
IPCS INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 18
ARSENIC
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organization, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Organization Geneva, 1981
ISBN 92 4 154078 8
(c) World Health Organization 1981
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. For rights of reproduction or
translation of WHO publications, in part or in toto, application
should be made to the Office of Publications, World Health
Organization, Geneva, Switzerland. The World Health Organization
welcomes such applications.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR ARSENIC
1. SUMMARY AND RECOMMENDATIONS FOR FURTHER RESEARCH
1.1. Summary
1.1.1. Properties, uses, and analytical procedures
1.1.1.1 Properties and uses
1.1.1.2 Analytical procedures
1.1.2. Environmental transport and distribution
1.1.3. Exposure
1.1.4. Metabolism
1.1.5. Normal levels in man and biological indicators of
exposure
1.1.6. Effects and evaluation of health risks
1.1.6.1 Inorganic arsenic compounds
1.1.6.2 Organic arsenic compounds
1.2. Recommendations for further research
1.2.1. Sampling and determination
1.2.2. Exposure
1.2.3. Metabolism and indicators of exposure
1.2.4. Effects
2. PROPERTIES AND ANALYTICAL PROCEDURES
2.1. Chemical and physical properties of arsenic compounds
2.1.1. Inorganic arsenic compounds
2.1.2. Organic arsenic compounds
2.2. Analytical procedures
2.2.1. Sampling and sample treatment
2.2.1.1 Natural waters
2.2.1.2 Air
2.2.1.3 Biological materials
2.2.2. Analytical methods
2.2.2.1 Methods for total arsenic
2.2.2.2 Analyses for specific arsenic compounds
3. SOURCES AND OCCURRENCE OF ARSENIC IN THE ENVIRONMENT
3.1. Natural occurrence
3.1.1. Rocks, soils, and sediments
3.1.2. Air
3.1.3. Water
3.1.4. Biota
3.2. Industrial production and uses of arsenic
3.2.1. Industrial production
3.2.2. Uses of arsenic compounds
3.2.3. Sources of environmental pollution
4. ENVIRONMENTAL TRANSPORT AND DISTRIBUTION
4.1. General
4.2. Aquatic systems
4.3. Air-soil systems
5. LEVELS OF EXPOSURE TO ARSENIC AND ITS COMPOUNDS
5.1. General population exposure through air, drinking water,
food, and beverages
5.1.1. Air
5.1.2. Drinking water
5.1.3. Food and beverages
5.1.4. Tobacco
5.1.5. Drugs
5.1.6. Total daily intake in the general population
5.2. Occupational exposure
6. METABOLISM OF ARSENIC
6.1. Inorganic arsenic
6.1.1. Absorption
6.1.1.1 Respiratory deposition and absorption
6.1.1.1.1 Animals
6.1.1.1.2 Man
6.1.1.2 Gastrointestinal absorption
6.1.1.2.1 Animals
6.1.1.2.2 Man
6.1.1.3 Skin absorption
6.1.1.3.1 Animals
6.1.1.3.2 Man
6.1.1.4 Placental transfer
6.1.1.4.1 Animals
6.1.1.4.2 Man
6.1.2. Distribution in organisms
6.1.2.1 Fate of arsenic in blood
6.1.2.1.1 Animals
6.1.2.1.2 Man
6.1.2.2 Tissue distribution
6.1.2.2.1 Animals
6.1.2.2.2 Man
6.1.3. Elimination
6.1.3.1 Animals
6.1.3.2 Man
6.1.4. Biotransformation
6.1.4.1 Animals
6.1.4.2 Man
6.2. Organic arsenic compounds
6.2.1. Absorption
6.2.1.1 Respiratory absorption
6.2.1.1.1 Animals
6.2.1.1.2 Man
6.2.1.2 Gastrointestinal absorption
6.2.1.2.1 Animals
6.2.1.2.2 Man
6.2.1.3 Skin absorption
6.2.1.4 Placental transfer
6.2.1.4.1 Animals
6.2.1.4.2 Man
6.2.2. Distribution in organisms
6.2.2.1 Fate of organic arsenic in blood
6.2.2.1.1 Animals
6.2.2.1.2 Man
6.2.2.2 Tissue distribution of organic arsenic
6.2.2.2.1 Animals
6.2.2.2.2 Man
6.2.3. Elimination
6.2.3.1 Animals
6.2.3.2 Man
6.2.4. Biotransformation
6.2.4.1 Animals
6.2.4.2 Man
7. NORMAL LEVELS IN MAN AND BIOLOGICAL INDICATORS OF EXPOSURE
7.1. Blood
7.2. Urine
7.3. Hair
7.4. Other tissues
8. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF INORGANIC ARSENIC
8.1. Acute and subacute effects after short-term exposure
8.1.1. Man
8.1.2. Animals
8.2. Effects on reproduction and teratogenicity
8.2.1. Man
8.2.2. Animals
8.3. Noncarcinogenic effects after long-term exposure and
sequelae of short-term exposure to inorganic arsenic
8.3.1. Effects on the respiratory system
8.3.1.1 Man
8.3.1.2 Animals
8.3.2. Effects on skin
8.3.2.1 Man
8.3.2.2 Animals
8.3.3. Effects on the liver
8.3.3.1 Man
8.3.3.2 Animals
8.3.4. Effects on the cardiovascular system
8.3.4.1 Man
8.3.4.2 Animals
8.3.5. Effects on the nervous system
8.3.5.1 Man
8.3.5.2 Animals
8.3.6. Effects on other organs
8.3.6.1 Man
8.3.6.2 Animals
8.4. Carcinogenicity
8.4.1. Man
8.4.1.1 Cancer of the respiratory system
8.4.1.2 Cancer of the skin
8.4.1.3 Cancer of the liver
8.4.1.4 Leukaemia and tumours of the
haematopoietic system
8.4.1.5 Cancer of other organs
8.4.2. Experimental animal studies
8.4.2.1 Cancer of the respiratory system
8.4.2.2 Skin application
8.4.2.3 Oral administration
8.4.2.4 Other experimental systems
8.5. Mutagenicity
8.6. Mechanisms of toxicity
9. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF ORGANIC ARSENIC
COMPOUNDS
9.1. Acute and chronic toxicity
9.1.1. Man
9.1.2. Animals
9.2. Teratogenicity
9.3. Carcinogenicity
9.3.1. Animals
9.4. Mutagenicity
9.5. Mechanisms of toxicity
10. INTERACTIONS WITH OTHER CHEMICALS
10.1. Thiol-compounds
10.2. Selenium
10.3. Cadmium and lead
11. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO ARSENIC
11.1. Introduction
11.2. Exposure
11.3. Inorganic arsenic compounds
11.3.1. Acute and subacute effects after short-term
exposure
11.3.2. Noncarcinogenic effects after long-term exposure
and sequelae of short-term exposure
11.3.2.1 Skin effects
11.3.2.2 Cardiovascular effects
11.3.2.3 Neurological effects
11.3.3. Carcinogenicity
11.3.3.1 Cancer of the respiratory system
11.3.3.2 Skin cancer
11.4. Organic arsenic compounds
11.5. Assessment of the cancer risk for man from exposure to
inorganic arsenic
REFERENCES
NOTE TO READERS OF THE CRITERIA DOCUMENTS
While every effort has been made to present information in the
criteria documents as accurately as possible without unduly delaying
their publication, mistakes might have occurred and are likely to
occur in the future. In the interest of all users of the environmental
health criteria documents, readers are kindly requested to communicate
any errors found to the Division of Environmental Health, World Health
Organization, Geneva, Switzerland, in order that they may be included
in corrigenda which will appear in subsequent volumes.
In addition, experts in any particular field dealt with in the
criteria documents are kindly requested to make available to the WHO
Secretariat any important published information that may have
inadvertently been omitted and which may change the evaluation of
health risks from exposure to the environmental agent under
examination, so that the information may be considered in the event of
updating and re-evaluation of the conclusions contained in the
criteria documents.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR ARSENIC
Members
Dr R. Albert, Institute of Environmental Medicine, New York University
Medical Center, New York, NY, USA (Chairman of Subgroup 2)
Dr V. Bencko, Department of General and Environmental Hygiene, Medical
Faculty of Hygiene, Charles University, Prague, Czechoslovakia
Dr G. Corey, Department of Environment Programs, Ministry of Health,
Santiago, Chile
Dr L. Friberg, Departments of Environmental Hygiene of the Karolinska
Institute and of the National Swedish Environment Protection
Board, Stockholm, Sweden (Chairman)
Professor Dr N. Ishinishi, Department of Hygiene, Faculty of Medicine,
Kyushu University, Fukuoka City, Japan (Vice Chairman)
Dr C. Maltoni, Institute of oncology, Bologna, Italya
Dr B. Ordóñez, Undersecretary for Environmental Improvement,
Secretariat for Health and Welfare, Mexico
Professor Dr R. Preussmann, German Cancer Research Center, Institute
of Toxicology and Chemotherapy, Heidelberg, Federal Republic of
Germany
Dr G. Samarawickrama, Department of Community Medicine, Faculty of
Medicine, Peradeniya, Sri Lanka
Dr E. Sandi, Bureau of Chemical Safety, Food Directorate, Health
Protection Branch, Department of National Health & Welfare,
Ottawa, Ontario, Canada (Chairman of Sub group 1)
Dr G. Pershagen, Department of Environmental Hygiene, Karolinska
Institute, Stockholm, Sweden (Rapporteur)
Ms M. Vahter, Department of Environmental Hygiene, National Swedish
Environment Protection Board, Stockholm, Sweden (Rapporteur)
a Unable to attend the Task Group meeting.
Representatives of other organizations
Dr A. Berlin, Commission of the European Communities, Luxembourg
Dr G. F. Nordberg, Commission of the European Communities, Department
of Environmental Hygiene, University of Umeå, Umeå, Sweden
Mr C. Satkunananthan, United Nations Environment Programme, Geneva,
Switzerland
Dr M. Stoeppler, International Union of Pure and Applied Chemistry,
Institute for Chemistry of the Jülich Nuclear Research Facility
Ltd, Jülich, Federal Republic of Germany
Observers
Mr K. W. Nelson, ASARCO Inc., New York, NY USA (representing the
International Center of Industry and Environment)
Mr H. Norin, Department of Environmental Hygiene, National Swedish
Environment Protection Board, Stockholm, Sweden
Dr S.S. Pinto, ASARCO Inc., Tacoma, WA, USA (representing the
International Center of Industry and Environment)
Dr M. Piscator, Department of Environmental Hygiene, Karolinska
Institute, Stockholm, Sweden
Mr R. Svedberg, Boliden Metall AB, Skelleftehamn, Sweden
Secretariat
Mr G. Ozolins, Associate Manager, Environmental Health Criteria and
Standards, Division of Environmental Health, World Health
Organization, Geneva, Switzerland
Mr J. D. Wilbourn, Division of Chemical and Biological Carcinogenesis,
International Agency for Research on Cancer, Lyons, France
CONSULTATION ON THE PREPARATION OF THE ENVIRONMENTAL HEALTH
CRITERIA DOCUMENT ON ARSENIC
STOCKHOLM FROM 4 TO 6 OCTOBER 1978
Participants
Dr V. Bencko, Department of General and Environmental Hygiene, Medical
Faculty of Hygiene, Charles University, Prague, Czechoslovakia
Dr A. Berlin, Commission of the European Communities, Luxembourg
Dr B. Fowler, Environmental Toxicology Branch, National Institute of
Environmental Health Sciences, Research Triangle Park, NC, USA
Dr L. Friberg, Departments of Environmental Hygiene of the Karolinska
Institute and of the National Environment Protection Board,
Stockholm, Sweden (Chairman)
Dr G. Lunde, Central Institute for Industrial Research, Oslo, Norway
Dr G. F. Nordberg, representative of the Commission of the European
Communities, Institute of Community Health and Environmental
Medicine, Odense University, Odense, Denmark
Mr G. Ozolins, Coordinator, Environmental Health Criteria and
Standards, Division of Environmental Health, World Health
Organization, Geneva, Switzerland
Dr G. Pershagen, Department of Environmental Hygiene, Karolinska
Institute, Stockholm, Sweden (Rapporteur)
Dr M. Piscator, Department of Environmental Hygiene, Karolinska
Institute, Stockholm, Sweden
Ms M. Vahter, Department of Environmental Hygiene, National Swedish
Environment Protection Board, Stockholm, Sweden (Rapporteur)
ENVIRONMENTAL HEALTH CRITERIA FOR ARSENIC
Members of the Task Group on Environmental Criteria for Arsenic
met in Stockholm from 28 January to 1 February 1980. The meeting was
opened on behalf of the Director-General by Mr G. Ozolins, Associate
Manager, Environmental Health Criteria and Standards. The Task Group
reviewed and revised the draft criteria document and made an
evaluation of the health risks from exposure to arsenic and its
compounds.
The meeting worked in two subgroups, one on chemical and
environmental aspects and metabolism (subgroup 1) and the other on
effects (subgroup 2). Comments of the subgroups were discussed in
plenary sessions and the conclusions were drawn by the whole group.
The first draft of the biomedical parts of the document was
prepared at the WHO Collaborating Centre for Environmental Health
Effects, Departments of Environmental Hygiene of the Karolinska
Institute and the National Environment Protection Board, Stockholm,
Sweden. Dr G. Pershagen and Ms M. Vahter were primarily responsible
for its preparation. Discussions were held with a group preparing a
report on arsenic for the Health Directorate of the Commission of the
European Communities, Luxembourg and the draft was reviewed and
revised at a consultation arranged by WHO at the Karolinska Institute
in Stockholm from 4 to 6 October, 1978.
The second draft, which was sent out to the national focal points
for environmental health criteria documents, included sections on the
chemical and environmental aspects of arsenic prepared by Dr. R. S.
Braman, Department of Chemistry, University of South Florida, Tampa,
FL, USA.
The third draft was prepared by Dr R. S. Braman and Dr G.
Pershagen based on comments from the national focal points in
Australia, Belgium, Canada, Chile, Finland, Federal Republic of
Germany, Greece, Japan, Mexico, New Zealand, Poland, the United
Kingdom, and the USA, and from the International Labour Office (ILO)
and the American Smelting and Refining Company (ASARCO).
The document, scientifically edited by Dr G. Pershagen and Ms M.
Vahter and reviewed by Dr V. B. Vouk, is based primarily on original
publications listed in the reference section. However, some
comprehensive reviews on the health effects of arsenic including
Fowler (1977), NAS (1977), IARC (1973, 1980), and Pershagen & Vahter
(1979) have also been used.
The possible role of arsenic as an essential element and the
effects of arsine have not been discussed in this document.
Details of the WHO Environmental Health Criteria Programme,
including some of the terms frequently used in the documents, may be
found in the introduction to the Environmental Health Criteria
Programme published together with the environmental health criteria
document on mercury (Environmental Health Criteria 1 -- Mercury, World
Health Organization, Geneva, 1976), and now available as a reprint.
Mrs M. Dahlquist at the WHO Collaborating Centre for Environmental
Health Effects, Stockholm, acted as technical and administrative
assistant and her work is greatly appreciated.
Financial support for the publication of this criteria document
was kindly provided by the Department of Health and Human Services
through a contract from the National Institute of Environmental Health
Sciences, Research Triangle Park, North Carolina, USA -- a WHO
Collaborating Centre for Environmental Health Effects.
1. SUMMARY AND RECOMMENDATIONS FOR FURTHER RESEARCH
1.1 Summary
1.1.1 Properties, uses and analytical procedures
1.1.1.1 Properties and uses
Arsenic is a ubiquitous element with metalloid properties. Its
chemistry is complex and there are many different compounds of both
inorganic and organic arsenic. In nature, it is widely distributed in
a number of minerals, mainly as the arsenides of copper, nickel, and
iron, or as arsenic sulfide or oxide. In water, arsenic is usually
found in the form of arsenate or arsenite. Methylated arsenic
compounds occur naturally in the environment as the result of
biological activity. The most important commercial compound,
arsenic(III) oxide, is produced as a by-product in the smelting of
copper and lead ores.
Arsenic compounds are mainly used in agriculture and forestry as
pesticides, herbicides, and silvicides; smaller amounts are used in
the glass and ceramics industries and as feed additives.
1.1.1.2 Analytical procedures
If total arsenic has to be determined, the first step usually
consists of complete mineralization. The arsenic can then be measured
directly by, for example, flame or graphite tube atomic absorption
spectrophotometry (AAS). In an ordinary flame, the detection limit is
0.5-1 mg/litre. Using a long-path cell, a detection limit of a few
µg/litre can be obtained.
The most commonly used techniques for the determination of arsenic
involve its transformation into arsine. Subsequent measurements of
arsine can be carried out using, spectrophotometry, flames and
electrothermal devices for AAS, atomic fluorescence (AFS), or atomic
emission spectroscopy (AES).
Spectrophotometry of the silver diethyldithiocarbamate complex of
arsine has been used for several years, and is suitable for
determining arsenic levels in the range of 1-100 µg. Passing the
arsine, generated, for instance, by sodium borohydride, into a heated
tube of an AAS or AES instrument gives an absolute detection limit of
about 0.5 ng. If oxidation can be avoided prior to the arsine
generation step, it is possible to differentiate between As(III) and
As(V) by changing the pH value at this step. Furthermore, cold
trapping of the arsines and separation upon heating can be used for
the separation and detection of inorganic and methylated arsenic
compounds present in natural waters and urine. Other separation
methods include ion exchange chromatography, gas chromatography, and
liquid chromatography.
Neutron activation analysis using radiochemical separation is a
very sensitive method for the determination of arsenic, with detection
limits near 1 ng.
1.1.2 Environmental transport and distribution
Arsenic is mainly transported in the environment by water.
Sedimentation of arsenic in association with iron and aluminium may
sometimes be considerable. In oxygenated water, arsenic usually occurs
as arsenate, but under reducing conditions, for instance, in deep well
waters, arsenite predominates. Methylation of inorganic arsenic to
methyl- and dimethylarsenic acids is associated with biological
activity in water. Some marine organisms have been shown to transform
inorganic arsenic into more complex organic compounds, such as
arsenobetaine, arsenocholine, and arsoniumphospholipids.
In oxygenated soil, inorganic arsenica is present in the
pentavalent form. Under reducing conditions, it is in the trivalent
form. Leaching of arsenate is slow, because of binding to hydrous
oxides of iron and aluminium. There is ample evidence of
biomethylation in the soil and of the release of methylarsines into
the air and high levels of methylated arsenic compound have been
detected in greenhouse air. However, airborne arsenic is mainly
inorganic.
1.1.3 Exposure
Because the metabolic fates and toxicities of arsenic compounds
differ, it is important to distinguish between them in the
environment. The forms of arsenic to which man is actually exposed
have not been considered in detail until recently, mainly because of a
lack of suitable analytical methods.
Airborne concentrations of arsenic in urban areas may range from a
few nanograms to a few tenths of a microgram per cubic metre. Near
point emissions of arsenic, such as smelters, airborne arsenic
concentrations have exceeded 1 µg/m3. Drinking water ordinarily
contains a few micrograms of arsenic per litre or less, mainly in the
form of inorganic compounds. Levels exceeding 1 mg/litre recorded in
some areas, have usually been naturally occurring, but have sometimes
been the result of industrial contamination.
a Abbreviations "inorganic arsenic" and "organic arsenic" mean
"arsenic and its inorganic compounds" and "organic arsenic
compounds", respectively.
Arsenic is present in most foodstuffs in concentrations of less
than 1 mg/kg. However, marine fish may contain arsenic concentrations
of up to 5 mg/kg wet weight and concentrations in some crustacea and
bottom-feeding fish may reach several tens of milligrams per kilogram,
predominantly in the form of organic arsenic. Accumulation of arsenic
in the tissues of poultry and swine can result from the use of some
organic arsenic compounds as feed additives.
Wine and mineral waters can sometimes contain several hundreds of
micrograms of arsenic per litre, probably as a result of the use of
arsenic-containing pesticides. Inorganic forms of arsenic have been
shown to predominate in wine.
The total daily intake of arsenic by man is greatly influenced by
the amount of seafood in the diet, but it is usually less than 0.2 mg
per day. Normally, the daily intake of inorganic arsenic will not
exceed 50 µg. Depending on the content of arsenic in tobacco, an
average smoker may inhale between a few micrograms and 20 µg of
arsenic daily. Some decades ago, when the arsenic content of tobacco
was higher, more than 100 µg might have been inhaled per day. The
chemical form of arsenic in tobacco smoke is not known.
Various arsenic compounds have been used in medicine for many
years. Inorganic trivalent arsenic, often in the form of sodium
arsenite (Fowler's solution) has been used for the treatment of
leukaemia, psoriasis, and as a tonic, frequently at a dose of several
milligrams daily. Some inorganic as well as organic arsenic compounds
are still used in drugs in a number of countries.
Occupational exposure to arsenic mainly occurs through the
inhalation of particles containing arsenic, i.e., among smelter
workers and workers engaged in the production and use of
arsenic-containing pesticides. Concentrations in air ranging from a
few micrograms to more than 1 mg/m3 have been reported.
1.1.4 Metabolism
Studies on animals and man have shown that both trivalent and
pentavalent inorganic arsenic compounds in solution are readily
absorbed after ingestion. Inhalation usually involves particles
containing inorganic arsenic. Most of the inhaled and deposited
arsenic will probably be absorbed from either the respiratory or the
gastrointestinal tract.
The biological half-time of arsenic in rats is long (60 days),
because of its accumulation in erythrocytes. In other animals and in
man, most inorganic arsenic is eliminated at a much higher rate,
mainly via the kidneys. As far as exposure to trivalent arsenic in a
single dose is concerned, both animal and human data indicate an
initial elimination of about 75% in the urine and a few percent in the
faeces during the first days or, at the most, the first week. As for
pentavalent arsenic, a few animal experiments have indicated that
80-90% of a single dose is eliminated during the first 2 days, while
available human data indicate a slower rate of elimination. Animal
data show a somewhat higher retention of arsenic in different organs
after exposure to trivalent arsenic than after exposure to the
pentavalent form. The differences increase with increasing dose
levels.
Placental transfer of inorganic arsenic has been demonstrated in
both experimental animal (rat and hamster) and human studies. In a
study on rats, dimethylarsinic acid was shown to pass through the
placental barrier, the blood values in the fetus being comparable with
those of the mother.
No data are available which indicate that long-term accumulation
of arsenic exists. Some data on mice and rabbits exposed for up to one
year to arsenic indicated that levels of arsenic in the body increased
during the first 2 weeks and then decreased. There are very few data
concerning accumulation in people heavily exposed to inorganic
arsenic, such as industrial populations or populations in areas where
the drinking water contains high levels of arsenic. However, some data
have indicated that arsenic levels in the lungs of smelter workers,
several years after exposure, were 6 times those of controls. The
concentrations of arsenic in human tissues seem to be log-normally
distributed and the highest levels are generally found in hair, skin,
and nails.
In vivo methylation of inorganic arsenic has been demonstrated
in both animals and man. Following ingestion or inhalation of
inorganic arsenic, the major forms of arsenic excreted in human urine
are dimethylarsinic acid and methylarsonic acid accounting for about
65% and 20% of excreted arsenic, respectively. In other species,
methylarsonic acid has only been observed in minimal amounts.
In both animals and man, organic arsenic compounds ingested via
fish and crustacea are readily absorbed from the gastrointestinal
tract and 70%-80% is eliminated within a week, mainly in the urine.
Some data indicate that these compounds are eliminated without being
converted to inorganic arsenic or simple methylated arsenic compounds.
Organic arsenic compounds from other sources show various degrees of
absorption, transformation, and retention.
1.1.5 Normal levels in man and biological indicators
of exposure
In subjects not known to have been exposed to arsenic, whole blood
arsenic levels are in the range of only a few micrograms per litre,
while in persons exposed to water containing high levels of arsenic,
whole blood levels exceeding 50 µg/litre have been reported. No data
are available on the influence of dietary habits on arsenic levels in
blood.
Studies on the metabolism of inorganic arsenic show that in most
animals and man arsenic is taken up readily by the blood and also
rapidly cleared. Arsenic in blood will therefore reflect exposure for
only a short period following absorption and will be highly
time-dependent. If exposure is continuous, as may be the case with
drinking water, it should be possible to find a relationship between
arsenic levels in blood and exposure. However, such studies have not
been carried out.
Effects of arsenic have been seen in a large number of organs in
both animals and man. However, data are not available from which it is
possible to correlate such effects with tissue concentrations, or with
the concentrations in blood. It has not been possible to define a
critical organ for arsenic in the way that the kidney is considered a
critical organ for chronic cadmium intoxication and the central
nervous system for methylmercury intoxication. The short half-time of
arsenic in the blood compared with those in the whole body and
individual organs makes it difficult to establish a relationship
between concentrations of arsenic in blood and the total body burden.
A metabolic model for arsenic has not yet been developed.
Arsenic concentrations in the urine of persons who have not been
excessively exposed to arsenic through, for example, occupation or
dietary habits, have been estimated to range from 10 to 50 µg/litre.
Excretion of up to a few milligrams of arsenic in the urine on the
first day following ingestion of fish with a high arsenic content has
been reported.
Smelter workers exposed to inorganic arsenic compounds may have
urine values of a few hundred micrograms per litre. One study
indicated that the major part of the arsenic was excreted as
dimethylarsinic acid. Increased urinary levels of arsenic have also
been observed in persons living around point sources emitting arsenic.
Urine is a suitable indicator medium for assessment of exposure to
inorganic arsenic, since most studies show that the elimination of
arsenic, in both animals and man, takes place mainly via the kidneys.
A method of assessment must be used that differentiates between the
organic arsenic compounds from sea food and the main metabolites of
inorganic arsenic.
Arsenic levels in the hair of unexposed human adults are usually
below 1 mg/kg. There are no published data to indicate whether
exposure to arsenic in sea food results in increased hair values.
Levels of up to about 80 mk/kg have been recorded in subjects with
chronic arsenic poisoning caused by ingestion of contaminated well
water.
The use of arsenic concentrations in hair as an indicator of
exposure to airborne arsenic is limited, as no reliable method exists
for distinguishing between arsenic from external contamination and
arsenic that has been absorbed and metabolized in the body.
1.1.6 Effects and evaluation of health risks
1.1.6.1 Inorganic arsenic compounds
Acute and subacute effects of arsenic may involve many organ
systems including the respiratory, gastrointestinal, cardiovascular,
nervous, and haematopoietic systems. Unfortunately, in most cases of
human intoxication, the doses and valence states of arsenic have not
been determined. Data from studies on experimental animals indicate
that trivalent inorganic arsenic is more toxic than pentavalent. It is
also evident that arsenic in solution is more toxic than undissolved
arsenic, probably because of better absorption. An ingested dose of
70-180 mg of arsenic (III) oxide has been reported to be fatal in man.
Long-term exposure to inorganic arsenic has been found to give
rise to effects in a large number of organs. However, in general, the
details of human exposure (e.g., type of arsenic compound), have been
inadequate for the establishment of dose-response relationships.
Lesions of the upper respiratory tract including perforation of
the nasal septum, laryngitis, pharyngitis, and bronchitis have
frequently been encountered in workers in the smelting industry
exposed to high levels of arsenic. In general, such lesions have been
reported in instances of prolonged exposure to several hundred
micrograms of arsenic per cubic metre of air and mostly with arsenic
in the trivalent inorganic form. In the case of lower respiratory
tract lesions in workers in the smelting industry, the influence of
concurrent exposure to high levels of sulfur dioxide should be
considered as well as interaction with tobacco smoking.
Inorganic arsenic in the trivalent state can give rise to skin
lesions in man, especially palmo-plantar hyperkeratosis which has a
characteristic appearance. It has been observed in patients under
prolonged medication with Fowler's solution, who have received daily
doses of arsenic of up to 10 mg. In one study, the incidence of
hyperkeratosis was reported to be over 50% in a group of patients,
each of whom had received a total dose of more than 3 g of arsenic.
Palmo-plantar hyperkeratosis has also been reported following
ingestion of arsenic in drinking water (oxidation state not
determined) in some parts of the world including Argentina, China
(Province of Taiwan) and Mexico. Other dermatological symptoms,
including hyperpigmentation, have also appeared in inhabitants of
these areas.
It should be noted that hyperkeratotic lesions of the palms and
soles and hyperpigmentation are very rare among smelter workers
exposed to inorganic arsenic, but have been reported in other
occupational situations. The reason for this discrepancy is not clear
but could be the result of differences in dose.
Disturbances of liver function have been observed in both man and
animals after chronic exposure to inorganic arsenic. An association
between medication with trivalent inorganic arsenic and the
development of portal hypertension in man has been suggested, though
this has not been reported in experimental animals. Indications of
severe hepatic damage resulting in cirrhosis, have come from both
epidemiological and toxicological data. The role of alcohol
consumption in situations of arsenic exposure has, unfortunately, not
been considered in most of the studies.
Evidence of effects on the heart, including minor ECG changes, has
been found in human subjects after exposure to comparatively high
doses of arsenic, which produced other symptoms and signs of
intoxication. These findings have been supported by animal data. A
moderate excess mortality attributed to cardiovascular lesions was
detected in 2 independent epidemiological studies on smelter workers
exposed to high levels of airborne, inorganic arsenic (exposure levels
not given). This finding has not been confirmed in other studies on
workers exposed to arsenic.
Peripheral vascular disturbances have been reported in some areas
of the world where heavy exposure due to ingestion of inorganic
arsenic has occurred, e.g., in Chile, China (Province of Taiwan) and
the Federal Republic of Germany. Exposures have been of the order of
several hundred micrograms to over one milligram daily; the valence
state is not known. Generally, peripheral vascular changes have not
been reported in connexion with occupational exposure to inorganic
arsenic, and, unfortunately, their possible existence in
arsenic-exposed animals has not been considered.
Inorganic arsenic can exert chronic effects on the peripheral
nervous system in man. The only information on these effects as far as
occupational exposure is concerned comes from case reports, and
exposure levels have not been given. It is obviously difficult to draw
any conclusions from such reports. Disturbances of CNS function were
reported in Japanese youths, 15 years after they had been exposed as
infants to inorganic arsenic in average daily doses of 3.5 mg for
about one month. The effects included severe hearing loss and
electroencephalographic abnormalities. CNS effects have also been
reproduced in animals. Children living near a coal-fired power plant,
which emitted large amounts of arsenic, were reported to have moderate
hearing losses, but such effects were not confirmed in another
instance of exposure to elevated levels of inorganic arsenic in
ambient air. Ingestion of moderate amounts of inorganic arsenic at
levels of a few hundred micrograms daily in drinking water (length of
exposure and valence state of arsenic unknown) has been associated
with abnormal electromyographic findings in one study. This effect
might serve as a sensitive indicator of arsenic intoxication, but the
association must be identified and evaluated elsewhere before any
definite conclusions can be drawn.
Because inorganic trivalent arsenic has an effect on the
haematopoietic system, it has been used for several decades as a
therapeutic agent for various forms of leukaemia, often in doses of
several milligrams dally. The impaired resistance to viral infections,
associated with arsenic exposure in some animal studies, should be
noted, when considering the high frequency of chronic cough,
bronchopulmonary disease, and lip herpes observed in persons exposed
to arsenic in water in Chile. The lack of evidence of these effects in
other studies on human subjects is worth noting. Animal data suggest
that arsenic exposure may have chronic effects on the kidneys, but
this has not been confirmed for human exposure situations.
There are both in vivo and in vitro studies indicating effects
of inorganic arsenic on human chromosomes. An increased frequency of
chromosomal aberrations has been found among persons exposed to
arsenic, mainly in the trivalent form, through medication. Similar
findings have been reported among workers exposed to arsenic. However,
the exposure of these workers to other toxic substances may have been
of importance.
Several studies have indicated that inorganic arsenic affects DNA
repair mechanisms.
Human data on the teratogenicity of inorganic arsenic are lacking.
One epidemiological study on the offspring of women working at a
copper smelter, where high levels of airborne arsenic were registered
in some workplaces, pointed towards an increased frequency of
malformations and spontaneous abortions. Since exposure to several
other toxic substances also took place, no conclusions can be drawn as
to the specific role of arsenic.
Results of studies on hamsters, rats, and mice have shown that
high doses of both trivalent and pentavalent inorganic arsenic induce
teratogenic effects. The high doses used in these studies make it
difficult to judge how significant such animal data are for man.
There is substantial epidemiological evidence of respiratory
carcinogenicity in association with exposure to mainly inorganic
arsenic in the manufacture of arsenic-containing insecticides.
However, conclusions cannot be drawn on the carcinogenic potential of
trivalent versus pentavalent inorganic compounds since exposure to
both forms occurred in these workplaces. A possible association
between the use of pesticides containing arsenic, often in the form of
arsenate, in vineyards and orchards, and in an increased risk of lung
cancer has been found, but the data are not conclusive.
The carcinogenic potential of inorganic arsenic in smelter
environments is evident from many epidemiological studies. One report
revealed a roughly linear relationship between cumulative arsenic
exposure and lung cancer risk. Although exposure data are uncertain,
it is estimated that exposure to airborne arsenic levels of about
50 µg/m3 (probably mostly arsenic (III) oxide) for more than 25 years
could result in a nearly 3-fold increase in the mortality rate of
cancer in the respiratory tract after the age of 65 years.
Exposure to inorganic arsenic can cause skin cancer, mainly
tumours of low malignancy. This has been observed following ingestion
of arsenic in drinking water or drugs resulting in a total intake of
several grams of arsenic over a number of decades. The form of arsenic
in drinking water has yet to be elucidated, but in medication it has
most often been inorganic trivalent arsenic.
The association between arsenic and tumours of other organs, most
notably the liver and lymphatic and haematopoietic system, needs
further confirmation.
At present, no definite evidence exists to show that inorganic
arsenic compounds are carcinogenic in animals. This holds true as far
as both tumour initiation and promotion are concerned. Results of four
studies on rats and mice, however, suggest that arsenic plays a role
in the development of tumours of the lung and the haemapoietic system.
An attempt has been made to assess the risk of cancer of the lung
and skin from low doses of arsenic by extrapolating data concerning
the risks from relatively high doses.
1.1.6.2 Organic arsenic compounds
Medication with some organic arsenic compounds such as
[4-[2-amino-2 oxoethyl]-amino]-phenyl] arsonic acid (tryparsamide),
has induced side-effects, mainly in the central nervous system. These
include encephalopathy and optic atrophy. Toxic effects on the nervous
system have been reproduced in experimental animals fed high doses of
arsanilic acid, which is commonly used as a feed additive for poultry
and swine. Limited data indicate that the toxicity of the organic
arsenic compounds present in seafood is low.
No conclusive evidence of carcinogenic activity has been reported
for any of the organoarsenic compounds tested in experimental animals.
1.2 Recommendations For Further Research
1.2.1 Sampling and analysis
A number of important problems remain to be solved in the
following areas:
(a) sampling of arsenic in air;
(b) pretreatment of samples with special attention to seafood
arsenic; and
(c) differentiation between the various arsenic species, including
the identification of the arsenic compounds in seafood.
The development of reference materials for biological specimens is
recommended and interlaboratory calibration exercises should be
performed.
1.2.2 Exposure
There are only a few dose-response relationships established for
the exposure of man to arsenic, mainly because of a lack of reliable
exposure data. More data are therefore needed on the exposure levels
of arsenic in both general and occupational environments. Continuous
monitoring of arsenic in foodstuffs, especially poultry and pork, is
required in view of the use of arsenic compounds as feed additives.
It is important not only to get quantitative measurements of the
dose but also to determine the chemical form of arsenic. Such
qualitative information is lacking for most foodstuffs as well as for
cigarette smoke. In fish and crustacea, most of the arsenic has been
reported to be in an organic form. However, data on the chemical form
of arsenic in seafood from water polluted with inorganic arsenic are
required, as fish probably cannot convert inorganic arsenic to organic
arsenic compounds.
More studies are needed to understand the volatilization of
arsenic into the air. The effect of naturally occurring oxidants such
as ozone and nitrogen oxides on volatilized arsines is of particular
interest. The oxidants may demethylate methylarsenic compounds and
convert them to inorganic forms. The arsenic forms found over the open
oceans and in remote regions also need to be determined to assess the
impact of volatilization on global transport.
1.2.3 Metabolism and indicators of exposure
In the evaluation of health effects, arsenic has generally been
treated as such without any reference to its chemical form, e.g.,
trivalent or pentavalent inorganic arsenic. Though it has been shown
that both forms are methylated in vivo, possible quantitative
differences have not been studied. Thus, the rate, extent, and
mechanism of biomethylation of different forms of arsenic should be
further investigated. Conversion of As(V) to the more toxic As(III) in
the body has been indicated in some studies, but data are not
conclusive. This needs to be further investigated together with the
possibility of in vivo oxidation. Data on the biotransformation of
arsenic compounds are also needed for the identification of biological
indicators of exposure to these compounds.
Further efforts should be made to establish a suitable animal
model for arsenic.
More data are required on the concentrations of arsenic in human
organs in high-exposure groups, including those heavily exposed to
seafood arsenic. It has been shown at autopsies that smelter workers
may retain arsenic in their lungs for several years after cessation of
exposure. It is important to study the nature of this arsenic.
More data are also needed on possible interactions between arsenic
and nutrients in the human diet as well as interactions between
arsenic and other pollutants in the human environment.
1.2.4 Effects
Dose-response data on various health effects caused by exposure to
arsenic are generally very scanty or nonexistent. Damage to the liver
and the cardiovascular and nervous systems, reported in some chronic
exposure situations, needs to be validated in future studies. In many
instances, animal models would be of use. Sensitive indicators of
arsenic exposure have been suggested such as the urinary excretion of
uroporphyrin or electromyographic abnormalities, but further
confirmation is needed.
It has been demonstrated recently that the two major metabolites
formed after exposure to inorganic arsenic compounds are methylarsonic
acid and dimethylarsinic acid. It is important to study the toxicity
of these compounds. Man is also exposed to large amounts of organic
arsenic compounds through consumption of some seafoods. Though the
acute toxicity of these compounds must be considered to be low, there
are very limited data on possible long-term effects. Studies should be
carried out on both human subjects and on animals.
Severe effects of exposure to arsenic have been demonstrated in
Japan in the form neurological effects and in Chile and China
(Province of Taiwan) in the form of severe vascular disorders. There
is a need to carry out followup studies using modern epidemiological
techniques. In order to elucidate the cardiovascular diseases,
including peripheral vascular disease in Chile and China (Province of
Taiwan), it is recommended that WHO should initiate an internationally
coordinated study.
Although there is strong epidemiological evidence that inorganic
arsenic is carcinogenic for man, more work is needed to determine if
this is true for both valence forms. Conclusive animal data are not
available. Further epidemiological investigations concerning the
relationship between exposure to arsenic and cancer of the lung and
skin should be undertaken in both ambient and occupational exposure
situations, because of the considerable uncertainty concerning
dose-response relationships.
Health effects of arsenic in industry have generally been seen
where exposure to arsenic has been combined with exposure to other
metals and irritating substances, such as sulfur dioxide. Possible
synergism in relation to the carcinogenic activity of arsenic should
be investigated in epidemiological studies as well as in experimental
systems.
Some data indicate that arsenic may induce effects in the human
reproductive system. More studies in this field are needed.
In this document, pulmonary cancer and skin cancer have been
regarded as the critical effects in man for long-term exposure to
inorganic arsenic through inhalation and oral exposure, respectively.
There is still considerable uncertainty regarding the effects of
different chemical forms of arsenic and dose-response relationships
and it is recommended that these questions should be studied further,
both in industry and in the general environment. Studies should
comprise both human epidemiological and experimental animal studies.
There is also a need for further investigation of the mutagenic
activity of different arsenic compounds.
2. PROPERTIES AND ANALYTICAL PROCEDURES
2.1 Chemical and Physical Properties of Arsenic Compounds
There are many different forms of inorganic and organic arsenic.
The most important forms for the evaluation of health effects are
shown in Table 1.
2.1.1 Inorganic arsenic compounds
The most important commercial compound is arsenic (III) oxide, the
molecular formula of which is generally accepted to be As4O6, at
temperatures up to 1073°C. This compound is recovered from copper
smelters as a by-product of copper production. The arsenic in
naturally occurring metal arsenides and arsenic sulfides is
volatilized and oxidized during the ore roasting process and condenses
as the trioxide in flues. Arsenic-containing coal also produces
chiefly arsenic(III) oxide, when it is combusted. Arsenic(III) oxide
has a reasonably low boiling point (465°C) and will sublime at lower
temperatures (Durrant & Durrant, 1966; Carapella, 1973). Its vapour
pressure at ambient temperatures is significant, a fact which is
important in its transport and distribution in the environment
(Lao et al., 1974). If data on the vapour pressure of arsenic(III)
oxide are extrapolated to 25°C, the saturating concentration of
arsenic(III) oxide is 0.6 µg/m3.
The solubility of arsenic(III) oxide in water is fairly low, about
2% at 25°C and 8.2% at 98°C (Durrant & Durrant, 1966). The resulting
solution is slightly acidic and contains arsenous acid (H3AsO3).
Arsenic(III) oxide is highly soluble in either hydrochloric acid or in
alkali. In aqueous solution, arsenic is usually in the form of the
arsenate or arsenite.
Alkali earth metals combine with arsenate anions to form salts
that are only slightly soluble; consequently arsenic tends to form a
precipitate frequently in association with phosphates.
Reported pKa values for arsenous and arsenic acids are: HAsO2,
pKa 9.23; H3AsO4, pKa1 2.20, pKa2 6.97, pKa3 11.53 (Flis et al.,
1959).
 Arsenates and arsenic acid are strong oxidants and may for
example, oxidize I-ion to I-3. In air saturated water, arsenic(V)
compounds should predominate, but arsenic(III) compounds have been
shown to exist under these conditions. Sulfides of arsenic predominate
under reducing systems in the presence of reduced forms of sulfur
(Ferguson & Gavis, 1972). Reduction by organic matter of arsenic(III)
and sulfate ions in the sediments of aquatic systems is likely to be
responsible for the formation of both metallic arsenic and arsenic
sulfides at the same location. Lead arsenate, copper arsenate,
copper(II) acetate meta-arsenate (Paris Green), and calcium arsenate,
all of which have been used as insecticides, are only slightly soluble
in water.
The halides of arsenic and arsine are not found in the environment
but are important in organoarsenic chemistry and in chemical analysis.
Arsenic(III) chloride, for example, is formed when arsenic(III) oxide
is treated with concentrated hydrochloric acid (Durrant & Durrant,
1966). It is easily hydrolyzed by water. Arsenic halides are rapidly
hydrolized and easily alkylated by a number of organic alkylating
agents, such as the Grignard reagents.
2.1.2 Organic arsenic compounds
The organic chemistry of arsenic is extensive. Carbon-arsenic
bonds are quite stable under a variety of environmental conditions of
pH and oxidation potential. Some methylarsenic compounds, such as di-
and trimethylarsines, occur naturally as a consequence of biological
activity. In water solutions, these may undergo oxidation to the
corresponding methylarsenic acids. These and other higher organic
arsenic compounds such as arsenobetaine and arsenocholine, which are
found in marine organisms, are very resistant to chemical degradation
(Lauwerys et al., 1979).
Methylarsonic acid is a difunctional acid, with pKa1 4.1, pKa2
8.7, that forms soluble salts with alkali metals. Dimethylarsinic
acid, which acts as a monofunctional weak acid, pKa 6.2, also forms
fairly soluble alkali metal salts. The alkylarsenic acids can undergo
reduction to the corresponding arsines, a reaction important in
analysis. They also react with hydrogen sulfide and alkanethiols to
produce sulfur derivatives such as (CH3)2AsS SH (NAS, 1977). It
appears likely that the reduction of dimethylarsinic acid and its
subsequent reaction with thiols may be a key to its involvement in
biological activity.
The alkylchloroarsines are reasonably stable with respect to
hydrolysis but quite reactive with reduced compounds of sulfur. One
such compound, 2-chlorovinylarsine dichloride (Lewisite), has been
used as a war gas.
An extensive review of the chemical and physical properties of
organoarsenic compounds has been made by Doak & Freedman (1970).
2.2 Analytical Procedures
2.2.1 Sampling and sample treatment
Arsenic poses some special problems in sampling not experienced in
the determination of other trace elements. Water, urine, and
biologically active samples should either be analysed within a few
hours or frozen and stored (Andreae, 1977; Feldman 1979). Low
concentrations of arsenic compounds found in natural waters slowly
decrease with time, unless stabilized in some manner to prevent
adsorptive losses. The biomethylation of inorganic arsenic in a
biologically active sample can cause a change in its composition.
Since environmental analyses often involve trace concentrations,
sample treatment frequently includes some type of preconcentration
prior to analysis. Conversion of arsenic to arsine, co-precipitation
with iron(III) hydroxide, distillation as arsenic(III) chloride, or
extraction are typical examples of the approaches used.
2.2.1.1 Natural waters
Sea water and fresh natural waters are generally analysed without
oxidative treatment prior to a preconcentration step, when the
molecular forms of arsenic are to be analysed. If the preconcentration
step or the final steps in the analytical method require the
conversion of organoarsenic compound to an inorganic form, oxidation
procedures may be necessary. An acid-potassium persulfate preoxidation
method (Pierce et al., 1976) and a method involving ultraviolet (UV)
have both been automated (Fishman & Spencer, 1977). Acid-permanganate
oxidation was found to be effective in the conversion of
dimethylarsinic acid to inorganic arsenic (Sandhu & Nelson, 1978).
Arsine generation followed by cold trapping in liquid nitrogen is
a technique that can be used with or without prior oxidation (Braman
et al., 1977; Siemer & Koteel, 1977). Arsine generation has long been
used as a first step in the determination of arsenic in water samples,
prior to spectrophotometric analysis of the complex formed with
silverdiethyldithiocarbamate (SDDC) (Skonieczny & Hahn, 1978). A
recent adaptation of this method is the analysis for the SDDC complex
by graphite tube furnace AAS which gives an improved detection limit
of about 10 ng (Shaikh & Tallman, 1977). Arsenic has been separated
from samples by volatilization as the trichloride or tribromide. A
recent application combines distillation as the chloride with anodic
stripping voltametry (Davis et al., 1978).
A number of coprecipitation methods have been reported for the
preconcentration of arsenic from water followed by different methods
of analysis. Iron(III) hydroxide (Portmann & Riley, 1964) and
hydroxides of zirconium and cerium (Plotnikov & Usatova, 1964) are
among the many coprecipitants that have been studied. Thionalide has
also been used in the coprecipitation of arsenic from sea water
(Portmann & Riley, 1964) with 95% efficiency, but the procedure is
slow, requiring much sample handling, a problem with all of the
precipitation methods.
2.2.1.2 Air
Air sampling for trace amounts of arsenic in the environment has
mainly been confined to sampling the particulate phase. It is likely
that many different types of particulate filters are satisfactory for
this type of sampling, though arsenic is usually associated with small
size particles.
As the estimated saturated concentration of arsenic(III) oxide in
air at 25°C is about 600 ng/m3, it is possible that, when air
concentrations are below this level, arsenic(III) oxide collected on a
filter may evaporate or may not be collected completely. Results of
laboratory work using filters and pure arsenic(III) oxide in air
support this theory (Lao et al., 1974; Walsh et al., 1977b).
Nevertheless, in studies using a filter impregnated with ethyleneimine
in glycerol which is 65% efficient in trapping arsenic(III) oxide
vapour, it was shown that the major portion of arsenic in air (78-99%)
could be collected on untreated, 0.4 micrometer pore size,
polycarbonate type filters (Nuclepore Co.). Millipore membrane filters
have also given satisfactory results (Walsh et al., 1977b). This was
found to be the case for both ambient air samples containing low
levels of arsenic and near-smelter air samples containing high levels.
Approximately 15% of the arsenic in the air collected was found to be
in a vapour form that was not collected on untreated filters. These
results agree with those of Johnson & Braman (1975a) who also found
that approximately 15% of the collected arsenic was volatile.
Vapour forms of arsenic in air, particularly the arsines, can be
preconcentrated from air onto silver-coated glass beads (Johnson &
Braman, 1975a). Even if oxidized after adsorption, the identity of the
compound is not lost. For example, dimethylarsine, if present, can
only be oxidized to dimethylarsinic acid. Adsorbed compounds can be
desorbed using dilute sodium hydroxide (Braman et al., 1977).
2.2.1.3 Biological materials
Samples of biological materials to be analysed for total arsenic
are generally completely oxidized prior to analysis. A number of
oxidation methods have been studied, the majority of which involved
the use of oxidizing acids or persulfates. The completeness of the
oxidation has however seldom been checked. Perhaps the best oxidizing
procedure involves the use of a mixture of sulfuric and nitric acids
(Chu et al., 1972), a mixture of sulfuric, nitric, and perchloric
acids (Christian & Feldman, 1970) or hydrogen peroxide (Samsahl,
1967). Dry ashing with magnesium oxide or magnesium nitrate has been
successfully applied in the analyses of a variety of biological
samples (Snell & Snell, 1945; Evans & Bandermer, 1954). Other methods
with fewer contamination problems or losses of arsenic are the Parr
bomb (Beamish & Collins, 1934) and the Carius oxidation (Day, 1964)
techniques. Schoeninger flask oxidation has been used in the oxidation
of dried tissue samples (Schwedt & Russel, 1972). The proper
preanalytical treatment for samples of certain marine organisms
containing compounds such as arsenobetaine has yet to be established
(Edmonds, et al., 1977).
There has been some success in analysing homogenized samples
without oxidation by treating them with hydrochloric acid (Kingsley &
Schaffert, 1951) or sodium hydroxide (Johnson & Braman, 1975b) prior
to analysis. This approach is particularly necessary if the molecular
forms of arsenic present are to be identified. In no case has the
accuracy of the analyses been unequivocally determined.
Various methylarsenic compounds have been determined in human
urine samples without pretreatment (Braman, et al., 1977; Crecelius,
1977b).
2.2.2 Analytical methods
2.2.2.1 Methods for total arsenic
One early very common method for the determination of total
arsenic was the Gutzeit method (Vogel, 1955).
Spectrophotometry using the silver diethyldithiocarbamate (SDDC)
complex of arsine is the classical method for determining arsenic in
the 1-100 microgram range (Vasak & Sedivec, 1952). Arsenic is reduced
to arsine by either granular zinc in hydrochloric acid or by sodium
borohydride. Arsine reacts with SDDC in pyridine and the absorption of
the red coloured complex is read at 533 nm. Methylarsine and
dimethylarsine, but not trimethylarsine, form SDDC complexes which
absorb at 533 nm, but their complexes have lower molar absorptivities.
A large number of studies can be found in the literature
concerning the use of the SDDC method as it is often designated a
standard method of analysis (Stratton & Whitehead, 1962). Some more
recent papers include one in which the somewhat disagreeable pyridine
solvent was replaced by L-erythro-2-(methylamine)-1-phenylpropan-1-ol
(L-ephedrine) in chloroform (Hundley & Underwood, 1970; Gastiner,
1972; Kopp, 1973). Ionic interference in the SDDC procedure has been
studied by Sandhu & Nelson (1978).
The arsenate ion reacts with ammonium molybdate to form a complex
which, when reduced, gives a blue colour (Portmann & Riley, 1964).
Under favourable conditions, the limit of detection is near 0.1 µg. An
adaptation of the method has been used to determine the amounts of
phosphate, arsenate, and arsenite in sea water (Johnson & Pilson,
1972). The method is applicable to sea water samples with arsenic
concentrations below 3 × 10-6 mol/litre. Precision is of the order of
± 0.015 × 10-6 mol/litre.
Atomic absorption spectrophotometry (AAS) is gaining in popularity
as a method for the determination of total arsenic. Sensitivity of the
ordinary flame type AAS for arsenic in solution is comparatively poor,
detection limits are in the 0.5-1 mg/litre range (Holak, 1969;
Kirkbright & Ranson, 1971). When an electrodeless discharge lamp and
an argon-air-hydrogen flame are used, the detection limit is reduced
to 0.1 mg/litre (Menis & Rains, 1969). With a long-path cell, the
detection limit is about 6 µg/litre (Ando et al. 1969). Arsine can
also be passed into a heated graphite or quartz furnace mounted in an
AAS instrument. The arsine can be continuously passed through the
atomizer (Smith, 1975; Siemer et al., 1976) or collected in a cold
trap and passed through rapidly when the cold trap in heated (Griffin
et al., 1975; McDaniel et al., 1976). This second technique provides
the best detection limits which are in the fraction of a ng range
(Siemer & Koteel, 1977).
Neutron activation analysis is one of the more sensitive
analytical methods. The arsenic-75 isotope is converted to arsenic-76
by thermal neutron absorption. Detection limits are near 1 ng, but the
method is susceptible to interference, particularly from sodium. There
have been many applications of this method in the analyses of
biological samples (Takeo & Shibuya, 1972; Heydorn & Damsgaard, 1973;
Maruyama & Komiya, 1973; Orvini, et al., 1974), water (Ray & Johnson,
1972) and particulate matter in air (Walsh et al., 1977b). Activated
sample solutions are frequently subjected to separation to eliminate
interfering radioisotopes (Gallorini, et al., 1978).
The determination of trace amounts of arsenic has also been
performed using differential pulse polarography and anodic stripping
voltametry (Arnold & Johnson, 1969; Myers & Osteryoung, 1973; Davis,
et al., 1978). The second of these methods was applied to biological
samples that were wet ashed with nitric, sulfuric, and perchloric
acids before distillation of arsenic as arsenic(III) chloride. The
detection limit was in the ng range. Some of the organoarsenic
compounds are also electroactive (Elton & Gieger, 1978) but no
practical methods for environmental analyses have appeared since mg/kg
concentrations are required to observe responses.
A variety of other analytical methods have been successfully used
for the determination of trace amounts of arsenic. Among these are:
atomic emission spectroscopy (Kirkbright et al., 1973; Braman et al.,
1977; Robbins et al., 1979), X-ray fluorescence (Thomson, 1975), and
isotope dilution mass spectrometry (Zeman et al., 1964).
Recently an electron spectroscopic method (ESCA) has been reported
in which arsine collected on filter surfaces was analysed (Carvalho &
Hercules, 1978). Detection limits were in the ng range so that
preconcentration resulted in further reduction of detection limits to
sub µg/kg.
A recent enzyme method gave reasonable results in the
0.02-2.0 mg/kg range (Goode & Matthews, 1978).
Very recently, a small interlaboratory comparison study on the
determination of total urinary arsenic was performed with the
participation of 4 laboratories using different analytical procedures
(NAA and AAS). Ten samples containing arsenic concentrations of
between 0.001 and 1 mg/litre were examined (Buchet et al., in press).
2.2.2.2 Analyses for specific arsenic compounds
Low concentrations of inorganic arsenic(III) and arsenic(V) in sea
water can be determined using the molybdenum blue method (Johnson &
Pilson, 1972). Inorganic arsenic(III) and (V) can be separated by
direct extraction with toluene of acidified aqueous solutions
containing, for example, cysteine (Lauwerys et al., 1979).
Differentiation between arsenic(III) and arsenic(V) is also possible
using pH sensitive, selective reduction with sodium borohydride
followed by atomic emission spectroscopy or AAS detection. Inorganic
and methylarsenic compounds are reduced according to the reactions
shown in Table 2. By buffering at pH 4, reduction of arsenic(V) is
avoided. At pH 1.5, all compounds are reduced. The methylarsine
compounds produced may be cold trapped, separated, and detected
individually. Cold trapping and separation on heating, with detection
by d.c. discharge in helium, has been used in the determination of
arsenic in natural water, human urine (Braman et al., 1977; Crecelius,
1977b) and sea water (Johnson & Braman, 1975b; Andreae, 1977) at µg/kg
and sub µg/kg concentrations. The detector cell has recently been
studied and improved (Feldman & Batistoni, 1977) as has the analysis
train (Crecelius, 1978).
Table 2. Reduction reactions of inorganic and methylarsenic compounds
Compound pKa1 pH Product B.P.
arsenous acid (meta) 9.23 < 7 AsH3 -55°C
(HAsO2)
arsenic acid (ortho) 2.20 > 4.0 no reaction
(H3AsO4) 1.5 AsH3 -55°C
methylarsonic acid 4.1 > 5.0 little reaction
(CH3AsO(OH)2) 1.5 CH3AsH2 2°C
dimethylarsinic acid 6.2 1.5 (CH3)2AsH 36°C
((CH3)AsO(OH))
trimethylarsine oxide -- 1.5 (CH3)3As 70°C
(CH3)3AsO
phenylarsonic acid 1.5 C6H5AsH2 148°C
(C6H5AsO(OH)2)
p-aminophenyl arsinic -- 1.5 H3+ N C6H4 AsH2 --
acid (arsanilic acid)
p-H2N-C6H4AsO(OH)2
Gas chromatographic detection of arsines trapped in cold toluene
solvent using a microwave stimulated plasma detector has been
developed by Talmi & Norvell (1975). The detection limits of this
method are excellent (about 20 pg).
The electrochemical reactions of dimethylarsinic acid and
trimethylarsine were studied by Elton & Geiger (1978). Dimethylarsinic
acid may be converted to its iodide and determined by gas
chromatography (Söderquist, et al., 1974) but the method is not
applicable to the same wide range of arsenic compounds as the
hydride-generating procedures.
Arsenic has been determined in marine organisms (Portmann & Riley,
1964). Substantial efforts have been made to identify the different
organic arsenic compounds and, only recently, arsenobetaine was
identified in rock lobsters (Edmonds et al., 1977) and
arsenophospholipids in algae (Cooney et al., 1978). Analytical methods
for the determination of these compounds are not well developed. Thin
layer chromatography was used in studies by Lunde (1977), the results
of which indicated the possible presence of several as yet
unidentified organic arsenic compounds.
Analytical methods for the determination of total arsenic and
different forms of arsenic in human biological materials have recently
been reviewed by Lauwerys et al. (1979).
3. SOURCES AND OCCURRENCE OF ARSENIC IN THE ENVIRONMENT
3.1 Natural Occurrence
3.1.1 Rocks, soils, and sediments
Arsenic is widely distributed in a large number of minerals. The
highest mineral concentrations generally occur as arsenides of copper,
lead, silver, or gold or as the sulfide. Major arsenic-containing
minerals are arsenopyrite (FeAsS), realgar (As4S4), and orpiment
(As2S3). The arsenic content of the earth's crust is 1.5-2 mg/kg; it
ranks 20th in abundance in relation to other elements (NAS, 1977).
Oxidized forms of arsenic are usually found in sedimentary deposits.
The elemental oxidation state, though stable in reducing environments,
is rarely found. Table 3 gives some ranges of the arsenic contents of
crustal materials. Although the values shown are generally low,
mineralized zones of sulfidic ores may contain much higher
concentrations of arsenic.
Table 3. Arsenic in crustal materialsa
Type Range
As (mg/kg)
Igneous rocks
ultrabasic 0.3-16
basalts 0.06-113
andesites 0.5-5.8
granitic 0.2-13.8
silicic, volcanic 0.2-12.2
Sedimentary rocks
limestones 0.1-20
sandstones 0.6-120
shales and clay 0.3-490
phosphorites 0.4-188
a From: NAS (1977).
High levels of arsenic may also occur in some coals. The average
arsenic content of coal in the USA was estimated at 1-10 mg/kg (Davis
& Associates, 1971). In some coal mined in Czechoslovakia, the
concentration of arsenic has been shown to be as high as 1500 mg/kg
(Cmarko, 1963).
Uncontaminated soils were found to contain arsenic levels between
0.2 and 40 mg/kg, while arsenic-treated soils contained up to
550 mg/kg (Walsh & Keeney, 1975). The soil in the city of Antofagasta,
Chile, contains natural levels of arsenic of about 3.2 mg/kg (Borgono
& Greiber, 1972). In the Comarca Lagunera, Mexico, values between 3
and 9 mg/kg were found at the soil surface and more than 20 mg/kg,
deep down (Gonzalez, 1977).
Peat may contain considerable quantities of arsenic. Minkkinen &
Yliruokanen (1978) found maximum arsenic concentrations in various
Finnish peat bogs of between 16 and 340 mg/kg dry peat.
The natural level of arsenic in sediments is usually below
10 mg/kg dry weight (Crecelius, 1974). Bottom sediments can become
substantially contaminated by arsenic from man-made sources. Levels of
up to 10 000 mg/kg dry weight were found in bottom sediments near a
copper smelter in Washington, USA (Crecelius, 1974).
3.1.2 Air
Airborne particulate matter has been shown to contain both
inorganic and organic arsenic compounds (Johnson & Braman, 1975a;
Attrep & Anirudahn, 1977). Crecelius (1974) showed that only 35% of
the inorganic arsenic in rain from an urban area was present as
arsenite; however, some post-sampling oxidation could not be excluded.
In studies by Johnson & Braman (1975a), methylarsines made up
approximately 20% of the total arsenic in ambient air from rural and
urban areas.
In unpolluted areas, airborne arsenic concentrations ranging from
less than one to a few nanograms per cubic metre have been reported
(Peirson, et al., 1974; Johnson & Braman, 1975a; Walsh, et al., 1977b;
Beavington & Cawse, 1978; Brimblecombe, 1979).
3.1.3 Water
Arsenic occurs in both inorganic and organic forms in water
(Braman & Foreback, 1973; Crecelius, 1974). The main organic arsenic
species, methylarsonic acid and dimethylarsinic acid, are generally
present in smaller amounts than the inorganic forms, arsenite and
arsenate. The chemistry of arsenic in the aqueous environment has been
reviewed by Ferguson & Gavis (1972).
The arsenic contents of surface waters in unpolluted areas vary
but typical values seem to be a few micrograms per litre or less. In a
study of river waters in the USA, about 80% of the samples contained
levels of less than 0.01 mg/litre (Durum et al., 1971). Quentin &
Winkler (1974) found an average value of 0.003 mg/litre in river water
and 0.004 mg/litre in lake water in the Federal Republic of Germany. A
mean arsenic concentration of 0.0025 mg/litre was reported in some
Norwegian rivers (Lenvik et al., 1978). Much higher values have been
reported from some areas including Antofagasta, Chile, where the
average arsenic level in a river water supply of drinking water
between 1958 and 1970 was 0.8 mg/litre (Borgono et al., 1977).
The oxidation state of arsenic in surface waters in various parts
of the world remains largely unknown. Braman & Foreback (1973) found
that the ratio of trivalent to pentavalent inorganic arsenic ranged
from < 0.06 to 6.7 in a few uncontaminated surface water samples
containing between 0.0025 and 0.0030 mg As/litre. About 8% of the
total arsenic in 2 samples of well-aerated stream water (0.014 and
0.06 mg/litre, respectively) was reported by Clement & Faust (1973) to
be in the trivalent form. In anaerobic reservoirs, all of the arsenic
present (0.14-1.3 mg As/litre) seemed to be in this form.
Penrose et al. (1977) reported that sea-water ordinarily contains
arsenic concentrations ranging from 0.001-0.008 mg/litre. Levels of
about 0.002 mg/litre have been reported by Onishi (1969) and Johnson &
Braman (1975b). The major chemical form of arsenic appears to be the
thermodynamically stable arsenate ion; even so, arsenite often
accounts for one third of the total arsenic (Johnson, 1972; Andreae,
1978).
Clement & Faust (1973) analysed water from 2 groundwater supplies
with very high levels of arsenic (224 and 280 mg/litre) and found that
about 50% was present as arsenic(III). In a groundwater-fed stream,
26% of the total arsenic (0.08 mg/litre) was in the form of trivalent
arsenic. Arsenic speciation has also been performed on well water
samples from an area in Alaska containing high levels of arsenic
(Harrington et al., 1978). In 5 samples containing arsenic
concentrations ranging from 0.52 to 3.6 mg/litre, between 3% and 39%
of the arsenic present was trivalent, the rest being pentavalent. No
methylated arsenic compounds could be detected.
High levels of arsenic have been found in waters from areas of
thermal activity. Thermal waters in New Zealand have been shown to
contain up to 8.5 mg/litre (Ritchie, 1961). Geothermal water in Japan
contained arsenic levels of 1.8-6.4 mg/litre and neighbouring streams
contained about 0.002 mg/litre (Nakahara et al., 1978).
The chemical forms of arsenic in thermal water from New Zealand
were investigated by Aggelt & Aspell (1978). In the geothermal bores,
more than 90% of the arsenic was present in the trivalent form.
However, in a river flowing through the area, the pentavalent form was
predominant but some seasonal variation in the ratio between the two
valence states was indicated.
3.1.4 Biota
The sorption of arsenate ions in the soil by iron and aluminum
components, greatly restricts the availability of arsenic to plants
(Walsh et al., 1977a). The arsenic content of plants grown on soils
that had never been treated with arsenic-containing pesticides varied
from 0.01 to about 5 mg/kg dry weight (NAS, 1977). Plants grown on
arsenic-contaminated soils may, however, contain considerably higher
levels, especially in the roots (Walsh & Keeney, 1975; Grant & Dobbs,
1977; Wauchope & McWhorter, 1977). Some grasses growing on soils
containing high levels of arsenic have been found to have elevated
arsenic contents (Porter & Peterson, 1975). Andersson & Nilsson (1972)
reported that arsenic in soils treated with sewage sludge was highly
available to plants, but only a few samples were analysed. In
contrast, Furr et al. (1976) claimed that soil arsenic is not readily
available to plants.
Marine algae and seaweed usually contain considerable amounts of
arsenic. Lunde (1970) showed values of 10-100 mg/kg dry weight in
marine algae from the Norwegian coast. The degree of enrichment was
found to be between 1500 and 5000 compared with the level of arsenic
in the growth medium (Lunde, 1973a). Similar and even higher
enrichment ratios were reported for fresh water plants in the Waikato
River, New Zealand (Reay, 1972). The elevated arsenic concentrations
in the water (0.03-0.07 mg/litre) gave rise to concentrations of up to
971 mg As/kg dry weight in aquatic plants.
3.2 Industrial Production and Uses of Arsenic
3.2.1 Industrial production
Based on the limited data available (US Bureau of Mines, 1975;
Nelson, 1977), it can be estimated that the total world production in
1975 was around 60 000 tonnes. This production seems to be stable. The
main producers are: China, France, Federal Republic of Germany,
Mexico, Namibia, Peru, Sweden, USA, and USSR. These countries account
for about 90% of the production. For a more detailed discussion of
production of arsenic and its compounds, see IARC (1980).
Arsenic(III) oxide, the major basic chemical of the arsenic
industry, is emitted as a by-product in smelting, mainly of copper and
lead ores. It is recovered from the flue dust in a reasonably pure
form.
3.2.2 Uses of arsenic compounds
Arsenic compounds are mainly used in agriculture and forestry
(NAS, 1977). Much smaller amounts are used in the glass and ceramics
industry and as feed additives and drugs. The use pattern for
arsenic(III) oxide in 1975-78 has been reported as follows:
manufacture of agricultural chemicals (pesticides), 82%; glass and
glassware, 8%; industrial chemicals, copper and lead alloys, and
pharmaceuticals, 10% (US Bureau of Mines, 1979).
In agriculture, compounds such as lead arsenate, copper
acetoarsenite, sodium arsenite, calcium arsenate, and organic arsenic
compounds are used as pesticides. Substantial amount of methylarsonic
acid and dimethylarsinic acid are used as selective herbicides. These
herbicides are particularly necessary for the control of Johnson grass
(Sorghum halepense) in cotton fields. They are also used to treat
other weeds such as sandbur ( Cenchrus sp.), cocklebur
( Xanthium sp.) and crabgrass in lawns (Weed Science Society of
America, 1974). Dimethylarsinic acid is used as a silvicide in forest
control and workers may be exposed to the compound and its volatile
reaction products in the soil (Wagner & Weswig, 1974). Dimethylarsinic
acid was the Agent Blue used in Viet Nam as a defoliant for military
purposes.
Chromated copper arsenate, sodium arsenate, and zinc arsenate are
used as wood preservatives (Lansche, 1965). When these compounds are
applied under pressure they react with the wood to create water
insoluble compounds. The preserved timber is resistant to both fungal
and insect attack (Dobbs et al., 1976). The use of arsenic in wood
preservatives is increasing.
Some phenylarsenic compounds such as arsanilic acid are used as
feed additives for poultry and swine and to combat certain diseases in
chickens.
Small amounts of arsenic compounds continue to be used as drugs in
some countries. Other applications of arsenic are found in metallurgy,
where it is used to dope germanium and silicon or in the production of
gallium arsenide or indium arsenide.
3.2.3 Sources of environmental pollution
The burning of coal and smelting of metals are major sources of
arsenic in air. A British study showed yearly average concentrations
in suspended matter in town air of 0.04-0.14 µg/m3 (Goulden et al.,
1952). In Prague, Vondracek (1963) found a winter mean concentration
in air of 0.56 µg/m3 and a summer mean of 0.07 µg/m3. In urban areas
in the USA, air concentrations of arsenic ranged from below the
detection limit (0.01 µg/m3) to 0.36 µg/m3 on a quarterly average
basis in 1964 (Sullivan, 1969). In 1974, about 200 of the 280 US
National Air Surveillance Network sites recorded quarterly average
concentrations below 0.001 µg/m3 (Thompson, 1977). Only 13 sites,
mainly highly urbanized areas and smelter locations, showed levels
exceeding 0.02 µg/m3.
In the vicinity of smelters, levels of arsenic in air exceeding
1 µg/m3 have been recorded. Rozenshtein (1970) found levels of
airborne arsenic, given as arsenic(III) oxide, of 0.7-2.5 µg/m3
(i.e., 0.5-1.9 µg As/m3) within 4 km of a copper smelter in the USSR.
Data were not given on the duration of sampling. In the USA, quarterly
average levels of up to 1.4 µg/m3 were reported in El Paso, Texas, at
the site of a large copper smelter (Sullivan, 1969). Near a copper
smelter in Tacoma, Washington, monthly averages of arsenic in air of
up to 1.46 µg/m3 were recorded (Nelson, 1977) and a maximum 24-h
concentration of 7.9 µg/m3 was reported by Roberts, et al. (1977).
Daily mean concentrations of up to 1.6 µg/m3 were found in the air
near a smelter in Romania (Gabor & Coldea, 1977). Auermann et al.
(1977) reported airborne arsenic concentrations in a polluted region
of the German Democratic Republic ranging from 0.9-1.5 µg/m3 (average
0.9 µg As/m3; duration of sampling not stated). In the vicinity of a
Canadian gold mine, where ore was roasted, annual mean arsenic
concentrations in ambient air ranged from 0.06 to 0.09 µg/m3 between
1973 and 1975 (Hazra & Prokupok, 1977). Individual 24-h arsenic
concentrations varied from less than 0.01 to 3.91 µg/m3. The
concentrations of arsenic in flue dust from a coal-fired power plant
in Czechoslovakia ranged from 43 to 110 mg/kg (Zdrazil & Picha, 1966).
In fly ash from 24 US coal-fired power plants, the arsenic
concentrations ranged between 2.3 and 312 mg/kg (Kaakinen et al.,
1975; Furr et al., 1977).
In the stack dust from nonferrous smelting operations, arsenic is
predominantly in the trivalent inorganic form (Crecelius, 1974,
Rosehart & Chu, 1975). No conclusive data on the extent of oxidation
of airborne trivalent arsenic are available at present.
A thorough study has been made of arsenic in the environs of a
copper smelter near Tacoma, WA, USA (Crecelius, 1974). Dated segments
of sediment cores showed that the arsenic buildup started with the
operation of the smelter. Less than 30% of the arsenic entering
neighbouring waterways accumulated in the sediments. The remaining 70%
presumably left the location in solution. Elevated concentrations of
arsenic were found in water in locations within 2-4 km of the smelter.
Analyses of air, rain water, and snow all indicated elevated arsenic
levels in the Tacoma, Washington area, attributable to the smelter
effluent. Levels of up to 380 mg/kg (dry weight) were found in top
soil in the vicinity of the plant.
A similar pattern, was observed in a study of the distribution of
arsenic from a copper smelter in Sweden (Lindau, 1977). The arsenic
concentrations in air a few kilometres from the smelter were higher
than normal, as were arsenic levels in the soil, moss, and nearby
natural water bodies.
Suzuki et al. (1974) reported concentrations of arsenic of up to
2470 mg/kg in soil near a smelter in Japan.
Attrep & Anirudhan (1977) found a quarterly average total arsenic
concentration in air of 0.08 µg/m3 in an area polluted by arsenic
from defoliants. About half of the airborne arsenic was in the form of
organic arsenic compounds. Four years later, during a season of low
arsenic use, a monthly average of 0.009 µg/m3 was detected in the
same area. At this time, only about 15% of total airborne arsenic was
in the form of organoarsenic compounds.
Burning of wood treated with arsenic-containing preservatives,
mainly inorganic pentavalent compounds, can result in the release of
arsenic into the atmosphere. The concentration of arsenic in the
combustion fumes is closely related to the temperature. Smouldering of
wood, treated with inorganic arsenic salts, at a temperature of 415°C
resulted in volatilization of 8.6% of the total arsenic in the wood
(Watson, 1958). When wood treated with a preservative containing
inorganic pentavalent arsenic salts was burned at temperatures of
700-800°C, about 50% of the arsenic was present in the ashes (the rest
was mainly in the smoke), while at 1000°C only about 15% remained in
the ashes (Öhman, 1960).
The use of geothermal energy can result in severe arsenic
contamination. Crecelius, et al. (1976) found that the natural arsenic
level 0.002 mg/litre had increased 1000 times in a water reservoir in
which some of the discharge from a Mexican geothermal power plant was
emitted. Between 6% and 51% of the total arsenic in this reservoir was
present as trivalent inorganic arsenic and the rest as pentavalent.
The emissions of arsenic into the environment from the plant totalled
about 60 kg/day. In El Salvador, water from a reservoir near a
geothermal power plant contained an arsenic level of 8.9 mg/litre
(Jernelöv et al., 1976).
Arsenic is also present in trace amounts in fertilizers. In a
recent study, it was reported that concentrations of up to several
hundred mg/kg were present in some instances (Senesi et al., 1979).
4. ENVIRONMENTAL TRANSPORT AND DISTRIBUTION
4.1 General
Most environmental transformations of arsenic appear to occur in
the soil, in sediments, in plants and animals, and in zones of
biological activity in the oceans. Biomethylation and bioreduction are
probably the most important environmental transformations of the
element, since they can produce organometallic species that are
sufficiently stable to be mobile in air and water. However, the
biomethylated forms of arsenic produced are subject to oxidation and
bacterial demethylation back to inorganic forms.
The biomethylation of arsenic was first recognized long ago when
arsines were produced from cultures of a fungus Scopulariopsis
brevicaulis (Challenger, 1945). This work was done in an
investigation of poisoning incidents attributed to arsenic-containing
wall paper -- thought to contain Paris Green colouring pigment. It was
eventually ascertained that methylarsines were the toxic agents. More
recently, the methylation of arsenic by methanogenic bacteria (McBride
& Wolfe, 1971) and by reaction with methylcobalamine (Schrauzer
et al., 1972) or L-methionine-methyl-d3 (Cullen et al., 1977) has
been demonstrated in laboratory work. McBride et al. (1978) reported
that dimethylarsine was mainly produced by anaerobic organisms, while
trimethylarsine resulted from aerobic methylation. The following
mechanism for the methylation of arsenate has been proposed by
Challenger (1945) and McBride et al. (1978).
2e CH3+ 2e CH3+
AsVO43- --->ÄsIIIO33- --->CH3AsVO32- --->CH3ÄsIIIO22- --->
--O2- --O2-
2e CH3+ 2e
(CH3)2AsVO2- --->(CH3)2ÄsIIIO- --->(CH3)3AsVO --->(CH3)3ÄsIII
--O2- --O2-
The proposed mechanism indicates that As(V) has to be reduced to
As(III) before being methylated.
4.2 Aquatic Systems
Studies on the molecular forms of arsenic compounds in sea water
have been reported. The concentration ratio As(III)/As(V) was found to
be 0.18 in some Sargasso sea water (Johnson & Braman, 1975b).
Fluctuations in the As(III)/As(V) ratio from 0.02 to 0.09 in the
saline water of Naragansett Bay appeared to be associated with
phytoplankton activity (Johnson & Burke, 1978). Sea water samples off
southern California also exhibited a variable As(III)/As(V) ratio,
again associated with biological activity (Andreae, 1977). In some
instances, the arsenic(III) concentrations exceeded those of
arsenic(V). The same type of biological activity was observed in
natural fresh waters (Braman & Foreback, 1973; Clement & Faust, 1973).
It is evident that the presence of arsenic(III) compounds is the
result of some reductive activity, which could be either biological or
a non-biological effect of dissolved organic matter on arsenic(V).
The oxidation rate of arsenic(III) in Sargasso seawater was
studied under carefully controlled laboratory conditions by Johnson &
Pilson (1975). Temperature, pH, salinity, and the presence of light
all influenced the rate of arsenite oxidation.
The finding of methylarsenic acids in seawater and fresh natural
water is evidence that arsenic goes through reactions other than
simple oxidation or reduction. In both sea and fresh water, the
occurrence of methylarsenic compounds is associated with phytoplankton
activity. In fresh water, the levels of methylarsenic compounds were
especially high in locations where nutrients from fertilizers
(presumably, also containing arsenic) had built up in lakes and ponds.
There is little evidence that sediments play a substantial role in the
methylation of arsenic (Braman, 1975). Sediment samples from two
natural water environments did not contain unusually large amounts of
methylarsenic compounds.
The analysis of biota associated with Sargassum weed indicated
that substantial amounts of arsenic were present in forms other than
the inorganic or methylarsenic forms (Johnson & Braman, 1975b). Only
small amounts of methylarsenic acid type compounds were present in the
organisms.
The involvement of arsenic in the biochemistry of marine organisms
through production of arsenobetaine, arsenocholine, and
arsenophospholipids is a new and only partially explored aspect of the
local cycle. Much work has been done in an effort to identify arsenic
compounds in marine organisms (Edmonds et al., 1977; Irgolic et al.,
1977; Lunde, 1977; Penrose et al., 1977; Cooney et al., 1978).
4.3 Air-soil Systems
It has already been mentioned (section 3) that large quantities of
arsenic compounds are used in agriculture and are initially
distributed in the soil. This is an important aspect of arsenic
distribution in the environment. The occurrence and distribution of
arsenic in soils and plants have been reviewed by Walsh et al.
(1977a). Arsenic is converted to arsenates except under highly
reducing conditions. Arsenate ions are readily sorbed by hydrous
oxides of iron and aluminum and thus leaching of arsenate is slow.
Absorption appears to be a major factor in the retention of arsenic in
soils.
Slow removal of arsenic from the soil is of concern, when old
orchards previously treated with arsenic are used for crop growing
(Bishop & Chisholm, 1962). High arsenic levels can cause a depression
in plant growth but the amounts required to produce this effect depend
on the plant species. Bioaccumulation of arsenic in food crops is not
particularly high.
Methylarsines are released into the air from soil treated with
various arsenic compounds. Dimethylarsine and trimethylarsine were
detected over grass areas treated with the methylarsenic compounds,
soon after application. Methylarsines evolved much more slowly from
grass treated with sodium arsenite (Braman, 1975). Despite these
observations in locations where arsenic was obviously volatilized into
the air following biomethylation, the amounts of methylarsenic
compounds actually found in unpolluted air appear to be small. In one
study, approximately 15% of the total arsenic in outdoor air was in a
methylated form (Johnson & Braman, 1975a). The total arsenic was much
greater in greenhouse air than in ambient air outside and the
methylarsenic forms were much in excess of inorganic forms.
A proposed model of an air-soil arsenic system is shown in
Fig. 1. The system has little chance of being in apparent equilibrium,
since air transport of transpired volatile arsenic is rapid, compared
with evolution rates. Because of lack of data concerning arsenic
compounds in air, especially in locations with arsenic-rich soils, the
rates of evolution and buildup of arsenic in air are not known. A
pseudo-equilibrium can be approached if the air transported into a
site is equivalent to the air transported away from a site. This cycle
is similar to one developed for an agronomic ecosystem, in which
arsenic pesticides were the input (Sandberg & Allen, 1975). The most
important translocation factors were absorption by soil and oxidation,
uptake by vegetation, and volatilization after biomethylation.
Arsenates and arsenic acid are strong oxidants and may for
example, oxidize I-ion to I-3. In air saturated water, arsenic(V)
compounds should predominate, but arsenic(III) compounds have been
shown to exist under these conditions. Sulfides of arsenic predominate
under reducing systems in the presence of reduced forms of sulfur
(Ferguson & Gavis, 1972). Reduction by organic matter of arsenic(III)
and sulfate ions in the sediments of aquatic systems is likely to be
responsible for the formation of both metallic arsenic and arsenic
sulfides at the same location. Lead arsenate, copper arsenate,
copper(II) acetate meta-arsenate (Paris Green), and calcium arsenate,
all of which have been used as insecticides, are only slightly soluble
in water.
The halides of arsenic and arsine are not found in the environment
but are important in organoarsenic chemistry and in chemical analysis.
Arsenic(III) chloride, for example, is formed when arsenic(III) oxide
is treated with concentrated hydrochloric acid (Durrant & Durrant,
1966). It is easily hydrolyzed by water. Arsenic halides are rapidly
hydrolized and easily alkylated by a number of organic alkylating
agents, such as the Grignard reagents.
2.1.2 Organic arsenic compounds
The organic chemistry of arsenic is extensive. Carbon-arsenic
bonds are quite stable under a variety of environmental conditions of
pH and oxidation potential. Some methylarsenic compounds, such as di-
and trimethylarsines, occur naturally as a consequence of biological
activity. In water solutions, these may undergo oxidation to the
corresponding methylarsenic acids. These and other higher organic
arsenic compounds such as arsenobetaine and arsenocholine, which are
found in marine organisms, are very resistant to chemical degradation
(Lauwerys et al., 1979).
Methylarsonic acid is a difunctional acid, with pKa1 4.1, pKa2
8.7, that forms soluble salts with alkali metals. Dimethylarsinic
acid, which acts as a monofunctional weak acid, pKa 6.2, also forms
fairly soluble alkali metal salts. The alkylarsenic acids can undergo
reduction to the corresponding arsines, a reaction important in
analysis. They also react with hydrogen sulfide and alkanethiols to
produce sulfur derivatives such as (CH3)2AsS SH (NAS, 1977). It
appears likely that the reduction of dimethylarsinic acid and its
subsequent reaction with thiols may be a key to its involvement in
biological activity.
The alkylchloroarsines are reasonably stable with respect to
hydrolysis but quite reactive with reduced compounds of sulfur. One
such compound, 2-chlorovinylarsine dichloride (Lewisite), has been
used as a war gas.
An extensive review of the chemical and physical properties of
organoarsenic compounds has been made by Doak & Freedman (1970).
2.2 Analytical Procedures
2.2.1 Sampling and sample treatment
Arsenic poses some special problems in sampling not experienced in
the determination of other trace elements. Water, urine, and
biologically active samples should either be analysed within a few
hours or frozen and stored (Andreae, 1977; Feldman 1979). Low
concentrations of arsenic compounds found in natural waters slowly
decrease with time, unless stabilized in some manner to prevent
adsorptive losses. The biomethylation of inorganic arsenic in a
biologically active sample can cause a change in its composition.
Since environmental analyses often involve trace concentrations,
sample treatment frequently includes some type of preconcentration
prior to analysis. Conversion of arsenic to arsine, co-precipitation
with iron(III) hydroxide, distillation as arsenic(III) chloride, or
extraction are typical examples of the approaches used.
2.2.1.1 Natural waters
Sea water and fresh natural waters are generally analysed without
oxidative treatment prior to a preconcentration step, when the
molecular forms of arsenic are to be analysed. If the preconcentration
step or the final steps in the analytical method require the
conversion of organoarsenic compound to an inorganic form, oxidation
procedures may be necessary. An acid-potassium persulfate preoxidation
method (Pierce et al., 1976) and a method involving ultraviolet (UV)
have both been automated (Fishman & Spencer, 1977). Acid-permanganate
oxidation was found to be effective in the conversion of
dimethylarsinic acid to inorganic arsenic (Sandhu & Nelson, 1978).
Arsine generation followed by cold trapping in liquid nitrogen is
a technique that can be used with or without prior oxidation (Braman
et al., 1977; Siemer & Koteel, 1977). Arsine generation has long been
used as a first step in the determination of arsenic in water samples,
prior to spectrophotometric analysis of the complex formed with
silverdiethyldithiocarbamate (SDDC) (Skonieczny & Hahn, 1978). A
recent adaptation of this method is the analysis for the SDDC complex
by graphite tube furnace AAS which gives an improved detection limit
of about 10 ng (Shaikh & Tallman, 1977). Arsenic has been separated
from samples by volatilization as the trichloride or tribromide. A
recent application combines distillation as the chloride with anodic
stripping voltametry (Davis et al., 1978).
A number of coprecipitation methods have been reported for the
preconcentration of arsenic from water followed by different methods
of analysis. Iron(III) hydroxide (Portmann & Riley, 1964) and
hydroxides of zirconium and cerium (Plotnikov & Usatova, 1964) are
among the many coprecipitants that have been studied. Thionalide has
also been used in the coprecipitation of arsenic from sea water
(Portmann & Riley, 1964) with 95% efficiency, but the procedure is
slow, requiring much sample handling, a problem with all of the
precipitation methods.
2.2.1.2 Air
Air sampling for trace amounts of arsenic in the environment has
mainly been confined to sampling the particulate phase. It is likely
that many different types of particulate filters are satisfactory for
this type of sampling, though arsenic is usually associated with small
size particles.
As the estimated saturated concentration of arsenic(III) oxide in
air at 25°C is about 600 ng/m3, it is possible that, when air
concentrations are below this level, arsenic(III) oxide collected on a
filter may evaporate or may not be collected completely. Results of
laboratory work using filters and pure arsenic(III) oxide in air
support this theory (Lao et al., 1974; Walsh et al., 1977b).
Nevertheless, in studies using a filter impregnated with ethyleneimine
in glycerol which is 65% efficient in trapping arsenic(III) oxide
vapour, it was shown that the major portion of arsenic in air (78-99%)
could be collected on untreated, 0.4 micrometer pore size,
polycarbonate type filters (Nuclepore Co.). Millipore membrane filters
have also given satisfactory results (Walsh et al., 1977b). This was
found to be the case for both ambient air samples containing low
levels of arsenic and near-smelter air samples containing high levels.
Approximately 15% of the arsenic in the air collected was found to be
in a vapour form that was not collected on untreated filters. These
results agree with those of Johnson & Braman (1975a) who also found
that approximately 15% of the collected arsenic was volatile.
Vapour forms of arsenic in air, particularly the arsines, can be
preconcentrated from air onto silver-coated glass beads (Johnson &
Braman, 1975a). Even if oxidized after adsorption, the identity of the
compound is not lost. For example, dimethylarsine, if present, can
only be oxidized to dimethylarsinic acid. Adsorbed compounds can be
desorbed using dilute sodium hydroxide (Braman et al., 1977).
2.2.1.3 Biological materials
Samples of biological materials to be analysed for total arsenic
are generally completely oxidized prior to analysis. A number of
oxidation methods have been studied, the majority of which involved
the use of oxidizing acids or persulfates. The completeness of the
oxidation has however seldom been checked. Perhaps the best oxidizing
procedure involves the use of a mixture of sulfuric and nitric acids
(Chu et al., 1972), a mixture of sulfuric, nitric, and perchloric
acids (Christian & Feldman, 1970) or hydrogen peroxide (Samsahl,
1967). Dry ashing with magnesium oxide or magnesium nitrate has been
successfully applied in the analyses of a variety of biological
samples (Snell & Snell, 1945; Evans & Bandermer, 1954). Other methods
with fewer contamination problems or losses of arsenic are the Parr
bomb (Beamish & Collins, 1934) and the Carius oxidation (Day, 1964)
techniques. Schoeninger flask oxidation has been used in the oxidation
of dried tissue samples (Schwedt & Russel, 1972). The proper
preanalytical treatment for samples of certain marine organisms
containing compounds such as arsenobetaine has yet to be established
(Edmonds, et al., 1977).
There has been some success in analysing homogenized samples
without oxidation by treating them with hydrochloric acid (Kingsley &
Schaffert, 1951) or sodium hydroxide (Johnson & Braman, 1975b) prior
to analysis. This approach is particularly necessary if the molecular
forms of arsenic present are to be identified. In no case has the
accuracy of the analyses been unequivocally determined.
Various methylarsenic compounds have been determined in human
urine samples without pretreatment (Braman, et al., 1977; Crecelius,
1977b).
2.2.2 Analytical methods
2.2.2.1 Methods for total arsenic
One early very common method for the determination of total
arsenic was the Gutzeit method (Vogel, 1955).
Spectrophotometry using the silver diethyldithiocarbamate (SDDC)
complex of arsine is the classical method for determining arsenic in
the 1-100 microgram range (Vasak & Sedivec, 1952). Arsenic is reduced
to arsine by either granular zinc in hydrochloric acid or by sodium
borohydride. Arsine reacts with SDDC in pyridine and the absorption of
the red coloured complex is read at 533 nm. Methylarsine and
dimethylarsine, but not trimethylarsine, form SDDC complexes which
absorb at 533 nm, but their complexes have lower molar absorptivities.
A large number of studies can be found in the literature
concerning the use of the SDDC method as it is often designated a
standard method of analysis (Stratton & Whitehead, 1962). Some more
recent papers include one in which the somewhat disagreeable pyridine
solvent was replaced by L-erythro-2-(methylamine)-1-phenylpropan-1-ol
(L-ephedrine) in chloroform (Hundley & Underwood, 1970; Gastiner,
1972; Kopp, 1973). Ionic interference in the SDDC procedure has been
studied by Sandhu & Nelson (1978).
The arsenate ion reacts with ammonium molybdate to form a complex
which, when reduced, gives a blue colour (Portmann & Riley, 1964).
Under favourable conditions, the limit of detection is near 0.1 µg. An
adaptation of the method has been used to determine the amounts of
phosphate, arsenate, and arsenite in sea water (Johnson & Pilson,
1972). The method is applicable to sea water samples with arsenic
concentrations below 3 × 10-6 mol/litre. Precision is of the order of
± 0.015 × 10-6 mol/litre.
Atomic absorption spectrophotometry (AAS) is gaining in popularity
as a method for the determination of total arsenic. Sensitivity of the
ordinary flame type AAS for arsenic in solution is comparatively poor,
detection limits are in the 0.5-1 mg/litre range (Holak, 1969;
Kirkbright & Ranson, 1971). When an electrodeless discharge lamp and
an argon-air-hydrogen flame are used, the detection limit is reduced
to 0.1 mg/litre (Menis & Rains, 1969). With a long-path cell, the
detection limit is about 6 µg/litre (Ando et al. 1969). Arsine can
also be passed into a heated graphite or quartz furnace mounted in an
AAS instrument. The arsine can be continuously passed through the
atomizer (Smith, 1975; Siemer et al., 1976) or collected in a cold
trap and passed through rapidly when the cold trap in heated (Griffin
et al., 1975; McDaniel et al., 1976). This second technique provides
the best detection limits which are in the fraction of a ng range
(Siemer & Koteel, 1977).
Neutron activation analysis is one of the more sensitive
analytical methods. The arsenic-75 isotope is converted to arsenic-76
by thermal neutron absorption. Detection limits are near 1 ng, but the
method is susceptible to interference, particularly from sodium. There
have been many applications of this method in the analyses of
biological samples (Takeo & Shibuya, 1972; Heydorn & Damsgaard, 1973;
Maruyama & Komiya, 1973; Orvini, et al., 1974), water (Ray & Johnson,
1972) and particulate matter in air (Walsh et al., 1977b). Activated
sample solutions are frequently subjected to separation to eliminate
interfering radioisotopes (Gallorini, et al., 1978).
The determination of trace amounts of arsenic has also been
performed using differential pulse polarography and anodic stripping
voltametry (Arnold & Johnson, 1969; Myers & Osteryoung, 1973; Davis,
et al., 1978). The second of these methods was applied to biological
samples that were wet ashed with nitric, sulfuric, and perchloric
acids before distillation of arsenic as arsenic(III) chloride. The
detection limit was in the ng range. Some of the organoarsenic
compounds are also electroactive (Elton & Gieger, 1978) but no
practical methods for environmental analyses have appeared since mg/kg
concentrations are required to observe responses.
A variety of other analytical methods have been successfully used
for the determination of trace amounts of arsenic. Among these are:
atomic emission spectroscopy (Kirkbright et al., 1973; Braman et al.,
1977; Robbins et al., 1979), X-ray fluorescence (Thomson, 1975), and
isotope dilution mass spectrometry (Zeman et al., 1964).
Recently an electron spectroscopic method (ESCA) has been reported
in which arsine collected on filter surfaces was analysed (Carvalho &
Hercules, 1978). Detection limits were in the ng range so that
preconcentration resulted in further reduction of detection limits to
sub µg/kg.
A recent enzyme method gave reasonable results in the
0.02-2.0 mg/kg range (Goode & Matthews, 1978).
Very recently, a small interlaboratory comparison study on the
determination of total urinary arsenic was performed with the
participation of 4 laboratories using different analytical procedures
(NAA and AAS). Ten samples containing arsenic concentrations of
between 0.001 and 1 mg/litre were examined (Buchet et al., in press).
2.2.2.2 Analyses for specific arsenic compounds
Low concentrations of inorganic arsenic(III) and arsenic(V) in sea
water can be determined using the molybdenum blue method (Johnson &
Pilson, 1972). Inorganic arsenic(III) and (V) can be separated by
direct extraction with toluene of acidified aqueous solutions
containing, for example, cysteine (Lauwerys et al., 1979).
Differentiation between arsenic(III) and arsenic(V) is also possible
using pH sensitive, selective reduction with sodium borohydride
followed by atomic emission spectroscopy or AAS detection. Inorganic
and methylarsenic compounds are reduced according to the reactions
shown in Table 2. By buffering at pH 4, reduction of arsenic(V) is
avoided. At pH 1.5, all compounds are reduced. The methylarsine
compounds produced may be cold trapped, separated, and detected
individually. Cold trapping and separation on heating, with detection
by d.c. discharge in helium, has been used in the determination of
arsenic in natural water, human urine (Braman et al., 1977; Crecelius,
1977b) and sea water (Johnson & Braman, 1975b; Andreae, 1977) at µg/kg
and sub µg/kg concentrations. The detector cell has recently been
studied and improved (Feldman & Batistoni, 1977) as has the analysis
train (Crecelius, 1978).
Table 2. Reduction reactions of inorganic and methylarsenic compounds
Compound pKa1 pH Product B.P.
arsenous acid (meta) 9.23 < 7 AsH3 -55°C
(HAsO2)
arsenic acid (ortho) 2.20 > 4.0 no reaction
(H3AsO4) 1.5 AsH3 -55°C
methylarsonic acid 4.1 > 5.0 little reaction
(CH3AsO(OH)2) 1.5 CH3AsH2 2°C
dimethylarsinic acid 6.2 1.5 (CH3)2AsH 36°C
((CH3)AsO(OH))
trimethylarsine oxide -- 1.5 (CH3)3As 70°C
(CH3)3AsO
phenylarsonic acid 1.5 C6H5AsH2 148°C
(C6H5AsO(OH)2)
p-aminophenyl arsinic -- 1.5 H3+ N C6H4 AsH2 --
acid (arsanilic acid)
p-H2N-C6H4AsO(OH)2
Gas chromatographic detection of arsines trapped in cold toluene
solvent using a microwave stimulated plasma detector has been
developed by Talmi & Norvell (1975). The detection limits of this
method are excellent (about 20 pg).
The electrochemical reactions of dimethylarsinic acid and
trimethylarsine were studied by Elton & Geiger (1978). Dimethylarsinic
acid may be converted to its iodide and determined by gas
chromatography (Söderquist, et al., 1974) but the method is not
applicable to the same wide range of arsenic compounds as the
hydride-generating procedures.
Arsenic has been determined in marine organisms (Portmann & Riley,
1964). Substantial efforts have been made to identify the different
organic arsenic compounds and, only recently, arsenobetaine was
identified in rock lobsters (Edmonds et al., 1977) and
arsenophospholipids in algae (Cooney et al., 1978). Analytical methods
for the determination of these compounds are not well developed. Thin
layer chromatography was used in studies by Lunde (1977), the results
of which indicated the possible presence of several as yet
unidentified organic arsenic compounds.
Analytical methods for the determination of total arsenic and
different forms of arsenic in human biological materials have recently
been reviewed by Lauwerys et al. (1979).
3. SOURCES AND OCCURRENCE OF ARSENIC IN THE ENVIRONMENT
3.1 Natural Occurrence
3.1.1 Rocks, soils, and sediments
Arsenic is widely distributed in a large number of minerals. The
highest mineral concentrations generally occur as arsenides of copper,
lead, silver, or gold or as the sulfide. Major arsenic-containing
minerals are arsenopyrite (FeAsS), realgar (As4S4), and orpiment
(As2S3). The arsenic content of the earth's crust is 1.5-2 mg/kg; it
ranks 20th in abundance in relation to other elements (NAS, 1977).
Oxidized forms of arsenic are usually found in sedimentary deposits.
The elemental oxidation state, though stable in reducing environments,
is rarely found. Table 3 gives some ranges of the arsenic contents of
crustal materials. Although the values shown are generally low,
mineralized zones of sulfidic ores may contain much higher
concentrations of arsenic.
Table 3. Arsenic in crustal materialsa
Type Range
As (mg/kg)
Igneous rocks
ultrabasic 0.3-16
basalts 0.06-113
andesites 0.5-5.8
granitic 0.2-13.8
silicic, volcanic 0.2-12.2
Sedimentary rocks
limestones 0.1-20
sandstones 0.6-120
shales and clay 0.3-490
phosphorites 0.4-188
a From: NAS (1977).
High levels of arsenic may also occur in some coals. The average
arsenic content of coal in the USA was estimated at 1-10 mg/kg (Davis
& Associates, 1971). In some coal mined in Czechoslovakia, the
concentration of arsenic has been shown to be as high as 1500 mg/kg
(Cmarko, 1963).
Uncontaminated soils were found to contain arsenic levels between
0.2 and 40 mg/kg, while arsenic-treated soils contained up to
550 mg/kg (Walsh & Keeney, 1975). The soil in the city of Antofagasta,
Chile, contains natural levels of arsenic of about 3.2 mg/kg (Borgono
& Greiber, 1972). In the Comarca Lagunera, Mexico, values between 3
and 9 mg/kg were found at the soil surface and more than 20 mg/kg,
deep down (Gonzalez, 1977).
Peat may contain considerable quantities of arsenic. Minkkinen &
Yliruokanen (1978) found maximum arsenic concentrations in various
Finnish peat bogs of between 16 and 340 mg/kg dry peat.
The natural level of arsenic in sediments is usually below
10 mg/kg dry weight (Crecelius, 1974). Bottom sediments can become
substantially contaminated by arsenic from man-made sources. Levels of
up to 10 000 mg/kg dry weight were found in bottom sediments near a
copper smelter in Washington, USA (Crecelius, 1974).
3.1.2 Air
Airborne particulate matter has been shown to contain both
inorganic and organic arsenic compounds (Johnson & Braman, 1975a;
Attrep & Anirudahn, 1977). Crecelius (1974) showed that only 35% of
the inorganic arsenic in rain from an urban area was present as
arsenite; however, some post-sampling oxidation could not be excluded.
In studies by Johnson & Braman (1975a), methylarsines made up
approximately 20% of the total arsenic in ambient air from rural and
urban areas.
In unpolluted areas, airborne arsenic concentrations ranging from
less than one to a few nanograms per cubic metre have been reported
(Peirson, et al., 1974; Johnson & Braman, 1975a; Walsh, et al., 1977b;
Beavington & Cawse, 1978; Brimblecombe, 1979).
3.1.3 Water
Arsenic occurs in both inorganic and organic forms in water
(Braman & Foreback, 1973; Crecelius, 1974). The main organic arsenic
species, methylarsonic acid and dimethylarsinic acid, are generally
present in smaller amounts than the inorganic forms, arsenite and
arsenate. The chemistry of arsenic in the aqueous environment has been
reviewed by Ferguson & Gavis (1972).
The arsenic contents of surface waters in unpolluted areas vary
but typical values seem to be a few micrograms per litre or less. In a
study of river waters in the USA, about 80% of the samples contained
levels of less than 0.01 mg/litre (Durum et al., 1971). Quentin &
Winkler (1974) found an average value of 0.003 mg/litre in river water
and 0.004 mg/litre in lake water in the Federal Republic of Germany. A
mean arsenic concentration of 0.0025 mg/litre was reported in some
Norwegian rivers (Lenvik et al., 1978). Much higher values have been
reported from some areas including Antofagasta, Chile, where the
average arsenic level in a river water supply of drinking water
between 1958 and 1970 was 0.8 mg/litre (Borgono et al., 1977).
The oxidation state of arsenic in surface waters in various parts
of the world remains largely unknown. Braman & Foreback (1973) found
that the ratio of trivalent to pentavalent inorganic arsenic ranged
from < 0.06 to 6.7 in a few uncontaminated surface water samples
containing between 0.0025 and 0.0030 mg As/litre. About 8% of the
total arsenic in 2 samples of well-aerated stream water (0.014 and
0.06 mg/litre, respectively) was reported by Clement & Faust (1973) to
be in the trivalent form. In anaerobic reservoirs, all of the arsenic
present (0.14-1.3 mg As/litre) seemed to be in this form.
Penrose et al. (1977) reported that sea-water ordinarily contains
arsenic concentrations ranging from 0.001-0.008 mg/litre. Levels of
about 0.002 mg/litre have been reported by Onishi (1969) and Johnson &
Braman (1975b). The major chemical form of arsenic appears to be the
thermodynamically stable arsenate ion; even so, arsenite often
accounts for one third of the total arsenic (Johnson, 1972; Andreae,
1978).
Clement & Faust (1973) analysed water from 2 groundwater supplies
with very high levels of arsenic (224 and 280 mg/litre) and found that
about 50% was present as arsenic(III). In a groundwater-fed stream,
26% of the total arsenic (0.08 mg/litre) was in the form of trivalent
arsenic. Arsenic speciation has also been performed on well water
samples from an area in Alaska containing high levels of arsenic
(Harrington et al., 1978). In 5 samples containing arsenic
concentrations ranging from 0.52 to 3.6 mg/litre, between 3% and 39%
of the arsenic present was trivalent, the rest being pentavalent. No
methylated arsenic compounds could be detected.
High levels of arsenic have been found in waters from areas of
thermal activity. Thermal waters in New Zealand have been shown to
contain up to 8.5 mg/litre (Ritchie, 1961). Geothermal water in Japan
contained arsenic levels of 1.8-6.4 mg/litre and neighbouring streams
contained about 0.002 mg/litre (Nakahara et al., 1978).
The chemical forms of arsenic in thermal water from New Zealand
were investigated by Aggelt & Aspell (1978). In the geothermal bores,
more than 90% of the arsenic was present in the trivalent form.
However, in a river flowing through the area, the pentavalent form was
predominant but some seasonal variation in the ratio between the two
valence states was indicated.
3.1.4 Biota
The sorption of arsenate ions in the soil by iron and aluminum
components, greatly restricts the availability of arsenic to plants
(Walsh et al., 1977a). The arsenic content of plants grown on soils
that had never been treated with arsenic-containing pesticides varied
from 0.01 to about 5 mg/kg dry weight (NAS, 1977). Plants grown on
arsenic-contaminated soils may, however, contain considerably higher
levels, especially in the roots (Walsh & Keeney, 1975; Grant & Dobbs,
1977; Wauchope & McWhorter, 1977). Some grasses growing on soils
containing high levels of arsenic have been found to have elevated
arsenic contents (Porter & Peterson, 1975). Andersson & Nilsson (1972)
reported that arsenic in soils treated with sewage sludge was highly
available to plants, but only a few samples were analysed. In
contrast, Furr et al. (1976) claimed that soil arsenic is not readily
available to plants.
Marine algae and seaweed usually contain considerable amounts of
arsenic. Lunde (1970) showed values of 10-100 mg/kg dry weight in
marine algae from the Norwegian coast. The degree of enrichment was
found to be between 1500 and 5000 compared with the level of arsenic
in the growth medium (Lunde, 1973a). Similar and even higher
enrichment ratios were reported for fresh water plants in the Waikato
River, New Zealand (Reay, 1972). The elevated arsenic concentrations
in the water (0.03-0.07 mg/litre) gave rise to concentrations of up to
971 mg As/kg dry weight in aquatic plants.
3.2 Industrial Production and Uses of Arsenic
3.2.1 Industrial production
Based on the limited data available (US Bureau of Mines, 1975;
Nelson, 1977), it can be estimated that the total world production in
1975 was around 60 000 tonnes. This production seems to be stable. The
main producers are: China, France, Federal Republic of Germany,
Mexico, Namibia, Peru, Sweden, USA, and USSR. These countries account
for about 90% of the production. For a more detailed discussion of
production of arsenic and its compounds, see IARC (1980).
Arsenic(III) oxide, the major basic chemical of the arsenic
industry, is emitted as a by-product in smelting, mainly of copper and
lead ores. It is recovered from the flue dust in a reasonably pure
form.
3.2.2 Uses of arsenic compounds
Arsenic compounds are mainly used in agriculture and forestry
(NAS, 1977). Much smaller amounts are used in the glass and ceramics
industry and as feed additives and drugs. The use pattern for
arsenic(III) oxide in 1975-78 has been reported as follows:
manufacture of agricultural chemicals (pesticides), 82%; glass and
glassware, 8%; industrial chemicals, copper and lead alloys, and
pharmaceuticals, 10% (US Bureau of Mines, 1979).
In agriculture, compounds such as lead arsenate, copper
acetoarsenite, sodium arsenite, calcium arsenate, and organic arsenic
compounds are used as pesticides. Substantial amount of methylarsonic
acid and dimethylarsinic acid are used as selective herbicides. These
herbicides are particularly necessary for the control of Johnson grass
(Sorghum halepense) in cotton fields. They are also used to treat
other weeds such as sandbur ( Cenchrus sp.), cocklebur
( Xanthium sp.) and crabgrass in lawns (Weed Science Society of
America, 1974). Dimethylarsinic acid is used as a silvicide in forest
control and workers may be exposed to the compound and its volatile
reaction products in the soil (Wagner & Weswig, 1974). Dimethylarsinic
acid was the Agent Blue used in Viet Nam as a defoliant for military
purposes.
Chromated copper arsenate, sodium arsenate, and zinc arsenate are
used as wood preservatives (Lansche, 1965). When these compounds are
applied under pressure they react with the wood to create water
insoluble compounds. The preserved timber is resistant to both fungal
and insect attack (Dobbs et al., 1976). The use of arsenic in wood
preservatives is increasing.
Some phenylarsenic compounds such as arsanilic acid are used as
feed additives for poultry and swine and to combat certain diseases in
chickens.
Small amounts of arsenic compounds continue to be used as drugs in
some countries. Other applications of arsenic are found in metallurgy,
where it is used to dope germanium and silicon or in the production of
gallium arsenide or indium arsenide.
3.2.3 Sources of environmental pollution
The burning of coal and smelting of metals are major sources of
arsenic in air. A British study showed yearly average concentrations
in suspended matter in town air of 0.04-0.14 µg/m3 (Goulden et al.,
1952). In Prague, Vondracek (1963) found a winter mean concentration
in air of 0.56 µg/m3 and a summer mean of 0.07 µg/m3. In urban areas
in the USA, air concentrations of arsenic ranged from below the
detection limit (0.01 µg/m3) to 0.36 µg/m3 on a quarterly average
basis in 1964 (Sullivan, 1969). In 1974, about 200 of the 280 US
National Air Surveillance Network sites recorded quarterly average
concentrations below 0.001 µg/m3 (Thompson, 1977). Only 13 sites,
mainly highly urbanized areas and smelter locations, showed levels
exceeding 0.02 µg/m3.
In the vicinity of smelters, levels of arsenic in air exceeding
1 µg/m3 have been recorded. Rozenshtein (1970) found levels of
airborne arsenic, given as arsenic(III) oxide, of 0.7-2.5 µg/m3
(i.e., 0.5-1.9 µg As/m3) within 4 km of a copper smelter in the USSR.
Data were not given on the duration of sampling. In the USA, quarterly
average levels of up to 1.4 µg/m3 were reported in El Paso, Texas, at
the site of a large copper smelter (Sullivan, 1969). Near a copper
smelter in Tacoma, Washington, monthly averages of arsenic in air of
up to 1.46 µg/m3 were recorded (Nelson, 1977) and a maximum 24-h
concentration of 7.9 µg/m3 was reported by Roberts, et al. (1977).
Daily mean concentrations of up to 1.6 µg/m3 were found in the air
near a smelter in Romania (Gabor & Coldea, 1977). Auermann et al.
(1977) reported airborne arsenic concentrations in a polluted region
of the German Democratic Republic ranging from 0.9-1.5 µg/m3 (average
0.9 µg As/m3; duration of sampling not stated). In the vicinity of a
Canadian gold mine, where ore was roasted, annual mean arsenic
concentrations in ambient air ranged from 0.06 to 0.09 µg/m3 between
1973 and 1975 (Hazra & Prokupok, 1977). Individual 24-h arsenic
concentrations varied from less than 0.01 to 3.91 µg/m3. The
concentrations of arsenic in flue dust from a coal-fired power plant
in Czechoslovakia ranged from 43 to 110 mg/kg (Zdrazil & Picha, 1966).
In fly ash from 24 US coal-fired power plants, the arsenic
concentrations ranged between 2.3 and 312 mg/kg (Kaakinen et al.,
1975; Furr et al., 1977).
In the stack dust from nonferrous smelting operations, arsenic is
predominantly in the trivalent inorganic form (Crecelius, 1974,
Rosehart & Chu, 1975). No conclusive data on the extent of oxidation
of airborne trivalent arsenic are available at present.
A thorough study has been made of arsenic in the environs of a
copper smelter near Tacoma, WA, USA (Crecelius, 1974). Dated segments
of sediment cores showed that the arsenic buildup started with the
operation of the smelter. Less than 30% of the arsenic entering
neighbouring waterways accumulated in the sediments. The remaining 70%
presumably left the location in solution. Elevated concentrations of
arsenic were found in water in locations within 2-4 km of the smelter.
Analyses of air, rain water, and snow all indicated elevated arsenic
levels in the Tacoma, Washington area, attributable to the smelter
effluent. Levels of up to 380 mg/kg (dry weight) were found in top
soil in the vicinity of the plant.
A similar pattern, was observed in a study of the distribution of
arsenic from a copper smelter in Sweden (Lindau, 1977). The arsenic
concentrations in air a few kilometres from the smelter were higher
than normal, as were arsenic levels in the soil, moss, and nearby
natural water bodies.
Suzuki et al. (1974) reported concentrations of arsenic of up to
2470 mg/kg in soil near a smelter in Japan.
Attrep & Anirudhan (1977) found a quarterly average total arsenic
concentration in air of 0.08 µg/m3 in an area polluted by arsenic
from defoliants. About half of the airborne arsenic was in the form of
organic arsenic compounds. Four years later, during a season of low
arsenic use, a monthly average of 0.009 µg/m3 was detected in the
same area. At this time, only about 15% of total airborne arsenic was
in the form of organoarsenic compounds.
Burning of wood treated with arsenic-containing preservatives,
mainly inorganic pentavalent compounds, can result in the release of
arsenic into the atmosphere. The concentration of arsenic in the
combustion fumes is closely related to the temperature. Smouldering of
wood, treated with inorganic arsenic salts, at a temperature of 415°C
resulted in volatilization of 8.6% of the total arsenic in the wood
(Watson, 1958). When wood treated with a preservative containing
inorganic pentavalent arsenic salts was burned at temperatures of
700-800°C, about 50% of the arsenic was present in the ashes (the rest
was mainly in the smoke), while at 1000°C only about 15% remained in
the ashes (Öhman, 1960).
The use of geothermal energy can result in severe arsenic
contamination. Crecelius, et al. (1976) found that the natural arsenic
level 0.002 mg/litre had increased 1000 times in a water reservoir in
which some of the discharge from a Mexican geothermal power plant was
emitted. Between 6% and 51% of the total arsenic in this reservoir was
present as trivalent inorganic arsenic and the rest as pentavalent.
The emissions of arsenic into the environment from the plant totalled
about 60 kg/day. In El Salvador, water from a reservoir near a
geothermal power plant contained an arsenic level of 8.9 mg/litre
(Jernelöv et al., 1976).
Arsenic is also present in trace amounts in fertilizers. In a
recent study, it was reported that concentrations of up to several
hundred mg/kg were present in some instances (Senesi et al., 1979).
4. ENVIRONMENTAL TRANSPORT AND DISTRIBUTION
4.1 General
Most environmental transformations of arsenic appear to occur in
the soil, in sediments, in plants and animals, and in zones of
biological activity in the oceans. Biomethylation and bioreduction are
probably the most important environmental transformations of the
element, since they can produce organometallic species that are
sufficiently stable to be mobile in air and water. However, the
biomethylated forms of arsenic produced are subject to oxidation and
bacterial demethylation back to inorganic forms.
The biomethylation of arsenic was first recognized long ago when
arsines were produced from cultures of a fungus Scopulariopsis
brevicaulis (Challenger, 1945). This work was done in an
investigation of poisoning incidents attributed to arsenic-containing
wall paper -- thought to contain Paris Green colouring pigment. It was
eventually ascertained that methylarsines were the toxic agents. More
recently, the methylation of arsenic by methanogenic bacteria (McBride
& Wolfe, 1971) and by reaction with methylcobalamine (Schrauzer
et al., 1972) or L-methionine-methyl-d3 (Cullen et al., 1977) has
been demonstrated in laboratory work. McBride et al. (1978) reported
that dimethylarsine was mainly produced by anaerobic organisms, while
trimethylarsine resulted from aerobic methylation. The following
mechanism for the methylation of arsenate has been proposed by
Challenger (1945) and McBride et al. (1978).
2e CH3+ 2e CH3+
AsVO43- --->ÄsIIIO33- --->CH3AsVO32- --->CH3ÄsIIIO22- --->
--O2- --O2-
2e CH3+ 2e
(CH3)2AsVO2- --->(CH3)2ÄsIIIO- --->(CH3)3AsVO --->(CH3)3ÄsIII
--O2- --O2-
The proposed mechanism indicates that As(V) has to be reduced to
As(III) before being methylated.
4.2 Aquatic Systems
Studies on the molecular forms of arsenic compounds in sea water
have been reported. The concentration ratio As(III)/As(V) was found to
be 0.18 in some Sargasso sea water (Johnson & Braman, 1975b).
Fluctuations in the As(III)/As(V) ratio from 0.02 to 0.09 in the
saline water of Naragansett Bay appeared to be associated with
phytoplankton activity (Johnson & Burke, 1978). Sea water samples off
southern California also exhibited a variable As(III)/As(V) ratio,
again associated with biological activity (Andreae, 1977). In some
instances, the arsenic(III) concentrations exceeded those of
arsenic(V). The same type of biological activity was observed in
natural fresh waters (Braman & Foreback, 1973; Clement & Faust, 1973).
It is evident that the presence of arsenic(III) compounds is the
result of some reductive activity, which could be either biological or
a non-biological effect of dissolved organic matter on arsenic(V).
The oxidation rate of arsenic(III) in Sargasso seawater was
studied under carefully controlled laboratory conditions by Johnson &
Pilson (1975). Temperature, pH, salinity, and the presence of light
all influenced the rate of arsenite oxidation.
The finding of methylarsenic acids in seawater and fresh natural
water is evidence that arsenic goes through reactions other than
simple oxidation or reduction. In both sea and fresh water, the
occurrence of methylarsenic compounds is associated with phytoplankton
activity. In fresh water, the levels of methylarsenic compounds were
especially high in locations where nutrients from fertilizers
(presumably, also containing arsenic) had built up in lakes and ponds.
There is little evidence that sediments play a substantial role in the
methylation of arsenic (Braman, 1975). Sediment samples from two
natural water environments did not contain unusually large amounts of
methylarsenic compounds.
The analysis of biota associated with Sargassum weed indicated
that substantial amounts of arsenic were present in forms other than
the inorganic or methylarsenic forms (Johnson & Braman, 1975b). Only
small amounts of methylarsenic acid type compounds were present in the
organisms.
The involvement of arsenic in the biochemistry of marine organisms
through production of arsenobetaine, arsenocholine, and
arsenophospholipids is a new and only partially explored aspect of the
local cycle. Much work has been done in an effort to identify arsenic
compounds in marine organisms (Edmonds et al., 1977; Irgolic et al.,
1977; Lunde, 1977; Penrose et al., 1977; Cooney et al., 1978).
4.3 Air-soil Systems
It has already been mentioned (section 3) that large quantities of
arsenic compounds are used in agriculture and are initially
distributed in the soil. This is an important aspect of arsenic
distribution in the environment. The occurrence and distribution of
arsenic in soils and plants have been reviewed by Walsh et al.
(1977a). Arsenic is converted to arsenates except under highly
reducing conditions. Arsenate ions are readily sorbed by hydrous
oxides of iron and aluminum and thus leaching of arsenate is slow.
Absorption appears to be a major factor in the retention of arsenic in
soils.
Slow removal of arsenic from the soil is of concern, when old
orchards previously treated with arsenic are used for crop growing
(Bishop & Chisholm, 1962). High arsenic levels can cause a depression
in plant growth but the amounts required to produce this effect depend
on the plant species. Bioaccumulation of arsenic in food crops is not
particularly high.
Methylarsines are released into the air from soil treated with
various arsenic compounds. Dimethylarsine and trimethylarsine were
detected over grass areas treated with the methylarsenic compounds,
soon after application. Methylarsines evolved much more slowly from
grass treated with sodium arsenite (Braman, 1975). Despite these
observations in locations where arsenic was obviously volatilized into
the air following biomethylation, the amounts of methylarsenic
compounds actually found in unpolluted air appear to be small. In one
study, approximately 15% of the total arsenic in outdoor air was in a
methylated form (Johnson & Braman, 1975a). The total arsenic was much
greater in greenhouse air than in ambient air outside and the
methylarsenic forms were much in excess of inorganic forms.
A proposed model of an air-soil arsenic system is shown in
Fig. 1. The system has little chance of being in apparent equilibrium,
since air transport of transpired volatile arsenic is rapid, compared
with evolution rates. Because of lack of data concerning arsenic
compounds in air, especially in locations with arsenic-rich soils, the
rates of evolution and buildup of arsenic in air are not known. A
pseudo-equilibrium can be approached if the air transported into a
site is equivalent to the air transported away from a site. This cycle
is similar to one developed for an agronomic ecosystem, in which
arsenic pesticides were the input (Sandberg & Allen, 1975). The most
important translocation factors were absorption by soil and oxidation,
uptake by vegetation, and volatilization after biomethylation.
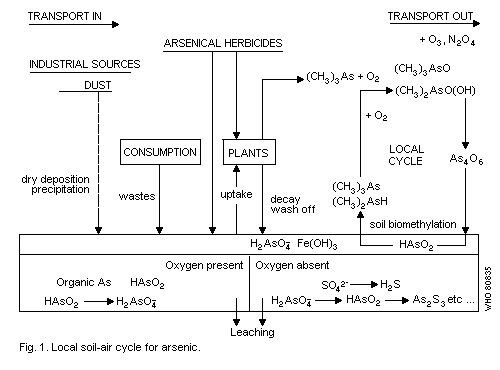 5. LEVELS OF EXPOSURE TO ARSENIC AND ITS COMPOUNDS
Identification of the form in which arsenic occurs has only
recently become part of the determination of arsenic in various
environmental media. Generally, only total arsenic concentrations have
been measured. In several reports however, the concentration of
arsenic has been expressed as arsenic(III) oxide, even though the
exact nature of the compound has not been determined. An attempt has
been made in this section to distinguish between the various forms of
arsenic, where sufficient knowledge exists. Unless specifically noted,
the concentrations given in this section refer to elemental arsenic.
The levels should be considered tentative as, in most instances, the
accuracy of the analytical methods has not been assured.
5.1 General Population Exposure through Air, Drinking-Water,
Food, and Beverages
5.1.1 Air
From data on air concentrations of arsenic in unpolluted areas
(section 3.1.2), it can be calculated that the amount of arsenic
inhaled per day is about 0.05 µg or less (assuming that about 20 m3
of air is inhaled per day). However, in areas where coal with a high
arsenic content is used in power plants, or in the vicinity of
smelters, the intake of arsenic may be considerably higher. Airborne
arsenic levels of about 1 µg/m3, have been detected in such areas,
(section 3.2.3), which would result in the inhalation of approximately
20 µg of arsenic per day.
The amount of arsenic absorbed from the lungs depends on particle
size and the chemical form of the arsenic. Analysis of arsenic in
airborne fly ash from coal-fired power plants indicated that the
highest concentration was associated with respirable particles. On a
mass basis, 76% of the arsenic present was recovered from particles
with a diameter of less than 7.3 µm (Natusch et al., 1974).
5.1.2 Drinking-water
The natural concentration of total arsenic in drinking-water
varies in different parts of the world. McCabe et al. (1970)
investigated more than 18 000 community water supplies in the USA and
found that less than 1% had arsenic levels exceeding 0.01 mg/litre. In
a report by Grantham & Jones (1977) on arsenic concentrations in water
from more than 800 wells in Nova Scotia, Canada, 13% had arsenic
levels exceeding 0.05 mg/litre. Apparently, some of these wells had
been contaminated by gold-mining activities in previous years. In some
areas where chronic arsenic poisoning has occurred, levels exceeding
1 mg/litre have been recorded in well water. In the region of Cordoba,
Argentina, Arguello et al. (1938) reported maximum levels of arsenic
of between 0.9 and 3.4 mg/litre. Artesian well water in the Tainan
county of the Province of Taiwan contained up to 1.8 mg/litre (Kuo,
1968). Well waters in Oregon also contained elevated levels of arsenic
(0.07-1.7 mg/litre) (Goldblatt, et al., 1963).
Drinking-water can be severely contaminated through industrial
operations. In the city of Torreon, Mexico, Espinosa González (1963)
reported that levels of arsenic in drinking-water from a deep well
ranged from 4 to 6 mg/litre. In Niigata, Japan, waste water from a
factory producing arsenic sulfide contaminated nearby well water, and
arsenic levels up to 3 mg/litre were recorded (Terada, 1960). Leaching
of arsenic from coal preparation wastes and fly ash from coal-fired
power plants may also result in the contamination of water (Williams,
et al., 1977; Chu, et al., 1978).
When considering exposure through drinking water, it is important
to ensure that exposures are assessed for water delivered from the
consumer's tap. Conventional flocculation treatment using either
aluminum or ferric salts removes a high proportion, at least, of
arsenic(V) (Gulledge & O'Connor, 1973).
5.1.3 Food and beverages
Arsenic levels in food, with the exception of some seafoods, are
generally well below 1 mg/kg wet weight (Westöö & Rydälv, 1974).
Marine fish on an average contain below 5 mg/kg wet weight (LeBlanc &
Jackson, 1973; Lunde, 1973b; Leatherland & Burton, 1974; Kennedy,
1976; Stoeppler & Mohl, 1980). Certain bottom feeding fish, crustacea
and shellfish may contain arsenic concentrations of several tens of
milligrams per kilo (Westöö & Rydälv, 1972; Crecelius, 1974; Munro
et al., 1974). Arsenic concentrations of between 0.6 and 58 mg/kg dry
weight, have been found in some food supplements prepared from kelp
(Walkiw & Douglas, 1975). Edible seaweed, a common product in Japan,
has been reported to contain arsenic levels ranging from 19 to
172 mg/kg dry weight with a mean concentration of 112 mg/kg (Watanabe
et al., 1979). The use of some organic arsenic compounds as feed
additives for poultry and swine may lead to accumulation of arsenic in
certain organs (Ledet et al., 1973; Calvert, 1975) (section 6.2.2.2)
and limits of tolerance have been established in the USA for edible
by-products from chickens, turkeys, and swine (Jelinek & Corneliussen,
1977).
Most of the arsenic in marine organisms occurs in the form of
either fat-soluble or water-soluble organoarsenic compounds (Lunde,
1975). The water-soluble compounds are characterized by high chemical
stability. Lunde (1973b) separated inorganic and organic arsenic in
some fish and crustacea from the Norwegian Atlantic coast. The
concentrations of inorganic arsenic (including organic-bound arsenic
degradable by 6.6 M hydrochloric acid) ranged from 1.0 to 2.5 mg/kg
and those of organoarsenic compounds from 3 to 37 mg/kg. Seafood
arsenic, i.e., the major organic arsenic compounds found in seafood,
is not degradable by this treatment. Crecelius (1977b) did not find
any increase in human urinary excretion of inorganic or of simple
methylated arsenic compounds, i.e., methylarsenic acid and
dimethylarsenic acid, following the ingestion of 2 mg of arsenic in
crab meat. This indicated that the inorganic arsenic content of the
crab meat was very low (<1% of the arsenic).
Wine may contain appreciable amounts of arsenic. Noble et al.
(1976) found concentrations between 0.02 and 0.11 mg/litre in 9 US
wines produced between 1949 and 1974. Crecelius (1977a) also
investigated the levels and forms of arsenic in some US table wines.
In over half of the samples, levels greatly exceeded 0.05 mg/litre
(tentative limit in the international drinking water standards
published by WHO). Most of the arsenic present was in the trivalent
form. Arsenate was also found, but no methylated species were
detected. This study indicated that considerable reduction from
arsenate to arsenite occurred during the fermentation of grape juice
by wine yeast. It is probable that the arsenic in the wines originated
mainly from the arsenic-containing insecticides used on the grapes.
Elevated arsenic levels have been found in some bottled mineral
waters. Zoeteman & Brinkmann (1976) reported a mean arsenic
concentration of 0.021 mg/litre (range <0.001-0.19 mg/litre) in
bottled mineral waters sold in countries within the European
Community. In an investigation on lager beers from various countries,
none of the samples contained more than 0.02 mg/litre (Binns et al.,
1978).
5.1.4 Tobacco
The content of arsenic in tobacco grown on soils not treated with
arsenic compounds is usually below 3 mg/kga a (Satterlee, 1956;
Bailey et al., 1957; Hjern, 1961; Griffin et al., 1975). During the
first part of this century, the use of arsenic insecticides, mainly in
the USA, brought about a steady increase in the content of arsenic in
a The weight of a cigarette is approximately 1 g.
tobacco products. In the 1950s, levels of up to 52 mg/kg, given as
As(III) oxide (40 mg As/kg) were found in American cigarettes (Holland
& Acevedo, 1966). However, during the last 20 years the concentrations
of arsenic have decreased to below 8 mg/kg, because of a great
reduction in the use of inorganic arsenic compounds in agriculture. Of
the total arsenic originally present in cigarettes, 10-15% was
recovered in the main stream smoke, the remainder mainly being
distributed in the ash and butt (Thomas & Collier, 1945). Cigarettes
in Japan have been reported to contain arsenic levels of less than
1 mg/kg (Maruyama et al., 1970). The chemical form of arsenic in the
smoke has yet to be elucidated.
5.1.5 Drugs
Both inorganic and organic arsenic compounds have been widely used
in medicine. Arsenical Solution, also called Liquor Arsenicalis,
Solutio Kalii Arsenitis or Fowler's Solution, contained arsenic(III)
oxide dissolved in potassium hydroxide, neutralized with hydrochloric
acid and diluted with chloroform water (Martindale, 1977). The arsenic
administered was thus in the form of arsenite. The drug ordinarily
contained an arsenic concentration of 7.6 g/litre and the daily dose
of arsenic was sometimes as high as 10 mg (Pearson & Ponds, 1971). It
was used for the treatment of leukaemia, psoriasis, chronic bronchial
asthma, and as a tonic. Other preparations described in the Extra
Pharmacopoeia by Martindale (1977) include various pastes containing
inorganic arsenic in combination with other drugs, such as cocaine or
procaine. Sodium arsenate was formerly used in the treatment of
chronic skin diseases, some parasitic diseases, and anaemia
(Martindale, 1977). Pearson's Arsenical Solution, which contained
about 0.5% arsenic in the form of arsenate, has been included in
several pharmacopoeias. The recommended dose was 1-10 mg of the
arsenate (0.2-2.4 mg As) with a maximum of 20 mg in 24 h. Drugs
containing inorganic arsenic compounds are being phased out and
replaced by more effective and less toxic drugs.
Salvarsan (arsphenamine), an organic arsenic compound containing
32% arsenic, was formerly used in the treatment of syphilis
(Martindale, 1977). Because of the difficulties in preparing it for
injection and because of its high toxicity, it was replaced by
neoarsphenamine. The recommended dose used to be 100-600 mg (32-192 mg
As) administered intravenously. Antibiotics have finally replaced
these drugs. Some organic arsenic compounds including carbarsone,
melarsoprol, and tryparsamide, are still in use in human medicine,
mainly as antiparasitic drugs.
5.1.6 Total daily intake in the general population
Daily intake of arsenic from ambient air and water will ordinarily
be of the order of a few micrograms, predominantly in the inorganic
form (section 5.1.1 and 5.1.2).
As mentioned previously, the total daily dietary intake of arsenic
depends, to a great extent, on the amount of seafood in the diet. A
seafood meal may lead to the ingestion of several milligrams of
arsenic, predominantly in organic forms. The daily intake of total
arsenic in Japan has been reported to be between 0.07 and 0.17 mg
(Nakao, 1960). The US Food and Drug Administration has monitored
arsenic in foodstuffs since 1967 (Jelinek & Corneliussen, 1977). Data
from this programme indicate that the total daily intake of arsenic
has decreased from about 0.05-0.1 mg per day in the late sixties to
0.01-0.02 mg per day in 1972-74. Most of the arsenic was found in the
group "meat, fish, and poultry". From analysis of composites of food
representing the Canadian diet during 1970-73, it was estimated that
the total intake of arsenic was 0.025-0.035 mg daily (Smith et al.,
1972, 1973, 1975). Hamilton & Minski (1973) estimated the total intake
of arsenic in the United Kingdom to be about 0.1 mg/day, based on
analysis of diets containing fish. The considerable variations in the
estimated dietary arsenic intake can be expected because of
differences in the amounts of seafood in the diets investigated.
Moreover, in neither of the reports was a distinction made between the
amount of inorganic and organic arsenic consumed. Because of
differences in metabolism and toxicity (sections 6, 7, and 8), it is
important to distinguish between inorganic and organic forms of
arsenic.
During the 1950s, the smoking of some tobacco, especially from the
US, may have led to inhalation of more than 0.1 mg of arsenic daily.
At present, the arsenic content of most tobacco is much lower and it
can be estimated that less than 0.02 mg may be inhaled by an average
smoker.
Data on the urinary excretion of various arsenic compounds in
individuals not excessively exposed to arsenic can be helpful for
deducing daily intake figures. Inorganic arsenic will be excreted
mainly as inorganic and simple methylated arsenic compounds
(Crecelius, 1977b). Smith et al. (1977) found an average urinary
concentration of these forms of arsenic of 17.5 µg/litre in 41 male
workers in the USA without known occupational exposure to arsenic.
This would correspond to an intake of 0.025-0.040 mg of inorganic
arsenic per day.
5.2 Occupational Exposure
Occupational exposure to arsenic compounds takes place mainly
among workers, especially those involved in the processing of copper,
gold, and lead ores. Occupational exposure may also occur among
workers using or producing arsenic-containing pesticides.
Unfortunately, very few data exist on the actual air levels of arsenic
to which persons in such occupations have been exposed. This is also
the case for wood treatment plant workers and carpenters, who may
become exposed to inorganic arsenic compounds (mainly pentavalent)
through their use as wood preservatives (section 3.2.2).
In a plant where sodium arsenite was being manufactured, Perry et
al. (1948) found mean air arsenic concentrations of between 0.078 and
1.034 mg/m3 around various workstations during sampling times of
"10 minutes or more". The respirable fraction (< 5 µm) of the airborne
arsenic ranged from 20% to 38% by mass. Ott et al. (1974) reported
airborne arsenic levels in 1943 of 0.18 to 18 mg/m3 in the packaging
department of a plant where lead arsenate and calcium arsenate
insecticides were produced. In 1952, airborne arsenic levels ranged
between 0.26 and 40.8 mg/m3 in another workplace at the plant. In the
workroom air of a factory producing lead arsenate, Horiguchi et al.
(1976) found levels ranging from 0.01 to 0.9 mg/m3 during the years
1959-70.
When the airborne arsenic in a Swedish copper smelter was
measured, the average concentrations near the roasters, reverberatory
furnaces, and in the converter hall ranged between 0.06 and 2 mg/m3
during sampling times of "several hours" (Lundgren, 1954). No data
were given on the size distribution of the airborne arsenic-containing
particles. At the same Swedish copper smelter Carlsson (1976) found
weighted 8-h average concentrations at different workplaces of between
0.002 mg and 0.23 mg/m3 in the air inhaled by the workers (i.e.,
after filtration in a respirator). The highest exposures were found
among the roasterworkers. Kodama et al. (1976) measured airborne
arsenic concentrations in a copper refinery, where arsenic(III) oxide
was being manufactured. They found levels of between 0.006 and
0.012 mg/m3 when the ventilation was normal, and up to 0.2 mg/m3
when the ventilation was shut off. Around the furnaces in the copper
smelter, average concentrations of between 0.001 and 0.012 mg/m3 were
reported, and around the furnaces in a ferronickel smelter, the
corresponding concentrations were between 0.002 and 0.005 mg/m3.
Smith et al. (1977) described a study at a US copper smelter where
airborne particulate matter was collected in personal exposure
samplers. The concentrations were found to be log-normally
distributed, with a geometric mean of 0.053 mg/m3 in a high exposure
group (i.e., workers in the baghouse, flue, cotterell, stack, and
reverberatory furnace areas). Workers in the concerter area were
exposed to 0.046 mg/m3 (geometric mean). In the high exposure area,
only 32% of the airborne arsenic was respirable (< 5 µm), compared
with over 80% in the converter area. Pinto et al. (1976) reported an
overall mean airborne arsenic concentration of 0.05 mg/m3 (range
0.003-0.3 mg/m3) in the working environment of 24 smelter workers
wearing personal air samplers on 5 consecutive days.
Airborne arsenic particulate matter in smelters is generally
assumed to consist primarily of arsenic(III) oxide. However, it is
probable that some of the arsenic is firmly bound to other metals,
especially in the reverberatory furnace. There is also evidence of the
presence of arsenic sulfides (Smith et al., 1976). The form in which
arsenic is present clearly depends, to a great extent, on the
characteristics of the industrial process involved, such as the
temperature, humidity, and other elements present. More work is
urgently needed to characterize the arsenic compounds by form and size
distribution.
Workers may be exposed to airborne arsenic in cutting and sawing
operations on wood treated with arsenic-containing preservatives.
Arsenault (1977) found concentrations of arsenic in air of 0.043-
0.36 mg/m3 originating from the sawing of wood treated with copper,
chromium, and arsenic salts. The duration of measurement was 100 min.
Only about 5% of the dust particles (on a mass basis) were less than
10 µm.
6. METABOLISM OF ARSENIC
The metabolism of arsenic in man is very complex since the fate of
arsenic compounds in the human body varies with the type of compound.
The metabolism of a compound also varies with animal species, for
example, the metabolism of arsenic in the rat is unique and quite
different from that in man or other mammals. The rat is therefore not
a suitable model for most metabolic pathways in man and the emphasis
in this document has been placed, as much as possible, on data
concerning other experimental animals.
6.1 Inorganic Arsenic
The metabolism of inorganic arsenic depends on its chemical form.
Possible changes in the different forms of inorganic arsenic before
the time of exposure should be considered. Even commercially available
isotopes of pentavalent arsenic have been shown to contain up to 98%
of trivalent arsenic (Lunde, 1973a; Reay & Asher, 1977). In many
studies on the metabolism of arsenic, the valence of the compound used
has not been under complete control. Throughout the following
description, an attempt has been made to assess the validity of
information concerning valence. In cases where the valence has been
checked before exposure, this has been stated. If no such statements
are made, it must be appreciated that substantial uncertainly exists.
6.1.1 Absorption
6.1.1.1 Respiratory deposition and absorption
Human exposure to inorganic arsenic through inhalation usually
occurs occupationally or during cigarette smoking. Information on the
respiratory deposition and clearance of different inorganic arsenic
compounds is very limited. Inhaled arsenic is mainly in the form of an
aerosol and it can be assumed that its deposition is the same as that
of other particulate matter. In many workplaces, the particles
containing arsenic are of relatively large size (Perry et al., 1948;
Pinto & McGill, 1953), resulting in deposition primarily in the upper
respiratory passages (i.e., nasal cavity, nasopharynx, larynx,
trachea, and bronchus). Subsequent absorption can then take place
either directly from the respiratory tract or gastrointestinally after
mucociliary clearance in the airways. Retention, deposition, and
absorption from the respiratory tract depend, furthermore, on the
solubility of the inhaled material.
6.1.1.1.1 Animals
Hairless mice exposed for several weeks to a solid aerosol of fly
ash (particle size less than 10 µm) containing arsenic at a
concentration of 0.18 mg/m3 showed increased tissue levels of arsenic
(Bencko & Symon, 1970). Hairless mice were used to minimize oral
intake of arsenic deposited on the fur. Despite this, it was not
possible to differentiate between the amount of arsenic absorbed after
inhalation and that absorbed after ingestion. A similar study has also
been performed on rats exposed to arsenic(III) oxide in the form of
condensation aerosols (arsenic concentrations of 0.001, 0.0037 and
0.046 mg/m3) for 3 months (Rozenshtein, 1970). Increased tissue
levels were found in the groups exposed to the 2 highest
concentrations. It was not possible to differentiate between inhaled
and ingested arsenic.
Rapid absorption of arsenic in rats following intratracheal
administration of a solution of sodium arsenate (0.1-4 mg As per kg
body weight) labelled with 74As has been reported by Dutkiewicz
(1977). The rapidity of the absorption was indicated by the relatively
high tissue levels (2.5 and 0.7% of the dose per gram tissue in liver
and spleen, respectively) found one hour after the administration.
6.1.1.1.2 Man
Holland et al. (1959) studied the uptake of inorganic arsenic in 8
terminal lung cancer patients who volunteered to smoke cigarettes
impregnated with 74As-labelled arsenic, reported to be in the form of
sodium arsenite. Between 5% and 8% of the arsenic originally present
in the cigarettes was deposited in the thoracic region. In 2 other
lung cancer patients inhaling a nebulized solution of 74As-arsenite,
the fractions of radioactive arsenic deposited were 32% and 62%,
respectively. Clearance from the lungs seemed to be fast. Four days
after exposure, only about 20% of the dose could be detected by
external scanning of the thoracic region. Since the study was
performed on terminal lung cancer patients, great care must be
exercised in extrapolating these data to healthy human subjects.
Some information on the absorption of arsenic following inhalation
is given in reports on urinary excretion among persons exposed to
arsenic occupationally. Pinto et al. (1976) studied the excretion of
arsenic in 24 workers exposed regularly to airborne arsenic in a
copper smelter (section 7.2). The workers wore personal air samplers
for 5 consecutive working days and the overall average concentration
of airborne arsenic was 0.053 mg/m3. Urine was collected for 2 days
prior to the working week, each day during the working week, and for 3
days afterwards. A correlation was found between urinary arsenic
levels and average airborne arsenic concentrations over the ranges
studied. This investigation seems to indicate a fair absorption of
inhaled arsenic, although no precise estimations can be made.
Arsenic-containing dust which enters the body orally may be
present in the gastrointestinal tract together with the arsenic
transported by mucociliary clearance from the respiratory tract. In
workers exposed to dust (cadmium and nickel dust) in a battery
factory, Adamsson et al. (1979) recently showed that, in some cases,
the amount of cadmium in the faeces was more than 10 times the amount
that could have been inhaled.
6.1.1.2 Gastrointestinal absorption
Absorption of inorganic arsenic from the gastrointestinal tract
can occur following the ingestion of food, water, beverages, or drugs,
containing arsenic or as a result of inhalation and subsequent
mucociliary clearance. The absorption of ingested arsenic will depend
on the solubility of the compound in question. Gastrointestinal
absorption will also depend on whether the arsenic compound is given
in solution or as undissolved particles.
6.1.1.2.1 Animals
Trivalent arsenic in the form of arsenic(III) oxide, suspended in
a gum solution was administered to rabbits and rats in single doses of
22 mg of arsenic per kg body weight. The recovery of arsenic in the
faeces in the 4 days following dosing was 59% for the rabbits and 69%
for the rats (Ariyoshi & Ikeda, 1974). Arsenic(III) oxide dissolved in
water and mixed in the food was administered to rats by Coulson et al.
(1935). Of the calculated average intake of arsenic (0.37 mg), only
about 14% was eliminated with faeces during the first 3 days. The
results of these 2 studies indicate that dissolved arsenic(III) oxide
is more rapidly absorbed than undissolved arsenic(III) oxide. However,
the differences in dose might have contributed to the differences in
absorption.
Pigs given 0.3 mg of arsenic per kg body weight as arsenic(III)
oxide with pig chow eliminated about 10% of this single dose with the
faeces during 10 days (Munro et al., 1974). Adult female Cynomolgus
monkeys given a single dose of arsenic(III) oxide (1 mg As/kg body
weight) by stomach tube eliminated only about 2% of the dose with the
faeces during 14 days indicating that essentially all the administered
dose had been absorbed from the gastrointestinal tract (Charbonneau et
al., 1978a).
Absorption of a water solution of arsenic(III) oxide infused into
a ligated loop of the ileocaecal part of the intestine of rabbits was
studied by Tsutsumi & Nozaki (1975). They found that about 30% of the
infused arsenic (total 15 mg As) was absorbed into the blood over 1 h.
Otani (1957) studied the absorption of arsenic(III) oxide
solutions from different parts of the digestive tract in cats and
rats. The concentration of arsenic in blood and tissues was measured
after administration of the arsenic solution (15 mg As/kg body weight)
directly into the mouth, stomach, small intestine, or colon. The
highest absorption took place in the small intestine; absorption from
the mouth and stomach was relatively low.
Dogs given a single dose of pentavalent arsenic in the form of
74As-arsenate (about 0.02 µg As/kg body weight) orally in a gelatin
capsule eliminated less than 5% of the dose with the faeces during the
first week, indicating almost complete absorption from the
gastrointestinal tract (Hollins et al., 1979). In Golden Syrian
hamsters given 74As-arsenic acid orally (0.01 µg As/hamster), as much
as 70% of the dose was recovered in the faeces (Charbonneau et al.,
1980).
The absorption of orally administered 74As-labelled trivalent and
pentavalent arsenic (checked as to valence state at the time of
exposure) has been studied in mice (Vahter & Norin, 1980). The
elimination of arsenic with faeces during the first 48 h was 6-9% of
the dose (0.4 or 4 mg/kg body weight) for both valence forms. As about
the same faecal elimination of arsenic was seen after subcutaneous
administration, the results indicate almost complete initial
absorption from the gastrointestinal tract following oral
administration.
The nature of the daily diet may effect the enteric absorption of
arsenic. Arsenic(III) oxide added to a milk diet (80% whole milk
powder and 20% dextrin) was eliminated with the faeces of rats in
greater amounts, after several weeks of feeding, than the same
substance added to a cereal diet (Tamura et al., 1972). When the
cereal diet of rats was supplemented with casein (20%), in addition to
As(III) oxide, the faeces contents of arsenic were higher than after
supplementation with cheese, butter, or whey powder (Nozaki, 1972). No
differences in the faecal elimination of arsenic were noted in rats
fed arsenic together with cereal and cereal supplemented with 20% egg
albumin, lactalbumin, polypeptone, or polyamine, or with 1%
methionine, taurine, or cysteine (Tamura et al., 1974a,b,c). The doses
in these experiments were very high (500 mg As per kg diet) and may
have injured the gastrointestinal mucosa. In a more recent study by
Tamura et al. (1977), rats were exposed to arsenic(III) oxide in both
milk and cereal diets (75 mg As per kg diet) for 6 months, but no
significant differences in the faecal elimination of arsenic were
observed. It is of interest to note that milk increases enteric
absorption of other metals, such as lead and cadmium (Kello & Kostial,
1973, 1977; Engström & Nordberg, 1978).
Enteric absorption of arsenic(III) oxide from the rabbit
intestine, ligated in the ileocaecal portion, was inhibited by casein
and a polypeptide from hydrolyzed casein with a relative mass of more
than 14 000 (Nozaki et al., 1975). When the 2 substances were examined
by dialyses or gel filtration, binding to arsenic was not detected.
The enteric absorption of arsenic(III) oxide was also inhibited by
phosphoric acid and potassium dihydrogen phosphate.
6.1.1.2.2 Man
As in animals, dissolved trivalent inorganic arsenic is readily
absorbed from the gastrointestinal tract in man. Bettley & O'Shea
(1975) gave 8.5 mg of arsenic as Liquor Arsenicalis (B.P. 1963;
arsenite solution; see section 5.1.5) to 7 patients in a skin ward.
The total amount of arsenic recovered in the faeces over a 10-day
period was, at the most, 3.5% of the total dose implying that by far
the major part of the dose was absorbed. A high absorption of
trivalent arsenic in solution, in man, is also evident from data on
high urinary levels of arsenic (section 6.1.3.2).
In an experiment on himself, Mappes (1977) took a single oral dose
of 12 mg of finely powdered arsenic selenide (equal to 4.8 mg As). The
urinary arsenic level did not increase, indicating that this compound,
which is almost insoluble in water or 0.1 mol/litre hydrochloric acid,
was poorly absorbed from the gastrointestinal tract. A low uptake of
arsenic would also be expected if the relatively poorly soluble
arsenic(III) oxide were ingested in the form of undissolved particles.
The fate of pentavalent arsenic, in the form of 74As-arsenate,
following ingestion by healthy human volunteers was studied by Tam
et al. (1979a). At the end of the 7th day, cumulative elimination with
the faeces equalled 6.1% of the dose.
6.1.1.3 Skin absorption
6.1.1.3.1 Animals
Results of studies by Dutkiewicz (1977) in which rat tails were
immersed in solutions containing different concentrations of sodium
arsenate (As levels of 750, 7500, and 15 000 mg/litre) labelled with
74As, showed that arsenic is absorbed through the intact skin of
rats.
6.1.1.3.2 Man
Human data concerning the uptake of arsenic through the skin are
extremely limited. Robinson (1975) reported a case of systemic
poisoning in a patient whose cheek had been treated with arsenical
paste.
6.1.1.4 Placental transfer
6.1.1.4.1 Animals
Arsenic was detected (no quantitative data given) in newborn rats
of dams given a diet containing 27 or 215 mg of arsenic/kg diet as
arsenic(III) oxide (Morris et al., 1938). Placental transfer of
arsenic has been shown in hamsters intravenously injected with high
doses (20 mg/kg body weight) of sodium arsenate labelled with 74As
(Ferm, 1977) or 4.5 mg of arsenic/kg body weight labelled with 74As
(Hanlon & Ferm, 1977). Examination of tissues, 24 or 96 h after
injection, showed that 74As crossed the placenta during the critical
stage of embryogenesis and entered the embryonic tissues. The arsenic
level in the embryo 24 h after dosing was comparable with that in the
maternal blood, i.e., about 0.05 mg As/kg tissue.
6.1.1.4.2 Man
In studies on tissue levels of arsenic in fetuses and newborn
babies in Japan (Kadowaki, 1960), the total amount of arsenic in the
fetus tended to increase with age (from 4 to 7 months). The origin of
the arsenic in the tissues was not known. It may have been
organoarsenic compounds present in the mother's food as well as
exposure to inorganic arsenic.
Further evidence of placental transfer of arsenic was presented in
a case of arsenic(III) oxide ingestion during the third trimester of
pregnancy (Lugo et al., 1969). A total of about 400 mg (as As) was
taken in a liquid preparation causing the death of the child. On the
fourth day after ingestion, concentrations in the infant of between
0.2 mg As/kg wet weight (brain) and 5.6 mg As/kg wet weight (liver)
were reported. In this case, destruction of normal placental function
by arsenic must also be considered.
In studies on 101 women in 2 southern cities in the USA (Kagey et
al., 1977), cord blood levels of arsenic were about as high as
maternal blood levels.
6.1.2 Distribution in organisms
6.1.2.1 Fate of arsenic in blood
6.1.2.1.1 Animals
It has long been recognized that rats accumulate arsenic in the
blood. The blood levels of arsenic in rats after oral or parenteral
administration of single doses of trivalent or pentavalent inorganic
arsenic have been measured in a considerable number of studies (Hunter
et al., 1942; Ducoff et al., 1948; Lanz et al., 1950; Ariyoshi &
Ikeda, 1974; Cikrt & Bencko, 1974; Klaassen, 1974; Tsutsumi & Kato,
1975; Dutkiewicz, 1977). From some of these studies, it can be
estimated that about half of the dose is accumulated in the blood,
mainly in the red blood cells. The half-life of arsenic, administered
as trivalent or pentavalent inorganic arsenic, in the blood of rats
varied from 60 to 90 days (Lanz et al., 1950; Ariyoshi & Ikeda, 1974).
Accumulation of arsenic in the blood does not occur in other
animals. In mice, guineapigs, rabbits, and monkeys, less than 1% of
the administered dose of trivalent inorganic arsenic (0.1-22 mg As/kg
body weight) was found in the blood, 1-2 days after dosing (Hunter
et al., 1942; Ducoff et al., 1948; Crema, 1955; Ariyoshi & Ikeda,
1974). The work of Crema (1955) indicated a multi-phase clearance of
trivalent arsenic from the blood in mice 1-48 h after intravenous
injection of 76As-arsenic(III) oxide (0.1-0.2 mg As/kg body weight).
It appears that in monkeys, dogs, and rabbits given trivalent arsenic
in the form of arsenite or arsenic(III) chloride, only part of the
blood arsenic is localized in the erythrocytes (Hunter et al., 1942;
Klaassen, 1974), the whole blood arsenic levels being 2-7 times higher
than the plasma levels.
When intramuscular injections of pentavalent arsenic (carrier-free
74As-arsenate oxidized with nitric acid to the pentavalent state)
were given to dogs, rabbits, guineapigs, chicks, and mice, less than
0.3% of the dose was found in the blood 48 h after dosing (Lanz et
al., 1950). The corresponding value for cats was 5.6% Lambs poisoned
with arsenic acid also showed very low arsenic levels in the blood
(Nelson et al., 1971).
Vahter & Norin (1980) examined the distribution of arsenic in the
blood of mice 0.5-24 h after a single oral administration of
74As-labelled arsenic(III) or arsenic(V) in doses of 0.4 mg and
4 mg/kg body weight. The oxidation states were checked at the time of
dosing. At the high dose level, the ratio between the arsenic
concentrations in the erythrocytes and plasma was about 2-3 after
exposure to arsenic(III) but close to 1 for arsenic(V). A similar
tendency was also evident in the low exposure groups. A higher
retention of arsenic in the blood of the arsenic(III)-treated animals
was observed, but because of the rapid elimination of arsenic from the
blood in all groups, this was of minor importance after 24 h.
Radioactive arsenic followed a 3-phase kinetic pattern in its
disappearance from the blood of chickens exposed to 74As-arsenate
(Overby & Frederickson, 1963). The half-times for the first 2 phases
were rapid (3 and 12 h, respectively), while the remaining 74As (less
than 0.1% of the dose) had a biological half-time in blood of 60 h.
The disappearance of 74As from the blood of dogs intravenously
administered carrier-free 74As-arsenic acid has been shown to fit a
3-phase model similar to that for man (Charbonneau et al., 1978b).
Most of the injected 74As left the blood at a very high rate
(half-times of the first 2 phases of 1 and 5 h, respectively), while
the remaining, minor amount was cleared with a half-time of about
35 h. The small amount of arsenic still present in the blood 3 h after
the injection was known to be equally distributed between plasma and
erythrocytes.
6.1.2.1.2 Man
Radioactive arsenic has been used for locating tumours in man.
Mealey et al. (1959) measured the plasma and erythrocyte levels of
radioactive arsenic following intravenous injections of labelled
arsenite. The rate of decline of arsenic in the erythrocytes was
comparable with that in plasma but the red cells contained about 3
times more arsenic than the plasma, 10 h after the injection. The
plasma curve, shown in Fig. 2, fits a 3-compartment model. The first
half-time seems to be very short and the bulk of the arsenic was
removed from the plasma at this high rate. Twenty-four hours after
dosing, less than 0.1% of the dose remained per litre of plasma. The
second phase of the curve shows a half-time of about 30 h, which is
comparable with that calculated from the data of Hunter et al. (1942).
The third phase of the curve, beginning about one week after the
injection, shows a very low rate of disappearance. The half-time can
be estimated to be over 200 h.
Among three healthy subjects, Bergström & Wester (1966) found a
mean arsenic level in serum of 0.0008 mg/litre. The corresponding
value for uraemic patients was 0.023 mg/litre. The arsenic
concentration in these patients decreased considerably following
dialysis, indicating that arsenic is not firmly bound to the high
relative molecular mass serum proteins. A much smaller decrease was
found in whole blood, indicating that arsenic is only slowly released
from the cells.
5. LEVELS OF EXPOSURE TO ARSENIC AND ITS COMPOUNDS
Identification of the form in which arsenic occurs has only
recently become part of the determination of arsenic in various
environmental media. Generally, only total arsenic concentrations have
been measured. In several reports however, the concentration of
arsenic has been expressed as arsenic(III) oxide, even though the
exact nature of the compound has not been determined. An attempt has
been made in this section to distinguish between the various forms of
arsenic, where sufficient knowledge exists. Unless specifically noted,
the concentrations given in this section refer to elemental arsenic.
The levels should be considered tentative as, in most instances, the
accuracy of the analytical methods has not been assured.
5.1 General Population Exposure through Air, Drinking-Water,
Food, and Beverages
5.1.1 Air
From data on air concentrations of arsenic in unpolluted areas
(section 3.1.2), it can be calculated that the amount of arsenic
inhaled per day is about 0.05 µg or less (assuming that about 20 m3
of air is inhaled per day). However, in areas where coal with a high
arsenic content is used in power plants, or in the vicinity of
smelters, the intake of arsenic may be considerably higher. Airborne
arsenic levels of about 1 µg/m3, have been detected in such areas,
(section 3.2.3), which would result in the inhalation of approximately
20 µg of arsenic per day.
The amount of arsenic absorbed from the lungs depends on particle
size and the chemical form of the arsenic. Analysis of arsenic in
airborne fly ash from coal-fired power plants indicated that the
highest concentration was associated with respirable particles. On a
mass basis, 76% of the arsenic present was recovered from particles
with a diameter of less than 7.3 µm (Natusch et al., 1974).
5.1.2 Drinking-water
The natural concentration of total arsenic in drinking-water
varies in different parts of the world. McCabe et al. (1970)
investigated more than 18 000 community water supplies in the USA and
found that less than 1% had arsenic levels exceeding 0.01 mg/litre. In
a report by Grantham & Jones (1977) on arsenic concentrations in water
from more than 800 wells in Nova Scotia, Canada, 13% had arsenic
levels exceeding 0.05 mg/litre. Apparently, some of these wells had
been contaminated by gold-mining activities in previous years. In some
areas where chronic arsenic poisoning has occurred, levels exceeding
1 mg/litre have been recorded in well water. In the region of Cordoba,
Argentina, Arguello et al. (1938) reported maximum levels of arsenic
of between 0.9 and 3.4 mg/litre. Artesian well water in the Tainan
county of the Province of Taiwan contained up to 1.8 mg/litre (Kuo,
1968). Well waters in Oregon also contained elevated levels of arsenic
(0.07-1.7 mg/litre) (Goldblatt, et al., 1963).
Drinking-water can be severely contaminated through industrial
operations. In the city of Torreon, Mexico, Espinosa González (1963)
reported that levels of arsenic in drinking-water from a deep well
ranged from 4 to 6 mg/litre. In Niigata, Japan, waste water from a
factory producing arsenic sulfide contaminated nearby well water, and
arsenic levels up to 3 mg/litre were recorded (Terada, 1960). Leaching
of arsenic from coal preparation wastes and fly ash from coal-fired
power plants may also result in the contamination of water (Williams,
et al., 1977; Chu, et al., 1978).
When considering exposure through drinking water, it is important
to ensure that exposures are assessed for water delivered from the
consumer's tap. Conventional flocculation treatment using either
aluminum or ferric salts removes a high proportion, at least, of
arsenic(V) (Gulledge & O'Connor, 1973).
5.1.3 Food and beverages
Arsenic levels in food, with the exception of some seafoods, are
generally well below 1 mg/kg wet weight (Westöö & Rydälv, 1974).
Marine fish on an average contain below 5 mg/kg wet weight (LeBlanc &
Jackson, 1973; Lunde, 1973b; Leatherland & Burton, 1974; Kennedy,
1976; Stoeppler & Mohl, 1980). Certain bottom feeding fish, crustacea
and shellfish may contain arsenic concentrations of several tens of
milligrams per kilo (Westöö & Rydälv, 1972; Crecelius, 1974; Munro
et al., 1974). Arsenic concentrations of between 0.6 and 58 mg/kg dry
weight, have been found in some food supplements prepared from kelp
(Walkiw & Douglas, 1975). Edible seaweed, a common product in Japan,
has been reported to contain arsenic levels ranging from 19 to
172 mg/kg dry weight with a mean concentration of 112 mg/kg (Watanabe
et al., 1979). The use of some organic arsenic compounds as feed
additives for poultry and swine may lead to accumulation of arsenic in
certain organs (Ledet et al., 1973; Calvert, 1975) (section 6.2.2.2)
and limits of tolerance have been established in the USA for edible
by-products from chickens, turkeys, and swine (Jelinek & Corneliussen,
1977).
Most of the arsenic in marine organisms occurs in the form of
either fat-soluble or water-soluble organoarsenic compounds (Lunde,
1975). The water-soluble compounds are characterized by high chemical
stability. Lunde (1973b) separated inorganic and organic arsenic in
some fish and crustacea from the Norwegian Atlantic coast. The
concentrations of inorganic arsenic (including organic-bound arsenic
degradable by 6.6 M hydrochloric acid) ranged from 1.0 to 2.5 mg/kg
and those of organoarsenic compounds from 3 to 37 mg/kg. Seafood
arsenic, i.e., the major organic arsenic compounds found in seafood,
is not degradable by this treatment. Crecelius (1977b) did not find
any increase in human urinary excretion of inorganic or of simple
methylated arsenic compounds, i.e., methylarsenic acid and
dimethylarsenic acid, following the ingestion of 2 mg of arsenic in
crab meat. This indicated that the inorganic arsenic content of the
crab meat was very low (<1% of the arsenic).
Wine may contain appreciable amounts of arsenic. Noble et al.
(1976) found concentrations between 0.02 and 0.11 mg/litre in 9 US
wines produced between 1949 and 1974. Crecelius (1977a) also
investigated the levels and forms of arsenic in some US table wines.
In over half of the samples, levels greatly exceeded 0.05 mg/litre
(tentative limit in the international drinking water standards
published by WHO). Most of the arsenic present was in the trivalent
form. Arsenate was also found, but no methylated species were
detected. This study indicated that considerable reduction from
arsenate to arsenite occurred during the fermentation of grape juice
by wine yeast. It is probable that the arsenic in the wines originated
mainly from the arsenic-containing insecticides used on the grapes.
Elevated arsenic levels have been found in some bottled mineral
waters. Zoeteman & Brinkmann (1976) reported a mean arsenic
concentration of 0.021 mg/litre (range <0.001-0.19 mg/litre) in
bottled mineral waters sold in countries within the European
Community. In an investigation on lager beers from various countries,
none of the samples contained more than 0.02 mg/litre (Binns et al.,
1978).
5.1.4 Tobacco
The content of arsenic in tobacco grown on soils not treated with
arsenic compounds is usually below 3 mg/kga a (Satterlee, 1956;
Bailey et al., 1957; Hjern, 1961; Griffin et al., 1975). During the
first part of this century, the use of arsenic insecticides, mainly in
the USA, brought about a steady increase in the content of arsenic in
a The weight of a cigarette is approximately 1 g.
tobacco products. In the 1950s, levels of up to 52 mg/kg, given as
As(III) oxide (40 mg As/kg) were found in American cigarettes (Holland
& Acevedo, 1966). However, during the last 20 years the concentrations
of arsenic have decreased to below 8 mg/kg, because of a great
reduction in the use of inorganic arsenic compounds in agriculture. Of
the total arsenic originally present in cigarettes, 10-15% was
recovered in the main stream smoke, the remainder mainly being
distributed in the ash and butt (Thomas & Collier, 1945). Cigarettes
in Japan have been reported to contain arsenic levels of less than
1 mg/kg (Maruyama et al., 1970). The chemical form of arsenic in the
smoke has yet to be elucidated.
5.1.5 Drugs
Both inorganic and organic arsenic compounds have been widely used
in medicine. Arsenical Solution, also called Liquor Arsenicalis,
Solutio Kalii Arsenitis or Fowler's Solution, contained arsenic(III)
oxide dissolved in potassium hydroxide, neutralized with hydrochloric
acid and diluted with chloroform water (Martindale, 1977). The arsenic
administered was thus in the form of arsenite. The drug ordinarily
contained an arsenic concentration of 7.6 g/litre and the daily dose
of arsenic was sometimes as high as 10 mg (Pearson & Ponds, 1971). It
was used for the treatment of leukaemia, psoriasis, chronic bronchial
asthma, and as a tonic. Other preparations described in the Extra
Pharmacopoeia by Martindale (1977) include various pastes containing
inorganic arsenic in combination with other drugs, such as cocaine or
procaine. Sodium arsenate was formerly used in the treatment of
chronic skin diseases, some parasitic diseases, and anaemia
(Martindale, 1977). Pearson's Arsenical Solution, which contained
about 0.5% arsenic in the form of arsenate, has been included in
several pharmacopoeias. The recommended dose was 1-10 mg of the
arsenate (0.2-2.4 mg As) with a maximum of 20 mg in 24 h. Drugs
containing inorganic arsenic compounds are being phased out and
replaced by more effective and less toxic drugs.
Salvarsan (arsphenamine), an organic arsenic compound containing
32% arsenic, was formerly used in the treatment of syphilis
(Martindale, 1977). Because of the difficulties in preparing it for
injection and because of its high toxicity, it was replaced by
neoarsphenamine. The recommended dose used to be 100-600 mg (32-192 mg
As) administered intravenously. Antibiotics have finally replaced
these drugs. Some organic arsenic compounds including carbarsone,
melarsoprol, and tryparsamide, are still in use in human medicine,
mainly as antiparasitic drugs.
5.1.6 Total daily intake in the general population
Daily intake of arsenic from ambient air and water will ordinarily
be of the order of a few micrograms, predominantly in the inorganic
form (section 5.1.1 and 5.1.2).
As mentioned previously, the total daily dietary intake of arsenic
depends, to a great extent, on the amount of seafood in the diet. A
seafood meal may lead to the ingestion of several milligrams of
arsenic, predominantly in organic forms. The daily intake of total
arsenic in Japan has been reported to be between 0.07 and 0.17 mg
(Nakao, 1960). The US Food and Drug Administration has monitored
arsenic in foodstuffs since 1967 (Jelinek & Corneliussen, 1977). Data
from this programme indicate that the total daily intake of arsenic
has decreased from about 0.05-0.1 mg per day in the late sixties to
0.01-0.02 mg per day in 1972-74. Most of the arsenic was found in the
group "meat, fish, and poultry". From analysis of composites of food
representing the Canadian diet during 1970-73, it was estimated that
the total intake of arsenic was 0.025-0.035 mg daily (Smith et al.,
1972, 1973, 1975). Hamilton & Minski (1973) estimated the total intake
of arsenic in the United Kingdom to be about 0.1 mg/day, based on
analysis of diets containing fish. The considerable variations in the
estimated dietary arsenic intake can be expected because of
differences in the amounts of seafood in the diets investigated.
Moreover, in neither of the reports was a distinction made between the
amount of inorganic and organic arsenic consumed. Because of
differences in metabolism and toxicity (sections 6, 7, and 8), it is
important to distinguish between inorganic and organic forms of
arsenic.
During the 1950s, the smoking of some tobacco, especially from the
US, may have led to inhalation of more than 0.1 mg of arsenic daily.
At present, the arsenic content of most tobacco is much lower and it
can be estimated that less than 0.02 mg may be inhaled by an average
smoker.
Data on the urinary excretion of various arsenic compounds in
individuals not excessively exposed to arsenic can be helpful for
deducing daily intake figures. Inorganic arsenic will be excreted
mainly as inorganic and simple methylated arsenic compounds
(Crecelius, 1977b). Smith et al. (1977) found an average urinary
concentration of these forms of arsenic of 17.5 µg/litre in 41 male
workers in the USA without known occupational exposure to arsenic.
This would correspond to an intake of 0.025-0.040 mg of inorganic
arsenic per day.
5.2 Occupational Exposure
Occupational exposure to arsenic compounds takes place mainly
among workers, especially those involved in the processing of copper,
gold, and lead ores. Occupational exposure may also occur among
workers using or producing arsenic-containing pesticides.
Unfortunately, very few data exist on the actual air levels of arsenic
to which persons in such occupations have been exposed. This is also
the case for wood treatment plant workers and carpenters, who may
become exposed to inorganic arsenic compounds (mainly pentavalent)
through their use as wood preservatives (section 3.2.2).
In a plant where sodium arsenite was being manufactured, Perry et
al. (1948) found mean air arsenic concentrations of between 0.078 and
1.034 mg/m3 around various workstations during sampling times of
"10 minutes or more". The respirable fraction (< 5 µm) of the airborne
arsenic ranged from 20% to 38% by mass. Ott et al. (1974) reported
airborne arsenic levels in 1943 of 0.18 to 18 mg/m3 in the packaging
department of a plant where lead arsenate and calcium arsenate
insecticides were produced. In 1952, airborne arsenic levels ranged
between 0.26 and 40.8 mg/m3 in another workplace at the plant. In the
workroom air of a factory producing lead arsenate, Horiguchi et al.
(1976) found levels ranging from 0.01 to 0.9 mg/m3 during the years
1959-70.
When the airborne arsenic in a Swedish copper smelter was
measured, the average concentrations near the roasters, reverberatory
furnaces, and in the converter hall ranged between 0.06 and 2 mg/m3
during sampling times of "several hours" (Lundgren, 1954). No data
were given on the size distribution of the airborne arsenic-containing
particles. At the same Swedish copper smelter Carlsson (1976) found
weighted 8-h average concentrations at different workplaces of between
0.002 mg and 0.23 mg/m3 in the air inhaled by the workers (i.e.,
after filtration in a respirator). The highest exposures were found
among the roasterworkers. Kodama et al. (1976) measured airborne
arsenic concentrations in a copper refinery, where arsenic(III) oxide
was being manufactured. They found levels of between 0.006 and
0.012 mg/m3 when the ventilation was normal, and up to 0.2 mg/m3
when the ventilation was shut off. Around the furnaces in the copper
smelter, average concentrations of between 0.001 and 0.012 mg/m3 were
reported, and around the furnaces in a ferronickel smelter, the
corresponding concentrations were between 0.002 and 0.005 mg/m3.
Smith et al. (1977) described a study at a US copper smelter where
airborne particulate matter was collected in personal exposure
samplers. The concentrations were found to be log-normally
distributed, with a geometric mean of 0.053 mg/m3 in a high exposure
group (i.e., workers in the baghouse, flue, cotterell, stack, and
reverberatory furnace areas). Workers in the concerter area were
exposed to 0.046 mg/m3 (geometric mean). In the high exposure area,
only 32% of the airborne arsenic was respirable (< 5 µm), compared
with over 80% in the converter area. Pinto et al. (1976) reported an
overall mean airborne arsenic concentration of 0.05 mg/m3 (range
0.003-0.3 mg/m3) in the working environment of 24 smelter workers
wearing personal air samplers on 5 consecutive days.
Airborne arsenic particulate matter in smelters is generally
assumed to consist primarily of arsenic(III) oxide. However, it is
probable that some of the arsenic is firmly bound to other metals,
especially in the reverberatory furnace. There is also evidence of the
presence of arsenic sulfides (Smith et al., 1976). The form in which
arsenic is present clearly depends, to a great extent, on the
characteristics of the industrial process involved, such as the
temperature, humidity, and other elements present. More work is
urgently needed to characterize the arsenic compounds by form and size
distribution.
Workers may be exposed to airborne arsenic in cutting and sawing
operations on wood treated with arsenic-containing preservatives.
Arsenault (1977) found concentrations of arsenic in air of 0.043-
0.36 mg/m3 originating from the sawing of wood treated with copper,
chromium, and arsenic salts. The duration of measurement was 100 min.
Only about 5% of the dust particles (on a mass basis) were less than
10 µm.
6. METABOLISM OF ARSENIC
The metabolism of arsenic in man is very complex since the fate of
arsenic compounds in the human body varies with the type of compound.
The metabolism of a compound also varies with animal species, for
example, the metabolism of arsenic in the rat is unique and quite
different from that in man or other mammals. The rat is therefore not
a suitable model for most metabolic pathways in man and the emphasis
in this document has been placed, as much as possible, on data
concerning other experimental animals.
6.1 Inorganic Arsenic
The metabolism of inorganic arsenic depends on its chemical form.
Possible changes in the different forms of inorganic arsenic before
the time of exposure should be considered. Even commercially available
isotopes of pentavalent arsenic have been shown to contain up to 98%
of trivalent arsenic (Lunde, 1973a; Reay & Asher, 1977). In many
studies on the metabolism of arsenic, the valence of the compound used
has not been under complete control. Throughout the following
description, an attempt has been made to assess the validity of
information concerning valence. In cases where the valence has been
checked before exposure, this has been stated. If no such statements
are made, it must be appreciated that substantial uncertainly exists.
6.1.1 Absorption
6.1.1.1 Respiratory deposition and absorption
Human exposure to inorganic arsenic through inhalation usually
occurs occupationally or during cigarette smoking. Information on the
respiratory deposition and clearance of different inorganic arsenic
compounds is very limited. Inhaled arsenic is mainly in the form of an
aerosol and it can be assumed that its deposition is the same as that
of other particulate matter. In many workplaces, the particles
containing arsenic are of relatively large size (Perry et al., 1948;
Pinto & McGill, 1953), resulting in deposition primarily in the upper
respiratory passages (i.e., nasal cavity, nasopharynx, larynx,
trachea, and bronchus). Subsequent absorption can then take place
either directly from the respiratory tract or gastrointestinally after
mucociliary clearance in the airways. Retention, deposition, and
absorption from the respiratory tract depend, furthermore, on the
solubility of the inhaled material.
6.1.1.1.1 Animals
Hairless mice exposed for several weeks to a solid aerosol of fly
ash (particle size less than 10 µm) containing arsenic at a
concentration of 0.18 mg/m3 showed increased tissue levels of arsenic
(Bencko & Symon, 1970). Hairless mice were used to minimize oral
intake of arsenic deposited on the fur. Despite this, it was not
possible to differentiate between the amount of arsenic absorbed after
inhalation and that absorbed after ingestion. A similar study has also
been performed on rats exposed to arsenic(III) oxide in the form of
condensation aerosols (arsenic concentrations of 0.001, 0.0037 and
0.046 mg/m3) for 3 months (Rozenshtein, 1970). Increased tissue
levels were found in the groups exposed to the 2 highest
concentrations. It was not possible to differentiate between inhaled
and ingested arsenic.
Rapid absorption of arsenic in rats following intratracheal
administration of a solution of sodium arsenate (0.1-4 mg As per kg
body weight) labelled with 74As has been reported by Dutkiewicz
(1977). The rapidity of the absorption was indicated by the relatively
high tissue levels (2.5 and 0.7% of the dose per gram tissue in liver
and spleen, respectively) found one hour after the administration.
6.1.1.1.2 Man
Holland et al. (1959) studied the uptake of inorganic arsenic in 8
terminal lung cancer patients who volunteered to smoke cigarettes
impregnated with 74As-labelled arsenic, reported to be in the form of
sodium arsenite. Between 5% and 8% of the arsenic originally present
in the cigarettes was deposited in the thoracic region. In 2 other
lung cancer patients inhaling a nebulized solution of 74As-arsenite,
the fractions of radioactive arsenic deposited were 32% and 62%,
respectively. Clearance from the lungs seemed to be fast. Four days
after exposure, only about 20% of the dose could be detected by
external scanning of the thoracic region. Since the study was
performed on terminal lung cancer patients, great care must be
exercised in extrapolating these data to healthy human subjects.
Some information on the absorption of arsenic following inhalation
is given in reports on urinary excretion among persons exposed to
arsenic occupationally. Pinto et al. (1976) studied the excretion of
arsenic in 24 workers exposed regularly to airborne arsenic in a
copper smelter (section 7.2). The workers wore personal air samplers
for 5 consecutive working days and the overall average concentration
of airborne arsenic was 0.053 mg/m3. Urine was collected for 2 days
prior to the working week, each day during the working week, and for 3
days afterwards. A correlation was found between urinary arsenic
levels and average airborne arsenic concentrations over the ranges
studied. This investigation seems to indicate a fair absorption of
inhaled arsenic, although no precise estimations can be made.
Arsenic-containing dust which enters the body orally may be
present in the gastrointestinal tract together with the arsenic
transported by mucociliary clearance from the respiratory tract. In
workers exposed to dust (cadmium and nickel dust) in a battery
factory, Adamsson et al. (1979) recently showed that, in some cases,
the amount of cadmium in the faeces was more than 10 times the amount
that could have been inhaled.
6.1.1.2 Gastrointestinal absorption
Absorption of inorganic arsenic from the gastrointestinal tract
can occur following the ingestion of food, water, beverages, or drugs,
containing arsenic or as a result of inhalation and subsequent
mucociliary clearance. The absorption of ingested arsenic will depend
on the solubility of the compound in question. Gastrointestinal
absorption will also depend on whether the arsenic compound is given
in solution or as undissolved particles.
6.1.1.2.1 Animals
Trivalent arsenic in the form of arsenic(III) oxide, suspended in
a gum solution was administered to rabbits and rats in single doses of
22 mg of arsenic per kg body weight. The recovery of arsenic in the
faeces in the 4 days following dosing was 59% for the rabbits and 69%
for the rats (Ariyoshi & Ikeda, 1974). Arsenic(III) oxide dissolved in
water and mixed in the food was administered to rats by Coulson et al.
(1935). Of the calculated average intake of arsenic (0.37 mg), only
about 14% was eliminated with faeces during the first 3 days. The
results of these 2 studies indicate that dissolved arsenic(III) oxide
is more rapidly absorbed than undissolved arsenic(III) oxide. However,
the differences in dose might have contributed to the differences in
absorption.
Pigs given 0.3 mg of arsenic per kg body weight as arsenic(III)
oxide with pig chow eliminated about 10% of this single dose with the
faeces during 10 days (Munro et al., 1974). Adult female Cynomolgus
monkeys given a single dose of arsenic(III) oxide (1 mg As/kg body
weight) by stomach tube eliminated only about 2% of the dose with the
faeces during 14 days indicating that essentially all the administered
dose had been absorbed from the gastrointestinal tract (Charbonneau et
al., 1978a).
Absorption of a water solution of arsenic(III) oxide infused into
a ligated loop of the ileocaecal part of the intestine of rabbits was
studied by Tsutsumi & Nozaki (1975). They found that about 30% of the
infused arsenic (total 15 mg As) was absorbed into the blood over 1 h.
Otani (1957) studied the absorption of arsenic(III) oxide
solutions from different parts of the digestive tract in cats and
rats. The concentration of arsenic in blood and tissues was measured
after administration of the arsenic solution (15 mg As/kg body weight)
directly into the mouth, stomach, small intestine, or colon. The
highest absorption took place in the small intestine; absorption from
the mouth and stomach was relatively low.
Dogs given a single dose of pentavalent arsenic in the form of
74As-arsenate (about 0.02 µg As/kg body weight) orally in a gelatin
capsule eliminated less than 5% of the dose with the faeces during the
first week, indicating almost complete absorption from the
gastrointestinal tract (Hollins et al., 1979). In Golden Syrian
hamsters given 74As-arsenic acid orally (0.01 µg As/hamster), as much
as 70% of the dose was recovered in the faeces (Charbonneau et al.,
1980).
The absorption of orally administered 74As-labelled trivalent and
pentavalent arsenic (checked as to valence state at the time of
exposure) has been studied in mice (Vahter & Norin, 1980). The
elimination of arsenic with faeces during the first 48 h was 6-9% of
the dose (0.4 or 4 mg/kg body weight) for both valence forms. As about
the same faecal elimination of arsenic was seen after subcutaneous
administration, the results indicate almost complete initial
absorption from the gastrointestinal tract following oral
administration.
The nature of the daily diet may effect the enteric absorption of
arsenic. Arsenic(III) oxide added to a milk diet (80% whole milk
powder and 20% dextrin) was eliminated with the faeces of rats in
greater amounts, after several weeks of feeding, than the same
substance added to a cereal diet (Tamura et al., 1972). When the
cereal diet of rats was supplemented with casein (20%), in addition to
As(III) oxide, the faeces contents of arsenic were higher than after
supplementation with cheese, butter, or whey powder (Nozaki, 1972). No
differences in the faecal elimination of arsenic were noted in rats
fed arsenic together with cereal and cereal supplemented with 20% egg
albumin, lactalbumin, polypeptone, or polyamine, or with 1%
methionine, taurine, or cysteine (Tamura et al., 1974a,b,c). The doses
in these experiments were very high (500 mg As per kg diet) and may
have injured the gastrointestinal mucosa. In a more recent study by
Tamura et al. (1977), rats were exposed to arsenic(III) oxide in both
milk and cereal diets (75 mg As per kg diet) for 6 months, but no
significant differences in the faecal elimination of arsenic were
observed. It is of interest to note that milk increases enteric
absorption of other metals, such as lead and cadmium (Kello & Kostial,
1973, 1977; Engström & Nordberg, 1978).
Enteric absorption of arsenic(III) oxide from the rabbit
intestine, ligated in the ileocaecal portion, was inhibited by casein
and a polypeptide from hydrolyzed casein with a relative mass of more
than 14 000 (Nozaki et al., 1975). When the 2 substances were examined
by dialyses or gel filtration, binding to arsenic was not detected.
The enteric absorption of arsenic(III) oxide was also inhibited by
phosphoric acid and potassium dihydrogen phosphate.
6.1.1.2.2 Man
As in animals, dissolved trivalent inorganic arsenic is readily
absorbed from the gastrointestinal tract in man. Bettley & O'Shea
(1975) gave 8.5 mg of arsenic as Liquor Arsenicalis (B.P. 1963;
arsenite solution; see section 5.1.5) to 7 patients in a skin ward.
The total amount of arsenic recovered in the faeces over a 10-day
period was, at the most, 3.5% of the total dose implying that by far
the major part of the dose was absorbed. A high absorption of
trivalent arsenic in solution, in man, is also evident from data on
high urinary levels of arsenic (section 6.1.3.2).
In an experiment on himself, Mappes (1977) took a single oral dose
of 12 mg of finely powdered arsenic selenide (equal to 4.8 mg As). The
urinary arsenic level did not increase, indicating that this compound,
which is almost insoluble in water or 0.1 mol/litre hydrochloric acid,
was poorly absorbed from the gastrointestinal tract. A low uptake of
arsenic would also be expected if the relatively poorly soluble
arsenic(III) oxide were ingested in the form of undissolved particles.
The fate of pentavalent arsenic, in the form of 74As-arsenate,
following ingestion by healthy human volunteers was studied by Tam
et al. (1979a). At the end of the 7th day, cumulative elimination with
the faeces equalled 6.1% of the dose.
6.1.1.3 Skin absorption
6.1.1.3.1 Animals
Results of studies by Dutkiewicz (1977) in which rat tails were
immersed in solutions containing different concentrations of sodium
arsenate (As levels of 750, 7500, and 15 000 mg/litre) labelled with
74As, showed that arsenic is absorbed through the intact skin of
rats.
6.1.1.3.2 Man
Human data concerning the uptake of arsenic through the skin are
extremely limited. Robinson (1975) reported a case of systemic
poisoning in a patient whose cheek had been treated with arsenical
paste.
6.1.1.4 Placental transfer
6.1.1.4.1 Animals
Arsenic was detected (no quantitative data given) in newborn rats
of dams given a diet containing 27 or 215 mg of arsenic/kg diet as
arsenic(III) oxide (Morris et al., 1938). Placental transfer of
arsenic has been shown in hamsters intravenously injected with high
doses (20 mg/kg body weight) of sodium arsenate labelled with 74As
(Ferm, 1977) or 4.5 mg of arsenic/kg body weight labelled with 74As
(Hanlon & Ferm, 1977). Examination of tissues, 24 or 96 h after
injection, showed that 74As crossed the placenta during the critical
stage of embryogenesis and entered the embryonic tissues. The arsenic
level in the embryo 24 h after dosing was comparable with that in the
maternal blood, i.e., about 0.05 mg As/kg tissue.
6.1.1.4.2 Man
In studies on tissue levels of arsenic in fetuses and newborn
babies in Japan (Kadowaki, 1960), the total amount of arsenic in the
fetus tended to increase with age (from 4 to 7 months). The origin of
the arsenic in the tissues was not known. It may have been
organoarsenic compounds present in the mother's food as well as
exposure to inorganic arsenic.
Further evidence of placental transfer of arsenic was presented in
a case of arsenic(III) oxide ingestion during the third trimester of
pregnancy (Lugo et al., 1969). A total of about 400 mg (as As) was
taken in a liquid preparation causing the death of the child. On the
fourth day after ingestion, concentrations in the infant of between
0.2 mg As/kg wet weight (brain) and 5.6 mg As/kg wet weight (liver)
were reported. In this case, destruction of normal placental function
by arsenic must also be considered.
In studies on 101 women in 2 southern cities in the USA (Kagey et
al., 1977), cord blood levels of arsenic were about as high as
maternal blood levels.
6.1.2 Distribution in organisms
6.1.2.1 Fate of arsenic in blood
6.1.2.1.1 Animals
It has long been recognized that rats accumulate arsenic in the
blood. The blood levels of arsenic in rats after oral or parenteral
administration of single doses of trivalent or pentavalent inorganic
arsenic have been measured in a considerable number of studies (Hunter
et al., 1942; Ducoff et al., 1948; Lanz et al., 1950; Ariyoshi &
Ikeda, 1974; Cikrt & Bencko, 1974; Klaassen, 1974; Tsutsumi & Kato,
1975; Dutkiewicz, 1977). From some of these studies, it can be
estimated that about half of the dose is accumulated in the blood,
mainly in the red blood cells. The half-life of arsenic, administered
as trivalent or pentavalent inorganic arsenic, in the blood of rats
varied from 60 to 90 days (Lanz et al., 1950; Ariyoshi & Ikeda, 1974).
Accumulation of arsenic in the blood does not occur in other
animals. In mice, guineapigs, rabbits, and monkeys, less than 1% of
the administered dose of trivalent inorganic arsenic (0.1-22 mg As/kg
body weight) was found in the blood, 1-2 days after dosing (Hunter
et al., 1942; Ducoff et al., 1948; Crema, 1955; Ariyoshi & Ikeda,
1974). The work of Crema (1955) indicated a multi-phase clearance of
trivalent arsenic from the blood in mice 1-48 h after intravenous
injection of 76As-arsenic(III) oxide (0.1-0.2 mg As/kg body weight).
It appears that in monkeys, dogs, and rabbits given trivalent arsenic
in the form of arsenite or arsenic(III) chloride, only part of the
blood arsenic is localized in the erythrocytes (Hunter et al., 1942;
Klaassen, 1974), the whole blood arsenic levels being 2-7 times higher
than the plasma levels.
When intramuscular injections of pentavalent arsenic (carrier-free
74As-arsenate oxidized with nitric acid to the pentavalent state)
were given to dogs, rabbits, guineapigs, chicks, and mice, less than
0.3% of the dose was found in the blood 48 h after dosing (Lanz et
al., 1950). The corresponding value for cats was 5.6% Lambs poisoned
with arsenic acid also showed very low arsenic levels in the blood
(Nelson et al., 1971).
Vahter & Norin (1980) examined the distribution of arsenic in the
blood of mice 0.5-24 h after a single oral administration of
74As-labelled arsenic(III) or arsenic(V) in doses of 0.4 mg and
4 mg/kg body weight. The oxidation states were checked at the time of
dosing. At the high dose level, the ratio between the arsenic
concentrations in the erythrocytes and plasma was about 2-3 after
exposure to arsenic(III) but close to 1 for arsenic(V). A similar
tendency was also evident in the low exposure groups. A higher
retention of arsenic in the blood of the arsenic(III)-treated animals
was observed, but because of the rapid elimination of arsenic from the
blood in all groups, this was of minor importance after 24 h.
Radioactive arsenic followed a 3-phase kinetic pattern in its
disappearance from the blood of chickens exposed to 74As-arsenate
(Overby & Frederickson, 1963). The half-times for the first 2 phases
were rapid (3 and 12 h, respectively), while the remaining 74As (less
than 0.1% of the dose) had a biological half-time in blood of 60 h.
The disappearance of 74As from the blood of dogs intravenously
administered carrier-free 74As-arsenic acid has been shown to fit a
3-phase model similar to that for man (Charbonneau et al., 1978b).
Most of the injected 74As left the blood at a very high rate
(half-times of the first 2 phases of 1 and 5 h, respectively), while
the remaining, minor amount was cleared with a half-time of about
35 h. The small amount of arsenic still present in the blood 3 h after
the injection was known to be equally distributed between plasma and
erythrocytes.
6.1.2.1.2 Man
Radioactive arsenic has been used for locating tumours in man.
Mealey et al. (1959) measured the plasma and erythrocyte levels of
radioactive arsenic following intravenous injections of labelled
arsenite. The rate of decline of arsenic in the erythrocytes was
comparable with that in plasma but the red cells contained about 3
times more arsenic than the plasma, 10 h after the injection. The
plasma curve, shown in Fig. 2, fits a 3-compartment model. The first
half-time seems to be very short and the bulk of the arsenic was
removed from the plasma at this high rate. Twenty-four hours after
dosing, less than 0.1% of the dose remained per litre of plasma. The
second phase of the curve shows a half-time of about 30 h, which is
comparable with that calculated from the data of Hunter et al. (1942).
The third phase of the curve, beginning about one week after the
injection, shows a very low rate of disappearance. The half-time can
be estimated to be over 200 h.
Among three healthy subjects, Bergström & Wester (1966) found a
mean arsenic level in serum of 0.0008 mg/litre. The corresponding
value for uraemic patients was 0.023 mg/litre. The arsenic
concentration in these patients decreased considerably following
dialysis, indicating that arsenic is not firmly bound to the high
relative molecular mass serum proteins. A much smaller decrease was
found in whole blood, indicating that arsenic is only slowly released
from the cells.
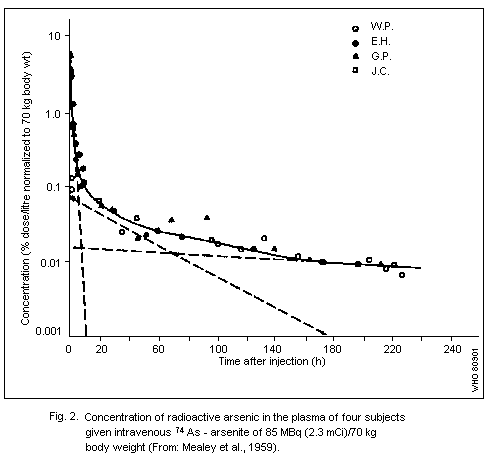 6.1.2.2 Tissue distribution
6.1.2.2.1 Animals
Mice, rabbits, guineapigs, hamsters, chickens, and monkeys given
radiolabelled arsenic in the trivalent form in parenteral doses of
0.1-4 mg of arsenic/kg body weight displayed highest levels of arsenic
in the liver, kidney, skin, lung, and spleen (Hunter et al., 1942;
Ducoff et al., 1948; Crema, 1955; Cikrt et al., 1980). The tissue
distribution of radioactive arsenic 10 min, and 1 h, 6 h, and 48 h
after intravenous administration of 76As-arsenic(III) oxide
(0.1-0.2 mg As/kg body weight) is shown in Table 4. As can be seen,
the highest activity of 76As per gram of tissue was found in the
liver and kidney of the mice. In most organs, the arsenic levels fell
fairly rapidly with time. In some organs, such as skin, brain, and
skeleton, arsenic levels decreased more slowly. The rate of decrease
of arsenic levels in the skin appeared to be especially slow as high
arsenic levels were still present 48 h after exposure.
Table 4. The distribution of arsenic in mice after intravenous
administration of 76As-arsenic(III) oxide (0.1-0.2 mg As/kg
body weight).a
activity per gram tissue/injected activity
per gram body weight
Organ 10 min 1 h 6 h 48 h
liver 6.1 2.47 0.64 0.10
kidney 3.55 3.90 1.40 0.12
spleen 1.37 1.14 0.48 0.08
blood 0.85 0.48 0.15 0.01
skin 0.56 0.91 0.72 0.30
muscle 0.50 0.62 0.40 0.08
small intestine 2.05 2.50 0.60 0.08
large intestine 0.76 0.68 0.26 0.13
skeleton 0.06 0.09 0.11 0.05
brain 0.002 0.02 0.08 0.01
lung 1.73 1.90 0.45 0.02
a From: Crema (1955).
Marked species differences in the biliary excretion of arsenic
were observed after intravenous administration of 1 mg of arsenic/kg
body weight as 74As-arsenic(III) chloride to rats, rabbits, and dogs
(Klaassen, 1974). The rate of excretion of arsenic into the bile in
rats was 40 times that in rabbits and 800 times that in dogs. Rats as
well as mice and hamsters given trivalent arsenic excreted arsenic
into the bile at a higher rate than when given pentavalent arsenic
(Cikrt & Bencko, 1974; Cikrt et al., 1980; Vahter & Norin, 1980).
Arsenic administered as arsenite or arsenic(III) oxide passes the
blood-brain barrier in mice, guineapigs, rabbits, hamsters, and
monkeys, although the levels found in the brain are low compared with
those in other tissues (Hunter et al., 1942; Ducoff et al., 1948;
Crema, 1955, Peoples, 1964; Vahter & North, 1980).
When arsenite was given to guineapigs, chimpanzees, and baboons,
the bulk of the tissue arsenic was shown to be in the protein fraction
and minor amounts in the acid-soluble and lipid fractions (Lowry et
al., 1942). Spontaneous tumours in mice (mammary carcinoma) did not
show any specific affinity for arsenic (Crema, 1955).
Du Pont et al. (1941) administered pentavalent arsenic in the form
of 76As-labelled sodium arsenate intravenously to rabbits (2 mg
As/rabbit, oxidation of As2S5 with hydrogen peroxide and addition of
sodium hydroxide to form arsenate prior to the dosing) and measured
the distribution at various times after the dosing. The percentage of
the dose per whole organ 1 h after dosing was 16.5% in muscle, 12.2%
in skin-fur, 9.5% in bone, 8.5% in blood serum, 5.9% in kidney, and
5.6% in liver. Intramuscular injections of pentavalent arsenic as
carrier-free 74As-arsenate (0.30-0.44 MBq/kg body weight, 8-12 µCi/kg
body weight) in cats, rabbits, guineapigs, chicks, and mice resulted
in a tissue content of radioactive arsenic of less than 0.2% of the
dose per gram wet weight, 48 h after dosing (Lanz et al., 1950).
Autoradiography of mice given carrier-free 74As-arsenic acid
intravenously showed a high affinity of arsenic for the intestinal
mucosa as well as for the kidney cortex, bone, and hair follicles
(Deak et al., 1976).
From the work of Vahter & Norin (1980), it is possible to compare
the tissue distribution of arsenic following administration of the
trivalent and pentavalent forms in the same animal species.
74As-labelled arsenite and arsenate were both administered to mice in
single oral doses of 0.4 or 4 mg of arsenic/kg body weight.
Concentrations of 74As-labelled arsenic in various organs 0.5-24 h
after dosing are given in Table 5 for both valence forms and dose
levels. Higher levels are seen in most tissues of animals receiving
arsenic(III), especially in the liver and bile and the differences are
more pronounced at the higher dose level. The high retention of
arsenic in the skin seen in the animals receiving arsenic(III) may be
explained by reaction of trivalent arsenic with sulfhydryl groups of
proteins, which are abundant in the skin (section 7.6).
Table 5. Arsenic in organs (µg As/g) of mice 0.5-24 h after single oral administration
of 10 µg As/mouse (0.4 mg/kg body weight) or 100 µg As/mouse (4 mg/kg body weight)
of 74As-labelled arsenate As(V), or arsenite, As(III). The figures represent the
mean for 6 animalsa
Dose level 0.5 h 2 h 6 h 24 h
Organ (mg/kg
body As(V) As(III) As(V) As(III) As(V) As(III) As(V) As(III)
weight)
kidney 0.4 1.17 0.74 0.97 1.01 0.72 0.61 0.03 0.05
4 7.24 7.41 8.73 7.44 4.33 4.37 0.06 0.78b
liver 0.4 0.93 2.02b 0.57 0.92b 0.26 0.31 0.02 0.04b
4 2.71 6.43b 3.52 6.72b 0.92 3.19b 0.04 0.39b
bile 0.4 1.86 3.31 0.51 5.30b 0.34 0.94b -- --
4 4.01 12.3ab 7.18 23.8b 2.16b 15.1ab <0.1 <0.1
brain 0.4 0.01 0.01 0.03 0.04b 0.05 0.06 <0.01 <0.01
4 0.09 0.06 0.54 0.17b 0.39 0.28b <0.01 0.05
skeleton 0.4 0.08 0.07 0.12 0.16 0.18 0.18 0.02 0.01
4 0.62 0.72 1.00 0.97 0.58 0.87 <0.01 0.03
skin 0.4 0.07 0.06 0.10 0.15b 0.11 0.13 0.02 0.06b
4 0.44 0.64b 0.94 1.10 0.37 1.10b 0.04 0.76b
a "From: Vahter & Norin (1980).
b p < 0.05.
c Based on 3 animals.
d Based on 2 animals.
Using considerably lower doses, Sabbioni et al. (1979) did not
find any major differences in tissue distribution, 16 h after
intraperitoneal injection of radiolabelled trivalent and pentavalent
inorganic arsenic in rabbits. Both oxidation states were administered
in doses of 0.5-1 µg or 50 µg of arsenic per animal. Intracellular
distribution was similar after exposure to either form of arsenic in
lung, liver, and kidney, where over 80% of the arsenic was found in
the nuclei and cytosol.
Several attempts have been made to demonstrate adaptation or
tolerance towards arsenic in experimental animals. In 2 studies, where
mice were given drinking water containing arsenic(III) oxide (32 days
at 250 mg As/litre and 256 days at 50 mg As/litre), the maximum
arsenic concentrations in skin and liver were reached on the 16th day
with a marked drop in concentrations (about 15-fold) during the rest
of the long-term experiment (Bencko & Symon, 1969a). Similar results
were obtained in a later experiment where mice were exposed to fly ash
with a mean concentration of arsenic of 180 µg/m3 for 6 h daily, 5
days a week, for up to 6 weeks (Bencko & Symon, 1970). Arsenic levels
in the liver and kidney reached a peak after 2 weeks' exposure, but,
by the 6th week, they were only slightly higher than those in the
unexposed controls. Similar dynamics in arsenic accumulation have been
found in rabbits exposed for up to one year to air near a power plant
emitting arsenic (Bencko et al., 1968) and in dogs given daily oral
doses of arsenious acid (0.2-0.4 mg As/kg body weight) for several
months (Katsura, 1958). The work by Bencko & Symon (1969b) and Bencko
et al. (1973) indicated increased tolerance towards parenterally
administered arsenic (5-18 mg As/kg body weight) in mice pretreated
with arsenite in the drinking water (50 mg As/litre).
6.1.2.2.2 Man
Following injection of radiolabelled arsenite in patients
terminally ill with malignant diseases, the isotope was found to be
widely distributed in the body, just as is the case in experimental
animals (Hunter et al., 1942; Ducoff et al., 1948; Mealey et al.,
1959). The highest concentrations were in the liver and kidney.
Twenty-four hours after subcutaneous injections of radiolabelled
arsenite (0.73-1.65 mg As) in patients who were to undergo
pneumo-encephalography, no arsenic could be detected in the spinal
fluid (Hunter et al., 1942). Measurements of radioactive arsenic in
biopsy samples of the normal brain tissue of brain-tumour patients
intravenously injected with 74As-arsenite 85 MBq (2.3 mCi/70 kg body
weight) showed an arsenic concentration of about 0.30% of the dose per
kg tissue during the first hour after injection (Mealey et al., 1959).
The arsenic levels decreased to about 0.25% during the second hour and
were down to about 0.16% by the seventh day. Intracranial tumours were
shown to contain much higher (2-30 times) arsenic concentrations than
normal brain tissue.
The concentrations of arsenic in different areas of the brains of
5 persons (15-81 years of age) were determined at autopsy using
neutron activation analysis (Larsen et al., 1979). Cerebral white
matter contained, on average, 2.7 times more arsenic than the grey
matter of the cerebral cortex which contained about 2 mg/kg wet
weight. A similar ratio was found between cerebellar white matter and
cerebellar cortex.
No data seem to be available on the biliary excretion of arsenic
in man. Data are also lacking concerning the distribution of
pentavalent inorganic arsenic in man.
Elimination
6.1.3.1 Animals
The elimination of arsenic in rats is very slow because of the
accumulation in red blood cells (section 6.1.2.1.1). In animals other
than the rat, absorbed arsenic is excreted from the body at a much
higher rate, mainly via the kidneys. Mice and rabbits excreted about
70% of injected trivalent inorganic arsenic via the kidneys in the
first 24 h following exposure (Ducoff et al., 1948; Crema, 1955). The
elimination of arsenic 10 min-48 h after intravenous injections of
76As-arsenic(III) oxide (0.1-0.2 mg As/kg body weight) in mice is
shown in Fig. 3 (Crema, 1955). As can be seen, almost 10% of the dose
was eliminated in the faeces during 2 days. It can also be seen that
the arsenic remaining in the body after the first day was eliminated
at a very low rate. About the same elimination pattern was shown in
pigs given a single test meal containing 0.3 mg As/kg body weight in
the form of arsenic(III) oxide; 75% of the absorbed dose was found in
urine collected over a 10-day period (Munro et al., 1974). Following
administration of arsenic(III) oxide (1 mg As/kg body weight) by
stomach intubation, the average urinary excretion in 3 adult female
Cynomologus monkeys was 57% of the absorbed dose during the first day
and a total of 73% over 14 days (Charbonneau et al., 1978a).
Whole body retention and elimination in dogs following intravenous
or oral administration of 74As-arsenic acid (0.02 µg As/kg body
weight) were studied by Hollins et al. (1979). It was concluded that
85% of the dose was cleared very rapidly with a half-time of about
6 h. The second phase of elimination, representing 14% of the dose,
has a half-time of 2.4 days. No significant differences were found
between intravenous and oral administration.
6.1.2.2 Tissue distribution
6.1.2.2.1 Animals
Mice, rabbits, guineapigs, hamsters, chickens, and monkeys given
radiolabelled arsenic in the trivalent form in parenteral doses of
0.1-4 mg of arsenic/kg body weight displayed highest levels of arsenic
in the liver, kidney, skin, lung, and spleen (Hunter et al., 1942;
Ducoff et al., 1948; Crema, 1955; Cikrt et al., 1980). The tissue
distribution of radioactive arsenic 10 min, and 1 h, 6 h, and 48 h
after intravenous administration of 76As-arsenic(III) oxide
(0.1-0.2 mg As/kg body weight) is shown in Table 4. As can be seen,
the highest activity of 76As per gram of tissue was found in the
liver and kidney of the mice. In most organs, the arsenic levels fell
fairly rapidly with time. In some organs, such as skin, brain, and
skeleton, arsenic levels decreased more slowly. The rate of decrease
of arsenic levels in the skin appeared to be especially slow as high
arsenic levels were still present 48 h after exposure.
Table 4. The distribution of arsenic in mice after intravenous
administration of 76As-arsenic(III) oxide (0.1-0.2 mg As/kg
body weight).a
activity per gram tissue/injected activity
per gram body weight
Organ 10 min 1 h 6 h 48 h
liver 6.1 2.47 0.64 0.10
kidney 3.55 3.90 1.40 0.12
spleen 1.37 1.14 0.48 0.08
blood 0.85 0.48 0.15 0.01
skin 0.56 0.91 0.72 0.30
muscle 0.50 0.62 0.40 0.08
small intestine 2.05 2.50 0.60 0.08
large intestine 0.76 0.68 0.26 0.13
skeleton 0.06 0.09 0.11 0.05
brain 0.002 0.02 0.08 0.01
lung 1.73 1.90 0.45 0.02
a From: Crema (1955).
Marked species differences in the biliary excretion of arsenic
were observed after intravenous administration of 1 mg of arsenic/kg
body weight as 74As-arsenic(III) chloride to rats, rabbits, and dogs
(Klaassen, 1974). The rate of excretion of arsenic into the bile in
rats was 40 times that in rabbits and 800 times that in dogs. Rats as
well as mice and hamsters given trivalent arsenic excreted arsenic
into the bile at a higher rate than when given pentavalent arsenic
(Cikrt & Bencko, 1974; Cikrt et al., 1980; Vahter & Norin, 1980).
Arsenic administered as arsenite or arsenic(III) oxide passes the
blood-brain barrier in mice, guineapigs, rabbits, hamsters, and
monkeys, although the levels found in the brain are low compared with
those in other tissues (Hunter et al., 1942; Ducoff et al., 1948;
Crema, 1955, Peoples, 1964; Vahter & North, 1980).
When arsenite was given to guineapigs, chimpanzees, and baboons,
the bulk of the tissue arsenic was shown to be in the protein fraction
and minor amounts in the acid-soluble and lipid fractions (Lowry et
al., 1942). Spontaneous tumours in mice (mammary carcinoma) did not
show any specific affinity for arsenic (Crema, 1955).
Du Pont et al. (1941) administered pentavalent arsenic in the form
of 76As-labelled sodium arsenate intravenously to rabbits (2 mg
As/rabbit, oxidation of As2S5 with hydrogen peroxide and addition of
sodium hydroxide to form arsenate prior to the dosing) and measured
the distribution at various times after the dosing. The percentage of
the dose per whole organ 1 h after dosing was 16.5% in muscle, 12.2%
in skin-fur, 9.5% in bone, 8.5% in blood serum, 5.9% in kidney, and
5.6% in liver. Intramuscular injections of pentavalent arsenic as
carrier-free 74As-arsenate (0.30-0.44 MBq/kg body weight, 8-12 µCi/kg
body weight) in cats, rabbits, guineapigs, chicks, and mice resulted
in a tissue content of radioactive arsenic of less than 0.2% of the
dose per gram wet weight, 48 h after dosing (Lanz et al., 1950).
Autoradiography of mice given carrier-free 74As-arsenic acid
intravenously showed a high affinity of arsenic for the intestinal
mucosa as well as for the kidney cortex, bone, and hair follicles
(Deak et al., 1976).
From the work of Vahter & Norin (1980), it is possible to compare
the tissue distribution of arsenic following administration of the
trivalent and pentavalent forms in the same animal species.
74As-labelled arsenite and arsenate were both administered to mice in
single oral doses of 0.4 or 4 mg of arsenic/kg body weight.
Concentrations of 74As-labelled arsenic in various organs 0.5-24 h
after dosing are given in Table 5 for both valence forms and dose
levels. Higher levels are seen in most tissues of animals receiving
arsenic(III), especially in the liver and bile and the differences are
more pronounced at the higher dose level. The high retention of
arsenic in the skin seen in the animals receiving arsenic(III) may be
explained by reaction of trivalent arsenic with sulfhydryl groups of
proteins, which are abundant in the skin (section 7.6).
Table 5. Arsenic in organs (µg As/g) of mice 0.5-24 h after single oral administration
of 10 µg As/mouse (0.4 mg/kg body weight) or 100 µg As/mouse (4 mg/kg body weight)
of 74As-labelled arsenate As(V), or arsenite, As(III). The figures represent the
mean for 6 animalsa
Dose level 0.5 h 2 h 6 h 24 h
Organ (mg/kg
body As(V) As(III) As(V) As(III) As(V) As(III) As(V) As(III)
weight)
kidney 0.4 1.17 0.74 0.97 1.01 0.72 0.61 0.03 0.05
4 7.24 7.41 8.73 7.44 4.33 4.37 0.06 0.78b
liver 0.4 0.93 2.02b 0.57 0.92b 0.26 0.31 0.02 0.04b
4 2.71 6.43b 3.52 6.72b 0.92 3.19b 0.04 0.39b
bile 0.4 1.86 3.31 0.51 5.30b 0.34 0.94b -- --
4 4.01 12.3ab 7.18 23.8b 2.16b 15.1ab <0.1 <0.1
brain 0.4 0.01 0.01 0.03 0.04b 0.05 0.06 <0.01 <0.01
4 0.09 0.06 0.54 0.17b 0.39 0.28b <0.01 0.05
skeleton 0.4 0.08 0.07 0.12 0.16 0.18 0.18 0.02 0.01
4 0.62 0.72 1.00 0.97 0.58 0.87 <0.01 0.03
skin 0.4 0.07 0.06 0.10 0.15b 0.11 0.13 0.02 0.06b
4 0.44 0.64b 0.94 1.10 0.37 1.10b 0.04 0.76b
a "From: Vahter & Norin (1980).
b p < 0.05.
c Based on 3 animals.
d Based on 2 animals.
Using considerably lower doses, Sabbioni et al. (1979) did not
find any major differences in tissue distribution, 16 h after
intraperitoneal injection of radiolabelled trivalent and pentavalent
inorganic arsenic in rabbits. Both oxidation states were administered
in doses of 0.5-1 µg or 50 µg of arsenic per animal. Intracellular
distribution was similar after exposure to either form of arsenic in
lung, liver, and kidney, where over 80% of the arsenic was found in
the nuclei and cytosol.
Several attempts have been made to demonstrate adaptation or
tolerance towards arsenic in experimental animals. In 2 studies, where
mice were given drinking water containing arsenic(III) oxide (32 days
at 250 mg As/litre and 256 days at 50 mg As/litre), the maximum
arsenic concentrations in skin and liver were reached on the 16th day
with a marked drop in concentrations (about 15-fold) during the rest
of the long-term experiment (Bencko & Symon, 1969a). Similar results
were obtained in a later experiment where mice were exposed to fly ash
with a mean concentration of arsenic of 180 µg/m3 for 6 h daily, 5
days a week, for up to 6 weeks (Bencko & Symon, 1970). Arsenic levels
in the liver and kidney reached a peak after 2 weeks' exposure, but,
by the 6th week, they were only slightly higher than those in the
unexposed controls. Similar dynamics in arsenic accumulation have been
found in rabbits exposed for up to one year to air near a power plant
emitting arsenic (Bencko et al., 1968) and in dogs given daily oral
doses of arsenious acid (0.2-0.4 mg As/kg body weight) for several
months (Katsura, 1958). The work by Bencko & Symon (1969b) and Bencko
et al. (1973) indicated increased tolerance towards parenterally
administered arsenic (5-18 mg As/kg body weight) in mice pretreated
with arsenite in the drinking water (50 mg As/litre).
6.1.2.2.2 Man
Following injection of radiolabelled arsenite in patients
terminally ill with malignant diseases, the isotope was found to be
widely distributed in the body, just as is the case in experimental
animals (Hunter et al., 1942; Ducoff et al., 1948; Mealey et al.,
1959). The highest concentrations were in the liver and kidney.
Twenty-four hours after subcutaneous injections of radiolabelled
arsenite (0.73-1.65 mg As) in patients who were to undergo
pneumo-encephalography, no arsenic could be detected in the spinal
fluid (Hunter et al., 1942). Measurements of radioactive arsenic in
biopsy samples of the normal brain tissue of brain-tumour patients
intravenously injected with 74As-arsenite 85 MBq (2.3 mCi/70 kg body
weight) showed an arsenic concentration of about 0.30% of the dose per
kg tissue during the first hour after injection (Mealey et al., 1959).
The arsenic levels decreased to about 0.25% during the second hour and
were down to about 0.16% by the seventh day. Intracranial tumours were
shown to contain much higher (2-30 times) arsenic concentrations than
normal brain tissue.
The concentrations of arsenic in different areas of the brains of
5 persons (15-81 years of age) were determined at autopsy using
neutron activation analysis (Larsen et al., 1979). Cerebral white
matter contained, on average, 2.7 times more arsenic than the grey
matter of the cerebral cortex which contained about 2 mg/kg wet
weight. A similar ratio was found between cerebellar white matter and
cerebellar cortex.
No data seem to be available on the biliary excretion of arsenic
in man. Data are also lacking concerning the distribution of
pentavalent inorganic arsenic in man.
Elimination
6.1.3.1 Animals
The elimination of arsenic in rats is very slow because of the
accumulation in red blood cells (section 6.1.2.1.1). In animals other
than the rat, absorbed arsenic is excreted from the body at a much
higher rate, mainly via the kidneys. Mice and rabbits excreted about
70% of injected trivalent inorganic arsenic via the kidneys in the
first 24 h following exposure (Ducoff et al., 1948; Crema, 1955). The
elimination of arsenic 10 min-48 h after intravenous injections of
76As-arsenic(III) oxide (0.1-0.2 mg As/kg body weight) in mice is
shown in Fig. 3 (Crema, 1955). As can be seen, almost 10% of the dose
was eliminated in the faeces during 2 days. It can also be seen that
the arsenic remaining in the body after the first day was eliminated
at a very low rate. About the same elimination pattern was shown in
pigs given a single test meal containing 0.3 mg As/kg body weight in
the form of arsenic(III) oxide; 75% of the absorbed dose was found in
urine collected over a 10-day period (Munro et al., 1974). Following
administration of arsenic(III) oxide (1 mg As/kg body weight) by
stomach intubation, the average urinary excretion in 3 adult female
Cynomologus monkeys was 57% of the absorbed dose during the first day
and a total of 73% over 14 days (Charbonneau et al., 1978a).
Whole body retention and elimination in dogs following intravenous
or oral administration of 74As-arsenic acid (0.02 µg As/kg body
weight) were studied by Hollins et al. (1979). It was concluded that
85% of the dose was cleared very rapidly with a half-time of about
6 h. The second phase of elimination, representing 14% of the dose,
has a half-time of 2.4 days. No significant differences were found
between intravenous and oral administration.
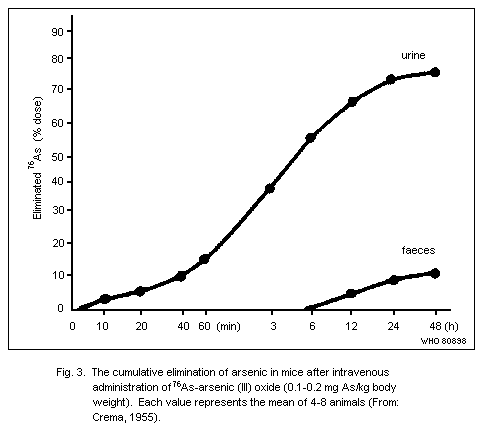 Whole body retention and elimination were studied in mice
following administration of 74As-labelled As(III) and As(V), with
strict control of valence state at the time of exposure (Vahter &
Norin, 1980). In mice given a single oral dose of 4 mg As/kg body
weight, whole body retention was 2-3 times higher after exposure to
As(III) than after exposure to As(V). A similar difference was seen
after subcutaneous injection of 0.4 mg/kg body weight but differences
in retention were not seen when this dose was given orally.
Daily doses of 0.03-0.66 mg As/kg body weight given to cows in the
form of arsenic acid for up to 8 weeks did not cause the arsenic
levels in milk to rise (Peoples, 1964). Following an outbreak of
arsenic poisoning in cattle in Mexico caused by the ingestion of feed
containing up to 0.28% arsenic, there was a very drastic reduction in
milk production. Levels of up to 0.5 mg As/litre were found in the
milk of cows still producing it (de Navarro, 1976; Gonzales, 1977). In
view of the high exposure, these levels of arsenic in milk indicate
that arsenic ingested in the form of inorganic arsenic does not
readily pass the blood-mammary barrier.
Two studies in rats on the elimination of arsenic via the lungs
have been reported (Lanz et al., 1950; Dutkiewicz, 1977). Both seem to
indicate that little, if any, is eliminated by this route. A study by
Overby & Fredrickson (1963) using chickens also indicated a very low
elimination of 74As-arsenic through the lungs, following exposure to
74As-arsenate.
6.1.3.2 Man
Mappes (1977) described a series of experiments in which he,
himself, ingested 2 mg of arsenic as arsenic(III) oxide in an aqueous
solution. About 30% of the ingested arsenic had been recovered in
urine (molybdenum blue method) 24 h after exposure. Mappes also took
daily doses of 0.8 mg of arsenic as arsenic(III) oxide in an aqueous
solution. The excretion rate of arsenic reached equilibrium after 5
days, by which time about 70% of the daily dose was being excreted in
the urine daily.
Crecelius (1977b) studied the excretion of arsenic in the urine of
a person who had ingested arsenic-rich wine (50 µg As(III) and
13 µg As(V)) and water containing 0.2 mg of arsenic mainly in the
pentavalent form. About 80% of the arsenic ingested with the wine was
excreted within 61 h. The biotransformation of the ingested arsenic
was also studied (section 6.1.4.2) and the apparent biological
half-time for the in vivo methylated arsenic, which was the major
species of arsenic excreted, was found to be of the order of 30 h,
compared with 10 h for arsenic eliminated in the inorganic form. Only
50% of the pentavalent arsenic ingested with the water was recovered
in the urine during the first 70 h following ingestion.
A single oral dose of carrier-free 74As was administered in a
gelatine capsule to each of 6 adult male volunteers (about 0.01 µg
As/man; over 90% As(V)) (Tam et al., 1979a; Pomroy et al., 1980).
During the first 24 h after dosing, 22.4% of the 74As dose was
excreted via the urine. After 5 days, a total of 58% had been
recovered in the urine. No data were presented on faecal elimination.
Data from whole body measurements were best represented by a
3-compartment exponential model. 65.9% of the dose was eliminated with
a half-time of 2.1 days, 30.4% with a half-time of 9.4 days, and 3.7%
with a half-time of 38.4 days.
Though absorbed arsenic is excreted mainly via the kidneys, a
small amount is removed by other routes. Studies on the constituents
of profuse sweat induced in a hot and humid environment have been
reported by Vellar (1969). The mean concentration of arsenic in the
sweat of 2 subjects was 1.5 µg As/litre (neutron activation analysis)
and the calculated hourly loss of arsenic was 2 µg. Data concerning
sweat levels under normal conditions were not reported. Desquamation
of skin will also result in the removal of arsenic, since arsenic has
a high affinity for skin (section 6.1.2.2). Molin & Wester (1976)
calculated the daily loss of arsenic through desquamation of normal
skin (10 male patients, apparently unexposed to arsenic) to be
0.1-0.2 µg based on their finding of a mean arsenic concentration in
skin of 0.18 mg/kg dry weight (neutron activation analysis). There are
some data on the role of the hair as a route of elimination of
arsenic. Several authors (section 7.3) have reported hair levels of
arsenic exceeding 100 mg/kg among occupationally exposed subjects.
Some of this arsenic may result from external exposure, however. Hair
arsenic levels in Japanese subjects poisoned by contaminated soy sauce
(3 mg of arsenic daily for 2 or 3 weeks) were between 1.8 and
13 mg/kg, 2 weeks after ingestion (Mitzuta et al., 1956). If a hair
weight of 20 g is assumed (Task Group on Reference Man, 1975), this
would account for 0.6% of the ingested arsenic, at the most.
Grimanis et al. (1979) determined the concentrations of arsenic
and some other trace metals in human milk using neutron activation
analysis. There were no differences between levels in human colostrum,
transitional, and mature milk, all of which were about 3 µg/litre
(range 0.6-6.3 µg/litre). Colostrum and transitional milk were
obtained from 15 healthy mothers living in the Athens area with a mean
age of 26 years. Mature milk was obtained from 5 of the 15 mothers.
6.1.4 Biotransformation
The form of arsenic present in most tissues is largely unknown
because of the analytical difficulties involved. However,
differentiation between the various forms of arsenic is more reliable
in plasma and urine, where arsenic can be measured without previous
digestion.
6.1.4.1 Animals
After intramuscular administration of arsenate (arsenic(V)) to
rats, about 10-15% of the total urinary arsenic was reported to be in
the form of arsenic(III) (Lanz et al., 1950). The different forms were
separated by precipitation of magnesium-ammonia-arsenate. In dogs,
given an intravenous infusion of 74As-arsenate, arsenic(III) was
reported to be present in the urine (about 14% of the total arsenic)
as well as in the plasma (about 5.5% of the total plasma arsenic)
(Ginsburg & Lotspeich, 1963). The method used for the separation of
the different forms of arsenic was that described by Crawford & Storey
(1944) in which arsenic(III) was extracted with ethyl xanthate. In
further work, Ginsburg (1965) reported that injection of arsenate in
dogs caused arsenic(III) to appear in the glomerular filtrate and, to
a still greater extent, in the urine.
When trivalent arsenic (arsenite) was intravenously infused, both
arsenate and arsenite were detected in the plasma, glomerulus
filtrate, and urine of dogs indicating an in vivo oxidation of
trivalent to pentavalent arsenic (Ginsburg, 1965). Bencko et al.
(1976) reported that in vivo oxidation of arsenite can occur in
mice. About one half of the intramuscularly administered arsenic
(74As-labelled sodium arsenite, 1.3 mg As/kg body weight) was
recovered as pentavalent arsenic in urine taken directly from the
bladder (analysed by paper chromatography). When the mice were exposed
to arsenite in drinking water before the injection, an even greater
part of the excreted arsenic was pentavalent.
The studies, just referred to, did not take into consideration
that in vivo methylation of arsenic occurs. It was therefore not
possible for the Task Group to evaluate to what extent such methylated
forms of arsenic may have interfered in the separation of the two
inorganic forms. In the absence of more detailed studies, no firm
conclusions can be drawn about the in vivo reduction or oxidation of
inorganic arsenic.
Methylated arsenic has been detected in the urine of cows and dogs
fed arsenate or arsenite (Lakso & Peoples, 1975). When the dogs were
fed doses of about 1.0 mg As/kg body weight of either valence form for
5 days, approximately equal amounts of inorganic and methylated
arsenic were excreted in the urine. The cows produced about 3 times as
much methylated arsenic as inorganic arsenic in their urine.
Following intravenous administration of carrier-free 74As-arsenic
acid to an adult male beagle (14.8 MBq, 0.4 mCi), 74As was present in
plasma or urine predominantly as inorganic arsenic and dimethylarsinic
acid, as revealed by separation on a cation-exchange column (Tam et
al., 1978, 1979b). When plasma, red blood cells, or urine from a
beagle were incubated in vitro with 74As-arsenic acid, no
methylated arsenic was found in the plasma or urine samples but a
small amount (0.2%) of dimethylarsinic acid did appear in the
erythrocyte samples (Tam et al., 1979c).
Ten minutes after intravenous administration of 74As-arsenic acid
to beagles (0.5 µg As/dog), about 8% of the total amount of 74As
present in the erythrocytes was in the form of dimethylarsinic acid
whereas no methylated arsenic could be found in the plasma
(Charbonneau et al., 1978b). Six hours after dosing, the small amount
of 74As still present in the erythrocytes and plasma was
predominantly in the form of dimethylarsinic acid as was 5-25% of the
total 74As present in urine 1 h after dosing and 90%, 6 h after. When
a single oral dose of 0.2-0.6 µg As was administered to beagles as
74As-arsenic acid, a similar metabolic pattern was revealed
(Charbonneau et al., 1979).
6.1.4.2 Man
Braman & Foreback (1973) reported the presence of methylated forms
of arsenic in the urine of 4 subjects. Dimethylarsinic acid
constituted on average 66% of the total urinary arsenic, while
methylarsonic acid, pentavalent arsenic, and trivalent arsenic
accounted for 8.0, 17.0, and 8.4%, respectively. Actual in vivo
methylation of inorganic arsenic was later indicated by the work of
Crecelius (1977b), who measured the different forms of arsenic in the
urine of a subject who had ingested wine containing 50 µg of
arsenic(III) and 13 µg of arsenic(V). Urinary dimethylarsinic acid
accounted for 50% of the ingested arsenic, methylarsonic acid for 14%
and inorganic arsenic for 8%. Dimethylarsinic acid was also the major
form of arsenic found in the urine of smelter workers occupationally
exposed to arsenic, chiefly in the form of arsenic(Ill) oxide (Smith
et al., 1977).
In a person who had ingested well water containing 0.2 mg of
pentavalent arsenic, the inorganic arsenic(V) concentration in urine
showed a marked increase (5-fold) the first 10 h after exposure,
indicating that some of the ingested arsenic was rapidly excreted
unchanged in the urine (Crecelius, 1977b). The urinary levels of
dimethylarsinic acid increased 5 to 10-fold between 10 and 70 h after
exposure. Only 50% of the arsenic ingested was excreted in the urine
within 70 h. Following oral ingestion of 74As (> 90% As(V)) by 6
adult males (about 0.01 µg As/man), 58% of the dose was excreted in
the urine during the first 5 days (Tam et al., 1979a). Of the excreted
arsenic, 51% was in the form of dimethylarsinic acid, 21%
monomethylarsenic compounds, and 27%, inorganic arsenic.
No methylation of 74As was found in human plasma or urine
incubated in vitro with 74As-arsenic acid (Tam et al., 1979c).
Further studies are required in other species to determine whether
the monomethylarsenic compound is a metabolite unique to man.
6.2 Organic Arsenic Compounds
6.2.1 Absorption
6.2.1.1 Respiratory absorption
6.2.1.1.1 Animals
One organic arsenic compound of interest is dimethylarsinic acid
(cacodylic acid), since it may be inhaled when it is used as a
herbicide. Absorption of this compound following intratracheal
administration was studied in rats by Stevens et al. (1977). He found
that a solution of 14C-dimethylarsinic acid was rapidly absorbed from
the lung. Less than 5% remained unabsorbed after 15 min and the
absorption half-time was calculated to be 2.2 min.
6.2.1.1.2 Man
No data are available concerning the respiratory absorption of
organic arsenic in man.
6.2.1.2 Gastrointestinal absorption
6.2.1.2.1 Animals
Absorption of seafood arsenic from the gastrointestinal tract was
investigated in rats by Coulson et al. (1935). When rats were given a
shrimp diet, only about 4% of the ingested organic arsenic
(approximately 0.5 mg As) was recovered in the faeces during the first
2 days following exposure, indicating almost complete absorption from
the gastrointestinal tract.
When fish containing arsenic was given to pigs in a single meal in
amounts corresponding to 0.3 mg As/kg body weight, 23% of the ingested
arsenic was recovered in the faeces collected over a period of 10 days
following exposure (Munro, 1976). About the same high absorption (80%
or more) was observed by Munro (1976) in adolescent monkeys fed
arsenic-containing fish corresponding to a dose of 1 mg As/kg body
weight and by Charbonneau et al. (1978a) in adult female cynomolgus
monkeys given a single test meal (via a stomach tube) of homogenized
fish containing arsenic (1 mg As/kg body weight).
Thirty-one percent of an oral dose of 0.5 ml of an aqueous
solution containing 40 µg of radiolabelled dimethylarsinic acid
(approximately 20 µg As/rat) was eliminated in the faeces of rats
during 24 h (Stevens et al., 1977).
From reports on the effects and distribution of other organic
arsenic compounds (pesticides, feed additives for poultry and swine,
chemotherapeutic agents), it is evident that uptake of these compounds
occurs, when they are given orally to laboratory animals. The amount
of the administered dose absorbed from the gastrointestinal tract
differs over a wide range, depending on the chemical properties of the
compound. A chemotherapeutic agent of low lipid solubility,
p-N-glycol-arsanilate, was shown to be poorly absorbed from the
gastrointestinal tract (McChesney et al., 1962). Other
chemotherapeutic agents, such as carbarsone ( p-ureidophenylarsonic
acid) and tryparsamide ( N-(carbamoylmethyl) arsanilic acid), and
pesticides such as sodium methane arsenate and dimethylarsinic acid,
were readily absorbed, when fed to rats and rabbits (Hwang & Schanker,
1973; Exon et al., 1974; Stevens et al., 1977). The absorption
half-times of carbarsone, tryparsamide, and dimethylarsinic acid from
the small intestine of the rat were 87, 184, and 201 min, respectively
(Hwang & Schanker, 1973). The absorption process did not show any
evidence of saturation when the concentrations of the compounds were
increased up to 100-fold. The absorption rates were ranked in the same
order as the chloroform-to-water partition coefficients indicating,
according to the author, that absorption takes place mainly by
diffusion.
Liver, from pigs fed arsanilic acid, which contained about 6 mg
As/kg, was administered to rats at a level of 30% of the diet. The
daily faecal elimination of the rats contained 70-85% of the intake of
arsenic, indicating that this form of arsenic is not as readily
absorbed as inorganic arsenic or "seafood arsenic" (Overby & Frost,
1962).
The same type of experiment was reported by Calvert (1975) who fed
wethers dried broiler manure containing 3-nitro-4-hydroxy-
phenylarsonic acid in concentrations of 3.4-5 mg As/kg of diet. He
collected urine and faeces during the last 5 days of a 15-day feeding
period and found 60-73% of the ingested arsenic in the faeces.
6.2.1.2.2 Man
A single meal of fish or crustacea containing high levels of
mainly organic arsenic may result in the ingestion of several
milligrams of arsenic, most of which is apparently absorbed from the
gastrointestinal tract. Coulson et al. (1935) reported an experiment
in which each of 2 human subjects ate boiled shrimps containing about
1 mg of arsenic. By the fourth day, approximately 5% had been
recovered in the faeces indicating that absorption from the
gastrointestinal tract was almost complete. Four persons who ate
plaice and cod with high arsenic levels excreted an average of 83% of
the ingested arsenic (0.5-2.2 mg/person) in the urine during the 2.5
days following exposure indicating that the fish arsenic was readily
absorbed (Westöö & Rydälv, 1972).
Arsanilic acid, used as a feed additive for poultry and swine, may
be ingested in trace amounts, when meat from these animals is eaten.
The availability to man of arsanilic acid or of arsenic in the tissues
of chicks fed arsanilic acid was studied by Calesnick et al. (1966).
Four adult male volunteers ingested single doses of 1.3-3.0 mg of
arsenic as 74As-arsanilic acid. The average faecal elimination within
6 days of exposure was 74% of the dose. Following ingestion of pâté
made from chicks fed 74As-arsanilic acid, approximately 64% of the
arsenic ingested was recovered in the faeces. Apparently, arsenic from
the flesh of animals fed with additives containing arsenic is not
absorbed from the gastrointestinal tract as readily as arsenic from
fish or crustacea.
6.2.1.3 Skin absorption
No data are available on the absorption of various organic arsenic
compounds through the skin of animals or man.
6.2.1.4 Placental transfer
6.2.1.4.1 Animals
There are no data on the placental transfer of organic arsenic
compounds present in seafood. Dimethylarsinic acid has been shown to
pass the placental barrier of rats, when administered intravenously
just prior to parturition (Stevens et al., 1977). The dose given to
the pregnant rats was 40 µg of radiolabelled dimethylarsinic acid per
rat (20 µg As/rat). Fetal whole blood levels were comparable with
those in the maternal blood.
Transfer of organic arsenic from hens to eggs has been reported.
Increased concentrations of arsenic were found in eggs of hens fed 50
or 100 mg As/kg diet as 3-nitro-4-hydroxyphenylarsonic acid (Daghir &
Hariri, 1977). The highest levels of arsenic (about 0.24 mg/kg) were
found after 4-6 weeks of feeding, after which the levels gradually
decreased, indicating that the hens developed a tolerance to organic
arsenic similar to that found towards inorganic arsenic (section
6.1.2.2.1).
6.2.1.4.2 Man
There are no human data available concerning the placental
transfer of organic arsenic compounds.
6.2.2 Distribution in organisms
6.2.2.1 Fate of organic arsenic in blood
6.2.2.1.1 Animals
Data are not available on blood arsenic levels following ingestion
of seafood arsenic.
Stevens et al. (1977) investigated the kinetics of dimethylarsinic
acid in the plasma of rats after intravenous administration of 200 mg
14C-dimethylarsinic acid/kg body weight
(108 mg As/kg body weight). After a single injection, the plasma
concentration followed a three-exponential equation with half-times of
0.014, 0.217, and 3.42 h. The retention of 14C-dimethylarsinic acid
in whole blood was high, about 10% of the dose 3 months after the
administration indicating that the rat differs from other animals with
regard to metabolism of this arsenic compound. As dimethylarsinic acid
is a major metabolite of inorganic arsenic, it might be expected to be
cleared from the blood fairly rapidly.
The clearance of the major part of the arsenic from the blood of
chickens given a single oral dose of arsanilic acid showed 2 rapid
phases, with half-times of about 90 min and 6 h, respectively (Overby
& Fredrickson, 1963). About 99.9% of the dose was cleared at these
rates. The remaining 0.1% was cleared with a half-time, of 36 h.
Less than 6% of doses of 4 different organo-arsenic drugs, given
intravenously to rabbits, remained in the blood 2 h after the
administration (Hogan & Eagle, 1944). The 4 arsenic compounds were
unsubstituted phenylarsenoxide, 2 substituted phenylarsenoxides, and
tryparsamide. Red blood cells and plasma showed marked differences in
clearance rate and distribution. In the case of the most toxic
compound, the unsubstituted phenylarsenoxide (trivalent arsenic),
almost all the injected arsenic was still in the blood 0.75-1.5 min
after the injection. More than 95% of the dose was found in the red
blood cells. More than 50% of the dose of the less toxic tryparsamide
(pentavalent arsenic) had left the blood within the same span of time,
and more than 95% of the remaining arsenic was in the plasma. The same
distribution among blood cells and plasma was obtained in in vitro
studies of the binding of arsenic compounds to red blood cells. The
amounts of the various phenylarsenoxides (acid-substituted compounds
excepted) bound to red blood cells corresponded very well to their
acute toxicities in white mice. The arsonic acids were bound only to a
minor degree to the red blood cells in vitro. They were also
relatively nontoxic in vivo, but the toxicity varied from one
compound to another.
6.2.2.1.2 Man
No human data are available concerning the fate of organo-arsenic
compounds in the blood.
6.2.2.2 Tissue distribution of organic arsenic
6.2.2.2.1 Animals
Data on the tissue distribution of seafood arsenic in experimental
animals and man are lacking. The only available report is that of
Lunde (1972), who studied the distribution of organic arsenic in fish
(rainbow trout) fed a marine diet containing about 15 mg As/kg in the
form of organoarsenic compounds to which inorganic 74As had been
added. The content of radioactive inorganic arsenic in the fish was
negligible, 6-10 days after the addition of radioactive arsenic was
stopped, but a small fraction of the inorganic arsenic was converted
to organoarsenic compounds. Autoradiography revealed that the 74As
was especially concentrated in the eyes, throat, gills, and pylorus
organ. The liver and kidney also contained much radioactivity, but
arsenic disappeared faster in these than in other organs, when the
feeding of radioactive arsenic was discontinued.
Administration of a diet containing 50 mg/kg of monosodium methane
arsonate (MSMA) (27.5 mg As/kg) for 52 weeks caused a rapid increase
in the arsenic contents in the liver and kidney of rabbits during the
first 2 weeks (Exon et al., 1974). Accumulation of arsenic in hair was
observed in cattle exposed to dietary MSMA or dimethylarsinic acid
(Dickinson, 1972). The animals were fed daily doses of dimethylarsinic
acid at 10 mg/kg body weight (5.4 mg As/kg body weight) and had
arsenic levels in hair of 2.0-4.3 mg/kg (three animals) after 10 days
and 13-33 mg/kg after 48 days.
Calvert (1975) fed wethers various amounts of arsanilic acid and
measured the arsenic levels in the liver, kidney, and muscle. The
results, shown in Table 6, suggest that arsenic given as arsanilic
acid is concentrated in the liver and kidney. When the animals were
placed on an arsenic-free diet, the tissue levels decreased rapidly,
as shown in Table 7, and had dropped to about 15% of the original
value by the sixth day.
Table 6. Arsenic levels in tissues of wethers, fed arsanilic acid
for 28 daysa
Arsenic Whole Liver Kidney Muscle
fed blood
(mg/kg of mg/kg dry
diet) tissue
0.0 < 0.01 < 0.01 < 0.01 < 0.01
26.8 0.063 3.1 3.2 0.2
144.4 0.270 26.8 12.2 1.1
273.3 0.536 29.2 23.6 1.2
a From: Calvert (1975).
Table 7. Depletion of arsenic in the liver of wethers fed
arsanilic acid for 28 daysa
Arsenic Withdrawal time (days)
fed 0 2 4 6
(mg/kg of mg/kg dry tissue
diet)
0.0 < 0.01 < 0.01 < 0.01 < 0.01
26.8 3.1 4.9 2.9 1.9
144.4 26.8 15.4 8.4 3.5
273.3 29.2 27.0 11.4 5.0
a From: Calvert (1975).
When pigs were fed arsanilic acid (1000 mg/kg diet, approximately
340 As mg/kg diet), the maximum arsenic level in most tissues was
reached on the 13th day (Ledet et al., 1973). However, maximum levels
in the nervous tissues (CNS and peripheral nerves) were not reached
until the 20th day. Clearance of arsenic was slower in these than in
the other tissues. Highest levels were found in liver and kidney.
Injections of the trivalent organoarsenic drug phenylarsenoxide in
rabbits resulted in 50-100 times higher liver arsenic levels than
injections of the relatively nontoxic 3-NH2-4-OH derivative of the
pentavalent compound tryparsamide (substituted phenylarsonic acid)
(Hogan & Eagle, 1944). After injections of these compounds at the
LD50-level, comparable amounts of arsenic were found in the tissues
despite the 500-fold difference in dose. The highly toxic
acid-substituted 4-COOH-phenylarsenoxide caused extraordinarily high
kidney levels in the first hours after injection (10-20% of the dose)
and levels comparable with the other substituted phenylarsonic acids
after 24-48 h.
6.2.2.2.2 Man
No data are available on the distribution of organic arsenic
compounds in man.
6.2.3 Elimination
6.2.3.1 Animals
A substantial fraction of seafood arsenic administered to animals
is rapidly eliminated from the body. About 75% of a dose of shrimp
arsenic (approximately 0.5 mg As) given to rats in food was eliminated
within the first day, and an additional 20% was eliminated during the
second day, predominantly in the urine (Coulson et al., 1935). Pigs
given a fish diet providing a dose of 0.3 mg As/kg body weight
eliminated 90% of the ingested arsenic within 3 days (Munro, 1976).
About 70% of the dose was recovered in the urine. When the same type
of diet was given to adolescent monkeys (Macaca irus) corresponding
to doses of 1 mg As/kg body weight, only 63% of the dietary arsenic
was recovered in the excreta within 10 days (18% in the faeces and 45%
in the urine). When a single oral dose of fish arsenic providing about
1 mg fish arsenic/kg body weight to adult female cynomolgus monkeys,
57-84% of the ingested arsenic was excreted in the urine within 3
days. The total recovery in the excreta was 66-85% during 14 days
(Charbonneau, et al., 1978a).
The feeding of organic arsenic in the form of arsanilic acid to
poultry and swine as a feed additive, may result in tissue residues.
Rats fed protein from swine liver containing 24.4 mg arsenic/kg
protein as arsanilic acid, at a level of 300 g/kg diet for 14 days,
eliminated almost all of the arsenic within 7 days of the end of the
feeding period (Overby & Frost, 1962). The amount of arsenic excreted
in the urine was one third of the amount of arsenic eliminated in the
faeces during this period. Experiments with pigs given a diet
containing 0.01% arsanilic acid for 31 days showed a rapid decrease in
the tissue levels of arsenic during the first days after the feeding
of arsanilic acid was discontinued (Ferslew & Edds, 1979). The livers
contained arsenic concentrations of 1.5-2 mg/kg on the 31st day of
feeding and about 0.2 mg/kg on the 7th day after the removal of
arsanilic acid from the diet.
When pentavalent arsenic compounds in the form of dimethylarsinic
acid, p-N-glycolylarsanilate, arsanilic acid, 4-nitrophenylarsonic
acid, and 3-nitro-4-hydroxyphenylarsonic acid were administered
parenterally to rats, some 50%-80% of the injected dose was excreted
in the urine within the first 48 h (McChesney, et al., 1962; Schreiber
& Brouwer, 1964; Stevens, et al., 1977). Only minor amounts were found
in the bile and faeces. Seventy percent of a dose of p-N-
glycolylarsanilate (used in veterinary medicine), intravenously
injected into cats, was excreted in the urine and 0.8% excreted in the
bile during the first 160 min following exposure (McChesney, et al.,
1962). Rabbits injected intravenously with another drug, tryparsamide,
excreted 68% of the dose in 24 h and 81% of the dose in 48 h (Hogan &
Eagle, 1944). Derivatives of phenylarsenoxide, a trivalent
organoarsenic compound, were excreted in the urine of rabbits at much
slower rates than the pentavalent arsenic compound, tryparsamide. The
elimination rates depended on the functional groups on the benzene
ring and varied from 5% in 24 h for unsubstituted phenylarsenoxide to
60% for the 3-NH2-4-OH-derivative.
Cristau et al. (1972, 1975) investigated the influence of the
molecular structure of some arsenic-containing drugs on the
elimination kinetics of rats and guineapigs. Acetarsol, tyrparsamide,
diphetarsone, and melarsonyl, all of which possess hydrophilic
functional groups that facilitate elimination without
biotransformation, were rapidly eliminated (65%-90% of the dose in
24 h). Acetarsol and tryparsamide were mainly excreted in the urine
and to a much lesser extent in the bile. Melarsonyl, which has a
higher relative molecular mass (532.5) than the other drugs tested,
was excreted to a greater extent in the bile in both species. About 10
times more diphetarsone, which has a relative molecular mass of 460,
was excreted in the bile of rats than in the bile of guineapigs. This
was reported to agree with previous observations that rats excrete
more of some drugs in the bile than guineapigs. Melarsoprol and
arsthinol are both hydrophobic, which indicates that they must undergo
biotransformation prior to elimination. Both drugs were excreted
slowly (20%-50% of the dose in 24 h), predominantly via the bile.
There are only a few reports on the excretion of arsenic in the
milk of animals. The arsenic level in cow's milk did not increase with
the blood concentration when the cows were fed methylarsonic acid or
dimethylarsinic acid (Peoples, 1975). However, the milk arsenic levels
did increase when cows were fed arsanilic acid or
3-nitro-4-hydroxyphenylarsonic acid (1.6-3.2 mg As/kg body weight)
(Calvert, 1973).
No radioactive arsenic was detected in air exhaled by chickens,
given an 74As-labelled arsanilic acid orally, indicating that this
arsenic compound was not eliminated via the lungs or metabolized to
form expirable products (Overby & Fredrickson, 1963).
6.2.3.2 Man
Only a few studies are available on the elimination of organic
arsenic compounds in man. As early as 1919, Bang stated that organic
arsenic in fish and other marine foods was readily excreted, mainly in
human urine. Chapman (1926) reported that a person, who had ingested
lobster containing 33 mg of arsenic, excreted 74% of the arsenic in
the urine within 48 h. In studies by Coulson, et al. (1935), 2
subjects were given sea food arsenic almost all of which (> 90% of
approximately 1 mg As) was recovered in the urine within 4 days of
ingestion. When the urinary excretion of arsenic was measured in 4
subjects after they had ingested plaice and cod containing high
arsenic levels, about 70% of the ingested arsenic (0.5, 2.0, 2.1 and
2.2 mg As, respectively) was excreted in the urine during the first 24
h after ingestion and 83% in 2.5 days (Westöö & Rydälv, 1972).
Freeman, et al. (1979) gave 6 men flounder containing high levels of
arsenic, corresponding to a total ingested quantity of 5 mg of arsenic
per man. During the first day, the urinary excretion of arsenic
corresponded to more than half of the ingested amount of arsenic and
the mean total arsenic excreted during 9 days was 77%.
Calesnick, et al. (1966) studied the recovery of 74As in the
urine and faeces of human subjects who had ingested radiolabelled
arsanilic acid or pâté made from chickens fed 74As-labelled arsanilic
acid. After 6 days, only 20% of the ingested dose was recovered in the
urine and between 64% and 74% was recovered in the faeces regardless
of whether the arsenic had been given as pure arsanilic acid or in the
form of pâté from arsenic-exposed chickens.
6.2.4 Biotransformation
6.2.4.1 Animals
Stevens, et al. (1977) investigated the in vivo
biotransformation of dimethylarsinic acid. They injected
14C-dimethylarsinic acid (18 µg As) and 74As-dimethylarsinic acid
(1.9 µg As) simultaneously into rats and measured the distribution in
different organs. Since the tissue distributions of 74As and 14C were
the same, dimethylarsinic acid was thought not to be converted to
inorganic arsenic in rats. In another experiment by Stevens, et al.
(1977), doses of 200 mg/kg body weight of 14C-dimethylarsinic acid
(108 mg As/kg body weight) were administered intravenously,
intratracheally, or orally to rats. Radioactive carbon dioxide was
measured in the air emitted from the animal chambers. Approximately
0.008%-0.13% of the dose was detected as 14CO2 during a 24-h period.
This result was interpreted as indicating that only a small fraction
of the dimethylarsinic acid was demethylated by the rats. The lowest
fraction (0.008%) of the dose was exhaled by the rats that had
received intravenous injections, while the highest fraction (0.13%)
was exhaled by the peroraly exposed rats. The in vivo transformation
of dimethylarsinic acid in other species is unclear.
Orally administered dimethylarsinic acid was found to be mainly
unchanged in the liver of rats (Winkler, 1962) and in the excreta of
chickens (Overby & Fredrickson, 1963; Moody & Williams, 1964a,b,c).
Dimethylarsinate was detected in the blood, urine, and faeces of rats
given oral doses of ferric methane arsonate (approximately 40 mg As/kg
body weight). This would point towards in vivo methylation of the
compound (Odanaka, et al., 1978). The major components in the urine
were dimethylarsinate and unchanged methane arsonate.
Investigations on the biotransformation of some of the organic
arsenic compounds used as feed additives and drugs indicate that they
are converted to more easily excretable and, in some cases, more toxic
substances (Hogan & Eagle, 1944; Cristau, et al., 1972, 1975; Calvert,
1975). These changes in the molecular structure of the arsenic
compound seldom affect its valence state; nor do they result in the
formation of inorganic arsenic compounds. Hogan & Eagle (1944) cited
considerable evidence of the reduction of arsonic acids in vivo to
the corresponding arsenoxides.
6.2.4.2 Man
Elevated levels of inorganic arsenic, methylarsonic acid, or
dimethylarsinic acid were not found in human urine following ingestion
of crab meat containing 2 mg of arsenic (Crecelius, 1977b). Digestion
of the urine with hot 2 N sodium hydroxide solution was reported to
convert the unknown organic arsenic compound to dimethylarsinic acid.
The author interpreted his results as indicating that organoarsenic
compounds originating from seafood are excreted without being
biotransformed in the body. Canon et al. (1979) demonstrated the
presence of arsenobetaine in the urine of 2 human subjects after
ingestion of rock lobster tails. This indicates that this compound is
not biotransformed in the body.
7. NORMAL LEVELS IN MAN AND BIOLOGICAL INDICATORS
OF EXPOSURE
This section will deal with concentrations of arsenic in
"unexposed" persons as well as in persons excessively exposed to
arsenic through food, drinking-water, drugs, or occupation. It will
also deal with concentrations of arsenic in biological indicator media
and the extent to which they reflect exposure, e.g., concentrations in
ambient or industrial air, drinking-water, or food. It would have been
desirable to discuss the extent to which concentrations in biological
indicator media reflect concentrations of arsenic in critical organs,
i.e., organs where the earliest toxic symptoms are manifested.
However, since no organ is generally accepted as the critical organ
for arsenic toxicity, such data are lacking. Some comments will be
made based on human and animal metabolic data. The arsenic levels in
biological media will be discussed separately.
7.1 Blood
The reported figures on arsenic levels in the blood vary. Using
neutron activation, Brune et al. (1966) found a mean arsenic
concentration of 0.004 mg/kg in the whole blood of 8 normal subjects.
Bergström & Wester (1966), also using neutron activation, noted a mean
level of 0.002 mg/kg in whole blood (3 samples), of which 0.0011 mg/kg
was present in the serum. A mean concentration of 0.0011 mg/litre was
also found in 11 samples of normal human serum by Damsgaard, et al.
(1973) using neutron activation analysis. Mean arsenic values (7-16
samples) of 0.0024 mg/litre in plasma, 0.0027 mg/litre in red cells
and 0.025 mg/litre in whole blood have been reported for healthy
Danish individuals (Heydorn, 1969) whereas corresponding values for
normal subjects (6-17 samples) from China (Province of Taiwan) were
0.0154 mg, 0.0327 mg, and 0.0216 mg/litre, respectively. All
measurements were performed using neutron activation analysis.
Blackfoot disease patients and members of their families living in the
endemic area of China (Province of Taiwan) (section 8.3.4) showed mean
values of about 0.03 mg/litre in plasma (Astrup, 1968; Heydorn, 1969),
0.093 mg/litre in red cells, and 0.060 mg/litre in whole blood
(Heydorn, 1969). The mean concentrations of arsenic in well water in
the endemic area ranged from 0.054 mg to 0.743 mg/litre (Kuo, 1968).
Kagey, et al. (1977) reported a mean blood arsenic level of
0.0023 mg/litre among 50 female smokers and a mean of 0.0015 mg/litre
among 49 nonsmokers in the USA (atomic absorption spectrophotometry).
A mean blood level of arsenic of 0.00145 mg/kg (range 0.0005-
0.032 mg/kg, neutron activation analysis) has been reported for
10-year-old children living in a country town in Czechoslovakia
(Bencko & Symon, 1977). A mean blood-arsenic level of 0.00188 mg/kg
(range 0.0005-0.0038 mg/kg) was found among children of the same age
in a metropolitan city and a mean of 0.00453 mg/kg (range
0.0025-0.0082 mg/kg) among children living near a coal-fired power
plant.
In a study by Wagner & Weswig (1974), levels of arsenic in the
blood ranging from 0.01 to 0.27 mg/kg were found in forestry workers
exposed to dimethylarsinic acid (SDDC method). Unexposed workers were
reported to have levels in the range of 0.01-0.13 mg/litre, which seem
to be high compared with normal values reported by others using
neutron activation analysis. An average arsenic level in whole blood
of 0.033 mg/kg (range 0.012-0.055 mg/kg; atomic absorption
spectrophotometry) was found among 23 workers in a workshop producing
wood preservatives (arsenic acid) from arsenic(III) oxide which
probably was the cause of most of the airborne arsenic (Yamamura &
Yamauchi, 1976). The average value for the controls was reported to be
0.007 mg/kg.
The major part of both inorganic and organic arsenic in blood is
cleared fairly rapidly in man. Blood arsenic will therefore reflect
exposure for only a short period following absorption and will be very
timedependent. Only if exposure is continuous and steady, as is
sometimes the case with exposure through drinking-water, will arsenic
reach a steady-state in the blood and, thus, make it possible to
arrive at a relationship between blood arsenic levels and exposure.
Even so, data are not available that indicate any quantitative
relationship relevant to man between arsenic exposure and
concentrations of arsenic in the blood.
The short half-time of arsenic in the blood compared with the
biological half-time in the whole body makes it difficult to establish
a relationship between the blood level of arsenic and total body
burden or concentrations in different organs. A metabolic model for
the different forms of arsenic has yet to be established.
7.2 Urine
Values given in the literature for normal background levels of
arsenic in urine cover quite a wide range, probably because of the
influence of dietary sources and differences in analytical methods.
When a person has ingested a seafood meal, the urinary arsenic
concentration can often rise to over 1 mg/litre during the subsequent
24 h (Schrenk & Schreibeis, 1958; Pinto et al., 1976). Westöö & Rydälv
(1972) found that up to 1.5 mg of arsenic was excreted in the urine
during the first day following ingestion of fish containing about 2 mg
of arsenic. Studies by Crecelius (1977b) and Cannon et al. (1979)
indicated that the organic arsenic compounds present in marine
organisms are probably not metabolized in the human body, but are
excreted in the urine in the form in which they are ingested (section
6.2.4). Determination of inorganic arsenic and methylated arsenic
acids in urine (the main metabolites of inorganic arsenic), using a
method described by Braman & Foreback (1973) and Crecelius (1977), is
not influenced by the presence of organic arsenic compounds in seafood
(Crecelius, 1977). Thus, it is possible to differentiate between
arsenic taken in the form of inorganic arsenic and that ingested in
the form of seafood organoarsenic compounds.
A mean urinary value of about 0.011 mg/litre (SDDC method) was
reported by Bencko & Symon (1977) for a group of 10-year-old boys with
no known exposure to arsenic. Seafood was probably only a minor
ingredient in the diet. The mean urinary arsenic values in groups of
children living within 7.5 km of a coal-fired power plant were
0.0189-0.0253 mg/litre (range < 0.001-0.105 mg/litre).
In preemployment examinations of over 200 men, Pinto et al.
(1976), using the arsine generation/spectrophotometric method, found a
background arsenic value in urine of 0.053 mg/litre. Samples were
collected without regard to prior seafood consumption. Using arsine
generation/plasma excitation emission spectrophotometry Smith et al.
(1977) reported a geometric mean of 0.021 mg/litre among 41 unexposed
workers. The total urinary arsenic concentrations as well as various
forms of arsenic (inorganic arsenic, dimethylarsinic acid,
methylarsonic acid) were log-normally distributed among the 41 male
workers. Braman & Foreback (1973) reported total arsenic levels
(arsine generation and atomic emission spectrophotometry) in the urine
of 4 subjects of between 0.010 and 0.030 mg/litre. They found that the
predominant form of arsenic was dimethylarsinic acid, which accounted
for between 40% and 87% of the total arsenic. Other forms of arsenic
in the urine were methylarsonic acid and inorganic trivalent and
pentavalent arsenic.
Increased urinary levels of arsenic have been detected among
people living in the vicinity of smelters. Holmqvist (1975) measured
arsenic in the urine of smelter workers with low exposure to arsenic
and women living in 2 villages, 1 within 3 km of the smelter and the
other approximately 5 km from it. The highest values, mean
0.05 mg/litre (analytical method not given), were found in women
living in the village 5 km from the plant. Women living over 70 km
from the plant showed a mean value of 0.03 mg/litre.
In a survey among children in 11 copper smelter towns in the USA,
a geometric mean of 0.019 mg Am/litre of urine (atomic absorption
spectroscopy) was found and 0.006 mg/litre in 3 control towns without
a smelter (Baker et al., 1977). The children in the town most heavily
contaminated with arsenic had a geometric mean of 0.018 mg As/litre of
urine.
Total arsenic in urine has been used traditionally to assess
occupational exposure to inorganic arsenic. It is obvious that
detailed records of dietary habits should accompany all studies in
which total urinary arsenic is used as an indicator of industrial or
other environmental exposure. Generally, this has not been the case.
There are many reports concerning total urinary arsenic excretion
following industrial exposure, some examples of which will be given
here. Lundgren (1954) found an average arsenic concentration of
0.54 mg/litre in the urine of smelter employees, as opposed to only
0.04 mg/litre in workers without arsenic exposure (analytical method
not given). An average urinary concentration of 0.82 mg/litre (median
0.58 mg/litre; titrimetric method) was reported by Pinto & McGill
(1953) in men exposed to arsenic in a smelter in the USA. In the same
smelter, more than 20 years later, Pinto, et al. (1976) found an
average urinary level of arsenic of 0.174 mg/litre (range
0.038-0.539 mg/litre). Kodama, et al. (1976) reported an average
urinary arsenic concentration of 0.056 mg/litre (standard deviation
0.045) among 42 workers in a copper smelter in Japan (molybdenum blue
method).
Using the silver diethyldithiocarbamate method, Tarrant & Allard
(1972) studied the urinary excretion of forest workers in thinning
operations whereby they were exposed mainly to dimethylarsinic acid
(cacodylic acid) and monosodium methane arsonate. The average urinary
arsenic value at the beginning of a working week (Monday morning) was
0.08 mg/litre compared with 0.32 mg/litre at the end of the week
(Friday afternoon).
Concentrations of arsenic in both air and urine have been reported
in only a few studies. Pinto et al. (1976) found a correlation between
urinary arsenic levels in smelter workers and concentrations of
arsenic in the air in the smelter (Fig. 4). Arsenic was determined by
an arsine generation/spectrophotometric method. The workers wore
personal monitors for 5 consecutive workdays to measure the levels of
arsenic in the air. The correlation was reported to be significant at
arsenic concentrations in air below 0.3 mg/m3 and urinary arsenic
levels below 0.5 mg/litre. However, as can be seen in Fig. 4, there is
considerable scatter in the values and this correlation is not evident
at air levels below 0.05 mg/m3.
Carlsson (1976) studied urinary arsenic excretion in workers at a
Swedish smelter. Where it was established that subjects had eaten
seafood on the days immediately prior to the collection of urine
samples, they were excluded from the study. The men used gas masks and
personal air samplers in such a way that only the arsenic present in
the inhaled air was measured. The 8-h mean arsenic concentration in
air was 0.057 mg/m3 (range 0.002-0.227 mg/m3; X-ray spectrometric
method) and the mean arsenic concentration in the morning urine,
collected the day following that of the exposure was 0.2 mg/litre
(Gutzeit method). No clear linear relationship emerged between
airborne arsenic exposure and the total urinary arsenic.
Only one report has been published on the different forms of
arsenic in the urine of exposed smelter workers. Smith et al. (1977)
measured the concentration of respirable (< 5 µ) and nonrespirable
arsenic in the air during one working day as well as the
concentrations in urine. Arsenic was mainly excreted as
dimethylarsinic acid and the proportion of the different forms of
arsenic in the urine was, according to the authors, independent of
exposure levels (arsine generation/plasma excitation emission
spectrophotometry). The correlation between total airborne arsenic and
total urinary arsenic and the correlation between airborne arsenic and
the different forms of urinary arsenic were reported to be
significant. However, the values showed substantial scatter and
furthermore, the data could not be used to arrive at a correlation
between airborne arsenic and urinary excretion, since the workers used
chemical cartridge respirators, and it was not stated how much of the
arsenic was captured on the filters.
Urinary arsenic levels have also been shown to be correlated with
intake of arsenic in drinking-water. A survey was conducted by
Harrington, et al. (1978) among a population in an area with elevated
arsenic levels in the well water. Drinkers of well water with an
arsenic content exceeding 0.1 mg/litre (mean 0.401 mg/litre and an
estimated total daily intake of 0.324 mg of arsenic) had an average
urinary concentration of 0.178 mg/litre (atomic absorption
spectroscopy). Drinkers of well water containing an average arsenic
concentration of 0.031 mg/litre (estimated daily intake of 0.046 mg of
arsenic) had a mean urinary arsenic concentration of 0.041 mg/litre.
Ingestion of arsenic in water was also found to be reflected in
urinary excretion in a smelter town in the USA (Morse et al., 1979).
Children drinking tap water containing arsenic at 0.09 mg/litre
excreted an average of 0.059 mg As/litre of urine, while children in a
control town, where the arsenic level in the water was 0.01 mg/litre,
excreted a mean concentration of 0.018 mg As/litre of urine. Children
in both towns, who drank bottled water (with undetectable arsenic
contents), excreted average urinary arsenic levels of 0.03 mg/litre
and 0.01 mg/litre, respectively.
Whole body retention and elimination were studied in mice
following administration of 74As-labelled As(III) and As(V), with
strict control of valence state at the time of exposure (Vahter &
Norin, 1980). In mice given a single oral dose of 4 mg As/kg body
weight, whole body retention was 2-3 times higher after exposure to
As(III) than after exposure to As(V). A similar difference was seen
after subcutaneous injection of 0.4 mg/kg body weight but differences
in retention were not seen when this dose was given orally.
Daily doses of 0.03-0.66 mg As/kg body weight given to cows in the
form of arsenic acid for up to 8 weeks did not cause the arsenic
levels in milk to rise (Peoples, 1964). Following an outbreak of
arsenic poisoning in cattle in Mexico caused by the ingestion of feed
containing up to 0.28% arsenic, there was a very drastic reduction in
milk production. Levels of up to 0.5 mg As/litre were found in the
milk of cows still producing it (de Navarro, 1976; Gonzales, 1977). In
view of the high exposure, these levels of arsenic in milk indicate
that arsenic ingested in the form of inorganic arsenic does not
readily pass the blood-mammary barrier.
Two studies in rats on the elimination of arsenic via the lungs
have been reported (Lanz et al., 1950; Dutkiewicz, 1977). Both seem to
indicate that little, if any, is eliminated by this route. A study by
Overby & Fredrickson (1963) using chickens also indicated a very low
elimination of 74As-arsenic through the lungs, following exposure to
74As-arsenate.
6.1.3.2 Man
Mappes (1977) described a series of experiments in which he,
himself, ingested 2 mg of arsenic as arsenic(III) oxide in an aqueous
solution. About 30% of the ingested arsenic had been recovered in
urine (molybdenum blue method) 24 h after exposure. Mappes also took
daily doses of 0.8 mg of arsenic as arsenic(III) oxide in an aqueous
solution. The excretion rate of arsenic reached equilibrium after 5
days, by which time about 70% of the daily dose was being excreted in
the urine daily.
Crecelius (1977b) studied the excretion of arsenic in the urine of
a person who had ingested arsenic-rich wine (50 µg As(III) and
13 µg As(V)) and water containing 0.2 mg of arsenic mainly in the
pentavalent form. About 80% of the arsenic ingested with the wine was
excreted within 61 h. The biotransformation of the ingested arsenic
was also studied (section 6.1.4.2) and the apparent biological
half-time for the in vivo methylated arsenic, which was the major
species of arsenic excreted, was found to be of the order of 30 h,
compared with 10 h for arsenic eliminated in the inorganic form. Only
50% of the pentavalent arsenic ingested with the water was recovered
in the urine during the first 70 h following ingestion.
A single oral dose of carrier-free 74As was administered in a
gelatine capsule to each of 6 adult male volunteers (about 0.01 µg
As/man; over 90% As(V)) (Tam et al., 1979a; Pomroy et al., 1980).
During the first 24 h after dosing, 22.4% of the 74As dose was
excreted via the urine. After 5 days, a total of 58% had been
recovered in the urine. No data were presented on faecal elimination.
Data from whole body measurements were best represented by a
3-compartment exponential model. 65.9% of the dose was eliminated with
a half-time of 2.1 days, 30.4% with a half-time of 9.4 days, and 3.7%
with a half-time of 38.4 days.
Though absorbed arsenic is excreted mainly via the kidneys, a
small amount is removed by other routes. Studies on the constituents
of profuse sweat induced in a hot and humid environment have been
reported by Vellar (1969). The mean concentration of arsenic in the
sweat of 2 subjects was 1.5 µg As/litre (neutron activation analysis)
and the calculated hourly loss of arsenic was 2 µg. Data concerning
sweat levels under normal conditions were not reported. Desquamation
of skin will also result in the removal of arsenic, since arsenic has
a high affinity for skin (section 6.1.2.2). Molin & Wester (1976)
calculated the daily loss of arsenic through desquamation of normal
skin (10 male patients, apparently unexposed to arsenic) to be
0.1-0.2 µg based on their finding of a mean arsenic concentration in
skin of 0.18 mg/kg dry weight (neutron activation analysis). There are
some data on the role of the hair as a route of elimination of
arsenic. Several authors (section 7.3) have reported hair levels of
arsenic exceeding 100 mg/kg among occupationally exposed subjects.
Some of this arsenic may result from external exposure, however. Hair
arsenic levels in Japanese subjects poisoned by contaminated soy sauce
(3 mg of arsenic daily for 2 or 3 weeks) were between 1.8 and
13 mg/kg, 2 weeks after ingestion (Mitzuta et al., 1956). If a hair
weight of 20 g is assumed (Task Group on Reference Man, 1975), this
would account for 0.6% of the ingested arsenic, at the most.
Grimanis et al. (1979) determined the concentrations of arsenic
and some other trace metals in human milk using neutron activation
analysis. There were no differences between levels in human colostrum,
transitional, and mature milk, all of which were about 3 µg/litre
(range 0.6-6.3 µg/litre). Colostrum and transitional milk were
obtained from 15 healthy mothers living in the Athens area with a mean
age of 26 years. Mature milk was obtained from 5 of the 15 mothers.
6.1.4 Biotransformation
The form of arsenic present in most tissues is largely unknown
because of the analytical difficulties involved. However,
differentiation between the various forms of arsenic is more reliable
in plasma and urine, where arsenic can be measured without previous
digestion.
6.1.4.1 Animals
After intramuscular administration of arsenate (arsenic(V)) to
rats, about 10-15% of the total urinary arsenic was reported to be in
the form of arsenic(III) (Lanz et al., 1950). The different forms were
separated by precipitation of magnesium-ammonia-arsenate. In dogs,
given an intravenous infusion of 74As-arsenate, arsenic(III) was
reported to be present in the urine (about 14% of the total arsenic)
as well as in the plasma (about 5.5% of the total plasma arsenic)
(Ginsburg & Lotspeich, 1963). The method used for the separation of
the different forms of arsenic was that described by Crawford & Storey
(1944) in which arsenic(III) was extracted with ethyl xanthate. In
further work, Ginsburg (1965) reported that injection of arsenate in
dogs caused arsenic(III) to appear in the glomerular filtrate and, to
a still greater extent, in the urine.
When trivalent arsenic (arsenite) was intravenously infused, both
arsenate and arsenite were detected in the plasma, glomerulus
filtrate, and urine of dogs indicating an in vivo oxidation of
trivalent to pentavalent arsenic (Ginsburg, 1965). Bencko et al.
(1976) reported that in vivo oxidation of arsenite can occur in
mice. About one half of the intramuscularly administered arsenic
(74As-labelled sodium arsenite, 1.3 mg As/kg body weight) was
recovered as pentavalent arsenic in urine taken directly from the
bladder (analysed by paper chromatography). When the mice were exposed
to arsenite in drinking water before the injection, an even greater
part of the excreted arsenic was pentavalent.
The studies, just referred to, did not take into consideration
that in vivo methylation of arsenic occurs. It was therefore not
possible for the Task Group to evaluate to what extent such methylated
forms of arsenic may have interfered in the separation of the two
inorganic forms. In the absence of more detailed studies, no firm
conclusions can be drawn about the in vivo reduction or oxidation of
inorganic arsenic.
Methylated arsenic has been detected in the urine of cows and dogs
fed arsenate or arsenite (Lakso & Peoples, 1975). When the dogs were
fed doses of about 1.0 mg As/kg body weight of either valence form for
5 days, approximately equal amounts of inorganic and methylated
arsenic were excreted in the urine. The cows produced about 3 times as
much methylated arsenic as inorganic arsenic in their urine.
Following intravenous administration of carrier-free 74As-arsenic
acid to an adult male beagle (14.8 MBq, 0.4 mCi), 74As was present in
plasma or urine predominantly as inorganic arsenic and dimethylarsinic
acid, as revealed by separation on a cation-exchange column (Tam et
al., 1978, 1979b). When plasma, red blood cells, or urine from a
beagle were incubated in vitro with 74As-arsenic acid, no
methylated arsenic was found in the plasma or urine samples but a
small amount (0.2%) of dimethylarsinic acid did appear in the
erythrocyte samples (Tam et al., 1979c).
Ten minutes after intravenous administration of 74As-arsenic acid
to beagles (0.5 µg As/dog), about 8% of the total amount of 74As
present in the erythrocytes was in the form of dimethylarsinic acid
whereas no methylated arsenic could be found in the plasma
(Charbonneau et al., 1978b). Six hours after dosing, the small amount
of 74As still present in the erythrocytes and plasma was
predominantly in the form of dimethylarsinic acid as was 5-25% of the
total 74As present in urine 1 h after dosing and 90%, 6 h after. When
a single oral dose of 0.2-0.6 µg As was administered to beagles as
74As-arsenic acid, a similar metabolic pattern was revealed
(Charbonneau et al., 1979).
6.1.4.2 Man
Braman & Foreback (1973) reported the presence of methylated forms
of arsenic in the urine of 4 subjects. Dimethylarsinic acid
constituted on average 66% of the total urinary arsenic, while
methylarsonic acid, pentavalent arsenic, and trivalent arsenic
accounted for 8.0, 17.0, and 8.4%, respectively. Actual in vivo
methylation of inorganic arsenic was later indicated by the work of
Crecelius (1977b), who measured the different forms of arsenic in the
urine of a subject who had ingested wine containing 50 µg of
arsenic(III) and 13 µg of arsenic(V). Urinary dimethylarsinic acid
accounted for 50% of the ingested arsenic, methylarsonic acid for 14%
and inorganic arsenic for 8%. Dimethylarsinic acid was also the major
form of arsenic found in the urine of smelter workers occupationally
exposed to arsenic, chiefly in the form of arsenic(Ill) oxide (Smith
et al., 1977).
In a person who had ingested well water containing 0.2 mg of
pentavalent arsenic, the inorganic arsenic(V) concentration in urine
showed a marked increase (5-fold) the first 10 h after exposure,
indicating that some of the ingested arsenic was rapidly excreted
unchanged in the urine (Crecelius, 1977b). The urinary levels of
dimethylarsinic acid increased 5 to 10-fold between 10 and 70 h after
exposure. Only 50% of the arsenic ingested was excreted in the urine
within 70 h. Following oral ingestion of 74As (> 90% As(V)) by 6
adult males (about 0.01 µg As/man), 58% of the dose was excreted in
the urine during the first 5 days (Tam et al., 1979a). Of the excreted
arsenic, 51% was in the form of dimethylarsinic acid, 21%
monomethylarsenic compounds, and 27%, inorganic arsenic.
No methylation of 74As was found in human plasma or urine
incubated in vitro with 74As-arsenic acid (Tam et al., 1979c).
Further studies are required in other species to determine whether
the monomethylarsenic compound is a metabolite unique to man.
6.2 Organic Arsenic Compounds
6.2.1 Absorption
6.2.1.1 Respiratory absorption
6.2.1.1.1 Animals
One organic arsenic compound of interest is dimethylarsinic acid
(cacodylic acid), since it may be inhaled when it is used as a
herbicide. Absorption of this compound following intratracheal
administration was studied in rats by Stevens et al. (1977). He found
that a solution of 14C-dimethylarsinic acid was rapidly absorbed from
the lung. Less than 5% remained unabsorbed after 15 min and the
absorption half-time was calculated to be 2.2 min.
6.2.1.1.2 Man
No data are available concerning the respiratory absorption of
organic arsenic in man.
6.2.1.2 Gastrointestinal absorption
6.2.1.2.1 Animals
Absorption of seafood arsenic from the gastrointestinal tract was
investigated in rats by Coulson et al. (1935). When rats were given a
shrimp diet, only about 4% of the ingested organic arsenic
(approximately 0.5 mg As) was recovered in the faeces during the first
2 days following exposure, indicating almost complete absorption from
the gastrointestinal tract.
When fish containing arsenic was given to pigs in a single meal in
amounts corresponding to 0.3 mg As/kg body weight, 23% of the ingested
arsenic was recovered in the faeces collected over a period of 10 days
following exposure (Munro, 1976). About the same high absorption (80%
or more) was observed by Munro (1976) in adolescent monkeys fed
arsenic-containing fish corresponding to a dose of 1 mg As/kg body
weight and by Charbonneau et al. (1978a) in adult female cynomolgus
monkeys given a single test meal (via a stomach tube) of homogenized
fish containing arsenic (1 mg As/kg body weight).
Thirty-one percent of an oral dose of 0.5 ml of an aqueous
solution containing 40 µg of radiolabelled dimethylarsinic acid
(approximately 20 µg As/rat) was eliminated in the faeces of rats
during 24 h (Stevens et al., 1977).
From reports on the effects and distribution of other organic
arsenic compounds (pesticides, feed additives for poultry and swine,
chemotherapeutic agents), it is evident that uptake of these compounds
occurs, when they are given orally to laboratory animals. The amount
of the administered dose absorbed from the gastrointestinal tract
differs over a wide range, depending on the chemical properties of the
compound. A chemotherapeutic agent of low lipid solubility,
p-N-glycol-arsanilate, was shown to be poorly absorbed from the
gastrointestinal tract (McChesney et al., 1962). Other
chemotherapeutic agents, such as carbarsone ( p-ureidophenylarsonic
acid) and tryparsamide ( N-(carbamoylmethyl) arsanilic acid), and
pesticides such as sodium methane arsenate and dimethylarsinic acid,
were readily absorbed, when fed to rats and rabbits (Hwang & Schanker,
1973; Exon et al., 1974; Stevens et al., 1977). The absorption
half-times of carbarsone, tryparsamide, and dimethylarsinic acid from
the small intestine of the rat were 87, 184, and 201 min, respectively
(Hwang & Schanker, 1973). The absorption process did not show any
evidence of saturation when the concentrations of the compounds were
increased up to 100-fold. The absorption rates were ranked in the same
order as the chloroform-to-water partition coefficients indicating,
according to the author, that absorption takes place mainly by
diffusion.
Liver, from pigs fed arsanilic acid, which contained about 6 mg
As/kg, was administered to rats at a level of 30% of the diet. The
daily faecal elimination of the rats contained 70-85% of the intake of
arsenic, indicating that this form of arsenic is not as readily
absorbed as inorganic arsenic or "seafood arsenic" (Overby & Frost,
1962).
The same type of experiment was reported by Calvert (1975) who fed
wethers dried broiler manure containing 3-nitro-4-hydroxy-
phenylarsonic acid in concentrations of 3.4-5 mg As/kg of diet. He
collected urine and faeces during the last 5 days of a 15-day feeding
period and found 60-73% of the ingested arsenic in the faeces.
6.2.1.2.2 Man
A single meal of fish or crustacea containing high levels of
mainly organic arsenic may result in the ingestion of several
milligrams of arsenic, most of which is apparently absorbed from the
gastrointestinal tract. Coulson et al. (1935) reported an experiment
in which each of 2 human subjects ate boiled shrimps containing about
1 mg of arsenic. By the fourth day, approximately 5% had been
recovered in the faeces indicating that absorption from the
gastrointestinal tract was almost complete. Four persons who ate
plaice and cod with high arsenic levels excreted an average of 83% of
the ingested arsenic (0.5-2.2 mg/person) in the urine during the 2.5
days following exposure indicating that the fish arsenic was readily
absorbed (Westöö & Rydälv, 1972).
Arsanilic acid, used as a feed additive for poultry and swine, may
be ingested in trace amounts, when meat from these animals is eaten.
The availability to man of arsanilic acid or of arsenic in the tissues
of chicks fed arsanilic acid was studied by Calesnick et al. (1966).
Four adult male volunteers ingested single doses of 1.3-3.0 mg of
arsenic as 74As-arsanilic acid. The average faecal elimination within
6 days of exposure was 74% of the dose. Following ingestion of pâté
made from chicks fed 74As-arsanilic acid, approximately 64% of the
arsenic ingested was recovered in the faeces. Apparently, arsenic from
the flesh of animals fed with additives containing arsenic is not
absorbed from the gastrointestinal tract as readily as arsenic from
fish or crustacea.
6.2.1.3 Skin absorption
No data are available on the absorption of various organic arsenic
compounds through the skin of animals or man.
6.2.1.4 Placental transfer
6.2.1.4.1 Animals
There are no data on the placental transfer of organic arsenic
compounds present in seafood. Dimethylarsinic acid has been shown to
pass the placental barrier of rats, when administered intravenously
just prior to parturition (Stevens et al., 1977). The dose given to
the pregnant rats was 40 µg of radiolabelled dimethylarsinic acid per
rat (20 µg As/rat). Fetal whole blood levels were comparable with
those in the maternal blood.
Transfer of organic arsenic from hens to eggs has been reported.
Increased concentrations of arsenic were found in eggs of hens fed 50
or 100 mg As/kg diet as 3-nitro-4-hydroxyphenylarsonic acid (Daghir &
Hariri, 1977). The highest levels of arsenic (about 0.24 mg/kg) were
found after 4-6 weeks of feeding, after which the levels gradually
decreased, indicating that the hens developed a tolerance to organic
arsenic similar to that found towards inorganic arsenic (section
6.1.2.2.1).
6.2.1.4.2 Man
There are no human data available concerning the placental
transfer of organic arsenic compounds.
6.2.2 Distribution in organisms
6.2.2.1 Fate of organic arsenic in blood
6.2.2.1.1 Animals
Data are not available on blood arsenic levels following ingestion
of seafood arsenic.
Stevens et al. (1977) investigated the kinetics of dimethylarsinic
acid in the plasma of rats after intravenous administration of 200 mg
14C-dimethylarsinic acid/kg body weight
(108 mg As/kg body weight). After a single injection, the plasma
concentration followed a three-exponential equation with half-times of
0.014, 0.217, and 3.42 h. The retention of 14C-dimethylarsinic acid
in whole blood was high, about 10% of the dose 3 months after the
administration indicating that the rat differs from other animals with
regard to metabolism of this arsenic compound. As dimethylarsinic acid
is a major metabolite of inorganic arsenic, it might be expected to be
cleared from the blood fairly rapidly.
The clearance of the major part of the arsenic from the blood of
chickens given a single oral dose of arsanilic acid showed 2 rapid
phases, with half-times of about 90 min and 6 h, respectively (Overby
& Fredrickson, 1963). About 99.9% of the dose was cleared at these
rates. The remaining 0.1% was cleared with a half-time, of 36 h.
Less than 6% of doses of 4 different organo-arsenic drugs, given
intravenously to rabbits, remained in the blood 2 h after the
administration (Hogan & Eagle, 1944). The 4 arsenic compounds were
unsubstituted phenylarsenoxide, 2 substituted phenylarsenoxides, and
tryparsamide. Red blood cells and plasma showed marked differences in
clearance rate and distribution. In the case of the most toxic
compound, the unsubstituted phenylarsenoxide (trivalent arsenic),
almost all the injected arsenic was still in the blood 0.75-1.5 min
after the injection. More than 95% of the dose was found in the red
blood cells. More than 50% of the dose of the less toxic tryparsamide
(pentavalent arsenic) had left the blood within the same span of time,
and more than 95% of the remaining arsenic was in the plasma. The same
distribution among blood cells and plasma was obtained in in vitro
studies of the binding of arsenic compounds to red blood cells. The
amounts of the various phenylarsenoxides (acid-substituted compounds
excepted) bound to red blood cells corresponded very well to their
acute toxicities in white mice. The arsonic acids were bound only to a
minor degree to the red blood cells in vitro. They were also
relatively nontoxic in vivo, but the toxicity varied from one
compound to another.
6.2.2.1.2 Man
No human data are available concerning the fate of organo-arsenic
compounds in the blood.
6.2.2.2 Tissue distribution of organic arsenic
6.2.2.2.1 Animals
Data on the tissue distribution of seafood arsenic in experimental
animals and man are lacking. The only available report is that of
Lunde (1972), who studied the distribution of organic arsenic in fish
(rainbow trout) fed a marine diet containing about 15 mg As/kg in the
form of organoarsenic compounds to which inorganic 74As had been
added. The content of radioactive inorganic arsenic in the fish was
negligible, 6-10 days after the addition of radioactive arsenic was
stopped, but a small fraction of the inorganic arsenic was converted
to organoarsenic compounds. Autoradiography revealed that the 74As
was especially concentrated in the eyes, throat, gills, and pylorus
organ. The liver and kidney also contained much radioactivity, but
arsenic disappeared faster in these than in other organs, when the
feeding of radioactive arsenic was discontinued.
Administration of a diet containing 50 mg/kg of monosodium methane
arsonate (MSMA) (27.5 mg As/kg) for 52 weeks caused a rapid increase
in the arsenic contents in the liver and kidney of rabbits during the
first 2 weeks (Exon et al., 1974). Accumulation of arsenic in hair was
observed in cattle exposed to dietary MSMA or dimethylarsinic acid
(Dickinson, 1972). The animals were fed daily doses of dimethylarsinic
acid at 10 mg/kg body weight (5.4 mg As/kg body weight) and had
arsenic levels in hair of 2.0-4.3 mg/kg (three animals) after 10 days
and 13-33 mg/kg after 48 days.
Calvert (1975) fed wethers various amounts of arsanilic acid and
measured the arsenic levels in the liver, kidney, and muscle. The
results, shown in Table 6, suggest that arsenic given as arsanilic
acid is concentrated in the liver and kidney. When the animals were
placed on an arsenic-free diet, the tissue levels decreased rapidly,
as shown in Table 7, and had dropped to about 15% of the original
value by the sixth day.
Table 6. Arsenic levels in tissues of wethers, fed arsanilic acid
for 28 daysa
Arsenic Whole Liver Kidney Muscle
fed blood
(mg/kg of mg/kg dry
diet) tissue
0.0 < 0.01 < 0.01 < 0.01 < 0.01
26.8 0.063 3.1 3.2 0.2
144.4 0.270 26.8 12.2 1.1
273.3 0.536 29.2 23.6 1.2
a From: Calvert (1975).
Table 7. Depletion of arsenic in the liver of wethers fed
arsanilic acid for 28 daysa
Arsenic Withdrawal time (days)
fed 0 2 4 6
(mg/kg of mg/kg dry tissue
diet)
0.0 < 0.01 < 0.01 < 0.01 < 0.01
26.8 3.1 4.9 2.9 1.9
144.4 26.8 15.4 8.4 3.5
273.3 29.2 27.0 11.4 5.0
a From: Calvert (1975).
When pigs were fed arsanilic acid (1000 mg/kg diet, approximately
340 As mg/kg diet), the maximum arsenic level in most tissues was
reached on the 13th day (Ledet et al., 1973). However, maximum levels
in the nervous tissues (CNS and peripheral nerves) were not reached
until the 20th day. Clearance of arsenic was slower in these than in
the other tissues. Highest levels were found in liver and kidney.
Injections of the trivalent organoarsenic drug phenylarsenoxide in
rabbits resulted in 50-100 times higher liver arsenic levels than
injections of the relatively nontoxic 3-NH2-4-OH derivative of the
pentavalent compound tryparsamide (substituted phenylarsonic acid)
(Hogan & Eagle, 1944). After injections of these compounds at the
LD50-level, comparable amounts of arsenic were found in the tissues
despite the 500-fold difference in dose. The highly toxic
acid-substituted 4-COOH-phenylarsenoxide caused extraordinarily high
kidney levels in the first hours after injection (10-20% of the dose)
and levels comparable with the other substituted phenylarsonic acids
after 24-48 h.
6.2.2.2.2 Man
No data are available on the distribution of organic arsenic
compounds in man.
6.2.3 Elimination
6.2.3.1 Animals
A substantial fraction of seafood arsenic administered to animals
is rapidly eliminated from the body. About 75% of a dose of shrimp
arsenic (approximately 0.5 mg As) given to rats in food was eliminated
within the first day, and an additional 20% was eliminated during the
second day, predominantly in the urine (Coulson et al., 1935). Pigs
given a fish diet providing a dose of 0.3 mg As/kg body weight
eliminated 90% of the ingested arsenic within 3 days (Munro, 1976).
About 70% of the dose was recovered in the urine. When the same type
of diet was given to adolescent monkeys (Macaca irus) corresponding
to doses of 1 mg As/kg body weight, only 63% of the dietary arsenic
was recovered in the excreta within 10 days (18% in the faeces and 45%
in the urine). When a single oral dose of fish arsenic providing about
1 mg fish arsenic/kg body weight to adult female cynomolgus monkeys,
57-84% of the ingested arsenic was excreted in the urine within 3
days. The total recovery in the excreta was 66-85% during 14 days
(Charbonneau, et al., 1978a).
The feeding of organic arsenic in the form of arsanilic acid to
poultry and swine as a feed additive, may result in tissue residues.
Rats fed protein from swine liver containing 24.4 mg arsenic/kg
protein as arsanilic acid, at a level of 300 g/kg diet for 14 days,
eliminated almost all of the arsenic within 7 days of the end of the
feeding period (Overby & Frost, 1962). The amount of arsenic excreted
in the urine was one third of the amount of arsenic eliminated in the
faeces during this period. Experiments with pigs given a diet
containing 0.01% arsanilic acid for 31 days showed a rapid decrease in
the tissue levels of arsenic during the first days after the feeding
of arsanilic acid was discontinued (Ferslew & Edds, 1979). The livers
contained arsenic concentrations of 1.5-2 mg/kg on the 31st day of
feeding and about 0.2 mg/kg on the 7th day after the removal of
arsanilic acid from the diet.
When pentavalent arsenic compounds in the form of dimethylarsinic
acid, p-N-glycolylarsanilate, arsanilic acid, 4-nitrophenylarsonic
acid, and 3-nitro-4-hydroxyphenylarsonic acid were administered
parenterally to rats, some 50%-80% of the injected dose was excreted
in the urine within the first 48 h (McChesney, et al., 1962; Schreiber
& Brouwer, 1964; Stevens, et al., 1977). Only minor amounts were found
in the bile and faeces. Seventy percent of a dose of p-N-
glycolylarsanilate (used in veterinary medicine), intravenously
injected into cats, was excreted in the urine and 0.8% excreted in the
bile during the first 160 min following exposure (McChesney, et al.,
1962). Rabbits injected intravenously with another drug, tryparsamide,
excreted 68% of the dose in 24 h and 81% of the dose in 48 h (Hogan &
Eagle, 1944). Derivatives of phenylarsenoxide, a trivalent
organoarsenic compound, were excreted in the urine of rabbits at much
slower rates than the pentavalent arsenic compound, tryparsamide. The
elimination rates depended on the functional groups on the benzene
ring and varied from 5% in 24 h for unsubstituted phenylarsenoxide to
60% for the 3-NH2-4-OH-derivative.
Cristau et al. (1972, 1975) investigated the influence of the
molecular structure of some arsenic-containing drugs on the
elimination kinetics of rats and guineapigs. Acetarsol, tyrparsamide,
diphetarsone, and melarsonyl, all of which possess hydrophilic
functional groups that facilitate elimination without
biotransformation, were rapidly eliminated (65%-90% of the dose in
24 h). Acetarsol and tryparsamide were mainly excreted in the urine
and to a much lesser extent in the bile. Melarsonyl, which has a
higher relative molecular mass (532.5) than the other drugs tested,
was excreted to a greater extent in the bile in both species. About 10
times more diphetarsone, which has a relative molecular mass of 460,
was excreted in the bile of rats than in the bile of guineapigs. This
was reported to agree with previous observations that rats excrete
more of some drugs in the bile than guineapigs. Melarsoprol and
arsthinol are both hydrophobic, which indicates that they must undergo
biotransformation prior to elimination. Both drugs were excreted
slowly (20%-50% of the dose in 24 h), predominantly via the bile.
There are only a few reports on the excretion of arsenic in the
milk of animals. The arsenic level in cow's milk did not increase with
the blood concentration when the cows were fed methylarsonic acid or
dimethylarsinic acid (Peoples, 1975). However, the milk arsenic levels
did increase when cows were fed arsanilic acid or
3-nitro-4-hydroxyphenylarsonic acid (1.6-3.2 mg As/kg body weight)
(Calvert, 1973).
No radioactive arsenic was detected in air exhaled by chickens,
given an 74As-labelled arsanilic acid orally, indicating that this
arsenic compound was not eliminated via the lungs or metabolized to
form expirable products (Overby & Fredrickson, 1963).
6.2.3.2 Man
Only a few studies are available on the elimination of organic
arsenic compounds in man. As early as 1919, Bang stated that organic
arsenic in fish and other marine foods was readily excreted, mainly in
human urine. Chapman (1926) reported that a person, who had ingested
lobster containing 33 mg of arsenic, excreted 74% of the arsenic in
the urine within 48 h. In studies by Coulson, et al. (1935), 2
subjects were given sea food arsenic almost all of which (> 90% of
approximately 1 mg As) was recovered in the urine within 4 days of
ingestion. When the urinary excretion of arsenic was measured in 4
subjects after they had ingested plaice and cod containing high
arsenic levels, about 70% of the ingested arsenic (0.5, 2.0, 2.1 and
2.2 mg As, respectively) was excreted in the urine during the first 24
h after ingestion and 83% in 2.5 days (Westöö & Rydälv, 1972).
Freeman, et al. (1979) gave 6 men flounder containing high levels of
arsenic, corresponding to a total ingested quantity of 5 mg of arsenic
per man. During the first day, the urinary excretion of arsenic
corresponded to more than half of the ingested amount of arsenic and
the mean total arsenic excreted during 9 days was 77%.
Calesnick, et al. (1966) studied the recovery of 74As in the
urine and faeces of human subjects who had ingested radiolabelled
arsanilic acid or pâté made from chickens fed 74As-labelled arsanilic
acid. After 6 days, only 20% of the ingested dose was recovered in the
urine and between 64% and 74% was recovered in the faeces regardless
of whether the arsenic had been given as pure arsanilic acid or in the
form of pâté from arsenic-exposed chickens.
6.2.4 Biotransformation
6.2.4.1 Animals
Stevens, et al. (1977) investigated the in vivo
biotransformation of dimethylarsinic acid. They injected
14C-dimethylarsinic acid (18 µg As) and 74As-dimethylarsinic acid
(1.9 µg As) simultaneously into rats and measured the distribution in
different organs. Since the tissue distributions of 74As and 14C were
the same, dimethylarsinic acid was thought not to be converted to
inorganic arsenic in rats. In another experiment by Stevens, et al.
(1977), doses of 200 mg/kg body weight of 14C-dimethylarsinic acid
(108 mg As/kg body weight) were administered intravenously,
intratracheally, or orally to rats. Radioactive carbon dioxide was
measured in the air emitted from the animal chambers. Approximately
0.008%-0.13% of the dose was detected as 14CO2 during a 24-h period.
This result was interpreted as indicating that only a small fraction
of the dimethylarsinic acid was demethylated by the rats. The lowest
fraction (0.008%) of the dose was exhaled by the rats that had
received intravenous injections, while the highest fraction (0.13%)
was exhaled by the peroraly exposed rats. The in vivo transformation
of dimethylarsinic acid in other species is unclear.
Orally administered dimethylarsinic acid was found to be mainly
unchanged in the liver of rats (Winkler, 1962) and in the excreta of
chickens (Overby & Fredrickson, 1963; Moody & Williams, 1964a,b,c).
Dimethylarsinate was detected in the blood, urine, and faeces of rats
given oral doses of ferric methane arsonate (approximately 40 mg As/kg
body weight). This would point towards in vivo methylation of the
compound (Odanaka, et al., 1978). The major components in the urine
were dimethylarsinate and unchanged methane arsonate.
Investigations on the biotransformation of some of the organic
arsenic compounds used as feed additives and drugs indicate that they
are converted to more easily excretable and, in some cases, more toxic
substances (Hogan & Eagle, 1944; Cristau, et al., 1972, 1975; Calvert,
1975). These changes in the molecular structure of the arsenic
compound seldom affect its valence state; nor do they result in the
formation of inorganic arsenic compounds. Hogan & Eagle (1944) cited
considerable evidence of the reduction of arsonic acids in vivo to
the corresponding arsenoxides.
6.2.4.2 Man
Elevated levels of inorganic arsenic, methylarsonic acid, or
dimethylarsinic acid were not found in human urine following ingestion
of crab meat containing 2 mg of arsenic (Crecelius, 1977b). Digestion
of the urine with hot 2 N sodium hydroxide solution was reported to
convert the unknown organic arsenic compound to dimethylarsinic acid.
The author interpreted his results as indicating that organoarsenic
compounds originating from seafood are excreted without being
biotransformed in the body. Canon et al. (1979) demonstrated the
presence of arsenobetaine in the urine of 2 human subjects after
ingestion of rock lobster tails. This indicates that this compound is
not biotransformed in the body.
7. NORMAL LEVELS IN MAN AND BIOLOGICAL INDICATORS
OF EXPOSURE
This section will deal with concentrations of arsenic in
"unexposed" persons as well as in persons excessively exposed to
arsenic through food, drinking-water, drugs, or occupation. It will
also deal with concentrations of arsenic in biological indicator media
and the extent to which they reflect exposure, e.g., concentrations in
ambient or industrial air, drinking-water, or food. It would have been
desirable to discuss the extent to which concentrations in biological
indicator media reflect concentrations of arsenic in critical organs,
i.e., organs where the earliest toxic symptoms are manifested.
However, since no organ is generally accepted as the critical organ
for arsenic toxicity, such data are lacking. Some comments will be
made based on human and animal metabolic data. The arsenic levels in
biological media will be discussed separately.
7.1 Blood
The reported figures on arsenic levels in the blood vary. Using
neutron activation, Brune et al. (1966) found a mean arsenic
concentration of 0.004 mg/kg in the whole blood of 8 normal subjects.
Bergström & Wester (1966), also using neutron activation, noted a mean
level of 0.002 mg/kg in whole blood (3 samples), of which 0.0011 mg/kg
was present in the serum. A mean concentration of 0.0011 mg/litre was
also found in 11 samples of normal human serum by Damsgaard, et al.
(1973) using neutron activation analysis. Mean arsenic values (7-16
samples) of 0.0024 mg/litre in plasma, 0.0027 mg/litre in red cells
and 0.025 mg/litre in whole blood have been reported for healthy
Danish individuals (Heydorn, 1969) whereas corresponding values for
normal subjects (6-17 samples) from China (Province of Taiwan) were
0.0154 mg, 0.0327 mg, and 0.0216 mg/litre, respectively. All
measurements were performed using neutron activation analysis.
Blackfoot disease patients and members of their families living in the
endemic area of China (Province of Taiwan) (section 8.3.4) showed mean
values of about 0.03 mg/litre in plasma (Astrup, 1968; Heydorn, 1969),
0.093 mg/litre in red cells, and 0.060 mg/litre in whole blood
(Heydorn, 1969). The mean concentrations of arsenic in well water in
the endemic area ranged from 0.054 mg to 0.743 mg/litre (Kuo, 1968).
Kagey, et al. (1977) reported a mean blood arsenic level of
0.0023 mg/litre among 50 female smokers and a mean of 0.0015 mg/litre
among 49 nonsmokers in the USA (atomic absorption spectrophotometry).
A mean blood level of arsenic of 0.00145 mg/kg (range 0.0005-
0.032 mg/kg, neutron activation analysis) has been reported for
10-year-old children living in a country town in Czechoslovakia
(Bencko & Symon, 1977). A mean blood-arsenic level of 0.00188 mg/kg
(range 0.0005-0.0038 mg/kg) was found among children of the same age
in a metropolitan city and a mean of 0.00453 mg/kg (range
0.0025-0.0082 mg/kg) among children living near a coal-fired power
plant.
In a study by Wagner & Weswig (1974), levels of arsenic in the
blood ranging from 0.01 to 0.27 mg/kg were found in forestry workers
exposed to dimethylarsinic acid (SDDC method). Unexposed workers were
reported to have levels in the range of 0.01-0.13 mg/litre, which seem
to be high compared with normal values reported by others using
neutron activation analysis. An average arsenic level in whole blood
of 0.033 mg/kg (range 0.012-0.055 mg/kg; atomic absorption
spectrophotometry) was found among 23 workers in a workshop producing
wood preservatives (arsenic acid) from arsenic(III) oxide which
probably was the cause of most of the airborne arsenic (Yamamura &
Yamauchi, 1976). The average value for the controls was reported to be
0.007 mg/kg.
The major part of both inorganic and organic arsenic in blood is
cleared fairly rapidly in man. Blood arsenic will therefore reflect
exposure for only a short period following absorption and will be very
timedependent. Only if exposure is continuous and steady, as is
sometimes the case with exposure through drinking-water, will arsenic
reach a steady-state in the blood and, thus, make it possible to
arrive at a relationship between blood arsenic levels and exposure.
Even so, data are not available that indicate any quantitative
relationship relevant to man between arsenic exposure and
concentrations of arsenic in the blood.
The short half-time of arsenic in the blood compared with the
biological half-time in the whole body makes it difficult to establish
a relationship between the blood level of arsenic and total body
burden or concentrations in different organs. A metabolic model for
the different forms of arsenic has yet to be established.
7.2 Urine
Values given in the literature for normal background levels of
arsenic in urine cover quite a wide range, probably because of the
influence of dietary sources and differences in analytical methods.
When a person has ingested a seafood meal, the urinary arsenic
concentration can often rise to over 1 mg/litre during the subsequent
24 h (Schrenk & Schreibeis, 1958; Pinto et al., 1976). Westöö & Rydälv
(1972) found that up to 1.5 mg of arsenic was excreted in the urine
during the first day following ingestion of fish containing about 2 mg
of arsenic. Studies by Crecelius (1977b) and Cannon et al. (1979)
indicated that the organic arsenic compounds present in marine
organisms are probably not metabolized in the human body, but are
excreted in the urine in the form in which they are ingested (section
6.2.4). Determination of inorganic arsenic and methylated arsenic
acids in urine (the main metabolites of inorganic arsenic), using a
method described by Braman & Foreback (1973) and Crecelius (1977), is
not influenced by the presence of organic arsenic compounds in seafood
(Crecelius, 1977). Thus, it is possible to differentiate between
arsenic taken in the form of inorganic arsenic and that ingested in
the form of seafood organoarsenic compounds.
A mean urinary value of about 0.011 mg/litre (SDDC method) was
reported by Bencko & Symon (1977) for a group of 10-year-old boys with
no known exposure to arsenic. Seafood was probably only a minor
ingredient in the diet. The mean urinary arsenic values in groups of
children living within 7.5 km of a coal-fired power plant were
0.0189-0.0253 mg/litre (range < 0.001-0.105 mg/litre).
In preemployment examinations of over 200 men, Pinto et al.
(1976), using the arsine generation/spectrophotometric method, found a
background arsenic value in urine of 0.053 mg/litre. Samples were
collected without regard to prior seafood consumption. Using arsine
generation/plasma excitation emission spectrophotometry Smith et al.
(1977) reported a geometric mean of 0.021 mg/litre among 41 unexposed
workers. The total urinary arsenic concentrations as well as various
forms of arsenic (inorganic arsenic, dimethylarsinic acid,
methylarsonic acid) were log-normally distributed among the 41 male
workers. Braman & Foreback (1973) reported total arsenic levels
(arsine generation and atomic emission spectrophotometry) in the urine
of 4 subjects of between 0.010 and 0.030 mg/litre. They found that the
predominant form of arsenic was dimethylarsinic acid, which accounted
for between 40% and 87% of the total arsenic. Other forms of arsenic
in the urine were methylarsonic acid and inorganic trivalent and
pentavalent arsenic.
Increased urinary levels of arsenic have been detected among
people living in the vicinity of smelters. Holmqvist (1975) measured
arsenic in the urine of smelter workers with low exposure to arsenic
and women living in 2 villages, 1 within 3 km of the smelter and the
other approximately 5 km from it. The highest values, mean
0.05 mg/litre (analytical method not given), were found in women
living in the village 5 km from the plant. Women living over 70 km
from the plant showed a mean value of 0.03 mg/litre.
In a survey among children in 11 copper smelter towns in the USA,
a geometric mean of 0.019 mg Am/litre of urine (atomic absorption
spectroscopy) was found and 0.006 mg/litre in 3 control towns without
a smelter (Baker et al., 1977). The children in the town most heavily
contaminated with arsenic had a geometric mean of 0.018 mg As/litre of
urine.
Total arsenic in urine has been used traditionally to assess
occupational exposure to inorganic arsenic. It is obvious that
detailed records of dietary habits should accompany all studies in
which total urinary arsenic is used as an indicator of industrial or
other environmental exposure. Generally, this has not been the case.
There are many reports concerning total urinary arsenic excretion
following industrial exposure, some examples of which will be given
here. Lundgren (1954) found an average arsenic concentration of
0.54 mg/litre in the urine of smelter employees, as opposed to only
0.04 mg/litre in workers without arsenic exposure (analytical method
not given). An average urinary concentration of 0.82 mg/litre (median
0.58 mg/litre; titrimetric method) was reported by Pinto & McGill
(1953) in men exposed to arsenic in a smelter in the USA. In the same
smelter, more than 20 years later, Pinto, et al. (1976) found an
average urinary level of arsenic of 0.174 mg/litre (range
0.038-0.539 mg/litre). Kodama, et al. (1976) reported an average
urinary arsenic concentration of 0.056 mg/litre (standard deviation
0.045) among 42 workers in a copper smelter in Japan (molybdenum blue
method).
Using the silver diethyldithiocarbamate method, Tarrant & Allard
(1972) studied the urinary excretion of forest workers in thinning
operations whereby they were exposed mainly to dimethylarsinic acid
(cacodylic acid) and monosodium methane arsonate. The average urinary
arsenic value at the beginning of a working week (Monday morning) was
0.08 mg/litre compared with 0.32 mg/litre at the end of the week
(Friday afternoon).
Concentrations of arsenic in both air and urine have been reported
in only a few studies. Pinto et al. (1976) found a correlation between
urinary arsenic levels in smelter workers and concentrations of
arsenic in the air in the smelter (Fig. 4). Arsenic was determined by
an arsine generation/spectrophotometric method. The workers wore
personal monitors for 5 consecutive workdays to measure the levels of
arsenic in the air. The correlation was reported to be significant at
arsenic concentrations in air below 0.3 mg/m3 and urinary arsenic
levels below 0.5 mg/litre. However, as can be seen in Fig. 4, there is
considerable scatter in the values and this correlation is not evident
at air levels below 0.05 mg/m3.
Carlsson (1976) studied urinary arsenic excretion in workers at a
Swedish smelter. Where it was established that subjects had eaten
seafood on the days immediately prior to the collection of urine
samples, they were excluded from the study. The men used gas masks and
personal air samplers in such a way that only the arsenic present in
the inhaled air was measured. The 8-h mean arsenic concentration in
air was 0.057 mg/m3 (range 0.002-0.227 mg/m3; X-ray spectrometric
method) and the mean arsenic concentration in the morning urine,
collected the day following that of the exposure was 0.2 mg/litre
(Gutzeit method). No clear linear relationship emerged between
airborne arsenic exposure and the total urinary arsenic.
Only one report has been published on the different forms of
arsenic in the urine of exposed smelter workers. Smith et al. (1977)
measured the concentration of respirable (< 5 µ) and nonrespirable
arsenic in the air during one working day as well as the
concentrations in urine. Arsenic was mainly excreted as
dimethylarsinic acid and the proportion of the different forms of
arsenic in the urine was, according to the authors, independent of
exposure levels (arsine generation/plasma excitation emission
spectrophotometry). The correlation between total airborne arsenic and
total urinary arsenic and the correlation between airborne arsenic and
the different forms of urinary arsenic were reported to be
significant. However, the values showed substantial scatter and
furthermore, the data could not be used to arrive at a correlation
between airborne arsenic and urinary excretion, since the workers used
chemical cartridge respirators, and it was not stated how much of the
arsenic was captured on the filters.
Urinary arsenic levels have also been shown to be correlated with
intake of arsenic in drinking-water. A survey was conducted by
Harrington, et al. (1978) among a population in an area with elevated
arsenic levels in the well water. Drinkers of well water with an
arsenic content exceeding 0.1 mg/litre (mean 0.401 mg/litre and an
estimated total daily intake of 0.324 mg of arsenic) had an average
urinary concentration of 0.178 mg/litre (atomic absorption
spectroscopy). Drinkers of well water containing an average arsenic
concentration of 0.031 mg/litre (estimated daily intake of 0.046 mg of
arsenic) had a mean urinary arsenic concentration of 0.041 mg/litre.
Ingestion of arsenic in water was also found to be reflected in
urinary excretion in a smelter town in the USA (Morse et al., 1979).
Children drinking tap water containing arsenic at 0.09 mg/litre
excreted an average of 0.059 mg As/litre of urine, while children in a
control town, where the arsenic level in the water was 0.01 mg/litre,
excreted a mean concentration of 0.018 mg As/litre of urine. Children
in both towns, who drank bottled water (with undetectable arsenic
contents), excreted average urinary arsenic levels of 0.03 mg/litre
and 0.01 mg/litre, respectively.
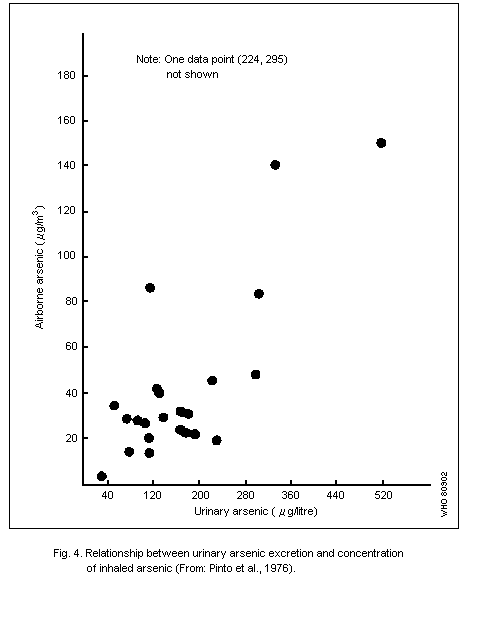 7.3 Hair
Arsenic is normally found in higher concentrations in human hair
and nails than in other parts of the body. This has been explained by
the high content of keratin in these tissues (Shapiro, 1967). The
SH-groups of keratin may bind trivalent arsenic.
The level of arsenic in hair was found to be less than 1 mg/kg in
more than 80% of 1000 persons examined in a study using neutron
activation analysis (Smith, 1964). The average level was 0.81 mg/kg
and the median 0.51 mg/kg. Liebscher & Smith (1968) found a log-normal
distribution of arsenic concentrations in over 1200 hair samples from
residents of the Glasgow area in Scotland. They performed neutron
activation analysis on the samples and found arsenic levels ranging
from 0.02 to 8.17 mg/kg dry weight with a geometric mean of
0.46 mg/kg. Geometric mean arsenic concentrations in hair of about
0.3 mg/kg were reported by Boylen & Hardy (1967) and Cornelis (1973).
The first authors used a colorimetric method with SDDC as a reagent
while Cornelis (1973) used neutron activation analysis. Leslie & Smith
(1978) also used neutron activation analysis and reported hair arsenic
levels of 0.01-0.40 mg/kg (median 0.11 mg/kg) in 52 persons with no
known exposure to arsenic.
Elevated arsenic levels in hair have been noted in persons exposed
to airborne arsenic in industrial or ambient air as well as in persons
exposed to arsenic-rich drinking water. Arsenic in the hair can arise
from 2 major sources: (a) the incorporation of absorbed arsenic into
the growing portion of the hair root; and (b) external contamination.
Lander et al. (1965) suggested that arsenic in the hair could also
originate from sweat in cases of acute poisoning. They found high
arsenic levels along the whole length of the hair at post-exposure
times too short to have allowed the incorporation of arsenic into the
hair root. Their findings are in contrast to those of other authors
(Shapiro, 1967; Pearson & Pounds, 1971), the reason probably being the
different methods and levels of arsenic administration. In acute
arsenic poisoning, profuse sweating usually occurs, which could
account for the data presented by Lander et al. (1965); this is not
the case with lower doses. It is also possible that sweat and water
can dissolve arsenic particles on the surface of the hair and in this
way augment the possible binding of arsenic to SH-groups in the hair.
It has been shown by Maes & Pate (1977) that the absorption of arsenic
in hair soaked in a solution of radiolabelled arsenite is highly
stratified on some subjects, showing zones of very high and very low
absorption. To say that peaks in arsenic concentration along the
length of the hair are indicative of days of high arsenic ingestion or
inhalation is therefore risky, at least in some subjects.
Much effort has been made to develop techniques to remove possible
external arsenic contamination from the hair. Atalla et al. (1965),
after trying several different washing methods, concluded that it was
not possible to distinguish between arsenic incorporated in the hair
after absorption and arsenic settling on the hair from external
contamination. The data seemed to indicate a saturation value of
70 mg/kg for linkage of arsenic to keratin in hair. When hair was
impregnated with arsenic (pentavalent) from a water solution, almost
all of the arsenic was easily washed off with dilute hydrochloric
acid. However, when workers' hair containing 50 and 400 mg As/kg was
washed, only about 50% of the arsenic was dissolved (van den Berg et
al., 1969). Smith (1976) suggested a method to distinguish between
arsenic in, and on the surface of human hair in which scanning was
used on hair cut perpendicularly to the long axis. He concluded that
most of the arsenic in the hair of residents in the vicinity of a
smelter emitting arsenic(III) oxide was in, rather than on, the hair.
Arsenic levels in the hair of members of the general population
exposed to arsenic-polluted air have been reported in a number of
studies. Bencko & Symon (1977) determined hair concentrations in boys
living in the vicinity of a power plant that burned local coal with a
high arsenic content. In the most highly exposed groups, the values
for arsenic in hair ranged from 0.6 to 10 mg/kg (SDDC method; hair
washed with detergent and 3% hydrochloric acid). At 36 km distance
from the source of the emission, a mean value of 0.3 mg/kg (range from
not detectable up to 0.9 mg/kg) was found. The value in a control
group residing in a metropolitan city was 0.15 mg/kg. Hammer, et al.
(1971) compared hair levels of arsenic in fourth grade boys living in
cities representing exposure dose gradients for arsenic. In the city
with the highest arsenic exposure (no data on air concentrations were
given), a geometric mean hair concentration of 9.1 mg As/kg was found
compared with 0.3 mg/kg in the city with the lowest arsenic exposure
(analytical method reported as spectrophotometry). Head hair samples
were collected from children aged between 5 and 12 years, who lived
near an open cast metal mine in Ireland (Corridan, 1974). The mean
arsenic concentration in the hair of these children was 2.1 mg/kg
(SDDC method), which was more than 17 times higher than that in a
comparable group of urban children. Suzuki, et al. (1974) measured the
arsenic contents of head hair from primary-school boys living near a
smelter in Japan. The concentrations showed a log-normal distribution
with a geometric mean of 1.87 mg/kg (SDDC method), which was about 9
times higher than that in hair from children in a control city. Higher
levels of arsenic in hair were also reported among children in various
copper smelter towns in the USA (Baker, et al., 1977) where the median
level was 0.38 mg As/kg (atomic absorption spectroscopy) compared with
a median level of 0.08 mg/kg in the hair of children from control
towns without copper smelters. Children, 5-9 years of age, in a
gold-mining town in Canada had mean hair arsenic concentrations
(neutron activation) of 1.76 mg/kg compared with 0.39 mg/kg in another
town without a mine (Canadian Public Health Association, 1978).
The contents of arsenic in the hair of occupationally exposed
persons can reach several hundred mg/kg (Smith, 1964; Atalla, et al.,
1965; Porazik, et al., 1966; Leslie & Smith, 1978). A survey
concerning the arsenic contents in hair was conducted on 703 residents
in Yellowknife, Canada (Canadian Public Health Association, 1977). Of
the 135 gold mine and mill workers participating in the survey, 33%
had hair arsenic levels exceeding 10 mg/kg (analytical method not
given). Among other residents, only 3.4% had hair levels of more than
10 mg/kg.
Reported studies do not include any information on the correlation
between arsenic exposure via air (industrial or general environment)
and arsenic concentrations in hair. The only conclusion that can be
drawn is that the hair of exposed persons contains higher levels.
Arsenic levels in the hair of persons exposed to inorganic arsenic
through ingestion are a more relevant indication of exposure,
providing that external contamination can be excluded. The influence
on hair arsenic levels of ingested organic arsenic from seafood and
drugs is not known.
A correlation between hair arsenic contents and the dose of
inorganic arsenic ingested has been reported by Pearson & Pounds
(1971) and Curry & Pounds (1977). It has also been shown that arsenic
concentrations along the length of the hair can serve to indicate
uptake over a period of time (Pearson & Pounds, 1971) (Fig. 5).
Hindmarsh, et al. (1977) measured arsenic concentrations in the
hair of 110 people in Nova Scotia, Canada, who used drinking-water
from wells with an arsenic content ranging from 0.01 to 1.4 mg/litre.
The relationship between arsenic concentrations in drinking water and
arsenic levels in hair (samples of hair cut close to the scalp) is
seen in Fig. 6. The spread is quite substantial and the correlation
was reported to be poor (without any quantification). Hair arsenic
concentrations were determined using neutron activation analysis in a
population in an area with elevated arsenic levels in the well water
(Harrington, et al., 1978). Drinkers of well water with an arsenic
concentration exceeding 0.1 mg/litre had a mean arsenic concentration
of 3.3 mg/kg in hair, while those who drank well water containing less
than 0.1 mg As/litre had a mean arsenic concentration in the hair of
0.46 mg/kg. There were indications of external contamination in the
high exposure group, as bottled water drinkers, with a substantially
lower intake of arsenic as shown by comparatively low urinary arsenic
concentrations, had hair arsenic concentrations similar to those of
well water drinkers with arsenic levels exceeding 0.1 mg/litre in the
drinking-water.
7.3 Hair
Arsenic is normally found in higher concentrations in human hair
and nails than in other parts of the body. This has been explained by
the high content of keratin in these tissues (Shapiro, 1967). The
SH-groups of keratin may bind trivalent arsenic.
The level of arsenic in hair was found to be less than 1 mg/kg in
more than 80% of 1000 persons examined in a study using neutron
activation analysis (Smith, 1964). The average level was 0.81 mg/kg
and the median 0.51 mg/kg. Liebscher & Smith (1968) found a log-normal
distribution of arsenic concentrations in over 1200 hair samples from
residents of the Glasgow area in Scotland. They performed neutron
activation analysis on the samples and found arsenic levels ranging
from 0.02 to 8.17 mg/kg dry weight with a geometric mean of
0.46 mg/kg. Geometric mean arsenic concentrations in hair of about
0.3 mg/kg were reported by Boylen & Hardy (1967) and Cornelis (1973).
The first authors used a colorimetric method with SDDC as a reagent
while Cornelis (1973) used neutron activation analysis. Leslie & Smith
(1978) also used neutron activation analysis and reported hair arsenic
levels of 0.01-0.40 mg/kg (median 0.11 mg/kg) in 52 persons with no
known exposure to arsenic.
Elevated arsenic levels in hair have been noted in persons exposed
to airborne arsenic in industrial or ambient air as well as in persons
exposed to arsenic-rich drinking water. Arsenic in the hair can arise
from 2 major sources: (a) the incorporation of absorbed arsenic into
the growing portion of the hair root; and (b) external contamination.
Lander et al. (1965) suggested that arsenic in the hair could also
originate from sweat in cases of acute poisoning. They found high
arsenic levels along the whole length of the hair at post-exposure
times too short to have allowed the incorporation of arsenic into the
hair root. Their findings are in contrast to those of other authors
(Shapiro, 1967; Pearson & Pounds, 1971), the reason probably being the
different methods and levels of arsenic administration. In acute
arsenic poisoning, profuse sweating usually occurs, which could
account for the data presented by Lander et al. (1965); this is not
the case with lower doses. It is also possible that sweat and water
can dissolve arsenic particles on the surface of the hair and in this
way augment the possible binding of arsenic to SH-groups in the hair.
It has been shown by Maes & Pate (1977) that the absorption of arsenic
in hair soaked in a solution of radiolabelled arsenite is highly
stratified on some subjects, showing zones of very high and very low
absorption. To say that peaks in arsenic concentration along the
length of the hair are indicative of days of high arsenic ingestion or
inhalation is therefore risky, at least in some subjects.
Much effort has been made to develop techniques to remove possible
external arsenic contamination from the hair. Atalla et al. (1965),
after trying several different washing methods, concluded that it was
not possible to distinguish between arsenic incorporated in the hair
after absorption and arsenic settling on the hair from external
contamination. The data seemed to indicate a saturation value of
70 mg/kg for linkage of arsenic to keratin in hair. When hair was
impregnated with arsenic (pentavalent) from a water solution, almost
all of the arsenic was easily washed off with dilute hydrochloric
acid. However, when workers' hair containing 50 and 400 mg As/kg was
washed, only about 50% of the arsenic was dissolved (van den Berg et
al., 1969). Smith (1976) suggested a method to distinguish between
arsenic in, and on the surface of human hair in which scanning was
used on hair cut perpendicularly to the long axis. He concluded that
most of the arsenic in the hair of residents in the vicinity of a
smelter emitting arsenic(III) oxide was in, rather than on, the hair.
Arsenic levels in the hair of members of the general population
exposed to arsenic-polluted air have been reported in a number of
studies. Bencko & Symon (1977) determined hair concentrations in boys
living in the vicinity of a power plant that burned local coal with a
high arsenic content. In the most highly exposed groups, the values
for arsenic in hair ranged from 0.6 to 10 mg/kg (SDDC method; hair
washed with detergent and 3% hydrochloric acid). At 36 km distance
from the source of the emission, a mean value of 0.3 mg/kg (range from
not detectable up to 0.9 mg/kg) was found. The value in a control
group residing in a metropolitan city was 0.15 mg/kg. Hammer, et al.
(1971) compared hair levels of arsenic in fourth grade boys living in
cities representing exposure dose gradients for arsenic. In the city
with the highest arsenic exposure (no data on air concentrations were
given), a geometric mean hair concentration of 9.1 mg As/kg was found
compared with 0.3 mg/kg in the city with the lowest arsenic exposure
(analytical method reported as spectrophotometry). Head hair samples
were collected from children aged between 5 and 12 years, who lived
near an open cast metal mine in Ireland (Corridan, 1974). The mean
arsenic concentration in the hair of these children was 2.1 mg/kg
(SDDC method), which was more than 17 times higher than that in a
comparable group of urban children. Suzuki, et al. (1974) measured the
arsenic contents of head hair from primary-school boys living near a
smelter in Japan. The concentrations showed a log-normal distribution
with a geometric mean of 1.87 mg/kg (SDDC method), which was about 9
times higher than that in hair from children in a control city. Higher
levels of arsenic in hair were also reported among children in various
copper smelter towns in the USA (Baker, et al., 1977) where the median
level was 0.38 mg As/kg (atomic absorption spectroscopy) compared with
a median level of 0.08 mg/kg in the hair of children from control
towns without copper smelters. Children, 5-9 years of age, in a
gold-mining town in Canada had mean hair arsenic concentrations
(neutron activation) of 1.76 mg/kg compared with 0.39 mg/kg in another
town without a mine (Canadian Public Health Association, 1978).
The contents of arsenic in the hair of occupationally exposed
persons can reach several hundred mg/kg (Smith, 1964; Atalla, et al.,
1965; Porazik, et al., 1966; Leslie & Smith, 1978). A survey
concerning the arsenic contents in hair was conducted on 703 residents
in Yellowknife, Canada (Canadian Public Health Association, 1977). Of
the 135 gold mine and mill workers participating in the survey, 33%
had hair arsenic levels exceeding 10 mg/kg (analytical method not
given). Among other residents, only 3.4% had hair levels of more than
10 mg/kg.
Reported studies do not include any information on the correlation
between arsenic exposure via air (industrial or general environment)
and arsenic concentrations in hair. The only conclusion that can be
drawn is that the hair of exposed persons contains higher levels.
Arsenic levels in the hair of persons exposed to inorganic arsenic
through ingestion are a more relevant indication of exposure,
providing that external contamination can be excluded. The influence
on hair arsenic levels of ingested organic arsenic from seafood and
drugs is not known.
A correlation between hair arsenic contents and the dose of
inorganic arsenic ingested has been reported by Pearson & Pounds
(1971) and Curry & Pounds (1977). It has also been shown that arsenic
concentrations along the length of the hair can serve to indicate
uptake over a period of time (Pearson & Pounds, 1971) (Fig. 5).
Hindmarsh, et al. (1977) measured arsenic concentrations in the
hair of 110 people in Nova Scotia, Canada, who used drinking-water
from wells with an arsenic content ranging from 0.01 to 1.4 mg/litre.
The relationship between arsenic concentrations in drinking water and
arsenic levels in hair (samples of hair cut close to the scalp) is
seen in Fig. 6. The spread is quite substantial and the correlation
was reported to be poor (without any quantification). Hair arsenic
concentrations were determined using neutron activation analysis in a
population in an area with elevated arsenic levels in the well water
(Harrington, et al., 1978). Drinkers of well water with an arsenic
concentration exceeding 0.1 mg/litre had a mean arsenic concentration
of 3.3 mg/kg in hair, while those who drank well water containing less
than 0.1 mg As/litre had a mean arsenic concentration in the hair of
0.46 mg/kg. There were indications of external contamination in the
high exposure group, as bottled water drinkers, with a substantially
lower intake of arsenic as shown by comparatively low urinary arsenic
concentrations, had hair arsenic concentrations similar to those of
well water drinkers with arsenic levels exceeding 0.1 mg/litre in the
drinking-water.
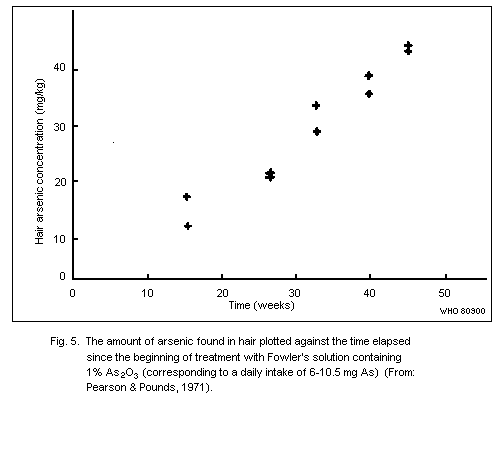
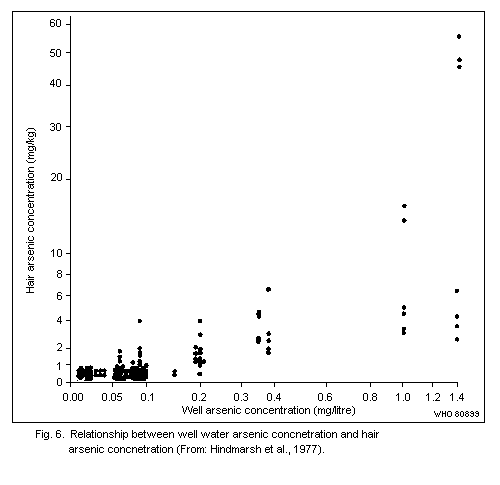 In conclusion, it can be stated that hair arsenic can be used on a
group basis as an indicator of arsenic exposure through ingestion,
providing that external contamination is only slight. The use of hair
arsenic as an indicator of exposure to airborne arsenic is limited, as
no reliable method exists of distinguishing arsenic from external
contamination from arsenic that has been absorbed and metabolized in
the body. However, it can be used on a group basis as an indicator of
possible exposure situations.
7.4 Other Tissues
The concentrations of arsenic in various human tissues determined
by neutron activation analysis and reported by Liebscher & Smith
(1968), Larsen et al. (1972), and Brune et al. (1980) are shown in
Table 8.
Arsenic levels in the lungs of 22 smelter workers, who had not
been exposed occupationally to arsenic for the last 2-19 years, were
reported by Brune et al. (1980). A median arsenic value of 0.048 mg/kg
wet weight (range 0.014-0.21 mg/kg) was found compared with a median
value of 0.008 mg/kg wet weight (range 0.001-0.018 mg/kg) in 9
controls. No such differences could be found in the arsenic contents
of the liver and kidneys of 10 workers and 8 controls.
Table 8. Arsenic concentrations in human organs and tissues
Arsenic concentration
(mg/kg)
Tissue or organ Dry weighta
(geometric Wet weightb Wet weightc
mean (mean values) (median values)
values)
adrenal 0.03
aorta 0.04
whole blood 0.04
brain 0.01
hair 0.46
heart 0.02
kidney 0.03 0.007 0.004
liver 0.03 0.011 0.003
lung 0.08 0.010 0.008
muscle 0.06 (pectoral) 0.004
nail 0.28
ovary 0.05
pancreas 0.05 0.005
prostate 0.04
skin 0.08
spleen 0.02 0.003
stomach 0.02
teeth 0.05
thymus 0.02
thyroid 0.04
uterus 0.04
a Compiled from Liebscher & Smith (1968).
b Compiled from Larsen et al. (1972).
c Compiled from Brune et al. (1980).
8. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF INORGANIC ARSENIC
Occupational exposure to inorganic arsenic occurs mainly in the
smelting industry, and in the manufacture and application of
arsenic-containing pesticides (section 5.2) It is generally considered
that smelter workers are exposed to trivalent inorganic arsenic
compounds, while workers handling pesticides are exposed primarily to
pentavalent arsenic. In the general environment, high levels of
arsenic may be found in drinking-water in various parts of the world,
including, Argentina, Chile, China (Province of Taiwan), Japan, and
Mexico. It is believed that arsenic occurs predominantly in an
inorganic form in water; however, the oxidation state of the arsenic
associated with adverse health effects is not known at present.
Substantial exposure to arsenic also results from medication with
inorganic (mainly trivalent) and organic arsenic compounds. The use of
inorganic arsenic in drugs is now limited in most countries.
8.1 Acute and Subacute Effects after Short-Term Exposure
8.1.1 Man
Acute effects caused by the ingestion of inorganic arsenic
compounds, mainly arsenic(III) oxide, are well documented in the
literature. The major lesion is profound gastrointestinal damage,
resulting in severe vomiting and diarrhoea, often with blood-tinged
stools. Other acute symptoms and signs include muscular cramps, facial
oedema, and cardiac abnormalities. Shock can develop rapidly as a
result of dehydration. Symptoms may occur within a few minutes of the
exposure if the arsenic compound is in a solution, but may be delayed
for several hours if it is solid or taken with a meal. When taken
orally, the toxicity of the arsenic compound largely depends on its
solubility (Done & Peart, 1971). Human data on the differences in
toxicity between trivalent and pentavalent arsenic are limited. The
fatal dose of ingested arsenic(III) oxide for man has been reported to
range from 70-180 mg (Vallee et al., 1960).
Effects resulting from exposure to quantities of arsenic
sufficient to cause acute symptoms and signs, but inadequate to
produce systemic collapse, are of particular interest. Unfortunately,
the doses that have resulted in such symptoms and signs have rarely
been reported. Subacute effects mainly involve the respiratory,
gastrointestinal, cardiovascular, nervous, and haematopoietic systems.
Exposure to irritant arsenic compounds, such as arsenic(III) oxide, in
air can acutely damage the mucous membranes of the respiratory system
and exposed skin. This can result in severe irritation of the nasal
mucosa, larynx, bronchi, and ear canal, as well as in conjunctivitis
and dermatitis (Holmqvist, 1951; Pinto & McGill, 1953). Nasal septum
perforation may appear within two weeks.
Peripheral nervous disturbances, primarily of a sensory type, are
frequently encountered in individuals surviving acute poisoning with
inorganic arsenic compounds (Heyman et al., 1956; Jenkins, 1966;
Nagamatsu & Igata, 1975; O'Shaughnessy & Kraft, 1976; Le Quesne &
McLeod, 1977). These disturbances usually become manifest 1-2 weeks
after ingestion. Recovery is slow, usually starting between 1 and 2
months after the onset of symptoms. The degree of recovery depends on
the severity of the symptoms. The lower extremities are often more
severely affected than the upper ones.
Histological examination of the peripheral nerves in a case of
arsenic poisoning showed Wallerian degeneration, especially in the
longest axons (Ohta, 1970). Clinical and electrodiagnostic recordings
made over an extended period of time were consistent with the
histological changes.
The haematopoietic system may also show effects, characterized by
anaemia and leukopenia, especially granulocytopenia (Hamamoto, 1955;
Heyman et al., 1956). These effects are usually reversible within 2-3
weeks.
Reversible changes in the electrocardiogram have frequently been
encountered following acute exposure to arsenic compounds (Hamamoto,
1955; Weinberg, 1960; Barry & Herndon, 1962; Chhuttani et al., 1967).
In these situations, the doses have generally been high enough to
produce other symptoms and signs of acute intoxication. The observed
effects usually include extensions of the QT-time and T-wave
abnormalities.
Two instances of mass poisoning by inorganic arsenic in Japan give
a good picture of the diversity of symptoms associated with acute and
subacute poisoning, though the nature of the clinical investigations
on the victims makes it difficult to interpret some of the findings.
The first episode occurred when over 12 000 infants were poisoned with
dried milk contaminated with inorganic arsenic (Hamamoto, 1955:
Nakagawa & Ibuchi, 1970). The milk powder contained 15-24 mg As/kg and
the arsenic was reported to be in the pentavalent state, although no
data exist on its form at the time of ingestion. It was estimated that
the infants ingested 1.3-3.6 mg of arsenic daily depending on age, and
130 deaths were reported. Symptoms usually appeared after a few weeks
of exposure and often included fever, insomnia, and anorexia. Liver
swelling and melanosis were present in all but one of 61 hospitalized
patients examined by Hamamoto. The blood picture showed anaemia and
leukopenia with relative lymphocytosis. Acute renal damage was
indicated by a high incidence of microscopic haematuria. Although
swelling of the liver was characteristic among the poisoned infants,
liver function tests were normal in all cases. Disturbance of the
heart function was another common finding, and was characterized by
rises in the ST, decreases in the T, and extensions of the QT-time.
Most symptoms were rapidly reversible upon cessation of exposure and
the beginning of therapy; however, the changes observed in the
electrocardiograms took longer to disappear than the other clinical
symptoms.
It must be emphasized that this group of patients constituted a
limited sample of the intoxicated population and was selected
according to certain diagnostic criteria, i.e., liver swelling and
melanosis. Consequently, little emphasis should be put on the
abundance of these symptoms; however, the coexistence of a high degree
of other symptoms is noteworthy.
Mizuta et al. (1956) examined 220 out of 417 patients, who had
been poisoned by soy sauce contaminated with inorganic arsenic at a
concentration of 100 mg/litre. The average estimated ingestion per
person was 3 mg of arsenic (valence state unknown), daily, for 2-3
weeks. The main findings were facial oedema, anorexia, and upper
respiratory symptoms followed by skin lesions and neuritic signs at a
later stage, i.e., after 10-20 days. Though the livers of most
patients were enlarged, relatively few abnormalities were found in the
liver function tests and liver size gradually decreased after
cessation of exposure. Abnormal electrocardiograms were found in 16
out of 20 cases tested. The arsenic content of hair in 5 patients
about 2 weeks after arsenic intake ceased was between 3.8 and 13 mg/kg
near the roots and 0-1.8 mg/kg at the ends. Levels in control subjects
ranged from 0.4 to 2.8 mg/kg.
In a clinical study of 13 cases of polyneuropathy connected with
arsenic poisoning, in Sri Lanka, Senanayake et al. (1972) found Mee's
lines, i.e., transverse white bands across finger nails, to be the
constant feature, at least 6 weeks after the onset of initial
symptoms. In 7 of these cases, the source of arsenic was contaminated
well water, 4 others had a long history of consuming illicit liquor.
Mee's lines are of value both in the diagnosis and in the
assessment of the approximate time of exposure to arsenic. The time
may be calculated by considering the distance of the line from the
base of the nail and the rate of nail growth, which is of the order of
about 0.3 cm per month or about 0.1 mm per day (Smith & Fiddes, 1955).
Appearance of polyneuropathy, 3 days to 3 weeks after acute
poisoning, was seen in all the cases reported by Senanayake et al.
(1972). The first symptom of neuropathy in these cases was numbness of
extremities. The lower limbs were affected earlier and more severely
than the upper limbs.
8.1.2 Animals
The oral LD50 (or "certain fatal dose") for arsenic ranged from
15-293 mg As/kg body weight in rats and 11-150 mg/kg in other
laboratory animals (Schwartze, 1922; Dieke & Richter, 1946; Harrison
et al., 1958; Done & Peart, 1971). The lower values are generally
found, when the arsenic is administered in solution. Sodium arsenite,
which is more soluble in water than arsenic(III) oxide, has been shown
to be 10 times as toxic as arsenic(III) oxide (Done & Peart, 1971).
Trivalent arsenic is generally more toxic than pentavalent arsenic.
Franke & Moxon (1936) obtained a minimal fatal dose (the smallest dose
which killed 75% of intraperitoneally exposed rats in 48 h) of 4-5 mg
As/kg body weight for sodium arsenite and 14-18 mg As/kg body weight
for sodium arsenate. The development of tolerance towards the acute
effects of arsenic(III) oxide following pretreatment with arsenic was
demonstrated by Bencko & Symon (1969b). The LD50 following
subcutaneous injection of arsenic(III) oxide was significantly higher
for hairless mice, pretreated for 15 weeks with inorganic arsenic in
the drinking water at a concentration of 50 mg/litre than for mice not
previously exposed to arsenic (14 and 11 mg/kg body weight,
respectively). A discussion of tolerance to arsenic appears in section
6.1.2.2.1. The effects observed in acutely intoxicated animals
resemble those found in human subjects and included gastroenteritis,
diarrhoea, lowered blood pressure, and ECG changes (Nelson et al.,
1971; Tsutsumi & Nozaki, 1973; Selby et al., 1977).
In addition to the experimental data, the results of a large
epizootic survey on cattle accidentally given feed containing high
levels of inorganic arsenic have been reported (González, 1977).
Nearly 6000 cattle were fed for 1-2 days with a mixture containing
arsenic(III) oxide at levels of between 490 and 2900 mg/kg. Acute
signs were observed immediately, and the first deaths occurred after 3
days. More than 50% of the 1464 animal deaths took place during the
first week following administration of the feed. The rest of the
deaths occurred over a 6-month period as the result of visceral
damage. The main acute signs observed were: drastic reduction in milk
production (85% reduction), diarrhoea, dehydration, dyspnoea,
cyanosis, abortion, and central nervous effects. Among the chronic
signs, the most frequently observed were: hyperkeratosis of the skin,
rigidity and inflammation of the joints, and blindness with opacity of
the cornea. With respect to the pathological observations the most
serious were: haemorrages and ulcers of the gastrointestinal tract,
fatty degeneration of the liver and kidney, nephritis, emphysema and
pulmonary oedema, and albuminar degeneration of the heart.
Animal data on subacute effects, will be discussed in section 8.3
together with chronic effects.
8.2 Effects on Reproductive Function and Teratogenicity
8.2.1 Man
Human data on the teratogenicity of inorganic arsenic are very
limited. Children born to women who worked during pregnancy at a
Swedish copper smelter and were exposed to airborne arsenic in some
workplaces, showed a significantly, higher frequency of congenital
malformations (Nordström et al., 1979). The frequency of all
malformations in the children of women at the smelter was twice as
high as that in the children of other women in the region. A 5-fold
higher frequency was noted for multiple malformations. Data were
collected from the records of the regional hospital, and included a
total of about 25 000 live births during the period 1955-76. The
exposure environment of the smelter was very complex, involving a
number of heavy metals and sulfur dioxide. No conclusions can be drawn
with regard to the specific cause of the observed excess of
malformations.
Nordström et al. (1978) studied the frequency of spontaneous
abortions during 4427 pregnancies in women living in the vicinity of
this copper smelter. The smelter had emitted many potentially
genotoxic substances in to the environment including arsenic and lead.
The frequency of abortions was significantly higher in women living
nearest the factory than in a reference population living more than
50 km from the plant. The abortion rates in the 2 areas were 11% and
7.6%, respectively. Obviously, no firm conclusions could be drawn with
regard to the role of the inorganic arsenic exposure in these cases;
but the results indicate that further research in this field is
needed.
8.2.2 Animals
Teratogenic effects of inorganic arsenic have frequently been
reported in laboratory animals. Ridgway & Karnofsky (1952) reported
that sodium arsenate gave rise to nonspecific effects in chick embryo
tests. Ferm & Carpenter (1968) were the first to describe clear
teratogenic effects of arsenic in laboratory animals. Pregnant golden
hamsters were given an intravenous injection of sodium arsenate on the
8th day of gestation and the embryos were examined 4 to 5 days later.
A dose of 2 mg As/kg body weight as sodium arsenate did not induce any
malformations, while 3 mg As/kg body weight caused an increased
incidence of resorption and malformation, especially exencephaly. A
level of 16 mg As/kg body weight resulted in the death of all embryos.
In a later study, Ferm et al. (1971) investigated the influence of the
time of injection on the teratogenic profile in hamsters. Intravenous
injections of 6-10 mg As/kg body weight as sodium arsenate at various
times on the eighth and ninth days of gestation caused different types
of lesions. The observed malformations included exencephaly,
anencephaly, renal agenesis, and rib and genitourinary abnormalities.
In addition, high resorption rates were present.
Results similar to those reported in hamsters were found in mice,
intraperitoneally injected with sodium arsenate at 45 mg/kg body
weight (11 mg As/kg body weight) on the 6th to 12th days of gestation
(Hood & Bishop, 1972). Offspring of mice treated with 6 mg As/kg body
weight as arsenate did not differ from those of the controls.
Administration of sodium arsenite in doses of 10 or 12 mg/kg body
weight (6 or 7 mg As/kg body weight) resulted in a lower incidence of
malformations and a higher incidence, of resorptions than sodium
arsenate at 11 mg As/kg body weight (Hood, 1972; Hood et al., 1977).
The teratogenicity of sodium arsenate in rats was investigated by
Beaudoin (1974). Doses of 5-12 mg As/kg body weight were given as a
single intraperitoneal injection on the 7th to 12th day of gestation.
All dose levels produced malformations such as eye defects,
exencephaly, renal agenesis, and gonadal agenesis.
The route of administration was shown to have a significant
influence on the teratogenic action of arsenic in mice (Thacker et
al., 1977). A much higher oral dose of an aqueous solution of arsenate
was needed to induce the same effects as found after intraperitoneal
injection. The doses given were 120 and 40 mg arsenate per kg body
weight, respectively.
In all the studies mentioned so far, single high doses of arsenic
were used to produce teratogenic effects. Schroeder & Mitchener (1971)
exposed 3 generations of mice to low doses of arsenite but did not
find any abnormalities other than reduced litter size. The mice were
exposed to arsenic in feed (5 mg As/kg diet) in the form of arsenite.
Without supplementary data on food consumption it is difficult to
estimate the actual dose level.
8.3 Noncarcinogenic Effects After Long-Term Exposure and Sequelae
of Short-Term Exposure
8.3.1 Effects on the respiratory system
8.3.1.1 Man
Effects of arsenic on the respiratory system have been reported
primarily as a result of occupational exposure. In the smelting
industry, where high levels of airborne inorganic arsenic are
frequently encountered, lesions of the mucous membranes in the
respiratory system, including perforation of the nasal septum, have
been observed (Pinto & McGill, 1953; Lundgren, 1954; Birmingham et
al., 1965; Ishinishi, 1973; Hine et al., 1977). Lundgren performed
medical examinations on 1276 workers at a copper smelter in Sweden.
The levels of airborne arsenic were as high as 7 mg/m3 in some
workplaces, but generally did not exceed 0.5 mg/m3. Two types of
respiratory syndromes were seen, each of them characteristic for a
certain group of workers. Symptoms of the upper respiratory passages
with septum perforation and rhino-pharyngo-laryngitis were found
chiefly among workers exposed to arsenic in the crude or refined form.
In some workplaces, over one third of the workers showed changes in
the nasal mucosa. The other syndrome included symptoms of
tracheobronchitis and signs of pulmonary insufficiency, often due to
emphysematous lesions. This picture was found especially among those
who had worked at the roasters, reverberatory furnaces, and in
converter halls, where mixed exposure to arsenic and sulfur dioxide
took place. This study did not include data on smoking habits, which
probably played a role in the symptoms and signs noted. No controls
were used.
It has been claimed that exposure to arsenic via routes other than
inhalation can affect the respiratory system. A high frequency of
chronic cough and a history of bronchopulmonary disease were reported
by Borgono et al. (1977) among 180 inhabitants of Antofagasta in Chile
who had abnormal skin pigmentation attributed to arsenic exposure in
the drinking water. From the same area, Rosenberg (1974) found diffuse
interstitial fibrosis of the lungs in 2 out of 5 children with
systemic arterial lesions. When evaluating these 2 reports, the
suspected role of arsenic as a suppressant of the immune response
should be kept in mind, since this could impair resistance to
infections (Gainer & Pry, 1972). It is also important to consider the
very low socioeconomic status of this population, which resulted in
various nutritional deficiencies.
8.3.1.2 Animals
There is a lack of data regarding the chronic effects of arsenic
on the respiratory system in experimental animals. In view of the many
different effects observed in workers exposed to airborne arsenic, and
the difficulties in controlling various confounding factors, it is of
great importance to develop animal model systems. The studies should
preferably involve exposure via inhalation over long periods.
8.3.2 Effects on skin
8.3.2.1 Man
A number of skin lesions have been attributed to chronic exposure
to inorganic arsenic compounds. Symmetric verrucous hyperkeratosis of
the palms and soles is a characteristic finding after long-term
ingestion of inorganic arsenic via drinking-water or drugs.
Hyperpigmentation (melanosis) of the skin, often associated with paler
spots (depigmentation), is also commonly encountered and occurs mainly
in the areas of the skin not exposed to the sun, i.e., axillae and
trunk. These lesions have been reported from regions in Argentina,
Chile, China (Province of Taiwan), Japan, and Mexico, where the
contents of arsenic in drinking-water were elevated (Arguello et al.,
1938; Yoshikawa et al., 1960; Alvarado et al., 1964; Tseng et al.,
1968; Borgono et al., 1977). Arguello et al. (1938) reported that the
keratodermia appeared insidiously between the second and third year of
intoxication and did not disappear after cessation of exposure. Some
individuals were followed for more than 30 years after termination of
exposure.
As stated earlier, substantial exposure has also resulted from the
ingestion of arsenic-containing drugs. The compound most often used
was sodium arsenite in daily doses of up to 10 mg of arsenic, and the
treatment could extend over decades. In a study by Fierz (1965), a
dose-response relationship was found between the amount of arsenic
ingested and the incidence of palmoplantar hyperkeratosis in 262
patients treated 6-26 years earlier for chronic dermatoses with
diluted (1:1) Fowler's Solution, containing 3.8 g As/litre. In the
patients who had received the equivalent of more than 400 ml of
Fowler's Solution (3 g of arsenic), the prevalence of hyperkeratosis
was more than 50%, and the author stated that as little as 60 ml of
Fowler's Solution (about 0.46 g of arsenic) resulted in keratosis in
one patient after 2.5 years of treatment. Melanosis was present in
only 5 of the patients at the time of examination, but 3 others
recalled that they looked "dirtyish" during the periods of arsenic
treatment and that this had regressed over the years. It should be
noted that this study did not include a control group and could have
had a substantial selection bias, but the findings with regard to
palmoplantar hyperkeratosis are worthy of note in view of the rarity
of this lesion.
Hyperkeratotic lesions of the palms and soles and melanosis are
uncommon among smelter workers exposed to airborne arsenic (Pinto &
McGill, 1953) the most common lesion in these situations being
dermatosis due to local irritation.
One third of a group of 31 workers manufacturing sodium arsenite
had "warts", which, unfortunately, were not further described (Perry
et al., 1948). All but 3 of these workers showed hyperpigmentation of
the skin. Less than 4% of the controls at a factory with low exposure
to arsenic had warts and 9 out of 56 showed melanosis of the skin.
Typical cutaneous manifestations of chronic arsenic poisoning were
detected in 7 out of 28 male Japanese workers, who had been exposed to
arsenic in the form of lead arsenate and calcium arsenate in the
manufacture of insecticides (Hamada & Horiguchi, 1976). The lesions
were symmetric punctuated palmo-plantar hyperkeratosis and "bronze"
hyperpigmentation. The authors did not find any correlation between
the intensity of cutaneous manifestations and the length of exposure
to arsenic. Similar conclusions were reached with regard to the skin
lesions found following arsenic exposure among German wine growers
(Wolf, 1974). Sixteen cases of typical arsenic-induced keratosis,
which appeared from 3 to 31 years after the beginning of exposure,
were reported. The total intake of arsenic through contaminated
beverages was estimated to be between 5.7 and 133 g.
Sensitization of the skin following exposure to inorganic arsenic
compounds, such as arsenic(III) oxide, has been reported among smelter
workers (Holmqvist, 1951).
8.3.2.2 Animals
Skin effects have been observed in rats given oral intubations of
aqueous solutions of arsenic(III) oxide in daily doses of 1.5 and
7.6 mg As/kg body weight (Ishinishi et al., 1976). The exposure was
started when the rats were 2 weeks old and lasted for 40 days with an
observation time of 30 weeks. Rats given the lower dose did not differ
from the controls, while the rats given 7.6 mg As/kg body weight, lost
the glossy appearance of their pelage, especially on the back and nape
of the neck. Moist eczema developed into severe skin changes with
ulcerations and crust formations in 9 out of 21 animals.
Histopathological findings included ulcers and scarring of the
epidermis and subcutaneous tissues, hyperkeratosis, and acanthosis.
Enlargement and hyperplasia of the hair bulbs were also observed.
8.3.3 Effects on the liver
8.3.3.1 Man
Exposure to inorganic arsenic compounds has been associated with
the development of chronic pathological liver changes. Several authors
have reported cases of liver damage following treatment with arsenic
in the trivalent inorganic form (Neale & Azzopardi, 1971; Knolle
et al., 1974; Morris et al., 1974; Huet et al., 1975; Szuler et al.,
1979). A common finding in these reports was portal hypertension
without signs of liver cirrhosis. All patients had been on the arsenic
medication, mostly Fowler's solution, for several years. Typical
cutaneous signs of long-term arsenic exposure were also observed in
some of the patients. There have also been case reports on liver
cirrhosis following medication with inorganic arsenic compounds (cf.
review in Franklin et al., 1950). It was claimed that alcohol could be
ruled out as the causative agent in most of the cases. Zachariae
et al. (1974) took liver biopsies from 44 psoriatic patients who had
received potassium arsenite and from 37 psoriatic patients who had
not. Histopathological changes were common in both groups; however, no
statistical differences could be established between the two. No cases
of cirrhosis were demonstrated.
Exposure to arsenic-containing pesticides and contaminated wine
was claimed to be the causative factor in the large number of cases of
liver cirrhosis among German vintners in the forties and fifties
(Roth, 1957). The number of cases diminished in the late fifties;
however, less severe liver changes continue to be found regularly
among the vintners (Lüchtrath, 1972; Wolf, 1974). It is probable that
the heavy wine consumption, often 3-4 litres daily, among the vintners
played an important role in the development of the lesions.
Kodama et al. (1976) made various biochemical determinations on
the blood of 42 copper smelter workers belonging to 3 different
occupational categories (14 subjects in each group, matched for sex,
age, and period of employment). The average urinary arsenic
concentrations in the 3 groups were 82.6, 40.6, and 45.2 µg/litre,
respectively. Workers with the highest arsenic concentrations in the
urine showed an increase in serum GOT and LDH, even though the levels
were within normal limits. It should be noted that exposure to arsenic
was fairly moderate in all the groups (< 13 µg/m3, as a 6-h
average).
An increased mortality from liver cirrhosis was reported in 2
studies on smelter workers heavily exposed to inorganic arsenic by Lee
& Fraumeni (1969) and Axelson et al. (1978) (section 8.4.1.1).
However, the total number of cases was quite small and a confounding
effect of alcohol consumption cannot be ruled out.
8.3.3.2 Animals
Liver lesions have frequently been observed in animals following
long-term exposure to trivalent or pentavalent inorganic arsenic.
Liver cirrhosis and necrosis as well as bile duct proliferation were
found in rabbits after administration of arsenate as lead, copper, or
sodium salts (von Glahn et al., 1938). The arsenic was mixed in the
feed and administered in daily doses of 1.4-9.3 mg of arsenic per
animal for 50-250 days. Liver damage has also been observed in
domestic animals exposed to arsenic (Selby et al., 1977).
Histopathological findings included fatty changes and necrosis.
Ishinishi et al. (1980b) gave 4 groups of male adult Wistar-King rats
distilled water, per os, containing arsenic(III) oxide at arsenic
levels of 0, 0.125, 12.5 or 62.5 mg/litre, respectively, for 7 months.
The animals were then given distilled water without the addition of
arsenic(III) oxide for 4 months. Though no differences were observed
in growth and in general physiological condition between the 4 groups,
light liver injuries and dose-dependent proliferation of the bile duct
with some chronic angitis in Glisson's capsules were found.
Ultrastructural changes were studied in the hepatocytes of mice in the
course of arsenic exposure via drinking water (50 mg As(III) per
litre) over 4-64 days (Mohelská et al., 1980). The results obtained
showed 2 types of response: the first (maximum on the 4th day) was
enlargement of inner membranous structures (invaginations of the
nuclear membrane and undulation of the mitochondrial structures) and
disappearance of glycogen. The second type of response was represented
by a gradual appearance of dense lamellar structures in the
peroxisomes that persisted till the end of the exposure.
Disturbances of liver function have also been seen in animals
exposed to arsenic. Impaired liver function, including delay in BSP
excretion and increase in serum transaminases, was noted in rabbits
given intravenous injections of arsenious acid in doses of 0.6 mg
As/kg body weight, 3 times a week for up to 3 months (Shibuya, 1971).
Mice exposed to arsenite in drinking water in daily doses of 12 mg
As/kg body weight showed a progressive decrease in relative liver
weight as well as a partially reversible decrease in liver oxygen
consumption (Bencko & Nemeckova, 1971). This finding was also noted in
animals receiving a daily dose of about 6 mg As/kg body weight in the
form of arsenic(III) oxide dissolved in drinking water. The oxygen
consumption of liver homogenates decreased somewhat during the first
month of exposure, but after 2 months it did not differ from that of
the controls (Bencko, 1972). No differences in the metabolic oxygen
consumption of liver homogenates were observed during 2 months in mice
exposed to about 0.8 mg As/kg body weight per day. In a later study on
liver dehydrogenase activity by Bencko et al. (1975), who used the
same exposure groups, a decrease was revealed in the highest exposure
group only, i.e., animals receiving a daily dose of 12 mg As/kg body
weight. The concentration of free SH-groups in the liver decreased to
a minimum level during the eighth day of exposure in all groups, and
again reached the level of the control group by two months of exposure
(Bencko et al., 1978a). The activity of liver glutathione reductase,
on the other hand, showed a tendency to increase during the treatment
period, especially in the highest exposure group.
Swollen mitochondria and biochemical changes in the form of
altered enzyme activity in the hepatocytes were reported in rats given
drinking water containing 20, 40 or 85 mg As/litre as arsenate for up
to 6 weeks (Fowler et al., 1977; Schiller et al., 1977; Woods &
Fowler, 1977). Inhibition of enzymes responsible for haem biosynthesis
was observed. An increased urinary level of uroporphyrin was suggested
to be a possible early indication of effects due to arsenic exposure
(Woods & Fowler, 1977).
8.3.4 Effects on the cardiovascular system
8.3.4.1 Man
Though reversible changes in the electrocardiogram have often been
encountered following acute exposure to inorganic arsenic (section
8.1), such effects have rarely been reported after chronic exposure.
However, Zettel (1943) described an incident of arsenic intoxication
among 170 German soldiers, who had ingested water contaminated with
inorganic trivalent arsenic for several months. Unfortunately, the
content of arsenic in the water was not reported, but was high enough
to produce gastrointestinal symptoms. A broadening of the QRS-complex
was observed on the electrocardiograms of 45 out of 80 soldiers
examined. Other, less frequent changes included ST-depressions and
flattening of the T-wave. In a check-up 3 months after cessation of
exposure, electrocardiograms were normal in all but 6 cases.
Myocardial damage as determined from electrocardiograms was described
by Butzengeiger (1949) in 28.7% of 192 persons exposed to
arsenic-containing insecticides before 1942. Marked electrocardiogram
changes were observed in 55 patients, 65% of which were ascribed to
arsenic. Neither of the 2 investigations related here included control
groups, which makes it difficult to assess the significance of the
findings. When considering effects on the heart, among vintners, their
heavy alcohol consumption must also be kept in mind.
An increased mortality from cardiovascular disease has been
observed in 2 epidemiological investigations on smelter workers
exposed to high levels of airborne arsenic (Lee & Fraumeni, 1969;
Axelson et al., 1978) (section 8.4.1.1). In the study by Axelson et
al., a dose-response relationship between arsenic exposure and
cardiovascular effects appeared. The excess mortality in both studies
was 2-fold or less, and has not been confirmed in other, similar
studies on workers occupationally exposed to arsenic.
Peripheral vascular lesions have been reported in some arsenic
exposure situations. Such effects were first described by Geyer (1898)
in residents of Reichenstein (now Silesia, Poland), who were exposed
to arsenic via contaminated drinking water. Butzengeiger (1940)
described peripheral vascular lesions in 23% of 180 vintners with
chronic arsenic intoxication. In 6 cases, the inadequate peripheral
circulation caused gangrene. In a study by Grobe (1976), the author
examined 100 vinedressers over 30 years after arsenic exposure had
been terminated (mode of selection not determined), the exposure
having lasted 20 years on an average. He reported distinct peripheral
vascular lesions, including symptoms and signs of endangiitis
obliterans and acrodermatitis atrophicans, in between 60% and 95% of
those in various age groups from 50 to 80 years of age. These symptoms
were found in only 1-2% of a control group that had not been exposed
to arsenic, but unfortunately was not described further. The authors
stated that the most important source of arsenic exposure was
contaminated wine.
In conclusion, it can be stated that hair arsenic can be used on a
group basis as an indicator of arsenic exposure through ingestion,
providing that external contamination is only slight. The use of hair
arsenic as an indicator of exposure to airborne arsenic is limited, as
no reliable method exists of distinguishing arsenic from external
contamination from arsenic that has been absorbed and metabolized in
the body. However, it can be used on a group basis as an indicator of
possible exposure situations.
7.4 Other Tissues
The concentrations of arsenic in various human tissues determined
by neutron activation analysis and reported by Liebscher & Smith
(1968), Larsen et al. (1972), and Brune et al. (1980) are shown in
Table 8.
Arsenic levels in the lungs of 22 smelter workers, who had not
been exposed occupationally to arsenic for the last 2-19 years, were
reported by Brune et al. (1980). A median arsenic value of 0.048 mg/kg
wet weight (range 0.014-0.21 mg/kg) was found compared with a median
value of 0.008 mg/kg wet weight (range 0.001-0.018 mg/kg) in 9
controls. No such differences could be found in the arsenic contents
of the liver and kidneys of 10 workers and 8 controls.
Table 8. Arsenic concentrations in human organs and tissues
Arsenic concentration
(mg/kg)
Tissue or organ Dry weighta
(geometric Wet weightb Wet weightc
mean (mean values) (median values)
values)
adrenal 0.03
aorta 0.04
whole blood 0.04
brain 0.01
hair 0.46
heart 0.02
kidney 0.03 0.007 0.004
liver 0.03 0.011 0.003
lung 0.08 0.010 0.008
muscle 0.06 (pectoral) 0.004
nail 0.28
ovary 0.05
pancreas 0.05 0.005
prostate 0.04
skin 0.08
spleen 0.02 0.003
stomach 0.02
teeth 0.05
thymus 0.02
thyroid 0.04
uterus 0.04
a Compiled from Liebscher & Smith (1968).
b Compiled from Larsen et al. (1972).
c Compiled from Brune et al. (1980).
8. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF INORGANIC ARSENIC
Occupational exposure to inorganic arsenic occurs mainly in the
smelting industry, and in the manufacture and application of
arsenic-containing pesticides (section 5.2) It is generally considered
that smelter workers are exposed to trivalent inorganic arsenic
compounds, while workers handling pesticides are exposed primarily to
pentavalent arsenic. In the general environment, high levels of
arsenic may be found in drinking-water in various parts of the world,
including, Argentina, Chile, China (Province of Taiwan), Japan, and
Mexico. It is believed that arsenic occurs predominantly in an
inorganic form in water; however, the oxidation state of the arsenic
associated with adverse health effects is not known at present.
Substantial exposure to arsenic also results from medication with
inorganic (mainly trivalent) and organic arsenic compounds. The use of
inorganic arsenic in drugs is now limited in most countries.
8.1 Acute and Subacute Effects after Short-Term Exposure
8.1.1 Man
Acute effects caused by the ingestion of inorganic arsenic
compounds, mainly arsenic(III) oxide, are well documented in the
literature. The major lesion is profound gastrointestinal damage,
resulting in severe vomiting and diarrhoea, often with blood-tinged
stools. Other acute symptoms and signs include muscular cramps, facial
oedema, and cardiac abnormalities. Shock can develop rapidly as a
result of dehydration. Symptoms may occur within a few minutes of the
exposure if the arsenic compound is in a solution, but may be delayed
for several hours if it is solid or taken with a meal. When taken
orally, the toxicity of the arsenic compound largely depends on its
solubility (Done & Peart, 1971). Human data on the differences in
toxicity between trivalent and pentavalent arsenic are limited. The
fatal dose of ingested arsenic(III) oxide for man has been reported to
range from 70-180 mg (Vallee et al., 1960).
Effects resulting from exposure to quantities of arsenic
sufficient to cause acute symptoms and signs, but inadequate to
produce systemic collapse, are of particular interest. Unfortunately,
the doses that have resulted in such symptoms and signs have rarely
been reported. Subacute effects mainly involve the respiratory,
gastrointestinal, cardiovascular, nervous, and haematopoietic systems.
Exposure to irritant arsenic compounds, such as arsenic(III) oxide, in
air can acutely damage the mucous membranes of the respiratory system
and exposed skin. This can result in severe irritation of the nasal
mucosa, larynx, bronchi, and ear canal, as well as in conjunctivitis
and dermatitis (Holmqvist, 1951; Pinto & McGill, 1953). Nasal septum
perforation may appear within two weeks.
Peripheral nervous disturbances, primarily of a sensory type, are
frequently encountered in individuals surviving acute poisoning with
inorganic arsenic compounds (Heyman et al., 1956; Jenkins, 1966;
Nagamatsu & Igata, 1975; O'Shaughnessy & Kraft, 1976; Le Quesne &
McLeod, 1977). These disturbances usually become manifest 1-2 weeks
after ingestion. Recovery is slow, usually starting between 1 and 2
months after the onset of symptoms. The degree of recovery depends on
the severity of the symptoms. The lower extremities are often more
severely affected than the upper ones.
Histological examination of the peripheral nerves in a case of
arsenic poisoning showed Wallerian degeneration, especially in the
longest axons (Ohta, 1970). Clinical and electrodiagnostic recordings
made over an extended period of time were consistent with the
histological changes.
The haematopoietic system may also show effects, characterized by
anaemia and leukopenia, especially granulocytopenia (Hamamoto, 1955;
Heyman et al., 1956). These effects are usually reversible within 2-3
weeks.
Reversible changes in the electrocardiogram have frequently been
encountered following acute exposure to arsenic compounds (Hamamoto,
1955; Weinberg, 1960; Barry & Herndon, 1962; Chhuttani et al., 1967).
In these situations, the doses have generally been high enough to
produce other symptoms and signs of acute intoxication. The observed
effects usually include extensions of the QT-time and T-wave
abnormalities.
Two instances of mass poisoning by inorganic arsenic in Japan give
a good picture of the diversity of symptoms associated with acute and
subacute poisoning, though the nature of the clinical investigations
on the victims makes it difficult to interpret some of the findings.
The first episode occurred when over 12 000 infants were poisoned with
dried milk contaminated with inorganic arsenic (Hamamoto, 1955:
Nakagawa & Ibuchi, 1970). The milk powder contained 15-24 mg As/kg and
the arsenic was reported to be in the pentavalent state, although no
data exist on its form at the time of ingestion. It was estimated that
the infants ingested 1.3-3.6 mg of arsenic daily depending on age, and
130 deaths were reported. Symptoms usually appeared after a few weeks
of exposure and often included fever, insomnia, and anorexia. Liver
swelling and melanosis were present in all but one of 61 hospitalized
patients examined by Hamamoto. The blood picture showed anaemia and
leukopenia with relative lymphocytosis. Acute renal damage was
indicated by a high incidence of microscopic haematuria. Although
swelling of the liver was characteristic among the poisoned infants,
liver function tests were normal in all cases. Disturbance of the
heart function was another common finding, and was characterized by
rises in the ST, decreases in the T, and extensions of the QT-time.
Most symptoms were rapidly reversible upon cessation of exposure and
the beginning of therapy; however, the changes observed in the
electrocardiograms took longer to disappear than the other clinical
symptoms.
It must be emphasized that this group of patients constituted a
limited sample of the intoxicated population and was selected
according to certain diagnostic criteria, i.e., liver swelling and
melanosis. Consequently, little emphasis should be put on the
abundance of these symptoms; however, the coexistence of a high degree
of other symptoms is noteworthy.
Mizuta et al. (1956) examined 220 out of 417 patients, who had
been poisoned by soy sauce contaminated with inorganic arsenic at a
concentration of 100 mg/litre. The average estimated ingestion per
person was 3 mg of arsenic (valence state unknown), daily, for 2-3
weeks. The main findings were facial oedema, anorexia, and upper
respiratory symptoms followed by skin lesions and neuritic signs at a
later stage, i.e., after 10-20 days. Though the livers of most
patients were enlarged, relatively few abnormalities were found in the
liver function tests and liver size gradually decreased after
cessation of exposure. Abnormal electrocardiograms were found in 16
out of 20 cases tested. The arsenic content of hair in 5 patients
about 2 weeks after arsenic intake ceased was between 3.8 and 13 mg/kg
near the roots and 0-1.8 mg/kg at the ends. Levels in control subjects
ranged from 0.4 to 2.8 mg/kg.
In a clinical study of 13 cases of polyneuropathy connected with
arsenic poisoning, in Sri Lanka, Senanayake et al. (1972) found Mee's
lines, i.e., transverse white bands across finger nails, to be the
constant feature, at least 6 weeks after the onset of initial
symptoms. In 7 of these cases, the source of arsenic was contaminated
well water, 4 others had a long history of consuming illicit liquor.
Mee's lines are of value both in the diagnosis and in the
assessment of the approximate time of exposure to arsenic. The time
may be calculated by considering the distance of the line from the
base of the nail and the rate of nail growth, which is of the order of
about 0.3 cm per month or about 0.1 mm per day (Smith & Fiddes, 1955).
Appearance of polyneuropathy, 3 days to 3 weeks after acute
poisoning, was seen in all the cases reported by Senanayake et al.
(1972). The first symptom of neuropathy in these cases was numbness of
extremities. The lower limbs were affected earlier and more severely
than the upper limbs.
8.1.2 Animals
The oral LD50 (or "certain fatal dose") for arsenic ranged from
15-293 mg As/kg body weight in rats and 11-150 mg/kg in other
laboratory animals (Schwartze, 1922; Dieke & Richter, 1946; Harrison
et al., 1958; Done & Peart, 1971). The lower values are generally
found, when the arsenic is administered in solution. Sodium arsenite,
which is more soluble in water than arsenic(III) oxide, has been shown
to be 10 times as toxic as arsenic(III) oxide (Done & Peart, 1971).
Trivalent arsenic is generally more toxic than pentavalent arsenic.
Franke & Moxon (1936) obtained a minimal fatal dose (the smallest dose
which killed 75% of intraperitoneally exposed rats in 48 h) of 4-5 mg
As/kg body weight for sodium arsenite and 14-18 mg As/kg body weight
for sodium arsenate. The development of tolerance towards the acute
effects of arsenic(III) oxide following pretreatment with arsenic was
demonstrated by Bencko & Symon (1969b). The LD50 following
subcutaneous injection of arsenic(III) oxide was significantly higher
for hairless mice, pretreated for 15 weeks with inorganic arsenic in
the drinking water at a concentration of 50 mg/litre than for mice not
previously exposed to arsenic (14 and 11 mg/kg body weight,
respectively). A discussion of tolerance to arsenic appears in section
6.1.2.2.1. The effects observed in acutely intoxicated animals
resemble those found in human subjects and included gastroenteritis,
diarrhoea, lowered blood pressure, and ECG changes (Nelson et al.,
1971; Tsutsumi & Nozaki, 1973; Selby et al., 1977).
In addition to the experimental data, the results of a large
epizootic survey on cattle accidentally given feed containing high
levels of inorganic arsenic have been reported (González, 1977).
Nearly 6000 cattle were fed for 1-2 days with a mixture containing
arsenic(III) oxide at levels of between 490 and 2900 mg/kg. Acute
signs were observed immediately, and the first deaths occurred after 3
days. More than 50% of the 1464 animal deaths took place during the
first week following administration of the feed. The rest of the
deaths occurred over a 6-month period as the result of visceral
damage. The main acute signs observed were: drastic reduction in milk
production (85% reduction), diarrhoea, dehydration, dyspnoea,
cyanosis, abortion, and central nervous effects. Among the chronic
signs, the most frequently observed were: hyperkeratosis of the skin,
rigidity and inflammation of the joints, and blindness with opacity of
the cornea. With respect to the pathological observations the most
serious were: haemorrages and ulcers of the gastrointestinal tract,
fatty degeneration of the liver and kidney, nephritis, emphysema and
pulmonary oedema, and albuminar degeneration of the heart.
Animal data on subacute effects, will be discussed in section 8.3
together with chronic effects.
8.2 Effects on Reproductive Function and Teratogenicity
8.2.1 Man
Human data on the teratogenicity of inorganic arsenic are very
limited. Children born to women who worked during pregnancy at a
Swedish copper smelter and were exposed to airborne arsenic in some
workplaces, showed a significantly, higher frequency of congenital
malformations (Nordström et al., 1979). The frequency of all
malformations in the children of women at the smelter was twice as
high as that in the children of other women in the region. A 5-fold
higher frequency was noted for multiple malformations. Data were
collected from the records of the regional hospital, and included a
total of about 25 000 live births during the period 1955-76. The
exposure environment of the smelter was very complex, involving a
number of heavy metals and sulfur dioxide. No conclusions can be drawn
with regard to the specific cause of the observed excess of
malformations.
Nordström et al. (1978) studied the frequency of spontaneous
abortions during 4427 pregnancies in women living in the vicinity of
this copper smelter. The smelter had emitted many potentially
genotoxic substances in to the environment including arsenic and lead.
The frequency of abortions was significantly higher in women living
nearest the factory than in a reference population living more than
50 km from the plant. The abortion rates in the 2 areas were 11% and
7.6%, respectively. Obviously, no firm conclusions could be drawn with
regard to the role of the inorganic arsenic exposure in these cases;
but the results indicate that further research in this field is
needed.
8.2.2 Animals
Teratogenic effects of inorganic arsenic have frequently been
reported in laboratory animals. Ridgway & Karnofsky (1952) reported
that sodium arsenate gave rise to nonspecific effects in chick embryo
tests. Ferm & Carpenter (1968) were the first to describe clear
teratogenic effects of arsenic in laboratory animals. Pregnant golden
hamsters were given an intravenous injection of sodium arsenate on the
8th day of gestation and the embryos were examined 4 to 5 days later.
A dose of 2 mg As/kg body weight as sodium arsenate did not induce any
malformations, while 3 mg As/kg body weight caused an increased
incidence of resorption and malformation, especially exencephaly. A
level of 16 mg As/kg body weight resulted in the death of all embryos.
In a later study, Ferm et al. (1971) investigated the influence of the
time of injection on the teratogenic profile in hamsters. Intravenous
injections of 6-10 mg As/kg body weight as sodium arsenate at various
times on the eighth and ninth days of gestation caused different types
of lesions. The observed malformations included exencephaly,
anencephaly, renal agenesis, and rib and genitourinary abnormalities.
In addition, high resorption rates were present.
Results similar to those reported in hamsters were found in mice,
intraperitoneally injected with sodium arsenate at 45 mg/kg body
weight (11 mg As/kg body weight) on the 6th to 12th days of gestation
(Hood & Bishop, 1972). Offspring of mice treated with 6 mg As/kg body
weight as arsenate did not differ from those of the controls.
Administration of sodium arsenite in doses of 10 or 12 mg/kg body
weight (6 or 7 mg As/kg body weight) resulted in a lower incidence of
malformations and a higher incidence, of resorptions than sodium
arsenate at 11 mg As/kg body weight (Hood, 1972; Hood et al., 1977).
The teratogenicity of sodium arsenate in rats was investigated by
Beaudoin (1974). Doses of 5-12 mg As/kg body weight were given as a
single intraperitoneal injection on the 7th to 12th day of gestation.
All dose levels produced malformations such as eye defects,
exencephaly, renal agenesis, and gonadal agenesis.
The route of administration was shown to have a significant
influence on the teratogenic action of arsenic in mice (Thacker et
al., 1977). A much higher oral dose of an aqueous solution of arsenate
was needed to induce the same effects as found after intraperitoneal
injection. The doses given were 120 and 40 mg arsenate per kg body
weight, respectively.
In all the studies mentioned so far, single high doses of arsenic
were used to produce teratogenic effects. Schroeder & Mitchener (1971)
exposed 3 generations of mice to low doses of arsenite but did not
find any abnormalities other than reduced litter size. The mice were
exposed to arsenic in feed (5 mg As/kg diet) in the form of arsenite.
Without supplementary data on food consumption it is difficult to
estimate the actual dose level.
8.3 Noncarcinogenic Effects After Long-Term Exposure and Sequelae
of Short-Term Exposure
8.3.1 Effects on the respiratory system
8.3.1.1 Man
Effects of arsenic on the respiratory system have been reported
primarily as a result of occupational exposure. In the smelting
industry, where high levels of airborne inorganic arsenic are
frequently encountered, lesions of the mucous membranes in the
respiratory system, including perforation of the nasal septum, have
been observed (Pinto & McGill, 1953; Lundgren, 1954; Birmingham et
al., 1965; Ishinishi, 1973; Hine et al., 1977). Lundgren performed
medical examinations on 1276 workers at a copper smelter in Sweden.
The levels of airborne arsenic were as high as 7 mg/m3 in some
workplaces, but generally did not exceed 0.5 mg/m3. Two types of
respiratory syndromes were seen, each of them characteristic for a
certain group of workers. Symptoms of the upper respiratory passages
with septum perforation and rhino-pharyngo-laryngitis were found
chiefly among workers exposed to arsenic in the crude or refined form.
In some workplaces, over one third of the workers showed changes in
the nasal mucosa. The other syndrome included symptoms of
tracheobronchitis and signs of pulmonary insufficiency, often due to
emphysematous lesions. This picture was found especially among those
who had worked at the roasters, reverberatory furnaces, and in
converter halls, where mixed exposure to arsenic and sulfur dioxide
took place. This study did not include data on smoking habits, which
probably played a role in the symptoms and signs noted. No controls
were used.
It has been claimed that exposure to arsenic via routes other than
inhalation can affect the respiratory system. A high frequency of
chronic cough and a history of bronchopulmonary disease were reported
by Borgono et al. (1977) among 180 inhabitants of Antofagasta in Chile
who had abnormal skin pigmentation attributed to arsenic exposure in
the drinking water. From the same area, Rosenberg (1974) found diffuse
interstitial fibrosis of the lungs in 2 out of 5 children with
systemic arterial lesions. When evaluating these 2 reports, the
suspected role of arsenic as a suppressant of the immune response
should be kept in mind, since this could impair resistance to
infections (Gainer & Pry, 1972). It is also important to consider the
very low socioeconomic status of this population, which resulted in
various nutritional deficiencies.
8.3.1.2 Animals
There is a lack of data regarding the chronic effects of arsenic
on the respiratory system in experimental animals. In view of the many
different effects observed in workers exposed to airborne arsenic, and
the difficulties in controlling various confounding factors, it is of
great importance to develop animal model systems. The studies should
preferably involve exposure via inhalation over long periods.
8.3.2 Effects on skin
8.3.2.1 Man
A number of skin lesions have been attributed to chronic exposure
to inorganic arsenic compounds. Symmetric verrucous hyperkeratosis of
the palms and soles is a characteristic finding after long-term
ingestion of inorganic arsenic via drinking-water or drugs.
Hyperpigmentation (melanosis) of the skin, often associated with paler
spots (depigmentation), is also commonly encountered and occurs mainly
in the areas of the skin not exposed to the sun, i.e., axillae and
trunk. These lesions have been reported from regions in Argentina,
Chile, China (Province of Taiwan), Japan, and Mexico, where the
contents of arsenic in drinking-water were elevated (Arguello et al.,
1938; Yoshikawa et al., 1960; Alvarado et al., 1964; Tseng et al.,
1968; Borgono et al., 1977). Arguello et al. (1938) reported that the
keratodermia appeared insidiously between the second and third year of
intoxication and did not disappear after cessation of exposure. Some
individuals were followed for more than 30 years after termination of
exposure.
As stated earlier, substantial exposure has also resulted from the
ingestion of arsenic-containing drugs. The compound most often used
was sodium arsenite in daily doses of up to 10 mg of arsenic, and the
treatment could extend over decades. In a study by Fierz (1965), a
dose-response relationship was found between the amount of arsenic
ingested and the incidence of palmoplantar hyperkeratosis in 262
patients treated 6-26 years earlier for chronic dermatoses with
diluted (1:1) Fowler's Solution, containing 3.8 g As/litre. In the
patients who had received the equivalent of more than 400 ml of
Fowler's Solution (3 g of arsenic), the prevalence of hyperkeratosis
was more than 50%, and the author stated that as little as 60 ml of
Fowler's Solution (about 0.46 g of arsenic) resulted in keratosis in
one patient after 2.5 years of treatment. Melanosis was present in
only 5 of the patients at the time of examination, but 3 others
recalled that they looked "dirtyish" during the periods of arsenic
treatment and that this had regressed over the years. It should be
noted that this study did not include a control group and could have
had a substantial selection bias, but the findings with regard to
palmoplantar hyperkeratosis are worthy of note in view of the rarity
of this lesion.
Hyperkeratotic lesions of the palms and soles and melanosis are
uncommon among smelter workers exposed to airborne arsenic (Pinto &
McGill, 1953) the most common lesion in these situations being
dermatosis due to local irritation.
One third of a group of 31 workers manufacturing sodium arsenite
had "warts", which, unfortunately, were not further described (Perry
et al., 1948). All but 3 of these workers showed hyperpigmentation of
the skin. Less than 4% of the controls at a factory with low exposure
to arsenic had warts and 9 out of 56 showed melanosis of the skin.
Typical cutaneous manifestations of chronic arsenic poisoning were
detected in 7 out of 28 male Japanese workers, who had been exposed to
arsenic in the form of lead arsenate and calcium arsenate in the
manufacture of insecticides (Hamada & Horiguchi, 1976). The lesions
were symmetric punctuated palmo-plantar hyperkeratosis and "bronze"
hyperpigmentation. The authors did not find any correlation between
the intensity of cutaneous manifestations and the length of exposure
to arsenic. Similar conclusions were reached with regard to the skin
lesions found following arsenic exposure among German wine growers
(Wolf, 1974). Sixteen cases of typical arsenic-induced keratosis,
which appeared from 3 to 31 years after the beginning of exposure,
were reported. The total intake of arsenic through contaminated
beverages was estimated to be between 5.7 and 133 g.
Sensitization of the skin following exposure to inorganic arsenic
compounds, such as arsenic(III) oxide, has been reported among smelter
workers (Holmqvist, 1951).
8.3.2.2 Animals
Skin effects have been observed in rats given oral intubations of
aqueous solutions of arsenic(III) oxide in daily doses of 1.5 and
7.6 mg As/kg body weight (Ishinishi et al., 1976). The exposure was
started when the rats were 2 weeks old and lasted for 40 days with an
observation time of 30 weeks. Rats given the lower dose did not differ
from the controls, while the rats given 7.6 mg As/kg body weight, lost
the glossy appearance of their pelage, especially on the back and nape
of the neck. Moist eczema developed into severe skin changes with
ulcerations and crust formations in 9 out of 21 animals.
Histopathological findings included ulcers and scarring of the
epidermis and subcutaneous tissues, hyperkeratosis, and acanthosis.
Enlargement and hyperplasia of the hair bulbs were also observed.
8.3.3 Effects on the liver
8.3.3.1 Man
Exposure to inorganic arsenic compounds has been associated with
the development of chronic pathological liver changes. Several authors
have reported cases of liver damage following treatment with arsenic
in the trivalent inorganic form (Neale & Azzopardi, 1971; Knolle
et al., 1974; Morris et al., 1974; Huet et al., 1975; Szuler et al.,
1979). A common finding in these reports was portal hypertension
without signs of liver cirrhosis. All patients had been on the arsenic
medication, mostly Fowler's solution, for several years. Typical
cutaneous signs of long-term arsenic exposure were also observed in
some of the patients. There have also been case reports on liver
cirrhosis following medication with inorganic arsenic compounds (cf.
review in Franklin et al., 1950). It was claimed that alcohol could be
ruled out as the causative agent in most of the cases. Zachariae
et al. (1974) took liver biopsies from 44 psoriatic patients who had
received potassium arsenite and from 37 psoriatic patients who had
not. Histopathological changes were common in both groups; however, no
statistical differences could be established between the two. No cases
of cirrhosis were demonstrated.
Exposure to arsenic-containing pesticides and contaminated wine
was claimed to be the causative factor in the large number of cases of
liver cirrhosis among German vintners in the forties and fifties
(Roth, 1957). The number of cases diminished in the late fifties;
however, less severe liver changes continue to be found regularly
among the vintners (Lüchtrath, 1972; Wolf, 1974). It is probable that
the heavy wine consumption, often 3-4 litres daily, among the vintners
played an important role in the development of the lesions.
Kodama et al. (1976) made various biochemical determinations on
the blood of 42 copper smelter workers belonging to 3 different
occupational categories (14 subjects in each group, matched for sex,
age, and period of employment). The average urinary arsenic
concentrations in the 3 groups were 82.6, 40.6, and 45.2 µg/litre,
respectively. Workers with the highest arsenic concentrations in the
urine showed an increase in serum GOT and LDH, even though the levels
were within normal limits. It should be noted that exposure to arsenic
was fairly moderate in all the groups (< 13 µg/m3, as a 6-h
average).
An increased mortality from liver cirrhosis was reported in 2
studies on smelter workers heavily exposed to inorganic arsenic by Lee
& Fraumeni (1969) and Axelson et al. (1978) (section 8.4.1.1).
However, the total number of cases was quite small and a confounding
effect of alcohol consumption cannot be ruled out.
8.3.3.2 Animals
Liver lesions have frequently been observed in animals following
long-term exposure to trivalent or pentavalent inorganic arsenic.
Liver cirrhosis and necrosis as well as bile duct proliferation were
found in rabbits after administration of arsenate as lead, copper, or
sodium salts (von Glahn et al., 1938). The arsenic was mixed in the
feed and administered in daily doses of 1.4-9.3 mg of arsenic per
animal for 50-250 days. Liver damage has also been observed in
domestic animals exposed to arsenic (Selby et al., 1977).
Histopathological findings included fatty changes and necrosis.
Ishinishi et al. (1980b) gave 4 groups of male adult Wistar-King rats
distilled water, per os, containing arsenic(III) oxide at arsenic
levels of 0, 0.125, 12.5 or 62.5 mg/litre, respectively, for 7 months.
The animals were then given distilled water without the addition of
arsenic(III) oxide for 4 months. Though no differences were observed
in growth and in general physiological condition between the 4 groups,
light liver injuries and dose-dependent proliferation of the bile duct
with some chronic angitis in Glisson's capsules were found.
Ultrastructural changes were studied in the hepatocytes of mice in the
course of arsenic exposure via drinking water (50 mg As(III) per
litre) over 4-64 days (Mohelská et al., 1980). The results obtained
showed 2 types of response: the first (maximum on the 4th day) was
enlargement of inner membranous structures (invaginations of the
nuclear membrane and undulation of the mitochondrial structures) and
disappearance of glycogen. The second type of response was represented
by a gradual appearance of dense lamellar structures in the
peroxisomes that persisted till the end of the exposure.
Disturbances of liver function have also been seen in animals
exposed to arsenic. Impaired liver function, including delay in BSP
excretion and increase in serum transaminases, was noted in rabbits
given intravenous injections of arsenious acid in doses of 0.6 mg
As/kg body weight, 3 times a week for up to 3 months (Shibuya, 1971).
Mice exposed to arsenite in drinking water in daily doses of 12 mg
As/kg body weight showed a progressive decrease in relative liver
weight as well as a partially reversible decrease in liver oxygen
consumption (Bencko & Nemeckova, 1971). This finding was also noted in
animals receiving a daily dose of about 6 mg As/kg body weight in the
form of arsenic(III) oxide dissolved in drinking water. The oxygen
consumption of liver homogenates decreased somewhat during the first
month of exposure, but after 2 months it did not differ from that of
the controls (Bencko, 1972). No differences in the metabolic oxygen
consumption of liver homogenates were observed during 2 months in mice
exposed to about 0.8 mg As/kg body weight per day. In a later study on
liver dehydrogenase activity by Bencko et al. (1975), who used the
same exposure groups, a decrease was revealed in the highest exposure
group only, i.e., animals receiving a daily dose of 12 mg As/kg body
weight. The concentration of free SH-groups in the liver decreased to
a minimum level during the eighth day of exposure in all groups, and
again reached the level of the control group by two months of exposure
(Bencko et al., 1978a). The activity of liver glutathione reductase,
on the other hand, showed a tendency to increase during the treatment
period, especially in the highest exposure group.
Swollen mitochondria and biochemical changes in the form of
altered enzyme activity in the hepatocytes were reported in rats given
drinking water containing 20, 40 or 85 mg As/litre as arsenate for up
to 6 weeks (Fowler et al., 1977; Schiller et al., 1977; Woods &
Fowler, 1977). Inhibition of enzymes responsible for haem biosynthesis
was observed. An increased urinary level of uroporphyrin was suggested
to be a possible early indication of effects due to arsenic exposure
(Woods & Fowler, 1977).
8.3.4 Effects on the cardiovascular system
8.3.4.1 Man
Though reversible changes in the electrocardiogram have often been
encountered following acute exposure to inorganic arsenic (section
8.1), such effects have rarely been reported after chronic exposure.
However, Zettel (1943) described an incident of arsenic intoxication
among 170 German soldiers, who had ingested water contaminated with
inorganic trivalent arsenic for several months. Unfortunately, the
content of arsenic in the water was not reported, but was high enough
to produce gastrointestinal symptoms. A broadening of the QRS-complex
was observed on the electrocardiograms of 45 out of 80 soldiers
examined. Other, less frequent changes included ST-depressions and
flattening of the T-wave. In a check-up 3 months after cessation of
exposure, electrocardiograms were normal in all but 6 cases.
Myocardial damage as determined from electrocardiograms was described
by Butzengeiger (1949) in 28.7% of 192 persons exposed to
arsenic-containing insecticides before 1942. Marked electrocardiogram
changes were observed in 55 patients, 65% of which were ascribed to
arsenic. Neither of the 2 investigations related here included control
groups, which makes it difficult to assess the significance of the
findings. When considering effects on the heart, among vintners, their
heavy alcohol consumption must also be kept in mind.
An increased mortality from cardiovascular disease has been
observed in 2 epidemiological investigations on smelter workers
exposed to high levels of airborne arsenic (Lee & Fraumeni, 1969;
Axelson et al., 1978) (section 8.4.1.1). In the study by Axelson et
al., a dose-response relationship between arsenic exposure and
cardiovascular effects appeared. The excess mortality in both studies
was 2-fold or less, and has not been confirmed in other, similar
studies on workers occupationally exposed to arsenic.
Peripheral vascular lesions have been reported in some arsenic
exposure situations. Such effects were first described by Geyer (1898)
in residents of Reichenstein (now Silesia, Poland), who were exposed
to arsenic via contaminated drinking water. Butzengeiger (1940)
described peripheral vascular lesions in 23% of 180 vintners with
chronic arsenic intoxication. In 6 cases, the inadequate peripheral
circulation caused gangrene. In a study by Grobe (1976), the author
examined 100 vinedressers over 30 years after arsenic exposure had
been terminated (mode of selection not determined), the exposure
having lasted 20 years on an average. He reported distinct peripheral
vascular lesions, including symptoms and signs of endangiitis
obliterans and acrodermatitis atrophicans, in between 60% and 95% of
those in various age groups from 50 to 80 years of age. These symptoms
were found in only 1-2% of a control group that had not been exposed
to arsenic, but unfortunately was not described further. The authors
stated that the most important source of arsenic exposure was
contaminated wine.
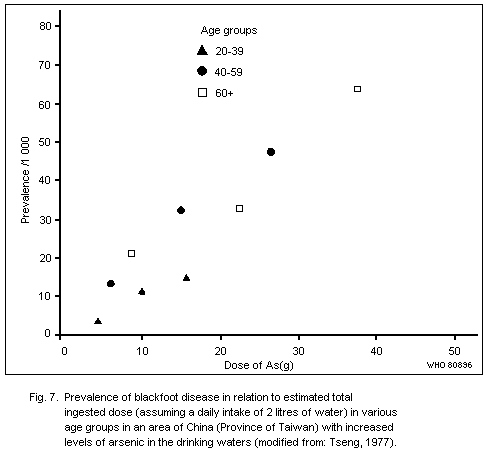 A high prevalence of a peripheral vascular disease called
"blackfoot disease" was found in a population living in China
(Province of Taiwan), where the arsenic levels in well water used for
drinking purposes ranged from 0.01-1.82 mg/litre but were mainly
between 0.4 and 0.6 mg/litre (Tseng et al., 1968; Tseng, 1977). The
overall prevalence rate of the disease of 8.9 per 1000 increased with
age and with the arsenic content of the water. From Fig. 7, it can be
seen that there is a roughly linear increase in the prevalence rate
with increasing total ingested dose of arsenic. Exposure for many
years resulting in a total ingested dose of about 20 g corresponds to
a prevalence rate of 3%. It should be noted that a control group was
not included in the study and that there would be a rise in prevalence
of peripheral gangrene with increasing age. If the most mildly exposed
group, i.e., the group that consumed well water with an arsenic
content below 0.3 mg/litre, is taken as a control group, a roughly
linear increase in excess morbidity expressed as prevalence rate of
between 9.6 and 41.1 per 1000 still exists in various age groups
(Fig. 8). Obviously this excess cannot be explained by the age factor
or other concurrent diseases, and appears to be the result of arsenic
exposure.
The severity of the disease was related to duration of water
intake. Furthermore, the average age at death was lower in the group
of "blackfoot disease" patients exposed to high arsenic levels in the
water than in those exposed to lower levels. Carcinomas were the cause
of 18.8% of the deaths in patients with "blackfoot disease" compared
with 13.1% of deaths in the general population of the endemic area and
7.9% of deaths in the whole population of the Province of Taiwan.
Further supporting evidence that arsenic in drinking water was a
causative factor in "blackfoot disease" is that no new cases of the
disease have appeared in children in the area, since the installation
of tap water systems with low levels of arsenic in the water.
The occurrence of various fluorescent compounds in well water
samples from the "blackfoot disease" area had been reported
(Lu et al., 1975). The authors suggest that an ergot-like action of
the fluorescent substances may have been a cause, or contributing
cause, of "blackfoot disease". Collectively, however, the data
presented are not adequate to provide firm support for the authors'
hypothesis.
A high prevalence of a peripheral vascular disease called
"blackfoot disease" was found in a population living in China
(Province of Taiwan), where the arsenic levels in well water used for
drinking purposes ranged from 0.01-1.82 mg/litre but were mainly
between 0.4 and 0.6 mg/litre (Tseng et al., 1968; Tseng, 1977). The
overall prevalence rate of the disease of 8.9 per 1000 increased with
age and with the arsenic content of the water. From Fig. 7, it can be
seen that there is a roughly linear increase in the prevalence rate
with increasing total ingested dose of arsenic. Exposure for many
years resulting in a total ingested dose of about 20 g corresponds to
a prevalence rate of 3%. It should be noted that a control group was
not included in the study and that there would be a rise in prevalence
of peripheral gangrene with increasing age. If the most mildly exposed
group, i.e., the group that consumed well water with an arsenic
content below 0.3 mg/litre, is taken as a control group, a roughly
linear increase in excess morbidity expressed as prevalence rate of
between 9.6 and 41.1 per 1000 still exists in various age groups
(Fig. 8). Obviously this excess cannot be explained by the age factor
or other concurrent diseases, and appears to be the result of arsenic
exposure.
The severity of the disease was related to duration of water
intake. Furthermore, the average age at death was lower in the group
of "blackfoot disease" patients exposed to high arsenic levels in the
water than in those exposed to lower levels. Carcinomas were the cause
of 18.8% of the deaths in patients with "blackfoot disease" compared
with 13.1% of deaths in the general population of the endemic area and
7.9% of deaths in the whole population of the Province of Taiwan.
Further supporting evidence that arsenic in drinking water was a
causative factor in "blackfoot disease" is that no new cases of the
disease have appeared in children in the area, since the installation
of tap water systems with low levels of arsenic in the water.
The occurrence of various fluorescent compounds in well water
samples from the "blackfoot disease" area had been reported
(Lu et al., 1975). The authors suggest that an ergot-like action of
the fluorescent substances may have been a cause, or contributing
cause, of "blackfoot disease". Collectively, however, the data
presented are not adequate to provide firm support for the authors'
hypothesis.
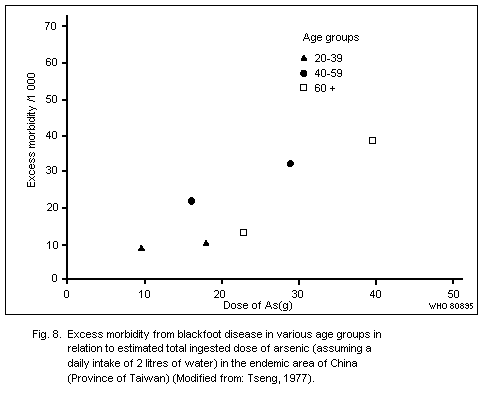 Peripheral vascular disease was also reported in inhabitants of
the Antofagasta region of northern Chile, who had been exposed to
arsenic levels of about 0.6 mg/litre in the drinking water for 15
years. A clinical investigation among 180 inhabitants revealed several
effects associated with chronic arsenic exposure, including
hyperkeratosis, and a high prevalence of cardiovascular disturbances
(Borgono et al., 1977). Most common were peripheral vascular
phenomena, i.e., Raynaud's syndrome and acrocyanosis, which were
present, respectively, in 38.8% and 24.3% of the persons with abnormal
skin pigmentation attributed to arsenic exposure, compared with 9.3%
and 12.5% of persons with normal skin. Infants and children showed
more pronounced symptoms than adults. Systemic arterial disease
resulting in myocardial infarction was reported in 2 children
(Rosenberg, 1974). The small and medium-sized arteries of most organs
showed marked thickening of the intima. Similar microscopic findings
have been reported in 2 men (32 and 36 years of age) from the same
area who had myocardial infarctions but lacked the risk factors
usually associated with coronary sclerosis (Moran et al., 1977).
It is noteworthy that effects on the peripheral vascular system
have not been reported among patients undergoing medication with
inorganic arsenic or among workers exposed to high levels of airborne
arsenic. It is not clear, however, whether investigators have looked
for such effects in these groups.
8.3.4.2 Animals
In order to study the chronic effects of arsenic, Massmann & Opitz
(1954) fed 12 cats about 1.5 mg As/kg body weight as sodium arsenite
and sodium arsenate mixed with the feed. T-wave abnormalities, mainly
flattening, were found in the electrocardiograms of 9 of the animals
and the QT-time was prolonged in some animals. No control group was
used, which makes it difficult to interpret these findings. The
animals were allowed to die spontaneously, at which time histological
analysis of the heart was performed. No marked changes were found. No
differences in effects were observed between the 2 forms of arsenic
used.
Data are lacking with regard to the extent to which experimental
animals develop the peripheral vascular lesions observed in human
subjects following chronic inorganic arsenic exposure.
8.3.5 Effects on the nervous system
8.3.5.1 Man
As noted in section 8.1, reversible peripheral neurological damage
has often followed acute and subacute exposure to inorganic arsenic.
Recovery is slow and may take several months or even years.
Some studies have shown that long-term exposure to arsenic in the
workplace and through drugs has resulted in peripheral neuropathy.
Very few studies have dealt with possible neurological effects
following occupational exposure to arsenic.
Heyman et al. (1956) reported 41 cases of suspected
arsenic-neuropathy in the USA. At least 7 of these cases were caused
by occupational exposure to arsenate sprays. Cases of occupational
arsenic polyneuropathy have also been reported from Japan (Oida, 1957;
Hara et al., 1968). The first report included 4 male workers, 24-54
years of age, employed for 11-32 years in the manufacture of arsenic
(form not stated) at a copper refinery. The symptoms and signs
included peripheral nervous disturbances and neuritis retrobulbaris as
well as chronic rhinitis combined with septum perforation. The other
report described 9 persons, aged 20-32 years, employed in the
desulfurization of coal gas and exposed to arsenic in both the
trivalent and pentavalent inorganic forms, who had developed symptoms
and signs of sensimotor polyneuropathy. The symptoms and prognosis of
chronic occupational arsenical polyneuropathy in these 2 studies
resembled those of the neuropathy following acute exposure; some
recovery was seen following cessation of exposure.
Tay & Seah (1975) examined 74 patients who had taken
anti-asthmatic herbal preparations containing inorganic arsenic at
levels of up to 107 g/kg. The recommended daily dose resulted in an
intake of 3.3 mg from a pill, which contained arsenic(III) oxide, and
an intake of 10.3 mg from a pill, which contained arsenic sulfide.
Arsenic levels in the hair of over 1 mg/kg were found in 45% of the
patients. Cutaneous manifestations of arsenic exposure were observed
in 92% of patients including 6 cases of skin cancer. Over half of the
patients presented neurological complications, the most common being
sensimotor polyneuropathy.
Two case reports attribute peripheral neuropathy of the sensimotor
type to cutaneous exposure to arsenic. In one case a worker was
splashed with arsenic acid in an industrial accident and visual
symptoms appeared (Garb & Hine, 1977). The source of the arsenic in
the other case was the topical application of a caustic paste in which
the form of arsenic was not specified (Robinson, 1975).
Severe hearing loss (> 30 dB) was observed in 18% of 415 children
examined in a follow-up study of the poisoning episode in Japan in
1955, where infants were given powdered milk containing inorganic
pentavalent arsenic (Yamashita et al., 1972). According to the local
health statistics, the portion of the population in the same age
groups with corresponding hearing losses was less than 1%. Moreover,
the percentage of brain wave abnormalities observed in the
arsenic-exposed children (14%) was twice as high as would be expected.
Children exposed during the first 6 months of their life showed a
higher rate of abnormalities (17%) than those exposed later (11%).
Another follow-up investigation of the same incident of arsenic
poisoning, made on a different part of the population and
independently of the first study, showed a statistically significant
increase in electroencephalographic abnormalities in children fed
arsenic-contaminated powdered milk compared with breastfed infants
(Ohira & Aoyama, 1972). This study also revealed a number of
pathological eye changes in the powdered milk group, including a case
of bilateral optic atrophy.
Hearing losses associated with exposure to arsenic have also been
reported from Czechoslovakia. Bencko et al. (1977) examined a group of
56 10-year old children living near a power plant burning local coal
that had a high arsenic content. When compared to a control group of
51 children in the same age group living outside the polluted area,
the exposed children showed significant hearing losses in both air and
bone conduction at a high frequency range, indicating inner ear
damage. A higher proportion of children who had suffered middle ear
inflammation was found in the control group, while the 2 groups did
not show any major differences as far as most other medical conditions
affecting hearing were concerned. A study carried out near a copper
smelter in the USA, which emitted considerable amounts of arsenic,
failed to show any impairment of hearing in children living in the
area (Milham, 1977).
Electromyographic examinations were made on 33 people living in an
area of Canada with a high content of arsenic in the well water (more
than 0.05 mg/litre) and 12 controls from the same community who used
water sources with a lower content of arsenic (Hindmarsh et al.,
1977). EMGs were abnormal in 33% of the exposed persons, but not in
any of the controls. Of persons using well water with an arsenic
content exceeding 0.1 mg/litre, 50% exhibited abnormal EMGs. A similar
relationship was found when hair concentrations of arsenic were
correlated with abnormal EMGs. Of persons with hair levels exceeding
1 mg/kg, 52.6% had abnormal EMG findings. The way the results in this
study are presented makes their interpretation somewhat difficult. It
is not clear how well the controls matched the exposed persons in the
various exposure groups with regard to such factors as age, alcohol
consumption, and diseases that would predispose them to have EMG
abnormalities.
Effects on the nervous system have not been reported in the
investigations from Argentina, Chile, or China (Province of Taiwan),
where the subjects studied used drinking-water containing appreciable
amounts of arsenic. Clinical and subclinical neurological effects do
not seem to have been looked for. It would be of great value to
conduct adequate neurological studies in these areas as well as in
occupational exposure situations.
8.3.5.2 Animals
A one-year toxicity study was performed in which rhesus monkeys
were given arsenic in the form of a complex arsenate salt
(2Na3(PO4AsO4VO4)NaF.18H2O) as a suspension in milk (Heywood &
Sortwell, 1979). This is the same arsenic compound that was reported
to be present in the dried milk resulting in massive infant poisoning
in Japan (Tsuchiya, 1977). Five out of 7 infant monkeys survived daily
oral doses of 2.8 mg of arsenic per kg body weight for one year and
did not show any neurological abnormalities or other signs of toxic
effects. It is, however, difficult to estimate the amount of arsenic
absorbed as the substance was given as a suspension, indicating that
it had a low solubility.
Dysfunction of the blood-brain barrier was indicated in rats fed
arsenite at a concentration of 500 mg/kg diet in a cereal diet for 35
days (Tamura & Nozaki, 1972).
Effects on the ear have been reported in experimental animals
treated with inorganic arsenic. Destruction of the Corti organ and
loss of Reissemer's membrane, causing deafness, were observed in
guineapigs given sodium arsenate intraperitoneally for 2 months (Aly
et al., 1975). The dose was reported as 0.2 mg sodium arsenate per kg
body weight. Further investigations by Aly et al. (1975) revealed
diminished acetyl cholinesterase [EG 3.1.1.7] activity in the temporal
lobe and decreased blood cholinesterase levels in exposed animals.
Inhibition of cholinesterase activity was also observed in rats
exposed for 3 months to arsenic(III) oxide in the form of condensation
aerosols containing 46 µg As/m3 (Rozenshtein, 1970). Disturbances in
the functional state of the CNS were reflected as changes in
conditioned reflexes and in chronaximetry. Histopathological changes
in the brain included pericellular oedema, plasmatic impregnation of
the vascular walls, plasmolysis, and karyolysis of the neurons.
Several of the effects mentioned, although less marked, were also
observed in a group of rats exposed to an aerosol containing 3.7 µg
As/m3. Osato (1977) gave suckling rats 2 and 10 mg of arsenic(III)
oxide through a stomach tube for 40 days. That the central nervous
system had been affected was indicated by a significantly poorer
performance in the avoidance conditioning test in both groups of
exposed animals. No histopathological changes of note were found in
the brains of these animals.
8.3.6 Effects on other organs
8.3.6.1 Man
Long-term exposure to inorganic arsenic, through drinking-water,
medication, or in occupational situations, has resulted in
disturbances of the haematopoietic system (Terada et al., 1960; Kyle &
Pease, 1965; Westhoff et al., 1975; Feussner et al., 1979) The blood
picture in these situations often resembles that in acute
intoxication. Bone marrow examination shows disturbed erythropoiesis,
and occasionally megaloblastic changes. Severe granulocytopenia may
also be present, with possible effects on resistance to bacterial
infections. As in acute intoxication, the blood picture has been
reported to return to normal, 2-3 weeks following cessation of
exposure.
8.3.6.2 Animals
Results of animal experiments show effects on the haematopoietic
system similar to those observed in man. A decrease in haematocrit and
in haemoglobin has been observed in female rats exposed to arsenite in
the feed (250 mg As/kg diet) for 2 years (Byron et al., 1967) and in
rats given sodium arsenate in the feed (50 mg As/kg diet) for 10 weeks
(Mahaffey & Fowler, 1977). The same effects were observed in cats
given arsenite or arsenate in the diet in doses of 1.5 mg As/kg body
weight (Massmann & Opitz, 1954).
Studies on laboratory animals indicate that arsenic can impair
resistance to viral infections. Increased mortality from viral
infections among mice exposed to arsenic was reported by Gainer & Pry
(1972). The mice were given arsenic subcutaneously at the time of
inoculation or in drinking water for 2 weeks before inoculation. The
subcutaneous doses were 2-4 mg As/kg body weight as sodium arsenite or
arsenic(III) oxide and the drinking water doses 75-150 mg As/litre as
sodium arsenite or sodium arsenate. The viruses used were
pseudorabies, encephalomyocarditis, and St. Louis encephalitis
viruses. Mice given intraperitoneal injections of sodium arsenite
(1.8 mg As/kg body weight) were less protected by poly I/poly C
(a synthetic homopolynucleotide complex) against encephalomyocarditis
virus than the controls (Gainer, 1972). The protective action of poly
I/poly C against viruses was reported to be associated with interferon
formation.
Minor changes in kidney function and histology have been reported
in laboratory animals. Rats exposed to calcium and lead arsenate in
daily doses of 2 mg/animal in the food for 2 years showed casts in the
straight collecting tubules and swollen cells with large vesicular
nuclei in groups of convoluted tubules (Fairhall & Miller, 1941). Rats
given arsenic in the drinking water (deionized water; 40, 85, or
125 mg As/litre as sodium arsenate), for 6 weeks, showed increased
kidney weights in relation to body weights (Brown et al., 1976). The
proximal tubular cells contained electron dense lysosome-like bodies
and swollen mitochondria. Indications of impaired kidney function,
including decreased urea clearance and increased serum creatinine have
been reported in rabbits given intravenous injections of arsenious
acid. The doses (0.6 mg As/kg body weight) were administered 3 times a
week for 2-12 weeks (Shibuya, 1971).
8.4 Carcinogenicity
Exposure to arsenic has been associated with the induction of
cancer for nearly a century. In 1888, Hutchinson discussed the
possibility that medication with inorganic arsenic was an aetiological
factor for skin cancer. Ever since, arsenic has been implicated as a
causative agent for cancer in other organs also. In 1979, an IARC
working group concluded that there was sufficient evidence that
inorganic arsenic compounds were skin and lung carcinogens in man, but
that the data for other sites were inadequate for evaluation (IARC,
1980).
8.4.1 Man
8.4.1.1 Cancer of the respiratory system
Several investigations concerning occupational populations exposed
to inorganic arsenic have indicated an association with lung cancer.
Many early reports in this field do not lend themselves to evaluation
of dose-response relationships and will not be discussed in detail.
An excess of deaths due to respiratory cancer has been observed
among workers exposed to inorganic arsenic in the production and use
of pesticides, gold mining, and in the smelting of nonferrous metals,
especially copper (Hill & Faning, 1948; Osburn, 1957, 1969; Roth,
1958; Lee & Fraumeni, 1969; Ott et al., 1974; Beatjer et al., 1975;
Tokudome & Kuratsune, 1976; Pershagen et al., 1977; Pinto et al.,
1977; Rencher et al., 1977; Axelson et al., 1978; Mabuchi et al.,
1979). The composition of the environment in most occupational
situations involving arsenic is very complex, but the extent of
exposure to other agents has rarely been reported. In the following
discussion, the various exposure situations will be dealt with
separately. The effects observed should be considered in the context
of the total exposure in these workplaces.
The mortality experience among workers engaged in the production
of insecticides containing inorganic arsenic compounds has been the
subject of 3 major epidemiological studies. Hill & Faning (1948) found
a significant excess proportion of deaths attributable to cancer among
workers producing sheep dip powder from sodium arsenite. Atmospheric
concentrations of arsenic in this plant in 1946 averaged between 78
and 1034 µg/m3 in different workplaces during sampling periods of
10 min or more (Perry et al., 1948). Analysis of 179 cancer deaths
showed that 31.8% of the factory workers had died from cancer of the
respiratory organs (larynx, lung, mediastinum, and bronchus), compared
with 15.9% of the deaths in other occupational groups from the
environs of the plant, i.e., agricultural workers, general labourers,
artisans, and shopworkers (Hill & Faning, 1948). Smoking habits, which
of course may have had a substantial influence on deaths from cancer
of the respiratory organs, were not recorded. If considerable
differences in smoking habits existed between the factory workers and
the other occupational groups, the findings might be invalidated.
Ott et al. (1974) examined the causes of death for the period
1940-72 with reference to proportionate mortality rates in nearly 2000
workers, who had been engaged in the production of insecticides
including lead arsenate, calcium arsenate, copper acetoarsenite, and
magnesium arsenite. Various forms of inorganic arsenic, including
trivalent compounds were used in the processes. Airborne arsenic
levels in 1943 ranged from 0.18 mg/m3 to 19 mg/m3 (time interval for
measurement not stated) in the packaging department. In 1952,
concentrations between 0.26 mg/m3 and 40.8 mg/m3 were recorded. On
the basis of work histories, 173 of the workers were classified as
workers who had been exposed to arsenic, i.e., engaged in formulating
and packaging arsenic-containing insecticides for one or more days.
"Respiratory malignancies" accounted for 16.2% of the deaths in the
exposed group compared with 5.7% in the rest of the workers. It should
be noted that 16 of the 28 deaths from "malignant neoplasms of the
respiratory system" in the exposed group occurred in individuals with
an arsenic exposure of less than 1 year. Only men who died during
employment or following retirement from the company were included in
the survey, and hence both the exposed and unexposed categories could
be biased samples of the actual population of employees. A positive
dose-response relationship between the degree of arsenic exposure and
lung cancer mortality was indicated (Fig. 9). The ratio of observed to
expected respiratory cancer deaths ranged from 0.6 in the lowest
exposure category to 7.0 in the highest. The degree of exposure was
based on available industrial hygiene data, annual personnel lists and
assessment by 2 experienced industrial hygienists.
Peripheral vascular disease was also reported in inhabitants of
the Antofagasta region of northern Chile, who had been exposed to
arsenic levels of about 0.6 mg/litre in the drinking water for 15
years. A clinical investigation among 180 inhabitants revealed several
effects associated with chronic arsenic exposure, including
hyperkeratosis, and a high prevalence of cardiovascular disturbances
(Borgono et al., 1977). Most common were peripheral vascular
phenomena, i.e., Raynaud's syndrome and acrocyanosis, which were
present, respectively, in 38.8% and 24.3% of the persons with abnormal
skin pigmentation attributed to arsenic exposure, compared with 9.3%
and 12.5% of persons with normal skin. Infants and children showed
more pronounced symptoms than adults. Systemic arterial disease
resulting in myocardial infarction was reported in 2 children
(Rosenberg, 1974). The small and medium-sized arteries of most organs
showed marked thickening of the intima. Similar microscopic findings
have been reported in 2 men (32 and 36 years of age) from the same
area who had myocardial infarctions but lacked the risk factors
usually associated with coronary sclerosis (Moran et al., 1977).
It is noteworthy that effects on the peripheral vascular system
have not been reported among patients undergoing medication with
inorganic arsenic or among workers exposed to high levels of airborne
arsenic. It is not clear, however, whether investigators have looked
for such effects in these groups.
8.3.4.2 Animals
In order to study the chronic effects of arsenic, Massmann & Opitz
(1954) fed 12 cats about 1.5 mg As/kg body weight as sodium arsenite
and sodium arsenate mixed with the feed. T-wave abnormalities, mainly
flattening, were found in the electrocardiograms of 9 of the animals
and the QT-time was prolonged in some animals. No control group was
used, which makes it difficult to interpret these findings. The
animals were allowed to die spontaneously, at which time histological
analysis of the heart was performed. No marked changes were found. No
differences in effects were observed between the 2 forms of arsenic
used.
Data are lacking with regard to the extent to which experimental
animals develop the peripheral vascular lesions observed in human
subjects following chronic inorganic arsenic exposure.
8.3.5 Effects on the nervous system
8.3.5.1 Man
As noted in section 8.1, reversible peripheral neurological damage
has often followed acute and subacute exposure to inorganic arsenic.
Recovery is slow and may take several months or even years.
Some studies have shown that long-term exposure to arsenic in the
workplace and through drugs has resulted in peripheral neuropathy.
Very few studies have dealt with possible neurological effects
following occupational exposure to arsenic.
Heyman et al. (1956) reported 41 cases of suspected
arsenic-neuropathy in the USA. At least 7 of these cases were caused
by occupational exposure to arsenate sprays. Cases of occupational
arsenic polyneuropathy have also been reported from Japan (Oida, 1957;
Hara et al., 1968). The first report included 4 male workers, 24-54
years of age, employed for 11-32 years in the manufacture of arsenic
(form not stated) at a copper refinery. The symptoms and signs
included peripheral nervous disturbances and neuritis retrobulbaris as
well as chronic rhinitis combined with septum perforation. The other
report described 9 persons, aged 20-32 years, employed in the
desulfurization of coal gas and exposed to arsenic in both the
trivalent and pentavalent inorganic forms, who had developed symptoms
and signs of sensimotor polyneuropathy. The symptoms and prognosis of
chronic occupational arsenical polyneuropathy in these 2 studies
resembled those of the neuropathy following acute exposure; some
recovery was seen following cessation of exposure.
Tay & Seah (1975) examined 74 patients who had taken
anti-asthmatic herbal preparations containing inorganic arsenic at
levels of up to 107 g/kg. The recommended daily dose resulted in an
intake of 3.3 mg from a pill, which contained arsenic(III) oxide, and
an intake of 10.3 mg from a pill, which contained arsenic sulfide.
Arsenic levels in the hair of over 1 mg/kg were found in 45% of the
patients. Cutaneous manifestations of arsenic exposure were observed
in 92% of patients including 6 cases of skin cancer. Over half of the
patients presented neurological complications, the most common being
sensimotor polyneuropathy.
Two case reports attribute peripheral neuropathy of the sensimotor
type to cutaneous exposure to arsenic. In one case a worker was
splashed with arsenic acid in an industrial accident and visual
symptoms appeared (Garb & Hine, 1977). The source of the arsenic in
the other case was the topical application of a caustic paste in which
the form of arsenic was not specified (Robinson, 1975).
Severe hearing loss (> 30 dB) was observed in 18% of 415 children
examined in a follow-up study of the poisoning episode in Japan in
1955, where infants were given powdered milk containing inorganic
pentavalent arsenic (Yamashita et al., 1972). According to the local
health statistics, the portion of the population in the same age
groups with corresponding hearing losses was less than 1%. Moreover,
the percentage of brain wave abnormalities observed in the
arsenic-exposed children (14%) was twice as high as would be expected.
Children exposed during the first 6 months of their life showed a
higher rate of abnormalities (17%) than those exposed later (11%).
Another follow-up investigation of the same incident of arsenic
poisoning, made on a different part of the population and
independently of the first study, showed a statistically significant
increase in electroencephalographic abnormalities in children fed
arsenic-contaminated powdered milk compared with breastfed infants
(Ohira & Aoyama, 1972). This study also revealed a number of
pathological eye changes in the powdered milk group, including a case
of bilateral optic atrophy.
Hearing losses associated with exposure to arsenic have also been
reported from Czechoslovakia. Bencko et al. (1977) examined a group of
56 10-year old children living near a power plant burning local coal
that had a high arsenic content. When compared to a control group of
51 children in the same age group living outside the polluted area,
the exposed children showed significant hearing losses in both air and
bone conduction at a high frequency range, indicating inner ear
damage. A higher proportion of children who had suffered middle ear
inflammation was found in the control group, while the 2 groups did
not show any major differences as far as most other medical conditions
affecting hearing were concerned. A study carried out near a copper
smelter in the USA, which emitted considerable amounts of arsenic,
failed to show any impairment of hearing in children living in the
area (Milham, 1977).
Electromyographic examinations were made on 33 people living in an
area of Canada with a high content of arsenic in the well water (more
than 0.05 mg/litre) and 12 controls from the same community who used
water sources with a lower content of arsenic (Hindmarsh et al.,
1977). EMGs were abnormal in 33% of the exposed persons, but not in
any of the controls. Of persons using well water with an arsenic
content exceeding 0.1 mg/litre, 50% exhibited abnormal EMGs. A similar
relationship was found when hair concentrations of arsenic were
correlated with abnormal EMGs. Of persons with hair levels exceeding
1 mg/kg, 52.6% had abnormal EMG findings. The way the results in this
study are presented makes their interpretation somewhat difficult. It
is not clear how well the controls matched the exposed persons in the
various exposure groups with regard to such factors as age, alcohol
consumption, and diseases that would predispose them to have EMG
abnormalities.
Effects on the nervous system have not been reported in the
investigations from Argentina, Chile, or China (Province of Taiwan),
where the subjects studied used drinking-water containing appreciable
amounts of arsenic. Clinical and subclinical neurological effects do
not seem to have been looked for. It would be of great value to
conduct adequate neurological studies in these areas as well as in
occupational exposure situations.
8.3.5.2 Animals
A one-year toxicity study was performed in which rhesus monkeys
were given arsenic in the form of a complex arsenate salt
(2Na3(PO4AsO4VO4)NaF.18H2O) as a suspension in milk (Heywood &
Sortwell, 1979). This is the same arsenic compound that was reported
to be present in the dried milk resulting in massive infant poisoning
in Japan (Tsuchiya, 1977). Five out of 7 infant monkeys survived daily
oral doses of 2.8 mg of arsenic per kg body weight for one year and
did not show any neurological abnormalities or other signs of toxic
effects. It is, however, difficult to estimate the amount of arsenic
absorbed as the substance was given as a suspension, indicating that
it had a low solubility.
Dysfunction of the blood-brain barrier was indicated in rats fed
arsenite at a concentration of 500 mg/kg diet in a cereal diet for 35
days (Tamura & Nozaki, 1972).
Effects on the ear have been reported in experimental animals
treated with inorganic arsenic. Destruction of the Corti organ and
loss of Reissemer's membrane, causing deafness, were observed in
guineapigs given sodium arsenate intraperitoneally for 2 months (Aly
et al., 1975). The dose was reported as 0.2 mg sodium arsenate per kg
body weight. Further investigations by Aly et al. (1975) revealed
diminished acetyl cholinesterase [EG 3.1.1.7] activity in the temporal
lobe and decreased blood cholinesterase levels in exposed animals.
Inhibition of cholinesterase activity was also observed in rats
exposed for 3 months to arsenic(III) oxide in the form of condensation
aerosols containing 46 µg As/m3 (Rozenshtein, 1970). Disturbances in
the functional state of the CNS were reflected as changes in
conditioned reflexes and in chronaximetry. Histopathological changes
in the brain included pericellular oedema, plasmatic impregnation of
the vascular walls, plasmolysis, and karyolysis of the neurons.
Several of the effects mentioned, although less marked, were also
observed in a group of rats exposed to an aerosol containing 3.7 µg
As/m3. Osato (1977) gave suckling rats 2 and 10 mg of arsenic(III)
oxide through a stomach tube for 40 days. That the central nervous
system had been affected was indicated by a significantly poorer
performance in the avoidance conditioning test in both groups of
exposed animals. No histopathological changes of note were found in
the brains of these animals.
8.3.6 Effects on other organs
8.3.6.1 Man
Long-term exposure to inorganic arsenic, through drinking-water,
medication, or in occupational situations, has resulted in
disturbances of the haematopoietic system (Terada et al., 1960; Kyle &
Pease, 1965; Westhoff et al., 1975; Feussner et al., 1979) The blood
picture in these situations often resembles that in acute
intoxication. Bone marrow examination shows disturbed erythropoiesis,
and occasionally megaloblastic changes. Severe granulocytopenia may
also be present, with possible effects on resistance to bacterial
infections. As in acute intoxication, the blood picture has been
reported to return to normal, 2-3 weeks following cessation of
exposure.
8.3.6.2 Animals
Results of animal experiments show effects on the haematopoietic
system similar to those observed in man. A decrease in haematocrit and
in haemoglobin has been observed in female rats exposed to arsenite in
the feed (250 mg As/kg diet) for 2 years (Byron et al., 1967) and in
rats given sodium arsenate in the feed (50 mg As/kg diet) for 10 weeks
(Mahaffey & Fowler, 1977). The same effects were observed in cats
given arsenite or arsenate in the diet in doses of 1.5 mg As/kg body
weight (Massmann & Opitz, 1954).
Studies on laboratory animals indicate that arsenic can impair
resistance to viral infections. Increased mortality from viral
infections among mice exposed to arsenic was reported by Gainer & Pry
(1972). The mice were given arsenic subcutaneously at the time of
inoculation or in drinking water for 2 weeks before inoculation. The
subcutaneous doses were 2-4 mg As/kg body weight as sodium arsenite or
arsenic(III) oxide and the drinking water doses 75-150 mg As/litre as
sodium arsenite or sodium arsenate. The viruses used were
pseudorabies, encephalomyocarditis, and St. Louis encephalitis
viruses. Mice given intraperitoneal injections of sodium arsenite
(1.8 mg As/kg body weight) were less protected by poly I/poly C
(a synthetic homopolynucleotide complex) against encephalomyocarditis
virus than the controls (Gainer, 1972). The protective action of poly
I/poly C against viruses was reported to be associated with interferon
formation.
Minor changes in kidney function and histology have been reported
in laboratory animals. Rats exposed to calcium and lead arsenate in
daily doses of 2 mg/animal in the food for 2 years showed casts in the
straight collecting tubules and swollen cells with large vesicular
nuclei in groups of convoluted tubules (Fairhall & Miller, 1941). Rats
given arsenic in the drinking water (deionized water; 40, 85, or
125 mg As/litre as sodium arsenate), for 6 weeks, showed increased
kidney weights in relation to body weights (Brown et al., 1976). The
proximal tubular cells contained electron dense lysosome-like bodies
and swollen mitochondria. Indications of impaired kidney function,
including decreased urea clearance and increased serum creatinine have
been reported in rabbits given intravenous injections of arsenious
acid. The doses (0.6 mg As/kg body weight) were administered 3 times a
week for 2-12 weeks (Shibuya, 1971).
8.4 Carcinogenicity
Exposure to arsenic has been associated with the induction of
cancer for nearly a century. In 1888, Hutchinson discussed the
possibility that medication with inorganic arsenic was an aetiological
factor for skin cancer. Ever since, arsenic has been implicated as a
causative agent for cancer in other organs also. In 1979, an IARC
working group concluded that there was sufficient evidence that
inorganic arsenic compounds were skin and lung carcinogens in man, but
that the data for other sites were inadequate for evaluation (IARC,
1980).
8.4.1 Man
8.4.1.1 Cancer of the respiratory system
Several investigations concerning occupational populations exposed
to inorganic arsenic have indicated an association with lung cancer.
Many early reports in this field do not lend themselves to evaluation
of dose-response relationships and will not be discussed in detail.
An excess of deaths due to respiratory cancer has been observed
among workers exposed to inorganic arsenic in the production and use
of pesticides, gold mining, and in the smelting of nonferrous metals,
especially copper (Hill & Faning, 1948; Osburn, 1957, 1969; Roth,
1958; Lee & Fraumeni, 1969; Ott et al., 1974; Beatjer et al., 1975;
Tokudome & Kuratsune, 1976; Pershagen et al., 1977; Pinto et al.,
1977; Rencher et al., 1977; Axelson et al., 1978; Mabuchi et al.,
1979). The composition of the environment in most occupational
situations involving arsenic is very complex, but the extent of
exposure to other agents has rarely been reported. In the following
discussion, the various exposure situations will be dealt with
separately. The effects observed should be considered in the context
of the total exposure in these workplaces.
The mortality experience among workers engaged in the production
of insecticides containing inorganic arsenic compounds has been the
subject of 3 major epidemiological studies. Hill & Faning (1948) found
a significant excess proportion of deaths attributable to cancer among
workers producing sheep dip powder from sodium arsenite. Atmospheric
concentrations of arsenic in this plant in 1946 averaged between 78
and 1034 µg/m3 in different workplaces during sampling periods of
10 min or more (Perry et al., 1948). Analysis of 179 cancer deaths
showed that 31.8% of the factory workers had died from cancer of the
respiratory organs (larynx, lung, mediastinum, and bronchus), compared
with 15.9% of the deaths in other occupational groups from the
environs of the plant, i.e., agricultural workers, general labourers,
artisans, and shopworkers (Hill & Faning, 1948). Smoking habits, which
of course may have had a substantial influence on deaths from cancer
of the respiratory organs, were not recorded. If considerable
differences in smoking habits existed between the factory workers and
the other occupational groups, the findings might be invalidated.
Ott et al. (1974) examined the causes of death for the period
1940-72 with reference to proportionate mortality rates in nearly 2000
workers, who had been engaged in the production of insecticides
including lead arsenate, calcium arsenate, copper acetoarsenite, and
magnesium arsenite. Various forms of inorganic arsenic, including
trivalent compounds were used in the processes. Airborne arsenic
levels in 1943 ranged from 0.18 mg/m3 to 19 mg/m3 (time interval for
measurement not stated) in the packaging department. In 1952,
concentrations between 0.26 mg/m3 and 40.8 mg/m3 were recorded. On
the basis of work histories, 173 of the workers were classified as
workers who had been exposed to arsenic, i.e., engaged in formulating
and packaging arsenic-containing insecticides for one or more days.
"Respiratory malignancies" accounted for 16.2% of the deaths in the
exposed group compared with 5.7% in the rest of the workers. It should
be noted that 16 of the 28 deaths from "malignant neoplasms of the
respiratory system" in the exposed group occurred in individuals with
an arsenic exposure of less than 1 year. Only men who died during
employment or following retirement from the company were included in
the survey, and hence both the exposed and unexposed categories could
be biased samples of the actual population of employees. A positive
dose-response relationship between the degree of arsenic exposure and
lung cancer mortality was indicated (Fig. 9). The ratio of observed to
expected respiratory cancer deaths ranged from 0.6 in the lowest
exposure category to 7.0 in the highest. The degree of exposure was
based on available industrial hygiene data, annual personnel lists and
assessment by 2 experienced industrial hygienists.
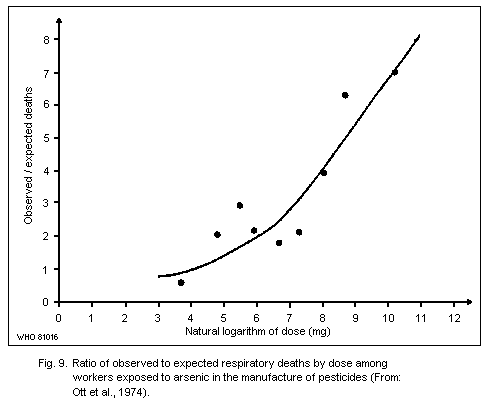 Blejer & Wagner (1976) calculated the daily 8-h time-weighted
average (TWA) airborne arsenic concentrations over a 40-year working
life that would correspond to the various exposure categories used by
Ott et al. (1974). The results are shown in Table 9. It was suggested
that a "no-effect level vis-à-vis an increased respiratory cancer
mortality risk might lie in the very low microgram range of arsenic
per m3." If, however, workers exposed to arsenic for less than one
year are excluded, no deaths due to "respiratory cancer" occurred in
the categories with daily TWA concentrations of up to 90 µg/m3 over
40 years. It should also be noted that a consistent dose-response
relationship was not observed below this level. Great care should be
taken not to draw too firm conclusions from these data in view of the
substantial unreliability involved in the computations of expected
"respiratory cancer deaths", which were based on proportionate
mortality rates. Furthermore, smoking histories could not be obtained
for the workers included in the study.
Table 9. Observed and expected deaths due to "respiratory cancer"
by exposure categorya
Daily TWA dose Respiratory cancer deaths
(µg/m3) Observed Expected Observed/
Expected
1 1 1.77 0.6
3 2 1.01 2.0
6 4 1.38 2.9
10 3 1.36 2.2
20 3 1.70 1.8
40 2 0.97 2.1
90 3 0.77 3.9
160 5 0.79 6.3
740 5 0.72 7.0
a Modified from: Ott et al. (1974) and Blejer & Wagner (1976).
Mortality rates were studied, according to cause, in 1393 workers
employed from 1946-74 in a factory where pesticides were manufactured
(Mabuchi et al., 1979). The workers were exposed to many arsenic
compounds including arsenic(III) oxide, lead arsenate, calcium
arsenate, and various arsenites as well as to copper sulfate,
chlorinated hydrocarbons, organic phosphates, and carbamates, and
other organic herbicides. In 1972, an atmospheric concentration of
0.5 mg As/m3 was reported; however, the concentrations at the
pertinent time of exposure were not known. By August 1977, 197 males
and 43 females had died (the vital status of 18% of the total cohort
was not available). The observed number of deaths from all or selected
causes were compared with the numbers expected from the death rates
for the general population in the city where the plant was located.
The overall standardized mortality ratiosa (SMR) were close to 100,
and the only statistically significant excesses of deaths were seen
for cancer of the trachea, bronchus, and lung in males (23 observed,
13.7 expected; SMR = 168) and anaemias in males (2 observed, 0.2
expected; SMR = 1000). Furthermore, though based on small numbers of
deaths, there was also an increase in the SMR for lung cancer with
increasing duration of exposure to arsenic compounds, but not with
nonarsenic products. No data were given on smoking habits.
Cases of lung cancer have also been reported among workers engaged
in the spraying of insecticides containing inorganic arsenic, in the
Federal Republic of Germany and France (Roth, 1958: Galy et al.,
1963). Among 47 vintners with signs of chronic arsenic intoxication,
Roth found bronchial cancer in 19 at autopsy. Nine of these also had
primary tumours of other organs, most notably the skin. A selection
bias cannot be excluded; however, the frequently observed simultaneous
development of primary tumours in the bronchus and skin should
represent an excess morbidity. When looking at the mortality
statistics of the region between 1950 and 1956, the same author noted
that the death rates of bronchial carcinomas was markedly higher in
the wine producing districts than in other areas i.e., 5.1% and below
1%, respectively. Besides exposure through inhalation, the consumption
of arsenic-contaminated wine must be considered. In 1938, 43% of 336
samples of wine from the area studied by Roth contained more than 5 mg
As/litre (Koelsch, 1958).
In a follow-up of more than 1200 orchard workers, who were
registered in a medical survey in the USA in 1938-39, no excess was
detected either for total deaths or for deaths from cancer (Nelson
et al., 1973). The male workers had been exposed to lead arsenate
resulting in an average urinary arsenic concentration of 0.14 mg/litre
in 1938. No data on exposure other than the duration of employment of
each individual as an orchardist could be obtained, when the follow-up
was made. The mortality data were analysed for the counties in which
the orchardists surveyed by Nelson et al. had lived, and a respiratory
cancer mortality 7% higher than that in the state as a whole was found
a The standardized mortality ratio is the ratio of observed
to expected deaths multiplied by 100.
(NIOSH, 1975). When looking at death records of the whole state from
1961-71, a significant increase in respiratory cancer mortality was
observed among decedents classified as orchardists. The data in the
NIOSH document cannot be evaluated in detail because of lack of
information concerning such factors as the methods and numbers of
people.
An increased respiratory cancer mortality has been observed in
many studies on smelter workers exposed to high levels of inorganic
arsenic, mainly in the form of arsenic(III) oxide. Lee & Fraumeni
(1969) compared the mortality rate among more than 8000 smelter
workers in the USA with that of the white male population of the
states in question. The mortality was followed from 1938-63 and a
total of 1877 deaths were recorded. About 10% of the original cohort
was lost in the follow-up. An excess mortality was found for cancer of
the respiratory system (lung and bronchus, larynx, and mediastinum)
with an overall standardized mortality ratio, SMR, equal to 329. A
positive relationship between SMR and estimated degree of exposure was
indicated, with an SMR of 800 in the highest arsenic exposure group. A
positive relationship was also found between exposure to sulfur
dioxide and respiratory cancer mortality with a SMR of over 700 in the
highest exposure groups. Unfortunately, data were not available
concerning the actual exposure levels and it was difficult to separate
the effects associated with arsenic from those associated with sulfur
dioxide, since both types of exposure occurred in most work areas.
Smoking histories were not obtained, and if large age distribution
differences were at hand between the different exposure groups the
comparison of SMRs would be unreliable.
The mortality among 2675 Japanese metal workers from 1949-1971 was
examined by Tokudome & Kuratsune (1976), using the cohort technique.
Workers were divided into 5 cohorts depending on their work histories,
i.e., copper smelting, ferronickel smelting, maintenance or transport,
copper or lead electrolysis or production of sulfuric acid, and
clerical work. Among the 839 copper smelters, an almost 12-fold
increase in the number of deaths was observed for cancer of the
trachea, bronchus, and lung in comparison with the expected number,
which was derived from the national rates for Japanese males during
the period at issue. A positive correlation was observed between
length of employment and excess of deaths from cancer of the trachea,
bronchi, and lung as well as between estimated levels of arsenic
exposure and excess mortality. The exposure was not described in
detail and smoking habits and age distributions in the various
exposure groups were not reported.
Pinto et al. (1977), in a follow-up of an earlier study in 1963,
analysed the cause-specific mortality experience above 65 years of age
among 527 retired workers at a large copper smelter in Washington,
USA. Expected deaths were computed from the mortality rates of the
male population in the state of Washington during the time period at
issue, i.e., 1949-73. Exposure occurred, mainly to arsenic(III) oxide,
and was assessed on the basis of the determination of arsenic
concentrations in the urine of workers in each department of the
factory in 1973. It was emphasized that the relevant exposure levels,
i.e., those during the active working life of the decedents, were
higher. Each worker was assigned an exposure index that was calculated
by multiplying the period of time spent in the various departments by
the 1973 urinary arsenic levels of workers in these departments.
A highly significant increase in mortality from cancer of the
respiratory system (ICD 160-164)a was found for the whole group of
former smelter workers under study (SMR = 304.8), which had a roughly
linear relationship with the estimated time-weighted average total
life-time exposure (Fig. 10). The comparison between the different
SMRs should be made with caution, as their magnitudes depend partly on
the age distribution of the index population for each SMR.
A group of workers with more than 25 years of exposure to airborne
arsenic at concentrations associated with urinary arsenic levels of
50-200 µg/litre, i.e., (according to Fig. 4) up to approximately
40 µg/m3 or a rough average of about 25 µg/m3, showed an SMR of
277.8. As previously stated, the exposure was probably underestimated
because of higher levels of airborne arsenic in earlier years. Urinary
arsenic data from Pinto et al. (1978) indicate that the exposures were
higher by perhaps a factor of 2 than those given in the previous
paragraph. This would mean that exposure to airborne arsenic at levels
of around 25 µg/m3, according to the original estimates, or perhaps
more appropriately 50 µg/m3 according to the revised value, would
lead to a nearly 3-fold (increase in) mortality from lung cancer. It
should be emphasized that the uncertainty in the estimated exposure
could well amount to a factor of 2.
Smoking habits were recorded on a sample of the cohort (Pinto et
al., 1978), i.e., directly from all men still living and from the
friends and relatives of men who died after 1961, a total of 377
subjects. Observed respiratory cancer deaths among smokers,
ex-smokers, and nonsmokers were compared with data on expected deaths
in these smoking categories. The SMR in the study population was
elevated in both the smokers and ex-smokers (245.1-506.5) indicating
that smoking habits in the study population as a whole were not
responsible for the increased mortality rate from cancer of the
respiratory system. In fact, the highest SMR was noted among the
nonsmokers, although it must be stressed that its magnitude is
unstable due to the fact that the number of observed respiratory
cancer deaths was small.
a WHO (1965) International Classification of Diseases
(8th revision).
Blejer & Wagner (1976) calculated the daily 8-h time-weighted
average (TWA) airborne arsenic concentrations over a 40-year working
life that would correspond to the various exposure categories used by
Ott et al. (1974). The results are shown in Table 9. It was suggested
that a "no-effect level vis-à-vis an increased respiratory cancer
mortality risk might lie in the very low microgram range of arsenic
per m3." If, however, workers exposed to arsenic for less than one
year are excluded, no deaths due to "respiratory cancer" occurred in
the categories with daily TWA concentrations of up to 90 µg/m3 over
40 years. It should also be noted that a consistent dose-response
relationship was not observed below this level. Great care should be
taken not to draw too firm conclusions from these data in view of the
substantial unreliability involved in the computations of expected
"respiratory cancer deaths", which were based on proportionate
mortality rates. Furthermore, smoking histories could not be obtained
for the workers included in the study.
Table 9. Observed and expected deaths due to "respiratory cancer"
by exposure categorya
Daily TWA dose Respiratory cancer deaths
(µg/m3) Observed Expected Observed/
Expected
1 1 1.77 0.6
3 2 1.01 2.0
6 4 1.38 2.9
10 3 1.36 2.2
20 3 1.70 1.8
40 2 0.97 2.1
90 3 0.77 3.9
160 5 0.79 6.3
740 5 0.72 7.0
a Modified from: Ott et al. (1974) and Blejer & Wagner (1976).
Mortality rates were studied, according to cause, in 1393 workers
employed from 1946-74 in a factory where pesticides were manufactured
(Mabuchi et al., 1979). The workers were exposed to many arsenic
compounds including arsenic(III) oxide, lead arsenate, calcium
arsenate, and various arsenites as well as to copper sulfate,
chlorinated hydrocarbons, organic phosphates, and carbamates, and
other organic herbicides. In 1972, an atmospheric concentration of
0.5 mg As/m3 was reported; however, the concentrations at the
pertinent time of exposure were not known. By August 1977, 197 males
and 43 females had died (the vital status of 18% of the total cohort
was not available). The observed number of deaths from all or selected
causes were compared with the numbers expected from the death rates
for the general population in the city where the plant was located.
The overall standardized mortality ratiosa (SMR) were close to 100,
and the only statistically significant excesses of deaths were seen
for cancer of the trachea, bronchus, and lung in males (23 observed,
13.7 expected; SMR = 168) and anaemias in males (2 observed, 0.2
expected; SMR = 1000). Furthermore, though based on small numbers of
deaths, there was also an increase in the SMR for lung cancer with
increasing duration of exposure to arsenic compounds, but not with
nonarsenic products. No data were given on smoking habits.
Cases of lung cancer have also been reported among workers engaged
in the spraying of insecticides containing inorganic arsenic, in the
Federal Republic of Germany and France (Roth, 1958: Galy et al.,
1963). Among 47 vintners with signs of chronic arsenic intoxication,
Roth found bronchial cancer in 19 at autopsy. Nine of these also had
primary tumours of other organs, most notably the skin. A selection
bias cannot be excluded; however, the frequently observed simultaneous
development of primary tumours in the bronchus and skin should
represent an excess morbidity. When looking at the mortality
statistics of the region between 1950 and 1956, the same author noted
that the death rates of bronchial carcinomas was markedly higher in
the wine producing districts than in other areas i.e., 5.1% and below
1%, respectively. Besides exposure through inhalation, the consumption
of arsenic-contaminated wine must be considered. In 1938, 43% of 336
samples of wine from the area studied by Roth contained more than 5 mg
As/litre (Koelsch, 1958).
In a follow-up of more than 1200 orchard workers, who were
registered in a medical survey in the USA in 1938-39, no excess was
detected either for total deaths or for deaths from cancer (Nelson
et al., 1973). The male workers had been exposed to lead arsenate
resulting in an average urinary arsenic concentration of 0.14 mg/litre
in 1938. No data on exposure other than the duration of employment of
each individual as an orchardist could be obtained, when the follow-up
was made. The mortality data were analysed for the counties in which
the orchardists surveyed by Nelson et al. had lived, and a respiratory
cancer mortality 7% higher than that in the state as a whole was found
a The standardized mortality ratio is the ratio of observed
to expected deaths multiplied by 100.
(NIOSH, 1975). When looking at death records of the whole state from
1961-71, a significant increase in respiratory cancer mortality was
observed among decedents classified as orchardists. The data in the
NIOSH document cannot be evaluated in detail because of lack of
information concerning such factors as the methods and numbers of
people.
An increased respiratory cancer mortality has been observed in
many studies on smelter workers exposed to high levels of inorganic
arsenic, mainly in the form of arsenic(III) oxide. Lee & Fraumeni
(1969) compared the mortality rate among more than 8000 smelter
workers in the USA with that of the white male population of the
states in question. The mortality was followed from 1938-63 and a
total of 1877 deaths were recorded. About 10% of the original cohort
was lost in the follow-up. An excess mortality was found for cancer of
the respiratory system (lung and bronchus, larynx, and mediastinum)
with an overall standardized mortality ratio, SMR, equal to 329. A
positive relationship between SMR and estimated degree of exposure was
indicated, with an SMR of 800 in the highest arsenic exposure group. A
positive relationship was also found between exposure to sulfur
dioxide and respiratory cancer mortality with a SMR of over 700 in the
highest exposure groups. Unfortunately, data were not available
concerning the actual exposure levels and it was difficult to separate
the effects associated with arsenic from those associated with sulfur
dioxide, since both types of exposure occurred in most work areas.
Smoking histories were not obtained, and if large age distribution
differences were at hand between the different exposure groups the
comparison of SMRs would be unreliable.
The mortality among 2675 Japanese metal workers from 1949-1971 was
examined by Tokudome & Kuratsune (1976), using the cohort technique.
Workers were divided into 5 cohorts depending on their work histories,
i.e., copper smelting, ferronickel smelting, maintenance or transport,
copper or lead electrolysis or production of sulfuric acid, and
clerical work. Among the 839 copper smelters, an almost 12-fold
increase in the number of deaths was observed for cancer of the
trachea, bronchus, and lung in comparison with the expected number,
which was derived from the national rates for Japanese males during
the period at issue. A positive correlation was observed between
length of employment and excess of deaths from cancer of the trachea,
bronchi, and lung as well as between estimated levels of arsenic
exposure and excess mortality. The exposure was not described in
detail and smoking habits and age distributions in the various
exposure groups were not reported.
Pinto et al. (1977), in a follow-up of an earlier study in 1963,
analysed the cause-specific mortality experience above 65 years of age
among 527 retired workers at a large copper smelter in Washington,
USA. Expected deaths were computed from the mortality rates of the
male population in the state of Washington during the time period at
issue, i.e., 1949-73. Exposure occurred, mainly to arsenic(III) oxide,
and was assessed on the basis of the determination of arsenic
concentrations in the urine of workers in each department of the
factory in 1973. It was emphasized that the relevant exposure levels,
i.e., those during the active working life of the decedents, were
higher. Each worker was assigned an exposure index that was calculated
by multiplying the period of time spent in the various departments by
the 1973 urinary arsenic levels of workers in these departments.
A highly significant increase in mortality from cancer of the
respiratory system (ICD 160-164)a was found for the whole group of
former smelter workers under study (SMR = 304.8), which had a roughly
linear relationship with the estimated time-weighted average total
life-time exposure (Fig. 10). The comparison between the different
SMRs should be made with caution, as their magnitudes depend partly on
the age distribution of the index population for each SMR.
A group of workers with more than 25 years of exposure to airborne
arsenic at concentrations associated with urinary arsenic levels of
50-200 µg/litre, i.e., (according to Fig. 4) up to approximately
40 µg/m3 or a rough average of about 25 µg/m3, showed an SMR of
277.8. As previously stated, the exposure was probably underestimated
because of higher levels of airborne arsenic in earlier years. Urinary
arsenic data from Pinto et al. (1978) indicate that the exposures were
higher by perhaps a factor of 2 than those given in the previous
paragraph. This would mean that exposure to airborne arsenic at levels
of around 25 µg/m3, according to the original estimates, or perhaps
more appropriately 50 µg/m3 according to the revised value, would
lead to a nearly 3-fold (increase in) mortality from lung cancer. It
should be emphasized that the uncertainty in the estimated exposure
could well amount to a factor of 2.
Smoking habits were recorded on a sample of the cohort (Pinto et
al., 1978), i.e., directly from all men still living and from the
friends and relatives of men who died after 1961, a total of 377
subjects. Observed respiratory cancer deaths among smokers,
ex-smokers, and nonsmokers were compared with data on expected deaths
in these smoking categories. The SMR in the study population was
elevated in both the smokers and ex-smokers (245.1-506.5) indicating
that smoking habits in the study population as a whole were not
responsible for the increased mortality rate from cancer of the
respiratory system. In fact, the highest SMR was noted among the
nonsmokers, although it must be stressed that its magnitude is
unstable due to the fact that the number of observed respiratory
cancer deaths was small.
a WHO (1965) International Classification of Diseases
(8th revision).
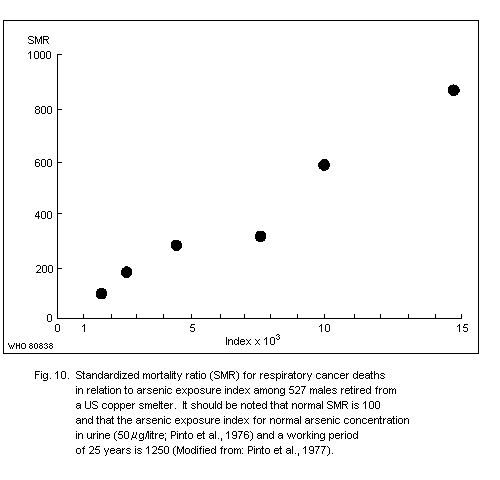 Rencher et al. (1977) examined the lung cancer mortality at a
smelter with high levels of airborne arsenic in the USA. A higher
proportional mortality was obtained for smelter workers than for mine
workers or for males in the state as a whole (7.0, 2.2 and 2.7%,
respectively). Estimated cumulative exposure indices for sulfur
dioxide, arsenic, and lead were higher among those dying from lung
cancer than among those dying of nonrespiratory causes. No indication
of a smoking synergism could be found.
An increased mortality from lung cancer has also been reported at
a Swedish copper smelter (Holmqvist, 1964; Pershagen et al., 1977;
Axelson et al., 1978). Employing the case control technique, Axelson
et al. examined the death records of 369 men aged 30-74, who had died
in the years 1960-1970 and who had been residing in the parish
surrounding the smelter at the time of death. A total of 44 subjects
were omitted because of vague diagnoses and diseases that might have
excluded them from employment at the smelter, e.g., mental deficiency
and diabetes mellitus.
"Cases" included men who had died from malignant tumours of the
lung, other malignancies, cardiovascular disease, cerebrovascular
disease, or cirrhosis of the liver. The "controls" were men who had
died from all remaining causes.
Assessment of exposure at the smelter was based on employment
registers that had been kept since the start of operations at the
smelter in 1928. These records included detailed information regarding
the time spent at various workplaces by all workers. The exposure
levels at these workplaces, from 1928 onwards, was estimated by an
experienced safety engineer. Using this information and a calculated
half median latency period for lung cancer of 17 years, the men were
divided into 4 different exposure categories. The 2 highest exposure
categories were chosen to include subjects exposed, before death, for
more than half of the latency period, to levels of airborne arsenic
exceeding 0.5 mg/m3. A rate ratio of about 5 for death due to lung
cancer was observed for workers exposed to arsenic compared with
unexposed workers and subjects never employed at the smelter. A
positive dose-response relationship was also indicated. Pershagen
(1978) showed that the excess mortality could not be explained by
smoking habits. It was not possible to establish detailed
dose-response relationships because of the rough measurements of
exposure and the small number of subjects in these studies.
In recent years, a number of reports have associated exposure to
arsenic in the ambient air near point emissions with an increased risk
of lung cancer. Blot & Fraumeni (1975) found an increased mortality
rate from lung cancer in the period 1950-69 for white males and
females (averaging 17% and 15%, respectively) in counties in the USA
with copper, lead, or zinc smelting and refining industries compared
with the rest of the country. The increase could not be explained by
differences in population density, urbanization, or socioeconomic
status. No data on the actual exposure levels of arsenic in ambient
air were given. Caution must be applied when associating arsenic
exposure with the excess of deaths, because of considerable variations
in emissions among the smelting and refining operations in the various
counties in question.
The vital statistics of 2 copper mining and smelting counties in
Montana, USA, were reviewed for the years 1969-71 by Newman et al.
(1976). Respiratory cancer death rates were significantly elevated in
both men and women in the mining as well as in the smelter town but
not in the counties as a whole, when compared with national rates
during the same period. Annual mortality rates for cancer of the
bronchus and lung among persons from the age of 21 years upwards were
20.4 and 3.2 per 10 000 among men and women, respectively, in the
smelting town and 13.8 and 6.0 per 10 000 in the mining town.
Unfortunately, the data obtained on smoking habits were not compared
with data on smoking habits for the nation as a whole. Furthermore,
the authors did not report which fraction of the population under
study was also employed at the mines or smelters. Atmospheric
concentrations of arsenic were reported to be markedly higher in the
smelter town, and a figure of 0.45 µg As/m3 was given (measurement
period not stated).
Matanoski et al. (1976), in analysing the mortality data from
1970-72 in white men who had lived in a heavily industrialized area
surrounding a plant producing pesticides containing arsenic, found an
excess of deaths from lung cancer which was between 3 and 4 times that
in men in adjacent areas. The populations in the areas were matched
for age, race, and sex. The observed excess in the number of deaths
remained statistically significant, when the pesticide plant workers
were excluded from the population. There were indications of a
decrease in lung cancer deaths with increasing distance from the
plant. No significant excess in lung cancer deaths was observed among
women in the area. No data were given on the levels of arsenic or
other contaminants in the ambient air and further information on
occupation and personal characteristics such as smoking habits is
needed to validate the findings.
A study has been reported by Pershagen et al. (1977) on the lung
cancer mortality between 1961 and 1975 in an area around a large
smelter in northern Sweden. During the period 1930-60, arsenic was
emitted into the air in amounts of 1-3 tonnes by day. Other pollutants
of interest were sulfur dioxide and, in lower amounts, lead, cadmium,
mercury, and nickel. No emission data were obtained. A total of 28
male cases of death from lung cancer were found, which constituted a 2
to 3-fold increase in the number of deaths (SMR = 250) compared with
control population with a similar degree of urbanization and a similar
occupational profile. The increase in the number of deaths was no
longer significant, when the occupational population exposed at the
smelter was excluded, though the tendency remained (SMR = 173). No
corresponding increases in death from lung cancer could be detected
among women.
Some reports have claimed an association between exposure to
inorganic arsenic as medication and lung cancer (Sommers & McManus,
1953; Robson & Jelliffe, 1963; Goldman, 1973). This association was
based on the occurrence of lung cancer in patients receiving trivalent
inorganic arsenic in daily doses of several milligrams, for decades.
Unfortunately, these are only case histories, and epidemiological
studies in this field are greatly needed.
The respiratory tract rumours associated with arsenic exposure
have been classified according to histological type in some studies
(Robson & Jelliffe, 1963; Newman et al., 1976; Axelson et al., 1978).
A clustering of poorly differentiated types is evident in all these
reports. Poorly differentiated or undifferentiated types were noted in
5 out of 6 patients with lung cancer, who had received medication with
inorganic arsenic (Robson & Jelliffe, 1963). Half of the patients were
nonsmokers. In the previously described report on lung cancer
mortality from 1954-72 in 2 mining and smelting counties in Montana,
USA (Newman et al., 1976), the histological types of 143 respiratory
tumours in men and women were presented. Microscopic slides were
examined independently by a panel of 4 experienced pathologists on a
"blind" basis, i.e., they did not know the residence or occupation of
the individual from whom the specimens were obtained. Information on
smoking habits, place of residence, and occupation was obtained from a
tumour registry, hospital records, or relatives. Smelter workers
showed a high number of poorly differentiated epidermoid carcinomas
(40.0%) that did not appear in mine workers or other men in the area.
The difference could not be explained by smoking habits, which were
very similar in all 3 groups, a high percentage (36.0%) of poorly
differentiated carcinomas also occurred among women from the mining
town in association with elevated lung cancer rates.
Out of the 24 persons with lung cancer at the Swedish copper
smelter investigated by Axelson et al. (1978) (reported earlier in
this section), 18 were classified as having been exposed to arsenic.
In this group, 22.2% of the rumours were classified as poorly
differentiated epidermoid carcinomas, whereas this type of tumour was
not found among lung cancer-stricken employees not exposed to arsenic.
There also appeared to be an increase in other types of epidermoid
carcinoma as well as of the small cell undifferentiated type; however,
the numbers in all groups were too small to draw any definite
conclusions.
8.4.1.2 Cancer of the skin
Several types of neoplastic changes of the skin, including Bowen's
disease and basal and squamous cell carcinomas, have been associated
with arsenic exposure. Neither of these lesions, when attributable to
arsenic, possesses any unique histological features (Hundeiker &
Petres, 1968; Sanderson, 1976; Deng & How, 1977). While the effects of
other skin carcinogens such as UV-radiation and polyaromatic
hydrocarbons are limited to areas of exposure, arsenic lesions, can
occur on every part of the body. Bowen's disease and basal cell
carcinomas of arsenical origin are usually multiple and located on the
trunk (Fierz, 1965; Yeh et al., 1968). Squamous cell carcinomas
develop primarily from the keratoses on the extremities.
Skin cancer has been associated with inorganic arsenic exposure in
reports from many parts of the world, i.e., Argentina, Canada, China
(Province of Taiwan), Czechoslovakia, France, Federal Republic of
Germany, Israel, Japan, South Africa, Switzerland, the United Kingdom,
and the USA (Hutchinson, 1888; Arguello et al., 1938; Neubauer, 1947;
Hill & Faning, 1948; Sommers & McManus, 1953; Berlin & Tager, 1962;
Fierz, 1965; Schulz, 1967; Thiers et al., 1967; Tseng et al., 1968;
Bartak & Kejda, 1972; Wolf, 1974; Jackson & Grainge, 1975; Hamada &
Horiguchi, 1976). The total number of cases reported is well over 1000
with exposure occurring most frequently via the oral route, either
through contaminated drinking-water or medication. Ingestion has
usually taken place over several decades, with daily doses of several
mg of arsenic. As a rule, the skin tumours appear earlier in life than
is ordinarily encountered. In the medication, the arsenic was mainly
in the form of arsenite, while the oxidation state in
arsenic-contaminated water is unknown.
In the following discussion, it is assumed that the response is
related to the total ingested close of arsenic. In the study by Fierz
(1965), mentioned in section 8.3.2, 8% of 262 patients treated with
arsenic compounds mainly in the form of arsenite, for up to 26 years,
showed various types of skin cancer, the most common being multiple
basal cell carcinoma. The prevalence of skin cancer increased as the
total ingested dose of Fowler's solution increased. In patients who
had ingested between 200 and 800 ml of Fowler's solution (= 1.5 and
6.0 g of arsenic), the prevalence rate ranged from 5% to 10%. No firm
conclusions can be drawn regarding dose-response relationships in view
of the possible selection bias in this study. A control group, which
would have enabled an estimation of the "background" morbidity in skin
cancer to be made, was also lacking. Obviously, however, an increased
morbidity did occur among patients who received a total of more than
1000 ml of Fowler's solution (= 7.6 g of arsenic). The prevalence rate
in this group was over 20%.
In the survey on more than 40 000 inhabitants in China, Province
of Taiwan, reported in section 8.3.4, Tseng (1977) established a
positive dose-response relationship between the contents of arsenic in
well water and the prevalence rate for skin cancer. The overall
prevalence was 10.6 per 1000, and the male to female ratio, 2.9. The
prevalence of skin cancer in relation to increasing doses of arsenic
is depicted in Fig. 11. Assuming a daily intake of 2 litres of water a
total ingested dose of about 20 g of arsenic over a life-time
corresponds to a prevalence of roughly 6%. This is an average between
the 2 lowest exposure groups for age 60 and over. The oldest age was
selected in order to ensure that the maximum tumour response was
achieved for a given dose. Because the tumours were of low malignancy,
they would be expected to persist for a long period and hence
prevalence would be a reasonable approximation of life-time cumulative
incidence. The increase in prevalence with increasing arsenic dose is
partly due to a higher background prevalence of skin cancer in older
age groups. Unfortunately, no control group was included, but if the
lowest exposure group using well water with an arsenic content below
0.3 mg/litre, is taken as a control group, a roughly linear increase
in excess morbidity expressed as prevalence rate of between 0.9 and
164.9 per 1000 is still seen in various age groups (Fig. 12). As was
concluded in section 8.3.4.1 with regard to "blackfoot disease", the
increase in age could not explain the observed increase in skin cancer
with estimated dose of arsenic.
The extrapolation to low doses is very uncertain, as indicated by
the discrepancies between prevalence rates in the reports of Fierz
(1965) and Tseng (1977). While the patients who had been on the
arsenic-containing medication showed a prevalence of 20% (after an
estimated total intake of arsenic of 7.6 g), the corresponding
prevalence among the Chinese in Taiwan was less than 3%. None of the
reports, however, included a control group, and at least the first one
could have had a substantial selection bias. The medicated patients
probably had a higher dose rate than the Taiwanese.
Rencher et al. (1977) examined the lung cancer mortality at a
smelter with high levels of airborne arsenic in the USA. A higher
proportional mortality was obtained for smelter workers than for mine
workers or for males in the state as a whole (7.0, 2.2 and 2.7%,
respectively). Estimated cumulative exposure indices for sulfur
dioxide, arsenic, and lead were higher among those dying from lung
cancer than among those dying of nonrespiratory causes. No indication
of a smoking synergism could be found.
An increased mortality from lung cancer has also been reported at
a Swedish copper smelter (Holmqvist, 1964; Pershagen et al., 1977;
Axelson et al., 1978). Employing the case control technique, Axelson
et al. examined the death records of 369 men aged 30-74, who had died
in the years 1960-1970 and who had been residing in the parish
surrounding the smelter at the time of death. A total of 44 subjects
were omitted because of vague diagnoses and diseases that might have
excluded them from employment at the smelter, e.g., mental deficiency
and diabetes mellitus.
"Cases" included men who had died from malignant tumours of the
lung, other malignancies, cardiovascular disease, cerebrovascular
disease, or cirrhosis of the liver. The "controls" were men who had
died from all remaining causes.
Assessment of exposure at the smelter was based on employment
registers that had been kept since the start of operations at the
smelter in 1928. These records included detailed information regarding
the time spent at various workplaces by all workers. The exposure
levels at these workplaces, from 1928 onwards, was estimated by an
experienced safety engineer. Using this information and a calculated
half median latency period for lung cancer of 17 years, the men were
divided into 4 different exposure categories. The 2 highest exposure
categories were chosen to include subjects exposed, before death, for
more than half of the latency period, to levels of airborne arsenic
exceeding 0.5 mg/m3. A rate ratio of about 5 for death due to lung
cancer was observed for workers exposed to arsenic compared with
unexposed workers and subjects never employed at the smelter. A
positive dose-response relationship was also indicated. Pershagen
(1978) showed that the excess mortality could not be explained by
smoking habits. It was not possible to establish detailed
dose-response relationships because of the rough measurements of
exposure and the small number of subjects in these studies.
In recent years, a number of reports have associated exposure to
arsenic in the ambient air near point emissions with an increased risk
of lung cancer. Blot & Fraumeni (1975) found an increased mortality
rate from lung cancer in the period 1950-69 for white males and
females (averaging 17% and 15%, respectively) in counties in the USA
with copper, lead, or zinc smelting and refining industries compared
with the rest of the country. The increase could not be explained by
differences in population density, urbanization, or socioeconomic
status. No data on the actual exposure levels of arsenic in ambient
air were given. Caution must be applied when associating arsenic
exposure with the excess of deaths, because of considerable variations
in emissions among the smelting and refining operations in the various
counties in question.
The vital statistics of 2 copper mining and smelting counties in
Montana, USA, were reviewed for the years 1969-71 by Newman et al.
(1976). Respiratory cancer death rates were significantly elevated in
both men and women in the mining as well as in the smelter town but
not in the counties as a whole, when compared with national rates
during the same period. Annual mortality rates for cancer of the
bronchus and lung among persons from the age of 21 years upwards were
20.4 and 3.2 per 10 000 among men and women, respectively, in the
smelting town and 13.8 and 6.0 per 10 000 in the mining town.
Unfortunately, the data obtained on smoking habits were not compared
with data on smoking habits for the nation as a whole. Furthermore,
the authors did not report which fraction of the population under
study was also employed at the mines or smelters. Atmospheric
concentrations of arsenic were reported to be markedly higher in the
smelter town, and a figure of 0.45 µg As/m3 was given (measurement
period not stated).
Matanoski et al. (1976), in analysing the mortality data from
1970-72 in white men who had lived in a heavily industrialized area
surrounding a plant producing pesticides containing arsenic, found an
excess of deaths from lung cancer which was between 3 and 4 times that
in men in adjacent areas. The populations in the areas were matched
for age, race, and sex. The observed excess in the number of deaths
remained statistically significant, when the pesticide plant workers
were excluded from the population. There were indications of a
decrease in lung cancer deaths with increasing distance from the
plant. No significant excess in lung cancer deaths was observed among
women in the area. No data were given on the levels of arsenic or
other contaminants in the ambient air and further information on
occupation and personal characteristics such as smoking habits is
needed to validate the findings.
A study has been reported by Pershagen et al. (1977) on the lung
cancer mortality between 1961 and 1975 in an area around a large
smelter in northern Sweden. During the period 1930-60, arsenic was
emitted into the air in amounts of 1-3 tonnes by day. Other pollutants
of interest were sulfur dioxide and, in lower amounts, lead, cadmium,
mercury, and nickel. No emission data were obtained. A total of 28
male cases of death from lung cancer were found, which constituted a 2
to 3-fold increase in the number of deaths (SMR = 250) compared with
control population with a similar degree of urbanization and a similar
occupational profile. The increase in the number of deaths was no
longer significant, when the occupational population exposed at the
smelter was excluded, though the tendency remained (SMR = 173). No
corresponding increases in death from lung cancer could be detected
among women.
Some reports have claimed an association between exposure to
inorganic arsenic as medication and lung cancer (Sommers & McManus,
1953; Robson & Jelliffe, 1963; Goldman, 1973). This association was
based on the occurrence of lung cancer in patients receiving trivalent
inorganic arsenic in daily doses of several milligrams, for decades.
Unfortunately, these are only case histories, and epidemiological
studies in this field are greatly needed.
The respiratory tract rumours associated with arsenic exposure
have been classified according to histological type in some studies
(Robson & Jelliffe, 1963; Newman et al., 1976; Axelson et al., 1978).
A clustering of poorly differentiated types is evident in all these
reports. Poorly differentiated or undifferentiated types were noted in
5 out of 6 patients with lung cancer, who had received medication with
inorganic arsenic (Robson & Jelliffe, 1963). Half of the patients were
nonsmokers. In the previously described report on lung cancer
mortality from 1954-72 in 2 mining and smelting counties in Montana,
USA (Newman et al., 1976), the histological types of 143 respiratory
tumours in men and women were presented. Microscopic slides were
examined independently by a panel of 4 experienced pathologists on a
"blind" basis, i.e., they did not know the residence or occupation of
the individual from whom the specimens were obtained. Information on
smoking habits, place of residence, and occupation was obtained from a
tumour registry, hospital records, or relatives. Smelter workers
showed a high number of poorly differentiated epidermoid carcinomas
(40.0%) that did not appear in mine workers or other men in the area.
The difference could not be explained by smoking habits, which were
very similar in all 3 groups, a high percentage (36.0%) of poorly
differentiated carcinomas also occurred among women from the mining
town in association with elevated lung cancer rates.
Out of the 24 persons with lung cancer at the Swedish copper
smelter investigated by Axelson et al. (1978) (reported earlier in
this section), 18 were classified as having been exposed to arsenic.
In this group, 22.2% of the rumours were classified as poorly
differentiated epidermoid carcinomas, whereas this type of tumour was
not found among lung cancer-stricken employees not exposed to arsenic.
There also appeared to be an increase in other types of epidermoid
carcinoma as well as of the small cell undifferentiated type; however,
the numbers in all groups were too small to draw any definite
conclusions.
8.4.1.2 Cancer of the skin
Several types of neoplastic changes of the skin, including Bowen's
disease and basal and squamous cell carcinomas, have been associated
with arsenic exposure. Neither of these lesions, when attributable to
arsenic, possesses any unique histological features (Hundeiker &
Petres, 1968; Sanderson, 1976; Deng & How, 1977). While the effects of
other skin carcinogens such as UV-radiation and polyaromatic
hydrocarbons are limited to areas of exposure, arsenic lesions, can
occur on every part of the body. Bowen's disease and basal cell
carcinomas of arsenical origin are usually multiple and located on the
trunk (Fierz, 1965; Yeh et al., 1968). Squamous cell carcinomas
develop primarily from the keratoses on the extremities.
Skin cancer has been associated with inorganic arsenic exposure in
reports from many parts of the world, i.e., Argentina, Canada, China
(Province of Taiwan), Czechoslovakia, France, Federal Republic of
Germany, Israel, Japan, South Africa, Switzerland, the United Kingdom,
and the USA (Hutchinson, 1888; Arguello et al., 1938; Neubauer, 1947;
Hill & Faning, 1948; Sommers & McManus, 1953; Berlin & Tager, 1962;
Fierz, 1965; Schulz, 1967; Thiers et al., 1967; Tseng et al., 1968;
Bartak & Kejda, 1972; Wolf, 1974; Jackson & Grainge, 1975; Hamada &
Horiguchi, 1976). The total number of cases reported is well over 1000
with exposure occurring most frequently via the oral route, either
through contaminated drinking-water or medication. Ingestion has
usually taken place over several decades, with daily doses of several
mg of arsenic. As a rule, the skin tumours appear earlier in life than
is ordinarily encountered. In the medication, the arsenic was mainly
in the form of arsenite, while the oxidation state in
arsenic-contaminated water is unknown.
In the following discussion, it is assumed that the response is
related to the total ingested close of arsenic. In the study by Fierz
(1965), mentioned in section 8.3.2, 8% of 262 patients treated with
arsenic compounds mainly in the form of arsenite, for up to 26 years,
showed various types of skin cancer, the most common being multiple
basal cell carcinoma. The prevalence of skin cancer increased as the
total ingested dose of Fowler's solution increased. In patients who
had ingested between 200 and 800 ml of Fowler's solution (= 1.5 and
6.0 g of arsenic), the prevalence rate ranged from 5% to 10%. No firm
conclusions can be drawn regarding dose-response relationships in view
of the possible selection bias in this study. A control group, which
would have enabled an estimation of the "background" morbidity in skin
cancer to be made, was also lacking. Obviously, however, an increased
morbidity did occur among patients who received a total of more than
1000 ml of Fowler's solution (= 7.6 g of arsenic). The prevalence rate
in this group was over 20%.
In the survey on more than 40 000 inhabitants in China, Province
of Taiwan, reported in section 8.3.4, Tseng (1977) established a
positive dose-response relationship between the contents of arsenic in
well water and the prevalence rate for skin cancer. The overall
prevalence was 10.6 per 1000, and the male to female ratio, 2.9. The
prevalence of skin cancer in relation to increasing doses of arsenic
is depicted in Fig. 11. Assuming a daily intake of 2 litres of water a
total ingested dose of about 20 g of arsenic over a life-time
corresponds to a prevalence of roughly 6%. This is an average between
the 2 lowest exposure groups for age 60 and over. The oldest age was
selected in order to ensure that the maximum tumour response was
achieved for a given dose. Because the tumours were of low malignancy,
they would be expected to persist for a long period and hence
prevalence would be a reasonable approximation of life-time cumulative
incidence. The increase in prevalence with increasing arsenic dose is
partly due to a higher background prevalence of skin cancer in older
age groups. Unfortunately, no control group was included, but if the
lowest exposure group using well water with an arsenic content below
0.3 mg/litre, is taken as a control group, a roughly linear increase
in excess morbidity expressed as prevalence rate of between 0.9 and
164.9 per 1000 is still seen in various age groups (Fig. 12). As was
concluded in section 8.3.4.1 with regard to "blackfoot disease", the
increase in age could not explain the observed increase in skin cancer
with estimated dose of arsenic.
The extrapolation to low doses is very uncertain, as indicated by
the discrepancies between prevalence rates in the reports of Fierz
(1965) and Tseng (1977). While the patients who had been on the
arsenic-containing medication showed a prevalence of 20% (after an
estimated total intake of arsenic of 7.6 g), the corresponding
prevalence among the Chinese in Taiwan was less than 3%. None of the
reports, however, included a control group, and at least the first one
could have had a substantial selection bias. The medicated patients
probably had a higher dose rate than the Taiwanese.
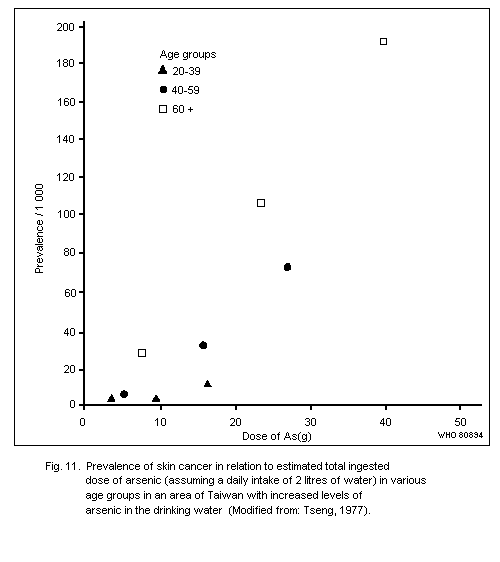
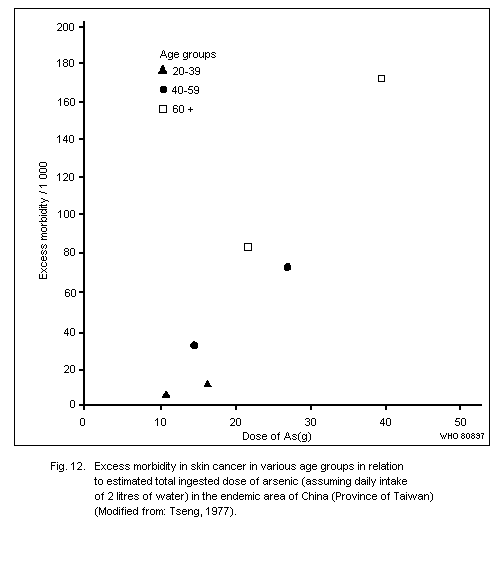 Morton et al. (1976) examined the incidence of skin cancer in Lane
County, Oregon, where high levels of arsenic had been found in water
supplies. No relationship was detected between arsenic levels in water
and the appearance of squamous or basal cell carcinomas. However,
these data do not necessarily contradict previous findings as drinking
water sources with high arsenic concentrations were fewer than
expected. The mean levels in the different regions of the community
ranged between 0.004 and 0.033 mg As/litre.
8.4.1.3 Cancer of the liver
Haemangioendothelioma of the liver has been associated with
exposure to inorganic arsenic in a number of cases. The exposure has
come from contaminated wine, drinking-water, or Fowler's solution.
Roth (1957) reported 5 cases of haemangioendothelioma of the liver
among former vintners, who died between 47 and 58 years of age. The
workers had been handling arsenic-containing insecticides (type not
specified) for a number of years and had typical cutaneous
manifestations of arsenic intoxication, i.e., palmo-plantar
hyperkeratosis and melanosis. All cases also suffered from liver
cirrhosis, which the author ascribed to arsenic ingestion (section
8.3.3). The latent period, from the beginning of arsenic exposure
ranged from 20 to 28 years. No data were given on the actual doses,
but it can be estimated that the total ingested dose of arsenic was
10-20 grams (Grobe, 1976).
Rennke et al. (1971) described a case of malignant
haemangioendothelioma in a 22-year old male from the Chilean province
of Antofagasta. Cutaneous signs of arsenic exposure were also present.
The decedent had been exposed to arsenic in drinking water for 12
years at an average level of 0.8 mg/litre, i.e., a total of
5.46 grams. An arsenic level of 20 mg/kg was found in the hair.
Some cases of haemangioendothelioma have also been reported
following prolonged ingestion of Fowler's solution. Liver
haemangioendothelioma was found in a patient who had been taking
Fowler's solution over 20 years for psoriasis (Rosset, 1958). Regelson
et al. (1968) described a psoriatic patient who had taken Fowler's
solution for 17 years, so that the total ingested dose amounted to
approximately 10 grams of inorganic trivalent arsenic. This patient
also displayed palmo-plantar hyperkeratosis. The tumour was diagnosed
24 years after initiation of treatment with the drug. A 43-year-old
psoriatic patient, who developed haemangioendothelioma 21 years after
the initiation of a 15-year-long treatment with Fowler's solution has
been reported by Lander et al. (1975). The total ingested dose was
estimated by the authors to have been 15 g (not stated whether this
was calculated as As or As(III) oxide). Popper et al. (1978) compiled
5 cases of haemangioendothelioma in the US each with a history of
ingestion of Fowler's solution. The duration of exposure ranged from
10 to 17 years. It is not clear, whether the cases reported by
Regelson et al. (1968) and Lander et al. (1975) were included in this
series.
Although only case reports of this extremely rare tumour exist, it
is noteworthy that the reported latency times seem to be similar,
i.e., 20-25 years. If trivalent arsenic plays a role in the
development of haemangioendothelioma, it can be concluded that large
doses, at least several grams, are required and that the effect is
rare in view of the large number of people who have been exposed
through drinking water or medication.
8.4.1.4 Leukaemia and tumours of the haematopoietic system
A carcinogenic effect on the haematopoietic system in situations
of occupational exposure to inorganic arsenic compounds cannot be
ruled out. In 2 studies on the mortality of workers exposed to high
levels of airborne arsenic, an increased mortality due to malignant
neoplasms of the lymphatic and haematopoietic tissues was indicated
compared with unexposed control groups (Ott et al., 1974; Axelson
et al., 1978). It should be pointed out that the numbers of deaths due
to these causes were small in both studies.
The data need further confirmation, especially in view of the lack
of supporting evidence from other instances of exposure to inorganic
arsenic, i.e., through drinking water and medication.
8.4.1.5 Cancer of other organs
Exposure to inorganic arsenic through medication, mostly as
Fowler's solution, has been associated with the development of various
malignant neoplasms. Sommers & McManus (1953) compiled a total of 27
cases of multiple primary tumours of the skin and internal organs. In
24 of the cases, there was a history of excessive arsenic exposure and
all but one of the patients had palmo-plantar hyperkeratosis. Nineteen
of the patients had been exposed through ingestion of Fowler's
solution, mostly for treatment of psoriasis. Unfortunately, exposure
doses were not given. Multiple skin cancers, predominantly basal cell
carcinomas, were observed in 17 patients (section 7.3.2). In another
10 cases, skin cancers were combined with primary tumours of other
organs, e.g., lung, oesophagus, and bladder. The average latency
between commencement of arsenic exposure and diagnosis of a tumour was
24 years. The data presented in this study do not lend themselves to
statistical analysis. The 10 cases with simultaneous skin cancer and
internal malignancy could have involved a selection artefact.
In a study by Reymann et al. (1978), a total of 389 patients were
followed, who had been on arsenic therapy, primarily with Fowler's
solution. Fifty-three of the patients had typical arsenical keratosis.
A total of 41 cases had developed cancer of internal organs during the
period 1943-74, as traced from the Danish Cancer Registry. It was
found that this was not statistically different, in terms of the life
table method, from the national average. Neither the estimated total
dose of arsenic ingested, nor the nature of the agent was correlated
with the incidence of internal cancer. In the group of patients with
arsenical keratosis, 9 died from cancer of internal organs (type not
specified) during the period under study, as opposed to 5.2 expected
(increase not statistically significant).
The data relating to arsenic exposure and malignancies other than
lung and skin cancer are inadequate. Further studies are needed before
any conclusions can be drawn.
8.4.2 Experimental animal studies
8.4.2.1 Cancer of the respiratory system
Only a few studies have been performed on the carcinogenic effects
of inorganic arsenic following exposure of the respiratory tract in
experimental animals. Ishinishi, et al. (1977) administered copper
ore, metal refinery flue dust, or arsenic(III) oxide, alone or
together with benzo (a)pyrene in a saline suspension, intratracheally
to male rats. Another group received benzo (a)pyrene alone. The doses
of arsenic in the various groups were 1.5, 3.0, and 3.0 mg per animal,
divided into 15 weekly instillations. A total of 6 mg of
benzo (a)pyrene was given. Altogether, 161 animals were treated,
including a control group of 23 animals that received saline only. The
mortality in the various groups during the exposure period ranged from
30% to 50%, the lower value being recorded in the control group. The
results indicated a positive interaction between benzo (a)pyrene and
arsenic(III) oxide in the induction of lung tumours, but the number of
animals in this study was too small to permit any firm conclusions.
Ishinishi, et al. (1980a) gave 30 male adult Wistar strain rats
intratracheal instillations of arsenic(III) oxide in a suspension once
a week for 15 weeks. Sixteen untreated rats acted as controls.
Observation of these rats over their life span revealed only one
malignant lung tumour (squamous cell carcinoma) in the 19 rats that
survived 15 instillations. No lung tumours were found in the control
group. This report does not provide definite evidence of arsenic
respiratory carcinogenicity in animals.
In a study by Ivankovic et al. (1979), 25 rats were given a single
intratracheal instillation of 0.1 ml of a mixture of calcium arsenate,
copper sulfate, and calcium oxide. The preparation was similar to that
of a pesticide used on vines between the 1920s and 1940s. The dose of
calcium arsenate administered was reported to correspond to 0.07 mg of
arsenic. During the first week after treatment, 10 of the animals died
due to pneumonia or lung necrosis. Of the 15 surviving animals, 9
developed lung tumours (7 bronchogenic adenocarcinomas and 2
bronchiolar-alveolar cell carcinomas). The average induction time was
470 days. No lung tumours were observed in 25 control animals,
instilled with 0.1 ml of 0.9% saline, and observed throughout their
life span (mean survival time: 670 days). The data in this report
indicate that the mixture given was carcinogenic, but no firm
conclusion can be reached as to the causative agent, because of the
design of the experiment. It seems, however, that arsenic might have
played an important role and further studies should clarify whether
calcium arsenate alone is sufficient to evoke a carcinogenic response
and also the possible modification of effects by the other components
in the mixture.
8.4.2.2 Skin application
Inorganic arsenic has been tested repeatedly in skin applications
and not been found to be carcinogenic. Leitch & Kennaway (1922)
produced one metastasizing squamous cell carcinoma of the skin in 100
mice painted 3 times weekly with a solution of sodium arsenite in
alcohol. The first skin applications contained 1.8% of arsenic(III)
oxide (about 13.7 g As/litre), but the concentration was reduced to
0.12% (about 0.9 g As/litre) because of the high mortality induced by
the higher dose. In 3 months, two thirds of the animals had died, and,
by that time, a tumour appeared in one of the survivors at the site of
application of the arsenic. At autopsy after 5 1/2 months, a lung
metastasis was observed. No control group was included in the study.
The cocarcinogenicity of sodium arsenite was tested in a series of
experiments by Boutwell (1963). Each of 20 female mice was given a
total of 1.24 mg potassium arsenite in 80% ethanol, in 8 skin
applications over a 5-day period. From 2 days after completion of the
arsenic treatment, the mice were treated twice weekly with croton oil.
After 18 weeks, it was observed that previous exposure to sodium
arsenite did not make the treated mice more responsive to croton oil
than the controls which had been treated with croton oil only. A
single skin application of 75 µg dimethylbenzanthracene and
subsequent, twice-daily applications potassium arsenite (2.2 mg
KAsO2/week) for the duration of the experiment (30 weeks) did not
result in tumour formation. Thus neither tumour initiating nor tumour
promoting activity could be shown for sodium arsenite.
In 1956, Salaman & Roe painted 14 mice with a total of 30 mg of
potassium arsenite (8.7 mg As), dissolved in methanol, once a week for
10 weeks. Croton oil was also applied weekly, starting 25 days later.
Three papillomas were observed in mice treated with potassium
arsenite, while 4 papillomas appeared in 19 animals receiving croton
oil only.
The skin of 10 rabbits was painted with 10% arsenic(III) oxide in
vaseline (about 76 g As/kg). After daily applications for 70 days, 1
epithelioma and 4 papillomas had appeared. A control group was not
used (Raposa, 1928). Friedewald & Rous (1944) repeated this study and
failed to produce any malignant tumours. They did observe verrucous
lesions in 3 out of 7 animals surviving a 22-month treatment. During
the first 6 months, 12 rabbits were painted with 10% arsenic(III)
oxide in vaseline on one ear and vaseline only on the other ear. For
the subsequent 13.5 months, the rabbits were painted with a 2%
solution of arsenic(III) oxide (15 g As/litre) in water mixed with
acetone in the proportion of 1:2. Three out of 6 lesions were excised
and proved to be papillomas. One of these appeared on a control ear.
Sodium arsenate at a concentration of 15.8 g/litre (about 3.8 g of
As) in a 2.5% solution of Tween 60 in water was applied twice weekly
to the skin of 54 male and 14 female mice. A total of 3 skin
papillomas appeared in the exposed animals, and 4 in 69 control
animals. Sodium arsenate applied to the skin in combination with
croton oil treatments twice weekly, failed to show
papilloma-initiating action. An absence of tumour promoting activity
was noted when exposure to arsenic followed a single skin application
of 200 µg dimethylbenzanthracene or a stomach tube dose of 60 mg
urethane (Baroni et al., 1963).
8.4.2.3 Oral administration
Fairhall & Miller (1941) could not find any evidence of
carcinogenicity of lead arsenate or sodium arsenate in 148 rats, fed
daily doses of approximately 2 mg of arsenic, over a period of 2
years. Groups of 50 mice or rats were given arsenic(III) oxide
dissolved in 12% ethyl alcohol or water at arsenic concentrations of
3.0 mg/litre at the beginning of an experiment by Hueper & Payne
(1962). The level was successively raised over a span of 15 months and
from then on kept at 25.5 mg As/litre. In the rats, these levels were
reported to result in daily intakes of between 0.08 and 0.6 mg per
animal. No differences in tumour incidence were found between the
arsenic-exposed animals and the controls.
Boutwell (1963) gave each of 20 female mice a total of 2.4 mg
potassium arsenite (corresponding to 0.7 mg As) by stomach tube. The
dose was administered in 7 portions over a 5 day period. Other groups
were given potassium arsenite mixed with the feed combined with
tumour-initiation treatments with 5 µg dimethylbenzanthracene and
tumour-promotion treatments with croton oil, both applied to the skin.
There were no differences in the production of papillomas and
carcinomas between the arsenic-fed animals and controls.
Arsenic (III) oxide was given in the drinking water in a
concentration of 76 mg As/litre to 15 female and 62 male mice. No skin
tumours were observed in the animals, 21 of which survived for 60
weeks. The rumour incidence in other groups, treated in addition with
croton oil, dimethylbenzanthracene, and urethane, respectively, as
described in section 8.4.2.2, did not differ from that in control
animals given these substances separately (Baroni et al., 1963).
In studies by Byron et al. (1967), groups of 50 rats each were fed
for up to 2 years with sodium arsenite at arsenic levels in the diet
of 0, 15.6, 31.3, 62.5, 125, and 250 mg/kg and sodium arsenate at
arsenic levels of 0, 31.3, 62.5, 125, 250 and 400 mg/kg. The arsenic
was mixed in the standard laboratory diet. After the 2 years, both the
incidence and type of tumours were similar in exposed animals (4-15
survivors/group) and controls (8-12 survivors/group). In the same
studies, groups of 6 dogs (3 male and 3 female) were fed sodium
arsenite or sodium arsenate at arsenic levels in the diet of 5, 25,
50, and 125 mg/kg. In the group receiving the highest dose of sodium
arsenite, no animal survived 2 years, while only one of the 6 dogs
given the highest level of sodium arsenate died during this time. All
of the other animals were killed at the end of 2 years. No tumours
were observed in any of the animals.
Kanisawa & Schroeder (1969) reported a study in which sodium
arsenite was administered in the drinking-water to 103 mice at a
concentration of 5 mg As/litre, throughout their lifetime. A total of
170 mice received double-deionized water. In 67 animals surviving 15
weeks in the exposed group, 11.9% subsequently died with tumours
compared with 34.5% in the control group. The death rates during this
period were similar in both groups.
Mice of 3 different strains were exposed by Milner (1969) to
arsenic(III) oxide in the drinking water at a concentration of
100 mg/litre (corresponding to 76 mg As/litre) over periods of 4 to 11
weeks. Cutaneous tumours were initiated by the topical application of
1,2-dihydro-3-methylbenz[j]accanthrylene (methylcholanthrene) and
promoted by transplantation. The results were contradictory, as
arsenic treatment seemed to increase the number of papillomas in one
strain, i.e., 5 papillomas in 16 arsenic-treated animals compared with
one papilloma in 10 controls, while it seemed to decrease the
incidence in another strain (9/28 in exposed versus 17/29 in
unexposed).
Schrauzer & Ishmael (1974) exposed 30 mice of a strain with a high
incidence of spontaneous mammary tumours to sodium arsenite in the
drinking-water at a concentration of 10 mg/litre (3.6 mg As/litre).
Tumours appeared between 6 and 9 months of age only and were seen in
27% of the animals. In control animals, tumours did not occur before
the eleventh month, but affected 82% of the animals after the
sixteenth month. The growth rate of the spontaneous tumours, as well
as of transplanted mammary tumours, was significantly enhanced in
arsenic-fed animals. When a similar experiment was performed on 30
mice exposed to 2 mg As/litre as arsenic(III) oxide in drinking-water,
mammary tumours started to appear at the age of 9 months, while in
control animals, the age of onset was 4.5 months (Schrauzer et al.,
1978). The tumour incidence was lower in the arsenic-exposed group,
i.e. 36% compared with 41% in the controls (not statistically
significant). As in the previous study, the growth rate of the tumours
was higher in the arsenic-treated animals. When the administration of
arsenite was accompanied by administration of 2 mg selenite per litre
of drinking water, a higher incidence of spontaneous mammary tumours
occurred (62%) than when selenite was administered alone (17%). The
tumour growth rate was similar in the arsenic/selenite and arsenic
groups and was significantly higher than in the selenite only and
control groups.
Lead arsenate was administered at levels of 463 and 1850 mg/kg
(100 and 400 mg As/kg diet)and sodium arsenate at 416 mg/kg (100 mg
As/kg diet), in both cases mixed with the feed, to rats in groups of
48-110 animals (Kroes et al., 1974). For 5 days per week, exposure to
arsenic was combined with oesophageal intubations of
diethylnitrosamine at a dose of 5 µg/day. Mortality was comparable in
all groups except the group given 1850 mg lead arsenate/kg, which
showed a marked increase after 26 weeks. After 120 weeks, all
survivors were killed and the animals were examined for tumours. In
the control groups, the incidence of malignant tumours ranged from 10%
to 17% and in the exposed groups from 0% to 13%. Similarly, there were
no differences in the development of benign tumours between the
groups.
8.4.2.4 Other experimental systems
Hueper (1954) injected metallic arsenic in lanolin into the femur
marrow of 25 male rats and 6 rabbits. The doses were 0.43 mg and
0.65 mg, respectively. Only 4 rats survived 18 months and one of these
developed a spindle cell sarcoma at the site of injection. None of the
rabbits showed any metaplastic reactions. No tumours were produced at
the site of injection in 25 rats injected intrapleurally once a month
for 6 months resulting in a total dose of 0.65 mg of arsenic. Similar
results were obtained after nasal sinus injection of 0.65 mg of
arsenic in 20 rats.
In studies by Osswald & Goerttler (1971), 24 pregnant mice were
injected daily with 0.5 mg of arsenic per kg body weight in the form
of sodium arsenate for 20 days. Half of the 22 animals that died
during a subsequent 24-month period of observation had developed
lymphocytic leukaemias or lymphomas. Two animals were still alive at
this time. None of 16 dead female controls had developed such lesions.
Four controls were still alive. Some of the offspring of the
arsenic-treated mothers (41 males and 56 females) received 20 weekly
subcutaneous injections of sodium arsenate in doses of 0.5 mg As/kg
body weight. Leukaemias and lymphomas had developed in 50% of the
males and 42.0% of the females that had died at the time of reporting.
Seven males were still alive at this time. In 71 untreated progeny,
leukaemias and lymphomas were present in 13.3% of males and 20.7% of
females that had died at the time of reporting. Four out of 34 males
and 8 out of 37 females were alive at this time. Lymphomas appeared in
11 out of 20 mice injected intravenously with 0.5 mg of arsenic in the
form of sodium arsenate once a week, for 20 weeks.
8.5 Mutagenicity
Several studies have indicated an effect of inorganic arsenic on
human chromosomes, both in vivo and in vitro. An increased
frequency of chromosomal aberrations has been found among persons
exposed to arsenic, mainly in inorganic trivalent form, both as
medication and in the workplace. Petres et al. (1977) examined
lymphocytes from 62 dermatology patients, 31 with a history of
extensive arsenic contact, displaying typical palmo-plantar
hyperkeratosis, and 31 controls. The exposed group consisted of 14
psoriasis patients and 17 wine-growers. The control group also
included 14 psoriasis patients but these did not have any history of
arsenic medication. The frequency of chromosome aberrations, both
structurally and numerically, in the arsenic-exposed group was
significantly higher than that in the controls, especially as regards
chromatid aberrations. An in vitro addition of sodium arsenite to
lymphocyte cultures from healthy subjects induced the same chromosomal
changes. Similar results were earlier obtained by Oppenheim & Fishbein
(1965) after adding potassium arsenite to a culture of human
leukocytes and by Paton & Allison (1972) following exposure of human
diploid cells to arsenic salts, including sodium arsenate.
The chromosomal aberrations in lymphocytes from 39 employees at a
Swedish copper smelter with high levels of airborne arsenic in some
workplaces were counted (Nordenson el al., 1978). The workers were
divided into 4 exposure categories based on type and duration of work
with arsenic compounds and presence or absence of septum perforation
and arsenic dermatitis. Controls were apparently healthy individuals
living about 100 km from the smelter. A total of 4106 cells from
arsenic-exposed workers and 1312 from the controls were examined. The
frequency of chromosomal aberrations was significantly higher in the
arsenic-exposed workers than in the controls. However, the correlation
between the chromosomal aberrations and the estimated arsenic exposure
was poor.
An interaction effect of tobacco smoking and arsenic exposure on
the frequency of chromosomal aberrations was indicated. The effect of
arsenic alone cannot be assessed in this study, as simultaneous
exposure to other agents occurred.
An elevated sister chromatid exchange rate was found by Burgdorf
et al. (1977) in the lymphocytes of 6 patients treated with Fowler's
solution (daily doses of up to 3 mg of arsenic as arsenite).
The mean sister chromatid exchange rate per mitosis in the
arsenic-treated patients was 14.0, and only 5.8 in healthy controls.
It should be noted that all 6 arsenic-treated patients had developed
skin cancer and that at least 2 of them had received X-ray treatment.
No information of this sort was given regarding the control subjects.
Several studies have indicated that inorganic arsenic affects DNA
repair mechanisms. Jung (1971) examined the effectiveness of the dark
repair enzyme system in human skin biopsies treated with sodium
arsenate after irradiation with a Xenon lamp. The repair activity,
which was determined in terms of incorporation of a radioactively
labelled nucleotide, was significantly reduced in arsenic-treated
cells. These results have been confirmed by Petres et al. (1977), who
showed that doses of about 1.0 mg sodium arsenate per litre of culture
medium, reduced the incorporation of radioactively labelled
nucleotides into RNA and DNA in lymphocytes. These 2 studies were
performed on dermal cells and lymphocytes, respectively, but other
cell systems could also be expected to be affected by arsenic in a
similar manner.
A significant increase was found in the frequency of dominant
lethals in the F3 generation of mice given sodium arsenite in the
drinking water at a level of 100 mg As/litre, when this exposure was
combined with intraperitoneal injection of tris-l-aziridinyl-phosphine
oxide (TEPA) in a dose of 1 mg/kg body weight prior to mating. No
increase was observed in animals exposed to 1 mg As/litre (Srám &
Bencko, 1974).
Rossman et al. (1975) showed that survival of Escherichia coli
following UV-radiation was decreased by arsenite. Studies on different
strains of bacteria, deficient in various repair mechanisms, indicated
that arsenic particularly affects post-replication repair.
The incidence of early fetal deaths is determined in the
dominant-lethal assay, and may be an indication of genetic damage.
Sodium arsenate (5 mg/kg bodyweight) and sodium arsenite (5 mg/kg
bodyweight), when given to male mice in a single dose
intraperitoneally, has been shown not to elicit a dominant-lethal
effect (Hodge & Embree, 1977). An effect on chromosomes by a toxic
agent might cause malformations or spontaneous abortion, if it occurs
at the germ cell level. Malformations can also result from
interference with fetal development without necessarily damaging the
genetic material (section 8.2).
Nishioka (1975) carried out mutagenicity tests in vitro for 56
inorganic metal compounds using the DNA recombination assay of
Bacillus subtilis. Sodium arsenite, sodium arsenate, and arsenic
(III) chloride gave positive results in the test. On the other hand,
Löfroth & Ames (1978) failed to show any mutagenic activity of
inorganic trivalent and pentavalent arsenic in the Salmonella plate
incorporation test.
8.6 Mechanisms of Toxicity
Inorganic arsenic has been shown to cause impaired tissue
respiration in vivo in the liver and kidneys of mice and rats
(Bencko & Nemeckova, 1971; Bencko, 1972; Brown et al., 1976; Fowler et
al., 1977). To affect the enzymatic activity responsible for
respiration, arsenic has to pass the mitochondrial membranes and
membrane damage appears to play a prominent role in the emergence of
some of the observed effects. In vitro studies have shown that rat
liver mitochondria can accumulate arsenite (Harris & Achenjang, 1977)
and arsenate through energy-dependent processes (Kagawa & Kagawa,
1969). Arsenic-bound components with properties similar to those of a
low relative molecular mass arsenate ester have been isolated from rat
liver mitochondria (Chan et al., 1969).
It was recognized many years ago that inorganic arsenic inhibits
enzyme activity and that trivalent inorganic arsenic reacts with the
sulfhydryl groups of proteins. Many enzymes containing such groups
have been shown to be affected by arsenite (Thompson, 1948; Webb,
1966). In particular, the marked inhibitory effects of As(III) on
mitochondrial respiration mediated by NAD-linked substrates, appear to
play a critical role in the toxicity of this agent. Suppression of
NAD-linked substrates (pyruvate, glutamate, and a-ketoglutarate, in
particular) in rat liver mitochondria is thought to occur through the
reaction between the arsenite ion and the dihydrolipoic acid cofactor,
necessary for oxidation of the substrate (Fluharty & Sanadi, 1960,
1961). A depression in the activity of succinic acid dehydrogenase
[EC I. 3.99.1] in various tissues has also been demonstrated (Tsutsumi
et al., 1974). Arsenite has been shown to decrease or uncouple
mitochondrial oxidative phosphorylation. This phenomenon is associated
with the stimulation of mitochondrial ATPase activity, which in turn,
is usually, a factor in the distortion of mitochondrial membranes. The
dithiol-containing compound, 2,3-dimercapto/propanol (BAL) has been
shown to potentiate the uncoupling action of arsenite in rat liver
mitochondria, suggesting that dithiols have a function in the
transport of arsenite to the enzyme site (Fluharty & Sanadi, 1960,
1961; Fletcher & Sanadi, 1962). Addition of BAL or dithiols in excess
was found to recouple the phosphorylation. At high, in vitro
concentrations, arsenite also inhibited respiration in rat thymus
nuclei (Konings, 1972).
In vitro studies on rat heart and liver mitochondria and
in vivo studies have shown that pentavalent and trivalent arsenic
exert similar effects in the inhibition of mitochondrial respiration
and uncoupling of oxidative phosphorylation (Crane & Lipmann, 1953;
Azzone & Ernster, 1961; Packer, 1961; Wadkins, 1961; Fowler, 1975).
The mechanism of this inhibition is not clear and several
possibilities exist. One is that arsenate is reduced by the
mitochondria to As(III) and that inhibition occurs through the
formation of a complex with the lipoic acid cofactor. The increasing
inhibitory effect on NAD-linked substrates with time found by Crane &
Lipmann (1953) indicates that this could be the case. From studies on
rat liver and beef heart mitochondria, Mitchell et al. (1971) proposed
that inhibition of mitochondrial energy-linked functions by arsenate
occurs in 2 ways: competition with phosphate during oxidative
phosphorylation and inhibition of energy-linked reduction of NAD.
9. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF ORGANIC ARSENIC
COMPOUNDS
9.1 Acute and Chronic Toxicity
The form of organic arsenic determines the tissue distribution and
hence affects the pattern of toxicity (section 6.2.2.2). Some organic
arsenic compounds have a high toxicity for certain organs, while the
organic arsenic compounds in seafood are apparently of low toxicity.
9.1.1 Man
Organic arsenic compounds are still used widely as antiparasitic
drugs (section 5.3.3). Side-effects have been reported in many organ
systems, most notably the central nervous system. Encephalopathy was
observed in 1.5% of 1066 patients treated with arsobal (tryparsamide)
for trypanosomiasis (Sina et al., 1977). The mortality was high
(62.5%) among those with encephalopathy. Encephalopathy also occurred
in patients treated with glycobiarsol, another organic arsenic
compound (Cole et al., 1966). A well-known side-effect of tryparsamide
treatment is optical atrophy. Visual effects arise in 3%-4% of
patients treated with this drug, according to Neujean et al. (1948).
Other, less frequent, side-effects include dermatitis, liver damage,
and disturbances of the haematopoietic system.
Organic arsenic compounds may be found in high concentrations in
some seafoods (section 5.2). Although there have not been any
scientific studies published on the acute and subacute toxicity of
these forms of arsenic for man, experience indicates that it is very
low. Chronic effects, however, cannot be ruled out. Walkiw & Douglas
(1975) described 2 women who had been on health food supplements
prepared from kelp (duration of intake not stated). The women had
developed neuropathy, one of them having foot drop and the other
peripheral neuropathy. At the time of hospitalization, the urinary
excretion of arsenic for the 2 patients was 138 µg and 293 µg/24 h,
respectively. No information was given on the course of the symptoms
following cessation of exposure. Clearly, this report cannot
incriminate organic arsenic in the seaweed, beyond doubt, as the cause
of the neuropathy.
9.1.2 Animals
The toxicity of the form of organic arsenic present in seafoods is
of special interest with regard to general human exposure. From the
very limited research performed to date it appears to be relatively
low. Coulson et al. (1935) reported that rats fed a diet containing
shrimp arsenic at a concentration of about 14 mg As/kg for 12 months
did not have any defects in growth or physical appearance. No
histological changes were found in the spleen, liver, or kidneys. In
studies by Lancaster et al. (1971), lakeweed with a high arsenic
content was given to sheep at a daily dose of 1.4 mg As/kg body weight
for 3 weeks. The animals remained healthy, and examination of organs
and tissues removed from the animals at slaughter (3 animals per week)
did not reveal any gross morphological changes.
Aliphatic arsenic compounds such as dimethylarsinic acid and mono-
and disodium methane arsenate, are used commercially as herbicides in
noncrop areas. These herbicides have been reported to be a source of
intoxication for domestic animals (Selby et al., 1977). The clinical
symptoms and histological findings seem to be the same as those
induced by inorganic arsenic, discussed in section 8.1.
Experimental research on the acute inhalation toxicity of
dimethylarsinic acid for rats was performed by Stevens et al. (1976).
No mortality was found among rats, exposed to an aerosol containing a
commercial product (Phytar 138) at a concentration of 2600 mg/m3 for
2 h. The particle size of the dust was 3 µm (MMD). No other details of
the product were specified in the report. Assuming a content of 65%
dimethylarsinic acid (Gosselin et al., 1976), it can be calculated
that the arsenic concentration in the aerosol was approximately
840 mg/m3. Exon et al. (1974) fed rabbits a diet containing
monosodium methane arsenate (MSMA) at a level of 50 mg/kg diet, equal
to 27.5 mg As/kg diet, for 7 or 8 weeks. Toxic hepatitis was found in
all of the 8 animals necropsied and reactive hyperplasia in 5 of them.
Steers and heifers, weighing 100-200 kg, were given daily doses of
pure MSMA at 10 mg/kg (5 mg As/kg body weight; aqueous solution) for
up to 10 days (Dickinson, 1972). Two steers died during the treatment
and the third, some days later. Of the 2 heifers, one was killed due
to morbidity. The other recovered, when the exposure was ended. All
the arsenic-exposed animals lost weight and developed severe
diarrhoea, haemorrhagic gastritis, and intense hyperaemia. Liver
necrosis and renal tubular degeneration were also found.
The short-term toxicity of phenoxarsine oxide (PXO) and
phenarsazine oxide (PZO), 2 organic arsenic compounds used as
industrial biocides, was studied by Ballantyne (1978). Acute oral
toxicities, determined as the LD50 for guineapigs and rats, were 24
and 40 mg/kg body weight (equal to 7 and 12 mg As/kg body weight) for
PXO and 77 and 83 mg/kg body weight (23 and 25 mg As/kg body weight)
for PZO. PZO was more hepatotoxic, producing cellular infiltration and
oedema of the portal tracts as well as periportal hepatocellular
necrosis in rats receiving a single oral dose of 30 mg/kg body weight
(10 mg As/kg body weight). Histological changes of the liver were not
observed in animals receiving the same dose of PXO. Inhalation of PXO
and PZO for 30 days in concentrations of 1-2 mg/m3 (0.3-0.6 mg As/m3
(MMD 4-5 µm), for 5 h a day, did not elicit any toxic signs except for
transient cellular infiltration of the portal tracts of the liver.
Both substances produced eye and skin irritation in guineapigs with a
local application of a 25% suspension in water. Increases in
intraocular pressure in rabbits were concentration dependent.
The toxicity of derivatives of phenylarsonic acid, such as
arsanilic acid, is a subject of great interest, since these compounds
are commonly used as feed additives for poultry and swine. Ledet
et al. (1973) studied the effects of a diet containing arsanilic acid
at a concentration of 1000 mg/kg (350 mg As/kg) in pigs. This was
reported to be 10 times the level recommended for growth stimulation.
Roughened haircoat and mild diarrhoea were the first signs of
toxicity. Cutaneous hyperaemia and hyperaesthesia usually occurred
after a few days, and the animals often stumbled and staggered about.
Histopathological lesions were confined to peripheral and optic
nerves. The same dietary level of arsanilic acid was found to cause
blindness and optic disc atrophy in pigs within 25-30 days (Witzel
et al., 1976). Sodium arsanilate injected subcutaneously into
guineapigs in doses exceeding 70 mg/kg body weight caused degeneration
of the sensory cells of the inner ear (Anniko & Wersäll, 1977).
Retention of arsenic in the cochlea and delayed elimination from the
inner ear compared with elimination from the blood have been reported
(Anniko & Plantin, 1977).
9.2 Teratogenicity
Teratogenic effects were not observed in 7 generations of rats fed
0.01, 0.02, or 0.05% arsanilic acid (3.5, 7 or 17.5 mg As/kg diet)
(Frost et al., 1964). On the contrary, both the litter sizes and the
survival of pups increased significantly.
9.3 Carcinogenicity
No human epidemiological investigations have been conducted on the
carcinogenicity of organic arsenic compounds. Consequently, only data
concerning laboratory animals can be considered.
9.3.1 Animals
Twelve mice belonging to a strain with a high incidence of
spontaneous mammary tumours were injected with neoarsphenamine in
doses of 6.7 mg/kg body weight, twice weekly, for up to 10 weeks
(Hueper & Itami, 1933). Exposed and control animals were found to have
similar average life spans. The arsenic-exposed animals showed an
increased growth rate, and histologically, a higher grade of
malignancy.
In an experiment in which 50 trout were fed a synthetic diet
containing carbarsone at 4.8 g/kg diet for 20 months, 5 trout
developed hepatomas (Halver, 1962; cited in Kraybill & Shimkin 1964).
A control group of 300 trout given the same diet but omitting the
carbasone, did not develop any hepatomas. The original data in this
report were not available, and consequently a thorough evaluation
could not be made of the findings.
Frost et al. (1962) fed groups of 60 rats (30 males and 30
females) diets containing arsanilic acid at 0, 0.1, 0.5 and 1 g/kg
diet for 106 weeks. Tumour incidence was similar in all the groups. In
studies by Boutwell (1963), arsanilic acid was given to 30 mice for a
2-week period at a level corresponding to 200 mg As/kg diet and to
another group for 48 weeks at a level of 100 mg/kg diet. The arsenic
exposures were combined with a skin application of the tumour
initiator 7,12-dimethylbenz (a)anthracene (5 µg) and the tumour
promoter, croton oil. No differences were noted in tumour incidence
between the arsenic-fed animals and controls.
3-Nitro-4-hydroxyphenyl-arsonic acid was incorporated into the
feed of groups of 100 rats and 100 mice (50 animals of each sex in
each group) at levels of 0, 50, and 200 mg/kg diet for the rats and
0, 50, and 100 mg/kg diet for the mice. Tumours occurred in
essentially similar numbers of animals in all groups over a 2-year
period (Prier et al., 1963). The same authors also exposed groups of 6
dogs (3 male and 3 female) to 4-hydroxy-3-nitrophenylarsonic acid
added to the feed at levels of 50 and 200 mg/kg. In this study also,
no differences were seen between exposed and control groups. A
solution of 4-hydroxy-3-nitrophenylarsonic acid in ethanol acetone
(1:4), at a concentration of 10 g/litre, was painted on the skin of
100 mice, 3 times weekly, for 1 year. No skin tumours developed during
this and the following year. Subcutaneous injections of 10 mg of
4-hydroxy-3-nitrophenylarsonic acid were given to 100 female mice and
5 mg to 100 male mice. Neither of the 2 groups differed from control
animals with regard to tumour incidence during a 2-year period of
observation.
The diets of 5 groups of 100 rats (equal numbers of each sex in
each group) were supplemented with carbarsone, ( p-ureidobenzene
arsonic acid) to provide daily intakes of 2.5, 5, 25, 50, and
100 mg/kg body weight, respectively. After 72 weeks, the group
receiving the highest level of carbarsone in the feed was given an
unsupplemented diet. Survival was similar in all groups except the one
with the highest exposure, in which there was a marked increase in the
death rate before the transfer to the unsupplemented diet. A total of
13 malignant tumours was observed in a control group of 200 rats
followed for 2 years. In the 3 groups receiving the highest levels of
carbarsone, the number of malignant neoplasms varied between 0 and 14
and was not related to dose (Oser et al., 1966).
9.4 Mutagenicity
No increase in the incidence of early fetal deaths was observed by
Hodge & Embree (1977) in the offspring of male mice given a single
dose intraperitoneally of [(dimethylarsino)oxy] sodium As-oxide
(sodium dimethylarsonate) (200 mg/kg body weight) arsenodiacetic acid
(arsenoacetic acid) (50 mg/kg body weight), and methanearsonic acid
(250 mg/kg body weight).
9.5 Mechanisms of Toxicity
The mechanisms by which arsenic compounds exert their action in
biological tissues have been thoroughly described and reviewed (Barron
& Singer, 1945; Barron et al., 1947; Thompson, 1946, 1948; Gordon &
Quastel, 1948; Stocken & Thompson, 1949; Peters, 1955; Vallee et al.,
1960; Johnstone, 1963). Arsenic compounds do not constitute a
homogeneous group and the effects in biological systems, caused by
reactions between the arsenic compounds and functional groups of
different enzymes, are highly dependent on the chemical character of
the compound involved in each particular case.
The first distinction to be made is that between trivalent and
pentavalent organic arsenic compounds. Pentavalent arsenic compounds
(R-AsO3H2) have little effect on enzyme activity but can be reduced
in vivo to more toxic trivalent compounds (Peters, 1955; Johnstone,
1963).
Trivalent organic arsenic compounds include, according to the
nomenclature used by Johnstone (1963), arseno and arsenoso compounds.
Arseno compounds, (R-As = As-R) are readily oxidized, even by trace
amounts of oxygen, and their action has been suggested to be due to
their conversion to the corresponding arsenoso derivatives. These can
be divided into monosubstituted and disubstituted compounds according
to their reaction with sulfhydryl groups (Peters, 1955). The
monosubstituted compounds, exemplified by R As = O, react with enzymes
containing sulfhydryl groups:
SR'
/
R - As = O + 2R'SH <==> R - As + H2O
\
SR'
Inhibition of different enzyme systems by these arsenic compounds
was shown to be reversed by addition of an excess of a monothiol,
e.g., glutathione. Some enzymes contain 2 thiol groups, which can
react with the monosubstituted arsenic compound, thereby yielding a
5-membered ring structure. This reaction is reversed by dithiols,
e.g., 2,3-dimercaptopropanol (BAL), but not by monothiols. Lipoic
acid, necessary for the initial stages in the oxidation of pyruvate,
is inhibited in this way by lewisite (used as a war gas), and the
reaction is successfully reversed by 2,3-dimercaptopropanol (BAL).
SH
/
protein + Cl2AsCH = CHCl <==>
\
SH
S
/ \
protein
\ /
S AsCH = CHCl + 2HCl
S
/ \
protein AsCH = CHCl + BAL <==>
\ /
S
SH H2C - S
/ ' \
protein + ' AsCH = CHCl
\ ' /
SH HC - S
'
'
'
HOH2C
R
\
The disubstituted arsenoso compounds, As - OH, exerts its
/
R
action by combining with enzymes containing monothiol groups. The
resulting enzyme poisoning can probably be reversed by the monothiol
defence mechanisms present in the body. One complication, however, is
the possible conversion of disubstituted to mono-substituted arseno
compounds. This is indicated by the finding that dithiols are needed
for reversing the reaction between diphenylchloroarsine and certain
enzyme systems in brain and kidney.
10. INTERACTIONS WITH OTHER CHEMICALS
Until recently, studies on the toxicity of different chemical
agents have been based almost exclusively on the administration of a
single substance as the point of departure. Considering that man is
exposed simultaneously to a wide variety of agents, it is urgent that
the interactions between these agents should be investigated. In the
case of arsenic, attention has been paid to its interaction with
dithiol-containing substances such as BAL, because of the introduction
of BAL as an antidote to the arsenic-containing war gas lewisite.
Investigations concerning combined exposures to arsenic and other
chemicals may, in the future, explain some of the data which appear
controversial at present. Interactions between arsenic and other
agents have been dealt with by the Scientific Committee on the
Toxicology of Metals under the Permanent Commission and International
Association on Occupational Health at its Stockholm meeting on
"Factors Influencing Metabolism and Toxicity of Metals", held in July,
1977. Among the items discussed were interactions between arsenic and
selenium, cadmium, and lead (Nordberg, 1978). Because of the lack of
data on human exposure situations, the following discussion will refer
exclusively to experimental animal studies.
10.1 Thiol Compounds
The toxicity of arsenic compounds, the influence of mono- and
dithiol-containing compounds, and the introduction of
2,3-dimercaptopropanol (BAL) as an antidote in arsenic poisoning have
been reviewed by Stocken & Thompson (1949). The mechanism for the
interaction was discussed in terms of the biochemical aspects of
arsenic toxicity (sections 8.6 and 9.5). It should be noted that
organic and inorganic arsenic react differently with dithiols. The
toxic effects of arsenite can be potentiated by dithiols, as these can
serve as vehicles for the transport of arsenite to the enzymes. The
distribution and excretion of inorganic arsenic in the form of
arsenate, given orally to rats, can be influenced by BAL or thioctic
acid (TA) (Tsutsumi & Kato, 1975). Five consecutive injections of
either substance decreased the tissue levels of arsenic and increased
its excretion more than a single dose.
In rabbits, given BAL or TA parenterally, the amount of
arsenic(III) oxide absorbed in the blood from a ligated loop of the
intestine was considerably lower than in control animals (Tsutsumi &
Nozaki, 1975). Furthermore, when BAL and TA were added directly into
the loop containing arsenic(III) oxide, the amount of arsenic absorbed
in the blood was lower than that of the control group. It was
suggested that BAL and TA, after being excreted into the intestine
with the bile, can inhibit the absorption of arsenic.
BAL, TA, and to some extent diisopropylaminodichloroacetate
(DADA), intraperitoneally injected in rats, increased the amounts of
74As in the stomach and small intestine following orally administered
74As-arsenate (Tsutsumi et al., 1976). When glucuronolacton (GL) and
glutathione (GT) were injected into the animals, the 74As-content in
these organs was comparable to that of the control group. BAL, TA and
DADA were also shown to suppress intestinal movements. The authors
suggested that the delayed uptake of arsenic might be because of the
inhibited transport of arsenic in the gastrointestinal canal, as it
had been shown earlier that arsenic is absorbed preferentially in the
small intestine.
BAL, subcutaneously injected in mice (50 mg/kg body weight),
reduced the teratogenic action of simultaneous intraperitoneal
injections of sodium arsenate at a level of 16 mg As/kg body weight
(Hood & Pike, 1972).
10.2 Selenium
The interaction between selenium and arsenic has been reviewed by
Levander (1977), who concluded that arsenic has a protective effect
against the toxicity of a variety of forms of selenium. He found that
this effect had been demonstrated in several species, including rats,
dogs, swine, cattle, and poultry. It was suggested that arsenic acts
by enhancing the biliary excretion of selenium. Sodium arsenite was
shown to be most potent, arsenate somewhat less potent, and various
organic arsenic compounds least potent. The opposite course of events
has also been observed, i.e., selenite can stimulate the excretion of
arsenite in the bile of rats. The experiments referred to have been
performed almost exclusively on rats. Taking into account what is
known about the metabolism of arsenic in rats and especially about the
partition of different forms of arsenic in the enterohepatic
circulation, it must be concluded that research on the mechanism of
interaction between selenium and arsenic is far from complete.
Holmberg & Ferm (1969) observed that selenite decreased the
teratogenic effects of arsenate when the 2 compounds were given to
pregnant hamsters in simultaneous intravenous injections.
The protective effect of arsenic against selenium has also been
noticed in studies on mouse fibroblasts (Rössner et al., 1977). A
study carried out on mice demonstrated that arsenic (III) was more
efficient in protecting against selenium toxicity than arsenic (V)
(Bencko et al., 1978b).
10.3 Cadmium and Lead
Mahaffey & Fowler (1977) examined the effects of dietary cadmium
(50 mg/kg diet) and lead (200 mg/kg) on the toxicity of arsenic
administered to rats as sodium arsenate or arsanilic acid in the food
(50 mg As/kg diet). The efficiency of food utilization was more
reduced by the combination of arsenic and cadmium than by each metal
alone. The combination of arsenic and cadmium also caused a greater
decrease in serum alkaline phosphatase levels than either metal alone.
Additive effects of arsenic and lead in coproporphyrin excretion were
also noted. Increased uroporphyrin excretion in rats was reported
(Fowler & Mahaffey, 1978), when lead, cadmium, and arsenic were
combined compared with excretion levels following exposure to arsenic
alone.
11. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO ARSENIC
11.1 Introduction
Arsenic can give rise to acute, subacute, and chronic effects. The
adverse health effects of arsenic may involve the respiratory,
gastrointestinal, cardiovascular, nervous, and haematopoietic systems.
Effects may be local and systemic.
In the discussions of effects in the following sections,
quantitative aspects on dose-response relationships are given,
whenever possible. Unfortunately, such data are generally very scanty
or nonexistent, and this makes risk evaluation difficult.
Various models have been developed for assessing the risk of
cancer at low doses; however, the simple linear non-threshold
dose-response extrapolation model is most often used. If this model is
adopted, the pulmonary carcinogenicity of inorganic arsenic should
constitute the basis for setting environmental standards for airborne
exposure. In the case of oral exposure, several effects, including
skin cancer, have to be taken into consideration.
Mutagenic and teratogenic effects will not be discussed separately
in this section, as the significance of such data as a basis for
environmental standards is not clearly recognizable.
11.2 Exposure
Exposure to arsenic may occur industrially as well as through
ambient air, tobacco smoke, water, food and beverages. In addition,
considerable exposure may take place through the ingestion of drugs,
still in use in certain countries.
Occupational exposure occurs through the inhalation of particulate
matter containing inorganic arsenic, which may be trivalent or
pentavalent. Concentrations varying from a few micrograms to more than
one milligram per cubic metre have been reported.
Most arsenic in particulate matter in ambient air is in the form
of inorganic arsenic compounds. Concentrations in urban areas may
range from a few nanograms per cubic metre to a few tenths of a
microgram. In the vicinity of point sources emitting arsenic,
concentrations exceeding 1 µg As/m3 have been reported.
In the past, tobacco contained high concentrations of arsenic
because of the widespread use of arsenic in pesticides and
concentrations of up to about 40 mg/kg have been found. However,
levels are now well below 10 mg/kg, in countries that restrict the use
of pesticides containing arsenic.
In both ground and surface waters, the total arsenic concentration
is usually below 10 µg/litre. In certain areas, levels of more than
1 mg/litre have been recorded. Some studies indicate that arsenic is
mainly present in inorganic forms. The oxidation state of the arsenic
in waters depends on the prevailing redox potential. In surface
waters, in the presence of dissolved oxygen, arsenic(V) is
predominant. Under reducing conditions, particularly in deep well
water, arsenic(III) is present.
Food and beverages are the most important sources of exposure to
arsenic for the general population. The total daily intake of arsenic
from the diet has been estimated to be less than 200 µg in the adult.
The figure is greatly influenced by the intake of seafood, which may
contain up to 100 mg As/kg. Most of the arsenic of marine origin is
organic. The total daily intake of inorganic arsenic from food and
water has been estimated to be under 50 µg.
11.3 Inorganic Arsenic Compounds
Exposure can occur to both trivalent and pentavalent inorganic
arsenic compounds, the solubilities of which vary. The nature of the
compounds present in different exposure situations has not been well
identified. Furthermore, the absorption and retention, in the lungs
for example, of compounds of different solubilities are virtually
unknown.
Both arsenic(III) and arsenic(V) are methylated to a great extent
after entering the body. The major metabolites in the urine are
methylarsonic acid and dimethylarsinic acid. The possibility of
dose-dependence in this respect should be considered. Furthermore, the
conversion of arsenic(V) to arsenic(III) in vivo has been suggested,
but data are not conclusive.
In view of these uncertainties, health evaluations, with few
exceptions, have to be confined to inorganic arsenic in general.
11.3.1 Acute and subacute effects after short-term exposure
A fatal dose of ingested arsenic(III) oxide for man has been
reported to range from 1 to 2.5 mg As/kg body weight. This is lower
than the LD50 values generally reported for animals. Trivalent
arsenic is considered to be more toxic than pentavalent.
Two recent mass outbreaks of arsenic poisoning due to the
ingestion of inorganic arsenic have been described. In the first
episode, more than 12 000 infants were poisoned with dried milk
contaminated with inorganic arsenic compounds (oxidation state
uncertain, although in one report it was given as pentavalent). The
average exposure level for a baby of 3 months had been about 3.5 mg of
arsenic daily and symptoms usually appeared after a few weeks. In all,
130 deaths occurred, and effects were observed in several organs.
The other episode concerned about 400 persons who ate contaminated
soy sauce and were thus exposed to an average of 3 mg of arsenic,
daily, for 2-3 weeks. Many acute symptoms were encountered but
neurological symptoms usually did not appear until the 10th-20th day
of illness.
Dose-response relationships are difficult to estimate from the
incidents just described, but it appears that ingestion of 3 mg of
inorganic arsenic per day, over a period of few weeks, may give rise
to severe poisoning in infants and symptoms of toxicity in adults. The
toxic effects cannot be related to any specific valence form of
arsenic.
11.3.2 Noncarcinogenic effects after long-term exposure and
sequelae of short-term exposure
Inhalation of inorganic arsenic compounds can result in local
damage to the respiratory system, including perforation of the nasal
septum. Systemic effects after inhalation and/or ingestion involve the
skin, liver, and the cardiovascular and nervous systems.
In general, details of exposure in human situations have been
inadequate, as a basis for dose-response evaluations. For certain
effects, however, some estimations of dose-response relationships can
be made and information about this is given in the following sections.
11.3.2.1 Skin effects
Skin effects in the form of hyperkeratosis, hyperpigmentation, and
depigmentation have been observed in different parts of the world
after exposure to drinking-water containing high levels of arsenic,
and after treatment with drugs containing inorganic arsenic. The
oxidation state of the arsenic in the water involved in these
incidents is not known but, in drugs, the arsenic component has
usually been in the trivalent state. Skin effects have also been
reported following exposure to arsenic during the manufacture of
insecticides and also among wine growers.
Very few of the data available are of much help in estimating
dose-response relationships. It seems, however, that several years of
exposure to approximately 1 mg of arsenic per day may give rise to
skin effects. It is not possible to state to what extent effects are
caused by exposure to trivalent arsenic alone or whether pentavalent
arsenic may also be of importance.
11.3.2.2 Cardiovascular effects
Cardiovascular effects, in the form of electrocardiographic
changes and peripheral vascular disorders have been observed in
persons exposed to arsenic. Peripheral vascular disorders have been
reported in German vintners, and, in Chile and China (Province of
Taiwan), in parts of the population consuming water containing
0.5-1 mg As/litre. In both the Taiwanese villagers and the German
vintners, the inadequate peripheral circulation caused gangrene,
referred to as "blackfoot disease" by the Taiwanese. A dose-response
relationship was arrived at on the basis of the data concerning the
Taiwanese villagers, in which a roughly linear increase in the
prevalence with increasing arsenic dose was indicated. Exposure to
arsenic for many years, resulting in a total ingested dose of about
20 g of arsenic, corresponded to a prevalence of "blackfoot disease"
of about 3%.
11.3.2.3 Neurological effects
Peripheral neurological damage has been observed in persons
consuming arsenic-containing antiasthmatic preparations on a long-term
basis. The exposure corresponded to 3-10 mg of arsenic per day in the
form of arsenic(III) oxide or arsenic sulfide.
Disturbances of the central nervous system function were noted in
a follow-up of Japanese infants, fifteen years after exposure to an
average daily arsenic dose of about 3.5 mg for one month. The
occurrence of severe hearing loss and brain wave abnormalities was
indicated. However, the data were considered not to be conclusive.
11.3.3 Carcinogenicity
It has already been mentioned that the pulmonary carcinogenicity
of inorganic arsenic should constitute the basis for setting
environmental standards for airborne exposure, and that in the case of
oral exposure, several effects, including skin cancer, have to be
taken into consideration. In reviewing available data in 1973, as well
as in 1980, IARC concluded that there was sufficient evidence to
associate exposure to inorganic arsenic with cancer of the lung and
skin.
11.3.3.1 Cancer of the respiratory system
Arsenic has been associated with pulmonary cancer in the
manufacture and use of arsenic-containing pesticides and in the
smelting of copper. The carcinogenic potential of inorganic arsenic,
mainly trivalent, in the smelter environment is evident from many
epidemiological studies in different countries. One report revealed a
roughly linear relationship between cumulative arsenic exposure and
the lung cancer risk. Though data are uncertain, it could be estimated
that exposure to airborne arsenic levels of about 50 µg/m3 (probably
mostly arsenic(III) oxide) for more than 25 years would result in a
nearly 3-fold increase in mortality due to lung cancer over the age of
65 years (section 8.4.1).
11.3.3.2 Skin cancer
Exposure to inorganic arsenic can cause skin cancer, mainly
tumours of low malignancy. This has been observed following ingestion
of arsenic-rich drinking-water and the consumption of
arsenic-containing drugs. A total dose of several grams has usually
been required for the development of cancer. The form of arsenic in
the different types of drinking water in question has yet to be
elucidated, while, in drugs, it has most often been inorganic
trivalent arsenic.
There are very few dose-response data on arsenic and skin cancer
that can be used for quantitative estimations. From one study on
exposure to arsenic via drinking-water in China (Province of Taiwan),
there was evidence that a total of about 20 g of arsenic over a
lifetime resulted in a prevalence of skin cancer of about 6%.
11.4 Organic Arsenic Compounds
The application of organic arsenic compounds in medicine, most
notably tryparsamide, has induced side effects, mainly in the central
nervous system, in the form of encephalopathy and optical atrophy.
Toxic effects on the nervous system have also been reported in
experimental animals given high doses of arsanilic acid. This is a
compound commonly used as a feed additive for poultry and swine in
some countries. No data on dose-response relationships, which would be
directly applicable to the long-term exposure of man, are available.
Human beings are exposed to organic arsenic compounds in seafood,
certain types of which may contain arsenic concentrations of
20-50 mg/kg wet weight or even more. The form of arsenic in seafood is
largely unknown, but it has been found to be readily absorbed (more
than 80%) from the gastrointestinal tract in both animals and man. It
is also rapidly excreted in the urine (70-80% within a week).
No adverse effects were found in a study in which rats were fed a
diet containing "seafood arsenic" in the form of shrimps at a
concentration of about 14 mg As/kg for 12 months. There are no data
concerning the toxicity of "seafood arsenic" for man, apart from the
fact that acute effects have not been reported. It seems obvious that
this question has not been studied sufficiently, especially
considering that a large number of people are exposed to this form of
arsenic. Another organic arsenic compound, dimethylarsinic acid, has
been shown to pass through the placental barrier of rats. Blood values
of the fetus were comparable to those of the mother. Placental
transfer should be investigated when the toxicity of "seafood arsenic"
is studied.
There is no conclusive evidence that any of the organoarsenic
compounds tested for carcinogenicity in laboratory animals are
carcinogenic. Epidemiological studies are greatly needed on
populations exposed to organic arsenic, especially in view of the
discrepancy between animal and human data with regard to the
carcinogenicity of inorganic arsenic compounds. Suitable groups for
study are workers exposed in the manufacture, handling, and use of
organoarsenic compounds as well as patients treated with such
compounds. Data have not been reported concerning the possible
carcinogenic effects of "seafood arsenic".
11.5 Assessment of the Cancer Risk for Man from Exposure to
Inorganic Arsenic
The purpose of this section is to provide guidance for the
estimation of the cancer risk to the lung and skin from the inhalation
and ingestion, respectively, of inorganic arsenic. For the purposes of
this risk assessment, it is assumed that both pentavalent and
trivalent arsenic are carcinogenic.
It must be recognized that the assessment of cancer risks by
currently available methods can provide only crude estimates and this
should be borne in mind particularly in making regulatory decisions
about permissible limits of exposure. The use of the linear
non-threshold model is recommended for extrapolation of risks from
relatively high dose levels, where cancer responses can be measured,
to relatively low dose levels, which are of concern in environmental
protection where such risks are too small to be measured directly
either through animal or human epidemiological studies.
The linear non-threshold model has been generally accepted amongst
regulatory bodies in the USA for chemical carcinogens (IRLG) and for
ionizing radiation on an international basis (ICRP). The linear
non-threshold philosophy was accepted by a Task Group on Air Pollution
and Cancer in Stockholm in 1977 (Task Group on Air Pollution and
Cancer, 1978). The scientific justification for the use of a linear
non-threshold extrapolation model stems from several sources: the
similarity between carcinogenesis and mutagenesis as processes which
both have DNA as target molecules, the strong evidence of the
linearity of dose-response relationships for mutagenesis, the evidence
for the linearity of the DNA binding of chemical carcinogens in the
liver and skin, the evidence for the linearity in the dose-response
relationship in the initiation stage of the mouse 2-stage
tumorigenesis model, and the rough consistency with the linearity of
the dose-response relationships for several epidemiological studies;
for example, aflatoxin and liver cancer, leukaemia and radiation. This
rationale for the linear non-threshold dose-response model is
strongest for the genotoxic carcinogens.
The mechanism of the carcinogenesis of arsenic is not clear at
present.
In the case of the lung cancer estimates for the inhalation of
inorganic arsenic, based on epidemiological data from an occupational
smelter population, it is assumed that the life-time cancer risk is a
function of the total dose of arsenic. This is a necessary assumption
because occupational exposures begin at maturity, whereas exposures to
airborne arsenic in the general environment begin at conception.
Furthermore, in the case of lung cancer risk estimates, it is assumed
that there are no age or sex differences in susceptibility to cancer
induced by arsenic. There is not much basis in scientific fact for
assuring the validity of these assumptions. It is not unreasonable to
assume that the cancer response is proportional to the total dose,
since the occupational smelter exposures extended over a substantial
portion of the life span. On the basis of animal data, it is possible
that children may generally be somewhat more susceptible than adults
to carcinogens, but it is not known whether this is the case for
arsenic and lung cancer. It is also not known whether males and
females are equally susceptible. It is possible that sulfur dioxide
and smelter dusts other than arsenic potentiate arsenic in the
workplace. Presumably there are more carcinogen co-factors in the
occupational setting than in the general environment, but this is not
certain.
Risk estimates for lung cancer from inorganic arsenic exposure can
be based on the study of Pinto et al. (1977) on smelter workers, in
which there was a standard mortality ratio of about 300 or a 200%
excess of lung cancer at an average air concentration of 50 µg/m3 for
an average duration of exposure of more than 25 years. It was assumed
that the 200% excess of lung cancer applied to the life-time risk even
though the Pinto study was limited to observations in men over 65
years of age. This assumption of relative-risk model has a sound
factual basis particularly for cancer response to protracted
exposures. The 200% excess needs to be extrapolated for the same total
dose to the average life span of the population under consideration;
i.e., if the average life span is 70 years, then a 200% excess of
cancer would be produced for the same total arsenic dose by a
life-time exposure to 8 µg/m3. It is assumed that the 200% excess of
lung cancer, observed in the Pinto study, corresponds to an exposure
level of 50 µg/m3 for 25 years. (This will lead to some
overestimation of the risk as the duration of exposure in the original
data was given as more than 25 years.) The calculation is based on an
occupational air intake of 8 m3 per day, for 240 days a year, over 25
years compared with an environmental air intake of 12 m3 per day for
365 days a year over 70 years. In other words, a life-time exposure to
8 µg/m3 would be expected to produce a 200% excess in lung cancer.
This excess risk can conveniently be expressed as a percentage
increase per unit concentration of arsenic in air. Thus 1 µg/m3 would
produce 200/8 or a 25% excess in lung cancer incidence. Knowing the
existing lifetime cancer incidence for the population under
consideration, the risk from the nominal concentration of 1 µg/m3 can
be expressed in terms of absolute excess risk. If the life-time risk
of lung cancer is for example 3%, then the excess risk is 3% × 25% or
0.03 × 0.25 or 0.0075 per microgram of airborne arsenic per cubic
metre or 0.8% per microgram of airborne arsenic per cubic metre.
There are relatively few assumptions that need to be made for the
estimation of the skin cancer risk from the ingestion of arsenic in
drinking water. These risk estimates can be based on the
epidemiological data from China (Province of Taiwan) (Tseng, 1977),
where the population at risk was exposed for at least 50 years and the
data were obtained from more than 40 000 men, women, and children of
all ages. Nevertheless, the response could have been affected by
socio-economic, cultural, and racial factors such as skin pigment,
which may not be comparable to other populations. The risk assessment
for skin cancer from the ingestion of inorganic arsenic can be based
on the data shown in Fig. 11. The skin tumour data in this study was
given in terms of prevalence. However, since these tumours were of low
malignancy and would be expected to persist for a very long time, it
has been assumed that the characterization of tumour yield in terms of
prevalence is equivalent to cumulative incidence. The middle dose
group shown in Fig. 11 for the age range of 60 years and over was
chosen as the point of departure for the downward linear non-threshold
extrapolation because it is the one that is the best description of
exposure; the choice of the oldest age group maximizes the tumour
yield for a given total dose. The observed prevalence data were not
modified to account for a control incidence, since Tseng stated that
actinic skin cancers were not included in this series of cases. The
slope of the resultant linear extrapolation is about 5% skin tumour
prevalence per 10 grams of total ingested arsenic. Assuming a 2
litre/day average intake of drinking water, a concentration of 0.2 mg
As/litre would result in a total dose of 10 grams in an assumed
average life span of 70 years. Thus the life-time risk from arsenic in
drinking-water is about 25% per mg As/litre (5%/0.2 mg As/litre).
REFERENCES
ADAMSSON, E., PISCATOR, M. & NOGAWA, K. (1979) Pulmonary and
gastrointestinal exposure to cadmium oxide dust in a battery
factory. Environ. Health Perspect., 28: 219-222.
AGGETT, J. & ASPELL, A. C. (1978) Release of arsenic from geothermal
sources. (New Zealand Energy Research and Development Committee
Report No. 35).
ALVARADO, L. C., VINIEGRA, G., GARCIA, R. E. & ACEVEDO, J. A. (1964)
[Arsenism in the Lagoon Region.] Salud Pública Méx.,
6 (3): 375-385 (in Spanish).
ALY, S., MOUSA, S., EL-KAHKY, M., SALEH, A. & EL-MOFTY, A. (1975)
Toxic deafness. I. Histological study of the effect arsenic,
salicylates and quinine, on the organ of Corti of guinea pigs.
J. Egypt. Med. Assoc., 58 (3-4): 144-157.
ANDERSSON, A. & NILSSON, K. O. (1972) Enrichment of trace elements
from sewage sludge fertilizer in soils and plants. Ambio,
1: 176-179.
ANDO, S., SUZUKI, M., FUWA, K. & VALLEE, B. L. (1969) Atomic
absorption of arsenic in nitrogen (entrained air) hydrogen flames.
Anal. Chem., 41: 1974-1979.
ANDREAE, M. O. (1977) Determination of arsenic species in natural
waters. Anal. Chem., 49: 820-825.
ANDREAE, M. O. (1978) Distribution and speciation of arsenic in
natural waters and some marine algae. Deep-sea Res.,
25: 391-402.
ANNIKO, M. & PLANTIN, L. -O. (1977) Delayed elimination of the
ototoxic compound atoxyl from the inner ear. Arch.
Oto-Rhino-Laryng., 215: 81-89.
ANNIKO, M. & WERSÄLL, J. (1977) Experimentally (atoxyl) induced
ampullar degeneration and damage to the maculae utriculi.
Acta Otolaryngol, 83: 429-440.
ARGÜELLO, R. A., CENGET, D. D. & TELLO, E. E. (1938) [Cancer and
endemic arsenism in the Cordoba Region.] Rev. Argent.
Dermatosifilogr., 22 (4th part): 461-487 (in Spanish).
ARIYOSHI, T. & IKEDA, T. (1974) On the tissue distribution and the
excretion of arsenic in rats and rabbits of administration with
arsenical compounds. J. Hyg. Chem., 20 (5): 290-295.
ARNOLD, J. P. & JOHNSON, R. M. (1969) Polarography of arsenic.
Talanta, 16: 1191-1207.
ARSENAULT, R. D. (1977) (6) Health aspects of C. C. A. wood
preservatives - a review of arsenates and cancer. Annual
convention, London, British Wood Preservers Association.
ASO, T. & ABIKO, Y. (1978) Tissue distribution of arsenic after
subcutaneous implantation of arsenic trioxide pellet in rats.
J. Toxicol. Sci., 3: 109-116.
ASTRUP, P. (1968) Blackfoot disease. Ugeskr. Laeg., 130: 1807-1815.
ATALLA, L. T., SILVA, C. M. & LIMA, F. W. (1965) Activation analysis
of arsenic in human hair -- some observations on the problem of
external contamination. An. Acad. Bras. Cienc., 37: 431-441.
ATTREP, M., JR. & ANIRUDHAN, M. (1977) Atmospheric inorganic and
organic arsenic. Trace Subst. Environ. Health, 11: 365-369.
AUERMANN, E., SEIDLER, G. & KNEUER, M. (1977) [The assessment of
arsenic in the air.]. Nahrung, 21: 799-806 (in German).
AXELSON, O., DAHLGREN, E., JANSSON, C. -D. & REHNLUND, S. O. (1978)
Arsenic exposure and mortality: a case-referent study from a
Swedish copper smelter. Br. J. ind. Med. 35: 8-15.
AZZONE, S. F. & ERNSTER, L. (1961) Compartmentation of mitochondrial
phosphorylation as disclosed by studies with arsenate. J. Biol.
Chem., 236: 1510-1517.
BAETJER, A. M., LILIENFELD, A. M. & LEVIN, M. L. (1975) Cancer and
occupational exposure to inorganic arsenic. In: Abstracts. 18th
International Congress on Occupational Health, Brighton, England,
14-19 September.
BAILEY, E. J., KENNAWAY, E. L. & URQUHART, M. E. (1957) Arsenic
content of cigarettes. Br. J. Cancer, 11: 49-53.
BAKER, E. L., HAYES, C. G., LANDRIGAN, P. J., HANDKE, J. L., LEGER, R.
T., HOUSWORTH, W. J. & HARRINGTON, J. M. (1977) A national-wide
survey of heavy metal absorption in children living near primary
copper, lead, and zinc smelters. Am. J. Epidemiol.,
106: 261-273.
BALLANTYNE, B. (1978) The comparative short-term mammalian toxicology
of phenarsazine oxide and phenoxarsine oxide. Toxicology,
10: 341-361.
BANG, I. (1919) [Report by the Swedish arsenic commission], volume 11.
In: Lund, H., ed. Ohlssons Boktryckeri, pp. 1-56 (in Swedish).
BARONI, C., VAN ESCH, G. J. & SAFFIOTTI, U. (1963) Carcinogenesis
tests of two inorganic arsenicals. Arch. Environ. Health,
7: 668-674.
BARRON, E. S. G. & SINGER, T. P. (1945) Studies on biological
oxidations. XIX. Sulfhydryl enzymes in carbohydrate metabolism.
J. Biol. Chem., 157: 221-253.
BARRON, E. S. G., MILLER, Z. B., BARTLETT, G. R., MEYER, J. & SINGER,
T. P. (1947) Reactivation by dithiols of enzymes inhibited by
Lewisite. Biochem. J., 41: 69-74.
BARRY, K. G. & HERNDON, E. G., JR. (1962) Electrocardiographic changes
associated with acute arsenic poisoning. Med. Ann. D.C.,
31: 25-27, 65-66.
BARTAK, P. & KEJDA, J. (1972) [Basaloma induced by arsenic in young
skin.] Hautarzt, 23 (10): 457-458.
BEAMISH, F. E. & COLLINS, H. L. (1934) Nickle microbomb for
microestimation of organic arsenic. Ind. Eng. Chem., Sept. 15
(6): 379-380.
BEAUDOIN, A. R. (1974) Teratogenicity of sodium arsenate in rats.
Teratology, 10: 153-158.
BEAVINGTON, F. & CAWSE, P. A. (1978) Comparative studies of trace
elements in air particulate in northern Nigeria. Sci. total
Environ., 10: 239-244.
BENCKO, V. (1972) Oxygen consumption by mouse liver homogenate during
drinking water arsenic exposure. Part II. J. Hyg. Epidemiol.
Microbiol. Immunol., 16: 42-46.
BENCKO, V. & NEMECKOVA, H. (1971) Oxygen consumption by mouse liver
homogenate during drinking water arsenic exposure. Part I.
J. Hyg. Epidemiol. Microbiol. Immunol., 15: 104-110.
BENCKO, V. & SYMON, K. (1969a) Dynamics of arsenic cumulation in
hairless mice after peroral administration. J. Hyg. Epidemiol.
Microbiol. Immunol., 13: 248-253.
BENCKO, V. & SYMON, K. (1969b) Suitability of hairless mice for
experimental work and their sensitivity to arsenic.
J. Hyg. Epidemiol. Microbiol. Immunol., 13: 1-6.
BENCKO, V. & SYMON, K. (1970) The cumulation dynamics in some tissue
of hairless mice inhaling arsenic. Atmos. Environ., 4: 157-162.
BENCKO, V. & SYMON, K. (1977) Health aspects of burning coal with a
high arsenic content. Environ. Res., 13: 378-385.
BENCKO, V., CMARKO, V. & PALAN, S. (1968) The cumulation dynamics of
arsenic in the tissues of rabbits exposed in the area of the ENO
plant. Cesk. Hyg., 13: 18-22.
BENCKO, V., DOBISOVA, A. & MACAJ, M. (1971) Arsenic in the hair of a
nonoccupationally exposed population. Atmos. Environ.,
5: 275-279.
BENCKO, V., DVORAK, V. & SYMON, K. (1973) Organ retention of
parenterally administered arsenic (labelled with 74As) in mice
preliminary exposed to the element in drinking water: A study in
arsenic tolerance. J. Hyg. Epidemiol. Microbiol. Immunol.,
17: 165-168.
BENCKO, V., RÖSSNER, P. & MOKRY, M. (1975) Dehydrogenase activity of
liver parenchyma in mice exposed to arsenic in drinking water.
J. Hyg. Epidemiol. Microbiol. Immunol., 19: 17-21.
BENCKO, V., BENES, B. & CIKRT, M. (1976) Biotransformation of As(III)
to As(V) and arsenic tolerance. Arch. Toxicol., 36: 159-162.
BENCKO, V., SYMON, K., CHLADEK, V. & PIHRT, J. (1977) Health aspects
of burning coal with a high arsenic content. II. Hearing changes
in exposed children. Environ. Res., 13: 386-395.
BENCKO, V., BENES, B. & HAJEK, V. (1978a) The content of free
sulfhydryl groups and the activity of glutathione reductase in the
liver of mice exposed to arsenic in drinking water. Cesk. Hyg.,
23: 209-214.
BENCKO, V., RÖSSNER, P., HAVRANKOVA, H., PUZANOVA, A. & TUCEK, M.
(1978b) Effects of the combined action of selenium and arsenic on
mice versus suspension culture of mice fibroblasts. In: Fouts, J.
R. & Gut, I., ed. Industrial and environmental xenobiotics,
Amsterdam, Excerpta Medica Foundation (International Congress
Series No. 440).
BERGSTRÖM, J. & WESTER, P.O. (1966) The effect of dialysis on the
arsenic content of blood and muscle tissue from uraemic patients.
In: Kerr, D. N.S., Fraeger, J., Frieds, D., & Elliott, R. W., ed.
Replacement of renal function, Amsterdam, Excerpta Medica
Foundation. (International Congress Series No. 131).
BERLIN, C. & TAGER, A. (1962) Psoriasis complicated by cutaneous and
visceral carcinomatosis due to ingestion of arsenic. Acta Derm.
Venereol., 42: 252-255.
BETTLEY, F. R. & O'SHEA, J. A. (1975) The absorption of arsenic and
its relation to carcinoma. Br. J. Dermatol., 92: 563-568.
BINNS, F., ENSOR, R. J. & MACPHERSON, A. L. (1978) Metal content of
United Kingdom and overseas lager beers. J. Sci. Food Agric.,
29: 71-74.
BIRMINGHAM, D. J., KEY, M. M., HOLADAY, D. A. & PERONE, V. B. (1965)
An outbreak of arsenical dermatoses in a mining community.
Arch. Dermatol., 91: 457-464.
BISHOP, R. F. & CHISHOLM, D. (1962) Arsenic accumulation in Annapolis
Valley orchard soils. Can. J. soil Sci., 42: 77.
BLEJER, H. P. & WAGNER, W. (1976) Case study 4: Inorganic arsenic --
ambient level approach to the control of occupational cancerigenic
exposures. Ann. N. Y. Acad. Sci., 271: 179-186.
BLOT, W. J. & FRAUMENI, J. F., Jr (1975) Arsenical air pollution and
lung cancer. Lancet, (July 26): 142-144.
BORGOÑO, J. M. & GREIBER, R. (1972) Epidemiological study of
arsenicism in the city of Antofagasta. In: Hemphill, D. D, ed.
Trace substances in environmental health -- V. A symposium,
Columbia, University of Missouri Press, pp. 13-24.
BORGONO, J. M., VICENT, P., VENTURINO, H. & INFANTE, A. (1977) Arsenic
in the drinking water of the city of Antofagasta: epidemiological
and clinical study before and after the installation of the
treatment plant. Environ. Health Perspect., 19: 103-105.
BOUTWELL, R. K. (1963) A carcinogenicity evaluation of potassium
arsenite and arsanilic acid. J. agric. Food Chem., 11: 381-385.
BOYLEN, G. W. Jr & HARDY, H. L. (1967) Distribution of arsenic in
nonexposed persons (hair, liver, and urine). Am. Ind. Hyg.
Assoc. J., 28: 148-150.
BRAMAN, R. S. (1975) Arsenic in the environment. In: Woolson, E. A.,
ed. Arsenical Pesticides, Washington, DC, American Chemical
Society (ACS Symp. Ser. No. 7).
BRAMAN, R. S. & FOREBACK, C. C. (1973) Methylated forms of arsenic in
the environment. Science, 182: 1247-1249.
BRAMAN, R. S., JOHNSON, D. L., FOREBACK, C. C., AMMONS, J. M. &
BRICKER, J. L. (1977) Separation and determination of nanogram
amounts of inorganic arsenic and methylarsenic compounds.
Anal. Chem., 49: 621-625.
BRIMBLECOMBE, P. (1979) Atmospheric arsenic. Nature (Lond.),
280: 104-105.
BROWN, M. M., RHYNE, B. C., GOYER, R. A. & FOWLER, B. A. (1976)
Intracellular effects of chronic arsenic administration on renal
proximal tubule cells. J. Toxicol. environ. Health, 1: 505-514.
BRUNE, D., SAMSAHL, K. & WESTER, P. O. (1966) A comparison between the
amounts of As, Au, Br, Cu, Fe, Mo, Se and Zn in normal and uraemic
human whole blood by means of neutron activation analysis.
Clin. Chim. Acta, 13: 285-291.
BRUNE, D., NORDBERG, G. & WESTER, P. O. (1980) Distribution of 23
elements in the kidney, liver and lungs of workers from a smeltery
and refinery in North Sweden exposed to a number of elements and
of a control group. Sci. total Environ., 16 (1): 13-35.
BUCHANAN, W. D. (1962) Toxicity of arsenic compounds. In: Browning,
E., ed. Elsevier monographs on toxic agents, New York, Elsevier
Publishing Company.
BUCHET, J. P., LAUWERYS, R. R., & BERLIN, A. (in press)
Interlaboratory comparison of biological parameters used in the
evaluation of occupational exposure to inorganic arsenic.
Report EUR.
BURGDORF, W., KURVINK, K. & CERVENKA, J. (1977) Elevated sister
chromatic exchange rate in lymphocytes of subjects treated with
arsenic. Hum. Genet., 36: 69-72.
BUTZENGEIGER, K. H. (1940) [Peripheral circulation disorders in
arsenism.] Klin. Wochenschr., 19:523-527 (in German).
BUTZENGEIGER, K. H. (1949) [Arsenism. I. ECG changes and other cardiac
and circulatory phenomena. II: Mucosal symptoms and pathogenesis.]
Dtsch. Arch. Klin. Med., 1974: 1-16 (in German).
BYRON, W. R., BIERBOWER, G. W., BROUWER, J. B. & HANSEN, W. H. (1967)
Pathologic changes in rats and dogs from two-year feeding of
sodium arsenite and sodium arsenate. Toxicol. appl. Pharmacol.,
10: 132-147.
CALESNICK, B., WASE, A. & OVERBY, L. R. (1966) Availability during
human consumption of the arsenic in tissues of chicks fed
arsanilic- 74As acid. Toxicol. appl. Pharmacol., 9: 27-30.
CALVERT, C. C. (1973) Feed additive residues in animal manure
processed for feed. Feedstuffs, 45 (17): 32-33.
CALVERT, C. C. (1975) Arsenicals in animal feeds and wastes. In:
Woolson, E. A., ed. Arsenical Pesticides, Washington, DC,
American Chemical Society (ACS Symp. Series No. 7).
CANADIAN PUBLIC HEALTH ASSOCIATION (1977) Task Force on Arsenic,
Yellowknife, Northwest Territories. Final report, Ottawa.
CANADIAN PUBLIC HEALTH ASSOCIATION (1978) Electromyography.
Yellowknife and Hay River, Northwest Territories, Ottawa.
CANNON, J. R., EDMONDS, J. S., FRANCESCONI, K. A. & LANGSFORD, J. B.
(1979) Arsenic in Marine Fauna. In: International Conference.
Management & Control of Heavy Metals in the Environment, London,
September, 1979. Edinburgh, CEP Consultants Ltd.
CARAPELLA, S.C. JR (1973) Arsenic and compounds. In: Hampel, C. A. &
Hawley, G. G., ed. The encyclopaedia of chemistry, third
edition, New York, Van Nostrand Reinhold Company.
CARLSSON, G. (1976) [Correlation between industrial arsenic exposure
and excretion in urine.] Examination report. Stockholm, The
National Board of Industrial Safety, Dept of Occupational Health
(In Swedish).
CARVALHO, M. B. & HERCULES, D. M. (1978) Trace arsenic determination
by volatilization and X-ray photoelectron spectroscopy.
Anal. Chem., 50: 2030-2034.
CHALLENGER, F. (1945) Biological methylation. Chem. Rev., 36: 315.
CHAN, T.-L., THOMAS, B. R. & WADKINS, C. L. (1969) The formation and
isolation of an arsenylated component of rat liver mitochondria.
J. Biol. Chem., 244: 2883-2890.
CHAPMAN, A. C. (1926) On the presence of compounds of arsenic in
marine crustaceans and shell fish. Analyst, 51: 548-563.
CHARBONNEAU, S. M., SPENCER, K., BRYCE, F. & SANDI, E. (1978a) Arsenic
excretion by monkeys dosed with arsenic-containing fish and with
inorganic arsenic. Bull. Environ. Contam. Toxicol., 20: 470-477.
CHARBONNEAU, S. M., TAM, G. K. H., BRYCE, F. & COLLINS, B. (1978b)
Pharmacokinetics and metabolism of inorganic arsenic in the dog.
In: Hemphill, D. D., ed. Trace substances in environmental health
-- XII. A symposium, Columbia, University of Missouri Press,
pp. 276-283.
CHARBONNEAU, S. M., TAM, G. K. H., BRYCE, F., ZAWIDZKA, Z. & SANDI, E.
(1979) Metabolism of orally administered inorganic arsenic in the
dog. Toxicol. Lett., 3: 107-113.
CHARBONNEAU, S. M., HOLLINS, J. G., TAM, G. K. H., BRYCE, F.,
RIDGEWAY, J. M. & WILLES, R. F. (1980) Whole-body retention,
excretion and metabolism of [74As] arsenic acid in the hamster.
Toxicol. Lett., 5 (3-4): 175-182.
CHHUTTANI, P. N., CHAWLA, L. S. & SHARMA, T. D. (1967) Arsenical
neuropathy. Neurology (Minneap.), 17: 269-274.
CHRISTIAN, G. D. & FELDMAN, F. J. (1970) Atomic absorption
spectroscopy; Applications in agriculture, biology, and medicine,
New York, Wiley-Interscience, pp. 188-195.
CHU, R. C., BARRON, G. P. & BAUMGARNER, P. A. W. (1972) Arsenic
determination at sub-microgram levels by arsine evolution and
flameless atomic absorption spectrophotometric technique.
Anal. Chem., 44: 1476-1479.
CHU, T.-Y. J., RUANE, R. J. & KRENKEL, P. A. (1978) Characterization
and reuse of ash pond effluents in coal-fired power plants.
J. Water Pollut. Control Fed., 50: 2494-2508.
CIKRT, M. & BENCKO, V. (1974) Fate of arsenic after parenteral
administration to rats, with particular reference to excretion via
bile. J. Hyg. Epidemiol. Microbiol. Immunol., 18: 129-136.
CIKRT, M., BENCKO, V., TICHY, M. & BENES, B. (1980) Biliary excretion
of 74As and its distribution in the golden hamster after
treatment with 74As(III) and 74As(V). J. Hyg. Epidemiol.
Microbiol. Immunol. 9 (24): 384-388.
CLEMENT, W. H. & FAUST, S. D. (1973) A new convenient method for
determining arsenic (+3) in natural waters. Environ. Lett.,
5: 155-164.
CMARKO, V. (1963) [Hygienic problems of arsenic exhalations of ENO
plant.] Cesk. Hyg., 8: 359-362 (In Slovak, with English
summary).
COLE, M., SCHEULEIN, M. & KERWIN, D. M. (1966) Arsenical
encephalopathy due to use of Milibis. Arch. intern. Med.,
177: 706-711.
COONEY, R. V., MUMMA, R. O. & BENSON, A. A. (1978)
Arsoniumphospholipid in algae. Proc. Natl Acad. Sci.,
75: 4262-4264.
CORNELIS, R. (1973) Neutron activation analysis of hair failure of a
mission. J. radioanal. Chem., 15: 305-316.
CORRIDAN, J. P. (1974) Head hair samples as indicators of
environmental pollution. Environ. Res., 8: 12-16.
COULSON, E. J., REMINGTON, R. E. & LYNCH, K. M. (1935) Metabolism in
the rat of the naturally occurring arsenic of shrimp as compared
with arsenic trioxide. J. Nutr., 10: 255-270.
CRANE, R. K. & LIPMANN, F. (1953) The effect of arsenate on aerobic
phosphorylation. J. biol. Chem., 201: 235-243.
CRAWFORD, T. B. B. & STOREY, I. D. E. (1944) Quantitative micro-method
for the separation of inorganic arsenite from arsenate in blood
and urine. Biochem. J., 38: 195-198.
CRECELIUS, E. A. (1974) The geochemistry of arsenic and antimony in
Puget Sound and Lake Washington, Washington, Thesis, Seattle,
Washington, University of Washington.
CRECELIUS, E. A. (1977a) Arsenite and arsenate levels in wine.
Bull. environ. Contam. Toxicol., 18: 227-230.
CRECELIUS, E. A. (1977b) Changes in the chemical speciation of arsenic
following ingestion by man. Environ. health Perspect.,
19: 147-150.
CRECELIUS, E. A. (1978) Modification of the arsenic speciation
technique using hydride generation. Anal. Chem., 50: 826-827.
CRECELIUS, E. A., ROBERTSON, D. E., FRUCHTER, J. S. & LUDWICK, J. D.
(1976) Chemical forms of mercury and arsenic emitted by a
geothermal power plant. In: Hemphill, D. D., ed. Trace substances
in environmental health -- X, Columbia, University of Missouri
Press, pp. 287-293.
CREMA, A. (1955) Distribution et élimination de l'arsenic 76 chez la
souris normale et cancéreuse. Arch. int. Pharmacodyn.,
103: 57-70.
CRISTAU, B., PLACIDI, M. & AUDIBERT, P. (1972) Voies et cinétiques
d'élimination de l'arsenic chez le rat après administration de
médicaments organoarséniés. Med. trop., 32(3): 275-283.
CRISTAU, B., CHABAS, E. & PLACIDI, M. (1975) Voies et cinétiques
d'excrétion de l'arsenic chez le Cobaye après injection de divers
médicaments organo-arséniés. Ann. Pharm. Fr., 33: 577-589.
CULLEN, W. R., FROESE, C. L., LUI, A., McBRIDE, B. C., PATMORE, D. J.
& REIMER, M. (1977) The aerobic methylation of arsenic by
micro-organisms in the presence of L-methionine-methyl-d3.
J. organomet. Chem., 139: 61-69.
CURRY, A. S. & POUNDS, C. A. (1977) Arsenic in hair. J. Forensic Sci.
Soc., 17: 37-44.
DAGHIR, N. J. & HARIRI, N. N. (1977) Determination of total arsenic
residues in chicken eggs. J. agric. Food Chem., 25: 1009-1010.
DAMSGAARD, E., HEYDORN, K., LARSEN, N. A. & NIELSEN, B. (1973)
Simultaneous determination of arsenic, manganese and selenium in
human serum by neutron activation analysis, Roskilde, Danish
Atomic Energy Commission (Riso Report N:o 271).
DAVIS, P. H., DULUDE, G. R., GRIFFIN, R. M., MATSON, W. R. & ZINK, E.
W. (1978) Determination of total arsenic at the nanogram level by
highspeed anodic stripping voltammetry. Anal. Chem, 50: 137-143.
DAVIS, W. E. et al. (1971) In: Medical and biologic effects of
environmental pollutants: Arsenic, Washington, DC, National
Academy of Sciences.
DAY, G. T. (1964) Analysis of tungstic oxide by the conducting briquet
technique. Can. Spectrom., 9: 118-120.
DEAK, S. T., CSAKY, K. G. & WADDELL, W. J. (1976) Localization and
histochemical correlation, of 73As by whole-body autoradiography
in mice. J. Toxicol. environ. Health, 1: 981-984.
DE NAVARRO, E. T. (1976) [Arsenic poisoning in cattle.] Salud publica
mex., 18(6): 1037-1044 (in Spanish).
DENG, J.-S. & HOW, S.-W. (1977) Comparison of the fine structure of
basal cell carcinomas in ordinary patients and patients with
chronic arsenicalism. J. Formosan Med. Assoc., 76: 685-692.
DICKINSON, J. O. (1972) Toxicity of the arsenical herbicide monosodium
acid methanearsonate in cattle. Am. J. vet. Res., 33: 1889-1892.
DIEKE, S. H. & RICHTER, C. P. (1946) Comparative assays of
rodenticides on wild Norway rats. I. Toxicity. Publ. Health Rep.,
61: 672-679.
DOAK, G. O. & FREEDMAN, L. D. (1970) Organometallic compounds of
arsenic, antimony, and bismuth. In: Seyferth, D., ed.
The chemistry of organometallic compounds. A series of
monographs, New York, John Wiley & Sons.
DOBBS, A. J., PHIL, D. & GRANT, C. (1976) Report on the burning of
wood treated with wood preservatives containing copper, chromium
and arsenic, Aylesbury, Buckinghamshire, UK, Building Research
Establishment (Report Cp 63/73).
DONE, A. K. & PEART, A. J. (1971) Acute toxicities of arsenical
herbicides. Toxicol., 4: 343-355.
DUCE, R. A., HOFFMAN, G. L. & ZOLLER, W. H. (1975) Atmospheric trace
metals at remote northern and southern hemisphere sites; pollution
or natural? Science, 187: 59-61.
DUCOFF, H. S., NEAL, W. B., STRAUBE, R. L., JACOBSON, L. O. & BRUES,
A. M. (1948) Biological studies with arsenic76. II. Excretion and
tissue localization. Proc. Soc. Exp. Biol. Med., 69: 548-554.
DU PONT, O., ARIEL, I. & WARREN, S. L. (1941) The distribution of
radioactive arsenic in the normal and tumor-bearing (Brown-Pearce)
rabbit. Am. J. Syph. Gon. vener. Dis., 26: 96-118.
DURRANT, P. J. & DURRANT, B. (1966) Introduction to advanced
inorganic chemistry, third edition, London, Longmans, Green and
Co. Ltd.
DURUM, W. H., HEM, J. D. & HEIDEL, S. G. (1971) Reconnaissance of
selected minor elements in surface waters of the United States,
October 1970, Washington, DC, US Department of Interior
(Geological Survey Circular 643).
DUTKIEWICZ, T. (1977) Experimental studies on arsenic absorption
routes in rates. Environ. health Perspect., 19: 173-177.
EDMONDS, J. S., FRANCESONI, K. A., CANNON, J. R., RASTON, C. L.,
SKELTON, B. W. & WHITE, A. H. (1977) Isolation, crystal structure
and synthesis of arsenobetaine, the arsenical constituent of the
western rock lobster Panulirus Longipes Cygnus George.
Tetrahedron Lett., 18: 1543-1546.
ELTON, R. K. & GEIGER, W. E. (1978) Analytical and mechanistic studies
of the electrochemical reduction of biologically active
organoarsenic acids. Anal Chem., 50: 712-717.
ENGSTRÖM, B. M. & NORDBERG, G. F. (1978) Effects of milk diet on
gastrointestinal absorption of cadmium in adult mice. Toxicology,
9: 195-203.
ESPINOSA GONZALEZ, E. (1963) [Mass arsenic poisoning in Terreón,
Coahuila State, Mexico.] Bol. Epidemiol., 27(4): 213-220
(in Spanish).
EVANS, R. J. & BANDEMER, S. L. (1954) Determination of arsenic in
biological materials. Anal. Chem., 26: 595.
EXON, J. H., HARR, J. R. & CLAEYS, R. R. (1974) The effects of long
term feeding of monosodium acid methanearsenate (MSMA) to rabbits.
Nutr. Rep. Int., 9: 351-357.
FAIRHALL, L. T. & MILLER, J. W. (1941) A study of the relative
toxicity of the molecular components of lead arsenate.
Publ. Health Rep., 56: 1610-1625.
FELDMAN, C. (1979) Improvements in the arsine accumulation -- helium
glow detector procedure for determining traces of arsenic.
Anal. Chem., 51: 664-669.
FELDMAN, C. & BATISTONI, D. A. (1977) Spectroscopic element detector
for gas chromatography. Anal Chem., 49: 2215-2221.
FERGUSON, J. F. & GAVIS, J. (1972) A review of the arsenic cycle in
natural waters. Water Res, 6: 1259-1274.
FERM, V. H. (1977) Arsenic as a teratogenic agent. Environ. health
Perspect., 19: 215-217.
FERM, V. H. & CARPENTER, S. J. (1968) Malformations induced by sodium
arsenate. J. Reprod. Fer., 17: 199-201.
FERM, V. H., SAXON, A. & SMITH, B. M. (1971) The teratogenic profile
of sodium arsenate in the golden hamster. Arch. environ. Health,
22: 557-560.
FERSLEW, K. E. & EDDS, G. T. (1979) Effects of arsanilic acid on
growth, serum enzymes, hematologic values, and residual arsenic in
young swine Am. J. vet. Res., 40: 1365-1369.
FEUSSNER, J. R., SHELBURNE, J. D., BREDEHOEFT, S., & COHEN, H. J.
(1979) Arsenic-induced bone marrow toxicity: Ultrastructural and
electron probe analysis. Blood, 53: 820-827.
FIERZ, U. (1965) [Catamnestic research into the side effects of
inorganic arsenotherapy in skin diseases.] Dermatologica,
131: 41-58 (in German).
FISHMAN, M. & SPENCER, R. (1977) Automated atomic absorption
spectrometric determination of total arsenic in water and
streambed materials. Anal. Chem., 49: 1599-1602.
FLETCHER, M. J. & SANADI, D. R. (1962) On the mechanism of oxidative
phosphorylation. III. Effects of arsenite and
2,3-dimercaptopropanol on coupled phosphorylation in heart
mitochondria. Arch. Biochem. Biophys., 96: 139-142.
FLIS, I. E., MISHCHENKO, K. P., TUMANOVA, T. A. & RUSS, J. (1959)
Dissociation of arsenic acid. J. inorg. Chem., 4: 120-124.
FLUHARTY, A. L. & SANADI, D. R. (1960) Evidence for a vicinal dithiol
in oxidative phosphorylation. Proc. Natl Acad. Sci.,
46: 608-616.
FLUHARTY, A. L. & SANADI, D. R. (1961) On the mechanism of oxidative
phosphorylation. II. Effects of arsenite alone and in combination
with 2,3-dimercaptopropanol. J. biol. Chem., 236: 2772-2778.
FOWLER, B. A. (1975) The ultrastructural and biochemical effects of
arsenate on the kidney. (Abstract) In: Proceedings of the XVIII
International Congress on Occupational Health, Brighton.
FOWLER, B. A. & MAHAFFEY, K. R. (1978) Interactions between lead,
cadmium and arsenic in relation to porphyrin excretion patterns.
Environ. health Perspect., 25: 87-90.
FOWLER, B. A., WOODS, J. S. & SCHILLER, C. M. (1977) Ultrastructural
and biochemical effects of prolonged oral arsenic exposure on
liver mitochondria of rats. Environ. health Perspect.,
19: 197-204.
FRANKE, K. W. & MOXON, A. L. (1936) A comparison of the minimum fatal
doses of selenium, tellurium, arsenic and vanadium. J. Pharmacol.
exp. Ther., 58: 454-459.
FRANKLIN, M., BEAN, W. B. & HARDIN, R. C. (1950) Fowler's solution as
an etiologic agent in cirrhosis. Am. J. med. Sci., 219: 589-596.
FREEMAN, H. C., UTHE, J. F., FLEMING, R. B., ODENSE, P. H., ACKMAN, R.
G., LANDRY, G. & MUSIAL, C. (1979) Clearance of arsenic ingested
by man from arsenic contaminated fish. Bull. environ. Contam.
Toxicol., 22: 224-229.
FRIBERG, L. & CEDERLÖF, R. (1978) Late effects of air pollution with
special reference to lung cancer. Environ. health Perspect.,
22: 45-66.
FRIBERG, L. F., PISCATOR, M. & NORDBERG, G. F. (1974) Cadmium in the
environment, 2nd edition, Cleveland, CRC Press.
FRIEDEWALD, W. F. & ROUS, P. (1944) The determining influence of tar,
benzpyrene, and methylcholanthrene on the character of the benign
tumors induced therewith in rabbit skin. J. exp. Med.,
80: 127-144.
FROST, D. V., PERDUE, H. S., MAIN, B. T., KOLAR, J. A., SMITH, I. D.,
STEIN, R. J. & OVERBY, L. R. (1962) Further considerations on the
safety of arsanilic acid for feed use. In: Proceedings of the
Twelfth World's Poultry Congress, Sydney, pp. 234-237.
FROST, D. V., MAIN, B. T., COLE, J., SANDERS, P. G. & PERDUE, H. S.
(1964) Reproduction studies in rats with arsanilic acid.
Fed. Proc. Fed. Am. Soc. Exp. Biol., 23: 291.
FURR, A. K., KELLY, W. C., BACHE, C. A., GUTENMANN, W. H. & LISK,
D. J. (1976) Multielement absorption by crops grown in pots on
municipal sludge-amended soil. J. agric. Food Chem.,
24: 889-892.
FURR, A. K., PARKINSON, T. F., HINRICHS, R. A., VAN CAMPEN, D. R.,
BACHE, C. A. GUTENMANN, W. H., T S. JOHN, L. E. JR, PAKKALA, I. S.
& LISK, D. J. (1977) National survey of elements and radioactivity
in fly ashes. Absorption of elements by cabbage grown in fly
ash-soil mixtures. Environ. Sci. Technol., 11: 1194-1201.
GABOR, S. & COLDEA, V. (1977) Some aspects of the environmental
exposure to arsenic in Romania. Environ. health Perspect.,
19: 107-108.
GAINER, J. H. (1972) Effects of arsenicals on interferon formation and
action. Am. J. vet. Res., 33: 2579-2586.
GAINER, J. H. & PRY, T. W. (1972) Effects of arsenicals on viral
infections in mice. Am. J. vet. Res., 33: 2299-2307.
GALLORINI, M., GREENBERG, R. R. & GILLS, T. E. (1978) Simultaneous
determination of arsenic, antimony, cadmium, chromium, copper, and
selenium in environmental material by radiochemical neutron
activation analysis. Anal. Chem., 50: 1479-1481.
GALY, P., TOURAINE, R., BRUNE, J., ROUDIER, P. & GALLOIS, P. (1963) Le
cancer pulmonaire d'origine arsénicale des vignerons du
Beaujolais. J. Fr. Med. Chir. thorac., 17: 303-311.
GARB, L. G. & HINE, C. H. (1977) Arsenical neuropathy: Residual
effects following acute industrial exposure. J. occup. Med.,
19(8): 567-568.
GASTINER, E. (1972) [On the spectrophotometric determination of
arsenic using silves diethyl-thiocarbamidate.] Mikrochim. Acta,
1972: 526-543 (in German).
GEYER, L. (1898) [Chronic skin changes in arsenism and reflections on
the great number of cases at Reichehnstein, Silesia.] Arch.
Dermatol. Syphilol., 43: 221-280 (in German).
GINSBURG, J. M. (1965) Renal mechanism for excretion and
transformation of arsenic in the dog. Am. J. Physiol.,
208: 832-840.
GINSBURG, J. M. & LOTSPEICH, W. D. (1963) Interrelations of arsenate
and phosphate transport in the dog kidney. Am. J. Physiol.,
205: 707-714.
GOLDBLATT, E. L., VANDENBURGH, A. S. & MARSLAND, R. A. (1963)
The usual and widespread occurrence of arsenic in well waters
of Lane county, Oregon, Oregon Department of Health.
GOLDMAN, A. L. (1973) Lung cancer in Bowen's disease. Am. Rev.
respir. Dis., 108: 1205-1207.
GOLDSMITH, J. R., DEANE, M., THOM, J. & GENTRY, G. (1972) Evaluation
of health implications of elevated arsenic in well water. Water
Res., 6: 1133-1136.
GONZALEZ, S. G. (1977) [Acute arsenic poisoning in dairy cattle in the
Lagoon Region.] In: Memorias del I Simposium International de
Laboratorios Veterinarios de Diagnósticos, México, Volume III,
pp. 551-560 (in Spanish).
GOODE, S. R. & MATTHEWS, R. J. (1978) Enzyme -- catalyzed
reaction-rate method for determination of arsenic in water.
Anal. Chem., 50: 1608-1610.
GORDON, J. J. & QUASTEL, J. H. (1948) Effects of organic arsenicals on
enzyme systems. Biochem. J., 42: 337-350.
GOSSELIN, R. E., HODGE, H. C., SMITH, R. P. & GLEASON, M. N. (1976)
Clinical toxicology of commercial products, Baltimore, The
Williams & Wilkins Co.
GOULDEN, F., KENNAWAY, E. L. & URQUHART, M. E. (1952) Arsenic in the
suspended matter of town air. Br. J. Cancer, 6: 1-7.
GRANT, C. & DOBBS, A. J. (1977) The growth and metal content of plants
grown in soil contaminated by a copper/chrome/arsenic wood
preservative. Environ. Pollut., 14: 213-226.
GRANTHAM, D. A. & JONES, J. F. (1977) Arsenic contamination of water
wells in Nova Scotia. J. Am. Water Works Assoc., 69: 653-657.
GRIFFIN, H. R., HOCKING, M. B. & LOWERY, D. G. (1975) Arsenic
determination in tobacco by atomic absorption spectrometry.
Anal. Chem., 47: 229.
GRIMANIS, A. P., VASSILAKI-GRIMANI, M., ALEXIOU, D. & PAPADATOS, C.
(1979) Determination of seven trace elements in human milk,
powdered cow's milk and infant foods In: Proceedings of a
Symposium. Nuclear Activation Techniques in the Life Sciences,
Vienna 1978, Vienna, International Atomic Energy Agency,
pp. 241-253.
GROBE. J.-W. (1976) [Peripheral circulatory disorders and acrocyanosis
in Moselle valley vineyard workers with arsenic poisoning.]
Berufsdermatosen, 24(3): 78-84.
GULLEDGE, J. H. & O'CONNOR, J. T. (1973) Removal of arsenic(V) from
water by adsorption on aluminum and ferric hydroxides. J. Am.
Water Works Assoc., 65: 548.
HALVER, J. E. (1962) Cited in: KRAYBILL, H. F. & SHIMKIN, M. B. (1964)
Carcinogenesis related to foods contaminated by processing and
fungal metabolites. Adv. Cancer Res., 8: 205.
HAMADA, T. & HORIGUCHI, S. (1976) Occupational chronic arsenical
poisoning. On the cutaneous manifestations. Jpn. J. ind. Health,
18: 103-115.
HAMAMOTO, E. (1955) Infant arsenic poisoning by powdered milk. Nihon
Iji Shimpo, No. 1649, pp. 3-12 (in Japanese).
HAMILTON, E. I. & MINSKI, M. J. (1973) Abundance of the chemical
elements in man's diet and possible relations with environmental
factors. Sci. total Environ., 1: 375-394.
HAMMER, D. I., FINKLEA, J. F., HENDRICKSON, R. H., SHY, C. M. &
HORTON, R. J. M. (1971) Hair trace metal levels and environmental
exposure. Am. J. Epidemiol., 93: 84-92.
HANLON, D. P. & FERM, V. H. (1977) Placental permeability of arsenate
ion during early embryogenesis in the hamster. Experientia
(Basle), 33: 1221-1222.
HARA, I., HASHIMOTO, K., MIYAZAKI, K. & SUNADA, K. (1968) A case
report on multiple neuritis arsenic poisoning. Saigai Igaku,
11: 84-90.
HARRINGTON, J. M., MIDDAUGH, J. P., MORSE, D. L., & HOUSWORTH, J.
(1978) A survey of a population exposed to high concentrations of
arsenic in well water in Fairbanks, Alaska. Am. J. Epidemiol.,
108: 377-385.
HARRIS, E. J. & ACHENJANG, F. M. (1977) Energy-dependent uptake of
arsenite by rat liver mitochondria. Biochem. J., 168: 129-132.
HARRISON, J. W. E., PACKMAN, E. W. & ABBOTT, D. D. (1958) Acute oral
toxicity and chemical and physical properties of arsenic
trioxides. A. M. A. Arch. ind. Health, 17: 118-123.
HAZRA, A. K. & PROKUPOK, R. (1977) A report on air quality in
Yellowknife, Northwest Territories, Environment, Canada
(Surveillance report EPS-5-NW-77-55).
HEYDORN, K. (1969) Environmental variation of arsenic levels in human
blood determined by neutron activation analysis. Clin. Chim.
Acta, 28: 349-357.
HEYDORN, K. & DAMSGAARD, E. (1973) Simultaneous determination of
arsenic, manganese and selenium in biological materials by
neutron-activation analysis. Talanta, 20: 1-11.
HEYMAN, A., PFEIFFER, J. B., JR., WILLETT, R. W. & TAYLOR, H. M.
(1956) Peripheral neuropathy caused by arsenical intoxication. A
study of 41 cases with observations on the effects of BAL
(2,3-dimercaptopropanol). New Engl. J. Med., 254: 401-409.
HEYWOOD, R. & SORTWELL, R. J. (1979) Arsenic intoxication in the
rhesus monkey. Toxicol. Lett., 3: 127-144.
HILL, A. B. & FANING, E. L. (1948) Studies in the incidence of cancer
in a factory handling inorganic compounds of arsenic. I. Mortality
experience in the factory. Br. J. ind. Med., 5: 1-6.
HINDMARSH, J. T., MCLETCHIE, O. R., HEFFERNAN, L. P. M., HAYNE, O. A.,
ELLENBERGER, H. A., MCCURDY, R. F. & THIEBAUX, H. J. (1977)
Electromyographic abnormalities in chronic environmental
arsenicalism. J. anal. Toxicol., 1: 270-276.
HINE, C. H., PINTO, S. S. & NELSON, K. W. (1977) Medical problems
associated with arsenic exposure. J. occup. Med., 19: 391-396.
HJERN, L. (1961) [The arsenic content in Swedish cigarettes.] Nord.
Hyg. Tidskr., 42: 272-276 (in Swedish).
HOCKING, D., KUCHAR, P., PLAMBECK, J. A. & SMITH, R. A. (1978) The
impact of gold smelter emissions on vegetation and soils of a
subarctic forest-tundra transition ecosystem. J. Air Pollut.
Control Assoc., 28: 133-137.
HODGE, H. C. & EMBREE, J. W. (1977) Estimation of the mutagenicity of
arsenic compounds utilizing dominant lethal mutations in mice,
San Francisco, University of California Toxicology Research
Laboratory (Final report, Contract No. SERA-HI-2).
HOGAN, R. B. & EAGLE, H. (1944) The pharmacologic basis for the widely
varying toxicity of arsenicals. J. Pharmacol. exp. Ther.,
80: 93-113.
HOLAK, W. (1969) Gas-sampling technique for arsenic determination by
atomic absorption spectroscopy. Anal. Chem., 41: 1712-1713.
HOLLAND, R. H. & ACEVEDO, A. R. (1966) Current status of arsenic in
American cigarettes. Cancer, 19: 1248-1250.
HOLLAND, R. H., MCCALL, M. S. & LANZ, H. C. (1959) A study of inhaled
arsenic-74 in man. Cancer Res., 19: 1154-1156.
HOLLINS, J. G., CHARBONNEAU, S. M., BRYCE, F., RIDGEWAY, J. M., TAM,
G. K. H. & WILLES, R. F. (1979) Whole body retention and excretion
of [74As] arsenic acid in the adult beagle dog. Toxicol. Lett.,
4: 7-13.
HOLMBERG, R. E. & FERM, V. H. (1969) Interrelationships of selenium,
cadmium, and arsenic in mammalian teratogenesis. Arch. environ.
Health, 18: 873-877.
HOLMQVIST, I. (1951) Occupational arsenical dermatitis. A study among
employees at a copper ore smelting work including investigations
of skin reactions to contact with arsenic compounds. Acta Derm.
Venereol., 31: (Suppl. 26): 1-214.
HOLMQVIST, I. (1964) [Causes of death among workers at
Rönnskärsverken.] Internal report to the management, Boliden AB,
Skelleftehamn, Sweden (in Swedish).
HOLMQVIST, I. (1975) Investigations of the absorption of some metals
among people in the surrounding area of a smelting plant. In:
Proceedings from Recent Advances in the Assessment of the Health
Effects of Environmental Pollution. Vol. II. Paris, June 24-28,
1974, Luxembourg, Commission of the European Communities
Directorate General for Dissemination of Knowledge, Center for
Information and Documentation CID.
HOOD, R. D. (1972) Effects of sodium arsenite on fetal development.
Bull. environ. Contam. Toxicol., 7: 216-222.
HOOD, R. D. & BISHOP, S. L. (1972) Teratogenic effects of sodium
arsenate in mice. Arch. environ. Health, 24: 62-65.
HOOD, R. D. & PIKE, C. T. (1972) BAL alleviation of arsenate-induced
teratogenesis in mice. Teratology, 6: 235-237.
HOOD, R. D., THACKER, G. T. & PATTERSON, B. L. (1977) Effects in the
mouse and rat of prenatal exposure to arsenic. Environ. health
Perspect., 19: 219-222.
HORIGUCHI, S., NAKANO, H., SHINAGAWA, K., TERAMOTO, K., & KIYOTA, I.
(1976) A long-term observation of the environmental conditions of
a factory manufacturing lead arsenate as an insecticide. (Studies
on lead arsenate poisoning, part I). Osaka City med. J.,
22(1): 43-46.
HOVE, E., ELVEHJEM, C. A. & HART, E. B. (1938) Arsenic in the
nutrition of the rat. Am. J. Physiol., 124: 205-212.
HUEPER, W. C. (1954) Experimental studies in metal cancerigenesis.
VI. Tissue reactions in rats and rabbits after parenteral
introduction of suspensions of arsenic, beryllium, or asbestos in
lanolin. J. Natl Cancer Inst., 15: 113-124.
HUEPER, W. C. & ITAMI, S. (1933) Effects of neoarsphenamine on
spontaneous breast tumors of mice. Am. J. Cancer., 17: 106-115.
HUEPER, W. C. & PAYNE, W. W. (1962) Experimental studies in metal
carcinogenesis. Chromium, nickel, iron, arsenic. Arch. environ.
Health, 5: 445-462.
HUET, P.-M., GUILLAUME, E., COTÉ, J., LÉGARÉ, A., LAVOIE, P. &
VIALLET, A. (1975) Noncirrhotic presinusoidal portal hypertension
associated with chronic arsenic intoxication. Gastroenterology,
68: 1270-1277.
HUNDEIKER, M. & PETRES, J. (1968) [Morphogenesis and variety of
procancerous lesions induced by arsenic.] Arch. klin. exp.
Dermatol., 231: 355-365 (in German).
HUNDLEY, H. K. & UNDERWOOD, J. C. (1970) Determination of total
arsenic in total diet samples. J. Assoc. Off. Anal. Chem.,
53: 1176-1178.
HUNTER, F. T., KIP, A. F. & IRVINE, J. W., Jr (1942) Radioactive
tracer studies on arsenic injected as potassium arsenite.
J. Pharmacol. exp. Ther., 76: 207-220.
HUTCHINSON, J. (1888) On some examples of arsenic-keratoses of the
skin and of arsenic-cancer. Trans. Pathol. Soc. (London),
39: 352-363.
HWANG, S. W. & SCHANKER, L. S. (1973) Absorption of organic arsenical
compounds from the rat small intestine. Xenobiotica, 3: 351-355.
IARC (1973) Monographs. Evaluation of carcinogenic risk of chemicals
to man. Some inorganic and organometallic compounds, Volume 2,
Lyons, International Agency for Research on Cancer.
IARC (1980) Monographs. Arsenic and its compounds, Volume 23, Lyons,
International Agency for Research on Cancer, pp. 39-141.
ICRP (1975) Task group on reference man. Oxford, Pergammon Press
(International Commission on Radiological Protection Publication
No. 23).
IRGOLIC, K. J., WOOLSON, E. A., STOCKTON, R. A., NEWMAN, R. D.,
BOTTINO, N. R., ZINGARO, R. A., KEARNEY, P. C., PYLES, R. A.,
MAEDA, S., MCSHANE, W. J. & COX, E. R. (1977) Characterization of
arsenic compounds formed by Daphnia magna and Tetraselmis chuii
from inorganic arsenate Environ. health Perspect., 19: 61-66.
IRLG, Work Group on Risk Assessment (1979) Scientific bases for
identifying potential carcinogens and estimating their risks,
Washington, DC, Interagency Regulatory Liaison Group (IRLG).
ISHINISHI, N. (1973) Review on toxicity of arsenic and arsenic
compounds. Nippon Rinsho, 31(6): 75-83.
ISHINISHI, N., KODAMA, Y. & INAMAZ. U. T. (1974) Clinical examination
of the effects of arsenic for the health. Rynsho-Kagaku,
3: 176-187.
ISHINISHI, N., OSATO, K., KODAMA, Y. & KUNITAKE, E. (1976) Skin
effects and carcinogenicity of arsenic trioxide: A preliminary
experimental study in rats. In: Nordberg, G. F., ed. Effects and
dose-response relationships of toxic metals, Amsterdam, Elsevier
Scientific Publishing Company, pp. 471-479.
ISHINISHI, N., KODAMA, Y., NOBUTOMO, K. & HISANAGA, A. (1977)
Preliminary experimental study on carcinogenicity of arsenic
trioxide in rat lung. Environ. health Perspect., 19: 191-196.
ISHINISHI, N., MIZUNOE, M., INAMASU, T. & HISANAGA, A. (1980a)
Experimental study on carcinogenicity of beryllium oxide and
arsenic trioxide to the lung of rats by an intratracheal
instillation. Fukuoka Acta Med., 71: 19-26.
ISHINISHI, N., TOMITA, M. & HISANAGA, A. (1980b) Study on chronic
toxicity of arsenic trioxide in rats with special reference to the
liver damages. Fukuoka Acta Med., 71: 27-40.
IVANKOVIC, S., EISENBRAND, G. & PREUSSMANN, R. (1979) Lung carcinoma
induction in BD rats after single intratracheal instillation of an
arsenic-containing pesticide mixture formerly used in vineyards.
Int. J. Cancer, 24: 786-789.
JACKSON, R. & GRAINGE, J. W. (1975) Arsenic and cancer. Can. Med.
Assoc. J., 113: 396-401.
JELINEK, C. F. & CORNELIUSSEN, P. E. (1977) Levels of arsenic in the
United States food supply. Environ. health Perspect., 19: 83-87.
JENKINS, R. B. (1966) Inorganic arsenic and the nervous system.
Brain, 89: 479-498.
JERNELÖV, A., HULTBERG, H. & ROSENBLUM, I. (1976) The geothermal
plant at Ahuachapan in El Salvador and its possible effects on
the water quality on the border river Rio Paz between El Salvador
and Guatemala, Stockholm, Institute for Water and Air Pollution
(Report No. IVL-B-297).
JILER, H., ed. (1975) Commodity year book 1975, New York, Commodity
Research Bureau.
JOHNSON, D. L. (1972) Bacterial reduction of arsenate in sea water.
Nature (Lond.), 240: 44-45.
JOHNSON, D. L. & BRAMAN, R. S. (1975a) Alkyl- and inorganic arsenic in
air samples. Chemosphere, 6: 333-338.
JOHNSON, D. L. & BRAMAN, R. S. (1975b) The speciation of arsenic and
the content of germanium and mercury in members of the Pelagic
Sargassum community. Deep-sea Res., 22: 503.
JOHNSON, D. L. & BURKE, R. M. (1978) Biological mediation of chemical
speciation II arsenate reduction during marine phytoplankton
bloom. Chemosphere, 8: 645-648.
JOHNSON, D. L. & PILSON, M. E. Q. (1972) Spectrophotometric
determination of arsenite, arsenate and phosphate in natural
waters. Anal. Chim. Acta, 48: 289.
JOHNSON, D. L. & PILSON, M. E. Q. (1975) The oxidation of arsenite in
seawater. Environ. Lett., 8: 157-171.
JOHNSTONE, R. M. (1963) Sulfhydryl agents: Arsenicals. In: Hochster,
R. M. & Quastel, J. H., ed. Metabolic inhibitors. A comprehensive
treatise, Volume 2, New York, Academic Press.
JUNG, E. (1971) [Research in the molecular biology of arsenism.]
Z. Haupt. Geschlechtskr., 46: 35-36 (in German).
KAAKINEN, J. W., JORDEN, R. M., LAWASANI, M. H. & WEST, R. E. (1975)
Trace element behavior in coal-fired power plant. Environ. Sci
Technol., 9: 862-869.
KADOWAKI, K. (1960) [Studies on the arsenic contents in organ-tissues
of the normal Japanese.] Osaka City med. J., 9: 2083-2099
(in Japanese with English summary).
KAGAWA, Y. & KAGAWA, A. (1969) Accumulation of arsenate-76 by
mitochondria. J. Biochem., 65(1): 105-112.
KAGEY, B. T., BUMGARNER, J. E. & CREASON, J. P. (1977) Arsenic levels
in maternal-fetal tissue sets. In: Hemphill, D. D., ed. Trace
substances in environmental health XI. A symposium. Columbia,
University of Missouri Press, pp. 252-256.
KANISAWA, M. & SCHROEDER, H. A. (1969) Life term studies on the effect
of trace elements on spontaneous tumors in mice and rats. Cancer
Res., 29: 892-895.
KATSURA, K. (1958) Medicolegal studies of arsenic poisoning. II.
Distribution of arsenic in visceral organs and arsenic
concentrations of bone and hair in arsenic poisoning. Shikoku
Igaku Zasshi, 12: 706-720.
KELLO, D. & KOSTIAL, K. (1973) The effect of milk diet on lead
metabolism in rats. Environ. Res., 6: 355-360.
KELLO, D. & KOSTIAL, K. (1977) Influence of age and milk diet on
cadmium absorption from the gut. Toxicol. appl. Pharmacol.,
40: 277-282.
KENNEDY, V. S. (1976) Arsenic concentrations in some coexisting marine
organisms from Newfoundland and Labrador. J. Fish. Res. Board
Can., 33: 1388-1393.
KINGSLEY, G. R. & SCHAFFERT, R. R. (1951) Microdetermination of
arsenic and its application to biological materials. Anal. Chem.,
23: 914.
KIRKBRIGHT, G. F. & RANSON, L. (1971) Use of the nitrous oxide --
acetylene flame for determination of arsenic and selenium by
atomic absorption spectrometry. Anal. Chem., 43: 1238-1241.
KIRKBRIGHT, G. F., WARD, A. F. & WEST, T. S. (1973) Atomic emission
spectrometry with an induction -- coupled high frequency plasma
source. The determination of iodine, mercury, arsenic and
selenium. Anal. Chim. Acta, 64: 353.
KLAASSEN, C. D. (1974) Biliary excretion of arsenic in rats, rabbits,
and dogs. Toxicol. appl. Pharmacol., 29: 447-457.
KNOLLE, J., FÖRSTER, E., RÖSSNER, A., THEMANN, H., HÖHN, P. & MEYER
ZUM BÜSCHENFELDE, K.-H. (1974) [Non-cirrhotic portal fibrosis
(hepatoportal sclerosis) following arsenism.] Dtsch. Med.
Wochenschr., 99: 903-908 (in German).
KODAMA, Y., ISHINISHI, N., KUNITAKE, E., INAMASU, T. & NOBUTOMO, K.
(1976) Subclinical signs of the exposure to arsenic in a copper
refinery. In: Nordberg, G. F., ed. Effects and dose-response
relationships of toxic metals, Amsterdam, Elsevier Scientific
Publishing Company, pp. 464-470.
KOELSCH, F. (1958) [Occupational arsenic lesions in vineyards and
other places of employment.] Arch. Gewerbepathol. Gewerbehyg.,
16: 405-438 (in German).
KONINGS, A. W. T. (1972) Inhibition of nuclear and mitochondrial
respiration by arsenite. Experientia (Basel), 28: 883-884.
KOPP, J. F. (1973) l-Ephedrine in chloroform as a solvent for silver
diethyldithiocarbamate in the determination of arsenic. Anal.
Chem., 45: 1786-1787.
KRAYBILL, H. F. & SHIMKIN, M. B. (1964) Carcinogenesis related to
foods contaminated by processing and fungal metabolites.
Adv. Cancer Res., 8: 205.
KROES, R., VAN LOGTEN, M. J., BERKVENS, J. M., DE VRIES, T. & VAN
ESCH, G. J. (1974) Study on the carcinogenicity of lead arsenate
and sodium arsenate and on the possible synergistic effect of
diethylnitrosamine. Food cosmet. Toxicol., 12: 671-679.
KUO, T. (1968) Arsenic content of artesian well water in endemic area
of chronic arsenic poisoning. Rep. Inst. Pathol. Natl Taiwan
Univ., 20: 7-13.
KYLE, R. A. & PEASE, G. L. (1965) Hematologic aspects of arsenic
intoxication. New. Engl. J. Med., 273: 18-23.
LAKSO, J. U. & PEOPLES, S. A. (1975) Methylation of inorganic arsenic
by mammals. J. agric. Food Chem., 23: 674-676.
LANCASTER, R. J., COUP, M. R. & HUGHES, J. W. (1971) Toxicity of
arsenic present in lakeweed. N.Z. vet. J., 19: 141-145.
LANDER, H., HODGE, P. R. & CRISP, C. S. (1965) Arsenic in hair and
nails. Its significance in acute arsenical poisoning. J. Forensic
Med., 12: 52-67.
LANDER, J. J., STANELY, R. J., SUMNER, H. W., BOSWELL, D. C. & AACH,
R. D. (1975) Angiosarcoma of the liver associated with Fowler's
solution (potassium arsenite). Gastroenterology, 68: 1582-1586.
LANSCHE, A. M. (1965) Arsenic. In: Mineral facts and problems,
Washington, DC, US, Department of the Interior (Bureau of Mines
Bulletin 630).
LANZ, H., Jr., WALLACE, P. C. & HAMILTON, J. G. (1950) The metabolism
of arsenic in laboratory animals using As74 as a tracer. Univ.
California Publ. Pharmacol., 2: 263-282.
LAO, R. C., THOMAS, R. S., TEICHMAN, T. & DUBOIS, L. (1974) Efficiency
of collection of arsenic trioxide in high volume sampling.
Sci. total Environ., 2: 373-379.
LARSEN, N. A., NIELSEN, B., PAKKENBERG, H., CHRISTOFFERSEN, P.,
DAMSGAARD, E. & HEYDORN, K. (1972) Neutron activation analysis of
arsenic, manganese and selenium concentrations in organs of
uraemic and normal persons. In: Kripper, M., ed. Proceedings of a
Symposium, Bled, Yugoslavia, pp. 561-568 (IAEA-SM-157/4).
LARSEN, N. A., PAKKENBERG, H., DAMSGAARD, E. & HEYDORN, K. (1979)
Topographical distribution of arsenic, manganese, and selenium in
the normal human brain. J. neurol. Sci., 42: 407-416.
LAUWERYS, R. R., BUCHET, J. P. & ROELS, H. (1979) The determination of
trace levels of arsenic in human biological materials.
Arch. Toxicol., 41: 239-247.
LEATHERLAND, T. M. & BURTON, J. D. (1974) The occurrence of some trace
metals in coastal organisms with particular reference to the
Solent region. J. Mar. Biol. Assoc. U.K., 54: 457-468.
LEBLANC, P. J. & JACKSON, A. L. (1973) Arsenic in marine fish and
invertebrates. Mar. pollut. Bull., 4: 88-90.
LEDET, A. E., DUNCAN, J. R., BUCK, W. B. & RAMSEY, F. K. (1973)
Clinical, toxicological, and pathological aspects of arsanilic
acid poisoning in swine. Clin. Toxicol., 6: 439-457.
LEE, A. M. & FRAUMENI, J. F., Jr (1969) Arsenic and respiratory cancer
in man: an occupational study. J. Natl Cancer Inst., 42:
1045-1052.
LEITCH, A. & KENNAWAY, E. L. (1922) Experimental production of cancer
by arsenic. Brit. med. J., 2: 1107-1108.
LENVIK, K., STEINNES, E. & PAPPAS, A. C. (1978) Contents of some heavy
metals in Norwegian rivers. Nord. Hydrol., 9: 197-206.
LE QUESNE, P. M. & MCLEOD, J. G. (1977) Peripheral neuropathy
following a single exposure to arsenic. J. neurol. Sci.,
32: 437-451.
LESLIE, A. C. D. & SMITH, H. (1978) Self-poisoning by the abuse of
arsenic containing tonics. Med. Sci. Law, 18: 159-162.
LEVANDER, O. A. (1977) Metabolic interrelationships between arsenic
and selenium. Environ. health Perspect., 19: 159-164.
LIEBHARDT, W. C. (1976) Nutrient concentrations of corn as affected by
poultry manure. Soil Sci. plant Anal., 7: 169.
LIEBSCHER, K. & SMITH, H. (1968) Essential and nonessential trace
elements. A method of determining an element is essential or
nonessential in human tissue. Arch. environ. Health,
17: 881-890.
LINDAU, L. (1977) Emissions of arsenic in Sweden and their reduction.
Environ. health Perspect., 19: 25-29.
LÖFROTH, G. & AMES, B. N. (1978) Mutagenicity of inorganic compounds
in Salmonella typhimurium: arsenic, chromium and selenium.
Mutat. Res., 54: 65-66.
LOWRY, O. H., HUNTER, F. T., KIP, A. F. & IRVINE, J. W., Jr (1942)
Radioactive tracer studies on arsenic injected as potassium
arsenite. II. Chemical distribution in tissues. J. Pharmacol.
exp. Ther., 76: 221-225.
LU, F.-J., YANG, C.-K. & LIN, K.-H. (1975) Physico-chemical
characteristics of drinking water in Blackfoot endemic areas in
Chia-I and Tainan Hsiens. J. Formosan Med. Assoc., 79: 596-605.
LÜCHTRATH, H. (1972) [Cirrhosis of the liver in the arsenism of
vineyard workers.] Dtsch. Med. Wochenschr., 97:21-22
(in German.)
LUGO, G., CASSADY, G. & PALMISANO, P. (1969) Acute maternal arsenic
intoxication with neonatal death. Am. J. Dis. Child.,
117: 328-330.
LUNDE, G. (1970) Analysis of trace elements in seaweed. J. Sci. Food
Agric., 21: 416-418.
LUNDE, G. (1972) The absorption and metabolism of arsenic in fish.
Teknologiske Undersokelser, 5: 1-16.
LUNDE, G. (1973a) The synthesis of fat and water soluble arseno
organic compounds in marine and limnetic algae. Acta Chem.
Scand., 27: 1586-1594.
LUNDE, G. (1973b) Separation and analysis of organic-bound and
inorganic arsenic in marine organisms. J. Sci. Food Agric,
24: 1021-1027.
LUNDE, G. (1975) Isolation of an organoarsenic compound present in cod
liver. J. Sci. Food Agric., 26: 1247-1255.
LUNDE, G. (1977) Occurrence and transformation of arsenic in the
marine environment. Environ. health Perspect., 19: 47-52.
LUNDGREN, K. D. (1954) [Damages in the respiratory organs of workers
at a smeltery.] Nord. Hyg. Tidskr., 3: 66-82 (in Swedish).
MABUCHI, K., LILIENFELD, A. M. & SNELL, L. M. (1979) Lung cancer among
pesticide workers exposed to inorganic arsenicals. Arch. environ.
Health, 34: 312-319.
MAES, D. & PATE, B. D. (1977) The absorption of arsenic into single
human head hairs. J. forensic Sci., 22: 89-94.
MAHAFFEY, K. R. & FOWLER, B. A. (1977) Effects of concurrent
administration of lead, cadmium, and arsenic in the rat. Environ.
health Perspect., 19: 165-171.
MAPPES, R. (1977) [Experiments on the excretion of arsenic in urine.]
Int. Arch. occup. environ. Health, 40: 267-272 (in German).
MARTINDALE, W. (1977) The extra pharmacopoeia, 27th edition, London,
The Pharmaceutical Press.
MARUYAMA, Y., KOMIYA, K. & MANRI, T. (1970) Determination of copper,
arsenic and mercury in cigarettes by neutron activation analysis.
Radioisotopes, 19: 44-46.
MARUYAMA, Y. & KOMIYA, K. (1973) Determination of copper, arsenic, and
mercury in tobacco leaves by neutron activation analysis.
Radioisotopes, 5: 279.
MASSMANN, W. & OPITZ, H. (1954) [Experimental research on ECG changes
in arsenism.] Z. Kreislaufforsch., 43:704-713 (in German).
MATANOSKI, G., LANDAU, E. & ELLIOTT, E. (1976) Epidemiology studies.
Task I. Pilot study of cancer mortality near an arsenical
pesticide plant in Baltimore, Washington, DC, US Environmental
Protection Agency (EPA 560/6-76-003).
McBRIDE, B. C. & WOLFE, R. S. (1971) Biosynthesis of dimethylarsine by
methanobacterium. Biochemistry, 10: 4312-4317.
McBRIDE, B. C., MERILEES, H., CULLEN, W. R. & PICKETT, W. (1978)
Anaerobic and aerobic alkylation of arsenic. In: Brinckman, F. E.
& Bellama, J. M., ed. Organometals and organometalloids,
Washington, DC, American Chemical Society, pp. 94-115
(ACS Symp. Ser. 82).
McCABE, L. J., SYMONS, J. M., LEE, R. D. & ROBECK, G. G. (1970) Survey
of community water supply systems. J. Am. Water Works Assoc.,
62: 670-687.
McCHESNEY, E. W., HOPPE, J. O., McAULIFF, J. P. & BANKS, W. F., Jr.
(1962) Toxicity and physiological disposition of sodium
p-N-glycolylarsanilate. I. Observations in the mouse, cat, rat,
and man. Toxicol. appl. Pharmacol., 4: 14-23.
McDANIEL, M., SHENDRIKAR, A. D., REISZNER, K. D. & WEST, P. W. (1976)
Concentration and determination of selenium from environmental
samples. Anal. Chem., 48: 2240-2243.
MEALEY, J., JR, BROWNELL, G. L. & SWEET, W. H. (1959) Radioarsenic in
plasma, urine, normal tissues, and intracranial neoplasms. Arch.
Neurol. Psychiatr., 81: 310-320.
MENIS, O. & RAINS, T. C. (1969) Determination of arsenic by atomic
absorption spectrometry with electrodeless discharge lamp as
source of radiation. Anal. Chem., 41: 952-954.
MILHAM, S., JR (1977) Studies of morbidity near a copper smelter.
Environ. health Perspect., 19: 131-132.
MILHAM, S., JR & STRONG, T. (1974) Human arsenic exposure in relation
to a copper smelter. Environ. Res., 7: 176-182.
MILNER, J. E. (1969) The effect of ingested arsenic on
methylcholantherene-induced skin tumors in mice. Arch. environ.
Health, 18: 7-11.
MINKKINEN, P. & YLIRUOKANEN, I. (1978) The arsenic distribution in
Finnish peat bogs. Kemia-Kemi, 7-8: 331-335.
MITCHELL, R. A., CHANG, B. F., HUANG, C. H. & DeMASTER, E. G. (1971)
Inhibition of mitochondrial energy-linked functions by arsenate.
Evidence for a nonhydrolytic mode of inhibitor action.
Biochemistry, 10 (11): 2049-2054.
MIZUTA, N., MIZUTA, M., ITO, F., ITO, T., UCHIDA, H., WATANABE, Y.,
AKAMA, H., MURAKAMI, T., HAYASHI, F., NAKAMURA, K., YAMAGUCHI, T.,
MIZUIA, W., OISHI, S. & MATSUMURA, H. (1956) An outbreak of acute
arsenic poisoning caused by arsenic contaminated soy-sauce
(shoyu): A clinical report of 220 cases. Bull. Yamaguchi Med.
Sch., 4: 131-150.
MOHELSKA, H., BENCKO, V., SMETANA, K. & HYNCICA, V. (1980)
Ultrastructural changes in hepatocytes of mice exposed to arsenic
in drinking water. Exp. Pathol. (Jena), 18 (5): 275-281.
MOLIN, L. & WESTER, P. O. (1976) The estimated daily loss of trace
elements from normal skin by desquamation. Scand. J. clin. lab.
Invest., 36: 679-682.
MOODY, J. P. & WILLIAMS, R. T. (1964a) The fate of arsanilic acid and
acetylarsanilic acid in hens. Food cosmet. Toxicol., 2: 687-693.
MOODY, J. P. & WILLIAMS, R. T. (1964b) The fate of
4-nitrophenylarsonic acid in hens. Food cosmet. Toxicol.,
2: 695-706.
MOODY, J. P. & WILLIAMS, R. T. (1964c) The metabolism of
4-hydroxy-3-nitrophenylarsonic acid in hens. Food cosmet.
Toxicol., 2: 707-715.
MORAN, S., MATURANA, G., ROSENBERG, H., CASANEGRA, P. & DUBERNET, J.
(1977) Occlusions coronariennes liées à une intoxication
arsenicale chronique. Arch. Mal. Coeur, 70 (10): 1115-1120.
MORRIS, H. P., LAUG, E. P., MORRIS, H. J. & GRANT, R. L. (1938) The
growth and reproduction of rats fed diets containing lead acetate
and arsenic trioxide and the lead and arsenic content of newborn
and suckling rats. J. Pharmacol. exp. Ther., 64: 420-445.
MORRIS, J. S., SCHMID, M., NEWMAN, S., SCHEUER, P. J. & SHERLOCK, S.
(1974) Arsenic and noncirrhotic portal hypertension.
Gastroenterology, 64: 86-94.
MORSE, D. L., HARRINGTON, J. M., HOUSWORTH, J. & LANDRIGAN, P. J.
(1979) Arsenic exposure in multiple environmental media in
children near a smelter. Clin. Toxicol., 14: 389-399.
MORTON, W., STARR, G., POHL, D., STONER, J., WAGNER, S. & WESWIG, P.
(1976) Skin cancer and water arsenic in Lane County, Oregon.
Cancer, 37: 2523-2532.
MUNRO, I. C. (1976) Naturally occurring toxicants in foods and their
significance. Clin. Toxicol., 9: 647-663.
MUNRO, I. C., CHARBONNEAU, S. M., SANDI, E., SPENCER, K., BRYCE, F. &
GRICE, H. C. (1974) Biological availability of arsenic in fish.
In: Proceedings at the 13th annual meeting of the Society of
Toxicology, Washington, March 1974, 9 pp.
MYERS, D. J. & OSTERYOUNG, J. (1973) Determination of arsenic (III) at
the parts-per-billion level by differential pulse polarography.
Anal. Chem., 45: 267-271.
NAGAMATSU, K. & IGATA, A. (1975) [Arsenical neuropathy. Report of two
cases.] Rinsho-Shinkei, 15: 1-4 (in Japanese with English
summary).
NAKAGAWA, Y. & IIBUCHI, Y. (1970) On the follow-up investigation of
Morinaga milk arsenic poisoning. Igaku no Ayumi, 74: 1-3.
NAKAHARA, H., YANOKURA, M. & MURAKAMI, Y. (1978) Environmental effects
of geothermal waste water on the near-by river system.
J. radioanal. Chem., 45: 25-36.
NAKAO, M. (1960) [A study on the arsenic content in daily food
consumption in Japan.] Osaka City Med. J., 9: 541-571 (in
Japanese with English summary).
NAS (1977) Medical and biologic effects of environmental pollutants:
Arsenic, Washington, DC, National Academy of Sciences.
NATUSCH, D. F. S., WALLACE, J. R. & EVANS, C. A. (1974) Toxic trace
elements: preferential concentration in respirable particles.
Science, 183: 202-204.
NEALE, G. & AZZOPARDI, J. G. (1971) Chronic arsenical poisoning and
noncirrhotic portal hypertension -- a case for diagnosis.
Br. med. J., 4: 725-730.
NELSON, K. W. (1977) Industrial contributions of arsenic to the
environment. Environ. health Perspect., 19: 31-34.
NELSON, H. A., CRANE, M. R. & TOMSON, K. (1971) Inorganic arsenic
poisoning in pastured feeder lambs. J. Am. Vet. Med. Assoc.,
158: 1943-1945.
NELSON, W. C., LYKINS, M. H., MACKEY, J., NEWILL, V. A., FINKLEA, J.
F. & HAMMER, D. I. (1973) Mortality among orchard workers exposed
to lead arsenate spray: A cohort study. J. chron. Dis.,
26: 105-118.
NEUBAUER, O. (1947) Arsenical cancer: A review. Br. J. Cancer,
1: 192-251.
NEUJEAN, G., WEYTS, E. & BACQ, Z. M. (1948) Action du B.A.L. sur les
accidents ophtalmologiques de la thérapeutique à la tryparsamide.
Bull. Acad. R. Med. Belg., 13: 341-350.
NEWMAN, J. A., ARCHER, V. E., SACCOMANNO, G., KUSCHNER, M., AUERBACH,
O., GRONDAHL, R. D. & WILSON, J. C. (1976) Occupational
carcinogenesis. Histologic types of bronchogenic carcinoma among
members of coppermining and smelting communities. Ann. N.Y. Acad.
Sci., 271: 260-268.
NIOSH (1975) Occupational exposure to inorganic arsenic, Cincinnati,
Department of Health, Education, and Welfare, National Institute
for Occupational Safety and Health.
NISHIOKA, H. (1975) Mutagenic activities of metal compounds in
bacteria. Mutat. Res., 31: 185-189.
NOBLE, A. C., ORR, B. H., COOK, W. B. & CAMPBELL, J. L. (1976) Trace
element analysis of wine by proton-induced X-ray fluorescence
spectrometry. J. agric. Food Chem., 24: 532-535.
NORDBERG, G. F., ed. (1976.) Effects and dose-response relationships
of toxic metals, Amsterdam, Elsevier Scientific Publishing
Company.
NORDBERG, G. F., ed. (1978) Factors influencing metabolism and
toxicity of metals. Environ. health Perspect., 25: 3-41.
NORDENSON, I., BECKMAN, G., BECKMAN, L. & NORDSTRÖM, S. (1978)
Occupational and environmental risks in and around a smelter in
northern Sweden. II. Chromosomal aberrations in workers exposed to
arsenic. Hereditas, 88: 47-50.
NORDSTRÖM, S., BECKMAN, L. & NORDENSON, I. (1978) Occupational and
environmental risks in and around a smelter in northern Sweden.
III. Frequencies of spontaneous abortion. Hereditas, 88: 51-54.
NORDSTRÖM, S., BECKMAN, L. & NORDENSON, I. (1979) Occupational and
environmental risks in and around a smelter in northern Sweden. VI
Congenital malformations. Hereditas, 90: 297-302.
NOZAKI, S. (1972) Studies on arsenic metabolism. VI. Effect of
components of milk on arsenic tolerance in the digestive tract.
Folia Pharmacol. Jpn, 68: 857-868.
NOZAKI, S. (1973) Studies on the arsenic metabolism. VII. Added
arsenite in diet and resulting content in each organ, as well as
the effect on diet. Folia Pharmacol. Jpn, 69: 201-212.
NOZAKI, S., TSUTSUMI, S., & TAMURA, S. (1975) Effect of casein on
enteral absorption of arsenic trioxide. Jpn. J. Pharmacol.,
25: 122-123.
ODANAKA, Y., MATANO, O. & GOTO, S. (1978) Identification of
dimethylated arsenic by gas chromatography -- mass spectrometry in
blood, urine, and feces of rats treated with ferric
methanearsonate. J. agric. Food Chem., 26: 505-507.
OHIRA, M. & AOYAMA, H. (1972) Epidemiological studies on the Morinaga
powdered milk poisoning incident. Jpn. J. Hyg., 27: 500-531
(translated for Information Sciences Division, EPA, by Leo Kanner
Associates, Redwood City, California 94063).
ÖHMAN, H. (1960) [Some investigations of wood impregnated with
arsenic.] Stockholm, The National (Swedish) Institute of Public
Health (in Swedish).
OHTA, M. (1970) Ultra-structure of sural nerve in a case of arsenical
neuropathy. Acta Neuropathol. (Berl.), 16: 233-242.
OIDA, O. (1957) [Chronic occupational arsenic poisoning.]
Rodo-Kagaku, 33: 74-81 (in Japanese with English summary).
ONISHI, H. (1969) Arsenic. In: Wedepohl, K.H., ed. Handbook of
geochemistry, Volume II-2, Chapter 33, Berlin, Springer-Verlag.
OPPENHEIM, J. J. & FISHBEIN, W. N. (1965) Induction of chromosome
breaks in cultured normal human leukocytes by potassium arsenite,
hydroxyurea and related compounds. Cancer Res., 25: 980-985.
ORVINI, E., GILLS, T. E. & LaFLEUR, P. D. (1974) Method for
determination of selenium, arsenic, zinc, cadmium, and mercury in
environmental matrices by neutron activation analysis. Anal.
Chem., 46: 1294-1297.
OSATO, K. (1977) [Effects of oral administration of arsenic trioxide
during the suckling stage of rats.] Fukuoka Acta Med.,
68 (10): 464-491 (in Japanese, with English summary).
OSBURN, H. S. (1957) Cancer of the lung in Gwanda. Cent. Afr. J.
Med., 3: 215-223.
OSBURN, H. S. (1969) Lung cancer in a mining district in Rhodesia.
South Afr. med. J., 43: 1307-1312.
OSER, B. L., MORGAREIDGE, K., WEINBERG, M. S. & OSER, M. (1966)
Carcinogenicity study of carbarsone. Toxicol. appl. Pharmacol.,
9: 528-535.
O'SHAUGHNESSY, E. & KRAFT, G. H. (1976) Arsenic poisoning: Long-term
follow-up of a nonfatal case. Arch. phys. Med. Rehabil.,
57: 403-406.
OSSWALD, H. & GOERTTLER, Kl. (1971) [Leukaemia in mice following the
post-natal diaplacental application of arsenic.] Verh. Dtsch.
Ges. Pathol., 55: 289-293 (in German).
OTANI, K. (1957) [Studies on the absorption and distribution of
arsenic.] Sapporo Igaku Zasshi, 11: 285-294 (in Japanese with
English summary).
OTT, M. G., HOLDER, B. B. & GORDON, H. L. (1974) Respiratory cancer
and occupational exposure to arsenicals. Arch. environ. Health,
29: 250-255.
OVERBY, L. R. & FREDRICKSON, R. L. (1963) Metabolic stability of
radioactive arsanilic acid in chickens. J. agric. Food Chem.,
11: 378-381.
OVERBY, L. R. & FROST, D. V. (1962) Nonavailability to the rat of the
arsenic in tissues of swine fed arsanilic acid. Toxicol. appl.
Pharmacol., 4: 38-43.
PACKER, L. (1961) Metabolic and structural states of mitochondria. II.
Regulation by phosphate. J. biol. Chem., 236: 214-220.
PATON, G. R. & ALLISON, A. C. (1972) Chromosome damage in human cell
cultures induced by metal salts. Mutat. Res., 16: 332-336.
PEARSON, E. F. & POUNDS, C. A. (1971) A case involving the
administration of known amounts of arsenic and its analysis in
hair. J. Forensic. Sci. Soc., 11: 229-234.
PEIRSON, D. H., CAWSE, P. A. & CAMBRAY, R. S. (1974) Chemical
uniformity of airborne particulate material, and a maritime
effect. Nature (Lond.), 251: 675-679.
PENROSE, W. R., CONACHER, H. B. S., BLACK, R., MÉRANGER, J. C., MILES,
W., CUNNINGHAM, H. M. & SQUIRES, W. R. (1977) Implications of
inorganic/organic interconversion on fluxes of arsenic in marine
food webs. Environ. health Perspect., 19: 53-59.
PEOPLES, S. A. (1964) Arsenic toxicity in cattle. Ann. N.Y. Acad.
Sci., 111: 644-649.
PEOPLES, S. A. (1975) Review of arsenical pesticides. In: Woolson, E.
A., ed. Arsenical pesticides, Washington, DC, American Chemical
Society, pp. 1-12 (ACS symp. ser. 7).
PERRY, K., BOWLER, R. G., BUCKELL, H. M., DRUETT, H. A. & SHILLING, R.
S. F. (1948) Studies in the incidence of cancer in a factory
handling inorganic compounds of arsenic. II. Clinical and
environmental investigations. Brit. J. ind. Med., 5: 6-15.
PERSHAGEN, G. (1978) Lung cancer mortality, occupational exposure and
smoking habits in a region surrounding a smeltery. In:
Proceedings from an International Symposium on the Control of Air
Pollution in the Working Environment, Stockholm, 6-8 September
1977, Geneva, International Labour Office.
PERSHAGEN, G. & VAHTER, M. (1979) Arsenic, Stockholm, National
Swedish Environment Protection Board (SNV PM 1128).
PERSHAGEN, G., ELINDER, C.-G. & BOLANDER, A.-M. (1977) Mortality in a
region surrounding an arsenic emitting plant. Environ. health
Perspect. 19: 133-137.
PETERS, R. A. (1955) Biochemistry of some toxic agents. I. Present
state of knowledge of biochemical lesions induced by trivalent
arsenical poisoning. Bull. Johns Hopkins Hosp., 97: 1-20.
PETRES, J., BARON, D. & HAGEDORN, M. (1977) Effects of arsenic cell
metabolism and cell proliferation: Cytogenic and biochemical
studies. Environ. health Perspect., 19: 223-227.
PIERCE, F. D., LAMOREAUX, T. C., BROWN, H. R. & FRASER, R. S. (1976)
Automated technique for submicrogram determination of selenium and
arsenic in surface waters by atomic absorption spectroscopy.
Appl. Spectrosc., 30: 38.
PINTO, S. S. & McGILL, C. M. (1953) Arsenic trioxide exposure in
industry. Ind. Med. Surg., 22: 281-287.
PINTO, S. S., VARNER, M. O., NELSON, K. W., LABBE, A. L. & WHITE, L.
D. (1976) Arsenic trioxide absorption and excretion in industry.
J. occup. Med., 18: 677-680.
PINTO, S. S., ENTERLINE, P. E., HENDERSON, V. & VARNER, M. O. (1977)
Mortality experience in relation to a measured arsenic trioxide
exposure. Environ. health Perspect., 19: 127-130.
PINTO, S. S., HENDERSON, V. & ENTERLINE, P. E. (1978) Mortality
experience of arsenic-exposed workers. Arch. environ. Health,
33: 325-331.
PLOTNIKOV, V. I. & USATOVA, L. P. (1964) Coprecipitation of small
amounts of arsenic with metal hydroxides. J. anal. Chem. USSR,
19: 1101-1104.
POMROY, C., CHARBONNEAU, S. M., McCULLOUGH, R. S. & TAM, G. K. H.
(1980) Human retention studies with 74As. Toxicol. appl.
Pharmacol., 53 (3): 550-556.
POPPER, H., THOMAS, L. B., TELLES, N. C., FALK, H. & SELIKOFF, I. J.
(1978) Development of hepatic angiosarcoma in man induced by vinyl
chloride, thorotrast, and arsenic. Am. J. Pathol., 92: 349-369.
PORAZIK, I., LEGATH, V., PUCHA, K. & KRATOCHVIL, I. (1966) [Evaluation
of exposure to atmospheric arsenic oxide from the content of
arsenic in the hair.] Prac. Lék., 18: 352-356 (in Czech with
English summary).
PORTER, E. K. & PETERSON, P. J. (1975) Arsenic accumulation by plants
on mine waste (United Kingdom). Sci. total Environ., 4: 365-371.
PORTER, E. K. & PETERSON, P. J. (1977) Biogeochemistry of arsenic on
polluted sites in S.W. England. In: Hemphill, D.D., ed. Trace
substances in environmental health -- XI. A symposium. Columbia,
University of Missouri Press, pp. 89-99.
PORTMANN, J. E. & RILEY, J. P. (1964) Determination of arsenic in sea
water, marine plants and silicate and carbonate sediments.
Anal. Chim. Acta, 31: 509-519.
PRIER, R. F., NEES, P. O. & DERSE, P. H. (1963) The toxicity of an
organic arsenical, 3-nitro-4-hydroxyphenylarsonic acid. II.
Chronic toxicity. Toxicol. appl. Pharmacol., 5: 526-542.
PUPP, C., LAO, R. C., MURRAY, J. J. & POTTIE, R. F. (1974) Equilibrium
vapour concentrations of some polycyclic aromatic hydrocarbons,
As406 and SeO2, and the collection efficiencies of these air
pollutants. Atmos. Environ., 8: 915-925.
QUENTIN, K.-E. & WINKLER, H. A. (1974) Occurrence and determination of
inorganic polluting agents. Zentralbl. Bakteriol. (Orig. B),
158: 514-523.
RAPOSO, L. S. (1928) Le cancer à l'arsenic. C.R. Soc. Biol. (Paris),
98: 86.
RAY, B. J. & JOHNSON, D. L. (1972) A method for the neutron activation
analysis of natural waters for arsenic. Anal. Chim. Acta,
62: 196-199.
REAY, P. F. (1972) The accumulation of arsenic from arsenic-rich
natural waters by aquatic plants. J. appl. Ecol., 9: 557-565.
REAY, P. F. & ASHER, C. J. (1977) Preparation and purification of
74As-labeled arsenate and arsenite for use in biological
experiments. Anal. Biochem., 78: 557-560.
REGELSON, W., KIM, U., OSPINA, J. & HOLLAND, J. F. (1968)
Hemangio-endothelial sarcoma of liver from chronic arsenic
intoxication by Fowler's solution. Cancer, 12: 514-522.
RENCHER, A. C, CARTER, M. W. & McKEE, D. W. (1977) A retrospective
epidemiological study of mortality at a large western copper
smelter. J. occup. Med., 19: 754-758.
RENNKE, H., PRAT, G. A., ETCHEVERRY, R. B., KATZ, R. U. & DONOSO, S.
(1971) [Malignant hemangioendothelioma of the liver and chronic
arsenicism.] Rev. Med. Chil., 99 (9): 664-668 (in Spanish with
abstract in English).
REYMANN, F., MOLLER, R. & NIELSEN, A. (1978) Relationship between
arsenic intake and internal malignant neoplasms. Arch. Dermatol.,
114: 378-381.
RIDGWAY, L. P. & KARNOFSKY, D. A. (1952) The effects of metals on the
chick embryo: Toxicity and production of abnormalities in
development. Ann. N.Y. Acad. Sci., 55: 203-215.
RITCHIE, J. A. (1961) Arsenic and antimony in some New Zealand thermal
waters. N.Z. J. Sci., 4: 218-229.
ROBBINS, W. B., CARUSO, J. A. & FRICKE, F. L. (1979) Determination of
germanium, arsenic, selenium, tin and antimony in complex samples
by hydride generation-microwave-induced plasma atomic-emission
spectrometry. Analyst, 104: 35-40.
ROBERTS, J. W., POLLOCK, R. D., SVOBODA, M. J. & WATTERS, H. A. (1977)
Ambient air sampling for total arsenic near the Tacoma copper
smelter. In: Proceedings of the 4th International Clean Air
Congress, Seattle, Puget Sound Air Pollution Control Agency,
pp. 424-426.
ROBINSON, T. J. (1975) Arsenical polyneuropathy due to caustic
arsenical paste. Brit. med. J., 3: 139.
ROBSON, A. O. & JELLIFFE, A. M. (1963) Medicinal arsenic poisoning and
lung cancer. Brit. med. J., 2: 207-209.
ROSEHART, R. G. & CHU, R. (1975) Methods for identification of arsenic
compounds. Water Air Soil Pollut., 4: 395-398.
ROSENBERG, H. G. (1974) Systemic arterial disease and chronic
arsenicism in infants. Arch. Pathol., 97: 360-365.
ROSSET, M. (1958) Arsenical keratoses associated with carcinomas of
the internal organs. Can. Med. Assoc. J., 78: 416-419.
ROSSMAN, T., MEYN, M. S. & TROLL, W. (1975) Effects of sodium arsenite
on the survival of UV-irradiated Escherichia coli: Inhibition of
a recA-dependent function. Mutat. Res., 30: 157-162.
RÖSSNER, P., BENCKO, V. & HAVRANKOVA, H. (1977) Effect of the combined
action of selenium and arsenic on suspension culture of mice
fibroblasts. Environ. health Perspect., 19: 235-237.
ROTH, F. (1957) [Arsenic-induced liver tumours
(haemangio-endothelioma).] Z. Krebsforsch., 61: 468-503
(in German).
ROTH, F. (1958) [Bronchial cancer in vineyard workers with arsenic
poisoning.] Virchows Arch., 331: 119-137 (in German).
ROZENSHTEIN, I. S. (1970) Sanitary toxicological assessment of low
concentrations of arsenic trioxide in the atmosphere.
Hyg. Sanit., 35 (1-3): 16-22.
SABBIONI, E., MARAFANTE, E., BERTOLERO, F. & FOA, V. (1979) Inorganic
arsenic: Metabolic patterns and identification of arsenic binding
components in the rabbit. In: International Conference.
Management & Control of Heavy Metals in the Environment, London,
September, 1979, Edinburgh, CEP Consultants Ltd.
SALAMAN, M. H. & ROE, F. J. C. (1956) Further tests for
tumour-initiating activity: N,N-di(2-chlorethyl)-
P-aminophenylbutyric acid (CB1348) as an initiator of skin tumour
formation in the mouse. Br. J. Cancer, 10: 363-377.
SAMSAHL, K. (1967) Radiochemical method for determination of arsenic,
bromine, mercury, antimony and selenium in neutron-irradiated
biological material. Anal. Chem., 39: 1480-1483.
SANDBERG, G. R. & ALLEN, I. K. (1975) A proposed arsenic cycle in an
agronomic ecosystem. In: Woolson, E. A., ed. Arsenical
pesticides, Washington, D.C., American Chemical Society,
(ACS Symp. Ser. No. 7).
SANDERSON, K. V. (1976) Arsenic and skin cancer. In: Andrade, R.,
Gumport, S. L., Popkin, G. L., & Rees, T. D., ed. Cancer of the
skin, Philadelphia, London, Toronto, W. B. Saunder Company,
pp. 473-491.
SANDHU, S. S. & NELSON, P. (1978) Ionic interference in the
determination of arsenic in water by the silver
diethyldithiocarbamate method. Anal. Chem., 50: 322-325.
SATTERLEE, H. S. (1956) The problem of arsenic in American cigarette
tobacco. New Engl. J. Med., 254: 1149-1154.
SCHILLER, C. M., FOWLER, B. A. & WOODS, J. S. (1977) Effects of
arsenic on pyruvate dehydrogenase activation. Environ. health
Perspect., 19: 205-207.
SCHRAUZER, G. N. & ISHMAEL, D. (1974) Effects of selenium and of
arsenic on the genesis of spontaneous mammary tumors in inbred
C3H mice. Ann. clin. lab. Sci., 4: 441-447.
SCHRAUZER, G. N., SECK, J. A., HOLLAND, R. J., BECKHAM, T. M., RUBIN,
E. M. & SIBERT, J. W. (1972) Reductive dealkylation of
alkylcobaloximes, alkylcobalamins, and related compounds:
Stimulation of corrin dependent reductase and methyl group
transfer reactions. Bioinorg. Chem., 2: 93-124.
SCHRAUZER, G. N., WHITE, D. A., McGINNESS, J. E., SCHNEIDER, C. J. &
BELL, L. J. (1978) Arsenic and cancer: Effects of joint
administration of arsenite and selenite on the genesis of mammary
adenocarcinoma in inbred female C3H/St mice. Bioinorg. Chem.,
9: 245-253.
SCHREIBER, M. & BROUWER, E. A. (1964) Metabolism and toxicity of
arsenicals. I. Excretion and distribution patterns in rats.
Fed. Proc., 23: 199.
SCHRENK, H. H. & SCHREIBEIS, L., Jr (1958) Urinary arsenic levels as
an index of industrial exposure. Am. Ind. Hyg. Assoc. J.,
19: 225-228.
SCHROEDER, H. A. & MITCHENER, M. (1971) Toxic effects of trace
elements on the reproduction of mice and rats. Arch. environ.
Health, 23: 102-106.
SCHROEDER, H. A., KANISAWA, M., FROST, D. V. & MITCHENER, M. (1968)
Germanium, tin, and arsenic in rats: Effects on growth, survival,
pathological lesions and life span. J. Nutr., 96: 37-45.
SCHULZ, E. J. (1967) Arsenic as a cause of skin cancer with notes on
its occurrence in Pretoria. South. A. med. J., 33: 819-822.
SCHWARTZE, E. (1922) The so-called habituation to "arsenic": Variation
in the toxicity of arsenous oxide. G. Pharmacol. ed. exp. Ther,
20: 181-203.
SCHWEDT, G. & RUSSEL, H. A. (1972) Gas chromatographic determination
of arsenic as triphenylarsine. Chromatographia, 5: 242.
SELBY, L. A., CASE, A. A., OSWEILER, G. D. & HAYES, H. M. Jr. (1977)
Epidemiology and toxicology of arsenic poisoning in domestic
animals. Environ. health Perspect., 19: 183-189.
SENANAYAKE, N., DE SILVA, W. A. S. & SALGADO, M. S. L. (1972)
Arsenical polyneuropathy -- a clinical study. Ceylon med. J.,
17: 195-203.
SENESI, N., POLEMIO, M. & LORUSSO, L. (1979) Content and distribution
of arsenic, bismuth, lithium and selenium in mineral and synthetic
fertilizers and their contribution to soil. Commun. soil Sci.
plant Anal., 10: 1109-1126.
SHAIKH, A. U. & TALLMAN, D. E. (1977) Determination of sub-microgram
per liter quantities of arsenic in water by arsine generation
followed by graphite furnace atomic absorption spectrometry.
Anal. Chem., 49: 1093-1096.
SHAPIRO, H. A. (1967) Arsenic content of human hair and nails. Its
interpretation. J. forensic Med., 14: 65-71.
SHIBUYA, Y. (1971) [Studies on experimental arsenious acid poisoning.]
Tokyo Jik. Daigaku Zasshi, 86:563-575 (in Japanese).
SIEMER, D. D. & KOTEEL, P. (1977) Comparisons of methods of hydride
generation atomic absorption spectrometric arsenic and selenium
determination. Anal. Chem., 49: 1096-1099.
SIEMER, D. D., KOTEEL, P. & JARIWALA, V. (1976) Optimization of arsine
generation in atomic absorption arsenic determinations. Anal.
Chem., 48: 836-840.
SINA, G., TRIOLO, N., TROVA, P. & CLABAUT, J. M. (1977)
L'encéphalopathie arsénicale lors du traitement de la
trypanosomiase humaine africaine à T. Gambiense (à propos de
16 cas). Ann. Soc. Belg. Méd. Trop., 57: 67-74.
SKONIECZNY, R. F. & HAHN, R. B. (1978) Arsenic. In: Koltheff, I. M. &
Elving, P. J., ed. Treatise on analytical chemistry. Part II:
Analytical chemistry of inorganic and organic compounds,
Volume 10, New York, John Wiley and Sons.
SMITH, A. E. (1975) Interferences in the determination of elements
that form volatile hydrides with sodium borohydride, using atomic
absorption spectrophotometry and the argon-hydrogen flame.
Analyst, 100: 300-306.
SMITH, H. (1964) The interpretation of the arsenic content of human
hair. Forensic Sci. Soc. J., 4: 192-199.
SMITH, R. A. (1976) A method to distinguish between arsenic in and on
human hair. Environ. Res., 12: 171-173.
SMITH, R. A. (1977) Dispersion of particulate arsenic waste from the
chimney of Giant Yellowknife Gold Mines Ltd. Sci. total Environ.,
7: 227-233.
SMITH, S. & FIDDES, F. S. (1955) Forensic medicine -- A textbook for
students and practitioners, London, J. & A. Churchill, p. 458.
SMITH, D. C., SANDI, E. & LEDUC, R. (1972) Pesticide residues in the
total diet in Canada. II. 1970. Pestic. Sci., 3: 207-210.
SMITH, D. C., LEDUC, R. & CHARBONNEAU, C. (1973) Pesticide residues in
the total diet in Canada. III. 1971. Pestic. Sci., 4: 211-214.
SMITH, D. C., LEDUC, R. & TREMBLAY, L. (1975) Pesticide residues in
the total diet in Canada. IV. 1972 and 1973. Pestic Sci.,
6: 75-82.
SMITH, T. J., EATOUGH, D. J., HANSEN, L. D. & MANGELSEN, N. F. (1976)
The chemistry of sulfur and arsenic in airborne copper smelter
particulates. Bull. environ. Contam. Toxicol., 15: 651-658.
SMITH, T. J., CRECELIUS, E. A. & READING, J. C. (1977) Airborne
arsenic exposure and excretion of methylated arsenic compounds.
Environ. health Perspect., 19: 89-93.
SNELL, F. D. & SNELL, C. T. (1945) Colorimetric methods of analysis,
New York, Van Nostrand Reinhold Company.
SÖDERQUIST, C. J., CROSBY, D. G. & BOWERS, J. B. (1974) Determination
of cacodylic acid (hydroxydimethylarsine oxide) gas
chromatography. Anal. Chem., 46: 155-157.
SOMMERS, S. C. & McMANUS, R. G. (1953) Multiple arsenical cancers of
skin and internal organs. Cancer, 6: 347-359.
SRAM, R. J. & BENCKO, V. (1974) [A contribution to the evaluation of
the genetic risk of exposure to arsenic.] Cesk. Hyg.,
19: 308-315 (in Czech with English summary).
STEVENS, J. T., DiPASQUALE, L. C. & FARMER, J. D. (1976) The acute
inhalation toxicology of cacodylic acid. Toxicol. appl.
Pharmacol., 37: 165-166.
STEVENS, J. T., HALLE, L. L., FARMER, J. D., DiPASQUALE, L. C.,
CHERNOFF, N. & DURHAM, W. F. (1977) Disposition of 14C and/or
74As-cacodylic acid in rats after intravenous, intratracheal, or
peroral administration. Environ. health Perspect., 19: 151-157.
STOCKEN, L. A. & THOMPSON, R. H. S. (1949) Reactions of British
anti-lewisite with arsenic and other metals in living systems.
Physiol. Rev., 29: 168-194.
STOEPPLER, M. & MOHL, C. (in press) [Research on the arsenic content
of foods chiefly of marine origin.] Lebensmittelchem. gerichtl.
Chem. (in German).
STRATTON, G. & WHITEHEAD, H. C. (1962) Colorimetric determination of
arsenic in water with silver diethyldithiocarbamate. J. Am. Water
Works Assoc., 54: 861-864.
SULLIVAN, R. J. (1969) Preliminary air pollution survey of arsenic
and its compounds. A literature review, Raleigh, NC,
US Department of Health, Education, and Welfare (APTD 69-26).
SUZUKI, Y., SUZUKI, Y., FUJII, N. & MOURI, T. (1974) [Environmental
contamination around a smeltery by arsenic.] Shikoku Igaku
Zasshi, 30: 213-218 (in Japanese).
SZULER, I. M., WILLIAMS, C. N., HINDMARSH, J. T. & PARK-DINCSOY, H.
(1979) Massive variceal hemorrhage secondary to presinusoidal
portal hypertension due to arsenic poisoning. Can. Med.
Assoc. J., 120: 168-171.
TAKEO, T. & SHIBUYA, M. (1972) Determination of arsenic in tea plant
by neutron activation analysis. Nippon Shokuhin Kogyo Gakkai-Shi,
19 (2): 91.
TALMI, Y. & NORVELL, V. E. (1975) Determination of arsenic and
antimony in environmental samples using gas chromatography with a
microwave emission spectrometric system. Anal. Chem,
47: 1510-1516.
TAM, G. K. H., CHARBONNEAU, S. M., BRYCE, F. & LACROIX, G. (1978)
Separation of arsenic metabolites in dog plasma and urine
following intravenous injection of 74As. Anal. Biochem.,
86: 505-511.
TAM, G. K. H., CHARBONNEAU, S. M., BRYCE, F., POMROY, C. & SANDI, E.
(1979a) Metabolism of inorganic arsenic (74As) in humans
following oral ingestion. Toxicol. appl. Pharmacol.,
50: 319-322.
TAM, G. K. H., CHARBONNEAU, S. M., LACROIX, G. & BRYCE, F. (1979b)
Confirmation of inorganic arsenic and dimethylarsinic acid in
urine and plasma of dog by ion-exchange and TLC. Bull. environ.
Contam. Toxicol., 21: 371-374.
TAM, G. K. H., CHARBONNEAU, S. M., LACROIX, G. & BRYCE, F. (1979c)
In vitro methylation of 74As in urine, plasma and red blood
cells of human and dog. Bull. environ. Contam. Toxicol.,
22: 69-71.
TAMURA, S. & NOZAKI, S. (1972) Studies on arsenic metabolism. V. Blood
brain barrier and arsenite in relation to foods. Folia Pharmacol.
Jpn, 68: 800-808.
TAMURA, S., NOZAKI, S. & TSUZUKI, S. (1972) Studies on arsenic
metabolism. IV. Effect of food on arsenic tolerance in the
digestion tract. Folia Pharmacol. Jpn, 68: 586-601.
TAMURA, S., MIZUKAMI, R. & TANAKA, I. (1974a) Studies on arsenic
metabolism. XIV. Effect of lactoalbumin, eggalbumin on arsenic
excretion and storage. Folia Pharmacol. Jpn, 70: 843-847.
TAMURA, S., MAEHASHI, H., NOZAKI, S. & MIZUKAMI, R. (1974b) Studies on
arsenic metabolism. XII. Effect of polypeptone an polytamine on
arsenic accumulation in certain organs and effect on excretion.
Folia Pharmacol. Jpn, 70: 831-836.
TAMURA, S., NOZAKI, S., MAEHASHI, H. & TANAKA, I. (1974c) Studies on
arsenic metabolism. XIII. Effects of methionine, taurine and
cysteine on arsenic accumulation in certain organs and the
excretion rates. Folia Pharmacol. Jpn, 70: 837-841.
TAMURA, S., MAEHASHI, H., OZAWA, R. & GOTO, K. (1977) Studies on
arsenic metabolism. XX. Arsenic accumulation in organs and
excretion into the feces and urine of rats chronically poisoned
with arsenic. Folia Pharmacol. Jpn, 73: 877-885.
TARRANT, R. F. & ALLARD, J. (1972) Arsenic levels in urine of forest
workers applying silvicides. Arch. environ. Health, 24: 277-280.
TASK GROUP ON AIR POLLUTION AND CANCER (1978) Air pollution and
cancer: Risk assessment methodology and epidemiological evidence.
Environ. health Perspect., 22: 1-12.
TAY, C. -H. & SEAH, C. -S. (1975) Arsenic poisoning from
anti-asthmatic herbal preparations. Med. J. Aust., 2: 424-428.
TERADA, H. (1960) Clinical observation of chronic toxicosis by
arsenic. Nihon Rinsho, 18 (10): 118-127.
TERADA, H., KATSUTA, K., SASAGAWA, T., SAITO, H., SHIRATA, H.,
FUKUCHI, K., SEKIYA, T., YOKOYAMA, Y., HIROKAWA, S., WATANABE, Y.,
HASEGAWA, K., OSHINA, T. & SEKIGUCHI, T. (1960) Clinical
observations of chronic toxicosis by arsenic. Nihon Rinsho,
118: 2394-2403 (EPA translation No. TR106-74).
THACKER, G. T., PATTERSON, B. L. & HOOD, R. D. (1977) Effects of
administration route on arsenate teratogenesis in mice.
Teratology, 15: 30-31.
THIERS, H., COLOMB, D., MOULIN, G. & COLIN, L. (1967) Le cancer cutané
arsenical des viticulteurs du Beaujolais. Ann. Dermatol.
Syphiligr., 94: 133-158.
THOMAS, M. D. & COLLIER, T. R. (1945) The concentration of arsenic in
tobacco smoke determined by a rapid titrimetric method. J. ind.
Hyg. Toxicol., 27: 201-206.
THOMPSON, R. H. S. (1946) The effect of arsenical vesicants on the
respiration of skin. Biochem. J., 40: 525-535.
THOMPSON, R. H. S. (1948) The reactions of arsenicals in living
tissues. Biochem. Soc. Symp, 2: 28-38.
THOMPSON, R. J. (1977) The collection and measurement of airborne
arsenic. In: Air pollution measuring techniques, Part 2, Geneva,
World Health Organization, pp. 126-131.
THOMSON, K. C. (1975) The atomic-fluorescence determination of
antimony, arsenic, selenium and tellurium by using the hydride
generation technique. Analyst, 100: 307-310.
TOKUDOME, S. & KURATSUNE, M. (1976) A cohort study on mortality from
cancer and other causes among workers at a metal refinery.
Int. J. Cancer, 17: 310-317.
TSENG, W.-P. (1977) Effects and dose-response relationships of skin
cancer and Blackfoot disease with arsenic. Environ. Health
Perspect., 19: 109-119.
TSENG, W.-P., CHU, H. M., HOW, S. W., FONG, J. M., LIN, C. S. & YEH,
S. (1968) Prevalence of skin cancer in an endemic area of chronic
arsenicism in Taiwan. J. Natl Cancer Inst., 40: 453-463.
TSUCHIYA, K. (1977) Various effects of arsenic in Japan depending on
type of exposure. Environ. health Perspect., 19: 35-42.
TSUTSUMI, S. & KATO, K. (1975) Effects of dimercaprol or thioctic acid
on the distribution and excretion of 74As in rats. Bull. Tokyo
Dent. Coll., 16: 69-74.
TSUTSUMI, S. & NOZAKI, S. (1975) Studies on arsenic metabolism. Report
9. On the general pharmacological actions of arsenic trioxide
(As203). Bull. Tokyo Dent. Coll., 14: 147-168.
TSUTSUMI, S. & NOZAKI, S. (1975) Studies on arsenic metabolism. Report
15. Influence of arsenic antidotes on enteral absorption of
arsenic trioxide (As203) in rabbits. Folia Pharmacol. Jpn,
71: 545-551.
TSUTSUMI, S., USUI, Y. & MATSUMOTO, Y. (1974) Studies on arsenic
metabolism. Report 8. Influence of different diets on inhibition
of succinic acid dehydrogenase activity in rats. Folia Pharmacol.
Jpn, 70: 515-522.
TSUTSUMI, S., HATTORI, K., SATO, H. & KAWAGUCHI, M. (1976) Studies on
the arsenic metabolism. Report 18. On effects of various antidotes
on the enteral absorption of arsenical. Bull. Tokyo Dent. Coll.,
2: 73-82.
URAKUBO, G., HASEGAWA, A. & NAKAURA, S. (1975) [Studies on the fate of
poisonous metals in experimental animal (V). Body retention and
excretion of arsenic.] J. Food Hyg. Soc. Jpn, 16: 334-336
(in Japanese with English summary).
US Bureau of Mines (1975) Metals, minerals, and fuels. In: Minerals
yearbook, Washington, DC, US, Department of the Interior,
Vol. I.
US Bureau of Mines (1979) Metals, minerals, and fuels. In: Minerals
yearbook, Washington, DC, US, Department of the Interior,
Vol. I.
VAHTER, M. & NORIN, H. (1980) Metabolism of 74As-labelled trivalent
and pentavalent inorganic arsenic in mice. Environ. Res.
21: 446-457.
VALLEE, B. L., ULMER, D. D. & WACKER, W. E. C. (1960) Arsenic
toxicology and biochemistry. A.M.A. Arch. ind. Health,
21: 132-151.
VAN DEN BERG, A. J., DE GOEIJ, J. J. M., HOUTMAN, J. P. W. & ZEGERS,
C. (1969) Arsenic content of human hair after washing as
determined by neutron activation analysis. National Bureau of
Standards, No. 312, pp. 272-282.
VASAK, V. & SEDIVEC, V. (1952) The colorimetric determination of
arsenic. Chem. Listy, 46: 341-344.
VELLAR, O. D. (1969) Nutrient losses through sweating, Thesis. Oslo,
Universitetsförlaget.
VOGEL, A. E. (1955) A text-book of macro and semimacro qualitative
inorganic analysis, 4th ed., London, Longmans.
VONDRACEK, V. (1963) Concentration of 3,4-benzpyrene and arsenic
compounds in the Prague atmosphere. Cesk. Hyg., 8: 333-339.
VON GLAHN, W. C., FLINN, F. B. & KEIM, W. F., JR (1938) Effect of
certain arsenates on the liver. Arch. Pathol., 25: 488-505.
WADKINS, C. L. (1961) The adenosine triphosphate-adenosine diphosphate
exchange reaction of intact rat liver mitochondria. J. biol.
Chem., 236 (1): 221-224.
WAGNER, S. L. & WESWIG, P. (1974) Arsenic in blood and urine of forest
workers. Arch. environ. Health, 28: 77-79.
WALKIW, O. & DOUGLAS, D. E. (1975) Health food supplements prepared
from kelp -- a source of elevated urinary arsenic. Clin.
Toxicol., 8 (3): 325-331.
WALSH, L. M. & KEENEY, D. R. (1975) Behavior and phytotoxicity of
inorganic arsenicals in soils. In: Woolson, E. A., ed. Arsenical
pesticides Washington, DC, American Chemical Society, (ACS Symp.
Ser. No. 7).
WALSH, L. M., SUMNER, M. E. & KEENEY, D. R. (1977a) Occurrence and
distribution of arsenic in soils and plants. Environ. health
Perspect., 19: 67-71.
WALSH, P. R., DUCE, R. A. & FASCHING, J. L. (1977b) Impregnated filter
sampling system for collection of volatile arsenic in the
atmosphere. Environ. Sci. Technol., 11: 163-166.
WATANABE, T., HIRAYAMA, T., TAKAHASHI, T., KOKUBO, T. & IKEDA, M.
(1979) Toxicological evaluation of arsenic in edible seaweed,
Hizikia species. Toxicology, 14: 1-22.
WATSON, C. C. (1958) The contamination of bacon by arsenic from smoke
derived from preservatized wood. N.Z. J. Sci., 1: 361-368.
WAUCHOPE, R. D. & McWHORTER, C. G. (1977) Arsenic residues in soybean
seed from simulated MSMA spray drift. Bull. environ. Contam.
Toxicol., 17: 165-167.
WEBB, J. L. (1966) Enzyme and metabolic inhibitors. New York,
Academic Press, Vol. 3, pp. 595-793.
WEED SCIENCE SOCIETY OF AMERICA (1974) Herbicide handbook, 3rd ed.,
Champaign, Ill., Weed Science Society of America.
WEINBERG, S. L. (1960) The electrocardiogram in acute arsenic
poisoning. Am. Heart J., 60: 971-975.
WESTHOFF, D. D., SAMAHA, R. J. & BARNES, A., Jr. (1975) Arsenic
intoxication as a cause of megaloblastic anemia. Blood, 45:
241-246.
WESTÖÖ, G. & RYDÄLV, M. (1972) [Arsenic levels in foods.] Vår föda,
24: 21-40 (in Swedish, with English summary).
WHANGER, P. D., WESWIG, P. H. & STONER, J. C. (1977) Arsenic levels in
Oregon waters. Environ. health Perspect., 19: 139-143.
WILLIAMS, J. M., WEWERKA, E. M., VANDERBORGH, N. E., WAGNER, P.,
WANEK, P. L. & OLSEN, J. D. (1977) Environmental pollution by
trace elements in coal preparation wastes. In: Proceedings of the
7th Symposium on Mine Drainage Control, at NCA/BRC Coal
Conference, Louisville, Oct. 19, 1977, Los Alamos, New Mexico,
Los Alamos Scientific Laboratory.
WINKLER, W. O. (1962) Identification and estimation of the arsenic
residue in livers of rats ingesting arsenicals. J. Assoc. Off.
Anal. Chem., 45: 80-91.
WITZEL, D. A., SMITH, E. L., BEERWINKLE, K. R. & JOHNSON, J. H. (1976)
Arsanilic acid-induced blindness in swine: Electroretinographic
and visually evoked responses. Am. J. vet. Res., 37: 521-524.
WOLF, R. (1974) [On the question of occupational arsenic poisoning in
vineyard workers.] Berufsdermatosen, 22 (1): 34-47 (in German).
WOODS, J. S. & FOWLER, B. A. (1977) Effects of chronic arsenic
exposure on hematopoietic in adult mammalian liver. Environ.
health Perspect., 19: 209-213.
YAMAMURA, Y. & YAMAUCHI, H. (1976) Arsenic in biological samples of
workers exposed to arsenic trioxide. Jpn J. ind. Health,
18: 530-531.
YAMASHITA, N., DOI, M., NISHIO, M., HOJO, H. & TANAKA, M. (1972)
[Current state of Kyoto children by arsenic tainted Morinaga dry
milk.] Jpn J. Hyg., 27: 364-399 (in Japanese).
YEH, S., HOW, S. W. & LIN, C. S. (1968) Arsenical cancer of skin.
Histologic study with reference to Bowen's disease. Cancer,
21: 312-339.
YOSHIKAWA, T., UTSUMI, J., OKADA, T., MORIUCHI, M., OZAWA, K. &
KANEKO, Y. (1960) Concerning the mass outbreak of chronic arsenic
toxicosis in Niigata Prefecture. Chiryo, 42: 1739-1749.
ZACHARIAE, H., SOGAARD, H. & NYFORS, A. (1974) Liver biopsy in
psoriatics previously treated with potassium arsenite. Acta Derm.
Venreol. (Stockh.). 54: 235-236.
ZDRAZIL, J. & PICHA, F. (1966) The occurrence of the carcinogenetic
compounds 3,4-benzpyrene and arsenic in the soil. Neoplasma,
13: 49-55.
ZEMAN, A., RUZICKA, J., STARY, J. & KLECKOVA, E. (1964) A new
principle of activation analysis separations. VII.
Substoichiometric determination of traces of arsenic. Talanta,
11: 1143.
ZETTEL, H. (1943) [Effects of arsenism on the heart and blood
vessels.] Z. klin. Med., 142: 689-703 (in German).
ZOETEMAN, B. C. J. & BRINKMANN, F. J. J. (1976) Human intake of
minerals from drinking water in the European Communities. In:
Amavis, R., Hunter, W. J., & Smeets, J. G. P. M., ed. Hardness of
drinking water and public health, Oxford, Pergamon Press,
pp. 173-202.
Morton et al. (1976) examined the incidence of skin cancer in Lane
County, Oregon, where high levels of arsenic had been found in water
supplies. No relationship was detected between arsenic levels in water
and the appearance of squamous or basal cell carcinomas. However,
these data do not necessarily contradict previous findings as drinking
water sources with high arsenic concentrations were fewer than
expected. The mean levels in the different regions of the community
ranged between 0.004 and 0.033 mg As/litre.
8.4.1.3 Cancer of the liver
Haemangioendothelioma of the liver has been associated with
exposure to inorganic arsenic in a number of cases. The exposure has
come from contaminated wine, drinking-water, or Fowler's solution.
Roth (1957) reported 5 cases of haemangioendothelioma of the liver
among former vintners, who died between 47 and 58 years of age. The
workers had been handling arsenic-containing insecticides (type not
specified) for a number of years and had typical cutaneous
manifestations of arsenic intoxication, i.e., palmo-plantar
hyperkeratosis and melanosis. All cases also suffered from liver
cirrhosis, which the author ascribed to arsenic ingestion (section
8.3.3). The latent period, from the beginning of arsenic exposure
ranged from 20 to 28 years. No data were given on the actual doses,
but it can be estimated that the total ingested dose of arsenic was
10-20 grams (Grobe, 1976).
Rennke et al. (1971) described a case of malignant
haemangioendothelioma in a 22-year old male from the Chilean province
of Antofagasta. Cutaneous signs of arsenic exposure were also present.
The decedent had been exposed to arsenic in drinking water for 12
years at an average level of 0.8 mg/litre, i.e., a total of
5.46 grams. An arsenic level of 20 mg/kg was found in the hair.
Some cases of haemangioendothelioma have also been reported
following prolonged ingestion of Fowler's solution. Liver
haemangioendothelioma was found in a patient who had been taking
Fowler's solution over 20 years for psoriasis (Rosset, 1958). Regelson
et al. (1968) described a psoriatic patient who had taken Fowler's
solution for 17 years, so that the total ingested dose amounted to
approximately 10 grams of inorganic trivalent arsenic. This patient
also displayed palmo-plantar hyperkeratosis. The tumour was diagnosed
24 years after initiation of treatment with the drug. A 43-year-old
psoriatic patient, who developed haemangioendothelioma 21 years after
the initiation of a 15-year-long treatment with Fowler's solution has
been reported by Lander et al. (1975). The total ingested dose was
estimated by the authors to have been 15 g (not stated whether this
was calculated as As or As(III) oxide). Popper et al. (1978) compiled
5 cases of haemangioendothelioma in the US each with a history of
ingestion of Fowler's solution. The duration of exposure ranged from
10 to 17 years. It is not clear, whether the cases reported by
Regelson et al. (1968) and Lander et al. (1975) were included in this
series.
Although only case reports of this extremely rare tumour exist, it
is noteworthy that the reported latency times seem to be similar,
i.e., 20-25 years. If trivalent arsenic plays a role in the
development of haemangioendothelioma, it can be concluded that large
doses, at least several grams, are required and that the effect is
rare in view of the large number of people who have been exposed
through drinking water or medication.
8.4.1.4 Leukaemia and tumours of the haematopoietic system
A carcinogenic effect on the haematopoietic system in situations
of occupational exposure to inorganic arsenic compounds cannot be
ruled out. In 2 studies on the mortality of workers exposed to high
levels of airborne arsenic, an increased mortality due to malignant
neoplasms of the lymphatic and haematopoietic tissues was indicated
compared with unexposed control groups (Ott et al., 1974; Axelson
et al., 1978). It should be pointed out that the numbers of deaths due
to these causes were small in both studies.
The data need further confirmation, especially in view of the lack
of supporting evidence from other instances of exposure to inorganic
arsenic, i.e., through drinking water and medication.
8.4.1.5 Cancer of other organs
Exposure to inorganic arsenic through medication, mostly as
Fowler's solution, has been associated with the development of various
malignant neoplasms. Sommers & McManus (1953) compiled a total of 27
cases of multiple primary tumours of the skin and internal organs. In
24 of the cases, there was a history of excessive arsenic exposure and
all but one of the patients had palmo-plantar hyperkeratosis. Nineteen
of the patients had been exposed through ingestion of Fowler's
solution, mostly for treatment of psoriasis. Unfortunately, exposure
doses were not given. Multiple skin cancers, predominantly basal cell
carcinomas, were observed in 17 patients (section 7.3.2). In another
10 cases, skin cancers were combined with primary tumours of other
organs, e.g., lung, oesophagus, and bladder. The average latency
between commencement of arsenic exposure and diagnosis of a tumour was
24 years. The data presented in this study do not lend themselves to
statistical analysis. The 10 cases with simultaneous skin cancer and
internal malignancy could have involved a selection artefact.
In a study by Reymann et al. (1978), a total of 389 patients were
followed, who had been on arsenic therapy, primarily with Fowler's
solution. Fifty-three of the patients had typical arsenical keratosis.
A total of 41 cases had developed cancer of internal organs during the
period 1943-74, as traced from the Danish Cancer Registry. It was
found that this was not statistically different, in terms of the life
table method, from the national average. Neither the estimated total
dose of arsenic ingested, nor the nature of the agent was correlated
with the incidence of internal cancer. In the group of patients with
arsenical keratosis, 9 died from cancer of internal organs (type not
specified) during the period under study, as opposed to 5.2 expected
(increase not statistically significant).
The data relating to arsenic exposure and malignancies other than
lung and skin cancer are inadequate. Further studies are needed before
any conclusions can be drawn.
8.4.2 Experimental animal studies
8.4.2.1 Cancer of the respiratory system
Only a few studies have been performed on the carcinogenic effects
of inorganic arsenic following exposure of the respiratory tract in
experimental animals. Ishinishi, et al. (1977) administered copper
ore, metal refinery flue dust, or arsenic(III) oxide, alone or
together with benzo (a)pyrene in a saline suspension, intratracheally
to male rats. Another group received benzo (a)pyrene alone. The doses
of arsenic in the various groups were 1.5, 3.0, and 3.0 mg per animal,
divided into 15 weekly instillations. A total of 6 mg of
benzo (a)pyrene was given. Altogether, 161 animals were treated,
including a control group of 23 animals that received saline only. The
mortality in the various groups during the exposure period ranged from
30% to 50%, the lower value being recorded in the control group. The
results indicated a positive interaction between benzo (a)pyrene and
arsenic(III) oxide in the induction of lung tumours, but the number of
animals in this study was too small to permit any firm conclusions.
Ishinishi, et al. (1980a) gave 30 male adult Wistar strain rats
intratracheal instillations of arsenic(III) oxide in a suspension once
a week for 15 weeks. Sixteen untreated rats acted as controls.
Observation of these rats over their life span revealed only one
malignant lung tumour (squamous cell carcinoma) in the 19 rats that
survived 15 instillations. No lung tumours were found in the control
group. This report does not provide definite evidence of arsenic
respiratory carcinogenicity in animals.
In a study by Ivankovic et al. (1979), 25 rats were given a single
intratracheal instillation of 0.1 ml of a mixture of calcium arsenate,
copper sulfate, and calcium oxide. The preparation was similar to that
of a pesticide used on vines between the 1920s and 1940s. The dose of
calcium arsenate administered was reported to correspond to 0.07 mg of
arsenic. During the first week after treatment, 10 of the animals died
due to pneumonia or lung necrosis. Of the 15 surviving animals, 9
developed lung tumours (7 bronchogenic adenocarcinomas and 2
bronchiolar-alveolar cell carcinomas). The average induction time was
470 days. No lung tumours were observed in 25 control animals,
instilled with 0.1 ml of 0.9% saline, and observed throughout their
life span (mean survival time: 670 days). The data in this report
indicate that the mixture given was carcinogenic, but no firm
conclusion can be reached as to the causative agent, because of the
design of the experiment. It seems, however, that arsenic might have
played an important role and further studies should clarify whether
calcium arsenate alone is sufficient to evoke a carcinogenic response
and also the possible modification of effects by the other components
in the mixture.
8.4.2.2 Skin application
Inorganic arsenic has been tested repeatedly in skin applications
and not been found to be carcinogenic. Leitch & Kennaway (1922)
produced one metastasizing squamous cell carcinoma of the skin in 100
mice painted 3 times weekly with a solution of sodium arsenite in
alcohol. The first skin applications contained 1.8% of arsenic(III)
oxide (about 13.7 g As/litre), but the concentration was reduced to
0.12% (about 0.9 g As/litre) because of the high mortality induced by
the higher dose. In 3 months, two thirds of the animals had died, and,
by that time, a tumour appeared in one of the survivors at the site of
application of the arsenic. At autopsy after 5 1/2 months, a lung
metastasis was observed. No control group was included in the study.
The cocarcinogenicity of sodium arsenite was tested in a series of
experiments by Boutwell (1963). Each of 20 female mice was given a
total of 1.24 mg potassium arsenite in 80% ethanol, in 8 skin
applications over a 5-day period. From 2 days after completion of the
arsenic treatment, the mice were treated twice weekly with croton oil.
After 18 weeks, it was observed that previous exposure to sodium
arsenite did not make the treated mice more responsive to croton oil
than the controls which had been treated with croton oil only. A
single skin application of 75 µg dimethylbenzanthracene and
subsequent, twice-daily applications potassium arsenite (2.2 mg
KAsO2/week) for the duration of the experiment (30 weeks) did not
result in tumour formation. Thus neither tumour initiating nor tumour
promoting activity could be shown for sodium arsenite.
In 1956, Salaman & Roe painted 14 mice with a total of 30 mg of
potassium arsenite (8.7 mg As), dissolved in methanol, once a week for
10 weeks. Croton oil was also applied weekly, starting 25 days later.
Three papillomas were observed in mice treated with potassium
arsenite, while 4 papillomas appeared in 19 animals receiving croton
oil only.
The skin of 10 rabbits was painted with 10% arsenic(III) oxide in
vaseline (about 76 g As/kg). After daily applications for 70 days, 1
epithelioma and 4 papillomas had appeared. A control group was not
used (Raposa, 1928). Friedewald & Rous (1944) repeated this study and
failed to produce any malignant tumours. They did observe verrucous
lesions in 3 out of 7 animals surviving a 22-month treatment. During
the first 6 months, 12 rabbits were painted with 10% arsenic(III)
oxide in vaseline on one ear and vaseline only on the other ear. For
the subsequent 13.5 months, the rabbits were painted with a 2%
solution of arsenic(III) oxide (15 g As/litre) in water mixed with
acetone in the proportion of 1:2. Three out of 6 lesions were excised
and proved to be papillomas. One of these appeared on a control ear.
Sodium arsenate at a concentration of 15.8 g/litre (about 3.8 g of
As) in a 2.5% solution of Tween 60 in water was applied twice weekly
to the skin of 54 male and 14 female mice. A total of 3 skin
papillomas appeared in the exposed animals, and 4 in 69 control
animals. Sodium arsenate applied to the skin in combination with
croton oil treatments twice weekly, failed to show
papilloma-initiating action. An absence of tumour promoting activity
was noted when exposure to arsenic followed a single skin application
of 200 µg dimethylbenzanthracene or a stomach tube dose of 60 mg
urethane (Baroni et al., 1963).
8.4.2.3 Oral administration
Fairhall & Miller (1941) could not find any evidence of
carcinogenicity of lead arsenate or sodium arsenate in 148 rats, fed
daily doses of approximately 2 mg of arsenic, over a period of 2
years. Groups of 50 mice or rats were given arsenic(III) oxide
dissolved in 12% ethyl alcohol or water at arsenic concentrations of
3.0 mg/litre at the beginning of an experiment by Hueper & Payne
(1962). The level was successively raised over a span of 15 months and
from then on kept at 25.5 mg As/litre. In the rats, these levels were
reported to result in daily intakes of between 0.08 and 0.6 mg per
animal. No differences in tumour incidence were found between the
arsenic-exposed animals and the controls.
Boutwell (1963) gave each of 20 female mice a total of 2.4 mg
potassium arsenite (corresponding to 0.7 mg As) by stomach tube. The
dose was administered in 7 portions over a 5 day period. Other groups
were given potassium arsenite mixed with the feed combined with
tumour-initiation treatments with 5 µg dimethylbenzanthracene and
tumour-promotion treatments with croton oil, both applied to the skin.
There were no differences in the production of papillomas and
carcinomas between the arsenic-fed animals and controls.
Arsenic (III) oxide was given in the drinking water in a
concentration of 76 mg As/litre to 15 female and 62 male mice. No skin
tumours were observed in the animals, 21 of which survived for 60
weeks. The rumour incidence in other groups, treated in addition with
croton oil, dimethylbenzanthracene, and urethane, respectively, as
described in section 8.4.2.2, did not differ from that in control
animals given these substances separately (Baroni et al., 1963).
In studies by Byron et al. (1967), groups of 50 rats each were fed
for up to 2 years with sodium arsenite at arsenic levels in the diet
of 0, 15.6, 31.3, 62.5, 125, and 250 mg/kg and sodium arsenate at
arsenic levels of 0, 31.3, 62.5, 125, 250 and 400 mg/kg. The arsenic
was mixed in the standard laboratory diet. After the 2 years, both the
incidence and type of tumours were similar in exposed animals (4-15
survivors/group) and controls (8-12 survivors/group). In the same
studies, groups of 6 dogs (3 male and 3 female) were fed sodium
arsenite or sodium arsenate at arsenic levels in the diet of 5, 25,
50, and 125 mg/kg. In the group receiving the highest dose of sodium
arsenite, no animal survived 2 years, while only one of the 6 dogs
given the highest level of sodium arsenate died during this time. All
of the other animals were killed at the end of 2 years. No tumours
were observed in any of the animals.
Kanisawa & Schroeder (1969) reported a study in which sodium
arsenite was administered in the drinking-water to 103 mice at a
concentration of 5 mg As/litre, throughout their lifetime. A total of
170 mice received double-deionized water. In 67 animals surviving 15
weeks in the exposed group, 11.9% subsequently died with tumours
compared with 34.5% in the control group. The death rates during this
period were similar in both groups.
Mice of 3 different strains were exposed by Milner (1969) to
arsenic(III) oxide in the drinking water at a concentration of
100 mg/litre (corresponding to 76 mg As/litre) over periods of 4 to 11
weeks. Cutaneous tumours were initiated by the topical application of
1,2-dihydro-3-methylbenz[j]accanthrylene (methylcholanthrene) and
promoted by transplantation. The results were contradictory, as
arsenic treatment seemed to increase the number of papillomas in one
strain, i.e., 5 papillomas in 16 arsenic-treated animals compared with
one papilloma in 10 controls, while it seemed to decrease the
incidence in another strain (9/28 in exposed versus 17/29 in
unexposed).
Schrauzer & Ishmael (1974) exposed 30 mice of a strain with a high
incidence of spontaneous mammary tumours to sodium arsenite in the
drinking-water at a concentration of 10 mg/litre (3.6 mg As/litre).
Tumours appeared between 6 and 9 months of age only and were seen in
27% of the animals. In control animals, tumours did not occur before
the eleventh month, but affected 82% of the animals after the
sixteenth month. The growth rate of the spontaneous tumours, as well
as of transplanted mammary tumours, was significantly enhanced in
arsenic-fed animals. When a similar experiment was performed on 30
mice exposed to 2 mg As/litre as arsenic(III) oxide in drinking-water,
mammary tumours started to appear at the age of 9 months, while in
control animals, the age of onset was 4.5 months (Schrauzer et al.,
1978). The tumour incidence was lower in the arsenic-exposed group,
i.e. 36% compared with 41% in the controls (not statistically
significant). As in the previous study, the growth rate of the tumours
was higher in the arsenic-treated animals. When the administration of
arsenite was accompanied by administration of 2 mg selenite per litre
of drinking water, a higher incidence of spontaneous mammary tumours
occurred (62%) than when selenite was administered alone (17%). The
tumour growth rate was similar in the arsenic/selenite and arsenic
groups and was significantly higher than in the selenite only and
control groups.
Lead arsenate was administered at levels of 463 and 1850 mg/kg
(100 and 400 mg As/kg diet)and sodium arsenate at 416 mg/kg (100 mg
As/kg diet), in both cases mixed with the feed, to rats in groups of
48-110 animals (Kroes et al., 1974). For 5 days per week, exposure to
arsenic was combined with oesophageal intubations of
diethylnitrosamine at a dose of 5 µg/day. Mortality was comparable in
all groups except the group given 1850 mg lead arsenate/kg, which
showed a marked increase after 26 weeks. After 120 weeks, all
survivors were killed and the animals were examined for tumours. In
the control groups, the incidence of malignant tumours ranged from 10%
to 17% and in the exposed groups from 0% to 13%. Similarly, there were
no differences in the development of benign tumours between the
groups.
8.4.2.4 Other experimental systems
Hueper (1954) injected metallic arsenic in lanolin into the femur
marrow of 25 male rats and 6 rabbits. The doses were 0.43 mg and
0.65 mg, respectively. Only 4 rats survived 18 months and one of these
developed a spindle cell sarcoma at the site of injection. None of the
rabbits showed any metaplastic reactions. No tumours were produced at
the site of injection in 25 rats injected intrapleurally once a month
for 6 months resulting in a total dose of 0.65 mg of arsenic. Similar
results were obtained after nasal sinus injection of 0.65 mg of
arsenic in 20 rats.
In studies by Osswald & Goerttler (1971), 24 pregnant mice were
injected daily with 0.5 mg of arsenic per kg body weight in the form
of sodium arsenate for 20 days. Half of the 22 animals that died
during a subsequent 24-month period of observation had developed
lymphocytic leukaemias or lymphomas. Two animals were still alive at
this time. None of 16 dead female controls had developed such lesions.
Four controls were still alive. Some of the offspring of the
arsenic-treated mothers (41 males and 56 females) received 20 weekly
subcutaneous injections of sodium arsenate in doses of 0.5 mg As/kg
body weight. Leukaemias and lymphomas had developed in 50% of the
males and 42.0% of the females that had died at the time of reporting.
Seven males were still alive at this time. In 71 untreated progeny,
leukaemias and lymphomas were present in 13.3% of males and 20.7% of
females that had died at the time of reporting. Four out of 34 males
and 8 out of 37 females were alive at this time. Lymphomas appeared in
11 out of 20 mice injected intravenously with 0.5 mg of arsenic in the
form of sodium arsenate once a week, for 20 weeks.
8.5 Mutagenicity
Several studies have indicated an effect of inorganic arsenic on
human chromosomes, both in vivo and in vitro. An increased
frequency of chromosomal aberrations has been found among persons
exposed to arsenic, mainly in inorganic trivalent form, both as
medication and in the workplace. Petres et al. (1977) examined
lymphocytes from 62 dermatology patients, 31 with a history of
extensive arsenic contact, displaying typical palmo-plantar
hyperkeratosis, and 31 controls. The exposed group consisted of 14
psoriasis patients and 17 wine-growers. The control group also
included 14 psoriasis patients but these did not have any history of
arsenic medication. The frequency of chromosome aberrations, both
structurally and numerically, in the arsenic-exposed group was
significantly higher than that in the controls, especially as regards
chromatid aberrations. An in vitro addition of sodium arsenite to
lymphocyte cultures from healthy subjects induced the same chromosomal
changes. Similar results were earlier obtained by Oppenheim & Fishbein
(1965) after adding potassium arsenite to a culture of human
leukocytes and by Paton & Allison (1972) following exposure of human
diploid cells to arsenic salts, including sodium arsenate.
The chromosomal aberrations in lymphocytes from 39 employees at a
Swedish copper smelter with high levels of airborne arsenic in some
workplaces were counted (Nordenson el al., 1978). The workers were
divided into 4 exposure categories based on type and duration of work
with arsenic compounds and presence or absence of septum perforation
and arsenic dermatitis. Controls were apparently healthy individuals
living about 100 km from the smelter. A total of 4106 cells from
arsenic-exposed workers and 1312 from the controls were examined. The
frequency of chromosomal aberrations was significantly higher in the
arsenic-exposed workers than in the controls. However, the correlation
between the chromosomal aberrations and the estimated arsenic exposure
was poor.
An interaction effect of tobacco smoking and arsenic exposure on
the frequency of chromosomal aberrations was indicated. The effect of
arsenic alone cannot be assessed in this study, as simultaneous
exposure to other agents occurred.
An elevated sister chromatid exchange rate was found by Burgdorf
et al. (1977) in the lymphocytes of 6 patients treated with Fowler's
solution (daily doses of up to 3 mg of arsenic as arsenite).
The mean sister chromatid exchange rate per mitosis in the
arsenic-treated patients was 14.0, and only 5.8 in healthy controls.
It should be noted that all 6 arsenic-treated patients had developed
skin cancer and that at least 2 of them had received X-ray treatment.
No information of this sort was given regarding the control subjects.
Several studies have indicated that inorganic arsenic affects DNA
repair mechanisms. Jung (1971) examined the effectiveness of the dark
repair enzyme system in human skin biopsies treated with sodium
arsenate after irradiation with a Xenon lamp. The repair activity,
which was determined in terms of incorporation of a radioactively
labelled nucleotide, was significantly reduced in arsenic-treated
cells. These results have been confirmed by Petres et al. (1977), who
showed that doses of about 1.0 mg sodium arsenate per litre of culture
medium, reduced the incorporation of radioactively labelled
nucleotides into RNA and DNA in lymphocytes. These 2 studies were
performed on dermal cells and lymphocytes, respectively, but other
cell systems could also be expected to be affected by arsenic in a
similar manner.
A significant increase was found in the frequency of dominant
lethals in the F3 generation of mice given sodium arsenite in the
drinking water at a level of 100 mg As/litre, when this exposure was
combined with intraperitoneal injection of tris-l-aziridinyl-phosphine
oxide (TEPA) in a dose of 1 mg/kg body weight prior to mating. No
increase was observed in animals exposed to 1 mg As/litre (Srám &
Bencko, 1974).
Rossman et al. (1975) showed that survival of Escherichia coli
following UV-radiation was decreased by arsenite. Studies on different
strains of bacteria, deficient in various repair mechanisms, indicated
that arsenic particularly affects post-replication repair.
The incidence of early fetal deaths is determined in the
dominant-lethal assay, and may be an indication of genetic damage.
Sodium arsenate (5 mg/kg bodyweight) and sodium arsenite (5 mg/kg
bodyweight), when given to male mice in a single dose
intraperitoneally, has been shown not to elicit a dominant-lethal
effect (Hodge & Embree, 1977). An effect on chromosomes by a toxic
agent might cause malformations or spontaneous abortion, if it occurs
at the germ cell level. Malformations can also result from
interference with fetal development without necessarily damaging the
genetic material (section 8.2).
Nishioka (1975) carried out mutagenicity tests in vitro for 56
inorganic metal compounds using the DNA recombination assay of
Bacillus subtilis. Sodium arsenite, sodium arsenate, and arsenic
(III) chloride gave positive results in the test. On the other hand,
Löfroth & Ames (1978) failed to show any mutagenic activity of
inorganic trivalent and pentavalent arsenic in the Salmonella plate
incorporation test.
8.6 Mechanisms of Toxicity
Inorganic arsenic has been shown to cause impaired tissue
respiration in vivo in the liver and kidneys of mice and rats
(Bencko & Nemeckova, 1971; Bencko, 1972; Brown et al., 1976; Fowler et
al., 1977). To affect the enzymatic activity responsible for
respiration, arsenic has to pass the mitochondrial membranes and
membrane damage appears to play a prominent role in the emergence of
some of the observed effects. In vitro studies have shown that rat
liver mitochondria can accumulate arsenite (Harris & Achenjang, 1977)
and arsenate through energy-dependent processes (Kagawa & Kagawa,
1969). Arsenic-bound components with properties similar to those of a
low relative molecular mass arsenate ester have been isolated from rat
liver mitochondria (Chan et al., 1969).
It was recognized many years ago that inorganic arsenic inhibits
enzyme activity and that trivalent inorganic arsenic reacts with the
sulfhydryl groups of proteins. Many enzymes containing such groups
have been shown to be affected by arsenite (Thompson, 1948; Webb,
1966). In particular, the marked inhibitory effects of As(III) on
mitochondrial respiration mediated by NAD-linked substrates, appear to
play a critical role in the toxicity of this agent. Suppression of
NAD-linked substrates (pyruvate, glutamate, and a-ketoglutarate, in
particular) in rat liver mitochondria is thought to occur through the
reaction between the arsenite ion and the dihydrolipoic acid cofactor,
necessary for oxidation of the substrate (Fluharty & Sanadi, 1960,
1961). A depression in the activity of succinic acid dehydrogenase
[EC I. 3.99.1] in various tissues has also been demonstrated (Tsutsumi
et al., 1974). Arsenite has been shown to decrease or uncouple
mitochondrial oxidative phosphorylation. This phenomenon is associated
with the stimulation of mitochondrial ATPase activity, which in turn,
is usually, a factor in the distortion of mitochondrial membranes. The
dithiol-containing compound, 2,3-dimercapto/propanol (BAL) has been
shown to potentiate the uncoupling action of arsenite in rat liver
mitochondria, suggesting that dithiols have a function in the
transport of arsenite to the enzyme site (Fluharty & Sanadi, 1960,
1961; Fletcher & Sanadi, 1962). Addition of BAL or dithiols in excess
was found to recouple the phosphorylation. At high, in vitro
concentrations, arsenite also inhibited respiration in rat thymus
nuclei (Konings, 1972).
In vitro studies on rat heart and liver mitochondria and
in vivo studies have shown that pentavalent and trivalent arsenic
exert similar effects in the inhibition of mitochondrial respiration
and uncoupling of oxidative phosphorylation (Crane & Lipmann, 1953;
Azzone & Ernster, 1961; Packer, 1961; Wadkins, 1961; Fowler, 1975).
The mechanism of this inhibition is not clear and several
possibilities exist. One is that arsenate is reduced by the
mitochondria to As(III) and that inhibition occurs through the
formation of a complex with the lipoic acid cofactor. The increasing
inhibitory effect on NAD-linked substrates with time found by Crane &
Lipmann (1953) indicates that this could be the case. From studies on
rat liver and beef heart mitochondria, Mitchell et al. (1971) proposed
that inhibition of mitochondrial energy-linked functions by arsenate
occurs in 2 ways: competition with phosphate during oxidative
phosphorylation and inhibition of energy-linked reduction of NAD.
9. EFFECTS AND DOSE-RESPONSE RELATIONSHIPS OF ORGANIC ARSENIC
COMPOUNDS
9.1 Acute and Chronic Toxicity
The form of organic arsenic determines the tissue distribution and
hence affects the pattern of toxicity (section 6.2.2.2). Some organic
arsenic compounds have a high toxicity for certain organs, while the
organic arsenic compounds in seafood are apparently of low toxicity.
9.1.1 Man
Organic arsenic compounds are still used widely as antiparasitic
drugs (section 5.3.3). Side-effects have been reported in many organ
systems, most notably the central nervous system. Encephalopathy was
observed in 1.5% of 1066 patients treated with arsobal (tryparsamide)
for trypanosomiasis (Sina et al., 1977). The mortality was high
(62.5%) among those with encephalopathy. Encephalopathy also occurred
in patients treated with glycobiarsol, another organic arsenic
compound (Cole et al., 1966). A well-known side-effect of tryparsamide
treatment is optical atrophy. Visual effects arise in 3%-4% of
patients treated with this drug, according to Neujean et al. (1948).
Other, less frequent, side-effects include dermatitis, liver damage,
and disturbances of the haematopoietic system.
Organic arsenic compounds may be found in high concentrations in
some seafoods (section 5.2). Although there have not been any
scientific studies published on the acute and subacute toxicity of
these forms of arsenic for man, experience indicates that it is very
low. Chronic effects, however, cannot be ruled out. Walkiw & Douglas
(1975) described 2 women who had been on health food supplements
prepared from kelp (duration of intake not stated). The women had
developed neuropathy, one of them having foot drop and the other
peripheral neuropathy. At the time of hospitalization, the urinary
excretion of arsenic for the 2 patients was 138 µg and 293 µg/24 h,
respectively. No information was given on the course of the symptoms
following cessation of exposure. Clearly, this report cannot
incriminate organic arsenic in the seaweed, beyond doubt, as the cause
of the neuropathy.
9.1.2 Animals
The toxicity of the form of organic arsenic present in seafoods is
of special interest with regard to general human exposure. From the
very limited research performed to date it appears to be relatively
low. Coulson et al. (1935) reported that rats fed a diet containing
shrimp arsenic at a concentration of about 14 mg As/kg for 12 months
did not have any defects in growth or physical appearance. No
histological changes were found in the spleen, liver, or kidneys. In
studies by Lancaster et al. (1971), lakeweed with a high arsenic
content was given to sheep at a daily dose of 1.4 mg As/kg body weight
for 3 weeks. The animals remained healthy, and examination of organs
and tissues removed from the animals at slaughter (3 animals per week)
did not reveal any gross morphological changes.
Aliphatic arsenic compounds such as dimethylarsinic acid and mono-
and disodium methane arsenate, are used commercially as herbicides in
noncrop areas. These herbicides have been reported to be a source of
intoxication for domestic animals (Selby et al., 1977). The clinical
symptoms and histological findings seem to be the same as those
induced by inorganic arsenic, discussed in section 8.1.
Experimental research on the acute inhalation toxicity of
dimethylarsinic acid for rats was performed by Stevens et al. (1976).
No mortality was found among rats, exposed to an aerosol containing a
commercial product (Phytar 138) at a concentration of 2600 mg/m3 for
2 h. The particle size of the dust was 3 µm (MMD). No other details of
the product were specified in the report. Assuming a content of 65%
dimethylarsinic acid (Gosselin et al., 1976), it can be calculated
that the arsenic concentration in the aerosol was approximately
840 mg/m3. Exon et al. (1974) fed rabbits a diet containing
monosodium methane arsenate (MSMA) at a level of 50 mg/kg diet, equal
to 27.5 mg As/kg diet, for 7 or 8 weeks. Toxic hepatitis was found in
all of the 8 animals necropsied and reactive hyperplasia in 5 of them.
Steers and heifers, weighing 100-200 kg, were given daily doses of
pure MSMA at 10 mg/kg (5 mg As/kg body weight; aqueous solution) for
up to 10 days (Dickinson, 1972). Two steers died during the treatment
and the third, some days later. Of the 2 heifers, one was killed due
to morbidity. The other recovered, when the exposure was ended. All
the arsenic-exposed animals lost weight and developed severe
diarrhoea, haemorrhagic gastritis, and intense hyperaemia. Liver
necrosis and renal tubular degeneration were also found.
The short-term toxicity of phenoxarsine oxide (PXO) and
phenarsazine oxide (PZO), 2 organic arsenic compounds used as
industrial biocides, was studied by Ballantyne (1978). Acute oral
toxicities, determined as the LD50 for guineapigs and rats, were 24
and 40 mg/kg body weight (equal to 7 and 12 mg As/kg body weight) for
PXO and 77 and 83 mg/kg body weight (23 and 25 mg As/kg body weight)
for PZO. PZO was more hepatotoxic, producing cellular infiltration and
oedema of the portal tracts as well as periportal hepatocellular
necrosis in rats receiving a single oral dose of 30 mg/kg body weight
(10 mg As/kg body weight). Histological changes of the liver were not
observed in animals receiving the same dose of PXO. Inhalation of PXO
and PZO for 30 days in concentrations of 1-2 mg/m3 (0.3-0.6 mg As/m3
(MMD 4-5 µm), for 5 h a day, did not elicit any toxic signs except for
transient cellular infiltration of the portal tracts of the liver.
Both substances produced eye and skin irritation in guineapigs with a
local application of a 25% suspension in water. Increases in
intraocular pressure in rabbits were concentration dependent.
The toxicity of derivatives of phenylarsonic acid, such as
arsanilic acid, is a subject of great interest, since these compounds
are commonly used as feed additives for poultry and swine. Ledet
et al. (1973) studied the effects of a diet containing arsanilic acid
at a concentration of 1000 mg/kg (350 mg As/kg) in pigs. This was
reported to be 10 times the level recommended for growth stimulation.
Roughened haircoat and mild diarrhoea were the first signs of
toxicity. Cutaneous hyperaemia and hyperaesthesia usually occurred
after a few days, and the animals often stumbled and staggered about.
Histopathological lesions were confined to peripheral and optic
nerves. The same dietary level of arsanilic acid was found to cause
blindness and optic disc atrophy in pigs within 25-30 days (Witzel
et al., 1976). Sodium arsanilate injected subcutaneously into
guineapigs in doses exceeding 70 mg/kg body weight caused degeneration
of the sensory cells of the inner ear (Anniko & Wersäll, 1977).
Retention of arsenic in the cochlea and delayed elimination from the
inner ear compared with elimination from the blood have been reported
(Anniko & Plantin, 1977).
9.2 Teratogenicity
Teratogenic effects were not observed in 7 generations of rats fed
0.01, 0.02, or 0.05% arsanilic acid (3.5, 7 or 17.5 mg As/kg diet)
(Frost et al., 1964). On the contrary, both the litter sizes and the
survival of pups increased significantly.
9.3 Carcinogenicity
No human epidemiological investigations have been conducted on the
carcinogenicity of organic arsenic compounds. Consequently, only data
concerning laboratory animals can be considered.
9.3.1 Animals
Twelve mice belonging to a strain with a high incidence of
spontaneous mammary tumours were injected with neoarsphenamine in
doses of 6.7 mg/kg body weight, twice weekly, for up to 10 weeks
(Hueper & Itami, 1933). Exposed and control animals were found to have
similar average life spans. The arsenic-exposed animals showed an
increased growth rate, and histologically, a higher grade of
malignancy.
In an experiment in which 50 trout were fed a synthetic diet
containing carbarsone at 4.8 g/kg diet for 20 months, 5 trout
developed hepatomas (Halver, 1962; cited in Kraybill & Shimkin 1964).
A control group of 300 trout given the same diet but omitting the
carbasone, did not develop any hepatomas. The original data in this
report were not available, and consequently a thorough evaluation
could not be made of the findings.
Frost et al. (1962) fed groups of 60 rats (30 males and 30
females) diets containing arsanilic acid at 0, 0.1, 0.5 and 1 g/kg
diet for 106 weeks. Tumour incidence was similar in all the groups. In
studies by Boutwell (1963), arsanilic acid was given to 30 mice for a
2-week period at a level corresponding to 200 mg As/kg diet and to
another group for 48 weeks at a level of 100 mg/kg diet. The arsenic
exposures were combined with a skin application of the tumour
initiator 7,12-dimethylbenz (a)anthracene (5 µg) and the tumour
promoter, croton oil. No differences were noted in tumour incidence
between the arsenic-fed animals and controls.
3-Nitro-4-hydroxyphenyl-arsonic acid was incorporated into the
feed of groups of 100 rats and 100 mice (50 animals of each sex in
each group) at levels of 0, 50, and 200 mg/kg diet for the rats and
0, 50, and 100 mg/kg diet for the mice. Tumours occurred in
essentially similar numbers of animals in all groups over a 2-year
period (Prier et al., 1963). The same authors also exposed groups of 6
dogs (3 male and 3 female) to 4-hydroxy-3-nitrophenylarsonic acid
added to the feed at levels of 50 and 200 mg/kg. In this study also,
no differences were seen between exposed and control groups. A
solution of 4-hydroxy-3-nitrophenylarsonic acid in ethanol acetone
(1:4), at a concentration of 10 g/litre, was painted on the skin of
100 mice, 3 times weekly, for 1 year. No skin tumours developed during
this and the following year. Subcutaneous injections of 10 mg of
4-hydroxy-3-nitrophenylarsonic acid were given to 100 female mice and
5 mg to 100 male mice. Neither of the 2 groups differed from control
animals with regard to tumour incidence during a 2-year period of
observation.
The diets of 5 groups of 100 rats (equal numbers of each sex in
each group) were supplemented with carbarsone, ( p-ureidobenzene
arsonic acid) to provide daily intakes of 2.5, 5, 25, 50, and
100 mg/kg body weight, respectively. After 72 weeks, the group
receiving the highest level of carbarsone in the feed was given an
unsupplemented diet. Survival was similar in all groups except the one
with the highest exposure, in which there was a marked increase in the
death rate before the transfer to the unsupplemented diet. A total of
13 malignant tumours was observed in a control group of 200 rats
followed for 2 years. In the 3 groups receiving the highest levels of
carbarsone, the number of malignant neoplasms varied between 0 and 14
and was not related to dose (Oser et al., 1966).
9.4 Mutagenicity
No increase in the incidence of early fetal deaths was observed by
Hodge & Embree (1977) in the offspring of male mice given a single
dose intraperitoneally of [(dimethylarsino)oxy] sodium As-oxide
(sodium dimethylarsonate) (200 mg/kg body weight) arsenodiacetic acid
(arsenoacetic acid) (50 mg/kg body weight), and methanearsonic acid
(250 mg/kg body weight).
9.5 Mechanisms of Toxicity
The mechanisms by which arsenic compounds exert their action in
biological tissues have been thoroughly described and reviewed (Barron
& Singer, 1945; Barron et al., 1947; Thompson, 1946, 1948; Gordon &
Quastel, 1948; Stocken & Thompson, 1949; Peters, 1955; Vallee et al.,
1960; Johnstone, 1963). Arsenic compounds do not constitute a
homogeneous group and the effects in biological systems, caused by
reactions between the arsenic compounds and functional groups of
different enzymes, are highly dependent on the chemical character of
the compound involved in each particular case.
The first distinction to be made is that between trivalent and
pentavalent organic arsenic compounds. Pentavalent arsenic compounds
(R-AsO3H2) have little effect on enzyme activity but can be reduced
in vivo to more toxic trivalent compounds (Peters, 1955; Johnstone,
1963).
Trivalent organic arsenic compounds include, according to the
nomenclature used by Johnstone (1963), arseno and arsenoso compounds.
Arseno compounds, (R-As = As-R) are readily oxidized, even by trace
amounts of oxygen, and their action has been suggested to be due to
their conversion to the corresponding arsenoso derivatives. These can
be divided into monosubstituted and disubstituted compounds according
to their reaction with sulfhydryl groups (Peters, 1955). The
monosubstituted compounds, exemplified by R As = O, react with enzymes
containing sulfhydryl groups:
SR'
/
R - As = O + 2R'SH <==> R - As + H2O
\
SR'
Inhibition of different enzyme systems by these arsenic compounds
was shown to be reversed by addition of an excess of a monothiol,
e.g., glutathione. Some enzymes contain 2 thiol groups, which can
react with the monosubstituted arsenic compound, thereby yielding a
5-membered ring structure. This reaction is reversed by dithiols,
e.g., 2,3-dimercaptopropanol (BAL), but not by monothiols. Lipoic
acid, necessary for the initial stages in the oxidation of pyruvate,
is inhibited in this way by lewisite (used as a war gas), and the
reaction is successfully reversed by 2,3-dimercaptopropanol (BAL).
SH
/
protein + Cl2AsCH = CHCl <==>
\
SH
S
/ \
protein
\ /
S AsCH = CHCl + 2HCl
S
/ \
protein AsCH = CHCl + BAL <==>
\ /
S
SH H2C - S
/ ' \
protein + ' AsCH = CHCl
\ ' /
SH HC - S
'
'
'
HOH2C
R
\
The disubstituted arsenoso compounds, As - OH, exerts its
/
R
action by combining with enzymes containing monothiol groups. The
resulting enzyme poisoning can probably be reversed by the monothiol
defence mechanisms present in the body. One complication, however, is
the possible conversion of disubstituted to mono-substituted arseno
compounds. This is indicated by the finding that dithiols are needed
for reversing the reaction between diphenylchloroarsine and certain
enzyme systems in brain and kidney.
10. INTERACTIONS WITH OTHER CHEMICALS
Until recently, studies on the toxicity of different chemical
agents have been based almost exclusively on the administration of a
single substance as the point of departure. Considering that man is
exposed simultaneously to a wide variety of agents, it is urgent that
the interactions between these agents should be investigated. In the
case of arsenic, attention has been paid to its interaction with
dithiol-containing substances such as BAL, because of the introduction
of BAL as an antidote to the arsenic-containing war gas lewisite.
Investigations concerning combined exposures to arsenic and other
chemicals may, in the future, explain some of the data which appear
controversial at present. Interactions between arsenic and other
agents have been dealt with by the Scientific Committee on the
Toxicology of Metals under the Permanent Commission and International
Association on Occupational Health at its Stockholm meeting on
"Factors Influencing Metabolism and Toxicity of Metals", held in July,
1977. Among the items discussed were interactions between arsenic and
selenium, cadmium, and lead (Nordberg, 1978). Because of the lack of
data on human exposure situations, the following discussion will refer
exclusively to experimental animal studies.
10.1 Thiol Compounds
The toxicity of arsenic compounds, the influence of mono- and
dithiol-containing compounds, and the introduction of
2,3-dimercaptopropanol (BAL) as an antidote in arsenic poisoning have
been reviewed by Stocken & Thompson (1949). The mechanism for the
interaction was discussed in terms of the biochemical aspects of
arsenic toxicity (sections 8.6 and 9.5). It should be noted that
organic and inorganic arsenic react differently with dithiols. The
toxic effects of arsenite can be potentiated by dithiols, as these can
serve as vehicles for the transport of arsenite to the enzymes. The
distribution and excretion of inorganic arsenic in the form of
arsenate, given orally to rats, can be influenced by BAL or thioctic
acid (TA) (Tsutsumi & Kato, 1975). Five consecutive injections of
either substance decreased the tissue levels of arsenic and increased
its excretion more than a single dose.
In rabbits, given BAL or TA parenterally, the amount of
arsenic(III) oxide absorbed in the blood from a ligated loop of the
intestine was considerably lower than in control animals (Tsutsumi &
Nozaki, 1975). Furthermore, when BAL and TA were added directly into
the loop containing arsenic(III) oxide, the amount of arsenic absorbed
in the blood was lower than that of the control group. It was
suggested that BAL and TA, after being excreted into the intestine
with the bile, can inhibit the absorption of arsenic.
BAL, TA, and to some extent diisopropylaminodichloroacetate
(DADA), intraperitoneally injected in rats, increased the amounts of
74As in the stomach and small intestine following orally administered
74As-arsenate (Tsutsumi et al., 1976). When glucuronolacton (GL) and
glutathione (GT) were injected into the animals, the 74As-content in
these organs was comparable to that of the control group. BAL, TA and
DADA were also shown to suppress intestinal movements. The authors
suggested that the delayed uptake of arsenic might be because of the
inhibited transport of arsenic in the gastrointestinal canal, as it
had been shown earlier that arsenic is absorbed preferentially in the
small intestine.
BAL, subcutaneously injected in mice (50 mg/kg body weight),
reduced the teratogenic action of simultaneous intraperitoneal
injections of sodium arsenate at a level of 16 mg As/kg body weight
(Hood & Pike, 1972).
10.2 Selenium
The interaction between selenium and arsenic has been reviewed by
Levander (1977), who concluded that arsenic has a protective effect
against the toxicity of a variety of forms of selenium. He found that
this effect had been demonstrated in several species, including rats,
dogs, swine, cattle, and poultry. It was suggested that arsenic acts
by enhancing the biliary excretion of selenium. Sodium arsenite was
shown to be most potent, arsenate somewhat less potent, and various
organic arsenic compounds least potent. The opposite course of events
has also been observed, i.e., selenite can stimulate the excretion of
arsenite in the bile of rats. The experiments referred to have been
performed almost exclusively on rats. Taking into account what is
known about the metabolism of arsenic in rats and especially about the
partition of different forms of arsenic in the enterohepatic
circulation, it must be concluded that research on the mechanism of
interaction between selenium and arsenic is far from complete.
Holmberg & Ferm (1969) observed that selenite decreased the
teratogenic effects of arsenate when the 2 compounds were given to
pregnant hamsters in simultaneous intravenous injections.
The protective effect of arsenic against selenium has also been
noticed in studies on mouse fibroblasts (Rössner et al., 1977). A
study carried out on mice demonstrated that arsenic (III) was more
efficient in protecting against selenium toxicity than arsenic (V)
(Bencko et al., 1978b).
10.3 Cadmium and Lead
Mahaffey & Fowler (1977) examined the effects of dietary cadmium
(50 mg/kg diet) and lead (200 mg/kg) on the toxicity of arsenic
administered to rats as sodium arsenate or arsanilic acid in the food
(50 mg As/kg diet). The efficiency of food utilization was more
reduced by the combination of arsenic and cadmium than by each metal
alone. The combination of arsenic and cadmium also caused a greater
decrease in serum alkaline phosphatase levels than either metal alone.
Additive effects of arsenic and lead in coproporphyrin excretion were
also noted. Increased uroporphyrin excretion in rats was reported
(Fowler & Mahaffey, 1978), when lead, cadmium, and arsenic were
combined compared with excretion levels following exposure to arsenic
alone.
11. EVALUATION OF HEALTH RISKS TO MAN FROM EXPOSURE TO ARSENIC
11.1 Introduction
Arsenic can give rise to acute, subacute, and chronic effects. The
adverse health effects of arsenic may involve the respiratory,
gastrointestinal, cardiovascular, nervous, and haematopoietic systems.
Effects may be local and systemic.
In the discussions of effects in the following sections,
quantitative aspects on dose-response relationships are given,
whenever possible. Unfortunately, such data are generally very scanty
or nonexistent, and this makes risk evaluation difficult.
Various models have been developed for assessing the risk of
cancer at low doses; however, the simple linear non-threshold
dose-response extrapolation model is most often used. If this model is
adopted, the pulmonary carcinogenicity of inorganic arsenic should
constitute the basis for setting environmental standards for airborne
exposure. In the case of oral exposure, several effects, including
skin cancer, have to be taken into consideration.
Mutagenic and teratogenic effects will not be discussed separately
in this section, as the significance of such data as a basis for
environmental standards is not clearly recognizable.
11.2 Exposure
Exposure to arsenic may occur industrially as well as through
ambient air, tobacco smoke, water, food and beverages. In addition,
considerable exposure may take place through the ingestion of drugs,
still in use in certain countries.
Occupational exposure occurs through the inhalation of particulate
matter containing inorganic arsenic, which may be trivalent or
pentavalent. Concentrations varying from a few micrograms to more than
one milligram per cubic metre have been reported.
Most arsenic in particulate matter in ambient air is in the form
of inorganic arsenic compounds. Concentrations in urban areas may
range from a few nanograms per cubic metre to a few tenths of a
microgram. In the vicinity of point sources emitting arsenic,
concentrations exceeding 1 µg As/m3 have been reported.
In the past, tobacco contained high concentrations of arsenic
because of the widespread use of arsenic in pesticides and
concentrations of up to about 40 mg/kg have been found. However,
levels are now well below 10 mg/kg, in countries that restrict the use
of pesticides containing arsenic.
In both ground and surface waters, the total arsenic concentration
is usually below 10 µg/litre. In certain areas, levels of more than
1 mg/litre have been recorded. Some studies indicate that arsenic is
mainly present in inorganic forms. The oxidation state of the arsenic
in waters depends on the prevailing redox potential. In surface
waters, in the presence of dissolved oxygen, arsenic(V) is
predominant. Under reducing conditions, particularly in deep well
water, arsenic(III) is present.
Food and beverages are the most important sources of exposure to
arsenic for the general population. The total daily intake of arsenic
from the diet has been estimated to be less than 200 µg in the adult.
The figure is greatly influenced by the intake of seafood, which may
contain up to 100 mg As/kg. Most of the arsenic of marine origin is
organic. The total daily intake of inorganic arsenic from food and
water has been estimated to be under 50 µg.
11.3 Inorganic Arsenic Compounds
Exposure can occur to both trivalent and pentavalent inorganic
arsenic compounds, the solubilities of which vary. The nature of the
compounds present in different exposure situations has not been well
identified. Furthermore, the absorption and retention, in the lungs
for example, of compounds of different solubilities are virtually
unknown.
Both arsenic(III) and arsenic(V) are methylated to a great extent
after entering the body. The major metabolites in the urine are
methylarsonic acid and dimethylarsinic acid. The possibility of
dose-dependence in this respect should be considered. Furthermore, the
conversion of arsenic(V) to arsenic(III) in vivo has been suggested,
but data are not conclusive.
In view of these uncertainties, health evaluations, with few
exceptions, have to be confined to inorganic arsenic in general.
11.3.1 Acute and subacute effects after short-term exposure
A fatal dose of ingested arsenic(III) oxide for man has been
reported to range from 1 to 2.5 mg As/kg body weight. This is lower
than the LD50 values generally reported for animals. Trivalent
arsenic is considered to be more toxic than pentavalent.
Two recent mass outbreaks of arsenic poisoning due to the
ingestion of inorganic arsenic have been described. In the first
episode, more than 12 000 infants were poisoned with dried milk
contaminated with inorganic arsenic compounds (oxidation state
uncertain, although in one report it was given as pentavalent). The
average exposure level for a baby of 3 months had been about 3.5 mg of
arsenic daily and symptoms usually appeared after a few weeks. In all,
130 deaths occurred, and effects were observed in several organs.
The other episode concerned about 400 persons who ate contaminated
soy sauce and were thus exposed to an average of 3 mg of arsenic,
daily, for 2-3 weeks. Many acute symptoms were encountered but
neurological symptoms usually did not appear until the 10th-20th day
of illness.
Dose-response relationships are difficult to estimate from the
incidents just described, but it appears that ingestion of 3 mg of
inorganic arsenic per day, over a period of few weeks, may give rise
to severe poisoning in infants and symptoms of toxicity in adults. The
toxic effects cannot be related to any specific valence form of
arsenic.
11.3.2 Noncarcinogenic effects after long-term exposure and
sequelae of short-term exposure
Inhalation of inorganic arsenic compounds can result in local
damage to the respiratory system, including perforation of the nasal
septum. Systemic effects after inhalation and/or ingestion involve the
skin, liver, and the cardiovascular and nervous systems.
In general, details of exposure in human situations have been
inadequate, as a basis for dose-response evaluations. For certain
effects, however, some estimations of dose-response relationships can
be made and information about this is given in the following sections.
11.3.2.1 Skin effects
Skin effects in the form of hyperkeratosis, hyperpigmentation, and
depigmentation have been observed in different parts of the world
after exposure to drinking-water containing high levels of arsenic,
and after treatment with drugs containing inorganic arsenic. The
oxidation state of the arsenic in the water involved in these
incidents is not known but, in drugs, the arsenic component has
usually been in the trivalent state. Skin effects have also been
reported following exposure to arsenic during the manufacture of
insecticides and also among wine growers.
Very few of the data available are of much help in estimating
dose-response relationships. It seems, however, that several years of
exposure to approximately 1 mg of arsenic per day may give rise to
skin effects. It is not possible to state to what extent effects are
caused by exposure to trivalent arsenic alone or whether pentavalent
arsenic may also be of importance.
11.3.2.2 Cardiovascular effects
Cardiovascular effects, in the form of electrocardiographic
changes and peripheral vascular disorders have been observed in
persons exposed to arsenic. Peripheral vascular disorders have been
reported in German vintners, and, in Chile and China (Province of
Taiwan), in parts of the population consuming water containing
0.5-1 mg As/litre. In both the Taiwanese villagers and the German
vintners, the inadequate peripheral circulation caused gangrene,
referred to as "blackfoot disease" by the Taiwanese. A dose-response
relationship was arrived at on the basis of the data concerning the
Taiwanese villagers, in which a roughly linear increase in the
prevalence with increasing arsenic dose was indicated. Exposure to
arsenic for many years, resulting in a total ingested dose of about
20 g of arsenic, corresponded to a prevalence of "blackfoot disease"
of about 3%.
11.3.2.3 Neurological effects
Peripheral neurological damage has been observed in persons
consuming arsenic-containing antiasthmatic preparations on a long-term
basis. The exposure corresponded to 3-10 mg of arsenic per day in the
form of arsenic(III) oxide or arsenic sulfide.
Disturbances of the central nervous system function were noted in
a follow-up of Japanese infants, fifteen years after exposure to an
average daily arsenic dose of about 3.5 mg for one month. The
occurrence of severe hearing loss and brain wave abnormalities was
indicated. However, the data were considered not to be conclusive.
11.3.3 Carcinogenicity
It has already been mentioned that the pulmonary carcinogenicity
of inorganic arsenic should constitute the basis for setting
environmental standards for airborne exposure, and that in the case of
oral exposure, several effects, including skin cancer, have to be
taken into consideration. In reviewing available data in 1973, as well
as in 1980, IARC concluded that there was sufficient evidence to
associate exposure to inorganic arsenic with cancer of the lung and
skin.
11.3.3.1 Cancer of the respiratory system
Arsenic has been associated with pulmonary cancer in the
manufacture and use of arsenic-containing pesticides and in the
smelting of copper. The carcinogenic potential of inorganic arsenic,
mainly trivalent, in the smelter environment is evident from many
epidemiological studies in different countries. One report revealed a
roughly linear relationship between cumulative arsenic exposure and
the lung cancer risk. Though data are uncertain, it could be estimated
that exposure to airborne arsenic levels of about 50 µg/m3 (probably
mostly arsenic(III) oxide) for more than 25 years would result in a
nearly 3-fold increase in mortality due to lung cancer over the age of
65 years (section 8.4.1).
11.3.3.2 Skin cancer
Exposure to inorganic arsenic can cause skin cancer, mainly
tumours of low malignancy. This has been observed following ingestion
of arsenic-rich drinking-water and the consumption of
arsenic-containing drugs. A total dose of several grams has usually
been required for the development of cancer. The form of arsenic in
the different types of drinking water in question has yet to be
elucidated, while, in drugs, it has most often been inorganic
trivalent arsenic.
There are very few dose-response data on arsenic and skin cancer
that can be used for quantitative estimations. From one study on
exposure to arsenic via drinking-water in China (Province of Taiwan),
there was evidence that a total of about 20 g of arsenic over a
lifetime resulted in a prevalence of skin cancer of about 6%.
11.4 Organic Arsenic Compounds
The application of organic arsenic compounds in medicine, most
notably tryparsamide, has induced side effects, mainly in the central
nervous system, in the form of encephalopathy and optical atrophy.
Toxic effects on the nervous system have also been reported in
experimental animals given high doses of arsanilic acid. This is a
compound commonly used as a feed additive for poultry and swine in
some countries. No data on dose-response relationships, which would be
directly applicable to the long-term exposure of man, are available.
Human beings are exposed to organic arsenic compounds in seafood,
certain types of which may contain arsenic concentrations of
20-50 mg/kg wet weight or even more. The form of arsenic in seafood is
largely unknown, but it has been found to be readily absorbed (more
than 80%) from the gastrointestinal tract in both animals and man. It
is also rapidly excreted in the urine (70-80% within a week).
No adverse effects were found in a study in which rats were fed a
diet containing "seafood arsenic" in the form of shrimps at a
concentration of about 14 mg As/kg for 12 months. There are no data
concerning the toxicity of "seafood arsenic" for man, apart from the
fact that acute effects have not been reported. It seems obvious that
this question has not been studied sufficiently, especially
considering that a large number of people are exposed to this form of
arsenic. Another organic arsenic compound, dimethylarsinic acid, has
been shown to pass through the placental barrier of rats. Blood values
of the fetus were comparable to those of the mother. Placental
transfer should be investigated when the toxicity of "seafood arsenic"
is studied.
There is no conclusive evidence that any of the organoarsenic
compounds tested for carcinogenicity in laboratory animals are
carcinogenic. Epidemiological studies are greatly needed on
populations exposed to organic arsenic, especially in view of the
discrepancy between animal and human data with regard to the
carcinogenicity of inorganic arsenic compounds. Suitable groups for
study are workers exposed in the manufacture, handling, and use of
organoarsenic compounds as well as patients treated with such
compounds. Data have not been reported concerning the possible
carcinogenic effects of "seafood arsenic".
11.5 Assessment of the Cancer Risk for Man from Exposure to
Inorganic Arsenic
The purpose of this section is to provide guidance for the
estimation of the cancer risk to the lung and skin from the inhalation
and ingestion, respectively, of inorganic arsenic. For the purposes of
this risk assessment, it is assumed that both pentavalent and
trivalent arsenic are carcinogenic.
It must be recognized that the assessment of cancer risks by
currently available methods can provide only crude estimates and this
should be borne in mind particularly in making regulatory decisions
about permissible limits of exposure. The use of the linear
non-threshold model is recommended for extrapolation of risks from
relatively high dose levels, where cancer responses can be measured,
to relatively low dose levels, which are of concern in environmental
protection where such risks are too small to be measured directly
either through animal or human epidemiological studies.
The linear non-threshold model has been generally accepted amongst
regulatory bodies in the USA for chemical carcinogens (IRLG) and for
ionizing radiation on an international basis (ICRP). The linear
non-threshold philosophy was accepted by a Task Group on Air Pollution
and Cancer in Stockholm in 1977 (Task Group on Air Pollution and
Cancer, 1978). The scientific justification for the use of a linear
non-threshold extrapolation model stems from several sources: the
similarity between carcinogenesis and mutagenesis as processes which
both have DNA as target molecules, the strong evidence of the
linearity of dose-response relationships for mutagenesis, the evidence
for the linearity of the DNA binding of chemical carcinogens in the
liver and skin, the evidence for the linearity in the dose-response
relationship in the initiation stage of the mouse 2-stage
tumorigenesis model, and the rough consistency with the linearity of
the dose-response relationships for several epidemiological studies;
for example, aflatoxin and liver cancer, leukaemia and radiation. This
rationale for the linear non-threshold dose-response model is
strongest for the genotoxic carcinogens.
The mechanism of the carcinogenesis of arsenic is not clear at
present.
In the case of the lung cancer estimates for the inhalation of
inorganic arsenic, based on epidemiological data from an occupational
smelter population, it is assumed that the life-time cancer risk is a
function of the total dose of arsenic. This is a necessary assumption
because occupational exposures begin at maturity, whereas exposures to
airborne arsenic in the general environment begin at conception.
Furthermore, in the case of lung cancer risk estimates, it is assumed
that there are no age or sex differences in susceptibility to cancer
induced by arsenic. There is not much basis in scientific fact for
assuring the validity of these assumptions. It is not unreasonable to
assume that the cancer response is proportional to the total dose,
since the occupational smelter exposures extended over a substantial
portion of the life span. On the basis of animal data, it is possible
that children may generally be somewhat more susceptible than adults
to carcinogens, but it is not known whether this is the case for
arsenic and lung cancer. It is also not known whether males and
females are equally susceptible. It is possible that sulfur dioxide
and smelter dusts other than arsenic potentiate arsenic in the
workplace. Presumably there are more carcinogen co-factors in the
occupational setting than in the general environment, but this is not
certain.
Risk estimates for lung cancer from inorganic arsenic exposure can
be based on the study of Pinto et al. (1977) on smelter workers, in
which there was a standard mortality ratio of about 300 or a 200%
excess of lung cancer at an average air concentration of 50 µg/m3 for
an average duration of exposure of more than 25 years. It was assumed
that the 200% excess of lung cancer applied to the life-time risk even
though the Pinto study was limited to observations in men over 65
years of age. This assumption of relative-risk model has a sound
factual basis particularly for cancer response to protracted
exposures. The 200% excess needs to be extrapolated for the same total
dose to the average life span of the population under consideration;
i.e., if the average life span is 70 years, then a 200% excess of
cancer would be produced for the same total arsenic dose by a
life-time exposure to 8 µg/m3. It is assumed that the 200% excess of
lung cancer, observed in the Pinto study, corresponds to an exposure
level of 50 µg/m3 for 25 years. (This will lead to some
overestimation of the risk as the duration of exposure in the original
data was given as more than 25 years.) The calculation is based on an
occupational air intake of 8 m3 per day, for 240 days a year, over 25
years compared with an environmental air intake of 12 m3 per day for
365 days a year over 70 years. In other words, a life-time exposure to
8 µg/m3 would be expected to produce a 200% excess in lung cancer.
This excess risk can conveniently be expressed as a percentage
increase per unit concentration of arsenic in air. Thus 1 µg/m3 would
produce 200/8 or a 25% excess in lung cancer incidence. Knowing the
existing lifetime cancer incidence for the population under
consideration, the risk from the nominal concentration of 1 µg/m3 can
be expressed in terms of absolute excess risk. If the life-time risk
of lung cancer is for example 3%, then the excess risk is 3% × 25% or
0.03 × 0.25 or 0.0075 per microgram of airborne arsenic per cubic
metre or 0.8% per microgram of airborne arsenic per cubic metre.
There are relatively few assumptions that need to be made for the
estimation of the skin cancer risk from the ingestion of arsenic in
drinking water. These risk estimates can be based on the
epidemiological data from China (Province of Taiwan) (Tseng, 1977),
where the population at risk was exposed for at least 50 years and the
data were obtained from more than 40 000 men, women, and children of
all ages. Nevertheless, the response could have been affected by
socio-economic, cultural, and racial factors such as skin pigment,
which may not be comparable to other populations. The risk assessment
for skin cancer from the ingestion of inorganic arsenic can be based
on the data shown in Fig. 11. The skin tumour data in this study was
given in terms of prevalence. However, since these tumours were of low
malignancy and would be expected to persist for a very long time, it
has been assumed that the characterization of tumour yield in terms of
prevalence is equivalent to cumulative incidence. The middle dose
group shown in Fig. 11 for the age range of 60 years and over was
chosen as the point of departure for the downward linear non-threshold
extrapolation because it is the one that is the best description of
exposure; the choice of the oldest age group maximizes the tumour
yield for a given total dose. The observed prevalence data were not
modified to account for a control incidence, since Tseng stated that
actinic skin cancers were not included in this series of cases. The
slope of the resultant linear extrapolation is about 5% skin tumour
prevalence per 10 grams of total ingested arsenic. Assuming a 2
litre/day average intake of drinking water, a concentration of 0.2 mg
As/litre would result in a total dose of 10 grams in an assumed
average life span of 70 years. Thus the life-time risk from arsenic in
drinking-water is about 25% per mg As/litre (5%/0.2 mg As/litre).
REFERENCES
ADAMSSON, E., PISCATOR, M. & NOGAWA, K. (1979) Pulmonary and
gastrointestinal exposure to cadmium oxide dust in a battery
factory. Environ. Health Perspect., 28: 219-222.
AGGETT, J. & ASPELL, A. C. (1978) Release of arsenic from geothermal
sources. (New Zealand Energy Research and Development Committee
Report No. 35).
ALVARADO, L. C., VINIEGRA, G., GARCIA, R. E. & ACEVEDO, J. A. (1964)
[Arsenism in the Lagoon Region.] Salud Pública Méx.,
6 (3): 375-385 (in Spanish).
ALY, S., MOUSA, S., EL-KAHKY, M., SALEH, A. & EL-MOFTY, A. (1975)
Toxic deafness. I. Histological study of the effect arsenic,
salicylates and quinine, on the organ of Corti of guinea pigs.
J. Egypt. Med. Assoc., 58 (3-4): 144-157.
ANDERSSON, A. & NILSSON, K. O. (1972) Enrichment of trace elements
from sewage sludge fertilizer in soils and plants. Ambio,
1: 176-179.
ANDO, S., SUZUKI, M., FUWA, K. & VALLEE, B. L. (1969) Atomic
absorption of arsenic in nitrogen (entrained air) hydrogen flames.
Anal. Chem., 41: 1974-1979.
ANDREAE, M. O. (1977) Determination of arsenic species in natural
waters. Anal. Chem., 49: 820-825.
ANDREAE, M. O. (1978) Distribution and speciation of arsenic in
natural waters and some marine algae. Deep-sea Res.,
25: 391-402.
ANNIKO, M. & PLANTIN, L. -O. (1977) Delayed elimination of the
ototoxic compound atoxyl from the inner ear. Arch.
Oto-Rhino-Laryng., 215: 81-89.
ANNIKO, M. & WERSÄLL, J. (1977) Experimentally (atoxyl) induced
ampullar degeneration and damage to the maculae utriculi.
Acta Otolaryngol, 83: 429-440.
ARGÜELLO, R. A., CENGET, D. D. & TELLO, E. E. (1938) [Cancer and
endemic arsenism in the Cordoba Region.] Rev. Argent.
Dermatosifilogr., 22 (4th part): 461-487 (in Spanish).
ARIYOSHI, T. & IKEDA, T. (1974) On the tissue distribution and the
excretion of arsenic in rats and rabbits of administration with
arsenical compounds. J. Hyg. Chem., 20 (5): 290-295.
ARNOLD, J. P. & JOHNSON, R. M. (1969) Polarography of arsenic.
Talanta, 16: 1191-1207.
ARSENAULT, R. D. (1977) (6) Health aspects of C. C. A. wood
preservatives - a review of arsenates and cancer. Annual
convention, London, British Wood Preservers Association.
ASO, T. & ABIKO, Y. (1978) Tissue distribution of arsenic after
subcutaneous implantation of arsenic trioxide pellet in rats.
J. Toxicol. Sci., 3: 109-116.
ASTRUP, P. (1968) Blackfoot disease. Ugeskr. Laeg., 130: 1807-1815.
ATALLA, L. T., SILVA, C. M. & LIMA, F. W. (1965) Activation analysis
of arsenic in human hair -- some observations on the problem of
external contamination. An. Acad. Bras. Cienc., 37: 431-441.
ATTREP, M., JR. & ANIRUDHAN, M. (1977) Atmospheric inorganic and
organic arsenic. Trace Subst. Environ. Health, 11: 365-369.
AUERMANN, E., SEIDLER, G. & KNEUER, M. (1977) [The assessment of
arsenic in the air.]. Nahrung, 21: 799-806 (in German).
AXELSON, O., DAHLGREN, E., JANSSON, C. -D. & REHNLUND, S. O. (1978)
Arsenic exposure and mortality: a case-referent study from a
Swedish copper smelter. Br. J. ind. Med. 35: 8-15.
AZZONE, S. F. & ERNSTER, L. (1961) Compartmentation of mitochondrial
phosphorylation as disclosed by studies with arsenate. J. Biol.
Chem., 236: 1510-1517.
BAETJER, A. M., LILIENFELD, A. M. & LEVIN, M. L. (1975) Cancer and
occupational exposure to inorganic arsenic. In: Abstracts. 18th
International Congress on Occupational Health, Brighton, England,
14-19 September.
BAILEY, E. J., KENNAWAY, E. L. & URQUHART, M. E. (1957) Arsenic
content of cigarettes. Br. J. Cancer, 11: 49-53.
BAKER, E. L., HAYES, C. G., LANDRIGAN, P. J., HANDKE, J. L., LEGER, R.
T., HOUSWORTH, W. J. & HARRINGTON, J. M. (1977) A national-wide
survey of heavy metal absorption in children living near primary
copper, lead, and zinc smelters. Am. J. Epidemiol.,
106: 261-273.
BALLANTYNE, B. (1978) The comparative short-term mammalian toxicology
of phenarsazine oxide and phenoxarsine oxide. Toxicology,
10: 341-361.
BANG, I. (1919) [Report by the Swedish arsenic commission], volume 11.
In: Lund, H., ed. Ohlssons Boktryckeri, pp. 1-56 (in Swedish).
BARONI, C., VAN ESCH, G. J. & SAFFIOTTI, U. (1963) Carcinogenesis
tests of two inorganic arsenicals. Arch. Environ. Health,
7: 668-674.
BARRON, E. S. G. & SINGER, T. P. (1945) Studies on biological
oxidations. XIX. Sulfhydryl enzymes in carbohydrate metabolism.
J. Biol. Chem., 157: 221-253.
BARRON, E. S. G., MILLER, Z. B., BARTLETT, G. R., MEYER, J. & SINGER,
T. P. (1947) Reactivation by dithiols of enzymes inhibited by
Lewisite. Biochem. J., 41: 69-74.
BARRY, K. G. & HERNDON, E. G., JR. (1962) Electrocardiographic changes
associated with acute arsenic poisoning. Med. Ann. D.C.,
31: 25-27, 65-66.
BARTAK, P. & KEJDA, J. (1972) [Basaloma induced by arsenic in young
skin.] Hautarzt, 23 (10): 457-458.
BEAMISH, F. E. & COLLINS, H. L. (1934) Nickle microbomb for
microestimation of organic arsenic. Ind. Eng. Chem., Sept. 15
(6): 379-380.
BEAUDOIN, A. R. (1974) Teratogenicity of sodium arsenate in rats.
Teratology, 10: 153-158.
BEAVINGTON, F. & CAWSE, P. A. (1978) Comparative studies of trace
elements in air particulate in northern Nigeria. Sci. total
Environ., 10: 239-244.
BENCKO, V. (1972) Oxygen consumption by mouse liver homogenate during
drinking water arsenic exposure. Part II. J. Hyg. Epidemiol.
Microbiol. Immunol., 16: 42-46.
BENCKO, V. & NEMECKOVA, H. (1971) Oxygen consumption by mouse liver
homogenate during drinking water arsenic exposure. Part I.
J. Hyg. Epidemiol. Microbiol. Immunol., 15: 104-110.
BENCKO, V. & SYMON, K. (1969a) Dynamics of arsenic cumulation in
hairless mice after peroral administration. J. Hyg. Epidemiol.
Microbiol. Immunol., 13: 248-253.
BENCKO, V. & SYMON, K. (1969b) Suitability of hairless mice for
experimental work and their sensitivity to arsenic.
J. Hyg. Epidemiol. Microbiol. Immunol., 13: 1-6.
BENCKO, V. & SYMON, K. (1970) The cumulation dynamics in some tissue
of hairless mice inhaling arsenic. Atmos. Environ., 4: 157-162.
BENCKO, V. & SYMON, K. (1977) Health aspects of burning coal with a
high arsenic content. Environ. Res., 13: 378-385.
BENCKO, V., CMARKO, V. & PALAN, S. (1968) The cumulation dynamics of
arsenic in the tissues of rabbits exposed in the area of the ENO
plant. Cesk. Hyg., 13: 18-22.
BENCKO, V., DOBISOVA, A. & MACAJ, M. (1971) Arsenic in the hair of a
nonoccupationally exposed population. Atmos. Environ.,
5: 275-279.
BENCKO, V., DVORAK, V. & SYMON, K. (1973) Organ retention of
parenterally administered arsenic (labelled with 74As) in mice
preliminary exposed to the element in drinking water: A study in
arsenic tolerance. J. Hyg. Epidemiol. Microbiol. Immunol.,
17: 165-168.
BENCKO, V., RÖSSNER, P. & MOKRY, M. (1975) Dehydrogenase activity of
liver parenchyma in mice exposed to arsenic in drinking water.
J. Hyg. Epidemiol. Microbiol. Immunol., 19: 17-21.
BENCKO, V., BENES, B. & CIKRT, M. (1976) Biotransformation of As(III)
to As(V) and arsenic tolerance. Arch. Toxicol., 36: 159-162.
BENCKO, V., SYMON, K., CHLADEK, V. & PIHRT, J. (1977) Health aspects
of burning coal with a high arsenic content. II. Hearing changes
in exposed children. Environ. Res., 13: 386-395.
BENCKO, V., BENES, B. & HAJEK, V. (1978a) The content of free
sulfhydryl groups and the activity of glutathione reductase in the
liver of mice exposed to arsenic in drinking water. Cesk. Hyg.,
23: 209-214.
BENCKO, V., RÖSSNER, P., HAVRANKOVA, H., PUZANOVA, A. & TUCEK, M.
(1978b) Effects of the combined action of selenium and arsenic on
mice versus suspension culture of mice fibroblasts. In: Fouts, J.
R. & Gut, I., ed. Industrial and environmental xenobiotics,
Amsterdam, Excerpta Medica Foundation (International Congress
Series No. 440).
BERGSTRÖM, J. & WESTER, P.O. (1966) The effect of dialysis on the
arsenic content of blood and muscle tissue from uraemic patients.
In: Kerr, D. N.S., Fraeger, J., Frieds, D., & Elliott, R. W., ed.
Replacement of renal function, Amsterdam, Excerpta Medica
Foundation. (International Congress Series No. 131).
BERLIN, C. & TAGER, A. (1962) Psoriasis complicated by cutaneous and
visceral carcinomatosis due to ingestion of arsenic. Acta Derm.
Venereol., 42: 252-255.
BETTLEY, F. R. & O'SHEA, J. A. (1975) The absorption of arsenic and
its relation to carcinoma. Br. J. Dermatol., 92: 563-568.
BINNS, F., ENSOR, R. J. & MACPHERSON, A. L. (1978) Metal content of
United Kingdom and overseas lager beers. J. Sci. Food Agric.,
29: 71-74.
BIRMINGHAM, D. J., KEY, M. M., HOLADAY, D. A. & PERONE, V. B. (1965)
An outbreak of arsenical dermatoses in a mining community.
Arch. Dermatol., 91: 457-464.
BISHOP, R. F. & CHISHOLM, D. (1962) Arsenic accumulation in Annapolis
Valley orchard soils. Can. J. soil Sci., 42: 77.
BLEJER, H. P. & WAGNER, W. (1976) Case study 4: Inorganic arsenic --
ambient level approach to the control of occupational cancerigenic
exposures. Ann. N. Y. Acad. Sci., 271: 179-186.
BLOT, W. J. & FRAUMENI, J. F., Jr (1975) Arsenical air pollution and
lung cancer. Lancet, (July 26): 142-144.
BORGOÑO, J. M. & GREIBER, R. (1972) Epidemiological study of
arsenicism in the city of Antofagasta. In: Hemphill, D. D, ed.
Trace substances in environmental health -- V. A symposium,
Columbia, University of Missouri Press, pp. 13-24.
BORGONO, J. M., VICENT, P., VENTURINO, H. & INFANTE, A. (1977) Arsenic
in the drinking water of the city of Antofagasta: epidemiological
and clinical study before and after the installation of the
treatment plant. Environ. Health Perspect., 19: 103-105.
BOUTWELL, R. K. (1963) A carcinogenicity evaluation of potassium
arsenite and arsanilic acid. J. agric. Food Chem., 11: 381-385.
BOYLEN, G. W. Jr & HARDY, H. L. (1967) Distribution of arsenic in
nonexposed persons (hair, liver, and urine). Am. Ind. Hyg.
Assoc. J., 28: 148-150.
BRAMAN, R. S. (1975) Arsenic in the environment. In: Woolson, E. A.,
ed. Arsenical Pesticides, Washington, DC, American Chemical
Society (ACS Symp. Ser. No. 7).
BRAMAN, R. S. & FOREBACK, C. C. (1973) Methylated forms of arsenic in
the environment. Science, 182: 1247-1249.
BRAMAN, R. S., JOHNSON, D. L., FOREBACK, C. C., AMMONS, J. M. &
BRICKER, J. L. (1977) Separation and determination of nanogram
amounts of inorganic arsenic and methylarsenic compounds.
Anal. Chem., 49: 621-625.
BRIMBLECOMBE, P. (1979) Atmospheric arsenic. Nature (Lond.),
280: 104-105.
BROWN, M. M., RHYNE, B. C., GOYER, R. A. & FOWLER, B. A. (1976)
Intracellular effects of chronic arsenic administration on renal
proximal tubule cells. J. Toxicol. environ. Health, 1: 505-514.
BRUNE, D., SAMSAHL, K. & WESTER, P. O. (1966) A comparison between the
amounts of As, Au, Br, Cu, Fe, Mo, Se and Zn in normal and uraemic
human whole blood by means of neutron activation analysis.
Clin. Chim. Acta, 13: 285-291.
BRUNE, D., NORDBERG, G. & WESTER, P. O. (1980) Distribution of 23
elements in the kidney, liver and lungs of workers from a smeltery
and refinery in North Sweden exposed to a number of elements and
of a control group. Sci. total Environ., 16 (1): 13-35.
BUCHANAN, W. D. (1962) Toxicity of arsenic compounds. In: Browning,
E., ed. Elsevier monographs on toxic agents, New York, Elsevier
Publishing Company.
BUCHET, J. P., LAUWERYS, R. R., & BERLIN, A. (in press)
Interlaboratory comparison of biological parameters used in the
evaluation of occupational exposure to inorganic arsenic.
Report EUR.
BURGDORF, W., KURVINK, K. & CERVENKA, J. (1977) Elevated sister
chromatic exchange rate in lymphocytes of subjects treated with
arsenic. Hum. Genet., 36: 69-72.
BUTZENGEIGER, K. H. (1940) [Peripheral circulation disorders in
arsenism.] Klin. Wochenschr., 19:523-527 (in German).
BUTZENGEIGER, K. H. (1949) [Arsenism. I. ECG changes and other cardiac
and circulatory phenomena. II: Mucosal symptoms and pathogenesis.]
Dtsch. Arch. Klin. Med., 1974: 1-16 (in German).
BYRON, W. R., BIERBOWER, G. W., BROUWER, J. B. & HANSEN, W. H. (1967)
Pathologic changes in rats and dogs from two-year feeding of
sodium arsenite and sodium arsenate. Toxicol. appl. Pharmacol.,
10: 132-147.
CALESNICK, B., WASE, A. & OVERBY, L. R. (1966) Availability during
human consumption of the arsenic in tissues of chicks fed
arsanilic- 74As acid. Toxicol. appl. Pharmacol., 9: 27-30.
CALVERT, C. C. (1973) Feed additive residues in animal manure
processed for feed. Feedstuffs, 45 (17): 32-33.
CALVERT, C. C. (1975) Arsenicals in animal feeds and wastes. In:
Woolson, E. A., ed. Arsenical Pesticides, Washington, DC,
American Chemical Society (ACS Symp. Series No. 7).
CANADIAN PUBLIC HEALTH ASSOCIATION (1977) Task Force on Arsenic,
Yellowknife, Northwest Territories. Final report, Ottawa.
CANADIAN PUBLIC HEALTH ASSOCIATION (1978) Electromyography.
Yellowknife and Hay River, Northwest Territories, Ottawa.
CANNON, J. R., EDMONDS, J. S., FRANCESCONI, K. A. & LANGSFORD, J. B.
(1979) Arsenic in Marine Fauna. In: International Conference.
Management & Control of Heavy Metals in the Environment, London,
September, 1979. Edinburgh, CEP Consultants Ltd.
CARAPELLA, S.C. JR (1973) Arsenic and compounds. In: Hampel, C. A. &
Hawley, G. G., ed. The encyclopaedia of chemistry, third
edition, New York, Van Nostrand Reinhold Company.
CARLSSON, G. (1976) [Correlation between industrial arsenic exposure
and excretion in urine.] Examination report. Stockholm, The
National Board of Industrial Safety, Dept of Occupational Health
(In Swedish).
CARVALHO, M. B. & HERCULES, D. M. (1978) Trace arsenic determination
by volatilization and X-ray photoelectron spectroscopy.
Anal. Chem., 50: 2030-2034.
CHALLENGER, F. (1945) Biological methylation. Chem. Rev., 36: 315.
CHAN, T.-L., THOMAS, B. R. & WADKINS, C. L. (1969) The formation and
isolation of an arsenylated component of rat liver mitochondria.
J. Biol. Chem., 244: 2883-2890.
CHAPMAN, A. C. (1926) On the presence of compounds of arsenic in
marine crustaceans and shell fish. Analyst, 51: 548-563.
CHARBONNEAU, S. M., SPENCER, K., BRYCE, F. & SANDI, E. (1978a) Arsenic
excretion by monkeys dosed with arsenic-containing fish and with
inorganic arsenic. Bull. Environ. Contam. Toxicol., 20: 470-477.
CHARBONNEAU, S. M., TAM, G. K. H., BRYCE, F. & COLLINS, B. (1978b)
Pharmacokinetics and metabolism of inorganic arsenic in the dog.
In: Hemphill, D. D., ed. Trace substances in environmental health
-- XII. A symposium, Columbia, University of Missouri Press,
pp. 276-283.
CHARBONNEAU, S. M., TAM, G. K. H., BRYCE, F., ZAWIDZKA, Z. & SANDI, E.
(1979) Metabolism of orally administered inorganic arsenic in the
dog. Toxicol. Lett., 3: 107-113.
CHARBONNEAU, S. M., HOLLINS, J. G., TAM, G. K. H., BRYCE, F.,
RIDGEWAY, J. M. & WILLES, R. F. (1980) Whole-body retention,
excretion and metabolism of [74As] arsenic acid in the hamster.
Toxicol. Lett., 5 (3-4): 175-182.
CHHUTTANI, P. N., CHAWLA, L. S. & SHARMA, T. D. (1967) Arsenical
neuropathy. Neurology (Minneap.), 17: 269-274.
CHRISTIAN, G. D. & FELDMAN, F. J. (1970) Atomic absorption
spectroscopy; Applications in agriculture, biology, and medicine,
New York, Wiley-Interscience, pp. 188-195.
CHU, R. C., BARRON, G. P. & BAUMGARNER, P. A. W. (1972) Arsenic
determination at sub-microgram levels by arsine evolution and
flameless atomic absorption spectrophotometric technique.
Anal. Chem., 44: 1476-1479.
CHU, T.-Y. J., RUANE, R. J. & KRENKEL, P. A. (1978) Characterization
and reuse of ash pond effluents in coal-fired power plants.
J. Water Pollut. Control Fed., 50: 2494-2508.
CIKRT, M. & BENCKO, V. (1974) Fate of arsenic after parenteral
administration to rats, with particular reference to excretion via
bile. J. Hyg. Epidemiol. Microbiol. Immunol., 18: 129-136.
CIKRT, M., BENCKO, V., TICHY, M. & BENES, B. (1980) Biliary excretion
of 74As and its distribution in the golden hamster after
treatment with 74As(III) and 74As(V). J. Hyg. Epidemiol.
Microbiol. Immunol. 9 (24): 384-388.
CLEMENT, W. H. & FAUST, S. D. (1973) A new convenient method for
determining arsenic (+3) in natural waters. Environ. Lett.,
5: 155-164.
CMARKO, V. (1963) [Hygienic problems of arsenic exhalations of ENO
plant.] Cesk. Hyg., 8: 359-362 (In Slovak, with English
summary).
COLE, M., SCHEULEIN, M. & KERWIN, D. M. (1966) Arsenical
encephalopathy due to use of Milibis. Arch. intern. Med.,
177: 706-711.
COONEY, R. V., MUMMA, R. O. & BENSON, A. A. (1978)
Arsoniumphospholipid in algae. Proc. Natl Acad. Sci.,
75: 4262-4264.
CORNELIS, R. (1973) Neutron activation analysis of hair failure of a
mission. J. radioanal. Chem., 15: 305-316.
CORRIDAN, J. P. (1974) Head hair samples as indicators of
environmental pollution. Environ. Res., 8: 12-16.
COULSON, E. J., REMINGTON, R. E. & LYNCH, K. M. (1935) Metabolism in
the rat of the naturally occurring arsenic of shrimp as compared
with arsenic trioxide. J. Nutr., 10: 255-270.
CRANE, R. K. & LIPMANN, F. (1953) The effect of arsenate on aerobic
phosphorylation. J. biol. Chem., 201: 235-243.
CRAWFORD, T. B. B. & STOREY, I. D. E. (1944) Quantitative micro-method
for the separation of inorganic arsenite from arsenate in blood
and urine. Biochem. J., 38: 195-198.
CRECELIUS, E. A. (1974) The geochemistry of arsenic and antimony in
Puget Sound and Lake Washington, Washington, Thesis, Seattle,
Washington, University of Washington.
CRECELIUS, E. A. (1977a) Arsenite and arsenate levels in wine.
Bull. environ. Contam. Toxicol., 18: 227-230.
CRECELIUS, E. A. (1977b) Changes in the chemical speciation of arsenic
following ingestion by man. Environ. health Perspect.,
19: 147-150.
CRECELIUS, E. A. (1978) Modification of the arsenic speciation
technique using hydride generation. Anal. Chem., 50: 826-827.
CRECELIUS, E. A., ROBERTSON, D. E., FRUCHTER, J. S. & LUDWICK, J. D.
(1976) Chemical forms of mercury and arsenic emitted by a
geothermal power plant. In: Hemphill, D. D., ed. Trace substances
in environmental health -- X, Columbia, University of Missouri
Press, pp. 287-293.
CREMA, A. (1955) Distribution et élimination de l'arsenic 76 chez la
souris normale et cancéreuse. Arch. int. Pharmacodyn.,
103: 57-70.
CRISTAU, B., PLACIDI, M. & AUDIBERT, P. (1972) Voies et cinétiques
d'élimination de l'arsenic chez le rat après administration de
médicaments organoarséniés. Med. trop., 32(3): 275-283.
CRISTAU, B., CHABAS, E. & PLACIDI, M. (1975) Voies et cinétiques
d'excrétion de l'arsenic chez le Cobaye après injection de divers
médicaments organo-arséniés. Ann. Pharm. Fr., 33: 577-589.
CULLEN, W. R., FROESE, C. L., LUI, A., McBRIDE, B. C., PATMORE, D. J.
& REIMER, M. (1977) The aerobic methylation of arsenic by
micro-organisms in the presence of L-methionine-methyl-d3.
J. organomet. Chem., 139: 61-69.
CURRY, A. S. & POUNDS, C. A. (1977) Arsenic in hair. J. Forensic Sci.
Soc., 17: 37-44.
DAGHIR, N. J. & HARIRI, N. N. (1977) Determination of total arsenic
residues in chicken eggs. J. agric. Food Chem., 25: 1009-1010.
DAMSGAARD, E., HEYDORN, K., LARSEN, N. A. & NIELSEN, B. (1973)
Simultaneous determination of arsenic, manganese and selenium in
human serum by neutron activation analysis, Roskilde, Danish
Atomic Energy Commission (Riso Report N:o 271).
DAVIS, P. H., DULUDE, G. R., GRIFFIN, R. M., MATSON, W. R. & ZINK, E.
W. (1978) Determination of total arsenic at the nanogram level by
highspeed anodic stripping voltammetry. Anal. Chem, 50: 137-143.
DAVIS, W. E. et al. (1971) In: Medical and biologic effects of
environmental pollutants: Arsenic, Washington, DC, National
Academy of Sciences.
DAY, G. T. (1964) Analysis of tungstic oxide by the conducting briquet
technique. Can. Spectrom., 9: 118-120.
DEAK, S. T., CSAKY, K. G. & WADDELL, W. J. (1976) Localization and
histochemical correlation, of 73As by whole-body autoradiography
in mice. J. Toxicol. environ. Health, 1: 981-984.
DE NAVARRO, E. T. (1976) [Arsenic poisoning in cattle.] Salud publica
mex., 18(6): 1037-1044 (in Spanish).
DENG, J.-S. & HOW, S.-W. (1977) Comparison of the fine structure of
basal cell carcinomas in ordinary patients and patients with
chronic arsenicalism. J. Formosan Med. Assoc., 76: 685-692.
DICKINSON, J. O. (1972) Toxicity of the arsenical herbicide monosodium
acid methanearsonate in cattle. Am. J. vet. Res., 33: 1889-1892.
DIEKE, S. H. & RICHTER, C. P. (1946) Comparative assays of
rodenticides on wild Norway rats. I. Toxicity. Publ. Health Rep.,
61: 672-679.
DOAK, G. O. & FREEDMAN, L. D. (1970) Organometallic compounds of
arsenic, antimony, and bismuth. In: Seyferth, D., ed.
The chemistry of organometallic compounds. A series of
monographs, New York, John Wiley & Sons.
DOBBS, A. J., PHIL, D. & GRANT, C. (1976) Report on the burning of
wood treated with wood preservatives containing copper, chromium
and arsenic, Aylesbury, Buckinghamshire, UK, Building Research
Establishment (Report Cp 63/73).
DONE, A. K. & PEART, A. J. (1971) Acute toxicities of arsenical
herbicides. Toxicol., 4: 343-355.
DUCE, R. A., HOFFMAN, G. L. & ZOLLER, W. H. (1975) Atmospheric trace
metals at remote northern and southern hemisphere sites; pollution
or natural? Science, 187: 59-61.
DUCOFF, H. S., NEAL, W. B., STRAUBE, R. L., JACOBSON, L. O. & BRUES,
A. M. (1948) Biological studies with arsenic76. II. Excretion and
tissue localization. Proc. Soc. Exp. Biol. Med., 69: 548-554.
DU PONT, O., ARIEL, I. & WARREN, S. L. (1941) The distribution of
radioactive arsenic in the normal and tumor-bearing (Brown-Pearce)
rabbit. Am. J. Syph. Gon. vener. Dis., 26: 96-118.
DURRANT, P. J. & DURRANT, B. (1966) Introduction to advanced
inorganic chemistry, third edition, London, Longmans, Green and
Co. Ltd.
DURUM, W. H., HEM, J. D. & HEIDEL, S. G. (1971) Reconnaissance of
selected minor elements in surface waters of the United States,
October 1970, Washington, DC, US Department of Interior
(Geological Survey Circular 643).
DUTKIEWICZ, T. (1977) Experimental studies on arsenic absorption
routes in rates. Environ. health Perspect., 19: 173-177.
EDMONDS, J. S., FRANCESONI, K. A., CANNON, J. R., RASTON, C. L.,
SKELTON, B. W. & WHITE, A. H. (1977) Isolation, crystal structure
and synthesis of arsenobetaine, the arsenical constituent of the
western rock lobster Panulirus Longipes Cygnus George.
Tetrahedron Lett., 18: 1543-1546.
ELTON, R. K. & GEIGER, W. E. (1978) Analytical and mechanistic studies
of the electrochemical reduction of biologically active
organoarsenic acids. Anal Chem., 50: 712-717.
ENGSTRÖM, B. M. & NORDBERG, G. F. (1978) Effects of milk diet on
gastrointestinal absorption of cadmium in adult mice. Toxicology,
9: 195-203.
ESPINOSA GONZALEZ, E. (1963) [Mass arsenic poisoning in Terreón,
Coahuila State, Mexico.] Bol. Epidemiol., 27(4): 213-220
(in Spanish).
EVANS, R. J. & BANDEMER, S. L. (1954) Determination of arsenic in
biological materials. Anal. Chem., 26: 595.
EXON, J. H., HARR, J. R. & CLAEYS, R. R. (1974) The effects of long
term feeding of monosodium acid methanearsenate (MSMA) to rabbits.
Nutr. Rep. Int., 9: 351-357.
FAIRHALL, L. T. & MILLER, J. W. (1941) A study of the relative
toxicity of the molecular components of lead arsenate.
Publ. Health Rep., 56: 1610-1625.
FELDMAN, C. (1979) Improvements in the arsine accumulation -- helium
glow detector procedure for determining traces of arsenic.
Anal. Chem., 51: 664-669.
FELDMAN, C. & BATISTONI, D. A. (1977) Spectroscopic element detector
for gas chromatography. Anal Chem., 49: 2215-2221.
FERGUSON, J. F. & GAVIS, J. (1972) A review of the arsenic cycle in
natural waters. Water Res, 6: 1259-1274.
FERM, V. H. (1977) Arsenic as a teratogenic agent. Environ. health
Perspect., 19: 215-217.
FERM, V. H. & CARPENTER, S. J. (1968) Malformations induced by sodium
arsenate. J. Reprod. Fer., 17: 199-201.
FERM, V. H., SAXON, A. & SMITH, B. M. (1971) The teratogenic profile
of sodium arsenate in the golden hamster. Arch. environ. Health,
22: 557-560.
FERSLEW, K. E. & EDDS, G. T. (1979) Effects of arsanilic acid on
growth, serum enzymes, hematologic values, and residual arsenic in
young swine Am. J. vet. Res., 40: 1365-1369.
FEUSSNER, J. R., SHELBURNE, J. D., BREDEHOEFT, S., & COHEN, H. J.
(1979) Arsenic-induced bone marrow toxicity: Ultrastructural and
electron probe analysis. Blood, 53: 820-827.
FIERZ, U. (1965) [Catamnestic research into the side effects of
inorganic arsenotherapy in skin diseases.] Dermatologica,
131: 41-58 (in German).
FISHMAN, M. & SPENCER, R. (1977) Automated atomic absorption
spectrometric determination of total arsenic in water and
streambed materials. Anal. Chem., 49: 1599-1602.
FLETCHER, M. J. & SANADI, D. R. (1962) On the mechanism of oxidative
phosphorylation. III. Effects of arsenite and
2,3-dimercaptopropanol on coupled phosphorylation in heart
mitochondria. Arch. Biochem. Biophys., 96: 139-142.
FLIS, I. E., MISHCHENKO, K. P., TUMANOVA, T. A. & RUSS, J. (1959)
Dissociation of arsenic acid. J. inorg. Chem., 4: 120-124.
FLUHARTY, A. L. & SANADI, D. R. (1960) Evidence for a vicinal dithiol
in oxidative phosphorylation. Proc. Natl Acad. Sci.,
46: 608-616.
FLUHARTY, A. L. & SANADI, D. R. (1961) On the mechanism of oxidative
phosphorylation. II. Effects of arsenite alone and in combination
with 2,3-dimercaptopropanol. J. biol. Chem., 236: 2772-2778.
FOWLER, B. A. (1975) The ultrastructural and biochemical effects of
arsenate on the kidney. (Abstract) In: Proceedings of the XVIII
International Congress on Occupational Health, Brighton.
FOWLER, B. A. & MAHAFFEY, K. R. (1978) Interactions between lead,
cadmium and arsenic in relation to porphyrin excretion patterns.
Environ. health Perspect., 25: 87-90.
FOWLER, B. A., WOODS, J. S. & SCHILLER, C. M. (1977) Ultrastructural
and biochemical effects of prolonged oral arsenic exposure on
liver mitochondria of rats. Environ. health Perspect.,
19: 197-204.
FRANKE, K. W. & MOXON, A. L. (1936) A comparison of the minimum fatal
doses of selenium, tellurium, arsenic and vanadium. J. Pharmacol.
exp. Ther., 58: 454-459.
FRANKLIN, M., BEAN, W. B. & HARDIN, R. C. (1950) Fowler's solution as
an etiologic agent in cirrhosis. Am. J. med. Sci., 219: 589-596.
FREEMAN, H. C., UTHE, J. F., FLEMING, R. B., ODENSE, P. H., ACKMAN, R.
G., LANDRY, G. & MUSIAL, C. (1979) Clearance of arsenic ingested
by man from arsenic contaminated fish. Bull. environ. Contam.
Toxicol., 22: 224-229.
FRIBERG, L. & CEDERLÖF, R. (1978) Late effects of air pollution with
special reference to lung cancer. Environ. health Perspect.,
22: 45-66.
FRIBERG, L. F., PISCATOR, M. & NORDBERG, G. F. (1974) Cadmium in the
environment, 2nd edition, Cleveland, CRC Press.
FRIEDEWALD, W. F. & ROUS, P. (1944) The determining influence of tar,
benzpyrene, and methylcholanthrene on the character of the benign
tumors induced therewith in rabbit skin. J. exp. Med.,
80: 127-144.
FROST, D. V., PERDUE, H. S., MAIN, B. T., KOLAR, J. A., SMITH, I. D.,
STEIN, R. J. & OVERBY, L. R. (1962) Further considerations on the
safety of arsanilic acid for feed use. In: Proceedings of the
Twelfth World's Poultry Congress, Sydney, pp. 234-237.
FROST, D. V., MAIN, B. T., COLE, J., SANDERS, P. G. & PERDUE, H. S.
(1964) Reproduction studies in rats with arsanilic acid.
Fed. Proc. Fed. Am. Soc. Exp. Biol., 23: 291.
FURR, A. K., KELLY, W. C., BACHE, C. A., GUTENMANN, W. H. & LISK,
D. J. (1976) Multielement absorption by crops grown in pots on
municipal sludge-amended soil. J. agric. Food Chem.,
24: 889-892.
FURR, A. K., PARKINSON, T. F., HINRICHS, R. A., VAN CAMPEN, D. R.,
BACHE, C. A. GUTENMANN, W. H., T S. JOHN, L. E. JR, PAKKALA, I. S.
& LISK, D. J. (1977) National survey of elements and radioactivity
in fly ashes. Absorption of elements by cabbage grown in fly
ash-soil mixtures. Environ. Sci. Technol., 11: 1194-1201.
GABOR, S. & COLDEA, V. (1977) Some aspects of the environmental
exposure to arsenic in Romania. Environ. health Perspect.,
19: 107-108.
GAINER, J. H. (1972) Effects of arsenicals on interferon formation and
action. Am. J. vet. Res., 33: 2579-2586.
GAINER, J. H. & PRY, T. W. (1972) Effects of arsenicals on viral
infections in mice. Am. J. vet. Res., 33: 2299-2307.
GALLORINI, M., GREENBERG, R. R. & GILLS, T. E. (1978) Simultaneous
determination of arsenic, antimony, cadmium, chromium, copper, and
selenium in environmental material by radiochemical neutron
activation analysis. Anal. Chem., 50: 1479-1481.
GALY, P., TOURAINE, R., BRUNE, J., ROUDIER, P. & GALLOIS, P. (1963) Le
cancer pulmonaire d'origine arsénicale des vignerons du
Beaujolais. J. Fr. Med. Chir. thorac., 17: 303-311.
GARB, L. G. & HINE, C. H. (1977) Arsenical neuropathy: Residual
effects following acute industrial exposure. J. occup. Med.,
19(8): 567-568.
GASTINER, E. (1972) [On the spectrophotometric determination of
arsenic using silves diethyl-thiocarbamidate.] Mikrochim. Acta,
1972: 526-543 (in German).
GEYER, L. (1898) [Chronic skin changes in arsenism and reflections on
the great number of cases at Reichehnstein, Silesia.] Arch.
Dermatol. Syphilol., 43: 221-280 (in German).
GINSBURG, J. M. (1965) Renal mechanism for excretion and
transformation of arsenic in the dog. Am. J. Physiol.,
208: 832-840.
GINSBURG, J. M. & LOTSPEICH, W. D. (1963) Interrelations of arsenate
and phosphate transport in the dog kidney. Am. J. Physiol.,
205: 707-714.
GOLDBLATT, E. L., VANDENBURGH, A. S. & MARSLAND, R. A. (1963)
The usual and widespread occurrence of arsenic in well waters
of Lane county, Oregon, Oregon Department of Health.
GOLDMAN, A. L. (1973) Lung cancer in Bowen's disease. Am. Rev.
respir. Dis., 108: 1205-1207.
GOLDSMITH, J. R., DEANE, M., THOM, J. & GENTRY, G. (1972) Evaluation
of health implications of elevated arsenic in well water. Water
Res., 6: 1133-1136.
GONZALEZ, S. G. (1977) [Acute arsenic poisoning in dairy cattle in the
Lagoon Region.] In: Memorias del I Simposium International de
Laboratorios Veterinarios de Diagnósticos, México, Volume III,
pp. 551-560 (in Spanish).
GOODE, S. R. & MATTHEWS, R. J. (1978) Enzyme -- catalyzed
reaction-rate method for determination of arsenic in water.
Anal. Chem., 50: 1608-1610.
GORDON, J. J. & QUASTEL, J. H. (1948) Effects of organic arsenicals on
enzyme systems. Biochem. J., 42: 337-350.
GOSSELIN, R. E., HODGE, H. C., SMITH, R. P. & GLEASON, M. N. (1976)
Clinical toxicology of commercial products, Baltimore, The
Williams & Wilkins Co.
GOULDEN, F., KENNAWAY, E. L. & URQUHART, M. E. (1952) Arsenic in the
suspended matter of town air. Br. J. Cancer, 6: 1-7.
GRANT, C. & DOBBS, A. J. (1977) The growth and metal content of plants
grown in soil contaminated by a copper/chrome/arsenic wood
preservative. Environ. Pollut., 14: 213-226.
GRANTHAM, D. A. & JONES, J. F. (1977) Arsenic contamination of water
wells in Nova Scotia. J. Am. Water Works Assoc., 69: 653-657.
GRIFFIN, H. R., HOCKING, M. B. & LOWERY, D. G. (1975) Arsenic
determination in tobacco by atomic absorption spectrometry.
Anal. Chem., 47: 229.
GRIMANIS, A. P., VASSILAKI-GRIMANI, M., ALEXIOU, D. & PAPADATOS, C.
(1979) Determination of seven trace elements in human milk,
powdered cow's milk and infant foods In: Proceedings of a
Symposium. Nuclear Activation Techniques in the Life Sciences,
Vienna 1978, Vienna, International Atomic Energy Agency,
pp. 241-253.
GROBE. J.-W. (1976) [Peripheral circulatory disorders and acrocyanosis
in Moselle valley vineyard workers with arsenic poisoning.]
Berufsdermatosen, 24(3): 78-84.
GULLEDGE, J. H. & O'CONNOR, J. T. (1973) Removal of arsenic(V) from
water by adsorption on aluminum and ferric hydroxides. J. Am.
Water Works Assoc., 65: 548.
HALVER, J. E. (1962) Cited in: KRAYBILL, H. F. & SHIMKIN, M. B. (1964)
Carcinogenesis related to foods contaminated by processing and
fungal metabolites. Adv. Cancer Res., 8: 205.
HAMADA, T. & HORIGUCHI, S. (1976) Occupational chronic arsenical
poisoning. On the cutaneous manifestations. Jpn. J. ind. Health,
18: 103-115.
HAMAMOTO, E. (1955) Infant arsenic poisoning by powdered milk. Nihon
Iji Shimpo, No. 1649, pp. 3-12 (in Japanese).
HAMILTON, E. I. & MINSKI, M. J. (1973) Abundance of the chemical
elements in man's diet and possible relations with environmental
factors. Sci. total Environ., 1: 375-394.
HAMMER, D. I., FINKLEA, J. F., HENDRICKSON, R. H., SHY, C. M. &
HORTON, R. J. M. (1971) Hair trace metal levels and environmental
exposure. Am. J. Epidemiol., 93: 84-92.
HANLON, D. P. & FERM, V. H. (1977) Placental permeability of arsenate
ion during early embryogenesis in the hamster. Experientia
(Basle), 33: 1221-1222.
HARA, I., HASHIMOTO, K., MIYAZAKI, K. & SUNADA, K. (1968) A case
report on multiple neuritis arsenic poisoning. Saigai Igaku,
11: 84-90.
HARRINGTON, J. M., MIDDAUGH, J. P., MORSE, D. L., & HOUSWORTH, J.
(1978) A survey of a population exposed to high concentrations of
arsenic in well water in Fairbanks, Alaska. Am. J. Epidemiol.,
108: 377-385.
HARRIS, E. J. & ACHENJANG, F. M. (1977) Energy-dependent uptake of
arsenite by rat liver mitochondria. Biochem. J., 168: 129-132.
HARRISON, J. W. E., PACKMAN, E. W. & ABBOTT, D. D. (1958) Acute oral
toxicity and chemical and physical properties of arsenic
trioxides. A. M. A. Arch. ind. Health, 17: 118-123.
HAZRA, A. K. & PROKUPOK, R. (1977) A report on air quality in
Yellowknife, Northwest Territories, Environment, Canada
(Surveillance report EPS-5-NW-77-55).
HEYDORN, K. (1969) Environmental variation of arsenic levels in human
blood determined by neutron activation analysis. Clin. Chim.
Acta, 28: 349-357.
HEYDORN, K. & DAMSGAARD, E. (1973) Simultaneous determination of
arsenic, manganese and selenium in biological materials by
neutron-activation analysis. Talanta, 20: 1-11.
HEYMAN, A., PFEIFFER, J. B., JR., WILLETT, R. W. & TAYLOR, H. M.
(1956) Peripheral neuropathy caused by arsenical intoxication. A
study of 41 cases with observations on the effects of BAL
(2,3-dimercaptopropanol). New Engl. J. Med., 254: 401-409.
HEYWOOD, R. & SORTWELL, R. J. (1979) Arsenic intoxication in the
rhesus monkey. Toxicol. Lett., 3: 127-144.
HILL, A. B. & FANING, E. L. (1948) Studies in the incidence of cancer
in a factory handling inorganic compounds of arsenic. I. Mortality
experience in the factory. Br. J. ind. Med., 5: 1-6.
HINDMARSH, J. T., MCLETCHIE, O. R., HEFFERNAN, L. P. M., HAYNE, O. A.,
ELLENBERGER, H. A., MCCURDY, R. F. & THIEBAUX, H. J. (1977)
Electromyographic abnormalities in chronic environmental
arsenicalism. J. anal. Toxicol., 1: 270-276.
HINE, C. H., PINTO, S. S. & NELSON, K. W. (1977) Medical problems
associated with arsenic exposure. J. occup. Med., 19: 391-396.
HJERN, L. (1961) [The arsenic content in Swedish cigarettes.] Nord.
Hyg. Tidskr., 42: 272-276 (in Swedish).
HOCKING, D., KUCHAR, P., PLAMBECK, J. A. & SMITH, R. A. (1978) The
impact of gold smelter emissions on vegetation and soils of a
subarctic forest-tundra transition ecosystem. J. Air Pollut.
Control Assoc., 28: 133-137.
HODGE, H. C. & EMBREE, J. W. (1977) Estimation of the mutagenicity of
arsenic compounds utilizing dominant lethal mutations in mice,
San Francisco, University of California Toxicology Research
Laboratory (Final report, Contract No. SERA-HI-2).
HOGAN, R. B. & EAGLE, H. (1944) The pharmacologic basis for the widely
varying toxicity of arsenicals. J. Pharmacol. exp. Ther.,
80: 93-113.
HOLAK, W. (1969) Gas-sampling technique for arsenic determination by
atomic absorption spectroscopy. Anal. Chem., 41: 1712-1713.
HOLLAND, R. H. & ACEVEDO, A. R. (1966) Current status of arsenic in
American cigarettes. Cancer, 19: 1248-1250.
HOLLAND, R. H., MCCALL, M. S. & LANZ, H. C. (1959) A study of inhaled
arsenic-74 in man. Cancer Res., 19: 1154-1156.
HOLLINS, J. G., CHARBONNEAU, S. M., BRYCE, F., RIDGEWAY, J. M., TAM,
G. K. H. & WILLES, R. F. (1979) Whole body retention and excretion
of [74As] arsenic acid in the adult beagle dog. Toxicol. Lett.,
4: 7-13.
HOLMBERG, R. E. & FERM, V. H. (1969) Interrelationships of selenium,
cadmium, and arsenic in mammalian teratogenesis. Arch. environ.
Health, 18: 873-877.
HOLMQVIST, I. (1951) Occupational arsenical dermatitis. A study among
employees at a copper ore smelting work including investigations
of skin reactions to contact with arsenic compounds. Acta Derm.
Venereol., 31: (Suppl. 26): 1-214.
HOLMQVIST, I. (1964) [Causes of death among workers at
Rönnskärsverken.] Internal report to the management, Boliden AB,
Skelleftehamn, Sweden (in Swedish).
HOLMQVIST, I. (1975) Investigations of the absorption of some metals
among people in the surrounding area of a smelting plant. In:
Proceedings from Recent Advances in the Assessment of the Health
Effects of Environmental Pollution. Vol. II. Paris, June 24-28,
1974, Luxembourg, Commission of the European Communities
Directorate General for Dissemination of Knowledge, Center for
Information and Documentation CID.
HOOD, R. D. (1972) Effects of sodium arsenite on fetal development.
Bull. environ. Contam. Toxicol., 7: 216-222.
HOOD, R. D. & BISHOP, S. L. (1972) Teratogenic effects of sodium
arsenate in mice. Arch. environ. Health, 24: 62-65.
HOOD, R. D. & PIKE, C. T. (1972) BAL alleviation of arsenate-induced
teratogenesis in mice. Teratology, 6: 235-237.
HOOD, R. D., THACKER, G. T. & PATTERSON, B. L. (1977) Effects in the
mouse and rat of prenatal exposure to arsenic. Environ. health
Perspect., 19: 219-222.
HORIGUCHI, S., NAKANO, H., SHINAGAWA, K., TERAMOTO, K., & KIYOTA, I.
(1976) A long-term observation of the environmental conditions of
a factory manufacturing lead arsenate as an insecticide. (Studies
on lead arsenate poisoning, part I). Osaka City med. J.,
22(1): 43-46.
HOVE, E., ELVEHJEM, C. A. & HART, E. B. (1938) Arsenic in the
nutrition of the rat. Am. J. Physiol., 124: 205-212.
HUEPER, W. C. (1954) Experimental studies in metal cancerigenesis.
VI. Tissue reactions in rats and rabbits after parenteral
introduction of suspensions of arsenic, beryllium, or asbestos in
lanolin. J. Natl Cancer Inst., 15: 113-124.
HUEPER, W. C. & ITAMI, S. (1933) Effects of neoarsphenamine on
spontaneous breast tumors of mice. Am. J. Cancer., 17: 106-115.
HUEPER, W. C. & PAYNE, W. W. (1962) Experimental studies in metal
carcinogenesis. Chromium, nickel, iron, arsenic. Arch. environ.
Health, 5: 445-462.
HUET, P.-M., GUILLAUME, E., COTÉ, J., LÉGARÉ, A., LAVOIE, P. &
VIALLET, A. (1975) Noncirrhotic presinusoidal portal hypertension
associated with chronic arsenic intoxication. Gastroenterology,
68: 1270-1277.
HUNDEIKER, M. & PETRES, J. (1968) [Morphogenesis and variety of
procancerous lesions induced by arsenic.] Arch. klin. exp.
Dermatol., 231: 355-365 (in German).
HUNDLEY, H. K. & UNDERWOOD, J. C. (1970) Determination of total
arsenic in total diet samples. J. Assoc. Off. Anal. Chem.,
53: 1176-1178.
HUNTER, F. T., KIP, A. F. & IRVINE, J. W., Jr (1942) Radioactive
tracer studies on arsenic injected as potassium arsenite.
J. Pharmacol. exp. Ther., 76: 207-220.
HUTCHINSON, J. (1888) On some examples of arsenic-keratoses of the
skin and of arsenic-cancer. Trans. Pathol. Soc. (London),
39: 352-363.
HWANG, S. W. & SCHANKER, L. S. (1973) Absorption of organic arsenical
compounds from the rat small intestine. Xenobiotica, 3: 351-355.
IARC (1973) Monographs. Evaluation of carcinogenic risk of chemicals
to man. Some inorganic and organometallic compounds, Volume 2,
Lyons, International Agency for Research on Cancer.
IARC (1980) Monographs. Arsenic and its compounds, Volume 23, Lyons,
International Agency for Research on Cancer, pp. 39-141.
ICRP (1975) Task group on reference man. Oxford, Pergammon Press
(International Commission on Radiological Protection Publication
No. 23).
IRGOLIC, K. J., WOOLSON, E. A., STOCKTON, R. A., NEWMAN, R. D.,
BOTTINO, N. R., ZINGARO, R. A., KEARNEY, P. C., PYLES, R. A.,
MAEDA, S., MCSHANE, W. J. & COX, E. R. (1977) Characterization of
arsenic compounds formed by Daphnia magna and Tetraselmis chuii
from inorganic arsenate Environ. health Perspect., 19: 61-66.
IRLG, Work Group on Risk Assessment (1979) Scientific bases for
identifying potential carcinogens and estimating their risks,
Washington, DC, Interagency Regulatory Liaison Group (IRLG).
ISHINISHI, N. (1973) Review on toxicity of arsenic and arsenic
compounds. Nippon Rinsho, 31(6): 75-83.
ISHINISHI, N., KODAMA, Y. & INAMAZ. U. T. (1974) Clinical examination
of the effects of arsenic for the health. Rynsho-Kagaku,
3: 176-187.
ISHINISHI, N., OSATO, K., KODAMA, Y. & KUNITAKE, E. (1976) Skin
effects and carcinogenicity of arsenic trioxide: A preliminary
experimental study in rats. In: Nordberg, G. F., ed. Effects and
dose-response relationships of toxic metals, Amsterdam, Elsevier
Scientific Publishing Company, pp. 471-479.
ISHINISHI, N., KODAMA, Y., NOBUTOMO, K. & HISANAGA, A. (1977)
Preliminary experimental study on carcinogenicity of arsenic
trioxide in rat lung. Environ. health Perspect., 19: 191-196.
ISHINISHI, N., MIZUNOE, M., INAMASU, T. & HISANAGA, A. (1980a)
Experimental study on carcinogenicity of beryllium oxide and
arsenic trioxide to the lung of rats by an intratracheal
instillation. Fukuoka Acta Med., 71: 19-26.
ISHINISHI, N., TOMITA, M. & HISANAGA, A. (1980b) Study on chronic
toxicity of arsenic trioxide in rats with special reference to the
liver damages. Fukuoka Acta Med., 71: 27-40.
IVANKOVIC, S., EISENBRAND, G. & PREUSSMANN, R. (1979) Lung carcinoma
induction in BD rats after single intratracheal instillation of an
arsenic-containing pesticide mixture formerly used in vineyards.
Int. J. Cancer, 24: 786-789.
JACKSON, R. & GRAINGE, J. W. (1975) Arsenic and cancer. Can. Med.
Assoc. J., 113: 396-401.
JELINEK, C. F. & CORNELIUSSEN, P. E. (1977) Levels of arsenic in the
United States food supply. Environ. health Perspect., 19: 83-87.
JENKINS, R. B. (1966) Inorganic arsenic and the nervous system.
Brain, 89: 479-498.
JERNELÖV, A., HULTBERG, H. & ROSENBLUM, I. (1976) The geothermal
plant at Ahuachapan in El Salvador and its possible effects on
the water quality on the border river Rio Paz between El Salvador
and Guatemala, Stockholm, Institute for Water and Air Pollution
(Report No. IVL-B-297).
JILER, H., ed. (1975) Commodity year book 1975, New York, Commodity
Research Bureau.
JOHNSON, D. L. (1972) Bacterial reduction of arsenate in sea water.
Nature (Lond.), 240: 44-45.
JOHNSON, D. L. & BRAMAN, R. S. (1975a) Alkyl- and inorganic arsenic in
air samples. Chemosphere, 6: 333-338.
JOHNSON, D. L. & BRAMAN, R. S. (1975b) The speciation of arsenic and
the content of germanium and mercury in members of the Pelagic
Sargassum community. Deep-sea Res., 22: 503.
JOHNSON, D. L. & BURKE, R. M. (1978) Biological mediation of chemical
speciation II arsenate reduction during marine phytoplankton
bloom. Chemosphere, 8: 645-648.
JOHNSON, D. L. & PILSON, M. E. Q. (1972) Spectrophotometric
determination of arsenite, arsenate and phosphate in natural
waters. Anal. Chim. Acta, 48: 289.
JOHNSON, D. L. & PILSON, M. E. Q. (1975) The oxidation of arsenite in
seawater. Environ. Lett., 8: 157-171.
JOHNSTONE, R. M. (1963) Sulfhydryl agents: Arsenicals. In: Hochster,
R. M. & Quastel, J. H., ed. Metabolic inhibitors. A comprehensive
treatise, Volume 2, New York, Academic Press.
JUNG, E. (1971) [Research in the molecular biology of arsenism.]
Z. Haupt. Geschlechtskr., 46: 35-36 (in German).
KAAKINEN, J. W., JORDEN, R. M., LAWASANI, M. H. & WEST, R. E. (1975)
Trace element behavior in coal-fired power plant. Environ. Sci
Technol., 9: 862-869.
KADOWAKI, K. (1960) [Studies on the arsenic contents in organ-tissues
of the normal Japanese.] Osaka City med. J., 9: 2083-2099
(in Japanese with English summary).
KAGAWA, Y. & KAGAWA, A. (1969) Accumulation of arsenate-76 by
mitochondria. J. Biochem., 65(1): 105-112.
KAGEY, B. T., BUMGARNER, J. E. & CREASON, J. P. (1977) Arsenic levels
in maternal-fetal tissue sets. In: Hemphill, D. D., ed. Trace
substances in environmental health XI. A symposium. Columbia,
University of Missouri Press, pp. 252-256.
KANISAWA, M. & SCHROEDER, H. A. (1969) Life term studies on the effect
of trace elements on spontaneous tumors in mice and rats. Cancer
Res., 29: 892-895.
KATSURA, K. (1958) Medicolegal studies of arsenic poisoning. II.
Distribution of arsenic in visceral organs and arsenic
concentrations of bone and hair in arsenic poisoning. Shikoku
Igaku Zasshi, 12: 706-720.
KELLO, D. & KOSTIAL, K. (1973) The effect of milk diet on lead
metabolism in rats. Environ. Res., 6: 355-360.
KELLO, D. & KOSTIAL, K. (1977) Influence of age and milk diet on
cadmium absorption from the gut. Toxicol. appl. Pharmacol.,
40: 277-282.
KENNEDY, V. S. (1976) Arsenic concentrations in some coexisting marine
organisms from Newfoundland and Labrador. J. Fish. Res. Board
Can., 33: 1388-1393.
KINGSLEY, G. R. & SCHAFFERT, R. R. (1951) Microdetermination of
arsenic and its application to biological materials. Anal. Chem.,
23: 914.
KIRKBRIGHT, G. F. & RANSON, L. (1971) Use of the nitrous oxide --
acetylene flame for determination of arsenic and selenium by
atomic absorption spectrometry. Anal. Chem., 43: 1238-1241.
KIRKBRIGHT, G. F., WARD, A. F. & WEST, T. S. (1973) Atomic emission
spectrometry with an induction -- coupled high frequency plasma
source. The determination of iodine, mercury, arsenic and
selenium. Anal. Chim. Acta, 64: 353.
KLAASSEN, C. D. (1974) Biliary excretion of arsenic in rats, rabbits,
and dogs. Toxicol. appl. Pharmacol., 29: 447-457.
KNOLLE, J., FÖRSTER, E., RÖSSNER, A., THEMANN, H., HÖHN, P. & MEYER
ZUM BÜSCHENFELDE, K.-H. (1974) [Non-cirrhotic portal fibrosis
(hepatoportal sclerosis) following arsenism.] Dtsch. Med.
Wochenschr., 99: 903-908 (in German).
KODAMA, Y., ISHINISHI, N., KUNITAKE, E., INAMASU, T. & NOBUTOMO, K.
(1976) Subclinical signs of the exposure to arsenic in a copper
refinery. In: Nordberg, G. F., ed. Effects and dose-response
relationships of toxic metals, Amsterdam, Elsevier Scientific
Publishing Company, pp. 464-470.
KOELSCH, F. (1958) [Occupational arsenic lesions in vineyards and
other places of employment.] Arch. Gewerbepathol. Gewerbehyg.,
16: 405-438 (in German).
KONINGS, A. W. T. (1972) Inhibition of nuclear and mitochondrial
respiration by arsenite. Experientia (Basel), 28: 883-884.
KOPP, J. F. (1973) l-Ephedrine in chloroform as a solvent for silver
diethyldithiocarbamate in the determination of arsenic. Anal.
Chem., 45: 1786-1787.
KRAYBILL, H. F. & SHIMKIN, M. B. (1964) Carcinogenesis related to
foods contaminated by processing and fungal metabolites.
Adv. Cancer Res., 8: 205.
KROES, R., VAN LOGTEN, M. J., BERKVENS, J. M., DE VRIES, T. & VAN
ESCH, G. J. (1974) Study on the carcinogenicity of lead arsenate
and sodium arsenate and on the possible synergistic effect of
diethylnitrosamine. Food cosmet. Toxicol., 12: 671-679.
KUO, T. (1968) Arsenic content of artesian well water in endemic area
of chronic arsenic poisoning. Rep. Inst. Pathol. Natl Taiwan
Univ., 20: 7-13.
KYLE, R. A. & PEASE, G. L. (1965) Hematologic aspects of arsenic
intoxication. New. Engl. J. Med., 273: 18-23.
LAKSO, J. U. & PEOPLES, S. A. (1975) Methylation of inorganic arsenic
by mammals. J. agric. Food Chem., 23: 674-676.
LANCASTER, R. J., COUP, M. R. & HUGHES, J. W. (1971) Toxicity of
arsenic present in lakeweed. N.Z. vet. J., 19: 141-145.
LANDER, H., HODGE, P. R. & CRISP, C. S. (1965) Arsenic in hair and
nails. Its significance in acute arsenical poisoning. J. Forensic
Med., 12: 52-67.
LANDER, J. J., STANELY, R. J., SUMNER, H. W., BOSWELL, D. C. & AACH,
R. D. (1975) Angiosarcoma of the liver associated with Fowler's
solution (potassium arsenite). Gastroenterology, 68: 1582-1586.
LANSCHE, A. M. (1965) Arsenic. In: Mineral facts and problems,
Washington, DC, US, Department of the Interior (Bureau of Mines
Bulletin 630).
LANZ, H., Jr., WALLACE, P. C. & HAMILTON, J. G. (1950) The metabolism
of arsenic in laboratory animals using As74 as a tracer. Univ.
California Publ. Pharmacol., 2: 263-282.
LAO, R. C., THOMAS, R. S., TEICHMAN, T. & DUBOIS, L. (1974) Efficiency
of collection of arsenic trioxide in high volume sampling.
Sci. total Environ., 2: 373-379.
LARSEN, N. A., NIELSEN, B., PAKKENBERG, H., CHRISTOFFERSEN, P.,
DAMSGAARD, E. & HEYDORN, K. (1972) Neutron activation analysis of
arsenic, manganese and selenium concentrations in organs of
uraemic and normal persons. In: Kripper, M., ed. Proceedings of a
Symposium, Bled, Yugoslavia, pp. 561-568 (IAEA-SM-157/4).
LARSEN, N. A., PAKKENBERG, H., DAMSGAARD, E. & HEYDORN, K. (1979)
Topographical distribution of arsenic, manganese, and selenium in
the normal human brain. J. neurol. Sci., 42: 407-416.
LAUWERYS, R. R., BUCHET, J. P. & ROELS, H. (1979) The determination of
trace levels of arsenic in human biological materials.
Arch. Toxicol., 41: 239-247.
LEATHERLAND, T. M. & BURTON, J. D. (1974) The occurrence of some trace
metals in coastal organisms with particular reference to the
Solent region. J. Mar. Biol. Assoc. U.K., 54: 457-468.
LEBLANC, P. J. & JACKSON, A. L. (1973) Arsenic in marine fish and
invertebrates. Mar. pollut. Bull., 4: 88-90.
LEDET, A. E., DUNCAN, J. R., BUCK, W. B. & RAMSEY, F. K. (1973)
Clinical, toxicological, and pathological aspects of arsanilic
acid poisoning in swine. Clin. Toxicol., 6: 439-457.
LEE, A. M. & FRAUMENI, J. F., Jr (1969) Arsenic and respiratory cancer
in man: an occupational study. J. Natl Cancer Inst., 42:
1045-1052.
LEITCH, A. & KENNAWAY, E. L. (1922) Experimental production of cancer
by arsenic. Brit. med. J., 2: 1107-1108.
LENVIK, K., STEINNES, E. & PAPPAS, A. C. (1978) Contents of some heavy
metals in Norwegian rivers. Nord. Hydrol., 9: 197-206.
LE QUESNE, P. M. & MCLEOD, J. G. (1977) Peripheral neuropathy
following a single exposure to arsenic. J. neurol. Sci.,
32: 437-451.
LESLIE, A. C. D. & SMITH, H. (1978) Self-poisoning by the abuse of
arsenic containing tonics. Med. Sci. Law, 18: 159-162.
LEVANDER, O. A. (1977) Metabolic interrelationships between arsenic
and selenium. Environ. health Perspect., 19: 159-164.
LIEBHARDT, W. C. (1976) Nutrient concentrations of corn as affected by
poultry manure. Soil Sci. plant Anal., 7: 169.
LIEBSCHER, K. & SMITH, H. (1968) Essential and nonessential trace
elements. A method of determining an element is essential or
nonessential in human tissue. Arch. environ. Health,
17: 881-890.
LINDAU, L. (1977) Emissions of arsenic in Sweden and their reduction.
Environ. health Perspect., 19: 25-29.
LÖFROTH, G. & AMES, B. N. (1978) Mutagenicity of inorganic compounds
in Salmonella typhimurium: arsenic, chromium and selenium.
Mutat. Res., 54: 65-66.
LOWRY, O. H., HUNTER, F. T., KIP, A. F. & IRVINE, J. W., Jr (1942)
Radioactive tracer studies on arsenic injected as potassium
arsenite. II. Chemical distribution in tissues. J. Pharmacol.
exp. Ther., 76: 221-225.
LU, F.-J., YANG, C.-K. & LIN, K.-H. (1975) Physico-chemical
characteristics of drinking water in Blackfoot endemic areas in
Chia-I and Tainan Hsiens. J. Formosan Med. Assoc., 79: 596-605.
LÜCHTRATH, H. (1972) [Cirrhosis of the liver in the arsenism of
vineyard workers.] Dtsch. Med. Wochenschr., 97:21-22
(in German.)
LUGO, G., CASSADY, G. & PALMISANO, P. (1969) Acute maternal arsenic
intoxication with neonatal death. Am. J. Dis. Child.,
117: 328-330.
LUNDE, G. (1970) Analysis of trace elements in seaweed. J. Sci. Food
Agric., 21: 416-418.
LUNDE, G. (1972) The absorption and metabolism of arsenic in fish.
Teknologiske Undersokelser, 5: 1-16.
LUNDE, G. (1973a) The synthesis of fat and water soluble arseno
organic compounds in marine and limnetic algae. Acta Chem.
Scand., 27: 1586-1594.
LUNDE, G. (1973b) Separation and analysis of organic-bound and
inorganic arsenic in marine organisms. J. Sci. Food Agric,
24: 1021-1027.
LUNDE, G. (1975) Isolation of an organoarsenic compound present in cod
liver. J. Sci. Food Agric., 26: 1247-1255.
LUNDE, G. (1977) Occurrence and transformation of arsenic in the
marine environment. Environ. health Perspect., 19: 47-52.
LUNDGREN, K. D. (1954) [Damages in the respiratory organs of workers
at a smeltery.] Nord. Hyg. Tidskr., 3: 66-82 (in Swedish).
MABUCHI, K., LILIENFELD, A. M. & SNELL, L. M. (1979) Lung cancer among
pesticide workers exposed to inorganic arsenicals. Arch. environ.
Health, 34: 312-319.
MAES, D. & PATE, B. D. (1977) The absorption of arsenic into single
human head hairs. J. forensic Sci., 22: 89-94.
MAHAFFEY, K. R. & FOWLER, B. A. (1977) Effects of concurrent
administration of lead, cadmium, and arsenic in the rat. Environ.
health Perspect., 19: 165-171.
MAPPES, R. (1977) [Experiments on the excretion of arsenic in urine.]
Int. Arch. occup. environ. Health, 40: 267-272 (in German).
MARTINDALE, W. (1977) The extra pharmacopoeia, 27th edition, London,
The Pharmaceutical Press.
MARUYAMA, Y., KOMIYA, K. & MANRI, T. (1970) Determination of copper,
arsenic and mercury in cigarettes by neutron activation analysis.
Radioisotopes, 19: 44-46.
MARUYAMA, Y. & KOMIYA, K. (1973) Determination of copper, arsenic, and
mercury in tobacco leaves by neutron activation analysis.
Radioisotopes, 5: 279.
MASSMANN, W. & OPITZ, H. (1954) [Experimental research on ECG changes
in arsenism.] Z. Kreislaufforsch., 43:704-713 (in German).
MATANOSKI, G., LANDAU, E. & ELLIOTT, E. (1976) Epidemiology studies.
Task I. Pilot study of cancer mortality near an arsenical
pesticide plant in Baltimore, Washington, DC, US Environmental
Protection Agency (EPA 560/6-76-003).
McBRIDE, B. C. & WOLFE, R. S. (1971) Biosynthesis of dimethylarsine by
methanobacterium. Biochemistry, 10: 4312-4317.
McBRIDE, B. C., MERILEES, H., CULLEN, W. R. & PICKETT, W. (1978)
Anaerobic and aerobic alkylation of arsenic. In: Brinckman, F. E.
& Bellama, J. M., ed. Organometals and organometalloids,
Washington, DC, American Chemical Society, pp. 94-115
(ACS Symp. Ser. 82).
McCABE, L. J., SYMONS, J. M., LEE, R. D. & ROBECK, G. G. (1970) Survey
of community water supply systems. J. Am. Water Works Assoc.,
62: 670-687.
McCHESNEY, E. W., HOPPE, J. O., McAULIFF, J. P. & BANKS, W. F., Jr.
(1962) Toxicity and physiological disposition of sodium
p-N-glycolylarsanilate. I. Observations in the mouse, cat, rat,
and man. Toxicol. appl. Pharmacol., 4: 14-23.
McDANIEL, M., SHENDRIKAR, A. D., REISZNER, K. D. & WEST, P. W. (1976)
Concentration and determination of selenium from environmental
samples. Anal. Chem., 48: 2240-2243.
MEALEY, J., JR, BROWNELL, G. L. & SWEET, W. H. (1959) Radioarsenic in
plasma, urine, normal tissues, and intracranial neoplasms. Arch.
Neurol. Psychiatr., 81: 310-320.
MENIS, O. & RAINS, T. C. (1969) Determination of arsenic by atomic
absorption spectrometry with electrodeless discharge lamp as
source of radiation. Anal. Chem., 41: 952-954.
MILHAM, S., JR (1977) Studies of morbidity near a copper smelter.
Environ. health Perspect., 19: 131-132.
MILHAM, S., JR & STRONG, T. (1974) Human arsenic exposure in relation
to a copper smelter. Environ. Res., 7: 176-182.
MILNER, J. E. (1969) The effect of ingested arsenic on
methylcholantherene-induced skin tumors in mice. Arch. environ.
Health, 18: 7-11.
MINKKINEN, P. & YLIRUOKANEN, I. (1978) The arsenic distribution in
Finnish peat bogs. Kemia-Kemi, 7-8: 331-335.
MITCHELL, R. A., CHANG, B. F., HUANG, C. H. & DeMASTER, E. G. (1971)
Inhibition of mitochondrial energy-linked functions by arsenate.
Evidence for a nonhydrolytic mode of inhibitor action.
Biochemistry, 10 (11): 2049-2054.
MIZUTA, N., MIZUTA, M., ITO, F., ITO, T., UCHIDA, H., WATANABE, Y.,
AKAMA, H., MURAKAMI, T., HAYASHI, F., NAKAMURA, K., YAMAGUCHI, T.,
MIZUIA, W., OISHI, S. & MATSUMURA, H. (1956) An outbreak of acute
arsenic poisoning caused by arsenic contaminated soy-sauce
(shoyu): A clinical report of 220 cases. Bull. Yamaguchi Med.
Sch., 4: 131-150.
MOHELSKA, H., BENCKO, V., SMETANA, K. & HYNCICA, V. (1980)
Ultrastructural changes in hepatocytes of mice exposed to arsenic
in drinking water. Exp. Pathol. (Jena), 18 (5): 275-281.
MOLIN, L. & WESTER, P. O. (1976) The estimated daily loss of trace
elements from normal skin by desquamation. Scand. J. clin. lab.
Invest., 36: 679-682.
MOODY, J. P. & WILLIAMS, R. T. (1964a) The fate of arsanilic acid and
acetylarsanilic acid in hens. Food cosmet. Toxicol., 2: 687-693.
MOODY, J. P. & WILLIAMS, R. T. (1964b) The fate of
4-nitrophenylarsonic acid in hens. Food cosmet. Toxicol.,
2: 695-706.
MOODY, J. P. & WILLIAMS, R. T. (1964c) The metabolism of
4-hydroxy-3-nitrophenylarsonic acid in hens. Food cosmet.
Toxicol., 2: 707-715.
MORAN, S., MATURANA, G., ROSENBERG, H., CASANEGRA, P. & DUBERNET, J.
(1977) Occlusions coronariennes liées à une intoxication
arsenicale chronique. Arch. Mal. Coeur, 70 (10): 1115-1120.
MORRIS, H. P., LAUG, E. P., MORRIS, H. J. & GRANT, R. L. (1938) The
growth and reproduction of rats fed diets containing lead acetate
and arsenic trioxide and the lead and arsenic content of newborn
and suckling rats. J. Pharmacol. exp. Ther., 64: 420-445.
MORRIS, J. S., SCHMID, M., NEWMAN, S., SCHEUER, P. J. & SHERLOCK, S.
(1974) Arsenic and noncirrhotic portal hypertension.
Gastroenterology, 64: 86-94.
MORSE, D. L., HARRINGTON, J. M., HOUSWORTH, J. & LANDRIGAN, P. J.
(1979) Arsenic exposure in multiple environmental media in
children near a smelter. Clin. Toxicol., 14: 389-399.
MORTON, W., STARR, G., POHL, D., STONER, J., WAGNER, S. & WESWIG, P.
(1976) Skin cancer and water arsenic in Lane County, Oregon.
Cancer, 37: 2523-2532.
MUNRO, I. C. (1976) Naturally occurring toxicants in foods and their
significance. Clin. Toxicol., 9: 647-663.
MUNRO, I. C., CHARBONNEAU, S. M., SANDI, E., SPENCER, K., BRYCE, F. &
GRICE, H. C. (1974) Biological availability of arsenic in fish.
In: Proceedings at the 13th annual meeting of the Society of
Toxicology, Washington, March 1974, 9 pp.
MYERS, D. J. & OSTERYOUNG, J. (1973) Determination of arsenic (III) at
the parts-per-billion level by differential pulse polarography.
Anal. Chem., 45: 267-271.
NAGAMATSU, K. & IGATA, A. (1975) [Arsenical neuropathy. Report of two
cases.] Rinsho-Shinkei, 15: 1-4 (in Japanese with English
summary).
NAKAGAWA, Y. & IIBUCHI, Y. (1970) On the follow-up investigation of
Morinaga milk arsenic poisoning. Igaku no Ayumi, 74: 1-3.
NAKAHARA, H., YANOKURA, M. & MURAKAMI, Y. (1978) Environmental effects
of geothermal waste water on the near-by river system.
J. radioanal. Chem., 45: 25-36.
NAKAO, M. (1960) [A study on the arsenic content in daily food
consumption in Japan.] Osaka City Med. J., 9: 541-571 (in
Japanese with English summary).
NAS (1977) Medical and biologic effects of environmental pollutants:
Arsenic, Washington, DC, National Academy of Sciences.
NATUSCH, D. F. S., WALLACE, J. R. & EVANS, C. A. (1974) Toxic trace
elements: preferential concentration in respirable particles.
Science, 183: 202-204.
NEALE, G. & AZZOPARDI, J. G. (1971) Chronic arsenical poisoning and
noncirrhotic portal hypertension -- a case for diagnosis.
Br. med. J., 4: 725-730.
NELSON, K. W. (1977) Industrial contributions of arsenic to the
environment. Environ. health Perspect., 19: 31-34.
NELSON, H. A., CRANE, M. R. & TOMSON, K. (1971) Inorganic arsenic
poisoning in pastured feeder lambs. J. Am. Vet. Med. Assoc.,
158: 1943-1945.
NELSON, W. C., LYKINS, M. H., MACKEY, J., NEWILL, V. A., FINKLEA, J.
F. & HAMMER, D. I. (1973) Mortality among orchard workers exposed
to lead arsenate spray: A cohort study. J. chron. Dis.,
26: 105-118.
NEUBAUER, O. (1947) Arsenical cancer: A review. Br. J. Cancer,
1: 192-251.
NEUJEAN, G., WEYTS, E. & BACQ, Z. M. (1948) Action du B.A.L. sur les
accidents ophtalmologiques de la thérapeutique à la tryparsamide.
Bull. Acad. R. Med. Belg., 13: 341-350.
NEWMAN, J. A., ARCHER, V. E., SACCOMANNO, G., KUSCHNER, M., AUERBACH,
O., GRONDAHL, R. D. & WILSON, J. C. (1976) Occupational
carcinogenesis. Histologic types of bronchogenic carcinoma among
members of coppermining and smelting communities. Ann. N.Y. Acad.
Sci., 271: 260-268.
NIOSH (1975) Occupational exposure to inorganic arsenic, Cincinnati,
Department of Health, Education, and Welfare, National Institute
for Occupational Safety and Health.
NISHIOKA, H. (1975) Mutagenic activities of metal compounds in
bacteria. Mutat. Res., 31: 185-189.
NOBLE, A. C., ORR, B. H., COOK, W. B. & CAMPBELL, J. L. (1976) Trace
element analysis of wine by proton-induced X-ray fluorescence
spectrometry. J. agric. Food Chem., 24: 532-535.
NORDBERG, G. F., ed. (1976.) Effects and dose-response relationships
of toxic metals, Amsterdam, Elsevier Scientific Publishing
Company.
NORDBERG, G. F., ed. (1978) Factors influencing metabolism and
toxicity of metals. Environ. health Perspect., 25: 3-41.
NORDENSON, I., BECKMAN, G., BECKMAN, L. & NORDSTRÖM, S. (1978)
Occupational and environmental risks in and around a smelter in
northern Sweden. II. Chromosomal aberrations in workers exposed to
arsenic. Hereditas, 88: 47-50.
NORDSTRÖM, S., BECKMAN, L. & NORDENSON, I. (1978) Occupational and
environmental risks in and around a smelter in northern Sweden.
III. Frequencies of spontaneous abortion. Hereditas, 88: 51-54.
NORDSTRÖM, S., BECKMAN, L. & NORDENSON, I. (1979) Occupational and
environmental risks in and around a smelter in northern Sweden. VI
Congenital malformations. Hereditas, 90: 297-302.
NOZAKI, S. (1972) Studies on arsenic metabolism. VI. Effect of
components of milk on arsenic tolerance in the digestive tract.
Folia Pharmacol. Jpn, 68: 857-868.
NOZAKI, S. (1973) Studies on the arsenic metabolism. VII. Added
arsenite in diet and resulting content in each organ, as well as
the effect on diet. Folia Pharmacol. Jpn, 69: 201-212.
NOZAKI, S., TSUTSUMI, S., & TAMURA, S. (1975) Effect of casein on
enteral absorption of arsenic trioxide. Jpn. J. Pharmacol.,
25: 122-123.
ODANAKA, Y., MATANO, O. & GOTO, S. (1978) Identification of
dimethylated arsenic by gas chromatography -- mass spectrometry in
blood, urine, and feces of rats treated with ferric
methanearsonate. J. agric. Food Chem., 26: 505-507.
OHIRA, M. & AOYAMA, H. (1972) Epidemiological studies on the Morinaga
powdered milk poisoning incident. Jpn. J. Hyg., 27: 500-531
(translated for Information Sciences Division, EPA, by Leo Kanner
Associates, Redwood City, California 94063).
ÖHMAN, H. (1960) [Some investigations of wood impregnated with
arsenic.] Stockholm, The National (Swedish) Institute of Public
Health (in Swedish).
OHTA, M. (1970) Ultra-structure of sural nerve in a case of arsenical
neuropathy. Acta Neuropathol. (Berl.), 16: 233-242.
OIDA, O. (1957) [Chronic occupational arsenic poisoning.]
Rodo-Kagaku, 33: 74-81 (in Japanese with English summary).
ONISHI, H. (1969) Arsenic. In: Wedepohl, K.H., ed. Handbook of
geochemistry, Volume II-2, Chapter 33, Berlin, Springer-Verlag.
OPPENHEIM, J. J. & FISHBEIN, W. N. (1965) Induction of chromosome
breaks in cultured normal human leukocytes by potassium arsenite,
hydroxyurea and related compounds. Cancer Res., 25: 980-985.
ORVINI, E., GILLS, T. E. & LaFLEUR, P. D. (1974) Method for
determination of selenium, arsenic, zinc, cadmium, and mercury in
environmental matrices by neutron activation analysis. Anal.
Chem., 46: 1294-1297.
OSATO, K. (1977) [Effects of oral administration of arsenic trioxide
during the suckling stage of rats.] Fukuoka Acta Med.,
68 (10): 464-491 (in Japanese, with English summary).
OSBURN, H. S. (1957) Cancer of the lung in Gwanda. Cent. Afr. J.
Med., 3: 215-223.
OSBURN, H. S. (1969) Lung cancer in a mining district in Rhodesia.
South Afr. med. J., 43: 1307-1312.
OSER, B. L., MORGAREIDGE, K., WEINBERG, M. S. & OSER, M. (1966)
Carcinogenicity study of carbarsone. Toxicol. appl. Pharmacol.,
9: 528-535.
O'SHAUGHNESSY, E. & KRAFT, G. H. (1976) Arsenic poisoning: Long-term
follow-up of a nonfatal case. Arch. phys. Med. Rehabil.,
57: 403-406.
OSSWALD, H. & GOERTTLER, Kl. (1971) [Leukaemia in mice following the
post-natal diaplacental application of arsenic.] Verh. Dtsch.
Ges. Pathol., 55: 289-293 (in German).
OTANI, K. (1957) [Studies on the absorption and distribution of
arsenic.] Sapporo Igaku Zasshi, 11: 285-294 (in Japanese with
English summary).
OTT, M. G., HOLDER, B. B. & GORDON, H. L. (1974) Respiratory cancer
and occupational exposure to arsenicals. Arch. environ. Health,
29: 250-255.
OVERBY, L. R. & FREDRICKSON, R. L. (1963) Metabolic stability of
radioactive arsanilic acid in chickens. J. agric. Food Chem.,
11: 378-381.
OVERBY, L. R. & FROST, D. V. (1962) Nonavailability to the rat of the
arsenic in tissues of swine fed arsanilic acid. Toxicol. appl.
Pharmacol., 4: 38-43.
PACKER, L. (1961) Metabolic and structural states of mitochondria. II.
Regulation by phosphate. J. biol. Chem., 236: 214-220.
PATON, G. R. & ALLISON, A. C. (1972) Chromosome damage in human cell
cultures induced by metal salts. Mutat. Res., 16: 332-336.
PEARSON, E. F. & POUNDS, C. A. (1971) A case involving the
administration of known amounts of arsenic and its analysis in
hair. J. Forensic. Sci. Soc., 11: 229-234.
PEIRSON, D. H., CAWSE, P. A. & CAMBRAY, R. S. (1974) Chemical
uniformity of airborne particulate material, and a maritime
effect. Nature (Lond.), 251: 675-679.
PENROSE, W. R., CONACHER, H. B. S., BLACK, R., MÉRANGER, J. C., MILES,
W., CUNNINGHAM, H. M. & SQUIRES, W. R. (1977) Implications of
inorganic/organic interconversion on fluxes of arsenic in marine
food webs. Environ. health Perspect., 19: 53-59.
PEOPLES, S. A. (1964) Arsenic toxicity in cattle. Ann. N.Y. Acad.
Sci., 111: 644-649.
PEOPLES, S. A. (1975) Review of arsenical pesticides. In: Woolson, E.
A., ed. Arsenical pesticides, Washington, DC, American Chemical
Society, pp. 1-12 (ACS symp. ser. 7).
PERRY, K., BOWLER, R. G., BUCKELL, H. M., DRUETT, H. A. & SHILLING, R.
S. F. (1948) Studies in the incidence of cancer in a factory
handling inorganic compounds of arsenic. II. Clinical and
environmental investigations. Brit. J. ind. Med., 5: 6-15.
PERSHAGEN, G. (1978) Lung cancer mortality, occupational exposure and
smoking habits in a region surrounding a smeltery. In:
Proceedings from an International Symposium on the Control of Air
Pollution in the Working Environment, Stockholm, 6-8 September
1977, Geneva, International Labour Office.
PERSHAGEN, G. & VAHTER, M. (1979) Arsenic, Stockholm, National
Swedish Environment Protection Board (SNV PM 1128).
PERSHAGEN, G., ELINDER, C.-G. & BOLANDER, A.-M. (1977) Mortality in a
region surrounding an arsenic emitting plant. Environ. health
Perspect. 19: 133-137.
PETERS, R. A. (1955) Biochemistry of some toxic agents. I. Present
state of knowledge of biochemical lesions induced by trivalent
arsenical poisoning. Bull. Johns Hopkins Hosp., 97: 1-20.
PETRES, J., BARON, D. & HAGEDORN, M. (1977) Effects of arsenic cell
metabolism and cell proliferation: Cytogenic and biochemical
studies. Environ. health Perspect., 19: 223-227.
PIERCE, F. D., LAMOREAUX, T. C., BROWN, H. R. & FRASER, R. S. (1976)
Automated technique for submicrogram determination of selenium and
arsenic in surface waters by atomic absorption spectroscopy.
Appl. Spectrosc., 30: 38.
PINTO, S. S. & McGILL, C. M. (1953) Arsenic trioxide exposure in
industry. Ind. Med. Surg., 22: 281-287.
PINTO, S. S., VARNER, M. O., NELSON, K. W., LABBE, A. L. & WHITE, L.
D. (1976) Arsenic trioxide absorption and excretion in industry.
J. occup. Med., 18: 677-680.
PINTO, S. S., ENTERLINE, P. E., HENDERSON, V. & VARNER, M. O. (1977)
Mortality experience in relation to a measured arsenic trioxide
exposure. Environ. health Perspect., 19: 127-130.
PINTO, S. S., HENDERSON, V. & ENTERLINE, P. E. (1978) Mortality
experience of arsenic-exposed workers. Arch. environ. Health,
33: 325-331.
PLOTNIKOV, V. I. & USATOVA, L. P. (1964) Coprecipitation of small
amounts of arsenic with metal hydroxides. J. anal. Chem. USSR,
19: 1101-1104.
POMROY, C., CHARBONNEAU, S. M., McCULLOUGH, R. S. & TAM, G. K. H.
(1980) Human retention studies with 74As. Toxicol. appl.
Pharmacol., 53 (3): 550-556.
POPPER, H., THOMAS, L. B., TELLES, N. C., FALK, H. & SELIKOFF, I. J.
(1978) Development of hepatic angiosarcoma in man induced by vinyl
chloride, thorotrast, and arsenic. Am. J. Pathol., 92: 349-369.
PORAZIK, I., LEGATH, V., PUCHA, K. & KRATOCHVIL, I. (1966) [Evaluation
of exposure to atmospheric arsenic oxide from the content of
arsenic in the hair.] Prac. Lék., 18: 352-356 (in Czech with
English summary).
PORTER, E. K. & PETERSON, P. J. (1975) Arsenic accumulation by plants
on mine waste (United Kingdom). Sci. total Environ., 4: 365-371.
PORTER, E. K. & PETERSON, P. J. (1977) Biogeochemistry of arsenic on
polluted sites in S.W. England. In: Hemphill, D.D., ed. Trace
substances in environmental health -- XI. A symposium. Columbia,
University of Missouri Press, pp. 89-99.
PORTMANN, J. E. & RILEY, J. P. (1964) Determination of arsenic in sea
water, marine plants and silicate and carbonate sediments.
Anal. Chim. Acta, 31: 509-519.
PRIER, R. F., NEES, P. O. & DERSE, P. H. (1963) The toxicity of an
organic arsenical, 3-nitro-4-hydroxyphenylarsonic acid. II.
Chronic toxicity. Toxicol. appl. Pharmacol., 5: 526-542.
PUPP, C., LAO, R. C., MURRAY, J. J. & POTTIE, R. F. (1974) Equilibrium
vapour concentrations of some polycyclic aromatic hydrocarbons,
As406 and SeO2, and the collection efficiencies of these air
pollutants. Atmos. Environ., 8: 915-925.
QUENTIN, K.-E. & WINKLER, H. A. (1974) Occurrence and determination of
inorganic polluting agents. Zentralbl. Bakteriol. (Orig. B),
158: 514-523.
RAPOSO, L. S. (1928) Le cancer à l'arsenic. C.R. Soc. Biol. (Paris),
98: 86.
RAY, B. J. & JOHNSON, D. L. (1972) A method for the neutron activation
analysis of natural waters for arsenic. Anal. Chim. Acta,
62: 196-199.
REAY, P. F. (1972) The accumulation of arsenic from arsenic-rich
natural waters by aquatic plants. J. appl. Ecol., 9: 557-565.
REAY, P. F. & ASHER, C. J. (1977) Preparation and purification of
74As-labeled arsenate and arsenite for use in biological
experiments. Anal. Biochem., 78: 557-560.
REGELSON, W., KIM, U., OSPINA, J. & HOLLAND, J. F. (1968)
Hemangio-endothelial sarcoma of liver from chronic arsenic
intoxication by Fowler's solution. Cancer, 12: 514-522.
RENCHER, A. C, CARTER, M. W. & McKEE, D. W. (1977) A retrospective
epidemiological study of mortality at a large western copper
smelter. J. occup. Med., 19: 754-758.
RENNKE, H., PRAT, G. A., ETCHEVERRY, R. B., KATZ, R. U. & DONOSO, S.
(1971) [Malignant hemangioendothelioma of the liver and chronic
arsenicism.] Rev. Med. Chil., 99 (9): 664-668 (in Spanish with
abstract in English).
REYMANN, F., MOLLER, R. & NIELSEN, A. (1978) Relationship between
arsenic intake and internal malignant neoplasms. Arch. Dermatol.,
114: 378-381.
RIDGWAY, L. P. & KARNOFSKY, D. A. (1952) The effects of metals on the
chick embryo: Toxicity and production of abnormalities in
development. Ann. N.Y. Acad. Sci., 55: 203-215.
RITCHIE, J. A. (1961) Arsenic and antimony in some New Zealand thermal
waters. N.Z. J. Sci., 4: 218-229.
ROBBINS, W. B., CARUSO, J. A. & FRICKE, F. L. (1979) Determination of
germanium, arsenic, selenium, tin and antimony in complex samples
by hydride generation-microwave-induced plasma atomic-emission
spectrometry. Analyst, 104: 35-40.
ROBERTS, J. W., POLLOCK, R. D., SVOBODA, M. J. & WATTERS, H. A. (1977)
Ambient air sampling for total arsenic near the Tacoma copper
smelter. In: Proceedings of the 4th International Clean Air
Congress, Seattle, Puget Sound Air Pollution Control Agency,
pp. 424-426.
ROBINSON, T. J. (1975) Arsenical polyneuropathy due to caustic
arsenical paste. Brit. med. J., 3: 139.
ROBSON, A. O. & JELLIFFE, A. M. (1963) Medicinal arsenic poisoning and
lung cancer. Brit. med. J., 2: 207-209.
ROSEHART, R. G. & CHU, R. (1975) Methods for identification of arsenic
compounds. Water Air Soil Pollut., 4: 395-398.
ROSENBERG, H. G. (1974) Systemic arterial disease and chronic
arsenicism in infants. Arch. Pathol., 97: 360-365.
ROSSET, M. (1958) Arsenical keratoses associated with carcinomas of
the internal organs. Can. Med. Assoc. J., 78: 416-419.
ROSSMAN, T., MEYN, M. S. & TROLL, W. (1975) Effects of sodium arsenite
on the survival of UV-irradiated Escherichia coli: Inhibition of
a recA-dependent function. Mutat. Res., 30: 157-162.
RÖSSNER, P., BENCKO, V. & HAVRANKOVA, H. (1977) Effect of the combined
action of selenium and arsenic on suspension culture of mice
fibroblasts. Environ. health Perspect., 19: 235-237.
ROTH, F. (1957) [Arsenic-induced liver tumours
(haemangio-endothelioma).] Z. Krebsforsch., 61: 468-503
(in German).
ROTH, F. (1958) [Bronchial cancer in vineyard workers with arsenic
poisoning.] Virchows Arch., 331: 119-137 (in German).
ROZENSHTEIN, I. S. (1970) Sanitary toxicological assessment of low
concentrations of arsenic trioxide in the atmosphere.
Hyg. Sanit., 35 (1-3): 16-22.
SABBIONI, E., MARAFANTE, E., BERTOLERO, F. & FOA, V. (1979) Inorganic
arsenic: Metabolic patterns and identification of arsenic binding
components in the rabbit. In: International Conference.
Management & Control of Heavy Metals in the Environment, London,
September, 1979, Edinburgh, CEP Consultants Ltd.
SALAMAN, M. H. & ROE, F. J. C. (1956) Further tests for
tumour-initiating activity: N,N-di(2-chlorethyl)-
P-aminophenylbutyric acid (CB1348) as an initiator of skin tumour
formation in the mouse. Br. J. Cancer, 10: 363-377.
SAMSAHL, K. (1967) Radiochemical method for determination of arsenic,
bromine, mercury, antimony and selenium in neutron-irradiated
biological material. Anal. Chem., 39: 1480-1483.
SANDBERG, G. R. & ALLEN, I. K. (1975) A proposed arsenic cycle in an
agronomic ecosystem. In: Woolson, E. A., ed. Arsenical
pesticides, Washington, D.C., American Chemical Society,
(ACS Symp. Ser. No. 7).
SANDERSON, K. V. (1976) Arsenic and skin cancer. In: Andrade, R.,
Gumport, S. L., Popkin, G. L., & Rees, T. D., ed. Cancer of the
skin, Philadelphia, London, Toronto, W. B. Saunder Company,
pp. 473-491.
SANDHU, S. S. & NELSON, P. (1978) Ionic interference in the
determination of arsenic in water by the silver
diethyldithiocarbamate method. Anal. Chem., 50: 322-325.
SATTERLEE, H. S. (1956) The problem of arsenic in American cigarette
tobacco. New Engl. J. Med., 254: 1149-1154.
SCHILLER, C. M., FOWLER, B. A. & WOODS, J. S. (1977) Effects of
arsenic on pyruvate dehydrogenase activation. Environ. health
Perspect., 19: 205-207.
SCHRAUZER, G. N. & ISHMAEL, D. (1974) Effects of selenium and of
arsenic on the genesis of spontaneous mammary tumors in inbred
C3H mice. Ann. clin. lab. Sci., 4: 441-447.
SCHRAUZER, G. N., SECK, J. A., HOLLAND, R. J., BECKHAM, T. M., RUBIN,
E. M. & SIBERT, J. W. (1972) Reductive dealkylation of
alkylcobaloximes, alkylcobalamins, and related compounds:
Stimulation of corrin dependent reductase and methyl group
transfer reactions. Bioinorg. Chem., 2: 93-124.
SCHRAUZER, G. N., WHITE, D. A., McGINNESS, J. E., SCHNEIDER, C. J. &
BELL, L. J. (1978) Arsenic and cancer: Effects of joint
administration of arsenite and selenite on the genesis of mammary
adenocarcinoma in inbred female C3H/St mice. Bioinorg. Chem.,
9: 245-253.
SCHREIBER, M. & BROUWER, E. A. (1964) Metabolism and toxicity of
arsenicals. I. Excretion and distribution patterns in rats.
Fed. Proc., 23: 199.
SCHRENK, H. H. & SCHREIBEIS, L., Jr (1958) Urinary arsenic levels as
an index of industrial exposure. Am. Ind. Hyg. Assoc. J.,
19: 225-228.
SCHROEDER, H. A. & MITCHENER, M. (1971) Toxic effects of trace
elements on the reproduction of mice and rats. Arch. environ.
Health, 23: 102-106.
SCHROEDER, H. A., KANISAWA, M., FROST, D. V. & MITCHENER, M. (1968)
Germanium, tin, and arsenic in rats: Effects on growth, survival,
pathological lesions and life span. J. Nutr., 96: 37-45.
SCHULZ, E. J. (1967) Arsenic as a cause of skin cancer with notes on
its occurrence in Pretoria. South. A. med. J., 33: 819-822.
SCHWARTZE, E. (1922) The so-called habituation to "arsenic": Variation
in the toxicity of arsenous oxide. G. Pharmacol. ed. exp. Ther,
20: 181-203.
SCHWEDT, G. & RUSSEL, H. A. (1972) Gas chromatographic determination
of arsenic as triphenylarsine. Chromatographia, 5: 242.
SELBY, L. A., CASE, A. A., OSWEILER, G. D. & HAYES, H. M. Jr. (1977)
Epidemiology and toxicology of arsenic poisoning in domestic
animals. Environ. health Perspect., 19: 183-189.
SENANAYAKE, N., DE SILVA, W. A. S. & SALGADO, M. S. L. (1972)
Arsenical polyneuropathy -- a clinical study. Ceylon med. J.,
17: 195-203.
SENESI, N., POLEMIO, M. & LORUSSO, L. (1979) Content and distribution
of arsenic, bismuth, lithium and selenium in mineral and synthetic
fertilizers and their contribution to soil. Commun. soil Sci.
plant Anal., 10: 1109-1126.
SHAIKH, A. U. & TALLMAN, D. E. (1977) Determination of sub-microgram
per liter quantities of arsenic in water by arsine generation
followed by graphite furnace atomic absorption spectrometry.
Anal. Chem., 49: 1093-1096.
SHAPIRO, H. A. (1967) Arsenic content of human hair and nails. Its
interpretation. J. forensic Med., 14: 65-71.
SHIBUYA, Y. (1971) [Studies on experimental arsenious acid poisoning.]
Tokyo Jik. Daigaku Zasshi, 86:563-575 (in Japanese).
SIEMER, D. D. & KOTEEL, P. (1977) Comparisons of methods of hydride
generation atomic absorption spectrometric arsenic and selenium
determination. Anal. Chem., 49: 1096-1099.
SIEMER, D. D., KOTEEL, P. & JARIWALA, V. (1976) Optimization of arsine
generation in atomic absorption arsenic determinations. Anal.
Chem., 48: 836-840.
SINA, G., TRIOLO, N., TROVA, P. & CLABAUT, J. M. (1977)
L'encéphalopathie arsénicale lors du traitement de la
trypanosomiase humaine africaine à T. Gambiense (à propos de
16 cas). Ann. Soc. Belg. Méd. Trop., 57: 67-74.
SKONIECZNY, R. F. & HAHN, R. B. (1978) Arsenic. In: Koltheff, I. M. &
Elving, P. J., ed. Treatise on analytical chemistry. Part II:
Analytical chemistry of inorganic and organic compounds,
Volume 10, New York, John Wiley and Sons.
SMITH, A. E. (1975) Interferences in the determination of elements
that form volatile hydrides with sodium borohydride, using atomic
absorption spectrophotometry and the argon-hydrogen flame.
Analyst, 100: 300-306.
SMITH, H. (1964) The interpretation of the arsenic content of human
hair. Forensic Sci. Soc. J., 4: 192-199.
SMITH, R. A. (1976) A method to distinguish between arsenic in and on
human hair. Environ. Res., 12: 171-173.
SMITH, R. A. (1977) Dispersion of particulate arsenic waste from the
chimney of Giant Yellowknife Gold Mines Ltd. Sci. total Environ.,
7: 227-233.
SMITH, S. & FIDDES, F. S. (1955) Forensic medicine -- A textbook for
students and practitioners, London, J. & A. Churchill, p. 458.
SMITH, D. C., SANDI, E. & LEDUC, R. (1972) Pesticide residues in the
total diet in Canada. II. 1970. Pestic. Sci., 3: 207-210.
SMITH, D. C., LEDUC, R. & CHARBONNEAU, C. (1973) Pesticide residues in
the total diet in Canada. III. 1971. Pestic. Sci., 4: 211-214.
SMITH, D. C., LEDUC, R. & TREMBLAY, L. (1975) Pesticide residues in
the total diet in Canada. IV. 1972 and 1973. Pestic Sci.,
6: 75-82.
SMITH, T. J., EATOUGH, D. J., HANSEN, L. D. & MANGELSEN, N. F. (1976)
The chemistry of sulfur and arsenic in airborne copper smelter
particulates. Bull. environ. Contam. Toxicol., 15: 651-658.
SMITH, T. J., CRECELIUS, E. A. & READING, J. C. (1977) Airborne
arsenic exposure and excretion of methylated arsenic compounds.
Environ. health Perspect., 19: 89-93.
SNELL, F. D. & SNELL, C. T. (1945) Colorimetric methods of analysis,
New York, Van Nostrand Reinhold Company.
SÖDERQUIST, C. J., CROSBY, D. G. & BOWERS, J. B. (1974) Determination
of cacodylic acid (hydroxydimethylarsine oxide) gas
chromatography. Anal. Chem., 46: 155-157.
SOMMERS, S. C. & McMANUS, R. G. (1953) Multiple arsenical cancers of
skin and internal organs. Cancer, 6: 347-359.
SRAM, R. J. & BENCKO, V. (1974) [A contribution to the evaluation of
the genetic risk of exposure to arsenic.] Cesk. Hyg.,
19: 308-315 (in Czech with English summary).
STEVENS, J. T., DiPASQUALE, L. C. & FARMER, J. D. (1976) The acute
inhalation toxicology of cacodylic acid. Toxicol. appl.
Pharmacol., 37: 165-166.
STEVENS, J. T., HALLE, L. L., FARMER, J. D., DiPASQUALE, L. C.,
CHERNOFF, N. & DURHAM, W. F. (1977) Disposition of 14C and/or
74As-cacodylic acid in rats after intravenous, intratracheal, or
peroral administration. Environ. health Perspect., 19: 151-157.
STOCKEN, L. A. & THOMPSON, R. H. S. (1949) Reactions of British
anti-lewisite with arsenic and other metals in living systems.
Physiol. Rev., 29: 168-194.
STOEPPLER, M. & MOHL, C. (in press) [Research on the arsenic content
of foods chiefly of marine origin.] Lebensmittelchem. gerichtl.
Chem. (in German).
STRATTON, G. & WHITEHEAD, H. C. (1962) Colorimetric determination of
arsenic in water with silver diethyldithiocarbamate. J. Am. Water
Works Assoc., 54: 861-864.
SULLIVAN, R. J. (1969) Preliminary air pollution survey of arsenic
and its compounds. A literature review, Raleigh, NC,
US Department of Health, Education, and Welfare (APTD 69-26).
SUZUKI, Y., SUZUKI, Y., FUJII, N. & MOURI, T. (1974) [Environmental
contamination around a smeltery by arsenic.] Shikoku Igaku
Zasshi, 30: 213-218 (in Japanese).
SZULER, I. M., WILLIAMS, C. N., HINDMARSH, J. T. & PARK-DINCSOY, H.
(1979) Massive variceal hemorrhage secondary to presinusoidal
portal hypertension due to arsenic poisoning. Can. Med.
Assoc. J., 120: 168-171.
TAKEO, T. & SHIBUYA, M. (1972) Determination of arsenic in tea plant
by neutron activation analysis. Nippon Shokuhin Kogyo Gakkai-Shi,
19 (2): 91.
TALMI, Y. & NORVELL, V. E. (1975) Determination of arsenic and
antimony in environmental samples using gas chromatography with a
microwave emission spectrometric system. Anal. Chem,
47: 1510-1516.
TAM, G. K. H., CHARBONNEAU, S. M., BRYCE, F. & LACROIX, G. (1978)
Separation of arsenic metabolites in dog plasma and urine
following intravenous injection of 74As. Anal. Biochem.,
86: 505-511.
TAM, G. K. H., CHARBONNEAU, S. M., BRYCE, F., POMROY, C. & SANDI, E.
(1979a) Metabolism of inorganic arsenic (74As) in humans
following oral ingestion. Toxicol. appl. Pharmacol.,
50: 319-322.
TAM, G. K. H., CHARBONNEAU, S. M., LACROIX, G. & BRYCE, F. (1979b)
Confirmation of inorganic arsenic and dimethylarsinic acid in
urine and plasma of dog by ion-exchange and TLC. Bull. environ.
Contam. Toxicol., 21: 371-374.
TAM, G. K. H., CHARBONNEAU, S. M., LACROIX, G. & BRYCE, F. (1979c)
In vitro methylation of 74As in urine, plasma and red blood
cells of human and dog. Bull. environ. Contam. Toxicol.,
22: 69-71.
TAMURA, S. & NOZAKI, S. (1972) Studies on arsenic metabolism. V. Blood
brain barrier and arsenite in relation to foods. Folia Pharmacol.
Jpn, 68: 800-808.
TAMURA, S., NOZAKI, S. & TSUZUKI, S. (1972) Studies on arsenic
metabolism. IV. Effect of food on arsenic tolerance in the
digestion tract. Folia Pharmacol. Jpn, 68: 586-601.
TAMURA, S., MIZUKAMI, R. & TANAKA, I. (1974a) Studies on arsenic
metabolism. XIV. Effect of lactoalbumin, eggalbumin on arsenic
excretion and storage. Folia Pharmacol. Jpn, 70: 843-847.
TAMURA, S., MAEHASHI, H., NOZAKI, S. & MIZUKAMI, R. (1974b) Studies on
arsenic metabolism. XII. Effect of polypeptone an polytamine on
arsenic accumulation in certain organs and effect on excretion.
Folia Pharmacol. Jpn, 70: 831-836.
TAMURA, S., NOZAKI, S., MAEHASHI, H. & TANAKA, I. (1974c) Studies on
arsenic metabolism. XIII. Effects of methionine, taurine and
cysteine on arsenic accumulation in certain organs and the
excretion rates. Folia Pharmacol. Jpn, 70: 837-841.
TAMURA, S., MAEHASHI, H., OZAWA, R. & GOTO, K. (1977) Studies on
arsenic metabolism. XX. Arsenic accumulation in organs and
excretion into the feces and urine of rats chronically poisoned
with arsenic. Folia Pharmacol. Jpn, 73: 877-885.
TARRANT, R. F. & ALLARD, J. (1972) Arsenic levels in urine of forest
workers applying silvicides. Arch. environ. Health, 24: 277-280.
TASK GROUP ON AIR POLLUTION AND CANCER (1978) Air pollution and
cancer: Risk assessment methodology and epidemiological evidence.
Environ. health Perspect., 22: 1-12.
TAY, C. -H. & SEAH, C. -S. (1975) Arsenic poisoning from
anti-asthmatic herbal preparations. Med. J. Aust., 2: 424-428.
TERADA, H. (1960) Clinical observation of chronic toxicosis by
arsenic. Nihon Rinsho, 18 (10): 118-127.
TERADA, H., KATSUTA, K., SASAGAWA, T., SAITO, H., SHIRATA, H.,
FUKUCHI, K., SEKIYA, T., YOKOYAMA, Y., HIROKAWA, S., WATANABE, Y.,
HASEGAWA, K., OSHINA, T. & SEKIGUCHI, T. (1960) Clinical
observations of chronic toxicosis by arsenic. Nihon Rinsho,
118: 2394-2403 (EPA translation No. TR106-74).
THACKER, G. T., PATTERSON, B. L. & HOOD, R. D. (1977) Effects of
administration route on arsenate teratogenesis in mice.
Teratology, 15: 30-31.
THIERS, H., COLOMB, D., MOULIN, G. & COLIN, L. (1967) Le cancer cutané
arsenical des viticulteurs du Beaujolais. Ann. Dermatol.
Syphiligr., 94: 133-158.
THOMAS, M. D. & COLLIER, T. R. (1945) The concentration of arsenic in
tobacco smoke determined by a rapid titrimetric method. J. ind.
Hyg. Toxicol., 27: 201-206.
THOMPSON, R. H. S. (1946) The effect of arsenical vesicants on the
respiration of skin. Biochem. J., 40: 525-535.
THOMPSON, R. H. S. (1948) The reactions of arsenicals in living
tissues. Biochem. Soc. Symp, 2: 28-38.
THOMPSON, R. J. (1977) The collection and measurement of airborne
arsenic. In: Air pollution measuring techniques, Part 2, Geneva,
World Health Organization, pp. 126-131.
THOMSON, K. C. (1975) The atomic-fluorescence determination of
antimony, arsenic, selenium and tellurium by using the hydride
generation technique. Analyst, 100: 307-310.
TOKUDOME, S. & KURATSUNE, M. (1976) A cohort study on mortality from
cancer and other causes among workers at a metal refinery.
Int. J. Cancer, 17: 310-317.
TSENG, W.-P. (1977) Effects and dose-response relationships of skin
cancer and Blackfoot disease with arsenic. Environ. Health
Perspect., 19: 109-119.
TSENG, W.-P., CHU, H. M., HOW, S. W., FONG, J. M., LIN, C. S. & YEH,
S. (1968) Prevalence of skin cancer in an endemic area of chronic
arsenicism in Taiwan. J. Natl Cancer Inst., 40: 453-463.
TSUCHIYA, K. (1977) Various effects of arsenic in Japan depending on
type of exposure. Environ. health Perspect., 19: 35-42.
TSUTSUMI, S. & KATO, K. (1975) Effects of dimercaprol or thioctic acid
on the distribution and excretion of 74As in rats. Bull. Tokyo
Dent. Coll., 16: 69-74.
TSUTSUMI, S. & NOZAKI, S. (1975) Studies on arsenic metabolism. Report
9. On the general pharmacological actions of arsenic trioxide
(As203). Bull. Tokyo Dent. Coll., 14: 147-168.
TSUTSUMI, S. & NOZAKI, S. (1975) Studies on arsenic metabolism. Report
15. Influence of arsenic antidotes on enteral absorption of
arsenic trioxide (As203) in rabbits. Folia Pharmacol. Jpn,
71: 545-551.
TSUTSUMI, S., USUI, Y. & MATSUMOTO, Y. (1974) Studies on arsenic
metabolism. Report 8. Influence of different diets on inhibition
of succinic acid dehydrogenase activity in rats. Folia Pharmacol.
Jpn, 70: 515-522.
TSUTSUMI, S., HATTORI, K., SATO, H. & KAWAGUCHI, M. (1976) Studies on
the arsenic metabolism. Report 18. On effects of various antidotes
on the enteral absorption of arsenical. Bull. Tokyo Dent. Coll.,
2: 73-82.
URAKUBO, G., HASEGAWA, A. & NAKAURA, S. (1975) [Studies on the fate of
poisonous metals in experimental animal (V). Body retention and
excretion of arsenic.] J. Food Hyg. Soc. Jpn, 16: 334-336
(in Japanese with English summary).
US Bureau of Mines (1975) Metals, minerals, and fuels. In: Minerals
yearbook, Washington, DC, US, Department of the Interior,
Vol. I.
US Bureau of Mines (1979) Metals, minerals, and fuels. In: Minerals
yearbook, Washington, DC, US, Department of the Interior,
Vol. I.
VAHTER, M. & NORIN, H. (1980) Metabolism of 74As-labelled trivalent
and pentavalent inorganic arsenic in mice. Environ. Res.
21: 446-457.
VALLEE, B. L., ULMER, D. D. & WACKER, W. E. C. (1960) Arsenic
toxicology and biochemistry. A.M.A. Arch. ind. Health,
21: 132-151.
VAN DEN BERG, A. J., DE GOEIJ, J. J. M., HOUTMAN, J. P. W. & ZEGERS,
C. (1969) Arsenic content of human hair after washing as
determined by neutron activation analysis. National Bureau of
Standards, No. 312, pp. 272-282.
VASAK, V. & SEDIVEC, V. (1952) The colorimetric determination of
arsenic. Chem. Listy, 46: 341-344.
VELLAR, O. D. (1969) Nutrient losses through sweating, Thesis. Oslo,
Universitetsförlaget.
VOGEL, A. E. (1955) A text-book of macro and semimacro qualitative
inorganic analysis, 4th ed., London, Longmans.
VONDRACEK, V. (1963) Concentration of 3,4-benzpyrene and arsenic
compounds in the Prague atmosphere. Cesk. Hyg., 8: 333-339.
VON GLAHN, W. C., FLINN, F. B. & KEIM, W. F., JR (1938) Effect of
certain arsenates on the liver. Arch. Pathol., 25: 488-505.
WADKINS, C. L. (1961) The adenosine triphosphate-adenosine diphosphate
exchange reaction of intact rat liver mitochondria. J. biol.
Chem., 236 (1): 221-224.
WAGNER, S. L. & WESWIG, P. (1974) Arsenic in blood and urine of forest
workers. Arch. environ. Health, 28: 77-79.
WALKIW, O. & DOUGLAS, D. E. (1975) Health food supplements prepared
from kelp -- a source of elevated urinary arsenic. Clin.
Toxicol., 8 (3): 325-331.
WALSH, L. M. & KEENEY, D. R. (1975) Behavior and phytotoxicity of
inorganic arsenicals in soils. In: Woolson, E. A., ed. Arsenical
pesticides Washington, DC, American Chemical Society, (ACS Symp.
Ser. No. 7).
WALSH, L. M., SUMNER, M. E. & KEENEY, D. R. (1977a) Occurrence and
distribution of arsenic in soils and plants. Environ. health
Perspect., 19: 67-71.
WALSH, P. R., DUCE, R. A. & FASCHING, J. L. (1977b) Impregnated filter
sampling system for collection of volatile arsenic in the
atmosphere. Environ. Sci. Technol., 11: 163-166.
WATANABE, T., HIRAYAMA, T., TAKAHASHI, T., KOKUBO, T. & IKEDA, M.
(1979) Toxicological evaluation of arsenic in edible seaweed,
Hizikia species. Toxicology, 14: 1-22.
WATSON, C. C. (1958) The contamination of bacon by arsenic from smoke
derived from preservatized wood. N.Z. J. Sci., 1: 361-368.
WAUCHOPE, R. D. & McWHORTER, C. G. (1977) Arsenic residues in soybean
seed from simulated MSMA spray drift. Bull. environ. Contam.
Toxicol., 17: 165-167.
WEBB, J. L. (1966) Enzyme and metabolic inhibitors. New York,
Academic Press, Vol. 3, pp. 595-793.
WEED SCIENCE SOCIETY OF AMERICA (1974) Herbicide handbook, 3rd ed.,
Champaign, Ill., Weed Science Society of America.
WEINBERG, S. L. (1960) The electrocardiogram in acute arsenic
poisoning. Am. Heart J., 60: 971-975.
WESTHOFF, D. D., SAMAHA, R. J. & BARNES, A., Jr. (1975) Arsenic
intoxication as a cause of megaloblastic anemia. Blood, 45:
241-246.
WESTÖÖ, G. & RYDÄLV, M. (1972) [Arsenic levels in foods.] Vår föda,
24: 21-40 (in Swedish, with English summary).
WHANGER, P. D., WESWIG, P. H. & STONER, J. C. (1977) Arsenic levels in
Oregon waters. Environ. health Perspect., 19: 139-143.
WILLIAMS, J. M., WEWERKA, E. M., VANDERBORGH, N. E., WAGNER, P.,
WANEK, P. L. & OLSEN, J. D. (1977) Environmental pollution by
trace elements in coal preparation wastes. In: Proceedings of the
7th Symposium on Mine Drainage Control, at NCA/BRC Coal
Conference, Louisville, Oct. 19, 1977, Los Alamos, New Mexico,
Los Alamos Scientific Laboratory.
WINKLER, W. O. (1962) Identification and estimation of the arsenic
residue in livers of rats ingesting arsenicals. J. Assoc. Off.
Anal. Chem., 45: 80-91.
WITZEL, D. A., SMITH, E. L., BEERWINKLE, K. R. & JOHNSON, J. H. (1976)
Arsanilic acid-induced blindness in swine: Electroretinographic
and visually evoked responses. Am. J. vet. Res., 37: 521-524.
WOLF, R. (1974) [On the question of occupational arsenic poisoning in
vineyard workers.] Berufsdermatosen, 22 (1): 34-47 (in German).
WOODS, J. S. & FOWLER, B. A. (1977) Effects of chronic arsenic
exposure on hematopoietic in adult mammalian liver. Environ.
health Perspect., 19: 209-213.
YAMAMURA, Y. & YAMAUCHI, H. (1976) Arsenic in biological samples of
workers exposed to arsenic trioxide. Jpn J. ind. Health,
18: 530-531.
YAMASHITA, N., DOI, M., NISHIO, M., HOJO, H. & TANAKA, M. (1972)
[Current state of Kyoto children by arsenic tainted Morinaga dry
milk.] Jpn J. Hyg., 27: 364-399 (in Japanese).
YEH, S., HOW, S. W. & LIN, C. S. (1968) Arsenical cancer of skin.
Histologic study with reference to Bowen's disease. Cancer,
21: 312-339.
YOSHIKAWA, T., UTSUMI, J., OKADA, T., MORIUCHI, M., OZAWA, K. &
KANEKO, Y. (1960) Concerning the mass outbreak of chronic arsenic
toxicosis in Niigata Prefecture. Chiryo, 42: 1739-1749.
ZACHARIAE, H., SOGAARD, H. & NYFORS, A. (1974) Liver biopsy in
psoriatics previously treated with potassium arsenite. Acta Derm.
Venreol. (Stockh.). 54: 235-236.
ZDRAZIL, J. & PICHA, F. (1966) The occurrence of the carcinogenetic
compounds 3,4-benzpyrene and arsenic in the soil. Neoplasma,
13: 49-55.
ZEMAN, A., RUZICKA, J., STARY, J. & KLECKOVA, E. (1964) A new
principle of activation analysis separations. VII.
Substoichiometric determination of traces of arsenic. Talanta,
11: 1143.
ZETTEL, H. (1943) [Effects of arsenism on the heart and blood
vessels.] Z. klin. Med., 142: 689-703 (in German).
ZOETEMAN, B. C. J. & BRINKMANN, F. J. J. (1976) Human intake of
minerals from drinking water in the European Communities. In:
Amavis, R., Hunter, W. J., & Smeets, J. G. P. M., ed. Hardness of
drinking water and public health, Oxford, Pergamon Press,
pp. 173-202.