
First draft prepared by
Diane Benford1, Catherine Boyle1, Wolfgang Dekant2, Radovan Fuchs3, David W. Gaylor4, Gordon Hard5, Douglas B. McGregor6, John I. Pitt7, Radovan Plestina3, Gordon Shephard8, Michelle Solfrizzo9, Philippe J.P.Verger10, Ronald Walker11
1 Food Standards Agency, London, United Kingdom
2
University of Würzburg, Würzburg, Germany3
Institute for Medical Research and Occupational Health, Zagreb, Croatia4
Science International, Little Rock, Arkansas, USA5
American Health Foundation, Valhalla, New York, USA6
Lyon, France7
Food Science Australia, North Ryde, New South Wales, Australia8
Medical Research Council, Tygerberg, South Africa9
Consiglio Nazionale delle Ricerche, Bari, Italy10
Institut National de la Recherche Agronomique, Paris, France11
University of Surrey, Guildford, United KingdomOchratoxin A was evaluated by the Committee at its thirty-seventh meeting (Annex 1, reference 94), when it established a provisional tolerable weekly intake (PTWI) of 112 ng/kg bw, on the basis of deterioration of renal function in pigs, for which the lowest-observed-effect level (LOEL) was 0.008 mg/kg bw per day, and a safety factor of 500. At that time, the Committee recommended that further studies be conducted to elucidate the role of ochratoxin A (and other mycotoxins) in causing nephropathy in pigs and humans, the mechanisms of induction of tumours, and the role of phenylalanine in antagonizing the adverse effects of ochratoxin A. (The present Committee noted that the adverse effects noted at the thirty-seventh meeting consisted of nephrotoxicity.) Ochratoxin A was re-evaluated by the Committee at its forty-fourth meeting (Annex 1, reference 116), when it considered toxicological data that had become available since the previous evaluation, including studies on the epidemiology of nephropathy, on genotoxicity and on experimental nephrotoxicity. At that meeting, the Committee reconfirmed the PTWI, rounding it to 100 ng/kg bw, and reiterated its request for further studies on ochratoxin A.
The Codex Committee on Food Additives and Contaminants at its Thirty-first Session requested the Expert Committee to perform a risk assessment of the consequences of establishing a maximum level of 5 or 20 µg/kg in cereals and cereal products.
Ochratoxin A is produced by a single Penicillium species, P. verrucosum, by Aspergillus ochraceus and several related Aspergillus species, and by A. carbonarius, with a small percentage of isolates of the closely related A. niger. These three groups of species differ in their ecological niches, in the commodities affected, and in the frequency of their occurrence in different geographical regions. P. verrucosum grows only at temperatures below 30 °C and at water activity as low as 0.8. It is therefore found only in cool temperate regions; it is the source of ochratoxin A in cereals and cereal products in Canada and Europe. As cereals are widely used in animal feeds in Europe, and ochratoxin A is relatively stable in vivo, this mycotoxin is also found in some animal products in that region, especially in pig kidney and liver. As P. verrucosum does not occur in the tropics and subtropics, cereals from these regions are unlikely to contain ochratoxin A from this source. A. ochraceus grows at moderate temperatures and at a water activity above 0.8. It is found sporadically in a wide range of stored food commodities, including cereals, but is seldom the cause of substantial concentrations of ochratoxin A. It may also infect coffee beans during sun-drying and is a source of ochratoxin A in green coffee beans. A. carbonarius grows at high temperatures and is associated with maturing fruits, especially grapes. Because of its black spores, it is highly resistant to sunlight and survives sun-drying. It is the source of ochratoxin A in fresh grapes, dried vine fruits, and wine; it is also one source of ochratoxin A in coffee.
The Committee considered several new studies that had become available since the previous evaluation of ochratoxin A. These included further studies of absorption, distribution (including secretion into the milk of experimental animals), metabolism, and excretion; biochemical studies; toxicological studies on genotoxicity, immunotoxicity, neurotoxicity, embryotoxicity, and hepatotoxicity; and studies on the mechanisms of cytotoxicity and nephrotoxicity. The results of epidemiological studies were also reviewed. New data from surveys of food commodities for ochratoxin A and of food consumption were also considered, and intakes were estimated for various countries and regions of the world.
It has been suggested that, in most species, ochratoxin A is absorbed from the stomach as a result of its acidic properties (pKa = 7.1) (Galtier, 1978; Roth et al., 1988). In studies of animals with ligated gastrointestinal loops, however, the small intestine was found to be the major site of absorption, with maximal absorption from the proximal jejunum. Absorption from the jejunum can take place against a concentration gradient and depends on the pH at the mucosal surface of the jejunum. Ochratoxin A that is so transferred is lipid-soluble and non-ionized (Kumagai & Aibara, 1982; Kumagai, 1988).
The results of studies in which a low dose of [3H]ochratoxin A was given by intubation to mice were interpreted by the authors as indicating rapid absorption from the stomach, but they could also be interpreted as showing that intestinal absorption is the major route, with rapid transit from the stomach to the intestine. Secondary peaks of ochratoxin A found in the intestinal contents and serum may have been a consequence of enterohepatic circulation, since the biliary excretion of this toxin is very efficient (Fuchs et al., 1988a; Roth et al., 1988).
The overall percentage of ochratoxin A absorbed was 66% in pigs, 56% in rats, 56% in rabbits, and 40% in chickens (Suzuki et al., 1977; Galtier et al., 1981).
In male Wistar rats that received a single intratracheal dose of crystalline ochratoxin A (purity unknown) at 50 ng/g bw, absorption from the lungs was found to be very efficient, the bioavailability being calculated as 98%. The biological half-life of ochratoxin A was estimated to be 127 h. The toxicokinetics of the toxin when given intratracheally, orally, or intravenously were comparable (Breitholtz-Emanuelsson et al., 1995).
Phenylalanine given to mice by gavage with ochratoxin A in a 10:1 molar ratio appeared to increase the absorption of ochratoxin A from the stomach and intestine and to increase gastrointestinal transit. This resulted in an eightfold higher concentration of ochratoxin A in serum and and a fourfold higher concentration in liver during the next 12 h (Roth et al., 1988).
The bioavailability of ochratoxin A, estimated from a comparison of the maximal serum concentration after oral and intravenous administration, was very low in fish but 44 and 97% for two mammalian species investigated (Hagelberg et al., 1989). Once it reaches the blood, ochratoxin A bound readily to serum albumin (Galtier et al., 1980) and other macromolecules (Hult & Fuchs, 1986). Erythrocytes contained only traces (Galtier, 1978).
The association constant for the binding of ochratoxin A to serum albumin was 7.1 × 104 per mol for pigs, 5.1 × 104 per mol for chickens, and 4.0 × 104 per mol for rats (Galtier et al., 1981). The fraction of ochratoxin A bound to serum albumin and other macromolecules constitutes a mobile reserve of mycotoxin that can be made available for release to the tissues for a long time (Galtier, 1978; Hult et al., 1982). Studies with albumin-deficient rats showed that the main effect of ochratoxin A binding to serum albumin is to retard its elimination by limiting its transfer from the bloodstream to hepatic and renal cells (Kumagai, 1985).
In studies of the stability of ochratoxin A bound to porcine albumin, it was displaced by the acidic drug phenylbutazone, so that more free toxin was available. In male rats, ochratoxin A was more toxic in the presence of phenylbutazone, with a significant decrease in the LD50 value from 33 to 21 mg/kg bw (Galtier et al., 1980).
Ochratoxin A had strong affinity for an unidentified serum macromolecule (relative molecular mass, 20 000), with association constants of 2.3 × 1010 per mol in human serum and 0.59 × 1010 per mol in porcine serum. The specific binding of this macromolecule was saturated at concentrations of ochratoxin A of 10–20 ng/ml serum. Significant amounts of serum albumin were bound at higher concentrations of ochratoxin A, with saturation above several hundred micrograms per millilitre of serum (Stojkovic et al., 1984; Hult & Fuchs, 1986).
The fraction of ochratoxin A that remained unbound to two identified plasma proteins was 0.02% in humans and rats, 0.08% in monkeys, 0.1% in mice and pigs, and 22% in fish (Hagelberg et al., 1989).
Once ochratoxin A has been absorbed, the concentrations of the toxin and its metabolites in tissues and plasma residues depend on the length of feeding, the dose, whether the ochratoxin A is naturally occurring or crystalline, the route, the degree of serum binding, the half-life of ochratoxin A, and the duration on an ochratoxin A-free diet before sacrifice. These factors are important in assessing the natural occurrence of residues in animal tissues (Kuiper-Goodman & Scott, 1989).
After a single oral dose, the maximum serum concentrations of ochratoxin A were found within 10–48 h in pigs and rats (Suzuki et al., 1977; Galtier, 1978; Galtier et al., 1981; Mortensen et al., 1983a), at 2–4 h in ruminant calves (Sreemannarayana et al., 1988), after 1 h in rabbits, and after 0.33 h in chickens (Galtier et al., 1981). Maximum concentrations in tissues were found within 48 h in rats.
Wide species differences have been reported in the serum half-life of ochratoxin A. The half-life after oral administration was found to be 510 h in Macaca mulata monkeys (Hagelberg et al., 1989), 72–120 h in pigs (Galtier et al., 1981; Mortensen et al., 1983a), 77 h in pre-ruminant calves (Sreemannarayana et al., 1988), 55–120 h in rats (Galtier et al., 1979; Ballinger et al., 1986; Hagelberg et al., 1989), 6.7 h in quail (Hagelberg et al., 1989), and 4.1 h in chickens (Galtier et al., 1981). In those species tested, the serum half-time was longer after intravenous administration (Hagelberg et al., 1989), perhaps due in part to differences in absorption (Galtier et al., 1981), differences in peak plasma concentrations (see above), and species differences in the degree of binding to serum macromolecules, including albumin.
The rate of disappearance of ochratoxin A was slower from blood than from kidney, liver, and other tissues in pigs (Hult et al., 1979).
Whole-body autoradiography of mice after a single intravenous dose of [14C]ochratoxin A at approximately 200 µg/kg bw showed that the toxin persisted for > 4 days in the blood, interpreted as showing that the toxin is present mainly in bound form at this dose (Fuchs et al., 1988a). In a similar experiment in rats, the distribution after 24 h was greatest in lung and, in decreasing order, in adrenal medulla, skin, liver, myocardium, kidney, salivary gland, adrenal cortex, muscle, gastric mucosa, and bone marrow (Breitholtz-Emanuelsson et al., 1992). The tissue distribution in pigs, rats, chickens, and goats generally followed the order kidney > liver > muscle > fat (Harwig et al., 1983) or kidney > muscle > liver > fat (Mortensen et al., 1983a; Madsen et al., 1982).
In hens fed ochratoxin A, none was found in eggs (Krogh et al., 1976). In another study, it was found in eggs when the birds were fed 10 mg/kg bw (Juszkiewicz et al., 1982). A study of the tissue distribution of [14C]ochratoxin A in laying Japanese quail showed specific retention of unidentified radiolabel as a ring-shaped deposition in eggs, indicating that the toxin could be deposited over a short period (Fuchs et al., 1988b). Egg-laying Japanese quail were given a single oral dose of 0, 1, 5, or 20 mg/kg bw. The concentrations of ochratoxin A in abdominal yolk of birds 6 h later were 13 µg/kg in those given 5 mg/kg bw and and 34 µg/kg in those given 20 mg/kg bw. The toxin was still present in abdominal yolks 4 days after administration, and the mean concentration was 10-fold higher than in whole eggs. No ochratoxin A was found in eggs of birds given 1 mg/kg bw (Piskorska-Pliszczynska & Juszkiewicz, 1990).
Lactating rats, treated orally with a single dose of ochratoxin A up to 250 µg/kg bw excreted the toxin in their milk. The milk:blood concentration ratio was 0.4 at 24 h and 0.7 at 72 h. A linear relationship was found between the concentration of ochratoxin A in the dam’s milk and that in the blood and kidneys of pups at 72 h. The pup milk:blood concentration ratio was approximately 1.7. At 72 h, the suckling pups had higher concentrations of ochratoxin A than their dams in both blood and kidney (Breitholtz-Emanuelsson et al., 1993a).
Whole-body autoradiography after intravenous administration of high doses of [14C]ochratoxin A showed that it could cross the placenta more readily when given on days 8 and 9 than day 10 of gestation, with radiolabel appearing within 20 min in the uterine wall, placenta, and fetal tissues. Ochratoxin A given to mice on day 17 of gestation resulted in very little radiolabel in fetuses (Appelgren & Arora, 1983a,b).
Differences in fetal uptake of ochratoxin A at different times during gestation were suggested to be due to differences in the placenta, which was considered to be completely developed by day 9 of gestation. After intraperitoneal injection of ochratoxin A on day 11 or 13 of gestation, residues appeared more slowly and reached maximum values 30–48 h after dosing. The concentrations in the placenta were high 2–6 h after injection and then decreased more slowly than from other tissues. The serum half-life of ochratoxin A was 29 h at day 11 and 24 h at day 13 of gestation. The authors considered the embryo to be a ‘deep compartment’ (Fukui et al., 1987).
A group of 39 female Sprague-Dawley rats received ochratoxin A orally at 50 µg/kg bw five times a week for 2 weeks before mating, during gestation, and then 7 days a week during lactation. Pups from ochratoxin A-treated dams were cross-fostered at birth to control dams and vice versa. Treatment did not affect maternal body weight nor alter the birthweight or development of pups. The concentrations of ochratoxin A in the blood and kidney of exposed pups were three to four times higher than those in the dams. No differences in weight gain or in body or kidney weight were seen between pups exposed in utero, via lactation, or both. The transfer of ochratoxin A to milk was very efficient (60% of the blood concentration at 8 weeks). The highest blood and kidney concentrations were found in offspring exposed in utero and via milk, but the most significant exposure was via milk (Hallén et al., 1998).
After subcutaneous administration of [3H]ochratoxin A to rats on day 12 of gestation, fetal uptake was delayed, with maximum concentrations 48–72 h after dosing, representing about 0.1% of the dose administered (Ballinger et al., 1986).
Four lactating Blanc de Termonde rabbits received ochratoxin A from feed naturally contaminated at 190 ng/g, equivalent to 16 µg/kg bw, on days 3–19 of lactation. The toxin was effectively transported from blood to milk and subsequently to the offspring. Higher concentrations were found in maternal plasma than in milk, and a linear relationship was found between the concentrations in milk and plasma of offspring. The plasma:kidney concentrations were much higher in offspring than in adults, perhaps due to slower detoxication in the former (Ferrufino-Guardia et al., 2000).
Ochratoxin A given at 0.38 mg/kg bw to pregnant sows on days 21–28 of pregnancy did not cross the placenta (Patterson et al., 1976). Similarly, no residues were found in piglets of sows fed diets containing ochratoxin A at 7–16 µg/kg bw per day throughout gestation (Mortensen et al., 1983b). In a more recent study, however, ochratoxin A was transmitted to six piglets in utero when the sow was fed naturally contaminated feed; the blood concentrations in the newborn piglets were 0.075–0.12 ng/ml, whereas that in the sow was 0.20 ng/ml (Barnikol & Thalmann, 1988).
Both biliary excretion and glomerular filtration play important roles in the plasma clearance of ochratoxin A in rats. This is related to its relative molecular mass of 403.8, since both pathways are used in this species for substances with relative molecular masses between 350 and 450. Thus, in rats, both the urinary and faecal excretory routes are important, the relative contribution of each depending on factors such as route of administration and dose (Kuiper-Goodman & Scott, 1989).
In all species, the relative contribution of each excretory route is also influenced by the degree of serum macromolecular binding and differences in the degree of enterohepatic recirculation of ochratoxin A (Hagelberg et al., 1989).
In rats, the major excretory products were ochratoxin alpha (in urine and faeces), ochratoxin A, and the 4R-OH-ochratoxin A epimer. In urine, these represented 25–27%, 6%, and 1–1.5% of the administered dose, respectively (Storen et al., 1982a).
Up to 33% of the radiolabel on an orally administered dose of ochratoxin A was excreted into the bile of rats up to 6 h after dosing; only trace amounts of ochratoxin alpha were detected in the bile (Suzuki et al., 1977).
Biliary excretion of ochratoxin A was increased and urinary excretion of ochratoxin A and ochratoxin alpha was decreased in mice pretreated with phenobarbital (Moroi et al., 1985).
When ochratoxin A was administered to rats intraperitoneally, only traces of ochratoxin A and ochratoxin alpha were identified in faeces, whereas after oral administration 12% ochratoxin A and 9% ochratoxin alpha were found in faeces (Storen et al., 1982a).
In pre-ruminant and ruminant calves, 85–90% of orally administered ochratoxin A was excreted as ochratoxin alpha, most of it in the urine (Sreemannarayana et al., 1988).
Ochratoxin A is hydrolysed to the non-toxic ochratoxin alpha at various sites. In rats, detoxication by hydrolysis to ochratoxin alpha is a function of the bacterial microflora of the caecum (Galtier, 1978). The enzymes responsible for hydrolysis to ochratoxin alpha in cows and rodents are carboxypeptidase A and chymotrypsin (Pitout, 1969a,b; Pitout & Nel, 1969). Other mycotoxins such as penicilloic acid inhibit this reaction (Parker et al., 1982). Inhibition of the flora of the lower gastrointestinal tract of rats by neomycin reduced hydrolysis of ochratoxin A to ochratoxin alpha and increased the blood concentration of ochratoxin A (Madhyastha et al., 1992).
Studies with rat tissue homogenate showed that the duodenum, ileum, and pancreas also have a high capacity to carry out this reaction, whereas the activity in the liver and kidney was low (Suzuki et al., 1977). It was non-existent in rat hepatocytes (Hansen et al., 1982) and rabbit and rat liver (Kanisawa et al., 1979; Stormer et al., 1983).
In rats given [14C]ochratoxin A, most of the radiolabel was attached to ochratoxin A, indicating that efficient metabolism of this toxin is lacking in most tissues other than the intestine (Galtier et al., 1979).
Incubation of the contents of the four stomachs of cows indicated effective hydrolysis of ochratoxin A to ochratoxin alpha by the ruminant protozoa. Assuming a similar reaction velocity in vivo, it was estimated that up to 12 mg/kg of feed could be degraded (Hult et al., 1976; Pettersson et al., 1982), so that this species is assumed to be relatively resistant to the effects of ochratoxin A in feed. Sheep also have a good capacity to detoxify ochratoxin A before it reaches the blood (Kiessling et al., 1984).
Studies in mice suggest that ochratoxin A circulates from the liver into the bile and into the intestine, where it is hydrolysed to ochratoxin alpha (Moroi et al., 1985).
About 25–27% of ochratoxin A, given either intraperitoneally or orally to rats, was present as ochratoxin alpha in the urine. Its presence in the urine can be explained by reabsorption from the intestine (Storen et al., 1982a). A similar mechanism of intestinal reabsorption of ochratoxin alpha has been suggested to occur in ruminant calves (Sreemannarayana et al., 1988).
Other minor urinary metabolites of ochratoxin A are 4-OH (4R-and 4S) epimers produced in rat and rabbit liver (Størmer et al., 1981) and rat kidney (Stein et al., 1985) by the action of cytochromes P450 (CYPs; Størmer et al., 1981, 1983). The 4R-OH epimer, which is considered less toxic than ochratoxin A, is the main one formed in human and rat liver microsomal systems (Størmer et al., 1981), whereas the 4S-OH epimer is more prevalent in pig liver microsomes. No data were available on its toxicity (Moroi et al., 1985).
The biotransformation of ochratoxin A has also been studied in various microsomal preparations and in recombinant human and rat CYP preparations (Gautier et al., 2001; Zepnik et al., 2001). Incubation of ochratoxin A with liver microsomes from rats and mice produced 4R- and 4S-hydroxyochratoxin A, but at very low rates, whereas oxidation of ochratoxin A was not observed in kidney microsomes from these species. 4R-Hydroxyochratoxin A was also formed at low rates by recombinant human CYP 3A4 (both studies), CYP 1A1 and CYP 2C9-1 (both in single studies), while conflicting results were obtained with CYP1A2. Oxidation was not observed with recombinant human CYP 2E1 or rat CYP 1A2 or the male rat-specific CYP 2C11 (all in one study). Prostaglandin H-synthase produced small amounts of a non-polar product.
The 10-OH derivative was formed from ochratoxin A in a rabbit liver microsomal system (Størmer et al., 1983). Ochratoxin C, a metabolite of ochratoxin A produced in rumenal fluid, is as toxic as ochratoxin A (cited by Galtier et al., 1981). Ochratoxin B, a dechloro derivative of ochratoxin A, may occur with ochratoxin A in cereal products. In rats, it is less toxic than ochratoxin A and is metabolized to 4-OH-ochratoxin B and ochratoxin beta (Størmer et al., 1985).
Ochratoxin B was not antagonistic to ochratoxin A with respect to effects on the formation of phenylalanyl-tRNA and protein synthesis (Roth et al., 1989).
Many researchers consider that the toxicity of ochratoxin A is due to one of its metabolites. The studies cited above indicate, however, that, in rats, ochratoxin A itself, rather than one of its metabolites, is the active toxic agent, since the known metabolites are less toxic than or as toxic as ochratoxin A. This conclusion concurs with findings in mice, in which the LD50 of ochratoxin A increased by 1.5- to 2-fold after the animals were fed phenobarbital at 500 mg/kg of diet for 1 week before oral or intraperitoneal administration (Moroi et al., 1985).
Similarly, pretreatment with sodium phenobarbital at 80 mg/kg bw per day by gavage for 5 days or 3-methylcholanthrene at 20 mg/kg bw per day by gavage for 2 days resulted in increased LD50 values for ochratoxin A. With phenobarbital, the difference was smaller 144 h after dosing with ochratoxin A than at 48 h. Administration of piperonyl butoxide, an inhibitor of microsomal monooxygenases, decreased the 144-h LD50 of ochratoxin A from 40 to 19 mg/kg bw (Chakor et al., 1988). In contrast, preliminary studies in mice showed that simultaneous feeding of phenobarbital slightly increased the incidence of liver tumours seen with ochratoxin A alone, and that the mice developed large, multiple hepatomas (Suzuki et al., 1986).
Few data are available on the metabolic disposition of ochratoxin A in humans. It has been suggested that it has a long serum half-life, on the basis of its strong binding to human serum macromolecules (Bauer & Gareis, 1987; Hagelberg et al., 1989).
The activities of glycolytic enzymes were reduced, whereas those of gluconeo-genic enzymes were increased. The diabetogenic effect of ochratoxin A was thought to be due to inhibited synthesis and/or release of insulin from pancreatic cells, thereby suppressing glycolysis and glycogenesis and enhancing gluconeogenesis and glycogenolysis (Subramanian et al., 1989).
Calcium homeostasis was studied in rats treated intraperitoneally with ochratoxin A at a single dose of 10 mg/kg bw or multiple doses of 0.5–2 mg/kg bw per day. An increase in renal endoplasmic reticulum calcium pump activity was observed, suggesting an association with ochratoxin A-induced renal cytotoxicity (Rahimtula & Chong, 1991).
Studies with pig renal cortical explants indicated that inhibition of the biosynthesis of macromolecules (protein, RNA and DNA) by ochratoxin A was not due to impairment of cellular respiration (Braunberg et al., 1992).
The biochemistry and molecular aspects of the action of ochratoxin A in both prokaryotes and eukaryotes have been reviewed (Röschenthaler et al., 1984). The findings are inconsistent, owing to differences and limitations in experimental models and procedures as well as interfering factors, especially in more complex organisms. In prokaryotes (Konrad & Röschenthaler, 1977), eukaryotic microorganisms (Creppy et al., 1979a), mammalian cells (Creppy et al., 1980a, 1983a), and experimental animals in vivo (Creppy et al., 1980b, 1984), the primary effect of ochratoxin A is inhibition of protein synthesis; secondarily, RNA and DNA synthesis may be inhibited.
The inhibition of protein synthesis is specific and occurs at the post-transcriptional level, ochratoxin A having a direct effect on the translation step in protein synthesis. This involves competitive inhibition of phenylalanine-tRNAPhe synthetase, so that amino-acylation and peptide elongation are stopped. This reaction is fundamental for all living organisms. In yeast, the first part of this reaction, phenylalanine-dependent pyrophosphate exchange, was inhibited five times more than transfer to tRNA, the second part. In this reaction ochratoxin A may be regarded as an analogue of phenylalanine, and in cell cultures the competitive inhibition could be reversed by an increase in phenylalanine concentration (Creppy et al., 1979a). Similarly, in mice, the lethality of a single dose of 0.8 mg of ochratoxin A injected intraperitoneally was completely prevented by simultaneous injection of 1 mg of phenylalanine (Creppy et al., 1980b).
In yeast, the effect on protein synthesis of the rR-OH-ochratoxin A epimer was similar to that of ochratoxin A, but ochratoxin alpha, which lacks the phenylalanine moiety, had no effect (Creppy et al., 1983a). Analogues of ochratoxin A in which phenylalanine has been replaced by other amino acids, such as tyrosine, inhibit the respective amino acid-specific tRNA synthetases similarly (Creppy et al., 1983b).
The binding affinity of phenylalanine-tRNAPhe synthetase for ochratoxin A is weaker than for phenylalanine and ranges from 1/300 in yeast (KM = 1.3 mmol/L for ochratoxin A; 3.3 µmol/L for phenylalanine) (Creppy et al., 1983a) to 1/20 in rat liver (Km = 0.28 mmol/L for ochratoxin A; 6 µmol/L for phenylalanine) (Röschenthaler et al., 1984). Despite these differences in binding affinity, the inhibition of phenylalanine-tRNAPhe by ochratoxin A is very effective, since the toxin is more readily concentrated by cells than phenylalanine. The concentration of ochratoxin A inside hepatoma cells was 200- to 300-fold that in the medium (Creppy et al., 1983a).
A dose-related inhibition of protein synthesis was found in mice given ochratoxin A intraperitoneally at a dose > 1 mg/kg bw. The degree of inhibition of protein synthesis 5 h after administration of ochratoxin A at 1 mg/kg bw was 26% in liver, 68% in kidney, and 75% in spleen as compared with controls (Creppy et al., 1984).
Ochratoxin A may also act on other enzymes that use phenylalanine as a substrate, although no direct effect on the activity of other isolated enzyme systems has been demonstrated (Röschenthaler et al., 1984). In kidney slices from rats 2 days after they had been fed ochratoxin A at 2 mg/kg bw, the activity of renal phosphoenolpyruvate carboxykinase, a key enzyme in the gluconeogenic pathway, was lowered by 50% (Meisner & Krogh, 1986). The inhibition was due indirectly to specific degradation of the mRNA coding for this enzyme. A similar effect was not seen in rat liver (Meisner et al., 1983).
The effect of ochratoxin A on phenylalanine metabolism was studied in isolated hepatocytes and in liver homogenates from male rats treated in vivo. Both the hydroxylation of phenylalanine to tyrosine and the subsequent metabolism of tyrosine, as measured by homogenate oxidation, were inhibited when ochratoxin A at a concentration of 0.12–1.4 mmol/L was incubated with isolated hepatocytes (Creppy et al., 1990).
Ochratoxin A enhanced NADPH- or ascorbate-dependent lipid peroxidation in rat liver microsomes and NADPH-dependent lipid peroxidation in kidney microsomes in vitro, as measured by malondialdehyde formation or oxygen uptake. It was suggested that ochratoxin A stimulates lipid peroxidation by complexing Fe3+ and facilitating its reduction. Subsequent to oxygen binding, an iron–oxygen complex initiates lipid peroxidation. Cytochrome P450, free active oxygen species, and free hydroxy radicals do not appear to be involved in Fe3+–ochratoxin A- stimulated lipid peroxidation. Oral administration of ochratoxin A at 6 mg/kg bw to rats appeared to increase lipid peroxidation in vivo, causing a sevenfold increase in ethane exhalation (Rahimtula et al., 1988; Omar et al., 1990).
In pig renal cortical tissue, ochratoxin A and citrinin added singly or in combination at a concentration of 10–6 or 10–3 mol/L did not elicit consistent or strong synergistic effects, as measured by transport of tetraethylammonium and paraaminohippurate ions, or protein synthesis measured with [3H]leucine (Braunberg et al., 1994).
The effects of superoxide dismutase and catalase on ochratoxin A-induced nephrotoxicity were studied. Superoxide removes oxygen by converting it to hydrogen peroxide; this enzyme works in conjunction with catalase, which removes hydrogen peroxide within cells. Rats were given 20 mg/kg bw of each enzyme by subcutaneous injection every 48 h, 1 h before gavage with ochratoxin A at 290 µg/kg bw every 48 h, for 3 weeks. Superoxide dismutase and catalase prevented most of the nephrotoxic effects induced by ochratoxin A, observed as enzymuria, proteinuria, and creatinaemia, and increased the urinary excretion of ochratoxin A. The results indicated that superoxide radicals and hydrogen peroxide are likely to be involved in the nephrotoxic effects of ochratoxin A in vivo (Baudrimont et al., 1994).
After short-term administration of ochratoxin A to rats, the renal proximal tubule did not appear to be the main target for nephrotoxicity, although decreased capacity to eliminate the toxin may result in a self-enhancing effect (Gekle & Silbernagl, 1994). The main renal effect of ochratoxin A in rats was found in the ‘postproximal’ nephron, as measured by a reduced glomerular filtration rate, increased fractional water, Na+, K+, and Cl– excretion, and increased dependence of osmol clearance on urine flow. In addition, ochratoxin A blocked membrane anion conductance in canine kidney cells in vitro (Gekle et al., 1993).
The LD50 values found in various species treated by various routes are shown in Table 1. Dogs and pigs were the most sensitive species and rats and mice the least sensitive. Simultaneous oral administration of phenylalanine at 100 mg/kg bw to mice increased the oral LD50 from 46 mg/kg bw to 71 mg/kg bw (Moroi et al., 1985). As is the case with many xenobiotics, neonatal rats were considerably more susceptible than adults.
Table 1. LD50 values for ochratoxin A in various species
|
Species |
LD50 (mg/kg bw) |
||
|
Oral |
Intraperitoneal |
Intravenous |
|
|
Mouse |
46–58 |
22–40 |
26–34 |
|
Rat |
20–30 |
13 |
13 |
|
Rat neonate |
3.9 |
|
|
|
Dog |
0.2 |
|
|
|
Pig |
1 |
|
|
|
Chicken |
3.3 |
|
|
Based on a literature compilation by Harwig et al. (1983)
Histopathological and electron microscopic studies were conducted with groups of 10 male Long-Evans and Sprague-Dawley rats given benzene-free ochratoxin A at a single dose of 0, 17, or 22 mg/kg bw in 0.1 mol/L sodium bicarbonate by gavage and examined for up to 48 h afterwards. The earliest changes were multifocal haemorrhages in many organs and fibrin thrombi in the spleen, the choroid plexus of the brain, liver, kidney and heart, suggesting disseminated intravascular coagulation. The effect was postulated by the authors to be due to activation of extrinsic and intrinsic systems of coagulation. Other changes were hepatic and lymphoid necrosis, enteritis with villous atrophy, affecting the jejunum most severely, and nephrosis. The myocardial changes were considered to be related to shock and subsequent ischaemic injuries (Albassam et al., 1987).
Ochratoxin A had nephrotoxic effects in all monogastric mammalian species tested so far (Kuiper-Goodman & Scott, 1989). The results of short-term studies with this toxin are shown in Table 2.
Table 2. Results of short-term studies of the toxicity of ochratoxin A
|
Species, strain, sex, age |
No . |
Route |
Dose |
Time |
NOEL |
Effects |
Reference |
|
Rat, Wistar, male, weanling |
10 |
Diet |
0.24–2.4 [2.4–24] |
14 |
~0.48 |
Growth retardation |
Munro et al. (1974) |
|
~0.48 |
Increased serum blood urea nitrogen |
||||||
|
~0.96 |
Increased kidney weight |
||||||
|
< 0.24 |
Decreased urine volume |
||||||
|
< 0.24 |
Renal lesions |
||||||
|
Rat Wistar, male, female, weanling |
15 |
Diet |
0.015–0.37 [0.2–5] |
90 |
~0.075 |
Reduced weight gain |
Munro et al. (1974) |
|
~0.016 |
Reduced kidney weight; no change in blood urea nitrogen, urinary or haematological parameters |
||||||
|
0.37 |
Desquamation; increase in smooth endoplasmic reticulum, changes in rough endoplasmic reticulum, basement membrane thickening of proximal convoluted tubule cells; increased eosinophilia and karyomegaly in proximal convoluted tubule cells |
||||||
|
Rat, Wistar, male, adult |
5 |
Gavage |
5 |
3 |
< 5 |
Reduced para-amino hippuric acid clearance, basement membrane thickening |
Suzuki et al. (1975) |
|
Rat, Wistar, male, adult |
10 |
Gavate |
0.5-2 |
10 |
1 |
Increased blood urea nitrogen |
Haley & Galtier (1977) |
|
< 0.5 |
Increased urine volume |
||||||
|
Rat, Sprague-Dawley and Wistar, male, female adult |
4-6 |
intraperi-toneal |
0.75, 2 |
5-7 |
< 0.75 |
Decreased body weight, increased urine flow; decreased urine osmolality; increased urinary protein; increased urinary glucose; impaired urinary transport of organic substances; Sprague-Dawley more sensitive than Wistar, females less sensitive than males |
Berndt & Hayes (1979) |
|
Rat, Wistar, male, adult |
14 |
Gavage |
4 |
4-10 |
< 4 |
Decreased factors II, VI, X; decreased plasma fibrinogen, decreased thrombocyte, megakaryocyte counts |
Gaultier et al. (1979) |
|
Rat, Wistar, male, adult |
9 |
Gavage |
4 |
10 |
< 4 |
Hypothermia, cachexia, tremors, diarrhoea |
Galtier et al. (1980) |
|
Rat, Wistar, male, adult |
3 |
Gavage |
0.14-2 |
56-84 |
< 0.14 |
Decreased kidney enzyme activity; increased urinary enzyme activity |
Kane et al. (1986a) |
|
Rat, Fischer 344/N, male, female, weanling |
5 |
Gavage |
1–6 |
16 (12 doses) |
1 |
Increased relative kidney, heart, and brain weight; thymus atrophy; forestomach necrosis; adrenal haemorrhage |
National Toxicology Program (1989) |
|
< 1 |
Bone-marrow hypoplasia |
||||||
|
< 1 |
Renal nephropathy |
||||||
|
Rat, Fischer 344/N, male, female, weanling |
10 |
Gavage |
0.06–1 |
91 |
0.12, males |
Growth retardation |
National Toxicology Program (1989) |
|
0.12, males |
Reduced relative kidney weight |
||||||
|
0.06 |
Kidney tubular necrosis |
||||||
|
< 0.06 |
Karyomegaly |
||||||
|
Dog, beagle, male, young |
3–6 |
Capsule |
0.1–0.2 |
14 |
0.2 |
No change in kidney function |
Kitchen et al. (1977a,b,c) |
|
< 0.1 |
Renal tubular necrosis |
||||||
|
< 0.1 |
Proximal tubule changes; thymus, lymphoid necrosis |
||||||
|
Pig, female, 8–12 weeks |
3–6 |
Diet |
0.008. 0.04, 0.2 |
5–90 |
< 0.008 |
Renal enzyme changes; changes in renal function |
Elling (1979a); Krogh et al. (1988) |
(a) Rats
Groups of 10 male weanling Wistar rats were fed semi-purified diets containing ochratoxin A at a concentration of 0, 2.4, 4.8 9.6, or 24 mg/kg, equivalent to 0, 0.24, 0.48, 0.96, and 2.4 mg/kg bw per day, for 14 days. At the two higher doses, growth retardation, reduced food consumption, and increased serum urea nitrogen were seen. At the highest dose, the relative kidney weight was increased. Renal lesions, involving degenerative changes in the entire tubular system, and a decrease in urine volume were seen at all doses. Increased eosinophilia and karyomegaly in cells of the proximal convoluted tubules were noted at all doses (Munro et al., 1974).
Semi-purified diets containing ochratoxin A at 0, 0.2, 1, or 5 mg/kg, equivalent to 0, 0.015, 0.075, or 0.37 mg/kg bw per day, were fed to groups of 15 weanling Wistar rats of each sex for 90 days. At that time, eight animals from each group were killed, and the remaining rats were fed control diet for an additional 90 days. No changes in blood urea nitrogen or urinary or haematological parameters were seen at any dose. After 90 days at the two higher dietary concentrations, the relative kidney weights were reduced in animals of each sex; these had returned to control values after the 90-day recovery period, except in males at the highest dose. Dose-related changes in morphological appearance were seen after 90 days of treatment at doses > 0.2 mg/kg of diet and involved karyomegaly and increased eosinophilia in cells of the proximal convoluted tubules. The authors considered the latter change to be a phenomenon of ageing which had been accelerated by administration of ochratoxin A. Desquamation of proximal tubular cells, autolysis, changes in the rough and smooth endoplasmic reticulum, and tubular basement membrane thickening up to 4 µm were noted at the highest dose. In animals at the highest dose that were subsequently given control diet for 90 days, the karyomegaly and tubular basement membrane thickening persisted, but otherwise the kidneys appeared normal (Munro et al., 1974).
Similar effects were seen when ochratoxin A was administered to groups of four to six adult Sprague-Dawley and Wistar rats by intraperitoneal injection for 5–7 days at a dose of 0, 0.75, or 2 mg/kg bw per day. Decreased body weight, increased urine flow, increased urinary protein, increased urinary glucose, and impaired urinary transport of organic substances were seen at all doses. Sprague-Dawley rats were more sensitive than Wistar rats, and males were more sensitive than females. It was suggested that the increased urinary protein indicated interference with protein reabsorption by cells of the convoluted tubules (Berndt & Hayes, 1979).
Ochratoxin A was administered by gavage in maize oil to groups of five weanling male and female Fischer 344/N rats at a dose of 0, 1, 4, or 16 mg/kg bw per day on 5 days per week for a total of 12 doses over 16 days. All rats that received the highest dose had diarrhoea and nasal discharge and died before the end of the study. Increased relative weights of kidneys, heart, and brain, thymus atrophy, forestomach necrosis and/or hyperplasia, and haemorrhage of adrenal glands were seen at the two higher doses. Bone-marrow hypoplasia and nephropathy were seen at all doses, involving renal tubular degenerative and regenerative changes (National Toxicology Program, 1989).
Ochratoxin A was administered by gavage in maize oil to groups of 10 male and female weanling Fischer 344/N rats at a dose of 0, 0.06, 0.12, 0.25, 0.5, or 1 mg/kg bw per day for 5 days/week for 91 days. Growth retardation and a reduced relative kidney weight were seen in males at the two higher doses. The NOEL for renal tubular necrosis was 0.062 mg/kg bw, but karyomegaly of dose-related severity was observed in the proximal tubules at all doses. Milder renal changes consisting of tubular atrophy were seen at lower doses (National Toxicology Program, 1989).
Groups of 15 weanling rats were given ochratoxin A in 0.1 mol/L sodium bicarbonate at a dose of 0 or 100 µg/rat (equivalent to 1.25 mg/kg bw per day) by gavage for 8 weeks. Blood samples from fasted treated rats contained about twice the amount of glucose as those of controls. In a glucose tolerance test, the insulin concentration did not reach that in control rats. Total carbohydrate and glycogen concentrations in liver of treated rats were reduced, as seen earlier (Suzuki et al., 1975; T. Kuiper-Goodman, personal observation).
(b) Dogs
Groups of three to six young beagle dogs were given ochratoxin A by capsule at a dose of 0, 0.1, or 0.2 mg/kg bw per day for 14 days. No changes were observed in renal function, but tubular necrosis and ultrastructural changes in the proximal tubules were observed at all doses. Necrosis of lymphoid tissues of the thymus and tonsils was also seen at all doses (Kitchen et al., 1977a,b,c).
(c) Pigs
In a series of experiments, groups of three to six sows were given feed containing ochratoxin A at a concentration of 0, 0.2, 1, or 5 mg/kg, equivalent to 0, 0.008, 0.04, and 0.2 mg/kg bw per day, for periods of 5 days, 8 or 12 weeks, or up to 2 years. Decreased renal function, nephropathy, and reduced renal enzyme activity were reported. Progressive nephropathy but no renal failure was seen in female pigs given feed containing 1 mg/kg for 2 years; no results were reported for male pigs (Krogh & Elling, 1977; Elling, 1979a,b, 1983; Elling et al., 1985; Krogh et al., 1988).
(d) Chickens
In groups of 10 broiler chicken given ochratoxin A at a dietary concentration of 4 mg/kg for 2 months, the mortality rate was 42%. When the feed was supplemented with 0.8 or 2.4% L-phenylalanine, the mortality rate decreased to 12 and 15%, respectively (Gibson et al., 1990).
Mice
Diets containing ochratoxin A at 0 or 40 mg/kg, equivalent to 5.6 mg/kg bw per day, were fed to groups of adult male 10 ddY mice for 44 weeks, followed by 5 weeks of basal diet. Of the nine surviving treated mice, five had hepatic-cell tumours, nine had renal cystic adenomas, and two had solid renal-cell tumours (terms used by the authors). No hepatic or renal tumours were observed in control mice, and no data on the incidence of these tumours in other control groups of this strain of mice were presented. It was not clear indicated whether the liver tumours were benign or malignant (Kanisawa & Suzuki, 1978).
In a second study from the same laboratory, diets containing ochratoxin A at 0 or 25 mg/kg, equivalent to 3.5 mg/kg bw per day, were fed to groups of 20 6-week-old male DDD mice for 70 weeks. All 20 surviving treated mice had renal cystic adenomas, six had solid renal tumours, and eight had hepatic-cell tumours. One of the 17 control mice had a hepatic-cell tumour (Kanisawa, 1984).
In a third study from the same laboratory, the mice were not exposed for life but for 70 weeks. Diets containing ochratoxin A at 0 or 50 mg/kg, equivalent to 7 mg/kg bw per day, were fed to groups of 16 adult male ddY mice for 5–30 weeks, followed by control diet for 40–65 weeks. No renal or liver tumours were observed in control mice or in mice fed ochratoxin A for 10 weeks. The incidences of renal-cell tumours were 3/15, 1/14, 2/15, and 4/17 after 15, 20, 25, and 30 weeks on the ochratoxin A-containing diet, respectively. The incidence of renal cystic adenomas was not indicated. A significant increase in the incidence of liver tumours was observed after mice had been fed ochratoxin A for 25 weeks (5/15) or 30 weeks (6/17). These results indicated that the renal and liver tumours persisted through subsequent feeding of control diet (Kanisawa, 1984).
In these studies, two types of renal tumour were distinguished by the authors: papillary cyst adenomas (benign) and solid renal-cell tumours, which contained atypical cells, displayed infiltrative growth, and were interpreted by the Committee as malignant. Preneoplastic renal lesions were frequent and multiple and consisted of distended tubules with atypical epithelial cells. No metastases attributable to the kidney or liver tumours were found.
Diets containing ochratoxin A at a concentration of 0, 1, or 40 mg/kg were fed to groups of 50 weanling B6C3F1 mice of each sex for 24 months. The test compound contained about 84% ochratoxin A, 7% ochratoxin B, and 9% benzene. Dead and moribund mice were identified daily. The mice were examined and weighed, and their food consumption was recorded weekly for the first 4 weeks, then monthly. Animals at the high dose showed decreased body weights, by 25% for females and 33% for males, indicating that the maximum tolerated dose was exceeded, although no other signs of toxicity were observed. Nephropathy, characterized by cystic dilatation of renal tubules often with hyperplasia of the lining epithelium, was seen only in mice fed diets containing the highest concentration and was more severe in males than in females. No nephropathy was found in males or females given a control diet or the lower concentration of ochratoxin A. Benign and malignant renal tumours were seen only in male mice fed diets containing the high concentration, at incidences of 53% and 29%, respectively (combined incidence, 63%). No metastases from the renal tumours were found.
When the combined incidence of hepatocellular adenomas and carcinomas in treated mice was compared with that in concurrent controls, the increase was statistically significant in both male and female mice given the high dose; however, the 20% incidence in males was within the range of past controls of 0–22% for this strain of mouse, but the 14% incidence in females was greater than the incidence of 0–3.9% in previous controls (Ward et al., 1979). The authors noted that the ochratoxin A used in their study contained 9% benzene, a proven carcinogen, and thus the possibility of synergism must be considered. The presence of renal tumours in males did not decrease their survival rate. In fact, the survival rates of males at 18 months were 75% in the controls and 65% among those at 1 mg/kg of diet, compared with 98% for those at 40 mg/kg of diet, owing to a high incidence of fatal obstructive urinary-tract disease among the controls and low-dose mice, with onset as early as 4 months (Bendele et al., 1985a). It was suggested that the apparent protective effect of ochratoxin A at 40 mg/kg of diet was due to inhibition of the growth of gram-positive bacteria and to the induction of polyuria as a result of renal proximal tubule damage (Bendele & Carlton, 1986). Group caging and fighting-related lesions of the prepuce and penis may have contributed to the chronic uropathy (Rao, 1987).
Rats
Groups of 80 male and female Fischer 344/N rats were given ochratoxin A by gavage in maize oil at a concentration of 0, 21, 70, or 210 µg/kg bw per day, 5 days/week for 9 months, 15 months, or 103 weeks. The rats were observed twice daily, and body weights and food consumption were recorded weekly for the first 13 weeks and then monthly. Feed and water were available ad libitum. Groups of 15 rats of each sex were killed after 9 and 15 months. The body weight of rats at the highest dose was decreased by 4–7% between 18 and 77 weeks for male rats and between 6 and 89 weeks for female rats. No compound-related clinical signs were seen, and the results of haematological and serum chemical analyses showed no effects of biological significance. Urinary analysis indicated a mild to moderate change in the ability to concentrate urine, with no other change in renal function.
The incidences of renal adenomas in males were 1/50, 1/51, 6/51, and 10/50 and those of renal carcinomas were 0/50, 0/51, 16/51, and 30/50, in the four groups, respectively. The combined incidences of renal tubule-cell adenomas and carcinomas were 20/51 and 36/50 at the two higher doses. At the highest dose, many of the renal adenomas and carcinomas were multiple or bilateral. There was a dose-related increase in the number of males found dead or moribund before the terminal sacrifice (7, 19, 23, and 26, respectively, at 0, 21, 70, and 210 µg/kg bw per day). The decreased survival rates among rats at the two higher doses were attributed by the authors to the presence of kidney tumours, since 15/23 and 18/26 rats that died at these two doses had kidney tumours. In addition. a larger proportion of animals that died before the terminal sacrifice had carcinomas that had become metastatic (3/8 and 11/15 at the intermediate and high doses, respectively) than of animals killed at terminal sacrifice (0/7 and 3/15 at the intermediate and high doses, respectively). In male rats given the low dose of ochratoxin A, only one kidney tumour was present, although the decrease in survival was similar to that of rats at the two higher doses. The reduced survival of this group must therefore be attributed to a non-neoplastic treatment-related effect. In females, the combined incidences of renal adenomas and carcinomas were 0/50, 0/51, 2/50, and 8/50 at 0, 21, 70, and 210 µg/kg bw per day, respectively. The significance of the ochratoxin A-induced renal carcinomas in rats is increased by the high frequency of metastases, attributed to renal-cell carcinomas, mainly in the lungs and lymph nodes. Females at the high dose also had a greater multiplicity of fibroadenomas in the mammary gland (14/50) than controls and rats at lower doses (4–5/50).
The non-neoplastic lesions involved mainly the kidney. Chronic diffuse nephropathy, common to old rats, was seen at about the same incidence in all groups, but the extent and grade were not reported. Karyomegaly or karyocytomegaly (large kidney epithelial cells with giant polyploid nuclei and prominent nucleoli) was seen in all males and females at the two higher doses, and it was the most consistent finding in these groups at the the 9- and 15-month interim sacrifices as well as in a preliminary 13-week study (National Toxicology Program, 1989).
In reviewing these data at its forty-fourth meeting, the Committee noted that renal carcinomas were found in 16/51 male rats at 70 µg/kg bw per day and in 30/50 at 210 µg/kg bw per day; no carcinomas were found at the lower doses. In female rats, renal carcinomas were less common, with 0/50, 1/50, and 3/50 animals showing carcinomas at the low, intermediate, and high doses, respectively. Renal adenomas were found in all groups of male rats, increasing in frequency with increasing dose. In the female rats, renal adenomas were found only at the two higher doses. Fibroadenomas in the mammary gland were found in 45–56% of treated females, a significantly higher percentage than in the control group (Annex 1, reference 117).
The slides of the kidneys from the National Toxicology Program study were reviewed subsequently (Hard, 2000). The review confirmed that the site of injury was the straight segment of proximal tubule S3 in the outer stripe of the outer medulla. In the 2-year bioassay, the lesion consisted of contraction and disorganization of the normal linear pattern of the S3 tubules due to marked development of karyomegaly and cytomegaly. This change showed a clear dose–response relationship in both males and females. The 16-day and 13-week studies showed that this response was preceded by focal tubule basophilia involving mainly the outer stripe of the outer medulla, associated with single-cell death, increased mitotic activity, and some simple tubule hyperplasia. Other non-neoplastic lesions involving only the outer stripe of the outer medulla in the 2-year bioassay were dilated atypical tubules, chromophobic tubules, and cystic tubules, the latter being more prominent in females than in males. The review also confirmed that low (microgram) concentrations of ochratoxin A induced a high incidence of renal tubule tumours (74% in males at the high dose), with carcinomas predominating over adenomas. The carcinomas had a relatively rapid onset, progressing with malignant and aggressive behaviour, some tumours showing a tendency towards an uncommon anaplastic phenotype. There was a relatively high incidence of metastasis, and some tumours were undoubtedly the cause of death. These various features of the ochratoxin A-induced tumours distinguish them from the kidney tumours induced by model non-genotoxic renal carcinogens such as d-limonene and chloroform. However, the tendency towards anaplasia and their aggressive nature were reminiscent of renal tubule tumours induced by fumonisin B1. Renal tumour development was clearly related to the site of ochratoxin A-induced tubule damage, in that preneoplastic atypical tubule hyperplasia, adenomas, and very early carcinomas developed within the outer stripe of the outer medulla. However, a mode of action of sustained cytotoxicity and compensatory cell regeneration coupled with simple tubule hyperplasia, although a possibility, could not be established within the limits of conventional histology alone. Nevertheless, the very high incidence of renal neoplasms, their relatively rapid onset and highly malignant behaviour, coupled with a tendency towards an aggressive anaplastic phenotype and their contribution to death all favour a conclusion that ochratoxin A-induced renal tumour development occurs via DNA reactivity.
The Committee noted that the long-term effects were preceded by evidence of renal toxicity in the 16-day and 13-week studies. It is unclear whether the malignancy and aggressive nature of the tumours is a secure indication that the mechanism of induction is via DNA reactivity. The analogy with tumours induced by fumonisin B1 is not evidence of a genotoxic mechanism, since it has been postulated that the mechanism by which fumonisins induce tumours may be indirect, involving altered sphingolipid metabolism.
The results of studies of genotoxicity with ochratoxin A are summarized in Table 3.
Table 3. Results of assays for genotoxicity with ochratoxin A
|
Test system |
Test object |
Concentration |
Results |
Reference |
|
In vitro |
||||
|
Reverse mutation |
S. typhimurium TA 98, TA100, TA1535, TA1537, TA1538 |
0.4–400 µg/plate |
Negative (highly variable TA100 controls, not tested to cytotoxicity) |
Wehner et al. (1978); Kuczuk et al. (1978) |
|
Reverse mutation |
S. typhimurium TA100, TA1538 |
~ 200 µg/plate |
Negative with mouse and rat liver activation |
Bartsch et al. (1980) |
|
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537, TA1538 |
50–600 µg/plate |
Negative |
Bendele et al. (1985b) |
|
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537, TA1538, G46, G3076, D3052 |
0.1–100 µg/ml |
Negative al. (1985b) |
Bendele et |
|
Reverse mutation |
S. typhimurium TA1538 |
0.1–500 µg/plate (mixture of ochratoxin A:ochratoxin B, 17) |
Positive > 100 µg/plate |
Kuczuk et al. (1978) |
|
Reverse mutation |
S. typhimurium TA97, TA98, TA100, TA1535 |
1–100 µg/plate |
Negative with hamster or rat liver activation |
National Toxicology Program (1989) |
|
Reverse mutation |
S. typhimurium TA98, TA1535, TA1538 |
0–1200 µg/plate |
Positive only after activation by mouse kidney microsomes |
Obrecht-Pflumio et al. (1999) |
|
Reverse mutation |
S. typhimurium TA100, TA2638 |
0–200 µg/plate |
Negative in preincubation assay with mouse liver and kidney, and isolated enzyme activation systems |
Zepnik et al. (2001) |
|
Reverse mutation |
S. typhimurium TA98, TA100, TA1535, TA1537, TA1538 |
|
Positive after activation by medium derived from hepatocytes exposed to ochratoxin A |
Hennig et al. (1991) |
|
Gene mutation |
S. cerevisiae D3 |
75, 200 µg/plate |
|
Kuczuk et al. (1978) |
|
Gene mutation |
B. subtilis rec |
20–100 µg/disc |
Negative |
Ueno & Kubota (1976) |
|
DNA repair |
E. coli, SOS assay |
1–2 mg/100 µl |
Negative |
Reiss (1986); Auffray & Boutibonnes (1986) |
|
DNA repair |
E. coli WP2 |
Gradient plate, not stated |
Negative |
Bendele et al. (1985b) |
|
Forward gene mutation |
Mouse lymphoma cells, Tk locus |
0.1–13 µg/ml |
Negative (>12 µg/ml cytotoxic) |
Bendele et al. (1985b) |
|
Gene mutation |
C3H mouse mammary cells |
5–10 µg/ml |
Negative (10 µg/ml cytotoxic) |
Umeda et al. (1977) |
|
Gene mutation (lacZ on shuttle vector) |
NIH 3T3 cells transfected with human cytochrome P450 |
25 µg/ml |
Positive |
De Groene et al. (1996) |
|
Unscheduled DNA synthesis |
Fischer 344 rat primary hepatocytes |
0.000025–500 µg/ml (2 lots tested at 15 doses) |
Negative (> 0.05 µg/ml cytotoxic) |
Bendele et al. (1985b) |
|
Unscheduled DNA synthesis |
ACI rat primary hepatocytes |
0.4, 4 µg/ml |
Weakly positive at 0.4, cytotoxic at 4.0 µg/ml |
Mori et al. (1984) |
|
Unscheduled DNA synthesis |
C3H mouse primary hepatocytes |
4, 40 µg/ml |
Weakly positive at 4.0, cytotoxic at 40 µg/ml |
Mori et al. (1984) |
|
Unscheduled DNA synthesis |
Rat hepatocytes; porcine urinary bladder epitheilial cells |
250 nmol/L–1 µmol/L |
Positive |
Dorrenhaus & Follmann (1997) |
|
Unscheduled DNA synthesis |
Cultured human urothelial cells |
0.005–0.05 µmol/L |
Positive |
Flieger et al. (1998) |
|
Unscheduled DNA synthesis |
Primary human urothelial cells |
10–2000 nmol/L |
Positive |
Dorrenhaus et al. (2000) |
|
DNA strand break, alkaline elution |
Chinese hamster ovary cells; rat fibroblasts |
200 µg/ml |
Positive (1.2 strand breaks/109 Da) |
Stetina & Votava (1986) |
|
DNA damage |
Mouse spleen, phytohaemagglutinin-stimulated |
1–10 µg/ml |
Positive (dose-related) |
Creppy et al. (1985) |
|
DNA damage, 32P-post-labelling assay |
Mouse kidney, liver, spleen |
0.6, 1.2, 2.5 mg/kg bw |
Positive (‘adducts’ not shown to contain bound ochratoxin A) |
Pfohl-Leszkowicz et al. (1991) |
|
DNA binding |
Rat kidney, liver, seminal vesicle; mouse kidney |
100 µmol/L incubated with S9 protein |
Negative |
Gautier et al. (2001) |
|
Sister chromatid exchange |
Human peripheral blood lymphocytes |
5–10 µg/ml |
Negative (mitotic inhibition at 10 µg/ml) |
Cooray (1984) |
|
Sister chromatid exchange |
Chinese hamster ovary cells, 26 h with ochratoxin A |
0.5–5 µg/ml |
Negative |
National Toxicology Program (1989) |
|
Sister chromatid exchange |
Chinese hamster ovary cells, 2 h with ochratoxin A |
5–160 µg/ml |
Positive (frequency 37% above control, weak dose–response relationship) |
National Toxicology Program (1989) |
|
Sister chromatid exchange |
Human lymphocytes |
|
Positive |
Hennig et al. (1991) |
|
Chromosomal aberration |
Chinese hamster ovary cells, 8–10 h with ochratoxin A |
30–160 µg/ml |
Negative |
National Toxicology Program (1989) |
|
2 h with ochratoxin |
100–300 µg/ml |
Negative |
||
|
Chromosomal aberration |
Human lymphocytes, 48 h with ochratoxin A |
4.5 µg/ml |
Positive (4.5–5-fold increase) |
Manolova et al. (1990) |
|
Micronucleus formation |
Ovine seminal vesicle cell cultures |
12–30 µmol/L |
Positivea |
Degen et al. (1997) |
|
Micronucleus formation |
Syrian hamster embryo fibroblasts |
|
Positiveb |
Dopp et al. (1999) |
|
In vivo |
||||
|
Chromosomal aberration |
Mouse |
1 µg/kg bw per day in diet, 45 days |
Positive (ameliorated by 10 mg/kg bw ascorbic acid) |
Bose & Sinha (1994) |
|
Chromosomal aberration |
Mouse |
1 µg/kg bw per day in diet, 14 days |
Positive (ameliorated by 130 IU vitamin A/kg bw) |
Kumari & Sinha (1994) |
|
Sister chromatid exchange |
Chinese hamster bone marrow |
25–400 mg/kg bw by gavage |
Negative (> 100 mg/kg bw cytotoxic) |
Bendele et al. (1985b) |
|
DNA damage (single-strand breaks) |
BALB/c mouse |
2500 µg/kg bw intraperitoneally |
|
Creppy et al. (1985) |
|
Spleen 4,16, 24 h after treatment |
Positive (max. response at 24 h) |
|||
|
Kidney 24, 48 h after treatment |
Positive (max. response at 24 h) |
|||
|
Liver 24, 48, 72 h after treatment |
Positive (max. resposne at 48 h; recovery at 72 h) |
|||
|
DNA damage |
Wistar rat kidney, liver |
290 µg/kg bw by gavage every 48 h for 6 or 12 weeks |
Positive, no recovery between treatments |
Kane et al. (1986b) |
a No inhibition by indomethacin, suggesting absence of activation by prostaglandin H synthase
b Clastogenic effects due to changes in intracellular calcium
(a) DNA adducts
Almost all the available studies in which DNA adducts were detected by 32P-postlabelling after exposure to ochratoxin A were from one laboratory (Pfohl-Leszkowicz et al., 1991, 1993; Grosse et al., 1995, 1997; Castegnaro et al., 1998; Pfohl-Leszkowicz et al., 1998). All showed positive results in rats and mice given 0.4–2.5 mg/kg bw for 1–16 days or even up to 2 years. The number of adducts ranged from 1 to 200/109 nucleotides in kidney DNA. However, the nonspecific postlabelling technique used may have resulted in adducts that did not contain an ochrotoxin A or ochratoxin A metabolite moiety. At least some of the adducts might have been due to ochratoxin A-induced cytotoxic effects that generate reactive oxygen species. Thus, Grosse et al. (1997) found that prior treatment of rats with superoxide dismutase or catalase, ascorbic acid, or alpha-tocopherol significantly decreased the number of adducts.
Indications that oxidative damage to DNA is not the only source of the presumed adducts are provided by the results of experiments in vitro with purified DNA and mononucleotides incubated with kidney or liver microsomes from mouse and rabbit, ochratoxin A, and either NADPH or arachidonic acid as cofactors (Obrecht-Pflumio & Dirheimer, 2000). Presumed adducts were obtained in all cases but particularly with mouse and rabbit kidney microsomes and arachidonic acid as the cofactor. Liver microsomes were much less active. With NADPH as the cofactor with mouse kidney microsomal enzymes, the adduct level was only 44% that obtained with arachidonic acid. When dAMP, dGMP, dTMP, and dCMP were used as substrates, three adducts were formed with dGMP, mouse kidney microsomes, and either cofactor. However, only one of these adducts was common to the two cofactors. Inhibition of lipid peroxidation and the generation of hydroxyl radicals with desferrioxamine B methanesulfonate did not change the adduct profile. The major adduct obtained with dGMP co-chromatographed with the major adduct obtained with purified DNA. No adducts were obtained with the other three mononucleotides.
In contrast to these results with 32P-postlabelling methods, Schlatter et al. (1996) and Rasonyi (1995) reported that the level of covalent binding of [3H]ochratoxin A to DNA was below the limit of detection (LOD) of scintillation counting in kidney and liver (< 1.3/1010 and 5.6/1011 DNA bases, respectively). In addition, Gautier et al. (2001), using scintillation counting, did not find covalent binding of [3H]ochratoxin A to the DNA of the kidneys of male Fischer 344 rats dosed by gavage 24 h earlier. The LOD was 2.7 adducts/109 purified DNA bases. The authors also used a 32P-postlabelling method with these rat kidney DNA samples and found adducts at levels ranging from 31 to 71/109 DNA bases 24 h after dosing, compared with 6–24/109 DNA bases in untreated controls. Since the adducts occurred at a level 3–17 times higher than the detection limit for scintillation counting and there was no evidence of tritium exchange, most, if not all, of the adducts observed by the 32P-postlabelling method would not have contained an ochratoxin A moiety.
Furthermore, no adducts were found (detection limit, 20 adducts/109 DNA bases) by scintillation counting when DNA and [3H]ochratoxin A were incubated in the presence of male rat kidney microsomes with NADPH, mouse kidney microsomes with NADPH, rat seminal vesicle microsomes with arachidonic acid, or horseradish peroxidase with hydrogen peroxide.
(b) DNA damage and repair
There was no evidence of DNA repair as a result of possible DNA damage in bacteria, whereas DNA single-strand breaks were consistently induced in cultured mammalian cells. DNA single-strand breaks were also observed in vivo in spleen, liver, and kidney cells of mice after intraperitoneal injection of ochratoxin A. DNA repair, manifested as unscheduled DNA synthesis, was observed in most studies with primary cultures of rat and mouse hepatocytes, porcine epithelial cells from bladder, and human urothelial cells.
(c) Gene mutation
Most tests for gene mutation induction in bacteria showed no effect of exposure to ochratoxin A. Two studies showed positive results. One was in S. typhimurium strains TA1535 and TA1538 treated in the presence of mouse kidney microsomes (Obrecht-Pflumio et al., 1999), while the other was in S. typhimurium strains TA1535, TA1538, and TA100 treated with the culture medium of rat hepatocytes exposed to ochratoxin A (Hennig et al., 1991). Both papers described preliminary results that required further investigation before they could be readily accepted. It should be noted, however, that Hennig et al. (1991) obtained negative results with the same bacterial strains when rat liver microsomes were used as the exogenous metabolic activation system. This portion of the results has been confirmed in independent studies in other laboratories.
Gene mutations were not induced in the yeast Saccharomyces cerevisiae D3 (Kuczuk et al., 1978). In mammalian cells, gene mutations were not induced in two studies, while positive results were observed in a third. The last study was performed with NIH 3T3 cells transfected with a human CYP gene (De Groene et al., 1996) at a concentration of 25 µg/ml. In the studies with negative results, concentrations of 10 µg/ml (C3H mouse mammary cells) and > 12 µg/ml (mouse lymphoma L5178Y cells) were cytotoxic. The positive result therefore requires confirmation. No studies of mutation in vivo have been reported.
(d) Chromosomal aberrations
Sister chromatid exchange was induced in two of four studies in vitro but not in a single study in vivo after gavage of a range of doses that included cytotoxic doses.
Chromosomal aberrations were not induced in Chinese hamster ovary cells (National Toxicology Program, 1989) but were induced in cultured human lymphocytes (Manolova et al., 1990), and micronuclei were induced in ovine seminal vesicle cells and Syrian hamster embryo fibroblasts. In vivo, chromosomal aberrations were induced in mouse cells, an effect that was reduced by treatment of the mice with either ascorbic acid (by gavage) or vitamin A (in the diet). These protective effects are consistent with the observation that the formation of 32P-postlabelling spots was prevented in some studies in which mice were treated with ochratoxin A (Grosse et al., 1997).
No adequate studies on the reproductive toxicity of ochratoxin A were available for review. Several studies of effects on developmental toxicity are summarized.
(a) Mice
Groups of 4–26 pregnant CBA mice were given a single dose of ochratoxin A in maize oil by gavage at 0, 1, 2, or 4 mg/kg bw on day 8 or 9 of gestation (day of vaginal plug considered to be day 1 after conception) or 4 mg/kg bw per day 2 days before mating and on days 2, 4, 6, 7, 10, and 14 of gestation, and observed until day 19. At this time, the numbers of viable and dead fetuses and the number of resorption sites were determined, and fetuses were weighed and examined for morphological changes. No mention was made of maternal toxicity. Prenatal survival was decreased in groups that had received 4 mg/kg bw on days 7 (24% deaths), 8 (17% deaths), and 9 (22% deaths) of gestation. Overt craniofacial anomalies were seen only after treatment on day 8 or 9; their incidence, multiplicity, and severity increased with increasing dose, the peak effect being seen on day 9. The incidences of malformed pups among surviving pups were 0%, 0%, 8.1%, and 16% of mice given ochratoxin A at 0, 1, 2, or 4 mg/kg bw on day 8 of gestation, and 0%, 29%, 42%, and 91% of mice given the same doses on day 9 of gestation. The mean number of malformations per fetus was 0.3 and 2.3 on days 8 and 9 at 4 mg/kg bw, and 1.7 and 3.9 in animals given 8 mg/kg bw in a separate study. The central nervous system, the eye, and the axial skeleton were the main systems affected. The most important malformations were those affecting the craniofacial structures, including aplasia and dysplasia of the upper facial structures, such as exencephaly, microcephaly, blunt jaws, anophthalmia, microphthalmia, and median cleft face. In animals treated on day 9 of gestation at 4 mg/kg bw, the incidences of the various major anomalies were exencephaly, 89%; anophthalmia, 45%; microphthalmia, 27%; open eyelids, 16%; agenesis of external nares, 21%; cleft lip, 7.1%; median cleft face, 8.9%; and malformed jaws or short maxilla with protruding tongue, 41%. The craniofacial anomalies were considered by the authors to have arisen from failure of closure of the neurocranium, resulting in abnormal configuration, position, and size of the bones of the base and lateral walls of the skull (Arora & Frölen, 1981).
The effects of protein deprivation on the teratogenic effects of ochratoxin A were studied in groups of 10–13 CD-1 mice, maintained on diets providing 26% (control), 16%, 8%, and 4% purified protein (casein), after mating and throughout gestation. A single dose of ochratoxin A in 0.1 N sodium bicarbonate was administered by gavage at a dose of 0, 2, or 3 mg/kg bw on day 8 of gestation (vaginal plug considered to be day 1), and the mice were killed on day 18 of gestation for examination. The dams were monitored twice daily, and food consumption was monitored. Diets and water were available ad libitum.
Ochratoxin A treatment did not affect maternal food consumption, but maternal deaths were significantly more frequent in the group receiving ochratoxin A at 3 mg/kg bw and 26% protein (five deaths), in that given the same dose and 4% protein (four deaths), and in that given 2 mg/kg bw and 4% protein (nine deaths), with no deaths in the untreated groups. The percentages of litters with grossly malformed fetuses and the percentages of malformed fetuses (in brackets) for each of the four diets (26, 16, 8, and 4% protein, respectively) were 58 (25), 50 (17), 75 (45), and 100 (81) at 3 mg/kg bw; 25 (5), 50 (21), 30 (13), and 100 (78) at 2 mg/kg bw; and 0 (0), 0 (0), 18 (3), and 31 (9.8) without ochratoxin A. The fetal weights were reduced as a result of treatment and protein deprivation. Cranofacial malformations were the commonest abnormality, but at lower protein concentrations gross malformations affecting the limbs and tail were also seen (Singh & Hood, 1985).
In microcephalic mice derived from females treated intraperitoneally with ochratoxin A at 3 mg/kg bw on day 10 of gestation, a quantitative assessment of neurons and synapses at 6 weeks of age showed that the somatosensory cortices of treated mice had fewer synapses per neuron than those of controls, indicating reduced dendritic growth (Fukui et al., 1992).
(b) Rats
Five groups of 12–20 pregnant Wistar rats were given ochratoxin A at a total dose of 5 mg/kg bw in 0.16 mol/L sodium bicarbonate by gavage, as follows: a single dose of 2.5 mg/kg bw on each of days 8 and 9 of gestation (vaginal plug considered to be day 1), a dose of 1.2 mg/kg bw on each of days 8–11 of gestation, a dose of 0.83 mg/kg bw on each of days 8–13 of gestation, or a dose of 0.63 mg/kg bw on each of days 8–15 of gestation. A control group was given the vehicle only. In a similar way, three groups of 20 rats were given ochratoxin A at a single dose of 2.5 mg/kg bw on each of days 8 and 9 of gestation or a dose of 1.7 mg/kg bw on each of days 8–10 of gestation. The rats were killed on day 20 of gestation. No significant difference was seen in the number of implantations per female in the various groups. Females that had received the same total amount of ochratoxin A but divided into fewer single doses and early in gestation were most affected. There was a dose-related increase in the number of resorptions per female and decreases in the mean number of fetuses per female, mean fetal weight, and mean placental weight. A high dose-related incidence of fetal haemorrhages (seen at 2, 2.5, and 4 times the 1.2 mg/kg dose) and coelosome with or without oedema were considered to be teratogenic responses (Moré & Galtier, 1974).
In a study from the same laboratory, a similar protocol for administration of ochratoxin A was used, but the rats were observed until 82 days after birth. Dose-related decreases in the mean number of newborn rats, the mean number of rats alive at 4 days, and the viability index were seen, but not in the lactation index. In the group given 2.5 mg/kg bw twice, the mean body weights of male and female offspring at 82 days were reduced by 12 and 8%, respectively. Hydrocephalus was observed on day 15 after birth in 26% of the male offspring at that dose, and 40% of these animals died by 20 days after birth. A second generation was bred to examine residual maternal or paternal effects, without further administration of ochratoxin A. No differences in reproductive parameters were noted, and few details were given (Moré & Galtier, 1975).
A dose of 0.5 mg/kg bw given by gavage to rats on days 11–14 of gestation caused learning deficits in pups tested for 26 weeks (Kihara et al., 1984).
Oral administration of ochratoxin A to pregnant rats at 1 mg/kg bw per day on days 6–15 of gestation resulted in decreased fetal weight and increased numbers of resorptions but no overt adverse effects on the dams. Skeletal and/or lung malformations were reported in up to 20% of the fetuses; the incidence of renal malformations was 40%. Concurrent administration of methionine at 43 mg/kg bw protected against these adverse effects (Abdel-Wahhab et al., 1999).
Other studies on the teratogenicity of ochratoxin A in mice and rats treated intraperitoneally or subcutaneously were reviewed by Kuiper-Goodman & Scott (1989).
(c) Chickens
The embryotoxic potential of ochratoxin A was tested in chicks by injecting hens’ eggs on day 3 and incubating them until day 13 or 18, when visible abnormalities, weight, and length of chicks were recorded. A dose-related increase in the mortality rate was seen after injection of 1–2 µg of ochratoxin A. An increased frequency of abnormalities was seen in one of the two reported experiments (Edrington et al., 1995). The Committee noted that this is not a validated method, and the results could not be used in risk assessment.
(d) In vitro
Prechondrogenic mesenchymal cells from the limb buds of 4-day-old chick embryos were cultured with ochratoxin A for 6 days. Ochratoxin A inhibited the accumulation of cartilage proteoglycans and general protein synthesis in a dose-related manner (Wiger & Stormer, 1990).
Rat embryos explanted on day 10 of gestation were cultured in a medium containing ochratoxin A at concentrations up to 300 µg/ml. Dose-dependent reductions in the protein and DNA content of the embryos were seen. The malformations induced included hypoplasia of the telencephalon, stunted limb bud development, and decreased size of mandibular and maxillary bones. Cellular necrosis of mesodermal and neuroectodermal structures was observed (Mayura et al., 1989).
(a) Covalent binding to nucleic acids and/or proteins
Preliminary observations indicated no specific binding of ochratoxin A to macromolecules in porcine kidney cytosol (Stojkovic et al., 1984).
Subcellular fractions of a number of kidney-derived cell lines and rat intestine, liver, spleen, kidney, and plasma were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then incubated with ochratoxin A coupled with horseradish peroxidase in order to locate the ochratoxin A-binding proteins. The toxin was shown to bind to virtually all rat blood serum proteins and to some proteins in rat intestine, liver, spleen, and kidney, particularly at 60, 40, and 27 kDa. Binding of ochratoxin A to the 60- and 27-kDa proteins, but not the 40-kDa protein, was inhibited by phenylalanine and aspartame in liver but not in the other organs. The binding of ochratoxin A to cytosolic or organelle proteins was comparable in all the kidney cell lines, which were derived from various species and from various regions of the kidney. Phenylalanine and aspartame had no effect on the binding. The authors concluded that ochratoxin A can bind to several cellular proteins, and that this accounts for its accumulation in cells, but that the results do not explain the protective effects of phenylalanine and aspartame described previously (Schwerdt et al., 1999a).
Mice
The size of the mouse thymus was reduced to 33% that of controls after four intraperitoneal injections of ochratoxin A at 20 mg/kg bw on alternate days, a dose which caused minimal nephrotoxicity. Bone marrow depression was shown as dose-related, significantly (p < 0.01) decreased marrow cellularity, including a reduction in bone marrow macrophage–granulocyte progenitors, a decreased number of haematopoietic stem cells and a significant decrease in erythropoiesis as measured by 59Fe uptake; increased phagocytosis by macrophages was also observed (Boorman et al., 1984).
Residual damage was seen 3 weeks after exposure as increased sensitivity to radiation, even though bone-marrow cellularity and the peripheral blood count had returned to normal (Hong et al., 1988; National Toxicology Program, 1989).
Ochratoxin A administered to 8–10-week-old Swiss mice at 5 mg/kg bw per day by intraperitoneal injection for 50 days reduced the antibody response to Brucella abortis, a cell-mediated immune response. This was postulated to be due to suppression of immunoglobulin (Ig)M synthesis. The same treatment also reduced mitogen (concanavalin A)-induced blast formation in lymphocytes derived from mouse spleen (Prior & Sisodia, 1982).
Groups of eight female BALB/c mice were fed diets containing ochratoxin A at a concentration of 6, 250, or 2600 µg/kg for 28 or 90 days, equivalent to 1, 40, and 400 µg/kg bw per day. Treatment did not cause changes in body or lymphoid organ weights. Kidney weights were reduced at the two higher doses at 28 days and at the highest dose at 90 days. The concentrations of ochratoxin A in kidney were clearly dose-related. No differences in leukocyte count were observed, but a significant reduction in the number of spleen cells (by about 20%) was observed at the highest dose after 90 days. No changes were observed in blood or thymic T lymphocytes at 28 days, but a decreased proportion of mature CD4+ and CD8+ cells was seen with a corresponding increase in the immature double-positive sub-population at the two higher doses after 90 days. After 28 days, the primary (humoral) antibody response to sheep red blood cells was significantly suppressed in a dose-dependent manner at the two higher doses. The antibody response to another T-cell-dependent antigen (viral antigen PR8) was not affected, suggesting that exposure to ochratoxin A alters certain immune functions in mice, and, as previously demonstrated, the spleen may be the most sensitive immune tissue to ochratoxin A. Differences in the proportions of mature and immature CD4+ and CD8+ populations suggest that ochratoxin A may affect late-stage differentiation of T cells (Thuvander et al., 1995).
Female BALB/c mice were given diets containing ochratoxin A to provide a calculated average dietary intake of 5–30 µg/kg bw per day for 2 weeks before mating. At birth, the pups were cross-fostered to unexposed dams. Exposed and control pups were killed at 14 and 28 days of age. Ochratoxin A did not effect the reproductive outcome or body weight of pups. No differences in spleen or thymus weight or cell numbers were observed on day 14, but significant increases were seen in both thymus weights (by 20%) and cell number (by 67%) in the offspring of dams at the high dose on day 28. Although the percentages of splenic CD4+ and CD8+ cells were decreased in pups at the high dose, there were no alterations in absolute numbers. No significant differences were observed in the proliferative responses of splenic or thymic lymphocytes to mitogens nor in the production of interleukin-2 in concanavalin A-stimulated cell cultures. No significant differences in the humoral antibody response to sheep red blood cells or viral antigen PR8 were found. Natural killer cell activity on day 28 was not affected by prenatal exposure to ochratoxin A. Thus, the treatment did not suppress immune function but altered the absolute and relative numbers of lymphocyte subpopulations in lymphoid organs (Thuvander et al., 1996a).
Groups of 10 Han-NMRI mice (sex not specified) received commercial (Serva) or ‘raw’ ochratoxin A at a dose of 1, 3, or 6 mg/kg bw per day by intraperitoneal injection for 8–17 days and were then monitored for up to 20 days. Animals receiving ‘raw’ ochratoxin A at 3 mg/kg bw per day had a significantly lower body weight than controls on days 5–17; however, this correlated with a reduction in feed consumption. No significant change in body weight was noted in the groups receiving crystalline ochratoxin A. The total leukocyte count was unchanged in all groups; however, lymphopenia, neutrophilia, and eosinophilia were observed at 3 and 6 mg/kg bw per day. The blood IgM titre was suppressed at these doses in a dose-dependent manner. The authors concluded that ochratoxin A has a nonselective suppressive effect on various immune reactions, but the paper contains inadequate detail to verify their conclusion (Müller et al., 1995).
Rats
Bone-marrow hypocellularity and a reduced thymic size were also seen in Fischer rats given ochratoxin A at 1 or 4 mg/kg bw per day by gavage for 16 days (National Toxicology Program, 1989).
Necrosis of germinal centres in the spleen and lymph nodes was seen in Wistar rats given a single dose of ochratoxin A at 5–50 mg/kg bw (Kanisaw et al., 1977) and in dogs given ochratoxin A by capsule at doses of 0.1–0.2 mg/kg bw per day for 14 days (Kitchen et al., 1977c).
The effects of ochratoxin A on the bone marrow and lymphatic cell population may reflect the sensitivity of these cells to the inhibition of protein synthesis induced by ochratoxin A. These effects on the structural components of the immune system indicated that ochratoxin A is likely to have an effect on immune function.
The immunotoxic effects of perinatal exposure to ochratoxin A were investigated in the offspring of Sprague-Dawley rats treated singly or repeatedly. In a short-term study, dams received a single oral dose of 10, 50, or 250 µg/kg bw on day 11 of lactation, and the pups were examined on day 14. Dose-dependent uptake of ochratoxin A was observed in both dams and pups. The toxin did not induce consistent changes in the weights of the lymphoid organs of pups. A small but significant increase in the number of thymocytes was observed in offspring of dams dosed at 50 µg/kg bw, but it was not dose-dependent. A small but significant decrease in the proliferative response of splenocytes to T-cell mitogen lipopolysaccharide was seen in pups of dams given 250 µg/kg bw. In contrast, exposure to 10–50 µg/kg bw per day resulted in significant increases in the proliferative responses of both splenocytes and thymocytes of pups to concanavalin A. This was not seen at the higher dose. The authors proposed that short-term exposure of suckling pups via the milk stimulates the immune response, measured as proliferation of lymphocytes in response to concanavalin A and lipopolysaccharide (Thuvander et al., 1996b).
In a long-term study, dams received repeated oral doses of ochratoxin A at 50 µg/kg bw on 5 days/week for 2 weeks before mating, during gestation, and then 7 days/week until weaning. At parturition, the number of pups was reduced to eight per litter and they were cross-fostered to produce groups of prenatally, postnatally, and pre- and postnatally exposed pups. The highest blood concentrations of ochratoxin A were detected in pups exposed both pre- and postnatally, but exposure via the milk appeared to account for most of the content. Long-term exposure to 50 µg/kg bw per day did not induce any consistent changes in body or lymphoid organ weights of pups, but prenatal exposure suppressed the lymphocyte response to both B- and T-cell mitogens at 14 days of age. The background proliferation of unstimulated cells was significantly suppressed in cultures from prenatally exposed pups. These effects were not observed in pups exposed during lactation, although the blood concentrations were higher in pups exposed postnatally. Prenatally exposed pups showed a significantly lower primary antibody response to PR8 viral antigen (± 0.36). No significant difference in the natural killer cell activity of splenocytes was measured in exposed pups at 13 weeks of age. The authors concluded that long-term prenatal exposure to ochratoxin A, but not postnatal exposure via milk, may cause immunosuppression; however, short-term postnatal exposure may stimulate proliferation of lymphocytes in response to mitogens (Thuvander et al., 1996b).
The Committee noted that no details were given about the ochratoxin A used. These authors previously used commercial ochratoxin A, but in this paper they quoted a 1984 reference for details of how the ochratoxin A was produced. The size of the groups was not given, but they seem to have consisted of four to five dams.
Pigs
Groups of six weanling hybrid pigs received either pure or crude ochratoxin A at doses of 7–50 µg/kg bw per day by subcutaneous injection for 19–39 days. The animals were immunized 8 days after ochratoxin A challenge with Pasteurella by inhalation. The authors stated that ochratoxin A had no effect on body-weight gain and that the serum concentrations were dose-dependent. The concentrations were reported to be lower after administration of crude ochratoxin A than pure material. A reduction in relative lymphocyte count and increases in total leukocyte, relative neutrophil, and eosinophil counts were seen. Crude toxin had a greater effect than pure toxin. Ochratoxin A decreased the phagocytosis index of individual cells and decreased expression of SWC1 (a lymphocyte cell surface marker) but did not change lymphocyte proliferation (Müller et al., 1999).
The Committee noted that many of the results were conflicting and the study was inadequately reported.
Chickens
In chickens fed diets containing ochratoxin A at a concentration of 2–4 mg/kg for 20 days, the lymphoid cell population of immune organs was decreased (Dwivedi & Burns, 1984a).
Several studies have shown that ochratoxin A affects both humoral and cell-mediated immunity. In chickens fed a diet containing ochratoxin A at 5 mg/kg for 56 days, the contents of alpha1-, alpha2-, beta-, and gamma-globulins in plasma were reduced (Rupic et al., 1978).
In chickens fed diets containing ochratoxin A at a concentration of 2–4 mg/kg for 20 days, immunoglobulin (Ig)G, IgA, and IgM in lymphoid tissues and serum were depressed (Dwivedi & Burns, 1984b), and complement activity was slightly affected in birds fed at diets containing 2 mg/kg for 5–6 weeks (Campbell et al., 1983).
Ochratoxin A also reduced IgG and increased IgM in the bursa of Fabricius in chick embryos that had been injected with 2.5 µg of the toxin on day 13. This did not affect their immunocompetence, however, as seen after challenge of the hatched chickens with E. coli at 1, 2, and 4 weeks of age, indicating that the effect on immunoglobulins may have been transient (Harvey et al., 1987).
Immunosuppression was observed in chickens fed diets containing ochratoxin A at 0.5 or 2 mg/kg for 21 days. When compared with controls, the treated animals had reduced total serum protein, lymphocyte counts, and weights of the thymus, bursa of Fabricius, and spleen (Singh et al., 1990).
In vitro
The effects of ochratoxin A on T-cell activation were investigated in purified (> 95%) human lymphocytes cultured in medium containing 1% bovine serum albumin. Intracellular free Ca2+ and activation of protein kinase C were measured as indicators of the early stages of activation; the effect on phytohaemagglutinin-induced proliferation was measured as a late event mediated by expression of functional interleukin-2 receptors. The early-stage events were not inhibited by ochratoxin A at a concentration of 12 µmol/L. In contrast, incubation of ochratoxin A with phytohaemagglutinin-stimulated lymphocytes resulted in inhibition of DNA synthesis at concentrations > 6.4 µmol/L. Protein synthesis in resting lymphocytes was markedly inhibited by 12 µmol/L but to a lesser extent in phytohaemagglutinin-stimulated lymphocytes. The authors concluded that ochratoxin A can block DNA synthesis at a late stage in lymphocyte activation and that this effect may be partially mediated by inhibition of protein synthesis (Størmer & Lea, 1995).
Ochratoxin A also inhibited the proliferative response of bovine peripheral blood mononuclear cells cultured in 10% fetal calf serum. The ID50 value varied from 0.1 to 4 µg/ml, depending on the mitogen used to stimulate the cells and the incubation time. The authors considered these results indicative of immunosuppressive potential (Charoenpornsook et al., 1998).
Rats
Three male Wistar rats received 1 nmol (about 400 ng) of ochratoxin A by intracerebral administration and four received a diet containing 290 µg/kg by oral gavage for 8 days. The animals were killed 24 h after dosing. Although ochratoxin A was detected in areas of the central nervous system after intracerebral injection, it was not detected in the periphery or blood, kidney, or urine, indicating that it is transferred poorly or not at all from the spinal fluid to blood, kidney, or urine. After administration in the diet, the ventral mesencephalon, hippocampus, striatum, and cerebellum were the main targets of cytotoxicity in rat brain (Belmadani et al., 1998a)
Four male Wistar rats received ochratoxin A at 290 µg/kg bw orally every 48 h for 1–6 weeks. The treated animals had a slight reduction in body weight after 4 weeks, but feed and water consumption were not significantly different from those of controls. Ochratoxin A accumulated in the brain in a linear time-dependent manner, to reach about 100 ng/g of brain after 6 weeks. The toxin was shown to change the concentrations of the amino acids tyrosine and phenanthrene and to damage tissues in the hippocampus (Belmadani et al., 1998b)
Ten adult female Fischer rats received ochratoxin A at 120 µg/kg bw per day by oral gavage for 10, 20, or 35 days. Treatment altered the activity of all enzymes tested. Significant increases in gamma-glutamyl transferase activity were observed in the three brain regions examined. The changes in the other enzyme activities were regionally selective, but most of the activities had returned to control levels by day 35 of dosing (Zanic-Grubisic et al., 1996).
(b) In vitro
The neurotoxicity of ochratoxin A has been investigated in nerve tissue cell cultures (embryonic chick neural retina and brain) and cultured meningeal fibroblasts. The cells were incubated with ochratoxin A in serum-free medium for 8 days. The median inhibitory concentration (IC50) for a number of parameters of cytotoxicity (cellular protein, 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide [MTT] reduction, neutral red uptake) were found to be about 170 nmol/L in all three culture systems, indicating that ochratoxin A did not have cell-specific effects. Ochratoxin B and the heat-induced 3S-epimer of ochratoxin A induced comparable effects at 19- and 10-fold higher concentrations, respectively (Bruinink et al., 1997).
In a study with a similar protocol, markers of neuritic outgrowth and differentiation (NF68 and 160 kDa, MAP2 and MAP5) were affected at significantly lower concentrations than the markers of cytotoxicity. Although the presentation of the data is unclear, the IC50 values for the most sensitive parameters appeared to be 20–50 nmol/L for the embryonic brain and neural retinal cultures. Binding of ochratoxin A to bovine serum albumin resulted in significantly decreased potency (IC50 values increased by 15–30-fold). Differences were noted between serum-free primary cultures and the cell lines. In these culture systems, phenylalanine did not decrease the effects of ochratoxin A and in contrast appeared to cause a concentration-related decrease in the IC50 [no statistical analysis presented]. The authors concluded that ochratoxin A specifically affected neurite formation and that its toxicity was decreased by protein binding but not by phenylalanine (Bruinink & Sidler, 1997).
These authors also investigated whether the effects of ochratoxin A could be attributed to its isocoumarin structure, by comparing the toxicity of ochratoxin A with that of ochratoxin alpha and ochracin in serum-free embryonic chick brain cultures. Ochratoxin A decreased the end-points at concentrations > 15 nmol/L, with a greater effect on neurite outgrowth (neurofilament 68 kD). Ochratoxin alpha and ochracin had minimal effects at concentrations up to 1 mmol/L. The isocoumarin structure was therefore considered not to be responsible for the toxicity of ochratoxin A in this brain cell culture model (Bruinink et al., 1998).
The regional selectivity of ochratoxin A was investigated in primary cultures of neurons and astrocytes isolated from embryonic or newborn rat brain ventral mesencephalon and cerebellum. The cultures were exposed to ochratoxin A in a medium containing 10% fetal calf serum for 46 h, before measurement of DNA and protein synthesis, lactate dehydrogenase leakage, and lipid peroxidation. Ochratoxin A inhibited protein and DNA synthesis in all cell types, with IC50 values ranging from 14 to 69 µmol/L. Neuronal cells were more sensitive than astrocytes, and the cells of the ventral mesencephalon were more sensitive than those of the cerebellum. Increases in lactate dehydrogenase leakage and lipid peroxidation were also seen, but the sensitivity of the cell types did not mirror that for DNA and protein synthesis. The authors concluded that ochratoxin A is neurotoxic and may affect particular structures of the brain (Bruinink et al., 1998).
In vivo
Renal function and morphology are greatly affected at high doses of ochratoxin A, as indicated by increased kidney weight, urine volume, blood urea nitrogen (Hatey & Galtier, 1977), urinary glucose, and proteinuria (Berndt & Hayes, 1979). The last two findings indicate that the site of reabsorption, i.e. the proximal convoluted tubules, is damaged. The NOELs for changes in renal function depend on the species and on the parameter tested. At low doses of ochratoxin A, no increase in blood urea nitrogen, creatinine, or glucose was found in the urine of male or female rats given 210 µg/kg bw per day by gavage for 6–12 months, but a mild to moderate decrease in the ability to concentrate urine was seen. The NOEL for this effect was 70 µg/kg bw per day for male rats and 21 µg/kg bw per day for female rats (National Toxicology Program, 1989).
Various groups of investigators have shown that this specific nephrotoxic effect is due to an ochratoxin A-induced defect of the organic anion transport mechanism located on the brush border of the proximal convoluted tubule cells and basolateral membranes (Endou et al., 1986; Sokol et al., 1988). The organic ion transport system is also the mechanism by which ochratoxin A enters proximal tubular cells (Friis et al., 1988; Sokol et al., 1988).
The middle (S2) and terminal (S3) segments of the proximal tubule of isolated nephron segments were found to be the most sensitive to the toxic effects of ochratoxin A (0.05 mmol/L), as shown by a significant decrease in cellular ATP and a dose-related decrease in mitochondrial ATP content (Jung & Endou, 1989).
Several investigators have measured the effect of ochratoxin A on the release of enzymes from the kidney into the urine. Changes in enzyme and protein patterns can be used to distinguish different types of renal injury (Stonard et al., 1987).
Subcutaneous doses of ochratoxin A at 10 mg/kg bw for 5 days decreased the activity of muramidase and then decreased the activities of lactate dehydrogenase, alkaline phosphatase, glutamate dehydrogenase, and acid phosphatase in the kidney (Ngaha, 1985). The activities of alanine peptidase, leucine amino peptidase, and alkaline phosphatase were decreased by 60%, 50%, and 35%, respectively in isolated kidney tubules in the presence of 0.1 mmol/L ochratoxin A (Endou et al., 1986).
In male rats given ochratoxin A at 0.1–2 mg/kg bw per day orally for 2–5 days, the phosphoenolpyruvate carboxykinase activity decreased by 50–70% at the highest dose (Meisner et al., 1983; Meisner & Krogh, 1986). The minimum effect level was 0.1 mg/kg bw per day (Meisner & Polsinelli, 1986); at 2 mg/kg bw per day, enzymes such as pyruvate carboxylase, malate dehydrogenase, hexokinase, and gamma-glutamyl transpeptidase were not affected (Meisner & Selanik, 1979).
In rats given ochratoxin A by gavage at a dose of 0.14 mg/kg bw every 48 h (equivalent to about 2 mg/kg diet) for 8–12 weeks, the activities of lactate dehydrogenase, alkaline phosphatase, leucine aminopeptidase, and gamma-glutamyl transferase decreased significantly. The last three enzymes are located in the brush border of the proximal convoluted tubules, indicating damage at that site. Concomitantly with the decrease of enzyme activity in the kidney, these enzymes appeared in the urine. A late event was a urinary increase in the activity of N-acetyl beta-D-glucosidase, a lysosomal enzyme. The activity of this enzyme in the kidney was not affected (Kane et al., 1986a). The late appearance of this enzyme may indicate active regeneration and exfoliation of necrotic proximal convoluted tubular cells, releasing lysosomal enzymes (Stonard et al., 1987). In this study, para-aminohippurate clearance was reduced initially by 56% at 2 weeks and 8% at 12 weeks of dosing, indicating damage followed by regeneration.
Pigs are very sensitive to the effect of ochratoxin A on renal enzyme activity. In the kidneys of pigs fed diets containing ochratoxin A at 0.2–1 mg/kg, equivalent to 0.008–0.041 mg/kg bw per day, a dose-related decrease in the activity of phosphoenolpyruvate carboxykinase and gamma-glutamyl transpeptidase was accompa-nied by a dose-related decrease in renal function, as indicated by a reduction in the maximal tubular excretion of para-aminohippurate per clearance of inulin and an increase in glucose excretion. Only cytosolic, and not mitochondrial, phosphoenol-pyruvate carboxykinase activity was inhibited (Meisner & Krogh, 1986; Krogh et al., 1988).
Subcutaneous administration of superoxide dismutase and catalase together was found to offset the nephrotoxic effects of ochratoxin A, leading to the suggestion that superoxide radicals and hydrogen peroxide are likely to be involved in the nephrotoxic effects of ochratoxin A in vivo (Baudrimont et al., 1994).
Male Wistar rats weighing 70–150 g were given ochratoxin A in order to determine its effects on the pH in the vasa recta of the renal papilla after a single intravenous injection pf 3 µmol/kg bw or six intraperitoneal injections of 1.2 µmol/kg bw per day. Both regimes increased the pH in the descending and ascending vasa recta, with no significant difference between the renal arterial and aortic pH values, in serum and urine osmolality, or in urinary flow rate. An increased pH over that before treatment was detected in the collecting ducts of individual animals after an intravenous dose, but the differences between groups were not significant. Treated rats had a lower body-weight gain (19%) than controls (34%) during the 6-day intraperitoneal treatment. The authors suggested that ochratoxin A upsets pH homeostasis in the interstitium of the renal papilla, leading to alkalinization, in addition to impairment of urinary acidification (Kuramochi et al., 1997a).
In a subsequent study in which male Wistar rats were treated intravenously with ochratoxin A at 3 µmol/kg bw, the pH in the proximal tubule, distal tubule, and collecting ducts and the descending and ascending vasa recta was increased. The concentration of bicarbonate ion increased significantly in the proximal tubule and collecting ducts of treated animals, but there were no significant differences between treated and control animals in any of the parameters in aortic blood. No significant differences were observed in pCO2 in any region, and there were no significant differences in serum or urine osmolality or urinary flow rate. The authors concluded that ochratoxin A increased the pH and bicarbonate ion concentration in the tubular fluid or vasa recta but did not alter pCO2. They hypothesized that this disturbance in pH homeostasis could contribute to alterations in acid–base status and hence to the nephrotoxicity of ochratoxin A (Kuramochi et al., 1997b).
Groups of Wistar rats (sex not specified) were given ochratoxin A at 0, 0.4, or 0.8 mg/kg bw intraperitoneally every 72 h for 90 days in order to investigate the relationship between the pathogenesis of nephropathy and the genotoxic and carcinogenic effects of ochratoxin A. Treatment resulted in significant, dose-related decreases in relative kidney weight (80 and 77% of control), average kidney length (83 and 78% of control), and creatinine clearance (90 and 76% of control) at 0.4 and 0.8 mg/kg bw, respectively. Severe renal atrophy was reported in animals at both doses, but these were not dose-related. Dose-dependent concentrations of ochratoxin A were found in the blood (920 and 1900 ng/ml) and kidney (30 and 170 ng/g) at the two doses, respectively. The urinary concentration was similar at both doses, consistent with the low urinary elimination of ochratoxin A. Histological examination of the kidneys of rats given 0.8 mg/kg bw every 72 h for 30 days showed giant karyomegalic tubule cells with limited degeneration of interstitial tissue and fewer apoptotic bodies in the tubule epithelium than in the kidneys of control animals. The authors also found abnormal mitoses after 30 days of dosing and suggested that regeneration would have occurred if treatment had been stopped (Maaroufi et al., 1999).
Administration of ochratoxin A at 1 or 3 mg/kg of diet and cholestyramine at 1 or 5% of diet for up to 14 days decreased the plasma concentrations of ochratoxin A and the urinary and biliary excretion of this toxin and its metabolites. Increased concentrations of ochratoxin A were found in the faeces. The authors suggested that cholestyramine may have decreased the absorption of ochratoxin A, either by interfering with bile acid secretion or by direct binding (Kerkadi et al., 1998).
These authors subsequently confirmed that cholestyramine can bind ochratoxin A and bile salts in vitro and that depletion of bile salts by interruption of enterohepatic circulation in rats resulted in decreased plasma concentrations of ochratoxin A (Kerkadi et al., 1999).
In vitro
When ochratoxin A was added to isolated rat renal proximal tubules in suspension, mitochondrial dysfunction was seen as an early event in the process of nephrotoxicity. Mitochondrial impairment apparently occurred at sites I and II of the respiratory chain. Although lipid oxidation occurred before cell death, it did not seem to be responsible for the toxic effect (Aleo et al., 1991).
The effects of ochratoxin A on cell growth, cell viability and transepithelial transport were investigated in male Wistar rat proximal tubule cells in serum-free primary culture. Biphasic effects were reported, depending on the concentration of ochratoxin A (0.1–10 µmol/L) and the incubation time (24–72 h). The authors considered that these effects could be explained by accelerated cell growth, followed by decreased DNA synthesis due to increased cell density. Addition of albumin or acidification of the culture medium reduced the effects of ochratoxin A in a concentration-dependent manner, whereas alkalinization to pH 7.7 had little effect. Transepithelial electrolyte transport was disrupted at 10 µmol/L but not at 0.1 µmol/L. The authors concluded that ochratoxin A at physiological (nanomolar) concentrations can stimulate proliferation of proximal tubule cells without exerting toxic effects or reducing cell viability, and that this effect may be mediated by ochratoxin A-induced changes of cellular pH homeostasis (Gekle et al., 1995).
Transport of ochratoxin A by the kidney-specific organic anion transporter 1 (ochratoxin AT1) was investigated in Xenopus oocytes, which transiently express ochratoxin AT1, and cultured S3 cells, with stable expression of ochratoxin AT1, in 5% fetal calf serum. Oocytes with ochratoxin AT1 took up significantly more ochratoxin A (4 µmol/L) than oocytes transfected with the vector alone. para-Aminohippurate, probenecid, prioxicam, octanoate, and citronin, which have been reported previously to inhibit the nephrotoxicity of ochratoxin A, inhibited uptake. Uptake of the compound was also greater in ochratoxin AT1-expressing S3 cells than in the mock-transfected parent cells. Cell proliferation was significantly decreased by ochratoxin A at 2 and 10 µmol/L, and their viability was decreased by 10 µmol/L, in ochratoxin AT1-expressing S3 cells but not in the mock-transfected cells. Incubation with para-aminohippurate suppressed the effects of ochratoxin A. The authors concluded that ochratoxin AT1 plays a pivotal role in the nephrotoxicity of mycotoxins (Tsuda et al., 1999).
The transport of ochratoxin A across the renal peritubular membrane was studied in suspensions of freshly isolated rabbit renal proximal tubules, in order to investigate whether the accumulation of ochratoxin A in proximal tubule cells is involved in its nephrotoxicity. The accumulation and kinetics were determined by fluorescence, which correlates linearly with the concentration of ochratoxin A over a range of 1–70 nmol/L. Accumulation of 10 µmol/L was approximately linear for 60 s and approached steady state after 5 min. The uptake was almost completely blocked by 2 mmol/L probenecid, which inhibits the organic anion transport pathway. para-Aminohippurate, which is the prototypic substrate for the peritubular organic anion transporter, also inhibited ochratoxin A uptake, but only by 40–50% at a concentration of 2.5 mmol/L, indicating that uptake occurs by a mechanism in addition to the organic anion transporter. Use of other inhibitors indicated that phenylalanine was not involved, but that a fatty acid transporter may also contribute to uptake of ochratoxin A. The overall results suggest that the peritubular membrane is a significant site for accumulation of ochratoxin A (Groves et al., 1998).
The potential of ochratoxin A to induce apoptosis was examined in human proximal tubule-derived cells (IHKE cells) and compared with that in renal-cell lines derived from opossum proximal tubule (OK cells) and from canine renal collecting duct (MDCK-C11 and MDCK-C7 cells), cultured in 19% fetal calf serum. Ochratoxin A induced a time- and concentration-dependent increase in caspase 3 activity in IHKE cells. Significant increases were seen at 5 nmol/L and a 7-day incubation and at 10 nmol/L with a 24 or 72-h incubation. DNA fragmentation and chromatin condensation confirmed the occurrence of apoptosis at concentrations of 30 and 100 nmol/L. The free-radical scavenger N-acetylcysteine and the intracellular calcium chelator BAPTA-AM, had no effect on ochratoxin A-induced caspase 3 activation, indicating that the mechanism did not involve free-radical production or disturbed calcium homeostasis. IKHE cells were more sensitive to low concentrations of ochratoxin A than MDCK-C11, MDCK-C7, or OK cells. The authors concluded that low concentrations of ochratoxin A led to caspase 3 activation and subsequently apoptosis in cultured human proximal tubule cells but that the mechanism was unclear (Schwerdt et al., 1999b).
The ability of ochratoxin A to activate c-Jun N-terminal kinase has also been investigated in two clones of the MDCK kidney-derived cell line (C7 resembling principal cells and C11 resembling intercalated cells), cultured in 10% fetal calf serum. Incubation with ochratoxin A for 8 h at 10 nmol/L, 100 nmol/L, or 1 µmol/L resulted in stimulation of c-Jun N-terminal kinase 1 in MDCK-C7 cells but not in MDCK-C11 cells. Apoptosis, as measured by caspase 3 activity and DNA fragmentation, was observed in MDCK-C7 cells treated with ochratoxin A at 100 nmol/L, which did not cause necrosis as measured by leakage of the cytosolic enzyme lactate dehydrogenase. MDCK-C11 cells were less responsive than MDCK-C7 cells, with a smaller increase in caspase 3 activity at 300 nmol/L ochratoxin A. Lactate dehydrogenase leakage was proportional to DNA fragmentation, indicating that ochratoxin A primarily caused necrosis in MDCK-11 cells. Ochratoxin A at 0.1–0.5 µmol/L also potentiated the pro-apoptotic action of tumour necrosis factor-alpha in a concentration-dependent manner, with a greater effect in MDCK-C7 cells than in MDCK-C11 cells. The cell specificity demonstrated in this study indicates that c-Jun N-terminal kinase signalling pathways may play a role in ochratoxin A-induced apoptosis. The authors suggested that this may explain some of the changes in renal function and teratogenicity induced by the toxin (Gekle et al., 2000).
Ochratoxin A inhibited protein synthesis and caused leakage of cytosolic enzymes in Vero monkey kidney cells cultured in the presence of 5% newborn calf serum. Incubation with ochratoxin A for 24 h in the presence of aspartame at 250 µmol/L increased the IC50 for inhibition of protein synthesis from 14 µmol/L to 22 µmol/L. An increase to 34 µmol/L was seen when the cells were incubated with the same concentration of aspartame for 24 h before addition of ochratoxin A. Aspartame was also shown to prevent binding of ochratoxin A to plasma proteins and to displace ochratoxin A already bound to plasma proteins. The authors concluded that aspartame could decrease the toxicity of ochratoxin A by affecting binding to plasma proteins as well as by preventing inhibition of protein synthesis (Baudrimont et al., 1997). The Committee noted that these effects required high concentrations of aspartame.
(e) Mechanism of tumorigenesis
The mechanisms of tumour induction in rodent kidney by ochratoxin A have been addressed in many studies, including investigations of the role of biotransformation and bioactivation and the formation of ochratoxin A-derived nucleic acid derivatives in target and non-target organs for toxicity. The results diverge, as do those of the studies on mutagenicity. Although no definite mechanism for the carcinogenicity of ochratoxin A to rodent kidney has been described, non-genotoxic events make a major contribution to the induction and progression of ochratoxin A-derived renal tumours.
Several studies have addressed the biotransformation of ochratoxin A and its role in its toxicity. Biotransformation has been postulated to be involved in the DNA binding and renal tumorigenicity of ochratoxin A, and a variety of CYPs, peroxidases, and glutathione S-transferases have been suggested to catalyse the transformation of ochratoxin A to reactive intermediates (Hietanen et al., 1991; Hennig et al., 1991; Würgler et al., 1991; Malaveille et al., 1994; Fink-Gremmels et al., 1995; Obrecht-Pflumio et al., 1996; Grosse et al., 1997; Pfohl-Leszkowicz et al., 1998; Obrecht-Pflumio et al., 1999; El Adlouni et al., 2000). However, none of these studies assessed the capacity of the respective enzymes to transform ochratoxin A to metabolites or suggested the structure(s) of a reactive metabolite (Castegnaro et al., 1998). Most studies assessed potentially relevant end-points in the toxicity of ochratoxin A and their modulation by changes in xenobiotic-metabolizing enzyme activities. Because of these limitations, no conclusions can be drawn about the mechanisms of ochratoxin A-induced tumour formation in rat kidney.
The possible biotransformation reactions of ochratoxin A have been postulated on the basis of rigorous analytical chemistry. Formation of an ochratoxin A-derived reactive quinone was suggested (Gillman et al., 1999), but this metabolite was formed only by a chemical system that mimics the CYP system. The ochratoxin A-derived reactive quinone was not detected by the use of isolated enzymes and microsomes with high activity for specific CYPs, and only 4R- and 4S-hydroxy-ochratoxin A were formed at very low yields (Gautier et al., 2001; Zepnik et al., 2001). Subcellular fractions rich in prostaglandin synthase activity or purified CYP enzymes also did not catalyse the formation of reactive ochratoxin A metabolites (Gautier et al., 2001).
The known mechanisms of formation of ochratoxin A metabolites (insertion of an oxygen into a carbon–hydrogen bond) do not suggest formation of reactive and toxic intermediates. The lack of involvement of CYP-mediated oxidation in the toxicity of ochratoxin A is supported by the observation that increasing the rates of biotransformation of the toxin by induction of CYP decreases its renal toxicity (Omar et al., 1996), and the observation of typical toxic effects of ochratoxin A in cell systems with very low or no CYP activity (Seegers et al., 1994; Hoehler et al., 1996; Xiao et al., 1996; Dopp et al., 1999). The formation of ochratoxin A-derived radicals capable of interacting with macromolecules is also not indicated. In contrast, the electron spin resonance spectra suggest the formation of hydroxy radicals (Hoehler et al., 1996, 1997).
Formation of DNA adducts has also been postulated as an important event in the tumorigenicity of ochratoxin A. The formation of spots interpreted as ochratoxin A-derived DNA adducts was observed in target tissues in rodents by the very sensitive 32P-postlabelling assay. The nature of the DNA damage and/or mutations caused by ochratoxin A is unknown (Pfohl-Leszkowicz et al., 1991; Würgler et al., 1991; Grosse et al., 1995, 1997; Obrecht-Pflumio & Dirheimer, 2000). The end-points in many of the studies on the mechanisms of tumorigenicity of ochratoxin A was the possible formation of DNA adducts (spots by 32P-postlabelling). However, a role of DNA binding of ochratoxin A is not supported by the results of studies of biotransformation cited above or of experiments to investigate the binding of radiolabelled ochratoxin A to nucleic acids (Gautier et al., 2001). Studies of DNA binding with [3H]ochratoxin A revealed no binding of ‘metabolically activated’ ochratoxin A to calf thymus DNA in vitro or to DNA from rat liver or kidney in vivo. The sensitivity of these experiments was similar to that of the postlabelling studies. Lack of DNA binding of ochratoxin A or its metabolites was observed in vivo after administration of a single dose of [3H]ochratoxin A (Rasonyi, 1995).
In summary, these data cast doubt on the hypothesis that ochratoxin A causes renal tumours by covalent binding of reactive intermediates to DNA. The hypothesis that DNA damage induced by ochratoxin A is due to oxidative stress represents an alternative explanation for the discrepant data and is more consistent with the observations. Several experimental observations support this hypothesis. An unusually large number of DNA adducts (up to 30 individual adducts) was formed from ochratoxin A in low yields in various experimental systems (Castegnaro et al., 1998; Pfohl-Leszkowicz et al., 1998). Patterns of modifications similar to those observed with ochratoxin A by postlabelling were observed in kidney DNA of rodents exposed to iron(III) nitrilotriacetate (Randerath et al., 1995), a renal carcinogen that acts through oxidative stress, or in DNA exposed to hydrogen peroxide (Randerath et al., 1996). Some of these results are consistent with a major role of oxidative stress in the toxicity of ochratoxin A. For example, antioxidants prevent the induction of DNA damage by ochratoxin A in mice (Grosse et al., 1997).
Induction of renal toxicity, oxidative stress due to mitochondrial dysfunction, and persistent cell proliferation represent an alternative mechanism for the renal carcinogenicity of ochratoxin A. The toxin is known to induce oxidative stress (Aleo et al., 1991) and the formation of hydrogen peroxides (Omar et al., 1990). In addition, mechanisms linked to long-term renal toxicity and oxidative stress are known to play an important role in tumour induction in rat kidney (Swenberg & Maronpot, 1991; Dietrich & Swenberg, 1993; Hard, 1998). Several non-genotoxic chemicals that do not undergo bioactivation reactions induce renal tumours in rodents. For example, DNA damage and cellular toxicity mediated by oxidative stress seem to be involved in the renal carcinogenicity of iron(III) nitrilotriacetate and potassium bromate in rodents. These compounds are potent renal carcinogens and induce renal tumours in rodents in high yields after short exposure (Li et al., 1987; Wolf et al., 1998). Sex differences in tumour incidences are also seen with these compounds. For example, as seen with ochratoxin A, male rats are more susceptible to renal tumour induction by potassium bromate (Kurokawa et al., 1983, 1990; Umemura et al., 1998).
(f) Mechanisms of cytotoxicity
Ochratoxin A induced apoptosis in the HL-60 human promyelotic leukaemia cell line as seen by a DNA fragmentation technique and ultrastructural observation. Incubation for 24 h with ochratoxin A at a concentration of 3–4 µg/ml resulted in both apoptosis and cytotoxicity, as measured by MTT reduction (Ueno et al., 1995).
A brief report on the possible etiology of Balkan endemic nephropathy noted that apoptosis was not observed in kidneys of rats given ochratoxin A in the diet at 0.8 mg/rat per day for 5 days, which was sufficient to cause extensive necrosis (Mantle et al., 1998). No other details were available.
The toxicity of ochratoxin A, three natural analogues, and 10 synthetic analogues was compared in vitro and in vivo in order to identify the active moiety of the ochratoxin A structure. The studies in vivo involved intraperitoneal injection of mice and intravenous injection of rats, with lethality as the end-point. The hydroxyl, carboxyl, chlorine, and lactone groups of ochratoxin A affected its bactericidal activity (Bacillus brevis), its cytotoxicity to HeLa cells, and its toxicity to mice and rats. Its biological reactivity may be partly associated with the lactone carbonyl group of the isocoumarin moiety. There appeared to be no direct relationship between toxicity and the extent of iron chelation. In addition, formation of a previously undescribed ring-opened metabolite of ochratoxin A was detected in the bile but not in the blood or urine of rats after instillation of 100 µg of ochratoxin A into the carotid artery (Xiao et al., 1996).
The Committee noted that these studies are not helpful for risk assessment, because high doses were given by injection and lethality was the only end-point. Furthermore, the studies were inadequately reported.
(g) Effects on the male reproductive system
Ochratoxin A inhibited testosterone secretion in isolated testicular interstitial cells of gerbils (Fenske & Fink-Gremmels, 1990).
Male rats treated by gavage with ochratoxin A at 290 µg/kg bw every second day for up to 8 weeks showed a twofold increase in the testicular content of testosterone and accumulation of premeiotic germinal cells, as measured by increases in alpha-amylase, alkaline phosphatase, and gamma-glutamyl transpeptidase activities in testis homogenate. All of these effects were indicative of a disturbance of spermatogenesis (Gharbi et al., 1993).
Groups of 20 Peterson x Hubbard broiler chickens were fed diets containing ochratoxin A alone at 0 or 2.5 mg/kg of diet or in combination with cyclopiazonic acid for 3 weeks. A significant reduction in body-weight gain was seen by the second week of feeding and was still present at the third week (by 19%). The relative kidney weight was increased in the group given ochratoxin A, and significant increases in serum uric acid and triglycerides but decreased total protein, albumin, and cholesterol were seen (Gentles et al., 1999).
Commercial ochratoxin A was administered orally at a dose of at 20 or 40 µg/day for 5 weeks to groups of two sexually mature male Hungarian large white and Dutch Landrace boars weighing 250 kg. Ochratoxin A was detected in serum and seminal plasma of both groups (Solti et al., 1996).
Ochratoxin A has a half-life of about 35 days in humans (Bauer & Gareis, 1987; Hagelberg et al., 1989; Studer-Rohr et al., 1995), and the blood concentrations are considered to represent a convenient biomarker of exposure during recent weeks. This biomarker has been used extensively in epidemiological studies (see below). Similar estimates of exposure have been derived from dietary surveys and from blood analyses, suggesting that the latter is a reliable biomarker.
The nephrotoxic effect of ochratoxin A is detectable by urinary analysis, but this is a relatively non-specific effect and late in onset. Anaemia is an early manifestation but is also non-specific, and early diagnosis is difficult.
Since ochratoxin A was suggested to be a possible determinant of endemic nephropathy, considerable efforts have been made to determine a correlation between human exposure to this toxin and the incidence of the disease. Endemic nephropathy is a fatal human renal disease, recognized as a specific entity and affecting predominantly rural populations in limited areas of the central Balkan peninsula. So far, the disease has been reported in Bosnia and Herzegovina, Bulgaria, Croatia, Romania, and Yugoslavia (Serbia). The disease was first recognized in the 1950s (Tancev et al., 1956), but there is evidence that it occurred even earlier (Belicza et al., 1979).
The disease starts without an acute episode. Onset is common between the ages of 30 and 50, although there have been reports of patients aged 10–19 (Stoyanov et al., 1978). Its progress is very slow, and after development of nonspecific signs and symptoms there is atypical manifestation of renal impairment (Radonic et al., 1966). The effect on the primary tubules is characterized by a decrease in tubular transport and becomes evident through proteinuria. As a rule, the proteinuria is very mild and is accompanied by the characteristic presence of low-relative-molecular-mass proteins (Hall & Vasiljevic, 1973). Anaemia of the normochromic type is among the first signs of the disease and precedes clinical manifestation of renal impairment (Radonic et al., 1966). The ultrasonic appearance of the kidney is normal at the early stage of the disease, but it becomes smaller as the disease progresses (Borso, 1996). Since there are neither characteristic clinical data nor pathognomonic laboratory indicators, the early diagnosis of endemic nephropathy is difficult and relies on repeated findings of proteinuria, creatininaemia, anaemia, and a family history of the disease.
The prevalence rate of the disease is reported to be 2–10%. In the endemic area of Croatia, a systematic field survey of cases between 1975 and 1990 revealed a prevalence of 0.5–4.4%. The average specific mortality (based on official statistics and documented cases) during the period 1957–84 was 1.5/1000 per year, although some studies have shown that the mortality rate is actually more than twice as high. The disease affects more women than men, and women die more frequently from endemic nephropathy (Ceovic et al., 1992).
A remarkable reduction in the size of the kidney is seen post mortem: in one extreme case, one organ weighed only 20 g. In almost all advanced cases, a characteristic pale dirty yellow discolouration of the skin was common, with a peculiar yellowish colouration of the adipose tissue. The shrinking is progressive, and the organs can be normal or rather small in the early stages of the disease. The kidneys are pale grey and hard to cut (Vukelic et al., 1991). Pathomorphologically, the disease can be described as interstitial, bilateral, non-inflammatory, and non-obstructive nephropathy with heavy damage to the tubular epithelium and extensive interstitial fibrosis starting in the cortex (Vukelic et al., 1992).
The reported incidence of epithelial tumours of the upper urinary tract is much higher in endemic than in non-endemic areas (Chernozemsky et al., 1977; Nicolov et al., 1978; Ceovic & Miletic-Medved, 1996). In the endemic region of Croatia, the prevalence of tumours of the pyelon and ureter is 11 times that in the non-endemic area (Vukelic et al., 1987). Of the malignant tumours, transitional-cell carcinomas were the most frequent (95%); squamous-cell carcinomas were seen in only 5% of cases. Generally, the differences in urothelial tumours between endemic and non-endemic regions include the following: the incidence of tumours is higher in the endemic region, and they affect younger people and women more frequently; the renal pelvis and urethra are the usual sites of tumours in the endemic region, whereas in non-endemic regions the most frequent site is the urinary bladder (Vukelic & Sostaric, 1991). A study of 766 patients treated at the Belgrade Department of Urology for upper urinary tract tumours in 1970–97 showed that the incidence of these tumours was 68% in patients from endemic and probably endemic regions and 32% in patients from non-endemic regions in Yugoslavia (Serbia). The tumours were more frequent in women. A much higher incidence of bilateral tumours was reported in patients from the endemic region (13%) than from non-endemic regions (2%) (Djokic et al., 1999; Table 4).
Table 4. Incidence, by anatomical location, of urothelial tumours among inhabitants of areas endemic and non-endemic for endemic nephropathy
|
Anatomical location |
Endemic area |
Non-endemic area |
||
|
(10 094 inhabitants) |
(96 306 inhabitants) |
|||
|
No . |
% |
No : |
% |
|
|
Pyelon |
29 |
0.286 |
20 |
0.021 |
|
Ureter |
9 |
0.089 |
13 |
0.013 |
|
Urinary bladder |
23 |
0.23 |
86 |
0.089 |
|
Combination |
6 |
0.059 |
7 |
0.007 |
|
Total |
67 |
0.66 |
126 |
0.13 |
From Vukelic et al. (1992)
Striking similarities between the changes in the renal structure and function found in endemic nephropathy and in ochratoxin A-induced porcine nephropathy suggested a common causal relationship (Krogh, 1974). Epidemiological similarities, in particular the endemic occurrence (Krogh, 1976), support the hypothesis that ochratoxin A is a causative agent of endemic nephropathy in humans.
Although ochratoxin A has been found as a contaminant of food and feed all over the world (Krogh, 1992), food samples collected in the endemic areas showed higher contamination. In 1979 in the endemic region of Croatia, ochratoxin A was found in 9.4% of food samples. In a 5-year study in Bulgaria in which 524 food samples from endemic and control villages were analysed, the frequency of positive samples from endemic villages was several times higher than that from non-endemic villages (Pavlovic et al., 1979).
Ochratoxin A was first detected in humans in blood samples from inhabitants of endemic villages (Hult et al., 1982), at a much higher concentration than in non-endemic villages. The prevalence rate was 17% in endemic and 6.0% in non-endemic villages, and similar rates were found in blood samples from endemic (18%) and non-endemic (7.7%) areas in Bulgaria (Petkova-Bocharova et al., 1988).
Low blood concentrations of ochratoxin A have been found in countries where endemic nephropathy has not been detected, such as Canada, the Czech Republic, Egypt, France, Germany, Italy, Sweden, Switzerland, and Tunisia (Bauer & Gareis, 1987; Hadlok, 1993; Breitholtz-Emanuelsson et al., 1994; Zimmerli & Dick, 1995; Malir et al., 1998; Wafa et al., 1998). Some regional differences in exposure to ochratoxin A have been found (Breitholtz et al., 1991; Creppy et al., 1993; Maaroufi et al., 1995a; Scott et al., 1998), and in Croatia in a study of blood from donors in five major cities (Peraica et al., 1999). The mean concentration in the 250 samples was 0.39 ng/ml of plasma, and 59% of samples contained the toxin (detection limit, 0.2 ng/ml). The highest frequency of positive samples (100%), the highest mean ochratoxin A concentration (0.68 ng/ml), and the largest number of samples with a concentration > 1.0 ng/ml (18%) were found in a city relatively near the endemic region. The concentrations reported in blood from healthy persons are shown in Table 5.
Table 5. Occurrence of ochratoxin A in blood samples from healthy persons
|
Country |
Period of collection |
Positive/analysed |
Concentration (ng/ml) |
Reference |
||
|
No . |
% |
Mean |
Range |
|||
|
Bulgaria |
1984–90 |
9/125 |
7 |
|
1.0–10 |
Petkova-Bocharova et al. (1991) |
|
Canada |
1994 |
144/144 |
100 |
0.88 |
0.29–2.4 |
Scott et al. (1998) |
|
Czechoslovakia |
1990 |
35/143 |
24 |
0.14 |
0.1–1.3 |
Fukal & Reisnerova (1990) |
|
Czech Republic |
1994 |
734/809 |
91 |
0.23 |
0.1–14 |
Malir et al. (1998) |
|
1995 |
404/413 |
98 |
0.24 |
0.1–1.9 |
||
|
Croatia |
1997 |
148/249 |
59 |
0.39 |
0.2–16 |
Peraica et al., (1999) |
|
Denmark |
1986 |
|
|
1.5 |
0.1–9.7 |
Hald (1991) |
|
1987 |
|
|
2.3 |
0.1–9.4 |
||
|
1988 |
|
|
1.6 |
0.1–13 |
||
|
France |
1991–92 |
|
|
|
|
Creppy et al. (1993) |
|
Alsace |
|
97/500 |
19 |
|
0.1–12 |
|
|
Aquitaine |
|
385/2055 |
19 |
|
0.1–160 |
|
|
Rhone-Alpe |
|
75/515 |
15 |
|
0.1–4.3 |
|
|
Germany |
1977 |
84/165 |
51 |
0.79 |
0.1–14 |
Bauer & Gareis (1987) |
|
1985 |
89/141/ |
63 |
0.42 |
0.1–1.8 |
||
|
1988 |
142/208 |
68 |
0.75 |
0.1–8.4 |
Hadlok (1993) |
|
|
Hungary |
1995 |
291/355 |
82 |
|
0.2–10 |
Solti et al. (1997) |
|
1997 |
213/277 |
77 |
|
0.1–1.4 |
Tapai et al. (1997) |
|
|
Italy |
1992 |
65/65 |
100 |
0.53 |
0.1–2.0 |
Breitholtz-Emanuelsson et al. (1994) |
|
Japan, Tokyo |
1992–96 |
156/184 |
85 |
0.068 |
0.004–0.28 |
Ueno et al. (1998) |
|
Poland |
1983–84 |
25/397 |
6 |
0.21 |
1.0–13 |
Golinski (1987) |
|
1984–85 |
52/668 |
8 |
0.31 |
1.0–40 |
||
|
Sierra Leone |
1996 |
12/36 |
33a |
|
1.5–18 |
Jonsyn (1996) |
|
Spain |
1996–98 |
40/75 |
53 |
0.71 |
0.5–4.0 |
Jimenez et al. (1998) |
|
Switzerland |
1992–93 |
|
|
|
|
Zimmerli & Dick (1995) |
|
North of Alps |
251/252 |
100 |
|
0.06–2.1 |
||
|
South of Alps |
116/116 |
100 |
|
0.11–6.0 |
||
|
Sweden |
1989 |
|
|
|
|
Breitholtz et al. (1991) |
|
Visby |
|
29/99 |
29 |
0.26 |
0.3–7.0 |
|
|
Uppsala |
|
3/99 |
3 |
0.02 |
0.3–0.8 |
|
|
Ostersund |
|
6/99 |
6 |
0.03 |
0.3–0.8 |
|
|
|
1990–91 |
39/39 |
100 |
0.17 |
0.09–0.94 |
Breitholtz-Emanuelsson et al. (1993b) |
|
Tunisia |
1993–95 |
73/140 |
52 |
1.2 |
0.1–8.8 |
Maaroufi et al. (1995a |
From Peraica et al. (1999). Mean concentration calculated for all samples, range given only for positive samples
a
Non-breastfed infants up to 5 years of ageOchratoxin A has been found in human milk. Nine of 50 samples of milk from women in various regions of Italy contained the toxin, at concentrations of 1.2–6.6 ng/ml of milk (Micco et al., 1991).
Ochratoxin A has been found in many commodities, including cereals, cereal products, coffee, grapes, dried vine fruit, grape juice, wine, cocoa and chocolate, beer, meat, pork products, pulses, milk and milk products, and spices. Several published analytical methods for the determination of ochratoxin A in maize, barley, wheat, wheat bran, wheat wholemeal, rye, wine, beer, and roasted coffee have been formally validated in collaborative studies. The methods are based on liquid chromatography (LC) with fluorescence detection, include a solid-phase extraction clean-up step with reversed-phase C18, silica gel 60, or immunoaffinity columns, and can guarantee detection of < 0.5 µg/kg. These methods have also been used successfully to analyse a number of other cereals, cereal products, and dried fruit.
The first LC method for determining ochratoxin A in maize and barley was validated in a collaborative study with materials spiked with ochratoxin A in the range of 10–50 ng/g. Ochratoxin A was extracted from grains with chloroform:aqueous phosphoric acid and isolated by liquid–liquid partitioning into aqueous bicarbonate solution that had been cleaned-up on a C18 (solid-phase extraction) cartridge. Identification and quantification were performed by reversed-phase LC with fluorescence detection. The identity of ochratoxin A in samples that contained it was confirmed by methyl ester derivatization followed by LC analysis (Nesheim et al., 1992). The performance characteristics achieved in an international collaborative study involving 16 laboratories are shown in Table 6. The method, which is quantitative for ochratoxin A at concentrations > 10 µg/kg in maize and barley, has been accepted as final-action AOAC International Official Method 991.44.
Table 6. Results of collaborative study for determination of ochratoxin A in maize and barley
|
Matrix |
Mean |
No. acceptable results |
Mean recovery |
RSDr |
RSDR |
|
Maize |
0.8 |
15 |
– |
– |
– |
|
8.2 |
15 |
82 |
– |
21 |
|
|
16 |
15 |
82 |
20 |
28 |
|
|
40 |
14 |
77 |
– |
32 |
|
|
Barley |
0.8 |
15 |
– |
– |
– |
|
7.4 |
15 |
74 |
– |
27 |
|
|
14 |
14 |
72 |
7.9 |
26 |
|
|
3 |
15 |
74 |
– |
28 |
RSDr, relative standard deviation for repeatability; RSDR, relative standard deviation for reproducibility
This method was successively validated for other cereals and at lower ochratoxin A concentrations (Larsson & Moeller, 1996). Spiked and naturally contaminated barley, wheat bran, and rye containing ochratoxin A at concentrations of 2–9 µg/kg were used. The performance characteristics achieved in the international collaborative study involving 12 laboratories are shown in Table 7. The European Committee for Standardisation (CEN, technical committee 275/WG5 ‘Biotoxins’), which uses specific criteria to select methods, has adopted this method as CEN standard (EN ISO 15141-2) for determination of ochratoxin A in barley, maize, and wheat bran.
Table 7. Results of collaborative study for determination of ochratoxin A in rye, barley, and wheat bran
|
Matrix |
Mean |
No. acceptable |
Mean recovery |
RSDr |
RSDR |
|
Rye |
2.8 |
12 |
64 |
22 |
29 |
|
4.8 |
12 |
65 |
16 |
23 |
|
|
Barley |
2.9a |
12 |
– |
17 |
22 |
|
3.0a |
12 |
– |
15 |
23 |
|
|
Wheat bran |
3.8 |
12 |
70 |
21 |
24 |
|
4.5 |
12 |
68 |
17 |
26 |
For abbreviations, see Table 6.
a
Naturally contaminated sampleThe second LC method, adopted as CEN standard (EN ISO 15141-1) for determination of ochratoxin A in cereals and cereal products, was validated in a collaborative study on wheat wholemeal containing ochratoxin A at 0.4 or 1.2 µg/kg (Majerus et al., 1994). Ochratoxin A was extracted from grains with toluene after addition of hydrochloric acid and magnesium chloride solution. The filtered extract was cleaned-up on a mini-silica gel column, and ochratoxin A was determined by reversed-phase LC with fluorescence detection. The performance characteristics achieved in the collaborative study involving 13 laboratories according to ISO 5725: 1986 are shown in Table 8. Laboratory experience has shown that this method is also applicable to cereals, dried fruits, oilseeds, pulses, wine, beer, fruit juices, and raw coffee (Jiao et al., 1992; Majerus et al., 1993; Jiao et al., 1994).
During the past few years, the use of antibody-based immunoaffinity columns in the clean-up step has improved the analysis of ochratoxin A. Two methods based on immunoaffinity clean-up for determination of ochratoxin A in barley and roasted coffee have been developed and validated in collaborative studies under the auspices of the European Commission, Standard and Measurement Testing programme (Entwisle et al., 2000a,b). Ochratoxin A was extracted from barley with acetonitrile: water solution, and the filtered sample extract was diluted with phosphate-buffered saline that had been cleaned-up by passage through an immunoaffinity column. For the determination of ochratoxin A in roasted coffee, phenyl silane solid-phase extraction clean-up before the immunoaffinity column stage was introduced to avoid any deleterious effects of caffeine on the immunoaffinity columns (Koch et al., 1996; Entwisle et al., 2000b). In both these methods, ochratoxin A was identified and quantified by reversed-phase LC with fluorescence detection. Spiked and naturally contaminated samples containing ochratoxin A at 1.2–3.7 µg/kg were used in the collaborative study. These two methods have been accepted by AOAC International as first-action methods and are being considered for adoption by the CEN as standards. The performance characteristics achieved in the collaborative studies involving 15 laboratories are shown in Tables 9 and 10 for barley and roasted coffee, respectively.
Table 8. Results of collaborative study for determination of ochratoxin A in wholemeal wheat
|
Matrix |
Mean |
No. acceptable results |
Mean recovery |
RSDr |
RSDR |
|
Wheat, |
0.41 |
13 |
80 |
15 |
26 |
|
wholemeal |
1.2 |
13 |
80 |
20 |
32 |
For abbreviations, see Table 6.
Table 9. Results of collaborative study for determination of ochratoxin A in barley
|
Matrix |
Mean |
No. of acceptable results |
Mean recovery |
RSDr |
RSDR |
|
Barley |
0.1a |
14 |
– |
26 |
72 |
|
1.3b |
15 |
– |
24 |
33 |
|
|
3.0b |
14 |
– |
12 |
17 |
|
|
4.5b |
12 |
– |
14 |
15 |
|
|
3.7 |
12 |
93 |
4 |
12 |
For abbreviations, see Table 6.
a
Blank sampleb
Naturally conataminated sampleTable 10. Results of collaborative study for determination of ochratoxin A in roasted coffee
|
Matrix |
Mean |
No. of acceptable results |
Mean recovery |
RSDr |
RSDR |
|
Roasted coffee |
0.1a |
13 |
– |
27 |
71 |
|
1.2a |
14 |
– |
22 |
26 |
|
|
2.6a |
15 |
– |
11 |
15 |
|
|
5.4a |
12 |
– |
2 |
14 |
|
|
3.5 |
13 |
85 |
6 |
13 |
For abbreviations, see Table 6.
a
Naturally contaminated sampleThe occurrence of ochratoxin A in wine, especially red wine, was first reported by Zimmerli and Dick (1995). The analytical method they used involved extraction of ochratoxin A by liquid–liquid partitioning with chloroform, clean-up on an immunoaffinity column, and determination by reversed-phase LC with fluorescence detection. A more accurate and precise method has since been developed for determination of ochratoxin A in red, rosé, and white wine (Visconti et al., 1999) and beer (Visconti et al., 2000a). After simple dilution with water containing polyethylene glycol and NaHCO3, wine or beer samples were cleaned-up on immunoaffinity columns and analysed by reversed-phase LC with fluorescence detection. The method was validated in a collaborative study. It has been adopted as a First Action Method by AOAC International and is being considered for adoption by the CEN as a standard (Visconti et al., 2000b). The performance characteristic obtained in the collaborative study are reported in Table 11.
Table 11. Results of collaborative study for determination of ochratoxin A in wine and beer
|
Matrix |
Mean |
No. of acceptable results |
Mean recovery |
RSDr |
RSDR |
|
White wine |
< 0.01a |
– |
– |
– |
– |
|
0.10 |
13 |
100 |
10 |
14 |
|
|
1.0 |
14 |
91 |
6.6 |
14 |
|
|
1.8 |
14 |
88 |
8.5 |
13 |
|
|
0.28b |
15 |
– |
11 |
15 |
|
|
Red wine |
< 0.01a |
– |
– |
– |
– |
|
0.19 |
12 |
93 |
5.5 |
9.9 |
|
|
0.81 |
14 |
90 |
9.9 |
12 |
|
|
2.5 |
15 |
85 |
8.9 |
13 |
|
|
1.7b |
14 |
– |
11 |
13 |
|
|
Beer |
< 0.01a |
– |
– |
– |
– |
|
0.19 |
13 |
95 |
10 |
18 |
|
|
0.70 |
15 |
87 |
7.2 |
18 |
|
|
1.4 |
13 |
94 |
4.6 |
16 |
|
|
0.069b |
14 |
– |
19 |
20 |
For abbreviations, see Table 6.
a Blank sample
b Naturally contaminated sample
The European Commission, Measurement and Testing Programme sponsored projects to improve the method and to prepare certified reference materials for determination of ochratoxin A in wheat and pig kidney (Hald et al., 1993; Wood et al., 1996; Entwisle et al., 1996; Wood et al., 1997; Williams et al., 1998). The study in wheat involved 26 European laboratories and was conducted in three phases: a first comparison of procedures, a second comparison of procedures, and certification of two reference materials. Various procedures were compared in the first step, including chloroform, methanol, toluene, or ethyl acetate for extraction, and silica, reversed-phase, or immunoaffinity columns for clean-up. LC was used for deter-mination in all laboratories except one, where thin-layer chromatography TLC) was used (Hald et al., 1993). In the second comparison of procedures, all laboratories used high-performance liquid chromatography (HPLC) for quantification of ochratoxin A, whereas acetonitrile, chloroform, dichloromethane, ethyl acetate, methanol, and toluene were used as extraction solvents. Various clean-up procedures were used, including silica, reversed-phase, diatomaceous earth, and immunoaffinity columns and liquid–liquid partitioning. Fifteen of 26 participants had results that fulfilled the agreed acceptance criteria and therefore participated in the certification study (Wood et al., 1996). The results of nine of these 15 laboratories were accepted for certification of the two wheat reference materials. The methods used by these nine laboratories for setting the certification value of ochratoxin A in blank and naturally contaminated wheat flour are summarized in Table 12. The two certified reference materials are useful for ensuring the quality of analyses and can be used to prepare in-house reference materials easily and inexpensively. Certified reference materials for mycotoxins, including ochratoxin A, are available from the European Union Joint Research Centre, Institute for Reference Materials and Measurements, Geel, Belgium.
Table 12. Methods used by laboratories accepted for setting the certification value for ochratoxin A in wheat flour
|
Extraction |
No. of laboratories |
Clean-up |
|
25-g test portion |
4 |
Silica column (1 laboratory) |
|
125 ml CHCl3 |
|
C18 column (2 laboratories) |
|
12.5 ml 0.1 mol/L H3PO4 |
|
Liquid–liquid defatting (1 laboratory) |
|
25-g test portion |
3 |
Silica column |
|
125 ml toluene |
|
|
|
30 ml 2 mol/L HCl |
|
|
|
25 ml 0.4 mol/L MgCl2 |
|
|
|
25-g test portion |
1 |
Immunoaffinity column |
|
24 ml acetonitrile |
|
|
|
16 ml phosphate-buffered saline |
|
|
|
25-g test portion |
1 |
C18 column |
|
80 ml ethyl acetate |
|
|
|
8 ml 5% acetic acid |
|
|
All laboratories used liquid chromatography with reversed-phase C18 and fluorescence detection
Two comparative studies of methods for the analysis of ochratoxin A in freeze-dried pig kidney were performed in order to determine the feasibility of preparing certified reference materials. In the first study, almost all of the 20 European laboratories reported lower than usual recoveries for the freeze-dried material (Entwisle et al., 1996). The methods used were similar to those used in the comparative studies for ochratoxin A in wheat (see above). The results of the second comparative study of methods for the analysis of ochratoxin A in freeze-dried pig kidney indicated the following: the reconstitution of freeze-dried material is crucial, as small lumps of powdered material may be formed; in recovery experiments, the spiking solution must be added to reconstituted material and not to powdered material in order to avoid formation of small lumps; immunoaffinity clean-up resulted in clearer extracts and chromatograms than reversed-phase or silica gel columns or liquid–liquid partitioning; the use of 1 mol/L instead of 0.1 mol/L phosphoric acid in the extraction step did not improve the recovery of ochratoxin A (Williams et al., 1998).
Screening methods based on TLC are available, and one has been collaboratively validated (Nesheim et al., 1973). These methods are used in only a few laboratories since they do not provide an adequate limit of quantification (LOQ). Enzyme-linked immunoabsorbent assays (ELISAs) have been developed for the detection of ochratoxin A in pig kidney, animal and human sera, cereals, and mixed feed. The results obtained with these methods require confirmation since the antibodies produced often show cross-reactivity to compounds similar to ochratoxin A. These methods were not used in the surveys considered by the Committee.
Because of the large number of commodities contaminated with ochratoxin A, several LC analytical methods have been proposed. Validated analytical methods are available for accurate and precise determination of ochratoxin A in maize, barley, rye, wheat, wheat bran, wheat wholemeal, roasted coffee, wine, and beer. The introduction of immunoaffinity columns has improved analytical methods for ochratoxin A. Use of these columns reduces the need for dangerous solvents, drastically improves the clean-up of extracts, improves detection, and simplifies sample preparation and clean-up.
The analytical methods used for the determination of ochratoxin A in foods are summarized in Table 13.
Table 13 (a). Analytical methods used to determine ochratoxin A in foods
|
Reference |
Commodity |
Test portion |
Extraction |
Clean-up |
Quantification |
|
Wolff et al. (2000) |
Wine, juices, oil, vinegar |
5 (ml) |
Diluted with 1 ml PBS |
Immunoaffinity column |
HPLC (RP18)/ fluorescence detection |
|
Wolff et al. (2000) |
Meat and meat products |
25 |
HCl–MgCl2 solution + CHCl3 |
Liquid–liquid partitioning with NaHCO3 + immunoaffinity |
HPLC (RP18)/ |
|
Wolff et al. (2000) |
Cereals |
40 |
30 ml HCl–MgCl2 + 125 ml toluene |
SiO2 column Sep pak |
HPLC (RP18)/ |
|
Wolff et al. (2000) |
Beer |
5 (ml) |
Diluted with 1 ml PBS |
Immunoaffinity column |
HPLC (RP18)/ |
|
Wolff et al. (2000) |
Coffee, tea |
25 |
500 ml H2O–NaHCO3 |
Dilution with PBS and clean-up on immunoaffinity column |
HPLC (RP18)/ |
|
Wolff et al. (2000) |
Milk products, sweets, oilseeds |
50 |
200 ml acetonitrile: water (6:4) 2-min blending |
Dilution with PBS and clean-up on immunoaffinity column |
HPLC (RP18)/ |
|
Langseth et al. (1989); Langseth (1999) |
Cereals |
25 |
125 ml CHCl3 + 12.5 ml 0.1 mol/L H3PO4; shaking 60 min |
Silica Sep-pack cartridge or immunoaffinity column |
HPLC (RP18)/ |
|
Larsson & Moeller (1996) |
Wheat, barley, rye |
50 |
25 ml 0.1 mol/L H3PO4 + 250 ml CHCl3 + 10 g diatomaceus earth; 3-min blending |
Liquid–liquid partitioning with 3% NaHCO3 + C18 (Sep-pack) |
HPLC (RP18)/ |
|
Soares et al. (1985) |
Corn, peanuts, beans, rice, cassava |
50 |
270 ml MeOH + 30 ml 4% KCl; 5 min blending |
Clarification with (NH4)2SO4 and Hyflo Super-Cel; dilution with water and liquid-liquid partitioning with CHCl3 |
Visual TLC (20 x 20 cm silica gel 60) |
|
Trucksess et al. (1999) |
Wheat, barley, coffee |
25 |
100 ml MeOH:1% NaHCO3 (7+3); 3-min blending |
Dilution with PBS and clean-up on Immunoaffinity |
HPLC (RP18)/ |
|
Solfrizzo et al. (1998) |
Wheat, oats |
10 |
60 ml CHCl3 + 5 ml 0.1 mol/L H3PO4 30 min shaking |
Dried extract dissolved in HPLC mobile phase and defatted with n- |
HPLC (RP18)/ |
|
Jorgensen et al. (1996) |
Wheat, rye, barley, oats, bran, pork, poultry meat and liver |
50 |
250 ml CH2Cl2:EtOH (4+1) + 25 ml 0.1 mol/L H3PO4 30-min shaking |
Liquid–liquid partitioning with 0.35 mol/L NaHCO3+EtOH (5+2) and CH2Cl2 |
HPLC (RP18)/ |
|
Jorgensen (1998) |
Beer |
150 (ml) |
Beer degassed for 60 min |
Degassed beer passed on immunoaffinity column |
HPLC (RP18)/ |
|
Jorgensen (1998) |
Roasted coffee |
50 |
200 ml 1% m/m NaHCO3; 2-min blending |
Dilution with PBS and clean-up on immunoaffinity column |
HPLC (RP18)/ |
|
Jorgensen (1998) |
Pulses |
50 |
200 ml acetonitrile: water (6:4); 2-min blending |
Dilution with PBS and clean-up on immunoaffinity column |
HPLC (RP18)/ |
|
Patel et al. (1996) |
Cereals, oils, nuts, seeds, herbs, pickles, canned food |
25 |
125 ml CHCl3 + 12.5 ml 0.1 mol/L H3PO4 30-min shaking |
Silica Sep-pack cartridge |
HPLC (RP18)/ |
|
Maaroufi et al. (1995) |
Olives, cereals and derived foods, vegetables |
100 |
120 ml CHCl3+HCl (100+1); shaking overnight |
Preparative TLC |
HPLC (RP18)/ |
|
Sharman et al. (1992) |
Cereal products |
10 |
40 ml PBS:MeOH (50:50); 3–5-min blending |
Immunoaffinity column |
HPLC (RP18)/ |
|
Sharman et al. (1992) |
Animal products |
10 |
100 ml CHCl3+ 0.6 g 85% H3PO4 3–5-min blending |
Immunoaffinity column |
HPLC (RP18)/ |
|
Rao (2000) |
Foods |
NR |
Acetonitrile:water or MeOH:water |
Immunoaffinity column |
HPLC (RP18)/ |
|
MAFF (1999a) |
Cereals |
25 |
CHCl3 + 0.1 mol/L H3PO4 |
Silica Sep-pack cartridge |
HPLC (RP18)/ |
|
Howell & Taylor (1981; modified) |
Maize |
25 |
250 ml CHCl3 + 25 ml 0.1 mol/L H3PO4 30-min shaking |
Silica Sep-Pak cartridge |
HPLC (RP18)/ |
|
Pineiro & Giribone (1994) |
Various foods and feeds |
NR |
NR |
NR |
TLC |
|
MAFF (1999b) |
Dried fruit, chocolate, cocoa, pulses |
NR |
NR |
Immunoaffinity column |
HPLC (RP18)/ |
|
MAFF (1999b) |
Wine, grape juice |
NR |
NR |
Immunoaffinity column |
HPLC (RP18)/ |
|
MAFF (1999c) |
Total diet |
NR |
CHCl3:H3PO4 |
Liquid–liquid partitioning with NaHCO3 solution and clean-up on immunoaffinity column |
HPLC (RP18)/ |
|
MAFF (1996b) |
Green coffee beans |
50 |
25 ml 0.1 mol/L H3PO4 + 250 ml CHCl3 + 10 g diatomaceus earth; 3-min blending |
Liquid–liquid partii- tioning with 3% NaHCO3, immunoaffinity and C18 (Sep-pack) |
HPLC (RP18)/ |
|
Stegen et al. (1997) |
Coffee products |
Cooperative study with 9 laboratories using different methods |
HPLC (RP18)/ |
||
|
Nesheim et al. (1973) |
Barley |
50 |
25 ml 0.1 mol/L H3PO4 + 250 ml CHCl3 + 10 g |
Liquid–liquid partitioning |
TLC sprayed with ammonia and scanned with fluoridensitometer |
|
Scott et al. (1991) |
Meat, kidney, liver |
25 |
100 ml CHCl3 + 50 ml 2 nmol/L NaCl + 50 ml 0.5 mol/L H3PO4 60-min shaking |
Silica gel column |
HPLC (RP18)/ |
|
Patel et al. (1997) |
Roasted and soluble coffee |
25 |
125 ml CHCl3 + 12.5 ml 0.1 mol/L H3PO4; 30-min shaking |
Silica Sep-Pak cartridge + immunoaffinity column |
HPLC (RP18)/ |
|
Burdaspal & Legarda (2000) |
Baby food |
25 |
100 ml 0.5 mol/L H3PO4 2 mol/L NaCl + 50 ml tert-butyl methyl ether; 2-min blending |
Liquid–liquid partitioning + immunoaffinity clean-up |
HPLC (RP18)/ |
|
Zimmerli & Dick (1995, 1996) |
Wine, grape juice |
5 (ml) |
Dilution with H3PO4: 2 mol/L NaCl (33.7: 966.3), extraction with 5 ml CHCl3 1-min vortex |
immunoaffinity column |
HPLC (RP18)/ |
|
Burdaspal & Legarda (1998a) |
Beer |
5 |
Dilution with 1 ml 1% NaHCO3 15% NaCl |
Immunoaffinity column |
HPLC (RP18)/ |
|
Ueno (1998) |
Coffee |
0.5 |
8 ml 1% Na2CO3 and diluted with PBS |
Immunoaffinity column |
HPLC (RP18)/ |
|
Canned coffee |
5 (ml) |
Filtration |
|||
|
Wine and beer |
5 (ml) |
Mixed with 1 ml 2.5% Na2CO3 1.5% NaCl |
|||
|
Visconti et al. (1999, 2000a) |
Wine, beer |
10 (ml) 10 ml 1% |
Dilution with PEG and 5% NaHCO3 |
Immunoaffinity column |
HPLC (RP18)/ |
|
Ottender & Majerus (2000) |
Wine |
25 (ml) |
Dilution with 25 ml PBS and pH 7.0–7.5 |
Immunoaffinity column |
HPLC (RP18)/ |
|
Koch et al. (1996) |
Roasted coffee |
15 |
150 ml MeOH + 3% NaHCO3 (1+1, v/v) 30-min shaking column |
Double clean-up: phenyl silane column and immunoaffinity |
HPLC (RP18)/ |
|
Majerus et al. (1994) |
Wheat wholemeal, cereals, dried fruits, pulses, wine, beer, fruit juice, raw coffee |
20 |
30 ml HCl+ 50 ml 0.4 mol/L MgCl2; extracted with 100 ml toluene by shaking 60 min |
Silica gel column |
HPLC (RP18)/ |
|
Leoni et al. (2000) |
Instant and roasted coffee |
10 |
200 ml 1% NaHCO3 2-min blending |
Immunoaffinity column |
HPLC (RP18)/ |
|
Sizoo & van Egmond |
Wheat and wheat products |
25 |
CH3CN/water |
Immunoaffinity column |
HPLC (RP18)/ |
|
Inspectorate of Health Protection |
Wheat and wheat products |
20 |
MeOH/water or CH3CN/water |
Immunoaffinity column |
HPLC (RP18)/ |
|
Inspectorate for Health Protection |
Wine |
5 (ml) |
Dilution with water |
Immunoaffinity column |
HPLC (RP18)/ |
|
Inspectorate for Health Protection |
Roasted coffee |
10 |
MeOH/3% NaHCO3 |
Phenyl silane column + immunoaffinity column |
HPLC (RP18)/ |
|
Inspectorate for Health Protection |
Paprika, pepper, nuts |
20 |
MeOH/water or CH3CN/water |
Immunoaffinity column |
HPLC (RP18)/ |
|
Entwisle et al. (2000b) |
Roasted coffee |
15 |
MeOH/3% NaHCO3 |
Phenyl silane column + immunoaffinity column |
HPLC (RP18)/ |
Table 13 (b). Analytical methods used to determine ochratoxin A in foods
|
Reference |
Commodity |
Test portion |
LOD/LOQ |
Accuracy |
|||
|
Spiking |
Recovery (%) |
RSDr |
RSDR |
||||
|
Wolff et al. (2000) |
Wine, juices, oil, vinegar |
5 (ml) |
0.01/ |
|
|
|
|
|
Wolff et al. (2000) |
Meat and meat products |
25 |
0.01/ |
0.05 –5.0 |
34–104 |
|
|
|
Wolff et al. (2000) |
Cereals |
40 |
0.01/ |
|
|
|
|
|
Wolff et al. (2000) |
Beer |
5 (ml) |
|
0.01/ |
|
|
|
|
Wolff et al. (2000) |
Coffee, tea |
25 |
|
0.3/ |
|
|
|
|
Wolff et al. (2000) |
Milk products, sweets, oilseeds |
50 |
|
0.01/ |
|
|
|
|
Langseth et al. (1989); Langseth (1999) |
Cereals |
25 |
0.01/ |
10 |
80–114 |
|
|
|
Larsson & Moeller (1996) |
Wheat, barley, rye |
50 |
0.1/ |
4.96 |
72 |
|
|
|
5.95 |
69 |
17–22 (5–7 µg/kg) |
24–28 (5–7 µg/kg) |
||||
|
7.44 |
66 |
||||||
|
Soares et al. (1985) |
Corn, peanuts, beans, rice, cassava |
50 |
10/ |
10–400 |
86–125 |
0–24 |
|
|
Trucksess et al. (1999) |
Wheat, barley, coffee |
25 |
0.03/ |
1–4 |
71–96 |
2–17 |
|
|
Solfrizzo et al. (1998) |
Wheat, oats |
10 |
/0.8 |
1–100 |
82–104 |
3–7 |
|
|
Jorgensen et al. (1996) |
Wheat, rye, barley, oats, bran, pork, poultry meat and liver |
50 |
0.05/ |
5 |
80-90 |
<15 |
|
|
0.5–4 (meat, liver) |
60–121 |
||||||
|
Jorgensen (1998) |
Beer |
150 (ml) |
|
0.001/ |
0.3 |
66–113 |
|
|
Jorgensen (1998) |
Roasted coffee |
50 |
|
0.1/ |
5 |
59–83 |
|
|
Jorgensen (1998) |
Pulses |
50 |
|
0.1/ |
5 |
78–103 |
|
|
Patel et al. (1996) |
Cereals, oils, nuts, seeds, herbs, pickles, canned food |
25 |
|
0.1/ |
10 |
90 |
|
|
Maaroufi et al. (1995) |
Olives, cereals and derived foods, vegetables |
100 |
0.1/ |
10–100 |
60–85 |
|
|
|
Sharman et al. (1992) |
Cereal products |
10 |
0.2/ |
10 |
74 |
4 |
|
|
Sharman et al. (1992) |
Animal products |
10 |
0.2/ |
10 |
74–79 |
3–6 |
|
|
Rao (2000) |
Foods |
NR |
0.2/0.5 |
1–5 |
70–126 |
8–13 |
|
|
MAFF (1999a) |
Cereals |
25 |
0.1/0.2 |
2 |
71–104 |
|
|
|
Howell & Taylor (1981; modified) |
Maize |
25 |
0.1/0.2 |
2 |
90–98 |
5 |
|
|
Pineiro & Giribone (1994) |
Various foods and feeds |
NR |
50/ |
|
|
|
( |
|
MAFF (1999b) |
Dried fruit, chocolate, cocoa, pulses |
NR |
0.1/0.2 |
|
79–92 |
1.7–11.1 |
|
|
MAFF (1999b) |
Wine, grape juice |
NR |
0.01/0.02 |
|
79–92 |
5.9–7.0 |
|
|
MAFF (1999c) |
Total diet |
NR |
|
/0.002 |
|
89 |
16.8 |
|
MAFF (1996b) |
Green coffee beans |
50 |
|
/0.26 |
|
73.3 |
18 |
|
Stegen et al. (1997) |
Coffee products |
Cooperative study with 9 laboratories using different methods |
0.2–1/ |
|
|
20 |
42 (CV) |
|
Nesheim et al. (1973) |
Barley |
50 |
2/ |
|
|
|
|
|
Scott et al. (1991) |
Meat, kidney, liver |
25 |
0.5/ |
1–10 |
93–106 |
9–12 |
|
|
Patel et al. (1997) |
Roasted and soluble coffee |
25 |
0.1/ |
2 |
91 |
5 |
|
|
Burdaspal & Legarda (2000) |
Baby food |
25 |
/0.008 |
< 1 |
91–109 |
18 |
|
|
Zimmerli & Dick (1995, 1996) |
Wine, grape juice |
5 (ml) |
0.003/0.005 |
0.055 |
74–91 |
6 |
|
|
Burdaspal & Legarda (1998a) |
Beer |
5 |
0.004/ |
0.114 |
100 |
2.4 |
|
|
Ueno (1998) |
Coffee |
0.5 |
0.003–0.06/ |
NR |
80–90 |
|
|
|
Canned coffee |
5 (ml) |
||||||
|
Wine and beer |
5 (ml) |
||||||
|
Visconti et al. (1999, 2000a) |
Wine, beer |
10 (ml) 10 ml 1% |
0.01/ |
0.1–3.0 |
85–102 |
5–19 |
10–20 |
|
Ottender & Majerus (2000) |
Wine |
25 (ml) |
0.01/ |
|
|
8.4 |
|
|
Koch et al. (1996) |
Roasted coffee |
15 |
0.1/ |
6 |
87–94 |
|
|
|
Majerus et al. (1994) |
Wheat wholemeal, cereals, dried fruits, pulses, wine, beer, fruit juice, raw coffee |
20 |
0.1/ |
0.41 |
80 |
15 |
26 |
|
1.2 |
80 |
20 |
32 |
|
|||
|
Leoni et al. (2000) |
Instant and roasted coffee |
10 |
0.2/ |
10 |
77–100 |
|
|
|
Sizoo & van Egmond |
Wheat and wheat products |
25 |
0.3/1 |
5 |
99 |
6 |
|
|
Inspectorate of Health Protection |
Wheat and wheat products |
20 |
0.1/0.25 |
0.51–54 |
86–106 |
8 |
NA |
|
Inspectorate for Health Protection |
Wine |
5 (ml) |
0.05/0.1 |
0.33 |
83–93 |
9 |
NA |
|
Inspectorate for Health Protection |
Roasted coffee |
10 |
0.13/0.25 |
0.2–1.0 |
76–104 |
33 |
NA |
|
Inspectorate for Health Protection |
Paprika, pepper, nuts |
20 |
0.13/0.25 |
NA |
> 70 |
NA |
NA |
|
Entwisle et al. (2000b) |
Roasted coffee |
15 |
0.2/ |
4 |
65–97 |
2.0–22 |
14–26 |
CV, coefficient of variation; HPLC, high-performance liquid chromatography; LOD, limit of detection; LOQ, limit of quantification; MAFF, Ministry of Agriculture, Fisheries and Food (United Kingdom); RSDr, relative standard deviation for repeatability; RSDR, relative standard deviation for reproducibility; NR, not reported; PBS, phosphate-buffered saline; PEG, polyethylene glycol; TLC, thin-layer chromatography
The main foods in which the effects of processing have been studied are cereals and coffee, although a little work has been carried out on other foods as well.
In flour manufacture, some parts of the wheat grain are removed, possibly reducing the concentrations of ochratoxin A in flour and subsequent products. This was investigated by Osborne et al. (1996), who allowed growth of a toxin-producing strain of P. verrucosum in two samples of clean hard and soft wheat and then produced both white and wholemeal flour and bread from them. The process greatly influenced the final concentration of ochratoxin A in the bread (Table 14). The scouring process used by Osborne et al. (1996) resulted in a reduction of more than 50% in the concentration of ochratoxin A, but this process is not standard milling practice. Milling hard wheat to produce white flour resulted in an approximately 65% reduction, and a further 10% decrease occurred during baking. The reduction in soft wheat was much smaller; explanations were offered, but these were somewhat academic because bread is usually made from hard wheat. Wholemeal flour and bread showed much smaller reductions in the concentration of ochratoxin A during processing, as might be expected, because less of the grain is discarded. The loss during milling was only 10%, with a further 4% loss during baking (Table 14).
Table 14. Effect of flour and bread manufacture on the concentration of ochratoxin A (fg/kg) in wheat
|
Wheat type |
Initial concentration |
Cleaning process |
After cleaning |
White flour |
White bread |
Wholemeal flour |
Wholemeal bread |
|
Hard |
618 |
Clean |
624 (0) |
209 (66) |
140 (77) |
555 (10) |
531 (14) |
|
Clean and scour |
256 (59) |
60 (90) |
72 (88) |
127 (80) |
226 (63) |
||
|
Soft |
643 |
Clean |
473 (27) |
389 (40) |
224 (65) |
553 (9) |
476 (26) |
|
Clean and scour |
179 (72) |
111 (83) |
94 (85) |
189 (71) |
160 (75) |
From Osborne et al. (1996). Percentage reductions are given in parentheses. All values are from duplicate experiments, with generally good agreement.
In a study of the stability of ochratoxin A in cereals during heating, a toxigenic strain of A. ochraceus on wheat, and dry and moistened samples were heated at several temperatures and times and the level of destruction recorded. From those figures, the times to 50% destruction of ochratoxin A under various heating conditions were calculated (Table 15; Boudra et al., 1995).
Table 15. Time to reduce ochratoxin A content in wheat by 50% under various conditions
|
Moisture content of wheat |
Temperature of heating (°C) |
Time to 50% destruction of ochratoxin A (min) |
|
Dry wheat |
100 |
700 |
|
150 |
200 |
|
|
200 |
12 |
|
|
250 |
6 |
|
|
Wet wheat |
100 |
140 |
|
150 |
60 |
|
|
200 |
19 |
From Boudra et al. (1995). The wheat samples had been ground; the dry wheat had been dried in a vacuum oven, the wet wheat was 50:50 dry wheat and water.
A reduction in ochratoxin A concentrations resulting from manufacture of retail products from contaminated raw materials was reported by the Ministry of Agriculture, Fisheries and Food (1996a). The processes involved in making breakfast cereals and biscuits caused substantial reductions in the ochratoxin A content; however manufacture of egg noodles and pasta caused little or no reduction (Table 16).
Table 16. Reduction in concentrations of ochratoxin A during food manufacture
|
Raw material |
Ochratoxin |
Rate of incorporation of raw material into food |
Retail food |
Ochratoxin |
Reduction (%) (allowing for incorporation rate) |
|
Breakfast cereal grain |
0.8 |
43 |
Breakfast cereal |
< 0.2 |
> 50 |
|
Breakfast cereal grain |
0.8 |
90 |
Breakfast cereal |
< 0.2 |
> 76 |
|
Biscuit flour |
6.4 |
40 |
Biscuits |
0.4 |
84 |
|
Durum wheat flour |
1.4 |
82.5 |
Egg noodles |
1.0 |
13 |
|
Durum wheat flour |
0.9 |
82.5 |
Egg noodles |
0.8 |
0 |
|
Durum wheat flour |
1.6 |
82.5 |
Egg noodles |
1.3 |
2 |
|
Semolina |
0.8 |
100 |
Pasta (dried) |
0.6 |
25 |
|
Semolina |
0.8 |
100 |
Pasta (dried) |
1.0 |
0 |
Modified from Ministry of Agriculture, Fisheries and Food (1996a)
The perception that coffee may be an important source of ochratoxin A led to much work on its formation, destruction during roasting, and presence in brewed coffee. It now seems clear, however, that coffee is not a major source of ochratoxin A in the normal diet (Ministry of Agriculture, Fisheries and Food, 1995).
Reductios in the concentration of ochratoxin A during cleaning and roasting of coffee have been studied, with variable results. Wilkens & Jörissen (1999) showed that cyclone cleaning of green coffee beans had little effect: although the ochratoxin A concentration in the discarded fraction was high, the dust comprised < 1% of the weight of the cleaned coffee. Sorting with colour sorters resulted in some reduction, and steaming caused a mean 25% reduction. Decaffeination is an effective process, resulting in 92% reduction (Heilmann et al., 1999).
Several reports on the effect of roasting on the concentration of ochratoxin A are summarized in Table 17. The reported reductions vary, but a major factor in such variation is that the natural concentration of ochratoxin A in coffee destined for retail markets, i.e. in coffee of sufficient quality for human consumption, seldom exceeds 10 µg/kg. Reports based on samples of such coffee usually showed high rates of reduction, e.g. 50–90% (Micco et al., 1989), 30–90% (Wilkens & Jörissen, 1999), and 81% (Blanc et al., 1998). Much less reduction (0–22%) was reported by Studer-Rohr et al. (1995); however, they studied green coffee beans that were naturally spoiled or on which a culture of A. ochraceus had been grown in the laboratory, creating artificially high concentrations (90–1300 µg/kg). It is unlikely that such high concentrations are relevant to the conditions found in commercial beans or that degradation of such high concentrations by natural chemical reactions during heating would resemble those that occur in the minute traces of ochratoxin A normally found in coffee beans.
Table 17. Reduction in concentrations of ochratoxin A during roasting of coffee
|
Sample origin |
Method of contamination |
Ochratoxin A concentration (fg/kg) |
Reduction |
Reference |
|
|
In green coffee |
After roasting |
||||
|
Zaire |
Natural |
8.6 |
0.2 |
98 |
Micco et al. (1989) |
|
Conillon |
Natural |
4.0 |
0.3 |
92 |
Micco et al. (1989) |
|
Santos |
Spiked |
46 |
6.1 |
87 |
Micco et al. (1989) |
|
Costa Rica |
Spiked |
42 |
20 |
49 |
Micco et al. (1989) |
|
Brazil/Ivory Coast |
Spiked |
47 |
11 |
80 |
Micco et al. (1989) |
|
Thailand |
Natural |
7.3 |
1.4 |
81 |
Blanc et al. (1998) |
|
Commercial |
Spiked |
780 |
890 |
0 |
Studer-Rohr et al. (1995) |
|
|
Spiked |
1300 |
1200 |
11 |
Studer-Rohr et al. (1995) |
|
|
Natural (spoiled) |
360 |
280 |
22 |
Studer-Rohr et al. (1995) |
|
|
Natural (spoiled) |
140 |
121 |
16 |
Studer-Rohr et al. (1995) |
|
|
Natural (spoiled) |
92 |
92 |
0 |
Studer-Rohr et al. (1995) |
|
Unknown |
Natural |
0.90 |
0.63 |
30 |
Wilkens & Jörissen (1999) |
|
|
Natural |
9.9 |
2.1 |
79 |
Wilkens & Jörissen (1999) |
|
|
Natural |
18 |
1.9 |
89 |
Wilkens % Jörissen (1999) |
|
Ivory Coast |
Natural |
4.9 |
1.5 |
69 |
Stegen et al. (2001) |
Ochratoxin A was originally described as a metabolite of Aspergillus ochraceus in laboratory experiments (van der Merwe et al., 1965). It was subsequently reported in several related Aspergillus species (Ciegler, 1972; Hesseltine et al., 1972), but the first report of its natural occurrence, and its potential importance, was in a different source, a Penicillium species (Scott et al., 1970; Krogh et al., 1973). Recently, A. carbonarius was identified as a third major source, with a low percentage of isolates of the closely related species A. niger (Abarca et al., 1994; Téren et al., 1996). It is now clear that ochratoxin A is produced by a single Penicillium species, P. verrucosum, and a rather remarkable range of Aspergillus species. The following sections deal with these species in more detail.
The latest information (Frisvad & Samson, 2000 and unpublished data) indicates that ochratoxin A is produced by only a few species related to A. ochraceus, all of which are classified in Aspergillus subgenus Circumdati section Circumdati. Apart from A. ochraceus, this group of ochratoxin A producers includes two ascosporic fungi, Neopetromyces muricatus (asexual state A. muricatus) and Petromyces alliaceus (asexual state A. alliaceus) plus Aspergillus sclerotiorum and A. sulphureus. N. muricatus is the correct name for isolates producing ochratoxin A that were previously identified as A. melleus. Both N. muricatus and P. alliaceus are uncommon species. P. sclerotiorum isolates make this toxin only rarely, and although isolates of A. suphureus are usually producers, this is a rare species (J.C. Frisvad, personal communication). Apart from A. ochraceus, all of these species are very uncommon in foods and are not known to cause food spoilage. Hence, the only species of any importance for ochratoxin A production in the Aspergillus section Circumdati is A. ochraceus.
A. carbonarius has been recognized as a source of ochratoxin A only recently (Horie, 1995; Téren et al., 1996; Wicklow et al., 1996). It is now known that most if not all isolates of A. carbonarius are producers when grown in pure culture (Heenan et al., 1998; Taniwaki et al., 1999; J.I. Pitt and colleagues, unpublished), although the extent of production is variable. The closely related species A. niger has also been reported reliably as a producer (Ueno et al., 1991; Abarca et al., 1994; Heenan et al., 1998; Taniwaki et al., 1999; J.I. Pitt and colleagues, unpublished). All reports agree, however, that ochratoxin A production by A. niger is very uncommon, being formed under pure culture conditions by only 1–2% of isolates. A. carbonarius and A. niger are classified in Aspergillus subgenus Circumdati section Nigri.
A number of other species of Aspergillus have been reported to produce ochratoxin A, but in the opinion of Dr J.C. Frisvad, Department of Biotechnology, Technical University of Denmark, an authority on Aspergillus secondary metabolism, none of these has been substantiated (personal communication to J.I. Pitt, 2000).
Soon after the isolation of ochratoxin A from A. ochraceus, the formation of ochratoxin A by a Penicillium species, P. viridicatum, was reported (van Walbeek et al., 1969), and its natural occurrence was confirmed (Krogh et al., 1973). The view that P. viridicatum was a major source of ochratoxin A in foods and feeds in some parts of the world was accepted for more than a decade. The species involved was later correctly identified as P. verrucosum (Pitt, 1987), as confirmed by others (Frisvad, 1989; Frisvad & Filtenborg, 1989). Although a number of more recent reports refer to ochratoxin A production by other, often unspecified, Penicillium species, this is known to be erroneous (Frisvad 1989; Frisvad & Filtenborg, 1989; Pitt & Hocking, 1997). It is now clear that P. verrucosum is the only Penicillium species that has been shown to produce ochratoxin A. It is classified in Penicillium subgenus Penicillium section Penicillium, along with many other mycotoxin-producing species.
Each of the three major producers of ochratoxin A, P. verrucosum, A. ochraceus, and A. carbonarius, has a quite different physiology and consequently quite different ecological habitat. To understand the kinds of foods in which ochratoxin A occurs and to predict the potential for its formation, it is necessary to understand the physiology and ecology of these species and the differences between them.
(a) Penicillium verrucosum
P. verrucosum is a slowly growing species under any conditions but is capable of growth at low water activity (aw) (down to 0.80) and at low temperature (range, 0–31 oC; optimum, 20 °C) (Pitt & Hocking, 1997).
A notable feature of the ecology of P. verrucosum is that it grows only at lower temperatures. This results in a distribution which is apparently confined to cool temperate regions. Its major food habitat is cereal crops grown in cool temperate climates, ranging across northern and central Europe and Canada. It also occurs in European meat products and in cheese. It appears to be uncommon, indeed almost unknown, in warm climates or in other kinds of foods. The occurrence of this species in European cereals has two consequences: ochratoxin A is present in many kinds of European cereal products, especially bread and flour-based foods, and in animals that eat cereals as a major dietary component. Ochratoxin A was detected in Danish pig meat 25 years ago (Krogh et al., 1973), and its implications for human and animal health were recognized at the same time. As bread, other cereal products, and pig meats are major components of the European diet, the further consequence is that most Europeans who have been tested had appreciable concentrations of ochratoxin A in their blood (Hald, 1991; Petkova-Bocharova & Castegnaro, 1991; Breitholtz-Emanuelsson et al., 1993b; Zimmerli & Dick, 1995; Burdaspal & Legarda, 1998; Jiménez et al., 1998b; Scott et al., 1998). There is no doubt that this results from the growth of P. verrucosum in cereals.
(b) Aspergillus ochraceus
A. ochraceus can be described as a mesophilic xerophile. Growth occurs between 8 and 37 °C, with the optimum at 24–31 °C. Optimal conditions for growth are 0.95–0.99 aw, while the lower limit for growth is 0.79 aw on media containing sugars and down to 0.81 aw on media based on NaCl. A. ochraceus grows slowly at pH 2.2 and well between pH 3 and 10 (Pitt & Hocking, 1997).
A. ochraceus has been isolated from a wide range of food products but is more common in dried and stored foods than elsewhere. Stored foods from which it has been isolated include smoked and salted dried fish, dried beans, biltong, soya beans, chickpeas, rapeseed, pepper, dried fruit, and sesame seeds. Nuts are also a major source, especially pecans and pistachios, and also peanuts, hazelnuts, and walnuts. It has been reported infrequently in cereals and cereal products, including rice, barley, maize, wheat, flour, and bran. A. ochraceus has also been reported in cheese, spices, black olives, cassava, and processed meats. However, this species rarely causes spoilage and is often found in foods only at low concentrations; its presence is therefore not a good indicator of significant mycotoxin production (Pitt & Hocking, 1997).
Several workers have detected A. ochraceus in green coffee beans (Levi et al., 1974; Cantafora et al., 1983; Tsubouchi et al., 1984; Micco et al., 1989; Studer-Rohr et al., 1994), and this species appears to be one source of ochratoxin A in coffee (Taniwaki et al., 1999).
A. ochraceus has been isolated from a variety of South-East Asian commodities, including maize, peanuts, soya beans and other beans, cashews, and sorghum. Its presence or absence in any sample was probably related to the length of storage rather than to geographical location or other factors (Pitt et al., 1993, 1994, 1998).
As noted above, a few species closely related to A. ochraceus can produce ochratoxin A. Little is known about the physiology and ecology of any of these species, but what information there is suggests that their important features are similar to those of A. ochraceus. The occurrence of any of them in foods or food commodities is very rare, however.
(c) Aspergillus carbonarius
Relatively little is known about the third major ochratoxin A producer, A. carbonarius. The ability of this species to produce ochratoxin A was reported only recently (Horie, 1995; Téren et al., 1996; Varga et al., 1996; Heenan et al., 1998). It resembles A. niger in many features, and indeed the two species are very closely related. A. carbonarius differs from A. niger most notably in the production of larger spores, although other minor morphological differences exist. The available information on its physiology indicates a broad similarity to A. niger. However, preliminary studies indicate that A. carbonarius grows at rather lower temperatures than A. niger, with a maximum around 40 °C and optimal conditions about 32–35 °C. The ability to grow at reduced aw is also more restricted: germination occurs down to 0.82 aw at 25 and 30 oC. Unlike A. niger, A. carbonarius failed to germinate at 0.82 aw and 37 °C (S.-L. Leong & J.I. Pitt, unpublished data).
A. carbonarius appears to be less common than A. niger, but many surveys of Aspergilli in foods have probably not differentiated the two species, calling all black Aspergilli A. niger.
As a result of the high resistance of the black Aspergilli, which include A. niger, A. carbonarius, and A. japonicus, to sunlight and ultra-violet light, a major habitat of these species is dried vine fruits, which in most producing countries are dried in the sun without preservatives. The incidence of the black Aspergilli in grapes at harvest and during drying has been studied in the major grape-growing region surrounding Mildura, Victoria, Australia, which is an irrigated area with a hot (35–42 °C) climate during the harvest season. The three major black species, A. niger, A. carbonarius, and A. japonicus, were all very common. The percentage of each species varied from season to season, presumably due to seasonal differences in climatic factors, especially average temperatures and rainfall patterns (S.-L. Leong & J.I. Pitt, unpublished data).
In that study, 470 A. niger isolates, 200 of A. japonicus, and 245 of A. carbonarius were isolated and identified. All were subsequently assayed for ochratoxin A production. The techniques included examination under ultra-violet light after growth on coconut cream agar (Heenan et al., 1998), TLC from agar plugs (Filtenborg & Frisvad, 1980), and, on some isolates, growth in culture, extraction, and HPLC. A. japonicus did not make ochratoxin A, A. niger made it rarely and at low concentrations, but all isolates of A. carbonarius were capable of producing ochratoxin A. These findings indicate that it is extremely likely that A. carbonarius is the major source of ochratoxin A in grapes and grape products, including table grapes, wines, and dried vine fruits.
(d) Aspergillus niger
A detailed account of the ecology of A. niger is included here on the basis that A. carbonarius probably occurs in most habitats in which A. niger has been found. However, this must be regarded as a hypothesis, not a factual statement, at present.
Like many Aspergilli, A. niger grows optimally at relatively high temperatures, with minimal growth at 6–8 °C, maximal growth at 45–47 °C, and optimal conditions of 35–37 °C. A. niger is a xerophile with germination reported at 0.77 aw at 35 °C. The growth rates vary only slightly on media based on sugars, NaCl, or glycerol or at pH 4.0 and 6.5, and at various water activities. Thus, the growth of A. niger appears to be little affected by food type. A. niger can grow down to pH 2.0 at high aw (Pitt & Hocking, 1997).
Among the fungi most commonly reported in foods, A. niger is prevalent in warmer climates, both in field situations and stored foods. It is by far the commonest Aspergillus species responsible for post-harvest decay of fresh fruit, including apples, pears, peaches, citrus, grapes, figs, strawberries, mangoes, tomatoes, and melons and some vegetables, especially onions, garlic, and yams (Snowdon, 1990, 1991). Most of these diseases are sporadic and of minor significance. A. niger sometimes causes kernel rot in cashews and can cause thread mould spoilage of cheese (Hocking & Faedo, 1992).
A. niger is among the commonest fungi isolated from nuts, especially peanuts, and has also been reported in pecans, pistachios, hazelnuts, walnuts, coconut, and copra. Cereals and oilseeds are also sources, especially maize and also barley, soya beans, canola, sorghum, stored and parboiled rice, and dried beans (Pitt & Hocking, 1997).
A. niger has commonly been isolated from South-East Asian foods (Pitt et al., 1993, 1994, 1998). The heaviest contamination was found in peanuts, maize, cashews, copra, pepper, and spices from Indonesia, the Philippines, and Thailand and in kemiri nuts from Indonesia.
In terms of mycotoxin production, A. niger is usually regarded as a benign fungus and has been widely used in food processing. It is categorized as ‘generally regarded as safe’ by the Government of the USA. However, 2 of 19 A. niger isolates were reported to produce ochratoxin A by Abarca et al. (1994) and 2 of 115 by Heenan et al. (1998). Ochratoxin A production by A. niger in commercially grown crops appears possible but is probably uncommon.
The results of surveys for ochratoxin A are shown in Appendix A. As contamination with ochratoxin-producing fungi is widespread, numerous commodities have been analysed, including cereal and cereal products, green and roasted coffee, dried fruits, wine, grape juice, cocoa and chocolate, herbs and spices, canned foods, oils, olive, pulses, chickpeas, lentils, soya products, sweets, milk and milk products, meat, kidney, liver, beer, tea, vinegar, mustard, baby food, and house dust.
Most of the information for the past 5 years was taken from the literature and one Internet site. Other data were submitted to the Committee for the current meeting. The natural occurrence of ochratoxin A before 1995 was reviewed by WHO (1990) and the Commission of the European Union (1997).
The total number of samples for which data are shown in Appendix A was 23 167, with 85% from Europe (Croatia, Denmark, Finland, France, Germany, Italy, the Netherlands, Norway, Spain, Sweden, Switzerland, and the United Kingdom), 7% from South America (Brazil and Uruguay), 6% from North America (Canada and the USA), 1% from Africa (Sierra Leone and Tunisia), and 1% from Asia (Dubai and Japan).
When mean values and 90th percentiles were not available, they were calculated from a single datum, if available, assuming 0 for those samples containing no detectable toxin. The parent reference (P), analytical method (A) and sampling method used (S) are shown for each entry. When available, details of the sampling procedure are reported. The analytical methods cited in Appendix A are described in Table 13.
Adequate sampling procedures should be used for future surveys of ochratoxin A in cereals and cereal products. For example, 10 of 22 submitted papers giving data on cereals described the sampling procedure, whereas no description was reported in the remaining 12 papers. When the collected sample was small, the number of total samples was adequately reduced in order to limit the impact of occasionally high values on the weighted mean concentration (Jorgensen et al., 1996; Solfrizzo et al., 1998; Jurcevic et al., 1999). No sampling plans for the determination of ochratoxin A in foods have been published, and details of sampling variation have not been reported.
Most of the samples reported in Appendix A (95%) were analysed by LC with fluorescence detection, and the LOD or LOQ was usually < 0.5 µg/kg. The remaining 5% of samples were analysed by TLC with limits of detection or quantification > 5 µg/kg. These data were not used to estimate intake as the analytical method used did not provide an LOQ down to 5 µg/kg, the lower level that the Committee was requested to evaluate by the Codex Committee on Food Additives and Contaminants. In fact, only one of the 1225 samples analysed by TLC, a sample of wheat, was found to be contaminated with 40 µg/kg of ochratoxin A (Pineiro & Giribone, 1994; Furlong et al., 1995a; Pineiro et al., 1996). When a more sensitive method was used (LOD = 0.2 µg/kg), most of the samples (78%) from the same geographical area were contaminated with ochratoxin A (Leoni et al., 2000).
The incidence of contaminated samples varied by commodity, and the incidence was higher in the same commodity when analytical methods with lower LOQs were used. For example, a survey of 300 samples of pig kidney in Denmark showed a high incidence of contamination (79%), and five samples were contaminated at > 5 µg/kg (Petersen, 2000). Similar results were obtained in an analysis of 61 samples of pig kidney collected in Germany (Wolff et al., 2000), whereas a low incidence of positive samples (6%) was found among 1010 samples collected in France (Dragacci et al., 1999). The different analytical methods used in these studies, with different LOQs (0.06 µg/kg and 0.5 µg/kg), could explain the discrepant results.
The concentrations of ochratoxin A in the various commodities were highly variable; 1.4% and 0.6% of all samples contained more than 5 µg/kg and 20 µg/kg, respectively. Within one type of cereal, 1.2% and 0.3% of samples contained more than 5 µg/kg and 20 µg/kg, respectively. Within cereal products, 0.3% and 0.05% of samples contained more than 5 µg/kg and 20 µg/kg, respectively. For this calculation, the cereal product samples from Tunisia were not considered, as they were selected samples from families of patients with nephropathy (Maaroufi et al., 1995b).
The weighted mean concentrations of ochratoxin A in cereals, cereal products, cocoa and chocolate, coffee, dried vine fruit, grape juice, pig kidney, other products of animal origin, and wine are shown in Table 18. Samples for which the mean concentration of ochratoxin A was lacking were not considered. High concentrations ( 50 µg/kg) were also reported in herbs and spices (Patel et al., 1996; Rao, 2000) as well as in house dust (< 1600 µg/kg), confirming the widespread presence of ochratoxin A (Richard et al., 1999). Low concentrations were found in all 50 samples of the total diet analysed in the United Kingdom by a very sensitive method capable of detecting down to 0.002 µg/kg (Ministry of Agriculture, Fisheries and Food, 1999b).
Table 18. Weighted mean concentrations of ochratoxin A in commodities evaluated
|
Commodity |
No. of samples |
Weighted mean concentration (µg/kg) |
|
Beer |
660 |
0.025 |
|
Cereals, all |
2700 |
0.94 |
|
Barley |
350 |
0.53 |
|
Maize |
95 |
7.5 |
|
Oats |
280 |
0.44 |
|
Rice |
45 |
0.06 |
|
Rye |
790 |
1.2 |
|
Wheat |
1200 |
0.38 |
|
Cereal products |
1500 |
0.19 |
|
Cocoa and chocolate |
270 |
0.18 |
|
Coffee, green and roasted |
1900 |
0.86 |
|
Green |
130 |
1.0 |
|
Roasted |
1700 |
0.76 |
|
Instant |
290 |
1.4 |
|
Dried vine fruit |
860 |
2.3 |
|
Grape juice |
68 |
0.44 |
|
Pig kidney |
380 |
0.12 |
|
Products of animal origin |
810 |
0.052 |
|
Wine, all |
1800 |
0.32 |
|
Red |
1300 |
0.4 |
|
White |
260 |
0.1 |
As most of the samples (85%) represented the European diet, it is difficult to evaluate the geographical distribution of ochratoxin A. The data indicate that it occurs in coffee regardless of the geographical origin of the samples. For example, high percentages of contaminated samples were found in all countries and regions where coffee has been analysed, i.e. Brazil, Canada, Dubai, Europe, including eastern Europe, Japan, and the USA (Pittet et al., 1996; Stegen et al., 1997; Ueno, 1998; Trucksess et al., 1999; Government of Canada; Leoni et al., 2000; Rao, 2000; Wolff et al., 2000). There is clear evidence for the occurrence of ochratoxin A in cereals and cereal products in the European diet, but little information was available for other diets. The limited data indicate that ochratoxin A is also found in cereals produced elsewhere; in particular, contamination was found in 1 of 10 wheat samples and 3 of 28 mixed cereal samples in Dubai (Rao, 2000) and 56 of 383 wheat samples and 11 of 103 barley samples in the USA (Trucksess et al., 1999).
Ochratoxin A was found in 13 of 25 oat samples and 8 of 22 rye samples imported into Denmark (Jorgensen et al., 1996), 15 of 108 wheat samples and 12 of 41 rye samples imported into Norway (Langseth, 1999), and 14 of 139 maize samples imported into the United Kingdom (Scudamore & Patel, 2000). The data from Brazil and Uruguay could not be used as they had been obtained by analytical methods with high LODs, ranging from 5 µg/kg to 50 µg/kg, which are inadequate to detect and measure ochratoxin A at 0.94 µg/kg and 0.19 µg/kg, the weighted mean concen-trations found in Europe and the USA in cereals and cereal products, respectively (Pineiro & Giribone, 1994; Furlong et al., 1995a,b; Soares & Furlani, 1996; Pineiro et al., 1996).
Indirect evidence for the occurrence of ochratoxin A in foodstuffs in Africa is provided by the high incidence of contaminated samples (35%) and the high mean ochratoxin A concentration (7.9 µg/kg) found in breast milk collected in Sierra Leone, where some infants were exposed to concentrations that far exceeded the permissible levels in animal feeds in developed countries (Jonsyn et al., 1995). These high concentrations indicate considerable human exposure to this toxin from food; however, no surveys have been reported from that area. More surveys are needed in regions of the world other than Europe in order that intake in those regions can be assessed.
An unexpectedly high mean concentration of ochratoxin A, < 33 000 µg/kg, was reported in foods (wheat, barley, mixed cereals, dried vegetables, and olives) collected in Tunisia (Maaroufi et al., 1995b). One of the authors (E. Creppy) of the paper was contacted in order to check whether results had erroneously been reported as ng/kg instead of µg/kg. The response confirmed the concentrations reported in the paper, and the information was provided that the samples had been taken from members of families with one or more patient with nephropathy. These results should be confirmed in further studies with validated methods for determination of ochratoxin A, and they were not considered for estimating intake.
Data on annual variations in contamination with ochratoxin A were available for wine (Pietri, 2000), wheat, barley (Scudamore, 1999), and maize (Jurcevic et al., 1999). Higher incidences of contaminated samples and higher mean concentrations of ochratoxin A were found in maize collected in Croatia in 1997 (35%, 57 µg/kg) than in 1996 (10%, 38 µg/kg) (Jurcevic et al., 1999). A survey of wheat and barley was carried out in the United Kingdom during the crop year 1993–94. In 1993, only two of 611 cereal samples (0.3%) were contaminated, each containing 15 µg/kg. In 1994, the incidence was much higher, with 22 of 450 samples (9%) containing ochratoxin A at concentrations ranging from 1 to 32 µg/kg (Scudamore, 1999). Limited data from Italy for red wine (169 samples) showed no substantial variation of incidence over the years 1996–99, ranging from 70% to100%, whereas the mean concentration was 0.54–0.76 µg/kg in 1996–98 (54 samples) and 2.1 µg/kg in 1999 (115 samples) (Pietri, 2000).
Ochratoxin A was first encountered as a natural contaminant in maize and to a lesser extent in some beans, including coffee and cocoa. Residues of ochratoxin A are not generally found in ruminants, because the toxin is cleaved in the rumen by protozoan and bacterial enzymes. Residues have been detected in a number of tissues of non-ruminant food animals, such as pigs, and in the muscle of hens and chickens but not in eggs.
Three types of data were submitted, the first on the occurrence of ochratoxin A in foods, the second on the intake of potentially contaminated foods, and the last on biomarkers of the exposure for use in epidemiological studies.
Data were submitted by 13 countries: Argentina, Brazil, Canada, China, Denmark, France, Germany, Italy, Norway, Sweden, the United Kingdom, Uruguay, and the USA. Additional data were submitted by the Commission of the European Union and the Coffee Science Information Centre. Most of the data referred to cereals and cereal products, of which a total of 7877 samples were analysed. More than 2000 analyses were provided by Germany, pooled into six categories; 485 were from the USA, 212 from Sweden, 117 from Canada, and 75 from France. More than 2500 analyses were provided by the Commission of the European Union on the contamination of cereals by ochratoxin A in Denmark, Italy, The Netherlands, Spain, and the United Kingdom.
Brazil submitted information about ochratoxin A contamination in the electronic submission format of GEMS/Food. The data related to peanuts, grapes, beans, coffee, and maize, with a total number of 806 samples. The results for most of the samples (624) were, however, expressed as a percentage of positive values or as a range.
Canada submitted the results of surveys conducted between 1997 and 1999 on ochratoxin A in coffee and cereals, in a total of 101 samples of coffee and 117 samples of cereals and cereal products.
China submitted analytical data for 1989–91. The LOQ was very high (10 µg/kg), and none of the samples had concentrations that exceeded this value.
A sample of kidney and a sample of muscle were taken from 300 pigs from all parts of Denmark in 1999 and were analysed for their content of ochratoxin A. The analyses were performed at the Division of Chemical Contaminants, Danish Veterinary and Food Administration.
France provided results on the occurrence of ochratoxin A in pork offal (1011 samples) and in various fruits and vegetables (333 samples). The mean, the median, and the distribution of the contamination in intervals were provided for each food category.
Germany provided information on a total of 6476 samples of various foods analysed between 1995 and 1998. The data were very detailed, giving the number of samples, the LOQ of the method of analysis, the number of samples with concentrations above the LOQ, the percentage of samples with amounts below the LOQ, and the range of ochratoxin A concentrations for each food category, and, in a majority of cases, the mean, median, and 90th percentile of the distribution of contamination.
Italy provided data on the occurrence of ochratoxin A in 280 samples of wine.
Norway provided results for more than 1000 samples of cereals and cereal products.
Sweden submitted information on ochratoxin A contamination in the GEMS/Food electronic submission format. The data were for wheat, oat, rye, beans, peas, and coffee (soluble, green, and roasted). Analytical results were also provided for each sample of wheat, pulses, and coffee, making it possible to calculale the standard deviation of the distribution.
More than 2000 analytical results for various commodities were submitted by the United Kingdom.
Uruguay submitted the abstracts of scientific publications presented during the last meeting of AOAC/IUPAC (Guaruja, May 2000). Only one publication was relevant to the occurrence of ochratoxin A and gave the percentage of positive values in 600 samples of maize collected between 1991 and 1998 in Argentina, Brazil, and Paraguay.
The USA submitted data from five laboratories, reflecting the concentrations of ochratoxin A in coffee, raisins, wheat, and barley. Some of the results for green coffee (180 samples) and soluble coffee (23 samples) were expressed as percentages of positive values.
The Commission of the European Union submitted an assessment of the dietary intake of ochratoxin A by the populations of Member States of the European Union, for which data on occurrence were collected from the 12 Member States and Norway. ‘Best estimates’ were made of the mean concentrations in foods and food groups, and these are included in this monograph except for countries that provided more recent and more accurate data directly to the Committee, i.e. France, Germany, and Sweden.
The Coffee Science Information Centre provided information on total exposure to ochratoxin A contained in the position paper of the Codex Alimentarius Commission (CX/FAC 99/14), the report of a survey by the Ministry of Agriculture, Fisheries and Food of the United Kingdom of human exposure to ochratoxin A, and several published papers on the occurrence of ochratoxin A in coffee in European Member States and the USA (Patel et al., 1997; Stegen et al., 1997; Burdaspal & Legarda, 1998b; Jorgensen, 1998; Trucksess et al., 1999).
National food consumption data were provided by France, Germany, Sweden, and the Commission of the European Union.
The distribution of consumption of foods in the relevant food categories was provided on the basis of individual data. A report in which data on food intake were compared with data on contamination by ochratoxin A indicated the distribution of intake and the probability of high intake.
The German approach consisted of combining the median and the 90th percentile of the distribution of contamination with various portion sizes (small, medium, and large) obtained in 3-day and 4-week studies of mean intake. The basis of the intake studies was not provided. Surprisingly, the intakes in the longer study were higher than those obtained in the shorter study.
A Swedish report provided an estimate of the intake of ochratoxin A from various foods and was based on data on food consumption from their study of dietary habits and nutrient intake in Sweden (Becher, 1992; Swedish National Food Administration, 1994).
Data on consumption were submitted in two reports from the European Union Scientific Cooperation (SCOOP) programme: from Task Group 3.2.2 (Commission of the European Union, 1997) and from Task Group 4.1 (Commission of the European Union, 1996). These reports provided a compilation of the mean intakes of various food categories across the European Union.
The results of intake assessments that included biomarkers were provided by Sweden and the Commission of the European Union. The results of a study in the United Kingdom, available on the Internet, were submitted by the Coffee Science Information Centre.
Sweden submitted a publication on the concentrations of ochratoxin A in blood from Norwegian and Swedish donors (Thuvander et al., 2000). The mean concentration of ochratoxin A in plasma ranged from 0.17 to 0.21 ng/ml, and the 95th percentile concentration was 0.38–0.57 ng/ml.
The Ministry of Agriculture, Fisheries and Food in the United Kingdom conducted a survey of intake of ochratoxin A in which samples of duplicate diets, plasma, and urine were collected each week from 50 volunteers living in one area of the United Kingdom. A statistical analysis of the results indicated a stronger correlation between the urinary concentration of ochratoxin A and the level of consumption than with the plasma concentration.
SCOOP report 3.2.2 contains data on the occurrence of ochratoxin A in plasma and milk collected from healthy persons between 1977 and 1994 in Denmark, France, Germany, Italy, and Sweden.. These data showed a mean concentration of ochratoxin A in plasma of 1.8 ng/ml in Denmark (1986–88), 0.4 ng/ml in France (1993), 0.45 ng/ml in Germany (1977–94), 0.53 ng/ml in Italy (1992), and 0.18 ng/ml in Sweden (1994).
Intake at the international level was assessed from data on mean consumption combined with the weighted mean of contamination. As ochratoxin A occurs mainly in Europe, data on food consumption in Europe obtained from the GEMS/Food programme were considered the most relevant for risk assessment. As no information was available on coffee, beer, wine, or fruit juices in the GEMS/Food database, the mean intakes of the food categories in Europe were obtained from a report of the Commission of the European Union (1997), and those for coffee from a published paper (Jorgensen, 1998).
The available data on levels of contamination were aggregated as per the recommendations of a FAO/WHO workshop (Geneva, June 2000). The national results were presented in an aggregated format, so that one figure could represent the mean of a large number of individual samples. The first step therefore consisted of weighting each result as a function of the number of samples it represented. Each result was then multiplied by the number of individual samples in the original survey. The sum was then divided by the total number of individual samples. The result of this operation provided a weighted mean level of contamination for the food category considered. Some data were not used because they were expressed only in terms of the presence or absence of ochratoxin A, with no quantification of the mean level of contamination. As it was not possible to identify analytical results for targeted samples, all the data were considered to be representative of the total contamination of foodstuffs.
Each weighted mean can therefore be multiplied by the mean consumption of the corresponding food category to derive the contribution of that food category to human intake. The results are presented in Table 19. With this approach, the mean total intake of ochratoxin A was about 45 ng/kg bw per week, assuming a body weight of 60 kg.
Table 19. Classification of food categories as a function of their relative contribution to human exposure
|
Food category |
Contamination |
Intake |
||
|
g |
µg/person per week |
ng/kg bw per week |
||
|
Cerealsa |
0.94 |
230 |
1.5 |
25 |
|
Beerb |
0.023 |
260 |
0.04 |
0.69 |
|
Winec |
0.32 |
240 |
0.54 |
8.9 |
|
Grape juiced |
0.39 |
69 |
0.19 |
3.1 |
|
Tea |
0.3 |
2.3 |
0.00 |
0.08 |
|
Cocoa |
0.55 |
6.3 |
0.02 |
0.40 |
|
Pork |
0.17 |
76 |
0.09 |
1.5 |
|
Poultry |
0.041 |
53 |
0.02 |
0.25 |
|
Dried fruits |
2.2 |
2.3 |
0.03 |
0.58 |
|
Pulses |
0.19 |
25 |
0.03 |
0.55 |
|
Roasted coffeee |
0.76 |
24 |
0.13 |
2.1 |
a
From GEMS/Food database, data for the Far Eastern dietb
From SCOOP report 4.1, data for Norwayc
From SCOOP report 4.1, data for the United Kingdomd
From SCOOP report 4.1, data for Portugale
From Jorgensen (1998)From these calculations, various food categories could be classified as a function of their potential impact in terms of public health. Cereals and wine contributed about 25 and 10 ng/kg bw per week, respectively, to average intake, whereas grape juice and coffee each contributed 2–3 ng/kg bw per week. Other food products (dried fruits, beer, tea, milk, cocoa, poultry, and pulses) contributed < 1 ng/kg bw per week. Most of the results submitted for pig meat and products were for pig liver and kidney, whereas the figure for food consumption was based on pig meat: the resulting estimate of 1.5 ng/kg bw per week can therefore be considered a gross overestimate of intake.
The data on ‘cereal products’ included several foods manufactured from cereals. In these products, the reported mean levels of contamination were about one-fifth those in the raw material. However, these data were not adequate for use in the intake assessment.
The second step, relating to contamination of the most relevant food categories for human intake (i.e. cereals, wine, grape juice, and coffee), consisted of simulating a worldwide distribution of ochratoxin A on the basis of several assumptions (WHO, 2000b). The first concerns the form of the distribution curve. It is generally considered that food contaminants follow a log-normal distribution. In order to construct a global distribution curve assuming log-normality, the mean and the standard deviation must be determined.
The results are presented in Table 20 and in Figures 1–4.
Table 20. Distribution of contamination of major food categories with ochratoxin A
|
Food category |
No. of samplees |
Weighted mean |
Standard deviation (ln) |
|
Cereals |
2714 |
0.94 |
1.23 |
|
Cereal products |
1536 |
0.19 |
1.96 |
|
Wine |
1834 |
0.32 |
1.26 |
|
Grape juice |
68 |
0.44 |
0.48 |
|
Green coffee |
127 |
1.02 |
6.12 |
|
Roasted coffee |
1726 |
0.76 |
0.29 |
|
Grapes |
857 |
2.29 |
0.79 |
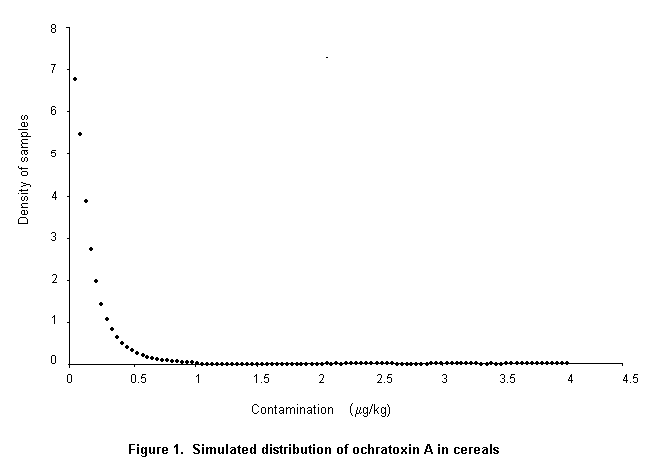
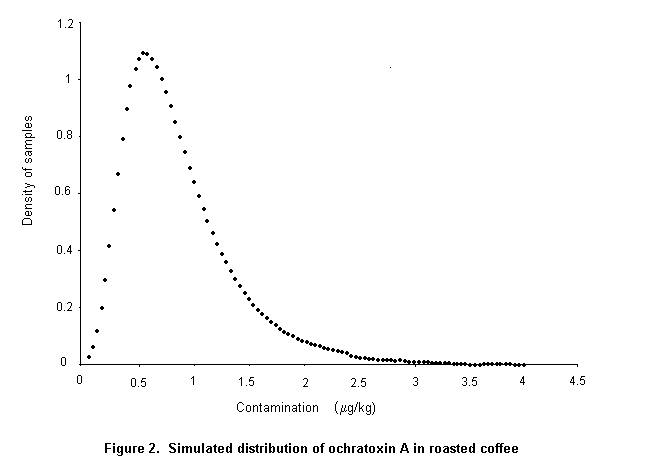
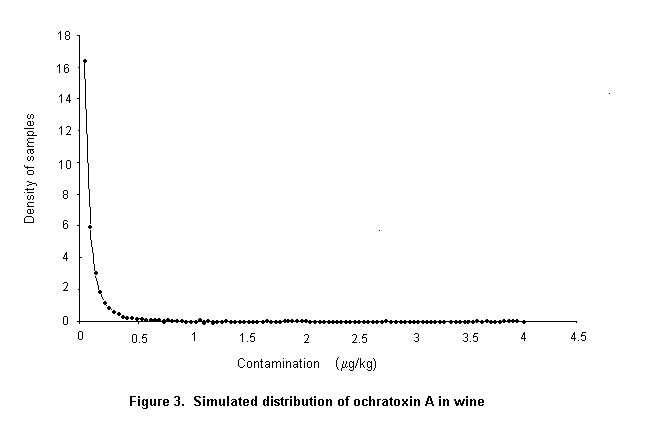
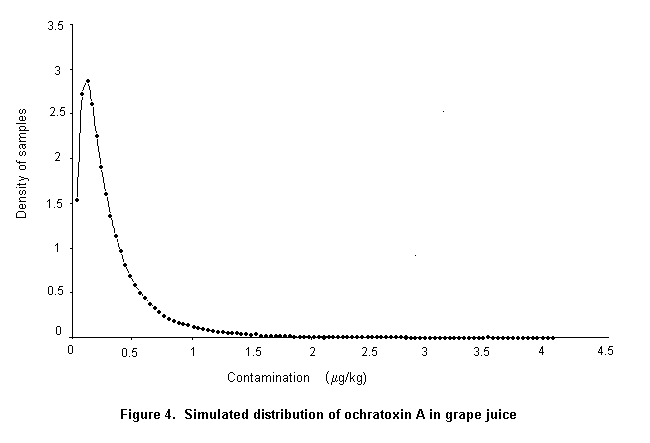
In order to assess intake from cereals, a probabilistic approach was used, in which a simulated distribution of contamination and the distribution of cereal consumption in France were used.
For cereal consumption, the results of a survey of food consumption by 1161 individuals were used, with individual body weights, assuming that 100% were consumers with a mean consumption of 28 g/week per kg bw and consumption at the 95th percentile of 61 g/week per kg bw.
For contamination of cereals, assuming that the distribution is log-normal, the model included the mean and the standard deviation and, for the specific task, the minimum and the maximum values. The maximum value was assumed to be 0 in all situations. Three simulations were made, the first with the maximum observed value (121 µg/kg), the second with the higher proposed maximum limit (20 µg/kg), and the third with the lower proposed maximum limit (5 µg/kg).
A probabilistic approach with a Monte-Carlo simulation made it possible to assess the intake of ochratoxin A by the population. The software used was @risk, and the equation for constructing the curves for contamination was riskTlognorm (mean, SD, min, max).
This simulation, which was considered to be realistic for European-type diets, showed that the intake of ochratoxin A by consumers of cereals at the 95th percentile would be 92 ng/kg bw per week. Use of the proposed maximum limit of 5 µg/kg, as opposed to 20 µg/kg, would have a statistically significant effect on intake only for consumers of cereals above the 95th percentile. However, the difference would be very small (84 vs 92 ng/kg of body weight per week) in view of the distribution of the level of contamination indicated by the available data.
Control of ochratoxin A in foods depends on the kind of crop and its geographical location, as those two factors are primarily responsible for determining which of the three major ochratoxin A-producing fungi is likely to grow and produce toxin. Moreover, as experience with other mycotoxins has shown, the likelihood and extent of toxin production by any particular species is greatly influenced by whether the fungus concerned has a particular affinity for a specific crop, i.e. can invade and grow in a crop before or during harvesting and drying. As these factors, and the physiology of the three species, differ, the control of ochratoxin A production by each species is considered separately.
As stated above, A. ochraceus occurs primarily in stored foods, and no association with plants before harvest is known. Its control therefore consists mainly of the standard methods used for controlling the growth of any fungus in dried foods.
The main commodities in which A. ochraceus may produce ochratoxin A are stored grains. The traditional way of avoiding microbial growth in grains is to dry them thoroughly and to keep them dry. Adequate ventilation in storage bins will remove moisture, prevent condensation, lower and equilibrate temperatures, and prevent heating. Over the past 20 years, bag stacks and manual handling of grain have given way to bulk handling and storage, with great improvements in the control of insect and fungal damage, even in some tropical areas (Champ & Highley, 1988). Drying techniques have made great advances, with sophisticated computer control of drying rates and temperatures now in use, at least in developed countries. Control of insect and fungal damage in grain stores is of particular importance in the tropics, where most grains are stored in sacks in warehouses unsuitable for sealing and fumigation. Recent approaches to sealing stacks of bags in such stores, fumigating and then maintaining the sealed stacks under controlled atmospheres have shown the potential to greatly reduce grain losses (Annis, 1990a; Graver, 1990).
The moisture content of grains must be reduced to below 0.8 aw (17–19% moisture; Iglesias & Chirife, 1982) to prevent ochratoxin A formation by A. ochraceus.
Modern approaches to grain storage rely on fumigation and sealed storage under controlled atmospheres, especially in tropical and subtropical regions where insect damage is a major problem (Champ et al., 1990). Fumigants, highly toxic gases or vapours added to grain stores specifically to kill insects, in some cases also achieve fungal destruction. Fumigants are usually used as a rapid method for killing insects and are subsequently removed by ventilation. A variety of gases have been used as fumigants, either singly or in combination, including ethylene dichloride, carbon tetrachloride, carbon disulfide, ethylene dibromide, chloropicrin, hydrogen cyanide, ethylene oxide, methyl chloride, methyl bromide, and phosphine. For various reasons, only methyl bromide and phosphine are in widespread use (Annis, 1990b). The concentrations recommended for use are given in Table 21. Environmental considerations are resulting in the phasing out of methyl bromide, and the search for alternative fumigants continues.
Table 21. Suggested target doses for gaseous treatments of grain at 25 °C
|
Gas |
Time (days)a |
Concentrationb |
Concentration x time |
|
Carbon dioxide |
15 |
> 35% |
– |
|
Oxygen |
20 |
< 1% |
– |
|
Phosphine |
7 |
100 mg/m3 |
– |
|
Methyl bromide |
1–2 |
– |
150 g h/m3 |
|
Hydrogen cyanide |
1 |
Not well defined |
– |
From Annis (1990b)
a In cases of slow gas introduction or poor gas distribution, longer exposure may be necessary.
b Minimum concentration achieved at end of exposure
Controlled atmospheres may be used for grain storage. This technique relies on continuous application of atmospheres low in O2 or with high CO2 concentrations. The recommended approach is to add such gas mixtures to sealed storage and maintain the grain in totally sealed systems. Where this is not practicable, continuous flow of such gas mixtures may be possible (Annis, 1990b). The recommended O2 and CO2 concentrations are given in Table 21.
Fumigation and controlled atmospheres help control mould growth on grains by directly destroying spores, by inhibiting growth, or by killing insects that damage kernels. Fumigants that merely destroy insects have no lasting effect on mould growth (Vandegraft et al., 1973); however, methyl bromide destroys fungi as well as insects (Majumder, 1974), and phosphine has some fungicidal properties (Hocking & Banks, 1993). Modified atmospheres that control insects may have a substantial effect in controlling fungi as well (Hocking, 1990).
Many investigators have suggested using heat or chlorine to destroy micro-organisms in grains; however, this technique has had little use, with the recognition that such remedial processes do not destroy mycotoxins and are not a substitute for clean grain. In the same way, ionizing radiation at 2–3 kGy destroys moulds that ordinarily spoil rice (Iizuka & Ito, 1968; Ito et al., 1971), but this process remains illegal or unacceptable to consumers in many countries.
Control of ochratoxin A production in coffee by either A. ochraceus or A. carbonarius is similar, but the control measures used are different from those for other commodities. Although some research is still required, it appears that ochratoxin A can be controlled in coffee by good manufacturing practice. Studies in Brazil by Taniwaki et al. (1999) and Taniwaki and Pitt (unpublished) have shown that mould growth and ochratoxin A production occur only during drying of green coffee beans, and that if drying is rapid and effective ochratoxin A will not be produced. Good sun-drying or a combination of sun-drying and mechanical dehydration provide effective control. No evidence has been found that either A. ochraceus or A. carbonarius invades coffee beans before harvest or has an association with the coffee tree.
The formation of ochratoxin A during drying of coffee was studied in Thailand by Bucheli et al. (2000), who showed that the toxin was normally produced during sun-drying of coffee in that country, that overripe cherries were more susceptible than green ones, and that defects, especially the inclusion of husks, were the most important source of ochratoxin A contamination. They agreed with Taniwaki et al. (1999), that better quality raw material, appropriate drying and dehulling procedures, and reduction of defects can substantially reduce the concentration of ochratoxin A in green coffee.
The formation of ochratoxin A did not increase during storage for 18 months in Thailand, even with bag storage at high humidity (Bucheli et al., 1998). However, as the initial aw of the beans stored in bags was 0.72 and did not exceed 0.75 even in the rainy season, these results are not surprising.
The suggestion by Mantle (1998) that ochratoxin A in coffee beans may result from uptake of ochratoxin A in soil by the roots of the coffee tree and then translocation is conjectural at best.
The available evidence indicates that A. carbonarius and A. niger are not pathogens on fruit, but saprophytes, and hence cannot gain entry to sound fruit. However, damage to fruit by any means, mechanical, chemical, or by disease microorganisms, may allow entry into fruit tissue, where the low pH, high sugar, and often warm temperature provide an ideal habitat for these species. This is especially true of grapes, which have very tough skins. When the skins are intact, they are resistant to attacks by these fungi, but ideal growth conditions prevail once the skin is disrupted. Control of growth of these species in grapes before harvest therefore relies on:
|
• |
control of pathogenic fungi, especially Rhizopus stolonifer, Botrytis cinerea, and powdery mildews such as Erysiphe species; |
|
• |
control of mechanical damage, from pruning, leaf reduction and, for dried fruit, harvesting equipment; and |
|
• |
control of splitting due to rain just before harvest. |
Growth of fungal pathogens is especially difficult to control on cultivars that have tight bunches or excessive leaf cover over bunches, or under rainy or misty conditions at harvest time. Control of Rhizopus stolonifer, Botrytis cinerea, and Erysiphe species relies primarily on vineyard hygiene, with removal of diseased plant tissue, thinning of tight bunches, and removal of excessively tight leaf clusters (Snowdon, 1990; Emmett et al., 1992). Fungicides are sometimes used against pathogenic fungi, but the effectiveness of such treatments is variable and depends on seasonal and geographic factors (Nair et al., 1987; Snowdon, 1990).
Control of mechanical damage relies on good farm management. In many countries, the importance of minimizing damage to grapes probably still needs emphasis. This is a serious problem when grapes are to be dried, as mechanical damage at harvest is difficult to avoid and the length of the drying process (2 weeks or more) provides ample time for growth of A. carbonarius and ochratoxin A production. This is much less of a problem with wine grapes, which are usually crushed within a few hours of picking, and it is a reasonable assumption that the rapid establishment of anaerobic conditions prevents further growth of A. carbonarius or ochratoxin A production.
Some cultivars, especially the sultana grapes favoured for drying, are susceptible to rain damage during the week preceding harvest, when turgor pressure inside the fruit is high and the skins often inflexible. Splitting around the neck of the grape below the stem provides an ideal environment for invasion by A. carbonarius. Control is very difficult. The only useful recommendation is to cut vines or bunches and commence drying as quickly as possible after rain, if damage is seen as a possibility.
Little has been published specifically about preharvest and postharvest control of ochratoxin A production in coffee by A. carbonarius, but see the section on A. ochraceus, above.
Little published information exists about the time of invasion of cereal crops by P. verrucosum. It is commonly stated that this species is a storage fungus, invading after harvest, but that does not explain the paradox that P. verrucosum is a slowly growing, not notably xerophilic species which should compete poorly against many other species known to spoil grains. There have been few reports on the incidence of P. verrucosum in Canadian or northern European grain. Frisvad & Vuif (1986) found P. viridicatum Group II (= P. verrucosum) in each of 70 samples of Danish grain containing ochratoxin A. Holmberg et al. (1991) found that the incidence of storage fungi, and particularly P. verrucosum, was significantly higher in feed samples (barley and oats or cereal mash feed) of pig herds infected with ochratoxin A: Ochratoxin A-producing P. verrucosum was found in 60% of feed samples of infected herds and in only 5% of feed of uninfected herds. Moreover, as ochratoxin A is known to occur in grain in cold climates and no Aspergillus species is likely to occur in such grain, and as P. verrucosum is the only Penicillium species that produces ochratoxin A, it is likely that P. verrucosum is the source of ochratoxin A in Canadian and northern European grains.
The occurrence of ochratoxin A in such grains is attributed to insufficient drying or over-long storage before drying. Jonsson et al. (1997) studied the effect of moisture content, temperature, and time on the growth of moulds and the production of ochratoxin A in winter wheat. The maximum storage time without mould growth appeared to be halved when the moisture content at harvest was increased by 2–3% or if the storage temperature was increased by 5 °C. Ochratoxin A could generally be detected quite soon after microbial growth had begun.
The occurrence of ochratoxin A in dried grain used for human food can be controlled by analysis and segregation of defective lots. Little or no information exists about whether this procedure is practised anywhere in the world.
Absorption, distribution, metabolism and excretion
Ochratoxin A is slowly absorbed from the gastrointestinal tract. It is distributed in a number of species via the blood, mainly to the kidneys, lower concentrations being found in liver, muscle, and fat. Transfer to milk has been demonstrated in rats, rabbits, and humans, but little is transferred to the milk of ruminants owing to metabolism of ochratoxin A by the rumenal microflora. The major metabolite of ochratoxin A in all species examined is ochratoxin alpha. This and minor metabolites that have been identified are all reported to be less toxic than ochratoxin A itself. Ochratoxin A is excreted in urine and faeces, and the relative contribution of each of these routes in different species is influenced by the extent of the enterohepatic recirculation of ochratoxin A and its binding to serum macromolecules. These factors are also important in the determination of the serum half-life of ochratoxin A, which varies widely among species. It has a long half-life in non-ruminant mammals, e.g. 24–39 h in mice, 55–120 h in rats, 72–120 h in pigs, 510 h in one macaque, and 840 h in a volunteer.
Toxicological studies
Ochratoxin A has been shown to be nephrotoxic in all mammalian species tested. Its main target is the renal proximal tubule, where it exerts cytotoxic and carcinogenic effects. Significant sex and species differences in sensitivity to nephrotoxicity were evident, in the order pig > rat > mouse. The doses at which carcinogenicity was observed in rodents were higher than those that caused nephrotoxicity. The Committee reconsidered the report of the study of carcinogenicity conducted by the National Toxicology Program (USA) in 1989 and noted the consistent presence and severity of karyomegaly in male and female rats and the aggressive nature of the renal tumours in this study. However, the biological and mechanistic significance of these observations was unclear.
Gene mutations were induced in bacteria and mammalian cells in a few studies of genotoxicity, but not in most. Ochratoxin A did, however, induce DNA damage, DNA repair, and chromosomal aberrations in mammalian cells in vitro and DNA damage and chromosomal aberrations in mice treated in vivo. Putative DNA adducts were found consistently with a 32P-postlabelling method in the kidneys of mice and rats dosed with ochratoxin A, but none of these adducts has been demonstrated to contain fragments of ochratoxin A. It was therefore uncertain whether ochratoxin A interacts directly with DNA or whether it acts by generating reactive oxygen species. There was no indication that a reactive metabolite of ochratoxin A is generated in vivo. Ochratoxin A is thus genotoxic both in vitro and in vivo, but the mechanism of genotoxicity is unclear and there was no evidence that it is mediated by direct interaction with DNA. The doses used in the studies of genetic toxicity were in the same range as those at which the incidence of renal tumours was increased in mice. In rats, however, the incidences of nephrotoxicity and renal tumours were increased at much lower doses; therefore the contribution of the genotoxicity of ochratoxin A to neoplasia in rats is unknown.
Ochratoxin A can cross the placenta and it is embryotoxic and teratogenic in rats and mice. It has been shown to have immunosuppressive effects in a number of species. Prenatal administration of ochratoxin A to rats caused immunosuppression, but perinatal administration stimulated certain aspects of the immune response in rats. Ochratoxin A inhibited the proliferation of B and T lymphocytes and affected the late stages of T-lymphocyte activation in vitro. However, both the immunological and teratogenic effects have been observed only at doses much higher than those that cause nephrotoxicity.
Observations in humans
Ochratoxin A has been found in human blood samples, most notably in a number of countries in the cool temperate climatic areas of the Northern Hemisphere; however, no cases of acute intoxication in humans have been reported. The Committee noted that ochratoxin A was found more frequently and at higher average concentrations in blood samples obtained from people living in regions where a fatal human kidney disease (known as Balkan endemic nephropathy) occurs and is associated with an increased incidence of tumours of the upper urinary tract. Nevertheless, similar average concentrations have been reported in several other European countries where this disease is not observed. The Committee concluded that the epidemiological and clinical data available do not provide a basis for calculating the likely carcinogenic potency in humans and that the etiology of Balkan endemic nephropathy may involve other nephrotoxic agents.
Analytical methods
Reliable, validated methods have been developed for the analysis of ochratoxin A in maize, barley, rye, wheat, wheat bran, wheat whole meal, roasted coffee, wine, and beer, which are based on liquid chromatography with fluorescence detection. The limit of quantification was 0.03 µg/kg for wine and beer and 0.3–0.6 µg/kg for other commodities. These methods have also been used successfully to analyse a number of other cereals, cereal products, and dried fruit. Two certified reference materials (blank and naturally contaminated wheat) are available, which improve quality assurance in laboratories. Screening methods based on TLC are available but have been used in only a few laboratories. Data obtained by these analytical methods, with a limit of quantification greater than 5 µg/kg, were not considered in this evaluation, as this was the lower concentration for which the Codex Committee on Food Additives and Contaminants requested a risk assessment. Furthermore, enzyme-linked immunosorbent assay (ELISA) techniques had not been used to produce the survey data considered by the Committee.
There are no formally validated methods for the analysis of ochratoxin A in human blood. The available methods are based on liquid chromatography with fluorescence detection and have different limits of quantification, ranging from about 0.1 to 2 ng/ml.
Sampling protocols
Adequate sampling procedures should be used in future surveys of cereals and cereal products for ochratoxin A. For example, an acceptable sampling procedure was used in 10 of 22 studies on cereals submitted to this Committee, whereas no description was reported in the remaining 12. No sampling plans for the determination of ochratoxin A in foods have been published, and details of sampling variability have not been reported.
Effects of processing
Milling has been reported to reduce substantially the concentration of ochratoxin A in white flour, but it had little effect on levels in wholemeal flour. Milling is a physical process: the ochratoxin A removed from the grain in the production of white flour remains in bran and other fractions, some of which may be used in foods. Ochratoxin A is relatively stable to heat: at 100 °C, a 50% reduction in the concentration was achieved after 2.3 h in wet wheat and 12 h in dry wheat. The process involved in the manufacture of breakfast cereals and biscuits resulted in substantial reductions in ochratoxin content, but little or no reduction was found in the manufacture of egg noodles and pasta. Decaffeination of coffee reduced the ochratoxin concentration by about 90%. The reduction obtained by roasting coffee varies but may also be as much as 90%.
Levels and patterns of contamination of food commodities
The data on ochratoxin A over the past 5 years that were reviewed by the Committee originated mainly from Europe (85%); 7% came from South America, 6% from North America, 1% from Africa, and 1% from Asia. The concentrations of ochratoxin A in the different commodities were highly variable; 1.4% and 0.6% of samples contained more than 5 µg/kg and 20 µg/kg, respectively. Within the cereals, 1.2% and 0.3% of samples contained more than 5 µg/kg and 20 µg/kg of ochratoxin A, respectively. Within cereal products, 0.3% and 0.05% of samples contained more than 5 µg/kg and 20 µg/kg of ochratoxin A, respectively. The weighted mean concentrations of ochratoxin A that were used for estimating intake were: 0.94 µg/kg for cereals, 0.19 µg/kg for cereal products, 0.32 µg/kg for wine, 0.86 µg/kg for coffee, 2.3 µg/kg for dried vine fruit, and 0.44 µg/kg for grape juice. The incidence of samples found to contain ochratoxin A depended on the commodity and was higher in the same commodity when analytical methods with lower limits of quantification were used.
Food consumption/intake assessment
Intake of ochratoxin A at the international level has been assessed on the basis of data on mean consumption combined with weighted mean levels of contamination. As ochratoxin A occurs mainly in the diet in European countries, data on food consumption in Europe obtained from the GEMS/Food database were considered the most relevant for risk assessment. The submitted data on levels of contamination were aggregated according to the recommendations of a FAO/WHO workshop to obtain a weighted mean. When this approach was used, the mean total intake of ochratoxin A was estimated to be 45 ng/kg bw per week, assuming a body weight of 60 kg.
Cereals and wine contributed about 25 and 10 ng/kg bw per week, respectively, to the mean intake, whereas grape juice and coffee each contributed 2–3 ng/kg bw per week. Other food products (dried fruits, beer, tea, milk, cocoa, poultry, and pulses) contributed less than 1 ng/kg bw per week. Most of the results submitted for pig meats and pig meat products were for samples of pig liver and kidney, whereas the figure for food consumption in the GEMS/Food database was based on pig meats. The resulting estimate of 1.5 ng/kg bw per week can therefore be considered a gross overestimate of intake.
A probabilistic approach was used to assess intake from cereals and cereal products, in which a simulated distribution of contamination and the distribution of cereal consumption in France were used. This example, which was considered to be realistic for European diets, showed that consumers of cereals at the 95th percentile would have an intake of ochratoxin A of 92 ng/kg bw per week. Use of a proposed maximum limit of 5 µg/kg as opposed to 20 µg/kg would have a statistically significant effect on intake of ochratoxin A only for consumers of quantities of cereals greater than the 95th percentile. However, the difference would be very small (84 vs 92 ng/kg bw per week at the 95th percentile) in view of the distribution of the level of contamination indicated by the available data.
Prevention and control
As formation of ochratoxin A depends on the fungal source, the type of crop, and its geographical location, control of ochratoxin A production by each fungal species was considered separately. Control of A. ochraceus, which occurs primarily in stored foods, consists of the standard methods for preventing growth of any fungus in dried foods. The major commodities in which A. ochraceus may produce ochratoxin A are stored grains. The traditional means of avoiding fungal growth in grains is to dry them rapidly and thoroughly and to keep them dry. Reduction of the moisture content of grains to provide a water activity below 0.8 is necessary to prevent formation of ochratoxin A by A. ochraceus. Further effective approaches to grain storage include fumigation, aeration and cooling, sealed storage, and controlled atmospheres, especially in tropical and subtropical regions where insect damage is a major problem. Controlled atmosphere storage is achieved by continuous application of atmospheres with a low oxygen or a high carbon dioxide concentration. Modified atmospheres to control insects may contribute to controlling fungi. Some fumigants used for insect control may also control fungi.
As ochratoxin A is apparently formed in green coffee beans after harvest, agricultural practice has little or no influence on the concentration of the toxin in dried beans. Control measures for ochratoxin A in coffee are therefore based on good manufacturing practice, i.e. rapid and effective drying, good storage practices, and, in some countries, colour sorting to reject defective beans.
The available evidence indicates that A. carbonarius and A. niger are not pathogens on fruit such as grapes and hence cannot gain entry to sound fruit. However, mechanical or chemical damage to fruit or damage caused by insects or microorganisms may permit fungal invasion of fruit tissue. Controlling the growth of these species in grapes before harvest therefore relies on controlling pathogenic fungi, mechanical damage, and splitting due to rain just before harvest. The occurrence of ochratoxin A from P. verrucosum in Canadian and European grains was attributed to insufficient drying or inadequate storage. Analysis and segregation of defective lots could be used to reduce the concentration of ochratoxin A in dried grain used for human food.
The Committee concluded that the new data raised further questions about the mechanisms by which ochratoxin A causes nephrotoxicity and renal carcinogenicity and the interdependence of these effects. The mechanism by which ochratoxin A causes carcinogenicity is unknown, although both genotoxic and non-genotoxic modes of action have been proposed. The Committee noted that studies to resolve these issues are in progress and would wish to review the results when they become available. The Committee retained the previously established PTWI of 100 ng/kg bw per week, pending the results of on-going studies on the mechanisms of nephrotoxicity and carcinogenicity, and recommended a further review of ochratoxin A in 2004. In reaching this conclusion, the Committee noted the large safety factor applied to the NOEL for nephrotoxicity in deriving the PTWI, which corresponds to a factor of 1500 applied to the NOEL for carcinogenicity in male rats, the most sensitive species and sex for this end-point.
The adverse effect at the lowest effective dose in several mammalian species is nephrotoxicity, and this is likely also to be true in humans. Although an association between the intake of ochratoxin A and nephropathy in humans has been postulated, causality has not been established. The Committee noted that the intake of ochratoxin A by 95th percentile consumers of cereals may approach the PTWI from this source alone. Given the distribution of ochratoxin A contamination of cereals, application of a limit of 5 or of 20 µg/kg would make no significant difference to the average intake. The estimated intake at the 95th percentile of cereal consumers on a European diet would be about 84 and 92 ng/kg bw per week, respectively. Intakes below the PTWI would not present an appreciable risk. The Committee was unable, on the basis of the available data, to arrive at a quantitative estimate of the risk for nephrotoxicity if the PTWI were to be exceeded. Efforts are needed to ensure that intakes of ochratoxin A do not exceed the PTWI, and this could best be achieved by lowering overall contamination by appropriate agricultural, storage, and processing practices.
Recommendations
|
• |
Studies should be conducted to clarify the mechanism by which ochratoxin A induces nephrotoxicity and carcinogenicity. |
|
• |
Appropriate sampling procedures should be developed for food commodities likely to be contaminated with ochratoxin A. |
|
• |
Better surveys are needed, particularly in regions of the world other than Europe, in order that intakes in those regions may be assessed. |
|
• |
Epidemiological investigations should be encouraged to explore the role of ochratoxin A in chronic renal disease. |
|
• |
Studies should be conducted to improve understanding of the occurrence and ecology of the fungi that produce ochratoxin A, especially in fresh produce. |
Abarca, M.L., Bragulat, M.R., Castellá, G. & Cabañes, F.J. (1994) Ochratoxin A production by strains of Aspergillus niger var. niger. Appl. Environ. Microbiol., 60, 2650–2652.
Abdel-Wahhab, M.A., Nada, S.A. & Arbid, M.S. (1999. Ochratoxicosis: Prevention of developmental toxicity by L-methionine in rats. J. Appl. Toxicol., 19, 7–12.
Albassam, M.A., Yong, S.I., Bhatnagar, R., Sharma, A.K. & Prior, M.G. (1987) Histopathologic and electron microscopic studies on the acute toxicity of ochratoxin A in rats. Vet. Pathol., 424, 427–435.
Aleo, M.D., Wyatt, R.D. & Schnellmann, R.G. (1991) Mitochondrial dysfunction is an early event in ochratoxin A but not oosporein toxicity to rats’ renal proximal tubules. Toxicol. Appl. Pharm.,107, 73–80.
Annis, P. (1990a) Sealed storage of bag stacks: Status of the technology. In: Champ, B.R., Highley, E. & Banks, H.J., eds, Fumigation and Controlled Atmosphere Storage of Grain, Canberra, ACT, Australian Centre for International Agricultural Research, pp. 203–210.
Annis, P. (1990b) Requirements for fumigation and controlled atmospheres as options for pest and quality control in stored grain. In: Champ, B.R., Highley, E. & Banks, H.J., eds, Fumigation and Controlled Atmosphere Storage of Grain, Canberra, ACT, Australian Centre for International Agricultural Research, pp. 20–28.
Appelgren, L.E. & Arora, R.G. (1983a) Distribution of 14C-labelled ochratoxin A in pregnant mice. Food Chem. Toxicol., 21, 563–568.
Appelgren, L.E. & Arora, R.G. (1983b) Distribution studies of 14C-labelled aflatoxin B1 and ochratoxin A in pregnant mice. Vet. Res. Commun., 7, 141–144.
Arora, R.G. & Frölen, H. (1981) Interference of mycotoxins with prenatal development of the mouse. 2. Ochratoxin A-induced teratogenic effects in relation to the dose and stage of gestation. Acta Vet. Scand., 22, 535–552.
Auffray, Y. & Boutibonnes, P. (1986) Evaluation of the genotoxic activity of some mycotoxins using Escherichia coli, in the SOS spot test. Mutat. Res., 171, 79–82.
Ballinger, M.B., Phillips, T.D. & Kubena, L.F. (1986) Assessment of the distribution and elimination of ochratoxin A in the pregnant rat. 1. Food Saf., 8, 11–24.
Barnikol, H. & Thalmann, A. (1988) [Clinical observations in the pig in relation to the mycotoxins ochratoxin A and zearalenone.] Tierärztl. Umsch., 43, 74–82 (in German).
Bartsch, H., Malaveille, C., Camus, A.M., Martel-Planche, G., Brun, G., Hautefeuille, A., Sabadie, N., Barbin, A., Kuroki, T., Drevon, C., Piccoli, A. & Montesano, R. (1980) Validation and comparative studies on 180 chemicals with S. typhimurium strains and V79 Chinese hamster cells in the presence of various metabolizing systems. Mutat. Res., 76, 1–50.
Baudrimont, I., Betbeder, A., Gharbi, A.M., Pfohl-Leszkowicz, A., Dirheimer, G. & Creppy, E.E. (1994) Effect of superoxide dismutase and catalase on the nephrotoxicity induced by subchronical administration of ochratoxin A in rats. Toxicology, 89, 101–111.
Baudrimont, I., Betbeder, A. & Creppy, E.E. (1997) Reduction of the ochratoxin A-induced cytotoxicity in Vero cells by aspartame. Arch Toxicol., 71, 290–298.
Bauer, J. & Gareis, M. (1987) [Ochratoxin A in the food chain.] Z. Veterinärmed. B., 34, 613–627 (in German).
Becher, W. (1992) Food habirs and nutrient intake in Sweden. Vår Föda, 349–362 (in Swedish with English summary).
Belicza, M., Radonic, M. & radosevic, Z. (1979) [Pathoanatomical findings in kidneys of persons who died from endemic nephropathy.] In: Danilovic, V., ed., Proceedings of the 2nd Symposium on Endemic Nephropathy, Belgrade: Serbian Academy of Science and Arts, pp. 103–108.
Belmadani, A, Tramu, G, Betbeder, A.M., Steyn, P.S. & Creppy, E.E. (1998a) Regional selectivity to ochratoxin A, distribution and cytotoxicity in rat brain. Arch Toxicol., 72, 656–562.
Belmadani, A, Tramu, G, Betbeder, A.M. & Creppy, E.E. (1998b) Subchronic effects of ochratoxin A on young adult rat brain and partial prevention by aspartame, a sweetener. Hum. Exp. Toxicol., 17, 380–386.
Bendele, A.M. & Carlton, W.W. (1986) Incidence of obstructive uropathy in male B6C3F1 mice on a 24-month carcinogenicity study and its apparent prevention by ochratoxin A. Lab. Anim. Sci., 36, 282–285.
Bendele, A.M., Carlton, W.W., Krogh, P. & Lillehoj, E.B. (1985a) Ochratoxin A carcinogenesis in the (C57BL/6J x C3H)F1 mouse. J. Natl Cancer Inst., 75, 733–742.
Bendele, A.M., Neal, S.B., Oberly, T.J., Thompson, C.Z., Bewsey, B.J., Hill, L.E., Rexroat, M.A., Carlton, W.W. & Probst, G.S. (1985b) Evaluation of ochratoxin A for mutagenicity in a battery of bacterial and mammalian cell assays. Food Chem. Toxicol., 23, 911–918.
Berndt, W.O. & Hayes, A.W. (1979) In vivo and in vitro changes in renal function caused by ochratoxin A in the rat. Toxicology, 12, 5–17.
Blanc, M., Pittet, A., Muñoz-Box, R. & Viani, R. (1998) Behavior of ochratoxin A during green coffee roasting and soluble coffee manufacture. J. Agric. Food Chem., 46, 673–675.
Boorman, G.A., Hong, H.L., Dieter, M.P., Hayes, H.T., Pohland, A.E., Stack, M. & Luster, M.I. (1984) Myelotoxicity and macrophage alteration in mice exposed to ochratoxin A. Toxicol. Appl. Pharmacol., 72, 304–312.
Borso, G. (1996) Characteristics of clinical data on endemic nephropathy. In: Cvorisec, D., Ceovic, S. & Stavljenic-Rukavina, A., eds, Endemic Nephropathy in Croatia, Zagreb: Academia Croatica Scientiarum Medicarum, pp. 73–75.
Bose, S. & Sinha, S.P. (1994) Modulation of ochratoxin-produced genotoxicity in mice by vitamin C. Food Chem. Toxicol., 32, 533–537.
Boudra, H., Le Bars, P. & Le Bars, J. (1995) Thermostability of ochratoxin A in wheat under two moisture conditions. Appl. Environ. Microbiol., 61, 1156–1158.
Braunberg, R.C., Gantt, O., Barton, C. & Friedman, L. (1992) In vitro effects of the nephrotoxin ochratoxin A and citrinin upon biochemical function of porcine kidney. Arch. Environ. Contam. Toxicol., 22, 464–470.
Braunberg, R.C., Barton, C., Gantt, O. & Friedman, L. (1994) Interaction of citrinin and ochratoxin A. Nat. Toxins., 2, 124–131.
Breitholtz, A., Olsen, M., Dahlback, A. & Hult, K. (1991) Plasma ochratoxin A levels in three Swedish populations surveyed using an ion-pair HPLC technique. Food Addit. Contam., 8, 183–192.
Breitholtz-Emanuelsson, A., Fuchs, R., Hult, K. & Appelgren, L-.E. (1992) Syntheses of 14C-ochratoxin A and 14C-ochratoxin B and a comparative study of their distribution in rats using whole body autoradiography. Pharmacol. Toxicol., 70, 255–261.
Breitholtz-Emanuelsson, A., Palminger-Hallen, I., Wohlin, P.O., Oskarsson, A., Hult, K. & Olsen, M. (1993a) Transfer of ochratoxin A from lactating rats to their offspring: A short-term study. Nat. Toxins, 1, 347–352.
Breitholtz-Emanuelsson, A., Olsen, M., Oskarsson, A., Palminger, I. & Hult, K. (1993b) Ochratoxin A in cow’s milk and in human milk with corresponding human blood samples. J. AOAC Int., 76, 842–846.
Breitholtz-Emanuelsson, A., Minervini, F., Hult, K. & Visconti, A. (1994) Ochratoxin A in human serum samples collected in southern Italy from healthy individuals and individuals suffering from different kidney disorders. In: Breitholtz-Emanuelsson, A., dissertation, Ochratoxin A. Analysis, Occurrence and Exposure. Stockholm: Royal Institute of Technology, Department of Biochemistry and Biotechnology.
Breitholtz-Emanuelsson, A., Fuchs, R. & Hult, K. (1995) Toxicokinetics of ochratoxin A in rat following intratracheal administration. Nat. Toxins, 3, 101–103.
Bruinink, A. & Sidler, C. (1997) The neurotoxic effects of ochratoxin-A are reduced by protein binding but are not affected by l-phenylalanine. Toxicol. Appl. Pharmacol., 146, 173–179.
Bruinink, A., Rasonyi, T. & Sidler, C. (1997) Reduction of ochratoxin A toxicity by heat-induced epimerisation. In vitro effects of ochratoxins on embryonic chick meningeal and other cell cultures. Toxicology, 118, 205–210.
Bruinink, A., Rasonyi, T. & Sidler, C. (1998) Differences in neurotoxic effects of ochratoxin A, ochracin and ochratoxin-alpha in vitro. Nat. Toxins, 6, 173–177.
Bucheli, P., Meyer, I., Pittet, A., Vuataz, G. & Viani, R. (1998) Industrial storage of green robusta coffee under tropical conditions and its impact on raw material quality and ochratoxin A content. J. Agric. Food Chem., 46, 4507–4511.
Bucheli, P., Kanchanomai, C., Meyer, I. & Pittet, A. (2000) Development of ochratoxin A during robusta (Coffea canephora) coffee cherry drying. J. Agric. Food Chem., 48, 1358–1362.
Burdaspal, P.A. & Legarda, T.M. (1998a) [Ochratoxin A in beer produced in Spain and other European countries.] Alimentaria, 291, 115–122 (in Spanish).
Burdaspal, P.A. & Legarda, T.M. (1998b) [Ochratoxin A in commercial coffee in Spain.] Alimentaria, 31–35 (in Spanish).
Burdaspal, P.A. & Legarda, T.M. (1999) [Ochratoxin A in wines, must and grape juice produced in Spain and other European countries.] Alimentaria, 299, 107–113 (in Spanish).
Burdaspal P.A. & Legarda T.M. (2000) Occurrence of ochratoxin A in bread marketed in Spain and other European countries. In: X International IUPAC Symposium on Mycotoxins and Phycotoxins, Guaruja, Brazil.
Campbell, M.L., Jr, May, J.D., Huff, W.E. & Doerr, J.A. (1983) Evaluation of immunity of young broiler chickens during simultaneous aflatoxicosis and ochratoxicosis. Poult. Sci., 62, 2138–2144.
Canada (2000) Submission of data for the 56th meeting of the Joint FAO//WHO Expert Committee on Food Additives.
Cantafora, A., Grossi, M., Miraglia, M. & Benelli, L. (1983) Determination of ochratoxin A in coffee beans using reversed-phase high performance liquid chromatography. Riv. Soc. Ital. Sci. Aliment., 12, 103–108.
Castegnaro, M., Mohr, U., Pfohl-Leszkowicz, A., Estève, J., Steinmann, J., Tillmann, T., Michelon, J. & Bartsch, H. (1998) Sex- and strain-specific induction of renal tumors by ochratoxin A in rats correlates with DNA adduction. Int. J. Cancer, 77, 70–75.
Ceovic, S., Hrabar, A. & Saric, M. (1992) Epidemiology of Balkan endemic nephropathy. Food Chem. Toxicol., 30, 183–188.
Ceovic, S. & Miletic-Medved, M. (1996) Epidemiological features of endemic nephropathy in the focal area of Brodska Posavina, Croatia. In: Cvorisec, D., Ceovic, S. & Stavljenic-Rukavina, A., eds, Endemic Nephropathy in Croatia. Zagreb: Academia Croatica Scientiarum Medicarum, pp. 7–21.
Chakor, K., Creppy, E.E., & Dirheimer, G. (1988) In vivo studies on the relationship between hepatic metabolism and toxicity of ochratoxin A. Arch. Toxicol., Suppl. 12, 201–204.
Champ, B.R. & Highley, E., eds (1988) Bulk Handling and Storage of Grain (ACIAR Proceedings No. 22), Canberra, ACT: Australian Centre for International Agricultural Research.
Champ, B.R., Highley, E. & Banks, H.J., eds (1990) Fumigation and Controlled Atmosphere Storage of Grain (ACIAR Proceedings No. 25), Canberra, ACT: Australian Centre for International Agricultural Research.
Charoenpornsook, K., Fitzpatrick, J.L. & Smith, J.E. (1998) The effects of four mycotoxins on the mitogen stimulated proliferation of bovine peripheral blood mononuclear cells in vitro. Mycopathologia, 143, 105–111
Chernozemsky, I.N., Stoyanov, I.S., Petkova-Bocharova, T.K., Nicolov, I.G., Draganov, I.V., Stoichev, I., Tanchev, Y., Naidenov, D. & Kalcheva, N.D. (1977) Geographic correlation between the occurrence of endemic nephropathy and urinary tract tumours in Vratza district, Bulgaria. J. Cancer, 19, 1–11.
Ciegler, A. (1972) Bioproduction of ochratoxin A and penicillic acid by members of the Aspergillus ochraceus group. Can. J. Microbiol., 18, 631–636.
Commission of the European Union (1996) Task 4.1, Improvement of knowledge of food consumption with a view to protection of public health by means of exchanges and collaboration between database managers (SCOOP report), Brussels.
Commission of the European Union (1997) Task 3.2.2, Assessment of dietary intake of ochratoxin A by the populations of EU Member States (EUR 17523) (SCOOP report), Brussels.
Cooray, R. (1984) Effects of some mycotoxins on mitogen-induced blastogenesis and SCE frequency in human lymphocytes. Food Chem. Toxicol., 22, 529–534.
Creppy, E.E., Lugnier, A.A.J., Fasolo, F., Heller, K., Röschenthaler, R. & Dirheimer, G. (1979a) In vitro inhibition of yeast phenylalanyl–tRNA synthetase by ochratoxin A. Chem. Biol. Interactions, 24, 257–262.
Creppy, E.E., Lugnier, A.A.J., Beck, G., Röschenthaler, R. & Dirheimer, G. (1979b) Action of ochratoxin A on cultured hepatoma cells reversion of inhibition by phenylalanine. FEBS Lett., 104, 287–290.
Creppy, E.E., Lorkowski, G., Beck, G., Röschenthaler, R. & Dirheimer, G. (1980a) Combined action of citrinin and ochratoxin A on hepatoma tissue culture cells. Toxicol. Lett., 5, 375–380.
Creppy, E.E., Schlegel, M., Röschenthaler, R. & Dirheimer, G. (1980b) Phenylalanine prevents acute poisoning by ochratoxin A in mice. Toxicol. Lett., 6, 77–80.
Creppy, E.E., Størmer, F.C., Kern, D., Röschenthaler, R. & Dirheimer, G. (1983a) Effect of ochratoxin A metabolites on yeast phenylalanyl–tRNA synthetase and on the growth and in vivo protein synthesis of hepatoma cells. Chem. Biol. Interactions, 47, 239–247.
Creppy, E.E., Kern, D., Steyn, P.S., Vleggaar, R., Röschenthaler, R. & Dirheimer, G. (1983b) Comparative study of the effect of ochratoxin A analogues on yeast aminoacyl–tRNA synthetases and on the growth and protein synthesis of hepatoma cells. Toxicol. Lett., 19, 217–224.
Creppy, E.E., Röschenthaler, R. & Dirheimer, G. (1984) Inhibition of protein synthesis in mice by ochratoxin A and its prevention by phenylalanine. Food Chem. Toxicol., 22, 883–886.
Creppy, E.E., Kane, A., Dirheimer, G., Lafarge-Frayssinet, C., Mousset, S. & Frayssinet, C. (1985) Genotoxicity of ochratoxin A in mice: DNA single-strand break evaluation in spleen, liver and kidney. Toxicol. Lett., 28, 29–35.
Creppy, E.E., Chakor, K., Fisher, M.J. & Dirheimer, G. (1990) The mycotoxin ochratoxin A is a substrate for phenylalanine hydroxylase in isolated rat hepatocytes and in vivo. Arch. Toxicol., 64, 279–284.
Creppy, E.E., Castegnaro, M., Grosse, Y., Meriaux, J., Manier, C., Montcharmont, P. & Waller, C. (1993) [Study of human ochratoxicosis in three regions of France: Alsace, Aquitaine, and the Rhone-Alpes region.] In: Creppy, E.E., Castegnaro, M. & Dirheimer, G., eds, Human Ochratoxicosis and its Pathologies, Montrouge: John Libbey Eurotext, INSERM, pp. 147–158 (in French).
Degen, G.H., Gerber, M.M., Obrecht-Pflumio, S. & Dirheimer, G. (1997) Induction of micronuclei with ochratoxin A in ovine seminal vesicle cell cultures. Arch. Toxicol., 71, 365–371.
De Groene, E.M., Hassing, I.G., Blonm, M.J., Seinen, W., Fink-Gremmels, J. & Horbach, G.J. (1996) Development of human cytochrome P450-expressing cell lines: Application in mutagenicity testing of ochratoxin A. Cancer Res., 56, 299–304.
Dietrich, D.R. & Swenberg, J.A. (1993) Renal carcinogenesis. In: Hook, J.B. & Goldstein, R.S., eds, Toxicology of the Kidney. New York: Raven Press, pp. 495–537.
Djokic, M., Hadzi-Djokic, J., Nikolic, J., Dragicevic, D. & Radivojevic, D. (1999) [Comparison of upper urinary tract tumours in the region of Balkan endemic nephropathy with those in other Yugoslav regions.] Prog. Urol., 9, 61–68.
Dopp, E., Muller, J., Hahnel, C. & Schiffmann, D. (1999) Induction of genotoxic effects and modulation of the intracellular calcium level in Syrian hamster embryo (SHE) fibroblasts caused by ochratoxin A. Food Chem. Toxicol., 37, 713–721.
Dorrenhaus, A. & Follmann, W. (1997) Effects of ochratoxin a on DNA repair in cultures of rat hepatocytes and porcine urinary bladder epithelial cells. Arch. Toxicol., 71, 709–713.
Dorrenhaus, A., Flieger, A., Golka, K., Schulze, H., Albrecht, M., Degen, G.H. & Follmann, W. (2000) Induction of unscheduled DNA synthesis in primary human urothelial cells by the mycotoxin ochratoxin A. Toxicol. Sci., 53, 271–277.
Dragacci, S., Grosso, F., Bire, R., Fremy, J.M. & Coulon, S. (1999) A French monitoring programme for determininon ochratoxin A occurrence in pig kidney. Nat. Toxins, 7, 167–173.
Dwivedi, P. & Burns, R.B. (1984a) Pathology of ochratoxicosis A in young broiler chicks. Res. Vet. Sci., 36, 92–103.
Dwivedi, P. & Burns, R.B. (1984b) Effect of ochratoxin A on immunoglobulins in broiler chicks. Res. Vet. Sci., 36, 117–121.
Edrington, T.S., Harvey, R.B. & Kubena, L.F. (1995) Toxic effects of aflatoxin B1 and ochratoxin A, alone and in combination, on chicken embryos. Bull. Environ. Contam. Toxicol., 54, 331–336
El Adlouni, C., Pinelli, E., Azemar, B., Zaoui, D., Beane, P. & Pfohl-Leszkowicz, A. (2000) Phenobarbital increases DNA adduct and metabolites formed by ochratoxin A: Role of CYP 2C9 and microsomal glutathione-S-transferase. Environ. Mol. Mutag., 35, 123–131.
Elling, F. (1979a) Ochratoxin A-induced mycotoxic porcine nephropathy: Alterations in enzyme activity in tubular cells. Acta Pathol. Microbiol. Scand., 87, 237–243.
Elling, F. (1979b) Enzyme histochemical studies of ochratoxin A-induced mycotoxic porcine nephropathy (abstract). In: 6th International Symposium Animal, Plant Microbial Toxins, Vol. 17.
Elling, F. (1983) Feeding experiments with ochratoxin A-contaminated barley to bacon pigs. IV. Renal lesions. Acta Agric. Scand., 33, 153–159.
Elling, F., Nielsen, J.P., Lillehoj, E.B., Thomassen, M.S. & Størmer, F.C. (1985) Ochratoxin A-induced porcine nephropathy: Enzyme and ultrastructure changes after short-term exposure. Toxicology, 23, 247–254.
Emmett, R.W., Harris, A.R., Taylor, R.H. & McGechan, J.K. (1992) Grape diseases and vineyard protection. In: Coombe, B.G. & Dry, P.R., eds, Viticulture. 2. Practices, Wine Titles, Adelaide, pp. 232–278.
Endou, H., Koseki, C., Yamada, H. & Obara, T. (1986) Evaluation of nephrotoxicity using isolated nephron segments. Dev. Toxicol. Environ. Sci., 14, 207–216.
Entwisle, A.C., Williams, A.C., Farnell, P.J., Jorgensen, K. & Boenke, A. (1996) The intercomparison of methods for the analysis of ochratoxin A in pig kidney (EUR report, EUR 16879 EN), Brussels.
Entwisle, A.C., Williams, A.C., Mann, P.J., Slack, P.T. & Gilbert, J. (2000a) Immunoaffinity column clean-up with liquid chromatography for the determination of ochratoxin A in barley: collaborative study. J. AOAC Int. (in press).
Entwisle, A.C., Williams, A.C., Mann, P.J., Russel, J., Slack, P.T. & Gilbert, J. (2000b) Combined phenyl silane and immunoaffinity column clean-up with liquid chromatography for the determination of ochratoxin A in roasted coffee: Collaborative study. J. AOAC Int. (in press).
Ferrufino-Guardia, E.V., Tangni, E.K., Larondelle, Y. & Ponchaut, S. (2000) Transfer of ochratoxin A during lactation: Exposure of suckling via the milk of rabbit does fed a naturally contaminated feed. Food Addit. Contam., 17, 167–175.
Fenske, M. & Fink Gremmels, J. (1990) Effects of fungal metabolites on testosterone secretion in vitro. Arch. Toxicol., 64, 72–75.
Filtenborg, O. & Frisvad, J.C. (1980) A simple screening-method for toxigenic moulds in pure cultures. Lebensm.Wiss. Technol., 13, 128–130.
Fink-Gremmels, J., John, A. & Blom, M.J. (1995) Toxicity and metabolism of ochratoxin A. Nat. Toxins, 3, 214–220.
Flieger, A., Dörrenhaus, A., Golka, K., Schulze, H. & Föllman, W. (1998) Genotoxic effect of the mycotoxin ochratoxin A in cultured human urothelial cells. Occup. Hyg., 4, 297–307.
Friis, C., Brinn, R. & Hald, B. (1988) Uptake of ochratoxin A by slices of pig kidney cortex. Toxicology, 52, 209–217.
Frisvad, J.C. (1989) The connection between the Penicillia and Aspergilli and mycotoxins with special emphasis on misidentified isolates. Arch. Environ. Contam. Toxicol., 18, 452–467.
Frisvad, J.C. & Filtenborg, O. (1989) Terverticillate Penicillia: Chemotaxonomy and mycotoxin production. Mycologia, 81, 837–861.
Frisvad, J.C. & Samson, R.A. (2000) Neopetromyces gen. nov. and an overview of teleomorphs of Aspergillus subgenus Circumdati. Stud. Mycol. (Baarn), 45, 201–207.
Frisvad, J.C. & Vuif, B.T. (1986) Comparison of direct and dilution plating for detection of Penicillium viridicatum in barley containing ochratoxin. In: King, A.D., Pitt, J.I., Beuchat, L.R. & Corry, J.E.L., eds, Methods for the Mycological Examination of Food, New York: Plenum Press, pp. 45– 47.
Fuchs, R., Radic, B., Peraica, M., Hult, K. & Plestina, R. (1988a) Enterohepatic circulation of ochratoxin A in rats. Period. Biol., 90, 39–42.
Fuchs, R., Appelgren, L., Hagelberg, S. & Hult, K. (1988b) Carbon-14-ochratoxin A distribution in the Japanese quail (Coturnix coturnix japonica) monitored by whole body autoradiography. Poult. Sci., 67, 707–714.
Fukal, L. & Reisnerova, H. (1990) Monitoring of aflatoxins and ochratoxin A in Czechoslovak human sera by immunoassay. Bull. Environ. Contam. Toxicol., 44, 345–349.
Fukui, Y., Hoshino, K., Kameyana, Y., Yasui, T., Toda, C. & Nagano, H. (1987) Placental transfer of ochratoxin A and its cytotoxic effect on the mouse embryonic brain. Food Chem. Toxicol., 25, 17–24.
Fukui, Y., Hayasaka, S., Itoh, M. & Tabenchi, Y. (1992) Development of neurons and synapses in ochratoxin A-induced microcephalic mice: A quantitative assessment of somatosensory cortex. Neurotox. Teratol., 14, 191–196.
Furlong, E.B., Soares, L.M.V., Lasca, C.C. & Kohara, E.Y. (1995a) Mycotoxins and fungi in wheat harvested during 1990 in test plots in the state of Sao Paulo, Brazil. Mycopathologia, 131, 185–190.
Furlong, E.B., Soares, L.M.V., Lasca, C.C. & Kohara, E.Y. (1995b) Mycotoxins and fungi in wheat stored in elevators in the state of Rio Grande do Sul, Brazil. Food Addit. Contam., 12, 683–688.
Galtier, P. (1978) Contribution of pharmacokinetic studies to mycotoxicology—Ochratoxin A. Vet. Sci. Commun., 1, 349–358.
Galtier, P., Charpenteau, J.L., Alvinerie, M. & Labouche, C. (1979) The pharmacokinetic profile of ochratoxin A in the rat after oral and intravenous administration. Drug Metabol. Disposition, 7, 429–434.
Galtier, P., Camguilhem, R. & Bodin, G. (1980) Evidence for in vitro and in vivo interaction between ochratoxin A and three acidic drugs. Food Cosmet. Toxicol., 18, 493–496.
Galtier, P., Alvinerie, M. & Charpenteau, J.L. (1981) The pharmacokinetic profiles of ochratoxin A in pigs, rabbits and chickens. Food Cosmet. Toxicol., 19, 735–738.
Gautier, J.-C., Richoz, J., Welti, D.H., Markovic, J., Gremaud, E., Guengerich, F.P. & Turesky, R.J. (2001) Metabolism of ochratoxin A: Absence of formation of genotoxic derivatives by human and rat enzymes. Chem. Res. Toxicol. (in press).
Gekle, M. & Silbernagl, S. (1994) The role of the proximal tubule in ochratoxin A nephrotoxicity in vivo: toxodynamic and toxokinetic aspects. Renal Physiol. Biochem., 17, 40–49.
Gekle, M., Oberleithner, H. & Silbernagl, S. (1993) Ochratoxin A impairs ‘postproximal’ nephron function in vivo and blocks plasma membrane anion conductance in Madin-Darby canine kidney cells in vitro. Pflügers Arch., 425, 401–408.
Gekle, M., Pollock, C.A. & Silbernagl, S. (1995) Time- and concentration-dependent biphasic effect of ochratoxin A on growth of proximal tubular cells in primary culture. J. Pharmacol. Exp. Ther., 275, 397–404. ,
Gekle, M., Schwerdt G., Freudinger, R., Mildenberger S., Wilflingseder D., Pollack, V., Dander, M. & Schramek, H. (2000) Ochratoxin A induces JNK activation and apoptosis in MDCK-C7 cells at nanomolar concentrations. J. Pharmacol. Exp. Ther., 293, 837–844.
Gentles, A., Smith, E.E., Kubena, L.F., Duffus, E., Johnson, P., Thompson, J., Harvey, R.B. & Edrington, T.S. (1999) Toxicological evaluations of cyclopiazonic acid and ochratoxin A in broilers. Poult Sci., 78, 1380–1384
Gharbi, A., Trillon, O., Betbeder, A.M., Counord, J., Gauret, M.F., Pfohl-Leszkowicz, A., Dirheimer, G. & Creppy, E.E. (1993) Some effects of ochratoxin A, a mycotoxin contaminating feeds and food, on rat testis. Toxicology, 83, 9–18.
Gibson, R., Bailey, C., Kuena, L., Huff, W. & Harvey, R. (1990) Impact of L-phenylalanine supplementation on the performance of three-week-old broilers fed diets containing ochratoxin A. 1. Effects on body weight, feed conversion, relative organ weight, and mortality. Poult. Sci., 69, 414–419.
Gillman, I.G., Clark, T.N. & Manderville, R.A. (1999) Oxidation of ochratoxin A by an Fe-porphyrin system: Model for enzymatic activation and DNA cleavage. Chem. Res. Toxicol., 12, 1066–1076.
Gloria, E.M., Fonseca, H. & Souza, I.M. (1997) Occurrence of mycotoxins in maize delivered to the food industry in Brazil. Trop. Sci., 37, 107–110.
Golinski, P. (1987) Ochratoxin A in human organism as a result of food and feed contamination. Rocz. AR Poznaniu, 168, 1–61.
Graver, J.E. van S. (1990) Fumigation and controlled atmospheres as components of integrated commodity management in the tropics. In: Champ, B.R., Highley, E. & Banks, H.J., eds, Fumigation and Controlled Atmosphere Storage of Grain. Canberra, ACT: Australian Centre for International Agricultural Research, pp. 38–52.
Grosse, Y., Baudrimont, I., Castegnaro, M., Betbeder, A.M., Creppy, E.E., Dirheimer, G. & Pfohl-Leszkowicz, A. (1995) Formation of ochratoxin A metabolites and DNA-adducts in monkey kidney cells. Chem.-Biol. Interactions, 95, 175–187.
Grosse, Y., Chekir-Ghedira, L., Huc, A., Obrecht-Pflumio, S., Dirheimer, G., Bacha, H., & Pfohl-Leszkowicz, A. (1997) Retinol, ascorbic acid and alpha-tocopherol prevent DNA adduct formation in mice treated with the mycotoxins ochratoxin A and zearalenone. Cancer Lett., 114, 225–229.
Groves, C.E., Morales, M. & Wright, S.H. (1998) Peritubular transport of ochratoxin A in rabbit renal proximal tubules. J. Pharmacol. Exp. Ther., 284, 943–948.
Hadlok, R.M. (1993) Human ochratoxicosis in Germany updating 1993. In: Creppy, E.E., Castegnaro, M. & Dirheimer, G., eds, Human Ochratoxicosis and its Pathologies, Montrouge: John Libbey Eurotext, INSERM, pp. 141–145.
Hagelberg, S., Hult, K. & Fuchs, R. (1989) Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol., 9, 91–96.
Hald, B. (1991) Ochratoxin A in human blood in European countries. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 159–164.
Hald, B., Wood, G.M., Boenke, A., Schurer, B. & Finglas, P. (1993) Ochratoxin A in wheat: An intercomparison of procedures. Food Addit. Contam., 10, 185–207.
Hall, P. & Vasiljevic, M. (1973) alpha-microglobulin excretion as an index of renal tubular disorders with special reference to endemic Balkan nephropathy. J. Lab. Clin. Med., 81, 897–904.
Hallén, I.P., Breitholtz-Emanuelsson, A., Hult, K., Olsen, M. & Oskarsson, A. (1998) Placental and lactational transfer of ochratoxin A in rats. Nat. Toxins, 6, 43–49.
Hansen, C.E., Dueland, S., Drevon, C.A. & Størmer, F.C. (1982) Metabolism of ochratoxin A by primary cultures of rat hepatocytes. Appl. Environ. Microbiol., 43, 1267–1271.
Hard, G.C. (1998) Mechanisms of chemically induced renal carcinogenesis in the laboratory rodent. Toxicol. Pathol., 26, 104–112.
Hard, G.C. (2000) Histopathologic evaluation of rat kidney from toxicity and carcinogenicity studies with ochratoxin A. Expert report submitted by the International Life Sciences Institute, Washington DC, USA.
Harvey, R.B., Kubena, L.F., Naqi, S.A., Gyimah, J.E., Corrier, D.E., Panigrahy, B. & Phillips, T.D. (1987) Immunologic effects of low levels of ochratoxin A in ovo: Utilization of a chicken embryo model. Avian Dis., 31, 787–791.
Harwig, J., Kuiper-Goodman, T. & Scott, P.M. (1983) Microbial food toxicants: Ochratoxins. In: Rechcigl, M., ed., Handbook of Foodborne Diseases of Biological Origin, Boca Raton, FL: CRC Press, pp. 193–238.
Hatey, F. & Galtier, P. (1977) [Short-term toxicity of ochratoxin A in rats; some biochemical manifestations of intoxication.] Ann. Rech. Vét., 8, 7–12 (in French).
Heenan, C.N., Shaw, K.J. & Pitt, J.I. (1998) Ochratoxin A production by Aspergillus carbonarius and A. niger isolates and detection using coconut cream agar. J. Food Mycol., 1, 67–72.
Heilmann, W., Rehfeldt, A.G. & Rotzoll, F. (1999) Behaviour and reduction of ochratoxin A in green coffee beans in response to various processing methods. Eur. Food Res. Technol., 209, 297–300.
Hennig, A., Fink-Gremmels, J. & Leistner, L. (1991) Mutagenicity and effects of ochratoxin A on the frequency of sister chromatid exchange after metabolic activation. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 255–260.
Hesseltine, C.W., Vandegraft, E.E., Fennell, D.I., Smith, M.L. & Shotwell, O.L. (1972) Aspergilli as ochratoxin producers. Mycologia, 64, 539–550.
Hietanen, E., Bartsch, H:, Béréziat, J.C., Castegnaro, M. & Michelon, J. (1991) Characterization of the cytochrome P450 isozyme that metabolizes ochratoxin A, using metabolic inducers, inhibitors, and antibodies. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp.297–304.
Hocking, A.D. (1990) Responses of fungi to modified atmospheres. In: Champ, B.R., Highley, E. & Banks, H.J., eds, Fumigation and Controlled Atmosphere Storage of Grain, Canberra, ACT: Australian Centre for International Agricultural Research, pp. 70–82.
Hocking, A.D. & Banks, H.J. (1993) The use of phosphine for inhibition of fungal growth in stored grains. In: Navarro, S. & Donayahe, E., eds, Controlled Atmosphere and Fumigation in Grain Storages, Jerusalem: Caspit Press, pp. 173–182.
Hocking, A.D. & Faedo, M. (1992) Fungi causing thread mould spoilage of vacuum packaged cheddar cheese during maturation. Int. J. Food Microbiol., 16, 123–130.
Hoehler, D., Marquardt, R.R., McIntosh, A.R. & Xiao, H. (1996) Free radical generation as induced by ochratoxin A and its analogs in bacteria (Bacillus brevis). J. Biol. Chem., 271, 27388–27394.
Hoehler, D., Marquardt, R.R., McIntosh, A.R. & Hatch, G.M. (1997) Induction of free radicals in hepatocytes, mitochondria and microsomes of rats by ochratoxin A and its analogs. Biochim. Biophys. Acta, 1357, 225–233.
Holmberg, T., Breitholtz-Emanuelsson, A., Häggblom, P., Schwan, O. & Hult, K. (1991) Penicillium verrucosum in feed of ochratoxin A positive swine herds. Mycopathologia, 116, 169–176.
Hong, H.H.L., Jameson, C.W. & Boorman, G.A. (1988) Residual haematopoietic effect in mice exposed to ochratoxin A prior to irradiation. Toxicology, 53, 57–67.
Horie, Y. (1995) Productivity of ochratoxin A of Aspergillus carbonarius in Aspergillus section Nigri. Nippon Kingakukai Kaiho, 36, 73–76.
Howell, M.V., & Taylor, W. (1981) Determination of aflatoxins, ochratoxin A, and zearalenone in mixed feeds, with detection by thin layer chromatography or high performance liquid chromatography. J. AOAC Int., 64, 1356–1363.
Hult, K. & Fuchs, R. (1986) Analysis and dynamics of ochratoxin A in biological systems. In: Steyn, P.S. & Vleggaar, R., eds, Mycotoxins and Phycotoxins, Amsterdam: Elsevier Science Publishers BV, pp. 365–376.
Hult, K., Teiling, A. & Gatenbeck, S. (1976) Degradation of ochratoxin A by a ruminant. Appl. Environ. Microbiol., 32, 443–444.
Hult, K., Hökby, E., Hägglund, U., Gatenbeck, S., Rutqvist, L. & Sellyey, G. (1979) Ochratoxin A in pig blood: method of analysis and use as a tool for feed studies. Appl. Environ. Microbiol., 38, 772–776.
Hult, K., Plestina, R., Habazin-Novak, V., Radic, B. & Ceovic, S. (1982) Ochratoxin A in human blood and Balkan endemic nephropathy. Arch. Toxicol., 51, 313–321.
Hult, K., Hokby, E., Sellyey, G., Rutqvist, L. & Gatenbeck, S. (1992) Ochratoxin A occurrence in slaughter-pigs in Sweden and its use us a tool for feed screening programs. J. Environ. Pathol. Toxicol.Oncol., 11, 39–40.
IARC (1976) Ochratoxin A. In: IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, Vol. 10, Lyon: IARCPress, pp. 191–197.
IARC (1993) Ochratoxin A. In: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 56, Lyon: IARCPress, pp. 489–521.
Iglesias, H.H. & Chirife, J. (1982) Handbook of Food Isotherms, New York: Academic Press.
Iizuka, H. & Ito, H. (1968) Effect of gamma-irradiation on the microflora of rice. Cereal Chem., 45, 503–511.
Ito, H., Shibabe, S. & Iizuka, H. (1971) Effect of storage studies of microorganisms on gamma-irradiated rice. Cereal Chem., 48, 140–149.
Jiao, Y., Blaas, W., Rühl, C., & Weber, R. (1992) Identification of ochratoxin A in food samples by chemical derivatization and gas chromatography–mass spectrometry. J. Chromatogr., 595, 364–367.
Jiao, Y., Blaas, W., Rühl, C. & Weber, R. (1994) [Ochratoxin A in foods of plant origin.] Dtsch. Lebensm. Rundsch., 90 (in German).
Jiménez, A.M., López de Cerain, A., González Peñas, E., Bello, J., Betbeder, A.M. & Creppy, E.E. (1998) Exposure to ochratoxin A in Europe: Comparison with a region of northern Spain. J. Toxicol.Toxin Rev., 17, 479–491.
Jonsson, N., Pettersson, H. & Schnürer, J. (1997) Study of the relationship between storage conditions and the growth of moulds and production of ochratoxin A in grain—Preliminary results. 1997 Annual ASAE International meeting, Paper No. 976037. St Joseph, MI.
Jonsyn, F.E. (1996) The intake of aflatoxins and ochratoxin A by infants in Sierra Leone. In: Miraglia, M., Brera, C. & Onori, R., eds, IX International IUPAC Symposium on Mycotoxins and Phycotoxins, Rome, p. 209.
Jonsyn, F.E., Maxwell, S.M. & Hendrickse, R.G. (1995) Ochratoxin A and aflatoxins in breast milk samples from Sierra Leone. Mycopathologia, 131, 121–126.
Jorgensen, K. (1998) Survey of pork, poultry, coffee, beer and pulses for ochratoxin A. Food Addit. Contam., 15, 550–554.
Jorgensen, K., Rasmussen, G. & Thorup, I. (1996) Ochratoxin A in Danish cereals 1986–1992 and daily intake by the Danish population. Food Addit. Contam., 13, 95–104.
Jung, K.Y. & Endou, H. (1989) Nephrotoxicity assessment by measuring cellular ATP content. II. Intranephron site of ochratoxin A nephrotoxicity. Toxicol. Appl. Pharmacol., 100, 383–390.
Jurcevic, Z., Solfrizzo, M., Cvjetkovic, B., Avantaggiato, G., & Visconti, A. (1999) Ochratoxin A and fumonisins (B1 and B2) in maize from Balkan nephropathy endemic and non endemic areas of Croatia. Mycotoxin Res., 15, 67–80.
Juszkiewicz, T., Piskorska-Pliszczynska, J & Wisniewska, H. (1982) Ochratoxin A in laying hens: Tissue deposition and passage into eggs. In: Mycotoxins and Phycotoxins. Proceedings of the V International IUPAC Symposium, Vienna, Technical University, 1–2 September, pp. 122–125.
Kane, A., Creppy, E.E., Röschenthaler, R. & Dirheimer, G. (1986a) Changes in urinary and renal tubular enzymes caused by subchronic administration of ochratoxin A in rats. Toxicology, 42, 233–243.
Kane, A., Creppy, E.E., Roth, A., Röschenthaler, R. & Dirheimer, G. (1986b) Distribution of the [H3]-label from low doses of radioactive ochratoxin A ingested by rats, and evidence for DNA single-strand breaks caused in liver and kidneys. Arch. Toxicol., 58, 219–224.
Kanisawa, M. (1984) Synergistic effect of citrinin on hepatorenal carcinogenesis of OA in mice. In: Kurata, H. & Ueno, Y., eds, Toxigenic Fungi—Their Toxins and Health Hazard, Tokyo: Kodansha and Amsterdam: Elsevier, pp. 245–254.
Kanisawa, M. & Suzuki, S. (1978) Induction of renal and hepatic tumors in mice by ochratoxin A; a mycotoxin. Gann., 69, 599–600.
Kanisawa, M., Suzuki, S., Kozuka, Y. & Yamazaki, M. (1977) Histopathological studies on the toxicity of ochratoxin A in rats 1. Acute oral toxicity. Toxicol. Appl. Pharmacol., 41, 55–64.
Kanisawa, M., Suzuki, S. & Moroi, K. (1979) Ochratoxin-alpha, is it an inducing factor of acute enteritis by ochratoxin A? In: 6th International Symposium Animal, Plant, Microbial Toxins, Uppsala, Sweden.
Kerkadi, A., Barriault, C., Tuchweber, B., Frohlich, A.A., Marquardt, R.R., Bouchard, G. & Yousef, I.M. (1998) Dietary cholestyramine reduces ochratoxin A-induced nephrotoxicity in the rat by decreasing plasma levels and enhancing fecal excretion of the toxin. J Toxicol Environ Health, 53, 237–250.
Kerkadi, A., Barriault, C., Marquardt, R.R., Frohlich, A.A., Yousef, I.M., Zhu, X.X. & Tuchweber, B. (1999) Cholestyramine protection against ochratoxin A toxicity: Role of ochratoxin A sorption by the resin and bile acid enterohepatic circulation. J Food Prot., 62, 1461–1465.
Kiessling, K.H., Pettersson, H., Sandholm, K. & Olsen, M. (1984) Metabolism of aflatoxin, ochratoxin, zearalenone, and three trichothecenes by intact rumen fluid, rumen protozoa, and rumen bacteria. Appl. Environ. Microbiol., 47, 1070–1073.
Kihara, T., Nakagawa, K., Yamamaoto, Y. & Tanimura, T. (1984) Behavioural teratological study of rat offspring exposed to ochratoxin A in utero by using cross-fostering (abstract). Teratology, 30, 10A.
Kitchen, D.N., Carlton, W.W. & Hinsman, E.J. (1977a) Ochratoxin A and citrinin induced nephrosis in beagle dogs. III. Terminal renal ultrastructural alterations. Vet. Pathol., 14, 392–406.
Kitchen, D.N., Carlton, W.W. & Tuite, J. (1977b) Ochratoxin A and citrinin induced nephrosis in beagle dogs. I. Clinical and clinicopathological features. Vet. Pathol., 14, 154–172.
Kitchen, D.N., Carlton, W.W. & Tuite, J. (1977c) Ochratoxin A and citrinin-induced nephrosis in beagle dogs. II. Pathology. Vet. Pathol., 14, 261–272.
Koch, M., Steinmeyer, S., Tiebach, G. & Weber, R. (1996) Improved determination of ochratoxin A in roasted coffee after separation of caffeine. Poster presented at the IX IUPAC international symposium on mycotoxins and phycotoxins, Rome, 27–31 May.
Konrad, I. & Röschenthaler, R. (1977) Inhibition of phenylalanine tRNA synthetase from Bacillus subtillis by ochratoxin A. FEBS Lett., 83, 341–347.
Krogh, P. (1974) Mycotoxic porcine neprophathy: A possible model for Balkan endemic nephropathy. In: Puhlev, A., ed., Endemic Nephropathy, Sofia: Publishing House of the Bulgarian Academy of Science, pp. 266–270.
Krogh, P. (1976) Epidemiology of mycotoxic porcine nephropathy. Nord Veterinaemedicin, 28, 452–458.
Krogh, P. (1992) Role of ochratoxin A in disease causation. Food Chem. Toxicol., 30, 213–224.
Krogh, P. & Elling, F. (1977) Mycotoxic nephropathy. Vet. Sci.Commun., 1, 51–63.
Krogh, P., Hald, B. & Pedersen, E.J. (1973) Occurrence of ochratoxin A and citrinin in cereals associated with mycotoxic porcine nephropathy. Acta Pathol. Microbiol. Scand., Sect. B, 81, 689–695.
Krogh, P., Elling, F., Hald. B., Jylling, B., Petersen, V.E., Skadhauge, E. & Svendsen, C.K. (1976) Experimental arian nephropathy. Acta Pathol. Microbiol., Scand. A., 84, 215–221.
Krogh, P., Gyrd-Hansen, N., Hald, B., Larsen, S., Neilsen, J.P., Smith, M., Ivanoff, C. & Meisner, H. (1988) Renal enzyme activities in experimental ochratoxin A-induced porcine nephropathy: Diagnostic potential of phosphoenolpyruvate carboxykinase and gamma-glutamyl transpeptidase activity. J. Toxicol. Environ. Health, 23, 1–14.
Kuczuk, M.H., Benson, P.M., Heath, H. & Hayes, W. (1978) Evaluation of the mutagenic potential of mycotoxins using Salmonella typhimurium and Saccharomyces cerevisiae. Mutat. Res., 53, 11–20.
Kuiper-Goodman, T. & Scott, P.M. (1989) Risk assessment of the mycotoxin ochratoxin A. Biomed. Environ. Sci., 2, 179–248.
Kumagai, S. (1985) Ochratoxin A: Plasma concentration and excretion into bile and urine in albumin-deficient rats. Food Chem. Toxicol., 23, 941–943.
Kumagai, S. (1988) Effects of plasma ochratoxin A and luminal pH on the jejunal absorption of ochratoxin A in rats. Food Chem. Toxicol., 26, 753–758.
Kumagai, S. & Aibara, K. (1982) Intestinal absorption and secretion of ochratoxin A in the rat. Toxicol. Appl. Pharmacol., 64, 94–102.
Kumari, D. & Sinha, S.P. (1994) Effect of retinol on ochratoxin-produced genotoxicity in mice. Food Chem. Toxicol., 32, 471–475.
Kuramochi, G., Gekle, M. & Silbernagl, S. (1997a) Derangement of pH homeostasis in the renal papilla: Ochratoxin A Increases pH in vasa recta blood. Nephron, 76, 472–476.
Kuramochi, G., Gekle, M. & Silbernagl, S. (1997b) Ochratoxin A disturbs pH homeostasis in the kidney: Increases in pH and HCO3– in the tubules and vasa recta. Pflügers Arch. Eur. J. Physiol., 434, 392–397.
Kurokawa, Y., Hayashi, Y., Maekawa, A., Takahashi, M., Kokubo, T. & Odashima, S. (1983) Carcinogenicity of potassium bromate administered orally to F344 rats. J. Natl Cancer Inst., 71, 965–671.
Kurokawa, Y., Maekawa, A., Takahashi, M. & Hayashi, Y. (1990) Toxicity and carcinogenicity of potassium bromate—A new renal carcinogen. Environ. Health Perspectives, 87, 309–335.
Langseth, W. (1999) Mycotoxins in Norwegian cereals for human consumption (SNT-rapport, mykotoksiner i norsk matkorn). Oslo: Norwegian Food Authorities.
Langseth, W., Ellingsen, Y., Nyomoen, U. & Okland, E.M. (1989) High performance liquid chromatographic determination of zearalenone and ochratoxin A in cereals and feed. J. Chromatogr., 478, 269–274
Larsson, K., & Moeller, T. (1996) Liquid chromatographic determination of ochratoxin A in barley, wheat bran, and rye by the AOAC/IUPAC/NMKL method: NMKL collaborative study. J. AOAC Int., 79, 1102–1105.
Lehtonen, P. (1999) Quantitative determination of ochratoxin A in wine. Office int. Vigne Vin., 1095.
Leoni, L.A.B., Soares, L.M.V. & Oliveira, P.L.C. (2000) Ochratoxin A in Brazilian roasted and instant coffees. Food Addit. Contam., 17, 867–870.
Levi, C., Trenk, H.L. & Mohr, H.K. (1974) Study of the occurrence of ochratoxin A in green coffee beans. J. Assoc. Off. Anal. Chem., 57, 866–870.
Li, J.L., Okada, S., Hamazaki, S., Ebina, Y. & Midorikawa, O. (1987) Subacute nephrotoxicity and induction of renal cell carcinoma in mice treated with ferric nitrilotriacetate. Cancer Res., 47, 1867–1869.
Maaroufi, K., Achour, A., Hammami, M., Betbeder, A.M., Ellouz, F., Creppy, E.E. & Bacha, H. (1995) Ochratoxin A in human blood in relation to nephropathy in Tunisia. Hum. Exp. Toxicol., 14, 609–615.
Maaroufi, K., Achour, A., Betbeder, A.M., Hammammi, M., Ellouz, F., Creppy, E.E. & Bacha, H. (1995a) Foodstuffs and human blood contamination by the mycotoxin ochratoxin A: Correlation with chronic interstitial nephropathy in Tunisia Arch. Toxicol., 69, 552–558
Maaroufi, K., Zakhama, A., Baudrimont, I., Achour, A., Abid, S., Ellouz, F., Dhouib, S., Creppy, E.E. & Bacha, H. (1999b) Karyomegaly of tubular cells as an early stage marker of nephrotoxicity induced by ochratoxin A in rats. Hum. Exp. Toxicol., 18, 410–415.
MacDonald, S., Wilson, P., Barnes, K., Daman, A., Massey, R., Mortby, E. & Sheperd, M.J. (1999) Ochratoxin A in dried vine fruit: Method development and survey Food Addit. Contam., 16, 253–260.
Madhyastha, M.S., Marquardt R.R. & Frohlich, A.A. (1992) Hydrolysis of ochratoxin A by the microbial activity of digesta in the gastrointestinal tract of rats. Arch. Environ. Contam. Toxicol., 23, 468–472.
Madsen, A., Mortensen, H.P. & Hald, B. (1982) Feeding experiments with ochratoxin A contaminated barley for bacon pigs. I. Influence on pig performance and residues. Acta Agric. Scand., 32, 225–239.
Majerus, P. & Ottender, H. (1996) [Identification and occurrence of ochratoxin A in wine and grape juice.] Dtsch. Lebensm.-Rundsch., 12, 388–390.
Majerus, P., Cutka, I., Dreyer, A., El-Dessouki, S., Eyrich, W., Reusch, H., Schurer, H. & Waiblinger, H.U. (1993) [The burden of ochratoxin A in foods of plant origin.] Dtsch. Lebensm. Rundsch., 89 (in German)
Majerus, P., Weber, R. & Wolff, J. (1994) Detection and determination of ochratoxin A in cereals and cereal products.J. Fed.Health Office, 37, 454–458.
Majumder, S.K. (1974) Control of Microflora and Related Production of Mycotoxins in Stored Sorghum, Rice and Groundnut, Mysore: Wesley Press.
Malaveille, C., Brun, G. & Bartsch, H. (1994) Structure–activity studies in E. coli strains on ochratoxin A (ochratoxin A) and its analogues implicate a genotoxic free radical and a cytotoxic thiol derivative as reactive metabolites. Mutat. Res., 307, 141–147.
Malir, F., Jergeova, Z., Severa, J., Cerna, M., Smid, J., Betbeder, A.M., Baudrimont, I. & Creppy, E.E.(1998) The level of ochratoxin A in blood serum of adults in the Czech Republic. Rev. Med. Vet., 149, 710.
Manolova, Y., Manolov, G., Parvanova, L., Petkova-Bocharova, T., Castegnaro, M. & Chernozemsky, I.N. (1990) Induction of characteristic chromosomal aberrations, particularly x-trisomy, in cultured human lymphocytes treated by ochratoxin A; a mycotoxin implicated in Balkan endemic nephropathy. Mutat. Res., in press.
Mantle, P.G. (1998) Ochratoxin A in coffee. Food Mycol., 1, 63–65.
Mantle, P.G., Miljkovic, A., Udupa, V. & Dobrota, M. (1998) Does apoptosis cause renal atrophy in Balkan endemic nephropathy? Lancet, 352, 1118–1119.
Mayura, K., Edwards, J.F., Maull, E.A. & Phillips, D.T. (1989) The effects of ochratoxin A on postimplantation rat embryos in culture. Arch. Environ. Contam. Toxicol., 18, 411–415.
Meisner, H. & Krogh, P. (1986) Phosphoenolpyruvate carboxykinase as a selective indicator of ochratoxin A induced nephropathy. Dev. Toxicol. Environ. Sci., 14, 199–206.
Meisner, H. & Polsinelli, L. (1986) Changes in renal mRNA species abundance by ochratoxin A. Biochem. Pharmacol., 35, 661–665.
Meisner, H. & Selanik, P. (1979) Inhibition of renal gluconeogenesis in rats by ochratoxin. Biochem. J., 180, 681–684.
Meisner, H., Cimbala, M.A. & Hanson, R.W. (1983) Decrease of renal phospho-enolpyruvate carboxykinase RNA and poly(A) RNA level by ochratoxin A. Arch. Biochem. Biophys., 223, 264–270.
van der Merwe, K.J., Steyn, P.S., Fourie, L., Scott, D.B. & Theron, J.J. (1965) Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature, 205, 1112–1113.
Micco, C., Grossi, M., Miraglia, M. & Brera, C. (1989) A study of the contamination of ochratoxin A of green and roasted coffee beans. Food Addit. Contam., 6, 333–339.
Micco, C., Ambruzzi, M.A., Miraglia, M., Brera, C., Onori, R. & Benelli, L. (1991) Contamination of human milk with ochratoxin A. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 105–108.
Ministry of Agriculture, Fisheries and Food (1995) Surveillance of Ochratoxin A in Retail Coffee Products (Food Surveillance Information Sheet No. 73), London: Joint Food Safety and Standard Group.
Ministry of Agriculture, Fisheries and Food (1996a) Ochratoxin A in Cereals and Flour, and Carry-over into Retail Processed Foods (Food Surveillance Information Sheet No. 95), London: Joint Food Safety and Standard Group.
Ministry of Agriculture, Fisheries and Food (1996b) Surveillance of Ochratoxin A in Green (Unroasted) Coffee Beans (Food Surveillance Information Sheet No. 80), London: Joint Food Safety and Standard Group.
Ministry of Agriculture, Fisheries and Food (1997) Survey of Aflatoxins and Ochratoxin A in Cereals and Retail Products (Food Surveillance Information Sheet No. 130), London: Joint Food Safety and Standard Group .
Ministry of Agriculture, Fisheries and Food (1999a) Survey of Ochratoxin A in Grain Traded by Central Deposits 1997–1998 (Food Surveillance Information Sheet No. 171), London: Joint Food Safety and Standard Group.
Ministry of Agriculture, Fisheries and Food (1999b) 1998 Survey of Retail Products for Ochratoxin A (Food Surveillance Information Sheet No. 185), London: Joint Food Safety and Standards Group.
Ministry of Agriculture, Fisheries and Food (1999c) A Survey of Human Exposure to Ochratoxin A (Food Surveillance Information Sheet No. 172), London: Joint Food Safety and Standard Group.
Moré, J. & Galtier, P. (1974) [Toxicity of ochratoxin A. I. Embryotoxic and teratogenic effect in rats.] Ann. Réch. Vet., 5, 167–178 (in French).
Moré, J. & Galtier, P. (1975) [Toxicity of ochratoxin A. II. Effects of treatment on the progeny (F1 and F2) of intoxicated rats.] Ann. Rech. Vét., 6, 379–389 (in French).
Mori, H., Kawai, K., Ohbayashi, F., Kuniyasu, T., Yamazaki, M., Hamasaki, T. & Williams, G.M. (1984) Genotoxicity of a variety of mycotoxins in the hepatocyte primary culture/DNA repair test using rat and mouse hepatocytes. Cancer Res., 44, 2918–2923.
Moroi, K., Suzuki, S., Kuga, T., Yamazaki, M. & Kanisawa, M. (1985) Reduction of ochratoxin A toxicity in mice treated with phenylalanine and phenobarbital. Toxicol. Lett., 25, 1–5.
Mortensen, H.P., Hald, B. & Madsen, A. (1983a) Feeding experiments with ochratoxin A contaminated barley for bacon pigs. 5. Ochratoxin A in pig blood. Acta Agric. Scand., 33, 235–239.
Mortensen, H.P., Hald, B., Larsen, A.E. & Madsen, A. (1983b) Ochratoxin A-contaminated barley for sows and piglets. Pig performance and residues in milk and pigs. Acta Agric. Scand., 33, 349–352.
Müller, G, Kielstein, P., Köhler, H., Berndt, A. & Rosner, H. (1995) Studies of the influence of ochratoxin A on immune and defence reactions in the mouse model. Mycoses, 38, 85–91.
Müller, G., Kielstein, P., Rosner, H., Berndt, A., Heller, M. & Köhler, H. (1999) Studies of the influence of ochratoxin A on immune and defence reactions in weaners. Mycoses, 42, 495–505
Munro, I.C., Moodie, C.A., Kuiper-Goodman, T., Scott, P.M. & Grice, H.C. (1974) Toxicologic changes in rats fed graded dietary levels of ochratoxin A. Toxicol. Appl. Pharmacol., 28, 180–188.
Nair, N.G., Emmett, R.W. & Parker, F.E. (1987) Programming applications of dicarboximides to control bunch rot of grapes caused by Botrytis cinerea. Plant Pathol., 36, 175–179.
Nesheim, S., Hardin, N.F., Francis, O.I. & Langham, W.S. (1973) Analysis of ochratoxin A and B and their esters in barley: Using partitions and thin-layer chromatography. I Development of the method J. AOAC Int., 56, 817–821.
Nesheim, S., Stack, M.E., Trucksess, W., Eppley, R. & Krogh, P. (1992) Rapid solvent-efficient method for liquid chromatographic determination of ochratoxin A in corn, barley and kidney: Collaborative study. J. AOAC Int., 75, 481–487.
Ngaha, E.O. (1985) Biochemical changes in the rat during experimentally induced acute ochratoxicosis. Enzyme, 33, 1–8.
Nicolov, I.G., Chernozemsky, I.N., Petkova-Bocharova, T., Stoyanov, I.S. & Stoichev, I.I. (1978) Epidemiological characteristics of urinary system tumours and Balkan nephropathy in an endemic region of Bulgaria. Eur. J. Cancer, 14, 1237–1242.
National Toxicology Program (1989) Technical Report on the Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303–47-9) in F344 Rats (Gavage Studies) (NIH Publication No. 89-2813), Research Triangle Park, NC: US Department of Health and Human Services, National Institutes of Health.
Obrecht-Pflumio, S., & Dirheimer, G. (2000) In vitro DNA and dGMP adducts formation caused by ochratoxin A. Chem.-Biol. Interactions, 127, 29–44.
Obrecht-Pflumio, S., Grosse, Y., Pfohl-Leszkowicz, A. & Dirheimer, G (1996) Protection by indomethacin and aspirin against genotoxicity of ochratoxin A, particulary in the urinary bladder and kidney. Arch. Toxicol., 70, 244–248.
Obrecht-Pflumio, S., Chassat, T., Dirheimer, G., & Marzin, D. (1999) Genotoxicity of ochratoxin A by Salmonella mutagenicity test after bioactivation by mouse kidney microsomes. Mutat. Res., 446, 95–102.
Olsen, M. (2000) Ochratoxin A in Sweden. Unpublished results from the National Food Administration, Uppsala.
Omar, R.F., Hasinoff, B.B.. Mejilla, F. & Rahimtula, A.D. (1990) Mechanism of ochratoxin A-stimulated lipid peroxidation. Biochem. Pharmacol., 40, 1183–1191.
Omar, R.F., Gelboin, H. V., & Rahimtula, A. D. (1996) Effect of cytochrome P450 induction on the metabolism and toxicity of ochratoxin A. Biochem. Pharmacol., 51, 207–216.
Osborne, B.G., Ibe, F., Brown, G.L., Petagine, F., Scudamore, K.A., Banks, J.N., Hetminski,, M.T. & Leonard, C.T. (1996) The effects of milling and processing on wheat contaminated with ochratoxin A. Food Addit. Contam., 13, 141–153.
Ospital, M., Cazabeil, J.M., Betbeder, A.M., Tricard, C., Creppy, E. & Medina, B. (1998) [Ochratoxin A in wones.] Rev. Fr. Oenolog., 169, 16–18 (in French).
Ottender, H. & Majerus, P. (2000) Occurrence of ochratoxin A (ochratoxin A) in wines: Influence of the type of wine and its geographical origin. Food Addit. Contam., 17, 793–798.
Ottender, H. & Majerus, P. (2001) Ochratoxin A (OTA) in coffee: Nation-wide evaluation of data collected by German food control 1995–1999. Food Addit. Contam. (in press).
Parker, R.W., Phillips, T.D., Kubena, L.F., Russell, L.H. & Heidelbaugh, N.D. (1982) Inhibition of pancreatic carboxypeptidase A: A possible mechanism of interaction between penicillic acid and ochratoxin A. J. Environ. Sci. Health, B17, 77–91.
Patel, S., Hazel, C.M., Winterton, A.G.M. & Mortby, E. (1996) Survey of ethnic foods for mycotoxins. Food Addit. Contam., 13, 833–841.
Patel, S., Hazel, C.M., Winterton, A.G.M. & Gleandle, A.E. (1997) Survey of ochratoxin A in UK retail coffee. Food Addit. Contam., 14, 217–222.
Patterson, D.S.P., Roberts, B.A. & Small, B.J. (1976) Metabolism of ochratoxins A and B in the pig during early pregnancy and the accumulation in body tissue of ochratoxin A only. Food Cosmet.Toxicol., 14, 439–442.
Pavlovic, M., Plestina, R. & Krogh, P. (1979) Ochratoxin A contamination of foodstuffs in an area with Balkan (endemic) nephropathy. Acta Pathol. Microbiol. Scand. B., 87, 243–246.
Peraica, M., Domijan, A.M., Fuchs, R., Lucic, A. & Radic, B. (1999) The occurrence of ochratoxin A in blood in general population of Croatia. Toxicol. Lett., 110, 105–112.
Petersen, A. (2000) Investigation of the content of ochratoxin A in kidney and muscle from healthy pigs. Unpublished results from Danish and Food Administration, Institute of Food Research and Nutrition, Soborg.
Petkova-Bocharova, T. & Castegnaro, M. (1991) Ochratoxin A in human blood in relation to Balkan endemic nephropathy and urinary tract tumours in Bulgaria. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 135–137.
Petkova-Bocharova, T., Chernozemsky, I.N. & Castegnaro, M. (1988) Ochratoxin A in human blood in relation to Balkan endemic nephropathy and urinary system tumours in Bulgaria. Food Addit. Contam., 5, 299–301.
Pettersson, H., Kiessling, K.H. & Ciszuk, P. (1982) Degradation of ochratoxin A in rumen. In: Proceedings, V International IUPAC Symposium Mycotoxins Phycotoxins, September 1–3, 1982, Vienna, Austria, Vienna: Austrian Chemical Society, pp. 313–316.
Pfohl-Leszkowicz, A., Chakor, K., Creppy, E. & Dirheimer, G. (1991) DNA adduct formation in mice treated with ochratoxin A. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 245–253.
Pfohl-Leszkowicz, A., Grosse, Y., Castegnaro, M., Nicolov, I.G., Chernozemsky, I.N., Bartsch, H., Betbeder, A.M., Creppy, E.E. & Dirheimer, G. (1993) Ochratoxin A-related DNA adducts in urinary tract tumours of Bulgarian subjects. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 141–148.
Pfohl-Leszkowicz, A., Pinelli, E., Bartsch, H., Mohr, U., & Castegnaro, M. (1998) Sex- and strain-specific expression of cytochrome P450s in ochratoxin A-induced genotoxicity and carcinogenicity in rats. Mol. Carcinog., 23, 76–85.
Pietri, A. (2000) University of Piacenza, Italy
Pineiro, M. & Giribone, A.G. (1994) [Evaluation of methods and quantification of aflatoxin, zearalenone and ochratoxin A in samples of Uruguayan meat products.] In: Proc. VI Congreso Aregentino de Ciencia y Tecnologia de Alimentos, p. 93 (in Spanish).
Pineiro, M., Dawson, R. & Lourdes Costarrica, M. (1996) Monitoring program for mycotoxin contamination in Uruguayan food and feeds. Nat. Toxins, 4, 242–245.
Piskorska-Pliszczynska, J. & Juszkiewicz (1990) Tissue deposition and passage into eggs of ochratoxin A in Japanese quail. J. Environ. Pathol. Toxicol. Oncol., 10, 8–10.
Pittet, A., Tornare, D., Hugget, A. & Viani, R. (1996) Liquid chromatographic determination of ochratoxin A in pure and adultered solubile coffee using an immunoaffinity column cleanup procedure. J. Agric. Food Chem., 44, 3564–3569.
Pitout, M.J. (1969a) The hydrolysis of ochratoxin A by some proteolytic enzymes. Biochem. Pharmacol., 18, 485–491.
Pitout, M.J. (1969b) A rapid spectrophotometric method for the assay of carboxypeptidase A. Biochem. Pharmacol., 18, 1829–1836.
Pitout, M.J. & Nel, W. (1969) The inhibitory effect of ochratoxin A on bovine carboxypeptidase A in vitro. Biochem. Pharmacol., 18, 1837–1843.
Pitt, J.I. (1987) Penicillium viridicatum, Penicillium verrucosum and production of ochratoxin A. Appl. Environ. Microbiol., 53, 266–269.
Pitt, J.I. & Hocking, A.D. (1997) Fungi and Food Spoilage, 2nd Ed., Gaitherburg, MD: Aspen Publishers.
Pitt, J.I., Hocking, A.D., Bhudhasamai, K., Miscamble, B.F., Wheeler, K.A. & Tanboon-Ek, P. (1993) The normal mycoflora of commodities from Thailand. 1. Nuts and oilseeds. Int. J. Food Microbiol., 20, 211–226.
Pitt, J.I., Hocking, A.D., Bhudhasamai, K., Miscamble, B.F., Wheeler, K.A. & Tanboon-Ek, P. (1994) The normal mycoflora of commodities from Thailand. 2. Beans, rice, small grains and other commodities. Int. J. Food Microbiol., 23, 35–53.
Pitt, J.I., Hocking, A.D., Miscamble, B.F., Dharmaputra, O.S., Kuswanto, K.R., Rahayu, E.S. & Sardjono. (1998) The mycoflora of food commodities from Indonesia. J. Food Mycol., 1, 41–60.
Pozzi, C.L., Correas, B., Gambale, W., Paula, C.R., Chacorn-Reche, N.O. & Meirelles, M.C.A. (1995) Postharvest and stored corn in Brazil: Mycoflora interaction, abiotic factors and mycotoxin occurrence Food Addit. Contam., 12, 313–319.
Prior, M.G. & Sisodia, C.S. (1982) The effects of ochratoxin A on the immune response of Swiss mice. Can. J. Comp. Med., 46, 91–96.
Radic, B., Fuchs, R., Peraica, M. & Lucic, A. (1997) Ochratoxin A in human sera in the area with endemic nephropathy in Croatia. Toxicol. Lett., 91, 105–109.
Radonic, M., Radosevic, Z. & Zupanic, V. (1966) Endemic nephropathy in Yugoslavia, In: The Kidney, Baltimore: Williams & Wilkins, pp. 503–522.
Radovanovic, Z (1989) Aetiology of Balkan nephropathy: A reappraisal after 30 years. Eur. J. Epidemiol., 5, 372–376.
Rahimtula, A., & Chong, X. (1991) Alterations in calcium homeostasis as a possible cause of ochratoxin A nephrotoxicity. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 207–214.
Rahimtula, A.D., Béreziat, J.C., Bussacchini-Griot, V. & Bartsch, H. (1988) Lipid peroxidation as a possible cause of ochratoxin A toxicity. Biochem. Pharmacol., 37, 4469–4477.
Randerath, E., Watson, W.P., Zhou, G.D., Chang, J. & Randerath, K. (1995) Intensification and depletion of specific bulky renal DNA adducts (I-compounds) following exposure of male F344 rats to the renal carcinogen ferric nitrilotriacetate (Fe-NTA). Mutat. Res., 341, 265–279.
Randerath, K., Randerath, E., Smith, C.V. & Chang, J. (1996) Structural origins of bulky oxidative DNA adducts (type II I-compounds) as deduced by oxidation of oligonucleotides of known sequence. Chem. Res. Toxicol., 9, 247–254.
Rao, C.N. (1987) Obstructive uropathy in group caged male B6C3F1 mice on a 24-month carcinogenicity study (letter). Lab. Anim. Sci., 37, 8–9.
Rao, M.V. (2000) A study on the validation of analytical methods and monitoring of mycotoxins in foods. Unpublished report from Dubai Municipality Food & Environment Laboratory. Submitted to WHO/FAO by Food & Environment Laboratory, Dubai Central Laboratory, Dubai Municipality.
Rasonyi, T. (1995) Mechanistic Investigations in Ochratoxin A Induced Nephrotoxicity and Their Relevance for the Sex Specific Renal Tumor Induction in Rats (Diss. ETH No. 11343), Thesis, University of Zürich.
Reiss, J. (1986) Detection of genotoxic properties of mycotoxins with the SOS chromotest. Naturwissenschaftern, 73, 677–678.
Richard, J.L., Platner, R.D., May, J. & Liska, S.L. (1999) The occurrence of ochratoxin A in dust collected from a problem household. Mycopathologia, 146, 99–103.
Röschenthaler, R., Creppy, E.E. & Dirheimer, G. (1984) Ochratoxin A: On the mode of action of a ubiquitous mycotoxin. J. Toxicol., 3, 53–86.
Roth, A., Chakor, K., Creppy, E.E., Kane, A., Röschenthaler, R. & Dirheimer, G. (1988) Evidence for an enterohepatic circulation of ochratoxin A in mice. Toxicology, 48, 293–308.
Roth, A., Creppy, E.E., Kane, A., Bacha, H., Steyn, P.S., Röschenthaler, R. & Dirheimer, G. (1989) Influence of ochratoxin B on the ochratoxin A inhibition of phenylalanyl–tRNA formation in vitro and protein synthesis in hepatoma tissue culture cells. Toxicol. Lett., 45, 307–313.
Rupic, V., Liker, B., Muzic, S., Bogdanic, I.C. & Balzer, I. (1978) [The effects of ochratoxin A in feed on the blood content of lipids and proteins in chickens.] Arh. Hig. Rada. Toxsikol., 29, 139–145 (in Serbo-Croat].
Schlatter, C., Studer, R.J. & Rasonyi, T. (1996) Carcinogenicity and kinetic aspects of ochratoxin A. Food Addit. Contam., 13 (Suppl.), 43–44.
Schwerdt G., Freudinger R., Silbernagl S. & Gekle M. (1999a) Ochratoxin A-binding proteins in rat organs and in different cell lines of the kidney. Toxicology, 135, 1–10.
Schwerdt G., Freudinger R., Mildenberger S., Silbernagl S. & Gekle M. (1999b) The nephrotoxin ochratoxin A induces apoptosis in cultured human proximal tubule cells. Cell Biol. Toxicol., 15, 405–415.
Scott, P.M. & Kanhere, S.R. (1995) Determination of ochratoxin A in beer. Food Addit. Contam., 12, 591–598.
Scott, P.M., Lawrence, J.W. & van Walbeek, W. (1970) Detection of mycotoxins by thin-layer chromatography: Application to screening of fungal extracts. Appl. Microbiol., 20, 839–842.
Scott, P.M., Kanhere, R.S., Canela, R., Lombaert, G.A. & Bacler, S. (1991) Determination of ochratoxin A in meat by liquid chromatography. Prehrambeno-tehnol. Biotehnol. Rev., 29, 61–64.
Scott, P.M., Kanhere, S.R., Lau, B.P.-Y., Lewis, D.A., Hayward, S., Ryan, J.J. & Kuiper-Goodman, T. (1998) Survey of Canadian human blood plasma for ochratoxin A. Food Addit. Contam., 15, 555–562.
Scudamore, K.A. (1999) Mycotoxins, an independent assessment of MAFF-funded applied research and surveillance 1993–1996. Report of studies commissioned by the Food Contaminants Division of the UK Ministry of Agriculture, Fisheries and Food, PB 4045.
Scudamore, K.A. & Patel, S. (2000) Survey for aflatoxins, ochratoxin A, zearalenone and fumonisins in maize imported into United Kingdom Food Addit. Contam., 17, 407–416
Seegers, J.C., Bohmer, L.H., Kruger, M.C., Lottering, M.L. & de Kock, M. (1994) A comparative study of ochratoxin A-induced apoptosis in hamster kidney and HeLa cells. Toxicol. Appl. Pharmacol., 129, 1–11.
Sharman, M., MacDonald, S. & Gilbert, J. (1992) Automated liquid chromatographic determination of ochratoxin A in cereals and animal products using immunoaffinity column clean-up. J. Chromatogr., 603, 285–289.
Singh, J. & Hood, R.D. (1985) Maternal protein deprivation enhances the teratogenicity of ochratoxin A in mice. Teratology, 32, 381–388.
Singh, G.S., Chauhan, H.V., Jha, G.J. & Singh, K.K. (1990) Immunosuppression due to chronic ochratoxicosis in broiler chicks. J. Comp. Pathol., 103, 399–410.
Sizoo, E.A. & van Egmond, H.P. (1997) Investigations on the occurrence of ochratoxin A in animal feeding-stuffs and cereals; samples drawn in the Netherlands in 1995 (RIVM Report No. 388802013), Bilthoven: National Institute of Public Health and the Environment (in Dutch with English abstract).
Skaug, M.A. (1999) Analysis of Norwegian milk and infant formulas for ochratoxin A. Food Addit. Contam., 16, 75–78.
Snowdon, A.L. (1990) A Colour Atlas of Post-harvest Diseases and Disorders of Fruits and Vegetables. 1. General Introduction and Fruits, London: Wolfe Scientific.
Snowdon, A.L. (1991) A Colour Atlas of Post-harvest Diseases and Disorders of Fruits and Vegetables. 2. Vegetables, London: Wolfe Scientific.
Soares, L.M.V. & Furlani, R.P.Z. (1996) Survey of mycotoxins in wheat and products sold in health food stores of the city of Campinas, State of Sao Paulo. Rev. Microbiol., 27, 41–45.
Soares, L.M.V. & Rodriguez-Amaya, D.B. (1985) Screening and quantitation of ochratoxin A in corn, peanuts, beans, rice, and cassava. J. AOAC Int., 68, 1128–1130.
Sokol, P.P., Ripich, G., Holohan, P.D. & Ross, C.R. (1988) Mechanism of ochratoxin A transport in kidney. J. Pharmacol. Exp. Ther., 246, 460–465.
Solfrizzo, M., Avantaggiato, G. & Visconti, A. (1998) Use of various clean-up procedures for the analysis of ochratoxin A in cereals. J. Chromatogr. A, 815, 67–73
Solti, L., Salamon, F., Barna-Vetró, I., Gyongyosi, A., Szabó, E. & Wolfling, A. (1997) Ochratoxin A content of human sera determined by a sensitive Elisa. J. Analyt. Toxicol. 21, 44–48.
Solti, L., Pécsi, T., Barna-Vetró, I., Szász, F., Jr, Biró, K. & Szabó, E. (1999) Analysis of serum and seminal plasma after feeding ochratoxin A with breeding boars. Anim. Reprod. Sci., 56, 123–132
Sreemannarayana, O., Frohlich, A.A., Vitti, T.G., Marquardt, R.R. & Abramson, D. (1988) Studies of the tolerance and disposition of ochratoxin A in young calves. Anim. Sci., 66, 1703–1711.
Stegen, G., Jorissen, U., Pittet, A., Saccon, M., Steiner, W., Vincenzi, M., Winkler, M., Zapp, J. & Schlatter, C. (1997) Screening of European coffee final products for occurrence of ochratoxin A (ochratoxin A). Food Addit. Contam., 14, 211–216
Stegen, G., Essens, P. & van der Lijn, J. (2001) Effect of roasting conditions on ochratoxin A in coffee. Unpublished manuscript.
Stein, A.F., Phillips, T.D., Kubena, L.F. & Harvey, R.B. (1985) Renal tubular secretion and reabsorption as factors in ochratoxicosis: Effects of probenecid on nephrotoxicity. J. Toxicol. Environ. Health, 16, 593–605.
Stetina, R. & Votava, M. (1986) Induction of DNA single-strand breaks and DNA synthesis inhibition by patulin, ochratoxin A, citrinin, and aflatoxin B, in cell lines CHO and AWRF. Folia Biol., 32, 128–144.
Stojkovic, R., Hult, K., Gamulin, S. & Plestina, R. (1984) High affinity binding of ochratoxin A to plasma constituents. Biochem. Int., 9, 33–38.
Stonard, M.D., Gore, C.W., Oliver, G.J.A. & Smith, I.K. (1987) Urinary enzymes and protein patterns as indicators of injury to different regions of the kidney. Fund. Appl. Toxicol., 9, 339–351.
Storen, O., Holm, H. & Størmer, F.C. (1982a) Metabolism of ochratoxin A by rats. Appl. Environ. Microbiol., 44, 785–789.
Storen, O., Helgerud, P., Holm, H. & Størmer, F.C. (1982b) Formation of (43)-4-hydroxyochratoxin A and ochratoxin B from ochratoxin A by rats. In: Proceedings, V International IUPAC Symposium Mycotoxins and Phycotoxins, September l–3, 1982, Vienna, Vienna: Austrian Chemical Society, pp. 321–324.
Størmer F.C. & Lea, T. (1995) Effects of ochratoxin A upon early and late events in human T-cell proliferation. Toxicology, 95, 45–50.
Størmer F.C., Hansen, C.E., Pedersen, J.I., Hvistendhal, G. & Aasen, A.J. (1981) Formation of (4R)- and (4S)-4-hydroxyochratoxin A from ochratoxin A by liver microsomes from various species. Appl. Environ. Microbiol., 42, 1051–1056.
Størmer F.C., Storen, O., Hansen, C.E., Pedersen, J.I. & Aasen, A.J. (1983) Formation of (4R)- and (4S)-4-hydroxchratoxin A and 10-hydroxyochratoxin A from ochratoxin A by rabbit liver microsomes. Appl. Environ. Microbiol., 45, 1183–1187.
Størmer, F.C., Kolsaker, P., Holm, H., Rogstad, S. & Elling, F. (1985) Metabolism of ochratoxin B and its possible effects upon the metabolism and toxicity of ochratoxin A in rats. Appl. Environ. Microbiol., 49, 1108–1112.
Stoyanov, I.S., Chernozemsky, I.N., Nicolov, I.G., Stoichev, I.I. & Petkova-Bocharova, T.K. (1978) Epidemiological association between endemic nephropathy and urinary system tumours in endemic region. J. Chron. Dis., 31, 721–724.
Studer-Rohr, I., Dietrich, D.R., Schlatter, J. & Schlatter, C. (1994) Ochratoxin A and coffee. Mitt. Geb- Lebesmittel. Hyg., 85, 719–727.
Studer-Rohr, I., Dietrich, D.R., Schlatter, J. & Schlatter, C. (1995) The occurrence of ochratoxin A in coffee. Food Chem. Toxicol., 33, 341–355.
Subramanian, S., Kanthasamy, A., Balasubramanian, N., Sekar, N. & Govindasamy, S. (1989) Ochratoxin A toxicity on carbohydrate metabolism in rats. Bull. Environ. Contam. Toxicol., 43, 180–184.
Suzuki, S., Kozuka, Y., Satoh, T. & Yamazaki, M. (1975) Studies on the nephrotoxicity of ochratoxin A in rats. Toxicol. Appl. Pharmacol., 34, 479–490.
Suzuki, S., Satoh, T. & Yamazaki, M. (1977) The pharmacokinetics of ochratoxin A in rats. Jpn. J. Pharmacol., 27, 735–744.
Suzuki, S., Moroi, K., Kanisawa, M. & Satoh, T. (1986) Effects of drug metabolizing enzyme inducers on carcinogenesis and toxicity of ochratoxin A in mice (abstract) Toxicol. Lett., 31 (Suppl.), 206.
Swedish National Food Administration (1994) Study of Food Habits and Nutrients in Sweden in 1989—Method and Result Analysis. Uppsala: Livsmedelsverkets Förlag, pp. 27–36 (in Swedish).
Swenberg, J.A. & Maronpot, R.R. (1991) Chemically induced cell proliferation as a criterion in selecting doses for long-term bioassays. In: Chemically Induced Cell Proliferation: Implications for Risk Assessment, New York: Wiley-Liss, pp. 245–251.
Tancev, I., Evstatiev, P., Dorosiev, D., Panceva, Z. & Cvetkov, G. (1956) [Proucavanje na nefrite v Vracanska okolija.] Savremena Med., 7, 14–29 (in Bulgarian).
Taniwaki, M.H., Pitt, J.I., Urbano, G.R., Teixeira, A.A. & Leitão, M.F.F. (1999) Fungi producing ochratoxin A in coffee. In: Proceedings of the 18th International Scientific Colloquium on Coffee, Helsinki, Finland, 2–6 August, 1999, pp. 239–247.
Tapai, K., Teren, J. & Mesterhazy, A. (1997) Ochratoxin A in the sera of blood donors and ill persons. Cereal Res. Commun., 25, 307–308.
Téren, J., Varga, J., Hamari, Z., Rinyu, E. & Kevei, F. (1996) Immunochemical detection of ochratoxin A in black Aspergillus strains. Mycopathologia, 134, 171–176.
Thuvander, A., Breitholtz-Emanuelsson, A. & Olsen, M. (1995) Effects of ochratoxin A on the mouse immune system after subchronic exposure. Food Chem. Toxicol., 33, 1005–1011.
Thuvander, A., Breitholtz-Emanuelsson, A., Brabencova, D. & Gadhasson, I. (1996a) Prenatal exposure of Balb/c mice to ochratoxin A: Effects on the immune system in the offspring. Food Chem. Toxicol., 34, 547–554.
Thuvander, A., Funseth, E., Breitholtz-Emanuelsson, A., Hallén, I.P. & Oskarsson, A. (1996b) Effects of ochratoxin A on the rat immune system after perinatal exposure. Nat. Toxins, 4, 141–147.
Thuvander, A., Moller, T., Enghardt Barbieri, H., Jansson, A., Salomonsson, A.C. & Olsen, M. (2000) Dietary intake of some important mycotoxins by the Swedish population. Unpublished study. Submitted to WHO by National Food Administration, Uppsala.
Trucksess, M.W., Giler, J., Young, K., White, K.D. & Page, S.W. (1999) Determination and survey of ochratoxin A in wheat, barley, and coffee—1997 J. AOAC Int., 82, 85–89.
Tsubouchi, H., Yamamoto, K., Hisada, K. & Sakabe, Y. (1984) A survey of occurrence of mycotoxins and toxigenic fungi in imported green coffee beans. Proc. Jpn. Assoc. Mycotox., 19, 14–21.
Tsuda, M., Sekine, T., Takeda, M., Cha, S.H, Kanai, Y., Kimura, M. & Endou, H. (1999) Transport of ochratoxin A by renal multispecific organic anion transporter 1. J. Pharmacol. Exp. Ther., 289, 1301–1305.
Ueno, Y. (1998) Residue and risk of ochratoxin A in human plasma and beverages in Japan Mycotoxins, 47, 1998
Ueno, Y. & Kubota, K. (1976) DNA-attacking ability of carcinogenic mycotoxins in recombination-deficient mutant cells of Bacillus subtilis. Cancer Res., 36, 445–451.
Ueno, Y., Kawamura, O., Sugiura, Y., Horiguchi, K., Nakajima, M., Yamamoto, K. & Sato, S. (1991) Use of monoclonal antibodies, enzyme-linked immunosorbent assay and immunoaffinity column chromatography to determine ochratoxin A in porcine sera, coffee products and toxin-producing fungi. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 71–75.
Ueno, Y., Umemori, K., Niime, E., Tanuma, S., Nagata, S., Sugamata, M., Ihara, T., Sekifima, M., Kawai, K., Ueno, I. & Tashiro, F. (1995) Induction of apoptosis by T-2 toxin and other natural toxins in HL-60 human promyelotic leukemia cells. Nat. Toxins, 3, 129–137.
Ueno, Y., Maki, S., Lin, J., Furuya, M., Suguira, Y. & Kawamura, O. (1998) A 4-year study of plasma ochratoxin A in a selected population in Tokyo by immunoassay and immunoaffinity column-linked HPLC. Food Chem. Toxicol., 36, 445–449.
Umeda, M., Tsutsui, T. & Saito, M. (1977) Mutagenicity and inducibility of DNA single-strand breaks and chromosome aberrations by various mycotoxins. Gann, 68, 619–625.
Umemura, T., Takagi, A., Sai, K., Hasegawa, R. & Kurokawa, Y. (1998) Oxidative DNA damage and cell proliferation in kidneys of male and female rats during 13-weeks exposure to potassium bromate (KBrO3). Arch. Toxicol., 72, 264–269.
Vandegraft, E.E., Shotwell, O.L., Smith, M.L. & Hesseltine, C.W. (1973) Mycotoxin formation affected by fumigation of wheat. Cereal Sci. Today, 18, 412–414.
Varga, J., Kevei, F., Rinyu, E., Téren, J. & Kozakiewicz, Z. (1996) Ochratoxin production by Aspergillus species. Appl. Environ. Microbiol., 60, 4461–4464.
Visconti, A., Pascale, M. & Centonze, G. (1999) Determination of ochratoxin A in wine by means of immunoaffinity column clean-up and high-performance liquid chromatography. J. Chromatogr. A, 864, 89–101
Visconti, A., Pascale, M. & Centonze, G. (2000a) Determination of ochratoxin A in domestic and imported beers in Italy by immunoaffinity clean-up and liquid chromatography. J. Chromatogr. A, 888, 321–326
Visconti, A., Pascale, M. & Centonze, G. (2000b) Determination of ochratoxin A in wine and beer by immunoaffinity column clean-up and HPLC analysis with fluorometric detection: Collaborative study. J. AOAC Int. (in press).
Vukelic, M. & Sostaric, B. (1991) Characteristics of urinary tract tumours in the area of Balkan endemic nephropathy in Croatia. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 29–35.
Vukelic, M., Belitza, M., Ceovic, S., Radonic, M., Sostaric, B. & Plestina, R. (1987) Urothelial tumours in the region of Balkan endemic nephropathy. In: Dirheimer, G. & Schlagel, M., eds, Abstracts, 28th Congress of the European Society of Toxicology, 17–19 September 1987, Strasbourg, p. 58.
Vukelic, M., Sostaric, B. & Fuchs, R., (1991) Some pathomorphological features of Balkan endemic nephropathy in Croatia. In: Castegnaro, M., Plestina, R., Dirheimer, G., Chernozemsky, I.N. & Bartsch, H., eds, Mycotoxins, Endemic Nephropathy and Urinary Tract Tumours (IARC Scientific Publications No. 115), Lyon: IARCPress, pp. 37–42.
Vukelic, M., Sostaric, B. & Belicza, M. (1992) Pathomorphology of Balkan endemic nephropathy. Food Chem. Toxicol., 30, 193–200.
Wafa, E.W., Yahya, R.S., Sobh, M.A., Eraky, I., El-Baz, M., El-Gayar, H.A.M., Betbeder, A.M. & Creppy, E.E. (1998) Human ochratoxicosis and nephropathy in Egypt: A preliminary study. Hum. Exp. Toxicol., 17, 124–129.
van Walbeek, W., Scott, P.M., Harwig, J., & Lawrence, J.W. (1969) Penicillium viridicatum Westling: A new source of ochratoxin A. Can. J. Microbiol., 15, 1281–1285.
Ward, J.M., Goodman, D.G., Squire, R.A., Chu, K.C. & Linhart, M.S. (1979) Neoplastic and nonneoplastic lesions in aging (C57BL/6N x C3H/HeN)F1 (B6C3F1) mice. J. Natl Cancer Inst., 63, 849–854.
Wehner, F.C., Thiel, P.G., van Rensburg, S.J. & Demasius, I.P.C. (1978) Mutagenicity to Salmonella typhimurium of some Aspergillus and Penicillium mycotoxins. Mutat. Res., 58, 193–203.
WHO (1990) Selected Mycotoxins: Ochratoxins, Trichothecenes, Ergot (Environmental Health Criteria 105), Geneva
Wicklow, D.T., Dowd, P.F., Alfatafta, A.A. & Gloer, J.B. (1996) Ochratoxin A: An antiinsectan metabolite from the sclerotia of Aspergillus carbonarius NRRL 369. Can. J. Microbiol., 42, 1100–1103.
Wiger, R. & Størmer, F.C. (1990) Effects of ochratoxin A and B on prechondrogenic mesenchymal cells from chick embryo limb buds. Toxicol. Lett., 54, 129–134.
Wilkens, J. & Jörissen, U. (1999) [Degradation of ochratoxin A during manufacture of roasted coffee.] In: 21st Mycotoxin Workshop, Jena (BgVV), June 7–9, 1999.
Williams, A.C., Slack, P.T., Jorgensen, K., Donnely, C. & Boenke, A. (1998) The Second Interlaboratory Study of Methods for the Analysis of Ochratoxin A in Pig Kidney (EUR report, EUR 18458 EN), Brussels.
Wolf, D.C., Crosby, L.M., George, M.H., Kilburn, S.R., Moore, T.M., Miller, R.T. & DeAngelo, A.B. (1998) Time- and dose-dependent development of potassium bromate-induced tumors in male Fischer 344 rats. Toxicol. Pathol., 26, 724–729.
Wolff, J., Bresch, H., Cholmakow-Bodechtel, C., Engel, G., Erhardt, S., Gareis, M., Majerus, P., Rosner, H. & Scheuer, R. (1997) [Burden of ochratoxin A in food and in the consumer. Final report. Institute for the Biochemistry of Cereals and Potatoes. Federal Institute for Research on Cereals, Potatoes and Fat (in German).
Wood, G.M., Patel, S., Entwisle, A.C. & Boenke, A. (1996) Ochratoxin A in wheat: A second intercomparison of procedures. Food Addit. Contam., 13, 519–539.
Wood, G.M., Patel, S., Entwisle, A.C., Williams, A.C., Boenke, A. & Farnell, P.J. (1997) Ochratoxin A in wheat: Certification of two reference materials. Food Addit. Contam., 14, 237–248.
Würgler, F.E., Friedrich, U. & Schlatter, J. (1991) Lack of mutagenicity of ochratoxin A and B, citrinin, patulin and conestine in Salmonella typhimurium TA102. Mutat. Res., 261, 209–216.
Xiao, H., Madhyastha, S., Marquardt, R.R., Li, S., Vodela, J.K., Frohlich A.A. & Kemppainen, B.W. (1996) Toxicity of ochratoxin A, its opened lactone form and several of its analogs: Structure–activity relationships. Toxicol. Appl. Pharmacol., 137, 182–92
Zanic-Grubisic, T., Santini, A., Cepelak, I., Barisic, K., Juretic, D., Pepeljnjak, S. (1996) Influence of ochratoxin A treatment on the activity of membrane bound enzymes in rat brain regions. Biol. Chem. Hoppe Seyler, 377, 121–127.
Zepnik, H., Pahler, A., Schauer, U. & Dekant, W (2000) Ochratoxin A induced tumour formation: Is there a role of reactive ochratoxin A metabolites? Toxicol. Sci. (in press).
Zimmerli, B. & Dick, R. (1995) Determination of ochratoxin A at the ppt level in human blood, serum, milk and some foodstuffs by high performance liquid chromatography with enhanced fluorescence detection and immunoaffinity column cleanup: Methodology and Swiss data. J. Chromatogr. B, 666, 85–99.
Zimmerli, B. & Dick, R. (1996) Ochratoxin A in table wine and grape-juice: Occurrence and risk assessment. Food Addit. Contam., 13, 655–668.
Results of surveys for ochratoxin A showing concentrations and distribution of
contamination in food commodities
|
Country/ Region |
Commodity |
Year/ Season |
No. of samples |
LOQ |
n < LOQ |
Mean/Max (µg/kg) |
|
|
Wheat |
|
|
|
|
|
|
Germany |
Wheat grain |
1995–98 |
35 |
0.01a |
21 |
0.11/0.65 |
|
Wheat flour < T550 |
1995–98 |
98 |
0.01a |
16 |
0.10/1 |
|
|
1995–98 |
83 |
0.01a |
6 |
0.20/1.73 |
||
|
Wheat wholemeal flour |
1995–98 |
18 |
0.01a |
0 |
0.20/1.2 |
|
|
Wheat semolina |
1995–98 |
25 |
0.01a |
15 |
0.41/2.58 |
|
|
Wheat bran |
1995–98 |
25 |
0.01a |
22 |
0.22/1.59 |
|
|
Wheat germ |
1995–98 |
19 |
0.01a |
13 |
0.11/0.45 |
|
|
Netherlands |
Wheat, domestic |
1995 |
7 |
1.000 |
7 |
0/0 |
|
Wheat, imported |
1995 |
24 |
1.000 |
23 |
0.36/8.7 |
|
|
White wheat flour |
1999 |
31 |
0.250 |
30 |
0.048/1.5 |
|
|
Whole-wheat meal |
1999 |
19 |
0.250 |
19 |
0/0 |
|
|
Norway |
Wheat |
1990 |
138 |
0.3a |
122 |
0.17/1.5 |
|
1993 |
7 |
0.2a |
6 |
0.77/4.7 |
||
|
1994 |
24 |
0.2a |
20 |
0.3/3.4 |
||
|
1995 |
32 |
0.25a |
20 |
0.15/0.57 |
||
|
1996 |
28 |
0.3a |
28 |
0/0 |
||
|
1997 |
25 |
0.01a |
22 |
0.22/3.5 |
||
|
1998 |
35 |
0.05a |
30 |
0.7/20 |
||
|
Total wheat samples |
1990–98 |
289 |
|
248 |
0.26/20 |
|
|
Wheat, imported |
1990 |
28 |
0.3a |
25 |
0.34/3.8 |
|
|
1993 |
11 |
0.2a |
10 |
0.52/4.6 |
||
|
Norway |
Wheat, imported |
1994 |
9 |
0.2a |
8 |
0.46/3.3 |
|
1995 |
13 |
0.25a |
10 |
0.89/8.2 |
||
|
1996 |
14 |
0.3a |
13 |
0.67/7.5 |
||
|
1997 |
10 |
0.01a |
7 |
0.14/0.56 |
||
|
1998 |
24 |
0.05a |
20 |
0.07/0.54 |
||
|
Total imported wheat |
1990–98 |
108 |
|
93 |
0.4/8.2 |
|
|
Sweden |
Wheat grain |
1996–98 |
57 |
0.1 |
41 |
0.24/2.3 |
|
1999 |
75 |
0.05 |
36 |
0.37/5.2 |
||
|
Brazil |
Wheat grain |
1988–90 |
16 |
5a |
15 |
2.5/40 |
|
1990 |
20 |
5a |
20 |
0/0 |
||
|
Wheat products |
1991 |
38 |
5a |
38 |
0/0 |
|
|
USA |
Wheat grain |
1997 |
383 |
0.03a |
327 |
NR/31.4 |
|
Finland |
Wheat grain |
1996 |
34 |
0.800 |
32 |
18.2/430 |
|
Denmark |
Wheat grain |
1986–92 |
402 |
0.05a |
283 |
0.7/51 |
|
Wheat grain, organic |
1986–92 |
73 |
0.05a |
44 |
1.2/36 |
|
|
Wheat grain |
1986–92 |
45 |
0.05a |
28 |
0.9/13 |
|
|
Wheat bran |
1986–92 |
120 |
0.05a |
46 |
0.8/12 |
|
|
Wheat bran, organic |
1986–92 |
22 |
0.05a |
7 |
0.6/2.6 |
|
|
Dubai |
Wheat flour |
NR |
11 |
0.500 |
10 |
0.023/0.25 |
|
United Kingdom |
Wheat noodles |
1995 |
4 |
0.1a |
3 |
0.1/0.4 |
|
Wheat grain |
1993 |
384 |
0.1a |
NR |
< 1/< 1 |
|
|
Wheat grain stored |
1992–93 |
25 |
0.1a |
NR |
< 1/ 1 |
|
|
Wheat from millers |
1993 |
129 |
0.1a |
NR |
NA/15 |
|
|
1994 |
250 |
0.1a |
NR |
NA/32 |
||
|
Wheat grain |
1997-98 |
148 |
0.200 |
126 |
0.3/9.2 |
|
|
1996 |
76 |
0.200 |
74 |
0.042/2.4 |
||
|
Cereals and flours |
1996 |
67 |
0.200 |
30 |
NA/6.4 |
|
|
Uruguay |
Wheat grain |
1993–95 |
123 |
50a |
123 |
0/0 |
|
|
Buckwheat |
|
|
|
|
|
|
Germany |
Buckwheat |
1995–98 |
23 |
0.01a |
13 |
0.07/0.59 |
|
Buckwheat flour |
1995–98 |
14 |
0.01a |
3a |
0.96/12.1 |
|
|
|
Barley |
|
|
|
|
|
|
Germany |
Barley |
1995–98 |
22 |
0.01a |
3 |
0.07/0.49 |
|
Pearl barley |
1995–98 |
31 |
0.01a |
10 |
0.094/0.95 |
|
|
Norway |
Barley |
1990 |
10 |
0.3a |
10 |
0/0 |
|
Barley groats |
1990 |
10 |
0.3a |
10 |
0/0 |
|
|
USA |
Barley |
1997 |
103 |
0.03a |
92 |
NA/17 |
|
United Kingdom |
Barley |
1997-98 |
131 |
0.1a |
96 |
0.7/17.8 |
|
Barley |
1996 |
37 |
0.2a |
34 |
0.20/6.4 |
|
|
Barley |
1993 |
73 |
0.1a |
NR |
NA/<1 |
|
|
stored in 1992 |
|
|
|
|
|
|
|
Barley |
1994 |
150 |
0.1a |
NR |
NA/33 |
|
|
Barley stored in 1993 |
1994 |
50 |
0.1a |
NR |
NA/14 |
|
|
Uruguay |
Barley and malt |
1993-95 |
137 |
50a |
137 |
0/0 |
|
Finland |
Barley kernel |
1996 |
45 |
0.800 |
42 |
0.55/12.3 |
|
Canada |
Barley cereals |
1998–99 |
20 |
0.500 |
NR |
NA/0.57 |
|
Barley based cereals |
1998–99 |
20 |
0.200 |
NR |
NA/6.92 |
|
|
|
Maize |
|
|
|
|
|
|
Germany |
Maize and popcorn |
1995–98 |
31 |
0.01a |
12 |
0.17/3.35 |
|
United Kingdom |
Raw maize, imported |
1998–99 |
139 |
0.200 |
125 |
NA/1.5 |
|
Croatia |
Raw maize |
1996 |
105 |
0.2a |
95 |
3.61/224 |
|
Raw maize |
1997 |
104 |
0.2a |
68 |
19.8/614 |
|
|
Brazil |
Raw maize |
1991 |
130 |
5a |
130 |
0/0 |
|
Brazil |
Raw maize |
1993–94 |
292 |
5a |
292 |
0/0 |
|
Uruguay |
Maize and by-products |
1993-95 |
147 |
50a |
147 |
0/0 |
|
United Kingdom |
Corn flour |
1995 |
4 |
0.1a |
3 |
0.15/0.6 |
|
|
Oats |
|
|
|
|
|
|
Sweden |
Oat grain |
1996–98 |
23 |
0.1 |
16 |
0.32/3.6 |
|
Oat grain |
1999 |
10 |
0.05 |
8 |
0.05/0.15 |
|
|
Norway |
Oat groats |
1990 |
20 |
0.3a |
14 |
0.26/0.9 |
|
Oats |
1990 |
20 |
0.3a |
17 |
0.44/5.8 |
|
|
Oats |
1993 |
3 |
0.2 |
2 |
0.17/0.26 |
|
|
Oats |
1994 |
3 |
0.2a |
2 |
3.47/10.2 |
|
|
Oats |
1995 |
21 |
0.25a |
20 |
0.32/4.2 |
|
|
Oats |
1996 |
14 |
0.3a |
14 |
0/0 |
|
|
Oats |
1997 |
14 |
0.01a |
? |
0.053/0.23 |
|
|
Oats |
1998 |
22 |
0.01a |
19 |
0.065/0.47 |
|
|
Total oats samples |
1990–98 |
97 |
|
|
0.46/10.2 |
|
|
Denmark |
Oat kernels |
1986–92 |
50 |
0.05a |
29 |
0.5/5.6 |
|
Oat kernels, organic |
1986–92 |
17 |
0.05a |
11 |
0.3/4.2 |
|
|
Oat kernels, imported |
1986–92 |
25 |
0.05a |
12 |
0.5/4.6 |
|
|
Finland |
Oat kernels |
1996 |
34 |
0.80 |
32 |
1.7/56.6 |
|
United Kingdom |
Oat kernels |
1997–98 |
21 |
0.1a |
15 |
0.1/2.2 |
|
1996 |
18 |
0.2a |
17 |
0.33/5.9 |
||
|
Germany |
Oats |
1995–98 |
30 |
0.01a |
6 |
0.06/0.14 |
|
|
Rice |
|
|
|
|
|
|
Uruguay |
Rice |
1993-95 |
62 |
50a |
62 |
0/0 |
|
United |
Basmati rice |
1995 |
4 |
0.1a |
4 |
0/0 |
|
Kingdom |
Chinese rice |
1995 |
4 |
0.1a |
4 |
0/0 |
|
Dubai |
Rice |
NR |
15 |
0.50 |
14 |
0.017/0.25 |
|
Germany |
Rice |
1995–98 |
22 |
0.1a |
18 |
0.11/0.28 |
|
Parboiled rice |
1995–98 |
21 |
0.1a |
21 |
|
|
|
Long-grain rice |
1995–98 |
24 |
0.1a |
24 |
|
|
|
Round-grain rice |
1995–98 |
15 |
0.1a |
15 |
|
|
|
|
Rye |
|
|
|
|
|
|
Sweden |
Rye grain |
1996–98 |
28 |
0.1 |
8 |
0.56/2.3 |
|
1999 |
19 |
0.05 |
6 |
1.8/27 |
||
|
Norway |
Rye, imported |
1990 |
18 |
0.3a |
13 |
0.22/0.8 |
|
1993 |
9 |
0.2a |
5 |
0.31/1.0 |
||
|
1994 |
6 |
0.2a |
4 |
0.28/1.0 |
||
|
1995 |
4 |
0.25a |
3 |
0.75/2.5 |
||
|
1996 |
4 |
0.3a |
4 |
0/0 |
||
|
Total |
1990–98 |
41 |
|
29 |
0.31/2.5 |
|
|
Denmark |
Rye kernels |
1986–92 |
503 |
0.05a |
326 |
1.2/121 |
|
Rye kernels, organic |
1986–92 |
91 |
0.05a |
20 |
5.4/120 |
|
|
Rye kernels, imported |
1986–92 |
22 |
0.05a |
14 |
0.1/0.7 |
|
|
United Kingdom |
Rye kernels |
1996 |
22 |
0.2a |
21 |
0.05/1.1 |
|
Germany |
Rye |
1995–98 |
37 |
0.01a |
23 |
0.11/0.8 |
|
Rye flour < T997 |
1995–98 |
26 |
0.01a |
6 |
0.42/6.4 |
|
|
1995–98 |
71 |
0.01a |
3 |
0.32/2.14 |
||
|
Rye wholemeal flour |
1995–98 |
43 |
0.01a |
11 |
0.11/1.46 |
|
|
|
Sorghum |
|
|
|
|
|
|
Germany |
Sorghum |
1995–98 |
26 |
0.01a |
3 |
0.11/0.83 |
|
|
Spelt |
|
|
|
|
|
|
Germany |
Spelt/spelt flour |
1995–98 |
21 |
0.01a |
3 |
0.66/9.43 |
|
|
Cereal products |
|
|
|
|
|
|
United Kingdom |
Pitta bread |
1995 |
4 |
0.1a |
NR |
NA/0.8 |
|
Chapatti |
1995 |
4 |
0.1a |
NR |
NA/0.9 |
|
|
Nan bread |
1995 |
4 |
0.1a |
4 |
0/0 |
|
|
Poppadoms |
1995 |
4 |
0.1a |
4 |
0/0 |
|
|
Tunisia |
Cereal-derived food |
1994 |
66 |
0.1a |
0 |
1715/12 770 |
|
Dubai |
Mixed cereals |
NR |
28 |
0.500 |
25 |
NA/4.3 |
|
Canada |
Mixed grain cereals |
1998–99 |
31 |
0.200 |
NR |
0.25/0.54 |
|
Mixed cereals |
1998–99 |
19 |
0.500 |
19 |
0/0 |
|
|
Europe, Tunisia |
Bread |
NR |
141 |
0.008 |
0 |
NA/6.66 |
|
Germany |
Bread, wheat mix |
1995–98 |
125 |
0.01a |
15 |
0.19/2.09 |
|
Germany |
Bread, rye mix |
1995–98 |
128 |
0.01a |
7 |
0.24/2.24 |
|
White bread |
1995–98 |
57 |
0.01a |
9 |
0.11/1.9 |
|
|
Toast bread |
1995–98 |
59 |
0.01a |
7 |
0.081/0.58 |
|
|
Rye meal bread |
1995–98 |
96 |
0.01a |
7 |
0.22/5.49 |
|
|
Milk–water bread roll |
1995–98 |
89 |
0.01a |
10 |
0.09/0.52 |
|
|
Various cereals, bread |
1995–98 |
49 |
0.01a |
1 |
0.24/1.76 |
|
|
Various cereals, bread +oilseed |
1995–98 |
101 |
0.01a |
3 |
0.17/2.44 |
|
|
Crispbread |
1995–98 |
87 |
0.01a |
25 |
0.076/0.44 |
|
|
Wheat whole bread |
1995–98 |
13 |
0.01a |
0 |
0.134/0.40 |
|
|
Special bread |
1995–98 |
64 |
0.01a |
9 |
0.145/2.23 |
|
|
Wholemeal bread roll |
1995–98 |
31 |
0.01a |
0 |
0.17/0.77 |
|
|
Various cereals and muesli bread |
1995–98 |
49 |
0.01a |
1 |
0.36/5.54 |
|
|
Rye bread roll |
1995–98 |
38 |
0.01a |
1 |
0.16/0.44 |
|
|
Pasta without egg |
1995–98 |
50 |
0.1a |
21 |
0.28/1.75 |
|
|
Pasta with egg |
1995–98 |
84 |
0.1a |
57 |
0.199/0.95 |
|
|
Wholemeal pasta |
1995–98 |
27 |
0.1a |
17 |
2.0/29.77 |
|
|
Oat flakes |
1995–98 |
66 |
0.01a |
40 |
0.07/0.25 |
|
|
Oats bran |
1995–98 |
26 |
0.01a |
12 |
0.089/0.33 |
|
|
Polenta |
1995–98 |
29 |
0.01a |
23 |
0.20/1.53 |
|
|
Green corn |
1995–98 |
17 |
0.01a |
15 |
0.07/0.10 |
|
|
Infant food |
1995–98 |
97 |
0.01a |
31 |
0.12/2.13 |
|
|
Peas, lentil, beans |
1995–98 |
103 |
0.01a |
102 |
ND/0.84 |
|
|
Soya beans |
1995–98 |
31 |
0.01a |
5 |
0.06/0.10 |
|
|
FSIS185 pulses |
1998 |
50 |
0.2a |
50 |
|
|
|
FSIS185 pulses |
1997 |
29 |
0.2a |
27 |
1.1/15.4 |
|
|
|
Seeds |
|
|
|
|
|
|
United Kingdom |
Fennel |
1995 |
3 |
0.1a |
3 |
0/0 |
|
Sesame seeds |
1995 |
3 |
0.1a |
3 |
0/0 |
|
|
Coriander |
1995 |
3 |
0.1a |
2 |
1.33/4.0 |
|
|
|
Herbs and spices |
|
|
|
|
|
|
Netherlands |
Paprika powder |
1996–98 |
12 |
0.250 |
3 |
1.7/9.8 |
|
Pepper |
1996–97 |
14 |
0.250 |
7 |
3.73/14.5 |
|
|
United Kingdom |
Chilli powder |
1995 |
4 |
0.1a |
NR |
NA/50.4 |
|
Curry powder |
1995 |
10 |
0.1a |
NR |
NA/21.3 |
|
|
Tandoori |
1995 |
3 |
0.1a |
NR |
NA/23.9 |
|
|
Ginger |
1995 |
4 |
0.1a |
NR |
NA/7.5 |
|
|
Garlic |
1995 |
4 |
0.1a |
4 |
0/0 |
|
|
Five spices powder |
1995 |
4 |
0.1a |
3 |
0.65/2.6 |
|
|
Tunisia |
Dried vegetables |
1994 |
6 |
0.1a |
0 |
2934/7444 |
|
Dubai |
Spices |
NR |
7 |
0.500 |
3 |
NA/3.56 |
|
|
Pickles and pastes |
|
|
|
|
|
|
United Kingdom |
Chilli pickle |
1995 |
4 |
0.1a |
NR |
NA/1.2 |
|
Garlic pickle |
1995 |
4 |
0.1a |
NR |
NA/2.5 |
|
|
United Kingdom |
Curry paste |
1995 |
4 |
0.1a |
NR |
NA/15.5 |
|
Chilli sauce |
1995 |
4 |
0.1a |
3 |
0.82/3.3 |
|
|
|
Canned foods |
|
|
|
|
|
|
United Kingdom |
Canned foods |
1995 |
8 |
0.1a |
NR |
NA/0.3 |
|
|
Oils |
|
|
|
|
|
|
United |
Sesame oil |
1995 |
3 |
0.1a |
2 |
0.13/0.4 |
|
Kingdom |
Chili, almond oils |
1995 |
4 |
0.1a |
4 |
0/0 |
|
Uruguay |
Oilseed |
1993-95 |
80 |
50a |
80 |
0/0 |
|
Dubai |
Oilseed |
NR |
5 |
0.500 |
5 |
0/0 |
|
|
Olive |
|
|
|
|
|
|
Tunisia |
Olives |
1994 |
6 |
0.1a |
0 |
7809/46 830 |
|
|
Beans |
|
|
|
|
|
|
Uruguay |
Soya beans |
1993–95 |
19 |
50a |
19 |
0/0 |
|
Sweden |
Brown beans |
1996–98 |
20 |
0.1 |
18 |
0.18/1.9 |
|
United Kingdom |
Baked beans |
1996 |
50 |
0.2a |
49 |
0.006/0.3 |
|
Butter beans |
1996 |
12 |
0.2a |
11 |
1.14/13.7 |
|
|
|
Pulses |
|
|
|
|
|
|
Denmark |
Pulses |
1993–94 |
22 |
0.1a |
22 |
0/0 |
|
United Kingdom |
Pulses |
1998 |
50 |
0.200 |
50 |
0/0 |
|
|
Chickpeas |
|
|
|
|
|
|
Sweden |
Peas, dry |
1996–98 |
30 |
0.1 |
28 |
0.12/1.2 |
|
United Kingdom |
Dried chickpeas |
1996 |
14 |
0.2a |
14 |
0/0 |
|
|
Lentils |
|
|
|
|
|
|
|
Dried lentils |
1996 |
21 |
0.2a |
21 |
0/0 |
|
|
Soya |
|
|
|
|
|
|
Canada |
Soya-based cereals |
1998–99 |
16 |
0.200 |
NR |
NA/0.92 |
|
Japan |
Soya sauce |
1996 |
5 |
0.003a |
0 |
0.0068/0.026 |
|
|
Coffee |
|
|
|
|
|
|
Netherlands |
Roasted coffee |
1999 |
22 |
0.1a |
13 |
0.45/4.5 |
|
USA |
Green coffee |
1997 |
19 |
0.03a |
10 |
NA/4.6 |
|
USA |
Green coffee, imported |
1995–99 |
180 |
4–10a |
174 |
0.353/19.2 |
|
Soluble coffee, imported |
1995–99 |
23 |
4–10a |
23 |
0/0 |
|
|
Roasted coffee |
1997 |
13 |
0.03a |
4 |
NA/1.2 |
|
|
Denmark |
Roasted coffee |
1993-94 |
11 |
0.1a |
0 |
0.51/3.2 |
|
United Kingdom |
Green coffee, imported |
NR |
291 |
0.260 |
181 |
NA/27.3 |
|
Coffee products |
1995 |
100 |
0.1a |
19 |
NA/8 |
|
|
Europe |
Coffee products |
1999 |
633 |
0.2–1a |
334 |
0.90/27.2 |
|
Eastern Europe |
Adulterated soluble coffee |
NR |
15 |
0.2a |
0 |
5.9/15.9 |
|
World |
Soluble coffee |
NR |
101 |
0.2a |
26 |
1.1/6.5 |
|
Canada |
Instant coffee |
1997–98 |
30 |
0.1a |
NR |
NA/3.1 |
|
Coffee, ground and beans |
1997–98 |
71 |
0.1a |
NR |
NA/2.3 |
|
|
Dubai |
Coffee beans |
NR |
8 |
0.500 |
5 |
NA/7.46 |
|
Sweden |
Green coffee |
1999 |
45 |
0.05 |
23 |
0.53/12.1 |
|
Roasted coffee |
1999 |
37 |
0.05 |
29 |
0.40/3.86 |
|
|
Coffee granulate |
1999 |
6 |
0.05 |
0 |
0.50/0.79 |
|
|
Spain |
Coffee, roasted and soluble |
1997 |
38 |
0.110 |
0 |
1.01/5.64 |
|
Decaffeinated coffee |
1997 |
8 |
0.110 |
0 |
0.55/1.29 |
|
|
Japan |
Canned coffee |
1996 |
10 |
0.003a |
1 |
0.028/0.133 |
|
Instant coffee |
1996 |
12 |
0.06a |
0 |
0.018/0.063 |
|
|
Regular coffee |
1996 |
10 |
0.06a |
10 |
0/0 |
|
|
European Union, |
Roasted coffee |
1995–96 |
86 |
NR |
NR |
0.8/NR |
|
Roasted coffee |
1995–97 |
504 |
NR |
NR |
0.8/NR |
|
|
Switzerland |
Roasted coffee |
1996–98 |
232 |
NR |
NR |
0.6/NR |
|
Roasted coffee |
1999 |
107 |
NR |
NR |
0.4/NR |
|
|
Brazil |
Roasted coffee |
2000 |
34 |
0.2a |
11 |
0.93/6.5 |
|
Instant coffee |
2000 |
16 |
0.2a |
0 |
2.17/5.10 |
|
|
Germany |
Roasted coffee |
1997 |
34 |
0.3a |
10 |
1.43/7.54 |
|
Germany |
Roasted coffee |
1995–98 |
113 |
0.3a |
61 |
0.61/6.32 |
|
Roasted coffee |
1995–98 |
60 |
0.3a |
39 |
0.45/4.75 |
|
|
Roasted coffee |
1995–98 |
67 |
0.3a |
35 |
0.56/3.34 |
|
|
Soluble coffee |
1995–98 |
52 |
0.3a |
6 |
1.83/9.47 |
|
|
Soluble coffee |
1995–98 |
32 |
0.3a |
13 |
0.59/1.8 |
|
|
Malt coffee |
1995–98 |
33 |
0.3a |
28 |
< 0.3/0.96 |
|
|
Green coffee |
1995–99 |
82 |
0.250 |
60 |
1.29/24.5 |
|
|
Roasted coffee |
1995–99 |
419 |
0.250 |
228 |
0.99/12.1 |
|
|
Decaffeinated coffee |
1995–99 |
71 |
0.250 |
45 |
0.49/2.7 |
|
|
Instant coffee |
1995–99 |
41 |
0.250 |
12 |
1.0/4.8 |
|
|
|
Cocoa |
|
|
|
|
|
|
Netherlands |
Cocoa products |
1996 |
19 |
0.250 |
19 |
0/0 |
|
Uruguay |
Cocoa beans and by-products |
1993-95 |
91 |
50a |
91 |
0/0 |
|
United Kingdom |
Cocoa powder |
1998 |
20 |
0.200 |
0 |
1.67/2.4 |
|
|
Cocoa powder |
1996 |
20 |
0.2a |
3 |
0.67/1.1 |
|
Germany |
Cocoa |
1995–98 |
40 |
0.01a |
0 |
NR/1.8 |
|
Cocoa powder |
1995–98 |
56 |
0.01a |
5 |
NR/0.63 |
|
|
Cocoa drinks |
1995–98 |
34 |
0.01a |
0 |
NR/0.05 |
|
|
FSIS185 powder |
1998 |
20 |
0.01a |
0 |
1.7/2.4 |
|
|
FSIS185 powder |
1997 |
20 |
0.01a |
5 |
0.68/1.1 |
|
|
|
Chocolate |
|
|
|
|
|
|
United Kingdom |
Chocolate |
1998 |
40 |
0.020 |
10 |
0.16/0.6 |
|
Germany |
Milk chocolate < 30% |
1995–98 |
39 |
0.01a |
3 |
NR/0.41 |
|
Germany |
Plain chocolate > 60% |
1995–98 |
78 |
0.01a |
0 |
NR/0.66 |
|
Germany |
Chocolate with nuts |
1995–98 |
35 |
0.01a |
4 |
NR/0.16 |
|
Germany |
Filled chocolate |
1995–98 |
58 |
0.01a |
3 |
NR/0.324 |
|
Germany |
FSIS185 milk chocolate |
1998 |
28 |
0.01a |
9 |
0.15/0.6 |
|
Germany |
FSIS185 plain chocolate |
1998 |
12 |
0.01a |
1 |
0.27/0.6 |
|
|
Dried fruits |
|
|
|
|
|
|
Uruguay |
Dried fruits |
1993–95 |
157 |
50a |
157 |
0/0 |
|
USA |
Raisins |
NR |
63 |
NR |
14 |
1.56/11.5 |
|
USA |
Raisins |
1998–99 |
133 |
NR |
43 |
1.27/29 |
|
Raisins |
1998 |
114 |
NR |
38 |
0.82/8.1 |
|
|
Raisins |
1997 |
69 |
NR |
19 |
0.42/3.1 |
|
|
United Kingdom |
Sultanas |
1998 |
100 |
0.200 |
8 |
3.42/25.1 |
|
Raisins |
1998 |
101 |
0.200 |
3 |
2.87/29.8 |
|
|
Currants |
1998 |
100 |
0.200 |
4 |
4.97/40.8 |
|
|
Currants |
1996 |
20 |
0.2a |
1 |
9.19/53.6 |
|
|
United Kingdom |
Apricots |
1996 |
20 |
0.2a |
20 |
0/0 |
|
Dried, fresh coconut |
1996 |
20 |
0.2a |
20 |
0/0 |
|
|
Dried dates |
1996 |
20 |
0.2a |
19 |
0.01/0.2 |
|
|
Raisins |
1996 |
20 |
0.2a |
3 |
2.79/20 |
|
|
Sultanas |
1996 |
20 |
0.2a |
3 |
4.86/18.1 |
|
|
Figs |
1998 |
20 |
0.200 |
18 |
0.05/0.8 |
|
|
Germany |
FSIS 185 currants |
1998 |
100 |
0.01a |
4 |
NA/40.8 |
|
FSIS 185 sultanas |
1998 |
100 |
0.01a |
8 |
NA/53.6 |
|
|
Dried raisins |
1995–98 |
117 |
0.01a |
5 |
0.90/7.74 |
|
|
FSIS 185 raisins |
1998 |
101 |
0.01a |
3 |
NA/29.8 |
|
|
Dried plums |
1995–98 |
31 |
0.01a |
5 |
NR/0.07 |
|
|
Other dried fruit |
1995–98 |
49 |
0.01a |
23 |
NR/0.09 |
|
|
Dried figs |
1995–98 |
34 |
0.01a |
7 |
NR/3.95 |
|
|
|
Sweets |
|
|
|
|
|
|
Germany |
Marmelade |
1995–98 |
42 |
0.01a |
42 |
0/0 |
|
Nut-nugget cream |
1995–98 |
33 |
0.01a |
2 |
0.06/0.27 |
|
|
Other puddings and creams |
1995–98 |
32 |
0.01a |
25 |
< 0.01/0.09 |
|
|
Cocoa cream |
1995–98 |
32 |
0.01a |
25 |
0.03/0.08 |
|
|
|
Milk and milk products |
|
|
|
|
|
|
Germany |
Milk and milk products |
1995–98 |
264 |
0.01a |
242 |
<0.01/0.86 |
|
Norway |
Milk |
1995–98 |
87 |
0.01a |
76 |
NA/0.058 |
|
Infant formula |
1995–98 |
20 |
0.01a |
20 |
0/0 |
|
|
Human milk |
1995–96 |
80 |
0.01a |
63 |
0.006/0.18 |
|
|
Sierra Leone |
Human milk |
NR |
113 |
0.2a |
73 |
7.9/337 |
|
|
Dried vegetables |
|
|
|
|
|
|
Uruguay |
Dried vegetables |
1993–95 |
100 |
50a |
100 |
0/0 |
|
|
Oil and oilseeds |
|
|
|
|
|
|
Germany |
Sunflower seed |
1995–98 |
34 |
0.01a |
14 |
NA/0.1 |
|
Sesame seed |
1995–98 |
24 |
0.01a |
15 |
NA/0.86 |
|
|
Germany |
Linseed |
1995–98 |
24 |
0.01a |
12 |
NA/1.79 |
|
Poppy seed |
1995–98 |
16 |
0.01a |
16 |
0/0 |
|
|
Edible oils |
1995–98 30 |
0.01a |
30 |
0/0 |
NA |
|
|
|
Meat |
|
|
|
|
|
|
Uruguay |
Meat products |
1993–95 |
59 |
10a |
59 |
0/0 |
|
Denmark |
Pig kidney |
1999 |
300 |
0.060 |
63 |
NA/14.72 |
|
Pig meat |
1999 |
300 |
0.090 |
227 |
NA/2.88 |
|
|
Pork |
1993–94 |
76 |
0.02a |
12 |
0.11/1.3 |
|
|
Pork, organic |
1993–94 |
7 |
0.02a |
3 |
0.05/0.12 |
|
|
Duck |
1993–94 |
19 |
0.03a |
8 |
0.02/0.09 |
|
|
Duck liver |
1993–94 |
7 |
0.03a |
3 |
0.06/0.16 |
|
|
Goose |
1993–94 |
12 |
0.03a |
7 |
0.03/0.10 |
|
|
Goose liver |
1993–94 |
12 |
0.03a |
8 |
0.02/0.06 |
|
|
Turkey |
1993–94 |
17 |
0.03a |
7 |
0.02/0.11 |
|
|
Turkey liver |
1993–94 |
17 |
0.03a |
14 |
0.04/0.28 |
|
|
Chicken |
1993–94 |
65 |
0.03a |
29 |
0.03/0.18 |
|
|
United Kingdom |
Pork liver |
1996 |
10 |
0.2a |
9 |
0.02/0.2 |
|
Pork salami |
1996 |
9 |
0.2a |
9 |
0/0 |
|
|
Germany |
Raw sausage |
1995–98 |
56 |
0.01a |
28 |
0.04/0.27 |
|
Sausage |
1995–98 |
40 |
0.01a |
26 |
0.02/0.18 |
|
|
Germany |
Sausage |
1995–98 |
45 |
0.01a |
24 |
0.04/0.38 |
|
Liver sausage |
1995–98 |
53 |
0.01a |
17 |
0.15/4.56 |
|
|
Blood sausage |
1995–98 |
57 |
0.01a |
13 |
0.16/3.16 |
|
|
Other meat products |
1995–98 |
21 |
0.01a |
18 |
0.01/0.04 |
|
|
Beef sausage |
1995–98 |
31 |
0.01a |
26 |
0.02/0.19 |
|
|
Poultry sausage |
1995–98 |
40 |
0.01a |
33 |
0.01/0.03 |
|
|
Beaf meat |
1995–98 |
58 |
0.01a |
57 |
0.01/0.03 |
|
|
Pig meat |
1995–98 |
58 |
0.01a |
48 |
0.02/0.14 |
|
|
Poultry meat |
1995–98 |
41 |
0.01a |
41 |
0/0 |
|
|
Pig kidney |
1995–98 |
61 |
0.01a |
34 |
0.43/9.33 |
|
|
Pig liver |
1995–98 |
59 |
0.01a |
49 |
0.07/2.72 |
|
|
France |
Pig kidney |
1997 |
300 |
1 |
297 |
0.01/1.4 |
|
Pig kidney |
1998 |
710 |
1 |
656 |
NA/5.0 |
|
|
Nephropathic pig kidneys |
1997 |
100 |
0 |
94 |
NA/0.48 |
|
|
|
Snacks |
|
|
|
|
|
|
Germany |
Bar |
1995–98 |
32 |
0.01a |
4 |
NR/0.11 |
|
Bar with nuts |
1995–98 |
47 |
0.01a |
7 |
NR/3.6 |
|
|
Muesli bar |
1995–98 |
67 |
0.01a |
28 |
NR/1.72 |
|
|
Muesli |
1995–98 |
115 |
0.01a |
44 |
NR/31.8 |
|
|
Breakfast cereals |
1995–98 |
85 |
0.01a |
20 |
NR/0.94 |
|
|
Corn flakes |
1995–98 |
38 |
0.01a |
26 |
NR/0.1 |
|
|
Biscuit |
1995–98 |
102 |
0.01a |
20 |
NR/3.81 |
|
|
Biscuit with chocolate |
1995–98 |
67 |
0.01a |
2 |
NR/0.39 |
|
|
Germany |
Rusk |
1995–98 |
37 |
0.01a |
5 |
NR/2.26 |
|
Rye biscuit |
1995–98 |
31 |
0.01a |
8 |
NR/0.92 |
|
|
Chips, popcorn |
1995–98 |
33 |
0.01a |
23 |
NR/2.1 |
|
|
|
Nuts |
|
|
|
|
|
|
Netherlands |
Roasted peanuts |
1996 |
12 |
0.250 |
12 |
0/0 |
|
Peanuts products |
1996 |
4 |
0.250 |
4 |
0/0 |
|
|
Pistachio nuts |
1996 |
3 |
0.250 |
3 |
0/0 |
|
|
Germany |
Hazelnuts |
1995–98 |
32 |
0.01a |
13 |
NR/0.08 |
|
Groundnuts |
1995–98 |
31 |
0.01a |
28 |
NR/0.08 |
|
|
Other nuts |
1995–98 |
125 |
0.01a |
99 |
NR/0.27 |
|
|
|
Beer |
|
|
|
|
|
|
Canada |
Beer |
NR |
41 |
0.1a |
15 |
0.04/0.65 |
|
Denmark |
Beer |
1993–94 |
21 |
0.001a |
0 |
0.049/0.16 |
|
United Kingdom |
Beer |
1996 |
20 |
0.2a |
20 |
0/0 |
|
Spain |
Beer |
1997 |
40 |
0.004a |
1 |
0.024/0.075 |
|
Europe |
Beer |
1997 |
40 |
0.004a |
0 |
0.025/0.121 |
|
Italy |
Beer, imported |
1999 |
61 |
0.01a |
31 |
0.017/0.135 |
|
Japan |
Beer |
1998 |
22 |
0.001a |
1 |
0.012/0.045 |
|
Beer, imported |
1998 |
94 |
0.001a |
8 |
0.01/0.066 |
|
|
Germany |
Pils beer |
1995–98 |
135 |
0.01a |
34 |
0.026/0.137 |
|
Export beer |
1995–98 |
31 |
0.01a |
6 |
0.027/0.123 |
|
|
Wheat beer |
1995–98 |
30 |
0.01a |
7 |
0.031/0.293 |
|
|
Strong beer |
1995–98 |
54 |
0.01a |
9 |
0.031/0.126 |
|
|
Beer, alcohol-free |
1995–98 |
24 |
0.01a |
11 |
0.013/0.035 |
|
|
Light beer |
1995–98 |
14 |
0.01a |
8 |
0.012/0.047 |
|
|
Malt beer |
1995–98 |
30 |
0.01a |
16 |
0.016/0.081 |
|
|
|
Teas |
|
|
|
|
|
|
Germany |
Black tea |
1995–98 |
32 |
0.3a |
32 |
0/0 |
|
Green tea |
1995–98 |
32 |
0.3a |
31 |
< 0.3/1.33 |
|
|
Fruit tea |
1995–98 |
32 |
0.3a |
32 |
0/0 |
|
|
|
Wine |
|
|
|
|
|
|
Netherlands |
Red wine |
1999 |
150 |
0.100 |
90 |
0.22/3.1 |
|
White wine |
1999 |
20 |
0.100 |
18 |
0.12/2.1 |
|
|
Sweden |
Wine |
1998–99 |
32 |
0.005 |
3 |
0.21/2.5 |
|
United Kingdom |
Red wine |
1998 |
50 |
0.020 |
22 |
0.074/0.46 |
|
1996 |
10 |
0.2a |
6 |
0.38/1.1 |
||
|
White wine |
1996 |
10 |
0.2a |
10 |
0/0 |
|
|
Switzerland |
Wine |
NR |
18 |
0.005 |
5 |
NA/0.11 |
|
Japan |
Wine |
1996 |
46 |
0.003a |
27 |
NA/0.245 |
|
Italy |
Red wine |
1992–94 |
8 |
0.001 |
1 |
0.54/1.29 |
|
Passito |
1990–94 |
5 |
0.001 |
3 |
0.009/0.04 |
|
|
Red wine |
1995 |
9 |
0.001 |
0 |
1.05/2.47 |
|
|
Passito |
1995 |
2 |
0.001 |
1 |
1.92/3.86 |
|
|
Red wine |
1996 |
23 |
0.001 |
7 |
0.54/1.78 |
|
|
Passito |
1996 |
2 |
0.001 |
1 |
0.007/0.01 |
|
|
Passito |
1997 |
5 |
0.001 |
1 |
1.42/3.48 |
|
|
Red wine |
1997 |
13 |
0.001 |
0 |
0.76/2.15 |
|
|
Red wine |
1998 |
18 |
0.001 |
4 |
0.66/3.17 |
|
|
Red wine |
1999 |
115 |
0.01a |
12 |
2.10/15.61 |
|
|
White wine |
1999 |
21 |
0.01a |
14 |
0.57/8.86 |
|
|
Rosé wine |
1999 |
4 |
0.01a |
2 |
0.13/0.28 |
|
|
Italy |
Red wine |
1997–98 |
38 |
0.01a |
1 |
1.21/7.63 |
|
Rosé wine |
1997–98 |
8 |
0.01a |
1 |
0.63/1.15 |
|
|
White wine |
1997–98 |
9 |
0.01a |
0 |
0.16/0.97 |
|
|
North Italy |
Red wine |
1997–99 |
8 |
0.01a |
4 |
0.102/0.54 |
|
South Italy |
Red wine |
1997–99 |
43 |
0.01a |
15 |
0.193/2.55 |
|
Red wine |
1997–99 |
20 |
0.01a |
1 |
1.153/3.31 |
|
|
North France |
Red wine |
1997–99 |
68 |
0.01a |
60 |
0.061/0.78 |
|
South France |
Red wine |
1997–99 |
40 |
0.01a |
19 |
0.07/0.47 |
|
Germany |
White wine |
1995–98 |
58 |
0.01a |
44 |
NA/1.4 |
|
Rosè wine |
1995–98 |
51 |
0.01a |
33 |
NA/2.4 |
|
|
Red wine |
1995–98 |
172 |
0.01a |
110 |
NA/7 |
|
|
FSIS 185 red wine |
1998 |
50 |
0.01a |
22 |
0.08/0.8 |
|
|
FSIS 185 red wine |
1997 |
10 |
0.01a |
6 |
0.44/1.1 |
|
|
North Germany |
Red wine |
1997–99 |
30 |
0.01a |
23 |
0.022/0.23 |
|
White wine |
1997–99 |
26 |
0.01a |
22 |
0.012/0.04 |
|
|
South Germany |
White wine |
1997–99 |
18 |
0.01a |
10 |
0.054/1.36 |
|
Germany |
Red wine |
|
40 |
|
20 |
0.17/1.90 |
|
Red wine |
|
48 |
|
28 |
0.14/1.10 |
|
|
White wine |
|
7 |
|
5 |
0.08/0.35 |
|
|
France |
Wine |
1998 |
29 |
0.01a |
15 |
0.038/0.19 |
|
World |
Red wine |
1997–99 |
305 |
0.01a |
140 |
0.20/3.31 |
|
Rosé wine |
1997–99 |
55 |
0.01a |
33 |
0.12/2.38 |
|
|
White wine |
1997–99 |
60 |
0.01a |
45 |
0.108/1.36 |
|
|
World |
White wine |
NR |
41 |
0.00 |
27 |
NA/1.2 |
|
Rosé wine |
NR |
14 |
0.00 |
8 |
NA/2.4 |
|
|
Red wine |
NR |
89 |
0.00 |
49 |
NA/7.0 |
|
|
Europe |
Wine |
1998 |
40 |
NR |
20 |
0.17/1.90 |
|
Europe |
Red wine |
1997 |
91 |
0.003a |
7 |
0.054/0.603 |
|
Rosé wine |
1997 |
32 |
0.003a |
3 |
0.031/0.161 |
|
|
White wine |
1997 |
69 |
0.003a |
24 |
0.020/0.267 |
|
|
Aperitif wine |
1997 |
47 |
0.003a |
12 |
0.04/0.254 |
|
|
Sparkling wine |
1997 |
12 |
0.003a |
2 |
0.012//0.037 |
|
|
Dessert wine |
1997 |
16 |
0.003a |
1 |
1.05/2.54 |
|
|
Europe |
Red wine |
1994–95 |
79 |
0.005 |
NR |
0.039/0.39 |
|
Rosé wine |
1994–95 |
15 |
0.005 |
NR |
0.025/0.12 |
|
|
White wine |
1994–95 |
24 |
0.005 |
NR |
0.011/0.18 |
|
|
Special wines |
1994–95 |
15 |
0.005 |
NR |
NA/0.45 |
|
|
|
Vinegar and mustard |
|
|
|
|
|
|
Germany |
Apple and fruit vinegar |
1995–98 |
18 |
0.01a |
17 |
NA/< 0.01 |
|
Wine vinegar |
1995–98 |
38 |
0.01a |
19 |
NA/1.9 |
|
|
Balsam vinegar |
1995–98 |
29 |
0.01a |
5 |
NA/4.35 |
|
|
Mustard |
1995–98 |
4 |
0.01a |
1 |
NA/0.34 |
|
|
|
Grape juice |
|
|
|
|
|
|
United Kingdom |
Grape juices |
1998 |
20 |
0.020 |
1 |
0.48/2.05 |
|
Germany |
White grape juice |
1995–98 |
27 |
0.01a |
6 |
NA/1.3 |
|
Red grape juice |
1995–98 |
64 |
0.01a |
8 |
NA/5.3 |
|
|
FSIS 185 white grape juice |
1998 |
11 |
0.01a |
1 |
0.27/0.6 |
|
|
FSIS 185 red grape juice |
1998 |
9 |
0.01a |
0 |
0.76/2.05 |
|
|
FSIS 185 grape juice |
1998 |
20 |
0.01a |
1 |
0.48/2.05 |
|
|
Europe |
Grape juice |
1994–95 |
8 |
0.005 |
3 |
0.137/0.31 |
|
Japan |
Grape juice |
1996 |
12 |
0.003a |
10 |
NA/0.006 |
|
World |
Grape juice |
NR |
20 |
000 |
6 |
NA/4.7 |
|
Other juices |
|
|
|
|
|
|
|
Germany |
Apple juice |
1995–98 |
33 |
0.01a |
33 |
0/0 |
|
Orange juice |
1995–98 |
30 |
0.01a |
30 |
0/0 |
|
|
Blackcurrant juice |
1995–98 |
19 |
0.01a |
16 |
NA/0.06 |
|
|
Tomato juice |
1995–98 |
30 |
0.01a |
27 |
NA/0.032 |
|
|
Carrot juice |
1995–98 |
18 |
0.01a |
17 |
NA/0.01 |
|
|
Other vegetable juice |
1995–98 |
30 |
0.01a |
30 |
0/0 |
|
|
|
Seasonings |
|
|
|
|
|
|
Germany |
Ketchup |
1995–98 |
57 |
0.01a |
41 |
NA/3.8 |
|
Herb sauce |
1995–98 |
15 |
0.01a |
13 |
NA/0.25 |
|
|
Pepper sauce |
1995–98 |
50 |
0.01a |
43 |
NA/0.72 |
|
|
|
Fermented beverages |
|
|
|
|
|
|
Japan |
Fermented beverages |
1996 |
15 |
0.003a |
15 |
0/0 |
|
|
Baby food |
|
|
|
|
|
|
Canada |
Baby food |
1998–99 |
11 |
0.500 |
11 |
0/0 |
|
|
Diet |
|
|
|
|
|
|
United Kingdom |
Normal diet |
NR |
32 |
0.002 |
0 |
0.025/0.073 |
|
Vegetarian diet |
NR |
11 |
0.002 |
0 |
0.045/0.114 |
|
|
Traditional diet |
NR |
7 |
0.002 |
0 |
0.029/0.066 |
|
|
|
Dust |
|
|
|
|
|
|
USA |
Dust |
2000 |
7 |
NR |
0 |
278.8/1581.8 |
|
Country/ Region |
Commodity |
Year/ Season |
90th %ile (µg/kg) |
n > 5 20(µg/kg |
n > 20µg/kg) |
References |
Sampling procedure |
|
|
Wheat |
|
|
|
|
|
|
|
Germany |
Wheat grain |
1995–98 |
0.26 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Wheat flour < T550 |
1995–98 |
0.26 |
0 |
0 |
|||
|
1995–98 |
0.59 |
0 |
0 |
||||
|
Wheat wholemeal flour |
1995–98 |
0.66 |
0 |
0 |
|||
|
Wheat semolina |
1995–98 |
1.49 |
0 |
0 |
|||
|
Wheat bran |
1995–98 |
0.72 |
0 |
0 |
|||
|
Wheat germ |
1995–98 |
0.27 |
0 |
0 |
|||
|
Netherlands |
Wheat, domestic |
1995 |
NR |
0 |
0 |
P,S,A, Sizoo & van Egmond (1997) P,S,A, Inspectorate for Health Protection (personal communication, 1999) |
|
|
Wheat, imported |
1995 |
NR |
1 |
0 |
|||
|
White wheat flour |
1999 |
NR |
0 |
0 |
|||
|
Whole-wheat meal |
1999 |
NR |
0 |
0 |
|||
|
Norway |
Wheat |
1990 |
NR |
0 |
0 |
P,S,A, Langseth (1999)* |
|
|
1993 |
NR |
0 |
0 |
||||
|
1994 |
NR |
0 |
0 |
||||
|
1995 |
NR |
0 |
0 |
||||
|
1996 |
NR |
0 |
0 |
||||
|
1997 |
NR |
0 |
0 |
||||
|
1998 |
NR |
1 |
0 |
||||
|
Total wheat samples |
1990–98 |
NR |
5 |
0 |
|||
|
Wheat, imported |
1990 |
NR |
0 |
0 |
|||
|
1993 |
NR |
0 |
0 |
||||
|
Norway |
Wheat, imported |
1994 |
NR |
0 |
0 |
||
|
1995 |
NR |
1 |
0 |
||||
|
1996 |
NR |
1 |
0 |
||||
|
1997 |
NR |
0 |
0 |
||||
|
1998 |
NR |
0 |
0 |
||||
|
Total imported wheat |
1990–98 |
NR |
2 |
0 |
|||
|
Sweden |
Wheat grain |
1996–98 |
0.84 |
0 |
0 |
P,S, Thuvander et al. (2000); A, Larsson & Møller (1996)* |
Mills; 1 kg sampled in national pesticide control programme/ |
|
1999 |
0.69 |
1 |
0 |
P, National Food Administration; S, Thuvander et al. (2000); A, Larsson (1996)b |
|
||
|
Brazil |
Wheat grain |
1988–90 |
0.00 |
0 |
1 |
P,S, Furlong et al. (1995a); A, Soares et al. (1985)c |
From experimental plots; all grain within 3 m x 6 rows/ 3.0–10 |
|
1990 |
0.00 |
0 |
0 |
P,S, Furlong et al. (1995); A, Soares et al. (1985)c |
|
||
|
Wheat products |
1991 |
0.00 |
0 |
0 |
P,S, Soares & Furlani (1996); A, Soares et al. (1985)c |
|
|
|
USA |
Wheat grain |
1997 |
NA |
3 |
1 |
P,S,A, Trucksess et al. (1999) |
Sampled by GIPSA by unspecified USDA sampling plan |
|
Finland |
Wheat grain |
1996 |
0.00 |
0 |
2 |
P,A, Solfrizzo et al. (1998)d |
Random 1-kg samples from farms |
|
Denmark |
Wheat grain |
1986–92 |
NR |
6 |
3 |
P,S,A, Jorgensen et al. (1996)d |
Random 1-kg samples from mills |
|
Wheat grain, organic |
1986–92 |
NR |
3 |
1 |
|||
|
Wheat grain |
1986–92 |
NR |
1 |
0 |
|||
|
Wheat bran |
1986–92 |
NR |
2 |
0 |
|||
|
Wheat bran, organic |
1986–92 |
NR |
0 |
0 |
|||
|
Dubai |
Wheat flour |
NR |
0.00 |
0 |
0 |
P,S,A, Rao (2000) |
|
|
United Kingdom |
Wheat noodles |
1995 |
0.28 |
0 |
0 |
P,S,A, Patel et al. (1996) |
Purchased in specialist food shops |
|
Wheat grain |
1993 |
NR |
0 |
0 |
P, Scudamore (1999) |
|
|
|
Wheat grain stored |
1992–93 |
NR |
0 |
0 |
A, Sharman et al. (1992) |
|
|
|
Wheat from millers |
1993 |
NR |
2 |
0 |
S, NR |
|
|
|
1994 |
NR |
NR |
3 |
|
|
||
|
Wheat grain |
1997-98 |
NR |
3 |
0 |
P,S,A, MAFF (1999a) |
|
|
|
1996 |
0.00 |
0 |
0 |
P,S, MAFF (1997) |
|
||
|
Cereals and flours |
1996 |
NR |
2 |
0 |
P,S, MAFF (1996b) |
|
|
|
Uruguay |
Wheat grain |
1993–95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996) |
|
|
|
Buckwheat |
|
|
|
|
|
|
|
Germany |
Buckwheat |
1995–98 |
0.02 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Buckwheat flour |
1995–98 |
0.04 |
1 |
0 |
|||
|
|
Barley |
|
|
|
|
|
|
|
Germany |
Barley |
1995–98 |
0.10 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Pearl barley |
1995–98 |
0.10 |
0 |
0 |
|||
|
Norway |
Barley |
1990 |
0.00 |
0 |
0 |
P,S,A, Langseth (1999)b |
|
|
Barley groats |
1990 |
0.00 |
0 |
0 |
|||
|
USA |
Barley |
1997 |
NR |
1 |
0 |
P,S,A, Trucksess et al. (1999) |
Sampled by GIPSA with unspecified USDA sampling plan |
|
United Kingdom |
Barley |
1997-98 |
NR |
5 |
0 |
P,S,A, MAFF (1999a) |
|
|
Barley |
1996 |
0.00 |
1 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
|
Barley |
1993 |
NR |
0 |
0 |
P, Scudamore (1999); A, Sharman et al. (1992); S, NR |
|
|
|
stored in 1992 |
|
|
|
|
|||
|
Barley |
1994 |
NR |
0 |
1 |
|||
|
Barley stored in 1993 |
1994 |
NR |
1 |
1 |
|||
|
Uruguay |
Barley and malt |
1993-95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994)c |
|
|
Finland |
Barley kernel |
1996 |
0.00 |
2 |
0 |
P,A, Solfrizzo et al. (1998)d |
Random 1-kg samples from farms |
|
Canada |
Barley cereals |
1998–99 |
NR |
0 |
0 |
P, Canada; S,A, NRb |
|
|
Barley based cereals |
1998–99 |
NR |
NR |
0 |
|||
|
|
Maize |
|
|
|
|
|
|
|
Germany |
Maize and popcorn |
1995–98 |
0.26 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
United Kingdom |
Raw maize, imported |
1998–99 |
NR |
0 |
0 |
P,S, Scudamore (2000), A, Howell & Taylor (1981) |
At ports; from conveyor between silos and mill or from ships' holds |
|
Croatia |
Raw maize |
1996 |
0.00 |
0 |
2 |
P,S, Jurjevic et al. (1999); A, Solfrizzo et al. (1998)d |
Random 1-kg samples from farms |
|
Raw maize |
1997 |
1.29 |
2 |
7 |
|||
|
Brazil |
Raw maize |
1991 |
0.00 |
0 |
0 |
P,S, Pozzi et al. (1995) A, Soares et al. (1985)c |
Samples from stored material collected from 60-kg sacks at monthly intervals |
|
Brazil |
Raw maize |
1993–94 |
0.00 |
0 |
0 |
P,S, Gloria et al. (1997) |
|
|
Uruguay |
Maize and by-products |
1993-95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996) |
|
|
United Kingdom |
Corn flour |
1995 |
0.42 |
0 |
0 |
P,S,A, Patel et al. (1996) |
|
|
|
Oats |
|
|
|
|
|
|
|
Sweden |
Oat grain |
1996–98 |
0.81 |
0 |
0 |
P,S, Thuvander et al. (2000); A, Larsson & Møller (1996)b |
|
|
|
Oat grain |
1999 |
0.11 |
0 |
0 |
P, National Food Administration (2000); S, Thuvander et al (2000), A, Larsson & Møller (1996)b |
|
|
Norway |
Oat groats |
1990 |
NR |
0 |
0 |
P,S,A, Langseth (1999)b |
|
|
Oats |
1990 |
NR |
1 |
0 |
|||
|
Oats |
1993 |
NR |
0 |
0 |
|||
|
Oats |
1994 |
NR |
1 |
0 |
|||
|
Oats |
1995 |
NR |
0 |
0 |
|||
|
Oats |
1996 |
0 |
0 |
0 |
|||
|
Oats |
1997 |
NR |
0 |
0 |
|||
|
Oats |
1998 |
NR |
0 |
0 |
|||
|
Total oats samples |
1990–98 |
NR |
2 |
0 |
|||
|
Denmark |
Oat kernels |
1986–92 |
NR |
1 |
0 |
P,S,A, Jorgensen et al. (1996)d |
Random 1-kg samples from mills |
|
Oat kernels, organic |
1986–92 |
NR |
0 |
0 |
|||
|
Oat kernels, imported |
1986–92 |
NR |
0 |
0 |
|||
|
Finland |
Oat kernels |
1996 |
0.00 |
0 |
1 |
P,A, Solfrizzo et al. (1998)d |
Random 1-kg samples from farms |
|
United Kingdom |
Oat kernels |
1997–98 |
NR |
0 |
0 |
P,S,A, MAFF (1999a) |
|
|
1996 |
0.00 |
1 |
0 |
P,S, MAFF (1997) A, Sharman et al. (1992) |
|
||
|
Germany |
Oats |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
|
Rice |
|
|
|
|
|
|
|
Uruguay |
Rice |
1993-95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996); A, Pineiro & Giribone (1994)c |
Stratified random sampling to obtain 5-kg samples |
|
United |
Basmati rice |
1995 |
0.00 |
0 |
0 |
P,S,A, Patel et al. (1996)b |
|
|
Kingdom |
Chinese rice |
1995 |
0.00 |
0 |
0 |
||
|
Dubai |
Rice |
NR |
0.00 |
0 |
0 |
P,S,A, Rao (2000) |
|
|
Germany |
Rice |
1995–98 |
0.01 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Parboiled rice |
1995–98 |
|
0 |
0 |
|||
|
Long-grain rice |
1995–98 |
|
0 |
0 |
|||
|
Round-grain rice |
1995–98 |
|
0 |
0 |
|||
|
|
Rye |
|
|
|
|
|
|
|
Sweden |
Rye grain |
1996–98 |
1.2 |
0 |
0 |
P,S, Thuvander et al. (2000); A, Larsson & Møller (1996)b |
|
|
1999 |
1.5 |
0 |
1 |
P, National Food Administration, S, Thuvander et al. (2000), A, Larsson & Møller (1996)b |
|
||
|
Norway |
Rye, imported |
1990 |
NR |
0 |
0 |
P,S,A, Langseth (1999)b |
|
|
1993 |
NR |
0 |
0 |
||||
|
1994 |
NR |
0 |
0 |
||||
|
1995 |
NR |
0 |
0 |
||||
|
1996 |
0.00 |
0 |
0 |
||||
|
Total |
1990–98 |
NR |
0 |
0 |
|||
|
Denmark |
Rye kernels |
1986–92 |
NR |
16 |
4 |
P,S,A, Jorgensen et al. (1996)d |
Random 1-kg samples from mills |
|
Rye kernels, organic |
1986–92 |
NR |
12 |
4 |
|||
|
Rye kernels, imported |
1986–92 |
NR |
0 |
0 |
|||
|
United Kingdom |
Rye kernels |
1996 |
0.00 |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
Germany |
Rye |
1995–98 |
0.01 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Rye flour < T997 |
1995–98 |
0.87 |
1 |
0 |
|||
|
1995–98 |
0.06 |
0 |
0 |
||||
|
Rye wholemeal flour |
1995–98 |
0.03 |
0 |
0 |
|||
|
|
Sorghum |
|
|
|
|
||
|
Germany |
Sorghum |
1995–98 |
0.01 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
|
Spelt |
|
|
|
|
|
|
|
Germany |
Spelt/spelt flour |
1995–98 |
0.06 |
NR |
0 |
P,S,A, Wolff et al. (2000) |
|
|
|
Cereal products |
|
|
|
|
|
|
|
United Kingdom |
Pitta bread |
1995 |
NR |
0 |
0 |
P,S,A, Patel et al. (1996)b |
|
|
Chapatti |
1995 |
NR |
0 |
0 |
|||
|
Nan bread |
1995 |
000' |
0 |
0 |
|||
|
Poppadoms |
1995 |
000' |
0 |
0 |
|||
|
Tunisia |
Cereal-derived food |
1994 |
4314.4 |
6 |
56 |
P,S,A, Maaroufi et al. (1995b) |
From homes of nephropathy patients |
|
Dubai |
Mixed cereals |
NR |
NR |
0 |
0 |
P,S,A, Rao (2000) |
|
|
Canada |
Mixed grain cereals |
1998–99 |
NR |
0 |
0 |
P, Canada (2000); S and A, NRb |
|
|
Mixed cereals |
1998–99 |
000' |
0 |
0 |
|||
|
Europe, Tunisia |
Bread |
NR |
NR |
NR |
0 |
P, Burdaspal & Legarda (2000); A, Burdaspal & Legarda (2001); S, NRb |
|
|
Germany |
Bread, wheat mix |
1995–98 |
000 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Germany |
Bread, rye mix |
1995–98 |
0.04 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
White bread |
1995–98 |
0.01 |
0 |
0 |
|||
|
Toast bread |
1995–98 |
0.20 |
0 |
0 |
|||
|
Rye meal bread |
1995–98 |
0.32 |
1 |
0 |
|||
|
Milk–water bread roll |
1995–98 |
0.21 |
0 |
0 |
|||
|
Various cereals, bread |
1995–98 |
0.73 |
0 |
0 |
|||
|
Various cereals, bread +oilseed |
1995–98 |
0.33 |
0 |
0 |
|||
|
Crispbread |
1995–98 |
0.01 |
0 |
0 |
|||
|
Wheat whole bread |
1995–98 |
0.02 |
0 |
0 |
|||
|
Special bread |
1995–98 |
0.02 |
0 |
0 |
|||
|
Wholemeal bread roll |
1995–98 |
0.02 |
0 |
0 |
|||
|
Various cereals and muesli bread |
1995–98 |
0.02 |
2 |
0 |
|||
|
Rye bread roll |
1995–98 |
0.03 |
0 |
0 |
|||
|
Pasta without egg |
1995–98 |
0.61 |
0 |
0 |
|||
|
Pasta with egg |
1995–98 |
0.04 |
0 |
0 |
|||
|
Wholemeal pasta |
1995–98 |
0.11 |
0 |
1 |
|||
|
Oat flakes |
1995–98 |
0.01 |
0 |
0 |
|||
|
Oats bran |
1995–98 |
0.01 |
0 |
0 |
|||
|
Polenta |
1995–98 |
0.03 |
0 |
0 |
|||
|
Green corn |
1995–98 |
0.01 |
0 |
0 |
|||
|
Infant food |
1995–98 |
0.01 |
0 |
0 |
|||
|
Peas, lentil, beans |
1995–98 |
|
0 |
0 |
|||
|
Soya beans |
1995–98 |
0.01 |
0 |
0 |
|||
|
FSIS185 pulses |
1998 |
|
0 |
0 |
|||
|
FSIS185 pulses |
1997 |
< 0.2 |
2 |
0 |
|||
|
|
Seeds |
|
|
|
|
|
|
|
United Kingdom |
Fennel |
1995 |
0.00 |
0 |
0 |
P,S,A, Patel et al. (1996) |
|
|
Sesame seeds |
1995 |
0.00 |
0 |
0 |
|||
|
Coriander |
1995 |
3.20 |
0 |
0 |
|||
|
|
Herbs and spices |
|
|
|
|
||
|
Netherlands |
Paprika powder |
1996–98 |
NR |
1 |
0 |
P,S,A, Inspectorate for Health Protection (personal communication, 1999) |
|
|
Pepper |
1996–97 |
NR |
4 |
0 |
|||
|
United Kingdom |
Chilli powder |
1995 |
NR |
NR |
NR |
P,S,A, Patel et al. (1996) |
|
|
Curry powder |
1995 |
NR |
NR |
NR |
|||
|
Tandoori |
1995 |
NR |
NR |
NR |
|||
|
Ginger |
1995 |
NR |
NR |
0 |
|||
|
Garlic |
1995 |
0.00 |
0 |
0 |
|||
|
Five spices powder |
1995 |
1.82 |
0 |
0 |
|||
|
Tunisia |
Dried vegetables |
1994 |
3426.6 |
0 |
6 |
P,S,A, Maaroufi et al. (1995b) |
From homes of nephropathy patients |
|
Dubai |
Spices |
NR |
NR |
0 |
0 |
P,S,A, Rao (2000) |
|
|
|
Pickles and pastes |
|
|
|
|
|
|
|
United Kingdom |
Chilli pickle |
1995 |
NR |
0 |
0 |
P,S,A, Patel et al. (1996) |
|
|
Garlic pickle |
1995 |
NR |
0 |
0 |
|||
|
United Kingdom |
Curry paste |
1995 |
NR |
NR |
0 |
P,S,A, Patel et al. (1996) |
|
|
Chilli sauce |
1995 |
2.31 |
0 |
0 |
|||
|
|
Canned foods |
|
|
|
|
|
|
|
United Kingdom |
Canned foods |
1995 |
NR |
0 |
0 |
P,S,A, Patel et al. (1996) |
|
|
|
Oils |
|
|
|
|
|
|
|
United |
Sesame oil |
1995 |
0.32 |
0 |
0 |
P,S,A, Patel et al. (1996) |
|
|
Kingdom |
Chili, almond oils |
1995 |
0.00 |
0 |
0 |
||
|
Uruguay |
Oilseed |
1993-95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994) |
|
|
Dubai |
Oilseed |
NR |
0.00 |
0 |
0 |
P,S,A, Rao (2000) |
|
|
|
Olive |
|
|
|
|
|
|
|
Tunisia |
Olives |
1994 |
32 782 |
2 |
1 |
P,S,A, Maaroufi et al. (1995b) |
From homes of nephropathy patients |
|
|
Beans |
|
|
|
|
|
|
|
Uruguay |
Soya beans |
1993–95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994) |
|
|
Sweden |
Brown beans |
1996–98 |
0.07 |
0 |
0 |
P,S, Thuvander et al. (2000), A, Larsson & Møller (1996) |
|
|
United Kingdom |
Baked beans |
1996 |
0.00 |
0 |
0 |
P,S, MAFF (1997) |
|
|
Butter beans |
1996 |
0.00 |
1 |
0 |
A, Sharman et al. (1992) |
|
|
|
|
Pulses |
|
|
|
|
|
|
|
Denmark |
Pulses |
1993–94 |
NA |
0 |
0 |
P,S,A, Jorgensen (1998) |
'Random samples' from retail shops |
|
United Kingdom |
Pulses |
1998 |
NA |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
|
Chickpeas |
|
|
|
|
|
|
|
Sweden |
Peas, dry |
1996–98 |
0.08 |
0 |
0 |
P,S, Thuvander et al. (2000); A, Larsson & Møller (1996) |
|
|
United Kingdom |
Dried chickpeas |
1996 |
NA |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
|
Lentils |
|
|
|
|
|
|
|
|
Dried lentils |
1996 |
NA |
0 |
0 |
|
|
|
|
Soya |
|
|
|
|
|
|
|
Canada |
Soya-based cereals |
1998–99 |
NR |
0 |
0 |
P, Canada (2000); S,A, NR |
|
|
Japan |
Soya sauce |
1996 |
NR |
0 |
0 |
P,A, Ueno (1998); S, NR |
|
|
|
Coffee |
|
|
|
|
|
|
|
Netherlands |
Roasted coffee |
1999 |
NR |
0 |
0 |
P,S,A, Inspectorate for Health Protection (personal communication, 1999) |
|
|
USA |
Green coffee |
1997 |
NR |
0 |
0 |
P,S,A, Trucksess et al. (1999) |
Sampled by GIPSA with unspecified USDA sampling plan |
|
USA |
Green coffee, imported |
1995–99 |
0.00 |
3 |
0 |
P, Ochratoxin A Monitoring Program, S,A, NRc |
|
|
Soluble coffee, imported |
1995–99 |
0.00 |
0 |
0 |
|
|
|
|
Roasted coffee |
1997 |
NR |
0 |
0 |
P,S,A, Trucksess et al. (1999) |
'Random samples' from retail shops |
|
|
Denmark |
Roasted coffee |
1993-94 |
NR |
0 |
0 |
P,S,A, Jorgensen (1998) |
'Random samples' from retail shops |
|
United Kingdom |
Green coffee, imported |
NR |
NR |
11 |
2 |
P,S,A, MAFF (1996a) |
|
|
Coffee products |
1995 |
NR |
NR |
0 |
P,S,A, Patel et al. (1997) |
|
|
|
Europe |
Coffee products |
1999 |
NR |
3 |
1 |
P,S,A, Stegen et al. (1997) |
|
|
Eastern Europe |
Adulterated soluble coffee |
NR |
1.40 |
6 |
0 |
P,S,A, Pittet et al. (1996) |
|
|
World |
Soluble coffee |
NR |
NA |
NR |
0 |
P,S,A, Pittet et al. (1996) |
|
|
Canada |
Instant coffee |
1997–98 |
NR |
0 |
0 |
P, Canada (2000);S,A, |
|
|
Coffee, ground and beans |
1997–98 |
NR |
0 |
0 |
NR |
|
|
|
Dubai |
Coffee beans |
NR |
NR |
1 |
0 |
P,S,A, Rao (2000) |
|
|
Sweden |
Green coffee |
1999 |
0.74 |
1 |
0 |
P,S,A, National Food Administration |
|
|
Roasted coffee |
1999 |
1.7 |
0 |
0 |
|
||
|
Coffee granulate |
1999 |
0.68 |
0 |
0 |
|
|
|
|
Spain |
Coffee, roasted and soluble |
1997 |
NR |
NR |
0 |
P, Burdaspal & Legarda (1998b), A, Pittet et al. (1996); S, NR |
|
|
Decaffeinated coffee |
1997 |
NR |
0 |
0 |
|||
|
Japan |
Canned coffee |
1996 |
NR |
0 |
0 |
P,A, Ueno (1998); S, NR |
|
|
Instant coffee |
1996 |
NR |
0 |
0 |
|||
|
Regular coffee |
1996 |
000' |
0 |
0 |
|||
|
European Union, |
Roasted coffee |
1995–96 |
NR |
NR |
NR |
Olsen (2000) |
|
|
Roasted coffee |
1995–97 |
NR |
NR |
NR |
|||
|
Switzerland |
Roasted coffee |
1996–98 |
NR |
NR |
NR |
||
|
Roasted coffee |
1999 |
NR |
NR |
NR |
|||
|
Brazil |
Roasted coffee |
2000 |
000 |
1 |
0 |
P,A, Leoni et al. (2000); S, NR |
|
|
Instant coffee |
2000 |
0'05 |
1 |
0 |
|||
|
Germany |
Roasted coffee |
1997 |
NR |
3 |
0 |
P,A, Koch et al. (1996); S, NR |
|
|
Germany |
Roasted coffee |
1995–98 |
2 |
1 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Roasted coffee |
1995–98 |
1 |
0 |
0 |
|||
|
Roasted coffee |
1995–98 |
2 |
0 |
0 |
|||
|
Soluble coffee |
1995–98 |
4 |
5 |
0 |
|||
|
Soluble coffee |
1995–98 |
2 |
0 |
0 |
|||
|
Malt coffee |
1995–98 |
1 |
0 |
0 |
|||
|
Green coffee |
1995–99 |
NR |
NR |
NR |
P,S, Ottender & Majerus (2001); A, Entwisle et al. (2000b) |
|
|
|
Roasted coffee |
1995–99 |
NR |
NR |
0 |
|||
|
Decaffeinated coffee |
1995–99 |
NR |
0 |
0 |
|||
|
Instant coffee |
1995–99 |
NR |
0 |
0 |
|||
|
|
Cocoa |
|
|
|
|
|
|
|
Netherlands |
Cocoa products |
1996 |
0.00 |
0 |
0 |
P,S,A, Inspectorate for Health Protection (personal communication, 1999) |
|
|
Uruguay |
Cocoa beans and by-products |
1993-95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994)c |
|
|
United Kingdom |
Cocoa powder |
1998 |
2.11 |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
|
Cocoa powder |
1996 |
1.00 |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
Germany |
Cocoa |
1995–98 |
0.93 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Cocoa powder |
1995–98 |
0.03 |
0 |
0 |
|||
|
Cocoa drinks |
1995–98 |
0.00 |
0 |
0 |
|||
|
FSIS185 powder |
1998 |
NR |
0 |
0 |
|||
|
FSIS185 powder |
1997 |
NR |
0 |
0 |
|||
|
|
Chocolate |
|
|
|
|
|
|
|
United Kingdom |
Chocolate |
1998 |
0.31 |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
Germany |
Milk chocolate < 30% |
1995–98 |
0.01 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Germany |
Plain chocolate > 60% |
1995–98 |
0.02 |
0 |
0 |
||
|
Germany |
Chocolate with nuts |
1995–98 |
0.01 |
0 |
0 |
||
|
Germany |
Filled chocolate |
1995–98 |
0.01 |
0 |
0 |
||
|
Germany |
FSIS185 milk chocolate |
1998 |
NR |
0 |
0 |
||
|
Germany |
FSIS185 plain chocolate |
1998 |
NR |
0 |
0 |
||
|
|
Dried fruits |
|
|
|
|
|
|
|
Uruguay |
Dried fruits |
1993–95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994)c |
|
|
USA |
Raisins |
NR |
3.48 |
5 |
0 |
P, Ochratoxin A Monitoring Program, S,A, NR |
|
|
USA |
Raisins |
1998–99 |
3.96 |
3 |
1 |
P, Ochratoxin A Monitoring Program; S,A, NR |
|
|
Raisins |
1998 |
1.87 |
6 |
0 |
|||
|
Raisins |
1997 |
0.92 |
0 |
0 |
|||
|
United Kingdom |
Sultanas |
1998 |
8.56 |
15 |
2 |
P,S,A, MAFF (1999b) |
|
|
Raisins |
1998 |
6.40 |
13 |
1 |
|||
|
Currants |
1998 |
11.47 |
20 |
5 |
|||
|
Currants |
1996 |
14.33 |
9 |
2 |
P,A, MacDonald et al. (1999); A, NR |
From retail shops with MAFF sampling scheme |
|
|
United Kingdom |
Apricots |
1996 |
NA |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
Dried, fresh coconut |
1996 |
NA |
0 |
0 |
|||
|
Dried dates |
1996 |
0.00 |
0 |
0 |
|||
|
Raisins |
1996 |
8.60 |
4 |
0 |
P,A, MacDonald et al. (1999); A, NR |
|
|
|
Sultanas |
1996 |
11.35 |
7 |
0 |
|||
|
Figs |
1998 |
0.15 |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
|
Germany |
FSIS 185 currants |
1998 |
NA |
29 (> 4) |
5 |
P,S,A, Wolff et al. (2000) |
|
|
FSIS 185 sultanas |
1998 |
NA |
18 (> 4) |
2 |
|||
|
Dried raisins |
1995–98 |
NA |
2 |
6 |
|||
|
FSIS 185 raisins |
1998 |
NA |
19 (> 4) |
1 |
|||
|
Dried plums |
1995–98 |
0.00 |
0 |
0 |
|||
|
Other dried fruit |
1995–98 |
0.00 |
0 |
0 |
|||
|
Dried figs |
1995–98 |
0.02 |
0 |
0 |
|||
|
|
Sweets |
|
|
|
|
|
|
|
Germany |
Marmelade |
1995–98 |
NA |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Nut-nugget cream |
1995–98 |
0.00 |
0 |
0 |
|||
|
Other puddings and creams |
1995–98 |
0.00 |
0 |
0 |
|||
|
Cocoa cream |
1995–98 |
0.00 |
0 |
0 |
|||
|
|
Milk and milk products |
|
|
|
|
|
|
|
Germany |
Milk and milk products |
1995–98 |
NA |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Norway |
Milk |
1995–98 |
NA |
0 |
0 |
P,S, Skaug (1999) |
|
|
Infant formula |
1995–98 |
NA |
0 |
0 |
A, Breitholtz-Emanuelsson (1993b) |
|
|
|
Human milk |
1995–96 |
NA |
0 |
0 |
|||
|
Sierra Leone |
Human milk |
NR |
NA |
NR |
4 |
P,S,A, Jonsyn et al. (1995) |
|
|
|
Dried vegetables |
|
|
|
|
|
|
|
Uruguay |
Dried vegetables |
1993–95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996), A, Pineiro & Giribone (1994)c |
|
|
|
Oil and oilseeds |
|
|
|
|
|
|
|
Germany |
Sunflower seed |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Sesame seed |
1995–98 |
1 |
0 |
0 |
|||
|
Germany |
Linseed |
1995–98 |
1 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Poppy seed |
1995–98 |
NA |
0 |
0 |
|||
|
Edible oils |
1995–98 30 |
0 |
0 |
|
|||
|
|
Meat |
|
|
|
|
|
|
|
Uruguay |
Meat products |
1993–95 |
0.00 |
0 |
0 |
P,S, Pineiro et al. (1996); A, Pineiro & Giribone (1994)c |
|
|
Denmark |
Pig kidney |
1999 |
1.5b |
5 |
0 |
P,S,A, Petersen (2000) |
|
|
Pig meat |
1999 |
0.3b |
0 |
0 |
|||
|
Pork |
1993–94 |
NR |
0 |
0 |
P,S, Jorgensen (1998); A, Jorgensen et al. (1996) |
'Random samples' from slaughterhouses |
|
|
Pork, organic |
1993–94 |
NR |
0 |
0 |
|||
|
Duck |
1993–94 |
NR |
0 |
0 |
|||
|
Duck liver |
1993–94 |
NR |
0 |
0 |
|||
|
Goose |
1993–94 |
NR |
0 |
0 |
|||
|
Goose liver |
1993–94 |
NR |
0 |
0 |
|||
|
Turkey |
1993–94 |
NR |
0 |
0 |
|||
|
Turkey liver |
1993–94 |
NR |
0 |
0 |
|||
|
Chicken |
1993–94 |
NR |
0 |
0 |
|||
|
United Kingdom |
Pork liver |
1996 |
0.02 |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
Pork salami |
1996 |
NA |
0 |
0 |
|||
|
Germany |
Raw sausage |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Sausage |
1995–98 |
0.00 |
0 |
0 |
|||
|
Germany |
Sausage |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Liver sausage |
1995–98 |
0.00 |
0 |
0 |
|||
|
Blood sausage |
1995–98 |
0.00 |
0 |
0 |
|||
|
Other meat products |
1995–98 |
0.00 |
0 |
0 |
|||
|
Beef sausage |
1995–98 |
0.00 |
0 |
0 |
|||
|
Poultry sausage |
1995–98 |
0.00 |
0 |
0 |
|||
|
Beaf meat |
1995–98 |
0.00 |
0 |
0 |
|||
|
Pig meat |
1995–98 |
0.00 |
0 |
0 |
|||
|
Poultry meat |
1995–98 |
NA |
0 |
0 |
|||
|
Pig kidney |
1995–98 |
0.00 |
3 |
0 |
|||
|
Pig liver |
1995–98 |
0.00 |
0 |
0 |
|||
|
France |
Pig kidney |
1997 |
< 0.5 |
0 |
0 |
P,S,A, Dragacci et al. (1999) |
|
|
Pig kidney |
1998 |
NA |
0 |
0 |
|||
|
Nephropathic pig kidneys |
1997 |
NA |
0 |
0 |
|||
|
|
Snacks |
|
|
|
|
|
|
|
Germany |
Bar |
1995–98 |
0.097 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Bar with nuts |
1995–98 |
0.02 |
0 |
0 |
|||
|
Muesli bar |
1995–98 |
0.01 |
0 |
0 |
|||
|
Muesli |
1995–98 |
0.03 |
1 |
1 |
|||
|
Breakfast cereals |
1995–98 |
0.02 |
0 |
0 |
|||
|
Corn flakes |
1995–98 |
0.01 |
0 |
0 |
|||
|
Biscuit |
1995–98 |
0.02 |
0 |
0 |
|||
|
Biscuit with chocolate |
1995–98 |
0.02 |
0 |
0 |
|||
|
Germany |
Rusk |
1995–98 |
0.03 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Rye biscuit |
1995–98 |
0.01 |
0 |
0 |
|||
|
Chips, popcorn |
1995–98 |
0.01 |
0 |
0 |
|||
|
|
Nuts |
|
|
|
|
|
|
|
Netherlands |
Roasted peanuts |
1996 |
0.00' |
0 |
0 |
P,S,A, Inspectorate for Health Protection (personal communication, 1999) |
|
|
Peanuts products |
1996 |
0.00 |
0 |
0 |
|||
|
Pistachio nuts |
1996 |
0.00 |
0 |
0 |
|||
|
Germany |
Hazelnuts |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Groundnuts |
1995–98 |
0.00 |
0 |
0 |
|||
|
Other nuts |
1995–98 |
0.024 |
0 |
0 |
|||
|
|
Beer |
|
|
|
|
|
|
|
Canada |
Beer |
NR |
NA |
0 |
0 |
P,S,A, Scott & Kanhere (1995) |
|
|
Denmark |
Beer |
1993–94 |
NR |
0 |
0 |
P,S,A, Jorgensen (1998) |
'Random samples' from retail shops |
|
United Kingdom |
Beer |
1996 |
NA |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
|
Spain |
Beer |
1997 |
NR |
0 |
0 |
P,A, Burdaspal & Legarda (1998a); S, NR |
|
|
Europe |
Beer |
1997 |
NR |
0 |
0 |
||
|
Italy |
Beer, imported |
1999 |
0.05 |
0 |
0 |
P,S,A, Visconti et al. (2000b) |
|
|
Japan |
Beer |
1998 |
NR |
0 |
0 |
P and A, Nakajima et al. (1999); S, NR |
|
|
Beer, imported |
1998 |
NR |
0 |
0 |
|||
|
Germany |
Pils beer |
1995–98 |
0.058 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Export beer |
1995–98 |
0.059 |
0 |
0 |
|||
|
Wheat beer |
1995–98 |
0.041 |
0 |
0 |
|||
|
Strong beer |
1995–98 |
0.082 |
0 |
0 |
|||
|
Beer, alcohol-free |
1995–98 |
0.030 |
0 |
0 |
|||
|
Light beer |
1995–98 |
0.044 |
0 |
0 |
|||
|
Malt beer |
1995–98 |
0.033 |
0 |
0 |
|||
|
|
Teas |
|
|
|
|
|
|
|
Germany |
Black tea |
1995–98 |
NA |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Green tea |
1995–98 |
NA |
0 |
0 |
|||
|
Fruit tea |
1995–98 |
NA |
0 |
0 |
|||
|
|
Wine |
|
|
|
|
|
|
|
Netherlands |
Red wine |
1999 |
1 |
0 |
0 |
P,S,A, Inspectorate for Health Protection and van Egmond (personal communication, 1999) |
|
|
White wine |
1999 |
0.00 |
0 |
0 |
P,S,A, van Egmond (personal communication, 1999) |
|
|
|
Sweden |
Wine |
1998–99 |
0.45 |
0 |
0 |
P,S, A, National Food Administration |
|
|
United Kingdom |
Red wine |
1998 |
0.22 |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
1996 |
1.10 |
0 |
0 |
P,S, MAFF (1997); A, Sharman et al. (1992) |
|
||
|
White wine |
1996 |
NA |
0 |
0 |
|||
|
Switzerland |
Wine |
NR |
NR |
0 |
0 |
P,A, Zimmerli & Dick (1995); S, NR |
|
|
Japan |
Wine |
1996 |
NR |
0 |
0 |
P,A, Ueno (1998), S, NR |
|
|
Italy |
Red wine |
1992–94 |
0.91 |
0 |
0 |
P, Pietri (2000); A, Zimmerli & Dick (1995), S, NR |
|
|
Passito |
1990–94 |
0.028 |
0 |
0 |
|||
|
Red wine |
1995 |
2.34 |
0 |
0 |
|||
|
Passito |
1995 |
3.47 |
0 |
0 |
|||
|
Red wine |
1996 |
1.55 |
0 |
0 |
|||
|
Passito |
1996 |
0.01 |
0 |
0 |
|||
|
Passito |
1997 |
3.07 |
0 |
0 |
|||
|
Red wine |
1997 |
1.41 |
0 |
0 |
|||
|
Red wine |
1998 |
0.09 |
0 |
0 |
|||
|
Red wine |
1999 |
7.12 |
18 |
0 |
P, Pietri (2001); A, Visconti et al. (1999);S, NR |
|
|
|
White wine |
1999 |
2.03 |
1 |
0 |
|||
|
Rosé wine |
1999 |
0.27 |
0 |
0 |
|||
|
Italy |
Red wine |
1997–98 |
2.28 |
1 |
0 |
P,S,A, Visconti et al. (1999) |
|
|
Rosé wine |
1997–98 |
1.09 |
0 |
0 |
|||
|
White wine |
1997–98 |
0.43 |
0 |
0 |
|||
|
North Italy |
Red wine |
1997–99 |
NR |
0 |
0 |
P,A, Ottender & Majerus (2000); S, NR |
|
|
South Italy |
Red wine |
1997–99 |
NR |
0 |
0 |
||
|
Red wine |
1997–99 |
NR |
0 |
0 |
|||
|
North France |
Red wine |
1997–99 |
NR |
0 |
0 |
||
|
South France |
Red wine |
1997–99 |
NR |
0 |
0 |
|
|
|
Germany |
White wine |
1995–98 |
0.00 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Rosè wine |
1995–98 |
0.00 |
0 |
0 |
|||
|
Red wine |
1995–98 |
1 |
1 |
0 |
|||
|
FSIS 185 red wine |
1998 |
NA |
0 |
0 |
|||
|
FSIS 185 red wine |
1997 |
NA |
0 |
0 |
|||
|
North Germany |
Red wine |
1997–99 |
NR |
0 |
0 |
P,A, Ottender & Majerus (2000); S, NR |
|
|
White wine |
1997–99 |
NR |
0 |
0 |
|||
|
South Germany |
White wine |
1997–99 |
NR |
0 |
0 |
||
|
Germany |
Red wine |
|
NR |
0 |
0 |
P,A, Lehtonen (1999); S, NR |
|
|
Red wine |
|
NR |
0 |
0 |
|||
|
White wine |
|
NR |
0 |
0 |
|||
|
France |
Wine |
1998 |
0.01 |
0 |
0 |
P,A, Ospital et al. (1998); S, NR |
|
|
World |
Red wine |
1997–99 |
NR |
0 |
0 |
P,A, Ottender & Majerus (2000); S, NR |
|
|
Rosé wine |
1997–99 |
NR |
0 |
0 |
|||
|
White wine |
1997–99 |
NR |
0 |
0 |
|||
|
World |
White wine |
NR |
NA |
0 |
0 |
P,S,A, Majerus & Ottender (1996) |
|
|
Rosé wine |
NR |
NA |
0 |
0 |
|||
|
Red wine |
NR |
NA |
|
0 |
|||
|
Europe |
Wine |
1998 |
0.02 |
0 |
0 |
P, www.elintarvikevirastofi/; A, S, NR |
|
|
Europe |
Red wine |
1997 |
NR |
0 |
0 |
P, Burdaspal & Legarda (1999);A, Zimmerli & Dick (1996);S, NR |
|
|
Rosé wine |
1997 |
NR |
0 |
0 |
|||
|
White wine |
1997 |
NR |
0 |
0 |
|||
|
Aperitif wine |
1997 |
NR |
0 |
0 |
|||
|
Sparkling wine |
1997 |
NR |
0 |
0 |
|||
|
Dessert wine |
1997 |
NR |
0 |
0 |
|||
|
Europe |
Red wine |
1994–95 |
NR |
0 |
0 |
P,A, Zimmerli & Dick (1996);S, NR |
|
|
Rosé wine |
1994–95 |
NR |
0 |
0 |
|||
|
White wine |
1994–95 |
NR |
0 |
0 |
|||
|
Special wines |
1994–95 |
NR |
0 |
0 |
|||
|
|
Vinegar and mustard |
|
|
|
|
|
|
|
Germany |
Apple and fruit vinegar |
1995–98 |
< 0.01 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Wine vinegar |
1995–98 |
0.00 |
0 |
0 |
|||
|
Balsam vinegar |
1995–98 |
3 |
0 |
0 |
|||
|
Mustard |
1995–98 |
0.00 |
0 |
0 |
|||
|
|
Grape juice |
|
|
|
|
|
|
|
United Kingdom |
Grape juices |
1998 |
0'02 |
0 |
0 |
P,S,A, MAFF (1999b) |
|
|
Germany |
White grape juice |
1995–98 |
1 |
0 |
0 |
P,S,A, Wolff et al. |
|
|
Red grape juice |
1995–98 |
3 |
2 |
0 |
(2000) |
|
|
|
FSIS 185 white grape juice |
1998 |
NA |
0 |
0 |
|
|
|
|
FSIS 185 red grape juice |
1998 |
NA |
0 |
0 |
|
|
|
|
FSIS 185 grape juice |
1998 |
1.73 |
0 |
0 |
|
|
|
|
Europe |
Grape juice |
1994–95 |
0.30 |
0 |
0 |
P,A, Zimmerli & Dick (1996); S, NR |
|
|
Japan |
Grape juice |
1996 |
NR |
0 |
0 |
P,A, Ueno (1998); S, NR |
|
|
World |
Grape juice |
NR |
NA |
0 |
0 |
P,S,A, Majerus & Ottender (1996) |
|
|
Other juices |
|
|
|
|
|||
|
Germany |
Apple juice |
1995–98 |
NA |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Orange juice |
1995–98 |
NA |
0 |
0 |
|||
|
Blackcurrant juice |
1995–98 |
0.048 |
0 |
0 |
|||
|
Tomato juice |
1995–98 |
000 |
0 |
0 |
|||
|
Carrot juice |
1995–98 |
< 0.01 |
0 |
0 |
|||
|
Other vegetable juice |
1995–98 |
NA |
0 |
0 |
|||
|
|
Seasonings |
|
|
|
|
|
|
|
Germany |
Ketchup |
1995–98 |
1 |
0 |
0 |
P,S,A, Wolff et al. (2000) |
|
|
Herb sauce |
1995–98 |
0 |
0 |
0 |
|||
|
Pepper sauce |
1995–98 |
0 |
0 |
0 |
|||
|
|
Fermented beverages |
|
|
|
|
|
|
|
Japan |
Fermented beverages |
1996 |
0.00 |
0 |
0 |
P,A, Ueno (1998); S, NR |
|
|
|
Baby food |
|
|
|
|
|
|
|
Canada |
Baby food |
1998–99 |
0.00 |
0 |
0 |
P, Canada (2000); S,A, NR |
|
|
|
Diet |
|
|
|
|
|
|
|
United Kingdom |
Normal diet |
NR |
0.03 |
0 |
0 |
P,S,A, MAFF (1999c) |
|
|
Vegetarian diet |
NR |
0.09 |
0 |
0 |
|||
|
Traditional diet |
NR |
0.05 |
0 |
0 |
|||
|
|
Dust |
|
|
|
|
|
|
|
USA |
Dust |
2000 |
816 |
0 |
3 |
P,A, Richard et al. (1999); S, NR |
|
NA, not analysed; NR, not reported; MAFF, Ministry of Agriculture, Fisheries and Food (United Kingdom); USDA, Department of Agriculture of the USA; GIPSA, Grain Inspection, Packers and Stockyards Administration (United Kingdom)
a
Limit of detectionb
Sampling not describedc
LOQ > 5 µg/kg; not used in calculation of weighted meand
The number of samples was divided by a factor of 3 for calculation of the weighted mean.
See Also:
Toxicological Abbreviations
Ochratoxin A (WHO Food Additives Series 28)
OCHRATOXIN A (JECFA Evaluation)
Ochratoxin A (IARC Summary & Evaluation, Volume 56, 1993)