costly to produce by the IPA process,
this process has no benzene contamination. Acetone produced through
the cumene process contains benzene at concentrations < 10 ppm
(SRI, 1996).
Some companies recover acetone as a by-product (SRI, 1996). For
example, in the United Kingdom there is a plant producing 52 000
tonnes per year that recovers acetone as a by-product of acetic acid
manufacture, and two Japanese manufacturers recover acetone from
cresol production.
3.2.2 Uses
Acetone is used primarily as an intermediate in chemical
production and as a solvent (SKI, I996). It is used as a solvent for
resins, paints, inks, varnishes and lacquers and in adhesives,
thinners and clean-up solvents. Pharmaceutical applications of acetone
include use as an intermediate and solvent for drags, vitamins and
cosmetics (Nelson & Webb, 1978). It has uses as an extraction solvent
for fats and oils and a precipitation agent in the purification of
starches and sugars (FAO/WHO, 1971).
Table 4: Major manufacturers of acetone in the USA in 1995a
Manufacturer Location Annual capacity
(thousands of tonnes)
Allied Signal, Inc, Philadelphia, PA 280
Aristech Chemical Corp. Ironton, OH 180
Dow Chemical USA Oyster Creek, TX 161
Eastman Chemical Co. Kingsport, TN 13
Mt. Vernon Partnership Mount Vernon, IN 191
Georgia Gulf Corp. Pasadena, TX 45
Plaquemine, LA 123
Goodyear Tire & Rubber Co. Bayport, TX 7
JLM Chemicals, Inc. Blue Island, IL 26
Shell Chemical Co. Deer Park, TX 182
Texaco, Inc. El Dorado, KS 26
Union Carbide Corp. Institute, WV 77
Total 1281
a SRI (1996)
Table 5: Production capacity of acetone in 1995 (excluding the USA)a
Country Annual capacity
(thousands of tonnes)
Germany 388
Italy 235
France 168
United Kingdom 97
Netherlands 80
Spain 75
Brazil 71
Finland 65
Mexico 22
Argentina 20
Venezuela 10
TOTAL 1185
a SRI (1996)
In 1995 the USA use pattern for acetone was as follows: acetone
cyanohydrin/methyl methacrylate, methacrylic acid and higher
methacrylates (45%); solvent applications (17%); bisphenol A (18%);
aldol chemicals/methyl isobutyl ketone and others (12%); and
pharmaceutical and other applications (8%) (SKI, 1996).
The largest solvent application for acetone is as a surface
coating, including use as a thinner and wash solvent. In 1995,
greatest use of acetone as a solvent was in automotive coatings, both
original equipment and automotive refinishing (SKI, 1996). The next
greatest use for acetone is the production of acetone cyanohydrin
which is used to produce an acrylic resin monomer, methyl
methacrylate. Bisphenol A is produced from acetone and used in
polycarbonate resins.
Acetone is also used in food processing as an extraction solvent
for oils and fats and as a precipitation agent in the purification of
starches and sugars.
3.2.3 Releases
3.2.3.1 Air
Atmospheric emissions are likely from the many consumer products
containing acetone (US EPA, 1989). Such products include nail polish
removers, particle board (Tichenor & Mason, 1988), carpet backing
(Hodgson et al., 1993), some paint removers, a number of liquid/paste
waxes or polishes, some detergents/cleansers, adhesives (Knöppel &
Schauenburg, 1989; Sack et al., 1992) and carburetor and choke
cleaners (US EPA, 1989).
Atmospheric emissions from the phenol/acetone production process
are approximately 0.44 g per kg of acetone produced (Sifniades, 1985).
3.2.3.2 Water
Acetone is released into surface water as wastewater from certain
chemical manufacturing industries (Jungclaus et al., 1978; Hites &
Lopez-Avila, 1980; Gordon & Gordon, 1981). It is also released in
water from energy-related industries, such as coal-gasification
(Pellizzari et al., 1979; Mohr & King, 1985) and oil shale processing
(Pellizzari et al., 1979; Hawthorne & Sievers, 1984). Acetone was
found in 27 of 63 effluent water samples from a wide range of chemical
industries in the USA (Perry et al., 1979). It has been detected in
effluents from various industrial production processes including
paper, plastic, pharmaceutical, specialty cleaning and polishing
products, paint and allied products, gum and wood chemicals, cyclic
intermediates, industrial organic chemicals, gypsum products, and
paper board products.
Acetone can be released to groundwater as a result of leaching
from municipal and industrial landfills (Gould et al., 1983; Steelman
& Ecker, 1984; Sawhney & Raabe, 1986; Brown & Donnelly, 1988). It may
also leach from solvent cement used in joining polyethylene and other
plastic pipes used in drinking-water distribution and domestic
plumbing (Anselme et al., 1985). One of the sources of acetone in
seawater is the sensitized photoreaction of dissolved organic matter
(Mopper & Stahovec, 1986).
3.2.3.3 Soil
Acetone leaches readily in soil. The US Agency for Toxic
Substances and Disease Registry (ATSDR, 1994) found the amount of
acetone released into soil from landfills in the USA accounted for
approximately 0.1% of the total environmental release of acetone.
Sources of acetone release into soil include disposal of agricultural
and food waste, animal wastes, and atmospheric wet deposition. Acetone
was detected in 43% of the soil from designated waste disposal sites
tested for acetone. Household septic tank effluents are another source
of acetone in soil (DeWalle et al., 1985).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution among media
Acetone is commonly found in air, water, soil and biological
samples, and these background levels can he from both human-made and
natural sources. Acetone occurs naturally in trees, plants, forest
fires and volcanic gases. When animals and humans catabolize body fat,
acetone is exhaled and metabolized. Human-made sources include tobacco
smoke, combustive engine exhaust and waste incineration. The exchange
of carbonyl compounds (including acetone) between air and natural
waters is governed by the appropriate partition coefficients, in
addition to production and loss processes in both media (Benkelberg et
al., 1995).
4.1.1 Air
The significant environmental fate processes for the degradation
of acetone in the ambient environment are photolysis and reaction with
hydroxyl radicals (Meyrahn et al., 1986; Kerr & Stocker, 1986).
Meyrahn et al. (1986) measured the quantum yields of acetone
photolysis at environmental wavelengths and projected the following
rate constants for the lower troposphere at 40°N latitude: in January,
3.3 × 10-8/sec; in July, 1.8 × 10-7/sec; yearly average,
1.0 × 10-7/sec. These rate constants correspond to half-lives of
243, 45 and 80 days for January, July and the yearly average,
respectively. These rate constants intentionally neglect reaction of
excited acetone molecules with oxygen. Based on the photodecomposition
data of Gairdner et al. (1984), the rate constants of Meyrahn et al.
(1986) would be about twice as great if the neglected reaction were
included. Using this factor of 2, the total yearly average photolysis
half-life (plus reaction of excited acetone molecules) is about 40
days. The rate constant for the reaction of hydroxyl radicals with
acetone at 25°C is in the range of 2.2-2.6 × 10-13 cm3/molecule-sec
(Kerr & Stocker, 1986; Wallington & Kurylo, 1987). Probable pathways
for the reaction of acetone with hydroxyl radicals in the troposphere
have been postulated, and methyl-glyoxal is the primary product of
this reaction (Altshuller, 1991). The primary products of acetone
photolysis in sunlight are carbon dioxide and acetylperoxynitrate
(Altshuller, 1991). The photochemical oxidation of acetone in the
presence of nitrogen oxides produces small amounts of peroxyacetic
acid and peroxyacetyl nitrate (Hanst & Gay, 1983).
The photolysis lifetimes of acetone under cloudless conditions at
40°N latitude, and at sea level during winter and summer were
estimated to be 83 and 19 days, respectively (Martinez et al., 1992).
Other investigators have estimated that the average atmospheric
lifetime of acetone due to photolysis at 40°N latitude is 80 days/year
and varies from 243 in January to 45 in July (Meyrahn et al., 1986).
Meyrahn et al. (1986) estimated the average lifetime of acetone at
40°N due to combined hydroxyl radical reaction and photolysis to be 32
days/year, corresponding to a half-life of approx. 22 days. The
decomposition rate showed a pronounced dependence on latitude, with
greater losses of acetone occurring near the equator compared to the
poles. In very polluted air, the hydroxyl radical concentration
increased by an order of magnitude, which would lower the half-life by
an order of magnitude.
The complete miscibility of acetone in water suggests that
physical removal from air by wet deposition (rainfall, dissolution in
clouds, etc.) is probable (Aneja, 1993). The reactions of acetone
vapour with nitrogen oxides, hydroxyl radicals (OH), singlet molecular
oxygen (1 Delta g), singlet atomic oxygen (O(3P)), and nitrate
radicals have been studied. Given the second order rate constants for
the reactions of acetone with 1 Delta g (Datta & Rao, 1979) and O(3P)
(Lee & Timmons, 1977), and the concentrations of singlet molecular and
atomic oxygen in the atmosphere (Graedel, 1978), these reactions are
insignificant in determining the fate of acetone in the atmosphere.
However, Grosjean & Wright (1983) detected acetone in rain, cloud,
mist and fog water that was collected in Southern California, USA. In
certain instances, physical removal by wet deposition may be
environmentally significant, especially since the degradation rate is
not very fast. The reaction of acetone with nitrate radicals in the
atmosphere was also determined to be insignificant (Boyd et al.,
199l). Smog chamber studies with acetone and nitrogen oxides have
shown that acetone has low reactivity in terms of ozone and nitrogen
dioxide formation and that the rate of disappearance of acetone by
this process is low (Altshuller & Cohen, 1963; Dimitriades & Joshi,
1977).
Using 72-h back trajectories, Aneja (1993) studied organic
compounds transported in cloud water whose origin was an industrial
valley. Acetone was found in cloud water at an average of 460 ng/litre
(range 0-4100 ng/litre), in clouds of low pH (2.78).
4.1.2 Water
The miscibility of acetone in water and the estimated low value
of 0.73 for log Koc (see Table 1) suggests that adsorption of acetone
to sediments and suspended solids is not significant. When water is
not present, acetone vapour adsorbs rather strongly to the clay
component of soil by hydrogen bonding (Goss, 1992; Steinberg &
Kreamer, 1993). The sorption is inversely dependent on relative
humidity, so increasing the humidity decreases sorption drastically.
In water-saturated soil or sediment, Koc values (organic carbon), and
not hydrogen bonding, may control the sorption of acetone (Steinberg &
Kreamer, 1993). The experimental adsorption studies with Kaolinite,
montmorillonite, and stream sediments showed very little or no loss of
acetone from water to the adsorbents (Rathbun et al., 1982).
The transport of acetone from the water column to the atmosphere
depends on the Henry's law constant. The Henry's law constant for
acetone is 4.26 × 10-5 atm-m3/mol (see Table 1), which suggests that
volatilization of acetone from water, although not very fast, could be
significant (Thomas, 1982), and likely to be important in determining
the fate of acetone in streams (Rathbun et al., 1982). The
volatilization rate of a chemical depends on the characteristics of
the chemical and the presence of water, and on other ambient
conditions (e.g., water depth, suspended solid concentration, water
current, wind speed, temperature). Based on an estimation method
(Thomas, 1982) and the Henry's law constant value, the volatilization
half-life of acetone from a model river 1 m deep, flowing at a current
of 1 m/second with a wind velocity of 3 m/sec is between 18 and 19 h.
The mean volatilization coefficient for acetone in a model outdoor
stream was found to be in the range of 7.15 × 10-4 to 14.8 × 10-4/min
(Rathbun et al., 1989, 1991). Therefore, the volatilization half-life
of acetone from the model stream is in the range of 8-16 h. It was
concluded that volatilization will control the fate of acetone in
water (Rathbun et al., 1989, 1991). Using a computer simulation model
the volatilization half-life from a model pond (2 m deep) was
estimated to be around 9 days.
The average of four experimentally determined rate constants for
the reaction of acetone with hydroxyl radicals in water (pH 6-7) is
1.1 × 10-8 litres/mol-sec (Buxton et al., 1988). Assuming the
hydroxyl radical concentration in brightly sunlit natural water is 1.0
× 10-17 mol/litre, the half-life for the reaction is almost 20 years.
Thus, photo-oxidation reactions of acetone in environmental waters do
not appear to be a significant removal process. Also, photolysis of
acetone in water, based on a rate constant for the reaction of acetone
with hydroxyl radicals in water at pH 7 of 5.8-7.7 × 107
litres/mol-sec and a concentration of hydroxyl radicals in eutrophic
waters of 3 × 10-17 M (Mill & Mabey, 1985), will not be significant.
Rathbun & Tai (1982) measured the mass transfer coefficient (KL for
acetone in water and reported values ranging from 0.310 to 0.537. When
distilled water or natural water containing acetone was exposed to
sunlight for 2-3 days, no photodecomposition of acetone was observed
(Rathbun et al., 1982). Experimental hydrolysis data for acetone have
not been found in the available literature. However, ketones generally
resist aqueous environmental hydrolysis (Harris, 1982) and hydrolysis
of acetone is not expected to be significant in the environment.
Bacterial degradation of acetone occurs, and the rate is
increased if acclimatization of the bacteria is achieved before higher
concentrations are present (see section 4.2.2.1). Both volatilization
and biodegradation are likely to play a part in the loss of acetone
from surface waters. The most significant process will depend on
particular circumstances, such as depth and amount of aeration.
4.1.3 Soil
The two significant transport properties for acetone in soil are
volatilization and leaching, and acetone is also expected to
biodegrade rapidly. Leaching transports acetone from soil to
groundwater, with the rate of leaching from soil by rainwater
depending on the sorption characteristics of acetone in the various
types of soil. Since acetone may be controlled by Koc in
water-saturated soil and has a low Koc value, sorption of acetone
in such soil will be weak. A sorption study with moist clay soils
indicated that aqueous acetone causes swelling in these soils (Green
et al., 1983), and this process may allow the retention of a small
fraction of acetone. Volatilization transports acetone from soil to
the atmosphere. The volatility rate of acetone from soil depends on
the soil characteristics (moisture content, soil porosity, etc.).
Since acetone is weakly sorbed to soil, the volatility depends
primarily on fire moisture content of the soil. In dry soil, the
volatilization rate from soil surfaces is high due to the high vapour
pressure of acetone. In moist soil, the rate of volatilization is
similar to that of acetone in water and depends on the Henry's law
constant. Acetone volatilizes moderately under these conditions. The
detection of acetone at higher concentrations in downwind air of a
landfill site, compared to upwind air (Militana & Mauch, 1989),
indicates that acetone can volatilize from soil.
No data regarding the transport or uptake of acetone from soil to
plants are available.
While acetone is expected to biodegrade readily in soil, no data
are available to suggest that any degradation process in soil, other
than biodegradation, is significant.
Acetone has been detected in leachates from municipal and
industrial landfills (Sabel & Clark, 1984; Sawhney & Kozloski, 1984;
Brown & Donnelly, 1988), demonstrating that leaching through soil can
occur. The presence of other leachate constituents can adversely
affect the biodegradation efficiency of microbes to use acetone.
Acetone has a relatively high vapour pressure (231.06 mmHg at
25°C) (Riddick et al., 1986) and is used as an evaporative solvent in
a variety of applications. Because of its volatile properties, acetone
can be expected to evaporate from dry surfaces, particularly in spills
on the soil surface. Although evaporation from dry surfaces should be
a significant process, sufficient data are not available to predict
the relative significance of evaporation from moist soils, where
biodegradation and leaching will compete with evaporation as a removal
process.
4.2 Biotransformation
4.2.1 Bioconcentration and biomagnification
The very low log Kow value of -0.24 (see Table 1) suggests that
bioconcentration (a process leading to a higher concentration of a
chemical in an organism relative to that in its environment) of
acetone in either aquatic or terrestrial organisms, and
biomagnification (series of processes in an ecosystem by which higher
concentrations of a chemical are attained in organisms at higher
trophic levels) of acetone from animals of lower to higher trophic
levels is unlikely.
4.2.2 Biodegradation
Many aerobic biodegradation screening studies with mixed
microorganisms from waste-treatment plant effluents, activated sludge,
or sewage have examined the biodegradability of acetone (Lamb &
Jenkins, 1952; Heukelekian & Rand, 1955; Stafford & Northup, 1955;
Ettinger, 1956; Hatfield, 1957; Gaudy et al., 1963; Price et al.,
1974; Thom & Agg, 1975; Bridie et al., 1979; Urano & Kato, 1986a,b;
Babeu & Vaishnav, 1987; Bhattacharya et al., 1990). These strutues
indicate that acetone is easily biodegradable with acclimatized
microorganisms or after a suitable lag period (approx. 1 day) (Urano &
Kato, 1986a,b), as long as the initial concentration of acetone is not
at a toxic level. For example, acetone at a concentration of 500
mg/litre was toxic to microorganisms when biooxidation of acetone by
activated sludge was attempted (Gerhold & Malaney, 1966).
Biodegradation of acetone was similar in seawater and fresh water
(Takemoto et al., 1981 ). The 20-day biochemical oxygen demand (BOD)
for acetone for fresh water and saltwater was 78% and 76%,
respectively (Lamb & Jenkins, 1952; Price et al., 1974). After a
suitable lag period (5 days), acetone biodegraded quantitatively under
anaerobic conditions with anaerobic acetate-enriched culture medium
(Chou et al., 1979). A biodegradation study of acetone in natural
water collected from Lago Lake near Athens, Georgia, determined that
the biodegradation kinetics were multiphasic in nature and depended on
the substrate concentration. The determined rate of degradation was
faster at higher initial concentrations (the maximum concentration
used was 0.5 mg/litre) (Hwang et al., 1989).
In a laboratory experiment with natural stream water and
sediment, no acetone was lost in 338 h under sterile conditions in
closed flasks. However, with non-sterile natural sediment, 100% of the
acetone was lost in 500 h following a lag period of 90 h. (Rathbun et
al., 1982). The authors of this study concluded that biodegradation
was one of the important processes for the loss of acetone in streams.
Rathbun et al. (1982) separated his study into two groups to observe
the effects of pre-exposure acclimatization. One group was pre-treated
with a small concentration of acetone overnight and the other did not
get pre-treatment. The treatment reduce the lag time, and degradation
coefficients were much lower for the pre-treated groups. First-order
rate coefficients for the bacterial degradation of acetone at 25°C
ranged from 0.43-0.9 days-1 (not pre-treated), giving half-lives of 2
days. Significant loss of acetone due to biodegradation was not
observed in a later study when acetone was injected continuously in an
outdoor model stream (Rathbun et al., 1988, 1989, 1991, 1993).
Attempts to induce biodegradation by adding glucose and a nutrient
solution containing bacteria acclimated to acetone were unsuccessful.
The authors concluded that the residence time of acetone in the model
stream (6 h) was too short for the bacteria to become acclimated in
the water before initiation of biodegradation. However, this
explanation may not be valid if attached bacteria, rather than
free-floating bacteria, dominate the biodegradation process. As an
alternative explanation, the authors indicated that the observed
limitation in the nitrate concentration in the stream may be
responsible for the lack of acetone biodegradation.
4.2.2.1 Microbial degradation
Many aerobic biological screening studies have examined the
biodegradability of acetone and have found it to be readily
biodegradable (Lamb & Jenkins, 1952; Heukelekian & Rand, 1955;
Stafford & Northrup, 1955; Ettinger, 1956; Hatfield, 1957; Ludzack &
Ettinger, 1960; Price et al., 1974; Bridie et al., 1979; Takemoto et
al., 1981; Urano & Kato, 1986a,b; Vaishnav et al., 1987; Hwang et al.,
1989). One of these studies examined acetone biodegradation in a
natural water experiment and found acetone to be readily biodegraded
in Lago Lake water collected near Athens, Georgia, USA (Hwang et al.,
1989).
Platen et al. (1990) studied the enrichment, isolation,
characterization and the stoichiometry of acetone and its degradation.
In their study, acetone was oxidized completely by
Desulfococcus biacutus, a gram negative, anaerobic sulfate-reducing
bacterium using acetone as its sole organic substrate. Enzyme studies
indicated that acetone was metabolized by condensation with carbon
dioxide to a C4 compound (possibly free acetoacetate) and moved into
intermediary metabolism as acetoacetyl-coenzyme A. Acetoacetyl-CoA is
cleared by a thiolase reaction to acetyl-CoA which is completely
oxidized by the carbon monoxide dehydrogenase pathway. In
acetone-amended slurries, 76% of the theoretically-expected sulfate
was depleted, and in nitrate-amended slurries > 100% of the
theoretically-expected amounts of nitrate were consumed after 85 days
of incubation. Chou et al. (1979) also showed that acetone can be
degraded by anaerobic biodegradation.
Waggy et al. (1994) compared a USA 20-day biochemical oxygen
demand (BOD) test with the Organization for Economic Cooperation and
Development (OECD) closed bottle biodegradation test (Test 301D)
(OECD, 1981). In the 20-day BOD test, the results were 56, 76, 83 and
84%, at 5, 10, 15 and 20 days, respectively, and in the OECD test were
68, 72 and 78% for 5, 15 and 28 days, respectively, indicating good
correlation (Waggy et al., 1994). These test results classify acetone
as readily biodegradable. In a laboratory study using a microbial
culture from domestic waste water without acclimation, Price et al.
(1974) measured fresh water BODs (% biooxidation) to be 76, 82, 85 and
96% for 5, 19, 15 and 20 days, respectively. In "synthetic" saltwater,
the values for the same periods were 66, 88, 88 and 100%.
4.3 Bioavailability from environmental media
Acetone is expected to be bioavailable.
4.4 Interaction with other physical, chemical or biological factors
The atmospheric degradation of volatile organic compounds (VOCs)
in the presence of nitrogen oxides (NOx) leads to the production of
ozone. During complete oxidation of the VOCs free radical reactions
occur in the presence of sunlight with acetone (and other ketones),
participating as an intermediate with ozone as a byproduct. One method
of measuring the contribution of acetone is by the reactivity of it
with the hydroxyl radical (OH*).
The degradation of acetone in the lower troposphere may be
initiated by photolysis or reaction with OH* radicals. The reactions
with ozone (OD) or NOx are too slow to be important under
tropospheric conditions (Johnson & Jenkin, 1991). The rate of the
initiating reaction of OH* with acetone is well established at
2.26 × 10-13cm3/ molecule per sec (Atkinson, 1985).
Accordingly, the tropospheric lifetime of acetone with respect to
removal by OH radicals is approximately one month; therefore, the loss
of acetone by photooxidation is the major removal process of acetone
in the troposphere (Johnson & Jenkin, 1991).
The mechanism for acetone photodissociation has been reviewed by
Gardner et al. (1984). At 40°C, using the Gardner equations, the
average tropospheric lifetime would be halved to about 15 days, In
summary, the ozone concentrations predicted by the model were not
significantly affected by removal of the acetone emissions (Johnson &
Jenkin, 1991). Chatfield et al. (1987) examined the effect of
atmospheric pressure on the photolytic lifetime of acetone, and then
compared the result with losses caused by hydroxyl radical reactivity.
Reactions with hydroxyl radicals were much higher at ground level than
at increasing altitude where photolysis was more important in
degrading acetone.
The formation of ground-level ozone has become an air pollution
problem, especially in crowded, urban areas. Ozone is formed from the
complex photochemical interaction of some VOCs and NOx compounds.
Andersson-Sköld et al. (1992) calculated photochemical ozone creation
potentials (POCP) for 75 organic compounds, while Carter (1994)
developed maximum incremental ozone reactivity (MIR) scales to measure
the potential of VOCs to create ozone. Both research groups found that
ketones are weak producers of ozone, with acetone having one of the
lowest ozone formation potentials. Derwent et al. (1996) calculated a
POCP for acetone using a European model, which takes into account the
difference in conditions between European and North American cities;
the MIR model is considered more appropriate for North American
conditions. Andersson-Sköld et al. (1992) found similar values to
Derwent et al. (1966), indicating that acetone has "a remarkably low
POCP". Because of the low POCP, acetone has been suggested as a
potential substitute for high POCP aromatic hydrocarbons or the
chlorine-containing solvents.
4.5 Ultimate fate following use
The environmental fate of acetone can be predicted, since many of
the major fate processes have been investigated. When released to the
atmosphere, acetone will degrade through a combination of photolysis
and reaction with hydroxyl radicals (Meyrahn et al., 1986). Acetone
can be removed from the air by rainfall (wet deposition), as shown by
its detection in rainwater samples (Grosjean & Wright, 1983), but this
does not appear to be a significant route most of the time. In soil,
many studies have shown that acetone is readily biodegradable.
However, leaching may occur, especially if other chemicals are present
that may destroy or hinder microorganisms from degrading acetone.
Acetone can volatilize from water, as well as soil surfaces (Rathbun
et al., 1982). Since acetone is miscible with water and has a low
Koc, it leaches rather than adsorbs to soil. Where biodegradation is
inhibited or limited, acetone may reach the groundwater as a result of
leaching from spills or landfills (Steelman & Ecker, 1984; Brown &
Donnelly, 1988). Manufacturing and processing facilities may also
release acetone to air and water through discharges, and through other
wastes transported to landfills.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
Acetone is a commonly found volatile contaminant. It is one of
the more long-lived intermediates that are produced in the oxidation
of light non-methane hydrocarbons (Henderson et al., 1989). Monitoring
data, covering rural, urban, remote and other areas, are available;
values depend upon where the sampling was done, as well as the time of
the year and sampling technique. Examples arc presented in Table 6.
Grosjean et al. (1989) collected samples in three large urban
areas in Brazil (Sao Paulo, Rio de Janeiro, Salvador) with populations
ranging from 2 to 13 million people. In Sao Paulo, acetone levels were
in the range of 0.5-7 µg/m3 (0.2-3 ppb), in Rio 1.2-9 µg/m3 (0.5-3.8
ppb), and in Salvador 0-49.9 µg/m3 (0-21 ppb). In 1975, Brazil
initiated a nationwide programme of production of ethanol from sugar
cane, and by 1988, when these samples were taken, approximately
one-third of the vehicles in use were ethanol-fuelled. Formaldehyde,
acetaldehyde and acetone were the three carbonyls with the highest
values, but these levels were still not higher than levels in other
parts of the world.
Acetone was one of the VOCs identified in the air of the storage
section of a municipal waste truck (Wilkins, 1994). Although the exact
measurement was not given in the study report, the author stated that
the concentration was below 1780 mg/m3 (750 ppm) (the TLV value).
Brosseau & Heitz (1994) measured the gases emitted from a municipal
landfill site and found acetone in two samples: one at 77 µg/m3 (32.5
ppbv) and the other at 16 µg/m3 (6.84 ppbv).
Chatfield et al. (1987) studied the behaviour of acetone in the
troposphere. Over the Atlantic Ocean (35°N), the mean concentration of
acetone in the lower troposphere is approximately 1.2 µg/m3 (0.5
ppb). Chatfield et al. (1987) stated that a significant amount of
carbon appeared to be cycled as acetone, with attack by hydroxyl
radicals and photolysis as the chief loss mechanisms, and that propane
may contribute nearly half of the acetone observed in the upper
atmosphere. Henderson et al. (1989) continued this work by showing
that the effects of surface sources of higher order alkanes, alkenes
and terpenes play a major role in the amount of acetone in the
troposphere.
Granby et al. (1997) measured acetone levels simultaneously in a
busy Copenhagen street (22 000 cars/day) and a semi-rural site 30 km
west and found little difference in mean concentrations (2.4 µg/m3
vs. 2.1 µg/m3; 1 ppb vs. 0.9 ppb). They found very weak correlations
with carbon monoxide and NOx; indicating sources other than
automobile exhaust, the most likely being oxidation of reactive
hydrocarbons from long-range transport of polluted air masses. Since
Table 6. Environmental air levels in various locations
Sampling Area Concentrationa Sampling dates Reference
µg/m3 ppb
City/Tucson, Arizona, USA 28.5 12 February-September 1982 Snider & Dawson (1985)
Urban/Tulsa, OK, USA 11.4-125.4 4.8-52.8 1978 Arnts & Meeks (1981)
Urban/South and Central America 0.5-49.9 0.2-21 1988 Grosjean et al. (1989)
Rural/Arizona, USA 6.7 2.8 February-September 1982 Snider & Dawson (1985)
Rural/Colorado, USA 12.1-56.3 5.1-23.7 1978 Arnts & Meeks (1981)
Rural/Egbert, Ontario 0.9-8.8 0.39-3.6 1989 Shepson et al. (1991)
9
Rural/Dorset, Ontario 1.5-10.2 0.65-6.3 1989 Shepson et al. (1991)
Forest/Texas, USA 6.9-46 2.9-19.4 January 1978 Seila (1979)
Remote/Alaska 0.7-6.9 0.3-2.9 1967 Cavanagh et al. (1969)
Mountains/Tennessee, USA 5-28.5 2.1-12 1978 Arnts & Meeks (1981)
Mountains/Bavaria, Germany approx. 1.2 0.54b August 1995 Leibrock & Slemr (1997)
Ocean/Atlantic 35°N 1.2 0.5 Chatfield et al. (1987)
a Some sampling in the above studies may have been conducted using Tedlar bags that are known to contaminate
air samples with acetone (Henderson et at., 1989). Non-range values are mean values.
b Measured as propylene equivalents of oxygenated hydrocarbons in ppbC (ppb of carbon).
the acetone concentrations in this study are only slightly higher than
those found in rural, remote and ocean atmospheres, it appears that
the acetone is probably not transported a great distance in the lower
troposphere.
Arnold et al. (1997) measured upper tropospheric concentrations
of acetone at 9000 m over the northeastern Atlantic, near Ireland in
1993. Measured acetone concentration was found to correlate positively
with that of sulfur dioxide (SO2), reaching a maximum abundance of
approx. 7 µg/m3 (3 ppb). This concentration is markedly higher than
the concentration of 1.2 µg/m3 (0.5 ppb) in the lower troposphere
reported by Chatfield et al. (1987). As the SO2 level decreased, so
did the acetone concentration. Either the acetone was transported from
direct emissions from the USA, or a photochemical hydrocarbon
conversion had occurred.
In a review of earlier studies, Singh et al. (1994) found acetone
at a range of approx. 0.9-5.2 µg/m3 (approx. 0.4-2.3 ppb), with a
mean of 3.1 µg/m3 (1.14 ppb), in the troposphere. Using a three
dimensional photochemical model, Singh et al. (1994) found that the
greatest source of acetone was the oxidation of precursor hydrocarbons
(51%); other sources were biomass burning (26%), biogenic emissions
(21%) and an anthropogenic emission (approx. 3%). Atmospheric removal
was mainly by photolysis (64%), followed by reaction with OH*
radicals (24%) and deposition (12%). Other important points were:
* there is substantial variability in atmospheric abundance
* the concentration of acetone appears to vary with altitude
* upper atmospheric transport is possible since the half-life is
>10 days
* acetone appears to be the most abundant non-methane organic
species in the atmosphere
* the geochemical background of acetone appears to be 1.2 µg/m3
(approx. 0.5 ppb)
5.1.1.1 Indoor air
Shah & Singh (1988) reported a concentration of 19 µg/m3 (8 ppb)
in household indoor air. These authors compiled available data to
calculate an average outdoor concentration of 16.4 µg/m3 (6.9 ppb).
Other investigators reported similar results (Jarke et al., 1981).
Tichenor & Mason (1988) measured acetone levels in the range of 37-41
µg/m3 (approx. 15-17 ppb) per hour being emitted from low-density
particle board used in home construction in the USA. The reason for
the higher indoor air concentration was the use of acetone-containing
consumer products inside homes. The potential for intrusion of acetone
present as Soil gas into a house adjacent to a landfill was
characterized by Hodgson et al. (1992), but the measurement was for
only a single house. The average concentration was 47.5 µg/m3 (20
ppb).
Hodgson et al. (1991) collected air samples in a
newly-constructed building at four different times over a period of
14 months. The major source of VOCs was not the new construction
materials, but the liquid-process copiers and plotters where acetone
concentrations ranged from 28.8 to 66.6 µg/ms (12-28 ppb).
5.1.2 Water
Acetone has been qualitatively detected in drinking-water in
various cities in the USA, including Miami, FL; Ottumwa, IO;
Philadelphia, PA; Cincinnati, OH; Calhoun, GA; Dalton, GA; Gastonia,
NC; Durham, NC; New Orleans, LA; Rome, GA; and Tuscaloosa, AL (Bertsch
et al., 1975; US EPA, 1975; Shackelford & Keith, 1976). In the US EPA
National Organics Reconnaissance Survey (NORS), involving
drinking-water supplies from 10 cities in the USA, acetone was
qualitatively detected in all the cities. An acetone concentration of
1 µg/litre was found in drinking-water samples from Seattle, WA (US
EPA, 1975).
Acetone was detected in 33/204 surface water samples collected
from sites near heavily industrialized areas in the USA during
1975-1976 (Ewing et al., 1977). It was detected in 12.4% of all
groundwater samples analysed from 178 USA hazardous waste (Superfund)
sites as part of a national programme to investigate and remedy
potential problems at these sites (Plumb, 1987).
Acetone is released to water in wastewater discharges from
industry and sewage treatment. It was found in 23/63 effluent waters
from a wide range of chemical manufacturers around the USA at
concentrations ranging from < 10 to 100 µg/litre (Perry et al.,
1978). A comprehensive survey of wastewater from 4000 industrial and
publicly owned treatment works detected acetone in a wide range of
wastewater from industries such as leather tanning, petroleum
refining, nonferrous metals, paint and ink, printing and publishing,
coal mining, organics and plastics, inorganic chemicals, textile
mills, pulp and paper, robber processing, pesticide manufacture,
photographic industries, pharmaceuticals, porcelain/enamels,
mechanical products and transportation equipment. The highest effluent
concentration of acetone from all industries was 37.7 mg/litre, which
was detected in the paint and ink industry; however, the median
acetone level was 0.89 mg/litre (NLM, 1992).
Acetone can be released to groundwater by leaching from municipal
and industrial landfills. Leachate collected from a Minnesota (USA)
municipal landfill contained as much as 13 mg acetone/litre (Sabel &
Clark, 1984). Levels of 2.94.8 mg/litre were detected in leachate
samples collected in the USA from an industrial landfill in
Connecticut in 1982-1983 (Sawhney & Kozloski, 1984) and from one in
Michigan that contained up to 62 mg acetone/litre (Brown & Donnelly,
1988).
Acetone has also been detected at 0.2-0.7 µg/litre in water from
several artesian wells adjacent to a landfill in Wilmington, Delaware,
USA (DeWalle & Chian, 1981). The concentration of acetone was up to 3
mg/litre in a drinking-water well in New Jersey (Burmaster, 1982;
Steelman & Ecker, 1984).
The concentration of acetone in open ocean water (Tongue of the
Ocean, Bahamas) was approx. 0.35 µg/litre (Kieber & Mopper, 1990).
Corwin (1969) measured VOCs in seawater and found acetone levels in
the Florida Straits (USA) of 14-52 µg/litre at depths ranging from 0
to 160 metres at approx. 35% salinity. Similar concentrations were
found in the Mediterranean where the measurements were 18-52 µg/litre
at slightly higher salinity (approx. 39%).
5.1.3 Soil and sediment
There are few data regarding the level of acetone in soil and
sediment. Acetone has been detected in 43% of the soil samples in
designated waste disposal sites in the USA for which acetone
determination has been made (ATSDR, 1994). The maximum concentration
of acetone in soils from Vega Alta Public Supply well sites in Puerto
Rico and the mean concentration of acetone in soil from Summit
National Site, Ohio, was 9.5 mg/kg (ATSDR, 1994). Because of its high
water solubility and low sediment adsorption coefficient, acetone in
an aquatic system is predominantly found in water, rather than in
sediment.
5.1.4 Food
Acetone has been qualitatively detected in blue cheese (Day &
Anderson, 1965), baked potatoes (Coleman et al., 1981), roasted
filbert nuts (Kinlin et al., 1972), chicken breast muscle (Grey &
Shrimpton, 1967) and nectarines (Takeoka et al., 1988). Acetone
concentrations of 795 mg/kg and 11 mg/kg were identified in
Czechoslovakian milk samples and milk cream culture, respectively
(Palo & Ilkova, 1970). Milk samples from Swedish dairy cattle were
found to contain acetone concentrations ranging from 18 to 226
mg/litre (0.32-3.89 µmol/litre) (Andersson & Lundstrom, 1984).
Pellizzari et al. (1982) qualitatively identified acetone in all 8
selected human milk samples collected from volunteers in Bayonne, NJ,
Jersey City, NJ, Bridgeville, PA, and Baton Rouge, LA. A variety of
bean types (common, lima, mung and soy) contained acetone levels
ranging from 260-2000 µg/kg, with a mean level of 880 µg/kg, and
levels of 530 and 230 µg/kg were detected in split peas and lentils,
respectively (Lovegren et al., 1979). Acetone has also been detected
in onions, grapes, cauliflower, tomatoes and wild mustard (NLM, 1992).
5.1.5 Other environmental levels
Acetone is ubiquitous in the environment and is found at a wide
range of concentrations.
5.2 General population exposure
Acetone is readily absorbed from the lung and gastrointestinal
tract following inhalation and ingestion (see chapter 6). It can also
be absorbed through the skin. The low values for Koc (see Table 1)
and a moderate value for Henry's law constant (Rathbun & Tai, 1987)
suggest that the bioavailability of acetone from contaminated water
and soil as a result of contact may be significant. However,
quantitative data regarding the rate and extent of dermal absorption
of acetone from contaminated water and soil are lacking. The high
water solubility and low Koc value for acetone suggest that
bioavailability from ingested soil (e.g., children playing at or near
contaminated sites) will be high, but, again, quantitative absorption
data are lacking. Data on bioavailability of acetone from ingested
plant food are not available.
Exposure to acetone occurs from both natural and anthropogenic
sources, and it is endogenously produced by all humans. The general
population is exposed to acetone by inhaling ambient air, ingesting
food, and drinking-water containing acetone. Dermal exposure to
acetone may result from skin contact with consumer products (e.g.,
certain nail polish removers, paint removers, and household cleaning
and waxing products). Assuming concentrations of acetone are 19 µg/m3
(8.0 ppb) in indoor air and 16.4 µg/m3 (6.9 ppb) in outdoor air (Shah
& Singh, 1988) and that an average person inhales 15 m3/day of indoor
air and 5 m3/day of outdoor air daily, the estimated exposure to
acetone by inhalation is 0.37 mg/day. This value is much lower than an
estimate based upon an earlier exposure level found by one of these
researchers. Singh & Hanst (1981) estimated that an acetone
concentration of 0.26 µg/m3 (0.111 ppb) will occur in the lower
troposphere as a result of atmospheric oxidation of naturally
occurring propane, with levels of 35 ng/m3 (15 ppt) in the upper
troposphere and 7 ng/m3 (3 ppt) in the stratosphere. Since the
sampled atmospheric concentrations of acetone are 0.723-127.25 µg/m3
(0.3-52.8 ppb), and maintaining that the average adult human inhales
20 m3 air/day, the average daily exposure of acetone from inhalation
can be estimated to be 14.5-2545.0 µg, or up to 2.5 mg/day.
Wang et al. (1994) measured acetone concentrations in 89
non-occupationally exposed subjects and found acetone mean values of
840 µg/litre in blood, 842 µg/litre in urine, 715 ng/litre in alveolar
air and 154 ng/litre in environmental air. The researchers found no
significant difference in blood levels between smokers (896 µg/litre)
and nonsmokers (792 µg/litre), and likewise between hospital staff
(719 µg/litre) and blood donors (966 µg/litre). The results are
similar to those of Pezzagno et al. (1986) who measured 760 µg
acetone/litre in urine.
The endogenous acetone level in the body at any instant reflects
acetoacetate production (Morgott, 1993). The concentration of acetone
in whole blood does not differ from that in plasma (Gavino et al.,
1986). Even in healthy subjects, the level of acetone in blood or
plasma varies with fasting or non-fasting conditions and depends on
the weight of the subject. Generally, the blood or plasma acetone
concentrations are higher in fasted than non-fasted subjects and
higher in subjects who are not obese, compared to obese subjects (Haff
& Reichard, 1977). It should be noted that normal and abnormal
physiological conditions and disease states may increase ketogenesis
and the body burden of acetone. Acetone levels in athletes and
pregnant women (among many groups) may be elevated because these
groups of people have greater energy requirements. Ashley et al.
(1994) measured blood concentrations in non-occupationally exposed
populations. The mean concentration in a control group in the USA was
3.1 mg/litre. In a group of nine volunteer subjects, the mean blood
concentration before entering a van designed for clinical examinations
for a health survey was 1.9 mg/litre and after 3 h in the van the mean
blood concentration was virtually unchanged at 2 mg/litre, although
the range before entry was 1 3.6 mg/litre and after 3 h was 0.9-5
mg/litre, i.e. the high end of the range was over 1.4 mg/litre higher
when the subjects were tested after breathing the same air for 3 h.
Individuals with uncontrolled diabetes mellitus or diabetic
ketoacidosis may have plasma acetone levels as high as 750 mg/litre
(Trotter et al., 1971). The acetone concentrations in body fluids and
expired air in studies of healthy individuals and diabetic patients
are shown in Table 7. Clinical findings in eases of acute acetone
intoxication suggest that acetone blood levels over 1000 mg/litre are
necessary to cause unconsciousness in humans (Ramu et al., 1978), but
lower levels may interrupt physiological processes in diabetics.
Approximate reference concentrations for human plasma acetone are
< 10 mg/litre for a "healthy" individual, < 100 mg/litre for an
occupationally exposed individual, 100-700 mg/litre for an individual
with diabetic ketoacidosis and > 200 mg/litre for an individual
showing symptoms of "toxic" exposure (Tietz, 1983).
5.3 Occupational exposure
Kiesswetter et al. (1994) investigated occupational acetone
exposure using two groups of eight healthy male workers on nine shift
days. Using personal sampling, exposure was higher in the first half
of the shift (2730 mg/m3) than in the second half (1720 mg/m3).
For monitoring purposes, the researchers studied the relationship of
acetone in air versus three urine parameters: (1) concentration of
acetone in urine; (2) concentration of urine related to creatinine
excretion; and (3) concentration of acetone in urine in relation to
time (sampling period) and excreted urine volume. The concentration of
acetone in urine was moderately correlated to that in air. Some of the
ratings of well-being in the workers con-elated with the acetone
concentrations in the urine but not with the acetone concentrations in
the workplace air.
Table 7. Concentrations of acetone in body fluids and expired air of humans
Medium Subject Concentration Reference
Blood Healthy (non-fasted) 0.93 mg/litre Gavino et al. (1986)
Blood Health (non-fasted) 0.84 mg/litre Brugnone et al. (1994)
Blood Healthy (non-fasted) 1.8 mg/litre (median) Ashley et al. (1994)
Plasma Healthy (3-day fasted) 46.5 mg/litre Haff & Reichard (1977)
Plasma Healthy (non-fasted) 1.74 mg/litre Trotter et al. (1971)
Plasma Obese (3-day fasted) 17.4 mg/litre Haff & Reichard (1977)
Plasma Ketoacidotic 424 mg/titre Trotter et al. (1971)
Plasma Ketoacidotic 290 mg/litre Haff & Reichard (1977)
Urine Healthy 0.23-0.41 mg/litre Kobayashi et al. (1983)
Urine Healthy 0.84 mg/litre Brugnone et al. (1994)
Urine Healthy (endogenous) 0.76 mg/litre Pezzagno et al. (1986)
Urine Diabetic 0.64-9.0 mg/litre Kobayashi et al. (1983)
Expired air Healthy 1.23 µg/litre Jansson & Larsson (1969)
Expired air Healthy 1.16 µg/litre Trotter et al. (1971)
Expired air Healthy 1.3 µg/litre Phillips & Greenberg (1987)
Wang et al. (1994) calculated a blood-air coefficient for acetone
of 146. On average, the blood acetone levels of workers were 56 times
higher than those of subjects only exposed environmentally. These
researchers calculated the half-life of acetone in blood to be 5.8 h
for the interval between the end of one shift and the beginning of the
next (approx. 16 h). Analyses were made of workers before the start of
their shift, and mean acetone levels were 3.5 mg/litre in blood and 13
mg/litre in urine. Wigaeus (1981) calculated the acetone half-life to
be 6.1 h. These values indicate that the 16-h period between
workshifts did not allow for complete elimination of acetone absorbed
from the previous workshift.
In a study of environmental tobacco smoke (ETS) and its
contribution to VOC concentrations, Heavner et al. (1996) measured
acetone levels in smoking and non-smoking workplaces and homes. The
mean levels were: non-smoking workplace, 59.77 µg/m3 (SD 79.78);
smoking workplace, 952.86 µg/m3 (SD 3988.25); non-smoking home, 50.12
µg/m3 (SD 58.5); smoking home, 71.19 µg/m3 (SD 118.17).
Approximately 6% of the acetone found in the air of smoking workplaces
and homes was attributed to ETS.
Workers in industries that manufacture and use acetone can be
exposed to much higher concentrations of acetone than the general
population. For example, the concentrations of acetone in the
breathing zone air in a paint factory, a plastics factory, and an
artificial fibre factory in Italy were > 3.48 mg/m3 (Pezzagno et
al., 1986). The concentration of acetone in a plastic plant in Japan,
where bathtubs were produced, was > 100 mg/m3 (Kawai et al., 1990a).
The inhalation exposure of workers to acetone in a shoe factory in
Finland ranged from 25.4-393.4 mg/m3 (Ahonen & Schimberg, 1988). The
concentration of acetone in the air of a solvent recycling plant was
as high as 42 mg/m3, the mean exposure being 1 mg/m3 (Kupfersehmid &
Perkins, 1986).
Exposure to acetone may also occur indirectly. For example,
isopropyl alcohol is known to oxidize in the liver and is converted to
acetone (Kawai et al., 1990b). Therefore, occupational exposure
(printing plants) or accidental ingestion of isopropyl alcohol can
produce acetone in expired air, blood and urine (Lacouture et al.,
1989).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
There is extensive information regarding acetone and its
metabolism within animals and humans. Because acetone has a low
relative molecular mass and is miscible with water, it is absorbed and
uniformly distributed throughout the non-adipose tissues of the body.
There are absorption and tissue distribution studies, radiolabelled
metabolic and kinetic studies and elimination/excretion data. Other
variables such as diet, exercise and alcohol consumption affect the
kinetics of acetone.
6.1 Absorption
6.1.1 Inhalation exposure
6.1.1.1 Human studies
In humans exposed to acetone concentrations of up to 2970 mg/m3
(1250 ppm) for < 7.5 h/day in a complex protocol for < 6 weeks,
the concentration of acetone in venous blood was directly related to
the vapour concentration and duration of exposure, and inversely
related to the time elapsed following exposure (Stewart et al., 1975).
Data on the actual distribution of acetone in the human body are
scarce. Digs et al. (1994) measured in vitro blood/air partition
coefficients (KB/A) in blood samples from 73 humans. The average
KB/A (±SD) for acetone was 301 ± 22, with a range of 250-410, and
was normally distributed. Since acetone is absorbed into the blood
from the respiratory tract and is highly water soluble, it is to be
expected that there will be distribution to tissues with high water
content.
Acetone vapour is rapidly taken up by the tissue of the
respiratory tract and absorbed into the bloodstream during inhalation
exposure. This rapid uptake was shown by Haggard and co-workers when
substantial concentrations of acetone appeared in the blood only 30
min after exposure (Haggard et al., 1944). This is probably due to its
high blood-air partition coefficient (167-330) (Haggard et al., 1944;
Sate & Nakajima, 1979; Fiserova-Bergerova & Diaz, 1986; Paterson &
Mackay, 1989; Dills et al., 1994). Studies in humans exposed to up to
10 940 mg/m3 (4607 ppm) for up to 4 h showed measured pulmonary
uptakes ranging widely from -30% to 80% (Landahl & Herrmarm, 1950;
DiVincenzo et al., 1973; Nomiyama & Nomiyama, 1974a; Wigaeus et al.,
1981; Pezzagno et al., 1986). The reason for the wide range in
reported values involves the aqueous wash-in/wash-out effect when
acetone is inhaled, which can lead to spurious results (Schrikker et
al., 1985, 1989). During this phase, acetone, which is highly water
soluble, will dissolve in epithelial cells during inspiration
(wash-in) and evaporate during expiration (wash-out). This could
account for the lower than expected pulmonary absorption based on the
high blood/air partition coefficient (Wigaeus et al., 1981).
There have been a number of studies on highly soluble vapours
(such as acetone) and expiration from the respiratory tract. Cander &
Forster (1959) stated that a substantial portion of the acetone
adsorbed into the pulmonary tissue during inspiration was removed
during expiration, thus mixing with any expired alveolar sample that
was used as a measurement for analysis. Schrikker et al. (1985, 1989)
performed a series of measurements and found several interesting
results. There was an appreciable difference in alveolar air expired
at the beginning and ending of any breath during the wash-in cycle. At
first, acetone level was greatest at the beginning of a breath, but
when work load (exercise) started, the levels of the end started
rising, until after approximately 8 breaths, acetone level was higher
at the end of a breath. Part of the explanation was that the upper
airway linings give off the acetone during the initial breaths, and
the deeper portions of the respiratory tract give off the acetone in
the later breaths. Schrikker et al. (1989) found that the pulmonary
excretion of acetone is dependent upon the exchange of vapour between
the respiratory tract tissues and the inspired air, and is a
substantial proportion of the excretion of acetone via the respiratory
tract.
The turnover of acetone is highly dependent upon the organism's
state. Schrikker et al. (1989) and Jakubowski & Wieczorek (1988)
showed, as exercise increased, so did the amount of acetone being
expired and the rate of acetone uptake by the body was also directly
proportional to the ventilation rate. Haggard et al. (1944) compared
resting and exercising males and found that a ninefold increase in the
metabolic rate was required to account for a less-than-expected rise
in acetone blood levels. They also approached the idea that there was
a limit to the amount of acetone that can be accumulated in the body.
Exhaled breath levels of acetone in humans rise during exposure
to acetone and reach a steady state within approximately 2 h
(DiVincenzo et al., 1973; Nomiyama & Nomiyama, 1974a; Brown et al.,
1987). Uptake is directly proportional to exposure concentration and
duration (DiVincenzo et al., 1973; Wigaeus et al., 1981). Uptake also
increases as the level of physical activity increases, i.e. during
exercise, due to increased pulmonary ventilation (Haggard et al.,
1944; DiVincenzo et al., 1973; Wigaeus et al., 1981; Jakubowski &
Wieczorek, 1988). Lungs (including the mouth and trachea) were shown
to retain a greater percentage of inspired acetone (55%) than the
nasal cavity (18%) in two humans exposed to acetone vapour
concentrations of 0.3-3.0 mg/litre and 0.8-11.0 mg/litre for nasal and
lung retention measurements, respectively, with a flow rate of 18
litres/min. This might indicate that the nasal cavity absorbs acetone
less readily than the rest of the respiratory system (Landahl &
Herrmann, 1950). Blood levels of acetone rose rapidly during exposure
for up to 4 h with no indication that a steady state was reached
(DiVincenzo et al., 1973; Brown et al., 1987; Dick et al., 1989),
suggesting that during exposure, the rate of absorption exceeded the
rate of distribution and elimination. During short-term (2-4 h)
exposure to 240 or 1190 mg acetone/m3 (100 or 500 ppm), 75-80% of
the amount of acetone inspired was absorbed by blood after 15 mm of
exposure, and 20-25% remained in the dead space volume (DiVincenzo et
al., 1973). Higher inspired amounts resulted in higher blood levels
(Haggard et al., 1944; Matsushita et al., 1969a; DiVincenzo et al.,
1973; Pezzagno et al., 1986). Pezzagno et al. (1986) similarly
reported that an approximate average of 53% of administered acetone
was retained during exposure of 15 humans to acetone vapour
concentrations of 964-8610 mol/m3 (56-500 mg/m3) for 2-4 h in an
exposure chamber. In contrast to other studies, moderate exercise did
little to change the net retention of acetone. The mean net retention
was 54% during exposure at rest and 53% during exposure with exercise.
A correlation between blood level at the end of exposure and exposure
concentration was found in humans exposed to 55-495 mg/m3 (23-208
ppm) for 24 h (Pezzagno et al., 1986). No significant difference in
uptake or retention was found between men and women (Brown et al.,
1987).
6.1.1.2 Experimental animal studies
Animals also absorb acetone rapidly during inhalation exposure.
Measurement of blood acetone levels in rats after 4-6 h of exposure to
various concentrations shows that blood levels correlate well with
exposure concentrations (Charbonneau et al., 1986a, 1991; NTP, 1988)
and are highest immediately after exposure (NTP, 1988). In rats
exposed to 355 mg/m3 (150 ppm) for 0.5-4 h, measurement of blood
acetone concentrations during exposure revealed that blood levels
increased steadily for 2 h and then remained constant for the next 2 h
of exposure (Geller et al., 1979). Blood acetone levels also
correlated well with exposure concentration in dogs exposed for 2 h
(DiVincenzo et al., 1973). Blood levels were 4, 12 and 25 mg/litre
after exposures to 240, 1190 and 2375 mg/m3 (100, 500 and 1000 ppm),
respectively. In anaesthetized dogs allowed to inhale concentrated
vapour of acetone spontaneously from a respirator at various
ventilation rates, uptake by the respiratory tract was 52% at flow
rates of 5-18 litres/min and 42% at ventilation rates of 21-44
litres/min (Egle, 1973). Retention in the lower respiratory tract was
48% at 5-18 litres/mm and 37.5% at 21-40 litres/min. Retention by the
upper respiratory tract was 57% at 4-18 litres/min. The effect of
exposure concentration on total uptake was studied at a range of
ventilation rates equated with exposure concentrations. Percentage
uptakes were 52.1% at a mean concentration of 503 mg/m3 (212 ppm),
52.9% at 672 mg/m3 (283 ppm), and 58.7% at 1553 mg/m3 (654 ppm).
These results indicate the respiratory uptake of acetone by dogs is
similar to human uptake values reported by Landahl & Herrmann (1950).
Comparison of data from dogs and humans by ATSDR (1994) revealed that
the amount absorbed in humans was greater in absolute quantity under
comparable exposure conditions, but when expressed in terms of kg body
weight, the amount absorbed in dogs was five times more than in
humans. The retention in the upper respiratory tract was higher than
in the lower respiratory tract of dogs (Egle, 1973). Exposure
concentration had little effect on retention. The absorption of
acetone by the nasal walls of anaesthetized dogs, in which the nasal
passage was isolated, increased when the airflow rate was increased
(Aharonson et al., 1974), but the percentage uptake actually
decreased.
In rats exposed continuously to 5250 mg/m3 (2210 ppm) for 9
days, peak acetone blood levels were approximately half the exposure
concentration, were reached in 3-4 days, and remained at this level
for the duration of exposure (Haggard et al., 1944) In rats exposed to
10 200 mg/m3 (4294 ppm) for 12 days, acetone blood levels slightly
greater than half the initial concentration were reached in 4 days and
continued to day 12. Blood levels in rats exposed to these
concentrations for 8 h/day were about half of those reached during
continuous exposure. The amount of acetone absorbed in the first 8 h
exceeded the amount eliminated in the next 16 h of exposure to fresh
air, meaning them was a small accumulation.
Studies with laboratory animals illustrate the absorption of
acetone by the nasal cavity (Morris et al., 1986). Absorption
efficiencies (i.e. relative net uptake) were measured in the upper
respiratory tracts of two strains of rat and a single strain of
guinea-pig. At flow rates approximately 3 times the normal minute
volumes, upper respiratory tract retention of acetone vapour averaged
7, 12 and 21% for guinea-pigs, Fischer-344 rats and Sprague-Dawley
rats, respectively.
As in the case of humans (Landahl & Herrmann, 1950) and dogs
(Egle, 1973), the disposition of acetone in the upper respiratory
tract of other animals, including rats, mice, guinea-pigs and
hamsters, indicates that relatively little acetone is absorbed from
the upper respiratory tract (Morris et al., 1986; Morris & Cavanagh,
1986, 1987; Morris, 1991). The absorption efficiency was greater in
Sprague-Dawley rats than in Fischer-344 rats, but there was no
difference between male and female Sprague-Dawley rats (Morris, 1991).
Deposition was similar in B6C3F1 mice and Fischer-344 rats, and was
greater than in Hartley guinea-pigs and Syrian golden hamster. The
differences between strains and species could not be attributed to
differences in metabolism, because acetone is not significantly
metabolized in the upper respiratory tract of these species. The
reason was thought to be differences in perfusion rates (Morris &
Cavanagh, 1987; Morris, 1991).
6.1.2 Oral exposure
6.1.2.1 Human studies
In a series of experiments conducted in male volunteers given
acetone orally at 40-80 mg/kg, an estimated 65-93% of the administered
dose was metabolized, with the remainder being eliminated in the urine
and expired air in about 2 h, indicating rapid and extensive
gastrointestinal absorption (Haggard et al., 1944). Early research
demonstrated that after oral exposure, acetone moved quickly through
the body. In a human who ingested 137 mg acetone/kg on an empty
stomach, the blood level of acetone rose sharply to a peak 10 min
after dosing (Widmark, 1919). In another example, a subject ingested
the same dose 10 or 12 min after eating porridge. The blood acetone
level rose slowly over 48-59 min to levels of about one-half to
two-thirds that achieved after taking acetone on an empty stomach. The
presence of food in the gastrointestinal tract led to a slower rate of
absorption.
Measurement of acetone in blood and urine of patients who
accidentally or intentionally ingested acetone indicated that acetone
was absorbed, but the percentage absorbed cannot be determined from
the data. A man ingested liquid cement (approximate acetone dose of
231 mg/kg) (Sakata et al., 1989) causing his plasma acetone level to
be approx. 110 mg/litre and his urinary level to be 123 mg/litre 5 h
after ingestion. In another study, a woman who had ingested nail
polish remover had a blood acetone level of 2.5 g/litre (Ramu et al.,
1978). The authors estimated that her body burden was 150 g acetone at
the time of admission to the hospital. The serum acetone level of a
30-month-old child was (4.45 g/litre) 1 h after ingestion of a 6-ounce
bottle of nail polish remover (65% acetone) (Gamis & Wasserman, 1988).
6.1.2.2 Experimental animal studies
Acetone appears to be absorbed by the gastrointestinal tract as
indicated by early studies of laboratory animals. Price & Rittenberg
(1950), administered orally a dose of 14C-acetone (0.22 mg/189 g
body weight, in 1 ml water) to a female rat and collected respiratory
14CO2: for 13.5 h. During this period, 47.4% of the administered
dose was accounted for by exhaled 14CO2. In a separate experiment,
a rat (152 g body weight) was given seven daily oral doses of
14C-acetone (1.08 mg), and radioactivity in carbon dioxide collected
during 24-h post-administration periods accounted for 67-76% of the
administered radioactivity. Radioactive acetone was detected in
expired air in the first 20-min period following the first
administration; however, radioactive acetone, exhaled during 24-h
post-administration periods, accounted for a maximum of 7% of a daily
dose. In an experiment in which a 210 g rat was given 1.30 mg of
radioactive acetone, the rate of exhalation of radioactivity as
acetone was maximal approx. 2 h after administration, but declined
thereafter, suggesting that gastrointestinal absorption was rapid
(Price & Rittenberg, 1950).
Experiments in rats indicate that acetone is rapidly and almost
completely absorbed from the gastrointestinal tract after oral
exposure. A rat expired 97.4% of a 1.16 mg/kg oral dose over a 13.5-h
period, while another rat given 7 mg/kg 14C-acetone expired 67-76%
of the administered dose over a 24-h period after the last dose (Price
& Rittenberg, 1950). From these data, absorption of least 74-83% of
the administered dose can be inferred. In other studies where rats
were given similar or higher doses of acetone, plasma acetone levels
rose proportionately with dose in rats given acetone as single doses
by gavage (Lewis et al., 1984; Charbonneau et al., 1986a) or in the
drinking-water for 7 days (Skutches et al., 1990).
In a study comparing the blood levels of acetone achieved after
fasting with those after oral dosing, peak blood levels of acetone of
approx. 35 and 110 mg/litre were reached within about 3 h after dosing
of rats with 78 and 196 mg acetone/kg, respectively (Miller & Yang,
1984). The levels declined to near background levels within the next
16 h. At an acetone dose of 20 mg/kg, the blood level increased to
about 5 mg/litre over 19 h, when the rats were sacrificed. In rats
fasted for 48 h, blood acetone levels increased continuously to about
13 mg/litre. While the maximal blood concentrations of the treated
rats differed considerably from those of the fasting group, the
calculated areas under the curve for the 78 and 196 mg/kg groups were
comparable to those of the fasting groups (ATSDR, 1994).
There are conflicting data regarding the effect of vehicle on the
gastrointestinal absorption of acetone. In the one study where water
was used as the vehicle, maximum blood levels were higher and achieved
earlier in rats given acetone by gavage as compared to those given
acetone by gavage in corn oil (Charbonneau et al., 1986a). The slower
absorption of acetone in corn oil may have resulted from a delayed
gastric emptying due to the presence of corn oil in the stomach. In a
later repetition of this study, very little difference in blood and
liver levels of acetone were found in rats given the same dose of
acetone in water or in corn oil (Charbonneau et al., 1991).
6.1.3 Dermal exposure
6.1.3.1 Human studies
Dermal absorption of acetone is known to occur in humans.
Application of cotton soaked in acetone to a 12.5 cm2 uncovered area
of skin of volunteers for 2 h/day for 4 days resulted in blood levels
of acetone of 5-12 mg/litre, alveolar air levels of 12-28 mg/m3 (512
ppm), and urinary concentrations of 8-14 mg/litre on each day
(Fukabori et al., 1979). When the daily exposure increased to 4 h/day,
body concentrations more than doubled with 26-44 mg/litre in blood,
59-81 mg/m3 (25-34 ppm) in alveolar air, and 29-41 mg/litre in
urine. The absorption was mediate, with peak blood levels appearing at
the end of each daily application. The authors noted that it was not
possible to completely prevent exposure from inhalation; the acetone
concentration in the breathing zone of one subject was found to be
0.95-1.4 mg/m3 (0.4-0.6 ppm). From the alveolar air and urine
concentrations, it was estimated that a 2-h dermal exposure was
equivalent to a 2-h inhalation exposure to 120-355 mg/m3 (50-150
ppm), and a 4-h dermal exposure was equivalent to a 2-h inhalation
exposure to 590-1190 mg/m3 (250-500 ppm).
6.1.3.2 Experimental animal studies
In a study using isolated perfused pig skin and 14C-labelled
acetone, 60% of the acetone evaporated in the first 2 min, but
approximately 33% remained after 2 h, and around 20% was still present
after 8 h (Williams et al., 1994). The findings of cataract formation
in guinea-pigs exposed dermally to acetone 0.5 ml applied daily for 5
days per week for 6 weeks to the back was attributed to dermal
absorption of acetone (Rengstorff et al., 1972; Rengstorff & Khafagy,
1985). These authors suggested an association between cataract
formation and significant depression of the antioxidant, ascorbate, in
the aqueous humour. To study this relationship in more depth, Taylor
et al. (1993) fed albino hairless guinea-pigs diets containing low
(4.9 mg/day) and high (55 mg/day) levels of ascorbate. Daily dermal
exposure to acetone occurred over the next 6 months, with a total
applied dose of 65 ml. No cataracts were found in any of the
guinea-pigs from either group even though Taylor et al. (1993) used
four times the dose of Rengstorff & Khafagy (1985).
6.1.4. Absorption summary
From the information presented, acetone is absorbed rapidly no
matter which route of exposure. It appears that acetone is absorbed by
all parts of the respiratory system, and that oral and dermal
absorption can also occur. However, absorption can be affected by the
stomach contents.
6.2 Distribution
6.2.1 Inhalation exposure
6.2.1.1 Human studies
Human in vitro tissue-blood partition coefficients of acetone
for cadaver muscle, kidney, lung and brain grey matter range from 0.74
to 0.82, indicating nearly complete tissue distribution
(Fiserova-Bergerova & Diaz, 1986). Dills et al. (1994) measured
in vitro blood/air partition coefficients (KB/A) of acetone in
blood samples from 73 subjects. The mean value was 301 ± 22; the range
was 250-410 and was normally distributed.
6.2.1.2 Experimental animal studies
The distribution of acetone has been studied in mice exposed to
acetone by inhalation (Wigaeus et al., 1982). Mice were exposed to
1190 mg/m3 (500 ppm) 14C-acetone for 1, 3, 6, 12 and 24 h or for 6
h/day for 1, 3 or 5 consecutive days and sacrificed. Radioactive
unmetabolized acetone and total radioactivity were found in blood,
heart, pancreas, spleen, kidney, brain, liver, thymus, testis, vas
deferens, lung, muscle, brown adipose tissue, subcutaneous adipose
tissue and intraperitoneal adipose tissue. Acetone levels generally
peaked from about 145.2 to 203.3 µg/g tissue except in adipose tissues
for total radioactivity. Peak levels of metabolized acetone were
generally < 58.0-75.5 µg/g tissue. Exposure for longer than 6 h
resulted in no further accumulation of total radioactivity except in
the liver and brown adipose tissue, where levels rose to 278.8 µg/g in
the liver and 151.0 µg/g in brown adipose tissue at 24 h. Only about
10% of the radioactivity in the liver at 24 h was unmetabolized
acetone. When file mice were exposed intermittently on 3 or 5
consecutive days, most tissues showed no or only a small additional
increase in radioactivity after more than 1 day of exposure; however,
the concentration in adipose tissue increased significantly with
increasing exposure duration up to 5 days. The ratio of acetone in the
tissues to that in blood was < 1 at all exposure times except for the
lungs (the site of exposure). However, the ratio of total
radioactivity in the tissues to that in the blood showed that after 1
and 3 h exposure, only the lung had a ratio > 1, whereas the ratios
in the kidneys and liver were > 1 after 6 h. Only the muscle and
subcutaneous and intraperitoneal adipose tissue levels rose
continuously. Elimination of acetone was complete in all tissues by
24 h after exposure, but total radioactivity, indicative of
metabolites, was still present in all tissues except blood and muscle.
These data indicate that acetone is not selectively distributed to any
tissues but is more evenly distributed in body water. It appears that
acetone does not accumulate with repeated exposure. The continued
accumulation of radioactivity in the liver and brown adipose tissue is
probably the result of high metabolic turnover in these tissues.
6.2.2 Oral exposure
No studies were located regarding the distribution of acetone or
its metabolites in humans or animals after oral exposure except that
acetone was found in the liver of rats after oral exposure
(Charbonneau et al., 1986a, 1991). Acetone is absorbed from the
gastrointestinal tract and is highly water soluble. Therefore,
distribution to tissues with high water content is to be expected.
6.2.3 Injection exposure
Intravenous injection of 100 mg acetone/kg body weight to
pregnant rats on gestational day 19 resulted in high levels of
1,2-propanediol and acetol in the fetuses (Peinado et al., 1986).
Whether these findings reflect transfer of the metabolites from the
dams or metabolism of transferred or endogenous acetone by the
fetuses was not resolved.
6.2.4 Distribution summary
There are few studies on the distribution of acetone. Since
acetone is highly water soluble, it has been shown to be widely
distributed to tissues with a high water content.
6.3 Metabolism
6.3.1 Human studies
Acetone is a normal product of intermediary metabolism. The
metabolism of acetone appears to be independent of route of
administration and involves at least three separate gluconeogenic
pathways, with ultimate incorporation of carbon atoms into glucose and
other products and substrates of intermediary metabolism with the
generation of carbon dioxide. As previously mentioned, acetone is one
of three ketone bodies produced by acetyl coenzyme A within the liver.
The metabolic pathways appear to be similar in humans and animals. The
primary pathway involves hepatic metabolism of acetone to acetol,
followed by metabolism of acetol to methylglyoxal, while two secondary
(minor) pathways are partially extrahepatic, involving the
extrahepatic reduction of acetol to L-1,2-propanediol. Some exogenous
acetone is unmetabolized and is excreted primarily in the expired air.
Little acetone is excreted in urine.
Metabolic studies in humans were conducted in normal fasted,
obese fasted, and diabetic patients (Reichard et al., 1979, 1986). The
involvement of gluconeogenesis was demonstrated in normal patients
fasted for 3 days, obese patients fasted for 3 days, and obese
patients fasted for 21 days before intravenous injection of
2-[14C]-acetone (Reichard et al., 1979). According to the
researchers, fasted individuals had a daily intake of one multivitamin
capsule and at least 1500 ml of water. The percentages of 14C-glucose
in plasma derived from 14C-acetone were 4.2, 3.1 and 11.0% in the
three respective groups, suggesting the involvement of gluconeogenesis.
Cumulative 14C-carbon dioxide excretion by the lungs during the 6-h
collection period accounted for 17.4, 21.5 and 4.9% in the three
respective groups. Radioactivity was also incorporated into plasma
lipids and plasma proteins. Unmetabolized acetone in the expired air
accounted for 14.7, 5.3 and 25.2%, urinary excretion of acetone
accounted for 1.4, 0.6 and 1.3%, respectively, and in vivo
metabolism accounted for 83, 94.1 and 73%, respectively, of the
radioactivity, Intravenous infusion of 2-[14C]-acetone into patients
with diabetic ketoacidosis resulted in a mean plasma acetone turnover
rate of 6.45 µmol/kg/min (Reichard et al., 1986). Analysis of glucose
in urine revealed a labelling pattern in five of the six patients
consistent with the involvement of pyruvate in the gluconeogenic
pathway, while a different pathway may have been followed in the other
patient. Acetol and 1,2-propanediol were also detected in the plasma
and the concentrations of these metabolites were directly related to
the plasma level of acetone. The results demonstrated high plasma
acetone levels in decompensated diabetic patients with moderate to
severe ketoacidosis. The proposed pathway of acetone metabolism in
these patients is acetone to acetol to 1,2-propanediol to pyruvate and
ultimately to glucose, but other pathways may exist.
Jones & Andersson (1995) reported on a man arrested and suspected
of drunken driving. Both isopropyl alcohol (0.17 mg/ml) and acetone
(0.45 mg/ml) were found in his blood, but one of the breath tests
identified that an interferant was present. The case history showed
that the man was being treated for hyperglycaemia with a controlled
diet.
Jones et al. (1993) examined blood concentrations of acetone in
three groups: drunk drivers, type-1 diabetic outpatients and healthy
blood donors. The median concentrations were 2.03 mg/litre, 1.90
mg/litre and 1.26 mg/litre, respectively. While previous research
indicated that during fasting and uncontrolled diabetes mellitus
acetone increases, Jones et al. (1993) indicated that controlled
diabetes results in near normal acetone concentrations.
Kundu et al. (1993) studied acetone and established a correlation
between rate of fat loss and breath acetone concentration. The study
consisted of 58 men and women, 10-30% above their ideal body weight on
special diets of 1000 and 1200 calories per day. Breath acetone
concentrations were taken immediately upon wakening and increased in
the first few days to a plateau at approx. 7 days.
6.3.2 Experimental animal studies
The metabolism of acetone has been studied extensively in
laboratory animals, primarily in rats, and three separate pathways of
gluconeogenesis have been elucidated (Fig. 1). In many experiments,
rats, mice or rabbits have been exposed by inhalation, gavage,
drinking-water, or by intravenous, subcutaneous or intraperitoneal
injection of non-radiolabelled acetone or acetone labelled with 14C
in the methyl groups, number 2 carbon atom, or all three carbon atoms
(Price & Rittenberg, 1950; Rudney, 1954; Mourkides et al., 1959;
Casazza et al., 1984; Hetenyi & Ferrarotto, 1985; Koop & Casazza,
1985; Johansson et al., 1986; Kosugi et al., 1986a,b; Puccini et al.,
1990; Skutches et al., 1990). In these experiments, identification of
metabolites in liver, plasma or urine, the labelling patterns of 14C
incorporation into metabolites from 14C-acetone in plasma or in
liver, or the results of enzyme reactions using microsomes from
acetone-treated animals have led to the known metabolic pathways
illustrated in Fig. 1. Initially, acetone is oxidized (hydroxylation
of a methyl group) to acetol by acetone monooxygenase, an activity
associated with the cytochrome P-450IIE1, and requiring oxygen and
NADPH (Casazza et al., 1984; Koop & Casazza, 1985; Johansson et al.,
1986; Puccini et al., 1990). Cytochrome P-450IIE1 can be induced by
fasting, experimental diabetes, or exposure to ethanol or acetone
(Patten et al., 1986; Johansson et al., 1988; Puccini et al., 1990).
When the rate of acetone oxidation was evaluated in microsomes with
acetone added to the incubation system, microsomes from rats
(Johansson et al., 1986) and mice (Puccini et al., 1990) pretreated
with acetone had a 7-8 times greater rate than microsomes from control
rats or mice, indicating that acetone induces its own metabolism.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR ACETONE
Members
Dr D. Anderson, British Industrial Biological Research Association
(BIBRA) Toxicology International, Carshalton, Surrey, United Kingdom
Dr Sin-Eng Chia, Department of Community, Occupational and Family
Medicine, National University of Singapore, Faculty of Medicine,
Singapore
Mr J. Fawell, National Centre for Environmental Toxicology, Medmenham,
United Kingdom
Dr L. Fishbein, Fairfax, Virginia, USA ( Chairman)
Dr H. Hansen, Division of Toxicology, Agency for Toxic
Substances and Disease Registry, Atlanta, Georgia, USA
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, United Kingdom ( Co-Rapporteur)
Dr M.V. Park, Edinburgh Centre for Toxicology, Edinburgh, United
Kingdom
Mr D.J. Reisman, National Center for Environmental Assessment, US
Environmental Protection Agency, Cincinnati, Ohio, USA
( Co-Rapporteur)
Dr A. Wibbertman, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany ( Vice-Chairman)
Observers
Dr D. Morgott, Toxicological Sciences Laboratory, Health, Safety and
Environment, Eastman Kodak Company, Rochester, New York, USA
(representing the American Industrial Health Council)
Dr D. Owen, Shell Chemicals Europe Limited, London, United Kingdom
(representing the European Centre for Ecotoxicology and Toxicology of
Chemicals)
Dr P. Montuschi, Department of Pharmacology, Catholic University of
the Sacred Heart, Rome, Italy (representing the International Union of
Pharmacology)
Secretariat
Dr E. Smith, International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR ACETONE
A WHO Task Group on Environmental Health Criteria for Acetone met
at the British Industrial Biological Research Association (BIBRA)
Toxicology International, Carshalton, Surrey, United Kingdom, from 1
to 5 December 1997. Dr S. Jaggers opened the meeting and welcomed the
participants on behalf of the host institute. Dr E. Smith, IPCS,
welcomed the participants on behalf of the Director, IPCS, and the
three IPCS cooperating organizations (UNEP/ILO/WHO). The Task Group
reviewed and revised the draft criteria monograph and made an
evaluation of the risks for human health and the environment from
exposure to acetone.
Mr D.J. Reisman, US Environmental Protection Agency, Cincinnati,
USA, prepared the first draft of this monograph. The second draft,
incorporating comments received following the circulation of the first
draft to the IPCS Contact Points for Environmental Health Criteria
monographs, was also prepared by Mr. Reisman.
Dr E.M. Smith and Dr P.G. Jenkins, both of the IPCS Central Unit,
were responsible for the overall scientific content and technical
editing, respectively.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
* * *
The US Environmental Protection Agency funded the preparation of
this Environmental Health Criteria monograph, financial support for
the Task Group meeting was provided by the United Kingdom Department
of Health, and the meeting was organized by the British Industrial
Biological Research Association (BIBRA).
ABBREVIATIONS
BOD biochemical oxygen demand
CAS Chemical Abstracts Services
DOT/UN/NA/IMCO Department of Transportation/United Nations/North
America/International Maritime Dangerous Goods Code
EINECS European Inventory of Existing Chemical Substances
EPA Environmental Protection Agency
FID flame ionization detector
GC gas chromatography
HPLC high performance liquid chromatography
HRGC high resolution gas chromatography
HSDB Hazardous Substances Data Bank
IC ion chromatography
LOEL lowest-observed-effect level
MS mass spectrometry
NCI National Cancer Institute
NIOSH National Institute for Occupational Safety and Health
NOEL no-observed-effect level
OHM/TADS Oil and Hazardous Materials/Technical Assistance Data
System
ppbv parts per billion (by volume)
RBC red blood cell
RCRA Resource Conservation and Recovery Act
RGD reduction gas detector
RTECS Registry of Toxic Effects of Chemical Substances
TWA time-weighted average
UV ultraviolet
v/v volume per volume
WBC white blood cell
1. SUMMARY
1.1 Properties
Acetone (relative molecular mass = 58.08) is a clear colourless
flammable liquid (flash point -17°C closed cup, -9°C open cup;
flammability limits in air at 25°C = 2.15-13% v/v). The explosive
limits in air are 2.6-12.8% v/v. It has a high evaporation rate
(vapour pressure 181.72 mmHg at 20°C) and a low viscosity (0.303 cP at
25°C). It is miscible with water and organic solvents.
1.2 Uses and sources of exposure
1.2.1 Production
Acetone is manufactured mainly by the cumene peroxidation or
isopropyl alcohol dehydrogenation processes. The cumene peroxidation
process produces trace quantities of benzene as a by-product.
1.2.2 Uses and emissions into the environment
Acetone is used mainly as a solvent and intermediate in chemical
production. Major uses are in the production of methyl methacrylate,
methacrylic acid and higher methacrylates, bisphenol A, methyl
isobutyl ketone, drug and pharmaceutical applications, and as a
solvent for coatings and for cellulose acetate. There are also food
uses as an extraction solvent for fats and oils, and as a
precipitation agent in sugar and starch purification.
Atmospheric emissions occur from consumer products including nail
polish removers, particle board, carpet backing, some paint removers,
and liquid/paste waxes or polishes. Certain detergents/cleansers,
adhesives, and automobile carburetor and choke cleaners also contain
acetone.
Acetone is released into surface water in wastewater effluents
from a wide range of manufacturing processes and industries, such as
paper, plastic, pharmaceuticals, specialty cleaning and polishing
products, paint and allied products, gum and wood chemicals, cyclic
intermediates, industrial organic chemicals, gypsum products, paper
board products, and energy-related industries, such as
coal-gasification and oil shale processing.
Sources of acetone release into soil include disposal of
agricultural and food waste, animal waste, atmospheric wet deposition,
household septic tank effluents and chemical waste disposal sites.
1.3 Environmental transport, distribution and transformation
Acetone released to the atmosphere is degraded by a combination
of photolysis and reaction with hydroxyl radicals. The average
half-life for acetone degradation in the atmosphere is approximately
30 days. Acetone can be physically removed from air by wet deposition.
The dominant degradation process for acetone in soil and water is
biodegradation, and acetone is readily biodegradable. Volatilization
of acetone from the aquatic environment can be a significant transport
process. Acetone is a volatile compound that will evaporate from dry
surfaces. Since acetone is miscible in water, it can leach readily in
most types of soil. Concurrent biodegradation may diminish the general
significance of leaching if biodegradation occurs fast enough.
1.4 Environmental levels and human exposure
Exposure to acetone results from both natural and anthropogenic
sources. Acetone also occurs as a metabolic component in blood, urine
and human breath. It occurs as a biodegradation product of sewage,
solid wastes and alcohols, and as an oxidation product of humic
substances. Acetone has been detected in a variety of plants and foods
including onions, grapes, cauliflower, tomatoes, morning glory, wild
mustard, milk, beans, peas, cheese and chicken breast. Natural
emissions from a variety of tree species contain acetone vapour. Human
sources of emissions to the aquatic environment include waste-water
discharges from many industries and leaching from industrial and
municipal landfills. A major source of human emission to air is
evaporation of acetone solvent from coating products such as paints,
cleaners, varnishes and inks. Acetone is an emission product from the
combustion of wood, refuse and plastics. It is also emitted in exhaust
from automobile, diesel and turbine engines. Concentrations of acetone
monitored in the atmosphere range from 0.5 to 125.4 µg/m3 (0.2-52.9
ppb).
1.5 Kinetics and metabolism
Acetone is one of three ketone bodies that occur naturally
throughout the body. It can be formed endogenously in the mammalian
body from fatty acid oxidation. Fasting, diabetes mellitus and
strenuous exercise increase endogenous generation of acetone. Under
normal conditions, the production of ketone bodies occurs almost
entirely within the liver and to a smaller extent in the lung and
kidney. The process is continuous, and the three products are excreted
into the blood and transported to all tissues and organs of the body
where they can be used as a source of energy. Two of these ketone
bodies, acetoacetate and ß-hydroxybutyrate, are organic acids that can
cause metabolic acidosis when produced in large amounts, as in
diabetes mellitus. Acetone, in contrast, is non-ionic and is derived
endogenously from the spontaneous and enzymatic breakdown of
acetoacetate. Endogenous acetone is eliminated from the body either by
excretion in urine and exhaled air or by enzymatic metabolism. Under
normal circumstances, metabolism is the predominant route of
elimination and handles 70-80% of the total body burden.
Acetone is rapidly absorbed via the respiratory and
gastrointestinal tracts of humans and laboratory animals, as indicated
by the detection of acetone in blood within 30 min of inhalation
exposure and 20 min of oral administration. Studies of rats indicate
that orally administered acetone is extensively absorbed, whereas
during inhalation exposures humans absorb approximately 50% of the
amount of inhaled acetone. However, lower and higher respiratory
absorption values have been reported. The nasal cavities of humans and
laboratory animals appear to have a limited ability to absorb and
excrete acetone vapour, compared with the remainder of the respiratory
tract.
Acetone is uniformly distributed among non-adipose tissues and
does not accumulate in adipose tissues. In mice, maximum acetone
concentrations in adipose tissues were reported to be about one-third
of those in non-adipose tissues following inhalation exposure. Acetone
is rapidly cleared from the body by metabolism and excretion.
Half-times for acetone in human alveolar air and venous and arterial
blood are -4, 6 and 4 h, respectively. Exhalation is the major route of
elimination for acetone and its terminal metabolite (CO2) and the
fraction of administered acetone that is exhaled as unchanged acetone
is dose-related. Urinary excretion of acetone and its metabolites
occurs, but this route of elimination is minor compared with
exhalation of acetone and respiratory CO2.
Exogenously supplied acetone enters into many metabolic reactions
in tissues throughout the body, but the liver appears to be the site
of most extensive metabolism. Carbon from orally administered acetone
has been detected in cholesterol, ammo acids, fatty acids and glycogen
in rat tissues, urea in urine and unchanged acetone and CO2 in
exhaled breath. Metabolically, acetone is degraded to acetate and
formate; this accounts for the entry of carbon from acetone into
cholesterol, fatty acids, urea and amino acids, and formation of
3-carbon gluconeogenic compounds.
Gluconeogenesis from acetone has been proposed to proceed by two
pathways. The first pathway proceeds through the initial catalytic
action of acetone monooxygenase and acetol monooxygenase, which
convert acetone to acetol and acetol to methylglyoxal, respectively.
Both of these enzymatic activities are induced by acetone and have
been identified as an isozyme of ethanol-inducible, hepatic eytochrome
P-450IIE1. The second gluconeogenic pathway involves the formation of
1,2-propanediol from acetone catalysed by acetone monooxygenase and a
non-characterized enzyme capable of converting acetol to
1,2-propanediol.
1.6 Effects on laboratory mammals and in vitro systems
Oral LD50 values in adult rats are in the range of 5800-7138
mg/kg. The 4-h inhalation LC50 value is 76 000 mg/m3 (32 000 ppm).
Acute exposure to acetone has been found to alter performance in
neurobehavioural tests in laboratory animals at concentrations greater
than 7765 mg/m3 (>3270 ppm).
Experimental animal data characterizing the effects of long-term
oral or inhalation exposure to acetone are not available, due probably
to its low toxicity and its endogenous characteristics.
Prolonged acetone inhalation exposure of rats to 45 100 mg/m3
(19 000 ppm), 3 h/day, 5 days/week for 8 weeks, produced a reversible
decrease in absolute brain weight. No consistent changes were noted in
weights of other organs or the whole body, in blood chemical indices,
in liver triglyceride levels or in the histology of the heart, lung,
kidney, brain or liver.
In a 90-day gavage study of rats, increased blood parameters
(increased haemoglobin, haematocrit) were observed at dose levels
>500 mg/kg per day, and a NOAEL of 500 mg/kg per day was identified.
In a 13-week drinking-water study, toxic effects were noted in male
rats exposed to concentrations >20 g/litre (approx. 1700 mg/kg body
weight per day), namely increased relative organ weights, altered
haematological indices and mild nephropathy. In female rats
administered the highest concentration, 50 g/litre (approx. 3400 mg/kg
body weight per day), the effects were increased organ relative
weights and altered haematological indices. In addition, a 13-week
exposure to 50 g/litre caused altered relative testis weight and
altered sperm motility and morphology in male rats. Female mice given
50 g/litre (approx. 11 298 mg/kg body weight per day) in
drinking-water had altered liver and spleen weights and a marginally
increased incidence of centrilobular hepatocellular hypertrophy. No
toxic effects were observed in male mice administered 20 g/litre (4858
mg/kg body weight per day), the highest acetone level administered to
male mice. Thirteen-week exposures to concentrations < 10 g/litre
(900 mg/kg body weight per day) in drinking-water were associated with
no toxic effects in male rats; concentrations < 20 g/litre were
NOELs for female rats (1600 mg/kg body weight per day) and mice (male
4858 mg/kg body weight per day; female 5945 mg/kg body weight per day)
of both sexes.
In a preliminary 14-day drinking-water study of rats and mice,
dose-related centrilobular hepatocellular hypertrophy was noted in
male mice exposed to concentrations of 20-100 g/litre.
Pretreatraent of rodents with acetone enhances the hepatotoxic
effects of a number of compounds, notably halogenated alkanes, It is
hypothesized that the potentiation of the hepatotoxicity is mediated
by acetone-induced elevations of enzymatic activities (hepatic
mixed-function oxidases) that are responsible for the generation of
toxic intermediates from administered halogenated alkanes.
Acetone has tested negatively for genetic toxicity in numerous
non-mammalian systems, as well as in in vitro and in vivo
mammalian systems. Positive results are restricted to a single test
for aneuploidy in a yeast species exposed to high concentrations of
acetone (6.82%) in its growth medium. Acetone is not considered to be
genotoxic or mutagenic.
In a study of pregnant rats and mice exposed to acetone vapour
during days 6-19 of gestation, slight developmental toxicity was
observed following exposures of rats to 26 100 mg/m3 (11 000 ppm) for
6 h/day (increased percentage of litters with at least one fetal
malformation) and following exposures of mice to 15 670 mg/m3 (6600
ppm) for 6 h/day (small decrease in fetal weight and small increase in
percentage incidence of late resorptions). An atmospheric
concentration of 5200 mg/m3 (2200 ppm) was identified as a NOAEL for
developmental toxicity in both mice and rats. In a gavage study,
treatment at 3500 mg/kg per day during organogenesis impaired
reproduction in a screening test in mice. Negative results in vivo
in two different species, using oral and intraperitoneal routes,
indicated that no mutagenic changes were produced in mammals exposed
to acetone.
Reports of other reproductive effects of acetone include
observations of testicular effects and changes in sperm quality in
rats administered drinking-water containing 50 g acetone/litre for 13
weeks. No investigations of the effect of oral doses of acetone on
fetal development (fetotoxicity and teratogenicity) were available.
Acetone has been used extensively as a solvent vehicle in skin
carcinogenicity studies and is not considered carcinogenic when
applied to the skin.
1.7 Effects on humans
Acetone is relatively less toxic than many other industrial
solvents; however, at high concentrations, acetone vapour can cause
CNS depression, cardiorespiratory failure and death. Acute exposures
of humans to atmospheric concentrations as high as approx. 4750 mg/m3
(approx. 2000 ppm) have been reported to produce either no gross toxic
effects or minor transient effects, such as eye irritation. More
severe transient effects (including vomiting and fainting) were
reported for workers exposed to acetone vapour concentrations >25 500
mg/m3 (>12 000 ppm) for approx. 4 h. Acute exposures to acetone have
also been reported to alter performance in neurobehavioural tests in
humans at 595 mg/m3 (250 ppm). Females exposed to atmospheric
concentrations of 2370 mg/m3 (1000 ppm) were reported to suffer
menstrual irregularities.
1.8 Effects on other organisms in the laboratory and field
For most freshwater and saltwater animal species, 48- and 96-h
LC50 and EC50 values are >5540 mg/litre.
Growth of the alga Chlorella pyrendoidosa exposed to acetone at
257.4 mg/litre for 76 h was inhibited. There was inhibition of growth
of Chlamydomonas eugametos exposed to acetone for 48 h at 790
mg/litre. Photosynthesis was increased in Scendesmus quadricauda and
C. pyrenoidosa exposed to 79.0 and 790 mg/litre.
The 7- to 8-day toxicity thresholds for the green alga
S. quadricauda and the cyanobaeterium (blue-green alga)
Microcystis aeruginosa were 7500 and 530 mg/litre, respectively,
indicating that the green alga was more resistant to the toxic action of
acetone. The diatom Nitzschia linearis also seemed very resistant,
with a 5-day EC50 of 11 493 to 11 727 mg/litre. Similarly, the saltwater
diatom Skeletonema costatum was very resistant with 5-day EC50
values of 11 798 and 14 440 mg/litre.
Bacteria appear more resistant to acetone than protozoans.
Photobacterium phosphoreum, Pseudomonas putida and a mixed
microbial culture had EC50 values of 1700 to 35 540 mg/litre, and the
protozoan Entosehon sulcatum had an EC50 of 28 mg/litre. This may
be related to cell wall differences.
Quails and pheasants had oral 5-day LC50 values > 40 g/kg
diet. Fertile mallard eggs were not affected when immersed in 10%
acetone for 30 seconds; however, immersion in 100% acetone resulted in
decreases in survival, embryonic weight and embryonic length, but it
is not clear if this was due to the toxic or the solvent properties of
acetone. White Leghorn chicken eggs injected with 5 µl acetone did not
appear to have any significant changes in mortality or malformations.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Chemical identity
Chemical name acetone
Synonym(s) dimethyl ketone; 2-propanone;
beta-ketopropane
Chemical formula C3H6O
Chemical structure
O
"
H3C - C - CH3
Identification numbers:
CAS registry 67-64-1
NIOSH RTECS AL3150000
EPA Hazardous Waste
(RCRA) U002; F003
OHM/TADS 7216568
DOT/UN/NA/IMCO shipping UN1090
HSDB 41
EINECS 200-662-2
Relative molecular mass 58.08
2.2 Physical and chemical properties
2.2.1 Physical properties
Acetone is a clear and colourless liquid with a strong "fruity"
odour. It is miscible with water and organic solvents such as ether,
methanol, ethanol and esters (Nelson & Webb, 1978). The physical
properties of acetone, such as high evaporation rate, low viscosity
and miscibility, make it suitable for use as a solvent (Krasavage et
al., 1982). The physical properties of acetone are shown in Table 1.
2.2.2 Chemical properties
Acetone shows reactions typical of saturated ketones (SRI, 1996).
These reactions include addition, oxidation-reduction and
condensation, and yield alcohols, ketals, acids and amines (Papa &
Sherman, 1981). The chemical reactivity of acetone is commercially
important for the synthesis of methyl methacrylate, diacetone alcohol,
bisphenol A and other derivatives (SRI, 1996).
Table 1. Physical and chemical properties of acetone
Property Value/descriptiona Reference
Relative molecular mass 58.08 Riddick et al, (1986)
Colour Clear colourless Sax & Lewis (1987)
Physical state Liquid Sax & Lewis (1987)
Melting point -95.35°C Weast (1987}
Freezing point -94.7°C at 1 atm Riddick et al. (1986)
Boiling point 56.2°C at 1 atm Weast (1987)
(760 torr)
Density:
at 20°C 0.78996 g/ml Riddick et al. (1986)
at 26°C 0.78440 g/ml Riddick et al. (1966)
at 30°C 0.78033 g/ml Riddick et al. (1986)
Odour threshold:
Acetone in water 20 mg/litre Amoore & Hautala
(1983)
Air (absolute) 30-48 mg/m3 Amoore & Hautala
(13-26 ppm (v/v)) (1983)
Air (detection) 9.5 mg/m3 (4 ppm) Wysocki et al. (1997)
100% odour recognition 237-332 mg/m3 Hellman & Small
(100-140 ppm) (1974); Leonardos et
al. (1969)
Table 1 (contd).
Property Value/descriptiona Reference
Solubility:
Water at 20°C Completely miscible Windholz (1963)
Organic solvent(s) Soluble in organic
solvents Windholz (1983)
Viscosity at 25°C 0.303 cP Riddick et al. (1986)
Partition coefficients:
Log Kow -0.24 Sangster (1989)
Log Koc 0.73b Lyman (1982)
KB/A 301 ± 22 Dills et al. (1994)
Vapour pressure 181.72 mmHg (at 20°C) Riddick et al. (1986)
231.06 mmHg (at 25°C) Riddick et al. (1986)
Henry's law constant: 4.26 × 10-5 atm-m3/mol Rathbun & Tai (1987)
at 25°C
Flashpoint (closed cup) -17°C Riddick et al. (1986)
(open cup) -9°C Riddick et al. (1986)
Flammability limits Lower, 2.2%; Clayton & Clayton
in air at 25°C upper, 13.0% (1982)
Minimum ignition 465°C Riddick et al. (1986)
temperature
Explosive limits Lower, 2.6% in air (v/v); Sax & Lewis (1987)
upper, 12.8% in air (v/v) Sax & Lewis (1987)
a w/v = weight per volume, v/v = volume per volume.
b Estimated by regression equation 4-13 in Lyman (1982).
2.3 Conversion factors
Conversion factors in air at 25°C:
1 ppm = 2.374 mg/m3
1 mg/m3 = 0.421 ppm
2.4 Analytical methods
A number of analytical methods is available for the detection,
sampling and monitoring of acetone and its metabolites in the various
media. Acetone is a well-studied chemical and is used frequently in
the laboratory. This section is a review of the more established and
standard practices in use today.
2.4.1 Biological media
Methods for determining the presence of acetone in biological
organisms are listed in Table 2. Acetone is found in almost every
tissue and organ in the human body. Acetone and two other chemicals,
beta hydroxybutyrate and acetoacetate, are collectively referred to as
"ketone bodies". In the last 30 years much has been learned of acetone
in biological tissue since the discovery that acetone levels in
diabetes mellitus patients with severe hyperketonaemia may be
significant (Trotter et al., 1971). Higher acetone levels may be found
in the blood levels of individuals or animals after strenuous exercise
or prolonged dieting. Acetone production is also increased in animals
in disease states such as diabetes and anorexia.
The development of biological analytical methods can be done to
measure, but this does not distinguish acetone from either endogenous
and exogenous sources or from acetone in ketone levels in body fluids,
since acetone is produced within the biological system by breaking
down lipids and stored fats. Most of the methods for measuring acetone
in expired air use gas chromatography (GC/FID) and involve the
breakdown of beta-hydoxybutyrate and acetoacetate into acetone, which
is isolated and quantified by any of the techniques listed in Table 2.
The differences between these methods have been mainly concerned with
the nature of the column packing and with the various methods of
sample collection.
The determination of acetone in blood is difficult because it is
a metabolite and the quantity produced depends on storage time, even
when the blood samples are stored at 4°C (Trotter et al., 1971). The
delay between sample collection and analysis could lead to spuriously
elevated acetone concentrations because of the spontaneous
decarboxylation of acetoacetate (Van Stekelenburg & Koorevaar, 1972).
One method for the determination of acetone in the clinical laboratory
involves deproteinizing with acetonitrile and derivitization of the
sample with 2,4-dinitrophenylhydrazine, followed by isolation and
quantification of the hydrazone by high pressure liquid chromatograph
Table 2. Analytical methods for determining acetone in biological media
Sample matrix Preparation method Analytical Sample detection Reference
method limit
Whole blood, urine Centrifuged and deproteinized GC-HPLC 33 µg/ml Gavino et al. (1986)
with acetonitdle and 2,4-DNPH
added
Whole blood Deproteinized with HClO4 and GC-FID 0.4 µmol/litre Mangani & Ninfali
subjected to purge-and-trap (1988)
Whole blood Purge-and-trap GC-MS 0.2 µg/ml Ashley et al. (1992)
Serum Deproteinized with sodium HRGC-FID <58 µg/ml Smith (1984)
tungstate and cupric sulfate (<1 nmol/litre)
Serum Sample centrifuged and clear GC-FID 5.8 mg/ml Cheung & Lin
filtrate injected (0.1 nmol/ml) (1987)
into GC
Urine Diluted sample derivatized with GC-FID 0.2 µg/ml Kobayashi et al.
pentafiuorobenzyloxyl ammonium (3.45 µmol/ml) (1983)
chloride and extracted with
hexane
Liver perfusate, Reduction to isopropanol using GC-HPLC 33 µmol/ml Gavino et al. (1987)
blood, urine sodium borohydrid and
separation by HPLC
Liver Liver perfusion medium reduced GC-FID 3.78 µg/ml in Holm & Lundgren
with NaBH4 and an aliquot of perfusate (1984)
reduced solution injected (65 µmol/litre)
into GC
Liver, kidney, lung Purge-and-trap GC-FID No data Holm & Lundgren
and adipose tissue (1984)
Breath Direct injection into GC GC-FID No data Trotter et al. (1971);
Jansson & Larsson (1969)
(HPLC) (Brega et al., 1991). This method prevents acetoacetate, which
is present in plasma, from being thermally degraded to acetone on the
column when using a GC method (Gavino et al., 1987). The HPLC method
can also be used to measure acetone in urine or liver perfusate. This
method can be used in experiments requiring multiple samples and thus
can be used for diabetic patient monitoring, as well as for
occupational exposure monitoring.
2.4.2. Environmental media
Analytical methods for determining acetone in air, water and soil
are presented in Table 3. The commonly used methods are direct GC/MS
of a sample concentrate or HPLC of the 2,4-dinitrophenyl-hydrazine
derivative. In the United Kingdom, the 2,4-dinitrophenyl-hydrazine
HPLC method is applied to the analysis of acetone in water and there
is a standardized validated method (UK SAC, 1988).
When sampling for acetone, the incorrect use of Tedlar bags and
activated carbon may lead to spurious results.
Table 3 Analytical methods for determining acetone in environmental samples
Sample matrix Preparation method Analytical method Sample detection Reference
limit
Air (occupational) Air passed through charcoal and components GC-FID (NIOSH 7 µg/litre NIOSH (1994)
desorbed with CS2 method 1300)
Air Air passed through a cryogenic trap and the GC-RGD 10 ppt O'Hara & Singh
(ambient) trapped component injected into GC (1988)
Air Air passed through HPLC-UV <3 µg/litre Risner (1995)
(ambient) 2,4-dinitrophenylhydrazine-coated cartridge and eluted reversed-phase
with acetonitrile and tetrahydrofuran column
Air (indoor) Diffusive sampler with silica gel tape impregnated with HPLC-UV 15 µg/litre Brown et al.
2,4-dinitrophenylhydrazine and eluted with acetonitrile (1994)
Air Air passed through a 1% sodium bisulfite solution and Spectrophotometry <0.5 ppm (in Amlathe & Gupta
absorbed acetone reacted with alkaline vanilla solution solution) (1990)
Rural air Air passed through silica gel coated with HPLC-UV No data Shepson et al.
2,4-dinitrophenylhydrazine and eluted with acetonitrile (1991)
Water Sample reacted with alkaline diazotized anthranilic Spectrophotometry 500 µg/litre Rahim & Bashir
acid solution (1981)
Fresh and seawater Sample derivatized with 2,4-dinitrophenylhydrazine HPLC-UV detection 0.5 nmol/litre Kieber & Mopper
passed through a C18 cartridge and absorbed (0.03 µg/litre) (1990)
compound eluted with acetonitrile
Waste water, soil or Sample or sample mixed with reagent water subjected GC-MS 100 µg/litre US EPA (1986b)
sediment to purge-and-trap (EPA method 8240) (water)
100 µg/k9
(sediment and
soil)
Fresh fruit Vacuum distillation followed by solvent extraction HRGC-MS No data Takeoka et al.
of pulp (1988)
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Acetone occurs as a metabolic component in blood, urine and human
breath (Conkle et al., 1975). Because endogenous acetone formation is
so closely linked with the utilization of stored fats as a source of
energy, background levels can fluctuate depending on an individual's
health, nutrition, and level of activity (Morgott, 1993). The acetone
level in the human body at any instant is reflective of acetoacetate
production and ketogenesis. It occurs naturally as a biodegradation
product of sewage, solid wastes and alcohols and as an oxidation
product of humic substances. Acetone has been detected in a variety of
plants and foods, including onions, grapes, cauliflower, tomatoes,
morning glories, wild mustard, milk, beans, peas, cheese and chicken
breast (Day & Anderson, 1965; Grey & Shrimpton, 1967; Palo & Ilkova,
1970; Lovegren et al., 1979). Natural emissions from a variety of tree
species contain acetone vapour (Isidorov et al., 1985) and another
source is direct emission from the ocean (Zhou & Mopper, i990).
3.2 Anthropogenic sources
There are many anthropogenic sources of acetone, with various
levels and concentrations that cover a broad range. Human sources of
emissions to the aquatic environment include wastewater discharges
from many industries (Perry et al., 1978; NLM, 1992) and leaching from
industrial and municipal landfills (Sabel & Clark, 1984; Brown &
Donnelly, 1988). A major source of emission to the air is from
evaporation of acetone solvent from coating products such as paints,
cleaners, varnishes and inks. Acetone is an emission product from the
combustion of wood, refuse and plastics (Lipari et al., 1984; Graedel
et al., 1986), and is emitted in exhaust from automobile, diesel and
turbine engines (Barber & Lodge, 1963; Lloyd, 1978; Jonsson ct al.,
1985; Graedel et al., 1986; Westerholm et al., 1988; Zweidinger et
al., 1988).
Other important anthropogenic sources of acetone in the air are
chemical manufacture (Graedel et al., 1986), tobacco smoke (Manning et
al., 1983), wood burning and pulping (Lipari et al., 1984; Graedel et
al., 1986; Kleindienst et al., 1986), polyethylene burning (Hodgkin et
al., 1982), refuse combustion (NAS, 1976), petroleum production
(Graedel, 1978), and certain landfill sites (LaRegina et al., 1986;
Militana & Mauch, 1989; Hodgson et al., 1992). Acetone is formed in
the atmosphere from the photochemical oxidation of propane (Singh &
Hanst, 1981; Arnold et al., 1986) and possibly from propylene oxide
and epichlorohydrin (Spicer et al., 1985).
In a US EPA-sponsored survey of household products analysed by
purge-and-trap GC/MS for volatile organic compounds, acetone was found
in 314 of 1005 products (31.2%). Of the eight product categories, the
highest categories were paint-related (51.5% contained acetone),
adhesive-related (24.3%) and automotive (22.7%) products (Sack et al.,
1992).
3.2.1 Production levels and processes
In 1994, world acetone capacity amounted to almost 3.83 million
tonnes (SRI, 1996). Since approximately 80% of acetone is produced as
a co-product of phenol, demand for phenol largely determines acetone
production levels. World production in 1994 was estimated to be 3.22
million tomes, and demand for acetone was expected to grow at an
average annual rate of 3.3% annually from 1994 to 1999 (SRI, 1996).
The USA is the largest producer of acetone. Table 4 depicts the
capacity of the largest manufacturers in the USA in 1995, while Table
5 shows the capacity of other countries. The annual capacity in the
European Union in 1992-1994 was 1.1-1.2 million tonnes.
Most acetone is manufactured by one of two processes, cumene
peroxidation (94% yield) or isopropyl alcohol dehydrogenation (IPA)
(95% yield) (SRI, 1996). In the peroxidation process, cumene is
oxidized to hydroperoxide, which is cleaved to yield acetone and
phenol. In the dehydrogenation process, isopropyl alcohol is
catalytically dehydrogenated to yield acetone and hydrogen (Nelson &
Webb, 1978). The cumene peroxidation process accounts for 96%; IPA
accounts for the other 4%. Production grade acetone is 99.5% acetone,
0.5% water. Fermentation of corn starch and molasses to produce
acetone, using Clostridium acetobutylicium, is utilized in several
countries, including Russia, Egypt, Brazil and India (Sifniades,
1985). Although acetone is more costly to produce by the IPA process,
this process has no benzene contamination. Acetone produced through
the cumene process contains benzene at concentrations < 10 ppm
(SRI, 1996).
Some companies recover acetone as a by-product (SRI, 1996). For
example, in the United Kingdom there is a plant producing 52 000
tonnes per year that recovers acetone as a by-product of acetic acid
manufacture, and two Japanese manufacturers recover acetone from
cresol production.
3.2.2 Uses
Acetone is used primarily as an intermediate in chemical
production and as a solvent (SKI, I996). It is used as a solvent for
resins, paints, inks, varnishes and lacquers and in adhesives,
thinners and clean-up solvents. Pharmaceutical applications of acetone
include use as an intermediate and solvent for drags, vitamins and
cosmetics (Nelson & Webb, 1978). It has uses as an extraction solvent
for fats and oils and a precipitation agent in the purification of
starches and sugars (FAO/WHO, 1971).
Table 4: Major manufacturers of acetone in the USA in 1995a
Manufacturer Location Annual capacity
(thousands of tonnes)
Allied Signal, Inc, Philadelphia, PA 280
Aristech Chemical Corp. Ironton, OH 180
Dow Chemical USA Oyster Creek, TX 161
Eastman Chemical Co. Kingsport, TN 13
Mt. Vernon Partnership Mount Vernon, IN 191
Georgia Gulf Corp. Pasadena, TX 45
Plaquemine, LA 123
Goodyear Tire & Rubber Co. Bayport, TX 7
JLM Chemicals, Inc. Blue Island, IL 26
Shell Chemical Co. Deer Park, TX 182
Texaco, Inc. El Dorado, KS 26
Union Carbide Corp. Institute, WV 77
Total 1281
a SRI (1996)
Table 5: Production capacity of acetone in 1995 (excluding the USA)a
Country Annual capacity
(thousands of tonnes)
Germany 388
Italy 235
France 168
United Kingdom 97
Netherlands 80
Spain 75
Brazil 71
Finland 65
Mexico 22
Argentina 20
Venezuela 10
TOTAL 1185
a SRI (1996)
In 1995 the USA use pattern for acetone was as follows: acetone
cyanohydrin/methyl methacrylate, methacrylic acid and higher
methacrylates (45%); solvent applications (17%); bisphenol A (18%);
aldol chemicals/methyl isobutyl ketone and others (12%); and
pharmaceutical and other applications (8%) (SKI, 1996).
The largest solvent application for acetone is as a surface
coating, including use as a thinner and wash solvent. In 1995,
greatest use of acetone as a solvent was in automotive coatings, both
original equipment and automotive refinishing (SKI, 1996). The next
greatest use for acetone is the production of acetone cyanohydrin
which is used to produce an acrylic resin monomer, methyl
methacrylate. Bisphenol A is produced from acetone and used in
polycarbonate resins.
Acetone is also used in food processing as an extraction solvent
for oils and fats and as a precipitation agent in the purification of
starches and sugars.
3.2.3 Releases
3.2.3.1 Air
Atmospheric emissions are likely from the many consumer products
containing acetone (US EPA, 1989). Such products include nail polish
removers, particle board (Tichenor & Mason, 1988), carpet backing
(Hodgson et al., 1993), some paint removers, a number of liquid/paste
waxes or polishes, some detergents/cleansers, adhesives (Knöppel &
Schauenburg, 1989; Sack et al., 1992) and carburetor and choke
cleaners (US EPA, 1989).
Atmospheric emissions from the phenol/acetone production process
are approximately 0.44 g per kg of acetone produced (Sifniades, 1985).
3.2.3.2 Water
Acetone is released into surface water as wastewater from certain
chemical manufacturing industries (Jungclaus et al., 1978; Hites &
Lopez-Avila, 1980; Gordon & Gordon, 1981). It is also released in
water from energy-related industries, such as coal-gasification
(Pellizzari et al., 1979; Mohr & King, 1985) and oil shale processing
(Pellizzari et al., 1979; Hawthorne & Sievers, 1984). Acetone was
found in 27 of 63 effluent water samples from a wide range of chemical
industries in the USA (Perry et al., 1979). It has been detected in
effluents from various industrial production processes including
paper, plastic, pharmaceutical, specialty cleaning and polishing
products, paint and allied products, gum and wood chemicals, cyclic
intermediates, industrial organic chemicals, gypsum products, and
paper board products.
Acetone can be released to groundwater as a result of leaching
from municipal and industrial landfills (Gould et al., 1983; Steelman
& Ecker, 1984; Sawhney & Raabe, 1986; Brown & Donnelly, 1988). It may
also leach from solvent cement used in joining polyethylene and other
plastic pipes used in drinking-water distribution and domestic
plumbing (Anselme et al., 1985). One of the sources of acetone in
seawater is the sensitized photoreaction of dissolved organic matter
(Mopper & Stahovec, 1986).
3.2.3.3 Soil
Acetone leaches readily in soil. The US Agency for Toxic
Substances and Disease Registry (ATSDR, 1994) found the amount of
acetone released into soil from landfills in the USA accounted for
approximately 0.1% of the total environmental release of acetone.
Sources of acetone release into soil include disposal of agricultural
and food waste, animal wastes, and atmospheric wet deposition. Acetone
was detected in 43% of the soil from designated waste disposal sites
tested for acetone. Household septic tank effluents are another source
of acetone in soil (DeWalle et al., 1985).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution among media
Acetone is commonly found in air, water, soil and biological
samples, and these background levels can he from both human-made and
natural sources. Acetone occurs naturally in trees, plants, forest
fires and volcanic gases. When animals and humans catabolize body fat,
acetone is exhaled and metabolized. Human-made sources include tobacco
smoke, combustive engine exhaust and waste incineration. The exchange
of carbonyl compounds (including acetone) between air and natural
waters is governed by the appropriate partition coefficients, in
addition to production and loss processes in both media (Benkelberg et
al., 1995).
4.1.1 Air
The significant environmental fate processes for the degradation
of acetone in the ambient environment are photolysis and reaction with
hydroxyl radicals (Meyrahn et al., 1986; Kerr & Stocker, 1986).
Meyrahn et al. (1986) measured the quantum yields of acetone
photolysis at environmental wavelengths and projected the following
rate constants for the lower troposphere at 40°N latitude: in January,
3.3 × 10-8/sec; in July, 1.8 × 10-7/sec; yearly average,
1.0 × 10-7/sec. These rate constants correspond to half-lives of
243, 45 and 80 days for January, July and the yearly average,
respectively. These rate constants intentionally neglect reaction of
excited acetone molecules with oxygen. Based on the photodecomposition
data of Gairdner et al. (1984), the rate constants of Meyrahn et al.
(1986) would be about twice as great if the neglected reaction were
included. Using this factor of 2, the total yearly average photolysis
half-life (plus reaction of excited acetone molecules) is about 40
days. The rate constant for the reaction of hydroxyl radicals with
acetone at 25°C is in the range of 2.2-2.6 × 10-13 cm3/molecule-sec
(Kerr & Stocker, 1986; Wallington & Kurylo, 1987). Probable pathways
for the reaction of acetone with hydroxyl radicals in the troposphere
have been postulated, and methyl-glyoxal is the primary product of
this reaction (Altshuller, 1991). The primary products of acetone
photolysis in sunlight are carbon dioxide and acetylperoxynitrate
(Altshuller, 1991). The photochemical oxidation of acetone in the
presence of nitrogen oxides produces small amounts of peroxyacetic
acid and peroxyacetyl nitrate (Hanst & Gay, 1983).
The photolysis lifetimes of acetone under cloudless conditions at
40°N latitude, and at sea level during winter and summer were
estimated to be 83 and 19 days, respectively (Martinez et al., 1992).
Other investigators have estimated that the average atmospheric
lifetime of acetone due to photolysis at 40°N latitude is 80 days/year
and varies from 243 in January to 45 in July (Meyrahn et al., 1986).
Meyrahn et al. (1986) estimated the average lifetime of acetone at
40°N due to combined hydroxyl radical reaction and photolysis to be 32
days/year, corresponding to a half-life of approx. 22 days. The
decomposition rate showed a pronounced dependence on latitude, with
greater losses of acetone occurring near the equator compared to the
poles. In very polluted air, the hydroxyl radical concentration
increased by an order of magnitude, which would lower the half-life by
an order of magnitude.
The complete miscibility of acetone in water suggests that
physical removal from air by wet deposition (rainfall, dissolution in
clouds, etc.) is probable (Aneja, 1993). The reactions of acetone
vapour with nitrogen oxides, hydroxyl radicals (OH), singlet molecular
oxygen (1 Delta g), singlet atomic oxygen (O(3P)), and nitrate
radicals have been studied. Given the second order rate constants for
the reactions of acetone with 1 Delta g (Datta & Rao, 1979) and O(3P)
(Lee & Timmons, 1977), and the concentrations of singlet molecular and
atomic oxygen in the atmosphere (Graedel, 1978), these reactions are
insignificant in determining the fate of acetone in the atmosphere.
However, Grosjean & Wright (1983) detected acetone in rain, cloud,
mist and fog water that was collected in Southern California, USA. In
certain instances, physical removal by wet deposition may be
environmentally significant, especially since the degradation rate is
not very fast. The reaction of acetone with nitrate radicals in the
atmosphere was also determined to be insignificant (Boyd et al.,
199l). Smog chamber studies with acetone and nitrogen oxides have
shown that acetone has low reactivity in terms of ozone and nitrogen
dioxide formation and that the rate of disappearance of acetone by
this process is low (Altshuller & Cohen, 1963; Dimitriades & Joshi,
1977).
Using 72-h back trajectories, Aneja (1993) studied organic
compounds transported in cloud water whose origin was an industrial
valley. Acetone was found in cloud water at an average of 460 ng/litre
(range 0-4100 ng/litre), in clouds of low pH (2.78).
4.1.2 Water
The miscibility of acetone in water and the estimated low value
of 0.73 for log Koc (see Table 1) suggests that adsorption of acetone
to sediments and suspended solids is not significant. When water is
not present, acetone vapour adsorbs rather strongly to the clay
component of soil by hydrogen bonding (Goss, 1992; Steinberg &
Kreamer, 1993). The sorption is inversely dependent on relative
humidity, so increasing the humidity decreases sorption drastically.
In water-saturated soil or sediment, Koc values (organic carbon), and
not hydrogen bonding, may control the sorption of acetone (Steinberg &
Kreamer, 1993). The experimental adsorption studies with Kaolinite,
montmorillonite, and stream sediments showed very little or no loss of
acetone from water to the adsorbents (Rathbun et al., 1982).
The transport of acetone from the water column to the atmosphere
depends on the Henry's law constant. The Henry's law constant for
acetone is 4.26 × 10-5 atm-m3/mol (see Table 1), which suggests that
volatilization of acetone from water, although not very fast, could be
significant (Thomas, 1982), and likely to be important in determining
the fate of acetone in streams (Rathbun et al., 1982). The
volatilization rate of a chemical depends on the characteristics of
the chemical and the presence of water, and on other ambient
conditions (e.g., water depth, suspended solid concentration, water
current, wind speed, temperature). Based on an estimation method
(Thomas, 1982) and the Henry's law constant value, the volatilization
half-life of acetone from a model river 1 m deep, flowing at a current
of 1 m/second with a wind velocity of 3 m/sec is between 18 and 19 h.
The mean volatilization coefficient for acetone in a model outdoor
stream was found to be in the range of 7.15 × 10-4 to 14.8 × 10-4/min
(Rathbun et al., 1989, 1991). Therefore, the volatilization half-life
of acetone from the model stream is in the range of 8-16 h. It was
concluded that volatilization will control the fate of acetone in
water (Rathbun et al., 1989, 1991). Using a computer simulation model
the volatilization half-life from a model pond (2 m deep) was
estimated to be around 9 days.
The average of four experimentally determined rate constants for
the reaction of acetone with hydroxyl radicals in water (pH 6-7) is
1.1 × 10-8 litres/mol-sec (Buxton et al., 1988). Assuming the
hydroxyl radical concentration in brightly sunlit natural water is 1.0
× 10-17 mol/litre, the half-life for the reaction is almost 20 years.
Thus, photo-oxidation reactions of acetone in environmental waters do
not appear to be a significant removal process. Also, photolysis of
acetone in water, based on a rate constant for the reaction of acetone
with hydroxyl radicals in water at pH 7 of 5.8-7.7 × 107
litres/mol-sec and a concentration of hydroxyl radicals in eutrophic
waters of 3 × 10-17 M (Mill & Mabey, 1985), will not be significant.
Rathbun & Tai (1982) measured the mass transfer coefficient (KL for
acetone in water and reported values ranging from 0.310 to 0.537. When
distilled water or natural water containing acetone was exposed to
sunlight for 2-3 days, no photodecomposition of acetone was observed
(Rathbun et al., 1982). Experimental hydrolysis data for acetone have
not been found in the available literature. However, ketones generally
resist aqueous environmental hydrolysis (Harris, 1982) and hydrolysis
of acetone is not expected to be significant in the environment.
Bacterial degradation of acetone occurs, and the rate is
increased if acclimatization of the bacteria is achieved before higher
concentrations are present (see section 4.2.2.1). Both volatilization
and biodegradation are likely to play a part in the loss of acetone
from surface waters. The most significant process will depend on
particular circumstances, such as depth and amount of aeration.
4.1.3 Soil
The two significant transport properties for acetone in soil are
volatilization and leaching, and acetone is also expected to
biodegrade rapidly. Leaching transports acetone from soil to
groundwater, with the rate of leaching from soil by rainwater
depending on the sorption characteristics of acetone in the various
types of soil. Since acetone may be controlled by Koc in
water-saturated soil and has a low Koc value, sorption of acetone
in such soil will be weak. A sorption study with moist clay soils
indicated that aqueous acetone causes swelling in these soils (Green
et al., 1983), and this process may allow the retention of a small
fraction of acetone. Volatilization transports acetone from soil to
the atmosphere. The volatility rate of acetone from soil depends on
the soil characteristics (moisture content, soil porosity, etc.).
Since acetone is weakly sorbed to soil, the volatility depends
primarily on fire moisture content of the soil. In dry soil, the
volatilization rate from soil surfaces is high due to the high vapour
pressure of acetone. In moist soil, the rate of volatilization is
similar to that of acetone in water and depends on the Henry's law
constant. Acetone volatilizes moderately under these conditions. The
detection of acetone at higher concentrations in downwind air of a
landfill site, compared to upwind air (Militana & Mauch, 1989),
indicates that acetone can volatilize from soil.
No data regarding the transport or uptake of acetone from soil to
plants are available.
While acetone is expected to biodegrade readily in soil, no data
are available to suggest that any degradation process in soil, other
than biodegradation, is significant.
Acetone has been detected in leachates from municipal and
industrial landfills (Sabel & Clark, 1984; Sawhney & Kozloski, 1984;
Brown & Donnelly, 1988), demonstrating that leaching through soil can
occur. The presence of other leachate constituents can adversely
affect the biodegradation efficiency of microbes to use acetone.
Acetone has a relatively high vapour pressure (231.06 mmHg at
25°C) (Riddick et al., 1986) and is used as an evaporative solvent in
a variety of applications. Because of its volatile properties, acetone
can be expected to evaporate from dry surfaces, particularly in spills
on the soil surface. Although evaporation from dry surfaces should be
a significant process, sufficient data are not available to predict
the relative significance of evaporation from moist soils, where
biodegradation and leaching will compete with evaporation as a removal
process.
4.2 Biotransformation
4.2.1 Bioconcentration and biomagnification
The very low log Kow value of -0.24 (see Table 1) suggests that
bioconcentration (a process leading to a higher concentration of a
chemical in an organism relative to that in its environment) of
acetone in either aquatic or terrestrial organisms, and
biomagnification (series of processes in an ecosystem by which higher
concentrations of a chemical are attained in organisms at higher
trophic levels) of acetone from animals of lower to higher trophic
levels is unlikely.
4.2.2 Biodegradation
Many aerobic biodegradation screening studies with mixed
microorganisms from waste-treatment plant effluents, activated sludge,
or sewage have examined the biodegradability of acetone (Lamb &
Jenkins, 1952; Heukelekian & Rand, 1955; Stafford & Northup, 1955;
Ettinger, 1956; Hatfield, 1957; Gaudy et al., 1963; Price et al.,
1974; Thom & Agg, 1975; Bridie et al., 1979; Urano & Kato, 1986a,b;
Babeu & Vaishnav, 1987; Bhattacharya et al., 1990). These strutues
indicate that acetone is easily biodegradable with acclimatized
microorganisms or after a suitable lag period (approx. 1 day) (Urano &
Kato, 1986a,b), as long as the initial concentration of acetone is not
at a toxic level. For example, acetone at a concentration of 500
mg/litre was toxic to microorganisms when biooxidation of acetone by
activated sludge was attempted (Gerhold & Malaney, 1966).
Biodegradation of acetone was similar in seawater and fresh water
(Takemoto et al., 1981 ). The 20-day biochemical oxygen demand (BOD)
for acetone for fresh water and saltwater was 78% and 76%,
respectively (Lamb & Jenkins, 1952; Price et al., 1974). After a
suitable lag period (5 days), acetone biodegraded quantitatively under
anaerobic conditions with anaerobic acetate-enriched culture medium
(Chou et al., 1979). A biodegradation study of acetone in natural
water collected from Lago Lake near Athens, Georgia, determined that
the biodegradation kinetics were multiphasic in nature and depended on
the substrate concentration. The determined rate of degradation was
faster at higher initial concentrations (the maximum concentration
used was 0.5 mg/litre) (Hwang et al., 1989).
In a laboratory experiment with natural stream water and
sediment, no acetone was lost in 338 h under sterile conditions in
closed flasks. However, with non-sterile natural sediment, 100% of the
acetone was lost in 500 h following a lag period of 90 h. (Rathbun et
al., 1982). The authors of this study concluded that biodegradation
was one of the important processes for the loss of acetone in streams.
Rathbun et al. (1982) separated his study into two groups to observe
the effects of pre-exposure acclimatization. One group was pre-treated
with a small concentration of acetone overnight and the other did not
get pre-treatment. The treatment reduce the lag time, and degradation
coefficients were much lower for the pre-treated groups. First-order
rate coefficients for the bacterial degradation of acetone at 25°C
ranged from 0.43-0.9 days-1 (not pre-treated), giving half-lives of 2
days. Significant loss of acetone due to biodegradation was not
observed in a later study when acetone was injected continuously in an
outdoor model stream (Rathbun et al., 1988, 1989, 1991, 1993).
Attempts to induce biodegradation by adding glucose and a nutrient
solution containing bacteria acclimated to acetone were unsuccessful.
The authors concluded that the residence time of acetone in the model
stream (6 h) was too short for the bacteria to become acclimated in
the water before initiation of biodegradation. However, this
explanation may not be valid if attached bacteria, rather than
free-floating bacteria, dominate the biodegradation process. As an
alternative explanation, the authors indicated that the observed
limitation in the nitrate concentration in the stream may be
responsible for the lack of acetone biodegradation.
4.2.2.1 Microbial degradation
Many aerobic biological screening studies have examined the
biodegradability of acetone and have found it to be readily
biodegradable (Lamb & Jenkins, 1952; Heukelekian & Rand, 1955;
Stafford & Northrup, 1955; Ettinger, 1956; Hatfield, 1957; Ludzack &
Ettinger, 1960; Price et al., 1974; Bridie et al., 1979; Takemoto et
al., 1981; Urano & Kato, 1986a,b; Vaishnav et al., 1987; Hwang et al.,
1989). One of these studies examined acetone biodegradation in a
natural water experiment and found acetone to be readily biodegraded
in Lago Lake water collected near Athens, Georgia, USA (Hwang et al.,
1989).
Platen et al. (1990) studied the enrichment, isolation,
characterization and the stoichiometry of acetone and its degradation.
In their study, acetone was oxidized completely by
Desulfococcus biacutus, a gram negative, anaerobic sulfate-reducing
bacterium using acetone as its sole organic substrate. Enzyme studies
indicated that acetone was metabolized by condensation with carbon
dioxide to a C4 compound (possibly free acetoacetate) and moved into
intermediary metabolism as acetoacetyl-coenzyme A. Acetoacetyl-CoA is
cleared by a thiolase reaction to acetyl-CoA which is completely
oxidized by the carbon monoxide dehydrogenase pathway. In
acetone-amended slurries, 76% of the theoretically-expected sulfate
was depleted, and in nitrate-amended slurries > 100% of the
theoretically-expected amounts of nitrate were consumed after 85 days
of incubation. Chou et al. (1979) also showed that acetone can be
degraded by anaerobic biodegradation.
Waggy et al. (1994) compared a USA 20-day biochemical oxygen
demand (BOD) test with the Organization for Economic Cooperation and
Development (OECD) closed bottle biodegradation test (Test 301D)
(OECD, 1981). In the 20-day BOD test, the results were 56, 76, 83 and
84%, at 5, 10, 15 and 20 days, respectively, and in the OECD test were
68, 72 and 78% for 5, 15 and 28 days, respectively, indicating good
correlation (Waggy et al., 1994). These test results classify acetone
as readily biodegradable. In a laboratory study using a microbial
culture from domestic waste water without acclimation, Price et al.
(1974) measured fresh water BODs (% biooxidation) to be 76, 82, 85 and
96% for 5, 19, 15 and 20 days, respectively. In "synthetic" saltwater,
the values for the same periods were 66, 88, 88 and 100%.
4.3 Bioavailability from environmental media
Acetone is expected to be bioavailable.
4.4 Interaction with other physical, chemical or biological factors
The atmospheric degradation of volatile organic compounds (VOCs)
in the presence of nitrogen oxides (NOx) leads to the production of
ozone. During complete oxidation of the VOCs free radical reactions
occur in the presence of sunlight with acetone (and other ketones),
participating as an intermediate with ozone as a byproduct. One method
of measuring the contribution of acetone is by the reactivity of it
with the hydroxyl radical (OH*).
The degradation of acetone in the lower troposphere may be
initiated by photolysis or reaction with OH* radicals. The reactions
with ozone (OD) or NOx are too slow to be important under
tropospheric conditions (Johnson & Jenkin, 1991). The rate of the
initiating reaction of OH* with acetone is well established at
2.26 × 10-13cm3/ molecule per sec (Atkinson, 1985).
Accordingly, the tropospheric lifetime of acetone with respect to
removal by OH radicals is approximately one month; therefore, the loss
of acetone by photooxidation is the major removal process of acetone
in the troposphere (Johnson & Jenkin, 1991).
The mechanism for acetone photodissociation has been reviewed by
Gardner et al. (1984). At 40°C, using the Gardner equations, the
average tropospheric lifetime would be halved to about 15 days, In
summary, the ozone concentrations predicted by the model were not
significantly affected by removal of the acetone emissions (Johnson &
Jenkin, 1991). Chatfield et al. (1987) examined the effect of
atmospheric pressure on the photolytic lifetime of acetone, and then
compared the result with losses caused by hydroxyl radical reactivity.
Reactions with hydroxyl radicals were much higher at ground level than
at increasing altitude where photolysis was more important in
degrading acetone.
The formation of ground-level ozone has become an air pollution
problem, especially in crowded, urban areas. Ozone is formed from the
complex photochemical interaction of some VOCs and NOx compounds.
Andersson-Sköld et al. (1992) calculated photochemical ozone creation
potentials (POCP) for 75 organic compounds, while Carter (1994)
developed maximum incremental ozone reactivity (MIR) scales to measure
the potential of VOCs to create ozone. Both research groups found that
ketones are weak producers of ozone, with acetone having one of the
lowest ozone formation potentials. Derwent et al. (1996) calculated a
POCP for acetone using a European model, which takes into account the
difference in conditions between European and North American cities;
the MIR model is considered more appropriate for North American
conditions. Andersson-Sköld et al. (1992) found similar values to
Derwent et al. (1966), indicating that acetone has "a remarkably low
POCP". Because of the low POCP, acetone has been suggested as a
potential substitute for high POCP aromatic hydrocarbons or the
chlorine-containing solvents.
4.5 Ultimate fate following use
The environmental fate of acetone can be predicted, since many of
the major fate processes have been investigated. When released to the
atmosphere, acetone will degrade through a combination of photolysis
and reaction with hydroxyl radicals (Meyrahn et al., 1986). Acetone
can be removed from the air by rainfall (wet deposition), as shown by
its detection in rainwater samples (Grosjean & Wright, 1983), but this
does not appear to be a significant route most of the time. In soil,
many studies have shown that acetone is readily biodegradable.
However, leaching may occur, especially if other chemicals are present
that may destroy or hinder microorganisms from degrading acetone.
Acetone can volatilize from water, as well as soil surfaces (Rathbun
et al., 1982). Since acetone is miscible with water and has a low
Koc, it leaches rather than adsorbs to soil. Where biodegradation is
inhibited or limited, acetone may reach the groundwater as a result of
leaching from spills or landfills (Steelman & Ecker, 1984; Brown &
Donnelly, 1988). Manufacturing and processing facilities may also
release acetone to air and water through discharges, and through other
wastes transported to landfills.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
5.1.1 Air
Acetone is a commonly found volatile contaminant. It is one of
the more long-lived intermediates that are produced in the oxidation
of light non-methane hydrocarbons (Henderson et al., 1989). Monitoring
data, covering rural, urban, remote and other areas, are available;
values depend upon where the sampling was done, as well as the time of
the year and sampling technique. Examples arc presented in Table 6.
Grosjean et al. (1989) collected samples in three large urban
areas in Brazil (Sao Paulo, Rio de Janeiro, Salvador) with populations
ranging from 2 to 13 million people. In Sao Paulo, acetone levels were
in the range of 0.5-7 µg/m3 (0.2-3 ppb), in Rio 1.2-9 µg/m3 (0.5-3.8
ppb), and in Salvador 0-49.9 µg/m3 (0-21 ppb). In 1975, Brazil
initiated a nationwide programme of production of ethanol from sugar
cane, and by 1988, when these samples were taken, approximately
one-third of the vehicles in use were ethanol-fuelled. Formaldehyde,
acetaldehyde and acetone were the three carbonyls with the highest
values, but these levels were still not higher than levels in other
parts of the world.
Acetone was one of the VOCs identified in the air of the storage
section of a municipal waste truck (Wilkins, 1994). Although the exact
measurement was not given in the study report, the author stated that
the concentration was below 1780 mg/m3 (750 ppm) (the TLV value).
Brosseau & Heitz (1994) measured the gases emitted from a municipal
landfill site and found acetone in two samples: one at 77 µg/m3 (32.5
ppbv) and the other at 16 µg/m3 (6.84 ppbv).
Chatfield et al. (1987) studied the behaviour of acetone in the
troposphere. Over the Atlantic Ocean (35°N), the mean concentration of
acetone in the lower troposphere is approximately 1.2 µg/m3 (0.5
ppb). Chatfield et al. (1987) stated that a significant amount of
carbon appeared to be cycled as acetone, with attack by hydroxyl
radicals and photolysis as the chief loss mechanisms, and that propane
may contribute nearly half of the acetone observed in the upper
atmosphere. Henderson et al. (1989) continued this work by showing
that the effects of surface sources of higher order alkanes, alkenes
and terpenes play a major role in the amount of acetone in the
troposphere.
Granby et al. (1997) measured acetone levels simultaneously in a
busy Copenhagen street (22 000 cars/day) and a semi-rural site 30 km
west and found little difference in mean concentrations (2.4 µg/m3
vs. 2.1 µg/m3; 1 ppb vs. 0.9 ppb). They found very weak correlations
with carbon monoxide and NOx; indicating sources other than
automobile exhaust, the most likely being oxidation of reactive
hydrocarbons from long-range transport of polluted air masses. Since
Table 6. Environmental air levels in various locations
Sampling Area Concentrationa Sampling dates Reference
µg/m3 ppb
City/Tucson, Arizona, USA 28.5 12 February-September 1982 Snider & Dawson (1985)
Urban/Tulsa, OK, USA 11.4-125.4 4.8-52.8 1978 Arnts & Meeks (1981)
Urban/South and Central America 0.5-49.9 0.2-21 1988 Grosjean et al. (1989)
Rural/Arizona, USA 6.7 2.8 February-September 1982 Snider & Dawson (1985)
Rural/Colorado, USA 12.1-56.3 5.1-23.7 1978 Arnts & Meeks (1981)
Rural/Egbert, Ontario 0.9-8.8 0.39-3.6 1989 Shepson et al. (1991)
9
Rural/Dorset, Ontario 1.5-10.2 0.65-6.3 1989 Shepson et al. (1991)
Forest/Texas, USA 6.9-46 2.9-19.4 January 1978 Seila (1979)
Remote/Alaska 0.7-6.9 0.3-2.9 1967 Cavanagh et al. (1969)
Mountains/Tennessee, USA 5-28.5 2.1-12 1978 Arnts & Meeks (1981)
Mountains/Bavaria, Germany approx. 1.2 0.54b August 1995 Leibrock & Slemr (1997)
Ocean/Atlantic 35°N 1.2 0.5 Chatfield et al. (1987)
a Some sampling in the above studies may have been conducted using Tedlar bags that are known to contaminate
air samples with acetone (Henderson et at., 1989). Non-range values are mean values.
b Measured as propylene equivalents of oxygenated hydrocarbons in ppbC (ppb of carbon).
the acetone concentrations in this study are only slightly higher than
those found in rural, remote and ocean atmospheres, it appears that
the acetone is probably not transported a great distance in the lower
troposphere.
Arnold et al. (1997) measured upper tropospheric concentrations
of acetone at 9000 m over the northeastern Atlantic, near Ireland in
1993. Measured acetone concentration was found to correlate positively
with that of sulfur dioxide (SO2), reaching a maximum abundance of
approx. 7 µg/m3 (3 ppb). This concentration is markedly higher than
the concentration of 1.2 µg/m3 (0.5 ppb) in the lower troposphere
reported by Chatfield et al. (1987). As the SO2 level decreased, so
did the acetone concentration. Either the acetone was transported from
direct emissions from the USA, or a photochemical hydrocarbon
conversion had occurred.
In a review of earlier studies, Singh et al. (1994) found acetone
at a range of approx. 0.9-5.2 µg/m3 (approx. 0.4-2.3 ppb), with a
mean of 3.1 µg/m3 (1.14 ppb), in the troposphere. Using a three
dimensional photochemical model, Singh et al. (1994) found that the
greatest source of acetone was the oxidation of precursor hydrocarbons
(51%); other sources were biomass burning (26%), biogenic emissions
(21%) and an anthropogenic emission (approx. 3%). Atmospheric removal
was mainly by photolysis (64%), followed by reaction with OH*
radicals (24%) and deposition (12%). Other important points were:
* there is substantial variability in atmospheric abundance
* the concentration of acetone appears to vary with altitude
* upper atmospheric transport is possible since the half-life is
>10 days
* acetone appears to be the most abundant non-methane organic
species in the atmosphere
* the geochemical background of acetone appears to be 1.2 µg/m3
(approx. 0.5 ppb)
5.1.1.1 Indoor air
Shah & Singh (1988) reported a concentration of 19 µg/m3 (8 ppb)
in household indoor air. These authors compiled available data to
calculate an average outdoor concentration of 16.4 µg/m3 (6.9 ppb).
Other investigators reported similar results (Jarke et al., 1981).
Tichenor & Mason (1988) measured acetone levels in the range of 37-41
µg/m3 (approx. 15-17 ppb) per hour being emitted from low-density
particle board used in home construction in the USA. The reason for
the higher indoor air concentration was the use of acetone-containing
consumer products inside homes. The potential for intrusion of acetone
present as Soil gas into a house adjacent to a landfill was
characterized by Hodgson et al. (1992), but the measurement was for
only a single house. The average concentration was 47.5 µg/m3 (20
ppb).
Hodgson et al. (1991) collected air samples in a
newly-constructed building at four different times over a period of
14 months. The major source of VOCs was not the new construction
materials, but the liquid-process copiers and plotters where acetone
concentrations ranged from 28.8 to 66.6 µg/ms (12-28 ppb).
5.1.2 Water
Acetone has been qualitatively detected in drinking-water in
various cities in the USA, including Miami, FL; Ottumwa, IO;
Philadelphia, PA; Cincinnati, OH; Calhoun, GA; Dalton, GA; Gastonia,
NC; Durham, NC; New Orleans, LA; Rome, GA; and Tuscaloosa, AL (Bertsch
et al., 1975; US EPA, 1975; Shackelford & Keith, 1976). In the US EPA
National Organics Reconnaissance Survey (NORS), involving
drinking-water supplies from 10 cities in the USA, acetone was
qualitatively detected in all the cities. An acetone concentration of
1 µg/litre was found in drinking-water samples from Seattle, WA (US
EPA, 1975).
Acetone was detected in 33/204 surface water samples collected
from sites near heavily industrialized areas in the USA during
1975-1976 (Ewing et al., 1977). It was detected in 12.4% of all
groundwater samples analysed from 178 USA hazardous waste (Superfund)
sites as part of a national programme to investigate and remedy
potential problems at these sites (Plumb, 1987).
Acetone is released to water in wastewater discharges from
industry and sewage treatment. It was found in 23/63 effluent waters
from a wide range of chemical manufacturers around the USA at
concentrations ranging from < 10 to 100 µg/litre (Perry et al.,
1978). A comprehensive survey of wastewater from 4000 industrial and
publicly owned treatment works detected acetone in a wide range of
wastewater from industries such as leather tanning, petroleum
refining, nonferrous metals, paint and ink, printing and publishing,
coal mining, organics and plastics, inorganic chemicals, textile
mills, pulp and paper, robber processing, pesticide manufacture,
photographic industries, pharmaceuticals, porcelain/enamels,
mechanical products and transportation equipment. The highest effluent
concentration of acetone from all industries was 37.7 mg/litre, which
was detected in the paint and ink industry; however, the median
acetone level was 0.89 mg/litre (NLM, 1992).
Acetone can be released to groundwater by leaching from municipal
and industrial landfills. Leachate collected from a Minnesota (USA)
municipal landfill contained as much as 13 mg acetone/litre (Sabel &
Clark, 1984). Levels of 2.94.8 mg/litre were detected in leachate
samples collected in the USA from an industrial landfill in
Connecticut in 1982-1983 (Sawhney & Kozloski, 1984) and from one in
Michigan that contained up to 62 mg acetone/litre (Brown & Donnelly,
1988).
Acetone has also been detected at 0.2-0.7 µg/litre in water from
several artesian wells adjacent to a landfill in Wilmington, Delaware,
USA (DeWalle & Chian, 1981). The concentration of acetone was up to 3
mg/litre in a drinking-water well in New Jersey (Burmaster, 1982;
Steelman & Ecker, 1984).
The concentration of acetone in open ocean water (Tongue of the
Ocean, Bahamas) was approx. 0.35 µg/litre (Kieber & Mopper, 1990).
Corwin (1969) measured VOCs in seawater and found acetone levels in
the Florida Straits (USA) of 14-52 µg/litre at depths ranging from 0
to 160 metres at approx. 35% salinity. Similar concentrations were
found in the Mediterranean where the measurements were 18-52 µg/litre
at slightly higher salinity (approx. 39%).
5.1.3 Soil and sediment
There are few data regarding the level of acetone in soil and
sediment. Acetone has been detected in 43% of the soil samples in
designated waste disposal sites in the USA for which acetone
determination has been made (ATSDR, 1994). The maximum concentration
of acetone in soils from Vega Alta Public Supply well sites in Puerto
Rico and the mean concentration of acetone in soil from Summit
National Site, Ohio, was 9.5 mg/kg (ATSDR, 1994). Because of its high
water solubility and low sediment adsorption coefficient, acetone in
an aquatic system is predominantly found in water, rather than in
sediment.
5.1.4 Food
Acetone has been qualitatively detected in blue cheese (Day &
Anderson, 1965), baked potatoes (Coleman et al., 1981), roasted
filbert nuts (Kinlin et al., 1972), chicken breast muscle (Grey &
Shrimpton, 1967) and nectarines (Takeoka et al., 1988). Acetone
concentrations of 795 mg/kg and 11 mg/kg were identified in
Czechoslovakian milk samples and milk cream culture, respectively
(Palo & Ilkova, 1970). Milk samples from Swedish dairy cattle were
found to contain acetone concentrations ranging from 18 to 226
mg/litre (0.32-3.89 µmol/litre) (Andersson & Lundstrom, 1984).
Pellizzari et al. (1982) qualitatively identified acetone in all 8
selected human milk samples collected from volunteers in Bayonne, NJ,
Jersey City, NJ, Bridgeville, PA, and Baton Rouge, LA. A variety of
bean types (common, lima, mung and soy) contained acetone levels
ranging from 260-2000 µg/kg, with a mean level of 880 µg/kg, and
levels of 530 and 230 µg/kg were detected in split peas and lentils,
respectively (Lovegren et al., 1979). Acetone has also been detected
in onions, grapes, cauliflower, tomatoes and wild mustard (NLM, 1992).
5.1.5 Other environmental levels
Acetone is ubiquitous in the environment and is found at a wide
range of concentrations.
5.2 General population exposure
Acetone is readily absorbed from the lung and gastrointestinal
tract following inhalation and ingestion (see chapter 6). It can also
be absorbed through the skin. The low values for Koc (see Table 1)
and a moderate value for Henry's law constant (Rathbun & Tai, 1987)
suggest that the bioavailability of acetone from contaminated water
and soil as a result of contact may be significant. However,
quantitative data regarding the rate and extent of dermal absorption
of acetone from contaminated water and soil are lacking. The high
water solubility and low Koc value for acetone suggest that
bioavailability from ingested soil (e.g., children playing at or near
contaminated sites) will be high, but, again, quantitative absorption
data are lacking. Data on bioavailability of acetone from ingested
plant food are not available.
Exposure to acetone occurs from both natural and anthropogenic
sources, and it is endogenously produced by all humans. The general
population is exposed to acetone by inhaling ambient air, ingesting
food, and drinking-water containing acetone. Dermal exposure to
acetone may result from skin contact with consumer products (e.g.,
certain nail polish removers, paint removers, and household cleaning
and waxing products). Assuming concentrations of acetone are 19 µg/m3
(8.0 ppb) in indoor air and 16.4 µg/m3 (6.9 ppb) in outdoor air (Shah
& Singh, 1988) and that an average person inhales 15 m3/day of indoor
air and 5 m3/day of outdoor air daily, the estimated exposure to
acetone by inhalation is 0.37 mg/day. This value is much lower than an
estimate based upon an earlier exposure level found by one of these
researchers. Singh & Hanst (1981) estimated that an acetone
concentration of 0.26 µg/m3 (0.111 ppb) will occur in the lower
troposphere as a result of atmospheric oxidation of naturally
occurring propane, with levels of 35 ng/m3 (15 ppt) in the upper
troposphere and 7 ng/m3 (3 ppt) in the stratosphere. Since the
sampled atmospheric concentrations of acetone are 0.723-127.25 µg/m3
(0.3-52.8 ppb), and maintaining that the average adult human inhales
20 m3 air/day, the average daily exposure of acetone from inhalation
can be estimated to be 14.5-2545.0 µg, or up to 2.5 mg/day.
Wang et al. (1994) measured acetone concentrations in 89
non-occupationally exposed subjects and found acetone mean values of
840 µg/litre in blood, 842 µg/litre in urine, 715 ng/litre in alveolar
air and 154 ng/litre in environmental air. The researchers found no
significant difference in blood levels between smokers (896 µg/litre)
and nonsmokers (792 µg/litre), and likewise between hospital staff
(719 µg/litre) and blood donors (966 µg/litre). The results are
similar to those of Pezzagno et al. (1986) who measured 760 µg
acetone/litre in urine.
The endogenous acetone level in the body at any instant reflects
acetoacetate production (Morgott, 1993). The concentration of acetone
in whole blood does not differ from that in plasma (Gavino et al.,
1986). Even in healthy subjects, the level of acetone in blood or
plasma varies with fasting or non-fasting conditions and depends on
the weight of the subject. Generally, the blood or plasma acetone
concentrations are higher in fasted than non-fasted subjects and
higher in subjects who are not obese, compared to obese subjects (Haff
& Reichard, 1977). It should be noted that normal and abnormal
physiological conditions and disease states may increase ketogenesis
and the body burden of acetone. Acetone levels in athletes and
pregnant women (among many groups) may be elevated because these
groups of people have greater energy requirements. Ashley et al.
(1994) measured blood concentrations in non-occupationally exposed
populations. The mean concentration in a control group in the USA was
3.1 mg/litre. In a group of nine volunteer subjects, the mean blood
concentration before entering a van designed for clinical examinations
for a health survey was 1.9 mg/litre and after 3 h in the van the mean
blood concentration was virtually unchanged at 2 mg/litre, although
the range before entry was 1 3.6 mg/litre and after 3 h was 0.9-5
mg/litre, i.e. the high end of the range was over 1.4 mg/litre higher
when the subjects were tested after breathing the same air for 3 h.
Individuals with uncontrolled diabetes mellitus or diabetic
ketoacidosis may have plasma acetone levels as high as 750 mg/litre
(Trotter et al., 1971). The acetone concentrations in body fluids and
expired air in studies of healthy individuals and diabetic patients
are shown in Table 7. Clinical findings in eases of acute acetone
intoxication suggest that acetone blood levels over 1000 mg/litre are
necessary to cause unconsciousness in humans (Ramu et al., 1978), but
lower levels may interrupt physiological processes in diabetics.
Approximate reference concentrations for human plasma acetone are
< 10 mg/litre for a "healthy" individual, < 100 mg/litre for an
occupationally exposed individual, 100-700 mg/litre for an individual
with diabetic ketoacidosis and > 200 mg/litre for an individual
showing symptoms of "toxic" exposure (Tietz, 1983).
5.3 Occupational exposure
Kiesswetter et al. (1994) investigated occupational acetone
exposure using two groups of eight healthy male workers on nine shift
days. Using personal sampling, exposure was higher in the first half
of the shift (2730 mg/m3) than in the second half (1720 mg/m3).
For monitoring purposes, the researchers studied the relationship of
acetone in air versus three urine parameters: (1) concentration of
acetone in urine; (2) concentration of urine related to creatinine
excretion; and (3) concentration of acetone in urine in relation to
time (sampling period) and excreted urine volume. The concentration of
acetone in urine was moderately correlated to that in air. Some of the
ratings of well-being in the workers con-elated with the acetone
concentrations in the urine but not with the acetone concentrations in
the workplace air.
Table 7. Concentrations of acetone in body fluids and expired air of humans
Medium Subject Concentration Reference
Blood Healthy (non-fasted) 0.93 mg/litre Gavino et al. (1986)
Blood Health (non-fasted) 0.84 mg/litre Brugnone et al. (1994)
Blood Healthy (non-fasted) 1.8 mg/litre (median) Ashley et al. (1994)
Plasma Healthy (3-day fasted) 46.5 mg/litre Haff & Reichard (1977)
Plasma Healthy (non-fasted) 1.74 mg/litre Trotter et al. (1971)
Plasma Obese (3-day fasted) 17.4 mg/litre Haff & Reichard (1977)
Plasma Ketoacidotic 424 mg/titre Trotter et al. (1971)
Plasma Ketoacidotic 290 mg/litre Haff & Reichard (1977)
Urine Healthy 0.23-0.41 mg/litre Kobayashi et al. (1983)
Urine Healthy 0.84 mg/litre Brugnone et al. (1994)
Urine Healthy (endogenous) 0.76 mg/litre Pezzagno et al. (1986)
Urine Diabetic 0.64-9.0 mg/litre Kobayashi et al. (1983)
Expired air Healthy 1.23 µg/litre Jansson & Larsson (1969)
Expired air Healthy 1.16 µg/litre Trotter et al. (1971)
Expired air Healthy 1.3 µg/litre Phillips & Greenberg (1987)
Wang et al. (1994) calculated a blood-air coefficient for acetone
of 146. On average, the blood acetone levels of workers were 56 times
higher than those of subjects only exposed environmentally. These
researchers calculated the half-life of acetone in blood to be 5.8 h
for the interval between the end of one shift and the beginning of the
next (approx. 16 h). Analyses were made of workers before the start of
their shift, and mean acetone levels were 3.5 mg/litre in blood and 13
mg/litre in urine. Wigaeus (1981) calculated the acetone half-life to
be 6.1 h. These values indicate that the 16-h period between
workshifts did not allow for complete elimination of acetone absorbed
from the previous workshift.
In a study of environmental tobacco smoke (ETS) and its
contribution to VOC concentrations, Heavner et al. (1996) measured
acetone levels in smoking and non-smoking workplaces and homes. The
mean levels were: non-smoking workplace, 59.77 µg/m3 (SD 79.78);
smoking workplace, 952.86 µg/m3 (SD 3988.25); non-smoking home, 50.12
µg/m3 (SD 58.5); smoking home, 71.19 µg/m3 (SD 118.17).
Approximately 6% of the acetone found in the air of smoking workplaces
and homes was attributed to ETS.
Workers in industries that manufacture and use acetone can be
exposed to much higher concentrations of acetone than the general
population. For example, the concentrations of acetone in the
breathing zone air in a paint factory, a plastics factory, and an
artificial fibre factory in Italy were > 3.48 mg/m3 (Pezzagno et
al., 1986). The concentration of acetone in a plastic plant in Japan,
where bathtubs were produced, was > 100 mg/m3 (Kawai et al., 1990a).
The inhalation exposure of workers to acetone in a shoe factory in
Finland ranged from 25.4-393.4 mg/m3 (Ahonen & Schimberg, 1988). The
concentration of acetone in the air of a solvent recycling plant was
as high as 42 mg/m3, the mean exposure being 1 mg/m3 (Kupfersehmid &
Perkins, 1986).
Exposure to acetone may also occur indirectly. For example,
isopropyl alcohol is known to oxidize in the liver and is converted to
acetone (Kawai et al., 1990b). Therefore, occupational exposure
(printing plants) or accidental ingestion of isopropyl alcohol can
produce acetone in expired air, blood and urine (Lacouture et al.,
1989).
6. KINETICS AND METABOLISM IN LABORATORY ANIMALS AND HUMANS
There is extensive information regarding acetone and its
metabolism within animals and humans. Because acetone has a low
relative molecular mass and is miscible with water, it is absorbed and
uniformly distributed throughout the non-adipose tissues of the body.
There are absorption and tissue distribution studies, radiolabelled
metabolic and kinetic studies and elimination/excretion data. Other
variables such as diet, exercise and alcohol consumption affect the
kinetics of acetone.
6.1 Absorption
6.1.1 Inhalation exposure
6.1.1.1 Human studies
In humans exposed to acetone concentrations of up to 2970 mg/m3
(1250 ppm) for < 7.5 h/day in a complex protocol for < 6 weeks,
the concentration of acetone in venous blood was directly related to
the vapour concentration and duration of exposure, and inversely
related to the time elapsed following exposure (Stewart et al., 1975).
Data on the actual distribution of acetone in the human body are
scarce. Digs et al. (1994) measured in vitro blood/air partition
coefficients (KB/A) in blood samples from 73 humans. The average
KB/A (±SD) for acetone was 301 ± 22, with a range of 250-410, and
was normally distributed. Since acetone is absorbed into the blood
from the respiratory tract and is highly water soluble, it is to be
expected that there will be distribution to tissues with high water
content.
Acetone vapour is rapidly taken up by the tissue of the
respiratory tract and absorbed into the bloodstream during inhalation
exposure. This rapid uptake was shown by Haggard and co-workers when
substantial concentrations of acetone appeared in the blood only 30
min after exposure (Haggard et al., 1944). This is probably due to its
high blood-air partition coefficient (167-330) (Haggard et al., 1944;
Sate & Nakajima, 1979; Fiserova-Bergerova & Diaz, 1986; Paterson &
Mackay, 1989; Dills et al., 1994). Studies in humans exposed to up to
10 940 mg/m3 (4607 ppm) for up to 4 h showed measured pulmonary
uptakes ranging widely from -30% to 80% (Landahl & Herrmarm, 1950;
DiVincenzo et al., 1973; Nomiyama & Nomiyama, 1974a; Wigaeus et al.,
1981; Pezzagno et al., 1986). The reason for the wide range in
reported values involves the aqueous wash-in/wash-out effect when
acetone is inhaled, which can lead to spurious results (Schrikker et
al., 1985, 1989). During this phase, acetone, which is highly water
soluble, will dissolve in epithelial cells during inspiration
(wash-in) and evaporate during expiration (wash-out). This could
account for the lower than expected pulmonary absorption based on the
high blood/air partition coefficient (Wigaeus et al., 1981).
There have been a number of studies on highly soluble vapours
(such as acetone) and expiration from the respiratory tract. Cander &
Forster (1959) stated that a substantial portion of the acetone
adsorbed into the pulmonary tissue during inspiration was removed
during expiration, thus mixing with any expired alveolar sample that
was used as a measurement for analysis. Schrikker et al. (1985, 1989)
performed a series of measurements and found several interesting
results. There was an appreciable difference in alveolar air expired
at the beginning and ending of any breath during the wash-in cycle. At
first, acetone level was greatest at the beginning of a breath, but
when work load (exercise) started, the levels of the end started
rising, until after approximately 8 breaths, acetone level was higher
at the end of a breath. Part of the explanation was that the upper
airway linings give off the acetone during the initial breaths, and
the deeper portions of the respiratory tract give off the acetone in
the later breaths. Schrikker et al. (1989) found that the pulmonary
excretion of acetone is dependent upon the exchange of vapour between
the respiratory tract tissues and the inspired air, and is a
substantial proportion of the excretion of acetone via the respiratory
tract.
The turnover of acetone is highly dependent upon the organism's
state. Schrikker et al. (1989) and Jakubowski & Wieczorek (1988)
showed, as exercise increased, so did the amount of acetone being
expired and the rate of acetone uptake by the body was also directly
proportional to the ventilation rate. Haggard et al. (1944) compared
resting and exercising males and found that a ninefold increase in the
metabolic rate was required to account for a less-than-expected rise
in acetone blood levels. They also approached the idea that there was
a limit to the amount of acetone that can be accumulated in the body.
Exhaled breath levels of acetone in humans rise during exposure
to acetone and reach a steady state within approximately 2 h
(DiVincenzo et al., 1973; Nomiyama & Nomiyama, 1974a; Brown et al.,
1987). Uptake is directly proportional to exposure concentration and
duration (DiVincenzo et al., 1973; Wigaeus et al., 1981). Uptake also
increases as the level of physical activity increases, i.e. during
exercise, due to increased pulmonary ventilation (Haggard et al.,
1944; DiVincenzo et al., 1973; Wigaeus et al., 1981; Jakubowski &
Wieczorek, 1988). Lungs (including the mouth and trachea) were shown
to retain a greater percentage of inspired acetone (55%) than the
nasal cavity (18%) in two humans exposed to acetone vapour
concentrations of 0.3-3.0 mg/litre and 0.8-11.0 mg/litre for nasal and
lung retention measurements, respectively, with a flow rate of 18
litres/min. This might indicate that the nasal cavity absorbs acetone
less readily than the rest of the respiratory system (Landahl &
Herrmann, 1950). Blood levels of acetone rose rapidly during exposure
for up to 4 h with no indication that a steady state was reached
(DiVincenzo et al., 1973; Brown et al., 1987; Dick et al., 1989),
suggesting that during exposure, the rate of absorption exceeded the
rate of distribution and elimination. During short-term (2-4 h)
exposure to 240 or 1190 mg acetone/m3 (100 or 500 ppm), 75-80% of
the amount of acetone inspired was absorbed by blood after 15 mm of
exposure, and 20-25% remained in the dead space volume (DiVincenzo et
al., 1973). Higher inspired amounts resulted in higher blood levels
(Haggard et al., 1944; Matsushita et al., 1969a; DiVincenzo et al.,
1973; Pezzagno et al., 1986). Pezzagno et al. (1986) similarly
reported that an approximate average of 53% of administered acetone
was retained during exposure of 15 humans to acetone vapour
concentrations of 964-8610 mol/m3 (56-500 mg/m3) for 2-4 h in an
exposure chamber. In contrast to other studies, moderate exercise did
little to change the net retention of acetone. The mean net retention
was 54% during exposure at rest and 53% during exposure with exercise.
A correlation between blood level at the end of exposure and exposure
concentration was found in humans exposed to 55-495 mg/m3 (23-208
ppm) for 24 h (Pezzagno et al., 1986). No significant difference in
uptake or retention was found between men and women (Brown et al.,
1987).
6.1.1.2 Experimental animal studies
Animals also absorb acetone rapidly during inhalation exposure.
Measurement of blood acetone levels in rats after 4-6 h of exposure to
various concentrations shows that blood levels correlate well with
exposure concentrations (Charbonneau et al., 1986a, 1991; NTP, 1988)
and are highest immediately after exposure (NTP, 1988). In rats
exposed to 355 mg/m3 (150 ppm) for 0.5-4 h, measurement of blood
acetone concentrations during exposure revealed that blood levels
increased steadily for 2 h and then remained constant for the next 2 h
of exposure (Geller et al., 1979). Blood acetone levels also
correlated well with exposure concentration in dogs exposed for 2 h
(DiVincenzo et al., 1973). Blood levels were 4, 12 and 25 mg/litre
after exposures to 240, 1190 and 2375 mg/m3 (100, 500 and 1000 ppm),
respectively. In anaesthetized dogs allowed to inhale concentrated
vapour of acetone spontaneously from a respirator at various
ventilation rates, uptake by the respiratory tract was 52% at flow
rates of 5-18 litres/min and 42% at ventilation rates of 21-44
litres/min (Egle, 1973). Retention in the lower respiratory tract was
48% at 5-18 litres/mm and 37.5% at 21-40 litres/min. Retention by the
upper respiratory tract was 57% at 4-18 litres/min. The effect of
exposure concentration on total uptake was studied at a range of
ventilation rates equated with exposure concentrations. Percentage
uptakes were 52.1% at a mean concentration of 503 mg/m3 (212 ppm),
52.9% at 672 mg/m3 (283 ppm), and 58.7% at 1553 mg/m3 (654 ppm).
These results indicate the respiratory uptake of acetone by dogs is
similar to human uptake values reported by Landahl & Herrmann (1950).
Comparison of data from dogs and humans by ATSDR (1994) revealed that
the amount absorbed in humans was greater in absolute quantity under
comparable exposure conditions, but when expressed in terms of kg body
weight, the amount absorbed in dogs was five times more than in
humans. The retention in the upper respiratory tract was higher than
in the lower respiratory tract of dogs (Egle, 1973). Exposure
concentration had little effect on retention. The absorption of
acetone by the nasal walls of anaesthetized dogs, in which the nasal
passage was isolated, increased when the airflow rate was increased
(Aharonson et al., 1974), but the percentage uptake actually
decreased.
In rats exposed continuously to 5250 mg/m3 (2210 ppm) for 9
days, peak acetone blood levels were approximately half the exposure
concentration, were reached in 3-4 days, and remained at this level
for the duration of exposure (Haggard et al., 1944) In rats exposed to
10 200 mg/m3 (4294 ppm) for 12 days, acetone blood levels slightly
greater than half the initial concentration were reached in 4 days and
continued to day 12. Blood levels in rats exposed to these
concentrations for 8 h/day were about half of those reached during
continuous exposure. The amount of acetone absorbed in the first 8 h
exceeded the amount eliminated in the next 16 h of exposure to fresh
air, meaning them was a small accumulation.
Studies with laboratory animals illustrate the absorption of
acetone by the nasal cavity (Morris et al., 1986). Absorption
efficiencies (i.e. relative net uptake) were measured in the upper
respiratory tracts of two strains of rat and a single strain of
guinea-pig. At flow rates approximately 3 times the normal minute
volumes, upper respiratory tract retention of acetone vapour averaged
7, 12 and 21% for guinea-pigs, Fischer-344 rats and Sprague-Dawley
rats, respectively.
As in the case of humans (Landahl & Herrmann, 1950) and dogs
(Egle, 1973), the disposition of acetone in the upper respiratory
tract of other animals, including rats, mice, guinea-pigs and
hamsters, indicates that relatively little acetone is absorbed from
the upper respiratory tract (Morris et al., 1986; Morris & Cavanagh,
1986, 1987; Morris, 1991). The absorption efficiency was greater in
Sprague-Dawley rats than in Fischer-344 rats, but there was no
difference between male and female Sprague-Dawley rats (Morris, 1991).
Deposition was similar in B6C3F1 mice and Fischer-344 rats, and was
greater than in Hartley guinea-pigs and Syrian golden hamster. The
differences between strains and species could not be attributed to
differences in metabolism, because acetone is not significantly
metabolized in the upper respiratory tract of these species. The
reason was thought to be differences in perfusion rates (Morris &
Cavanagh, 1987; Morris, 1991).
6.1.2 Oral exposure
6.1.2.1 Human studies
In a series of experiments conducted in male volunteers given
acetone orally at 40-80 mg/kg, an estimated 65-93% of the administered
dose was metabolized, with the remainder being eliminated in the urine
and expired air in about 2 h, indicating rapid and extensive
gastrointestinal absorption (Haggard et al., 1944). Early research
demonstrated that after oral exposure, acetone moved quickly through
the body. In a human who ingested 137 mg acetone/kg on an empty
stomach, the blood level of acetone rose sharply to a peak 10 min
after dosing (Widmark, 1919). In another example, a subject ingested
the same dose 10 or 12 min after eating porridge. The blood acetone
level rose slowly over 48-59 min to levels of about one-half to
two-thirds that achieved after taking acetone on an empty stomach. The
presence of food in the gastrointestinal tract led to a slower rate of
absorption.
Measurement of acetone in blood and urine of patients who
accidentally or intentionally ingested acetone indicated that acetone
was absorbed, but the percentage absorbed cannot be determined from
the data. A man ingested liquid cement (approximate acetone dose of
231 mg/kg) (Sakata et al., 1989) causing his plasma acetone level to
be approx. 110 mg/litre and his urinary level to be 123 mg/litre 5 h
after ingestion. In another study, a woman who had ingested nail
polish remover had a blood acetone level of 2.5 g/litre (Ramu et al.,
1978). The authors estimated that her body burden was 150 g acetone at
the time of admission to the hospital. The serum acetone level of a
30-month-old child was (4.45 g/litre) 1 h after ingestion of a 6-ounce
bottle of nail polish remover (65% acetone) (Gamis & Wasserman, 1988).
6.1.2.2 Experimental animal studies
Acetone appears to be absorbed by the gastrointestinal tract as
indicated by early studies of laboratory animals. Price & Rittenberg
(1950), administered orally a dose of 14C-acetone (0.22 mg/189 g
body weight, in 1 ml water) to a female rat and collected respiratory
14CO2: for 13.5 h. During this period, 47.4% of the administered
dose was accounted for by exhaled 14CO2. In a separate experiment,
a rat (152 g body weight) was given seven daily oral doses of
14C-acetone (1.08 mg), and radioactivity in carbon dioxide collected
during 24-h post-administration periods accounted for 67-76% of the
administered radioactivity. Radioactive acetone was detected in
expired air in the first 20-min period following the first
administration; however, radioactive acetone, exhaled during 24-h
post-administration periods, accounted for a maximum of 7% of a daily
dose. In an experiment in which a 210 g rat was given 1.30 mg of
radioactive acetone, the rate of exhalation of radioactivity as
acetone was maximal approx. 2 h after administration, but declined
thereafter, suggesting that gastrointestinal absorption was rapid
(Price & Rittenberg, 1950).
Experiments in rats indicate that acetone is rapidly and almost
completely absorbed from the gastrointestinal tract after oral
exposure. A rat expired 97.4% of a 1.16 mg/kg oral dose over a 13.5-h
period, while another rat given 7 mg/kg 14C-acetone expired 67-76%
of the administered dose over a 24-h period after the last dose (Price
& Rittenberg, 1950). From these data, absorption of least 74-83% of
the administered dose can be inferred. In other studies where rats
were given similar or higher doses of acetone, plasma acetone levels
rose proportionately with dose in rats given acetone as single doses
by gavage (Lewis et al., 1984; Charbonneau et al., 1986a) or in the
drinking-water for 7 days (Skutches et al., 1990).
In a study comparing the blood levels of acetone achieved after
fasting with those after oral dosing, peak blood levels of acetone of
approx. 35 and 110 mg/litre were reached within about 3 h after dosing
of rats with 78 and 196 mg acetone/kg, respectively (Miller & Yang,
1984). The levels declined to near background levels within the next
16 h. At an acetone dose of 20 mg/kg, the blood level increased to
about 5 mg/litre over 19 h, when the rats were sacrificed. In rats
fasted for 48 h, blood acetone levels increased continuously to about
13 mg/litre. While the maximal blood concentrations of the treated
rats differed considerably from those of the fasting group, the
calculated areas under the curve for the 78 and 196 mg/kg groups were
comparable to those of the fasting groups (ATSDR, 1994).
There are conflicting data regarding the effect of vehicle on the
gastrointestinal absorption of acetone. In the one study where water
was used as the vehicle, maximum blood levels were higher and achieved
earlier in rats given acetone by gavage as compared to those given
acetone by gavage in corn oil (Charbonneau et al., 1986a). The slower
absorption of acetone in corn oil may have resulted from a delayed
gastric emptying due to the presence of corn oil in the stomach. In a
later repetition of this study, very little difference in blood and
liver levels of acetone were found in rats given the same dose of
acetone in water or in corn oil (Charbonneau et al., 1991).
6.1.3 Dermal exposure
6.1.3.1 Human studies
Dermal absorption of acetone is known to occur in humans.
Application of cotton soaked in acetone to a 12.5 cm2 uncovered area
of skin of volunteers for 2 h/day for 4 days resulted in blood levels
of acetone of 5-12 mg/litre, alveolar air levels of 12-28 mg/m3 (512
ppm), and urinary concentrations of 8-14 mg/litre on each day
(Fukabori et al., 1979). When the daily exposure increased to 4 h/day,
body concentrations more than doubled with 26-44 mg/litre in blood,
59-81 mg/m3 (25-34 ppm) in alveolar air, and 29-41 mg/litre in
urine. The absorption was mediate, with peak blood levels appearing at
the end of each daily application. The authors noted that it was not
possible to completely prevent exposure from inhalation; the acetone
concentration in the breathing zone of one subject was found to be
0.95-1.4 mg/m3 (0.4-0.6 ppm). From the alveolar air and urine
concentrations, it was estimated that a 2-h dermal exposure was
equivalent to a 2-h inhalation exposure to 120-355 mg/m3 (50-150
ppm), and a 4-h dermal exposure was equivalent to a 2-h inhalation
exposure to 590-1190 mg/m3 (250-500 ppm).
6.1.3.2 Experimental animal studies
In a study using isolated perfused pig skin and 14C-labelled
acetone, 60% of the acetone evaporated in the first 2 min, but
approximately 33% remained after 2 h, and around 20% was still present
after 8 h (Williams et al., 1994). The findings of cataract formation
in guinea-pigs exposed dermally to acetone 0.5 ml applied daily for 5
days per week for 6 weeks to the back was attributed to dermal
absorption of acetone (Rengstorff et al., 1972; Rengstorff & Khafagy,
1985). These authors suggested an association between cataract
formation and significant depression of the antioxidant, ascorbate, in
the aqueous humour. To study this relationship in more depth, Taylor
et al. (1993) fed albino hairless guinea-pigs diets containing low
(4.9 mg/day) and high (55 mg/day) levels of ascorbate. Daily dermal
exposure to acetone occurred over the next 6 months, with a total
applied dose of 65 ml. No cataracts were found in any of the
guinea-pigs from either group even though Taylor et al. (1993) used
four times the dose of Rengstorff & Khafagy (1985).
6.1.4. Absorption summary
From the information presented, acetone is absorbed rapidly no
matter which route of exposure. It appears that acetone is absorbed by
all parts of the respiratory system, and that oral and dermal
absorption can also occur. However, absorption can be affected by the
stomach contents.
6.2 Distribution
6.2.1 Inhalation exposure
6.2.1.1 Human studies
Human in vitro tissue-blood partition coefficients of acetone
for cadaver muscle, kidney, lung and brain grey matter range from 0.74
to 0.82, indicating nearly complete tissue distribution
(Fiserova-Bergerova & Diaz, 1986). Dills et al. (1994) measured
in vitro blood/air partition coefficients (KB/A) of acetone in
blood samples from 73 subjects. The mean value was 301 ± 22; the range
was 250-410 and was normally distributed.
6.2.1.2 Experimental animal studies
The distribution of acetone has been studied in mice exposed to
acetone by inhalation (Wigaeus et al., 1982). Mice were exposed to
1190 mg/m3 (500 ppm) 14C-acetone for 1, 3, 6, 12 and 24 h or for 6
h/day for 1, 3 or 5 consecutive days and sacrificed. Radioactive
unmetabolized acetone and total radioactivity were found in blood,
heart, pancreas, spleen, kidney, brain, liver, thymus, testis, vas
deferens, lung, muscle, brown adipose tissue, subcutaneous adipose
tissue and intraperitoneal adipose tissue. Acetone levels generally
peaked from about 145.2 to 203.3 µg/g tissue except in adipose tissues
for total radioactivity. Peak levels of metabolized acetone were
generally < 58.0-75.5 µg/g tissue. Exposure for longer than 6 h
resulted in no further accumulation of total radioactivity except in
the liver and brown adipose tissue, where levels rose to 278.8 µg/g in
the liver and 151.0 µg/g in brown adipose tissue at 24 h. Only about
10% of the radioactivity in the liver at 24 h was unmetabolized
acetone. When file mice were exposed intermittently on 3 or 5
consecutive days, most tissues showed no or only a small additional
increase in radioactivity after more than 1 day of exposure; however,
the concentration in adipose tissue increased significantly with
increasing exposure duration up to 5 days. The ratio of acetone in the
tissues to that in blood was < 1 at all exposure times except for the
lungs (the site of exposure). However, the ratio of total
radioactivity in the tissues to that in the blood showed that after 1
and 3 h exposure, only the lung had a ratio > 1, whereas the ratios
in the kidneys and liver were > 1 after 6 h. Only the muscle and
subcutaneous and intraperitoneal adipose tissue levels rose
continuously. Elimination of acetone was complete in all tissues by
24 h after exposure, but total radioactivity, indicative of
metabolites, was still present in all tissues except blood and muscle.
These data indicate that acetone is not selectively distributed to any
tissues but is more evenly distributed in body water. It appears that
acetone does not accumulate with repeated exposure. The continued
accumulation of radioactivity in the liver and brown adipose tissue is
probably the result of high metabolic turnover in these tissues.
6.2.2 Oral exposure
No studies were located regarding the distribution of acetone or
its metabolites in humans or animals after oral exposure except that
acetone was found in the liver of rats after oral exposure
(Charbonneau et al., 1986a, 1991). Acetone is absorbed from the
gastrointestinal tract and is highly water soluble. Therefore,
distribution to tissues with high water content is to be expected.
6.2.3 Injection exposure
Intravenous injection of 100 mg acetone/kg body weight to
pregnant rats on gestational day 19 resulted in high levels of
1,2-propanediol and acetol in the fetuses (Peinado et al., 1986).
Whether these findings reflect transfer of the metabolites from the
dams or metabolism of transferred or endogenous acetone by the
fetuses was not resolved.
6.2.4 Distribution summary
There are few studies on the distribution of acetone. Since
acetone is highly water soluble, it has been shown to be widely
distributed to tissues with a high water content.
6.3 Metabolism
6.3.1 Human studies
Acetone is a normal product of intermediary metabolism. The
metabolism of acetone appears to be independent of route of
administration and involves at least three separate gluconeogenic
pathways, with ultimate incorporation of carbon atoms into glucose and
other products and substrates of intermediary metabolism with the
generation of carbon dioxide. As previously mentioned, acetone is one
of three ketone bodies produced by acetyl coenzyme A within the liver.
The metabolic pathways appear to be similar in humans and animals. The
primary pathway involves hepatic metabolism of acetone to acetol,
followed by metabolism of acetol to methylglyoxal, while two secondary
(minor) pathways are partially extrahepatic, involving the
extrahepatic reduction of acetol to L-1,2-propanediol. Some exogenous
acetone is unmetabolized and is excreted primarily in the expired air.
Little acetone is excreted in urine.
Metabolic studies in humans were conducted in normal fasted,
obese fasted, and diabetic patients (Reichard et al., 1979, 1986). The
involvement of gluconeogenesis was demonstrated in normal patients
fasted for 3 days, obese patients fasted for 3 days, and obese
patients fasted for 21 days before intravenous injection of
2-[14C]-acetone (Reichard et al., 1979). According to the
researchers, fasted individuals had a daily intake of one multivitamin
capsule and at least 1500 ml of water. The percentages of 14C-glucose
in plasma derived from 14C-acetone were 4.2, 3.1 and 11.0% in the
three respective groups, suggesting the involvement of gluconeogenesis.
Cumulative 14C-carbon dioxide excretion by the lungs during the 6-h
collection period accounted for 17.4, 21.5 and 4.9% in the three
respective groups. Radioactivity was also incorporated into plasma
lipids and plasma proteins. Unmetabolized acetone in the expired air
accounted for 14.7, 5.3 and 25.2%, urinary excretion of acetone
accounted for 1.4, 0.6 and 1.3%, respectively, and in vivo
metabolism accounted for 83, 94.1 and 73%, respectively, of the
radioactivity, Intravenous infusion of 2-[14C]-acetone into patients
with diabetic ketoacidosis resulted in a mean plasma acetone turnover
rate of 6.45 µmol/kg/min (Reichard et al., 1986). Analysis of glucose
in urine revealed a labelling pattern in five of the six patients
consistent with the involvement of pyruvate in the gluconeogenic
pathway, while a different pathway may have been followed in the other
patient. Acetol and 1,2-propanediol were also detected in the plasma
and the concentrations of these metabolites were directly related to
the plasma level of acetone. The results demonstrated high plasma
acetone levels in decompensated diabetic patients with moderate to
severe ketoacidosis. The proposed pathway of acetone metabolism in
these patients is acetone to acetol to 1,2-propanediol to pyruvate and
ultimately to glucose, but other pathways may exist.
Jones & Andersson (1995) reported on a man arrested and suspected
of drunken driving. Both isopropyl alcohol (0.17 mg/ml) and acetone
(0.45 mg/ml) were found in his blood, but one of the breath tests
identified that an interferant was present. The case history showed
that the man was being treated for hyperglycaemia with a controlled
diet.
Jones et al. (1993) examined blood concentrations of acetone in
three groups: drunk drivers, type-1 diabetic outpatients and healthy
blood donors. The median concentrations were 2.03 mg/litre, 1.90
mg/litre and 1.26 mg/litre, respectively. While previous research
indicated that during fasting and uncontrolled diabetes mellitus
acetone increases, Jones et al. (1993) indicated that controlled
diabetes results in near normal acetone concentrations.
Kundu et al. (1993) studied acetone and established a correlation
between rate of fat loss and breath acetone concentration. The study
consisted of 58 men and women, 10-30% above their ideal body weight on
special diets of 1000 and 1200 calories per day. Breath acetone
concentrations were taken immediately upon wakening and increased in
the first few days to a plateau at approx. 7 days.
6.3.2 Experimental animal studies
The metabolism of acetone has been studied extensively in
laboratory animals, primarily in rats, and three separate pathways of
gluconeogenesis have been elucidated (Fig. 1). In many experiments,
rats, mice or rabbits have been exposed by inhalation, gavage,
drinking-water, or by intravenous, subcutaneous or intraperitoneal
injection of non-radiolabelled acetone or acetone labelled with 14C
in the methyl groups, number 2 carbon atom, or all three carbon atoms
(Price & Rittenberg, 1950; Rudney, 1954; Mourkides et al., 1959;
Casazza et al., 1984; Hetenyi & Ferrarotto, 1985; Koop & Casazza,
1985; Johansson et al., 1986; Kosugi et al., 1986a,b; Puccini et al.,
1990; Skutches et al., 1990). In these experiments, identification of
metabolites in liver, plasma or urine, the labelling patterns of 14C
incorporation into metabolites from 14C-acetone in plasma or in
liver, or the results of enzyme reactions using microsomes from
acetone-treated animals have led to the known metabolic pathways
illustrated in Fig. 1. Initially, acetone is oxidized (hydroxylation
of a methyl group) to acetol by acetone monooxygenase, an activity
associated with the cytochrome P-450IIE1, and requiring oxygen and
NADPH (Casazza et al., 1984; Koop & Casazza, 1985; Johansson et al.,
1986; Puccini et al., 1990). Cytochrome P-450IIE1 can be induced by
fasting, experimental diabetes, or exposure to ethanol or acetone
(Patten et al., 1986; Johansson et al., 1988; Puccini et al., 1990).
When the rate of acetone oxidation was evaluated in microsomes with
acetone added to the incubation system, microsomes from rats
(Johansson et al., 1986) and mice (Puccini et al., 1990) pretreated
with acetone had a 7-8 times greater rate than microsomes from control
rats or mice, indicating that acetone induces its own metabolism.
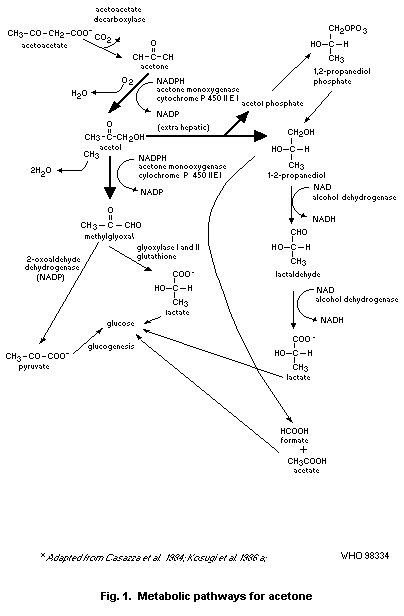 The relative importance of the three pathways in the metabolism
of acetone may depend upon the amount of acetone administered. When a
trace amount of [2-14C]-acetone was administered intravenously to
rats, the pattern of incorporation of 14C into glucose was
consistent with the production of glucose via the
methylglyoxal/lactate pathway (Kosugi et al., 1986a). When a higher
dose of [2-14C]-acetone (325 mg/kg) was injected, the pattern of
incorporation was more consistent with the 1,2-propanediol pathway.
These results suggest that at low doses of acetone or endogenous
acetone, the methylglyoxal and lactate pathways predominate, but at
higher doses, these pathways become saturated and metabolism is
shunted to the formate-acetate branch of the 1,2-propanediol pathway.
Methylglyoxal can then be converted to D-glucose by an
unidentified pathway (Fig. 1), and/or possibly by catalysis by
glyoxalase I and II and glutathione to D-lactate, which is converted
to D-glucose (Casazza et al., 1984). This formation of D-lactate from
acetone provides the body with a mechanism for recovering a portion of
the energy that is lost when acetone is formed from acetoacetate
(Morgott, 1993). The conversion of methylglyoxal to D-lactate by the
actions of glyoxalase I and II is well established (Racker, 1951), but
may represent a minor pathway in the metabolism of acetone (Casazza et
al., 1984; Kosugi et al., 1986a; Thomalley, 1990). In addition,
methyl-glyoxal is converted to D-glucose via conversion of
methylglyoxal to pyruvate by 2-oxoaldehyde dehydrogenase, an activity
identified using aqueous extracts of sheep liver acetone powders
(Monder, 1967).
In the second and third pathways, acetol is converted to
L-1,2-propanediol by an extrahepatic mechanism that has not been
characterized (Rudney, 1954; Casazza et al., 1984; Kosugi et al.,
1986a,b; Sktuches et al., 1990). The two pathways then diverge from
the point of production of 1,2-propanediol. In the second pathway,
1,2-propanediol formed extra-hepatically returns to the liver where it
is converted to L-lactaldehyde by nicotinamide adenine dinucleotide
(NADH)-dependent alcohol dehydrogenase (Casazza et al., 1984; Kosugi
et al., 1986a,b), and L-lactaldehyde, in mm, is converted to L-lactate
(Rudney, 1954; Ruddick, 1972; Casazza et al., 1984) by NADH-dependent
aldehyde dehydrogenase (Casazza et al., 1984). L-lactate can then be
converted to D-glucose (Casazza et al., 1984). In the third pathway,
the L-1,2-propanediol formed extra-hepatically returns to the liver
where it is degraded by other mechanisms to acetate and formate
(Sakami & Rudney, 1952; Ruddick, 1972; Casazza et al., 1984),
Several studies have traced the labelling patterns of 14C from
[2-14C]-acetone or [1,3-14C]-acetone to gluconeogenic precursors
and formate to incorporation of 14C into glycogen, glycogenic amino
acids, fatty acids, haem, cholesterol, choline and urea (Price &
Rittenherg, 1950; Sakami & LaFaye, 1950; Mourkides et al., 1959). The
pattern of labelling suggested the involvement of the pathway to
acetate and formate. Fatty acids, amino acids and glycogen may also
enter stages of intermediary metabolism. Factors affecting the
formation and release of long-chain fatty acids from adipose tissue
during lipolysis can affect the circulatory levels of acetone
(Morgott, 1993).
Although the liver is the primary site of acetone metabolism,
radioactive unmetabolized acetone and total radioactivity have been
found in many other mice tissue and organs after inhalation exposure
to 14C-acetone (Wigaeus et al., 1982). The fraction of total
radioactivity that was not unchanged acetone represented metabolites.
Elimination of acetone was complete in all tissues by 24 h after
exposure, but total radioactivity, indicative of metabolites, was
still present in all tissues except blood and muscle. Whether these
tissues (other than the liver) were capable of metabolizing acetone or
whether the metabolites themselves were distributed to the tissues was
unclear. However, microsomes from the lungs of hamsters exposed to
acetone in drinking-water for 7 days had a 500% increased activity of
aniline hydroxylase activity, an enzyme associated with cytochrome
P-450IIE1 (Ueng et al., 1991). The level of cytochrome P-450IIE1
increased 6-fold in microsomes from the nasal mucosa of rabbits
exposed to acetone in drinking-water for 1 week (Ding & Coon, 1990).
In hamsters given drinking-water containing acetone for 7 days (Ueng
et al., 1991) or 10 days (Menicagli et al., 1990), the microsomes
prepared from kidneys had increased levels of cytochrome P-450 and
cytochrome b5. These results suggest that acetone metabolism, which
involves cytochrome P-450IIE1, may occur in the lungs and kidneys of
hamsters and the nasal mucosa of rabbits. Incubation of acetone with
homogenates of nasal mucosa from mice indicated that acetone was
metabolized via a NADPH-dependent pathway in vitro, but no evidence
of in vivo metabolism of acetone by the upper respiratory tract was
found in mice, rats, guinea-pigs or hamsters (Morris, 1991 ).
Diet as well as physiological or genetic status may alter the
metabolism of acetone. When non-diabetic and diabetic rats were
treated by gavage with acetone at doses of 1000, 2000 or 4000 mg/kg,
isopropyl alcohol was detected in the blood (Lewis et al., 1984). The
levels of isopropyl alcohol and acetone increased with higher doses in
the diabetic rats, although with plateaus for both acetone and
isopropyl alcohol at 1000 and 2000 mg/kg doses, but levelled off in
the non-diabetic rats, indicating either saturation of the metabolic
pathway from acetone to isopropyl alcohol or a reversibility of the
conversion at high doses. It was suggested that in the diabetic rats,
acetone and NADH+, both needed for isopropyl alcohol production from
acetone, presumably by alcohol dehydrogenase, may be diverted to
gluco-neogenic pathways to meet the diabetic rat's need for glucose,
resulting in the short plateau. The subsequent rises of both compounds
at the high dose of acetone in the diabetic rats could be accounted
for by greater generation of NADH+ from fatty acid oxidation in the
diabetic rat, which reduces acetone to isopropyl alcohol, accounting
for the rising level of isopropyl alcohol. Liver homogenates from mice
heterozygous for the obesity gene treated with acetone were more
effective in converting acetone to lactate than liver homogenates from
normal homozygous mice treated with acetone (Coleman, 1980).
In pregnant or virgin rats (either fed or fasted) injected
intravenously with acetone, plasma acetol levels were not
significantly different between fasted and non-fasted rats, but
pregnant rats had significantly lower levels than virgin rats (Peinado
et al., 1986). Liver levels of acetol were also significantly lower in
pregnant rats than in virgin rats, Methylglyoxal levels were very high
in the livers and plasma of non-fasted rats (pregnant or virgin), but
fasting resulted in much lower levels. In contrast, no major
differences were found in the expiration of carbon dioxide between
fasted and diabetic rats injected intraperitoneally with acetone
(Mourkides et al., 1959) or in the labelling pattern of 14C derived
from 14C-acetone into glucose among non-fasted diabetic, fasted
diabetic, normal non-fasted and normal fasted rats injected
intravenously with 14C-acetone (Kosugi et al., 1986a,b).
6.3.3 Metabolism summary
As shown in Fig. 1, acetone is oxidised to acetol, which is then
metabolized by at least three different pathways and is independent of
the route of exposure. Glycogenesis is a driving force and acetone is
one of three ketone bodies produced by acetyl coenzyme A in the liver.
The relative importance of each pathway is probably dependent on
both the amount and route of exposure. Diet, physiological and genetic
status, and diabetes may affect the metabolism of acetone. Acetone is
either metabolized or excreted in both humans and animals, and the
time taken for elimination depends on the amount of acetone absorbed.
6.4 Elimination and excretion
Exhalation accounts for the elimination of about 20% as breath
acetone at low plasma acetone concentrations. Approximately 75% is
metabolized, only small amounts appearing in the urine. However, at
high plasma acetone concentrations, 80% can be accounted for by breath
acetone with only 20% being metabolised in vivo. The main route of
excretion of acetone is by the lung regardless of the route of
exposure (Owen et al., 1982).
6.4.1 Human studies
The clearance of acetone from blood was constant regardless of
blood acetone concentration (DiVincenzo et al., 1973). Halftimes for
blood elimination of 3-3.9 h have been estimated in humans exposed to
240-590 mg/m3 (100-500 ppm) for 2-4 h (DiVincenzo et al., 1973;
Wigaeus et al., 1981; Brown et al., 1987). Elimination half-times
between 3.9 and 6.2 h have been estimated for blood (Wigaeus et al.,
1981, Wang et al., 1994), and no differences in elimination half-times
were found between men and women (Brown et al., 1987). Because the
elimination rate depends on the metabolism, the effect of induction of
P450-dependant monooxygenase hydroxylase by acetone itself (Puccini et
al., 1989) or other specific inducers of the isozymes involved could
change the half-time. Longo et al. (1993) have shown that the set of
isozymes differs among animal species, and some have not been isolated
and characterized (Puccini et al., 1989). The elimination from blood
was found to be complete in 24 h after a 6-h exposure in subjects
exposed to 470 mg/m3 (250 ppm), in 32 h in subjects exposed to 1190
mg/m3 (500 ppm), and in 48 h in subjects exposed to 2370 mg/m3
(1000 ppm) (Matsushita et al., 1969b). When volunteers were exposed
for 6 h/day for 6 days, the blood levels of acetone rose each day and
declined to background levels by the following morning each day when
the exposure concentration was 470 mg/m3 (250 ppm) (Matsushita et
al., 1969a). At an exposure concentration of 1190 mg/m3 (500 ppm),
however, the blood levels declined each day, but not to pre-exposure
levels. At the end of the 6-day exposure, blood acetone levels
declined to pre-exposure levels within 2 days for the 470 mg/m3
group and declined within 3 days for the 1190 mg/m3 group. From the
half-life and the data on time for decline to pre-exposure levels, it
appears that at higher concentrations, acetone may accumulate slightly
in the blood during daily intermittent exposure, as would be
experienced by workers.
The rate and pattern of respiratory excretion of acetone is
influenced by exposure concentration, duration, the level of physical
activity during exposure and gender. In humans exposed to acetone
concentrations of < 2970 mg/m3 (< 1250 ppm) for < 7.5 h/day
in a complex protocol for < 6 weeks, the rate of respiratory
excretion was a function of the duration, and the concentration of
acetone in breath after exposure was directly related to the
time-average concentration during exposure, with constant duration
(Stewart et al., 1975). The length of time after exposure in which
acetone could be detected in the expired air was related to the
magnitude of exposure, with acetone still readily detectable 16 h after
exposure to 2370 or 2970 mg/m3 (1000 or 1250 ppm) for 7.5 h.
Excretion of acetone by the lungs was complete within 20 h post-exposure
in humans exposed to 560 mg/m3 (237 ppm) for 4 h (Dick et al.,
1989). During exposure for 2 h, the acetone concentration in expired
air rose to 50 mg/m3 (20 ppm) in humans exposed to 240 mg/m3 (100
ppm) and to 215-240 mg/m3 (90-100 ppm) in those exposed to 1190 mg/m3
(500 ppm) (DiVincenzo et al., 1973). After exposure to 240 mg/m3, the
expired air concentration of acetone declined biphasically over the
next 7 h to 12 mg/m3 (5 ppm). However, after exposure to 1190 mg/m3
(500 ppm), the expired air concentration dropped sharply to 5 mg/m3
(2 ppm) and declined to 2 mg/m3 (1 ppm) over the next 7 h. Prolonging
the exposure duration to 4 h resulted in less than a 2-fold increase
in acetone levels in post-exposure expired air, which may reflect a
greater loss of acetone through metabolism and urinary excretion.
Exercise during the exposure period increased the elimination almost
2-fold. In humans exposed to acetone at rest, during exercise at a
constant workload, or during exercise with step-wise increments in
workload, expiration of acetone via the lungs amounted to 70, 80 and
200 nag, respectively, at 4 h post-exposure and to 50, 80 and 200 mg,
respectively, over the next 4-20 h (Wigaeus et al., 1981). Excretion
of acetone from the lungs and kidneys (combined) amounted to 16, 20
and 27% of the amount absorbed in the three respective groups of
subjects. Urinary excretion amounted to only 1% of the total uptake.
Women expired acetone more slowly than men after a 4-h exposure to
300-310 mg/m3 (127-131 ppm), but the percentages excreted by the
lungs were not statistically significantly different between men and
women (17.6% for men, 15.0% for women) (Nomiyama & Nomiyama, 1974b).
Very little acetone is excreted in the urine (DiVincenzo et al.,
1973; Wigaeus et al., 1981; Vangala et al., 1991; Kawai et al., 1992).
Urinary excretion is biphasic (Pezzagno et al., 1986). Peak urinary
excretion occurred between 1 and 3.5 h after exposure (Matsushita et
al., 1969b; Wigaeus et al., 1981). In male volunteers exposed by
inhalation to 1180-2350 mg/m3 (497 or 990 ppm) acetone for 4 h,
cumulative acetone excretion in urine at 18 h after cessation of
exposure was 89.5 mg, suggesting slow excretion of acetone in the
urine (Vangala et al., 1991). Again, the amount of acetone excreted in
the urine will be influenced by the exposure concentration, the
duration of exposure, and the level of physical activity during
exposure. The acetone concentration in the urine ranged from 0 to 17.5
mg/litre at the end of the 8-h work shift in 45 workers exposed to
0-165 mg/m3 (0-70 ppm) acetone (background urinary concentration in
343 non-exposed subjects averaged 1.5 mg/litre) (Kawai et al., 1992).
Acetone levels in the preshift urine samples were significantly higher
than background levels when acetone exposure on file previous day was
above 35 mg/m3 (15 ppm). There was no significant difference between
background urine levels and preshift urine levels when the previous
day's exposure was < 35 mg/m3 (< 15 ppm). Total 24-h urine
content of acetone was 1.25 mg in subjects exposed to 240 mg/m3 (100
ppm) for 2 h and 3.51 mg in subjects exposed to 1190 mg/m3 (500 ppm)
for 2 h (DiVincenzo et al., 1973). Prolonging the duration to 4 h in
the 240 mg/m3 group resulted in a total of 1.99 mg acetone in the
urine. A slight increase in the urinary content of acetone (1.39 nag)
was found when humans exposed to 240 mg/m3 for 2 h exercised during
the exposure. The nature of physical activity during exposure also
influenced the urinary excretion. At 3-3.5 h after exposure, 8.5, 8.5
and 13.4 mg were excreted by the kidney in subjects exposed at rest,
during exercise at a constant workload, and during exercise with
step-wise increments in workload, respectively (Wigaeus et al., 1981).
Urinary excretion amounted to only 1% of the total uptake.
In volunteers who ingested 40-60 mg/kg acetone, the elimination
of acetone in expired air and urine was determined 2 h later, and a
rate of metabolism of 1.82 mg/kg per h along with the excretion data
was used to calculate that 3.54-7.38 mg/kg had been excreted and
metabolized (Haggard et al., 1944). The authors estimated that 65-93%
of the administered dose was metabolized, the remainder being
eliminated in the urine and expired air.
The only other information regarding excretion of acetone in
humans after oral exposure is from case reports of accidental or
intentional ingestion of materials containing acetone plus other
components that may have influenced the elimination of acetone. In a
man who ingested liquid cement containing 18% acetone (231 mg/kg), 28%
2-butanone and 29% cyclohexanone, and 720 ml sake, the plasma level of
acetone was approx. 1120 mg/litre 5 h after ingestion and declined to
65 mg/litre at 18 h, 60 mg/litre at 24 h and < 5 mg/litre at 48 h
(Sakata et al., 1989). A first-order plasma elimination rate constant
of 0.038/h and a half-time of 18.2 h were calculated. The urinary
level of acetone decreased gradually from about 123 mg/litre at 5 h
after ingestion to about 61 mg/litre at 19 h. In a case of a known
alcoholic who had ingested nail polish remover and whose blood acetone
level was 2.5 g/litre) upon admission to the hospital, the blood level
of acetone declined in a log-linear manner to about 0.6 g/litre about
86 h after admission, with a half-life of 31 h (Ramu et al., 1978).
The calculated clearance of acetone from the lungs was 29 ml/min or
0.39 ml/min per kg. A half-time of 25 h for lung clearance was
calculated, which is in agreement with the observed plasma elimination
half-tune of 31 h. The serum acetone level of a 30-month-old child was
4.45 g/litre 1 h after ingestion of a 6-ounce bottle of nail polish
remover (65% acetone) and declined to 2.65 g/litre at 117 h, to 0.42
g/litre at 48 h, and to 0.04 g/litre at 72 h (Gamis & Wasserman,
1988). The half-time of acetone in this patient was 19 h in the severe
early stage and 13 h in later stages of intoxication, which suggested
to the authors greater metabolism and/or excretion in children,
compared with adults.
Information regarding excretion of acetone after dermal exposure
of humans is limited, but the main route of excretion is via the
lungs, with little excreted in the urine. Application of an
unspecified quantity of acetone to a 12.5 cm2 area of skin of
volunteers for 2 h/day for 4 days resulted in alveolar air levels of
12-30 mg/m3 (5-12 ppm) and urinary concentrations of 8-14 mg/litre
on each day (Fukabori et al., 1979). These levels declined to
background levels by the next day after each exposure. Higher alveolar
air and urinary levels were obtained when the daily exposure increased
to 4 h/day: 60-80 mg/m3 (25-34 ppm) in alveolar air and 29-41
mg/litre in urine. However, these levels also returned to background
each day.
As determined in humans (Reichard et al., 1979), physiological
status may influence the disposition of endogenous and exogenous
acetone. In groups of non-obese patients fasted for 3 days, obese
patients fasted for 3 days, and obese patients fasted for 21 days and
injected intravenously with 14C-acetone, 8-29% of the urinary
acetone was derived from plasma radioactive acetone (Reichard et al.,
1979). The concentrations of urinary acetone were 1.2, 0.4 and 151
mg/litre in 3-day-fasted non-obese, 3-day-fasted obese, and
21-day-fasted obese patients, respectively. The rates of urinary
acetone excretion were 2962, 1800 and 3542 µg/min, respectively,
suggesting marked renal reabsorption or back-diffusion. The percentages
of measured acetone production that could be accounted for by excretion
via the lungs were 14.7, 5.3 and 25.2%, respectively. The percentages
that could be accounted for by urinary excretion were 1.4, 0.6 and
1.3%, respectively. Cumulative excretion of 14C-carbon dioxide
during the 6-h turnover study periods accounted for 17.4, 21.5 and
4.9%, respectively. Thus, non-obese subjects fasted for 3 days
excreted more acetone at higher rates than did obese subjects fasted
for 3 days. However, excretion by the obese patients fasted for 21
days exceeded that by both 3-day-fasted groups. These differences are
probably related to the effect that the degree of starvation ketosis
has on the metabolism and overall disposition of acetone.
6.4.1.1 Occupational exposure studies
There are many reports in the literature regarding exposure to
solvents, many of which are confounded for risk assessment purposes by
exposure to a mix of several solvents. The following information takes
the essence of these studies to give the reader an understanding of
the various exposure scenarios and the measured acetone
concentrations.
Satoh et al. (1995) studied male shift workers at a Japanese
acetate fibre manufacturing facility. The first group was 110
acetone-exposed (AE) workers, while the control groups (CG) consisted
of 67 workers in the same factory area but not engaged in acetone
manufacturing (background concentration < 12 µg/m3; < 5 ppb).
Levels were measured in the breathing zone and in samples of alveolar
air, blood and urine. Mean levels were reported as 857 mg/m3 (361
ppm), 216 mg/m3 (91 ppm), 67 mg/litre and 37 mg/litre, respectively.
They evaluated whether exposure to acetone during the previous day
affects the biological monitoring value at the end of a work day, i.e.
urinary values of acetone from monitoring do not revert to levels seen
with normal background exposure. Matsushita et al. (1969a,b) observed
that humans exposed to 1190 mg/m3 (500 ppm) of acetone had higher
than background levels the next morning. According to Satoh et al.
(1995), the extent of previous exposure and the sampling time
influenced the biological monitoring value and were very critical.
Additionally, other factors such as alcohol consumption, could give
rise to more than 30 mg/litre of acetone in the urine. When the
exposure concentration of the workers was 595 mg/m3 (250 ppm), the
urinary level of acetone returned to background the next morning. When
the exposure concentration was raised to 1190 mg/m3 (500 ppm), the
following morning's acetone levels were higher than background. Wang
et al. (1994) reported similar results. The next morning, 16 h after
the end of a workshift, with a mean occupational exposure to acetone
of 336 mg/m3, blood and urinary concentrations of 3.5 mg/litre and
13.0 mg/litre, respectively, were much higher than levels in
non-exposed subjects.
Fujino et al. (1992) also investigated the relationship between
the environmental concentration and the concentration in urine,
alveolar air and blood. The environmental air concentration to which
each of the 110 subjects were exposed was closely correlated with all
body concentrations. The authors also found that urinary acetone
concentration is probably the best biological index of exposure
because it shows the strongest correlation.
Kawai et al. (1992) studied 45 acetone-exposed male workers
together with 343 non-exposed men to examine the quantitative
relationship between the levels of acetone vapour exposure and the
concentration of acetone in urine. Up to a vapour concentration of 35
mg/m3 (15 ppm), there was no increase in acetone concentration in
end-of-shift urine, but above this concentration there was an increase
in urinary acetone concentration in proportion to the acetone vapour
concentration. The authors reported that urinary acetone
concentrations collected at the shift-end and those before the shift
of the next morning were similar among those exposed to < 35
mg/m3 (15 ppm) acetone. However, among those exposed to acetone at
more than 35 mg/m3, acetone levels in the shift-end samples were
significantly higher than those in the pre-shift samples.
Mizunuma et al. (1993) and DeRosa et al. (1993) reported
different results in factory workers exposed to styrene and acetone.
Mizunuma et al. (1993) studied 41 workers in a factory making
fibre-reinforced plastics who were exposed to mixed styrene and
acetone vapour and 20 non-exposed workers. Acetone and styrene
concentrations in blood and urine correlated significantly with
intensity of exposure. Acetone was found to distribute evenly in blood
and urine, and was also distributed evenly between the cellular and
non-cellular fractions of the blood. DeRosa et al. (1993) monitored 22
workers exposed to styrene and acetone in two fibreglass industries to
determine if excretion of styrene metabolites differed over a working
week and if it was modified by simultaneous exposure to acetone.
Acetone exposure had no affect on styrene metabolism.
6.4.2 Experimental animal studies
As in humans, acetone is excreted mainly by the lungs of
experimental animals. Studies in animals have followed the elimination
of acetone from blood and tissues, excretion of acetone and carbon
dioxide in expired air, and the urinary excretion of formic acid.
Blood levels of acetone were highest immediately after a 4-h
exposure of rats to acetone (Charbonneau et al., 1986a). In rats
exposed to 23 700 mg/m3 (10 000 ppm), the blood level dropped from
2114 to 5 mg/litre in 25 h. In rats exposed to 35 600 mg/m3 (15 000
ppm), the blood level dropped from 3263 mg/litre to 50 mg/litre after
25 h. Elimination from blood was biphasic in rats exposed to 23 700
and 35 600 mg/m3. Elimination from blood was triphasic in rats
exposed to 2370, 5940 or 11 900 mg/m3 (1000, 2500 or 5000 ppm) and
was complete within 17-25 h. In dogs exposed to 240, 1190 or 2370
mg/m3 (100, 500 or 1000 ppm) acetone for 2 h, blood levels declined
in a log-linear manner with a half-time of 3 h, similar to that
observed m humans (DiVincenzo et al., 1973). Blood levels declined
from 25 mg/litre immediately after exposure to 10 mg/litre at 5 h
post-exposure for the 2370 mg/m3 (1000 ppm) group, from 12 to 3
mg/litre for the 1190 mg/m3 (500 ppm) group, and from 4 to 1.5
mg/litre for the 240 mg/m3 (100 ppm) group. Elimination of
radioactivity and 14C-acetone was fastest from blood, kidney, lungs,
brain and muscle tissues of mice exposed to 1190 mg/m3 (500 ppm)
14C-acetone for 6 h, with half-times of 2-3 h during 6 h
post-exposure (Wigaeus et al., 1982). Elimination of acetone was
complete in 24 h in all tissues, but radioactivity (indicative of
metabolites) was still present in all tissues except blood and muscle.
When rats were exposed for 5 days, acetone tended to slowly attain
steady state in adipose tissue.
Excretion of acetone in air followed pseudo-first-order kinetics
in rats exposed to up to 50 mg/m3 (< 20 ppm) acetone for 1-7 days,
while at higher concentration saturation kinetics were observed
(Hallier et al., 1981). In rats exposed to 1190 mg/m3 (500 ppm)
14C-acetone for 6 h, 42 µmol of radioactive acetone and 37 µmol
14C-carbon dioxide were excreted in the expired air during a 12-h
post-exposure period, 95% and 85%, respectively, being recovered in
the first 6 h post-exposure (Wigaeus et al., 1982). Radioactive
acetone accounted for 52% and radioactive carbon dioxide accounted for
48% of the expired radioactivity. The concentration of acetone in the
expired breath of dogs exposed to 240, 1190 or 2370 mg/m3 (100, 500
or 1000 ppm) acetone for 2 h declined in a log-linear manner
(DiVincenzo et al., 1973). The breath levels were directly related to
the magnitude of exposure. Breath levels declined from 3.8 mg/m3
(1.6 ppm) at 30 min after exposure to 0.7 mg/m3 (0.3 ppm) at 300 mm
in the 240 mg/m3 (100 ppm group), from 16.1 to 3.5 mg/m3 (6.8 to
1.5 ppm) in the 1190 mg/m3 (500 ppm) group, and from 35.6 to 9.5
mg/m3 (15 to 4 ppm) in the 2370 mg/m3 (1000 ppm) group.
Urinary excretion of formic acid was followed for 7 days in rats
exposed by inhalation to 147 200 mg/m3 (62 000 ppm) acetone for 2
days. The rate of formic acid excretion was 344 µg/h compared with 144
µg/h in controls (Hallier et al., 1981).
For experimental animals, information regarding the excretion of
acetone after oral exposure is available only for rats. As is the case
after inhalation exposure, acetone, mainly as carbon dioxide, is
excreted primarily by the lungs. In a rat given 1.16 mg/kg
14C-acetone by gavage in water, expiration of 14C-carbon dioxide
totalled 47.4% of the administered radioactivity over the 13.5-h
collection period (Price & Rittenberg, 1950). In another experiment, a
rat was given 7.11 mg/kg radioactive acetone. A small amount of
radioactive acetone (10%) was found in the expired air. Radioactive
carbon dioxide and acetate were also detected. In a rat made diabetic
by alloxan and given 6.15 mg/kg 14C-acetone, a total of 7.29% of the
administered radioactivity was expired as acetone and 51.78% as carbon
dioxide. Radioactive acetate was detected in the urine. These data
indicate that very little acetone (< 10%) was excreted by the lungs
after small doses of acetone. A major fraction was oxidized to carbon
dioxide and some of the derived carbon was used for acetylation. The
diabetic rat was also able to oxidize acetone, but only to approx. 70%
of that in the normal rat.
The dose of acetone influences the elimination of acetone from
blood (Plaa et al., 1982). At a dose of 78.44 mg/kg, the maximum blood
level of 200 mg/litre at 3 h declined to 10 mg/litre at 12 h, where it
remained for the next 12 h (data inadequate to evaluate total body
clearance). At a dose of 196.1 mg/kg body weight, the maximum blood
level of 400 mg/litre at 6 h declined biphasically to 50 mg/litre at
12 h and to 30 mg/litre at 18 h where it remained until 24 h (total
body clearance = 64 ml/h). At a dose of 784.4 mg/kg body weight, the
maximum blood level of 900 mg/litre at 1 h declined to 300 mg/litre at
12 h, to 110 mg/litre at 18 h, and to 50 mg/litre at 24 h (total body
clearance = 86 ml/h). At a dose of 1961 mg/kg body weight, the maximum
blood level of 1900 mg/litre at 3 h declined slowly to 400 mg/litre at
24 h (total body clearance = 75 ml/h). Thus total body clearance was
independent of dose, but the half-time for elimination increased from
2.4 h for 196.1 mg/kg to 4.9 h for 784.4 mg/kg, and 7.2 h for 1961
mg/kg.
The vehicle (corn oil or water) in which acetone is administered
has little influence on the elimination of acetone from blood
(Charbonneau et al., 1986a). After gavage treatment of rats with 78
196, 392, 784 or 1177 mg/kg acetone in corn oil or water, elimination
was biphasic for the two higher doses and triphasic for the lower
doses. Acetone elimination from blood declined to <5 to <10 µg/ml by
18-26 h at all dose levels, but minor differences were found between
water and corn oil as vehicle. The blood concentration curves from
rats given acetone in water more closely resembled those from rats
exposed by inhalation.
6.4.3 Elimination/excretion summary
Following single exposure, endogenous acetone is eliminated from
the body at low doses via metabolic pathways and at high doses via the
lungs with the urine as a minor pathway. In general, acetone is
eliminated from the body within 24 h, unless exposure is continuous or
intermittent.
At low exposure levels, acetone is primarily eliminated by
metabolism, while at higher exposure levels it is mainly eliminated by
the lungs. Very little acetone is excreted in the urine. The
elimination rate is highly dependent upon exposure level, duration of
exposure, metabolism, genetic and pathophysiological conditions, and
physical activity.
6.4.4 Physiologically based pharmacokinetic model
A physiologically based pharmacokinetic model has been developed
to study the kinetic behaviour of acetone (Kumagai & Matsunaga, 1995).
In this model, acetone can be distributed into eight tissue groups:
the mucous layer of the inhaled air tract, the mucous layer of the
exhaled air tract, a compartment for gas exchange (alveolus of the
lung), a group of blood-vessel-rich tissues including the brain and
heart, a group of tissues including muscles and skin that have low
blood perfusion rates, a group of fatty tissues, an organ for
metabolism (liver), and a compartment for urinary excretion (kidney).
With an appropriate value for the volume of mucous layer, the
simulated acetone concentration in arterial blood, and exhaled air,
urine, and fatty tissue were found to agree well with the experimental
data. The volume of mucous layer and rate of respiration were critical
for the appropriate simulation.
This model is suitable for occupational exposure assessments.
6.5 Retention and turnover
Reichard et al. (1979) studied acetone production in obese and
non-obese patients during starvation-induced ketonaemia. The
elimination and metabolism of acetone were measured, as well as urine
and plasma concentrations by radioactive labels in order to calculate
a plasma turnover rate for acetone. This was 20-77 µmol/m2 body
surface area per min. Owen et al. (1982) and Reichard et al. (1986)
furthered the original study in patients with severe diabetic
ketoacidosis. Owen et al. (1982) measured rates of acetone production
ranging from 68 to 581 µmol/min (using a standard human body surface
area of 1.73 m2), with plasma concentrations ranging from 1.55 to
8.91 mmol/litre. In the Reichard et al. (1986) study, the plasma
concentrations ranged from 0.50 to 6.02 mmol/litre, and the acetone
remover rate was linearly related to the plasma concentration up to a
level of 7.61 mmol/litre. The two studies found opposite relationships
between acetone turnover rate and plasma concentration.
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Short-term toxicity
A summary of short-term inhalation toxicity studies of acetone is
given in Table 8, acute oral studies are in Table 9, and dermal
studies in Table 10. In the studies on rats and guinea-pigs, very high
levels of acetone were required to cause death. In rats, a 4-h LC50
of 76 mg/m3 and a 8-h LC50 of 50.1 mg/m3 have been reported
(Pozzani et al., 1959). A range of LD50 values was found for rats.
Kimura et al. (1971) stated that the lethality of acetone decreases
with age to a certain point in the rat life cycle. However, the
difference in values from older to younger adult rats was not found to
be statistically significant.
In acute oral studies, signs of narcosis usually precede death
(ATSDR, 1994). In the EHRT (1987) study, mice that were dosed by
gavage at 4800 mg/kg per day or more for 10 days displayed wheezing
and rapid/laboured breathing. Narcosis was present before death.
Savage treated rats were found to have increased haematocrit and
haemoglobin in high-dose males (2500 mg/kg per day), but not in
females. As will be seen below, these effects change with increasing
dose and duration. Considering all the studies, it appears that there
are sex and species differences for haematological effects from
exposure to acetone.
Acetone is known to be only mildly toxic to the liver, unless
physiological processes are compromised (e.g., diabetes mellitus). It
is known to potentiate and antagonize the effects of other chemicals
by inducing microsomal enzymes that metabolize other chemicals to
reactive intermediates. Acetone has been shown to increase the
activity of glutathione S-transferase (Sippel et al., 1991 ).
Induction of microsomal enzymes has been considered as a normal
physiological response to xenobiotics when it does not cause other
adverse effects such as increased liver weight, enzyme changes or
other hepatic effects.
In the NTP (1991) study, Dietz and co-workers found increased
liver weights at doses > 965 mg/kg per day, and in higher dose
groups (> 3896 mg/kg per day) hepatocellular changes accompanied
this effect. A 14-day drinking-water study of F344/N rats and B6C3F1
mice served as a preliminary study to the 13-week NTP (1991) study
discussed in section 7.2. Five animals/sex of each species were
administered drinking-water at concentrations of 0, 5, 10, 20, 50 or
100 g/litre (NTP, 1991; Dietz et al., 1991). Based upon water
consumption data, the authors estimated doses as follows: 0, 714,
1616, 2559, 4312 and 6942 mg/kg per day for male rats; 0, 751, 1485,
2328, 4350 and 8560 mg/kg per day for female rats; 0, 965, 1579, 3896,
6348 and 10 314 mg/kg per day for male mice; and 0, 1569, 3023, 5481,
8804 and 12 725 mg/kg per day for female mice.
At necropsy, body weights were decreased, compared with controls,
in male rats exposed to concentrations of > 50 g/litre and female
rats exposed to 100 g/litre. The report for the 14-day study did not
provide the collected organ weight data but stated that increased
relative weights of kidneys and liver were measured for exposed rats
and mice (dose level was not specified). No histopathological changes
were observed in the kidneys or livers of exposed rats or in the
kidneys of mice. Centrilobular hepatocellular hypertrophy was noted in
ali male mice at concentrations > 20 g/litre and in female mice at
concentrations of > 50 g/litre (NTP, 1991; Dietz et al., 1991).
The hypertrophy was described as "minimal" in female mice, but
increased incidence was noted with increasing concentration 2/5 and
5/5 for 50 g/litre and 100 g/litre, respectively. The incidences of
male mice with more severe hypertrophy (classified by the authors as
either "mild" or "moderate") increased with increasing concentrations
> 20 g/litre. The authors hypothesized that male mice may develop
a tolerance to acetone because hepatic changes were not observed in
male mice in the second study, the 13-week NTP study presented in the
next section. Thus, in evaluating oral studies of the effects of
acetone, the data indicated that changes occurred in the liver. The
determination of whether or not these were adverse depends more on the
determination of the severity of the effects. Acetone induced
microsomal enzymes and increased liver weights. It caused liver injury
with increased serum levels of liver enzymes and hepatocellular
hypertrophy. As will be seen later, acetone potentiates other
chemicals, which may increase these adverse effects or potentiate
adverse effects caused by other chemicals. Oral exposure to acetone
has also caused effects on the kidney in mice and rats, as shown in
the NTP (1991) study (see also section 7.2). The best-known effects of
acetone ingestion are the diabetes-like symptoms of hyper-glycaemia
and glycosuria.
7.1.1 Skin and eye irritation
Smyth et al. (1962) applied 1.0 ml acetone to the shaved dorsal
skin of rabbits without occlusion. After 24 h there was no evidence of
irritation. In CD-1 mice, a single application of 0.2 ml acetone to
shaved skin produced increased DNA synthesis and moderate hyperplasia
of the epidermis that was considered to be evidence of slight
irritation, In hairless mice treated with twice weekly application of
0.1 ml acetone for 18 weeks, moderate hyperplasia of the epidermis was
noted (Iversen et al., 1988)
In rabbits, severe eye inflammation and corneal necrosis followed
instillation of 0.005 ml acetone into the eye (Carpenter & Smyth,
1946; Smyth et al., 1962). A 3-min application of 3.9 ml caused
conjunctival oedema (Larson et al., 1956).
Table 8. Short-term animal inhalation exposure to acetone
Species Exposure NOAELb LOAELb Critical effect Reference
duration/frequencya mg/m3(ppm) mg/m3(ppm)
Guinea-pig 2 days 23 700 (10 000) 5/5 died; spleen and lung congestion; Specht et al.
24 h/day fatty liver; renal tubular distension (1939)
Guinea-pig 22-26 h 47 500 (20 000) 8/9 died; congestion and haemorrhage Specht et al.
of spleen and lung (1939)
Guinea-pig 25 min-23.4 h 51 750 (21 800) 2/10 died; narcosis, paralysis Specht et al.
(1939)
Guinea-pig 3-8.75 h 118 700 (50 000) 8/8 died; pulmonary congestion, Specht et al.
oedema, glomerular distension (1939)
Rat 2 h 120 120 (50 600) 5/5 died Bruckner &
Peterson (1981)
Rat 3 h 29 900 (12 600) CNS depression Bruckner &
Peterson (1981)
Rat 4 h 76 000 (32 000) LC50 Pozzani et al.
(1959)
Rat 4 h 38 000 (16 000) 1/6 died Smyth et al.
(1962)
Rat 4h 2850 (1200) 7100 (3000) Audiogenic seizures depressed Frantik et al.
(1988)
Rat 8 h 50 100 (21 050) Pozzani et al.
(1959)
Rat 14 days 5200 (2200) 26 100 (11 000) Reduced body weight, uterine weight, NTP (1988)
7 days/week decreased fetal weight;
6 h/day
GD 6-19
Rat 2 weeks 7100 (3000) 14 240 (6000) inhibition of avoidance behaviour Goldberg et al.
5 days/week in over 1/3 of rats (1964)
4 h/day
Mouse 6h 26100(11000) Severe narcosis NTP (1988)
Mouse 1 day 2370 (1000) 7100 (3000) Decreased behavioura[ response Glowa & Dews
(1987)
Table 8. (Continued)
Species Exposure NOAELb LOAELb Critical effect Reference
duration/frequencya mg/m3(ppm) mg/m3(ppm)
Mouse 12 days 5200 (2200) 15 650 (6600) Decreased fetal weight, reduced NTP (1988)
7 days/week sternal ossification
6 h/day
GD 6-17
a GD = gestation day.
b Dose levels for all studies are included in this table or the text only.
Table 9. Short-term oral exposure to acetone
Species Route Exposure NOAEL LOAEL Critical effect Reference
duration/frequency (mg/kg) (mg/kg)
Rat drinking-water 3-7 days 3214 reduced insulin production, less Skutches et al.
serious systemic problems (1990)
Rat drinking-water 14 days 4312 (male) 6942 (male) bone marrow hypoplasia NTP (1991)
8560 (female)
Rat drinking-water 10 days 3500 induction of hepatic cytochrome Puccini et al.
P450, aminopyrineN-demethylase, (1989)
ethoxycourmarin O-deethylase,
p-nitrophenol hydroxylase, acetone
hydroxylase, diethyl-ngrosamine
deethylase
Rat drinking-water 14 days 3500 induction of dealkylation of Fiodo & Bronzetti
amino-pyrine; induction of dealkylation (1995)
of anisole, hydroxylation of aniline;
decreased survival of tissue culture
cells treated with
dimethylnitros-amine; increased mutation
rate of tissue culture cells treated with
dimethylnitrosamine
Rat gavage once -- 15 mmol/kg induction of hepatic P450EI, Barnett et al.
(871 mg/kg) P450IB, P45OIIB, P45OIA2 (1992)
Rat gavage once 1726 LD50 Kimura et al.
(newborn) (1971)
Rat gavage once 4393 LD50 Kimura et al.
(14 day) (1971)
Table 9. (Continued)
Species Route Exposure NOAEL LOAEL Critical effect Reference
duration/frequency (mg/kg) (mg/kg)
Rat(young gavage once 7138 LD50 Kimura et al.
adult) (1971)
Rat gavage once 6667 LD50 Kimura et al.
(older adult) (1971)
Rat gavage once 5806 LD50 15% body weight loss, Freeman &
prostration Hayes (1985)
Mouse drinking-water 10-12 days 3500 induction of hepatic cytochrome Puccini et al.
P450, cytochrome b5, (1990)
ethoxy-courmarin O-deethylase,
p-nitrophenol, hydroxylase,
diethylnitrosamine deethylase
Mouse drinking-water 14 days 1579 (male) 3896 (male) mild hepatocellular hypertrophy NTP (1991)
12 725 (female)
Mouse gavage with 10 days 3500 reduced maternal body weight; EHRT (1987)
water (GD 6-15) reduced reproduction index;
once/day decreased pup survival
Hamster water 10 days 2100 induction of cytochrome P450 Puccini et al,
(1992)
a GD = gestation day.
Dose calculated on the assumption taken from Puccini et al. (1992) that 1% acetone in drinking-water is equivalent to 700 mg/kg per day.
7.2 Longer term toxicity
A limited number of oral animal studies are reviewed in Table 10.
Only one longer-term dermal study was available.
Bruckner & Peterson (1981) exposed male ARS/Sprague-Dawley rats
to acetone vapour concentrations of 0 or 45 100 mg/m3 (0 or 19 000
ppm), 3 h/day, 5 days/week for < 8 weeks. Groups of five rats were
killed and examined at 2, 4 and 8 weeks and at 2 weeks after
termination of exposure. No significant (p<0.05) effects were
noted in blood chemistry (blood urea nitrogen (BUN), serum
glutamic-oxaloacetic transaminase (SGOT), lactate dehydrogenase (LDH),
liver triglyceride levels and histology of heart, lung, kidney, brain
and liver. Small, statistically significant (p<0.02) decreases in
absolute weights of the brain at 4 and 8 weeks and kidney at 4 weeks
(but not at 8 weeks) were observed compared with controls. Without
providing quantitative data, the authors stated that the organ-to-body
weight ratios were slightly increased, suggesting that the effect on
absolute organ weight reflected a slight but statistically
insignificant reduction in rate of body weight gain. Two weeks after
termination of exposure, no significant differences in organ weights
were observed between treated and control rats.
Sollman (1921) exposed three rats to 2.5% acetone in
drinking-water for 18 weeks and reported that body weights and
consumption of food and water were decreased, compared with normal
values; the report cited normal values for consumption of food and
fluid and for body weight, but did not indicate whether these were
historical control values or values from concurrent control animals.
No deaths were noted during the experimental period, No other
end-points were examined.
Acetone was administered by gavage (as an aqueous solution) to
groups of 30 Sprague-Dawley rats/sex for 90 days at dose levels of 0,
100, 500 or 2500 mg/kg per day (US EPA, 1986a). At the highest dose
level, significantly increased values of RBC parameters were measured
at 46 and 90 days for males (haemoglobin, haematocrit, corpuscular
volume) and at 90 days for females (haemoglobin, haematocrit), Females
in the 500 and 2500 mg/kg per day groups had significantly increased
absolute kidney weights. Both sexes showed increased relative weights
of kidney and brain and increased absolute and relative liver weights
at the 2500 mg/kg per day dose. Body weights were unaffected in males,
but transient, significantly elevated body weights were observed in
females in the 500 and 2500 mg/kg per day groups. Statistically
significant clinical chemistry alterations, possibly attributable to
acetone, included increased alanine aminotransferase activities in
both sexes at 2500 mg/kg per day and decreased platelet counts, serum
glucose and potassium levels in males at 2500 mg/kg per day.
Table 10. Longer-term oral exposure to acetone
Species Route Exposure NOAEL LOAEL Critical effect Reference
duratron/frequency (mg/kg/day) (mg/kg/day)
Rat gavage with water 46-47 days 500 (male) 2500 excessive salivation; US EPA (1986a)
(once/day) increased haemoglobin,
haematocrit and mean cell
volume
Rat gavage with water 93-95 days 500 (female) 2500 decreased brain weight; US EPA (1986a)
(once/day) increased blood parameters
(as above)
Rat water 13 weeks 900 (male) 1700 increased incidence/severity NTP (1991)
1600 (female) 3100 of nephropathy in males;
haematological effects
Mouse water 13 weeks 4858 (male) - - NTP (1991)
11 298 (female)
Histological examinations were made of all major tissues and organs
from control and high-dose rats. For the low-dose (100 mg/kg) and
middle-dose (500 mg/kg) rats, histological examinations were
restricted to the heart, liver, kidneys and any tissues with gross
changes. Increasing doses were associated with increasing intensity of
tubular degeneration of the kidneys and hyaline droplet accumulation.
The Task Group noted that these changes were observed in all groups,
including controls. These changes were accentuated in males at the 500
and 2500 mg/kg per day dose levels and in females at the 2500 mg/kg
per day dose level. No other chemical-related histological changes
were noted. There were no effects on urinalysis or ophthalmological
parameters examined before sacrifice. No acetone-related effects were
seen at the 100 mg/kg per day dose level.
In a 13-week drinking-water study (NTP, 1991; Dietz et al.,
1991), groups of 10 F344/N rats of each sex and 10 female B6C3F1
mice were administered drinking-water containing 0, 2.5, 5, 10, 20 or
50 g/litre acetone. Groups of 10 male mice were exposed to
drinking-water containing 0, 1.25, 2.5, 5, 10 or 20 g/litre.
Corresponding TWA doses in treated animals were estimated by the
authors as 200, 400, 900, 1700 and 3400 mg/kg per day for male rats;
300, 600, 1200, 1600 and 3100 mg/kg per day for female rats; 892,
2007, 4156, 5945 and 11 298 mg/kg per day for female mice; and 380,
611, 1353, 2258 and 4858 mg/kg per day for male mice.
Necropsy was performed on all animals and histological
examinations were conducted on all major tissues and organs of animals
from control and high-dose groups. For lower dose groups, tissues
examined included heart, kidneys and spleen of male rats at the 10 and
20 g/litre dose levels; kidneys of male rats at 5 g/litre; bone marrow
of male rats at 20 g/litre; nasal cavity and turbinates of all groups
of female rats; mandibular lymph nodes of female rats at 20 g/litre;
and liver of all female mice. At necropsy, organ weights were obtained
and haematological examinations and examinations of sperm morphology
and vaginal cytology were performed.
No mortalities or overt clinical signs of toxicity were observed
for any of the rats within the experimental period. Depressed water
consumption and body weight gain were noted in rats of both sexes at
the highest concentration. Significantly increased (p<0.05) relative
weights of kidney, liver and lung were noted in rats of both sexes
exposed to 50 g/litre. Relative testis weights were also increased at
the highest concentration. Also noted at 50 g/litre were significant
decreases in caudal and epididymal weights and in sperm motility and
increased counts of morphologically abnormal sperm. Administration of
20 g/litre was associated with increased relative liver weights in
both sexes and increased relative kidney weights in female rats,
compared with controls.
"Mild but statistically significant" (p<0.05) alterations in
haematological parameters (haematocrit, haemoglobin and mean
corpuscular haemoglobin) were noted in rats of both sexes given 50
g/litre (increased leukocytes and lymphocyte counts, mean corpuscular
haemoglobin and mean cell volume, and decreased platelet counts) (NTP,
1991; Dietz et al., 1991). At 20 g/litre, haematological alterations
were restricted to male rats with the exception that females also
showed decreased platelet counts. The haematological alterations in
males were accompanied by increased pigmentation in the splenic red
pulp of all males exposed to concentrations >20 g/litre, compared with
controls.
"Mild" nephropathy was noted in male rats, including controls.
The incidence and intensity of the kidney lesions increased with
increasing drinking-water concentrations of acetone, particularly at
20 g/litre. However, the criteria for distinguishing between "minimal"
and "mild severity" for the observed nephropathy were not described.
Based on accentuated nephropathy in male rats, the LOEL was considered
by the Task Group to be 1700 mg/kg per day.
During the 13-week exposure of mice to drinking-water containing
acetone, no deaths or clinical signs of toxicity were recorded (NIP,
1991; Dietz et al., 1991). Body weights, growth, sperm morphology and
vaginal cytology also were unaffected in exposed mice. Significant
organ weight changes (p<0.05) were restricted to female mice
administered 50 g/litre; absolute and relative weights were increased
for liver and decreased for spleen. Small changes in a few
haematological parameters were noted in female mice (increased
haematocrit and haemoglobin) and male mice (increased haemoglobin and
mean corpuscular haemoglobin) exposed to their highest respective
concentrations. The authors did not consider the changes to be
toxicologically significant. The only histopathological alteration
noted for acetone-exposed mice was centrilobular hepatocellular
hypertrophy (liver cells with abundant eosinophilic cytoplasm and
slightly enlarged nuclei) in 2 of the 10 female mice administered 50
g/litre.
7.3 Reproductive toxicity, embryotoxicity and teratogenicity
NTP (1988) examined the in vivo developmental toxicity of inhaled
acetone in mice and rats. Groups of pregnant Sprague-Dawley rats
(26-29 rats/group) were exposed to 0, 1045, 5200 or 26 100 mg/m3 (0,
440, 2200 or 11 000 ppm) acetone vapour, 6 h/day, 7 days/week for 14
days (days 6-19 of gestation). Groups of pregnant Swiss CD-1 mice
(28-31 mice/group) were exposed to 0, 1045, 5200 or 15 670 mg/m3 (0,
440, 2200 or 6600 ppm) with the same protocol for 12 days (days 6-17
of gestation). Maternal body weights were obtained throughout the
experimental period. Uterine and fetal body weights were recorded at
sacrifice (gestation days 20 and 18 for rats and mice, respectively).
Live fetuses were examined for gross, visceral, skeletal and soft
tissue craniofacial defects.
No signs of maternal toxicity were noted in the pregnant rats
except that statistically significant reductions in body weight at 14,
17 and 20 days gestation, in cumulative body weight gain and in
uterine weight were observed for the 26 100 mg/m3 group. The
following were unaffected by exposure to acetone: absolute and
relative liver and kidney weights of the dams; number of
implantations; percentage of live pups/litter; percentage of
resorptions/litter; and the fetal sex ratio. Fetal weights were
significantly decreased in the 26 100 mg/m3 group compared with the
control group. The incidences of fetal malformations were not
significantly increased in any of the acetone-exposed groups compared
with controls, although the percentage of litters with at least one
malformed pup was greater in the 26 100 mg/m3 group (11.5%) compared
with the control group (3.8%). NTP (1988) concluded that acetone had
not caused a teratogenic effect in the rats in this study.
The only significant acetone-related effect in the pregnant mice
was increased relative liver weights in the 15 670 mg/m3 group
compared with controls. Developmental and reproductive indices were
unaffected in the acetone-treated groups of mice with the exceptions
of a statistically significant decrease in fetal weight and a slight,
but statistically significant, increase in the percentage incidence of
late resorptions, both in the 15 670 mg/m3 group of mice. The
authors concluded that slight developmental toxicity occurred at the
respective highest exposure levels in the studies of Swiss mice and
Sprague-Dawley rats, and that 5200 mg/m3 (2200 ppm) was the
no-observed-effect level (NOEL) for maternal and developmental
toxicity in both species (NTP, 1988).
Ten male Wistar rats were exposed to 11 870 mg/m3 (5000 ppm)
acetone in drinking-water for 8 weeks (Larsen et al., 1991). During
the sixth week, the treated males were mated with untreated females.
Numbers of pregnancies, numbers of fetuses and testes weights were
recorded and histopathology of the seminiferous tubules and testes was
undertaken. Acetone appeared to have no effects on these parameters.
Negative and positive results have been obtained in in vitro
tests of the teratogenic potential of acetone. Guntakatta et al.
(1984) reported that acetone at concentrations as high as 100 mg/ml
did not alter the cellular incorporation of radiolabelled sulfate or
thymidine in an in vitro mouse embryo limb bud culture system.
In vitro growth of rat embryos also was unaffected by the presence
of 0.1 or 0.5% acetone (v/v), but the presence of 0.5% caused
increased incidence of morphological abnormalities in the embryos;
2.5% caused embryo death (Kitchin & Ebron, 1984).
In a 13-week drinking-water study (NTP, 1991 Dietz et al., 1991),
small changes in sperm motility and incidence of morphologically
abnormal sperm were noted in rats exposed to concentrations of 50
g/litre (3400 mg/kg per day). Reproductive performance of these
animals, however, was not examined.
EHRT (1987) evaluated acetone in a reproduction screening test
using mice. Acetone in distilled water was administered by gavage to
groups of 50 mated CD-1 mice on days 6-15 of gestation at doses of 0
or 3500 mg/kg per day. Parameters of toxicity evaluated included
maternal toxicity (mortality, body weight and clinical signs), number
of live and dead offspring, pup body weight at birth, survival and
litter weight at postpartum day 3. Although two treated dams exhibited
clinical signs and eventually died, the authors did not consider that
maternal mortality had been increased by treatment with acetone.
Survivors exhibited no clinical signs or effects on body weight.
Effects attributed to acetone included decreased reproductive index,
increased gestation length, reduced birth weights, decreased neonatal
survival and increased neonatal weight gain.
7.4 Mutagenicity
Table 11 presents relevant genotoxicity studies and their
corresponding data for acetone. The data are almost entirely negative;
only one report of positive genetic activity was located. Exposure of
Saccharomyces cerevisiae to approx. 7% concentrations caused
aneuploidy (Zimmermann et al., 1985). Abbondandolo et al. (1980)
reported no forward mutations in a test on
Schizosaccharomyces pombe. In prokaryotes, acetone did not induce
reverse mutations in Salmonella typhiraurium (McCann et al., 1975;
NTP, 1991). Acetone did not induce DNA-cell binding with
Escherichia coli or ascites cells (Kubinski et al., 1981). Negative
results were reported for assays in animal systems in vitro and
in vivo, including mutation at the TK locus in mouse lymphoma cells
(Amacher et al., 1980), sister chromatic exchange or chromosome
aberrations in Chinese hamster ovary cells and human lymphocytes
(Norppa et al., 1981; NTP, 1991), cell transformation in Fischer rat
embryo cells (Freeman et al., 1973) and micronucleated erythrocytes in
mice and Chinese hamsters (Basler, 1986; NTP, 1991).
Table 11. Genotoxicity testing of acetone
Assay Indicator organism Application Purity Concentration Activating Response Comment Reference
(%) or dose system
Reverse Salmonella typhimurium plate NR 10, 100, 1000, +/- Aroclor-induced -/- NC McCann et al.
mutation TA1535, TA1537, TA98, incorporation 10 000 µg/plate rat liver S9 (1975)
TA100
Reverse S. typhimurium, TA97, liquid NR <10mg/plate +/-Aroclor-induced -/- NC NTP (1991);
mutation TA98, TA100, TA1535, suspension rat, Syrian hamster Dietz et al.
TA1537 liver S9 (1991)
Forward Schizosaccharomyces liquid NR 3.75% +/-phenobarbital- -/- 3ml 5% Abbondandolo
mutation pombe suspension induced mouse acetone et al. (1980)
plus 1 ml
yeast
culture
Aneuploidy Saccaharomyces liquid >97 6.98, 7.41, - + Expression Zimmermann
cerevisiae diploid suspension 7.83% of et al. (1985)
D61.M aneuploidy
required
overnight
incubation
on ice
Forward mouse lymphoma cell culture NR 0.134-0.421 - - Cytotoxicity Amacher
mutation L5178Y TK +/- cells mol/litre prevented et al. (1980)
use of
higher
concentrations
Sister Chinese hamster ovary cell culture NR <5000 +/- Aroclor-induced - NC NTP (1991)
chromatid cells mg/litre rat liver S9 Dietz et al.
exchange (1991)
Table 11. (Continued)
Assay Indicator organism Application Purity Concentration Activating Response Comment Reference
(%) or dose system
Chromosomal Chinese hamster ovary cell culture NR <5000 mg/lltre +/- Aroclor-induced NTP (1991);
aberrations cells rat liver S9 - NC Dietz
et al.(1991)
Cell Rauscher leukaemia cell culture NR 0.001-100 - - NC Freeman et al.
transformation virus/Fischer rat mg/litre (1973)
embryo cells
DNA-cell binding of E. coli liquid NR 50, 100 mg/litre +/- S9 - Radiolabelled Kubinski
binding DNA to suspension DNA is et al
assay Ehrlich ascites or incubated (1981)
E. coli cells with cells in
the presence
of test
substance
Sister cultured human whole blood 99.5 2.7, 12.8 - - NC Norppa et al.
chromatid lymphocytes cell culture mmol/litre (1981)
exchange
Chromosomal cultured human whole blood 99.5 12.8 - - NC Norppa
aberrations lymphocytes cell culture mmol/litre et al. (1981)
Micronucleus mouse peripheral drinking-water NR 5-20 g/litreb NA - NC NTP (1991)
test blood normochromatic Dietz et al
or polychromatic (1991)
erythrocytes
Micronucleus Chinese hamster intraperitoneal NR 865mg/kgc NA - NC Basler (1986)
test polychromatic injection
erythrocytes
a NA = Not applicable; NC = no comment; NR = not reported.
b For 13 weeks.
c Single dose.
The negative results in the in vivo bone marrow micronucleus
tests in mice and Chinese hamsters suggest that the aneuploidy which
was detected in yeast is not expressed in vivo in mammalian cells.
As all of the other mutagenicity tests (covering a range of end-points
including gene mutations and clastogenicity) were negative, it is
concluded that acetone presents no mutagenic risk to humans.
Acetone is widely used as a solvent in genotoxicity studies.
There are no indications that acetone interacts with other chemicals
to alter their genotoxic potential (ATSDR, 1994).
7.5 Carcinogenicity
There are no studies available on the carcinogenicity of acetone
by either inhalation or oral dosing. Acetone has been used extensively
as a solvent vehicle in skin carcinogenicity studies (NTP, 1991).
Generally, acetone is not considered to cause or promote tumours when
applied to the skin, but comprehensive examinations of tissue sites
remote from the dermal site of application are not murine in these
studies (Ward et al., 1986; NTP, 1991).
In cultured Syrian hamster embryonic cells, there was no evidence
of cellular transformation when the cells were cultured in the
presence of 0.02% acetone (DiPaolo et al., 1969).
7.6 Immunotoxicity
In an investigation of acetone as a solvent vehicle for skin
studies, Singh et al. (1996) examined the effects of topically applied
acetone on immune function in SSIN mice. Acetone (200 µl) was applied
to the dorsal trunk four or eight times. Responses in the sheep red
blond cell plaque-forming assay were measured at the end of treatment.
Responses were depressed in the 4x group but not in the 8x group,
suggesting a temporary effect on humoral immunity. Plaque-forming
response was further investigated using 50, 100, 200 and 300 µl
acetone with one, four or eight applications. Response was depressed
in mice treated one, four or eight times with 300 µl acetone. There
were no changes in spleen cellularity, the CD4+ to CD8+ T cell
ratio, or the alloantigen induced mixed lymphocytic response.
In vitro proliferative responses to the mitogen concanavalin A were
increased in the 200 µl 4x or 8x groups. The authors concluded that
acetone can modulate humoral immunity.
The effects of topically applied acetone on systemic immune
function were analysed by Singh et al. (1996). SSIN mice, derived by
inbreeding Sencar mice, were exposed to four or eight topical
applications of 200 µl acetone. Plaque-forming assays were used to
assess the effects of solvents on the development of humoral immunity.
The responses in the 8x-treated group were indistinguishable from the
controls, while in the 4x-treated group a retardation in the
development of splenic B cells secreting IgM against SRBC occurred,
The rate of loss of plaque-forming cells (PFCs) was not affected. A
functional suppression of the development of humoral immunity had
occurred. The researchers then assessed plaque-forming ability using
50, 100, 200 and 300 µl of acetone in order to determine the effects
of solvent dose and duration of treatment on the development of
humoral immunity. In the 300 µl group, plaque formation 4 days after
immunization was statistically suppressed in mice treated one, four or
eight times. At the other concentrations, suppression of PFC
development was schedule-dependent. The suppression was seen more
after the first or fourth application, but the suppressive activity
was lost following the eighth dose. While the reason is not known,
Singh et al. (1996) speculated that it may be an adaptive response by
the skin.
Singh et al. (1996) showed that the modulation of humoral
immunity was also dependent upon the nature of the antigen used for
immunization. The capacity of the T cells to respond and to function
following different stimuli does not appear to he suppressed in
acetone-treated mice.
7.7 Special studies
Vodicková et al. (1995) studied the inhibition of electrically
evoked seizures in male albino SPF Wistar rats (ages 0.5 to 1 year)
and female albino H-strain mice (ages 24 months) exposed to acetone
concentrations of 3630 mg/m3 (1530 ppm) (mice) and 4035 mg/m3 (1700
ppm) (rats) for 2 h (mice) and 4 h (rats). The central nervous system
(CNS) effect of inhibition of electrically evoked seizure discharge
was measured immediately after exposure. Blood levels were also
monitored, Frantik et al, (1994) used the same methodology and
measured blood concentrations of 1190 mg/m3 (500 ppm) for mice and
8300 mg/m3 (3500 ppm) for rats. In another study Frantik et al.
(1996) exposed rats to constant or fluctuating air concentrations of
acetone and measured inhibition of electrically evoked seizures.
Four-hour exposures to air concentrations of 4 and 10 mg/litre
resulted in blood levels of 183 and 520 mg acetone/line blood,
respectively. Inhibition of seizures at these blood levels was 10% and
50%, respectively. There were no significant differences between
constant or fluctuating inhalation exposures. If a concentration
depresses seizure discharge, then generally it will inhibit behaviour
in higher doses and provoke sleep and narcosis in still higher doses.
In these studies, it appears that the acetone levels were below the
concentration evoking behavioural inhibition. Furthermore, Vodicková
et al. (1995) found that when mice were exposed to acetone and toluene
in a binary mixture, the inhibiting neurotropic effect of the exposure
was not increased, and when rats were exposed, the effect
significantly decreased. The decline of blood toluene and xylene
levels was slowed down by a simultaneous exposure to acetone. The
authors of the studies concluded that the effects of solvents appear
to be less than additive. The significance of these studies is
uncertain.
7.8 Factors modifying toxicity; toxicity of metabolites
Primarily, factors that may affect endogenously produced acetone
toxicity are alterations in carbohydrate metabolism, accumulation of
ketone bodies, interaction with ketone body metabolism, and
interaction with ketone bodies. There are many secondary factors, such
as an imbalance of hormones, e.g., insulin, that can effect the body
burden and hence the toxicity of acetone (Ramu et al., 1978).
Acetone can induce the enzymatic activity of a cytochrome P450
isozyme, one which plays a role in the metabolism of endogenous and
exogenous substrates. Acetone is metabolized by the same P450 isozyme
that is induced during higher doses, thus making a homeostatic-type
mechanism for decreasing acetone levels when higher body burdens
develop.
Because acetone is non-ionic and miscible with water, it can
passively diffuse across cell membranes. Normally, metabolism is the
principal route of elimination from the body, and this metabolic
breakdown is either through an intrahepatic or extrahepatic pathway.
The metabolites of acetone include carbon dioxide, acetate, formate,
glucose and 1,2-propanediol, with pyruvate and other compounds as
intermediates. It does not appear that any of these compounds affect
the toxicity of acetone. However, as shown in section 7.8, acetone can
affect the toxicity of other compounds.
Ruddick (1972) reviewed the toxicology, biochemistry and
metabolism of 1,2-propanediol. The compound is used as a solvent for
flavouring material in baking and candy production, as well as a
humectant and preserver to keep packaged foods fresh. Oral LD50
values of 1,2-propanediol in the rat are 21-30 mg/kg body weight, in
the mouse 23.9, in the rabbit 18.0-19.0, in the guinea-pig 18.9, and
in the dog 18.9-20.0.
Another possible and relatively non-toxic metabolite is isopropyl
alcohol. Lewis et al. (1984) showed that metabolism of acetone was
different in normal and diabetic rats. These experiments indicated
that high levels of blood acetone resulted in transformation to
isopropyl alcohol.
7.9 Mechanisms of toxicity - mode of action
Within the liver, acetone is metabolized by three separate
gluconeogenic pathways through several intermediates, but most of its
intermediate or final metabolites are not considered toxic.
Unmetabolized acetone does not appear to accumulate in any tissue, but
is excreted mainly in the expired breath following high (> 1180
mg/m3, > 500 ppb) exposures. Acetone is irritating to mucous
membranes, possibly due to its lipid solvent properties, resulting ha
eye, nose, throat and lung irritation following exposure to the
vapour, and skin irritation upon dermal contact.
Systemically, acetone is moderately toxic to the liver and
produces haematological effects. The mechanism by which acetone
produces these effects is unknown. The renal toxicity may be due to
the metabolite, formate, which is known to be nephrotoxic (NTP, 1991)
and is excreted by the kidneys (Hallier et al., 1981). Furthermore,
the renal toxicity, which appears to be specific for male rats, may
involve the alpha 2u-globulin syndrome, as hyaline droplet formation
was associated with the nephropathy observed in male rats in the US
EPA (1986a) study. Acetone also causes increases in liver and kidney
weight, probably through the induction of microsomal enzymes, which
would increase the weight of the organs by virtue of the increased
protein content. Acetone causes effects in the testes of male rats and
is fetotoxic at high (approx. 15 670 mg/m3, approx. 6600 ppm)
concentrations. Although the exact mechanism for many of the effects
of acetone is not known, distribution studies in mice indicate that
acetone and metabolites arc found in all of the target organs (Wigaeus
et al., 1982). Acetone and some of its metabolites were also
transferred to rat fetuses after the dams were exposed to acetone
(Peinado et al., 1986).
One of the major effects of acetone is the potentiation of the
toxicity of other chemicals. Pretreatment with acetone has been shown
to potentiate the hepatotoxicity and nephrotoxicity of carbon
tetrachloride and chloroform (Plaa & Traiger, 1972; Traiger & Plaa,
1972, 1974; Sipes et al., 1973; Plaa et al., 1973, 1982; Folland et
al., 1976; Hewitt et al., 1980, 1987; Brown & Hewitt, 1984;
Charbonneau et al., 1985, 1986a,b, 1988, 1991) by inducing particular
forms of cytochrome P-450, especially cytochrome P-450IIE1, and
associated enzyme activities (Johansson et al., 1988; Brady et al.,
1989; Kobusch et al., 1989). The induction of these enzymes leads to
the enhanced metabolism of carbon tetrachloride (CCl4) and chloroform
to reactive intermediates capable of causing liver and kidney injury.
Acetone enhances the formation of carboxyhemoglobin by dichloromethane
via induction of cytochrome P-450IIE1, leading to enhanced metabolism
of dichloromethane to carbon monoxide (Pankow & Hoffmann, 1989).
Acetone, methyl ethyl ketone and methyl isobutyl ketone pretreatment
was made in rats at a dosage of 6.8 mmol/kg given daily for 3 days.
Acetone markedly potentiated CCl4-induced liver toxicity as indicated
by a decrease in the CC14 ED50 to 3.4 mmol/kg compared to
vehicle-pretreated rats (17.1 mmol ketone/kg). Pretreatment with
acetone also potentiated chloroform kidney toxicity but to a lower
degree; chloroform ED50 values for vehicle- and acetone-pretreated
rats were 3.4 and 1.6 mmol/kg, respectively (Raymond & Plaa, 1995a).
In a subsequent study, Raymond & Plaa (1995b) examined the role
of monooxygenases induced by ketones as a mechanism for potentiating
the CCI4 hepatotoxicity and chloroform nephrotoxicity. Hepatic and
renal monooxygenase activities (aminopyrine and benzo-phetamine
N-demethylase, aniline, hydroxylase) from rats pretreated with
acetone or other ketones (6.8 or 13.6 mmoL/kg) were increased. This
profile of induction was consistent with the ketone potentiation
potency ranking profile observed in vivo for liver but not kidney
injury (Raymond & Plaa, 1995a).
Acetone also potentiates the hepatotoxicity of acetaminophen
(Moldetts & Gergely, 1980; Jeffery et al., 1991; Lin et al., 1991),
N-nitrosodimethylamine and N-nitrosodiethylamme (Sipes et al.,
1978; Lorr et al., 1984; Hong & Yang, 1985), thiobenzamide (Chieli et
al., 1990), oxygen (Tindberg & Ingelman-Sundberg, 1989), chromate
(Ct[VI]) (Mikalsen et al., 1991), and benzene (Johansson et al., 1988;
Johansson & Ingelman-Sundberg, 1988; Schnier et al., 1989); the
genotoxicity of N-nitrosodimethylamine (Glatt et al., 1981; Yoo &
Yang, 1985; Yoo et al., 1990); and the lethality of acetonitrile
(Freeman & Hayes, 1985, 1988) by inducing cytochrome P-450IIE1. The
hepatotoxic and nephrotoxic effects of dibromochloromethane and
bromodichloromethane (Hewitt & Plaa, 1983) and the hepatotoxic effects
of 1,1,2-trichloroethane (MacDonald et al., 1982a,b),
1,1-dichloroethene (Jaeger et al., 1975; Hewitt & Plaa, 1983) and
dichlorobenzene (Brondean et al., 1989) are also enhanced by acetone.
The details of the mechanisms for these interactions are not clear,
hut the involvement of mixed-function oxidases has been implicated.
The renal toxicity of N-(3,5-dichlorophenyl)succinimide (a
fungicide) is potentiated by acetone via the induction of cytochrome
P-450IIE1 (Lo et al., 1987). Acetone in drinking-water increased lung
toxicity of inhaled styrene (Elovaara et al., 1990). Ethanol at 10% in
drinking-water had no adverse effect on mice, hut when 5% acetone was
added, the mice lost weight and there were lipid droplets in
hepatocytes (Forkert et al., 1991).
In other interactions, acetone enhances the neurotoxicity of
ethanol by a proposed mechanism whereby acetone inhibits the activity
of alcohol dehydrogenase, a reaction responsible for 90% of the
elimination of ethanol (Cunningham et al., 1989). Acetone also
potentiates the neurotoxieity and reproductive toxicity of
2,5-hexanedione (Ladefoged et al., 1989; Lam et al., 1991; Larsen et
al., 1991). The exact mechanism for these interactions is not clear
but appears to involve decreased body clearance of 2,5-hexanedione by
acetone (Ladefoged & Perbellini, 1986). Acetone also antagonizes in
that it decreases the incidence of liver necrosis in male Long-Evans
rats treated with acetaminophen (Price & Jollow, 1983). The antagonism
is possibly due to increased glutathione conjugation. No reasonable
explanation was offered for the apparent ability of acetone to
potentiate acetaminophen toxicity in vitro and antagonize the
hepatotoxicity in vivo (Morgott, 1993).
It appears that the potentiating effects of acetone may be
occurring by any of three different mechanisms:
a) interference with uptake or elimination;
b) induction of microsomal enzymes, particularly cytochrome
P450IIE1;
c) additive interactions at the target site or toxic receptor
protein.
The uneventful use of acetone as a carrier solvent in many
in vitro genotoxicity assays that utilize microsomal activating
enzymes suggests the third mechanism does not operate in vitro.
8. EFFECTS ON HUMANS
8.1 Effects on humans
Acetone is produced endogenously within the human body during
metabolism. It is released into the atmosphere by soil and water from
a very wide range of natural and anthropogenic sources and is present
in many household products. All humans will, therefore, be exposed to
additional exogenous acetone in varying quantities, depending on
specific circumstances (see sections 3 and 5).
8.1.1 Non-occupational exposure
The toxicity from inhalation and dermal/ocular exposure is listed
in Tables 12 and 13.
The 1991 Annual Report of the American Association of Poison
Control Centers National Data Collection System documented 1137
incidents of human exposure to acetone (Litovitz et al., 1992). This
same report listed 1001 cases for 1988. Of these incidents, 1124 were
due to accidental or intentional ingestion (the others were not
clearly specified). No fatalities were reported, only three cases had
a major acetone-related medical problem, 364 were treated in a health
care facility, 233 cases were referred to hospitals but had no
effects, 367 cases suffered minor effects, and 39 suffered from
moderate effects. According to the classification of the AAPCC, none
of the major, minor or moderate effects were further described, and
the outcomes of the remainder of the incidents were not reported.
Matsushita et al. (1969b) exposed 25 male volunteer students to
acetone vapour for 3 h in the morning and 3 h in the afternoon during
1 day at concentrations of 240, 590, 1190 and 2400 mg/m3 (100, 250,
500 and 1000 ppm). The subjects were asked to describe any subjective
symptoms, and most reported that exposure to concentration of 1190 and
2400 mg/m3 for 6 h was irritating to the nose, eyes, throat and
trachea. At concentrations of 240 and 590 mg/m3 only a few of the
subjects complained of symptoms. Subjective symptoms also included the
loss of the ability to smell acetone as exposure proceeded. In another
controlled experiment, volunteer subjects were exposed to acetone
concentrations of 475, 720 and 1190 mg/m3 (200, 300 and 500 ppm) for
3-5 min in a chamber. Eye and throat irritation was experienced at 720
and 1190 mg/m3, but the subjects estimated that they could tolerate
an exposure level of 475 mg/m3 for an 8-h workshift (Nelson et al.,
1943). Volunteers exposed to 1000 ppm acetone for 4 h reported more
subjective complaints of throat irritation and "annoyance" (Seeber et
al., 1992). However, the perceived irritation or annoyance associated
with occupational and experimental exposures to acetone may be
influenced by the perceived odour of acetone and unrelated to
irritation and symptoms (Dalton et al., 1997; Wysocki et al., 1997).
Pulmonary function testing of volunteers exposed to < 2970 mg/m3
(< 1250 ppm) intermittently for various durations in a complex
protocol revealed no abnormalities caused by the exposure (Stewart et
al., 1975). The volunteers experienced sporadic throat irritation.
Table 12. Short-term human inhalation exposure to acetone
Exposure NOEL LOEL Critical effect Reference
duration/frequency mg/m3 (ppm) mg/m3 (ppm)
3-5 min 475 (200) 1190 (500) eye, nose and throat Nelson et al. (1943)
irritation
5.25 h 240 (100) 590 (250) eye, nose and throat Matsushita et al.
(6 h/day with 45-min break irritation (1969b)
at 590 mg/m3
5.25 h 1190 (500) increased WBC and Matsushita et al.
(6 h/day with 45-min break) eosinophil count, (1969b)
decreased phagocytic
activity of neutrophils
4 h approx 560 behavioural changes Dick et al. (1989)
(approx 237)
7 days (8 h/day) 2370 (1000) headache, dizziness, Raleigh & McGee
confusion (1972)
5 days (7.5 h/day) 2370 (1000) 2970 (1250) visual evoked response Stewart et al. (1975)
changes
Table 13. Short-term human skin and eye exposure to acetone
Exposure Target NOEL LOEL Critical effect Reference
duration/frequency mg/m3 (ppm)
vapour: 3-5 min skin/eyes 475 (200) 1190 (500) eye irritation Nelson et al. (1943)
liquid: 30 or 90 min skin 1 ml degenerative changes Lupulescu et al.
in epidermis (1973)
liquid: 90 min skin 1 ml decreased protein Lupulescu &
synthesis Birmingham (1975)
vapour: 2-3 days skin/eyes 2140 mg/m3 (901) eye irritation Raleigh & McGee
(8 h/day) (1972)
vapour: 7 days skin/eyes 2390 mg/m3 (1006) eye irritation Raleigh & McGee
(8 h/day) (1972)
vapour: 4 and 8 h skin/eyes 2375 (1000) no complaints of Seeber et al. (1993)
skin or eye
irritation
vapour: 4 h skin/eyes 2350 (990) perceived irritation Seeber et al. (1992)
of nose and throat
and "annoyance"
vapour: 4 and 8 h CNS 2375 (1000) mood change Seeber et al. (1993)
In the USA, the National Institute for Occupational Safety and
Health (Stewart et al., 1975) sponsored a research study to determine
physiological responses by humans exposed to acetone in air, and to
develop a biological test to indicate the magnitude of exposure.
Twenty adults of both sexes were exposed to acetone vapour
concentrations of 0, 475, 2370 and 2970 mg/m3 (0, 200, 1000 and 1250
ppm) for periods of 3 or 7.5 h for various durations up to 5 days. The
results of this study showed that a predictable excretion pattern
resulted for each of the tested vapour concentrations, and the rate of
excretion of acetone in the breath was a function of the duration of
exposure. After 3 h of exposure, the majority of the subjects could no
longer detect the odour of acetone when breathing normally. No
significant neurological abnormalities were noted, but there was a
statistically significant amplitude change in the visual evoked
response test. It was also noted that three out of four of the women
had premature menstruation, early by one week or more, after four days
of exposure to 2370 mg/m3 (1000 ppm) for 7.5 h/day, which the authors
saw as worrisome.
High pulse rates (120-160/min) were commonly found in patients
exposed to acetone by inhalation and/or dermally, in most cases at
concentrations sufficient to cause acute intoxications, after
application of casts for which acetone was used in the setting
solution (Chatterton & Elliott, 1946; Pomerantz, 1950; Renshaw &
Mitchell, 1956; Hift & Patel, 1961). In a controlled laboratory study
using a complex protocol, electrocardiography of volunteers exposed to
atmospheric concentrations of 2970 mg/m3 (1250 ppm) for 6 weeks (2-5
days; 7 h/day) revealed no alterations, compared with their
pre-exposure electrocardiograms (Stewart et al., 1975).
The acetone concentrations in the body fluids and expired air of
healthy and diabetic patients can be very different. Even in healthy
subjects, the level of acetone in blood/plasma varies according to
fasting or non-fasting conditions and depends on the weight of the
subject. Generally, the blood/plasma acetone concentrations are higher
in fasted than non-fasted subjects and higher in subjects who are not
obese, compared to obese subjects (Haff & Reichard, 1977). The
narcotic effects of acetone occur after oral as well as inhalation
exposure. Several case reports describe patients in minimally
responsive, lethargic, or comatose conditions after ingesting acetone,
but some of these cases are confounded by co-exposure to other
possible narcotic agents. For example, a 30-mouth-old child ingested
most of a 180 ml (6 ounce) bottle of nail polish remover containing
65% acetone and 10% isopropyl alcohol (Gamis & Wasserman, 1988); a
known alcoholic woman ingested nail polish remover (Ramu et al.,
1978); and a man ingested 200 ml of sake prior to intentionally
ingesting liquid cement containing a mixture of polyvinyl chloride,
acetone, 2-butanone, and cyclohexanone (Sakata et al., 1989). Blood
levels of acetone in some of these patients were 2.5 g/litre (Ramu et
al., 1978) and 4.45 g/litre (Gamis & Wasserman, 1988). In the case
reported by Sakata et al. (1989), the blood level of acetone was 110
mg/litre and the urine level was 123 mg/litre 5 h after the ingestion,
but the patient had been subjected to gastric lavage. A man who
intentionally ingested about 200 ml of pure acetone (about 2241
mg/kg) subsequently became deeply comatose, but responded to
treatment (Gitelson et al., 1966). Six days later, he was ambulatory,
but a marked disturbance of gait was observed. This condition had
improved upon follow-up examination 2 months later.
Acetone is reported to be irritating to mucous membranes. Raleigh
& McGee (1972) listed eye irritation as a common complaint of workers
exposed to acetone, and Matsushita et al. (1969a) reported the same
finding for volunteers in their study. However, the complaints of
irritation associated with exposure to acetone may be influenced by
the strong odour of acetone (Dalton et al., 1997; Wysocki et al.,
1997).
Lupulescu & Birmingham (1975, 1976) found that application of 0.1
ml directly to the skin resulted in histological and degenerative
epidermis changes in volunteers after 60 or 90 min of exposure. These
changes were denoted by reduction as well as disorganization of the
horny layers, intercellular oedema and vacuolization of the stratum
spinosum. Haematological effects have been observed in humans after
inhalation exposure to acetone in controlled laboratory studies of
volunteers. A statistically significant increase in white blood cell
counts and decrease in phagocytic activity of neutrophils, compared
with controls, were observed in the volunteers after a single 6-h
inhalation exposure or repeated 6-h exposures for 6 days to 1190
mg/m3 (500 ppm) (Matsushita et al., 1969a,b). No significant
difference was seen in haematological parameters in volunteers exposed
to 590 mg/m3 (250 ppm) compared with controls. In contrast,
haematological findings were within normal limits in volunteers
exposed to 1190 mg/m3 (500 ppm) for 2 h (DiVincenzo et al., 1973) or
to < 2970 mg/m3 (< 1250 ppm) repeatedly for 1-7.5 h/day for
as long as 6 weeks (Stewart et al., 1975). Exposure to acetone vapour
can also lead to increased pulse rates, gastrointestinal irritation,
nausea, vomiting, and haemorrhage. However, the odour threshold of
acetone (240-330 mg/m3, 100-140 ppm) and the feelings of irritancy
are excellent warning properties that generally preclude serious
inhalation overexposure. Accidental or intentional ingestion of
acetone can cause erosions in the mouth, coma and diabetes-like
symptoms.
8.1.2 Occupational exposure
Most occupational exposure standards are in the range 1780-2370
mg/m3 (750-1000 ppm). Occupations in which workers may be exposed to
higher levels of acetone include paint manufacturing, plastics
manufacturing, artificial fibre industries, shoe factories,
professional painting, and commercial cleaning. As with the general
population, there are case studies of accidental poisonings and
inhalation of acetone fumes. Many of these situations are mixed
exposures and not deemed relevant to this discussion, except where
acetone potentiates or antagonizes the effects of another chemical
present (see also section 8.3).
Workers in industries that manufacture or use acetone are
potentially exposed to higher concentrations of acetone than the
general population. For example, the concentrations of acetone in the
breathing-zone air in a paint factory, a plastics factory, and an
artificial fibre factory in Italy were >3.48 mg/m3 (Pezzagno et al.,
1986). The concentration of acetone in the breathing-zone air of a
fibre-reinforced plastic plant in Japan, where bathtubs were produced,
was < 108 mg/m3 (Kawai et al., 1990a). The inhalation exposure for
workers to acetone in a shoe factory in Finland ranged from 25.4 to
393.4 mg/m3 (Ahonen & Schimberg, 1988), and concentrations were
similar in the breathing-zone air in shoe factories in Italy (Brugone
et al., 1978). The concentration of acetone in the breathing-zone air
of a solvent recycling plant in the USA ranged from not detectable to
43 mg/m3 (Kupferschmid & Perkins, 1986). High levels of acetone were
detected in the occupational air in other industries, including the
chemical, plastic button, and paint manufacturing industries in Italy
(Ghittori et al., 1987). Isopropyl alcohol is known to oxidize in the
liver and is converted to acetone (Kawai et al., 1990b); therefore,
occupational exposure (e.g., printing plants) or accidental ingestion
of isopropyl alcohol also produces acetone in expired air, blood and
urine.
Satoh et al. 0996) carried out a cross-sectional study on 110
male (age range 18.7 to 56.8 years) acetone-exposed and 67 male (age
range 20.7 to 57.5 years) non-exposed shift workers. The personal
passive monitors and biological monitoring indices measured at the end
of the workshift were 46.5-2583 mg/m3 (19.6-1088 ppm) (mean 864
mg/m3, 364 ppm) in breathing-zone, 5.9-1002 mg/m3 (2.5-422 ppm)
(mean 231 mg/m3, 97.3 ppm) in alveolar air, 4-220 mg/litre (mean 66.8
mg/litre) in blood, and 0.75-170 mg/litre (mean 37.8 mg/litre) in
urine. Symptoms at the end of the workshift showing good exposure
response relationships were eye irritation, tear production and
complaints of acetone odour. In acetone workers in the 30-44 year
range, simple reaction time and digit span scores were significantly
lower in acetone-exposed workers but exposure-relationships were not
clear. Manifest Anxiety Scale Scores, Self-rating Depression Scale
Scores, R-R internal variation on the ECG, haematological examinations
and liver function tests did not show any significant differences
between the two groups.
In a retrospective mortality study of 948 employees (697 men, 251
women) in a cellulose fibre plant where acetone was used as the only
solvent, there was no significant excess risk of death from any cause
(all causes, malignant neoplasm, circulatory system disease, ischaemic
heart disease) compared with rates for the general population in the
USA (Ott et al., 1983a,b). The workers had been employed at the plant
for at least 3 months to 23 years. Industrial hygiene surveys found
that median TWA acetone concentrations were 902, 1678 and 2540 mg/m3
(380, 770 and 1070 ppm) based on job categories. All haematologieal
parameters and all clinical blood chemistry parameters (aspartate
aminotrausferase, alanine aminotransferase, lactic dehydrogenase,
alkaline phosphatase, total bilirubin, and albumin) were within normal
limits.
The only effect on the respiratory system observed in humans
exposed to acetone vapour is irritation of the nose, throat, trachea
and longs. The irritant properties of acetone in humans have been
noted both in workers who were exposed to acetone occupationally
(Raleigh & McGee, 1972; Ross, 1973) and in volunteers under controlled
laboratory conditions (Nelson et al., 1943; Matsushita et al.,
1969a,b). Complaints of irritation were reported by workers with
average exposures to acetone in the workroom of 2140 mg/m3 (901 ppm)
(Raleigh & McGee, 1972; Ross, 1973). Sallee & Sappington (1949), in a
report of experience at the Tennessee Eastman Corporation on acetone
concentrations not associated with injury, noted that acetone is
mildly irritating to the eyes at 4750-7120 mg/m3 (2000-3000 ppm), but
that no irritation persisted after exposure ceased.
In an on-site medical appraisal of nine workers, in which the
exposure concentration was 2390 mg/m3 (1006 ppm), three of the
workers mentioned headache and lightheadedness as subjective symptoms
(Raleigh & McGee, 1972). In another on-site medical appraisal of four
workers, in which the TWA exposure concentration was 2140 mg/m3 (901
ppm), none of the workers complained of neurological effects (Raleigh
& McGee, 1972). The medical examinations included the Romberg test,
finger-to-nose test, and observations for nystagmus (involuntary rapid
movement of the eyeball). These tests revealed no neurobehavioural
effects in either study. Such symptoms as unconsciousness, dizziness,
unsteadiness, confusion and headache were experienced by seven workers
exposed to > 28 500 mg/m3 (> 12 000 ppm) while clearing out a pit
containing acetone that had escaped from nearby tanks (Ross, 1973).
The degree of the symptoms varied depending on the length of time that
the workers had spent in the pit (2 min to 4 h).
8.2 Subpopulations at special risk
Because of higher exposure, workers in industries that
manufacture or use acetone are one segment of the population at higher
risk of acetone exposure compared to the general population (see
section 5.3). Professional painters and commercial and household
cleaners, using detergents, cleansers, waxes or polishes containing
acetone, are also likely to be exposed to acetone at higher
concentrations than the general population, although experimental data
regarding the extent of exposures for this segment of workers are not
available. Among the general population, high exposure to acetone may
occur among several subgroups. Cigarette smoke contains < 0.54 mg
acetone/cigarette (Manning et al., 1983); therefore, smokers are
exposed to higher concentrations of acetone than non-smokers. The
content of acetone in certain nail polish removers is high, and so
individuals who frequently use nail polish removers, such as
manicurists, are exposed to higher levels of acetone than the general
population. People who live near landfill sites that emit acetone or
those who live near industrial sources of emission (e.g., refinery,
incinerator, close to high vehicular traffic areas) are also
susceptible to higher exposure concentrations of acetone than the
general population that does not reside near these sites.
There is also a small possibility of ketone formation by
ketoacidosis in humans. The most common form is diabetic ketoacidosis
(DKA), which appears to require insulin deficiency coupled with a
relative or absolute increase in glycogen concentration. Dieting has
also been shown to cause starvation ketosis. Apart from these two
types, the other common ketoacidotic state is alcoholic ketoacidosis.
Presumably the liver is activated by kctogenesis as a result of
starvation in cases of alcoholic ketoacidosis and driven to maximal
rates of ketone formation by the high fatty acid levels (Wilson et
al., 1991).
The intrinsic toxicity of acetone and the results of the US EPA
(1986a) animal studies suggest that male animals are more susceptible
than female animals. Acetone at higher concentrations may exacerbate
preexisting haematological, liver, kidney or reproductive disorders in
humans. As with other chemicals, neonates and the elderly may be more
susceptible to acetone because of immaturity or decrease function of
their metabolic systems, respectively. Physiological disorders are
exacerbated by higher exposure. Fasting and diabetes increases
endogenous levels of acetone in humans, suggesting that dieters and
diabetics may have a higher body burden, and additional exposure to
acetone may make them more at risk. Results in animals suggest that
the rate of acetone metabolism is slower during pregnancy (Peinado et
al., 1986).
One of the most-studied effects of acetone is the induction of
mierosomal enzymes, particularly of cytochrome P-450IIE1. Acetone
thereby induces its own metabolism, and it potentiates the toxicity of
numerous other chemicals by enhancing the metabolism, which depends on
cytochrome P-450IIE1, to reactive intermediates, The indnction of
cytochrome P-450IIE1 by acetone has been documented in many species,
and therefore poses a concern for humans exposed to acetone or to
those chemicals whose toxicity is potentiated or antagonized by
acetone. The observed potentiation following acetone ingestion
generally causes a quantitative increase in the extent of damage to
the affected tissues or organs.
Acetone pretreatment has been shown to potentiate halogenated
solvent hepatotoxicity and nephrotoxicity, and is related to the
induction of microsomal enzymes that metabolize these solvents to
reactive intermediates (Morgott, 1993), People with altered
physiological states can have permanent potentiating effects of
acetone. These states include starvation, alcoholism, diabetes
mellitus, hypoglycaemia, eating disorders, prolonged vomiting or
inborn errors of metabolism (Morgott, 1993). This potentiation has
been shown to occur with carbon tetrachloride, chloroform and
1,1,2-trichloroethane (Traiger & Plaa, 1974), 1,1-dichloroethylene
(Hewitt & Plaa, 1983), bromodichloromethane, dibromomethane (Hewitt &
Plaa, 1983), 1,2-dichlorobenzene (Brondeau et al., 1989), and several
other solvents.
9. EFFECT ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Aquatic organisms
9.1.1 Acute toxic effects on aquatic fauna
Data on the effects of acute exposure of aquatic organisms to
acetone are summarized in Table 14. The four freshwater fishes tested,
Salmo gairdneri, Lepomis macrochirus, Pimephales promelas and
Gambusia affinis, had 96-h LC50s between 5540 and 13 000 mg/litre,
indicating low toxicity for these species (Wallen et al., 1957; Cairns
& Scheier, 1968; Patrick et al., 1968; Mayer & Ellersieck, 1980; Veith
et al., 1983; Brooke et al., 1984). Similarly, the freshwater Asiatic
clam ( Corbiculo manilensis) had a 96-h LC50 of 20 000 mg/litre
(Chandler & Marking, 1979). Various species of the water flea
( Daphnia magna, D, pulex and D. cuculata) had 48-h LC50s and
EC50s of 7460 to 13 500 mg/litre; however, test conditions for these
studies were scantily reported (Canton & Adema, 1978; Randall & Knopp,
1980). Bringmann & Kühn (1982) found a 24-h EC50 (immobilization) of
10 000 mg/litre for Daphnia magna. Dowden & Bennett (1965) reported a
48-h LC50 of 10 mg/litre for D. magna, but they noted that tests
had been done by several different investigators and that values might
be erroneous. Ewell et al. (1986) also tested several freshwater
species under static conditions for 96 h. All of the species; pillbug
( Asellus intermedius), waterflea ( Daphnia magna), flatworm
( Dugesia tigrina), sideswimmer ( Gammarus fasciatus), snail
( Helisoma trivolvis), segmented worm ( Lumbriculus variegatus) and
fathead minnow ( Pimephales promelas) had 96-h LC50s >100 mg/litre,
the highest concentration tested.
Two saltwater species were tested by Linden et al. (1979). The
copepod, Nitocra spinipes, and the bleak ( Alburnus alburnus,
tested under static conditions had 96-h LC50s of 16 700 and 11 000
mg/litre, respectively.
Slooff & Baerselman (1980) studied groups of 10 amphibians,
axolotls ( Ambystoma mexicanus) and clawed toads ( Xenopus laevis),
34 weeks after hatching. The animals were exposed to varying
concentrations of acetone in standardized medium and 48-h LC50 values
for the respective species were 20 and 24 mg/litre.
Table 14 Acute toxicity of acetone to aquatic fauna
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Water flea 48-h LC50 9218 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
(Daphniamagna) 24-h LC50 4068 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
NOEL <403 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
Water flea 48-h LC50 12 100 to 13 300 nominal Canton & Aderna (1978)
(Daphnia magna)
Water flea 48-h EC50 13 500 7.7 22 154.5 nominal Randall & Knepp (1980)
(Daphnia magna)
Water flea 24-h LC50 10 nominal Dowden & Bennett (1965)
(Daphnia magna) 48-h LC50 10 Dowden & Bennett (1965)
Water flea 96-h LC50 >100 6.5-8.5 20 nominal Ewell et al. (1986)
(Daphnia magna)
Water flea 24-h EC50 >10 000 immobilization Bringmann & Kühn (1982)
(Daphnia magna)
Water flea 48-h LC50 8800 nominal Canton & Adema (1978)
(Daphnia pulex)
Water flea 48-h LC50 7460-7810 nominal Canton & Adema (1978)
(Daphnia cuculata)
Water flea 48-h LC50 8098 8.2 25 90-110 nominal Cowgill & Milazzo (1991)
(Cesiodaphnia dubia)
240-h LC50 6693 8.2 25 90-110 nominal Cowgill & Milazzo (1991)
Pillbug 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Asellus intermedius)
Table 14 (contd).
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Flatworm 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1966)
(Dugesia tigrina)
Side swimmer 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Gammarus fasciatus)
Segmented worm 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Lumbriculus)
variegatus
Snail 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Helisoma trivolvis)
Asiatic clam 96-h LC50 >20 000 16 16-26 static, nominal Chandler & Marking (1979)
(Corbicula
manilensis)
Crustacean 24-h LC50 64 300 7.8 25 250 static, nominal Crisinel et al (1994)
(Streptocephalus
rubricandatus)
Copepod 96-h LC50 16 700 7.8 10 static, nominal Lindén et al (1979)
(Nitocra spinipes)
Brine shrimp 24-h LC50 2100 24.5 saltwater, Price et al. (1974)
(Artemia salina) static, nominal
24-h LC50 6010 7.4 12 40 static, nominal Mayer & Ellersieck (1980)
96-h LC50 5540 7.4 12 40 static, nominal Mayer & Ellersieck (1980)
Rainbow trout 24-h LC50 6100 8.0 10 90 flow-through, Majewski et al. (1977)
(Salmo gairdnen) nominal
Table 14 (contd).
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Rainbow trout 24-h CV50 36 500 18 CV50 is the Segner & Lenz (1993)
(Salmo gairdnen) midpoint
cytotoxicity of
the R1 cell line
Bluegill 96-h LC50 8300 18 10 static, nominal Cairns & Scheier (1968)
(Lepomis macrochirus)
Fathead minnow 96-h LC50 8120 7.58 25 48.5 measured Brooke et al (1984)
(Pimephales promelas) exposure
concentration
96-h LC50 6880 6.93 25 535 measured Brooke et al. (1984)
exposure
concentration
96-h LC50 6290 7.62 24 44.0 measured Brooke et al. (1984)
exposure
concentration
Fathead minnow 96-h LC50 >100 6.5-8.5 static, nominal Ewell et al. (1986)
(Pimephales promelas)
Mosquito fish 24-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal Wallen et al. (1957)
(Gambusia affinis)
48-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal Wallen et al. (1957)
96-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal WaIlen et al. (1957)
Bleak 96-h LC50 11 000 7.8 10 static, nominal, Linden et al (1979)
(Alburnus alburnus) saltwater
9.1.2 Chronic effects on aquatic fauna
Cowgill & Milazzo (1991) calculated EC50 values for two species
of Daphnia exposed to acetone using the three-brood test. For
Daphnia magna, EC50 values for number of progeny, number of broods
and mean brood size were 6369, 6406 and 6714 mg/litre, and the
corresponding NOEL values were 3110, 5184 and 3110 mg/litre. The EC50
values for Ceriodaphnia dubia were 6469, 5908 and 6928 mg/litre, and
the corresponding NOEL values were 5184 mg/litre for all three
parameters.
9.1.3 Effects on aquatic plants
Freshwater diatoms and algae were tested with acetone.
Chlorella pyrendoidosa, a green alga, was exposed to acetone at
concentrations of 2574 and 25 740 mg/litre (3.3 and 33 ml/litre
assuming an acetone density of approx. 790 mg/ml at 30°C) in culture
tubes with 300 ml nutrient solution at 30°C under continuous light for
76 h (Parasher et al., 1978). The lowest concentration bad a slight
negative effect on algal growth; the high concentration caused
disintegration of the cell membranes and cytoplasm.
Hess (1980) examined the effects of acetone on
Chlamydomonas eugametus, a green alga. The alga was exposed for 48 h
at 25°C; pH and hardness were not reported. An acetone concentration
of 0.5% (v/v; 3950 mg/litre) resulted in no inhibition of growth.
Significant inhibition (a 15% growth reduction) occurred at 1.0% (7900
mg/litre) and growth was only 24% of control values at 2.5% acetone
(1975 mg/litre); at 5.0% (39 500 mg/litre), there was complete
inhibition. Bringmann & Kühn (1978, 1980a) examined the effects of
acetone on the green alga, Scendesmus quadricauda. The alga was
exposed at 27°C for 7-8 days. The toxicity threshold was 7500
mg/litre. Bringmann & Kühn (1978) determined that the toxicity
threshold for the cyanobacterium (blue-green alga)
Microcystis aeruginosa exposed under similar conditions was only 530
mg/litre. Stratton & Corke (1981) noted that acetone (0.1-1.0% v/v;
790-7900 mg/litre; length of exposure not reported) actually
stimulated photosynthesis in S. quadricauda and C. pyrendoidosa,
perhaps by increasing the rate of CO2 diffusion into the cells.
The freshwater diatom, Nitzchia linearis, was tested in soft
water (100 mg CaCO3/litre) for 5 days but temperature and pH were not
reported (Patrick et al., 1968). The EC50 for cell multiplication was
11 493 to 11 727 mg/litre.
The marine diatom Skeletonema costatum was also tested with
acetone (Kleppel & McLaughlin, 1980; Cowgill et al., 1989). Following
exposure to acetone for 5 days, EC50 values for decreases in total
cell count and total cell volume were 11 798 and 14 440 mg/litre,
respectively (Cowgill et al., 1989). Kleppel & McLaughlin (1980)
exposed skeletonema costatum (at cell densities of 3.8 × 102,
3.8 × 103 and 3.8 × 104) to 1 ml acetone per litre of medium for 5
days. There were no significant changes in cell growth and
reproduction in cells exposed to acetone, compared with controls.
Cowgill et al. (1991) estimated the NOEL and the EC50 (50%
reduction in the number of plants or fronds as compared to controls)
of acetone to Lemna sp. (duckweed). Plants belonging to this genus
have become "aquatic test organisms" because they are a part of the
food chain of fish, water fowl and aquatic organisms. For
Lemna gibba G-3, the EC50 for the 7-day test was 12.4 g/litre for
plants and 10.2 g/litre for fronds. For Lemna minor 6591, it was
13.4 g/litre for plants and 11.4 g/litre for fronds. Three other
Lemna species had EC50 values greater than 10 g/litre.
9.2 Effects on bacteria and protozoa
The effects of acetone have been studied with bacteria and
protozoans. Protozoans seemed more sensitive to toxicological effects
than bacteria. Bringmann & Kühn (1980a) exposed the bacterium
Pseudomonas putida (for 16 h) and the protozoan
Entosiphon sulcatum (for 72 h), to acetone at a temperature of 25°C
in a cell multiplication test to determine toxicity. The toxicity
thresholds for these two organisms were 1700 and 28 mg/litre,
respectively. In another study, Bringmann & Kühn (1980b) determined a
toxicity threshold for the protozoan Uronema porduczi of 1710
mg/litre. A mixed microbial culture from wastewater (specific biota
not reported) tested with acetone for an unreported length of time
resulted in an EC50 (decrease in biodegradation) of 0.612 mol/litre
(35 540 mg/litre), indicating very low toxicity (Vaishnav, 1986). The
bioluminescent bacterium Photobacterium phosphoreum had an EC50
(inhibition of luminescence) of 0.363 and 0.372 mol/litre (21 088 and
21 579 mg/litre), also indicating very low toxicity (Kamlet et al.,
1986). McFeters et al. (1983) reported a similar EC50 of 18 250
mg/litre for P. phosphoreum.
Stratton (1987) estimated EC50 values in several species of
cyanobacteria (blue-green algae). Acetone stimulated growth at
concentrations less than 790-7900 mg/litre (0.1 to 1.0%), except for
Anabaena sp. where levels above 5500 mg/litre (0.7%) caused total
inhibition. Total growth inhibition occurred at acetone concentrations
of 569 g/litre (72%) with A. cylindrica, 31.6 g/litre (>4%) with
A. inaegualis and > 63.2 g/litre (8%) with A. variabilis and
Nostoc sp. The estimated EC50 for these species ranged from 2.84 to
34.6 g/litre (0.36 to 4.38% v/v).
Ecotoxicological testing was done using the Microtox test, which
is based upon the toxicant-based diminution of light emission of
Photobacterium phosphoreum (Bulich, 1986). The parameter used to
measure toxicity was the EC50, i.e. the toxicant concentration that
diminishes light emission by 50% at 15°C. The EC50 for acetone has
been measured at 14.7 g/litre (254 mmol/litre) for 5 min, 14.2 g/litre
(245 mmol/litre) for 15 min and 14.1 g/litre (243 mmol/litre) for 25
min (Chen & Que Hee, 1994). These EC50 values are very high compared
to other compounds, indicating that acetone toxicity as reported by
this test is low. These EC50 values can be compared to the results of
a study by De Zwart & Slooff (1983), who determined an EC50 of 18.3
g/litre (316 mmol/litre).
In a series of studies on the toxicity of acetone to bacteria,
Nirmalakhandan et al. (1994a,h) determined the IC50 in activated
sludge cultures from a municipal wastewater treatment plan to be
almost 4.9 g/litre.
Blum & Speece (1991) found low inhibition, with IC50 values of
1.2 g/litre for Nitrosomonas, 50 g/litre in methanogens and 16
g/litre for aerobic heterotrophs. Methanogens are generally the most
sensitive bacteria that convert organic matter to carbon dioxide and
methane in anaerobic environments.
Rajini et al. (1989) examined the cytotoxicity to the protozoan
Paramecium caudatum. The 10-min LC100 was 22.9 g/litre (2.9% v/v)
with a concentration of 22.6 g/litre, and the 4-h LC50 was 5.37
g/litre (0.68% v/v) with a concentration of 5.2 g/litre. The
researchers also found that insecticides were more toxic to
P. caudatum when they were dissolved in acetone.
9.3 Terrestrial organisms
9.3.1 Effects on fauna
Hill et al. (1975) administered acetone for 5 days to both quail
and pheasants. The LC50 for both of these birds was 40 g/kg diet, the
highest dose administered. Acetone was also tested with mallard eggs
(Hoffman & Eastin, 1981). Fertile eggs were immersed in 0, 10 or 100%
acetone for 30 seconds at room temperature on days 3 or 8 of
incubation. There were no significant effects with 10% acetone;
however, 100% acetone caused a significant decrease in survival,
embryonic weight and embryonic length for both exposure groups. It is
unknown whether the mortality was due to the toxicity of acetone or to
its solvent capabilities. White Leghorn chick embryos were also tested
with acetone (Korhonen et al., 1983). The test article was injected
into the eggs at 5 µl/egg. Statistical analysis was not performed and
controls were not used; however, it appeared that acetone did not
affect mortality or malformation of the embryos.
9.3.2 Effects on flora
Pertinent data regarding the effects of exposure of terrestrial
flora to acetone have not been found. Acetone did not cause a
significant decrease in seed germination or the percentage of normal
seedlings of sweet corn (5h2 cultivars). However, with longer
immersion times (8 h), it was detrimental (Hung et al., 1992).
Gorsuch et al. (1990) tested the potential of acetone to affect
germination and early growth of terrestrial plants. Plants were
exposed to the chemical for 7 days and then germination, root length
and plant heights were determined and compared with controls. Acetone
had a no-observed-effect concentration of 100 mg/litre (nominal
concentration) for ryegrass, radish and lettuce.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health effects
Acetone is of a low order of acute toxicity. However a
significant number of poisonings have occurred in humans following
accidental or intentional misuse.
Acetone can produce neurobehavioural and other changes, including
headache, dizziness, confusion and, at high vapour concentrations, CNS
depression and narcosis. Exposures to acetone vapour will cause
irritation of eyes, nose and throat. Continuous exposure to vapour can
lead to adaptation to the odour.
Liquid acetone is an eye irritant and repeated exposure of skin
will cause defatting, drying and cracking.
It is considered that acetone is neither a skin nor a respiratory
tract sensitizer.
Acetone is formed endogenously from fatty acid oxidation and is
uniformly distributed throughout the body among non-adipose tissues.
It is rapidly cleared from the body by metabolism and excretion,
mainly through the lungs. Acetone induces the hepatic mixed-function
oxidase enzymes that bring about its own metabolism, and so the body
has a homeostatic mechanism that has evolved to maintain acetone
levels in the body at a "baseline" level. Induction of hepatic
mixed-function oxidase enzymes can potentiate (and in some instances
antagonise) the effects of other chemicals. People at most risk to
potentiation include diabetics, alcoholics and those undergoing
prolonged fasting. In common with other chemicals, metabolism of
acetone may be reduced in neonates, the elderly and in hepatic
diseases.
In one study on human volunteers, increases in leucocyte count
were reported. However, this has not been found in other studies, in
an inhalation study, human female volunteers reported menstrual
irregularities (delayed menstruation).
Mild haematological effects were found in two strains of rats
(F344 and Sprague-Dawley). The mean cell haemoglobin concentration and
mean cell volume were elevated in both, but blood haemoglobin
concentration was raised in Sprague-Dawley rats and decreased in F344
rats. At an acetone dose of 50 000 mg/litre in the drinking-water,
effects on sperm quality were observed in rats. At lower doses only
small changes in sperm motility were seen. In a reproductive study in
which males were exposed to 5000 mg/litre in drinking-water, there
were no changes in reproductive index. High doses in animals produce
minimal fetal toxicity although only at doses causing maternal
effects. Acetone produced an increase in nephropathy in treated male
rats over the levels found in controls: this is of uncertain
significance to humans, Acetone is not genotoxic nor has it been shown
to be carcinogenic in dermal bioassays in mice.
No long-term experimental studies have been conducted. From the
available shorter-term studies, a no-observed-effect level in a
13-week drinking-water study in rats of 900 mg/kg body weight per day
(male rats), based on parameters including changes in organ weights,
was selected. Applying an uncertainty factor of 100 gives a guidance
value of 9 mg/kg body weight per day.
The relevance to humans of the liver, reproductive and
developmental effects observed in animal studies is not known, and
these end-points have not been sufficiently examined in humans.
However, because few species differences exist in the toxicokinetics
of acetone, these effects might be of concern for humans. The renal
effects may be specific for male rats, and the cataract formation may
be specific for guinea-pigs. The relevance of amyloidosis in
completely unknown. Acetone appears to have no delayed toxic effects.
The majority of genotoxicity assays on acetone were negative;
therefore, acetone can be considered to present no potential genotoxic
hazard to humans.
It should be noted that the perception of "irritation" from
acetone vapour by humans may be at a concentration in air as low as
23.7 mg/m3 (100 ppm), which is at or near the odour threshold.
10.2 Evaluation of effects on the environment
Acetone is of low toxicity to both aquatic and terrestrial
organisms. It is readily biodegraded in the environment and does not
bioaccumulate or magnify through the food chain. Even if acetone is
spilt in water, it is unlikely to have a major or lasting effect on
the ecosystem. Owing to evaporation and dispersal, spills on land are
likewise not expected to have any major or lasting effects on
terrestrial organisms.
11. FURTHER RESEARCH
a) Reproductive effects need to be examined in animals and/or in
humans. Clarification of the dose-response relationship is
required with special reference to male reproductive effects at
doses where abnormal sperm are found and to determine if there
are complications during menstration, pregnancy and childbirth,
as existing data are not conclusive.
b) Longer-term studies are required to determine whether the kidney
effects are attributable to acetone or are exacerbating an
existing condition. If they are acetone-related, the mechanism
should be determined.
c) Clarification of the potentiation and antagonism mechanisms in
humans is needed.
d) Clarification of the mechanisms of potential immunotoxic effects
is required.
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
Acetone was evaluated in 1970 as an extraction solvent for fats
and oils and a precipitation agent in the purification of starches and
sugars by the Joint FAO/WHO Expert Committee on Food Additives. The
Committee recommended that its use as an extraction solvent should be
restricted to that determined by good manufacturing practice, which is
expected to result in minimum residues. Within these limits residues
are unlikely to have any significant toxicological effect (FAO/WHO,
1971; WHO, 1971).
REFERENCES
Abbondandolo A, Bonatti S, Corsi C, Corti G, Fiorio R, Leporini C,
Mazzaccaro A, Nieri R, Barale R, & Loprieno N (1980) The use of
organic solvents in mutagenicity testing. Mutat Res, 79: 141-150.
Aharonson EF, Menkes H, Guntner G, Swift DL, & Proctor DF (1974)
Effect of respiratory airflow rate on removal of soluble vapors by the
nose, J Appl Physiol, 37: 654-657.
Ahonen I & Schimberg RW (1989) 2,5-Hexanedione excretion after
occupational exposure to n-hexane. Br J Ind Med, 45: 133-136.
Altshuller AP (1991) Chemical reactions and transport of alkanes and
their products in the troposphere. J Atmos Chem, 12: 19-61.
Altshuller AP & Cohen IR (1963) Structural effects of the rate of
nitrogen dioxide formation in the photooxidation of organic
compound-nitric oxide mixtures in air. Int J Air Water Pollut, 7:
789-797.
Amacher DE, Paillet SC, Turner GN, Verne V, & Salsburg DS (1980) Point
mutations at the thymidine kinase locus in L5178Y mouse lymphoma
cells: 2. Test validation and interpretation. Mutat Res, 72: 447-474.
Amlathe S & Gupta VK (1990) Spectrophotometric determination of
acetone using vanillin. Analyst, 115:1385-1387.
Amoore JE & Hautala E (1983) Odor as an aid to chemical safety: odor
thresholds compared with threshold limit values and volatilities for
214 industrial chemicals in air and water dilution. J Appl Toxicol, 3:
272-290.
Andersson L & Lundstrom K (1984) Milk and blood ketone bodies, blood
isopropanol and plasma glucose in dairy cows; methodological studies
and diurnal variations. Zent. bl Vet. med, A31: 340-349.
Andersson-Sköld Y, Grennfelt P, & Pleijel K (1992) Photochemical ozone
creation potentials: a study of different concepts. J Air Waste Manage
Assoc, 42:1152-1158.
Aneja VP (1993) Organic compounds in cloud water and their deposition
at a remote continental site. J Air Waste Manage Assoc, 43: 1239-1244.
Anselme C, Nguyen K, Burchet A, & Mallevialle J (1985)
Characterization of low molecular weight products desorbed from
polyethylene tubings. Sci Total Environ, 47:371-384.
Arnold F, Knop G, & Ziereis H (1986) Acetone measurements in the upper
troposphere and lower stratosphere-implications for hydroxyl radical
abundances. Nature (Lend), 321: 505-507.
Arnold F, Schneider J, Gollinger K, Schlager H, Schulte P, Hagen DE,
Whitefield PD, & van Velthoven P (1997) Observation of upper
tropospheric sulfur dioxide- and acetone-pollution: potential
implications for hydroxyl radical and aerosol formation. Geophys Res
Lett, 24(1): 57-60.
Arnts RR & Meeks SA (1981) Biogenic hydrocarbon contribution to the
ambient air of selected areas. Atmos Environ, 15: 1643-1651.
Ashley DL, Bonin MA, Cardinali FL, McCraw JM, Holler JS, Needham LL, &
Patterson DG Jr (1992) Determining volatile organic compounds in human
blood from a large population using purge and trap gas
chromatography/mass spectrometry. Anal Chem, 64:1021-1029.
Ashley DL, Bonin MA, Cardinali FL, McCraw JM, & Wooten JV (1994) Blood
concentrations of volatile organic compounds in a nonoccupationally
exposed US population and in groups with suspected exposure. Clin
Chemistry, 40(7): 1401-1404.
Atkinson R (1985) Kinetics and mechanisms of the gas-phase reactions
of hydroxyl radical with organic compounds under atmospheric
conditions, Chem Rev, 85: 69-201.
ATSDR (1994) Toxicological profile for acetone. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry, 257 pp.
Babeu L & Vaishnav DD (1987) Prediction of biodegradability for
selected organic chemicals. J Ind Microbial, 2: 107-115.
Barber ED & Lodge JP Jr (1963) Paper chromatographic identification of
carbonyl compounds as their 2,4-dinitrophenyl hydrazones in automobile
exhaust. Anal Chem, 35: 348-350.
Barnett CR, Petrides L, Wilson J, Flatt PR, & Ioannides C (1992)
Induction of rat hepatic mixed-function oxidases by acetone and other
physiological ketones: Their role in diabetes-induced changes in
cytochrome P450 proteins. Xenobiotica, 22:1411-1450.
Basler A (1986) Aneuploidy-inducing chemicals in yeast evaluated by
the micronucleus test. Mutat Res, 174(1): 11-13.
Benkelberg H-J, Hamm S, & Warneck P (1995) Henry's law coefficients for
aqueous solutions of acetone, aldehyde and acetonitrile and
equilibrium constants for the additional compounds and acetaldehyde
with bisulfite. J Atmos Chem, 20:17-34.
Bertsch W, Anderson E, & Holzer G (1975) Trace analysis of organic
volatiles in water by gas chromatography-mass spectrometry. J Chromat,
112: 701 - 718.
Bhattacharya SK, Angara RVR, Bishop DF Jr, Dobbs PA, & Austern BM
(1990) Removal and fate of RCRA (Resource Conservation and Recovery
Act) and CERCLA (Comprehensive Environmental Response Compensation and
Liability Act) toxic organic pollutants in wastewater treatment.
Cincinnati, Ohio, US Environmental Protection Agency, Risk Reduction
Engineering Laboratory, 150 pp (EPA 600/S2-89-026).
Blum DJW & Speeca RE (1991) A database of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Poison Control Fed, 63(3): 198-207.
Boyd AA, Canosa-Mas CE, & King AD (1991) Use of a stopped-flow
technique to measure the rate constants at room temperature for
reactions between the nitrate radical and various organic species. J
Chem Bec Faraday Trans, 87:2913-2919.
Brady JF, Li D, Ishizaki H, Lee M, Ning SM, Xiao F, & Yang CS (1989)
Induction of cytochrome P45011E1 and P450IIE1 by secondary acetones
and the role of P450IIE1 in chloroform metabolism. Toxicol Appl
Pharmacol, 100: 342-349.
Brega A, Villa P, Quadrini G, Quadri A, & Lucarella C (1991)
High-performance liquid chromatographic determination of acetone in
blood and urine in the clinical diagnostic laboratory. J Chromatogr,
553: 249-254.
Bridie AL, Wolff CJM & Winter M (1979) BOD and COD of some
petrochemicals. Water Res, 13: 627-630.
Bringmann G & Kühn R (1978) Testing of substances for their toxicity
threshold: Model organisms Microcystis (Diplocystis) aeruginosa and
Scenedesmus quadricauda. Mitt Int Verein Limnol, 21; 275-284.
Bringmann G & Kühn R (1980a) Comparison of the toxicity thresholds of
water pollutants to bacteria, algae, and protozoa in the cell
multiplication inhibition test. Water Res, 14(3): 231-241.
Bringmann G & Kühn R (1980b) [Determination of the harmful biological
effect of water pollutants on protozoa: II. Bacteriovorous ciliates.] Z
Wasser Abwasser Forsch, 13(1): 26-31 (in German).
Bringmann G & Kühn R (1982) [The toxic action of water pollutants on
Daphnia magna tested by an improved standardized procedure.] Z
Wasser Abwasser Forsch, 15(1): 1-6 (in German).
Brondeau MT, Ban M, Bonnet P, Guenier JP, & De Ceaurriz J (1989)
Acetone compared to other ketones in modifying the hepatotoxicity of
inhaled 1,2-dichlorobenzene in rats and mice. Toxicol Lett, 49: 69-78.
Brooke LT, Call DJ, Geiger DL, & Northcott CE ed. (1984) Acute
toxicities of organic chemicals to fathead minnows ( Pimephales
promelas). Superior, Wisconsin, University of Wisconsin,
Center for Lake Superior Environmental Studies, 24 pp.
Brosseau J & Heitz M (1994) Trace gas compound emissions from
municipal landfill sanitary sites. Atmos Environ, 28(2): 285-293.
Brown KW & Donnelly KC (1988) An estimation of the risk associated
with the organic constituents of hazardous and municipal waste
landfill leachates. Hazard Waste Hazard Mater, 5: 1-30.
Brown EM & Hewitt WR (1984) Dose-response relationships in
ketone-induced potentiation of chloroform hepato- and nephrotoxicity.
Toxicol Appl Pharmacol, 78: 437-53.
Brown WD, Setzer JV, Dick RB, Phipps FC, & Lowry LK (1987) Body burden
profiles of single and mixed solvent exposures. J Occup Mod, 29:
877-883.
Brown VM, Crump DR, Gardiner D, & Gavin M (1994) Assessment of a
passive sampler for the determination of aldehydrates and ketones in
indoor air. Environ Technol, 15: 679-85.
Brown BL, Allis JW, Simmons JE, & House DE (1995) Fasting for less
than 24 h induces cytochrome P4502E1 and 2B1/2 activities in rats.
Toxicol Lett, 81: 39-44.
Bruckner JV & Peterson RJ (1981) Evaluation of toluene and acetone
inhalant abuse: II. Model development and toxicology. Toxicol Appl
Pharmacol, 61(3): 302-312.
Brugnone F, Perbellini L, & Grigolini L (1978) Solvent exposure in a
shoe upper factory. Int Arch Occup Environ Health, 42: 51-62.
Brugnone F, Perbellini L, Giuliari C, Cerpelloni M, & Suave M (1994)
Blood and urine concentrations of chemical pollutants in the general
population. Med Lav, 85(5): 370-369.
Bulich AA (1986) Bioluminescence assays. In: Bitton G & Dutka BJ ed.
Toxicity testing using microorganisms, Boca Raton, Florida, CRC Press,
vol 1, pp 57-74.
Burmaster DE (1982) The new pollution-groundwater contamination.
Environment, 24: 6-13, 33-36.
Buxton GV, Greenstock CL, Heiman WP, & Ross AB (1988) Critical review
of rate constants for reactions of hydrated electrons, hydrogen atoms
and hydroxyl radicals ("OH/"O-) in aqueous solution. J Phys Chem Ref
Data, 17: 513.
Cairns J Jr & Scheier A (1998) A comparison of the toxicity of some
common industrial waste components tested individually and combined.
Prog Fish Cult, 30(1): 3-6.
Cander L & Forster RE (1959) Determination of pulmonary parenchymal
volume and pulmonary capillary blood flow in man. J Appl Physol, 59:
541-651.
Canton JH & Adema DMM (1978) Reproducibility of short-term and
reproduction toxicity experiments with Daphnia magna and comparison
of the sensitivity of Daphnia magna with Daphnia pulex and
Daphnia cucullata in short-term experiments. Hydrobiologia, 59(2):
135-140.
Carpenter CP & Smyth HF Jr (1946) Chemical burns of the rabbit cornea.
Am J Opthalmol, 29: 1363-1372.
Carter WPL (1994) Development of ozone reactivity scales for volatile
organic compounds. J Air Waste Manage Assoc, 44: 881-899.
Casazza JP, Felver ME, & Veech RL (1984) The metabolism of acetone in
rat. J Biol Chem, 259: 231-236.
Cavanagh LA, Schadt CF, & Robinson E (1969) Atmospheric hydrocarbon
and carbon monoxide measurements at Point Barrow, Alaska. Environ Sci
Technol, 3: 251-257.
Chandler JH Jr & Marking LL (1979) Toxicity of fishery chemicals to
the Asiatic clam, Curbicula manilensis. Prog Fish Cult, 41(3):
148-151.
Charbonneau M, Iijima M, Côté MG, & Plaa GL (1985) Temporal analysis
of rat liver injury following potentiation of carbon tetrachloride
hepatotoxicity with ketonic or ketogenic compounds. Toxicology,
35:95-111.
Charbonneau M, Brodeur J, DuSouich P, & Plaa GL (1985a) Correlation
between acetone-potentiated CCl4-induced liver injury and blood
concentrations after inhalation or oral administration. Toxicol Appl
Pharmacol, 84: 286-294.
Charbonneau M, Oleskevich S, Brodeur J, Plaa GL (1986b) Acetone
potentiation of rat liver injury induced by trichloroethylene-carbon
tetrachloride mixtures. Fundam Appl Toxicol, 6: 654-661.
Charbonneau M, Perreault F, Greselin E, Brodeur J, & Plaa GL (1988)
Assessment of the minimal effective dose of acetone for potentiation
of the hepatotoxicity induced by trichloroethylene-carbon
tetrachloride mixtures. Fundam Appl Toxicol, 10: 431-438.
Charbonneau M, Couture J, & Plaa GL (1991) Inhalation versus oral
administration of acetone: effect of the vehicle on the potentiation
of CCI4-induced liver injury. Toxicol Lett, 57: 47-54.
Chatfield RS, Gardner EP, & Calvert JG (1987) Sources and sinks of
acetone in the troposphere: behavior of reactive hydrocarbons and a
stable product. J Geophys Res, 92: 4208-4216.
Chatterton CC & Elliott RB (1946) Acute acetone poisoning from leg
casts of a synthetic plaster substitute. J Am Med Assoc,
130: 1222-1223.
Chen H-F & Que Hee SS (1994) Ketone EC50 in the Microtox test.
Ecotoxicol Environ Saf, 30: 120-123.
Cheung ST & Lin WN (1967) Simultaneous determination of methanol,
ethanol, acetone, isopropanol and ethylene glycol in plasma by gas
chromatography. J Chromatogr Biomed Appl, 414: 248-250.
Chieli E, Puccini P, Longo V, & Gervasi PG (1990) Possible role of the
acetone-inducible cytachrome P-450IIE1 in the metabolism and
hepatotoxicity of thiobenzamide. Arch Toxicol, 64(2): 122-127.
Chou WL, Speece RE, & Siddiqi RH (1979) Acclimation and degradation of
petrochemical wastewater components by methane fermentation.
Biotechnol Bioeng Syrup, 8: 391-414.
Clayton GD & Clayton FE ed, (1982) Patty's industrial hygiene and
toxicology: Volume 2C, 3rd ed. New York, John Wiley & Sons, pp
4722-4723.
Coleman DL (1980) Acetone metabolism in mice: increased activity in
mice heterozygous for obesity genes. Proc Natl Acad Sci (USA), 77(1);
290-293.
Coleman EC, Ho C-T, & Chang SS (1981) Isolation and identification of
volatile compounds from baked potatoes. J Agric Food Chem, 29(1);
42-48.
Conkle JP, Camp B J, & Welch BE (1975) Trace composition of human
respiratory gas. Arch Environ Health, 30: 290-295.
Corwin JF (1969) Volatile oxygen-containing organic compounds in sea
water determination. Bull Mar Sci, 19(3): 504-509.
Cowgill UM & Milazzo DP (1991) The sensitivity of Ceriodaphnia dubia
and Daphnia magna to seven chemicals utilizing the three-brood test.
Arch Environ Contam Toxicol, 20: 211-217.
Cowgill UM, Milazzo DP, & Laudenberger BD (1989) Toxicity of nine
benchmark chemicals to Skeletonema costatum, a marine diatom.
Environ Toxicol Chem, 8: 451-455.
Cowgill UM, Mirazzo DP, & Laudenberger BD (1991) The sensitivity of
Lemna gibba G3 and four clones of Lemna minor to eight common
chemicals using a 7-day test. Res J Water Pollut Control Fed, 63:
991-998.
Crisinel A, Delannay L, Rossel D, Tarradellas J, Meyer H, Saïah H,
Vogel P, Delisle C, & Blaise C (1994) Cyst-based ecotoxicological
tests using Anostracans: comparison of two species of
Streptocephalus. Environ Toxicol Water Qual, 9: 317-326.
Cunningham J, Sharkawi M, & Plaa GL (1989) Pharmacological and
metabolic interactions between ethanol and methyl n-butyl ketone,
methyl isobutyl ketone, methyl ethyl ketone, or acetone in mice.
Fundam Appl Toxicol, 13: 102-109.
Dalton P, Wysocki CJ, Brody MJ, & Lawley HJ (1997) The influence of
cognitive bias on the perceived odor, irritation and health symptoms
from chemical exposure. Int Arch Occup Environ Health, 69: 407-417.
Datta RK & Rao KN (1979) Kinetics of reactions of single molecular
oxygen (1 Delta g) with organic compounds. Indian J Chem, 18A:
102-105.
Day EA & Anderson DF (1965) Gas chromatographic and mass spectral
identification of natural components of the aroma fraction of blue
cheese. J Agric Food Chem, 13: 2-4.
De Rosa E, Cellini M, Sessa G, Saletti C, Raus G, Maracuzzo G, &
Bartolucci GB (1993) Biological monitoring of workers exposed to
styrene and acetone, Int Arch Occup Environ Health, 65: S107-S110.
Derwent RG, Jenkin ME, & Saunders SM (1996) Photochemical ozone
creation potentials for a large number of reactive hydrocarbons under
European conditions. Atmos Environ, 30(2): 181-199.
DeWalle FB & Chian ESK (1981) Detection of trace organics in well
water near a solid waste landfill. J Am Water Works Assoc, 73;
205-211.
De Walle FB, Katman DA, Norman D, Sung J, & Plews G (1985)
Determination of toxic chemicals in effluent from household septic
tanks. Cincinnati, Ohio, US Environmental Protection Agency, 25 pp
(EPA 600/S2-85-050).
De Zwart D & Slooff W (1983) The Microtox as an alternative assay in
the acute toxicity assessment of water pollutants. Aquat Toxicol,
4:129-138.
Dick RB, Setzer JV, Taylor BJ, & Shukla R (1989) Neurobehavioral
effects of short duration exposures to acetone and methyl ethyl
ketone. Br J Ind Med, 46(2): 111-121.
Dietz DD, Leininger JR, Rauckman EJ, Thompson MB, Chapin RE, Morrissey
EJ, & Levine DS (1991) Toxicity studies of acetone administered in the
drinking water of rodents. Fundam Appl Toxicol, 17: 347-360.
Dills RJ, Ackerlund WS, Kalman DA, & Morgan MS (1994) Inter-individual
variability in blood/air partitioning of volatile organic compounds
and correlation with blood chemistry. J Expo Anal Environ Epidemiol,
4(2); 229-245
Dimitriades B & Joshi SB (1977) Application of reactivity criteria in
oxidant-related emission control in the USA. In: Proceedings of
International Conference on Photochemical Oxidant Pollution and its
Control, Raleigh, North Carolina, 12-17 September 1976. Research
Triangle Park, North Carolina, US Environmental Protection Agency,
Environmental Sciences Research Laboratory, pp 705-711
(EPA 600/3-77-001b).
Ding X & Coon MJ (1990) Induction of cytochrome P-450 isozyme 3a
(P-450IIE1) in rabbit olfactory mucosa by ethanol and acetone. Drug
Metab Dispos, 18: 742-745.
DiPaolo JA, Donovon AP, & Nelson R (1969) Quantitative studies of
in vitro transformation by chemical carcinogens. J Natl Cancer Inst,
42: 867-874.
DiVincenzo GD, Yanno FJ, & Astill BD (1973) Exposure of man and dog to
low concentrations of acetone vapor. Am Ind Hyg Assoc J, 34: 329-336.
Dowden BF & Bennett HJ (1965) Toxicity of selected chemicals to
certain animals. J Water Pollut Control Fed, 37(9): 1308-1316.
Egle JL Jr (1973) Retention of inhaled acetone and ammonia in the dog.
Am Ind Hyg Assoc J, 34: 533-639.
EHRT (Environmental Health Research and Testing, Inc.) (1987)
Screening of priority chemicals for reproductive hazards: benzethonium
chloride (CAS No. 121-54-0); 3-ethoxy-1-propanol (CAS No. 111-35-3);
acetone (CAS No. 67-94-1). Cincinnati, Ohio, National Institute for
Occupational Safety and Health, 50 pp (NTIS P889-139083).
Elovaara E, Vainio H, & Aitio A (1990) Pulmonary toxicity of inhaled
styrene in acetone-phenobarbital - and 3-methylcholanthrene-treated
rats. Arch Toxicol, 64: 365-369.
Ettinger MB (1956) Biochemical oxidation characteristics of stream
pollutant organics. Ind Eng Chem, 48: 255-259.
Ewell WS, Gorsuch JW, Kringle RO, Robilliard KA, & Spiegel RC (1986)
Simultaneous evaluation of the acute effects of chemicals on seven
aquatic species. Environ Toxicol Chem, 5(9): 631-840.
Ewing BB, Chian ESK, Cook JC, Evans CA, Hopke PK, & Perkins EG (1977)
Monitoring to detect previously unrecognized pollutants in surface
waters - Appendix: Organic analysis data. Washington, DC, US
Environmental Protection Agency, pp 1, 3, 72 (EPA-560/6-77-015).
FAO/WHO (1971) Toxicological evaluation of some extraction solvents
and certain other substances. Report of the Joint FAO/WHO Expert
Committee on Food Additives, 24 June-2 July 1970 Rome, Food and
Agriculture Organization, pp 86-8.
Fiorio R & Bronzetti G (1995) Diallyl sulfide inhibits the induction
of HPRT-deficient mutants in Chinese hamster V79 cells treated with
dimethylnitrosamine in the presence of S-9 of rats induced with
acetone. Environ Mol Mutagen, 25(4): 344-346.
Fiserova-Bergerova V & Diaz ML (1986) Determination and prediction of
tissue-gas partition coefficients. Int Arch Occup Environ Health, 58:
75-87.
Folland DS, Shaffner W, Grinn HE, Crafford OB, & McMurray DR (1976)
Carbon tetrachloride toxicity potentiated by isopropyl alcohol:
Investigation of an industrial outbreak. J Am Med Assoc, 26:1853 1856.
Forkert PG, Massey TE, Jones AB, Park SS, Gelboin HV, & Anderson LM
(1991) Distribution of cytochrome CYP2E1 in murine liver after ethanol
and acetone administration. Carcinogenesis, 12(12): 2259-2268.
Frantik E, Horvárth M, & Vodicková L (1988) Effects of solvent
mixtures on behaviour and seizure characteristics at the utmost
additive. Activ Nerv Super, 30: 260-264.
Frantik E, Hornychová M, & Horvárth M (1994) Relative acute
neurotoxicity of solvents: isoeffective air concentrations of 48
compounds evaluated in rats and mice. Environ Res, 66: 173-185.
Frantik E, Vodicková L, Hornychová M, & Nosek M (1996) Pattern of
inhalation exposure: blood levels and acute subnarcotic effects of
toluene and acetone in rats. Cent Eur J Public Health, 4: 226-232.
Freeman JJ & Hayes EP (1985) Acetone potentiation of acute
acetonitrile toxicity in rats. J Toxicol Environ Health, 15: 809-621.
Freeman JJ & Hayes EP (1986) Microsomal metabolism of acetonitrile to
cyanide. Biochem Pharmicol, 37:1153-1159.
Freeman AE, Weisburger EK, Weisburger JH, Wolford RG, Maryak JM, &
Huebner RJ (1973) Transformation of cell cultures as an indication of
the carcinogenic potential of chemicals. J Natl Cancer Inst, 51(3):
799-807.
Fujino A, Satoh T, Takebayashi T, Nakashima H, Sakurai H, Higashi T,
Matumura H, Minaguchi H, & Kawai T (1992) Biological monitoring of
workers exposed to acetone in acetate fibre plants. Br J Ind Med, 4.9;
654-657.
Fukabori S, Nakaaki K, & Tada O (1979) [The cutaneous absorption of
acetone.] Jpn J Sci Labour, 55: 525-532 (in Japanese).
Gamis AS & Wasserman GS (1988) Acute acetone intoxication in a
pediatric patient. Pediatr Emerg Care, 4: 24-26.
Gardner EP, Wijayaratne RD, & Calvert JG (1984) Primary quantum yields
of the photodecomposition of acetone in air under tropospheric
conditions. J Phys Chem, 88: 5069-5076.
Gaudy AF Jr, Turner BG, & Pusztaszeri S (1968) Biological treatment of
volatile waste components. J Water Pollut Control Fed, 35: 75-93.
Gavino VC, Vinet B, David F, Garneau M, & Brunengraber H (1985)
Determination of the concentration and specific activity of acetone in
biological fluids. Anal Biochem, 152: 256-261.
Gavino VC, Somma J, Philbert L, David F, Garneau M, Bélair J, &
Brunengraber H (1987) Production of acetone and conversion of acetone
to acetate in the perfused rat liver. J Biol Chem, 262: 6735-6740.
Geller I, Hartmann RJ, Randle SR, & Gause EM (1979) Effects of acetone
and toluene vapors on multiple schedule performance of rats. Pharmacol
Biochem Behav, 11: 395-399.
Gerhold RM & Malaney GW (1966) Structural determinants in the
oxidation of aliphatic compounds by activated sludge. J Water Pollut
Control Fed, 38: 562-579.
Ghittori S, Imbriani M, Pezzagno G, & Capodaglio E (1987) The urinary
concentration of solvents as a biological indicator of exposure:
Proposal for the biological equivalent exposure limit for nine
solvents. Am Ind Hyg Assoc J, 48: 786.
Gitelson S, Werczberger A, & Herman JR (1966) Coma and hyperglycemia
following drinking of acetone. Diabetes, 15:816-811.
Glatt H, de Balle L, & Oesch F (1981) Ethanol- or acetone-pretreatment
of mice strongly enhances the bacterial mutagenicity of
dimethylnitrosamine in assays mediated by liver subcellular fraction,
but not in host-mediated assays. Carcinogenesis, 2: 1057-1067.
Glowa JR & Dews PB (1987) Behavioral toxicology of volatile organic
solvents: IV. Comparisons of the rate-decreasing effects of acetone,
ethyl acetate, methyl ethyl ketone, toluene, and carbon disulfide on
schedule-controlled behavior of mice. J Am Coll Toxicol, 6(4):
461-469.
Goldberg ME, Johnson HE, Pozzani UC, & Smyth HF Jr (1964) Effect of
repeated inhalation vapors of industrial solvents on animal behaviour.
Am Ind Hyg Assoc J, 25: 369-375.
Gordon AW & Gordon M (1981) Analysis of volatile organic compounds in
a textile finishing plant effluent. Trans Ky Acad Sci, 42: 149-157.
Gorsuch JW, Kringle RO, & Robillard KC, (1990) Chemical effects on the
germination and early growth of terrestrial plants. In: Wang W,
Gorsuch JW, & Lower WR ed. Plants for toxicity assessment.
Philadelphia, Pennsylvania, American Society for Testing and
Materials, pp 49-58 (ASTM STP 1091).
Goss K-U (1992) Effects of temperature and relative humidity on the
sorption of organic vapors on quartz sand. Environ Sci Technol, 26:
2287-2294.
Gould JP, Ramsay RE, Giabbai M, & Pohland FG (1983) Formation of
volatile haloorganic compounds in the chlorination of municipal
landfill leachates. In: Jolley RL, Brungs WA, Cotruvo JA, Cumming RB,
Mattice JS, & Jacobs VA ed. Water chlorination: Environmental impact
and health effects. Ann Arbor, Michigan, Ann Arbor Science Publishers,
vol 4, book 1, chapter 36, pp 525-539.
Graedel TE (1978) Chemical compounds in the atmosphere. New York,
London, San Francisco, Academic Press, pp 7, 182.
Graedel TE, Hawkins DT, & Claxton LD (1986) Atmospheric chemical
compounds: Sources, occurrence, and bioassay. Orlando, Florida,
Academic Press, p 263.
Granby K, Christensen CS, & Lohse C (1997) Urban and semi-rural
observations of carboxylic acids and carbonyls. Atmos Environ
31: 1403-1415.
Green WJ, Lee GF, Jones RA, & Palit T (1963) Interaction of clay
soils with water and organic solvents; Implications for the disposal
of hazardous wastes. Environ Sci Technol, 17: 278-282.
Grey TC & Shrimpton H (1967) Volatile components of raw chicken breast
muscle. Br Poult Sci, 8: 23-33.
Grosjean D & Wright 8 (1983) Carbonyls in urban fog, ice fog,
cloudwater and rainwater. Atmos Environ, 17: 2093-2096.
Grosjean D, Miguer AH, & Tavares TM (1989) Urban air pollution in
Brazil: Acetaldehyde and other carbonyls. Atmos Environ, 248: 101-106.
Guntakatta ME, Matthews EJ, & Rundell JO (1984) Development of a mouse
embryo limb bud cell culture system for the estimation of chemical
teratogenic potential. Teratogen Carcinog Mutagen, 4:349-364
Haff AC & Reichard GA Jr (1977) A method for estimation of acetone
radioactivity and concentration in blood and urine. Biochem Med, 18;
308-314.
Haggard HW, Greenberg LA, & Turner JM (1944) The physiological
principles governing the action of acetone together with determination
of toxicity. J Ind Hyg Toxicol, 26: 133-151.
Hallier E, Filser JG, & Bolt HM (1981) Inhalation pharmacokinetics
based on gas uptake studies. II. Pharmacokinetics of acetone in rats.
Arch Toxicol, 47: 293-304.
Hanst PL & Gay BW Jr (1983) Atmospheric oxidation of hydrocarbons:
Formation of hydroperoxides and peroxyacids. Atmos Environ, 17:
2259-2265.
Harris JC (1962) Fate of hydrolysis. In: Lyman WJ, Reehl WF, &
Hosenblett DH ed. Handbook of chemical property estimation methods.
New York, McGraw-Hill, pp 7/2-7/9.
Hatfield R (1957) Biological oxidation of some organic compounds. Ind
Eng Chem, 49:192-198.
Hawthorne SB & Sievers RE (1984) Emission of organic air pollutants
from shale oil wastewaters. Environ Sci Technol, 18: 483-490.
Heavner DL, Morgan WT, & Ogden MW (1996) Determination of volatile
organic compounds and respirable suspended particulate matter in New
Jersey and pennsylvania homes and workplaces. Environ Int, 22(2):
159-183.
Hellman TM & Small FH (1974) Characterization of the odor properties
of 101 petrochemicals using sensory methods. J Air Pollut Control
Assoc, 24: 979-982.
Henderson GS, McConnell JC, & Evans WFJ (1989) A comparison of model
calculations and measurements of acetone in the troposphere and
stratosphere, J Atmos Chem, 9: 277-298.
Hess FD (1980) A Chlamydomonas algal bioassay for detecting growth
inhibitor herbicides. Weed Sci, 28(5): 515-520.
Hetenyi G Jr & Ferrarotto C (1985) Gluconeogenesis from acetone in
starved rats. Biochem J, 231; 151-155.
Heukelekian H & Rand MC (1955) Biochemical oxygen demand of pure
organic compounds. J Water Pollut Control Assoc, 29:1040-1053.
Hewitt WR & Plaa GL (1983) Dose-dependent modification of
1,1-dichloroethylene toxicity by acetone, Toxicol Lett, 16: 145-152.
Hewitt WR, Miyajima H, Côté MG, & Plaa GL (1980) Acute alteration of
chloroform-induced hepato- and nephrotoxicity by n-hexane, methyl
n-butyl ketone and 2,5-hexanedione, Toxicol Appl Pharmacol, 53:
230-248.
Hewitt LA, Valiquette C, & Plaa GL (1987) The role of
biotransformation-detoxication in acetone-, 2-butanone-, and
2-hexanone-potentiated chloroform-induced hepatotoxicity. Can J
Physiol Pharmacol, 65: 2315-2318.
Hift W & Patel PL (1981) Acute acetone poisoning due to a synthetic
plaster cast with a discussion of acetone metabolism. S Afr Med J, 35;
245-250.
Hill EF, Heath RG, Spamm JW, & Williams JD (1975) Lethal dietary
toxicities of environmental pollutants to birds. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, pp 1-2, 8, 35
(Special Scientific Report Wildlife No. 191).
Hites RA & Lopez-Avila V (1980) Sedimentary accumulations of
industrial organic compounds discharged into a river system. In Baker
RA ed. Contaminants and sediments - Volume 1: Fate and transport case
studies, modeling, toxicity. Ann Arbor, Michigan, Ann Arbor Science,
chapter 3, pp 53-66.
Hodgkin JH, Galbraith MN, & Chong YK (1982) Combustion products from
burning polyethylene. J Macromol Sci Chem, 17: 35-44.
Hodgson AT, Daisey JM, & Grot RA (1991) Sources and source strengths
of volatile organic compounds in a new office building. J Air Waste
Manage Assoc, 41: 1461-1468.
Hodgson AT, Garbesi K, Sextro RG, & Daisey JM (1992) Soil-gas
contamination and entry of volatile organic compounds into a house
near a landfill. J Air Waste Manage Assoc, 42; 277-283.
Hodgson AT, Wooley JD, & Daisey JM (1993) Emissions of volatile
organic compounds from new carpets measured in a large-scale
environmental chamber. J Air Waste Manage Assoc, 42: 316-324.
Hoffman DJ & Eastin WC (1981) Effects of industrial effluents, heavy
metals and organic solvents on mallard embryo development, Toxicol
Lett, 9: 35-40.
Holm S & Lundgron E (1984) A purge-and-tap method for the analysis of
acetone in biological tissues. Anal Biochem, 136: 157-180.
Hong J & Yang CS (1985) The nature of microsomal
N-nitrosodimethylamine demethylase and its role in carcinogen
activation. Carcinogenesis, 6: 1805-1809.
Hung PE, Fritz VA, & Waters L Jr (1992) Infusion of shrunken-2 sweet
corn seed with organic solvents: Effects on germination and vigor.
Hortic Sci, 27(5): 467-470.
Hwang H-M, Hodson RE, & Lewis DL (1989) Microbial degradation kinetics
of toxic organic chemicals over a wide range of concentrations in
natural aquatic systems. Environ Toxicol Chem, 8: 65-74.
Isidorov VA, Zenkevich IG, & Ioffe BV (1985) Volatile organic
compounds in the atmosphere of forests. Atmos Environ, 19: 1-6.
Iversen OH, Ljunggren S, & Olsen WM (1988) The early effects of a
single application of acetone and various doses of
7,12-dimethylbenz(alpha)anthracene on CD-1 and hairless mouse
epidermis: A cell kinetic study of so-called initiation and complete
carcinogenesis (initiation plus promotion) in chemical skin tumor
induction. Acta Pathol Microbial Immunol Scand, 27(suppl): 7-80
Jaeger RJ, Conolly RB, Reynolds ES, & Murphy SO (1975) Biochemical
toxicology of unsaturated halogenated monomers. Environ Health
Perspect, 11: 121-128.
Jakubowski M & Wieczorek H (1988) The effects of physical effort on
pulmonary uptake of selected organic compound vapours. Pol J Occup
Med, I: 62-71.
Jansson BO & Lareson BT (1969) Analysis of organic compounds in human
breath by gas chromatography-mass-spectrometry. J Lab Clin Med, 74:
961-966.
Jarke FH, Dravnieks A, & Gordon SM (1981) Organic contaminants in
indoor air and their relation to outdoor contamination. Trans Am Soc
Heating Refrig Air.-Cond Eng. 87: 153-16.
Jeffery EH, Arndt K, & Haschek WM (1991) The role of cytochrome
P45011E1 in bioactivation of acetaminophen in diabetic and
acetone-treated mice. In: Witmer CM, Snyder RR, & Jollow DJ ed.
Biological reactive intermediates- IV: Molecular and cellular effects
and their impact on human health. New York, Plenum Publishing
Corporation, pp 249-251.
Johansson I & Ingelman-Sundberg M (1988) Benzene metabolism by
ethanol-, acetone- and benzene-inducible cytochrome P-450 (IIE1) in
rat and rabbit liver microsomes. Cancer Res, 48: 5837-5390.
Johansson I, Eliasson E, Norsten C, & Ingelman-Sundberg M (1986)
Hydroxylation of acetone by ethanol- and acetone-inducible cytochrome
P-450 in liver microsomes and reconstituted membranes. FEBS Lett, 196:
59-64.
Johanason I, Ekström E, Scholte B, Puzycki D, Jörnvall H,
& Ingelman-Sundberg M (1988) Ethanol-, fasting-, and acetone-inducible
cytochromes P-450 in rat liver: regulation and characteristics of
enzymes belonging to the IIB and IIE gene subfamilies. Biochemistry,
27(6): 1925-1934.
Johnson CE & Jenkin ME (1991) Modelling the photochemistry of acetone.
Harwell, Atomic Energy Agency, Environment and Energy, 29 pp.
Jones AW & Andersson L (1995) Biotransformation of acetone to
isopropanol observed in a motorist involved in a sobriety check. J
Forensic Sci, 40(4): 586487.
Jones AW, Sagarduy A, Ericsson E, & Arnqvist HJ (1993) Concentrations
of acetone in venous blood samples from drunk drivers, Type-I diabetic
outpatients, and healthy blood donors. J Appl Toxicol, 17; 182-185.
Jonssen A, Persson KA, & Grigoriadis V (1985) Measurements of some
low-molecular -weight oxygenated, aromatic and chlorinated
hydrocarbons in ambient air and in vehicle emissions. Environ Int, 11:
383-392.
Jungclaus GA, Lopez-Avila V, & Hites RA (1976) Organic compounds in an
industrial waste water: A case study of their environmental impact.
Environ Sci Technol, 12: 88-96.
Kamlet M J, Doherty RM, Veith GD, Taft RW, & Abraham MH (1986)
Solubility properties in polymers and biological media: 7. An analysis
of toxicant properties that influence inhibition of bioluminescence in
Photobacterium phosphoroum (the Microtox test). Environ Sci Technol,
20(7): 690-695.
Kawai T, Yasugi T, Uchida Y, Iwami O, & Ikeda M (1990a) Urinary
excretion of unmetabolized acetone as aa indicator of Occupational
exposure to acetone. Int Arcb Occup Environ Health, 62: 165-170.
Kawai T, Yasugi T, Horiguchi S, Uchida Y, Iwami O, Iguchi H, Inoue O,
Watanabe T, Nakatsuka H, & Ikeda M (1990b) Biological monitoring of
occupational exposure to isopropy alcohol vapor by urinalysis for
acetone. Int Arch Occup Environ Health, 62: 409-13.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Iguchi H, & Ikeda M (1992)
Curvi-linear relation between acetone in breathing zone air and
acetone in urine among workers exposed to acetone vapor. Toxicol Lett,
62: 85-91.
Kerr JA & Stocker DW (1986) Kinetics of the reactions of hydroxyl
radicals with alkyl nitrates and with some oxygen-containing organic
compounds studied under simulated atmospheric conditions. J Atmos
Chem, 4: 253-262.
Kieber RJ & Mopper K (1990) Determination of picomolar concentrations
of carbonyl compounds in natural waters, including seawater, by liquid
chromatography. Environ Sci Technol, 24: 1477-1481.
Kiesswetter E, Blaszekewicz M, Vangala RR, & Seeber A (1994) Acute
exposure to acetone in a factory and ratings of well-being.
Neurotoxicology, 15: 597-602.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl Pharmacol,
19: 699-704.
Kinlin TE, Muralidhara R, Pittet AO, Sanderson A, & Walradt JP (1972)
Volatile components in roasted filberts. J Agric Food Chem, 20:
1021-1028.
Kitchin KT & Ebron MT (1984) Further development of rodent whole
embryo culture: Solvent toxicity and water insoluble compound delivery
system. Toxicology, 30: 45-57.
Kleindienst TE, Shepson PB, Edney EO, Claxton LD, & Cupitt LT (1986)
Wood smoke: measurement of the radiogenic activities of its gas and
particulate-phase photo-oxidation products. Environ Sci Technol, 20:
493-501.
Kleppel GS & McLaughlin JJA (1980) PCB toxicity to phytoplankton:
Effects of dose and density-dependent recovery responses. Bull Environ
Contam Toxicol, 24(5): 696-703.
Knöppel H & Schauenburg H (1969) Screening of household products for
the emission of volatile organic compounds. Environ Int, 15: 413-418.
Kobayashi K, Okada M, Yasuda Y, & Kawai S (1983) A gas chromatographic
method for the determination of acetone and acetoacetic acid in urine.
Clin Chim Acta, 133: 223-226.
Kobusch AB, Plaa GL, & DuSonich P (1989) Effects of acetone and methyl
n-butyl ketone on hepatic mixed function oxidase. Biochem Pharmacol,
38: 3461-3467.
Koop DR & Casazza PJ (1985) Identification of ethanol-inducible
p-450 isozyme 3a as the acetone and acetol monooxygenase of rabbit
microsomes. J Biol Chem, 260:13607-13612.
Korhonen A, Hemminki K, & Vainio H (1983) Embryotoxic effects of
acrolein, methacrylates, guanidines and resorcinol on three day chicken
embryos. Acta Pharmacol Toxicol, 52: 95-99.
Kosugi K, Chandramouli V, Kumaran K, Schumann WC, & Landau BR (1986a)
Determinants in the pathways followed by the carbons of acetone in
their conversion to glucose. J Biol Chem, 261: 13179-13181.
Kosugi K, Scofield RF, Chandramouli V, Kumaren K, Schumann W, & Landau
BR (1986b) Pathways of acetone's metabolism in the rat. J Biol Chem,
162: 3952-3957.
Krasavage WJ, O'Donoghue JL, & DiVincenzo GD (1982) Ketones. In:
Clayton GD & Clayton FE ed. Patty's industrial hygiene and toxicology,
3rd ed. New York, John Wiley & Sons, vol II, pp 4709, 4720-4721,
4724-4727, 4780-4800.
Kubinski H, Gutzke GE, & Kubinski ZO (1981) DNA-cell binding (DCB)
assay for suspected carcinogens and mutagens. Mutat Res, 89: 95-136.
Kumagai S & Matsunaga I (1995) Physiologically based pharmacokinetic
model for acetone, Occup Environ Med, 52: 344-352.
Kundu SK, Bruzek JA, Nair R, & Judilla AM (1993) Breath acetone
analyzer: diagnostic tool to monitor dietary fat loss. Clin Chem,
39(1): 87-92.
Kupferschmid LL & Perkins JL (1986) Organic solvent recycling plant
exposure levels. Appl Ind Hyg, 1: 122-124.
Lacouture PG, Heldreth DD, Shannon M, & Lovejoy FH (1989) The
generation of acetonemia/acetonuria following ingestion of a subtoxic
dose of isopropyl alcohol. Am J Emerg Med. 7(1): 38-40.
Ladefoged O & Perbellini L (1986) Acetone-induced changes in the
toxicokinetics of 2,5-hexanedione in rabbits. Scand J Work Environ
Health, 122: 627-29.
Ladefoged O, Hass U, & Simonsen L (1999) Neurophysiological and
behavioural effects of combined exposure to 2,5-hexanedione and
acetone or ethanol in rats. Pharmacol Toxicol, 65: 372-375.
Lam HR, Larsan JJ, Ladefoged O, Mollor A, Strange P, & Arlien-Soborg P
(1991) Effects of 2,4-hexanedione alone and in combination with
acetone on radial arm maze behavior, the "brain-swelling" reaction and
synaptosomal functions. Neurotoxicol Teratol, 13: 407-412.
Lamb CB & Jenkins GF (1952) BOD of synthetic organic chemicals, in:
Proceedings of the 8th Industrial Waste Conference, Purdue University,
Lafayette, Indiana, USA, 7-9 May 1952. Lafayette, Purdue University,
pp 326-331.
Landahl HD & Herrmann RG (1950) Retention of vapors and gases in the
human nose and lung. Am Med Assoc Arch Ind Health, 1: 36-45.
LaRegina J, Bozzelli JW, Harkov R, & Gianti S (1986) Volatile organic
compounds at hazardous waste sites and a sanitary landfill in New
Jersey: An up-to-date review of the present situation. Environ Prog,
8: 18-27.
Larsen J-J, Lykkegeard M, & Ladefoged O (1991) Infidelity in rats
induced by 2,4-hexanedione in combination with acetone. Pharmacol
Toxicol, 69: 43-46.
Larson TS, Finnegan JK, & Haag HD (1956) Observations on the effect of
the edema-producing potency of acids, aldehydes, ketones and alcohols.
J Pharmacol Exp Ther, 116; 119.
Lee JH & Timmons RB (1977) Kinetics and mechanism of the gas phase
reaction of O(3) atoms with acetone. Intl Chem Kinet, 9: 133-139.
Leibrock E & Slemr J (1997) Method for measurement of volatile
oxygenated hydrocarbons in ambient air. Atmos Environ, 31 (20):
3329-3339.
Leonardos G, Kendall D, & Barnard N (1969) Odor threshold determination
of 53 odorant chemicals. J Air Pollut control Assoc, 19: 91-95.
Lewis GD, Laufman AK, McAnalley BH, & Garriott JC (1984) Metabolism
of acetone to isopropyl alcohol in rats and humans. J Forensic Sci,
29: 541-549.
Lindén E, Bengtsson B-E, Svanberg O, & Sundström G (1979) The acute
toxicity of 78 chemicals and pesticide formulations against two
brackish water organisms, the Bleak ( Alburnus alburnus) and the
Harpacticoid Nitocra spinipes. Chemosphere, 11/12: 843-851.
Lipari F, Dasch JM, & Screggs WF (1984) Aldehyde emissions from
wood-burning fireplaces. Environ Sci Technol, 18: 326-330.
Litovitz TL, Holm KC, Bailey KM, & Schmitz BF (1992) 1991 Annual
report of the American Association of Poison Control Centers National
Data Collection System. Am J Emerg Med, 10: 452-505.
Liu J, Sato C, & Marumo F (1991) Characterization of the
acetaminophen-glutathione conjugation reaction by liver microsomes:
Species difference in the effects of acetone. Toxicol Lett, 56:
260-274.
Lloyd AC (1978) Trophospheric chemistry of aldehydes. Washington, DC,
National Bureau of Standards, pp 27-48.
LO H-H, Teets VJ, Yang DJ, Brown PI, & Rankin GO (1987) Acetone
effects on N-(3,5-dichlorophenyl) succinimide-induced
nephrotoxicity. Toxicol Lett, 38: 161-166.
Longo V, Menicagli S, Minks M, Santucci A, & Gervasi PG (1993)
Purification and Characterization of hepatic P-450IIE1 from
acetone-treated mice. Toxicol and Health, 9: 539-546.
Lorr NA, Miller KW, Chung HR, & Yang CS (1984) Potentiation of the
hepatotoxicity of n-nitrosodimethylamine by fasting, diabetes,
acetone and isopropanol. Toxicol Appl Pharmacol, 73: 423-431.
Lovegren NV, Fisher GS, Legendre MG, & Schuller WH (1979) Volatile
constituents of dried legumes. J Agric Food Chem, 27: 851-853.
Ludzack FJ & Ettinger MB (1960) Chemical structures resistant to
aerobic biochemical stabilization. J Water Pollut Control Fed,
32:1173-1200.
Lupulescu AP & Birmingham DJ (1975) Effect of lipid solvents on
protein, DNA, and collagen synthesis in human skin: An electron
microscopic autoradiographic study. J Invest Dermatol, 65: 419-422.
Lupulescu AP & Birmingham DJ (1979) The effect of protective agent
against lipid solvent induced damages: ultrastructural and scanning
electromicroscopical study of human epidermis. Arch Environ Health,
31: 33-36.
Lupulescu AP, Birmingham DJ, & Pinkus H (1973) An electron microscopic
study of human epidermis after acetone and kerosene administration. J
Invest Dermatol, 65: 419-422.
Lyman WJ (1982) Adsorption coefficient for soils and sediments. In:
Handbook of chemical property estimation methods. New York,
McGraw-Hill, pp 4-9.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Aced Sci (USA), 72:5135-5139.
MacDonald JR, Gandolfi AJ, & Sipes IG (1962a) Acetone potentiation of
1,1,2-trichloroethane hepatotoxicity. Toxicol Lett, 13: 57-69.
MacDonald JR, Gandolfi AJ, & Sipes IG (1962b) Covalent binding of
14C-1,1,2-trichloroethane to hepatic proteins following acetone
pretreatment, Drug Chem Toxicol, 5: 233-247.
McFeters GA, Bond JP, Olson SB, & Tchan TY (1983) A comparison of
microbial bioassays for the detection of aquatic toxicants. Water Res,
17(12): 1757-1762.
Majewski HS, Klaverkamp JF, & Scott DP (1977) Acute lethality, and
sub-lethal effects of acetone, ethanol, and propylene glycol on the
cardiovascular and respiratory systems of rainbow trout ( Salmo
galrdneri). Water Res, 13: 217-221.
Mangani F & Ninfali P (1986) Gas chromatographic determination of
acetaldehyde and acetone in human blood by purge and trap, using
permeation tubes for calibration. J Chromatogr, 437(1): 294-300.
Manning DL, Maskarinec MP, Jenkins PA, & Marshall AH (1983) High
performance liquid chromatographic determination of selected gas-phase
carbonyls in tobacco smoke. J Asset Off Anal Chem, 66: 5-12.
Martinez RD, Buitraga AA, Howell NW, Hearn CH, & Joens JA (1992) The
near U.V. absorption spectra of several aliphatic aldehydes and
ketones at 300 K. Atmos Environ, 26A; 785-792.
Matsushita T, Yoshimune A, Inoue T, Yamaka S, & Suzuki H (1969a)
Experimental studies for determining the maximum permissible
concentrations of acetone: 2. Biological reactions in six-day exposure
to acetone. Sangyo Igaku, 11: 507-511.
Matsushita T, Yoshimune A, Inoue T, Yamaka S, & Suzuki H (1969b)
Experimental studies for determining the maximum permissible
concentrations of acetone: I. Biological reactions in one-day exposure
to acetone. Sangyo Igaku, 11: 477-485.
Mayer FL & Ellersieck MR (1980) Manual of acute toxicity:
Interpretation and data base for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior,
Fish and Wildlife Service, pp 1,4 (Resource Publication No. 160).
Menicagli S, Puccini P, Longo V, & Gervasi PG (1990) Effect of acetone
administration on renal, pulmonary and hepatic monooxygenase
activities in hamster. Toxicology, 4: 141-153.
Meyrahn H, Pauly J, Schneider W, & Warneck P (1986) Quantum yields for
the photodissociation of acetone in air and an estimate for the life
time of acetone in the lower troposphere. J Atmos Chem, 4: 277-291.
Mikalsen A, Alexander J, Andersen PA, & Ingelman-Sundberg M (1991)
Effect of in vivo chromate, acetone and combined treatment on rat
liver in vitro microsomal chromium(VI) reductive activity and on
cytochrome P450 expression. Pharmacol Toxicol, 68: 456-463.
Miltiana LM & Mauch SC (1989) Statistical modeling of ambient air
toxics impact during remedial investigation at a landfiil site. In:
Proceedings of the 10th National Conference "Superfund '89",
Washington, DC, 27-29 November 1989. Washington, DC, The Hazardous
Materials Control Research Institute, pp 157-162.
Mill T & Mabey W (1985) Photochemical transformations. In: Neely WB &
Blau GE ed. Environmental exposure from chemicals. Boca Raton,
Florida, CRC Press, vol 1, pp 175-216.
Miller KW & Yang CS (1984) Studies on the mechanisms of induction of
N-nitroso-diethylamine demethylase by fasting, acetone, and ethanol.
Arch Biochem Biophys, 229: 483-491.
Mizunuma K, Yasugi T, Kawai T, Horiguchi S, & Ikeda M (1993)
Exposure-excretion relationship of styrene and acetone in factory
workers; a comparison of a lipophilic solvent and a hydrophilic
solvent. Arch Environ Contain Toxicol, 25(1): 129-133.
Mohr DH & King CJ (1985) Identification of polar organic compounds in
coal-gasification condensate water by gas chromatography-mass
spectrometry analysis of high pressure liquid chromatography
fractions. Environ Sci Technol, 19: 929-935.
Moldeus P & Gergely V (1980) Effect of acetone on the activation of
acetaminophen. Toxicol Appl Pharmacol, 53: 9-13.
Monder C (1997) alpha-Keto aldehyde dehydrogenase, an enzyme that
catalyzes the enzymic oxidation of methygiyoxal to pyruvate. J Biol
Chem, 242: 4603-4609.
Mopper K & Stahovec WL (1986) Sources and sinks of low molecular
weight organic carbonyl compounds in seawater. Mar Chem, 19: 305-321.
Morgott D (1993) Acetone. In: Clayton GE & Clayton FE ed. Patty's
industrial hygiene and toxicology, 4th ed. New York, John Wiley &
Sons, vol II, part A, chapter 5, pp 149-281.
Morris JB (1991) Deposition of acetone vapor in the upper respiratory
tract of the B6C3F1 mouse. Toxicol Lett, 56: 187-196.
Morris JB & Cavanagh DG (1986) Deposition of ethanol and acetone
vapors in the upper respiratory tract of the rat. Fundam Appl ToxicoI,
6: 78-88.
Morris JB & Cavanagh DG (1987) Metabolism and deposition of propanol
and acetone vapors in the upper respiratory tract of the hamster.
Fundam Appl Toxicol, 9: 34-40.
Morris JB, Clay RJ, & Cavanagh DG (1986) Species differences in upper
respiratory tract deposition of acetone and ethanol vapors. Fundam
Appl Toxicol, 7(4): 671480.
Mourkides GA, Hobbs DC, & Koeppe RE (1959) The metabolism of
acetone-2-C14 by intact rats. J Biol Chem, 234: 27-30.
NAS (1976) Vapor-phase organic pollutants: Volatile hydrocarbons and
oxidation products. Washington, DC, National Academy of Sciences,
Committee on Medical and Biologic Effects of Environmental Pollutants,
pp 23-24, 26-27.
Nelson DL & Webb BP (1978) Acetone. In: Greyson M & Eckroth D ed.
Kirk-Othmer encyclopedia of chemical technology, 3rd ed. New York,
John Wiley & Sons, vol 1, pp 179-191.
Nelson KW, Ege JF Jr, Ross M, Woodman LE, & Silverman L (1943) Sensory
response to certain industrial solvent vapors. J Ind Hyg Toxicol 25:
282-285.
NIOSH (1994) Manual of analytical methods - Ketones 1 (acetone);
method 1300, 4th ed. Cincinnati, Ohio, National Institute for
Occupational Safety and Health, 5 pp.
Nirmalakhandan N, Sun B, Arulgnandran VJ, Mohsin M, Wang XH, Prakash J,
& Hall N (1994a) Analyzing and modeling toxicity of mixtures of
organic chemicals to micro-organisms. Water Sci Technol, 30(10): 87-90
Nimalakhandan N, Arulgnanendran V, Mohsin M, Sun B, & Cadena F (1994b)
Toxicity of mixtures of organic chemicals to microorganisms. Water
Res, 28(3): 543-551.
NLM (1992) Hazardous substances databank. Washington, DC, National
Library of Medicine, vol 11,45 pp.
Nomiyama K & Nomiyama H (1974a) Respiratory retention, uptake and
excretion of organic solvents in man: Benzene, toluene, n-hexane,
trichloroethylene, acetone, ethyl acetate and ethyl alcohol. Int Arch
Arbeitsmed, 32; 75-83.
Nomiyama K & Nomiyama H (1974b) Respiratory elimination of organic
solvents in man: Benzene, toluene, n-hexane, trichloroethylene,
acetone, ethyl acetate and ethyl alcohol. Int Arch Arbeitsmed, 32:
65-91.
Norppa H, Hemminki K, Sorsa M, & Vainio H (1981) Effect of
monosubstituted epoxides on chromosome aberrations and SCE in cultured
human lymphocytea, Mutat Res, 91: 243-250.
NTP (1988) Inhalation developmental toxicology studies: teratology
study of acetone in mice and rats: Final report. Research Triangle
Park, North Carolina, US Department of Health and Human Services,
National Toxicology Program, 260 pp (Report No. PNL-6T68, prepared by
Pacific Northwest Laboratory).
NTP (1991) Toxicity studies of acetone in E344/N rats and S6C3F1 mice
(drinking water studies). Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program,
38 pp (Technical Report No. 3; NIH Publication No. 91-3122).
OECD (1981) Guidelines for testing of chemicals 301 D. Ready
biodegradability: Closed bottle test. Paris, Organisation for Economic
Co-operation and Development, 13 pp.
O'Hara D & Singh HB (1988) Sensitive gas chromatographic detection of
acetaldehyde and acetone using a reduction gas detector. Atmos
Environ, 22(11): 2613-2616.
Ott MG, Skory LK, Holder BB, Bronson JM, & Williams PR (1983a) Health
evaluation of employees occupationally exposed to methylene chloride:
general study design and environmental considerations. Scand J Work
Environ Health, 9(suppl 1): 1-7.
Ott MG, Skory LK, Holder BB, Bronson JM, & Williams PR (1963b) Health
evaluation of employees occupationally exposed to methylene chloride:
mortality, Scand J Work Environ Health, 9(suppl 1); 8-16.
Owen OE, Trapp VE, Skutches CL, Mozzoli MA, Hoeldtke RD, Boden G, &
Reichard CA Jr (1962) Acetone metabolism during diabetic ketoacidosis.
Diabetes, 31: 242-248.
Palo V & Ilkova H (1970) Direct gas chromatographic estimation of
lower alcohols, acetaldehyde, acetone and diacetyl in milk products. J
Chromatogr, 53: 363-367.
Pankow D & Hoffmann P (1989) Dichloromethane metabolism to carbon
monoxide can be induced by isoniazid, acetone and fasting. Arch
Toxicol, 13(suppl); 302-303.
Papa AJ & Sherman PD Jr (1981) Ketones. In: Grayson M & Eckroth D ed.
Kirk-Othmer encyclopedia of chemical technology, 3rd ed. New York,
John Wiley & Sons, vol 13, p 895.
Parasher CD, Ozel M, & Geike F (1978) Effect of hexachlorobenzene and
acetone on algal growth: Physiology and ultrastructure. Chem-Biol
Interact, 20: 89-95.
Paterson S & Mackay D (1989) Correlation of tissue, blood, and air
partition coefficients of volatile organic chemicals. Br J Ind Med,
46: 321-328.
Patrick R, Cairns J Jr, & Schier A (1968) The relative sensitivity of
diatoms, snails and fish to twenty common constituents of industrial
wastes, prog Fish-Cult, 30(3): 137-140.
Patten CJ, Ning SM, Lu AYH, & Yang CS (1986) Acetone-inducible
cytochrome P-450 Purification, catalytic activity, and interaction
with cytochrome b5. Arch Biochem Biophys, 251: 629-38.
Peinado J, Lopez-Sodano FJ, & Argiles JM (1986) The metabolism of
acetone in the pregnant rat. Biosci Rep, 6: 983-989.
Pellizzari ED, Castillo NP, Willis S, Smith M, & Burney JT (1979)
Identification of organic components in aqueous effluents from
energy-related processes. In: Van Hall CE ed. Measurement of organic
pollutants in water and wastewater. Washington, DC, American Society
for Testing and Materials, pp 256-274 (Publication STP 666).
Pellizzari ED, Hartwell TD, Harris BSH, Waddell RD, Whitaker DA, &
Erickson MD (1962) Purgeable organic compounds in mother's milk. Bull
Environ Contain Toxicol, 28: 322-28.
Perry DL, Chuang CC, Jungclaus GA, & Warner JS (1978) Identification
of organic compounds in industrial effluent discharges (Prepared by
Battelle Columbus Laboratory). Athens, Georgia, US Environmental
Protection Agency, Environmental Research Laboratory, pp 44, 46
(EPA-560/678-009; NTIS PB-291900).
Perry DL, Chuang CC, Jungclaus GA, & Warner JS (1979) Identification
of organic compounds in industrial effluent discharges. Washington,
DC, US Environmental Protection Agency, 34 pp (NTIS PB 294794).
Pezzagno G, Imbriani M, Ghittori S, Capodaglio E, & Huang J (1986)
Urinary elimination of acetone in experimental and occupational
exposure. Scand J Work Environ Health, 12(6): 603608.
Phillips M & Greenberg J (1987) Detection of endogenous acetone in
normal human breath. J Chromatogr, 422: 235-238.
Plaa GL & Traiger GJ (1972) Mechanism of potentiation of CCI4-induced
hepatotoxicity. In: Loomis TA ed. Pharmacology and the future of
man - Volume 3: Toxicological problems. Basel, Switzerland, Larger, pp
100-113.
Plaa GL, Traiger GJ, Hanasono GK, & Witschi H (1973) Effect of
alcohols on various forms of chemically induced liver injury. In:
Khanna JM, Israel Y, & Kalant H ed. Alcoholic liver pathology.
Toronto, Ontario, Canada, Alcoholism and Drug Addiction Research
Foundation of Ontario, pp 225-244.
Plaa GL, Hewitt WR, Du Souich P, Caillè G, & Lock S (1982) Isopropanol
and acetone potentiation of carbon tetrachloride-induced
hepatotoxicity: Single versus repetitive pretreatments in rats. J
Toxicol Environ Health, 9: 235-250.
Platen H, Temmes A, & Schink B (1990) Anaerobic degradation of acetone
by Desulfococcas biacutus spec. nov. Arch Microbiol, 154: 335-361.
Plumb RH Jr (1987) A comparison of ground water monitoring data from
CERCLA and RCRA sites. Ground Water Monit Rev, 7:94-100.
Pomerantz RB (1950) Acute acetone poisoning from castex. Am J Surg,
80:117-118.
Pozzani UC, Weil CS, & Carpenter CP (1959) The toxicological basis of
threshold limit values: 5. The experimental inhalation of vapor
mixtures by rats, with notes upon the relationship between single dose
inhalation and single dose oral data. Am Ind Hyg Assoc J, 20: 364-369.
Price VF & Jollow DJ (1983) Mechanism of ketone-induced protection
from acetaminophen hepatotoxicity in the rat. Drug Metab Dispos, 11:
451-457.
Price TD & Rittenberg D (1950) The metabolism of acetone: I. Gross
aspects of catabolism and excretion. J Biol Chem, 185: 449-459.
Price KS, Waggy GT, & Conway RA (1974) Brine shrimp bioaasay and
seawater BOD of petrochemicals. J Water Pollut Control Fed, 46: 65-77.
Puccini P, Fiorio R, Longo V, & Gervasi PG (1989) High affinity
diethylnitrosamine-deethylase in liver microsomes from
acetone-induced rats. Carcinogenesis, 19: 1629-1634.
Puccini P, Fiorio R, Longa V, & Gervasi PG (1990) Effects of acetone
administration on drug-metabolizing enzyme in mice: Presence of a
high-affinity diethylnitrosamine deethyiase. Toxicol Lett, 54:
143-150.
Puccini P, Menicagli S, Longo V, Santucci A, & Gervasi PG (1992)
Purification and characterization of an acetone-inducible cytochrome
P-450 from hamster liver microsomes. Biochem J, 287: 863-870.
Racker E (1951) The mechanism of action of glyoxalase. J Biol Chem,
190: 685-698.
Rahim SA & Bashir WA (1981) Microdetermination of acetone in aqueous
solution. Microchem J, 26(3): 329-333.
Rajini PS, Krishnakumari MK, & Majumder SK (1989) Cytotoxicity of
certain organic solvents and organophosphorus insecticides to the
ciliated protozoan Paramecium caudatum. Microbios, 59: 157-163.
Raleigh RL & McGee WA (1972) Effects of short, high-concentration
exposures to acetone as determined by observation in the work area. J
Occup Med, 14(8): 607-610.
Ramu A, Rosenbaum J, & Blaschke TF (1978) Disposition of acetone
following acute acetone intoxication. West J Mad, 129: 429-32.
Randall TL & Knopp PV (1980) Detoxification of specific organic
substances by wet oxidation. J Water Pollut Control Fed, 52(8):
2117-2130.
Rathbun RE & Tai DY (1982) Volatilization of ketones from water. Water
Air Soil Pollut, 17: 281-293.
Rathbun RE & Tai DY (1887) Vapor pressures and gas-film coefficients
for ketones. Chemosphere, 18: 69-78.
Rathbun RE, Stephens DW, Shultz DJ, & Tai DY (1982) Fate of acetone in
water. Chemosphere, 11: 1097-1114.
Rathbun RE, Stephens DW, Schultz DJ, & Tai DY (1988) Fate of acetone
in an outdoor model stream in southern Mississippi. J Hydrol, 104:
181-209.
Rathbun RE, Shultz DJ, Stephens DW, & Tai DY (1989) Transport and fate
of acetone in an outdoor model stream, Stennis Space Center Near Bay
St. Louis, Mississippi. Denver, Colorado, US Geological Survey, 191 pp
(Water-Resources Investigations Report No. 894141).
Rathbun RE, Stephens DW, & Tai DY (1991) Fate of acetone in an outdoor
model stream with a nitrate supplement, southern Mississippi, USA. J
Hydrol, 123: 225-242.
Rathbun RE, Stephens DW, & Tai DY (1993) Bacterial degradation of
acetone in an outdoor model stream. Environ Pollut, 79: 153-182.
Raymond P & Plaa GL (1995a) Ketone potentiation of haloalkane-induced
hepatic- and nephrotoxicity: I. Dose-response relationships. J Toxicol
Environ Health, 45: 455-480.
Raymond P & Plaa GL (1995b) Ketone potentiation of haloalkane-induced
hepoto- and nephrotoxicity: I. Implication of monooxygenases. J
Toxicol Environ Health, 46; 317-378.
Reichard GA Jr, Haff AC, Skutches CL, Holroyde CP, & Owen OE (1979)
Plasma acetone metabolism in the fasting human. J Clin Invest, 63:
619-628.
Reichard GA Jr, Skutches CL, Holroyde CP, & Owen OE (1986) Acetone
metabolism in humans during diabetic ketoacidosis. Diabetes, 35:
668-674.
Rengstorff RH & Khafagy HI (1985) Cutaneous acetone depresses aqueous
humor ascorbate in guinea pigs. Arch Toxicol, 58: 64-66.
Rengstorff RH, Petrali JP, & Sim VM (1972) Cataracts induced in guinea
pigs by acetone, cyclohexanone, and dimethyl sulfoxide. Am J Optom
Arch Acad Optom, 49: 308-319.
Renshaw PK & Mitchell RM (1956) Acute poisoning following the
application of a lightweight cast. Br Mad J, 1:615.
Riddick JA, Bunger WB, & Sakano TK (1986) Organic solvents: Physical
properties and methods of purification. In: Techniques of chemistry,
4th ed. New York, Wiley-Interscience, vol 2, pp 336-337.
Risner CH (1995) High performance liquid chromatographic determination
of major carbonyl compounds from various sources in ambient air. J
Chromatogr Sci, 33; 168-176.
Ross DG (1973) Acute acetone intoxication involving eight male
workers, Am Occup Hyg J, 16: 73-75.
Ruddick JA (1972) Toxicology, metabolism, and biochemistry of
1,2-propanediol. Toxicol Appl Pharmacol, 21; 102-111.
Rudney H (1954) Propanediol phosphate as a possible intermediate in
the metabolism of acetone. J Stol Chain, 210: 361-371.
Sabel GV & Clark TP (1984) Volatile organic compounds as indicators of
municipal solid waste leachete contamination. Waste Manage Res,
2:119-130.
Sack TM, Steele DH, Hammerstrom K, & Remers JA (1992) A survey of
household products for volatile organic compounds. Atmos Environ,
26A(6): 1063-1070.
Sakami W & LaFaye JM (1950) Formation of formate and labile methyl
groups from acetone in the intact rat. J Biol Chem, 187: 369-378.
Sakami W & Rodney H (1952) The metabolism of acetone and acetoacetate
in the mammalian organism. Brookhaven Syrup Biol, 5: 176-191.
Sakata M, Kikuchi J, Haga M, Ishiyama N, Maeda T, Isa T, & Hikita N
(1989) Disposition of acetone, methyl ethyl ketone and cyclohexanone
in acute poisoning. J Toxicol Clin Toxicol, 27: 67-77.
Sallee ED & Sappington CO (1949) Eleventh Annual Meeting of the
American Conference of Governmental Industrial Hygienists / Tenth
Annual Meeting of the American Industrial Hygiene Association,
Detroit. Michigan, 2-7 April 1949. Ind Hyg Q, 10: 36-44.
Sangster J (1989) Octanol-water partition coefficients of simple
organic compounds. J Phys Chem Ref Data, 18:1111-1229.
Sato A & Nakajima T (1979) Partition coefficients of some aromatic
hydrocarbons and ketones in water, blood and oil. Br J Ind Med,
36: 231-234.
Satoh T, Omae K, Takebayashi T, Nakashima H, Higashi T, & Sakurai H
(1995) Acetone excretion into urine of workers exposed to acetone in
acetate fiber plants. Int Arch Occup Environ Health, 67: 131-134.
Satoh T, Omae K, Nakashima H, Takebayashi T, Matsumura H, Kawat T,
Nakaza M, & Sakurai H (1996) Relationship between acetone exposure
concentration and health effects in acetate fiber plant workers. Int
Arch Occup Environ Health. 68:147-153.
Sawhney BL & Kozloski RP (1984) Organic pollutants in leachates from
landfill sites. J Environ Qual, 13; 349-352.
Sawhney BL & Raabe JA (1986) Ground water contamination: Movement of
organic pollutants in the Granby landfill, New Haven, Connecticut, The
Connecticut Agricultural Experiment Station, 9 pp (Connecticut
Agricultural Experiment Station BulLetin No. 833).
Sax NI & Lewis RJ Sr (1987) Acetone. In: Hawley's condensed chemical
dictionary, 11th ed. New York, Van Nostrand Reinhold Co., p 8.
Schnier GS, Laethem CL, & Koop DR (1989) Identification and induction
of cytochromes P450, P450IIE1 and P45IA1 in rabbit bone marrow. J
Pharmacol Exp Ther, 251: 790-796.
Schrikker ACM, de Vries WR, Zwart A, & Luijendjk CM (1985) Uptake of
highly soluble gases in the epithelium of the conducting airways.
Pflügers Arch/Eur J Physiol, 405: 989-394.
Schrikker ACM, de Vries WR, Zwart A, & Luijendjk CM (1989) The
excretion of highly soluble gases by the lung in man. Pflügers
Archiv/Eur J Physiol, 415: 214-219.
Seeber A, Kiesswetter E, Vangala RR, Blaszkewicz M, & Golka K (1992)
Combined exposure to organic solvents: An experimental approach using
acetone and ethyl acetate. Appl Psychol Int Rev, 41: 281-292.
Seeber A, Kiesswetter E, Giller D, Blaszkewicz M, Golka K, Vangala RR,
& Bolt HM (1993) [Acute effects of acetone and ethylacetate:
comparison of 4 and 8 hour exposure.] in: [Occupational health for a
healthy environment.] Stuttgart, Gentner Vedag, pp 145-148 (in
German).
Segner H & Lenz D (1993) Cytotoxicity assays with the rainbow trout
R1 cell line. Toxicol in vitro, 7(4): 537-540.
Seila RL (1979) Non-urban hydrocarbon concentrations in ambient air
north of Houston, Texas. Research Triangle Park, North Carolina, US
Environmental Protection Agency, 39 pp (EPA-500/3-79-010).
Shackelford WM & Keith LH (1978) Frequency of organic compounds
identified in water. Athens, Georgia, US Environmental Protection
Agency, Environmental Research Laboratory, pp 49-52
(EPA-600/4-76-062).
Shah JJ & Singh HB (1988) Distribution of volatile organic chemicals
in outdoor and indoor air. Environ Sci Technol, 22: 1381-1388.
Shepson PB, Hastie DR, Schiff HI, Polizzi M, Bottenheim JW, Anlauf K,
Mackay GI, & Karecki DR (1991) Atmospheric concentrations and temporal
variations of C1-C3 carbonyl compounds at two rural sites in central
Ontario. Atmos Environ, 25A: 2001-2015.
Sifniades S (1985) Acetone. In: Gerhartz W, Yamamoto YS, Campbell FT,
Pfefferkorn R, & Rousaville JF ed. Ullman's encyclopedia of industrial
chemistry, 5th ed. Weinheim, VCH Vedag, vol 1A, pp 79-95.
Singh HB & Hanst PL (1981) Peroxyacetyl nitrate (PAN) in the
unpolluted atmosphere: an important reservoir for nitrogen oxides.
Geophys Res Lett, 8: 941-944.
Singh HB, O'Hara D, Herlth D, Sachse W, Blake DR, Bradshaw JD,
Kanakidou M, & Crutzen PJ (1994) Acetone in the atmosphere:
distribution, sources, and sinks. J Geophys Res, 99: 1805-1819.
Singh KP, Yoon HL Ratner S, & Reiners JJ Jr (1996) Modulation of the
development of humoral immunity by topically applied acetone, ethanol
and 12- o-tetradecanoylphorbol-13-acetate. Fundam Appl Toxicol, 33:
129-139.
Sipes IG, Stripp B, Krishna G, Maling HM, & Gillette JR (1973)
Enhanced hepatic microsomal activity by pretreatment of rats with
acetone or isopropanol. Proc Soc Exp Biol Med, 142: 237.
Sipes IG, Slocumb ML, & Holtzman G (1978) Stimulation of microsomal
dimethyl nitrosamine- n-demethylase by pretreatment of mice with
acetone. Chem-Biol Interact, 21:155-166.
Sippel H, Penttila KE, & Lindros KO (1991) Regioselective induction of
liver glutathione transferase by ethanol and acetone. Pharmacol
Toxicol, 68: 391-393.
Skutches CL, Owen OE, & Reichard GA Jr (1990) Acetone and acetol
inhibition of insulin-stimulated glucose oxidation in adipose tissue
and isolated adipocytes. Diabetes, 39; 450-455.
Slooff W & Baerselman R (1980) Comparison of the usefulness of the
Mexican axolotl ( Ambystoma mexicanum) and the clawed toad
( Xenopus laevis) in toxicological bioassays. Bull Environ Contam
Toxicol, 24: 439-443.
Smith NB (1984) Determination of volatile alcohols and acetone in
serum by non-polar capillary gas chromatography after direct sample
injection. Clin Chem, 10: 1672-1674.
Smyth HF Jr, Carpenter CP, Well CS, & West JS (1962) Range-finding
toxicity data: List VI. Am Ind Hyg Assoc J, 23: 95-107.
Snider JR & Dawson GA (1985) Tropospheric light alcohols, carbonyls,
and acetonitrile concentrations in the southwestern United States and
Henry's law data. J Geophys Res, 90: 3797-3805.
Sollman T (1921) Studies of chronic intoxications in albino rats: II.
Alcohols (ethyl methyl and "wood") and acetone, J Pharmacol Exp Ther,
16: 291-309.
Specht H, Miller JW, & Valaer PJ (1939) Acute response of guinea pigs
to the inhalation of dimethyl ketone (acetone) vapor in air. Public
Health Rep, 54: 944-954.
Spicer CW, Riggin RM, Holdren MW, De Roos FL, & Lee RN (1985)
Atmospheric reaction products from hazardous air pollutant
degradation. Research Triangle Park, North Carolina, US Environmental
Protection Agency, 78 pp (EPA-60013-85-025).
SRI (1996) Directory of chemical producers, United States of America.
Menlo Park, California, Stanford Research institute International, 430
pp.
Stafford W & Northup HJ (1955) The BOD of textile chemicals. Am
Dyestuff Rep, 44: 355-359.
Steelman BL & Ecker RM (1984) Organic contamination of groundwater: An
open literature review. Seattle, Washington, Battelle Pacific
Northwest Laboratory, 66 pp (Report No. DE-AC05-76RLO 1830 13).
Steinberg SM & Kreamer DK (1993) Evaluation of the sorption of
volatile organic compounds by unsaturated calcareous soil from
southern Nevada using inverse gas chromatography. Environ Sci Technol,
27: 883-888.
Stewart RD, Hake CL, Wu A, Graft SA, Forster AV, Keeler WH, Lebrun AJ,
Newton PE, & Soto RJ (1975) Acetone: development of a biologic
standard for the industrial worker by breath analysis. Cincinnati,
Ohio, National Institute for Occupational Safety and Health, 80 pp
(NTIS P582-172917).
Strange P, Moller A, Ladefoged O, Lam HR, Laraen JJ, & Arlien-Soborg P
(1991) Total number and mean cell volume of neocortical neurons in
rats exposed to 2.5-hexaeedione with and without acetone. Neurotoxicol
Teratol, 13: 401-406.
Stratton GW (1937) Toxic effects of organic solvents on the growth of
blue-green algae. Bull Environ Contam Toxicol, 38:1012-1019.
Stratton GW & Corke GT (1981) Interaction between acetone and two
pesticides towards several unicellular green algae. Bull Environ
Contam Toxicol, 27(1): 13-16.
Takemoto S, Kuge Y, & Nakamoto M (1981) [The measurement of BOD in sea
water.] Suishitsu Odaku Kenkyu, 4:80-90 (in Japanese with English
summary).
Takeoka GR, Flath PA, Guntert M, & Jennings W (1989) Nectarine
volatiles: Vacuum steam distillation versus headspace sampling. J Agric
Food Chem, 36: 553-560.
Taylor A, Smith DE, Palmer VJ, Shepard D, Padhye N, Theriault C, &
Morrow F (1993) Relationships between acetone, cataracts and ascorbate
in hairless guinea pigs. Ophthalmol Res, 25: 30-35.
Thom NS & Agg AR (1975) The breakdown of synthetic organic compounds
in biological processes. Proc R Sac Land, B189: 347-37.
Thomas RG (1982) Volatilization from water. In: Lyman WJ, Reehl WF, &
Rosenblatt DH ed. Handbook of chemical property estimation methods.
New York, McGraw-Hill, chapter 15, pp 15/1-15/34.
Thornalley PJ (1990) The glyoxalase system: New developments towards
functional characterization of a metabolic pathway fundamental to
biological life. Biochem J, 269: 1-11.
Tichenor BA & Mason MA (1968) Organic emissions from consumer products
and building materials to the indoor environment. J Air Pollut Control
Assoc. 38: 284-268.
Tietz NW (1983) Clinical guide in laboratory tests. Philadelphia,
Pennsylvania, WB Saunders Company, pp 2-3.
Tindberg N & Ingelman-Sundberg M (1989) Cytochrome P-450 and oxygen
toxicity: Oxygen-dependent induction of ethanol-inducible cytochrome
P-450 (IIE1) in rat liver and lung. Biochemistry, 28: 4499-4504.
Traiger GJ & Plaa GL (1972) Relationship of alcohol metabolism to the
potentiation of CCI4 hepatotoxicity induced by aliphatic alcohols. J
Pharmacol Exp Ther, 183: 481-488.
Traiger GJ & Plaa GL (1974) Chlorinated hydrocarbon toxicity:
Potentiation by isopropyl alcohol and acetone. Arch Environ Health,
28: 276-278.
Trotter MD, Sulway MJ, & Trotter E (1971) The rapid determination of
acetone in breath and plasma. Clin Chim Acta, 35: 137-143.
Ueng T-H, Tsai J-n, Ju J-M, Ueng Y-F, Iwasaki M, & Guengerich FP
(1991) Effects of acetone administration of cytochrome P-450-dependent
monooxygenases in hamster liver, kidney and lung. Arch Toxicol, 65:
45-51.
UK SAC (UK Standing Committee of Analysts) (1988) The determination of
formaldehyde, other volatile aldehydes, ketones and alcohols in water:
Methods for the examination of waters and associated materials.
London, Her Majesty's Stationery Office, 12 pp.
Urano K & Kato Z (1986a) A method to classify biodegradabilities of
organic compounds. J Hazard Mater, 13: 135-145.
Urano K & Kate Z (1986b) Evaluation of biodegradation ranks of
priority organic compounds. J Hazard Mater, 13: 147-159, 203.
US EPA (1975) preliminary assessment of suspected carcinogens in
drinking water: Interim report to Congress (June 1975). Washington,
DC, US Environmental Protection Agency, 9 pp.
US EPA (1986a) Ninety-day gavage study in albino rats using acetone.
Washington, DC, US Environmental Protection Agency, Office of Solid
Waste, 950 pp.
US EPA (1986b) Health and environmental effects document, Cincinnati,
Ohio, US Environmental Protection Agency, Office of Research and
Development, 186 pp.
US EPA (1989) Compilation and speciation of national emissions factors
for consumer/commercial solvent use information compiled to support
urban air toxics assessment studies. Research Triangle Park, North
Carolina, US Environmental Protection Agency, 209 pp
(EPA/450/2-89/008; NTIS PB89-207203).
Vaishnav DD (1986) Chemical structure biodegradation inhibition and
fish acute toxicity relationships for narcotic industrial chemicals.
Toxicol Assess, 1(2): 227-240.
Vaishnav DD, Boethling RS, & Babeu L (1987) Quantitative
structure-biodegradability relationships for alcohols, ketches and
alicyclic compounds. Chemosphere, 16: 695-703.
Vangala RR, Blaszkewicz M, Bolt HM, Golka K, Kiesswetter E, & Seeber A
(1991) Acute experimental exposures to acetone and ethyl acetate, Arch
Toxicol, 14(suppl): 259-282.
Van Stekelenburg GJ & Koorevaar G (1972) Evidence for the existence of
mammalian acetoacetate decarboxylase: with special reference to human
blood serum. Clin Chim Acta, 39: 191-199.
Veith GD, Call DJ, & Brooke LT (1983) Structure-toxicity relationships
for the fathead minnow, Pimephales promelas: Narcotic industrial
chemicals. Can J Fish Aquat Sci, 40(6): 743-748.
Vodickova L, Frantik E, & Vodikova A (1995) Neurotropic effects and
blood levels of solvents at combined exposures: binary mixtures of
toluene, o-xylene and acetone in rats and mice. Cent Eur J Public
Health, 2: 57-64.
Waggy GT, Conway PA, Hansen JL, & Blessing RL (1994) Comparison of
20-d BOD and OECD closed-bottle biodegradation tests. Environ Toxicol
Chem, 13(8): 1277-1280.
Wallen IE, Greer WC, & Lasater R (1957) Toxicity to Gambusia affinis
of certain pure chemicals in turbid waters. Sewage Ind Wastes, 29(6):
695-711.
Wallington TJ & Kurylo MJ (1987) Flash photolysis resonance
fluorescence investigation of the gas-phase reactions of hydroxyl
radicals with a series of aliphatic ketones over the temperature range
240-440 K. J Phys Chem, 91: 5050-5054.
Wang G, Maranelli G, Perbellini L, Raineri E, & Brugnone F (1994) Blood
acetone concentration in "normal people" and in exposed workers 16 h
after the end of the workshift. Int Arch Occup Environ Health, 65(5):
285-289.
Ward JM, Quander R, Devor D, Wenk ML, & Spangler EF (1986) Pathology
of aging female SENCAR mice used as controls in skin two-stage
carcinogenesis studies. Environ Health Perspect, 68: 81-89.
Weast RC ed. (1987) Handbook of chemistry and physics, 67th ed. Boca
Raton, Florida, CRC press, p 51.
Westerholm RN, Alsberg TE, Frommelin AB, Strandell ME, Ranung U,
Winsquist L, & Grigoriadis V (1988) Effect of fuel polycyclic aromatic
hydrocarbon content on the emissions of polycyclic aromatic
hydrocarbons and other mutagenic substances from a gasoline fueled
automobile. Environ Sci Technol, 22: 925-930.
WHO (1971) Evaluation of food additives. Fourteenth report of the
Joint FAO/WHO Expert Committee on Food Additives. Geneva, World Health
Organization, p 21 (WHO Technical Report Series, NO. 462).
Widmark EMP (1919) Studies in the concentration of indifferent
narcotics in blood and tissues. Acta Med Scand, 52: 87-164.
Wigaeus E, Holm S, & Astrend I (1981) Exposure to acetone: Uptake and
elimination in man. Scand J Work Environ Health, 7(2): 84-94.
Wigaeus E, Löf A, & Nordqvist M (1982) Distribution and elimination of
2-[14C]-acetone in mice after inhalation exposure. Scand J Work
Environ Health, 8(2): 121-128.
Wilkins K (1994) Volatile organic compounds from household waste.
Chemosphere, 29(1): 47-53.
Williams PL, Brooks JD, Inman AG, Monteiro-Riviere NA, & Riviera DE
(1994) Determination of physicochemical properties of phenol,
p-nitrophenol, acetone and ethanol relevant to quantitating their
percutaneous absorption in porcine skin. Res Commun Chem Pathol
Pharmacol, 83(1): 61-75.
Wilson JE, Brunwald E, Isselbacher K, Petersdorf RG, Martin JB, Favei
AS, & Root RK (1991) Principles of internal medicine, 12th ed. New
York, McGraw-Hill, 525 pp.
Windholz M ed. (1983) Acetone. In: The Merck index, 10th ed. Rahway,
New Jersey, Merck & Co., p8.
Wysocki CJ, Dalton P, Brody M, & Hanley HJ (1997) Odor and irritation
thresholds for acetone in acetone-exposed factory workers and control
(occupationally-non exposed) subjects. Am Ind Hyg Assoc J, 58:
704-712.
Yoo J-S & Yang CS (1985) Enzyme specificity in the metabolic
activation of N-nitrosodimethylamine to a mutagen for Chinese
hamster V79 cells. Cancer Res, 45; 5569-5574.
Yoo J-SH, Ishizaki H, & Yang CS (1990) Roles of cytochrome-P450IIE1 in
the dealkylation and denitrosation of N-nitrosodiethylamine in rat
liver mircosomes. Carcinogenesis, 11: 2239-2243.
Zhou X & Mopper K (1990) Apparent partition coefficients of 15
carbonyl compounds between air end seawater and between air and
freshwater: Implications for air-sea exchange. Environ Sci TechnoL
24(12): 1864-1869.
Zimmermann FK, Mayer VW, Scheel I, & Resnick MA (1985) Acetone, methyl
ethyl ketone, ethyl acetate, acetenitrile and other polar aprotic
solvents are strong inducers of aneuploidy in Saccharomyces
cerevisiae. Mutat Res, t49(3); 339-351.
Zweidinger RB, Sigsby JE Jr, Tajada SB, Stump FD, Dropkin DL, Ray WD,
& Duncan JW (1988) Detailed hydrocarbon and aldehyde mobile source
emissions from roadway studies. Environ Sci Technol, 22: 956-962.
RÉSUMÉ
1. Propriétés
L'acétone (masse moléculaire relative = 58,08) est un liquide
inflammable, limpide et incolore (point d'éclair - 17°C en coupe
fermée, -9°C en coupe ouverte; limites d'inflammabilité dans l'air à
25°C = 2,15-13% v/v). Les limites d'explosion dans l'air sont de:
2,6-12,8% v/v). L'acétone s'évapore rapidement (tension de vapeur
181,72 mm Hg à 20°C) et présente une faible viscosité (0,303 cP à
25°C). Elle est miscible à l'eau et aux solvants organiques.
2. Usages et sources d'exposition
2.1 Production
Industriellement, l'acétone se prépare principalement par
peroxydation du cumène ou déshydrogénation de l'alcool isopropylique.
La peroxydation du eumène produit également des traces de benzène.
2.2 Usages et émissions dans l'environnement
L'acétone est principalement utilisée comme solvant et comme
intermédiaire dans la préparation de divers produits chimiques. Ses
principaux usages consistent dans la production de méthacrylate de
méthyle, acide méthacrylique et méthacrylates supérieurs, bisphénol A,
méthylisobutylcétone, médicaments et dans des applications
pharmaceutiques diverses. On l'utilise aussi comme solvant de
l'acétate de cellulose et de divers enduits. Dans l'industrie
alimentaire, on l'emploie comme solvant d'extraction des graisses et
des huiles et comme agent de précipitation au cours de la purification
du sucre et de l'amidon.
Les émissions d'acétone dans l'atmosphère proviennent de produits
de consommation qui en contiennent: dissolvants pour vernis à ongles,
panneaux de particules, doublures de tapis, décapants pour peintures,
cires et encaustiques liquides ou pâteux. Un certain nombre de
détergents, produits de nettoyage (notamment pour certains éléments de
moteurs d'automobile tels que carburateurs et starters) et adhésifs
contiennent aussi de l'acétone.
De nombreuses industries polluent les eaux superficielles par
leurs rejets d'acétone dans les eaux résiduaires: papier, plastiques,
produits pharmaceutiques, produits d'entretien, peintures et vernis,
produits pour le traitement des gommes, colles et bois, intermédiaires
cycliques, dérivés organiques industriels, produits de gypseuse,
cartonnages, production d'énergie (gazéification de la houille et
traitement de l'huile de schiste).
La pollution des sols par l'acétone trouve son origine dans le
rejet de déchets agricoles et alimentaires, les excréments d'animaux,
les précipitations, les effluents de fosses septiques et les
infiltrations provenant de décharges de produits chimiques.
3. Transport, distribution et transformation dans l'environnement
L'acétone libérée dans l'atmosphère se décompose par voie
photolytique et par réaction avec des radicaux hydroxyles. Sa demi-vie
atmosphérique est dans ce cas d'environ 30 jours. Elle peut être
éliminée physiquement de l'atmosphère par les précipitations. Dans les
sols et dans l'eau, c'est la biodégradation qui constitue le principal
mode de décomposition de l'acétone, ce composé étant facilement
biodégradable, Sa volatilisation à partir de l'eau peut constituer une
voie de transport environnemental non négligeable. En raison de sa
volatilité, elle s'évapore des surfaces sèches sur lesquelles elle est
déposée. En outre, sa miscibilité à l'eau en facilite le lessivage à
partir de la plupart des sols. En cas de biodégradation suffisamment
rapide, ce lessivage perd de son importance.
4. Concentrations dans l'environnement et exposition humaine
L'exposition à l'acétone peut être d'origine naturelle ou
artificielle. Elle est présente dans le sang, l'urine et l'haleine
humaines par suite du métabolisme normal. Dans l'environnement, sa
présence résulte entre autres de la biodégradation des effluents, des
déchets solides et des alcools ainsi que de l'oxydation des substances
humiques. On l'a mise en évidence dans diverses plantes et denrées
alimentaires comme les oignons, le raisin, les choux-fleurs, les
tomates, les volubilis, la moutarde sauvage, le lait, les haricots,
les pois, le fromage et le blanc de poulet. Diverses espèces d'arbres
émettent naturellement des vapeurs d'acétone. La pollution du milieu
aquatique imputable à l'activité humaine est due au déversement des
eaux résiduaires d'un grand nombre d'industries et au lessivage des
décharges industrielles ou municipales. L'évaporation de l'acétone
utilisée comme solvant dans les peintures, vernis, produits
d'entretien et encres constitue une source importante de pollution de
l'air. Sa présence dans l'atmosphère résulte également de la
combustion du bois, de plastiques et de déchets divers. Elles est
également émises avec les gaz d'échappement des moteurs à explosion et
des moteurs diesel ou encore des turbines à gaz. Les concentrations
relevées dans l'atmosphère vont de 0,5 à 125,4 µg/m3 (0,2-52,9
parties par milliard).
5. Cinétique et métabolisme
L'acétone est l'une des trois cétones qui sont présentes à l'état
naturel dans l'organisme. Elle peut se former de manière endogène dans
l'organisme des mammifères par oxydation des acides gras. Le jeûne, le
diabète sucré et un exercice physique intense accroissent la formation
endogène d'acétone. Dans les conditions normales, la production de
corps cétoniques s'effectue presque exclusivement dans le foie et,
dans une moindre proportion, dans les reins et les poumons. Il s'agit
d'un processus continu et les trois produits, après être passés dans
le sang, gagnent la totalité des tissus et des organes où ils peuvent
être utilisés comme source d'énergie. Deux d'entre eux, l'acide
acétoacétique et l'acide ß-hydroxybutyrique, sont des acides
organiques susceptibles de provoquer une acidose lorsqu'ils sont
produits en grande quantité, comme dans le cas du diabète sucré.
L'acétone, en revanche, n'est pas ionisée et elle est produite de
manière endogène par le clivage enzymatique ou spontané de
l'acétoacétate. L'acétone endogène est éliminée de l'organisme soit
par la voie urinaire, soit dans l'air expiré, soit par l'action
d'enzymes. Dans les conditions normales, c'est la voie métabolique qui
constitue le mode principal d'élimination de l'acétone (70 à 80% de la
quantité totale présente dans l'organisme).
L'acétone est rapidement résorbée au niveau des voies
respiratoires et digestives de l'Homme et de l'animal, comme on peut
le constater à la présence d'acétone dans le sang 30 minutes après
inhalation et 20 minutes après ingestion. Les études effectuées sur
des rats montant qu'après ingestion, l'acétone est absorbée dans une
forte proportion, alors qu'après inhalation chez l'Homme, la
proportion absorbée est d'environ 50% de la quantité initiale. Il est
vrai cependant, que des valeurs plus fortes et plus faibles ont été
relevées après inhalation. Il semblerait que les fosses nasales de
l'Homme et des animaux de laboratoire n'aient qu'une capacité limitée
à absorber et à excréter les vapeurs d'acétone, comparativement aux
autres parties des voies respiratoires.
L'acétone se répartit uniformément dans les tissus non adipeux et
ne s'accumule pas dans les graisses. Chez la souris, la concentration
maximale d'acétone dans les tissus adipeux s'est révélée égale au
tiers de ce qu'elle était dans les autres tissus, après exposition des
animaux par la voie respiratoire. L'acétone s'élimine rapidement de
l'organisme par métabolisation et excrétion. Sa demi-vie dans l'air
alvéolaire, dans le sang veineux et dans le sang artériel est
respectivement égale à approx. 4, 6 et 4 h. La principale voie
d'élimination de l'acétone et de son métabolite terminal, le CO2, est
l'air expiré et la fraction du composé qui est rejetée inchangée
dépend de la dose. Il y a aussi excrétion de l'acétone et de ses
métabolites par la voie urinaire, mais il s'agit d'une voie mineure
par rapport à l'expiration.
L'acétone d'origine endogène prend part à nombre des réactions
métaboliques qui se produisent dans les diverses parties de
l'organisme, mais c'est le foie qui semble constituer le site
métabolique le plus important. Le carbone provenant de l'acétone
administrée par voie orale à des rat se retrouve dans le cholestérol,
les acides aminés, les acides gras et le glycogène fistulaires ainsi
que dans l'urée unanime. On le retrouve aussi dans l'acétone inchangée
et dans le CO2 présents dans l'air expiré. L'acétone est métabolisée
en acétate et en formiate; cela explique que le carbone acétonique
entre dans le cholestérol, les acides gras, l'urée et les acides
aminés et qu'il se forme des composés tricarbonés par gluconéogénèse.
On a avancé que la gluconéogénèse à partir de l'acétone
s'effectuait selon deux voies métaboliques. La première comporte
l'action catalytique initiale de l'acétone-monooxygénase et de
l'acétol-monooxygénase qui transforme respectivement l'acétone en
acétol et l'acétol en méthylglyoxal. Ces deux enzymes sont induites
par l'acétone et constituent des isozymes du cytochrome hépatique
P-450IIE1 industriel par l'éthanol. Le deuxième mode de néoglucogénèse
consiste dans la formation de 1,2-propanediol à partir de l'acétone,
sous l'action de l'acétone-monooxygénase et d'une autre enzyme non
caractérisée, capable de convertir l'acétol en 1,2-propanediol.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Chez le rat, la valeur de la DL50 par voie orale varie entre
5800 et 7138 mg/kg. La CL50 par inhalation à 4 h est égale à 76 000
mg/m3 (32 000 ppm).
On a constaté, chez l'animal de laboratoire exposé à l'acétone,
une moindre performance dans les tests neurocomportementaux, aux
concentrations supérieures à 7765 mg/m3 (> 3270 ppm).
On ne dispose pas de données sur les effets d'une exposition de
longue durée à l'acétone par la voie orale ou respiratoire, sans doute
parce que sa toxicité est faible et qu'il s'agit d'un composé
endogène.
En faisant inhaler de l'acétone à des rats pendant une longue
période (45 000 mg/m3, soit 19 000 ppm, 3 h par jour, 5 jours par
semaine, pendant 8 semaines), on a observé une diminution réversible
du poids absolu du cerveau. Aucune modification systématique n'a été
notée dans le poids du corps ou celui d'antres organes, au niveau des
constantes hématologiques, dans le taux des triglycérides hépatiques
ou dans l'aspect histologique du coeur, du poumon, du rein, du cerveau
ou du foie.
Lors d'une étude de 90 jours comportant l'administration
d'acétone à des rats par gavage, on a observé une augmentation de
certains paramètres hématologiques (hémoglobine, hématocrite) aux
doses supérieures à 500 mg/kg par jour et on a déterminé une NOAEL
(dose sans effet nocif observable) de 500 mg/kg par jour. Dans une
autre étude, qui a duré 13 semaines et qui a consisté à ajouter de
l'acétone à l'eau de boisson des rats, on a noté la présence d'effets
toxiques chez les mâles exposés à des concentrations supérieures à 20
g/litre (env. 1700 mg/kg p.c. par.jour), à savoir une augmentation du
poids relatif des organes et une modification des constantes
hématologiques, avec en outre une légère néphropathie. Chez les
femelles soumises à la dose la plus forte, soit 50 g/litre (env. 3400
mg/kg p.c. par jour), les effets consistaient en une augmentation du
poids relatif des organes et une modification des constantes
hématologiques. En outre, l'exposition à 50 g/litre pendant 13
semaines a provoqué chez les mâles une modification du poids relatif
des testicules et ainsi qu'une baisse de la motilité des
spermatozoïdes accompagnée d'anomalies morphologiques. Des souris
femelles exposées à la même concentration de 50 g/litre (env. 11 298
mg/kg p.c. par jour) dans leur eau de boisson, présentaient une
réduction du poids du foie et de la rate ainsi qu'une hypertrophie
hépatique touchant les cellules centrilobulaires, mais dont
l'augmentation d'incidence restait marginale. Aucun effet toxique n'a
été relevé chez les souris mâles qui avaient reçu la plus forte dose
d'acétone (20 g/litre, soit env. 4858 mg/kg p.c. par jour). Une
exposition de 13 semaines à des doses inférieures ou égales à 10
g/litre (900 mg/kg p.c. par jour), toujours par ingestion d'acétone
mélangée à l'eau de boisson, n'a pas produit d'effets toxiques chez
des rats mâles; les doses sans effets observables étaient < 20
g/litre chez les rats (1600 mg/kg p.c. par jour) et les souris des
deux sexes (mâles: 4858 mg/kg p.c. par jour; femelles: 5945 mg/kg p.c.
par jour).
Lors d'une étude préliminaire de 14 jours consistant à faire
boire de l'eau additionnée d'acétone à des rats et à des souris, on a
noté chez les mâles exposés aux concentrations de 20 à 100 g/litre,
une hypertrophie hépatique affectant les cellules centrilobulaires.
Chez les rongeurs, un prétraitement par l'acétone accroît les
effets hépatotoxiques de certains composés, notamment ceux des dérivés
halogénés des alcanes. On suppose que cette potentialisation de
l'hépatotoxicité est due à l'augmentation, sous l'action de l'acétone,
de l'activité de certaines enzymes (les oxydases hépatiques â fonction
mixte), qui sont responsables de la production d'intermédiaires
toxiques à partir des alcanes halogénés.
Les épreuves de génotoxicité ont donné des résultats négatifs sur
de nombreux systèmes mammaliens, tant in vivo qu' in vitro. Il n'y
a qu'une seule épreuve qui ait donné un résultat positif, à savoir la
présence d'aneuploïdies chez une espèce de levure exposée à une forte
concentration d'acétone (6,82%) dans son milieu de croissance. On
considère que l'acétone n'est ni génotoxique, ni mutagène.
Lors d'une étude portant sur des rats et des souris gravides, on
a exposé les animaux à des vapeurs d'acétone du 6ème au 19ème jour de
la gestation. De légers effets toxiques ont été observés sur le
développement de la progéniture lorsque les rats étaient exposées à
une concentration de 26 100 mg/m3 (11 000 ppm) à raison de 6 h par
jour (augmentation de la proportion de portées comportant au moins une
malformation foetale) et les souris à une concentration de 15 670
mg/m3 (6600 ppm) à raison également de 6 h par jour (légère
diminution du poids des foetus et petite augmentation de l'incidence
relative des résorptions tardives). On a fixé à 5200 mg/m3 (2200 ppm)
la concentration atmosphérique sans effet nocif observable sur le
développement de la souris et du rat. Dans une étude par gavage, un
traitement par l'acétone au cours de l'organogénèse à la dose
quotidienne de 3500 mg/kg, a eu un effet négatif sur la reproduction
lors d'un test de criblage sur des souris. Les résultats négatifs
obtenus in vivo chez deux espèces différentes, l'administration se
faisant par la voie buccale et par la voie intrapéritonéale, montrent
que l'exposition de mammifères à l'acétone n'a pas d'effets mutagènes.
Les observations relatives aux effets de l'acétone sur la
reproduction consistent en anomalies testiculaires et modification de
la qualité des spermatozoïdes, qui ont été mises en évidence chez des
rats dont l'eau de boisson avait contenu pendant 13 semaines 50 g
d'acétone par litre. On n'a pu trouver aucune étude consacrée aux
effets que pourrait avoir l'ingestion d'acétone sur le développement
foetal (foetotoxicité et tératogénicité).
L'acétone est largement utilisée comme solvant dans les études
sur le pouvoir cancérogène cutané et on estime qu'en application sur
la peau, elle est dénuée d'activité cancérogène.
7. Effets sur l'Homme
L'acétone est relativement moins toxique que nombre d'autres
solvants industriels; cependant, sous forte concentration, les vapeurs
d'acétone peuvent provoquer une dépression du système nerveux central,
un collapsus cardiorespiratoire et la mort. On connaît des cas
d'exposition humaine aiguë à des concentrations d'acétone atteignant
environ 4750 mg/m3 (soit à peu près 2000 ppm) qui n'ont été
accompagnés d'aucun effet toxique majeur ou tout au plus d'effets
mineurs et passagers, tels qu'une irritation oculaire. Des effets
passagers plus sérieux (notamment des vomissement et une perte de
conscience) ont le observés chez des travailleurs exposés à des
vapeurs d'acétone dont la concentration dépassait 25 500 mg/m3 (>
12 000 ppm) pendant environ 4 h. On a également fait état d'une baisse
dans les résultats des tests neurocomportementaux chez l'Homme à la
concentration de 595 mg/m3 (250 ppm). Des femmes exposées à une
concentration atmosphérique de 2370 mg/m3 (1000 ppm) ont présenté des
irrégularités du cycle menstruel.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Pour la plupart des espaces animales dulçaquicoles ou marines,
les valeurs de la CL50 et de la CE50 à 48 et 96 h se sont révélées >
5540 mg/litre.
Une exposition de 76 h à de l'acétone à la concentration de 257,4
mg/litre a provoqué une inhibition de la croissance de l'algue
Chlorella pyrendoidosa. Il en a été de même pour
Chlamydomonas eugametos après 48 h d'exposition à 790 mg/litre. En
exposant Scendensemus quadricauda et C. pyrendoidosa à de
l'acétone aux concentrations respectives de 79,0 et 790 mg/litre, on a
observé un accroissement de la photosynthèse.
Les seuils de toxicité respectifs à 7 et 8 jours pour l'algue
verte S. quadricauda et pour la cyanobactérie (algue bleue)
Microcystis aeruginosa se situent à 7500 et 530 mg/litre, ce qui
traduit la plus grande résistance de l'algue verte à l'action toxique
de l'acétone. La diatomée Nitzschia linearis a également semblé
faire preuve d'une grande résistance, avec une CE50 à 5 jours
comprise entre 11 493 et 11 727 mg/litre. De même la diatomée marine
Skelatonema costatum s'est aussi révélée lès résistante, avec une
CE50 comprise entre 11 798 et 14 440 mg/litre.
Les bactéries se révèlent plus résistantes à l'acétone que les
protozoaires. Pour Photobacterium phosphoreum, Pseudomonas putida
et une culture mixte, on a obtenu une valeur de la CE50 comprise
entre 1700 et 35 540 mg/litre, contre 28 mg/litre dans le cas du
protozoaire Entosiphon sulcatum. Ce résultat pourrait s'expliquer
par le fait que ces deux types de microorganismes ont une paroi
cellulaire différente.
Chez des cailles et des faisans, on a trouvé des valeurs de la
CL50 à 5 jours supérieures ou égaies à 40 g par kg de nourriture. Des
oeufs fécondés de colvert n'ont pas souffert d'une immersion de 30
secondes dans de l'acétone à 10%; en revanche, l'immersion dans de
l'acétone pneu a entraîné une diminution de la survie, du poids et de
la taille des embryons sans que l'on puisse dire avec certitude si e'
est la toxicité de l'acétone qui était en cause ou son caractère de
solvant. L'injection de 5 µl d'acétone dans des oeufs de poules
Leghorn ne semble pas avoir entraîné de mortalité ni de malformations
chez les embryons.
RESUMEN
1. Propiedades
La acetona (masa molecular relativa = 58,08) es un liquido
transparente, incoloro e inflamable (punto de inflamación - 17°C en
crisol cerrado, -9°C en crisol abierto; limites de inflamabilidad en
el aire a 25°C = 2,15-13% v/v). Los límites de explosión en el aire
son 2,6-12,8% v/v. Tiene una elevada tasa de evaporación (presión de
vapor 181,72 mmHg a 20°C) y baja viscosidad (0,303 cP a 25°C). Es
miscible con el agua y con disolventes orgánicos.
2. Usos y fuentes de exposición
2.1 Producción
La acetona se fabrica principalmente mediante los procesos de
peroxidación del cumeno o la deshidrogenación del isopropil alcohol.
En el primer proceso se producen cantidades infamas de benceno como
subproducto.
2.2 Usos y emisiones al medio ambiente
La acetona se utiliza principalmente como disolvente y como
intermedio en la producción de sustancias químicas. Sus principales
aplicaciones son la producción de metil metacrilato, ácido metacrílico
y metacrilatos de mayor tamaño, bisfenol A, metil isobutil cetona,
aplicaciones farmacéuticas y medicamentosas, y como disolvente para
revestimientos y para el acetato de celulosa. También tiene usos
alimentarios como disolvente de extracción para grasas y aceites, y
como agente de precipitación en la purificación del azúcar y el
almidón.
Las emisiones a la atmósfera proceden de los productos de
consumo, como quitaesmalte para las uñas, tableros de conglomerado,
revestimientos inferiores de moquetas, algunos decapantes de pinturas
y ceras o abrillantadores líquidos o sólidos. Ciertos detergentes y
limpiadores, adhesivos y limpiadores del carburador y el estrangulador
en automóviles también contienen acetona.
La acetona se vierte a las aguas superficiales en los efluentes
de aguas residuales de una amplia gama de procesos e industrias de
fabricación, como el papel, el plástico, productos farmacéuticos,
limpiadores y abrillantadores químicos elaborados, pinturas y
productos conexos, productos químicos del caucho y la madera,
intermedios cíclicos, productos orgánicos industriales, productos del
yeso, productos de cartón de papel e industrias de la energía, como la
gasificación del carbón y el tratamiento de esquistos bituminosos.
Entre las fuentes de incorporación de acetona al suelo figuran el
vertido de residuos agrícolas y alimentarios, residuos animales,
deposición húmeda desde la atmósfera, efluentes de fosas sépticas
domésticas y vertederos de residuos químicos.
3. Transporte, distribución y transformación en el medio ambiente
La acetona que ingresa en la atmósfera es degradada por una
combinación de fotolisis y reacción con radicales hidroxilo. La
semivida media para la degradación de la acetona en la atmósfera es de
unos 30 días. La acetona puede ser eliminada por medios fiscos del
aire por deposición húmeda. El principal proceso de degradación de la
acetona en el suelo y el agua es la biodegradación; la acetona es
fácilmente biodegradable. La volatilización de la acetona desde el
medio acuático puede ser un proceso de transporte significativo. La
acetona es un compuesto volátil que se evapora fácilmente de las
superficies secas. Puesto que es miscible en agua, puede ser objeto de
lixiviación en la mayoría de los tipos de suelo. La biodegradación
concurrente puede reducir la importancia global de la lixiviación si
aquélla se produce con la rapidez suficiente.
4. Concentraciones en el medio ambiente y exposición humana
La exposición a la acetona procede de fuentes tanto naturales
como antropogénicas. La acetona también aparece como compuesto
metabólico en la sangre, la orina y el aire pulmonar del ser humano.
Se produce en la biodegradación de las aguas residuales, los residuos
sólidos y los alcoholes, así como por la oxidación de sustancias
húmicas. Se ha detectado en muy diversas plantas y alimentos, como las
cebollas, las uvas, la coliflor, los tomates, las convolvuláceas, la
mostaza silvestre, la leche, las indias, los guisantes, el queso y la
pechuga de pollo. Las emisiones naturales de varias especies de
árboles contienen vapores de acetona. Las fuentes antropogénicas de
emisión al medio acuático comprenden los vertidos de aguas residuales
de muchas industrias y la lixiviación que tiene lugar en vertederos
industriales y municipales. Una de las principales fuentes de emisión
humana al aire es la evaporación de la acetona utilizada como
disolvente en productos de revestimiento como pinturas, limpiadores,
barnices y tintas. La acetona es un producto de emisión de la
combustión de madera, basuras y plásticos. También se emite en los
escapes de automóviles y de motores diesel y de turbina. Las
concentraciones de acetona detectadas en la atmósfera varían entre 0,5
y 125,4 µg/m3 (0,2-52,9 ppmm).
5. Cinética y metabolismo
La acetona es uno de los tres cuerpos cetónicos que se producen
naturalmente en el organismo humano. Puede formarse de modo endógeno
en el organismo de los mamíferos por oxidación de los ácidos grasos.
El ayuno, la diabetes mellitus y el ejercicio físico vigoroso
incrementan la generación endógena de acetona. En condiciones
normales, prácticamente todos los cuerpos cetónicos se producen en el
hígado, y en menor medida en el pulmón y el riñón. El proceso es
continuo y los tres productos son excretados a la sangre y
transportados a todos los tejidos y órganos del cuerpo, donde pueden
ser utilizados como fuente de energía. Dos de esos cuerpos cetónicos,
el acetoacetato y el ß-hidroxibutirato, son ácidos orgánicos que
pueden provocar acidosis metabólica cuando se producen en grandes
cantidades, como en la diabetes mellitus. La acetona, en cambio, tiene
carácter no iónico y se deriva de forma endógena de la degradación
enzimática espontánea del acetoacetato. La acetona endógena se elimina
del organismo por excreción en la orina y el aire exhalado o por
metabolismo enzimático. En circunstancias normales, el metabolismo es
la vía principal de eliminación y procesa el 70-80% de la carga total
del organismo.
La acetona es absorbida rápidamente por los tractos respiratorio
y gastrointestinal del ser humano y los animales de laboratorio, como
lo indica la detección de acetona en la sangre a los 30 minutos de la
exposición por inhalación y a los 20 minutos de la exposición por vía
oral. Los estudios efectuados en ratas indican que la acetona
administrada por vía oral se absorbe fácilmente, mientras que en la
exposición por inhalación el ser humano absorbe aproximadamente el 50%
de la cantidad de acetona inhalada. Sin embargo, se han comunicado
valores tanto menores como mayores de absorción por vía respiratoria.
Las cavidades nasales de los seres humanos y los animales de
laboratorio parecen tener una capacidad limitada de absorción y
excreción de vapores de acetona, en comparación con el resto del
tracto respiratorio.
La acetona se distribuye uniformemente por los tejidos no
adiposos y no se acumula en los tejidos adiposos. En el ratón, se
comunicó que las concentraciones máximas de acetona en los tejidos
adiposos eran aproximadamente la tercera parte de las observadas en
los tejidos no adiposos tras la exposición por inhalación. La acetona
se elimina rápidamente del organismo por vía metabólico y por
excreción. Las semividas de la acetona en el aire alveolar y la sangre
venosa y arterial en el ser humano son approx. 4, 6 y 4 horas,
respectivamente. La exhalación es la principal vía de eliminación de
la acetona y su metabolito terminal, el CO2, y la fracción de la
acetona adminisáada que se exhala sin modificar depende de la dosis.
También se produce excreción de acetona y sus metabolitos en la orina,
pero esta vía de eliminación es menos importante que la exhalación de
acetona y CO2 por vía respiratoria.
La acetona de origen exógeno se incorpora a numerosas reacciones
metabólicas en tejidos de todo el organismo, pero el hígado parece ser
el órgano en el que se metabólico más intensamente. El carbono de la
acetona administrada por veta oral se ha detectado en el colesterol,
los amanecidos, los ácidos grasos y el glucógeno en tejidos de rata,
la urea en la orina y acetona no modificada y CO2 en el aire
exhalado. Desde el punto de vista metabólico, la acetona se degrada a
acetato y formato; por esta vía se produce la incorporación de los
átomos de carbono de la acetona al colesterol, los ácidos grasos, la
urea y los amanecidos, así como la formación de compuestos
gluconeogénicos de tres átomos de carbono.
Se han propuesto dos posibles vías de gluconeogénesis a partir de
la acetona. La primera vía comienza por la acción catalítica inicial
de la acetona-monooxigenesa y la acetol-monooxigenesa, que convierten
la acetona en acetol y el acetol en metilglioxal, respectivamente.
Ambas actividades enzimáticas son inducidas por la acetona y han sido
identificadas como isoenzimas del citocromo hepático P-450IIE1,
inducible por el etanol. La segunda ruta gluconeogénica posible
entraña la formación de 1,2-propanodiol a partir de la acetona,
catalizada por la acetona monooxigenasa y una enzima no caracterizada
capaz de convertir el acetol en 1,2-propanodiol.
6. Efectos en mamíferos de experimentación y en sistemas in vitro
Los valores de la DL50 en ratas adultas se encuentran en el
intervalo 5800-7138 mg/kg. El valor de la CL50 a las 4 horas de la
inhalación es de 76 000 mg/m3 (32 000 ppm).
Se ha observado que la exposición aguda a la acetona altera los
resultados de las pruebas necrológicas de conducta en animales de
laboratorio cuando las concentraciones superan los 7765 mg/m3 (>3270
ppm).
No se dispone de datos en animales de experimentación para
caracterizar los efectos de la exposición oral o por inhalación a
largo plazo de la acetona, probablemente a causa de su escasa
toxicidad y sus características endógenas.
La exposición prolongada de ratas a la inhalación de acetona, a
razón de 45 100 mg/m3 (19 000 ppm), durante 3 horas al día, 5 días a
la semana durante 8 semanas, provocó una reducción reversible del peso
cerebral absoluto. No se observaron cambios uniformes en el peso de
otros órganos ni del organismo en conjunto, ni tampoco en los índices
químicos de la sangre, en los niveles de triglicéridos hepáticos o en
las características histológicas del corazón, el pulmón, el riñón, el
cerebro o el higado.
En un estudio de alimentación forzada de ratas durante 90 días,
se determinó un aumento en los parámetros sanguíneos (aumento de la
hemoglobina y el hematócrito) con concentraciones superioras a 500
mg/kg al día, y una concentración sin observación de efectos adversos
de 500 mg/kg al día. En un estudio de administración de acetona en el
agua de bebida durante 13 semanas, se observaron efectos tóxicos en
las ratas macho expuestas a concentraciones superioras a 20 g/litro
(approx. 1700 mg/kg de peso corporal al día), a saber, aumento del
peso relativo de algunos órganos, y alteración de los índices
hematológicos y leve nefropatía. En hembras de rata a las que se
administró la concentración más elevada, 50 g/litro (approx. 3400
mg/kg de peso corporal al día), los efectos observados fueron un
aumento de los pesos relativos de ciertos órganos y la alteración de
los índices hematológicos. Además, la exposición durante 13 semanas a
50 g/litro provocó una alteración del peso relativo de los testículos
y de la mutualidad y morfología de los espermatozoides en ratas macho.
Las hembras de ratón a las que se administraron 50 g/litro (approx. 11
298 mg/kg de peso corporal al día) en el agua de bebida presentaron
alteraciones del peso del hígado y el bazo y una incidencia
ligeramente mayor de hipertrofia hepatocelular centrilobular. No se
observaron efectos tóxicos en ratones macho a los que se administraron
20 g/litro (4858 mg/kg de peso corporal al tila), la concentración de
acetona más alta que se ha administrado a ratones macho. La exposición
durante 13 semanas a concentraciones < 10 g/litro (900 mg/kg de
peso corporal al día) administradas en el agua de bebida no se vio
acompañada de efectos tóxicos en ratas macho; las concentraciones
< 20 g/litro no produjeron efectos observables en hembras de rata
(1600 mg/kg de peso corporal al día) ni en ratones de ambos sexos
(machos: 4858 mg/kg de peso corporal al día; hembras: 5945 mg/kg de
peso corporal al día).
En un estudio preliminar en el que se administró acetona en el
agua de bebida a ratas y ratones durante 14 alias, se observó
hipertrofia hepatocelular centrilobular relacionada con la dosis en
ratones machos expuestos a concentraciones de 20-100 g/litro.
El tratamiento previo de roedores con acetona acentúa los efectos
hepatotóxicos de varios compuestos, particularmente los alcanos
halogenados. Una hipótesis es que la potenciación de la
hepatotoxicidad está mediada por el aumento de las actividades
enzimáticas mediadas por la acetona (oxidabas hepáticas de funcion
mixta) que son responsables de la generación de productos intermedios
tóxicos originados a partir de los alcanos halogenados administrados.
La acetona ha dado resultados negativos en relación con la
toxicidad genética en numerosos sistemas no mamíferos, así como en
sistemas mamíferos in vitro e in vivo. Los resultados positivos se
limitan a una sola prueba de aneuploidía en una especie de levadura
expuesta a concentraciones elevadas de acetona (6,82%) en su medio de
cultivo. No se considera que la acetona sea genotóxica ni mutagénica.
En un estudio de hembras gestantes de ratón y rata expuestas a
vapores de acetona durante los días ó a 19 de la gestación, se observó
una ligera toxicidad para el desarrollo tras la exposición de las
ratas a 26 100 mg/m3 (11 000 ppm) durante 6 horas al día (pequeña
disminución en el peso del feto y pequeño aumento en la incidencia
porcentual de retorciones tardías). Se determinó que una concentración
atmosférica de 5200 mg/m3 (2200 ppm) era el nivel sin observación de
efectos adversos respecto de la toxicidad para el desarrollo tanto en
ratones como en ratas. En un estudio de alimentación forzada, el
tratamiento con 3500 mg/kg al día durante la organogénesis
obstaculizaba la reproducción en una prueba de detección en ratones.
Los resultados negativos obtenidos in vivo en dos especies
diferentes, utilizando las vías oral e intraperitoneal, indicó que no
se producían cambios mutagénicos en mamíferos expuestos a la acetona.
Entre los datos comunicados acerca de otros efectos reproductivos
de la acetona figuran observaciones de efectos testiculares y
alteraciones de la calidad espermática en ratas a las que se
administró agua de bebida con 50 g de acetona por litro durante 13
semanas. No se dispuso de datos sobre investigaciones del efecto de
dosis de acetona administradas por vía oral en el desarrollo fetal
(fetotoxicidad y teratogenicidad).
La acetona se ha utilizado ampliamente como vehículo disolvente en
estudios de carcinogenicidad en la piel y no se considera
carcinogénico cuando se aplica a la piel.
7. Efectos en el ser humano
La acetona es relativamente menos tóxica que muchos otros
disolventes industriales; sin embargo, en concentraciones altas, el
vapor de acetona puede provocar depresión del sistema nervioso
central, fallo cardiorrespiratorio y la muerte. Se ha comunicado que
la exposición aguda del ser humano a concentraciones atmosféricas tan
altas como approx. 4750 mg/m3 (approx 2000 ppm) no produce grandes
efectos tóxicos ni efectos transitorios leyes, como irritación ocular.
Se comunicaron efectos transitorios más graves (inclusive vómitos y
desmayos) en trabajadores expuestos a concentraciones de vapor de
acetona > 25 500 mg/m3 (250 ppm). Las mujeres expuestas a
concentraciones atmosféricas de 2370 mg/m3 (1000 ppm) padecieron
trastornos menstruales.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
En la mayoría de las especies animales de agua tanto dulce como
salada, los valores de la CL50 y la CE50 a las 48 y las 96 horas son
superiores a 5540 mg/litro.
La exposición a una concentración de acetona de 257,4 mg/litro
durante 76 horas inhibió el crecimiento del alga
Chlorella pyrendoidosa. También se observó inhibición del
crecimiento de Chlamydomonas eugametos expuesta a acetona durante 48
horas en una concentración de 790 mg/litro. En
Scendesmus quadricauda y C pyrenoidosa expuestas a 79,0 y 790
mg/litro de acetona se observo un aumento de la fotosíntesis.
Los umbrales de toxicidad a 7 y 8 días para el alga verde
S. quadricauda y la cianobacteria (alga verdeazulada)
Microcystis aeruginosa fueron 7500 y 530 mg/litro, respectivamente,
lo que indica que el alga verde era más resistente a la actividad
tóxica de la acetona. La diatomea Nitzschia linearis también resultó
ser muy resistente, con una EC50 en cinco días de 11 493 a 11 727
mg/litro. De modo similar, la diatomea de agua salada
Skeletonema costatum resultó muy resistente, con valores de EC50
a tos cinco días de 11 798 y 14 440 mg/litro.
Las bacterias parecen ser más resistentes a la acetona que los
protozoos. Photobacterium phosphoreum, Pseudomonas putida y un
cultivo microbiano mixto presentaron valores de la CE50 de 1706 a 35
540 mg/litro, mientras que el protozoo Entosiphon sulcatum presentó
una CE50 de 28 mg/litro. Esto puede guardar relación con las
diferencias en la pared celular.
En codornices y faisanes se observaron valores de la CL50 por
vía oral a los cinco días > 40 g/kg de dieta. Los huevos
fecundados de pato silvestre no se vieron afectados por la inmersión
en acetona al 10% durante 30 segundos; en cambio, la inmersión en
acetona pura dio lugar a un descenso de la supervivencia, el peso y la
talla del embrión, aunque no está claro si ello se debía a las
propiedades tóxicas o a las propiedades disolventes de la acetona. Los
huevos de gallina Leghorn blanca inyectados con 5 µl de acetona no
mostraron cambios significativos en la mortalidad ni malformaciones.
The relative importance of the three pathways in the metabolism
of acetone may depend upon the amount of acetone administered. When a
trace amount of [2-14C]-acetone was administered intravenously to
rats, the pattern of incorporation of 14C into glucose was
consistent with the production of glucose via the
methylglyoxal/lactate pathway (Kosugi et al., 1986a). When a higher
dose of [2-14C]-acetone (325 mg/kg) was injected, the pattern of
incorporation was more consistent with the 1,2-propanediol pathway.
These results suggest that at low doses of acetone or endogenous
acetone, the methylglyoxal and lactate pathways predominate, but at
higher doses, these pathways become saturated and metabolism is
shunted to the formate-acetate branch of the 1,2-propanediol pathway.
Methylglyoxal can then be converted to D-glucose by an
unidentified pathway (Fig. 1), and/or possibly by catalysis by
glyoxalase I and II and glutathione to D-lactate, which is converted
to D-glucose (Casazza et al., 1984). This formation of D-lactate from
acetone provides the body with a mechanism for recovering a portion of
the energy that is lost when acetone is formed from acetoacetate
(Morgott, 1993). The conversion of methylglyoxal to D-lactate by the
actions of glyoxalase I and II is well established (Racker, 1951), but
may represent a minor pathway in the metabolism of acetone (Casazza et
al., 1984; Kosugi et al., 1986a; Thomalley, 1990). In addition,
methyl-glyoxal is converted to D-glucose via conversion of
methylglyoxal to pyruvate by 2-oxoaldehyde dehydrogenase, an activity
identified using aqueous extracts of sheep liver acetone powders
(Monder, 1967).
In the second and third pathways, acetol is converted to
L-1,2-propanediol by an extrahepatic mechanism that has not been
characterized (Rudney, 1954; Casazza et al., 1984; Kosugi et al.,
1986a,b; Sktuches et al., 1990). The two pathways then diverge from
the point of production of 1,2-propanediol. In the second pathway,
1,2-propanediol formed extra-hepatically returns to the liver where it
is converted to L-lactaldehyde by nicotinamide adenine dinucleotide
(NADH)-dependent alcohol dehydrogenase (Casazza et al., 1984; Kosugi
et al., 1986a,b), and L-lactaldehyde, in mm, is converted to L-lactate
(Rudney, 1954; Ruddick, 1972; Casazza et al., 1984) by NADH-dependent
aldehyde dehydrogenase (Casazza et al., 1984). L-lactate can then be
converted to D-glucose (Casazza et al., 1984). In the third pathway,
the L-1,2-propanediol formed extra-hepatically returns to the liver
where it is degraded by other mechanisms to acetate and formate
(Sakami & Rudney, 1952; Ruddick, 1972; Casazza et al., 1984),
Several studies have traced the labelling patterns of 14C from
[2-14C]-acetone or [1,3-14C]-acetone to gluconeogenic precursors
and formate to incorporation of 14C into glycogen, glycogenic amino
acids, fatty acids, haem, cholesterol, choline and urea (Price &
Rittenherg, 1950; Sakami & LaFaye, 1950; Mourkides et al., 1959). The
pattern of labelling suggested the involvement of the pathway to
acetate and formate. Fatty acids, amino acids and glycogen may also
enter stages of intermediary metabolism. Factors affecting the
formation and release of long-chain fatty acids from adipose tissue
during lipolysis can affect the circulatory levels of acetone
(Morgott, 1993).
Although the liver is the primary site of acetone metabolism,
radioactive unmetabolized acetone and total radioactivity have been
found in many other mice tissue and organs after inhalation exposure
to 14C-acetone (Wigaeus et al., 1982). The fraction of total
radioactivity that was not unchanged acetone represented metabolites.
Elimination of acetone was complete in all tissues by 24 h after
exposure, but total radioactivity, indicative of metabolites, was
still present in all tissues except blood and muscle. Whether these
tissues (other than the liver) were capable of metabolizing acetone or
whether the metabolites themselves were distributed to the tissues was
unclear. However, microsomes from the lungs of hamsters exposed to
acetone in drinking-water for 7 days had a 500% increased activity of
aniline hydroxylase activity, an enzyme associated with cytochrome
P-450IIE1 (Ueng et al., 1991). The level of cytochrome P-450IIE1
increased 6-fold in microsomes from the nasal mucosa of rabbits
exposed to acetone in drinking-water for 1 week (Ding & Coon, 1990).
In hamsters given drinking-water containing acetone for 7 days (Ueng
et al., 1991) or 10 days (Menicagli et al., 1990), the microsomes
prepared from kidneys had increased levels of cytochrome P-450 and
cytochrome b5. These results suggest that acetone metabolism, which
involves cytochrome P-450IIE1, may occur in the lungs and kidneys of
hamsters and the nasal mucosa of rabbits. Incubation of acetone with
homogenates of nasal mucosa from mice indicated that acetone was
metabolized via a NADPH-dependent pathway in vitro, but no evidence
of in vivo metabolism of acetone by the upper respiratory tract was
found in mice, rats, guinea-pigs or hamsters (Morris, 1991 ).
Diet as well as physiological or genetic status may alter the
metabolism of acetone. When non-diabetic and diabetic rats were
treated by gavage with acetone at doses of 1000, 2000 or 4000 mg/kg,
isopropyl alcohol was detected in the blood (Lewis et al., 1984). The
levels of isopropyl alcohol and acetone increased with higher doses in
the diabetic rats, although with plateaus for both acetone and
isopropyl alcohol at 1000 and 2000 mg/kg doses, but levelled off in
the non-diabetic rats, indicating either saturation of the metabolic
pathway from acetone to isopropyl alcohol or a reversibility of the
conversion at high doses. It was suggested that in the diabetic rats,
acetone and NADH+, both needed for isopropyl alcohol production from
acetone, presumably by alcohol dehydrogenase, may be diverted to
gluco-neogenic pathways to meet the diabetic rat's need for glucose,
resulting in the short plateau. The subsequent rises of both compounds
at the high dose of acetone in the diabetic rats could be accounted
for by greater generation of NADH+ from fatty acid oxidation in the
diabetic rat, which reduces acetone to isopropyl alcohol, accounting
for the rising level of isopropyl alcohol. Liver homogenates from mice
heterozygous for the obesity gene treated with acetone were more
effective in converting acetone to lactate than liver homogenates from
normal homozygous mice treated with acetone (Coleman, 1980).
In pregnant or virgin rats (either fed or fasted) injected
intravenously with acetone, plasma acetol levels were not
significantly different between fasted and non-fasted rats, but
pregnant rats had significantly lower levels than virgin rats (Peinado
et al., 1986). Liver levels of acetol were also significantly lower in
pregnant rats than in virgin rats, Methylglyoxal levels were very high
in the livers and plasma of non-fasted rats (pregnant or virgin), but
fasting resulted in much lower levels. In contrast, no major
differences were found in the expiration of carbon dioxide between
fasted and diabetic rats injected intraperitoneally with acetone
(Mourkides et al., 1959) or in the labelling pattern of 14C derived
from 14C-acetone into glucose among non-fasted diabetic, fasted
diabetic, normal non-fasted and normal fasted rats injected
intravenously with 14C-acetone (Kosugi et al., 1986a,b).
6.3.3 Metabolism summary
As shown in Fig. 1, acetone is oxidised to acetol, which is then
metabolized by at least three different pathways and is independent of
the route of exposure. Glycogenesis is a driving force and acetone is
one of three ketone bodies produced by acetyl coenzyme A in the liver.
The relative importance of each pathway is probably dependent on
both the amount and route of exposure. Diet, physiological and genetic
status, and diabetes may affect the metabolism of acetone. Acetone is
either metabolized or excreted in both humans and animals, and the
time taken for elimination depends on the amount of acetone absorbed.
6.4 Elimination and excretion
Exhalation accounts for the elimination of about 20% as breath
acetone at low plasma acetone concentrations. Approximately 75% is
metabolized, only small amounts appearing in the urine. However, at
high plasma acetone concentrations, 80% can be accounted for by breath
acetone with only 20% being metabolised in vivo. The main route of
excretion of acetone is by the lung regardless of the route of
exposure (Owen et al., 1982).
6.4.1 Human studies
The clearance of acetone from blood was constant regardless of
blood acetone concentration (DiVincenzo et al., 1973). Halftimes for
blood elimination of 3-3.9 h have been estimated in humans exposed to
240-590 mg/m3 (100-500 ppm) for 2-4 h (DiVincenzo et al., 1973;
Wigaeus et al., 1981; Brown et al., 1987). Elimination half-times
between 3.9 and 6.2 h have been estimated for blood (Wigaeus et al.,
1981, Wang et al., 1994), and no differences in elimination half-times
were found between men and women (Brown et al., 1987). Because the
elimination rate depends on the metabolism, the effect of induction of
P450-dependant monooxygenase hydroxylase by acetone itself (Puccini et
al., 1989) or other specific inducers of the isozymes involved could
change the half-time. Longo et al. (1993) have shown that the set of
isozymes differs among animal species, and some have not been isolated
and characterized (Puccini et al., 1989). The elimination from blood
was found to be complete in 24 h after a 6-h exposure in subjects
exposed to 470 mg/m3 (250 ppm), in 32 h in subjects exposed to 1190
mg/m3 (500 ppm), and in 48 h in subjects exposed to 2370 mg/m3
(1000 ppm) (Matsushita et al., 1969b). When volunteers were exposed
for 6 h/day for 6 days, the blood levels of acetone rose each day and
declined to background levels by the following morning each day when
the exposure concentration was 470 mg/m3 (250 ppm) (Matsushita et
al., 1969a). At an exposure concentration of 1190 mg/m3 (500 ppm),
however, the blood levels declined each day, but not to pre-exposure
levels. At the end of the 6-day exposure, blood acetone levels
declined to pre-exposure levels within 2 days for the 470 mg/m3
group and declined within 3 days for the 1190 mg/m3 group. From the
half-life and the data on time for decline to pre-exposure levels, it
appears that at higher concentrations, acetone may accumulate slightly
in the blood during daily intermittent exposure, as would be
experienced by workers.
The rate and pattern of respiratory excretion of acetone is
influenced by exposure concentration, duration, the level of physical
activity during exposure and gender. In humans exposed to acetone
concentrations of < 2970 mg/m3 (< 1250 ppm) for < 7.5 h/day
in a complex protocol for < 6 weeks, the rate of respiratory
excretion was a function of the duration, and the concentration of
acetone in breath after exposure was directly related to the
time-average concentration during exposure, with constant duration
(Stewart et al., 1975). The length of time after exposure in which
acetone could be detected in the expired air was related to the
magnitude of exposure, with acetone still readily detectable 16 h after
exposure to 2370 or 2970 mg/m3 (1000 or 1250 ppm) for 7.5 h.
Excretion of acetone by the lungs was complete within 20 h post-exposure
in humans exposed to 560 mg/m3 (237 ppm) for 4 h (Dick et al.,
1989). During exposure for 2 h, the acetone concentration in expired
air rose to 50 mg/m3 (20 ppm) in humans exposed to 240 mg/m3 (100
ppm) and to 215-240 mg/m3 (90-100 ppm) in those exposed to 1190 mg/m3
(500 ppm) (DiVincenzo et al., 1973). After exposure to 240 mg/m3, the
expired air concentration of acetone declined biphasically over the
next 7 h to 12 mg/m3 (5 ppm). However, after exposure to 1190 mg/m3
(500 ppm), the expired air concentration dropped sharply to 5 mg/m3
(2 ppm) and declined to 2 mg/m3 (1 ppm) over the next 7 h. Prolonging
the exposure duration to 4 h resulted in less than a 2-fold increase
in acetone levels in post-exposure expired air, which may reflect a
greater loss of acetone through metabolism and urinary excretion.
Exercise during the exposure period increased the elimination almost
2-fold. In humans exposed to acetone at rest, during exercise at a
constant workload, or during exercise with step-wise increments in
workload, expiration of acetone via the lungs amounted to 70, 80 and
200 nag, respectively, at 4 h post-exposure and to 50, 80 and 200 mg,
respectively, over the next 4-20 h (Wigaeus et al., 1981). Excretion
of acetone from the lungs and kidneys (combined) amounted to 16, 20
and 27% of the amount absorbed in the three respective groups of
subjects. Urinary excretion amounted to only 1% of the total uptake.
Women expired acetone more slowly than men after a 4-h exposure to
300-310 mg/m3 (127-131 ppm), but the percentages excreted by the
lungs were not statistically significantly different between men and
women (17.6% for men, 15.0% for women) (Nomiyama & Nomiyama, 1974b).
Very little acetone is excreted in the urine (DiVincenzo et al.,
1973; Wigaeus et al., 1981; Vangala et al., 1991; Kawai et al., 1992).
Urinary excretion is biphasic (Pezzagno et al., 1986). Peak urinary
excretion occurred between 1 and 3.5 h after exposure (Matsushita et
al., 1969b; Wigaeus et al., 1981). In male volunteers exposed by
inhalation to 1180-2350 mg/m3 (497 or 990 ppm) acetone for 4 h,
cumulative acetone excretion in urine at 18 h after cessation of
exposure was 89.5 mg, suggesting slow excretion of acetone in the
urine (Vangala et al., 1991). Again, the amount of acetone excreted in
the urine will be influenced by the exposure concentration, the
duration of exposure, and the level of physical activity during
exposure. The acetone concentration in the urine ranged from 0 to 17.5
mg/litre at the end of the 8-h work shift in 45 workers exposed to
0-165 mg/m3 (0-70 ppm) acetone (background urinary concentration in
343 non-exposed subjects averaged 1.5 mg/litre) (Kawai et al., 1992).
Acetone levels in the preshift urine samples were significantly higher
than background levels when acetone exposure on file previous day was
above 35 mg/m3 (15 ppm). There was no significant difference between
background urine levels and preshift urine levels when the previous
day's exposure was < 35 mg/m3 (< 15 ppm). Total 24-h urine
content of acetone was 1.25 mg in subjects exposed to 240 mg/m3 (100
ppm) for 2 h and 3.51 mg in subjects exposed to 1190 mg/m3 (500 ppm)
for 2 h (DiVincenzo et al., 1973). Prolonging the duration to 4 h in
the 240 mg/m3 group resulted in a total of 1.99 mg acetone in the
urine. A slight increase in the urinary content of acetone (1.39 nag)
was found when humans exposed to 240 mg/m3 for 2 h exercised during
the exposure. The nature of physical activity during exposure also
influenced the urinary excretion. At 3-3.5 h after exposure, 8.5, 8.5
and 13.4 mg were excreted by the kidney in subjects exposed at rest,
during exercise at a constant workload, and during exercise with
step-wise increments in workload, respectively (Wigaeus et al., 1981).
Urinary excretion amounted to only 1% of the total uptake.
In volunteers who ingested 40-60 mg/kg acetone, the elimination
of acetone in expired air and urine was determined 2 h later, and a
rate of metabolism of 1.82 mg/kg per h along with the excretion data
was used to calculate that 3.54-7.38 mg/kg had been excreted and
metabolized (Haggard et al., 1944). The authors estimated that 65-93%
of the administered dose was metabolized, the remainder being
eliminated in the urine and expired air.
The only other information regarding excretion of acetone in
humans after oral exposure is from case reports of accidental or
intentional ingestion of materials containing acetone plus other
components that may have influenced the elimination of acetone. In a
man who ingested liquid cement containing 18% acetone (231 mg/kg), 28%
2-butanone and 29% cyclohexanone, and 720 ml sake, the plasma level of
acetone was approx. 1120 mg/litre 5 h after ingestion and declined to
65 mg/litre at 18 h, 60 mg/litre at 24 h and < 5 mg/litre at 48 h
(Sakata et al., 1989). A first-order plasma elimination rate constant
of 0.038/h and a half-time of 18.2 h were calculated. The urinary
level of acetone decreased gradually from about 123 mg/litre at 5 h
after ingestion to about 61 mg/litre at 19 h. In a case of a known
alcoholic who had ingested nail polish remover and whose blood acetone
level was 2.5 g/litre) upon admission to the hospital, the blood level
of acetone declined in a log-linear manner to about 0.6 g/litre about
86 h after admission, with a half-life of 31 h (Ramu et al., 1978).
The calculated clearance of acetone from the lungs was 29 ml/min or
0.39 ml/min per kg. A half-time of 25 h for lung clearance was
calculated, which is in agreement with the observed plasma elimination
half-tune of 31 h. The serum acetone level of a 30-month-old child was
4.45 g/litre 1 h after ingestion of a 6-ounce bottle of nail polish
remover (65% acetone) and declined to 2.65 g/litre at 117 h, to 0.42
g/litre at 48 h, and to 0.04 g/litre at 72 h (Gamis & Wasserman,
1988). The half-time of acetone in this patient was 19 h in the severe
early stage and 13 h in later stages of intoxication, which suggested
to the authors greater metabolism and/or excretion in children,
compared with adults.
Information regarding excretion of acetone after dermal exposure
of humans is limited, but the main route of excretion is via the
lungs, with little excreted in the urine. Application of an
unspecified quantity of acetone to a 12.5 cm2 area of skin of
volunteers for 2 h/day for 4 days resulted in alveolar air levels of
12-30 mg/m3 (5-12 ppm) and urinary concentrations of 8-14 mg/litre
on each day (Fukabori et al., 1979). These levels declined to
background levels by the next day after each exposure. Higher alveolar
air and urinary levels were obtained when the daily exposure increased
to 4 h/day: 60-80 mg/m3 (25-34 ppm) in alveolar air and 29-41
mg/litre in urine. However, these levels also returned to background
each day.
As determined in humans (Reichard et al., 1979), physiological
status may influence the disposition of endogenous and exogenous
acetone. In groups of non-obese patients fasted for 3 days, obese
patients fasted for 3 days, and obese patients fasted for 21 days and
injected intravenously with 14C-acetone, 8-29% of the urinary
acetone was derived from plasma radioactive acetone (Reichard et al.,
1979). The concentrations of urinary acetone were 1.2, 0.4 and 151
mg/litre in 3-day-fasted non-obese, 3-day-fasted obese, and
21-day-fasted obese patients, respectively. The rates of urinary
acetone excretion were 2962, 1800 and 3542 µg/min, respectively,
suggesting marked renal reabsorption or back-diffusion. The percentages
of measured acetone production that could be accounted for by excretion
via the lungs were 14.7, 5.3 and 25.2%, respectively. The percentages
that could be accounted for by urinary excretion were 1.4, 0.6 and
1.3%, respectively. Cumulative excretion of 14C-carbon dioxide
during the 6-h turnover study periods accounted for 17.4, 21.5 and
4.9%, respectively. Thus, non-obese subjects fasted for 3 days
excreted more acetone at higher rates than did obese subjects fasted
for 3 days. However, excretion by the obese patients fasted for 21
days exceeded that by both 3-day-fasted groups. These differences are
probably related to the effect that the degree of starvation ketosis
has on the metabolism and overall disposition of acetone.
6.4.1.1 Occupational exposure studies
There are many reports in the literature regarding exposure to
solvents, many of which are confounded for risk assessment purposes by
exposure to a mix of several solvents. The following information takes
the essence of these studies to give the reader an understanding of
the various exposure scenarios and the measured acetone
concentrations.
Satoh et al. (1995) studied male shift workers at a Japanese
acetate fibre manufacturing facility. The first group was 110
acetone-exposed (AE) workers, while the control groups (CG) consisted
of 67 workers in the same factory area but not engaged in acetone
manufacturing (background concentration < 12 µg/m3; < 5 ppb).
Levels were measured in the breathing zone and in samples of alveolar
air, blood and urine. Mean levels were reported as 857 mg/m3 (361
ppm), 216 mg/m3 (91 ppm), 67 mg/litre and 37 mg/litre, respectively.
They evaluated whether exposure to acetone during the previous day
affects the biological monitoring value at the end of a work day, i.e.
urinary values of acetone from monitoring do not revert to levels seen
with normal background exposure. Matsushita et al. (1969a,b) observed
that humans exposed to 1190 mg/m3 (500 ppm) of acetone had higher
than background levels the next morning. According to Satoh et al.
(1995), the extent of previous exposure and the sampling time
influenced the biological monitoring value and were very critical.
Additionally, other factors such as alcohol consumption, could give
rise to more than 30 mg/litre of acetone in the urine. When the
exposure concentration of the workers was 595 mg/m3 (250 ppm), the
urinary level of acetone returned to background the next morning. When
the exposure concentration was raised to 1190 mg/m3 (500 ppm), the
following morning's acetone levels were higher than background. Wang
et al. (1994) reported similar results. The next morning, 16 h after
the end of a workshift, with a mean occupational exposure to acetone
of 336 mg/m3, blood and urinary concentrations of 3.5 mg/litre and
13.0 mg/litre, respectively, were much higher than levels in
non-exposed subjects.
Fujino et al. (1992) also investigated the relationship between
the environmental concentration and the concentration in urine,
alveolar air and blood. The environmental air concentration to which
each of the 110 subjects were exposed was closely correlated with all
body concentrations. The authors also found that urinary acetone
concentration is probably the best biological index of exposure
because it shows the strongest correlation.
Kawai et al. (1992) studied 45 acetone-exposed male workers
together with 343 non-exposed men to examine the quantitative
relationship between the levels of acetone vapour exposure and the
concentration of acetone in urine. Up to a vapour concentration of 35
mg/m3 (15 ppm), there was no increase in acetone concentration in
end-of-shift urine, but above this concentration there was an increase
in urinary acetone concentration in proportion to the acetone vapour
concentration. The authors reported that urinary acetone
concentrations collected at the shift-end and those before the shift
of the next morning were similar among those exposed to < 35
mg/m3 (15 ppm) acetone. However, among those exposed to acetone at
more than 35 mg/m3, acetone levels in the shift-end samples were
significantly higher than those in the pre-shift samples.
Mizunuma et al. (1993) and DeRosa et al. (1993) reported
different results in factory workers exposed to styrene and acetone.
Mizunuma et al. (1993) studied 41 workers in a factory making
fibre-reinforced plastics who were exposed to mixed styrene and
acetone vapour and 20 non-exposed workers. Acetone and styrene
concentrations in blood and urine correlated significantly with
intensity of exposure. Acetone was found to distribute evenly in blood
and urine, and was also distributed evenly between the cellular and
non-cellular fractions of the blood. DeRosa et al. (1993) monitored 22
workers exposed to styrene and acetone in two fibreglass industries to
determine if excretion of styrene metabolites differed over a working
week and if it was modified by simultaneous exposure to acetone.
Acetone exposure had no affect on styrene metabolism.
6.4.2 Experimental animal studies
As in humans, acetone is excreted mainly by the lungs of
experimental animals. Studies in animals have followed the elimination
of acetone from blood and tissues, excretion of acetone and carbon
dioxide in expired air, and the urinary excretion of formic acid.
Blood levels of acetone were highest immediately after a 4-h
exposure of rats to acetone (Charbonneau et al., 1986a). In rats
exposed to 23 700 mg/m3 (10 000 ppm), the blood level dropped from
2114 to 5 mg/litre in 25 h. In rats exposed to 35 600 mg/m3 (15 000
ppm), the blood level dropped from 3263 mg/litre to 50 mg/litre after
25 h. Elimination from blood was biphasic in rats exposed to 23 700
and 35 600 mg/m3. Elimination from blood was triphasic in rats
exposed to 2370, 5940 or 11 900 mg/m3 (1000, 2500 or 5000 ppm) and
was complete within 17-25 h. In dogs exposed to 240, 1190 or 2370
mg/m3 (100, 500 or 1000 ppm) acetone for 2 h, blood levels declined
in a log-linear manner with a half-time of 3 h, similar to that
observed m humans (DiVincenzo et al., 1973). Blood levels declined
from 25 mg/litre immediately after exposure to 10 mg/litre at 5 h
post-exposure for the 2370 mg/m3 (1000 ppm) group, from 12 to 3
mg/litre for the 1190 mg/m3 (500 ppm) group, and from 4 to 1.5
mg/litre for the 240 mg/m3 (100 ppm) group. Elimination of
radioactivity and 14C-acetone was fastest from blood, kidney, lungs,
brain and muscle tissues of mice exposed to 1190 mg/m3 (500 ppm)
14C-acetone for 6 h, with half-times of 2-3 h during 6 h
post-exposure (Wigaeus et al., 1982). Elimination of acetone was
complete in 24 h in all tissues, but radioactivity (indicative of
metabolites) was still present in all tissues except blood and muscle.
When rats were exposed for 5 days, acetone tended to slowly attain
steady state in adipose tissue.
Excretion of acetone in air followed pseudo-first-order kinetics
in rats exposed to up to 50 mg/m3 (< 20 ppm) acetone for 1-7 days,
while at higher concentration saturation kinetics were observed
(Hallier et al., 1981). In rats exposed to 1190 mg/m3 (500 ppm)
14C-acetone for 6 h, 42 µmol of radioactive acetone and 37 µmol
14C-carbon dioxide were excreted in the expired air during a 12-h
post-exposure period, 95% and 85%, respectively, being recovered in
the first 6 h post-exposure (Wigaeus et al., 1982). Radioactive
acetone accounted for 52% and radioactive carbon dioxide accounted for
48% of the expired radioactivity. The concentration of acetone in the
expired breath of dogs exposed to 240, 1190 or 2370 mg/m3 (100, 500
or 1000 ppm) acetone for 2 h declined in a log-linear manner
(DiVincenzo et al., 1973). The breath levels were directly related to
the magnitude of exposure. Breath levels declined from 3.8 mg/m3
(1.6 ppm) at 30 min after exposure to 0.7 mg/m3 (0.3 ppm) at 300 mm
in the 240 mg/m3 (100 ppm group), from 16.1 to 3.5 mg/m3 (6.8 to
1.5 ppm) in the 1190 mg/m3 (500 ppm) group, and from 35.6 to 9.5
mg/m3 (15 to 4 ppm) in the 2370 mg/m3 (1000 ppm) group.
Urinary excretion of formic acid was followed for 7 days in rats
exposed by inhalation to 147 200 mg/m3 (62 000 ppm) acetone for 2
days. The rate of formic acid excretion was 344 µg/h compared with 144
µg/h in controls (Hallier et al., 1981).
For experimental animals, information regarding the excretion of
acetone after oral exposure is available only for rats. As is the case
after inhalation exposure, acetone, mainly as carbon dioxide, is
excreted primarily by the lungs. In a rat given 1.16 mg/kg
14C-acetone by gavage in water, expiration of 14C-carbon dioxide
totalled 47.4% of the administered radioactivity over the 13.5-h
collection period (Price & Rittenberg, 1950). In another experiment, a
rat was given 7.11 mg/kg radioactive acetone. A small amount of
radioactive acetone (10%) was found in the expired air. Radioactive
carbon dioxide and acetate were also detected. In a rat made diabetic
by alloxan and given 6.15 mg/kg 14C-acetone, a total of 7.29% of the
administered radioactivity was expired as acetone and 51.78% as carbon
dioxide. Radioactive acetate was detected in the urine. These data
indicate that very little acetone (< 10%) was excreted by the lungs
after small doses of acetone. A major fraction was oxidized to carbon
dioxide and some of the derived carbon was used for acetylation. The
diabetic rat was also able to oxidize acetone, but only to approx. 70%
of that in the normal rat.
The dose of acetone influences the elimination of acetone from
blood (Plaa et al., 1982). At a dose of 78.44 mg/kg, the maximum blood
level of 200 mg/litre at 3 h declined to 10 mg/litre at 12 h, where it
remained for the next 12 h (data inadequate to evaluate total body
clearance). At a dose of 196.1 mg/kg body weight, the maximum blood
level of 400 mg/litre at 6 h declined biphasically to 50 mg/litre at
12 h and to 30 mg/litre at 18 h where it remained until 24 h (total
body clearance = 64 ml/h). At a dose of 784.4 mg/kg body weight, the
maximum blood level of 900 mg/litre at 1 h declined to 300 mg/litre at
12 h, to 110 mg/litre at 18 h, and to 50 mg/litre at 24 h (total body
clearance = 86 ml/h). At a dose of 1961 mg/kg body weight, the maximum
blood level of 1900 mg/litre at 3 h declined slowly to 400 mg/litre at
24 h (total body clearance = 75 ml/h). Thus total body clearance was
independent of dose, but the half-time for elimination increased from
2.4 h for 196.1 mg/kg to 4.9 h for 784.4 mg/kg, and 7.2 h for 1961
mg/kg.
The vehicle (corn oil or water) in which acetone is administered
has little influence on the elimination of acetone from blood
(Charbonneau et al., 1986a). After gavage treatment of rats with 78
196, 392, 784 or 1177 mg/kg acetone in corn oil or water, elimination
was biphasic for the two higher doses and triphasic for the lower
doses. Acetone elimination from blood declined to <5 to <10 µg/ml by
18-26 h at all dose levels, but minor differences were found between
water and corn oil as vehicle. The blood concentration curves from
rats given acetone in water more closely resembled those from rats
exposed by inhalation.
6.4.3 Elimination/excretion summary
Following single exposure, endogenous acetone is eliminated from
the body at low doses via metabolic pathways and at high doses via the
lungs with the urine as a minor pathway. In general, acetone is
eliminated from the body within 24 h, unless exposure is continuous or
intermittent.
At low exposure levels, acetone is primarily eliminated by
metabolism, while at higher exposure levels it is mainly eliminated by
the lungs. Very little acetone is excreted in the urine. The
elimination rate is highly dependent upon exposure level, duration of
exposure, metabolism, genetic and pathophysiological conditions, and
physical activity.
6.4.4 Physiologically based pharmacokinetic model
A physiologically based pharmacokinetic model has been developed
to study the kinetic behaviour of acetone (Kumagai & Matsunaga, 1995).
In this model, acetone can be distributed into eight tissue groups:
the mucous layer of the inhaled air tract, the mucous layer of the
exhaled air tract, a compartment for gas exchange (alveolus of the
lung), a group of blood-vessel-rich tissues including the brain and
heart, a group of tissues including muscles and skin that have low
blood perfusion rates, a group of fatty tissues, an organ for
metabolism (liver), and a compartment for urinary excretion (kidney).
With an appropriate value for the volume of mucous layer, the
simulated acetone concentration in arterial blood, and exhaled air,
urine, and fatty tissue were found to agree well with the experimental
data. The volume of mucous layer and rate of respiration were critical
for the appropriate simulation.
This model is suitable for occupational exposure assessments.
6.5 Retention and turnover
Reichard et al. (1979) studied acetone production in obese and
non-obese patients during starvation-induced ketonaemia. The
elimination and metabolism of acetone were measured, as well as urine
and plasma concentrations by radioactive labels in order to calculate
a plasma turnover rate for acetone. This was 20-77 µmol/m2 body
surface area per min. Owen et al. (1982) and Reichard et al. (1986)
furthered the original study in patients with severe diabetic
ketoacidosis. Owen et al. (1982) measured rates of acetone production
ranging from 68 to 581 µmol/min (using a standard human body surface
area of 1.73 m2), with plasma concentrations ranging from 1.55 to
8.91 mmol/litre. In the Reichard et al. (1986) study, the plasma
concentrations ranged from 0.50 to 6.02 mmol/litre, and the acetone
remover rate was linearly related to the plasma concentration up to a
level of 7.61 mmol/litre. The two studies found opposite relationships
between acetone turnover rate and plasma concentration.
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Short-term toxicity
A summary of short-term inhalation toxicity studies of acetone is
given in Table 8, acute oral studies are in Table 9, and dermal
studies in Table 10. In the studies on rats and guinea-pigs, very high
levels of acetone were required to cause death. In rats, a 4-h LC50
of 76 mg/m3 and a 8-h LC50 of 50.1 mg/m3 have been reported
(Pozzani et al., 1959). A range of LD50 values was found for rats.
Kimura et al. (1971) stated that the lethality of acetone decreases
with age to a certain point in the rat life cycle. However, the
difference in values from older to younger adult rats was not found to
be statistically significant.
In acute oral studies, signs of narcosis usually precede death
(ATSDR, 1994). In the EHRT (1987) study, mice that were dosed by
gavage at 4800 mg/kg per day or more for 10 days displayed wheezing
and rapid/laboured breathing. Narcosis was present before death.
Savage treated rats were found to have increased haematocrit and
haemoglobin in high-dose males (2500 mg/kg per day), but not in
females. As will be seen below, these effects change with increasing
dose and duration. Considering all the studies, it appears that there
are sex and species differences for haematological effects from
exposure to acetone.
Acetone is known to be only mildly toxic to the liver, unless
physiological processes are compromised (e.g., diabetes mellitus). It
is known to potentiate and antagonize the effects of other chemicals
by inducing microsomal enzymes that metabolize other chemicals to
reactive intermediates. Acetone has been shown to increase the
activity of glutathione S-transferase (Sippel et al., 1991 ).
Induction of microsomal enzymes has been considered as a normal
physiological response to xenobiotics when it does not cause other
adverse effects such as increased liver weight, enzyme changes or
other hepatic effects.
In the NTP (1991) study, Dietz and co-workers found increased
liver weights at doses > 965 mg/kg per day, and in higher dose
groups (> 3896 mg/kg per day) hepatocellular changes accompanied
this effect. A 14-day drinking-water study of F344/N rats and B6C3F1
mice served as a preliminary study to the 13-week NTP (1991) study
discussed in section 7.2. Five animals/sex of each species were
administered drinking-water at concentrations of 0, 5, 10, 20, 50 or
100 g/litre (NTP, 1991; Dietz et al., 1991). Based upon water
consumption data, the authors estimated doses as follows: 0, 714,
1616, 2559, 4312 and 6942 mg/kg per day for male rats; 0, 751, 1485,
2328, 4350 and 8560 mg/kg per day for female rats; 0, 965, 1579, 3896,
6348 and 10 314 mg/kg per day for male mice; and 0, 1569, 3023, 5481,
8804 and 12 725 mg/kg per day for female mice.
At necropsy, body weights were decreased, compared with controls,
in male rats exposed to concentrations of > 50 g/litre and female
rats exposed to 100 g/litre. The report for the 14-day study did not
provide the collected organ weight data but stated that increased
relative weights of kidneys and liver were measured for exposed rats
and mice (dose level was not specified). No histopathological changes
were observed in the kidneys or livers of exposed rats or in the
kidneys of mice. Centrilobular hepatocellular hypertrophy was noted in
ali male mice at concentrations > 20 g/litre and in female mice at
concentrations of > 50 g/litre (NTP, 1991; Dietz et al., 1991).
The hypertrophy was described as "minimal" in female mice, but
increased incidence was noted with increasing concentration 2/5 and
5/5 for 50 g/litre and 100 g/litre, respectively. The incidences of
male mice with more severe hypertrophy (classified by the authors as
either "mild" or "moderate") increased with increasing concentrations
> 20 g/litre. The authors hypothesized that male mice may develop
a tolerance to acetone because hepatic changes were not observed in
male mice in the second study, the 13-week NTP study presented in the
next section. Thus, in evaluating oral studies of the effects of
acetone, the data indicated that changes occurred in the liver. The
determination of whether or not these were adverse depends more on the
determination of the severity of the effects. Acetone induced
microsomal enzymes and increased liver weights. It caused liver injury
with increased serum levels of liver enzymes and hepatocellular
hypertrophy. As will be seen later, acetone potentiates other
chemicals, which may increase these adverse effects or potentiate
adverse effects caused by other chemicals. Oral exposure to acetone
has also caused effects on the kidney in mice and rats, as shown in
the NTP (1991) study (see also section 7.2). The best-known effects of
acetone ingestion are the diabetes-like symptoms of hyper-glycaemia
and glycosuria.
7.1.1 Skin and eye irritation
Smyth et al. (1962) applied 1.0 ml acetone to the shaved dorsal
skin of rabbits without occlusion. After 24 h there was no evidence of
irritation. In CD-1 mice, a single application of 0.2 ml acetone to
shaved skin produced increased DNA synthesis and moderate hyperplasia
of the epidermis that was considered to be evidence of slight
irritation, In hairless mice treated with twice weekly application of
0.1 ml acetone for 18 weeks, moderate hyperplasia of the epidermis was
noted (Iversen et al., 1988)
In rabbits, severe eye inflammation and corneal necrosis followed
instillation of 0.005 ml acetone into the eye (Carpenter & Smyth,
1946; Smyth et al., 1962). A 3-min application of 3.9 ml caused
conjunctival oedema (Larson et al., 1956).
Table 8. Short-term animal inhalation exposure to acetone
Species Exposure NOAELb LOAELb Critical effect Reference
duration/frequencya mg/m3(ppm) mg/m3(ppm)
Guinea-pig 2 days 23 700 (10 000) 5/5 died; spleen and lung congestion; Specht et al.
24 h/day fatty liver; renal tubular distension (1939)
Guinea-pig 22-26 h 47 500 (20 000) 8/9 died; congestion and haemorrhage Specht et al.
of spleen and lung (1939)
Guinea-pig 25 min-23.4 h 51 750 (21 800) 2/10 died; narcosis, paralysis Specht et al.
(1939)
Guinea-pig 3-8.75 h 118 700 (50 000) 8/8 died; pulmonary congestion, Specht et al.
oedema, glomerular distension (1939)
Rat 2 h 120 120 (50 600) 5/5 died Bruckner &
Peterson (1981)
Rat 3 h 29 900 (12 600) CNS depression Bruckner &
Peterson (1981)
Rat 4 h 76 000 (32 000) LC50 Pozzani et al.
(1959)
Rat 4 h 38 000 (16 000) 1/6 died Smyth et al.
(1962)
Rat 4h 2850 (1200) 7100 (3000) Audiogenic seizures depressed Frantik et al.
(1988)
Rat 8 h 50 100 (21 050) Pozzani et al.
(1959)
Rat 14 days 5200 (2200) 26 100 (11 000) Reduced body weight, uterine weight, NTP (1988)
7 days/week decreased fetal weight;
6 h/day
GD 6-19
Rat 2 weeks 7100 (3000) 14 240 (6000) inhibition of avoidance behaviour Goldberg et al.
5 days/week in over 1/3 of rats (1964)
4 h/day
Mouse 6h 26100(11000) Severe narcosis NTP (1988)
Mouse 1 day 2370 (1000) 7100 (3000) Decreased behavioura[ response Glowa & Dews
(1987)
Table 8. (Continued)
Species Exposure NOAELb LOAELb Critical effect Reference
duration/frequencya mg/m3(ppm) mg/m3(ppm)
Mouse 12 days 5200 (2200) 15 650 (6600) Decreased fetal weight, reduced NTP (1988)
7 days/week sternal ossification
6 h/day
GD 6-17
a GD = gestation day.
b Dose levels for all studies are included in this table or the text only.
Table 9. Short-term oral exposure to acetone
Species Route Exposure NOAEL LOAEL Critical effect Reference
duration/frequency (mg/kg) (mg/kg)
Rat drinking-water 3-7 days 3214 reduced insulin production, less Skutches et al.
serious systemic problems (1990)
Rat drinking-water 14 days 4312 (male) 6942 (male) bone marrow hypoplasia NTP (1991)
8560 (female)
Rat drinking-water 10 days 3500 induction of hepatic cytochrome Puccini et al.
P450, aminopyrineN-demethylase, (1989)
ethoxycourmarin O-deethylase,
p-nitrophenol hydroxylase, acetone
hydroxylase, diethyl-ngrosamine
deethylase
Rat drinking-water 14 days 3500 induction of dealkylation of Fiodo & Bronzetti
amino-pyrine; induction of dealkylation (1995)
of anisole, hydroxylation of aniline;
decreased survival of tissue culture
cells treated with
dimethylnitros-amine; increased mutation
rate of tissue culture cells treated with
dimethylnitrosamine
Rat gavage once -- 15 mmol/kg induction of hepatic P450EI, Barnett et al.
(871 mg/kg) P450IB, P45OIIB, P45OIA2 (1992)
Rat gavage once 1726 LD50 Kimura et al.
(newborn) (1971)
Rat gavage once 4393 LD50 Kimura et al.
(14 day) (1971)
Table 9. (Continued)
Species Route Exposure NOAEL LOAEL Critical effect Reference
duration/frequency (mg/kg) (mg/kg)
Rat(young gavage once 7138 LD50 Kimura et al.
adult) (1971)
Rat gavage once 6667 LD50 Kimura et al.
(older adult) (1971)
Rat gavage once 5806 LD50 15% body weight loss, Freeman &
prostration Hayes (1985)
Mouse drinking-water 10-12 days 3500 induction of hepatic cytochrome Puccini et al.
P450, cytochrome b5, (1990)
ethoxy-courmarin O-deethylase,
p-nitrophenol, hydroxylase,
diethylnitrosamine deethylase
Mouse drinking-water 14 days 1579 (male) 3896 (male) mild hepatocellular hypertrophy NTP (1991)
12 725 (female)
Mouse gavage with 10 days 3500 reduced maternal body weight; EHRT (1987)
water (GD 6-15) reduced reproduction index;
once/day decreased pup survival
Hamster water 10 days 2100 induction of cytochrome P450 Puccini et al,
(1992)
a GD = gestation day.
Dose calculated on the assumption taken from Puccini et al. (1992) that 1% acetone in drinking-water is equivalent to 700 mg/kg per day.
7.2 Longer term toxicity
A limited number of oral animal studies are reviewed in Table 10.
Only one longer-term dermal study was available.
Bruckner & Peterson (1981) exposed male ARS/Sprague-Dawley rats
to acetone vapour concentrations of 0 or 45 100 mg/m3 (0 or 19 000
ppm), 3 h/day, 5 days/week for < 8 weeks. Groups of five rats were
killed and examined at 2, 4 and 8 weeks and at 2 weeks after
termination of exposure. No significant (p<0.05) effects were
noted in blood chemistry (blood urea nitrogen (BUN), serum
glutamic-oxaloacetic transaminase (SGOT), lactate dehydrogenase (LDH),
liver triglyceride levels and histology of heart, lung, kidney, brain
and liver. Small, statistically significant (p<0.02) decreases in
absolute weights of the brain at 4 and 8 weeks and kidney at 4 weeks
(but not at 8 weeks) were observed compared with controls. Without
providing quantitative data, the authors stated that the organ-to-body
weight ratios were slightly increased, suggesting that the effect on
absolute organ weight reflected a slight but statistically
insignificant reduction in rate of body weight gain. Two weeks after
termination of exposure, no significant differences in organ weights
were observed between treated and control rats.
Sollman (1921) exposed three rats to 2.5% acetone in
drinking-water for 18 weeks and reported that body weights and
consumption of food and water were decreased, compared with normal
values; the report cited normal values for consumption of food and
fluid and for body weight, but did not indicate whether these were
historical control values or values from concurrent control animals.
No deaths were noted during the experimental period, No other
end-points were examined.
Acetone was administered by gavage (as an aqueous solution) to
groups of 30 Sprague-Dawley rats/sex for 90 days at dose levels of 0,
100, 500 or 2500 mg/kg per day (US EPA, 1986a). At the highest dose
level, significantly increased values of RBC parameters were measured
at 46 and 90 days for males (haemoglobin, haematocrit, corpuscular
volume) and at 90 days for females (haemoglobin, haematocrit), Females
in the 500 and 2500 mg/kg per day groups had significantly increased
absolute kidney weights. Both sexes showed increased relative weights
of kidney and brain and increased absolute and relative liver weights
at the 2500 mg/kg per day dose. Body weights were unaffected in males,
but transient, significantly elevated body weights were observed in
females in the 500 and 2500 mg/kg per day groups. Statistically
significant clinical chemistry alterations, possibly attributable to
acetone, included increased alanine aminotransferase activities in
both sexes at 2500 mg/kg per day and decreased platelet counts, serum
glucose and potassium levels in males at 2500 mg/kg per day.
Table 10. Longer-term oral exposure to acetone
Species Route Exposure NOAEL LOAEL Critical effect Reference
duratron/frequency (mg/kg/day) (mg/kg/day)
Rat gavage with water 46-47 days 500 (male) 2500 excessive salivation; US EPA (1986a)
(once/day) increased haemoglobin,
haematocrit and mean cell
volume
Rat gavage with water 93-95 days 500 (female) 2500 decreased brain weight; US EPA (1986a)
(once/day) increased blood parameters
(as above)
Rat water 13 weeks 900 (male) 1700 increased incidence/severity NTP (1991)
1600 (female) 3100 of nephropathy in males;
haematological effects
Mouse water 13 weeks 4858 (male) - - NTP (1991)
11 298 (female)
Histological examinations were made of all major tissues and organs
from control and high-dose rats. For the low-dose (100 mg/kg) and
middle-dose (500 mg/kg) rats, histological examinations were
restricted to the heart, liver, kidneys and any tissues with gross
changes. Increasing doses were associated with increasing intensity of
tubular degeneration of the kidneys and hyaline droplet accumulation.
The Task Group noted that these changes were observed in all groups,
including controls. These changes were accentuated in males at the 500
and 2500 mg/kg per day dose levels and in females at the 2500 mg/kg
per day dose level. No other chemical-related histological changes
were noted. There were no effects on urinalysis or ophthalmological
parameters examined before sacrifice. No acetone-related effects were
seen at the 100 mg/kg per day dose level.
In a 13-week drinking-water study (NTP, 1991; Dietz et al.,
1991), groups of 10 F344/N rats of each sex and 10 female B6C3F1
mice were administered drinking-water containing 0, 2.5, 5, 10, 20 or
50 g/litre acetone. Groups of 10 male mice were exposed to
drinking-water containing 0, 1.25, 2.5, 5, 10 or 20 g/litre.
Corresponding TWA doses in treated animals were estimated by the
authors as 200, 400, 900, 1700 and 3400 mg/kg per day for male rats;
300, 600, 1200, 1600 and 3100 mg/kg per day for female rats; 892,
2007, 4156, 5945 and 11 298 mg/kg per day for female mice; and 380,
611, 1353, 2258 and 4858 mg/kg per day for male mice.
Necropsy was performed on all animals and histological
examinations were conducted on all major tissues and organs of animals
from control and high-dose groups. For lower dose groups, tissues
examined included heart, kidneys and spleen of male rats at the 10 and
20 g/litre dose levels; kidneys of male rats at 5 g/litre; bone marrow
of male rats at 20 g/litre; nasal cavity and turbinates of all groups
of female rats; mandibular lymph nodes of female rats at 20 g/litre;
and liver of all female mice. At necropsy, organ weights were obtained
and haematological examinations and examinations of sperm morphology
and vaginal cytology were performed.
No mortalities or overt clinical signs of toxicity were observed
for any of the rats within the experimental period. Depressed water
consumption and body weight gain were noted in rats of both sexes at
the highest concentration. Significantly increased (p<0.05) relative
weights of kidney, liver and lung were noted in rats of both sexes
exposed to 50 g/litre. Relative testis weights were also increased at
the highest concentration. Also noted at 50 g/litre were significant
decreases in caudal and epididymal weights and in sperm motility and
increased counts of morphologically abnormal sperm. Administration of
20 g/litre was associated with increased relative liver weights in
both sexes and increased relative kidney weights in female rats,
compared with controls.
"Mild but statistically significant" (p<0.05) alterations in
haematological parameters (haematocrit, haemoglobin and mean
corpuscular haemoglobin) were noted in rats of both sexes given 50
g/litre (increased leukocytes and lymphocyte counts, mean corpuscular
haemoglobin and mean cell volume, and decreased platelet counts) (NTP,
1991; Dietz et al., 1991). At 20 g/litre, haematological alterations
were restricted to male rats with the exception that females also
showed decreased platelet counts. The haematological alterations in
males were accompanied by increased pigmentation in the splenic red
pulp of all males exposed to concentrations >20 g/litre, compared with
controls.
"Mild" nephropathy was noted in male rats, including controls.
The incidence and intensity of the kidney lesions increased with
increasing drinking-water concentrations of acetone, particularly at
20 g/litre. However, the criteria for distinguishing between "minimal"
and "mild severity" for the observed nephropathy were not described.
Based on accentuated nephropathy in male rats, the LOEL was considered
by the Task Group to be 1700 mg/kg per day.
During the 13-week exposure of mice to drinking-water containing
acetone, no deaths or clinical signs of toxicity were recorded (NIP,
1991; Dietz et al., 1991). Body weights, growth, sperm morphology and
vaginal cytology also were unaffected in exposed mice. Significant
organ weight changes (p<0.05) were restricted to female mice
administered 50 g/litre; absolute and relative weights were increased
for liver and decreased for spleen. Small changes in a few
haematological parameters were noted in female mice (increased
haematocrit and haemoglobin) and male mice (increased haemoglobin and
mean corpuscular haemoglobin) exposed to their highest respective
concentrations. The authors did not consider the changes to be
toxicologically significant. The only histopathological alteration
noted for acetone-exposed mice was centrilobular hepatocellular
hypertrophy (liver cells with abundant eosinophilic cytoplasm and
slightly enlarged nuclei) in 2 of the 10 female mice administered 50
g/litre.
7.3 Reproductive toxicity, embryotoxicity and teratogenicity
NTP (1988) examined the in vivo developmental toxicity of inhaled
acetone in mice and rats. Groups of pregnant Sprague-Dawley rats
(26-29 rats/group) were exposed to 0, 1045, 5200 or 26 100 mg/m3 (0,
440, 2200 or 11 000 ppm) acetone vapour, 6 h/day, 7 days/week for 14
days (days 6-19 of gestation). Groups of pregnant Swiss CD-1 mice
(28-31 mice/group) were exposed to 0, 1045, 5200 or 15 670 mg/m3 (0,
440, 2200 or 6600 ppm) with the same protocol for 12 days (days 6-17
of gestation). Maternal body weights were obtained throughout the
experimental period. Uterine and fetal body weights were recorded at
sacrifice (gestation days 20 and 18 for rats and mice, respectively).
Live fetuses were examined for gross, visceral, skeletal and soft
tissue craniofacial defects.
No signs of maternal toxicity were noted in the pregnant rats
except that statistically significant reductions in body weight at 14,
17 and 20 days gestation, in cumulative body weight gain and in
uterine weight were observed for the 26 100 mg/m3 group. The
following were unaffected by exposure to acetone: absolute and
relative liver and kidney weights of the dams; number of
implantations; percentage of live pups/litter; percentage of
resorptions/litter; and the fetal sex ratio. Fetal weights were
significantly decreased in the 26 100 mg/m3 group compared with the
control group. The incidences of fetal malformations were not
significantly increased in any of the acetone-exposed groups compared
with controls, although the percentage of litters with at least one
malformed pup was greater in the 26 100 mg/m3 group (11.5%) compared
with the control group (3.8%). NTP (1988) concluded that acetone had
not caused a teratogenic effect in the rats in this study.
The only significant acetone-related effect in the pregnant mice
was increased relative liver weights in the 15 670 mg/m3 group
compared with controls. Developmental and reproductive indices were
unaffected in the acetone-treated groups of mice with the exceptions
of a statistically significant decrease in fetal weight and a slight,
but statistically significant, increase in the percentage incidence of
late resorptions, both in the 15 670 mg/m3 group of mice. The
authors concluded that slight developmental toxicity occurred at the
respective highest exposure levels in the studies of Swiss mice and
Sprague-Dawley rats, and that 5200 mg/m3 (2200 ppm) was the
no-observed-effect level (NOEL) for maternal and developmental
toxicity in both species (NTP, 1988).
Ten male Wistar rats were exposed to 11 870 mg/m3 (5000 ppm)
acetone in drinking-water for 8 weeks (Larsen et al., 1991). During
the sixth week, the treated males were mated with untreated females.
Numbers of pregnancies, numbers of fetuses and testes weights were
recorded and histopathology of the seminiferous tubules and testes was
undertaken. Acetone appeared to have no effects on these parameters.
Negative and positive results have been obtained in in vitro
tests of the teratogenic potential of acetone. Guntakatta et al.
(1984) reported that acetone at concentrations as high as 100 mg/ml
did not alter the cellular incorporation of radiolabelled sulfate or
thymidine in an in vitro mouse embryo limb bud culture system.
In vitro growth of rat embryos also was unaffected by the presence
of 0.1 or 0.5% acetone (v/v), but the presence of 0.5% caused
increased incidence of morphological abnormalities in the embryos;
2.5% caused embryo death (Kitchin & Ebron, 1984).
In a 13-week drinking-water study (NTP, 1991 Dietz et al., 1991),
small changes in sperm motility and incidence of morphologically
abnormal sperm were noted in rats exposed to concentrations of 50
g/litre (3400 mg/kg per day). Reproductive performance of these
animals, however, was not examined.
EHRT (1987) evaluated acetone in a reproduction screening test
using mice. Acetone in distilled water was administered by gavage to
groups of 50 mated CD-1 mice on days 6-15 of gestation at doses of 0
or 3500 mg/kg per day. Parameters of toxicity evaluated included
maternal toxicity (mortality, body weight and clinical signs), number
of live and dead offspring, pup body weight at birth, survival and
litter weight at postpartum day 3. Although two treated dams exhibited
clinical signs and eventually died, the authors did not consider that
maternal mortality had been increased by treatment with acetone.
Survivors exhibited no clinical signs or effects on body weight.
Effects attributed to acetone included decreased reproductive index,
increased gestation length, reduced birth weights, decreased neonatal
survival and increased neonatal weight gain.
7.4 Mutagenicity
Table 11 presents relevant genotoxicity studies and their
corresponding data for acetone. The data are almost entirely negative;
only one report of positive genetic activity was located. Exposure of
Saccharomyces cerevisiae to approx. 7% concentrations caused
aneuploidy (Zimmermann et al., 1985). Abbondandolo et al. (1980)
reported no forward mutations in a test on
Schizosaccharomyces pombe. In prokaryotes, acetone did not induce
reverse mutations in Salmonella typhiraurium (McCann et al., 1975;
NTP, 1991). Acetone did not induce DNA-cell binding with
Escherichia coli or ascites cells (Kubinski et al., 1981). Negative
results were reported for assays in animal systems in vitro and
in vivo, including mutation at the TK locus in mouse lymphoma cells
(Amacher et al., 1980), sister chromatic exchange or chromosome
aberrations in Chinese hamster ovary cells and human lymphocytes
(Norppa et al., 1981; NTP, 1991), cell transformation in Fischer rat
embryo cells (Freeman et al., 1973) and micronucleated erythrocytes in
mice and Chinese hamsters (Basler, 1986; NTP, 1991).
Table 11. Genotoxicity testing of acetone
Assay Indicator organism Application Purity Concentration Activating Response Comment Reference
(%) or dose system
Reverse Salmonella typhimurium plate NR 10, 100, 1000, +/- Aroclor-induced -/- NC McCann et al.
mutation TA1535, TA1537, TA98, incorporation 10 000 µg/plate rat liver S9 (1975)
TA100
Reverse S. typhimurium, TA97, liquid NR <10mg/plate +/-Aroclor-induced -/- NC NTP (1991);
mutation TA98, TA100, TA1535, suspension rat, Syrian hamster Dietz et al.
TA1537 liver S9 (1991)
Forward Schizosaccharomyces liquid NR 3.75% +/-phenobarbital- -/- 3ml 5% Abbondandolo
mutation pombe suspension induced mouse acetone et al. (1980)
plus 1 ml
yeast
culture
Aneuploidy Saccaharomyces liquid >97 6.98, 7.41, - + Expression Zimmermann
cerevisiae diploid suspension 7.83% of et al. (1985)
D61.M aneuploidy
required
overnight
incubation
on ice
Forward mouse lymphoma cell culture NR 0.134-0.421 - - Cytotoxicity Amacher
mutation L5178Y TK +/- cells mol/litre prevented et al. (1980)
use of
higher
concentrations
Sister Chinese hamster ovary cell culture NR <5000 +/- Aroclor-induced - NC NTP (1991)
chromatid cells mg/litre rat liver S9 Dietz et al.
exchange (1991)
Table 11. (Continued)
Assay Indicator organism Application Purity Concentration Activating Response Comment Reference
(%) or dose system
Chromosomal Chinese hamster ovary cell culture NR <5000 mg/lltre +/- Aroclor-induced NTP (1991);
aberrations cells rat liver S9 - NC Dietz
et al.(1991)
Cell Rauscher leukaemia cell culture NR 0.001-100 - - NC Freeman et al.
transformation virus/Fischer rat mg/litre (1973)
embryo cells
DNA-cell binding of E. coli liquid NR 50, 100 mg/litre +/- S9 - Radiolabelled Kubinski
binding DNA to suspension DNA is et al
assay Ehrlich ascites or incubated (1981)
E. coli cells with cells in
the presence
of test
substance
Sister cultured human whole blood 99.5 2.7, 12.8 - - NC Norppa et al.
chromatid lymphocytes cell culture mmol/litre (1981)
exchange
Chromosomal cultured human whole blood 99.5 12.8 - - NC Norppa
aberrations lymphocytes cell culture mmol/litre et al. (1981)
Micronucleus mouse peripheral drinking-water NR 5-20 g/litreb NA - NC NTP (1991)
test blood normochromatic Dietz et al
or polychromatic (1991)
erythrocytes
Micronucleus Chinese hamster intraperitoneal NR 865mg/kgc NA - NC Basler (1986)
test polychromatic injection
erythrocytes
a NA = Not applicable; NC = no comment; NR = not reported.
b For 13 weeks.
c Single dose.
The negative results in the in vivo bone marrow micronucleus
tests in mice and Chinese hamsters suggest that the aneuploidy which
was detected in yeast is not expressed in vivo in mammalian cells.
As all of the other mutagenicity tests (covering a range of end-points
including gene mutations and clastogenicity) were negative, it is
concluded that acetone presents no mutagenic risk to humans.
Acetone is widely used as a solvent in genotoxicity studies.
There are no indications that acetone interacts with other chemicals
to alter their genotoxic potential (ATSDR, 1994).
7.5 Carcinogenicity
There are no studies available on the carcinogenicity of acetone
by either inhalation or oral dosing. Acetone has been used extensively
as a solvent vehicle in skin carcinogenicity studies (NTP, 1991).
Generally, acetone is not considered to cause or promote tumours when
applied to the skin, but comprehensive examinations of tissue sites
remote from the dermal site of application are not murine in these
studies (Ward et al., 1986; NTP, 1991).
In cultured Syrian hamster embryonic cells, there was no evidence
of cellular transformation when the cells were cultured in the
presence of 0.02% acetone (DiPaolo et al., 1969).
7.6 Immunotoxicity
In an investigation of acetone as a solvent vehicle for skin
studies, Singh et al. (1996) examined the effects of topically applied
acetone on immune function in SSIN mice. Acetone (200 µl) was applied
to the dorsal trunk four or eight times. Responses in the sheep red
blond cell plaque-forming assay were measured at the end of treatment.
Responses were depressed in the 4x group but not in the 8x group,
suggesting a temporary effect on humoral immunity. Plaque-forming
response was further investigated using 50, 100, 200 and 300 µl
acetone with one, four or eight applications. Response was depressed
in mice treated one, four or eight times with 300 µl acetone. There
were no changes in spleen cellularity, the CD4+ to CD8+ T cell
ratio, or the alloantigen induced mixed lymphocytic response.
In vitro proliferative responses to the mitogen concanavalin A were
increased in the 200 µl 4x or 8x groups. The authors concluded that
acetone can modulate humoral immunity.
The effects of topically applied acetone on systemic immune
function were analysed by Singh et al. (1996). SSIN mice, derived by
inbreeding Sencar mice, were exposed to four or eight topical
applications of 200 µl acetone. Plaque-forming assays were used to
assess the effects of solvents on the development of humoral immunity.
The responses in the 8x-treated group were indistinguishable from the
controls, while in the 4x-treated group a retardation in the
development of splenic B cells secreting IgM against SRBC occurred,
The rate of loss of plaque-forming cells (PFCs) was not affected. A
functional suppression of the development of humoral immunity had
occurred. The researchers then assessed plaque-forming ability using
50, 100, 200 and 300 µl of acetone in order to determine the effects
of solvent dose and duration of treatment on the development of
humoral immunity. In the 300 µl group, plaque formation 4 days after
immunization was statistically suppressed in mice treated one, four or
eight times. At the other concentrations, suppression of PFC
development was schedule-dependent. The suppression was seen more
after the first or fourth application, but the suppressive activity
was lost following the eighth dose. While the reason is not known,
Singh et al. (1996) speculated that it may be an adaptive response by
the skin.
Singh et al. (1996) showed that the modulation of humoral
immunity was also dependent upon the nature of the antigen used for
immunization. The capacity of the T cells to respond and to function
following different stimuli does not appear to he suppressed in
acetone-treated mice.
7.7 Special studies
Vodicková et al. (1995) studied the inhibition of electrically
evoked seizures in male albino SPF Wistar rats (ages 0.5 to 1 year)
and female albino H-strain mice (ages 24 months) exposed to acetone
concentrations of 3630 mg/m3 (1530 ppm) (mice) and 4035 mg/m3 (1700
ppm) (rats) for 2 h (mice) and 4 h (rats). The central nervous system
(CNS) effect of inhibition of electrically evoked seizure discharge
was measured immediately after exposure. Blood levels were also
monitored, Frantik et al, (1994) used the same methodology and
measured blood concentrations of 1190 mg/m3 (500 ppm) for mice and
8300 mg/m3 (3500 ppm) for rats. In another study Frantik et al.
(1996) exposed rats to constant or fluctuating air concentrations of
acetone and measured inhibition of electrically evoked seizures.
Four-hour exposures to air concentrations of 4 and 10 mg/litre
resulted in blood levels of 183 and 520 mg acetone/line blood,
respectively. Inhibition of seizures at these blood levels was 10% and
50%, respectively. There were no significant differences between
constant or fluctuating inhalation exposures. If a concentration
depresses seizure discharge, then generally it will inhibit behaviour
in higher doses and provoke sleep and narcosis in still higher doses.
In these studies, it appears that the acetone levels were below the
concentration evoking behavioural inhibition. Furthermore, Vodicková
et al. (1995) found that when mice were exposed to acetone and toluene
in a binary mixture, the inhibiting neurotropic effect of the exposure
was not increased, and when rats were exposed, the effect
significantly decreased. The decline of blood toluene and xylene
levels was slowed down by a simultaneous exposure to acetone. The
authors of the studies concluded that the effects of solvents appear
to be less than additive. The significance of these studies is
uncertain.
7.8 Factors modifying toxicity; toxicity of metabolites
Primarily, factors that may affect endogenously produced acetone
toxicity are alterations in carbohydrate metabolism, accumulation of
ketone bodies, interaction with ketone body metabolism, and
interaction with ketone bodies. There are many secondary factors, such
as an imbalance of hormones, e.g., insulin, that can effect the body
burden and hence the toxicity of acetone (Ramu et al., 1978).
Acetone can induce the enzymatic activity of a cytochrome P450
isozyme, one which plays a role in the metabolism of endogenous and
exogenous substrates. Acetone is metabolized by the same P450 isozyme
that is induced during higher doses, thus making a homeostatic-type
mechanism for decreasing acetone levels when higher body burdens
develop.
Because acetone is non-ionic and miscible with water, it can
passively diffuse across cell membranes. Normally, metabolism is the
principal route of elimination from the body, and this metabolic
breakdown is either through an intrahepatic or extrahepatic pathway.
The metabolites of acetone include carbon dioxide, acetate, formate,
glucose and 1,2-propanediol, with pyruvate and other compounds as
intermediates. It does not appear that any of these compounds affect
the toxicity of acetone. However, as shown in section 7.8, acetone can
affect the toxicity of other compounds.
Ruddick (1972) reviewed the toxicology, biochemistry and
metabolism of 1,2-propanediol. The compound is used as a solvent for
flavouring material in baking and candy production, as well as a
humectant and preserver to keep packaged foods fresh. Oral LD50
values of 1,2-propanediol in the rat are 21-30 mg/kg body weight, in
the mouse 23.9, in the rabbit 18.0-19.0, in the guinea-pig 18.9, and
in the dog 18.9-20.0.
Another possible and relatively non-toxic metabolite is isopropyl
alcohol. Lewis et al. (1984) showed that metabolism of acetone was
different in normal and diabetic rats. These experiments indicated
that high levels of blood acetone resulted in transformation to
isopropyl alcohol.
7.9 Mechanisms of toxicity - mode of action
Within the liver, acetone is metabolized by three separate
gluconeogenic pathways through several intermediates, but most of its
intermediate or final metabolites are not considered toxic.
Unmetabolized acetone does not appear to accumulate in any tissue, but
is excreted mainly in the expired breath following high (> 1180
mg/m3, > 500 ppb) exposures. Acetone is irritating to mucous
membranes, possibly due to its lipid solvent properties, resulting ha
eye, nose, throat and lung irritation following exposure to the
vapour, and skin irritation upon dermal contact.
Systemically, acetone is moderately toxic to the liver and
produces haematological effects. The mechanism by which acetone
produces these effects is unknown. The renal toxicity may be due to
the metabolite, formate, which is known to be nephrotoxic (NTP, 1991)
and is excreted by the kidneys (Hallier et al., 1981). Furthermore,
the renal toxicity, which appears to be specific for male rats, may
involve the alpha 2u-globulin syndrome, as hyaline droplet formation
was associated with the nephropathy observed in male rats in the US
EPA (1986a) study. Acetone also causes increases in liver and kidney
weight, probably through the induction of microsomal enzymes, which
would increase the weight of the organs by virtue of the increased
protein content. Acetone causes effects in the testes of male rats and
is fetotoxic at high (approx. 15 670 mg/m3, approx. 6600 ppm)
concentrations. Although the exact mechanism for many of the effects
of acetone is not known, distribution studies in mice indicate that
acetone and metabolites arc found in all of the target organs (Wigaeus
et al., 1982). Acetone and some of its metabolites were also
transferred to rat fetuses after the dams were exposed to acetone
(Peinado et al., 1986).
One of the major effects of acetone is the potentiation of the
toxicity of other chemicals. Pretreatment with acetone has been shown
to potentiate the hepatotoxicity and nephrotoxicity of carbon
tetrachloride and chloroform (Plaa & Traiger, 1972; Traiger & Plaa,
1972, 1974; Sipes et al., 1973; Plaa et al., 1973, 1982; Folland et
al., 1976; Hewitt et al., 1980, 1987; Brown & Hewitt, 1984;
Charbonneau et al., 1985, 1986a,b, 1988, 1991) by inducing particular
forms of cytochrome P-450, especially cytochrome P-450IIE1, and
associated enzyme activities (Johansson et al., 1988; Brady et al.,
1989; Kobusch et al., 1989). The induction of these enzymes leads to
the enhanced metabolism of carbon tetrachloride (CCl4) and chloroform
to reactive intermediates capable of causing liver and kidney injury.
Acetone enhances the formation of carboxyhemoglobin by dichloromethane
via induction of cytochrome P-450IIE1, leading to enhanced metabolism
of dichloromethane to carbon monoxide (Pankow & Hoffmann, 1989).
Acetone, methyl ethyl ketone and methyl isobutyl ketone pretreatment
was made in rats at a dosage of 6.8 mmol/kg given daily for 3 days.
Acetone markedly potentiated CCl4-induced liver toxicity as indicated
by a decrease in the CC14 ED50 to 3.4 mmol/kg compared to
vehicle-pretreated rats (17.1 mmol ketone/kg). Pretreatment with
acetone also potentiated chloroform kidney toxicity but to a lower
degree; chloroform ED50 values for vehicle- and acetone-pretreated
rats were 3.4 and 1.6 mmol/kg, respectively (Raymond & Plaa, 1995a).
In a subsequent study, Raymond & Plaa (1995b) examined the role
of monooxygenases induced by ketones as a mechanism for potentiating
the CCI4 hepatotoxicity and chloroform nephrotoxicity. Hepatic and
renal monooxygenase activities (aminopyrine and benzo-phetamine
N-demethylase, aniline, hydroxylase) from rats pretreated with
acetone or other ketones (6.8 or 13.6 mmoL/kg) were increased. This
profile of induction was consistent with the ketone potentiation
potency ranking profile observed in vivo for liver but not kidney
injury (Raymond & Plaa, 1995a).
Acetone also potentiates the hepatotoxicity of acetaminophen
(Moldetts & Gergely, 1980; Jeffery et al., 1991; Lin et al., 1991),
N-nitrosodimethylamine and N-nitrosodiethylamme (Sipes et al.,
1978; Lorr et al., 1984; Hong & Yang, 1985), thiobenzamide (Chieli et
al., 1990), oxygen (Tindberg & Ingelman-Sundberg, 1989), chromate
(Ct[VI]) (Mikalsen et al., 1991), and benzene (Johansson et al., 1988;
Johansson & Ingelman-Sundberg, 1988; Schnier et al., 1989); the
genotoxicity of N-nitrosodimethylamine (Glatt et al., 1981; Yoo &
Yang, 1985; Yoo et al., 1990); and the lethality of acetonitrile
(Freeman & Hayes, 1985, 1988) by inducing cytochrome P-450IIE1. The
hepatotoxic and nephrotoxic effects of dibromochloromethane and
bromodichloromethane (Hewitt & Plaa, 1983) and the hepatotoxic effects
of 1,1,2-trichloroethane (MacDonald et al., 1982a,b),
1,1-dichloroethene (Jaeger et al., 1975; Hewitt & Plaa, 1983) and
dichlorobenzene (Brondean et al., 1989) are also enhanced by acetone.
The details of the mechanisms for these interactions are not clear,
hut the involvement of mixed-function oxidases has been implicated.
The renal toxicity of N-(3,5-dichlorophenyl)succinimide (a
fungicide) is potentiated by acetone via the induction of cytochrome
P-450IIE1 (Lo et al., 1987). Acetone in drinking-water increased lung
toxicity of inhaled styrene (Elovaara et al., 1990). Ethanol at 10% in
drinking-water had no adverse effect on mice, hut when 5% acetone was
added, the mice lost weight and there were lipid droplets in
hepatocytes (Forkert et al., 1991).
In other interactions, acetone enhances the neurotoxicity of
ethanol by a proposed mechanism whereby acetone inhibits the activity
of alcohol dehydrogenase, a reaction responsible for 90% of the
elimination of ethanol (Cunningham et al., 1989). Acetone also
potentiates the neurotoxieity and reproductive toxicity of
2,5-hexanedione (Ladefoged et al., 1989; Lam et al., 1991; Larsen et
al., 1991). The exact mechanism for these interactions is not clear
but appears to involve decreased body clearance of 2,5-hexanedione by
acetone (Ladefoged & Perbellini, 1986). Acetone also antagonizes in
that it decreases the incidence of liver necrosis in male Long-Evans
rats treated with acetaminophen (Price & Jollow, 1983). The antagonism
is possibly due to increased glutathione conjugation. No reasonable
explanation was offered for the apparent ability of acetone to
potentiate acetaminophen toxicity in vitro and antagonize the
hepatotoxicity in vivo (Morgott, 1993).
It appears that the potentiating effects of acetone may be
occurring by any of three different mechanisms:
a) interference with uptake or elimination;
b) induction of microsomal enzymes, particularly cytochrome
P450IIE1;
c) additive interactions at the target site or toxic receptor
protein.
The uneventful use of acetone as a carrier solvent in many
in vitro genotoxicity assays that utilize microsomal activating
enzymes suggests the third mechanism does not operate in vitro.
8. EFFECTS ON HUMANS
8.1 Effects on humans
Acetone is produced endogenously within the human body during
metabolism. It is released into the atmosphere by soil and water from
a very wide range of natural and anthropogenic sources and is present
in many household products. All humans will, therefore, be exposed to
additional exogenous acetone in varying quantities, depending on
specific circumstances (see sections 3 and 5).
8.1.1 Non-occupational exposure
The toxicity from inhalation and dermal/ocular exposure is listed
in Tables 12 and 13.
The 1991 Annual Report of the American Association of Poison
Control Centers National Data Collection System documented 1137
incidents of human exposure to acetone (Litovitz et al., 1992). This
same report listed 1001 cases for 1988. Of these incidents, 1124 were
due to accidental or intentional ingestion (the others were not
clearly specified). No fatalities were reported, only three cases had
a major acetone-related medical problem, 364 were treated in a health
care facility, 233 cases were referred to hospitals but had no
effects, 367 cases suffered minor effects, and 39 suffered from
moderate effects. According to the classification of the AAPCC, none
of the major, minor or moderate effects were further described, and
the outcomes of the remainder of the incidents were not reported.
Matsushita et al. (1969b) exposed 25 male volunteer students to
acetone vapour for 3 h in the morning and 3 h in the afternoon during
1 day at concentrations of 240, 590, 1190 and 2400 mg/m3 (100, 250,
500 and 1000 ppm). The subjects were asked to describe any subjective
symptoms, and most reported that exposure to concentration of 1190 and
2400 mg/m3 for 6 h was irritating to the nose, eyes, throat and
trachea. At concentrations of 240 and 590 mg/m3 only a few of the
subjects complained of symptoms. Subjective symptoms also included the
loss of the ability to smell acetone as exposure proceeded. In another
controlled experiment, volunteer subjects were exposed to acetone
concentrations of 475, 720 and 1190 mg/m3 (200, 300 and 500 ppm) for
3-5 min in a chamber. Eye and throat irritation was experienced at 720
and 1190 mg/m3, but the subjects estimated that they could tolerate
an exposure level of 475 mg/m3 for an 8-h workshift (Nelson et al.,
1943). Volunteers exposed to 1000 ppm acetone for 4 h reported more
subjective complaints of throat irritation and "annoyance" (Seeber et
al., 1992). However, the perceived irritation or annoyance associated
with occupational and experimental exposures to acetone may be
influenced by the perceived odour of acetone and unrelated to
irritation and symptoms (Dalton et al., 1997; Wysocki et al., 1997).
Pulmonary function testing of volunteers exposed to < 2970 mg/m3
(< 1250 ppm) intermittently for various durations in a complex
protocol revealed no abnormalities caused by the exposure (Stewart et
al., 1975). The volunteers experienced sporadic throat irritation.
Table 12. Short-term human inhalation exposure to acetone
Exposure NOEL LOEL Critical effect Reference
duration/frequency mg/m3 (ppm) mg/m3 (ppm)
3-5 min 475 (200) 1190 (500) eye, nose and throat Nelson et al. (1943)
irritation
5.25 h 240 (100) 590 (250) eye, nose and throat Matsushita et al.
(6 h/day with 45-min break irritation (1969b)
at 590 mg/m3
5.25 h 1190 (500) increased WBC and Matsushita et al.
(6 h/day with 45-min break) eosinophil count, (1969b)
decreased phagocytic
activity of neutrophils
4 h approx 560 behavioural changes Dick et al. (1989)
(approx 237)
7 days (8 h/day) 2370 (1000) headache, dizziness, Raleigh & McGee
confusion (1972)
5 days (7.5 h/day) 2370 (1000) 2970 (1250) visual evoked response Stewart et al. (1975)
changes
Table 13. Short-term human skin and eye exposure to acetone
Exposure Target NOEL LOEL Critical effect Reference
duration/frequency mg/m3 (ppm)
vapour: 3-5 min skin/eyes 475 (200) 1190 (500) eye irritation Nelson et al. (1943)
liquid: 30 or 90 min skin 1 ml degenerative changes Lupulescu et al.
in epidermis (1973)
liquid: 90 min skin 1 ml decreased protein Lupulescu &
synthesis Birmingham (1975)
vapour: 2-3 days skin/eyes 2140 mg/m3 (901) eye irritation Raleigh & McGee
(8 h/day) (1972)
vapour: 7 days skin/eyes 2390 mg/m3 (1006) eye irritation Raleigh & McGee
(8 h/day) (1972)
vapour: 4 and 8 h skin/eyes 2375 (1000) no complaints of Seeber et al. (1993)
skin or eye
irritation
vapour: 4 h skin/eyes 2350 (990) perceived irritation Seeber et al. (1992)
of nose and throat
and "annoyance"
vapour: 4 and 8 h CNS 2375 (1000) mood change Seeber et al. (1993)
In the USA, the National Institute for Occupational Safety and
Health (Stewart et al., 1975) sponsored a research study to determine
physiological responses by humans exposed to acetone in air, and to
develop a biological test to indicate the magnitude of exposure.
Twenty adults of both sexes were exposed to acetone vapour
concentrations of 0, 475, 2370 and 2970 mg/m3 (0, 200, 1000 and 1250
ppm) for periods of 3 or 7.5 h for various durations up to 5 days. The
results of this study showed that a predictable excretion pattern
resulted for each of the tested vapour concentrations, and the rate of
excretion of acetone in the breath was a function of the duration of
exposure. After 3 h of exposure, the majority of the subjects could no
longer detect the odour of acetone when breathing normally. No
significant neurological abnormalities were noted, but there was a
statistically significant amplitude change in the visual evoked
response test. It was also noted that three out of four of the women
had premature menstruation, early by one week or more, after four days
of exposure to 2370 mg/m3 (1000 ppm) for 7.5 h/day, which the authors
saw as worrisome.
High pulse rates (120-160/min) were commonly found in patients
exposed to acetone by inhalation and/or dermally, in most cases at
concentrations sufficient to cause acute intoxications, after
application of casts for which acetone was used in the setting
solution (Chatterton & Elliott, 1946; Pomerantz, 1950; Renshaw &
Mitchell, 1956; Hift & Patel, 1961). In a controlled laboratory study
using a complex protocol, electrocardiography of volunteers exposed to
atmospheric concentrations of 2970 mg/m3 (1250 ppm) for 6 weeks (2-5
days; 7 h/day) revealed no alterations, compared with their
pre-exposure electrocardiograms (Stewart et al., 1975).
The acetone concentrations in the body fluids and expired air of
healthy and diabetic patients can be very different. Even in healthy
subjects, the level of acetone in blood/plasma varies according to
fasting or non-fasting conditions and depends on the weight of the
subject. Generally, the blood/plasma acetone concentrations are higher
in fasted than non-fasted subjects and higher in subjects who are not
obese, compared to obese subjects (Haff & Reichard, 1977). The
narcotic effects of acetone occur after oral as well as inhalation
exposure. Several case reports describe patients in minimally
responsive, lethargic, or comatose conditions after ingesting acetone,
but some of these cases are confounded by co-exposure to other
possible narcotic agents. For example, a 30-mouth-old child ingested
most of a 180 ml (6 ounce) bottle of nail polish remover containing
65% acetone and 10% isopropyl alcohol (Gamis & Wasserman, 1988); a
known alcoholic woman ingested nail polish remover (Ramu et al.,
1978); and a man ingested 200 ml of sake prior to intentionally
ingesting liquid cement containing a mixture of polyvinyl chloride,
acetone, 2-butanone, and cyclohexanone (Sakata et al., 1989). Blood
levels of acetone in some of these patients were 2.5 g/litre (Ramu et
al., 1978) and 4.45 g/litre (Gamis & Wasserman, 1988). In the case
reported by Sakata et al. (1989), the blood level of acetone was 110
mg/litre and the urine level was 123 mg/litre 5 h after the ingestion,
but the patient had been subjected to gastric lavage. A man who
intentionally ingested about 200 ml of pure acetone (about 2241
mg/kg) subsequently became deeply comatose, but responded to
treatment (Gitelson et al., 1966). Six days later, he was ambulatory,
but a marked disturbance of gait was observed. This condition had
improved upon follow-up examination 2 months later.
Acetone is reported to be irritating to mucous membranes. Raleigh
& McGee (1972) listed eye irritation as a common complaint of workers
exposed to acetone, and Matsushita et al. (1969a) reported the same
finding for volunteers in their study. However, the complaints of
irritation associated with exposure to acetone may be influenced by
the strong odour of acetone (Dalton et al., 1997; Wysocki et al.,
1997).
Lupulescu & Birmingham (1975, 1976) found that application of 0.1
ml directly to the skin resulted in histological and degenerative
epidermis changes in volunteers after 60 or 90 min of exposure. These
changes were denoted by reduction as well as disorganization of the
horny layers, intercellular oedema and vacuolization of the stratum
spinosum. Haematological effects have been observed in humans after
inhalation exposure to acetone in controlled laboratory studies of
volunteers. A statistically significant increase in white blood cell
counts and decrease in phagocytic activity of neutrophils, compared
with controls, were observed in the volunteers after a single 6-h
inhalation exposure or repeated 6-h exposures for 6 days to 1190
mg/m3 (500 ppm) (Matsushita et al., 1969a,b). No significant
difference was seen in haematological parameters in volunteers exposed
to 590 mg/m3 (250 ppm) compared with controls. In contrast,
haematological findings were within normal limits in volunteers
exposed to 1190 mg/m3 (500 ppm) for 2 h (DiVincenzo et al., 1973) or
to < 2970 mg/m3 (< 1250 ppm) repeatedly for 1-7.5 h/day for
as long as 6 weeks (Stewart et al., 1975). Exposure to acetone vapour
can also lead to increased pulse rates, gastrointestinal irritation,
nausea, vomiting, and haemorrhage. However, the odour threshold of
acetone (240-330 mg/m3, 100-140 ppm) and the feelings of irritancy
are excellent warning properties that generally preclude serious
inhalation overexposure. Accidental or intentional ingestion of
acetone can cause erosions in the mouth, coma and diabetes-like
symptoms.
8.1.2 Occupational exposure
Most occupational exposure standards are in the range 1780-2370
mg/m3 (750-1000 ppm). Occupations in which workers may be exposed to
higher levels of acetone include paint manufacturing, plastics
manufacturing, artificial fibre industries, shoe factories,
professional painting, and commercial cleaning. As with the general
population, there are case studies of accidental poisonings and
inhalation of acetone fumes. Many of these situations are mixed
exposures and not deemed relevant to this discussion, except where
acetone potentiates or antagonizes the effects of another chemical
present (see also section 8.3).
Workers in industries that manufacture or use acetone are
potentially exposed to higher concentrations of acetone than the
general population. For example, the concentrations of acetone in the
breathing-zone air in a paint factory, a plastics factory, and an
artificial fibre factory in Italy were >3.48 mg/m3 (Pezzagno et al.,
1986). The concentration of acetone in the breathing-zone air of a
fibre-reinforced plastic plant in Japan, where bathtubs were produced,
was < 108 mg/m3 (Kawai et al., 1990a). The inhalation exposure for
workers to acetone in a shoe factory in Finland ranged from 25.4 to
393.4 mg/m3 (Ahonen & Schimberg, 1988), and concentrations were
similar in the breathing-zone air in shoe factories in Italy (Brugone
et al., 1978). The concentration of acetone in the breathing-zone air
of a solvent recycling plant in the USA ranged from not detectable to
43 mg/m3 (Kupferschmid & Perkins, 1986). High levels of acetone were
detected in the occupational air in other industries, including the
chemical, plastic button, and paint manufacturing industries in Italy
(Ghittori et al., 1987). Isopropyl alcohol is known to oxidize in the
liver and is converted to acetone (Kawai et al., 1990b); therefore,
occupational exposure (e.g., printing plants) or accidental ingestion
of isopropyl alcohol also produces acetone in expired air, blood and
urine.
Satoh et al. 0996) carried out a cross-sectional study on 110
male (age range 18.7 to 56.8 years) acetone-exposed and 67 male (age
range 20.7 to 57.5 years) non-exposed shift workers. The personal
passive monitors and biological monitoring indices measured at the end
of the workshift were 46.5-2583 mg/m3 (19.6-1088 ppm) (mean 864
mg/m3, 364 ppm) in breathing-zone, 5.9-1002 mg/m3 (2.5-422 ppm)
(mean 231 mg/m3, 97.3 ppm) in alveolar air, 4-220 mg/litre (mean 66.8
mg/litre) in blood, and 0.75-170 mg/litre (mean 37.8 mg/litre) in
urine. Symptoms at the end of the workshift showing good exposure
response relationships were eye irritation, tear production and
complaints of acetone odour. In acetone workers in the 30-44 year
range, simple reaction time and digit span scores were significantly
lower in acetone-exposed workers but exposure-relationships were not
clear. Manifest Anxiety Scale Scores, Self-rating Depression Scale
Scores, R-R internal variation on the ECG, haematological examinations
and liver function tests did not show any significant differences
between the two groups.
In a retrospective mortality study of 948 employees (697 men, 251
women) in a cellulose fibre plant where acetone was used as the only
solvent, there was no significant excess risk of death from any cause
(all causes, malignant neoplasm, circulatory system disease, ischaemic
heart disease) compared with rates for the general population in the
USA (Ott et al., 1983a,b). The workers had been employed at the plant
for at least 3 months to 23 years. Industrial hygiene surveys found
that median TWA acetone concentrations were 902, 1678 and 2540 mg/m3
(380, 770 and 1070 ppm) based on job categories. All haematologieal
parameters and all clinical blood chemistry parameters (aspartate
aminotrausferase, alanine aminotransferase, lactic dehydrogenase,
alkaline phosphatase, total bilirubin, and albumin) were within normal
limits.
The only effect on the respiratory system observed in humans
exposed to acetone vapour is irritation of the nose, throat, trachea
and longs. The irritant properties of acetone in humans have been
noted both in workers who were exposed to acetone occupationally
(Raleigh & McGee, 1972; Ross, 1973) and in volunteers under controlled
laboratory conditions (Nelson et al., 1943; Matsushita et al.,
1969a,b). Complaints of irritation were reported by workers with
average exposures to acetone in the workroom of 2140 mg/m3 (901 ppm)
(Raleigh & McGee, 1972; Ross, 1973). Sallee & Sappington (1949), in a
report of experience at the Tennessee Eastman Corporation on acetone
concentrations not associated with injury, noted that acetone is
mildly irritating to the eyes at 4750-7120 mg/m3 (2000-3000 ppm), but
that no irritation persisted after exposure ceased.
In an on-site medical appraisal of nine workers, in which the
exposure concentration was 2390 mg/m3 (1006 ppm), three of the
workers mentioned headache and lightheadedness as subjective symptoms
(Raleigh & McGee, 1972). In another on-site medical appraisal of four
workers, in which the TWA exposure concentration was 2140 mg/m3 (901
ppm), none of the workers complained of neurological effects (Raleigh
& McGee, 1972). The medical examinations included the Romberg test,
finger-to-nose test, and observations for nystagmus (involuntary rapid
movement of the eyeball). These tests revealed no neurobehavioural
effects in either study. Such symptoms as unconsciousness, dizziness,
unsteadiness, confusion and headache were experienced by seven workers
exposed to > 28 500 mg/m3 (> 12 000 ppm) while clearing out a pit
containing acetone that had escaped from nearby tanks (Ross, 1973).
The degree of the symptoms varied depending on the length of time that
the workers had spent in the pit (2 min to 4 h).
8.2 Subpopulations at special risk
Because of higher exposure, workers in industries that
manufacture or use acetone are one segment of the population at higher
risk of acetone exposure compared to the general population (see
section 5.3). Professional painters and commercial and household
cleaners, using detergents, cleansers, waxes or polishes containing
acetone, are also likely to be exposed to acetone at higher
concentrations than the general population, although experimental data
regarding the extent of exposures for this segment of workers are not
available. Among the general population, high exposure to acetone may
occur among several subgroups. Cigarette smoke contains < 0.54 mg
acetone/cigarette (Manning et al., 1983); therefore, smokers are
exposed to higher concentrations of acetone than non-smokers. The
content of acetone in certain nail polish removers is high, and so
individuals who frequently use nail polish removers, such as
manicurists, are exposed to higher levels of acetone than the general
population. People who live near landfill sites that emit acetone or
those who live near industrial sources of emission (e.g., refinery,
incinerator, close to high vehicular traffic areas) are also
susceptible to higher exposure concentrations of acetone than the
general population that does not reside near these sites.
There is also a small possibility of ketone formation by
ketoacidosis in humans. The most common form is diabetic ketoacidosis
(DKA), which appears to require insulin deficiency coupled with a
relative or absolute increase in glycogen concentration. Dieting has
also been shown to cause starvation ketosis. Apart from these two
types, the other common ketoacidotic state is alcoholic ketoacidosis.
Presumably the liver is activated by kctogenesis as a result of
starvation in cases of alcoholic ketoacidosis and driven to maximal
rates of ketone formation by the high fatty acid levels (Wilson et
al., 1991).
The intrinsic toxicity of acetone and the results of the US EPA
(1986a) animal studies suggest that male animals are more susceptible
than female animals. Acetone at higher concentrations may exacerbate
preexisting haematological, liver, kidney or reproductive disorders in
humans. As with other chemicals, neonates and the elderly may be more
susceptible to acetone because of immaturity or decrease function of
their metabolic systems, respectively. Physiological disorders are
exacerbated by higher exposure. Fasting and diabetes increases
endogenous levels of acetone in humans, suggesting that dieters and
diabetics may have a higher body burden, and additional exposure to
acetone may make them more at risk. Results in animals suggest that
the rate of acetone metabolism is slower during pregnancy (Peinado et
al., 1986).
One of the most-studied effects of acetone is the induction of
mierosomal enzymes, particularly of cytochrome P-450IIE1. Acetone
thereby induces its own metabolism, and it potentiates the toxicity of
numerous other chemicals by enhancing the metabolism, which depends on
cytochrome P-450IIE1, to reactive intermediates, The indnction of
cytochrome P-450IIE1 by acetone has been documented in many species,
and therefore poses a concern for humans exposed to acetone or to
those chemicals whose toxicity is potentiated or antagonized by
acetone. The observed potentiation following acetone ingestion
generally causes a quantitative increase in the extent of damage to
the affected tissues or organs.
Acetone pretreatment has been shown to potentiate halogenated
solvent hepatotoxicity and nephrotoxicity, and is related to the
induction of microsomal enzymes that metabolize these solvents to
reactive intermediates (Morgott, 1993), People with altered
physiological states can have permanent potentiating effects of
acetone. These states include starvation, alcoholism, diabetes
mellitus, hypoglycaemia, eating disorders, prolonged vomiting or
inborn errors of metabolism (Morgott, 1993). This potentiation has
been shown to occur with carbon tetrachloride, chloroform and
1,1,2-trichloroethane (Traiger & Plaa, 1974), 1,1-dichloroethylene
(Hewitt & Plaa, 1983), bromodichloromethane, dibromomethane (Hewitt &
Plaa, 1983), 1,2-dichlorobenzene (Brondeau et al., 1989), and several
other solvents.
9. EFFECT ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1 Aquatic organisms
9.1.1 Acute toxic effects on aquatic fauna
Data on the effects of acute exposure of aquatic organisms to
acetone are summarized in Table 14. The four freshwater fishes tested,
Salmo gairdneri, Lepomis macrochirus, Pimephales promelas and
Gambusia affinis, had 96-h LC50s between 5540 and 13 000 mg/litre,
indicating low toxicity for these species (Wallen et al., 1957; Cairns
& Scheier, 1968; Patrick et al., 1968; Mayer & Ellersieck, 1980; Veith
et al., 1983; Brooke et al., 1984). Similarly, the freshwater Asiatic
clam ( Corbiculo manilensis) had a 96-h LC50 of 20 000 mg/litre
(Chandler & Marking, 1979). Various species of the water flea
( Daphnia magna, D, pulex and D. cuculata) had 48-h LC50s and
EC50s of 7460 to 13 500 mg/litre; however, test conditions for these
studies were scantily reported (Canton & Adema, 1978; Randall & Knopp,
1980). Bringmann & Kühn (1982) found a 24-h EC50 (immobilization) of
10 000 mg/litre for Daphnia magna. Dowden & Bennett (1965) reported a
48-h LC50 of 10 mg/litre for D. magna, but they noted that tests
had been done by several different investigators and that values might
be erroneous. Ewell et al. (1986) also tested several freshwater
species under static conditions for 96 h. All of the species; pillbug
( Asellus intermedius), waterflea ( Daphnia magna), flatworm
( Dugesia tigrina), sideswimmer ( Gammarus fasciatus), snail
( Helisoma trivolvis), segmented worm ( Lumbriculus variegatus) and
fathead minnow ( Pimephales promelas) had 96-h LC50s >100 mg/litre,
the highest concentration tested.
Two saltwater species were tested by Linden et al. (1979). The
copepod, Nitocra spinipes, and the bleak ( Alburnus alburnus,
tested under static conditions had 96-h LC50s of 16 700 and 11 000
mg/litre, respectively.
Slooff & Baerselman (1980) studied groups of 10 amphibians,
axolotls ( Ambystoma mexicanus) and clawed toads ( Xenopus laevis),
34 weeks after hatching. The animals were exposed to varying
concentrations of acetone in standardized medium and 48-h LC50 values
for the respective species were 20 and 24 mg/litre.
Table 14 Acute toxicity of acetone to aquatic fauna
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Water flea 48-h LC50 9218 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
(Daphniamagna) 24-h LC50 4068 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
NOEL <403 8.2 25 90-110 static, nominal Cowgill & Milazzo (1991)
Water flea 48-h LC50 12 100 to 13 300 nominal Canton & Aderna (1978)
(Daphnia magna)
Water flea 48-h EC50 13 500 7.7 22 154.5 nominal Randall & Knepp (1980)
(Daphnia magna)
Water flea 24-h LC50 10 nominal Dowden & Bennett (1965)
(Daphnia magna) 48-h LC50 10 Dowden & Bennett (1965)
Water flea 96-h LC50 >100 6.5-8.5 20 nominal Ewell et al. (1986)
(Daphnia magna)
Water flea 24-h EC50 >10 000 immobilization Bringmann & Kühn (1982)
(Daphnia magna)
Water flea 48-h LC50 8800 nominal Canton & Adema (1978)
(Daphnia pulex)
Water flea 48-h LC50 7460-7810 nominal Canton & Adema (1978)
(Daphnia cuculata)
Water flea 48-h LC50 8098 8.2 25 90-110 nominal Cowgill & Milazzo (1991)
(Cesiodaphnia dubia)
240-h LC50 6693 8.2 25 90-110 nominal Cowgill & Milazzo (1991)
Pillbug 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Asellus intermedius)
Table 14 (contd).
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Flatworm 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1966)
(Dugesia tigrina)
Side swimmer 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Gammarus fasciatus)
Segmented worm 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Lumbriculus)
variegatus
Snail 96-h LC50 >100 6.5-8.5 20 static, nominal Ewell et al. (1986)
(Helisoma trivolvis)
Asiatic clam 96-h LC50 >20 000 16 16-26 static, nominal Chandler & Marking (1979)
(Corbicula
manilensis)
Crustacean 24-h LC50 64 300 7.8 25 250 static, nominal Crisinel et al (1994)
(Streptocephalus
rubricandatus)
Copepod 96-h LC50 16 700 7.8 10 static, nominal Lindén et al (1979)
(Nitocra spinipes)
Brine shrimp 24-h LC50 2100 24.5 saltwater, Price et al. (1974)
(Artemia salina) static, nominal
24-h LC50 6010 7.4 12 40 static, nominal Mayer & Ellersieck (1980)
96-h LC50 5540 7.4 12 40 static, nominal Mayer & Ellersieck (1980)
Rainbow trout 24-h LC50 6100 8.0 10 90 flow-through, Majewski et al. (1977)
(Salmo gairdnen) nominal
Table 14 (contd).
Species Test Result pH Temperature Hardness Comments Reference
(mg/litre) (°C) (mg/litre)
Rainbow trout 24-h CV50 36 500 18 CV50 is the Segner & Lenz (1993)
(Salmo gairdnen) midpoint
cytotoxicity of
the R1 cell line
Bluegill 96-h LC50 8300 18 10 static, nominal Cairns & Scheier (1968)
(Lepomis macrochirus)
Fathead minnow 96-h LC50 8120 7.58 25 48.5 measured Brooke et al (1984)
(Pimephales promelas) exposure
concentration
96-h LC50 6880 6.93 25 535 measured Brooke et al. (1984)
exposure
concentration
96-h LC50 6290 7.62 24 44.0 measured Brooke et al. (1984)
exposure
concentration
Fathead minnow 96-h LC50 >100 6.5-8.5 static, nominal Ewell et al. (1986)
(Pimephales promelas)
Mosquito fish 24-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal Wallen et al. (1957)
(Gambusia affinis)
48-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal Wallen et al. (1957)
96-h LC50 13 000 8.0-8.5 23-27 <100 static, nominal WaIlen et al. (1957)
Bleak 96-h LC50 11 000 7.8 10 static, nominal, Linden et al (1979)
(Alburnus alburnus) saltwater
9.1.2 Chronic effects on aquatic fauna
Cowgill & Milazzo (1991) calculated EC50 values for two species
of Daphnia exposed to acetone using the three-brood test. For
Daphnia magna, EC50 values for number of progeny, number of broods
and mean brood size were 6369, 6406 and 6714 mg/litre, and the
corresponding NOEL values were 3110, 5184 and 3110 mg/litre. The EC50
values for Ceriodaphnia dubia were 6469, 5908 and 6928 mg/litre, and
the corresponding NOEL values were 5184 mg/litre for all three
parameters.
9.1.3 Effects on aquatic plants
Freshwater diatoms and algae were tested with acetone.
Chlorella pyrendoidosa, a green alga, was exposed to acetone at
concentrations of 2574 and 25 740 mg/litre (3.3 and 33 ml/litre
assuming an acetone density of approx. 790 mg/ml at 30°C) in culture
tubes with 300 ml nutrient solution at 30°C under continuous light for
76 h (Parasher et al., 1978). The lowest concentration bad a slight
negative effect on algal growth; the high concentration caused
disintegration of the cell membranes and cytoplasm.
Hess (1980) examined the effects of acetone on
Chlamydomonas eugametus, a green alga. The alga was exposed for 48 h
at 25°C; pH and hardness were not reported. An acetone concentration
of 0.5% (v/v; 3950 mg/litre) resulted in no inhibition of growth.
Significant inhibition (a 15% growth reduction) occurred at 1.0% (7900
mg/litre) and growth was only 24% of control values at 2.5% acetone
(1975 mg/litre); at 5.0% (39 500 mg/litre), there was complete
inhibition. Bringmann & Kühn (1978, 1980a) examined the effects of
acetone on the green alga, Scendesmus quadricauda. The alga was
exposed at 27°C for 7-8 days. The toxicity threshold was 7500
mg/litre. Bringmann & Kühn (1978) determined that the toxicity
threshold for the cyanobacterium (blue-green alga)
Microcystis aeruginosa exposed under similar conditions was only 530
mg/litre. Stratton & Corke (1981) noted that acetone (0.1-1.0% v/v;
790-7900 mg/litre; length of exposure not reported) actually
stimulated photosynthesis in S. quadricauda and C. pyrendoidosa,
perhaps by increasing the rate of CO2 diffusion into the cells.
The freshwater diatom, Nitzchia linearis, was tested in soft
water (100 mg CaCO3/litre) for 5 days but temperature and pH were not
reported (Patrick et al., 1968). The EC50 for cell multiplication was
11 493 to 11 727 mg/litre.
The marine diatom Skeletonema costatum was also tested with
acetone (Kleppel & McLaughlin, 1980; Cowgill et al., 1989). Following
exposure to acetone for 5 days, EC50 values for decreases in total
cell count and total cell volume were 11 798 and 14 440 mg/litre,
respectively (Cowgill et al., 1989). Kleppel & McLaughlin (1980)
exposed skeletonema costatum (at cell densities of 3.8 × 102,
3.8 × 103 and 3.8 × 104) to 1 ml acetone per litre of medium for 5
days. There were no significant changes in cell growth and
reproduction in cells exposed to acetone, compared with controls.
Cowgill et al. (1991) estimated the NOEL and the EC50 (50%
reduction in the number of plants or fronds as compared to controls)
of acetone to Lemna sp. (duckweed). Plants belonging to this genus
have become "aquatic test organisms" because they are a part of the
food chain of fish, water fowl and aquatic organisms. For
Lemna gibba G-3, the EC50 for the 7-day test was 12.4 g/litre for
plants and 10.2 g/litre for fronds. For Lemna minor 6591, it was
13.4 g/litre for plants and 11.4 g/litre for fronds. Three other
Lemna species had EC50 values greater than 10 g/litre.
9.2 Effects on bacteria and protozoa
The effects of acetone have been studied with bacteria and
protozoans. Protozoans seemed more sensitive to toxicological effects
than bacteria. Bringmann & Kühn (1980a) exposed the bacterium
Pseudomonas putida (for 16 h) and the protozoan
Entosiphon sulcatum (for 72 h), to acetone at a temperature of 25°C
in a cell multiplication test to determine toxicity. The toxicity
thresholds for these two organisms were 1700 and 28 mg/litre,
respectively. In another study, Bringmann & Kühn (1980b) determined a
toxicity threshold for the protozoan Uronema porduczi of 1710
mg/litre. A mixed microbial culture from wastewater (specific biota
not reported) tested with acetone for an unreported length of time
resulted in an EC50 (decrease in biodegradation) of 0.612 mol/litre
(35 540 mg/litre), indicating very low toxicity (Vaishnav, 1986). The
bioluminescent bacterium Photobacterium phosphoreum had an EC50
(inhibition of luminescence) of 0.363 and 0.372 mol/litre (21 088 and
21 579 mg/litre), also indicating very low toxicity (Kamlet et al.,
1986). McFeters et al. (1983) reported a similar EC50 of 18 250
mg/litre for P. phosphoreum.
Stratton (1987) estimated EC50 values in several species of
cyanobacteria (blue-green algae). Acetone stimulated growth at
concentrations less than 790-7900 mg/litre (0.1 to 1.0%), except for
Anabaena sp. where levels above 5500 mg/litre (0.7%) caused total
inhibition. Total growth inhibition occurred at acetone concentrations
of 569 g/litre (72%) with A. cylindrica, 31.6 g/litre (>4%) with
A. inaegualis and > 63.2 g/litre (8%) with A. variabilis and
Nostoc sp. The estimated EC50 for these species ranged from 2.84 to
34.6 g/litre (0.36 to 4.38% v/v).
Ecotoxicological testing was done using the Microtox test, which
is based upon the toxicant-based diminution of light emission of
Photobacterium phosphoreum (Bulich, 1986). The parameter used to
measure toxicity was the EC50, i.e. the toxicant concentration that
diminishes light emission by 50% at 15°C. The EC50 for acetone has
been measured at 14.7 g/litre (254 mmol/litre) for 5 min, 14.2 g/litre
(245 mmol/litre) for 15 min and 14.1 g/litre (243 mmol/litre) for 25
min (Chen & Que Hee, 1994). These EC50 values are very high compared
to other compounds, indicating that acetone toxicity as reported by
this test is low. These EC50 values can be compared to the results of
a study by De Zwart & Slooff (1983), who determined an EC50 of 18.3
g/litre (316 mmol/litre).
In a series of studies on the toxicity of acetone to bacteria,
Nirmalakhandan et al. (1994a,h) determined the IC50 in activated
sludge cultures from a municipal wastewater treatment plan to be
almost 4.9 g/litre.
Blum & Speece (1991) found low inhibition, with IC50 values of
1.2 g/litre for Nitrosomonas, 50 g/litre in methanogens and 16
g/litre for aerobic heterotrophs. Methanogens are generally the most
sensitive bacteria that convert organic matter to carbon dioxide and
methane in anaerobic environments.
Rajini et al. (1989) examined the cytotoxicity to the protozoan
Paramecium caudatum. The 10-min LC100 was 22.9 g/litre (2.9% v/v)
with a concentration of 22.6 g/litre, and the 4-h LC50 was 5.37
g/litre (0.68% v/v) with a concentration of 5.2 g/litre. The
researchers also found that insecticides were more toxic to
P. caudatum when they were dissolved in acetone.
9.3 Terrestrial organisms
9.3.1 Effects on fauna
Hill et al. (1975) administered acetone for 5 days to both quail
and pheasants. The LC50 for both of these birds was 40 g/kg diet, the
highest dose administered. Acetone was also tested with mallard eggs
(Hoffman & Eastin, 1981). Fertile eggs were immersed in 0, 10 or 100%
acetone for 30 seconds at room temperature on days 3 or 8 of
incubation. There were no significant effects with 10% acetone;
however, 100% acetone caused a significant decrease in survival,
embryonic weight and embryonic length for both exposure groups. It is
unknown whether the mortality was due to the toxicity of acetone or to
its solvent capabilities. White Leghorn chick embryos were also tested
with acetone (Korhonen et al., 1983). The test article was injected
into the eggs at 5 µl/egg. Statistical analysis was not performed and
controls were not used; however, it appeared that acetone did not
affect mortality or malformation of the embryos.
9.3.2 Effects on flora
Pertinent data regarding the effects of exposure of terrestrial
flora to acetone have not been found. Acetone did not cause a
significant decrease in seed germination or the percentage of normal
seedlings of sweet corn (5h2 cultivars). However, with longer
immersion times (8 h), it was detrimental (Hung et al., 1992).
Gorsuch et al. (1990) tested the potential of acetone to affect
germination and early growth of terrestrial plants. Plants were
exposed to the chemical for 7 days and then germination, root length
and plant heights were determined and compared with controls. Acetone
had a no-observed-effect concentration of 100 mg/litre (nominal
concentration) for ryegrass, radish and lettuce.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1 Evaluation of human health effects
Acetone is of a low order of acute toxicity. However a
significant number of poisonings have occurred in humans following
accidental or intentional misuse.
Acetone can produce neurobehavioural and other changes, including
headache, dizziness, confusion and, at high vapour concentrations, CNS
depression and narcosis. Exposures to acetone vapour will cause
irritation of eyes, nose and throat. Continuous exposure to vapour can
lead to adaptation to the odour.
Liquid acetone is an eye irritant and repeated exposure of skin
will cause defatting, drying and cracking.
It is considered that acetone is neither a skin nor a respiratory
tract sensitizer.
Acetone is formed endogenously from fatty acid oxidation and is
uniformly distributed throughout the body among non-adipose tissues.
It is rapidly cleared from the body by metabolism and excretion,
mainly through the lungs. Acetone induces the hepatic mixed-function
oxidase enzymes that bring about its own metabolism, and so the body
has a homeostatic mechanism that has evolved to maintain acetone
levels in the body at a "baseline" level. Induction of hepatic
mixed-function oxidase enzymes can potentiate (and in some instances
antagonise) the effects of other chemicals. People at most risk to
potentiation include diabetics, alcoholics and those undergoing
prolonged fasting. In common with other chemicals, metabolism of
acetone may be reduced in neonates, the elderly and in hepatic
diseases.
In one study on human volunteers, increases in leucocyte count
were reported. However, this has not been found in other studies, in
an inhalation study, human female volunteers reported menstrual
irregularities (delayed menstruation).
Mild haematological effects were found in two strains of rats
(F344 and Sprague-Dawley). The mean cell haemoglobin concentration and
mean cell volume were elevated in both, but blood haemoglobin
concentration was raised in Sprague-Dawley rats and decreased in F344
rats. At an acetone dose of 50 000 mg/litre in the drinking-water,
effects on sperm quality were observed in rats. At lower doses only
small changes in sperm motility were seen. In a reproductive study in
which males were exposed to 5000 mg/litre in drinking-water, there
were no changes in reproductive index. High doses in animals produce
minimal fetal toxicity although only at doses causing maternal
effects. Acetone produced an increase in nephropathy in treated male
rats over the levels found in controls: this is of uncertain
significance to humans, Acetone is not genotoxic nor has it been shown
to be carcinogenic in dermal bioassays in mice.
No long-term experimental studies have been conducted. From the
available shorter-term studies, a no-observed-effect level in a
13-week drinking-water study in rats of 900 mg/kg body weight per day
(male rats), based on parameters including changes in organ weights,
was selected. Applying an uncertainty factor of 100 gives a guidance
value of 9 mg/kg body weight per day.
The relevance to humans of the liver, reproductive and
developmental effects observed in animal studies is not known, and
these end-points have not been sufficiently examined in humans.
However, because few species differences exist in the toxicokinetics
of acetone, these effects might be of concern for humans. The renal
effects may be specific for male rats, and the cataract formation may
be specific for guinea-pigs. The relevance of amyloidosis in
completely unknown. Acetone appears to have no delayed toxic effects.
The majority of genotoxicity assays on acetone were negative;
therefore, acetone can be considered to present no potential genotoxic
hazard to humans.
It should be noted that the perception of "irritation" from
acetone vapour by humans may be at a concentration in air as low as
23.7 mg/m3 (100 ppm), which is at or near the odour threshold.
10.2 Evaluation of effects on the environment
Acetone is of low toxicity to both aquatic and terrestrial
organisms. It is readily biodegraded in the environment and does not
bioaccumulate or magnify through the food chain. Even if acetone is
spilt in water, it is unlikely to have a major or lasting effect on
the ecosystem. Owing to evaporation and dispersal, spills on land are
likewise not expected to have any major or lasting effects on
terrestrial organisms.
11. FURTHER RESEARCH
a) Reproductive effects need to be examined in animals and/or in
humans. Clarification of the dose-response relationship is
required with special reference to male reproductive effects at
doses where abnormal sperm are found and to determine if there
are complications during menstration, pregnancy and childbirth,
as existing data are not conclusive.
b) Longer-term studies are required to determine whether the kidney
effects are attributable to acetone or are exacerbating an
existing condition. If they are acetone-related, the mechanism
should be determined.
c) Clarification of the potentiation and antagonism mechanisms in
humans is needed.
d) Clarification of the mechanisms of potential immunotoxic effects
is required.
12. PREVIOUS EVALUATION BY INTERNATIONAL BODIES
Acetone was evaluated in 1970 as an extraction solvent for fats
and oils and a precipitation agent in the purification of starches and
sugars by the Joint FAO/WHO Expert Committee on Food Additives. The
Committee recommended that its use as an extraction solvent should be
restricted to that determined by good manufacturing practice, which is
expected to result in minimum residues. Within these limits residues
are unlikely to have any significant toxicological effect (FAO/WHO,
1971; WHO, 1971).
REFERENCES
Abbondandolo A, Bonatti S, Corsi C, Corti G, Fiorio R, Leporini C,
Mazzaccaro A, Nieri R, Barale R, & Loprieno N (1980) The use of
organic solvents in mutagenicity testing. Mutat Res, 79: 141-150.
Aharonson EF, Menkes H, Guntner G, Swift DL, & Proctor DF (1974)
Effect of respiratory airflow rate on removal of soluble vapors by the
nose, J Appl Physiol, 37: 654-657.
Ahonen I & Schimberg RW (1989) 2,5-Hexanedione excretion after
occupational exposure to n-hexane. Br J Ind Med, 45: 133-136.
Altshuller AP (1991) Chemical reactions and transport of alkanes and
their products in the troposphere. J Atmos Chem, 12: 19-61.
Altshuller AP & Cohen IR (1963) Structural effects of the rate of
nitrogen dioxide formation in the photooxidation of organic
compound-nitric oxide mixtures in air. Int J Air Water Pollut, 7:
789-797.
Amacher DE, Paillet SC, Turner GN, Verne V, & Salsburg DS (1980) Point
mutations at the thymidine kinase locus in L5178Y mouse lymphoma
cells: 2. Test validation and interpretation. Mutat Res, 72: 447-474.
Amlathe S & Gupta VK (1990) Spectrophotometric determination of
acetone using vanillin. Analyst, 115:1385-1387.
Amoore JE & Hautala E (1983) Odor as an aid to chemical safety: odor
thresholds compared with threshold limit values and volatilities for
214 industrial chemicals in air and water dilution. J Appl Toxicol, 3:
272-290.
Andersson L & Lundstrom K (1984) Milk and blood ketone bodies, blood
isopropanol and plasma glucose in dairy cows; methodological studies
and diurnal variations. Zent. bl Vet. med, A31: 340-349.
Andersson-Sköld Y, Grennfelt P, & Pleijel K (1992) Photochemical ozone
creation potentials: a study of different concepts. J Air Waste Manage
Assoc, 42:1152-1158.
Aneja VP (1993) Organic compounds in cloud water and their deposition
at a remote continental site. J Air Waste Manage Assoc, 43: 1239-1244.
Anselme C, Nguyen K, Burchet A, & Mallevialle J (1985)
Characterization of low molecular weight products desorbed from
polyethylene tubings. Sci Total Environ, 47:371-384.
Arnold F, Knop G, & Ziereis H (1986) Acetone measurements in the upper
troposphere and lower stratosphere-implications for hydroxyl radical
abundances. Nature (Lend), 321: 505-507.
Arnold F, Schneider J, Gollinger K, Schlager H, Schulte P, Hagen DE,
Whitefield PD, & van Velthoven P (1997) Observation of upper
tropospheric sulfur dioxide- and acetone-pollution: potential
implications for hydroxyl radical and aerosol formation. Geophys Res
Lett, 24(1): 57-60.
Arnts RR & Meeks SA (1981) Biogenic hydrocarbon contribution to the
ambient air of selected areas. Atmos Environ, 15: 1643-1651.
Ashley DL, Bonin MA, Cardinali FL, McCraw JM, Holler JS, Needham LL, &
Patterson DG Jr (1992) Determining volatile organic compounds in human
blood from a large population using purge and trap gas
chromatography/mass spectrometry. Anal Chem, 64:1021-1029.
Ashley DL, Bonin MA, Cardinali FL, McCraw JM, & Wooten JV (1994) Blood
concentrations of volatile organic compounds in a nonoccupationally
exposed US population and in groups with suspected exposure. Clin
Chemistry, 40(7): 1401-1404.
Atkinson R (1985) Kinetics and mechanisms of the gas-phase reactions
of hydroxyl radical with organic compounds under atmospheric
conditions, Chem Rev, 85: 69-201.
ATSDR (1994) Toxicological profile for acetone. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry, 257 pp.
Babeu L & Vaishnav DD (1987) Prediction of biodegradability for
selected organic chemicals. J Ind Microbial, 2: 107-115.
Barber ED & Lodge JP Jr (1963) Paper chromatographic identification of
carbonyl compounds as their 2,4-dinitrophenyl hydrazones in automobile
exhaust. Anal Chem, 35: 348-350.
Barnett CR, Petrides L, Wilson J, Flatt PR, & Ioannides C (1992)
Induction of rat hepatic mixed-function oxidases by acetone and other
physiological ketones: Their role in diabetes-induced changes in
cytochrome P450 proteins. Xenobiotica, 22:1411-1450.
Basler A (1986) Aneuploidy-inducing chemicals in yeast evaluated by
the micronucleus test. Mutat Res, 174(1): 11-13.
Benkelberg H-J, Hamm S, & Warneck P (1995) Henry's law coefficients for
aqueous solutions of acetone, aldehyde and acetonitrile and
equilibrium constants for the additional compounds and acetaldehyde
with bisulfite. J Atmos Chem, 20:17-34.
Bertsch W, Anderson E, & Holzer G (1975) Trace analysis of organic
volatiles in water by gas chromatography-mass spectrometry. J Chromat,
112: 701 - 718.
Bhattacharya SK, Angara RVR, Bishop DF Jr, Dobbs PA, & Austern BM
(1990) Removal and fate of RCRA (Resource Conservation and Recovery
Act) and CERCLA (Comprehensive Environmental Response Compensation and
Liability Act) toxic organic pollutants in wastewater treatment.
Cincinnati, Ohio, US Environmental Protection Agency, Risk Reduction
Engineering Laboratory, 150 pp (EPA 600/S2-89-026).
Blum DJW & Speeca RE (1991) A database of chemical toxicity to
environmental bacteria and its use in interspecies comparisons and
correlations. Res J Water Poison Control Fed, 63(3): 198-207.
Boyd AA, Canosa-Mas CE, & King AD (1991) Use of a stopped-flow
technique to measure the rate constants at room temperature for
reactions between the nitrate radical and various organic species. J
Chem Bec Faraday Trans, 87:2913-2919.
Brady JF, Li D, Ishizaki H, Lee M, Ning SM, Xiao F, & Yang CS (1989)
Induction of cytochrome P45011E1 and P450IIE1 by secondary acetones
and the role of P450IIE1 in chloroform metabolism. Toxicol Appl
Pharmacol, 100: 342-349.
Brega A, Villa P, Quadrini G, Quadri A, & Lucarella C (1991)
High-performance liquid chromatographic determination of acetone in
blood and urine in the clinical diagnostic laboratory. J Chromatogr,
553: 249-254.
Bridie AL, Wolff CJM & Winter M (1979) BOD and COD of some
petrochemicals. Water Res, 13: 627-630.
Bringmann G & Kühn R (1978) Testing of substances for their toxicity
threshold: Model organisms Microcystis (Diplocystis) aeruginosa and
Scenedesmus quadricauda. Mitt Int Verein Limnol, 21; 275-284.
Bringmann G & Kühn R (1980a) Comparison of the toxicity thresholds of
water pollutants to bacteria, algae, and protozoa in the cell
multiplication inhibition test. Water Res, 14(3): 231-241.
Bringmann G & Kühn R (1980b) [Determination of the harmful biological
effect of water pollutants on protozoa: II. Bacteriovorous ciliates.] Z
Wasser Abwasser Forsch, 13(1): 26-31 (in German).
Bringmann G & Kühn R (1982) [The toxic action of water pollutants on
Daphnia magna tested by an improved standardized procedure.] Z
Wasser Abwasser Forsch, 15(1): 1-6 (in German).
Brondeau MT, Ban M, Bonnet P, Guenier JP, & De Ceaurriz J (1989)
Acetone compared to other ketones in modifying the hepatotoxicity of
inhaled 1,2-dichlorobenzene in rats and mice. Toxicol Lett, 49: 69-78.
Brooke LT, Call DJ, Geiger DL, & Northcott CE ed. (1984) Acute
toxicities of organic chemicals to fathead minnows ( Pimephales
promelas). Superior, Wisconsin, University of Wisconsin,
Center for Lake Superior Environmental Studies, 24 pp.
Brosseau J & Heitz M (1994) Trace gas compound emissions from
municipal landfill sanitary sites. Atmos Environ, 28(2): 285-293.
Brown KW & Donnelly KC (1988) An estimation of the risk associated
with the organic constituents of hazardous and municipal waste
landfill leachates. Hazard Waste Hazard Mater, 5: 1-30.
Brown EM & Hewitt WR (1984) Dose-response relationships in
ketone-induced potentiation of chloroform hepato- and nephrotoxicity.
Toxicol Appl Pharmacol, 78: 437-53.
Brown WD, Setzer JV, Dick RB, Phipps FC, & Lowry LK (1987) Body burden
profiles of single and mixed solvent exposures. J Occup Mod, 29:
877-883.
Brown VM, Crump DR, Gardiner D, & Gavin M (1994) Assessment of a
passive sampler for the determination of aldehydrates and ketones in
indoor air. Environ Technol, 15: 679-85.
Brown BL, Allis JW, Simmons JE, & House DE (1995) Fasting for less
than 24 h induces cytochrome P4502E1 and 2B1/2 activities in rats.
Toxicol Lett, 81: 39-44.
Bruckner JV & Peterson RJ (1981) Evaluation of toluene and acetone
inhalant abuse: II. Model development and toxicology. Toxicol Appl
Pharmacol, 61(3): 302-312.
Brugnone F, Perbellini L, & Grigolini L (1978) Solvent exposure in a
shoe upper factory. Int Arch Occup Environ Health, 42: 51-62.
Brugnone F, Perbellini L, Giuliari C, Cerpelloni M, & Suave M (1994)
Blood and urine concentrations of chemical pollutants in the general
population. Med Lav, 85(5): 370-369.
Bulich AA (1986) Bioluminescence assays. In: Bitton G & Dutka BJ ed.
Toxicity testing using microorganisms, Boca Raton, Florida, CRC Press,
vol 1, pp 57-74.
Burmaster DE (1982) The new pollution-groundwater contamination.
Environment, 24: 6-13, 33-36.
Buxton GV, Greenstock CL, Heiman WP, & Ross AB (1988) Critical review
of rate constants for reactions of hydrated electrons, hydrogen atoms
and hydroxyl radicals ("OH/"O-) in aqueous solution. J Phys Chem Ref
Data, 17: 513.
Cairns J Jr & Scheier A (1998) A comparison of the toxicity of some
common industrial waste components tested individually and combined.
Prog Fish Cult, 30(1): 3-6.
Cander L & Forster RE (1959) Determination of pulmonary parenchymal
volume and pulmonary capillary blood flow in man. J Appl Physol, 59:
541-651.
Canton JH & Adema DMM (1978) Reproducibility of short-term and
reproduction toxicity experiments with Daphnia magna and comparison
of the sensitivity of Daphnia magna with Daphnia pulex and
Daphnia cucullata in short-term experiments. Hydrobiologia, 59(2):
135-140.
Carpenter CP & Smyth HF Jr (1946) Chemical burns of the rabbit cornea.
Am J Opthalmol, 29: 1363-1372.
Carter WPL (1994) Development of ozone reactivity scales for volatile
organic compounds. J Air Waste Manage Assoc, 44: 881-899.
Casazza JP, Felver ME, & Veech RL (1984) The metabolism of acetone in
rat. J Biol Chem, 259: 231-236.
Cavanagh LA, Schadt CF, & Robinson E (1969) Atmospheric hydrocarbon
and carbon monoxide measurements at Point Barrow, Alaska. Environ Sci
Technol, 3: 251-257.
Chandler JH Jr & Marking LL (1979) Toxicity of fishery chemicals to
the Asiatic clam, Curbicula manilensis. Prog Fish Cult, 41(3):
148-151.
Charbonneau M, Iijima M, Côté MG, & Plaa GL (1985) Temporal analysis
of rat liver injury following potentiation of carbon tetrachloride
hepatotoxicity with ketonic or ketogenic compounds. Toxicology,
35:95-111.
Charbonneau M, Brodeur J, DuSouich P, & Plaa GL (1985a) Correlation
between acetone-potentiated CCl4-induced liver injury and blood
concentrations after inhalation or oral administration. Toxicol Appl
Pharmacol, 84: 286-294.
Charbonneau M, Oleskevich S, Brodeur J, Plaa GL (1986b) Acetone
potentiation of rat liver injury induced by trichloroethylene-carbon
tetrachloride mixtures. Fundam Appl Toxicol, 6: 654-661.
Charbonneau M, Perreault F, Greselin E, Brodeur J, & Plaa GL (1988)
Assessment of the minimal effective dose of acetone for potentiation
of the hepatotoxicity induced by trichloroethylene-carbon
tetrachloride mixtures. Fundam Appl Toxicol, 10: 431-438.
Charbonneau M, Couture J, & Plaa GL (1991) Inhalation versus oral
administration of acetone: effect of the vehicle on the potentiation
of CCI4-induced liver injury. Toxicol Lett, 57: 47-54.
Chatfield RS, Gardner EP, & Calvert JG (1987) Sources and sinks of
acetone in the troposphere: behavior of reactive hydrocarbons and a
stable product. J Geophys Res, 92: 4208-4216.
Chatterton CC & Elliott RB (1946) Acute acetone poisoning from leg
casts of a synthetic plaster substitute. J Am Med Assoc,
130: 1222-1223.
Chen H-F & Que Hee SS (1994) Ketone EC50 in the Microtox test.
Ecotoxicol Environ Saf, 30: 120-123.
Cheung ST & Lin WN (1967) Simultaneous determination of methanol,
ethanol, acetone, isopropanol and ethylene glycol in plasma by gas
chromatography. J Chromatogr Biomed Appl, 414: 248-250.
Chieli E, Puccini P, Longo V, & Gervasi PG (1990) Possible role of the
acetone-inducible cytachrome P-450IIE1 in the metabolism and
hepatotoxicity of thiobenzamide. Arch Toxicol, 64(2): 122-127.
Chou WL, Speece RE, & Siddiqi RH (1979) Acclimation and degradation of
petrochemical wastewater components by methane fermentation.
Biotechnol Bioeng Syrup, 8: 391-414.
Clayton GD & Clayton FE ed, (1982) Patty's industrial hygiene and
toxicology: Volume 2C, 3rd ed. New York, John Wiley & Sons, pp
4722-4723.
Coleman DL (1980) Acetone metabolism in mice: increased activity in
mice heterozygous for obesity genes. Proc Natl Acad Sci (USA), 77(1);
290-293.
Coleman EC, Ho C-T, & Chang SS (1981) Isolation and identification of
volatile compounds from baked potatoes. J Agric Food Chem, 29(1);
42-48.
Conkle JP, Camp B J, & Welch BE (1975) Trace composition of human
respiratory gas. Arch Environ Health, 30: 290-295.
Corwin JF (1969) Volatile oxygen-containing organic compounds in sea
water determination. Bull Mar Sci, 19(3): 504-509.
Cowgill UM & Milazzo DP (1991) The sensitivity of Ceriodaphnia dubia
and Daphnia magna to seven chemicals utilizing the three-brood test.
Arch Environ Contam Toxicol, 20: 211-217.
Cowgill UM, Milazzo DP, & Laudenberger BD (1989) Toxicity of nine
benchmark chemicals to Skeletonema costatum, a marine diatom.
Environ Toxicol Chem, 8: 451-455.
Cowgill UM, Mirazzo DP, & Laudenberger BD (1991) The sensitivity of
Lemna gibba G3 and four clones of Lemna minor to eight common
chemicals using a 7-day test. Res J Water Pollut Control Fed, 63:
991-998.
Crisinel A, Delannay L, Rossel D, Tarradellas J, Meyer H, Saïah H,
Vogel P, Delisle C, & Blaise C (1994) Cyst-based ecotoxicological
tests using Anostracans: comparison of two species of
Streptocephalus. Environ Toxicol Water Qual, 9: 317-326.
Cunningham J, Sharkawi M, & Plaa GL (1989) Pharmacological and
metabolic interactions between ethanol and methyl n-butyl ketone,
methyl isobutyl ketone, methyl ethyl ketone, or acetone in mice.
Fundam Appl Toxicol, 13: 102-109.
Dalton P, Wysocki CJ, Brody MJ, & Lawley HJ (1997) The influence of
cognitive bias on the perceived odor, irritation and health symptoms
from chemical exposure. Int Arch Occup Environ Health, 69: 407-417.
Datta RK & Rao KN (1979) Kinetics of reactions of single molecular
oxygen (1 Delta g) with organic compounds. Indian J Chem, 18A:
102-105.
Day EA & Anderson DF (1965) Gas chromatographic and mass spectral
identification of natural components of the aroma fraction of blue
cheese. J Agric Food Chem, 13: 2-4.
De Rosa E, Cellini M, Sessa G, Saletti C, Raus G, Maracuzzo G, &
Bartolucci GB (1993) Biological monitoring of workers exposed to
styrene and acetone, Int Arch Occup Environ Health, 65: S107-S110.
Derwent RG, Jenkin ME, & Saunders SM (1996) Photochemical ozone
creation potentials for a large number of reactive hydrocarbons under
European conditions. Atmos Environ, 30(2): 181-199.
DeWalle FB & Chian ESK (1981) Detection of trace organics in well
water near a solid waste landfill. J Am Water Works Assoc, 73;
205-211.
De Walle FB, Katman DA, Norman D, Sung J, & Plews G (1985)
Determination of toxic chemicals in effluent from household septic
tanks. Cincinnati, Ohio, US Environmental Protection Agency, 25 pp
(EPA 600/S2-85-050).
De Zwart D & Slooff W (1983) The Microtox as an alternative assay in
the acute toxicity assessment of water pollutants. Aquat Toxicol,
4:129-138.
Dick RB, Setzer JV, Taylor BJ, & Shukla R (1989) Neurobehavioral
effects of short duration exposures to acetone and methyl ethyl
ketone. Br J Ind Med, 46(2): 111-121.
Dietz DD, Leininger JR, Rauckman EJ, Thompson MB, Chapin RE, Morrissey
EJ, & Levine DS (1991) Toxicity studies of acetone administered in the
drinking water of rodents. Fundam Appl Toxicol, 17: 347-360.
Dills RJ, Ackerlund WS, Kalman DA, & Morgan MS (1994) Inter-individual
variability in blood/air partitioning of volatile organic compounds
and correlation with blood chemistry. J Expo Anal Environ Epidemiol,
4(2); 229-245
Dimitriades B & Joshi SB (1977) Application of reactivity criteria in
oxidant-related emission control in the USA. In: Proceedings of
International Conference on Photochemical Oxidant Pollution and its
Control, Raleigh, North Carolina, 12-17 September 1976. Research
Triangle Park, North Carolina, US Environmental Protection Agency,
Environmental Sciences Research Laboratory, pp 705-711
(EPA 600/3-77-001b).
Ding X & Coon MJ (1990) Induction of cytochrome P-450 isozyme 3a
(P-450IIE1) in rabbit olfactory mucosa by ethanol and acetone. Drug
Metab Dispos, 18: 742-745.
DiPaolo JA, Donovon AP, & Nelson R (1969) Quantitative studies of
in vitro transformation by chemical carcinogens. J Natl Cancer Inst,
42: 867-874.
DiVincenzo GD, Yanno FJ, & Astill BD (1973) Exposure of man and dog to
low concentrations of acetone vapor. Am Ind Hyg Assoc J, 34: 329-336.
Dowden BF & Bennett HJ (1965) Toxicity of selected chemicals to
certain animals. J Water Pollut Control Fed, 37(9): 1308-1316.
Egle JL Jr (1973) Retention of inhaled acetone and ammonia in the dog.
Am Ind Hyg Assoc J, 34: 533-639.
EHRT (Environmental Health Research and Testing, Inc.) (1987)
Screening of priority chemicals for reproductive hazards: benzethonium
chloride (CAS No. 121-54-0); 3-ethoxy-1-propanol (CAS No. 111-35-3);
acetone (CAS No. 67-94-1). Cincinnati, Ohio, National Institute for
Occupational Safety and Health, 50 pp (NTIS P889-139083).
Elovaara E, Vainio H, & Aitio A (1990) Pulmonary toxicity of inhaled
styrene in acetone-phenobarbital - and 3-methylcholanthrene-treated
rats. Arch Toxicol, 64: 365-369.
Ettinger MB (1956) Biochemical oxidation characteristics of stream
pollutant organics. Ind Eng Chem, 48: 255-259.
Ewell WS, Gorsuch JW, Kringle RO, Robilliard KA, & Spiegel RC (1986)
Simultaneous evaluation of the acute effects of chemicals on seven
aquatic species. Environ Toxicol Chem, 5(9): 631-840.
Ewing BB, Chian ESK, Cook JC, Evans CA, Hopke PK, & Perkins EG (1977)
Monitoring to detect previously unrecognized pollutants in surface
waters - Appendix: Organic analysis data. Washington, DC, US
Environmental Protection Agency, pp 1, 3, 72 (EPA-560/6-77-015).
FAO/WHO (1971) Toxicological evaluation of some extraction solvents
and certain other substances. Report of the Joint FAO/WHO Expert
Committee on Food Additives, 24 June-2 July 1970 Rome, Food and
Agriculture Organization, pp 86-8.
Fiorio R & Bronzetti G (1995) Diallyl sulfide inhibits the induction
of HPRT-deficient mutants in Chinese hamster V79 cells treated with
dimethylnitrosamine in the presence of S-9 of rats induced with
acetone. Environ Mol Mutagen, 25(4): 344-346.
Fiserova-Bergerova V & Diaz ML (1986) Determination and prediction of
tissue-gas partition coefficients. Int Arch Occup Environ Health, 58:
75-87.
Folland DS, Shaffner W, Grinn HE, Crafford OB, & McMurray DR (1976)
Carbon tetrachloride toxicity potentiated by isopropyl alcohol:
Investigation of an industrial outbreak. J Am Med Assoc, 26:1853 1856.
Forkert PG, Massey TE, Jones AB, Park SS, Gelboin HV, & Anderson LM
(1991) Distribution of cytochrome CYP2E1 in murine liver after ethanol
and acetone administration. Carcinogenesis, 12(12): 2259-2268.
Frantik E, Horvárth M, & Vodicková L (1988) Effects of solvent
mixtures on behaviour and seizure characteristics at the utmost
additive. Activ Nerv Super, 30: 260-264.
Frantik E, Hornychová M, & Horvárth M (1994) Relative acute
neurotoxicity of solvents: isoeffective air concentrations of 48
compounds evaluated in rats and mice. Environ Res, 66: 173-185.
Frantik E, Vodicková L, Hornychová M, & Nosek M (1996) Pattern of
inhalation exposure: blood levels and acute subnarcotic effects of
toluene and acetone in rats. Cent Eur J Public Health, 4: 226-232.
Freeman JJ & Hayes EP (1985) Acetone potentiation of acute
acetonitrile toxicity in rats. J Toxicol Environ Health, 15: 809-621.
Freeman JJ & Hayes EP (1986) Microsomal metabolism of acetonitrile to
cyanide. Biochem Pharmicol, 37:1153-1159.
Freeman AE, Weisburger EK, Weisburger JH, Wolford RG, Maryak JM, &
Huebner RJ (1973) Transformation of cell cultures as an indication of
the carcinogenic potential of chemicals. J Natl Cancer Inst, 51(3):
799-807.
Fujino A, Satoh T, Takebayashi T, Nakashima H, Sakurai H, Higashi T,
Matumura H, Minaguchi H, & Kawai T (1992) Biological monitoring of
workers exposed to acetone in acetate fibre plants. Br J Ind Med, 4.9;
654-657.
Fukabori S, Nakaaki K, & Tada O (1979) [The cutaneous absorption of
acetone.] Jpn J Sci Labour, 55: 525-532 (in Japanese).
Gamis AS & Wasserman GS (1988) Acute acetone intoxication in a
pediatric patient. Pediatr Emerg Care, 4: 24-26.
Gardner EP, Wijayaratne RD, & Calvert JG (1984) Primary quantum yields
of the photodecomposition of acetone in air under tropospheric
conditions. J Phys Chem, 88: 5069-5076.
Gaudy AF Jr, Turner BG, & Pusztaszeri S (1968) Biological treatment of
volatile waste components. J Water Pollut Control Fed, 35: 75-93.
Gavino VC, Vinet B, David F, Garneau M, & Brunengraber H (1985)
Determination of the concentration and specific activity of acetone in
biological fluids. Anal Biochem, 152: 256-261.
Gavino VC, Somma J, Philbert L, David F, Garneau M, Bélair J, &
Brunengraber H (1987) Production of acetone and conversion of acetone
to acetate in the perfused rat liver. J Biol Chem, 262: 6735-6740.
Geller I, Hartmann RJ, Randle SR, & Gause EM (1979) Effects of acetone
and toluene vapors on multiple schedule performance of rats. Pharmacol
Biochem Behav, 11: 395-399.
Gerhold RM & Malaney GW (1966) Structural determinants in the
oxidation of aliphatic compounds by activated sludge. J Water Pollut
Control Fed, 38: 562-579.
Ghittori S, Imbriani M, Pezzagno G, & Capodaglio E (1987) The urinary
concentration of solvents as a biological indicator of exposure:
Proposal for the biological equivalent exposure limit for nine
solvents. Am Ind Hyg Assoc J, 48: 786.
Gitelson S, Werczberger A, & Herman JR (1966) Coma and hyperglycemia
following drinking of acetone. Diabetes, 15:816-811.
Glatt H, de Balle L, & Oesch F (1981) Ethanol- or acetone-pretreatment
of mice strongly enhances the bacterial mutagenicity of
dimethylnitrosamine in assays mediated by liver subcellular fraction,
but not in host-mediated assays. Carcinogenesis, 2: 1057-1067.
Glowa JR & Dews PB (1987) Behavioral toxicology of volatile organic
solvents: IV. Comparisons of the rate-decreasing effects of acetone,
ethyl acetate, methyl ethyl ketone, toluene, and carbon disulfide on
schedule-controlled behavior of mice. J Am Coll Toxicol, 6(4):
461-469.
Goldberg ME, Johnson HE, Pozzani UC, & Smyth HF Jr (1964) Effect of
repeated inhalation vapors of industrial solvents on animal behaviour.
Am Ind Hyg Assoc J, 25: 369-375.
Gordon AW & Gordon M (1981) Analysis of volatile organic compounds in
a textile finishing plant effluent. Trans Ky Acad Sci, 42: 149-157.
Gorsuch JW, Kringle RO, & Robillard KC, (1990) Chemical effects on the
germination and early growth of terrestrial plants. In: Wang W,
Gorsuch JW, & Lower WR ed. Plants for toxicity assessment.
Philadelphia, Pennsylvania, American Society for Testing and
Materials, pp 49-58 (ASTM STP 1091).
Goss K-U (1992) Effects of temperature and relative humidity on the
sorption of organic vapors on quartz sand. Environ Sci Technol, 26:
2287-2294.
Gould JP, Ramsay RE, Giabbai M, & Pohland FG (1983) Formation of
volatile haloorganic compounds in the chlorination of municipal
landfill leachates. In: Jolley RL, Brungs WA, Cotruvo JA, Cumming RB,
Mattice JS, & Jacobs VA ed. Water chlorination: Environmental impact
and health effects. Ann Arbor, Michigan, Ann Arbor Science Publishers,
vol 4, book 1, chapter 36, pp 525-539.
Graedel TE (1978) Chemical compounds in the atmosphere. New York,
London, San Francisco, Academic Press, pp 7, 182.
Graedel TE, Hawkins DT, & Claxton LD (1986) Atmospheric chemical
compounds: Sources, occurrence, and bioassay. Orlando, Florida,
Academic Press, p 263.
Granby K, Christensen CS, & Lohse C (1997) Urban and semi-rural
observations of carboxylic acids and carbonyls. Atmos Environ
31: 1403-1415.
Green WJ, Lee GF, Jones RA, & Palit T (1963) Interaction of clay
soils with water and organic solvents; Implications for the disposal
of hazardous wastes. Environ Sci Technol, 17: 278-282.
Grey TC & Shrimpton H (1967) Volatile components of raw chicken breast
muscle. Br Poult Sci, 8: 23-33.
Grosjean D & Wright 8 (1983) Carbonyls in urban fog, ice fog,
cloudwater and rainwater. Atmos Environ, 17: 2093-2096.
Grosjean D, Miguer AH, & Tavares TM (1989) Urban air pollution in
Brazil: Acetaldehyde and other carbonyls. Atmos Environ, 248: 101-106.
Guntakatta ME, Matthews EJ, & Rundell JO (1984) Development of a mouse
embryo limb bud cell culture system for the estimation of chemical
teratogenic potential. Teratogen Carcinog Mutagen, 4:349-364
Haff AC & Reichard GA Jr (1977) A method for estimation of acetone
radioactivity and concentration in blood and urine. Biochem Med, 18;
308-314.
Haggard HW, Greenberg LA, & Turner JM (1944) The physiological
principles governing the action of acetone together with determination
of toxicity. J Ind Hyg Toxicol, 26: 133-151.
Hallier E, Filser JG, & Bolt HM (1981) Inhalation pharmacokinetics
based on gas uptake studies. II. Pharmacokinetics of acetone in rats.
Arch Toxicol, 47: 293-304.
Hanst PL & Gay BW Jr (1983) Atmospheric oxidation of hydrocarbons:
Formation of hydroperoxides and peroxyacids. Atmos Environ, 17:
2259-2265.
Harris JC (1962) Fate of hydrolysis. In: Lyman WJ, Reehl WF, &
Hosenblett DH ed. Handbook of chemical property estimation methods.
New York, McGraw-Hill, pp 7/2-7/9.
Hatfield R (1957) Biological oxidation of some organic compounds. Ind
Eng Chem, 49:192-198.
Hawthorne SB & Sievers RE (1984) Emission of organic air pollutants
from shale oil wastewaters. Environ Sci Technol, 18: 483-490.
Heavner DL, Morgan WT, & Ogden MW (1996) Determination of volatile
organic compounds and respirable suspended particulate matter in New
Jersey and pennsylvania homes and workplaces. Environ Int, 22(2):
159-183.
Hellman TM & Small FH (1974) Characterization of the odor properties
of 101 petrochemicals using sensory methods. J Air Pollut Control
Assoc, 24: 979-982.
Henderson GS, McConnell JC, & Evans WFJ (1989) A comparison of model
calculations and measurements of acetone in the troposphere and
stratosphere, J Atmos Chem, 9: 277-298.
Hess FD (1980) A Chlamydomonas algal bioassay for detecting growth
inhibitor herbicides. Weed Sci, 28(5): 515-520.
Hetenyi G Jr & Ferrarotto C (1985) Gluconeogenesis from acetone in
starved rats. Biochem J, 231; 151-155.
Heukelekian H & Rand MC (1955) Biochemical oxygen demand of pure
organic compounds. J Water Pollut Control Assoc, 29:1040-1053.
Hewitt WR & Plaa GL (1983) Dose-dependent modification of
1,1-dichloroethylene toxicity by acetone, Toxicol Lett, 16: 145-152.
Hewitt WR, Miyajima H, Côté MG, & Plaa GL (1980) Acute alteration of
chloroform-induced hepato- and nephrotoxicity by n-hexane, methyl
n-butyl ketone and 2,5-hexanedione, Toxicol Appl Pharmacol, 53:
230-248.
Hewitt LA, Valiquette C, & Plaa GL (1987) The role of
biotransformation-detoxication in acetone-, 2-butanone-, and
2-hexanone-potentiated chloroform-induced hepatotoxicity. Can J
Physiol Pharmacol, 65: 2315-2318.
Hift W & Patel PL (1981) Acute acetone poisoning due to a synthetic
plaster cast with a discussion of acetone metabolism. S Afr Med J, 35;
245-250.
Hill EF, Heath RG, Spamm JW, & Williams JD (1975) Lethal dietary
toxicities of environmental pollutants to birds. Washington, DC, US
Department of the Interior, Fish and Wildlife Service, pp 1-2, 8, 35
(Special Scientific Report Wildlife No. 191).
Hites RA & Lopez-Avila V (1980) Sedimentary accumulations of
industrial organic compounds discharged into a river system. In Baker
RA ed. Contaminants and sediments - Volume 1: Fate and transport case
studies, modeling, toxicity. Ann Arbor, Michigan, Ann Arbor Science,
chapter 3, pp 53-66.
Hodgkin JH, Galbraith MN, & Chong YK (1982) Combustion products from
burning polyethylene. J Macromol Sci Chem, 17: 35-44.
Hodgson AT, Daisey JM, & Grot RA (1991) Sources and source strengths
of volatile organic compounds in a new office building. J Air Waste
Manage Assoc, 41: 1461-1468.
Hodgson AT, Garbesi K, Sextro RG, & Daisey JM (1992) Soil-gas
contamination and entry of volatile organic compounds into a house
near a landfill. J Air Waste Manage Assoc, 42; 277-283.
Hodgson AT, Wooley JD, & Daisey JM (1993) Emissions of volatile
organic compounds from new carpets measured in a large-scale
environmental chamber. J Air Waste Manage Assoc, 42: 316-324.
Hoffman DJ & Eastin WC (1981) Effects of industrial effluents, heavy
metals and organic solvents on mallard embryo development, Toxicol
Lett, 9: 35-40.
Holm S & Lundgron E (1984) A purge-and-tap method for the analysis of
acetone in biological tissues. Anal Biochem, 136: 157-180.
Hong J & Yang CS (1985) The nature of microsomal
N-nitrosodimethylamine demethylase and its role in carcinogen
activation. Carcinogenesis, 6: 1805-1809.
Hung PE, Fritz VA, & Waters L Jr (1992) Infusion of shrunken-2 sweet
corn seed with organic solvents: Effects on germination and vigor.
Hortic Sci, 27(5): 467-470.
Hwang H-M, Hodson RE, & Lewis DL (1989) Microbial degradation kinetics
of toxic organic chemicals over a wide range of concentrations in
natural aquatic systems. Environ Toxicol Chem, 8: 65-74.
Isidorov VA, Zenkevich IG, & Ioffe BV (1985) Volatile organic
compounds in the atmosphere of forests. Atmos Environ, 19: 1-6.
Iversen OH, Ljunggren S, & Olsen WM (1988) The early effects of a
single application of acetone and various doses of
7,12-dimethylbenz(alpha)anthracene on CD-1 and hairless mouse
epidermis: A cell kinetic study of so-called initiation and complete
carcinogenesis (initiation plus promotion) in chemical skin tumor
induction. Acta Pathol Microbial Immunol Scand, 27(suppl): 7-80
Jaeger RJ, Conolly RB, Reynolds ES, & Murphy SO (1975) Biochemical
toxicology of unsaturated halogenated monomers. Environ Health
Perspect, 11: 121-128.
Jakubowski M & Wieczorek H (1988) The effects of physical effort on
pulmonary uptake of selected organic compound vapours. Pol J Occup
Med, I: 62-71.
Jansson BO & Lareson BT (1969) Analysis of organic compounds in human
breath by gas chromatography-mass-spectrometry. J Lab Clin Med, 74:
961-966.
Jarke FH, Dravnieks A, & Gordon SM (1981) Organic contaminants in
indoor air and their relation to outdoor contamination. Trans Am Soc
Heating Refrig Air.-Cond Eng. 87: 153-16.
Jeffery EH, Arndt K, & Haschek WM (1991) The role of cytochrome
P45011E1 in bioactivation of acetaminophen in diabetic and
acetone-treated mice. In: Witmer CM, Snyder RR, & Jollow DJ ed.
Biological reactive intermediates- IV: Molecular and cellular effects
and their impact on human health. New York, Plenum Publishing
Corporation, pp 249-251.
Johansson I & Ingelman-Sundberg M (1988) Benzene metabolism by
ethanol-, acetone- and benzene-inducible cytochrome P-450 (IIE1) in
rat and rabbit liver microsomes. Cancer Res, 48: 5837-5390.
Johansson I, Eliasson E, Norsten C, & Ingelman-Sundberg M (1986)
Hydroxylation of acetone by ethanol- and acetone-inducible cytochrome
P-450 in liver microsomes and reconstituted membranes. FEBS Lett, 196:
59-64.
Johanason I, Ekström E, Scholte B, Puzycki D, Jörnvall H,
& Ingelman-Sundberg M (1988) Ethanol-, fasting-, and acetone-inducible
cytochromes P-450 in rat liver: regulation and characteristics of
enzymes belonging to the IIB and IIE gene subfamilies. Biochemistry,
27(6): 1925-1934.
Johnson CE & Jenkin ME (1991) Modelling the photochemistry of acetone.
Harwell, Atomic Energy Agency, Environment and Energy, 29 pp.
Jones AW & Andersson L (1995) Biotransformation of acetone to
isopropanol observed in a motorist involved in a sobriety check. J
Forensic Sci, 40(4): 586487.
Jones AW, Sagarduy A, Ericsson E, & Arnqvist HJ (1993) Concentrations
of acetone in venous blood samples from drunk drivers, Type-I diabetic
outpatients, and healthy blood donors. J Appl Toxicol, 17; 182-185.
Jonssen A, Persson KA, & Grigoriadis V (1985) Measurements of some
low-molecular -weight oxygenated, aromatic and chlorinated
hydrocarbons in ambient air and in vehicle emissions. Environ Int, 11:
383-392.
Jungclaus GA, Lopez-Avila V, & Hites RA (1976) Organic compounds in an
industrial waste water: A case study of their environmental impact.
Environ Sci Technol, 12: 88-96.
Kamlet M J, Doherty RM, Veith GD, Taft RW, & Abraham MH (1986)
Solubility properties in polymers and biological media: 7. An analysis
of toxicant properties that influence inhibition of bioluminescence in
Photobacterium phosphoroum (the Microtox test). Environ Sci Technol,
20(7): 690-695.
Kawai T, Yasugi T, Uchida Y, Iwami O, & Ikeda M (1990a) Urinary
excretion of unmetabolized acetone as aa indicator of Occupational
exposure to acetone. Int Arcb Occup Environ Health, 62: 165-170.
Kawai T, Yasugi T, Horiguchi S, Uchida Y, Iwami O, Iguchi H, Inoue O,
Watanabe T, Nakatsuka H, & Ikeda M (1990b) Biological monitoring of
occupational exposure to isopropy alcohol vapor by urinalysis for
acetone. Int Arch Occup Environ Health, 62: 409-13.
Kawai T, Yasugi T, Mizunuma K, Horiguchi S, Iguchi H, & Ikeda M (1992)
Curvi-linear relation between acetone in breathing zone air and
acetone in urine among workers exposed to acetone vapor. Toxicol Lett,
62: 85-91.
Kerr JA & Stocker DW (1986) Kinetics of the reactions of hydroxyl
radicals with alkyl nitrates and with some oxygen-containing organic
compounds studied under simulated atmospheric conditions. J Atmos
Chem, 4: 253-262.
Kieber RJ & Mopper K (1990) Determination of picomolar concentrations
of carbonyl compounds in natural waters, including seawater, by liquid
chromatography. Environ Sci Technol, 24: 1477-1481.
Kiesswetter E, Blaszekewicz M, Vangala RR, & Seeber A (1994) Acute
exposure to acetone in a factory and ratings of well-being.
Neurotoxicology, 15: 597-602.
Kimura ET, Ebert DM, & Dodge PW (1971) Acute toxicity and limits of
solvent residue for sixteen organic solvents. Toxicol Appl Pharmacol,
19: 699-704.
Kinlin TE, Muralidhara R, Pittet AO, Sanderson A, & Walradt JP (1972)
Volatile components in roasted filberts. J Agric Food Chem, 20:
1021-1028.
Kitchin KT & Ebron MT (1984) Further development of rodent whole
embryo culture: Solvent toxicity and water insoluble compound delivery
system. Toxicology, 30: 45-57.
Kleindienst TE, Shepson PB, Edney EO, Claxton LD, & Cupitt LT (1986)
Wood smoke: measurement of the radiogenic activities of its gas and
particulate-phase photo-oxidation products. Environ Sci Technol, 20:
493-501.
Kleppel GS & McLaughlin JJA (1980) PCB toxicity to phytoplankton:
Effects of dose and density-dependent recovery responses. Bull Environ
Contam Toxicol, 24(5): 696-703.
Knöppel H & Schauenburg H (1969) Screening of household products for
the emission of volatile organic compounds. Environ Int, 15: 413-418.
Kobayashi K, Okada M, Yasuda Y, & Kawai S (1983) A gas chromatographic
method for the determination of acetone and acetoacetic acid in urine.
Clin Chim Acta, 133: 223-226.
Kobusch AB, Plaa GL, & DuSonich P (1989) Effects of acetone and methyl
n-butyl ketone on hepatic mixed function oxidase. Biochem Pharmacol,
38: 3461-3467.
Koop DR & Casazza PJ (1985) Identification of ethanol-inducible
p-450 isozyme 3a as the acetone and acetol monooxygenase of rabbit
microsomes. J Biol Chem, 260:13607-13612.
Korhonen A, Hemminki K, & Vainio H (1983) Embryotoxic effects of
acrolein, methacrylates, guanidines and resorcinol on three day chicken
embryos. Acta Pharmacol Toxicol, 52: 95-99.
Kosugi K, Chandramouli V, Kumaran K, Schumann WC, & Landau BR (1986a)
Determinants in the pathways followed by the carbons of acetone in
their conversion to glucose. J Biol Chem, 261: 13179-13181.
Kosugi K, Scofield RF, Chandramouli V, Kumaren K, Schumann W, & Landau
BR (1986b) Pathways of acetone's metabolism in the rat. J Biol Chem,
162: 3952-3957.
Krasavage WJ, O'Donoghue JL, & DiVincenzo GD (1982) Ketones. In:
Clayton GD & Clayton FE ed. Patty's industrial hygiene and toxicology,
3rd ed. New York, John Wiley & Sons, vol II, pp 4709, 4720-4721,
4724-4727, 4780-4800.
Kubinski H, Gutzke GE, & Kubinski ZO (1981) DNA-cell binding (DCB)
assay for suspected carcinogens and mutagens. Mutat Res, 89: 95-136.
Kumagai S & Matsunaga I (1995) Physiologically based pharmacokinetic
model for acetone, Occup Environ Med, 52: 344-352.
Kundu SK, Bruzek JA, Nair R, & Judilla AM (1993) Breath acetone
analyzer: diagnostic tool to monitor dietary fat loss. Clin Chem,
39(1): 87-92.
Kupferschmid LL & Perkins JL (1986) Organic solvent recycling plant
exposure levels. Appl Ind Hyg, 1: 122-124.
Lacouture PG, Heldreth DD, Shannon M, & Lovejoy FH (1989) The
generation of acetonemia/acetonuria following ingestion of a subtoxic
dose of isopropyl alcohol. Am J Emerg Med. 7(1): 38-40.
Ladefoged O & Perbellini L (1986) Acetone-induced changes in the
toxicokinetics of 2,5-hexanedione in rabbits. Scand J Work Environ
Health, 122: 627-29.
Ladefoged O, Hass U, & Simonsen L (1999) Neurophysiological and
behavioural effects of combined exposure to 2,5-hexanedione and
acetone or ethanol in rats. Pharmacol Toxicol, 65: 372-375.
Lam HR, Larsan JJ, Ladefoged O, Mollor A, Strange P, & Arlien-Soborg P
(1991) Effects of 2,4-hexanedione alone and in combination with
acetone on radial arm maze behavior, the "brain-swelling" reaction and
synaptosomal functions. Neurotoxicol Teratol, 13: 407-412.
Lamb CB & Jenkins GF (1952) BOD of synthetic organic chemicals, in:
Proceedings of the 8th Industrial Waste Conference, Purdue University,
Lafayette, Indiana, USA, 7-9 May 1952. Lafayette, Purdue University,
pp 326-331.
Landahl HD & Herrmann RG (1950) Retention of vapors and gases in the
human nose and lung. Am Med Assoc Arch Ind Health, 1: 36-45.
LaRegina J, Bozzelli JW, Harkov R, & Gianti S (1986) Volatile organic
compounds at hazardous waste sites and a sanitary landfill in New
Jersey: An up-to-date review of the present situation. Environ Prog,
8: 18-27.
Larsen J-J, Lykkegeard M, & Ladefoged O (1991) Infidelity in rats
induced by 2,4-hexanedione in combination with acetone. Pharmacol
Toxicol, 69: 43-46.
Larson TS, Finnegan JK, & Haag HD (1956) Observations on the effect of
the edema-producing potency of acids, aldehydes, ketones and alcohols.
J Pharmacol Exp Ther, 116; 119.
Lee JH & Timmons RB (1977) Kinetics and mechanism of the gas phase
reaction of O(3) atoms with acetone. Intl Chem Kinet, 9: 133-139.
Leibrock E & Slemr J (1997) Method for measurement of volatile
oxygenated hydrocarbons in ambient air. Atmos Environ, 31 (20):
3329-3339.
Leonardos G, Kendall D, & Barnard N (1969) Odor threshold determination
of 53 odorant chemicals. J Air Pollut control Assoc, 19: 91-95.
Lewis GD, Laufman AK, McAnalley BH, & Garriott JC (1984) Metabolism
of acetone to isopropyl alcohol in rats and humans. J Forensic Sci,
29: 541-549.
Lindén E, Bengtsson B-E, Svanberg O, & Sundström G (1979) The acute
toxicity of 78 chemicals and pesticide formulations against two
brackish water organisms, the Bleak ( Alburnus alburnus) and the
Harpacticoid Nitocra spinipes. Chemosphere, 11/12: 843-851.
Lipari F, Dasch JM, & Screggs WF (1984) Aldehyde emissions from
wood-burning fireplaces. Environ Sci Technol, 18: 326-330.
Litovitz TL, Holm KC, Bailey KM, & Schmitz BF (1992) 1991 Annual
report of the American Association of Poison Control Centers National
Data Collection System. Am J Emerg Med, 10: 452-505.
Liu J, Sato C, & Marumo F (1991) Characterization of the
acetaminophen-glutathione conjugation reaction by liver microsomes:
Species difference in the effects of acetone. Toxicol Lett, 56:
260-274.
Lloyd AC (1978) Trophospheric chemistry of aldehydes. Washington, DC,
National Bureau of Standards, pp 27-48.
LO H-H, Teets VJ, Yang DJ, Brown PI, & Rankin GO (1987) Acetone
effects on N-(3,5-dichlorophenyl) succinimide-induced
nephrotoxicity. Toxicol Lett, 38: 161-166.
Longo V, Menicagli S, Minks M, Santucci A, & Gervasi PG (1993)
Purification and Characterization of hepatic P-450IIE1 from
acetone-treated mice. Toxicol and Health, 9: 539-546.
Lorr NA, Miller KW, Chung HR, & Yang CS (1984) Potentiation of the
hepatotoxicity of n-nitrosodimethylamine by fasting, diabetes,
acetone and isopropanol. Toxicol Appl Pharmacol, 73: 423-431.
Lovegren NV, Fisher GS, Legendre MG, & Schuller WH (1979) Volatile
constituents of dried legumes. J Agric Food Chem, 27: 851-853.
Ludzack FJ & Ettinger MB (1960) Chemical structures resistant to
aerobic biochemical stabilization. J Water Pollut Control Fed,
32:1173-1200.
Lupulescu AP & Birmingham DJ (1975) Effect of lipid solvents on
protein, DNA, and collagen synthesis in human skin: An electron
microscopic autoradiographic study. J Invest Dermatol, 65: 419-422.
Lupulescu AP & Birmingham DJ (1979) The effect of protective agent
against lipid solvent induced damages: ultrastructural and scanning
electromicroscopical study of human epidermis. Arch Environ Health,
31: 33-36.
Lupulescu AP, Birmingham DJ, & Pinkus H (1973) An electron microscopic
study of human epidermis after acetone and kerosene administration. J
Invest Dermatol, 65: 419-422.
Lyman WJ (1982) Adsorption coefficient for soils and sediments. In:
Handbook of chemical property estimation methods. New York,
McGraw-Hill, pp 4-9.
McCann J, Choi E, Yamasaki E, & Ames BN (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc Natl Aced Sci (USA), 72:5135-5139.
MacDonald JR, Gandolfi AJ, & Sipes IG (1962a) Acetone potentiation of
1,1,2-trichloroethane hepatotoxicity. Toxicol Lett, 13: 57-69.
MacDonald JR, Gandolfi AJ, & Sipes IG (1962b) Covalent binding of
14C-1,1,2-trichloroethane to hepatic proteins following acetone
pretreatment, Drug Chem Toxicol, 5: 233-247.
McFeters GA, Bond JP, Olson SB, & Tchan TY (1983) A comparison of
microbial bioassays for the detection of aquatic toxicants. Water Res,
17(12): 1757-1762.
Majewski HS, Klaverkamp JF, & Scott DP (1977) Acute lethality, and
sub-lethal effects of acetone, ethanol, and propylene glycol on the
cardiovascular and respiratory systems of rainbow trout ( Salmo
galrdneri). Water Res, 13: 217-221.
Mangani F & Ninfali P (1986) Gas chromatographic determination of
acetaldehyde and acetone in human blood by purge and trap, using
permeation tubes for calibration. J Chromatogr, 437(1): 294-300.
Manning DL, Maskarinec MP, Jenkins PA, & Marshall AH (1983) High
performance liquid chromatographic determination of selected gas-phase
carbonyls in tobacco smoke. J Asset Off Anal Chem, 66: 5-12.
Martinez RD, Buitraga AA, Howell NW, Hearn CH, & Joens JA (1992) The
near U.V. absorption spectra of several aliphatic aldehydes and
ketones at 300 K. Atmos Environ, 26A; 785-792.
Matsushita T, Yoshimune A, Inoue T, Yamaka S, & Suzuki H (1969a)
Experimental studies for determining the maximum permissible
concentrations of acetone: 2. Biological reactions in six-day exposure
to acetone. Sangyo Igaku, 11: 507-511.
Matsushita T, Yoshimune A, Inoue T, Yamaka S, & Suzuki H (1969b)
Experimental studies for determining the maximum permissible
concentrations of acetone: I. Biological reactions in one-day exposure
to acetone. Sangyo Igaku, 11: 477-485.
Mayer FL & Ellersieck MR (1980) Manual of acute toxicity:
Interpretation and data base for 410 chemicals and 66 species of
freshwater animals. Washington, DC, US Department of the Interior,
Fish and Wildlife Service, pp 1,4 (Resource Publication No. 160).
Menicagli S, Puccini P, Longo V, & Gervasi PG (1990) Effect of acetone
administration on renal, pulmonary and hepatic monooxygenase
activities in hamster. Toxicology, 4: 141-153.
Meyrahn H, Pauly J, Schneider W, & Warneck P (1986) Quantum yields for
the photodissociation of acetone in air and an estimate for the life
time of acetone in the lower troposphere. J Atmos Chem, 4: 277-291.
Mikalsen A, Alexander J, Andersen PA, & Ingelman-Sundberg M (1991)
Effect of in vivo chromate, acetone and combined treatment on rat
liver in vitro microsomal chromium(VI) reductive activity and on
cytochrome P450 expression. Pharmacol Toxicol, 68: 456-463.
Miltiana LM & Mauch SC (1989) Statistical modeling of ambient air
toxics impact during remedial investigation at a landfiil site. In:
Proceedings of the 10th National Conference "Superfund '89",
Washington, DC, 27-29 November 1989. Washington, DC, The Hazardous
Materials Control Research Institute, pp 157-162.
Mill T & Mabey W (1985) Photochemical transformations. In: Neely WB &
Blau GE ed. Environmental exposure from chemicals. Boca Raton,
Florida, CRC Press, vol 1, pp 175-216.
Miller KW & Yang CS (1984) Studies on the mechanisms of induction of
N-nitroso-diethylamine demethylase by fasting, acetone, and ethanol.
Arch Biochem Biophys, 229: 483-491.
Mizunuma K, Yasugi T, Kawai T, Horiguchi S, & Ikeda M (1993)
Exposure-excretion relationship of styrene and acetone in factory
workers; a comparison of a lipophilic solvent and a hydrophilic
solvent. Arch Environ Contain Toxicol, 25(1): 129-133.
Mohr DH & King CJ (1985) Identification of polar organic compounds in
coal-gasification condensate water by gas chromatography-mass
spectrometry analysis of high pressure liquid chromatography
fractions. Environ Sci Technol, 19: 929-935.
Moldeus P & Gergely V (1980) Effect of acetone on the activation of
acetaminophen. Toxicol Appl Pharmacol, 53: 9-13.
Monder C (1997) alpha-Keto aldehyde dehydrogenase, an enzyme that
catalyzes the enzymic oxidation of methygiyoxal to pyruvate. J Biol
Chem, 242: 4603-4609.
Mopper K & Stahovec WL (1986) Sources and sinks of low molecular
weight organic carbonyl compounds in seawater. Mar Chem, 19: 305-321.
Morgott D (1993) Acetone. In: Clayton GE & Clayton FE ed. Patty's
industrial hygiene and toxicology, 4th ed. New York, John Wiley &
Sons, vol II, part A, chapter 5, pp 149-281.
Morris JB (1991) Deposition of acetone vapor in the upper respiratory
tract of the B6C3F1 mouse. Toxicol Lett, 56: 187-196.
Morris JB & Cavanagh DG (1986) Deposition of ethanol and acetone
vapors in the upper respiratory tract of the rat. Fundam Appl ToxicoI,
6: 78-88.
Morris JB & Cavanagh DG (1987) Metabolism and deposition of propanol
and acetone vapors in the upper respiratory tract of the hamster.
Fundam Appl Toxicol, 9: 34-40.
Morris JB, Clay RJ, & Cavanagh DG (1986) Species differences in upper
respiratory tract deposition of acetone and ethanol vapors. Fundam
Appl Toxicol, 7(4): 671480.
Mourkides GA, Hobbs DC, & Koeppe RE (1959) The metabolism of
acetone-2-C14 by intact rats. J Biol Chem, 234: 27-30.
NAS (1976) Vapor-phase organic pollutants: Volatile hydrocarbons and
oxidation products. Washington, DC, National Academy of Sciences,
Committee on Medical and Biologic Effects of Environmental Pollutants,
pp 23-24, 26-27.
Nelson DL & Webb BP (1978) Acetone. In: Greyson M & Eckroth D ed.
Kirk-Othmer encyclopedia of chemical technology, 3rd ed. New York,
John Wiley & Sons, vol 1, pp 179-191.
Nelson KW, Ege JF Jr, Ross M, Woodman LE, & Silverman L (1943) Sensory
response to certain industrial solvent vapors. J Ind Hyg Toxicol 25:
282-285.
NIOSH (1994) Manual of analytical methods - Ketones 1 (acetone);
method 1300, 4th ed. Cincinnati, Ohio, National Institute for
Occupational Safety and Health, 5 pp.
Nirmalakhandan N, Sun B, Arulgnandran VJ, Mohsin M, Wang XH, Prakash J,
& Hall N (1994a) Analyzing and modeling toxicity of mixtures of
organic chemicals to micro-organisms. Water Sci Technol, 30(10): 87-90
Nimalakhandan N, Arulgnanendran V, Mohsin M, Sun B, & Cadena F (1994b)
Toxicity of mixtures of organic chemicals to microorganisms. Water
Res, 28(3): 543-551.
NLM (1992) Hazardous substances databank. Washington, DC, National
Library of Medicine, vol 11,45 pp.
Nomiyama K & Nomiyama H (1974a) Respiratory retention, uptake and
excretion of organic solvents in man: Benzene, toluene, n-hexane,
trichloroethylene, acetone, ethyl acetate and ethyl alcohol. Int Arch
Arbeitsmed, 32; 75-83.
Nomiyama K & Nomiyama H (1974b) Respiratory elimination of organic
solvents in man: Benzene, toluene, n-hexane, trichloroethylene,
acetone, ethyl acetate and ethyl alcohol. Int Arch Arbeitsmed, 32:
65-91.
Norppa H, Hemminki K, Sorsa M, & Vainio H (1981) Effect of
monosubstituted epoxides on chromosome aberrations and SCE in cultured
human lymphocytea, Mutat Res, 91: 243-250.
NTP (1988) Inhalation developmental toxicology studies: teratology
study of acetone in mice and rats: Final report. Research Triangle
Park, North Carolina, US Department of Health and Human Services,
National Toxicology Program, 260 pp (Report No. PNL-6T68, prepared by
Pacific Northwest Laboratory).
NTP (1991) Toxicity studies of acetone in E344/N rats and S6C3F1 mice
(drinking water studies). Research Triangle Park, North Carolina, US
Department of Health and Human Services, National Toxicology Program,
38 pp (Technical Report No. 3; NIH Publication No. 91-3122).
OECD (1981) Guidelines for testing of chemicals 301 D. Ready
biodegradability: Closed bottle test. Paris, Organisation for Economic
Co-operation and Development, 13 pp.
O'Hara D & Singh HB (1988) Sensitive gas chromatographic detection of
acetaldehyde and acetone using a reduction gas detector. Atmos
Environ, 22(11): 2613-2616.
Ott MG, Skory LK, Holder BB, Bronson JM, & Williams PR (1983a) Health
evaluation of employees occupationally exposed to methylene chloride:
general study design and environmental considerations. Scand J Work
Environ Health, 9(suppl 1): 1-7.
Ott MG, Skory LK, Holder BB, Bronson JM, & Williams PR (1963b) Health
evaluation of employees occupationally exposed to methylene chloride:
mortality, Scand J Work Environ Health, 9(suppl 1); 8-16.
Owen OE, Trapp VE, Skutches CL, Mozzoli MA, Hoeldtke RD, Boden G, &
Reichard CA Jr (1962) Acetone metabolism during diabetic ketoacidosis.
Diabetes, 31: 242-248.
Palo V & Ilkova H (1970) Direct gas chromatographic estimation of
lower alcohols, acetaldehyde, acetone and diacetyl in milk products. J
Chromatogr, 53: 363-367.
Pankow D & Hoffmann P (1989) Dichloromethane metabolism to carbon
monoxide can be induced by isoniazid, acetone and fasting. Arch
Toxicol, 13(suppl); 302-303.
Papa AJ & Sherman PD Jr (1981) Ketones. In: Grayson M & Eckroth D ed.
Kirk-Othmer encyclopedia of chemical technology, 3rd ed. New York,
John Wiley & Sons, vol 13, p 895.
Parasher CD, Ozel M, & Geike F (1978) Effect of hexachlorobenzene and
acetone on algal growth: Physiology and ultrastructure. Chem-Biol
Interact, 20: 89-95.
Paterson S & Mackay D (1989) Correlation of tissue, blood, and air
partition coefficients of volatile organic chemicals. Br J Ind Med,
46: 321-328.
Patrick R, Cairns J Jr, & Schier A (1968) The relative sensitivity of
diatoms, snails and fish to twenty common constituents of industrial
wastes, prog Fish-Cult, 30(3): 137-140.
Patten CJ, Ning SM, Lu AYH, & Yang CS (1986) Acetone-inducible
cytochrome P-450 Purification, catalytic activity, and interaction
with cytochrome b5. Arch Biochem Biophys, 251: 629-38.
Peinado J, Lopez-Sodano FJ, & Argiles JM (1986) The metabolism of
acetone in the pregnant rat. Biosci Rep, 6: 983-989.
Pellizzari ED, Castillo NP, Willis S, Smith M, & Burney JT (1979)
Identification of organic components in aqueous effluents from
energy-related processes. In: Van Hall CE ed. Measurement of organic
pollutants in water and wastewater. Washington, DC, American Society
for Testing and Materials, pp 256-274 (Publication STP 666).
Pellizzari ED, Hartwell TD, Harris BSH, Waddell RD, Whitaker DA, &
Erickson MD (1962) Purgeable organic compounds in mother's milk. Bull
Environ Contain Toxicol, 28: 322-28.
Perry DL, Chuang CC, Jungclaus GA, & Warner JS (1978) Identification
of organic compounds in industrial effluent discharges (Prepared by
Battelle Columbus Laboratory). Athens, Georgia, US Environmental
Protection Agency, Environmental Research Laboratory, pp 44, 46
(EPA-560/678-009; NTIS PB-291900).
Perry DL, Chuang CC, Jungclaus GA, & Warner JS (1979) Identification
of organic compounds in industrial effluent discharges. Washington,
DC, US Environmental Protection Agency, 34 pp (NTIS PB 294794).
Pezzagno G, Imbriani M, Ghittori S, Capodaglio E, & Huang J (1986)
Urinary elimination of acetone in experimental and occupational
exposure. Scand J Work Environ Health, 12(6): 603608.
Phillips M & Greenberg J (1987) Detection of endogenous acetone in
normal human breath. J Chromatogr, 422: 235-238.
Plaa GL & Traiger GJ (1972) Mechanism of potentiation of CCI4-induced
hepatotoxicity. In: Loomis TA ed. Pharmacology and the future of
man - Volume 3: Toxicological problems. Basel, Switzerland, Larger, pp
100-113.
Plaa GL, Traiger GJ, Hanasono GK, & Witschi H (1973) Effect of
alcohols on various forms of chemically induced liver injury. In:
Khanna JM, Israel Y, & Kalant H ed. Alcoholic liver pathology.
Toronto, Ontario, Canada, Alcoholism and Drug Addiction Research
Foundation of Ontario, pp 225-244.
Plaa GL, Hewitt WR, Du Souich P, Caillè G, & Lock S (1982) Isopropanol
and acetone potentiation of carbon tetrachloride-induced
hepatotoxicity: Single versus repetitive pretreatments in rats. J
Toxicol Environ Health, 9: 235-250.
Platen H, Temmes A, & Schink B (1990) Anaerobic degradation of acetone
by Desulfococcas biacutus spec. nov. Arch Microbiol, 154: 335-361.
Plumb RH Jr (1987) A comparison of ground water monitoring data from
CERCLA and RCRA sites. Ground Water Monit Rev, 7:94-100.
Pomerantz RB (1950) Acute acetone poisoning from castex. Am J Surg,
80:117-118.
Pozzani UC, Weil CS, & Carpenter CP (1959) The toxicological basis of
threshold limit values: 5. The experimental inhalation of vapor
mixtures by rats, with notes upon the relationship between single dose
inhalation and single dose oral data. Am Ind Hyg Assoc J, 20: 364-369.
Price VF & Jollow DJ (1983) Mechanism of ketone-induced protection
from acetaminophen hepatotoxicity in the rat. Drug Metab Dispos, 11:
451-457.
Price TD & Rittenberg D (1950) The metabolism of acetone: I. Gross
aspects of catabolism and excretion. J Biol Chem, 185: 449-459.
Price KS, Waggy GT, & Conway RA (1974) Brine shrimp bioaasay and
seawater BOD of petrochemicals. J Water Pollut Control Fed, 46: 65-77.
Puccini P, Fiorio R, Longo V, & Gervasi PG (1989) High affinity
diethylnitrosamine-deethylase in liver microsomes from
acetone-induced rats. Carcinogenesis, 19: 1629-1634.
Puccini P, Fiorio R, Longa V, & Gervasi PG (1990) Effects of acetone
administration on drug-metabolizing enzyme in mice: Presence of a
high-affinity diethylnitrosamine deethyiase. Toxicol Lett, 54:
143-150.
Puccini P, Menicagli S, Longo V, Santucci A, & Gervasi PG (1992)
Purification and characterization of an acetone-inducible cytochrome
P-450 from hamster liver microsomes. Biochem J, 287: 863-870.
Racker E (1951) The mechanism of action of glyoxalase. J Biol Chem,
190: 685-698.
Rahim SA & Bashir WA (1981) Microdetermination of acetone in aqueous
solution. Microchem J, 26(3): 329-333.
Rajini PS, Krishnakumari MK, & Majumder SK (1989) Cytotoxicity of
certain organic solvents and organophosphorus insecticides to the
ciliated protozoan Paramecium caudatum. Microbios, 59: 157-163.
Raleigh RL & McGee WA (1972) Effects of short, high-concentration
exposures to acetone as determined by observation in the work area. J
Occup Med, 14(8): 607-610.
Ramu A, Rosenbaum J, & Blaschke TF (1978) Disposition of acetone
following acute acetone intoxication. West J Mad, 129: 429-32.
Randall TL & Knopp PV (1980) Detoxification of specific organic
substances by wet oxidation. J Water Pollut Control Fed, 52(8):
2117-2130.
Rathbun RE & Tai DY (1982) Volatilization of ketones from water. Water
Air Soil Pollut, 17: 281-293.
Rathbun RE & Tai DY (1887) Vapor pressures and gas-film coefficients
for ketones. Chemosphere, 18: 69-78.
Rathbun RE, Stephens DW, Shultz DJ, & Tai DY (1982) Fate of acetone in
water. Chemosphere, 11: 1097-1114.
Rathbun RE, Stephens DW, Schultz DJ, & Tai DY (1988) Fate of acetone
in an outdoor model stream in southern Mississippi. J Hydrol, 104:
181-209.
Rathbun RE, Shultz DJ, Stephens DW, & Tai DY (1989) Transport and fate
of acetone in an outdoor model stream, Stennis Space Center Near Bay
St. Louis, Mississippi. Denver, Colorado, US Geological Survey, 191 pp
(Water-Resources Investigations Report No. 894141).
Rathbun RE, Stephens DW, & Tai DY (1991) Fate of acetone in an outdoor
model stream with a nitrate supplement, southern Mississippi, USA. J
Hydrol, 123: 225-242.
Rathbun RE, Stephens DW, & Tai DY (1993) Bacterial degradation of
acetone in an outdoor model stream. Environ Pollut, 79: 153-182.
Raymond P & Plaa GL (1995a) Ketone potentiation of haloalkane-induced
hepatic- and nephrotoxicity: I. Dose-response relationships. J Toxicol
Environ Health, 45: 455-480.
Raymond P & Plaa GL (1995b) Ketone potentiation of haloalkane-induced
hepoto- and nephrotoxicity: I. Implication of monooxygenases. J
Toxicol Environ Health, 46; 317-378.
Reichard GA Jr, Haff AC, Skutches CL, Holroyde CP, & Owen OE (1979)
Plasma acetone metabolism in the fasting human. J Clin Invest, 63:
619-628.
Reichard GA Jr, Skutches CL, Holroyde CP, & Owen OE (1986) Acetone
metabolism in humans during diabetic ketoacidosis. Diabetes, 35:
668-674.
Rengstorff RH & Khafagy HI (1985) Cutaneous acetone depresses aqueous
humor ascorbate in guinea pigs. Arch Toxicol, 58: 64-66.
Rengstorff RH, Petrali JP, & Sim VM (1972) Cataracts induced in guinea
pigs by acetone, cyclohexanone, and dimethyl sulfoxide. Am J Optom
Arch Acad Optom, 49: 308-319.
Renshaw PK & Mitchell RM (1956) Acute poisoning following the
application of a lightweight cast. Br Mad J, 1:615.
Riddick JA, Bunger WB, & Sakano TK (1986) Organic solvents: Physical
properties and methods of purification. In: Techniques of chemistry,
4th ed. New York, Wiley-Interscience, vol 2, pp 336-337.
Risner CH (1995) High performance liquid chromatographic determination
of major carbonyl compounds from various sources in ambient air. J
Chromatogr Sci, 33; 168-176.
Ross DG (1973) Acute acetone intoxication involving eight male
workers, Am Occup Hyg J, 16: 73-75.
Ruddick JA (1972) Toxicology, metabolism, and biochemistry of
1,2-propanediol. Toxicol Appl Pharmacol, 21; 102-111.
Rudney H (1954) Propanediol phosphate as a possible intermediate in
the metabolism of acetone. J Stol Chain, 210: 361-371.
Sabel GV & Clark TP (1984) Volatile organic compounds as indicators of
municipal solid waste leachete contamination. Waste Manage Res,
2:119-130.
Sack TM, Steele DH, Hammerstrom K, & Remers JA (1992) A survey of
household products for volatile organic compounds. Atmos Environ,
26A(6): 1063-1070.
Sakami W & LaFaye JM (1950) Formation of formate and labile methyl
groups from acetone in the intact rat. J Biol Chem, 187: 369-378.
Sakami W & Rodney H (1952) The metabolism of acetone and acetoacetate
in the mammalian organism. Brookhaven Syrup Biol, 5: 176-191.
Sakata M, Kikuchi J, Haga M, Ishiyama N, Maeda T, Isa T, & Hikita N
(1989) Disposition of acetone, methyl ethyl ketone and cyclohexanone
in acute poisoning. J Toxicol Clin Toxicol, 27: 67-77.
Sallee ED & Sappington CO (1949) Eleventh Annual Meeting of the
American Conference of Governmental Industrial Hygienists / Tenth
Annual Meeting of the American Industrial Hygiene Association,
Detroit. Michigan, 2-7 April 1949. Ind Hyg Q, 10: 36-44.
Sangster J (1989) Octanol-water partition coefficients of simple
organic compounds. J Phys Chem Ref Data, 18:1111-1229.
Sato A & Nakajima T (1979) Partition coefficients of some aromatic
hydrocarbons and ketones in water, blood and oil. Br J Ind Med,
36: 231-234.
Satoh T, Omae K, Takebayashi T, Nakashima H, Higashi T, & Sakurai H
(1995) Acetone excretion into urine of workers exposed to acetone in
acetate fiber plants. Int Arch Occup Environ Health, 67: 131-134.
Satoh T, Omae K, Nakashima H, Takebayashi T, Matsumura H, Kawat T,
Nakaza M, & Sakurai H (1996) Relationship between acetone exposure
concentration and health effects in acetate fiber plant workers. Int
Arch Occup Environ Health. 68:147-153.
Sawhney BL & Kozloski RP (1984) Organic pollutants in leachates from
landfill sites. J Environ Qual, 13; 349-352.
Sawhney BL & Raabe JA (1986) Ground water contamination: Movement of
organic pollutants in the Granby landfill, New Haven, Connecticut, The
Connecticut Agricultural Experiment Station, 9 pp (Connecticut
Agricultural Experiment Station BulLetin No. 833).
Sax NI & Lewis RJ Sr (1987) Acetone. In: Hawley's condensed chemical
dictionary, 11th ed. New York, Van Nostrand Reinhold Co., p 8.
Schnier GS, Laethem CL, & Koop DR (1989) Identification and induction
of cytochromes P450, P450IIE1 and P45IA1 in rabbit bone marrow. J
Pharmacol Exp Ther, 251: 790-796.
Schrikker ACM, de Vries WR, Zwart A, & Luijendjk CM (1985) Uptake of
highly soluble gases in the epithelium of the conducting airways.
Pflügers Arch/Eur J Physiol, 405: 989-394.
Schrikker ACM, de Vries WR, Zwart A, & Luijendjk CM (1989) The
excretion of highly soluble gases by the lung in man. Pflügers
Archiv/Eur J Physiol, 415: 214-219.
Seeber A, Kiesswetter E, Vangala RR, Blaszkewicz M, & Golka K (1992)
Combined exposure to organic solvents: An experimental approach using
acetone and ethyl acetate. Appl Psychol Int Rev, 41: 281-292.
Seeber A, Kiesswetter E, Giller D, Blaszkewicz M, Golka K, Vangala RR,
& Bolt HM (1993) [Acute effects of acetone and ethylacetate:
comparison of 4 and 8 hour exposure.] in: [Occupational health for a
healthy environment.] Stuttgart, Gentner Vedag, pp 145-148 (in
German).
Segner H & Lenz D (1993) Cytotoxicity assays with the rainbow trout
R1 cell line. Toxicol in vitro, 7(4): 537-540.
Seila RL (1979) Non-urban hydrocarbon concentrations in ambient air
north of Houston, Texas. Research Triangle Park, North Carolina, US
Environmental Protection Agency, 39 pp (EPA-500/3-79-010).
Shackelford WM & Keith LH (1978) Frequency of organic compounds
identified in water. Athens, Georgia, US Environmental Protection
Agency, Environmental Research Laboratory, pp 49-52
(EPA-600/4-76-062).
Shah JJ & Singh HB (1988) Distribution of volatile organic chemicals
in outdoor and indoor air. Environ Sci Technol, 22: 1381-1388.
Shepson PB, Hastie DR, Schiff HI, Polizzi M, Bottenheim JW, Anlauf K,
Mackay GI, & Karecki DR (1991) Atmospheric concentrations and temporal
variations of C1-C3 carbonyl compounds at two rural sites in central
Ontario. Atmos Environ, 25A: 2001-2015.
Sifniades S (1985) Acetone. In: Gerhartz W, Yamamoto YS, Campbell FT,
Pfefferkorn R, & Rousaville JF ed. Ullman's encyclopedia of industrial
chemistry, 5th ed. Weinheim, VCH Vedag, vol 1A, pp 79-95.
Singh HB & Hanst PL (1981) Peroxyacetyl nitrate (PAN) in the
unpolluted atmosphere: an important reservoir for nitrogen oxides.
Geophys Res Lett, 8: 941-944.
Singh HB, O'Hara D, Herlth D, Sachse W, Blake DR, Bradshaw JD,
Kanakidou M, & Crutzen PJ (1994) Acetone in the atmosphere:
distribution, sources, and sinks. J Geophys Res, 99: 1805-1819.
Singh KP, Yoon HL Ratner S, & Reiners JJ Jr (1996) Modulation of the
development of humoral immunity by topically applied acetone, ethanol
and 12- o-tetradecanoylphorbol-13-acetate. Fundam Appl Toxicol, 33:
129-139.
Sipes IG, Stripp B, Krishna G, Maling HM, & Gillette JR (1973)
Enhanced hepatic microsomal activity by pretreatment of rats with
acetone or isopropanol. Proc Soc Exp Biol Med, 142: 237.
Sipes IG, Slocumb ML, & Holtzman G (1978) Stimulation of microsomal
dimethyl nitrosamine- n-demethylase by pretreatment of mice with
acetone. Chem-Biol Interact, 21:155-166.
Sippel H, Penttila KE, & Lindros KO (1991) Regioselective induction of
liver glutathione transferase by ethanol and acetone. Pharmacol
Toxicol, 68: 391-393.
Skutches CL, Owen OE, & Reichard GA Jr (1990) Acetone and acetol
inhibition of insulin-stimulated glucose oxidation in adipose tissue
and isolated adipocytes. Diabetes, 39; 450-455.
Slooff W & Baerselman R (1980) Comparison of the usefulness of the
Mexican axolotl ( Ambystoma mexicanum) and the clawed toad
( Xenopus laevis) in toxicological bioassays. Bull Environ Contam
Toxicol, 24: 439-443.
Smith NB (1984) Determination of volatile alcohols and acetone in
serum by non-polar capillary gas chromatography after direct sample
injection. Clin Chem, 10: 1672-1674.
Smyth HF Jr, Carpenter CP, Well CS, & West JS (1962) Range-finding
toxicity data: List VI. Am Ind Hyg Assoc J, 23: 95-107.
Snider JR & Dawson GA (1985) Tropospheric light alcohols, carbonyls,
and acetonitrile concentrations in the southwestern United States and
Henry's law data. J Geophys Res, 90: 3797-3805.
Sollman T (1921) Studies of chronic intoxications in albino rats: II.
Alcohols (ethyl methyl and "wood") and acetone, J Pharmacol Exp Ther,
16: 291-309.
Specht H, Miller JW, & Valaer PJ (1939) Acute response of guinea pigs
to the inhalation of dimethyl ketone (acetone) vapor in air. Public
Health Rep, 54: 944-954.
Spicer CW, Riggin RM, Holdren MW, De Roos FL, & Lee RN (1985)
Atmospheric reaction products from hazardous air pollutant
degradation. Research Triangle Park, North Carolina, US Environmental
Protection Agency, 78 pp (EPA-60013-85-025).
SRI (1996) Directory of chemical producers, United States of America.
Menlo Park, California, Stanford Research institute International, 430
pp.
Stafford W & Northup HJ (1955) The BOD of textile chemicals. Am
Dyestuff Rep, 44: 355-359.
Steelman BL & Ecker RM (1984) Organic contamination of groundwater: An
open literature review. Seattle, Washington, Battelle Pacific
Northwest Laboratory, 66 pp (Report No. DE-AC05-76RLO 1830 13).
Steinberg SM & Kreamer DK (1993) Evaluation of the sorption of
volatile organic compounds by unsaturated calcareous soil from
southern Nevada using inverse gas chromatography. Environ Sci Technol,
27: 883-888.
Stewart RD, Hake CL, Wu A, Graft SA, Forster AV, Keeler WH, Lebrun AJ,
Newton PE, & Soto RJ (1975) Acetone: development of a biologic
standard for the industrial worker by breath analysis. Cincinnati,
Ohio, National Institute for Occupational Safety and Health, 80 pp
(NTIS P582-172917).
Strange P, Moller A, Ladefoged O, Lam HR, Laraen JJ, & Arlien-Soborg P
(1991) Total number and mean cell volume of neocortical neurons in
rats exposed to 2.5-hexaeedione with and without acetone. Neurotoxicol
Teratol, 13: 401-406.
Stratton GW (1937) Toxic effects of organic solvents on the growth of
blue-green algae. Bull Environ Contam Toxicol, 38:1012-1019.
Stratton GW & Corke GT (1981) Interaction between acetone and two
pesticides towards several unicellular green algae. Bull Environ
Contam Toxicol, 27(1): 13-16.
Takemoto S, Kuge Y, & Nakamoto M (1981) [The measurement of BOD in sea
water.] Suishitsu Odaku Kenkyu, 4:80-90 (in Japanese with English
summary).
Takeoka GR, Flath PA, Guntert M, & Jennings W (1989) Nectarine
volatiles: Vacuum steam distillation versus headspace sampling. J Agric
Food Chem, 36: 553-560.
Taylor A, Smith DE, Palmer VJ, Shepard D, Padhye N, Theriault C, &
Morrow F (1993) Relationships between acetone, cataracts and ascorbate
in hairless guinea pigs. Ophthalmol Res, 25: 30-35.
Thom NS & Agg AR (1975) The breakdown of synthetic organic compounds
in biological processes. Proc R Sac Land, B189: 347-37.
Thomas RG (1982) Volatilization from water. In: Lyman WJ, Reehl WF, &
Rosenblatt DH ed. Handbook of chemical property estimation methods.
New York, McGraw-Hill, chapter 15, pp 15/1-15/34.
Thornalley PJ (1990) The glyoxalase system: New developments towards
functional characterization of a metabolic pathway fundamental to
biological life. Biochem J, 269: 1-11.
Tichenor BA & Mason MA (1968) Organic emissions from consumer products
and building materials to the indoor environment. J Air Pollut Control
Assoc. 38: 284-268.
Tietz NW (1983) Clinical guide in laboratory tests. Philadelphia,
Pennsylvania, WB Saunders Company, pp 2-3.
Tindberg N & Ingelman-Sundberg M (1989) Cytochrome P-450 and oxygen
toxicity: Oxygen-dependent induction of ethanol-inducible cytochrome
P-450 (IIE1) in rat liver and lung. Biochemistry, 28: 4499-4504.
Traiger GJ & Plaa GL (1972) Relationship of alcohol metabolism to the
potentiation of CCI4 hepatotoxicity induced by aliphatic alcohols. J
Pharmacol Exp Ther, 183: 481-488.
Traiger GJ & Plaa GL (1974) Chlorinated hydrocarbon toxicity:
Potentiation by isopropyl alcohol and acetone. Arch Environ Health,
28: 276-278.
Trotter MD, Sulway MJ, & Trotter E (1971) The rapid determination of
acetone in breath and plasma. Clin Chim Acta, 35: 137-143.
Ueng T-H, Tsai J-n, Ju J-M, Ueng Y-F, Iwasaki M, & Guengerich FP
(1991) Effects of acetone administration of cytochrome P-450-dependent
monooxygenases in hamster liver, kidney and lung. Arch Toxicol, 65:
45-51.
UK SAC (UK Standing Committee of Analysts) (1988) The determination of
formaldehyde, other volatile aldehydes, ketones and alcohols in water:
Methods for the examination of waters and associated materials.
London, Her Majesty's Stationery Office, 12 pp.
Urano K & Kato Z (1986a) A method to classify biodegradabilities of
organic compounds. J Hazard Mater, 13: 135-145.
Urano K & Kate Z (1986b) Evaluation of biodegradation ranks of
priority organic compounds. J Hazard Mater, 13: 147-159, 203.
US EPA (1975) preliminary assessment of suspected carcinogens in
drinking water: Interim report to Congress (June 1975). Washington,
DC, US Environmental Protection Agency, 9 pp.
US EPA (1986a) Ninety-day gavage study in albino rats using acetone.
Washington, DC, US Environmental Protection Agency, Office of Solid
Waste, 950 pp.
US EPA (1986b) Health and environmental effects document, Cincinnati,
Ohio, US Environmental Protection Agency, Office of Research and
Development, 186 pp.
US EPA (1989) Compilation and speciation of national emissions factors
for consumer/commercial solvent use information compiled to support
urban air toxics assessment studies. Research Triangle Park, North
Carolina, US Environmental Protection Agency, 209 pp
(EPA/450/2-89/008; NTIS PB89-207203).
Vaishnav DD (1986) Chemical structure biodegradation inhibition and
fish acute toxicity relationships for narcotic industrial chemicals.
Toxicol Assess, 1(2): 227-240.
Vaishnav DD, Boethling RS, & Babeu L (1987) Quantitative
structure-biodegradability relationships for alcohols, ketches and
alicyclic compounds. Chemosphere, 16: 695-703.
Vangala RR, Blaszkewicz M, Bolt HM, Golka K, Kiesswetter E, & Seeber A
(1991) Acute experimental exposures to acetone and ethyl acetate, Arch
Toxicol, 14(suppl): 259-282.
Van Stekelenburg GJ & Koorevaar G (1972) Evidence for the existence of
mammalian acetoacetate decarboxylase: with special reference to human
blood serum. Clin Chim Acta, 39: 191-199.
Veith GD, Call DJ, & Brooke LT (1983) Structure-toxicity relationships
for the fathead minnow, Pimephales promelas: Narcotic industrial
chemicals. Can J Fish Aquat Sci, 40(6): 743-748.
Vodickova L, Frantik E, & Vodikova A (1995) Neurotropic effects and
blood levels of solvents at combined exposures: binary mixtures of
toluene, o-xylene and acetone in rats and mice. Cent Eur J Public
Health, 2: 57-64.
Waggy GT, Conway PA, Hansen JL, & Blessing RL (1994) Comparison of
20-d BOD and OECD closed-bottle biodegradation tests. Environ Toxicol
Chem, 13(8): 1277-1280.
Wallen IE, Greer WC, & Lasater R (1957) Toxicity to Gambusia affinis
of certain pure chemicals in turbid waters. Sewage Ind Wastes, 29(6):
695-711.
Wallington TJ & Kurylo MJ (1987) Flash photolysis resonance
fluorescence investigation of the gas-phase reactions of hydroxyl
radicals with a series of aliphatic ketones over the temperature range
240-440 K. J Phys Chem, 91: 5050-5054.
Wang G, Maranelli G, Perbellini L, Raineri E, & Brugnone F (1994) Blood
acetone concentration in "normal people" and in exposed workers 16 h
after the end of the workshift. Int Arch Occup Environ Health, 65(5):
285-289.
Ward JM, Quander R, Devor D, Wenk ML, & Spangler EF (1986) Pathology
of aging female SENCAR mice used as controls in skin two-stage
carcinogenesis studies. Environ Health Perspect, 68: 81-89.
Weast RC ed. (1987) Handbook of chemistry and physics, 67th ed. Boca
Raton, Florida, CRC press, p 51.
Westerholm RN, Alsberg TE, Frommelin AB, Strandell ME, Ranung U,
Winsquist L, & Grigoriadis V (1988) Effect of fuel polycyclic aromatic
hydrocarbon content on the emissions of polycyclic aromatic
hydrocarbons and other mutagenic substances from a gasoline fueled
automobile. Environ Sci Technol, 22: 925-930.
WHO (1971) Evaluation of food additives. Fourteenth report of the
Joint FAO/WHO Expert Committee on Food Additives. Geneva, World Health
Organization, p 21 (WHO Technical Report Series, NO. 462).
Widmark EMP (1919) Studies in the concentration of indifferent
narcotics in blood and tissues. Acta Med Scand, 52: 87-164.
Wigaeus E, Holm S, & Astrend I (1981) Exposure to acetone: Uptake and
elimination in man. Scand J Work Environ Health, 7(2): 84-94.
Wigaeus E, Löf A, & Nordqvist M (1982) Distribution and elimination of
2-[14C]-acetone in mice after inhalation exposure. Scand J Work
Environ Health, 8(2): 121-128.
Wilkins K (1994) Volatile organic compounds from household waste.
Chemosphere, 29(1): 47-53.
Williams PL, Brooks JD, Inman AG, Monteiro-Riviere NA, & Riviera DE
(1994) Determination of physicochemical properties of phenol,
p-nitrophenol, acetone and ethanol relevant to quantitating their
percutaneous absorption in porcine skin. Res Commun Chem Pathol
Pharmacol, 83(1): 61-75.
Wilson JE, Brunwald E, Isselbacher K, Petersdorf RG, Martin JB, Favei
AS, & Root RK (1991) Principles of internal medicine, 12th ed. New
York, McGraw-Hill, 525 pp.
Windholz M ed. (1983) Acetone. In: The Merck index, 10th ed. Rahway,
New Jersey, Merck & Co., p8.
Wysocki CJ, Dalton P, Brody M, & Hanley HJ (1997) Odor and irritation
thresholds for acetone in acetone-exposed factory workers and control
(occupationally-non exposed) subjects. Am Ind Hyg Assoc J, 58:
704-712.
Yoo J-S & Yang CS (1985) Enzyme specificity in the metabolic
activation of N-nitrosodimethylamine to a mutagen for Chinese
hamster V79 cells. Cancer Res, 45; 5569-5574.
Yoo J-SH, Ishizaki H, & Yang CS (1990) Roles of cytochrome-P450IIE1 in
the dealkylation and denitrosation of N-nitrosodiethylamine in rat
liver mircosomes. Carcinogenesis, 11: 2239-2243.
Zhou X & Mopper K (1990) Apparent partition coefficients of 15
carbonyl compounds between air end seawater and between air and
freshwater: Implications for air-sea exchange. Environ Sci TechnoL
24(12): 1864-1869.
Zimmermann FK, Mayer VW, Scheel I, & Resnick MA (1985) Acetone, methyl
ethyl ketone, ethyl acetate, acetenitrile and other polar aprotic
solvents are strong inducers of aneuploidy in Saccharomyces
cerevisiae. Mutat Res, t49(3); 339-351.
Zweidinger RB, Sigsby JE Jr, Tajada SB, Stump FD, Dropkin DL, Ray WD,
& Duncan JW (1988) Detailed hydrocarbon and aldehyde mobile source
emissions from roadway studies. Environ Sci Technol, 22: 956-962.
RÉSUMÉ
1. Propriétés
L'acétone (masse moléculaire relative = 58,08) est un liquide
inflammable, limpide et incolore (point d'éclair - 17°C en coupe
fermée, -9°C en coupe ouverte; limites d'inflammabilité dans l'air à
25°C = 2,15-13% v/v). Les limites d'explosion dans l'air sont de:
2,6-12,8% v/v). L'acétone s'évapore rapidement (tension de vapeur
181,72 mm Hg à 20°C) et présente une faible viscosité (0,303 cP à
25°C). Elle est miscible à l'eau et aux solvants organiques.
2. Usages et sources d'exposition
2.1 Production
Industriellement, l'acétone se prépare principalement par
peroxydation du cumène ou déshydrogénation de l'alcool isopropylique.
La peroxydation du eumène produit également des traces de benzène.
2.2 Usages et émissions dans l'environnement
L'acétone est principalement utilisée comme solvant et comme
intermédiaire dans la préparation de divers produits chimiques. Ses
principaux usages consistent dans la production de méthacrylate de
méthyle, acide méthacrylique et méthacrylates supérieurs, bisphénol A,
méthylisobutylcétone, médicaments et dans des applications
pharmaceutiques diverses. On l'utilise aussi comme solvant de
l'acétate de cellulose et de divers enduits. Dans l'industrie
alimentaire, on l'emploie comme solvant d'extraction des graisses et
des huiles et comme agent de précipitation au cours de la purification
du sucre et de l'amidon.
Les émissions d'acétone dans l'atmosphère proviennent de produits
de consommation qui en contiennent: dissolvants pour vernis à ongles,
panneaux de particules, doublures de tapis, décapants pour peintures,
cires et encaustiques liquides ou pâteux. Un certain nombre de
détergents, produits de nettoyage (notamment pour certains éléments de
moteurs d'automobile tels que carburateurs et starters) et adhésifs
contiennent aussi de l'acétone.
De nombreuses industries polluent les eaux superficielles par
leurs rejets d'acétone dans les eaux résiduaires: papier, plastiques,
produits pharmaceutiques, produits d'entretien, peintures et vernis,
produits pour le traitement des gommes, colles et bois, intermédiaires
cycliques, dérivés organiques industriels, produits de gypseuse,
cartonnages, production d'énergie (gazéification de la houille et
traitement de l'huile de schiste).
La pollution des sols par l'acétone trouve son origine dans le
rejet de déchets agricoles et alimentaires, les excréments d'animaux,
les précipitations, les effluents de fosses septiques et les
infiltrations provenant de décharges de produits chimiques.
3. Transport, distribution et transformation dans l'environnement
L'acétone libérée dans l'atmosphère se décompose par voie
photolytique et par réaction avec des radicaux hydroxyles. Sa demi-vie
atmosphérique est dans ce cas d'environ 30 jours. Elle peut être
éliminée physiquement de l'atmosphère par les précipitations. Dans les
sols et dans l'eau, c'est la biodégradation qui constitue le principal
mode de décomposition de l'acétone, ce composé étant facilement
biodégradable, Sa volatilisation à partir de l'eau peut constituer une
voie de transport environnemental non négligeable. En raison de sa
volatilité, elle s'évapore des surfaces sèches sur lesquelles elle est
déposée. En outre, sa miscibilité à l'eau en facilite le lessivage à
partir de la plupart des sols. En cas de biodégradation suffisamment
rapide, ce lessivage perd de son importance.
4. Concentrations dans l'environnement et exposition humaine
L'exposition à l'acétone peut être d'origine naturelle ou
artificielle. Elle est présente dans le sang, l'urine et l'haleine
humaines par suite du métabolisme normal. Dans l'environnement, sa
présence résulte entre autres de la biodégradation des effluents, des
déchets solides et des alcools ainsi que de l'oxydation des substances
humiques. On l'a mise en évidence dans diverses plantes et denrées
alimentaires comme les oignons, le raisin, les choux-fleurs, les
tomates, les volubilis, la moutarde sauvage, le lait, les haricots,
les pois, le fromage et le blanc de poulet. Diverses espèces d'arbres
émettent naturellement des vapeurs d'acétone. La pollution du milieu
aquatique imputable à l'activité humaine est due au déversement des
eaux résiduaires d'un grand nombre d'industries et au lessivage des
décharges industrielles ou municipales. L'évaporation de l'acétone
utilisée comme solvant dans les peintures, vernis, produits
d'entretien et encres constitue une source importante de pollution de
l'air. Sa présence dans l'atmosphère résulte également de la
combustion du bois, de plastiques et de déchets divers. Elles est
également émises avec les gaz d'échappement des moteurs à explosion et
des moteurs diesel ou encore des turbines à gaz. Les concentrations
relevées dans l'atmosphère vont de 0,5 à 125,4 µg/m3 (0,2-52,9
parties par milliard).
5. Cinétique et métabolisme
L'acétone est l'une des trois cétones qui sont présentes à l'état
naturel dans l'organisme. Elle peut se former de manière endogène dans
l'organisme des mammifères par oxydation des acides gras. Le jeûne, le
diabète sucré et un exercice physique intense accroissent la formation
endogène d'acétone. Dans les conditions normales, la production de
corps cétoniques s'effectue presque exclusivement dans le foie et,
dans une moindre proportion, dans les reins et les poumons. Il s'agit
d'un processus continu et les trois produits, après être passés dans
le sang, gagnent la totalité des tissus et des organes où ils peuvent
être utilisés comme source d'énergie. Deux d'entre eux, l'acide
acétoacétique et l'acide ß-hydroxybutyrique, sont des acides
organiques susceptibles de provoquer une acidose lorsqu'ils sont
produits en grande quantité, comme dans le cas du diabète sucré.
L'acétone, en revanche, n'est pas ionisée et elle est produite de
manière endogène par le clivage enzymatique ou spontané de
l'acétoacétate. L'acétone endogène est éliminée de l'organisme soit
par la voie urinaire, soit dans l'air expiré, soit par l'action
d'enzymes. Dans les conditions normales, c'est la voie métabolique qui
constitue le mode principal d'élimination de l'acétone (70 à 80% de la
quantité totale présente dans l'organisme).
L'acétone est rapidement résorbée au niveau des voies
respiratoires et digestives de l'Homme et de l'animal, comme on peut
le constater à la présence d'acétone dans le sang 30 minutes après
inhalation et 20 minutes après ingestion. Les études effectuées sur
des rats montant qu'après ingestion, l'acétone est absorbée dans une
forte proportion, alors qu'après inhalation chez l'Homme, la
proportion absorbée est d'environ 50% de la quantité initiale. Il est
vrai cependant, que des valeurs plus fortes et plus faibles ont été
relevées après inhalation. Il semblerait que les fosses nasales de
l'Homme et des animaux de laboratoire n'aient qu'une capacité limitée
à absorber et à excréter les vapeurs d'acétone, comparativement aux
autres parties des voies respiratoires.
L'acétone se répartit uniformément dans les tissus non adipeux et
ne s'accumule pas dans les graisses. Chez la souris, la concentration
maximale d'acétone dans les tissus adipeux s'est révélée égale au
tiers de ce qu'elle était dans les autres tissus, après exposition des
animaux par la voie respiratoire. L'acétone s'élimine rapidement de
l'organisme par métabolisation et excrétion. Sa demi-vie dans l'air
alvéolaire, dans le sang veineux et dans le sang artériel est
respectivement égale à approx. 4, 6 et 4 h. La principale voie
d'élimination de l'acétone et de son métabolite terminal, le CO2, est
l'air expiré et la fraction du composé qui est rejetée inchangée
dépend de la dose. Il y a aussi excrétion de l'acétone et de ses
métabolites par la voie urinaire, mais il s'agit d'une voie mineure
par rapport à l'expiration.
L'acétone d'origine endogène prend part à nombre des réactions
métaboliques qui se produisent dans les diverses parties de
l'organisme, mais c'est le foie qui semble constituer le site
métabolique le plus important. Le carbone provenant de l'acétone
administrée par voie orale à des rat se retrouve dans le cholestérol,
les acides aminés, les acides gras et le glycogène fistulaires ainsi
que dans l'urée unanime. On le retrouve aussi dans l'acétone inchangée
et dans le CO2 présents dans l'air expiré. L'acétone est métabolisée
en acétate et en formiate; cela explique que le carbone acétonique
entre dans le cholestérol, les acides gras, l'urée et les acides
aminés et qu'il se forme des composés tricarbonés par gluconéogénèse.
On a avancé que la gluconéogénèse à partir de l'acétone
s'effectuait selon deux voies métaboliques. La première comporte
l'action catalytique initiale de l'acétone-monooxygénase et de
l'acétol-monooxygénase qui transforme respectivement l'acétone en
acétol et l'acétol en méthylglyoxal. Ces deux enzymes sont induites
par l'acétone et constituent des isozymes du cytochrome hépatique
P-450IIE1 industriel par l'éthanol. Le deuxième mode de néoglucogénèse
consiste dans la formation de 1,2-propanediol à partir de l'acétone,
sous l'action de l'acétone-monooxygénase et d'une autre enzyme non
caractérisée, capable de convertir l'acétol en 1,2-propanediol.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
Chez le rat, la valeur de la DL50 par voie orale varie entre
5800 et 7138 mg/kg. La CL50 par inhalation à 4 h est égale à 76 000
mg/m3 (32 000 ppm).
On a constaté, chez l'animal de laboratoire exposé à l'acétone,
une moindre performance dans les tests neurocomportementaux, aux
concentrations supérieures à 7765 mg/m3 (> 3270 ppm).
On ne dispose pas de données sur les effets d'une exposition de
longue durée à l'acétone par la voie orale ou respiratoire, sans doute
parce que sa toxicité est faible et qu'il s'agit d'un composé
endogène.
En faisant inhaler de l'acétone à des rats pendant une longue
période (45 000 mg/m3, soit 19 000 ppm, 3 h par jour, 5 jours par
semaine, pendant 8 semaines), on a observé une diminution réversible
du poids absolu du cerveau. Aucune modification systématique n'a été
notée dans le poids du corps ou celui d'antres organes, au niveau des
constantes hématologiques, dans le taux des triglycérides hépatiques
ou dans l'aspect histologique du coeur, du poumon, du rein, du cerveau
ou du foie.
Lors d'une étude de 90 jours comportant l'administration
d'acétone à des rats par gavage, on a observé une augmentation de
certains paramètres hématologiques (hémoglobine, hématocrite) aux
doses supérieures à 500 mg/kg par jour et on a déterminé une NOAEL
(dose sans effet nocif observable) de 500 mg/kg par jour. Dans une
autre étude, qui a duré 13 semaines et qui a consisté à ajouter de
l'acétone à l'eau de boisson des rats, on a noté la présence d'effets
toxiques chez les mâles exposés à des concentrations supérieures à 20
g/litre (env. 1700 mg/kg p.c. par.jour), à savoir une augmentation du
poids relatif des organes et une modification des constantes
hématologiques, avec en outre une légère néphropathie. Chez les
femelles soumises à la dose la plus forte, soit 50 g/litre (env. 3400
mg/kg p.c. par jour), les effets consistaient en une augmentation du
poids relatif des organes et une modification des constantes
hématologiques. En outre, l'exposition à 50 g/litre pendant 13
semaines a provoqué chez les mâles une modification du poids relatif
des testicules et ainsi qu'une baisse de la motilité des
spermatozoïdes accompagnée d'anomalies morphologiques. Des souris
femelles exposées à la même concentration de 50 g/litre (env. 11 298
mg/kg p.c. par jour) dans leur eau de boisson, présentaient une
réduction du poids du foie et de la rate ainsi qu'une hypertrophie
hépatique touchant les cellules centrilobulaires, mais dont
l'augmentation d'incidence restait marginale. Aucun effet toxique n'a
été relevé chez les souris mâles qui avaient reçu la plus forte dose
d'acétone (20 g/litre, soit env. 4858 mg/kg p.c. par jour). Une
exposition de 13 semaines à des doses inférieures ou égales à 10
g/litre (900 mg/kg p.c. par jour), toujours par ingestion d'acétone
mélangée à l'eau de boisson, n'a pas produit d'effets toxiques chez
des rats mâles; les doses sans effets observables étaient < 20
g/litre chez les rats (1600 mg/kg p.c. par jour) et les souris des
deux sexes (mâles: 4858 mg/kg p.c. par jour; femelles: 5945 mg/kg p.c.
par jour).
Lors d'une étude préliminaire de 14 jours consistant à faire
boire de l'eau additionnée d'acétone à des rats et à des souris, on a
noté chez les mâles exposés aux concentrations de 20 à 100 g/litre,
une hypertrophie hépatique affectant les cellules centrilobulaires.
Chez les rongeurs, un prétraitement par l'acétone accroît les
effets hépatotoxiques de certains composés, notamment ceux des dérivés
halogénés des alcanes. On suppose que cette potentialisation de
l'hépatotoxicité est due à l'augmentation, sous l'action de l'acétone,
de l'activité de certaines enzymes (les oxydases hépatiques â fonction
mixte), qui sont responsables de la production d'intermédiaires
toxiques à partir des alcanes halogénés.
Les épreuves de génotoxicité ont donné des résultats négatifs sur
de nombreux systèmes mammaliens, tant in vivo qu' in vitro. Il n'y
a qu'une seule épreuve qui ait donné un résultat positif, à savoir la
présence d'aneuploïdies chez une espèce de levure exposée à une forte
concentration d'acétone (6,82%) dans son milieu de croissance. On
considère que l'acétone n'est ni génotoxique, ni mutagène.
Lors d'une étude portant sur des rats et des souris gravides, on
a exposé les animaux à des vapeurs d'acétone du 6ème au 19ème jour de
la gestation. De légers effets toxiques ont été observés sur le
développement de la progéniture lorsque les rats étaient exposées à
une concentration de 26 100 mg/m3 (11 000 ppm) à raison de 6 h par
jour (augmentation de la proportion de portées comportant au moins une
malformation foetale) et les souris à une concentration de 15 670
mg/m3 (6600 ppm) à raison également de 6 h par jour (légère
diminution du poids des foetus et petite augmentation de l'incidence
relative des résorptions tardives). On a fixé à 5200 mg/m3 (2200 ppm)
la concentration atmosphérique sans effet nocif observable sur le
développement de la souris et du rat. Dans une étude par gavage, un
traitement par l'acétone au cours de l'organogénèse à la dose
quotidienne de 3500 mg/kg, a eu un effet négatif sur la reproduction
lors d'un test de criblage sur des souris. Les résultats négatifs
obtenus in vivo chez deux espèces différentes, l'administration se
faisant par la voie buccale et par la voie intrapéritonéale, montrent
que l'exposition de mammifères à l'acétone n'a pas d'effets mutagènes.
Les observations relatives aux effets de l'acétone sur la
reproduction consistent en anomalies testiculaires et modification de
la qualité des spermatozoïdes, qui ont été mises en évidence chez des
rats dont l'eau de boisson avait contenu pendant 13 semaines 50 g
d'acétone par litre. On n'a pu trouver aucune étude consacrée aux
effets que pourrait avoir l'ingestion d'acétone sur le développement
foetal (foetotoxicité et tératogénicité).
L'acétone est largement utilisée comme solvant dans les études
sur le pouvoir cancérogène cutané et on estime qu'en application sur
la peau, elle est dénuée d'activité cancérogène.
7. Effets sur l'Homme
L'acétone est relativement moins toxique que nombre d'autres
solvants industriels; cependant, sous forte concentration, les vapeurs
d'acétone peuvent provoquer une dépression du système nerveux central,
un collapsus cardiorespiratoire et la mort. On connaît des cas
d'exposition humaine aiguë à des concentrations d'acétone atteignant
environ 4750 mg/m3 (soit à peu près 2000 ppm) qui n'ont été
accompagnés d'aucun effet toxique majeur ou tout au plus d'effets
mineurs et passagers, tels qu'une irritation oculaire. Des effets
passagers plus sérieux (notamment des vomissement et une perte de
conscience) ont le observés chez des travailleurs exposés à des
vapeurs d'acétone dont la concentration dépassait 25 500 mg/m3 (>
12 000 ppm) pendant environ 4 h. On a également fait état d'une baisse
dans les résultats des tests neurocomportementaux chez l'Homme à la
concentration de 595 mg/m3 (250 ppm). Des femmes exposées à une
concentration atmosphérique de 2370 mg/m3 (1000 ppm) ont présenté des
irrégularités du cycle menstruel.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Pour la plupart des espaces animales dulçaquicoles ou marines,
les valeurs de la CL50 et de la CE50 à 48 et 96 h se sont révélées >
5540 mg/litre.
Une exposition de 76 h à de l'acétone à la concentration de 257,4
mg/litre a provoqué une inhibition de la croissance de l'algue
Chlorella pyrendoidosa. Il en a été de même pour
Chlamydomonas eugametos après 48 h d'exposition à 790 mg/litre. En
exposant Scendensemus quadricauda et C. pyrendoidosa à de
l'acétone aux concentrations respectives de 79,0 et 790 mg/litre, on a
observé un accroissement de la photosynthèse.
Les seuils de toxicité respectifs à 7 et 8 jours pour l'algue
verte S. quadricauda et pour la cyanobactérie (algue bleue)
Microcystis aeruginosa se situent à 7500 et 530 mg/litre, ce qui
traduit la plus grande résistance de l'algue verte à l'action toxique
de l'acétone. La diatomée Nitzschia linearis a également semblé
faire preuve d'une grande résistance, avec une CE50 à 5 jours
comprise entre 11 493 et 11 727 mg/litre. De même la diatomée marine
Skelatonema costatum s'est aussi révélée lès résistante, avec une
CE50 comprise entre 11 798 et 14 440 mg/litre.
Les bactéries se révèlent plus résistantes à l'acétone que les
protozoaires. Pour Photobacterium phosphoreum, Pseudomonas putida
et une culture mixte, on a obtenu une valeur de la CE50 comprise
entre 1700 et 35 540 mg/litre, contre 28 mg/litre dans le cas du
protozoaire Entosiphon sulcatum. Ce résultat pourrait s'expliquer
par le fait que ces deux types de microorganismes ont une paroi
cellulaire différente.
Chez des cailles et des faisans, on a trouvé des valeurs de la
CL50 à 5 jours supérieures ou égaies à 40 g par kg de nourriture. Des
oeufs fécondés de colvert n'ont pas souffert d'une immersion de 30
secondes dans de l'acétone à 10%; en revanche, l'immersion dans de
l'acétone pneu a entraîné une diminution de la survie, du poids et de
la taille des embryons sans que l'on puisse dire avec certitude si e'
est la toxicité de l'acétone qui était en cause ou son caractère de
solvant. L'injection de 5 µl d'acétone dans des oeufs de poules
Leghorn ne semble pas avoir entraîné de mortalité ni de malformations
chez les embryons.
RESUMEN
1. Propiedades
La acetona (masa molecular relativa = 58,08) es un liquido
transparente, incoloro e inflamable (punto de inflamación - 17°C en
crisol cerrado, -9°C en crisol abierto; limites de inflamabilidad en
el aire a 25°C = 2,15-13% v/v). Los límites de explosión en el aire
son 2,6-12,8% v/v. Tiene una elevada tasa de evaporación (presión de
vapor 181,72 mmHg a 20°C) y baja viscosidad (0,303 cP a 25°C). Es
miscible con el agua y con disolventes orgánicos.
2. Usos y fuentes de exposición
2.1 Producción
La acetona se fabrica principalmente mediante los procesos de
peroxidación del cumeno o la deshidrogenación del isopropil alcohol.
En el primer proceso se producen cantidades infamas de benceno como
subproducto.
2.2 Usos y emisiones al medio ambiente
La acetona se utiliza principalmente como disolvente y como
intermedio en la producción de sustancias químicas. Sus principales
aplicaciones son la producción de metil metacrilato, ácido metacrílico
y metacrilatos de mayor tamaño, bisfenol A, metil isobutil cetona,
aplicaciones farmacéuticas y medicamentosas, y como disolvente para
revestimientos y para el acetato de celulosa. También tiene usos
alimentarios como disolvente de extracción para grasas y aceites, y
como agente de precipitación en la purificación del azúcar y el
almidón.
Las emisiones a la atmósfera proceden de los productos de
consumo, como quitaesmalte para las uñas, tableros de conglomerado,
revestimientos inferiores de moquetas, algunos decapantes de pinturas
y ceras o abrillantadores líquidos o sólidos. Ciertos detergentes y
limpiadores, adhesivos y limpiadores del carburador y el estrangulador
en automóviles también contienen acetona.
La acetona se vierte a las aguas superficiales en los efluentes
de aguas residuales de una amplia gama de procesos e industrias de
fabricación, como el papel, el plástico, productos farmacéuticos,
limpiadores y abrillantadores químicos elaborados, pinturas y
productos conexos, productos químicos del caucho y la madera,
intermedios cíclicos, productos orgánicos industriales, productos del
yeso, productos de cartón de papel e industrias de la energía, como la
gasificación del carbón y el tratamiento de esquistos bituminosos.
Entre las fuentes de incorporación de acetona al suelo figuran el
vertido de residuos agrícolas y alimentarios, residuos animales,
deposición húmeda desde la atmósfera, efluentes de fosas sépticas
domésticas y vertederos de residuos químicos.
3. Transporte, distribución y transformación en el medio ambiente
La acetona que ingresa en la atmósfera es degradada por una
combinación de fotolisis y reacción con radicales hidroxilo. La
semivida media para la degradación de la acetona en la atmósfera es de
unos 30 días. La acetona puede ser eliminada por medios fiscos del
aire por deposición húmeda. El principal proceso de degradación de la
acetona en el suelo y el agua es la biodegradación; la acetona es
fácilmente biodegradable. La volatilización de la acetona desde el
medio acuático puede ser un proceso de transporte significativo. La
acetona es un compuesto volátil que se evapora fácilmente de las
superficies secas. Puesto que es miscible en agua, puede ser objeto de
lixiviación en la mayoría de los tipos de suelo. La biodegradación
concurrente puede reducir la importancia global de la lixiviación si
aquélla se produce con la rapidez suficiente.
4. Concentraciones en el medio ambiente y exposición humana
La exposición a la acetona procede de fuentes tanto naturales
como antropogénicas. La acetona también aparece como compuesto
metabólico en la sangre, la orina y el aire pulmonar del ser humano.
Se produce en la biodegradación de las aguas residuales, los residuos
sólidos y los alcoholes, así como por la oxidación de sustancias
húmicas. Se ha detectado en muy diversas plantas y alimentos, como las
cebollas, las uvas, la coliflor, los tomates, las convolvuláceas, la
mostaza silvestre, la leche, las indias, los guisantes, el queso y la
pechuga de pollo. Las emisiones naturales de varias especies de
árboles contienen vapores de acetona. Las fuentes antropogénicas de
emisión al medio acuático comprenden los vertidos de aguas residuales
de muchas industrias y la lixiviación que tiene lugar en vertederos
industriales y municipales. Una de las principales fuentes de emisión
humana al aire es la evaporación de la acetona utilizada como
disolvente en productos de revestimiento como pinturas, limpiadores,
barnices y tintas. La acetona es un producto de emisión de la
combustión de madera, basuras y plásticos. También se emite en los
escapes de automóviles y de motores diesel y de turbina. Las
concentraciones de acetona detectadas en la atmósfera varían entre 0,5
y 125,4 µg/m3 (0,2-52,9 ppmm).
5. Cinética y metabolismo
La acetona es uno de los tres cuerpos cetónicos que se producen
naturalmente en el organismo humano. Puede formarse de modo endógeno
en el organismo de los mamíferos por oxidación de los ácidos grasos.
El ayuno, la diabetes mellitus y el ejercicio físico vigoroso
incrementan la generación endógena de acetona. En condiciones
normales, prácticamente todos los cuerpos cetónicos se producen en el
hígado, y en menor medida en el pulmón y el riñón. El proceso es
continuo y los tres productos son excretados a la sangre y
transportados a todos los tejidos y órganos del cuerpo, donde pueden
ser utilizados como fuente de energía. Dos de esos cuerpos cetónicos,
el acetoacetato y el ß-hidroxibutirato, son ácidos orgánicos que
pueden provocar acidosis metabólica cuando se producen en grandes
cantidades, como en la diabetes mellitus. La acetona, en cambio, tiene
carácter no iónico y se deriva de forma endógena de la degradación
enzimática espontánea del acetoacetato. La acetona endógena se elimina
del organismo por excreción en la orina y el aire exhalado o por
metabolismo enzimático. En circunstancias normales, el metabolismo es
la vía principal de eliminación y procesa el 70-80% de la carga total
del organismo.
La acetona es absorbida rápidamente por los tractos respiratorio
y gastrointestinal del ser humano y los animales de laboratorio, como
lo indica la detección de acetona en la sangre a los 30 minutos de la
exposición por inhalación y a los 20 minutos de la exposición por vía
oral. Los estudios efectuados en ratas indican que la acetona
administrada por vía oral se absorbe fácilmente, mientras que en la
exposición por inhalación el ser humano absorbe aproximadamente el 50%
de la cantidad de acetona inhalada. Sin embargo, se han comunicado
valores tanto menores como mayores de absorción por vía respiratoria.
Las cavidades nasales de los seres humanos y los animales de
laboratorio parecen tener una capacidad limitada de absorción y
excreción de vapores de acetona, en comparación con el resto del
tracto respiratorio.
La acetona se distribuye uniformemente por los tejidos no
adiposos y no se acumula en los tejidos adiposos. En el ratón, se
comunicó que las concentraciones máximas de acetona en los tejidos
adiposos eran aproximadamente la tercera parte de las observadas en
los tejidos no adiposos tras la exposición por inhalación. La acetona
se elimina rápidamente del organismo por vía metabólico y por
excreción. Las semividas de la acetona en el aire alveolar y la sangre
venosa y arterial en el ser humano son approx. 4, 6 y 4 horas,
respectivamente. La exhalación es la principal vía de eliminación de
la acetona y su metabolito terminal, el CO2, y la fracción de la
acetona adminisáada que se exhala sin modificar depende de la dosis.
También se produce excreción de acetona y sus metabolitos en la orina,
pero esta vía de eliminación es menos importante que la exhalación de
acetona y CO2 por vía respiratoria.
La acetona de origen exógeno se incorpora a numerosas reacciones
metabólicas en tejidos de todo el organismo, pero el hígado parece ser
el órgano en el que se metabólico más intensamente. El carbono de la
acetona administrada por veta oral se ha detectado en el colesterol,
los amanecidos, los ácidos grasos y el glucógeno en tejidos de rata,
la urea en la orina y acetona no modificada y CO2 en el aire
exhalado. Desde el punto de vista metabólico, la acetona se degrada a
acetato y formato; por esta vía se produce la incorporación de los
átomos de carbono de la acetona al colesterol, los ácidos grasos, la
urea y los amanecidos, así como la formación de compuestos
gluconeogénicos de tres átomos de carbono.
Se han propuesto dos posibles vías de gluconeogénesis a partir de
la acetona. La primera vía comienza por la acción catalítica inicial
de la acetona-monooxigenesa y la acetol-monooxigenesa, que convierten
la acetona en acetol y el acetol en metilglioxal, respectivamente.
Ambas actividades enzimáticas son inducidas por la acetona y han sido
identificadas como isoenzimas del citocromo hepático P-450IIE1,
inducible por el etanol. La segunda ruta gluconeogénica posible
entraña la formación de 1,2-propanodiol a partir de la acetona,
catalizada por la acetona monooxigenasa y una enzima no caracterizada
capaz de convertir el acetol en 1,2-propanodiol.
6. Efectos en mamíferos de experimentación y en sistemas in vitro
Los valores de la DL50 en ratas adultas se encuentran en el
intervalo 5800-7138 mg/kg. El valor de la CL50 a las 4 horas de la
inhalación es de 76 000 mg/m3 (32 000 ppm).
Se ha observado que la exposición aguda a la acetona altera los
resultados de las pruebas necrológicas de conducta en animales de
laboratorio cuando las concentraciones superan los 7765 mg/m3 (>3270
ppm).
No se dispone de datos en animales de experimentación para
caracterizar los efectos de la exposición oral o por inhalación a
largo plazo de la acetona, probablemente a causa de su escasa
toxicidad y sus características endógenas.
La exposición prolongada de ratas a la inhalación de acetona, a
razón de 45 100 mg/m3 (19 000 ppm), durante 3 horas al día, 5 días a
la semana durante 8 semanas, provocó una reducción reversible del peso
cerebral absoluto. No se observaron cambios uniformes en el peso de
otros órganos ni del organismo en conjunto, ni tampoco en los índices
químicos de la sangre, en los niveles de triglicéridos hepáticos o en
las características histológicas del corazón, el pulmón, el riñón, el
cerebro o el higado.
En un estudio de alimentación forzada de ratas durante 90 días,
se determinó un aumento en los parámetros sanguíneos (aumento de la
hemoglobina y el hematócrito) con concentraciones superioras a 500
mg/kg al día, y una concentración sin observación de efectos adversos
de 500 mg/kg al día. En un estudio de administración de acetona en el
agua de bebida durante 13 semanas, se observaron efectos tóxicos en
las ratas macho expuestas a concentraciones superioras a 20 g/litro
(approx. 1700 mg/kg de peso corporal al día), a saber, aumento del
peso relativo de algunos órganos, y alteración de los índices
hematológicos y leve nefropatía. En hembras de rata a las que se
administró la concentración más elevada, 50 g/litro (approx. 3400
mg/kg de peso corporal al día), los efectos observados fueron un
aumento de los pesos relativos de ciertos órganos y la alteración de
los índices hematológicos. Además, la exposición durante 13 semanas a
50 g/litro provocó una alteración del peso relativo de los testículos
y de la mutualidad y morfología de los espermatozoides en ratas macho.
Las hembras de ratón a las que se administraron 50 g/litro (approx. 11
298 mg/kg de peso corporal al día) en el agua de bebida presentaron
alteraciones del peso del hígado y el bazo y una incidencia
ligeramente mayor de hipertrofia hepatocelular centrilobular. No se
observaron efectos tóxicos en ratones macho a los que se administraron
20 g/litro (4858 mg/kg de peso corporal al tila), la concentración de
acetona más alta que se ha administrado a ratones macho. La exposición
durante 13 semanas a concentraciones < 10 g/litro (900 mg/kg de
peso corporal al día) administradas en el agua de bebida no se vio
acompañada de efectos tóxicos en ratas macho; las concentraciones
< 20 g/litro no produjeron efectos observables en hembras de rata
(1600 mg/kg de peso corporal al día) ni en ratones de ambos sexos
(machos: 4858 mg/kg de peso corporal al día; hembras: 5945 mg/kg de
peso corporal al día).
En un estudio preliminar en el que se administró acetona en el
agua de bebida a ratas y ratones durante 14 alias, se observó
hipertrofia hepatocelular centrilobular relacionada con la dosis en
ratones machos expuestos a concentraciones de 20-100 g/litro.
El tratamiento previo de roedores con acetona acentúa los efectos
hepatotóxicos de varios compuestos, particularmente los alcanos
halogenados. Una hipótesis es que la potenciación de la
hepatotoxicidad está mediada por el aumento de las actividades
enzimáticas mediadas por la acetona (oxidabas hepáticas de funcion
mixta) que son responsables de la generación de productos intermedios
tóxicos originados a partir de los alcanos halogenados administrados.
La acetona ha dado resultados negativos en relación con la
toxicidad genética en numerosos sistemas no mamíferos, así como en
sistemas mamíferos in vitro e in vivo. Los resultados positivos se
limitan a una sola prueba de aneuploidía en una especie de levadura
expuesta a concentraciones elevadas de acetona (6,82%) en su medio de
cultivo. No se considera que la acetona sea genotóxica ni mutagénica.
En un estudio de hembras gestantes de ratón y rata expuestas a
vapores de acetona durante los días ó a 19 de la gestación, se observó
una ligera toxicidad para el desarrollo tras la exposición de las
ratas a 26 100 mg/m3 (11 000 ppm) durante 6 horas al día (pequeña
disminución en el peso del feto y pequeño aumento en la incidencia
porcentual de retorciones tardías). Se determinó que una concentración
atmosférica de 5200 mg/m3 (2200 ppm) era el nivel sin observación de
efectos adversos respecto de la toxicidad para el desarrollo tanto en
ratones como en ratas. En un estudio de alimentación forzada, el
tratamiento con 3500 mg/kg al día durante la organogénesis
obstaculizaba la reproducción en una prueba de detección en ratones.
Los resultados negativos obtenidos in vivo en dos especies
diferentes, utilizando las vías oral e intraperitoneal, indicó que no
se producían cambios mutagénicos en mamíferos expuestos a la acetona.
Entre los datos comunicados acerca de otros efectos reproductivos
de la acetona figuran observaciones de efectos testiculares y
alteraciones de la calidad espermática en ratas a las que se
administró agua de bebida con 50 g de acetona por litro durante 13
semanas. No se dispuso de datos sobre investigaciones del efecto de
dosis de acetona administradas por vía oral en el desarrollo fetal
(fetotoxicidad y teratogenicidad).
La acetona se ha utilizado ampliamente como vehículo disolvente en
estudios de carcinogenicidad en la piel y no se considera
carcinogénico cuando se aplica a la piel.
7. Efectos en el ser humano
La acetona es relativamente menos tóxica que muchos otros
disolventes industriales; sin embargo, en concentraciones altas, el
vapor de acetona puede provocar depresión del sistema nervioso
central, fallo cardiorrespiratorio y la muerte. Se ha comunicado que
la exposición aguda del ser humano a concentraciones atmosféricas tan
altas como approx. 4750 mg/m3 (approx 2000 ppm) no produce grandes
efectos tóxicos ni efectos transitorios leyes, como irritación ocular.
Se comunicaron efectos transitorios más graves (inclusive vómitos y
desmayos) en trabajadores expuestos a concentraciones de vapor de
acetona > 25 500 mg/m3 (250 ppm). Las mujeres expuestas a
concentraciones atmosféricas de 2370 mg/m3 (1000 ppm) padecieron
trastornos menstruales.
8. Efectos en otros organismos en el laboratorio y sobre el terreno
En la mayoría de las especies animales de agua tanto dulce como
salada, los valores de la CL50 y la CE50 a las 48 y las 96 horas son
superiores a 5540 mg/litro.
La exposición a una concentración de acetona de 257,4 mg/litro
durante 76 horas inhibió el crecimiento del alga
Chlorella pyrendoidosa. También se observó inhibición del
crecimiento de Chlamydomonas eugametos expuesta a acetona durante 48
horas en una concentración de 790 mg/litro. En
Scendesmus quadricauda y C pyrenoidosa expuestas a 79,0 y 790
mg/litro de acetona se observo un aumento de la fotosíntesis.
Los umbrales de toxicidad a 7 y 8 días para el alga verde
S. quadricauda y la cianobacteria (alga verdeazulada)
Microcystis aeruginosa fueron 7500 y 530 mg/litro, respectivamente,
lo que indica que el alga verde era más resistente a la actividad
tóxica de la acetona. La diatomea Nitzschia linearis también resultó
ser muy resistente, con una EC50 en cinco días de 11 493 a 11 727
mg/litro. De modo similar, la diatomea de agua salada
Skeletonema costatum resultó muy resistente, con valores de EC50
a tos cinco días de 11 798 y 14 440 mg/litro.
Las bacterias parecen ser más resistentes a la acetona que los
protozoos. Photobacterium phosphoreum, Pseudomonas putida y un
cultivo microbiano mixto presentaron valores de la CE50 de 1706 a 35
540 mg/litro, mientras que el protozoo Entosiphon sulcatum presentó
una CE50 de 28 mg/litro. Esto puede guardar relación con las
diferencias en la pared celular.
En codornices y faisanes se observaron valores de la CL50 por
vía oral a los cinco días > 40 g/kg de dieta. Los huevos
fecundados de pato silvestre no se vieron afectados por la inmersión
en acetona al 10% durante 30 segundos; en cambio, la inmersión en
acetona pura dio lugar a un descenso de la supervivencia, el peso y la
talla del embrión, aunque no está claro si ello se debía a las
propiedades tóxicas o a las propiedades disolventes de la acetona. Los
huevos de gallina Leghorn blanca inyectados con 5 µl de acetona no
mostraron cambios significativos en la mortalidad ni malformaciones.