Pesticide residues in food - 2003 - Joint FAO/WHO Meeting on Pesticide Residues
First draft prepared by
D.B. McGregor
Toxicology Evaluation Consultants, Lyon, France
|
Reversibility of effects on erythrocyte mass parameters in rats |
|
Significance of lens effects in studies in dogs treated with repeated doses |
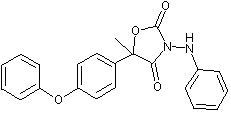
Famoxadone is the ISO approved common name for 5-methyl-5-(4-phenoxyphenyl)-3-phenylamino-2,4-oxazolidinedione. It is a racemic mixture containing two enantiomers in a 50:50 ratio.
Famoxadone has not been evaluated previously by the JMPR. Consequently, famoxadone was reviewed at the present Meeting in the context of the JMPR New Compounds Review Programme.
Famoxadone is used in agriculture, viticulture and horticulture for the control of a wide range of key fungal diseases of grapes, tomatoes and cereals. It inhibits mycelial growth and zoospore survival of various Oomycete fungi, e.g. Plasmopara viticola that causes grape downy mildew and Phytophthora infestans that causes tomato late blight, including mating types A1 and A2 that are resistant to phenylamide fungicides. Mycelial growth and/or spore germination of non-Oomycetes are also inhibited. Sensitive species encountered as cereal pathogens include Septoria spp., eyespot, brown rust, yellow rust and powdery mildew in wheat and net blotch, Rhynchosporium, brown rust and powdery mildew in barley.
The mechanism of antifungal action of famoxidone is inhibition of the mitochondrial respiratory chain at complex III, which results in decreased production of ATP.
The positions of the radiolabel in the compounds used in the studies of absorption, distribution and excretion are shown in Figure 1 and the metabolic pathways of famoxadone in rats are shown in Figure 2.
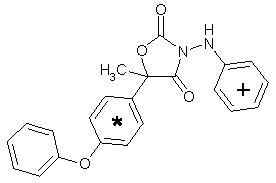
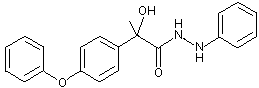
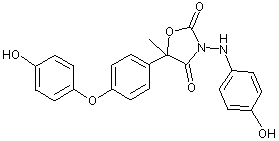
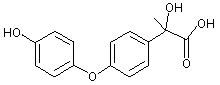
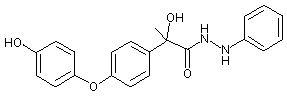
Figure 1. Positions of radiolabel on famoxadone used in studies of metabolism
* denotes [ C-phenoxyphenyl]famoxadone
+ denotes [14C-phenylamino]famoxadone
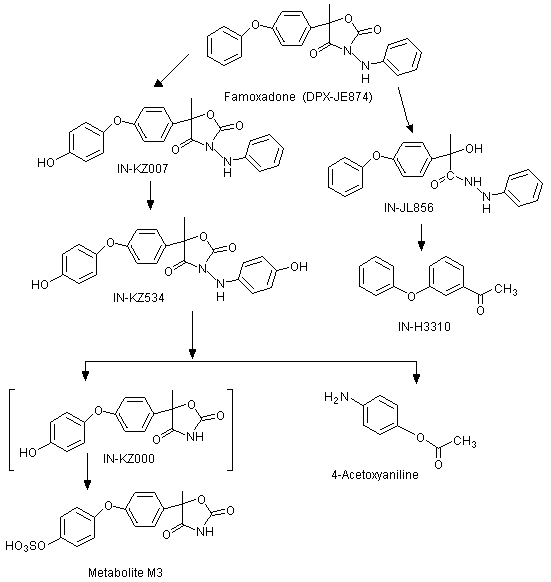
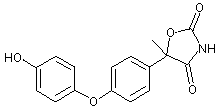
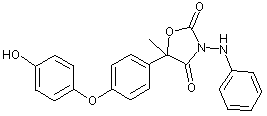
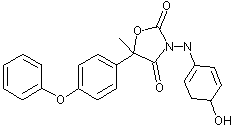
Figure 2. Proposed metabolic pathway of famoxadone in rats
Rats
The absorption, distribution, and excretion of famoxadone [5-methyl-5-(4-phenoxyphenyl)-3-phenylamino-2,4-oxazolidinedione] was evaluated in male and female Crl: CD BR (Sprague-Dawley) rats given famoxadone as a single oral dose at either 5 or 100 mg/kg bw, or as repeated doses at 5 mg/kg bw per day for 14 days. Pharmacokinetic and metabolic parameters were examined using [14C-phenoxyphenyl (POP)]famoxadone and [14C-phenylamino (PA)]famoxadone. The stereoselective metabolism of the two enantiomers of famoxadone was also investigated at the two doses (Savides et al., 1995; Savides et al., 1996; Himmelstein, 1999a).
Male and female Crl: CD®BR (Sprague-Dawley) rats (four or five rats of each sex per treatment, except in a pilot study in which two rats of each sex were used) were given the radiolabelled test material orally by gavage as a suspension in sodium carboxymethyl cellulose (1%) and ammonium acetate (0.01 mmol/l). Rats received either [14C-POP]famoxadone (radiochemical purity, >99%) or [14C-PA]famoxadone (radiochemical purity, >98%). In addition, some rats were treated with unlabelled famoxadone for 14 days before receiving [14C-PA]famoxadone. Pilot studies showed that a termination time of 120 h was sufficient to ensure that 90% of the administered radioactive dose was excreted. The initial absorption of famoxadone was rapid. The mean apparent absorption half-lives of total radioactive residues in the whole blood and plasma were approximately 0.8-1.4 h after an oral dose of 5 mg/kg bw of [14C-PA]famoxadone, or 100 mg/kg bw of [14C-POP]famoxadone. At a dose of 100 mg/kg bw, the mean apparent absorption time for [14C-PA] was 3.9-6.9 h. This difference suggests rapid enteric metabolism involving cleavage of the phenylamino group from the remainder of the molecule, and more rapid absorption of the phenoxyphenyl residues. Also, the rate of absorption of the phenylamino residue was saturable at a high dose. Excretion was rapid, with almost all the administered dose being recovered from the faeces (>75% within 24 h after dosing, and >90% within 120 h). In general, urinary elimination accounted for <10% of the administered dose. Pretreatment of male and female rats with unlabelled famoxadone had no significant effect on the rates of excretion of [14C-PA]famoxadone residues in either the faeces or urine. [14C-POP]famoxadone was not examined in this way in rats (but was in mice, see below). Expired air did not contain 14CO2. The ratios of radiolabel in tissue:blood were <1. Although there was rapid elimination of [14C-PA]famoxadone from plasma, the half-lives for elimination of [14C-PA]famoxadone equivalent residues from erthrocytes were approximately two- to three-fold longer. It appeared, therefore, that the radiolabelled residues from the phenylamino moiety were binding to erythrocytes. No binding of [14C-POP]famoxadone equivalent residues was observed in either whole blood or plasma. Plasma half-lives were about 7 and 22 h in rats given [14C-PA]- and [14C-POP]famoxadone, respectively, at 100 mg/kg bw (there was no significant difference between males and females). At a dose of 5 mg/kg bw, the plasma half-life of [14C-PA] residues was about 10.5 h. There were no significant differences in the overall fate of famoxadone in male and female rats.
The kinetic data described in the original report (Savides et al., 1995) were re-analysed Himmelstein (1999a). The justification offered for this re-analysis was that compartmental (model-dependent) analysis had been used, whereas a non-compartmental analysis would give a more accurate interpretation of terminal elimination. In so far as the calculation of the terminal elimination half-life, T1/2, is concerned, the difference between these methods is that the non-compartmental method uses the terminal elimination constant, K epsilon, (= -2.3 slope, calculated by plotting the log of the blood concentration over linear time and calculating the slope of the terminal linear portion of the time, limited to 12-72 h in this case) in the equation T1/2 = 0.693/K epsilon. The compartmental method, on the other hand, is more strongly influenced by the earlier time-points on the plasma concentration-time curve for deriving an elimination rate constant. This had the effect of overestimating the T1/2 value. T1/2 for rats given [14C-POP]famoxadone at a dose of 100 mg/kg bw was reduced from about 22 h (compartmental method) to 15.1 ± 2.2 h and 14.5 ± 2.5 h in male and female rats, respectively (non-compartmental method). The Tmax, Cmax and AUC(0-infinity) values were not changed significantly by the re-analysis, as indicated by the ranges of means in Table 1.
Table 1. Ranges of mean kinetic parameters obtained using compartmental and non-compartmental analysis
|
Radiolabel |
Dose |
Parameter |
Plasma |
Whole blood |
||
|
Male |
Female |
Male |
Female |
|||
|
[14C-PA] |
5 mg/kg |
Tmax |
2.3-3.3 |
3.8-4.8 |
4.8 |
6.7-8.0 |
|
Cmax |
0.9-1.1 |
1.0-1.1 |
0.7 |
0.8-0.9 |
||
|
AUC(0-inifinity) |
18.9-19.0 |
20.9-21.0 |
29.0-29.6 |
44.0-46.4 |
||
|
[14C-POP] |
100 mg/kg |
Tmax |
3.3-5.6 |
3.6-3.7 |
4.6-7.0 |
5.6-14.5 |
|
Cmax |
15.4-16.4 |
13.4-15.6 |
9.9-10.3 |
9.4-10.9 |
||
|
AUC(0-inifinity) |
507-515 |
430-435 |
359-368 |
336-345 |
||
|
[14C-PA] |
100 mg/kg |
Tmax |
9.5-10.0 |
7.0-7.5 |
9.5-13.9 |
13.3-18.0 |
|
Cmax |
18.6-24.7 |
13.5-17.5 |
18.3-22.2 |
13.3-15.7 |
||
|
AUC(0-inifinity) |
509-511 |
295-296 |
1010-1118 |
1031-1082 |
||
From Savides et al. (1995) and Himmelstein (1999a)
Tmax, time to maximal concentration
Cmax, maximal concentration
AUC(0-infinity), area under the concentration-time curve
The absorption, metabolism, and excretion of famoxadone was investigated in groups of five male and five female bile duct-cannulated Crl: CD BR (Sprague-Dawley) rats given a single oral dose of [14C-POP]- or [14C-PA]famoxadone at 5 mg/kg bw. Urine, bile, and faeces were collected continuously for up to 48 h after dosing. Absorption was calculated as the sum of the radioactivity in the bile, urine, cage wash, blood and carcass. The average amount of radiolabel excreted in the bile ranged from 30-39% of the administered dose. Faecal extracts contained 56-65% of the administered dose, while only 2-6% was excreted in urine. At the end of the experiment (48 h), only 0.4-3.0% of the administered dose remained in the carcass. The proportion of the administered dose that was absorbed (as indicated by the amount of radiolabel found in bile, urine, blood, carcass and case-wash) was: in males and females treated with [14C-PA]famoxadone, 38% and 37% respectively; in males and females treated with [14C-POP]famoxadone, 41% and 37% respectively. These values were not statistically significantly different from each other. There was no difference between males or females in terms of absorption, elimination in the bile, or excretion (Savides et al., 1997).
Mice
A study was undertaken to evaluate the absorption kinetics of famoxadone in groups of 40 male Crl: CD®-1(ICR)BR mice. The time course for total radioactive equivalents in plasma was evaluated in mice given a single oral dose of [14C-POP]-labelled famoxadone (radiochemical purity, 99.4%) at 50 mg/kg bw. The plasma values for Tmax, Cmax and area under the curve (AUC) were calculated for mice fed diets containing unlabelled famoxadone at a concentration of 50, 700, 2000, 3500, or 7000 mg/kg for 14 days, followed by a single oral dose of [14C-POP]famoxadone. Based on the daily dietary intakes on days 7-11, these dietary concentrations provided actual doses of famoxadone of 7, 142, 367, 804, and 1500 mg/kg bw per day, respectively. At each of 10 time-points (0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h after dosing), four mice per dose were killed and their blood was collected. No unchanged famoxadone was found in plasma after 14 days of dosing (limits of detection, about 0.5 µg/ml of plasma). In general, maximum concentrations of radiolabel in plasma occurred between 1 and 4 h after dosing. The Cmax values for each dose were 1.6, 55, 103, 398 and 406 µg of 14C-equivalents/g of plasma, respectively. The corresponding AUC values were 27, 478, 875, 2176 and 2486 µg of 14C-equivalents/g of plasma per h. Thus, for doses of up to 804 mg/kg bw, the kinetic parameters increased proportionally with dose, but there were no statistically significant differences in Cmax and AUC between animals at 804 or at 1500 mg/kg bw. It was concluded that at dietary concentrations providing famoxadone at a dose of more than about 800 mg/kg bw, absorption from the gastrointestinal tract becomes the limiting factor for internal exposure (Himmelstein, 1999b).
Dogs
The absorption, distribution, and excretion of [14C-PA]-labelled famoxadone (radiochemical purity, 97.4%) were studied in two groups of three male beagle dogs, each of which received a single oral dose at 15 mg/kg bw. One additional dog was killed four days after receiving carrier solution only. In the first group, urine, faeces, and blood were collected at specified time-points, and the dogs were killed 4 days after dosing. Dogs in the second group were killed at the time that the maximum concentration of radioactivity in plasma was observed in the first group (2 h after dosing). Erythrocytes, plasma, liver, fat, one whole eye, and the aqueous humour and the remainder of the other eye from each of these dogs were analysed for radioactivity.
In the first group, the highest mean concentration of radiolabel derived from [14C-PA]famoxadone and its residues in plasma (Cmax) was 1.5 µg equivalents/g at a Tmax of 2 h. The highest mean concentration of radiolabel in erythrocytes was 0.626 µg equivalents/g at 4 h after dosing. The range for terminal half-lives (T1/2) was 67-75 h in plasma and 146-159 h in erythrocytes. The area under the concentration-time curve (AUC(0-infinity) ranged from 96-109 µg/g per h in plasma and from 125-135 µg/g per h in erythrocytes. The rate of elimination was relatively slow from plasma and tissues: approximately 65% of the administered dose was eliminated in faeces and urine within 24 h, but high concentrations of radio-label remained both in plasma and erythrocytes at 96 h after dosing. The kinetic profile for one of the three dogs in the first group was somewhat different from those obtained for the other two dogs in this group. During the first 12 h after dosing, concentrations of radioactivity in plasma and erythrocytes for this dog were similar to those for the other two dogs, but from 18 h onwards concentrations of radioactivity in plasma and erythrocytes for this dog were more than three times higher than those observed in its companions. Speculative reasons for the different kinetics in this dog were offered, such as ingestion of some of its own faeces and aspiration of its own vomit (with subsequent increased absorption from the lungs). Neither suggestion appears likely, since the concentrations of radioactivity were actually higher in plasma in the period 18-48 h and in erythrocytes in the period 18-96 h. Furthermore, no cage-side observations were recorded that would support either of these suggestions.
Urine, faeces, cage washes, and cage wipes accounted for 78.8% (range, 65.8-86.0%) of the administered dose. Of the administered dose, 70.3% was recovered from the faeces and 7.67% was found in the urine. The overall recovery of radioactivity from tissues was 0.45% of the administered dose, with a range of 0.23-0.86%. The highest concentration of radioactive residues was found in the liver (equivalents, 1.34 µg/g), followed by mesenteric fat (equivalents, 0.945 µg/g).
In the second group of dogs, killed 2 h after receiving a single dose of 15 mg/kg bw, the highest mean concentrations of residue were also found in liver (equivalents, 4.45 µg/g) and mesenteric fat (equivalents, 2.80 µg/g). The concentrations in plasma and erythrocytes were 0.999 and 0.413 µg equivalents/g, respectively. Two h after dosing, residues in the whole eye, aqueous humour and the remainder of the eye averaged 0.106, 0.061 and 0.131 µg equivalents/g, respectively (Thalacker, 1996).
The CAS names and structures for the metabolites (referred to below by IN numbers) are given in the Appendix of this monograph. The proposed metabolic pathway in rats is shown in Figure 2.
In groups of five male and five female Crl: CD BR (Sprague-Dawley) rats given either [14C-POP]famoxadone or [14C-PA]famoxadone (radiochemical purities, >99% and >98%, respectively) by oral administration, unmetabolized famoxadone was the major component recovered from the faeces. The isomeric ratio of recovered parent was similar to that in the dosing material (R:S ratio, approximately 1) indicating that there was no stereoselective metabolism of enantiomers in the rat.
The primary metabolic pathway involved the hydroxylation of the intact parent molecule to the corresponding mono- and dihydroxylated derivatives (these being, respectively, IN-KZ007 and IN-KZ534), which were only recovered from the faeces. Metabolites resulting from the cleavage of the oxazolidinedione ring moiety were recovered from the urine. IN-KZ000 sulfate (metabolite M3) was the major urinary metabolite containing the [14C-POP] moiety, while 4-acetoxyaniline (IN-BY759) was the major urinary metabolite containing the [14C-PA] moiety. No parent famoxadone was detected in the urine. Several minor urinary metabolites were also observed. They were identified as the hydrolysis product of famoxadone (IN-JL856) and 4-phenoxyacetophenone (IN-H3310) from the [14C-POP] moiety. There were no significant quantitative differences in the chemical nature of the metabolites according to sex and/or treatment.
In a comparison of rats receiving a single dose of [14C-PA]famoxadone at 5 mg/kg bw with rats receiving repeated doses of [14C-PA]famoxadone at 5 mg/kg bw per day for 14 days, recovery of the administered dose from the urine and faeces was essentially the same, ranging from 10.4% to 11.2% in urine and from 85.7% to 89.3% in faeces. The major urinary metabolite was 4-acetoxyaniline, which constituted 4.9-8.3% of the administered dose. In faeces, unmetabolized famoxadone accounted for approximately 51-59% of the administered dose. Other metabolites identified in faeces were IN-KZ007 (males: 10.3% and 7.4% of the single and multiple doses administered, respectively; females: 13.0% and 2.8% of the single and multiple doses administered, respectively) and IN-KZ534 (males: 10.7% and 10.0% of the single and multiple doses administered, respectively; females: 7.7% and 13.4% of the single and multiple doses administered, respectively). The higher proportions of IN-KZ534 in females receiving multiple doses than in females receiving a single dose suggests self-induction of oxidative metabolism of the phenylamino moiety.
In rats receiving a single dose of [14C-PA]famoxadone at either 5 or 100 mg/kg bw, the same metabolites were recovered, but it was clear that a greater proportion of the administered dose was eliminated in faeces and a smaller proportion was eliminated in urine. Recovery of the administered dose in the urine was: males, 11.2% and 4.7% at the lower and higher dose, respectively; females, 10.5% and 3.4% at the lower and higher dose, respectively. The corresponding recoveries for 4-acetoxyaniline were: males, 7.1% and 3.4%; females, 4.9% and 1.9%. Recovery of the administered dose in the faeces was: males, 87.5% and 94.6% at the lower and higher dose, respectively; females, 87.8% and 88.4% at the lower and higher dose, respectively. Of the other metabolites in faeces, IN-KZ007 in males constituted 10.3% and 2.7% of the administered dose at the lower and higher dose, respectively, and, in females constituted 13.0% and 4.3% at the lower and higher dose, respectively, and IN-KZ534 in males constituted 10.7% and 3.1% of the administered dose at the lower and higher dose, respectively, and, in females constituted 7.7% and 1.6% at the lower and higher dose, respectively (Savides et al., 1995; Savides et al., 1996).
The biliary excretion and metabolism of both the phenoxyphenyl and phenylamino radiolabels of famoxadone were examined in Crl: CD BR (Sprague-Dawley) rats. Faecal and biliary extracts were examined, but the urine was not analysed owing to the small quantities of the administered dose present (2-6%). Unmetabolized famoxadone was the only radiolabelled component detected in the faeces. The major biliary metabolites were conjugates of IN-KZ007 and catechol (IN-03492) in rats treated with [14C-PA]-labelled test material, and conjugates of KZ007 and IN-ML436 in rats treated with [14C-POP]-labelled test material. Parent famoxadone was not detected in the bile samples. Metabolism of famoxadone occurred via the hydroxylation of the phenoxyphenyl and phenylamino rings, hydrolysis of the oxazolidinedione moiety, cleavage of the phenylamino ring, and combinations of these pathways. Further conjugation of primary metabolites also occurred (Savides et al., 1997).
The nature of the radiolabelled metabolites present in samples of erythrocytes, plasma, liver, fat, and aqueous humour was assessed in male beagle dogs 2 h after administration of [14C-PA]-labelled famoxadone as a single oral dose at 15 mg/kg bw. In addition, faeces and urine were examined for metabolites at intervals over the 96 h after dosing, and four plasma components (famoxadone, IN-KZ007, IN-JL856, and IN-ML815) were quantified at intervals up to the 96 h time-point.
The high-performance liquid chromatography (HPLC) profiles for extracts of plasma and erythrocytes contained 11 and 12 regions of radioactivity, respectively. The identified components in these profiles of both plasma and erythrocytes were famoxadone and its metabolites IN-KZ007, IN-JL856, and IN-ML815. The major identified component in plasma was IN-KZ007, the hydroxylated derivative of famoxadone. The concentration of famoxadone was lower in plasma than in erythrocytes, suggesting that the partitioning equilibrium favoured distribution into the erythrocytes. The analysed materials that were quantified accounted for only a small proportion of the total radioactivity. The additional metabolites in plasma could not be identified.
Extracts of liver and fat were shown to contain predominantly parent compound, with lesser amounts of IN-KZ007. No further metabolites were noted in fat, but liver contained several other unidentified components. It was not possible to extract radioactivity from the aqueous humour owing to very low concentrations and small sample sizes.
A complex pattern of radiolabelled metabolites was observed in the urine and faeces. Up to eight radioactive regions were assigned to each HPLC profile for urine samples, none of which corresponded to famoxadone. These were mainly polar components, none of which corresponded to famoxadone or could be identified with reference to known metabolites of the phenylamino moiety of famoxadone. Enzyme hydrolysis provided no evidence for the presence of either glucuronide or sulfate conjugates. Faecal extracts contained primarily famoxadone at early collection times, but at later times more metabolites were formed, including IN-ML815, IN-KZ007, IN-KZ532, IN-KZ534, and IN-JL856.
Plasma samples collected at intervals up to 96 h from three dogs that had received single oral doses of [14C-PA]famoxadone were analysed for famoxadone, KZ007, JL856 and ML815, using validated methods. Bimodal absorption profiles were observed for radioactivity and this was very pronounced in one dog. The metabolite KZ007 was present in the highest concentrations and its absorption profile showed a similar bimodal form. In addition, some components showed some degree of recycling, leading to multi-modal concentration/time profiles, which precluded pharmacokinetic analysis (Harrison, 1998).
An assessment was made of the potential of famoxadone (purity, 97.28%) to alter hepatic cytochrome (CYP) P450 content in Crl: CD®(SD)IGS BR rats and Crl:CD-1®(ICR)BR mice after 2 weeks of dietary exposure. Groups of five male and five female rats were given famoxadone at a dietary concentration of 0 or 20 000 mg/kg, and groups of five male and five female mice were given famoxadone at a dietary concentration of 0 or 7000 mg/kg. Mean daily intakes of famoxadone over the two weeks were 1540 and 1543 mg/kg bw per day for male and female rats, respectively. For mice, the mean daily intakes were 1559 and 1633 mg/kg bw per day in males and females, respectively. After approximately 2 weeks, the rats and mice were killed, their livers were weighed, and hepatic microsomes were prepared for evaluation of total P450 content and quantification of isozymes CYP1A1, CYP2B1/2, CYP3A, and CYP4A.
In both male and female rats, treatment with famoxadone caused reductions in body-weight gain. These decrements were associated with decreased food consumption and food use efficiency. Famoxadone caused a decrease in absolute and relative liver weights in male rats (means of 63% and 85% of the mean values for controls, respectively) and an increase in relative liver weights in female rats (a mean of 166% of the mean value for controls). The total concentration of cytochrome P450 in the livers was increased in male and female rats to 138% and 174% of the control values, respectively. There were also changes in the concentrations of specific isozymes. In male rats, concentrations of CYP2B1/2, CYP3A, and CYP4A were increased to 2452%, 228%, and 142% of control values, respectively. In female rats, concentrations of CYP2B1/2, CYP3A, and CYP4A were increased to 2759%, 363%, and 208% of control values, respectively. No alterations were observed in concentration of CYP1A1 in either male or female rats.
In mice, treatment with famoxadone did not result in alterations in body-weight gain in either sex. There were, however, increases in absolute and relative liver weights in male mice (155% and 161% of the control values, respectively) and in female mice (174% and 167% of the control values, respectively). Total hepatic concentration of cytochrome P450 was significantly increased by treatment with famoxadone (211% and 260% of the control values in male and female mice, respectively). Specific cytochrome P450 isozymes were also affected by treatment with famoxadone. In male mice, concentrations of CYP2B1/2 and CYP4A were increased to 1379% and 254% of the control values, respectively. In female mice, concentrations of CYP2B1/2 and CYP4A were increased to 940% and 401% of the control values, respectively. No alterations were observed in concentrations of CYP1A1 or CYP3A in either male or female mice (O'Connor, 1999).
These effects are attributed to a pharmacological response of the liver to exposure to a xenobiotic, which causes an induction of smooth endoplasmic reticulum and its associated enzymes. For this reason, the alterations in relative liver weight and cytochrome P450 content were considered to be adaptive responses and not adverse toxicological responses. The effects on hepatic cytochrome P450 concentrations and relative liver weight are consistent with the hepatocellular hypertrophy observed in long-term studies of toxicity in rats and mice (MacKenzie, 1996c, 1996d, 2002).
In an evaluation of acute oral toxicity, famoxadone (purity, 97.4%) in a suspension containing corn oil and acetone (85:15) was administered by gavage to fasted five male and five female Crl:CD-1®(ICR)BR mice (Finlay, 1994a) and five male and five female Crl: CD®BR rats (Sarver, 1994a) at a dose of 5000 mg/kg bw. The animals were observed for 14 days after dosing. There were no deaths or clinical signs of toxicity in either species and there were no statistically significant reductions in body-weight gain. No gross pathological findings were observed at autopsy. The oral median lethal dose (LD50) was >5000 mg/kg bw in male and female rats and mice (Finlay, 1994a; Sarver, 1994a).
Acute percutaneous (dermal) toxicity was studied in five male and five female New Zealand white rabbits. Famoxadone (purity, 97.4%) was mixed with approximately 0.5 ml of deionized water and applied to an area of approximately 190 cm2 (equivalent to approximately 10% of the total body surface) of the shaved intact skin at a dose of 2000 mg/kg bw. The application site was covered with an occlusive bandage for 24 h, then washed with soap and water. Observations for mortality and clinical signs were made approximately 3 h after dosing and then once daily for 14 days. Body weights were measured on days 1, 7 and 14 after treatment. Autopsies were performed on all rabbits after 14 days. No deaths occurred in this study. Body-weight losses of up to 6% of the initial values were observed in some rabbits 1 day after dosing. Two rabbits showed weight losses (up to approximately 2% of the previous body weight) on day 14. Slight to mild erythema was noted in four male and four female rabbits 1 day after application. On day 2, slight erythema was still observed in six rabbits. All dermal irritation had cleared by day 6 after exposure. No oedema was observed in any animal during this study. The dermal LD50 for famoxadone was >2000 mg/kg bw in both male and female rabbits (Sarver, 1994b).
A study of acute toxicity after administration by inhalation was conducted to determine the median lethal concentration (LC50) of famoxadone (purity, 96.1%) in five male and five female Crl: CD®BR rats. The test material was a milled particulate with a volume mean diameter of 2.5 µm suspended in air at a concentration of 5.3 mg/l air. Before the start of the study, samples of air were taken from several locations inside the exposure chamber. No statistically significant differences were observed, thereby indicating that homogeneous test atmospheres were being generated. During the 4 h exposure, the facial fur of the rats was coated with the test substance. Upon removal of rats from the restrainers immediately after the exposure, clinical signs observed included compound-stained fur and nasal discharge. Clinical signs observed on days 1-4 of the 14-day recovery period included stained perineum, ocular discharge, diarrhoea, and hunched posture. All clinical signs had resolved by day 5. There were no deaths during the study. All rats showed moderate to severe weight loss on the day after exposure to famoxadone (losses ranged from 5% to 11% of initial body weight). On day 2, four female rats experienced further, slight weight losses (losses ranged from 1% to 2% of the body weight from the previous day). All rats gained weight by the end of the 14-day recovery period, although two male rats and all female rats had instances of transient body-weight loss on one or more days. The LC50 of famoxadone was >5.3 mg/l air in both male and female rats (O'Neill, 1994).
The potential of famoxadone (purity, 97.4%) to cause acute ocular irritation was evaluated in six male young adult New Zealand white rabbits. Approximately 20 mg of a white solid milled to a fine powder was administered to one eye of each rabbit. The eyes remained unwashed after treatment and observation for effects was made 1, 24, 48 and 72 h after treatment, according to the method of Draize. Biomicroscopic examinations were also made at 24 and 48 h. Initial and final body weights were recorded. There were no deaths and no adverse clinical signs. Famoxadone produced transient ocular irritation in all six rabbits. Conjunctival redness (score of 1 or 2) and chemosis (score of 1) was observed in all treated eyes 1 h after exposure. One rabbit also had iritis (score of 1), while another had occult blood in the ocular discharge. All ocular irritation was resolved by 72 h. All animals gained weight during the study (Finlay, 1994b).
The potential of famoxadone (purity, 97.4%) to cause acute skin irritation was evaluated in four male and two female New Zealand white rabbits. Approximately 0.5 g of the test material moistened with deionized water was applied, under an occlusive dressing, to the shaved back of the rabbits. After 4 h, the dressing was removed and the skin was washed with soap and warm water. There were no deaths and no significant weight loss, or clinical signs in the treated animals. No oedema occurred. Treatment with famoxadone produced very slight erythema (score 1 or 2) in four of the six rabbits within 1 h after removal of the test substance. After 72 h, the erythema had disappeared in most rabbits. All irritation cleared by day 7 (Sarver, 1994c; Finlay, 1998).
The potential of famoxadone (purity, 97.4%) to produce delayed contact hypersensitivity in male Hartley guinea-pigs was assessed by the Magnusson-Kligman maximization test. Concentrations of famoxadone used were selected on the basis of preliminary screens for irritation. Group I (20 guinea-pigs) received famoxadone, group II (20 guinea-pigs) received the famoxadone vehicle only, group III (6 guinea-pigs) served as a positive control, and group IV (6 guinea-pigs) served as a positive control for the vehicle. On the day before intradermal induction treatments, the hair of the suprascapular area was clipped. On the first day (day 1) of the experiment, three pairs of intradermal injections (each of volume 0.05 ml) were made, one of each pair on either side of the dorsal midline. The treatments administered to each group at this stage are shown in Table 2.
Table 2. Skin sensitization assay: intradermal induction
|
Group |
Intradermal injection no. |
Treatment |
|
I |
1 |
FCA emulsified in deionized water (1:1) |
|
2 |
5% v/v famoxadone in white mineral oil |
|
|
3 |
5% famoxadone in white mineral oil emulsified in FCA (1:1) |
|
|
II |
1 |
FCA emulsified in deionized water (1:1) |
|
2 |
White mineral oil |
|
|
3 |
White mineral oil emulsified in FCA (1:1) |
|
|
III |
1 |
FCA emulsified in deionized water (1:1) |
|
2 |
0.1% DNCB in 50% ethanol: saline |
|
|
3 |
0.1% DNCB in 50% ethanol: saline emulsified in FCA (1:1) |
|
|
IV |
1 |
FCA emulsified in deionized water (1:1) |
|
2 |
50% ethanol in saline |
|
|
3 |
50% ethanol: saline emulsified in FCA (1:1) |
From Moore (1994)
FCA, Freund complete adjuvant
DNCB, 1 -chloro-2,4-dinitrobenzene
On day 7, the hair was again clipped and, for groups I, II and IV, the test site was treated with 3% sodium lauryl sulfate in petrolatum. On day 8, a 45 × 20 mm patch containing the topical induction dose was applied, which was covered with occlusive wrap and held in place with tape for 48 h. After unwrapping, any residual dose was removed using gauze soaked in deionized water (groups I and II) or 50% ethanol followed by deionized water (groups III and IV). The treatments used are shown in Table 3.
Table 3. Skin sensitization assay: topical induction
|
Group |
Treatment |
|
I |
0.4 g famoxadone mixed with 0.4 ml white mineral oil |
|
II |
0.4 ml white mineral oil |
|
III |
0.4 ml 0.1% DNCB in 50% ethanol: saline |
|
IV |
0.4 ml 50% ethanol: saline |
From Moore (1994)
DNCB, 1-chloro-2,4-dinitrobenzene
After clipping of both flanks of each guinea-pig on day 21, the guinea-pigs were challenged topically on day 22. Occluded topical applications were made using three chambers (diameter, 19 mm), one of which was positioned on a flank and contained vehicle only, while the other two chambers were positioned on the opposite flank and contained high and low doses of the appropriate material. The chambers were covered and strapped in place for 24 h. After removal of the chambers, the sites were wiped with the appropriate vehicle. These treatments are described in Table 4.
Table 4. Skin sensitization assay: challenge
|
Group |
Application site |
Treatment |
|
I and II |
Right flank |
0.1 g white mineral oil |
|
Left flank |
0.1 g famoxadone and 0.1 g white mineral oil |
|
|
Left flank |
0.1 g of 33% (w/w) famoxadone in white mineral oil |
|
|
III and IV |
Right flank |
0.1 ml acetone |
|
Left flank |
0.1 ml 0.1% DNCB in acetone |
|
|
Left flank |
0.1 ml 0.03% DNCB in acetone |
From Moore (1994)
DNCB, 1-chloro-2,4-dinitrobenzene
One guinea-pig was found dead on day 14. At autopsy, blood was found in the pericardial sac. All other guinea-pigs appeared to be normal. Skin reactions were scored at 24 and 48 h after removal of the chambers. No dermal irritation was observed at either time-point in the groups etreated with famoxadone or in the vehicle control group, while all of the guinea-pigs treated with 1-chloro-2,4-dinitrobenzene (DNCB) showed moderate redness at both doses. It was concluded that famoxadone did not produce delayed contact hypersensitivity (skin sensitization) (Moore, 1994).
Mice
In a 14-day feeding study, groups of five male and five female Crl: CD-1®(ICR)BR mice received diets containing famoxadone (purity, 97.4%) at a concentration of 0, 100, 1250, 3500, or 7000 mg/kg (equal to 0, 15.7, 204, 553 or 1094 mg/kg bw, respectively, in males and 0, 18.1, 236, 647 or 1291 mg/kg bw, respectively, in female). At the end of the period of feeding, all mice were killed and autopsied. Only the liver was examined microscopically. No deaths occurred and there were no compound-related clinical signs of toxicity. There were no statistically significant effects on mean body-weight gain, food consumption, or food use efficiency in either sex. No compound-related gross lesions were observed at necropsy. There was, however, a dose-related increase in mean absolute and mean relative liver weights at all doses. Mean absolute liver weights were statistically increased at 1250, 3500, and 7000 mg/kg in males and at 3500 and 7000 mg/kg in females. The increase in mean relative liver weights were statistically significant at 1250, 3500, and 7000 mg/kg in both male and female mice. Mean absolute and relative liver weights at 100 mg/kg were greater than those of controls, but were not statistically significant. Compound-related centrilobular hepatocellular hypertrophy was noted at 1250, 3500, and 7000 mg/kg in both males and females. Slight centrilobular fatty changes occurred in some mice of both sexes at 3500 and 7000 mg/kg. Some single-cell necrosis of hepatocytes was seen in females at 3500 and 7000 mg/kg. The hypertrophy found at 1250 mg/kg was considered to be a pharmacological response to exposure to the compound, while the fatty change and single-cell necrosis observed at 3500 and 7000 mg/kg were suggestive of slight hepatotoxicity. The NOAEL in male and female mice treated with famoxadone in the diet for 14 days was 1250 mg/kg (equal to 204 and 236 mg/kg bw per day, respectively) on the basis of slight hepatotoxicity observed at 3500 mg/kg (Ghantous, 1999).
The effect of famoxadone (purity, 97.7%) on blood chemistry and hepatic biochemical parameters was evaluated in Crl: CD-1®(ICR)BR mice treated for either 14 or 28 days. In the 14-day study, groups of 10 male and 10 female mice were fed diets containing famoxadone at a concentration of 0 or 3500 mg/kg. In the 28-day study, groups of 10 male and 10 female mice were fed diets containing famoxadone at a concentration of 0, 100, 500, 1000, 2000, 2500 or 3000 mg/kg. No compound-related effects on body weight, body-weight gain, or clinical signs of toxicity were produced by dietary concentrations of 3500 mg/kg for 14 days, or <3000 mg/kg for 28 days. Absolute and relative liver weights were increased in male mice at >1000 mg/kg and in female mice at >500 mg/kg. After 14 and 28 days of treatment, increases in liver-specific serum enzymes (alkaline phosphatase, alanine aminotransferase, and sorbital dehydrogenase) were observed at >1000 mg/kg. These increases over the values of the control group were generally small (less than twofold) and were not dose-dependent. Serum concentrations of triglyceride were not affected at any dose. Compound-related increases in hepatic beta-oxidation activity were also observed in males and females at >1000 mg/kg. Total hepatic cytochrome P450 content was increased in all mice treated with famoxadone. The NOAEL in male and female mice receiving diets containing famoxadone for 28 days was 500 mg/kg on the basis of increases in the activities of serum enzymes derived from hepatic cytosol and in peroxisomal beta-oxidation activity at >1000 mg/kg (MacKenzie, 1996a).
Groups of 20 male and 20 female Crl:CD-1®BR mice were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 35, 350, 3500 or 7000 mg/kg (equal to 0, 5.89, 62.4, 534 or 1149 mg/kg bw, respectively, for male mice and 0, 8.21, 79.7, 757 or 1552 mg/kg bw, respectively, for female mice) for approximately 90 days. There were no treatment-related deaths, clinical signs, or ophthalmic changes. Ophthalmology revealed only a few cases of phthisis bulbi (males: 2/10, 1/10, 0/10, 2/10 and 1/10, respectively; females: 1/10 only at 7000 mg/kg) and one complete cataract in one male at 350 mg/kg.
It was reported that there was a mild regenerative, Heinz body-associated, haemolytic anaemia in both sexes at 3500 and 7000 mg/kg and that this was characterized by decreases in erythrocyte count and increases in reticulocyte count, mean corpuscular volume, mean corpuscular haemoglobin and mean corpuscular haemoglobin concentration. However, the variations in erythrocyte count were almost random in male mice, being significantly elevated at 35 and 350 mg/kg after both 45 and 90 days and significantly depressed at 7000 mg/kg only after 45 days. Similarly, the concentrations of haemoglobin in males did not fit a diagnosis of anaemia in males, these concentrations being significantly elevated at 3500 and 7000 mg/kg after 45 days and at 35, 350 and 7000 mg/kg after 90 days. Erythrocyte volume fractions were increased at 35 and 350 mg/kg after 45 and 90 days and at 3500 mg/kg after 45 days. On the other hand, there was clearly a treatment-related increase in circulating reticulocytes that reached significance at 3500 and 7000 mg/kg after 45 and 90 days, while mean corpuscular volume, mean corpuscular haemoglobin and mean corpuscular haemoglobin concentrations were significantly elevated at 7000 mg/kg after both 45 and 90 days. A somewhat similar confusing combination of haematological data was obtained in female mice, in which erythrocyte counts were depressed while haemoglobin concentrations were increased at 7000 mg/kg after both 45 and 90 days. Erythrocyte volume fractions were depressed at 7000 mg/kg after 45 days, but there were no significant changes after 90 days. On the other hand, there was again a clearly dose-related increase in circulating reticulocytes that reached significance at 3500 and 7000 mg/kg after 45 and 90 days, while mean corpuscular volume, mean corpuscular haemoglobin and mean corpuscular haemoglobin concentrations were significantly elevated at 3500 and 7000 mg/kg after both 45 and 90 days. A mild leukocytosis in both sexes at 7000 mg/kg was considered to be secondary to haemolysis. Platelet counts were significantly depressed in all treated groups of males after 45 days and in females after 90 days. Increased spleen weights at 3500 (females only) and 7000 mg/kg and the microscopic finding of increased red pulp (i.e. congestion) in the spleen (females, 3500 mg/kg; males and females, 7000 mg/kg) correlated with the haematological findings. Increased haemosiderin pigment in the spleens and bile pigment in the livers of both sexes at >3500 mg/kg were also considered to be secondary to the haemolysis. Blood chemistry was not evaluated in this study.
In both male and female mice at 3500 mg/kg and 7000 mg/kg, liver weights were increased and there was centrilobular necrosis, diffuse fatty change and increased bile pigment. In addition, there were increases in total cytochrome P450 content and beta-oxidation activity. Hepatic cell proliferation indices, as measured by the incorporation of 5-bromo-2'-deoxyuridine delivered by implanted osmotic pumps were increased in female, but not male mice in both of these groups. At 350 mg/kg, there was an increase in total cytochrome P450 content in females, which was considered to be a non-adverse, pharmacological response. The NOAEL in male and female mice receiving diets containing famox-adone for 90 days was 350 mg/kg, equal to 62.4 and 79.7 mg/kg bw per day, respectively, on the basis of mild haemolytic anaemia and mild hepatotoxicity at 3500 mg/kg (Biegel, 1994; Saik, 1994).
Rats
Groups of five male and five female Crl: CD®BR rats were fed diets containing famoxadone (purity, >98%) at a concentration of 0, 100, 1000, 6000, or 20 000 mg/kg for 14 days. These dietary concentrations provided doses equal to 0, 8.97, 85.8, 428 or 1629 mg/kg bw, respectively, for males and 0, 8.85, 80.9, 440 or 1589 mg/kg bw, respectively, for females. After 14 days, all rats were killed and subjected to examination post mortem. No deaths occurred during the study. Piloerection was observed in rats of both sexes at 6000 mg/kg and 20 000 mg/kg, and hyperactivity, abnormal gait or mobility was observed in a few females at the same concentrations. Mean daily food consumption was significantly decreased in males and females at 1000, 6000, and 20 000 mg/kg. In males at 1000 mg/kg, this decrease was only statistically significant during the first week and was not considered to be an adverse effect as it did not result in decreased weight gain or food use efficiency. Body weights and body-weight gains were statistically significantly decreased in males at 6000 and 20 000 mg/kg and in females at 1000, 6000, and 20 000 mg/kg. There were no compound-related gross lesions. Statistically significant increases in mean relative liver weights were seen in females at 1000, 6000, and 20 000 mg/kg and in males at 20 000 mg/kg. Microscopic compound-related lesions were observed in the livers of males and females at 1000, 6000, and 20 000 mg/kg and consisted of hepatocellular hypertrophy, hepatocellular degeneration and single-cell necrosis, and an increase in hepatocellular mitotic figures. While the hepatocellular hypertrophy and the associated increases in relative liver weights were considered to be pharmacological (i.e. non-adverse) responses, the single-cell necrosis and increase in mitotic figures represented slight hepatotoxicity. The NOAEL in male and female rats receiving diets containing famoxadone for 14 days was 100 mg/kg, equal to 8.97 and 8.85 mg/kg bw per day, respectively, on the basis of the slight hepatotoxicity observed at 1000 mg/kg (MacKenzie, 1992; Slone, 1991).
Groups of 10 male and 10 female Crl: CD®BR rats were fed diets containing famoxadone (purity, 97.7%) at a concentration of 0, 100, 200, 300, 400, 500, or 600 mg/kg, for 28 days. Food consumption was not measured in this study. These dietary concentrations were equivalent to doses of 0, 10, 20, 30, 40, 50 or 60 mg/kg bw per day. Blood samples were collected and evaluated for effects on markers of hepatotoxicity after 14 and 28 days. No compound-related effects on body-weight gain or clinical signs of toxicity were produced at any concentration. At >400 mg/kg, minimal increases in liver-specific enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and sorbitol dehydrogenase) were observed in males, while sorbitol dehydrogenase activity was significantly increased in females. The latter enzyme provides the most sensitive indication of hepatotoxicity. In addition, decreases in concentrations of serum triglycerides were observed in females at >400 mg/kg, but only after 28 days. The results of this study suggest that administration of famoxadone in the diet causes minimal hepatocellular toxicity at dietary concentrations of >400 mg/kg in male and female rats. The NOAEL was 300 mg/kg in both male and females rats receiving diets containing famoxadone for 28 days, on the basis of increased activities of liver cytosolic enzymes released into serum, indicative of minimal hepatotoxicity at 400 mg/kg (MacKenzie, 1996b).
Groups of 20 male and 20 female Crl: CD®BR rats were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 50, 200, 800 or 1600 mg/kg, for approximately 90 days. These dietary concentrations provided doses equal to 0, 3.34, 13.0, 52.1 and 106 mg/kg bw, respectively, for males and 0, 4.24, 16.6, 65.7, and 130 mg/kg bw, respectively, for females. Cell proliferation was evaluated in the livers of five rats of each sex per group after approximately 14 days of treatment and blood samples were taken after 45 and 90 days. At the end of the 90-day period of feeding, all rats were killed and subjected to examination post mortem. There were no compound-related deaths, clinical signs, or ophthalmic effects. One male rat at 200 mg/kg was accidentally killed on day 11 and another male from the same group was found dead with septicaemia on day 42. Compound-related and biologically relevant decreases in body-weight gain were observed in females at 200 mg/kg (to 91% of the controls) and in both males and females at 800 mg/kg (to 91% and 87% of the controls, respectively) and at 1600 mg/kg (to 85% and 85% of the controls, respectively). Although the reductions in body-weight gain were not statistically significant in females at 200 mg/kg or in males at 800 mg/kg, they were considered to be biologically significant, since they represented part of a treatment-related response. There were few ophthalmic changes and these were not treatment-related. They consisted of one case of phthisis bulbi in a male at 800 mg/kg and one case of radial linear retinal atrophy in a female at 1600 mg/kg.
Mild haemolytic anaemia was observed in both sexes after both 45 and 90 days at 800 and 1600 mg/kg. The diagnosis was much clearer than was the case for mice (see above). It was characterized by decreases in erythrocyte counts and haemoglobin in males and females after both 45 and 90 days at 200, 800 and 1600 mg/kg, and in erythrocyte volume fractions in males and females at after both 45 and 90 days 800 and 1600 mg/kg, as well as in females after 90 days at 200 mg/kg. There were clearly dose-related increases in reticulocyte counts that reached statistical significance in males and females after both 45 and 90 days at 800 and 1600 mg/kg, while mean corpuscular volume and mean corpuscular haemoglobin were increased in males and females after both 45 and 90 days at 800 and 1600 mg/kg, and mean corpuscular volume was additionally increased in females at 200 mg/kg at both of these sampling times. Mean corpuscular haemoglobin concentrations were unaltered in either males or females at any dose or sampling time. On the basis of the reticulocytosis, the anaemia was described as regenerative. Decreases in erythrocyte counts and haemoglobin in male and female rats at 200 mg/kg and erythrocyte volume fractions in females at 200 mg/kg were not accompanied by reticulocytosis. The authors stated that the changes observed at 200 mg/kg were not biologically important because they were not accompanied by reticulocytosis. It is noted, however, that the reticulocyte counts were higher in both males and females at 200 mg/kg than at 0 and 50 mg/kg. Increased spleen weights and microscopic findings in the spleen (haemosiderin deposition, extramedullary haematopoiesis, and congestion) and bone marrow (hyperplasia) correlated with the haematological findings.
Hepatotoxicity occurred in both sexes at 800 and 1600 mg/kg, but it was more severe in males, being characterized by statistically significant, increased serum concentrations of the liver enzymes alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase and sorbitol dehydrogenase. Concentrations of bilirubin were also increased in males in these two groups. In females, only concentrations of the most sensitive marker of hepatotoxicity, sorbitol dehydrogenase, were increased. These effects were considered to be evidence of hepatocellular injury or necrosis and cholestasis. Scattered white foci in the livers were found during gross examination at autopsy in males at 1600 mg/kg, which correlated with the prominent microscopic focal degeneration found in this group. Mean absolute liver weights were decreased in the males at 800 mg/kg and 1600 mg/kg, while they were increased in the females at these dietary concentrations. Liver weights relative to body weights were significantly increased in females receiving dietary concentrations of >200 mg/kg. In addition to the focal degeneration mentioned above, other liver pathology observed in males and females at 800 mg/kg and 1600 mg/kg were centrilobular hypertrophy, single-cell necrosis (apoptosis), an associated increase in mitotic figures and bile duct hyperplasia. Cell proliferation tests showed that the 5-bromo-2'-deoxyuridine labelling indices in the groups receiving famoxadone at dietary concentrations of 800 and 1600 mg/kg was approximately 18-19 times higher than the value for male controls and approximately two-fold and five-fold higher than the control value in these two groups, respectively, in females. The incidence and severity of these lesions was greatest in males at 1600 mg/kg. Centrilobular hypertrophy, the most common lesion found, often results from hypertrophy of the smooth endoplasmic reticulum and increases in the enzymes associated with this organelle, and may be associated with proliferation of peroxisomes. While biochemical tests showed no treatment-related increase in total cytochrome P450 in the liver, hepatic beta-oxidation was increased approximately two-fold in males and females at 800 mg/kg and 1600 mg/kg and approximately 1.5-fold in females at 200 mg/kg, these increases probably representing an adaptive response.
Other blood chemistry findings were considered to be secondary to hepatotoxicity rather than direct effects of treatment. These included increased serum concentrations of cholesterol in females, slightly lowered serum concentrations of glucose and globulin in males and females and decreased concentrations of total protein in males. The increased cholesterol in females may have been due to alterations in lipid metabolism, rather than hepatotoxicity, since a similar effect was not observed in males in spite of evidence of more severe hepatotoxicity in this sex. Increased mean liver weights in females at 800 and 1600 mg/kg were attributed to hepatocellular hypertrophy, while decreased liver weights in males at 800 and 1600 mg/kg were attributed to the more severe hepatotoxicity observed in this sex.
The NOAEL in male and female rats receiving diets containing famoxadone for 90 days was 200 mg/kg and 50 mg/kg, respectively, equal to 13.0 and 4.24 mg/kg bw per day, respectively, on the basis of hepatotoxicity and haemolytic anaemia at 800 mg/kg, equal to 52.1 mg/kg bw in males (and to 65.7 mg/kg bw in females) and decreased body-weight gain at 200 mg/kg, equal to 16.6 mg/kg bw in females (MacKenzie, 1995; Sykes, 1995; MacKenzie, 1999). It is noted, however, that the reduction in body-weight gain in female rats at 200 mg/kg is not supported by observations made in three other studies of comparable or longer duration in rats. These are the 24-month study of toxicity/carcinogenicity, a two-generation study of reproduction and a 3-month study of neurotoxicity. The same strain of rat was used in all four studies. Consequently, the NOAEL identified on the basis of body-weight gain reduction from this 3-month study toxicity was not used in the evaluation made by the Meeting.
Groups of 10 male and 10 female Crl:CD (SD)®IGS BR rats received daily applications of famoxadone (purity, 97.3%) to the shaved, intact skin at a dose of 0, 250, 500, or 1000 mg/kg bw per day for approximately 28 days. The test site was covered with gauze dressing under layers of stretch gauze and self-adhesive bandages. The period of exposure was approximately 6 h/day. After treatment, the bandages were removed, and excess test substance was washed from the skin using water and mild soap. Body weight, food consumption, and clinical signs were evaluated throughout the study. Blood samples were collected before the rats were killed and subjected to examination post mortem.
There were no test substance-related effects on mortality, clinical signs of toxicity, body-weight gain or food consumption. Females in the groups receiving a dose of 500 or 1000 mg/kg bw per day had slight decreases in erythrocyte counts and haemoglobin concentration, but in the absence of other haematological effects, these small changes were not considered to be biologically significant. Clinical chemistry parameters were unaffected by treatment with famoxadone. Statistically significant increases in alkaline phosphatase, alanine aminotransferase, and sorbitol dehydrogenase activities were present at 500 and 1000 mg/kg bw per day and were considered to be indicative of minimal hepatocellular toxicity. These effects were not observed in females.
Liver weights were increased in all treated male and female groups. Relative to body weight, the liver weight increases at 250, 500 and 1000 mg/kg bw were, in male rats, 10%, 12% and 21%, respectively, and, in female rats, 11%, 14% and 9%, respectively. These increases were statistically significant in males at 1000 mg/kg bw per day in absolute and relative (to body and to brain weights) terms, and in males at 500 mg/kg bw per day group in relative (to body weight) terms. At 500 and 1000 mg/kg bw per day, the principal microscopic change observed in males and females was minimal hypertrophy of centrilobular hepatocytes, which may have been responsible for the weight changes. In the absence of more definitive microscopic evidence of hepatocellular toxicity, these changes were judged not to be adverse. In males at 500 and 1000 mg/kg bw per day, hypertrophy was associated with low incidences of apoptosis in the liver. Apoptosis has been reported to occur after the administration of substances that induce liver enzymes, perhaps as a homeostatic mechanism to eliminate excess cells, rather than as a result of primary cytotoxicity (Bursch et al., 1985). Additionally, incidences of minimal focal necrosis of the liver were increased in females receiving the highest dose (0/10, 0/10, 1/10 and 4/10 at 0, 250, 500 and 1000 mg/kg bw per day, respectively), but not in males (1/10, 0/10, 2/10 and 2/10 at 0, 250, 500 and 1000 mg/kg bw per day, respectively). The fact that male rats appear to be more sensitive than female rats to the hepatotoxic effects of famoxadone administered orally would suggest that the slightly higher incidence of focal necrosis observed in females at the highest dose in this study of dermal administration is not related to treatment.
In the treated males, increased incidences of extramedullary haematopoiesis were observed in the spleen, but neither the incidences nor the severity of the effect were dose-related and there were no associated haematological effects in males (the reductions in erythrocyte counts and haemoglobin, mentioned above, were in female rats).
The NOAEL for famoxadone administered dermally was 250 mg/kg bw per day in male rats, on the basis of slight increases in liver enzymes, which were suggestive of minimal hepatotoxicity. The NOAEL in female rats was 1000 mg/kg bw per day, the highest dose tested (Ladics, 1998)
Dogs
In a study designed to assess palatability and toxic potential, groups of two male and two female outbred beagle dogs were fed diets containing famoxadone (purity, 97.4%) for 5 weeks. An initial dietary concentration of 250 mg/kg was selected, which was increased to 500 mg/kg after the first week of dosing, to 1000 mg/kg after week 2, and to 2000 mg/kg after week 3. Famoxadone was administered at a dietary concentration of 2000 mg/kg for the remainder of the study. A concurrent control group was included in the study. Survival was unaffected by the administration of famoxadone. Transitory myotonic twitches in all four treated dogs were observed on the last 1-2 days of dosing at 2000 mg/kg. Slight decreases in body-weight gain and food consumption were noted in males and/or females receiving a dietary concentration of 500 mg/kg, which became marked in both males and females when the dietary concentration was increased to 1000 and 2000 mg/kg. Gross examination post mortem and measurements for liver enzyme activity in the serum revealed no effects of treatment (Tompkins, 1994).
Groups of four male and four female outbred beagle dogs were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 40, 300, or 1000 mg/kg (the last concentration was reduced to 600 mg/kg during week 4), for 90 days. These dietary concentrations provided doses equal to 0, 1.3, 10.0 and 23.8 (reduced to 21.2) mg/kg bw, respectively, for males and 0, 1.4, 10.1 and 23.3 (reduced to 20.1) mg/kg bw, respectively, for females. The reduction in dose was made because of the occurrence of myotonic twitches in both sexes and convulsions in one female. These twitches, which usually began approximately 4 h after feeding, and were first observed in all males and females in week 3, continued until the end of the study. The only other treatment-related clinical sign was soft stools in males and females at the highest dose. There were no deaths during the period of treatment. Mean body weights in the groups treated with 1000-600 mg/kg were lower than those of the controls by 11% in males and 14% in females after 13 weeks. These reductions were attributable to the effects of the higher dose experienced during the first 4 weeks.
A mild regenerative haemolytic anaemia was observed in both sexes at 1000-600 mg/kg. The effect was greater at week 5 (1000 mg/kg) than it was at week 12 (1000-600 mg/kg), as would be expected following a reduction in the dietary concentration (although the reduction in dose appears to have been quite small). This was characterized by decreases in erythrocyte counts, erythrocyte volume fraction, haemoglobin and mean corpuscular haemoglobin concentration. At the same time there were increases in reticulocyte count, Heinz body count, mean corpuscular volume, mean corpuscular haemoglobin concentration, and mean platelet count. A few statistically significant changes in haematological parameters were observed in females at 40 mg/kg and in males and females at 300 mg/kg, which comprised decreases in erythrocytes, erythrocyte volume fraction and haemoglobin. However, since reticulocyte counts were not elevated and most erythrocyte values were within the ranges for historical controls and/or were similar to the values measured before treatment began, the effect was not considered to be toxicologically significant at 40 and 300 mg/kg. Increased haemosiderin deposition in the liver of males at 300 mg/kg and males and females at 1000-600 mg/kg and in the bone marrow of females at 300 mg/kg and males and females at 1000-600 mg/kg was probably a consequence of the haemolytic anaemia.
The only treatment-related effect on blood chemistry parameters was a statistically significant increase in mean potassium concentrations at week 5 (fasted and non-fasted) and/or week 12. It was suggested that these elevations in potassium concentration were responsible for the myotonic twitching seen in the group receiving the highest dose. However, while the twitching persisted through the study, the concentrations of potassium at week 12 were no different to those for males in the control group (4.59 ± 0.163 versus 4.77 ± 0.227 milliequivalents per litre (meq/l)), although they did remain statistically significantly elevated in females.
Hepatotoxicity was not observed, and the activities of liver enzymes in serum, mean liver weights, and liver histology were not affected by administration of famoxadone in the diet.
Bilateral posterior cortical lens opacities—cataracts—(graded as slight) were observed at week 12 in males (2/4) and females (1/4) at 300 mg/kg and in males (2/4) and females (2/4) at 1000-600 mg/kg. None of the animals showed signs of visual impairment. Microscopically, minimal to mild treatment-related lenticular degeneration was seen in males (4/4) and females (4/4) at 300 mg/kg and males (3/4) and females (4/4) at 1000-600 mg/kg. One female in the group receiving famoxadone at a dietary concentration of 40 mg/kg had a unilateral lens lesion that was graded as minimal. The lenticular degeneration was characterized by a small focal zone of swollen lens fibers present at the Y suture of the posterior lens capsule. In some animals, these fibres formed morgagnian corpuscles, a change commonly associated with clinical lens opacities.
In this 90-day feeding study in dogs, the NOAEL for famoxadone in males was 40 mg/kg, equal to 1.3 mg/kg bw per day, and undetermined in females, on the basis of clinical and microscopic evidence of slight posterior subcortical lenticular opacities in males and females at 300 mg/kg, equal to 10.0 and 10.1 mg/kg bw per day, respectively. No NOAEL was identified for females because there was minimal microscopic unilateral lens degeneration in one out of four dogs at 40 mg/kg, equal to 1.4 mg/kg bw per day, the lowest dose tested (Saik, 1995; Tompkins, 1995).
Groups of four male and four female outbred beagle dogs were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 10, 20, 40, 300, or 300 mg/kg (the second high dose group being used as a recovery group, receiving famoxadone for the first 3 months and basal diet for the remaining 9 months), for 1 year. These dietary concentrations provided doses equal to 0, 0.3, 0.6, 1.2, 8.8 and 10.1 mg/kg bw per day, respectively, for males and 0, 0.3, 0.6, 1.2, 9.3 and 9.9 mg/kg bw per day, respectively, for females. There were no deaths during the study and there were no treatment-related effects on body weights, clinical signs of toxicity, organ weights, haematology, blood chemistry, urine analysis, or gross pathology. Microscopically, no evidence of hepatotoxicity was observed.
Treatment-related effects were limited to the lenses (i.e. occurrence of cataracts) of males and females at 300 mg/kg, and at 300 mg/kg followed by a recovery period. Ophthalmological examinations were conducted during study weeks -1 and 2, 8, 12, 16, 20, 25, 40 and 50. Posterior subcapsular lens opacities were observed in 2/4 males and 2/4 females at 300 mg/kg, and in 4/4 males and 4/4 females in the group receiving 300 mg/kg followed by a recovery period. Most of the lesions first appeared between weeks 8 and 12. The extent and progression of these lesions were variable. In no dog did the entire lens become opaque and no dog became clinically blind during the study. Regression of the lesion did not occur in any dog exposed to famoxadone for the entire year, however, regression was noted in some dogs in the group receiving 300 mg/kg followed by a recovery period. Clinical resolution of small opacities was noted in one eye of each of two dogs in the group receiving 300 mg/kg followed by a recovery period, although most of the posterior capsular opacities did not completely regress. During the period that dogs in the group receiving 300 mg/kg followed by a recovery period were receiving the control diet, no new ocular lesions developed, and most of the existing ocular lesions did not progress in severity. During this period, two dogs developed prolapsed third eyelids and one female showed progression of anterior Y lens opacities. Equatorial lens opacities, occasionally extending into the cortical regions of the lens, were observed in two out of four males and two out of four females at 300 mg/kg. These lesions developed after 6-12 months of exposure to the test substance and were not dependent on the previous development of a posterior subcapsular lesion. No other treatment-related ocular changes were observed. Intraocular pressure, measured at 3 months, did not differ among test groups.
Microscopic examination of the eyes demonstrated treatment-related lenticular degeneration in males and females at 300 mg/kg and in the group receiving 300 mg/kg followed by a recovery period. These lesions were characterized by fibre swelling with formation of morgagnian corpuscles and clefts within the lens cortex. Lenticular degeneration (including posterior subcapsular and equatorial degeneration) was observed in three out of four males and two out of four females at 300 mg/kg, and in two out of four males, and four out of four females in the group receiving 300 mg/kg followed by a recovery period. Microscopic findings in the lenses of dogs at 300 mg/kg and at 300 mg/kg followed by recovery were highly correlated, both in incidence and location, with the results of clinical ophthalmology.
In this 1-year feeding study in dogs, the NOAEL for famoxadone was 40 mg/kg in males and females, equivalent to 1.2 mg/kg bw per day in both sexes, on the basis of clinical and microscopic evidence of ocular lesions in both sexes at 300 mg/kg, equivalent to 8.8 and 9.3 mg/kg bw per day in males and females, respectively (Mertens, 1996; Frame, 1998).
Monkeys
Groups of four male and four female cynomolgus monkeys were given famoxadone (purity, 97.4%) orally by gavage at a dose of 0, 1, 100, or 1000 mg/kg bw per day for 52 weeks. The monkeys were observed for signs of toxicity, body-weight changes, and food consumption. Ophthalmic examinations were performed before treatment began and during weeks 5, 13, 26, 39, and 52, while haematology, blood chemistry, and urine analysis were performed before treatment began and during weeks 5, 13, 26, 39, and 53. After 53 weeks, the monkeys were killed and subjected to gross and microscopic examinations and organ weight analysis. Four pathologists examined the eyes of all of the animals.
Two monkeys died during the study (one male on day 14 at 1 mg/kg bw per day and one female on day 77 at 1000 mg/kg bw per day. It was concluded by the examining veterinarian that the causes of death were not treatment-related. Body weight, body-weight gain, and food consumption were unaffected by treatment. During the course of the study, there was the intermittent occurrence of white faeces in some monkeys at 1000 mg/kg bw per day, which was not considered adverse. The only adverse treatment-related effect observed was a mild haemolytic anaemia in both sexes at 1000 mg/kg bw per day. A slightly lower erythrocyte count, haemoglobin concentration, and erythrocyte volume fraction with secondary microscopic changes in the spleen, liver and kidney were observed in males and females at this dose. Blood chemistry parameters, organ weights (absolute and relative to body weight and to brain) showed no compound-related changes at any dose.
Throughout the course of the study, there was no clinical or microscopic evidence of lenticular opacities in any monkey at any dose. It is noted that the highest dose in this 1-year study (1000 mg/kg bw per day) is approximately 700-fold greater than the LOAEL (1.2 mg/kg bw per day) for ocular effects observed in the 1-year study in dogs (see above). These results indicate that primates either do not develop cataracts induced by famoxadone or that they are significantly less sensitive than dogs. The NOAEL for famoxadone was 100 mg/kg bw per day in male and female cynomolgus monkeys, on the basis of a mild haemolytic anaemia noted in both sexes at 1000 mg/kg bw per day) (Williams, 1997; Sykes, 1998).
Mice
Groups of 80 male and 80 female Crl: CD-1®(ICR)BR mice were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 5, 50, 700, or 2000 mg/kg for approximately 18 months. These dietary concentrations provided doses equal to 0, 0.70, 6.78, 95.6 or 274 mg/kg bw per day for males, respectively, and 0, 0.96, 9.84, 130 or 392 mg/kg bw per day for females, respectively. The study complied with European Commission (EC) directive 87/302/EEC and was conducted according to the data requirements of the United States Environmental Protection Agency (EPA) pesticide assessment guidelines subdivision F 83-2, OECD test guideline 451 and the Ministry of Agriculture, Forestry and Fisheries (MAFF) of Japan Nohsan No. 4200. Ophthalmology was performed on all mice before treatment, after 9 months of treatment and at the end of the study. Blood samples were taken from 10 mice of each sex per group after approximately 3, 6, 12 and 18 months of treatment. Cell proliferation in the liver was measured on five mice of each sex per group after 2 weeks and 9 months and, additionally, five mice of each sex per group were killed at each of these same times for measurement of total hepatic cytochrome P450 content and the activity of peroxisomal beta-oxidation enzymes.
There were no compound-related effects on survival, clinical signs, body weights, body-weight gains, or haematology. Ophthalmology performed on all surviving mice after approximately 9 months and near the end of the study revealed no treatment-related abnormalities. Observations recorded consisted mainly of a diffuse retinal degeneration that showed no dose-related response in either males or females; three cases of phthisis bulbi (one in each of three different groups of males) and focal cataracts (one male and one female at 50 mg/kg and one male and one female at 700 mg/kg). Blood chemistry was limited to plasma protein concentration measurements, which were not significantly different across the groups. Urine analysis was not performed in this study.
Mean absolute and relative (to body weight and brain weight) liver weights were observed in male and female mice at 700 and 2000 mg/kg and mean absolute liver weight was also significantly increased in female mice at 50 mg/kg. These liver weight changes were associated with mainly centrilobular hepatocellular hypertrophy in both sexes at 700 and 2000 mg/kg and with an equivocal level of this change in females at 50 mg/kg. The hypertrophy observed in these two groups was associated with increases in smooth endoplasmic reticulum and peroxisomes. Increases in total cytochrome P450 concentration to approximately 2-2.5-fold the control values were observed after 2 weeks and 9 months. Hepatic beta-oxidation was significantly increased in male mice at 700 and 2000 mg/kg after 2 weeks (1.8-fold and 2.3-fold, respectively) and in male mice at 2000 mg/kg after 9 months (1.8-fold). The rates of hepatic beta-oxidation were significantly increased in female mice at 2000 mg/kg after 2 weeks (2.0-fold) and in female mice at 700 and 2000 mg/kg after 9 months (1.9-fold and 2.0-fold, respectively). Microscopic lesions that were collectively considered to be indicative of liver toxicity included increases in diffuse fatty change, focal hepatocellular necrosis in males at 2000 mg/kg, eosinophilic foci in males at 700 and 2000 mg/kg, apoptosis (females), and the accumulation of haemosiderin and lipofuscin in Kupffer cells of both sexes. The biological significance of some of these observations is not clear. Thus, haemolysis was not observed, which might have explained the haemosiderin deposition and the eosinophilic foci showed no dose-response relationship and were not associated with neoplasia. Lipofuscin would be indicative of lipid oxidation. No statistically significant increases in cell proliferation in the livers of male or female mice were observed at either 2 weeks or 9 months.
An increase in the incidence of systemic amyloidosis and amyliodosis as a cause of death was observed in female mice at 2000 mg/kg. The toxicological significance of this increased incidence is unclear as it most likely is indicative of altered homeostasis, but it is considered to be evidence of treatment-related toxicity in the female mice at 2000 mg/kg.
There were no statistically significant increases in tumour incidence in the groups treated with famoxadone. Of the neoplasms observed, the only one requiring comment is malignant lymphoma, found at higher incidence in female mice at 2000 mg/kg. The incidences were 1/60, 2/60, 2/61, 0/60 and 6/60 at 0, 5, 50, 700 and 2000 mg/kg, respectively. The higher incidence was not statistically significant and was within the range for historical controls for the laboratory over a relevant period (1992-1994), when the numbers of mice with lymphomas in control groups in seven experiments were 0/80, 2/80, 9/80, 19/80, 7/80, 3/80 and 0/80. The NOAEL for famoxadone in mice after 18 months of dietary exposure was 700 mg/kg in males and females, equal to 95.6 and 130 mg/kg bw per day, respectively, on the basis of an increased incidence of hepatotoxicity in male and female mice and amyloidosis in female mice at 2000 mg/kg, equal to 274 and 392 mg/kg bw per day, respectively (MacKenzie, 1996c; Slone, 1997).
Groups of 50 male and 50 female Crl: CD-1®(ICR)BR mice were fed diets containing famoxadone (purity, 97.3%) at a concentration of 0, 2000, or 7000 mg/kg for approximately 18 months. These dietary concentrations provided doses equal to 0, 246 or 887 mg/kg bw per day for males, respectively, and 0, 348 or 1298 mg/kg bw per day for females, respectively. The study complied with EC Directive 87/302/EEC and was conducted according to the data requirements of the United States EPA pesticide assessment guidelines OPPTS 870-4200, OECD test guideline 451 and MAFF Japan Nohsan No. 4200. The objective of this study was to evaluate the tumourigenic potential of famoxadone in mice and not to establish an NOAEL in mice.
There were no treatment-related effects upon survival and the few clinical observations that were recorded were sometimes increased (incidence of pale mice and stained fur or skin), sometimes decreased (hair loss) in male and female mice at 7000 mg/kg, and consequently were not considered to be related to treatment. Compound-related reductions in mean body weight, body-weight gain, and food use efficiency were observed in males at 7000 mg/kg. Effects were primarily observed after the first three months of exposure. No adverse, treatment-related effects were observed on any of these parameters in males at 2000 mg/kg and no effects on food consumption were observed in any group of males. There were no adverse, treatment-related effects in either mean body weight or body-weight gain in females of any group. Mean food use efficiency in females at 7000 mg/kg, however, was below that for controls over the 18-month feeding period.
There were no compound-related effects on differential blood count observed in blood smears collected at 18 months from animals in the control group and in the group receiving the highest dose.
Ophthalmological examination was not performed, but microscopy of the eyes revealed no dose-related effects on the incidence of cataracts, these being of either mild or moderate severity in animals in the groups receiving famoxadone at 0, 2000 and 7000 mg/kg, respectively: males, 9/50, 1/14 and 0/50; and females, 2/50, 0/15 and 0/50.
Treatment-related increases in liver weight (absolute and relative to body weight and brain weight) and hepatocellular hypertrophy were observed in almost all males and females at 2000 and 7000 mg/kg, but these changes were considered to be non-adverse, physiologically adaptive responses to exposure to a xenobiotic. In males and females at 2000 or 7000 mg/kg, there was also microscopic evidence of hepatotoxicity, including diffuse fatty change (females only: 4% and 16%, respectively, versus 0% in controls), increased lipofuscin pigment in Kupffer cells (males: 10% and 42%, respectively, versus 6% in controls; females: 22% and 26%, respectively, versus 8% in controls), individual hepatocellular necrosis; (males only: 4% and 14%, respectively, versus 0% in controls), erythrocytic inclusions in hepatocytes and increased mitotic figures (females only: 8% and 14%, respectively, versus 0% in controls).
A NOAEL was not identified in this study owing to effects on body weight, body-weight gain and food use efficiency in male mice and hepatotoxicity in mice of both sexes receiving famoxadone at a dietary concentration of 7000 mg/kg (equal to 887 and 1298 mg/kg bw per day, respectively, for males and females, respectively), the highest dose tested (MacKenzie, 2002).
The Meeting concluded that famoxadone is not carcinogenic in male or female mice.
Rats
Groups of 92 male and 92 female Crl: CD®BR rats were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 10, 40, 200 or 400 mg/kg for approximately 23 months (males) or 24 months (females). These dietary concentrations provided doses equal to 0, 0.42, 1.62, 8.37 or 16.8 mg/kg bw per day for males, respectively, and 0, 0.53, 2.15, 10.7 or 23.0 mg/kg bw per day for females, respectively. The study complied with EC directive 87/302/EEC and was conducted according to the data requirements of the United States EPA pesticide assessment guidelines OPPTS 870-4200, OECD test guideline 451 and MAFF Japan Nohsan No. 4200. The objective of this study was to evaluate chronic toxicity and the tumourigenic potential of famoxadone in rats and to identify an NOAEL in rats. Ophthalmic observations were made on all rats before the start of the study and after approximately 12 and 22 months. Blood chemistry measurements and haematology were performed on 10 rats of each sex per group at approximately 3, 6, 12, 18 and 22 (males) or 23 (females) months. Studies of cell proliferation were made on livers of five rats of each sex per group at approximately 2 weeks and 12 months. beta-Oxidation enzyme measurements were made on an additional five rats of each sex per group at these two time-points. Full pathological evaluation was carried out on 10 rats of each sex per group that were killed after approximately 12 months.
There were no treatment-related deaths and clinical signs were limited to an increase in the incidence of ruffled fur in males at 400 mg/kg. Biologically significant decreases in mean body weight and mean body-weight gain were observed in females at 400 mg/kg; final mean body weights were 82% of those of the control group, and body-weight gain over the 713 days of treatment was 75% of that of the control group. No body-weight effects were observed in the male rats.
There were indications of a mild, treatment-related induction of cataracts, although indirect ophthalmoscopy alone gave little indication of any problem. In males at 0, 10, 40, 200 and 400 mg/kg, respectively, the incidences of cataracts (Ca) and corneal opacities (Op) after 12 months were: 0/82, 0/79, 0/76, 0/76 and 1Ca + 1 Op/78, while in males of the same groups after 23 months the incidences were: 1 Op/24, 0/22, 1Ca + 1 Op/31, 1 Op/27 and 2Ca + 1Op/21. In females after 12 months the respective incidences were: 0/79, 1Op/82, 2Ca/77, 0/81, 1Ca/78, while after 24 months the incidences were: 1Ca/31, 0/29, 1Ca/23, 0/27, 2Ca/32. Microscopic examination of the eyes suggested more strongly that there might be an increase in cataracts. The incidences of diagnosed cataracts in males at 0,10, 40, 200 and 400 mg/kg, respectively were: 3/62, 0/43, 1/34, 1/38 and 7/62. Of the seven microscopically diagnosed cataracts in the group receiving famoxadone at 400 mg/kg, three had been diagnosed ophthalmoscopically as such and one had been diagnosed as an opacity; the remaining three were not recognized in vivo. A subsequent re-evaluation of the eyes of the male rats concluded that the cataracts were not in fact cataracts but were lenticular degenerations (either unilateral or bilateral) and the total frequencies were 5/59, 4/56, 10/58, 4/60, 9/60 (Frame & Sykes, 1999).
No differences judged to be of any biological significance were observed in data on blood chemistry or urine analysis from any group. There were a few statistically significant changes in these parameters, but they were not toxicologically important, either because they were of small magnitude or because they failed to follow any monotonic response with increasing dose.
A mild regenerative, macrocytic, haemolytic anaemia was observed in both sexes at 400 mg/kg. In male rats of this group there were statistically significant reductions in erythrocyte counts, and increases in reticulocyte counts at 3, 6 and 12 months. At these same times, there were statistically significant increases in mean corpuscular volume and mean corpuscular haemoglobin. In female rats at 400 mg/kg, there were statistically significant reductions in erythrocyte counts, erythrocyte volume fraction and haemoglobin concentrations at 3, 6, 12 and 18 months, but increases in reticulocyte counts were observed only at 3 months. There was no evidence for a haemolytic effect in males at 18 or 22 months or in females at 23 months. No treatment-related effects on haematology were observed in rats at <200 mg/kg. Morphological evidence of extravascular haemolysis and a regenerative response was observed microscopically at 400 mg/kg in both sexes at 1 year and included increases in splenic macrophage pigment (in males), hepatic Kupffer cell pigment (in females), splenic haematopoiesis (in males), and bone-marrow hyperplasia (in males). After 2 years, microscopic evidence of haemolysis was no longer evident; but a small increase in large spleens in males and females at 400 mg/kg was probably related to the haemolysis.
No significant effects on liver weight or weights of other organs were noted in this study. Treatment-related gross liver gross liver foci were observed in some males at 400 mg/kg. Histological evidence of hepatocellular toxicity was also found at this dose, in both male and female rats. At 1 year, there was an increase in focal cystic degeneration and focal hepatocellular degeneration in male rats at 400 mg/kg. After 2 years, there were increases in focal hepatocellular degeneration in both sexes at this dose. This degeneration, however, was less severe and the foci were of smaller size than was observed at the higher doses used in the 90-day study (800 and 1600 mg/kg), and consequently it was not associated with any increases in liver enzymes in the serum. A compound-related increase in the incidence, size, and severity of eosinophilic foci of hepatocellular alteration was also observed in male rats at 400 mg/kg at 2 years. These eosinophilic foci occasionally included or were associated with the aforementioned focal hepatocellular degeneration. The biological significance of eosinophilic foci is not clear, since it appears that many such foci do not progress to neoplasia and, indeed, they may regress if exposure to the inducing compound is stopped. A retrospective analysis of six 2-year studies of carcinogenicity, previously reported by the United States National Toxicology Program, concluded that the common, spontaneously occurring eosinophilic foci in liver appeared not to be generally useful as predictive indicators of liver carcinogenesis (Harada et al., 1989).
Non-adverse liver changes observed in males and females at 400 mg/kg included hepatocellular hypertrophy and apoptosis (in females only). The hypertrophy was often associated with increases in some cytoplasmic organelles. In this case, hepatic total cytochrome P450, which was measured at 2 weeks and 1 year, was only increased in females at 400 mg/kg at the 1-year time-point and is, therefore, indicative of proliferation of smooth endoplasmic reticulum. Liver cell proliferation was measured using the 5-bromo-2'-deoxyuridine labelling technique, which showed that, at 2 weeks in male rats at 400 mg/kg, labelling was increased to 9.6-fold the control value, but values for both males and females for this group were similar to those of controls at the 1-year evaluation. Hepatic beta-oxidation activity, increases in which indicates peroxisomal proliferation, was elevated in males at 400 mg/kg only at the 1-year evaluation. After 2 years, hepatocellular apoptosis was increased slightly in female rats at 400 mg/kg. The increases in these liver effects at 400 mg/kg were all considered to be non-adverse effects related to the pharmacological response to a xenobiotic.
Neoplasms do not appear to have been induced by famoxadone. At 1 year, there was a statistically significant increase in adenomas of the pars distalis of the male pituitary at 400 mg/kg (0 mg/kg, 1/10; 400 mg/kg, 4/10). This result was not, however, substantiated by the data from the 2-year study in which the incidences for the same, very common tumour in this strain of rat were not increased by treatment (0 mg/kg, 49/62; 400 mg/kg, 41/62). Only for Leydig cell adenoma was a statistically significant increased incidence (according to a Cochran-Armitage test for trend) observed in the 2-year study. Incidences in the groups receiving famoxadone at 0, 10, 40, 200 and 400 mg/kg were: Leydig cell adenomas, 0/62, 0/62, 1/62, 1/61 and 3/62, respectively; and Leydig cell hyperplasia, 23/62, 21/62, 15/62, 20/62 and 19/62, respectively. A pair-wise comparison using the Fisher exact test (which is acceptable in this case, given the very similar patterns of mortality) of the groups receiving 0 and 400 mg/kg was not significant. Also, comparison by the Fisher exact test between the incidence for historical controls pooled from the six most appropriate studies (incidence, 9/351, range, 0-4.9%) and that for the group at 400 mg/kg was not significant (p = 0.26). Furthermore, there was no treatment-related increase in the incidence of hyperplasia of Leydig cells, from which adenomas are derived.
The NOAEL in male and female rats treated with diets containing famoxadone for 2 years was 200 mg/kg, equal to 8.4 and 10.7 mg/kg bw per day, respectively, on the basis of clinical signs (males), decreased body weights (females), mild hepatotoxicity, and mild regenerative haemolytic anaemia at 400 mg/kg, equal to 16.8 and 23.0 mg/kg bw per day, respectively, the highest dose tested (MacKenzie, 1996d; Sykes & Frame, 1997; Frame & Sykes, 1999).
The Meeting concluded that famoxadone is not carcinogenic in either rats or mice.
Famoxadone was tested for genotoxicity in a range of assays, both in vitro and in vivo (Table 5). There was no evidence of genotoxicity in vitro according to tests for gene mutation in bacteria (S. typhimurium and E. coli), gene mutation in Chinese hamster ovary cells and unscheduled DNA synthesis (two studies in primary cultures of rat hepatocytes). In a single study of chromosomal aberrations in cultures of human lymphocytes taken from two donors, significant increases in the proportion of abnormal cells were observed in two experiments conducted in the absence of an exogenous metabolic activation system, after incubation for 3 h. Significant responses were observed at a concentration of 20 and 25 µg/ml in the first experiment and at 15, 20 and 25 µg/ml in the second experiment. Most of the damage consisted of chromatid gaps, but chromatid breaks were also frequent. The frequency of chromosomal damage was not significantly elevated, but occasional chromatid triradials and quadriradials (i.e. complex exchanges) were observed in treated cells, whereas they did not occur in untreated cells of either experiment (although these rare exchange figures were also found in the vehicle control culture from one donor in the presence of a metabolic activation (S9, 9000 × g supernatant fraction of rat liver). In the presence of a metabolic activation system, again with an incubation of 3 h, there were no indications of clastogenic activity with famoxadone at concentrations of up to 30 µg/ml. This loss of activity could be caused either by metabolism to an inactive product or by competitive binding to the added proteins in the incubation mixture, thereby reducing the available concentration of famoxadone available for reaction with the target cells. No genotoxic activity was observed in three experiments conducted in vivo. One of these was a study of unscheduled DNA synthesis in vivo/in vitro in the liver of male Crl: CD®BR rats either 2-4 or 14-16 h after administration of famoxadone as a single oral dose at 0, 800 or 2000 mg/kg bw by gavage. The other two studies were tests for micronucleus formation in bone-marrow cells of mice. In the first of these, groups of Crl: CD-1®(ICR)BR mice were given famoxadone as a single oral dose at 0, 1250, 2500 or 5000 mg/kg bw by gavage. Mice receiving the highest dose (six of each sex per time-point) and the vehicle control (five of each sex per time-point) were killed at approximately 24, 48 and 72 h after treatment. Mice in the remaining two groups (five of each sex) were killed 24 h after dosing. No increases in the frequency of micronucleated polychromatic erythrocytes were observed in either sex at any dose. The second test for micronucleus formation was an addendum to a 14-day study of toxicity described above. Groups of Crl: CD®BR rats (five of each sex per group) were fed diets containing famoxadone (purity, >98%) at a concentration of 0, 6000, or 20 000 mg/kg, for 14 days. Bone-marrow smears were prepared from four rats of each sex per group and examined for micronuclei. No statistically significant increases in micronucleated polychromatic erythrocytes were observed and there was no change in the ratio of polychromatic erythrocytes to normochromatic erythrocytes.
Table 5. Results of studies of genotoxicity with famoxadone
|
End-point |
Test object |
Dose (LED/HID) |
Purity (%) |
Result |
Reference |
|
In vitro |
|||||
|
Gene mutation |
S. typhimurium strains TA100, TA1535, TA1537, TA1538, TA98; E. coli WP2 uvrA |
5000 µg/plate |
97.4 |
Negativeb |
Bentley (1995) |
|
Unscheduled DNA synthesis |
Male Crl: CD®BR rat primary hepatocytes |
7.5 µg/ml |
97.4 |
Negativeb |
Bentley (1994) |
|
Unscheduled DNA synthesis |
Male Crl: CD®BR rat primary hepatocytes |
5.0 µmg/ml |
97.4 |
Negativeb |
Cifone (1999b) |
|
Gene mutation |
Chinese hamster ovary cells, Hprt locus |
600 µg/ml |
97.3 |
Negativeb |
Cifone (1999a) |
|
Chromosomal aberration |
Human lymphocytes |
15 µg/ml ± S9 |
97.4 |
Positive |
Gerber (1995) |
|
In vivo |
|||||
|
Unscheduled DNA synthesis in vivo/in vitro |
Male Crl :CD®BR rat liver cells |
2000 mg/kg bw per os × 1 |
97.4 |
Negative |
Fellows (1998) |
|
Micronucleus formation |
Male Crl: CD®BR rat, bone-marrow cells |
20 000 mg/kg in diet, 14 days |
>98 |
Negative |
MacKenzie (1992) |
|
Micronucleus formation |
Male Crl :CD-1®(ICR)BR mouse, bone-marrow cells |
5000 mg/kg bw, per os × 1 |
97.4 |
Negativeb |
Kuykendall (1994) |
LED, lowest effective dose; HID, highest ineffective dose
S9, 9000 × g supernatant fraction of rat liver
Thus, the single indication of clastogenic activity observed in vitro was not confirmed in vivo. The Meeting concluded that famoxadone is unlikely to be genotoxic.
Rats
Groups of 30 male and 30 female Crl: CD®BR rats were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 20, 200 or 800 mg/kg, equal to 0, 1.14, 11.3 and 44.7 mg/kg bw per day in males and 0, 1.45, 14.2 and 53.3 mg/kg bw per day in females in a two-generation study of reproduction, which was performed according to EC directive 87/302/EEC and the data requirements of the United States EPA pesticide assessment guidelines subdivision F 83-4, OECD guidelines for testing of chemicals section 4, No. 416 and MAFF testing guidelines for toxicology testing NohSan 59 No. 4200. After receiving the test diets for at least 70 days, the animals were mated. Thirty offspring (F1) of each sex per group were fed diets containing the same concentrations of famoxadone for at least 105 days after weaning, before mating to produce a second generation (F2). Twenty F1 and F2 weanlings of each sex per group that had not been selected to produce the next generation were selected for gross examination post mortem. The remaining weanlings were killed without examination. Before mating, measurements of blood chemistry were made on selected adult rats (10 of each sex per group) in both generations. After the litters were weaned, all parental rats were killed and gross examinations were conducted post mortem. In particular, livers of all adults were weighed and peroxisomal beta-oxidation activities measured in livers from five rats of each sex per group. Some liver samples were lost, so these numbers were reduced to four per group in P0 males and F1 males and females at 200 mg/kg. Tissues from rats of both generations receiving famoxadone at 0 and 800 mg/kg were examined microscopically, as were any gross lesions and target organs from adult rats in all groups.
No adverse treatment related effects were observed among rats at <200 mg/kg.
At 800 mg/kg, a number of treatment-related and statistically significant effects were recorded. There were, however, no treatment-related effects on survival. There were increased incidences of diarrhoea in P1 males and of alopecia in F1 females during the period before mating, and in P1 and F1 females during gestation. Mean body weights, overall mean body-weight gains and overall mean food consumption was reduced in P1 and F1 males and females. Overall mean food use efficiency was also reduced for P1 males and females during the period before mating. During gestation, mean body weight and overall mean food consumption was reduced for P1 and F1 females. Mean body weight was also reduced on days 0, 7 and 14 of lactation for P1 and F1 females.
There were no compound-related gross lesions, and the histology of the reproductive organs was unremarkable. There were, however, increases in the activities of hepatic enzymes measured in serum, as well as increases in serum bilirubin concentrations, which was indicative of hepatocellular injury and cholestasis in P1 and F1 males. Significant reductions in mean triglyceride concentrations in serum of P1 and F1 males and females and significant increases in mean cholesterol concentrations in serum of P1 males and F1 males and females suggested altered lipid metabolism. Within samples of liver itself, the hepatic beta-oxidation rate was increased for P1 and F1 males and females. It is noted that these liver effects were similar to those observed in females at 800 mg/kg in the 90-day feeding study in rats, described above. Also, as in the 90-day study, the increase in serum liver enzymes was much greater in male than in female rats, correlating with a decrease in mean liver weights in male rats, rather than an increase as seen in female rats. Histological examination did not indicate hepatotoxicity.
Reproductive indices were unaffected by treatment at any dose. These indices included mating, fertility and gestation indices as well as the indices that describe pup viability at parturition, and on postnatal days 4 and 21. The only recorded effect of treatment on the pups was a reduction in mean pup weight throughout the 21-day period of lactation for F1 litters (average reductions, 7-9%) and from postnatal day 4 until the end of lactation for F2 litters (average reductions, 6-8%), these effects occurring at 800 mg/kg.
In the multigeneration study of reproduction in rats treated with famoxadone, the NOAEL for adult rats and their offspring was 200 mg/kg, equal to 11.3-14.8 and 14.2-17.5 mg/kg bw per day in adult males and females, respectively, on the basis of maternal and paternal systemic toxicity (decreased body-weight gains and food consumption, and hepatotoxicity) at 800 mg/kg, equal to 44.7-62.1 mg/kg bw per day. The NOAEL for offspring toxicity was 200 mg/kg, on the basis of reduced pup weight gain at 800 mg/kg. There were no effects on reproductive indices at concentrations up to and including 800 mg/kg, the highest dose tested (Kreckmann, 1995). The Meeting concluded that famoxadone is not a reproductive toxicant in rats.
Rat
In a study of developmental toxicity, groups of 25 time-mated, female Crl: CD®BR rats were given famoxadone (purity, 97.4%) in corn oil by gavage at a dose of 0, 125, 250, 500, and 1000 mg/kg bw per day on days 7-16 of gestation. All rats survived to the end of the study and no internal lesions were found at autopsy. Dams were killed on day 22 of gestation and subjected to gross examination. Fetuses were removed, weighed, sexed, and subjected to external, visceral, and skeletal examination.
Maternal effects were limited to a decrease in mean food consumption and body-weight gains at 500 and 1000 mg/kg bw per day during days 7-9 of gestation. At 1000 mg/kg bw per day, there was a recovery in food consumption on days 17-22 of gestation, when it was significantly increased. No compound-related effects were observed in any reproductive or developmental end-points at any dose.
The NOAELs in the study of developmental toxicity in rats were 250 mg/kg bw per day for the dams, on the basis of decreases in food consumption and body-weight gains at 500 mg/kg bw per day, and 1000 mg/kg bw per day, the highest dose tested, for the fetuses (Murray, 1994; Munley, 1999a). The Meeting concluded that famoxadone is neither a teratogen nor a developmental toxicant in rats.
Rabbits
In a study of developmental toxicity, groups of 20 time-mated, female Hra: (NZW)SPF rabbits were given famoxadone (purity, 97.4%) suspended in 0.5% Tween 80 at an oral dose of 0, 100, 350, or 1000 mg/kg bw per day by gavage on days 7-19 of gestation. Dams were killed on day 29 of gestation, or after aborting, and subjected to gross examination. Fetuses were removed, weighed, sexed, and subjected to external, visceral, and skeletal examinations.
Four out of the 17 pregnant rabbits receiving a dose of 1000 mg/kg bw per day suffered severe weight loss, decreased food consumption, faecal impaction, and subsequent abortions that occurred during days 19-23 of gestation. All these effects were considered to be secondary to gastrointestinal impaction with the dosing suspension, due to its high viscosity. In one of these four rabbits receiving the highest dose, trichobezoar (ball of hair in the stomach or intestines) was indicated by examination post mortem and may have exacerbated the impaction. Rabbits without signs of impaction had neither signs of maternal toxicity nor abortions; consequently, famoxadone was considered not to have produced systemic toxicity at up to and including a dose of 1000 mg/kg bw per day. One rabbit receiving a dose of 100 mg/kg bw per day aborted without showing any of the signs seen at a dose of 1000 mg/kg bw per day. This abortion was considered to be a chance occurrence; this conclusion was supported by the absence of abortions at 350 mg/kg bw per day. In a second case of abortion at 100 mg/kg bw per day, the animal experienced similar reductions in body-weight gain and food consumption as recorded for those cases in animals receiving a dose of 1000 mg/kg bw per day. There appears to be some doubt regarding the cause of this abortion, but examination post mortem did reveal white fibrous material (probably hair) in the stomach contents, which perhaps suggests that trichobezoar was also a preliminary cause of this abortion.
No compound-related effects were observed in any reproductive or developmental end-point at any dose. The maternal and fetal NOAEL in this study of developmental toxicity in rabbits was 1000 mg/kg bw per day, the highest dose tested (Munley, 1999b; Munley, 1999c).
The Meeting concluded that famoxadone is not teratogenic and does not present a developmental hazard in rabbits.
In a study of neurotoxic potential, groups of 12 male and 12 female Crl: CD®BR rats were given famoxadone (purity, 97.4%) as a single oral dose at 0, 500, 1000 or 2000 mg/kg bw, and were observed for 15 days. Neurobehavioural testing, consisting of FOB and motor activity monitoring, was conducted on all rats before treatment (baseline), on day 1 approximately 1-3 h after treatment, and on days 8 and 15. At the end of this period, six rats of each sex per group were perfused with fixative, and samples were taken of nervous and muscle tissue (brain, spinal cord, sciatic/tibial nerves gasserian ganglia, cervical and lumbar dorsal and ventral root fibres and ganglia and gastrocnemius muscle). Only the tissues from the controls and rats in the group receiving the highest dose were examined histologically.
No treatment-related general toxicological effects were observed in the groups given famoxadone at a dose of 500 or 1000 mg/kg bw. Male rats (but not females) at 2000 mg/kg bw group had significantly lower body-weight gain and food consumption over days 1-2. During the remainder of the test period, there were no treatment-related effects on body-weight gain or food consumption values in any group. There were no adverse effects on survival, clinical signs or indicators of neurotoxicity at any dose. An increase in the incidence of palpebral (eyelid) closure during day 1 of FOB monitoring, together with decreased body-weight gain and food consumption in males at 2000 mg/kg bw was interpreted as general malaise. Also, a significant reduction in the number of movements in the motor activity assessment on day 15 in males at 1000 mg/kg bw was not considered to be treatment-related, in view of the absence of any dose-response relationship. No treatment-related pathological effects were observed in this study.
The NOAELs in this assessment of acute neurotoxicity were 1000 mg/kg bw in males and 2000 mg/kg bw in females, on the basis of lower body-weight gain and food consumption and higher incidence of palpebral closure in males at the limit dose of 2000 mg/kg bw and the absence of any effects in females (Malley, 1995a).
In a study of neurotoxic potential, groups of 12 male and 12 female Crl: CD®BR rats were given diets containing famoxadone (purity, 97.4%) at a concentration of 0, 50, 200 or 800 mg/kg for 90 days. These dietary concentrations provided doses equal to 0, 2.9, 11.7 and 46.9 mg/kg bw per day in male rats and 0, 3.7, 14.4 and 59.3 mg/kg bw per day in female rats. Clinical signs, body weights, and food consumption were recorded during the treatment period. Neurobehavioural testing, consisting of FOB and motor activity monitoring, was conducted on all rats before treatment (baseline), on day 1 approximately 1-3 h after compound administration, and in weeks 4, 8 and 13. At the end of this period, six rats of each sex per group were perfused with fixative and samples were taken of nervous and muscle tissue (brain, spinal cord, sciatic/tibial nerves gasserian ganglia, cervical and lumbar dorsal and ventral root fibres and ganglia and gastrocnemius muscle). Only the tissues from rats in the control group and the group receiving the highest dose were examined histologically.
Both male and female rats at 800 mg/kg had decreased body-weight gains, food consumption, and food use efficiency. There were no compound-related effects detected in clinical observations, neurobehavioural evaluations, motor activity, or nervous system morphology. Therefore, evidence of neurotoxicity was not observed at any dose. The NOAEL for male and female rats in this 90-day study of neurotoxicity was 200 mg/kg, equal to 11.7 and 14.4 mg/kg bw per day, respectively, on the basis of general toxicity observed at 800 mg/kg, equal to 46.9 and 59.3 mg/kg bw per day, respectively (Malley, 1995b).
Studies were conducted in mice and rats to evaluate the potential of famoxadone to suppress the primary humoural immune response to sheep erythrocytes. Groups of 10 male and 10 female Crl:CD-1®(ICR)BR mice were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 50, 350, 2000, or 7000 mg/kg for 28 days. These dietary concentrations provided doses equal to 0, 8, 55, 327 or 1186 mg/kg bw per day in male mice and 0, 11, 72, 417 or 1664 mg/kg bw per day in female mice. Groups of 10 male and 10 female Crl: CD®(SD)IGS BR rats were fed diets containing famoxadone (purity, 97.4%) at a concentration of 0, 50, 100, 200, or 800 mg/kg. These dietary concentrations provided doses equal to 0, 4, 7, 14 or 55 mg/kg bw per day in male rats and 0, 4, 8, 16 or 57 mg/kg bw per day in female rats. Body weights, food consumption, and clinical observations were recorded during the test period. To evaluate the primary humoural immune response, all animals were injected intravenously on day 23 (mice) or day 22 (rats) with sheep erythrocytes and killed on test day 28. The spleen and thymus were weighed, and serum was analysed for sheep erythrocyte-specific immunoglobulin M (IgM) antibody.
In mice, there were no effects on body weight, body-weight gain, food consumption, food use efficiency, clinical signs of toxicity, or mortality at any dietary concentration. There were also no treatment-related effects on spleen weights of male mice or thymus weights of male and female mice. Increased spleen weights (29%) were observed in females at 7000 mg/kg, but these were considered secondary to the haematological effects, which although not a subject of this study, had been observed in previous studies. A small but statistically significant decrease in the primary humoural immune response to sheep erythrocytes occurred in male mice at 7000 mg/kg. Although the biological significance of the lowered humoural response was equivocal, the effect was considered to be indicative of minimal immunotoxicity. Famoxadone did not affect the primary humoural response to sheep erythrocytes in female mice at any dietary concentration tested. The NOAEL for immunotoxicity in mice was 2000 mg/kg for males, equal to 327 mg/kg bw per day, and 7000 mg/kg for females, equal to 1664 mg/kg bw per day. The NOAEL for systemic toxicity, other than immunotoxicity, was 7000 mg/kg for males, equal to 1186 mg/kg bw per day, the highest dose tested, and 2000 mg/kg for females, equal to 417 mg/kg bw per day, on the basis of increased spleen weight at 7000 mg/kg, equal to 1664 mg/kg bw per day.
In rats, there was no evidence of immunotoxicity observed in either males or females at any dietary concentration. Decreased body-weight gain (males, 24%; females, 49%), food consumption (males, 13%; females, 18%) and food use efficiency and increased spleen weights (males, 17%; females, 8%) were observed in rats at 800 mg/kg. The increased spleen weights were considered to be secondary to the haematological effects, which, although not a subject of this study, had been observed in previous studies. The NOAEL for immunotoxicity in rats was 800 mg/kg in males and females, equal to 55 and 57 mg/kg bw per day, respectively, the highest dose tested. The NOAEL for systemic toxicity, other than immunotoxicity, was 200 mg/kg in males and females, equal to 14 and 16 mg/kg bw per day, respectively, on the basis of decreases in body-weight gain, food consumption and food use efficiency and increased spleen weights in rats fed famoxadone at a dietary concentration of 800 mg/kg, equal to 55 and 57 mg/kg bw per day (Ladics, 1999a, 1999b).
(c) Pharmacological study in mice and rats
A study was conducted to evaluate the potential pharmacological activity of famoxadone. Groups of 3-10 male Crj: CD-1(ICR) mice or male Crj: Wistar rats were given a single oral dose of famoxadone (purity, 97.0%) at 0, 500, 1500 or 5000 mg/kg bw in corn oil and acetone (85:15). The study was conducted according to Japanese guidelines on agricultural chemicals (1985) and, although the study was not conducted to any particular GLP guidelines, the standard operating procedures of the performing laboratory (Mitsubishi Chemical Safety Institute) were followed.
The animals were observed for effects on the central nervous system (general behaviour assessment), hexobarbital sleeping time, synergistic effect on convulsions following electrical shock, body temperature, cardiovascular system (heart rate, blood pressure), autonomic nervous system (pupil size), gastrointestinal tract (intestinal propulsion), skeletal muscle (traction test), and blood coagulation (prothrombin and activated partial thromboplastin times).
A decrease in locomotor activity during the general behavioural observation period was noted. No significant effects were observed on hexobarbital sleeping time, subthreshold electrical shock, or body temperature. Soft faeces were noted at 500 and 1500 mg/kg bw. As the main component of the vehicle was corn oil, which often induces soft faeces, this effect was attributed to the solvent; however, animals in the control group received the same vehicle. Blood pressure and heart rate, pupil size, intestinal propulsion, traction, and blood coagulation times were not affected by treatment with famoxadone. The conclusion of the study was that there was no clear pharmacological effect produced by famoxadone at up to and including the highest dose tested, 5000 mg/kg bw (Horii, 1997).
(d) Reversibility of effects on erythrocyte mass parameters in rats
In studies in rats receiving repeated doses, described above, exposure to famoxadone at dietary concentrations of >400 mg/kg produced a mild haemolytic anaemia, characterized by reductions in erythrocyte count, haemoglobin concentration, and erythrocyte volume fraction. On the basis of the reticulocytosis and bone-marrow hyperplasia observed in these studies, the anaemia is considered regenerative. In an attempt to confirm the regenerative nature of the observed anaemia, a study was conducted to evaluate the potential reversal of the effects when exposure to famoxadone ceases.
Groups of 10 female Crl: CD®(SD)IGS BR rats were given diets containing famoxadone (purity, 97.3%) at a concentration of 0 or 800 mg/kg for 35 days, and then basal diet only until day 58, when the study was terminated. These dietary concentrations provided doses equal to 0 or 61.6 mg/kg bw per day. Body weight, food consumption, and clinical signs were evaluated weekly. Erythrocyte mass parameters (erythrocyte counts, erythrocyte volume fraction and haemoglobin concentrations) were evaluated at 16 days after first exposure to famoxadone, and every 2 weeks thereafter. Once toxicologically significant reductions were observed, the diet of the rats receiving famaxadone was replaced with control diet. Evaluation of erythrocyte counts, erythrocyte volume fraction and haemoglobin concentrations continued until all values returned to control levels. This study complied with United States EPA FIFRA (40 CFR, part 160) and/or EPA TSCA (40 CFR, part 792) standards for GLP, which are consistent with the OECD principles of GLP (as revised in 1997) published in ENV/MC/CHEM(98)17, and MAFF Japan good laboratory practice standards (59 NohSan No.3850).
After 30 days of exposure to famoxadone at 800 mg/kg, a mild anaemia was produced. The three erythrocyte-mass parameters were reduced by 11-16% below control values. The diet of the rats receiving famoxadone was replaced with the control diet on day 35. Nine days later, the mean erythrocyte volume fractions and haemoglobin concentrations in this group were slightly higher than those for the control rats. By the next time-point at which evaluation of blood parameters was made, after 23 days of exposure to control diet, full recovery from the anaemia had occurred. Erythrocyte counts, erythrocyte volume fractions and haemoglobin concentrations were similar to or greater than the values for the control group at this time.
No compound-related effects on mortality or clinical signs were produced. Lower body weight, body-weight gain, food consumption, and food use efficiency were observed by day 7 in female rats at 800 mg/kg. Once exposure to famoxadone was stopped, no further reductions in these parameters occurred and, during this recovery phase, body-weight gain and food use efficiency values rose above those of the controls (MacKenzie, 2000).
(e) Test for cytotoxicity in lens epithelial cells in vitro
A test for cytotoxicity in vitro with famoxadone (purity, 97.4%) was conducted using primary cultures of canine and primate (mostly rhesus monkey, but not always identified) lenticular epithelial cells, an immortalized mouse lens epithelial cell line (NK-35), and an immortalized human corneal epithelial cell line (SV-40). Cultures were exposed to famoxadone at eight concentrations, ranging from 100 pg/ml to 1 mg/ml, for 3, 24 or 48 h. Cytotoxicity was measured using a dual colour viability assay, which simultaneously assessed intracellular esterase activity and plasma membrane integrity.
Cytotoxicity was demonstrated in all cell cultures, irrespective of the species from which they were derived, when they were exposed to famoxadone at 1 mg/ml for 3, 24, or 48 h. No biologically significant effects were noted below this dose. Overall there were no clear differences observed in the susceptibility of the four cell cultures to cytotoxicity induced by famoxadone (Murphy, 1997).
(f) Other toxicological information
(i) Significance of haematological effects
Multiple exposures to famoxadone produced mild haematological changes, consistent with haemolysis, that were observed in rats, dogs and cynomolgus monkeys, but were less obvious in mice, particularly males. Haematological changes were frequently the basis for the NOAELs.
Functionally, anaemia is defined as a decrease in erythrocyte mass parameters (erythrocyte count, haemoglobin concentration, and erythrocyte volume fraction) to below normal levels (Farver, 1989; Erslev, 1990; Duncan et al., 1994; Sodikoff, 1995; Sasse, 1996). Typically, anaemia is classified within two broad categories—regenerative or non-regenerative—on the basis of the bone-marrow response. In regenerative anaemia, the bone marrow is actively responding by increasing the production of erythrocytes. Evidence of a bone-marrow response is seen as an increase in circulating young erythrocytes (reticulocytes) and an increase in erythropoiesis in bone marrow. In a non-regenerative (aplastic) anaemia, on the other hand, the bone marrow is unable to respond to the anaemic state, owing to reduced or defective erythropoiesis.
The consequence of anaemia is reduced delivery of oxygen to the tissues, an indicator of which is a change in the oxygen-haemoglobin dissociation curve. In humans, this change occurs when the concentration of haemoglobin decreases by about 30-40% (Stehling & Simon, 1994). Similarly, in dogs with experimentally-induced anaemia, transport of oxygen to tissues is not significantly altered until the erythrocyte volume fraction is decreased by about 30% (Fan et al., 1980). Therefore, adverse effects on the organism do not occur until erythrocyte parameters are depressed to a significant degree. Critical decreases in the concentration of haemoglobin and/or erythrocyte volume fraction were not reached in the studies reported here. It could be suggested, therefore, that the NOAELs proposed are conservative. However, it is well known that there are species differences in the sensitivity of erythrocytes to potentially haemolytic agents. While comparisons of the effects in vitro of a chemical (and its identified mammalian or plant metabolites) on erythrocytes from different species, including humans, are relatively easy to perform, such comparisons have not been performed with famoxadone. Consequently, at this stage, it is not possible to conclude with any confidence that the NOAELs identified on the basis of anaemia are indeed conservative when making the interspecies extrapolation to humans.
(ii) Significance of lens effects in studies in dogs treated with repeated doses
The lens does not contain pigment or blood vessels, which would decrease its transparency. Consequently, the lens derives almost all its metabolic requirements from the aqueous humour, which is also the medium through which systemically available xenobiotics are delivered to the lens. Any opacity of the lens and its capsule is termed a cataract. Lenticular opacities may result from numerous different mechanisms (Brown & Bron, 1996), but for the vast majority of chemicals that induce cataracts these are unknown. Chemicals with widely differing structures and pharmacological activities have been reported in the literature to cause cataract in laboratory animals. In a survey of ocular toxicological profiles, the correlation between toxicity in rodent and in non-rodents was not established; few compounds are known to cause cataracts in both rats and non-rodents (Heywood, 1981, 1983). Since the toxic effects of various substances on the lens are quantitatively very different in different species, extrapolation of experimental animal data to humans must be done with caution.
Mechanisms leading to lens opacity include: changes in osmotic pressure leading to an increase in water content, denaturation of lens proteins such as crystallins (which are critical for lens clarity), and generation of reactive oxygen radicals resulting in oxidative damage (Basher & Roberts, 1995; Clang & Aleo, 1997). The normal lens is in a "dehydrated" state. This state strongly depends on the proper functioning of the ion and water pumps that derive their energy from ATP delivered by lens epithelium and superficial lens fibres. The lens osmolarity is maintained by cations (sodium, Na+ and potassium, K+) and anions (chloride, bicarbonate, sulfate, ascorbate, glutathione, acidic groups of lens proteins and glycoproteins). The observation that the study in dogs fed with high dietary concentrations of famoxadone (1000 reduced to 600 mg/kg) resulted in an increase in serum K+, indicating an effect on ion homeostasis at toxic doses, could therefore have a bearing on cataract formation. However, the increase in K+ was significant only at this high dose, while lenticular damage was also found in dogs given diets containing faoxadone at 300 mg/kg. Another general mechanism implicated in cataract formation is activation of calcium-dependent proteases (calpains) by various agents (Clang & Aleo, 1997). Alterations in calcium homeostasis by a variety of mechanisms could, therefore, produce cataracts. There are no indications, however, that this happens in dogs treated with famoxadone.
The known biochemical mechanism of action of famoxadone is inhibition of the mitochondrial respiratory chain at complex III (ubiquinol: cytochrome c oxidoreductase), resulting in decreased production of ATP by the cell. ATP is essential for maintenance of cellular metabolism and the function of ATP-dependent cellular enzymes, in particular the Na+-K+-ATPase that is critical for maintenance of normal cellular hydration. Therefore, reduction in cellular ATP could result in an increase in lens hydration and subsequent cataract formation. It is likely that this mechanism would require famoxadone to be present in the eye at above a certain threshold concentration. However, studies of distribution indicate that the concentrations likely to be found in the eye of a dog are low (Thalacker, 1996), while comparative data from other species are not available. Whether the concentrations would be sufficient to uncouple oxidative phophorylation in the dog eye seems unlikely, since there is no evidence of mitochondrial toxicity in vivo in other organs in which the concentration of famoxadone may be 30-40-fold higher. However, metabolic studies conducted with famoxadone in rats and dogs demonstrated a longer half-life for radioactivity (parent and/or metabolite) in the dog, but no major qualitative differences in metabolic profile were found and the quantitative differences were small.
In addition, ATP is required to maintain the reducing power of the cell, thus a decrease in ATP (unlikely as this may be) would increase susceptibility to oxidative stress, which is also implicated in cataract formation. Dogs have been demonstrated to be relatively deficient (compared with primates) in numerous mechanisms that protect against oxidative stress. Thus, in an interspecies comparison of glutathione (GSH) reductase activity in the lens, carnivores were found to have the lowest GSH reductase activities, while non-human primates (including cynomolgus monkeys) and humans had the highest activities (Rathbun et al., 1986). GSH reductase is the rate-limiting enzyme in the GSH redox cycle, which can protect cells against oxidative damage, particularly from hydroperoxides. Hence cells with lower activities of this enzyme will be in a more vulnerable position, should such substances be produced. These lower activities in dogs may explain the observed increased sensitivity of this species to lens toxicity after exposure to famoxadone, but they do not render the dog unique.
Ascorbic acid can also act as a reducing agent, thereby protecting the cell against oxidative reactions initiated by oxygen and free radicals. It is present at unusually high concentrations in primate, including human, eyes and can be found in the cornea, aqueous humour, lens, vitreous humour and retina. Humans and monkeys contain identical concentrations of ascorbate in their aqueous humour and lens, (1.0 and 1.25mmol/l, respectively). In comparison, the concentrations of ascorbate in the aqueous humour and lens of dogs are 0.31 and 0.2 mmol/l, respectively. In one study, the concentration of ascorbate in the aqueous humour of human patients with cortical cataracts was significantly lower than in that of unaffected patients, suggesting that low ascorbate might predispose to cataract formation by reducing the antioxidant function in the lens (Varma & Richards, 1988).
Exposure to famoxadone resulted in lens opacities and cataracts in outbred beagle dogs. These lesions (generally bilateral in occurrence) were observed after exposure to low doses of famoxadone in 90-day and 1-year studies. Exposures of similar and longer durations did not produce cataracts in mice or cynomolgus monkeys, despite exposure at much higher doses. Microscopically, however, cataracts were identified in the 18-month study in mice treated orally and in the 24-month study in rats treated orally. In mice there was clearly no dose-response relationship and in rats the excess incidence among males occurred at 40 and 400 mg/kg, but not at 200 mg/kg.
An additional review of the animal data and microscopic examination of eye sections from the studies in dogs treated with famoxadone was undertaken (Heywood, 1999); this review reached the same conclusions as those made by the original investigators, namely, that cataracts were induced by famoxadone in beagle dogs, but not in mice, rats or cynomolgus monkeys.
There are no qualitative differences between mammalian species and strains with respect to the differentiation process of lens fibres. Thus, there is not sufficient support at the moment to be able to conclude that the observed posterior subcapsular cataracts, most of which developed after 2-3 months treatment, and equatorial lens opacities that developed after 6-12 months treatment, are species-specific and restricted to beagle dogs. Such a conclusion would require a full understanding of the mechanism of action of famoxadone on the eyes in the various species and knowledge of the concentration of famoxadone in the aqueous humour and the lens.
The absence of effects in monkeys may be due in part to the relatively short period of exposure relative to the lifespan of monkeys, or to use of non-toxic doses of the test substance. The period of exposure might have been too short to lead to manifest equatorial and posterior subcapsular changes that were detectable using routine clinical and histopathological methods. It is noted that cynomolgus monkeys seem generally to be less sensitive to the toxic effects of famoxadone; the only observed effect was a mild anaemia at 1000 mg/kg bw per day. With regard to the presence or absence of treatment-related increases in cataracts in rats, it is noted that there is evidence that albino and pigmented rats have different sensitivities to cataractogenic factors, albino rats being less sensitive (Wegener & Eiben, 1992; Eiben & Wegener, 1995).
No information was available.
Studies in rats show that about 40% of the administered dose of radiolabelled famoxadone is absorbed and rapidly eliminated from the body in the faeces (>75% in 24 h) and urine (about 10% in 24 h). Most of the administered dose found in the faeces is unmetabolized famoxadone. In rats, absorption from the gastrointestinal tract becomes the limiting factor for internal exposure at doses greater than about 800 mg/kg bw. It appears that there are no important differences in the metabolism of famoxadone between dogs and rats, within the limits imposed by the different doses used, and that there were no significant differences between male and female rats (only males having been used in the experiments with dogs). The primary metabolic pathway involved the hydroxylation of the parent molecule to the corresponding mono- and dihydroxylated derivatives, which were only recovered from the faeces. Metabolites resulting from the cleavage of the oxazolidinedione ring moiety were recovered from the urine. A sulfate was the major urinary metabolite containing the phenoxyphenyl moiety, whereas 4-acetoxyaniline was the major urinary metabolite containing the phenylamino moiety. No parent famoxadone was detected in the urine.
Famoxadone has low acute toxicity when administered by oral, dermal, and inhalation routes. The acute LD50 after oral administration is >5000 mg/kg bw in rats and the LD50 after dermal administration is >2000 mg/kg bw in rabbits. The LC50 in rats after 4 h is >5300mg/m3, the only concentration tested. Famoxadone produces transient mild dermal irritation and transient mild ocular irritation, but does not cause skin sensitization.
In short-term studies of oral administration in rodents, dogs and non-human primates, and in long-term studies of oral administration in rodents, NOAELs for famoxadone were identified on the basis of effects on body weight and nutrition, mild haemolytic anaemia, and/or mild to moderate liver toxicity. Mild regenerative haemolytic anaemia was found in rats, mice, dogs and monkeys, as indicated by decreased erythrocyte counts, haemoglobin and/or erythrocyte volume fraction, increased reticulocyte counts, or other related changes in haematological parameters. Methaemoglobin formation was not measured. Secondary effects of anaemia were also found in the spleen (e.g. increased spleen weight, deposition of haemosiderin pigment, extra-medullary haematopoiesis), in the bone marrow (compensatory erythropoiesis), and in the liver (increased Kupffer cell pigment, increased bile pigment). In studies involving repeated dosing, anaemia was found to occur early in the study and often appeared to be compensated for later. In an experiment in which rats were fed famoxadone as a single dose at 800 mg/kg, equal to 61.6 mg/kg bw per day, blood samples were taken at multiple time-points. Mild anaemia was observed after 30 days, but not after 16 days. Famoxadone also induced hepatocellular responses that are normally considered to be adaptive (e.g. enlarged livers, increased liver weights and liver:body-weight ratios, hepatocellular hypertrophy). These adaptive responses were characterized by increased quantities of cytochrome P450 and/or increased rates of peroxisomal beta-oxidation. Hepatotoxicity, which was mild, was observed only at higher doses and was characterized by mild histopathological lesions (e.g. single cell or focal necrosis, hepatocellular degeneration, diffuse fatty change, eosinophilic foci) and marginally elevated concentrations of blood enzymes suggestive of liver damage. The NOAELs after short-term oral administration were 62.4 mg/kg bw per day in mice treated for 3 months, 13 mg/kg bw per day in rats treated for 3 months, 1.2 mg/kg bw per day in dogs treated for 1 year and 100 mg/kg bw per day in cynomolgus monkeys treated for 1 year.
Long-term studies in rats (2 years) and mice (18 months) show little evidence of irreversible organ toxicity, although there was an increased incidence of generalized amyloidosis among female mice receiving famoxadone in the diet. Other effects that were observed, some of which formed the basis for the NOAELs, were reductions in body-weight gain, hepatotoxicity and mild regenerative anaemia. The NOAELs for long-term toxicity were 700 mg/kg, equal to 96 mg/kg bw per day, in mice, and 200 mg/kg, equal to 8.4 mg/kg bw per day, in rats. There was no evidence of carcinogenic potential with famoxadone at doses up to the highest tested, which was 400 mg/kg, equal to 17 mg/kg bw per day, in rats and 7000 mg/kg, equal to 96 mg/kg bw per day, in mice.
Famoxadone was tested for genotoxicity in an adequate range of studies, both in vitro and in vivo. The results observed were largely negative. Although in one study famoxadone produced a weak clastogenic effect in vitro, the Meeting did not consider this to be toxicologically significant.
The Meeting concluded that famoxadone is unlikely to pose a genotoxic risk to humans.
Because the results of the studies of carcinogenicity were negative, the Meeting concluded that famoxadone is unlikely to pose a carcinogenic risk to humans.
In a two-generation study of reproductive toxicity in rats, the NOAEL for adult rats and their offspring was 200 mg/kg, equal to 11.3 mg/kg bw per day in adults, on the basis of systemic toxicity in the parental rats and reduced body-weight gain in the offspring at a dose of 800 mg/kg, equal to 45 mg/kg bw per day; no other signs of reproductive toxicity were observed at this dose, the highest tested. In studies of developmental toxicity in rats and rabbits, no effects were observed in fetuses at doses of 1000 mg/kg bw per day, the highest dose tested. The results from the two studies of developmental toxicity and the study of reproductive toxicity did not reveal any increased susceptibility of fetuses or pups to famoxadone.
In 28-day studies of immunotoxicity in rats and mice, no evidence of immunotoxic-ity was found in rats receiving diets containing famoxadone at a concentration of 800 mg/kg, equal to 55 and 57 mg/kg bw per day in males and females respectively, or in mice receiving diets containing famoxadone at a concentration of 7000 mg/kg, equal to 1664 mg/kg bw per day in females, the highest doses tested. In male mice, there was a minimal but significant reduction in the primary humoural response to sheep erythrocytes at a dose of 7000 mg/kg; the NOAEL for this activity in male mice was thus 2000 mg/kg, equal to 327 mg/kg bw per day. The toxicological significance of this effect was considered to be minimal.
Clinical and microscopic evidence of lens opacities was clearly observed in female and male dogs (in both the 3-month and 1-year studies), at doses below those at which any other effects were observed in any other species. The mechanism by which these effects are induced is not understood.
Famoxadone does not appear to be neurotoxic. Some observations of minor effects made in an experiment investigating acute neurotoxicity were attributed to general malaise. Other than some clinical observations in males and females fed famoxadone at the highest dose in the 3-month study in dogs (myotonic twitching, possibly a result of high concentrations of serum potassium), no evidence for neurotoxicity was found in any other studies of toxicity, including a short-term study of neurotoxicity in rats.
The Meeting concluded that the existing database on famoxadone was adequate to characterize the potential hazard to fetuses, infants and children.
An ADI of 0-0.006 mg/kg bw was established for famoxadone based on the NOAEL of 1.2 mg/kg bw per day in a 1-year study in dogs treated by gavage, with a safety factor of 200; an extra safety factor was added because this study in dogs is not viewed as a long-term study. The critical effect was the occurrence of cataracts in dogs at 300 mg/kg, equal to 8.8 mg/kg bw per day; some of these cataracts developed late in the study, indicating that progression might have been possible, had a long-term study been conducted.
The Meeting established an acute reference dose (RfD) of 0.6 mg/kg bw for famoxadone on the basis of a NOAEL of 61.6 mg/kg bw per day, the only dose tested, in a study of haematotoxicity in rats treated for 16 days and a safety factor of 100.
Levels relevant to risk assessment
|
Species |
Study |
Effect |
NOAEL |
LOAEL |
|
Mouse |
18-month study of toxicity and carcinogenicitya |
Toxicity |
700 mg/kg, equal to |
2000 mg/kg, equal to |
|
Carcinogenicity |
7000 mg/kg, equal to |
— |
||
|
Rat |
2-year study of toxicity and carcinogenicitya |
Toxicity |
200 mg/kg, equal to |
400 mg/kg, equal to |
|
Carcinogenicity |
400 mg/kg, equal to |
— |
||
|
Two-generation study of reproductive toxicitya |
Parental toxicity |
200 mg/kg, equal to |
800 mg/kg, equal to |
|
|
Offspring toxicity |
800 mg/kg, equal to |
800 mg/kg, equal to |
||
|
Study of developmental toxicityb |
Maternal toxicity |
250 mg/kg bw per day |
500 mg/kg bw per day |
|
|
Offspring toxicity |
1000 mg/kg bw per dayc |
— |
||
|
Special study of haematotoxicitya |
Anaemia |
800 mg/kg, equal to |
800 mg/kg, equal to |
|
|
Single-dose study of neurotoxicityb |
Neurotoxicity |
2000 mg/kg bwc |
— |
|
|
3-month study of neurotoxicitya |
Neurotoxicity |
800 mg/kg, equal to |
— |
|
|
Rabbit |
Study of developmental toxicityb |
Maternal toxicity |
1000 mg/kg bw per dayc |
|
|
Offspring toxicity |
1000 mg/kg bw per dayc |
— |
||
|
Dog |
1 -year study of toxicitya |
Toxicity |
40 mg/kg, equal to |
300 mg/kg, equal to |
a Diet
b Gavage
cHighest dose tested
Estimate of acceptable daily intake for humans
0-0.006 mg/kg bw
Estimate of acute reference dose
0.6 mg/kg bw
Studies that would provide information useful for continued evaluation of the compound
Summary of critical end-points for famoxadone
|
Absorption, distribution, excretion and metabolism |
|
|
Rate and extent of oral absorption |
About 40% absorbed and >75% of the administered dose eliminated in faeces in 24 h |
|
Dermal absorption |
No study of direct dermal absorption available |
|
Distribution |
Distributed throughout the body; tissue residues generally very low; highest concentrations in liver and fat |
|
Potential for accumulation |
Low, due to rapid excretion |
|
Rate and extent of excretion |
>75% excretion within 24 h |
|
Metabolism in animals |
Extensive |
|
Toxicologically significant compounds |
Parent |
|
Acute toxicity |
|
|
Rat, LD50, oral |
>5000 mg/kg bw |
|
Rat, LD50, dermal |
No data |
|
Rat, LC50, inhalation |
5.3 mg/l (4 h) |
|
Rabbit, LD50, dermal |
>2000 mg/kg bw |
|
Rabbit, dermal irritation |
Mild irritant |
|
Rabbit, ocular irritation |
Mild irritant |
|
Skin sensitization |
Not sensitizing (Magnusson and Kligman) |
|
Short-term studies of toxicity |
|
|
Target/critical effect |
Body-weight gain decrement, hepatotoxicity, regenerative haemolytic anaemia and lens opacities |
|
Lowest relevant oral NOAEL |
1.2 mg/kg bw per day (1-year study in dogs) |
|
Lowest relevant dermal NOAEL |
250 mg/kg bw per day (28-day study in rats) |
|
Lowest relevant inhalation NOAEC |
No data available |
|
Genotoxicity |
No genotoxic potential |
|
Long-term toxicity and carcinogenicity |
|
|
Target/critical effect |
Decreased body-weight gain, hepatotoxicity and regenerative haemolytic anaemia |
|
Lowest relevant NOAEL |
8.4 mg/kg bw per day: (2-year study in rats) |
|
Carcinogenicity |
No carcinogenic potential |
|
Reproductive toxicity |
|
|
Reproductive target/critical effect |
Reduced parental and offspring body weight, clinical signs |
|
Lowest relevant reproductive NOAEL |
11 mg/kg bw per day |
|
Developmental target/critical effect |
Not teratogenic embryotoxic or fetotoxic |
|
Lowest relevant developmental NOAEL |
>1000 mg/kg bw per day (rats) |
|
Neurotoxicity/delayed neurotoxicity |
|
|
Target/critical effect |
None |
|
Lowest relevant NOAEL |
>1000 mg/kg bw |
|
90-day study of neurotoxicity |
|
|
Target/critical effect |
None |
|
Lowest relevant NOAEL |
>47 mg/kg bw per day |
|
Other toxicological studies |
None available |
|
Medical data |
None available |
|
Summary |
Value |
Study |
Safety factor |
|
ADI |
0-0.006 mg/kg bw |
Dog, 1 -year study, cataracts |
200 |
|
Acute RfD |
0.6 mg/kg bw |
Rat, study of haematotoxicity in rats, haemolytic anaemia |
100 |
Basher, A.W.P. & Roberts, S.M. (1995) Ocular manifestations of diabetes mellitus: diabetic cataracts in dogs. Vet. Clin. North Amer., 25, 661-676.
Bentley, K.S. (1994) Assessment of DPX-JE874-221 in the in vitro unscheduled DNA synthesis assay in primary rat hepatocytes. Unpublished report No. HLR 533-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Bentley, K.S. (1995) Mutagenicity testing of DPX-JE874-221 in the Salmonella typhimurium and Escherichia coli plate incorporation assay. Unpublished report No. HLR 707-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Biegel, L.B. (1994) Subchronic oral toxicity: 90-day study with DPX-JE874-65 feeding study in mice. Unpublished report No. HLR 73-93 (two volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Brown, N.A.P. & Bron, A.J. (1996) Lens disorders: a clinical manual of cataract diagnosis. Chapter 12, Oxford: Butterworth-Heinemann Ltd.
Bursch, W., Taper, H.S., Lauer, B. & Schulte-Hermann, R. (1985) Quantitative and histochemical studies on the occurrence and stages of controlled cell death (apoptosis) during regression of rat liver hyperplasia. VirchowArch. [Cell Pathol.], 50, 153-166.
Cifone, M.A. (1999a) Famoxadone technical (DPX-JE874): in vitro mammalian cell gene mutation test (CHO/HGPRT). Unpublished report No. DuPont-1821 from Covance, Vienna, Virginia, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Cifone, M.A. (1999b) Famoxadone technical (DPX-JE874): unscheduled DNA synthesis in mammalian cells in vitro. Revison No.1. Unpublished report No. DuPont-1822 from Covance, Vienna, Virginia, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA
Clang, C.M.W. & Aleo, M.D. (1997) Mechanistic analysis of S-(1,2-dichlorovinyl)-L-cysteine-induced cataractogenesis in vitro. Toxicol. Appl., Pharmacol., 146, 144-155.
Duncan, J.R., Prasse, K.W. & Mahaffey, E.A. (1994) Erythrocytes. In: Veterinary Laboratory Medicine: Clinical Pathology, Ames, Iowa: Iowa State University Press, pp. 3-36.
Eiben, R. & Wegener, A. (1995) Comparative investigations on the cataractogenic effect of a triazin-derivate in albino and pigmented rats: I. Effects detected with a slitlamp. In: Weisse, I., et al., eds, Ocular Toxicology, New York: Plenum Press, pp. 211-218.
Erslev, A.J. (1990) Clinical manifestation and classification of erythrocyte disorders. In: Williams W.J., Beutler, E., Erslev, A.J. & Lichtman, M.A., eds, Hematology, 4th Ed., New York: McGraw-Hill Inc., pp. 423-429.
Fan, F-C., Chen, R.Y.Z, Schuessler, G.B. & Chien. S. (1980) Effects of hematocrit variations on regional hemodynamics and oxygen transport in the dog. Am. J. Physiol., 238, H545-H552.
Farver, T.B. (1989) Concepts of normality in clinical chemistry. In: Kaneko, J.J., ed., Clinical Biochemistry of Domestic Animals, 4th Ed., San Diego: Academic Press Inc., pp. 1-20.
Fellows, M. (1998) DPX-JE874-221: Measurement of unscheduled DNA synthesis in rat liver using an in vivo/in vitro procedure. Revision No. 2. Unpublished report No. HLO-1998-01212 from Covance, Vienna, Virginia, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Finlay, C. (1994a) Acute oral toxicity study with DPX-JE874-221 in male and female mice. Unpublished report No. HLR 128-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Finlay, C. (1994b) Primary eye irritation study with DPX-JE874-221 in rabbits. Unpublished report No. HLR 60-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Finlay, C. (1998) Famoxadone Technical (DPX-JE874): Primary dermal irritation study in rabbits. Unpublished report No. DuPont-1800 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Frame, S.R. (1998) Chronic toxicity study with DPX-JE874-221 one year feeding study in dogs. Supplement No. 1 to unpublished report No. HLO 820-95 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Frame, S.R. & Sykes, G.P. (1999) Combined chronic toxicity/oncogenicity study with DPX-JE874-221. Two-year feeding study in rats. Supplement No. 2 to unpublished report No. HLR 527-95 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Gerber, K.M. (1995) In vitro evaluation of DPX-JE874-221 for chromosome aberrations in human lymphocytes. Unpublished report No. HLR 25-95 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Ghantous, H.N. (1999) Repeated dose oral toxicity: two-week feeding study with DPX-JE874-65 in mice. Revision No. 1. Unpublished report No. HLR 563-92 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Harada, T., Maronpot, R.R., Morris, R.W. & Boorman, G.A. (1989) Observations on altered hepatocellular foci in National Toxicology Program two-year carcinogenicity studies in rats. Toxicol. Pathol., 17, 690-706.
Harrison R. (1998) [14C-PA]DPX-JE874: determination of plasma and investigation of the number and nature of radiolabelled metabolites in excreta and selected tissues from male beagle dogs. Revision No. 1. Unpublished report No. HLO-1997-00761 from Covance, Vienna, Virginia, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Heywood R. (1981) Target organ toxicity. Toxicol. Lett., 8, 349-358.
Heywood, R. (1983) Target organ toxicity II, Toxicol. Lett., 18, 83-88.
Heywood, R. (1999) Famoxadone (DPX-JE874): relevance of ocular findings in animals for human risk. Unpublished report No. DuPont-3351 from R. Heywood. The Larches, The Lanes, Houghton, Huntingdon, UK. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Himmelstein, M.W. (1999a) Absorption, excretion, distribution and metabolism of [14C] DPX-JE874 in rats. Supplement No. 2 to unpublished report No. AMR 2440-92 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Himmelstein, M.W. (1999b) Famoxadone technical and 14C-DPX-JE874: the effect of increasing dietary feeding concentration and subsequent radiolbelled oral gavage dosing on plasma concentrations in mice. Unpublished report No. DuPont-3114 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Horii, D. (1997) General pharmacological study of famoxadone. Unpublished Study No. 7L518 [English Translation] from Mitsubishi Chemical Safety Institute Ltd., Kashima Laboratory, Ibaraki, Japan. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Kreckmann, K.H. (1995) Reproductive and fertility effects with DPX-JE874-221 multigeneration reproduction study in rats. Unpublished report No. HLR 238-95 (five volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Kuykendall, J.R. (1994) Mouse bone marrow micronucleus assay of DPX-JE874-221. Unpublished report No. HLR 96-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Ladics, G.S. (1998) Famoxadone technical (DPX-JE874): 28-day repeated dose dermal toxicity study in rats. Unpublished report No. DuPont-1549 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Ladics, G.S. (1999a) Famoxadone technical (DPX-JE874): 28-day immunotoxicology study in mice. Unpublished report No. DuPont-2125 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Ladics, G.S. (1999b) Famoxadone technical (DPX-JE874): 28-day immunotoxicology study in rats. Unpublished report No. DuPont-1798 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1992) Repeated dose oral toxicity: 14-day feeding study with IN JE874-36 in male and female rats. Unpublished report No. HLR 737-91 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1995) Subchronic oral toxicity: 90-day study with DPX-JE874-65 feeding study in rats. Unpublished report No. HLR 123-93 (two volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1996a) Pilot studies with DPX-JE874-133 in mice effects on clinical chemistry and biochemistry parameters. Unpublished report No. HLR 244-96 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1996b) Pilot study with DPX-JE874-133 in rats effects on clinical chemistry. Unpublished report No. HLR 305-96 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1996c) Oncogenicity study with DPX-JE874-221. Eighteen-month feeding study in mice. Unpublished report No. HLR 526-95 (five volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1996d) Combined chronic toxicity/oncogenicity study with DPX-JE874-221. Two-year feeding study in rats. Unpublished report No. HLR 527-95 (nine volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (1999) Subchronic oral toxicity: 90-day study with DPX-JE874-65; feeding study in rats. Supplement No. 2 to unpublished report No. HLR 123-93 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (2000) Famoxadone technical: reversibility study of red blood cell mass effects in female rats. Unpublished report No. DuPont-3863 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mackenzie, S.A. (2002) Famoxadone (DPX-JE874) technical: oncogenicity eighteen-month feeding study in mice. Unpublished report No. DuPont-4294 (five volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Malley, L.A. (1995a) Acute neurotoxicity study of DPX-JE874-221 in rats. Unpublished report No. HLR 513-94 (two volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Malley, L.A. (1995b) Subchronic oral neurotoxicity study of DPX-JE874-221 in rats. Revision No. 1. Unpublished report No. HLR 239-95 RV1 (two volumes) from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Mertens, J.J.W.M. (1996) Chronic toxicity study with DPX-JE874-221. One-year feeding study in dogs. Unpublished report No. HLO 820-95 (four volumes) from WIL. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Moore, G.E. (1994) Delayed contact hypersensitivity test (maximization method) with DPX-JE874-221 in guinea pigs. Revision No. 1. Unpublished report No. HLO 33-94 from Biosearch Incorporated, Philadelphia, PA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Munley, S.M. (1999a) Developmental toxicity of DPX-JE874-221 in rats. Supplement No. 1 to unpublished report No. HLR 375-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Munley, S.M. (1999b) Developmental toxicity of DPX-JE874-221 in rabbits. Revision No. 1. Unpublished report No. HLR 479-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Munley, S.M. (1999c) Developmental toxicity of DPX-JE874-221 in rabbits. Supplement No. 1 to unpublished report No. DuPont HLR 479-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Murphy, C.J. (1997) In vitro lens epithelial cell toxicity study with DPX-JE874-221. Unpublished report No. HLO-1997-01010 from Comparative Ophthalmic Research Laboratories, College of Veterinary Medicine, University of Wisconsin, Wisconsin, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Murray, S.M. (1994) Developmental toxicity of DPX-JE874-221 in rats. Unpublished report No. HLR 375-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
O'Connor, J.C. (1999) Famoxadone: investigation of cytochrome P-450 induction in a 14-day feeding study using rats and mice. Unpublished report No. DuPont-3280 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
O'Neill, A.J. (1994) Inhalation median lethal concentration (LC50) study with DPX-JE874-158 in rats. Unpublished report No. HLR 791-93 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Rathbun, W.B., Bovis, M.G. & Holleschau, A.M. (1986). Species survey of glutathione peroxidase and glutathione reductase: search for an animal model of the human lens. Ophthalmic Res., 18, 282-287.
Saik, J.E. (1995) Subchronic oral toxicity: 90-day study with DPX-JE874-221 study in dogs. Supplement No. 1 to unpublished report No. HLO 500-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Saik, J.E. (1994) Subchronic oral toxicity: 90 day study with DPX-JE874-65 feeding study in mice. Supplement No. 1 to unpublished report No. HLR 73-93 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sarver, J.W. (1994a) Acute oral toxicity study with DPX-JE874-221 in male and female rats. Revision No. 1. Unpublished report No. HLR 97-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sarver, J.W. (1994b) Acute dermal toxicity study with DPX-JE874-221 in rabbits. Unpublished report No. HLR 91-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sarver, J.W. (1994c) Primary dermal irritation study with DPX-JE874-221 in rabbits. Unpublished report No. HLR 67-94 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sasse, E.A. (1996). Reference intervals and clinical decision limits. In: Kaplan, L.A. & A. J. Pesce, A.J., eds, Clinical Chemistry, Theory, Analysis, Correlation, 3rd Ed., St Louis: Mosby, pp. 365-381.
Savides, M.C., Lee, D.Y., Laveglia, J. & Lee, P.W. (1995) Absorption, excretion, distribution and metabolism of [14C]DPX-JE874 in rats. Unpublished report No. AMR 2440-92 from Ricerca Inc., Painesville, Ohio, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Savides, M.C., Lee, D.Y., Laveglia, J. & Lee, P.W. (1996) Absorption, excretion, distribution and metabolism of [14C] DPX-JE874 in rats. Supplement No. 1 to unpublished report No. AMR 2440-92 from Ricerca, Inc, Painesville, Ohio. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Savides, M.C., McClanahan, R.H. & Delisio, P.L. (1997) Biliary excretion of [14C]DPX-JE874 in rats. Unpublished report No. AMR-3707-95 from Ricerca Inc., Painesville, Ohio, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Slone, T.W. (1997) Oncogenicity study with DPX-JE874-221. Eighteen-month feeding study in mice. Supplement No. 1 to unpublished report No. HLR 526-95 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Slone, T.W. (1991) Repeated dose oral toxicity: 14-day feeding study with In JE874-36 in rats. Unpublished report No. (Pathology Report) 77-91 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sodikoff, C.H. (1995) Diagnosis by hormonal and hematologic findings. In: Laboratory Profiles of Small Animal Diseases, St Louis: Mosby, pp. 207-231.
Stehling, L. & Simon, T.L. (1994) The red cell transfusion trigger: Physiology and clinical studies. Arch. Pathol. Lab. Med., 118, 429-434.
Sykes, G.P. (1998) 52-week oral gavage toxicity study with DPX-JE874-221 in cynomolgus monkeys (final report). Supplement No. 1 to unpublished report No. HLO-1997-00583 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sykes, G.P. & Frame, S.R. (1997) Combined chronic toxicity/oncogenicity study with DPX-JE874-221. Two-year feeding study in rats. Supplement No. 1 to unpublished report No. HLR 527-95 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Sykes, G.P. (1995) Subchronic oral toxicity: 90-day study with DPX-JE874-65; feeding study in rats. Supplement No. 1 to unpublished report No. HLR 123-93 from DuPont Haskell Laboratory, Newark, Delaware, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Thalacker, F. (1996) Absorption, metabolism, and excretion of [14C-PA]DPX-JE874 following single oral doses in male beagle dogs. Unpublished report No. HLO 247-96 from Corning Hazleton Inc., Madison, Wisconsin, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Tompkins, E.C. (1994) Four-week palatability and toxicity study with DPX-JE874-221. feeding study in dogs. Unpublished report No. HLO 372-94 from WIL Research Laboratories Inc. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Tompkins, E.C. (1995) Subchronic oral toxicity: 90-day study with DPX-JE874-221. Study in dogs. Unpublished report No. HLO 500-94 (three volumes) from WIL Research Laboratories Inc. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Varma, S.D. and Richards, R.D. (1988). Ascorbic Acid and the Eye Lens. Ophthalmic Res., 20, 164-173.
Wegener, A. & Eiben, R. (1992) Comparative investigations on the cataractogenic effect of a triazin-derivative in albino and pigmented rats: II. Effects documented by Scheimpflug photography. Lens Eye Toxic. Res., 9, 321-328.
Williams, K.D. (1997) 52-week oral gavage toxicity study with DPX-JE874-221 in cynomolgus monkeys. Unpublished report No. HLO-1997-00583 (two volumes) from Corning Hazleton Inc., Madison, Wisconsin, USA. Submitted to WHO by E.I. du Pont de Nemours and Company, Wilmington, Delaware, USA.
Famoxadone and its metabolites in rats and dogs
The compounds shown below were found in one or more studies of the metabolism of DPX-JE874 (famaxadone) in rats and dogs. Compounds are organized by ascending IN code, which is given in the upper left-hand corner for each molecule.
|
IN-03492 |
CAS name: |
1,2-benzenediol |
||
|
|
||||
|
CAS number: |
120-80-9 |
Relative molecular mass: |
110.11 |
|
|
Structural formula: |
C6H6O2 |
Observed in: |
rats |
|
|
IN-BY759 |
CAS name: |
4-aminophenyl acetate |
||
|
|
||||
|
CAS number: |
13871-68-6 |
Relative molecular mass: |
151.17 |
|
|
Structural formula: |
C8H9NO2 |
Observed in: |
rats |
|
|
IN-H3310 |
CAS name: |
1 -(4-phenoxyphenyl)ethanone |
||
|
|
||||
|
CAS number: |
5031-78-7 |
Relative molecular mass: |
212.25 |
|
|
Structural formula: |
C14H12O2 |
Observed in: |
rats |
|
|
IN-JE874 |
CAS name: |
5-methyl-5-(4-phenoxyphenyl)-3-(phenylamino)-2,4-oxazolidinedione |
||
|
|
||||
|
CAS number: |
131807-57-3 |
Relative molecular mass: |
374.40 |
|
|
Structural formula: |
C22H18N2O4 |
Observed in: |
active ingredient |
|
|
IN-JL856 |
CAS name: |
alpha-hydroxy-alpha-methyl-4-phenoxybenzeneacetic acid 2-phenylhydrazide |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
348.41 |
|
|
Structural formula: |
C21H2ON2O3 |
Observed in: |
rats, dogs |
|
|
IN-KZ000 |
CAS name: |
5-[4-(4-hydroxyphenoxy)phenyl]-5-methyl-2,4-oxazolidinedione |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
299.29 |
|
|
Structural formula: |
C16H13NO5 |
Observed in: |
rats |
|
|
IN-KZ007 |
CAS name: |
5-[4-(4-hydroxyphenoxy)phenyl]-5-methyl-3-(phenylamino)-2, 4-oxazolidinedione |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
390.40 |
|
|
Structural formula: |
C22H18N2O5 |
Observed in: |
rats, dogs |
|
|
IN-KZ532 |
CAS name: |
3-[(4-hydroxyphenyl)amino]-5-methyl-5-(4-phenoxyphenyl)-2, 4-oxazolidinedione |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
390.40 |
|
|
Structural formula: |
C22H18N2O5 |
Observed in: |
dogs |
|
|
IN-KZ534 |
CAS name: |
5-[4-(4-hydroxyphenoxy)phenyl]-3-[(4-hydroxyphenyl)amino]-5-methyl-2,4-oxazolidinedione |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
406.40 |
|
|
Structural formula: |
C22H18N2O6 |
Observed in: |
rats, dogs |
|
|
IN-ML436 |
CAS name: |
alpha-hydroxy-4-(4-hydroxyphenoxy)-alpha-methyl-4- phenoxybenzeneaceticacid |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
274.28 |
|
|
Structural formula: |
C15H14O5 |
Observed in: |
rat |
|
|
IN-ML815 |
CAS name: |
alpha-hydroxy-4-(4-hydroxyphenoxy)-alpha-methylbenzeneacetic acid 2-phenylhydrazide |
||
|
|
||||
|
CAS number: |
NA |
Relative molecular mass: |
364.40 |
|
|
Structural formula: |
C21H2ON2O4 |
Observed in: |
dogs |
|
See Also:
Toxicological Abbreviations