
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 100
VINYLIDENE CHLORIDE
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Organization
Geneva, 1990
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
WHO Library Cataloguing in Publication Data
Vinylidene Chloride.
(Environmental health criteria ; 100)
1.Dichloroethylenes
I.Series
ISBN 92 4 154300 0 (NLM Classification: QV 633)
ISSN 0250-863X
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1990
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR VINYLIDENE CHLORIDE
1. SUMMARY AND CONCLUSIONS
1.1. Properties, uses, and analytical methods
1.2. Sources and levels of exposure
1.3. Absorption, distribution, metabolism, and excretion
1.4. Effects on experimental animals and cellular systems
1.4.1. Covalent binding to tissues
1.4.2. Acute toxicity
1.4.3. Short-term studies
1.4.4. Long-term studies
1.4.5. Genotoxicity and carcinogenicity
1.4.6. Reproductive toxicity
1.5. Effects on human beings
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Analytical methods
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Production
3.3. Uses
3.4. Storage and transport
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media; degradation
4.1.1. Air
4.1.2. Water
4.1.3. Soils and sediments
4.2. Biodegradation
4.3. Bioaccumulation
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Air
5.1.1. Ambient air
5.1.2. Occupational exposure
5.2. Water
5.3. Soil
5.4. Food and food packaging
6. KINETICS AND METABOLISM
6.1. Animals
6.1.1. Absorption
6.1.1.1 Inhalation exposure
6.1.1.2 Oral exposure
6.1.2. Distribution and storage
6.1.3. Elimination
6.1.3.1 Elimination of unchanged vinylidene chloride
6.1.3.2 Elimination of metabolites
6.1.4. Metabolic transformation
6.1.5. Reaction with cellular macromolecules
6.1.6. Transformation by non-mammalian species
6.2. Human beings
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Effects on the stratospheric ozone layer
7.2. Aquatic organisms
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single exposures
8.1.1. Inhalation
8.1.1.1 Rats
8.1.1.2 Mice
8.1.1.3 Other animal species
8.1.2. Oral
8.1.2.1 Rats
8.1.2.2 Mice
8.1.3. Other routes
8.1.3.1 Intraperitoneal
8.1.3.2 Eyes and skin
8.1.4. Summary of acute toxicity
8.2. Short-term exposures
8.2.1. Inhalation
8.2.2. Oral
8.3. Long-term exposure
8.3.1. Inhalation
8.3.2. Oral
8.4. Toxicity in vitro
8.5. Mutagenicity and other genotoxicity assays
8.5.1. Interaction with DNA
8.5.2. Genotoxicity in bacteria
8.5.3. Genotoxicity in yeast
8.5.4. Genotoxicity in plants
8.5.5. Genotoxicity in mammalian cells in vitro
8.5.6. Genotoxicity in mammalian cells in vivo
8.5.7. Summary
8.6. Reproduction, embryotoxicity, and teratogenicity
8.7. Carcinogenicity
8.7.1. Inhalation
8.7.2. Oral
8.7.3. Other routes
8.7.4. Summary of carcinogenicity
9. EFFECTS ON HUMAN BEINGS
9.1. Single and short-term exposures
9.2. Long-term exposure
10. EVALUATION OF EFFECTS ON THE ENVIRONMENT AND HUMAN HEALTH RISKS
10.1. Evaluation of effects on the environment
10.2. Evaluation of human health risks
10.2.1. Levels of exposure
10.2.2. Acute effects
10.2.3. Long-term effects and genotoxicity
11. RECOMMENDATIONS
11.1. Recommendations for future work
11.2. Personal protection and treatment of poisoning
11.2.1. Personal protection
11.2.2. Treatment of poisoning in human beings
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME ET CONCLUSIONS, EVALUATION ET RECOMMANDATIONS
RESUMEN Y CONCLUSIONES, EVALUACION Y RECOMENDACIONES
WHO TASK GROUP ON VINYLIDENE CHLORIDE
Members
Dr M. Bignami, Laboratory of Ecotoxicology, Istituto Superiore di
Sanita, Rome, Italy
Mr J.F. Howlett, Food Science Division, Ministry of Agriculture,
Fisheries & Food, London, England ( Chairman)
Professor C.L. Galli, Institute of Pharmacological Sciences,
University of Milan, Milan, Italy
Professor E. Malizia, Emergency Toxicological Service, Antivenom
Centre, Umberto the First Polyclinic, La Sapienza University,
Rome, Italy
Dr K. Chipman, Department of Biochemistry, University of
Birmingham, Birmingham, England
Dr Patricia S. Schwartz, Center for Food Safety and Applied
Nutrition, Food & Drug Administration, Washington, DC, USA
Professor I.V. Sanotsky, Research Institute of Industrial Hygiene &
Occupational Diseases, USSR Academy of Medical Sciences, Moscow,
USSR ( Vice-Chairman)
Dr R. Frentzel-Beyme, Institute for Documentation Information
and Statistics, DKFZ, Heidelberg, Federal Republic of Germany
( Rapporteur)
Dr J.F. Payne, Department of Fisheries and Oceans, St Johns,
Newfoundland, Canada
Dr J.C. Parker, Office of Health & Environmental Assessment, US
Environmental Protection Agency, Washington, DC, USA
Observers
Dr M.G. Penman, ICI Central Toxicology Laboratory, Macclesfield,
Cheshire, England
Dr Chr. de Rooij, Solvay & Cie SA, Brussels, Belgium
Dr A. Mocchi, Centro Italiano Studi e Indagini (CISI), Rome, Italy
Secretariat
Mr J. Wilbourn, Unit of Carcinogen Identification and Evaluation,
International Agency for Research on Cancer, Lyons, France
Dr E. Smith, International Programme on Chemical Safety, Division
of Environmental Health, World Health Organization, Geneva,
Switzerland ( Secretary)
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the criteria
documents as accurately as possible without unduly delaying their
publication. In the interest of all users of the environmental
health criteria documents, readers are kindly requested to
communicate any errors that may have occurred to the Manager of the
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland, in order that they may be
included in corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Palais des
Nations, 1211 Geneva 10, Switzerland (Telephone no. 7988400/
7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR VINYLIDENE CHLORIDE
A WHO Task Group on Environmental Health Criteria for Vinylidene
Chloride met in Rome, Italy, from 3 October to 7 October 1988. Dr
E. Smith opened the meeting on behalf of the Director-General. The
Task Group reviewed and revised the draft criteria document and
made an evaluation of the health risks of exposure to vinylidene
chloride.
The drafts of this document were prepared by Dr J.K. CHIPMAN,
University of Birmingham, England. Dr E. SMITH, a member of the
IPCS Central Unit, was responsible for the overall scientific
content and Mrs M.O. HEAD, Oxford, England, for the editing.
The efforts of all who helped in the preparation and finalization
of the document are gratefully acknowledged.
* * *
Financial support for the meeting was provided by the Ministry of
the Environment of Italy; the Centro Italiano Studi e Indagini and
the Istituto Superiore di Sanita, Rome, contributed to the
organization and provision of meeting facilities.
Partial financial support for the publication of this criteria
document was kindly provided by the United States Department of
Health and Human Services, through a contract from the National
Institute of Environmental Health Sciences, Research Triangle Park,
North Carolina, USA-a WHO Collaborating Centre for Environmental
Health Effects.
1. SUMMARY AND CONCLUSIONS
1.1. Properties, Uses, and Analytical Methods
Vinylidene chloride (C2H2Cl2) is a volatile, colourless
liquid with a "sweet" odour. It is stabilized with p- methoxyphenol
to prevent the formation of explosive peroxides. Vinylidene
chloride is used for the production of 1,1,1-trichloroethane and
to form modacrylic fibres and copolymers (with vinyl chloride or
acrylonitrile). Gas chromatographic methods have been developed for
the determination of vinylidene chloride in air, water, packaging
films, body tissues, food, and soil. The most sensitive method of
detection is by electron capture.
1.2. Sources and Levels of Exposure
Up to approximately 5% of manufactured vinylidene chloride
(representing an approximate maximum of 23 000 tonnes) is emitted
into the atmosphere annually. The high vapour pressure and low
water solubility favour relatively high concentrations in the
atmosphere compared with those in other environmental
"compartments". Vinylidene chloride in the atmosphere is expected
to have a half-life of approximately 2 days.
Environmental levels in water are very low. Even in raw
industrial waste water, the concentrations rarely exceed the
µg/litre range, which is well below the mg/litre range of toxicity
for aquatic organisms. The level in untreated drinking-water is
generally not detectable. In treated, potable water, the levels of
vinylidene chloride have generally been found to be < 1 µg/litre,
though levels of up to 20 µg/litre have been detected. Levels of
vinylidene chloride in food are usually not detectable, the
maximum observed concentration being 10 µg/kg.
Occupational exposure to vinylidene chloride is mainly
through inhalation, though skin or eye contamination can occur.
Depending on the country, the maximum recommended or regulated
time-weighted average (TWA) exposure limit is in the range of 8 to
500 mg/m3, or else is the lowest reliably detectable
concentration, depending on the country. Short-term exposure
limits range from 16 to 80 mg/m3 and ceiling values range from 50
to 700 mg/m3.
1.3. Absorption, Distribution, Metabolism, and Excretion
Vinylidene chloride can be readily absorbed via the
respiratory and oral routes in mammals, but data are not available
on dermal absorption. Vinylidene chloride is widely distributed
within the rodent body with concentrations reaching maximal
levels in the liver and kidneys. The pulmonary elimination of
unchanged vinylidene chloride is at least biphasic and dose
dependent, being of greater importance at dose levels that saturate
metabolism (approximately 600 mg/m3 (150 ppm) via inhalation in
the rat). Fasting of rats led to a reduction in the metabolism of
an oral dose and a consequent higher level of exhaled vinylidene
chloride.
The major routes of metabolism in the rat have been
characterized. The predominant phase I metabolism involves
cytochrome P-450 and the formation (possibly but not necessarily
via an epoxide) of mono-chloroacetic acid. Cytochrome P-450
activity can be induced by vinylidene chloride. A number of phase
I metabolites are conjugated with glutathione and/or with
phosphatidyl ethanol-amine prior to further conversions.
Metabolism occurs at a greater rate in the mouse than in the rat
resulting in a similar metabolic profile with a relatively higher
proportion of glutathione conjugate derivatives. It has been shown
that vinylidene chloride is also metabolized by human microsomal
cytochrome P-450. Metabolism of vinylidene chloride in rodents
leads to depletion of glutathione and inhibition of the activity of
glutathione- S -transferase.
1.4. Effects on Experimental Animals and Cellular Systems
1.4.1. Covalent binding to tissues
Covalent binding of [14C]-vinylidene chloride-derived
radiolabel occurs in the liver, kidney, and lung of rodents
and is associated with toxicity in these organs. Covalent binding
and toxicity are exacerbated by glutathione depletion and occur
in the liver and kidney at a lower dose level in mice than in rats.
A number of vinylidene chloride metabolites covalently bind to
thiols in vitro .
1.4.2. Acute toxicity
Acute LC50 estimations for vinylidene chloride vary
considerably, but this variation does not mask the fact that mice
are much more susceptible to vinylidene chloride than rats or
hamsters. Estimations of 4-h LC50 values ranged from
approximately 8000 to 128 000 mg/m3 (2000-32 000 ppm) in fed rats,
460-820 mg/m3 (115-205 ppm) in fed mice, and 6640-11 780 mg/m3
(1660-2945 ppm) in fed hamsters. Inaccuracies in LC50
estimations may arise because of a non-linear concentration-
mortality relationship. Males of all species tended to have lower
LC50 values than females, and fasting (which causes depletion of
glutathione) increased toxicity in all three species. LD50 values
following oral administration were approximately 1500 and 200 mg/kg
in fed rats and mice, respectively. Acute inhalation toxicity
in experimental animals was manifested as irritation of the
mucous membranes, depression of the central nervous system, and
progressive cardiotoxicity (sinus bradycardia and arrhythmias).
Damage was caused to the liver, kidney, and lungs. In mice, which
are more susceptible than rats to the hepatotoxicity and renal
toxicity of vinylidene chloride, kidney damage and increased DNA
replication were induced by exposure to as little as 40 mg
vinylidene chloride/m3 (10 ppm) for 6 h. As with inhalation, the
principal organs affected by oral administration of vinylidene
chloride are the liver, kidney, and lungs. The sequelae of events
leading to hepatotoxicity appear to involve an early change in the
bile canaliculi, which is followed by signs of mitochondrial
damage. This precedes damage to the endoplasmic reticulum and cell
death. Vinylidene chloride-induced liver and renal toxicity are
apparently not caused by lipid peroxidation. Raised intracellular
Ca++ concentrations may play a role in toxicity for the
hepatocyte.
The toxic effects of vinylidene chloride are at least
partially dependent on cytochrome P-450 activity (which may also be
involved in detoxification) and can be exacerbated by glutathione
depletion. Hepatotoxicity may be enhanced by ethanol and
thyroxine, inhibited by dithiocarb and (+)-catechin, and modulated
by acetone.
1.4.3. Short-term studies
Hepatic, renal, and, to a lesser extent, pulmonary damage have
been observed in rodents exposed through inhalation to vinylidene
chloride at 40-800 mg/m3 for 48 h/day, 4 or more days/week, in
short-term studies. Mice were more susceptible than rats, guinea-
pigs, rabbits, dogs, and squirrel monkeys, and toxicity varied
between different strains of mice. In general, female mice were
less susceptible than males. Hepatotoxicity was reported in rats
and mice exposed intermittently to vinylidene chloride
concentrations of > 800 mg/m3 (> 200 ppm) or 220 mg/m3 (55
ppm), respectively. The levels required to produce
hepatotoxicity through continuous exposure for several days were
240 mg/m3 (60 ppm) for rats and 60 mg/m3 (15 ppm) for mice. These
intermittent and continuous treatments also caused nephrotoxicity
in mice. The male Swiss mouse was particularly susceptible to
vinylidene chloride-induced kidney toxicity. Male mice did not
survive continuous short-term exposure to 200 mg vinylidene
chloride/m3 (50 ppm). The apparent no-observed-effect level for
hepatotoxicity in dogs, squirrel monkeys, and rats was
approximately 80 mg/m3 (20 ppm) given as a continuous 90-day
exposure. Short-term (approximately 3 months) oral dosing studies
in rats (up to 20 mg/kg daily) and dogs (up to 25 mg/kg daily) did
not show any evidence of toxicity other than minimal reversible
hepatic damage in rats.
1.4.4. Long-term studies
Long-term studies of intermittent inhalation exposure to
vinylidene chloride revealed that 300 mg/m3 (75 ppm) caused only
mild reversible hepatic changes in rats. At 600 mg/m3 (150 ppm),
the highest tolerable dose for long-term exposure in rats, liver
damage with necrosis was evident. A high mortality rate with
evidence of liver damage was observed in mice at 200 mg/m3 (50
ppm). Kidney toxicity was evident following long-term treatment of
mice at 100 mg/m3 (25 ppm). Oral dosing of rats for one year with
up to 30 mg vinylidene chloride/kg daily also produced minimal
hepatic changes. These data do not provide a clear no-observed-
effect level. There was some evidence from a separate study that
renal inflammation and liver necrosis could be induced in rats
and mice, respectively, following long-term oral administration of
vinylidene chloride at daily dose levels of 5 mg/kg and 2 mg/kg,
respectively.
1.4.5. Genotoxicity and carcinogenicity
Vinylidene chloride was found to be mutagenic for bacteria and
yeast, only in the presence of a mammalian microsomal metabolic
activation system (S9). The compound induced unscheduled DNA
synthesis in isolated rat hepatocytes and increased the frequency
of sister chromatid exchanges and chromosomal aberrations in cell
cultures with S9 included. In contrast, no increase in mammalian
gene mutations was seen. A small, but statistically significant,
increase in DNA binding after in vivo exposure has been reported.
DNA binding was greater in mouse than in rat cells and greater in
the kidneys than in the liver following 6-h exposures to 40 and 200
mg vinylidene chloride/m3 (10 and 50 ppm). Furthermore,
vinylidene chloride slightly increased unscheduled DNA synthesis
in mouse kidney. There was no evidence of a dominant lethal
effect or cytogenetic effects after in vivo exposure of rodents,
with the exception of one study showing the induction of
chromosomal aberrations in the bone marrow of the Chinese hamster.
Carcinogenicity studies have been carried out on 3 animal
species (rats, mice, and hamsters). In male Swiss mice, there was
a clear indication of carcinogenicity (kidney adenocarcinoma)
following long-term intermittent exposure to 100 or 200 mg
vinylidene chloride/m3 (25 or 50 ppm) but not to 0 or 40 mg/m3
(0 or 10 ppm).
The kidney tumours may be related in some way to observed
kidney cytotoxicity and it is possible that repeated kidney
damage either leads directly to the carcinogenic response by a
non-genotoxic mechanism or facilitates the expression of the
genotoxic potential of metabolites in this particular species, sex,
and organ. However, this conclusion is uncertain in the light of
the limited available data on genetic effects in vivo and the
findings that vinylidene chloride may have acted as an initiator.
In the same study, statistically increased incidences of lung
tumours (mainly adenomas in mice of both sexes) and mammary
carcinomas (in females) were observed, but no dose-response
relationships were found. In adult rats exposed through inhalation,
a slight non-dose-related increase in mammary tumours was
reported as well as a slight increase in leukaemia when rats
were exposed in utero and then postnatally. These observations
could not be evaluated.
1.4.6. Reproductive toxicity
No evidence was found of effects on fertility in rats
continuously exposed to vinylidene chloride (up to 200 mg/litre,
200 ppm) in drinking-water. Inhalation of up to 1200 mg vinylidene
chloride/m3 (300 ppm), for 22-23 h, by rats and mice during
various periods of organogenesis did not induce fetal
abnormalities, other than those attributable to maternal
toxicity.
Inhalation of up to 640 mg vinylidene chloride/m3 (160 ppm)
for 7 h/day in rats and rabbits or oral intake of approximately
40 mg/kg per day in rats during critical periods of gestation did
not have any effects on embryos or fetuses at a level below that
which produced maternal toxicity, but embryo and fetal toxicity
and fetal abnormalities were seen at levels producing maternal
toxicity, as evidenced by decreased weight gain.
1.5. Effects on Human Beings
Concentrations of vinylidene chloride of 16 000 mg/m3
(4000 ppm) cause intoxication that may lead to unconsciousness.
Stabilized vinylidene chloride is also an irritant for the
respiratory tract, eyes, and skin. Kidney and liver damage have
been reported for sub-anaesthetic, prolonged or repeated short-term
exposures. Evaluation of epidemiological studies was hampered by
limited cohort sizes, co-exposure to vinyl chloride, and
insufficient attention to smoking habits. No statistically
significant increased incidence of cancer was found in human
beings exposed to vinylidene chloride, but the epidemiological
studies were inadequate and it is not possible to conclude that
there is no carcinogenic risk. No information is available on the
effects of vinylidene chloride on reproduction in human beings.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
Vinylidene chloride is a halogenated aliphatic hydrocarbon.
Chemical Cl H
structure: | |
C = C
| |
Cl H
Molecular C2H2Cl2
formula:
Relative
molecular mass: 96.95
Common 1,1-dichloroethylene; 1,1-dichloroethene;
synonyms: 1,1-dichloro; VDC; 1,1-DCE; VC; vinylidene
dichloride; chlorure de vinylidene (France);
asym-dichloroethylene; NCI-C54262
Common trade
name: Sconatex
IUPAC systematic
name: 1,1-dichloroethylene
NCI number: C54262
CAS registry
number: 75-35-4
RTECS number: KV9275000
EEC number: 602-025-00-8
Conversion 1 ppm vinylidene chloride = 4 mg/m3
factors: 1 mg vinylidene chloride/m3 = 0.25 ppm
at 25 °C, 1 atm.
Commercial vinylidene chloride
The technical product (which is > 99.6% pure) can contain
impurities (Table 1).
Table 1. Maximum levels of impurities found in
commercial vinylidene chloridea
----------------------------------------------------
Dichloroacetylene 10 mg/kg
Monochloroacetylene 1 mg/kg
Vinyl chloride 20 mg/kg
Water 100 mg/kg
Acidity (as HCL) 15 mg/kg
Iron 0.5 mg/kg
Peroxides (as H202) 1 mg/kg
Other halogenated impurities 500 mg/kg (total)
----------------------------------------------------
a From: ECETOC [45].
Note: Hydroquinone monomethyl ether ( p- methoxy-
phenol) is the most commonly used inhibitor, which is
added at a level of 50-200 mg/kg. The carcinogen
dichloroacetylene may also occur as an impurity, as it
is a by-product of vinylidene chloride synthesis [185].
2.2. Physical and Chemical Properties
The principal physical and chemical properties of
vinylidene chloride are shown in Table 2.
Table 2. Some physical and chemical properties of vinylidene chloridea
_________________________________________________________________
Physical form volatile, clear, colourless liquid; it
polymerizes readily in the presence of
oxygen above 0 °C
Odour "sweet" odour; apparent detection limit
for human beings, approximately 2000-4000
mg/m3b
Boiling point (°C) 31.56
Freezing point (°C) -122.5
Relative density
(20 °C/40 °C) 1.213
Vapour density
(air = 1, 20 °C) 3.34
Density in saturated 2.8
air (air = 1)
_________________________________________________________________
Table 2 (contd).
_________________________________________________________________
Vapour pressure (mmHg)
at: -20 °C 7
0 °C 215
20 °C 495
25 °C 591
Refractive index (ND)
(20 °C) 1.4247
Viscosity (P x s) (20 °C) 0.3302
Critical temperature (°C) 220.8
Critical pressure (atm) 51.3
Heat of combustion 261.9 (liquid monomer)
(kcal/mol) (25 °C)
Heat of formation -25.1 (liquid monomer)
(kcal/g) 1.26 (gaseous monomer)
Solubility in water
(21 °C) 2.5 g/kg
Solubility in organic very soluble-diethyl ether, chloroform
solvents: soluble-benzene, acetone, ethanol
Calculated log n -octanol/
water partition
coefficient 1.66c
Flash point (open cup) -15 °C
(closed cup) -19 °C
Flammability limits in
air (% vol) 5.6-16
Saturation concentration
in air (20 °C) 2640 g/m3
Autoignition
temperature (°C) 513
Heat of evaporation
(31.6 °C) (cal/mol) 6.3
Heat of polymerization
(25 °C) (kcal/mol) 18
_________________________________________________________________
a From: Buckingham [22], Gibbs & Wessling [59], Hushon & Kornreich
[84], Shelton et al. [201], Weast [241], and Wessling & Edwards [243],
unless stated otherwise.
b From: Torkelson & Rowe [222].
c From: Rekker [187].
In the absence of a stabilizer and in the presence of oxygen,
vinylidene chloride forms an explosive peroxide at temperatures
as low as -40 °C [22]. The decomposition products of vinylidene
chloride peroxides include phosgene, formaldehyde, and
hydrochloric acid [59]. Vinylidene chloride also reacts vigorously
with oxidizing materials and is highly dangerous when exposed to
heat or flame [197]. It undergoes addition reactions as in the
formation of 1,1,1-trichloroethane when it is reacted with hydrogen
chloride. Alcohols and halides react with vinylidene chloride to
give carboxylic acids [22]. Vinylidene chloride will react with
aluminium to form reactive aluminium chloroalkyls, and copper can
form reactive acetylides from its interaction with acetylenic
impurities. In the presence of a polymerization initiator,
vinylidene chloride forms homopolymers and copolymers with other
vinyl monomers [59].
2.3. Analytical Methods
Some spectral features of vinylidene chloride are shown in
Table 3. Vinylidene chloride is well suited to liquid and
headspace sampling and determination by gas chromatography. Details
of sampling, preparation, and the determination of vinylidene
chloride in different media are given in Table 4. The major
analytical limitation is interference by other constituents of the
media.
Table 3. Ultraviolet absorption and mass spectroscopic
characteristics of vinylidene chloride
------------------------------------------------------
UV absorption maximum 200 vapa
Mass spectrum 61 (100) 96 (61)b
98 (38) 63 (32)
26 (16) 60 (15)
25 (7) 35 (6)
------------------------------------------------------
a From: Weast [241].
b From: Grasselli & Ritchey [63].
Table 4. Sampling, preparation, and determination of vinylidene chloridea
________________________________________________________________________________________________________
Medium Sampling Analytical Detection Comments Refer-
method methodb limit ence
________________________________________________________________________________________________________
Air Cold trap (liquid 02) Introduction of a sorbent trap [212]
using column of allows a large sample; however,
glass beads; desorb cumbersome for handling
thermally by purge
Trap with pyridine Colourimetric 10 mg/m3 [66]
in cooled impinger measurement of
derivative
(cyanine)
Trap with charcoal; GC/FID 1 µg/m3 Well suited for monitoring [50]
desorb with CS2 (7 µg/sample occupational exposure levels; [205]
tube) trapped vinylidene chloride is [80]
stable for at least 16 days;
when desorbed in CS2, analysis
should be within 4 days;
humidity dramatically reduces
the breakthrough volume
Trap with adsorbent GC/FID 4 µg/m3 [191]
column; desorb (see
thermally also
[199])
Trap with charcoal GC/FID working range [226]
desorb with CS2 2-20 mg/m3 for a
5- to 7-litre sample
Trap with Tenax GC/MS 0.12 µg/m3 Suitable for monitoring [233]
polymer; desorb (0.6 µg/m3, environmental samples
thermally quantifiable
limit)
Human Spirometer used for GC/MS 0.16 µg/m3 [233]
breath sampling; trap and (0.82 µg/m3,
desorb as above quantifiable
limit)
________________________________________________________________________________________________________
Table 4 (contd).
________________________________________________________________________________________________________
Medium Sampling Analytical Detection Comments Refer-
method methodb limit ence
________________________________________________________________________________________________________
Human As above using GC/MS 0.16 µg/m3 [233,
breath liquid nitrogen (0.82 µg/m3 234]
cryogenic trap quantifiable
limit)
Vinyl Distillates GC/FID 5 mg/kg [107]
chloride
Waterc Direct injection Steam-modified approx. Obvious advantages of [67]
G-solid C/FID 5 µg/litre direct injection but rapid
deterioration of column
Direct headspace GC/FID 2 µg/litre [171]
analysis (purge and confirmation
trap between water by MS
columns)
Dynamic headspace (a) GC/FID (a) 0.5 µg/litre [162]
technique (b) GC/ECD (b) 0.1 µg/litre
Static headspace GC/FID 5 µg/litre [164]
technique (quantifiable
limit)
GC/ECD 10 µg/litre
(quantifiable
limit)
Purge with inert GC/ECD 0.13 µg/ A microcoulometric detector [223]
gas; trap (Tenax); litre can also be used; direct
desorb as vapour aqueous injection above
0.13 mg/litre
GC/FID and 1 µg/litre Linearity shown for response [17]
EC versus concentration between
10 µg/litre and 1 mg/litred [234]
________________________________________________________________________________________________________
Table 4 (contd).
________________________________________________________________________________________________________
Medium Sampling Analytical Detection Comments Refer-
method methodb limit ence
________________________________________________________________________________________________________
Water As above with isotope- GC/MS 10 µg/litre Internal standard corrects for [224]
(contd.) labelled vinylidene variability in recovery
chloride as internal
standard
Headspace transfer GC/EC 0.03 µg/ Sensitive and inexpensive- [31]
(vacuum distillation) litre recommended for field
to cryogenic trap conditions
Purge-closed loop GC/EC, EDC, 0.2 µg/litre Combines gas-stripping and [236]
method or FID (most (20-ml static headspace methods -
efficient sample) recommended as an effective,
not indicated) reliable and rapid method for
routine sample analysis
Packaging Films dissolved in G-solid C/EC 5 mg/kg Injection port needs cleaning [18]
materials tetrahydrofuran or confirmation regularly
(Saran carbon tetrachloride; by MS 1 mg/kg [78]
films) can be injected with
solvent-flush
technique
GC/MS 1 mg/kg [144,
218]
Vinylidene chloride GC/FID Requires internal standard of [62]
released thermally; polymer with known content
sample by headspace of vinylidene chloride
technique
Headspace technique GC/EC 1 µg/m2 [60]
Food Headspace technique G-solid C/EC 5-20 µg/kg [78]
simulating confirmation
solvents by MS
exposed to
Saran films GC/EC 1 µg/kg [238]
(corn oil,
heptane, and
water)
Table 4 (contd).
________________________________________________________________________________________________________
Medium Sampling Analytical Detection Comments Refer-
method methodb limit ence
________________________________________________________________________________________________________
Food Headspace technique GC/EC 5 µg/kg [60]
Body Minced tissue added GC/ECD approximately Well suited for pharmaco- [125]
tissues to iso-octane/water; 10 µg/kg kinetic studies; specific
(various) purge (helium); (limit of purging method avoids
trap (Tenax); detection of foaming
desorb as vapour injected
material,
50 pg)
Body Tissue homogenized; GC/MS 10 µg/kg Recovery reported better by [44]
tissue purge and trap vacuum distillation method
(fish) procedure [76]
Soil Extract ( n- hexa- GC/EC 10 µg/kg [37]
decane); add internal
standard
Sediment Sealed in vial with GC/EC 5 µg/kg Minimum recovery observed [214]
internal standard; = 67%
purge with inert
gas; trap (Tenax);
desorb
________________________________________________________________________________________________________
a Methods for grab sampling of air are not included, since these do not allow estimations of
time-weighted average values; laser Stark spectroscopy [215] or portable infrared analysers [50] give
poor sensitivity and specificity due to interference by other halohydrocarbons.
b GC = gas chromatography; FID = flame ionization detection; EC = electron capture;
EDC = electrolytic conductivity; MS = mass spectroscopy.
c If the sample contains chlorine, sodium thiosulfate should be added to prevent chlorination of
hydrocarbons [17].
d From: Ramstad et al.[181].
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural Occurrence
Vinylidene chloride is not known to occur naturally.
3.2. Production
Crude vinylidene chloride is produced by the treatment of
1,1,2-trichloroethane with sodium hydroxide or calcium hydroxide.
Fractional distillation of the washed and dried crude product
provides the commercial vinylidene chloride to which a stabilizer
(usually p- methoxyphenol) is added to prevent polymerization [59,
201].
Vinylidene chloride has also been shown to be produced in
substantial quantities from the thermal decomposition of methyl
chloroform [61]. Methyl chloroform vapours (1910 mg/m3)
decomposed to vinylidene chloride at temperatures above 350 °C and
180 °C in the absence and presence of copper, respectively. The
extent to which this dehydrohalogenation occurs in work
environments leading to human exposure to vinylidene chloride is
not known. It has been demonstrated [220] that 1,1,1,2-tetrachloro-
ethane is readily converted to vinylidene chloride in vivo in the
rat by reductive metabolism. Thus, 1,1,1,2-tetrachloroethane is a
potential source of bioavailable vinylidene chloride. In addition,
vinylidene chloride is a major aqueous abiotic degradation
product of a frequent contaminant of ground water, namely 1,1,1-
trichloroethane [169, 230].
In 1967, world production of vinylidene chloride was estimated at
220 000-330 000 tonnes [201]. The following annual world production
rates of vinylidene chloride (in thousands of tonnes) have been
reported for the early 1980s [86]: the Federal Republic of
Germany, 100; France, 50; Japan, 23; the Netherlands, 12; the
United Kingdom, 30; and the USA, 90.7. This totals 306 000 tonnes.
The estimates should be taken as very approximate and it is likely
that the production level has now decreased [86]. A recent
estimate of worldwide production is 290 000 tonnes annually: in
Western Europe, approximately 80% of the vinylidene chloride
produced is for internal use by the companies concerned (personal
communication: European Chemical Industry Ecology and Toxicology
Centre).
3.3. Uses
Vinylidene chloride is used for the production of
1,1,1-trichloroethane and to form modacrylic fibres and
copolymers (Saran(R)) with alkyl acrylates, methacrylates,
acrylonitrile, vinyl acetate, or vinyl chloride [59]. Vinylidene
chloride/vinyl chloride copolymers (Saran(R) B) are used for the
packaging of foods, as metal coatings in storage tanks, building
structures, and tapes, and as moulded filters, valves, and pipe
fittings. These copolymers are also used to reinforce polyesters,
inks, and composites for furniture upholstery and other
constructions. Polyvinylidene chloride or vinylidene chloride
copolymerized with acrylic esters or with acrylonitrile and
acrylic esters (Diofane(R)) are used for coating paper and board
and as flame-retardant binders in other coatings. It is prohibited
in the EEC to include vinylidene chloride in cosmetics [88].
Regulations in the USA [88] restrict the vapour concentration of
vinylidene chloride to 25% of the lower explosive limit when used
in spray finishing operations.
3.4. Storage and Transport
The storage and transport of vinylidene chloride may be sources
of exposure; however, reports of such exposure have not been found
in the literature. Vinylidene chloride should not be stored for
more than a day without a stabilizer [59], which does not need to
be removed prior to use in polymer syntheses. The monomer should be
blanketed with inert gas, stored (e.g., hermetically sealed steel
containers) at a maximum of -10 °C and protected from light, air,
free radical initiators, copper, and aluminium [201] (section 2.2).
Under these conditions, inhibited vinylidene chloride can be
transported and stored, though the length of the storage period
should be minimal. A water-spray system should be available for
cooling the tanks in the event of fire. Containers of vinylidene
chloride must be appropriately labelled. In the EEC, the following
labels apply: extremely flammable, harmful by inhalation, possible
risk of irreversible effects, keep container tightly closed, keep
away from sources of ignition, no smoking, do not empty into drains
[88]. Any industrial waste containing this substance must be listed
as hazardous and is therefore subject to handling, transport,
treatment, storage, and disposal regulation. Disposal should be by
incineration and not by discharge into sewers. Complete combustion
should be ensured to prevent the formation of phosgene. An acid
scrubber should be used to remove the halo-acids produced.
Vinylidene chloride-derived peroxides can be detected by the
liberation of iodine following the addition of acidified
isopropanol saturated with sodium iodide and can be destroyed by
contact with water at room temperature [201].
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and Distribution Between Media, Degradation
4.1.1. Air
The high vapour pressure and low water solubility of
vinylidene chloride favour relatively high atmospheric
concentrations compared with other environmental "compartments".
Atmospheric radicals will play a major role in the degradation of
vinylidene chloride. The rate constant for oxidation of
vinylidene chloride with hydroxyl radicals (the major reacting
radical) was reported to be 4 x 10-12 cm3/mol per second [34].
Judging by this and the half-lives of related chlorinated ethenes
reacting with hydroxy radicals, the half-life of vinylidene
chloride reacting with tropospheric hydroxyl radicals (assumed to
be 10-6 radicals/cm3) is expected to be approximately 2 days.
Degradation by reaction with other atmospheric radicals will
also take place. Vinylidene chloride may react with chlorine
atoms derived from chloro-olefins, peroxy radicals (estimated half-
life for this reaction in the atmosphere is 22 years; [20]) and
ozone (estimated half-life is 219 days; [225]). The gas-phase
ozonolysis of vinylidene chloride at 25 °C follows second-order
kinetics and appears to involve a chain mechanism the chain carrier
being C Cl2O. The products of the reaction are C Cl2O, HCOOH,
CH2ClCCl(O), CO, O2, HCl, and H2O [83]. The chloroacetyl chloride
is most likely formed by a rearrangement of 1,1-dichloroethylene
oxide [58]. The measured half-life of vinylidene chloride within
sealed quartz flasks exposed outdoors in the northwest of England
[169] was higher than expected from the information given above (56
days). However, the relevance of these data to the environmental
persistence of vinylidene chloride is difficult to interpret
considering the high concentration used (80 mg/m3) and the specific
conditions of exposure.
The very long half-lives estimated for removal by rain droplets
(1.1 x 105 years) or by adsorption on aerosol particles (1.5 x 108
years) indicates that these processes are insignificant [34].
4.1.2. Water
Consideration of the physical and chemical properties of
vinylidene chloride (section 2.2) suggests that volatilization is
the major transport process from water [46]. Dilling [40] measured
the half-life for the evaporation of vinylidene chloride (1 mg/ml)
from a stirred aqueous solution at 25 °C and with a depth of 6.5
cm. The value obtained (27.2 min) was remarkably close to the
calculated value (20.1 min). Using the calculated re-aeration rate
constant for vinylidene chloride and oxygen [127], a half-life can
be calculated of between approximately 6 days (static pond water)
and approximately 1 day (mobile river water).
Photolysis and hydrolysis are not likely to be significant
[127], though degradation of vinylidene chloride in water contained
in sealed bottles in the dark was apparently measurable (albeit
slow) in the study by Pearson & McConnell [169]. The dispersal
of vinylidene chloride was monitored by Wang et al. [236]
following its discharge and mixing into a drainage canal that led
to a river 1.5 km downstream. The maximum discharge water
concentration of vinylidene chloride was 36.7 µg/litre. Midway
canal water concentrations of vinylidene chloride were not only
dependent on the concentration in the discharged water but were
also inversely related to the canal flow rate. The highest midway
canal water concentration was 1.4 µg/litre, which arose from a
discharged concentration of 16.7 µg/litre with a canal flow
rate of about 200 litres/second. At the site of confluence of
the canal and river, the levels of vinylidene chloride were
consistently less than 0.2 µg/litre (detection limit).
4.1.3. Soils and sediments
Few data are available on the transport or persistence of
vinylidene chloride in soils and sediments.
The transformation of vinylidene chloride was studied in
anoxic microcosms containing organic sediment collected from the
Everglades in Southern Florida [13]. The first order rate constant
of dehalogenation was 3.57 x 10-4 h-1 for surficial sediment and
1.67 x 10-4 h-1 for bottom sediments. Transformation products
included low levels of vinylidene chloride but mechanisms of
transformation other than reductive dechlorination occurred. The
log n- octanol/water partition coefficient of 1.66 [187] and the
significant solubility of vinylidene chloride in water (2.5
g/litre) suggest that some leaching from soils may occur. As with
water, volatilization is expected to be a major process of removal.
Relatively high concentrations of vinylidene chloride (1600±
400 µg/litre) have been reported in municipal wastewaters (primary
treatment waters) in Orange Country, California [246]. However,
quantifiable concentrations of vinylidene chloride (> 5 µg/kg)
were not found in sediments in the outfall area.
4.2. Biodegradation
Tabak et al. [217] measured a microbial degradation of 78% of
vinylidene chloride (5 mg/litre) following 7 days incubation at
25 °C in a static culture flask, in the dark, with settled domestic
waste water as microbial inoculum. With subsequent incubations
(after adaptation), 100% loss of compound occurred. At 10 mg
vinylidene chloride/litre, 45% loss was found in the first 7 days
incubation. Volatilization losses over 7 days at 25 °C were 24
and 15% at 5 and 10 mg/litre, respectively. Activated sludge
treatment of waste water resulted in 97% removal of vinylidene
chloride at an inflow concentration of 0.04 mg/litre [168]. These
data suggest a possible role of biodegradation; however, the
evidence is not conclusive and volatilization may be responsible
for some of the measured losses from the hydrosphere (inadvertent
in the former study).
Recently, a mixed culture of methane-utilizing bacteria was
found to degrade vinylidene chloride from 630 to 200 µg/litre
following incubation in sealed culture bottles for 48 h. The
products were non-volatile chlorinated substances and the
corresponding amount of degradation using a dead culture was from
520 to 350 µg/litre [51]. Vogel & McCarty [230] have reported
that anaerobic microorganisms can completely convert vinylidene
chloride to vinyl chloride by reductive dehalogenation. Vinyl
chloride can subsequently be mineralized to carbon dioxide.
4.3. Bioaccumulation
Bioaccumulation is expected to be low, based on the
n- octanol/water partition coefficient and the water solubility
(Table 2). A bioconcentration factor of 4 and a bioaccumulation
factor of 6.9 were reported for fish in a review by Atri [9].
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Air
5.1.1. Ambient air
In an assessment of vinylidene chloride emission in the USA
[84], an annual release of a total of 599 tonnes was estimated
from production and polymerization operations. A more recent
estimate of this emission is placed at a much lower level of 93.5
tonnes (personal communication, US Chemical Manufacturers
Association, 1987). It was estimated that between 2 and 5% of
vinylidene chloride manufactured in the USA was emitted into the
air (20-50 tonnes per 1000 tonnes produced) [212] but, on the
basis of current experience, the emissions into the air are of the
order of 1%. With an annual global production of around 300 000
tonnes, the total emissions would be 3000 tonnes per year
(personal communication, European Chemical Industry Ecology and
Toxicology Centre).
Vinylidene chloride levels detected in ambient air from a
petrochemical manufacturing area and a non-industrial centre,
respectively, ranged from 0.06 to 416.07 µg/m3 (mean, 46.84 µg/m3)
and from 3.53 to 27.29 µg/m3 (mean, 11.21 µg/m3) [233]. There was,
therefore, marked variability. The concentrations in the breath
of human individuals in the respective areas were in the ranges of
0.08-25.17 µg/m3 and 3.94-14.12 µg/m3. The data suggested a log-
linear relationship between air and breath concentrations of
vinylidene chloride (section 6.2). Hushon & Kornreich [84]
reported the results of a US EPA ambient air sampling programme.
Concentrations of vinylidene chloride ranged from not
detectable to a maximum of 0.010 mg/m3. In the rural northwest of
the USA, in the mid 1970s [65], vinylidene chloride levels in air
were non-detectable (< 20 ng/m3; < 5 ppt), which is in
accordance with the short half-life estimated for vinylidene
chloride in the atmosphere (section 4.1.1). At the perimeters of
industrial sites in the USA, air levels ranged from non-
detectable up to 52 µg/m3 [62] 0.6 miles from the site being the
maximum distance for the detection of vinylidene chloride. In urban
environments in the USA, mean air concentrations were found to be
19.6, 50.4, and 119.2 ng/m3 (4.9, 12.6, and 29.8 ppt) [212]. These
authors estimated average daily doses of 0.4, 1.1, and 2.5 µg/day,
respectively, at these sites, based on a total air intake of 23
m3/day. In a subsequent study [213] of seven additional cities in
the USA, concentrations ranged from below the detection limit (20
ng/m3; 5 ppt) to 0.224 µg/m3, with arithmetic averages ranging
from 0 to 0.123 µg/m3. The median concentration of vinylidene
chloride in the seven cities was 0.036 µg/m3. Wallace et al. [235]
reported a 5-year US EPA study in urban populations of personal
exposures to vinylidene chloride amongst many other pollutants. A
total of nearly 5000 air, breath, and drinking-water samples were
collected for 400 respondents in New Jersey, North Carolina, and
North Dakota. The median coefficients of variance for the analysis
of air and breath samples was 20-40%. Vinylidene chloride was
quantifiable, exceeding approximately 1 mg/m3 only occasionally
(< 10% measurable).
Vinylidene chloride concentrations in the 1.640-4.08 µg/m3
(0.35-1.02 ppb) range were measured at urban sites in New Jersey as
part of the Airborne Trace Element and Organic Substances (ATEOS)
project [68, 69]. However, the authors considered that the
relatively high concentrations of vinylidene chloride found may be
an artifact of 1,1,1-trichloroethane dehydrochlorination on the
particular adsorption traps (Tenax GC) used in the study. US EPA
[225] estimated the ambient airborne level of vinylidene chloride
to be 8.7 µg/m3and 20 ng/m3 in industrial-source and non-
industrial areas of the USA, respectively. In the Federal Republic
of Germany, vinylidene chloride is classed among a group of organic
compounds, the total emission of which must not exceed a
concentration of 20 mg/m3 at a mass flow of 0.1 kg/h or more [88].
5.1.2. Occupational exposure
Industrial air concentrations of vinylidene chloride should be
restricted. Ott et al. [166] reported peak air exposure levels as
high as 7600 mg/m3 in a polymer production plant with operators
being exposed to estimated 8-h time-weighted average (TWA)
concentrations of between < 20 and 280 mg/m3. In a more recent
survey in the USA [225], levels of vinylidene chloride in monomer
and polymer plants were reported of 90-100 µg/m3 and 25-50 µg/m3,
respectively. Thus, exposures are generally within the time-
weighted average threshold limit value (TLVR) of 20 mg/m3 (5 ppm),
recommended by the ACGIH (Table 5). Similarly, vinylidene chloride
levels in air in other manufacturing plants [91, 111,165] where
exposure to vinylidene chloride was involved have been reported to
be below 40 mg/m3. This was also the case for occupational
exposures in confined atmospheres (submarines and spacecraft) [1]
and in certain USA telephone offices [155] where the airborne
levels of vinylidene chloride were found not to exceed 8 mg/m3 and
256 µg/m3, respectively.
Some national occupational exposure limits are listed in
Table 5.
5.2. Water
Vinylidene chloride has been measured in raw and treated
effluents discharged from various industrial plants in the USA
[225]. The mean levels detected in raw waste water in the USA
ranged from 18 to 760 µg/litre. Vinylidene chloride (isomer not
specified) has been detected in effluent discharged from chemical
manufacturing plants in the Netherlands at a concentration of
32 µg/litre [47]. Going & Spigarelli [62] reported water levels at
plant sites ranging from non-detectable to 550 µg/litre, the
highest level being detected in an industrial waste water canal.
Vinylidene chloride has also been detected in well and river water
from various areas in the USA [200]. Discharge into a sewer is
not an acceptable method of disposal for vinylidene chloride.
Waste water is therefore injected with steam to allow the
vaporization and recovery of vinylidene chloride. Determination of
the amount of vinylidene chloride in treated waste water [225]
indicated that treatment effected a removal of between 40 and 97%.
In urban storm-water runoff samples in the USA, Cole et al.
[30] reported a frequency of detection of vinylidene chloride of
3% when determined among other priority pollutants. The range
of detected concentrations was 1.5-4 µg/litre.
Wegman et al., [242] investigated the level of vinylidene
chloride present in water from 4 sampling sites in or around a
chemical dump that had been used, in particular, for the disposal
of by-products from a pesticide production plant. The levels
detected ranged from < 0.01 to 2.8 µg/litre, which compared with
levels of 0.3-80 µg/litre reported by the authors to have been
detected in the river Rhine.
Concentrations of up to 180 µg/litre were found in ground
water beneath a major landfill site in Ottawa, Canada [121].
Low levels of vinylidene chloride have also been observed in other
selected contaminated ground waters in Ontario, Canada (Lesage,
personal communication, Environment Canada).
Lake Ontario receives the largest burden of industrial and
municipal effluents in the Great Lakes Basin with the highly
polluted Niagara river contributing 80% of its total inflow.
Water samples from 95 stations in Lake Ontario were analysed for a
suite of volatile hydrocarbons [104]. Quantifiable amounts of
vinylidene chloride were found at 4 stations in the lake (80-190
ng/litre) and a relatively high value of 3500 ng/litre at the
fifth station.
In the USSR, the maximum allowable concentration (MAC) of
vinylidene chloride in surface water is 0.6 µg/litre [88].
In the study by Lao et al. [115], grab samples of raw sewage
and effluents from a sewage treatment plant were found to contain
only trace levels of vinylidene chloride (not more than 1
µg/litre). In the same study, ground water from the vicinity
of an abandoned waste dump contained a vinylidene chloride
(isomer not stated) concentration of 138 µg/litre. The levels of
vinylidene chloride in drinking-water were generally non-
detectable, the highest detected level being 0.06 µg/litre.
Table 5. Some occupational air exposure limits used in various countriesa
____________________________________________________________________________________
Country/ Exposure limit descriptionb Value Effective
Organization (mg/m3) datec
____________________________________________________________________________________
Belgium Threshold limit value (TLV) 1987 (r)
- Time-weighted average (TWA) 20
- Short-term exposure limit (STEL) 80
Brazil Acceptable limit (AC) (48 h/week) 31 1982 (r)
Finland Time-weighted average (TWA) 40 Not given
- Short-term exposure limit (STEL) 80
Germany, Federal Maximum work-site concentration (MAK) 1987 (r)
Republic of - Time-weighted average (TWA) 8
- Short-term exposure limit (STEL 30 min) 16
Netherlands Maximum limit (MXL) 1987 (r)
- Time-weighted average (TWA) 40
(notice of intended change) 20
Poland Maximum permissible concentration (MPC) 1985 (r)
- Ceiling value (CLV) 50
Romania Maximum permissible concentration (MPC) 1985 (r)
- Time-weighted average (TWA) 500
- Ceiling value (CLV) 700
Sweden Threshold limit value (TLV) 1985
- Time-weighted average (TWA) 20
- Short-term exposure limit (STEL) 40
Switzerland Maximum work-site concentration (MAK) 1987 (r)
- Time-weighted average (TWA) 8
United Kingdom Recommended limit (RECL) 1987 (r)
- Time-weighted average (TWA) 40
USA (ACGIH) Permissible exposure limit (PEL) 1987 (r)
- Time-weighted average (TWA) 20
- Short-term exposure limit (STEL) 80
Table 5. (contd.)
____________________________________________________________________________________
Country/ Exposure limit descriptionb Value Effective
Organization (mg/m3) datec
____________________________________________________________________________________
USA (NIOSH) Recommended exposure limit (REL) lowest 1987 (r)
reliably
detectable
concentration
USSR Maximum allowable concentration (MAC) 1977
- Ceiling value (CLV) 50
____________________________________________________________________________________
a From: IRPTC [88].
b TWA = A maximum mean exposure limit based generally over the period of a
working day (generally 8 or 12 h, except 15 min (Finland) and 30 min (FRG).
STEL = A maximum concentration of exposure for a specified time duration
(generally 15 or 30 min).
c When no effective date appears in the IRPTC legal file, the year of the reference
from which the data are taken is indicated by (r).
Levels of vinylidene chloride in tap water from
Philadelphia and Miami, USA were reported to be 0.1 µg/litre or
less [47]. Vinylidene chloride levels were also < 1 µg/litre in
raw water supplies at 30 potable water treatment facilities in
Canada [163, 164]. In treated water, vinylidene chloride was
detected in 1/30 supplies at an average concentration of < 1
µg/litre. The maximum concentration detected in potable water was
20 µg/litre. In the study by Wallace et al., [235] discussed in
section 5.1.1, drinking-water samples were also analysed for
vinylidene chloride. The median coefficient of variance for
analyses was < 10%. Vinylidene chloride was detected
occasionally; the percentage of samples that showed measurable
concentrations was 26-43 (New Jersey), 10 (North Carolina), and 0
(North Dakota). The mean concentration in New Jersey drinking-
water was measured as 0.1 or 0.2 µg/litre, depending on the year of
sampling, and did not exceed 2.5 µg/litre.
Otson [161] reported that vinylidene chloride was
detectable in treated (but not untreated) water in only one out of
ten municipal water supplies in the lower Great Lakes area of
Canada. At this one site, the concentration of vinylidene
chloride in treated water was < 0.1 µg/litre. Daily exposure
of individuals via the drinking-water in the USA has been
estimated at < 0.01 µg, though the maximum could exceed 1 µg
[225]. The World Health Organization recommends a maximum
concentration of 0.3 µg/litre of drinking-water [88]. The US EPA
has proposed a Maximum Contaminant Level (MCL) for vinylidene
chloride in drinking-water of 28 µg/litre (7 ppb) [42].
Trace levels of 2 commonly used solvents (trichloroethylene
and tetrachloroethylene) have been found in the marine and
freshwater environment [104,130, 231] as well as in ground water.
There is an indication that vinylidene chloride may be produced in
the degradation of these particular compounds [167, 174], but
insufficient data are available to assess the overall importance
of trichloroethylene and tetrachloroethylene as sources of
vinylidene chloride in the environment.
5.3. Soil
Contamination of the soil may arise through municipal solid
waste disposal. It has been estimated that, in the USA, the
maximum level of total monomer in the soil does not exceed 81.7 kg
(180 pounds) per year [150]. DeLeon et al. [37] investigated the
levels of vinylidene chloride in samples from 100 waste-disposal
sites. Only one out of three samples from a single site contained
a detectable level of vinylidene chloride (21.9 mg/kg dry weight
of soil). All remaining samples contained < 10 mg/kg (not
detectable).
5.4. Food and Food Packaging
Food may be contaminated by the migration of residual
vinylidene chloride monomer from packaging materials containing
vinylidene chloride co-polymers.
A number of authors have measured residual levels of vinylidene
chloride in commercial food packaging films. A survey of food
packaging materials carried out in 1975 in the United Kingdom
indicated levels of residual monomer ranging from < 0.001 to 3.8
mg/m2 [135]. The same group reported residual levels ranging from
0.0003 to 0.4 mg/m2 from a survey for the period 1977-78. Gilbert
et al., [60] reported concentrations of vinylidene chloride
monomer ranging from non-detectable (< 0.001 mg/m2) to 0.022
mg/m2 in packaging films used for retail foods. Going &
Spigarelli [62] reported 4.9 to 58 mg vinylidene chloride/kg
(4.9 and 58 ppm) in two samples of Saran wrap while Birkel et al.
[18] reported levels of 6.5-26.2 mg/kg (6.5-26.2 ppm). In a study
by Hollifield & McNeal [78], vinylidene chloride monomer was
detected in commercial food packaging films at 1.68.1 mg/kg.
However, Tan & Okada [218] and Motegi et al. [144] did not
detect vinylidene chloride (< 1 mg/kg) in polyvinylidene chloride
film used for fish jelly products, fish sausage, processed cheese,
or in household wraps.
Levels of vinylidene chloride reported to migrate into food
or food simulants have been quite low. This is consistent with
the high barrier properties of vinylidene chloride co-polymers.
These co-polymers require little or no added plasticizers to
produce flexible films. This has an important effect on their
migration characteristics since added plasticizers reduce the
barrier properties of polymers. As with any migrant, the amount
of vinylidene chloride migration from food packaging depends on
the duration of contact, the temperature, and the original
concentration in the polymer [142, 170].
Levels of vinylidene chloride were non-detectable (< 5 µg/kg,
i.e., < 0.005 ppm) in a range of film packaged foodstuffs
except for certain cooked meat products in which the maximum
observed concentration was 10 µg/kg (0.001 ppm) [60]. Levels
reported for a variety of foods packaged in vinylidene chloride-
containing materials ranged from < 1 to 6 µg/kg [135].
Studies on the migration into food simulants carried out by
Dow [41] did not show any vinylidene chloride migrating from a
vinylidene chloride/vinyl chloride co-polymer film into water (1 h;
212 °F-detection limit 7.5 µg/kg, 7.5 ppb) or into peanut oil (1 h;
212 °F-detection limit 2.5 µg/kg, 2.5 ppb). Migration into
heptane (a very efficient extraction solvent) was 13 µg/kg (13 ppb)
after 1 h at 180 °F (from a film containing 12 mg residual
monomer/kg). A vinylidene chloride/methyl acrylate co-polymer
containing residual vinylidene chloride at 9 mg/kg (9 ppm) was
extracted with cooking oil at 250 °F for 2 h and then at 120 °F for
15 days. The amount of vinylidene chloride measured in the oil was
18 µg/kg (18 ppb). Extraction with water under the same
conditions resulted in 6 µg/litre (6 ppb) migration.
Gas chromatographic determination of sorption isotherms of
vinylidene chloride on vinylidene chloride co-polymers by
Demertzis et al. [38] was consistent with a strong thermodynamic
polymer monomer interaction leading to a low level of monomer
migration from a polymeric package into a food-contacting medium.
It has been estimated that, in the United Kingdom, the
maximum possible intake of vinylidene chloride from food as a
result of the use of packaging materials is no more than 1
µg/person per day [135].
Proposals for controls on the presence of vinylidene
chloride in food-contact materials in the EEC seek to restrict
residual vinylidene chloride levels to 5 mg/kg maximum in packaging
material and to impose a maximum limit of 50 µg/kg on vinylidene
chloride in foods [25]. There is currently no formal regulatory
limit on residual vinylidene chloride monomer in food packaging in
the USA. The current major USA producer of Saran(R) has a quality
control limit for residual vinylidene chloride in their food
packaging film of 10 mg/kg (10 ppm) [41].
Aquatic organisms are a further possible source of
contaminated foods. While methods have been developed for analysis
of fish tissue [44, 76], no reports of studies on vinylidene
chloride levels in fish could be found. However, Ferrario et al.
[48] have measured the concentration of vinylidene chloride in
biota samples from three passes of Lake Pontchartrain, USA, which
serve as a source of aquatic food for human consumption.
Vinylidene chloride was not detected in oysters from the Inner
Harbour Navigation Canal nor in clams from the Chef Menteur Pass.
Clams from the Rigolets Pass contained vinylidene chloride at a
concentration of 4.4 µg/kg wet weight.
6. KINETICS AND METABOLISM
6.1. Animals
6.1.1. Absorption
Vinylidene chloride has been shown to be well absorbed via the
respiratory and oral routes in mammals. No data are available on
dermal absorption.
6.1.1.1 Inhalation exposure
Uptake through inhalation in anaesthetized adult male Sprague-
Dawley rats was very rapid, substantial levels being found in
venous blood within 2 min of exposure [35]. In this study,
calculations of the amount of vinylidene chloride taken up in the
body revealed that the cumulative uptake and metabolism of the
inhaled chemical was linear for exposures ranging from 100 to 600
mg/m3 (25 to 150 ppm). There was a trend towards the establishment
of equilibrium with saturation of metabolism in the rats exposed to
1200 mg/m3 (300 ppm), evidenced by levels of vinylidene chloride in
the blood and breath, which rose progressively during the last hour
of the 3-h exposure. This is in agreement with the approximate
saturation of metabolism occurring at the inhalation concentration
of 600 mg/m3 (150 ppm) that Filser & Bolt [49] determined by
indirect measurement of vinylidene chloride uptake in male Wistar
rats. Andersen et al. [6] found that uptake of vinylidene chloride
from a closed chamber occurred in two phases in starved male
Holtzmann rats. The rate constant (2.2/h) of the initial rapid
phase was independent of initial concentration (40-8000 mg/m3;
10-2000 ppm) and appeared to represent tissue distribution.
Metabolism became non-linear as dose levels increased (800-4000
mg/m3; 200-1000 ppm), which is consistent with the findings of
Dallas et al. [35] and Filser & Bolt [49] mentioned above.
6.1.1.2 Oral exposure
Jones & Hathaway [102] demonstrated that vinylidene chloride
given intragastrically (0.5-350 mg/kg) was completely absorbed
from the gastrointestinal tract of Alderley Park (Wistar-derived)
male rats. Peak arterial blood levels of orally administered
vinylidene chloride (50 mg/kg) were observed within 8 min in male
Sprague-Dawley rats [175] and the dose was completely absorbed.
Complete absorption of an oral dose of 200 mg/kg to male Sprague-
Dawley rats was independent of dose vehicle (aqueous Tween 80,
corn oil, or mineral oil) [27]. The vehicle did not affect the
half-time for the initial rapid phase of exhalation of vinylidene
chloride but the later half-time values were dependent on the
rates of absorption, which decreased according to the vehicle in
the following order (Tween 80 >corn oil >mineral oil).
6.1.2. Distribution and storage
Whole-body autoradiography revealed that an intragastric
dose of [14C]-vinylidene chloride administered to 80-g Alderley
Park strain male rats was distributed throughout the tissues of the
body within 1 h after initial concentration of the radiolabel in
the liver and kidneys, which retained 14C for the longest times
after dosing [102]. McKenna et al. [133] also studied the tissue
distribution of [14C]-vinylidene chloride following oral dosing
(1 or 50 mg/kg) in male Sprague-Dawley rats. Tissue residues, 72 h
after dosing, were found in descending order in the liver, kidneys,
and other tissues including lung, muscle, skin, blood, and fat.
Similarly, 14C activity derived from inhaled [14C]-vinylidene
chloride (exposure concentrations of 40 or 800 mg/m3 (10 or 200
ppm) for 6 h) in fed male Sprague-Dawley rats (4 animals per group)
was also highest in the liver and kidneys, 72 h after termination
of exposure. The levels in other tissues at this time showed the
following trend: lung >skin >plasma >carcass >muscle and fat
[132].
6.1.3. Elimination
The elimination of vinylidene chloride administered
intravenously (10-100 mg/kg in 50% polyethylene glycol 400) to fed
and fasted male Sprague-Dawley rats followed a tri-exponential
pattern corresponding to different half-times and redistribution
among tissue compartments. The biological half-life ranged from
approximately 4045 min (10 mg/kg iv) to approximately 55-70 min
(100 mg/kg iv). In orally dosed animals, the half-life values were
significantly reduced by fasting suggesting delayed absorption in
fed animals [175].
6.1.3.1 Elimination of unchanged vinylidene chloride
As the capacity for the metabolism of vinylidene chloride is
subject to saturation (section 6.1.4), the pulmonary elimination
of unchanged vinylidene chloride is dose dependent. Male rats
given an oral dose of 1 mg/kg, excreted <3% of the dose unchanged
via the lung [133]. A similar finding that only 1% of an oral dose
(0.5 mg/kg) to rats was eliminated via the pulmonary route was
reported by Jones & Hathaway [102]. However, at higher oral dose
levels, for example at 350 mg/kg, nearly 70% of the dose was
eliminated unchanged via the lungs within 72 h [102]. In a
separate study, almost 50% of an oral dose of 200 mg/kg was
eliminated via the lungs in rats [27]. The non-linear dose
dependency of pulmonary elimination following oral administration,
in addition to being influenced by saturable metabolism, is also
determined by an efficient transfer of vinylidene chloride from the
arteries to the alveoli. Thus, 80% of an intravenous dose of 0.5
mg/kg was eliminated unchanged via the lungs in 1 h. Hence,
vinylidene chloride that escapes first pass hepatic metabolism is
largely removed by pulmonary excretion [102]. Very similar results
were obtained by Reichert et al. [184] using female rats. In this
study, 1.3, 9.7, or 16.5% of the dose was exhaled within 72 h as
unchanged vinylidene chloride after single oral doses of 0.5, 5, or
50 mg/kg, respectively. Fasting of rats for 18 h prior to
vinylidene chloride administration (50 mg/kg, oral) led to a
reduction in metabolism and consequently a higher level of exhaled
unchanged compound [133].
The biphasic pulmonary elimination of [14C]-vinylidene chloride
in rats displayed half-lives of 21 and 66 min at an oral dose of 50
mg/kg and approximately 25 and 117 min at an oral dose of 1 mg/kg
[133]. When an oral dose of 200 mg/kg was administered to
Sprague-Dawley rats, the half-life values for the initial rapid
phase ranged from 15 to 21 min and from 10 to 13 min for fasted and
fed rats, respectively. The later slow phase of vinylidene
chloride exhalation was most prolonged when the compound was
administered in mineral oil (respective half-life values of 257 and
280 min), intermediate when it was given in corn oil (73 and 103
min), and shortest (22 and 42 min) when the vehicle was aqueous
Tween-80 [27]. Following inhalation of 40 mg [14C]-vinylidene
chloride/m3 (10 ppm), the biphasic pulmonary elimination in rats
displayed half-life values of 20 and 217 min for the rapid and slow
phases, respectively [132]. As with the oral route, rats exposed to
a relatively high dose (800 mg/m3; 200 ppm) excreted a greater
percentage (fed rats 4.7% and fasted rats 8.3%) of their body
burden via the lungs than rats exposed to a low exposure of 40
mg/m3 (10 ppm) (fed rats 1.63% and fasted rats 1.60%). The
pharmacokinetics of orally dosed and inhaled vinylidene chloride in
fed and fasted rats are illustrated in Fig. 1.
6.1.3.2 Elimination of metabolites
In the studies reported above [ 102, 133], in which fed rats
were dosed orally with 0.5 or 1 mg [14C]-vinylidene chloride/kg,
the major route of excretion of metabolites was the urine (63-80%
of dose in 3 days). In bile duct-cannulated rats, urinary
excretion of 14C was markedly reduced by approximately the same
extent as biliary 14C secretion [102]. Hence, approximately half of
the urinary metabolites appeared to be derived from the bile
following enterohepatic circulation. The lungs were a minor route
of elimination of metabolites, 5-14% of the dose was exhaled as
carbon dioxide (CO2). Urine was also the major excretory route
for vinylidene chloride metabolites in mice following oral
administration [103], only 8-16% of the dose appearing as
metabolites in the faeces. This was also the case following
intraperitoneal administration. At a higher oral dose level (350
mg/kg), as expected from the saturation of metabolism, a lower
level of approximately 30% of the dose appeared as urinary
metabolites with less than 1% as CO2 in expired air and 1.3% in the
faeces [102]. Following an intermediate oral dose of 50 mg/kg
[133], urinary and faecal elimination were 47% and 4%,
respectively, with only 4% exhaled as CO2. Urinary elimination was
biphasic at oral doses of 1 and 50 mg/kg and the initial rapid and
terminal phases of urinary elimination displayed half-lives of
approximately 6 and 17 h, respectively [133]. Results in female
rats were similar to those in males in that 43.6, 53.9, and 42.1%
of the dose appeared in the urine following oral administration of
0.5, 5, and 50 mg/kg [14C]-vinylidene chloride, respectively. The
respective values for CO2 in expired air were 13.6, 11.4, and 6.1%
[184]. Female rats also eliminated metabolites via the faeces
(15.7, 14.5, and 7.7% of the dose, respectively), presumably via
the biliary route.
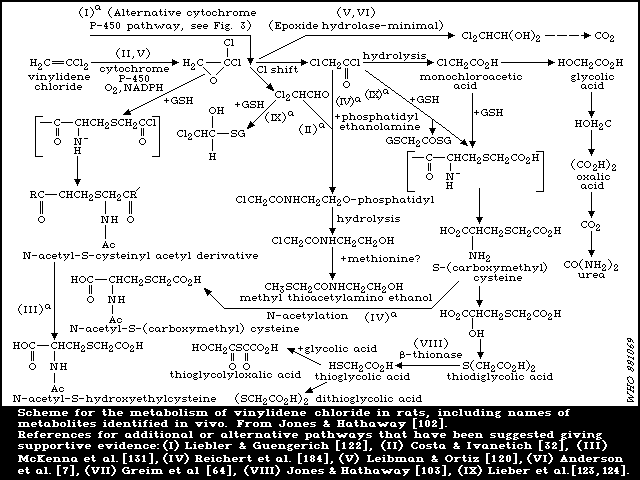 Following inhalation of [14C]-vinylidene chloride [132] at 40
mg/m3 (10 ppm), fed rats excreted 75% of the body burden as urinary
metabolites, 8.7% as CO2 from the lungs, and 9.7% in the faeces. At
800 mg/m3 (200 ppm), these percentages were slightly lower because
a greater proportion of the dose was expired unchanged.
In conclusion, irrespective of the route of administration,
the urine is the major route of excretion of metabolites of
vinylidene chloride. The extent of elimination by the urine is
dependent on dose, since a larger proportion of the dose is
eliminated unchanged via the lung at relatively high dose levels,
because of the saturation of the metabolism. The efficiency of
elimination is such that vinylidene chloride is not expected to
accumulate in animals.
6.1.4. Metabolic transformation
The profile of metabolites of vinylidene chloride produced in
rats is shown in Fig. 2. This pathway is based on the study by
Jones & Hathaway [102] with supportive and, in some cases,
conflicting or additional data superimposed. The proportion of
vinylidene chloride that is not eliminated unchanged in exhaled air
undergoes initial oxidation catalysed by the cytochrome P-450
system. The postulated transient product of this reaction,
vinylidene chloride oxide, has escaped isolation because of its
instability. This intermediate may be directly conjugated with
glutathione or, following an intramolecular rearrangement,
conjugated with mono- or bis-glutathione or with phosphatidyl
ethanolamine. Monochloroacetic acid may also be formed by
hydrolysis and this may be conjugated with glutathione and further
metabolized via a pathway involving beta-thionase activity or
alternatively may be degraded to CO2 via glycolic and oxalic acids.
Following inhalation of [14C]-vinylidene chloride [132] at 40
mg/m3 (10 ppm), fed rats excreted 75% of the body burden as urinary
metabolites, 8.7% as CO2 from the lungs, and 9.7% in the faeces. At
800 mg/m3 (200 ppm), these percentages were slightly lower because
a greater proportion of the dose was expired unchanged.
In conclusion, irrespective of the route of administration,
the urine is the major route of excretion of metabolites of
vinylidene chloride. The extent of elimination by the urine is
dependent on dose, since a larger proportion of the dose is
eliminated unchanged via the lung at relatively high dose levels,
because of the saturation of the metabolism. The efficiency of
elimination is such that vinylidene chloride is not expected to
accumulate in animals.
6.1.4. Metabolic transformation
The profile of metabolites of vinylidene chloride produced in
rats is shown in Fig. 2. This pathway is based on the study by
Jones & Hathaway [102] with supportive and, in some cases,
conflicting or additional data superimposed. The proportion of
vinylidene chloride that is not eliminated unchanged in exhaled air
undergoes initial oxidation catalysed by the cytochrome P-450
system. The postulated transient product of this reaction,
vinylidene chloride oxide, has escaped isolation because of its
instability. This intermediate may be directly conjugated with
glutathione or, following an intramolecular rearrangement,
conjugated with mono- or bis-glutathione or with phosphatidyl
ethanolamine. Monochloroacetic acid may also be formed by
hydrolysis and this may be conjugated with glutathione and further
metabolized via a pathway involving beta-thionase activity or
alternatively may be degraded to CO2 via glycolic and oxalic acids.
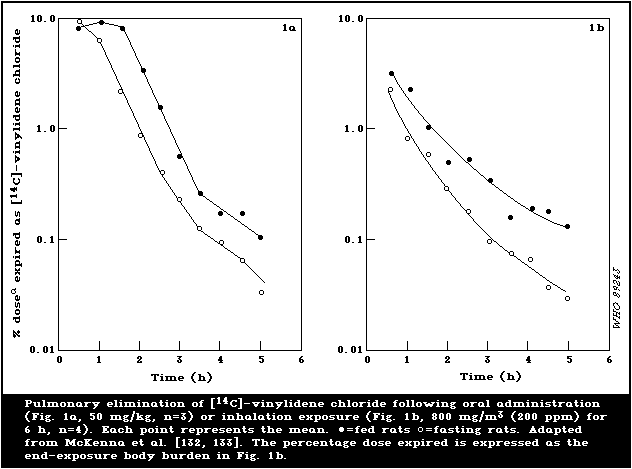 In the study by Jones & Hathaway [102], the metabolic fate of
[14C]-vinylidene chloride was investigated in groups of 4 Alderley
Park (Wistar derived) male rats. Excreta were analysed for
radiolabel following a single intragastric dose of either [1-14C]-
or [2-14C]-vinylidene chloride (350 mg/kg). Urinary metabolites
were separated by gas chromatography and mass spectra were obtained
for major metabolites. The metabolic fate of a single oral dose of
[14C]-vinylidene chloride was studied in female Wistar rats by
Reichert et al. [184], who also used gas chromatography and mass
spectroscopy for the identification of metabolites. The results
confirmed the thiodiglycolic acid pathway (Fig. 2) reported by
Jones & Hathaway [102]. However, these authors were unable to
detect any hydroxyethyl mercapturic acid (Fig. 2), which McKenna et
al. [133] had proposed to be a metabolite in male Sprague-Dawley
rats on the basis of analysis of the methylated product by mass
spectroscopy. A metabolic pathway involving phosphatidyl
ethanolamine was uncovered by Reichert et al. [184] by the
isolation of the ethanolamine derivative of chloroacetic acid (12%
of urinary 14C) (Fig. 2). While this derivative may stem from the
reaction of phosphatidyl ethanolamine with chloroacetic acid
chloride, it has been proposed by Costa & Ivanetich [32] (see
below) that the reaction may be with dichloroacetaldehyde (Fig. 2).
The metabolic fate of vinylidene chloride (50 mg/kg, oral) in
Alderley Park male mice is qualitatively very similar to that
described for the rat [103]. An exception is the excretion by the
mouse (but not the rat) of a small amount of N- acetyl- S- (2-
carboxymethyl) cysteine, derived either from the N- acetyl- S-
cysteinyl acetyl derivative or from S- (carboxymethyl) cysteine,
which are common to both species. Quantitative differences between
the metabolites formed in the two species are shown in Table 6. It
is seen that mice metabolized 22% more of the administered dose
than rats and, consequently, released less unchanged vinylidene
chloride in the expired air. The species difference was attributed
to higher cytochrome P-450-mediated epoxidation in mice. The
quantitative difference in the proportion of the N- acetyl- S-
cysteinyl acetyl derivative correlates with that expected on the
basis of hepatic glutathione- S- epoxide transferase activity in
these species, and may also be influenced by the lower extent of
chloroacetic acid metabolism in mice. Exposure to 40 mg [14C]-
vinylidene chloride/m3 (10 ppm) for 6 h resulted in a body burden
of 5.3 mg equivalents/kg in male Ha (ICR) mice compared with 2.9
mg equivalents/kg in male Sprague-Dawley rats [131]. Since only
0.65 and 1.63% of the dose were recovered respectively, as
unchanged vinylidene chloride, it was concluded that total
metabolism was more efficient in the mouse, as reported by Jones &
Hathaway [103].
Table 6. Relative proportion of 14C excretory products
after oral administration of [1-14C]-vinylidene chloride
to rodents at 50 mg/kga
____________________________________________________________
[14C] Excretory products % of 14C dose
_____________
Mice Rats
____________________________________________________________
Pulmonary excretion
Unchanged vinylidene chloride 6 28
CO2 3 3.5
Urinary excretion
Chloroacetic acid 0 1
Thiodiglycollic acid 3 22
Thioglycollic acid 5 3
Dithioglycollic acid 23 5
Thioglycollyloxalic acid 3 2
N-Acetyl- S-cysteinyl
acetyl derivative 50 28
N-Acetyl- S-(2-carboxymethyl)
cysteine 4
Urea 3 3.5
____________________________________________________________
a From: Jones & Hathway [103].
Recent studies by Liebler & Guengerich [122] confirmed the
hydrolytic lability of vinylidene chloride oxide, which was
chemically synthesized and characterized by nuclear magnetic
resonance and mass spectroscopy. Selectivity was observed
between purified rat liver cytochrome P-450s in the production of
Cl2CHCHO and P-450 inactivation but not in glycolic acid
(ClCH2CO2H) production. Further, aqueous decomposition of
vinylidene chloride did not produce Cl2CHCHO and yielded glycolic
acid only at low pH. These data, coupled with kinetic studies of
vinylidene chloride oxidation, suggest that vinylidene chloride
oxide is not an obligate intermediate in Cl2CHCHO and ClCH2CO2H
production. A proposed scheme for the role of cytochrome P-450 is
shown in Fig. 3. Whether or not this scheme operates in vivo , is
uncertain.
In the study by Jones & Hathaway [102], the metabolic fate of
[14C]-vinylidene chloride was investigated in groups of 4 Alderley
Park (Wistar derived) male rats. Excreta were analysed for
radiolabel following a single intragastric dose of either [1-14C]-
or [2-14C]-vinylidene chloride (350 mg/kg). Urinary metabolites
were separated by gas chromatography and mass spectra were obtained
for major metabolites. The metabolic fate of a single oral dose of
[14C]-vinylidene chloride was studied in female Wistar rats by
Reichert et al. [184], who also used gas chromatography and mass
spectroscopy for the identification of metabolites. The results
confirmed the thiodiglycolic acid pathway (Fig. 2) reported by
Jones & Hathaway [102]. However, these authors were unable to
detect any hydroxyethyl mercapturic acid (Fig. 2), which McKenna et
al. [133] had proposed to be a metabolite in male Sprague-Dawley
rats on the basis of analysis of the methylated product by mass
spectroscopy. A metabolic pathway involving phosphatidyl
ethanolamine was uncovered by Reichert et al. [184] by the
isolation of the ethanolamine derivative of chloroacetic acid (12%
of urinary 14C) (Fig. 2). While this derivative may stem from the
reaction of phosphatidyl ethanolamine with chloroacetic acid
chloride, it has been proposed by Costa & Ivanetich [32] (see
below) that the reaction may be with dichloroacetaldehyde (Fig. 2).
The metabolic fate of vinylidene chloride (50 mg/kg, oral) in
Alderley Park male mice is qualitatively very similar to that
described for the rat [103]. An exception is the excretion by the
mouse (but not the rat) of a small amount of N- acetyl- S- (2-
carboxymethyl) cysteine, derived either from the N- acetyl- S-
cysteinyl acetyl derivative or from S- (carboxymethyl) cysteine,
which are common to both species. Quantitative differences between
the metabolites formed in the two species are shown in Table 6. It
is seen that mice metabolized 22% more of the administered dose
than rats and, consequently, released less unchanged vinylidene
chloride in the expired air. The species difference was attributed
to higher cytochrome P-450-mediated epoxidation in mice. The
quantitative difference in the proportion of the N- acetyl- S-
cysteinyl acetyl derivative correlates with that expected on the
basis of hepatic glutathione- S- epoxide transferase activity in
these species, and may also be influenced by the lower extent of
chloroacetic acid metabolism in mice. Exposure to 40 mg [14C]-
vinylidene chloride/m3 (10 ppm) for 6 h resulted in a body burden
of 5.3 mg equivalents/kg in male Ha (ICR) mice compared with 2.9
mg equivalents/kg in male Sprague-Dawley rats [131]. Since only
0.65 and 1.63% of the dose were recovered respectively, as
unchanged vinylidene chloride, it was concluded that total
metabolism was more efficient in the mouse, as reported by Jones &
Hathaway [103].
Table 6. Relative proportion of 14C excretory products
after oral administration of [1-14C]-vinylidene chloride
to rodents at 50 mg/kga
____________________________________________________________
[14C] Excretory products % of 14C dose
_____________
Mice Rats
____________________________________________________________
Pulmonary excretion
Unchanged vinylidene chloride 6 28
CO2 3 3.5
Urinary excretion
Chloroacetic acid 0 1
Thiodiglycollic acid 3 22
Thioglycollic acid 5 3
Dithioglycollic acid 23 5
Thioglycollyloxalic acid 3 2
N-Acetyl- S-cysteinyl
acetyl derivative 50 28
N-Acetyl- S-(2-carboxymethyl)
cysteine 4
Urea 3 3.5
____________________________________________________________
a From: Jones & Hathway [103].
Recent studies by Liebler & Guengerich [122] confirmed the
hydrolytic lability of vinylidene chloride oxide, which was
chemically synthesized and characterized by nuclear magnetic
resonance and mass spectroscopy. Selectivity was observed
between purified rat liver cytochrome P-450s in the production of
Cl2CHCHO and P-450 inactivation but not in glycolic acid
(ClCH2CO2H) production. Further, aqueous decomposition of
vinylidene chloride did not produce Cl2CHCHO and yielded glycolic
acid only at low pH. These data, coupled with kinetic studies of
vinylidene chloride oxidation, suggest that vinylidene chloride
oxide is not an obligate intermediate in Cl2CHCHO and ClCH2CO2H
production. A proposed scheme for the role of cytochrome P-450 is
shown in Fig. 3. Whether or not this scheme operates in vivo , is
uncertain.
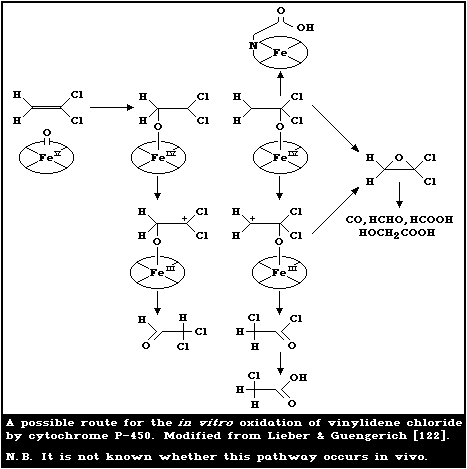 The role of cytochrome P-450 in the metabolism of vinylidene
chloride was confirmed by the measurement of a Type I difference
spectrum with the bound substrate in hepatic microsomes from male
Long-Evans rats [32]. Addition of vinylidene chloride to
microsomes also stimulated carbon monoxide-inhibitable NADPH
oxidation. NADPH was required for the conversion of vinylidene
chloride to monochloroacetic acid and dichloroacetaldehyde (not
found in the in vivo studies reported above), and these
metabolites were not formed in the presence of cytochrome P-450
inhibitors SKF-525A and carbon monoxide. Pretreatment of rats with
the cytochrome P-450-inducing agent beta-naphthoflavone did not
elevate the hepatic microsomal metabolism of vinylidene chloride,
and the cytochrome P-450-inducing agent phenobarbital gave a
slight enhancement of metabolism per mg microsomal protein. More
noticeable was the marked enhancement of the ability of liver
preparations to produce mutagenic metabolites of vinylidene
chloride in vitro following treatment of female BD-VI rats with the
selective inducing agents phenobarbital and 3-methylcholanthrene
[15].
In a study by Sato et al. [195], male Wistar rats were given
ethanol in the diet (2% ethanol, increased by 1% daily to a final
concentration of 5%, equivalent to 30% of total calorie intake).
Hepatic microsomes derived from the ethanol-treated rats
metabolized vinylidene chloride at a rate of 100.6 nmol/g liver per
min compared with 31.1 nmol/g liver per min in control rat
microsomes, indicating induction of microsomal enzymes involved in
vinylidene chloride metabolism.
As shown in Fig. 2, conjugation with glutathione required
prior microsomal metabolism of vinylidene chloride [131]. The
importance of glutathione in vinylidene chloride metabolism was
studied by Andersen et al. [7]. Pretreatment of rats with agents
that deplete hepatic non-protein sulfhydryl concentrations caused a
marked inhibition of vinylidene chloride metabolism as measured by
gas uptake (which is determined by metabolism). In particular,
treatment with cyclohexene oxide and dimethylmaleate resulted in
76 and 54% inhibition, respectively. Reichert et al. [183] also
noted an 18% reduction in the metabolism of vinylidene chloride
(given as 20 000 mg/m3 (5000 ppm) in the gaseous phase) in
isolated perfused rat livers following diethylmaleate
administration. This was considered to be due to an 85% reduction
in glutathione levels. The finding that the glutathione conjugate
ClCH2COSG is able to S- alkylate a second glutathione molecule to
yield GSCH2COSG [123, 124] suggests that the monoglutathione
conjugate may have the ability to interact with proteins, such as
those involved in the transport of glutathione conjugates.
The rate of metabolism of vinylidene chloride by isolated
hepatocytes from phenobarbital-pretreated male Long-Evans rats was
studied by Costa & Ivanetich [33] using the maximum dose of
vinylidene chloride that was not cytotoxic (2.1 mmol vinylidene
chloride/litre). The metabolites detected were dichloroacetic
acid (0.15 nmol/106 cells per 10 min), monochloroacetic acid
(0.068 nmol/106 cells per 60 min), and dichloroethanol (0.01
nmol/106 cells per 10 min). 2-Chloroethanol and
chloroacetaldehyde were not detected (<12 and <4 nmol/106 cells
per 30 min, respectively). No attempt was made to hydrolyse
conjugates.
In summary, the phase I metabolism of vinylidene chloride in
rodents involves the action of cytochrome P-450 and the production
of monochloroacetic acid. This, and its precursors, may undergo
conjugation with glutathione and/or phosphatidyl ethanolamine
prior to further conversions. Metabolism in the mouse occurs at a
greater rate than in the rat and results in a similar metabolic
profile with a relatively higher proportion of glutathione
conjugate derivatives.
6.1.5. Reaction with cellular macromolecules
The specific interaction of vinylidene chloride metabolite(s)
with DNA is covered in section 8.5.1.
In the studies by McKenna et al. [132, 133] reported in section
6.1.3.1, fasted rats showed a reduced capacity to metabolize
relatively high doses of both orally administered and inhaled
[14C]-vinylidene chloride. Fasted rats exposed to 800 mg
vinylidene chloride/m3 (200 ppm) sustained liver and kidney
damage, which was not found in fed rats. This toxicity was
associated with a greater level of covalently bound radiolabel in
the liver of the fasted animals. An elevated level of covalent
binding in the liver was also seen as a result of depriving rats of
food prior to administration of an oral dose of 50 mg vinylidene
chloride/kg. The results can be explained by the binding of
reactive metabolites (presumed to be vinylidene chloride oxide
and/or chloroacetyl chloride) to nucleophilic sites in tissue
macromolecules. This is thought to be enhanced by the depletion of
glutathione during fasting [93], which operates as an alternative
nucleophile [131]. Following exposure of male rats to 20800 mg
[14C]-vinylidene chloride/m3 (5200 ppm) for 6 h [131], hepatic non-
protein sulfhydryl levels fell in a dose-dependent saturable
manner. Appreciable covalent binding of radiolabel to hepatic
protein occurred when 30% or more glutathione was depleted.
Covalent binding of radiolabel to hepatic and kidney tissue in mice
occurred at the rate of 22 and 80-µg equivalents/g protein,
respectively, compared with 5 and 13 µg equivalents/g protein,
respectively, in rat tissue following exposure to 40 mg [14C]-
vinylidene chloride/m3 (10 ppm)for 6 h.
In a separate study on rats by Reichert et al. [183], the rate
of depletion of glutathione after oral doses of vinylidene chloride
was also found to be exponentially dependent on the concentration.
However, the authors questioned the correlation between a low
glutathione level and relatively high toxicity in fasted rats,
since the fall in glutathione levels after oral administration of
vinylidene chloride (1000 mg/kg) was identical in fasted rats and
fed rats. Furthermore, the rate of metabolism of vinylidene
chloride by isolated perfused livers (20 000 mg vinylidene
chloride/m3 (5000 ppm) in the gaseous phase) was not affected by an
18-h fast. The interpretation of these data is difficult in the
light of other results reported in this section.
Jaeger et al. [93] reported a diurnal variation in the levels
of glutathione in male Holtzmann rats. The animals were most
sensitive to the lethal and hepatotoxic effects of vinylidene
chloride when glutathione levels were at a minimum. Jaeger et
al. [96] also correlated susceptibility to the hepatotoxicity
of vinylidene chloride (exposure for 4 h at 4000 mg/m3 (1000 ppm))
to decreased hepatic glutathione concentrations in male Holtzmann
rats that had been fasted for 18 h or had been treated with
diethylmaleate. In a further study [97], the hepatic glutathione
concentration was decreased by the administration of
trichloropropane epoxide (0.1 ml of a 10% solution/kg) to fasted
rats. This treatment was also associated with elevated toxicity of
vinylidene chloride. A qualitative, but not a quantitative,
difference in the metabolism of vinylidene chloride (8000 mg/m3
(2000 ppm) initial exposure) resulted from the fasting of rats.
Thirty minutes after exposure to [14C]-vinylidene chloride at the
same concentration, the radioactivity in liver mitochondria and
microsomes was largely TCA-insoluble and was greater in fasted than
in control rats. Judging from the turn-over time of TCA-insoluble
14C, it was suggested that this 14C had entered the metabolic pool
rather than being covalently bound to macromolecules. However,
the demonstration of labile thiol adducts [123] could explain this
finding.
Covalent binding of [1,2-14C]-vinylidene chloride to tissues
peaked 6 h after an intraperitoneal dose of 125 mg/kg in male
C57B1/6N mice [159]. Pretreatment with diethylmaleate, which
depletes glutathione, enhanced covalent binding in the liver,
lung, and kidney, and also enhanced lethal toxicity. In accordance
with evidence for the formation of reactive metabolites of
vinylidene chloride by cytochrome P-450, covalent binding was
increased in liver and lung tissue by pretreatment with the P-450
inducers, phenobarbital and 3-methylcholanthrene. Inhibitors
of cytochrome P-450, piperonyl butoxide, and SKF-525A all decreased
covalent binding in the liver and lung. However, binding to kidney
tissue was not affected by P-450-inducing agents, was decreased by
piperonyl butoxide and was increased by SKF-525A. Covalent binding
occurred in hepatic and lung microsomes from these mice following
incubation of microsomes with NADPH. Surprisingly, however, oxygen
did not appear to be necessary. Kidney microsomes could not
metabolize vinylidene chloride to products that covalently bound to
tissue macromolecules, unless the mice had been pretreated with
cytochrome P-450-inducing agents suggesting that, in the absence
of enzyme induction, covalent binding in the kidney in vivo was
mediated by hepatic metabolites [157, 158]. Covalent binding of
[14C]-vinylidene chloride to lung and liver tissue accompanied
bronchiolar necrosis in CD-1 mice given an intraperitoneal dose
(125 mg/kg); only mild hepatic necrosis was observed ([56]section
8.1.3.1). The effects of various inducers and inhibitors of
cytochrome P-450 activity on toxicity and covalent binding were
variable in line with the evidence for microsomal-mediated
activation and deactivation (section 8.1.2) and in accordance
with multiple reactive metabolites (see below). Vinylidene chloride
epoxide, 2-chloroacetyl chloride, 2,2-dichloro-acetaldehyde, 2-
chloroacetic acid and S- (2-chloroacetyl)-glutathione all bind
covalently to thiols in vitro [123, 124]. Covalent binding of
radiolabel to microsomal protein occurred following incubation of
[14C]-vinylidene chloride with rat and human liver microsomes. When
rat microsomes were used, binding was inhibited by alcohol
dehydrogenase + NADH, suggesting that 2,2-dichloroacetaldehyde
played a role in the binding process. Metabolites of [14C]-
vinylidene chloride bound to microsomes from isolated lung as
well as from the liver of CD-1 mice [54]. By the use of specific
inhibitors and agents that induce cytochrome P-450 isoenzymes,
these authors were able to demonstrate the role of cytochrome P-450
isoenzymes in the production of reactive metabolites, though some
non-specific covalent binding was also seen.
In summary, there are a number of reactive metabolites of
vinylidene chloride, the production of which is dependent on the
activity of cytochrome P-450. In rodents, each of these products
may contribute to the depletion of glutathione and to covalent
binding to tissue macromolecules, which is greater in the liver
than in the kidney. The greater covalent binding of reactive
metabolites to tissue macromolecules in the mouse compared with the
rat is correlated with a relatively higher rate of metabolism
(section 6.1.4) and higher toxicity (section 8.1.1.2).
6.1.6. Transformation by non-mammalian species
No data were available to the Task Group on vinylidene chloride
metabolism in non-mammalian species, other than bacteria.
6.2. Human Beings
No data have been reported on the kinetics and metabolism of
vinylidene chloride in human beings, other than some very limited
information obtained indirectly through the following studies.
Liver 9000-g supernatants (S9) from four adults, who did not show
any pathological lesions, were capable of catalysing the formation
of products that were mutagenic to Salmonella typhimurium [15]
(section 8.5.2), suggesting that human cytochrome P-450 can
metabolize vinylidene chloride. The rate of conversion of
vinylidene chloride to dichloroacetaldehyde in hepatic microsomes
from two human organ donors (one of each sex) was 0.0340-0.038
nmol/min per nmol cytochrome P-450. The conversion was shown (by
lack of significant antibody inhibition) not to be mediated by
debrisoquine hydroxylase (a form of cytochrome P-450, which is
polymorphic in human beings) [245]. This rate of microsomal
metabolism to dichloroacetaldehyde is similar to that reported by
Costa & Ivanetich [33] for the rat (0.028 nmol dichloroacetaldehyde
and 0.035 nmol chloroacetate produced/min per nmol cytochrome
P-450).
Vinylidene chloride is exhaled in human breath following
inhalation exposure [233]. Breath from student volunteers, in
Texas and North Carolina, USA, was sampled using a spirometer as
the subjects inhaled pure air. The ratio of vinylidene chloride in
the breath to that in pre-exposure air was 0.78 ± 0.86 (n = 15). A
significant Spearman correlation coefficient of 0.77 was determined
between air and breath levels of vinylidene chloride in 17 human
subjects. The following log-linear model was capable of giving a
reasonable prediction of breath levels from the preceding 8-h air
exposure levels: log concentration in breath (µg/m3) = 0.24 ± 0.67
+ (0.71 ± 0.17) log concentration in air (µg/m3). The authors
suggested that, if these observations were confirmed, recent
exposures and body burdens of individuals could be estimated from
breath analysis. However, this may be hampered by biphasic
elimination as reported for animals (section 6.1.3).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Effects on the Stratospheric Ozone Layer
The rapid destruction of vinylidene chloride in the troposphere
by hydroxyl radicals (section 4.1.1) indicates that the substance
is unlikely to participate in the depletion of the stratospheric
ozone layer.
7.2. Aquatic Organisms
Studies on the impact of vinylidene chloride on living
organisms have concentrated on the aquatic environment and include
discussions on the levels of vinylidene chloride detected (section
5.2).
According to Leblanc [117], the acute toxicity for the water
flea ( Daphnia magna), under static conditions, is of a similar
magnitude to that reported by Buccafusco et al. [21] for the
bluegill fish (see below). The median LC50 values were 98 mg/litre
(95% confidence interval; range, 71-130 mg/litre) and 79 mg/litre
(95% confidence interval; range, 61-110 mg/litre) for 24 h and 48
h, respectively. The "no discernible effect" level for the water
flea was less than 2.4 mg/litre.
Dawson et al. [36] treated fresh-water bluegill sunfish
( Lepomis macrochiras) and marine tidewater silverside fish
( Menidia beryllina) with vinylidene chloride (132-750 mg/litre
and 180-320 mg/litre, respectively) for up to 96 h under static
conditions. No attempt was made to prevent loss of vinylidene
chloride by evaporation. The best-fit median lethal concentrations
(LC50) for 96 h were 220 and 250 mg/litre for bluegill sunfish and
tidewater silverside fish, respectively. Since these values were
less than 500 mg/litre (500 ppm), vinylidene chloride was
designated a hazardous substance. The 96-h static LC50 of
vinylidene chloride in juvenile marine sheepshead minnows
( Cyprinodon variegatus) was very similar to the above values (250
mg/litre; range, 200-340 mg/litre, 95% confidence limits), and was
the same when measured at 24 h [72]. The no-observed-effect
concentration was 80 mg/litre. The acute toxicity in bluegill fish
( L. macrochirus) was also investigated by Buccafusco et al. [2]
under static conditions, in capped jars to minimize volatilization.
The recorded LC50 value was 74 mg/litre (95% confidence interval,
57-91 mg/litre) at 96 h. This LC50, which was identical at 24 h,
is somewhat lower than those reported in the studies by Dawson et
al. [36] and Heitmuller et al. [72]. Toxicity values similar to
those noted above for fish and Daphnia were reported in a review by
Atri [9].
The assays under static uncapped conditions reported here are
relevant to acute spill conditions. One-week flow-through studies
have also been carried out by Dill et al. [39]. The LC50 value for
fathead minnows ( Pimephales promelas Rafinesque) in flowing water
was 29 mg/litre (range, 23-34 mg/litre) after 7 days exposure,
whereas the 96-h LC50 was 108 mg/litre (range, 85-117 mg/litre)
under flow-through conditions. Swimming disorientation was
observed to be the major sublethal toxic effect of vinylidene
chloride. A bioconcentration factor of 4 and a bioaccumulation
factor of 6.9 have been reported for fish in a review by Atri [9].
Few data are available on the sublethal effects of vinylidene
chloride on aquatic organisms. Preliminary studies demonstrated
that hepatic neoplastic lesions were not produced in guppy
( Poecilia reticulata) and Japanese medaka ( Oryzias latipes)
exposed to vinylidene chloride concentrations of up to 40 mg/litre
(40 ppm) for 3 months [71] (personal communication, Hawkins Gulf
Coast Research Laboratory, Mississippi, USA).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single Exposures
Acute toxicity data (LC50s and LD50s) for common laboratory
animals are shown in Table 7.
8.1.1. Inhalation
8.1.1.1 Rats
In an early study by Carpenter et al. [24], 3 groups of 6
Sherman rats were exposed to vinylidene chloride vapour for 4 h at
various concentrations. Within a 14-day post-exposure observation
period, a concentration of 128 000 mg/m3 (32 000 ppm) was lethal
for 2/6, 3/6, and 4/6 animals, respectively. Later, Siegel et al.
[206] estimated a 4-h LC50 of 25 400 mg vinylidene chloride/m3
(6350 ppm) for groups of 16 male Sprague-Dawley-derived rats.
Siletchnik & Carlson [211] considered that lethality from
vinylidene chloride might be related to cardiotoxicity. They
exposed male Charles River albino rats to 102 400 mg vinylidene
chloride/m3 (25 600 ppm) for 10 min or more and noted progressive
sinus bradycardia and arrhythmias (AV-block), multiple
continuous ventricular contractions, and ventricular fibrillation.
This treatment with vinylidene chloride also produced a marked
increase in sensitivity to epinephrine-induced cardiac arrhythmias.
Phenobarbital pretreatment enhanced the cardiac-sensitizing
properties of vinylidene chloride.
However, Jaeger et al. [95] reported that death from vinylidene
chloride inhalation was associated with bloody ascites in all
animals, with no signs of cardiac failure, and was therefore
thought to be due to vascular collapse and shock.
In the studies by Zeller et al. [248, 249], the estimated
LC50s of vinylidene chloride (Table 7) were lowered by fasting in
both male and female Sprague-Dawley rats. Females were less
susceptible than males to the lethal effect. Post-mortem
examination of animals dying from vinylidene chloride exposure
revealed acute contraction of heart blood vessels, acute swellings
and localized bloody oedema in the lung, greyish enlarged liver
lobules, and pale kidneys. Ascites and hydrothorax were also seen.
Table 7. Acute toxicity of vinylidene chloride for laboratory animals
______________________________________________________________________________________________________
Species Sex Nutri- Estimated LC50/LD50 Dosing criteria Limit of Reference
tional observation
status time
______________________________________________________________________________________________________ statustion time
Rat male, fed approximately 128 000 mg/m3 Inhalation, 4 h 14 days [24]
female (32 000 ppm)
Rat male fed 25 400 mg/m3 (6350 ppm) Inhalation, 4 h 14 days [206]
Rat male fed 60 000 mg/m3 (15 000 ppm) Inhalation, 4 h 24 h [96]
Rat male fed approximately 8000 mg/m3 inhalation, 4 h 23 h [93]
(2000 ppm) (pm but not am)
Rat male fed 28 400 mg/m3 (7100 ppm) Inhalation, 4 h 14 days [249]
Rat male 18-h fasted 2400 mg/m3 (600 ppm) Inhalation, 4 h 24 h [96]
Rat male fasted not measurable because of inhalation, 4 h [5]
(overnight) non-linear concentration-
mortality relationship
Rat male 16-h fasted 1660 mg/m3 (415 ppm) Inhalation, 4 h 14 days [248]
Rat female fed 41 200 mg/m3 (10 300 ppm) Inhalation, 4 h 14 days [249]
Rat female 16-h fasted 26 260 mg/m3 (6565 ppm) Inhalation, 4 h 14 days [248]
Mouse male fed 392 mg/m3 (98 ppm) inhalation, 1 day [204]
female 420 mg/m3 (105 ppm)
Mouse male fed 140 mg/m3 (35 ppm) Inhalation, 2 days [204]
Mouse male fed 460 mg/m3 (115 ppm) Inhalation, 4 h 14 days [251]
Mouse male fasted 200 mg/m3 (50 ppm) Inhalation, 4 h 14 days [250]
Mouse female fed 820 mg/m3 (205 ppm) Inhalation, 4 h 14 days [251]
Mouse female fasted 500 mg/m3 (125 ppm) Inhalation, 4 h 14 days [250]
Table 7. (contd.)
______________________________________________________________________________________________________
Species Sex Nutri- Estimated LC50/LD50 Dosing criteria Limit of Reference
tional observation
status time
______________________________________________________________________________________________________ statustion time
Hamster male fed 6640 mg/m3(1660 ppm) Inhalation, 4 h 14 days [109]
Hamster male fasted 600 mg/m3 (150 ppm) Inhalation, 4 h 14 days [108]
Hamster female fed 11 780 mg/m3 (2945 ppm) Inhalation, 4 h 14 days [109]
Hamster female fasted 1780 mg/m3 (445 ppm) Inhalation, 4 h 14 days [108]
Rat male fed 1550 mg/kg gavage 24 h [100]
Rat male fed 1510 mg/kg gavage 96 h [100]
Rat male fed 1800 mg/kg gavage not stated [172]
Rat female fed 1500 mg/kg gavage not stated [172]
Rat male fed 800-2000 mg/kg gavage 14 days [2]
Mouse male fed 201-235 mg/kg gavage not stated [103]
Mouse female fed 171-221 mg/kg gavage not stated [103]
______________________________________________________________________________________________________
a This report noted that mortality was consistently observed at doses as low as 50 mg/kg. In 73-g
male rats, dose-mortality curves showed a maximum mortality (10/10) at 300 mg/kg. Percent
mortality then decreased as dose was increased to 800 mg/kg.
Jaeger et al. [96] showed that the toxicity of vinylidene
chloride in male Holtzman rats was enhanced as a result of fasting.
The estimated 24-h LC50 for fed rats was 60 000 mg/m3 (15 000
ppm) (4-h exposure), while the corresponding value for 18-h fasted
rats was 2400 mg/m3 (600 ppm) (n = 5 or 6). The minimum lethal
concentrations were 40 000 and 800 mg/m3 (10 000 and 200 ppm),
respectively. At levels of 600 mg/m3 (150 ppm) or more, serum-
alanine-alpha-ketoglutarate transaminase rose rapidly in fasted
rats (within 2 h of termination of a 4-h exposure at 8000 mg/m3
(2000 ppm)). This indication of liver damage did not arise at
levels below 8000 mg/m3 (2000 ppm) in rats provided with food.
The increased susceptibility of fasted rats was considered to be
due to decreased availability of hepatic glutathione and this was
supported by the potentiation of hepatotoxicity by treatment of
fed rats with the agent diethylmaleate, which depletes glutathione
(section 6.1.5). This was further supported by the observation
[93] that a 4-h exposure of male Holtzman rats to 8000 mg
vinylidene chloride/m3 (2000 ppm) in the morning produced a
3-fold increase in serum alanine-alpha-ketoglutarate transaminase
activity with no deaths. Conversely, the same treatment in the
afternoon resulted in an almost 10-fold increase in serum
alanine-alpha-ketoglutarate transaminase, and 2 out of 5 treated
rats died within 23 h. The relative susceptibilities were
inversely related to hepatic glutathione levels.
In fasted male Sprague-Dawley rats exposed to 800 mg vinylidene
chloride/m3 (0.02%) , liver toxicity was observed within 2 h
[188]. Toxicity in liver parenchyma was characterized by
retraction of cell borders and the formation of pericellular
"lacunae". Nuclei showed segregation of chromatin towards the
margins of the nuclear envelope and mitochondria were swollen with
ruptured outer membranes. Midzonal hepatic necrosis led to
haemorrhagic centrilobular necrosis within 6 h. This liver
necrosis, along with raised levels of serum-alanine-alpha-
ketoglutarate transaminase and an associated fall in hepatic
glutathione levels, were all minimized by pretreatment of rats
with the cytochrome P-450-inducing agents phenobarbital and
Aroclor-1254.
In an earlier study [23], male Sprague-Dawley rats were exposed
to 5760 mg vinylidene chloride/m3 (1440 ppm) for 1 h. Twenty-four
hours later, liver damage was detected by measured elevations in
serum glutamic oxalacetic transaminase and glutamic pyruvic
transaminase. Pretreatment of these animals with phenobarbital and
3-methylcholanthrene did not alter the extent of the elevation of
serum hepatic enzyme levels. Furthermore, the activity of glucose-
6-phosphatase in the liver was also unaffected 24 h after exposure
to 9080 or 11 960 mg vinylidene chloride/m3 (2270 ppm or 2990 ppm)
in both 3-methylcholanthrene and phenobarbital-treated animals.
In contrast, exposure to 80 000 or 130 000 mg vinylidene
chloride/m3 (20 000 or 32 500 ppm) for 1 h was lethal for most of
the rats (groups of 4 rats) pretreated with the inducing agents,
despite the fact that these concentrations did not produce
deaths in similar groups of control rats. Treatment of rats with
the cytochrome P-450 inhibitors "SKF-525A" and "Lilly 18947" reduced
the survival time after inhalation of 168 000 or 212 000 mg
vinylidene chloride/m3 (42 000 ppm or 53 000 ppm). Because of the
differential effects of inducing agents on hepatotoxicity and
lethality, the results suggest that the lethal effects of inhaled
vinylidene chloride are distinct from the hepatotoxic effects.
Since cytochrome P-450 has been implicated in the activation of
vinylidene chloride to form toxic metabolite(s) (section 6.1.4) the
protective or lack of potentiating effect of inducing agents on
hepatotoxicity is not understood, but may result from multiple
roles of microsomal enzymes (section 8.1.2.1).
Subsequently, Reynolds et al. [189] exposed fasted male rats
to 800 mg vinylidene chloride/m3 (200 ppm). Glutathione levels in
the liver were rapidly depleted during the first and second hours
of exposure and were replenished during the third and fourth hours,
when toxicity was determined by analysis of tissue sections by
light microscopy. The rebound of glutathione levels was not
observed in the mitochondria, which might indicate a role of this
organelle in toxicity. Inactivation of the microsomal enzyme
cytochrome P-450 was not appreciable prior to histological
alterations in the liver and therefore did not appear to be an
early event in cytotoxicity. The toxicity of vinylidene chloride
(2000 or 4000 mg/m3 (500 or 1000 ppm)) inhaled for 3 or 24 h was
exacerbated by the glutathione-depleting agent phorone, as
evidenced by an increase in the levels of serum-aminotransferases
and sorbitol dehydrogenase at 3 h and mortality at 24 h in male
Wistar rats [207]. Phorone (250 mg/kg) also had the effect of
increasing the half-life of the terminal elimination phase of
vinylidene chloride from 0.89 to 2.33 h and from 1.55 to 4.21 h at
levels of 2000 and 4000 mg vinylidene chloride/m3 (500 and 1000
ppm), respectively, in a closed exposure chamber. Therefore,
increased toxicity was associated with decreased metabolism.
Since there was no evidence for an effect of phorone on the
activity of the mixed-function oxidase, the authors proposed that
phorone produced a quantitative change in vinylidene chloride
metabolism leading to intermediates of greater toxicity. However,
the results might also be explained by a build up of intermediates
(due to reduced ability to conjugate with glutathione) that inhibit
their own formation.
Andersen et al. [5] studied the dependence of inhalation
toxicity (as measured by plasma aspartate transaminase) on both
exposure concentration and duration in fasted male HOT:SD(BR)
Holtzman rats. Plasma-enzyme levels increased markedly after
exposure to 800 mg vinylidene chloride/m3 (200 ppm) for 1.25 h.
After this time, no further increase in plasma aspartate
transaminase was recorded. The concentrationmortality curve
increased rapidly between 400 and 800 mg/m3 (100-200 ppm) and
reached a plateau between 800 and 4000 mg/m3 (200-1000 ppm), making
it impossible to make a meaningful estimate of the LC50. Thus, a
concentration x time relationship for toxicity is not apparent,
and this is in agreement with saturable metabolism (section
6.1.3.1) to toxic intermediates. Thus, estimates by other workers
of the LC50 under similar conditions cannot be considered accurate.
After inhalation of 800 mg vinylidene chloride/m3 (200 ppm)
for 0.5 h, immature rats showed significant prolongation of
pentobarbital-induced sleeping times, suggesting an inhibition of
pentobarbital metabolism [5]. This increase in sleeping times was
maintained for at least 3 days after a 2-h exposure. In mature
rats, treated with 1600 mg/m3 (400 ppm) for 2 h, there was no
evidence of an altered sleeping time within the exposure period,
though this was elevated 2 and 24 h later.
Exposure of fasted male rats to 8000 mg vinylidene chloride/m3
(2000 ppm) for 4 h led not only to elevation of serum alanine
alpha-ketoglutarate transaminase levels but also to increased
levels of hepatic sodium and calcium with a concomitant decrease in
potassium and magnesium levels and diminished histochemical
glucose-6-phosphatase activity. These changes were associated
with centrilobular necrosis and haemorrhagic necrosis of the entire
hepatic lobule. Thyroidectomized, fasted rats, exposed under the
same conditions, showed significantly less change in hepatic
electrolyte concentrations. Morphological injury was also
minimized and was similar to that seen in non-pretreated fed
animals. Mortality was also inhibited by thyroidectomy. In
contrast, thyroxine pretreatment potentiated the toxicity of
vinylidene chloride and restored the susceptibility of
thyroidectomized rats. It was suggested that the protective
effect of thyroidectomy was at least partially mediated by the
observed elevation of hepatic glutathione levels [216].
However, differences were observed between fed, fasted, and
hyperthyroid Sprague-Dawley rats in the effects of orally
administered vinylidene chloride (50 mg/kg body weight) on body
temperature, serum glucose concentrations, hepatic glutathione,
and glutathione transferase [106]. The different patterns of
response in the three groups suggest different mechanisms of
toxicity in fasted and hyperthyroid rats.
Similar findings were reported by Jaeger et al. [98]. In a
study on male Sprague-Dawley rats, a raised serum sorbitol
dehydrogenase level (a cytoplasmic marker) coincided with an
elevation in serum ornithine carbamoyl transaminase from the
mitochondria. This finding suggests that mitochondrial damage is an
early event in the hepatotoxicity of vinylidene chloride. The data
support the authors' theory that the metabolite monochloracetic
acid is toxic to mitochondria via chlorocitric acid and "lethal
synthesis" leading to accumulation of citric acid [97].
The kidney is also affected by vinylidene chloride. At
sublethal concentrations (800 mg/m3 (200 ppm) for 6 h), male
Sprague-Dawley rats given food ad libitum did not show any signs of
an adverse response [132]. In contrast, rats previously fasted for
18 h, were found to have haemoglobinuria, which persisted for 12-
24 h after exposure. As well as seeing multiple foci of hepatic
centrilobular degeneration and necrosis, marked degeneration
of kidney proximal tubular epithelia was observed after this
exposure in the fasted rats, but not in those that were fed. These
changes were associated with an increase in the level of 14C
covalently bound in the liver following exposure to [14C]-
vinylidene chloride (section 6.1.5). Hepatotoxic effects were not
noted in either fed or fasted rats exposed to 40 mg vinylidene
chloride/m3 (10 ppm). In a more recent study, inhalation of
vinylidene chloride produced acute nephrotoxicity (which was not
associated with calcium oxalate formation) in male Sprague-Dawley
rats [90]. Twenty-four hours after a 4-h exposure to 1000 mg
vinylidene chloride/m3 (250 ppm) or more, kidney/body weight
ratios, serum urea nitrogen, and creatinine levels were
significantly increased. This was associated with moderate
cellular swelling in the renal cortex (800 mg/m3 (200 ppm)) and
severe tubular necrosis (>1200 mg/m3 (>300 ppm)). Aroclor-1254
and phenobarbital pretreatment antagonized renal toxicity.
8.1.1.2 Mice
A marked individual variation in the lethal concentration
of vinylidene chloride (ranging from 500 to 10 000 mg/m3; 125 to
2500 ppm) was noted by Lazarev [116].
In the study by Short et al. [204], the toxicity of inhaled
vinylidene chloride was investigated in CD-1 mice. The 1- and 2-day
LC50 values for groups of 10 mice were approximately 400 mg/m3 (100
ppm) (males and females) and 140 mg/m3 (35 ppm) (males),
respectively. These values are considerably lower than those
reported for rats and, in contrast to the data on mice, no deaths
were seen in male rats exposed to 240 mg vinylidene chloride/m3
(60 ppm). In addition, up to 240 mg vinylidene chloride/m3 (60
ppm) produced a dose-dependent histopathological change in mouse
liver and an increase in both serum glutamic oxaloacetic
transaminase and serum glutamic pyruvic transaminase. The serum
enzymes were also elevated in male rats but not to the same extent
and only with a longer exposure period. Mice exposed for 1 day to
60 mg vinylidene chloride/m3 (15 ppm) showed hepatocellular
degeneration and increased mitotic figures of hepatocytes together
with severe kidney tubular nephrosis. At 120 mg/m3 (30 ppm),
midzonal hepatic necrosis was also seen, the severity of which was
increased at 240 mg/m3 (60 ppm). In contrast, rats exposed to 240
mg/m3 (60 ppm) for 1 day showed only mild hepatic centrilobular
degeneration and/or necrosis and mild bileduct hyperplasia. The
inhibition of these toxic effects and of covalent binding of
[14C]-vinylidene chloride-derived radioactivity in the mouse liver
and kidney by disulfiram was described. The mechanism of this
protection was not established but may be via modulation of
metabolism.
The studies of Zeller et al. [250] on NMRI mice showed that,
as with rats, males were more susceptible than females (Table 7)
and that fasting potentiated lethality. Symptoms included apathy,
narcosis, dyspnoea, and immobility. Postmortem examination of
mice dying from vinylidene chloride exposure particularly showed
acute emphysema and congestion of lungs.
The effects of vinylidene chloride on DNA following acute
inhalation (40 or 200 mg/m3 (10 or 50 ppm) for 6 h) have been
studied in male CD-1 mice as well as Sprague-Dawley rats [186].
These exposures gave rise to tissue damage (nephrosis, increased
mitotic figures, and regeneration) and increased DNA semi-
conservative replication (25-fold) in mouse kidney, but not in
mouse liver. In contrast, DNA semiconservative replication in rat
kidney was increased only 2.2 fold and was slightly decreased in
rat liver following exposure to 40 mg/m3 (10 ppm). DNA repair and
DNA alkylation in these organs were minimal in both rats and mice
(section 8.5.1). In the light of these minimal effects, the
authors suggested that the carcinogenicity of vinylidene chloride
in the mouse kidney (section 8.7.1) might be via an epigenetic
mechanism.
8.1.1.3 Other animal species
Effects of vinylidene chloride on the respiratory system have
also been reported in cats, rabbits, and guinea-pigs [192].
Pulmonary irritation and lung oedema, haemorrhage, and pneumonia
were seen following exposure to vinylidene chloride at 2000 or 6000
mg/m3 (500 or 1500 ppm) (cats) and 5000 or 8000 mg/m3 (1250 or 2000
ppm) (guinea-pigs) for 2 h. Exposure to concentrations ranging from
500 to 2000 mg/m3 (125 to 500 ppm) for 40 min inhibited spinal
reflexes in rabbits. This species survived after exposure for 40
min to a concentration of 30 000 mg/m3 (7500 ppm). Klimisch &
Freisberg [108, 109] studied the inhalation toxicity of vinylidene
chloride in fed and fasted Chinese striped hamsters. The LC50
values are given in Table 7. As with rats and mice, males were
more susceptible to lethality than females, and fasting potentiated
the toxicity markedly. Hamsters that died showed acute dilation
and passive hyperaemia of the heart, congested lungs, and
lobulation of the liver.
8.1.2. Oral
As with inhalation, the principal organs affected by oral
administration of vinylidene chloride are the liver, kidneys, and
lungs.
The LD50 values for orally administered vinylidene chloride in
rats and mice are shown in Table 7. It can again be seen that mice
are more susceptible than rats, but few sex differences in response
are observed following administration of vinylidene chloride by
gavage.
8.1.2.1 Rats
Hepatic damage produced by intubation of 400 mg vinylidene
chloride/kg to fasted rats was indicated by elevation of serum
alanine alpha-ketoglutarate transaminase activity within 4 h [94].
The level of glucose-6-phosphatase activity was reduced in the
liver at 8 h, but not after 4 h, suggesting (in line with the
above discussion) that initial toxicity was not associated with the
endoplasmic reticulum membrane. Treatment of rats with a
relatively high dose of 12.5 mmol vinylidene chloride/kg did not
lead to elevation of malondialdehyde or conjugated dienes in
incubated liver homogenates taken 1 h after dosing. This
contrasts with the effect of a hepatotoxic dose of carbon
tetrachloride and suggests that lipid peroxidation is not involved
in the hepatotoxicity of vinylidene chloride.
Jenkins et al. [100] also reported, that pretreatment of rats
with the microsomal enzyme-inducing agent phenobarbital offered
protection against the hepatotoxic effects of orally administered
vinylidene chloride. The acute 24-h toxicity of vinylidene
chloride was enhanced 18-fold by adrenalectomy, suggesting that the
adrenals were involved in protection against lethal effects. The
mechanisms of protection were not understood.
The effects of induction and inhibition of microsomal enzymes
in fasted male Holtzman rats were studied in more detail by
Andersen et al. [4]. Pretreatment of rats with phenobarbital
markedly reduced the lethality of a 100 mg/kg oral dose of
vinylidene chloride in immature rats (140 g), but not in large
adult (331 g) rats.
These results suggest that a microsomal detoxification system,
inducible in immature rats, was operative. This is supported by
the finding that the cytochrome P-450 inhibitor SKF-525A
exacerbated the lethal effects of a dose of vinylidene chloride of
200 mg/kg in rats (260-270 g) but did not have any effect on
mortality in immature (80-100 g) 2,3-epoxy-propan-1-ol, rats. One
of a range of epoxides that exacerbate the toxicity of orally
administered vinylidene chloride, which is particularly potent, was
a relatively poor substrate for glutathione- S- transferase and
styrene oxide hydrolase. Thus 2,3-epoxypropan-1-ol, rather than
inhibiting the protective effects of epoxide hydrolase or
glutathione conjugation (section 6.1.4) appeared to inhibit a
further (uncharacterized) detoxification pathway [3]. The role of a
further microsomal enzyme system in the production of a toxic
intermediate was indicated by the protective effects of
pretreatment of rats of all sizes with pyrazole, 3-aminotriazole,
and carbon tetrachloride against the lethality of an oral dose of
200 mg/kg [4].
Andersen & Jenkins [2] noted that female Holtzman HOT:(SD)BR
rats were much less susceptible to the hepatotoxic effect than
male rats, the threshold oral dose for the elevation of plasma
transaminase activity being approximately 100 mg/kg in the females.
When mature, male rats were given 400 mg vinylidene chloride/kg,
levels of plasma-aspartate transaminase were 4-5 times greater in
18-h fasted rats than in control rats. The effects of fasting are
thought to be due to glutathione depletion (section 6.1.5).
Chieco et al. [28] studied hepatotoxicity in fasted male
Sprague-Dawley rats given vinylidene chloride as an oral dose in
mineral oil. Two hours after dosing with 200 mg vinylidene
chloride/kg or 6 h after a lower dose of 50 mg/kg, early damage to
plasma and mitochondrial membranes was indicated by raised hepatic
sodium levels and decreased central area histochemical staining
of bile canaliculi membrane Mg2+-ATPase, outer membrane
mitochondrial monoamine oxidase, and inner membrane mitochondrial
succinate dehydrogenase and cytochrome oxidase. The extent of
injury (indicated by raised serum transaminase activity and
decreased histochemical staining of membrane components) increased
with time after dosing or with increased dose. Four and 6 h after
administration of a 200 mg/kg dose, necrosis occurred around the
central vein of the liver. There were no histochemical alterations
in the kidneys at 6 h. The same investigators [27] noted that
this treatment produced increased plasma-haemoglobin levels and
granular haem casts in the loop of Henlé. No pathological changes
were seen in the heart, lungs, spleen, adrenals, or duodenum.
Hepatic damage was more severe with Tween 80 used as a dose vehicle
than with corn oil or mineral oil, reflecting a relatively high
rate of absorption when administered in Tween 80 (section 6.1.1.2).
Eight hours after oral administration of 40 mg vinylidene
chloride/kg body weight to rats, centrilobular hepatocytes showed a
dilated endoplasmic reticulum and swollen mitochondria and
perinuclear cisternae. The nucleosplasm was homogeneous suggesting
chromatinolysis [5].
In unanaesthetized, freely moving fed and fasted Sprague-
Dawley male rats [148], at least a 2-fold increase in inulin
excretion was observed within 2 h of oral administration of 200 mg
vinylidene chloride/kg. Bile flow decreased in treated rats (up to
40% and 65% in the fed and fasted rats, respectively). Thus,
vinylidene chloride alters hepatobiliary permeability and causes
cholestasis.
Kanz & Reynolds [105] investigated the occurrence of
morphological changes in the liver in relation to time after oral
administration of vinylidene chloride (25, 50, or 100 mg/kg in
mineral oil) (see also Kanz et al. [106], reported in section
8.1.1.1). One, 2, or 3 h after administration to fasted male
Sprague-Dawley rats, the liver was examined microscopically
following in situ perfusion fixation. Dilation of the bile
canaliculi with an increase in the number of microvilli or membrane
fragments in canaliculi was seen with the formation of canalicular
diverticuli in centrilobular hepatocytes within 1-2 h.
Subsequently, microvilli on the sinusoidal surfaces were lost, and
cytoplasmic vacuolation occurred. These early changes were seen
without morphological alteration of the endoplasmic reticulum or
mitochondria. Not until 4-6 h after an oral dose of 200 mg
vinylidene chloride/kg was a decrease seen in the activity of
enzymes in the sinusoidal plasma, mitochondrial matrix,
endoplasmic reticulum, lysosomes and cytosol, and then only in
regions of gross injury. At 2 h, scattered hepatocytes showed
nuclear and cell surface anomalies that were characteristic of
apoptosis [190].
In agreement with the findings of Reynolds et al. [189] in
inhalation studies (section 8.1.1.1), Moslen & Reynolds [147] found
that loss of activity of microsomal cytochrome P-450 was not an
early event in the toxicity of vinylidene chloride (200 mg/kg)
orally administered to fasted male Sprague-Dawley rats.
Cytochrome P-450 deactivation was concomitant with an elevation in
the activities of serum glutamate oxalacetate transaminase and
serum glutamate pyruvate transaminase and did not occur until
between 2 and 3 h after administration of vinylidene chloride.
These effects were preceded by a marked inhibition (within 1 h) of
the activity of glutathione- S- transferase towards
dichloronitrobenzene, chlorodinitrobenzene, and 1,2-epoxy-3-( p-
nitrophenoxy)-propane (but not towards ethacrynic acid) and a
concomitant reduction of hepatic glutathione levels. A correlation
was found between the dose-dependency of inhibition of
glutathione- S- transferase activity and of cytotoxicity. Thus,
both glutathione depletion and inhibition of specific glutathione-
S- transferase(s) precede toxicity.
Simultaneous treatment of 8 male Wistar rats with ethanol (4.8
g/kg, oral) protected against the hepatotoxicity of vinylidene
chloride (0.125 g/kg, oral) as measured by the elevation of the
activities of serum aminotransferases and sorbitol dehydrogenase
[208]. Conversely, pretreatment of rats with ethanol (5% in
drinking-water for 7 days) exacerbated the hepatotoxicity of orally
administered vinylidene chloride (0.125 or 0.2 g/kg). Dithiocarb or
(+)-catechin (0.2 g/kg) administered simultaneously with vinylidene
chloride also reduced vinylidene chloride-induced hepatotoxicity.
Evidence was provided that the simultaneous treatment with ethanol
and dithiocarb may lead to depression of the metabolism of
vinylidene chloride. It was postulated that (+)-catechin might act
as a scavenger of reactive intermediate(s), which may also be the
mechanism of protection afforded by (+)-cyanidanol-3 [207].
Acetone is another agent that modifies the hepatotoxicity
of orally administered vinylidene chloride [75]. Administration of
5 or 10 mmol acetone/kg orally to male Sprague-Dawley rats
potentiated liver injury (elevated plasma glutamic pyruvic
transaminase and ornithine carbamyl transferase activity and liver
total bilirubin content) caused by a single oral dose of 50 mg
vinylidene chloride/kg. However, acetone given at 1, 15, or 30
mmol/kg, did not potentiate hepatotoxicity. The biphasic effect
of acetone could not be explained but was considered by the authors
to be related to dose-dependent changes in more than one
biotransformation process.
A further effect of vinylidene chloride was the prolongation of
barbiturate sleeping times [92]. Two to 4 h after an oral dose of
400 mg vinylidene chloride/kg to male Holtzman rats, the
pentobarbital sleeping time was elevated (136% of control), (prior
to hepatotoxicity as indicated by loss of glucose-6-phosphatase
activity). Both hexobarbital and pentobarbital sleeping times were
elevated at 17-22 h, concomitant with hepatic injury. The early
effects on pentobarbital sleeping times were due, not to decreased
metabolism of the barbiturate, but to an elevation of its
concentration in serum through altered absorption or distribution.
Kidney toxicity was studied by Jenkins & Andersen [99] in
NMRI:0(SD), Sprague-Dawley-derived rats. Within 24 h of oral
administration of 400 mg vinylidene chloride/kg , fasted male rats
showed raised levels of plasma urea nitrogen and creatinine.
Within 48 h, tubular dilation was observed with necrosis and
vacuolation of tubular epithelium. Some tubules contained a blue-
black amorphous material. These histopathological effects were
preceded by inflammation in some animals. Elevation of plasma urea
nitrogen and creatinine was not observed in fed rats with identical
treatment and was less evident in fasted females than in males.
However, the histopathological effects were no less severe in the
female rats. The time-course for the nephrotoxic response (maximum
at 48 h) in male and female fasted rats was slightly preceded by
hepatotoxicity as indicated by the appearance in the plasma of
aspartate transaminase, alanine transaminase lactate dehydrogenase,
and sorbitol dehydrogenase (maximum at 8-24 h). This finding is
in agreement with that reported above [28].
8.1.2.2 Mice
Orally administered vinylidene chloride also caused pulmonary
injury in male C57B1/6 mice [52]. Following administration of
100 mg/kg, peribronchial and perivascular oedema were seen
in the lungs. Histopathology revealed dilation of Clara cell
cisternae and degeneration of the endoplasmic reticulum. A 200
mg/kg dose caused severe necrosis of ciliated and Clara cells and
exfoliation of the bronchial lining within 6 h. Both doses caused
concurrent hepatotoxicity as evidenced by raised levels of serum
glutamic oxalacetic transaminase and glutamic pyruvic transaminase.
By 24 h, pulmonary oedema, haemorrhage, and focal atelectasis
were also observed in association with hypoxia. Recovery was seen
within 7 days. Following a higher dose of 200 mg/kg, injury was
seen to be followed by cellular proliferation as indicated by
incorporation of a pulse of [3H]-thymidine into total
pulmonary DNA [53]. Proliferative activity reached a peak
between 3 and 5 days after treatment with vinylidene chloride.
The majority of the 3H was incorporated into non-ciliated
bronchiolar epithelial cells.
8.1.3. Other routes
8.1.3.1 Intraperitoneal
When given by intraperitoneal (ip) injection to male ddY strain
mice at a dose level of 120 mg/kg (0.1 ml/kg), vinylidene chloride
produced hypothermia within 30 min and severe renal damage at 24 h
(as shown by elevated plasma urea nitrogen and kidney calcium
levels) [143]. Renal tubular necrosis was much more severe than
hepatic damage. Pretreatment of mice with diethyldithiocarbamate or
carbon disulfide protected against renal and hepatic toxicity,
possibly via an inhibitory effect on metabolic activation.
Vinylidene chloride at 605 mg/kg (0.5 ml/kg, ip) caused liver
damage in male Sprague-Dawley rats as evidenced by raised serum
glutamate pyruvate transaminase and increased bile duct pancreatic
fluid flow at 24 h [70]. Hepatic microsomal glucose-6-phosphatase
and ATP-dependent calcium pump were both inhibited 24 h after
intraperitoneal administration of vinylidene chloride (1 mg/kg) to
male Sprague-Dawley rats [145]. The level of conjugated dienes in
microsomes obtained 2 h after vinylidene chloride injection was
not significantly different from that measured in control rat
microsomes, suggesting that the toxic effects were not mediated by
lipid peroxidation.
The results of Siegers et al. [209] also suggest that lipid
peroxidation is not a mechanism involved in the early stages of
vinylidene chloride hepatotoxicity. Up to 2 h after an
intraperitoneal injection of 0.5 g vinylidene chloride/kg to male
Wistar rats, ethane exhalation was only slightly higher than the
control levels and was not affected by hypoxia.
As was observed in the short-term inhalation studies on
vinylidene chloride (section 8.2.1), the compound was found to
induce cytochrome P-450 activity following a single administration
to C57B1/6N mice by intraperitoneal injection. Over the range of
50-150 mg/kg, a dose-dependent induction of microsomal 7-
ethoxyresorufin and 7-ethoxycoumarin O- deethylation (but not total
cytochrome P-450 content or benzo (alpha)pyrene hydroxylase) was found
in the kidney [112].
Male C57BL/6J mice given 125 mg vinylidene chloride/kg (ip)
developed extensive necrosis of the Clara cells in the lung within
24 h, but no necrosis was observed in the liver or kidney at this
time. The loss of Clara cells was associated with a significant
decrease in pulmonary cytochrome P-450 content and activity
[113]. The susceptibility of the Clara cells was considered to be
due to activation of vinylidene chloride by cytochrome P-450 in
these cells. This conclusion was also reached by Forkert et al.
[55] who showed that covalent binding of vinylidene chloride
products to cellular macromolecules in the lung accompanied lung
toxicity in CD-1 mice given an identical dose to that given in the
study described above (section 6.1.5). Degenerative changes
occurred in Clara cells as early as 1 h following treatment with
vinylidene chloride and were characterized by mitochondrial
swelling and aggregation of chromatin against the nuclear membrane.
Cell death was apparent at 2 h and, by 24 h, the majority of Clara
cells were exfoliated [56]. However, these authors concluded that
there was a lack of correlation between the extent of covalent
binding and either lung or liver toxicity [55].
8.1.3.2 Eyes and skin
Little information is available on the effects of vinylidene
chloride on the eyes and skin.
It is moderately irritating to the eyes of rabbits causing
transient corneal injury and is also a skin irritant in the rabbit
[222]. Rylova, [192] reported that vinylidene chloride was an
irritant for the eyes of rats, mice, guinea-pigs, and cats.
Transient redness was observed following application to shaved
rabbit skin. The stabilizer ( p- methoxyphenol) in vinylidene
chloride preparations may contribute to irritation.
8.1.4. Summary of acute toxicity
Vinylidene chloride may cause irritation of the skin and eye,
depression of the central nervous system, and acute toxic effects
on the heart, lung, liver, and kidney. The variation in estimations
of the acute LC50 for vinylidene chloride is considerable. This
can be explained partially by inaccuracies that arise because of a
non-linear concentration-mortality relationship. Generally, mice
are more susceptible than rats and males are more susceptible than
females. The toxic effects are dependent on cytochrome P-450
activity (which may also be involved in detoxification) and can be
exacerbated by glutathione depletion. Hepatotoxicity may be
enhanced by ethanol and thyroxine, inhibited by dithiocarb and
(+)- catechin, and modulated by acetone.
8.2. Short-Term Exposures
8.2.1. Inhalation
In the study by Prendergast et al. [173], groups of various
animal species were continuously or repeatedly exposed to
vinylidene chloride through inhalation for 90 days. The results
are shown in Table 8. In survivors of continuous exposure at a
concentration of 101 mg/m3 or less, no histopathological changes
could be attributed to vinylidene chloride. At 189 mg/m3, livers
from surviving dogs, monkeys, and rats showed morphological
changes consisting of fatty metamorphosis, focal necrosis,
haemosiderin deposition, lymphocytic infiltration, bile-duct
proliferation, fibrosis, and pseudolobule formation, particularly
in dogs. All rats displayed nuclear hypertrophy of the kidney
tubular epithelium. Non-specific inflammatory changes were seen in
the lungs of a majority of the animals. Liver alkaline phosphatase
activity and serum glutamic-pyruvic transaminase activity were
measured in rats and guinea-pigs and found to be elevated. Dogs,
monkeys, and rats showed reduced weight gain. The repeated
exposures are considered to be more relevant to human
occupational exposure. Gross examination of survivors showed a
high incidence of lung congestion in rabbits, monkeys, rats, and
guinea-pigs. Some fatty infiltration and several cases of focal and
sub-massive necrosis were observed in guinea-pig liver sections.
Table 8. Mortality of animals exposed to vinylidene chloride by inhalationa
__________________________________________________________________________
Mortality ratiob
_______________________________________________
Concentration Extent of Rat (Long Guinea- Rabbit Beagle Squirrel
of vinylidene exposurec Evans or pig (New dog monkey
chloride Sprague- (Hartley) Zealand
(mg/m3) Dawley) White)
_________________________________________________________________________
395 ± 32 A 0/15 0/15 0/3 0/2 0/3
189 ± 6.2 B 0/15 7/15 - 0/2 3/9
101 ± 4.4 B 0/15 3/15 0/3 0/2 2/3
61 ± 5.7 B 0/15 3/15 - 0/2 0/9
20 ± 2.1 B 2/45 2/45 - 0/6 1/21
no treatment B 7/304 2/314 2/48 0/34 1/57
_________________________________________________________________________
a From: Prendergast et al. [173].
b Mortality ratio shows the number of animals that died divided by the
number with which the study commenced.
c A = 30 exposures, 8 h/day, 5 days/week.
B = continuous 90-day exposure.
Shortly after this study, Gage [57] investigated the toxicity
of vinylidene chloride in Alderley Park rats. In this case, groups
of 4 male and 4 female rats were exposed to 2000 mg vinylidene
chloride/m3 (500 ppm) for 6 h per day over a period of 20 days.
Nose irritation and retarded weight gain were observed, and, at
autopsy, liver cell degeneration was detected by histology. At 800
mg/m3 (200 ppm) (4 male and 4 female rats), slight nose
irritation was seen, but the organs were normal on autopsy.
Inhalation studies [152, 176] demonstrated minimal recoverable
liver cell cytoplasmic vacuolation in Sprague-Dawley rats (20
animals per sex) after 90 days exposure for 6 h per day, 5
days/week, to vinylidene chloride at 100 or 300 mg/m3 (25 or 75
ppm). At both dose levels, body weight, haematology, urinalyses,
blood urea nitrogen, serum alkaline phosphatase and glutamic
pyruvic transaminase activities, gross pathology, organ weights,
kidneys, heart, testes, and brain were normal.
Maltoni & Patella [138] noted a greater toxicity in rats than
that reported by Gage [57]. The effects of 4 h exposure, 4-5 days
per week, for 28 days, were studied in Sprague-Dawley rats and
Swiss mice (40-800 mg/m3 (10-200 ppm)), Balb/c, C3H, and C57B1 mice
(600-800 mg/m3 (150-200 ppm)), and Chinese hamsters (100 mg/m3 (25
ppm) only). The exposure periods were reduced at or above 200
mg/m3 (50 ppm) in mice because of severe acute toxicity, and the
800 mg/m3 (200 ppm) treatment of rats was reduced to 600 mg/m3 (150
ppm) after 2 days because of toxic effects. A minimum of 30
animals was used in all groups exposed to vinylidene chloride.
The weight of animals, clinical signs of toxicity, mortality,
and histopathological changes were monitored at, or up to, 28 days,
except in the case of mice exposed to 400-800 mg/m3 (100-200 ppm),
when the observation period was 9 days. Histopathological studies
indicated that the liver and kidneys were the major target organs.
The toxicity varied with animal species and strain, susceptibility
being in the following order: Swiss mice >Balb/c mice >C3H mice
>C57B1 mice >rats. In general, females were less responsive than
males, with the exception of female C3H mice, which were more
susceptible than the females of the other strains tested. An
association between the occurrence of acute toxicity and the
reported carcinogenicity in the Swiss mouse was noted (section
8.7.4). Another comparison study also indicated marked strain and
sex differences in the response of mice to vinylidene chloride
[153]. The mice (10 males and 10 females per dose level) were
exposed to 220, 400, or 800 mg vinylidene chloride/m3 (55, 100, or
200 ppm) for 6 h/day, 5 days/week, for a total of 10 exposures.
Only at 800 mg/m3 (200 ppm) were exposure-related deaths observed
and the rates of mortality were greater in the males than in the
females in Ha (ICR), CD-1 and CF-W mice, but no sex-related
differences in mortality were observed in B6C3F mice. Gross and
histopathological examinations indicated that nephrotoxicity
accounted for mortality in the male mice, but this was not the case
in female mice. Hepatotoxicity was considered to be the cause of
death in Ha (ICR) and B6C3F1 mice. The greatest sensitivity to
renal toxicity was seen in male CF-W mice (a strain derived from
the Swiss-Webster strain, believed to be genetically comparable to
Maltoni's Swiss mouse).
The greater susceptibility to vinylidene chloride of mice
compared with rats was also shown by Short et al. [204], agreeing
with the findings from acute single exposures (section 8.1). After
2 days exposure to 240 mg/m3 (60 ppm), 8/10 and 0/10 deaths were
recorded in male CD-1 mice and male CD rats, respectively. No
mice survived a longer period of exposure at 240 mg/m3 (60 ppm).
Serum glutamic oxaloacetic transaminase and glutamic pyruvic
transaminase levels were raised in both rats and mice, but more so
in the latter. Severe hepatotoxicity and nephrotoxicity were
observed in mice at autopsy.
A comparative study on the short-term toxicity of vinylidene
chloride in male Sprague-Dawley rats and in both sexes of Swiss-
Webster mice was reported by Oesch et al. [156]. Animals (minimum
of 10 per group) were exposed to an atmosphere containing
vinylidene chloride at 40 or 200 mg/m3 (10 or 50 ppm) (mice) or
800 mg/m3 (200 ppm) (rats) for 6 h, on 1, 3, or 8 days, and killed
one day after the last treatment. The majority of male mice
exposed to 200 mg/m3 (50 ppm) for 8 days did not survive. However,
female mice survived this treatment as did rats exposed to 800
mg/m3 (200 ppm). Various changes in the activities of
monooxygenase, epoxide hydrolase, and glutathione transferase
enzymes occurred in animals treated with vinylidene chloride. The
enzyme changes could contribute to the relative susceptibility of
the animals, according to the balance of activating and detoxifying
activity. In particular, cytosolic glutathione transferase
activity towards the substrate 2,4-dinitrochlorobenzene was
decreased in the kidneys of male mice (an organ susceptible to
carcinogenicity (section 8.7.1)), but not in the kidney of rats or
female mice or in the liver of either of these species (where
activity was either unchanged or enhanced).
A study on rabbits has also been reported by Lazarev [116].
Bronchitis, degenerative changes in the liver and kidney, and an
increase in the rate of proliferation of lymphoid tissue in the
spleen were observed in animals exposed to concentrations of 500-
2000 mg vinylidene chloride/m3 (125-500 ppm) for 3 h/day over a
period of 4 months.
In summary, a number of studies have indicated the particular
susceptibility of the male Swiss mouse to kidney toxicity. This
strain, sex, and species selectivity has important implications
regarding the specificity of the carcinogenic action of vinylidene
chloride (section 8.7.4).
8.2.2. Oral
In a 90-day study, vinylidene chloride was incorporated in
the drinking-water of male and female Sprague-Dawley rats at
nominal concentrations of 0, 60, 100 or 200 mg/litre [152,176].
Even at the highest concentrations administered (equivalent to 19-
26 mg/kg body weight daily), only minimal, reversible liver
cytoplasmic vacuolation was observed, with no abnormalities in
any of the other parameters investigated (section 8.2.1). Maltoni
& Patella [138] also noted a lack of lethality and clinical signs
of toxicity in Sprague-Dawley rats (50 of each sex) orally dosed by
gavage with 0, 0.5, 5, 10, or 20 mg vinylidene chloride/kg for 28
consecutive days. Four female and 4 male beagle dogs were given
6.25, 12.5, or 25 mg vinylidene chloride/kg in peanut oil
incorporated in gelatin capsules, daily for 97 days [177]. No
significant differences were seen between these animals and
controls with respect to appearance and demeanor, mortality, body
weight, food consumption, haematology, urinalysis, clinical
chemistry, and organ weights. There was also no depletion in
hepatic non-protein sulfhydryl levels in the liver or kidneys.
Some evidence for hepatotoxicity was provided by Siegers et al.
[208] using higher levels of orally administered vinylidene
chloride. Male Wistar rats were given 0.125 g vinylidene
chloride/kg in olive oil, by gavage, twice weekly for 2 weeks,
followed by a similar treatment at 0.2 g/kg for 2 weeks.
Hepatotoxicity was evidenced by mild increases in serum
sorbitol dehydrogenase and aminotransferases. Ethanol co-treatment
(5% in drinking-water) with the 0.2 g/kg dose of vinylidene
chloride enhanced toxicity leading to 6 deaths out of 10 animals.
Simultaneous application of dithiocarb or (+)-catechin with
vinylidene chloride led to total protection against lethal effects.
The effects of these agents have been discussed elsewhere (section
8.1.2.1).
8.3. Long-Term Exposure
A number of studies described in this section are also
discussed in section 8.7 in relation to carcinogenicity.
8.3.1. Inhalation
Sprague-Dawley rats, 84-86 of each sex, 6-7 weeks of age at the
start of the study, were exposed to vinylidene chloride vapour at
0, 40, or 160 mg/m3 (0, 10, or 40 ppm) for, 6 h/day, 5 days/week,
for 5 weeks, after which the levels of vinylidene chloride were
changed to 0, 100, and 300 mg/m3(0, 25, and 75 ppm) [178,179,180].
After exposure for a total of 18 months, the rats were observed for
a further 6 months. Additional animals were used for interim kills
at 1, 6, and 12 months. No clinical signs of toxicity were seen in
the exposed groups. Mean body weight gain was reduced at both dose
levels during the period of 8-13 months in males, but was not
reduced in females. Mortality in exposed groups was only slightly
higher than that in the controls (not different at the 40-100 mg/m3
(10-25 ppm) dose levels in males) and only in the latter part of
the study. Histopathological studies indicated increased
cytoplasmic vacuolation in the livers of exposed animals at 6 and
12 months of exposure. During the 6-month postexposure period of
the study, the hepatic changes were no longer discernible,
indicating reversibility.
There was also evidence of liver damage in CD-1 mice and CD
rats (groups of 36 males and 36 females) exposed to 220 mg
vinylidene chloride/m3 (55 ppm) (6 h/day, 5 days/week); the
animals were about 2 months old at the start of the study. Four
animals of each species and sex were killed for examination after
1, 2, 3, 6, and 9 months of treatment. The remaining animals were
killed at 12 months [118]. Groups of 100 animals were used as
controls. No effects were seen regarding haematology, clinical
blood chemistry, pulmonary macrophage count, cytogenic analysis of
bone marrow, X-ray examination of extremities, serum alpha-
fetoprotein, collagen content of liver, and lung and serum or
urinary aminolevulinic acid (ALA) (collagen and ALA were not
measured in the mice). However, mice exposed to vinylidene
chloride for 6-12 months had enlarged and basophilic hepatocytes
with enlarged nuclei, focal degeneration, and necrosis. A mild to
markedly severe focal disseminated vacuolization of the liver was
seen in the treated rats.
A similar long-term toxicity study was carried out by Hong et
al. [79] on male and female CD-1 mice and CD rats exposed to 220 mg
vinylidene chloride /m3 (55 ppm) or filtered air (controls), for 6
h/day, 5 days/week. The numbers of rats used per sex group were 4,
8, 8, and 14-16, and these were exposed for 1, 3, 6, and 10 months,
respectively. Mice (8, 8, and 12 animals per sex group) were
exposed for 1, 3, and 6 months, respectively. Animals were aged 2
months at the start of the study and were observed for 12 months
after treatment. No histopathological changes were observed as a
result of these treatments with vinylidene chloride. A total of
11/24 mice exposed for 6 months died or were terminated in a
moribund condition (compared with 11/56 controls). Only 3 rats died
following exposure for 6 months or less (20/30 rats died in the
group exposed for 10 months compared with 13/32 controls).
Long-term inhalation toxicity was also studied in rats, mice,
and hamsters by Maltoni et al. [141]. Groups of male and female
Sprague-Dawley rats (60 of each) were exposed to 40, 100, 200, 400,
600, or 800 mg vinylidene chloride/m3 (10, 25, 50, 100, 150, or 200
ppm), for 4 h/day, 4-5 days/week, for 52 weeks. Unexposed control
groups consisted of 100 rats of each sex. The rats were 16 weeks
old at the start of the study. At spontaneous death, hepatocyte
vacuolization, cloudy swelling, fatty degeneration, necrobiosis,
and necrosis were more frequent (57.6%) in rats exposed to 800-600
mg/m3 (200-150 ppm) than in control animals (20.5%). The highest
tolerable dose for long-term exposure in rats was 600 mg/m3 (150
ppm). In Swiss mice (aged either 9 or 16 weeks at the start of the
study) exposed to 40 or 100 mg vinylidene chloride/m3 (10 or 25
ppm) for the same exposure periods (minimum of 30 mice per sex per
dose level), changes were seen in the liver and kidneys that were
compatible with changes seen in long-term studies on control
animals. However, a higher incidence of regressive or phlogistic
changes was seen in the kidneys with renal adenocarcinoma (section
8.7.1) at 100 mg/m3 (25 ppm). A high mortality was seen at 200
mg/m3 (50 ppm). Those that survived only 4 exposures had hepatic
fibrosis, which was considered to be due to repair of necrosis.
Chinese hamsters (30 male and 30 female) exposed to a
concentration of 100 mg vinylidene chloride/m3 (25 ppm) for
periods identical to those given above for rats and mice, showed no
signs of altered histology compared with controls (18 male and 17
female) at spontaneous death.
Thus, the most significant observation on toxicity from long-
term inhalation studies is that of dose-dependent kidney damage in
male Swiss mice at dose levels that were not nephrotoxic in female
mice or in other species tested. The findings have important
implications in the light of similar tissue, sex, and species
specificities in carcinogenicity (section 8.7.1).
8.3.2. Oral
The long-term oral toxicity of vinylidene chloride was
investigated by Maltoni et al. [141] in Sprague-Dawley rats at 0.5,
5, 10, or 20 mg/kg (given once daily by stomach tube, 4-5
days/week, for 52 weeks). Animals were aged 9 or 10 weeks at the
start of the study. Fifty animals of each sex were used per dose
group with 100 controls (except for the 0.5 mg/kg group where there
were 160 controls). No signs of toxicity were reported from a
complete autopsy after 147 weeks (or 136 weeks, 0.5 mg/kg) except
that "hepatocyte vacuolization, cloudy swelling, fatty degeneration,
necrobiosis, and necrosis were found in some animals in treated as
well as in control groups".
In a separate study, 48 Sprague-Dawley rats of each sex (aged
6-7 weeks at the start of the study) were given vinylidene chloride
in the drinking-water at the following dose levels for 2 years: 7,
10, or 20 mg/kg body weight for males and 9, 14, or 30 mg/kg for
females (nominally 50, 100, and 200 mg/litre drinking-water).
Eighty rats per sex were dosed as control groups without vinylidene
chloride treatment [177, 179, 180]. Various parameters were
monitored, as indicated for the 90-day study (section 8.2.1).
Slightly increased cytoplasmic vacuolation of hepatocytes and
hepatocellular fatty change were the only evidence of toxicity and
occurred at all dose levels in the females, but only at 20 mg/kg
body weight (200 mg/litre drinking-water) in the males.
A US National Toxicology Programme study [154] indicated
chronic renal inflammation in male and female F344/N rats (50 per
group) given 5 mg vinylidene chloride/kg in corn oil, by gavage, 5
times/week, for 104 weeks. At 1 mg/kg, no renal toxicity was
observed. In all treated rats, the clinical signs and histopathology
of other organs were the same as in control rats. This group also
studied long-term oral toxicity in male and female B6C3F1/N mice
(50 per group) administered 2 or 10 mg/kg in corn oil. At 10
mg/kg, necrosis of the liver was evident in male but not in female
mice, but the reverse was true at the 2 mg/kg dose level. However,
the sponsors found defects in the conduct of the study and it could
not be satisfactorily evaluated.
Ponomarkov & Tomatis [172] gave rats an oral dose of vinylidene
chloride (50 mg/kg) weekly for the life span of the animal
following an initial in utero exposure (section 8.7.2 for details).
Rats that died up to 30 weeks after the start of oral dosing (7/89
males and 7/90 females) showed congestion of the lungs and
kidneys. At up to 80-90 weeks, haemorrhages and multiple lobular
necrosis of the liver were observed. The numbers of animals that
survived for 90 weeks were 71/89 (males) and 75/90 (females)
compared with 49/50 and 47/53 male and female controls,
respectively. Some of the treated survivors of 90 weeks showed
degenerative lesions of liver parenchymal cells.
8.4. Toxicity in vitro
As revealed in in vivo studies ([145] section 8.1.3.1),
in vitro studies with rat liver microsomes showed that calcium pump
activity was inhibited in a dose-dependent manner by vinylidene
chloride in the presence of NADPH. Malonic dialdehyde production
(a consequence of lipid peroxidation) was not associated with
inhibition of the calcium pump [182].
These findings stimulated Long & Moore [126] to test whether
vinylidene chloride treatment of hepatocytes could raise cytosolic
Ca2+ concentrations. Isolated hepatocytes from Sprague-Dawley rats
were exposed to vinylidene chloride (4 mmol/litre). Within 5 min,
the concentration of Ca2+ (estimated from the activity of
glycogen phosphorylase a) was raised to 0.3 µmol/litre compared
with 0.04 µmol/litre in vehicle-treated controls. In separate
studies, the concentration of Ca2+ was measured in hepatocytes
loaded with quin 2 (a sensitive fluorescent indicator of
cytoplasmic concentrations of ionized calcium). Within 20 seconds
of addition of vinylidene chloride at 4 mmol/litre, Ca2+ levels
were increased from 0.26 ± 0.05 µmol/litre to 0.59 ± 0.06 µmol/litre.
Disruption of intracellular Ca2+ homeostasis may prove to be a
mechanism that can contribute to vinylidene chloride
hepatotoxicity.
8.5. Mutagenicity and Other Genotoxicity Assays
8.5.1. Interaction with DNA
The fact that a number of metabolites of vinylidene chloride
are reactive and covalently bind to cellular macromolecules and the
nucleophile glutathione has already been discussed in section
6.1.5. The specific covalent interaction with DNA has been little
studied. As reported in section 8.1.1.2, DNA binding in vivo in
rats and mice exposed to [14C]-vinylidene chloride at 40 mg/m3 (10
ppm) (rats) and 40 or 200 mg/m3 (10 or 50 ppm) (mice) for 6 h was
minimal, though DNA adducts were evident from the covalently bound
radiolabel [186]. Adduct formation was greater in mice than in
rats and greater in the kidneys than in the liver. Binding was
dose dependent in the kidneys of mice (equivalent to 30 adducts/106
nucleotides at 200 mg vinylidene chloride/m3 (50 ppm) and 11
adducts/106 nucleotides at 40 mg/m3 (10 ppm). Incorporation of
radiolabel into DNA during synthesis or contamination of DNA
with other radiolabelled macromolecules cannot be excluded.
It was not possible to trap any alkylating metabolites in
vitro with 4-(4-nitrobenzyl)-pyridine, following incubation of
vinylidene chloride with a mouse liver microsomal system [15].
8.5.2. Genotoxicity in bacteria
The mutagenicity of vinylidene chloride in bacteria has been
demonstrated by a number of research workers and has been related
to the asymmetry of the putative epoxide metabolite [73]. The
positive effect was not seen in every study (Table 9) and in some
cases this may have been due to volatilization of the compound.
Vinylidene chloride (of unspecified purity) in air at 0.2, 2, or
20% for 4 h induced revertants (gene mutations) in the TA1530 and
TA100 strains of Salmonella typhimurium in the presence of NADPH-
supplemented 9000-g liver supernatant (S9) from phenobarbital
pretreated male OF-1 mice. Mutagenicity towards TA100 was also
catalysed by mouse kidney and lung 9000-g supernatants [14]. These
authors demonstrated that mutagenicity was not due to the
stabilizer 4-methoxyphenol. The role of cytochrome P-450 in
metabolic activation was evident from the lack of mutagenicity in
the absence of NADPH and from the observed greater mutagenicity of
vinylidene chloride towards S. typhimurium TA100 when mice used
for S9 donation were pretreated with phenobarbital. The role of
reactive electrophiles in mutagenicity was supported by the marked
inhibition of mutagenicity by the nucleophiles N- acetyl-cysteine
and N- acetyl-methionine. Protection against bacterial
mutagenesis was also afforded by pretreatment of rats (which
provided the hepatic S9) with pregnenolone-16 alpha-carnonitrile,
amino-cetonitrile, or disulfiram [15]. The mechanism(s) of these
effects are not known but may be via increased detoxification of
vinylidene chloride or its metabolites. In this study, 3-
methylcholanthrene as well as phenobarbital pretreatment of rats
provided S9 that had up to 2-fold greater capacity than untreated
rat liver preparations to activate vinylidene chloride (99% pure)
to bacterial mutagens. Liver specimens showing no pathological
lesions, obtained from 4 adult human patients for diagnostic
purposes provided S9 fractions that activated vinylidene chloride
to mutagens detected by S. typhimurium TA100 at approximately
one-fifth the activity of untreated mouse liver S9.
Jones & Hathaway [101] found vinylidene chloride (unspecified
purity) to be only very weakly mutagenic in S. typhimurium strain
TA1535 in the presence of liver (1.6 x background) and kidney (2.3
x background) S9 from untreated male Alderley Park Swiss-derived
albino mice. Exposure was via the atmosphere (5% vinylidene
chloride for 72 h) inside gas-tight culture vessels. Use of S9
from mice pretreated with Aroclor 1254 enhanced mutagenicity in
the test. In contrast, liver S9 preparations from male Sprague-
Dawley rats were able to mediate mutagenicity only after Aroclor
pretreatment and at a mutation frequency approximately 25% of that
seen with the corresponding mouse S9. These data are in agreement
with the greater oxidative metabolism observed in the mouse
compared with the rat (section 6.1.4). In this study, liver S9
from uninduced marmoset or from a single, apparently uninduced,
human subject was not able to mediate the bacterial mutagenicity of
vinylidene chloride, though a positive result was obtained using a
liver S9 from a human subject who had received long-term
phenobarbital medication. Thus, human beings appear to be more
similar to rats than to mice with respect to hepatic metabolic
activation of vinylidene chloride.
Exposure of S. typhimurium strain TA100 to 2% vinylidene
chloride (99% pure) in air in the presence of a hepatic S9 fraction
from untreated or phenobarbital-pretreated male OF-1 mice caused a
linear increase in the mutagenic response up to 4 h of exposure
[137]. This was in agreement with the dependence of bacterial
mutagenicity on duration of exposure shown by Waskell [239].
Again, phenobarbital pretreatment enhanced the metabolic
activation.
Oesch et al. [156] detected the mutagenicity of vinylidene
chloride (99.996% pure) with S. typhimurium strains TA1535,
TA1537, TA92, TA100, TA98, and Escherichia coli strain WP2 uvrA
following exposure to vinylidene chloride in the atmosphere for 4
h at 1500-90 000 mg/m3 (375-22 500 ppm) and with an NADPH-
fortified liver S9 fraction from untreated male Swiss-Webster mice
as an activation system. Again, it was established that the
inhibitor, methoxyphenol, was not mutagenic. A comparative study
was made of kidney and liver S9 fractions from different species
regarding their ability to activate vinylidene chloride to
mutagens detected by S. typhimurium strain TA100. The order of
ability to mediate mutagenesis was as follows: male and female
Swiss-Webster and C57BL/6J Han mouse liver and Chinese hamster
(Fue:FUST) liver >Sprague-Dawley rat liver >human liver
>Chinese hamster kidney >male mouse kidney (both strains) >rat
kidney and female mouse kidney. Metabolic activation was not
accomplished or was very weak in the last two tissue samples. As
with the study by Bartsch et al. [14], mutagenicity was inhibited
by a nucleophile (in this case glutathione) and it was also found
not to be affected by the addition of purified microsomal epoxide
hydrolase to the test. Despite changes in drug-metabolizing
enzymes as a result of pretreatment of rats and mice with
vinylidene chloride (section 8.2.1), S9 fractions derived from
treated animals were not found to have a greater metabolic
activation capacity in the mutagenicity assay.
The mutagenicity of vinylidene chloride towards S. typhimurium
TA100 was detected by Baden et al. [10, 11, 12] not only following
gaseous exposure for 8 h at a level of 3% but also after incubation
in suspension at 3% for up to 2 h. In agreement with the data
given above, Aroclor pretreatment of rats enhanced the ability of
liver S9 to mediate vinylidene chloride mutagenicity and human
liver S9 was also capable of catalysing the formation of mutagens.
The mutagenicity of vinylidene chloride (unspecified purity) in
liquid suspension assays (2.5 mmol vinylidene chloride/litre; 2 h
incubation) was also shown to be positive by Greim et al. [64], as
evidenced by reverse gene mutation at one locus in E. coli strain
K12. When monitored for reverse gene mutation at 2 other loci, or
forward mutation at 1 locus, a negative result was obtained, but
only one dose level was used in this study.
Results of spot tests with S. typhimurium strains TA1950,
TA1951, TA1952, TA1535, TA1538, TA100, and TA98 were reported
briefly by Cerna & Kypenova [26]. Vinylidene chloride
(unspecified purity) was added to plates at 1, 10, and 100% in DMSO
(0.05 ml). At 100%, vinylidene chloride produced reversion of
both base substitution and frame-shift mutations in the absence of
a metabolic activation system. When tested in a host-mediated
assay (female ICR mice) at doses quoted as LD50 and 50% LD50, a
significant increase in the revertants of S. typhimurium TA1950,
TA1951, and TA1952 was reported that was inversely related to dose.
A further brief report was given by McCarroll et al. [129] in
which vinylidene chloride (1.6 and 3%, "metabolically activated
doses") was shown to be markedly mutagenic to S. typhimurium TA1535
and TA100 in a microfluctuation assay involving a 72-h exposure.
Chloroacetic acid, a metabolite of vinylidene chloride, was
found not to be mutagenic to S. typhimurium strains TA100, TA1535,
TA1537, TA1538, and TA98 using a plate incorporation protocol
without the addition of an S9 system [128]. Chloroacetic acid
did not cause mutagenicity in S. typhimurium strain TA1530 in
the presence or absence of S9 from phenobarbital-pretreated mice
[136].
8.5.3. Genotoxicity in yeast
Bronzetti et al. [19] studied the mutagenicity of vinylidene
chloride (99.57% pure) in yeast. Vinylidene chloride (0-50
mmol/litre for 2 h preincubation in suspension) did not induce
reverse gene point mutation or produce mitotic gene conversion in a
diploid strain (D7) of Saccharomyces cerevisiae in the absence of a
microsomal activation system. However, in the presence of a post-
mitochondrial supernatant from mice pretreated with Aroclor 1254,
a dose-related induction of revertants and convertants was seen
between 30 and 50 mmol vinylidene chloride/litre. Genotoxicity was
also examined in yeast exposed to vinylidene chloride in an
intrasanguinous host-mediated assay. The host species i.e., male
Swiss albino CD mice, were given vinylidene chloride orally in an
acute single dose study (400 mg/kg) or in a short-term study (100
mg/kg, 5 days per week followed by 200 mg/kg on the day of the
assay). The yeast cells (4 x 108) were injected via the retro-
orbital sinus immediately before the administration of vinylidene
chloride on the day of the assay and were recovered from various
organs 4 h later. A positive result for the induction of revertants
and convertants was found for yeast cells recovered from the liver
and kidneys, but the results were negative or only very weakly
positive for both parameters in cells from the lung.
8.5.4. Genotoxicity in plants
After exposure to 5152 mg vinylidene chloride/m3 (1288 ppm)
for 6 h, inactivation of a dominant gene (forward mutation) in
Tradescantia (hybrid clone 4430) was not observed [228]. However,
at 88 mg vinylidene chloride/m3 (22 ppm) for 24 h, a positive
result was recorded. Although this finding indicates the
potential for mutagenicity in plants, the dose-response
relationship needs to be clarified.
8.5.5. Genotoxicity in mammalian cells in vitro
Costa & Ivanetich [33] investigated the effects of vinylidene
chloride (unspecified purity) on DNA repair (unscheduled DNA
synthesis) in isolated hepatocytes from male Long-Evans rats that
had been pretreated with phenobarbital. When administered to
hepatocytes at a maximum subtoxic dose level (2.1 mmol/litre),
vinylidene chloride stimulated unscheduled DNA synthesis as shown
by enhanced incorporation of deoxy-[5-3H]-cytidine into DNA.
However, the mutagenicity of vinylidene chloride was found to
be negative when tested at two loci (induction of resistance to 8-
azaguanine and ouabain) in V79 Chinese hamster cells in the
presence of a post-mitochondrial supernatant from phenobarbital
pretreated rats and mice [43]. In this test, the V79 cells were
exposed to 2 and 10% vinylidene chloride (unspecified purity) in
air for 5 h. This treatment led to dose-dependent cytotoxicity in
the presence of rat but not mouse liver post-mitochondrial
fractions. Using a similar protocol, Huberman et al. [82]
demonstrated that the vinylidene chloride metabolite, mono-
chloroacetic acid, in accordance with negative mutagenicity in
bacteria (section 8.5.2), did not show any activity in inducing 8-
azaguanine and ouabain resistant mutants in Chinese hamster V79
cells, when tested up to a level of 2.5 mmol/litre.
In a survey of the cytogenetic effects of 60 chemicals on
cultured mammalian cells, Sasaki et al. [193] reported that
vinylidene chloride (3 x 10-2 and 3 x 10-3 mol/litre) failed to
produce chromosomal breaks in Chinese hamster (Don 6) cells. A
microsomal metabolic system was not included in the assay. Other
research workers have also carried out cytogenetic studies on
mammalian cells in vitro. McCarroll et al. [129] analysed Chinese
hamster ovary cells (CHO) for sister chromatid exchange following
exposure to vinylidene chloride (unspecified purity). Consistent
and dose-related increases resulted from a 24-h exposure to
atmospheres containing 1.8, 3.6, 5.4, or 7% vinylidene chloride.
Only at 7% was the effect significant and shorter exposure
periods provided negative results. In this brief report it was not
stated whether a mammalian hepatic microsomal fraction was included
in the study. Sawada et al. [196] also investigated the ability of
vinylidene chloride (99% pure) to induce chromosomal aberrations
and sister chromatid exchange in a Chinese hamster cell line (CHL).
Cells were treated for 6 h with a range of dose levels between 0
and 2 mg/ml, at which toxicity was observed. In the presence of a
liver S9 fraction from PCB (KC-400)-pretreated male F344 rats,
vinylidene chloride gave a relatively weak, but significant,
increase in the incidence of sister chromatid exchanges. The
result was negative in the absence of S9. The findings were similar
when chromosomal aberrations were used as an end point for genetic
toxicity. In the presence (but not in the absence) of S9, a dose-
dependent induction of chromosomal aberrations was seen (14%
aberrant cells at 0.25 mg/ml and 54% at 1.5 mg/ml). These effects
were not elicited by p- methoxyphenol (the inhibitor). The role
of cytochrome P-450 in the metabolic activation of vinylidene
chloride was shown by the inhibition of the enhancement of
aberrations with metyrapone and a protective effect was seen by
the addition of glutathione. Two metabolites of vinylidene
chloride, chloroacetyl chloride and chloroacetic acid, were
negative in these tests in support of the theory that vinylidene
chloride oxide may be the active genotoxic metabolite.
8.5.6. Genotoxicity in mammalian cells in vivo
Evidence for the detectable but minimal covalent binding of
[14C]-vinylidene chloride in vivo has already been discussed [186]
(sections 8.1.1.2 and 8.5.1). As part of this study, the ability
of vinylidene chloride to induce unscheduled DNA synthesis (DNA
repair) was also investigated in mice. The measurement of DNA
repair by the uptake of [3H]-thymidine into DNA was hampered by
incomplete inhibition of replicative DNA synthesis by hydroxyurea.
Though unscheduled DNA synthesis was minimal, the slight
increase was statistically significant in mouse kidney at the 200
mg vinylidene chloride/m3 (50 ppm) exposure level.
Short et al. [203] investigated the incidence of germinal
mutations of the dominant lethal type in 11 male CD rats exposed
through inhalation of 220 mg vinylidene chloride/m3 (55 ppm) for 6
h/day and 5 days/week. During week 11 of exposure, the treated
animals were housed with 2 virgin females until mating had taken
place. Neither pre-implantation nor post-implantation losses
were observed in the pregnancies that resulted from mating with
vinylidene chloride-exposed males; thus, dominant lethal mutations
were not produced. Anderson et al. [3] also did not find any
evidence for a dominant lethal effect. Male CD-1 mice were exposed
by inhalation to 40, 120, or 200 mg vinylidene chloride/m3 (10, 30,
and 50 ppm), for 6 h/day, over 5 days. Fifteen or 16 days after
caging the males with untreated females (over a 2-month period),
the females were killed for examination of the uteri. As a result
of toxicity, only 6/20 and 18/20 male mice survived exposure to
vinylidene chloride at 200 and 120 mg/m3 (50 and 30 ppm),
respectively. No effects were seen in the frequency of pregnancy
or in the number of post-implantational early fetal deaths. There
was also no evidence of pre-implantational egg losses as indicated
by the total implants/pregnant female.
A number of in vivo cytogenetic studies reported on vinylidene
chloride have been unable to show a significant positive response.
In the inhalation study by Lee et al. [118] (details given in
sections 8.3.1 and 8.7.1), cytogenetic analysis of bone marrow
revealed no change following exposure of CD-1 mice or CD rats to
220 mg vinylidene chloride/m3 (55 ppm) , for 6 h/day, 5
days/week, for up to 12 months. Cerna & Kypenova [26] gave single
and repeated (l dose per day for 5 days) ip doses of vinylidene
chloride to female ICR mice. Neither a single dose (quoted as 1/2
LD50) nor repeated doses (quoted as 1/6 LD50) induced chromosomal
aberrations. In the long-term study by Quast et al. [178] (section
8.3.1 for details), cytogenetic evaluations on 4 rats/sex exposed
to 0, 100, or 300 mg vinylidene chloride/m3 (0, 25, or 75 ppm) for
6 months did not show any adverse effects. A negative in vivo
cytogenetic effect was also borne out by the work of Sawada et al.
[196]. In this study, 6 male ddY mice per group were given
vinylidene chloride (99% pure) by gavage, either as a single dose
(0200 mg/kg) or as 4 doses (1100 mg/kg each) given at 24-h
intervals. No increase in the frequency of micronucleated
erythrocytes was observed in the bone marrow. A micronucleus
test in the liver and blood of fetuses in vinylidene chloride-
treated mice was also negative (section 8.6). These authors
suggest that, in contrast to the in vitro studies where positive
genotoxicity has been found, the life time of the reactive
metabolites of vinylidene chloride may be too short for a
sufficient amount to have reached the target cells analysed in vivo.
However, this conclusion conflicts with the findings of Hofmann
& Peh [77] who studied chromosome aberrations in 50 metaphase bone
marrow cells of 4 male and 5 female Chinese hamsters after short-
term inhalation (6 h/day, 5 days/week for 6 weeks) of either 120 or
400 mg vinylidene chloride/m3 (30 or 100 ppm). In comparison with
control groups (fresh air), there was no effect on mortality but 78
times more aberrations were observed in the animals exposed to 120
mg vinylidene chloride/m3 (30 ppm) and 910 times more aberrations
were seen at the 400 mg/m3 (100 ppm) dose level. Using the same
technique for analysis of chromosomal aberrations, Zeller & Peh
[247] studied the effects of a single oral dose (216 mg/kg) in
Chinese hamsters. Animals that were killed 6 h after dosing showed
a higher number of aberrations (1.6%) than animals given the same
doses and killed after 24 h (1.2%) and 48 h (1.4%). The untreated
control animals showed the lowest rate of aberrations (0.6%). No
statistical test was carried out.
8.5.7. Summary
Data on mutagenicity and other short-term tests for
carcinogenicity are summarized in Table 9. Genotoxicity has been
observed in prokaryotic (in the presence of mammalian enzymes)
and eukaryotic cells in vitro. However, genotoxicity was not
observed in the majority of tests carried out on mammals in vivo .
Reports of an effect in the latter are restricted to the observation
of chromosomal aberrations in the bone marrow cells of Chinese
hamsters and the finding of a slight increase in DNA repair in the
mouse kidney.
8.6. Reproduction, Embryotoxicity, and Teratogenicity
Details of studies on dominant lethality in rats and mice are
given in section 8.5.6. These results provide no evidence for an
adverse effect on male reproduction. The fertility of male and
female Sprague-Dawley rats and neonatal toxicity were investigated
in a 3-generation, 2-litter study [151] in which test animals were
continuously given drinking-water containing 0, 50, 100, or 200 mg
vinylidene chloride/litre (99.5% minimum purity). Ten male and 20
female F0 rats were treated and 15 male and 30 female rats were
used in the control F0 groups. These animals were mated after 100
days exposure (to provide F1a litters) and again 10 days after
weaning of the first litter (to provide F1blitters). Parents for
the F2 and F3 generations were selected randomly from the F1b and
F2 litters, respectively. They were mated at 110 days of age to
produce the F2 and F3a litters, respectively. The F2 rats were
then remated 10 days after weaning of the F3a and F3b litters to
produce the F3b and F3c litters, respectively. No evidence was
found for an effect of vinylidene chloride on fertility, though
marked fluctuations in the fertility index in all groups
including the controls made interpretation of the results
difficult. Neonatal survival was lower in the F2 and F3a litters of
dams ingesting vinylidene chloride than in the respective control
groups but not lower than that of the historical data for the
laboratory. Furthermore, reduced survival in some litters was
followed by normal survival in subsequent litters from the same
adults. It was concluded that the decreased survival was due to
chance. Necropsies were carried out on rats found dead or moribund,
all weanlings not selected for future matings, and F1 and F2 adults
after the litters were weaned.
Table 9. Summary of genotoxicity data for vinylidene chloride
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
PROKARYOTES IN VITRO
Salmonella typhimurium
(reverse gene mutation)
Strains TA 100, TA 1530 + phenobarbital-induced 0.2-20% in air + ve [14,15]
mouse kidney, liver, and for 4h
lung S9 mix
Strain TA 100 + phenobarbital-induced 0.2-20% in air - ve [14,15]
mouse liver S9 (-NADPH) for 4h
Strain TA 100 + human liver S9 mix 2% in air for 4h + ve [15]
Strain TA 1530 + phenobarbital or 2% or 20% in air + ve [15]
3-methyl cholanthrene- for 4 h
induced rat liver S9
Strains TA 100, TA 1535 ± rat or hamster S9 mix 0-3333 µg/plate - ve [146]
TA 98, TA 1537 "blind (Aroclor induced)
study", preincubation
Strain TA 100 ± Aroclor-induced 3% for 2 or 8 h + ve [10, 11,
rat liver S9 mix 12]
Strains TA 1950, TA 1951, None 1, 10, and 100% + ve at [26]
TA 1952, TA 1535, TA 1538 in DMSO 100% dose
TA 100, TA 98 (spot test) (0.05 ml/plate)
Strains TA 1535, TA 1537, + mouse liver S9 mix 1500-90 000 mg/m3 + ve [156]
TA 92, TA 100, TA 98 for 4 h
Strain TA 100 + liver and kidney S9 mix 360-50 000 mg/m3 + ve [156]
from mouse, rat, Chinese for 4 h (except where
hamster. Also human S9 obtained
liver S9 mix from female
mouse kidney
and rat kidney)
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
Strain TA 1535 + mouse liver and kidney 5% for 72 h + ve [103]
S9 mix (induction of
S9 increased
response
+ uninduced rat liver 5% for 72 h - ve [103]
S9 mix or marmoset or
human S9 mix
+ induced rat liver 5% for 72 h + ve [103]
S9 mix
+ uninduced marmoset 5% for 72 h - ve [103]
liver S9 mix
+ uninduced human liver 5% for 72 h - ve [103]
S9 mix
+ phenobarbital-induced 5% for 72 h + ve [103]
human liver S9 mix
Escherichia coli
(Reverse gene mutation)
Strain WP2 uvr A + mouse liver S9 mix 1500-90 000 mg/m3 + ve [156]
for 4 h
Strain K12 2.5 mmol/litre + ve [64]
(Forward and reverse gene (for 1 locus
liquid suspension only)
mutation)
PROKARYOTES IN VIVO
Salmonella typhimurium
Strains TA 1950, TA 1951 at LD50 and half- + ve [26]
TA 1952 host mediated LD50 dose
in ICR mice
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
EUKARYOTES IN VITRO
Saccharomyces (yeast) ± Aroclor-induced rat 0-50 mmol/litre + ve [19]
(reverse gene mutation liver S9 mix for 2 h (- ve without
and gene conversion) preincubation S9)
Chinese hamster V79 cells ± Aroclor-induced rat 2, 20% for 2 h - ve [43]
(forward gene mutation) liver S9 mix
Chinese hamster V79 cells 3x10-2 or 3x10-3 - ve [193]
(chromosomal breaks) mmol/litre
Chinese hamster ovary cells 1.8-7% atmosphere + ve [129]
(sister chromatid exchange) for 24 h
Chinese hamster ovary ± Aroclor-induced rat 0-2 mg/ml + ve [196]
cells (sister chromatid liver S9 mix (weak
exchange) response)
Primary hepatocytes from 2.1 mmol/litre + ve [32]
phenobarital-treated rats (maximum subtoxic
(unscheduled DNA dose)
synthesis)
EUKARYOTES IN VIVO
Tradescantia (flower) 88 and 5152 mg/m3 + ve [228]
(forward gene mutation) (at lower
dose only)
Saccharomyces (host 400 mg/kg or + ve for [19]
mediated assay in Swiss 5x100 mg/kg daily yeast cells
mice plus 200 mg/kg on recovered
last day from liver and
kidneys, but not
from lung
________________________________________________________________________________________________
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
CD-1 mice and Sprague- rats: 40 mg/m3 + ve (but [186]
Dawley rats (DNA adduct mice: 40 0r 200 minimal binding
formation in liver and mg/m3 for 6 h detected)
kidney) mouse > rat
kidney > liver
CD-1 mice (unscheduled 200 mg/m3 + ve (but [186]
DNA synthesis in liver for 6 h minimal); effect
and kidney) observed in
kidneys only
CD-1 mice (bone marrow 220 mg/m3 - ve [118]
cytogenetics for 6 h, 5 days/week
for 12 months
CD rats (bone marrow 220 mg/m3 - ve [118]
cytogenetics for 6 h, 5 days/week
for 12 months
Rats (male and female) 0, 100, or 300 - ve [178]
(bone marrow cytogenetics) mg/m3 for 6 months
ICR mice (bone marrow One-half LD50 - ve [26]
cytogenetics one-sixth LD50
x 5 days
Male ddY mice (bone 0-200 mg/kg or - ve [196]
marrow micronucleus 0-100 mg/kg x 4
assay) by gavage
Chinese hamster 120 or 400 mg/m3 + ve [77]
(bone marrow (30 or 100 ppm)
cytogenetics) 6 h/day, 5 days/week
for 6 weeks
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
CD-1 mice (male) 40, 120, 200 mg/m3 - ve [3]
(dominant lethal assay) 6 h/day for 5 days
CD rats 220 mg/m3, 6 h/day - ve [203]
(dominant lethal assay) 5 days/week for
11 weeks
_________________________________________________________________________________________________
a +ve = positive response.
-ve = negative response.
Absolute and relative kidney weights of weanling rats were
comparable with controls. No organ weight changes related to
treatment were seen in F1 adults but elevated relative liver
weights of female rats ingesting 200 mg vinylidene chloride/litre
were seen in the F2 generation. An increase in serum glutamic
pyruvic transaminase (25% above control mean) was observed in
female F2 rats at the 200 mg/litre level.
Signs of mild hepatotoxicity (fatty liver) were seen at
treatment levels of 100 mg vinylidene chloride/litre and 200
mg/litre in F1 and F2 rats and the incidence of chronic renal
disease (though high in controls) was greater in male rats treated
with 200 mg vinylidene chloride/litre than in control animals.
Nitschke et al. [151] concluded that vinylidene chloride treatment
did not significantly affect the reproductive capacity of rats.
Sawada et al. [196] investigated the incidence of micronuclei
in fetal liver and fetal erythrocytes 24 h following exposure of
pregnant ICR mice to 0, 25, 50, or 100 mg vinylidene chloride/kg
(given by intraperitoneal injection). No significant increase in
micronuclei in these cells was observed as a result of treatment,
and the results did not show any evidence of a transplacental
passage of genotoxic metabolites or precursors.
In a study on Charles River CD rats (18-20 per dose group) and
CD-1 mice (7-24 per dose group), Short et al. [202] exposed animals
to vinylidene chloride through inhalation at 60-1796 mg/m3 (15-449
ppm), for 22-23 h/day during various periods of organogenesis.
On day 20 (rats) or day 17 (mice) of gestation, fetal abnormalities
were seen at all dose levels in rats and a high incidence of early
and complete resorptions was seen in mice exposed to vinylidene
chloride concentrations >120 mg/m3 (30 ppm). However, these
effects were associated with a decreased weight gain and increased
mortality in the dams. Since similar fetal abnormalities of the
soft tissues and skeleton occurred in a feed-restricted group of
mice, the defects may be attributed to maternal toxicity. In rats,
malformations were seen in all groups and hydrocephalus was
significantly increased in a dose-related manner from 2.5% in
controls, 7.3% at 60 mg/m3 (15 ppm), 15.1% at 228 mg/m3 (57 ppm) to
33.3% at 1200 mg/m3 (300 ppm). Retarded ossification was seen in
all treated groups. Early resorptions were significantly increased
from 2% in controls to 49% at 228 mg/m3 (57 ppm) and 64% at 1796
mg/m3 (449 ppm). Fetal weight was reduced in a dose-related
manner and the reductions were significant at 228, 1200, and 1796
mg/m3 (57, 300, and 449 ppm). Food-restricted controls showed
reduced fetal weight and retarded ossification but no significant
increase in resorption rate or in the frequency of hydrocephalus.
Aspects of maternal toxicity may be responsible for these effects.
Part of this study involved an investigation of behavioural changes
in Charles River CD rats (19 or 20 per dose group). The rats were
exposed to 224 or 1132 mg vinylidene chloride/m3 (56 or 283 ppm)
for 22-23 h/day, on days 8-20 of gestation. A dose-related weight
loss over this period indicated maternal toxicity and the body
weight of pups from the rats treated with 1132 mg/m3 (283 ppm)
were lower than control weights (as with pups from feed-restricted
rats). Two groups of 3 pups/dose level and per sex were observed
for activity in a maze for 2-4 months after birth. Also one animal
of each sex per dose level from each litter was subjected to pre-
weaning behavioural and physical maturation tests. Maze activity
was not affected by the vinylidene chloride treatment, nor were
startle response, bar holding or swimming ability. However, surface
righting ability was delayed in pups from treated rats. Tooth
production was delayed in pups from the rats treated with 1132
mg/m3 (283 ppm) (as in feed-restricted rats) but opening of the
external ear was more rapid than in control pups. In conclusion,
the only adverse effects bserved could be attributed to maternal
toxicity. Murray et al. [1979] investigated embryonic and fetal
development in rats and rabbits following inhalation or oral
ingestion (rats only) of vinylidene chloride (minimum purity
99.5%) during gestation. In the inhalation studies, Sprague-
Dawley rats (30-44 per dose group) and New Zealand White rabbits
(18-20 per dose group) were exposed to 640, 320, or 80 (rats only)
mg vinylidene chloride/m3 (160, 80, or 20 ppm), for 7 h/day, from
days 6 to 15 (rats) and days 6 to18 (rabbits) of gestation. Groups
of 20-47 pregnant rats and 16 pregnant rabbits served as controls
for each dose level. For the ingestion study, 26 pregnant rats (24
controls) received drinking-water containing vinylidene chloride at
200 mg/litre (approximately 40 mg/kg per day) from days 6 to 15 of
gestation. Cesarean section was carried out on day 21 and day 29
for rats and rabbits, respectively.
Although a teratogenic effect was not observed, some evidence
of embryotoxicity and fetotoxicity was seen in rats and rabbits.
In the rat inhalation study, there was delayed ossification and a
dose-related increased incidence of wavy ribs at 320 and 640 mg/m3
(80 and 160 ppm), concentrations that were toxic to the dams. At
80 mg/m3 (20 ppm), a concentration that was not maternally toxic,
no embryo- or fetotoxic effects were seen. In the rabbits, a dose
of 640 mg/m3 (160 ppm) produced weight loss in the dams, increased
resorptions, increased incidence of 13 pairs of ribs and delayed
ossification of the fifth sternebra. At 320 mg/m3 (80 ppm), there
was no effect on dams or fetuses.
8.7. Carcinogenicity
Details of a number of the studies described here have been
provided in section 8.4. In such cases, only an outline of the
experimental protocol is given.
8.7.1. Inhalation
In a long-term inhalation study (220 mg vinylidene chloride/m3
(55 ppm)) carried out by Lee et al. [118, 119] (section 8.3.1
for details of treatments), haemangiosarcomas in the mesenteric
lymph node or subcutaneous tissue were reported in 2 treated male
rats and hepatic haemangiosarcomas were found in 2 male and 1
female mice in the treatment group. No haemangiosarcomas were seen
in control animals. Small bronchioalveolar adenomas were also
reported in 6 male mice (17%) compared with 1 in the control group
of male mice (4%). The significance of this finding was considered
questionable since, according to other reports, such adenomas,
which occurred relatively late in the study, were common in
untreated mice. The numbers of animals used in the treatment and
control groups in this study were very low (16 or less per group
after 9 months) and the study was limited to 12 months, which is
inadequate. Furthermore, the authors referred to the spontaneous
occurrence of hepatomas in mice at a similar age in other reports.
In a follow-up study, it was not possible to repeat the finding of
an increase in tumour incidence [79]. Intermittent exposure was
limited to up to 10 months (rats) and up to 6 months (mice) at a
vinylidene chloride concentration of 220 mg/m3 (55 ppm) (details
given in section 8.3.1), and was followed by a 12-month observation
period. In contrast to the results of Lee et al. [119], no tumours
arose in the treated animals other than spontaneous tumours
expected on the basis of their incidence in control animals. In
this study also, a small number of animals were used (14-16 rats
and 12 mice per sex for 10- and 6-month treatments, respectively)
and exposures were limited to periods of less than 12 months for
both species.
The results of a number of other long-term inhalation studies
on rats suggest a lack of carcinogenicity of vinylidene chloride in
this species, but, as explained, these reports are not conclusive.
Viola & Caputo [229] investigated the incidence of tumours in 51
male and 23 female Wistar rats exposed to 800 mg vinylidene
chloride/m3 (200 ppm) for 4 h/day, 5 days/week, for 5 months. For
the following 7 months, the concentration was reduced to 400 mg/m3
(100 ppm) because of toxicity. The animals were then given a
complete autopsy at spontaneous death or after being killed when
moribund. Sprague-Dawley rats were also examined after exposure to
400 mg/m3 (100 ppm) (30 of each sex) and 300 mg/m3 (75 ppm) (16
and 21 male and female rats, respectively). Thirty control rats of
each sex were used for each strain. No grossly observable
correlation between tumour formation and vinylidene chloride
inhalation was seen but a final report following the completion of
microscopic examination of tissues and organs has not been
released. A more substantial study has been reported in several
stages [134, 178, 179, 180]. The details of the long-term
inhalation study on rats are given in section 8.3.1. In outline,
following the first month of treatment, animals of a relatively
large group (minimum of 84 rats/sex per dose group) were treated
intermittently with levels of up to 300 mg vinylidene chloride/m3
(75 ppm) for the substantial period of 18 months and were
sacrificed at 24 months. The total incidence of tumours was
similar in control and dosed animals, though the incidences of
several tumours and/or tumour types were found to be statistically
increased compared with the controls (P < 0.05, Fisher's Exact
Probability Test). These were not attributed to vinylidene
chloride exposure on the basis of comparable historical control
data.
The carcinogenicity of vinylidene chloride by inhalation
was studied by Maltoni et al. [139, 141] in rats, mice, and
hamsters. The experimental details for the long-term study are
given in section 8.3.1. The intermittent exposure levels were up
to 100 mg/m3 (up to 25 ppm) (hamsters), 600 reduced from 800 mg/m3,
(150 reduced from 200 ppm) (rats), and 100 mg/m3 (25 ppm) (mice)
and were given over a period of 52 weeks. The dose levels in rats
and mice were limited by toxicity (section 8.3.1). Animals were
then observed until spontaneous death. Tumour incidence in hamsters
was not increased by treatment with vinylidene chloride. The only
type of tumour in treated rats for which the incidence was greater
than that in the controls was in the mammary gland, but a dose-
response relationship was not observed and the authors suspected
non-specific factors (linked to inhalation) to be responsible.
As part of the same project, Maltoni et al. [140] specifically
investigated the ability of vinylidene chloride to produce brain
tumours in Sprague-Dawley rats. Groups of 30 rats of each sex were
exposed to 40, 100, 200, or 400 mg vinylidene chloride/m3 (10, 25,
50, or 100 ppm), and 60 rats per sex to 600 mg/m3 (150 ppm), for 4
h/day, 4-5 days weekly, for 52 weeks (100 control rats were used
per sex). No evidence for the induction of brain tumours
(ependymomas, gliomas, or meningiomas) was found.
A number of tumour-types were observed in mice [139, 141]
including kidney adenocarcinomas, mammary tumours, pulmonary
adenomas, and leukaemias. The incidence of both mammary and
pulmonary tumours (mainly adenomas) was statistically higher in
treated mice compared with the controls (tested by the rank test of
Krauth, Fisher's Exact Probability Test, Logrank test and probit
analysis [142]. As with rats, non-specific factors may have been
responsible. However, a dose-response relationship was not
observed in either case. Kidney adenocarcinoma (a rare tumour in
mice) was observed in 29 (28 male) out of 257 mice (300 at start)
treated with 100 mg vinylidene chloride/m3 (25 ppm) and in 2 out of
the 18 male surviving mice treated with 200 mg/m3 (50 ppm).
Kidney adenocarcinomas were not seen in the 14 surviving females at
200 mg/m3 (50 ppm), in mice treated with 40 mg/m3 (10 ppm) (0/60),
or in control mice (0/380).
Maltoni et al. [142] exposed two groups of approximately
60 male and 60 female Sprague-Dawley rats to vinylidene chloride
transplacentally, continuing the exposure by inhalation at birth
(see Table 10). Treatment of dams with vinylidene chloride at 400
mg/m3 (100 ppm) through inhalation for 4 h/day, 5 days/week,
was started when embryos were 12 days of age. The inhalation
treatment of dams and offspring was continued after birth with
exposure to 400 mg vinylidene chloride/m3 (100 ppm) for 7 h/day, 5
days/week over 8 or 97 weeks, giving a total exposure period of 15
or 104 weeks. Offspring may also have been exposed via ingestion of
milk at the suckling stage.
An increased incidence of malignant neoplasias of the
haemolymphoreticular system, generally classified as leukaemia, was
observed in both males and females of both groups exposed to
vinylidene chloride. A slight decrease in body weight was also
reported in these animals. Although a quadrupling of leukaemia
incidence appeared to occur in female rats and a doubling of these
tumours occurred in the male rats, very little other information
was reported. It is possible that a few of the tumours called
leukaemia are histiocytic sarcomas and should be classified
separately from leukaemias. Unless this is known, the
contribution of this finding to the overall data base is
compromised. The authors pointed out that a considerable increase
in total malignant tumours occurred in the rats exposed to
vinylidene chloride for 104 weeks, when the treatment was started
prenatally. In contrast, a 52-week exposure of rats beginning in
young adulthood (see above), showed no increased incidence of
leukaemia at exposures as high as 600 mg/m3 (150 ppm) for 52 weeks,
and only a borderline increase in total malignant tumours. Both age
at the start of exposure and the length of exposure appear to be
important factors influencing tumour development in Sprague-Dawley
rats exposed to vinylidene chloride.
Laib et al. [114] investigated the occurrence of putative pre-
neoplastic nodules in the liver of rats treated with vinylidene
chloride. Neonate Wistar rats were exposed, together with
mothers, to vinylidene chloride in the air at 440 ± 60 mg/m3 (110
± 15 ppm) (8 h per day, 5 days per week). Exposure to this
concentration, did not result in increased lethality or reduction
in body and liver weights. Histochemical analysis of frozen liver
sections, produced immediately after 6 weeks exposure, revealed
ATPase-free islets, postulated to indicate preneoplasia.
8.7.2. Oral
There have been three studies on the oral carcinogenicity
of vinylidene chloride in which a reasonable number of rats were
given a range of dose levels for an adequate length of time. In the
first [177, 179], vinylidene chloride was included in the
drinking-water at a level up to 200 mg/litre (200 ppm) over a 2-
year period (details given in section 8.3.2). No exposure-related
neoplasms were detected. Although the incidence of mammary gland
fibroadenomas/ adenofibromas was greater in rats exposed to 50
mg/litre than in the control animals, this increase was not dose
dependent and was within the range of the historical control data
for untreated rats.
In the second study [139, 141], vinylidene chloride was given
by gavage in olive oil (details of animals and dosing regimen are
given in section 8.3.2). The intermittent dosing was over a
period of 52 weeks and the animals were examined at spontaneous
death. No increase in tumours was observed in treated rats and,
in particular, there was no increased incidence of mammary tumours
(these being considered due to non-specific factors when found in
the corresponding inhalation study (section 8.7.1)).
In a third gavage study [154], doses of 1 and 5 mg/kg were
given daily to rats for 2 years (details in section 8.3.2). No
vinylidene chloride-related tumours were reported. In mice
(section 8.3.2), given 2 or 10 mg/kg daily for 2 years, though the
incidence of lymphomas was increased at the lower dose level (P
>0.05), this was not found at 10 mg/ kg and thus does not appear
to be related to vinylidene chloride exposure. However, the
sponsors have recently found defects in the conduct of the study
and it could not be satisfactorily evaluated.
A further study [172] was restricted to a single oral dose
level given to BDIV rats throughout the life span from the time of
weaning. The study also included oral dosing of the mothers during
pregnancy. Twenty-four female pregnant rats were given vinylidene
chloride orally (150 mg/kg) on the 17th day of gestation and
their offspring (81 males and 64 females) were then treated weekly
with 50 mg/kg (orally in olive oil) from the time of weaning. All
survivors were killed at 120 weeks or when moribund and all major
organs, as well as those that showed gross abnormalities, were
examined histologically. Treatment did not increase the incidence
of tumour-bearing animals, though liver tumours were increased in
rats of both sexes (1/81 males and 3/80 females compared with 0/49
and 0/47 in male and female controls, respectively), and
meningiomas were increased in males (6/81 compared with 1/49 in
controls). The latter was found statistically to be not
significant and, since a dose-response analysis was not possible,
the results are inconclusive. In addition, hyperplastic nodules
were found in the livers of 2/23 females given the single dose of
vinylidene chloride during pregnancy and also in 2/81 males and
6/80 females among the progeny. There was a significant difference
( P =0.04) between treated and vehicle-treated control animals, no
hyperplastic nodules being observed in the latter.
8.7.3. Other routes
Van Duuren et al. [227] studied the carcinogenicity of
vinylidene chloride in groups of 30 female Ha: 1 CR Swiss mice
following percutaneous and subcutaneous application compared with
100 untreated control animals. Following the application of
vinylidene chloride at 121 mg/kg or 40 mg/kg in acetone to shaved
dorsal skin, 3 times per week for between 440 and 594 days,
animals were given a complete autopsy (except the cranial region)
and abnormal-appearing tissues and organs were examined
histologically. Routine sections of skin, liver, stomach, and
kidney were also taken. Autopsies and additional sections from the
injection site and liver were taken following the once weekly
subcutaneous administration of 2 mg vinylidene chloride per mouse
for the life span (78 weeks). Although in the percutaneous study,
benign lung papillomas (19/30, 12/30 and 30/100) were seen at 121
mg/kg, 40 mg/kg, and 0 mg/kg, respectively, and stomach tumours
were also observed at frequencies of 2/30, 0/30 and 5/100,
respectively, the tumour incidence were not significantly elevated
( P >0.05; Chi-square analysis) in any of the treatment groups.
However, when vinylidene chloride was given as an initiating agent
(single dermal dose of 121 mg/kg) followed 14 days later with 5 µg
of the promoting agent phorbol myristate acetate three times
weekly, for 428-576 days, 8/30 mice developed papillomas compared
with 9/120 and 6/90 in control (phorbol myristate acetate-treated)
mice and 1/30 treated mice developed a squamous cell carcinoma
compared with 1/120 and 2/90 in control mice treated with phorbol
ester (2.5 and 5.0 µg, respectively). The incidence of papillomas
in vinylidene chloride-treated mice was significantly greater
than in the control groups ( P <0.005; Chi-square analysis). The
authors concluded that "initiating" activity was shown with
vinylidene chloride but that it was not a complete carcinogen.
However, the finding of papillomas in the phorbol myristate
acetate-treated control mice and the low number of animals used in
the treatment groups make the interpretation of the results
difficult.
8.7.4. Summary of carcinogenicity
A number of studies on rodents have been conducted that provide
information on the potential carcinogenic action of vinylidene
chloride (see Table 10). In these studies, vinylidene chloride
was administered by inhalation, orally by gavage and in drinking-
water, and by skin application and subcutaneous injection.
Unfortunately, most of these studies were inadequate for the
conclusive evaluation of carcinogenicity because of less than
lifetime exposure regimens, insufficient numbers of animals, and an
inadequate number of dose levels. Only some of the studies were
designed and conducted as cancer biossays.
Increased tumour incidence was not found, in most of the
studies, but there were the following exceptions. Kidney
adenocarcinomas occurred in male Swiss mice exposed via inhalation
to 100 or 200 mg vinylidene chloride/m3 (25 or 50 ppm) but not to
40 mg/m3 (10 ppm). This carcinogenic response may be related to
the ability of vinylidene chloride to cause cytotoxic effects in
the target organ (section 8.1.2 and 8.2.1). In addition to kidney
adenocarcinomas, statistically significant excesses of mammary
carcinomas were observed in female mice and pulmonary adenomas in
mice of both sexes, but in these cases there was no dose-response
relationship. In a 2-stage skin carcinogenicity assay in mice,
there was some evidence that vinylidene chloride may have acted as
an initiating agent.
In rats, an increase in mammary tumours that was not dose
related was observed when adult animals were exposed through
inhalation. In separate study groups, a slight increase in
leukaemias was observed in rats exposed through inhalation in utero
and then post-natally.
Table 10. Animal carcinogenicity studiesa
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, inhalation 100 100 0 mg/m3 (0 ppm) 4 h/day, to - -
Sprague- 30 30 40 mg/m3 (10 ppm) 4-5 days/ spontaneous - -
Dawley 30 30 100 mg/m3 (25 ppm) week, death - -
[139,141] 30 30 200 mg/m3 (50 ppm) 52 weeks - -
60 60 600 mg/m3 (150 ppm) - -
Leukemia
Rat, inhalation 158 149 0 mg/m3 (0 ppm) 4/h day, 6 months 12/156 1/148
Sprague- ( in utero - 60c 0 mg/m3 (0 ppm) 5 days per
Drawley and post - 54b 400 mg/m3 (100 ppm) week, 7 weeks,
[142] natal 62 61 400 mg/m3 (100 ppm) then 7 h/day, 10/61 4/61
exposure) 5 days per
week, 97 weeks
60 60 400 mg/m3 (100 ppm) 4 h/day, 5 6 months 8/59 2/60
days per week,
7 weeks, then
7 h/day, 5
days per week,
8 weeks
Rat, CD inhalation 36 36 0 mg/m3 (0 ppm) 6 h/day, 5 none - -
[118, 119] 36 36 220 mg/m3 (55 ppm) days per none - -
week
Rat, CD 6 h/day, 5 52 weeks
[79] days per
week for:
inhalation 4 4 0 mg/m3 (0 ppm) 1 month
8 8 0 mg/m3 (0 ppm) 3 months
8 8 0 mg/m3 (0 ppm) 6 months
16 16 0 mg/m3 (0 ppm) 10 months
4 4 220 mg/m3 (55 ppm) 1 month - -
8 8 220 mg/m3 (55 ppm) 3 months - -
8 8 220 mg/m3 (55 ppm) 6 months - -
16 16 220 mg/m3 (55 ppm) 10 months - -
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, inhalation 86 86 0 mg/m3 (0 ppm)
Sprague-Dawley
[179, 180]
[134, 178] 86 86 40 mg/m3 (10 ppm) 6 h/day, 5 6 months - -
first 5 weeks, then days per week,
100 mg/m3 (25 ppm) 18 months
86 86 160 mg/m3 (40 ppm) - -
first 5 weeks, then
300 mg/m3 (75 ppm)
Rat, Wistar inhalation 30 30 0 mg/m3 (0 ppm) 4 h/day, 5 to
[229] days per spontaneous
week, for death or
51 23 800 mg/m3 (200 ppm) 12 months moribund state - -
first 5 months, -
400 mg/m3 (100 ppm)
subsequently
Rat, inhalation 30 30 0 mg/m3 (0 ppm) 4 h/day, 5 to only gross
Sprague- 16 16 300 mg/m3 (75 ppm) days per spontaneous pathology
Dawley 30 30 400 mg/m3 (100 ppm) week, 12 death or performed
[229] months moribund
state (22-
24 months)
Kidney adeno-
carcinomas
Mouse, Swiss inhalation 190 190 0 mg/m3 (0 ppm) 4 h/day, 5 up to 121 0/120 0/155
[139, 141] 30 30 40 mg/m3 (10 ppm) days per weeks 0/24 0/26
150 150 100 mg/m3 (25 ppm) week, 52 28/119 1/138
weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, Swiss 30 30 200 mg/m3 (50 ppm) 4 h/day, 4 to 2/18 0/14
(contd.) daysd spontaneous
[139, 141] death
Mammary gland
adenomas
0 mg/m3 (0 ppm) 1/180 3/187
40 mg/m3 (10 ppm) 0/30 6/30
100 mg/m3 (25 ppm) 1/148 16/148
200 mg/m3 (50 ppm) 0/52 6/54
Lung (mainly
adenomas)
0 mg/m3 (0 ppm) 6/154 7/178
40 mg/m3 (10 ppm) 6/28 3/30
100 mg/m3 (25 ppm) 23/141 12/147
200 mg/m3 (50 ppm) 4/51 6/59
Tumours of the
mammary gland
and lung were
not dose-
dependent
Mouse, CD-1 inhalation 36 36 0 mg/m3 (0 ppm) 6 h/day, 5 none - -
[118, 119] 36 36 220 mg/m3 (55 ppm) days per none
week, 52
weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, CD-1 Incidence of
(contd.) hepatic
[118, 119] haemangio-
sarcoma and
possibly
bronchiolo-
alveolar
adenoma
increased, but
this is not
thought to
have been
induced by
vinylidene
chloride [79]
Mouse, CD-1 inhalation 6 h/day, 5 52 weeks
[79] days per
week for:
16 16 0 mg/m3 (0 ppm) 1 month
16 16 0 mg/m3 (0 ppm) 3 months
28 28 0 mg/m3 (0 ppm) 6 months
8 8 220 mg/m3 (55 ppm) 1 month - -
8 8 220 mg/m3 (55 ppm) 3 months - -
12 12 220 mg/m3 (55 ppm) 6 months - -
Chinese inhalation 18 17 0 mg/m3 (0 ppm) 4 h/day, to - -
Hamster 30 30 100 mg/m3 (25 ppm) 4 - 5 days spontaneous
[139, 141] per week, death
for 52 weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, oral 80 80 0 mg/litre (0 ppm) daily for none
Sprague- (drinking- 24 months
Dawley water)
[179, 180]
[177] 48 48 50 mg/litre (50 ppm) none - -
(M = 7 mg/kgbw)
(F = 9 mg/kgbw)
48 48 100 mg/litre (100 ppm) none - -
(M = 10 mg/kgbw)
(F = 14 mg/kgbw)
48 48 200 mg/litre (200 ppm) none - -
(M = 20 mg/kgbw)
(F = 30 mg/kgbw)
Rat, gavage 100 100 olive oil daily to
Sprague- (olive oil) 82 77 (0 mg/kgbw) 4 - 5 days/ spontaneous
Dawley 50 50 0.5 mg/kgbw week, for death - -
[139, 141] 50 50 5 mg/kgbw 52 weeks - -
50 50 10 mg/kgbw - -
50 50 20 mg/kgbw - -
Mouse, Swiss dermal 100 0 mg 3 x/week to none -
Ha: ICR; (in 0.2 ml 30 0.1 ml acetone spontaneous none -
[227] acetone) 30 40 mg/mouse death or none -
30 121 mg/mouse moribund none -
state
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, Swiss subcutaneous 100 0 mg once per week
Ha: ICR; (in 0.05 ml 649 days none
[227] trioctanoin) 30 0.05 ml water 636 days none
30 0.05 ml 631 days none -
trioctanoin
30 2 mg/mouse 548 days none -
Mouse, Swiss dermal 30 121 mg/mouse once, to none 8/30
HA: ICR; (initiation then 5 mg PMAe spontaneous papillomas
[227] test on the per animal death or 1/30 skin
skin) moribund carcinoma
state; PMA
3 x/week
--------------------------------------------------------------------------------------------------------
a Modified from: ECETOC [45].
b Only 2 treatments, because of the high toxicity.
c Pregnant females.
d Only 4 treatments, because of the high toxicity and mortality.
e PMA = phorbol myristate acetate.
9. EFFECTS ON HUMAN BEINGS
9.1. Single and Short-term Exposures
According to Gibbs & Wessling, [59], exposure to a high
concentration of vinylidene chloride, e.g., 16 000 mg/m3 (4000
ppm), rapidly causes intoxication that can lead to unconsciousness.
The anaesthetic effects from short-term exposure are short-lived.
At unspecified sub-anaesthetic doses, prolonged exposure and
repeated short-term exposures may produce kidney and liver damage
[221].
Some adverse effects associated with vinylidene chloride
exposure have been attributed to contaminants or to the stabilizer
( p- methoxyphenol). Henschler et al. [74] reported the occurrence
of persistent cranial nerve disorders in two individuals who had
attempted to clean out tanks that had contained vinylidene chloride
co-polymers. The chemical responsible for this effect appeared,
however, to be a contaminant of vinylidene chloride (either mono-
or dichloroacetylene). Chivers [29] reported the incidence of
leukoderma in two subjects following skin contamination with
p- methoxyphenol. The irritant effect of vinylidene chloride [84,
192] on the eye, upper respiratory tract (at levels as low as 100
mg/m3 i.e., 25 ppm) [192], and skin may be at least partially due
to the stabilizer p- methoxyphenol and, in the case of the study
by Rylova [192], other impurities. Dermatitis was reported in an
individual whose skin was directly exposed to Saran film
(vinylidene chloride/ vinyl chloride co-polymer in the absence of a
stabilizer) [160].
9.2. Long-Term Exposure
A quantitative risk estimate based on the best available set
of data (mouse-kidney adenocarcinoma) from the animal tumour
assays, using a non-threshold mathematical model, linear at low
doses, provides an estimate of an upper limit of human risk [225].
The true risk is not likely to be greater than this estimate and
may be lower. The upper limit for human risk thus estimated was
5.0 x 10-5 for a continuous lifetime exposure to 1 µg/m3 in air and
3.3 x 10-5 for ingestion of drinking-water containing 1 µg/litre.
However, in the light of the discussion in section 8.6.4, the
limited evidence for carcinogenicity in animal models is not
sufficient to reach a firm conclusion on the carcinogenic risk of
vinylidene chloride for human beings.
Interpretation of epidemiological studies on the effects of
vinylidene chloride in human beings has been confounded by
concomitant exposure to vinyl chloride. A mortality study on 629
workers exposed to vinylidene chloride, for 6-10 h/day, 42 h/per
week, for various lengths of time in a vinylidene chloride
production and polymerization plant in the Federal Republic of
Germany was reported by Thiess et al. [219]. Individuals were
exposed to an estimated (unmeasured) average plant concentration
of 200 mg vinylidene chloride/m3 (50 ppm) from 1955 to 1965 and
subsequently to an average level of approximately 40 mg/m3 (10
ppm) up to 1975, based on measurements of airborne contamination
after 1975. All had also been exposed to vinyl chloride (measured
as <13 mg/m3 ( <5 ppm) since 1975) and acrylonitrile (measured as
<2 mg/m3 (<1 ppm) since 1975). A 97% tracing was obtained for
follow-up analysis of the majority (447) of the cohort that was
exposed for more than 6 months. The incidence of exposure for >1
year in the remainder, was 36% and tracing for follow-up analysis
was only 24%. The mortality rate of the cohort was compared with
that of two populations of 180 000 (local) and 3 700 000 (regional)
for the period 1969-75. The expected number of deaths was
calculated by applying age-specific mortality rates to the person-
years of observation of 7 age groups within the cohort.
Statistical evaluation was based on the Poisson distribution. The
distribution of deaths according to age and decade (total 39
deaths) indicated that workers in the exposure groups did not have
an elevated mortality rate (total 57 and 36 expected from the data
of the two reference populations). The expected numbers of deaths
resulting from cancers, infectious diseases, cardiovascular
diseases, other natural causes, and external causes were compared
with the observed incidence.
Although the number of deaths from cardiovascular diseases in
general was not different from that expected, a peculiar
distribution of deaths caused by cerebral haemorrhage in the young
age groups deserves mention. The occurrence of several deaths
attributable to cerebral haemorrhage, cerebral sclerosis/apoplexia,
and acute coronary failure in age-groups below 50 years was beyond
chance ( P- values far below 0.05). But this study did not verify
the validity of diagnoses on death certificates and adequately
designed studies are needed to ascertain this part of the mortality
findings.
Five deaths (out of 39) through suicide compares with 2.5-3.0
expected. This cause of death does not rely on diagnostic validity
and indicates the need for further investigations, because suicide
may relate to mental depression. Bronchial carcinomas were seen
in 5 individuals compared with expected numbers of 3.9 and 2.2
from the data on the two control populations. It was noted that 3
of the subjects with bronchial carcinoma were heavy smokers, the
remaining 2 cases (observed at age 37) were clearly in excess of
the expected incidence in the age-group below 40 (0.08 and 0.07 for
the 2 reference populations, P = 0.003). The incidence of
oesophageal cancer was within the range of age-specific expectation
for the larger (district) control population but not for the
smaller city population. The authors concluded that the overall
malignant tumour incidence was not statistically different from
the expected rate.
More detailed information was provided in a follow-up
investigation of this exposed population (535 persons exposed for
more than 6 months) [110]. In this extended analysis with an
estimated average exposure in the years before 1965 of 200 mg/m3
(50 ppm), the observed total number of deaths (48) was
significantly greater than that expected from the reference
populations (43.2-46.5) due to a greater incidence of
cardiovascular disease (20 observed deaths versus about 15
expected). Eleven deaths from myocardial infarction were
statistically significantly in excess of the 6.8 expected. The
number of malignant tumours was 12 compared with 9.8 expected and
this was reflected in the incidence of bronchial carcinomas (6
compared with 2.68-2.96 expected). In comparison with internal
reference groups not exposed to vinylidene chloride, the cancer
deaths were statistically in excess (3 of the lung-cancer cases
were aged under 50). However, this statistically increased
incidence was not considered to be related to vinylidene chloride
exposure since, in 2 cases, exposure was limited to 2 years
duration. The interpretation of the epidemiological study is
hampered by a low cohort number, while co-exposure to other
chemicals, such as vinyl chloride, was only considered in part by
the inclusion of internal reference populations.
In a further epidemiological study [240], employees had been
exposed to different extents to a range of substances in synthetic
chemical plants. These investigators combined detailed work
histories of 4806 individuals with exposure ratings for each of 19
chemicals during each calendar year from 1942 to 1973. After
construction of a serially additive expected dose model, the
authors tested whether vinylidene chloride was responsible for the
observed excess risk of lung cancer in the cohort by using the
one-sided t-test of the observed minus expected cumulative doses
over all years and for ten or more years before death. No
relationship was found between vinylidene chloride exposure and
lung cancers.
The only epidemiological study of individuals exposed to
vinylidene chloride where vinyl chloride was not used as a
co-polymer (ethyl acrylate was the co-polymer) was carried out by
Ott et al. [166]. Employees (138) were exposed to time-weighted
average (TWA) concentrations ranging from 20 to 280 mg vinylidene
chloride/m3 (5 to 70 ppm) for a minimum of one year, within the
period 1942-65. No association was found between exposure and
mortality ascertained in 1974 among this low cohort number, when
compared with US national statistics. Two employees suffered
hepatic damage, but, in both cases, alcohol consumption was known
to prevail. The size of the cohort having a long duration of
exposure or a long latency period since initial exposure was small.
No internal comparison was made, comparable to the approach by
Klimish et al. [110]. Although the results of the epidemiological
studies do not provide convincing evidence for an increased risk
of cancer in human beings exposed to vinylidene chloride, it is not
possible to conclude that there is no carcinogenic effect. It
should be remembered that inadequate studies often tend to
underestimate rather than to overestimate an association between
exposure and cancer.
Schmitz et al. [198] assessed serum glutamic oxaloacetic
transaminase, glutamic pyruvic transaminase, and gamma-glutamyl
transpeptidase in 133 human subjects exposed to vinylidene
chloride, as a test for liver damage. Serum enzyme levels changed
less in two comparison groups than in the exposed group but,
according to Fisher's Exact Probability Test, the duration of
exposure to vinylidene chloride did not influence the serum enzyme
levels.
10. EVALUATION OF EFFECTS ON THE ENVIRONMENT AND HUMAN
HEALTH RISKS
10.1. Evaluation of Effects on the Environment
As a result of volatilization, the atmosphere is the major
environmental compartment for vinylidene chloride. The half-life of
vinylidene chloride in the troposphere is expected to be
approximately 2 days and therefore the compound is unlikely to
participate in the depletion of the stratospheric ozone layer.
Leaching and volatilization render soil and sediments minor
compartments for vinylidene chloride in the environment and the
level of this chlorinated hydrocarbon in the aqueous environment is
also minimized by rapid volatilization. It is not known whether
the degradation of compounds, such as trichloroethylene and
perchloroethylene, which are often found in water, contributes in a
significant manner to the levels of vinylidene chloride found in
the environment.
The concentrations of vinylidene chloride found in
environmental waters and the acute toxicity levels for fish and
Daphnia indicate that acute toxic risks for the aquatic environment
are minimal. Available data on long-term toxicity are insufficient
to assess sub-lethal effects on any aquatic organisms residing near
point sources of relatively high levels of vinylidene chloride
contamination, such as contaminated ground water and municipal and
industrial outfalls.
10.2. Evaluation of Human Health Risks
10.2.1. Levels of exposure
The general population is exposed to very low levels of
vinylidene chloride. The maximum level reported in drinking-water
is 20 µg/litre, though the average daily individual exposure of USA
citizens via drinking-water has been estimated to be <0.01 µg.
The levels of vinylidene chloride in food are generally not
detectable and levels above 10 µg/kg have not been reported. The
levels in food derived from aquatic organisms are not known, but
are likely to be insignificant (section 10.1). Ambient air levels
of vinylidene chloride have been reported of up to 52 µg/m3 (at the
perimeter of an industrial site). Median urban air concentrations
in the USA of 20 ng/m3 and 8.7 µg/m3 have been reported for non-
industrial and industrial-source areas, respectively.
Occupational exposure occurs particularly in production and
polymerization processes. Respiration is the major route of uptake
and the maximum recommended or regulated mean exposure limits over
the period of a working day range from 8 to 500 mg/m3 (or the
lowest reliably detectable concentration) depending on the
country. Short-term exposure limits range from 16 to 80 mg/m3 and
ceiling values range from 50 to 700 mg/m3. Airborne levels of
vinylidene chloride in the confined atmospheres to which workers in
certain occupations are exposed have been found not to exceed 8
mg/m3.
10.2.2. Acute effects
In human beings, inhalation of high concentrations of
vinylidene chloride (very approximately, at or above the maximum
olfactory threshold of 4000 mg/m3) are likely to cause depression
of the central nervous system and could lead to coma. On the basis
of acute toxicity in animals, toxic effects of vinylidene chloride
may occur in the liver, kidneys, or lungs at well below the
minimum olfactory threshold of approximately 2000 mg/m3.
Vinylidene chloride exposure can lead to irritation of the eye, the
upper respiratory tract (at 100 mg/m3 in human beings (section
9.1), and the skin, and this is thought to be partially due to the
stabilizer p- methoxyphenol.
In mice, which are more susceptible than rats to the
hepatotoxic and renal toxic effects of vinylidene chloride,
kidney damage was induced by exposure to as little as 40 mg
vinylidene chloride/m3 (10 ppm) for 6 h. Marked hepatotoxicity and
renal toxicity were also seen in rats. After fasting, which
exacerbated toxicity, exposure to vinylidene chloride at 600 mg/m3
(150 ppm) and 800 mg/m3 (200 ppm) for 6 h caused toxicity in rat
liver and kidney, respectively. Studies on rats indicate that
alcohol ingestion prior to exposure can enhance the metabolism and
exacerbate the toxicity of vinylidene chloride. Acute toxicity is
dependent on species, sex, strain, and the dietary status of
animals. Species susceptibility is correlated with the activity
of oxidative metabolism of vinylidene chloride in rats and mice.
While it is not possible to predict whether the rat or the mouse
provides the more suitable model for human beings, the activity of
hepatic microsomal metabolism by human beings is quantitatively
similar to that of the rat, a species of relatively low
susceptibility. There is no evidence of a qualitative difference
in the oxidative metabolism of vinylidene chloride in human beings
and rodents.
It is apparent that the margin between the concentrations
capable of producing toxicity in animals (40 mg/m3 in mice) and the
occupational exposure limit set by some countries may not be
sufficient or may be non-existent.
10.2.3. Long-term effects and genotoxicity
Prolonged and repeated short-term exposures at sub-anaesthetic
doses may produce kidney and liver damage. On the basis of long-
term studies on animals, under conditions that simulated
occupational exposure, hepatic changes were reported at an exposure
level of 300 mg /m3 (75 ppm) in rats. In mice, kidney and liver
damage were seen at 100 mg/m3 (25 ppm) and 200 mg/m3 (50 ppm),
respectively. There was considerable variation in the sensitivity
to toxic effects observed in the different studies.
Vinylidene chloride does not appear to affect reproductive
capacity or to pose an embryotoxic or teratogenic risk at dose
levels below those required for maternal toxicity in animals, but
this has not been studied in human beings. Embryo and fetal
toxicity and fetal abnormalities were seen at levels producing
maternal toxicity, as evidenced by reduced weight gain.
Vinylidene chloride is mutagenic for bacteria and yeast
provided that a mammalian metabolic system is present. Some
mammalian cells are also receptive to DNA damage and mutagenicity
in vitro . Genotoxicity was not evident in the majority of in vivo
studies on rodents, as measured by dominant lethality and
cytogenetics, but aberrations in bone marrow cells of Chinese
hamsters have been reported. DNA binding and repair in vivo in
rodents, though detectable, was minimal. The data on in vivo
genetic studies therefore suggest some evidence for genetic
toxicity, but, in the majority of studies, the effects were
minimal or negative.
Several carcinogencity tests have been carried out on three
species of experimental animals (mouse, rat, and hamster) using
various routes of administration. Unfortunately, most of these
studies suffered from severe limitations in design or conduct for
carcinogenicity evaluation. No significant carcinogenic effects
were observed in rats dosed orally. In adult rats exposed through
inhalation, an increase in mammary tumours, which was not dose-
related, was reported. A slight increase in leukaemia was observed,
when rats were exposed both in utero and post-natally. These
observations could not be evaluated. In one study on mice,
increased incidence of kidney adenocarcinomas were observed in
males at exposure levels of 200 and 100 mg/m3 (50 and 25 ppm) but
not at 40 and 0 mg/m3 (10 and 0 ppm). In the same study,
statistically increased incidences of lung tumours (mainly adenomas
in both sexes) and mammary carcinomas (in females) were observed,
but no dose-response relationships were found.
The kidney tumours may be related in some way to observed
kidney cytotoxicity and it is possible that repeated kidney damage
either leads directly to the carcinogenic response via a non-
genotoxic mechanism or facilitates the expression of the genotoxic
potential of metabolites in this particular species, sex, and
organ. However, this conclusion is uncertain in the absence of
adequate dose-response data on genetic effects in vivo and the
findings that vinylidene chloride may have acted as an initiator in
a two-stage skin assay in mice.
Epidemiological studies, while not providing any statistically
significant evidence for an increased cancer risk from vinylidene
chloride exposure under occupational conditions, are not adequate
to permit a proper evaluation of the carcinogenic risk of
vinylidene chloride for human beings.
Although the evaluations of individual authors dismiss the
finding of excess cancer deaths as a chance occurrence (due to
small numbers and cohort sizes), the consistency of the higher than
expected values is worth mentioning. In the two cohort studies
reported, lung cancer was observed in 7 cases, whereas 3.16 deaths
would have been expected. The result cannot be dismissed, but co-
existent exposure to vinyl chloride (in one study) has to be borne
in mind. Since the cohorts were identified according to their
exposure to vinylidene chloride, it may be impossible to exclude
additional confounding exposures.
The morbidity findings reported (including one case of
testicular carcinoma) have some informatory value. The
interpretation by the authors that higher liver morbidity was
related to the alcohol consumption of the individuals is invalid,
since the alcohol intake by all members of the study (not only
that of identified cases) was not assessed.
11. RECOMMENDATIONS
11.1. Recommendations for future work
There is a need for better estimates of the global annual
production of vinylidene chloride and of the amounts of vinylidene
chloride entering the environment from all sources, whether arising
from the release of vinylidene chloride as such or from the
degradation of other chemical products.
The predicted environmental fate is based on little
experimental evidence. Information is required on rates of
degradation and on transformation products in the air, soil,
water, and sediment, and metabolism in representative non-
mammalian species.
Long-term toxicity studies investigating a variety of
pathological endpoints should be carried out on representative
aquatic species (fish, crustacea, and molluscs).
Thresholds for, and mechanisms of, toxic effects from short-
and long-term exposure to vinylidene chloride need to be defined
more accurately in animals and human beings, as a basis for
establishing safe levels of exposure.
More exhaustive use should be made of existing data on
carcinogenicity. If further carcinogenicity studies are carried
out, they should be conducted according to an accepted lifetime
bioassay protocol specifically designed to cater for the particular
properties of vinylidene chloride. Such studies should take into
consideration the short half-life of the chemical in the body,
the importance of age at onset of exposure, the daily exposure
duration, and other relevant information that might be related to
determining the dosing regimen. Species and strains of animals for
testing need to be carefully selected. Toxicity data as well as
metabolic and pharmacokinetic data for these animals would also be
extremely useful.
Epidemiological studies are needed to enable an assessment to
be made of the effects of exposure to vinylidene chloride
(including prolonged low-level exposure) on human populations.
Thus, long-term follow-up studies on morbidity and mortality on
whole, unselected populations exposed to vinylidene chloride
should be conducted. Information on effects, such as premature
cerebrovascular disease and cancer, is particularly necessary and
studies should take into account confounding factors, such as
smoking and alcohol consumption (ideally on a case-referent basis).
To overcome the problem of small numbers in individual production
sites, multicentre studies with pooling of data may provide a
valuable approach in both ongoing and future investigations.
Historical data should be used as a reference basis for comparison
with results from ongoing investigations to enable an assessment to
be made of the protective effects of regulatory action over recent
years.
There is a need to compare the in vivo/in vitro pharmacokinetics
and metabolism of vinylidene chloride, especially in the kidney,
liver, and lungs, in experimental animals of different species
and in human beings, in order to better understand the results
obtained in in vivo toxicity studies. Parallel data are required
on the potential genotoxicity of vinylidene chloride at the target
site for carcinogenesis in different species, to examine the
possible role of a genetic mechanism.
In the light of the neurotoxicological findings reported in
this review, there is a need to investigate the role of modulator
systems in the pathogenesis of vinylidene chloride intoxication.
The value of the use of a sulfydryl agent, such as
N- acetylcysteine, in the treatment of vinylidene chloride
poisoning in human beings should be investigated in experimental
animal studies.
11.2. Personal Protection and Treatment of Poisoning
11.2.1. Personal protection
In industrial situations where short-term inhalation exposures
above the recommended limits are possible, full face masks with
filters for organic vapours should be used and, where necessary for
emergency use, masks with air-line supply systems should be
provided. Properly maintained protective clothing including
safety goggles should be worn by those handling vinylidene
chloride, to prevent contact with the body. A constant air flow
should be maintained within industrial plants with adequate
filtered vents at points where spills or leaks are likely to occur.
The monitoring of vinylidene chloride emissions during distribution
operations is recommended. In the event of a leak, the vinylidene
chloride should be evaporated either directly in the case of small
leaks, or by controlled evaporation using an expansion synthetic
foam. Water spray curtains can be used to disperse the vapour from
the foam.
11.2.2. Treatment of poisoning in human beings
In cases of over-exposure or ingestion, medical advice should
be obtained. Because of the irritant properties of vinylidene
chloride, particular attention should be given to the lungs, skin,
and eyes. The functions of the heart, liver, kidney, and central
nervous system should be monitored. Since the animal data have
indicated that vinylidene chloride produces a marked increase in
sensitivity to epinephrine-induced cardiac arrhythmias, this drug
should be avoided. Severe hypotension may be treated by
transfusion, with whole blood or plasma expanders. There is no
known antidote.
A patient poisoned through inhalation of vinylidene chloride
should be kept warm in a semi-prone position, in fresh air. The
airway should be kept clear and oxygen should be administered, if
the subject is in a stupor or coma. Artificial respiration should
be provided, if necessary.
Following ingestion of vinylidene chloride, the mouth should be
rinsed with water. Vomiting should not be induced, because of the
risk of aspiration of vinylidene chloride into the larynx and
lungs. Gastric lavage and/or the oral administration of activated
charcoal or liquid paraffin may help to reduce the bioavailability
of vinylidene chloride, if given within approximately 1 h of
ingestion, and may prove of benefit up to 4 h after ingestion.
Eyes exposed to vinylidene chloride should be immediately
irrigated with water for at least 15 minutes and medical advice
should be sought.
In the case of dermal exposure, contaminated clothing should be
removed and the affected area of skin washed with soap and water.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Vinylidene chloride was evaluated by WHO in 1984 [244] in the
Guidelines for drinking-water quality. It was concluded that:
"Dichloroethenes have been detected in drinking-
water, generally at levels less than 1 µg/litre.
The isomers have not always been differentiated.
1,1-dichloroethene is the isomer that causes most
concern because of evidence that it is carcinogenic
in experimental animals. It is a chemical commonly
used in the synthesis of various polymers; for
example, food wrappers are often made of 1,1-
dichloroethene co-polymers. 1,1-Dichloroethene
produces mammary tumours in both mice and rats, and
kidney adenocarcinomas in mice (13). It has also
been shown to be mutagenic in the Ames assay. A
linear multi-stage extrapolation model was applied
to data concerning the incidence of kidney
adenocarcinomas in Swiss mice in order to calculate
the recommended guideline value of 0.3 µg/litre."
Vinylidene chloride was evaluated by IARC Working Groups in
1978 [85], 1985 [86], and 1987 [87]. In 1987, the following
conclusions were reported:
"VINYLIDENE CHLORIDE (Group 3)
"A. Evidence for carcinogenicity to humans ( inadequate)
In one epidemiological study of 138 US workers
exposed to vinylidene chloride, no excess of
cancer was found, but follow-up was incomplete,
and nearly 40% of the workers had less than 15
years' latency since first exposures. In a study
in the Federal Republic of Germany of 629 workers
exposed to vinylidene chloride, seven deaths from
cancer (five bronchial carcinomas) were reported;
this number was not in excess of the expected
value. Two cases of bronchial carcinoma were
found in workers, both of whom were 37 years old,
whereas 0.07 were expected for persons aged 35-39
years. The limitations of these two studies do
not permit assessment of carcinogenicity of the
agent to humans. No specific association was
found between exposure to vinylidene chloride and
the excess of lung cancer noted previously in a US
synthetic chemicals plant.
"B. Evidence for carcinogenicity to animals ( limited)
Vinylidene chloride was tested for carcinogenicity
in mice and rats by oral administration and by
inhalation, in mice by subcutaneous
administration and by topical application, and
in hamsters by inhalation. Studies in mice and
rats by oral administration gave negative results.
In inhalation studies, no treatment-related
neoplasm was observed in rats or hamsters. In
mice, a treatment-related increase in the
incidence of kidney adenocarcinomas was observed
in male mice, as were increases in the incidence
of mammary carcinomas in females and of pulmonary
adenomas in male and female mice. In skin-painting
studies in female mice, vinylidene chloride
showed activity as an initiator, but, in a study
of repeated skin application, no skin tumour
occurred. No tumour at the injection site was seen
in mice given repeated subcutaneous administrations.
"C. Other relevant data
"No data were available on the genetic and related
effects of vinylidene chloride in humans.
"Vinylidene chloride did not induce dominant lethal
mutations in mice or rats and did not induce
chromosomal aberrations in bone-marrow cells of
rats treated in vivo ; however, it induced
unscheduled DNA synthesis in treated mice. It did
not induce chromosomal aberrations or mutation in
Chinese hamster cells in vitro but did induce
unscheduled DNA synthesis in rat hepatocytes.
Vinylidene chloride was mutagenic to plant cells
and induced mutation and gene conversion in yeast.
It was mutagenic to bacteria."
REFERENCES
1 ALTMAN, P.L. & DITTMER, D.S. (1966) Environmental biology,
Bethesda, Maryland, Federation of American Societies for
Experimental Biology, pp. 326-328.
2 ANDERSEN, M.E. & JENKINS, L.J., Jr (1977) Oral toxicity of
1,1-dichloroethylene in the rat: effects of sex, age and
fasting. Environ. Health Perspect. 21: 157-163.
3 ANDERSEN, M.E., JONES, R.A., & JENKINS, L.J., Jr (1977)
Enhancement of 1,1-dichloroethylene toxicity by pretreatment
of fasted male rats with 2,3-epoxy-propan-1-ol. Drug Chem.
Toxicol., 1: 63-74.
4 ANDERSEN, M.E, JONES, R.A., & JENKINS, L.J., Jr (1978) The
acute toxicity of single, oral doses of 1,1-dichloroethylene
in the fasted, male rat: effect of induction and inhibition of
microsomal enzyme activities on mortality. Toxicol. appl.
Pharmacol., 46: 227-234.
5 ANDERSEN, M.E., FRENCH, J.E, GARGAS, M.L., JONES, R.A., &
JENKINS, L.J., Jr (1979a) Saturable metabolism and the acute
toxicity of 1,1-dichloroethene. Toxicol. appl. Pharmacol., 47:
385-393.
6 ANDERSEN, M.E., GARGAS, M.L., JONES, R.A., & JENKINS, L.J., Jr
(1979b) The use of inhalation techniques to assess the kinetic
constants of 1,1-dichloroethylene metabolism. Toxicol. appl.
Pharmacol., 47: 395-409.
7 ANDERSEN, M.E., THOMAS, O.E., GARGAS, M.L., JONES, R.A., &
JENKINS, L.J., Jr (1980) The significance of multiple
detoxification pathways for reactive metabolites in the
toxicity of 1,1-dichloroethylene. Toxicol. appl. Pharmacol, 52:
422-432.
8 ANDERSON, D., HODGE, M.C.E., & PURCHASE, I.F.H. (1977)
Dominant lethal studies with the halogenated olefins vinyl
chloride and vinylidene dichloride in male CD-1 mice. Environ.
Health Perspect., 21: 71-78.
9 ATRI, F.R. (1985) [Chlorinated compounds in the environment.]
Schriftenr. Ver. Wasser-Boden-Lufthyg., 60: 309-317 (in German)
10 BADEN, J.M., BRINKENHOFF, M., WHARTON, R.S., HITT, B.A.,
SIMMON, V.F., & MAZZE, R.I. (1976) Mutagenicity of volatile
anesthetics. Anesthesiology, 45: 311-318.
11 BADEN, J.M., KELLEY, M., SIMMON, V.F., RICE, S.A., & MAZZE,
R.I. (1978) Fluroxene mutagenicity. Mutat. Res., 58: 183-191.
12 BADEN, J.M., KELLEY, M., & MAZZE, R.I. (1982) Mutagenicity of
experimental inhalational anesthetic agents: sevofluorane,
synthane, dioxychlorane and dioxyfluorane. Anesthesiology, 56:
462-463.
13 BARRIO-LAGE, G., PARSONS, F.Z., NASSAR, R.S. & LORENZO, P.A.
(1986) Sequential dehalogenation of chlorinated ethenes.
Environ. Sci. Technol.,20, 96-99.
14 BARTSCH, H., MALAVEILLE, C., MONTESANO, R., & TOMATIS, L.
(1975) Tissue-mediated mutagenicity of vinylidene chloride and
2-chlorobutadiene in Salmonella typhimurium. Nature (Lond.),
255: 641-643.
15 BARTSCH, H., MALAVEILLE, C., BARBIN, A., & PLANCHE, G. (1979)
Mutagenic and alkylating metabolites of halo-ethylenes,
chloro-butadienes and dichlorobutenes produced by rodent or
human liver tissues. Evidence for oxirane formation by P450-
linked microsomal monooxygenases. Arch. Toxicol., 41: 249-277.
16 BATTELLE (1983) Study of discharges of certain chloroethylenes
into the aquatic environment and the best technical means for
the reduction of water pollution from such discharges, Geneva,
Battelle Institute (Contract U/82/-176(537).
17 BELLAR, T.A., BUDDE, W.L., & EICHELBERGER, J.W. (1979) The
identification and measurement of volatile organic compounds
in aqueous environmental samples. In: 94th ACS Symposium
Series on Monitoring of Toxic Substances, Washington, DC,
American Chemical Society, pp. 49-62.
18 BIRKEL, T.J., ROACH, J.A.G., & SPHON, J.A. (1977)
Determination of vinylidene chloride in saran films by
electron capture gas-solid chromatography and confirmation by
mass spectrometry. J. Assoc. Off. Anal. Chem., 60: 1210-1213.
19 BRONZETTI, G., BAUER, C., CORSI, C., LEPORINI, C., NIERI, R.,
& DEL CARRATORE, R. (1981) Genetic activity of vinylidene
chloride in yeast. Mutat. Res., 89: 179-185.
20 BROWN, S.L., CHAN, F.Y., JONES, J.L., LIU, D.H., MCCALEB, K.E,
MILL, T., SAPIO, K.N., & SCHENDEL, D.E. (1975) Research
program on hazard priority ranking of priority chemicals.
Phase II: Final Report Menlo Park, California, Stanford
Research Institute (NSF-RA-E-75-190A; NTIS PB-263-161).
21 BUCCAFUSCO, R.J., ELLS, S.J., & LEBLANC, G.A. (1981) Acute
toxicity of priority pollutants to bluegill (Lepomis
macrochirus). Bull. environ. Contam. Toxicol., 26: 446-452.
22 BUCKINGHAM, J., ed. (1982) Dictionary of organic compounds,
5th ed., New York, Chapman and Hall, Vol. 2, p. 1733.
23 CARLSON, G.P. & FULLER, G.C. (1972) Interaction of modifiers
of hepatic microsomal drug metabolism and the inhalation
toxicity of 1,1-dichloroethylene. Res. Commun. chem. Pathol.
Pharmacol., 4: 553-560.
24 CARPENTER, C.P., SMYTH, H.F., Jr, & POZZANI, U.C. (1949) The
assay of acute vapor toxicity and the grading and
interpretation of results of 96 chemical compounds. J. ind.
Hyg. Toxicol., 31: 343-346.
25 CEC (1988) Draft proposal for a Council Directive on the
approximation of the laws of the Member States relating to
plastic materials and articles intended to come into contact
with foodstuffs, Brussels, Commission of the European
Communities, p. 43.
26 CERNA, M. & KYPENOVA, H. (1977) Mutagenic activity of
chloroethylenes analysed by screening system tests. Mutat.
Res., 46: 214-215.
27 CHIECO, P., MOSLEN, M.T., & REYNOLDS, E.S. (1981) Effect of
administrative vehicle on oral 1,1-dichloroethylene toxicity.
Toxicol. appl. Pharmacol., 57: 146-155.
28 CHIECO, P., MOSLEN, M.T., & REYNOLDS, E.S. (1982)
Histochemical evidence that plasma and mitochondrial membranes
are primary foci of hepatocellular injury caused by 1,1-
dichloroethylene. Lab. Invest., 46: 413-421.
29 CHIVERS, C.P. (1972) Two cases of occupational leucoderma
following contact with hydroquinone monomethyl ether. Br. J.
ind. Med., 29: 105-107.
30 COLE, R.H., FREDERICK, R.E., HEALY, R.P., & ROLAN, R.G. (1984)
Preliminary findings of the priority pollutant monitoring
project of the nationwide urban runoff program. J. Water
Pollut. Control Fed., 56: 898-908.
31 COMBA, M.E. & KAISER, K.L.E. (1983) Determination of volatile
contaminants at the ng.1-1 level in water by capillary gas
chromatography with electron capture detection. Int. J.
environ. anal. Chem., 16: 17-31.
32 COSTA, A.K. & IVANETICH, K.M. (1982) Vinylidene chloride: its
metabolism by hepatic microsomal cytochrome P-450 in vitro .
Biochem. Pharmacol., 31: 2083-2092.
33 COSTA, A.K. & IVANETICH, K.M. (1984) Chlorinated ethylenes:
their metabolism and effect on DNA repair in rat hepatocytes.
Carcinogenesis, 5: 1629-1636.
34 CUPITT, L.T. (1980) Fate of toxic and hazardous materials in
the air, Washington, DC, US Environmental Protection Agency
(EPA 600/83-80-084; PB 80-221948).
35 DALLAS, C.E., WEIR, F.W., FELDMAN, S., PUTCHA, L., & BRUCKNER,
J.V. (1983) The uptake and disposition of 1,1-dichloroethylene
in rats during inhalation exposure. Toxicol. appl. Pharmacol.,
68: 140-151.
36 DAWSON, G.W., JENNINGS, A.L., DROZDOWSKI, D., & RIDER, E.
(1975/77) The acute toxicity of 47 industrial chemicals to
fresh and saltwater fishes. J. hazard. Mater., 1: 303-318.
37 DELEON, I.R., MABERRY, M.A., OVERTON, E.B., RASCHKE, C.K.,
REMELE, P.C., STEELE, C.F., WARREN, V.L., & LASETER, J.L.
(1980) Rapid gas chromatographic method for the determination
of volatile and semivolatile organochlorine compounds in soil
and chemical waste disposal site samples. J. chromatogr. Sci.,
18: 85-88.
38 DEMERTZIS, P.G., KONTOMINAS, M.G., & GILBERT, S.G. (1987) Gas
chromatographic determination of sorption isotherms of
vinylidene chloride n vinylidene chloride copolymers. J. Food.
Sci., 52 (3): 747-750.
39 DILL, D.C., MCCARTY, W.M., ALEXANDER, H.C., & BARTLETT, E.A.
(1980) Toxicity of 1,1-dichloroethylene (vinylidene chloride)
to aquatic organisms, Midland, Michigan, Dow Chemical Company
(PB 81-111098).
40 DILLING, W.L. (1977) Interphase transfer processes. II.
Evaporation rates of chloromethanes, ethanes, ethylenes,
propanes and propylenes from dilute aqueous solution.
Comparison with theoretical predictions. Environ. Sci.
Technol., 11: 405-409.
41 DOW (1988) Migration of vinylidene chloride monomer into food
simulating solvents from various vinylidene chloride
copolymers. Report, Midland, Michigan, Dow Chemical Company,
p. 6.
42 DOWD, R.M. (1985) EPA drinking-water proposals: round one.
Environ. Sci. Technol., 19: 1156.
43 DREVON, C. & KUROKI, T. (1979) Mutagenicity of vinyl chloride,
vinylidene chloride and chloroprene in V79 Chinese hamster
cells. Mutat. Res., 67: 173-182.
44 EASLEY, D.M., KLEOPFER, R.D., & CARASEA, A.M. (1981) Gas
chromatographic-mass spectrometric determination of volatile
organic compounds in fish. J. Assoc. Off. Anal. Chem., 64:
653-656.
45 ECETOC (1985) Joint Assessment of Commodity Chemicals No.5:,
vinylidene chloride, Brussels, European Chemical Industry
Ecology and Toxicology Centre., 54 pp.
46 EISENREICH, S.J., LOONEY, B.B., & THORNTON, J.D. (1981)
Airborne organic contaminants in the great lakes ecosystem.
Environ. Sci. Technol., 15: 30-38.
47 EUROCOP-COST (1976) Analysis of organic micropollutants in
water, Luxembourg, Commission of the European Communities
(Cost Project 64b; EUCO/MDV/73/76, XII/476/76).
48 FERRARIO, J.B., LAWLER, G.C., DELEON, I.R., & LASETER, J.L.
(1985) Volatile organic pollutants in biota and sediments of
Lake Pontchartrain. Bull. environ. Contam. Toxicol. 34:
246-255.
49 FILSER, J.G. & BOLT, H.M. (1979) Pharmacokinetics of
halogenated ethylenes in rats. Arch. Toxicol., 42: 123-136.
50 FOERST, D. (1979) A sampling and analytical method for
vinylidene chloride in air. Am. Ind. Hyg. Assoc. J., 40:
888-893.
51 FOGEL, M.M., TADDEO, A.R., & FOGEL, S. (1986) Biodegradation
of chlorinated ethenes by a methane-utilizing mixed culture.
Appl. environ. Microbiol., 51: 720-724.
52 FORKERT, P.G. & REYNOLDS, E.S. (1982) 1,1-dichloroethylene-
induced pulmonary injury. Exp. lung Res., 3: 57-68.
53 FORKERT, P.G., FORKERT, L., FAROOQUI, M., & REYNOLDS, E.S.
(1985) Lung injury and repair: DNA synthesis following 1,1-
dichloroethylene. Toxicology, 36: 199-214.
54 FORKERT, P.G., HOFLEY, M., & RACZ, W.J. (1986a) Metabolic
activation of 1,1-dichloroethylene by mouse lung and liver
microsomes. Can. J. Physiol. Pharmacol., 65: 1496-1499.
55 FORKERT, P.G., STRINGER, V., & RACZ, W.J. (1986b) Effects of
administration of metabolic inducers and inhibitors on
pulmonary toxicity and covalent binding by 1,1-
dichloroethylene in CD-1 mice. Exp. mol. Pathol., 45: 44-58.
56 FORKERT, P.G., STRINGER, S., & TROUGHTON, K.M. (1986c)
Pulmonary toxicity of 1,1-dichloroethylene correlation of
early changes with covalent binding. Can. J. Physiol.
Pharmacol., 64: 112-121.
57 GAGE, J.C. (1970) The subacute inhalation toxicity of 109
industrial chemicals. Br. J. ind. Med., 27: 1-18.
58 GAY, B.W., HANST, P.L., BUFALINI, J.J., & NOONAN, R.C. (1976)
Atmospheric oxidation of chlorinated ethylenes. Environ. Sci.
Technol., 10: 58-67.
59 GIBBS, D.S. & WESSLING, R.A. (1983) Vinylidene chloride and
polyvinylidene chloride. In: Mark, H.F., Othmer, D.F.,
Overberger, C.G., & Seaborg, G.T., ed. Kirk-Othmer
encyclopedia of chemical technology, 3rd ed., New York, John
Wiley and Sons, Vol. 23, pp. 764- 798.
60 GILBERT, J., SHEPHERD, M.J., STARTIN, J.R., & MCWEENY, D.J.
(1980) Gas chromatographic determination of vinylidene
chloride monomer in packaging films and in foods. J.
Chromatogr., 197: 71-78.
61 GLISSON, B.T., CRAFT, B.F., NELSON, J.H., & MEUZELAAR, H.L.C.
(1986) Production of vinylidene chloride from the thermal
decomposition of methyl chloroform. Am. Ind. Hyg. Assoc. J.,
47: 427-435.
62 GOING, J.E. & SPIGARELLI, J. (1977) Environmental monitoring
near industrial sites - vinylidene chloride, Washington, DC,
US Environmental Protection Agency (EPA 560/6-77-026; NTIS PB-
273358).
63 GRASSELLI, J.R. & RITCHEY, W.M., ed. (1975) CRC Atlas of
spectral data and physical constraints for organic compounds,
Cleveland, Ohio, CRC Press, Vol. 3.
64 GREIM, H., BONSE, G., RADWAN, Z., REICHERT, D., & HENSCHLER,
D. (1975) Mutagenicity in vitro and potential carcinogenicity
of chlorinated ethylenes as a function of metabolic oxirane
formation. Biochem. Pharmacol., 24: 2013-2017.
65 GRIMSRUD, E.P. & RASMUSSEN, R.A. (1975) Survey and analysis of
halocarbons in the atmosphere by gas chromatography-mass
spectroscopy. Atmos. Environ., 9: 1014-1017.
66 GRONSBERG, E.S. (1975) [Determination of vinylidene chloride
in the air.] Gig. i Sanit., 7: 77-79 (in Russian).
67 GUILLEMIN, C.L., MARTINEZ, R., & THIAULT, S. (1979) Steam-
modified gas-solid chromatography: a complementary technique
for organic pollutant survey. J. chromatogr. Sci., 17: 677-681.
68 HARKOV, R., KEBBEKUS, B., BOZZELLI, J.W., & LIOY, P.J. (1983)
Measurement of selected volatile organic compounds at three
locations in New Jersey during the summer season. J. Air
Pollut Control Assoc., 33: (12); 1177-1183.
69 HARKOV, R., KEBBEKUS, B., BOZZELLI, J.W., LIOY, P.J., &
DAISEY, J. (1984) Comparison of selected volatile organic
compounds during the summer and winter at urban sites in New
Jersey. Sci. total Environ., 38: 259-274.
70 HARMS, M.S., PETERSON, R.E., FUJIMOTO, J.M., & ERWIN, C.P.
(1976) Increased "bile duct-pancreatic fluid" flow in
chlorinated hydrocarbon-treated rats. Toxicol. appl.
Pharmacol., 35: 41-49.
71 HAWKINS, W.E., OVERSTREET, R.M., WALKER, W.W., & MANNING, C.S.
(1985) Tumour induction in several small fish species by
classical carcinogens and related compounds. In: Proceedings
of the Fifth Conference on Water Chlorination (Chemical,
Environmental Impact and Health Effects), pp. 429-438.
72 HEITMULLER, P.T., HOLLISTER, T.A., & PARRISH, P.R. (1981)
Acute toxicity of 54 industrial chemicals to sheepshead
minnows (Cyprinodon variegatus). Bull. environ. Contam.
Toxicol., 27: 596-604.
73 HENSCHLER, D. (1977) Metabolism and mutagenicity of
halogenated olefins. A comparison of structure and activity.
Environ. Health Perspect., 21: 61-64.
74 HENSCHLER, D., BROSER, F., & HOPF, H.C. (1970) ["Polyneuritis
cranialis" caused by poisoning with chlorinated acetylenes in
working with vinylidene chloride copolymers.] Arch. Toxicol.,
26: 62-75 (in German).
75 HEWITT, W.R. & PLAA, G.L. (1983) Dose-dependent modification
of 1,1-dichloroethylene toxicity by acetone. Toxicol. Lett.,
16: 145-152.
76 HIATT, M.H. (1983) Determination of volatile organic compounds
in fish samples by vacuum distillation and fused silica
capillary gas chromatography/mass spectrometry. Anal. Chem.,
55: 506-516.
77 HOFMANN, H.T. & PEH, J. (1976) [Report on the test of
vinylidene chloride for mutagenic effects in Chinese Hamsters
after subacute inhalation,] Ludwigshafen, BASF
Aktiengesellschaft, 22 pp. (in German).
78 HOLLIFIELD, H.C. & MCNEAL, T. (1978) Gas-solid chromatographic
determination of vinylidene chloride in saran film and three
simulating solvents. J. Assoc. Off. Anal. Chem., 61: 537-544.
79 HONG, C.B., WINSTON, J.M., THORNBURG, L.P., LEE, C.C., &
WOODS, J.S. (1981) Follow-up study on the carcinogenicity of
vinyl chloride and vinylidene chloride in rats and mice: tumor
incidence and mortality subsequent to exposure. J. Toxicol.
environ. Health, 7: 909-924.
80 HSE (1983) Methods for the determination of hazardous
substances 28: Chlorinated hydrocarbon solvent vapours in air,
London, Health and Safety Executive.
81 HSE (1985) Toxicity Review 13: Vinylidene chloride, London,
Health and Safety Executive, 59 pp.
82 HUBERMAN, E., BARTSCH, H., & SACHS, L. (1975) Mutation
induction in Chinese hamster V79 cells by two vinyl chloride
metabolites, chloroethylene oxide and 2-chloroacetaldehyde.
Int. J. Cancer, 16: 639-644.
83 HULL, L.A., HISATSUNE, I.C., & HEICKLEN, J. (1973) The
reaction of O3 with CCl2CH2. Can. J. Chem., 51:
1504-1510.
84 HUSHON, J. & KORNREICH, M. (1978) Air pollution assessment of
vinylidene chloride, Washington, DC, US Environmental
Protection Agency (EPA 450/3-78-015) (Prepared by the Metrek
Division of the Mitre Corporation, McLean, Virginia; Contract
No. 68-02-1495).
85 IARC (1979) Some monomers, plastics and synthetic elastomers,
acrolein, Lyons, International Agency for Research on Cancer,
pp. 439-459 (IARC Monograph on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol.19).
86 IARC (1986) Some chemicals used in plastics and elastomers:
vinylidene chloride, Lyons, International Agency for Research
on Cancer, pp. 195-226 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Vol. 39).
87 IARC (1987) Overall evaluations of carcinogenicity: An
updating of IARC Monographs Vols. 1 - 42, Lyons, International
Agency for Research on Cancer, pp. 376-377 (IARC Monographs on
the Evaluation of Carcinogenic Risks to Humans, Supplement 7).
88 IRPTC (1988) IRPTC Legal file, Geneva, International Register
of Potentially Toxic Chemicals, United Nations Environment
Programme.
89 ISHIDATE, M., Jr, ed. (1983) The data book of chromosomal
tests in vitro on 587 chemical substances using a Chinese
hamster fibroblast cell line (Chl cells), Tokyo, The Realize
Inc., p. 582.
90 JACKSON, N.M. & CONOLLY, R.B. (1985) Acute nephrotoxicity of
1,1-dichloroethylene in the rat after inhalation exposure.
Toxicol. Lett., 29: 191-199.
91 JAEGER, R.J. (1975) Vinyl chloride monomer: comments on its
hepatotoxicity and interaction with 1,1-dichloroethylene. Ann.
N.Y. Acad. Sci., 246: 150-151.
92 JAEGER, R.J. & MURPHY, S.D. (1973) Alterations of barbiturate
action following 1,1-dichloroethylene, corticosterone or
acrolein. Arch. int. Pharmacodyn., 205: 281-292.
93 JAEGER, R.J., CONOLLY, R.B., & MURPHY, S.D. (1973a) Diurnal
variation of hepatic glutathione concentration and its
correlation with 1,1-dichloroethylene inhalation toxicity in
rats. Res. Commun. chem. Pathol. Pharmacol., 6: 465-471.
94 JAEGER, R.J., TRABULUS, M.J., & MURPHY, S.D. (1973b)
Biochemical effects of 1,1-dichloroethylene in rats:
dissociation of its hepatotoxicity from a lipoperoxidative
mechanism. Toxicol. appl. Pharmacol., 24: 457-467.
95 JAEGER, R.J., TRABULUS, M.J., & MURPHY, S.D. (1973c) The
interaction of adrenalectomy, partial adrenal replacement
therapy, and starvation with hepatotoxicity and lethality of
1,1- dichloroethylene intoxication. Toxicol. appl. Pharmacol.,
25: 491 (Abstract No. 133).
96 JAEGER, R.J., CONOLLY, R.B., & MURPHY, S.D. (1974) Effect of
18-hr fast and glutathione depletion on 1,1-dichloroethylene-
induced hepatotoxicity and lethality in rats. Exp. mol.
Pathol., 20: 187-198.
97 JAEGER, R.J., SHONER, L.G., & COFFMAN, L. (1977a)
1,1-Dichloroethylene hepatotoxicity: proposed mechanism of
action and distribution and binding of 14C radioactivity
following inhalation exposure in rats. Environ. Health
Perspect., 21: 113-119.
98 JAEGER, R.J., SZABO, S., & COFFMAN, L.J. (1977b) 1,1-
Dichloroethylene hepatotoxicity. Effect of altered thyroid
funtion and evidence for the subcellular site of injury. J.
Toxicol. environ. Health, 3: 545-555.
99 JENKINS, L.J., Jr & ANDERSEN, M.E. (1978) 1,1-Dichloroethylene
nephrotoxicity in the rat. Toxicol. appl. Pharmacol., 46:
131-141.
100 JENKINS, L.J., Jr, TRABULUS, M.J., & MURPHY, S.D. (1972)
Biochemical effects of 1,1-dichloroethylene in rats:
comparison with carbon tetrachloride and 1,2-dichloroethylene.
Toxicol. appl. Pharmacol., 23: 501-510.
101 JONES, B.K. & HATHWAY, D.E. (1978a) Tissue-mediated
mutagenicity of vinylidene chloride in Salmonella typhimurium
TA1535. Cancer Lett., 5: 1-6.
102 JONES, B.K. & HATHWAY, D.E. (1978b) The biological fate of
vinylidene chloride in rats. Chem.-biol. Interact., 20: 27-41.
103 JONES, B.K. & HATHWAY, D.E. (1978c) Differences in metabolism
of vinylidene chloride between mice and rats. Br. J. Cancer,
37: 411- 417.
104 KAISER, K.L.E., COMBA, M.E., & HUNEAULT, H. (1983) Volatile
halocarbon contaminants in the Niagara River and in Lake
Ontario. J. Great Lakes Res., 9(2): 212-223.
105 KANZ, M.F. & REYNOLDS, E.S. (1986) Early effects of 1,1-
dichloroethylene on canalicular and plasma membranes:
ultrastructure and stereology. Exp. mol. Pathol., 44: 93-110.
106 KANZ, M.F., WHITEHEAD, R.F., FERGUSON, A.E., & MOSLEN, M.T.
(1988) Potentiation of 1,1-dichloroethylene hepatotoxicity:
comparative effects of hyperthyreodism and fasting. Toxicol.
appl. Pharmacol., 95: 93-103.
107 KIEZEL, L., LISZKA, M., & RUTKOWSKI, M. (1975) [Gas
chromatographic determination of trace impurities in
distillates of vinyl chloride monomer.] Chem. Anal. (Warsaw),
20: 555-562 (in Polish).
108 KLIMISCH, J.H. & FREISBERG, K.O. (1979a) [Report on the
determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in Chinese striped hamsters (fasting)
during a 4-hour exposure period.], Ludwigshafen, BASF
Aktiengesellschaft, 11 pp (in German).
109 KLIMISCH, J.H. & FREISBERG, K.O. (1979b) [Report on the
determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in Chinese striped hamsters (fed) during a
4-hour exposure period.], Ludwigshafen, BASF
Aktiengesellschaft, 14 pp (in German).
110 KLIMISCH, J. H., LINK, R., STOCKER, W.G., & THIESS, A.M.
(1982) Investigation of the mortality of workers predominantly
exposed to vinylidene chloride. In: Proceedings of the
Medichem Congress, Paris, 1982.
111 KRAMER, C.G. & MUTCHLER, J.E. (1972) The correlation of
clinical and environmental measurements for workers exposed to
vinyl chloride. Am. Ind. Hyg. Assoc. J., 33: 19-30.
112 KRIJGSHELD, K.R. & GRAM, T.E. (1984) Selective induction of
renal microsomal cytochrome P-450-linked monooxygenases by
1,1-dichloroethylene in mice. Biochem. Pharmacol., 33:
1951-1956.
113 KRIJGSHELD, K.R., LOWE, M.C, MIMNAUGH, E.G., TRUSH, M.A,
GINSBURG, E., & GRAM, T.E. (1983) Lung-selective impairment of
cytochrome P-450-dependent monooxygenases and cellular injury
by 1,1-dichloroethylene in mice. Biochem. biophys. Res.
Commun., 110: 675-681.
114 LAIB, R.J., KELIN, K.P., KAUFMANN, I., & BOLT, H.M. (1981) [On
the problem of carcinogenicity of vinylidene chloride
(1,1-dichloroethylene).] In: [ Epidemiological approaches in
occupational medicine], Stuttgart, Gentner Verlag, pp. 277-281
(in German).
115 LAO, R.C., THOMAS, R.S., BASTIEN, P., HALMAN, R.A., &
LOCKWOOD, J.A. (1982) Analysis of organic priority and non-
priority pollutants in environmental samples by GC/MS/computer
systems. Pergamon Ser. environ. Sci., 7: 107-118.
116 LAZAREV, N.V., ed. (1960) [Vinylidene chloride.] In: [ Harmful
substances in industry,] Leningrad, Chemia, pp. 215-216 (in
Russian).
117 LEBLANC, G.A. (1980) Acute toxicity of priority pollutants to
water flea ( Daphnia magna). Bull. environ. Contam. Toxicol.,
24: 684-691.
118 LEE, C.C., BHANDARI, J.C., WINSTON, J.M., HOUSE, W.B., PETERS,
P.J., DIXON, R.L., & WOODS, J.S. (1977) Inhalation toxicity of
vinyl chloride and vinylidene chloride. Environ. Health
Perspect., 21: 25-32.
119. LEE, C.C., BHANDARI, J.C., WINSTON, J.M., HOUSE, W.B., DIXON,
R.L., & WOODS, J.S. (1978) Carcinogenicity of vinyl chloride
and vinylidene chloride. J. Toxicol. environ. Health, 4: 15-30.
120 LEIBMAN, K.C. & ORTIZ, E. (1977) Metabolism of halogenated
ethylenes. Environ. Health Perspect., 21: 91-97.
121 LESAGE, S., PRIDDLE, M.W., & JACKSON, R.E. (1988) Organic
contaminants in ground water at the Gloucester landfill.
Report, National Water Research Institute, Burlington,
Ontario, p.13.
122 LIEBLER, D.C. & GUENGERICH, F.P. (1983) Olefin oxidation by
cytochrome P-450: evidence for group migration in catalytic
intermediates formed with vinylidene chloride and trans-1-
phenyl-1-butene. Biochemistry, 22: 5482-5489.
123 LIEBLER, D.C., MEREDITH, M.J., & GUENGERICH, F.P. (1985)
Formation of glutathione conjugates by reactive metabolites of
vinylidene chloride in microsomes and isolated hepatocytes.
Cancer Res., 45: 186-193.
124 LIEBLER, D.C., LATWESEN, D.G. & REEDER, T.C. (1988). S-(2-
chloroacetyl) glutathione, a reactive glutathione thiol ester
and a putative metabolite of 1,1-dichloroethylene.
Biochemistry, 27: 3652-3657.
125 LIN, S.-N., FU, F.W.-Y., BRUCKNER, J.V., & FELDMAN, S. (1982)
Quantitation of 1,1- and 1,2-dichloroethylene in body tissues
by purge-and-trap gas chromatography. J. Chromatogr., 244:
311-320.
126 LONG, R.M. & MOORE, L. (1987) Cytosolic calcium after carbon
tetrachloride, 1,1-dichloroethylene, and phenylephrine
exposure. Studies in rat hepatocytes with phosphorylase a
and quin 2. Biochem. Pharmacol., 36: 1215-122
127 MABEY, W.R., SMITH, J.H., PUDOLL, R.T., JOHNSON, H.L., MILL,
T., CHOU, T.W., GATES, J., PARTRIDGE, I.W., & VANDENBURG, D.
(1981) Aquatic fate process. Data for organic priority
pollutants, Washington, DC, US Environmental Protection
Agency (EPA 440/4-81-014).
128 MCCANN, J., SIMMON, V., STREITWIESER, D., & AMES, B.N. (1975)
Mutagenicity of chloroacetaldehyde, a possible product of 1,2-
dichloroethane (ethylene dichloride), chloroethanol (ethylene
chlorohydrin), vinyl chloride and cyclo-phosphamide. Proc.
Natl Acad. Sci. (USA), 72: 3190-3193.
129 MCCARROLL, N.E., CORTINA, T.A., ZITO, M.J., & FARROW, M.G.
(1983) Evaluation of methylene chloride and vinylidene
chloride in mutational assays. Environ. Mutagen., 5: 426-427.
130 MCDONALD, T.J., KENNICUTT, M.C., & BROOKS, J.M. (1988)
Volatile organic compounds at a coastal Gulf of Mexico site.
Chemosphere, 17, 123-136.
131 MCKENNA, M.J., WATANABE, P.G., & GEHRING, P.J. (1977)
Pharmacokinetics of vinylidene chloride in the rat. Environ.
Health Perspect., 21: 99-105.
132 MCKENNA, M.J., ZEMPEL, J.A, MADRID, E.O., & GEHRING, P.J.
(1978a) The pharmacokinetics of [14C]vinylidene chloride in
rats following inhalation exposure. Toxicol. appl. Pharmacol.,
45: 599-610.
133 MCKENNA, M.J., ZEMPEL, J.A, MADRID, E.O., BRAUN, W.H., &
GEHRING, P.J. (1978b) Metabolism and pharmacokinetic profile
of vinylidene chloride in rats following oral administration.
Toxicol. appl. Pharmacol., 45: 821-835.
134 MCKENNA, M.J., QUAST, J.F., YAKEL, H.O., BALMER, M.F., &
RAMPY, L.W. (1982) Vinylindene chloride: a chronic inhalation
toxicity and carcinogenicity study in rats. Final Report,
Midland, Michigan, Dow Chemical, 100 pp.
135 MAFF (1980) Survey of vinylidene chloride levels in food
contact materials and in foods. Third Report of the Steering
Group on Food Surveillance: Working Party on Vinylidene
Chloride, London, Ministry of Agriculture, Fisheries and Food,
23 pp (Food Surveillance Paper No. 3).
136 MALAVEILLE, C., BARTSCH, H., BARBIN, A., CAMUS, A.M.,
MONTESANO, R., CROISY, A., & JACQUIGNON, P. (1975)
Mutagenicity of vinyl chloride, chloroethylene-oxide,
chloroacetylaldehyde and chloroethanol. Biochem. biophys. Res.
Commun., 63: 363-370.
137 MALAVEILLE, C., PLANCHE, G., & BARTSCH, H. (1977) Factors for
efficiency of the Salmonella microsome, mutagenicity assay.
Chem.-biol. Interact., 17: 129-136.
138 MALTONI, C. & PATELLA, V. (1983) Comparative acute toxicity of
vinylidene chloride. The role of species, strain and sex. Acta
oncol., 4: 239-256.
139 MALTONI, C., COTTI, G., MORISI, L., & CHIECO, P. (1977)
Carcinogenicity biosassays of vinylidene chloride. Research
plan and early results. Med. Lav., 68: 241-262.
140 MALTONI, C., CILIBERTI, A., & CARRETTI, D. (1982) Experimental
contributions in identifying brain potential carcinogens in
the petro-chemical industry. Ann. N.Y. Acad. Sci., 381:
216-249.
141 MALTONI, C., COTTI, G., & CHIECO, P. (1984) Chronic toxicity
and carcinogenicity bioassays of vinylidene chloride. Acta
oncol., 5: 91-146.
142 MALTONI, C., LEFEMINE, G., COTTI, G., CHIECO, P., & PATELLA,
V. (1985) Experimental research on vinylidene chloride
carcinogenesis. In: Maltoni, C. & Mehlinan, M.A., ed. Archives
of research in industrial carcinogenesis, Princeton, New
Jersey, Princeton Scientific Publishers, Vol. 3, p.95.
143 MASUDA, Y. & NAKAYAMA, N. (1983) Protective action of
diethyldithiocarbamate and carbon disulfide against acute
toxicities induced by 1,1-dichloroethylene in mice. Toxicol.
appl. Pharmacol., 71: 42-53.
144 MOTEGI, S., UEDA, K., TANAKA, H., & OHTA, M. (1976)
Determination of residual vinylidene chloride monomer in
polyvinylidene chloride films used for fish jelly products.
Bull. Jpn. Soc. Sci. Fish., 42: 1387-1394.
145 MOORE, L. (1980) Inhibition of liver microsome calcium pump by
in vivo administration of CCl4, CHCl3 and 1,1-dichloroethylene
(vinylidene chloride). Biochem. Pharmacol., 29: 2505-2511.
146 MORTELMANS, K., HAWORTH, S., LAWLOR, T., SPECK, W., TAINER,
B., & ZEIGER, E. (1986) Salmonella mutagenicity tests III.
Results from the testing of 270 chemicals, Environ. Mutat.,
8, (Suppl. 7): 1-119.
147 MOSLEN, M.T. & REYNOLDS, E.S. (1985) Rapid, substrate-specific
and dose-dependent deactivation of liver cytosolic glutathione
S- transferases in vivo by 1,1-dichloro-ethylene. Res. Commun.
chem. Pathol. Pharmacol., 47: 59-72.
148 MOSLEN, M.T., POISSON, L.R., & REYNOLDS, E.S. (1985)
Cholestasis and increased biliary excretion of inulin in rats
given 1,1-dichloroethylene. Toxicology, 34: 201-209.
149 MURRAY, F.J., NITSCHKE, K.D., RAMPY, L.W., & SCHWETZ, B.A.
(1979) Embryotoxicity and fetotoxicity of inhaled or ingested
vinylidene chloride in rats and rabbits. Toxicol. appl.
Pharmacol., 49: 189- 202.
150 NEUFELD, M.L., SITTENFIELD, M., WOLK, K.F., & BOYD, R.E.
(1977) Market input/output studies. Task 1: vinylidene
chloride, Washington, DC, US Environmental Protection Agency
(EPA 560/6-77-003; NTIS PB-273-205) (Prepared by Auerbach
Associates; Contract No. 68- 01-1996).
151 NITSCHKE, K.D, SMITH, F.A., QUAST, J.F., NORRIS, J.M., &
SCHWETZ, B.A. (1983) A three-generation rat reproductive
toxicity study of vinylidene chloride in the drinking water.
Fundam. appl. Toxicol., 3: 75-79.
152 NORRIS, J.M. (1977) Toxicological and pharmacokinetic studies
on inhaled and ingested vinylidene chloride in laboratory
animals. In: Proceedings of the Technical Association of the
Pulp and Paper Industry (TAPPI) Paper Synthetics Conference,
Chicago, Illinois, 1977, Atlanta, Georgia, Technical
Association of the Pulp and Paper Industry, pp. 45-50.
153 NORRIS, J.M. & REITZ, R.H. (1984) Interpretative review of the
animal toxicological, pharmacokinetic/metabolism, biomolecular
and in vitro mutagenicity studies on vinylidene chloride and
the significance of the findings for man, Midland, Michigan,
Dow Chemical Co., p. 24.
154 NTP (1982) Carcinogenesis bioassay of vinylidene chloride (CAS
No. 75-35-4) in F344 rats and B6C3F1 mice (gavage study),
Research Triangle Park, North Carolina, National Toxicology
Program (Technical Report Series No. 228; PB 82-258393).
155 OBLAS, D.W., DUGGER, D.L., & LIEBERMAN, S.I. (1980) The
determination of organic species in the telephone central
office ambient. IEEE Trans. Compnents Hybrids Manuf. Technol.,
CHMT-3 (1): 17-20.
156 OESCH, F., PROTIC-SABLJIC, M., FRIEDBERG, T., KLIMISCH, H.J.,
& GLATT, H.R. (1983) Vinylidene chloride: changes in drug
metabolising enzyme, mutagenicity and relation to its targets
for carcinogenesis. Carcinogenesis, 4: 1031-1038.
157 OKINE, L.K.N. & GRAM, T.E. (1986a) Tissue distribution and
covalent binding of [14C]1,1-dichloroethylene in mice. In vivo
and in vitro studies. Adv. exp. Med. Biol., 197: 903-910.
158 OKINE, L.K.N. & GRAM, T.E. (1986b) In vitro studies on the
metabolism and covalent binding of [14C]1,1-dichloroethylene
by mouse liver, kidney and lung. Biochem. Pharmacol., 35:
2789-2795.
159 OKINE, L.K.N., GOOCHEE, J.M., & GRAM, T.E. (1985) Studies on
the distribution and covalent binding of 1,1-dichloroethylene
in the mouse. Effects of various pretreatments on covalent
binding in vivo. Biochem. Pharmacol., 34: 4051-4057.
160 OSBOURNE, R.A. (1964) Contact dermatitis caused by saran wrap.
J. Am. Med. Assoc., 188: 1159.
161 OTSON, R. (1987) Purgeable organics in Great Lakes raw and
treated water. Int. J. Environ. anal. Chem., 31: 41-53.
162 OTSON, R. & WILLIAMS, D.T. (1982) Headspace chromatographic
determination of water pollutants. Anal. Chem., 54: 942-946.
163 OTSON, R., WILLIAMS, D.T., & BIGGS, D.C. (1982a) Relationships
between raw water quality, treatment and occurrence of
organics in Canadian potable water. Bull. environ. Contam.
Toxicol., 28: 396-403.
164 OTSON, R., WILLIAMS, D.T., & BOTHWELL, P.D. (1982b) Volatile
organic compounds in water at thirty Canadian potable water
treatment facilities. J. Assoc. Off. Anal. Chem., 65:
1370-1374.
165 OTT, M.G., LANGNER, R.R., & HOLDER, B.B. (1975) Vinyl chloride
exposure in a controlled industrial environment. A long-term
mortality experience in 594 employees. Arch. environ. Health,
30: 333-339.
166 OTT, M.G., FISHBECK, W.A, TOWNSEND, J.C., & SCHNEIDER, E.J.
(1976) A health study of employees exposed to vinylidene
chloride. J.. occup. Med., 18: 735-738.
167 PARSONS, F., WOOD, P.R., & DEMARCO, J. (1984) Transformations
of tetrochloroethene and trichloroethene in microcosms and
groundwater. J. Am. Water Works Assoc., February: 56-59.
168 PATTERSON, J.W. & KODUKALA, P.S. (1981) Biodegradation of
hazardous organic pollutants. CEP, April: 48-55.
169 PEARSON, C.R. & MCCONNELL, G. (1975) Chlorinated C1 and C2
hydrocarbons in the marine environment. Proc. R. Soc. Lond.
Ser. B, 189: 305-332.
170 PFAB, W., VON & MUCKE, G. (1977) [On the migration of selected
monomers in foodstuffs and simulations.] Dtsch. Lebensm.
Rundschau, 73: 1-5 (in German).
171 PIET, G.J., SLINGERLAND, P., DE GRUNT, F.E., VAN DEN HEUVEL,
M.P.M., & ZOETEMAN, B.C.J. (1978) Determination of very
volatile halogenated organic compounds in water by means of
direct head-space analysis. Anal. Lett., A11(5): 437-448.
172 PONOMARKOV, V. & TOMATIS, L. (1980) Long-term testing of
vinylidene chloride and chloroprene for carcinogenicity in
rats. Oncology, 37: 136-141.
173 PRENDERGAST, J.A., JONES, R.A, JENKINS, L.J., Jr, & SIEGEL, J.
(1967) Effects on experimental animals of long-term inhalation
of trichloroethane, carbon tetrachloride, 1,1,1-trichloro-
ethylene, dichlorodifluoromethane and 1,1-dichloroethylene.
Toxicol. appl. Pharmacol., 10: 270-289.
174 PRICE, P.S. (1985) Volatile organochlorine compounds (VOC)
degradation. Technical Memorandum, Washington, DC, US
Environmental Protection Agency, p. 19.
175 PUTCHA, L., BRUCKNER, J.V., D'SOUZA, R., DESAI, F., & FELDMAN,
S. (1986) Toxicokinetics and bioavailability of oral and
intravenous 1,1-dichloroethylene. Fundam. appl. Toxicol., 6:
240-250.
176 QUAST, J.F., HUMISTON, C.G., SCHWETZ, B.A., BALMER, M.F.,
RAMPY, L.W., NORRIS, J.M., & GEHRING, P.J. (1977) Results of
90-day toxicity study in rats given vinylidene chloride in
their drinking water or exposed to VDC vapour by inhalation.
Toxicol. appl. Pharmacol., 4: 187.
177 QUAST, J.F., HUMISTON, C.G., WADE, C.E., BALLARD, J., BEYER,
J.E, SCHWETZ, R.W., & NORRIS, J.M. (1983) A chronic toxicity
and oncogenicity study in rats and subchronic toxicity study
in dogs on ingested vinylidene chloride. Fundam. appl.
Toxicol., 3: 55-62.
178 QUAST, J.F., MCKENNA, M.J., RAMPY, L.W., & NORRIS, J.M. (1986)
Chronic toxicity and oncogenicity study on inhaled vinylidene
chloride in rats. Fundam. appl. Toxicol., 6: 105-144.
179 RAMPY, L.W., QUAST, J.F., HUMISTON, C.G., BALMER, M.F., &
SCHWETZ, B.A. (1977) Interim results of two-year toxicological
studies in rats of vinylidene chloride incorporated in the
drinking water or administered by repeated inhalation.
Environ. Health Perspect., 21: 33-43.
180 RAMPY, L.W., QUAST, J.F., HUMISTON, C.G., BALMER, M.F., &
SCHWETZ, B.A. (1978) Results of two-year toxicological studies
in rats of vinylidene chloride incorporated in the drinking
water or administered by repeated inhalation. Toxicol. appl.
Pharmacol., 45: 244-245.
181 RAMSTAD, T., NESTRICK, T.J., & PETERS, T.L. (1981)
Applications of the purge-and-trap technique. Am. Lab., 13:
65-73.
182 RAY, P. & MOORE, L. (1982) 1,1-Dichloroethylene inhibition of
liver microsomal calcium pump in vitro . Arch. Biochem.
Biophys., 218: 26-30.
183 REICHERT, D., WERNER, H.W., & HENSCHLER, D. (1978) Role of
liver glutathione in 1,1-dichloroethylene metabolism and
hepatotoxicity in intact rats and isolated perfused rat liver.
Arch. Toxicol., 41: 169-178.
184 REICHERT, D., WERNER, H.W., METZLER, M., & HENSCHLER, D.
(1979) Molecular mechanism of 1,1-dichloroethylene toxicity:
excreted metabolites reveal different pathways of reactive
intermediates. Arch. Toxicol., 42: 159-169.
185 REICHERT, D., SPENGLER, U ., ROMEN, W., & HENSCHLER, D. (1984)
Carcinogenicity of dichloroacetylene: an inhalation study.
Carcinogenesis, 5: 1411-1420.
186 REITZ, R.H., WATANABE, P.G., MCKENNA, M.J., QUAST, J.F., &
GEHRING, P.J. (1980) Effects of vinylidene chloride on DNA
synthesis and DNA repair in the rat and mouse: a comparative
study with dimethylnitrosamine. Toxicol. appl. Pharmacol., 52:
357-370.
187 REKKER, R.F. (1977) The hydrophobic fragment constant, its
derivation and application. A means of characterizing membrane
systems. In: Nauta, W.Th. & Rekker, R.F., ed. Pharmacochemistry
library, Amsterdam, Elsevier Scientific Publishers, Vol. 1.
188 REYNOLDS, E.S, MOSLEN, M.T., SZABO, S., JAEGER, R.J., &
MURPHY, S.D. (1975) Hepatotoxicity of vinyl chloride and 1,1-
dichloroethylene. Role of mixed function oxidase system. Am.
J. Pathol., 81: 219-236.
189 REYNOLDS, E.S, MOSLEN, M.T., BOOR, P.J., & JAEGER, R.J. (1980)
1,1-Dichloroethylene hepatotoxicity. Time course of GSH
changes and biochemical aberrations. Am. J. Pathol., 101:
331-344.
190 REYNOLDS, E.S., KANZ, M.F., CHIECO, P., & MOSLEN, M.T. (1984)
1,1-Dichloroethylene: an apoptotic hepatotoxin. Environ.
Health Perspect., 57: 313-320.
191 RUSSELL, M.J. (1975) Analysis of air pollutants using sampling
tubes and gas chromatography. Environ. Sci. Technol., 9:
1175-1178.
192 RYLOVA, M.L. (1953) [Toxicity of vinylidene chloride.] Farmakol.
Toksikol., 16(1): 47-50 (in Russian).
193 SASAKI, M., SUGIMURA, K., YOSHIDA, M.A., & ABE, S. (1980)
Cytogenetic effects of 60 chemicals on cultured human and
Chinese hamster cells. Kromosomo II,20: 574-584.
194 SASSU, G.M, ZILIO-GRANDI, F., & CONTE, A. (1968) Gas
chromatographic determination of impurities in vinyl
chloride. J. Chromatogr., 34: 394-398.
195 SATO, A., NAKAJIMA, T., & KOYAMA, Y. (1980) Effects of chronic
ethanol consumption on hepatic metabolism of aromatic and
chlorinated hydrocarbons in rats. Br. J. ind. Med., 37:
382-386.
196 SAWADA, M., SOFUNI, T., & ISHIDATE, M., Jr (1987) Cytogenetic
studies on 1,1-dichloroethylene and its two isomers in
mammalian cells in vitro and in vivo. Mutat. Res., 187:
157-163.
197 SAX, N.I. (1984) Dangerous properties of industrial materials,
6th ed., New York, Van Nostrand Reinhold, p. 2730.
198 SCHMITZ, TH., THIESS, A.M., & PENNING, E. (1979) [Inquiry into
morbidity among workers exposed to vinylidene chloride (VDC)
and polyvinylidene chloride (PVDC).] In: [ Report on the Tenth
Annual Meeting of the German Occupational Medicine Society
together with the Federation of Industrial Employers
Associations, Munster, 2-5 May, 1979, ] Stuttgart, Gentner
Verlag (in German).
199 SEVERS, L.W. & SKORY, L.K. (1975) Monitoring personnel
exposure to vinyl chloride, vinylidene chloride and methyl
chloride in an industrial work environment. Am. Ind. Hyg.
Assoc. J., 39: 669-676.
200 SHACKELFORD, W.M. & KEITH, L.H. (1976) Frequency of organic
compounds identified in water, Washington, DC, US
Environmental Protection Agency, Office of Research and
Development, Environmental Research Laboratory (EPA 600/4-76-
062; PB-265-470).
201 SHELTON, L.G., HAMILTON, D.E., & FISACKERLY, R.H. (1971) Vinyl
and vinylidene chloride. In: Leonard, E.C., ed. Vinyl and
diene monomers. Part 3, New York, Wiley Interscience,
pp. 1505-1289.
202 SHORT, R.D., MINOR, J.L., PETERS, P., WINSTON, J.M., FERGUSON,
B., UNGER, T., SAWYER, M., & LEE, C.C. (1977a) The
developmental toxicity of vinylidene chloride inhaled by rats
and mice during gestation, Washington, DC, US Environmental
Protection Agency (EPA 560/6-77-022; PB-281-713) (Prepared by
Midwest Research International, Kansas City, Missouri).
203 SHORT, R.D., MINOR, J.L., WINSTON, J.M., & LEE, C.C. (1977b) A
dominant lethal study in male rats after repeated exposures to
vinyl chloride or vinylidene chloride. J. Toxicol. environ.
Health, 3: 965-968.
204 SHORT, R.D., WINSTON, J.M., MINOR, J.L., HONG, C.B., SEIFTER,
J., & LEE, C.C. (1977c) Toxicity of vinylidene chloride in
mice and rats and its alteration by various treatments. J.
Toxicol. environ. Health, 3: 913-921.
205 SIDHU, K.S. (1980) A gas-chromatographic method for the
determination of vinylidene chloride in air. J. anal.
Toxicol., 4: 266-268.
206 SIEGEL, J., JONES, R.A., COON, R.A., & LYON, J.P. (1971)
Effects on experimental animals of acute, repeated and
continuous inhalation exposures to dichloroacetylene mixtures.
Toxicol. appl. Pharmacol., 18: 168-174.
207 SIEGERS, C.-P., YOUNES, M., & SCHMITT, G. (1979) Effects of
dithiocarb and (+)-cyanidanol-3 on the hepatotoxicity and
metabolism of vinylidene chloride in rats. Toxicology, 15:
55-64.
208 SIEGERS, C.-P., HEIDBUCHEL, K., & YOUNES, M. (1983) Influence
of alcohol, dithiocarb, or (+)-catechin on the hepatotoxicity
and metabolism of vinylidene chloride in rats. J. appl.
Toxicol., 3: 90-95.
209 SIEGERS, C.-P., HORN, W., & YOUNES, M. (1985a) Effect of
hypoxia on the metabolism and hepatoxicity of carbon
tetrachloride and vinylidene chloride in rats. Acta pharmacol.
toxicol., 56: 81-86.
210 SIEGERS, C.-P., HORN, W., & YOUNES, M. (1985b) Effect of
phorone-induced glutathione depletion on the metabolism and
hepatotoxicity of carbon tetrachloride and vinylidene
chloride. J. appl. Toxicol., 5: 352-356.
211 SILETCHNIK, L.M. & CARLSON, G.P. (1974) Cardiac sensitizing
effects of 1,1-dichloroethylene: enhancement by phenobarbital
pretreatment. Arch. int. Pharmacodyn., 210: 359-364.
212 SINGH, H.B., SALAS, L.J., SMITH, A.J., & SHIGEISHI, M. (1981)
Measurements of some potentially hazardous organic chemicals
in urban environments. Atmos. Environ., 15: 601-612.
213 SINGH, H.B., SALAS, L .J., & STILES, R.E. (1982) Distribution
of selected gaseous organic mutagens and suspect carcinogens
in ambient air. Environ. Sci. Technol., 16: 872-880.
214 SPEIS, D.N. (1980) Determination of purgeable organics in
sediments. Environ. Sci. Res., 16: 201-206.
215 SWEGER, D.M. & TRAVIS, J.C. (1979) An application of infrared
lasers to the selective detection of trace organic gases.
Appl. Spectrosc., 33: 46-51.
216 SZABO, S., JAEGER, R.J., MOSLEN, M.T., & REYNOLDS, E.S. (1977)
Modification of 1,1-dichloroethylene hepatotoxicity by
hypothyroidism. Toxicol. appl. Pharmacol., 42: 367-376.
217 TABAK, H.H., QUAVE, S.A., ·MASHNI, C.I., & BARTH, E.F. (1981)
Biodegradability studies with organic priority pollutant
compounds. J. Water Pollut. Control Fed., 53: 1503-1518.
218 TAN, S. & OKADA, T. (1979) Determination of residual
vinylidene chloride monomer in polyvinylidene chloride.
Hygienic studies on plastic containers and packages. III. J.
Food Hyg. Soc. Jpn, 20: 223-227.
219 THIESS, A.M., FRENTZEL-BEYME, R., & PENNING, E. (1979)
Mortality study of vinylidene chloride exposed persons. In:
Heim, C. & Kilian, D.J.., ed. Proceedings of the 5th Medichem
Congress, San Francisco, September 1977, pp. 270-278.
220 THOMPSON, J.A, HO, B., & MASTOVICH, S.L. (1984) Reductive
metabolism of 1,1,1,2-tetrachloroethane and related chloro-
ethanes by rat liver microsomes. Chem-biol. Interact., 51:
321-333.
221 TIERNEY, D.R., BLACKWOOD, T.R., & PIANA, M.R. (1979) Status
assessment of toxic chemicals: vinylidene chloride,
Cincinnati, Ohio, US Environmental Protection Agency (EPA
600/2-79-2100; PB 80- 146442).
222 TORKELSON, T.R. & ROWE, V.K. ed. (1982) Vinylidene chloride.
In: Clayton, G.D. & Clayton, F.E., Patty's industrial hygiene
and toxicology, 3rd ed., New York, John Wiley and Sons, pp.
3545-3550.
223 US EPA (1984a) Method 601. Guidelines establishing test
procedures for the analysis of pollutants under the Clean
Water Act (40 CFR 136). Purgeable halocarbons. Fed. Reg., 49:
43261-43271.
224 US EPA (1984b) Method 1624, Revision B. Guidelines
establishing test procedures for the analysis of pollutants
under the Clean Water Act (40 CFR 136). Volatile organic
compounds by isotope dilution GC/MS. Fed. Reg., 49:
43407-43415.
225 US EPA (1985) Health assessment document for vinylidene
chloride, Washington, DC, US Environmental Protection Agency,
Office of Health and Environmental Assessment (EPA 600/8-83-
031F).
226 US NIOSH (1987) Manual of analytical methods, 3rd ed.,
Cincinnati, Ohio, National Institute of Health and Human
Services, pp.1-3 (Method 1015).
227 VAN DUUREN, B.L., GOLDSCHMIDT, B.M., LOEWENGART, G., SMITH,
A.C., MELCHIONNE, S., SEIDMAN, I., & ROTH, D. (1979)
Carcinogenicity of halogenated olefinic and aliphatic
hydrocarbons in mice. J. Natl Cancer Inst., 63:
1433-1439.
228 VAN'T HOF, J. & SCHAIRER, L.A. (1982) Tradescantia assay
system for gaseous mutagens. A report of the US Environmental
Protection Agency Gene-Tox Program. Mutat. Res., 99:
303-315.
229 VIOLA, P.L. & CAPUTO, A. (1977) Carcinogenicity studies on
vinylidene chloride. Environ. Health Perspect., 21: 45-47.
230 VOGEL, T.M. & MCCARTY, P.L. (1987) Abiotic and biotic
transformations of 1,1,1-trichloroethane under methanogenic
conditions. Environ. Sci. Technol., 21: 1208-1213.
231 WAKEHAM, S.G., GOODWIN, J.T., & DAVIS, A.C. (1983)
Distributions and fate of volatile organic compounds in
Narragansett Bay, Rhode Island. Can. J. Fish. Aquat. Sci., 40
(Suppl.2): 304-321.
232 WALKER, W.W., MANNING, C.S., OVERSTREET, R.M., & HAWKINS, W.E.
(1985) Development of aquarium fish models for environmental
carcinogenesis: an intermittent-flow exposure system for
volatile, hydrophobic chemicals. J. appl. Toxicol., 5:
255-260.
233 WALLACE, L., ZWEIDINGER, R., ERICKSON, M., COOPER, S.,
WHITAKER, D., & PELLIZZARI, E. (1982) Monitoring individual
exposure. Measurements of volatile organic compounds in
breathing-zone air, drinking water and exhaled breath.
Environ. Int., 8: 269-282.
234 WALLACE, L., PELLIZZARI, E., HARTWELL, T., ROSENZWEIG, M.,
ERICKSON, M., SPARACINO, C., & ZELON, H. (1984) Personal
exposure to volatile organic compounds. I. Direct measurements
in breathing-zone air, drinking water, food, and exhaled
breath. Environ. Res., 35: 293- 319.
235 WALLACE, L.A, PELLIZZARI, E., SHELDON, S., HARTWELL, T.,
SPARACINO, C., & ZELON, H. (1986) The total exposure
assessment methodology (TEAM) study: direct measurements of
personal exposures through air and water for 600 residents of
several US cities. In: Cohen, Y., ed. Pollutants in a
multimedia environment, New York, London, Plenum Publishing
Corporation. pp. 289-315.
236 WANG, T. & LENAHAN, R. (1984) Determination of volatile
halocarbons in water by purge-closed loop gas chromatography.
Bull. environ. Contam. Toxicol., 32: 429-438.
237 WANG, T., LENAHAN, R., & KANIK, M. (1985) Impact of
trichloroethylene contaminated groundwater discharged to the
main canal and Indian river lagoon, Vero Beach, Florida. Bull.
environ. Contam. Toxicol., 34: 578-586.
238 WARNER, C., MODDERMAN, J., FAZIO, T., BEROZA, M., SCHWARTZMAN,
G., FOMINAYA, K., & SHERMA, J. (1983) Food additives
analytical manual, Arlington, Virginia, Association of
Official Analytical Chemists, Vol. 1, pp. 348-357.
239 WASKELL, L. (1978) Study of the mutagenicity of anesthetics
and their metabolites. Mutat. Res., 57: 141-153.
240 WAXWEILER, R.J., SMITH, A.H., FALK, H., & TYROLER, H.A. (1981)
Excess lung cancer risk in a synthetic chemicals plant.
Environ. Health Perspect., 41: 159-165.
241 WEAST, R.C., ed. (1984) CRC Handbook of chemistry and physics,
65th ed., Boca Raton, Florida, CRC Press, p. C-295.
242 WEGMAN, R.C.C., BANK, C.A., & GREVE, P.A. (1981) Environmental
pollution by a chemical waste dump. Stud. environ. Sci., 17:
349-357.
243 WESSLING, R.A. & EDWARDS, F.G. (1971) Vinylidene chloride
polymers. In: Bikales, N.M., ed. Encyclopedia of polymer
science and technology, New York, Wiley Interscience, Vol. 14,
pp. 540-579.
244 WHO (1984) Guidelines for drinking-water quality, Vol. 1 and
2, Geneva, World Health Organization.
245 WOLFF, T., DISTLERATH, L.M., WORTHINGTON, M.T., GROOPMAN,
J.D., HAMMONS, G.J., KADLUBAR, F.F., PROUGH, R.A., MARTIN,
M.V., & GUENGERICH, F.P. (1985) Substrate specificity of human
liver cytochrome P-450 debrisoquine 4-hydroxylase probed using
immunochemical inhibition and chemical modeling. Cancer Res.,
45: 2116-2122.
246 YOUNG, D.R., GOSSETT, R.W., BAIRD, R.B., BROWN, D.A., TAYLOR,
P.A., & MIILLE, M.J. (1981) Wastewater inputs and marine
bioaccumulation of priority pollutant organics off Southern
California; In: Jolley, R.L., Brungs, W.A., Cotrivo, J.A.
Cumming, R.B., Mattice, J.S., & Jacobs, V.A., ed. Proceedings
of the Fourth Conference on Water Chlorination (Environmental
Impact and Health Effects), Pacific Grove, California, 18-23
October, 1981, Ann Arbor, Michigan, Ann Arbor Science
Publishers, Chapter 60, pp. 871-884.
247 ZELLER, H. & PEH, J. (1975) [ Report on the tests of vinylidene
chloride for mutagenic effects in Chinese Hamsters after
single oral application (chromosomal study)], Ludwigshafen,
BASF Aktiengesellschaft, 12 pp (in German).
248 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979a) [ Report
on the determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in vapour form in Sprague-Dawley rats
(fasting) during a 4-hour exposure period], Ludwigshafen, BASF
Aktiengesellschaft, 14 pp (in German).
249 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979b) [ Report
on the determination of acute toxicity (LC50) of vinylidene
chloride in Sprague-Dawley rats (fed) during a 4-hour exposure]
Ludwigshafen, BASF Aktiengesellschaft, 14 pp. (in German).
250 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979c) [ Report
on the determination of acute toxicity (LC50) by inhalation
of vinylidene chloride in NMRI mice (fasting) during a 4-hour
exposure] Ludwigshafen, BASF Aktiengesellschaft, 12 pp. (in
German).
251 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979d) [ Report
on the determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in NMRI mice (fed) during a 4-hour
exposure] Ludwigshafen, BASF Aktiengesellschaft, 12 pp. (in
German).
RESUME ET CONCLUSIONS, EVALUATION ET RECOMMANDATIONS
1. Résumé et conclusions
1.1 Propriétés, usages et méthodes d'analyse
Le chlorure de vinylidène (C2H2Cl2) est un liquide volatil et
incolore d'odeur douceâtre. On le stabilise au moyen de
p- méthoxyphénol afin d'éviter la formation de peroxydes explosifs.
Le chlorure de vinylidène est utilisé pour la production de
trichloro-1,1,1-éthane, de fibres et de copolymères modacryliques
(avec du chlorure de vinyle ou de l'acrylonitrile). On a mis au
point des méthodes de chromatographie en phase gazeuse pour la
recherche et le dosage du chlorure de vinylidène dans l'air, l'eau
ou les emballages, les tissus de l'organisme, les denrées
alimentaires et le sol. Le détecteur le plus sensible est le
détecteur à capture d'électrons.
1.2 Sources et niveaux d'exposition
Chaque année on libère dans l'atmosphère une quantité de
chlorure de vinylidène qui correspond à une proportion allant
jusqu'à 5 % de la production totale (soit environ 23 000 tonnes au
maximum). La forte tension de vapeur et la faible solubilité dans
l'eau de ce produit font qu'il est relativement abondant dans
l'atmosphère par rapport aux autres compartiments du milieu. On
pense que le chlorure de vinylidène présent dans l'atmosphère a une
demi-vie d'environ deux jours.
Dans l'eau, les concentrations sont très faibles. Même dans les
eaux résiduaires industrielles, les concentrations sont de l'ordre
du µg/litre, c'est-à-dire bien inférieures aux concentrations
toxiques pour la faune aquatique, concentrations qui sont de
l'ordre du mg/litre. Dans l'eau de boisson non traitée, les
concentrations ne sont généralement pas décelables. Dans l'eau
potable traitée, la teneur en chlorure de vinylidène est
généralement inférieure à 1 µg/litre encore qu'on ait trouvé des
échantillons qui en contenaient 20 < g/litre. Dans les denrées
alimentaires, les concentrations ne sont généralement pas
décelables, le maximum observé étant de 10 µg/kg.
L'exposition professionnelle au chlorure de vinylidène peut se
produire soit par inhalation, soit par contamination de la peau ou
des yeux. Selon les pays, la dose maximale recommandée ou
l'exposition moyenne pondérée en fonction du temps (TWA) se situent
dans les limites de 8 à 500 mg/m3; quelquefois, la dose maximale
correspond à la concentration la plus faible qui soit décelable de
façon certaine. Les limites d'exposition à court terme vont de 16
à 80 mg/m3 et les valeurs plafond de 500 à 700 mg/m3.
1.3 Absorption, distribution, métabolisme et excrétion
Le chlorure de vinylidène peut être absorbé facilement par les
voies respiratoires ou digestives chez les mammifères; en revanche
on de dispose pas de renseignements sur l'absorption percutanée.
Administré à des rongeurs, le chlorure de vinylidène se répartit
largement dans l'organisme de l'animal, les concentrations étant
maximales dans le foie et les reins. L'élimination par la voie
pulmonaire du chlorure de vinylidène inchangé s'effectue selon un
processus au moins biphasé qui dépend de la dose; elle est plus
importante aux doses qui provoquent une saturation du métabolisme
(c'est-à-dire 600 mg/m3 environ (150 ppm) chez le rat). Des rats à
qui l'on avait administré une dose de chlorure de vinylidène par
voie orale et que l'on avait ensuite fait jeûner ont exhalé
davantage de cette substance.
Les principales voies du métabolisme ont été identifiées chez
le rat. Dans la voie prédominante (phase I), intervient le
cytochrome P-450 et il y a formation (vraisemblablement, mais pas
forcément par l'intermédiaire d'un époxyde), d'acide
monochloracétique. Le chlorure de vinylidène peut stimuler
l'activité du cytochrome P-450. Un certain nombre de métabolites de
la Phase I peuvent se conjuguer au glutathion ou à la phosphatidyl-
éthanolamine avant de subir d'autres transformations. Le
métabolisme est plus rapide chez la souris que chez le rat, avec un
profil analogue où les dérivés conjugués au glutathion sont
relativement plus abondants. On a montré que le chlorure de
vinylidène était également métabolisé par le cytochrome P-450 des
microsomes humains.
Chez les rongeurs, le métabolisme du chlorure de vinylidène
conduit à la déplétion du glutathion et à l'inhibition de la
glutathion- S- transférase.
1.4 Effets sur les animaux d'expérience et les systèmes
cellulaires
1.4.1 Fixation aux tissus par liaison covalente
Le chlorure de vinylidène radio-marqué se fixe aux tissus
hépatiques, rénaux et pulmonaires des rongeurs par liaison
covalente et c'est ce phénomène qui déclenche le processus toxique.
Les liaisons par covalence et par conséquent la toxicité sont
accrues par la déplétion en glutathion et se produisent au niveau
du foie et du rein à plus faible dose chez la souris que chez le
rat. Un certain nombre de métabolites du chlorure de vinylidène se
fixent par liaison covalente aux thiols in vitro .
1.4.2 Toxicité aigue
Les estimations de la CL50 aiguë du chlorure de vinylidène
varient considérablement, mais cette variation ne masque pas le
fait que les souris sont beaucoup plus sensibles à cette substance
que les rats ou les hamsters. Les valeurs estimatives de la CL50
orale à 4-h varient d'environ 8000 à 128 000 mg/m3 (2000-32 000
ppm) chez le rat, de 450 à 820 mg/m3 (115-205 ppm) chez la souris et
de 6640-11 780 mg/m3 (1660-2945 ppm) chez le hamster.
Du fait que la relation entre la concentration et la mortalité
n'est pas linéaire, les estimations de la CL50 peuvent être
entachées d'erreurs. Chez toutes les espèces, la CL50 a tendance à
être plus faible pour les mâles que pour les femelles et le jeûne
(qui provoque une déplétion en glutathion) accroît la toxicité dans
tous les cas. Après administration par voie orale les valeurs de
la DL50 s'établissaient approximativement à 1500 et 100 mh/kilo
respectivement chez les rats et les souris. Aprés inhalation, la
toxicité s'est manifestée par une irritation des muqueuses, une
dépression du système nerveux central et une cardiotoxicité
progressive (bradycardie sinusale et arrythmies). On a noté des
lésions au niveau du foie, des reins et des poumons. Chez les
souris, qui sont plus sensibles que les rats à l'hépatotoxicité et
à la néphrotoxicité du chlorure de vinylidène, on a constaté
qu'une exposition à des concentrations ne dépassant pas 40 mg/m3
(10 ppm) pendant 6 heures accroissait les lésions rénales et la
réplication de l'ADN. Comme dans le cas de l'inhalation, les
principaux organes affectés par l'administration de chlorure de
vinylidène par voie orale, sont le foie, les reins et les poumons.
Le processus toxique au niveau du foie commence par des altérations
au niveau des canaux biliaires et se poursuit par l'apparition de
signes d'atteinte mitochondriale. Après quoi il y a lésion du
réticulum endoplasmique et mort de la cellule. Il ne semble pas que
la toxicité du chlorure de vinylidène pour le foie et le rein soit
due à la peroxydation des lipides. Il semblerait plutôt que
l'augmentation de la concentration intra-cellulaire des ions
calcium soit à l'origine de la toxicité de ce produit pour les
hépatocytes.
Les effets toxiques du chlorure de vinylidène dépendent, au
moins partiellement, de l'activité du cytochrome P-450 (qui peut
également intervenir dans le détoxication) et peuvent être
exacerbés par une déplétion en glutathion. L'éthanol et la
thyroxine peuvent accroître l'hépatotoxicité; en revanche celle-ci
est inhibée par le dithiorcarbe et la (+)-catéchine et modulée par
l'acétone.
1.4.3 Etudes à court terme
Des lésions hépatiques et rénales accompagnées, dans une
moindre proportion, de lésions pulmonaires ont été observées chez
des rongeurs exposés par inhalation à du chlorure de vinylidène à
raison de 40 à 800 mg/m3, 4 à 8 heures par jour, pendant 4 jours ou
plus par semaine. Les souris se sont révélées plus sensibles que
les rats, les cobayes, les lapins, les chiens et les saïmiris alors
que chez les souris, la toxicité variait selon la souche utilisée.
En général, les souris femelles étaient moins sensibles que mâles.
On a observé une hépatotoxicité chez des rats et des souris exposés
de façon intermittente à des concentrations de chlorure de
vinylidène respectivement >800 mg/m3 (>200 ppm) ou égales à 220
mg/m3 (55 ppm). Pour induire une hépatotoxicité par exposition
continue durant plusieurs jours, il fallait 240 mg/m3 (60 ppm) pour
les rats et 60 mg/m3 (15 ppm) pour les souris. Ce traitement
intermittant ou continu a également provoqué une néphrotoxicité
chez les souris. Les souris mâles de la race Swiss se sont montrées
particulièrement sensibles à la néphrotoxocité induite par le
chlorure de vinylidène. Les mâles n'ont pas survécu à une
exposition continue de courte durée à 200 mg de chlorure de
vinylidène par m3 (50 ppm). Chez les chiens, les saïmiris et les
rats, le seuil d'hépatotoxicité se situait à environ 80 mg/m3 (20
ppm) administrés de façon continue sur 90 jours. Des études à court
terme (d'une durée d'environ 3 mois) au cours desquelles du
chlorure de vinylidène a été administré par voie orale à des rats à
des doses allant jusqu'à 20 mg/kg par jour et á des chiens à des
doses allant jusqu'à 25 mg/kg, n'ont pas révélé de signes de
toxicité, si ce n'est quelques lésions hépatiques infimes et
réversibles chez les rats.
1.4.4 Etudes à long terme
Des études à long terme comportant l'inhalation
intermittente de chlorure de vinylidène ont révélé que la dose de
300 mg/m3 (75 ppm) ne produisait que des modifications bénignes et
réversibles au niveau du foie chez les rats. A 600 mg/m3
(150 ppm) c'est-à-dire la dose la plus forte qui soit supportable
au cours d'une exposition à long terme, on constatait de nettes
lésions hépatiques avec nécrose. On a observé une forte mortalité
avec des signes de lésion hépatique chez des souris soumises à une
dose de 200 mg/m3 (50 ppm). La néphrotoxicité était évidente
chez la souris après un traitement à long terme à la dose de 100
mg/m3 (25 ppm). L'administration de chlorure de vinylidène par voie
orale à des rats pendant un an à des doses quotidiennes allant
jusqu'à 300 mg/kg n'a produit que de minimes anomalies hépatiques.
Il n'est pas possible de tirer de ces données une valeur précise de
la dose sans effet observable. Une autre étude a révélé des signes
d'inflammation rénale et de nécrose hépatique chez des rats et des
souris soumis à une administration orale prolongée de chlorure de
vinylidène à des doses quotidiennes respectives de 5 mg/kg et 2
mg/kg.
1.4.5 Génotoxicité et cancérogénicité
On a constaté que le chlorure de vinylidène était mutagène pour
les bactéries et les levures, mais seulement en présence d'un
système mammalien d'activation métabolique microsomique (S9). Le
composé a provoqué une synthèse anarchique de l'ADN dans des
hépatocytes isolés de rats ainsi qu'une augmentation de la
fréquence des échanges entre chromatides soeurs et des aberrations
chromosomiques dans des cultures cellulaires additionnées de S9.
Par contre, on n'a pas observé d'accroissement des mutations
géniques chez les mammifères. Il a été fait état d'une augmentation
légère mais statistiquement significative de la liaison à l'ADN
après exposition in vivo . Cette liaison était plus importante dans
les cellules de souris que dans celles de rats et également plus
importante dans les reins que dans le foie après une exposition de
6 heures à des concentrations de 40 et 200 mg de chlorure de
vinylidène/m3 (10 et 50 ppm). En outre, le chlorure de vinylidène
augmentait légèrement la synthèse anarchique de l'ADN dans le rein
de la souris. On a relevé aucun signe de mutation létale dominante
ou d'effets cytogénétiques après exposition in vivo de rongeurs,
sauf dans le cas d'une étude au cours de laquelle on a observé des
aberrations chromosomiques dans la moëlle osseuse de hamsters
chinois.
Des études de cancérogénicité ont été effectuées sur trois
espèces animales (rats, souris et hamsters). Chez des souris mâles
de race Swiss, on a relevé un net effet cancérogène (adénocarcinome
rénal) après exposition prolongée intermittente à des
concentrations de 100 ou de 200 mg/m3 de chlorure de vinylidène (25
ou 50 ppm), mais pas aux concentrations de 0 ou 40 mg/m3 (0 ou 10
ppm).
Il est possible que ces tumeurs rénales soient liées d'une
manière ou d'une autre à la néphrotoxicité observée et que des
atteintes rénales répétées puissent conduire directement à une
réaction cancérogène selon un mécanisme non génotoxique ou qu'elles
facilitent l'expression de l'activité génotoxique de certains
métabolites chez cette espèce, pour ce sexe et au niveau de cet
organe. Toutefois, cette conclusion reste hypothétique du fait que
les données disponibles sur les effets génétiques in vivo sont
limitées et que le chlorure de vinylidène a pu jouer le rôle
d'initiateur.
Dans la même étude, on a constaté une augmentation statistique
de l'incidence des tumeurs pulmonaires (principalement des adénomes
chez les souris des deux sexes) et des cancers mammaires (chez les
femelles) mais on a pas observé de relation entre la dose et la
réponse. Chez des rats adultes exposés par inhalation au chlorure
de vinylidène, on a signalé une légère augmentation des tumeurs
mammaires sans relation avec la dose ainsi qu'une augmentation
modérée des leucémies lorsque les rats étaient exposés à la
substance in utero puis après leur naissance. Il n'a pas été
possible d'évaluer les résultats.
1.4.6 Toxicité pour la fonction de reproduction
Aucun effet n'a été observé sur la fécondité de rats exposés en
permanence à du chlorure de vinylidène (jusqu'à 200 mg/litre ou 200
ppm) ajouté à leur eau de boisson. Des rats et des souris qui
avaient inhalé jusqu'à 1200 mg de chlorure de vinylidène par m3
(300 ppm) pendant 22 à 23 heures, à différents stades de
l'organogenèse, n'ont pas produit de foetus présentant des
anomalies autres que celles qui peuvent être attribuées à une
action toxique sur la mère.
Des rats et des lapins ont inhalé 7 h/jour 640 mg de chlorure
de vinylidène par m3 (160 ppm) ou absorbé par voie orale environ 40
mg/kilo de cette substance au cours des stades critiques de la
gestation sans que les embryons ou les foetus présentent
d'anomalies à ces doses, inférieures aux doses toxiques pour la
mère. Toutefois, des anomalies ont été constatées sur les embryons
et les foetus aux doses toxiques pour la mère, comme l'a montré la
réduction du gain de poids.
1.5 Effets sur l'homme
Des concentrations de chlorure de vinylidène de l'ordre de 16
000 mg/m3 (4000 ppm) provoquent une intoxication pouvant entraîner
une perte de connaissance. Additionné de stabilisateur, le chlorure
de vinylidène est également irritant pour les voies respiratoires,
les yeux et la peau. A la suite d'expositions prolongées ou
répétées à des doses infra-anesthésiques, on a signalé l'apparition
de lésions rénales et hépatiques. Il est difficile d'évaluer les
résultats obtenus par les études épidémiologiques en raison de
l'effectif limité des cohortes, d'une exposition simultanée au
chlorure de vinyle et du fait qu'on n'a pas suffisamment tenu
compte du tabagisme. On n'a pas constaté d'augmentation
statistiquement significative dans l'incidence des cancers chez les
personnes exposées au chlorure de vinylidène, mais il est vrai que
les études épidémiologiques présentaient des insuffisances; aussi
n'est-il pas possible d'en conclure qu'il n'existe aucun risque de
cancérogénicité. On ne dispose d'aucun renseignement concernant les
effets du chlorure de vinylidène sur la fonction de reproduction
humaine.
2. Evaluation des effets sur l'environnement et des risques pour la
santé humaine
2.1 Evaluation des effets sur l'environnement
Par suite de la volatilité du chlorure de vinylidène, c'est
l'atmosphère qui est le compartiment du milieu où il est le plus
abondant. La demi-vie du chlorure de vinylidène dans la troposphère
est vraisemblablement d'environ deux jours, aussi ce composé ne
contribue-t-il probablement pas à la réduction de la couche d'ozone
stratosphérique. Lessivage et volatilisation font du sol et des
sédiments des compartiments où le chlorure de vinylidène n'est
présent qu'en petites quantités et cet hydrocarbure chloré
n'apparaît qu'en quantité minime dans le milieu aquatique du fait
de sa volatilisation rapide. On ignore si la dégradation de
composés tel que le trichloréthylène et le perchloréthylène,
souvent présents dans l'eau, contribuent de manière notable à la
concentration du chlorure de vinylidène dans l'environnement.
La concentration du chlorure de vinylidène dans l'environnement
et les valeurs de la toxicité aiguë pour les poissons et la daphnie
montrent que les risques d'intoxication aiguë sont minimes pour la
faune aquatique. On ne dispose pas de données suffisantes sur la
toxicité à long terme pour évaluer les effets sublétaux sur les
organismes aquatiques qui vivent à proximité de sources
relativement importantes de contamination par le chlorure de
vinylidène, qu'il s'agisse d'eaux souterraines contaminées ou
d'eaux résiduaires municipales ou industrielles.
2.2 Evaluation des risques pour la santé humaine
2.2.1 Niveau d'exposition
La population générale n'est exposée qu'à de très faibles
teneurs de chlorure de vinylidène. La concentration maximale qui
ait été signalée dans l'eau de boisson est de 20 µg par litre,
encore que l'exposition individuelle moyenne pour les citoyens des
Etats-Unis d'Amérique par l'intermédiaire de l'eau de boisson soit
estimée à moins de 0,01 µg par jour. Il n'y a pas de chlorure de
vinylidène en concentrations décelables dans les denrées
alimentaires et en tout état de cause on n'a pas signalé de teneurs
supérieures à 10 µg/kg. On ignore quelles sont les concentrations
dans les denrées alimentaires constituées d'organismes aquatiques
mais elles sont vraisemblablement insignifiantes (section 10.1).
Dans l'air ambiant on a signalé des concentrations en chlorure de
vinylidène allant jusqu'à 52 µg/m3 (dans le périmètre d'une zone
industrielle). Des concentrations médianes dans l'air urbain de 20
ng/m3 et de 8,7 µg/m3 ont été signalées aux Etats-Unis,
respectivement dans des zones non industrielles et dans des zones
industrielles.
L'exposition professionnelle se produit notamment lors de la
production et de la polymérisation du chlorure de vinylidène. C'est
principalement par la voie respiratoire que cette substance pénètre
dans l'organisme et les limites maximales recommandées ou
réglementées pendant une journée de travail vont de 8 à 500 mg/m3
(ou la concentration la plus faible qui soit décelable par une
méthode fiable), selon les pays. Les limites d'exposition à court
terme vont de 16 à 80 mg/m3 et les valeurs plafonds de 50 à 700
mg/m3. La concentration atmosphérique de chlorure de vinylidène en
atmosphère confinée à laquelle certains travailleurs peuvent être
exposés, ne dépasse pas 8 mg/m3.
2.2.2 Effets aigus
Chez l'homme, l'inhalation de fortes concentrations de chlorure
de vinylidène (très approximativement, supérieures ou égales au
seuil olfactif maximal de 4000 mg/m3) peuvent vraisemblablement
provoquer une dépression du système nerveux central susceptible
d'évoluer vers le coma. En se basant sur la toxicité aiguë de ce
composé chez l'animal, on pense que les effets toxiques du
chlorure de vinylidène peuvent se manifester au niveau du foie, des
reins ou des poumons bien en dessous du seuil olfactif minimum qui
se situe aux environs de 2000 mg/m3. L'exposition au chlorure de
vinylidène peut provoquer une irritation des yeux, des voies
respiratoires supérieures (à la concentration de 100 mg/m3 chez
l'homme), et de la peau, encore que cet effet irritant soit,
semble-t-il, dû en partie au para-méthoxyphénol utilisé comme
stabilisateur.
Chez la souris, qui est plus sensible que le rat aux effets
hépatotoxiques et néphrotoxiques du chlorure de vinylidène, on a
constaté une atteinte rénale après exposition à des concentrations
ne dépassant pas 40 mg de chlorure de vinylidène par m3 (soit 10
ppm) pendant 6 heures. On a également observé une hépatotoxicité
et une néphrotoxicité notables chez le rat. Lorsque les animaux
sont à jeun, ce qui a pour effet d'exacerber la toxicité,
l'exposition au chlorure de vinylidène à des teneurs de 600 mg/m3
(150 ppm) et de 800 mg/m3 (200 ppm) pendant 6 heures a provoqué
chez le rat des effets toxiques, respectivement au niveau du foie
et du rein. Des études sur le rat ont montré que l'ingestion
d'alcool avant l'exposition peut stimuler le métabolisme et
exacerber la toxicité du chlorure de vinylidène. La toxicité agiuë
dépend de l'espèce, du sexe, de la souche et de l'état alimentaire
de l'animal. Chez le rat et la souris, les différences de
sensibilité interspécifiques sont liées à l'activité du métabolisme
oxydatif. S'il n'est pas possible de déterminer qui du rat ou de
la souris constitue le meilleur modèle pour l'être humain, toujours
est-il que le métabolisme des microsomes hépatiques est, chez
l'homme, quantitativement analogue à celui du rat, espèce
relativement peu sensible aux effets du chlorure de vinylidène.
Rien n'indique qu'il existe une différence de nature entre l'homme
et les rongeurs pour ce qui est du métabolisme oxidatif du chlorure
de vinylidène.
Il semblerait que la marge entre la concentration toxique chez
l'animal (40 mg/m3 pour la souris) et les limites d'exposition
professionnelle fixées par certains pays puisse être insuffisante,
voire nulle.
2.2.3 Effets à long terme et génotoxicité
Une exposition prolongée ou des expositions de courte durée
répétées à des doses infra-anesthésiques peuvent provoquer des
lésions rénales ou hépatiques. Sur la base d'études à long terme
chez l'animal, dans des conditions reproduisant une exposition
professionnelle, on a observé chez le rat, des altérations au
niveau du foie à une dose de 300 mg/m3 (75 ppm). Chez la souris,
des lésions rénales et hépatiques ont été observées respectivement
à 100 mg/m3 (25 ppm) et 200 mg/m3 (50 ppm). La sensibilité aux
effets toxiques observée au cours des différentes études présente
des variations considérables.
Le chlorure de vinylidène n'affecte pas, semble-t-il, la
capacité de reproduction chez l'animal et ne semble pas non plus
comporter de risques d'embryotoxicité ou de tératogénicité aux
doses inférieures à celles qui seraient toxiques pour la mère, mais
on n'a pas effectué d'études de ce type chez l'homme. Aux doses
toxiques pour la mère-à en juger par une réduction du gain de
poids-on a observé des effets toxiques sur l'embryon et le foetus
et des anomalies foetales.
Le chlorure de vinylidène est mutagène pour les bactéries et
les levures en présence d'un système métabolique de mammifère.
Certaines cellules mammaliennes se révèlent également sensibles in
vitro aux effets mutagènes et aux lésions de l'ADN. La plupart des
études in vivo effectuées sur des rats n'ont pas permis d'observer
d'effets génotoxiques manifestes, à en juger par les tests de
létalité dominante et certains critères cytogénétiques mais on a
tout de même signalé la présence d'aberrations chromosomiques dans
les cellules de la moelle osseuse de hamsters chinois. La liaison
à l'ADN et sa réparation sont décelables in vivo chez les rongeurs
mais en proportion minime. Les études génétiques in vivo incitent
donc à penser qu'il existe une certaine toxicité génique mais, dans
la majorité des cas, il s'agit d'effets non décelables ou minimes.
Plusieurs épreuves de cancérogénicité ont été effectuées sur
trois espèces d'animaux d'expérience (souris, rats et hamsters)
selon différentes voies d'administration. Malheureusement, la
plupart de ces études laissent beaucoup à désirer tant au niveau de
leur conception que de l'évaluation du risque cancérogène. Aucun
effet cancérogène significatif n'a été observé chez des rats qui
recevaient ce composé par la voie orale. Chez des rats adultes
exposés par inhalation, on a signalé une augmentation de la
fréquence des tumeurs mammaires qui n'était toutefois pas liée à la
dose. Une légère augmentation de la fréquence des leucémies a été
également observée, lorsque les rats étaient exposés in utero ou
après leur naissance. Ces observations n'ont pas pu être évaluées.
Lors d'une étude sur la souris, on a observé chez les mâles une
augmentation de l'incidence des adénocarcinome du rein aux doses
de 200 et 100 mg/m3 (50 et 25 ppm), mais aucun effet de ce genre
aux doses de 40 et 0 mg/m3 (10 et 0 ppm). Au cours de la même
étude, une augmentation statistiquement significative de
l'incidence des tumeurs pulmonaires (essentiellement des adénomes
chez les deux sexes) et des carcinomes mammaires (chez les
femelles) a été observée, sans toutefois qu'il y ait de relation
dose-réponse.
Il est possible que ces tumeurs rénales soient liées d'une
manière ou d'une autre à la néphrotoxicité observée et que des
atteintes rénales répétées puissent conduire directement à une
réaction cancérogène selon un mécanisme non-génotoxique ou qu'elle
facilite l'expression de l'activité génotoxique de certains
métabolites chez cette espèce, pour ce sexe et au niveau de cet
organe. Toutefois, cette conclusion reste hypothétique du fait que
l'on ne possède pas suffisamment de résultats sur la relation dose-
réponse pour ce qui est des effets génétiques in vivo; en outre, le
chlorure de vinylidène a pu jouer le rôle d'initiateur lors d'un
test cutané en deux phases sur la souris.
Les études épidémiologiques ne fournissent pas de résultats
statistiquement significatifs qui puissent permettre de conclure à
un accroissement du risque de cancer à la suite d'une exposition
professionnelle au chlorure de vinylidène, toutefois ces études
présentent des insuffisances telles qu'on ne peut procéder à une
évaluation convenable du risque de cancérogénicité pour l'homme.
Même si les estimations effectuées par divers auteurs écartent
l'idée d'une surmortalité par cancer en arguant qu'il s'agit d'une
pure coincidence(en raison du petit nombre de cas et du faible
effectif des cohortes), il n'est pas inutile de préciser que les
résultats obtenus sont systématiquement supérieurs aux prévisions.
Ainsi, dans les deux études de cohorte dont il est fait état, on a
observé un cancer du poumon dans 7 cas, alors qu'on aurait dû avoir
3,16 décès. On ne peut pas écarter ce résultat, mais il ne faut pas
oublier l'existence, dans une étude, d'une exposition concomitante
au chlorure de vinylidène. Etant donné que les cohortes ont été
constituées sur la base d'une exposition au chlorure de vinylidène,
on peut se trouver dans l'impossibilité d'éliminer d'autres
expositions parasites.
Les données de morbidité indiquées (y compris un cas de cancer
du testicule) ne sont pas dénuées d'intérêt. Selon les auteurs, la
forte morbidité hépatique serait imputable à la consommation
d'alcool. Cette hypothèse ne tient pas, puisque la consommation
d'alcool de l'ensemble des personnes étudiées (pas seulement les
cas identifiés) n'a pas été évaluée.
3. Recommandations
3.1 Recommandations en vue de travaux futurs
Il faudrait disposer d'une meilleure estimation de la production
annuelle mondiale de chlorure de vinylidène ainsi que des quantités
de cette substance qui pénètrent dans l'environnement à partir de
l'ensemble des sources de pollution, que le composé soit libéré tel
quel ou qu'il résulte de la décomposition d'autres produits
chimiques.
Les prévisions relatives à sa destiné dans l'environnement
reposent sur un nombre limité de données expérimentales. Il
faudrait de nouvelles données sur les produits de dégradation et de
transformation de ce composé dans l'air, le sol, l'eau et les
sédiments ainsi que sur son métabolisme chez des espèces non-
mammaliennes représentatives.
Il conviendrait d'effectuer des études de toxicité à long terme
chez diverses espèces aquatiques représentatives (poissons,
crustacés et mollusques), selon divers critères pathologiques.
Il faut également définir de manière plus précise, afin
d'établir des critères de sécurité en matière d'exposition, quels
sont, chez l'animal et chez l'homme, les mécanismes des effets
toxiques résultant d'une exposition de brève ou prolongée au
chlorure de vinylidène.
Il faudrait exploiter de manière plus complète les données
existantes sur la cancérogénicité. Si l'on envisage d'autres études
de cancérogénicité, elles devront être menées selon un protocole
expérimental reconnu, pendant toute l'existence des sujets, ce
protocole étant conçu de manière à tenir compte des propriétés
particulières du chlorure de vinylidène. Ces études doivent
notamment prendre en considération la courte demi-vie du produit
dans l'organisme, l'importance de l'âge au début de l'exposition,
la durée de l'exposition quotidienne et autres données susceptibles
d'aider à la détermination des doses à administrer. Les espèces et
les souches d'animaux de laboratoire devront être soigneusement
sélectionnées. Il serait également très utile de disposer de
données de toxicité ainsi que de données métaboliques et pharmaco-
cinétiques sur ces animaux.
Il faudrait effectuer des études longitudinales à long terme
sur la morbidité et la mortalité au sein de populations prises au
hasard et qui sont exposées au chlorure de vinylidène.
Des études épidémiologiques sont nécessaires pour permettre
l'évaluation des effets de l'exposition au chlorure de vinylidène
(notamment une exposition prolongée à de faibles doses) dans les
populations humaines. Il est tout particulièrement important de
disposer de données sur des effets tels que les affections
cérébrovasculaires prématurées et le cancer. En outre, les études
qui seront effectuées devront tenir dûment compte de facteurs de
confusion tels que le tabagisme et la consommation d'alcool
(éventuellement selon un système cas/témoin).
Afin d'apprécier l'effet de l'action réglementaire menée au
cours des dernières années, il conviendrait de confronter les
résultats des études en cours aux données rétrospectives.
Pour résoudre le problème que posent les faibles effectifs du
personnel sur les lieux de production, on pourrait, pour les
investigations en cours comme pour les investigations futures,
recourir à des études multicentriques avec regroupement des
données. Il faudra également étudier sur l'animal d'expérience si
la N- acétylcystéine, un agent sulfhydrilé présente un intérêt
pour le traitement des intoxications par le chlorure de vinylidène.
Il est nécessaire de comparer la pharmacocinétique et le
métabolisme du chlorure de vinylidène tant in vivo qu' in vitro
spécialement au niveau du rein, du foie et des poumons chez
diverses espèces d'animaux d'expérience et chez l'homme, afin de
pouvoir mieux interpréter les résultats fournis par les études de
toxicité in vivo. Il faut également obtenir des données sur la
génotoxicité potentielle du chlorure de vinylidène au site de la
cancérogénèse, parallèlement sur plusieurs espèces, afin de voir si
un mécanisme génétique est en cause.
Compte tenu des observations neurotoxicologiques dont il est
fait état dans la présente analyse, il paraît nécessaire d'étudier
le rôle des systèmes de modulation dans la pathogénèse de
l'intoxication par le chlorure de vinylidène.
3.2 Protection personnelle et traitement des intoxications
3.2.1 Protection personnelle
Dans l'industrie, où peuvent se produire des expositions de
brèves durées par inhalation au dessus des limites recommandées, il
conviendrait d'utiliser des masques faciaux avec cartouche
filtrante pour se protéger des vapeurs organiques et, si nécessaire
en cas d'urgence, des masques respiratoires avec arrivée d'air. Les
personnes qui manipulent du chlorure de vinylidène devront porter
des vêtements protecteurs ainsi que des lunettes spéciales; cet
équipement devra être correctement entretenu afin de protéger le
corps contre tout contact. Dans les ateliers, on assurera une
ventilation permanente et l'on disposera des grilles d'aération
munies de filtres là où des déversements accidentels ou des fuites
risquent de se produire. Il est recommandé de surveiller les
émissions de chlorure de vinylidène au cours des opérations de
remplissage. En cas de fuite, on procédera à l'évaporation directe
du produit s'il s'agit d'une fuite mineure ou à son évaporation
controlée en présence d'une mousse synthétique s'il s'agit d'une
fuite plus importante. On pourra disperser la vapeur de chlorure de
vinylidène à partir de cette mousse par pulvérisation d'eau.
3.2.2 Traitement des intoxications humaines
En cas d'exposition excessive ou d'ingestion, il faut
s'adresser à un médecin. Il faut veiller particulièrement aux
poumons, à la peau et aux yeux du fait des propriétés irritantes du
chlorure de vinylidène. Il importe de surveiller les fonctions
cardiaque, hépatique et rénale ainsi que le système nerveux
central. Les données obtenues sur l'animal d'expérience montrent
que le chlorure de vinylidène accroît notablement la sensibilité
aux arythmies cardiaques induites par l'adrénaline, de sorte que ce
produit est à éviter. En cas d'hypotension grave, on pourra
procéder à une transfusion de sang total ou d'un succédané du
plasma. Il n'existe aucun antidote.
En cas d'intoxication par inhalation de chlorure de vinylidène,
il faut maintenir le malade à l'air libre en semi-décubitus
ventral. On dégagera les voies aériennes et l'on placera le malade
sous oxygène en cas de stupeur ou de coma. Si nécessaire, on
procédera à la respiration artificielle.
En cas d'ingestion de chlorure de vinylidène, rincer la bouche
avec de l'eau. Ne pas faire vomir le malade car il y a risque
d'aspiration du chlorure de vinylidène dans le larynx et les
poumons. Un lavage d'estomac ou l'administration par voie orale de
charbon actif ou de paraffine liquide peut contribuer à réduire la
biodisponibilité du chlorure de vinylidène si on procède à ce
traitement dans l'heure qui suit l'ingestion, l'effet bénéfique
étant encore sensible aprés 4 heures.
Si les yeux ont été atteints par du chlorure de vinylidène, les
rincer immédiatement à l'eau pendant plus de 15 minutes et
consulter un médecin.
En cas d'exposition cutanée, ôter les vêtements souillés et
laver la peau à l'eau et au savon.
RESUMEN Y CONCLUSIONES, EVALUACION Y RECOMENDACIONES
1. Resumen y conclusiones
1.1. Propiedades, usos y métodos analíticos
El cloruro de vinilideno (C2H2Cl2) es un líquido volátil e
incoloro con un olor dulzón. Se estabiliza con p- metoxifenol para
impedir la formación de peróxidos explosivos. El cloruro de
vinilideno se usa para producir 1,1,1-tricloroetano y para formar
fibras modacrílicas y copolímeros (con cloruro de vinilo o
acrilonitrilo). Se han puesto a punto métodos de cromatografía de
gases para analizar el cloruro de vinilideno en el aire, el agua,
las películas para envoltorios, los tejidos orgánicos, los
alimentos y el suelo. El método más sensible de detección es la
captura electrónica.
1.2. Fuentes y niveles de exposición
Todos los años ingresa en la atmósfera alrededor del 5% del
cloruro de vinilideno producido (que representa un máximo cercano a
23 000 toneladas). La elevada presión de vapor y la baja
solubilidad en agua favorecen concentraciones atmosféricas
relativamente elevadas en comparación con otros "compartimentos"
ambientales. Se cree que el cloruro de vinilideno tiene una
semivida en la atmósfera de aproximadamente dos días.
Los niveles ambientales en el agua son sumamente bajos. Incluso
en aguas residuales industriales sin tratar, las concentraciones
pasan raras veces del orden de los µg/litro, lo que está muy por
debajo del margen de mg/litro de toxicidad para los organismos
acuáticos. Los niveles en el agua de bebida sin tratar no suelen
ser detectables. En las aguas potables tratadas, se ha encontrado
que el nivel de cloruro de vinilideno suele ser < de 1 µg/litro,
si bien se han encontrado muestras que contienen hasta 20 µg/litro.
Los niveles de cloruro de vinilideno en los alimentos normalmente
no son detectables, siendo la concentración máxima observada de 10
µg/kg.
La exposición profesional al cloruro de vinilideno se da
principialmente por inhalación, aunque también puede producirse
contaminación por la piel o los ojos. Según los países, el límite
de exposición máximo recomendado o promedio ponderado en función
del tiempo se encuentra entre 8 y 500 mg/m3, o es la concentración
más baja detectable con cierto margen de confianza. Los límites de
exposición a corto plazo varían entre 16 y 80 mg/m3 y los valores
máximos varían entre 50 y 700 mg/m3.
1.3. Absorción, distribución, metabolismo y excreción
El cloruro de vinilideno puede absorberse rápidamente por la
vía respiratoria y oral en mamíferos; no se dispone de datos sobre
la absorción cutánea. El cloruro de vinilideno se distribuye por
todo el organismo del roedor y alcanza concentraciones máximas en
el hígado y el riñón. La eliminación pulmonar de cloruro de
vinilideno sin modificar es cuando menos bifásica y dependiente de
la dosis, siendo de mayor importancia en el caso de dosis que
saturan el metabolismo (>unos 600 mg/m3 (<150 ppm) por inhalación
en la rata). En la rata sometida a ayuno se observó una reducción
en el metabolismo de la dosis oral y un nivel consiguiente mayor de
cloruro de vinilideno exhalado.
Se han caracterizado las principales vías metabólicas en la
rata. El metabolismo predominante de la fase I entraña la
participación del citocromo P-450 y la formación (posible pero no
necesariamente por medio de un epóxido) de ácido monocloroacético.
El cloruro de vinilideno puede inducir actividad de citocromo P-
450. Varios metabolitos de la fase I se conjugan con glutatión y/o
con fosfatidil etanolamina antes de sufrir ulteriores
modificaciones. El metabolismo es más rápido en el ratón que en la
rata, lo que origina un perfil metabólico semejante con una
proporción relativamente mayor de derivados del glutatión por
conjugación. Se ha demostrado que el citocromo P-450 de microsomas
humanos también metaboliza el cloruro de vinilideno.
El metabolismo del cloruro de vinilideno en roedores lleva al
agotamiento de las reservas de glutatión y a la inhibición de la
actividad de la glutatión- S- transferasa.
1.4. Efectos en animales de experimentación y sistemas celulares
1.4.1 Enlaces covalentes en tejidos
Los marcadores radiactivos derivados del [14C]-cloruro de
vinilideno forman enlaces covalentes en el hígado, el riñón y el
pulmón de los roedores, lo que va asociado a toxicidad en esos
órganos. El enlace covalente y la toxicidad se agravan con el
agotamiento del glutatión y se producen en el hígado y el riñón a
dosis inferiores en ratones que en ratas. Se ha observado in vitro
que varios metabolitos del cloruro de vinilideno establecen enlaces
covalentes con tioles.
1.4.2 Toxicidad aguda
Aunque las estimaciones de la CL50 aguda para el cloruro de
vinilideno varían considerablemente, esta variación no enmascara el
hecho de que el ratón es mucho más sensible a la sustancia que la
rata o el criceto. Los valores estimados de la CL50 a las 4 h
variaron desde aproximadamente 8000 hasta 128 000 mg/m3 (2000-32
000 ppm) en ratas alimentadas, 460-820 mg/m3 (115-205 ppm) en
ratones alimentados y 6640-11 780 mg/m3 (1660-2945 ppm) en cricetos
alimentados. Pueden existir imprecisiones en los cálculos de la
CL50 debido a que la relación concentración-mortalidad no es de
carácter lineal. En todas las especies, los machos parecían tener
valores de CL50 más bajos que las hembras, y el ayuno (que agota
las reservas de glutatión) aumentó la toxicidad en las tres
especies. Tras la administración oral, los valores de la DL50
fueron aproximadamente 1500 y 200 mg/kg en ratas y ratones
alimentados, respectivamente. La toxicidad aguda por inhalación en
animales de laboratorio se manifestó en forma de irritación de las
mucosas, depresión del sistema nervioso central y cardiotoxicidad
progresiva (bradicardia sinusal y arritmias). Se observaron
lesiones en el hígado, el riñón y el pulmón. En el ratón, que es
más sensible que la rata a la toxicidad hepática y renal del
cloruro de vinilideno, la exposición a dosis tan reducidas como 40
mg de cloruro de vinilideno/m3 (10 ppm) durante 6 h indujo lesiones
renales y aumento de la replicación del ADN. Como en el caso de la
inhalación, los principales órganos afectados por la ingestión de
cloruro de vinilideno son el hígado, el riñón y el pulmón. La
cadena de procesos que llevan a la hepatotoxicidad parece comenzar
por un cambio temprano en los canalículos biliares, que se ve
seguido por síntomas de lesiones mitocondriales. A continuación se
producen lesiones en el retículo endoplasmático y la muerte
celular. La toxicidad hepática y renal inducida por el cloruro de
vinilideno no parece estar causada por peroxidación lipídica. El
aumento de las concentraciones intracelulares de Ca++ puede ser en
parte responsable de la toxicidad para el hepatocito.
Los efectos tóxicos del cloruro de vinilideno dependen, al
menos parcialmente, de la actividad del citocromo P-450 (que
también puede participar en la detoxificación) y pueden agravarse
por el agotamiento de las reservas de glutatión. La hepatotoxicidad
puede ser intensificada por el etanol y la tiroxina, inhibida por
el ditiocarbo y la (+)-catequina y modulada por la acetona.
1.4.3 Estudios a corto plazo
En estudios a corto plazo se han observado lesiones hepáticas,
renales y, en menor grado, pulmonares en roedores expuestos por
inhalación al cloruro de vinilideno a una concentración de 40-800
mg/m3 durante 4-8 h/día, 4 o más días/semana. El ratón demostró ser
más sensible que la rata, el cobayo, el conejo, el perro y el mono
ardilla, y la toxicidad fue distinta de unas familias de ratones a
otras. En general, las hembras eran menos sensibles que los machos.
Se ha comunicado la aparición de hepatotoxicidad en la rata y el
ratón expuestos intermitentemente a concentraciones de cloruro de
vinilideno >800 mg/m3 (>200 ppm) y 220 mg/m3 (55 ppm),
respectivamente. Los niveles necesarios para producir
hepatotoxicidad por exposición continua durante varios días fueron
240 mg/m3 (60 ppm) para la rata y 60 mg/m3 (15 ppm) para el ratón.
Estos tratamientos intermitentes y continuos también provocaron
nefrotoxicidad en el ratón. El ratón suizo macho resultó ser
especialmente propenso a la toxicidad renal inducida por cloruro de
vinilideno. El ratón macho no sobrevivió a una exposición continua
a corto plazo a 200 mg de cloruro de vinilideno/m3 (50 ppm). El
nivel aparente de no observación de efectos de hepatotoxicidad en
el perro, el mono ardilla y la rata fue de aproximadamente 80 mg/m3
(20 ppm) administrados en exposición continua durante 90 días. En
estudios de dosificación oral a corto plazo (aproximadamente 3
meses) en la rata (hasta 20 mg/kg diarios) y el perro (hasta 25
mg/kg diarios) no se observó prueba alguna de toxicidad aparte de
una mínima lesión hepática reversible en la rata.
1.4.4 Estudios a largo plazo
Los estudios a largo plazo de exposición intermitente al
cloruro de vinilideno por inhalación revelaron que 300 mg/m3 (75
ppm) sólo causaban ligeras lesiones hepáticas reversibles en la
rata. A 600 mg/m3 (150 ppm), la dosis más alta de exposición a
largo plazo que puede tolerar la rata, se apreció lesión hepática
con necrosis. En el ratón se observó una elevada tasa de mortalidad
con signos de lesión hepática a 200 mg/m3 (50 ppm). Se observó
toxicidad para el riñón en el tratamiento a largo plazo de ratones
con 100 mg/m3 (25 ppm). La administración de hasta 30 mg/kg al día
de cloruro de vinilideno a la rata durante un año volvió a producir
cambios hepáticos mínimos. A partir de estos datos no se puede
determinar claramente el nivel de no observación de efectos. En
otro estudio se observaron ciertas pruebas de que podía inducirse
inflamación renal y necrosis hepática en la rata y el ratón,
respectivamente, tras la administración oral a largo plazo de
cloruro de vinilideno a dosis diarias de 5 mg/kg y 2 mg/kg,
respectivamente.
1.4.5 Genotoxicidad y carcinogenicidad
Se observó que el cloruro de vinilideno es mutagénico para
bacterias y levaduras sólo en presencia de un sistema de activación
metabólica de microsomas de mamíferos (S9). El compuesto indujo
síntesis no programada de ADN en hepatocitos aislados de rata y
aumentó la frecuencia de intercambio de cromátidas hermanas y de
aberraciones cromosómicas en cultivos celulares con S9. En cambio,
no se observó aumento en la mutación de genes de mamíferos. Se ha
comunicado un aumento pequeño pero estadísticamente significativo
del enlace al ADN después de la exposición in vivo. El enlace al
ADN fue más frecuente en células de ratón que de rata y mayor en el
riñón que en el hígado tras exposiciones de 6 h a 40 y 200 mg de
cloruro de vinilideno/m3 (10 y 50 ppm). Además, el cloruro de
vinilideno aumentaba ligeramente la síntesis no programada de ADN
en el riñón de ratón. No se observó prueba alguna de efectos
letales dominantes o de efectos citogenéticos tras la exposición in
vivo de roedores a excepción de un estudio que demuestra la
inducción de aberraciones cromosómicas en la médula ósea del
criceto chino.
Se han llevado a cabo estudios de carcinogenicidad en tres
especies animales (rata, ratón y criceto). En el ratón suizo macho,
se vieron signos claros de carcinogenicidad (adenocarcinoma del
riñón) tras una exposición intermitente a largo plazo a 100 ó 200
mg de cloruro de vinilideno/m3 (25 ó 50 ppm) pero no a 0 ó 40 mg/m3
(0 ó 10 ppm).
Los tumores de riñón pueden guardar alguna relación con la
citotoxicidad renal observada y es posible que la lesión renal
repetida lleve directamente a la respuesta carcinogénica por un
mecanismo no genotóxico o bien que facilite la expresión del
potencial genotóxico de los metabolitos en esta especie, sexo y
órgano en concreto. No obstante, esta conclusión es dudosa a la luz
de los escasos datos disponibles sobre los efectos genéticos in
vivo y el descubrimiento de que el cloruro de vinilideno podía
haber actuado como iniciador.
En el mismo estudio, se observaron incidencias estadísticamente
mayores de tumor del pulmón (principalmente adenomas en el ratón de
ambos sexos) y carcinomas mamarios (en hembras), pero no se
encontraron relaciones dosis-respuesta. En rata adulta expuesta por
inhalación, se comunicó un ligero aumento independiente de la dosis
de tumores de la mama, así como un pequeño aumento de la leucemia
cuando se exponía a la rata in utero y recién nacida. Estas
observaciones no pudieron evaluarse.
1.4.6 Efectos sobre la reproducción
No se encontró efecto alguno sobre la fecundidad de la rata
continuamente expuesta al cloruro de vinilideno (hasta 200
mg/litro, 200 ppm) en el agua de bebida. La inhalación de hasta
1200 mg de cloruro de vinilideno/m3 (300 ppm), durante 22-23 horas,
por la rata y el ratón durante diversos periodos de la
organogénesis no indujo anomalías fetales que no pudieran
atribuirse a la toxicidad materna.
La inhalación de hasta 640 mg de cloruro de vinilideno/m3 (160
ppm) durante 7 h/día en ratas y conejos o la ingestión de
aproximadamente 40 mg/kg al día en la rata durante periodos
críticos de la gestación no ejercieron efecto alguno sobre los
embriones o los fetos en niveles inferiores al que produce la
toxicidad materna, pero se observaron toxicidad embrionaria y fetal
y anomalías fetales cuando se alcanzó el nivel que produce
toxicidad en la madre, como lo demostró la menor velocidad de
aumento de peso.
1.5. Efectos en el hombre
Una concentración de cloruro de vinilideno de 16 000 mg/m3
(4000 ppm) provoca una intoxicación que puede llevar a la pérdida
del conocimiento. El cloruro de vinilideno estabilizado es también
irritante para el tracto respiratorio, los ojos y la piel. Se han
comunicado lesiones renales y hepáticas correspondientes a
exposiciones subanestésicas, prolongadas o repetidas a corto plazo.
La evaluación de los estudios epidemiológicos se vio dificultada
por el pequeño tamaño de las cohortes, la coexposición a cloruro de
vinilo y la insuficiente atención al hábito de fumar. Aunque no se
encontró una incidencia mayor en grado estadísticamente
significativo del cáncer en el hombre expuesto al cloruro de
vinilideno, los estudios epidemiológicos fueron insuficientes y no
se puede concluir que no entrañe un riesgo carcinogénico. No se
dispone de información sobre los efectos del cloruro de vinilideno
en la reproducción humana.
2. Evaluación de los efectos en el medio ambiente y riesgos
para la salud humana
2.1. Evaluación de los efectos en el medio ambiente
A consecuencia de la volatilización, la atmósfera constituye el
principal compartimento ambiental del cloruro de vinilideno. Puesto
que la semivida de este compuesto en la troposfera es de unos dos
días aproximadamente, parece poco probable que el cloruro de
vinilideno contribuya a agotar la capa de ozono de la estratosfera.
La lixiviación y la volatilización hacen que el suelo y los
sedimentos sean compartimentos ambientales de menor importancia
para el cloruro de vinilideno; el nivel de este hidrocarburo
clorado en el medio acuoso se ve también reducido al mínimo por la
rápida volatilización. No se sabe si la degradación de compuestos
como el tricloroetileno y el percloroetileno, que a menudo se
encuentran en el agua, contribuye a aumentar apreciablemente los
niveles de cloruro de vinilideno en el medio ambiente.
Las concentraciones de cloruro de vinilideno que se observan en
las colecciones naturales de agua y los niveles de toxicidad aguda
para peces y Daphnia indican que los riesgos de toxicidad aguda
para el medio acuático son mínimos. No se dispone de suficientes
datos sobre toxicidad a largo plazo para evaluar los efectos
subletales sobre los organismos acuáticos que viven en las
proximidades de fuentes importantes de contaminación por cloruro de
vinilideno, como aguas subterráneas contaminadas y puntos de
vertido municipal e industrial.
2.2. Evaluación de los riesgos para la salud humana
2.2.1 Niveles de exposición
La población general está expuesta a niveles muy bajos de
cloruro de vinilideno. El nivel máximo notificado en el agua de
bebida es de 20 µg/litro, si bien se ha calculado que, en los
Estados Unidos de América, la exposición individual diaria del
ciudadano medio a través del agua de bebida es de <0,01 µg. Los
niveles de cloruro de vinilideno en los alimentos no suelen ser
detectables; no se han notificado niveles superiores a 10 µg/kg.
Los niveles en alimentos derivados de organismos acuáticos se
desconocen, pero es probable que sean insignificantes (sección
10.1). Se han comunicado niveles de cloruro de vinilideno en el
aire de hasta 52 µg/m3 (en el perímetro de una zona industrial). En
los Estados Unidos se han comunicado valores medios de
concentración en el aire urbano de 20 ng/m3 en zonas no
industriales y 8,7 µg/m3 en zonas industriales.
La exposición profesional tiene lugar especialmente en los
procesos de producción y polimerización. La respiración es la vía
principal de entrada en el organismo y los límites de exposición
máximos recomendados o promedios regulados a lo largo de un día de
trabajo varían entre 8 y 500 mg/m3 (o la concentración más baja
detectable con un margen de confianza), según el país. Los límites
de exposición a corto plazo varían entre 16 y 80 mg/m3 y los
valores máximos entre 50 y 700 mg/m3. Se ha encontrado que los
niveles de cloruro de vinilideno en las atmósferas cerradas a las
que algunos trabajadores se ven expuestos no superan los 8 mg/m3.
2.2.2 Efectos agudos
En el ser humano, es probable que la inhalación de
concentraciones elevadas de cloruro de vinilideno (muy
aproximadamente iguales o superiores al umbral olfativo máximo de
4000 mg/m3) provoque depresión del sistema nervioso central y pueda
llevar al coma. Basándose en la toxicidad aguda para animales, el
cloruro de vinilideno puede ejercer efectos tóxicos en el hígado,
el riñón o el pulmón a concentraciones muy inferiores al umbral
olfativo mínimo de aproximadamente 2000 mg/m3. La exposición al
cloruro de vinilideno puede producir irritación en los ojos, el
tracto respiratorio superior (a 100 mg/m3 en el hombre, Rylova
1953) y la piel, lo cual se ha atribuido en parte a un agente
estabilizante, el p- metoxifenol.
En el ratón, más sensible que la rata a los efectos
hapatotóxicos y nefrotóxicos del cloruro de vinilideno, se
indujeron lesiones renales por exposición a cantidades tan
reducidas como 40 mg de cloruro de vinilideno/m3 (10 ppm) durante 6
h. También se observaron hepatotoxicidad y nefrotoxicidad notables
en la rata. Tras el ayuno, que aumenta la toxicidad, la exposición
de la rata a concentraciones de cloruro de vinilideno de 600 mg/m3
(150 ppm) y 800 mg/m3 (200 ppm) durante 6 h provocó toxicidad en el
hígado y el riñón, respectivamente. Los estudios realizados en la
rata indican que la ingestión de alcohol antes de la exposición
puede acelerar el metabolismo y exacerbar la toxicidad del cloruro
de vinilideno. La toxicidad aguda depende de la especie, el sexo,
la estirpe y el régimen de alimentación de los animales. La
distinta sensibilidad del ratón y la rata guarda relación con la
diferente actividad del metabolismo oxidativo del cloruro de
vinilideno en una y otra especie. Aunque no se puede predecir si la
rata o el ratón constituyen el modelo más adecuado para el ser
humano, la actividad del metabolismo microsómico hepático del
hombre es cuantitativamente semejante al de la rata, cuya
susceptibilidad es relativamente baja. No hay pruebas de que
existan diferencias cualitativas en el metabolismo oxidativo del
cloruro de vinilideno en el ser humano y el roedor.
Está claro que el margen entre las concentraciones capaces de
producir toxicidad en animales (40 mg/m3 en el ratón) y los límites
de exposición profesional establecidos por algunos países es
insuficiente o inexistente.
2.2.3 Efectos a largo plazo y genotoxicidad
La exposición prolongada o repetida a corto plazo a dosis
subanestésicas puede producir lesiones renales y hepáticas.
Basándose en estudios a largo plazo realizados en animales, en
condiciones que simulaban la exposición profesional, se comunicó la
aparición de cambios hepáticos a un nivel de exposición de 300
mg/m3 (75 ppm) en la rata. En el ratón, se observaron lesiones en
el riñón y el hígado con 100 mg/m3 (25 ppm) y 200 mg/m3 (50 ppm),
respectivamente. Los datos sobre sensibilidad a los efectos tóxicos
varían considerablemente de unos estudios a otros.
En los animales, el cloruro de vinilideno no parece influir en
la capacidad reproductiva ni constituir un riesgo embriotóxico o
teratogénico a dosis inferiores a las que producen toxicidad
materna, pero este extremo no se ha estudiado en el hombre. Cuando
se utilizaron concentraciones capaces de producir toxicidad materna
se observaron toxicidad embrionaria y fetal y anomalías fetales,
reflejadas en la menor velocidad de aumento de peso.
El cloruro de vinilideno tiene efecto mutagénico en las
bacterias y las levaduras siempre que actúe en presencia de un
sistema metabólico de mamíferos. También algunas células de
mamíferos pueden sufrir lesiones del ADN y efectos mutagénicos in
vitro . En la mayoría de los estudios realizados en roedores in
vivo no se observaron efectos genotóxicos medidos por la letalidad
dominante ni desde el punto de vista citogenético, pero se ha
comunicado la observación de aberraciones en células de la médula
ósea del hámster chino. El enlace al ADN y la reparación de éste in
vivo en roedores fueron detectables pero mínimos. Así pues, los
estudios genéticos in vivo sugieren algunos signos de toxicidad
genética, pero, en la mayoría de los casos, los efectos fueron
mínimos o negativos.
Se han llevado a cabo varios ensayos de carcinogenicidad en
tres especies de animales de experimentación (ratones, ratas y
hámsters) utilizando diversas vías de administración.
Lamentablemente, la mayoría de estos estudios adolecían de graves
limitaciones de diseño o de método para la evaluación de la
carcinogenicidad. Por vía oral no se observaron efectos
carcinogénicos significativos en la rata. En la rata adulta
expuesta por inhalación, se notificó un aumento de los tumores de
la mama que no guardaba relación con la dosis. Se observó un ligero
aumento de la leucemia en las ratas expuestas tanto in utero como
recién nacidas. Estas observaciones no pudieron evaluarse. En un
estudio realizado en el ratón, se observó una mayor incidencia de
adenocarcinomas de riñón en los machos con niveles de exposición de
200 y 100 mg/m3 (50 y 25 ppm) pero no con 40 y 0 mg/m3 (10 y 0
ppm). En el mismo estudio, se observaron incidencias
estadísticamente mayores de tumores del pulmón (principalmente
adenomas en ambos sexos) y carcinomas mamarios (en hembras), pero
no se descubrieron relaciones entre la dosis y la respuesta.
Los tumores del riñón pueden estar relacionados de algún modo
con la citotoxicidad renal observada; puede ser que las lesiones
repetidas del riñón lleven directamente a la respuesta
carcinogénica por medio de un mecanismo no genotóxico o bien que
faciliten la expresión del potencial genotóxico de los metabolitos
en esta especie, este sexo y este órgano en particular. No
obstante, esta conclusión es dudosa en la ausencia de datos de
dosis-respuesta suficientes sobre los efectos genéticos in vivo,
así como ante el descubrimiento de que el cloruro de vinilideno
puede haber actuado como iniciador en un ensayo cutáneo en dos
etapas en el ratón.
Los estudios epidemiológicos, aunque no dan pruebas
estadísticamente significativas de que la exposición profesional al
cloruro de vinilideno entrañe un riesgo mayor de cáncer no son
adecuados para evaluar debidamente el riesgo carcinogénico del
cloruro de vinilideno para el ser humano.
Aunque en las evaluaciones de algunos autores el exceso de
defunciones por cáncer se atribuye al azar (a causa del reducido
número de sujetos y del tamaño de las cohortes), el hecho de que
aparezcan repetidamente valores más altos de lo esperado es digno
de mención. En los dos estudios de cohortes comunicados, se observó
cáncer del pulmón en 7 casos, cuando cabía esperar 3,16
defunciones. El resultado no puede desecharse, aunque hay que tener
presente la coexistencia de la exposición al cloruro de vinilideno
(en uno de los estudios). Puesto que las cohortes se determinaron
según su exposición al cloruro de vinilideno, puede ser imposible
excluir otras exposiciones que induzcan a error. Las conclusiones
comunicadas en materia de morbilidad (incluido un caso de carcinoma
testicular) pueden ser útiles a título informativo. La
interpretación por parte de los autores de que la mayor morbilidad
hepática guardaba relación con el consumo de alcohol por los
sujetos no es válida, puesto que no se evaluó la ingestión de
alcohol por todos los sujetos del estudio (y no sólo por los casos
identificados).
3. Recomendaciones
3.1 Recomendaciones para trabajos futuros
Es preciso disponer de mejores estimaciones de la producción
mundial anual de cloruro de vinilideno y de las cantidades de
cloruro de vinilideno que ingresan en el medio ambiente de todas
las procedencias, ya sea en forma de cloruro de vinilideno como tal
o por la degradación de otros productos químicos.
El destino ambiental previsto se basa en escasas pruebas
experimentales. Se necesita más información sobre las tasas de
degradación y sobre los productos de transformación en el aire, el
suelo, el agua y los sedimentos, y el metabolismo en especies no
mamíferas representativas.
Deben llevarse a cabo estudios de toxicidad a largo plazo en
los que se investiguen los diversos punto finales patológicos en
especies acuáticas representativas (peces, crustáceos y moluscos).
Deben definirse con más precisión los umbrales y los mecanismos de
los efectos tóxicos que tiene la exposición al cloruro de
vinilideno a corto y a largo plazo en el animal y el ser humano,
como base para establecer niveles seguros de exposición.
Conviene hacer un uso más exhaustivo de los datos existentes en
materia de carcinogenicidad. Los nuevos estudios sobre
carcinogenicidad deben hacerse según un protocolo aceptado de
bioensayo durante un lapso de vida entera que tenga en cuenta
específicamente las propiedades particulares del cloruro de
vinilideno. Esos estudios deben tener presentes la brevedad de la
semivida de la sustancia en el organismo, la importancia de la edad
al comienzo de la exposición, la duración de la exposición diaria y
otros datos pertinentes que puedan estar relacionados con el
establecimiento del régimen de dosificación. Hay que seleccionar
cuidadosamente las especies y estirpes de los animales de
experimentación. Los datos de toxicidad así como los datos
metabólicos y farmacocinéticos correspondientes a estos animales
también serían sumamente útiles.
Deben llevarse a cabo estudios de seguimiento a largo plazo
sobre la morbilidad y la mortalidad en poblaciones enteras y no
seleccionadas expuestas al cloruro de vinilideno.
Se necesitan estudios epidemiológicos que permitan evaluar los
efectos de la exposición al cloruro de vinilideno (incluida la
exposición prolongada a niveles reducidos) en poblaciones humanas.
Es particularmente necesario disponer de información sobre efectos
como la aparición precoz de enfermedades cerebrovasculares y
cáncer; los estudios deben tener en cuenta los factores que
introducen errores, como el hábito de fumar y el consumo de alcohol
(posiblemente en un sistema de referencia de casos).
Debe recurrise a datos históricos como base de comparación con
los resultados de las investigaciones en curso para poder evaluar
los efectos protectores que han tenido las medidas de reglamentación
durante los últimos años.
Para las investigaciones tanto en curso como futuras, un medio
valioso de salvar el problema del reducido número de sujetos que
hay en cada lugar de producción por separado sería aunar todos los
datos de éstos y realizar estudios multicéntricos. Debe
investigarse en animales de experimentación el valor de la
utilización de un agente con grupo sulfhidrilo como la
N- acetilcisteína en el tratamiento de la intoxicación por cloruro
de vinilideno.
Es necesario comparar la farmacocinética y el metabolismo in
vivo/in vitro del cloruro de vinilideno, especialmente en el
riñón, el hígado y el pulmón, en animales de experimentación de
distintas especies y en el ser humano, con el fin de comprender
mejor los resultados obtenidos en estudios de toxicidad in vivo . Se
precisan datos paralelos sobre la genotoxicidad potencial del
cloruro de vinilideno en el lugar escogido para estudiar la
carcinogénesis en distintas especies a fin de examinar el posible
papel de un mecanismo genético.
A la luz de las conclusiones neurotoxicológicas comunicadas en
el presente análisis, es necesario investigar el papel de los
sistemas moduladores en la patogénesis de la intoxicación por
cloruro de vinilideno.
3.2 Protección personal y tratamiento de la intoxicación
3.2.1 Protección personal
En los lugares de trabajo en la industria donde pueden
producirse exposiciones a corto plazo por inhalación superiores a
los límites recomendados, deben utilizarse mascarillas faciales
completas con filtro para los vapores orgánicos y, en previsión de
una emergencia, deben proporcionarse mascarillas con sistema de
abastecimiento de aire. Para evitar el contacto con el cuerpo, los
operarios que manejen cloruro de vinilideno deben llevar ropa
protectora, en buen estado, que incluya gafas de seguridad. Debe
mantenerse una ventilación constante dentro de las plantas
industriales mediante respiraderos con filtros en los lugares donde
puedan producirse derrames o fugas. Se recomienda vigilar las
emisiones de cloruro de vinilideno durante las operaciones de
distribución. En caso de una fuga, debe forzarse la evaporación del
cloruro de vinilideno ya sea directamente si se trata de una
cantidad pequeña o por evaporación controlada utilizando una espuma
sintética de expansión. Para disperar los vapores de la espuma
pueden utilizarse aspersores de agua en cortinas.
3.2.2 Tratamiento de la intoxicación en el hombre
En casos de exposición excesiva o de ingestión, debe
consultarse a un médico. Dadas las propiedades irritantes del
cloruro de vinilideno, debe prestarse especial atención a los
pulmones, la piel y los ojos. Deben vigilarse las funciones del
corazón, el hígado, el riñón y el sistema nervioso central. Puesto
que los datos correspondientes a animales indican que el cloruro de
vinilideno produce un aumento notable de la sensibilidad a las
arritmias cardiacas inducidas por la adrenalina, debe evitarse el
empleo de este fármaco. La hipotensión grave puede tratarse por
transfusión, con sangre entera o sustitutos del plasma. No se
conoce antídoto alguno.
Un paciente intoxicado por inhalación de cloruro de vinilideno
debe mantenerse abrigado en posición semiprona y en una atmósfera
bien ventilada y fresca. Las vías respiratorias deben mantenerse
despejadas y debe administrarse oxígeno si el sujeto se encuentra
en estado de estupor o de coma. En caso necesario debe aplicarse
respiración artificial.
Tras la ingestión de cloruro de vinilideno debe enjuagarse la
boca con agua. No debe provocarse el vómito por el riesgo de
aspiración de cloruro de vinilideno hacia la laringe y los
pulmones. El lavado gástrico y/o la administración oral de carbono
activado o de parafina líquida pueden ayudar a reducir la
biodisponibilidad de cloruro de vinilideno si se administran dentro
de la hora que sigue a la ingestión, pero pueden beneficiar al
paciente hasta 4 horas después de la ingestión.
Los ojos expuestos a cloruro de vinilideno deben irrigarse
inmediatamente con agua durante más de l5 minutos y debe acudirse
al médico.
En caso de exposición cutánea, las ropas contaminadas deben
retirarse y debe lavarse la zona afectada con agua y jabón.
The role of cytochrome P-450 in the metabolism of vinylidene
chloride was confirmed by the measurement of a Type I difference
spectrum with the bound substrate in hepatic microsomes from male
Long-Evans rats [32]. Addition of vinylidene chloride to
microsomes also stimulated carbon monoxide-inhibitable NADPH
oxidation. NADPH was required for the conversion of vinylidene
chloride to monochloroacetic acid and dichloroacetaldehyde (not
found in the in vivo studies reported above), and these
metabolites were not formed in the presence of cytochrome P-450
inhibitors SKF-525A and carbon monoxide. Pretreatment of rats with
the cytochrome P-450-inducing agent beta-naphthoflavone did not
elevate the hepatic microsomal metabolism of vinylidene chloride,
and the cytochrome P-450-inducing agent phenobarbital gave a
slight enhancement of metabolism per mg microsomal protein. More
noticeable was the marked enhancement of the ability of liver
preparations to produce mutagenic metabolites of vinylidene
chloride in vitro following treatment of female BD-VI rats with the
selective inducing agents phenobarbital and 3-methylcholanthrene
[15].
In a study by Sato et al. [195], male Wistar rats were given
ethanol in the diet (2% ethanol, increased by 1% daily to a final
concentration of 5%, equivalent to 30% of total calorie intake).
Hepatic microsomes derived from the ethanol-treated rats
metabolized vinylidene chloride at a rate of 100.6 nmol/g liver per
min compared with 31.1 nmol/g liver per min in control rat
microsomes, indicating induction of microsomal enzymes involved in
vinylidene chloride metabolism.
As shown in Fig. 2, conjugation with glutathione required
prior microsomal metabolism of vinylidene chloride [131]. The
importance of glutathione in vinylidene chloride metabolism was
studied by Andersen et al. [7]. Pretreatment of rats with agents
that deplete hepatic non-protein sulfhydryl concentrations caused a
marked inhibition of vinylidene chloride metabolism as measured by
gas uptake (which is determined by metabolism). In particular,
treatment with cyclohexene oxide and dimethylmaleate resulted in
76 and 54% inhibition, respectively. Reichert et al. [183] also
noted an 18% reduction in the metabolism of vinylidene chloride
(given as 20 000 mg/m3 (5000 ppm) in the gaseous phase) in
isolated perfused rat livers following diethylmaleate
administration. This was considered to be due to an 85% reduction
in glutathione levels. The finding that the glutathione conjugate
ClCH2COSG is able to S- alkylate a second glutathione molecule to
yield GSCH2COSG [123, 124] suggests that the monoglutathione
conjugate may have the ability to interact with proteins, such as
those involved in the transport of glutathione conjugates.
The rate of metabolism of vinylidene chloride by isolated
hepatocytes from phenobarbital-pretreated male Long-Evans rats was
studied by Costa & Ivanetich [33] using the maximum dose of
vinylidene chloride that was not cytotoxic (2.1 mmol vinylidene
chloride/litre). The metabolites detected were dichloroacetic
acid (0.15 nmol/106 cells per 10 min), monochloroacetic acid
(0.068 nmol/106 cells per 60 min), and dichloroethanol (0.01
nmol/106 cells per 10 min). 2-Chloroethanol and
chloroacetaldehyde were not detected (<12 and <4 nmol/106 cells
per 30 min, respectively). No attempt was made to hydrolyse
conjugates.
In summary, the phase I metabolism of vinylidene chloride in
rodents involves the action of cytochrome P-450 and the production
of monochloroacetic acid. This, and its precursors, may undergo
conjugation with glutathione and/or phosphatidyl ethanolamine
prior to further conversions. Metabolism in the mouse occurs at a
greater rate than in the rat and results in a similar metabolic
profile with a relatively higher proportion of glutathione
conjugate derivatives.
6.1.5. Reaction with cellular macromolecules
The specific interaction of vinylidene chloride metabolite(s)
with DNA is covered in section 8.5.1.
In the studies by McKenna et al. [132, 133] reported in section
6.1.3.1, fasted rats showed a reduced capacity to metabolize
relatively high doses of both orally administered and inhaled
[14C]-vinylidene chloride. Fasted rats exposed to 800 mg
vinylidene chloride/m3 (200 ppm) sustained liver and kidney
damage, which was not found in fed rats. This toxicity was
associated with a greater level of covalently bound radiolabel in
the liver of the fasted animals. An elevated level of covalent
binding in the liver was also seen as a result of depriving rats of
food prior to administration of an oral dose of 50 mg vinylidene
chloride/kg. The results can be explained by the binding of
reactive metabolites (presumed to be vinylidene chloride oxide
and/or chloroacetyl chloride) to nucleophilic sites in tissue
macromolecules. This is thought to be enhanced by the depletion of
glutathione during fasting [93], which operates as an alternative
nucleophile [131]. Following exposure of male rats to 20800 mg
[14C]-vinylidene chloride/m3 (5200 ppm) for 6 h [131], hepatic non-
protein sulfhydryl levels fell in a dose-dependent saturable
manner. Appreciable covalent binding of radiolabel to hepatic
protein occurred when 30% or more glutathione was depleted.
Covalent binding of radiolabel to hepatic and kidney tissue in mice
occurred at the rate of 22 and 80-µg equivalents/g protein,
respectively, compared with 5 and 13 µg equivalents/g protein,
respectively, in rat tissue following exposure to 40 mg [14C]-
vinylidene chloride/m3 (10 ppm)for 6 h.
In a separate study on rats by Reichert et al. [183], the rate
of depletion of glutathione after oral doses of vinylidene chloride
was also found to be exponentially dependent on the concentration.
However, the authors questioned the correlation between a low
glutathione level and relatively high toxicity in fasted rats,
since the fall in glutathione levels after oral administration of
vinylidene chloride (1000 mg/kg) was identical in fasted rats and
fed rats. Furthermore, the rate of metabolism of vinylidene
chloride by isolated perfused livers (20 000 mg vinylidene
chloride/m3 (5000 ppm) in the gaseous phase) was not affected by an
18-h fast. The interpretation of these data is difficult in the
light of other results reported in this section.
Jaeger et al. [93] reported a diurnal variation in the levels
of glutathione in male Holtzmann rats. The animals were most
sensitive to the lethal and hepatotoxic effects of vinylidene
chloride when glutathione levels were at a minimum. Jaeger et
al. [96] also correlated susceptibility to the hepatotoxicity
of vinylidene chloride (exposure for 4 h at 4000 mg/m3 (1000 ppm))
to decreased hepatic glutathione concentrations in male Holtzmann
rats that had been fasted for 18 h or had been treated with
diethylmaleate. In a further study [97], the hepatic glutathione
concentration was decreased by the administration of
trichloropropane epoxide (0.1 ml of a 10% solution/kg) to fasted
rats. This treatment was also associated with elevated toxicity of
vinylidene chloride. A qualitative, but not a quantitative,
difference in the metabolism of vinylidene chloride (8000 mg/m3
(2000 ppm) initial exposure) resulted from the fasting of rats.
Thirty minutes after exposure to [14C]-vinylidene chloride at the
same concentration, the radioactivity in liver mitochondria and
microsomes was largely TCA-insoluble and was greater in fasted than
in control rats. Judging from the turn-over time of TCA-insoluble
14C, it was suggested that this 14C had entered the metabolic pool
rather than being covalently bound to macromolecules. However,
the demonstration of labile thiol adducts [123] could explain this
finding.
Covalent binding of [1,2-14C]-vinylidene chloride to tissues
peaked 6 h after an intraperitoneal dose of 125 mg/kg in male
C57B1/6N mice [159]. Pretreatment with diethylmaleate, which
depletes glutathione, enhanced covalent binding in the liver,
lung, and kidney, and also enhanced lethal toxicity. In accordance
with evidence for the formation of reactive metabolites of
vinylidene chloride by cytochrome P-450, covalent binding was
increased in liver and lung tissue by pretreatment with the P-450
inducers, phenobarbital and 3-methylcholanthrene. Inhibitors
of cytochrome P-450, piperonyl butoxide, and SKF-525A all decreased
covalent binding in the liver and lung. However, binding to kidney
tissue was not affected by P-450-inducing agents, was decreased by
piperonyl butoxide and was increased by SKF-525A. Covalent binding
occurred in hepatic and lung microsomes from these mice following
incubation of microsomes with NADPH. Surprisingly, however, oxygen
did not appear to be necessary. Kidney microsomes could not
metabolize vinylidene chloride to products that covalently bound to
tissue macromolecules, unless the mice had been pretreated with
cytochrome P-450-inducing agents suggesting that, in the absence
of enzyme induction, covalent binding in the kidney in vivo was
mediated by hepatic metabolites [157, 158]. Covalent binding of
[14C]-vinylidene chloride to lung and liver tissue accompanied
bronchiolar necrosis in CD-1 mice given an intraperitoneal dose
(125 mg/kg); only mild hepatic necrosis was observed ([56]section
8.1.3.1). The effects of various inducers and inhibitors of
cytochrome P-450 activity on toxicity and covalent binding were
variable in line with the evidence for microsomal-mediated
activation and deactivation (section 8.1.2) and in accordance
with multiple reactive metabolites (see below). Vinylidene chloride
epoxide, 2-chloroacetyl chloride, 2,2-dichloro-acetaldehyde, 2-
chloroacetic acid and S- (2-chloroacetyl)-glutathione all bind
covalently to thiols in vitro [123, 124]. Covalent binding of
radiolabel to microsomal protein occurred following incubation of
[14C]-vinylidene chloride with rat and human liver microsomes. When
rat microsomes were used, binding was inhibited by alcohol
dehydrogenase + NADH, suggesting that 2,2-dichloroacetaldehyde
played a role in the binding process. Metabolites of [14C]-
vinylidene chloride bound to microsomes from isolated lung as
well as from the liver of CD-1 mice [54]. By the use of specific
inhibitors and agents that induce cytochrome P-450 isoenzymes,
these authors were able to demonstrate the role of cytochrome P-450
isoenzymes in the production of reactive metabolites, though some
non-specific covalent binding was also seen.
In summary, there are a number of reactive metabolites of
vinylidene chloride, the production of which is dependent on the
activity of cytochrome P-450. In rodents, each of these products
may contribute to the depletion of glutathione and to covalent
binding to tissue macromolecules, which is greater in the liver
than in the kidney. The greater covalent binding of reactive
metabolites to tissue macromolecules in the mouse compared with the
rat is correlated with a relatively higher rate of metabolism
(section 6.1.4) and higher toxicity (section 8.1.1.2).
6.1.6. Transformation by non-mammalian species
No data were available to the Task Group on vinylidene chloride
metabolism in non-mammalian species, other than bacteria.
6.2. Human Beings
No data have been reported on the kinetics and metabolism of
vinylidene chloride in human beings, other than some very limited
information obtained indirectly through the following studies.
Liver 9000-g supernatants (S9) from four adults, who did not show
any pathological lesions, were capable of catalysing the formation
of products that were mutagenic to Salmonella typhimurium [15]
(section 8.5.2), suggesting that human cytochrome P-450 can
metabolize vinylidene chloride. The rate of conversion of
vinylidene chloride to dichloroacetaldehyde in hepatic microsomes
from two human organ donors (one of each sex) was 0.0340-0.038
nmol/min per nmol cytochrome P-450. The conversion was shown (by
lack of significant antibody inhibition) not to be mediated by
debrisoquine hydroxylase (a form of cytochrome P-450, which is
polymorphic in human beings) [245]. This rate of microsomal
metabolism to dichloroacetaldehyde is similar to that reported by
Costa & Ivanetich [33] for the rat (0.028 nmol dichloroacetaldehyde
and 0.035 nmol chloroacetate produced/min per nmol cytochrome
P-450).
Vinylidene chloride is exhaled in human breath following
inhalation exposure [233]. Breath from student volunteers, in
Texas and North Carolina, USA, was sampled using a spirometer as
the subjects inhaled pure air. The ratio of vinylidene chloride in
the breath to that in pre-exposure air was 0.78 ± 0.86 (n = 15). A
significant Spearman correlation coefficient of 0.77 was determined
between air and breath levels of vinylidene chloride in 17 human
subjects. The following log-linear model was capable of giving a
reasonable prediction of breath levels from the preceding 8-h air
exposure levels: log concentration in breath (µg/m3) = 0.24 ± 0.67
+ (0.71 ± 0.17) log concentration in air (µg/m3). The authors
suggested that, if these observations were confirmed, recent
exposures and body burdens of individuals could be estimated from
breath analysis. However, this may be hampered by biphasic
elimination as reported for animals (section 6.1.3).
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Effects on the Stratospheric Ozone Layer
The rapid destruction of vinylidene chloride in the troposphere
by hydroxyl radicals (section 4.1.1) indicates that the substance
is unlikely to participate in the depletion of the stratospheric
ozone layer.
7.2. Aquatic Organisms
Studies on the impact of vinylidene chloride on living
organisms have concentrated on the aquatic environment and include
discussions on the levels of vinylidene chloride detected (section
5.2).
According to Leblanc [117], the acute toxicity for the water
flea ( Daphnia magna), under static conditions, is of a similar
magnitude to that reported by Buccafusco et al. [21] for the
bluegill fish (see below). The median LC50 values were 98 mg/litre
(95% confidence interval; range, 71-130 mg/litre) and 79 mg/litre
(95% confidence interval; range, 61-110 mg/litre) for 24 h and 48
h, respectively. The "no discernible effect" level for the water
flea was less than 2.4 mg/litre.
Dawson et al. [36] treated fresh-water bluegill sunfish
( Lepomis macrochiras) and marine tidewater silverside fish
( Menidia beryllina) with vinylidene chloride (132-750 mg/litre
and 180-320 mg/litre, respectively) for up to 96 h under static
conditions. No attempt was made to prevent loss of vinylidene
chloride by evaporation. The best-fit median lethal concentrations
(LC50) for 96 h were 220 and 250 mg/litre for bluegill sunfish and
tidewater silverside fish, respectively. Since these values were
less than 500 mg/litre (500 ppm), vinylidene chloride was
designated a hazardous substance. The 96-h static LC50 of
vinylidene chloride in juvenile marine sheepshead minnows
( Cyprinodon variegatus) was very similar to the above values (250
mg/litre; range, 200-340 mg/litre, 95% confidence limits), and was
the same when measured at 24 h [72]. The no-observed-effect
concentration was 80 mg/litre. The acute toxicity in bluegill fish
( L. macrochirus) was also investigated by Buccafusco et al. [2]
under static conditions, in capped jars to minimize volatilization.
The recorded LC50 value was 74 mg/litre (95% confidence interval,
57-91 mg/litre) at 96 h. This LC50, which was identical at 24 h,
is somewhat lower than those reported in the studies by Dawson et
al. [36] and Heitmuller et al. [72]. Toxicity values similar to
those noted above for fish and Daphnia were reported in a review by
Atri [9].
The assays under static uncapped conditions reported here are
relevant to acute spill conditions. One-week flow-through studies
have also been carried out by Dill et al. [39]. The LC50 value for
fathead minnows ( Pimephales promelas Rafinesque) in flowing water
was 29 mg/litre (range, 23-34 mg/litre) after 7 days exposure,
whereas the 96-h LC50 was 108 mg/litre (range, 85-117 mg/litre)
under flow-through conditions. Swimming disorientation was
observed to be the major sublethal toxic effect of vinylidene
chloride. A bioconcentration factor of 4 and a bioaccumulation
factor of 6.9 have been reported for fish in a review by Atri [9].
Few data are available on the sublethal effects of vinylidene
chloride on aquatic organisms. Preliminary studies demonstrated
that hepatic neoplastic lesions were not produced in guppy
( Poecilia reticulata) and Japanese medaka ( Oryzias latipes)
exposed to vinylidene chloride concentrations of up to 40 mg/litre
(40 ppm) for 3 months [71] (personal communication, Hawkins Gulf
Coast Research Laboratory, Mississippi, USA).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Single Exposures
Acute toxicity data (LC50s and LD50s) for common laboratory
animals are shown in Table 7.
8.1.1. Inhalation
8.1.1.1 Rats
In an early study by Carpenter et al. [24], 3 groups of 6
Sherman rats were exposed to vinylidene chloride vapour for 4 h at
various concentrations. Within a 14-day post-exposure observation
period, a concentration of 128 000 mg/m3 (32 000 ppm) was lethal
for 2/6, 3/6, and 4/6 animals, respectively. Later, Siegel et al.
[206] estimated a 4-h LC50 of 25 400 mg vinylidene chloride/m3
(6350 ppm) for groups of 16 male Sprague-Dawley-derived rats.
Siletchnik & Carlson [211] considered that lethality from
vinylidene chloride might be related to cardiotoxicity. They
exposed male Charles River albino rats to 102 400 mg vinylidene
chloride/m3 (25 600 ppm) for 10 min or more and noted progressive
sinus bradycardia and arrhythmias (AV-block), multiple
continuous ventricular contractions, and ventricular fibrillation.
This treatment with vinylidene chloride also produced a marked
increase in sensitivity to epinephrine-induced cardiac arrhythmias.
Phenobarbital pretreatment enhanced the cardiac-sensitizing
properties of vinylidene chloride.
However, Jaeger et al. [95] reported that death from vinylidene
chloride inhalation was associated with bloody ascites in all
animals, with no signs of cardiac failure, and was therefore
thought to be due to vascular collapse and shock.
In the studies by Zeller et al. [248, 249], the estimated
LC50s of vinylidene chloride (Table 7) were lowered by fasting in
both male and female Sprague-Dawley rats. Females were less
susceptible than males to the lethal effect. Post-mortem
examination of animals dying from vinylidene chloride exposure
revealed acute contraction of heart blood vessels, acute swellings
and localized bloody oedema in the lung, greyish enlarged liver
lobules, and pale kidneys. Ascites and hydrothorax were also seen.
Table 7. Acute toxicity of vinylidene chloride for laboratory animals
______________________________________________________________________________________________________
Species Sex Nutri- Estimated LC50/LD50 Dosing criteria Limit of Reference
tional observation
status time
______________________________________________________________________________________________________ statustion time
Rat male, fed approximately 128 000 mg/m3 Inhalation, 4 h 14 days [24]
female (32 000 ppm)
Rat male fed 25 400 mg/m3 (6350 ppm) Inhalation, 4 h 14 days [206]
Rat male fed 60 000 mg/m3 (15 000 ppm) Inhalation, 4 h 24 h [96]
Rat male fed approximately 8000 mg/m3 inhalation, 4 h 23 h [93]
(2000 ppm) (pm but not am)
Rat male fed 28 400 mg/m3 (7100 ppm) Inhalation, 4 h 14 days [249]
Rat male 18-h fasted 2400 mg/m3 (600 ppm) Inhalation, 4 h 24 h [96]
Rat male fasted not measurable because of inhalation, 4 h [5]
(overnight) non-linear concentration-
mortality relationship
Rat male 16-h fasted 1660 mg/m3 (415 ppm) Inhalation, 4 h 14 days [248]
Rat female fed 41 200 mg/m3 (10 300 ppm) Inhalation, 4 h 14 days [249]
Rat female 16-h fasted 26 260 mg/m3 (6565 ppm) Inhalation, 4 h 14 days [248]
Mouse male fed 392 mg/m3 (98 ppm) inhalation, 1 day [204]
female 420 mg/m3 (105 ppm)
Mouse male fed 140 mg/m3 (35 ppm) Inhalation, 2 days [204]
Mouse male fed 460 mg/m3 (115 ppm) Inhalation, 4 h 14 days [251]
Mouse male fasted 200 mg/m3 (50 ppm) Inhalation, 4 h 14 days [250]
Mouse female fed 820 mg/m3 (205 ppm) Inhalation, 4 h 14 days [251]
Mouse female fasted 500 mg/m3 (125 ppm) Inhalation, 4 h 14 days [250]
Table 7. (contd.)
______________________________________________________________________________________________________
Species Sex Nutri- Estimated LC50/LD50 Dosing criteria Limit of Reference
tional observation
status time
______________________________________________________________________________________________________ statustion time
Hamster male fed 6640 mg/m3(1660 ppm) Inhalation, 4 h 14 days [109]
Hamster male fasted 600 mg/m3 (150 ppm) Inhalation, 4 h 14 days [108]
Hamster female fed 11 780 mg/m3 (2945 ppm) Inhalation, 4 h 14 days [109]
Hamster female fasted 1780 mg/m3 (445 ppm) Inhalation, 4 h 14 days [108]
Rat male fed 1550 mg/kg gavage 24 h [100]
Rat male fed 1510 mg/kg gavage 96 h [100]
Rat male fed 1800 mg/kg gavage not stated [172]
Rat female fed 1500 mg/kg gavage not stated [172]
Rat male fed 800-2000 mg/kg gavage 14 days [2]
Mouse male fed 201-235 mg/kg gavage not stated [103]
Mouse female fed 171-221 mg/kg gavage not stated [103]
______________________________________________________________________________________________________
a This report noted that mortality was consistently observed at doses as low as 50 mg/kg. In 73-g
male rats, dose-mortality curves showed a maximum mortality (10/10) at 300 mg/kg. Percent
mortality then decreased as dose was increased to 800 mg/kg.
Jaeger et al. [96] showed that the toxicity of vinylidene
chloride in male Holtzman rats was enhanced as a result of fasting.
The estimated 24-h LC50 for fed rats was 60 000 mg/m3 (15 000
ppm) (4-h exposure), while the corresponding value for 18-h fasted
rats was 2400 mg/m3 (600 ppm) (n = 5 or 6). The minimum lethal
concentrations were 40 000 and 800 mg/m3 (10 000 and 200 ppm),
respectively. At levels of 600 mg/m3 (150 ppm) or more, serum-
alanine-alpha-ketoglutarate transaminase rose rapidly in fasted
rats (within 2 h of termination of a 4-h exposure at 8000 mg/m3
(2000 ppm)). This indication of liver damage did not arise at
levels below 8000 mg/m3 (2000 ppm) in rats provided with food.
The increased susceptibility of fasted rats was considered to be
due to decreased availability of hepatic glutathione and this was
supported by the potentiation of hepatotoxicity by treatment of
fed rats with the agent diethylmaleate, which depletes glutathione
(section 6.1.5). This was further supported by the observation
[93] that a 4-h exposure of male Holtzman rats to 8000 mg
vinylidene chloride/m3 (2000 ppm) in the morning produced a
3-fold increase in serum alanine-alpha-ketoglutarate transaminase
activity with no deaths. Conversely, the same treatment in the
afternoon resulted in an almost 10-fold increase in serum
alanine-alpha-ketoglutarate transaminase, and 2 out of 5 treated
rats died within 23 h. The relative susceptibilities were
inversely related to hepatic glutathione levels.
In fasted male Sprague-Dawley rats exposed to 800 mg vinylidene
chloride/m3 (0.02%) , liver toxicity was observed within 2 h
[188]. Toxicity in liver parenchyma was characterized by
retraction of cell borders and the formation of pericellular
"lacunae". Nuclei showed segregation of chromatin towards the
margins of the nuclear envelope and mitochondria were swollen with
ruptured outer membranes. Midzonal hepatic necrosis led to
haemorrhagic centrilobular necrosis within 6 h. This liver
necrosis, along with raised levels of serum-alanine-alpha-
ketoglutarate transaminase and an associated fall in hepatic
glutathione levels, were all minimized by pretreatment of rats
with the cytochrome P-450-inducing agents phenobarbital and
Aroclor-1254.
In an earlier study [23], male Sprague-Dawley rats were exposed
to 5760 mg vinylidene chloride/m3 (1440 ppm) for 1 h. Twenty-four
hours later, liver damage was detected by measured elevations in
serum glutamic oxalacetic transaminase and glutamic pyruvic
transaminase. Pretreatment of these animals with phenobarbital and
3-methylcholanthrene did not alter the extent of the elevation of
serum hepatic enzyme levels. Furthermore, the activity of glucose-
6-phosphatase in the liver was also unaffected 24 h after exposure
to 9080 or 11 960 mg vinylidene chloride/m3 (2270 ppm or 2990 ppm)
in both 3-methylcholanthrene and phenobarbital-treated animals.
In contrast, exposure to 80 000 or 130 000 mg vinylidene
chloride/m3 (20 000 or 32 500 ppm) for 1 h was lethal for most of
the rats (groups of 4 rats) pretreated with the inducing agents,
despite the fact that these concentrations did not produce
deaths in similar groups of control rats. Treatment of rats with
the cytochrome P-450 inhibitors "SKF-525A" and "Lilly 18947" reduced
the survival time after inhalation of 168 000 or 212 000 mg
vinylidene chloride/m3 (42 000 ppm or 53 000 ppm). Because of the
differential effects of inducing agents on hepatotoxicity and
lethality, the results suggest that the lethal effects of inhaled
vinylidene chloride are distinct from the hepatotoxic effects.
Since cytochrome P-450 has been implicated in the activation of
vinylidene chloride to form toxic metabolite(s) (section 6.1.4) the
protective or lack of potentiating effect of inducing agents on
hepatotoxicity is not understood, but may result from multiple
roles of microsomal enzymes (section 8.1.2.1).
Subsequently, Reynolds et al. [189] exposed fasted male rats
to 800 mg vinylidene chloride/m3 (200 ppm). Glutathione levels in
the liver were rapidly depleted during the first and second hours
of exposure and were replenished during the third and fourth hours,
when toxicity was determined by analysis of tissue sections by
light microscopy. The rebound of glutathione levels was not
observed in the mitochondria, which might indicate a role of this
organelle in toxicity. Inactivation of the microsomal enzyme
cytochrome P-450 was not appreciable prior to histological
alterations in the liver and therefore did not appear to be an
early event in cytotoxicity. The toxicity of vinylidene chloride
(2000 or 4000 mg/m3 (500 or 1000 ppm)) inhaled for 3 or 24 h was
exacerbated by the glutathione-depleting agent phorone, as
evidenced by an increase in the levels of serum-aminotransferases
and sorbitol dehydrogenase at 3 h and mortality at 24 h in male
Wistar rats [207]. Phorone (250 mg/kg) also had the effect of
increasing the half-life of the terminal elimination phase of
vinylidene chloride from 0.89 to 2.33 h and from 1.55 to 4.21 h at
levels of 2000 and 4000 mg vinylidene chloride/m3 (500 and 1000
ppm), respectively, in a closed exposure chamber. Therefore,
increased toxicity was associated with decreased metabolism.
Since there was no evidence for an effect of phorone on the
activity of the mixed-function oxidase, the authors proposed that
phorone produced a quantitative change in vinylidene chloride
metabolism leading to intermediates of greater toxicity. However,
the results might also be explained by a build up of intermediates
(due to reduced ability to conjugate with glutathione) that inhibit
their own formation.
Andersen et al. [5] studied the dependence of inhalation
toxicity (as measured by plasma aspartate transaminase) on both
exposure concentration and duration in fasted male HOT:SD(BR)
Holtzman rats. Plasma-enzyme levels increased markedly after
exposure to 800 mg vinylidene chloride/m3 (200 ppm) for 1.25 h.
After this time, no further increase in plasma aspartate
transaminase was recorded. The concentrationmortality curve
increased rapidly between 400 and 800 mg/m3 (100-200 ppm) and
reached a plateau between 800 and 4000 mg/m3 (200-1000 ppm), making
it impossible to make a meaningful estimate of the LC50. Thus, a
concentration x time relationship for toxicity is not apparent,
and this is in agreement with saturable metabolism (section
6.1.3.1) to toxic intermediates. Thus, estimates by other workers
of the LC50 under similar conditions cannot be considered accurate.
After inhalation of 800 mg vinylidene chloride/m3 (200 ppm)
for 0.5 h, immature rats showed significant prolongation of
pentobarbital-induced sleeping times, suggesting an inhibition of
pentobarbital metabolism [5]. This increase in sleeping times was
maintained for at least 3 days after a 2-h exposure. In mature
rats, treated with 1600 mg/m3 (400 ppm) for 2 h, there was no
evidence of an altered sleeping time within the exposure period,
though this was elevated 2 and 24 h later.
Exposure of fasted male rats to 8000 mg vinylidene chloride/m3
(2000 ppm) for 4 h led not only to elevation of serum alanine
alpha-ketoglutarate transaminase levels but also to increased
levels of hepatic sodium and calcium with a concomitant decrease in
potassium and magnesium levels and diminished histochemical
glucose-6-phosphatase activity. These changes were associated
with centrilobular necrosis and haemorrhagic necrosis of the entire
hepatic lobule. Thyroidectomized, fasted rats, exposed under the
same conditions, showed significantly less change in hepatic
electrolyte concentrations. Morphological injury was also
minimized and was similar to that seen in non-pretreated fed
animals. Mortality was also inhibited by thyroidectomy. In
contrast, thyroxine pretreatment potentiated the toxicity of
vinylidene chloride and restored the susceptibility of
thyroidectomized rats. It was suggested that the protective
effect of thyroidectomy was at least partially mediated by the
observed elevation of hepatic glutathione levels [216].
However, differences were observed between fed, fasted, and
hyperthyroid Sprague-Dawley rats in the effects of orally
administered vinylidene chloride (50 mg/kg body weight) on body
temperature, serum glucose concentrations, hepatic glutathione,
and glutathione transferase [106]. The different patterns of
response in the three groups suggest different mechanisms of
toxicity in fasted and hyperthyroid rats.
Similar findings were reported by Jaeger et al. [98]. In a
study on male Sprague-Dawley rats, a raised serum sorbitol
dehydrogenase level (a cytoplasmic marker) coincided with an
elevation in serum ornithine carbamoyl transaminase from the
mitochondria. This finding suggests that mitochondrial damage is an
early event in the hepatotoxicity of vinylidene chloride. The data
support the authors' theory that the metabolite monochloracetic
acid is toxic to mitochondria via chlorocitric acid and "lethal
synthesis" leading to accumulation of citric acid [97].
The kidney is also affected by vinylidene chloride. At
sublethal concentrations (800 mg/m3 (200 ppm) for 6 h), male
Sprague-Dawley rats given food ad libitum did not show any signs of
an adverse response [132]. In contrast, rats previously fasted for
18 h, were found to have haemoglobinuria, which persisted for 12-
24 h after exposure. As well as seeing multiple foci of hepatic
centrilobular degeneration and necrosis, marked degeneration
of kidney proximal tubular epithelia was observed after this
exposure in the fasted rats, but not in those that were fed. These
changes were associated with an increase in the level of 14C
covalently bound in the liver following exposure to [14C]-
vinylidene chloride (section 6.1.5). Hepatotoxic effects were not
noted in either fed or fasted rats exposed to 40 mg vinylidene
chloride/m3 (10 ppm). In a more recent study, inhalation of
vinylidene chloride produced acute nephrotoxicity (which was not
associated with calcium oxalate formation) in male Sprague-Dawley
rats [90]. Twenty-four hours after a 4-h exposure to 1000 mg
vinylidene chloride/m3 (250 ppm) or more, kidney/body weight
ratios, serum urea nitrogen, and creatinine levels were
significantly increased. This was associated with moderate
cellular swelling in the renal cortex (800 mg/m3 (200 ppm)) and
severe tubular necrosis (>1200 mg/m3 (>300 ppm)). Aroclor-1254
and phenobarbital pretreatment antagonized renal toxicity.
8.1.1.2 Mice
A marked individual variation in the lethal concentration
of vinylidene chloride (ranging from 500 to 10 000 mg/m3; 125 to
2500 ppm) was noted by Lazarev [116].
In the study by Short et al. [204], the toxicity of inhaled
vinylidene chloride was investigated in CD-1 mice. The 1- and 2-day
LC50 values for groups of 10 mice were approximately 400 mg/m3 (100
ppm) (males and females) and 140 mg/m3 (35 ppm) (males),
respectively. These values are considerably lower than those
reported for rats and, in contrast to the data on mice, no deaths
were seen in male rats exposed to 240 mg vinylidene chloride/m3
(60 ppm). In addition, up to 240 mg vinylidene chloride/m3 (60
ppm) produced a dose-dependent histopathological change in mouse
liver and an increase in both serum glutamic oxaloacetic
transaminase and serum glutamic pyruvic transaminase. The serum
enzymes were also elevated in male rats but not to the same extent
and only with a longer exposure period. Mice exposed for 1 day to
60 mg vinylidene chloride/m3 (15 ppm) showed hepatocellular
degeneration and increased mitotic figures of hepatocytes together
with severe kidney tubular nephrosis. At 120 mg/m3 (30 ppm),
midzonal hepatic necrosis was also seen, the severity of which was
increased at 240 mg/m3 (60 ppm). In contrast, rats exposed to 240
mg/m3 (60 ppm) for 1 day showed only mild hepatic centrilobular
degeneration and/or necrosis and mild bileduct hyperplasia. The
inhibition of these toxic effects and of covalent binding of
[14C]-vinylidene chloride-derived radioactivity in the mouse liver
and kidney by disulfiram was described. The mechanism of this
protection was not established but may be via modulation of
metabolism.
The studies of Zeller et al. [250] on NMRI mice showed that,
as with rats, males were more susceptible than females (Table 7)
and that fasting potentiated lethality. Symptoms included apathy,
narcosis, dyspnoea, and immobility. Postmortem examination of
mice dying from vinylidene chloride exposure particularly showed
acute emphysema and congestion of lungs.
The effects of vinylidene chloride on DNA following acute
inhalation (40 or 200 mg/m3 (10 or 50 ppm) for 6 h) have been
studied in male CD-1 mice as well as Sprague-Dawley rats [186].
These exposures gave rise to tissue damage (nephrosis, increased
mitotic figures, and regeneration) and increased DNA semi-
conservative replication (25-fold) in mouse kidney, but not in
mouse liver. In contrast, DNA semiconservative replication in rat
kidney was increased only 2.2 fold and was slightly decreased in
rat liver following exposure to 40 mg/m3 (10 ppm). DNA repair and
DNA alkylation in these organs were minimal in both rats and mice
(section 8.5.1). In the light of these minimal effects, the
authors suggested that the carcinogenicity of vinylidene chloride
in the mouse kidney (section 8.7.1) might be via an epigenetic
mechanism.
8.1.1.3 Other animal species
Effects of vinylidene chloride on the respiratory system have
also been reported in cats, rabbits, and guinea-pigs [192].
Pulmonary irritation and lung oedema, haemorrhage, and pneumonia
were seen following exposure to vinylidene chloride at 2000 or 6000
mg/m3 (500 or 1500 ppm) (cats) and 5000 or 8000 mg/m3 (1250 or 2000
ppm) (guinea-pigs) for 2 h. Exposure to concentrations ranging from
500 to 2000 mg/m3 (125 to 500 ppm) for 40 min inhibited spinal
reflexes in rabbits. This species survived after exposure for 40
min to a concentration of 30 000 mg/m3 (7500 ppm). Klimisch &
Freisberg [108, 109] studied the inhalation toxicity of vinylidene
chloride in fed and fasted Chinese striped hamsters. The LC50
values are given in Table 7. As with rats and mice, males were
more susceptible to lethality than females, and fasting potentiated
the toxicity markedly. Hamsters that died showed acute dilation
and passive hyperaemia of the heart, congested lungs, and
lobulation of the liver.
8.1.2. Oral
As with inhalation, the principal organs affected by oral
administration of vinylidene chloride are the liver, kidneys, and
lungs.
The LD50 values for orally administered vinylidene chloride in
rats and mice are shown in Table 7. It can again be seen that mice
are more susceptible than rats, but few sex differences in response
are observed following administration of vinylidene chloride by
gavage.
8.1.2.1 Rats
Hepatic damage produced by intubation of 400 mg vinylidene
chloride/kg to fasted rats was indicated by elevation of serum
alanine alpha-ketoglutarate transaminase activity within 4 h [94].
The level of glucose-6-phosphatase activity was reduced in the
liver at 8 h, but not after 4 h, suggesting (in line with the
above discussion) that initial toxicity was not associated with the
endoplasmic reticulum membrane. Treatment of rats with a
relatively high dose of 12.5 mmol vinylidene chloride/kg did not
lead to elevation of malondialdehyde or conjugated dienes in
incubated liver homogenates taken 1 h after dosing. This
contrasts with the effect of a hepatotoxic dose of carbon
tetrachloride and suggests that lipid peroxidation is not involved
in the hepatotoxicity of vinylidene chloride.
Jenkins et al. [100] also reported, that pretreatment of rats
with the microsomal enzyme-inducing agent phenobarbital offered
protection against the hepatotoxic effects of orally administered
vinylidene chloride. The acute 24-h toxicity of vinylidene
chloride was enhanced 18-fold by adrenalectomy, suggesting that the
adrenals were involved in protection against lethal effects. The
mechanisms of protection were not understood.
The effects of induction and inhibition of microsomal enzymes
in fasted male Holtzman rats were studied in more detail by
Andersen et al. [4]. Pretreatment of rats with phenobarbital
markedly reduced the lethality of a 100 mg/kg oral dose of
vinylidene chloride in immature rats (140 g), but not in large
adult (331 g) rats.
These results suggest that a microsomal detoxification system,
inducible in immature rats, was operative. This is supported by
the finding that the cytochrome P-450 inhibitor SKF-525A
exacerbated the lethal effects of a dose of vinylidene chloride of
200 mg/kg in rats (260-270 g) but did not have any effect on
mortality in immature (80-100 g) 2,3-epoxy-propan-1-ol, rats. One
of a range of epoxides that exacerbate the toxicity of orally
administered vinylidene chloride, which is particularly potent, was
a relatively poor substrate for glutathione- S- transferase and
styrene oxide hydrolase. Thus 2,3-epoxypropan-1-ol, rather than
inhibiting the protective effects of epoxide hydrolase or
glutathione conjugation (section 6.1.4) appeared to inhibit a
further (uncharacterized) detoxification pathway [3]. The role of a
further microsomal enzyme system in the production of a toxic
intermediate was indicated by the protective effects of
pretreatment of rats of all sizes with pyrazole, 3-aminotriazole,
and carbon tetrachloride against the lethality of an oral dose of
200 mg/kg [4].
Andersen & Jenkins [2] noted that female Holtzman HOT:(SD)BR
rats were much less susceptible to the hepatotoxic effect than
male rats, the threshold oral dose for the elevation of plasma
transaminase activity being approximately 100 mg/kg in the females.
When mature, male rats were given 400 mg vinylidene chloride/kg,
levels of plasma-aspartate transaminase were 4-5 times greater in
18-h fasted rats than in control rats. The effects of fasting are
thought to be due to glutathione depletion (section 6.1.5).
Chieco et al. [28] studied hepatotoxicity in fasted male
Sprague-Dawley rats given vinylidene chloride as an oral dose in
mineral oil. Two hours after dosing with 200 mg vinylidene
chloride/kg or 6 h after a lower dose of 50 mg/kg, early damage to
plasma and mitochondrial membranes was indicated by raised hepatic
sodium levels and decreased central area histochemical staining
of bile canaliculi membrane Mg2+-ATPase, outer membrane
mitochondrial monoamine oxidase, and inner membrane mitochondrial
succinate dehydrogenase and cytochrome oxidase. The extent of
injury (indicated by raised serum transaminase activity and
decreased histochemical staining of membrane components) increased
with time after dosing or with increased dose. Four and 6 h after
administration of a 200 mg/kg dose, necrosis occurred around the
central vein of the liver. There were no histochemical alterations
in the kidneys at 6 h. The same investigators [27] noted that
this treatment produced increased plasma-haemoglobin levels and
granular haem casts in the loop of Henlé. No pathological changes
were seen in the heart, lungs, spleen, adrenals, or duodenum.
Hepatic damage was more severe with Tween 80 used as a dose vehicle
than with corn oil or mineral oil, reflecting a relatively high
rate of absorption when administered in Tween 80 (section 6.1.1.2).
Eight hours after oral administration of 40 mg vinylidene
chloride/kg body weight to rats, centrilobular hepatocytes showed a
dilated endoplasmic reticulum and swollen mitochondria and
perinuclear cisternae. The nucleosplasm was homogeneous suggesting
chromatinolysis [5].
In unanaesthetized, freely moving fed and fasted Sprague-
Dawley male rats [148], at least a 2-fold increase in inulin
excretion was observed within 2 h of oral administration of 200 mg
vinylidene chloride/kg. Bile flow decreased in treated rats (up to
40% and 65% in the fed and fasted rats, respectively). Thus,
vinylidene chloride alters hepatobiliary permeability and causes
cholestasis.
Kanz & Reynolds [105] investigated the occurrence of
morphological changes in the liver in relation to time after oral
administration of vinylidene chloride (25, 50, or 100 mg/kg in
mineral oil) (see also Kanz et al. [106], reported in section
8.1.1.1). One, 2, or 3 h after administration to fasted male
Sprague-Dawley rats, the liver was examined microscopically
following in situ perfusion fixation. Dilation of the bile
canaliculi with an increase in the number of microvilli or membrane
fragments in canaliculi was seen with the formation of canalicular
diverticuli in centrilobular hepatocytes within 1-2 h.
Subsequently, microvilli on the sinusoidal surfaces were lost, and
cytoplasmic vacuolation occurred. These early changes were seen
without morphological alteration of the endoplasmic reticulum or
mitochondria. Not until 4-6 h after an oral dose of 200 mg
vinylidene chloride/kg was a decrease seen in the activity of
enzymes in the sinusoidal plasma, mitochondrial matrix,
endoplasmic reticulum, lysosomes and cytosol, and then only in
regions of gross injury. At 2 h, scattered hepatocytes showed
nuclear and cell surface anomalies that were characteristic of
apoptosis [190].
In agreement with the findings of Reynolds et al. [189] in
inhalation studies (section 8.1.1.1), Moslen & Reynolds [147] found
that loss of activity of microsomal cytochrome P-450 was not an
early event in the toxicity of vinylidene chloride (200 mg/kg)
orally administered to fasted male Sprague-Dawley rats.
Cytochrome P-450 deactivation was concomitant with an elevation in
the activities of serum glutamate oxalacetate transaminase and
serum glutamate pyruvate transaminase and did not occur until
between 2 and 3 h after administration of vinylidene chloride.
These effects were preceded by a marked inhibition (within 1 h) of
the activity of glutathione- S- transferase towards
dichloronitrobenzene, chlorodinitrobenzene, and 1,2-epoxy-3-( p-
nitrophenoxy)-propane (but not towards ethacrynic acid) and a
concomitant reduction of hepatic glutathione levels. A correlation
was found between the dose-dependency of inhibition of
glutathione- S- transferase activity and of cytotoxicity. Thus,
both glutathione depletion and inhibition of specific glutathione-
S- transferase(s) precede toxicity.
Simultaneous treatment of 8 male Wistar rats with ethanol (4.8
g/kg, oral) protected against the hepatotoxicity of vinylidene
chloride (0.125 g/kg, oral) as measured by the elevation of the
activities of serum aminotransferases and sorbitol dehydrogenase
[208]. Conversely, pretreatment of rats with ethanol (5% in
drinking-water for 7 days) exacerbated the hepatotoxicity of orally
administered vinylidene chloride (0.125 or 0.2 g/kg). Dithiocarb or
(+)-catechin (0.2 g/kg) administered simultaneously with vinylidene
chloride also reduced vinylidene chloride-induced hepatotoxicity.
Evidence was provided that the simultaneous treatment with ethanol
and dithiocarb may lead to depression of the metabolism of
vinylidene chloride. It was postulated that (+)-catechin might act
as a scavenger of reactive intermediate(s), which may also be the
mechanism of protection afforded by (+)-cyanidanol-3 [207].
Acetone is another agent that modifies the hepatotoxicity
of orally administered vinylidene chloride [75]. Administration of
5 or 10 mmol acetone/kg orally to male Sprague-Dawley rats
potentiated liver injury (elevated plasma glutamic pyruvic
transaminase and ornithine carbamyl transferase activity and liver
total bilirubin content) caused by a single oral dose of 50 mg
vinylidene chloride/kg. However, acetone given at 1, 15, or 30
mmol/kg, did not potentiate hepatotoxicity. The biphasic effect
of acetone could not be explained but was considered by the authors
to be related to dose-dependent changes in more than one
biotransformation process.
A further effect of vinylidene chloride was the prolongation of
barbiturate sleeping times [92]. Two to 4 h after an oral dose of
400 mg vinylidene chloride/kg to male Holtzman rats, the
pentobarbital sleeping time was elevated (136% of control), (prior
to hepatotoxicity as indicated by loss of glucose-6-phosphatase
activity). Both hexobarbital and pentobarbital sleeping times were
elevated at 17-22 h, concomitant with hepatic injury. The early
effects on pentobarbital sleeping times were due, not to decreased
metabolism of the barbiturate, but to an elevation of its
concentration in serum through altered absorption or distribution.
Kidney toxicity was studied by Jenkins & Andersen [99] in
NMRI:0(SD), Sprague-Dawley-derived rats. Within 24 h of oral
administration of 400 mg vinylidene chloride/kg , fasted male rats
showed raised levels of plasma urea nitrogen and creatinine.
Within 48 h, tubular dilation was observed with necrosis and
vacuolation of tubular epithelium. Some tubules contained a blue-
black amorphous material. These histopathological effects were
preceded by inflammation in some animals. Elevation of plasma urea
nitrogen and creatinine was not observed in fed rats with identical
treatment and was less evident in fasted females than in males.
However, the histopathological effects were no less severe in the
female rats. The time-course for the nephrotoxic response (maximum
at 48 h) in male and female fasted rats was slightly preceded by
hepatotoxicity as indicated by the appearance in the plasma of
aspartate transaminase, alanine transaminase lactate dehydrogenase,
and sorbitol dehydrogenase (maximum at 8-24 h). This finding is
in agreement with that reported above [28].
8.1.2.2 Mice
Orally administered vinylidene chloride also caused pulmonary
injury in male C57B1/6 mice [52]. Following administration of
100 mg/kg, peribronchial and perivascular oedema were seen
in the lungs. Histopathology revealed dilation of Clara cell
cisternae and degeneration of the endoplasmic reticulum. A 200
mg/kg dose caused severe necrosis of ciliated and Clara cells and
exfoliation of the bronchial lining within 6 h. Both doses caused
concurrent hepatotoxicity as evidenced by raised levels of serum
glutamic oxalacetic transaminase and glutamic pyruvic transaminase.
By 24 h, pulmonary oedema, haemorrhage, and focal atelectasis
were also observed in association with hypoxia. Recovery was seen
within 7 days. Following a higher dose of 200 mg/kg, injury was
seen to be followed by cellular proliferation as indicated by
incorporation of a pulse of [3H]-thymidine into total
pulmonary DNA [53]. Proliferative activity reached a peak
between 3 and 5 days after treatment with vinylidene chloride.
The majority of the 3H was incorporated into non-ciliated
bronchiolar epithelial cells.
8.1.3. Other routes
8.1.3.1 Intraperitoneal
When given by intraperitoneal (ip) injection to male ddY strain
mice at a dose level of 120 mg/kg (0.1 ml/kg), vinylidene chloride
produced hypothermia within 30 min and severe renal damage at 24 h
(as shown by elevated plasma urea nitrogen and kidney calcium
levels) [143]. Renal tubular necrosis was much more severe than
hepatic damage. Pretreatment of mice with diethyldithiocarbamate or
carbon disulfide protected against renal and hepatic toxicity,
possibly via an inhibitory effect on metabolic activation.
Vinylidene chloride at 605 mg/kg (0.5 ml/kg, ip) caused liver
damage in male Sprague-Dawley rats as evidenced by raised serum
glutamate pyruvate transaminase and increased bile duct pancreatic
fluid flow at 24 h [70]. Hepatic microsomal glucose-6-phosphatase
and ATP-dependent calcium pump were both inhibited 24 h after
intraperitoneal administration of vinylidene chloride (1 mg/kg) to
male Sprague-Dawley rats [145]. The level of conjugated dienes in
microsomes obtained 2 h after vinylidene chloride injection was
not significantly different from that measured in control rat
microsomes, suggesting that the toxic effects were not mediated by
lipid peroxidation.
The results of Siegers et al. [209] also suggest that lipid
peroxidation is not a mechanism involved in the early stages of
vinylidene chloride hepatotoxicity. Up to 2 h after an
intraperitoneal injection of 0.5 g vinylidene chloride/kg to male
Wistar rats, ethane exhalation was only slightly higher than the
control levels and was not affected by hypoxia.
As was observed in the short-term inhalation studies on
vinylidene chloride (section 8.2.1), the compound was found to
induce cytochrome P-450 activity following a single administration
to C57B1/6N mice by intraperitoneal injection. Over the range of
50-150 mg/kg, a dose-dependent induction of microsomal 7-
ethoxyresorufin and 7-ethoxycoumarin O- deethylation (but not total
cytochrome P-450 content or benzo (alpha)pyrene hydroxylase) was found
in the kidney [112].
Male C57BL/6J mice given 125 mg vinylidene chloride/kg (ip)
developed extensive necrosis of the Clara cells in the lung within
24 h, but no necrosis was observed in the liver or kidney at this
time. The loss of Clara cells was associated with a significant
decrease in pulmonary cytochrome P-450 content and activity
[113]. The susceptibility of the Clara cells was considered to be
due to activation of vinylidene chloride by cytochrome P-450 in
these cells. This conclusion was also reached by Forkert et al.
[55] who showed that covalent binding of vinylidene chloride
products to cellular macromolecules in the lung accompanied lung
toxicity in CD-1 mice given an identical dose to that given in the
study described above (section 6.1.5). Degenerative changes
occurred in Clara cells as early as 1 h following treatment with
vinylidene chloride and were characterized by mitochondrial
swelling and aggregation of chromatin against the nuclear membrane.
Cell death was apparent at 2 h and, by 24 h, the majority of Clara
cells were exfoliated [56]. However, these authors concluded that
there was a lack of correlation between the extent of covalent
binding and either lung or liver toxicity [55].
8.1.3.2 Eyes and skin
Little information is available on the effects of vinylidene
chloride on the eyes and skin.
It is moderately irritating to the eyes of rabbits causing
transient corneal injury and is also a skin irritant in the rabbit
[222]. Rylova, [192] reported that vinylidene chloride was an
irritant for the eyes of rats, mice, guinea-pigs, and cats.
Transient redness was observed following application to shaved
rabbit skin. The stabilizer ( p- methoxyphenol) in vinylidene
chloride preparations may contribute to irritation.
8.1.4. Summary of acute toxicity
Vinylidene chloride may cause irritation of the skin and eye,
depression of the central nervous system, and acute toxic effects
on the heart, lung, liver, and kidney. The variation in estimations
of the acute LC50 for vinylidene chloride is considerable. This
can be explained partially by inaccuracies that arise because of a
non-linear concentration-mortality relationship. Generally, mice
are more susceptible than rats and males are more susceptible than
females. The toxic effects are dependent on cytochrome P-450
activity (which may also be involved in detoxification) and can be
exacerbated by glutathione depletion. Hepatotoxicity may be
enhanced by ethanol and thyroxine, inhibited by dithiocarb and
(+)- catechin, and modulated by acetone.
8.2. Short-Term Exposures
8.2.1. Inhalation
In the study by Prendergast et al. [173], groups of various
animal species were continuously or repeatedly exposed to
vinylidene chloride through inhalation for 90 days. The results
are shown in Table 8. In survivors of continuous exposure at a
concentration of 101 mg/m3 or less, no histopathological changes
could be attributed to vinylidene chloride. At 189 mg/m3, livers
from surviving dogs, monkeys, and rats showed morphological
changes consisting of fatty metamorphosis, focal necrosis,
haemosiderin deposition, lymphocytic infiltration, bile-duct
proliferation, fibrosis, and pseudolobule formation, particularly
in dogs. All rats displayed nuclear hypertrophy of the kidney
tubular epithelium. Non-specific inflammatory changes were seen in
the lungs of a majority of the animals. Liver alkaline phosphatase
activity and serum glutamic-pyruvic transaminase activity were
measured in rats and guinea-pigs and found to be elevated. Dogs,
monkeys, and rats showed reduced weight gain. The repeated
exposures are considered to be more relevant to human
occupational exposure. Gross examination of survivors showed a
high incidence of lung congestion in rabbits, monkeys, rats, and
guinea-pigs. Some fatty infiltration and several cases of focal and
sub-massive necrosis were observed in guinea-pig liver sections.
Table 8. Mortality of animals exposed to vinylidene chloride by inhalationa
__________________________________________________________________________
Mortality ratiob
_______________________________________________
Concentration Extent of Rat (Long Guinea- Rabbit Beagle Squirrel
of vinylidene exposurec Evans or pig (New dog monkey
chloride Sprague- (Hartley) Zealand
(mg/m3) Dawley) White)
_________________________________________________________________________
395 ± 32 A 0/15 0/15 0/3 0/2 0/3
189 ± 6.2 B 0/15 7/15 - 0/2 3/9
101 ± 4.4 B 0/15 3/15 0/3 0/2 2/3
61 ± 5.7 B 0/15 3/15 - 0/2 0/9
20 ± 2.1 B 2/45 2/45 - 0/6 1/21
no treatment B 7/304 2/314 2/48 0/34 1/57
_________________________________________________________________________
a From: Prendergast et al. [173].
b Mortality ratio shows the number of animals that died divided by the
number with which the study commenced.
c A = 30 exposures, 8 h/day, 5 days/week.
B = continuous 90-day exposure.
Shortly after this study, Gage [57] investigated the toxicity
of vinylidene chloride in Alderley Park rats. In this case, groups
of 4 male and 4 female rats were exposed to 2000 mg vinylidene
chloride/m3 (500 ppm) for 6 h per day over a period of 20 days.
Nose irritation and retarded weight gain were observed, and, at
autopsy, liver cell degeneration was detected by histology. At 800
mg/m3 (200 ppm) (4 male and 4 female rats), slight nose
irritation was seen, but the organs were normal on autopsy.
Inhalation studies [152, 176] demonstrated minimal recoverable
liver cell cytoplasmic vacuolation in Sprague-Dawley rats (20
animals per sex) after 90 days exposure for 6 h per day, 5
days/week, to vinylidene chloride at 100 or 300 mg/m3 (25 or 75
ppm). At both dose levels, body weight, haematology, urinalyses,
blood urea nitrogen, serum alkaline phosphatase and glutamic
pyruvic transaminase activities, gross pathology, organ weights,
kidneys, heart, testes, and brain were normal.
Maltoni & Patella [138] noted a greater toxicity in rats than
that reported by Gage [57]. The effects of 4 h exposure, 4-5 days
per week, for 28 days, were studied in Sprague-Dawley rats and
Swiss mice (40-800 mg/m3 (10-200 ppm)), Balb/c, C3H, and C57B1 mice
(600-800 mg/m3 (150-200 ppm)), and Chinese hamsters (100 mg/m3 (25
ppm) only). The exposure periods were reduced at or above 200
mg/m3 (50 ppm) in mice because of severe acute toxicity, and the
800 mg/m3 (200 ppm) treatment of rats was reduced to 600 mg/m3 (150
ppm) after 2 days because of toxic effects. A minimum of 30
animals was used in all groups exposed to vinylidene chloride.
The weight of animals, clinical signs of toxicity, mortality,
and histopathological changes were monitored at, or up to, 28 days,
except in the case of mice exposed to 400-800 mg/m3 (100-200 ppm),
when the observation period was 9 days. Histopathological studies
indicated that the liver and kidneys were the major target organs.
The toxicity varied with animal species and strain, susceptibility
being in the following order: Swiss mice >Balb/c mice >C3H mice
>C57B1 mice >rats. In general, females were less responsive than
males, with the exception of female C3H mice, which were more
susceptible than the females of the other strains tested. An
association between the occurrence of acute toxicity and the
reported carcinogenicity in the Swiss mouse was noted (section
8.7.4). Another comparison study also indicated marked strain and
sex differences in the response of mice to vinylidene chloride
[153]. The mice (10 males and 10 females per dose level) were
exposed to 220, 400, or 800 mg vinylidene chloride/m3 (55, 100, or
200 ppm) for 6 h/day, 5 days/week, for a total of 10 exposures.
Only at 800 mg/m3 (200 ppm) were exposure-related deaths observed
and the rates of mortality were greater in the males than in the
females in Ha (ICR), CD-1 and CF-W mice, but no sex-related
differences in mortality were observed in B6C3F mice. Gross and
histopathological examinations indicated that nephrotoxicity
accounted for mortality in the male mice, but this was not the case
in female mice. Hepatotoxicity was considered to be the cause of
death in Ha (ICR) and B6C3F1 mice. The greatest sensitivity to
renal toxicity was seen in male CF-W mice (a strain derived from
the Swiss-Webster strain, believed to be genetically comparable to
Maltoni's Swiss mouse).
The greater susceptibility to vinylidene chloride of mice
compared with rats was also shown by Short et al. [204], agreeing
with the findings from acute single exposures (section 8.1). After
2 days exposure to 240 mg/m3 (60 ppm), 8/10 and 0/10 deaths were
recorded in male CD-1 mice and male CD rats, respectively. No
mice survived a longer period of exposure at 240 mg/m3 (60 ppm).
Serum glutamic oxaloacetic transaminase and glutamic pyruvic
transaminase levels were raised in both rats and mice, but more so
in the latter. Severe hepatotoxicity and nephrotoxicity were
observed in mice at autopsy.
A comparative study on the short-term toxicity of vinylidene
chloride in male Sprague-Dawley rats and in both sexes of Swiss-
Webster mice was reported by Oesch et al. [156]. Animals (minimum
of 10 per group) were exposed to an atmosphere containing
vinylidene chloride at 40 or 200 mg/m3 (10 or 50 ppm) (mice) or
800 mg/m3 (200 ppm) (rats) for 6 h, on 1, 3, or 8 days, and killed
one day after the last treatment. The majority of male mice
exposed to 200 mg/m3 (50 ppm) for 8 days did not survive. However,
female mice survived this treatment as did rats exposed to 800
mg/m3 (200 ppm). Various changes in the activities of
monooxygenase, epoxide hydrolase, and glutathione transferase
enzymes occurred in animals treated with vinylidene chloride. The
enzyme changes could contribute to the relative susceptibility of
the animals, according to the balance of activating and detoxifying
activity. In particular, cytosolic glutathione transferase
activity towards the substrate 2,4-dinitrochlorobenzene was
decreased in the kidneys of male mice (an organ susceptible to
carcinogenicity (section 8.7.1)), but not in the kidney of rats or
female mice or in the liver of either of these species (where
activity was either unchanged or enhanced).
A study on rabbits has also been reported by Lazarev [116].
Bronchitis, degenerative changes in the liver and kidney, and an
increase in the rate of proliferation of lymphoid tissue in the
spleen were observed in animals exposed to concentrations of 500-
2000 mg vinylidene chloride/m3 (125-500 ppm) for 3 h/day over a
period of 4 months.
In summary, a number of studies have indicated the particular
susceptibility of the male Swiss mouse to kidney toxicity. This
strain, sex, and species selectivity has important implications
regarding the specificity of the carcinogenic action of vinylidene
chloride (section 8.7.4).
8.2.2. Oral
In a 90-day study, vinylidene chloride was incorporated in
the drinking-water of male and female Sprague-Dawley rats at
nominal concentrations of 0, 60, 100 or 200 mg/litre [152,176].
Even at the highest concentrations administered (equivalent to 19-
26 mg/kg body weight daily), only minimal, reversible liver
cytoplasmic vacuolation was observed, with no abnormalities in
any of the other parameters investigated (section 8.2.1). Maltoni
& Patella [138] also noted a lack of lethality and clinical signs
of toxicity in Sprague-Dawley rats (50 of each sex) orally dosed by
gavage with 0, 0.5, 5, 10, or 20 mg vinylidene chloride/kg for 28
consecutive days. Four female and 4 male beagle dogs were given
6.25, 12.5, or 25 mg vinylidene chloride/kg in peanut oil
incorporated in gelatin capsules, daily for 97 days [177]. No
significant differences were seen between these animals and
controls with respect to appearance and demeanor, mortality, body
weight, food consumption, haematology, urinalysis, clinical
chemistry, and organ weights. There was also no depletion in
hepatic non-protein sulfhydryl levels in the liver or kidneys.
Some evidence for hepatotoxicity was provided by Siegers et al.
[208] using higher levels of orally administered vinylidene
chloride. Male Wistar rats were given 0.125 g vinylidene
chloride/kg in olive oil, by gavage, twice weekly for 2 weeks,
followed by a similar treatment at 0.2 g/kg for 2 weeks.
Hepatotoxicity was evidenced by mild increases in serum
sorbitol dehydrogenase and aminotransferases. Ethanol co-treatment
(5% in drinking-water) with the 0.2 g/kg dose of vinylidene
chloride enhanced toxicity leading to 6 deaths out of 10 animals.
Simultaneous application of dithiocarb or (+)-catechin with
vinylidene chloride led to total protection against lethal effects.
The effects of these agents have been discussed elsewhere (section
8.1.2.1).
8.3. Long-Term Exposure
A number of studies described in this section are also
discussed in section 8.7 in relation to carcinogenicity.
8.3.1. Inhalation
Sprague-Dawley rats, 84-86 of each sex, 6-7 weeks of age at the
start of the study, were exposed to vinylidene chloride vapour at
0, 40, or 160 mg/m3 (0, 10, or 40 ppm) for, 6 h/day, 5 days/week,
for 5 weeks, after which the levels of vinylidene chloride were
changed to 0, 100, and 300 mg/m3(0, 25, and 75 ppm) [178,179,180].
After exposure for a total of 18 months, the rats were observed for
a further 6 months. Additional animals were used for interim kills
at 1, 6, and 12 months. No clinical signs of toxicity were seen in
the exposed groups. Mean body weight gain was reduced at both dose
levels during the period of 8-13 months in males, but was not
reduced in females. Mortality in exposed groups was only slightly
higher than that in the controls (not different at the 40-100 mg/m3
(10-25 ppm) dose levels in males) and only in the latter part of
the study. Histopathological studies indicated increased
cytoplasmic vacuolation in the livers of exposed animals at 6 and
12 months of exposure. During the 6-month postexposure period of
the study, the hepatic changes were no longer discernible,
indicating reversibility.
There was also evidence of liver damage in CD-1 mice and CD
rats (groups of 36 males and 36 females) exposed to 220 mg
vinylidene chloride/m3 (55 ppm) (6 h/day, 5 days/week); the
animals were about 2 months old at the start of the study. Four
animals of each species and sex were killed for examination after
1, 2, 3, 6, and 9 months of treatment. The remaining animals were
killed at 12 months [118]. Groups of 100 animals were used as
controls. No effects were seen regarding haematology, clinical
blood chemistry, pulmonary macrophage count, cytogenic analysis of
bone marrow, X-ray examination of extremities, serum alpha-
fetoprotein, collagen content of liver, and lung and serum or
urinary aminolevulinic acid (ALA) (collagen and ALA were not
measured in the mice). However, mice exposed to vinylidene
chloride for 6-12 months had enlarged and basophilic hepatocytes
with enlarged nuclei, focal degeneration, and necrosis. A mild to
markedly severe focal disseminated vacuolization of the liver was
seen in the treated rats.
A similar long-term toxicity study was carried out by Hong et
al. [79] on male and female CD-1 mice and CD rats exposed to 220 mg
vinylidene chloride /m3 (55 ppm) or filtered air (controls), for 6
h/day, 5 days/week. The numbers of rats used per sex group were 4,
8, 8, and 14-16, and these were exposed for 1, 3, 6, and 10 months,
respectively. Mice (8, 8, and 12 animals per sex group) were
exposed for 1, 3, and 6 months, respectively. Animals were aged 2
months at the start of the study and were observed for 12 months
after treatment. No histopathological changes were observed as a
result of these treatments with vinylidene chloride. A total of
11/24 mice exposed for 6 months died or were terminated in a
moribund condition (compared with 11/56 controls). Only 3 rats died
following exposure for 6 months or less (20/30 rats died in the
group exposed for 10 months compared with 13/32 controls).
Long-term inhalation toxicity was also studied in rats, mice,
and hamsters by Maltoni et al. [141]. Groups of male and female
Sprague-Dawley rats (60 of each) were exposed to 40, 100, 200, 400,
600, or 800 mg vinylidene chloride/m3 (10, 25, 50, 100, 150, or 200
ppm), for 4 h/day, 4-5 days/week, for 52 weeks. Unexposed control
groups consisted of 100 rats of each sex. The rats were 16 weeks
old at the start of the study. At spontaneous death, hepatocyte
vacuolization, cloudy swelling, fatty degeneration, necrobiosis,
and necrosis were more frequent (57.6%) in rats exposed to 800-600
mg/m3 (200-150 ppm) than in control animals (20.5%). The highest
tolerable dose for long-term exposure in rats was 600 mg/m3 (150
ppm). In Swiss mice (aged either 9 or 16 weeks at the start of the
study) exposed to 40 or 100 mg vinylidene chloride/m3 (10 or 25
ppm) for the same exposure periods (minimum of 30 mice per sex per
dose level), changes were seen in the liver and kidneys that were
compatible with changes seen in long-term studies on control
animals. However, a higher incidence of regressive or phlogistic
changes was seen in the kidneys with renal adenocarcinoma (section
8.7.1) at 100 mg/m3 (25 ppm). A high mortality was seen at 200
mg/m3 (50 ppm). Those that survived only 4 exposures had hepatic
fibrosis, which was considered to be due to repair of necrosis.
Chinese hamsters (30 male and 30 female) exposed to a
concentration of 100 mg vinylidene chloride/m3 (25 ppm) for
periods identical to those given above for rats and mice, showed no
signs of altered histology compared with controls (18 male and 17
female) at spontaneous death.
Thus, the most significant observation on toxicity from long-
term inhalation studies is that of dose-dependent kidney damage in
male Swiss mice at dose levels that were not nephrotoxic in female
mice or in other species tested. The findings have important
implications in the light of similar tissue, sex, and species
specificities in carcinogenicity (section 8.7.1).
8.3.2. Oral
The long-term oral toxicity of vinylidene chloride was
investigated by Maltoni et al. [141] in Sprague-Dawley rats at 0.5,
5, 10, or 20 mg/kg (given once daily by stomach tube, 4-5
days/week, for 52 weeks). Animals were aged 9 or 10 weeks at the
start of the study. Fifty animals of each sex were used per dose
group with 100 controls (except for the 0.5 mg/kg group where there
were 160 controls). No signs of toxicity were reported from a
complete autopsy after 147 weeks (or 136 weeks, 0.5 mg/kg) except
that "hepatocyte vacuolization, cloudy swelling, fatty degeneration,
necrobiosis, and necrosis were found in some animals in treated as
well as in control groups".
In a separate study, 48 Sprague-Dawley rats of each sex (aged
6-7 weeks at the start of the study) were given vinylidene chloride
in the drinking-water at the following dose levels for 2 years: 7,
10, or 20 mg/kg body weight for males and 9, 14, or 30 mg/kg for
females (nominally 50, 100, and 200 mg/litre drinking-water).
Eighty rats per sex were dosed as control groups without vinylidene
chloride treatment [177, 179, 180]. Various parameters were
monitored, as indicated for the 90-day study (section 8.2.1).
Slightly increased cytoplasmic vacuolation of hepatocytes and
hepatocellular fatty change were the only evidence of toxicity and
occurred at all dose levels in the females, but only at 20 mg/kg
body weight (200 mg/litre drinking-water) in the males.
A US National Toxicology Programme study [154] indicated
chronic renal inflammation in male and female F344/N rats (50 per
group) given 5 mg vinylidene chloride/kg in corn oil, by gavage, 5
times/week, for 104 weeks. At 1 mg/kg, no renal toxicity was
observed. In all treated rats, the clinical signs and histopathology
of other organs were the same as in control rats. This group also
studied long-term oral toxicity in male and female B6C3F1/N mice
(50 per group) administered 2 or 10 mg/kg in corn oil. At 10
mg/kg, necrosis of the liver was evident in male but not in female
mice, but the reverse was true at the 2 mg/kg dose level. However,
the sponsors found defects in the conduct of the study and it could
not be satisfactorily evaluated.
Ponomarkov & Tomatis [172] gave rats an oral dose of vinylidene
chloride (50 mg/kg) weekly for the life span of the animal
following an initial in utero exposure (section 8.7.2 for details).
Rats that died up to 30 weeks after the start of oral dosing (7/89
males and 7/90 females) showed congestion of the lungs and
kidneys. At up to 80-90 weeks, haemorrhages and multiple lobular
necrosis of the liver were observed. The numbers of animals that
survived for 90 weeks were 71/89 (males) and 75/90 (females)
compared with 49/50 and 47/53 male and female controls,
respectively. Some of the treated survivors of 90 weeks showed
degenerative lesions of liver parenchymal cells.
8.4. Toxicity in vitro
As revealed in in vivo studies ([145] section 8.1.3.1),
in vitro studies with rat liver microsomes showed that calcium pump
activity was inhibited in a dose-dependent manner by vinylidene
chloride in the presence of NADPH. Malonic dialdehyde production
(a consequence of lipid peroxidation) was not associated with
inhibition of the calcium pump [182].
These findings stimulated Long & Moore [126] to test whether
vinylidene chloride treatment of hepatocytes could raise cytosolic
Ca2+ concentrations. Isolated hepatocytes from Sprague-Dawley rats
were exposed to vinylidene chloride (4 mmol/litre). Within 5 min,
the concentration of Ca2+ (estimated from the activity of
glycogen phosphorylase a) was raised to 0.3 µmol/litre compared
with 0.04 µmol/litre in vehicle-treated controls. In separate
studies, the concentration of Ca2+ was measured in hepatocytes
loaded with quin 2 (a sensitive fluorescent indicator of
cytoplasmic concentrations of ionized calcium). Within 20 seconds
of addition of vinylidene chloride at 4 mmol/litre, Ca2+ levels
were increased from 0.26 ± 0.05 µmol/litre to 0.59 ± 0.06 µmol/litre.
Disruption of intracellular Ca2+ homeostasis may prove to be a
mechanism that can contribute to vinylidene chloride
hepatotoxicity.
8.5. Mutagenicity and Other Genotoxicity Assays
8.5.1. Interaction with DNA
The fact that a number of metabolites of vinylidene chloride
are reactive and covalently bind to cellular macromolecules and the
nucleophile glutathione has already been discussed in section
6.1.5. The specific covalent interaction with DNA has been little
studied. As reported in section 8.1.1.2, DNA binding in vivo in
rats and mice exposed to [14C]-vinylidene chloride at 40 mg/m3 (10
ppm) (rats) and 40 or 200 mg/m3 (10 or 50 ppm) (mice) for 6 h was
minimal, though DNA adducts were evident from the covalently bound
radiolabel [186]. Adduct formation was greater in mice than in
rats and greater in the kidneys than in the liver. Binding was
dose dependent in the kidneys of mice (equivalent to 30 adducts/106
nucleotides at 200 mg vinylidene chloride/m3 (50 ppm) and 11
adducts/106 nucleotides at 40 mg/m3 (10 ppm). Incorporation of
radiolabel into DNA during synthesis or contamination of DNA
with other radiolabelled macromolecules cannot be excluded.
It was not possible to trap any alkylating metabolites in
vitro with 4-(4-nitrobenzyl)-pyridine, following incubation of
vinylidene chloride with a mouse liver microsomal system [15].
8.5.2. Genotoxicity in bacteria
The mutagenicity of vinylidene chloride in bacteria has been
demonstrated by a number of research workers and has been related
to the asymmetry of the putative epoxide metabolite [73]. The
positive effect was not seen in every study (Table 9) and in some
cases this may have been due to volatilization of the compound.
Vinylidene chloride (of unspecified purity) in air at 0.2, 2, or
20% for 4 h induced revertants (gene mutations) in the TA1530 and
TA100 strains of Salmonella typhimurium in the presence of NADPH-
supplemented 9000-g liver supernatant (S9) from phenobarbital
pretreated male OF-1 mice. Mutagenicity towards TA100 was also
catalysed by mouse kidney and lung 9000-g supernatants [14]. These
authors demonstrated that mutagenicity was not due to the
stabilizer 4-methoxyphenol. The role of cytochrome P-450 in
metabolic activation was evident from the lack of mutagenicity in
the absence of NADPH and from the observed greater mutagenicity of
vinylidene chloride towards S. typhimurium TA100 when mice used
for S9 donation were pretreated with phenobarbital. The role of
reactive electrophiles in mutagenicity was supported by the marked
inhibition of mutagenicity by the nucleophiles N- acetyl-cysteine
and N- acetyl-methionine. Protection against bacterial
mutagenesis was also afforded by pretreatment of rats (which
provided the hepatic S9) with pregnenolone-16 alpha-carnonitrile,
amino-cetonitrile, or disulfiram [15]. The mechanism(s) of these
effects are not known but may be via increased detoxification of
vinylidene chloride or its metabolites. In this study, 3-
methylcholanthrene as well as phenobarbital pretreatment of rats
provided S9 that had up to 2-fold greater capacity than untreated
rat liver preparations to activate vinylidene chloride (99% pure)
to bacterial mutagens. Liver specimens showing no pathological
lesions, obtained from 4 adult human patients for diagnostic
purposes provided S9 fractions that activated vinylidene chloride
to mutagens detected by S. typhimurium TA100 at approximately
one-fifth the activity of untreated mouse liver S9.
Jones & Hathaway [101] found vinylidene chloride (unspecified
purity) to be only very weakly mutagenic in S. typhimurium strain
TA1535 in the presence of liver (1.6 x background) and kidney (2.3
x background) S9 from untreated male Alderley Park Swiss-derived
albino mice. Exposure was via the atmosphere (5% vinylidene
chloride for 72 h) inside gas-tight culture vessels. Use of S9
from mice pretreated with Aroclor 1254 enhanced mutagenicity in
the test. In contrast, liver S9 preparations from male Sprague-
Dawley rats were able to mediate mutagenicity only after Aroclor
pretreatment and at a mutation frequency approximately 25% of that
seen with the corresponding mouse S9. These data are in agreement
with the greater oxidative metabolism observed in the mouse
compared with the rat (section 6.1.4). In this study, liver S9
from uninduced marmoset or from a single, apparently uninduced,
human subject was not able to mediate the bacterial mutagenicity of
vinylidene chloride, though a positive result was obtained using a
liver S9 from a human subject who had received long-term
phenobarbital medication. Thus, human beings appear to be more
similar to rats than to mice with respect to hepatic metabolic
activation of vinylidene chloride.
Exposure of S. typhimurium strain TA100 to 2% vinylidene
chloride (99% pure) in air in the presence of a hepatic S9 fraction
from untreated or phenobarbital-pretreated male OF-1 mice caused a
linear increase in the mutagenic response up to 4 h of exposure
[137]. This was in agreement with the dependence of bacterial
mutagenicity on duration of exposure shown by Waskell [239].
Again, phenobarbital pretreatment enhanced the metabolic
activation.
Oesch et al. [156] detected the mutagenicity of vinylidene
chloride (99.996% pure) with S. typhimurium strains TA1535,
TA1537, TA92, TA100, TA98, and Escherichia coli strain WP2 uvrA
following exposure to vinylidene chloride in the atmosphere for 4
h at 1500-90 000 mg/m3 (375-22 500 ppm) and with an NADPH-
fortified liver S9 fraction from untreated male Swiss-Webster mice
as an activation system. Again, it was established that the
inhibitor, methoxyphenol, was not mutagenic. A comparative study
was made of kidney and liver S9 fractions from different species
regarding their ability to activate vinylidene chloride to
mutagens detected by S. typhimurium strain TA100. The order of
ability to mediate mutagenesis was as follows: male and female
Swiss-Webster and C57BL/6J Han mouse liver and Chinese hamster
(Fue:FUST) liver >Sprague-Dawley rat liver >human liver
>Chinese hamster kidney >male mouse kidney (both strains) >rat
kidney and female mouse kidney. Metabolic activation was not
accomplished or was very weak in the last two tissue samples. As
with the study by Bartsch et al. [14], mutagenicity was inhibited
by a nucleophile (in this case glutathione) and it was also found
not to be affected by the addition of purified microsomal epoxide
hydrolase to the test. Despite changes in drug-metabolizing
enzymes as a result of pretreatment of rats and mice with
vinylidene chloride (section 8.2.1), S9 fractions derived from
treated animals were not found to have a greater metabolic
activation capacity in the mutagenicity assay.
The mutagenicity of vinylidene chloride towards S. typhimurium
TA100 was detected by Baden et al. [10, 11, 12] not only following
gaseous exposure for 8 h at a level of 3% but also after incubation
in suspension at 3% for up to 2 h. In agreement with the data
given above, Aroclor pretreatment of rats enhanced the ability of
liver S9 to mediate vinylidene chloride mutagenicity and human
liver S9 was also capable of catalysing the formation of mutagens.
The mutagenicity of vinylidene chloride (unspecified purity) in
liquid suspension assays (2.5 mmol vinylidene chloride/litre; 2 h
incubation) was also shown to be positive by Greim et al. [64], as
evidenced by reverse gene mutation at one locus in E. coli strain
K12. When monitored for reverse gene mutation at 2 other loci, or
forward mutation at 1 locus, a negative result was obtained, but
only one dose level was used in this study.
Results of spot tests with S. typhimurium strains TA1950,
TA1951, TA1952, TA1535, TA1538, TA100, and TA98 were reported
briefly by Cerna & Kypenova [26]. Vinylidene chloride
(unspecified purity) was added to plates at 1, 10, and 100% in DMSO
(0.05 ml). At 100%, vinylidene chloride produced reversion of
both base substitution and frame-shift mutations in the absence of
a metabolic activation system. When tested in a host-mediated
assay (female ICR mice) at doses quoted as LD50 and 50% LD50, a
significant increase in the revertants of S. typhimurium TA1950,
TA1951, and TA1952 was reported that was inversely related to dose.
A further brief report was given by McCarroll et al. [129] in
which vinylidene chloride (1.6 and 3%, "metabolically activated
doses") was shown to be markedly mutagenic to S. typhimurium TA1535
and TA100 in a microfluctuation assay involving a 72-h exposure.
Chloroacetic acid, a metabolite of vinylidene chloride, was
found not to be mutagenic to S. typhimurium strains TA100, TA1535,
TA1537, TA1538, and TA98 using a plate incorporation protocol
without the addition of an S9 system [128]. Chloroacetic acid
did not cause mutagenicity in S. typhimurium strain TA1530 in
the presence or absence of S9 from phenobarbital-pretreated mice
[136].
8.5.3. Genotoxicity in yeast
Bronzetti et al. [19] studied the mutagenicity of vinylidene
chloride (99.57% pure) in yeast. Vinylidene chloride (0-50
mmol/litre for 2 h preincubation in suspension) did not induce
reverse gene point mutation or produce mitotic gene conversion in a
diploid strain (D7) of Saccharomyces cerevisiae in the absence of a
microsomal activation system. However, in the presence of a post-
mitochondrial supernatant from mice pretreated with Aroclor 1254,
a dose-related induction of revertants and convertants was seen
between 30 and 50 mmol vinylidene chloride/litre. Genotoxicity was
also examined in yeast exposed to vinylidene chloride in an
intrasanguinous host-mediated assay. The host species i.e., male
Swiss albino CD mice, were given vinylidene chloride orally in an
acute single dose study (400 mg/kg) or in a short-term study (100
mg/kg, 5 days per week followed by 200 mg/kg on the day of the
assay). The yeast cells (4 x 108) were injected via the retro-
orbital sinus immediately before the administration of vinylidene
chloride on the day of the assay and were recovered from various
organs 4 h later. A positive result for the induction of revertants
and convertants was found for yeast cells recovered from the liver
and kidneys, but the results were negative or only very weakly
positive for both parameters in cells from the lung.
8.5.4. Genotoxicity in plants
After exposure to 5152 mg vinylidene chloride/m3 (1288 ppm)
for 6 h, inactivation of a dominant gene (forward mutation) in
Tradescantia (hybrid clone 4430) was not observed [228]. However,
at 88 mg vinylidene chloride/m3 (22 ppm) for 24 h, a positive
result was recorded. Although this finding indicates the
potential for mutagenicity in plants, the dose-response
relationship needs to be clarified.
8.5.5. Genotoxicity in mammalian cells in vitro
Costa & Ivanetich [33] investigated the effects of vinylidene
chloride (unspecified purity) on DNA repair (unscheduled DNA
synthesis) in isolated hepatocytes from male Long-Evans rats that
had been pretreated with phenobarbital. When administered to
hepatocytes at a maximum subtoxic dose level (2.1 mmol/litre),
vinylidene chloride stimulated unscheduled DNA synthesis as shown
by enhanced incorporation of deoxy-[5-3H]-cytidine into DNA.
However, the mutagenicity of vinylidene chloride was found to
be negative when tested at two loci (induction of resistance to 8-
azaguanine and ouabain) in V79 Chinese hamster cells in the
presence of a post-mitochondrial supernatant from phenobarbital
pretreated rats and mice [43]. In this test, the V79 cells were
exposed to 2 and 10% vinylidene chloride (unspecified purity) in
air for 5 h. This treatment led to dose-dependent cytotoxicity in
the presence of rat but not mouse liver post-mitochondrial
fractions. Using a similar protocol, Huberman et al. [82]
demonstrated that the vinylidene chloride metabolite, mono-
chloroacetic acid, in accordance with negative mutagenicity in
bacteria (section 8.5.2), did not show any activity in inducing 8-
azaguanine and ouabain resistant mutants in Chinese hamster V79
cells, when tested up to a level of 2.5 mmol/litre.
In a survey of the cytogenetic effects of 60 chemicals on
cultured mammalian cells, Sasaki et al. [193] reported that
vinylidene chloride (3 x 10-2 and 3 x 10-3 mol/litre) failed to
produce chromosomal breaks in Chinese hamster (Don 6) cells. A
microsomal metabolic system was not included in the assay. Other
research workers have also carried out cytogenetic studies on
mammalian cells in vitro. McCarroll et al. [129] analysed Chinese
hamster ovary cells (CHO) for sister chromatid exchange following
exposure to vinylidene chloride (unspecified purity). Consistent
and dose-related increases resulted from a 24-h exposure to
atmospheres containing 1.8, 3.6, 5.4, or 7% vinylidene chloride.
Only at 7% was the effect significant and shorter exposure
periods provided negative results. In this brief report it was not
stated whether a mammalian hepatic microsomal fraction was included
in the study. Sawada et al. [196] also investigated the ability of
vinylidene chloride (99% pure) to induce chromosomal aberrations
and sister chromatid exchange in a Chinese hamster cell line (CHL).
Cells were treated for 6 h with a range of dose levels between 0
and 2 mg/ml, at which toxicity was observed. In the presence of a
liver S9 fraction from PCB (KC-400)-pretreated male F344 rats,
vinylidene chloride gave a relatively weak, but significant,
increase in the incidence of sister chromatid exchanges. The
result was negative in the absence of S9. The findings were similar
when chromosomal aberrations were used as an end point for genetic
toxicity. In the presence (but not in the absence) of S9, a dose-
dependent induction of chromosomal aberrations was seen (14%
aberrant cells at 0.25 mg/ml and 54% at 1.5 mg/ml). These effects
were not elicited by p- methoxyphenol (the inhibitor). The role
of cytochrome P-450 in the metabolic activation of vinylidene
chloride was shown by the inhibition of the enhancement of
aberrations with metyrapone and a protective effect was seen by
the addition of glutathione. Two metabolites of vinylidene
chloride, chloroacetyl chloride and chloroacetic acid, were
negative in these tests in support of the theory that vinylidene
chloride oxide may be the active genotoxic metabolite.
8.5.6. Genotoxicity in mammalian cells in vivo
Evidence for the detectable but minimal covalent binding of
[14C]-vinylidene chloride in vivo has already been discussed [186]
(sections 8.1.1.2 and 8.5.1). As part of this study, the ability
of vinylidene chloride to induce unscheduled DNA synthesis (DNA
repair) was also investigated in mice. The measurement of DNA
repair by the uptake of [3H]-thymidine into DNA was hampered by
incomplete inhibition of replicative DNA synthesis by hydroxyurea.
Though unscheduled DNA synthesis was minimal, the slight
increase was statistically significant in mouse kidney at the 200
mg vinylidene chloride/m3 (50 ppm) exposure level.
Short et al. [203] investigated the incidence of germinal
mutations of the dominant lethal type in 11 male CD rats exposed
through inhalation of 220 mg vinylidene chloride/m3 (55 ppm) for 6
h/day and 5 days/week. During week 11 of exposure, the treated
animals were housed with 2 virgin females until mating had taken
place. Neither pre-implantation nor post-implantation losses
were observed in the pregnancies that resulted from mating with
vinylidene chloride-exposed males; thus, dominant lethal mutations
were not produced. Anderson et al. [3] also did not find any
evidence for a dominant lethal effect. Male CD-1 mice were exposed
by inhalation to 40, 120, or 200 mg vinylidene chloride/m3 (10, 30,
and 50 ppm), for 6 h/day, over 5 days. Fifteen or 16 days after
caging the males with untreated females (over a 2-month period),
the females were killed for examination of the uteri. As a result
of toxicity, only 6/20 and 18/20 male mice survived exposure to
vinylidene chloride at 200 and 120 mg/m3 (50 and 30 ppm),
respectively. No effects were seen in the frequency of pregnancy
or in the number of post-implantational early fetal deaths. There
was also no evidence of pre-implantational egg losses as indicated
by the total implants/pregnant female.
A number of in vivo cytogenetic studies reported on vinylidene
chloride have been unable to show a significant positive response.
In the inhalation study by Lee et al. [118] (details given in
sections 8.3.1 and 8.7.1), cytogenetic analysis of bone marrow
revealed no change following exposure of CD-1 mice or CD rats to
220 mg vinylidene chloride/m3 (55 ppm) , for 6 h/day, 5
days/week, for up to 12 months. Cerna & Kypenova [26] gave single
and repeated (l dose per day for 5 days) ip doses of vinylidene
chloride to female ICR mice. Neither a single dose (quoted as 1/2
LD50) nor repeated doses (quoted as 1/6 LD50) induced chromosomal
aberrations. In the long-term study by Quast et al. [178] (section
8.3.1 for details), cytogenetic evaluations on 4 rats/sex exposed
to 0, 100, or 300 mg vinylidene chloride/m3 (0, 25, or 75 ppm) for
6 months did not show any adverse effects. A negative in vivo
cytogenetic effect was also borne out by the work of Sawada et al.
[196]. In this study, 6 male ddY mice per group were given
vinylidene chloride (99% pure) by gavage, either as a single dose
(0200 mg/kg) or as 4 doses (1100 mg/kg each) given at 24-h
intervals. No increase in the frequency of micronucleated
erythrocytes was observed in the bone marrow. A micronucleus
test in the liver and blood of fetuses in vinylidene chloride-
treated mice was also negative (section 8.6). These authors
suggest that, in contrast to the in vitro studies where positive
genotoxicity has been found, the life time of the reactive
metabolites of vinylidene chloride may be too short for a
sufficient amount to have reached the target cells analysed in vivo.
However, this conclusion conflicts with the findings of Hofmann
& Peh [77] who studied chromosome aberrations in 50 metaphase bone
marrow cells of 4 male and 5 female Chinese hamsters after short-
term inhalation (6 h/day, 5 days/week for 6 weeks) of either 120 or
400 mg vinylidene chloride/m3 (30 or 100 ppm). In comparison with
control groups (fresh air), there was no effect on mortality but 78
times more aberrations were observed in the animals exposed to 120
mg vinylidene chloride/m3 (30 ppm) and 910 times more aberrations
were seen at the 400 mg/m3 (100 ppm) dose level. Using the same
technique for analysis of chromosomal aberrations, Zeller & Peh
[247] studied the effects of a single oral dose (216 mg/kg) in
Chinese hamsters. Animals that were killed 6 h after dosing showed
a higher number of aberrations (1.6%) than animals given the same
doses and killed after 24 h (1.2%) and 48 h (1.4%). The untreated
control animals showed the lowest rate of aberrations (0.6%). No
statistical test was carried out.
8.5.7. Summary
Data on mutagenicity and other short-term tests for
carcinogenicity are summarized in Table 9. Genotoxicity has been
observed in prokaryotic (in the presence of mammalian enzymes)
and eukaryotic cells in vitro. However, genotoxicity was not
observed in the majority of tests carried out on mammals in vivo .
Reports of an effect in the latter are restricted to the observation
of chromosomal aberrations in the bone marrow cells of Chinese
hamsters and the finding of a slight increase in DNA repair in the
mouse kidney.
8.6. Reproduction, Embryotoxicity, and Teratogenicity
Details of studies on dominant lethality in rats and mice are
given in section 8.5.6. These results provide no evidence for an
adverse effect on male reproduction. The fertility of male and
female Sprague-Dawley rats and neonatal toxicity were investigated
in a 3-generation, 2-litter study [151] in which test animals were
continuously given drinking-water containing 0, 50, 100, or 200 mg
vinylidene chloride/litre (99.5% minimum purity). Ten male and 20
female F0 rats were treated and 15 male and 30 female rats were
used in the control F0 groups. These animals were mated after 100
days exposure (to provide F1a litters) and again 10 days after
weaning of the first litter (to provide F1blitters). Parents for
the F2 and F3 generations were selected randomly from the F1b and
F2 litters, respectively. They were mated at 110 days of age to
produce the F2 and F3a litters, respectively. The F2 rats were
then remated 10 days after weaning of the F3a and F3b litters to
produce the F3b and F3c litters, respectively. No evidence was
found for an effect of vinylidene chloride on fertility, though
marked fluctuations in the fertility index in all groups
including the controls made interpretation of the results
difficult. Neonatal survival was lower in the F2 and F3a litters of
dams ingesting vinylidene chloride than in the respective control
groups but not lower than that of the historical data for the
laboratory. Furthermore, reduced survival in some litters was
followed by normal survival in subsequent litters from the same
adults. It was concluded that the decreased survival was due to
chance. Necropsies were carried out on rats found dead or moribund,
all weanlings not selected for future matings, and F1 and F2 adults
after the litters were weaned.
Table 9. Summary of genotoxicity data for vinylidene chloride
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
PROKARYOTES IN VITRO
Salmonella typhimurium
(reverse gene mutation)
Strains TA 100, TA 1530 + phenobarbital-induced 0.2-20% in air + ve [14,15]
mouse kidney, liver, and for 4h
lung S9 mix
Strain TA 100 + phenobarbital-induced 0.2-20% in air - ve [14,15]
mouse liver S9 (-NADPH) for 4h
Strain TA 100 + human liver S9 mix 2% in air for 4h + ve [15]
Strain TA 1530 + phenobarbital or 2% or 20% in air + ve [15]
3-methyl cholanthrene- for 4 h
induced rat liver S9
Strains TA 100, TA 1535 ± rat or hamster S9 mix 0-3333 µg/plate - ve [146]
TA 98, TA 1537 "blind (Aroclor induced)
study", preincubation
Strain TA 100 ± Aroclor-induced 3% for 2 or 8 h + ve [10, 11,
rat liver S9 mix 12]
Strains TA 1950, TA 1951, None 1, 10, and 100% + ve at [26]
TA 1952, TA 1535, TA 1538 in DMSO 100% dose
TA 100, TA 98 (spot test) (0.05 ml/plate)
Strains TA 1535, TA 1537, + mouse liver S9 mix 1500-90 000 mg/m3 + ve [156]
TA 92, TA 100, TA 98 for 4 h
Strain TA 100 + liver and kidney S9 mix 360-50 000 mg/m3 + ve [156]
from mouse, rat, Chinese for 4 h (except where
hamster. Also human S9 obtained
liver S9 mix from female
mouse kidney
and rat kidney)
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
Strain TA 1535 + mouse liver and kidney 5% for 72 h + ve [103]
S9 mix (induction of
S9 increased
response
+ uninduced rat liver 5% for 72 h - ve [103]
S9 mix or marmoset or
human S9 mix
+ induced rat liver 5% for 72 h + ve [103]
S9 mix
+ uninduced marmoset 5% for 72 h - ve [103]
liver S9 mix
+ uninduced human liver 5% for 72 h - ve [103]
S9 mix
+ phenobarbital-induced 5% for 72 h + ve [103]
human liver S9 mix
Escherichia coli
(Reverse gene mutation)
Strain WP2 uvr A + mouse liver S9 mix 1500-90 000 mg/m3 + ve [156]
for 4 h
Strain K12 2.5 mmol/litre + ve [64]
(Forward and reverse gene (for 1 locus
liquid suspension only)
mutation)
PROKARYOTES IN VIVO
Salmonella typhimurium
Strains TA 1950, TA 1951 at LD50 and half- + ve [26]
TA 1952 host mediated LD50 dose
in ICR mice
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
EUKARYOTES IN VITRO
Saccharomyces (yeast) ± Aroclor-induced rat 0-50 mmol/litre + ve [19]
(reverse gene mutation liver S9 mix for 2 h (- ve without
and gene conversion) preincubation S9)
Chinese hamster V79 cells ± Aroclor-induced rat 2, 20% for 2 h - ve [43]
(forward gene mutation) liver S9 mix
Chinese hamster V79 cells 3x10-2 or 3x10-3 - ve [193]
(chromosomal breaks) mmol/litre
Chinese hamster ovary cells 1.8-7% atmosphere + ve [129]
(sister chromatid exchange) for 24 h
Chinese hamster ovary ± Aroclor-induced rat 0-2 mg/ml + ve [196]
cells (sister chromatid liver S9 mix (weak
exchange) response)
Primary hepatocytes from 2.1 mmol/litre + ve [32]
phenobarital-treated rats (maximum subtoxic
(unscheduled DNA dose)
synthesis)
EUKARYOTES IN VIVO
Tradescantia (flower) 88 and 5152 mg/m3 + ve [228]
(forward gene mutation) (at lower
dose only)
Saccharomyces (host 400 mg/kg or + ve for [19]
mediated assay in Swiss 5x100 mg/kg daily yeast cells
mice plus 200 mg/kg on recovered
last day from liver and
kidneys, but not
from lung
________________________________________________________________________________________________
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
CD-1 mice and Sprague- rats: 40 mg/m3 + ve (but [186]
Dawley rats (DNA adduct mice: 40 0r 200 minimal binding
formation in liver and mg/m3 for 6 h detected)
kidney) mouse > rat
kidney > liver
CD-1 mice (unscheduled 200 mg/m3 + ve (but [186]
DNA synthesis in liver for 6 h minimal); effect
and kidney) observed in
kidneys only
CD-1 mice (bone marrow 220 mg/m3 - ve [118]
cytogenetics for 6 h, 5 days/week
for 12 months
CD rats (bone marrow 220 mg/m3 - ve [118]
cytogenetics for 6 h, 5 days/week
for 12 months
Rats (male and female) 0, 100, or 300 - ve [178]
(bone marrow cytogenetics) mg/m3 for 6 months
ICR mice (bone marrow One-half LD50 - ve [26]
cytogenetics one-sixth LD50
x 5 days
Male ddY mice (bone 0-200 mg/kg or - ve [196]
marrow micronucleus 0-100 mg/kg x 4
assay) by gavage
Chinese hamster 120 or 400 mg/m3 + ve [77]
(bone marrow (30 or 100 ppm)
cytogenetics) 6 h/day, 5 days/week
for 6 weeks
Table 9 (contd).
________________________________________________________________________________________________
Test system/End point of Metabolizing Dose Resulta Reference
analysis system range
________________________________________________________________________________________________
CD-1 mice (male) 40, 120, 200 mg/m3 - ve [3]
(dominant lethal assay) 6 h/day for 5 days
CD rats 220 mg/m3, 6 h/day - ve [203]
(dominant lethal assay) 5 days/week for
11 weeks
_________________________________________________________________________________________________
a +ve = positive response.
-ve = negative response.
Absolute and relative kidney weights of weanling rats were
comparable with controls. No organ weight changes related to
treatment were seen in F1 adults but elevated relative liver
weights of female rats ingesting 200 mg vinylidene chloride/litre
were seen in the F2 generation. An increase in serum glutamic
pyruvic transaminase (25% above control mean) was observed in
female F2 rats at the 200 mg/litre level.
Signs of mild hepatotoxicity (fatty liver) were seen at
treatment levels of 100 mg vinylidene chloride/litre and 200
mg/litre in F1 and F2 rats and the incidence of chronic renal
disease (though high in controls) was greater in male rats treated
with 200 mg vinylidene chloride/litre than in control animals.
Nitschke et al. [151] concluded that vinylidene chloride treatment
did not significantly affect the reproductive capacity of rats.
Sawada et al. [196] investigated the incidence of micronuclei
in fetal liver and fetal erythrocytes 24 h following exposure of
pregnant ICR mice to 0, 25, 50, or 100 mg vinylidene chloride/kg
(given by intraperitoneal injection). No significant increase in
micronuclei in these cells was observed as a result of treatment,
and the results did not show any evidence of a transplacental
passage of genotoxic metabolites or precursors.
In a study on Charles River CD rats (18-20 per dose group) and
CD-1 mice (7-24 per dose group), Short et al. [202] exposed animals
to vinylidene chloride through inhalation at 60-1796 mg/m3 (15-449
ppm), for 22-23 h/day during various periods of organogenesis.
On day 20 (rats) or day 17 (mice) of gestation, fetal abnormalities
were seen at all dose levels in rats and a high incidence of early
and complete resorptions was seen in mice exposed to vinylidene
chloride concentrations >120 mg/m3 (30 ppm). However, these
effects were associated with a decreased weight gain and increased
mortality in the dams. Since similar fetal abnormalities of the
soft tissues and skeleton occurred in a feed-restricted group of
mice, the defects may be attributed to maternal toxicity. In rats,
malformations were seen in all groups and hydrocephalus was
significantly increased in a dose-related manner from 2.5% in
controls, 7.3% at 60 mg/m3 (15 ppm), 15.1% at 228 mg/m3 (57 ppm) to
33.3% at 1200 mg/m3 (300 ppm). Retarded ossification was seen in
all treated groups. Early resorptions were significantly increased
from 2% in controls to 49% at 228 mg/m3 (57 ppm) and 64% at 1796
mg/m3 (449 ppm). Fetal weight was reduced in a dose-related
manner and the reductions were significant at 228, 1200, and 1796
mg/m3 (57, 300, and 449 ppm). Food-restricted controls showed
reduced fetal weight and retarded ossification but no significant
increase in resorption rate or in the frequency of hydrocephalus.
Aspects of maternal toxicity may be responsible for these effects.
Part of this study involved an investigation of behavioural changes
in Charles River CD rats (19 or 20 per dose group). The rats were
exposed to 224 or 1132 mg vinylidene chloride/m3 (56 or 283 ppm)
for 22-23 h/day, on days 8-20 of gestation. A dose-related weight
loss over this period indicated maternal toxicity and the body
weight of pups from the rats treated with 1132 mg/m3 (283 ppm)
were lower than control weights (as with pups from feed-restricted
rats). Two groups of 3 pups/dose level and per sex were observed
for activity in a maze for 2-4 months after birth. Also one animal
of each sex per dose level from each litter was subjected to pre-
weaning behavioural and physical maturation tests. Maze activity
was not affected by the vinylidene chloride treatment, nor were
startle response, bar holding or swimming ability. However, surface
righting ability was delayed in pups from treated rats. Tooth
production was delayed in pups from the rats treated with 1132
mg/m3 (283 ppm) (as in feed-restricted rats) but opening of the
external ear was more rapid than in control pups. In conclusion,
the only adverse effects bserved could be attributed to maternal
toxicity. Murray et al. [1979] investigated embryonic and fetal
development in rats and rabbits following inhalation or oral
ingestion (rats only) of vinylidene chloride (minimum purity
99.5%) during gestation. In the inhalation studies, Sprague-
Dawley rats (30-44 per dose group) and New Zealand White rabbits
(18-20 per dose group) were exposed to 640, 320, or 80 (rats only)
mg vinylidene chloride/m3 (160, 80, or 20 ppm), for 7 h/day, from
days 6 to 15 (rats) and days 6 to18 (rabbits) of gestation. Groups
of 20-47 pregnant rats and 16 pregnant rabbits served as controls
for each dose level. For the ingestion study, 26 pregnant rats (24
controls) received drinking-water containing vinylidene chloride at
200 mg/litre (approximately 40 mg/kg per day) from days 6 to 15 of
gestation. Cesarean section was carried out on day 21 and day 29
for rats and rabbits, respectively.
Although a teratogenic effect was not observed, some evidence
of embryotoxicity and fetotoxicity was seen in rats and rabbits.
In the rat inhalation study, there was delayed ossification and a
dose-related increased incidence of wavy ribs at 320 and 640 mg/m3
(80 and 160 ppm), concentrations that were toxic to the dams. At
80 mg/m3 (20 ppm), a concentration that was not maternally toxic,
no embryo- or fetotoxic effects were seen. In the rabbits, a dose
of 640 mg/m3 (160 ppm) produced weight loss in the dams, increased
resorptions, increased incidence of 13 pairs of ribs and delayed
ossification of the fifth sternebra. At 320 mg/m3 (80 ppm), there
was no effect on dams or fetuses.
8.7. Carcinogenicity
Details of a number of the studies described here have been
provided in section 8.4. In such cases, only an outline of the
experimental protocol is given.
8.7.1. Inhalation
In a long-term inhalation study (220 mg vinylidene chloride/m3
(55 ppm)) carried out by Lee et al. [118, 119] (section 8.3.1
for details of treatments), haemangiosarcomas in the mesenteric
lymph node or subcutaneous tissue were reported in 2 treated male
rats and hepatic haemangiosarcomas were found in 2 male and 1
female mice in the treatment group. No haemangiosarcomas were seen
in control animals. Small bronchioalveolar adenomas were also
reported in 6 male mice (17%) compared with 1 in the control group
of male mice (4%). The significance of this finding was considered
questionable since, according to other reports, such adenomas,
which occurred relatively late in the study, were common in
untreated mice. The numbers of animals used in the treatment and
control groups in this study were very low (16 or less per group
after 9 months) and the study was limited to 12 months, which is
inadequate. Furthermore, the authors referred to the spontaneous
occurrence of hepatomas in mice at a similar age in other reports.
In a follow-up study, it was not possible to repeat the finding of
an increase in tumour incidence [79]. Intermittent exposure was
limited to up to 10 months (rats) and up to 6 months (mice) at a
vinylidene chloride concentration of 220 mg/m3 (55 ppm) (details
given in section 8.3.1), and was followed by a 12-month observation
period. In contrast to the results of Lee et al. [119], no tumours
arose in the treated animals other than spontaneous tumours
expected on the basis of their incidence in control animals. In
this study also, a small number of animals were used (14-16 rats
and 12 mice per sex for 10- and 6-month treatments, respectively)
and exposures were limited to periods of less than 12 months for
both species.
The results of a number of other long-term inhalation studies
on rats suggest a lack of carcinogenicity of vinylidene chloride in
this species, but, as explained, these reports are not conclusive.
Viola & Caputo [229] investigated the incidence of tumours in 51
male and 23 female Wistar rats exposed to 800 mg vinylidene
chloride/m3 (200 ppm) for 4 h/day, 5 days/week, for 5 months. For
the following 7 months, the concentration was reduced to 400 mg/m3
(100 ppm) because of toxicity. The animals were then given a
complete autopsy at spontaneous death or after being killed when
moribund. Sprague-Dawley rats were also examined after exposure to
400 mg/m3 (100 ppm) (30 of each sex) and 300 mg/m3 (75 ppm) (16
and 21 male and female rats, respectively). Thirty control rats of
each sex were used for each strain. No grossly observable
correlation between tumour formation and vinylidene chloride
inhalation was seen but a final report following the completion of
microscopic examination of tissues and organs has not been
released. A more substantial study has been reported in several
stages [134, 178, 179, 180]. The details of the long-term
inhalation study on rats are given in section 8.3.1. In outline,
following the first month of treatment, animals of a relatively
large group (minimum of 84 rats/sex per dose group) were treated
intermittently with levels of up to 300 mg vinylidene chloride/m3
(75 ppm) for the substantial period of 18 months and were
sacrificed at 24 months. The total incidence of tumours was
similar in control and dosed animals, though the incidences of
several tumours and/or tumour types were found to be statistically
increased compared with the controls (P < 0.05, Fisher's Exact
Probability Test). These were not attributed to vinylidene
chloride exposure on the basis of comparable historical control
data.
The carcinogenicity of vinylidene chloride by inhalation
was studied by Maltoni et al. [139, 141] in rats, mice, and
hamsters. The experimental details for the long-term study are
given in section 8.3.1. The intermittent exposure levels were up
to 100 mg/m3 (up to 25 ppm) (hamsters), 600 reduced from 800 mg/m3,
(150 reduced from 200 ppm) (rats), and 100 mg/m3 (25 ppm) (mice)
and were given over a period of 52 weeks. The dose levels in rats
and mice were limited by toxicity (section 8.3.1). Animals were
then observed until spontaneous death. Tumour incidence in hamsters
was not increased by treatment with vinylidene chloride. The only
type of tumour in treated rats for which the incidence was greater
than that in the controls was in the mammary gland, but a dose-
response relationship was not observed and the authors suspected
non-specific factors (linked to inhalation) to be responsible.
As part of the same project, Maltoni et al. [140] specifically
investigated the ability of vinylidene chloride to produce brain
tumours in Sprague-Dawley rats. Groups of 30 rats of each sex were
exposed to 40, 100, 200, or 400 mg vinylidene chloride/m3 (10, 25,
50, or 100 ppm), and 60 rats per sex to 600 mg/m3 (150 ppm), for 4
h/day, 4-5 days weekly, for 52 weeks (100 control rats were used
per sex). No evidence for the induction of brain tumours
(ependymomas, gliomas, or meningiomas) was found.
A number of tumour-types were observed in mice [139, 141]
including kidney adenocarcinomas, mammary tumours, pulmonary
adenomas, and leukaemias. The incidence of both mammary and
pulmonary tumours (mainly adenomas) was statistically higher in
treated mice compared with the controls (tested by the rank test of
Krauth, Fisher's Exact Probability Test, Logrank test and probit
analysis [142]. As with rats, non-specific factors may have been
responsible. However, a dose-response relationship was not
observed in either case. Kidney adenocarcinoma (a rare tumour in
mice) was observed in 29 (28 male) out of 257 mice (300 at start)
treated with 100 mg vinylidene chloride/m3 (25 ppm) and in 2 out of
the 18 male surviving mice treated with 200 mg/m3 (50 ppm).
Kidney adenocarcinomas were not seen in the 14 surviving females at
200 mg/m3 (50 ppm), in mice treated with 40 mg/m3 (10 ppm) (0/60),
or in control mice (0/380).
Maltoni et al. [142] exposed two groups of approximately
60 male and 60 female Sprague-Dawley rats to vinylidene chloride
transplacentally, continuing the exposure by inhalation at birth
(see Table 10). Treatment of dams with vinylidene chloride at 400
mg/m3 (100 ppm) through inhalation for 4 h/day, 5 days/week,
was started when embryos were 12 days of age. The inhalation
treatment of dams and offspring was continued after birth with
exposure to 400 mg vinylidene chloride/m3 (100 ppm) for 7 h/day, 5
days/week over 8 or 97 weeks, giving a total exposure period of 15
or 104 weeks. Offspring may also have been exposed via ingestion of
milk at the suckling stage.
An increased incidence of malignant neoplasias of the
haemolymphoreticular system, generally classified as leukaemia, was
observed in both males and females of both groups exposed to
vinylidene chloride. A slight decrease in body weight was also
reported in these animals. Although a quadrupling of leukaemia
incidence appeared to occur in female rats and a doubling of these
tumours occurred in the male rats, very little other information
was reported. It is possible that a few of the tumours called
leukaemia are histiocytic sarcomas and should be classified
separately from leukaemias. Unless this is known, the
contribution of this finding to the overall data base is
compromised. The authors pointed out that a considerable increase
in total malignant tumours occurred in the rats exposed to
vinylidene chloride for 104 weeks, when the treatment was started
prenatally. In contrast, a 52-week exposure of rats beginning in
young adulthood (see above), showed no increased incidence of
leukaemia at exposures as high as 600 mg/m3 (150 ppm) for 52 weeks,
and only a borderline increase in total malignant tumours. Both age
at the start of exposure and the length of exposure appear to be
important factors influencing tumour development in Sprague-Dawley
rats exposed to vinylidene chloride.
Laib et al. [114] investigated the occurrence of putative pre-
neoplastic nodules in the liver of rats treated with vinylidene
chloride. Neonate Wistar rats were exposed, together with
mothers, to vinylidene chloride in the air at 440 ± 60 mg/m3 (110
± 15 ppm) (8 h per day, 5 days per week). Exposure to this
concentration, did not result in increased lethality or reduction
in body and liver weights. Histochemical analysis of frozen liver
sections, produced immediately after 6 weeks exposure, revealed
ATPase-free islets, postulated to indicate preneoplasia.
8.7.2. Oral
There have been three studies on the oral carcinogenicity
of vinylidene chloride in which a reasonable number of rats were
given a range of dose levels for an adequate length of time. In the
first [177, 179], vinylidene chloride was included in the
drinking-water at a level up to 200 mg/litre (200 ppm) over a 2-
year period (details given in section 8.3.2). No exposure-related
neoplasms were detected. Although the incidence of mammary gland
fibroadenomas/ adenofibromas was greater in rats exposed to 50
mg/litre than in the control animals, this increase was not dose
dependent and was within the range of the historical control data
for untreated rats.
In the second study [139, 141], vinylidene chloride was given
by gavage in olive oil (details of animals and dosing regimen are
given in section 8.3.2). The intermittent dosing was over a
period of 52 weeks and the animals were examined at spontaneous
death. No increase in tumours was observed in treated rats and,
in particular, there was no increased incidence of mammary tumours
(these being considered due to non-specific factors when found in
the corresponding inhalation study (section 8.7.1)).
In a third gavage study [154], doses of 1 and 5 mg/kg were
given daily to rats for 2 years (details in section 8.3.2). No
vinylidene chloride-related tumours were reported. In mice
(section 8.3.2), given 2 or 10 mg/kg daily for 2 years, though the
incidence of lymphomas was increased at the lower dose level (P
>0.05), this was not found at 10 mg/ kg and thus does not appear
to be related to vinylidene chloride exposure. However, the
sponsors have recently found defects in the conduct of the study
and it could not be satisfactorily evaluated.
A further study [172] was restricted to a single oral dose
level given to BDIV rats throughout the life span from the time of
weaning. The study also included oral dosing of the mothers during
pregnancy. Twenty-four female pregnant rats were given vinylidene
chloride orally (150 mg/kg) on the 17th day of gestation and
their offspring (81 males and 64 females) were then treated weekly
with 50 mg/kg (orally in olive oil) from the time of weaning. All
survivors were killed at 120 weeks or when moribund and all major
organs, as well as those that showed gross abnormalities, were
examined histologically. Treatment did not increase the incidence
of tumour-bearing animals, though liver tumours were increased in
rats of both sexes (1/81 males and 3/80 females compared with 0/49
and 0/47 in male and female controls, respectively), and
meningiomas were increased in males (6/81 compared with 1/49 in
controls). The latter was found statistically to be not
significant and, since a dose-response analysis was not possible,
the results are inconclusive. In addition, hyperplastic nodules
were found in the livers of 2/23 females given the single dose of
vinylidene chloride during pregnancy and also in 2/81 males and
6/80 females among the progeny. There was a significant difference
( P =0.04) between treated and vehicle-treated control animals, no
hyperplastic nodules being observed in the latter.
8.7.3. Other routes
Van Duuren et al. [227] studied the carcinogenicity of
vinylidene chloride in groups of 30 female Ha: 1 CR Swiss mice
following percutaneous and subcutaneous application compared with
100 untreated control animals. Following the application of
vinylidene chloride at 121 mg/kg or 40 mg/kg in acetone to shaved
dorsal skin, 3 times per week for between 440 and 594 days,
animals were given a complete autopsy (except the cranial region)
and abnormal-appearing tissues and organs were examined
histologically. Routine sections of skin, liver, stomach, and
kidney were also taken. Autopsies and additional sections from the
injection site and liver were taken following the once weekly
subcutaneous administration of 2 mg vinylidene chloride per mouse
for the life span (78 weeks). Although in the percutaneous study,
benign lung papillomas (19/30, 12/30 and 30/100) were seen at 121
mg/kg, 40 mg/kg, and 0 mg/kg, respectively, and stomach tumours
were also observed at frequencies of 2/30, 0/30 and 5/100,
respectively, the tumour incidence were not significantly elevated
( P >0.05; Chi-square analysis) in any of the treatment groups.
However, when vinylidene chloride was given as an initiating agent
(single dermal dose of 121 mg/kg) followed 14 days later with 5 µg
of the promoting agent phorbol myristate acetate three times
weekly, for 428-576 days, 8/30 mice developed papillomas compared
with 9/120 and 6/90 in control (phorbol myristate acetate-treated)
mice and 1/30 treated mice developed a squamous cell carcinoma
compared with 1/120 and 2/90 in control mice treated with phorbol
ester (2.5 and 5.0 µg, respectively). The incidence of papillomas
in vinylidene chloride-treated mice was significantly greater
than in the control groups ( P <0.005; Chi-square analysis). The
authors concluded that "initiating" activity was shown with
vinylidene chloride but that it was not a complete carcinogen.
However, the finding of papillomas in the phorbol myristate
acetate-treated control mice and the low number of animals used in
the treatment groups make the interpretation of the results
difficult.
8.7.4. Summary of carcinogenicity
A number of studies on rodents have been conducted that provide
information on the potential carcinogenic action of vinylidene
chloride (see Table 10). In these studies, vinylidene chloride
was administered by inhalation, orally by gavage and in drinking-
water, and by skin application and subcutaneous injection.
Unfortunately, most of these studies were inadequate for the
conclusive evaluation of carcinogenicity because of less than
lifetime exposure regimens, insufficient numbers of animals, and an
inadequate number of dose levels. Only some of the studies were
designed and conducted as cancer biossays.
Increased tumour incidence was not found, in most of the
studies, but there were the following exceptions. Kidney
adenocarcinomas occurred in male Swiss mice exposed via inhalation
to 100 or 200 mg vinylidene chloride/m3 (25 or 50 ppm) but not to
40 mg/m3 (10 ppm). This carcinogenic response may be related to
the ability of vinylidene chloride to cause cytotoxic effects in
the target organ (section 8.1.2 and 8.2.1). In addition to kidney
adenocarcinomas, statistically significant excesses of mammary
carcinomas were observed in female mice and pulmonary adenomas in
mice of both sexes, but in these cases there was no dose-response
relationship. In a 2-stage skin carcinogenicity assay in mice,
there was some evidence that vinylidene chloride may have acted as
an initiating agent.
In rats, an increase in mammary tumours that was not dose
related was observed when adult animals were exposed through
inhalation. In separate study groups, a slight increase in
leukaemias was observed in rats exposed through inhalation in utero
and then post-natally.
Table 10. Animal carcinogenicity studiesa
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, inhalation 100 100 0 mg/m3 (0 ppm) 4 h/day, to - -
Sprague- 30 30 40 mg/m3 (10 ppm) 4-5 days/ spontaneous - -
Dawley 30 30 100 mg/m3 (25 ppm) week, death - -
[139,141] 30 30 200 mg/m3 (50 ppm) 52 weeks - -
60 60 600 mg/m3 (150 ppm) - -
Leukemia
Rat, inhalation 158 149 0 mg/m3 (0 ppm) 4/h day, 6 months 12/156 1/148
Sprague- ( in utero - 60c 0 mg/m3 (0 ppm) 5 days per
Drawley and post - 54b 400 mg/m3 (100 ppm) week, 7 weeks,
[142] natal 62 61 400 mg/m3 (100 ppm) then 7 h/day, 10/61 4/61
exposure) 5 days per
week, 97 weeks
60 60 400 mg/m3 (100 ppm) 4 h/day, 5 6 months 8/59 2/60
days per week,
7 weeks, then
7 h/day, 5
days per week,
8 weeks
Rat, CD inhalation 36 36 0 mg/m3 (0 ppm) 6 h/day, 5 none - -
[118, 119] 36 36 220 mg/m3 (55 ppm) days per none - -
week
Rat, CD 6 h/day, 5 52 weeks
[79] days per
week for:
inhalation 4 4 0 mg/m3 (0 ppm) 1 month
8 8 0 mg/m3 (0 ppm) 3 months
8 8 0 mg/m3 (0 ppm) 6 months
16 16 0 mg/m3 (0 ppm) 10 months
4 4 220 mg/m3 (55 ppm) 1 month - -
8 8 220 mg/m3 (55 ppm) 3 months - -
8 8 220 mg/m3 (55 ppm) 6 months - -
16 16 220 mg/m3 (55 ppm) 10 months - -
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, inhalation 86 86 0 mg/m3 (0 ppm)
Sprague-Dawley
[179, 180]
[134, 178] 86 86 40 mg/m3 (10 ppm) 6 h/day, 5 6 months - -
first 5 weeks, then days per week,
100 mg/m3 (25 ppm) 18 months
86 86 160 mg/m3 (40 ppm) - -
first 5 weeks, then
300 mg/m3 (75 ppm)
Rat, Wistar inhalation 30 30 0 mg/m3 (0 ppm) 4 h/day, 5 to
[229] days per spontaneous
week, for death or
51 23 800 mg/m3 (200 ppm) 12 months moribund state - -
first 5 months, -
400 mg/m3 (100 ppm)
subsequently
Rat, inhalation 30 30 0 mg/m3 (0 ppm) 4 h/day, 5 to only gross
Sprague- 16 16 300 mg/m3 (75 ppm) days per spontaneous pathology
Dawley 30 30 400 mg/m3 (100 ppm) week, 12 death or performed
[229] months moribund
state (22-
24 months)
Kidney adeno-
carcinomas
Mouse, Swiss inhalation 190 190 0 mg/m3 (0 ppm) 4 h/day, 5 up to 121 0/120 0/155
[139, 141] 30 30 40 mg/m3 (10 ppm) days per weeks 0/24 0/26
150 150 100 mg/m3 (25 ppm) week, 52 28/119 1/138
weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, Swiss 30 30 200 mg/m3 (50 ppm) 4 h/day, 4 to 2/18 0/14
(contd.) daysd spontaneous
[139, 141] death
Mammary gland
adenomas
0 mg/m3 (0 ppm) 1/180 3/187
40 mg/m3 (10 ppm) 0/30 6/30
100 mg/m3 (25 ppm) 1/148 16/148
200 mg/m3 (50 ppm) 0/52 6/54
Lung (mainly
adenomas)
0 mg/m3 (0 ppm) 6/154 7/178
40 mg/m3 (10 ppm) 6/28 3/30
100 mg/m3 (25 ppm) 23/141 12/147
200 mg/m3 (50 ppm) 4/51 6/59
Tumours of the
mammary gland
and lung were
not dose-
dependent
Mouse, CD-1 inhalation 36 36 0 mg/m3 (0 ppm) 6 h/day, 5 none - -
[118, 119] 36 36 220 mg/m3 (55 ppm) days per none
week, 52
weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, CD-1 Incidence of
(contd.) hepatic
[118, 119] haemangio-
sarcoma and
possibly
bronchiolo-
alveolar
adenoma
increased, but
this is not
thought to
have been
induced by
vinylidene
chloride [79]
Mouse, CD-1 inhalation 6 h/day, 5 52 weeks
[79] days per
week for:
16 16 0 mg/m3 (0 ppm) 1 month
16 16 0 mg/m3 (0 ppm) 3 months
28 28 0 mg/m3 (0 ppm) 6 months
8 8 220 mg/m3 (55 ppm) 1 month - -
8 8 220 mg/m3 (55 ppm) 3 months - -
12 12 220 mg/m3 (55 ppm) 6 months - -
Chinese inhalation 18 17 0 mg/m3 (0 ppm) 4 h/day, to - -
Hamster 30 30 100 mg/m3 (25 ppm) 4 - 5 days spontaneous
[139, 141] per week, death
for 52 weeks
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Rat, oral 80 80 0 mg/litre (0 ppm) daily for none
Sprague- (drinking- 24 months
Dawley water)
[179, 180]
[177] 48 48 50 mg/litre (50 ppm) none - -
(M = 7 mg/kgbw)
(F = 9 mg/kgbw)
48 48 100 mg/litre (100 ppm) none - -
(M = 10 mg/kgbw)
(F = 14 mg/kgbw)
48 48 200 mg/litre (200 ppm) none - -
(M = 20 mg/kgbw)
(F = 30 mg/kgbw)
Rat, gavage 100 100 olive oil daily to
Sprague- (olive oil) 82 77 (0 mg/kgbw) 4 - 5 days/ spontaneous
Dawley 50 50 0.5 mg/kgbw week, for death - -
[139, 141] 50 50 5 mg/kgbw 52 weeks - -
50 50 10 mg/kgbw - -
50 50 20 mg/kgbw - -
Mouse, Swiss dermal 100 0 mg 3 x/week to none -
Ha: ICR; (in 0.2 ml 30 0.1 ml acetone spontaneous none -
[227] acetone) 30 40 mg/mouse death or none -
30 121 mg/mouse moribund none -
state
Table 10. (contd.)
--------------------------------------------------------------------------------------------------------
Species/ Route Number of Dose Duration of Post-exposure Result
strain (Vehicle) animals in administ- period ------------
[Reference] each group ration male female
------------
male female
--------------------------------------------------------------------------------------------------------
Mouse, Swiss subcutaneous 100 0 mg once per week
Ha: ICR; (in 0.05 ml 649 days none
[227] trioctanoin) 30 0.05 ml water 636 days none
30 0.05 ml 631 days none -
trioctanoin
30 2 mg/mouse 548 days none -
Mouse, Swiss dermal 30 121 mg/mouse once, to none 8/30
HA: ICR; (initiation then 5 mg PMAe spontaneous papillomas
[227] test on the per animal death or 1/30 skin
skin) moribund carcinoma
state; PMA
3 x/week
--------------------------------------------------------------------------------------------------------
a Modified from: ECETOC [45].
b Only 2 treatments, because of the high toxicity.
c Pregnant females.
d Only 4 treatments, because of the high toxicity and mortality.
e PMA = phorbol myristate acetate.
9. EFFECTS ON HUMAN BEINGS
9.1. Single and Short-term Exposures
According to Gibbs & Wessling, [59], exposure to a high
concentration of vinylidene chloride, e.g., 16 000 mg/m3 (4000
ppm), rapidly causes intoxication that can lead to unconsciousness.
The anaesthetic effects from short-term exposure are short-lived.
At unspecified sub-anaesthetic doses, prolonged exposure and
repeated short-term exposures may produce kidney and liver damage
[221].
Some adverse effects associated with vinylidene chloride
exposure have been attributed to contaminants or to the stabilizer
( p- methoxyphenol). Henschler et al. [74] reported the occurrence
of persistent cranial nerve disorders in two individuals who had
attempted to clean out tanks that had contained vinylidene chloride
co-polymers. The chemical responsible for this effect appeared,
however, to be a contaminant of vinylidene chloride (either mono-
or dichloroacetylene). Chivers [29] reported the incidence of
leukoderma in two subjects following skin contamination with
p- methoxyphenol. The irritant effect of vinylidene chloride [84,
192] on the eye, upper respiratory tract (at levels as low as 100
mg/m3 i.e., 25 ppm) [192], and skin may be at least partially due
to the stabilizer p- methoxyphenol and, in the case of the study
by Rylova [192], other impurities. Dermatitis was reported in an
individual whose skin was directly exposed to Saran film
(vinylidene chloride/ vinyl chloride co-polymer in the absence of a
stabilizer) [160].
9.2. Long-Term Exposure
A quantitative risk estimate based on the best available set
of data (mouse-kidney adenocarcinoma) from the animal tumour
assays, using a non-threshold mathematical model, linear at low
doses, provides an estimate of an upper limit of human risk [225].
The true risk is not likely to be greater than this estimate and
may be lower. The upper limit for human risk thus estimated was
5.0 x 10-5 for a continuous lifetime exposure to 1 µg/m3 in air and
3.3 x 10-5 for ingestion of drinking-water containing 1 µg/litre.
However, in the light of the discussion in section 8.6.4, the
limited evidence for carcinogenicity in animal models is not
sufficient to reach a firm conclusion on the carcinogenic risk of
vinylidene chloride for human beings.
Interpretation of epidemiological studies on the effects of
vinylidene chloride in human beings has been confounded by
concomitant exposure to vinyl chloride. A mortality study on 629
workers exposed to vinylidene chloride, for 6-10 h/day, 42 h/per
week, for various lengths of time in a vinylidene chloride
production and polymerization plant in the Federal Republic of
Germany was reported by Thiess et al. [219]. Individuals were
exposed to an estimated (unmeasured) average plant concentration
of 200 mg vinylidene chloride/m3 (50 ppm) from 1955 to 1965 and
subsequently to an average level of approximately 40 mg/m3 (10
ppm) up to 1975, based on measurements of airborne contamination
after 1975. All had also been exposed to vinyl chloride (measured
as <13 mg/m3 ( <5 ppm) since 1975) and acrylonitrile (measured as
<2 mg/m3 (<1 ppm) since 1975). A 97% tracing was obtained for
follow-up analysis of the majority (447) of the cohort that was
exposed for more than 6 months. The incidence of exposure for >1
year in the remainder, was 36% and tracing for follow-up analysis
was only 24%. The mortality rate of the cohort was compared with
that of two populations of 180 000 (local) and 3 700 000 (regional)
for the period 1969-75. The expected number of deaths was
calculated by applying age-specific mortality rates to the person-
years of observation of 7 age groups within the cohort.
Statistical evaluation was based on the Poisson distribution. The
distribution of deaths according to age and decade (total 39
deaths) indicated that workers in the exposure groups did not have
an elevated mortality rate (total 57 and 36 expected from the data
of the two reference populations). The expected numbers of deaths
resulting from cancers, infectious diseases, cardiovascular
diseases, other natural causes, and external causes were compared
with the observed incidence.
Although the number of deaths from cardiovascular diseases in
general was not different from that expected, a peculiar
distribution of deaths caused by cerebral haemorrhage in the young
age groups deserves mention. The occurrence of several deaths
attributable to cerebral haemorrhage, cerebral sclerosis/apoplexia,
and acute coronary failure in age-groups below 50 years was beyond
chance ( P- values far below 0.05). But this study did not verify
the validity of diagnoses on death certificates and adequately
designed studies are needed to ascertain this part of the mortality
findings.
Five deaths (out of 39) through suicide compares with 2.5-3.0
expected. This cause of death does not rely on diagnostic validity
and indicates the need for further investigations, because suicide
may relate to mental depression. Bronchial carcinomas were seen
in 5 individuals compared with expected numbers of 3.9 and 2.2
from the data on the two control populations. It was noted that 3
of the subjects with bronchial carcinoma were heavy smokers, the
remaining 2 cases (observed at age 37) were clearly in excess of
the expected incidence in the age-group below 40 (0.08 and 0.07 for
the 2 reference populations, P = 0.003). The incidence of
oesophageal cancer was within the range of age-specific expectation
for the larger (district) control population but not for the
smaller city population. The authors concluded that the overall
malignant tumour incidence was not statistically different from
the expected rate.
More detailed information was provided in a follow-up
investigation of this exposed population (535 persons exposed for
more than 6 months) [110]. In this extended analysis with an
estimated average exposure in the years before 1965 of 200 mg/m3
(50 ppm), the observed total number of deaths (48) was
significantly greater than that expected from the reference
populations (43.2-46.5) due to a greater incidence of
cardiovascular disease (20 observed deaths versus about 15
expected). Eleven deaths from myocardial infarction were
statistically significantly in excess of the 6.8 expected. The
number of malignant tumours was 12 compared with 9.8 expected and
this was reflected in the incidence of bronchial carcinomas (6
compared with 2.68-2.96 expected). In comparison with internal
reference groups not exposed to vinylidene chloride, the cancer
deaths were statistically in excess (3 of the lung-cancer cases
were aged under 50). However, this statistically increased
incidence was not considered to be related to vinylidene chloride
exposure since, in 2 cases, exposure was limited to 2 years
duration. The interpretation of the epidemiological study is
hampered by a low cohort number, while co-exposure to other
chemicals, such as vinyl chloride, was only considered in part by
the inclusion of internal reference populations.
In a further epidemiological study [240], employees had been
exposed to different extents to a range of substances in synthetic
chemical plants. These investigators combined detailed work
histories of 4806 individuals with exposure ratings for each of 19
chemicals during each calendar year from 1942 to 1973. After
construction of a serially additive expected dose model, the
authors tested whether vinylidene chloride was responsible for the
observed excess risk of lung cancer in the cohort by using the
one-sided t-test of the observed minus expected cumulative doses
over all years and for ten or more years before death. No
relationship was found between vinylidene chloride exposure and
lung cancers.
The only epidemiological study of individuals exposed to
vinylidene chloride where vinyl chloride was not used as a
co-polymer (ethyl acrylate was the co-polymer) was carried out by
Ott et al. [166]. Employees (138) were exposed to time-weighted
average (TWA) concentrations ranging from 20 to 280 mg vinylidene
chloride/m3 (5 to 70 ppm) for a minimum of one year, within the
period 1942-65. No association was found between exposure and
mortality ascertained in 1974 among this low cohort number, when
compared with US national statistics. Two employees suffered
hepatic damage, but, in both cases, alcohol consumption was known
to prevail. The size of the cohort having a long duration of
exposure or a long latency period since initial exposure was small.
No internal comparison was made, comparable to the approach by
Klimish et al. [110]. Although the results of the epidemiological
studies do not provide convincing evidence for an increased risk
of cancer in human beings exposed to vinylidene chloride, it is not
possible to conclude that there is no carcinogenic effect. It
should be remembered that inadequate studies often tend to
underestimate rather than to overestimate an association between
exposure and cancer.
Schmitz et al. [198] assessed serum glutamic oxaloacetic
transaminase, glutamic pyruvic transaminase, and gamma-glutamyl
transpeptidase in 133 human subjects exposed to vinylidene
chloride, as a test for liver damage. Serum enzyme levels changed
less in two comparison groups than in the exposed group but,
according to Fisher's Exact Probability Test, the duration of
exposure to vinylidene chloride did not influence the serum enzyme
levels.
10. EVALUATION OF EFFECTS ON THE ENVIRONMENT AND HUMAN
HEALTH RISKS
10.1. Evaluation of Effects on the Environment
As a result of volatilization, the atmosphere is the major
environmental compartment for vinylidene chloride. The half-life of
vinylidene chloride in the troposphere is expected to be
approximately 2 days and therefore the compound is unlikely to
participate in the depletion of the stratospheric ozone layer.
Leaching and volatilization render soil and sediments minor
compartments for vinylidene chloride in the environment and the
level of this chlorinated hydrocarbon in the aqueous environment is
also minimized by rapid volatilization. It is not known whether
the degradation of compounds, such as trichloroethylene and
perchloroethylene, which are often found in water, contributes in a
significant manner to the levels of vinylidene chloride found in
the environment.
The concentrations of vinylidene chloride found in
environmental waters and the acute toxicity levels for fish and
Daphnia indicate that acute toxic risks for the aquatic environment
are minimal. Available data on long-term toxicity are insufficient
to assess sub-lethal effects on any aquatic organisms residing near
point sources of relatively high levels of vinylidene chloride
contamination, such as contaminated ground water and municipal and
industrial outfalls.
10.2. Evaluation of Human Health Risks
10.2.1. Levels of exposure
The general population is exposed to very low levels of
vinylidene chloride. The maximum level reported in drinking-water
is 20 µg/litre, though the average daily individual exposure of USA
citizens via drinking-water has been estimated to be <0.01 µg.
The levels of vinylidene chloride in food are generally not
detectable and levels above 10 µg/kg have not been reported. The
levels in food derived from aquatic organisms are not known, but
are likely to be insignificant (section 10.1). Ambient air levels
of vinylidene chloride have been reported of up to 52 µg/m3 (at the
perimeter of an industrial site). Median urban air concentrations
in the USA of 20 ng/m3 and 8.7 µg/m3 have been reported for non-
industrial and industrial-source areas, respectively.
Occupational exposure occurs particularly in production and
polymerization processes. Respiration is the major route of uptake
and the maximum recommended or regulated mean exposure limits over
the period of a working day range from 8 to 500 mg/m3 (or the
lowest reliably detectable concentration) depending on the
country. Short-term exposure limits range from 16 to 80 mg/m3 and
ceiling values range from 50 to 700 mg/m3. Airborne levels of
vinylidene chloride in the confined atmospheres to which workers in
certain occupations are exposed have been found not to exceed 8
mg/m3.
10.2.2. Acute effects
In human beings, inhalation of high concentrations of
vinylidene chloride (very approximately, at or above the maximum
olfactory threshold of 4000 mg/m3) are likely to cause depression
of the central nervous system and could lead to coma. On the basis
of acute toxicity in animals, toxic effects of vinylidene chloride
may occur in the liver, kidneys, or lungs at well below the
minimum olfactory threshold of approximately 2000 mg/m3.
Vinylidene chloride exposure can lead to irritation of the eye, the
upper respiratory tract (at 100 mg/m3 in human beings (section
9.1), and the skin, and this is thought to be partially due to the
stabilizer p- methoxyphenol.
In mice, which are more susceptible than rats to the
hepatotoxic and renal toxic effects of vinylidene chloride,
kidney damage was induced by exposure to as little as 40 mg
vinylidene chloride/m3 (10 ppm) for 6 h. Marked hepatotoxicity and
renal toxicity were also seen in rats. After fasting, which
exacerbated toxicity, exposure to vinylidene chloride at 600 mg/m3
(150 ppm) and 800 mg/m3 (200 ppm) for 6 h caused toxicity in rat
liver and kidney, respectively. Studies on rats indicate that
alcohol ingestion prior to exposure can enhance the metabolism and
exacerbate the toxicity of vinylidene chloride. Acute toxicity is
dependent on species, sex, strain, and the dietary status of
animals. Species susceptibility is correlated with the activity
of oxidative metabolism of vinylidene chloride in rats and mice.
While it is not possible to predict whether the rat or the mouse
provides the more suitable model for human beings, the activity of
hepatic microsomal metabolism by human beings is quantitatively
similar to that of the rat, a species of relatively low
susceptibility. There is no evidence of a qualitative difference
in the oxidative metabolism of vinylidene chloride in human beings
and rodents.
It is apparent that the margin between the concentrations
capable of producing toxicity in animals (40 mg/m3 in mice) and the
occupational exposure limit set by some countries may not be
sufficient or may be non-existent.
10.2.3. Long-term effects and genotoxicity
Prolonged and repeated short-term exposures at sub-anaesthetic
doses may produce kidney and liver damage. On the basis of long-
term studies on animals, under conditions that simulated
occupational exposure, hepatic changes were reported at an exposure
level of 300 mg /m3 (75 ppm) in rats. In mice, kidney and liver
damage were seen at 100 mg/m3 (25 ppm) and 200 mg/m3 (50 ppm),
respectively. There was considerable variation in the sensitivity
to toxic effects observed in the different studies.
Vinylidene chloride does not appear to affect reproductive
capacity or to pose an embryotoxic or teratogenic risk at dose
levels below those required for maternal toxicity in animals, but
this has not been studied in human beings. Embryo and fetal
toxicity and fetal abnormalities were seen at levels producing
maternal toxicity, as evidenced by reduced weight gain.
Vinylidene chloride is mutagenic for bacteria and yeast
provided that a mammalian metabolic system is present. Some
mammalian cells are also receptive to DNA damage and mutagenicity
in vitro . Genotoxicity was not evident in the majority of in vivo
studies on rodents, as measured by dominant lethality and
cytogenetics, but aberrations in bone marrow cells of Chinese
hamsters have been reported. DNA binding and repair in vivo in
rodents, though detectable, was minimal. The data on in vivo
genetic studies therefore suggest some evidence for genetic
toxicity, but, in the majority of studies, the effects were
minimal or negative.
Several carcinogencity tests have been carried out on three
species of experimental animals (mouse, rat, and hamster) using
various routes of administration. Unfortunately, most of these
studies suffered from severe limitations in design or conduct for
carcinogenicity evaluation. No significant carcinogenic effects
were observed in rats dosed orally. In adult rats exposed through
inhalation, an increase in mammary tumours, which was not dose-
related, was reported. A slight increase in leukaemia was observed,
when rats were exposed both in utero and post-natally. These
observations could not be evaluated. In one study on mice,
increased incidence of kidney adenocarcinomas were observed in
males at exposure levels of 200 and 100 mg/m3 (50 and 25 ppm) but
not at 40 and 0 mg/m3 (10 and 0 ppm). In the same study,
statistically increased incidences of lung tumours (mainly adenomas
in both sexes) and mammary carcinomas (in females) were observed,
but no dose-response relationships were found.
The kidney tumours may be related in some way to observed
kidney cytotoxicity and it is possible that repeated kidney damage
either leads directly to the carcinogenic response via a non-
genotoxic mechanism or facilitates the expression of the genotoxic
potential of metabolites in this particular species, sex, and
organ. However, this conclusion is uncertain in the absence of
adequate dose-response data on genetic effects in vivo and the
findings that vinylidene chloride may have acted as an initiator in
a two-stage skin assay in mice.
Epidemiological studies, while not providing any statistically
significant evidence for an increased cancer risk from vinylidene
chloride exposure under occupational conditions, are not adequate
to permit a proper evaluation of the carcinogenic risk of
vinylidene chloride for human beings.
Although the evaluations of individual authors dismiss the
finding of excess cancer deaths as a chance occurrence (due to
small numbers and cohort sizes), the consistency of the higher than
expected values is worth mentioning. In the two cohort studies
reported, lung cancer was observed in 7 cases, whereas 3.16 deaths
would have been expected. The result cannot be dismissed, but co-
existent exposure to vinyl chloride (in one study) has to be borne
in mind. Since the cohorts were identified according to their
exposure to vinylidene chloride, it may be impossible to exclude
additional confounding exposures.
The morbidity findings reported (including one case of
testicular carcinoma) have some informatory value. The
interpretation by the authors that higher liver morbidity was
related to the alcohol consumption of the individuals is invalid,
since the alcohol intake by all members of the study (not only
that of identified cases) was not assessed.
11. RECOMMENDATIONS
11.1. Recommendations for future work
There is a need for better estimates of the global annual
production of vinylidene chloride and of the amounts of vinylidene
chloride entering the environment from all sources, whether arising
from the release of vinylidene chloride as such or from the
degradation of other chemical products.
The predicted environmental fate is based on little
experimental evidence. Information is required on rates of
degradation and on transformation products in the air, soil,
water, and sediment, and metabolism in representative non-
mammalian species.
Long-term toxicity studies investigating a variety of
pathological endpoints should be carried out on representative
aquatic species (fish, crustacea, and molluscs).
Thresholds for, and mechanisms of, toxic effects from short-
and long-term exposure to vinylidene chloride need to be defined
more accurately in animals and human beings, as a basis for
establishing safe levels of exposure.
More exhaustive use should be made of existing data on
carcinogenicity. If further carcinogenicity studies are carried
out, they should be conducted according to an accepted lifetime
bioassay protocol specifically designed to cater for the particular
properties of vinylidene chloride. Such studies should take into
consideration the short half-life of the chemical in the body,
the importance of age at onset of exposure, the daily exposure
duration, and other relevant information that might be related to
determining the dosing regimen. Species and strains of animals for
testing need to be carefully selected. Toxicity data as well as
metabolic and pharmacokinetic data for these animals would also be
extremely useful.
Epidemiological studies are needed to enable an assessment to
be made of the effects of exposure to vinylidene chloride
(including prolonged low-level exposure) on human populations.
Thus, long-term follow-up studies on morbidity and mortality on
whole, unselected populations exposed to vinylidene chloride
should be conducted. Information on effects, such as premature
cerebrovascular disease and cancer, is particularly necessary and
studies should take into account confounding factors, such as
smoking and alcohol consumption (ideally on a case-referent basis).
To overcome the problem of small numbers in individual production
sites, multicentre studies with pooling of data may provide a
valuable approach in both ongoing and future investigations.
Historical data should be used as a reference basis for comparison
with results from ongoing investigations to enable an assessment to
be made of the protective effects of regulatory action over recent
years.
There is a need to compare the in vivo/in vitro pharmacokinetics
and metabolism of vinylidene chloride, especially in the kidney,
liver, and lungs, in experimental animals of different species
and in human beings, in order to better understand the results
obtained in in vivo toxicity studies. Parallel data are required
on the potential genotoxicity of vinylidene chloride at the target
site for carcinogenesis in different species, to examine the
possible role of a genetic mechanism.
In the light of the neurotoxicological findings reported in
this review, there is a need to investigate the role of modulator
systems in the pathogenesis of vinylidene chloride intoxication.
The value of the use of a sulfydryl agent, such as
N- acetylcysteine, in the treatment of vinylidene chloride
poisoning in human beings should be investigated in experimental
animal studies.
11.2. Personal Protection and Treatment of Poisoning
11.2.1. Personal protection
In industrial situations where short-term inhalation exposures
above the recommended limits are possible, full face masks with
filters for organic vapours should be used and, where necessary for
emergency use, masks with air-line supply systems should be
provided. Properly maintained protective clothing including
safety goggles should be worn by those handling vinylidene
chloride, to prevent contact with the body. A constant air flow
should be maintained within industrial plants with adequate
filtered vents at points where spills or leaks are likely to occur.
The monitoring of vinylidene chloride emissions during distribution
operations is recommended. In the event of a leak, the vinylidene
chloride should be evaporated either directly in the case of small
leaks, or by controlled evaporation using an expansion synthetic
foam. Water spray curtains can be used to disperse the vapour from
the foam.
11.2.2. Treatment of poisoning in human beings
In cases of over-exposure or ingestion, medical advice should
be obtained. Because of the irritant properties of vinylidene
chloride, particular attention should be given to the lungs, skin,
and eyes. The functions of the heart, liver, kidney, and central
nervous system should be monitored. Since the animal data have
indicated that vinylidene chloride produces a marked increase in
sensitivity to epinephrine-induced cardiac arrhythmias, this drug
should be avoided. Severe hypotension may be treated by
transfusion, with whole blood or plasma expanders. There is no
known antidote.
A patient poisoned through inhalation of vinylidene chloride
should be kept warm in a semi-prone position, in fresh air. The
airway should be kept clear and oxygen should be administered, if
the subject is in a stupor or coma. Artificial respiration should
be provided, if necessary.
Following ingestion of vinylidene chloride, the mouth should be
rinsed with water. Vomiting should not be induced, because of the
risk of aspiration of vinylidene chloride into the larynx and
lungs. Gastric lavage and/or the oral administration of activated
charcoal or liquid paraffin may help to reduce the bioavailability
of vinylidene chloride, if given within approximately 1 h of
ingestion, and may prove of benefit up to 4 h after ingestion.
Eyes exposed to vinylidene chloride should be immediately
irrigated with water for at least 15 minutes and medical advice
should be sought.
In the case of dermal exposure, contaminated clothing should be
removed and the affected area of skin washed with soap and water.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
Vinylidene chloride was evaluated by WHO in 1984 [244] in the
Guidelines for drinking-water quality. It was concluded that:
"Dichloroethenes have been detected in drinking-
water, generally at levels less than 1 µg/litre.
The isomers have not always been differentiated.
1,1-dichloroethene is the isomer that causes most
concern because of evidence that it is carcinogenic
in experimental animals. It is a chemical commonly
used in the synthesis of various polymers; for
example, food wrappers are often made of 1,1-
dichloroethene co-polymers. 1,1-Dichloroethene
produces mammary tumours in both mice and rats, and
kidney adenocarcinomas in mice (13). It has also
been shown to be mutagenic in the Ames assay. A
linear multi-stage extrapolation model was applied
to data concerning the incidence of kidney
adenocarcinomas in Swiss mice in order to calculate
the recommended guideline value of 0.3 µg/litre."
Vinylidene chloride was evaluated by IARC Working Groups in
1978 [85], 1985 [86], and 1987 [87]. In 1987, the following
conclusions were reported:
"VINYLIDENE CHLORIDE (Group 3)
"A. Evidence for carcinogenicity to humans ( inadequate)
In one epidemiological study of 138 US workers
exposed to vinylidene chloride, no excess of
cancer was found, but follow-up was incomplete,
and nearly 40% of the workers had less than 15
years' latency since first exposures. In a study
in the Federal Republic of Germany of 629 workers
exposed to vinylidene chloride, seven deaths from
cancer (five bronchial carcinomas) were reported;
this number was not in excess of the expected
value. Two cases of bronchial carcinoma were
found in workers, both of whom were 37 years old,
whereas 0.07 were expected for persons aged 35-39
years. The limitations of these two studies do
not permit assessment of carcinogenicity of the
agent to humans. No specific association was
found between exposure to vinylidene chloride and
the excess of lung cancer noted previously in a US
synthetic chemicals plant.
"B. Evidence for carcinogenicity to animals ( limited)
Vinylidene chloride was tested for carcinogenicity
in mice and rats by oral administration and by
inhalation, in mice by subcutaneous
administration and by topical application, and
in hamsters by inhalation. Studies in mice and
rats by oral administration gave negative results.
In inhalation studies, no treatment-related
neoplasm was observed in rats or hamsters. In
mice, a treatment-related increase in the
incidence of kidney adenocarcinomas was observed
in male mice, as were increases in the incidence
of mammary carcinomas in females and of pulmonary
adenomas in male and female mice. In skin-painting
studies in female mice, vinylidene chloride
showed activity as an initiator, but, in a study
of repeated skin application, no skin tumour
occurred. No tumour at the injection site was seen
in mice given repeated subcutaneous administrations.
"C. Other relevant data
"No data were available on the genetic and related
effects of vinylidene chloride in humans.
"Vinylidene chloride did not induce dominant lethal
mutations in mice or rats and did not induce
chromosomal aberrations in bone-marrow cells of
rats treated in vivo ; however, it induced
unscheduled DNA synthesis in treated mice. It did
not induce chromosomal aberrations or mutation in
Chinese hamster cells in vitro but did induce
unscheduled DNA synthesis in rat hepatocytes.
Vinylidene chloride was mutagenic to plant cells
and induced mutation and gene conversion in yeast.
It was mutagenic to bacteria."
REFERENCES
1 ALTMAN, P.L. & DITTMER, D.S. (1966) Environmental biology,
Bethesda, Maryland, Federation of American Societies for
Experimental Biology, pp. 326-328.
2 ANDERSEN, M.E. & JENKINS, L.J., Jr (1977) Oral toxicity of
1,1-dichloroethylene in the rat: effects of sex, age and
fasting. Environ. Health Perspect. 21: 157-163.
3 ANDERSEN, M.E., JONES, R.A., & JENKINS, L.J., Jr (1977)
Enhancement of 1,1-dichloroethylene toxicity by pretreatment
of fasted male rats with 2,3-epoxy-propan-1-ol. Drug Chem.
Toxicol., 1: 63-74.
4 ANDERSEN, M.E, JONES, R.A., & JENKINS, L.J., Jr (1978) The
acute toxicity of single, oral doses of 1,1-dichloroethylene
in the fasted, male rat: effect of induction and inhibition of
microsomal enzyme activities on mortality. Toxicol. appl.
Pharmacol., 46: 227-234.
5 ANDERSEN, M.E., FRENCH, J.E, GARGAS, M.L., JONES, R.A., &
JENKINS, L.J., Jr (1979a) Saturable metabolism and the acute
toxicity of 1,1-dichloroethene. Toxicol. appl. Pharmacol., 47:
385-393.
6 ANDERSEN, M.E., GARGAS, M.L., JONES, R.A., & JENKINS, L.J., Jr
(1979b) The use of inhalation techniques to assess the kinetic
constants of 1,1-dichloroethylene metabolism. Toxicol. appl.
Pharmacol., 47: 395-409.
7 ANDERSEN, M.E., THOMAS, O.E., GARGAS, M.L., JONES, R.A., &
JENKINS, L.J., Jr (1980) The significance of multiple
detoxification pathways for reactive metabolites in the
toxicity of 1,1-dichloroethylene. Toxicol. appl. Pharmacol, 52:
422-432.
8 ANDERSON, D., HODGE, M.C.E., & PURCHASE, I.F.H. (1977)
Dominant lethal studies with the halogenated olefins vinyl
chloride and vinylidene dichloride in male CD-1 mice. Environ.
Health Perspect., 21: 71-78.
9 ATRI, F.R. (1985) [Chlorinated compounds in the environment.]
Schriftenr. Ver. Wasser-Boden-Lufthyg., 60: 309-317 (in German)
10 BADEN, J.M., BRINKENHOFF, M., WHARTON, R.S., HITT, B.A.,
SIMMON, V.F., & MAZZE, R.I. (1976) Mutagenicity of volatile
anesthetics. Anesthesiology, 45: 311-318.
11 BADEN, J.M., KELLEY, M., SIMMON, V.F., RICE, S.A., & MAZZE,
R.I. (1978) Fluroxene mutagenicity. Mutat. Res., 58: 183-191.
12 BADEN, J.M., KELLEY, M., & MAZZE, R.I. (1982) Mutagenicity of
experimental inhalational anesthetic agents: sevofluorane,
synthane, dioxychlorane and dioxyfluorane. Anesthesiology, 56:
462-463.
13 BARRIO-LAGE, G., PARSONS, F.Z., NASSAR, R.S. & LORENZO, P.A.
(1986) Sequential dehalogenation of chlorinated ethenes.
Environ. Sci. Technol.,20, 96-99.
14 BARTSCH, H., MALAVEILLE, C., MONTESANO, R., & TOMATIS, L.
(1975) Tissue-mediated mutagenicity of vinylidene chloride and
2-chlorobutadiene in Salmonella typhimurium. Nature (Lond.),
255: 641-643.
15 BARTSCH, H., MALAVEILLE, C., BARBIN, A., & PLANCHE, G. (1979)
Mutagenic and alkylating metabolites of halo-ethylenes,
chloro-butadienes and dichlorobutenes produced by rodent or
human liver tissues. Evidence for oxirane formation by P450-
linked microsomal monooxygenases. Arch. Toxicol., 41: 249-277.
16 BATTELLE (1983) Study of discharges of certain chloroethylenes
into the aquatic environment and the best technical means for
the reduction of water pollution from such discharges, Geneva,
Battelle Institute (Contract U/82/-176(537).
17 BELLAR, T.A., BUDDE, W.L., & EICHELBERGER, J.W. (1979) The
identification and measurement of volatile organic compounds
in aqueous environmental samples. In: 94th ACS Symposium
Series on Monitoring of Toxic Substances, Washington, DC,
American Chemical Society, pp. 49-62.
18 BIRKEL, T.J., ROACH, J.A.G., & SPHON, J.A. (1977)
Determination of vinylidene chloride in saran films by
electron capture gas-solid chromatography and confirmation by
mass spectrometry. J. Assoc. Off. Anal. Chem., 60: 1210-1213.
19 BRONZETTI, G., BAUER, C., CORSI, C., LEPORINI, C., NIERI, R.,
& DEL CARRATORE, R. (1981) Genetic activity of vinylidene
chloride in yeast. Mutat. Res., 89: 179-185.
20 BROWN, S.L., CHAN, F.Y., JONES, J.L., LIU, D.H., MCCALEB, K.E,
MILL, T., SAPIO, K.N., & SCHENDEL, D.E. (1975) Research
program on hazard priority ranking of priority chemicals.
Phase II: Final Report Menlo Park, California, Stanford
Research Institute (NSF-RA-E-75-190A; NTIS PB-263-161).
21 BUCCAFUSCO, R.J., ELLS, S.J., & LEBLANC, G.A. (1981) Acute
toxicity of priority pollutants to bluegill (Lepomis
macrochirus). Bull. environ. Contam. Toxicol., 26: 446-452.
22 BUCKINGHAM, J., ed. (1982) Dictionary of organic compounds,
5th ed., New York, Chapman and Hall, Vol. 2, p. 1733.
23 CARLSON, G.P. & FULLER, G.C. (1972) Interaction of modifiers
of hepatic microsomal drug metabolism and the inhalation
toxicity of 1,1-dichloroethylene. Res. Commun. chem. Pathol.
Pharmacol., 4: 553-560.
24 CARPENTER, C.P., SMYTH, H.F., Jr, & POZZANI, U.C. (1949) The
assay of acute vapor toxicity and the grading and
interpretation of results of 96 chemical compounds. J. ind.
Hyg. Toxicol., 31: 343-346.
25 CEC (1988) Draft proposal for a Council Directive on the
approximation of the laws of the Member States relating to
plastic materials and articles intended to come into contact
with foodstuffs, Brussels, Commission of the European
Communities, p. 43.
26 CERNA, M. & KYPENOVA, H. (1977) Mutagenic activity of
chloroethylenes analysed by screening system tests. Mutat.
Res., 46: 214-215.
27 CHIECO, P., MOSLEN, M.T., & REYNOLDS, E.S. (1981) Effect of
administrative vehicle on oral 1,1-dichloroethylene toxicity.
Toxicol. appl. Pharmacol., 57: 146-155.
28 CHIECO, P., MOSLEN, M.T., & REYNOLDS, E.S. (1982)
Histochemical evidence that plasma and mitochondrial membranes
are primary foci of hepatocellular injury caused by 1,1-
dichloroethylene. Lab. Invest., 46: 413-421.
29 CHIVERS, C.P. (1972) Two cases of occupational leucoderma
following contact with hydroquinone monomethyl ether. Br. J.
ind. Med., 29: 105-107.
30 COLE, R.H., FREDERICK, R.E., HEALY, R.P., & ROLAN, R.G. (1984)
Preliminary findings of the priority pollutant monitoring
project of the nationwide urban runoff program. J. Water
Pollut. Control Fed., 56: 898-908.
31 COMBA, M.E. & KAISER, K.L.E. (1983) Determination of volatile
contaminants at the ng.1-1 level in water by capillary gas
chromatography with electron capture detection. Int. J.
environ. anal. Chem., 16: 17-31.
32 COSTA, A.K. & IVANETICH, K.M. (1982) Vinylidene chloride: its
metabolism by hepatic microsomal cytochrome P-450 in vitro .
Biochem. Pharmacol., 31: 2083-2092.
33 COSTA, A.K. & IVANETICH, K.M. (1984) Chlorinated ethylenes:
their metabolism and effect on DNA repair in rat hepatocytes.
Carcinogenesis, 5: 1629-1636.
34 CUPITT, L.T. (1980) Fate of toxic and hazardous materials in
the air, Washington, DC, US Environmental Protection Agency
(EPA 600/83-80-084; PB 80-221948).
35 DALLAS, C.E., WEIR, F.W., FELDMAN, S., PUTCHA, L., & BRUCKNER,
J.V. (1983) The uptake and disposition of 1,1-dichloroethylene
in rats during inhalation exposure. Toxicol. appl. Pharmacol.,
68: 140-151.
36 DAWSON, G.W., JENNINGS, A.L., DROZDOWSKI, D., & RIDER, E.
(1975/77) The acute toxicity of 47 industrial chemicals to
fresh and saltwater fishes. J. hazard. Mater., 1: 303-318.
37 DELEON, I.R., MABERRY, M.A., OVERTON, E.B., RASCHKE, C.K.,
REMELE, P.C., STEELE, C.F., WARREN, V.L., & LASETER, J.L.
(1980) Rapid gas chromatographic method for the determination
of volatile and semivolatile organochlorine compounds in soil
and chemical waste disposal site samples. J. chromatogr. Sci.,
18: 85-88.
38 DEMERTZIS, P.G., KONTOMINAS, M.G., & GILBERT, S.G. (1987) Gas
chromatographic determination of sorption isotherms of
vinylidene chloride n vinylidene chloride copolymers. J. Food.
Sci., 52 (3): 747-750.
39 DILL, D.C., MCCARTY, W.M., ALEXANDER, H.C., & BARTLETT, E.A.
(1980) Toxicity of 1,1-dichloroethylene (vinylidene chloride)
to aquatic organisms, Midland, Michigan, Dow Chemical Company
(PB 81-111098).
40 DILLING, W.L. (1977) Interphase transfer processes. II.
Evaporation rates of chloromethanes, ethanes, ethylenes,
propanes and propylenes from dilute aqueous solution.
Comparison with theoretical predictions. Environ. Sci.
Technol., 11: 405-409.
41 DOW (1988) Migration of vinylidene chloride monomer into food
simulating solvents from various vinylidene chloride
copolymers. Report, Midland, Michigan, Dow Chemical Company,
p. 6.
42 DOWD, R.M. (1985) EPA drinking-water proposals: round one.
Environ. Sci. Technol., 19: 1156.
43 DREVON, C. & KUROKI, T. (1979) Mutagenicity of vinyl chloride,
vinylidene chloride and chloroprene in V79 Chinese hamster
cells. Mutat. Res., 67: 173-182.
44 EASLEY, D.M., KLEOPFER, R.D., & CARASEA, A.M. (1981) Gas
chromatographic-mass spectrometric determination of volatile
organic compounds in fish. J. Assoc. Off. Anal. Chem., 64:
653-656.
45 ECETOC (1985) Joint Assessment of Commodity Chemicals No.5:,
vinylidene chloride, Brussels, European Chemical Industry
Ecology and Toxicology Centre., 54 pp.
46 EISENREICH, S.J., LOONEY, B.B., & THORNTON, J.D. (1981)
Airborne organic contaminants in the great lakes ecosystem.
Environ. Sci. Technol., 15: 30-38.
47 EUROCOP-COST (1976) Analysis of organic micropollutants in
water, Luxembourg, Commission of the European Communities
(Cost Project 64b; EUCO/MDV/73/76, XII/476/76).
48 FERRARIO, J.B., LAWLER, G.C., DELEON, I.R., & LASETER, J.L.
(1985) Volatile organic pollutants in biota and sediments of
Lake Pontchartrain. Bull. environ. Contam. Toxicol. 34:
246-255.
49 FILSER, J.G. & BOLT, H.M. (1979) Pharmacokinetics of
halogenated ethylenes in rats. Arch. Toxicol., 42: 123-136.
50 FOERST, D. (1979) A sampling and analytical method for
vinylidene chloride in air. Am. Ind. Hyg. Assoc. J., 40:
888-893.
51 FOGEL, M.M., TADDEO, A.R., & FOGEL, S. (1986) Biodegradation
of chlorinated ethenes by a methane-utilizing mixed culture.
Appl. environ. Microbiol., 51: 720-724.
52 FORKERT, P.G. & REYNOLDS, E.S. (1982) 1,1-dichloroethylene-
induced pulmonary injury. Exp. lung Res., 3: 57-68.
53 FORKERT, P.G., FORKERT, L., FAROOQUI, M., & REYNOLDS, E.S.
(1985) Lung injury and repair: DNA synthesis following 1,1-
dichloroethylene. Toxicology, 36: 199-214.
54 FORKERT, P.G., HOFLEY, M., & RACZ, W.J. (1986a) Metabolic
activation of 1,1-dichloroethylene by mouse lung and liver
microsomes. Can. J. Physiol. Pharmacol., 65: 1496-1499.
55 FORKERT, P.G., STRINGER, V., & RACZ, W.J. (1986b) Effects of
administration of metabolic inducers and inhibitors on
pulmonary toxicity and covalent binding by 1,1-
dichloroethylene in CD-1 mice. Exp. mol. Pathol., 45: 44-58.
56 FORKERT, P.G., STRINGER, S., & TROUGHTON, K.M. (1986c)
Pulmonary toxicity of 1,1-dichloroethylene correlation of
early changes with covalent binding. Can. J. Physiol.
Pharmacol., 64: 112-121.
57 GAGE, J.C. (1970) The subacute inhalation toxicity of 109
industrial chemicals. Br. J. ind. Med., 27: 1-18.
58 GAY, B.W., HANST, P.L., BUFALINI, J.J., & NOONAN, R.C. (1976)
Atmospheric oxidation of chlorinated ethylenes. Environ. Sci.
Technol., 10: 58-67.
59 GIBBS, D.S. & WESSLING, R.A. (1983) Vinylidene chloride and
polyvinylidene chloride. In: Mark, H.F., Othmer, D.F.,
Overberger, C.G., & Seaborg, G.T., ed. Kirk-Othmer
encyclopedia of chemical technology, 3rd ed., New York, John
Wiley and Sons, Vol. 23, pp. 764- 798.
60 GILBERT, J., SHEPHERD, M.J., STARTIN, J.R., & MCWEENY, D.J.
(1980) Gas chromatographic determination of vinylidene
chloride monomer in packaging films and in foods. J.
Chromatogr., 197: 71-78.
61 GLISSON, B.T., CRAFT, B.F., NELSON, J.H., & MEUZELAAR, H.L.C.
(1986) Production of vinylidene chloride from the thermal
decomposition of methyl chloroform. Am. Ind. Hyg. Assoc. J.,
47: 427-435.
62 GOING, J.E. & SPIGARELLI, J. (1977) Environmental monitoring
near industrial sites - vinylidene chloride, Washington, DC,
US Environmental Protection Agency (EPA 560/6-77-026; NTIS PB-
273358).
63 GRASSELLI, J.R. & RITCHEY, W.M., ed. (1975) CRC Atlas of
spectral data and physical constraints for organic compounds,
Cleveland, Ohio, CRC Press, Vol. 3.
64 GREIM, H., BONSE, G., RADWAN, Z., REICHERT, D., & HENSCHLER,
D. (1975) Mutagenicity in vitro and potential carcinogenicity
of chlorinated ethylenes as a function of metabolic oxirane
formation. Biochem. Pharmacol., 24: 2013-2017.
65 GRIMSRUD, E.P. & RASMUSSEN, R.A. (1975) Survey and analysis of
halocarbons in the atmosphere by gas chromatography-mass
spectroscopy. Atmos. Environ., 9: 1014-1017.
66 GRONSBERG, E.S. (1975) [Determination of vinylidene chloride
in the air.] Gig. i Sanit., 7: 77-79 (in Russian).
67 GUILLEMIN, C.L., MARTINEZ, R., & THIAULT, S. (1979) Steam-
modified gas-solid chromatography: a complementary technique
for organic pollutant survey. J. chromatogr. Sci., 17: 677-681.
68 HARKOV, R., KEBBEKUS, B., BOZZELLI, J.W., & LIOY, P.J. (1983)
Measurement of selected volatile organic compounds at three
locations in New Jersey during the summer season. J. Air
Pollut Control Assoc., 33: (12); 1177-1183.
69 HARKOV, R., KEBBEKUS, B., BOZZELLI, J.W., LIOY, P.J., &
DAISEY, J. (1984) Comparison of selected volatile organic
compounds during the summer and winter at urban sites in New
Jersey. Sci. total Environ., 38: 259-274.
70 HARMS, M.S., PETERSON, R.E., FUJIMOTO, J.M., & ERWIN, C.P.
(1976) Increased "bile duct-pancreatic fluid" flow in
chlorinated hydrocarbon-treated rats. Toxicol. appl.
Pharmacol., 35: 41-49.
71 HAWKINS, W.E., OVERSTREET, R.M., WALKER, W.W., & MANNING, C.S.
(1985) Tumour induction in several small fish species by
classical carcinogens and related compounds. In: Proceedings
of the Fifth Conference on Water Chlorination (Chemical,
Environmental Impact and Health Effects), pp. 429-438.
72 HEITMULLER, P.T., HOLLISTER, T.A., & PARRISH, P.R. (1981)
Acute toxicity of 54 industrial chemicals to sheepshead
minnows (Cyprinodon variegatus). Bull. environ. Contam.
Toxicol., 27: 596-604.
73 HENSCHLER, D. (1977) Metabolism and mutagenicity of
halogenated olefins. A comparison of structure and activity.
Environ. Health Perspect., 21: 61-64.
74 HENSCHLER, D., BROSER, F., & HOPF, H.C. (1970) ["Polyneuritis
cranialis" caused by poisoning with chlorinated acetylenes in
working with vinylidene chloride copolymers.] Arch. Toxicol.,
26: 62-75 (in German).
75 HEWITT, W.R. & PLAA, G.L. (1983) Dose-dependent modification
of 1,1-dichloroethylene toxicity by acetone. Toxicol. Lett.,
16: 145-152.
76 HIATT, M.H. (1983) Determination of volatile organic compounds
in fish samples by vacuum distillation and fused silica
capillary gas chromatography/mass spectrometry. Anal. Chem.,
55: 506-516.
77 HOFMANN, H.T. & PEH, J. (1976) [Report on the test of
vinylidene chloride for mutagenic effects in Chinese Hamsters
after subacute inhalation,] Ludwigshafen, BASF
Aktiengesellschaft, 22 pp. (in German).
78 HOLLIFIELD, H.C. & MCNEAL, T. (1978) Gas-solid chromatographic
determination of vinylidene chloride in saran film and three
simulating solvents. J. Assoc. Off. Anal. Chem., 61: 537-544.
79 HONG, C.B., WINSTON, J.M., THORNBURG, L.P., LEE, C.C., &
WOODS, J.S. (1981) Follow-up study on the carcinogenicity of
vinyl chloride and vinylidene chloride in rats and mice: tumor
incidence and mortality subsequent to exposure. J. Toxicol.
environ. Health, 7: 909-924.
80 HSE (1983) Methods for the determination of hazardous
substances 28: Chlorinated hydrocarbon solvent vapours in air,
London, Health and Safety Executive.
81 HSE (1985) Toxicity Review 13: Vinylidene chloride, London,
Health and Safety Executive, 59 pp.
82 HUBERMAN, E., BARTSCH, H., & SACHS, L. (1975) Mutation
induction in Chinese hamster V79 cells by two vinyl chloride
metabolites, chloroethylene oxide and 2-chloroacetaldehyde.
Int. J. Cancer, 16: 639-644.
83 HULL, L.A., HISATSUNE, I.C., & HEICKLEN, J. (1973) The
reaction of O3 with CCl2CH2. Can. J. Chem., 51:
1504-1510.
84 HUSHON, J. & KORNREICH, M. (1978) Air pollution assessment of
vinylidene chloride, Washington, DC, US Environmental
Protection Agency (EPA 450/3-78-015) (Prepared by the Metrek
Division of the Mitre Corporation, McLean, Virginia; Contract
No. 68-02-1495).
85 IARC (1979) Some monomers, plastics and synthetic elastomers,
acrolein, Lyons, International Agency for Research on Cancer,
pp. 439-459 (IARC Monograph on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol.19).
86 IARC (1986) Some chemicals used in plastics and elastomers:
vinylidene chloride, Lyons, International Agency for Research
on Cancer, pp. 195-226 (IARC Monographs on the Evaluation of
the Carcinogenic Risk of Chemicals to Humans, Vol. 39).
87 IARC (1987) Overall evaluations of carcinogenicity: An
updating of IARC Monographs Vols. 1 - 42, Lyons, International
Agency for Research on Cancer, pp. 376-377 (IARC Monographs on
the Evaluation of Carcinogenic Risks to Humans, Supplement 7).
88 IRPTC (1988) IRPTC Legal file, Geneva, International Register
of Potentially Toxic Chemicals, United Nations Environment
Programme.
89 ISHIDATE, M., Jr, ed. (1983) The data book of chromosomal
tests in vitro on 587 chemical substances using a Chinese
hamster fibroblast cell line (Chl cells), Tokyo, The Realize
Inc., p. 582.
90 JACKSON, N.M. & CONOLLY, R.B. (1985) Acute nephrotoxicity of
1,1-dichloroethylene in the rat after inhalation exposure.
Toxicol. Lett., 29: 191-199.
91 JAEGER, R.J. (1975) Vinyl chloride monomer: comments on its
hepatotoxicity and interaction with 1,1-dichloroethylene. Ann.
N.Y. Acad. Sci., 246: 150-151.
92 JAEGER, R.J. & MURPHY, S.D. (1973) Alterations of barbiturate
action following 1,1-dichloroethylene, corticosterone or
acrolein. Arch. int. Pharmacodyn., 205: 281-292.
93 JAEGER, R.J., CONOLLY, R.B., & MURPHY, S.D. (1973a) Diurnal
variation of hepatic glutathione concentration and its
correlation with 1,1-dichloroethylene inhalation toxicity in
rats. Res. Commun. chem. Pathol. Pharmacol., 6: 465-471.
94 JAEGER, R.J., TRABULUS, M.J., & MURPHY, S.D. (1973b)
Biochemical effects of 1,1-dichloroethylene in rats:
dissociation of its hepatotoxicity from a lipoperoxidative
mechanism. Toxicol. appl. Pharmacol., 24: 457-467.
95 JAEGER, R.J., TRABULUS, M.J., & MURPHY, S.D. (1973c) The
interaction of adrenalectomy, partial adrenal replacement
therapy, and starvation with hepatotoxicity and lethality of
1,1- dichloroethylene intoxication. Toxicol. appl. Pharmacol.,
25: 491 (Abstract No. 133).
96 JAEGER, R.J., CONOLLY, R.B., & MURPHY, S.D. (1974) Effect of
18-hr fast and glutathione depletion on 1,1-dichloroethylene-
induced hepatotoxicity and lethality in rats. Exp. mol.
Pathol., 20: 187-198.
97 JAEGER, R.J., SHONER, L.G., & COFFMAN, L. (1977a)
1,1-Dichloroethylene hepatotoxicity: proposed mechanism of
action and distribution and binding of 14C radioactivity
following inhalation exposure in rats. Environ. Health
Perspect., 21: 113-119.
98 JAEGER, R.J., SZABO, S., & COFFMAN, L.J. (1977b) 1,1-
Dichloroethylene hepatotoxicity. Effect of altered thyroid
funtion and evidence for the subcellular site of injury. J.
Toxicol. environ. Health, 3: 545-555.
99 JENKINS, L.J., Jr & ANDERSEN, M.E. (1978) 1,1-Dichloroethylene
nephrotoxicity in the rat. Toxicol. appl. Pharmacol., 46:
131-141.
100 JENKINS, L.J., Jr, TRABULUS, M.J., & MURPHY, S.D. (1972)
Biochemical effects of 1,1-dichloroethylene in rats:
comparison with carbon tetrachloride and 1,2-dichloroethylene.
Toxicol. appl. Pharmacol., 23: 501-510.
101 JONES, B.K. & HATHWAY, D.E. (1978a) Tissue-mediated
mutagenicity of vinylidene chloride in Salmonella typhimurium
TA1535. Cancer Lett., 5: 1-6.
102 JONES, B.K. & HATHWAY, D.E. (1978b) The biological fate of
vinylidene chloride in rats. Chem.-biol. Interact., 20: 27-41.
103 JONES, B.K. & HATHWAY, D.E. (1978c) Differences in metabolism
of vinylidene chloride between mice and rats. Br. J. Cancer,
37: 411- 417.
104 KAISER, K.L.E., COMBA, M.E., & HUNEAULT, H. (1983) Volatile
halocarbon contaminants in the Niagara River and in Lake
Ontario. J. Great Lakes Res., 9(2): 212-223.
105 KANZ, M.F. & REYNOLDS, E.S. (1986) Early effects of 1,1-
dichloroethylene on canalicular and plasma membranes:
ultrastructure and stereology. Exp. mol. Pathol., 44: 93-110.
106 KANZ, M.F., WHITEHEAD, R.F., FERGUSON, A.E., & MOSLEN, M.T.
(1988) Potentiation of 1,1-dichloroethylene hepatotoxicity:
comparative effects of hyperthyreodism and fasting. Toxicol.
appl. Pharmacol., 95: 93-103.
107 KIEZEL, L., LISZKA, M., & RUTKOWSKI, M. (1975) [Gas
chromatographic determination of trace impurities in
distillates of vinyl chloride monomer.] Chem. Anal. (Warsaw),
20: 555-562 (in Polish).
108 KLIMISCH, J.H. & FREISBERG, K.O. (1979a) [Report on the
determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in Chinese striped hamsters (fasting)
during a 4-hour exposure period.], Ludwigshafen, BASF
Aktiengesellschaft, 11 pp (in German).
109 KLIMISCH, J.H. & FREISBERG, K.O. (1979b) [Report on the
determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in Chinese striped hamsters (fed) during a
4-hour exposure period.], Ludwigshafen, BASF
Aktiengesellschaft, 14 pp (in German).
110 KLIMISCH, J. H., LINK, R., STOCKER, W.G., & THIESS, A.M.
(1982) Investigation of the mortality of workers predominantly
exposed to vinylidene chloride. In: Proceedings of the
Medichem Congress, Paris, 1982.
111 KRAMER, C.G. & MUTCHLER, J.E. (1972) The correlation of
clinical and environmental measurements for workers exposed to
vinyl chloride. Am. Ind. Hyg. Assoc. J., 33: 19-30.
112 KRIJGSHELD, K.R. & GRAM, T.E. (1984) Selective induction of
renal microsomal cytochrome P-450-linked monooxygenases by
1,1-dichloroethylene in mice. Biochem. Pharmacol., 33:
1951-1956.
113 KRIJGSHELD, K.R., LOWE, M.C, MIMNAUGH, E.G., TRUSH, M.A,
GINSBURG, E., & GRAM, T.E. (1983) Lung-selective impairment of
cytochrome P-450-dependent monooxygenases and cellular injury
by 1,1-dichloroethylene in mice. Biochem. biophys. Res.
Commun., 110: 675-681.
114 LAIB, R.J., KELIN, K.P., KAUFMANN, I., & BOLT, H.M. (1981) [On
the problem of carcinogenicity of vinylidene chloride
(1,1-dichloroethylene).] In: [ Epidemiological approaches in
occupational medicine], Stuttgart, Gentner Verlag, pp. 277-281
(in German).
115 LAO, R.C., THOMAS, R.S., BASTIEN, P., HALMAN, R.A., &
LOCKWOOD, J.A. (1982) Analysis of organic priority and non-
priority pollutants in environmental samples by GC/MS/computer
systems. Pergamon Ser. environ. Sci., 7: 107-118.
116 LAZAREV, N.V., ed. (1960) [Vinylidene chloride.] In: [ Harmful
substances in industry,] Leningrad, Chemia, pp. 215-216 (in
Russian).
117 LEBLANC, G.A. (1980) Acute toxicity of priority pollutants to
water flea ( Daphnia magna). Bull. environ. Contam. Toxicol.,
24: 684-691.
118 LEE, C.C., BHANDARI, J.C., WINSTON, J.M., HOUSE, W.B., PETERS,
P.J., DIXON, R.L., & WOODS, J.S. (1977) Inhalation toxicity of
vinyl chloride and vinylidene chloride. Environ. Health
Perspect., 21: 25-32.
119. LEE, C.C., BHANDARI, J.C., WINSTON, J.M., HOUSE, W.B., DIXON,
R.L., & WOODS, J.S. (1978) Carcinogenicity of vinyl chloride
and vinylidene chloride. J. Toxicol. environ. Health, 4: 15-30.
120 LEIBMAN, K.C. & ORTIZ, E. (1977) Metabolism of halogenated
ethylenes. Environ. Health Perspect., 21: 91-97.
121 LESAGE, S., PRIDDLE, M.W., & JACKSON, R.E. (1988) Organic
contaminants in ground water at the Gloucester landfill.
Report, National Water Research Institute, Burlington,
Ontario, p.13.
122 LIEBLER, D.C. & GUENGERICH, F.P. (1983) Olefin oxidation by
cytochrome P-450: evidence for group migration in catalytic
intermediates formed with vinylidene chloride and trans-1-
phenyl-1-butene. Biochemistry, 22: 5482-5489.
123 LIEBLER, D.C., MEREDITH, M.J., & GUENGERICH, F.P. (1985)
Formation of glutathione conjugates by reactive metabolites of
vinylidene chloride in microsomes and isolated hepatocytes.
Cancer Res., 45: 186-193.
124 LIEBLER, D.C., LATWESEN, D.G. & REEDER, T.C. (1988). S-(2-
chloroacetyl) glutathione, a reactive glutathione thiol ester
and a putative metabolite of 1,1-dichloroethylene.
Biochemistry, 27: 3652-3657.
125 LIN, S.-N., FU, F.W.-Y., BRUCKNER, J.V., & FELDMAN, S. (1982)
Quantitation of 1,1- and 1,2-dichloroethylene in body tissues
by purge-and-trap gas chromatography. J. Chromatogr., 244:
311-320.
126 LONG, R.M. & MOORE, L. (1987) Cytosolic calcium after carbon
tetrachloride, 1,1-dichloroethylene, and phenylephrine
exposure. Studies in rat hepatocytes with phosphorylase a
and quin 2. Biochem. Pharmacol., 36: 1215-122
127 MABEY, W.R., SMITH, J.H., PUDOLL, R.T., JOHNSON, H.L., MILL,
T., CHOU, T.W., GATES, J., PARTRIDGE, I.W., & VANDENBURG, D.
(1981) Aquatic fate process. Data for organic priority
pollutants, Washington, DC, US Environmental Protection
Agency (EPA 440/4-81-014).
128 MCCANN, J., SIMMON, V., STREITWIESER, D., & AMES, B.N. (1975)
Mutagenicity of chloroacetaldehyde, a possible product of 1,2-
dichloroethane (ethylene dichloride), chloroethanol (ethylene
chlorohydrin), vinyl chloride and cyclo-phosphamide. Proc.
Natl Acad. Sci. (USA), 72: 3190-3193.
129 MCCARROLL, N.E., CORTINA, T.A., ZITO, M.J., & FARROW, M.G.
(1983) Evaluation of methylene chloride and vinylidene
chloride in mutational assays. Environ. Mutagen., 5: 426-427.
130 MCDONALD, T.J., KENNICUTT, M.C., & BROOKS, J.M. (1988)
Volatile organic compounds at a coastal Gulf of Mexico site.
Chemosphere, 17, 123-136.
131 MCKENNA, M.J., WATANABE, P.G., & GEHRING, P.J. (1977)
Pharmacokinetics of vinylidene chloride in the rat. Environ.
Health Perspect., 21: 99-105.
132 MCKENNA, M.J., ZEMPEL, J.A, MADRID, E.O., & GEHRING, P.J.
(1978a) The pharmacokinetics of [14C]vinylidene chloride in
rats following inhalation exposure. Toxicol. appl. Pharmacol.,
45: 599-610.
133 MCKENNA, M.J., ZEMPEL, J.A, MADRID, E.O., BRAUN, W.H., &
GEHRING, P.J. (1978b) Metabolism and pharmacokinetic profile
of vinylidene chloride in rats following oral administration.
Toxicol. appl. Pharmacol., 45: 821-835.
134 MCKENNA, M.J., QUAST, J.F., YAKEL, H.O., BALMER, M.F., &
RAMPY, L.W. (1982) Vinylindene chloride: a chronic inhalation
toxicity and carcinogenicity study in rats. Final Report,
Midland, Michigan, Dow Chemical, 100 pp.
135 MAFF (1980) Survey of vinylidene chloride levels in food
contact materials and in foods. Third Report of the Steering
Group on Food Surveillance: Working Party on Vinylidene
Chloride, London, Ministry of Agriculture, Fisheries and Food,
23 pp (Food Surveillance Paper No. 3).
136 MALAVEILLE, C., BARTSCH, H., BARBIN, A., CAMUS, A.M.,
MONTESANO, R., CROISY, A., & JACQUIGNON, P. (1975)
Mutagenicity of vinyl chloride, chloroethylene-oxide,
chloroacetylaldehyde and chloroethanol. Biochem. biophys. Res.
Commun., 63: 363-370.
137 MALAVEILLE, C., PLANCHE, G., & BARTSCH, H. (1977) Factors for
efficiency of the Salmonella microsome, mutagenicity assay.
Chem.-biol. Interact., 17: 129-136.
138 MALTONI, C. & PATELLA, V. (1983) Comparative acute toxicity of
vinylidene chloride. The role of species, strain and sex. Acta
oncol., 4: 239-256.
139 MALTONI, C., COTTI, G., MORISI, L., & CHIECO, P. (1977)
Carcinogenicity biosassays of vinylidene chloride. Research
plan and early results. Med. Lav., 68: 241-262.
140 MALTONI, C., CILIBERTI, A., & CARRETTI, D. (1982) Experimental
contributions in identifying brain potential carcinogens in
the petro-chemical industry. Ann. N.Y. Acad. Sci., 381:
216-249.
141 MALTONI, C., COTTI, G., & CHIECO, P. (1984) Chronic toxicity
and carcinogenicity bioassays of vinylidene chloride. Acta
oncol., 5: 91-146.
142 MALTONI, C., LEFEMINE, G., COTTI, G., CHIECO, P., & PATELLA,
V. (1985) Experimental research on vinylidene chloride
carcinogenesis. In: Maltoni, C. & Mehlinan, M.A., ed. Archives
of research in industrial carcinogenesis, Princeton, New
Jersey, Princeton Scientific Publishers, Vol. 3, p.95.
143 MASUDA, Y. & NAKAYAMA, N. (1983) Protective action of
diethyldithiocarbamate and carbon disulfide against acute
toxicities induced by 1,1-dichloroethylene in mice. Toxicol.
appl. Pharmacol., 71: 42-53.
144 MOTEGI, S., UEDA, K., TANAKA, H., & OHTA, M. (1976)
Determination of residual vinylidene chloride monomer in
polyvinylidene chloride films used for fish jelly products.
Bull. Jpn. Soc. Sci. Fish., 42: 1387-1394.
145 MOORE, L. (1980) Inhibition of liver microsome calcium pump by
in vivo administration of CCl4, CHCl3 and 1,1-dichloroethylene
(vinylidene chloride). Biochem. Pharmacol., 29: 2505-2511.
146 MORTELMANS, K., HAWORTH, S., LAWLOR, T., SPECK, W., TAINER,
B., & ZEIGER, E. (1986) Salmonella mutagenicity tests III.
Results from the testing of 270 chemicals, Environ. Mutat.,
8, (Suppl. 7): 1-119.
147 MOSLEN, M.T. & REYNOLDS, E.S. (1985) Rapid, substrate-specific
and dose-dependent deactivation of liver cytosolic glutathione
S- transferases in vivo by 1,1-dichloro-ethylene. Res. Commun.
chem. Pathol. Pharmacol., 47: 59-72.
148 MOSLEN, M.T., POISSON, L.R., & REYNOLDS, E.S. (1985)
Cholestasis and increased biliary excretion of inulin in rats
given 1,1-dichloroethylene. Toxicology, 34: 201-209.
149 MURRAY, F.J., NITSCHKE, K.D., RAMPY, L.W., & SCHWETZ, B.A.
(1979) Embryotoxicity and fetotoxicity of inhaled or ingested
vinylidene chloride in rats and rabbits. Toxicol. appl.
Pharmacol., 49: 189- 202.
150 NEUFELD, M.L., SITTENFIELD, M., WOLK, K.F., & BOYD, R.E.
(1977) Market input/output studies. Task 1: vinylidene
chloride, Washington, DC, US Environmental Protection Agency
(EPA 560/6-77-003; NTIS PB-273-205) (Prepared by Auerbach
Associates; Contract No. 68- 01-1996).
151 NITSCHKE, K.D, SMITH, F.A., QUAST, J.F., NORRIS, J.M., &
SCHWETZ, B.A. (1983) A three-generation rat reproductive
toxicity study of vinylidene chloride in the drinking water.
Fundam. appl. Toxicol., 3: 75-79.
152 NORRIS, J.M. (1977) Toxicological and pharmacokinetic studies
on inhaled and ingested vinylidene chloride in laboratory
animals. In: Proceedings of the Technical Association of the
Pulp and Paper Industry (TAPPI) Paper Synthetics Conference,
Chicago, Illinois, 1977, Atlanta, Georgia, Technical
Association of the Pulp and Paper Industry, pp. 45-50.
153 NORRIS, J.M. & REITZ, R.H. (1984) Interpretative review of the
animal toxicological, pharmacokinetic/metabolism, biomolecular
and in vitro mutagenicity studies on vinylidene chloride and
the significance of the findings for man, Midland, Michigan,
Dow Chemical Co., p. 24.
154 NTP (1982) Carcinogenesis bioassay of vinylidene chloride (CAS
No. 75-35-4) in F344 rats and B6C3F1 mice (gavage study),
Research Triangle Park, North Carolina, National Toxicology
Program (Technical Report Series No. 228; PB 82-258393).
155 OBLAS, D.W., DUGGER, D.L., & LIEBERMAN, S.I. (1980) The
determination of organic species in the telephone central
office ambient. IEEE Trans. Compnents Hybrids Manuf. Technol.,
CHMT-3 (1): 17-20.
156 OESCH, F., PROTIC-SABLJIC, M., FRIEDBERG, T., KLIMISCH, H.J.,
& GLATT, H.R. (1983) Vinylidene chloride: changes in drug
metabolising enzyme, mutagenicity and relation to its targets
for carcinogenesis. Carcinogenesis, 4: 1031-1038.
157 OKINE, L.K.N. & GRAM, T.E. (1986a) Tissue distribution and
covalent binding of [14C]1,1-dichloroethylene in mice. In vivo
and in vitro studies. Adv. exp. Med. Biol., 197: 903-910.
158 OKINE, L.K.N. & GRAM, T.E. (1986b) In vitro studies on the
metabolism and covalent binding of [14C]1,1-dichloroethylene
by mouse liver, kidney and lung. Biochem. Pharmacol., 35:
2789-2795.
159 OKINE, L.K.N., GOOCHEE, J.M., & GRAM, T.E. (1985) Studies on
the distribution and covalent binding of 1,1-dichloroethylene
in the mouse. Effects of various pretreatments on covalent
binding in vivo. Biochem. Pharmacol., 34: 4051-4057.
160 OSBOURNE, R.A. (1964) Contact dermatitis caused by saran wrap.
J. Am. Med. Assoc., 188: 1159.
161 OTSON, R. (1987) Purgeable organics in Great Lakes raw and
treated water. Int. J. Environ. anal. Chem., 31: 41-53.
162 OTSON, R. & WILLIAMS, D.T. (1982) Headspace chromatographic
determination of water pollutants. Anal. Chem., 54: 942-946.
163 OTSON, R., WILLIAMS, D.T., & BIGGS, D.C. (1982a) Relationships
between raw water quality, treatment and occurrence of
organics in Canadian potable water. Bull. environ. Contam.
Toxicol., 28: 396-403.
164 OTSON, R., WILLIAMS, D.T., & BOTHWELL, P.D. (1982b) Volatile
organic compounds in water at thirty Canadian potable water
treatment facilities. J. Assoc. Off. Anal. Chem., 65:
1370-1374.
165 OTT, M.G., LANGNER, R.R., & HOLDER, B.B. (1975) Vinyl chloride
exposure in a controlled industrial environment. A long-term
mortality experience in 594 employees. Arch. environ. Health,
30: 333-339.
166 OTT, M.G., FISHBECK, W.A, TOWNSEND, J.C., & SCHNEIDER, E.J.
(1976) A health study of employees exposed to vinylidene
chloride. J.. occup. Med., 18: 735-738.
167 PARSONS, F., WOOD, P.R., & DEMARCO, J. (1984) Transformations
of tetrochloroethene and trichloroethene in microcosms and
groundwater. J. Am. Water Works Assoc., February: 56-59.
168 PATTERSON, J.W. & KODUKALA, P.S. (1981) Biodegradation of
hazardous organic pollutants. CEP, April: 48-55.
169 PEARSON, C.R. & MCCONNELL, G. (1975) Chlorinated C1 and C2
hydrocarbons in the marine environment. Proc. R. Soc. Lond.
Ser. B, 189: 305-332.
170 PFAB, W., VON & MUCKE, G. (1977) [On the migration of selected
monomers in foodstuffs and simulations.] Dtsch. Lebensm.
Rundschau, 73: 1-5 (in German).
171 PIET, G.J., SLINGERLAND, P., DE GRUNT, F.E., VAN DEN HEUVEL,
M.P.M., & ZOETEMAN, B.C.J. (1978) Determination of very
volatile halogenated organic compounds in water by means of
direct head-space analysis. Anal. Lett., A11(5): 437-448.
172 PONOMARKOV, V. & TOMATIS, L. (1980) Long-term testing of
vinylidene chloride and chloroprene for carcinogenicity in
rats. Oncology, 37: 136-141.
173 PRENDERGAST, J.A., JONES, R.A, JENKINS, L.J., Jr, & SIEGEL, J.
(1967) Effects on experimental animals of long-term inhalation
of trichloroethane, carbon tetrachloride, 1,1,1-trichloro-
ethylene, dichlorodifluoromethane and 1,1-dichloroethylene.
Toxicol. appl. Pharmacol., 10: 270-289.
174 PRICE, P.S. (1985) Volatile organochlorine compounds (VOC)
degradation. Technical Memorandum, Washington, DC, US
Environmental Protection Agency, p. 19.
175 PUTCHA, L., BRUCKNER, J.V., D'SOUZA, R., DESAI, F., & FELDMAN,
S. (1986) Toxicokinetics and bioavailability of oral and
intravenous 1,1-dichloroethylene. Fundam. appl. Toxicol., 6:
240-250.
176 QUAST, J.F., HUMISTON, C.G., SCHWETZ, B.A., BALMER, M.F.,
RAMPY, L.W., NORRIS, J.M., & GEHRING, P.J. (1977) Results of
90-day toxicity study in rats given vinylidene chloride in
their drinking water or exposed to VDC vapour by inhalation.
Toxicol. appl. Pharmacol., 4: 187.
177 QUAST, J.F., HUMISTON, C.G., WADE, C.E., BALLARD, J., BEYER,
J.E, SCHWETZ, R.W., & NORRIS, J.M. (1983) A chronic toxicity
and oncogenicity study in rats and subchronic toxicity study
in dogs on ingested vinylidene chloride. Fundam. appl.
Toxicol., 3: 55-62.
178 QUAST, J.F., MCKENNA, M.J., RAMPY, L.W., & NORRIS, J.M. (1986)
Chronic toxicity and oncogenicity study on inhaled vinylidene
chloride in rats. Fundam. appl. Toxicol., 6: 105-144.
179 RAMPY, L.W., QUAST, J.F., HUMISTON, C.G., BALMER, M.F., &
SCHWETZ, B.A. (1977) Interim results of two-year toxicological
studies in rats of vinylidene chloride incorporated in the
drinking water or administered by repeated inhalation.
Environ. Health Perspect., 21: 33-43.
180 RAMPY, L.W., QUAST, J.F., HUMISTON, C.G., BALMER, M.F., &
SCHWETZ, B.A. (1978) Results of two-year toxicological studies
in rats of vinylidene chloride incorporated in the drinking
water or administered by repeated inhalation. Toxicol. appl.
Pharmacol., 45: 244-245.
181 RAMSTAD, T., NESTRICK, T.J., & PETERS, T.L. (1981)
Applications of the purge-and-trap technique. Am. Lab., 13:
65-73.
182 RAY, P. & MOORE, L. (1982) 1,1-Dichloroethylene inhibition of
liver microsomal calcium pump in vitro . Arch. Biochem.
Biophys., 218: 26-30.
183 REICHERT, D., WERNER, H.W., & HENSCHLER, D. (1978) Role of
liver glutathione in 1,1-dichloroethylene metabolism and
hepatotoxicity in intact rats and isolated perfused rat liver.
Arch. Toxicol., 41: 169-178.
184 REICHERT, D., WERNER, H.W., METZLER, M., & HENSCHLER, D.
(1979) Molecular mechanism of 1,1-dichloroethylene toxicity:
excreted metabolites reveal different pathways of reactive
intermediates. Arch. Toxicol., 42: 159-169.
185 REICHERT, D., SPENGLER, U ., ROMEN, W., & HENSCHLER, D. (1984)
Carcinogenicity of dichloroacetylene: an inhalation study.
Carcinogenesis, 5: 1411-1420.
186 REITZ, R.H., WATANABE, P.G., MCKENNA, M.J., QUAST, J.F., &
GEHRING, P.J. (1980) Effects of vinylidene chloride on DNA
synthesis and DNA repair in the rat and mouse: a comparative
study with dimethylnitrosamine. Toxicol. appl. Pharmacol., 52:
357-370.
187 REKKER, R.F. (1977) The hydrophobic fragment constant, its
derivation and application. A means of characterizing membrane
systems. In: Nauta, W.Th. & Rekker, R.F., ed. Pharmacochemistry
library, Amsterdam, Elsevier Scientific Publishers, Vol. 1.
188 REYNOLDS, E.S, MOSLEN, M.T., SZABO, S., JAEGER, R.J., &
MURPHY, S.D. (1975) Hepatotoxicity of vinyl chloride and 1,1-
dichloroethylene. Role of mixed function oxidase system. Am.
J. Pathol., 81: 219-236.
189 REYNOLDS, E.S, MOSLEN, M.T., BOOR, P.J., & JAEGER, R.J. (1980)
1,1-Dichloroethylene hepatotoxicity. Time course of GSH
changes and biochemical aberrations. Am. J. Pathol., 101:
331-344.
190 REYNOLDS, E.S., KANZ, M.F., CHIECO, P., & MOSLEN, M.T. (1984)
1,1-Dichloroethylene: an apoptotic hepatotoxin. Environ.
Health Perspect., 57: 313-320.
191 RUSSELL, M.J. (1975) Analysis of air pollutants using sampling
tubes and gas chromatography. Environ. Sci. Technol., 9:
1175-1178.
192 RYLOVA, M.L. (1953) [Toxicity of vinylidene chloride.] Farmakol.
Toksikol., 16(1): 47-50 (in Russian).
193 SASAKI, M., SUGIMURA, K., YOSHIDA, M.A., & ABE, S. (1980)
Cytogenetic effects of 60 chemicals on cultured human and
Chinese hamster cells. Kromosomo II,20: 574-584.
194 SASSU, G.M, ZILIO-GRANDI, F., & CONTE, A. (1968) Gas
chromatographic determination of impurities in vinyl
chloride. J. Chromatogr., 34: 394-398.
195 SATO, A., NAKAJIMA, T., & KOYAMA, Y. (1980) Effects of chronic
ethanol consumption on hepatic metabolism of aromatic and
chlorinated hydrocarbons in rats. Br. J. ind. Med., 37:
382-386.
196 SAWADA, M., SOFUNI, T., & ISHIDATE, M., Jr (1987) Cytogenetic
studies on 1,1-dichloroethylene and its two isomers in
mammalian cells in vitro and in vivo. Mutat. Res., 187:
157-163.
197 SAX, N.I. (1984) Dangerous properties of industrial materials,
6th ed., New York, Van Nostrand Reinhold, p. 2730.
198 SCHMITZ, TH., THIESS, A.M., & PENNING, E. (1979) [Inquiry into
morbidity among workers exposed to vinylidene chloride (VDC)
and polyvinylidene chloride (PVDC).] In: [ Report on the Tenth
Annual Meeting of the German Occupational Medicine Society
together with the Federation of Industrial Employers
Associations, Munster, 2-5 May, 1979, ] Stuttgart, Gentner
Verlag (in German).
199 SEVERS, L.W. & SKORY, L.K. (1975) Monitoring personnel
exposure to vinyl chloride, vinylidene chloride and methyl
chloride in an industrial work environment. Am. Ind. Hyg.
Assoc. J., 39: 669-676.
200 SHACKELFORD, W.M. & KEITH, L.H. (1976) Frequency of organic
compounds identified in water, Washington, DC, US
Environmental Protection Agency, Office of Research and
Development, Environmental Research Laboratory (EPA 600/4-76-
062; PB-265-470).
201 SHELTON, L.G., HAMILTON, D.E., & FISACKERLY, R.H. (1971) Vinyl
and vinylidene chloride. In: Leonard, E.C., ed. Vinyl and
diene monomers. Part 3, New York, Wiley Interscience,
pp. 1505-1289.
202 SHORT, R.D., MINOR, J.L., PETERS, P., WINSTON, J.M., FERGUSON,
B., UNGER, T., SAWYER, M., & LEE, C.C. (1977a) The
developmental toxicity of vinylidene chloride inhaled by rats
and mice during gestation, Washington, DC, US Environmental
Protection Agency (EPA 560/6-77-022; PB-281-713) (Prepared by
Midwest Research International, Kansas City, Missouri).
203 SHORT, R.D., MINOR, J.L., WINSTON, J.M., & LEE, C.C. (1977b) A
dominant lethal study in male rats after repeated exposures to
vinyl chloride or vinylidene chloride. J. Toxicol. environ.
Health, 3: 965-968.
204 SHORT, R.D., WINSTON, J.M., MINOR, J.L., HONG, C.B., SEIFTER,
J., & LEE, C.C. (1977c) Toxicity of vinylidene chloride in
mice and rats and its alteration by various treatments. J.
Toxicol. environ. Health, 3: 913-921.
205 SIDHU, K.S. (1980) A gas-chromatographic method for the
determination of vinylidene chloride in air. J. anal.
Toxicol., 4: 266-268.
206 SIEGEL, J., JONES, R.A., COON, R.A., & LYON, J.P. (1971)
Effects on experimental animals of acute, repeated and
continuous inhalation exposures to dichloroacetylene mixtures.
Toxicol. appl. Pharmacol., 18: 168-174.
207 SIEGERS, C.-P., YOUNES, M., & SCHMITT, G. (1979) Effects of
dithiocarb and (+)-cyanidanol-3 on the hepatotoxicity and
metabolism of vinylidene chloride in rats. Toxicology, 15:
55-64.
208 SIEGERS, C.-P., HEIDBUCHEL, K., & YOUNES, M. (1983) Influence
of alcohol, dithiocarb, or (+)-catechin on the hepatotoxicity
and metabolism of vinylidene chloride in rats. J. appl.
Toxicol., 3: 90-95.
209 SIEGERS, C.-P., HORN, W., & YOUNES, M. (1985a) Effect of
hypoxia on the metabolism and hepatoxicity of carbon
tetrachloride and vinylidene chloride in rats. Acta pharmacol.
toxicol., 56: 81-86.
210 SIEGERS, C.-P., HORN, W., & YOUNES, M. (1985b) Effect of
phorone-induced glutathione depletion on the metabolism and
hepatotoxicity of carbon tetrachloride and vinylidene
chloride. J. appl. Toxicol., 5: 352-356.
211 SILETCHNIK, L.M. & CARLSON, G.P. (1974) Cardiac sensitizing
effects of 1,1-dichloroethylene: enhancement by phenobarbital
pretreatment. Arch. int. Pharmacodyn., 210: 359-364.
212 SINGH, H.B., SALAS, L.J., SMITH, A.J., & SHIGEISHI, M. (1981)
Measurements of some potentially hazardous organic chemicals
in urban environments. Atmos. Environ., 15: 601-612.
213 SINGH, H.B., SALAS, L .J., & STILES, R.E. (1982) Distribution
of selected gaseous organic mutagens and suspect carcinogens
in ambient air. Environ. Sci. Technol., 16: 872-880.
214 SPEIS, D.N. (1980) Determination of purgeable organics in
sediments. Environ. Sci. Res., 16: 201-206.
215 SWEGER, D.M. & TRAVIS, J.C. (1979) An application of infrared
lasers to the selective detection of trace organic gases.
Appl. Spectrosc., 33: 46-51.
216 SZABO, S., JAEGER, R.J., MOSLEN, M.T., & REYNOLDS, E.S. (1977)
Modification of 1,1-dichloroethylene hepatotoxicity by
hypothyroidism. Toxicol. appl. Pharmacol., 42: 367-376.
217 TABAK, H.H., QUAVE, S.A., ·MASHNI, C.I., & BARTH, E.F. (1981)
Biodegradability studies with organic priority pollutant
compounds. J. Water Pollut. Control Fed., 53: 1503-1518.
218 TAN, S. & OKADA, T. (1979) Determination of residual
vinylidene chloride monomer in polyvinylidene chloride.
Hygienic studies on plastic containers and packages. III. J.
Food Hyg. Soc. Jpn, 20: 223-227.
219 THIESS, A.M., FRENTZEL-BEYME, R., & PENNING, E. (1979)
Mortality study of vinylidene chloride exposed persons. In:
Heim, C. & Kilian, D.J.., ed. Proceedings of the 5th Medichem
Congress, San Francisco, September 1977, pp. 270-278.
220 THOMPSON, J.A, HO, B., & MASTOVICH, S.L. (1984) Reductive
metabolism of 1,1,1,2-tetrachloroethane and related chloro-
ethanes by rat liver microsomes. Chem-biol. Interact., 51:
321-333.
221 TIERNEY, D.R., BLACKWOOD, T.R., & PIANA, M.R. (1979) Status
assessment of toxic chemicals: vinylidene chloride,
Cincinnati, Ohio, US Environmental Protection Agency (EPA
600/2-79-2100; PB 80- 146442).
222 TORKELSON, T.R. & ROWE, V.K. ed. (1982) Vinylidene chloride.
In: Clayton, G.D. & Clayton, F.E., Patty's industrial hygiene
and toxicology, 3rd ed., New York, John Wiley and Sons, pp.
3545-3550.
223 US EPA (1984a) Method 601. Guidelines establishing test
procedures for the analysis of pollutants under the Clean
Water Act (40 CFR 136). Purgeable halocarbons. Fed. Reg., 49:
43261-43271.
224 US EPA (1984b) Method 1624, Revision B. Guidelines
establishing test procedures for the analysis of pollutants
under the Clean Water Act (40 CFR 136). Volatile organic
compounds by isotope dilution GC/MS. Fed. Reg., 49:
43407-43415.
225 US EPA (1985) Health assessment document for vinylidene
chloride, Washington, DC, US Environmental Protection Agency,
Office of Health and Environmental Assessment (EPA 600/8-83-
031F).
226 US NIOSH (1987) Manual of analytical methods, 3rd ed.,
Cincinnati, Ohio, National Institute of Health and Human
Services, pp.1-3 (Method 1015).
227 VAN DUUREN, B.L., GOLDSCHMIDT, B.M., LOEWENGART, G., SMITH,
A.C., MELCHIONNE, S., SEIDMAN, I., & ROTH, D. (1979)
Carcinogenicity of halogenated olefinic and aliphatic
hydrocarbons in mice. J. Natl Cancer Inst., 63:
1433-1439.
228 VAN'T HOF, J. & SCHAIRER, L.A. (1982) Tradescantia assay
system for gaseous mutagens. A report of the US Environmental
Protection Agency Gene-Tox Program. Mutat. Res., 99:
303-315.
229 VIOLA, P.L. & CAPUTO, A. (1977) Carcinogenicity studies on
vinylidene chloride. Environ. Health Perspect., 21: 45-47.
230 VOGEL, T.M. & MCCARTY, P.L. (1987) Abiotic and biotic
transformations of 1,1,1-trichloroethane under methanogenic
conditions. Environ. Sci. Technol., 21: 1208-1213.
231 WAKEHAM, S.G., GOODWIN, J.T., & DAVIS, A.C. (1983)
Distributions and fate of volatile organic compounds in
Narragansett Bay, Rhode Island. Can. J. Fish. Aquat. Sci., 40
(Suppl.2): 304-321.
232 WALKER, W.W., MANNING, C.S., OVERSTREET, R.M., & HAWKINS, W.E.
(1985) Development of aquarium fish models for environmental
carcinogenesis: an intermittent-flow exposure system for
volatile, hydrophobic chemicals. J. appl. Toxicol., 5:
255-260.
233 WALLACE, L., ZWEIDINGER, R., ERICKSON, M., COOPER, S.,
WHITAKER, D., & PELLIZZARI, E. (1982) Monitoring individual
exposure. Measurements of volatile organic compounds in
breathing-zone air, drinking water and exhaled breath.
Environ. Int., 8: 269-282.
234 WALLACE, L., PELLIZZARI, E., HARTWELL, T., ROSENZWEIG, M.,
ERICKSON, M., SPARACINO, C., & ZELON, H. (1984) Personal
exposure to volatile organic compounds. I. Direct measurements
in breathing-zone air, drinking water, food, and exhaled
breath. Environ. Res., 35: 293- 319.
235 WALLACE, L.A, PELLIZZARI, E., SHELDON, S., HARTWELL, T.,
SPARACINO, C., & ZELON, H. (1986) The total exposure
assessment methodology (TEAM) study: direct measurements of
personal exposures through air and water for 600 residents of
several US cities. In: Cohen, Y., ed. Pollutants in a
multimedia environment, New York, London, Plenum Publishing
Corporation. pp. 289-315.
236 WANG, T. & LENAHAN, R. (1984) Determination of volatile
halocarbons in water by purge-closed loop gas chromatography.
Bull. environ. Contam. Toxicol., 32: 429-438.
237 WANG, T., LENAHAN, R., & KANIK, M. (1985) Impact of
trichloroethylene contaminated groundwater discharged to the
main canal and Indian river lagoon, Vero Beach, Florida. Bull.
environ. Contam. Toxicol., 34: 578-586.
238 WARNER, C., MODDERMAN, J., FAZIO, T., BEROZA, M., SCHWARTZMAN,
G., FOMINAYA, K., & SHERMA, J. (1983) Food additives
analytical manual, Arlington, Virginia, Association of
Official Analytical Chemists, Vol. 1, pp. 348-357.
239 WASKELL, L. (1978) Study of the mutagenicity of anesthetics
and their metabolites. Mutat. Res., 57: 141-153.
240 WAXWEILER, R.J., SMITH, A.H., FALK, H., & TYROLER, H.A. (1981)
Excess lung cancer risk in a synthetic chemicals plant.
Environ. Health Perspect., 41: 159-165.
241 WEAST, R.C., ed. (1984) CRC Handbook of chemistry and physics,
65th ed., Boca Raton, Florida, CRC Press, p. C-295.
242 WEGMAN, R.C.C., BANK, C.A., & GREVE, P.A. (1981) Environmental
pollution by a chemical waste dump. Stud. environ. Sci., 17:
349-357.
243 WESSLING, R.A. & EDWARDS, F.G. (1971) Vinylidene chloride
polymers. In: Bikales, N.M., ed. Encyclopedia of polymer
science and technology, New York, Wiley Interscience, Vol. 14,
pp. 540-579.
244 WHO (1984) Guidelines for drinking-water quality, Vol. 1 and
2, Geneva, World Health Organization.
245 WOLFF, T., DISTLERATH, L.M., WORTHINGTON, M.T., GROOPMAN,
J.D., HAMMONS, G.J., KADLUBAR, F.F., PROUGH, R.A., MARTIN,
M.V., & GUENGERICH, F.P. (1985) Substrate specificity of human
liver cytochrome P-450 debrisoquine 4-hydroxylase probed using
immunochemical inhibition and chemical modeling. Cancer Res.,
45: 2116-2122.
246 YOUNG, D.R., GOSSETT, R.W., BAIRD, R.B., BROWN, D.A., TAYLOR,
P.A., & MIILLE, M.J. (1981) Wastewater inputs and marine
bioaccumulation of priority pollutant organics off Southern
California; In: Jolley, R.L., Brungs, W.A., Cotrivo, J.A.
Cumming, R.B., Mattice, J.S., & Jacobs, V.A., ed. Proceedings
of the Fourth Conference on Water Chlorination (Environmental
Impact and Health Effects), Pacific Grove, California, 18-23
October, 1981, Ann Arbor, Michigan, Ann Arbor Science
Publishers, Chapter 60, pp. 871-884.
247 ZELLER, H. & PEH, J. (1975) [ Report on the tests of vinylidene
chloride for mutagenic effects in Chinese Hamsters after
single oral application (chromosomal study)], Ludwigshafen,
BASF Aktiengesellschaft, 12 pp (in German).
248 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979a) [ Report
on the determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in vapour form in Sprague-Dawley rats
(fasting) during a 4-hour exposure period], Ludwigshafen, BASF
Aktiengesellschaft, 14 pp (in German).
249 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979b) [ Report
on the determination of acute toxicity (LC50) of vinylidene
chloride in Sprague-Dawley rats (fed) during a 4-hour exposure]
Ludwigshafen, BASF Aktiengesellschaft, 14 pp. (in German).
250 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979c) [ Report
on the determination of acute toxicity (LC50) by inhalation
of vinylidene chloride in NMRI mice (fasting) during a 4-hour
exposure] Ludwigshafen, BASF Aktiengesellschaft, 12 pp. (in
German).
251 ZELLER, H., KLIMISCH, J.H., & FREISBERG, K.O. (1979d) [ Report
on the determination of acute toxicity (LC50) by inhalation of
vinylidene chloride in NMRI mice (fed) during a 4-hour
exposure] Ludwigshafen, BASF Aktiengesellschaft, 12 pp. (in
German).
RESUME ET CONCLUSIONS, EVALUATION ET RECOMMANDATIONS
1. Résumé et conclusions
1.1 Propriétés, usages et méthodes d'analyse
Le chlorure de vinylidène (C2H2Cl2) est un liquide volatil et
incolore d'odeur douceâtre. On le stabilise au moyen de
p- méthoxyphénol afin d'éviter la formation de peroxydes explosifs.
Le chlorure de vinylidène est utilisé pour la production de
trichloro-1,1,1-éthane, de fibres et de copolymères modacryliques
(avec du chlorure de vinyle ou de l'acrylonitrile). On a mis au
point des méthodes de chromatographie en phase gazeuse pour la
recherche et le dosage du chlorure de vinylidène dans l'air, l'eau
ou les emballages, les tissus de l'organisme, les denrées
alimentaires et le sol. Le détecteur le plus sensible est le
détecteur à capture d'électrons.
1.2 Sources et niveaux d'exposition
Chaque année on libère dans l'atmosphère une quantité de
chlorure de vinylidène qui correspond à une proportion allant
jusqu'à 5 % de la production totale (soit environ 23 000 tonnes au
maximum). La forte tension de vapeur et la faible solubilité dans
l'eau de ce produit font qu'il est relativement abondant dans
l'atmosphère par rapport aux autres compartiments du milieu. On
pense que le chlorure de vinylidène présent dans l'atmosphère a une
demi-vie d'environ deux jours.
Dans l'eau, les concentrations sont très faibles. Même dans les
eaux résiduaires industrielles, les concentrations sont de l'ordre
du µg/litre, c'est-à-dire bien inférieures aux concentrations
toxiques pour la faune aquatique, concentrations qui sont de
l'ordre du mg/litre. Dans l'eau de boisson non traitée, les
concentrations ne sont généralement pas décelables. Dans l'eau
potable traitée, la teneur en chlorure de vinylidène est
généralement inférieure à 1 µg/litre encore qu'on ait trouvé des
échantillons qui en contenaient 20 < g/litre. Dans les denrées
alimentaires, les concentrations ne sont généralement pas
décelables, le maximum observé étant de 10 µg/kg.
L'exposition professionnelle au chlorure de vinylidène peut se
produire soit par inhalation, soit par contamination de la peau ou
des yeux. Selon les pays, la dose maximale recommandée ou
l'exposition moyenne pondérée en fonction du temps (TWA) se situent
dans les limites de 8 à 500 mg/m3; quelquefois, la dose maximale
correspond à la concentration la plus faible qui soit décelable de
façon certaine. Les limites d'exposition à court terme vont de 16
à 80 mg/m3 et les valeurs plafond de 500 à 700 mg/m3.
1.3 Absorption, distribution, métabolisme et excrétion
Le chlorure de vinylidène peut être absorbé facilement par les
voies respiratoires ou digestives chez les mammifères; en revanche
on de dispose pas de renseignements sur l'absorption percutanée.
Administré à des rongeurs, le chlorure de vinylidène se répartit
largement dans l'organisme de l'animal, les concentrations étant
maximales dans le foie et les reins. L'élimination par la voie
pulmonaire du chlorure de vinylidène inchangé s'effectue selon un
processus au moins biphasé qui dépend de la dose; elle est plus
importante aux doses qui provoquent une saturation du métabolisme
(c'est-à-dire 600 mg/m3 environ (150 ppm) chez le rat). Des rats à
qui l'on avait administré une dose de chlorure de vinylidène par
voie orale et que l'on avait ensuite fait jeûner ont exhalé
davantage de cette substance.
Les principales voies du métabolisme ont été identifiées chez
le rat. Dans la voie prédominante (phase I), intervient le
cytochrome P-450 et il y a formation (vraisemblablement, mais pas
forcément par l'intermédiaire d'un époxyde), d'acide
monochloracétique. Le chlorure de vinylidène peut stimuler
l'activité du cytochrome P-450. Un certain nombre de métabolites de
la Phase I peuvent se conjuguer au glutathion ou à la phosphatidyl-
éthanolamine avant de subir d'autres transformations. Le
métabolisme est plus rapide chez la souris que chez le rat, avec un
profil analogue où les dérivés conjugués au glutathion sont
relativement plus abondants. On a montré que le chlorure de
vinylidène était également métabolisé par le cytochrome P-450 des
microsomes humains.
Chez les rongeurs, le métabolisme du chlorure de vinylidène
conduit à la déplétion du glutathion et à l'inhibition de la
glutathion- S- transférase.
1.4 Effets sur les animaux d'expérience et les systèmes
cellulaires
1.4.1 Fixation aux tissus par liaison covalente
Le chlorure de vinylidène radio-marqué se fixe aux tissus
hépatiques, rénaux et pulmonaires des rongeurs par liaison
covalente et c'est ce phénomène qui déclenche le processus toxique.
Les liaisons par covalence et par conséquent la toxicité sont
accrues par la déplétion en glutathion et se produisent au niveau
du foie et du rein à plus faible dose chez la souris que chez le
rat. Un certain nombre de métabolites du chlorure de vinylidène se
fixent par liaison covalente aux thiols in vitro .
1.4.2 Toxicité aigue
Les estimations de la CL50 aiguë du chlorure de vinylidène
varient considérablement, mais cette variation ne masque pas le
fait que les souris sont beaucoup plus sensibles à cette substance
que les rats ou les hamsters. Les valeurs estimatives de la CL50
orale à 4-h varient d'environ 8000 à 128 000 mg/m3 (2000-32 000
ppm) chez le rat, de 450 à 820 mg/m3 (115-205 ppm) chez la souris et
de 6640-11 780 mg/m3 (1660-2945 ppm) chez le hamster.
Du fait que la relation entre la concentration et la mortalité
n'est pas linéaire, les estimations de la CL50 peuvent être
entachées d'erreurs. Chez toutes les espèces, la CL50 a tendance à
être plus faible pour les mâles que pour les femelles et le jeûne
(qui provoque une déplétion en glutathion) accroît la toxicité dans
tous les cas. Après administration par voie orale les valeurs de
la DL50 s'établissaient approximativement à 1500 et 100 mh/kilo
respectivement chez les rats et les souris. Aprés inhalation, la
toxicité s'est manifestée par une irritation des muqueuses, une
dépression du système nerveux central et une cardiotoxicité
progressive (bradycardie sinusale et arrythmies). On a noté des
lésions au niveau du foie, des reins et des poumons. Chez les
souris, qui sont plus sensibles que les rats à l'hépatotoxicité et
à la néphrotoxicité du chlorure de vinylidène, on a constaté
qu'une exposition à des concentrations ne dépassant pas 40 mg/m3
(10 ppm) pendant 6 heures accroissait les lésions rénales et la
réplication de l'ADN. Comme dans le cas de l'inhalation, les
principaux organes affectés par l'administration de chlorure de
vinylidène par voie orale, sont le foie, les reins et les poumons.
Le processus toxique au niveau du foie commence par des altérations
au niveau des canaux biliaires et se poursuit par l'apparition de
signes d'atteinte mitochondriale. Après quoi il y a lésion du
réticulum endoplasmique et mort de la cellule. Il ne semble pas que
la toxicité du chlorure de vinylidène pour le foie et le rein soit
due à la peroxydation des lipides. Il semblerait plutôt que
l'augmentation de la concentration intra-cellulaire des ions
calcium soit à l'origine de la toxicité de ce produit pour les
hépatocytes.
Les effets toxiques du chlorure de vinylidène dépendent, au
moins partiellement, de l'activité du cytochrome P-450 (qui peut
également intervenir dans le détoxication) et peuvent être
exacerbés par une déplétion en glutathion. L'éthanol et la
thyroxine peuvent accroître l'hépatotoxicité; en revanche celle-ci
est inhibée par le dithiorcarbe et la (+)-catéchine et modulée par
l'acétone.
1.4.3 Etudes à court terme
Des lésions hépatiques et rénales accompagnées, dans une
moindre proportion, de lésions pulmonaires ont été observées chez
des rongeurs exposés par inhalation à du chlorure de vinylidène à
raison de 40 à 800 mg/m3, 4 à 8 heures par jour, pendant 4 jours ou
plus par semaine. Les souris se sont révélées plus sensibles que
les rats, les cobayes, les lapins, les chiens et les saïmiris alors
que chez les souris, la toxicité variait selon la souche utilisée.
En général, les souris femelles étaient moins sensibles que mâles.
On a observé une hépatotoxicité chez des rats et des souris exposés
de façon intermittente à des concentrations de chlorure de
vinylidène respectivement >800 mg/m3 (>200 ppm) ou égales à 220
mg/m3 (55 ppm). Pour induire une hépatotoxicité par exposition
continue durant plusieurs jours, il fallait 240 mg/m3 (60 ppm) pour
les rats et 60 mg/m3 (15 ppm) pour les souris. Ce traitement
intermittant ou continu a également provoqué une néphrotoxicité
chez les souris. Les souris mâles de la race Swiss se sont montrées
particulièrement sensibles à la néphrotoxocité induite par le
chlorure de vinylidène. Les mâles n'ont pas survécu à une
exposition continue de courte durée à 200 mg de chlorure de
vinylidène par m3 (50 ppm). Chez les chiens, les saïmiris et les
rats, le seuil d'hépatotoxicité se situait à environ 80 mg/m3 (20
ppm) administrés de façon continue sur 90 jours. Des études à court
terme (d'une durée d'environ 3 mois) au cours desquelles du
chlorure de vinylidène a été administré par voie orale à des rats à
des doses allant jusqu'à 20 mg/kg par jour et á des chiens à des
doses allant jusqu'à 25 mg/kg, n'ont pas révélé de signes de
toxicité, si ce n'est quelques lésions hépatiques infimes et
réversibles chez les rats.
1.4.4 Etudes à long terme
Des études à long terme comportant l'inhalation
intermittente de chlorure de vinylidène ont révélé que la dose de
300 mg/m3 (75 ppm) ne produisait que des modifications bénignes et
réversibles au niveau du foie chez les rats. A 600 mg/m3
(150 ppm) c'est-à-dire la dose la plus forte qui soit supportable
au cours d'une exposition à long terme, on constatait de nettes
lésions hépatiques avec nécrose. On a observé une forte mortalité
avec des signes de lésion hépatique chez des souris soumises à une
dose de 200 mg/m3 (50 ppm). La néphrotoxicité était évidente
chez la souris après un traitement à long terme à la dose de 100
mg/m3 (25 ppm). L'administration de chlorure de vinylidène par voie
orale à des rats pendant un an à des doses quotidiennes allant
jusqu'à 300 mg/kg n'a produit que de minimes anomalies hépatiques.
Il n'est pas possible de tirer de ces données une valeur précise de
la dose sans effet observable. Une autre étude a révélé des signes
d'inflammation rénale et de nécrose hépatique chez des rats et des
souris soumis à une administration orale prolongée de chlorure de
vinylidène à des doses quotidiennes respectives de 5 mg/kg et 2
mg/kg.
1.4.5 Génotoxicité et cancérogénicité
On a constaté que le chlorure de vinylidène était mutagène pour
les bactéries et les levures, mais seulement en présence d'un
système mammalien d'activation métabolique microsomique (S9). Le
composé a provoqué une synthèse anarchique de l'ADN dans des
hépatocytes isolés de rats ainsi qu'une augmentation de la
fréquence des échanges entre chromatides soeurs et des aberrations
chromosomiques dans des cultures cellulaires additionnées de S9.
Par contre, on n'a pas observé d'accroissement des mutations
géniques chez les mammifères. Il a été fait état d'une augmentation
légère mais statistiquement significative de la liaison à l'ADN
après exposition in vivo . Cette liaison était plus importante dans
les cellules de souris que dans celles de rats et également plus
importante dans les reins que dans le foie après une exposition de
6 heures à des concentrations de 40 et 200 mg de chlorure de
vinylidène/m3 (10 et 50 ppm). En outre, le chlorure de vinylidène
augmentait légèrement la synthèse anarchique de l'ADN dans le rein
de la souris. On a relevé aucun signe de mutation létale dominante
ou d'effets cytogénétiques après exposition in vivo de rongeurs,
sauf dans le cas d'une étude au cours de laquelle on a observé des
aberrations chromosomiques dans la moëlle osseuse de hamsters
chinois.
Des études de cancérogénicité ont été effectuées sur trois
espèces animales (rats, souris et hamsters). Chez des souris mâles
de race Swiss, on a relevé un net effet cancérogène (adénocarcinome
rénal) après exposition prolongée intermittente à des
concentrations de 100 ou de 200 mg/m3 de chlorure de vinylidène (25
ou 50 ppm), mais pas aux concentrations de 0 ou 40 mg/m3 (0 ou 10
ppm).
Il est possible que ces tumeurs rénales soient liées d'une
manière ou d'une autre à la néphrotoxicité observée et que des
atteintes rénales répétées puissent conduire directement à une
réaction cancérogène selon un mécanisme non génotoxique ou qu'elles
facilitent l'expression de l'activité génotoxique de certains
métabolites chez cette espèce, pour ce sexe et au niveau de cet
organe. Toutefois, cette conclusion reste hypothétique du fait que
les données disponibles sur les effets génétiques in vivo sont
limitées et que le chlorure de vinylidène a pu jouer le rôle
d'initiateur.
Dans la même étude, on a constaté une augmentation statistique
de l'incidence des tumeurs pulmonaires (principalement des adénomes
chez les souris des deux sexes) et des cancers mammaires (chez les
femelles) mais on a pas observé de relation entre la dose et la
réponse. Chez des rats adultes exposés par inhalation au chlorure
de vinylidène, on a signalé une légère augmentation des tumeurs
mammaires sans relation avec la dose ainsi qu'une augmentation
modérée des leucémies lorsque les rats étaient exposés à la
substance in utero puis après leur naissance. Il n'a pas été
possible d'évaluer les résultats.
1.4.6 Toxicité pour la fonction de reproduction
Aucun effet n'a été observé sur la fécondité de rats exposés en
permanence à du chlorure de vinylidène (jusqu'à 200 mg/litre ou 200
ppm) ajouté à leur eau de boisson. Des rats et des souris qui
avaient inhalé jusqu'à 1200 mg de chlorure de vinylidène par m3
(300 ppm) pendant 22 à 23 heures, à différents stades de
l'organogenèse, n'ont pas produit de foetus présentant des
anomalies autres que celles qui peuvent être attribuées à une
action toxique sur la mère.
Des rats et des lapins ont inhalé 7 h/jour 640 mg de chlorure
de vinylidène par m3 (160 ppm) ou absorbé par voie orale environ 40
mg/kilo de cette substance au cours des stades critiques de la
gestation sans que les embryons ou les foetus présentent
d'anomalies à ces doses, inférieures aux doses toxiques pour la
mère. Toutefois, des anomalies ont été constatées sur les embryons
et les foetus aux doses toxiques pour la mère, comme l'a montré la
réduction du gain de poids.
1.5 Effets sur l'homme
Des concentrations de chlorure de vinylidène de l'ordre de 16
000 mg/m3 (4000 ppm) provoquent une intoxication pouvant entraîner
une perte de connaissance. Additionné de stabilisateur, le chlorure
de vinylidène est également irritant pour les voies respiratoires,
les yeux et la peau. A la suite d'expositions prolongées ou
répétées à des doses infra-anesthésiques, on a signalé l'apparition
de lésions rénales et hépatiques. Il est difficile d'évaluer les
résultats obtenus par les études épidémiologiques en raison de
l'effectif limité des cohortes, d'une exposition simultanée au
chlorure de vinyle et du fait qu'on n'a pas suffisamment tenu
compte du tabagisme. On n'a pas constaté d'augmentation
statistiquement significative dans l'incidence des cancers chez les
personnes exposées au chlorure de vinylidène, mais il est vrai que
les études épidémiologiques présentaient des insuffisances; aussi
n'est-il pas possible d'en conclure qu'il n'existe aucun risque de
cancérogénicité. On ne dispose d'aucun renseignement concernant les
effets du chlorure de vinylidène sur la fonction de reproduction
humaine.
2. Evaluation des effets sur l'environnement et des risques pour la
santé humaine
2.1 Evaluation des effets sur l'environnement
Par suite de la volatilité du chlorure de vinylidène, c'est
l'atmosphère qui est le compartiment du milieu où il est le plus
abondant. La demi-vie du chlorure de vinylidène dans la troposphère
est vraisemblablement d'environ deux jours, aussi ce composé ne
contribue-t-il probablement pas à la réduction de la couche d'ozone
stratosphérique. Lessivage et volatilisation font du sol et des
sédiments des compartiments où le chlorure de vinylidène n'est
présent qu'en petites quantités et cet hydrocarbure chloré
n'apparaît qu'en quantité minime dans le milieu aquatique du fait
de sa volatilisation rapide. On ignore si la dégradation de
composés tel que le trichloréthylène et le perchloréthylène,
souvent présents dans l'eau, contribuent de manière notable à la
concentration du chlorure de vinylidène dans l'environnement.
La concentration du chlorure de vinylidène dans l'environnement
et les valeurs de la toxicité aiguë pour les poissons et la daphnie
montrent que les risques d'intoxication aiguë sont minimes pour la
faune aquatique. On ne dispose pas de données suffisantes sur la
toxicité à long terme pour évaluer les effets sublétaux sur les
organismes aquatiques qui vivent à proximité de sources
relativement importantes de contamination par le chlorure de
vinylidène, qu'il s'agisse d'eaux souterraines contaminées ou
d'eaux résiduaires municipales ou industrielles.
2.2 Evaluation des risques pour la santé humaine
2.2.1 Niveau d'exposition
La population générale n'est exposée qu'à de très faibles
teneurs de chlorure de vinylidène. La concentration maximale qui
ait été signalée dans l'eau de boisson est de 20 µg par litre,
encore que l'exposition individuelle moyenne pour les citoyens des
Etats-Unis d'Amérique par l'intermédiaire de l'eau de boisson soit
estimée à moins de 0,01 µg par jour. Il n'y a pas de chlorure de
vinylidène en concentrations décelables dans les denrées
alimentaires et en tout état de cause on n'a pas signalé de teneurs
supérieures à 10 µg/kg. On ignore quelles sont les concentrations
dans les denrées alimentaires constituées d'organismes aquatiques
mais elles sont vraisemblablement insignifiantes (section 10.1).
Dans l'air ambiant on a signalé des concentrations en chlorure de
vinylidène allant jusqu'à 52 µg/m3 (dans le périmètre d'une zone
industrielle). Des concentrations médianes dans l'air urbain de 20
ng/m3 et de 8,7 µg/m3 ont été signalées aux Etats-Unis,
respectivement dans des zones non industrielles et dans des zones
industrielles.
L'exposition professionnelle se produit notamment lors de la
production et de la polymérisation du chlorure de vinylidène. C'est
principalement par la voie respiratoire que cette substance pénètre
dans l'organisme et les limites maximales recommandées ou
réglementées pendant une journée de travail vont de 8 à 500 mg/m3
(ou la concentration la plus faible qui soit décelable par une
méthode fiable), selon les pays. Les limites d'exposition à court
terme vont de 16 à 80 mg/m3 et les valeurs plafonds de 50 à 700
mg/m3. La concentration atmosphérique de chlorure de vinylidène en
atmosphère confinée à laquelle certains travailleurs peuvent être
exposés, ne dépasse pas 8 mg/m3.
2.2.2 Effets aigus
Chez l'homme, l'inhalation de fortes concentrations de chlorure
de vinylidène (très approximativement, supérieures ou égales au
seuil olfactif maximal de 4000 mg/m3) peuvent vraisemblablement
provoquer une dépression du système nerveux central susceptible
d'évoluer vers le coma. En se basant sur la toxicité aiguë de ce
composé chez l'animal, on pense que les effets toxiques du
chlorure de vinylidène peuvent se manifester au niveau du foie, des
reins ou des poumons bien en dessous du seuil olfactif minimum qui
se situe aux environs de 2000 mg/m3. L'exposition au chlorure de
vinylidène peut provoquer une irritation des yeux, des voies
respiratoires supérieures (à la concentration de 100 mg/m3 chez
l'homme), et de la peau, encore que cet effet irritant soit,
semble-t-il, dû en partie au para-méthoxyphénol utilisé comme
stabilisateur.
Chez la souris, qui est plus sensible que le rat aux effets
hépatotoxiques et néphrotoxiques du chlorure de vinylidène, on a
constaté une atteinte rénale après exposition à des concentrations
ne dépassant pas 40 mg de chlorure de vinylidène par m3 (soit 10
ppm) pendant 6 heures. On a également observé une hépatotoxicité
et une néphrotoxicité notables chez le rat. Lorsque les animaux
sont à jeun, ce qui a pour effet d'exacerber la toxicité,
l'exposition au chlorure de vinylidène à des teneurs de 600 mg/m3
(150 ppm) et de 800 mg/m3 (200 ppm) pendant 6 heures a provoqué
chez le rat des effets toxiques, respectivement au niveau du foie
et du rein. Des études sur le rat ont montré que l'ingestion
d'alcool avant l'exposition peut stimuler le métabolisme et
exacerber la toxicité du chlorure de vinylidène. La toxicité agiuë
dépend de l'espèce, du sexe, de la souche et de l'état alimentaire
de l'animal. Chez le rat et la souris, les différences de
sensibilité interspécifiques sont liées à l'activité du métabolisme
oxydatif. S'il n'est pas possible de déterminer qui du rat ou de
la souris constitue le meilleur modèle pour l'être humain, toujours
est-il que le métabolisme des microsomes hépatiques est, chez
l'homme, quantitativement analogue à celui du rat, espèce
relativement peu sensible aux effets du chlorure de vinylidène.
Rien n'indique qu'il existe une différence de nature entre l'homme
et les rongeurs pour ce qui est du métabolisme oxidatif du chlorure
de vinylidène.
Il semblerait que la marge entre la concentration toxique chez
l'animal (40 mg/m3 pour la souris) et les limites d'exposition
professionnelle fixées par certains pays puisse être insuffisante,
voire nulle.
2.2.3 Effets à long terme et génotoxicité
Une exposition prolongée ou des expositions de courte durée
répétées à des doses infra-anesthésiques peuvent provoquer des
lésions rénales ou hépatiques. Sur la base d'études à long terme
chez l'animal, dans des conditions reproduisant une exposition
professionnelle, on a observé chez le rat, des altérations au
niveau du foie à une dose de 300 mg/m3 (75 ppm). Chez la souris,
des lésions rénales et hépatiques ont été observées respectivement
à 100 mg/m3 (25 ppm) et 200 mg/m3 (50 ppm). La sensibilité aux
effets toxiques observée au cours des différentes études présente
des variations considérables.
Le chlorure de vinylidène n'affecte pas, semble-t-il, la
capacité de reproduction chez l'animal et ne semble pas non plus
comporter de risques d'embryotoxicité ou de tératogénicité aux
doses inférieures à celles qui seraient toxiques pour la mère, mais
on n'a pas effectué d'études de ce type chez l'homme. Aux doses
toxiques pour la mère-à en juger par une réduction du gain de
poids-on a observé des effets toxiques sur l'embryon et le foetus
et des anomalies foetales.
Le chlorure de vinylidène est mutagène pour les bactéries et
les levures en présence d'un système métabolique de mammifère.
Certaines cellules mammaliennes se révèlent également sensibles in
vitro aux effets mutagènes et aux lésions de l'ADN. La plupart des
études in vivo effectuées sur des rats n'ont pas permis d'observer
d'effets génotoxiques manifestes, à en juger par les tests de
létalité dominante et certains critères cytogénétiques mais on a
tout de même signalé la présence d'aberrations chromosomiques dans
les cellules de la moelle osseuse de hamsters chinois. La liaison
à l'ADN et sa réparation sont décelables in vivo chez les rongeurs
mais en proportion minime. Les études génétiques in vivo incitent
donc à penser qu'il existe une certaine toxicité génique mais, dans
la majorité des cas, il s'agit d'effets non décelables ou minimes.
Plusieurs épreuves de cancérogénicité ont été effectuées sur
trois espèces d'animaux d'expérience (souris, rats et hamsters)
selon différentes voies d'administration. Malheureusement, la
plupart de ces études laissent beaucoup à désirer tant au niveau de
leur conception que de l'évaluation du risque cancérogène. Aucun
effet cancérogène significatif n'a été observé chez des rats qui
recevaient ce composé par la voie orale. Chez des rats adultes
exposés par inhalation, on a signalé une augmentation de la
fréquence des tumeurs mammaires qui n'était toutefois pas liée à la
dose. Une légère augmentation de la fréquence des leucémies a été
également observée, lorsque les rats étaient exposés in utero ou
après leur naissance. Ces observations n'ont pas pu être évaluées.
Lors d'une étude sur la souris, on a observé chez les mâles une
augmentation de l'incidence des adénocarcinome du rein aux doses
de 200 et 100 mg/m3 (50 et 25 ppm), mais aucun effet de ce genre
aux doses de 40 et 0 mg/m3 (10 et 0 ppm). Au cours de la même
étude, une augmentation statistiquement significative de
l'incidence des tumeurs pulmonaires (essentiellement des adénomes
chez les deux sexes) et des carcinomes mammaires (chez les
femelles) a été observée, sans toutefois qu'il y ait de relation
dose-réponse.
Il est possible que ces tumeurs rénales soient liées d'une
manière ou d'une autre à la néphrotoxicité observée et que des
atteintes rénales répétées puissent conduire directement à une
réaction cancérogène selon un mécanisme non-génotoxique ou qu'elle
facilite l'expression de l'activité génotoxique de certains
métabolites chez cette espèce, pour ce sexe et au niveau de cet
organe. Toutefois, cette conclusion reste hypothétique du fait que
l'on ne possède pas suffisamment de résultats sur la relation dose-
réponse pour ce qui est des effets génétiques in vivo; en outre, le
chlorure de vinylidène a pu jouer le rôle d'initiateur lors d'un
test cutané en deux phases sur la souris.
Les études épidémiologiques ne fournissent pas de résultats
statistiquement significatifs qui puissent permettre de conclure à
un accroissement du risque de cancer à la suite d'une exposition
professionnelle au chlorure de vinylidène, toutefois ces études
présentent des insuffisances telles qu'on ne peut procéder à une
évaluation convenable du risque de cancérogénicité pour l'homme.
Même si les estimations effectuées par divers auteurs écartent
l'idée d'une surmortalité par cancer en arguant qu'il s'agit d'une
pure coincidence(en raison du petit nombre de cas et du faible
effectif des cohortes), il n'est pas inutile de préciser que les
résultats obtenus sont systématiquement supérieurs aux prévisions.
Ainsi, dans les deux études de cohorte dont il est fait état, on a
observé un cancer du poumon dans 7 cas, alors qu'on aurait dû avoir
3,16 décès. On ne peut pas écarter ce résultat, mais il ne faut pas
oublier l'existence, dans une étude, d'une exposition concomitante
au chlorure de vinylidène. Etant donné que les cohortes ont été
constituées sur la base d'une exposition au chlorure de vinylidène,
on peut se trouver dans l'impossibilité d'éliminer d'autres
expositions parasites.
Les données de morbidité indiquées (y compris un cas de cancer
du testicule) ne sont pas dénuées d'intérêt. Selon les auteurs, la
forte morbidité hépatique serait imputable à la consommation
d'alcool. Cette hypothèse ne tient pas, puisque la consommation
d'alcool de l'ensemble des personnes étudiées (pas seulement les
cas identifiés) n'a pas été évaluée.
3. Recommandations
3.1 Recommandations en vue de travaux futurs
Il faudrait disposer d'une meilleure estimation de la production
annuelle mondiale de chlorure de vinylidène ainsi que des quantités
de cette substance qui pénètrent dans l'environnement à partir de
l'ensemble des sources de pollution, que le composé soit libéré tel
quel ou qu'il résulte de la décomposition d'autres produits
chimiques.
Les prévisions relatives à sa destiné dans l'environnement
reposent sur un nombre limité de données expérimentales. Il
faudrait de nouvelles données sur les produits de dégradation et de
transformation de ce composé dans l'air, le sol, l'eau et les
sédiments ainsi que sur son métabolisme chez des espèces non-
mammaliennes représentatives.
Il conviendrait d'effectuer des études de toxicité à long terme
chez diverses espèces aquatiques représentatives (poissons,
crustacés et mollusques), selon divers critères pathologiques.
Il faut également définir de manière plus précise, afin
d'établir des critères de sécurité en matière d'exposition, quels
sont, chez l'animal et chez l'homme, les mécanismes des effets
toxiques résultant d'une exposition de brève ou prolongée au
chlorure de vinylidène.
Il faudrait exploiter de manière plus complète les données
existantes sur la cancérogénicité. Si l'on envisage d'autres études
de cancérogénicité, elles devront être menées selon un protocole
expérimental reconnu, pendant toute l'existence des sujets, ce
protocole étant conçu de manière à tenir compte des propriétés
particulières du chlorure de vinylidène. Ces études doivent
notamment prendre en considération la courte demi-vie du produit
dans l'organisme, l'importance de l'âge au début de l'exposition,
la durée de l'exposition quotidienne et autres données susceptibles
d'aider à la détermination des doses à administrer. Les espèces et
les souches d'animaux de laboratoire devront être soigneusement
sélectionnées. Il serait également très utile de disposer de
données de toxicité ainsi que de données métaboliques et pharmaco-
cinétiques sur ces animaux.
Il faudrait effectuer des études longitudinales à long terme
sur la morbidité et la mortalité au sein de populations prises au
hasard et qui sont exposées au chlorure de vinylidène.
Des études épidémiologiques sont nécessaires pour permettre
l'évaluation des effets de l'exposition au chlorure de vinylidène
(notamment une exposition prolongée à de faibles doses) dans les
populations humaines. Il est tout particulièrement important de
disposer de données sur des effets tels que les affections
cérébrovasculaires prématurées et le cancer. En outre, les études
qui seront effectuées devront tenir dûment compte de facteurs de
confusion tels que le tabagisme et la consommation d'alcool
(éventuellement selon un système cas/témoin).
Afin d'apprécier l'effet de l'action réglementaire menée au
cours des dernières années, il conviendrait de confronter les
résultats des études en cours aux données rétrospectives.
Pour résoudre le problème que posent les faibles effectifs du
personnel sur les lieux de production, on pourrait, pour les
investigations en cours comme pour les investigations futures,
recourir à des études multicentriques avec regroupement des
données. Il faudra également étudier sur l'animal d'expérience si
la N- acétylcystéine, un agent sulfhydrilé présente un intérêt
pour le traitement des intoxications par le chlorure de vinylidène.
Il est nécessaire de comparer la pharmacocinétique et le
métabolisme du chlorure de vinylidène tant in vivo qu' in vitro
spécialement au niveau du rein, du foie et des poumons chez
diverses espèces d'animaux d'expérience et chez l'homme, afin de
pouvoir mieux interpréter les résultats fournis par les études de
toxicité in vivo. Il faut également obtenir des données sur la
génotoxicité potentielle du chlorure de vinylidène au site de la
cancérogénèse, parallèlement sur plusieurs espèces, afin de voir si
un mécanisme génétique est en cause.
Compte tenu des observations neurotoxicologiques dont il est
fait état dans la présente analyse, il paraît nécessaire d'étudier
le rôle des systèmes de modulation dans la pathogénèse de
l'intoxication par le chlorure de vinylidène.
3.2 Protection personnelle et traitement des intoxications
3.2.1 Protection personnelle
Dans l'industrie, où peuvent se produire des expositions de
brèves durées par inhalation au dessus des limites recommandées, il
conviendrait d'utiliser des masques faciaux avec cartouche
filtrante pour se protéger des vapeurs organiques et, si nécessaire
en cas d'urgence, des masques respiratoires avec arrivée d'air. Les
personnes qui manipulent du chlorure de vinylidène devront porter
des vêtements protecteurs ainsi que des lunettes spéciales; cet
équipement devra être correctement entretenu afin de protéger le
corps contre tout contact. Dans les ateliers, on assurera une
ventilation permanente et l'on disposera des grilles d'aération
munies de filtres là où des déversements accidentels ou des fuites
risquent de se produire. Il est recommandé de surveiller les
émissions de chlorure de vinylidène au cours des opérations de
remplissage. En cas de fuite, on procédera à l'évaporation directe
du produit s'il s'agit d'une fuite mineure ou à son évaporation
controlée en présence d'une mousse synthétique s'il s'agit d'une
fuite plus importante. On pourra disperser la vapeur de chlorure de
vinylidène à partir de cette mousse par pulvérisation d'eau.
3.2.2 Traitement des intoxications humaines
En cas d'exposition excessive ou d'ingestion, il faut
s'adresser à un médecin. Il faut veiller particulièrement aux
poumons, à la peau et aux yeux du fait des propriétés irritantes du
chlorure de vinylidène. Il importe de surveiller les fonctions
cardiaque, hépatique et rénale ainsi que le système nerveux
central. Les données obtenues sur l'animal d'expérience montrent
que le chlorure de vinylidène accroît notablement la sensibilité
aux arythmies cardiaques induites par l'adrénaline, de sorte que ce
produit est à éviter. En cas d'hypotension grave, on pourra
procéder à une transfusion de sang total ou d'un succédané du
plasma. Il n'existe aucun antidote.
En cas d'intoxication par inhalation de chlorure de vinylidène,
il faut maintenir le malade à l'air libre en semi-décubitus
ventral. On dégagera les voies aériennes et l'on placera le malade
sous oxygène en cas de stupeur ou de coma. Si nécessaire, on
procédera à la respiration artificielle.
En cas d'ingestion de chlorure de vinylidène, rincer la bouche
avec de l'eau. Ne pas faire vomir le malade car il y a risque
d'aspiration du chlorure de vinylidène dans le larynx et les
poumons. Un lavage d'estomac ou l'administration par voie orale de
charbon actif ou de paraffine liquide peut contribuer à réduire la
biodisponibilité du chlorure de vinylidène si on procède à ce
traitement dans l'heure qui suit l'ingestion, l'effet bénéfique
étant encore sensible aprés 4 heures.
Si les yeux ont été atteints par du chlorure de vinylidène, les
rincer immédiatement à l'eau pendant plus de 15 minutes et
consulter un médecin.
En cas d'exposition cutanée, ôter les vêtements souillés et
laver la peau à l'eau et au savon.
RESUMEN Y CONCLUSIONES, EVALUACION Y RECOMENDACIONES
1. Resumen y conclusiones
1.1. Propiedades, usos y métodos analíticos
El cloruro de vinilideno (C2H2Cl2) es un líquido volátil e
incoloro con un olor dulzón. Se estabiliza con p- metoxifenol para
impedir la formación de peróxidos explosivos. El cloruro de
vinilideno se usa para producir 1,1,1-tricloroetano y para formar
fibras modacrílicas y copolímeros (con cloruro de vinilo o
acrilonitrilo). Se han puesto a punto métodos de cromatografía de
gases para analizar el cloruro de vinilideno en el aire, el agua,
las películas para envoltorios, los tejidos orgánicos, los
alimentos y el suelo. El método más sensible de detección es la
captura electrónica.
1.2. Fuentes y niveles de exposición
Todos los años ingresa en la atmósfera alrededor del 5% del
cloruro de vinilideno producido (que representa un máximo cercano a
23 000 toneladas). La elevada presión de vapor y la baja
solubilidad en agua favorecen concentraciones atmosféricas
relativamente elevadas en comparación con otros "compartimentos"
ambientales. Se cree que el cloruro de vinilideno tiene una
semivida en la atmósfera de aproximadamente dos días.
Los niveles ambientales en el agua son sumamente bajos. Incluso
en aguas residuales industriales sin tratar, las concentraciones
pasan raras veces del orden de los µg/litro, lo que está muy por
debajo del margen de mg/litro de toxicidad para los organismos
acuáticos. Los niveles en el agua de bebida sin tratar no suelen
ser detectables. En las aguas potables tratadas, se ha encontrado
que el nivel de cloruro de vinilideno suele ser < de 1 µg/litro,
si bien se han encontrado muestras que contienen hasta 20 µg/litro.
Los niveles de cloruro de vinilideno en los alimentos normalmente
no son detectables, siendo la concentración máxima observada de 10
µg/kg.
La exposición profesional al cloruro de vinilideno se da
principialmente por inhalación, aunque también puede producirse
contaminación por la piel o los ojos. Según los países, el límite
de exposición máximo recomendado o promedio ponderado en función
del tiempo se encuentra entre 8 y 500 mg/m3, o es la concentración
más baja detectable con cierto margen de confianza. Los límites de
exposición a corto plazo varían entre 16 y 80 mg/m3 y los valores
máximos varían entre 50 y 700 mg/m3.
1.3. Absorción, distribución, metabolismo y excreción
El cloruro de vinilideno puede absorberse rápidamente por la
vía respiratoria y oral en mamíferos; no se dispone de datos sobre
la absorción cutánea. El cloruro de vinilideno se distribuye por
todo el organismo del roedor y alcanza concentraciones máximas en
el hígado y el riñón. La eliminación pulmonar de cloruro de
vinilideno sin modificar es cuando menos bifásica y dependiente de
la dosis, siendo de mayor importancia en el caso de dosis que
saturan el metabolismo (>unos 600 mg/m3 (<150 ppm) por inhalación
en la rata). En la rata sometida a ayuno se observó una reducción
en el metabolismo de la dosis oral y un nivel consiguiente mayor de
cloruro de vinilideno exhalado.
Se han caracterizado las principales vías metabólicas en la
rata. El metabolismo predominante de la fase I entraña la
participación del citocromo P-450 y la formación (posible pero no
necesariamente por medio de un epóxido) de ácido monocloroacético.
El cloruro de vinilideno puede inducir actividad de citocromo P-
450. Varios metabolitos de la fase I se conjugan con glutatión y/o
con fosfatidil etanolamina antes de sufrir ulteriores
modificaciones. El metabolismo es más rápido en el ratón que en la
rata, lo que origina un perfil metabólico semejante con una
proporción relativamente mayor de derivados del glutatión por
conjugación. Se ha demostrado que el citocromo P-450 de microsomas
humanos también metaboliza el cloruro de vinilideno.
El metabolismo del cloruro de vinilideno en roedores lleva al
agotamiento de las reservas de glutatión y a la inhibición de la
actividad de la glutatión- S- transferasa.
1.4. Efectos en animales de experimentación y sistemas celulares
1.4.1 Enlaces covalentes en tejidos
Los marcadores radiactivos derivados del [14C]-cloruro de
vinilideno forman enlaces covalentes en el hígado, el riñón y el
pulmón de los roedores, lo que va asociado a toxicidad en esos
órganos. El enlace covalente y la toxicidad se agravan con el
agotamiento del glutatión y se producen en el hígado y el riñón a
dosis inferiores en ratones que en ratas. Se ha observado in vitro
que varios metabolitos del cloruro de vinilideno establecen enlaces
covalentes con tioles.
1.4.2 Toxicidad aguda
Aunque las estimaciones de la CL50 aguda para el cloruro de
vinilideno varían considerablemente, esta variación no enmascara el
hecho de que el ratón es mucho más sensible a la sustancia que la
rata o el criceto. Los valores estimados de la CL50 a las 4 h
variaron desde aproximadamente 8000 hasta 128 000 mg/m3 (2000-32
000 ppm) en ratas alimentadas, 460-820 mg/m3 (115-205 ppm) en
ratones alimentados y 6640-11 780 mg/m3 (1660-2945 ppm) en cricetos
alimentados. Pueden existir imprecisiones en los cálculos de la
CL50 debido a que la relación concentración-mortalidad no es de
carácter lineal. En todas las especies, los machos parecían tener
valores de CL50 más bajos que las hembras, y el ayuno (que agota
las reservas de glutatión) aumentó la toxicidad en las tres
especies. Tras la administración oral, los valores de la DL50
fueron aproximadamente 1500 y 200 mg/kg en ratas y ratones
alimentados, respectivamente. La toxicidad aguda por inhalación en
animales de laboratorio se manifestó en forma de irritación de las
mucosas, depresión del sistema nervioso central y cardiotoxicidad
progresiva (bradicardia sinusal y arritmias). Se observaron
lesiones en el hígado, el riñón y el pulmón. En el ratón, que es
más sensible que la rata a la toxicidad hepática y renal del
cloruro de vinilideno, la exposición a dosis tan reducidas como 40
mg de cloruro de vinilideno/m3 (10 ppm) durante 6 h indujo lesiones
renales y aumento de la replicación del ADN. Como en el caso de la
inhalación, los principales órganos afectados por la ingestión de
cloruro de vinilideno son el hígado, el riñón y el pulmón. La
cadena de procesos que llevan a la hepatotoxicidad parece comenzar
por un cambio temprano en los canalículos biliares, que se ve
seguido por síntomas de lesiones mitocondriales. A continuación se
producen lesiones en el retículo endoplasmático y la muerte
celular. La toxicidad hepática y renal inducida por el cloruro de
vinilideno no parece estar causada por peroxidación lipídica. El
aumento de las concentraciones intracelulares de Ca++ puede ser en
parte responsable de la toxicidad para el hepatocito.
Los efectos tóxicos del cloruro de vinilideno dependen, al
menos parcialmente, de la actividad del citocromo P-450 (que
también puede participar en la detoxificación) y pueden agravarse
por el agotamiento de las reservas de glutatión. La hepatotoxicidad
puede ser intensificada por el etanol y la tiroxina, inhibida por
el ditiocarbo y la (+)-catequina y modulada por la acetona.
1.4.3 Estudios a corto plazo
En estudios a corto plazo se han observado lesiones hepáticas,
renales y, en menor grado, pulmonares en roedores expuestos por
inhalación al cloruro de vinilideno a una concentración de 40-800
mg/m3 durante 4-8 h/día, 4 o más días/semana. El ratón demostró ser
más sensible que la rata, el cobayo, el conejo, el perro y el mono
ardilla, y la toxicidad fue distinta de unas familias de ratones a
otras. En general, las hembras eran menos sensibles que los machos.
Se ha comunicado la aparición de hepatotoxicidad en la rata y el
ratón expuestos intermitentemente a concentraciones de cloruro de
vinilideno >800 mg/m3 (>200 ppm) y 220 mg/m3 (55 ppm),
respectivamente. Los niveles necesarios para producir
hepatotoxicidad por exposición continua durante varios días fueron
240 mg/m3 (60 ppm) para la rata y 60 mg/m3 (15 ppm) para el ratón.
Estos tratamientos intermitentes y continuos también provocaron
nefrotoxicidad en el ratón. El ratón suizo macho resultó ser
especialmente propenso a la toxicidad renal inducida por cloruro de
vinilideno. El ratón macho no sobrevivió a una exposición continua
a corto plazo a 200 mg de cloruro de vinilideno/m3 (50 ppm). El
nivel aparente de no observación de efectos de hepatotoxicidad en
el perro, el mono ardilla y la rata fue de aproximadamente 80 mg/m3
(20 ppm) administrados en exposición continua durante 90 días. En
estudios de dosificación oral a corto plazo (aproximadamente 3
meses) en la rata (hasta 20 mg/kg diarios) y el perro (hasta 25
mg/kg diarios) no se observó prueba alguna de toxicidad aparte de
una mínima lesión hepática reversible en la rata.
1.4.4 Estudios a largo plazo
Los estudios a largo plazo de exposición intermitente al
cloruro de vinilideno por inhalación revelaron que 300 mg/m3 (75
ppm) sólo causaban ligeras lesiones hepáticas reversibles en la
rata. A 600 mg/m3 (150 ppm), la dosis más alta de exposición a
largo plazo que puede tolerar la rata, se apreció lesión hepática
con necrosis. En el ratón se observó una elevada tasa de mortalidad
con signos de lesión hepática a 200 mg/m3 (50 ppm). Se observó
toxicidad para el riñón en el tratamiento a largo plazo de ratones
con 100 mg/m3 (25 ppm). La administración de hasta 30 mg/kg al día
de cloruro de vinilideno a la rata durante un año volvió a producir
cambios hepáticos mínimos. A partir de estos datos no se puede
determinar claramente el nivel de no observación de efectos. En
otro estudio se observaron ciertas pruebas de que podía inducirse
inflamación renal y necrosis hepática en la rata y el ratón,
respectivamente, tras la administración oral a largo plazo de
cloruro de vinilideno a dosis diarias de 5 mg/kg y 2 mg/kg,
respectivamente.
1.4.5 Genotoxicidad y carcinogenicidad
Se observó que el cloruro de vinilideno es mutagénico para
bacterias y levaduras sólo en presencia de un sistema de activación
metabólica de microsomas de mamíferos (S9). El compuesto indujo
síntesis no programada de ADN en hepatocitos aislados de rata y
aumentó la frecuencia de intercambio de cromátidas hermanas y de
aberraciones cromosómicas en cultivos celulares con S9. En cambio,
no se observó aumento en la mutación de genes de mamíferos. Se ha
comunicado un aumento pequeño pero estadísticamente significativo
del enlace al ADN después de la exposición in vivo. El enlace al
ADN fue más frecuente en células de ratón que de rata y mayor en el
riñón que en el hígado tras exposiciones de 6 h a 40 y 200 mg de
cloruro de vinilideno/m3 (10 y 50 ppm). Además, el cloruro de
vinilideno aumentaba ligeramente la síntesis no programada de ADN
en el riñón de ratón. No se observó prueba alguna de efectos
letales dominantes o de efectos citogenéticos tras la exposición in
vivo de roedores a excepción de un estudio que demuestra la
inducción de aberraciones cromosómicas en la médula ósea del
criceto chino.
Se han llevado a cabo estudios de carcinogenicidad en tres
especies animales (rata, ratón y criceto). En el ratón suizo macho,
se vieron signos claros de carcinogenicidad (adenocarcinoma del
riñón) tras una exposición intermitente a largo plazo a 100 ó 200
mg de cloruro de vinilideno/m3 (25 ó 50 ppm) pero no a 0 ó 40 mg/m3
(0 ó 10 ppm).
Los tumores de riñón pueden guardar alguna relación con la
citotoxicidad renal observada y es posible que la lesión renal
repetida lleve directamente a la respuesta carcinogénica por un
mecanismo no genotóxico o bien que facilite la expresión del
potencial genotóxico de los metabolitos en esta especie, sexo y
órgano en concreto. No obstante, esta conclusión es dudosa a la luz
de los escasos datos disponibles sobre los efectos genéticos in
vivo y el descubrimiento de que el cloruro de vinilideno podía
haber actuado como iniciador.
En el mismo estudio, se observaron incidencias estadísticamente
mayores de tumor del pulmón (principalmente adenomas en el ratón de
ambos sexos) y carcinomas mamarios (en hembras), pero no se
encontraron relaciones dosis-respuesta. En rata adulta expuesta por
inhalación, se comunicó un ligero aumento independiente de la dosis
de tumores de la mama, así como un pequeño aumento de la leucemia
cuando se exponía a la rata in utero y recién nacida. Estas
observaciones no pudieron evaluarse.
1.4.6 Efectos sobre la reproducción
No se encontró efecto alguno sobre la fecundidad de la rata
continuamente expuesta al cloruro de vinilideno (hasta 200
mg/litro, 200 ppm) en el agua de bebida. La inhalación de hasta
1200 mg de cloruro de vinilideno/m3 (300 ppm), durante 22-23 horas,
por la rata y el ratón durante diversos periodos de la
organogénesis no indujo anomalías fetales que no pudieran
atribuirse a la toxicidad materna.
La inhalación de hasta 640 mg de cloruro de vinilideno/m3 (160
ppm) durante 7 h/día en ratas y conejos o la ingestión de
aproximadamente 40 mg/kg al día en la rata durante periodos
críticos de la gestación no ejercieron efecto alguno sobre los
embriones o los fetos en niveles inferiores al que produce la
toxicidad materna, pero se observaron toxicidad embrionaria y fetal
y anomalías fetales cuando se alcanzó el nivel que produce
toxicidad en la madre, como lo demostró la menor velocidad de
aumento de peso.
1.5. Efectos en el hombre
Una concentración de cloruro de vinilideno de 16 000 mg/m3
(4000 ppm) provoca una intoxicación que puede llevar a la pérdida
del conocimiento. El cloruro de vinilideno estabilizado es también
irritante para el tracto respiratorio, los ojos y la piel. Se han
comunicado lesiones renales y hepáticas correspondientes a
exposiciones subanestésicas, prolongadas o repetidas a corto plazo.
La evaluación de los estudios epidemiológicos se vio dificultada
por el pequeño tamaño de las cohortes, la coexposición a cloruro de
vinilo y la insuficiente atención al hábito de fumar. Aunque no se
encontró una incidencia mayor en grado estadísticamente
significativo del cáncer en el hombre expuesto al cloruro de
vinilideno, los estudios epidemiológicos fueron insuficientes y no
se puede concluir que no entrañe un riesgo carcinogénico. No se
dispone de información sobre los efectos del cloruro de vinilideno
en la reproducción humana.
2. Evaluación de los efectos en el medio ambiente y riesgos
para la salud humana
2.1. Evaluación de los efectos en el medio ambiente
A consecuencia de la volatilización, la atmósfera constituye el
principal compartimento ambiental del cloruro de vinilideno. Puesto
que la semivida de este compuesto en la troposfera es de unos dos
días aproximadamente, parece poco probable que el cloruro de
vinilideno contribuya a agotar la capa de ozono de la estratosfera.
La lixiviación y la volatilización hacen que el suelo y los
sedimentos sean compartimentos ambientales de menor importancia
para el cloruro de vinilideno; el nivel de este hidrocarburo
clorado en el medio acuoso se ve también reducido al mínimo por la
rápida volatilización. No se sabe si la degradación de compuestos
como el tricloroetileno y el percloroetileno, que a menudo se
encuentran en el agua, contribuye a aumentar apreciablemente los
niveles de cloruro de vinilideno en el medio ambiente.
Las concentraciones de cloruro de vinilideno que se observan en
las colecciones naturales de agua y los niveles de toxicidad aguda
para peces y Daphnia indican que los riesgos de toxicidad aguda
para el medio acuático son mínimos. No se dispone de suficientes
datos sobre toxicidad a largo plazo para evaluar los efectos
subletales sobre los organismos acuáticos que viven en las
proximidades de fuentes importantes de contaminación por cloruro de
vinilideno, como aguas subterráneas contaminadas y puntos de
vertido municipal e industrial.
2.2. Evaluación de los riesgos para la salud humana
2.2.1 Niveles de exposición
La población general está expuesta a niveles muy bajos de
cloruro de vinilideno. El nivel máximo notificado en el agua de
bebida es de 20 µg/litro, si bien se ha calculado que, en los
Estados Unidos de América, la exposición individual diaria del
ciudadano medio a través del agua de bebida es de <0,01 µg. Los
niveles de cloruro de vinilideno en los alimentos no suelen ser
detectables; no se han notificado niveles superiores a 10 µg/kg.
Los niveles en alimentos derivados de organismos acuáticos se
desconocen, pero es probable que sean insignificantes (sección
10.1). Se han comunicado niveles de cloruro de vinilideno en el
aire de hasta 52 µg/m3 (en el perímetro de una zona industrial). En
los Estados Unidos se han comunicado valores medios de
concentración en el aire urbano de 20 ng/m3 en zonas no
industriales y 8,7 µg/m3 en zonas industriales.
La exposición profesional tiene lugar especialmente en los
procesos de producción y polimerización. La respiración es la vía
principal de entrada en el organismo y los límites de exposición
máximos recomendados o promedios regulados a lo largo de un día de
trabajo varían entre 8 y 500 mg/m3 (o la concentración más baja
detectable con un margen de confianza), según el país. Los límites
de exposición a corto plazo varían entre 16 y 80 mg/m3 y los
valores máximos entre 50 y 700 mg/m3. Se ha encontrado que los
niveles de cloruro de vinilideno en las atmósferas cerradas a las
que algunos trabajadores se ven expuestos no superan los 8 mg/m3.
2.2.2 Efectos agudos
En el ser humano, es probable que la inhalación de
concentraciones elevadas de cloruro de vinilideno (muy
aproximadamente iguales o superiores al umbral olfativo máximo de
4000 mg/m3) provoque depresión del sistema nervioso central y pueda
llevar al coma. Basándose en la toxicidad aguda para animales, el
cloruro de vinilideno puede ejercer efectos tóxicos en el hígado,
el riñón o el pulmón a concentraciones muy inferiores al umbral
olfativo mínimo de aproximadamente 2000 mg/m3. La exposición al
cloruro de vinilideno puede producir irritación en los ojos, el
tracto respiratorio superior (a 100 mg/m3 en el hombre, Rylova
1953) y la piel, lo cual se ha atribuido en parte a un agente
estabilizante, el p- metoxifenol.
En el ratón, más sensible que la rata a los efectos
hapatotóxicos y nefrotóxicos del cloruro de vinilideno, se
indujeron lesiones renales por exposición a cantidades tan
reducidas como 40 mg de cloruro de vinilideno/m3 (10 ppm) durante 6
h. También se observaron hepatotoxicidad y nefrotoxicidad notables
en la rata. Tras el ayuno, que aumenta la toxicidad, la exposición
de la rata a concentraciones de cloruro de vinilideno de 600 mg/m3
(150 ppm) y 800 mg/m3 (200 ppm) durante 6 h provocó toxicidad en el
hígado y el riñón, respectivamente. Los estudios realizados en la
rata indican que la ingestión de alcohol antes de la exposición
puede acelerar el metabolismo y exacerbar la toxicidad del cloruro
de vinilideno. La toxicidad aguda depende de la especie, el sexo,
la estirpe y el régimen de alimentación de los animales. La
distinta sensibilidad del ratón y la rata guarda relación con la
diferente actividad del metabolismo oxidativo del cloruro de
vinilideno en una y otra especie. Aunque no se puede predecir si la
rata o el ratón constituyen el modelo más adecuado para el ser
humano, la actividad del metabolismo microsómico hepático del
hombre es cuantitativamente semejante al de la rata, cuya
susceptibilidad es relativamente baja. No hay pruebas de que
existan diferencias cualitativas en el metabolismo oxidativo del
cloruro de vinilideno en el ser humano y el roedor.
Está claro que el margen entre las concentraciones capaces de
producir toxicidad en animales (40 mg/m3 en el ratón) y los límites
de exposición profesional establecidos por algunos países es
insuficiente o inexistente.
2.2.3 Efectos a largo plazo y genotoxicidad
La exposición prolongada o repetida a corto plazo a dosis
subanestésicas puede producir lesiones renales y hepáticas.
Basándose en estudios a largo plazo realizados en animales, en
condiciones que simulaban la exposición profesional, se comunicó la
aparición de cambios hepáticos a un nivel de exposición de 300
mg/m3 (75 ppm) en la rata. En el ratón, se observaron lesiones en
el riñón y el hígado con 100 mg/m3 (25 ppm) y 200 mg/m3 (50 ppm),
respectivamente. Los datos sobre sensibilidad a los efectos tóxicos
varían considerablemente de unos estudios a otros.
En los animales, el cloruro de vinilideno no parece influir en
la capacidad reproductiva ni constituir un riesgo embriotóxico o
teratogénico a dosis inferiores a las que producen toxicidad
materna, pero este extremo no se ha estudiado en el hombre. Cuando
se utilizaron concentraciones capaces de producir toxicidad materna
se observaron toxicidad embrionaria y fetal y anomalías fetales,
reflejadas en la menor velocidad de aumento de peso.
El cloruro de vinilideno tiene efecto mutagénico en las
bacterias y las levaduras siempre que actúe en presencia de un
sistema metabólico de mamíferos. También algunas células de
mamíferos pueden sufrir lesiones del ADN y efectos mutagénicos in
vitro . En la mayoría de los estudios realizados en roedores in
vivo no se observaron efectos genotóxicos medidos por la letalidad
dominante ni desde el punto de vista citogenético, pero se ha
comunicado la observación de aberraciones en células de la médula
ósea del hámster chino. El enlace al ADN y la reparación de éste in
vivo en roedores fueron detectables pero mínimos. Así pues, los
estudios genéticos in vivo sugieren algunos signos de toxicidad
genética, pero, en la mayoría de los casos, los efectos fueron
mínimos o negativos.
Se han llevado a cabo varios ensayos de carcinogenicidad en
tres especies de animales de experimentación (ratones, ratas y
hámsters) utilizando diversas vías de administración.
Lamentablemente, la mayoría de estos estudios adolecían de graves
limitaciones de diseño o de método para la evaluación de la
carcinogenicidad. Por vía oral no se observaron efectos
carcinogénicos significativos en la rata. En la rata adulta
expuesta por inhalación, se notificó un aumento de los tumores de
la mama que no guardaba relación con la dosis. Se observó un ligero
aumento de la leucemia en las ratas expuestas tanto in utero como
recién nacidas. Estas observaciones no pudieron evaluarse. En un
estudio realizado en el ratón, se observó una mayor incidencia de
adenocarcinomas de riñón en los machos con niveles de exposición de
200 y 100 mg/m3 (50 y 25 ppm) pero no con 40 y 0 mg/m3 (10 y 0
ppm). En el mismo estudio, se observaron incidencias
estadísticamente mayores de tumores del pulmón (principalmente
adenomas en ambos sexos) y carcinomas mamarios (en hembras), pero
no se descubrieron relaciones entre la dosis y la respuesta.
Los tumores del riñón pueden estar relacionados de algún modo
con la citotoxicidad renal observada; puede ser que las lesiones
repetidas del riñón lleven directamente a la respuesta
carcinogénica por medio de un mecanismo no genotóxico o bien que
faciliten la expresión del potencial genotóxico de los metabolitos
en esta especie, este sexo y este órgano en particular. No
obstante, esta conclusión es dudosa en la ausencia de datos de
dosis-respuesta suficientes sobre los efectos genéticos in vivo,
así como ante el descubrimiento de que el cloruro de vinilideno
puede haber actuado como iniciador en un ensayo cutáneo en dos
etapas en el ratón.
Los estudios epidemiológicos, aunque no dan pruebas
estadísticamente significativas de que la exposición profesional al
cloruro de vinilideno entrañe un riesgo mayor de cáncer no son
adecuados para evaluar debidamente el riesgo carcinogénico del
cloruro de vinilideno para el ser humano.
Aunque en las evaluaciones de algunos autores el exceso de
defunciones por cáncer se atribuye al azar (a causa del reducido
número de sujetos y del tamaño de las cohortes), el hecho de que
aparezcan repetidamente valores más altos de lo esperado es digno
de mención. En los dos estudios de cohortes comunicados, se observó
cáncer del pulmón en 7 casos, cuando cabía esperar 3,16
defunciones. El resultado no puede desecharse, aunque hay que tener
presente la coexistencia de la exposición al cloruro de vinilideno
(en uno de los estudios). Puesto que las cohortes se determinaron
según su exposición al cloruro de vinilideno, puede ser imposible
excluir otras exposiciones que induzcan a error. Las conclusiones
comunicadas en materia de morbilidad (incluido un caso de carcinoma
testicular) pueden ser útiles a título informativo. La
interpretación por parte de los autores de que la mayor morbilidad
hepática guardaba relación con el consumo de alcohol por los
sujetos no es válida, puesto que no se evaluó la ingestión de
alcohol por todos los sujetos del estudio (y no sólo por los casos
identificados).
3. Recomendaciones
3.1 Recomendaciones para trabajos futuros
Es preciso disponer de mejores estimaciones de la producción
mundial anual de cloruro de vinilideno y de las cantidades de
cloruro de vinilideno que ingresan en el medio ambiente de todas
las procedencias, ya sea en forma de cloruro de vinilideno como tal
o por la degradación de otros productos químicos.
El destino ambiental previsto se basa en escasas pruebas
experimentales. Se necesita más información sobre las tasas de
degradación y sobre los productos de transformación en el aire, el
suelo, el agua y los sedimentos, y el metabolismo en especies no
mamíferas representativas.
Deben llevarse a cabo estudios de toxicidad a largo plazo en
los que se investiguen los diversos punto finales patológicos en
especies acuáticas representativas (peces, crustáceos y moluscos).
Deben definirse con más precisión los umbrales y los mecanismos de
los efectos tóxicos que tiene la exposición al cloruro de
vinilideno a corto y a largo plazo en el animal y el ser humano,
como base para establecer niveles seguros de exposición.
Conviene hacer un uso más exhaustivo de los datos existentes en
materia de carcinogenicidad. Los nuevos estudios sobre
carcinogenicidad deben hacerse según un protocolo aceptado de
bioensayo durante un lapso de vida entera que tenga en cuenta
específicamente las propiedades particulares del cloruro de
vinilideno. Esos estudios deben tener presentes la brevedad de la
semivida de la sustancia en el organismo, la importancia de la edad
al comienzo de la exposición, la duración de la exposición diaria y
otros datos pertinentes que puedan estar relacionados con el
establecimiento del régimen de dosificación. Hay que seleccionar
cuidadosamente las especies y estirpes de los animales de
experimentación. Los datos de toxicidad así como los datos
metabólicos y farmacocinéticos correspondientes a estos animales
también serían sumamente útiles.
Deben llevarse a cabo estudios de seguimiento a largo plazo
sobre la morbilidad y la mortalidad en poblaciones enteras y no
seleccionadas expuestas al cloruro de vinilideno.
Se necesitan estudios epidemiológicos que permitan evaluar los
efectos de la exposición al cloruro de vinilideno (incluida la
exposición prolongada a niveles reducidos) en poblaciones humanas.
Es particularmente necesario disponer de información sobre efectos
como la aparición precoz de enfermedades cerebrovasculares y
cáncer; los estudios deben tener en cuenta los factores que
introducen errores, como el hábito de fumar y el consumo de alcohol
(posiblemente en un sistema de referencia de casos).
Debe recurrise a datos históricos como base de comparación con
los resultados de las investigaciones en curso para poder evaluar
los efectos protectores que han tenido las medidas de reglamentación
durante los últimos años.
Para las investigaciones tanto en curso como futuras, un medio
valioso de salvar el problema del reducido número de sujetos que
hay en cada lugar de producción por separado sería aunar todos los
datos de éstos y realizar estudios multicéntricos. Debe
investigarse en animales de experimentación el valor de la
utilización de un agente con grupo sulfhidrilo como la
N- acetilcisteína en el tratamiento de la intoxicación por cloruro
de vinilideno.
Es necesario comparar la farmacocinética y el metabolismo in
vivo/in vitro del cloruro de vinilideno, especialmente en el
riñón, el hígado y el pulmón, en animales de experimentación de
distintas especies y en el ser humano, con el fin de comprender
mejor los resultados obtenidos en estudios de toxicidad in vivo . Se
precisan datos paralelos sobre la genotoxicidad potencial del
cloruro de vinilideno en el lugar escogido para estudiar la
carcinogénesis en distintas especies a fin de examinar el posible
papel de un mecanismo genético.
A la luz de las conclusiones neurotoxicológicas comunicadas en
el presente análisis, es necesario investigar el papel de los
sistemas moduladores en la patogénesis de la intoxicación por
cloruro de vinilideno.
3.2 Protección personal y tratamiento de la intoxicación
3.2.1 Protección personal
En los lugares de trabajo en la industria donde pueden
producirse exposiciones a corto plazo por inhalación superiores a
los límites recomendados, deben utilizarse mascarillas faciales
completas con filtro para los vapores orgánicos y, en previsión de
una emergencia, deben proporcionarse mascarillas con sistema de
abastecimiento de aire. Para evitar el contacto con el cuerpo, los
operarios que manejen cloruro de vinilideno deben llevar ropa
protectora, en buen estado, que incluya gafas de seguridad. Debe
mantenerse una ventilación constante dentro de las plantas
industriales mediante respiraderos con filtros en los lugares donde
puedan producirse derrames o fugas. Se recomienda vigilar las
emisiones de cloruro de vinilideno durante las operaciones de
distribución. En caso de una fuga, debe forzarse la evaporación del
cloruro de vinilideno ya sea directamente si se trata de una
cantidad pequeña o por evaporación controlada utilizando una espuma
sintética de expansión. Para disperar los vapores de la espuma
pueden utilizarse aspersores de agua en cortinas.
3.2.2 Tratamiento de la intoxicación en el hombre
En casos de exposición excesiva o de ingestión, debe
consultarse a un médico. Dadas las propiedades irritantes del
cloruro de vinilideno, debe prestarse especial atención a los
pulmones, la piel y los ojos. Deben vigilarse las funciones del
corazón, el hígado, el riñón y el sistema nervioso central. Puesto
que los datos correspondientes a animales indican que el cloruro de
vinilideno produce un aumento notable de la sensibilidad a las
arritmias cardiacas inducidas por la adrenalina, debe evitarse el
empleo de este fármaco. La hipotensión grave puede tratarse por
transfusión, con sangre entera o sustitutos del plasma. No se
conoce antídoto alguno.
Un paciente intoxicado por inhalación de cloruro de vinilideno
debe mantenerse abrigado en posición semiprona y en una atmósfera
bien ventilada y fresca. Las vías respiratorias deben mantenerse
despejadas y debe administrarse oxígeno si el sujeto se encuentra
en estado de estupor o de coma. En caso necesario debe aplicarse
respiración artificial.
Tras la ingestión de cloruro de vinilideno debe enjuagarse la
boca con agua. No debe provocarse el vómito por el riesgo de
aspiración de cloruro de vinilideno hacia la laringe y los
pulmones. El lavado gástrico y/o la administración oral de carbono
activado o de parafina líquida pueden ayudar a reducir la
biodisponibilidad de cloruro de vinilideno si se administran dentro
de la hora que sigue a la ingestión, pero pueden beneficiar al
paciente hasta 4 horas después de la ingestión.
Los ojos expuestos a cloruro de vinilideno deben irrigarse
inmediatamente con agua durante más de l5 minutos y debe acudirse
al médico.
En caso de exposición cutánea, las ropas contaminadas deben
retirarse y debe lavarse la zona afectada con agua y jabón.