
First draft prepared by Mike Bolger1, Raymond D. Coker2, Michael DiNovi1, David Gaylor3, Wentzel Gelderblom4, Monica Olsen5, Nachman Paster6, Ronald T. Riley7, Gordon Shephard4 and Gerrit J.A. Speijers8
1 Food and Drug Administration, Washington DC, USA
2
University of Greenwich, Kent, United Kingdom3
Science International, Little Rock, Arkansas, USA4
Medical Research Council, Programme on Mycotoxins and Experimental Carcinogenesis, Tygerberg, South Africa5
National Food Administration, Uppsala, Sweden6
Department of Stored Products, The Vocani Centre, Bet-Dagan, Israel7
Department of Agriculture, Athens, Georgia, USA8
National Institute of Public Health and the Environment, Bilthoven, The NetherlandsThe Committee evaluated fumonisins B1, B2, and B3 at the request of the Codex Committee on Food Additives and Contaminants; these toxins had not been evaluated previously by the Committee. In 2000, a monograph on fumonisin B1 was published (WHO, 2000a), which provided much of the information used in this evaluation.
Fumonisins are fungi produced by fungi of the genus Fusarium. The only species that produce significant quantities of fumonisins are Fusarium verticillioides (Sacc.) Nirenberg (= F. moniliforme (Sheldon)) and the related F. proliferatum (Matsushima) Nirenberg. At least 10 other Fusarium species also produce these toxins. F. verticillioides and F. proliferatum are among the most common fungi associated with maize, the most frequently contaminated food, and can be recovered from both damaged and undamaged maize kernels. These species cause Fusarium kernel rot of maize, an important disease in hot climates. A strong relationship also exists between insect damage and Fusarium kernel rot due to other Fusarium species, such as F. graminearum. Temperature stress may also play a role, especially in cultivars grown outside their area of adaptation. As F. verticillioides and F. proliferatum grow over a wide range of temperatures but only at relatively high water activities (above about 0.9), fumonisins are formed in maize only before harvest or during the early stage of drying. Except under extreme conditions, the concentrations of fumonisins do not increase during grain storage. Formation of fumonisins in the field correlates with the occurrence of F. verticillioides and F. proliferatum, which predominate during late maturity. Fumonisins are widely distributed geographically, and their natural occurrence in maize has been reported in many areas of the world. Of particular concern are the high concentrations found in maize produced and consumed by particular subpopulations, such as subsistence farmers. Considerable annual variations in contamination have been noted. Fumonisins occur infrequently in other foods, such as sorghum, asparagus, rice, beer, and mung beans.
Fumonisins are a group of structurally related compounds. Fumonisin B1 is the diester of propane-1,2,3-tricarboxylic acid and 2S-amino-12S,16R-dimethyl-3S,5R,10R,14S,15R-pentahydroxyeicosane in which the C-14 and C-15 hydroxy groups are esterified with the terminal carboxy group of propane-1,2,3-tricarboxylic acid. Fumonisin B2 is the C-10 deoxy analogue of fumonisin B1 in which the corresponding stereogenic units on the eicosane backbone have the same configuration. The full stereochemistry of fumonisin B3 and B4 is unknown, but the amino terminal of fumonisin B3 has the same absolute configuration as that of fumonisin B1.
As most biological data were available on fumonisin B1, and maize is the major source of intake, the Committee focused its evaluation on toxicological studies of fumonisin B1 and on studies of intake of contaminated maize and maize products. In many studies, culture materials and naturally contaminated maize were used, which can contain several other fumonisins, primarily fumonisins B2 and B3. The toxicological profiles of these toxins are very similar to that of fumonisin B1. Various chemical derivatives of fumonisins have been tested in a number of biological systems to gain insight into structure–activity relationships. Briefly, the fumonisins of the B series that have been investigated are more toxic in vivo than their hydrolysed or N-acetylated counterparts. The free amino group appears to play a specific role in the biological activity of fumonisin B1.
A thorough review of the biochemical aspects of fumonisin B1 is contained in Fumonisin B1 (Environmental Health Criteria 219), published by WHO (2000a), and much of what follows is derived from that review, with relevant new publications. With a few exceptions, only references that were not included in that monograph, recent or critical reviews, or studies with stated doses are cited here.
In many studies on fumonisins in animals in vivo, culture materials or naturally contaminated maize were used in which not only fumonisin B1 but also other fumonisins (primarily fumonisin B2 and B3) can be found. Various chemical derivatives of fumonisins have been studied in several biological systems to gain insight into the structural requirements of fumonisin-induced toxicity and biochemical mechanisms in vivo (see WHO, 2000a, pp. 79–80 for additional details). Briefly, the fumonisins of the B series that have been investigated are more toxic in vivo than their hydrolysed or N-acetylated counterparts. The free amino group has an important role, as the N-acetyl derivatives are less toxic in primary hepatocytes and hydrolysed fumonisin B1 is more toxic. The lesser toxicity of hydrolysed fumonisin B1 in rat liver is not due to reduced absorption. The fungus previously known as F. moniliforme Sheldon is referred to in this monograph as F. verticillioides (Sacc.) Nirenberg.
In rats and most other animals, the kinetics of absorption of fumonisin B1 indicates rapid distribution and elimination that is adequately described by a two- or three-compartment model (most recently, Martinez-Larranaga et al., 1999). Little fumonisin B1 was detected in plasma and tissues after oral administration, indicating that absorption is negligible ( 4% of the dose). Fumonisin B2 may be less bioavailable than fumonisin B1, and proportionally less fumonisin B2 is excreted in bile (Shephard & Snijman, 1999). In rats treated by gavage, more hydrolysed [14C]fumonisin B1 was excreted in urine than [14C]fumonisin B1 or the [14C]fumonisin B1–fructose adduct. For example, in female Fischer 344NHsd rats, 17% of a dose of hydrolysed [14C]fumonisin B1 and 0.7% of a dose of [14C]fumonisin B1 was recovered in urine after administration by gavage. The authors concluded that hydrolysed fumonisin B1 was better absorbed than fumonisin B1, although the biliary excretion of the two fumonisins was similar (Dantzer et al., 1999). While fumonisin B1 was distributed to most tissues, the liver and kidney retained most of the absorbed material, the liver retaining more toxin than the kidney in some studies and the inverse in others. In a study in male Wistar rats given pure fumonisin B1 by gavage, kidney contained > 10 times as much fumonisin B1 as liver (Martinez-Larranaga et al., 1999). This study confirmed previous reports that fumonisin B1 persists in rat liver and kidney much longer than in plasma. It was estimated that, after administration to pigs of fumonisin B1 in the diet at 2–3 mg/kg bw, a withdrawal period of at least 2 weeks would be required to eliminate the toxin from liver and kidney. The material retained in liver and kidney is primarily unmetabolized fumonisin B1, as shown in several studies of the persistence in kidney of free sphinganine (a biomarker for fumonisin). Thus, while fumonisin B1 is eliminated rapidly, the concentration of the biomarker in rat kidney (but not liver) is more persistent (most recently, Enongene et al., 2000; Garren et al., 2000). Similar kinetics was seen in the livers of non-human primates given a single dose of pure fumonisin B1 at 10 mg/kg bw by gavage, which increased the serum concentrations of sphingoid bases, cholesterol, and enzymes indicative of liver function, and these remained elevated for several weeks after dosing (van der Westhuizen et al., 2001).
In pregnant rats, low concentrations of fumonisin B1 were recovered in uteri (0.24–0.44%), individual placentae (0–0.4%), and fetuses ( 0.015%), indicating the absence of placental transfer of fumonisin B1. Similar results were reported in rabbits. There is also little evidence of significant transfer during lactation. For example, no fumonisin B1 was detected in the milk of lactating sows fed diets containing non-lethal concentrations of fumonisin B1, and there was no evidence of toxicosis in their suckling piglets. In a study in which lactating cows were given fumonisin B1 intravenously, the carry-over rate into the milk reached a maximum of 0.11%. In other studies, no fumonisins were detected in cows’ milk, and fumonisin B1 was found in only 1 of 165 samples of milk in the USA at a concentration of 5 ng/ml. The finding that little fumonisin B1 is retained in tissues, milk, or eggs has led to the conclusion that fumonisin residues in food products derived from animals are insufficient to make them injurious to consumers.
After intraperitoneally or intravenously administered fumonisin B1 has been distributed, its initial elimination is rapid, with no evidence of metabolism. In vervet monkeys (Cercopithecus aethiops) treated intraperitoneally with fumonisin B2, elimination was rapid and followed a bi-exponential pattern (half-time, 18 min), similar to that of the elimination of fumonisin B1 (half-time, 40 min). The elimination kinetics in non-human primates after oral dosing has not been determined, but peak plasma concentrations of fumonisin B1 and B2 occurred 1 to several hours after a dose of 7.5 mg/kg bw by gavage, and the plasma fumonisin concentrations ranged from < 20 ng/ml to nearly 210 ng/ml. Thus, the elimination kinetics after oral dosing is not easily described, unlike that of intraperitoneal or intravenous dosing. Furthermore, an oral dose of fumonisin cannot be fully accounted for (Shephard & Snijman, 1999). As the rate of elimination of fumonisin B1 is a function of body weight, elimination is rapid in mice but would be much longer in humans (Delongchamp & Young, 2001).
There is little or no evidence that fumonisins are metabolized in vitro or in vivo in animals, even though they are clearly excreted in bile. A study in which primary hepatocytes were exposed to [14C]fumonisin B1 showed that the toxin is associated with both the soluble and the insoluble membrane compartments of the cell, and no metabolites were detected after a 44-h incubation. Incubation with rat liver microsomal preparations also showed no metabolism by cytochrome P450 (CYP), microsomal esterase, or any other microsomal enzyme. Incubation with a triglyceride hepatic endothelial lipase or a porcine pancreatic lipase also did not result in hydrolysis of the tricarboxylic acid moieties of fumonisin B1.
Several studies in which different routes of exposure and different animal species were used showed that fumonisins are excreted primarily in the faeces, either unchanged or with loss of one or both of the tricarboxylic acid side-chains. The material excreted in bile is still biologically active, since fumonisin B1 given subcutaneously to mice rapidly entered the small intestine, where it inhibited ceramide synthase (Enongene et al., 2000). Loss of the tricarboxylic acid side-chains probably occurs in the gut, since after partial hydrolysis resulting in removal of only one of the two side-chains and full hydrolysis fumonisin B1 is recovered in the faeces but not in the bile. This finding was confirmed in a study in vervet monkeys (Shephard & Snijman, 1999). Most of the hydrolysed fumonisin B2 was present as a mixture of the two possible partially hydrolysed forms, while fully hydrolysed fumonisin B2 was a minor constituent. No hydrolysis products were found in urine, confirming that fumonisin was hydrolysed in the gut, probably by microbial degradation. While there is no evidence that fumonisin is metabolized by CYP enzymes, some studies have shown that fumonisins can alter their activity, and this observation was confirmed in vitro and in vivo (Merrill et al., 1999a; Spotti et al., 2000). In some studies, the effects on CYP activity have been shown to be the result of fumonisin-induced alterations in sphingolipid metabolism (Merrill et al., 1999b). For example, in HepG2 cells, fumonisin B1 inhibited the induction of CYP 1A1 (which metabolizes aryl hydrocarbons such as methylcholanthrene) (Merrill et al., 1999a). In rats given fumonisin B1 by gavage at 2 mg/kg bw, there was inhibition of CYP 2C11 and to a lesser extent of CYP 1A2 (Spotti et al., 2000). The inhibition of CYP 2C11 was attributed to suppression of protein kinase activity due to inhibition of sphingolipid biosynthesis. Sphingosine, a sphingolipid that accumulates in animals exposed to fumonisin B1, was also shown to inhibit CYP 2C11 in rat hepatocytes (Merrill et al., 1999a). Feeding rainbow trout a diet containing fumonisin B1 at a concentration of 104 mg/kg had no effect on acetylated fumonisin B1–DNA adduct formation (Carlson et al., 2001).
While there is little evidence that absorbed fumonisins are metabolized in animals, removal of the tricarboxylic acid side-chains (producing hydrolysed fumonisin B1) converts this inhibitor of ceramide synthase into a substrate for the enzyme. The product of this reaction, N-palmitoyl-hydrolysed fumonisin B1, also inhibits ceramide synthase in vitro. It is not known whether this product is formed in vivo, but it is more toxic than fumonisin B1 or hydrolysed fumonisin B1 for HT-29 cells (Merrill et al., 2001). Since hydrolysed fumonisin B1 and hydrolysed fumonisin B2 are major breakdown products in nixtamalized maize products and are also produced in the gut from fumonisin B1 and B2, the toxicity of the hydrolysed toxins should be addressed.
(a) Biochemical modes of action
Several biochemical reactions have been proposed to explain all or some of the toxic effects of fumonisins in animals. Two of them invoke disruption of lipid metabolism as the initial site of action, and they are similar in other respects (Gelderblom et al., 2001a; Merrill et al., 2001; Riley et al., 2001). Both of these hypothesized mechanisms are supported by data obtained in in vivo (Table 1) and in vitro (Table 2), in short-term and long-term studies in rodents (National Toxicology Program, 1999; Delongchamp & Young, 2001; Gelderblom et al., 2001a; Riley et al., 2001; Voss et al., 2001), long-term studies of carcinogenicity in trout (Carlson et al., 2001), and short-term studies of toxicity in other animals (WHO, 2000a). The first proposed lipid-based mechanism involves inhibition of ceramide synthase, a key enzyme in the biosynthesis of sphingolipids (reviewed extensively in WHO, 2000a). The second mechanism involves changes in the polyunsaturated fatty acid and phospholipid pools. Both lead ultimately to lipid-mediated alterations in signalling and metabolic pathways crucial to cell growth, death, and differentiation. Several studies in vitro indicate that fumonisin-induced changes in key enzymes involved in cell cycle regulation, differentiation, and/or apoptosis are initial or secondary triggers (most recently, Pinelli et al., 1999; Mobio et al., 2000a; Table 2).
Table 1. Selected biochemical mechanisms for the toxicity of fumonisin B1 in animal models in which the proposed biochemical action has been shown to be related to specific effects
|
Description |
Model |
Action |
Biochemical effects [lowest oral dose that caused an effector to change] |
Correlated adverse effects and associated molecular events |
|
Altered lipid metabolism |
|
|
|
|
|
Disruption of sphingolipid metabolism |
Rat/L,K, H,S,U |
Ceramide synthase inhibition |
Increased sphingoid bases and decreased complex sphingolipids [kidney, 1 mg/kg FB1, equivalent to 0.1 mg/kg bw per day (Wang et al., 1999)] |
Increased apoptotic and oncotic necrosis in liver and kidney, mitogenesis, decreased heart weight, kidney tumours, liver tumour promotion |
|
|
Mouse/L,K,GI,S |
|
Increased sphingoid bases and decreased complex sphingolipids [kidney, 0.3 mg/kg bw per day (Enongene et al., 2001)] |
Increased apoptotic and oncotic necrosis in liver and kidney, altered TNFalpha expression, liver tumours |
|
|
Rabbit/L,K, S,U |
|
Increased sphingoid bases [kidney, 0.1 mg/kg bw per day (LaBorde et al., 1997)] |
Nephrotoxicity |
|
|
Pig/L, K, Ln, H, S |
|
Increased sphingoid bases and decreased complex sphingolipids [serum, 5 mg/kg FB1, equivalent to 0.2 mg/kg bw per day (Riley et al., 1993)] |
Hepatotoxicity, cardiovascular toxicity, pulmonary oedema syndrome |
|
|
Monkey/L,K,S,U |
|
Increased sphingoid bases [serum, 1.0 mg/kg bw per day (van der Westhuizen et al., 2001)] |
Hepatotoxicity, nephrotoxicity |
|
|
Horse/L, K, S |
Ceramide synthase inhibition |
Increased sphingoid bases and decreased complex sphingolipids [serum, 22 mg/kg total fumonisins, equivalent to 0.44 mg/kg bw per day (Wang et al, 1992)] |
Hepatotoxicity, cardiovascular toxicity, leukoencephalo-malacia |
|
|
Trout, L, K, S |
|
Increased sphingoid bases and decreased complex sphingolipids [liver, 25 mg/kg FB1, equivalent to 3.75 mg/kg bw per day on basis of study in mice (Carlson et al., 2001)] |
Promotion of liver tumours induced by direct or indirect carcinogens |
|
Disruption of fatty acid and phospholipid metabolism |
Rat/L, S |
Impairment of N-6 fatty acid metabolism, phospho-lipid metabolism, and ceramide synthase inhibition |
Alterations in absolute and relative amounts of phosphatidylcholine and ethanolamine and in degree of saturation of fatty acids in phosphatidylcholine and ethanolamine in microsomal, mitochondrial, plasma, nuclear cell membranes and membranes associated with hepatic nodules; in particular, relative and absolute amounts of fatty acid products of N-6 and N-3 pathway. Also, increased free sphingoid bases and decreased sphingomyelin. [Liver, 10 mg/kg FB1, equivalent to 0.5 mg/kg bw per day (Gelderblom et al., 1997)] |
Increased lipid peroxidation, mitoinhibition, hepatotoxicity, growth of hepatocyte nodules, increased expression of hepatocyte growth factor and tumour growth factor-alpha, c-myc, alterations in retinoblastoma pathway, deregulation of cell cycle control by overexpression of cyclin D1, liver tumour promotion and hepatocarcinogenicity |
|
Increased oxidative stress |
Rat/L, Sl |
Lipid oxidation |
Increases superoxide radicals, increased lipid radicals [Liver, 16 mg/kg bw per day Lemmer et al., 1999a)] |
Increased iron-induced lipid peroxidation, oxidative DNA damage, protection from toxicity by antioxidants |
Abbreviations: FB1, fumonisin B1; L, liver; K, kidney; Ln, lung; H, heart; GI, digestive epithelia; M, muscle; Sl, spleen; S, serum; U, urine; TNF, tumour necrosis factor
Table 2. Selected biochemical mechanisms for the toxicity of fumonisin B1 in vitro in models in which the proposed biochemical action has been shown to be related to specific molecular events or physiological or toxic effects
|
Description |
Model |
Action |
Biochemical effectors |
Molecular targets |
Correlated adverse effect |
|
Altered lipid metabolism |
|||||
|
Disruption of sphingolipid metabolism |
Microsomes: Rat liver and mouse cerebellar neurons |
Ceramide synthase inhibition |
Competitive inhibition with fatty acids or sphingoid bases as substrates [IC50 0.075 µmol/L FB1 (Merrill et al., 1993)] |
NA |
NA |
|
|
Primary cultures: Rat hepatocytes, liver and kidney slices, hippocampal neurons, cerebellar Purkinje cells, fetal glial cells, sympathetic neurons; mouse neuronal cells and spinal cord cultures; chick embryos; pig endothelial cells; human keratinocytes |
|
Decreased biosynthesis of N-acetylated sphingoid bases (ceramides), increased sphingoid bases, increased sphingoid base 1-phosphates, |
Altered amounts of sphingolipid and glycerophospholipid second messengers; decreased expression of glycosphingolipid receptors. |
In hepatocytes, cytotoxicity and mitoinhibition reported but only at concentrations (> 75 µmol/L) far in excess of those that cause maximal inhibition of ceramide synthase (~1 µmol/L). In other primary cultures, sphingolipid-dependent growth inhibition, apoptosis, and functional effects have been found; e.g. sphinganine-dependent apoptosis, glycosphingolipid-dependent, growth factor-stimulated axonal growth, cytokine-induced adhesion molecule expression and bacterial toxin binding. |
|
|
Cell lines: Pig renal; mouse fibroblast, macrophage, and melanoma; hamster ovary; monkey kidney; human colon |
Ceramide synthase inhibition |
Increased sphingoid bases, decreased complex sphingolipids, and/or other specific glycosphingolipids, increased phosphatidylethanolamine, sphingoid-base-1-phosphates [1 µmol/L FB1; 10 µmol/L hydrolysed FB1 (Schmetz et al., 1998)]. |
Altered amounts of sphingolipid second messengers, activity of protein kinases, expression of cell cycle proteins, adhesion molecules (ICAM-1, integrins), and bacterial toxin and vitamin receptors |
Increased sphinganine-dependent apoptotic and/or oncotic necrosis, and/or altered proliferation. Decreased glycosphingolipid-dependent cell adhesion, growth, altered cell morphology, or differentiation, and altered vitamin transport |
|
Disruption of fatty acid and phospholipid metabolism |
Primary cultures: Rat hepatocytes |
Impaired N-6 fatty acid metabolism, phospholipid metabolism, and ceramide synthase inhibition |
Decreased biosynthesis of neutral lipids, triglycerides, and cholesterol; increased phosphatidylcholine and ethanolamine, decreased sphingomyelin, increased free sphinganine. Altered fatty acid saturation profiles in various lipid pools, in particular accumultion of C18: 2omega6 and C20:4omega6 [other than increase in free sphinganine: 150 µmol/L FB, (Gelderblom et al., 1996a)] |
Altered amounts of lipids required for maintaining membrane fluidity and as substrates for signalling pathways that regulate the epidermal growth factor-induced mitogenic response in hepatocytes. In particular, disruption of prostaglandin-mediated responses |
Increased cytotoxicity and inhibition of epidermal growth factor-induced mitogenesis. Altered prostaglandin and arachidonic acid-induced cytotoxicity. |
|
Oxidative stress |
Artificial membranes |
Altered redox state |
Increased membrane disorder, increased oxygen transport, increased free radicals [250 µmol/L FB1 (Yin et al., 1998)] |
Decreased membrane integrity |
Increased membrane permeability |
|
|
Isolated organelles: |
|
Lipid radicals |
Decreased membrane integrity |
Increased lipid peroxidation |
|
|
Primary cultures: |
|
Superoxide radicals and hydrogen peroxide, lipid radicals [5 µmol/L FB1 (Lee & Lee, submitted)] |
Increased oxidative damage to macromolecules and membrane lipids |
Increased DNA fragmentation, lipid peroxidation, cytotoxicity, and protection by antioxidants |
|
|
Cell lines: Rat brain glioma; pig renal; monkey kidney |
|
Oxidants [0.14 µmol/L FB1 (Abado-Becongnee et al., 1998)] |
Increased oxidative damage to macromolecules, glutathione depletion |
Increased DNA fragmentation, hypermethylation of DNA, lipid peroxidation, cytotoxicity, cell growth, protection by antioxidants |
|
Other mechanisms |
Isolated enzymes: |
Protein phosphatase inhibition |
Direct inhibition [80 µmol/L FB1 (Fukuda et al., 1996)] |
Dephosphorylation of proteins |
None |
|
|
Isolated organelles: |
Altered GTP binding and GTPase activity |
FB1, hydrolysed FB1, and sphingoid bases [IC50 = 75 µmol/L FB1 (Ho et al., 1996)] |
GTP binding proteins |
None |
|
|
Cell lines: Mouse fibroblasts, macrophages; monkey kidney; human epithelium |
Activation or inhibition of specific protein activities |
FB1 or unidentified effector (hypothesized sphingolipid effect) [1 µmol/L FB1 (Huang et al., 1995; Rotter & Oh, 1996)] |
Repressed protein kinase C; stimulated nitric oxide-synthase, mitogen-activated protein kinases, and cytoplasmic phospholipase A2; altered expression of cyclins and cytokine signalling pathways |
None or altered cell cycle progression, altered growth, mitogenesis, or apoptosis |
|
|
Primary cultures: Rat cerebrocortical slices |
Protein kinase C trans-location |
Direct inhibition [1 nmol/L FB1 (Yeung et al., 1996)] |
Activated protein kinase C and increased membrane association |
None |
|
|
Isolated tissues: Frog atrial muscle |
Calcium blockade |
Inhibition of calcium entry [100 µmol/L FB1 (Sauviat et al., 1991)] |
Calcium channels |
Altered muscle contractility |
Abbreviations: FB1, fumonisin B1; IC50, median inhibitory concentration; GTP, guanosine triphosphate; NA, not applicable
(i) Inhibition of ceramide synthase
The structural similarity between sphinganine and fumonisin B1 led to the hypothesis that this mycotoxin acts by disrupting the metabolism or a function of sphingolipids. There is considerable support for the hypothesis that fumonisin-induced disruption of sphingolipid metabolism is an important event in the cascade of events leading to altered cell growth, differentiation, and cell injury observed both in vitro and in vivo (Tables 1–3). A complete review of the literature on this subject is beyond the scope of this monograph, and the interested reader is referred to WHO (2000a) and reviews by Merrill et al. (2001) and Riley et al. (2001)
Table 3. Use of fumonisin B1 (FB1) in research: sphingosine-, ceramide-, and glycosylceramide-mediated processes in vitro are sensitive to fumonisin-inhibited ceramide synthase. Models or processes not summarized previously (WHO, 2000a) are shown in bold.
|
Description |
Model |
Process affected |
Correlated effects andassociated molecular events |
|
Inhibition of ceramide- or glucosylceramide-induced apoptotic or oncotic cell death |
Primary cultures: Hen granulosa; rat pancreas, hippocampal neurons; bovine cerebral endothelial, aortic endothelial |
Inhibition of free fatty acid-induced DNA fragmentation, TNF-alpha-cycloheximide-induced cell death, direct DNA damage-induced apoptosis, glucocerebrosidase-induced apoptosis [0.01 µmol/L FB1 (Xu et al., 1998)] |
Inhibition of apoptotic or oncotic necrosis induced by antineoplastic agents or other therapeutic agents designed to kill cancer cells selectively, treatments designed to introduce lethal double-strand breaks in DNA, glucocere-broside accumulation (Gaucher disease) |
|
|
Cell lines: Human breast cancer, prostate cancer, monocytic leukaemia, promyelocytic, immortalized B cells; mouse fibroblast, haematopoietic, oligo-dendrocyte; pig renal; HaCaT; PC12W cells |
Inhibition of poly(ADP-ribose) polymerase processing, modulation of multidrug resistance, inhibition of interleukin converting enzyme-like proteinase activity, inhibition of cell death induced by tetraphorbolacetate, mitochondrial-derived palmitate, chemical hypoxia, hexadecylphosphocholine, taxol, TNFalpha/PKC-beta, lymphotoxin, angiotensin II) [10 µmol/L FB1 (Charles et al., 2000)] |
|
|
Altered cell cycle progression |
Primary cultures: Human peripheral T cells; frog oocytes |
Inhibition of Fas (CD95)-induced proliferation, induction of oocyte maturation |
Altered cell growth or differentiation |
|
|
Cell lines: Human diploid fibroblasts |
Inhibition of Rb protein dephosphorylation |
|
|
Arachidonic acid release |
Cell lines: Mouse macrophage |
Inhibition of arachadonic acid release |
Inhibition of endotoxin/platelet activating factor-induced arachidonate mobilization |
|
Lipid raft function or processes involving uptake or release of toxins or other chemicals |
Primary cultures: Mouse spinal cord |
Reduced expression of tetanus and cholera toxin receptors [20 µmol/L FB1 (Williamson et al., 1999)] |
Prevention of toxin-induced neurotransmitter release and cell death, folate, Shiga toxin, and saposin transport, toxin-induced cell death, altered protein processing |
|
|
Cell lines: Human colon, epidermoid carcinoma; rat kidney; ScN2a; hamster ovary |
Reduced receptor function or expression (vitamin receptors, lipopolysaccharide |
|
|
Cell matrix and cell–cell adhesion |
Primary cultures: Human keratinocytes |
Modulation of adhesion molecule and antigen expression |
Disruption of cell–substrate and cell–cell contact |
|
|
Cell line: Mouse melanoma; pig kidney |
Inhibition of fibronectin binding ; reduced junctional integrity [1 µmol/L FB1 (Pelagalli et al., 1999)] |
|
|
Other mechanisms |
Primary culture: Rat cortical neurons |
Attenuation of ischaemic tolerance |
Prevention of protective effects of hypoxic preconditioning |
|
|
Cell line: Monkey kidney SV40, transformed |
Increased p21-activated serine/ threonine kinases activity [5 µmol/L FB1 (Bokoch et al., 1998)] |
Unknown |
PKC, phosphatase kinase C; TNF, tumour necrosis factor
Fumonisin B1 strongly inhibited the acylation of sphinganine and sphingosine in all cell lines and all animals, plants, and fungi in which it has been tested. Ceramide synthase recognizes both the amino group (sphingoid-binding domain) and the tricarboxylic acid side-chains (fatty acyl-coenzyme A domain) of fumonisin B1. While removal of the tricarboxylic acid side-chains reduces the ability of fumonisin B1 to inhibit ceramide synthase, N-acetylation completely abolishes the inhibitory activity (Norred et al., 2001). Complete inhibition of ceramide synthase by fumonisins causes a rapid increase in the intracellular concentration of sphinganine and sometimes of sphingosine, both in vivo and in vitro. In vivo there is a close relationship between sphinganine accumulation and the expression of toxicity in liver and kidney (Delongchamp & Young, 2001). Once accumulated, free sphingoid bases can persist in tissues (especially kidney) much longer than fumonisin B1 (Shephard & Snijman, 1999; Enongene et al., 2000). In the urine of rats fed fumonisin B1, > 95% of the free sphinganine was recovered in dead cells. An oral dose of fumonisin B1 insufficient to increase the concentration of free sphinganine (1 mg/kg of diet, equivalent to 0.1 mg/kg bw per day) can prolong the half-life of free sphinganine in urine of rats after they have been taken off diets that contained a dose sufficient to cause free sphinganine (10 mg/kg of diet, equivalent to 1 mg/kg bw per day) to accumulate in urine (Wang et al., 1999). This observation that a sub-threshold dose can prolong the increase in free sphinganine caused by a higher dose has been confirmed in mice treated by oral gavage (Enongene et al., 2001). Fumonisin B1-induced increases in free sphingoid bases and toxicity are both reversible, although elimination of free sphinganine from the liver is faster than from the kidney (Enongene et al., 2000).
A portion of the accumulated sphinganine is metabolized to sphinganine 1-phosphate and then cleaved into a fatty aldehyde and ethanolamine phosphate, both of which can be redirected into other biosynthetic pathways, such as increased biosynthesis of phosphatidylethanolamine. Free sphinganine that is not degraded can be acylated to form C-2 dihydroceramide (Merrill et al., 2001). Disrupted sphingolipid metabolism leads to imbalances in phosphoglycerolipid, fatty acid, and cholesterol metabolism by alterating phosphatidic acid phosphatase and monoacyl-glycerol acyltransferase. Thus, inhibition of ceramide synthase by fumonisin B1 can cause a wide spectrum of changes in lipid metabolism and associated lipid-dependent signalling pathways (reviewed by Merrill et al., 2001).
In short-term studies with mice, rats, and rabbits, disruption of sphingolipid metabolism, as shown by statistically significant increases in the free sphinganine concentration, occurred at doses at or below those that cause liver or kidney lesions (Table 1; for review, see Riley et al., 2001). In a long-term study in which Fischer 344/N Nctr BR rats were fed diets containing pure fumonisin B1, the increase in the ratio of sphinganine:sphingosine in kidney and urine correlated with increased incidences of non-neoplastic and neoplastic lesions in the kidney (National Toxicology Program, 1999). In the livers of female B6C3F1/Nctr BR mice, the concentrations of free sphinganine and the ratio of sphinganine:sphingosine were increased after 3 and 9 weeks on a diet containing fumonisin B1 at 50 or 80 mg/kg, which doses induced liver adenoma and carcinoma (National Toxicology Program, 1999). In rainbow trout, fumonisin B1 was not a complete carcinogen, but there was a close correlation between promotion of hepatocarcinogenicity caused by aflatoxin B1 and the concentration of free sphinganine in liver (Carlson et al., 2001; Riley et al., 2001). A mathematical model based on the data from the study of the National Toxicology Program confirmed that the concentration of sphinganine in mouse liver and rat kidney was a dose-related biomarker of fumonisin B1-induced cell death (Delongchamp & Young, 2001). In Sprague-Dawley rats fed AIN-76 diets containing pure fumonisin B1, the LOEL for an increased urinary concentration of free sphinganine was 5 mg/kg of diet (equivalent to 0.5 mg/kg bw per day) (Wang et al., 1999). In rats fed a diet containing a mixture of fumonisins from culture material for 13 days, the NOEL was 1–2 mg/kg of diet (equivalent to 0.1–0.2 mg/kg bw per day) (Solfrizzo et al., 1997).
The potential problems of using increased free sphinganine as a functional biomarker of human exposure to fumonisin B1 have been reviewed (Turner et al., 1999). In a study in men in China, the ratio of free sphinganine to free sphingosine in urine was significantly greater for men in households where the estimated daily intake of fumonisin B1 was > 110 µg/kg bw per day (Qui et al., 2001). In several other studies, an increase in free sphinganine in human urine or blood did not appear to be associated with fumonisin intake (e.g. van der Westhuizen et al., 1999).
In cultured cells, the sphingolipid-dependent mechanisms for inducing apoptosis include accumulation of excess ceramide, glucosylceramide (Korkotian et al., 1999), or sphingoid bases and depletion of ceramide, or more complex sphingolipids (Table 2). Conversely, the balance between sphingosine 1-phosphate and ceramide is critical for signalling proliferation or cell survival (Spiegel, 1998). A diversity of alterations in cellular regulation resulting from disruption of sphingolipid metabolism by fumonisin B can also be expected. This is demonstrated in numerous studies of the identify of cell processes mediated by ceramides (Table 3). Many of these processes are relevant to understanding the toxicity and carcinogenicity of fumonisin B1, in particular, the ability of this toxin to protect oxidant-damaged cells from apoptosis and to alter the proliferative response (Table 3).
There is no doubt that loss of complex sphingolipids also plays a role in the abnormal behaviour, altered morphology, and altered proliferation of fumonisin-treated cells (Tables 1 and 2), in particular the ability of fumonisin B1 to alter the function of specific glycosphingolipids and lipid rafts (membrane associations of sphingolipids, ceramide-anchored proteins, and other lipids). Examples are functions such as vitamin and toxin transport and cell–cell and cell–substratum contact (Table 3).
Inhibition of ceramide biosynthesis by fumonisins also protects cells from oxidant-induced cell death (Table 3). As the metabolism of ceramides is sensitive to the redox state of the cell, it is of particular interest that mice, pigs, and horses treated with fumonisin have increased amounts of complex sphingolipids containing sphinganine as the long-chain sphingoid-base backbone. The ceramide generated from these complex sphingolipids is dihydroceramide, the form of ceramide that is inactive in ceramide signalling and does not induce apoptosis of oxidant-damaged hepatocytes (Arora et al., 1997). Dihydroceramide also occurs in greater amounts in mouse hepatoma cells, in which 37% of the ceramides contain sphinganine, as compared with 5% in normal rat liver (Rylova et al., 1999).
(ii) Altered fatty acid metabolism in liver
Essential fatty acids are major constituents of all cell membrane glycerophospholipids, sphingolipids, and triglycerides. Apart from being structural components of all membranes, they are precursors of eicosanoids, prostaglandins, leukotrienes, and other oxygenated derivatives. In addition, the regulated turnover of membrane phospholipids is important in many intracellular signalling systems known to regulate cell growth, death, and differentiation. In rat liver, fumonisin B1 induced changes in phospholipids and their fatty acid composition that markedly affected the many cell functions that may contribute to its toxicity and carcinogenicity. The following summary is from the review of Gelderblom et al. (2001a).
In primary hepatocytes treated with fumonisin B1, incorporation of [14C]palmitic acid into total lipids, neutral lipids, triacylglycerol, and cholesterol esters were decreased, whereas it was increased in phospholipids (Gelderblom et al., 1996a). There was a concomitant increase in the cellular concentrations of phosphatidyl-choline and phosphatidylethanolamine, while the total cholesterol concentration decreased. The concentration and labelling of sphingomyelin was also decreased. Changes in the concentrations of specific polyunsaturated fatty acids were attributed to disruption of the Delta6 desaturase and cyclo-oxygenase metabolic pathways. These could be important in fumonisin B1-induced toxicity in primary hepatocytes.
Fumonisin B1 disrupted fatty acid and phospholipid biosynthesis in rat liver in vivo. In contrast to the situation in vitro, the main changes were associated with the phosphatidylethanolamine and phosphatidylcholine fractions, while the concentration of cholesterol was increased in both serum and liver (Tables 1 and 2). A characteristic fatty acid pattern was seen in the livers of rats given diets containing purified fumonisin B1 at concentrations that caused preneoplastic hepatic lesions in rats treated only with fumonisin B1 (> 1.6 mg/kg bw per day) or in rats fed fumonisin B1 after treatment with known liver cancer initiators such as N-nitrosodiethylamine (> 0.8 mg/kg bw per day). The pattern includes:
|
• |
increased amounts of saturated and monounsaturated fatty acids (C18:1omega9) in phosphatidylcholine and phosphatidylethanolamine; |
|
• |
increased relative amount of C18:2omega6 in phosphatidylcholine and increased absolute amount in phosphatidylcholine and phosphatidylethanolamine; |
|
• |
decreased relative and absolute amounts of C20:4omega6 in phosphatidylcholine and increases in phosphatidylethanolamine; |
|
• |
decreased relative and absolute amounts of C22:4omega6 and C22:5omega6 in phosphatidylcholine, increased relative amount of C22:5omega6 in phosphatidyl-ethanolamine; decreased amount of C22:6omega3 in phosphatidylcholine and increase in phosphatidylethanolamine; |
|
• |
decreased relative and absolute amounts of total polyunsaturated fatty acids in phosphatidylcholine and decreased relative amounts in phosphatidyl-ethanolamine; |
|
• |
decreased ratio of polyunsaturated to saturated fatty acids in phosphatidylcholine and phosphatidylethanolamine. |
As glycerophospholipids are important components of many cellular signalling pathways, perturbation of the phospholipid and fatty acid composition of cellular membranes could have a strong effect on processes that control cell growth and cell death (Tables 1 and 2). For example, increased expression of hepatocyte growth factor, transforming growth factor (TGF)-alpha) and especially TGF-beta1 and c-myc was seen in rat liver during short-term feeding with fumonisin B1 (Lemmer et al., 1999b). Overexpression of TGF-beta1 may contribute to the increased apoptosis seen in the livers of rodents fed fumonisin B1. The proto-oncogene c-myc is a positive regulator of cell proliferation that is involved in tumour progression (Nagy et al., 1988); it has also been implicated in TGF-beta1 signalling (Alexandrow & Moses, 1995). Increased expression of c-myc and TGF-beta1 may cooperate in the promotion of liver tumours during feeding of fumonisin B1, possibly by providing an environment that selects for the growth of TGF-beta1-resistant transformed liver cells. Overexpression of c-myc, depletion of growth factors and/or disruption of growth signalling pathways could result in imbalances in cell cycle progression and hence the induction of apoptosis (Steiner et al., 1996). In this regard, fumonisin B1 overexpressed c-myc in rat liver (Lemmer et al., 1999b), while it disrupted growth-related responses in cell types such as primary hepatocytes and in the liver in vivo (reviewed by Gelderblom et al., 2001a).
In male BDIX rats fed diets containing purified fumonisin B1 for 2 years, cyclin D1 accumulated in the nuclei of altered hepatocytes in foci, nodules, adenomas, and carcinomas (Ramljak et al., 2000). In male Fischer rats fed diets containing fumonisin B1 for 21 days, the concentration of cyclin D1 protein in liver was increased up to fivefold in a dose-dependent manner, with no simultaneous increase in mRNA. A fumonisin B1-induced increase in cyclin-dependent kinase 4 was confirmed by increased phosphorylation of the retinoblastoma protein; the accumulation of cyclin D1 appeared to result from stabilization of the protein associated with activation of protein kinase B (Akt), followed by inhibition of glycogen synthase kinase 3beta activity. Akt is part of the anti-apoptotic phosphatidylinositol 3 kinase cell survival pathway (Dudek et al., 1997) and can be activated by stimuli involving growth factors and cytokines and inhibited by the pro-apototic molecule, ceramide (Franke et al., 1997; Zhou et al., 1998). Therefore, the modulating effects of fumonisin B1 on both sphingolipids and phospholipids could play a major role in molecular events involving the stability of cyclin D1 protein (Ramljak et al., 2000).
It has been proposed that glycerophospholipids and the sphingolipid cycle interact to control a variety of cellular processes, including apoptosis. For example, C20:4omega6 generated by cytosolic phospholipase A2 initiated sphingomyelin hydrolysis, whereas ceramide generated de novo during ceramide synthase stimulated C20:4omega6 release via secretory phospholipase A2 (Jayadev et al., 1994; Balsinde et al., 1997). Ceramide has also been shown to regulate transcription of cyclooxygenase-2 (Subbaramaiah et al., 1998). A similar interactive pathway is likely to exist for fumonisins in the liver to regulate processes related to cell proliferation and apoptosis. Fumonisin B1 induces similar changes in phospholipids and in the profile of fatty acids in both the liver and hepatocyte nodules (Abel et al., 2001). However, subsequent effects on sphingolipid and/or prostaglandin production appear to inhibit the growth of normal hepatocytes, which, with the overexpression of TGFbeta-1 and c-myc, could affect apoptosis. Oxidative damage and the resultant lipid peroxidation products may further enhance apoptosis in the liver (Chen et al., 1997). Conversely, the increased C18:1omega9, the decreased long-chain polyunsaturated fatty acid concentration, and a phosphatidyl-ethanolamine-associated increase in C20:4omega6 fatty acids are critical for cell proliferation (Tang et al., 1993; Horribin, 1994), especially in initiated cell populations.
(b) Other biochemical changes
Several studies of fumonisins in vitro showed changes in cellular regulation and function that were attributed to actions independent of altered lipid metabolism (Tables 1 and 2). Many of these effects might be relevant to the toxicity of fumonisins.
Thus, fumonisin-induced disruption of lipid metabolism is observed both in vitro and in vivo. The biochemical consequences of the disruption of sphingolipid metabolism that are most likely to alter cell regulation are increased concentrations of free sphingoid bases and their 1-phosphates, alterations in complex sphingolipids, and decreased ceramide biosynthesis. Because free sphingoid bases and ceramide can induce cell death, inhibition of ceramide synthase can inhibit cell death induced by ceramide but can promote cell death induced by free sphingoid bases. The kinetics of the increases and decreases in the various bioactive sphingolipid pools in liver, kidney, lung, and heart is also important in the toxicity of these toxins. Fumonisins also induced changes in fatty acids and phospholipids in primary rat hepatocytes and rat liver in vivo, which closely reflected those expected from disruption of the Delta6 desaturase enzyme, the rate-limiting enzyme in fatty acid metabolism, and disruption of prostaglandin biosynthesis. The changes in fatty acid and phospholipid metabolism that probably alter cell regulation are changes in the degree of saturation of fatty acids in the phospholipid pools, increases in the ratio of phosphatidylcholine to phosphatidylethanolamine, changes in prostaglandin biosynthesis, and altered ceramide production.
Fumonisins also affect sites of cellular regulation that are apparently independent of the disruption of lipid metabolism. Nevertheless, disruption of lipid metabolism, membrane structure, and signal transduction pathways mediated by lipid second messengers appear to be important in all the proposed mechanisms of action.
Two cellular modes of action for the toxicity and carcinogenicity of fumonisin B1 have been proposed that are well supported by results obtained in vivo. In both hypotheses, altered lipid metabolism is the initial biochemical mechanism. In one hypothesis, the initial biochemical lesion is presumed to be inhibition of ceramide synthase (Merrill et al., 2001; Riley et al., 2001), and in the other, the biochemical lesion is attributed to disruption of the Delta6 desaturase and cyclooxygenase metabolic pathways (Gelderblom et al., 2001a). In both hypotheses, it is assumed that other initial sites of action could contribute to the observed cellular responses. The two invoke similar cellular mechanisms, to the extent that fumonisin B1-induced imbalances in the rates of cell death and proliferation in target tissues are considered to contribute to cancer development (Dragan et al., 2001; Howard et al., 2001a).
Fumonisin-induced disruption of sphingolipid metabolism in target tissues has been demonstrated in many independent studies. Nonetheless, the way in which disrupted sphingolipid metabolism contributes to toxicity in rodents is unclear. Current understanding of the sphingolipid signalling pathways (reviewed by Merrill et al., 2001; Riley et al., 2001) indicates that the balance between the intracellular concentrations of sphingolipid effectors that protect cells from apoptosis (decreased ceramide, increased sphingosine 1-phosphate) and the effectors that induce apoptosis (increased ceramide, increased free sphingoid bases, increased fatty acids) determines the cellular response. Cells sensitive to the proliferative effect of decreased ceramide and increased sphingosine 1-phosphate will be selected to survive and proliferate. Conversely, when the rate of increase in free sphingoid bases exceeds a cell’s ability to convert sphinganine or sphingosine to dihydro-ceramide or ceramide or their sphingoid base 1-phosphate, then free sphingoid bases will accumulate to toxic levels. In this case, cells that are sensitive to sphingoid base-induced growth arrest will cease growing, and insensitive cells will survive. Thus, the kinetics of fumonisin elimination (rapid), the affinity of fumonisin B1 for ceramide synthase (competitive and reversible), and the kinetics of sphinganine elimination (persistent but reversible) could affect the time course, amplitude, and frequency of peaks in the intracellular concentrations of ceramide, sphingoid base-1 phosphates, and free sphinganine in tissues of animals given diets containing fumonisins. This is important, because the balance between the rates of apoptosis and proliferation is a critical determinant in hepato- and nephrotoxicity and tumorigenesis in animal models (Dragan et al., 2001; Howard et al., 2001a; Voss et al., 2001). At the cellular level, apoptotic necrosis should be considered to be similar to oncotic necrosis (as defined by Levin et al., 1999), in that both lead to a regenerative process involving sustained cell proliferation (Dragan et al., 2001; Hard et al., 2001). Numerous endogenous processes can cause DNA damage; while most are repaired, an unrepaired mutation in DNA can occur. Increased DNA replication can thus increase the risk that damaged DNA is also replicated, resulting in an increased cancer risk. With respect to fumonisin B1, this hypothesis is best supported by observations in rat kidney, as in liver apoptotic and oncotic necrosis occur concurrently (Dragan et al., 2001). Nonetheless, regeneration after either apoptotic or oncotic necrosis is observed at the same doses that cause cancer development.
Fumonisin B1 alters normal cell proliferation in rat liver in vivo (Gelderblom et al., 1994, 1996a; Li et al., 2000) and in primary rat hepatocytes and many other cell lines in vitro (Gelderblom et al., 1994; Tolleson et al., 1996). Differential inhibition of cell proliferation is a possible mechanism: hepatocytes resistant to fumonisin B1-induced inhibition of cell growth are selectively stimulated to grow by creating an environment in which the growth of normal cells is impaired. Selective inhibition of normal cell growth could increase the chances of survival of cells initiated by processes such as free-radical damage, leading ultimately to manifestations of cancer initiation in liver, such as glutathione S-transferase, placental form (GST-P)+ foci and hepatocyte nodules. Fumonisin B1 may be not only a complete carcinogen but also promote cancer, and this should be considered in models for risk assessment.
Disruption of the Delta6 desaturase and cyclooxygenase metabolic pathways in the livers of male rats has been well documented. Disruption of lipid-mediated growth stimulation in the liver could be important in establishing a growth differential that results in clonal expansion of certain cell types associated with neoplastic development. For example, disruption of C20:4omega6 metabolism altered the mitogenic response to epidermal growth factor in primary rat hepatocytes, a known property of many hepatocarcinogens (Gelderblom et al., 1999a). Three lines of evidence support the hypothesis that fumonisin B1-induced alteration of lipid metabolism contributes to establishing a growth differential in rat liver. First, the lipid parameters associated with increased cell proliferation in hepatocyte nodules closely mimic those of normal regeneration in the liver; one major difference is that the changes in the nodules are persistent, whereas they are reversed in regenerating liver (Abel et al., 2001). The increased concentrations of phosphatidylethanolamine and C20:4omega6 are of special interest, as this fatty acid is known to regulate many processes related to cell growth, such as proliferation and apoptosis (Khan et al., 1995). Several studies indicated that fumonisin B1 interacts with C20:4omega6 metabolism in normal and cancer cell lines in vitro (Gelderblom et al., 1999a; Pinelli et al., 1999; Seegers et al., 2000). Second, alterations in the N-6 fatty acid desaturase pathway and the subsequent decrease in long-chain polyunsaturated fatty acids would result in a more rigid membrane structure. This could provide a survival advantage for hepatocytes under stress, since the membranes will be resistant to oxidative damage. Such membrane changes occur preferentially in hepatocyte nodules, creating an environment for differential growth relative to the surrounding normal tissue. Changes in membrane fluidity could also alter the response of membrane receptors and enzymes by affecting their mobility in the bilayer. Third, lipid metabolites, and in particular glycerophospho-lipids, are important components of many cellular signalling systems that control the balance between cell growth and cell death.
In the classical model of cancer initiation by genotoxic carcinogens, fumonisins did not increase the incidence of hepatocellular foci after single or multiple doses (Gelderblom et al., 1992). Subsequent studies indicated that the ‘effective dose’ for induction of preneoplastic lesions in liver, such as GST-P+ foci and hepatocyte nodules, in male Fischer rats depends on the dose and duration of exposure (Gelderblom et al., 1994). The toxicity of fumonisin B1 in rat liver appears to play an important role in cancer development (Abel & Gelderblom, 1998). Induction of oxidative damage and lipid peroxidation as a consequence of toxicity (Tables 1 and 2) could lead to DNA damage. Changes in the balance of the various cell regulatory molecules, such as those found in the livers of rats fed diets containing fumonisin B1, are likely to be involved in induction of a growth differential in which the growth of initiated cells is selectively stimulated and cancer develops.
Many of the studies summarized in this section are also described in the IPCS monograph (WHO, 2000a). The pathological findings and doses in pertinent studies are given in Tables 4–6. The diets used in most of the studies summarized in this section differed markedly in nutritional composition. As the proposed mechanisms of action involve alterations in de novo biosynthesis, nutritional factors might be important in toxic end-points. The liver was the target for fumonisin B1 in all animals in which toxicity was observed, and the kidney was also a target in many animals. In both liver and kidney, fumonisin B-induced toxicity is often characterized initially by increased apoptotic and oncotic necrosis, regeneration, and, in the case of liver, bile-duct hyperplasia (Tables 4 and 5). In rodents, the toxicity of fumonisin B1 depends on the strain and sex. For example, male BD IX rats appeared to be more resistant to the nephrotoxic effects of fumonisin B1 than male Fischer 344N, male Sprague-Dawley, and male RIVM:WU rats.
F. verticillioides culture material and naturally contaminated maize can contain various fumonisins and other mycotoxins. However, naturally contaminated maize and culture material of the F. verticillioides isolate known as MRC 826 contain predominantly fumonisins of the B series. Therefore, the results of studies with these materials corroborate those of studies in which pure fumonisin B1 was used. Although the results were not used for hazard characterization or risk assessment, when possible, the NOEL or LOEL was calculated for comparison with the results of studies with pure fumonisin B1.
No studies have been published on the lethality of single doses of pure fumonisin B1 in laboratory animals. The few available studies indicate that fumonisins are not acutely toxic. For example, mice given a single dose of 25 mg/kg bw by gavage or subcutaneous injection showed reversible alterations in cytokine expression, serum enzymes activity, and blood cell counts (Bhandari et al., 2001).
(a) BALB/c mice
Male mice were given a subcutaneous dose of fumonisin B1 at 0.3, 0.8, 2.3, or 6.8 mg/kg bw per day for 5 days. Apoptosis was detected in the livers of mice at doses > 0.8 mg/kg bw per day and in the kidney at all doses (Sharma et al., 1997; Tsunoda et al., 1998). If it is assumed that 10% of an oral dose would be absorbed in mice, the calculated LOEL for oral administration would be 8 mg/kg bw per day in liver and 3 mg/kg bw per day in kidney. The relative weight of the kidney was decreased at all doses except 0.8 mg/kg bw per day; no effect was observed on the relative weight of the liver. Increased apoptosis in liver was also seen in subsequent studies with various strains of mice and transgenic mouse models. The response to fumonisin differed in some transgenic mouse models in comparison with the wild type (Sharma et al., 2000a,b,c). The results of studies with mice that overexpress or lack tumour necrosis factor (TNF)-alpha suggest that this pathway plays a role in the hepatic toxicity of fumonisin B1 in mice.
(b) B6C3F1 mice
Male and female mice were fed diets containing fumonisin B1 at a concentration of 1, 3, 9, 27, or 81 mg/kg for 90 days, providing mean intakes of 0.3, 0.9, 2.5, 7.4, and 23 mg/kg bw per day for males and 0.3, 1, 3, 9.7, and 29 mg/kg bw per day for females. The serum concentrations of cholesterol and total bilirubin and the activities of alanine and aspartate aminotransferases, alkaline phosphatase, and lactate dehydrogenase were significantly increased in female mice at the high dose, with no effect in males. The changes were parallelled by histological alterations in the livers of the female mice, mainly restricted to the centrilobular zone. No lesions were reported in the kidney, but some were detected in the adrenal cortex (presumably a normal physiological reaction to stress induced by the treatment) in all females given the highest dose and two females given the next lowest dose (Table 4; Voss et al., 1995a).
Adult male and female mice were given fumonisin B1 at a daily dose of 1, 5, 15, 35, or 75 mg/kg bw per day by gavage for 14 days. Hepatotoxicity was observed in animals of each sex, but renal toxicity was seen only in females. Females were more sensitive than males to these effects. Single-cell necrosis was found in the liver at a dose of 35 mg/kg bw per day in males and 15 mg/kg bw per day in females. Increased hepatocyte mitosis was seen at 75 mg/kg bw per day in males and > 5 mg/kg bw per day in females. Mild single-cell necrosis was detected in the cortical and medullary tubules only in female mice at 15–75 mg/kg bw per day. Males (at doses > 35 mg/kg bw per day) and females (at doses > 15 mg/kg bw per day) had moderate diffuse vacuolization of adrenal cortical-cell cytoplasm. Mild thymic cortical lymphocytolysis was noted in a few female mice that received doses > 35 mg/kg bw per day (Table 4; Bondy et al., 1997).
Male and female mice were fed diets containing fumonisin B1 at a concentration of 99, 160, 230, or 480 mg/kg for 28 days in order to establish the doses for a 2-year bioassay. Males at the highest concentration developed liver lesions. Changes in clinical chemical end-points and cell cycle progression indicative of increased proliferation paralleled the pathological changes. Similar changes were seen in females but at all doses. Thus, female mice were more sensitive than males to the hepatic toxicity of fumonisin B1. No NOEL could be identified, as liver lesions were seen in females at all doses (Table 4; National Toxicology Program, 1999).
Table 4. Results of studies in male and female B6C3F1 mice given diets containing purified fumonisin B1
|
Sex |
Treatment |
Fumonisin B1 intake |
Main pathological lesions or effects |
Reference |
|
|
Liver |
Kidney |
||||
|
Males and females |
NIH open formula 07 diet, 90 days |
Voss et al. (1995a) |
|||
|
Males |
1 |
0.3 |
No lesions |
No lesions |
|
|
3 |
0.8 |
||||
|
9 |
2.4 |
||||
|
27 |
7.4 |
||||
|
81 |
23 |
||||
|
Females |
1 |
0.3 |
Highest dose only: Necrosis, mitotic figures, hepatocyte collapse of centrilobular zone; infiltration of inflammatory cells |
No lesions |
|
|
3 |
1 |
||||
|
9 |
3 |
||||
|
27 |
9.7 |
||||
|
81 |
29 |
||||
|
Males and females |
Gavage, 14 days |
1 |
Males: Increased cholesterol, alanine aminotransferase activity, single-cell necrosis (> 35 mg/kg bw per day). |
No lesions |
Bondy et al. (1997) |
|
5 |
|||||
|
15 |
|||||
|
35 |
Increased mitosis at 75 mg/kg bw per day |
||||
|
75 |
Females: Increased cholesterol, alanine aminotransferase activity, single-cell necrosis (> 15 mg/kg bw per day). |
Females: single-cell necrosis and vacuolization of cytoplasm in the cortical and medullary tubules at >15 mg/kg bw per day |
|||
|
Males and females |
NIH 31 diet, 28 days |
National Toxicology Program (1999) |
|||
|
Males |
99 |
19 |
Highest dose only: Hepatocellular necrosis, periportal hypertrophy, diffuse centrilobular hyperplasia, Kupffer cell hyperplasia |
No lesions in any group |
|
|
160 |
31 |
||||
|
230 |
44 |
||||
|
480 |
93 |
||||
|
Females |
99 |
24 |
All treated animals: Hepatocellular necroses, periportal hypertrophy, diffuse centrilobular hyperplasia, Kupffer cell hyperplasia |
No lesions in any group |
|
|
160 |
41 |
||||
|
230 |
62 |
||||
|
480 |
100 |
||||
|
Males and females |
NIH 31 diet, 728 days |
National Toxicology Program (1999); Howard et al. (2001b) |
|||
|
Males |
5 |
0.5 |
Hepatocyte hypertrophy (10/47, 9/47, 24/48, 25/48, 30/48), Multifocal necrosis (1/47, 1/47, 0/48, 4/48, 14/48) Hepatocellular adenomas (9/47, 7/47, 7/48, 6/48, 8/48), Hepatocellular carcinomas (4/47, 3/47,4/48, 3/48, 2/48) |
No lesions in any group |
|
|
15 |
1.6 |
||||
|
50 |
9.0 |
||||
|
150 |
15 |
||||
|
Females |
5 |
0.7 |
Hepatocyte hypertrophy (0/47, 0/48, 0/48, 7/47, 31/45), apoptosis (0/47, 0/48, 0/48, 7/47, 14/45), multifocal necrosis (1/47, 1/48, 1/48, 29/47, 26/45). Hepatocellular adenomas (5/47, 3/48, 1/48, 16/47, 31/45), hepatocellular carcinomas (0/47, 0/48,0/48, 10/47, 9/45) |
No lesions in any group |
|
|
15 |
1.9 |
||||
|
50 |
6.6 |
||||
|
80 |
13 |
||||
(c) BD IX rats
Male rats were fed diets containing fumonisin B1 at a concentration of 0.1% for 33 days. Major changes were reported in the liver and mild changes in kidney (Table 5). After 3 days of treatment of male rats with fumonisin B1 at 240 mg/kg bw per day by gavage, major pathological lesions were observed in the liver and only minor changes in kidney. Severe disseminated acute myocardial necrosis and severe pulmonary oedema were observed in two rats. At lower doses but longer treatment (9–12 days), pathological changes were observed only in liver. Early signs of bile-duct proliferation and fibrosis radiating from the portal areas were noted, and the nuclei of a few hepatocytes were enlarged (Gelderblom et al., 1988).
Table 5. Results of studies in rats given diets containing purified fumonisin B1
|
Strain and sex |
Treatment |
Fumonisin B1 intake |
Main pathological lesions or effects |
Reference |
|
|
Liver |
Kidney |
||||
|
BD IX, male |
Gavage |
240, 3 days |
75% of rats died; toxic hepatitis, myocardial necrosis, pulmonary oedema, bile-duct proliferation |
ND |
Gelderblom et al. (1988) |
|
70, 9 days |
Single-cell necrosis; hydrophic degeneration |
ND |
|||
|
48, 12 days |
Fibrosis |
||||
|
Epol 1000 |
70, 33 days |
Bile-duct proliferation; fibrosis; hepatocyte nodules |
Proximal convoluted tubule: fatty changes; scant necrosis |
||
|
BD IX, male |
Semi-purified diet, 780 days |
Gelderblom et al. (1991) |
|||
|
50 |
1.6 |
Cirrhosis (15/15); regenerative nodules (15/15); cholangio-fibrosis (15/15); hepatocellular carcinoma (10/15) |
Mild changes in proximal convoluted tubule towards end of experiment |
||
|
BD IX, male |
Semi-purified diet, 690 days |
Gelderblom et al. (2001a) |
|||
|
1 |
0.03 |
No lesions |
No lesions |
||
|
10 |
0.3 |
Mild changes |
At 10 and 25 mg/kg: |
||
|
25 |
0.8 |
Anisokaryosis (13/17); hepatocyte nodules (9/17); oval-cell proliferation (2/17); bile-duct hyperplasia (3/17); portal fibrosis (5/17); ground-glass foci (5/17); GST-P+ foci (11/11) |
|||
|
Fischer 344, male |
101, Dyetts Inc. diet, |
60 |
Bile -duct proliferation, hepatocyte nodules, hepatocyte necrosis |
ND |
Gelderblom et al. (1992) |
|
Fischer 344, male |
AIN-76 diet, 21 days |
Gelderblom et al. (1994) |
|||
|
25 |
1.7 |
No lesions |
|
||
|
50 |
3.5 |
No lesions |
|||
|
100 |
7.2 |
A few necrotic cells |
|||
|
250 |
15 |
Bile-duct proliferation |
|||
|
500 |
25 |
Apoptosis |
|||
|
750 |
31 |
Degenerative changes |
|||
|
Gavage, 14 days |
|||||
|
59 |
4 |
As above; hepatocyte nodules after promotion with acetylaminofluorene and partial hepatectomy |
ND |
||
|
120 |
8.5 |
||||
|
230 |
16 |
||||
|
320 |
23 |
||||
|
Fischer 344, male |
AIN-76 diet, 21 days |
Gelderblom et al. (1996c) |
|||
|
10 |
0.7 |
No lesions |
All treated groups: Nephrosis, necrotic epithelial cells, apoptosis, hypereosinophilia, sloughing of epithelial cells |
||
|
50a |
3.5 |
Scattered necrotic cells |
|||
|
100 |
6.8 |
Apoptosis, ductile endothelial cell proliferation, mitotic figures |
|||
|
250 |
15 |
Nodular regeneration, fibrosis, ductile endothelial cell proliferation |
|||
|
500 |
25 |
Distortion of lobular structure |
|||
|
Fischer 344, male |
AIN-76 diet, 35 days, 250 |
15 |
Hepatocyte necrosis, apoptosis, stellate-cell proliferation, fibrosis, regenerative nodules, foci and nodules, oval-cell proliferation |
ND |
Lemmer et al. (1999a,b) |
|
Fischer 344, male |
35 days at 15 mg/kg bw, 150 days at 100 mg/kg bw, 185 days on AIN-76 diet without FB1 |
|
|
|
Lemmer (2000) |
|
250 |
15 |
GST-P+ foci and nodules, fibrosis, oval-cell proliferation, cholangiofibrosis |
ND |
||
|
100 |
7 |
||||
|
"Stop model" |
|
Dysplastic liver nodules (6/6), cholangiofibrosis (1/6), hepatocellular carcinoma (1/6) |
|||
|
Fischer 344 Malesb |
NIH 31 diet, 28 days |
Tolleson et al. (1996); National Toxicology Program (1999); Howard et al. (2001b) |
|||
|
99 |
12 |
No lesions |
All treated groups: Increased apoptosis, degeneration, and mitosis; decreased relative kidney weights |
||
|
160 |
20 |
No lesions |
|||
|
230 |
28 |
Increased apoptosis and degeneration at doses > 20 mg/kg bw per day. Increased, mitosisc, bile-duct hyperplasia, decreased relative liver weight at doses > 28 mg/kg bw per day |
|||
|
480 |
56 |
||||
|
Fischer 344 Femalesb |
NIH 31 diet, 28 days |
Tolleson et al. (1996); National Toxicology Program (1999); Howard et al. (2001b) |
|||
|
99 |
12 |
Increased apoptosis in all groups; increased mitosis and degeneration at doses > 12 mg/kg bw per day; bile-duct hyperplasia and decreased relative liver weights at doses > 20 mg/kg bw per day |
Decreased relative kidney weights and increased mitosis in all groups; increased apoptosis, degeneration at doses > 12 mg/kg bw per day |
||
|
160 |
20 |
||||
|
230 |
28 |
||||
|
480 |
56 |
||||
|
Fischer 344 Males |
NIH open formula 07, 90 days |
Voss et al. (1995a) |
|||
|
1 |
0.1 |
No lesions |
No lesions |
||
|
3 |
0.2 |
No lesions |
|||
|
9 |
0.6 |
No lesions |
|||
|
27 |
1.9 |
Single-cell necrosis, necrotic tubule epithelial cells, eosinophilic cytoplasm, sloughing of tubule epithelia |
|||
|
81 |
5.7 |
||||
|
Females |
1 |
0.1 |
No lesions |
No lesions |
|
|
3 |
0.3 |
No lesions |
|||
|
9 |
0.7 |
No lesions |
|||
|
27 |
2.2 |
No lesions |
|||
|
91 |
6.4 |
Lesions |
|||
|
Fischer 344 Males |
Diet, 728 days |
Weeks 51–104 |
|
|
National Toxicology Program (1999); Hard et al. (2001); Howard et al. (2001b) |
|
5 |
0.22 |
No lesions |
(Data from Hard et al., 2001) Adenoma (0/48, 0/40, 0/48, 4/48,6/48); carcinoma (0/48, 0/40, 0/48,8/48, 10/48); atypical tubule (0/48, 0/40, 0/48, 4/48, 9/48); apoptosis and regeneration sustained throughout study at doses > 0.67 mg/kg bw |
||
|
15 |
0.67 |
||||
|
50 |
2.2 |
||||
|
150 |
6.6 |
||||
|
Females |
5 |
0.27 |
No lesions |
No lesions |
|
|
15 |
0.78 |
||||
|
50 |
2.6 |
||||
|
100 |
5.2 |
||||
|
Sprague-Dawley, male and female |
RMH 3000 diet, 28 days |
|
|
Voss et al. (1993, 1995b) |
|
|
15 |
1.4 |
No lesions |
Single-cell necrosis, sloughing of tubular epithelia, epithelial hyperplasia, cytoplasmic basophilia |
||
|
50 |
4.1 |
Mild changes |
|||
|
150 |
13 |
Single-cell necrosis, cell and nuclear polymorphism, bile-duct proliferation (?); lesions more severe in females |
|||
|
Sprague-Dawley, male and female |
Gavage, 11 days |
1 |
Males: single-cell necrosis, increased alanine and aspartate aminotransferases (15 mg/kg bw per day), mitosis (35 mg/kg bw per day) |
Necrosis of tubular epithelia, anisokaryosis, cytoplasmic basophilia, atrophy of tubular epithelia |
Bondy et al. (1996, 1998) |
|
5 |
|||||
|
15 |
|||||
|
35 |
|||||
|
75 |
|||||
|
Sprague-Dawley, male and female |
Gavage, 11 days |
1 |
Males: GST-P+ foci and PCNA+ nuclei at doses > 35 mg /kg bw per day. Females: GST-P+ foci at 75 mg/kg bw per day. PCNA+ nuclei at 35 and 75 mg /kg bw per day |
Not reported, but experimental design similar to Bondy et al. (1996) |
Mehta et al. (1998) |
|
5 |
|||||
|
15 |
|||||
|
35 |
|||||
|
75 |
|||||
|
RIVM:WU, male |
Gavage, 28 days |
0.2 |
No lesions at any dose |
Dose-dependent increase in tubular cell death at all doses; increased apoptosis and mitosis at 0.75 and 3 mg/kg bw per day |
de Nijs (1997) |
|
0.8 |
|||||
|
3.0 |
|||||
ND, not determined; GST-P+, positive for glutathione S-transferase. placental form; PCNA+, positive for proliferating cell nuclear antigen
a
50 mg/kg of diet; promotion in rats initiated with N-nitrosodiethylamineb
Final body weight significantly lower after 28 days in males and females at 484 mg/kg of dietc
Mitosis based on morphological evaluation was less than that measured in the PCNA assay.Culture material from F. verticillioides, strain MRC 826, was fed to male rats for 77 days. All developed cirrhosis, intraventricular cardiac thrombosis and nephrosis (Kriek et al., 1981a).
(d) Fischer rats
When male rats were fed diets containing fumonisin B1 at a concentration of 0.1% for 55 days, the pathological lesions reported in the livers were similar to those described in male BD IX rats. Hepatocyte nodules were induced when the fumonisin B1-fed rats were subjected to promotion by 2-acetylaminofluorene and partial hepatectomy (Gelderblom et al., 1992). In subsequent studies, in which rats were given diets containing 25–750 mg/kg, histopathological lesions were observed in the livers of animals at 500 and 750 mg/kg of diet after 21 days. Similar changes, although less pronounced, were observed in the rats that received 100 and 250 mg/kg of diet. A few necrotic cells were detected in the livers of rats at 50 mg/kg of diet, but none were observed at 25 mg/kg of diet. Measurements indicated that the mean daily intake of fumonisin B1 at these six doses was 1.7, 3.5, 7.2, 15, 25, and 31 mg/kg bw per day. Twenty-one days after the promoting treatment, GST-P+ foci and nodules were induced at 250 mg/kg of diet or a daily intake > 15 mg/kg bw. The dose-dependency for inducing the putative preneoplastic lesions in liver was also seen after daily treatment by gavage for 14 days. After the promoting treatment, hepatocyte nodules and toxic effects in the liver were seen only in rats that received a daily dose > 8.5 mg/kg bw. These studies showed that fumonisin B1-induced hepatoxicity was a prerequisite for development of the preneoplastic lesions in rat liver (Table 5; Gelderblom et al., 1994).
In a study of the cancer promoting potential of fumonisin B1, similar dietary concentrations were fed to male rats over 21 days. Pathological changes were reported in the livers of rats receiving 50 mg/kg of diet (3.5 mg/kg bw per day) but not in those that received 10 mg/kg of diet (0.7 mg/kg bw per day), and cancer promotion was seen in N-nitrosodiethylamine-initiated rats at the higher concentration (Table 5; Gelderblom et al., 1996b). Microscopic lesions were seen in the kidneys of rats that received the low dose (W.C.A. Gelderblom, unpublished data) and in the inner cortex and outer medulla of rats at the high dose. The changes were common in animals at the high dose and only mild at 10 mg/kg of diet (0.7 mg/kg bw per day). These results indicate that similar lesions are induced by comparable doses of fumonisin B1 in the livers of male BD IX and Fischer rats, but in Fischer rats lesions were detected in the livers at a dietary concentration as low as 50 mg/kg and in the kidneys at a concentration as low as low as 10 mg/kg.
The kinetics of hepatocyte injury in liver was investigated in male rats fed a diet containing fumonisin B1 at 250 mg/kg for 5 weeks. Hepatocyte necrosis and apoptosis were found mainly in zone 3 of the liver lobule. Hepatocyte injury and death were mirrored by desmin-positive hepatic stellate-cell proliferation and marked fibrosis, with progressive disturbance of the architecture and formation of regenerative nodules. Oval-cell proliferation was seen in the presence of hepatocyte mitotic activity. Oval-cell (OV-6 positive) proliferation was noted from week 2; glutathione S-transferase-positive hepatic foci and nodules developed; and, later, oval cells were seen inside some of the ‘atypical’ nodules (Table 5; Lemmer et al., 1999b).
In a subsequent experiment, groups of 12 male Fischer rats were fed diets containing fumonisin B1 at a concentration of 250 mg/kg for 5 weeks followed by 100 mg/kg of diet for the remainder of the experiment, up to 6 months, when exposure was discontinued (‘stop model’). Liver biopsy samples were collected from a subpopulation of the rats for another 6 months. The body-weight gain of treated animals was significantly lower after 6 months. A variety of preneoplastic and neoplastic hepatic lesions were observed in the samples collected. The intake of fumonisin B1 was estimated to be 15 mg/kg bw per day (Table 5; Vessey et al., 1999; Lemmer, 2000).
Male and female rats were fed diets containing fumonisin B1 at a concentration of 99, 160, 230, or 480 mg/kg for 28 days, in order to set the doses for a 2-year bioassay, providing average daily doses of the toxin of 12, 20, 28, and 56 mg/kg bw per day. The body weights of both males and females were at the highest dose were decreased. Similar pathological lesions and effects were seen in the livers and kidneys of males and females. The male kidney was the most sensitive target for fumonisin B1, and the liver was more severely affected in females than in males. The earliest cellular response in both liver and kidney was increased apoptosis, which was accompanied by increased cell proliferation. Structural degeneration as a result of apoptosis was seen in both liver and kidney. In females, the NOEL for bile-duct hyperplasia and decreased liver weight was 28 mg/kg bw per day, and that for liver degeneration and increased hepatocellular mitosis was 20 mg/kg bw per day. The NOEL for increased hepatocellular apoptosis and for decreased kidney weight, increased mitosis, increased structural degeneration, and increased mitosis in males was < 12 mg/kg bw per day (Table 5; Tolleson et al., 1996; National Toxicology Program, 1999; Howard et al., 2001b).
Male and female rats were fed diets containing fumonisin B1 at a concentration of 1, 3, 9, 27, or 81 mg/kg for 90 days. No hepatotoxic effects were seen in male or female rats, whereas renal toxicity was found in male rats fed diets containing > 9 mg/kg. Female rats were more resistant, as renal changes were observed only at 81 mg/kg of diet (5.7 mg/kg bw per day); however, when kidney weight was used as a marker, a significant difference was found between control females and females fed 9 mg/kg of diet (0.7 mg/kg bw per day). The NOEL for renal toxicity in male rats was 0.2 mg/kg bw per day (Table 5; Voss et al., 1995a).
A maize sample naturally contaminated with F. verticillioides that had been involved in an epizootic of equine leukoencephalomalacia in the USA was fed to male rats for 5–6 months. The liver was the major organ affected (Table 6; Wilson et al., 1985). Retrospective analyses indicated the the total concentration of fumonisin B1 and B2 was 33 mg/kg, and only trace amounts of aflatoxin B1 and B2 were present. Dietary deficiencies, especially those related to methionine and choline, were suggested to have accelerated and/or promoted the liver lesions. The mean fumonisin B intake was calculated to be equivalent to 2.3–3.2 mg/kg bw per day (Gelderblom et al., 2001a).
(e) Sprague-Dawley rats
Male and female rats were fed diets containing fumonisin B1 at a concentration of 15, 50, or 150 mg/kg for 4 weeks, providing estimated daily intakes of 1.4, 4.7, and 14 mg/kg bw per day for males and 1.4, 4.1, and 13 mg/kg bw per day for females. Only mild changes were observed by light microscopy in the livers of rats fed the high dose. The NOEL for effects on the liver was thus 4.1–13 mg/kg bw per day. The nephrotoxic changes were localized in the proximal convoluted tubules of males fed diets containing > 15 mg/kg and of females at > 50 mg/kg. The NOEL for effects on the kidney was < 1.4 mg/kg bw per day in males and 1.4 mg/kg bw per day in females (Table 5; Voss et al., 1993, 1995b). Ultrastructural changes were observed in the livers in males and females at doses > 15 and > 50 mg/kg of diet, respectively. The activity of serum enzymes and the concentrations of cholesterol and triglycerides were increased at 150 mg/kg of diet (Riley et al., 1994). In a subsequent study in male rats dosed intraperitoneally with fumonisin B1 at 2 mg/kg bw per day for 4 days, alanine aminotransferase activity, serum cholesterol concentration, and apoptosis (on the basis of morphology and the terminal UTP end-labelling assay) were increased in liver on day 3. Although increased proliferation was also seen on day 3, it was not statistically significant until day 5 (only liver was examined on days 1, 3, and 5) (Li et al., 2000).
In male and female rats given fumonisin B1 at 1, 5, 15, 35, or 75 mg/kg bw for 11 days by gavage, the histopathological changes in the kidneys were similar to those described in other studies, and males were more sensitive to the renal effects (< 1.0 mg/kg bw per day for males and 5 mg/kg bw per day for females). Primary changes associated with bone-marrow toxicity were also found in both sexes. Hepatotoxicity, reduced liver weight, and increased vacuolization of adrenal cortical cells were found in female rats treated with doses > 15 mg/kg bw per day. Elevated cholesterol concentrations were observed in female rats from 5 mg/kg bw per day. Changes in serum enzyme activity and cholesterol concentration in males were markedly increased from 15 mg/kg bw per day, while single-cell necrosis and mitosis were seen at 15–75 mg/kg bw per day. Mild lymphocytosis in the thymic cortex was seen in rats treated at 5 mg/kg bw per day and persisted in rats at the highest dose. It was concluded that male rats are marginally more sensitive than female rats to fumonisin B1 (Table 5; Bondy et al., 1996, 1998).
The number of GST-P+ hepatocytes was increased in males and females given 35 and 75 mg/kg bw per day by gavage, respectively, for 11 days. In the same study, the area occupied by GST-P+ minifoci was increased significantly in both sexes at 75 mg/kg bw per day. The hepatocyte proliferation rate, measured as the expression of proliferating cell nuclear antigen (PCNA), was significantly enhanced in the livers of rats in which the number of GST-P+ cells was increased ((Mehta et al., 1998). Pathological changes occurred at the same doses in males and females, although they appeared to be more severe in males (Bondy et al., 1996, 1998). After intraperitoneal administration of a single dose of 7.5 or 10 mg/kg bw to male rats on four consecutive days, only the higher dose significantly enhanced the induction of GST-P+ minifoci. The authors suggested that, like known genotoxic carcinogens, fumonisin B1 can induce GST-P+ hepatocytes and their subsequent development into minifoci in the presence of enhanced hepatocyte proliferation, presumably in response to toxicity.
Maize samples associated with outbreaks of equine leukoencephalomalacia fed to male rats for 28 days caused degenerative changes in the liver and kidney similar to those seen with purified fumonisin B1 (Table 6; Voss et al., 1989). The toxicity was probably exacerbated by the nutritionally imbalanced diet. On the basis of the estimated feed intake and the estimated fumonisin B content of the two samples (150 and 20 mg/kg of feed), the mean daily intake of fumonisin B1 and B2 was equivalent to 13 and 1.7 mg/kg bw, respectively (Plattner et al., 1990).
Table 6. Results of studies in rats given diets containing samples naturally contaminated with fumonisin B1 or fungal cultures
|
Strain and sex |
Dietary addition, duration |
Fumonisin B intake |
Main pathological lesions or effects |
Reference |
|
|
Liver |
Kidney |
||||
|
Sprague-Dawley, male |
Maize screeningsa,28 days |
13 |
Single-cell necrosis,fibrosis (mild), bile-duct hyperplasia, adenofibrosis, mitotic figures, disruption of lobular structure |
Tubular basophilia, cytoplasmic vacuolization, sloughing of tubular epithelia, single-cell necrosis, epithelial hyperplasia |
Voss et al. (1989) |
|
1.7 |
Mild changes |
||||
|
Sprague-Dawley, male |
MRC 826 diet, 28 days |
18 |
Bile-duct proliferation, single-cell necrosis, minimal fibrosis, mitotic figures, inflammatory cells |
Retrospective examination revealed lesions similar to those described above. |
Voss et al. (1990) |
|
Sprague-Dawley, male |
Fungal culture, 21 days) |
0.4–0.6 |
No lesions |
All groups: basophilic epithelial cells, apoptosis, sloughing of tubular epithelia |
Voss et al. (1998) |
|
2.7–4.5 |
Mild changes |
||||
|
14–20 |
Single-cell necrosis and bile-duct hyperplasia |
||||
|
Fischer 344, male |
Contaminated maize, 123–176 days |
2.3–3.2 |
Neoplastic nodules (12/12); adenofibrosis (12/12); cholangiocarcinoma (12/12) |
No lesions |
Wilson et al. (1985) |
|
BD IX |
Mouldy maize in Transkei diet, |
0.65–1.4 |
Proliferating bile-duct oval cells (grade 3; 17/23), hyperplastic nodules (grade 3; 8/23) |
Not reported |
Purchase & Joubert (1970); Purchase et al. (1975) |
|
BD IX, male |
MRC 826 in Epol diet; 288 days on 4%, 606 days on 2%; total, 894 days |
6.9 (4%) |
Cirrhosis (20/20); adenofibrosis (19/20); ductular carcinoma (10/20); hepatocellular carcinoma(12/20); basal-cell hyperplasia (11/15) |
No lesions |
Marasas et al. (1984) |
|
3.2 (2%) |
|||||
|
BD IX, male |
MRC 826 in semi-purified diet; 211 days on 0.25%, 311 days on 0.5%, 81 days on 0.75%, 266 days on 0.5%; total, 869 days |
0.4 (0.25%) |
Neoplastic nodules (18/21); GGT+ foci (18/21); fatty change (21/21); hepatocellular carcinoma (2/21); ductular hyperplasia (21/21); adenofibrosis (19/21); cholangiocarcinoma (8/21) |
No lesions reported |
Jaskiewicz et al. (1987a) |
|
0.9 (0.5%) |
|||||
|
1.3 (0.75%) |
|||||
GGT+, positive for gamma-glutamyl transpeptidase
a
Naturally contaminated maize involved in field outbreaks of equine leukoencephalomalacia in the USAMale rats were fed diets containing F. verticilioides MRC 826 culture material at a concentration of 270 mg/kg for 28 days, providing an intake of fumonisin B equivalent to 18 mg/kg bw per day. Pathological lesions were found in the liver (Table 5; Voss et al., 1990). Retrospective examination (K. Voss, personal communication) revealed renal lesions similar to those described by Voss et al. (1989), who reported on the effects of three batches of F. verticillioides that produced mainly fumonisin B1, B2, or B3. Concentrations of fumonisin B of 4.6–6.9, 32–53, and 220–300 mg/kg were incorporated into the diets, resulting in estimated intakes of 0.4–0.6, 2.7–4.5, and 14–20 mg/kg bw per day. Liver lesions were reported at the two higher doses, whereas renal lesions were observed at all doses (Table 6; Voss et al., 1998). The toxicological effects of fumonisin B2- and fumonisin B3-containing cultures, fed at similar doses, mimicked those of the fumonisin B1 culture material. The toxicity was reversed after 3 weeks on fumonisin B-free diets.
(f) RIVM rats
Male rats were given pure fumonisin B1 at a dose of 0.19, 0.75, or 3 mg/kg bw per day by gavage for 28 days. Treatment had no effect on body weight, but the kidney weight was significantly reduced in animals given the highest dose, and increased apoptosis and renal tubular-cell death were seen at the two higher doses. The serum concentrations of urea and creatinine were not altered. There was no histological indication of liver toxicity, but serum gamma-glutamyl transferase activity was significantly increased at 0.75 and 3 mg/kg bw per day; no change was observed in the activity of alanine and aspartate transaminases. The NOEL for increased renal tubular-cell death was 0.19 mg/kg bw per day (Table 5; de Nijs, 1997).
(g) Other species
As maize is an important component of many animal feeds, numerous studies have been conducted with agriculturally important species and pure fumonisins, contaminated maize screenings, or maize culture material of F. verticillioides. Examples of these studies include those in catfish, cattle (Mathur et al., 2000), goats (Gurung et al., 1998), lambs, mink, poultry, and rabbits. In all cases in which toxicity was seen, it involved the liver and/or kidney, heart, or homologous organs, and, when measured, increased concentrations of free sphinganine in tissues, serum, or urine. Of all the agriculturally important species studied, fetal male rabbit appeared to be the most sensitive to fumonisin-induced toxicity, on the basis of decreased absolute kidney weight (LOEL, 0.1 mg/kg bw per day by gavage); however, when the kidney weight was normalized to the fetal body weight, the difference was not significant (LaBorde et al., 1997).
(a) B6C3F1 mice
Male and female mice were fed diets containing pure fumonisin B1 at a concentration of 0, 5, 15, 50, or 80 mg/kg for 2 years, providing doses of 0, 0.65, 1.9, 6.6, and 13 mg/kg bw per day for females and 0, 0.53, 1.6, 9.0, and 15 mg/kg bw per day for males during weeks 51–104 of the study. No differences were found in the body weights of treated and control animals; however, the body weights of females were 30% lower and those of males about 15% lower than those of B6C3F1/NCTR BR mice in other studies at the laboratory. Analysis of feed consumption rates showed that the mice were consuming about 30% less feed than mice in other studies at the laboratory, and this was found to be due to reduced availability of feed through the screen feeders, although the particle size of the powdered feed was not altered by the addition of fumonisin B1. As a result, the tumour incidences were about 12% lower than expected (Howard et al., 2001b). The survival of female mice at the highest dose was shorter than that of mice on control diets or diets containing 50 mg/kg. The decrease in survival began at about 1 year of age and continued until the end of the study. The survival rates of females at other doses were similar to those of female controls. Treatment had no effect on the survival of male mice.
Hepatocellular adenomas were found in 12% of female mice given control diet and in 6.5% at 5 mg/kg, 2.1% at 15 mg/kg (not statistically significant), 36% at 50 mg/kg, and 74% at 80 mg/kg of diet. Hepatocellular carcinomas were not found in females receiving 0, 5, or 15 mg/kg of diet but occurred in 22% of those at 50 mg/kg of diet and 23% of those at 80 mg/kg of diet. The combined incidence of hepatocellular adenomas and carcinomas was thus 12% in female controls, 43% at 50 mg/kg, and 88% at 80 mg/kg of diet. The increases in the incidence rates of adenomas and carcinomas at 50 and 80 mg/kg of diet were statistically significant. The hepatocellular adenomas were characterized by distinct foci of eosinophilic or basophilic cells and routinely compressed the adjacent normal parenchymal cells. The carcinomas were characterized by poorly differentiated anaplastic cells. The increased incidences were accompanied by an increased prevalence of hepatocellular hypertrophy and multifocal necrosis. Male mice showed no increase in the incidence of neoplasia of any type, including the liver: about 25% of the mice had hepatocellular adenomas or carcinomas. Although hypertrophy was correlated with tumour incidence in the female mice, it was also present in the livers of the male mice at 80 and 150 mg/kg of diet in the absence of an increased tumour incidence. The NOEL for carcinogenicity in these feed-restricted female mice was 15 mg/kg of diet, equal to 1.9 mg/kg bw per day (Table 4; National Toxicology Program, 1999).
(b) BD IX rats
Male rats received a semi-purified diet marginally deficient in vitamins, lipotropes, and some minerals containing fumonisin B1 at a concentration of 50 mg/kg, equivalent to 1.6 mg/kg bw per day. From 18 months onwards, some hepatocyte nodules showed dysplasia and nuclear atypia characteristic of preneoplastic changes, a few of which were transformed into hepatocellular carcinoma. Of the 15 rats that were killed between 18 and 26 months, 10 developed hepatocellular carcinoma, of which two metastasized to the lung and heart and one to the kidneys. Another lesion that was present consistently from 6 months onwards was cholangiofibrosis, which developed into cholangiocarcinoma in some rats towards the end of the experiment. Histopathological changes in the kidney were observed rarely, although fumonisin B1 tended to cause some lesions towards the end of the experiment. These lesions were not specific, although scant necrosis and other degenerative changes were found in the proximal convoluted tubules (Table 5; Gelderblom et al., 1991).
The dose–response relationship between fumonisin B1 and hepatocarcinogenesis was studied in BD IX rats given the same diet as used in the previous study but containing fumonisin B1 at a concentration of 1, 10, or 25 mg/kg. Minor changes were found in some rats given 10 mg/kg of diet and only minimal changes in those given 1 mg/kg of diet. The rats that received 25 mg/kg of diet had apoptosis, proliferation of duct epithelial cells, and mild fibrosis, which in some cases caused bridging between the portal tracts, resulting in a slight distortion of the architecture of the liver in some rats. One rat in this group showed a large focal area of adenofibrosis. In rats killed at 24 months, hepatocyte nodules were found in 9/17 rats given 25 mg/kg of diet. Other pathological changes in the liver included mild periacinar fatty changes, mild-to-prominent anisonucleosis, ground-glass foci, and a few apoptotic bodies, mainly in rats given 10 or 25 mg/kg of diet. Lesions in the kidneys were restricted to the tubular epithelium and included the presence of granular casts, necrosis, apoptosis, calcification, and regenerative foci (which may also be interpreted as hyperplastic foci) of the tubular epithelium in the proximal convoluted tubules. These lesions were found mainly at 25 mg/kg of diet and to a lesser extent at 10 mg/kg of diet, representing mean daily intakes of 0.8 and 0.3 mg/kg bw, respectively (Table 5; Gelderblom et al., 2001b).
Lesions were induced in the livers of rats fed food samples intended for human consumption for 610–691 days. The food samples were obtained from an area with a high incidence of oesophageal cancer in the Kentani district of the Transkei, South Africa. Much of the maize used was damaged by mould growth (Purchase & Joubert, 1970). The diet that caused the most pronounced liver lesions consisted of maize, beans, a salt mixture, and imifino. Several rats also showed myocardial fibrosis, while some treatments induced epithelial-cell dysplasia of the oesophagus. As no aflatoxin was detected in the samples, it was suggested that the food contained other toxic principles that were responsible for the lesions. No detailed mycological analyses were performed on the samples, but a retrospective analysis of maize collected in the same region revealed relatively high concentrations of fumonisins (Rheeder et al., 1992). The mean intake of fumonisin B was calculated to be equivalent to 0.65–1.4 mg/kg bw per day (Table 6; Purchase et al., 1975).
Commercial rat feed containing freeze-dried maize cultures of F. verticillioides MRC 826 was fed to male rats for 763 days. None of the rats survived the highest dietary concentration, calculated to provide 8% fumonisin B. Of rats fed diets containing 2% and 4% (equivalent to 3.2 and 6.9 mg/kg bw per day), 80% developed hepatocellular carcinoma and 63% developed ductular carcinoma in the liver. These tumours invariably developed in severely cirrhotic livers showing nodular hyperplasia and other changes. Pulmonary metastases developed in three rats at each dietary concentration. Adenofibrosis developed in all the animals and progressed to neoplastic lesions, referred to as ‘cholangiocarcinoma’, that were seen macroscopically to extend above the surface of the liver. Other pathological changes included endothelial hyperplasia of the ventricular endocardium and/or intraventricular thrombosis and oesophageal basal-cell hyperplasia in about 50% of rats that received the culture material (Table 6; Marasas et al., 1984). The mild basal-cell hyperplasia in the oesophagus and the cardiac changes were not reproduced with fumonisin B1 (Gelderblom et al., 2001a).
The same freeze-dried culture material at concentrations of 0.25–0.75% was given to male rats in a semi-synthetic diet marginally deficient in certain vitamins and minerals. After a feeding period of 23–27 months, two of 21 rats developed hepatocellular carcinoma with lung metastases and eight of 21 animals developed cholangiocarcinoma. Other changes in the livers are summarized in Table 6. No lesions were found in the kidney. Basal-cell hyperplasia was prominent in the oesophageal epithelium; myocardial disseminated fibrosis occurred frequently; and endocardial and subcardial fibrosis were seen in three rats. The low incidence of liver tumours was due to the minimal hepatotoxicity induced by the relatively low dietary concentrations of culture material. The estimated intake of fumonisin B was 0.4–1.3 mg/kg bw per day (Table 6; Jaskiewicz et al., 1987a).
(c) Fischer 344N rats
Male and female rats were fed diets containing pure fumonisin B1 at a concentration of 0, 5, 15, 50, or 100 mg/kg for 2 years, equal to mean doses of 0, 0.27, 0.78, 2.6, and 5.2 mg/kg bw per day for females and 0, 0.22, 0.67, 2.2, and 6.6 mg/kg bw per day for males between weeks 51 and 104 (Howard et al., 2001b). There was no dose-related difference in the survival of rats at 104 weeks. Females had decreased weight only at the highest dose, and males showed no treatment-related changes in body weight. Serum from male rats killed at 6, 10, 14, or 26 weeks showed no treatment-related changes in cholesterol or triglyceride concentration or alanine aminotransferase activity. The relative weight of the liver in males, but not females, was significantly decreased at all doses, and the relative weight of the kidney was decreased in in males and females at concentrations > 15 mg/kg of diet.
Necropsy and microscopic evaluation showed an increase in the number of basophilic foci in the livers of males at 150 mg/kg of diet. The incidence of renal tubular adenomas and carcinomas was increased in males at 50 and 150 mg/kg of diet, but no tumours were found in the kidneys of males at lower doses. A re-evaluation of the renal tumours (Hard et al., 2001) showed that tubular adenomas were present in four and renal tubular carcinomas in eight of 48 male rats at 50 mg/kg of diet; a total of 10 rats bore tumours. At the highest dose, six of 48 rats developed adenomas and 10 of 48 developed carcinomas. These increases were statistically significant (Howard et al., 2001a). The increased incidence of renal tumours was accompanied by an increased incidence of foci of atypical tubular hyperplasia, in 4/48 and 9/48 rats at 50 and 150 mg/kg of diet, respectively (Hard et al., 2001). Howard et al. (2001b) also reported renal tubular epithelial-cell hyperplasia at these doses at 2 years, in 2/48, 1/40, 4/48, 14/48, and 8/48 of male rats receiving 0, 5, 15, 50, and 150 mg/kg of diet, respectively. Similarly, increased renal tubular epithelial-cell apoptosis and proliferation were detected at the two higher doses in male rats killed after 6, 10, 14, and 26 weeks on fumonisin B1-containing diets.
In a detailed review of the renal histopathology, nephrotoxicity, including apoptosis and regeneration, was found in male rats throughout treatment at concentrations > 15 mg/kg of diet. The single-cell death observed was usually preceded by loss of cell anchorage to the basement membrane and detachment into the lumen. The renal tubular adenomas were characterized by a defined focus of expansive tubular cells, and the nuclear and cell volumes were greater in the adenoma cells than in normal adjacent cells. The cytoplasm of the adenoma cells stained clear to basophilic. The renal tubular carcinomas were a rare, highly malignant varient characterized as growths of abnormal and atypical cells (anaplastic) that compressed and invaded neighbouring normal tissue. The cells within the growing boundary of the carcinoma contained basophilic cytoplasm with typically increased volume and hyperchromatic nuclei. Necrosis was evident within the larger carcinomas. In many of the carcinomas, small renal tubule-like structures were evident, and these carcinomas metastasized to the lung and lymphatic tissues. Female rats showed no significant increase in the incidence of fumonisin B1-induced tumours. One renal adenoma was detected in a female rat at 50 mg/kg of diet, and one renal tubular carcinoma was found in a female that consumed 100 mg/kg of diet. The NOEL for induction of renal tumours in male rats was 0.67 mg/kg bw per day, whereas the NOEL for renal toxicity was 0.22 mg/kg bw per day (Hard et al., 2001).
(d) Trout
Rainbow trout were fed Oregon test diet containing pure fumonisin B1 at a concentration of 0, 3.2, 23, or 100 mg/kg for 34 weeks, in the absence or presence of an initiator, equivalent to doses of 0.2, 1.3, and 3.5 mg/kg bw per day on the basis of the consumption of this diet by trout of 5.6% of body weight per day. In trout fed the treated diet, in the absence of initiation, no tumours or lesions were found after 60 weeks in any tissue examined (liver, kidney, stomach, swim bladder). Feeding of diets containing fumonisin B1 for 42 weeks increased the incidence of liver tumours in trout fry initiated by immersion for 30 min in a bath containing either aflatoxin B1 (100 ng/ml) or N-methyl-N´-nitro-N-nitrosoguanidine (35 µg/ml). The calculated NOEL for promotion of liver tumours was 0.2 mg/kg bw per day (Carlson et al., 2001).
(e) Baboons
The major pathological effects in baboons fed diets containing culture material of F. verticillioides included acute congestive heart failure in two baboons after 143 and 248 days. One baboon developed cirrhosis after being fed the diet for 720 days (Kriek et al., 1981b).
(f) Vervet monkeys
Culture material of F. verticillioides strain MRC 826 was fed to male and female vervet monkeys (Cercopithecus aethiops) for 13.5 years (WHO, 2000a; Gelderblom et al., 2001c). Analysis of feed intake and degree of fungal contamination indicated a fumonisin B content of 8.2-13 mg/kg of diet. Toxicity was monitored by bimonthly clinical chemical analyses throughout treatment, and liver biopsy samples were taken at regular intervals up to 4.5 years. The threshold dose of fumonisin B for renal and hepatic damage was calculated to be 0.11-0.18 mg/kg bw per day. Typical lesions observed in the liver of animals at the high dose included portal-to-portal fibrosis, hepatocyte nodules, bile-duct proliferation, apoptosis, and an increased sphinganine:sphingosine ratio (Fincham et al., 1992; Shephard et al., 1996a). The kidneys of these monkeys have not yet been examined for histopathological changes. The LOEL for sphingolipid changes in the serum was 22-48 mg/kg of diet, equivalent to 0.29-0.64 mg/kg bw per day. Other parameters that were also affected throughout the study as a result of the treatment included lipid parameters associated with hypercholesterolaemia and significantly decreased white and red blood cell and platelet counts.
A working group convened by IARC (IARC, 1993) reached the following conclusions about the carcinogenicity of toxins derived from F. verticillioides, including fumonisin B1:
|
• |
There is inadequate evidence in humans for the carcinogenicity of toxins derived from F. verticillioides. |
|
• |
There is sufficient evidence in experimental animals for the carcinogenicity of cultures of F. verticillioides that contain significant amounts of fumonisins. |
|
• |
There is limited evidence in experimental animals for the carcinogenicity of fumonisin B1. |
Overall evaluation: Toxins derived from Fusarium verticillioides are possibly carcinogenic to humans (Group 2B).
2.2.4 Genotoxicity
The possible genotoxicity of fumonisins has been assessed in vivo and in vitro (reviewed in WHO, 2000a; Table 7). The results of several independent studies showed that fumonisin B1 and other fumonisins were not mutagenic in several strains of Salmonella typhimurium, with and without addition of microsomal activation from rat liver. Tests for gene mutation in Escherichia coli PQ37, differential DNA repair in E. coli K12, and DNA repair in vivo and in vitro in two laboratories also gave negative results.
Table 7. Results of assays for the genotoxicity of fumonisins B1(FB)
|
End-point |
Test object |
Concentration |
Results |
Reference |
|
in vitro |
||||
|
Reverse mutation |
S typhimurium TA97a, TA98, TA100, TA102 |
FB1, FB2 (1-10 mg/plate), and FB3 (5 mg/plate) |
Negativea |
Gelderblom & Snyman (1991) |
|
Reverse mutation |
S typhimurium TA98, TA100 |
FB1 (0.7-500 µg/plate) |
Negativea |
Knasmüller et al. (1997) |
|
Unscheduled DNA synthesis |
Primary rat hepatocytes |
FB1 (80 µmol/ plate); FB2 (40 µmol/plate) |
Negative |
Gelderblom et al. (1992) |
|
Unscheduled DNA synthesis |
Primary rat hepatocytes |
FB1 (0.5-250 µmol/plate) |
Negative |
Norred et al. (1992) |
|
Chromosomal aberrations |
Primary rat hepatocytes |
FB1 (0.01-100 µg/ml) |
Positiveb |
Knasmüller et al. (1997) |
|
Micronucleus formation |
Negative |
|||
|
Gene mutation |
E. coli |
FB1 (5-500 µg/plate) |
Negativea |
|
|
DNA repair |
E. coli |
0.7-500 µg/plate |
Negativec |
|
|
DNA binding |
Oligonucleotides |
2.5 nmol/µl |
Negative |
Pocsfalvi et al. (2000) |
|
Transformation |
Mouse embryo cells |
500 µg/ml |
Positivec |
Sheu et al. (1996) |
|
Hypermethylation of DNA |
C6 glioma cells |
9-18 µmol/L |
Positive |
Mobio et al. (2000b) |
|
Lipid peroxidation |
Rat liver nuclei |
FB1 (40-300 µmol/L) |
Positive |
Sahu et al. (1998) |
|
Lipid peroxidation |
Phosphatidylcholine bilayers |
FB1 (10 mmol/L) |
Positive |
Yin et al. (1998) |
|
Lipid peroxidation |
Vero cells |
0.14-70 µmol/L |
Positive |
Abado-Becongnee et al. (1998) |
|
Lipid peroxidation |
C6 glioma cells |
3-27 µmol/L |
Positive |
Mobio et al. (2000a) |
|
Lipid peroxidation |
Primary hepatocytes |
75-500 µmol/L |
Positive |
Abel & Gelderblom (1998) |
|
Adduct formation |
32 P-Postlabelling |
Not reported |
Negative |
P. Howard (personal communication) |
|
In vivo |
||||
|
Unscheduled DNA synthesis |
Rat liver (single dose by gavage) |
FB1 (100 mg/kg bw) |
Negative |
Gelderblom et al. (1992) |
|
FB2 (100 mg/kg bw) |
||||
|
Lipid peroxidation |
Rat liver |
250 mg/kg of diet (21 days) |
Positived |
Abel & Gelderblom (1998) |
|
Lipid peroxidation |
Rat liver |
250 mg/kg of diet plus 1-2% carbonyl iron (35 days) |
Positived |
Lemmer et al. (1999a) |
|
Lipid peroxidation |
Mouse liver |
2.25 mg/kg bw per day (subcutaneous, 5 days) |
Negativee |
Riley et al. (2001) |
a
With and without added rat liver microsomal fractionb
The European Commission (2000) considered this report unreliable owing to methodological limitations.c
No activity at lower or higher concentrationd
Dose shown to initiate cancer in rat livere
Hepatoxic doseIn one unconfirmed report, fumonisin B1 was active in a commercial biolumines-cent bacterial assay. It increased the micronucleus frequency in primary rat hepatocytes in a non-concentration-dependent manner and caused a significant, concentration-dependent increase in the frequency of chromosomal aberrations. The Commission of the European Union (2000) considered that this report was unreliable owing to methodological limitations. Fumonisin B1 transformed mouse embryo cells only at a concentration of 500 µg/ml and not at lower or higher concentrations.
Evidence in vitro and in vivo indicates that fumonisin B1 can damage DNA indirectly by increasing oxidative stress (Atroshi et al., 1999; Mobio et al., 2000b). Oxidative damage was closely associated with fumonisin B1-induced hepatotoxicity and induction of putative preneoplastic lesions in vivo, while the plasma and microsomal membranes, and to some extent the mitochondria and nuclei, appeared to be significantly affected by lipid peroxidation (Abel & Gelderblom, 1998).
Studies in primary hepatocytes showed a similar relationship between cytotoxicity and lipid peroxidation. Although lipid peroxidation was prevented by the addition of alpha-tocopherol, cytotoxicity was reduced, but not to baseline levels, suggesting that the cytotoxicity was due not only to oxidative damage but also to fumonisin B1-induced toxic effects. Fumonisin B1 potentiated the effect of iron on lipid peroxidation and was toxic to the liver, independently of its effect on lipid peroxidation (Lemmer et al., 1999a). The toxin induced lipid peroxidation in cell membrane preparations (Yin et al., 1998) and isolated rat liver nuclei. Nuclear membrane lipid peroxidation with concomitant DNA strand breaks was found in isolated rat liver nuclei treated with fumonisin B1 in vitro at concentrations of 40-300 µmol/L (Sahu et al., 1998). It was suggested that the formation of hydroxy and peroxyl radicals in the close vicinity of nuclear material could induce DNA strand breaks. It is not known whether the induction of chromosomal aberrations in primary hepatocytes (Knasmüller et al., 1997) is related to oxidative damage. Oxidative damage has been reported in Vero cells (Abado-Becongnee et al., 1998) treated with fumonisin B1 at a concentration (0.14 µmol/L) below that which inhibits protein and DNA synthesis (> 14 µmol/L). However, in mice, fumonisin B1-induced liver toxicity occurred in the absence of evidence of increased lipid peroxidation (Riley et al., 2001). In numerous studies in vitro, fumonisin was used to protect cells from oxidant-induced cell death (for examples, see Table 3). Whether this occurs in vivo is unknown.
Although fumonisin B1 caused oxidative damage to DNA in vitro, there is no compelling evidence to suggest that it binds covalently to DNA. Extracts of Fusarium fungi have been shown to form DNA adducts (Lu et al., 1988; Bever et al., 2001), as detected by the sensitive 32P-postlabelling method. However, DNA adducts did not form after incubation of fumonisin B1 with DNA in the presence or absence of microsomal protein (rat liver microsomes), and are probably formed by other mycotoxins produced by the fungus (P. Howard, personal communication). Using mild electrospray ionization mass spectrometry, Pocsfalvi et al. (2000) found no evidence of a specific interaction between fumonisin B1 and single- or double-stranded oligonucleotides. Specific non-covalent interactions were noted with fusaproliferin and beauvercin, two mycotoxins produced by F. proliferatum and F. subglutinans.
2.5">2.2.5 Reproductive toxicityWhile fumonisins are embryotoxic in vitro, there were no published data to support the conclusion that they are developmental or reproductive toxicants in farm animals or humans (WHO, 2000a). Fumonisin B1 inhibits the biosynthesis of the glycosylphosphatidylinositol-anchored folate transporter in vitro, and folate deficiency is associated with an increased risk for neural tube defects. Inhibition of the folate transporter in vivo has not been confirmed in feeding studies. There is no evidence of neonatal toxicity in laboratory animals. Except in one study in Syrian hamsters, embryotoxicity occurred secondary to maternal toxicity. In Syrian hamsters, administration of fumonisin B1 by gavage at a dose of 18 mg/kg bw per day on days 8 to 10 or 12 of gestation resulted in a statistically significant increase in the number of fetal deaths. The fact that there was no maternal toxicity at this dose indicates that the Syrian hamster is extremely resistant to the effects of fumonisin. In rats and mice, reproductive and developmental effects occurred at doses that were also maternally toxic, suggesting that the reproductive and developmental toxicity of fumonisin is mediated through maternal toxicity. However, in rabbits, maternal toxicity was observed at a daily dose (given by gavage in water) as low as 0.25 mg/kg bw on days 3–19 of gestation, but there was no increase in the frequency of fetal loss or of gross visceral or skeletal abnormalities at any maternal dose of fumonisin B1 (0–1.75 mg/kg bw per day); however, slight decreases in fetal weight did occur at maternally toxic doses.
Because fumonisins are known to cause field outbreaks of equine leukoen-cephalomalacia and porcine pulmonary oedema, many studies have been undertaken to better understand the physiological and biochemical mechanisms responsible for these brain and lung diseases. The cardiovascular changes in equids are similar to those seen in pigs during onset of the syndrome (Smith et al., 1999; Constable et al., 2000a; Smith et al., 2000), and the results of studies suggest a common underlying mechanism for the two diseases involving sphingosine-mediated L-type calcium channel blockade (Constable et al., 2000b).
A thorough review of studies on these two diseases is contained in the IPCS monograph (WHO, 2000a). The following is a brief summary of that review.
Equine leukoencephalomalacia syndrome is a sporadic condition characterized by the presence of liquefactive necrotic lesions in the cerebrum. The disease appears to be unique to equids, although brain lesions have also been reported in rabbits (Bucci et al., 1996), pigs (Fazekas et al., 1998), and carp fed fumonisins (Pepeljnjak et al., 2000). The brain lesions in rabbits and pigs could not be reproduced in subsequent studies (T. Bucci, personal communication; Zomborszky et al., 2000), and the study in carp has not been repeated.
Analysis of feeds from confirmed cases of equine leukoencephalomalacia in the USA indicated that consumption of feed with a fumonisin B1 concentration > 10 mg/kg of diet (equivalent to 0.2 mg/kg bw per day) was associated with an increased risk for developing the disease, whereas a concentration < 6 mg/kg of diet (equivalent to 0.12 mg/kg bw per day) was not. The minimum oral dose sufficient to induce equine leukoencephalomalacia appeared to be 15–22 mg/kg of diet (equivalent to 0.30 mg/kg bw per day for 150 days to 0.44 mg/kg bw per day for 241 days) in studies of naturally contaminated maize screenings or culture material (F. proliferatum) containing fumonisins. The minimum oral dose of pure fumonisin B1 that will induce equine leukoencephalomalacia is unknown. The minimum intravenous dose of pure fumonisin B1 that induced neurological abnormalities was 0.01–0.05 mg/kg bw per day. If the intravenous dose is assumed to represents 5% of the oral dose, the equivalent oral dose would be 0.2–1.0 mg/kg bw per day. The NOEL for cardiovascular abnormalities was 0.2 mg/kg bw per day, but the NOEL for serum biochemical abnormalities was equivalent to < 0.2 mg/kg bw per day (Constable et al., 2000b).
Equine leukoencephalomalacia has been reproduced by giving fumonisin B1 orally or intravenously, giving naturally contaminated maize screenings, or giving diets containing F. proliferatum maize culture material with predominantly fumonisin B1 or fumonisin B2. In addition to the brain lesions, histopathological abnormalities are usually found in the liver, and recent studies suggest that fumonisin B1 is also nephrotoxic in horses. Changes in serum enzymes indicative of liver damage and behavioural changes are usually preceded by increased concentrations of free sphingoid bases and cholesterol in serum or plasma. It has been hypothesized that equine leukoencephalomalacia is a result of cerebral oedema due to an inability to shut down the blood flow to the brain when the horse lowers its head to eat and drink (Constable et al., 2000b).
Evidence of disruption of sphingolipid metabolism is an early indicator of exposure of horses to fumonisins. For example, all ponies fed diets containing maize screenings naturally contaminated with fumonisins at > 22 mg/kg (primarily fumonisin B1) had large increases in the serum concentration of free sphinganine. The increase was reversible. This and the increased sphinganine:sphingosine ratio occur before increases in serum transaminase activity and clinical signs of equine leukoencephalomalacia.
Porcine pulmonary oedema is also believed to be induced by cardiovascular dysfunction. Significant changes in oxygen consumption and in several haemodynamic parameters are seen before the onset of clinical signs in pigs fed diets containing fumonisins, suggesting that pulmonary hypertension caused by hypoxic vasoconstriction might contribute to the syndrome. Fumonisin B1 has been shown to reduce the mechanical efficiency of the left ventricle (Constable et al., 2000a), suggesting that pulmonary oedema in pigs is due primarily to acute left-sided heart failure (Smith et al., 1999; Constable et al., 2000a; Smith et al., 2000). Hepatotoxicity is usually observed at doses lower than those that induce the pulmonary oedema. Liver lesions were induced by maize screenings providing a dose of fumonisin of 1.1 mg/kg bw per day (17 mg/kg of diet). In a study in weaned piglets, mild pulmonary oedema was induced in three of four animals fed diets that contained 10 mg/kg (equivalent to 0.4 mg/kg bw per day) of fumonisin B1 from F. verticillioides culture material for 4 weeks (Zomborszky et al., 2000). Subtle changes in performance were reported at a concentration of fumonisin B1 (1 mg/kg), which does not cause overt toxicity. Erratic growth was reported at 0.1 mg/kg (Rotter et al., 1996).
In 1989–90, outbreaks of this disease were reported in various parts of the USA. Maize screenings obtained from farms where pigs had died of porcine pulmonary oedema were contaminated predominantly with F. verticillioides. The concentrations of fumonisin B1 in the suspected feeds ranged from 20 to 360 mg/kg, while those in other feeds was < 8 mg/kg (Ross et al., 1991). The clinical signs of porcine pulmonary oedema include dyspnoea, weakness, and cyanosis. At necropsy, the animals have varying degrees of interstitial and interlobular oedema, with pulmonary oedema and hydrothorax. Toxic hepatosis occurs concurrently. The concentrations of fumonisin B1 in feed associated with this disease are usually much greater than those associated with outbreaks of equine leukoencephalomalacia. Purified fumonisin B1 has been shown to reproduce the disease when administered intravenously. Porcine pulmonary oedema has not yet been reproduced by oral administration of pure fumonisins, although it has been induced many times with culture material containing fumonisin B1.
In pigs fed diets prepared from naturally contaminated maize screenings, there was a dose–response relationship between the dose of fumonisin, pathological lesions in the liver, and the ratio of free sphinganine to free sphingosine in serum or liver. After 14 days, statistically significant increases in the serum ratio of free sphinganine to free sphingosine were observed at a concentration of total fumonisins in feed as low as 5 mg/kg (equivalent to 0.2 mg/kg bw per day). The concentrations of free sphingoid bases were also significantly elevated in kidney and lung at doses that induced no signs of toxicity in these organs. In pigs fed fumonisin in maize culture materials, significant effects on cardiovascular function were associated with significant increases in free sphingoid base concentrations in heart tissue. Subsequent studies showed that damage to pig alveolar endothelial cells in vivo was preceded by accumulation of free sphingoid bases in lung tissue. Increased activity of serum enzymes and increased concentrations of free sphingoid bases in serum were seen at all doses in the study of Zomborszky et al. (2000). It has been hypothesized that the cardiovascular alterations leading to acute left-sided heart failure are a consequence of sphingoid-base-induced inhibition of L-type calcium channels (Smith et al., 1999; Constable et al., 2000a; Smith et al., 2000). The minimum oral dose needed to induce porcine pulmonary oedema has not been clearly established; however, Smith et al. (1999) predicted that when the concentra-tions of free sphinganine and free sphingosine in plasma are > 2.2 and 1 µmol/L, respectively, haemodynamic changes will occur.
The association between fumonisins and other fungal toxins and risk factors for various human diseases are summarized in Table 8.
Table 8. Interaction of fumonisins with other fungal toxins and risk factors in the development of disease in humans
|
Geographic region/ country |
Age-standardized incidence |
Fungal infection of major dietary staples |
Mycotoxin contamination |
Dietary and other risk factors |
|
Oesophageal cancer |
||||
|
Transkei, Southern Africa (rural) |
Males: Lusikisiki, 51; Bizana, 37; Butterworth, 43; Centane, 56 (Makaula et al., 1996) |
Maize: F. verticillioides and F. graminearum (Marasas et al., 1981; Gelderblom et al., 1984; Marasas et al., 1988; Sydenham et al., 1990; Van Rensburg et al., 1990; Rheeder et al., 1992; Bever et al., 2001) |
Healthy maize: fumonisin B (2.0–2.1 mg/kg) |
Vitamin A, E, and B12, folate, selenium deficiencies (Van Helden et al., 1987; Jaskiewicz et al., 1987c, 1988a,b) |
|
Henan, Hebei, Linxian, and Shanxi provinces, northern China |
Yancheng, 135; Hebei, 140; Linxian, 108 (Yang, 1980) |
Wheat, maize, dried sweet potato, rice, soya bean; Penicillium spp., Aspergillus spp., F. verticillioides predominant fungi (Zhen, 1984; Luo et al., 1990; Chu & Li, 1994; Yoshizawa et al., 1994; Gao & Yoshizawa, 1997; Zhang et al., 1997) |
Healthy maize: fumonisin B1 (0.7–3.5 mg/kg). One study: fumonisin B1 (35 mg/kg) |
Low intake of vitamins A and C. Inverse relationship with molybdenum, manganese, zinc; no relationship with pickled vegetables |
|
Mazandaran, Province, Gonbad region, Caspian littoral of Iran |
Females: 262 |
Aspergillus, Fusarium , Penicillium spp. on maize. F. verticillioides and F. proliferatum important spp. Alternaria alternata (Kmet & Mahboubi, 1972; Hormozdiari et al., 1975; Chen et al., 1992; Bujari & Ershad, 1993; Shephard et al., 2000) |
Healthy maize: fumonisin B (1.6–6.1 mg/kg) |
Micronutrient deficiencies: iron, manganese, copper, zinc, vitamins A, C, riboflavin. |
|
Friuli–Venezia Giullia, northeast Italy |
Pordenone Province Males: 17 |
Fumonisin-producing Fusarium species |
Fumonisins: fumonisin B1 (0.15–0.38 mg/kg), fumonisin B2 (0.06–0.91 mg/kg) |
Consumption of polenta. Low intake of micronutrients such as riboflavin and niacin; interactive role of alcohol |
|
Western and central Kenya |
45% of cases |
Maize: F. verticillioides |
Healthy maize: fumonisin B |
Dietary patterns and tribal customs vary; alcohol consumption; geographical and ethnic variations (Gatei et al., 1978) |
|
Zimbabwe |
Males: Harare: 30 |
No studies |
Breakfast cereals: |
No studies |
|
Charleston County, South Carolina, USA |
Black males, 170 (death rate) |
No studies |
Maize-based human foods |
Low socioeconomic status, tobacco and alcohol (‘moonshine’ distilled from fermented maize meal); low intake of fresh fruits (Fraumeni & Blot, 1977; Brown et al., 1988) |
|
Southern Brazil |
Santa Catarina, Paranà, Rio Grande do Sul |
Maize: F. verticillioides, A. flavus |
Animal mycotoxicosis: |
Farm workers, smoking and drinking: regional variation; hot beverages (maté and chimarrao) (Victoria et al., 1987; Dietz et al., 1998; Scaff & Scussel, 1999a,b) |
|
Liver cancer |
||||
|
Jiangsu County, China |
Haimen: 52–65 (mortality rate) |
No studies |
Maize: |
Microcystins |
|
Transkei, South Africa |
Kentani: 2.4–7.7 |
Maize-based food and home-grown maize |
Aflatoxin B1 (16 ng/kg bw) Mouldy maize: fumonisin B |
Low socieconomic status; nutritional deficiencies |
|
Neural tube defects |
||||
|
Southern Texas, USA |
Lower Rio Grande valley: |
Case–control study, 1995–99, |
Maize-based foods: 1.2 mg/kg (Sydenham et al., 1991) |
Tortillas; sphingonine: |
|
China |
Hebei and Shanxi Provinces: |
Penicillium, Aspergillus spp., |
Healthy maize: fumonisin B1 |
Low socioeconomic status; nutritional deficiencies (Yang, 1980) |
|
Transkei and Mpumulanga South Africa |
Umzimkulu: 38/10 000; |
F. verticillioides, F. proliferatum, |
Healthy maize: fumonisin B1 |
Low socioeconomic status; |
|
Foodborne disease outbreak |
||||
|
Southern India |
Deccan Plateau; gastrointestinal disease |
Mouldy sorghum and maize Fusarium, Aspergillus, Alternaria spp. (Bhat et al., 1997; Prathapkumar et al., 1997) |
Sorghum: fumonisin B1 (0.14–7.8 mg/kg); aflatoxin B1 (trace–0.08 mg/kg) |
Low socioeconomic status; lack of access to other foods such as rice (Bhat et al., 1997) |
An association has been established between the occurrence of the fungus, Fusarium verticillioides (Sacc.) Nirenberg (= Fusarium moniliforme Sheldon) on maize and the incidence of oesophageal cancer in various regions of the world. Geographi-cal differences in demography, ethnic groups, genetic susceptibility, culture, economy and nutritional status all affect the rates of disease; however, some common risk factors are emerging, such as having maize as the main dietary staple and, to some extent, a low socioeconomic status. Thus, high incidences of oesophageal cancer have been associated with limited diets consisting mainly of wheat or maize and low contents of certain minerals and vitamins (Blot, 1994). Fungal contamination of maize and wheat attracted the interest of many investigators, who have characterized the toxic, mutagenic, and carcinogenic metabolites of the major fungal contaminants of these grains. The possible involvement of F. verticillioides and fumonisins in the development of oesophageal cancer in various regions of the world is evaluated on the basis of incidence rates, the presence of Fusarium spp. in maize, the presence of fumonisins and other Fusarium mycotoxins, and nutritional deficiencies and other dietary risk factors. Those aspects that were addressed by WHO (2000) are not discussed in detail.
(a) South Africa
A dramatic increase in the incidence of oesophageal cancer in the Transkei region of Eastern Cape Province, South Africa, was first described by Burrell (1957, 1962; Burrell et al., 1966). Subsequent reports were published for the periods 1955–57 and 1965–69 covering all districts of the Transkei (Rose, 1965, 1973; Rose & Fellingham, 1981) and for the periods 1981–84 (Jaskiewicz et al., 1987b) and 1985–90 (Makaula et al., 1996) focusing on cancer incidence in four selected districts: two areas of high incidence (Centane or Kentani and Butterworth in the south-west) and two areas of low incidence (Lisikisiki and Bizana in the north-east). Data for 1991–95 indicate that the age-standardized incidence rate has decreased in Butterworth to 51 per 100 000 but increased in Lusikisiki and Bizana to 37 per 100 000. Conversely, the rate in Centane has remained consistently high in males, at 56 per 100 000.
A comparative study of the incidence of Fusarium spp. in maize in regions of low (Bizana and Lusikisiki) and high (Butterworth and Centane) incidence of oesophageal cancer in the Transkei indicated that three Fusarium species (F. graminearum, F. verticillioides, and F. subglutinans) were the predominant fungal contaminants of home-grown maize during the 1976–77 season (Marasas et al., 1979). The association between the occurrence of F. verticillioides in maize and the incidence of oesophageal cancer was established in a detailed mycological study of home-grown maize in the four districts, carried out for six seasons in 1976–79, 1985–86, and 1989 (Marasas et al., 1981, 1988; Sydenham et al., 1990; Rheeder et al., 1992). The incidences of F. graminearum and F. subglutinans and the concentrations of toxins produced by these Fusarium species, i.e. deoxynivalenol, nivalenol, and zearalenone and moniliformin, respectively, were significantly higher in home-grown maize from the area of low incidence of oesophageal cancer than that of high incidence (Sydenham et al., 1990; Rheeder et al., 1992). Significantly higher concentrations of fumonisin B1 and fumonisin B2 were detected in visibly uncontaminated (‘healthy’) and mouldy samples of maize from the high-incidence areas than the low-incidence areas. The environmental conditions in the high-incidence area favour the colonization of maize ears and toxin production by F. verticillioides, whereas the conditions in the low-incidence area favour F. graminearum and F. subglutinans. This interaction was clear from a mycological analysis of maize harvested during 1985 (Sydenham et al., 1991), while variable results were obtained in maize samples collected during the 1976 and 1977 seasons (Marasas et al., 1979). The population in these regions is exposed not only to the known Fusarium toxins but to various other mycotoxins that may play a role in the development of cancer in humans, and specifically cancers of the oesophagus and the liver. These toxins include aflatoxin B1, present in food and traditional beer (Van Rensburg et al., 1990), the mutagen fusarin C (Gelderblom et al., 1984), and other compounds produced by F. verticilioides (Bever et al., 2001).
People in the high-incidence area for oesophageal cancer in the Transkei were exposed not only to mycotoxin contamination of maize but also had nutritional deficiencies, such as of vitamins A, E, and B12, selenium, and folate, when compared with people in the low-incidence areas (Van Helden et al., 1987; Jaskiewicz et al., 1987b, 1988a,b). In contrast to the study of Van Rensburg et al. (1983), a study by Jaskiewicz et al. (1988b) showed no difference in the plasma concentrations of zinc, copper, and magnesium in people in the low- and high-incidence areas. Deficiencies in these micronutrients and in manganese and molybdenum have been implicated as risk factors for oesophageal cancer in populations that subsist on either maize or wheat (Van Rensburg, 1985). A case–control study in Zulu men with oesophageal cancer carried out in Durban, South Africa, indicated that consumption of purchased maize meal is one of the major risk factors for development of the disease (Van Rensburg et al., 1985). Mineral deficiencies in the soil were implicated as another possible risk factor in the high-risk areas in the Transkei (Burrell et al., 1966); however, a study of the elemental content of soil and maize leaves (Rheeder et al., 1994) showed no deficiency in mineral elements and no association with the risk for oesophageal cancer.
Alcohol consumption and tobacco smoking are widely regarded as major factors in the development of oesophageal cancer in developed countries (Blot, 1994). In the endemic areas in the Transkei, a number of the patients did not use tobacco or consume alcohol, hence ruling out these factors as the sole causative agents (Rose, 1973; Sammon, 1992). In Zulu men, however, smoking was identified as a risk factor, while alcohol consumption had no appreciable effect (Van Rensburg et al., 1985).
(b) China
A survey in Henan, Hebei, and Shanxi, three provinces in northern China, showed that the highest mortality rates from oesophageal cancer were those in two counties, Yancheng and Hebei, with crude rates of 135 and 140 per 100 000, respectively (Yang, 1980). Data for 12 years (1959–70) in a cancer registry in Linxian County showed an average incidence rate of 108 per 100 000, while the lowest rates were recorded in Hunyan County (1.4 per 100 000) and Tatong County (2.8 per 100 000). The male:female ratio ranged from 1.44:1 to 2.63:1.
The fungal contamination of 1121 food samples of wheat, maize, dried sweet potato, rice, and soya bean obtained from five counties of high incidence and three of low incidence for oesophageal cancer in Henan Province indicated that, in addition to Penicillium and Aspergillus species, the incidence of F. verticilioides, was significantly higher in the high-incidence area (Zhen, 1984). An increased risk was associated with a high intake of wheat and maize, further emphasizing the importance of performing detailed studies on the fungal contamination of these food commodities (Li et al., 1989).
Analysis of 31 maize samples collected from households in Linxian and Cixian counties showed high concentrations of fumonisin B1 in both mouldy and healthy samples; low concentrations of aflatoxin B1 and various type A and B trichothecenes were also found in the mouldy samples (Chu & Li, 1994). In studies on maize (Yoshizawa et al., 1994) and on wheat and maize (Gao & Yoshizawa, 1997), the frequency of fumonisin contamination was higher in Linxian County, while the mean concentrations of fumonisin B mycotoxins were similar to those in Shangqiu County, a low-incidence area of oesophageal cancer in Henan Province. The samples were also frequently co-contaminated with trichothecenes, consisting mainly of deoxy-nivalenol and to some extent nivalenol. The concentrations of these mycotoxins were significantly higher in Linxian, and zearalenone was detected only in the maize samples from this County.
Aflatoxin B1 and fumonisin B contamination was measured in 246 samples of healthy maize kernels collected from villages in the high-risk counties of Cixian, Linxian, and Anyang and the low-risk counties of Fanxian and Yanqui, during 1995–96. Once again, the frequency of fumonisin contamination was higher in the high-incidence areas (65%) than in the low-incidence areas (28%). Although no clear relationship could be detected between fumonisin contamination and oesophageal cancer incidence, people in the high-incidence area were exposed to higher mean concentrations of fumonisin B in maize than those in the low-incidence areas (Zhang et al., 1997). Significantly greater contamination with deoxynivalenol, nivalenol, and zearalenone was found in the main staple foods (maize and wheat) in the high-incidence county of Linxian than in the low-incidence area, Shangui (Luo et al., 1990).
A nutritionally inadequate diet appears to play an important role in the incidence of oesophageal cancer and the development of the precancerous dysplastic state. The diet in Linxian consisted mainly of maize, wheat, millet, rice, and some seasonal vegetables. Up to 80% of the calorie intake was obtained from grains, while consumption of fruit and vegetables was low, resulting in deficiencies in vitamins A and C during certain times of the year. Detailed analyses of food and drinking-water indicated an inverse correlation between the rate of mortality from oesophageal cancer and the mineral content of the diet, including molybdenum, manganese, and zinc. Analyses of hair, serum, and urine indicated that the molybdenum content was significantly lower in men from Linxian than from the low-incidence areas of Yuxian and Xinyangxian. Similar studies carried out in oesophageal cancer patients in Henan indicated lower zinc concentrations than in normal subjects. Analysis of resected oesophagi from patients in Linxian showed significantly lower molybdenum concentrations in cancerous tissue. The consumption of pickled vegetables heavily infested with various Aspergillus and Fusarium spp. and mouldy food is common in the high-incidence areas, whereas the intake in the areas of lower incidence is less common (Yang, 1980).
Li et al. (1989) found no association with the intake of pickled vegetables, however, while the intake of wheat and maize was associated with an increased risk and that of fresh vegetables and fruit with a decreased risk for oesophageal cancer. No association was found with alcohol intake, while smoking was a mild risk factor. The presence of nitrosamine precursors in drinking-water and food and the formation of nitrosamines in the stomach have been associated with the development of oesophageal cancer (Yang, 1980). Various nitrosamines (N-nitrosodimethylamine, N-nitrosodiethylamine, and N-nitrosomethylbenzylamine) were formed in maize bread inoculated with F. verticillioides in the presence of low concentrations of sodium nitrate, and secondary nitrosamines were detected with some Aspergillus and Penicillium species (Li et al., 1979). These findings have not, however, been confirmed. Maize meal infested with F verticillioides induced tumours in the oesophagus and stomach of rats and mice (Li et al., 1980, 1982).
(c) Islamic Republic of Iran
The incidence of oesophageal cancer varied by up to 30-fold in men and sixfold in women along a 300-mile stretch of the Caspian littoral of Iran. Gonbad, an eastern region of Mazandaran Province, was regarded as one of the world’s ‘hotspots’ for oesophageal cancer in 1960–61, with truncated age-standardized rates of 206 per 100 000 for men and 262 per 100 000 for women (Kmet & Mahboubi, 1972; Hormozdiari et al., 1975). The incidence was higher among women than men in the high-incidence area. Lower incidences were recorded in the western part of the Caspian littoral.
A survey of the mycoflora of maize kernels collected from seed production centres in Sari, Moghan, and Karaj indicated that isolates of Aspergillus, Fusarium, and Penicillium were dominant (Bujari & Ershad, 1993). These include the fumonisin-producing species F. verticillioides and F. proliferatum. Shephard et al. (2000) reported that maize in Mazandaran Province contained higher concentrations of fumonisins than maize in Isfahan Province to the south of Mazandaran, a low-incidence area for oesophageal cancer. This report of the occurrence of fumonisins in maize in a high-risk area for oesophageal cancer in Iran must be followed up by studies of the maize consumption pattern in this region, the fungal contamination, and the presence of other Fusarium mycotoxins in maize. Furthermore, the samples were intended for animal consumption.
Heavy fungal contamination was also found in wheat stored in underground pits in Iran, the most prevalent fungus being Alternaria alternata (Kmet & Mahboubi, 1972). The toxins associated with this fungus have a similar structure to fumonisins and have similar biological effects in plants and cultured mammalian cells (van der Westhuizen et al., 1998). It is not known whether they also mimic the characteristic biological effects of fumonisins in animals. It has been reported that A. alternata can produce fumonisins (Chen et al., 1992).
In a detailed study of the distribution of exogenous factors related to the differences in the incidence of oesophageal cancer in the Caspian littoral, no single causative agent could be identified. Bread and sheep and goat milk or yoghurt were the main dietary staples in high-incidence areas, while rice was the mainly staple in low-incidence regions (Hormozdiari et al., 1975). A low intake of vitamins A and C, riboflavin, animal protein, fresh vegetables and fruit and a high intake of bread and tea were common in the high-incidence areas. The micronutrient deficiencies are typical of those among persons of low socioeconomic status (Joint Iran–ARC Study Group, 1977; Cook-Mozaffari et al., 1979). Food samples from both the low- and the high-incidence areas contained low concentrations of aflatoxins, polycyclic aromatic hydrocarbons, and nitrosamines (Hormozdiari et al., 1975). Alcohol and tobacco smoking were reported to have no role, as intake was negligible and the women abstained from both alcohol and nass, a mixture of opium, lime, and ash favoured by some male inhabitants. Studies performed by the Joint Iran–IARC Study Group (1977) and Ghadirian et al. (1985) provided some support for the hypothesis that pyrolysate products of opium are involved in the etiology of oesophageal cancer. Subsequent studies by Kmet & Mahboubi (1972), Ghadirian (1987), and O’Neil et al. (1980) implicated deficiencies of micronutrients such as iron, manganese, copper, and zinc, thermal irritation from hot tea, contamination of bread with silica fibre, and consumption by women of a diet consisting of sour promegranate seeds, black pepper, and garlic.
(d) Northern Italy
The standardized mortality rate for oesophageal cancer among men in Pordenone Province in the Friuli–Venezia Giullia region of northeastern Italy was 17 per 100 000 (Franceschi et al., 1990). Fumonsin-producing Fusarium species were shown to be present in maize produced in northern Italy (Logrieco et al., 1995). One study showed the presence of fumonisin B in 20 samples of polenta at concentrations of 0.15–3.8 mg/kg (Pascale et al., 1995).
Two studies showed a correlation between consumption of maize, particularly polenta, and the incidence of cancers of the upper digestive track and oesophagus in the Fruili–Venezia Giulia region (Rossi et al., 1982; Franceschi et al., 1990). In both studies, deficiencies of several micronutrients in refined maize, including riboflavin and niacin, were implicated, especially in conjunction with alcohol consumption, which may aggravate the nutritional deficiency induced by maize-based diets.
(e) Kenya
A retrospective study in Kenya of 667 cases of oesophageal cancer from the major hospitals and the Kenya Cancer Registry during the period 1968–75 showed a male:female ratio of 8:1. About 45% of the cases were from western Kenya and 45% from the centre, which are considered to be high-incidence areas as compared with the coastal and northern regions and the Rift Valley (Gatei et al., 1978).
A survey of fungal contamination of maize showed that F. verticillioides was the most frequent contaminant in maize kernels from western and central Kenya (Macdonald & Chapman, 1996), and the finding was confirmed on screening the fungal contamination of 150 maize kernel samples collected in the tropical highlands of western Kenya. Chemical analyses of 197 samples collected in this region showed little fumonisin contamination: 46% of the samples contained concentrations above the limit of detection (100 µg/kg), 5% contained > 1 mg/kg, and a few samples (of poor quality) contained fumonisin B1 at 3.6–12 mg/kg (Kedera et al., 1999). A low concentration (0.06 mg/kg) of total fumonisins was also detected in only one of seven samples collected in western Kenya (Van der Westhuizen et al., 1999).
Consumption of alcohol does not explain the geographical and ethnic variations, as dietary patterns and tribal customs in Kenya vary considerably (Gatei et al., 1978). No significant correlation was observed between the presence of nitrosamine-like compounds and the incidence of oesophageal cancer. The spatial distribution corresponds to the annual rainfall patterns, with high-incidence areas in regions of heavy rainfall.
(f) Zimbabwe
The population-based cancer registry in Zimbabwe showed cancer incidence rates similar to those in other countries of sub-Saharan Africa, with high rates of cancers of the liver, prostate, cervix, and oesophagus (Bassett et al., 1995). The age-standardized incidence rates of oesophageal cancer among men were 30 per 100 000 in Harare in 1990–92 and 59 per 100 000 Bulawayo, further to the south, in 1963–72, and the rates in females were about 8 per 100 000 in both locations. Only three maize-based samples were analysed for fumonisins; one sample, a breakfast cereal intended for human consumption, contained fumonisin B at up to 4.9 mg/kg (Sydenham et al., 1993).
(g) USA
A survey of the mortality rates from oesophageal cancer in the USA between 1950 and 1969 indicated a clustering of cases among African–Americans in a narrow region along the southeastern coast of the Atlantic (Fraumeni & Blot, 1977; O’Brien et al., 1982; Brown et al., 1988). A small area of the Sea Islands and the coastal mainland in the vicinity of Charleston, South Carolina, was found to have a particularly high rate of death from this cancer. In Charleston County, the death rate among black males was about 170 per 100 000, and that among black females was 12 per 100 000, which is significantly higher than the State rate of 6.2 per 100 000.
Seven maize-based human foods purchased in retail outlets in Charleston in 1989 contained mean concentrations of fumonisin B1 and fumonisin B2 of 0.64 and 0.18 mg/kg, respectively (Sydenham et al., 1991).
Mortality rates in the USA tended to be inversely proportional to the socioeconomic indices of income and education (Fraumeni & Blot, 1977). The increased risk among black men in coastal South Carolina was associated with use of tobacco and alcohol, including ‘moonshine’ distilled from fermented maize meal and a low intake of fresh fruits (Brown et al., 1988).
(h) Brazil
Cancer of the oesophagus is the seventh most important cause of death from cancer among males in Brazil. The southern regions of Santa Catarina, Paranà, and Rio Grande do Sul have the highest incidence rate for oesophageal cancer in the country, 18 per 100 000 (Instituto Nacional do Cancer, 1989). The incidence is three to four times higher in men than in women, and marked differences in incidence are found in small geographical areas and as a function of time.
The south and western regions of Santa Catarina State, where there is a high volume of maize production and heavy consumption of maize by-products (especially polenta in rural areas), also have the highest incidence of oesophageal cancer. F. verticillioides was the predominant fungal contaminant in feed samples associated with mycotoxicoses in animals in Paranà (Sydenham et al., 1992a), and F. verticillioides and A. flavus were the most prevalent fungal contaminants in maize samples during the 1990–91 crop year in various regions of Paranà and the tropical regions, including Mate Grosso do Sul and Goias (Hirooka et al., 1996). A survey of the mycoflora in postharvested and stored maize in São Paulo indicated that Fusarium, Penicillium, and Aspergillus spp. were most common (Pozzi et al., 1995).
Fumonisin B1 and B2 concentrations < 38 and 12 mg/kg, respectively, were found in feed samples associated with outbreaks of mycotoxicoses in the State of Paranà (Sydenham et al., 1992a). Concentrations of up to 10 mg/kg of the two toxins were found in samples collected in the tropical regions of Mate Grosso do Sul, with lower concentrations in Paranà: fumonisin B1 at 4 mg/kg and fumonisin B2 at 3 mg/kg (Hirooka et al., 1996). Maize and maize-based food samples from Paranà and the southeast (São Paulo) contained fumonisin B1 and fumonisin B2 at concentrations up to 12 mg/kg and 10 mg/kg, respectively (Scaff & Scussel, 1999a). In São Paulo, maize cultivars grown in various regions contained up to 7 mg/kg of fumonisin B1 and 2 mg/kg of fumonisin B2 (Camargos et al., 2000). A high concentration of fumonisin B1 (5 mg/kg) was found in maize meal samples from markets and supermarkets in Campiñas, São Paulo (Machinski & Valente Soares, 2000). Fumonisin B1 was detected at up to 32 mg/kg in maize intended for human consumption collected in the western part of Santa Catarina (Hermans et al., 2000).
No information is available about the nutritional status of the populations of the regions of high incidence of oesophageal cancer in Brazil. Smoking and alcohol drinking have been implicated, although the prevalence of these habits does not differ from those in regions with lower incidence rates (Scaff & Scussel, 1999b). The consumption of hot beverages such as maté and chimarrao has also been implicated as a possible risk factor for oesophageal cancer (Victoria et al., 1987). An epidemiolgical study in Rio Grande do Sul indicated that smoking, alcohol and maté drinking, farm work, and having a father with cancer were more frequent in cases of oesophageal cancer (Dietz et al. (1998). Other studies suggested that consumption of maize, especially polenta, is a risk factor for this cancer in rural areas in southern and western Santa Catarina (Scaff & Scussel, 1999b).
The role of fumonisins in the causation of liver cancer was evaluated in Haimen (Jiangsu County) and Penlai (Shandong Province), China, the mortality rate in Haimen (52–65 per 100 000) being about fourfold higher than that in Penlai. In a 3-year survey of 240 maize samples, a 10–50-fold higher fumonisin B content was found in Haimen. Most of the samples also contained aflatoxin B1, but there was no significant difference between the two regions. However, deoxynivalenol was detected mainly in maize samples in Haimen. The authors suggested that a synergistic interaction between fumonisins, aflatoxins, and trichothecenes and the algal toxins, microcystins (Ueno et al., 1996), might contribute to the development of liver cancer (Ueno et al., 1997).
In Transkei, South Africa, the intake of aflatoxin B1 correlated with the incidence of liver cancer (Van Rensburg et al., 1990), the rates varying between 3.8 per 100 000 in Butterworth and 7.7 per 100 000 in Kentani (Jaskiewicz et al., 1987b) and 2.4 per 100 000 in Kentani and 13 per 100 000 in Lusikisiki (Makaula et al., 1996), with no apparent association with the concentration of fumonisins in maize.
Experimental evidence for synergistic interactions between aflatoxin B1 and fumonisin B1 (Gelderblom et al., 1999a; Carlson et al., 2001) and between aflatoxin B1 and nivalenol (Ueno et al., 1992) in inducing hepatic cancer in rats was reported.
In South Africa. high incidence rates of neural tube defects were recorded in Mpumulanga Province (3.6 per 1000) and Umzimkulu District (3.8 per 1000) in the Transkei (Venter et al., 1995; Ncayiyana, 1986). The rate in rural Transkei is approximately 5–10 times higher than that for blacks in Cape Town (Cornell et al., 1983). High rates of neural tube defects (5.7 per 1000) were also recorded in Hebei Province, China (Moore et al., 1997), and in northeastern USA between 1920 and 1949, peaking between 1929 and 1932 (2.3–4.3 per 1000). The latter epidemic seemed to correspond to two major socioeconomic events: the great depression and alcohol prohibition, although there are some discrepancies (Machon & Yen, 1971). A high rate of neural tube defects (2.7 per 1000) was also recorded in the lower Rio Grande valley in southern Texas, USA, among the offspring of women who had conceived during 1990–91 (Hendricks, 1999). An association between the epizootics of equine leukoencephalomalacia and porcine pulmonary oedema that occurred late in 1989 in the USA has been postulated (Ross et al., 1991). The concentrations of fumonisins in maize-based foods were high in both the Transkei and Hebei Province (Sydenham et al., 1990; Chu & Li, 1994), and maize-meal foods obtained in the USA during 1990–91 also had a relatively high concentration (1.2 mg/kg) of fumonisin B mycotoxins (Sydenham et al., 1991).
Interference with folate metabolism has been related to the development of neural tube defects, as supplementation with folate decreased the risk (Missmer et al., 2000). The blockage of folate uptake, a critical requirement during organogenesis (Lucock et al., 1998), by fumonisins was also implicated in the induction of neural tube defects (Stevens & Tang, 1997; Hendricks, 1999).
Consumption of rain-damaged, mouldy sorghum and maize by the inhabitants of 27 villages in the Deccan Plateau in southern India resulted in an episode of human mycotoxicosis in 1995. Diarrhoea was reproduced in 1-day-old cockerels fed contaminated grain from the affected households. An epidemiological survey in India indicated that consumption of unleavened bread prepared from mouldy sorghum or maize resulted in a gastrointestinal disease characterized by abdominal pain, borborymi, and diarrhoea. The victims were of low socioeconomic status and ate the mouldy grains mainly because of lack of access to other foods, such as rice.
The dominant mycoflora in the sorghum were Aspergillus, Fusarium, and Alternaria spp., while the first two fungal species were dominant in maize. Fumonisin B1 was the most common mycotoxin in both sorghum and maize samples, and a relatively high concentration of aflatoxin B1 was also detected in the maize. Fumonisin B1 concentrations up to 8.5 mg/kg were associated with diarrhoea in laying hens and 1-day-old cockerels, and addition of fumonisin B1 at 8 or 16 mg/kg of normal diet induced a similar response (Shetty & Bhat, 1997). The control of mycotoxins in human foods in India, especially as related to intake of aflatoxins and fumonisins, has been reviewed (Vasanthi & Bhat, 1998).
The chemical structures of fumonisin B1, fumonisin B2, fumonisin B3, and fumonisin B4 are given in Figure 1. Fumonisin B1 is the diester of propane-1,2,3-tricarboxylic acid and 2S-amino-12S,16R-dimethyl-3S,5R,10R,14S,15R-pentahy-droxyeicosane in which the C-14 and C-15 hydroxy groups are esterified with the terminal carboxy group of propane-1,2,3-tricarboxylic acid (Bezuidenhout et al., 1988; Hoye et al., 1994). Fumonisin B2 is the C-10 deoxy analogue of fumonisin B1 in which the corresponding stereogenic centres on the eicosane backbone have the same configuration (Bezuidenhout et al., 1988; Harmange et al., 1994). The full stereochemical configurations of fumonisin B3 and fumonisin B4 are unknown, although the amino terminal of fumonisin B3 has the same absolute configuration as that of fumonisin B1 (i.e. 2S, 3S) (Hartl & Humpf, 1998). The configuration of the chiral centre on the tricarboxylic acid moieties has been determined by three groups, but with conflicting results. In the initial publication of the stereochemistry of fumonisin B1, the S configuration was assigned to this centre (Shier et al., 1995), but subsequently both fumonisin B1 and fumonisin B2 were reported to have an R configuration (Boyle & Kishi, 1995a,b). A later study with a synthetic, optically active gamma-lactone related to the tricarboxylic acid correlated this with the same lactone in fumonisin B1 and confirmed the R configuration for the chiral site on the side-chains (Edwards et al., 1999).
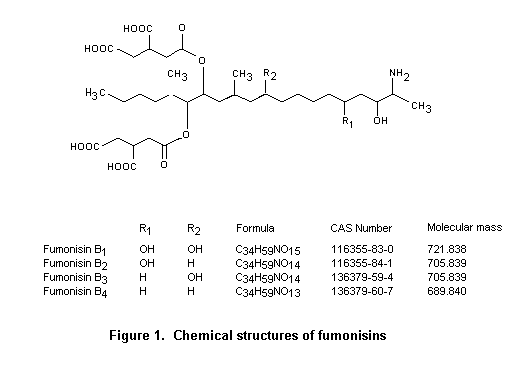
The most abundant fumonisins in naturally contaminated maize and maize-based products are fumonisins B1 and B2. Hence, analytical methods (and surveys) have been developed mainly for these two toxins. In general, methods developed for fumonisins B1 and B2 have been found to be valid for fumonisin B3 as well (Sydenham et al., 1992b), although use of these methods is limited by problems in the supply of analytical standards for fumonisin B3. No specific analytical methods have been developed for fumonisin B4, and little is known about its natural occurrence.
Screening tests for fumonisins are based either on thin-layer chromatography (TLC) separation after appropriate clean-up of maize extracts or on commercially available enzyme-linked immunosorbent assays (ELISAs). Other immunologically based methods, such as dipstick (Schneider et al., 1995) and biosensor methods (Thompson & Maragos, 1996; Maragos, 1997; Mullett et al., 1998), have been described but have not found general use. Immunoaffinity columns have been designed to purify extracts before high-performance liquid chromatography (HPLC) separation and quantification of fumonisins B1, B2, and B3 analogues, and have also been used in a direct fluorimetric method for rapid determination of ‘total fumonisin’ (Duncan et al., 1998).
The TLC and other chromatographic methods for fumonisins have been reviewed (Shephard, 1998). The reversed-phase technique developed by Rottinghaus et al. (1992) has been used in surveys of contamination of maize with fumonisins (Shelby et al., 1994a). When combined with an efficient extract clean-up procedure based on use of immunoaffinity columns and detection by densitometry, TLC can be considered quantitative (Preis & Vargas, 2000).
The performance characteristics of screening tests for fumonisins in maize, based on interlaboratory collaborative studies, have not been reported in the literature. However, in-house comparisons between HPLC methods and the various screening tests have been described. The TLC method of Rottinghaus et al. (1992) has been compared with HPLC over a contamination range of fumonisin B1 of 1–250 mg/kg (correlation coefficient, r = 0.953; p < 0.0005; Schaafsma et al., 1998). The results obtained with a fibre-optic immunosensor in a direct competitive monoclonal antibody format with a fumonisin B1–fluorescein isothiocyanate conjugate compare favourably with those obtained with HPLC (Maragos, 1997).
The commercial availability of ELISA methods has made them popular for screening for fumonisin contamination. Although the antibodies used in ELISAs are raised against fumonisin B1, they generally have significant (but lower) cross-reactivity with fumonisins B2 and B3. The performance of ELISAs is generally assessed by comparison with HPLC determination of fumonisins and has been found to depend on the antibody used (Pestka et al., 1994; Usleber et al., 1994; Sydenham et al., 1996a,b; Kulisek & Hazebroek, 2000). The correlation between the results of HPLC and ELISA for naturally contaminated samples has been reported to vary from 0.51 (p < 0.05; Pestka et al., 1994) to 0.97 (p < 0.001; Sydenham et al., 1996a). However, such comparisons have generally shown an overall trend for the concentrations of ‘total fumonisins’ with ELISA to be greater than those determined in the same samples by HPLC.
Quantitative analytical methods for fumonisins have been reviewed (Sydenham & Shephard, 1996; Wilson et al., 1998; Shephard, 1998). Almost all these methods involve HPLC separation of fumonisins B1, B2, and B3 after solvent extraction from maize or maize-based food matrices and purification by solid-phase extraction. In most surveys and studies on the natural occurrence of fumonisins involved use of variations or minor modifications of a few basic analytical methods.
The first quantitative HPLC method for determining fumonisins B1 and B2 in naturally contaminated maize involved extraction with methanol:water (3:1), clean-up on strong anion exchange solid-phase extraction cartridges, and quantification by reversed-phase HPLC after precolumn derivatization with the fluorigenic reagent, ortho-phthaldialdehyde (Shephard et al., 1990). The reproducibility of this method when used for naturally contaminated maize was studied collaboratively under the sponsorship of the Commission on Food Chemistry of IUPAC (Thiel et al., 1993). The method was subsequently improved and extended to include the determination of fumonisin B3 (Sydenham et al., 1992b). A further collaborative study of its accuracy and reproducibility was conducted under the auspices of both the Commission on Food Chemistry of IUPAC and AOAC International and resulted in adoption of the method by the latter (Sydenham et al., 1996c). It has now been accepted as official AOAC method 995.15 for the determination of fumonisins B1, B2, and B3 in maize (Trucksess, 2000). The performance characteristics in the international collaborative trials are given in Tables 9–12.
Table 9. Results of a collaborative study by 11 laboratories of the repeatability and reproducibility of the method of Shephard et al. (1990) for the determination of fumonisins B1 and B2 in naturally contaminated maize
|
Mean (µg/kg) |
Sr (µg/kg) |
RSDr (%) |
SR (µg/kg) |
RSDR (%) |
No. of outliers |
HORRAT value |
|
Fumonisin B1 |
||||||
|
< 50 |
– |
– |
– |
– |
|
|
|
226 |
33 |
15 |
56 |
25 |
1 |
1.2 |
|
340 |
84 |
26 |
86 |
26 |
1 |
1.4 |
|
560 |
44 |
7.7 |
100 |
18 |
1 |
1.0 |
|
1200 |
170 |
15 |
310 |
27 |
1 |
1.7 |
|
1983 |
260 |
13 |
460 |
23 |
1 |
1.6 |
|
Fumonisin B2 |
||||||
|
< 50 |
– |
– |
– |
– |
|
|
|
74 |
13 |
17 |
28 |
38 |
1 |
1.6 |
|
100 |
37 |
37 |
46 |
46 |
1 |
2.0 |
|
260 |
32 |
12 |
71 |
28 |
1 |
1.4 |
|
420 |
66 |
16 |
140 |
34 |
1 |
1.9 |
|
740 |
130 |
18 |
270 |
36 |
1 |
2.2 |
From Thiel et al. (1993)
Sr, standard deviation for repeatability; SR, standard deviation for reproducibility; RSDr, relative Sr; RSDR, relative SR; HORRAT, ratio of relative SD for reproducibility in the trial to that predicted. A HORRAT of 1 indicates a relative SD for reproducibility corresponding exactly to the Horwitz equation (Horwitz, 1989); a HORRAT < 1.0 ± 0.5 indicates normal reproducibility; a HORRAT > 1.5 indicates that reproducibility is higher than expected, whereas a HORRAT > 2.0 indicates problematic reproducibility (AOAC International, 2000).
Table 10. Results of collaborative study by 12 laboratories of the accuracy and reproducibility of the method of Sydenham et al. (1992a) for the determination of fumonisins B1, B2, and B3 in spiked and naturally contaminated maize
|
Spiking concentration (µg/kg) |
Mean (µg/kg) |
Mean recovery (%) |
Sr |
RSDr |
SR |
RSDR |
No. of outliers |
HORRAT value |
|
Fumonisin B1 |
||||||||
|
< 50 |
– |
– |
– |
– |
– |
– |
– |
– |
|
500 |
400 |
81 |
29 |
7.1 |
56 |
14 |
1 |
0.75 |
|
1000 |
810 |
81 |
47 |
5.8 |
130 |
16 |
– |
0.95 |
|
2000 |
1600 |
81 |
120 |
7.7 |
260 |
16 |
– |
1.1 |
|
4000 |
3200 |
81 |
200 |
6.2 |
490 |
15 |
– |
1.1 |
|
8000 |
6700 |
84 |
740 |
11 |
1100 |
16 |
– |
1.4 |
|
Naturally contaminated |
4200 |
– |
560 |
13 |
940 |
22 |
– |
1.7 |
|
Fumonisin B2 |
||||||||
|
< 50 |
– |
– |
– |
– |
– |
– |
– |
– |
|
200 |
150 |
76 |
13 |
8.4 |
25 |
16 |
1 |
0.77 |
|
400 |
310 |
78 |
27 |
8.5 |
49 |
16 |
– |
0.83 |
|
800 |
620 |
77 |
73 |
12 |
120 |
19 |
– |
1.1 |
|
1600 |
1300 |
81 |
93 |
7.2 |
220 |
17 |
– |
1.1 |
|
3200 |
2600 |
82 |
320 |
12 |
470 |
18 |
– |
1.3 |
|
Naturally contaminated |
1200 |
– |
220 |
18 |
330 |
27 |
– |
1.7 |
|
Fumonisin B3 |
||||||||
|
< 50 |
– |
– |
– |
– |
– |
– |
– |
– |
|
100 |
76 |
76 |
7.7 |
10 |
15 |
20 |
1 |
0.83 |
|
200 |
160 |
81 |
28 |
17 |
35 |
22 |
– |
1.0 |
|
400 |
320 |
80 |
49 |
15 |
71 |
22 |
– |
1.2 |
|
800 |
660 |
82 |
53 |
8.0 |
140 |
21 |
– |
1.2 |
|
1600 |
1400 |
87 |
200 |
14 |
270 |
20 |
– |
1.3 |
|
Naturally contaminated |
370 |
– |
63 |
17 |
93 |
25 |
– |
1.3 |
From Sydenham et al. (1996c). For abbreviations, see footnote to Table 9.
Table 11. Results of comparative study in 24 European laboratories of a maize sample containing fumonisin B1 at approximately 2000 µg/kg and fumonisin B2 at 1000 µg/kg prepared by spiking a naturally contaminated, ground maize sample
|
Result |
Fumonisin B1 |
Fumonisin B2 |
|
No. acceptable data sets |
25 |
25 |
|
No. outliers |
1 |
1 |
|
Overall mean (corrected for recovery) |
2300 µg/kg |
1200 µg/kg |
|
RSDr |
10% |
11% |
|
RSDR |
11% |
13% |
|
Overall mean recovery |
70 ± 14% |
69 ± 16% |
|
Recovery SD (within laboratories) |
6% |
7% |
|
Recovery SD (between laboratories) |
16% |
15% |
|
Recovery: Extraction by shaking (n = 9) |
85 ± 12% |
86 ± 14% |
|
Recovery: Extraction by blending (n = 16) |
62 ± 6% |
60 ± 6% |
From Visconti & Boenke (1995); Visconti et al., 1996a). All laboratories used methods derived from that of Shephard et al. (1990), with modifications. For abbreviations, see footnote to Table 9.
Table 12. Results of collaborative study by 23 laboratories of the reproducibility of high-performance liquid chromatography with immunoaffinity column clean-up for determining fumonisins B1 and B2 in maize and cornflakes
|
|
Mean |
Mean |
Sr |
RSDr |
SR |
RSDR |
No. of outliers |
HORRAT value |
|
Analysis of spiked maize and naturally contaminated maize |
||||||||
|
Fumonisin B1 |
||||||||
|
< 0.05 µg/kg |
0.04 |
– |
– |
– |
– |
– |
– |
– |
|
0.80 µg/kg |
0.65 |
76 |
0.14 |
21 |
0.16 |
26 |
– |
1.5 |
|
Naturally contaminated |
0.37 |
– |
0.09 |
24 |
0.10 |
28 |
– |
1.5 |
|
Naturally contaminated |
0.78 |
– |
0.15 |
19 |
0.20 |
26 |
1 |
1.5 |
|
Naturally contaminated |
1.4 |
– |
0.28 |
20 |
0.31 |
22 |
– |
1.4 |
|
Fumonisin B2 |
||||||||
|
< 0.05 µg/kg |
0.01 |
– |
– |
– |
– |
– |
– |
– |
|
0.40 µg/kg |
0.30 |
72 |
0.06 |
18 |
0.07 |
23 |
– |
1.2 |
|
Naturally contaminated |
0.09 |
– |
0.02 |
22 |
0.02 |
22 |
4 |
0.96 |
|
Naturally contaminated |
0.20 |
– |
0.05 |
27 |
0.06 |
30 |
– |
1.5 |
|
Naturally contaminated |
0.56 |
– |
0.13 |
22 |
0.14 |
26 |
– |
1.5 |
|
Analysis of spiked cornflakes and cornflakes with added naturally contaminated maize |
||||||||
|
Fumonisin B1 |
||||||||
|
< 0.05 µg/kg |
0.04 |
– |
– |
– |
– |
– |
– |
– |
|
0.80 µg/kg |
0.92 |
110 |
0.09 |
9.2 |
0.27 |
30 |
1 |
1.8 |
|
Naturally contaminateda |
0.32 |
– |
0.07 |
21 |
0.10 |
32 |
1 |
1.7 |
|
Naturally contaminateda |
0.57 |
– |
0.09 |
15 |
0.16 |
28 |
– |
1.6 |
|
Naturally contaminateda |
1.0 |
– |
0.12 |
11 |
0.29 |
27 |
– |
1.7 |
|
Fumonisin B2 |
||||||||
|
< 0.05 µg/kg |
0.00 |
– |
– |
– |
– |
– |
– |
– |
|
0.40 µg/kg |
0.39 |
97 |
0.03 |
7.7 |
0.12 |
31 |
1 |
1.7 |
|
Naturally contaminateda |
0.13 |
– |
0.03 |
22 |
0.04 |
35 |
– |
1.6 |
|
Naturally contaminateda |
0.24 |
– |
0.04 |
15 |
0.07 |
28 |
– |
1.4 |
|
Naturally contaminateda |
0.46 |
– |
0.05 |
10 |
0.12 |
26 |
– |
1.4 |
From Visconti et al. (2001). For abbreviations, see footnote to Table 9.
a
Spiked with minimal amounts (0.8–3.0%) of ground, naturally contaminated maizeModifications to the above method led to the development of other analytical methods. Acetonitrile:water (1:1) was formulated as an alternative extraction solvent, while reversed-phase C18 solid-phase extraction cartridges have been used to purify extracts (Bennett & Richard, 1994; Rice et al., 1995). Although these cartridges yield less pure extracts, they are necessary for determination of the hydrolysis products of fumonisins and for cases in which strong anion exchange columns give poor recovery (Shephard, 1998). The introduction of immunoaffinity columns containing antibodies reactive towards fumonisins has greatly improved the clean-up step in analytical methods (Ware et al., 1994; Duncan et al., 1998). Most researchers report ed quantification by means of precolumn derivatization with ortho-phthaldialdehyde, despite its limited stability. Use of naphthalene-2,3-dicarbox-aldeyde has been proposed as an alternative (Bennet & Richard, 1994). Recent advances in analytical instrumentation have resulted in the introduction of bench-top liquid chromatography–mass spectrometers, which are more sensitive and specific for the detection and quantification of fumonisins (Shephard, 1998).
The official AOAC International method for fumonisins in maize has also been used for maize-based foods (Stack & Eppley, 1992), although problems have been reported in the recovery of fumonisins from certain matrices (Scott & Lawrence, 1994). In order to improve the recovery from these matrices, other extraction solvents have been investigated, while retaining the solid-phase extraction clean-up steps and ortho-phthaldialdehyde derivatization for HPLC quantification. The alternative solvents include methanol:borate buffer (pH 9.2; 3:1), methanol:water:hydrochloric acid (2 or 5 N; 3:1:0.3), methanol:0.1 mol/L HCl (3:1); acetonitrile:methanol:water (1:1:2), and acetonitrile:phosphate buffer (0.1 mol/L; pH 3.0; 1:1) (Scott & Lawrence, 1994; Zoller et al., 1994; Scott & Lawrence, 1996; Scott et al., 1999; Solfrizzo et al., 2000; De Girolamo et al., 2001).
As part of a programme to develop analytical methods for mycotoxins in foods at concentrations of interest for future legislation for the European Union, methods for fumonisins have been investigated within the Measurements and Testing Programme and the Community Bureau of Reference. A comparative study was conducted by 24 European laboratories on a contaminated maize sample containing fumonisin B1 at approximately 2000 µg/kg and fumonisin B2 at 1000 µg/kg (Visconti & Boenke, 1995; Visconti et al., 1996a). The results are summarized in Table 11.
Further improvements have been made in the analytical methods for fumonisins in maize, cornflakes, maize muffins, extruded maize, and infant formula, resulting in adoption of acetonitrile:methanol:water (1:1:2) as the extraction solvent and use of immunoaffinity columns for clean-up (Visconti et al., 1999; Solfrizzo et al., 2000). These extraction and clean-up techniques and HPLC quantification of preformed ortho-phthaldialdehyde derivatives of fumonisins B1 and B2 were used in a collaborative study of maize and cornflakes, which resulted in the adoption of the method by AOAC International (Visconti et al., 2001). The performance characteristics achieved in the collaborative study are shown in Table 12.
A collaborative study for determination of total fumonisins with a commercially available ELISA kit is under consideration by the methods programme of AOAC International (2000).
Whitaker and co-workers have studied the sampling variance associated with the testing of shelled maize for fumonisin (Whitaker et al., 1998). A bulk sample of about 45 kg (100 lbs) was taken from each of 24 batches of shelled maize which had been harvested from 24 fields in North Carolina, USA. Each bulk sample was riffle-divided into 32 1.1-kg test samples, and these were comminuted in a Romer mill. A nested design was used to determine the variation associated with sampling, sample preparation, and analysis. Briefly, 10 batches with a wide range of fumonisin concentrations were selected. From each batch, 10 comminuted test samples were taken randomly, and two 25-g portions were taken from each by riffle division. Finally, the concentrations of fumonisins B1, B2, and B3 were determined by AOAC official method 995.15 (Sydenham et al., 1996c). At a batch contamination concentration of 2 mg/kg, the coefficient of variation associated with sampling (1.1-kg sample) was 17%, that associated with sample preparation (Romer mill and 25-g analytical portion) was 9.1%, and that for analysis was 9.7%. These values were independent of the fumonisin type (B1, B2, B3, or total). The coefficient of variation associated with the total test procedure (sampling, sample preparation, and analysis) was 45%, which was of the same order of magnitude as that for measuring aflatoxin in shelled maize by a similar test procedure.
The proposed sampling plans are shown in Table 13. Some general precepts are:
|
• |
A minimum of 30 batches of food should be sampled from each region (i.e. country, agroclimatic region within a country). |
|
• |
‘Samples’ should refer to initial samples collected from a batch, which may be large bulk samples or smaller composite samples. |
|
• |
‘Sub-samples’ should refer to samples produced by riffle division of unground bulk samples or samples produced by comminuting and subdividing smaller composite samples. |
|
• |
‘Analytical samples’ should refer to samples subjected to analysis, which are usually small portions drawn from subsamples. |
|
• |
The coefficient of variation of the sampling plan should be no more than 30%. |
|
• |
The coefficient of variation of the complete analytical method (extraction, clean-up, quantification) should be no more than 10%. |
Table 13. Proposed sampling plans
|
Commodity |
Increments |
Subsample |
Notes |
|
Fumonisins |
|||
|
Maize |
|||
|
Whole corn |
50 x 100 |
5.0 |
Whitaker et al. (1998): Sampling variation for fumonisins in maize similar to that reported for aflatoxins |
|
Corn-on-the-cob |
50 cobs |
7.5 |
Assuming that core of cob contributes about 30% of total weight of cob and that a cob yields about 100 g of kernels |
|
Cornflour |
Assumed that sampling variance for these commodities was similar to that associated with aflatoxin in comminuted feeds; suggested sampling plan associated with sampling precision of 12.5% for aflatoxin in comminuted feeds |
||
|
Maize meal |
|||
|
Maize grits |
|||
|
Bran |
10 x 100 |
1.0 |
|
|
Processed maize foods, e.g. cornflakes, tortilla chips, popcorn, muffin mix, starch |
|||
|
Deoxynivalenol and T-2 and HT-2 toxins |
|
|
No data on sampling variance for T-2 or HT-2 toxins; assumed to be similar to that for deoxynivalenol |
|
Maize |
|||
|
Whole corn |
20 x 100 |
2.0 |
Sample size for maize set at double the sample weight required for estimating deoxynivalenol in wheat and barley |
|
Corn-on-the-cob |
20 cobs |
3.0 |
Assuming that core of cob contributes about 30% of total weight of cob and that a cob yields about 100 g of kernels |
|
Maize grits |
Sample size for deoynivalenol in cereal products arbitrarily set at half that required for fumonisins and ochratoxin A |
||
|
Processed maize foods, e.g. cornflakes, tortilla chips, popcorn, muffin mix, starch |
10 x 50 |
0.5 |
|
|
Wheat |
Whitaker et al. (2000) |
||
|
Barley |
20 x 50 |
1.0 |
Freese et al. (2000) |
|
Oats |
|||
|
Rye |
|||
|
Flour, meal, and bran of all origins |
10 x 50 |
0.5 |
Sample size for deoxynivalenol in cereal products arbitrarily set at half that required for fumonisins and ochratoxin A |
|
Bread |
|||
|
Ochratoxin A |
|||
|
Maize |
|||
|
Whole maize |
50 x 100 |
5.0 |
Sampling variability for ochratoxin A assumed to be similar to that for fumonisins (and aflatoxins; see above) |
|
Corn-on-the-cob |
50 cobs |
7.5 |
Assuming that core of cob contributes about 30% of total weight of cob and that a cob yields about 100 g of kernels |
|
Maize grits |
|||
|
Processed maize foods, e.g. cornflakes, tortilla chips, popcorn, muffin mix, starch |
10 x 100 |
1.0 |
Coker et al. (in press): Assumed that sampling variance for these commodities was similar to that associated with aflaitoxin n comminuted feeds; suggested sampling plan associated with sampling precision of 12.5% for aflatoxin in comminuted feeds |
|
Wheat |
|||
|
Barley |
|||
|
Rice (including de-husked and polished) |
|||
|
Peas and beans (including coffee beans) |
50 x 100 |
5.0 |
Sampling variability for ochratoxin A assumed to be similar to that of fumonisins (and aflatoxins; see above) |
|
Dried fruit, (e.g. raisins, currants, sultanas, figs, dates, apricots) |
|||
|
Flour, meal, and bran of all origins |
|||
|
Bread |
10 x 100 |
1.0 |
Coker et al. (in press): Assumed that sampling variance for these commodities was similar to that associated with iaflatoxin n comminuted feeds; suggested sampling plan associated with sampling precision of 12.5% for aflatoxin in comminuted feeds |
|
Ground and instant coffee |
|||
|
Cocoa powder |
|||
|
Beverages (e.g. coffee, wine, grape juice) |
5 x 100 |
0.5 |
Commission of the European Union (1998a): Assumed that sampling variance of mycotoxins in beverages was similar to that for aflatoxin M1 in milk |
|
Aflatoxin M1 |
|||
|
Milk (liquid and dry), e.g. raw, pasteurized, homogenized, UHT, skimmed, semi-skimmed, evaporated, infant formula |
5 x 100 |
0.5 |
Commission of the European Union (1998a): Assumed that sampling variance of mycotoxins in beverages was similar to that for aflatoxin M1 in milk |
|
Milk products |
|||
Fumonisins are primarily produced by the fungi F. verticillioides (Sacc.) Nirenberg and F. proliferatum (Matsushima) Nirenberg, which are major pathogens of maize (Zea mays L.) around the world. The commodities most often contaminated with fumonisins are therefore maize and maize-based foods. Data on contamination of maize by fumonisins was submitted to the Committee by Argentina, Brazil, Canada, China, Denmark, Sweden, the United Kingdom, and the USA. Further data were derived from the literature published between 1995 and early 2000. Studies of the natural occurrence of fumonisins conducted before 1995 were reviewed by Shephard et al. (1996b).
Data on the occurrence of fumonisins B1, B2, and B3 are presented in Appendices A, B, and C, respectively. The information is often incomplete: most authors reported the limit of detection (LOD), rather than the limit of quantification (LOQ), of the analytical method; the mean value in samples is frequently given, and in these cases (or when its use implied), the data were recalculated so that all data represent the means of samples, those below the limit of detection being taken as zero. When possible, the numbers of samples containing > 1000 and > 2000 µg/kg are also given. The former level represents the legislative limit for fumonisins B1 and B2 in Switzerland (FAO, 1997). The references in the appendices show the primary reference, which usually includes details on sampling and some information on analytical method; further references to methods are recorded as ‘A =’. Few references were given for sampling methods.
Little is known about the natural occurrence of fumonisin B4. It is produced by strains of F. verticillioides, generally at lower concentrations than fumonisin B1, B2, or B3 (Abbas et al., 1992; Plattner et al., 1996). Fumonisin B4 was identified in 23 of 44 mouldy maize samples in the Republic of Korea (mean, 1600 µg/kg; range, 80–11 000 µg/kg) at concentrations lower than those of fumonisins B1, B2, and B3 in these samples (Seo & Lee, 1999).
Some surveys have been undertaken of contamination of food commodities other than maize products. Analysis of 41 local and imported beers in Canada by HPLC showed a low incidence of contamination, with only four samples containing > 2 ng/ml fumonisin B1 (maximum, 59 ng/ml) (Scott & Lawrence, 1995). Of these, three contained fumonisin B2 at a concentration > 2 ng/ml (maximum, 12 ng/ml). Of the rest, seven contained 1 ng/ml or traces of fumonisin B1. A survey of 46 imported beer samples in Canada by ELISA showed that 11 samples contained > 1 ng/ml (maximum, 25 ng/ml), and a further 11 contained 1 ng/ml or traces > 0.15 ng/ml (Scott et al., 1997). A similar survey of 32 Spanish beer samples by ELISA showed 14 positive samples (LOD, 3 ng/ml) with a range of 4.8–86 ng/ml (Torres et al., 1998). Another survey by HPLC of 29 domestic and imported beers in the USA showed 25 positive samples (> 0.3 ng/ml), with 17 samples containing > 1 ng/ml (maximum, 13 ng/ml) for fumonisins B1 plus B2 (Hlywka & Bullerman, 1999).
Investigation of milk and beef for contamination by fumonisins failed to raise any concern. Fumonisins were detected in beef muscle only after continuous exposure to highly contaminated feed (Smith & Thakur, 1996). In a survey of 165 milk samples in the USA, only one was found to be contaminated with fumonisin B1 at a concentration of 1.3 ng/ml by gas chromatography–mass spectrometry and > 5 ng/ml by HPLC (Maragos & Richard, 1994). Studies in which cows were given feed contaminated with culture material equivalent to 75 mg/kg of feed or pure fumonisin B1 at a dose equivalent to contamination of feed with 125 mg/kg showed no detectable carry-over of fumonisins into milk (LOD, approximately 5 ng/ml) (Scott et al., 1994; Richard et al., 1996). Re-analysis of these milk samples by ELISA (LOD, 0.5 ng/ml) also showed no carry-over into the milk of treated cows (Prelusky et al., 1996a). Similar studies in lactating sows given a diet containing fumonisin B1 at 100 mg/kg for 14 days showed no toxin in the milk at a LOD of 30 µg/kg (Becker et al., 1995). Pigs fed diets containing the toxin at 2–3 mg/kg also did not show accumulation of fumonisin residues in muscle, although residues did accumulate in kidneys and liver while the contaminated feed was being consumed (Prelusky et al., 1994, 1996a,b). Replacement of the contaminated feed by clean feed resulted in a rapid decline in the concentrations. Studies in laying hens also showed no fumonisin residues in most tissue samples or in eggs (Vudathala et al., 1994; Prelusky et al., 1996a).
Contamination of rice with fumonisins was reported in the USA in samples collected from fields known to have plants with symptoms of Fusarium sheath rot (Abbas et al., 1998). Fumonisins B1, B2, and B3 were detected (LOD, 500 µg/kg by HPLC) in 8 of 20 samples at concentrations < 5600 µg/kg. The appendices give the results of determinations in rice samples in Argentina, in which four of five imported samples showed contamination at maximum concentrations of 229 µg/kg fumonisin B1, 126 µg/kg fumonisin B2, and 16 µg/kg fumonisin B3. A further 11 samples of locally produced rice showed no fumonisin contamination (LOQ, 8 µg/kg), and five rice samples included in a survey of foods in the United Kingdom also contained no detectable fumonisin (LOD, 10 µg/kg) (Patel et al., 1997). Fumonisin B1 has been reported in rice in China (Appendix A). It has also been found on Fusarium-infected mouldy navy, adzuki, and mung beans (Tseng et al., 1995; Tseng & Tu, 1997) and in F. proliferatum-infected asparagus plants (Logrieco et al., 1998). In a survey of 35 sorghum syrup samples in the USA, one contained fumonisin B1 (at 120 µg/kg; LOQ, 100 µg/kg; LOD, 10 µg/kg) (Trucksess et al., 2000). A single analysis of a sorghum meal sample in Burundi showed a very high concentration (28 200 µg/kg) of fumonisin B1, but five sorghum samples were not contaminated (LOD not given) (Munimbazi & Bullerman, 1996). In an analysis of 20 sorghum and sorghum meal samples in Botswana, three contained fumonisin B1 at a concentration > 20 µg/kg (mean, 43 µg/kg; maximum, 60 µg/kg; LOD, 20 µg/kg) (Siame et al., 1998). A further two samples of sorghum meal in Botswana contained traces of fumonisin B1 (20 µg/kg) (LOD, < 20 µg/kg) (Doko et al., 1996), and five samples collected from control households during an epidemiological study in India contained fumonisin B1 at a concentration of 70–360 µg/kg (Bhat et al., 1997). In an extensive study of 43 normal sorghum samples in India, only two had concentrations above the LOD of 25 µg/kg (range, 150–510 µg/kg) (Shetty & Bhat, 1997). A survey of the effects of storage on sorghum in Brazil over 1 year showed mean concentrations of 110–150 µg/kg (Da Silva et al., 2000).
When numerical data on the distribution of fumonisins in harvested maize and maize-based products were available in the surveyed literature, the numbers are given in Appendix A. These data include the combined results from surveys in the USA on a wide range of maize-based products sampled between 1990 and 1994 (Pohland, 1996). A number of authors presented distributions in the form of histograms, from which it was not always possible to extract unambiguous, relevant numerical information.
Limited data were available on annual variation in fumonisin concentrations in maize harvested in consecutive years, although it is clear that considerable variation can occur. The most extensive data on annual variation was collected for maize sampled in Iowa, USA, between 1988 and 1996 (Appendices A–C; Murphy et al., 1993; Rice & Ross, 1994; P.A. Murphy, personal communication). During the initial years of the survey (1988–91), the average concentration of fumonisin B1 was > 2000 µg/kg each year, whereas in 1992–96 the concentration was significantly lower, with a mean < 450 µg/kg. A 5-year survey (1989–93) was conducted in South Africa on fumonisin concentrations in white and yellow maize. The concentrations of fumonisin B1 in white maize over the 5 years ranged from 320 to 570 µg/kg; those in yellow maize ranged from 170 to 190 µg/kg during the first 4 years and rose to 680 µg/kg in the last year (Shephard et al., 1996b). Contamination of maize in Croatia with fumonisins B1and B2 over two consecutive seasons (1996 and 1997) has also been reported (Appendix A; Jurjevic et al., 1999). The data for Brazil drawn from the GEMS/Food programme is poor (Appendices A and B). Similar data are available for Argentina over a number of years (Appendices A–C), although the geographical origin of the samples included in the survey varied slightly.
The annual variation in total fumonisin concentrations over the 5-year period 1994–98 in various maize-based food products in the USA is shown in Appendix A, with data on contamination of maize meal (degerminated, partially degerminated, and whole grain) from the 1997 and 1998 US crops.
The stability of mycotoxins during food processing is affected by many factors, including: the moisture of the product, the toxin concentration and its location, the presence of additives, and the type of food matrix (Scott, 1984). These factors should therefore be considered with respect to each type of processing (e.g. milling, roasting, canning, oil extraction) when estimating the fate of the mycotoxin. After decompositon or destruction of mycotoxins, the resulting compound(s) should be identified to ensure that undesirable materials (with harmful biological effects) do not contaminate the food.
Dry milling of maize is a physical process by which the components of the grain are separated. The primary products derived from the process are grits, bran, germ, meal, and flour (Alexander, 1987). Since fumonisins are not expected to be destroyed during this process, their distribution has been evaluated in the fractions obtained from commercially and experimentally dry-milled maize. After commercial dry milling, fumonisins were found in flaking grits, flour, germ, and bran. The pattern of distribution after experimental dry milling varied slightly for different types of maize, but in general the concentrations were lower in grits and higher in germ, bran, and fines. As all the fractions found to contain fumonisins are used in the production of animal or human food, maize- based ingredients should be monitored for the presence of fumonisins (Katta et al., 1997).
Wet milling is used to obtain products such as germ (for oil and feed preparation), fibres and gluten (used in animal feed), and starch (for e.g. syrup). Analysis of the fractions obtained from laboratory-scale wet milling of maize contaminated with fumonisins revealed that the starch fractions did not contain measurable amounts of fumonisin B1; 22% of the toxin was found in the steeping and process water, while the fumonisin content of the other fractions diminished in the order gluten > fibre > germ and represented 10–40% of the concentration found in the original maize. The presence of fumonisin in gluten, fibres, and germ could be a hazard to animal health, since these products are used as feed ingredients (Bennett et al., 1996).
As maize contaminated with fumonisin B1 has a low density, 86% of the toxin can be removed in the buoyant fraction after treatment with saturated sodium chloride solution (Shetty & Bhat, 1999). Steeping maize in water, which is a step in wet milling, was also effective in reducing the concentrations of fumonisins B1 and B2 in maize, but sulfur dioxide delayed toxin extraction from the contaminated grain (Canela et al., 1996). Nevertheless, steeping maize in 0.2% sulfur dioxide solution containing fumonisin B1 significantly decreased the amount of toxin in the solution, indicating that maize may contain fumonisin-binding constituents (Pujol et al., 1999).
The effects of heating on the stability of fumonisins during various food processing procedures have been studied. The temperatures required to achieve reductions > 50% in the concentration of fumonisin B1 and/or fumonisin B2 varied according to the process: for example, 190 °C for dry or moist maize meal (Scott & Lawrence, 1994) or frying maize chips (Jackson et al., 1997); extrusion temperatures for batter made from maize flour (Pineiro et al., 1999); 218 °C in roasting maize meal (Castelo et al., 1998a), and 160 °C for extrusion cooking of maize grits (Katta et al., 1999). The results indicate that fumonisins are fairly stable to heat and that significant removal occurs only during processes that reach temperatures > 150 °C. At each temperature tested in a specific food-processing system, the stability of the fumonisin was time-dependent (Jackson et al., 1996) and was strongly affected by other factors, some of which are mentioned above. The efficiency with which fumonisins are extracted from heated foods may decrease with time (Bordson et al., 1993), probably because of binding to the food matrix (Scott & Lawrence, 1994). As a consequence, controlled recovery experiments must be conducted for each type of processing system to ensure that the most appropriate methods of detection are used (Scott & Lawrence, 1994). Methods are needed to differentiate between binding and removal of fumonisins (Bullerman & Tsai, 1994).
Populations who consume large amounts of maize and maize products (e.g. in countries in South and Central America) are at high risk of exposure to fumonisins. The technique for preparing staple foods in these countries, such as tortillas, involves alkaline cooking and heating (nixtamalization), during which hydrolysed fumonisins are formed. Hydrolysed fumonisin B has been detected in commercial masa and tortilla chips, probably due to formation during nixtamalization (Murphy et al., 1996). In studies of the fate of fumonisins during processing and toxicological studies of the products, nixtamalized F. moniliforme culture (Voss et al., 1996a) and a maize-based diet containing F. proliferatum remained toxic after processing (Hendrich et al., 1993). Although the main toxic product formed from fumonisin during nixtama-lization is hydrolysed fumonisin, other products may be formed (Hendrich et al., 1993).
Treatment of fumonisin–contaminated, ground maize with calcium hydroxide resulted in transfer of the aminopentol moiety and, possibly, also the tricarballylic acid moiety to the aqueous fraction; these could easily be separated. Maize kernels from which the pericarp had been partially removed contained significantly greater amounts of fumonisin B1 and its aminopentol moiety, indicating that this treatment would reduce the concentration of fumonisins in maize (Sydenham et al., 1995). Use of a modified nixtamalization procedure on maize contaminated with fumonisin B1 resulted in a 100% reduction in the fumonisin content and removed the mutagenic potential of the maize extracts (Park et al., 1996).
Although it was expected that the fumonisin in nixtamalized maize products would be in the hydrolysed form, the content of that form in masa and tortillas in Mexico was lower than the content of fumonisins B1 and B2. Several explanations were proposed, including incomplete nixtamalization, a high initial concentration of fumonisin, and only partial removal of the pericarp, in which most of the fumonisin is located (Dombrink-Kurtzman & Dvorak, 1999).
Better control of fumonisins can thus be achieved by monitoring the degree of pericarp removal and analysing samples from each stage of production. Nixtamalization alone does not ensure complete detoxication of fumonisin-contaminated maize, and the process should be carefully controlled to ensure at least a significant reduction in fumonisins and other toxic products in the processed material.
The fermentation of naturally contaminated maize to produce ethanol led to only limited degradation of fumonisin B1, most of which was recovered in distillers’ dried grain, thin stillage, and distillers’ soluble fractions (Bothast et al., 1992). Fumonisins have also been found in beer, indicating that the toxins persist under the conditions (temperature, pH) prevailing during the brewing process (Scott & Lawrence, 1995; Scott et al., 1997; Hlywka & Bullerman, 1999).
Surveys of the frequency of occurrence and concentrations in maize and maize products throughout the world are summarized in Appendices A–C. Over 60% of the surveyed products contained detectable amounts of fumonisins. Unfortunately, most of the reports did not contain sufficient information to allow determination of the representativeness of the data, i.e. they lacked information on sampling methods and involved insufficient numbers of samples to allow statistical evaluation of the fumonisin contamination of the diet.
The highest concentrations of fumonisins are found in visibly damaged or mouldy maize. The data show that the concentrations and incidence of contamination vary considerably in relation to the commodity tested and the source. The highest frequency was recorded in maize feeds, followed by ground maize products such as flour, grits, polenta, semolina, and gluten, maize kernels, and miscellaneous maize foods. The list of commercial retail foods that may be contaminated with fumonisins includes maize flour, grits, polenta, semolina, snacks, cornflakes, sweet, canned, and frozen maize, extruded maize, maize bread, maize-extruded bread, biscuits, cereals, chips, pastes, starch, infant foods, gruel, purée, noodles, popcorn, porridge, tortillas, tortilla chips, masa, popped maize, soup, tacos, and tostadas. Generally, processed maize foods have lower concentrations and a lower frequency of contamination than untreated foods. These differences may be the result of dilution of maize in food commodities or may depend on differences in maize cultivars or in the quality requirements for different destinations. Additionally, fumonisins are water-soluble, and processes that involve washing or water treatment may result in their partial or complete removal from the final food product.
In this evaluation, only the consumption of contaminated maize or maize-containing food products was considered, as the contributions of other commodities to the intake of fumonisins are too low and too variable to affect overall long-term exposure significantly.
Dietary intake of fumonisins was assessed in accordance with the recommen-dations of an FAO/WHO workshop on methods for assessing exposure to contaminants and toxins (WHO, 2000b). That workshop recommended that the best available data on concentrations in foods be used to estimate intake. For commodities that contribute significantly to intake, distribution curves should be generated to provide options to governments and national and international regulatory agencies for use in risk management. The Workshop further recommended that international estimates of dietary intake be generated by multiplying mean or median concentrations by the values for consumption of a commodity in the five GEMS/Food regional diets (WHO, 1998), which were derived from food balance sheets compiled by FAO. Since these sheets are available for most countries, they provide a set of data that are comparable across countries and regions of the world (WHO, 2000b). The five regions represented by the diets are Africa, Europe (which includes Australia, Canada, New Zealand, and the USA), the Far East, Latin America, and the Middle East. The regional diets represent the average per-capita availability of food commodities rather than actual food consumption, and data on availability generally result in an overestimate of consumption by about 15% (WHO, 1998). The workshop noted that, if available, national intake estimates should also be reported, as they may provide information about the intake of specific population subgroups or consumers of large amounts, which cannot be derived from GEMS/Food regional diets.
As the toxic effects in humans attributed to consumption of fumonisins in food are primarily of a long-term nature, this assessment is concerned solely with long-term intake.
Nine countries, Argentina, Brazil, Canada, China, Denmark, Sweden, the United Kingdom, the United States, and Uruguay, submitted information on the concentra-tions of fumonisins in maize and maize-derived foods. Fumonisins were detected in over 60% of all food products tested. The rate of detection was much lower in sound maize than in mouldy maize, and processed maize-containing foods generally contained lower concentrations of fumonisins than maize grain, flour, or grits.
A frequency distribution of the concentrations of fumonisins in maize was derived from available data in 1997 and published as part of an assessment of human intake of fumonisins in the Netherlands (de Nijs et al., 1998a). All maize consumed in the Netherlands is imported, and most was from Europe, South America, and the USA, although some was imported from Asia and Africa. As the concentrations of fumonisins and the incidences of fumonisin contamination reflected those found in the submitted data, for the purposes of this analysis, they were taken as representative of the maize available in trade throughout the world. Analysis of data available since 1997 showed little change in the patterns of incidence and concentration of fumonisins in maize and maize-based foods.
The concentrations of fumonisins were shown by the least-squares method to be distributed log-normally. The arithmetic mean concentration in the 349 samples used in the distribution was 1.36 mg/kg, and this distribution was combined with appropriate food intakes for assessing intake.
Data on food intake from the GEMS/Food regional diets (Table 14; WHO, 1998) were combined with the distribution of fumonisin concentrations in maize in commerce described above to yield estimates of fumonisin intake. A commercially available software product, @Risk (Palisade Corp.) was used with Microsoft Excel 97 to derive the distributions. Each simulation was run with Latin Hypercube sampling and 25 000 iterations.
Table 14. Intake of foods containing fumonisins in the GEMS/Food regional diets
|
Commodity |
GEMS code |
Diet (g of food per person per day) |
||||
|
Middle Eastern |
Far Eastern |
African |
Latin American |
European |
||
|
Maize, all |
GC 645 |
48 |
31 |
110 |
42 |
8.8 |
|
Maize |
|
16 |
0 |
0 |
1.5 |
0 |
|
Maize, flour |
CF 1255 |
32 |
31 |
110 |
40 |
8.8 |
|
Sweet maize (kernels) |
VO 1275 |
0 |
0 |
3.3 |
0 |
6.2 |
|
Sweet maize (cob) |
VO 447 |
0 |
0 |
4.4 |
0 |
8.3 |
|
Popcorn |
GC 656 |
0.2 |
0.2 |
0.2 |
0.2 |
0.2 |
|
All cereals |
GC 80 |
430 |
450 |
330 |
250 |
230 |
Three scenarios were examined. In the first scenario, the per-capita consumtpion of maize in the GEMS/Food diets was combined with the distribution of concentrations of fumonisins to yield a distribution of fumonisin intake (Table 15). In the second scenario, a hypothetical distribution of maize consumption was estimated by assuming that it is log-normally distributed in each diet, with a standard deviation equal to 66% of the mean consumption. This standard deviation was based on empirical observations of various distributions of commonly eaten foods in the USA (Table 16). This scenario provides a better evaluation of the high end of the intake distribution of fumonisins; however, the mean intake in this scenario is the same as that in the first. The third scenario was intended to mimic the worst case, in which the only grain that a person consumes is maize (Table 17). This extremely conservative scenario could describe the intake of fumonisins by individuals who either have no access to grains other than maize (e.g. subsistence farmers) or are allergic to other grains, such as wheat and rice. In this scenario, the per-capita consumption of all grains was combined with the distribution of fumonisin intakes.
Table 15. Scenario 1: Distribution of intake of fumonisins
|
Intake of fumonisins (%) |
Intake of fumonisins (µg/day per person) |
||||
|
Middle Eastern |
Far Eastern |
African |
Latin American |
European |
|
|
Minimum |
0.2 |
0.1 |
0.4 |
0.2 |
0.0 |
|
Maximum |
1100 |
740 |
2500 |
990 |
210 |
|
Mean |
66 |
43 |
140 |
57 |
12 |
|
Standard deviation |
120 |
76 |
260 |
100 |
21 |
|
5 |
0.3 |
0.2 |
0.7 |
0.3 |
0.1 |
|
10 |
0.6 |
0.4 |
1.4 |
0.6 |
0.1 |
|
15 |
1.4 |
0.9 |
3.1 |
1.2 |
0.3 |
|
20 |
2.0 |
1.3 |
4.4 |
1.7 |
0.4 |
|
25 |
3.0 |
1.9 |
6.6 |
2.6 |
0.5 |
|
30 |
4.4 |
2.9 |
9.7 |
3.8 |
0.8 |
|
35 |
6.3 |
4.1 |
14 |
5.5 |
1.2 |
|
40 |
9.6 |
6.2 |
21 |
8.3 |
1.7 |
|
45 |
14 |
9.1 |
31 |
12 |
2.6 |
|
50 |
20 |
13 |
44 |
17 |
3.7 |
|
55 |
26 |
17 |
57 |
22 |
4.7 |
|
60 |
33 |
22 |
74 |
29 |
6.1 |
|
65 |
43 |
28 |
94 |
37 |
7.8 |
|
70 |
54 |
35 |
120 |
47 |
9.9 |
|
75 |
70 |
45 |
150 |
61 |
13 |
|
80 |
97 |
63 |
210 |
84 |
18 |
|
85 |
140 |
89 |
300 |
120 |
25 |
|
90 |
200 |
130 |
440 |
170 |
36 |
|
95 |
300 |
190 |
660 |
260 |
54 |
|
97.5 |
370 |
240 |
810 |
320 |
67 |
Table 16. Scenario 2: Distribution of intake of fumonisins
|
Intake of fumonisins (%) |
Intake of fumonisins (µg/day per person) |
||||
|
Middle Eastern |
Far Eastern |
African |
Latin American |
European |
|
|
Minimum |
0.0 |
0.0 |
0.1 |
0.0 |
0.0 |
|
Maximum |
4300 |
2200 |
9000 |
3000 |
490 |
|
Mean |
65 |
42 |
140 |
57 |
12 |
|
Standard deviation |
140 |
93 |
340 |
130 |
26 |
|
5 |
0.3 |
0.2 |
0.6 |
0.2 |
0.0 |
|
10 |
0.6 |
0.3 |
1.2 |
0.5 |
0.1 |
|
15 |
1.0 |
0.6 |
2.1 |
0.8 |
0.2 |
|
20 |
1.7 |
1.0 |
3.4 |
1.4 |
0.3 |
|
25 |
2.5 |
1.6 |
5.2 |
2.1 |
0.4 |
|
30 |
3.7 |
2.3 |
7.8 |
3.0 |
0.7 |
|
35 |
5.4 |
3.4 |
11 |
4.4 |
1.0 |
|
40 |
7.9 |
4.9 |
17 |
6.5 |
1.4 |
|
45 |
11 |
7.0 |
24 |
9.1 |
1.9 |
|
50 |
15 |
9.7 |
33 |
13 |
2.7 |
|
55 |
21 |
13 |
45 |
17 |
3.6 |
|
60 |
28 |
17 |
59 |
23 |
4.9 |
|
65 |
36 |
23 |
77 |
30 |
6.5 |
|
70 |
47 |
30 |
100 |
40 |
8.6 |
|
75 |
63 |
40 |
130 |
53 |
12 |
|
80 |
84 |
55 |
180 |
72 |
16 |
|
85 |
120 |
77 |
260 |
100 |
22 |
|
90 |
180 |
110 |
400 |
1530 |
33 |
|
95 |
300 |
190 |
670 |
260 |
55 |
|
97.5 |
440 |
300 |
980 |
390 |
82 |
Table 17. Scenario 3: Distribution of intake of fumonisins
|
Intake of fumonisins (%) |
Intake of fumonisins (µg/day per person) |
||||
|
Middle Eastern |
Far Eastern |
African |
Latin American |
European |
|
|
Minimum |
0.4 |
0.3 |
0.2 |
0.2 |
0.2 |
|
Maximum |
30 000 |
33 000 |
24 000 |
17 000 |
13 000 |
|
Mean |
590 |
610 |
430 |
350 |
310 |
|
Standard deviation |
1 200 |
1 300 |
920 |
770 |
660 |
|
5 |
2.5 |
2.6 |
1.8 |
1.4 |
1.3 |
|
10 |
5.0 |
5.3 |
3.7 |
2.9 |
2.7 |
|
15 |
9.7 |
9.9 |
7.0 |
5.4 |
5.0 |
|
20 |
15 |
16 |
11 |
8.7 |
8.0 |
|
25 |
23 |
24 |
17 |
13 |
12 |
|
30 |
34 |
35 |
25 |
20 |
18 |
|
35 |
50 |
51 |
36 |
29 |
26 |
|
40 |
72 |
75 |
53 |
41 |
37 |
|
45 |
100 |
110 |
76 |
59 |
54 |
|
50 |
140 |
150 |
100 |
82 |
75 |
|
55 |
190 |
200 |
140 |
110 |
100 |
|
60 |
250 |
260 |
190 |
140 |
130 |
|
65 |
330 |
340 |
240 |
190 |
170 |
|
70 |
440 |
450 |
320 |
250 |
220 |
|
75 |
580 |
600 |
420 |
330 |
300 |
|
80 |
780 |
830 |
580 |
440 |
410 |
|
85 |
1 100 |
1 200 |
800 |
630 |
580 |
|
90 |
1 600 |
1 700 |
1 200 |
940 |
860 |
|
95 |
2 700 |
2 800 |
2 000 |
1 600 |
1 400 |
|
97.5 |
4 000 |
4 000 |
2 900 |
2 300 |
2 100 |
The mean intake of fumonisins in scenarios 1 and 2 ranged from 12 µg/day per person in the European diet to 140 µg/day per person in the African diet. These estimates were based on the assumption that an individual consumes randomly contaminated maize over a lifetime and will consume maize at a daily rate equal to the per-capita disappearance of maize. Use of the presumed distribution of maize consumption, as in the second scenario, allows examination of fumonisin intake by people who consume larger amounts of maize over a lifetime as well as those who consume maize with higher concentrations of fumonisins in the long term. Over a lifetime, a person would be expected to consume maize with concentrations of fumonisins at approaching the mean of the distribution. Only those individuals in areas where maize is regularly and heavily contaminated would fall into the high end of the distribution of intake of fumonisins in this scenario.The intake of fumonisins at the 97.5th percentile in this scenario ranges from 82 µg/day per person in the European diet to 980 µg/day per person in the African diet. Below this percentile, the predicted intakes in the two scenarios are not appreciably different.
The predicted intake of fumonisins in the third scenario, which describes the potential intake of fumonisins by persons who eat maize in place of all other grains, is appreciably higher than those in the first two scenarios. It must be emphasized that the number of individuals covered by this scenario is extremely small on a global basis and consists primarily of rural subsistence farmers, who are not representative of national or GEMS/Food regional populations. The mean intake in this scenario ranged from 310 µg/day per person in the European diet to 610 µg/day per person in the Far Eastern diet (in which the diet would typically be dominated by rice). The 95th percentile intake ranged from 1400 µg/day per person in the European diet to 2800 µg/day per person in the Far Eastern diet.
Although nine countries submitted information on the concentrations of fumonisins in maize and maize-based foods, only the United Kingdom presented information that allowed estimation of national intake of fumonisins. Nevertheless, several national estimates have been published, from Argentina, Canada, the Netherlands, South Africa, Switzerland, and the USA (Kuiper-Goodman et al., 1996; Humphreys et al., 2000; Marasas, 1997; de Nijs, 1997; Solovey et al., 1999). The mean daily intake of persons aged 15–55 in Argentina was estimated to be 0.2 µg/kg bw per day. Intake in Canada between 1991 and early 1995 was estimated to be 0.017–0.089 µg/kg bw per day (Kuiper-Goodman et al., 1996).
On the basis of daily average consumption of maize and maize products of 3 g for the general population, 42 g for regular eaters of maize products, and 160 g for persons intolerant to gluten in the Netherlands, the estimated daily intakes of fumonisin B1 were 0.06, 1.0, and 3.7 µg/kg bw per day, respectively, assuming a mean fumonisin B1 content of 1.36 mg/kg of maize produce. It was estimated conservatively that 97% of individuals with gluten intolerance had a daily intake of fumonisin B1 of at least 1 µg, and 37% had an intake of at least 100 µg, while the proportions of the general population exposed to these concentrations of fumonisin B1 were 49% and 1%, respectively (de Nijs, 1997; de Nijs et al., 1998b).
The intakes of fumonisin B1 in the Transkei, South Africa, were estimated to be 14 and 440 µg/kg bw per day from healthy and mouldy maize, respectively (Thiel et al., 1992). The probable daily intake of rural blacks in the Transkei consuming home-grown mouldy maize was estimated to vary from 1.2 to 355 µg/kg bw per day (Marasas, 1997).
The mean daily intake of fumonisins in Switzerland was estimated to be 0.030 µg/kg bw per day (Zoller et al., 1994).
In the United Kingdom, various maize-based foods are consumed at levels of 0.6–12 g/day (Gregory et al., 1990). Polenta was found to contain the highest concentration of fumonisins, with a mean of 530 µg/kg and a maximum of 2100 µg/kg. All other retail foods contained < 100 µg/kg. On the basis of the mean consumption of polenta (maize meal), breakfast cereals, popcorn, and maize-based snacks and the mean fumonisin concentrations in these foods, the intake of fumonisins would be 1.8 mg/day per person, or approximately 0.03 µg/kg bw per day. Intake of fumonisin B1 at the 90th percentile of intake can be approximated by tripling the mean.
A preliminary estimate of the intake of fumonisins by maize eaters in the USA was 0.08 µg/kg bw per day (Humphreys et al., 2000).
National estimates of intake vary considerably according to the source and amount of maize in the diet as well as the prevalence of Fusarium kernel rot in the harvested crop. The national estimates are summarized in Table 18, in which intakes on a body-weight basis have been converted to micrograms per day by assuming an average body weight of 60 kg for an adult over a lifetime.
Table 18. National estimates of fumonisin intake
|
Country |
Intake (µg/kg bw per day) |
|
|
Mean or median |
High |
|
|
Argentina |
0.2 |
|
|
Canada |
0.02 |
0.08 |
|
Netherlands |
0.06, 1.0 |
|
|
Switzerland |
0.03 |
|
|
United Kingdoma |
0.03 |
0.1 |
|
USA |
0.08 |
|
a
Calculated for this risk assessment from data submitted to WHOAll the national estimates of fumonisin intake are appreciably lower than the international estimates prepared from the GEMS/Food diets. A number of factors may contribute to these differences. First, many of the national estimates were based on intakes of maize-containing food products, which are generally less heavily contaminated with fumonisins than the maize or maize flour used to make them. Secondly, the intakes of individual food products used in the national estimates were typically lower than the overall consumption of maize in the GEMS/Food diets. Finally, exported maize appears to be more heavily contaminated with fumonisins than maize used domestically in food products (Rheeder et al., 1994), due perhaps to lengthy storage before transport or a lack of strict regulation of exported maize consignments.
Imposition of limits on the concentration of fumonisins in maize in international trade would probably affect the intake of fumonisins, especially at the high end of the distribution. Table 19 shows the potential intakes of fumonisins in the GEMS/Food African diet if a limit of 1, 2, 5, or 10 mg/kg was imposed and enforced or with the default assumption of no limit. The model and data used to produce the estimates in Table 15 were used. The main assumption in this model is that maize would not be consumed at a concentration above the maximum limit. Additionally, it was assumed that the existence of a limit would have no effect on the distribution of fumonisins in maize below that limit, i.e. that no producer would seek to improve the overall distribution of fumonisins in maize to ensure that the maximal number of samples was acceptable. An approximation of the percentage of maize samples that would be rejected at each limit, assuming no change in production practices, is included.
Table 19. Potential intake of fumonisins from maize in the African diet when various limits are imposed and enforced
|
Limit (mg/kg) |
Intake of fumonisins (µg/day per person) |
||||||
|
Mean |
Minimum |
Maximum |
50th %ile |
90th %ile |
95th %ile |
% excluded |
|
|
None |
140 |
0.4 |
2500 |
44 |
440 |
660 |
0 |
|
1 |
27 |
0.4 |
110 |
13 |
77 |
90 |
32 |
|
2 |
46 |
0.4 |
210 |
21 |
130 |
160 |
20 |
|
5 |
86 |
0.4 |
530 |
34 |
260 |
370 |
7.6 |
|
10 |
120 |
0.4 |
1100 |
42 |
400 |
580 |
1.6 |
At least 12 fumonisins and structurally related analogues have been recognized and are categorized into five groups, A, B, C, P, and H ( Musser & Plattner, 1997). Of these, the fumonisins of the B series (B1, B2, B3) are the most widespread in nature (Sydenham et al., 1991; Nelson et al., 1993). The fumonisins are produced only by Fusarium spp. In a survey of the production of fumonisins B1 and B2 by 40 toxic Fusarium isolates, the toxins were produced only by F. moniliforme, F. proliferatum (both belong to the section Liseola), and F. nygami (Thiel et al., 1991). Strains of F. anthophilum, F. dlamini, and F. napiforme also produced fumonisins, but F. subglutinans and F. beomiforme did not (Nelson et al., 1992).
The fumonisin-producing fungi are common in grain. F. moniliforme is considered to be the main cause of Fusarium kernel rot, which occurs especially during warm, dry weather. F. moniliforme, F. proliferatum, F. nygamai, and F. napiforme are the most important producers of fumonisin B1 because of their association with food grains such as maize, millet, and sorghum (Nelson et al., 1993). Strains of F. moniliforme collected from various substrates and geographical areas were all found to produce fumonisins (Nelson et al., 1991), and the natural occurrence of fumonisins in foods and grains, especially maize, throughout the world is well documented (reviewed by Marasas, 1996; Shephard et al., 1996b; Munkvold & Desjardins, 1997). The potential presence of fumonisins in agricultural produce and processed foods is therefore a serious threat to public health, and efforts have been made to find ways of preventing or reducing the accumulation of fumonisins.
The prevalence of Fusarium and the subsequent production of fumonisins are enhanced in warm climates and under drought conditions, factors that cannot be controlled, although growers and consumers should be made aware that high concentrations of fumonisins are to be expected under such conditions. Insect damage also affects the accumulation of fumonisins, and both the prevalence and degree of insect damage are significantly correlated with the concentrations of the toxins. Maintenance of rigorous insect control may assist in reducing fumonisin contamination.
Maize hybrids that differ with respect to fumonisin accumulation have been identified, some of which contained only low concentrations of fumonisins (Shetty & Bhat, 1977; King & Scott, 1981; Shelby et al., 1994b; Doko et al., 1995; Visconti, 1996). However, hybrids grown in areas outside those to which they were adapted had enhanced fumonisins contents (Shelby et al., 1994b) as a result of the different environmental conditions. Hybrids that accumulate lower concentrations of fumonisins should therefore be screened in each growing area as part of a selection programme. Several genetic mechanisms may be responsible for the low incidence of toxins in some plants. These include factors related to inhibition of fungal invasion (e.g. hardness and composition of kernels; silk composition and viability; inhibitory compounds such as phytoalexins), the presence of enzymes that can degrade fumonisins (Duvick, 1999), and mechanical barriers (e.g. tight husks) or induced resistance to insect penetration. In maize hybrids genetically engineered for insect resistance (by insertion of Bacillus thuringiensis genes encoding the delta-endotoxin CryIA(b) expressed in kernels), the kernels consistently had less Fusarium ear rot and Fusarium infection than kernels from normal plants (Munkvold et al., 1997). Furthermore, the concentrations of fumonisins in the transgenic hybrids were lower than those in their normal counterparts (Munkvold et al., 1999). Thus, engineering of maize for insect resistance may reduce Fusarium infection in the kernels and the subsequent accumulation of fumonisin. Several mechanisms might be involved in the reduced concentrations of fumonisins in certain varieties, however. Their elucidation would provide a basis for planning breeding or genetic engineering strategies designed to develop host resistance.
The strategies for coping with mycotoxins after harvest include inhibition of fungal growth by methods including chemical and physical means, natural products, and biological control; removal of grains or particles suspected to contain toxins (segregation); and removal or destruction of existing mycotoxins by e.g. physical means, chemical treatment, adsorption, or biological degradation. Different methods can be combined to achieve a synergistic effect, expressed as enhanced activity.
As fungi will not grow if the water activity (aw) is lower than 0.65–0.70, drying of freshly harvested kernels is an elementary step towards reducing the accumulation of mycotoxins after harvest. The safe moisture content of maize is 14–15%.
The effect of propionate preservatives on the growth of F. verticillioides and F. proliferatum on irradiated maize and their production of fumonisin B1 was affected by the aw, temperature, and the concentration and source of propionate. Fumonisin production by F. verticilloides was not affected by this treatment, but increasing the dose of propionate decreased fumonisin production by F. proliferatum at 15 °C, regardless of the aw. It was concluded that environmental factors, fungal colonization, and preservatives interact in the grain ecosystem. Fumonisin production did not correlate with the fungal growth rate but was affected by interactions between growth conditions and propionate concentration (Marin et al., 1999).
Maize screenings can contain significantly higher concentrations of fumonisin than whole grain (Ross et al., 1991; Murphy et al., 1993), but no major size-associated segregation of fumonisin in screenings was found (Murphy et al., 1993). Feeding animals maize screenings has been correlated directly with diseases caused by fumonisins (equine leukoencephalomacia and porcine pulmonary oedema) (Harrison et al., 1990; Ross et al., 1990; Wilson et al., 1990). Thus, removal of all dockage constituents and bulk cleaning might reduce the concentrations of fumonisins (Sydenham et al., 1994; Malone et al., 1998) and, in turn, the incidence of animal and human intoxications. However, since fumonisins can occur in whole, undamaged grain, methods for removing fumonisin-containing materials must be improved before the grain enters food-processing operations (Bullerman, 1996; FAO/WHO, 2000).
As ammoniation is known to detoxify aflatoxins, its effect on fumonisins was also studied. Under atmospheric pressure and ambient temperature, ammoniation did not significantly reduce the concentration of fumonisins in maize (Norred et al., 1991; Sydenham et al., 1995), but ammoniation under high temperature or pressure reduced the concentration by 79%, leaving no mutagenic effect, indicating that ammoniation under the conditions described can be regarded as safe (Park et al., 1992). Several parameters, such as temperature, length of application, moisture content, ammonia concentration, and toxin concentration, could affect the efficacy of ammoniation, and the roles of these parameters in the process should be studied. Furthermore, the toxicity of the resulting compounds should be further analysed.
Another approach to the detoxication of fumonisin B1 involves a reaction with fructose to block the amine group, which is critical for its toxicity. The toxicity of fumonisin B1 to rats was indeed eliminated by such treatment, suggesting a new mechanism for fumonisin detoxication (Lu et al., 1997).
Exposure of maize to 15 kGy of gamma-irradiation, which is known to destroy mycotoxins, sterilized the maize but reduced the concentrations of fumonisins B1 and B2 by only 20% (Visconti et al., 1996b).
The extent to which a food contaminant is judged to represent a potential public health risk is usually assessed by comparing likely intake with a measure such as the tolerable daily intake (TDI). The TDI is derived by applying safety factors to the NOEL in the study considered to be critical, whether it was conducted in laboratory animals or humans. These factor account for extrapolation from animals to humans, variation in the human response, and, sometimes, for less-than-adequate data. This approach was followed by Kuiper-Goodman et al. (1996) in an assessment of fumonisin in maize products, noting that the point estimate of the intake of fumonisin by the group with the highest intake, 0.089 µg/day, was more than three orders of magnitude lower than the NOEL in studies in animals. A similar assessment was made with similar conclusions by the Nordic Council (TemaNord, 1998). However, de Nijs (1997) found no margin of safety for a sensitive sub-population, patients with coeliac disease, and the approach could not be used to derive an estimate of the probability of harm. These assessments are summarized in Table 20.
Table 20. Results of previous safety assessments
|
End-point |
NOEL or LOEL |
Safety factor |
ADI or TDI |
Reference |
|
Renal apoptosis |
LOEL: 190 |
100 |
0.50 |
de Nijs (1997) |
|
Rat; cancer |
|
100 |
8 |
Marasas (1997) |
|
|
|
1000 |
0.8 |
|
|
|
|
5000 |
0.16 |
|
|
Rat: kidney |
NOEL: 0.2 |
100 |
2 |
Commission of the European Union (1998b) |
Tables 21–24 show the LOELs and NOELs for renal and hepatic effects in studies in which experimental animals were fed fumonisin B-contaminated maize or culture material.
Table 21. NOEL or LOEL for hepatic effects in studies of rodents given purified fumonisin B1 by oral administration
|
Species and strain |
Duration |
Route |
NOEL/LOELa |
Reference |
|
B6C3F1 mouse |
90 |
Diet |
9.7/29 |
Voss et al. (1995a) |
|
B6C3F1 mouse |
14 |
Gavage |
5/15c |
Bondy et al. (1997) |
|
B6C3F1 mouse |
28 |
Diet |
< 24 |
National Toxicology Program (1999) |
|
B6C3F1 mouse |
728 |
Diet |
0.7/1.9d |
National Toxicology Program (1999) |
|
1.9/6.6 (tumours) |
||||
|
BD IX rat |
780/690 |
Diet |
0.03/0.3 |
Gelderblom et al. (1991, 2001b) |
|
0.8/1.6 (tumours) |
||||
|
Fischer 344 rat |
21 |
Diet |
1.7/3.5 |
Gelderblom et al. (1994) |
|
Fischer 344 rat |
14 |
Gavage |
4/8.5 |
Gelderblom et al. (1994) |
|
Fischer 344 rat |
21 |
Diet |
0.7/3.5 |
Gelderblom et al. (1996c and unpublished) |
|
Fischer 344 rat |
28 |
Diet |
< 12 |
National Toxicology Program (1999) |
|
Fischer 344 rat |
90 |
Diet |
> 6.4 |
Voss et al. (1995a) |
|
Fischer 344 rat |
728 |
Diet |
> 6.6 |
National Toxicology Program (1999) |
|
Sprague-Dawley rat |
28 |
Diet |
1.4/4.1 |
Voss et al. (1993) |
|
Sprague-Dawley rat |
11 |
Gavage |
5/15 |
Bondy et al. (1996, 1998) |
|
Sprague-Dawley rat |
11 |
Gavage |
15/35b |
Mehta et al. (1998) |
|
RIVM:WU rat |
28 |
Gavage |
> 3.0 |
de Nijs (1997) |
|
a |
Values preceded by ‘<’ indicates the NOEL was less than the value shown; ‘>’ indicates that the LOEL would be greater than the value shown; ‘/’ indicates that the first number is the NOEL and the second is the LOEL. |
|
b |
Based on foci positive for glutathione-S-transferase, placental form |
|
c |
Increased mitosis observed in females at 5 mg/kg bw per day |
|
d |
Based on hepatocellular hypertrophy |
Table 22. NOEL or LOEL for renal effects in studies of rodents given purified fumonisin B1 by oral administration
|
Species and strain |
Duration |
Route |
NOEL/LOELa |
Reference |
|
B6C3F1 mouse |
90 |
Diet |
> 29 |
Voss et al. (1995a) |
|
B6C3F1 mouse |
14 |
Gavage |
5/15 |
Bondy et al. (1997) |
|
B6C3F1 mouse |
28 |
Diet |
> 105 |
National Toxicology Program (1999) |
|
B6C3F1 mouse |
728 |
Diet |
> 15 |
National Toxicology Program (1999) |
|
BD IX rat |
780/690 |
Diet |
0.03/0.3 |
Gelderblom et al. 1991, 2001b) |
|
Fischer 344 rat |
21 |
Diet |
< 0.7 |
Gelderblom et al. (1996c and unpublished) |
|
Fischer 344 rat |
28 |
Diet |
< 12 |
National Toxicology Program (1999) |
|
Fischer 344 rat |
90 |
Diet |
0.2/0.6 |
Voss et al. (1995a) |
|
Fischer 344 rat |
728 |
Diet |
0.22/0.67 |
National Toxicology Program (1999) |
|
0.67/2.2 (tumours) |
||||
|
Sprague-Dawley rat |
28 |
Diet |
< 1.4 |
Voss et al. (1993) |
|
Sprague-Dawley rat |
11 |
Gavage |
< 1 |
Bondy et al. (1996, 1998) |
|
RIVM:WU rat |
28 |
Gavage |
< 0.2 |
de Nijs (1997) |
a
Values preceded by ‘<’ indicates the NOEL was less than the value shown; ‘>’ indicates that the LOEL would be greater than the value shown; ‘/’ indicates that the first number is the NOEL and the second is the LOEL.
Table 23. NOEL or LOEL for renal and hepatic effects in studies of rats given diets containing naturally contaminated maize or fungal culture material containing fumonisins
|
Strain |
Duration |
Site |
NOEL/LOELa |
Reference |
|
Sprague-Dawley |
21 |
Kidney |
< 0.6 |
Voss et al. (1998) |
|
Liver |
0.6/4.5 |
|||
|
Fischer 344 |
176 |
Kidney |
> 3.2 |
Wilson et al. (1985) |
|
Liver (tumours) |
< 2.3 |
|||
|
BDIX |
610–691 |
Liver |
< 0.65 |
Purchase & Joubert (1970); Purchase et al. (1975) |
|
BDIX |
894 |
Kidney |
> 6.9 |
Marasas et al. (1984) |
|
Liver (tumours) |
< 3.2 |
|||
|
BDIX |
869 |
Kidney |
> 1.3 |
Jaskiewicz et al. (1987a) |
|
Liver (tumours) |
< 0.4 |
a
Values preceded by ‘<’ indicates the NOEL was less than the value shown; ‘>’ indicates that the LOEL would be greater than the value shown; ‘/’ indicates that the first number is the NOEL and the second is the LOEL.
Table 24. NOEL or LOEL for various end-points in studies of animals other than rodents given diets containing purified fumonisin B1, MRC 826, other fungal cultures, or naturally contaminated maize
|
Species |
Treatment |
Duration |
Site |
NOEL/LOELa |
Reference |
|
Vervet monkey |
MRC 826 |
13.5 years |
Kidney |
0.11/0.18 |
Gelderblom et al. (2001c) |
|
Liver |
0.11/0.18 |
||||
|
Equid |
Contaminated maize and fungal culture |
150 and |
Equine |
0.30/0.44 |
Wang et al. (1992) |
|
241 days |
leukoencephalo-malacia |
||||
|
Pig |
Fungal culture |
28 days |
Liver and lung |
< 0.4 |
Zomborszky et al. (2000) |
|
Pig |
Contaminated maize |
14 days |
Liver |
0.2/.92 |
Riley et al. (1993) |
|
Lung |
4/7 |
||||
|
Rabbit |
Purified fumonisin B1 by gavage |
17 days |
Kidney |
< 0.1 |
LaBorde et al. (1997) |
a
Values preceded by ‘<’ indicates the NOEL was less than the value shown; ‘>’ indicates that the LOEL would be greater than the value shown; ‘/’ indicates that the first number is the NOEL and the second is the LOEL.Quantitative assessments were made of the magnitude of the adverse effects, variations in intake of fumonisins among populations, and the uncertainties of correlations, expressed as a range of possible outcomes. The most sensitive adverse response in rats, nephrotoxicity, was used for the dose–response analysis (Voss et al., 1995a). Dietary intake of fumonisin in maize products was estimated from a 3-day survey of 15 368 persons (US Department of Agriculture, 1989–90). The proportion of maize in each product consumed was estimated from standard recipes. The concentrations of fumonisins in maize products were estimated from surveys by the US Department of Agriculture, and the distribution of fumonisin consumption was modelled for each individual in the surveyed population. The uncertainties associated with the predictions made from each model and with model selection are described. The results of the analyses of the dose–response relationship and intake were assimilated in a two-dimensional Monte-Carlo simulation. The distributions representing variation and uncertainty were selected iteratively to form a two-dimensional array of estimates of the magnitude of harm to individuals and to the population as a whole.
The level of risk associated with fumonisin intake is described first, and then the reductions in risk achieved with two alternative risk management options (reduced concentration and reduced consumption) to gauge their relative effectiveness are described. Nephrotoxicity was used as the end-point because data on the dose–response relationship in individual animals were available (Voss et al., 1995a). The scenarios were designed to show how risk changes when exposure was altered by (1) limiting the intake of fumonisins in maize products and (2) decreasing the consumption of maize by frequent consumers.
(a) Analytical method
Three software routines were written in Microsoft Excel macro language (Visual Basic for Applications) for fitting curves and for Monte-Carlo simulations.
|
(1) |
ParamFit generates a two-dimensional description of the data from a number of alternative frequency (variability) models to represent uncertainty. Uncertainty associated with model selection is represented by distributing the frequency of use of each of the alternative models according to a specified weighting criterion (i.e. a balance between goodness-of-fit and the number of parameters). |
|
(2) |
The Quantitative Risk and Response program models the relationship between dose and response. It models both the magnitude of individual subject response and population variability, and it accounts for model uncertainty associated with the predictions. The program requires a data set of toxicological observations for individual subjects in which the dose and magnitude of response are specified. These data are used to derive a cumulative response distribution for each dose. The program generates two Excel functions: one that predicts response as a function of dose and another that predicts frequency as a function of response. These functions can be used as part of a Monte-Carlo simulation. Two distributions indicate uncertainty, in addition to the distributions of population variation: One is a frequency-based reflection of deviation of the data set from the individual models used to describe the data, while the other is a model probability distribution representing the uncertainty due to model selection. |
|
(3) |
MC2D runs two-dimensional Monte-Carlo simulations. The distribution of the model output is estimated by sampling the input distributions repeatedly. MC2D integrates the sources of variability and the sources of uncertainty into separate distributional dimensions. A Boolean variable on the worksheet controls the behaviour of random number generators in the model by regulating whether a given distribution is re-sampled for every calculation (for variability distributions) or re-sampled only at the start of a new uncertainty iteration (for uncertainty statements). Results from up to 10 output cells are collected. In the present analysis, one output cell was used for each of the four risk management scenarios. After the simulation, MC2D can generate a standard table of specific percentiles for each of the output cells chosen. The net difference across the entire simulation for two output cells can also be calculated. |
In both the Paramfit and Quantitative Risk and Response programs, the goodness of fit of each model was optimized by nonlinear regression (Excel Solver). Model weights were calculated from an algorithm that rewards models for goodness-of-fit and penalizes them for use of extra parameters.
(b) Data and model
Data from surveys of the US Department of Agriculture conducted in 1994 and 1995 were used to estimate the amount of fumonisin in maize, and these are summarized in Table 25. These data did not distinguish between sweet and field maize which are reported to have different concentrations of fumonisins (Trucksess et al., 1995); these differences may be real or be artefacts, since the sugars of sweet maize may hinder fumonisin extraction. Sweet maize constitutes only 5% of the maize produced in the USA. Both fumonisin B1 and fumonisin B2 were assayed. Because the concentration of fumonisin B2 was often below the LOD, the concentrations of fumonisin B1 were modelled, and those of total fumonisins were estimated to be 1.25 times the fumonisin B1 concentration. The distributions for shelled maize, maize meal, maize flour, and popcorn were modelled with ParamFit, and the distributions in corn flakes, maize grits, maize chips, and maize tortillas were described as a series of percentiles corresponding to actual values; values below the LOD were considered to be uniformly distributed between 0 and the LOD of 20 mg/kg.
Table 25. Concentrations of fumonisin B1 in maize products (µg/kg)
|
Maize product |
No. of samples |
Minimum |
Maximum |
Average |
|
Shelled maize |
78 |
ND |
3100 |
310 |
|
Maize meal |
64 |
ND |
1900 |
160 |
|
Maize flour |
15 |
ND |
150 |
37 |
|
Popcorn |
15 |
ND |
72 |
22 |
|
Maize grits |
15 |
ND |
180 |
30 |
|
Tortillas |
12 |
ND |
24 |
12 |
|
Maize chips |
6 |
ND |
ND |
ND |
|
Cereal |
5 |
ND |
ND |
ND |
ND, below the LOD of about 20 µg/kg
The concentrations of fumonisin B1 in maize products were fitted with 10 alternative distributions: exponential, normal, gamma, log-normal, logistic, Cauchy, beta, triangular, retangular, and Weibull. The best model for each product, as judged by the same criteria used for weighting models (goodness-of-fit and number of parameters), were used to describe the distribution of concentrations of fumonisin B1 in each commodity.
The consumption of maize products was evaluated from a US Department of Agriculture survey conducted in 1989–91, in which the 3-day eating habits of 15 368 people were recorded. The amount and type of maize product in each food consumed was calculated from a standard recipe. Maize starch and maize syrup were not included in the analysis as they are minor components in many recipes and because most fumonisin is removed from these products during processing.
Some form of maize product was consumed by 2655 persons in this population during the 3-day survey period. As a 90-day experiment would correspond to about 1/16th of the rats’ lifespan, a human exposure period of 360 days was simulated by randomly re-sampling (bootstrapping) each of the 3-day exposure records 360 times. The simulation was repeated 10 times to represent the uncertainty resulting from this sampling exercise.
As the US Department of Agriculture survey contains information about the age of each subject but not their body weights, intakes were calculated on the basis of the mean weights (National Acadaemy of Sciences, 1989) for the age group corresponding to each individual in the survey.
(c) Dose–response relationship for fumonisin and nephrotoxicity
A dose–response relationship for fumonisin and nephrotoxicity was based on data from the study of Voss et al. (1995a) in which rats were given feed containing fumonisin B1 at a concentration of 0, 1, 3, 9, 27, or 81 mg/kg. The end-point was lesions on a rating scale of 0–3, the smallest observable lesion being given a score of 1. Because the females had lesions only at the highest dose, no dose–response relationship could be derived. This dose was about 10 times that which caused adverse effects in males, and the females were estimated to be about 10 times less sensitive than males. A rectangular distribution with a range of 5–15 was used to represent the uncertainty of the relative dose in females. Thus, only the dose–response relationship in males was derived directly from the data.
The data were fitted into 273 models that differed in the dose–effect function (e.g. linear, sigmoidal, exponential), the population model (e.g. normal, log-normal), and the presence of dose-independent (background) factors. The uncertainty of the model was represented by weighting; no one model was chosen, and the validity of each model was given a relative probability (see discussion of quantitative risk and response).
Differences between rats and humans in fumonisin-induced nephrotoxicity would best be approximated by using data specific for toxicity induced by fumonisin-like compounds in the two species; however, such information was not available. In this analysis, the interspecies comparison was based on the ratio of the maximal tolerated dose in humans to the LD10 value in rats in their responses to alkylating agents, a class of compounds for which data on both rats and humans are available (Travis & White, 1988). The resulting frequency distribution of the toxicity ratios was used to extrapolate and represent the uncertainty associated with this inference. Human variation, based on a compilation of pharmacokinetics (Hattis et al., 1987), was accounted for by a log10 geometric standard deviation.
(d) Simulations
Simulations were conducted in two steps. The first was an analysis of intake with a minimal number of uncertainty iterations (10) but which simulated a 360-day intake for each of the 2655 maize-eating individuals. The second step was a human response simulation which contained 500 uncertainty iterations (outer loop) and 2655 variability iterations (inner loop). The second step involved four procedures: One of the 10 intake runs was selected randomly at the start of each uncertainty iteration. The human dose was adjusted to an equivalent rodent dose from the species extrapolation ratio, which was re-sampled at the start of each uncertainty iteration. The dose was adjusted to reflect human variation (the distribution of variation was re-sampled at each iteration). The response was calculated from the male dose–response mode. For females in the consumption survey, the dose was adjusted downwards by a factor of 10 to reflect the lower sensitivity of female rats.
Intake simulation: The scenarios designed to simulate concentration limits were generated by truncating the distributions of fumonisins in maize products (e.g. limit of 1 mg/kg). The effects on the intake of fumonisin B of varying the concentration limit of fumonisin in maize are summarized in Table 26. The reduction in intake of fumonisin at any percentile is not proportional to the reduction in the limit. Eaters in the lower percentile of consumption are not affected. Even at the restrictive limit of 0.5 mg/kg, which would eliminate much of the maize supply, the fumonisin intake of 50th percentile eaters would vary by less than 1.5 µg/day with the six concentration limits and no limit (the concentration of fumonisins at the time of the survey). The 0.5 mg/kg limit resulted in a predicted reduction of intake of about 40% for people in the highest percentiles.
Table 26. Effects of different concentration limits on intake of fumonisin B
|
Consumption |
Intake of fumonisin B (µg/day per person) at limits of : |
||||||
|
0.5 mg/kg |
1 mg/kg |
1.5 mg/kg |
2 mg/kg |
2.5 mg/kg |
3 mg/kg |
None |
|
|
Minimum |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
0.01 |
0.04 |
0.04 |
0.04 |
0.04 |
0.04 |
0.04 |
0.04 |
|
0.05 |
0.09 |
0.09 |
0.09 |
0.09 |
0.09 |
0.09 |
0.09 |
|
0.10 |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
|
0.25 |
0.30 |
0.30 |
0.31 |
0.31 |
0.31 |
0.31 |
0.31 |
|
0.50 |
2.2 |
2.6 |
2.8 |
3.0 |
3.0 |
3.1 |
3.6 |
|
0.75 |
6.6 |
9.0 |
10 |
11 |
11 |
11 |
11 |
|
0.90 |
12 |
17 |
19 |
20 |
20 |
21 |
21 |
|
0.95 |
17 |
24 |
27 |
29 |
29 |
30 |
31 |
|
0.99 |
28 |
38 |
43 |
46 |
47 |
48 |
48 |
|
Maximum |
59 |
83 |
95 |
100 |
100 |
100 |
100 |
Each value is the mean of 10 uncertainty iterations. The reduction in exposure at any percentile is not proportional to the reduction in the limit; even the draconian limit of 0.5 mg/kg would result in a predicted reduction in exposure of only 50% at the upper percentiles.
The scenarios that simulate consumption limits were generated by truncating the daily consumption (e.g. a limit of 100 g/day). Table 27 summarizes the effect of varying the consumption limit on intake. Again, eaters in the lower percentile are not affected. Eaters in the 50th percentile are affected only by about 1 µg/day at the 25 g/day limit. Eaters in the upper percentile are greatly affected; the fumonisin intake of eaters in the 95th percentile is reduced by about two-thirds, or 20 µg/day, and that of eaters in the 99th percentile is reduced by over sevenfold, or > 37 µg/day. In this assessment, reducing intake of fumonisin by lowering maize consumption was more effective than lowering the concentration of fumonisin in maize.
Table 27. Effects of different consumption limits of maize on intake of fumonisin B
|
Consumption |
Intake of fumonisin B (µg/day per person) at maize |
||||
|
25 g/day |
50 g/day |
100 g/day |
300 g/day |
No limit |
|
|
Minimum |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
0.01 |
0.04 |
0.04 |
0.04 |
0.04 |
0.04 |
|
0.05 |
0.09 |
0.09 |
0.09 |
0.09 |
0.09 |
|
0.10 |
0.12 |
0.12 |
0.12 |
0.12 |
0.12 |
|
0.25 |
0.30 |
0.31 |
0.31 |
0.31 |
0.31 |
|
0.50 |
2.6 |
3.0 |
3.2 |
3.6 |
3.6 |
|
0.75 |
8.0 |
11 |
11 |
11 |
11 |
|
0.90 |
9.5 |
18 |
21 |
21 |
21 |
|
0.95 |
9.7 |
19 |
28 |
30 |
30 |
|
0.99 |
10 |
20 |
38 |
47 |
47 |
|
Maximum |
15 |
29 |
41 |
100 |
100 |
Each value is the mean of 10 uncertainty iterations. Limiting maize intake alters the exposure of persons with a high intake . For instance, in the consumption survey only two individuals ate more than 300 g/day. This limit therefore changes the position only of those two individuals in the distribution relative to the other consumers.
Response simulation for fumononisin-induced nephrotoxicity: The simulation of fumonisin B-induced human nephrotoxicity consisted of integration of the intake assessment, the dose–response function, and the functions accounting for human variation and species extrapolation. Table 28 shows the predicted severity of human nephrotoxicity when maize is consumed as described in the survey and for each of the scenarios (concentration and consumption). The units in the table represent a rating scale for nephrotoxicity of 0–3, 1 representing the lowest observable effect. As all the predicted values are below 1, no observable effect is anticipated. The predicted nephrotoxicity represents the background rate and is largely independent of maize consumption, and therefore of the concentration of fumonisin. This is shown more clearly in Tables 29 and 30, which show little difference in the predicted rate of nephrotoxicity with different scenerios. The tables give a comparison of the concentration and consumption limit scenarios by subtracting one array (simulation) of variability-by-uncertainty from the other. Table 29 shows the result of subtracting the background (no maize) rate from the concentration limit scenario, and Table 30 shows the result of subtracting the background (no maize) rate from the consumption limit scenario.
Table 28. Distributions of pathologist’s rating of nephrotoxicity with limits on concentration of fumonisin B and consumption of maize
|
Simulation |
Uncertainty/variability |
|||||
|
Average/ median |
Average/ 0.95 |
Median/ median |
Median/ 0.95 |
0.95/median |
0.95/0.95 |
|
|
Null limit, fumonisin B |
0.264535 |
0.264978 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
Null limit, maize |
0.264536 |
0.264978 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
1 mg/kg limit, fumonisin B |
0.264533 |
0.264886 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
3 mg/kg limit, fumonisin B |
0.264535 |
0.264960 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
20 g/day limit, maize consumtion |
0.264529 |
0.264712 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
100 g/day limit, maize consumption |
0.264535 |
0.264950 |
0.311075 |
0.311078 |
0.397879 |
0.397879 |
|
No maize |
0.264522 |
0.264522 |
0.311075 |
0.311075 |
0.397879 |
0.397879 |
Pathologist’s rating of nephrotoxicity, from 0 to 3, with 1 representing the lowest observable effect. As all the predicted values are < 1, no observable effect is anticipated. The first two simulations are identical; the slight difference in the average–median value reflects random variation. The predicted response values are largely independent of dose and therefore of maize consumption.
Table 29. Effects of limiting the concentration of fumonisin B on a pathologist’s rating of nephrotoxicity
|
Simulation |
Uncertainty/variability |
|||||
|
Average/ median |
Average/ 0.95 |
Median/ median |
Median/0.95 |
0.95/median |
0.95/0.95 |
|
|
Null limit, fumonisin B |
0.000013 |
0.000456 |
0.000000 |
0.000156 |
0.000036 |
0.001542 |
|
1 mg/kg limit, fumonisin B |
0.000012 |
0.264960 |
0.000000 |
0.000150 |
0.000034 |
0.001492 |
|
3 mg/kg limit, fumonisin B |
0.000011 |
0.000364 |
0.000000 |
0.000121 |
0.000028 |
0.001204 |
Pathologist’s rating of nephrotoxicity, from 0 to 3, with 1 representing the lowest observable effect. The background level of nephrotoxicity associated with each fumonisin concentration limit has been subtracted.
Table 30. Effects of limiting the consumption of maize on a pathologist’s rating of nephrotoxicity
|
Simulation |
Uncertainty/variability |
|||||
|
Average/ median |
Average/ 0.95 |
Median/ median |
Median/ 0.95 |
0.95/median |
0.95/0.95 |
|
|
Null limit, maize |
0.000013 |
0.000455 |
0.000000 |
0.000154 |
0.000036 |
0.001528 |
|
20 g/day limit, maize consumption |
0.000013 |
0.000428 |
0.000000 |
0.000144 |
0.000035 |
0.001446 |
|
100 g/day limit, maize consumption |
0.000007 |
0.000190 |
0.000000 |
0.000056 |
0.000017 |
0.000602 |
Pathologist’s rating of nephrotoxicity, form 0 to 3, with 1 representing the lowest observable effect. The background level of nephrotoxicity at each limit of maize intake has been subtracted. As all the predicted values are <1, no observable effect is anticipated. The percentiles do not reflect the same individual values for uncertainty or variability as in Table 28, as the values were sorted after subtraction of the background nephrotoxicity.
The assessment of dietary intake of fumonisins presented here provides a quantitative estimate of the risk at concentrations above the ADI or TDI. In particular, it provides a quantitative description of the level of risk reduction achieved by two alternative risk management options. It shows incremental risk with consumption of a toxin. Dietary intake of fumonisins is a useful case study for this type of assessment, since, like many environmental contaminants, whether of natural or anthropogenic origin, it is widespread. Efforts to avoid or reduce intake and the resulting risk become major considerations in assessing the public health significance of an environmental contaminant like fumonisin. Safety assessments serve to screen out contaminants that present little risk. They may not be adequate when the intake of a contaminant, such as fumonisin, already exceeds a safe level and when reasonable, achievable measures for reducing intake remain to be identified.
The model presented here suggests that renal toxicity would not be expected to be detected at current levels of intake. Table 28 shows that the predicted values for renal lesions are lower than those that could be observed (i.e. > 1). The results presented in Tables 29 and 30 show that reducing intake by reducing consumption of fumonisin-containing products would be more effective in reducing human risk of kidney damage than would lowering the concentration of fumonisin permitted in maize by a similar factor. Limiting maize consumption alters only the intake of fumonisin by persons with a high maize intake.
The risks and the uncertainties of the models associated with human intake of fumonisin have thus been described. For each input, a range of values could be derived, depending on which model is used. Use of multiple models yields the model uncertainty, which is measured by the range of values for a given input from a series of weighted models. Some uncertainties were not represented in the analysis, such as the uncertainty due to unknown relative proportions of field maize to sweet maize eaten and whether maize consumption would vary over a longer period than represented in the 3-day survey. Other uncertainties include whether components of maize other than fumonisin are also toxic and the appropriateness of extrapolating from animals to humans. The kidney, the most sensitive target organ in rats, a species known to have renal problems, may not be the most sensitive target for fumonisins in humans. Knowledge of the mechanisms of toxicity, perhaps through L-calcium channels (Smith et al., 1999, 2000) or disruption of sphingolipid metabolism (Riley et al., 1994, 1996, 1997), could be used quantitatively in species extrapolation and to describe variation. Either of these mechanisms, which are not exclusive, could allow a signalling event in a variety of responses. The data used here are, however, unique in that they provide dose–response relationships for individual animals, a feature necessary for modelling with Monte Carlo analysis. Data of this type for other end-points would make other analyses possible.
On the basis of this analysis, the current intake of fumonisin in the USA would not be expected to result in renal lesions, even for eaters at upper percentiles of consumption. Even if dietary intake doubled, no measurable toxicity would be expected in the population of the USA. Limiting maize consumption would be more effective, would disrupt the eating habits of fewer people, and would eliminate less of the maize crop than would limiting the concentration of fumonisins in maize.
(a) Liver
Data from the National Toxicology Program study of the carcinogenicity of fumonisin B1 in B6C3F1 mice were used to fit the Moolgavkar–Venzon–Knudson (MVK) two-stage, clonal expansion model of carcinogenesis (Moolgavkar & Venzon, 1979; Moolgavkar & Knudson, 1981; Kodell et al., 2001). The data included tissue weight, cell proliferation, cell death, and sphingolipid metabolism in the primary target organ. This model was used to predict 2-year liver tumour rates in female and male mice on the basis of the effects of different doses of fumonisin B1 on the growth of normal cells and on the proliferation of preneoplastic cells as a compensatory response to sphinganine-induced cell death. The model reproduced the observed tumour rates reasonably well. Increased rates above background in females were predicted only at the highest dose.
The MVK model allows for birth and death of preneoplastic cells. It includes the assumption that a normal cell can undergo an initial mutation to become a preneoplastic cell at a certain rate (µ1) and that this cell can in turn undergo a second mutation to generate a malignant cell at another specified rate (µ2).
Preneoplastic cells can divide to produce two daughter cells at a rate beta(t), or they can die at death rate delta(t). In this implementation of the MVK model, µ1 and µ2 were assumed to be constant with respect to time, while P(t) and delta(t) were assumed to be time-dependent. Data on liver weights were supplemented with data on body weights to estimate the net growth rate of the liver over time. Data on PCNA were used to estimate the cell birth rate, beta(t), in the liver over time, while the cell death rate, delta(t),was estimated as the difference between the cell birth rate and the net growth rate of the liver. The differential effect of fumonisin B1 on delta(t) and, consequently, on beta(t) is proportional to the concentration of sphinganine in the liver. The mutation rates, µ1, are assumed to be unaffected by fumonisin B1, consistent with evidence indicating that fumonisin B1 is not genotoxic (Cohen et al., 2000). The model decribes the probability of such an occurrence at any time (Zheng, 1997), according to the following formula. The programming code for implementation of the model was provided by Zheng (1998).
The liver weights of four animals at each interim sacrifice (3, 7, 9, and 24 weeks) and of all animals killed at 104 weeks were recorded. The body weight of each animal was recorded each week. As the average liver weights throughout the 104-week study were needed to implement the MVK model, liver weights for the weeks
with no observations were estimated from an allometric relationship with body weight, according to the equation:
LW(t) = a[BW(t)]b
where BW(t) and LW(t) represent, respectively, the body weight and liver weight of a female mouse at time t. This equation may be expressed as:
loge[LW(t)] = loge(a) + b x loge[BW(t)].
A similar fit was preformed for each dose group, and the liver weights for intermediate times were inferred from each dose-specific allometric equation, on the basis of observed weekly body weights. The curves for 0, 5, and 15 mg/kg of diet are similar, while those for 50 mg/kg of diet were about 25% higher and those for 80 mg/kg of diet were 2–2.5 times higher. For each dose group, a locally linear smoothing technique (Fan & Gijbels, 1996) was used to approximate the derivative of the loge-transformed liver weight:
d[logeLW(t)]/dt
over the range 0–104 weeks. This derivative was used as an estimate of the net growth rate of the liver over time. During the first 12 months, the growth rate at 5 mg/kg of diet was roughly 10% more than that of the controls, 20% higher at 15 mg/kg of diet, 50% higher at 50 mg/kg of diet, and 2.5–3 times higher at 80 mg/kg of diet. The liver growth rates were somewhat erratic after 12 months. The number of cells in the liver each week, N(t),was estimated from:
N(t) = LW(t)/CW
where CW represents the average weight of a mouse liver cell, which has been reported to be approximately 6.6 x 10-6 (Altman & Dittmer, 1962).
The percentages of PCNA-labelled cells in actively replicating cells were used to estimate the cell birth rates.
As the PCNA values for individual animals were somewhat erratic, the raw data were considered unreliable for regression modelling. Although the regression fit to the weekly data was quite satisfactory, the somewhat erratic, non-monotone nature of the data for dose groups made these results difficult to fit. It was decided that a simple cubic (monotone) curve was a reasonable compromise between the increasing PCNA responses at 5 and 15 mg/kg of diet and the inverted response at 50 mg/kg of diet. The birth rate, beta(t), for dose d and time t was estimated from:
beta(t) = (1/cell cycle) x [0.31154 + 3.35 x 10–7d3 – 0.05562 x loge(t)],
where the cell cycle was taken to be 33.6 h. The plot of beta(t) for the dose groups is shown in Figure 2. The values for 0, 5, and 15 mg/kg of diet were almost indistinguishable.
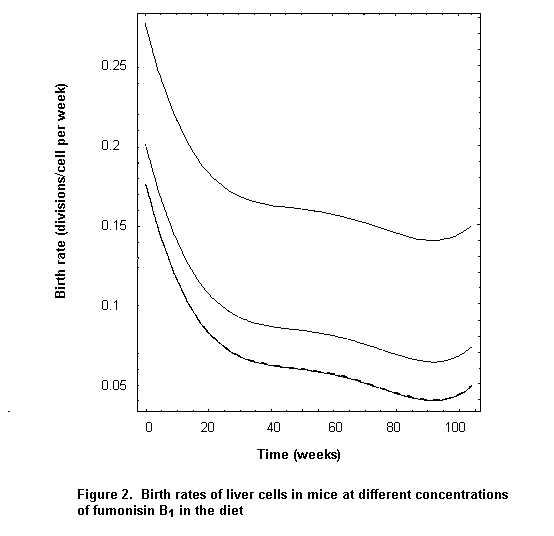
The study included data on apoptosis in the liver at the 3-, 7-, 9-, and 24-week interim kills and at terminal sacrifice. However, these data reflect only incidence and severity, not rates of cell death. Hence, an indirect approach had to be devised to estimate the cell death rate, delta(t). This was done by using the estimated net growth rate of the liver with the estimated cell birth rate. This approach proved to be a key factor in using the model. Specifically, the relationship
delta(t) = beta(t) – d[logeLW(t)]/dt
was used to estimate the cell death rate for each dose group. The shapes of the death-rate curves are similar to the birth-rate curves, but slightly less steep (Figure 3).
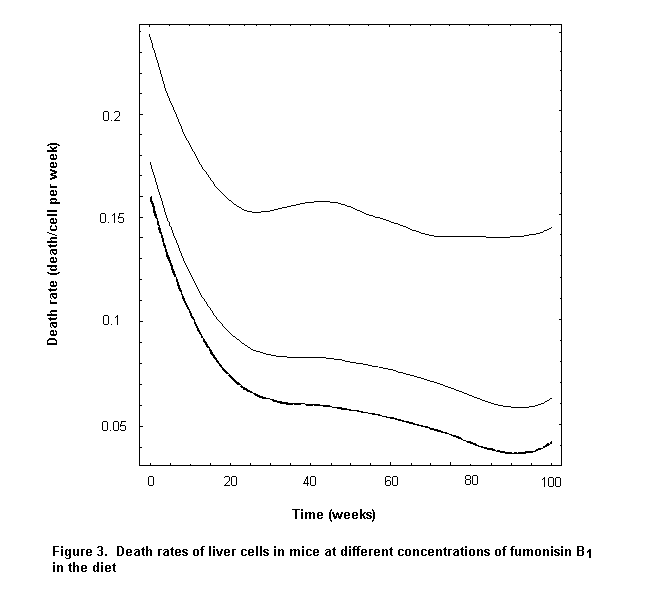
The average sphinganine concentrations in the livers of female mice were 2.3, 1.8, 3.1, 5.1, and 5.3 nmol/g at 0, 5, 15, 50, and 80 mg/kg of diet, respectively. These concentrations were used to model the effect of fumonisin B1 on the cell death rate according to the relationship
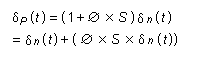
where the subscripts P and N denote, respectively, preneoplastic and normal cells, S denotes the average concentration of sphinganine (mmol/g) in the liver, and Ø is a constant. Thus, fumonisin B1, was assumed to increase the rate of apoptosis of preneoplastic liver cells over that of normal cells by an amount Ø.S.deltaN(t). The compensatory proliferation of preneoplastic cells was modelled according to the relationship:
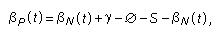
which indicates that the birth rate of preneoplastic cells is increased above that of normal cells by a constant gamma times the increase in the cell death rate. The constants Ø and gamma were assigned values of 0.2 and 1.38, respectively, to obtain representative predictions of the tumour incidences at 104 weeks.
Although not a key assumption for model fitting, it was considered plausible that the second mutation rate, µ2, might be higher than the first mutation rate, µ1. Hence, the relationship
µ1 = k µ2
was adopted, where k is a constant (0 < k < l). A reasonably good fit to the data on tumours at 104 weeks was obtained with µ1 = 1.0 x 10–7 and k = 0.715.
Putting all the model components together, i.e. cell growth rate, cell number, cell birth and death rates, liver sphinganine concentration, and rates of mutation, gave the predicted tumour probabilities shown in Figure 4. The plots reflect the fact that the MVK model is used to predict the tumour incidence over the entire interval between 0 and 104 weeks, even though the tumour incidences at 104 weeks were the only ones used to infer representative mutation rates.
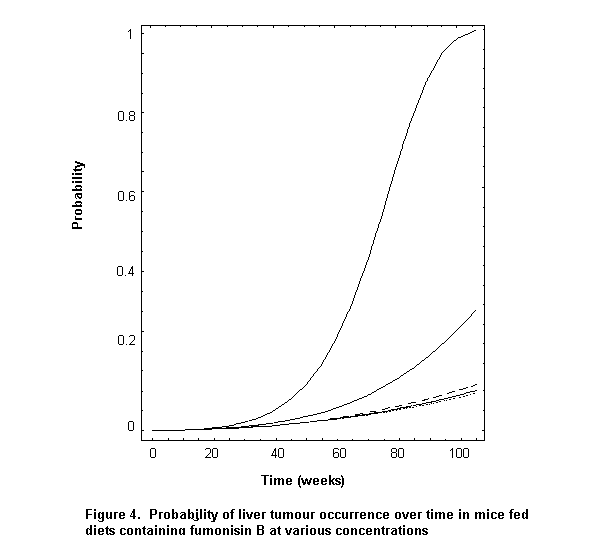
The predicted incidences were 0.091, 0.084, 0.105, 0.284, and 0.992 at 0, 5, 15, 50, and 80 mg/kg of diet, respectively whereas the observed incidences (corrected for survival) were 0.117, 0.065, 0.021, 0.427, and 0.883 at 104 weeks. The predicted incidence at 5 mg/kg of diet is slightly lower than that at 0 mg/kg of diet, in keeping with the observed data, but the pronounced decrease at 15 mg/kg of diet in the observed data is not reproduced. Although there is both over- and under-prediction of dose-specific incidences, the predictions are reasonably representative of the observed values. It should be remembered that predictions were made for each dose group separately, i.e. there was no mathematical dose–response relationship. Clearly, the model predicts little or no risk for liver tumour development at low doses of fumonisin B1 but a marked increase in risk at the higher doses.
In order to validate the predictions of tumour risk in females, the model was implemented a second time with data on the mechanism of action in males. Thus, liver and body weights, cell birth rates (PCNA), and sphinganine concentrations in the liver were used in the same way for males as was done for females. The predictions for males were 0.199, 0.201, 0.198, 0.233, and 0.237 at 0, 5, 15, 80, and 150 mg/kg of diet, respectively, whereas the observed values were 0.268, 0.211, 0.190, 0.213, and 0.213. The predictions are slightly higher at 80 and 150 mg/kg of diet than at 0, 5, and 15 mg/kg of diet, but they are all within a 4% range of the observed values and are consistent with a lack of a dose–response relationship. The value at 15 mg/kg of diet was actually slightly below that of controls. The prediction of little or no risk for males at low doses of fumonisin B1 is the same as the prediction for females. The hypothesis of cell death and compensatory proliferation on which the model for females was based appears to be validated by the excellent agreement of the fitted and observed values for males.
Although the range of measured concentrations of sphinganine in liver was not markedly different in males and females, the cell birth and death rates were estimated to be much higher in females than in males on the basis of the observed PCNA and liver-weight data. Hence, elevated sphinganine levels at higher doses had a much greater effect on the tumour incidence in females than in males because of the magnitude of the difference in estimated cell birth and death rates that sphinganine was assumed to affect. Thus, the presence of a dose–response relationship for tumours in females and the absence of such a relationship in males appears to be due primarily to a difference in estimated cell turnover in the liver.
How fumonisin B1 increases apoptosis by interfering with sphingolipid metabolism is not well understood (Riley et al., 1996; Merrill et al., 1997; Tsunoda et al., 1998). The MVK model assumes that sphinganine concentrations in target organs provide the best biomarker for this mechanism (Delongchamp & Young, 2001), although other assumptions are equally plausible. The model is an oversimplification of the biological mechanism by which fumonisin B1 causes liver tumours in mice. In fact, the assumption of a direct effect of sphinganine on the death rates only of preneo-plastic cells is an inaccurate reflection of the apoptosis hypothesis. Fumonisin B1 is believed to affect the death rates of normal cells also because of sphinganine build-up. The MVK model does not account for this directly. A more complex model could be considered, but increased complexity would be accompanied by increasing difficulty in resolving the model with the data. Direct measurements of the rates of apoptosis in both normal and preneoplastic cells of mice exposed to fumonisin B1 would help resolve these issues. The model used here is a reasonable representation of the tumour incidence in mice of each sex. It is based on insertion of mechanistic data into a mathematical model of cancer development, which embodies a plausible hypothesis for how fumonisin B1 increases the incidence of liver tumours.
(b) Kidney cancer
Dose–response models for tumour incidence can provide estimates of risk at the PMTDI which is based on effects in the kidney. These models can also provide a PMTDI for cancer for comparison with the PMTDI for other effects. A dose–response curve represents the cumulative probability of a tumour occurring on the basis of an underlying distribution of interindividual differences in susceptibility to a chemical.
For example, many biological effects are approximately described by a log-normal distribution (Mantel & Bryan, 1961). That is, the logarithm of doses that produce a biological effect is approximately normally distributed. The log-normal distribution is described by the mean (median) and standard deviation. Hence, estimates of the mean and standard deviation in bioassays provide the data necessary for estimating by how much the dose must be reduced below the NOEL to achieve a low level of cancer risk. Typically, a default safety factor of 10 is used to account for interindividual differences in susceptibility. Alternatively, the distribution of doses that cause tumours (or, equivalently, the resulting log-probit dose–response curve) can provide a safety factor for interindividual variation to replace the default factor of 10.
A log-normal distribution was estimated for renal tumours (adenomas and carcinomas) in male Fischer 344 rats observed in the study conducted for the National Toxicology Program (1999) in the USA. Two other commonly used models, the log-logistic and Weibull, were also fit to these data to indicate the effect of the choice of dose–response model (or equivalent underlying distribution of interindividual variation). All three dose–response models have shallow slopes at low doses, i.e. little increase in tumour incidence as the dose increases. At higher doses, these dose–response models curve upwards sharply as detoxication processes become saturated. Further, the log-logistic model is based on how a chemical affects the toxicokinetics of the formation and reduction of a chemically active agent, and the Weibull distribution is based on the assumption that multiple hits at a target site (cell) are required to affect the carcinogenic process.
As it is unlikely that a low tumour incidence would be observed in a sample of 48 animals, the zero incidence at the two lowest doses was replaced by a conservative Bayesian estimated incidence of 1/2n, where n is the number of animals. Hence, an incidence of 1/(2 x 48) = 0.01 was assigned to the group receiving 0.67 mg/kg bw per day. Since no renal tumours were induced at that dose, it was assumed that the animals would not have had tumours at 0.22 mg/kg bw per day. The estimated incidence for the 48 animals without tumours at this dose was 1/(2 x 96) = 0.005.
The estimated dose–response relationship for the log-probit model is:
P = phi (ln median = 2.114, standard deviation of ln d = 1.26)
where P is the estimated probability of tumour occurrence, d is dose expressed as mg/kg bw per day, and phi denotes the cumulative normal distribution.
The estimated dose–response curves for the other models are:
Log-logistic
P = 1/[1+ e–(-3.258+1.686 ln d)]
Weibull
P = 1– e-(0.0283 d (E) 1.62)
where d(E) 1.62 is d raised to the 1.62 power.
The slope of the dose–response curve accounts for intraspecies variation in the sensitivity of rats to fumonisin. In addition, allowing for a safety factor of 10 for animal to human extrapolation, the cancer risks of individuals exposed for a lifetime at the PMTDI/10, PMTDI/2, PMTDI, and 2 x PMTDI, where the PMTDI = 0.002 mg/kg bw per day, for each of the models are presented in Table 31. Since the Bayesian procedure increased the tumour incidence at the two lower doses, the cancer risks at low doses are likely to be overestimated.
Table 31. Conservative (probably over-) estimates of the cancer risk associated with lifetime exposure to fumonisin B1 relative to the PMTDI of 0.002 mg/kg bw
|
Model |
PMTDI/10 |
PMTDI/2 |
PMTDI |
2 X PMTDI |
|
Log-probit |
0 |
4.8 x 10–8 |
0.9 x 10–6 |
1.2 x 10–5 |
|
Log-logistic |
1.1 x 10–6 |
1.6 x 10–5 |
5.3 x 10–5 |
1.7 x 10–4 |
|
Weibull |
1.2 x 10–6 |
1.6 x 10–5 |
5.0 x 10–5 |
1.5 x 10–4 |
Absorption, distribution, metabolism and excretion
In all animal species studied, fumonisins are poorly absorbed from the digestive tract and are rapidly distributed and eliminated. The liver and kidney retain most of the absorbed material, and fumonisin B1 persists longer in rat liver and kidney than in plasma. In pregnant rats and rabbits, very low concentrations of fumonisin B1 were recovered in the uterus and placenta. No fumonisin B1 was found in fetuses, indicating the absence of placental transfer. There was little evidence of significant transfer during lactation, and fumonisins do not appear to be metabolized in vitro or in vivo. Although fumonisins are not metabolized by cytochrome P450 enzymes, fumonisin B1 can alter the activity of these enzymes through mechanisms that alter sphingolipid biosynthesis. Fumonisins are structurally related to sphingoid bases. Removal of the tricarballylic acid side-chains, presumably by the microbial flora of the gut, converts fumonisin B1 into a substrate for ceramide synthase. The product of the enzyme reaction, like fumonisin B1, is an inhibitor of the enzyme in vitro.
Toxicological studies
In all animal species studied, the liver was a target for fumonisin B1; the kidney was also a target in many species. In kidney, the early effects are often increases in free sphingoid bases, renal tubule-cell apoptosis, and cell regeneration. In the liver, apoptotic and oncotic necrosis, oval-cell proliferation, bile-duct hyperplasia, and regeneration are early signs of toxicity. In studies in rats and trout fed known cancer initiators and with various initiation and promotion protocols, purified fumonisin B1 enhanced liver cancer development. Brief administration of high doses or longer administration of lower doses that cause significant hepatotoxicity resulted in the appearance of foci positive for glutathione-S-transferase (placental form), hepatocellular nodules, and other precursors of liver tumour development. In rodents, the toxicity of fumonisin B1 was strain- and sex-dependent. For example, male BDIX rats appeared to be more sensitive to the hepatotoxic effects of fumonisin B1 than male Fischer 344N, male Sprague-Dawley, and male RIVM:WU rats, in which nephrotoxicity was observed at lower doses than hepatotoxicity. In mice, the liver is more sensitive than the kidney to the toxicity of fumonisin B1. Female mice were more sensitive than males. In long-term feeding studies, purified fumonisin B1 caused both liver and kidney tumours in rodents. The kidney carcinomas induced in male Fischer 344N rats by fumonisin B1 were a highly malignant variant of renal tubule tumour, but the significance of their aggressive nature was unclear. The NOEL for renal cancer in Fischer 344N rats was 0.67 mg/kg bw per day (Table 32), and the NOEL for renal toxicity was 0.2 mg/kg bw per day (Table 33). The NOEL for liver cancer in male BD IX rats was 0.8 mg/kg bw per day, and the NOEL in feed-restricted female B6C3F1 mice was 1.9 mg/kg bw per day.
Table 32. Dose–response relationship for renal toxicity and tumours in male Fischer 344N rats fed diets containing purified fumonisin B1 for 2 years
|
Dose of fumonisin B1 |
No. of animals showing signs of renal toxicity and tumours |
||
|
Cytotoxic or regenerative lesions |
Atypical tubulehyperplasia |
Renal tumours |
|
|
Untreated controls |
0/42 |
0/48 |
0/48 |
|
0.22 |
0/40 |
0/40 |
0/40 |
|
0.67 |
23/33 |
0/48 |
0/48 |
|
2.2 |
42/42 |
4/48 |
10/48 |
|
6.6 |
43/43 |
9/48 |
16/48 |
Table 33. Dose–response relationship for renal toxicity in male Fischer 344N rats fed diets containing purified fumonisin B1 for 90 days
|
Dose of fumonisin B1 |
No. of animals showing signs of renal toxicity |
|
Untreated controls |
0/10 |
|
0.1 |
0/10 |
|
0.2 |
0/10 |
|
0.6 |
9/10 |
|
1.9 |
10/10 |
|
5.7 |
10/10 |
Studies in rodents, non-human primates, and other animal species given F. verticillioides culture material from an isolate that produces predominantly fumonisin B1 (isolate MRC 826) or maize naturally contaminated with fumonisins showed toxic effects in the liver and kidney that were similar to those in studies with purified fumonisin B1. Both MRC 826 and naturally contaminated maize caused liver tumours in rats at doses similar to those that caused liver tumours in rodents fed purified fumonisin B1. The NOEL for the renal and hepatic toxicity of all fumonisins in vervet monkeys fed a diet containing MRC 826 culture material was 0.11 mg/kg bw per day. Purified fumonisin B1, F. verticillioides culture material, and naturally contaminated maize all induced not only hepatic toxicity but also leukoencephalo-malacia in equids and pulmonary oedema and hydrothorax in pigs. Both diseases appeared to occur secondarily to cardiovascular dysfunction. Cardiovascular effects have also been seen in other species. Field outbreaks of equine leukoencephalo-malacia and porcine pulmonary oedema associated with consumption of fumonisin-contaminated maize have been reported in the USA and elsewhere. The NOEL for fumonisin B1 in equine leukoencephalomalacia was equivalent to 0.3 mg/kg bw per day for animals fed diets containing Fusarium culture material. In pigs fed Fusarium culture material, evidence of pulmonary oedema was detected at a concentration of fumonisin B1 equivalent to 0.4 mg/kg bw per day. For pigs fed naturally contaminated maize, the concentration of fumonisin B1 required to induce pulmonary oedema was much higher, although the NOEL for liver toxicity was similar (equivalent to 0.2 mg/kg bw per day).
Several biochemical modes of action have been postulated to explain the induction by fumonisins of disease in animals. Two hypotheses involve disruption of lipid metabolism as the initial step. The first proposed mechanism involves disruption of sphingolipid metabolism through inhibition of ceramide synthase. The demonstrated consequences of inhibition of this enzyme in liver and kidney are changes in all the major pools of sphingolipids, including increased concentrations of free sphingoid bases and free sphingoid-base metabolites and decreased biosynthesis of ceramide and other sphingolipids containing ceramide. Glycerophos-pholipid metabolism is also affected. Clear evidence of fumonisin-induced disruption of sphingolipid metabolism has been obtained in all target tissues except brain and in all species tested. The second proposed mechanism involves disruption of fatty acid and glycerophospholipid metabolism. Fumonisin-induced changes in fatty acid profiles and prostaglandins have been demonstrated in vivo in rat liver. These two proposed lipid-based mechanisms of action are similar in many respects with regard to their ultimate effects on cell physiology and are consistent with data obtained in vitro, in short-term studies of toxicity, and in long-term studies of carcinogenicity in rodents. Fumonisins also affect sites of cellular regulation that are apparently independent of the disruption of lipid metabolism, but cancer and the other toxic effects observed in animals appear to depend on disruption of various aspects of lipid metabolism, membrane structure, and signal transduction pathways mediated by lipid second messengers. The demonstrated cellular effects include altered cell proliferation, altered rates of apoptosis, altered intracellular communication and cell adhesion, induction of oxidative stress, and modulation of gene expression. Since the proposed biochemical mechanisms of action involve alterations in de novo biosynthetic pathways, nutritional factors could play an important role in determining the potency of fumonisin B1 and the observed toxicological effects in rodents.
The available in-vivo observations are consistent with a proposed mode of action for fumonisin B1-induced toxicity that is dependent on perturbed lipid metabolism. The resulting increase in cell death coupled with regenerative cell proliferation, possibly by generation of oxidative damage, could in turn lead to increased incidences of tumours in target tissues. The primary evidence for sustained cell loss and regeneration is the observations of such effects in rat kidneys.
In a small number of studies in vitro and a single study in vivo, neither fumonisin B1 nor any other fumonisin was shown unequivocally to be genotoxic. Similarly, no adducts of fumonisin with DNA have been found.
While there was evidence that fumonisins are embryotoxic in vitro, no published data exist to support the conclusion that fumonisins cause developmental or reproductive toxicity in farm animals. Except in one study in hamsters, embryotoxicity occurred in laboratory animals (rats, mice, and rabbits) secondarily to maternal toxicity.
Consumption of mouldy sorghum or maize containing fumonisin B1 at up to 64 mg/kg was associated with an outbreak of human disease in India involving gastrointestinal symptoms. The grain was also reported to be contaminated with other toxigenic fungi.
The available evidence for an association between the intake of fumonisins and human cancer was limited to a few correlation studies. Typically, these involved a few regions in which populations were broadly classified with regard to their risk for oesophageal or liver cancer. The regions were then compared with respect to the proportions of contaminated samples and the level of contamination. In some studies, the measures of intake of fumonisins were indirect, and the incidence of disease was related to consumption of certain foods, notably maize. Taken together, the results of these studies could be interpreted as indicating an association between fungal contamination of foodstuffs and oesophageal cancer or liver cancer. However, bias, chance, or confounding could not be excluded, and hence there was only limited evidence of an independent carcinogenic effect of fumonisins.
A specific role for fumonisins in the development of neural tube defects has been proposed. The hypothesis includes a critical role of fumonisins in disruption of folate membrane transport, but no specific studies have been designed or performed to confirm this mechanism.
Analytical methods
Two validated analytical methods based on liquid chromatography have been developed for fumonisins. The first method, based on strong anion exchange clean-up of a solvent extract, was validated for fumonisins B1, B2, and B3 in maize. Although this method has been used to determine fumonisins in maize-based foods, recovery from certain food matrices can be problematic. A second method with improved extraction efficiency and immunoaffinity column clean-up has been validated for fumonisins B1 and B2 in maize and cornflakes. Although methods for unequivocal detection based on liquid chromatography with mass spectrometric detection are also available, their high cost prohibits their routine use. Screening tests based on thin-layer chromatography and, for the combined B-series fumonisins, enzyme-linked immunosorbent assays, have also been developed. No methods specific for fumonisin B4 have been described, and little is known about its occurrence, although the limited evidence suggests that it occurs at lower concentrations than fumonisin B1, B2, or B3, which were the subject of the present evaluation. The absence of a method to determine the concentration of fumonisin calibrant solutions remains a problem. In the laboratories that provided data on the natural occurrence of fumonisins in maize and maize-based foods for the current assessment, liquid chromatography was used predominantly, with solvent extraction, solid phase extraction clean-up, and quantification by pre-column formation of fluorescent ortho-phthaldialdehyde derivatives. The limits of detection were generally equal to or below 50 µg/kg and the analytical recovery greater than 70%.
Sampling protocols
The sampling variance in the testing of shelled maize for fumonisins was studied after collection of a large bulk sample and riffle-division into 1.1-kg test samples. At a batch contamination concentration of 2 mg/kg, the coefficients of variation were 17% associated with sampling (1.1-kg sample), 9.1% with sample preparation (Romer® mill and 25-g analytical portion), and 9.7% with analysis; they were independent of the fumonisin(s) tested (fumonisin B1, B2, B3, or all fumonisins). The coefficient of variation associated with the whole test procedure (sampling, sample preparation, and analysis) was 21%, which was of the same order of magnitude as that for the measurement of aflatoxin in shelled maize with a similar test procedure.
Effects of processing
The effects of various food processing procedures on the levels of fumonisin contamination have been studied. For example, maize screenings contain higher concentrations of fumonisins than whole grain. Separation and removal of screenings is a useful method for reducing the amount of fumonisins entering storage. Steeping maize in aqueous solutions during wet milling results in extraction of fumonisins and is thus effective in reducing the concentration in maize products. Fumonisins are fairly heat-stable, and the toxin content is significantly reduced only during processes in which the temperature exceeds 150 °C. Dry milling of maize results in distribution of fumonisin into the various maize constituents. In wet milling, some fumonisin is extracted into the steeping water. There is little degradation of fumonisins during fermentation. Alkaline cooking and heating (nixtamalization), which result in the production of hydrolysis products, does not completely detoxify fumonisin-contaminated maize. In each process, many parameters affect the fate of the fumonisins. In addition, toxic compounds resulting from the conversion of fumonisins may appear during processing.
Food consumption and dietary intake assessment
Distributions of estimates of the intake of fumonisin B1 around the world were based on the GEMS/Food regional diets and a published distribution of the concentrations of fumonisin B1 in maize. Data that supported use of the published distribution were submitted by Argentina, Brazil, Canada, China, Denmark, Sweden, the United Kingdom, Uruguay, and the USA. The international estimates were made on the assumption that all maize consumed contains fumonisin B1 at the concentration found in the unprocessed maize samples that were used to construct the distribution curve. While the mean concentration of fumonisin B1 used was 1.4 mg/kg of unprocessed maize (median, 0.42 mg/kg), surveys of fumonisin B1 over several years have shown that the median or mean concentration in maize varies greatly. The mean in sound maize in international trade in any given year could be expected to be between 0.2 and 2.5 mg/kg. Use of these concentrations of fumonisin B1 with the maize intakes in the GEMS/Food regional diets would alter the mean of the expected distribution of intake of fumonisin B1 by one-seventh to twice that reported here.
The estimated mean intake of fumonisin B1 ranged from 0.2 µg/kg bw per day in the European-type diet to 2.4 µg/kg bw per day in the African diet (Table 34).
Table 34. Estimated intake of fumonisin B1 based on GEMS/Food regional diets
|
|
Intake (µg/kg bw per day) |
||||
|
Middle Eastern |
Far Eastern |
African |
Latin American |
European |
|
|
Mean |
1.1 |
0.7 |
2.4 |
1.0 |
0.2 |
|
90th percentile |
3.3 |
2.1 |
7.3 |
2.9 |
0.6 |
The Committee also considered published national estimates of fumonisin B1 intake from Argentina, Canada, the Netherlands, Switzerland, and the USA. Additionally, fumonisin B1 intake was estimated on the basis of consumption of food containing maize and associated fumonisin B1 concentrations submitted by the United Kingdom. The estimates of national intake of fumonisin B1 were lower than the international estimates presented here because they took into account the effects of processing, and because the national estimates were prepared from more specific data, i.e. the intakes of foods as consumed were considered rather than raw agricultural commodities. The mean estimates of national intake of fumonisin B1 ranged from 0.02 µg/kg bw per day to 1.0 µg/kg bw per day (Table 35). These estimates also included certain assumptions that ensure conservatism, such as the assumption that all persons consume food containing fumonisins B1 at the default concentration. Finally, the Committee noted that subsistence farmers, who grow and eat their own maize, might consume larger amounts of fumonisin B1 thn those reported here.
Table 35. National estimates of intake of fumonisin B1
|
Country |
Intake (µg/kg bw per day) |
|
|
Mean or median |
90th percentile |
|
|
Argentina |
0.2 |
NR |
|
Canada |
0.02 |
0.08 |
|
Netherlands |
0.06, 1.0a |
NR |
|
Switzerland |
0.03 |
NR |
|
United Kingdom |
0.03 |
0.1 |
|
USA |
0.08 |
NR |
NR, not reported or calculated
a
The first value is for the whole population, the second for regular maize eaters.When they were quantified in the same sample, the ratio of fumonisin B1:B2:B3 was approximately 10:3:1. To approximate the intake of all three fumonisins, therefore, the intake figures for fumonisin B1 in this evaluation should be increased by 40%.
Prevention and control
The strategies for pre-harvest reduction of fumonisin contamination include agricultural practices, plant breeding, and genetic engineering. However, any breeding programme should take into account the growth conditions in specific regions to ensure full adaptation of the variety(ies) developed. The main means for preventing fumonisin contamination after harvest is immediate drying of the grain. Treatments with chemical preservatives before storage or by physical means, such as temperature reduction or modified atmospheres, during storage can also prevent fungal growth and subsequent mycotoxin formation.
Nephrotoxicity, which was observed in several strains of rat, was the most sensitive toxic effect of pure fumonisin B1. Since the available studies clearly indicated that long-term renal toxicity is a prerequisite for renal carcinogenesis, the potential for the latter is subsumed by the dose–response relationship for renal toxicity. Therefore, the pivotal studies that could serve as the basis for a tolerable intake of fumonisin B1 were the short-term and long-term studies of toxicity in rodents (see Tables 32 and 33). On the basis of these studies, the overall NOEL for renal toxicity was 0.2 mg/kg bw per day.
The Committee allocated a group provisional maximum tolerable daily intake (PMTDI) for fumonisins B1, B2, and B3, alone or in combination, of 2 µg/kg bw per day on the basis of the NOEL of 0.2 mg/kg bw per day and a safety factor of 100. All of the estimates of intake of fumonisin B1 based on the available data on national consumption were well below the group PMTDI. This remains true even when intake estimates for fumonisin B1 are increased by 40% to account for the presence of fumonisins B2 and B3.
The Committee was aware of an unpublished risk assessment in which the data on renal tumours had been used, and noted that the estimated risk was negligible at intakes below the group PMTDI established at the present meeting.
Recommendations
The Committee acknowledged the need for research in areas recommended in WHO Environmental Health Criteria (WHO, 1999). The Committee identified the following additional recommendations:
|
• |
As renal toxicity is the critical effect required for fumonisin-induced renal carcinogenesis in male rats, the tumour incidence in the male rat kidney should be modelled with biologically-based procedures. |
|
• |
The biochemical and physiological mechanism(s) underlying the highly aggressive behaviour of fumonisin-induced renal tubular carcinomas in Fischer 344N rats should be investigated, including studies on the effects of fumonisin B1 on the expression of cell adhesion molecules. |
|
• |
The biochemical and physiological mechanism for the apparently different sensitivities of Fischer 344N and BDIX rats to fumonisin-induced liver toxicity should be investigated. |
|
• |
Investigations should be conducted to determine whether dietary factors such as folate, vitamin E, and choline modify renal or hepatic toxicity induced by fumonisin B1 in laboratory animals. |
|
• |
The ability of fumonisin B1 to alter the transport of folate at the cellular level and placental transport to the fetus in vivo should be investigated. |
|
• |
Investigations should be conducted to determine the role of inhibition by fumonisins of ceramide biosynthesis in protection of cells from ceramide-mediated apoptosis induced by mitochondrial dysfunction. |
|
• |
The relationship between the intake of fumonisin and human disease in areas where nixtamalized maize-products comprise a large portion of the diet should be investigated. Particular emphasis should be placed on diseases of the liver and kidney and other diseases suspected of being associated with the intake of fumonisin B1, such as nasopharyngeal and oesophageal cancers and neural tube defects. |
|
• |
The ability of fumonisins to modify the expression of receptors for microbial pathogens and toxins that are associated with renal and hepatic disease in humans should be investigated. |
Abado-Becongnee, K., Mobio, T.A., Ennamany, R., Fleurat-Lessard, F., Shier, W.T., Badria, F. & Creppy, E.E. (1998) Cytotoxicity of fumonisin B1: Implication of lipid peroxidation and inhibition of protein and DNA synthesis. Arch Toxicol., 72, 233–236.
Abbas, H.K., Vesonder, R.F., Boyette, C.D., Hoagland, R.E. & Krick, T. (1992) Production of fumonisins by Fusarium moniliforme cultures isolated from jimsonweed in Mississippi. J. Phytopathol., 136, 199–203.
Abbas, H.K., Cartwright, R.D, Shier, W.T., Abouzied, M.M., Bird, C.B., Rice, L.G., Ross, P.F., Sciumbato, G.L. & Meredith, F.I. (1998) Natural occurrence of fumonisins in rice with Fusarium sheath rot disease. Plant Dis. 82, 22–25.
Abel, S. & Gelderblom, W.C.A. (1998) Oxidative primary rat hepatocytes and rat liver in vivo. Toxicology, 131, 121–131.
Abel, S., Smuts, C.M. & Gelderblom, W.C.A. (2001) Changes in essential fatty acid patterns associated with normal liver regeneration and the progression of hepatocyte nodules in rat hepatocarcinogenesis. Carcinogenesis (in press).
Alexander, R.J. (1987) Corn dry milling: Processes, products, and applications. In: Watson, S.A. & Ramsted, P.E., eds, Corn: Chemistry and Technology, St Paul, MN: American Association of Cereal Chemists, pp. 351–376.
Alexandrow, M.G. & Moses, H.L. (1995) Transforming growth factor and cell cycle regulation. Cancer Res., 55, 1452–1457.
Ali, N., Sardjono, Yamashita, A. & Yoshizawa, T. (1998) Natural co-occurrence of aflatoxins and Fusarium mycotoxins (fumonisins, deoxynivalenol, nivalenol and zearalenone) in corn from Indonesia. Food Addit. Contam., 15, 377–384.
Altman, P.L. & Dittmer, D.S. (1962) Growth Including Reproduction and Morphological Development. Unpublished document, prepared under the auspices of the Committee on Biological Handbooks, Federation of American Societies for Experimental Biology, Washington DC.
AOAC International (2000) Minutes of meeting official methods board meetings, Phoenix, AZ; Los Angeles, CA; Philadelphia, PA, USA. Arlington, VA, USA.
Arora, A., Jones, B.J., Patel, T.C. Bronk, S.F. & Gores, G.J. (1997) Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology, 25, 958–963.
Atroshi, F., Rizzo, A., Biese, I., Veojalainen, P., Saloniemi, H., Sankari, S. & Andersson, K. (1999) Fumonisin B1-induced DNA damage in rat liver and spleen: Effects of pretreatment with co-enzyme Q10, L-carnitine, alpha-tocopherol and selenium. Pharmacol. Res., 40, 398–407.
Balsinde, J., Balboa, M.A. & Dennis, E.A, (1997) Inflammatory activation of arachidonic acid signaling in murine P388D1 macrophages via sphingomyelin synthesis. J. Biol. Chem., 272, 20373–20377.
Bassett, M.T., Chokunonga, E., Mauchaza, B., Levy, L., Ferlay, J. & Parkin, D.M. (1995) Cancer in the African population of Harare, Zimbabwe, 1990–1992. Int. J. Cancer, 63, 29–36.
Becker, B.A., Pace, L., Rottinghaus, G.E., Shelby, R., Misfeldt, M. & Ross, P.F. (1995) Effects of feeding fumonisin B1 in lactating sows and their suckling pigs. Am. J. Vet. Res., 56, 1253–1258.
Bennett, G.A. & Richard, J.L. (1994) Liquid chromatographic method for analysis of the naphthalene dicarboxaldehyde derivative of fumonisins. J. AOAC Int., 77, 501–506.
Bennett, G.A. & Richard, J.L. (1996) Influence of processing on Fusarium mycotoxins in contaminated grains. Food Technol., May, 235–238.
Bennett, G.A., Richard, J. L. & Eckhoff, S. R. (1996) Distribution of fumonisins in food and feed products prepared from contaminated corn. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 317–322.
Bever, R.J., Couch, L.H., Suterland, J.B., Williams, A.J., Beger, R.D., Churchwell, J.B., Doerge, D.R. & Howard, P.C. (2001) DNA adduct formation by Fusarium culture extracts: Lack of role of fusarin C. Chem–Biol Interactions (in press).
Bezuidenhout, S.C., Gelderblom, W.C.A., Gorst-Allman, C.P., Horak, R.M., Marasas, W.F.O., Spiteller, G. & Vleggaar, R. (1988) Structure elucidation of the fumonisins, mycotoxins from Fusarium moniliforme. J. Chem. Soc. Chem. Commun., 1988, 743–745.
Bhandari, N., Enongene, E.N., Riley, R.T., Meredith, F.I. & Sharma, R.P. (2001) Temporal expression of fumonisin B1-induced tumor necrosis factor- and interferon- in mice. Arch Toxicol. (in press)
Bhat, R.V., Prathaphumar, H.S., Rao, P.A. & Rao, V.S. (1997) A foodborne disease outbreak due to the consumption of moldy sorghum and maize containing fumonisin mtcotoxins. Clin. Toxicol., 35, 249–255.
Blot, W.J. (1994) Esophageal cancer trends and risk factors. Semin. Oncol., 21, 403–410.
Bokoch, G.M., Reilly, A.M., Daniels, R.H., King, C.C., Olivera, A., Spiegel, S. & Knaus, U.G. (1998) A GPTase-independent mechanism of p21-activated kinase activation regulation by sphingosine and other biologically active lipids. J. Biol. Chem., 273, 8137–8144.
Bondy, G., Barker, M., Mueller, R., Fernie, S., Miller, J.D., Armstrong C., Hierlihy, S.L., Rowsell, P. & Suzuki, C. (1996) Fumonisin B1 toxicity in male Sprague-Dawley rats. Adv. Exp. Med. Biol., 392, 251–264.
Bondy, G.S., Suzuki, C.A.M., Fernie, S.M., Armstrong, C.L., Hierlihy, S.L., Savard, M.E. & Barker, M.G. (1997) Toxicity of fumonisin B1 to B6C3F1 mice: A 14-day gavage study. Food Chem. Toxicol., 35, 981–989.
Bondy, G., Suzuki, C.A., Mueller, R., Fernie, S M., Armstrong C.L., Hierlihy, S.L., Savard, M.E. & Barker, M.G. (1998) Gavage administration of the fungal toxin fumonisin B1 to female Sprague-Dawley rats. J. Toxicol. Environ. Health, 53, 135–151.
Bordson, G., Meerdink, G., Bauer, K. & Tumbleson, M. (1993) Fumonisin recovery from samples dried at various temperatures. In: Midwest Section meeting of AOAC International, Des Moines, Iowa.
Bothast, R.J., Bennett, G.A., Vancauwenberge, J.E. & Richard, J.L. (1992) Fate of fumonisin B1 in naturally contaminated corn during ethanol fermentation. Appl. Environ. Microbiol., 58, 233–236.
Boyle, C.D. & Kishi, Y. (1995a) Absolute configuration at the tricarballylic acid moieties of fumonisin B2. Tetrahedron Lett., 36, 4579–4582.
Boyle, C.D. & Kishi, Y. (1995b) Absolute configuration at the tricarballylic acid moieties of fumonisin B1 and AAL toxin TA. Tetrahedron Lett., 36, 5695–5698.
Brown, L.M., Blot, W.J., Schumann, S.H., Smith, V.M., Ershow, A.G., Marks, R.D. & Fraumeni, J.F. (1988) Environmental factors and high risk of esophageal cancer among men in coastal South Carolina. J. Natl Cancer Inst., 80,1120–1125.
Bryden, W.L., Ravindran, G., Amba, M.T., Gill, R.J., Benyon, F.H.L., Tri, N.D. & Burgess, L.W. (1998) Mycotoxin contamination of maize grown in Australia, the Philippines and Vietnam. In: Miraglia, M., Van Egmond, H., Brera, C. & Gilbert, J., eds, Mycotoxins and Phycotoxins—Developments in Chemistry, Toxicology and Food Safety, Fort Collins, Colorado: Alaken, pp. 115–119.
Bucci, T.J., Hansen, D.K. & LaBorde, J.B. (1996) Leukoencephalomalacia and hemorrhage in the brain of rabbits gavaged with mycotoxin fumonisin B1. Nat. Toxins, 4, 51–52.
Bujari, J. & Ershad, D. (1993) An investigation on corn-seed mycoflora. Iran J. Plant Pathol., 29,13–17.
Bullerman, L.B. (1996) Occurrence of Fusarium and fumonisins on food grain and in foods. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 27–37.
Bullerman, L.B. & Tsai, W.Y. (1994) Incidence and levels of Fusarium moniliforme, Fusarium proliferatum and fumonisins in corn and corn-based foods and feeds. J. Food Prot., 57, 541–546.
Burrell, R.J.W. (1957) Oesophageal cancer in the Bantu. S. Afr. Med. J., 31, 401–409.
Burrell, R.J.W. (1962) Esophageal cancer among Bantu in the Transkei. J. Natl Cancer Inst., 28, 495–514.
Burrell, R.J., Roach, W.A. & Shadwell, A. (1966) Esophageal cancer in the Bantu of the Transkei associated with mineral deficiency in garden plants. J. Natl Cancer Inst., 36, 201–214.
Camargos, S.M., Valente-Soares, L.M., Sawazaki, E., Bolonhezi, D., Castro, J.L. & Bortollento, N. (2000) Fumonisins in corn cultivars grown during the 94/95 season in the State of Sao Paulo, Brazil. In: X International Symposium on Mycotoxins and Phycotoxins, Instituto Adolfo Lutz, Guaruja, Mexico, p. 142 (abstract).
Campbell, A.D., Whitaker, T.B., Pohland, A.E., Dickens, J.W. & Park, D.L. (1986) Sampling, sample preparation, and sampling plans for mycotoxin analysis. Pure Appl. Chem., 58, 305–314.
Canela, R., Pujol, R., Sala, N. & Sanchis, V. (1996) Fate of fumonisin B1 and B2 in steeped corn kernels. Food Addit. Contam., 13, 511–517.
Carlson, D.B., Williams, D.E., Spitsbergen, J.M., Ross, P.F., Bacon, C.W., Meredith, F.I. & Riley, R.T. (2001) Fumonisin B1 promotes aflatoxin B1 and N-methyl-N’-nitro-nitrosoguanidine initiated liver tumors in rainbow trout. Toxicol. Appl. Pharmacol. (in press).
Castella, G., Bragulat, M.R. & Cabanes, F.J. (1999) Surveillance of fumonisins in maize-based feeds and cereals from Spain. J. Agric. Food Chem., 47, 4707–4710.
Castelo, M.M., Sumner, S.S. & Bullerman, L.B. (1998a) Stability of fumonisins in thermally processed corn products. J. Food Prot., 61, 1030–1033.
Castelo, M.M., Sumner, S.S. & Bullerman, L.B. (1998b) Occurrence of fumonisins in corn-based food products. J. Food Prot., 61, 704–707.
Charles, A.G., Hansen, N., Giuliano, A.E. & Cabot, M.C. (2000) Taxol-induced ceramide generation and apoptosis in human breast cancer cell lines. St John’s Health Center, Santa Monica, CA. FASEB J., 14, 1147 (abstract).
Chen, J., Mirocha, C.J., Xie, W., Hogge, L. & Olson, D., (1992) Production of the mycotoxin FB1 by Alternaria alternata f.sp. lycopersici. Appl. Environ. Microbiol., 58, 3928–3931.
Chen, Q., Galleano, M. & Cederbaum, A.I. (1997) Cytotoxicity and apoptosis produced by arachidonic acid in Hep G2 cells overexpressing human cytochrome P4502E1. J. Biol. Chem., 272, 14532–14541.
Chu, F.S. & Li, G.Y. (1994) Simultaneous occurrence of fumonisin B1 and other mycotoxins in moldy corn collected from the People‘s Republic of China in regions with high incidences of esophageal cancer. Appl. Environ. Microbiol., 60, 847–852.
Cohen, S.M., Bidlack, W.R., Dragan, Y., Goldsworthy, T., Hard, G., Howard, P.C., Uey, R. & Voss, K. (2000) Apoptosis and its implications for toxicity, carcinogenicity and risk: Fumonisin B, as an example. In: Fumonisins Risk Assessment Workshop (JIFSAN, WHO, USFDA, USDA), College Park, Maryland, 10–12 January 2000.
Coker, R., Nagler, M., Defize, P., Derksen, G., Buchholz, H., Putzka, H., Hoogland, H. & Roos, A. (2001) The development of sampling plans for the determination of aflatoxin B1 in animal feedingstuffs. J. AOAC Int. (in press).
Comision Venezolana de Normas Industriales (COVENIN) (1982) Cereales, Leguminosas, Oleaginosas y Productos Derivados (Norma 612-82), Caracas: Ministerio de Fomento.
Commission of the European Union (1998a) Commission Directive 98/53/EC of 16 July 1998 laying down the sampling methods and the methods of analysis for the official control of the levels of certain contaminants in foodstuffs. Off. J. Eur. Communities, L201, 93–101.
Commission of the European Union (1998b) Commission Regulation (EC) No. 1525/98 of 16 July 1998 amending Regulation (EC) No. 194/97 of 31 January 1997 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Communities, L201, 43–45
Commission of the European Union (2000) Opinion of the scientific committee on food on fusarium toxins part 3: fumonisin B1 (FB1), Health & Consumer Protection Directorate. Expressed on 17 October 2000.
Constable, P.D., Foreman, J.H., Waggoner, A.L., Smith, G.W., Eppley, R.M., Tumbleson, M.E & Haschek, W.M. (2000a) The mechanism of fumonisin mycotoxicosis in horses. Draft report on USDA–CSREES Grant #928-39453, pp. 1–28.
Constable, P.D., Smith, G.W., Rottinghaus, G.E. & Haschek, W.M. (2000b) Ingestion of fumonisin B1-containing culture material decreases cardiac contractility and mechanical efficiency in swine. Toxicol. Appl. Pharmacol., 162, 151–160.
Cook-Mozaffari, P.J., Azordegan, F., Day, N.E., Ressicaud, A., Sabai, C. & Aramesch, B. (1979) Oesophageal cancer studies in the Caspian littoral of Iran: Results of a case–control study. Br. J. Cancer , 39, 293–309.
Cornell, J., Nelson, M.M. & Beighton, P. (1983) Neural tube defects in the Cape Town area, 1975–1980. S. Afr. Med. J., 64, 83–84.
Dantzer, W.R., Mullin, J., Hendrich, K. & Murphy, P.A. (1999) Excretion of [14C]fumonisin B1, [14C]hydrolyzed fumonisin B1, and [14C]fumonisin B1-fructose in rats. J. Agric. Food Chem., 47, 4291–4296.
Da Silva, J.B., Pozzi, C.R., Mallozzi, A.B., Ortega, E.M. & Correa, B. (2000) Mycoflora and occurrence of aflatoxin B1 and fumonisin B1 during storage of Brazilian sorghum. J. Agric. Food Chem., 48, 4352–4356.
De Girolamo, A., Solfrizzo, M., Von Holst, C. & Visconti, A. (2001) Comparison of different extraction solvents and clean-up procedures for the determination of fumonisins in maize and maize-based food products. Food Addit. Contam., 18, 59–67.
Delongchamp, R.R. & Young, J.F. (2001) Tissue sphinganine as a biomarker of fumonisin-induced apoptosis. Food Addit. Contam. (in press).
Desjardins, A.E., Manandhar, G., Plattner, R.D., Maragos, C.M., Shrestha, K. & McCormick, S.P. (2000) Occurrence of Fusarium species and mycotoxins in Nepalese maize and wheat and the effect of traditional processing methods on mycotoxin levels. J. Agric. Food Chem., 48, 1377–1383.
Dietz, J., Pardo, S.H., Furtado, D., Harzheim, E. & Furtado, A.D. (1998) Risk factors related to esophageal cancer in the Rio Grande do Sul. Rev. Assoc. Med. Bras., 44, 269–272.
Doko, M.B. & Visconti, A. (1994) Occurrence of fumonisins B1 and B2 in corn and corn-based human foodstuffs in Italy. Food Addit. Contam., 11, 433–439.
Doko, M.B., Rapior, S., Visconti, A. & Schjoth, J. E. (1995) Incidence and level of fumonisin contamination in maize genotypes grown in Europe and Africa. J. Agric. Food Chem., 43, 429–434.
Doko, M.B., Canet, C., Brown, N., Sydenham, E.W., Mpuchane, S. & Siame, B.A. (1996) Natural occurrence of fumonisins and zearalenone in cereals and cereal-based foods from eastern and southern Africa. J. Agric. Food Chem., 44, 3240–3243.
Dombrink-Kurtzman, M.A. & Dvorak, T.J. (1999) Fumonisin content in masa and tortillas from Mexico. J. Agric. Food Chem., 47, 622–627.
Dragan, Y.P., Bidlack, W.R., Cohen, S.M., Goldsworthy, T.L., Hard, G.C., Howard, P.C., Riley, R.T. & Voss, K.A. (2001) Implications of apoptosis for toxicity, carcinogenicity and risk assessment: fumonisin B1 as an example. Toxicol. Sci. (In press).
Dudek, H., Datta, S.R., Franke, T.F., Birnbaum, M.J., Yao, R., Cooper, G.M., Segal, R.A., Kaplan, D.R. & Greenberg, M.E. (1997) Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science, 275, 661–665.
Duncan, K., Kruger, S., Zabe, N., Kohn, B. & Prioli, R. (1998) Improved fluorometric and chromatographic methods for the quantification of fumonisins B1, B2 and B3. J. Chromatogr. A, 815, 41–47.
Duvick, J. (1999) Prospects for reducing fumonisin contamination through genetic modification of maize. In: International Conference on the Toxicology of Fumonisin, 28–30 June 1999, Arlington, Virginia, p. 24 (abstract).
Edwards, O.E., Blackwell, B.A., Driega, A.B., Bensimon, C. & Apsimon, J.W. (1999) The absolute stereochemistry of the ester functions of fumonisin B1. Tetrahedron Lett., 40, 4515–4518.
Enongene, E.N., Sharma, R.P., Voss, K.A. & Riley, R.T. (2000) Subcutaneous fumonisin administration disrupts sphingolipid metabolism in mouse digestive epithelia, liver, and kidney. Food Chem. Toxicol., 38, 793–799.
Enongene, E.N., Sharma, R.P., Neetesh, B., Meredith, F.I., Voss, K.A. & Riley, R.T. (2001) Time- and dose-related changes in mouse sphingolipid metabolism following gavage fumonisin B1 administration. Food Chem. Toxicol. (submitted).
Fan, J. & Gijbels, I. (1996) Local Polynomial Modelling and Its Applications, New York: Chapman & Hall.
FAO (1997) Worldwide Regulations for Mycotoxins 1995. A Compendium (FAO Food and Nutrition Paper 64), Rome.
FAO/WHO (2000) Position paper on fumonisins. Joint FAO/WHO Food Standards Programme, Codex Committee on Food Additives and Contaminants, Thirty-second Session, Beijing, 20–24 March 2000.
Fazekas, B., Bajmócy, E., Glávits, R. & Fenyvesi, A. & Tanyi, J. (1998) Fumonisin B1 contamination of maize and experimental acute fumonisin toxicosis in pigs. J. Vet. Med B., 45, 171–181.
Fincham, J.E., Marasas, W.F.O., Taljaard, J.J.F., Kriek, N.P.J., Badenhorst, C.J., Gelderblom, W.C.A., Seier, J.V., Smuts, C.M., Faber, M., Weight, M.J., Slazus, W., Woodroof, C.W., van Wyk, M.J., Kruger, M. & Thiel, P.G. (1992) Atherogenic effects in a non-human primate of Fusarium moniliforme cultures added to a carbohydrate diet. Atherosclerosis, 94, 13–25.
Food Standards Agency (2000) Ochratoxin A—Comparison of Sampling Plans. Food Saf. Bull., 124, 11–12.
Franceschi, S., Bidoli, E., Barón, A.E. & La Vecchia, C. (1990) Maize and risk of cancer of the oral cavity, pharynx and esophagus in northeastern Italy. J. Natl Cancer Inst., 82, 1407–1411.
Franke, T.F., Kaplan, D.R. & Cantley, L.C. (1997) PI3K: Downstream AKTion blocks apoptosis. Cell, 88, 435–437.
Fraumeni, J.F. & Blot, W.J. (1977) Geographic variation in esophageal cancer mortality in the United States. J. Chron. Dis., 30, 759–767.
Freese, L., Friedrich, R., Kendall, D. & Tanner, S. (2000) Variability of deoxynivalenol measurements in barley. J. AOAC Int., 83, 1259–1263.
Fukuda, H., Shima, H., Vesonder, R.F., Tokuda, H., Nishino, H., Katoh, S., Tamura, S., Sugimura, T. & Nagao, M. (1996) Inhibition of protein serine/threonine phosphatases by fumonisin B1, a mycotoxin. Biochem. Biophys. Res. Commun., 220, 160–165.
Gao, H-P. & Yoshizawa, T. (1997) Further study on Fusarium mycotoxins in corn and wheat from a high-risk area for human esophageal cancer in China. Mycotoxins, 45, 51–55.
Garren, L., Galendo, D., Wild, C.P. & Castegnaro, M. (2001) The induction and persistence of altered sphingolipid biosynthesis in rats treated with fumonisin B1. Food Addit. Contam., 18, 850–856.
Gatei, D.G., Odhiambo, P.A., Orinda, D.A.O., Muruka, F.J. & Wasunnna, A. (1978) Retrospective study of carcinoma of the esophagus in Kenya. Cancer Res., 38, 303–307.
Gelderblom, W.C.A. & Snyman, S.D. (1991) Mutagenicity of potentially carcinogenic mycotoxins produced by Fusarium moniliforme. Mycotoxin Res., 7, 46–52.
Gelderblom, W.C.A., Thiel, P.G., Marasas, W.F.O. & Van der Merwe, K.J. (1984) Natural occurrence of fusarin C, a mutagen produced by Fusarium moniliforme, on corn. J. Agric. Food Chem., 32,1064–1067
Gelderblom, W.C.A., Jaskiewicz, K., Marasas, W.F.O., Thiel, P.G., Horak, M.J., Vleggaar, R. & Kriek, N.P.J. (1988) Fumonisins—Novel mycotoxins with cancer promoting activity produced by Fusarium moniliforme. Appl. Environ. Microbiol., 54, 1806–1811.
Gelderblom, W.C.A., Kriek, N.P.J., Marasas, W.F.O. & Thiel, P.G. (1991) Toxicity and carcinogenicity of the Fusarium moniliforme metabolite, fumonisin B1, in rats. Carcinogenesis, 12, 1247–1251.
Gelderblom, W.C.A., Semple, E., Marasas, W.F.O. & Farber, E. (1992) The cancer initiating potential of the fumonisin mycotoxins. Carcinogenesis, 13, 433– 437.
Gelderblom, W.C.A., Cawood, M.E., Snyman, S.D. & Marasas, W.F.O. (1994) Fumonisin B1 dosimetry in relation to cancer initiation in rat liver. Carcinogenesis, 15, 209–214.
Gelderblom, W.C.A., Smuts, C.M., Abel, S., Snyman, S.D., Cawood, M.E. & Van der Westhuizen, L. (1996a) Effect of fumonisin B1 on protein and lipid synthesis in primary rat hepatocytes. Food Chem. Toxicol., 34, 361–369.
Gelderblom, W.C.A., Snyman, S.D., Lebepe-Mazur, S., Van der Westhuizen, L., Kriek, N.P.J. & Marasas, W.F.O. (1996b) The cancer promoting potential of fumonisin B1 in rat liver using diethylnitrosamine as cancer initiator. Cancer Lett., 109, 101–108.
Gelderblom, W.C.A., Snyman, S.D., Abel, S., Lebepe-Mazur, S., Smuts, C.M., van der Westhuizen, L., Marasas, W.F.O., Victor, T.C., Knasmüller, S. & Huber, W. (1996c) Hepatotoxicity and carcinogenicity of the fumonisins in rats: A review regarding mechanistic implications for establishing risk in humans. Adv. Exp. Med. Biol., 392, 279–296.
Gelderblom, W.C.A., Smuts, C.M., Abel, S., Snyman, S.D., Van der Westhuizen L., Huber, W.W. & Swanevelder, S. (1997) The effect of fumonisin B1 on the levels and fatty acid composition of selected lipids in rat liver in vivo. Food Chem. Toxicol., 35, 647–656.
Gelderblom, W.C.A., Abel, S., Smuts, C.M., Swanevelder, S. & Snyman, S.D. (1999a) Regulation of fatty acid biosynthesis as a possible mechanism for the mitoinhibitory effect of fumonisin B1 in primary rat hepatocytes. Prostaglandins Leukotrienes Essent. Fatty Acids, 61, 225–234.
Gelderblom, W.C.A., Vessey, C.J., Lebepe-Mazur, S., Marasas, W.F. & Hall, P.D. (1999b) Interaction of aflatoxin B1 and fumonisin B1 in the generation of oval cells and hepatocyte nodules in a rat model of hepatocarcinogenesis. Hepatology, 30 (Suppl.), 517A–517A.
Gelderblom, W.C.A., Abel, S., Smuts, C.M., Marnewick, J., Marasas, W.F.O., Lemmer, E.R. & Ramljak, D. (2001a) Fumonisin-induced hepatocarcinogenesis: Mechanisms related to cancer initiation and promotion. Environ. Health Perspectives (in press).
Gelderblom, W.C.A., Lebepe-Mazur, S., Snijman, P.W., Abel, S., Swanevelder, S., Kriek, N.P.J. & Marasas, W.F.O. (2001b) Toxicological effects in rats chronically fed low dietary levels of fumonisin B1. Toxicology (in press).
Gelderblom, W.C.A., Seier, J.V., Snijman, P.W., Van Schalkwyk, D.J., Shephard, G.S. & Marasas, W.F.O. (2001c) Toxicity of culture material of Fusarium verticillioides strain MRC 826 to non-human primates. Environ. Health Perspectives (in press).
Ghadarian, P. (1987) Food habits of the people of the Caspian littoral of Iran in relation to esophageal cancer. Nutr Cancer, 9, 147–157.
Ghadarian, P., Stein, G.F., Gorodetzkym C., Robetfroid, M.B., Mahon, G.A.T., Bartsch, H. & Day, N.E. (1985) Oesophageal cancer studies in the Caspian littoral of Iran: Some residual results, including opium use as a risk factor. Int. J. Cancer, 35, 593–597.
Gregory, J.R., Foster, K., Tyler, H. & Wiseman, M. (1990) Dietary and Nutritional Survey of British Adults: Further Analysis (ISBN 0112429661), London, Her Majesty’s Stationery Office.
Groves, F.D., Zhang, L., Chang, Y.-S., Ross, P.F., Casper, H., Norred, W.P., You, W.-C. & Fraumeni, J.F., Jr (1999) Fusarium mycotoxins in corn and corn products in a high-risk area for gastric cancer in Shandong Province, China. J. AOAC Int., 82, 657–662.
Gurung, N.K., Rankins, D.L., Shelby, R.A. & Goel, S. (1998) Effects of fumonsin B1-contaminated feeds on weaning Angora goats. J. Anim. Sci., 76, 2863–2870.
Hard, G.C., Howard, P.C., Kovatch, R.M. & Bucci, T.J. (2001) Rat kidney pathology induced by chronic exposure to fumonisin B1 includes rare varients of renal tubule tumor. Toxicol. Pathol. (in press)
Harmange, J.-C., Boyle, C.D. & Kishi, Y. (1994) Relative and absolute stereochemistry of the fumonisin B2 backbone. Tetrahedron Lett., 35, 6819–6822.
Harrison, L.R., Colvin, B.M., Greene, J.T., Newman, L.E. & Cole, J.R. (1990) Pulmonary edema and hydrothorax in swine produced by fumonisin B1, a toxic metabolite of Fusarium moniliforme. J.Vet. Diagn. Invest., 2, 217–221.
Hartl, M. & Humpf, H.U. (1998) Assigning the absolute configuration of fumonisins by circular dichroism exciton chirality method. Tetrahedron-Asymmetry, 9, 1549–1556.
Hartl, M., Herderich, M. & Humpf, H.-U. (1999) Rapid determination of fumonisin FB1 in corn products by high-performance liquid chromatography–electrospray mass spectrometry. Eur. Food Res. Technol., 209, 348–351.
Hattis, D., Erdreich, L. & Ballew, M. (1987) Human variability in susceptibility to toxic chemicals—A preliminary analysis of pharmacokinetic data from normal volunteers. Risk Anal., 7, 415–426.
Hendrich, S., Miller, K.A., Wilson, T.M. & Murphy, P.A. (1993) Toxicity of Fusarium proliferatum-fermented nixtamalized corn-based diets fed to rats: Effect of nutritional status. J. Agric. Food Chem., 41, 1649–1654.
Hendricks, K. (1999) Fumonisins and neural tube defects in south Texas. Epidemiology, 10, 198–200.
Hennigen, M.R., Sanchez, S., Di Benedetto, N.M., Longhi, A., Torroba, J.E. & Valente Soares, L.M. (2000) Fumonisin levels in commercial corn products in Buenos Aires, Argentina. Food Addit. Contam., 17, 55–58.
Hermans, G., Costa, L.L.F. & Scussel, V.M. (2000) Evaluation of fumonsin contamination of corn (Zea mays L.) produced at the western region of Santa Catarina. In: X International Symposium on Mycotoxins and Phycotoxins. Instituto Adolfo Lutz, Guaruja, Mexico, p. 147.
Hirooka, E.Y., Yamaguchi, M.M., Aoyama, S., Sugiura, Y. & Ueno, Y. (1996) The natural occurrence of fumonisins in Brazilian corn kernels. Food Addit. Contam., 13, 173–183.
Hlywka, J.J. & Bullerman, L.B. (1999) Occurrence of fumonisin B1 and B2 in beer. Food Addit. Contam., 16, 319–324.
Ho, A.K., Peng, R., Ho, A.A., Duffield, R. & Dombrink-Kurtzman, M.A. (1996) Interactions of fumonisins and sphingoid bases with GTP-binding proteins. Biochem. Arch., 12, 249–260.
Hormozdiari, H., Day, N.E., Aramesh, B. & Mahboubi, E. (1975) Dietary factors and esophageal cancer in the Caspian littoral of Iran. Cancer Res., 35, 3493–3498.
Horribin, DF. (1994) Unsaturated lipids and photodynamic therapy. In: Horribin, D.F., ed., New Approaches to Cancer, London: Churchil Communucations International, pp. 3–66 .
Horwitz, W. (1989) Guidelines for collaborative study of procedure to validate characteristics of a method of analysis. J. Assoc. Off. Anal. Chem., 72, 649–704.
Howard, P.C., Warbritton, A., Voss, K.A., Lorentzen, R.J., Thurman, J.D., Kovach, R.M. & Bucci, T.J. (2001a) Compensatory regeneration as a mechanism for renal tubule carcinogenesis of fumonisin B1 in the F344/N/Nctr BR rat. Environ. Health Perspectives (in press).
Howard, P.C., Eppley, R.M., Stack, M.E., Warbritton, A., Voss, K.A., Lorentzen, R.J., Kovach, R.M. & Bucci, T.J., (2001b) Fumonisin B1, carcinogenicity in a two-year feeding study using F344 rats and B6C3F1. Environ. Health Perspectives (in press).
Hoye, T.R., Jimenez, J.I. & Shier, W.T. (1994) Relative and absolute configuration of the fumonisin B1 backbone. J. Am. Chem. Soc., 116, 9409–9410.
Huang, C., Dickman, M., Henderson, G. & Jones, C. (1995) Repression of protein kinase C and stimulation of cyclic AMP response elements by fumonisin, a fungal encoded toxin which is a carcinogen. Cancer Res., 55, 1655–1659.
Humphreys, S.H., Carrington, C. & Bolger, P.M. (2000) A quantitative risk assessment for fumonisin B1 and B2 in US corn. Food Addit. Contam. (in press).
IARC (1993) IARC Monographs on the Evaluation of Carcinogenic Risk to Humans, Vol. 56, Lyon: IARC Press, pp. 445–466.
Instituto Nacional do Cancer (1989) [Estimates of Cancer Metabolites in Brazil], Rio de Janeiro: Ministerio da Saude (in Portugese).
Jackson, L.S., Hlywka, J.J., Senthil, K.R. & Bullerman, L.B. (1996) Effect of thermal processing on the stability of fumonisins. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 345–353.
Jackson, L.S., Katta, S.K., Fingerhut, D.D., DeVries, J.W. & Bullerman, L.B. (1997) Effect of baking and frying on the fumonisin B1 content of corn–based foods. J. Agric. Food Chem., 45, 4800–4805.
Jaskiewicz, K., Van Rensburg, S.J., Marasas, W.F.O. & Gelderblom, W.C.A. (1987a) Carcinogenicity of Fusarium moniliforme culture material in rats. J. Natl Cancer Inst., 78, 321–325.
Jaskiewicz, K., Marasas, W.F.O. & Van der Walt, F.E. (1987b) Oesophageal and other main cancer patterns in four districts of Transkei, 1981–1984. S. Afr. Med. J., 72, 27–30.
Jaskiewicz, K., Van Rensburg, S.J., Venter, F.S. & Marais C.W. (1987c) Oesophageal cytological abnormalities in Transkei and possible nutritional influences. S. Afr. Med. J., Suppl., 3–4.
Jaskiewicz, K., Marasas, W.F.O., Lazarus, C., Beyers, A.D. & Van Helden, P.D. (1988a) Association of oesophageal cytological abnormalities with vitamin and lipotrope deficiencies in populations at risk for esophageal cancer. Anticancer Res., 8, 711–716.
Jaskiewicz, K., Marasas, W.F.O., Rossouw, J.E., Van Niekerk, F.E. & Heine, E.W.P. (1988b) Selenium and other mineral elements in populations at risk for esophageal cancer. Cancer, 62, 2635–2639.
Jayadev, S., Linardic, C.M. & Hannun, Y.A. (1994) Identification of arachidonic acid as a mediator of sphingomyelin hydroysis in response to tumor necrosis factor alpha. J. Biol. Chem., 269, 5757–5763.
Joint Iran–IARC Study Group (1977) Esophageal cancer studies in the Caspian littoral of Iran. Results of population studies—A prodome. J. Natl Cancer Inst., 59, 1127–1138.
Julian, A.M., Wareing, P.W., Phillips. S.I., Medlock, V.F.P., MacDonald, M.V. & Del Rio, L.E. (1995) Fungal contamination and selected mycotoxins in pre- and post-harvest maize in Honduras. Mycopathologia, 129, 5–16.
Jurjevic, Z., Solfrizzo, M., Cvjetkovic, B., Avantaggiato, G. & Visconti, A. (1999) Ochratoxin A and fumonisins (B1 and B2) in maize from Balkan nephropathy endemic and non endemic areas of Croatia. Mycotoxin Res., 15, 67–80.
Katta, S.K., Cagampang, A.E., Jackson, L.S. & Bullerman, L.B. (1997) Distribution of Fusarium molds and fumonisins in dry-milled corn fractions. Cereal Chem., 74, 858–863.
Katta, S.K., Jackson, L.S., Sumner, S.S., Hanna, M.A. & Bullerman, L.B. (1999) Effect of temperature and screw speed on stability of fumonisin B1 in extrusion-cooked corn grits. Cereal Chem., 76, 16–20.
Kedera, C.J., Plattner, R.D. & Desjardins, A.E. (1999) Incidence of Fusarium spp. and levels of fumonisin B1 in maize in western Kenya. Appl. Environ. Microbiol., 65, 41–44.
Khan, W.A., Blobe, G.C. & Hannun, Y.A. (1995) Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell Signalling, 7, 171–184.
King, S.B. & Scott, G.E. (1981) Genotypic differences in maize kernel infection by Fusarium moniliforme. Phytopathology, 71, 1245–1247.
Kmet, J. & Mahbuobi, E. (1972) Esophageal cancer in the Caspian littoral of Iran: Initial studies. Science, 175, 846–853.
Knasmüller, S., Bresgenm, N., Fekadu, K., Mersch-Sundermann, V., Gelderblom, W., Zöhrer, E. & Eckl, H. (1997) Genotoxic effects of three Fusarium mycotoxins, fumonisins B1, moniliformin and vomitoxin in bacteria and in primary cultures of rat hepatocytes. Mutat. Res., 391, 39–48.
Kodell, R., Young, J., Delongchamp, R., Turturro, A., Chen, J., Gaylor, D., Howard, P. & Zheng, Q. (2001) A mechanistic approach to modeling the risk of liver tumors in mice exposed to fumonisin B1 in the diet, Food Addit. Contam. (in press).
Korkotian, E., Schwartz, A., Pelled, D., Schwarzmann, G., Segal, M. & Futerman, A.H. (1999) Elevation of intercellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem., 274, 21673–21678.
Kriek, N.P.J., Kellerman, T.S. & Marasas, W.F.O. (1981a) A comparative study of the toxicity of Fusarium verticillioides (= F. moniliforme) to horses, primates, pigs, sheep and rats. Onderstepoort J. Vet. Res., 48, 129–131.
Kriek, N.P.J., Marasas, W.F.O. & Thiel, P.G. (1981b) Hepato- and cardiotoxicity of Fusarium verticillioides (F. moniliforme) isolates from southern African maize. Food Cosmet. Toxicol., 19, 447–456.
Kuiper-Goodman, T., Scott, P.M., McEwen, N.P., Lombaert, G.A. & Ng, W. (1996) Approaches to the risk assessment of fumonisins in corn-based foods in Canada. Adv. Exp. Med. Biol., 392, 369–393.
Kulisek, E.S. & Hazebroek, J.P. (2000) Comparison of extraction buffers for the detection of fumonisin B1 in corn by immunoassay and high-performance liquid chromatography. J. Agric. Food Chem., 48, 65–69.
LaBorde, J.B., Terry, K.K., Howard, P.C., Chen, J.J., Collins, F.X., Shackelford, M.E. & Hansen, D.K. (1997) Lack of embryotoxicity of fumonisin B1 in New Zealand white rabbits. Fundam. Appl. Toxicol., 40, 120–128.
Lee, J.Y., Leonhardt, L.G. & Obeid, L.M. (1998) Cell-cycle-dependent changes in ceramide levels preceding retinoblastoma protein dephoshorylation in G2/M. Biochem. J., 344, 457–461.
Lemmer, E.R. (2000) The Fumonisin B1-fed Rat as a Model for Liver Injury, Oval (‘Progenitor’) Cell Proliferation, and Carcinogenesis. PhD Thesis, University of Cape Town.
Lemmer, E.R., Gelderblom, W.C.A., Shephard, E.G., Abel, S., Seymour, B.L., Cruse, J.P., Kirsch, R.E., Marasas, W.F.O. & Hall, P. de la M. (1999a) The effects of dietary iron overload on fumonisin B1-induced cancer induction in rat liver. Cancer Lett., 146, 207–215.
Lemmer, E.R., Hall, P. de la M., Omori, N., Omori, M., Shephard, E.G., Gelderblom, W.C.A., Cruse, J.P. Barnard, R.A., Marasas, W.F.O., Kirsch, R.E. & Thorgeirsson, S.S. (1999b) Histopathology and gene expression changes in rat liver during feeding of fumonisin B1, a carcinogenic mycotoxin produced by Fusarium moniliforme. Carcinogenesis, 20, 817–824.
Levin, S., Bucci, T.J., Cohen, S.M., Fix, A.S., Hardistry, J.F., LeGrand, E.K., Maronpot, R.R. & Trump, B.F. (1999) The nomenclature of cell death: Recommendations of an ad hoc committee of the Socirty of Toxicological Pathologists. Toxicol. Pathol., 27, 484–490.
Li, M.X., Lu, S., Ji, C., Wang, M.Y., Cheng, S.J. & Jin, C.L. (1979) Formation of carcinogenic N-nitroso compounds in corn bread inoculated with fungi. Sci. Sin., 22, 471–477.
Li, M.-H., Lu, S.-H., Ji, C., Wang, Y., Wang, M., Cheng, S. & Tian, G. (1980) Experimental studies on the carcinogenicity of fungus-contaminated food from Lixian county. In: Gelboin, H.V. et al., eds, Genetic and Environmental Factors in Experimental and Human Cancer, Tokyo: Japan Scientific Society Press, pp. 139–148.
Li, M., Tain, G., Lu, S., Guo, S., Jin, C. & Wang, Y. (1982) Forestomach carcinoma induced in rats by cornbread inoculated with Fusarium moniliforme. Zhonghua Zongliu Sazhi, 4, 241–244.
Li, J.-Y., Ershow, A.G., Chen, Z.-J., Wacholder, S., Li, G.-Y., Li, B. & Blot, W.J. (1989) A case–control study of cancer of the esophagus and gastric cardia in Linxian. Int. J. Cancer, 43, 755–761.
Li, W., Riley, R.T., Voss, K.A. & Norred, W.P. (2000) Role of proliferation in the toxicity of fumonisin B1: Enhanced hepatotoxic response in partially hepatectomized rat. J. Toxicol. Environ. Health, 60, 441–457.
Logrieco, A., Moretti, A., Ritieni, A., Bottalico, A. & Corda, P. (1995) Occurrence and toxigenicity of Fusarium proliferatum from preharvest maize ear rot, and associated mycotoxin, in Italy. Plant Dis., 79, 727–731.
Logrieco, A., Doko, B., Moretti, A., Frisullo, S. & Visconti, A. (1998) Occurrence of fumonisin B1 and B2 in Fusarium proliferatum infected asparagus plants. J. Agric. Food Chem., 46, 5201–5204.
Lu, S.J., Ronai, Z.A., Li, M.H. & Jeffrey, A.M. (1988) Fusarium moniliforme metabolites: genotoxicity of culture extracts. Carcinogenesis, 9, 1523–1527.
Lu, Z., Dantzer, W.R., Hopmans, E.C., Prisk, V., Cunnik, J.E., Murphy, P.A. & Hendrick, S. (1997) Reaction with fructose detoxifies fumonisin B1 while stimulating liver-associated natural killer cell activity in rats. J. Agric. Food Chem., 45, 803–809.
Lucock, M.D., Daskalkis, I., Lumd, C.H., Schorah, C.J. & Levene, M.I. (1998) Impaired regeneration of monoglutamyl tetrahydrofolate leads to cellular folate depletion in mothers affected by a spina bifida pregnancy. Mol. Genet. Metab., 65, 18–30.
Lukacs, Z., Schaper, S., Herderich, M., Schreier, P. & Humpf, H-U. (1996) Identification and determination of fumonisins FB1 and FB2 in corn and corn products by high-performance liquid chromatography–electrospray–ionization tandem mass spectrometry (HPLC–ESI–MS–MS). Chromatographia, 43, 124–128.
Luo, Y., Yoshizawa, T. & Katayama, T. (1990) Comparative study on the natural occurrence of Fusarium mycotoxins (trichothecenes and zearalenone) in corn and wheat from high- and low-risk areas from human esophageal cancer in China. Appl. Environ. Microbiol., 56, 3723–3726.
Macdonald, M.V. & Chapman, R. (1996) The incidence of Fusarium moniliforme on maize from Central America, Africa and Asia during 1992–1995. Plant Pathol., 46, 112–125.
Machinski, M., Jr & Valente Soares, L.M. (2000) Fumonisins B1 and B2 in Brazilian corn-based food products. Food Addit. Contam., 17, 875–879.
Machon, B. & Yen, S. (1971) Unrecognized epidemic of anencephaly and spina bifida. Lancet, ii, 31–33.
Makaula, N.A., Marasas, W.F.O., Venter, F.S., Badenhorst, C.J., Bradshaw, D. & Swanevelder, S. (1996) Oesophageal and other cancer patterns in four selected districts of Transkei, Southern Africa: 1985–1990. Afr. J. Health Sci., 3, 11–15.
Malone, B.M., Richard, J.L., Romer, T., Johansson, A.S. & Whitaker, T. (1998) Fumonisin reduction in corn by cleaning during storage discharge. In: O’Brian, L., Blakeney, A.B., Ross, A.S. & Wrigley, C.W., eds, Proceedings of the 48th Australian Cereal Chemistry Conference, Cairns, Australia, pp. 372–379.
Mantel, N. & Bryan, W.R. (1961) ‘Safety’ testing of carcinogenic agents. J. Natl Cancer Inst., 27, 455–470.
Maragos, C.M. (1997) Measurement of mycotoxins in food with a fiber-optic immunosensor. J. Clin. Lig. Assay, 20, 136–140.
Maragos, C.M. & Richard, J.L. (1994) Quantitation and stability of fumonisins B1 and B2 in milk. J. AOAC Int., 77, 1162–1167.
Maragos, C.M., Bennett, G.A. & Richard, J.L. (1997) Affinity column clean-up for the analysis of fumonisins and their hydrolysis products in corn. Food Agric. Immunol., 9, 3–12.
Marasas, W.F.O. (1996) Fumonisins: History, world-wide occurrence and impact. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 1–17.
Marasas, W.F.O. (1997) Risk assessment of fumonisins produced by Fusarium moniliforme in corn. Cereal Res. Communications, 25, 399–406.
Marasas, W.F.O., Van Rensburg, S.J. & Mirocha, C.J. (1979) Incidence of Fusarium species and the mycotoxins, deoxynivalenol and zearalenone, in corn produced in esophageal cancer areas in Transkei. J. Agric. Food Chem., 27, 1108–1112.
Marasas, W.F.O., Wehner, F.C., Van Rensburg, S.J. & Van Schalkwyk, D.J. (1981) Mycoflora of corn produced in human esophageal cancer areas in Transkei, Southern Africa. Phytopathology, 71, 792–796.
Marasas, W.F.O., Kriek, N.P.J., Fincham, J.E. & Van Rensburg, S.J. (1984) Primary liver cancer and oesophageal basal cell hyperplasia in rats caused by Fusarium moniliforme. Int. J. Cancer, 34, 383–387.
Marasas, W.F.O., Jaskiewicz, K., Venter, F.S. & Van Schalkwyk, D.J. (1988) Fusarium moniliforme contamination of maize in oesophageal cancer areas in Transkei. S. Afr. Med. J., 74, 110–114.
Marin, S., Sanchis, V., Sanz, D., Castel, I., Ramos, A.J., Canela, R. & Magtan, N. (1999) Control of growth and fumonisin B1 production by Fusarium verticillioides and Fusarium proliferatum isolates in moist maize with propionates preservatives. Food Addit. Contam., 16, 555–563.
Martinez-Larranaga, M.R., Anadon, A., Diaz, M.J., Fernandez-Cruz, M.L., Martinez, M.A., Frejo, M.T., Martinez, M., Fernandez, R., Anton, R.M., Morales, M.E. & Tafur, M. (1999) Toxicokinetic and oral bioavailability of fumonisin B1. Vet. Hum. Toxicol., 41, 357–362.
Mathur, S., Constable, P.D., Eppley, R.M., Tumbleson, M.E., Smith, G.W., Tranquilli, W.J., Morin, D.E. & Haschek, W.M. (2000) Cardiovascular effects of fumonisin B1 in milk-fed calves. Toxicol. Sci., 54, 346.
Medina-Martinez, M.S. & Martinez, A.J. (2000) Mold occurrence and aflatoxin B1 and fumonisin B1 determination in corn samples in Venezuela. J. Agric. Food Chem., 48, 2833–2836.
Mehta, R., Lok, E., Rowsel, P., Miller, J., Suzuki, C. & Bondy, G. (1998) Glutathione S-transferase-placental form expression and proliferation of hepatocytes in fumonisin B1-treated male and female Sprague-Dawley rats. Cancer Lett., 128, 31–39.
Meredith, F.I., Torres, O.R., De Tejada, S.S., Riley, R.T. & Merrill, A.H., Jr (1999) Fumonisin B1 and hydrolyzed fumonisin B1 (AP1) in tortillas and nixtamalized corn (Zea mays L.) from two different geographic locations in Guatamala. J. Food Prot., 62, 1218–1222.
Merrill, A.H., Jr, van Echten, G., Wang, E. & Sandhoff, K. (1993) Fumonisin Bl inhibits sphingosine (sphinganine) N-acetyltransferase and de novo sphingolipid biosynthesis in cultured neurons in situ. J. Biol. Chem., 268, 27299–27306.
Merrill, A.H., Jr, Schmelz, E.M., Dillehay, D.L., Spiegel, S., Shayman, J.A., Schroeder, J.J., Mey, R.T., Voss, K.A. & Wang, E. (1997) Sphingolipids—The enigmatic lipid class: Biochemistry, physiology, and pathophysiology. Toxicol. Appl. Pharmacol., 142, 208–225.
Merrill, A.H., Morgan, E.T., Nikolova-Karakashian, M. & Stewart, J. (1999a) Sphingomyelin hydrolysis and regulation of the expression of the gene for cytochrome P450. Biochem. Soc. Trans., 27, 383–387.
Merrill, A.H., Nikolova-Karakashian, M., Schmelz, E.M., Morgan, E.T. & Stewart, J. (1999b) Regulation of cytochrome P450 expression by sphingolipids. Chem. Phys. Lipids, 102, 131–139.
Merrill, A.H., Jr, Sullards, M.C., Wang, E., Voss, K.A. & Riley, R.T. (2001) Sphingolipid metabolism:roles in signal transduction and disruption by fumonisins. Environ. Health Perspectives (in press).
Miller, J.D., Savard, M.E., Schaafsma, A.W., Seifert, K.A. & Reid, L.M. (1995) Mycotoxin production by Fusarium moniliforme and Fusarium proliferatum from Ontario and occurrence of fumonisin in the 1993 corn crop. Can. J. Plant Pathol., 17, 233–239.
Missmer, S.A., Suarez, L., Larsen, R.D., Rothman, K.J. & Hendricks, K.A. (2000) Fumonisins and neural tube defects—Texas Department of Health preliminary results. Presented at the International Society of Environmental Epidemiology meeting, 22 August 2000, Buffalo, NY.
Mobio, T.A., Baudrimont, I., Sanni, A., Shier, T.W., Sabourneau, D., Dano, S.D., Ueno,, Y., Steyn, P.S. & Creppy, E.E. (2000a) Prevention of vitamin E of DNA fragmentation and apoptosis induced by fumonisin B1 in C6 glioma cells. Arch. Toxicol., 74, 112–119.
Mobio, T.A., Anane, R., Baudrimont, I., Carratu, M.-R., Shier, T.W., Sabourneau, D., Dano, S.D., Ueno, Y. & Creppy, E.E. (2000b) Epigenetic properties of Fumonisin B1: Cell cycle arrest and DNA base modification in C6 glioma cells. Toxicol. Appl. Pharmacol., 164, 91–96.
Moller, T.E. & Gustavsson, H.F. (2000) Determination of fumonisins B1 and B2 in various maize products by a combined SAX + C18 column and immunoaffinity column. J. AOAC Int., 83, 99–103.
Moolgavkar, S.H. & Knudson, A.G. (1981) Mutation and cancer: A model of human carcinogenesis. J. Natl Cancer Inst., 66, 1037–1052.
Moolgavka,r, S.H. & Venzon, D.J. (1979) Two-event models for carcinogenesis incidence curves for childhood and adult tumors. Math. Biosci., 47, 55–77.
Moore, C.A., Li, S., Li, Z., Hong, S., Gu, H., Berry, R.J., Mulinare, J. & Erickson, J.D. (1997) Elevated rates of severe neural tube defects in a high-prevalence area in northern China. Am. J. Med. Genet., 73, 113–118.
Mullett, W., Lai, E.P.C. & Yeung, J.M., (1998) Immunoassay of fumonisins by a surface plasmon resonance biosensor. Anal. Biochem., 258, 161–167.
Munimbazi, C. & Bullerman, L.B. (1996) Molds and mycotoxins in foods from Burundi. J. Food Prot., 59, 869–875.
Munkvold, G.A. & Desjardins, A.E. (1997) Fumonisins in maize: Can we reduce their occurrence? Plant Dis., 81, 556–565.
Munkvold, G.P., Hellmich, R.L. & Showers, W.B. (1997) Reduced Fusarium ear rot and symptomless infection in kernels of maize genetically engineered for European corn borer resistance. Phytopathology, 87, 1071–1077.
Munkvold, G. P., Hellmich, R.L. & Rice, L.G. (1999) Comparison of fumonisin concentrations in kernels of transgenic Bt maize hybrids and nontransgenic hybrids. Plant Dis., 83, 130–138.
Murphy, P.A., Rice, L.G. & Ross, P.F. (1993) Fumonisin B1, B2, and B3 content of Iowa, Wisconsin, and Illinois corn and corn screenings. J. Agric. Food Chem., 41, 263–266.
Murphy, P.A., Hendrich, S., Hopmans, E.C., Hauck, C.C., Lu, Z., Buseman, G. & Munkvold, G. (1996) Effect of processing on fumonisin content of corn. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 323–334.
Musser, S.M. & Plattner, R.D. (1997) Fumonisin composition in cultures of Fusarium moniliforme, Fusarium proliferatum, and Fusarium nygami. J. Agric. Food Chem., 45, 1169–1173.
Nagy, P., Evarts, R.P., Marsden, E.R., Roach, J. & Thorgeirsson, S.S. (1988) Cellular distribution of c-myc transcripts during chemical hepatocarcinogenesis in rats. Cancer Res., 48, 5522–5527.
Nair, M.G. (1998) Fumonisins and human health. Ann. Trop. Paediatr., 18, S47–S52.
National Academy of Sciences (1989) Recommended Allowances, Washington DC: US Food and Nutrition Board.
National Toxicology Program (1999) Toxicology and Carcinogenesis Studies of Fumonisin B1 (CAS No 116355-83-0) in F344/N Rats and B6C3F, Mice (Feed Studies) (NTP Technical Report TR 496; NIH Publication No 99-3955), Research Triangle Park, North Carolina.
Ncayiyana, D.J. (1986) Neural tube defects among rural blacks in a Transkei district. S. Afr. Med. J., 69, 618–620.
Nelson, P.E., Plattner, R.D., Shackelford, D.D. & Desjardins, A.E. (1991) Production of fumonisins by Fusarium moniliforme strains from various substrates and geographic areas. Appl. Environ. Microbiol., 57, 2410–2412.
Nelson, P.E., Plattner, R.D., Shackelford, D.D. & Desjardins, A.E. (1992) Fumonisin B1 production by Fusarium species other than F. moniliforme in the section Liseola and by some related species. Appl. Environ. Microbiol., 58, 984–989.
Nelson, P.E., Desjardins, A.E. & Plattner, R.D. (1993) Fumonisins, mycotoxins produced by Fusarium species: Biology, chemistry, and significance. Annu. Rev. Phytopathol., 31, 233–252.
de Nijs, M. (1997) Public Health Aspects of Fusarium Mycotoxins in Food in The Netherlands: A Risk Assessment, Dissertation, Wageningen Agricultural University, Wageningen.
de Nijs, M., van Egmond, H.P., Nauta, M., Rombouts, F.M. & Notermans, S.H.W. (1998a) Assessment of human exposure to fumonisin B1. J. Food Prot., 61, 879–884.
de Nijs, M., Sizoo, E.A., Vermunt, A.E.M., Notermans, S.H.W. & van Egmond, H.P. (1998c) The occurrence of fumonisin B1 in maize-containing foods in The Netherlands. Food Addit. Contam., 15, 385–388.
de Nijs, M., Sizoo, E.A., Rombouts, F.M., Notermans, S.H.W. & van Egmond, H.P. (1998b) Fumonisin B1 in maize for food production imported in The Netherlands. Food Addit Contam., 15, 389–392.
Norred, W.P., Voss, K.A., Bacon, C.W. & Riley, R.T. (1991) Effectiveness of ammonia treatement in detoxification of fumonisin-contaminated corn. Food Chem. Toxicol., 29, 815–819.
Norred, W.P., Plattner, R.D., Vesonder, R.F., Bacon, C.W. & Voss, K.A. (1992) Effects of selected secondary metabolites of Fusarium moniliforme on unscheduled synthesis of DNA by rat primary hepatocytes. Food Chem. Toxicol., 30, 233–237.
Norred, W.P., Riley, R.T., Meredith, F.I., Poling, S.M. & Plattner, R.D. (2001) Instability of N-acetylated fumonisin B1 (FA1) and the impact on inhibition of ceramide synthase in rat liver slices. Food Chem. Toxicol., 39, 1071–1078.
O’Brien, P.H., Parker, E.F. & Gregory, H.B. (1982) Epidemiology and treatment of carcinoma of the esophagus in South Carolina, a high-risk area in the US. In: Pfeiffer, C.J., ed., Cancer of the Esophagus, Vol. I, Boca Baton, Florida: CRC Press, pp. 65–80.
O’Neil, C.H., Hodges, G.M., Riddle, P.N., Jordan, P.W., Newmans, R.H., Flood, R.J. & Toulson, E.C. (1980) A fine silica contaminant of flour in the high oesophageal cancer area of north-east Iran. Int. J. Cancer, 26, 617–628.
Ostry, V. & Ruprich, J. (1998) Determination of the mycotoxin fumonisins in gluten-free diet (corn-based commodities) in the Czech Republic. Cent. Eur. J. Public Health, 6, 57–60.
Park, D.L. & Pohland, A.E. (1989) Sampling and sample preparation for detection and quantitation of natural toxicants in food and feed. J. Assoc. Off. Anal. Chem., 72, 399–404.
Park, D.L., Rua, S.M., Mirocha, C.J., El-Sayad, A.M.A. & Weng, C.Y. (1992) Mutagenic potentials of fumonisin contaminated corn following ammonia decontamination procedure. Mycopathologia, 117, 105–108.
Park, D.L., Lopez-Garcia, R., Trujillo-Ppreciado, S. & Price, R.I. (1996) Reduction of risks associated with fumonisin contamination in corn. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 335– 344.
Pascale, M., Doko, M.B. & Visconti, A. (1995) [Determination of fumonisins in polenta by high performance liquid chromatography.] In: Proceedings of the 2nd National Congress on Food Chemistry, Giardini-Naxos, 24–27 May 1995, Messina: La Grafica Editoriale, pp. 1067–1071 (in Italian).
Patel, S., Hazel, C.M., Winterton, A.G.M. & Gleadle, A.E. (1997) Surveillance of fumonisins in UK maize-based foods and other cereals. Food Addit. Contam., 14, 187–191.
Pelagalli, A., Belisario, M.A., Squillacioti, C., Morte, R.D., d’Angelo, D., Tafuri, S., Lucisano, A. & Staiano, N. (1999) The mycotoxin fumonisin B1 inhibits integrin-mediated cell–matrix adhesion. Biochimie, 81, 1003–1008.
Pepeljnjak, S., Petrinec, Z., Drca, S., Segvic, M., Ferencic, Z. & Jurisic, B. (2000) Evaluation of fumonisin B1 toxicity to carp (Cyprinus carpio L.). In: X International IUPAC Symposium on Mycotoxins and Phycotoxins, p. 70 (abstract).
Perilla, N.S. & Diaz, G.J. (1998) Incidence and levels of fumonisin contamination in Colombian corn and corn products. Mycotoxin Res., 14, 74–82.
Pestka, J.J., Azcona-Olivera, J.I., Plattner, R.D., Minervini, F., Doko, M.B. & Visconti, A. (1994) Comparative assessment of fumonisin in grain-based foods by ELISA, GC–MS, and HPLC. J. Food Prot., 57, 169–172.
Petersen, A. (2000) Submitted to FAO from Danish Veterinary and Food Administration.
Pineiro, M.S., Miller, J., Silva, G. & Musser, S. (1999) Effect of commercial processing on fumonisin concentrations of maize-based foods. Mycotoxin Res., 15, 2–11.
Pineiro, M.S., Silva, G.E., Scott, P.M., Lawrence, G.A. & Stack, M.E. (1997) Fumonisin levels in Uruguayan corn products. J. AOAC Int., 80, 825–828.
Pinelli, E., Poux, N., Garren, L., Pipy, B., Castegnaro, M., Miller, D.J. & Pfohl-Leszkowicz (1999) Activation of mitogen-activated protein kinase by fumonisin B1 stimulates cPLA2 phosphorylation, the arachidonic acid cascade and camp production. Carcinogenesis, 20, 1683–1688.
Plattner, R.D., Norred, W.P., Bacon, C.W., Voss, K.A., Peterson, R., Shackelford, D.D. & Weisleder, D. (1990) A method of detection of fumonisin in corn samples associated with field cases of equine leukoencephalomalacia. Mycologia, 82, 698–702.
Plattner, R.D., Weisleder, D. & Poling, S.M. (1996) Analytical determination of fumonisins and other metabolites produced by Fusarium moniliforme and related species on corn. Adv. Exp. Med. Biol., 392, 57–64.
Pocsfalvi, G., Ritieni, A., Randazzo, G., Dobo, A. & Malorni, A. (2000) Interaction of Fusarium mycotoxins, fusaproliferin and fumonisin B1, with DNA studied by electrospray ionization mass spectrometry. J. Agric. Food Chem., 48, 5795–5801.
Pohland, A.E. (1996) Occurrence of fumonisins in the US food supply. Adv. Exp. Med. Biol., 392, 19–26.
Pozzi, C.R., Correa, B., Gambale, W., Paula, C.R., Chacon-Reche, N.O. & Meirelles, M.C.A (1995) Postharvest and stored corn in Brazil: Mycoflora interaction, abiotic and mycotoxin occurrence. Food Addit. Contam., 12, 313–319.
Prathapkumar, S.H., Rao, V.S., Paramkishan, R.J. & Bhat, R.V. (1997) Disease outbreak in laying hens arising from the consumption of fumonisin-contaminated food. Br. Poult. Sci., 38, 475–479.
Preis, R.A. & Vargas, E.A. (2000) A method for determining fumonisin B1 in corn using immunoaffinity column clean-up and thin layer chromatography/densitometry. Food Addit. Contam., 17, 463–468.
Prelusky, D.B., Trenholm, H.L. & Savard, M.E. (1994) Pharmacokinetic fate of 14C-labelled fumonisin B1 in swine. Nat. Toxins, 2, 73–80
Prelusky, D.B., Trenholm, H.L., Rotter, B.A., Miller, J.D., Savard, M.E., Yeung, J.M. & Scott, P.M. (1996a) Biological fate of fumonisin B1 in food-producing animals. Adv. Exp. Med. Biol., 392, 265–279.
Prelusky, D.B., Miller, J.D. & Trenholm, H.L. (1996b) Disposition of 14C-derived residues in tissues of pigs fed radiolabelled fumonisin B1. Food Addit. Contam., 13, 155–162.
Pujol, R., Torres, M., Sanchis, V. & Canela, R. (1999) Fate of fumonisin B1 in corn kernel steeping water containing SO2. J. Agric. Food Chem., 47, 276–278.
Purchase, I.F.H & Joubert, H.J.B. (1970) Biological screening as a laboratory aid in determining cancer aetiology. In: Purchase, I.F.H., ed., Symposium on Mycotoxins in Human Health, London. Macmillan, pp. 291–297.
Purchase, I.F.H., Tustin, R.C. & Van Rensburg, S.J. (1975) Biological testing of food grown in the Transkei. Food Chem. Toxicol., 13, 639–647.
Qui, M., Liu, X. & Wang, Y. (2001) Determination of sphinganine, sphingosine and Sa/So ratio in urine of humans exposed to dietary fumonisin B1. Food Addit. Contam. (in press).
Ramljak, D., Calvert, R.J., Wiesenfeld, P.W., Diwan, B.A., Catipovic, B., Marasas, W.F.O., Victor, T.C., Anderson, L.M. & Gelderblom, W.C.A. (2000) A potential mechanism for fumonisin B1-mediated hepatocarcinogenesis: Cyclin D1 stabilization associated with activation of Akt and inhibition of GSK-3_ activity. Carcinogenesis, 21, 1537–1546.
Rava, E. (1996) Mycotoxins in maize products of the 1994/95 marketing season. Mycotoxin Res., 12, 25–30.
Rava, E., Viljoen, J.H., Kallmeyer, H. & De Jager, A. (1996) Fungi and mycotoxins in South African maize of the 1993 crop. Mycotoxin Res., 12, 15–24.
Rheeder, J.P., Marasas, W.F.O., Thiel, P.G., Sydenham, E.W., Shephard, G.S. & van Schalkwyk, D.J. (1992) Fusarium moniliforme and fumonisins in corn in relation to human esophageal cancer in Transkei. Phytopathology, 82, 353–357.
Rheeder, J.P., Marasas, W.F.O., Farina, M.P.W., Thompson, G.R. & Nelson, P.E. (1994) Soil fertility factors in relation to oesophageal cancer risk areas in Transkei, South Africa. Eur. J. Cancer Prev., 3, 49–56.
Rice, L.G. & Ross, P.F. (1994) Methods for detection and quantitation of fumonisins in corn, cereal products and animal excreta. J. Food Prot., 57, 536–540.
Rice, L.G., Ross, P.F., Dejong, J., Plattner, R.D. & Coats, J.R. (1995) Evaluation of a liquid chromatographic method for the determination of fumonisins in corn, poultry feed, and Fusarium culture material. J. AOAC Int., 78, 1002–1009.
Richard, J.L., Meerdink, G., Maragos, C.M., Tumbleson, M., Bordson, G., Rice, L.G. & Ross, P.F. (1996) Absence of detectable fumonisins in the milk of cows fed Fusarium proliferatum (Matsushima) Nirenberg culture material. Mycopathologia, 133, 123–126.
Riley, R.T., An, N.-H., Showker, J.L., Yoo, H.-S., Norred, W.P., Chamberlain, W.J., Wang, E., Merrill, A.H., Jr, Motelin, G., Beasley, V.R. & Haschek, W.M. (1993) Alteration of tissue and serum sphinganine to sphingosine ratio: An early biomarker of exposure to fumonisin-containing feeds in pigs. Toxicol. Appl. Pharmacol., 118, 105–112.
Riley, R.T., Hinton, D.M., Chamberlain, W.J., Bacon, C.W., Wang, E., Merrill, A.H., Jr & Voss, K.A. (1994) Dietary fumonisin B1 induces disruption of sphingolipid metabolism in Sprague-Dawley rats: A new mechanism of nephrotoxicity. J. Nutr., 124, 594–603.
Riley, R.T., Wang, E., Schroeder, J.J., Smith, E.R., Plattner, R.D., Abbas, H., Yoo, H.S. & Merrill, A.H., Jr (1996) Evidence for disruption of sphingolipid metabolism as a contributing factor in the toxicity and carcinogenicity of fumonisins. Nat. Toxins, 4, 3 –15.
Riley, R.T., Showker, J.L., Owens, D.L. & Ross, P.F. (1997) Disruption of sphingolipid metabolism and and induction of equine leudoencephalomacia by Fusarium proliferatum culture material containing FB2 or FB3. Environ. Toxicol. Pharmacol., 3, 221–228.
Riley, R.T., Enongene, E., Voss, K.A., Norred, W.P., Meredith, F.I., Sharma, R.P., Williams, D.E., Carlson, D.B., Spitsbergen, J. & Merrill, A.H., Jr (2001) Sphingolipid perturbations as mechanisms for fumonisin carcinogenesis. Environ. Health Perspectives (in press).
Ritieni, A., Moretti, A., Logrieco, A., Bottalico, A., Randazzo, G., Monti, S.M., Ferracane, R. & Fogliano, V. (1997) Occurrence of fusaproliferin, fumonisin B1, and beauvericin in maize from Italy. J. Agric. Food Chem., 45, 4011–4016.
Rose, E.F. (1965) A study of esophageal cancer in the Transkei. Natl Cancer Inst. Monogr., 25, 84–96.
Rose, E.F. (1973) Esophageal cancer in the Transkei. J. Natl Cancer Inst., 51, 7–16.
Rose, E.F. & Fellingham, S.A. (1981) Cancer patterns in the Transkei. S. Afr. J. Sci., 77, 555–561.
Ross, P.F., Nelson, P.E., Richard, J.L., Osweiler, G.D., Rice, L.G., Plattner, R.D. & Wilson, T.M. (1990) Production of fumonisins by Fusarium moniliforme and Fusarium proliferatum isolates associated with equine leukoencephalomalacia and a pulmonary edema syndrome in swine. Appl. Environ. Microbiol., 56, 3225–3226.
Ross, P.F., Rice, L.G., Plattner, R.D., Osweiller, G.D., Wilson, T.M., Owens, D.L., Nelson, H.A. & Richard, J.L. (1991) Concentrations of fumonisin B1 in feeds associated with animal health problems. Mycopathologia, 114, 129–135.
Rossi, M., Ancona, E., Mastrangelo, G., Solimbergo, D., Paruzzolo, P., Assarini, G., Sorrentina, P. & Peracchia, A. (1982) Epidemiologic findings in esophageal cancer in the Veneto region. Minerva Med., 73,1531–1540.
Rotter, B.A. & Oh, Y.-N. (1996) Mycotoxin fumonisin B1 stimulates nitric oxide production in a murine macrophage cell line. Nat. Toxins, 4, 291–294.
Rotter, B.A., Thompson, B.K., Prelusky, D.B., Trenholm, H.L., Stewart, B., Miller, J.D. & Savard, M.E. (1996) Response of growing swine to dietary exposure pure fumonisin B1 during an eight-week period: Growth and clinical parameters. Nat. Toxins, 4, 42–50.
Rottinghaus, G.E., Coatney, C.E. & Minor, H.C. (1992) A rapid, sensitive thin layer chromatographic procedure for the detection of fumonisin B1 and B2. J. Vet. Diagn. Invest., 4, 326–329.
Rumbeiha, W.K. & Oehme, F.W. (1997) Fumonisin exposure to Kansans through consumption of corn-based market foods. Vet. Hum. Toxicol., 39, 220–225.
Rylova, S.N., Somova, E.S., Dudnik, L.B., Kogtev, L.S., Kozlov, A.M., Alesenko, A.V. & Dyatlovitskaya, E.V. (1999) Content and structure of ceramide and sphingomyelin and sphingomyelinase activity in mouse hepatoma-22. Biochemistry (Moscow), 64, 437–441.
Sahu, S.C., Eppley, R.M., Page, S.W., Gray, G.C., Barton, C.N. & O’Donnell, M.W. (1998) Peroxidation of membrane lipids and oxidative DNA damage by fumonisin B1 in isolated rat liver nuclei. Cancer Lett., 125, 117–121.
Sakata, K., Sakata, A., Vela-Roch, N., Espinosa, R., Escalante, A., Kong, L., Nakabayashi, T., Cheng, J., Talal, N. & Dang, H. (1998) Fas (CD95)-transduced signal preferentially stimulates lupus peripheral T lymphocytes. Eur. J. Immunol., 28, 2648–2660.
Sammon, A.M. (1992) A case–control study of diet and social factors in cancer of the oesophagus in Transkei. Cancer, 69, 860–865.
Sandvig, K., Garred, O., Van Helvoort, A., Van Meer, G. & Van Deurs, B. (1996) Importance of glycolipid synthesis for butryic acid-induced sensitization to Shiga toxin and intracellular sorting of toxin in A431 cells. Mol. Biol. Cell, 7, 1391–1404.
Sauviat, M.P., Laurent, D., Kohler, F. & Pellegrin, F. (1991) Fumonisin, a toxin from the fungus Fusarium moniliforme Sheld, blocks both the calcium current and the mechanical activity in frog atrial muscle. Toxicon, 29, 1025–1031.
Scaff, R.M.C. & Scussel, V. (1999a) Esophageal cancer in southern region of Brazil. Mycotoxins (Suppl.), 226–230.
Scaff, R.M.C. & Scussel, V. (1999b) Esophageal cancer and its relationship to diet and cultural habits. In: X International Symposium on Mycotoxins and Phycotoxins, Guaruja, Mexico, Brazil: Instituto Adolfo Lutz, p. 86.
Schaafsma, A.W., Nicol, R.W., Savard, M.E., Sinha, R.C., Reid, L.M. & Rottinghaus, G. (1998) Analysis of Fusarium toxins in maize and wheat using thin layer chromatography. Mycopathologia, 142, 107–113.
Schmelz, E.-M., Dombrink-Kurtzman, M.A., Roberts, P.C., Kozutsumi, Y., Kawasaki, T. & Merrill, A.H., Jr (1998) Induction of apoptosis by fumonisin B1 in HT29 cells is mediated by the accumulation of endogenous free sphingoid bases. Toxicol. Appl. Pharmacol., 148, 252–260.
Schmitt, S.C. & Hurburgh, C.R., Jr (1989) Distribution and measurement of aflatoxin in 1983 in Iowa corn. Cereal Chem., 66, 165–168.
Schneider, E., Usleber, E. & Martlbauer, E. (1995) Rapid detection of fumonisin B1 in corn-based food by competitive direct dipstick enzyme immunoassay/enzyme-linked immunofiltration assay with integrated negative control reaction. J. Agric. Food Chem., 43, 2548–2552.
Scott, P.M. (1984) Effect of food processing on mycotoxins. J. Food Prot., 47, 489–499.
Scott, P.M. & Lawrence, G.A. (1994) Stability and problems in recovery of fumonisins added to corn-based foods. J. AOAC Int., 77, 541–545.
Scott, P.M. & Lawrence, G.A. (1995) Analysis of beer for fumonisins. J. Food Prot., 58, 1379–1382.
Scott, P.M. & Lawrence, G.A. (1996) Determination of hydrolysed fumonisin B1 in alkali-processed corn foods. Food Addit. Contam., 13, 823–832.
Scott, P.M., Delgado, T., Prelusky, D.B., Trenholm, H.L. & Miller, J.D. (1994) Determination of fumonisins in milk. J. Environ. Sci. Health, B29, 989–998.
Scott, P.M., Yeung, J.M., Lawrence, G.A. & Prelusky, D.B. (1997) Evaluation of enzyme-linked immunosorbent assay for the analysis of beer for fumonisins. Food Addit. Contam., 14, 445–450.
Scott, P.M., Lawrence, G.A. & Lombaert, G.A. (1999) Studies on extraction of fumonisins from rice, corn-based foods and beans. Mycotoxin Res., 15, 50–60.
Scudamore, K.A. & Patel, S. (2000) Survey of aflatoxins, ochratoxin A, zearalenone and fumonisins in maize imported into the United Kingdom. Food Addit. Contam., 17, 407–416.
Seegers, J.C., Joubert, A.M., Panzer, A., Lottering, M.-L., Jordan, C.A., Joubert, F., Mareem, J.-L., Bianchi, P., de Kock, M. & Gelderblom, W.C.A. (2000) Fumonisin B1 influenced the effects of arachidonic acid, prostaglandins E2 and A2 on cell cycle progression, apoptosis induction, tyrosine- and CDC2-kinase activity in oesophageal cancer cells. Prostaglandins Leukotrienes Essent. Fatty Acids, 62, 75–84.
Seo, J.-A. & Lee, Y.-W. (1999) Natural occurrence of the C series of fumonisins in moldy corn. Appl. Environ. Microbiol., 65, 1331–1334.
Sharma, R.P., Dugyala, R.R. & Voss, K.A. (1997) Demonstration of in situ apoptosis in mouse liver and kidney after short-term repeated exposure to fumonisin B1. J. Comp. Pathol., 117, 371–381.
Sharma, R.P., Bhandari, N., Riley, R.T., Voss, K.A. & Meredith, F.I. (2000a) Tolerance to fumonisin toxicity in a mouse strain lacking the P75 tumor necrosis factor receptor. Toxicology, 143, 183–194.
Sharma, R.P., Bhandari, N., He, Q., Riley, R.T. & Voss, K.A. (2000b) Decreased fumonisin hepatoxicity in mice with a targeted deletion of tumor necrosis factor receptor 1. Toxicology (in press).
Sharma, R.P., Bhandari, N., Tsunoda, M., Riley, R.T. & Voss, K.A. (2000c) Fumonisin hepatotoxicity is reduced in mice carrying the human tumor necrosis factor transgene. Arch. Toxicol., 74, 238–248.
Shelby, R.A., Rottinghaus, G.E. & Minor, H.C. (1994a) Comparison of thin layer chromatography and competitive immunoassay methods for detecting fumonisin on maize. J. Agric. Food Chem., 42, 2064–2067.
Shelby, R.S., White, D.G. & Bauske, E.M. (1994b) Differential fumonisin production in maize hybrids. Plant Dis., 78, 582–584.
Shephard, G.S. (1998) Chromatographic determination of fumonisin mycotoxins. J. Chromatogr. A, 815, 31–39.
Shephard, G.S. & Snijman, P.W. (1999) Elimination and excretion of a single dose of the mycotoxin fumonisin B2 in a non-human primate. Food Chem. Toxicol., 37, 111–116.
Shephard, G.S., Sydenham, E.W., Thiel, P.G. & Gelderblom, W.C.A. (1990) Quantitative determination of fumonisins B1 and B2 by high-performance liquid chromatography with fluorescence detection. J. Liq. Chromatogr., 13, 2077–2087.
Shephard, G.S., Thiel, P.G., Stockenström, S. & Sydenham, E.W. (1996a) Worldwide survey of fumonisin contamination of corn and corn-based products. J. AOAC Int., 79, 671–687.
Shephard, G.S., van der Westhuizen, L., Thiel, P.G., Gelderblom, W.C.A., Marasas, W.F.O. & Van Schalkwyk, D.J. (1996b) Disruption of sphingolipid metabolism in non-human primates consuming diets of fumonisin-containing Fusarium moniliforme culture material. Toxicon, 34, 527–534.
Shephard, G.S., Marasas, W.F.O., Leggott, N.L., Yazdanpanah, H. & Safavi, N. (2000) Natural occurrence of fumonisins in corn in Iran. J. Agric. Food Chem., 48,1860–1864.
Shetty, P.H. & Bhat, R.V. (1977) Differential production of fumonisin B1 in maize inbreds and hybrids in laboratory. Cereal Res. Communications, 25, 1011–1015.
Shetty, P.H. & Bhat, R.V. (1997) Natural occurrence of fumonisin B1 and its co-occurrence with aflatoxin B1 in Indian sorghum, maize and poultry feeds. J. Agric. Food Chem., 45, 2170–2173.
Shetty, P.H. & Bhat, R.V. (1999) Physical method for segregation of fumonisin-contaminated maize. Food Chem., 66, 371–374.
Sheu, C.W., Rodriguez, I., Eppley, R.M. & Lee, J.K. (1996) Lack of transforming activity of fumonisin B1 in BALB/3T3 A31-1-1 mouse embryo cells. Food Chem. Toxicol., 34, 751–753.
Shier, W.T., Abbas, H.K. & Badria, F.A. (1995) Complete structures of the sphingosine analog mycotoxins fumonisin B1 and AAL toxin TA: Absolute configuration of the side chains. Tetrahedron Lett., 36, 1571–1574.
Siame, B.A., Mpuchane, S.F., Gashe, B.A., Allotey, J. & Teffera, G. (1998) Occurrence of aflatoxins, fumonisin B1, and zearalenone in foods and feeds in Botswana. J. Food Prot., 61, 1670–1673.
Smith, J.S. & Thakur, R.A. (1996) Occurrence and fate of fumonisins in beef. Adv. Exp. Med. Biol., 392, 39–55.
Smith, G.W., Constable, P.D., Bacon, C.W., Meredith, F.I. & Haschek, W.M. (1996) Cardiovascular effects of fumonisins in swine. Fundam. Appl.. Toxicol., 31,169–172.
Smith, G.W., Constable, P.D., Tumbleson, M.E., Rottinghaus, G.E. & Haschek, W.M. (1999) Sequence of cardiovascular changes leading to pulmonary edema in swine fed fumonisin-containing culture material. Am. J. Vet. Res., 60, 1292–1300.
Smith, G.W., Constable, P.D., Eppley, R.M., Tumbleson, M.E., Gumprecht, L.A. & Haschek-Hock, W.M. (2000) Purified fumonisin B1 decreases cardiovascular function but does not alter pulmonary capillary permeability in swine. Toxicol. Sci., 56, 240–249.
Sohn, H.-B., Seo, J.-A. & Lee, Y.-W. (1999) Co-occurrence of Fusarium mycotoxins in mouldy and healthy corn from Korea. Food Addit. Contam., 16, 153–158.
Solfrizzo, M., Avantaggiato, G. & Visconti, A. (1997) In vivo validation of the sphinganine/sphingosine ratio as a biomarker to display fumonisin ingestion. Cereal Res. Communications, 25, 437–441.
Solfrizzo, M., De Girolamo, A., Visconti, A., Sizoo, E. & Van Egmond, H. (2000) Determination of fumonisins B1 and B2 in corn based foodstuffs by high performance liquid chromatography after immunoaffinity clean-up. In: X International IUPAC Symposium on Mycotoxins and Phycotoxins, Guaruja, Brazil, 21–25 May, p. 52 (abstract).
Solovey, M.M.S., Somoza, C., Cano, G., Pacin, A. & Resnik, S. (1999) A survey of fumonisins, deoxynivalenol, zearalenone and aflatoxins contamination in corn-based food products in Argentina. Food Addit. Contam., 16, 325–329.
Spiegel, S. (1998) Sphingosine 1-phosphate: A prototype of a new class of second messenger. J. Leuk Biol., 65, 341–344.
Spotti, M., Maas, R.F.M., de Nijs, C.M. & Fink-Gremmels, J. (2000) Effect of fumonisin B1 on rat hepatic P450 systems. Environ. Toxicol. Phamacol., 8, 197–204.
Stack, M.E. (1998) Analysis of fumonisin B1 and its hydrolysis product in tortillas. J. AOAC Int., 81, 737–740.
Stack, M.E. & Eppley, R.M. (1992) Liquid chromatographic determination of fumonisins B1 and B2 in corn and corn products. J. AOAC Int., 75, 834–837.
Steiner, P., Rudolph, B., Müller, D. & Eilers, M. (1996)The functions of myc in cell cycle progression and apoptosis. In: Guidet, M.L. & Vogel, L., eds, Progress in Cell Cycle Research, New York: Plenum Press, Vol. 2, pp. 73–82.
Stevens, V.L. & Tang, J. (1997) Fumonisin B1-induced sphingolipid depletion inhibits vitamin uptake via the glycosylphosphatidylinositol-ancored folate uptake. J. Biol. Chem., 272, 18020–18025.
Subbaramaiah, K., Chung, W.J. & Dannenberg, A.J. (1998) Ceramide regulates the transcription of cyclooxygenase-2. J. Biol. Chem., 273, 32943–32949.
Sydenham, E.W. & Shephard, G.S. (1996) Chromatographic and allied methods of analysis for selected mycotoxins. In: Gilbert, J., ed., Progress in Food Contaminant Analysis, London: Blackie Academic & Professional, pp. 65–146.
Sydenham, E.W., Thiel, P.G., Marasas, W.F.O., Shephard, G.S., Van Schalkwyk, D.J. & Koch, K.R. (1990) Natural occurrence of some Fusarium mycotoxins in corn from low and high esophageal cancer prevalence in areas of the Transkei, Southern Africa. J. Agric. Food Chem., 38, 1900–1903.
Sydenham, E.W., Shephard, G.S., Thiel, P.G. & Marasas, W.F.O. (1991) Fumonisin contamination of commercial corn-based human foodstuffs. J. Agric. Food Chem., 39, 2014–2018.
Sydenham, E.W., Marasas, W.F.O., Shephard, G.S., Thiel, P.G. & Hirooka, E.Y. (1992a) Fumonisin concentrations in Brazilian feeds associated with field outbreaks of confirmed and suspected animal mycotoxicosis. J. Agric. Food Chem., 40, 994–997.
Sydenham, E.W., Shephard, G.S. & Thiel, P.G. (1992b) Liquid chromatographic determination of fumonisin B1, B2, and B3 in foods and feeds. J. AOAC Int., 75, 313–318.
Sydenham, E.W., Shephard, G.S., Gelderblom, W.C.A., Thiel, P.G. & Marasas, W.F.O. (1993) Fumonisins: Their implications for human and animal health. In: Scudamore, K.A., ed., Occurrence and Significance of Mycotoxins, London: Brunel University, pp. 42–48.
Sydenham, E.W., Van der Westhuizen, L., Stockenström, S., Shephard, G.S. & Thiel, P.G. (1994) Fumonisin-contaminated maize: physical treatment for the partial decontamination of bulk shipment. Food Addit. Contam., 11, 25–32.
Sydenham, E.W., Stockenström, S., Thiel, P.G., Shephard, G.S., Koch, K. & Marasas, W.F.O. (1995) Potential of alkaline hydrolysis for the removal of fumonisins from contaminated corn. J. Agric. Food Chem., 43, 1198–1201.
Sydenham, E.W., Stockenström, S., Thiel, P.G., Rheeder, J.P., Doko, M.B., Bird, C. & Miller, B.M. (1996a) Polyclonal antibody-based ELISA and HPLC methods for the determination of fumonisins in corn: A comparative study. J. Food Prot., 59, 893–897.
Sydenham, E.W., Shephard, G.S., Thiel, P.G., Bird, C. & Miller, B.M. (1996b) Determination of fumonisins in corn: Evaluation of competitive immunoassay and HPLC techniques. J. Agric. Food Chem., 44, 159–164.
Sydenham, E.W., Shephard, G.S., Thiel, P.G., Stockenström, S., Snijman, P.W. & Van Schalkwyk, D.J. (1996c) Liquid chromatographic determination of fumonisins B1, B2, and B3 in corn: AOAC–IUPAC collaborative study. J. AOAC Int., 79, 688–696.
Tang, Q., Denda, A., Tsujiuchi, T., Tsutsumi, M., Amanuma, T., Murata, Y., Maruyama, H. & Konisihi, Y. (1993) Inhibitory effects of inhibitors of arachidonic acid metabolism on the evolution of rat liver preneoplastic foci into nodules and hepatocellular carcinomas with or without phenobarbital exposure. Jpn. J. Cancer Res., 84, 120–127.
TemaNord (1998) 502. Copenhagen K: Nordisk Ministerraad.
Thiel, P.G., Marasas, W.F.O., Sydenham, E.W., Shephard, G.S., Gelderblom, W.C.A. & Nieuwenhuis, J.J. (1991) Survey of fumonisin production by Fusarium species. Appl. Environ. Microbiol., 57, 1089–1093.
Thiel, P.G., Marasas, W.F.O., Sydenham, E.W., Shephard, G.S. & Gelderblom, W.C.A. (1992) The implications of naturally occurring levels of fumonisins in corn for human and animal health. Mycopathologia, 117, 3–9.
Thiel, P.G., Sydenham, E.W., Shephard, G.S. & Van Schalkwyk, D.J. (1993) Study of the reproducibility characteristics of a liquid chromatographic method for the determination of fumonisins B1 and B2 in corn: IUPAC collaborative study. J. AOAC Int., 76, 361–366.
Thompson, V.S. & Maragos, C.M. (1996) Fiber-optic immunosensor for the detection of fumonisin B1. J. Agric. Food Chem., 44, 1041–1046.
Tolleson, W.H., Dooley, K.L., Sheldon, W.G., Thurman, J.D., Bucci, T.J. & Howard, P.C. (1996) The mycotoxin fumonisin induces apoptosis in cultured human cells and in livers and kidneys of rats. Adv. Exp. Med. Biol., 392, 237–250.
Torres, M.R., Sanchis, V. & Ramos, A.J. (1998) Occurrence of fumonisins in Spanish beers analyzed by an enzyme-linked immunosorbent assay method. Int. J. Food Microbiol., 39, 139–143.
Travis, C.C. & White, R.K. (1988) Interspecific scaling of toxicity data. Risk Anal., 8, 119–125.
Trucksess, M.W. (2000) Natural toxins. In: Horwitz, W., ed., Official Methods of Analysis of AOAC International, 17th Ed., AOAC Int., Gaithersberg, Florida: AOAC International, Chapter 49.
Trucksess, M.W., Stack, M.E., Allen, S. & Barrion, N. (1995) Immunoaffinity column coupled with liquid chromatography for determination of fumonisin B1 in canned and frozen sweet corn. J. AOAC Int., 78, 705–710.
Trucksess, M.W., Cho, T.-H. & Ready, D.E. (2000) Liquid chromatographic method for fumonisin B1 in sorghum syrup and corn-based breakfast cereals. Food Addit. Contam., 17, 161–166.
Tseng, T.C. & Liu, C-Y. (1997) Occurrence of fumonisin B1 and B2 in corn-based foodstuffs in Taiwan market. Mycopathologia, 137, 57–61.
Tseng, T.C. & Liu, C-Y. (1999) Natural occurrence of fumonisins B1 and B2 in domestic maize of Taiwan. J. Agric. Food Chem., 47, 4799–4801.
Tseng, T.C. & Tu, J.C. (1997) Mycoflora and mycotoxins in adzuki and mung beans produced in Ontario, Canada. Microbios, 90, 87–95.
Tseng, T.C., Tu, J.C. & Soo, L.C. (1995) Natural occurrence of mycotoxins in Fusarium infected beans. Microbios, 84, 21–28.
Tsunoda, M., Shanna, R.P. & Riley, R.T. (1998) Early fumonisin B, toxicity in relation to disrupted sphingolipid metabolism in male BALB/c mice. J. Biochem. Mol. Toxicol., 12, 281–289.
Turner, P.C., Nikiema, P. & Wild, C.P. (1999) Fumonisin contamination of food: progress in development of biomarkers to better assess human health risks. Mutat. Res., 443, 81–93.
Ueno, Y, Yabe, T., Hashimoto, H., Sekijima, M., Masuda, T., Kim, D.J., Hasegawe, R. & Ito, N. (1992) Enhancement of GST-P positive liver cell foci development by nivalenol, a trichothecene mycotoxin. Carcinogenesis, 13, 787–791.
Ueno, Y., Aoyama, S., Sugiura, Y., Wang, D-S., Lee, U.-S., Hirooka, E.Y., Hara, S., Karki, T., Chen, G. & Yu, S.-Z. (1993) A limited survey of fumonisins in corn and corn-based products in Asian countries. Mycotoxin Res., 9, 27–34.
Ueno, Y., Nagata, S., Tsutsumi, T., Hasega, A., Watanabe, F., Park, H., Chen, G.-C., Chen, G. & Yu, S.-Z. (1996) Detection of microsystins, a blue–green algal hepatotoxin, in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis, 17, 1317–1321.
Ueno, Y., Iijima, K., Wang, S.D., Sugiura, Y., Sekijima, M., Tanaka, T., Chen, C. & Yu S.Z. (1997) Fumonisins as a possible contributory risk factor for primary liver cancer: A 3-year study of corn harvested in Haimen, China by HPLC and ELISA. Food Chem. Toxicol., 35, 1143–1150
US Department of Agriculture (1989–90) Consumer Food Survey of the Intake of Individuals, Fiscal Year 1989–1990, Washington DC.
Usleber, E., Straka, M. & Terplan, G. (1994) Enzyme immunoassay for fumonisin B1 applied to corn-based food. J. Agric. Food Chem., 42, 1392–1396.
Van Helden, P.D., Beyers, A.D., Bester, A.J. & Jaskiewicz, K. (1987) Esophageal cancer: Vitamin and lipotrope deficiencies in an at-risk South African population. Nutr. Cancer, 10, 247–255.
Van Rensburg, S.J. (1985) Recent studies on the etiology of oesophageal cancer. S. Afr. Cancer Bull., 29, 22–31.
Van Rensburg, S.J., Benade, A.S., Rose, E.F. & du Plessis, J.P. (1983) Nutritional status of African populations predisposed to oesophageal cancer. Nutr. Cancer, 4, 206–216.
Van Rensburg, S.J., Bradshaw, E.S., Bradshwa, D. & Rose, E.F. (1985) Oesophageal cancer in Zulu men, South Africa: A case control study. Br. J. Cancer, 51, 399–405.
Van Rensburg, S.J., van Schalkwyk, G.C. & van Schalkwyk, D.J. (1990) Primary liver cancer and aflatoxin intake in Transkei. J. Environ. Pathol. Toxicol., 10, 11–16.
Vasanthi, S. & Bhat, R.V. (1998) Mycotoxins in foods—Occurrence, health and economic significance and food control measurements. Indian J. Med. Res., 108, 212–214.
Venter, P.A., Christianson, A.L., Humato, C.M., Makhura, M.P. & Gericke, G.S. (1995) Congenital anomalies in rural black South African neonates—A silent epidemic. S. Afr. Med. J., 85, 15–20.
Vessey, C.J., Lemmer, E.R., Gelderblom, W.C., Shephard, E.G., McCleod, H.A., Barnard, R.A., Cruse, J.P., Marasas, W.F., Kirsch, R.E. & Hall, P.D. (1999) Generation of hepatic nodules and hepatocellular carcinoma after long-term feeding of the mycotoxin fumonisin B1 in male Fisher rats. Hepatology, 30 (Suppl.), 517.
Victoria, V.C., Muñoz, N., Day, N.E., Barcelos, L.B., Peccin, D.A. & Braga, N.M. (1987) Hot beverages and oesophageal cancer in southern Brazil: A case–control study. Int. J. Cancer, 39, 710–716.
Viquez, O.M., Castell-Perez, M.E. & Shelby, R.A. (1996) Occurrence of fumonisin B1 in maize grown in Costa Rica. J. Agric. Food Chem., 44, 2789–2791.
Visconti, A. (1996) Fumonisins in maize genotypes grown in various geographic areas. In: Jackson, L.S., Devries, J.W. & Bullerman, L.B., eds, Fumonisins in Food, New York: Plenum Press, pp. 193–204.
Visconti, A. & Boenke, A. (1995) Improvement of the determination of fumonisins (FB1 and FB2) in maize and maize based feeds. Final report (EUR 16617 EN) Contract No. MAT1-CT92-0029, Brussels: European Commission.
Visconti, A., Boenke, A., Solfrizzo, M., Pascale, M. & Doko, M.B. (1996a) European intercomparison study for the determination of fumonisins content in two maize materials. Food Addit. Contam., 13, 909–927.
Visconti, A., Solfrizzo, M., Doko, M.B., Boenke, A. & Pascale, M. (1996b) Stability of fumonisins at different storage periods and temperatures in gamma-irradiated maize. Food Addit. Contam., 13, 929–938.
Visconti, A., Solfrizzo, M., De Girolamo, A. & Boenke, A. (1999) Development/optimisation of analytical methods for the measurement of fumonisins at levels of interest for future EU legislation. Progress report (EUR 19096 EN) Contract No. SMT4-CT97-2194, Brussels: European Commission.
Visconti, A., Solfrizzo, M., De Girolamo, A., (2001) Determination of fumonisins B1 and B2 in corn and corn flakes by high performance liquid chromatography and immunoaffinity column clean-up: Collaborative study. J. AOAC Int. (in press).
Voss, K.A., Norred, W.P., Plattner, R.D. & Bacon, C.W. (1989) Hepatotoxicity and renal toxicity in rats of corn samples associated with field cases of equine leukoencephalomalacia. Food Chem. Toxicol., 27, 89–96.
Voss, K.A., Plattner, R.D., Bacon, C.W. & Norred, W.P. (1990) Comparative studies of hepatotoxicity and fumonisins B1 and B2 content of water and chloroform/methanol extracts of Fusarium moniliforme strain MRC 826 culture material. Mycopathologia, 112, 81–92.
Voss, K.A., Chamlerlain, W.J., Bacon, C.W. & Norred, W.P. (1993) A preliminary investigation on hepatic toxicity in rats fed purified fumonisin B1. Nat. Toxins, 1, 222–228.
Voss, K.A., Chamberlain, W.J., Bacon, C.W., Herbert, R.A., Walters, D.B. & Norred, W.P. (1995a) Subchronic feeding study of the mycotoxin fumonisin B1 in B6C3F1 mice and Fischer 344 rats. Fundam. Appl. Toxicol., 24, 102–110.
Voss, K.A., Chamlerlain, W.J., Bacon, C.W., Riley, R.T. & Norred, W.P. (1995b) Subchronic toxicity of fumonisin B1 to male and female rats. Food Addit. Contam., 121, 473–478.
Voss, K.A., Bacon, C.W., Meredith, F.I. & Norred, W.P. (1996a) Comparative subchronic toxicity studies of nixtamalized and water-extracted Fusarium moniliforme culture material. Food Chem. Toxicol., 34, 623–632.
Voss, K.A., Riley, R.T., Bacon, C.W., Chamberlain, W.J. & Norred, W.P. (1996b) Subchronic toxic effects of Fusarium moniliforme and fumonisin B1 in rats and mice. Nat. Toxins, 4, 16–23.
Voss, K.A., Plattner, R.D., Riley, R.T., Meredith, F.I. & Norred, W.P. (1998) In vivo effects of fumonisin B1-producing and fumonisin B1-non-producing Fusarium moniliforme isolates are similar: Fumonisins B2 and B3 cause hepato- and nephrotoxicity in rats. Mycopathologia, 141, 45–58.
Voss, K.A., Riley, R.T., Norred, W.P., Bacon, C.W., Meredith, F.I., Howard, P.C., Plattner, R.D., Collins, T., Hansen, D.K. & Porter, J.K. (2001) An overview of rodent toxicities: Liver, and kidney effects of fusarium moniliforme and fumonisins. Environ. Health Perspectives (in press).
Vudathala, D.K., Prelusky, D.B., Ayroud, M., Trenholm, H.L. & Miller, J.D. (1994) Pharmacokinetic fate and pathological effects of 14C-fumonisin B1 in laying hens. Nat. Toxins, 2, 81–88.
Wang, E., Norred, W.P., Bacon, C.W., Riley, R.T. & Merrill, A.H., Jr (1991) Inhibition of sphingolipid biosynthesis by fumonisins: Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem., 266, 14486–14490.
Wang, E., Ross, P.F., Wilson, T.M., Riley, R.T. & Merrill, A.H., Jr (1992) Increases in serum sphingosine and sphinganine and decreases in complex sphingolipids in ponies given feed containing fumonisins, mycotoxins produced by Fusarium moniliforme. J. Nutr., 122, 1706–1716.
Wang, D.-S., Liang, Y.-X., Chau, N.T., Dien, L.D., Tanaka, T. & Ueno, Y. (1995) Natural co-occurrence of Fusarium toxins and aflatoxin B1 in corn for feed in north Vietnam. Nat. Toxins, 3, 445–449.
Wang, E., Riley, R.T., Meredith, F.I. & Merrill, A.H., Jr (1999) Fumonisin B1 consumption by rats causes reversible, dose-dependent increases in urinary sphinganine and sphingosine. J. Nutr., 129, 214–220.
Ware, G.M., Umrigar, P.P., Carman, A.S., Jr & Kuan, S.S. (1994) Evaluation of Fumonitest immunoaffinity columns. Anal. Lett., 27, 693–715.
van der Westhuizen, L., Shephard, G.S., Snyman, S.D., Abel, S., Swanevelder, S. & Gelderblom, W.C.A. (1998) Inhibition of sphingolipid biosynthesis in rat primary hepatocyte cultures by fumonisin B1 and other related structurally related compounds. Food Chem. Toxicol., 36, 497–503.
van der Westhuizen, L., Brown, N.L., Marasas, W.F.O., Swanevelder, S. & Shephard, G.S. (1999) Sphinganine/sphingosine ratio in plasma and urine as a possible biomarker for fumonisins exposure in humans in rural areas of Africa. Food Chem. Toxicol., 37, 1153–1158.
van der Westhuizen, L. , Shephard, G.S. & van Schalkwyk, D.J. (2001) The effect of a single gavage dose of fumonisin B1 on the sphinganine and sphingosine levels in vervet monkeys. Toxicon, 39, 273–281.
Whitaker, T.B., Truckess, M.W., Johansson, A.S., Giesbrecht, F.G., Hagler, W.M., Jr & Bowman, D.T. (1998) Variability associated with testing shelled corn for fumonisin. J. AOAC Int., 81, 1162–1168.
Whitaker, T.B., Hagler, W.M., Jr, Giesbrecht, F.G. & Johansson, A.S. (2000) Sampling, sample preparation, and analytical variability associated with testing wheat for deoxynivalenol. J. AOAC Int., 83, 1285–1292
WHO (1998) GEMS/Food Regional Diets. Regional Per Capita Consumption of Raw and Semi-processed Agricultural Commodities. WHO/FSF/FOS/98.3. Geneva.
WHO (2000a) Fumonisin B1 (Environmental Health Criteria 219), Geneva, 150 pp.
WHO (2000b) Report on the Joint FAO/WHO Workshop on Methodology for Exposure Assessment of Contaminants and Toxins, 7–8 June 2000. Geneva.
Williamson, L.C., Bateman, K.E., Clifford, J.C. & Neale, E.A. (1999) Neuronal sensitivity to tetanus toxin requires gangliosides. J. Biol. Chem., 274, 25173–25180.
Wilson, T.M., Nelson, P.E. & Knepp, C.R. (1985) Hepatic neoplastic nodules, adenofibrosis, and cholangiocarcinomas in male Fischer rats fed corn naturally contaminated with Fusarium moniliforme. Carcinogenesis, 6, 155–1160.
Wilson, T.M., Ross, P.F., Rice, L.G., Osweiler, G.D., Nelson, H.A., Owens, D.L., Plattner, R.D., Reggiardo, C., Noon, T.H. & Pickrell, J.W. (1990) Fumonisin B1 levels associated with an epizootic of equine leukoencephalomalacia. J. Vet. Diagn. Invest., 2, 213–216.
Wilson, D.M., Sydenham, E.W., Lombaert, G.A., Trucksess, M.W., Abramson, D. & Bennett, G.A. (1998) Mycotoxin analytical techniques. In: Sinha, K.K. & Bhatnagar, D., eds, Mycotoxins in Agriculture and Food Safety, New York: Marcel Dekker, pp. 135–182.
Xie, W., Mirocha, C.J. & Chen, J. (1997) Detection of two naturally occurring structural isomers of partially hydrolysed fumonisin B1 in corn by on-line capillary liquid chromatography–fast atom bombardment mass spectrometry. J. Agric. Food Chem., 45, 1251–1255.
Xu, J., Yeh, C.-H., Chen, S., He, L., Sensi, S.L., Canzoniero, L.M.T., Choi, D.W. & Hsu, C.Y. (1998) Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J. Biol. Chem., 273, 16521–16526.
Yamashita, A., Yoshizawa, T., Aiura, Y., Sanchez, P.C., Dizon, E.I., Arim, R.H. & Sardjono (1995) Fusarium mycotoxins (fumonisins, nivalenol, and zearalenone) and aflatoxins in corn from southeast Asia. Biosci. Biotech. Biochem., 59, 1804–1807.
Yang, C.S. (1980) Research on esophageal cancer in China: A review. Cancer Res., 40, 2633–2644.
Yeung, J.M., Wang, H.-Y. & Prelusky, D.B. (1996) Fumonisin B1 induces protein kinase C translocation via direct interaction with diacylglycerol binding site. Toxicol. Appl. Pharmacol., 141, 178–184.
Yin, J.-J., Smith, M.J., Eppley, R.M., Page, S.W. & Sphon, J.A. (1998) Effects of fumonisin B1 on lipid peroxidation in membranes. Biochim. Biophys. Acta, 1371, 134–142.
Yoshizawa, T. & Yamashita, A. (1995) Natural occurrence of fumonisins and aflatoxins, carcinogenic mycotoxins, in corn and corn products in the Philippines. In: Occurrence and Microbiological Control of Mycotoxins in Foodstuffs and Other Agricultural Commodities, pp. 25–36.
Yoshizawa, T., Yamashita, A. & Luo, Y. (1994) Fumonisin occurrence in corn from high- and low-risk areas for human esophageal cancer in China. Appl. Environ. Microbiol., 60, 1626–1629.
Yoshizawa, T., Yamashita, A. & Chokethaworn, N. (1996) Occurrence of fumonisins and aflatoxins in corn from Thailand. Food Addit. Contam., 13, 163–168.
Zhang, H., Nagashima, H. & Goto, T. (1997) Natural occurrence of mycotoxins in corn, samples from high and low risk areas for human esophageal cancer in China. Mycotoxins, 44, 29–35.
Zhen, Y.Z. (1984) Isolation and culture of fungi from the cereals in five high and three low incidence counties of esophageal cancer in Henan Province (China). Zhonghua Zhonglia Zashi, 6, 27–29.
Zheng, Q. (1997) A unified approach to a class of stochastic carcinogenesis models. Risk Anal., 17, 617–624.
Zheng, Q. (1998) CarcinoMod: A tool for stochastic carcinogenesis modeling. In: Proceedings of the Worldwide Mathematica Conference (CD-ROM).
Zhou, H., Summers, S.A., Birnbaum, M.J. & Pittman, R.N. (1998) Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J. Biol. Chem., 273, 16568–16575.
Zoller, O., Sager, F. & Zimmerli, B. (1994) [Occurrence of fumonisins in food.] Mitt. Geb. Lebensm. Hyg., 85, 81–99 (in German).
Zomborszky, M.K., Vetesi, F., Repa, I., Kovacs, F., Bata, A., Horn, P., Toth, A. & Romvari, R. (2000) Experiment to determine limits of tolerance for fumonsin B1 in weaned piglets. J. Vet Med. Ser. B Infect. Dis. Vet. Public Health, 47, 277.
Results of surveys for fumonisin B1, showing concentrations and distribution of
contamination in food commodities
|
Country/ Region |
Commodity |
Year/ Season |
No. of samples |
LOQ |
n < LOQ |
Mean/Max |
|
Unprocessed maize |
||||||
|
North America |
||||||
|
Canada |
Domestic |
1993 |
98 |
No data |
89 |
69/1800 |
|
USA |
Iowa State |
1988 |
22 |
250 |
No data |
2500/14 900 |
|
1989 |
44 |
250 |
No data |
2900/37 900 |
||
|
1990 |
59 |
250 |
No data |
3300/19 100 |
||
|
1991 |
50 |
250 |
No data |
2900/15 800 |
||
|
1992 |
80 |
250 |
No data |
50/1600 |
||
|
1993 |
43 |
250 |
No data |
340/1900 |
||
|
1994 |
37 |
250 |
No data |
240/2200 |
||
|
1995 |
85 |
250 |
No data |
440/6500 |
||
|
1996 |
93 |
250 |
No data |
130/3300 |
||
|
South and Central America |
||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
34 |
8 |
20 |
104/744 |
|
Domestic |
Jan–Oct 1999 |
186 |
8 |
2 |
3299/15 560 |
|
|
Domestic |
May–Jun 1999 |
66 |
8 |
17 |
840/9377 |
|
|
Domestic |
Jan–Aug 2000 |
56 |
8 |
10 |
545/4982 |
|
|
Rice, imported |
Nov–Dec 1999 |
5 |
8 |
1 |
60/229 |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
6 |
8 |
6 |
—/— |
|
|
Rice, polished, domestic |
Apr–Jun 1999 |
5 |
8 |
5 |
—/— |
|
|
Brazil |
Domestic |
1997–98 |
110 |
20* |
0 |
No data/44 |
|
Domestic |
1994-95 |
105 |
20* |
0 |
No data/6.6 |
|
|
Domestic |
No data |
150 |
No data |
No data |
4.6/13(?) |
|
|
Domestic |
4/1995–4/1996 |
150 |
0.05(?) |
1 |
580/13 460 |
|
|
Domestic |
4/1995–4/1996 |
150 |
0.093(?) |
3 |
1140/22600 |
|
|
Domestic |
1998 |
214 |
100* |
2 |
No data/6000 |
|
|
Distribution: |
||||||
|
Domestic |
1990–91 |
48 |
No data |
1 |
5380/18 520 |
|
|
Colombia |
Domestic |
Feb–Aug 1999 |
15 |
20* |
7 |
259/2170 |
|
Costa Rica |
Domestic |
1992–93 |
64 |
No data |
4 |
2650/6320 |
|
Honduras |
Domestic |
10/1992, |
23 |
No data |
0 |
1357/6555 |
|
Uruguay |
Maize kernels |
1995–96 |
22 |
50* |
11 |
938/5787 |
|
Venezuela |
Domestic |
Oct 1993 |
37 |
10* |
6 |
1459/15 050 |
|
Europe |
||||||
|
Croatia |
(Fumonisins B1 and B2) |
|||||
|
Domestic |
1996 |
105 |
10* |
3 |
627/11661 |
|
|
Domestic |
1997 |
104 |
10* |
7 |
125/2524 |
|
|
Hungary |
Non-mouldy |
|
28 |
50* |
26 |
7/151 |
|
Mouldy |
|
24 |
50* |
7 |
1842/19 800 |
|
|
Random |
1994 |
23 |
50* |
16 |
463/5100 |
|
|
Mouldy |
1994 |
23 |
50* |
7 |
4619/52 400 |
|
|
Italy |
Visibly mouldy |
1994 |
22 |
5000* |
2 |
67 000/ |
|
Netherlands |
Imported |
1994–96 |
62 |
25 |
1 |
620/3350 |
|
Spain |
Domestic |
1994–96 |
55 |
60* |
7 |
4200/19 200 |
|
Sweden |
Imported |
Jan–Dec 1996 |
42 |
5 |
23 |
45/393 |
|
United Kingdom |
(Data corrected for analytical recovery) |
|||||
|
Imported |
6/1998–4/1999 |
139 |
20 |
5 |
795/3406 |
|
|
Distribution: |
||||||
|
Imported |
6/1998–4/1999 |
All fumonisins |
|
|
||
|
139 |
20 |
5 |
1160/5007 |
|||
|
Distribution: |
||||||
|
Africa |
||||||
|
Botswana |
Domestic |
1/1996–12/1997 |
33 |
20* |
5 |
210/1270 |
|
Burundi |
Domestic |
No data |
6 |
No data |
0 |
12 200-75 200 |
|
Kenya |
Smallholder farms |
1996 |
197 |
100* |
104 |
316/12 000 |
|
Kenya |
Kernels |
1994 |
1 |
20* |
0 |
780/780 |
|
8 |
20* |
1 |
59/115 |
|||
|
3 |
20* |
0 |
260/295 |
|||
|
South Africa |
White maize |
1994–95 |
143 |
20* |
No data |
637/12 963 |
|
Yellow maize |
1994–95 |
148 |
20* |
No data |
664/5062 |
|
|
White grade 1 |
1993 |
No data |
20* |
No data |
329/5637 |
|
|
White grade 2 |
1993 |
No data |
20* |
No data |
311/2078 |
|
|
White grade 3 |
1993 |
No data |
20* |
No data |
161/1128 |
|
|
Yellow grade 1 |
1993 |
No data |
20* |
No data |
589/11773 |
|
|
Yellow grade 2 |
1993 |
No data |
20* |
No data |
767/4991 |
|
|
Yellow grade 3 |
1993 |
No data |
20* |
No data |
849/5629 |
|
|
Tanzania |
Kernels |
1994 |
9 |
20* |
1 |
71/165 |
|
1 |
20* |
0 |
605/605 |
|||
|
2 |
20* |
1 |
63/125 |
|||
|
Asia |
||||||
|
China |
Maize |
Spring 1996 |
177 |
10 |
83 |
8880/78 370 |
|
Rice |
155 |
10 |
130 |
1410/31830 |
||
|
China |
|
1996 |
16 |
500* |
14 |
288/2400 |
|
China |
(Total fumonisins by ELISA) |
|||||
|
|
Autumn 1995 |
164 |
500* |
58 |
700/16000 |
|
|
Fanxian & Yanqing counties |
|
Autumn 1995 |
82 |
500* |
59 |
200/1500 |
|
Linxian County, Henan Province |
|
1994 |
34 |
No data |
7 |
2168/21000 |
|
Shangqiu County, Henan Province |
|
1994 |
20 |
No data |
10 |
1351/8470 |
|
Haimen, Jiangsu County |
|
Apr–Jul 1993 |
40 |
50* |
3 |
4727/25 970 |
|
Penlai, Shandong Province |
|
Apr–Jul 1993 |
40 |
50* |
24 |
260/3190 |
|
India |
Domestic |
1995 |
6 |
No data |
No data |
Range, 50–240 |
|
Domestic |
No data |
35 |
10* |
9 |
Geometric mean of positives, 620/4740 |
|
|
Distribution: |
|
|
||||
|
Indonesia |
Domestic |
Nov 1995 |
16 |
50* |
0 |
788/2440 |
|
Domestic |
1992–94 |
12 |
50* |
5 |
492/1780 |
|
|
Iran |
Mazandaran |
Sept 1998 |
11 |
10* |
0 |
2269/3980 |
|
Iran |
Isfahan |
Oct 1998 |
8 |
10* |
0 |
169/590 |
|
Korea, Republic of |
Mouldy |
Nov 1997 |
36 |
50* |
3 |
21 300/ |
|
Healthy |
Nov 1997 |
35 |
50* |
25 |
910/12 500 |
|
|
Nepal |
(Total fumonisins by HPLC) |
|
|
|
|
|
|
Domestic |
Feb–Jul 1997 |
58 |
100* |
6 |
493/8400 |
|
|
Philippines |
(Total fumonisins by ELISA) |
|
|
|
|
|
|
Domestic |
Jun–Nov 1995 |
10 |
200* |
1 |
2000/10 000 |
|
|
Domestic |
1992–94 |
50 |
50* |
24 |
218/1820 |
|
|
Philippines |
Seed maize |
1992–94 |
24 |
50* |
14 |
148/970 |
|
Feed maize |
1992-1994 |
22 |
50* |
7 |
320/1820 |
|
|
Food maize |
1992-1994 |
23 |
50* |
16 |
41/268 |
|
|
Taiwan |
Domestic |
1996–97 |
110 |
40* |
61 |
79/1148 |
|
Distribution: |
||||||
|
Thailand |
Visibly mouldy human food |
1992 |
5 |
50* |
1 |
4277/18800 |
|
Domestic |
1992–94 |
27 |
50* |
8 |
1112/18 800 |
|
|
Viet Nam |
(Total fumonisins by ELISA) |
|
|
|
|
|
|
Domestic |
Jun–Nov 1995 |
12 |
200* |
0 |
6600/9100 |
|
|
Viet Nam |
Maize, feed |
1993 |
15 |
50* |
7 |
587/3447 |
|
Maize powder, feed |
1993 |
17 |
50* |
2 |
688/1516 |
|
|
Australia |
||||||
|
Australia |
(Total fumonisins by ELISA) |
|||||
|
|
Domestic |
Jun–Nov 1995 |
70 |
200* |
3 |
6700/40 600 |
|
Processed maize-based human food |
||||||
|
North America |
||||||
|
Canada |
(* number and mean of positive samples) |
|||||
|
Maize kernel, meal, flour |
1996–97 |
21* |
No data |
No data |
190*/850 |
|
|
Maize snacks (alkali-processed) |
20* |
No data |
No data |
203*/970 |
||
|
Maize snacks (non-alkali-processed) |
6* |
No data |
No data |
65*/100 |
||
|
Canned |
9* |
No data |
No data |
48*/90 |
||
|
Frozen |
2* |
No data |
No data |
35*/40 |
||
|
Maize bread |
1* |
No data |
No data |
30*/30 |
||
|
Maize-based breakfast cereals |
8* |
No data |
No data |
80*/170 |
||
|
Fresh tortillas |
1* |
No data |
No data |
750*/750 |
||
|
Maize-based infant foods: |
1997–98 |
|
|
|
|
|
|
Mixed cereals |
16* |
No data |
No data |
26*/110 |
||
|
Cereal with formula |
6* |
No data |
No data |
<10*/<10 |
||
|
Creamed maize or maize / vegetables |
6* |
No data |
No data |
<10*/<10 |
||
|
Infant cereals: 1998–99 |
|
|
|
|
||
|
Mixed cereals with fruit / formula |
13* |
No data |
No data |
<10*/<10 |
||
|
Rice-based |
16* |
No data |
No data |
<10*/<10 |
||
|
Soya-based |
14* |
No data |
No data |
20*/60 |
||
|
Mixed cereals |
20* |
No data |
No data |
20*/70 |
||
|
Tortilla, nacho, maize chips, taco shells |
1996 |
17 |
20* |
12 |
23/216 |
|
|
Hydrolysed fumonisin B1 |
17 |
10* |
16 |
2/40 |
||
|
Maize tortilla, dried |
10 |
20* |
2 |
202/612 |
||
|
Hydrolysed fumonisin B1 |
10 |
10* |
5 |
20/60 |
||
|
USA |
Maize meal, degermed |
1997 |
602 |
100* |
278 |
130/1080 |
|
Maize meal, partly degermed |
20 |
100* |
6 |
230/1130 |
||
|
Maize meal, whole grain |
50 |
100* |
5 |
910/4820 |
||
|
Maize meal, degermed |
1998 |
561 |
100* |
439 |
130/7010 |
|
|
Maize meal, partly degermed |
1998 |
20 |
100* |
6 |
730/3030 |
|
|
Maize meal, whole grain |
39 |
100* |
2 |
950/5500 |
||
|
Canned maize |
Spring 1993 |
70 |
25 |
42 |
11/235 |
|
|
Frozen maize |
27 |
25 |
18 |
18/350 |
||
|
Maize meal |
No data |
10 |
75* |
<75–5916 |
||
|
Muffin mix |
No data |
6 |
75* |
<75–417 |
||
|
Maize bread |
No data |
4 |
75* |
<75–1020 |
||
|
Tortilla chips |
No data |
19 |
75* |
<75–1565 |
||
|
Maize tortilla |
No data |
5 |
75* |
<75–330 |
||
|
Cornflakes |
No data |
6 |
75* |
<75–88 |
||
|
Maize starch |
No data |
1 |
75* |
<75 |
||
|
Popcorn |
No data |
1 |
75* |
<75 |
||
|
Infant foods |
No data |
2 |
75* |
<75 |
||
|
(Total fumonisins by HPLC) |
||||||
|
Maize, shelled |
1994 |
41 |
20* |
6 |
179/1100 |
|
|
1995 |
78 |
20* |
12 |
406/4400 |
||
|
1996 |
76 |
20* |
32 |
562/14900 |
||
|
1997 |
43 |
20* |
6 |
516/5100 |
||
|
1998 |
134 |
20* |
85 |
497/7300 |
||
|
Maize meal |
1994 |
39 |
20* |
2 |
266/1300 |
|
|
1995 |
64 |
20* |
14 |
188/2500 |
||
|
Maize meal |
1996 |
35 |
20* |
28 |
142/2700 |
|
|
1997 |
40 |
20* |
9 |
295/1800 |
||
|
1998 |
69 |
20* |
45 |
177/1600 |
||
|
Maize flour |
1994 |
18 |
20* |
2 |
178/630 |
|
|
1995 |
15 |
20* |
7 |
37/190 |
||
|
1996 |
17 |
20* |
13 |
80/860 |
||
|
1997 |
19 |
20* |
1 |
332/1300 |
||
|
1998 |
15 |
20* |
13 |
113/1300 |
||
|
Maize grits |
1994 |
8 |
20* |
4 |
60/231 |
|
|
1995 |
6 |
20* |
2 |
60/215 |
||
|
1996 |
6 |
20* |
5 |
17/100 |
||
|
1997 |
9 |
20* |
3 |
127/300 |
||
|
1998 |
5 |
20* |
5 |
—/— |
||
|
Hominy grits |
1994 |
0 |
20* |
— |
—/— |
|
|
1995 |
7 |
20* |
6 |
9/60 |
||
|
1996 |
2 |
20* |
1 |
110/220 |
||
|
1997 |
3 |
20* |
1 |
333/600 |
||
|
1998 |
10 |
20* |
10 |
—/— |
||
|
Maize chips, tortilla |
1994 |
17 |
20* |
13 |
19/164 |
|
|
1995 |
6 |
20* |
5 |
8/50 |
||
|
1996 |
5 |
20* |
5 |
—/— |
||
|
1997 |
27 |
20* |
19 |
83/740 |
||
|
1998 |
32 |
20* |
30 |
33/1000 |
||
|
Maize muffin mix |
1994 |
5 |
20* |
5 |
—/— |
|
|
1995 |
8 |
20* |
6 |
15/93 |
||
|
1996 |
3 |
20* |
3 |
—/— |
||
|
1997 |
6 |
20* |
4 |
33/100 |
||
|
1998 |
16 |
20* |
15 |
25/400 |
||
|
Cornflakes, dry cereals |
1994 |
9 |
20* |
9 |
—/— |
|
|
1995 |
4 |
20* |
4 |
—/— |
||
|
1996 |
12 |
20* |
12 |
—/— |
||
|
1997 |
15 |
20* |
14 |
2/30 |
||
|
1998 |
27 |
20* |
27 |
—/— |
||
|
Starch |
1994 |
5 |
20* |
5 |
—/— |
|
|
1995 |
5 |
20* |
4 |
3/13 |
||
|
1996 |
3 |
20* |
3 |
—/— |
||
|
1997 |
9 |
20* |
9 |
—/— |
||
|
1998 |
9 |
20* |
9 |
—/— |
||
|
Popcorn |
1994 |
17 |
20* |
6 |
71/280 |
|
|
1995 |
15 |
20* |
10 |
17/90 |
||
|
1996 |
14 |
20* |
12 |
49/570 |
||
|
1997 |
11 |
20* |
4 |
235/1800 |
||
|
1998 |
25 |
20* |
22 |
47/960 |
||
|
Tortillas |
No data |
52 |
10* |
2 |
187/750 |
|
|
Hydrolysed fumonisin B1 |
|
10* |
4 |
82/204 |
||
|
Masa |
No data |
8 |
10* |
0 |
262/689 |
|
|
Hydrolysed fumonisin B1 |
|
10* |
2 |
64/178 |
||
|
Starch |
No data |
4 |
10* |
2 |
158/335 |
|
|
Grits |
No data |
4 |
10* |
1 |
4889/10110 |
|
|
Masa |
No data |
2 |
10* |
1 |
55/110 |
|
|
Popcorn |
Feb-May 1993/ |
18 |
40* |
17 |
4/69 |
|
|
Breakfast cereals |
Feb–May 1993 |
16 |
40* |
16 |
—/— |
|
|
Snacks |
40 |
40* |
40 |
—/— |
||
|
Maize flour |
25 |
40* |
12 |
82/349 |
||
|
Tortilla shells |
5 |
40* |
5 |
—/— |
||
|
Sweet maize |
8 |
40* |
8 |
—/— |
||
|
Maize oil |
4 |
40* |
4 |
—/— |
||
|
Total fumonisins |
1990–94 |
|
|
|
0–250 |
|
|
Canned maize |
70 |
No data |
42 |
28 |
||
|
Frozen maize |
27 |
No data |
18 |
8 |
||
|
Maize bran cereal |
7 |
No data |
0 |
4 |
||
|
Maize bread mix |
13 |
No data |
2 |
3 |
||
|
Maize cereal |
3 |
No data |
3 |
|
||
|
Maize chips |
8 |
No data |
7 |
1 |
||
|
Cornflakes |
13 |
No data |
11 |
2 |
||
|
Maize grits |
21 |
No data |
4 |
4 |
||
|
Maize hominy |
1 |
No data |
0 |
1 |
||
|
Maize meal |
98 |
No data |
2 |
34 |
||
|
Maize muffin mix |
9 |
No data |
5 |
2 |
||
|
Maize pops cereal |
5 |
No data |
5 |
|
||
|
Maize tortillas |
15 |
No data |
9 |
6 |
||
|
Maize tortilla chips |
1990–94 |
2 |
No data |
1 |
1 |
|
|
Shelled maize |
41 |
No data |
6 |
19 |
||
|
Maize starch |
5 |
No data |
5 |
|
||
|
Fibre cereal |
3 |
No data |
3 |
|
||
|
Popcorn |
20 |
No data |
6 |
13 |
||
|
Spoon bread mix |
1 |
No data |
0 |
|
||
|
South and Central America |
||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
11 |
8 |
0 |
188/540 |
|
Popcorn |
May–Jun 1999/ |
42 |
8 |
1 |
1084/14241 |
|
|
Maize flour |
10/1996–1/1997 |
15 |
11* |
1 |
341/1860 |
|
|
Maize grits |
10/1996–1/1997 |
4 |
11* |
2 |
147/494 |
|
|
Maize flour |
Jan 1998 |
14 |
11* |
0 |
882/4987 |
|
|
Maize grits |
Jan 1998 |
2 |
11* |
0 |
577/832 |
|
|
Maize meal |
1997 |
21 |
1.5* |
2 |
503/2860 |
|
|
Cornflakes |
1997 |
17 |
1.5* |
1 |
11/38 |
|
|
Maize meal: |
Distribution of total fumonisins: |
|||||
|
Brazil |
Canned sweet maize |
Mar–May 1999 |
11 |
20* |
9 |
10/80 |
|
Cornflakes |
4 |
20* |
3 |
170/660 |
||
|
Maize flour |
11 |
20* |
2 |
610/1460 |
||
|
Maize flour, baby cereal |
2 |
20* |
1 |
220/440 |
||
|
Maize grits |
2 |
20* |
0 |
700/1230 |
||
|
Maize meal |
9 |
20* |
0 |
2290/4930 |
||
|
Corn-on-the-cob |
7 |
20* |
7 |
—/— |
||
|
Curau |
2 |
20* |
2 |
—/— |
||
|
Maize, degermed |
11 |
20* |
3 |
840/4520 |
||
|
Pamonha |
7 |
20* |
7 |
—/— |
||
|
Popcorn |
1 |
9 |
20* |
5 |
||
|
Pre-cooked maize meal |
6 |
20* |
2 |
840/1790 |
||
|
Colombia |
Pre-cooked maize meal |
Feb-Aug 1998 |
15 |
20* |
2 |
102/230 |
|
Popcorn |
8 |
20* |
4 |
84/246 |
||
|
Maize meal |
7 |
20* |
3 |
123/408 |
||
|
"Arepas" |
6 |
20* |
4 |
18/61 |
||
|
Maize snacks |
6 |
20* |
4 |
25/127 |
||
|
Maize starch |
3 |
20* |
3 |
—/— |
||
|
Guatemala |
Tortillas |
Aug–Sep 1995 |
|
|
|
|
|
Santa Maria de Jesus |
FumonisinB1 |
38 |
400* |
31 |
850/6500 |
|
|
Hydrolysed fumonisinB1 |
38 |
400* |
12 |
26 100/ 185 100 |
||
|
Tortillas |
Aug–Sep 1995 |
|
|
|
|
|
|
Patzicia |
FumonisinB1 |
35 |
400* |
24 |
2200/11 600 |
|
|
Hydrolysed fumonisinB1 |
35 |
400* |
24 |
5700/31 700 |
||
|
Nixtamal |
|
|
|
|
|
|
|
Santa Maria |
FumonisinB1 |
46 |
400* |
0 |
13600/77200 |
|
|
de Jesus |
Hydrolysed fumonisinB1 |
46 |
400* |
46 |
—/— |
|
|
Patzicia |
FumonisinB1 |
50 |
400* |
33 |
4400(?)/5000 |
|
|
Hydrolysed fumonisinB1 |
50 |
400* |
50 |
—/— |
||
|
Mexico |
Tortillas |
No data |
7 |
10* |
0 |
601/1070 |
|
Hydrolysed fumonisinB1 |
7 |
10* |
2 |
16/50 |
||
|
Masa |
No data |
2 |
10* |
0 |
1195/1800 |
|
|
Hydrolysed fumonisinB1 |
2 |
10* |
1 |
50/100 |
||
|
Uruguay |
Polenta |
1/1995–4/1996 |
8 |
50* |
5 |
101/427 |
|
Maize starch |
4 |
50* |
4 |
—/— |
||
|
Popcorn |
2 |
50* |
1 |
100/199 |
||
|
Snacks |
5 |
50* |
3 |
93/314 |
||
|
Breakfast cereal |
3 |
50* |
2 |
73/218 |
||
|
Canned maize |
4 |
50* |
4 |
—/— |
||
|
Frozen maize |
2 |
50* |
1 |
78/155 |
||
|
Maize grits |
1 |
50* |
1 |
—/— |
||
|
Europe |
||||||
|
Czech Republic |
(Total fumonisins by ELISA; * mean calculated with samples <LOQ taken as 4.5 µg/kg) |
|||||
|
Maize-extruded bread |
Autumn 1995– |
35 |
9 |
5 |
270*/1808 |
|
|
Maize-extruded products |
Spring 1996 |
26 |
9 |
0 |
301*/1178 |
|
|
Maize flour |
22 |
9 |
0 |
187*/487 |
||
|
Maize instant porridge |
19 |
9 |
1 |
124*/788 |
||
|
Maize paste |
11 |
9 |
5 |
75*/511 |
||
|
Polenta |
7 |
9 |
1 |
559*/1243 |
||
|
Denmark |
Cornflakes |
1996 |
10 |
1* |
4 |
110/1030 |
|
Maize flour |
8 |
1* |
2 |
33/86 |
||
|
Maize grits |
4 |
1* |
3 |
2/7 |
||
|
Maize starch |
6 |
1* |
6 |
0/0 |
||
|
Corn-on-the-cob |
4 |
1* |
4 |
0/0 |
||
|
Maize snacks |
10 |
1* |
4 |
16/65 |
||
|
Polenta |
1 |
1* |
0 |
84/84 |
||
|
Popcorn |
9 |
1* |
5 |
54/474 |
||
|
Sweet maize |
16 |
1* |
16 |
0/0 |
||
|
Germany |
Maize grits |
No data |
6 |
0.8 |
0 |
92/202 |
|
Maize meal |
2 |
0.8 |
0 |
86/138 |
||
|
Sweet maize |
6 |
0.8 |
6 |
—/— |
||
|
Popcorn |
2 |
0.8 |
0 |
<1/— |
||
|
Cornflakes |
2 |
0.8 |
0 |
<2/— |
||
|
Infant foods |
4 |
0.8 |
2 |
9/19 |
||
|
Maize grits |
No data |
11 |
20 |
1 |
208/712 |
|
|
Maize meal |
No data |
6 |
20 |
0 |
2272/7647 |
|
|
Italy |
Polenta |
No data |
20 |
20* |
0 |
1380/3760 |
|
Netherlands |
(Corrected for analytical recovery) |
|||||
|
Minimally treated maize: |
||||||
|
For bread |
1996 |
19 |
25 |
11 |
98/380 |
|
|
For popcorn |
1995 |
|
10 |
25 |
9 |
|
|
For flour |
1995 |
7 |
25 |
2 |
41/90 |
|
|
Polenta |
1995 |
3 |
25 |
1 |
No data/40 |
|
|
Processed maize: |
||||||
|
Tostada |
1995 |
1 |
25 |
1 |
—/— |
|
|
Canned |
1995 |
6 |
25 |
6 |
—/— |
|
|
Starch |
1995 |
5 |
25 |
5 |
—/— |
|
|
Bread |
1995 |
2 |
25 |
1 |
40/80 |
|
|
Popped |
1995 |
5 |
25 |
2 |
no data/300 |
|
|
Flour mixes |
1995 |
6 |
25 |
6 |
—/— |
|
|
Maize chips |
1995 |
9 |
25 |
6 |
no data/160 |
|
|
Cornflakes |
1995 |
5 |
25 |
4 |
286/1430 |
|
|
Sweden |
Cornflakes |
No data |
6 |
1* |
3 |
14/34 |
|
Fresh, frozen, canned maize |
13 |
1* |
13 |
—/— |
||
|
Maize chips |
7 |
1* |
0 |
143/252 |
||
|
Maize grits, flour, gruel |
No data |
8 |
1* |
4 |
16/52 |
|
|
Popcorn |
No data |
8 |
1* |
2 |
70/393 |
|
|
United Kingdom |
(Total fumonisins by HPLC; corrected for recovery) |
|||||
|
Polenta |
1994–95 |
20 |
10* |
4 |
529/2124 |
|
|
Breakfast cereals |
50 |
10* |
38 |
22/194 |
||
|
Popcorn, ready-made |
9 |
10* |
9 |
—/— |
||
|
Popcorn |
13 |
10* |
7 |
76/784 |
||
|
Maize snacks |
40 |
10* |
9 |
42/220 |
||
|
Tortilla, taco, enchilada |
20 |
10* |
14 |
6/31 |
||
|
Maize thickener |
21 |
10* |
17 |
15/110 |
||
|
Maize syrup |
11 |
10* |
11 |
—/— |
||
|
Maize oil |
20 |
10* |
20 |
—/— |
||
|
Corn-on- the-cob |
20 |
10* |
20 |
—/— |
||
|
Sweet maize |
22 |
10* |
21 |
<1/11 |
||
|
Africa |
||||||
|
Botswana |
Maize meal |
1994 |
4 |
20* |
0 |
185/255 |
|
South Africa |
Maize meal |
1994 |
2 |
20* |
0 |
65/70 |
|
Samp |
1994–95 |
13 |
20* |
No data |
461/1994 |
|
|
Maize rice |
11 |
20* |
No data |
295/991 |
||
|
Maize grits |
5 |
20* |
No data |
554/1800 |
||
|
Maize flour |
2 |
20* |
No data |
532/549 |
||
|
Super meal |
25 |
20* |
No data |
134/871 |
||
|
Special meal |
36 |
20* |
No data |
378/1400 |
||
|
Sifted meal |
47 |
20* |
No data |
562/4482 |
||
|
Unsifted meal |
19 |
20* |
No data |
827/3929 |
||
|
Germ meal |
8 |
20* |
No data |
437/1288 |
||
|
Maize bran |
32 |
20* |
No data |
1324/8180 |
||
|
Screenings |
7 |
20* |
No data |
6651/15716 |
||
|
Zambia |
Maize meal |
1994 |
1 |
20* |
0 |
740/740 |
|
Zimbabwe |
Maize meal |
1994 |
4 |
20* |
0 |
625/1910 |
|
Asia |
||||||
|
China (Linqu County, Shandong) |
Maize meal |
1996 |
14 |
500* |
7 |
1064/8800 |
|
Batter |
1996 |
32 |
500* |
26 |
494/7200 |
|
|
Pancake |
1996 |
16 |
500* |
10 |
400/2200 |
|
|
Nepal |
(Total fumonisins by HPLC) |
|||||
|
Maize flour |
Feb–Jul |
8 |
100* |
2 |
600/2400 |
|
|
Cornflakes |
1997 |
2 |
100* |
2 |
—/— |
|
|
Taiwan |
Maize snacks |
8/1994–12/1995 |
78 |
40* |
52 |
152/2395 |
|
Sweet canned maize |
24 |
40* |
12 |
200/1089 |
||
|
Popcorn |
22 |
40* |
15 |
111/1003 |
||
|
Cornflakes |
17 |
40* |
13 |
117/1281 |
||
|
Maize grits |
4 |
40* |
4 |
—/— |
||
|
Maize flour |
2 |
40* |
1 |
304/608 |
||
|
Country/ Region |
Commodity |
Year/ Season |
90th %ile |
n > 1000µg/kg |
n > 2000µg/kg |
References |
Sampling procedure |
|
Unprocessed maize |
|||||||
|
North America |
|||||||
|
Canada |
Domestic |
1993 |
< 200 |
3 |
0 |
Miller et al., 1995 |
Frequent small samples from grain handling equipment. Samples Pooled, mixed and 2.2-kg subsample ground |
|
USA |
Iowa State |
1988 |
No data |
No data |
No data |
Murphy et al., 1993; A = Ross et al., 1991 |
Random samples from trucks at grain elevators; 400-g subsample ground before analysis |
|
1989 |
No data |
No data |
No data |
||||
|
1990 |
No data |
No data |
No data |
||||
|
1991 |
No data |
No data |
No data |
||||
|
1992 |
No data |
No data |
0 |
Rice & Ross, 1994; L.G. Rice, P.F. Ross, USDA Veterinary Services Laboratory; P.A. Murphy, Iowa State University |
|||
|
1993 |
No data |
No data |
0 |
||||
|
1994 |
No data |
No data |
No data |
||||
|
1995 |
No data |
No data |
No data |
||||
|
1996 |
No data |
No data |
No data |
||||
|
South and Central America |
|||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
499 |
0 |
0 |
GEMS/Food programme |
Statistically based and representative for part of country |
|
Domestic |
Jan–Oct 1999 |
8558 |
132 |
89 |
|||
|
Domestic |
May–Jun 1999 |
1940 |
10 |
6 |
|||
|
Domestic |
Jan–Aug 2000 |
1647 |
9 |
3 |
|||
|
Rice, imported |
Nov–Dec 1999 |
No data |
0 |
0 |
GEMS/Food programme |
Not statistically based, samples from whole country |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
— |
— |
— |
GEMS/Food programme |
Not statistically based, samples from part of country |
|
|
Rice, polished, domestic |
Apr–Jun 1999 |
— |
— |
— |
|||
|
Brazil |
Domestic |
1997–98 |
No data |
|
|
GEMS/Food programme |
Statistically based, representative for part of country |
|
Domestic |
1994-95 |
No data |
|
|
|||
|
Domestic |
No data |
No data |
|
|
|||
|
Domestic |
4/1995–4/1996 |
No data |
|
|
|||
|
Domestic |
4/1995–4/1996 |
No data |
|
|
|||
|
Domestic |
1998 |
No data |
172 |
48>3000 |
Preis & Vargas, 2000; |
Maize collected in various regions |
|
|
Domestic |
1990–91 |
8750 |
46 |
42 |
Hirooka et al., 1996; |
Samples collected from silos and warehouses, mixed, reduced to 2 kg, and ground |
|
|
Colombia |
Domestic |
Feb–Aug 1999 |
No data |
No data |
No data |
Perilla & Diaz, 1998; |
Samples (minimum, 500 g) purchased at random from most popular retail outlets, ground and sub-sampled in grinding–sub-sampling mill (Romer series II) |
|
Costa Rica |
Domestic |
1992–93 |
5350 |
58 |
39 |
Viquez et al., 1996; |
Sampling by agricultural agency: representative daily samples (5 kg) reduced to 8-kg sub-sample/week, which was milled and divided to yield 2 kg/week; over 3 weeks, 6-kg sample reduced to 1 kg |
|
Honduras |
Domestic |
10/1992, |
2248 |
11 |
6 |
Julian et al., 1995; |
Samples collected by systematic sampling of ears in field (2 transects/field, ears taken at 10-m intervals (total, 25 ears per sample) or from farm stores (25 ears from different areas of store); samples ground, mixed in vane mixer, and test portions taken |
|
Uruguay |
Maize kernels |
1995–96 |
3025 |
7 |
3 |
Pineiro et al., 1997; A = Scott & Lawrence, 1996; Sydenham et al., 1996c |
Samples purchased from local shops; 1-kg sub-samples ground and mixed well |
|
Venezuela |
Domestic |
Oct 1993 |
2626 |
11 |
5 |
Medina-Martinez & Martinez, 2000; A = Stack & Eppley, 1992; |
Yellow maize collected as single random purchases from retail outlets in Caracas. White maize withdrawn from trucks at grain elevators; sub-samples prepared by grinding in a disk mill (Quaker model 4E) |
|
Europe |
|||||||
|
Croatia |
(Fumonisins B1 and B2) |
||||||
|
Domestic |
1996 |
No data |
7 |
5>5000 |
Jurjevic et al., 1999 |
Samples (whole ear, 1.5–2 kg) collected randomly from several farms in 14 counties. 270–320 g stored and ground beforre analysis |
|
|
Domestic |
1997 |
No data |
1 |
0>5000 |
|||
|
Hungary |
Non-mouldy |
|
No data |
0 |
0 |
Fazekas et al., 1996; A = Shephard et al., 1990 |
Storage samples from private producers and feed mixing plants: harvested autumn 1993; collected 12/93–2/94 |
|
Mouldy |
|
No data |
No data |
No data |
|||
|
Random |
1994 |
No data |
No data |
No data |
Fazekas et al., 1996; |
Harvest samples: random samples of 50 ears taken on each maize field |
|
|
Mouldy |
1994 |
No data |
No data |
No data |
|||
|
Italy |
Visibly mouldy |
1994 |
180 000 |
No data |
No data |
Ritieni et al., 1997; |
Ears collected before harvest from different fields |
|
Netherlands |
Imported |
1994–96 |
1218 |
11 |
3 |
de Nijs et al., 1998b; |
Kernels obtained from bulk ship loads or trailers; samples ground before analysis |
|
Spain |
Domestic |
1994–96 |
No data |
No data |
No data |
Castella et al., 1999; |
Samples (I kg representative) obtained from agricultural cooperatives and factories; sub-sampled into 200-g aliquot for analysis |
|
Sweden |
Imported |
Jan–Dec 1996 |
159 |
No data |
No data |
GEMS/Food programme |
Statistically based, representative for part of country |
|
United Kingdom |
(Data corrected for analytical recovery) |
||||||
|
Imported |
6/1998–4/1999 |
No data |
39 |
0>5000 |
Scudamore & Patel, 2000; |
Manchester: 20 incremental samples (0.5 kg) taken from conveyer going to mill over 2 h and bulked. |
|
|
Imported |
6/1998–4/1999 |
|
|
|
|
|
|
|
|
|
No data |
48 |
1>5000 |
Scudamore & Patel, 2000; |
|
|
|
Africa |
|
||||||
|
Botswana |
Domestic |
1/1996–12/1997 |
No data |
No data |
0 |
Siame et al., 1998; |
Samples (2–5 kg) collected from storage depots or purchased from retail outlets; ground and mixed beforealiquots taken for analysis |
|
Burundi |
Domestic |
No data |
No data |
No data |
No data |
Munimbazi & Bullerman, 1996; |
Samples collected from markets |
|
Kenya |
Smallholder farms |
1996 |
No data |
10 |
No data |
Kedera et al., 1999; Sydenham et al., 1992b |
Shelled maize kernels (0.5–1.0 kg) collected from randomly selected farms. 50-g sample ground, 5 g extracted |
|
Kenya |
Kernels |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground, and mixed before aliquots taken for analysis |
|
No data |
0 |
0 |
|||||
|
No data |
0 |
0 |
|||||
|
South Africa |
White maize |
1994–95 |
No data |
No data |
No data |
Rava, 1996; |
Samples collected from mills throughout country and ground before analysis |
|
Yellow maize |
1994–95 |
No data |
No data |
No data |
|||
|
White grade 1 |
1993 |
No data |
No data |
No data |
Rava et al., 1996; |
Representative samples (3 kg) collected at harvest from silos in main production zones; 500-g sample obtained from riffle splitter and ground |
|
|
White grade 2 |
1993 |
No data |
No data |
No data |
|||
|
White grade 3 |
1993 |
No data |
No data |
0 |
|||
|
Yellow grade 1 |
1993 |
No data |
No data |
No data |
|||
|
Yellow grade 2 |
1993 |
No data |
No data |
No data |
|||
|
Yellow grade 3 |
1993 |
No data |
No data |
No data |
|||
|
Tanzania |
Kernels |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground, and mixed before aliquots taken for analysis |
|
|
|
No data |
0 |
0 |
|||
|
|
|
No data |
0 |
0 |
|||
|
Asia |
|||||||
|
China |
Maize |
Spring 1996 |
25 260 |
91 |
77>5000 |
China |
No data |
|
Rice |
|
5990 |
25 |
17>5000 |
|
|
|
|
China |
1996 |
1100 |
2 |
2 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen and 5-g portions cut for analysis |
|
|
China |
(Total fumonisins by ELISA) |
||||||
|
Autumn 1995 |
No data |
No data |
No data |
Zhang et al., 1997 |
Samples collected from farmers 10/1995–3/1996 and milled |
||
|
Fanxian & Yanqing counties |
Autumn 1995 |
No data |
No data |
0 |
|||
|
Linxian County, Henan Province |
1994 |
No data |
No data |
No data |
Gao & Yoshizawa, 1997; |
Samples from 1994 harvest collected from peasant families during Jan–Feb 1995 |
|
|
Shangqiu County, Henan Province |
1994 |
No data |
No data |
No data |
|||
|
Haimen, Jiangsu County |
Apr–Jul 1993 |
11 074 |
26 |
20 |
Ueno et al., 1997; |
Kernels collected randomly from agricultural stocks. 25-g sample milled and 5 g extracted |
|
|
Penlai, Shandong Province |
Apr–Jul 1993 |
704 |
4 |
1 |
|||
|
India |
Domestic |
1995 |
No data |
0 |
0 |
Bhat et al., 1997; |
Samples (0.25–5 kg) collected from households |
|
Domestic |
No data |
No data |
9 |
7 |
Shetty & Bhat, 1997; |
Samples collected from households and retail shops |
|
|
Indonesia |
Domestic |
Nov 1995 |
1450 |
6 |
1 |
Ali et al., 1998; A = Yoshizawa et al., 1994, 1996 |
Samples ground and aliquots taken from 200 g |
|
Domestic |
1992–94 |
No data |
No data |
No data |
Yamashita et al., 1995; |
Samples collected at random from stores of wholesalers, retailers, university farms, and local farmers; ground and stored |
|
|
Iran |
Mazandaran |
Sept 1998 |
3360 |
11 |
4 |
Shephard et al., 2000; |
Farmers' maize lots collected at random from consignments sold to Iranian Agriculture Office; total sample ground |
|
Iran |
Isfahan |
Oct 1998 |
559 |
0 |
0 |
Shephard et al., 2000; |
Maize ears bought at different periods from local retail market; total sample ground |
|
Korea, Republic of |
Mouldy |
Nov 1997 |
No data |
No data |
No data |
Sohn et al., 1999; |
Samples collected from households and milled |
|
Healthy |
Nov 1997 |
No data |
No data |
No data |
|||
|
Nepal |
(Total fumonisins by HPLC) |
||||||
|
Domestic |
Feb–Jul 1997 |
No data |
No data |
No data |
Desjardin et al., 2000 |
Samples (0.25–0.5 kg) collected from farms and markets in 10 districts; 100 g ground and 10 g extracted |
|
|
Philippines |
(Total fumonisins by ELISA) |
||||||
|
Domestic |
Jun–Nov 1995 |
No data |
No data |
No data |
Bryden et al., 1998 |
Samples (1 kg) collected from major storage sites, subsampled, and ground |
|
|
Domestic |
1992–94 |
No data |
No data |
0 |
Yamashita et al., 1995; |
Samples collected at random from stores of wholesalers, retailers, university farms, and local farmers; ground and stored |
|
|
Philippines |
Seed maize |
1992–94 |
No data |
0 |
0 |
Yoshizawa & Yamashita, 1995; |
At least 100 g collected, 50 g ground, and 20 g extracted |
|
Feed maize |
1992-1994 |
No data |
No data |
0 |
|||
|
Food maize |
1992-1994 |
No data |
0 |
0 |
|||
|
Taiwan |
Domestic |
1996–97 |
No data |
No data |
0 |
Tseng & Liu, 1999; |
Samples (1 kg) collected from 8 districts; sub- |
|
Thailand |
Visibly mouldy human food |
1992 |
No data |
3 |
1 |
Yoshizawa et al., 1996; |
Samples collected before harvest; random 100-g aliquots analysed; 50 g milled and 20-g sub-sample extracted |
|
Domestic |
1992–94 |
No data |
No data |
No data |
Yamashita et al., 1995; |
Samples collected at random from stores of wholesalers, retailers, university farms, and local farmers; ground and stored |
|
|
Viet Nam |
(Total fumonisins by ELISA) |
||||||
|
Domestic |
Jun–Nov 1995 |
No data |
No data |
No data |
Bryden et al., 1998 |
Samples (1 kg) collected from major storage sites, subsampled, and ground |
|
|
Viet Nam |
Maize, feed |
1993 |
1506 |
4 |
1 |
Wang et al., 1995; |
Random samples from various locations. 25 g milled, and 5 g extracted |
|
Maize powder, feed |
1993 |
1179 |
4 |
0 |
|||
|
Australia |
|||||||
|
Australia |
(Total fumonisins by ELISA) |
||||||
|
|
Domestic |
Jun–Nov 1995 |
No data |
No data |
No data |
Bryden et al., 1998 |
Samples (1 kg) collected from major storage sites, subsampled, and ground |
|
Processed maize-based human food |
|||||||
|
North America |
|||||||
|
Canada |
(* number and mean of positive samples) |
||||||
|
Maize kernel, meal, flour |
1996–97 |
No data |
0 |
0 |
Canada |
No data |
|
|
Maize snacks (alkali-processed) |
|
No data |
0 |
0 |
|
No data |
|
|
Maize snacks (non-alkali-processed) |
|
No data |
0 |
0 |
|
No data |
|
|
Canned |
|
No data |
0 |
0 |
|
No data |
|
|
Frozen |
|
No data |
0 |
0 |
|
No data |
|
|
Maize bread |
|
No data |
0 |
0 |
|
No data |
|
|
Maize-based breakfast cereals |
|
No data |
0 |
0 |
|
No data |
|
|
Fresh tortillas |
|
No data |
0 |
0 |
|
No data |
|
|
Maize-based infant foods: |
1997–98 |
|
|
|
Canada |
|
|
|
Mixed cereals |
|
No data |
0 |
0 |
|
No data |
|
|
Cereal with formula |
|
No data |
0 |
0 |
|
No data |
|
|
Creamed maize or maize / vegetables |
|
No data |
0 |
0 |
|
No data |
|
|
Infant cereals: 1998–99 |
|
|
|
|
Canada |
|
|
|
Mixed cereals with fruit / formula |
|
No data |
0 |
0 |
|
No data |
|
|
Rice-based |
|
No data |
0 |
0 |
|
No data |
|
|
Soya-based |
|
No data |
0 |
0 |
|
No data |
|
|
Mixed cereals |
|
No data |
0 |
0 |
|
No data |
|
|
Tortilla, nacho, maize chips, taco shells |
1996 |
65 |
0 |
0 |
Scott & Lawrence, 1996 |
Samples purchased from retail outlets, mostly in Ottawa; sample sizes125–900 g; ground |
|
|
Hydrolysed fumonisin B1 |
|
<10 |
0 |
0 |
|
|
|
|
Maize tortilla, dried |
|
425 |
0 |
0 |
|
|
|
|
Hydrolysed fumonisin B1 |
|
51 |
0 |
0 |
|
|
|
|
USA |
Maize meal, degermed |
1997 |
300 |
1 |
0 |
USA |
Sampled 1998 |
|
Maize meal, partly degermed |
|
550 |
1 |
0 |
|
|
|
|
Maize meal, whole grain |
|
2852 |
10 |
6 |
|
|
|
|
Maize meal, degermed |
1998 |
260 |
21 |
8 |
USA |
Sampled 1999 |
|
|
Maize meal, partly degermed |
1998 |
2050 |
5 |
2 |
USA |
Sampled 1999 |
|
|
Maize meal, whole grain |
|
2414 |
11 |
5 |
|
|
|
|
Canned maize |
Spring 1993 |
No data |
0 |
0 |
Trucksess et al., 1995 |
Samples collected by 10 FDA district offices from retail establishments representing, where possible, different manufacturers and distributors |
|
|
Frozen maize |
|
No data |
0 |
0 |
|||
|
Maize meal |
No data |
No data |
No data |
No data |
Castelo et al., 1998b; |
Samples purchased in Maryland, Nebraska, and Arizona; ground before analysis |
|
|
Muffin mix |
No data |
No data |
0 |
0 |
|||
|
Maize bread |
No data |
No data |
No data |
0 |
|||
|
Tortilla chips |
No data |
No data |
No data |
0 |
|||
|
Maize tortilla |
No data |
No data |
0 |
0 |
|||
|
Cornflakes |
No data |
No data |
0 |
0 |
|||
|
Maize starch |
No data |
No data |
0 |
0 |
|||
|
Popcorn |
No data |
No data |
0 |
0 |
|||
|
Infant foods |
No data |
No data |
0 |
0 |
|||
|
(Total fumonisins by HPLC) |
|||||||
|
Maize, |
1994 |
No data |
No data |
0 |
USA |
No data |
|
|
shelled |
1995 |
No data |
No data |
No data |
|
No data |
|
|
|
1996 |
No data |
No data |
No data |
|
No data |
|
|
|
1997 |
No data |
No data |
No data |
|
No data |
|
|
|
1998 |
No data |
No data |
No data |
|
No data |
|
|
Maize meal |
1994 |
No data |
No data |
0 |
|
No data |
|
|
|
1995 |
No data |
No data |
No data |
|
No data |
|
|
Maize meal |
1996 |
No data |
No data |
No data |
USA |
No data |
|
|
|
1997 |
No data |
No data |
0 |
|
No data |
|
|
|
1998 |
No data |
No data |
0 |
|
No data |
|
|
Maize flour |
1994 |
No data |
0 |
0 |
USA |
No data |
|
|
|
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
0 |
0 |
|
No data |
|
|
|
1997 |
No data |
No data |
0 |
|
No data |
|
|
|
1998 |
No data |
No data |
0 |
|
No data |
|
|
Maize grits |
1994 |
No data |
0 |
0 |
USA |
No data |
|
|
|
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
0 |
0 |
|
No data |
|
|
|
1997 |
No data |
0 |
0 |
|
No data |
|
|
|
1998 |
No data |
— |
— |
|
No data |
|
|
Hominy grits |
1994 |
No data |
— |
— |
USA |
No data |
|
|
|
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
0 |
0 |
|
No data |
|
|
|
1997 |
No data |
0 |
0 |
|
No data |
|
|
|
1998 |
No data |
— |
— |
|
No data |
|
|
Maize chips, |
1994 |
No data |
0 |
0 |
USA |
No data |
|
|
tortilla |
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
— |
— |
|
No data |
|
|
|
1997 |
No data |
0 |
0 |
|
No data |
|
|
|
1998 |
No data |
No data |
0 |
|
No data |
|
|
Maize muffin |
1994 |
No data |
— |
— |
USA |
No data |
|
|
mix |
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
— |
— |
|
No data |
|
|
|
1997 |
No data |
0 |
0 |
|
No data |
|
|
|
1998 |
No data |
0 |
0 |
USA |
No data |
|
|
Cornflakes, |
1994 |
No data |
-- |
— |
USA |
No data |
|
|
dry cereals |
1995 |
No data |
— |
— |
|
No data |
|
|
|
1996 |
No data |
— |
— |
|
No data |
|
|
|
1997 |
No data |
0 |
0 |
|
No data |
|
|
|
1998 |
No data |
— |
— |
|
No data |
|
|
Starch |
1994 |
No data |
— |
— |
USA |
No data |
|
|
|
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
— |
— |
|
No data |
|
|
|
1997 |
No data |
— |
— |
|
No data |
|
|
|
1998 |
No data |
— |
— |
|
No data |
|
|
Popcorn |
1994 |
No data |
0 |
0 |
USA |
No data |
|
|
|
1995 |
No data |
0 |
0 |
|
No data |
|
|
|
1996 |
No data |
0 |
0 |
|
No data |
|
|
|
1997 |
No data |
No data |
0 |
|
No data |
|
|
|
1998 |
No data |
0 |
0 |
|
No data |
|
|
Tortillas |
No data |
326 |
0 |
0 |
Stack, 1998; |
Samples from Texas–Mexico border |
|
|
|
Hydrolysed fumonisin B1 |
104 |
0 |
0 |
|||
|
Masa |
No data |
418 |
0 |
0 |
|||
|
|
Hydrolysed fumonisin B1 |
127 |
0 |
0 |
|||
|
Starch |
No data |
No data |
0 |
0 |
Maragos et al., 1997; |
No data |
|
|
Grits |
No data |
No data |
No data |
No data |
|||
|
Masa |
No data |
No data |
0 |
0 |
|||
|
Popcorn |
Feb-May 1993/ |
<40 |
0 |
0 |
Rumbeiha & Oehme, 1997; |
Purchased in Manhattan, Kansas; 500 g blended and 25 g extracted |
|
|
Breakfast cereals |
Feb–May 1993 |
— |
— |
— |
Rumbeiha & Oehme, 1997; |
Purchased in Manhattan, Kansas; 500 g blended and 25 g extracted |
|
|
Snacks |
|
— |
— |
— |
|||
|
Maize flour |
|
168 |
— |
— |
|||
|
Tortilla shells |
|
— |
— |
— |
|||
|
Sweet maize |
8 |
— |
— |
|
|||
|
Maize oil |
|
— |
— |
— |
|||
|
Total fumonisins |
1990–94 |
251-500 |
501-1000 |
>1000 |
Pohland, 1996 |
No data |
|
|
Canned maize |
|
|
|
|
|||
|
Frozen maize |
|
1 |
|
|
|||
|
Maize bran cereal |
|
3 |
|
|
|||
|
Maize bread mix |
|
1 |
3 |
4 |
|||
|
Maize cereal |
|
|
|
|
|||
|
Maize chips |
|
|
|
|
|||
|
Cornflakes |
|
|
|
|
|||
|
Maize grits |
|
9 |
4 |
|
|||
|
Maize hominy |
|
|
|
|
|||
|
Maize meal |
|
15 |
19 |
28 |
|||
|
Maize muffin mix |
|
1 |
1 |
|
|||
|
Maize pops cereal |
|
|
|
|
|||
|
Maize tortillas |
|
|
|
|
|||
|
Maize tortilla chips |
1990–94 |
|
|
|
Pohland, 1996 |
No data |
|
|
Shelled maize |
|
8 |
7 |
1 |
|||
|
Maize starch |
|
|
|
|
|||
|
Fibre cereal |
|
|
|
|
|||
|
Popcorn |
|
1 |
|
|
|||
|
Spoon bread mix |
|
|
1 |
|
|||
|
South and Central America |
|||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
540 |
0 |
0 |
GEMS/Food programme |
Statistically based and representative for part of country |
|
Popcorn |
May–Jun 1999/ |
4311 |
6 |
4 |
|||
|
Maize flour |
10/1996–1/1997 |
638 |
1 |
0 |
Hennigen et al., 2000; |
Samples (1 kg) purchased from food shops and supermarkets in Buenos Aires, ground and well mixed by hand before analysis |
|
|
Maize grits |
10/1996–1/1997 |
373 |
0 |
0 |
|||
|
Maize flour |
Jan 1998 |
1435 |
5 |
1 |
|||
|
Maize grits |
Jan 1998 |
No data |
0 |
0 |
|||
|
Maize meal |
1997 |
1357 |
2 |
0 |
Solovey et al., 1999; |
Randomly purchased from comercial outlets; total sample ground |
|
|
Cornflakes |
1997 |
32 |
0 |
0 |
|||
|
Brazil |
Canned sweet maize |
Mar–May 1999 |
No data |
0 |
0 |
Machinski & Valente Soares, 2000; |
Samples (minimum 500 g) purchased from retail stores in Campinas and ground, homogenized, or both |
|
Cornflakes |
|
No data |
0 |
0 |
|||
|
Maize flour |
|
No data |
No data |
0 |
|||
|
Maize flour, baby cereal |
|
No data |
0 |
0 |
|||
|
Maize grits |
|
No data |
No data |
0 |
|||
|
Maize meal |
|
No data |
No data |
No data |
|||
|
Corn-on-the-cob |
|
— |
— |
— |
— |
||
|
Curau |
|
— |
— |
— |
|
||
|
Maize, degermed |
|
No data |
No data |
No data |
|
||
|
Pamonha |
|
— |
— |
— |
|
||
|
Popcorn |
|
330/1720 |
No data |
No data |
0 |
||
|
Pre-cooked maize meal |
|
No data |
No data |
0 |
|
||
|
Colombia |
Pre-cooked maize meal |
Feb-Aug 1998 |
No data |
0 |
0 |
Perilla & Diaz, 1998; |
Samples (minimum 500 g) purchased at random from most popular retail stores; ground and subsampled in grinding–subsampling mill (Romer series II) |
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Maize meal |
|
No data |
0 |
0 |
|||
|
"Arepas" |
|
No data |
0 |
0 |
|||
|
Maize snacks |
|
No data |
0 |
0 |
|||
|
Maize starch |
|
— |
— |
— |
|||
|
Guatemala |
Tortillas |
Aug–Sep 1995 |
|
|
|
Meredith et al., 1999 |
Samples collected from households, frozen, freeze-dried, and ground; 5–10 g extracted |
|
Santa Maria de Jesus |
FumonisinB1 |
No data |
No data |
No data |
|||
|
Hydrolysed fumonisinB1 |
No data |
No data |
No data |
||||
|
Tortillas |
Aug–Sep 1995 |
|
|
|
Meredith et al., 1999 |
Samples collected from households, frozen, freeze-dried, and ground; 5–10 g extracted |
|
|
Patzicia |
FumonisinB1 |
No data |
No data |
No data |
|||
|
Hydrolysed fumonisinB1 |
No data |
No data |
No data |
||||
|
Nixtamal |
|
|
|
|
|
|
|
|
Santa Maria |
FumonisinB1 |
No data |
No data |
No data |
|
|
|
|
de Jesus |
Hydrolysed fumonisinB1 |
— |
— |
— |
|
|
|
|
Patzicia |
FumonisinB1 |
No data |
No data |
No data |
|
|
|
|
Hydrolysed fumonisinB1 |
— |
— |
— |
|
|
||
|
Mexico |
Tortillas |
No data |
No data |
1 |
0 |
Dombrink-Kurtzman & Dvorak, 1999; A = Sydenham et al., 1995; Bennett & Richard, 1994 |
No data |
|
Hydrolysed fumonisinB1 |
No data |
0 |
0 |
||||
|
Masa |
No data |
No data |
1 |
0 |
|||
|
Hydrolysed fumonisinB1 |
No data |
0 |
0 |
||||
|
Uruguay |
Polenta |
1/1995–4/1996 |
322 |
0 |
0 |
Pineiro et al., 1997; |
Samples purchased from local shops; 1-kg subsamples ground and mixed well |
|
Maize starch |
|
— |
— |
— |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Snacks |
|
249 |
0 |
0 |
|||
|
Breakfast cereal |
|
No data |
0 |
0 |
|||
|
Canned maize |
|
— |
— |
— |
|||
|
Frozen maize |
|
No data |
0 |
0 |
|||
|
Maize grits |
|
— |
— |
— |
|||
|
Europe |
|||||||
|
Czech Republic |
(Total fumonisins by ELISA; * mean calculated with samples <LOQ taken as 4.5 µg/kg) |
|
|
|
|
|
|
|
Maize-extruded bread |
Autumn 1995– |
910 |
No data |
0 |
Ostry & Ruprich, 1998 |
Samples purchased in shopping network in 13 areas |
|
|
Maize-extruded products |
Spring 1996 |
1062 |
No data |
0 |
|
|
|
|
Maize flour |
|
355 |
0 |
0 |
|
|
|
|
Maize instant porridge |
|
273 |
0 |
0 |
|
|
|
|
Maize paste |
|
111 |
0 |
0 |
|
|
|
|
Polenta |
|
1221 |
No data |
0 |
|
|
|
|
Denmark |
Cornflakes |
1996 |
No data |
No data |
No data |
Petersen, 2000; A = Sydenham et al., 1992b |
Samples purchased in retail shops, homogenised in a mixer before analysis |
|
Maize flour |
|
No data |
No data |
No data |
|||
|
Maize grits |
|
No data |
0 |
0 |
|||
|
Maize starch |
|
No data |
0 |
0 |
|||
|
Corn-on-the-cob |
|
No data |
0 |
0 |
|||
|
Maize snacks |
|
No data |
0 |
0 |
|||
|
Polenta |
|
No data |
0 |
0 |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Sweet maize |
|
— |
0 |
|
|||
|
Germany |
Maize grits |
No data |
No data |
0 |
0 |
Lukacs et al., 1996; |
Samples from market |
|
Maize meal |
|
No data |
0 |
0 |
|||
|
Sweet maize |
|
— |
— |
— |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Cornflakes |
|
No data |
0 |
0 |
|||
|
Infant foods |
|
No data |
0 |
0 |
|||
|
Maize grits |
No data |
519 |
0 |
0 |
Hartl et al., 1999 |
Commercial samples blended; 5 g extracted |
|
|
Maize meal |
No data |
5343 |
4 |
2 |
|||
|
Italy |
Polenta |
No data |
3595 |
9 |
4 |
Pascale et al., 1995; |
Samples purchased in local retail market in Apulia |
|
Netherlands |
(Corrected for analytical recovery) |
|
|
|
|
de Nijs et al., 1998b; Shephard et al., 1990 |
Samples purchased in local retail stores and ground when necessary |
|
Minimally treated maize: |
|
|
|
|
|||
|
For bread 1996 |
|
246 |
0 |
|
0 |
||
|
For popcorn |
1995 |
11/110 |
11 |
0 |
0 |
||
|
For flour |
1995 |
72 |
0 |
0 |
|
||
|
Polenta |
1995 |
No data |
0 |
0 |
|
||
|
Processed maize: |
|
|
|
|
|
||
|
Tostada |
1995 |
— |
— |
— |
|
||
|
Canned |
1995 |
— |
— |
— |
|
||
|
Starch |
1995 |
— |
— |
— |
|
||
|
Bread |
1995 |
No data |
0 |
0 |
|
||
|
Popped |
1995 |
No data |
0 |
0 |
|
||
|
Flour mixes |
1995 |
— |
— |
— |
|
||
|
Maize chips |
1995 |
No data |
0 |
0 |
|
||
|
Cornflakes |
1995 |
No data |
0 |
0 |
|
||
|
Sweden |
Cornflakes |
No data |
No data |
0 |
0 |
Moller & Gustavsson, 2000 |
Samples taken randomly |
|
Fresh, frozen, canned maize |
|
— |
— |
— |
|||
|
Maize chips |
|
No data |
0 |
0 |
|||
|
Maize grits, flour, gruel |
No data |
No data |
0 |
0 |
Moller & Gustavsson, 2000 |
Samples taken randomly |
|
|
Popcorn |
No data |
No data |
0 |
0 |
|||
|
United Kingdom |
(Total fumonisins by HPLC; corrected for recovery) |
Patel et al., 1997; |
Samples (minimum 0.5 kg) purchased from retail outlets, ground or homogenized, and sub-sampled before analysis |
||||
|
Polenta |
1994–95 |
No data |
4 |
No data |
|||
|
Breakfast cereals |
|
No data |
0 |
0 |
|||
|
Popcorn, ready-made |
|
— |
— |
— |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Maize snacks |
|
No data |
0 |
0 |
|||
|
Tortilla, taco, enchilada |
|
No data |
0 |
0 |
|||
|
Maize thickener |
|
No data |
0 |
0 |
|||
|
Maize syrup |
|
— |
— |
— |
|||
|
Maize oil |
|
— |
— |
— |
|||
|
Corn-on- the-cob |
|
— |
— |
— |
|||
|
Sweet maize |
|
No data |
0 |
0 |
|||
|
Africa |
|||||||
|
Botswana |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground, and mixed before aliquots taken for analysis |
|
South Africa |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground .and mixed before aliquots taken for analysis |
|
Samp |
1994–95 |
No data |
No data |
0 |
Rava, 1996; |
Samples collected from mills throughout country and ground before analysis |
|
|
Maize rice |
|
No data |
0 |
0 |
|||
|
Maize grits |
|
No data |
No data |
0 |
|||
|
Maize flour |
|
No data |
0 |
0 |
|||
|
Super meal |
|
No data |
0 |
0 |
|||
|
Special meal |
|
No data |
No data |
0 |
|||
|
Sifted meal |
|
No data |
No data |
No data |
|||
|
Unsifted meal |
|
No data |
No data |
No data |
|||
|
Germ meal |
|
No data |
No data |
0 |
|||
|
Maize bran |
|
No data |
No data |
No data |
|||
|
Screenings |
|
No data |
No data |
No data |
|||
|
Zambia |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground, and mixed before aliquots taken for analysis |
|
Zimbabwe |
Maize meal |
1994 |
No data |
1 |
0 |
||
|
Asia |
|||||||
|
China (Linqu County, Shandong) |
Maize meal |
1996 |
1840 |
3 |
2 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen and 5 g portion cut for analysis |
|
Batter |
1996 |
780 |
2 |
2 |
|||
|
Pancake |
1996 |
1050 |
3 |
2 |
|||
|
Nepal |
(Total fumonisins by HPLC) |
|
|
|
|
Desjardins et al., 2000 |
Samples (0.25–0.5 kg) collected from farms and markets in 10 districts; 100 g ground and 10 g extracted |
|
Maize flour |
Feb–Jul |
No data |
No data |
No data |
|||
|
Cornflakes |
1997 |
— |
— |
— |
|||
|
Taiwan |
Maize snacks |
8/1994–12/1995 |
No data |
No data |
No data |
Tseng & Liu, 1997; |
Random purchases from retail markets in various districts; 150-g sub-sample ground and 25 g extracted |
|
Sweet canned maize |
|
No data |
No data |
0 |
|||
|
Popcorn |
|
No data |
No data |
0 |
|||
|
Cornflakes |
|
No data |
No data |
0 |
|||
|
Maize grits |
|
— |
— |
— |
|||
|
Maize flour |
|
No data |
0 |
0 |
|||
LOQ, limit of quantification; *, limit of detection; mean: true mean (for n analytical values, the true mean is the sum Xi / n, where Xi is the value of each analytical result; for not detected, Xi = 0); max: maximum concentration; COVENIN, Comision Venezolana de Normas Industriale; FDA, Food and Drug Administration; ELISA, enzyme-linked immunosorbent assay; HPLC, high-performance liquid chromatography
References: P, parent reference, S, sampling method, A, analytical method
Results of surveys for fumonisin B2, showing concentrations and distribution of
contamination in food commodities
|
Country/ Region |
Commodity |
Year/ Season |
No. of samples |
LOQ |
n < LOQ |
Mean/Max |
|
Unprocessed maize |
||||||
|
North America |
||||||
|
Canada |
Domestic |
1993 |
98 |
No data |
89 |
54/1000 |
|
USA |
Iowa State |
1988 |
22 |
500 |
No data |
700/5700 |
|
1989 |
44 |
500 |
No data |
800/12300 |
||
|
1990 |
59 |
500 |
No data |
900/6100 |
||
|
1991 |
50 |
500 |
No data |
800/4400 |
||
|
1992 |
80 |
500 |
No data |
10/600 |
||
|
1993 |
43 |
500 |
No data |
200/1100 |
||
|
1994 |
37 |
500 |
No data |
90/1100 |
||
|
1995 |
85 |
500 |
No data |
60/1800 |
||
|
1996 |
93 |
500 |
No data |
30/— |
||
|
South and Central America |
||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
34 |
10 |
28 |
52/446 |
|
Domestic |
Jan–Oct 1999 |
186 |
10 |
5 |
1451/6944 |
|
|
Domestic |
May–Jun 1999 |
66 |
10 |
34 |
208/4622 |
|
|
Domestic |
Jan–Aug 2000 |
56 |
10 |
40 |
103/995 |
|
|
Rice, imported |
Nov–Dec 1998 |
5 |
10 |
3 |
33/126 |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
6 |
10 |
6 |
—/— |
|
|
Rice, polished domestic |
Apr–Jun 1999 |
5 |
10 |
5 |
—/— |
|
|
Brazil |
Domestic |
4/1995–4/1996 |
150 |
0.08 |
11 |
200/6920 |
|
Domestic |
1990–91 |
48 |
No data |
2 |
4620/19 130 |
|
|
Colombia |
Domestic |
Feb–Aug 1998 |
15 |
20* |
10 |
102/833 |
|
Europe |
||||||
|
Spain |
Domestic |
1994–96 |
55 |
100* |
33 |
760/5900 |
|
Africa |
||||||
|
Kenya |
Kernels |
1994 |
1 |
20* |
0 |
275/275 |
|
Malawi |
Kernels |
1994 |
8 |
20* |
7 |
4/30 |
|
Mozambique |
Kernels |
1994 |
3 |
20* |
0 |
90/110 |
|
South Africa |
White maize |
1994–95 |
143 |
20* |
No data |
118/4187 |
|
Yellow maize |
1994–95 |
148 |
20* |
No data |
148/2000 |
|
|
White grade 1 |
1993 |
No data |
20* |
No data |
54/1430 |
|
|
White grade 2 |
1993 |
No data |
20* |
No data |
70/842 |
|
|
White grade 3 |
1993 |
No data |
20* |
No data |
30/200 |
|
|
Yellow grade 1 |
1993 |
No data |
20* |
No data |
198/5690 |
|
|
Yellow grade 2 |
1993 |
No data |
20* |
No data |
257/2039 |
|
|
Yellow grade 3 |
1993 |
No data |
20* |
No data |
227/1800 |
|
|
Tanzania |
Kernels |
1994 |
9 |
20* |
8 |
7/60 |
|
Uganda |
Kernels |
1994 |
1 |
20* |
0 |
155/155 |
|
Zimbabwe |
Kernels |
1994 |
2 |
20* |
1 |
20/40 |
|
Asia |
||||||
|
China |
Linqu County, Shandong Province |
1996 |
16 |
500* |
12 |
200/1000 |
|
Linxian County, Henan Province |
1994 |
34 |
No data |
14 |
409/4350 |
|
|
Shangqiu County, Henan Province |
1994 |
20 |
No data |
12 |
236/1220 |
|
|
Haimen, Jiangsu counties |
Apr–Jul 1993 |
40 |
100* |
15 |
1263/6770 |
|
|
Penlai, Shandong Province |
Apr–Jul 1993 |
40 |
100* |
33 |
87/1190 |
|
|
Indonesia |
Domestic |
Nov 1995 |
16 |
50* |
9 |
80/376 |
|
Domestic |
1992–94 |
12 |
50* |
9 |
111/556 |
|
|
Iran |
Mazandaran |
Sep 1998 |
11 |
10* |
0 |
512/1175 |
|
Isfahan |
Oct 1998 |
8 |
10* |
6 |
16/75 |
|
|
Korea Republic of |
Mouldy |
Nov 1997 |
36 |
50* |
5 |
6500/48400 |
|
Healthy |
1997/Nov |
35 |
50* |
27 |
250/5400 |
|
|
Philippines |
Domestic |
1992–94 |
50 |
50* |
44 |
34/1210 |
|
Philippines |
Seed maize |
1992–94 |
24 |
50* |
22 |
7/118 |
|
Feed maize |
|
22 |
50* |
18 |
70/1210 |
|
|
Food maize |
|
23 |
50* |
23 |
—/— |
|
|
Taiwan |
Domestic |
1996–97 |
110 |
80* |
108 |
4/255 |
|
Thailand |
Visibly mouldy human food |
1992 |
5 |
50* |
2 |
379/1400 |
|
Domestic |
1992–94 |
27 |
50* |
15 |
112/1400 |
|
|
Viet Nam |
Maize (feed) |
1993 |
15 |
100* |
11 |
74/560 |
|
Maize powder, feed |
1993 |
17 |
100* |
5 |
204/401 |
|
|
Processed maize-based human food |
||||||
|
North America |
|
|
|
|
|
|
|
Canada |
(* number and mean of samples positive for fumonisin B1 (not fumonisin B2)) |
|||||
|
Maize products: 1996–97 |
||||||
|
Maize kernel, meal, flour |
21* |
No data |
No data |
34*/60 (n=16) |
No data |
|
|
|
|
|
77*/200 (n=5) |
|
||
|
Maize snacks (alkali -processed) |
20* |
No data |
No data |
57*/240 (n=13) |
No data |
|
|
|
|
|
76*/270 (n=7) |
|
||
|
Maize snacks (not alkali-processed) |
6* |
No data |
No data |
29*/40 (n=4) |
No data |
|
|
|
|
|
<38*/<50 (n=2) |
|
||
|
Canned maize |
9* |
No data |
No data |
<25*/<25 (n=8) |
No data |
|
|
|
|
|
<30*/<30 (n=1) |
|
||
|
Frozen maize |
2* |
No data |
No data |
<25*/<25 |
No data |
|
|
Maize bread |
1* |
No data |
No data |
<25*/<25 |
No data |
|
|
Maize-based breakfast cereals |
8* |
No data |
No data |
33*/40 (n=2) |
No data |
|
|
|
|
|
<28*/<40 (n=6) |
|
||
|
Fresh tortillas |
1* |
No data |
No data |
190*/190 |
No data |
|
|
Maize-based infant foods: |
1997–98 |
|
|
|
|
|
|
Mixed cereals |
16* |
No data |
No data |
11*/20 |
No data |
|
|
Cereal with formula |
6* |
No data |
No data |
<10*/<10 |
No data |
|
|
Creamed maize or maize/vegetables |
6* |
No data |
No data |
<10*/<10 |
No data |
|
|
Infant cereals: 1998–99 |
||||||
|
Mixed cereals with fruit/formula |
13* |
No data |
No data |
<10*/<10 |
No data |
|
|
Rice-based |
16* |
No data |
No data |
<10*/<10 |
No data |
|
|
Soya-based |
14* |
No data |
No data |
11*/20 |
No data |
|
|
Mixed cereals |
20* |
No data |
No data |
11*/20 |
No data |
|
|
Processed maize-based human food |
||||||
|
USA |
Maize meal, degermed |
1997 |
602 |
100* |
575 |
10/450 |
|
Maize meal, partially de-germed |
|
20 |
100* |
17 |
40/360 |
|
|
Maize meal, whole grain |
|
50 |
100* |
25 |
250/1420 |
|
|
Maize meal, degermed |
1998 |
561 |
100* |
525 |
30/2480 |
|
|
Maize meal, partially de-germed |
|
20 |
100* |
14 |
170/1130 |
|
|
Maize meal, whole grain |
|
39 |
100* |
21 |
240/1830 |
|
|
Starch |
No data |
4 |
10* |
2 |
32/84 |
|
|
Grits |
|
4 |
10* |
2 |
915/1910 |
|
|
South and Central America |
||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
11 |
10 |
2 |
67/341 |
|
Popcorn |
May–Jun 1999 |
42 |
10 |
3 |
575/8008 |
|
|
Cornflour |
10/1996–1/1997 |
15 |
11* |
4 |
113/768 |
|
|
Maize grits |
10/1996–1/1997 |
4 |
11* |
2 |
30/100 |
|
|
Maize flour |
Jan 1998 |
14 |
11* |
1 |
308/1818 |
|
|
Maize grits |
Jan 1998 |
2 |
11* |
0 |
275/324 |
|
|
Maize meal |
1997 |
21 |
1.5* |
9 |
133/1090 |
|
|
Maize flakes |
1997 |
17 |
1.5* |
17 |
—/— |
|
|
Brazil |
Canned sweet maize |
Mar–May 1999 |
11 |
20* |
5 |
80/210 |
|
Cornflakes |
|
4 |
20* |
3 |
80/30 (?) |
|
|
Maize flour |
|
11 |
20* |
2 |
220/510 |
|
|
Cornflour, baby cereal |
|
2 |
20* |
1 |
30/50 |
|
|
Maize grits |
|
2 |
20* |
0 |
180/300 |
|
|
Maize meal |
|
9 |
20* |
0 |
600/1380 |
|
|
Corn-on-the-cob |
7 |
20* |
7 |
—/— |
— |
|
|
Curau |
|
2 |
20* |
2 |
—/— |
|
|
Dergermed maize |
11 |
20* |
3 |
150/640 |
No data |
|
|
Pamonha |
|
7 |
20* |
7 |
—/— |
|
|
Popcorn |
|
9 |
20* |
5 |
80/300 |
|
|
Pre-cooked maize meal |
6 |
20* |
2 |
200/420 |
No data |
|
|
Colombia |
Pre-cooked maize meal |
Feb–Aug 1998 |
15 |
20* |
8 |
20/81 |
|
Popcorn |
|
8 |
20* |
5 |
27/78 |
|
|
Maize meal |
|
7 |
20* |
5 |
24/105 |
|
|
"Arepas" |
|
6 |
20* |
3 |
30/93 |
|
|
Maize snacks |
|
6 |
20* |
5 |
12/73 |
|
|
Maize starch |
|
3 |
20* |
3 |
—/— |
|
|
Mexico |
Tortillas |
No data |
7 |
10* |
1 |
76/180 |
|
Masa |
No data |
2 |
10* |
0 |
745/1380 |
|
|
Europe |
||||||
|
Denmark |
Cornflakes |
1996 |
10 |
3* |
8 |
25/243 |
|
Maize flour |
|
8 |
3* |
2 |
10/24 |
|
|
Maize grits |
|
4 |
3* |
4 |
—/— |
|
|
Maize starch |
|
6 |
3* |
6 |
—/— |
|
|
Corn-on-the-cob |
4 |
3* |
4 |
—/— |
No data |
|
|
Maize snacks |
10 |
3* |
7 |
2/8 |
No data |
|
|
Polenta |
|
1 |
3* |
0 |
22/22 |
|
|
Popcorn |
|
9 |
3* |
8 |
7/59 |
|
|
Sweet maize |
16 |
3* |
16 |
—/— |
No data |
|
|
Germany |
Maize grits |
No data |
6 |
0.8 |
0 |
40/112 |
|
Maize meal |
|
2 |
0.8 |
0 |
39/62 |
|
|
Sweet maize |
6 |
0.8 |
0 |
—/— |
— |
|
|
Popcorn |
|
2 |
0.8 |
0 |
<1/— |
|
|
Cornflakes |
|
2 |
0.8 |
0 |
<2/— |
|
|
Infant foods |
|
4 |
0.8 |
2 |
8/10 |
|
|
Italy |
Polenta |
No data |
20 |
20* |
0 |
366/910 |
|
Sweden |
Cornflakes |
No data |
6 |
1* |
6 |
—/— |
|
Fresh, frozen, canned maize |
|
13 |
1* |
13 |
—/— |
|
|
Maize chips |
|
7 |
1* |
2 |
27/56 |
|
|
Maize grits, flour, gruel |
|
8 |
1* |
7 |
1/11 |
|
|
Popcorn |
|
8 |
1* |
5 |
7/40 |
|
|
Africa |
||||||
|
Botswana |
Maize meal |
1994 |
4 |
20* |
2 |
40/85 |
|
South Africa |
Maize meal |
1994 |
2 |
20* |
2 |
—/— |
|
Samp |
1994–95 |
13 |
20* |
No data |
3/41 |
|
|
Maize rice |
|
11 |
20* |
No data |
—/— |
|
|
Maize grits |
|
5 |
20* |
No data |
13/63 |
|
|
Maize flour |
|
2 |
20* |
No data |
—/— |
|
|
Super meal |
|
25 |
20* |
No data |
—/— |
|
|
Special meal |
36 |
20* |
No data |
32/507 |
No data |
|
|
Sifted meal |
|
47 |
20* |
No data |
87/1223 |
|
|
Unsifted meal |
19 |
20* |
No data |
148/1100 |
No data |
|
|
Germ meal |
|
8 |
20* |
No data |
25/200 |
|
|
Maize bran |
|
32 |
20* |
No data |
338/2368 |
|
|
Screenings |
|
7 |
20* |
No data |
1628/3718 |
|
|
Zambia |
Maize meal |
1994 |
1 |
20* |
0 |
380/380 |
|
Maize meal |
1994 |
4 |
20* |
2 |
193/620 |
|
|
Asia |
||||||
|
China (Linqu County, Shandong) |
Maize meal |
1996 |
14 |
500* |
10 |
350/2800 |
|
Batter |
1996 |
32 |
500* |
30 |
31/500 |
|
|
Pancake |
1996 |
16 |
500* |
14 |
113/1100 |
|
|
Taiwan |
Maize snacks |
8/1994–12/1995 |
78 |
80* |
62 |
30/715 |
|
Sweet, canned maize |
|
24 |
80* |
18 |
16/658 |
|
|
Popcorn |
|
22 |
80* |
15 |
37/273 |
|
|
Cornflakes |
|
17 |
80* |
14 |
29/466 |
|
|
Maize grits |
|
4 |
80* |
4 |
—/— |
|
|
Maize flour |
|
2 |
80* |
2 |
—/— |
|
|
Country/ Region |
Commodity |
Year/ Season |
90th %ile |
n > 1000µg/kg |
n > 2000µg/kg |
References |
Sampling procedure |
|
Unprocessed maize |
|||||||
|
North America |
|||||||
|
Canada |
Domestic |
1993 |
<200 |
1 |
0 |
Miller et al., 1995 |
Frequent small samples taken from grain handling equipment; samples pooled, mixed, and 2.2-kg subsample ground |
|
USA |
Iowa State |
1988 |
No data |
No data |
No data |
Murphy et al., 1993; |
Random samples taken from trucks at grain elevators; 400-g sub-sample ground before analysis |
|
1989 |
No data |
No data |
No data |
||||
|
1990 |
No data |
No data |
No data |
||||
|
1991 |
No data |
No data |
No data |
||||
|
1992 |
No data |
0 |
0 |
Rice & Ross, 1994; L.G. Rice, P.F. Ross, USDA Veterinary Services Laboratory; P.A. Murphy, Iowa State University |
|||
|
1993 |
No data |
No data |
0 |
||||
|
1994 |
No data |
No data |
0 |
||||
|
1995 |
No data |
No data |
0 |
||||
|
1996 |
No data |
No data |
0 |
||||
|
South and Central America |
|||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
279 |
0 |
0 |
GEMS/Food programme |
Statistically based, representative for part of country |
|
Domestic |
Jan–Oct 1999 |
3847 |
81 |
47 |
|||
|
Domestic |
May–Jun 1999 |
264 |
3 |
2 |
|||
|
Domestic |
Jan–Aug 2000 |
442 |
0 |
0 |
|||
|
Rice, imported |
Nov–Dec 1998 |
No data |
0 |
0 |
GEMS/Food programme |
Not statistically based, samples from whole country |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
— |
— |
— |
|||
|
Rice, polished domestic |
Apr–Jun 1999 |
— |
— |
— |
|||
|
Brazil |
Domestic |
4/1995–4/1996 |
No data |
No data |
No data |
GEMS/Food programme |
Statistically based, representative of part of country |
|
Domestic |
1990–91 |
9060 |
46 |
39 |
Hirooka et al., 1996; |
Samples collected from silos and warehouses, mixed, reduced to 2 kg, and ground |
|
|
Colombia |
Domestic |
Feb–Aug 1998 |
No data |
0 |
0 |
Perilla & Diaz, 1998; |
Samples (minimum 500 g) purchased at random from most popular retail shops; ground, sub-sampled in grinding–subsampling mill (Romer series II) |
|
Europe |
|||||||
|
Spain |
Domestic |
1994–96 |
No data |
No data |
No data |
Castella et al., 1999; |
Samples (I kg representative) obtained from agricultural cooperatives and factories; subsampled into 200-g aliquot |
|
Africa |
|||||||
|
Kenya |
Kernels |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground and mixed before aliquots taken for analysis |
|
Malawi |
Kernels |
1994 |
No data |
0 |
0 |
||
|
Mozambique |
Kernels |
1994 |
No data |
0 |
0 |
||
|
South Africa |
White maize |
1994–95 |
No data |
No data |
No data |
||
|
Yellow maize |
1994–95 |
No data |
No data |
No data |
|||
|
White grade 1 |
1993 |
No data |
No data |
0 |
Rava et al., 1996; |
Representative samples (3 kg) collected at harvest from silos in main production zones; 500-g sample obtained from riffle splitter and ground |
|
|
White grade 2 |
1993 |
No data |
0 |
0 |
|||
|
White grade 3 |
1993 |
No data |
0 |
0 |
|||
|
Yellow grade 1 |
1993 |
No data |
No data |
No data |
|||
|
Yellow grade 2 |
1993 |
No data |
No data |
No data |
|||
|
Yellow grade 3 |
1993 |
No data |
No data |
0 |
|||
|
Tanzania |
Kernels |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground and mixed before aliquots taken for analysis |
|
Uganda |
Kernels |
1994 |
No data |
0 |
0 |
||
|
Zimbabwe |
Kernels |
1994 |
No data |
0 |
0 |
||
|
Asia |
|||||||
|
China |
Linqu County, Shandong Province |
1996 |
800 |
1 |
0 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen and 5-g portion cut for analysis |
|
Linxian County, Henan Province |
1994 |
No data |
No data |
No data |
Gao & Yoshizawa, 1997; |
Samples from 1994 harvest collected from peasant families during Jan–Feb 1995 |
|
|
Shangqiu County, Henan Province |
1994 |
No data |
No data |
0 |
|||
|
Haimen, Jiangsu counties |
Apr–Jul 1993 |
2748 |
17 |
11 |
Ueno et al., 1997; |
Kernels collected randomly from agricultural stocks. 25-g sample milled and 5 g extracted |
|
|
Penlai, Shandong Province |
Apr–Jul 1993 |
280 |
1 |
0 |
|||
|
Indonesia |
Domestic |
Nov 1995 |
266 |
0 |
0 |
Ali et al., 1998; |
Samples ground and aliquots taken from 200 g |
|
Domestic |
1992–94 |
No data |
0 |
0 |
Yamashita et al., 1995; |
Samples collected at random from stores of wholesalers, retailers, university farms, and local farmers; ground and stored |
|
|
Iran |
Mazandaran |
Sep 1998 |
940 |
1 |
0 |
Shephard et al., 2000; |
Farmers' maize lots collected at random from total sample ground consignments sold to Iranian Agriculture Office; |
|
Isfahan |
Oct 1998 |
58 |
0 |
0 |
|||
|
Korea Republic of |
Mouldy |
Nov 1997 |
No data |
No data |
No data |
Sohn et al., 1999; |
Samples collected from households and milled |
|
Healthy |
1997/Nov |
No data |
No data |
No data |
|||
|
Philippines |
Domestic |
1992–94 |
No data |
No data |
0 |
Yamashita et al., 1995; |
Samples collected at random from stores of whole salers, retailers, university farms, and local farmers; ground and stored |
|
Philippines |
Seed maize |
1992–94 |
No data |
0 |
0 |
Yoshizawa & Yamashita, 1995; A = Shephard et al., 1990 |
At least 100-g sample collected, 50 g ground, and 20 g extracted |
|
Feed maize |
|
No data |
No data |
0 |
|||
|
Food maize |
|
— |
— |
— |
|||
|
Taiwan |
Domestic |
1996–97 |
No data |
0 |
0 |
Tseng & Liu, 1999; |
Samples (1 kg) collected from 8 districts; subsamples (200 g) ground and 25 g extracted |
|
Thailand |
Visibly mouldy human food |
1992 |
No data |
1 |
0 |
Yoshizawa et al., 1996; |
Samples collected before harvest; random 100-g aliquots analysed; 50 g milled and 20-g subsample extracted |
|
Domestic |
1992–94 |
No data |
No data |
0 |
Yamashita et al., 1995; |
Samples collected at random from stores of wholesalers, retailers, university farms, and local farmers; ground and stored |
|
|
Viet Nam |
Maize (feed) |
1993 |
184 |
0 |
0 |
Wang et al., 1995; |
Random samples from various locations; 25 g milled and 5 g extracted |
|
Maize powder, feed |
1993 |
375 |
0 |
0 |
|||
|
Processed maize-based human food |
|||||||
|
North America |
|||||||
|
Canada |
(* number and mean of samples positive for fumonisin B1 (not fumonisin B2)) |
||||||
|
Maize products: 1996–97 |
|||||||
|
Maize kernel, meal, flour |
21* |
0 |
0 |
Canada |
No data |
|
|
|
Maize snacks (alkali -processed) |
20* |
0 |
0 |
Canada |
No data |
|
|
|
Maize snacks (not alkali-processed) |
6* |
0 |
0 |
Canada |
No data |
|
|
|
Canned maize |
9* |
0 |
0 |
Canada |
No data |
|
|
|
Frozen maize |
2* |
0 |
0 |
Canada |
No data |
|
|
|
Maize bread |
1* |
0 |
0 |
Canada |
No data |
|
|
|
Maize-based breakfast cereals |
8* |
0 |
0 |
Canada |
No data |
|
|
|
Fresh tortillas |
1* |
0 |
0 |
Canada |
No data |
|
|
|
Maize-based infant foods:1997–98 |
|||||||
|
Mixed cereals |
16* |
0 |
0 |
Canada |
No data |
|
|
|
Cereal with formula |
6* |
0 |
0 |
Canada |
No data |
|
|
|
Creamed maize or maize/vegetables |
6* |
0 |
0 |
Canada |
No data |
|
|
|
Infant cereals: 1998–99 |
|||||||
|
Mixed cereals with fruit/formula |
13* |
0 |
0 |
Canada |
No data |
|
|
|
Rice-based |
16* |
0 |
0 |
Canada |
No data |
|
|
|
Soya-based |
14* |
0 |
0 |
Canada |
No data |
|
|
|
Mixed cereals |
20* |
0 |
0 |
Canada |
No data |
|
|
|
Processed maize-based human food |
|||||||
|
USA |
Maize meal, degermed |
1997 |
<10 |
0 |
0 |
USA |
Sampled 1998 |
|
Maize meal, partially de-germed |
|
193 |
0 |
0 |
|||
|
Maize meal, whole grain |
|
1176 |
6 |
0 |
|||
|
Maize meal, degermed |
1998 |
<10 |
2 |
1 |
USA |
Sampled 1999 |
|
|
Maize meal, partially de-germed |
|
569 |
1 |
0 |
|||
|
Maize meal, whole grain |
|
786 |
4 |
0 |
|||
|
Starch |
No data |
No data |
0 |
0 |
Maragos et al., 1997; |
No data |
|
|
Grits |
|
No data |
No data |
0 |
|||
|
South and Central America |
|||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
311 |
0 |
0 |
GEMS/Food programme GEMS/Food programme Hennigen et al., 2000; |
Statistically based, representative of part of country |
|
Popcorn |
May–Jun 1999 |
2045 |
5 |
4 |
|||
|
Cornflour |
10/1996–1/1997 |
190 |
0 |
0 |
|||
|
Maize grits |
10/1996–1/1997 |
76 |
0 |
0 |
|||
|
Maize flour |
Jan 1998 |
649 |
1 |
0 |
|||
|
Maize grits |
Jan 1998 |
No data |
0 |
0 |
|||
|
Maize meal |
1997 |
376 |
1 |
0 |
Solovey et al., 1999; |
Randomly purchased from comercial outlets; total sample ground |
|
|
Maize flakes |
1997 |
— |
— |
— |
|||
|
Brazil |
Canned sweet maize |
Mar–May 1999 |
No data |
0 |
0 |
Machinski & Valente Soares, 2000; |
Samples (minimum 500 g) purchased from retail shops in Campinas and ground. homogenized, or both |
|
Cornflakes |
|
No data |
0 |
0 |
|||
|
Maize flour |
|
No data |
0 |
0 |
|||
|
Cornflour, baby cereal |
|
No data |
0 |
0 |
|||
|
Maize grits |
|
No data |
0 |
0 |
|||
|
Maize meal |
|
No data |
No data |
0 |
|||
|
Corn-on-the-cob |
7 |
— |
— |
|
|||
|
Curau |
|
— |
— |
— |
|||
|
Dergermed maize |
11 |
0 |
0 |
|
|||
|
Pamonha |
|
— |
— |
— |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Pre-cooked maize meal |
6 |
0 |
0 |
|
|||
|
Colombia |
Pre-cooked maize meal |
Feb–Aug 1998 |
No data |
0 |
0 |
Perilla & Diaz, 1998; |
Samples (minimum 500 g) purchased at random from most popular retail shops; samples ground and subsampled in grinding–subsampling mill (Romer series II) |
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Maize meal |
|
No data |
0 |
0 |
|||
|
"Arepas" |
|
No data |
0 |
0 |
|||
|
Maize snacks |
|
No data |
0 |
0 |
|||
|
Maize starch |
|
— |
— |
— |
|||
|
Mexico |
Tortillas |
No data |
No data |
0 |
0 |
Dombrink-Kurtzman & Dvorak, 1999 |
No data |
|
Masa |
No data |
No data |
1 |
0 |
|||
|
Europe |
|||||||
|
Denmark |
Cornflakes |
1996 |
No data |
0 |
0 |
Petersen, 2000; |
Samples purchased in retail stores, homogenized in mixer before analysis |
|
|
Maize flour |
|
No data |
0 |
0 |
|
|
|
|
Maize grits |
|
No data |
0 |
0 |
|
|
|
Maize starch |
|
No data |
0 |
0 |
|||
|
Corn-on-the-cob |
4 |
0 |
0 |
|
|||
|
Maize snacks |
10 |
0 |
0 |
|
|||
|
Polenta |
|
No data |
0 |
0 |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Sweet maize |
16 |
0 |
0 |
|
|||
|
Germany |
Maize grits |
No data |
No data |
0 |
0 |
Lukacs et al., 1996; A = Zoller et al., 1994 |
Samples from market |
|
Maize meal |
|
No data |
0 |
0 |
|||
|
Sweet maize |
6 |
— |
— |
|
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Cornflakes |
|
No data |
0 |
0 |
|||
|
Infant foods |
|
No data |
0 |
0 |
|||
|
Italy |
Polenta |
No data |
840 |
0 |
0 |
Pascale et al., 1995; |
Samples purchased in local retail market in Apulia |
|
Sweden |
Cornflakes |
No data |
— |
— |
— |
Moller & Gustavsson, 2000 |
Samples taken randomly |
|
Fresh, frozen, canned maize |
|
— |
— |
— |
|||
|
Maize chips |
|
No data |
0 |
0 |
|||
|
Maize grits, flour, gruel |
|
No data |
0 |
0 |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Africa |
|||||||
|
Botswana |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground and mixed before aliquots taken for analysis |
|
South Africa |
Maize meal |
1994 |
— |
— |
— |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground and mixed before aliquots taken for analysis |
|
Samp |
1994–95 |
No data |
0 |
0 |
Rava, 1996; |
Samples collected from mills throughout country, ground before analysis |
|
|
Maize rice |
|
— |
— |
— |
|||
|
Maize grits |
|
No data |
0 |
0 |
|||
|
Maize flour |
|
— |
— |
— |
|||
|
Super meal |
|
— |
— |
— |
|||
|
Special meal |
36 |
0 |
0 |
|
|||
|
Sifted meal |
|
No data |
No data |
0 |
|||
|
Unsifted meal |
19 |
No data |
0 |
|
|||
|
Germ meal |
|
No data |
0 |
0 |
|||
|
Maize bran |
|
No data |
No data |
No data |
|||
|
Screenings |
|
No data |
No data |
No data |
|||
|
Zambia |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn Zimbabwe from sample lots, ground and mixed before aliquots taken for analysis |
|
Maize meal |
1994 |
No data |
0 |
0 |
|||
|
Asia |
|||||||
|
China (Linqu County, Shandong) |
Maize meal |
1996 |
880 |
2 |
1 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen, 5-g portion cut for analysis |
|
Batter |
1996 |
<500 |
0 |
0 |
|||
|
Pancake |
1996 |
<500 |
1 |
0 |
|||
|
Taiwan |
Maize snacks |
8/1994–12/1995 |
No data |
0 |
0 |
Tseng & Liu, 1997; |
Random purchases from retail markets in various districts; 150-g subsample ground and 25 g extracted |
|
Sweet, canned maize |
|
No data |
0 |
0 |
|||
|
Popcorn |
|
No data |
0 |
0 |
|||
|
Cornflakes |
|
No data |
0 |
0 |
|||
|
Maize grits |
|
— |
— |
— |
|||
|
Maize flour |
|
— |
— |
— |
|||
LOQ, limit of quantification; *, limit of detection; mean: true mean (for n analytical values, the true mean is the sum Xi / n, where Xi is the value of each analytical result; for not detected, Xi = 0); max: maximum concentration
References: P, parent reference, S, sampling method, A, analytical method
Results of surveys for fumonisin B3, showing concentrations and distribution of
contamination in food commodities
|
Country/ Region |
Commodity |
Year/ Season |
No. of |
LOQ |
n < LOQ |
Mean/Max |
|
Unprocessed maize |
||||||
|
North America |
||||||
|
USA |
Iowa State |
1988 |
22 |
500 |
No data |
200/2100 |
|
1989 |
44 |
500 |
No data |
200/4000 |
||
|
1990 |
59 |
500 |
No data |
300/2800 |
||
|
1991 |
50 |
500 |
No data |
400/2300 |
||
|
1992 |
80 |
500 |
No data |
—/— |
||
|
1993 |
43 |
500 |
No data |
30/700 |
||
|
1995 |
85 |
500 |
No data |
20/600 |
||
|
1994 |
37 |
500 |
No data |
30/600 |
||
|
1996 |
93 |
500 |
No data |
—/— |
||
|
South and Central America |
||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
34 |
8 |
29 |
8/61 |
|
Domestic |
Jan–Oct 1999 |
186 |
8 |
8 |
341/1684 |
|
|
Domestic |
May–Jun 1999 |
66 |
8 |
39 |
51/867 |
|
|
Domestic |
Jan–Aug 2000 |
56 |
8 |
43 |
39/442 |
|
|
Rice, imported |
Nov–Dec 1998 |
5 |
8 |
4 |
3/16 |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
6 |
8 |
6 |
—/— |
|
|
Rice, polished domestic |
Apr–Jun 1999 |
5 |
8 |
5 |
—/— |
|
|
Africa |
||||||
|
Kenya |
Kernels |
1994 |
1 |
20* |
0 |
130/130 |
|
Malawi |
Kernels |
1994 |
8 |
20* |
8 |
—/— |
|
Mozambique |
Domestic |
1994 |
3 |
20* |
0 |
40/50 |
|
South Africa |
White maize |
1994–95 |
143 |
20* |
No data |
83/3110 |
|
Yellow maize |
1994–95 |
148 |
20* |
No data |
54/1431 |
|
|
White grade 1 |
1993 |
No data |
20* |
No data |
25/355 |
|
|
White grade 2 |
1993 |
No data |
20* |
No data |
38/400 |
|
|
White grade 3 |
1993 |
No data |
20* |
No data |
—/— |
|
|
Yellow grade 1 |
1993 |
No data |
20* |
No data |
71/1570 |
|
|
Yellow grade 2 |
1993 |
No data |
20* |
No data |
167/1963 |
|
|
Yellow grade 3 |
1993 |
No data |
20* |
No data |
112/729 |
|
|
Tanzania |
Kernels |
1994 |
9 |
20* |
9 |
—/— |
|
Uganda |
Kernels |
1994 |
1 |
20* |
0 |
85/85 |
|
Zimbabwe |
Kernels |
1994 |
2 |
20* |
2 |
—/— |
|
China |
Linqui County, Shandong Province |
1996 |
16 |
500* |
15 |
31/500 |
|
|
Linxian County, Henan Province |
1994 |
34 |
No data |
19 |
169/1660 |
|
|
Shangqiu County, Henan Province |
1994 |
20 |
No data |
13 |
109/576 |
|
|
Haimen, Jiangsu counties |
Apr–Jul 1993 |
40 |
100* |
15 |
700/4130 |
|
|
Penlai, Shandong Province |
Apr–Jul 1993 |
40 |
100* |
36 |
17/270 |
|
Indonesia |
Domestic |
Nov 1995 |
16 |
50* |
12 |
27/222 |
|
Iran |
Mazandaran |
Sep 1998 |
11 |
10* |
0 |
361/960 |
|
Isfahan |
Oct 1998 |
8 |
10* |
6 |
16/75 |
|
|
Korea, Republic of |
Mouldy |
Nov 1997 |
36 |
50* |
5 |
5400/10600 |
|
Healthy |
Nov 1997 |
35 |
50* |
28 |
60/500 |
|
|
Viet Nam |
Maize, feed |
1993 |
15 |
100* |
12 |
46/432 |
|
Maize powder, feed |
1993 |
17 |
100* |
7 |
104/268 |
|
|
Processed maize-based human food |
||||||
|
North America |
||||||
|
USA |
Maize meal, degermed |
1997 |
602 |
100* |
602 |
—/— |
|
Maize meal, partially degermed |
|
20 |
100* |
20 |
—/— |
|
|
Maize meal, whole grain |
1997 |
50 |
100* |
40 |
40/290 |
|
|
Maize meal, degermed |
1998 |
561 |
100* |
552 |
4/610 |
|
|
Maize meal, partially degermed |
|
20 |
100* |
18 |
20/270 |
|
|
Maize meal, whole grain |
|
39 |
100* |
34 |
40/480 |
|
|
South and Central America |
||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
11 |
8 |
2 |
42/312 |
|
Popcorn |
May–Jun 1999 |
42 |
8 |
6 |
160/2027 |
|
|
Maize meal |
1997 |
21 |
1.5* |
10 |
78/1015 |
|
|
Cornflakes |
1997 |
17 |
1.5* |
17 |
—/— |
|
|
Africa |
||||||
|
Botswana |
Maize meal |
1994 |
4 |
20* |
3 |
8/30 |
|
South Africa |
Maize meal |
1994 |
2 |
20* |
2 |
—/— |
|
Samp |
1994–95 |
13 |
20* |
No data |
—/— |
|
|
Maize rice |
|
11 |
20* |
No data |
—/— |
|
|
Maize grit |
|
5 |
20* |
No data |
—/— |
|
|
Maize flour |
|
2 |
20* |
No data |
—/— |
|
|
Super meal |
|
25 |
20* |
No data |
—/— |
|
|
Special meal |
36 |
20* |
No data |
4/100 |
No data |
|
|
Sifted meal |
|
47 |
20* |
No data |
23/603 |
|
|
Unsifted meal |
19 |
20* |
No data |
64/522 |
No data |
|
|
Germ meal |
|
8 |
20* |
No data |
6/48 |
|
|
Maize bran |
|
32 |
20* |
No data |
126/2008 |
|
|
Screenings |
|
7 |
20* |
No data |
599/1604 |
|
|
Zambia |
Maize meal |
1994 |
1 |
20* |
0 |
85/85 |
|
Zimbabwe |
Maize meal |
1994 |
4 |
20* |
2 |
65/205 |
|
Asia |
||||||
|
China (Linqui County, Shandong Province) |
Maize meal |
1996 |
14 |
500* |
12 |
107/900 |
|
Batter |
1996 |
32 |
500* |
31 |
28/900 |
|
|
Pancake |
1996 |
16 |
500* |
14 |
81/700 |
|
|
Country/ Region |
Commodity |
Year/ Season |
90th %ile |
n > 1000µg/kg |
n > 2000µg/kg |
References |
Sampling procedure |
|
Unprocessed maize |
|
|
|
|
|
|
|
|
North America |
|
|
|
|
|
|
|
|
USA |
Iowa State |
1988 |
No data |
No data |
No data |
Murphy et al., 1993; |
Random samples taken from trucks at grain elevators; 400-g sub-sample ground before analysis |
|
1989 |
No data |
No data |
No data |
||||
|
1990 |
No data |
No data |
No data |
||||
|
1991 |
No data |
No data |
No data |
||||
|
1992 |
— |
— |
— |
Rice & Ross, 1994; L.G. Rice, P.F. Ross, USDA Veterinary Services Laboratory; P.A. |
|||
|
1993 |
No data |
0 |
0 |
||||
|
1995 |
No data |
0 |
0 |
||||
|
1994 |
No data |
0 |
0 |
||||
|
1996 |
No data |
— |
— |
||||
|
South and Central America |
|||||||
|
Argentina |
Domestic |
Apr–Nov 1998 |
44 |
0 |
0 |
GEMS/Food programme |
Statistically based, representative of part of country |
|
|
Domestic |
Jan–Oct 1999 |
874 |
15 |
0 |
||
|
|
Domestic |
May–Jun 1999 |
155 |
0 |
0 |
||
|
|
Domestic |
Jan–Aug 2000 |
202 |
0 |
0 |
||
|
|
Rice, imported |
Nov–Dec 1998 |
— |
0 |
0 |
GEMS/Food programme |
Not statistically based, samples from whole country |
|
|
Rice, husked, domestic |
Apr–Jun 1999 |
— |
— |
— |
GEMS/Food programme |
Not statistically based, samples from part of country |
|
|
Rice, polished domestic |
Apr–Jun 1999 |
— |
— |
— |
GEMS/Food programme |
Not statistically based, samples from part of country |
|
Africa |
|||||||
|
Kenya |
Kernels |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground, and mixed before aliquots taken for analysis |
|
Malawi |
Kernels |
1994 |
— |
— |
— |
||
|
Mozambique |
Domestic |
1994 |
No data |
0 |
0 |
||
|
South Africa |
White maize |
1994–95 |
No data |
No data |
No data |
Rava, 1996; |
Samples collected from mills throughout country, ground before analysis |
|
Yellow maize |
1994–95 |
No data |
No data |
No data |
|||
|
White grade 1 |
1993 |
No data |
0 |
0 |
Rava et al., 1996; |
Representative samples (3 kg) collected at harvest from silos in main production zones; 500-g sample obtained from riffle splitter and ground |
|
|
White grade 2 |
1993 |
No data |
0 |
0 |
|||
|
White grade 3 |
1993 |
— |
— |
— |
|||
|
Yellow grade 1 |
1993 |
No data |
No data |
0 |
|||
|
Yellow grade 2 |
1993 |
No data |
No data |
0 |
|||
|
Yellow grade 3 |
1993 |
No data |
0 |
0 |
|||
|
Tanzania |
Kernels |
1994 |
— |
— |
— |
Doko et al., 1996; |
Samples (1–5 kg) drawn from sample lots, ground, and mixed before aliquots taken for analysis |
|
Uganda |
Kernels |
1994 |
No data |
0 |
0 |
||
|
Zimbabwe |
Kernels |
1994 |
— |
— |
— |
||
|
China |
Linqui County, Shandong Province |
1996 |
<500 |
0 |
0 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen and 5-g portion cut for analysis |
|
Linxian County, Henan Province |
1994 |
No data |
No data |
0 |
Gao & Yoshizawa, 1997; A = Yoshizawa et al., 1994 |
Samples from 1994 harvest collected from peasant families during Jan–Feb 1995 |
|
|
Shangqiu County, Henan Province |
1994 |
No data |
0 |
0 |
|||
|
Haimen, Jiangsu counties |
Apr–Jul 1993 |
1950 |
10 |
4 |
Ueno et al., 1997; |
Kernels collected randomly from agricultural stocks; 25-g sample milled and 5 g extracted |
|
|
Penlai, Shandong Province |
Apr–Jul 1993 |
14 |
0 |
0 |
|||
|
Indonesia |
Domestic |
Nov 1995 |
76 |
0 |
0 |
Ali et al., 1998; A = Yoshizawa et al., 1994, 1996 |
Samples ground and aliquots taken from 200 g |
|
Iran |
Mazandaran |
Sep 1998 |
550 |
0 |
0 |
Shephard et al., 2000; A = Sydenham et al., 1996c |
Farmers' maize lots collected at random from consignments sold to Iranian Agriculture Office; total sample ground |
|
Isfahan |
Oct 1998 |
58 |
0 |
0 |
|
Maize ears bought at various periods from l local retail market; total sample ground |
|
|
Korea, Republic of |
Mouldy |
Nov 1997 |
No data |
No data |
No data |
Sohn et al., 1999; |
Samples collected from households and milled |
|
Healthy |
Nov 1997 |
No data |
0 |
0 |
|||
|
Viet Nam |
Maize, feed |
1993 |
135 |
0 |
0 |
Wang et al., 1995; |
Random samples from various locations; 25 g milled, 5 g extracted |
|
Maize powder, feed |
1993 |
203 |
0 |
0 |
|||
|
Processed maize-based human food |
|||||||
|
North America |
|||||||
|
USA |
Maize meal, degermed |
1997 |
— |
— |
— |
USA |
Sampled 1998 |
|
Maize meal, partially degermed |
|
— |
— |
— |
|||
|
Maize meal, whole grain |
1997 |
180 |
0 |
0 |
USA |
Sampled 1998 |
|
|
Maize meal, degermed |
1998 |
<10 |
0 |
0 |
USA |
Sampled 1999 |
|
|
Maize meal, partially degermed |
|
11 |
0 |
0 |
|||
|
Maize meal, whole grain |
|
18 |
0 |
0 |
|||
|
South and Central America |
|||||||
|
Argentina |
Maize flour |
Apr–Dec 1999 |
260 |
0 |
0 |
GEMS/Food programme |
Statistically based; representative of part of country |
|
Popcorn |
May–Jun 1999 |
658 |
2 |
1 |
|||
|
Maize meal |
1997 |
132 |
1 |
0 |
Solovey et al., 1999; |
Randomly purchased |
|
|
Cornflakes |
1997 |
— |
— |
— |
A = Sydenham et al., 1996c |
from commercial outlets; total sample ground |
|
|
Africa |
|||||||
|
Botswana |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground, and mixed before aliquots taken for analysis |
|
South Africa |
Maize meal |
1994 |
— |
— |
— |
Doko et al., 1996; |
|
|
Samp |
1994–95 |
— |
— |
— |
Rava, 1996; |
Samples collected from mills throughout country and ground before analysis |
|
|
Maize rice |
|
— |
— |
— |
|||
|
Maize grit |
|
— |
— |
— |
|||
|
Maize flour |
|
— |
— |
— |
|||
|
Super meal |
|
— |
— |
— |
|||
|
Special meal |
36 |
0 |
0 |
|
|||
|
Sifted meal |
|
No data |
0 |
0 |
|||
|
Unsifted meal |
19 |
0 |
0 |
|
|||
|
Germ meal |
|
No data |
0 |
0 |
|||
|
Maize bran |
|
No data |
No data |
No data |
|||
|
Screenings |
|
No data |
No data |
0 |
|||
|
Zambia |
Maize meal |
1994 |
No data |
0 |
0 |
Doko et al., 1996; |
Samples (1–5 kg) purchased from retail outlets, ground, and mixed before aliquots taken for analysis |
|
Zimbabwe |
Maize meal |
1994 |
No data |
0 |
0 |
||
|
Asia |
|||||||
|
China (Linqui County, Shandong Province) |
Maize meal |
1996 |
< 500 |
0 |
0 |
Groves et al., 1999; |
Random selection of households (3) in random selection of villages (7); samples frozen and 5-g portion cut for analysis |
|
Batter |
1996 |
<500 |
0 |
0 |
|||
|
Pancake |
1996 |
<500 |
0 |
0 |
|||
LOQ, limit of quantification; *, limit of detection; mean: true mean (for n analytical values, the true mean is the sum Xi / n, where Xi is the value of each analytical result; for not detected, Xi = 0); max: maximum concentration
References: P, parent reference, S, sampling method, A, analytical method
See Also:
Toxicological Abbreviations
FUMONISINS (JECFA Evaluation)