
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
WORLD HEALTH ORGANIZATION
SAFETY EVALUATION OF CERTAIN
FOOD ADDITIVES AND CONTAMINANTS
WHO FOOD ADDITIVES SERIES 40
Prepared by:
The forty-ninth meeting of the Joint FAO/WHO Expert
Committee on Food Additives (JECFA)
World Health Organization, Geneva 1998
AFLATOXINS
First draft prepared by
S. Henry1, F.X. Bosch2, J.C. Bowers1, C.J. Portier3,
B.J. Petersen4 and L. Barraj4
1 US Food and Drug Administration, Washington, DC
2 Institut d'Oncologia, Unitat d'Epidemiologia, Hospitalet del
Llobregat, Barcelona, Spain
3 National Institute of Environmental Health Sciences, Research
Triangle Park, NC, USA
4Novigen Sciences Inc., Washington, DC, USA (authors of section 4)
1. Explanation
2. Biological data
2.1 Biochemical aspects
2.1.1 Metabolism of aflatoxins
2.2 Toxicological studies
2.2.1 Acute toxicity
2.2.2 Special studies on reproductive toxicity
2.2.3 Special studies on genotoxicity
2.2.4 Special studies on immunosuppression
2.2.5 Factors modifying carcinogenicity of aflatoxins
2.2.6 Special studies on covalent binding of aflatoxin
residues with nucleic acids and proteins
2.2.7 Special studies on glucose tolerance
2.2.8 Special studies on effect of ammoniation of AFBI in
contaminated cottonseed
2.2.9 Special studies on aflatoxin and hepatitis B virus
infection in woodchucks, ducks, ground squirrels and
tree shrews
2.2.10 Observations in humans
2.2.10.1 Biomarkers of aflatoxin exposure
2.2.10.2 Mutations in p53 tumour-suppressor gene in
human hepatocellular carcinoma
2.2.10.3 Epidemiology of primary liver cancer
2.2.11 Summary of information on other aflatoxins
2.2.11.1 Aflatoxin B2
2.2.11.2 Aflatoxin G1
2.2.11.3 Aflatoxin G2
2.2.11.4 Aflatoxin M1
3. Estimating carcinogenic risks from the intake of aflatoxins
3.1 Information from various scientific disciplines and its
contribution to aflatoxin carcinogenic risk
3.1.1 Laboratory animal, mutagenicity and metabolic studies
3.1.2 Studies on the p53 gene
3.1.3 Epidemiological studies
3.1.4 Aflatoxin biomarker studies
3.2 General modelling issues
3.2.1 Choice of data
3.2.2 Measure of exposure
3.2.3 Measure of response
3.2.4 Choice of mathematical model
3.3 Potency estimates
3.3.1 Potency estimates based upon epidemiological data
3.3.2 Potency estimates not accounting for HBV infection
3.3.3 Potency estimates accounting for HBV infection
3.3.4 Potency estimates based on biomarker studies
3.3.5 Potency estimates from test species
4. Aflatoxin dietary intake estimates
4.1 Introduction
4.2 Background
4.3 Methods
4.3.1 Period of intake of relevance
4.3.2 Estimated levels of aflatoxins in foodstuff
4.3.3 Estimated intakes
4.4 Results
4.4.1 Aflatoxin levels in foods: general
4.4.2 Aflatoxin levels in foodstuffs: Occurrence data by
commodity
4.4.2.1 Amount of commodity imported
4.4.2.2 Accounting for the change in aflatoxin levels
during processing
4.4.3 National estimates of aflatoxin intake
4.4.3.1 Australia
4.4.3.2 China
4.4.3.3 European Union
4.4.3.4 USA
4.4.3.5 Zimbabwe
4.4.4 Relative impact of establishing maximum limits on
estimate of intake
4.4.4.1 Average aflatoxin concentrations using four
possible scenarios
4.4.4.2 Intake of total aflatoxins using four scenarios
4.4.4.3 Intake of aflatoxin b1 within four scenarios
4.4.5 Summary
5. Comments and evaluation
5.1 Aflatoxin potencies
5.2 Population risks
5.3. Conclusions
6. References
List of abbreviations
AAT alpha-1-antitrypsin
ADA aflatoxin-DNA adduct
AF aflatoxin (general)
AF-alb aflatoxin-albumin (adduct)
AFB1 aflatoxin B1
AFB2 aflatoxin B2
AFG1 aflatoxin G1
AFG2 aflatoxin G2
AFL aflatoxicol
AFM1 aflatoxin M1
AFP alpha-fetoprotein
AL ad libitum
ALT alanine aminotransferase
AM alveolar macrophage
APAT ambient temperature ammoniation procedure
BNF beta-naphthoflavone
CMI cell-mediated immunity
CR calorically restricted
CYP cytochrome P450
DHBV duck hepatitis B virus
DTH delayed type hypersensitivity
eAAIR estimated age adjusted incidence rate
EPHX epoxide hydrolase
GGT gamma-glutamyltranspeptidase
GHIS Gambia hepatitis intervention trial
GSHV ground squirrel hepatitis virus
GST glutathione S-transferase
GSTM1 glutathione S-transferase M1
HBV hepatitis B virus
HC high carbohydrate (diet)
HCC hepatocellular carcinoma
HCV hepatitis C virus
HF hypercaloric fat-containing (diet)
HPHT high temperature ammoniation procedure
IC isocaloric fat-containing (diet)
I3C indole-3-carbinol
LC liver cancer
LDH lactate dehydrogenase
MDA malonaldehyde
OECD Organisation for Economic Co-operation and Development
Orm matched odds ratio
PCR polymerase chain reaction
PHC primary hepatocellular carcinoma
PLC primary liver cancer
ROS reactive oxygen species
SeY selenium-enriched yeast extract
WHV woodchuck hepatitis virus
1. EXPLANATION
Aflatoxins B1, B2, G1, and G2 are mycotoxins that may be
produced by three moulds of the Aspergillus species: A. flavus,
A. parasiticus and A. nomius, which contaminate plants and plant
products. Aflatoxins M1 and M2, the hydroxylated metabolites of
aflatoxin B1 and B2, may be found in milk or milk products obtained
from livestock that has ingested contaminated feed. Of these
six aflatoxins, aflatoxin B1 is the most frequent one present in
contaminated samples and aflatoxins B2, G1 and G2 are generally not
reported in the absence of aflatoxin B1. Most of the toxicological
data relate to aflatoxin B1. Dietary intake of aflatoxins arises
mainly from contamination of maize and groundnuts and their products.
Aflatoxins were evaluated at the thirty-first meeting of the
Committee (Annex 1, reference 77), at which time the Committee
considered aflatoxin to be a potential human carcinogen. Sufficient
information was not available to establish a figure for a tolerable
level of intake. The Committee urged that the intake of dietary
aflatoxin be reduced to the lowest practicable levels so as to reduce,
as far as possible, the potential risk. A working group convened by
the International Agency for Research on Cancer also concluded that
naturally occurring aflatoxins are carcinogenic to humans1.
At the forty-sixth meeting (Annex 1, reference 122), potency
evaluations and population risk estimates were considered, and the
Committee recommended that these analyses be completed and presented
in an updated toxicological review.
At its present meeting, the Committee reviewed a wide range of
studies in both animals and humans that provided qualitative and
quantitative information on the hepatocarcinogenicity of the
aflatoxins. This monograph reviews the experimental evidence
concerning the carcinogenicity of the aflatoxins, evaluates the
potencies of these contaminants, links these potencies to intake
estimates, and discusses the impact of hypothetical standards on
sample populations and their overall risks.
The scientific literature on aflatoxins in the past thirty years
includes more than 3000 research articles. In 1971 aflatoxins were
reviewed in Volume 1 of the International Agency for Research on
Cancer (IARC) Monographs on the Evaluation of Carcinogenic Risk and
again in Volume 56 of the IARC monographs in 1993. Aflatoxins were
last reviewed by the JECFA in 1987. A key recent publication in the
aflatoxin field was the review by Eaton & Groopman (1994). Eaton &
1 Some naturally occurring substances: food items and constituents,
heterocyclic aromatic amines and mycotoxins. Lyon, International
Agency for Research on Cancer, 1993 (IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans, Vol. 56): 245-395.
Gallagher (1994) wrote a review of the mechanisms of aflatoxin
carcinogenesis. This review for JECFA will focus on key reports that
have appeared in the literature since the publication of the Eaton &
Groopman review and the 1993 IARC review.
2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Metabolism of aflatoxins
An excellent review on the cellular interactions and metabolism of
aflatoxin has been produced by McLean & Dutton (1995). Gorelick (1990)
compared metabolism of aflatoxin by different species. Guengerich
et al. (1996) discussed the involvement of cytochrome P450,
glutathione-S-transferase and epoxide hydrolase in the metabolism of
aflatoxin B1 (AFB1) and the relevance to risk of human liver cancer. A
wide variety of vertebrates, invertebrates, plants, bacteria and fungi
are sensitive to aflatoxins, but the range of sensitivity is wide for
reasons not yet fully understood (Cullen & Newberne, 1994). Two
important factors in species and strain variation of sensitivity are
1) the proportion of AFB1 that is metabolized to the 8,9-epoxide,
relative to other metabolites that are considerably less toxic, and 2)
the relative activity of phase II metabolism, which forms non-toxic
conjugates and inhibits cytotoxicity1. The 8,9-epoxide of AFB1 is
short-lived but highly reactive, and is believed to be the principal
mediator of cellular injury (McLean & Dutton, 1995).
Formation of DNA adducts of AFB1-epoxide is well characterized
(Cullen & Newberne, 1994). The primary site of adduct formation is the
N7 position of the guanine nucleotide.
It has been hypothesized that viral infection and associated
liver injury alter expression of carcinogen-metabolizing enzymes.
Kirby et al. (1994) tested this hypothesis in a hepatitis B virus
(HBV)-transgenic mouse model in which a synergistic interaction occurs
between AFB1 and HBV in the induction of hepatocellular carcinoma
(HCC). In this transgenic mouse lineage, overproduction of the HBV
large envelope protein results in progressive liver cell injury,
inflammation, and regenerative hyperplasia. Initially, two cytochrome
P450s important in AFB1 metabolism in the mice were identified -
CYP2a-5 and CYP3a, using specific antibodies and chemical inhibitors.
The expression of these P450 isoenzymes and an alpha-class glutathione
1 Phase I enzymes of major importance to carcinogen metabolism are
certain members of the superfamily (primarily within families 1-3) of
CYPs. In general, P450 enzymes catalyse the formation of more polar,
non-toxic products; however, bioactivation is sometimes a sequela. The
phase II enzymes of primary importance are the GST, which catalyse
conjugation of potentially toxic electrophiles to the tripeptide GSH,
generally rendering them non-toxic.
S-transferase (GST) isoenzyme, YaYa, was examined. Increased
expression and altered distribution of CYP2a-5 were demonstrated, by
immunohistochemical analysis, to be associated with the development of
liver injury in mice and to increase with age between 1 and 12 months.
CYP3a expression was also increased in HBV-transgenic mice, but the
increase was not as clearly related to age. GST YaYa levels were the
same in HBV-transgenic mice and their non-transgenic littermates of
all ages.
These results show that expression of specific cytochrome P450s
is altered in association with over-expression of HBV large envelope
protein and liver injury in this model. These findings may have
general relevance to human HCC, which is associated with a diverse
range of liver-damaging agents.
Judah et al. (1993) studied an aldehyde reductase in the rat,
which, in contrast with fractions from control animals, catalysed the
reduction of AFB1-dihydrodiol, in the dialdehyde form at physiological
pH values, to AFB1-dialcohol. This aldehyde reductase was expressed in
cytosolic fractions prepared from rat livers bearing pre-neoplastic
lesions, or following treatment with the anti-oxidant ethoxyquin. This
expression paralleled the development of resistance to the toxin. This
enzymatic mechanism might also have relevance in terms of the
development of resistance to other cytotoxic agents, the mechanism of
which involves metabolism to a reactive aldehyde. The authors
suggested that other systems, in particular human, be examined to
determine if this enzyme activity is expressed, and if so in what
circumstances, before its potential significance in the carcinogenic
process can be evaluated. For example, do the livers of humans
consuming diets contaminated with aflatoxins express such enzymes?
The monkey CYP1A1 has been expressed in BALB 3T3 A31-1-1 cells
and the expressed proteins were assayed for their capacity to activate
AFB1 and benzo[a]pyrene (B[a]P (Itoh et al., 1993). The transformed
cells were approximately 5.4- to 4.7-fold more sensitive to AFB1 and
B[a]P than the parental cells, respectively. The authors concluded
that monkey CYP1A1-cDNA encoded a functional protein and that the
expressed CYP1A1 enzyme is active in the activation of B[a]P as well
as AFB1 to produce toxic metabolites.
The combined presence of CYP1A2 and 3A4, both of which oxidize
AFB1 to the reactive AFB1-8,9-epoxide and to hydroxylated inactivation
products aflatoxin M1 (AFM1) and aflatoxin Q1 (AFQ1), substantially
complicates the kinetic analysis of AFB1 oxidation in human liver
microsomes. Gallagher et al. (1996) examined the reaction kinetics
of AFB1 oxidation in human liver microsomes (N = 3) and in human
CYP3A4 and CYP1A2 cDNA-expressed lymphoblastoid microsomes for the
purpose of identifying the CYP isoform(s) responsible for AFB1
oxidation at low substrate concentrations approaching those
potentially encountered in the diet. CYP3A4 with AFB1 was found to
have sigmoidal kinetics such that the rate of product formation fell
off quickly as the substrate concentration was reduced. CYP1A2 obeyed
Michaelis-Menten kinetics. Thus, at the low substrate concentrations
that probably occur in vivo, the formation of AFB oxide, as well as
AFB1 clearance, were predicted to be dominated by CYP1A2. Even at a
relatively higher substrate concentration, CYP1A2 formed approximately
three times as much AFB-exo-epoxide and generated three times as much
DNA binding as an equivalent amount of cDNA-expressed CYP3A4.
The authors pointed out that because AFB1 is highly lipophilic,
it is difficult to know how nominal concentrations of AFB in in
vitro microsomal preparations relate to concentrations in vivo.
The authors also discussed the work of Ueng et al. (1995), who
reported that CYP1A2 formed less AFB oxide than CYP3A4, using human
CYP1A2 and 3A4 proteins that were expressed in a bacterial expression
system. The discrepancy between the two studies, according to
Gallagher et al. (1996), may have been due to different sources of
P450s used in the two experiments.
Gallagher et al. (1996) concluded that the dominant route for
in vivo AFB1 activation at dietary concentrations obtained in human
liver is primarily thorough CYP1A2. Evidence that both CYP1A2 and 3A4
are involved in AFB1 metabolism in vivo is substantiated by
biomarker studies indicating the presence of AFM1 and AFQ1 in the
urine of individuals exposed to dietary AFB1 (Ross et al., 1992;
Qian et al., 1994). The ratio of activation:inactivation products
catalysed by CYP1A2 (roughly 2.5:1, AFBO:AFM1) and CYP3A4 (1:10;
AFBO:AFQ1) is likely to be a key determinant of the pathway and
biological consequences of in vivo AFB1 exposure. Unfortunately, the
actual urinary and faecal levels of these two metabolites, (in
particular, AFQ1 and possible secondary metabolites) following
exposure to AFB1 are not known. Thus, the relative ratio of these two
metabolites in individuals exposed to dietary AFB1, a key ratio, is
also unknown.
Sawada et al. (1993) show that human placental microsomes
activated AFB1, AFB1 showed relatively high mutagenic activity in the
Ames test when incubated with human placental microsomes. Addition of
alpha-naphthoflavone or aminoglutethimide, known inhibitors of
cytochrome P450 1A and P450 19, respectively, into the test system
partially inhibited the mutagen-producing activity.
Induction of glutathione-S-transferase placental form (GST-P)
positive hepatic foci has been examined by immunohistochemical
analysis in young male Fischer rats 3 weeks after a single i.p.
injection of AFB1 (Gopalan et al., 1993). Pretreatment of rats with
L-buthionine sulfoximine (BSO), a GSH depleter, at a dose of 4 mmol/kg
bw 4 and 2 hours before 1.0 mg AFB1 treatment enhanced both the number
of AFB1-induced hepatic foci and the area occupied by these foci by
approximately 400 and 575% above their respective controls without
affecting the mean diameter of these foci. Pretreatment of rats with
0.1% phenobarbital (PB) in their drinking water for 1 week before AFB1
(1 mg) treatment, inhibited AFB1-induced foci almost completely.
However, the number of AFB1-induced foci in PB-treated rats was not
significantly increased by BSO pretreatment.
Fetal rat liver has been shown to possess substantial levels of
glutathione-S-transferase (GST) activity toward AFB1-8,9-epoxide. The
enzyme responsible for this activity was an alpha-class GST
heterodimer comprising Yc1 and Yc2 subunits (Hayes et al., 1994).
The cDNAs encoding these polypeptides have been cloned and shown to
share approximately 91% identity over 920 base pairs, extending from
nucleotide -23 to the AATAAA polyadenylation signal. GST Yc2Yc2
expressed in Escherichia coli was found to exhibit 150-fold greater
activity toward AFB1-8,9-epoxide than GST Yc1Yc1. Comparison between
the structures of alpha-class GST suggested that tyrosine at residue
108 and/or aspartate at residue 208 is responsible for the high AFB1
detoxification capacity of Yc2. Immunoblotting and enzyme assays have
shown that liver from adult female rats contains about 10-fold greater
levels of Yc2 than is found in liver from adult male rats. This
sex-specific expression of Yc2 in adult rat liver may contribute to
the relative insensitivity of female rats to AFB1. Dietary
administration of oltipraz, a synthetic antioxidant which protects
against aflatoxin-hepatocarcinogenesis served as an inducer of GST
Yc2.
Gallagher & Eaton (1995) have investigated the biotransformation
of AFB1 in hepatic microsomal and cytosolic fractions from channel
catfish, an aquatic species shown to be refractory to AFB1 toxicity
and reported to be resistant to AFB1 hepatocarcinogenesis, and in
rainbow trout, a species sensitive to AFB1 toxicity and
hepatocarcinogenesis. AFB1 was poorly oxidized by channel catfish
microsomes, suggesting that the lack of microsomal AFB1 activation
together with the rapid conversion of AFB1 to aflatoxicol (AFL)
contributes to the apparent resistance of channel catfish to AFB1
toxicity and hepatocarcinogenesis.
Oltipraz is currently under evaluation as a possible
chemopreventive agent in humans. Primiano et al. (1995) investigated
the chemopreventive efficacy achieved by administration of
intermittent doses of oltipraz in rats. Fischer 344 rats were treated
with oltipraz (0.5 mmol/kg, p.o.) once weekly, twice weekly, or daily
over a 5-week period. After the first week, all rats were gavaged with
20 µg/kg AFB1 for 28 consecutive days. Livers were analysed 2 months
after the last AFB1 dose, and the volume of liver occupied by
glutathione-S-transferase (GST)-P positive foci, a presumptive marker
of neoplasia, was observed to be decreased by >95%, >97% or >99% in
livers of rats receiving once-, twice-weekly or daily oltipraz
treatments, respectively. The chemopreventive actions of oltipraz have
been associated with increases in the levels of phase 2 detoxifying
isozymes. Accordingly, GST conjugation activity measured with
1-chloro-2,4-dinitrobenzene as a substrate increased 1.5, 1.8 or
2.4-fold for the once-weekly, twice-weekly or daily treatments,
respectively, throughout a 7-day period. The authors suggested that
the protracted pharmacodynamic actions of oltipraz on enzyme induction
may account from the marked reduction in the hepatic burden of
AFB1-induced putative preneoplastic tumours after intermittent dosing.
Consequently, scheduling of intermittent dosing protocols may sustain
efficacy while improving drug tolerance and patient compliance over
long-term treatments. These properties of oltipraz increase its
attractiveness for clinical chemopreventive interventions, the authors
emphasized.
Langouet et al. (1995) investigated metabolism of AFB1 in
primary human hepatocytes with or without pretreatment by oltipraz.
AFM1, glutathione conjugates of AFB1 oxides and unchanged AFB1 were
quantified in cultures derived from eight human liver donors.
Parenchymal cell obtained from the three GST M1-positive livers
metabolized AFB1 to AFM1 and to AFB1 oxides derived from the isomeric
exo and endo-8,9-oxides, whereas no AFB1 oxides were formed in the GST
M1-null cells. Pretreatment of the cells with 3-methylcholanthrene or
rifampicin, inducers of CYP1A2 and CYP3A4 respectively, caused a
significant increase in AFB1 metabolism. Although oltipraz induced GST
A2, and to a lesser extent GST A1 and GST M1, it decreased formation
of AFM1 and AFB1 oxides, which involves CYP1A2 and CYP1A2. Inhibition
by oltipraz of AFB1 metabolism through a reduction in CYP1A2 and
CYP3A4 was also shown by decreased activity of their monooxygenase
activities toward ethooxyresorufin and nifedipine, respectively. The
significant inhibition by oltipraz of human recombinant yeast CYP1A2
and CYP3A4 was also shown. These results demonstrated that AFB1 oxides
can be formed by GST M1-positive human hepatocytes only, and suggested
that chemoprotection with oltipraz is due to an inhibition of
activation of AFB1 in addition to a GST-dependent inactivation of the
carcinogenic exo-epoxide.
AFB1-induced carcinogenesis has been shown to be both inhibited
and promoted by indole-3-carbinol (I3C), found in cruciferous
vegetables. Stresser et al. (1994a) examined the influence of
dietary treatment with I3C and the well-known Ah receptor agonist
beta-naphthoflavone (BNF) on the relative levels of different
cytochrome P-450 (CYP) isoforms known to metabolize AFB1 in male
Fischer 344 rats. After 7 days of feeding 0.3% I3C or 0.04% BNF alone
or in combination, the relative levels of hepatic CYP1A1, 1A2, 2B1/2
2C11 and 3A were assessed by laser densitometry of Western blots. Both
diets containing I3C markedly increased band densities of CYP1A1, 1A2,
and 3A1/2 with less effects on 2B1/2 and no effect on CYP2C11. BNF
also strongly increased band densities of CYP2C11, but had no effect
on CYP2C11 or 3A1/2, and repressed CYP2C11. In addition the in
vitro hepatic microsomal metabolism of AFB1 was examined at 16, 124,
and 512 TM substrate levels. The authors' results suggested that BNF
inhibits AFB1 carcinogenesis, in part by enhancing net production of
less toxic hydroxylated metabolites of AFB1, as a result of elevated
levels of P450, and that I3C may share this mechanism. However, other
mechanisms, such as direct inhibition of P450 bioactivation by I3C
oligomers, or induction of phase II enzymes, also appeared to
contribute.
Stresser et al. (1994a) also examined the influence of I3C and
BNF on the AFB1 glutathione detoxication pathway and AFB1-DNA
induction in rat liver. After 7 days of feeding approximately equally
inhibitory doses of I3C (0.2%) or BNF (0.04%) alone or in combination,
male Fischer 344 rats were administered [3H]AFB1 (0.5 mg/kg, 480
TCi/kg) i.p. and killed 2 hours later. All three diets inhibited
in vivo AFB1-DNA adduction. Using an improved HPLC method for
separation of the two isomeric forms of AFB1 8,9-epoxide-glutathione,
both I3C diets were shown to induce GST activities strongly toward
AFB1 exo-epoxide, whereas BNF alone induced activity weakly. Data
suggest that enhanced detoxication of AFB1 via increased glutathione
conjugation efficiency, as a result of elevated levels of the Yc2 GST
subunit, is one mechanism that contributes to a protective effect of
I3C against AFB1-induced preneoplastic lesions in the rat, and that
this mechanism also participates to a lesser degree in protection by
BNF.
The role of reactive oxygen species (ROS) in AFB1-induced cell
injury was investigated using cultured rat (male Fischer 344)
hepatocytes (Shen et al., 1995). Malonaldehyde (MDA) generation and
lactate dehydrogenase (LDH) release were determined as indices of
lipid peroxidation and cell injury, respectively. Exposure to AFB1 for
up to 72 hours resulted in significantly elevated levels of LDH being
released into the medium as well as the MDA generation in cultured
hepatocytes. These effects were dose-dependent, indicating that AFB1
was capable of inducing oxidative damages in the cell. Further, MDA
generation and LDH release were effectively inhibited by the addition
of the following: 1) superoxide dismutase (500 units/ml); 2) catalase
(1500 units/ml); 3) 10 mM desferrioxamine (a specific iron chelator),
or 4) 260 mM dimethyl sulfoxide (a hydroxyl radical scavenger). This
evidence therefore suggests that ROS, such as superoxide radicals,
hydroxyl radicals and hydrogen peroxides, are involved in AFB1-induced
cell injury in cultured rat hepatocytes, the authors concluded.
Kirby et al. (1993) examined liver tissues from 20 liver cancer
patients from Thailand, an area where the incidence of this tumour is
high and where exposure to aflatoxin occurs. The expression of hepatic
cytochrome P450s and GST was examined and this expression was compared
to the in vitro metabolism of AFB1. There was a >10-fold
inter-individual variation in expression of the various P450s
including CYP3A4 (57 fold), CYP2B6 (56-fold), and CYP2A6 (120 fold).
Microsomal metabolism of AFB1 to AFB1 8,9-epoxide and AFQ1, the major
metabolite produced, was statistically significantly correlated with
CYP3A3/4 expression and, to a lesser extent, with CYP2B6 expression.
There was a significantly reduced expression of major P450 proteins in
microsomes from liver tumours compared to microsomes from the paired
normal liver when analysed by Western immunoblot analysis.
The immunoreactive expression of the major human classes of
cytosolic GSTs (alpha, mu and pi) was also analysed in normal and
tumorous liver tissue. The expression of GSTA (alpha) and GSTM
(mu) class proteins was markedly decreased and GSTP (pi) increased
in the majority of tumour cytosols compared to normal liver. Cytosolic
GST activity was significantly lower in liver tumours compared to
normal liver. There was no detectable conjugation of AFB1 8,9-epoxide
to glutathione by cytosol either from tumorous or normal liver. Thus,
capacity of human cytosols to conjugate reactive AFB1 metabolites to
GSH resembled AFB1-sensitive species such as rat, trout and duck
rather than resistant species such as mouse and hamster. These data
indicate a strong capacity of multiple forms of human hepatic P450s to
metabolize AFB1 to both the reactive intermediate AFB1-8,9-epoxide and
the detoxification product AFQ1. The authors suggested, that, in view
of the lack of significant GST-mediated protection against AFB1 in
human liver, variations in expression of hepatic P450, due either to
genetic polymorphisms or to modulation by environmental factors, may
be important determinants in the risk of liver cancer development in
AFB1-exposed populations.
Liu et al. (1991) evaluated the functional significance of the
glutathione transferase (GST) mu polymorphism by measuring its
effect on AFB1-DNA adduct formation in vitro. Human liver cytosols
prepared from persons having low or high glutathione transferase
toward trans-stilbene oxide were incubated with human liver
microsomes, calf thymus DNA, and AFB1. AFB1-DNA binding was inhibited
to a greater extent in high conjugators than low conjugators; the
correlation between AFB1-DNA adduct concentrations and GST mu
activity was highly statistically significant. The authors suggested
that GST mu plays an important role in detoxifying DNA reactive
metabolites of AFB1, and this enzyme may be a susceptibility marker
for AFB1-related liver cancer.
Heinonen et al. (1996) studied the profile of AFB1 metabolism
and the extent of AFB1 binding to cell macromolecules in human liver
slices under experimental conditions that would allow direct
comparison to similar end-points in the rat, a species sensitive to
the carcinogenic actions of AFB1. Liver slices were prepared from
three individual human liver samples with a Krumdieck tissue slicer
and incubated with 0.5 µM [3H]AFB1 for 2 hours. Significant
inter-individual variations were observed in the rates of oxidative
metabolite formation and in specific binding to cell macromolecules.
The rates of oxidative metabolism of AFB1 to AFQ1, AFP1 and AFM1 in
the human liver samples were similar to those previously observed in
rat liver slices. AFB1-GSH conjugate formation was not detected in any
of the human liver samples, and yet specific binding of AFB1 to cell
macromolecules was considerably lower in the human liver slices
relative to that in rat liver slices. The authors postulated that
these studies suggest that an as yet unidentified protective pathway
may exit in human liver. These studies support the hypothesis that
humans do not form as much aflatoxin B1-8,9-epoxide as the rat, but
humans do not possess GST isozymes with high specific activity toward
the epoxide. Significant interindividual differences in AFB1
metabolism and binding between humans suggest the presence of genetic
and/or environmental factors that may make some humans more or less
susceptible to AFB1.
Gallagher et al. (1994) studied the metabolism of AFB1 in
microsomes derived from human lymphoblastoid cell lines expressing
transfected CYP1A2 or CYP3A4 (cytochrome P450) and in microsomes
prepared from human liver donors (n=4). The authors summarized their
findings as follows. Both CYP3A4 and CYP1A2 were involved in the
activation of AFB1 to the AFB1-8,9-epoxide; 1A2 appeared to have a
higher affinity for AFB1 and produced a higher ratio of activation
(AFB1-8,9-epoxide) to detoxification (AFM1) products, relative to 3A4.
3A4 may be expressed in human liver at a much higher level than 1A2,
such that in some individuals, 3A4 may be the predominant source of
AFB1-8,9-epoxide at low substrate concentrations, even though 3A4
produces AFQ1 predominantly. Such differences in the apparent kinetics
of these two P450s toward AFB1 indicate that the most important
determinant of individual susceptibility to AFB may well be the level
of expression of 1A2. Individuals with relatively high 1A2 expression
may be at particular risk for AFB1-induced DNA damage, since human
GSTs are relatively ineffective in detoxifying AFB1-8,9-epoxide.
Inhibition of 1A2 may prove to be an effective means of
chemointervention in AFB1-exposed populations. Of course, in vivo
human toxicity is ultimately determined by a complex set of processes.
In experiments by Ueng et al. (1995), human cytochromes P450
1A2 and 3A4 were expressed in Escherichia coli, purified, and used
in reconstituted oxidation systems. Relatively high catalytic
activities were obtained with such a system for AFB1
3 alpha-hydroxylation and 8,9-epoxidation. P450 3A4 was more active
than P450 1A2 in forming genotoxic AFB1 oxidation products; P450 3A4
formed AFQ1 and the exo-8,9-epoxide; P450 1A2 formed AFB1, some
AFQ1, and both the exo- and endo-8,9-epoxides. Plots of AFB1
3 alpha-hydroxylation and 8,9-epoxidation vs. AFB1 concentration
were sigmoidal in both human liver microsomes and the reconstituted
P450 3A4 system. The results were consistent, the authors
hypothesized, with the view that P450 3A4 is a major human liver P450
enzyme involved in AFB1 activation, although the in vivo situation
may be more complex due to the presence of the enzyme in the
gastrointestinal tract.
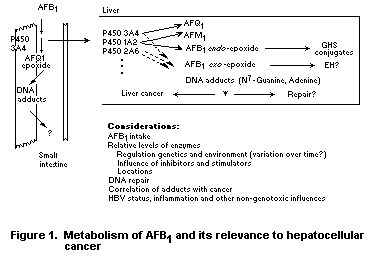
Guengerich et al. (1996) have reviewed a series of studies to show
the complexities encountered with metabolism of AFB1; the complexity
demonstrates the difficulties in doing molecular epidemiology studies,
even when a single chemical carcinogen has been identified. Figure 1
shows these metabolism complexities. With all the enzymes,
stereochemistry of the epoxide must be considered. In addition, the
P450s both activate and detoxify AFB1, and the effect of inducing
individual P450s is not easy to predict. P450 3A4 is expressed in the
small intestine, the site of absorption of orally ingested AFB1, where
the extent of detoxification is unknown. Even activation of AFB1 and
DNA alkylation in the small intestine may be considered to be a
detoxification process since the cells are sloughed rapidly, and
cancers of the small intestine are very rare.
 2.2 Toxicological studies
2.2.1 Acute toxicity
No additional acute toxicity studies have been reported in the
literature since the review by Eaton & Groopman (1994).
2.2.2 Special studies on reproductive toxicity
Ankrah et al. (1993) exposed ddy mice to AFB1 and AFG1 via
their feed (4.8 ng AFG1, 0.8 ng AFB1 (or both) per kg bw per day while
in utero. Levels of aflatoxin used were realistic relative to the
level of human exposure currently seen in Ghanaian foods. Offspring of
these animals (control and aflatoxin-fed) were continued on the
respective diets received by the parent stock until sacrifice at six
months of age. Blood obtained by cardiac puncture was used to
determine haema-tological indices and the sera were used to determine
glucose, triglyceride, total protein and albumin. AFG1 caused
significant accumulation of only neutral fat in the liver, a slight
rise in serum triglyceride and intensified hepatorenal inflammation,
necrosis and bile duct proliferation. AFB1 caused the accumulation of
both neutral fat and fatty acids in the liver, and was cytotoxic to
the liver and kidney. Iron storage in the liver, haematological
indices, serum total protein and albumin levels were not affected by
the aflatoxins. At the level used, AFG1 was six times in excess of
AFB1, but the latter was more severe in the observed hepatorenal
effects.
The authors pointed out that the mouse liver has been shown to
metabolize aflatoxin in a manner similar in some ways to the human
liver, although not all investigators would agree on this point. Hence
they postulated that the action of aflatoxin on mouse organs may shed
light on aflatoxin cytotoxicity in humans; results of this study are
of particular relevance to population groups that ingest foods known
to contain mainly AFG1 and to some extent AFB1.
2.2.3 Special studies on genotoxicity
AFB1 covalently binds to DNA and efficiently induces G to T
trans-versions; codon 249, one site in p53, is a striking hot spot for
AFB1 mutagenesis (Sengstag & Wurgler, 1994). Often, such mutations are
followed by the loss of the second functional alleles of tumour
suppressor genes, a phenomenon called loss of heterozygosity. To test
whether mitotic recombination leading to loss of heterozygosity is
induced by certain carcinogens, the authors genetically engineered a
Saccharomyces cerevisiae tester strain so that it metabolized two
important classes of carcinogens including AFB1. Human cDNAs coding
for the cytochrome P450 (CYP) enzymes CYP1A1 or CYP1A2 in combination
with NADPH-CYP oxidoreductase in a strain heterozygous for two
mutations in the trp5 gene were inserted. AFB1, when activated
intracellularly in microsomes isolated from the yeast strains
containing either human CYP enzyme, significantly induced mitotic
recombination. The authors concluded that activated AFB1 is a potent
inducer of DNA recombination in S. cerevisiae strains harbouring
various heterologous xenobiotic-metabolizing systems.
Young weanling Swiss albino mice were orally administered crude
AFB1 in a dose mimicking human exposure, i.e., at 0.05 µg/kg bw per
day for 14 weeks (Sinha & Dharmshila, 1994). Vitamin A (retinol) was
orally administered along with the toxin at double (132 IU/kg bw per
day) the human equivalent therapeutic dose. The authors concluded that
vitamin A minimized the frequency of toxin-induced clastogeny in both
mitotic and meiotic chromosomes. The decreases in sperm count, as well
as increases in abnormality in the gross morphology of the sperm head,
as observed upon toxin treatment, was ameliorated by the vitamin A.
Marquez-Marquez et al. (1993) evaluated the effects of an AFB1
inactivating system with ammonia on the genotoxicity of AFB1 measuring
micronucleus (MN) and sister chromatid exchange (SCE) analyses. Four
groups of CD1 male mice were fed for 8 weeks with a special diet
mainly composed of maize: 1) uncontaminated; 2) uncontaminated/
inactivated; 3) contaminated/ inactivated; and 4) contaminated. The
inactivating treatment was performed with ammonium hydroxide by
homogeneously impregnating the grain and leaving it for 20 days in
hermetically closed plastic bags and then heating in an oven for 24
hours to eliminate the residual ammonia. AFB1 was quantified before
and after inactivation. MN was evaluated at weekly intervals in
peripheral blood; SCE was quantified in bone marrow cells at weeks 4
and 8. The results showed that mice fed with AFB1 contaminated/
inactivated maize had a 45% lower level of induced cytogenetic damage
than those animals fed with AFB1 contaminated (but not inactivated)
maize. A residual amount of AFB1 remaining after the inactivating
treatment and the reconversion back to AFB1 in the organism may
account for the remaining increased levels of SCE and MN.
Marquez-Marquez et al. (1995) evaluated the efficiency of the
AFB1 inactivating system with ammonia, as described above, and using
mice (male CD-1) and micronucleus (MN) and sister chromatid exchange
(SCE) analysis. Apparently this study was the same as that published
earlier.
Occupational exposures to respirable dusts contaminated with the
mycotoxin AFB1 have been associated with an increased incidence of
upper airway tumours. To investigate this possible etiology Ball
et al. (1995) compared the abilities of tracheal and lung S9 from
rabbit (male, New Zealand white), hamster (male Syrian Golden) and rat
(male Sprague-Dawley) to activate AFB1 to mutagens in Salmonella
typhimurium TA98. These species differ in airway morphology with
respect to numbers of metabolically active non-ciliated tracheal
epithelial cells. Tracheas from hamster and rabbit and lung from
rabbit were active in converting AFB1 to bacterial mutagens. Tracheas
from hamster were more efficient in activating AFB1 to mutagens than
lung, while rabbit lung was over 4 times more active in converting
AFB1 to mutagens than that from trachea. In all cases, AFB1 was more
mutagenic than B[a]P. The relative capabilities of trachea to activate
AFB1 were in agreement with the ability of cultured tracheas from
these species to form AFB1-DNA adducts. These results demonstrate that
AFB1 is activated more efficiently than B[a]P in the lung, and that
the metabolic capabilities of airway epithelium to activate AFB1 are
not predictable by airway morphology.
A study by Shi et al. (1995a) examined the effect of two
selenium compounds, namely, sodium selenite and selenium-enriched
yeast extract (SeY) on the cytotoxicity, DNA binding, and mutagenicity
of AFB1 in cultured Chinese hamster ovary (CHO) cells. CHO cells,
after treatment with 2 µg/ml selenite or 80 µg/ml SeY, exhibited
increased resistance to AFB1-induced cell killing. At a concentration
of 50 µg/ml AFB1, cell survival, measured by the clonogenicity assay,
was increased by 21- and 10-fold in selenite- and SeY-treated cells,
respectively. However, selenium treatment did not appear to affect
AFB1-DNA binding. Similarly, no effect was observed on AFB1
mutagenicity, as determined by the hypoxanthine-guanine phosphoribosyl
transferase (HPRT) gene mutation assay. The results showed that
selenium could effectively protect cells from AFB1 cytotoxicity in
cultured cells, but had no effect on AFB1-DNA adduct formation or
mutagenesis. The authors suggested that there are multiple pathways of
AFB1 toxicity and that selenium can modulate AFB1-induced cell killing
independent of its genotoxicity.
Rats and mice differ markedly in sensitivity to AFB1
hepatocarcinogenicity, the former being sensitive and the latter
resistant. The purpose of this study was to determine whether the
formation of AFB1-albumin (AF-alb) adducts was related to the
induction of cytogenetic changes in vivo as a step to understanding
whether such markers of exposure may be informative with respect to
genetic alterations important in the carcinogenic process (Anwar
et al., 1994). The comparison was made at two levels: between
species and between individuals within a species. Animals (male Helwan
Wistar albino rats and Swiss albino mice) were treated with single
doses of different concentrations of AFB1 between 0.01 and 1.0 µg
AFB1/g bw. The frequency of chromosomal aberrations and micronuclei in
the bone marrow was measured and compared to the level of AFB1 bound
covalently to albumin in the peripheral blood. Both chromosomal
aberrations and micronuclei were significantly increased in treated
rats compared to the control group at doses above 0.1 µg/g. In
contrast, in mice, a slight increase in chromosome aberrations was
seen in the highest dose group (1.0 µg/g), but no increase in
micronuclei was observed at any of the doses. The level of chromosomal
aberrations was about 10 times higher in rats than in mice at the
highest dose of AFB1. AFB1-albumin increased linearly with dose of
AFB1, and there were strong statistically significant correlations at
the individual rat level with both chromosomal aberrations.
The level of AF-alb adducts was higher for a given dose in rats
than mice, as has been seen for the level of liver DNA adducts in the
two species. The metabolic basis of these differences has been
investigated and has been shown to be associated with the expression
of a specific glutathione-S-transferase isoenzyme in mice, which
efficiently conjugates the AFB1-epoxide to glutathione.
In rats, the level of AF-alb adducts was strongly correlated with
the frequency of both micronuclei and chromosomal aberrations in the
bone marrow. An increase in adduct levels was seen with exposures as
low as a few ng AFB1/g bw, whereas the genetic alterations were only
increased above control levels at doses around 0.1 µg/g.
The authors suggested two considerations for interpretation of
the present studies. First, the cells in which the micronuclei and
chromosomal aberrations were examined are not the target cells for
AFB1 hepatocarcinogenesis, and second, that this type of genetic
marker is relatively non-specific. Thus, the genetic alterations being
measured are not directly relevant to the carcinogenic process; this
limitation may be overcome as sensitive molecular techniques are
developed to measure mutation induced by aflatoxin in specific gene
sequences in somatic cells (See Aguilar et al., 1993). Recent
studies by these authors (Wild et al., in preparation) suggest that
AF-alb adducts reflect the differing species sensitivity to AFB1
carcinogenesis. This peripheral blood marker could be an indicator of
risk of liver cancer development in addition to being a marker of
exposure, the authors suggest, as has been further supported by the
study of Ross et al. (1992), in which the level of AFB1-N7 guanine
adduct in the urine was related to the subsequent risk of developing
hepatocellular carcinoma in a Chinese cohort. This study will be
discussed in section 2.2.10.
2.2.4 Special studies on immunosuppression
The immunosuppressive potential of AFB1 was evaluated in growing
rats (Raisuddin et al., 1993). The weanling rats (species
unspecified) were sub-chronically exposed to 60, 300 or 600 µg AFB1/kg
bw for four weeks on alternate days by oral feeding. Various
parameters of cell-mediated immunity (CMI) and humoral immunity were
assessed in control and treated animals. CMI was evaluated by
measuring delayed type of hypersensitivity (DTH) response and humoral
immunity was measured by plaque forming (PFC) assay. The
lympho-proliferative response assay for T- and B-cells was also
performed. It was observed that AFB1 selectively suppressed
cell-mediated immunity in growing rats. AFB1 suppressed CMI at the 300
and 600 µg dose levels only as measured by DTH response assay. The
authors concluded that continuous low level exposure of aflatoxin to
the growing host may enhance its susceptibility to infection and
tumorigenesis.
Jakab et al. (1994) conducted experiments to demonstrate the
immunosuppressive effects of AFB1 ingestion, in this case respiratory
tract exposure to AFB1. Rats (male Fischer 344) and mice (female
Swiss) were exposed either by aerosol inhalation or intratrachael
instillation to AFB1. Nose-only inhalation exposure of rats to AFB1
aerosols suppressed alveolar macrophage (AM) phagocytosis at an
estimated dose of 16.8 µg/kg with the effect of persisting for
approximately 2 weeks. To determine whether another mode of
respiratory tract exposure, intratrachael instillation, reflected
inhalation exposure, animals were treated with increasing
concentrations of AFB1, which also suppressed AM phagocytosis in a
dose-related manner, albeit at doses at least an order of magnitude
more than that obtained by aerosol inhalation. Intratrachael
administration of AFB1 also suppressed the release of tumour necrosis
factor-alpha from AMs and impaired systemic innate and acquired immune
defences as shown, respectively, by suppression of peritoneal
macrophage phagocytosis and the primary splenic antibody response. The
authors concluded that experimental respiratory tract exposure to AFB1
suppressed pulmonary and systemic host defences; they indicated that
inhalation exposure to AFB1 is an occupational hazard where exposure
to AFB1-laden dust is common, such as in grain dust.
2.2.5 Factors modifying carcinogenicity of aflatoxins
Young adult male Fischer rats maintained on a reduced calory diet
(60% of ad libitum food consumption) for 6 weeks showed a decrease
in the binding of AFB1 to hepatic or renal nuclear DNA and a reduction
of AFB-induced hepatocellular damage (Chou et al., 1993). Repeated
dosing of rats with AFB1 resulted in the inhibition of hepatic and
renal DNA synthesis as measured by [3H]thymidine incorporation.
However, the rate of DNA synthesis was greater in ad libitum (AL)
rats than in calorically restricted (CR) animals. Three days after
AFB1 dosing, the rate of DNA synthesis had recovered to the control
level. Cell cycle analyses measured by a flow cytometric method on
kidney cells of both AL and CR rats showed that there were no
significant changes in cell populations in the S phase between these
two groups of rats. AFB1 inhibited the cell proliferation by 33% (on
average). The restoration of the cell proliferation in kidney cells
was found on the third day after AFB1 dosing. The rate of regenerative
cell proliferation was found to be slightly greater in AL rats than in
CR animals. The AFB1-induced regenerative DNA synthesis in both liver
and kidney was retarded by CR.
Youngman & Campbell (1992) demonstrated that with young Fischer
344 rats the post-initiation development of AFB1-induced
gamma-glutamyltranspeptidase-positive (GGT+) hepatic foci was
markedly inhibited by low protein feeding, even though the energy
intake was greater. These investigators also studied this dietary
effect upon the development of hepatic tumours and the correlation of
foci development with tumour development. Following AFB1 dosing (15
daily doses of 0.3 mg/kg each), animals were fed diets containing 6,
14 or 22% casein (5.2, 12.2 or 19.1% protein) for 6, 12, 40, 58 or 100
weeks. Foci at 12 weeks and tumours at 40, 58 and 100 weeks developed
dose-dependently to protein intake. Foci development, tumour
incidence, tumour size and the number of tumours per animal were
markedly reduced, while the time to tumour emergence was increased
with low-protein feeding. Non-hepatic tumour incidence also was lower
in the animals fed the lowest protein diet. Foci development indices
(foci number, per cent liver volume occupied) were highly correlated
with tumour incidence at 58 and 100 weeks (r = 0.90-1.00). Tumour and
foci inhibition occurred in spite of the greater energy intake.
Previous results from a large ecological study in 65 rural
counties in China suggested that primary liver cancer in humans
primarily is associated with chronic HBV infection, coupled with
nutritional factors (e.g., animal protein) that elevate plasma
cholesterol level and encourage cancer growth (Campbell et al.,
1990). To test this hypothesis, the authors investigated the effect of
dietary animal protein on tumour development in HBV transgenic mice.
Male F2 offspring of 50-4 HBV transgenic mice were randomly assigned
to 6, 14 and 22% dietary casein. Serum was collected from the retro-
orbital vein and was analysed for the level of hepatitis B virus
antigen (HBsAg), the products of the S-transgene. The increases from
baseline in S-gene product observed for the normal protein animal
(22%) at 3 months was inhibited in the mid- and low-protein animals by
42% and 72%, respectively, with a highly significant dose-response
relationship (P<0.001). Serum glutamic-pyruvic transaminase activity
was not affected by diet treatment. The authors concluded that their
results strongly suggest that dietary casein controls, in a
dose-response manner, S-transgene expression in these experimental
animals.
Hasler et al. (1994) fed Fischer 344 rats a low-fat high
carbohydrate (HC) diet, an isocaloric fat-containing (IC) diet, a
hypercaloric fat-containing (HF) diet or a commercial rodent chow. The
effects of these diets were studied on the binding of AFB1 to
exogenous DNA and on the activities of hepatic glutathione
transferases (GSTs), cytochromes 2B1 and 1A1. Microsome-mediated
binding of [3H]AFB1 to exogenous DNA was significantly lower in the
HC rats than in the chow- and IC-fed rats. No significant differences
were noted between HF and either HC or IC rats. There was no
significant difference in hepatic GST activity of rats fed the
different diets. The authors suggested that high carbohydrate/low fat
diets may reduce microsome-mediated epoxidation of AFB1 to a larger
extent than high-fat diets. In general, high-fat diets increased
cytochrome 1A1 and 2B1 activities relative to chow and
high-carbohydrate diet. This suggested greater detoxification of AFB1,
thus reducing the amount of AFB1 available for hepatic macromolecular
binding, the authors concluded.
An excellent review by Massey et al. (1995) covers the
biochemical and molecular aspects of mammalian susceptibility to AFB1
carcinogenicity. Important considerations include: 1) different
mechanisms for bioactivation of AFB1 to its ultimate carcinogenic
epoxide metabolite; 2) the balance between bioactivation to and
detoxification of the epoxide; 3) the interaction of AFB1 epoxide with
DNA and the mutational events leading to neoplastic transformation; 4)
the role of cyto-toxicity in AFB1 carcinogenesis; 5) the significance
of non-epoxide metabolites in toxicity; and 6) the contribution of
mycotoxin-unrelated disease processes.
2.2.6 Special studies on covalent binding of aflatoxin residues with
nucleic acids and proteins
Shi et al. (1994) studied the effect of selenium on AFB1-DNA
binding and adduct formation. Male Fischer 344 rats, fed with up to 8
mg/litre sodium selenite in drinking-water for 8 weeks, were given a
single i.p. dose of AFB1. The rats were killed 24 hours later and the
amount of AFB1 bound to hepatic DNA and the amount of DNA adducts
formed were determined. Selenium pretreatment resulted in a
dose-dependent inhibition of AFB1-DNA binding as well as adduct
formation. This was accompanied by an increase of reduced glutathione
(GSH) in the liver of selenium-treated animals. These results
suggested that selenium could effectively inhibit AFB1-induced DNA
damage, which may be partially responsible for its anticarcinogenic
effect against AFB1.
Choy (1993) has reviewed the dose-response induction of DNA
adducts by AFB1 and its implication to quantitative cancer risk
assessment. Dose-response curves of DNA adduct formation after
ingestion or injection treatments in the rat were reviewed; a linear
dose-response relationship was observed in both injection and
ingestion studies at low doses. The author concluded that this
observation is consistent with the assumption of the linear
dose-response risk assessment model for genotoxic agents and justifies
the use of this model for quantitative cancer risk assessment for
aflatoxins. The author also concluded that although AFB1-DNA adducts
generated in rats, mice and humans reflect the "molecular dose" and
DNA damage in the target organ, bypassing the need for interspecies
pharmacokinetic dose adjustments, it is not possible to extrapolate
from rodents to humans at this time because human DNA adduct data are
incomplete.
2.2.7 Special studies on glucose tolerance
Glyoxalase-1 activity plays an important role in glucose
metabolism and has been reported to be depressed in mice fed low
levels of AFB1 (Ankrah, 1995). In the present study, glyoxalase-1
activity, glucose tolerance and pancreatic beta cell sensitivity were
examined in mice (male and female ddy) fed 0.045 ng AFB1 plus 0.450 ng
ABG1/g feed prenatally and for 6 months after birth. After glucose
challenge, the ratios between 0- and 2-hour serum glucose levels were
significantly higher than controls, indicating an increase in
tolerance of glucose in the aflatoxin-fed mice with lower glyoxalase-1
activity. Pancreatic beta cell sensitivity to stimulation by
tolbutamide was similar in both groups. However, liver malonic
dialdehyde was significantly higher in the aflatoxin-fed mice,
suggesting that the altered tolerance for glucose in the aflatoxin-fed
mice might be a consequence of aflatoxin-mediated peroxidative action
in the liver, the authors suggested.
2.2.8 Special studies on effect of ammoniation of AFB1 in
contaminated cottonseed
The effectiveness of ammonia in inactivating aflatoxin in
contaminated cottonseed was investigated (Bailey et al., 1994). Two
aflatoxin-contaminated cottonseed lots were treated separately using
atmospheric pressure, ambient temperature ammoniation procedure (APAT)
or a high pressure, high temperature ammoniation procedure (HPHT), and
incorporated into dairy cow rations. Isocalorific diets containing 25%
defatted, dried milk from cows fed aflatoxin-contaminated cottonseed
without or with APAT or HPHT treatment, or an aflatoxin-free human
grade commercial milk powder, were then fed for 12 months to rainbow
trout (Oncorhynchus mykiss). AFM1 concentrations in milk powders
without and with seed treatment were: APAT, 85 and <0.05 µg/kg; HPHT,
32 and <0.05 µg/kg. In the APAT experiment, trout consuming the diet
containing milk from cows fed the aflatoxin-contaminated cottonseed
had a 42% incidence of hepatic tumours; APAT cottonseed treatment
reduced this to 2.5%. Positive controls were included to demonstrate
trout responsiveness. AFB1 fed continuously for 12 months at 4 µg/kg
resulted in a 34% tumour incidence, whereas positive controls fed 20
µg AFB1/kg, 80 µg AFM1/kg, or 800 µg AFM1/kg for 2 weeks and killed 9
months later had a 37, 5.7 and 50% incidence of tumours, respectively.
The authors concluded that APAT ammonia treatment of
aflatoxin-contaminated dairy cattle cottonseed feedstock abolished the
detectable transfer of AFM1 or AFB1 into milk powder, and greatly
reduced the carcinogenic risk posed by any carry-over of aflatoxins or
their derivatives into milk.
In addition, the results confirm AFM1 to be a lower level
hepatocarcinogen in comparison with AFB1 in the trout carcinogenicity
assay. In the separate HPHT experiment, no tumours were observed in
the livers of trout fed diets containing milk from either the
ammonia-treated or untreated source, or the control diet containing 8
µg AFM1/kg. Positive controls fed 64 µg AFB1/kg for 2 weeks exhibited
a 29% tumour incidence 12 months later. Thus in this experiment,
neither AFM1 at 8 µg/kg nor any HPHT-derived aflatoxin derivatives
that might have been carried over into milk represented a detectably
carcinogenic hazard to trout, the authors conclude.
2.2.9 Special studies on aflatoxin and hepatitis B virus infection in
woodchucks, ducks, ground squirrels and tree shrews
Interactive hepadnavirus and chemical hepatocarcinogenesis has
been studied in woodchucks inoculated as newborns with woodchuck
hepatitis virus (WHV), which is closely related to the human hepatitis
B virus (Bannasch et al., 1995). When the woodchucks reached 12
months of age, AFB1 was administered in the diet at dose levels of 40
µg/kg bw per day for 4 months and subsequently 20 µg/kg bw per day
(5 days/week) for a lifetime. WHV DNA was demonstrated by Southern
blot hybridization in the serum and by PCR in the serum and/or liver
tissue. The histomorphology and cytomorphology of the liver were
investigated by light and electron microscopy. WHV carriers with and
without AFB1 treatment developed a high incidence of preneoplastic
foci or altered hepatocytes, hepatocellular adenoma and hepatocellular
carcinomas that appeared 6-26 months after the beginning of the
combination experiment. Administration of AFB1 to WHV carriers
resulted in a significantly earlier appearance of hepatocellular
neoplasms and a higher incidence of hepatocellular carcinomas compared
to WHV carriers not treated with AFB1. Neither hepatocellular adenomas
nor carcinomas (but preneoplastic foci of altered hepatocytes) were
detected in woodchucks receiving AFB1 alone, and no preneoplastic or
neoplastic lesions were found in untreated controls.
The authors pointed out that these results provide conclusive
evidence of a synergistic hepatocarcinogenic effect of hepadnaviral
infection and dietary AFB1. The striking similarities in altered
cellular phenotypes of preneoplastic hepatic foci similarities in
altered cellular phenotypes of preneoplastic hepatic foci emerging
after both hepadnaviral infection and exposure to AFB1 suggested
closely related underlying molecular mechanisms that may be mainly
responsible for the synergistic hepatocarcinogenic effect of these
oncogenic agents.
In addition, the authors observed that the decisive role of the
chronic WHV infection for hepatocarcinogenesis became particularly
evident in those animals that seroconverted after 1 year and showed
neither a chronic active hepatitis nor hepatocellular neoplasms, no
matter when AFB1 was given. From this observation, the authors
concluded that chronic hepatitis is not an absolutely necessary
condition for the development of HCC in WHV carriers.
To determine whether p53 mutations are common to HCCs of hosts
infected with related hepadnaviruses with and without treatment with
aflatoxin, Rivkina et al. (1994) studied the occurrence of mutations
in the p53 gene in HCCs of ground squirrels and woodchucks with a
history of infection with ground squirrel hepatitis virus (GSHV) and
woodchuck hepatitis virus (WHV). Sequencing of wild type p53 genes
from ground squirrels and woodchucks revealed remarkable homology
between the two species; using direct polymerase chain reaction
sequencing, the investigators analysed the state of the p53 gene in 20
HCCs from ground squirrels (2 uninfected, 7 with past and 11 with
ongoing infection with GSHV) and in 11 HCCs from woodchucks
persistently infected with WHV. Five GSHV carrier and two uninfected
ground squirrels received i.p. administration of AFB1. Only one
mutation - located in codon 176 of exon 5 - in the p53 gene of the
tested animals was detected and that in a GSHV-positive ground
squirrel treated with AFB1. The investigators suggested that in view
of the considerably lower apparent rate of mutations in comparison to
human HCCs, other etiological factors may be of greater significance
in the development of HCC in ground squirrels and woodchucks.
The unique mutation from G to T at the third base in codon 249
observed in human hepatocellular carcinoma has been suggested to be
linked to aflatoxin exposure. Imazeki et al. (1995) studied six
ducks with HCC, three of which were infected with duck hepatitis B
virus and five of which were fed a diet containing AFB1 for 1-2 years.
Liver tissues were analysed for the presence of point mutations at
this codon of the p53 gene by polymerase chain reaction and direct
nucleotide sequencing. None of the six ducks with HCC showed the
change at this codon regardless of duck hepatitis B virus infection.
Integration of duck hepatitis B virus DNA into the host genome was not
observed in two ducks that were chronically infected with the virus
and treated with AFB1. A third duck from Qitong Province in China,
where HBV and AFB1 are risk factors for HCC in humans, did show viral
integration. This suggested, in the opinion of the authors, that AFB1
itself might not be involved in the unique mutation at codon 249 in
hepatocarcinogenesis, or that other factors coincident with aflatoxin
may be responsible for this unique mutation.
Cova et al. (1996) used a Pekin duck model to examine the
effect of congenital duck hepatitis B virus (DHBV) infection and AFB1
exposure in the induction and development of liver cancer. The study
of the two major risk factors in the development of HCC, i.e.,
persistent hepatitis virus infection and exposure to dietary
aflatoxins, has been hampered by lack of an animal model, and these
experiments were undertaken to this end. AFB1 was administered to
groups of 13 DHBV infected or non-infected ducks at two doses (0.08
and 0.02 mg/kg) by i.p. injection once a week from the third month
posthatch until they were sacrificed 2.3 years later. Two control
groups of ducks not treated with AFB1 (one of which was infected with
DHBV) were observed for the same period. Higher mortality was observed
in ducks infected with DHBV and treated with AFB1 compared to
non-infected ducks treated with AFB1 and other control ducks. In the
groups of non-infected ducks treated with high and low doses of AFB1,
liver tumours developed in 3 out of 10 and 2 of 10 ducks,
respectively. In infected ducks treated with the high dose, 3 of 6
showed liver tumours; there were none with the low dose of AFB1. No
liver tumours were observed in the two control groups. Ducks infected
with DHBV and treated with AFB1 showed more pronounced periportal
inflammatory change, fibrosis and focal necrosis compared to other
groups. All DHBV carrier ducks showed persistent viraemia throughout
the observation period. An increase of viral DNA titres in livers and
sera of AFB1-treated animals compared to infected controls was
frequently observed.
No DHBV DNA integration into the host genome was observed,
although in one hepatocellular carcinoma from an AFB1-treated duck, an
accumulation of viral multimer DNA forms was detected. Unlike the
situation observed for woodchuck and ground squirrel, HCC has rarely
been associated with DHBV infection or integration of viral DNA in the
duck. HCC has to date been reported only in Chinese ducks from
Chi-tung County, not always associated with detectable virus, and with
only a single reported case of integrated DHBV. Colonies of
DHBV-infected ducks from other parts of the world do not develop HCC.
Prevalence of liver tumours observed in Chi-tung County ducks
reportedly correlated with the AFB1 food contamination and with the
incidence of primary liver cancer in these areas.
The authors observed a lower level of AFB1 binding to liver DNA
and plasma protein in the DHBV-infected ducks compared to non-infected
ducks after a single dose of AFB1; this finding appeared inconsistent
with the hypothesis that DHBV infection could increase the metabolic
activation of AFB1, as has been observed in woodchucks and in some
human data. The investigators noted that their observations were made
at a single dose at a single exposure and using one specific age of
ducks; all of these factors could have influenced the AFB1-DNA adduct
level.
Yan et al. (1996) reported the successful establishment of an
animal model in tree shrews (Tupaia belangeri chinensis) captured
from the wild and experimentally infected with human hepatitis B
virus. In animals exposed to AFB1 and infected with HBV, the incidence
of HCC was significantly higher than in the animals solely infected
with HBV or exposed to AFB1. AFB1-exposed animals received a total
dosage of 15-16 mg/animal. No HCC or precancerous lesions were found
in the controls that were neither HBV-infected nor AFB1-exposed. HBV
DNA and the protein it encodes were detected in the cancer cells
and/or the surrounding hepatocytes. Integration of HBV DNA into the
host liver genome was found during hepatocarcinogenesis among the
animals infected by HBV.
The investigators pointed out that the cumulative dose of AFB
used in their experiment was much lower than those (24-66 mg/animal)
used in previous experiments on tree shrews where HCC was seen. This
suggested that HBV infection might increase the hepatocarcinogenic
effect of AFB1. The occurrence of precancerous GT foci in the tree
shrews exposed only to AFB1 was much more frequent than in those
infected by HBV alone. Among the animals exposed to the same dose of
AFB1, the gamma-glutamyltranspeptidase (GGT) foci were more numerous
and larger in HBV-infected than in uninfected animals during the late
state (after the 83rd week), but not at the early state. This suggests
that, although both AFB1 and HBV may induce GGT foci and have a
synergistic effect, the effect of HBV is weaker and slower than that
of AFB1.
2.2.10 Observations in humans
2.2.10.1 Biomarkers of aflatoxin exposure
A key issue in the use of aflatoxin biomarkers is whether the
ratio of AFB1-albumin adduct to DNA adduct suggested in rodent
experiments is the same in humans (Wild et al., 1996). Direct
evidence for this is not available due to the limitation of measuring
DNA adducts in human liver. The human populations where AFB1 intake
vs. AFB1-albumin adduct relationship has been examined are populations
in which aflatoxin intake is relatively high. Examination of the
relationship in low-exposure populations would be important to test
whether the linear dose-response relationships seen in rats at
exposures as low as 1 ng AFB1/kg bw are also observed in man. There
are some data discussed in Wild et al. (1996) indicating that the
amounts of AFB1 intake bound to albumin are similar for rats and
humans; assuming that the majority of AFB1-DNA adducts are formed in
liver, then the initial ratio between the serum albumin and liver DNA
adducts would be expected also to be similar in humans and Fischer
rats. However, the capacity of human intestine to metabolize AFB1 must
be further explored to clarify this point.
It would appear that the AFB1-albumin adduct in peripheral blood
is a reliable marker of AFB1-DNA adducts in the liver in rodents (Wild
et al., 1996). Both of these parameters are at least qualitatively
associated with species susceptibility to AFB1 hepatocarcinogenesis.
Cross-species extrapolation to man suggests that the amount of
AFB1-albumin formed for a given exposure more closely approximates
that in the sensitive species rather than the resistant, and indicates
that the Fischer rat may be a more appropriate model than the mouse
for molecular dosimetry studies of AFB1 when, for example, validating
approaches for chemoprevention studies.
However, carcinogenesis is a multistep process; as pointed out in
Wild et al. (1996) AFB1-albumin adduct is acting as a surrogate
marker only for one critical step, the formation of AFB1-DNA adducts
in the target cell. The relationship between this marker and the
genetic consequences of exposure as well as the quantitative
association with HCC risk in man remain to be determined. In addition,
HBV and possibly HCC infection, are major risk factors. The
availability of more reliable markers of biologically effective dose
of AFB1 should contribute to improving attempts to understand the
mechanism of interaction between these two and other risk factors.
2.2.10.2 Mutations in p53 tumour-suppressor gene in human
hepatocellular carcinoma
Molecular epidemiological studies have found that a G to T
mis-sense mutation at the third base of codon 249 of the p53 gene,
effecting an arginine to serine substitution, occurs in high frequency
(up to 67%) in human liver tumours in regions with high risk of
aflatoxin exposure, but not in regions of low aflatoxin exposure
(Ozturk, 1991). Hsieh & Atkinson (1995) performed experiments to
confirm this, using liver tissue from liver cancer patients in Taiwan
and Japan. This was analysed for the presence of aflatoxin-DNA adducts
(ADA) as a marker for aflatoxin exposure and an AGG to AGT
transversion at codon 249 of the p53 gene. Ten per cent of samples
containing ADA, indicating definite exposure of the subjects to
aflatoxin, were found to harbour the codon 249 mutation, whereas 18%
of the samples with no detectable adducts also contained the mutation.
Since the presence of ADA in the liver tissue samples is an indication
of definite recent exposure of the liver cancer patients to aflatoxin,
these data indicated that the codon 249 mutation is not a high
frequency event associated with recent aflatoxin exposure. If recent
exposure to aflatoxin is indeed involved in the late stage
hepatocarcinogenesis, these data suggested that it is through some
mechanism other than codon 249 mutation. If either mutation at codon
249 of the p53 gene or exposure to aflatoxin is involved in earlier
stages of hepatocarcinogenesis, whether codon 249 of the p53 gene is a
"hot spot" for aflatoxin attack could be shown by the present
experiment, the authors concluded.
The tumour suppressor p53 exerts important protective functions
towards DNA-damaging agents (Gerbes & Caselmann, 1993). Its
inactivation by allelic deletions or point mutations within the p53
gene as well as complex formation of wildtype p53 with cellular or
viral proteins is a common and crucial event in carcinogenesis.
Mutations increase the half-life of the p53 protein allowing the
immunohistochemical detection and anti-p53 antibody formation.
Distinct G to T mutations in codon 249 leading to a substitution
of the basic amino acid arginine by the neutral amino acid serine are
responsible for the altered functionality of the mutation gene product
and were originally identified in 8 of 16 Chinese and 5 of 10 African
HCC patients, both groups living in regions with traditionally high
exposure to mycotoxins. None of these mutations was detectable in 20
patients with HCCs recently studied in the United Kingdom; only two of
13 HCC DNAs from Germany displayed a C to T and a T to A transversion,
respectively, in codons 257 or 273, but not in codon 249. An average
p53 gene nutation rate of 25% is currently assumed for high-AFB1
exposure regions; this is double the rate observed in low-AFB1
exposure countries. The authors concluded that although many HCC
patients displaying P53 mutations also suffer from HBV infection,
which itself can lead to rearrangement of P53 coding regions or induce
the synthesis of viral proteins possibly interacting with p53, the
specific G to T transversion within codon 249 of the P53 gene seems to
directly reflect the extent of AFB1 exposure and is not pathognomonic
for all HCCs.
Yap et al. (1993) analysed 24 HCC liver biopsy samples from
patients in Durban, South Africa, for p53 mutations and HBV infection.
One patient was negative for HBV (Hbsag, anti-HBcAb, anti-HBsAb) and
possessed the p53 249 mutation (which results in an arginine to serine
substitution). The authors suggested that HBV infection or integration
increases the likelihood of, but is not essential for, this p53
"hot-spot" mutation in HCC. AFB1 or other as yet unidentified
environmental carcinogens and cofactors are implicated; the mechanisms
by which cells exposed to these agents acquire such a specific
mutation and then expand clonally remains to be elucidated.
The subject of the mutation at codon 249 of the p53 tumour
suppressor gene has continued to be the subject of much research.
Fifty-eight per cent of HCCs from Quidong, China, contain this
mutation which is rarely seen in HCCs from Western countries (Aguilar
et al., 1994). The population of Qidong is exposed to high levels of
AFB1 and this toxin has been shown to induce the same mutation in
cultured human HCC cells. To investigate the role of AFB1 and of these
p53 mutations in hepatocarcinogenesis, normal liver samples from the
USA (5), Thailand (3), and Qidong (14) (where AFB1 exposures are
negligible, low, and high, respectively), were examined for p53
mutations. The frequency of the AGG to AGT mutation at codon 249
paralleled the level of AFB1 exposure, which supports the hypothesis
that this toxin has a causative - and probably early - role in
hepatocarcinogenesis. However, a role for other carcinogens cannot be
ruled out, the authors point out; bulky heterocyclic amines in cooked
foods and oxidants released by inflammatory leukocytes possess the
same specificity for G to T transversion and HBV infection is
associated with inflammation. All of the liver samples from Qidong and
Thailand were from HBV-infected individuals.
The presence of elevated frequencies of codon 249 AGT mutations
in the non-malignant tissue of HCC patents from Qidong suggested that
the mutagenic event occurred early in hepatocarcinogenesis. In
contrast, p53 mutations in HCCs from geographic areas with low
exposure to AFB1 could be late events. For example, p53 mutations have
been observed more frequently in large tumours and in advanced grades
of malignancy in HCCs from Japan. In other organs, such as the colon
and the bladder, p53 mutations are thought to occur late in
tumorigenesis. However, the methods used in previous work may not have
been sensitive enough to detect mutations at early stages of
tumorigenesis.
Fujimoto et al. (1994) tested the hypothesis that exposure to
AFB1 alone or coincident with other environmental carcinogens might be
associated with allelic losses occurring during development of human
hepatocarcinogenesis (HCC) in China. The HCCs were obtained from two
different areas in China: Qidong, where exposure to HBV and AFB1 is
high; and Beijing, where exposure to HBV is high, but that to AFB1 is
low. Tumours were analysed for mutations in the p53 gene and loss of
heterozygosity for the p53, Rb and APC genes and at marker loci on
chromosomes 4, 13 and 16. The data indicated that mutation and/or loss
of heterozygosity in the p53 gene, independent of the 249 mutation,
played a critical role in the development of HBV-associated HCCs in
China. The authors postulated that different mechanisms appeared to be
responsible for the development of HCC in Beijing and may have
resulted from exposure to unknown environmental carcinogens or a
different subtype of HBV. Also, the results demonstrated that multiple
alterations in DNA located on different chromosomes may be involved in
the development of HCC.
Additional support for the etiological role of AFB1 in
hepatocarcinogenesis in regions of the world with AFB1-contaminated
food has come from the studies of Aguilar et al. (1993). These
investigators studied the mutagenesis of codons 247-250 of p53 by rat
liver microsome-activated AFB1 in human HCC cells HepG2 by restriction
fragment length polymorphism/polymerase chain reaction genotypic
analysis. AFB1 preferentially induced the transversion of G to T in
the third position of codon 249, and also induced G to T and C to A
transversions into adjacent codons, albeit at lower frequencies. Since
the latter mutations are not observed in HCC, the investigators
concluded that both mutability on the DNA level and altered function
of the mutant serine 249 p53 protein are responsible for the observed
mutational hot spot in p53 HCC from AFB1-contaminated areas. The fact
that this mutation is only rarely found in HCC from low AFB1 regions
indicates that it is not a prerequisite for hepatocarcinogenesis;
perhaps HBV and the mutant serine 240 p53 protein play a synergistic
role.
In a later study, Aguilar et al. (1995) examined normal liver
samples from the USA, Thailand and Qidong, where AFB1 exposures are
negligible, low and high, respectively, for p53 mutations. The
frequency of the AGG to AGT mutation at codon 249 paralleled the level
of AFB1 exposure, which, according to the authors, provides additional
support for the hypothesis that this toxin has a causative and
probably early role in hepatocarcinogenesis.
Hulla et al. (1993) analysed the p53 gene at the site
corresponding to codon 249 of the human gene in AFB-induced
preneoplastic hepatic nodules from rats. No mutations were detected in
the tissues examined. Thus, at least in the rat, the authors suggested
that AFB exposure alone may not be sufficient for the specificity of
p53 mutations observed in HCC. The selective mutations have been
identified only in populations at risk for hepatitis B; it is possible
that both AFB1 and chronic hepatitis are essential for mutation at
codon 249 in the human p53 gene.
In another study in rats, Liu et al. (1996) looked at the
effects of AFB1 on the p53 locus at the preneoplastic stage of rat
liver oncogenesis. Male Wistar rats received a single dose of 1.5 mg
AFB1/kg bw by a gastric tube. Liver biopsies over a period of one year
were examined for aberrations of the p53 gene together with the
expression of placental GST, a marker for preneoplasia.
Immunohistochemistry, Western blot, polymerase chain
reaction-single-strand conformation polymorphism and DNA sequencing
techniques were used. AFB1 induction resulted in GST overexpression,
forming GST-positive multi-foci and nodules of hepatocytes but no
aberrations in the p53 expression and the microstructure of exons 5-8
of the p53 gene. Thus, the authors concluded that p53 mutations might
not occur at this early stage of AFB1-induced hepatocarcinogenesis.
Shi et al. (1995b) characterized p53 mutations in 44
hepatocellular carcinomas from Chinese patients residing in a
high-incidence area. In contrast to HCCs from other high HCC incidence
areas with endemic aflatoxin exposures, in which codon 249 is a
mutational hotspot, no mutations were observed at codon 249. The
authors concluded that risk factors other that dietary exposure to
aflatoxin may contribute to the high HCC incidence in Singapore.
Liang (1995) recently reviewed the relationship of p53 proteins
and AFB1. He pointed out firstly that the murine mutant p53 gene
p53Ser249 appears to have a hepatocyte-specific phenotype, which
suggests that this gene may interact with cellular factors(s) in a
liver-specific manner to alter the growth property of hepatocytes. It
is not known if the human form of p53Ser249 exhibits the same
properties. Secondly, cooperative interaction of this p53 mutation and
viral-induced cellular changes are probably involved in the
transformation of hepatocytes in situations where aflatoxin exposure
and hepatitis viral infection are evident. Recent studies of
non-aflatoxin-associated HCC showed that p53 mutations are not as
common as other human malignancies. This difference could be explained
by the relatively low proliferation rate of hepatocytes as compared
with other epithelial cells, such as colonic mucosa and mammary gland.
Because p53 plays a critical role in "damage control" of proliferating
cells and in regulation of abnormal proliferation, it is reasonable to
speculate that p53 mutations may play a lesser role in
hepatocarcinogenesis. However, dysregulated p53 function may still be
an important step in this process, in view of the recent observation
that HBX protein encoded by HBV appears to interact with p53 and
inhibit its function.
Thirdly, it has not been possible to induce the same p53 mutation
with aflatoxin exposure in a murine model, which casts a shadow of
doubt on the applicability of studies in the murine model to human
hepatocarcinogenesis. Liang (1995) recommended using human p53 genes
to perform parallel experiments.
Harris (1995) has also reviewed this subject. He reiterated that
in high-incidence liver cancer areas such as China and Mozambique, the
high frequency of G:C to T:A transversions in human hepatocellular
carcinomas in this region could be due to the high mutability of the
third base of codon 249 by AFB1 or a selective growth advantage of
hepatocyte clones carrying this specific p53 mutant in liver
chronically infected with HBV. The third base of codon 240 in a human
liver cell line exposed to AFB1 has been shown to be preferentially
mutated, and transfected 240Serp53 mutant enhances the growth rate of
the p53 null hepatocellular carcinoma cell line Hep3B.
The hypothesis that some of the mutations observed in the p53
tumour-suppressor gene may be specific markers of exposure to
aflatoxin may represent a real breakthrough in the field of liver
cancer epidemiology. In particular, the confirmation of the
specificity of the p53/aflatoxin association could be useful in
assessing and quantifying the responsibility of aflatoxin as an
independent cause of liver cancer and in evaluating the likely
interactions with the hepatitis viruses in humans. A word of caution
should be raised regarding the interpretation of the early studies
because of: 1) the small sample size and limited methodology as to the
criteria of specimen inclusion; 2) inadequate adjustment of the
correlations for exposures to other viral and non-viral risk factors
at the individual level; 3) limited information on the sensitivity and
specificity of the proposed genetic markers; in particular, some
animal data and cell system data are inconsistent in showing a
specific association between p53 codon 249 mutations and previous
exposure to aflatoxin; and 4) insufficient knowledge of the additional
genetic changes in p53 and other genes (i.e., N-ras, C-myc, c-fos,
alpha-TGF) associated with liver cancer development.
2.2.10.3 Epidemiology of primary liver cancer
(a) Descriptive epidemiology.
Liver cancer is a disease prevalent in some of the developing
parts of the world. It is frequent in China, South East Asia and
subsaharan Africa. In some of these regions, like the Qidong area in
Southern China, liver cancer is the major cause of death to cancer
among men. It is relatively common in Japan and in the countries in
the Mediterranean basin and it is rare in the Americas and Northern
Europe. Pockets of high risk populations have been described in the
Amazonian basin, among Eskimos and in special populations like the
renal transplant patients. The incidence of liver cancer is
consistently higher in men than in women with a sex ratio ranging from
2 to 3 in most countries. Within countries, further variation in
incidence rates is observed across cancer registries, men showing
greater variation than women. Worldwide, the incidence of liver cancer
in men and women shows a strong correlation.
Migration from high risk areas to lower risk areas tends to
reduce the risk to the levels of the host country, and this is
observable within first and second generations.
(b) Etiology
The etiology of primary liver cancer is nowadays largely
understood. Table 1 summarizes the range and the point estimates of
the attributable fractions in two different settings, the low-risk
areas in Europe and the USA and the high-risk areas in Africa and
Asia.
In both scenarios, viral infections to hepatitis B or C virus are
associated with liver cancer in a range from 65% to 100% of cases. In
low-risk countries HBV predominates and the other relevant factors are
alcohol, tobacco and oral contraceptives. In high-risk areas HBV
predominates and aflatoxins play a role, although quantification has
been difficult.
The evidence points to a synergistic interaction between HBV and
AF in the etiology of liver cancer and some debate exists as to the
independency of AF as an etiologic agent in humans.
It is noteworthy that the large majority of the available
epidemiological studies including data on aflatoxin exposure are based
on high-risk countries where both HBV and AF are highly prevalent.
Since the nature of the interaction at low levels of exposure is
unknown, extrapolation of results from available studies to other
settings is questionable.
In addition to these established factors, studies have identified
other factors that may modulate the incidence of the disease. Risk
factors identified are the use of contaminated drinking-water, liver
flukes and severe malnourishment. Protection from liver cancer has
Table 1. Causal factors of liver cancer and estimates of the attributable fractions
Factor Low-risk countries High-risk countries
Japan Europe and the USA Africa and Asia
Estimate Range Estimate Range Estimate Range
Hepatitis B <15% 4-50% 20% 18-44% 60% 40-90%
Hepatitis C3 60% 12-64% 50% 40-80% <10% NE
Aflatoxin limited exposure limited exposure important exposure1
Alcohol <15%4 <20% 11-30%5 NE
Tobacco <12%4 40% 38-51%5 NE
Oral contraceptive 10-50%2 NE NE
Other <5% <5%
1 Attributable risk not quantified. One study suggested attributable fraction close
to 50%.
2 Restricted to liver cancer in women. Likely to increase in future generations.
Uncertain if hepatitis infections (notably HCV) are necessary co-factors.
3 Not including double infections with HBV and HCV. Very few studies available using
second-generation assays.
4 Estimates for the USA
5 Estimates from three studies of LC in men
NE non evaluated.
Note: attributable fractions do not necessarily add to 100% due to multiple exposures and
possible interactions between risk factors.
Adapted from CDC, 1989; Bosch & Muñoz, 1991; Thomas, 1991; Tanaka et al., 1993; IARC,
1994; Bosch, 1995
been reported in the case of diets rich in retinol and protein.
Associations have been reported between liver cancer and blood
testosterone levels, HLA types, and predisposition due to
polimorphisms in some of the SGT and CYT metabolic regulatory genes.
(c) Vaccination against HBV as a preventive measure against liver
cancer
In 1983, the World Health Organization proposed as a medium-term
objective trials of immunization against hepatitis B to prevent liver
cancer. Since then more than 70 countries have introduced HBV
vaccination into their routine vaccination schemes. A recently
published study in Taiwan (Chang et al., 1997) has described the
rigorous application of universal immunization against hepatitis B and
the prevention of the carrier state in children; these data provide
further evidence of a direct causal relationship between HBV and liver
cancer.
The immunization programme against hepatitis B in Taiwan, an area
of hyperendemic infection and moderate to high aflatoxin exposure,
reduced the rate of HBV carriage in six-year-old children from about
10% in the period from 1981 to 1986 to between 0.9 and 0.8% in the
period from 1990 to 1994. The drop in the rate of carriage occurred as
the proportion of infants immunized against hepatitis B increased from
15% of children born to mothers at high risk during the earlier period
to 84-94% of all newborn infants during the later period. This
significant reduction in the prevalence of hepatitis B surface antigen
was accompanied by a decline in the average annual incidence of
hepatocellular carcinoma in children 6 to 14 years of age, from 0.7
per 100 000 between 1981 and 1986 to 0.57 between 1986 and 1989 and
0.36 between 1990 and 1994. The incidence of hepatocellular carcinoma
in children 6 to 19 years old fell even more dramatically, from 0.52
among those born between 1974 and 1984 to 0.13 among those born
between 1984 and 1986. As the investigators pointed out, since the
incidence of hepatocellular carcinoma in Taiwan peaks in the sixth
decade of life, it may take 40 years or longer to see an overall
decrease in the rate of hepatocellular carcinoma as a result of the
vaccination programme.
The Committee noted that studies like this one need to be observed
carefully in coming years for the light they may shed on the
relationship between aflatoxin, HBV and liver cancer.
(d) Effects of exposure to aflatoxins
Ahmed et al.(1995) undertook two prospective studies to
determine a possible relationship between perinatal aflatoxin exposure
and neonatal jaundice. First, cord blood samples from 37 neonates who
subsequently developed jaundice and from 40 non-jaundiced (control)
babies were analysed for six major aflatoxins and aflatoxicol.
Peripheral blood samples of both groups were also analysed postnatally
for aflatoxins. In a second study, serum aflatoxin levels of 64
jaundiced neonates admitted from outside the hospital were compared
with levels in 60 non-jaundiced control babies. Aflatoxins were
detected in 14 (38%) cord blood samples of jaundiced neonates and in
nine (23%) of the controls. The mean cord aflatoxin concentration was
highest in jaundiced neonates with septicaemia, but the difference was
not statistically significant. The frequency of detection of
aflatoxins in peripheral blood was not significantly different in
jaundiced and non-jaundiced babies. Aflatoxins were detected in the
blood of over 50% of neonates with jaundice of unknown etiology. There
was no correlation between severity of hyperbilirubinaemia and serum
aflatoxin levels. Further studies are needed to determine the extent
of pre-and postnatal exposure to aflatoxin in Nigerian infants and the
effects of such exposure on fetal and neonatal health, the authors
concluded.
In October 1988, 13 Chinese children died of acute hepatic
encephalopathy in the northwestern state of Perak in peninsular
Malaysia (Lye et al., 1995). Symptoms included vomiting,
haematemesis, seizures, diarrhoea, fever and abdominal pain. All had
liver dysfunction with increased aspartate aminotransferase and
alanine aminotransferase levels greater than 100 IU/litre. The
acuteness of the illness differed from previously reported outbreaks
described in Kenya, India and Thailand; median incubation period for
this outbreak was 8 hours, whereas the exposure was over a period of
days to weeks of consumption of highly contaminated food such as maize
in outbreaks in Kenya and India. Epidemiological investigations
determined that the children had eaten a Chinese noodle, loh see
fun, hours before they died. The attack rates among those who had
eaten the noodles were significantly higher than those who had not
(P < 0.0001). The cases were geographically scattered in six towns in
two districts along the route of distribution of the noodle supplied
by one factory in Kampar town. Aflatoxins were confirmed in the
postmortem samples from patients, but the noodles or their ingredients
were not analysed for aflatoxins. The authors questioned the etiology
of the outbreak.
Ibeh et al. (1994) examined the relationship between aflatoxin
levels in serum of infertile men in comparison with random controls
from the community. The subjects were 100 adult males, yielding 50
semen samples, from men attending infertility clinics at a university
teaching hospital and 50 normal men in the same community. The staple
foods of the men were assayed for aflatoxin content. Aflatoxin was
found in 20 semen samples from the infertile group (40 %) with a mean
concentration of 1.7 µg/ml and four samples from the fertile group
(8%) with a mean concentration of 1.0 µg/ml. Infertile men showed a
higher percentage of spermatozoa abnormality (50%) than the fertile
men (10-15 %).
In a parallel experiment, adult male rats were given an aflatoxin-
contaminated diet (8.5 µg purified AFB1/g of feed) for 14 days while
10 control age-matched rats were fed a normal aflatoxin-free diet
during the same period. Seven days after the withdrawal of aflatoxin
from the diet of test rats, two rats were randomly selected from the
test and control groups, killed and their semen harvested from the
epididymis and vasa deferentia and analysed. The process was repeated
at weekly intervals until four rats were left in the test and control
groups. Thereafter, four fertile adult female rats were introduced to
mate with test and control rats, and rats were observed for 90 days
with an adequate, non-aflatoxin-contaminated diet. Results
showed that rats exposed to dietary aflatoxin experienced changes in
spermatozoal profile which differed in a statistically significant
manner from the control rats; test rats showed depression in the
motility, viability and number of sperm cells which resembled features
seen in semen of infertile men exposed to aflatoxin. The four test
rats who were mated in the conclusion of the study were unable to
effect conception of fertile female rats, while the four control rats
were able to do so.
The authors hypothesized that aflatoxin may affect the
reproductive system by its toxic effect on the liver, leading to the
desquamation of the membranes of hepatocytes, the mitochondria, the
cytosol and the endoplasmic reticulum. This cellular damage could
include inhibition of enzyme synthesis and/or enzyme activities or
inhibition of lipid metabolism or fatty acid synthesis, which may
derail the capacity of the hepatocytes to handle the conversion of
intermediate biomolecules, such as precursor molecules for hormones,
e.g., testosterone and progesterone. Depression or absence of normal
hormone levels could cause a wide range of degenerative changes in
sexual organs. Aflatoxin may also affect the male reproductive system
by causing lysis of sperm cells as a result of constant reversible
reaction with the mycotoxin, binding of the toxin to free and/or bound
amino acids in the seminal fluid, depressing the motility of
spermatozoa and the formation of aflatoxin adducts with nucleic acids,
giving a risk of mutations of the spermatogonia.
El-Nazami et al. (1995) examined the exposure of infants to
aflatoxin M1 (AFM1) and of lactating mothers to AFB1, using AFM1 in
breast milk as a biomarker for exposure to AFB1. Prevalence of AFM1 in
breast milk samples from 73 women from Victoria, Australia
(low-exposure area) and 11 women from Thailand (high-exposure area)
was also compared. Assays were done by both HPLC and by ELISA. AFM1
was detected in 11 samples from Victoria and five samples from
Thailand at median concentrations of 0.071 mg/ml (range 0.028 - 1.031)
and 0.664 ng/ml (range 0.039 - 1.736). Levels of AFM1 were
significantly higher in milk samples from Thailand than in milk
samples from Victoria.
Ankrah et al. (1994) attempted to ascertain if the presumed
intake of dietary aflatoxins (AFB1 and AFG1) has adverse effect on the
liver; aflatoxins were measured in serum, urine and faecal specimens
obtained from a group of 40 apparently healthy adults (11 females and
29 males) from the Greater Accra region of Ghana. Liver status of the
subjects was monitored with serum alpha-fetoprotein (AFP),
alpha-1-antitrypsin (AAT) and direct: total bilirubin ratio. Aliquots
of serum were tested for HBsAg. AFG1, AFB1, AFQ1, and AFM1 were
detected in one or more of the body specimens in 35% of the subjects
(AFB1+ group). Sixty-five per cent of the subjects had only AFG1 in
their body specimens (AFB-group). Serum levels of AFP (greater than 20
ng/ml), AAT (greater than 170 ng/dl) and direct: total bilirubin ratio
(greater than 0.5), which indicated absence of predisposition to liver
cancer in all the subjects but were suggestive of liver inflammation,
were noted in both the AFB+ and AFB1-subjects. None of the subjects
had malaria or hepatitis B virus infection. The authors suggested that
the pattern of distribution of the aflatoxins in the subjects
indicates that the suspected liver inflammation may involve other
factors and may not be only due to present intake levels of
aflatoxins.
(e) Epidemiological studies on dietary aflatoxins and liver cancer
A number of important epidemiological studies have been published
since the Committee's last review of aflatoxins at its thirty-first
meeting (Annex 1, reference 77).
Yeh et al. (1989) examined the roles of the hepatitis B virus
and AFB1 in the development of primary hepatocellular carcinoma (PHC)
in a cohort of 7917 men aged 25 to 64 years old in southern Guangxi,
China, where the incidence of PHC is among the highest in the world.
After accumulating 30 188 person-years of observation, 149 deaths were
observed, 76 (51%) of which were due to PHC. Ninety-one per cent (69
of 76) were HBsAg+ at enrollment into the study in contrast to 23% of
all members of the cohort. Three of the four patients who died of
liver cirrhosis were also HBsAg+ at enrollment. There was no
association between HBsAg positivity and other causes of death. Within
the cohort, there was a 3.5-fold difference in PHC mortality by place
of residence.
To estimate AFB1 exposure, between 1978 and 1984, the Fusui Liver
Cancer Institute regularly sampled and tested staple foods consumed in
the counties of southern Guangxi for contamination by AFB1. Twice a
year, samples of raw foods were collected from all over the region and
analysed for AFB1 content by TLC. An estimated mean level was computed
for each commune as follows. The yearly amount consumed of a given raw
foodstuff was multiplied by the average AFB1 content as determined
from tested samples of raw foodstuffs. These cross-product terms were
then summed over all staple foods, and the resultant figure was
divided by the total population to obtain an estimated intake per
person per year. These population-based levels of AFB1 were correlated
with mortality rates of PHC among members of the cohort by the
communes from which the subjects were derived.
When estimated AFB1 levels in the subpopulations were plotted
against the corresponding mortality rates of PHC, a positive and
almost perfectly linear relationship was observed. On the other hand
the prevalence of HBsAg was very high and homogeneous across the study
areas (range 21.6%-24.7%) and therefore, no significant association
was observed when the prevalence of HBsAg positivity in the
subpopulations was compared with their corresponding rates of PHC
mortality. The authors conclude that despite the "crudeness" of their
exposure estimate, (i.e., population-based instead of personal
exposure assessments), it is reasonable to conclude that AFB1 seems to
play a role in the unusually high rates of PHC in southern Guangxi.
The population prevalence of HBsAg is extraordinarily high in this
study population, almost one in every four adult men being a
positive carrier of HBV. Primary infection occurs very early in this
high-risk population, possibly through vertical transmission from
carrier mothers to infants during the perinatal period, based on a
survey of serum HBsAg in children ages 1 to 9 years in a county
adjacent to Fusui. Even though most cases of liver cancer in this
study did not have histopathological confirmation, the authors
indicate that probably all were PHCs.
The Yeh et al. (1989) report is an early important study showing
that, in a region where HBV is highly prevalent and PLC is common, the
HBV carriers are at very high risk. It further indicates that in an
area of high AFB1 exposure, the PLC mortality rates are higher than in
areas of lower AFB1 exposure. This study provides the basic
information for most potency estimates. Most of the early correlation
studies (with or without HBV consideration) are consistent with the
basic conclusion of the study of Yeh et al. (1989), but other
studies are not (Campbell et al., 1990; Hsing et al., 1991).
However, the study has the general limitations of correlation studies
in which: i) exposure to AFB1 is estimated from raw foodstuffs
available to populations and attributed to individuals; ii) the
correlation between PLC and AFB1 was not adjusted for any of the
possible confounders such as HCV, alcohol, tobacco or nutritional
status as shown in Taiwan by Yu et al. (1995); iii) HBV exposure may
have been underestimated due to lack of use of PCR methodology; iv)
HBsAg prevalence was measured in a 25% sample of the cohort and
attributed to the region.
Campbell et al. (1990) conducted a comprehensive cross-sectional
survey in the People's Republic of China of possible risk factors for
primary liver cancer (PLC) to include 48 survey sites, an
approximately 600-fold aflatoxin exposure range, a 39-fold range of
HCC mortality rates, a 28-fold range of hepatitis B virus surface
antigen (HBsAg+) carrier prevalence, and estimation of exposures for a
large number of other nutritional, dietary and life-style features
(Campbell et al., 1990). PLC mortality was unrelated to aflatoxin
intake, but was positively correlated with HBsAg+ prevalence, plasma
cholesterol, frequency of liquor consumption, and mean daily intake of
cadmium from foods of plant origin. Multiple regression analysis for
various combinations of risk factors showed that aflatoxin exposure
consistently remained unassociated with PLC mortality regardless of
variable adjustment. In contrast, associations of PLC mortality with
HBsAg+, plasma cholesterol, and cadmium intake remained, regardless of
model specifications, while the association with liquor consumption
was markedly attenuated (nonsignificant) with adjustment for plasma
cholesterol.
The authors commented on the lack of an association between
aflatoxin exposure and PLC mortality in this study, in view of the
findings of most previous investigations. The absence of an
aflatoxin-PLC association was consistent with a similar lack of
association of PLC mortality with the consumption of the two foods
most commonly contaminated with aflatoxin i.e. maize and mouldy
groundnuts. In contrast to the lack of an association with aflatoxin,
PLC mortality was highly correlated with HBsAg+ prevalence and not
with past HBV infection, as assessed by the prevalence of antibody to
the HBV core protein. In this study, the association of plasma
cholesterol with PLC mortality was even more consistent than the
association of PLC mortality with HBsAg+ prevalence. Mortalities from
colon cancer, rectal cancer, lung cancer, leukemia, brain cancer and
total aggregate cancer are also known to be associated with plasma
cholesterol. This association was even more surprising in China where
plasma cholesterol in this cohort ranged up to about 190 mg/dl, which
is near the low end of the range for comparable Western subjects (Chen
et al., 1990).
The authors offered several explanations for the lack of an
association between aflatoxin intake and PLC mortality, which
contrasts with the finding of other previous studies. First, Chinese
people might respond differently to aflatoxin, perhaps because of
unique genetic or environmental characteristics. This is unlikely
given the previously shown positive association between aflatoxin
intake and PLC mortality in Chinese subjects (Yeh et al., 1985; Yeh
et al., 1989) and because major ethnic differences in risk for other
cancers are greatly reduced or eliminated after migration to new
environments.
A second argument could be that the lack of an effect in this
study may have been because measurement of aflatoxin exposure during
the survey period was not representative of past intakes when the
cancers were forming. However, a similar limitation existed for all
other Chinese studies; this study is more reliable, in the opinion of
the authors, because it is based on urinary aflatoxin metabolite
excretion which directly represents and integrates over a day or so
actual consumption. In addition, aflatoxin contamination rates in a
county in the Guangxi Autonomous Region were relatively stable during
the years 1972-1983.
A third line of reasoning suggests that aflatoxin may not be a
significant human carcinogen, in the opinion of the authors. The
present study has greater statistical power and more comprehensive
range, diversity and inclusiveness of risk factors than other previous
studies. Humans may also be resistant to aflatoxin carcinogenesis, a
finding which is supported by in vitro aflatoxin studies on species
of varying resistance (Booth et al., 1981). Humans may also be more
refractory when consuming lower protein diets; whereas acute toxicity
of aflatoxin is increased in protein-malnourished children (Hendrickse
et al., 1982).
The authors pointed out that data from animal studies have shown
that when animals were fed either lower levels of animal protein (5-
or the same level (20%) of plant protein after completion of aflatoxin
dosing, development or preneoplastic lesions and tumours was markedly
inhibited (Appleton & Campbell, 1983; Schulsinger et al., 1989).
Protein in the Chinese diet is primarily of plant or fish origin, as
compared to protein in the USA diet which is primarily of animal
origin (Food and Nutrition Board, 1989; Chen et al., 1990).
The authors continued with a critique of previous aflatoxin
epidemiology studies and offered the following model to explain the
etiology of PLC. The vast majority of individuals who are susceptible
to PLC are those who are persistently infected with HBV. Within this
HBsAg+ population, additional risk is contributed chiefly by
nutritional and dietary practices that enhance liver cell
proliferation, such as diets containing significant amounts of animal
protein. Aflatoxin may act as a carcinogenic initiator, but
contributes only a very small proportion of the initiating activity
routinely exposing the liver. Therefore, HBsAg+ is a necessary but
insufficient cause of PLC, aflatoxin is an unnecessary and
insufficient cause, and sustained nourishment causing liver cell
proliferation (and elevated plasma cholesterol) is a necessary and
insufficient cause for HBsAg negative carriers, but a necessary and
sufficient cause for HBsAg positive carriers. Why is PLC so much more
common in undernourished and impoverished societies? The authors
concluded that PLC is more common because HBsAg+ carriers are more
common.
In evaluating the significance of this study by Campbell et
al. (1990), a number of issues, both statistical and
non-statistical, should be considered. For example, PLC rates were
determined for the years 1973-1975 and the biochemical analyses
(covariate ascertainment) was conducted in 1983. With regard to the
statistical analysis presented in the paper, there is some indication
that the sample data do not adequately satisfy the normality
assumptions upon which the univariate correlation and multiple
regression analyses are based. Finally, the urinary aflatoxin
measurements were of total aflatoxin metabolites, which have been
shown not to correlate well with levels of AFB1 consumed (Wild
et al., 1992; Groopman et al., 1993).
(f) What can we learn from epidemiological studies that considered
HBV, HCV and AFB in relation to liver cancer?
Viral hepatitis is a major worldwide public health problem. It is
estimated that over 300 million individuals are chronically infected
with HBV and perhaps 100 million with HCV. Chronic infection with
either virus has been linked to cirrhosis and liver cancer. HBV is
prevalent in the developing parts of the world, and HCV is emerging as
a major cause of hepatocellular cancer in Japan and western societies
(Table 1).
Tests to detect HBV markers have increased in sensitivity, largely
due to the use of the polymerase chain reaction (PCR) to amplify HBV
DNA in serum and liver tissues. HBV infection has been shown to
persist in the serum (49.7%) or in the liver cancer tissue (24.9%) in
a number of patients with liver cancer that are at the same time HBSAg
negative (Bosch & Muñoz, 1991). This pattern has been documented in
cases from areas at low and high risk for HBV infection.
Although the significance of detecting low levels of HBV DNA in a
patient with liver cancer is not fully understood, from an
epidemiological viewpoint, these subjects could easily be classified
as persistently exposed to HBV and grouped with the HBsAg carriers in
computing risk estimates. Although data are sparse on the prevalence
of equivalent markers in the general population, it is likely that
among controls the prevalence of PCR-detected HBV DNA in the absence
of any other HBV marker is extremely low. If this is the case, the
case control studies that use PCR would increase the Risk Ratio
estimates for HBV as well as the estimates of the Attributable
Fraction. There are no case control studies that have used PCR methods
(in serum or liver tissue) to detect HBV exposure.
Ramesh & Panda (1993) have questioned the hypothesis that HBV
causes chronic liver disease and liver cell carcinoma in
HBsAg-positive individuals only. The presence of HBV in patients with
HCC who are seropositive for the envelope antigen (HBsAg) is well
established. Epidemiological studies have shown a small percentage of
patients with HCC with past HBV infection, positive for anti-HBsAb or
HBcAb. However, the role of HBV in HCC cases who seroconverted from
HBsAg to HBsAb is unclear. The authors described a study of 36 HCC
cases where four cases negative for HBsAg and with underlying
cirrhosis were found. Biopsy tissue was investigated by polymerase
chain reaction; all four samples tested were positive for a portion of
the surface region (nucleotide position: 636-735), but were negative
for the "X" and the "C" regions of HBV genome. Since hepatitis C virus
(HCV) has been associated with HCC, the authors tested serum samples
of the four cases for anti-HCV; one out of four was positive for
anti-HCV. The authors concluded that these observations indicate that
parts of the HBV genome can persist in liver cells of individuals who
have recovered from clinical illness and seroconverted to HBsAg
positive. However, the significance of these sub-genomic fragments in
the development of HCC is not clear.
The identification of HCV in the last decade has been a major step
forward in the understanding of the origins of liver cancer and in the
quantification of the proportion of cases related directly to viral
infections (IARC, 1994). Epidemiological studies are largely
consistent in showing a strong association between carriers of
anti-HCV and liver cancer. The specific potential to induce PLC by
each of the HVC types and variants of types as well as the impact of
other factors from the host and the environment still requires further
research. Likewise, few studies are available exploring the role of
aflatoxin in the presence of HBV and HCV. High estimates of the
Relative Risk for carriers of anti-HCV have also been reported in
areas of HBV endemicity. The risk linked to HCV is independent of HBV
and persons who are carriers of both HBsAg and anti-HCV are at a very
high risk of developing liver cancer. HCV is likely to be the major
cause of liver cancer in countries at low/intermediate risk like the
USA and Europe.
(g) Epidemiological studies including aflatoxins in countries where
the risk of liver cancer is low
In countries where liver cancer is rare and aflatoxin exposure is
low, most etiological studies on liver cancer have not considered
aflatoxins as a risk factor. The populations with higher exposures are
the workers occupationally exposed to grain dust in the animal feed
processing plants. In studies conducted in the Nordic countries in
Europe, Sweden, Denmark, the Netherlands and in the USA, aflatoxins
have been isolated from dust samples and an excess of mortality of
several such cohorts has been documented for liver cancer (risk, 2.4
times the expected rates) liver and biliary tract cancer (risk, 2.5
times the expected rates), lung cancer and lymphomas (risk, 1.5-3
times the expected rates (Hayes et al., 1984; Alavanja et al.,
1987; Olsen et al., 1988). It should be noted that some of these
studies did not evaluate other relevant exposures such as hepatitis
infections and alcohol.
It is of interest that few studies are available on liver cancer
in Latin America. In this extensive region of the world, agricultural
products are prone to mould growth, and consumption of maize is part
of the staple food in many countries. Yet liver cancer is rare in
these populations as is HBV infection. If that is the case, Latin
America would be potentially a very informative field to investigate
the occurrence of liver cancer in populations exposed to AF as the
central risk factor.
(h) Epidemiological studies that used biomarkers of exposure to
aflatoxins including studies on genetic susceptibility to aflatoxins
Biomarkers have been developed and are being introduced in
epidemiological studies with the purpose of increasing the accuracy of
the assessment of exposure to aflatoxins. Various biomarkers have been
developed, including urinary total aflatoxins, aflatoxin adducts in
urine, aflatoxin albumin adduct in serum, aflatoxin adducts in liver
cancer tissue and more recently p53 specific mutations in liver cancer
specimens. Other studies are investigating genetic polymorphisms in
some key genes involved in the metabolism of aflatoxin that may
introduce some variability in the response to aflatoxin. (Groopman
et al., 1994; Wild et al., 1996; IARC, 1997).
The importance of the major aflatoxin-nucleic acid adduct,
AFB-N7-guanine, in urine as a biomarker was enhanced by the finding
that this metabolite is excreted exclusively in urine of exposed rats,
thus simplifying pharmacokinetic considerations. The aflatoxin-albumin
adduct in serum has also been examined as a biomarker of exposure;
because of the longer half-life in vivo of albumin compared to the
urinary AFB-N7-guanine, the serum albumin adduct can integrate
exposures over longer time periods. Data from human exposure studies
have shown that the excretion of the urinary aflatoxin nucleic acid
adduct and formation of the serum albumin adduct are highly
correlated. In the rat, validation studies for the dose-dependent
excretion of urinary aflatoxin biomarkers were conducted in rats
following a single exposure to AFB1; excellent linear correspondence
between oral AFB1 dose and excretion of AFB-N7-guanine in urine was
shown (Scholl et al., 1995).
Aflatoxin metabolites in urine or adducts in serum can be a useful
tool to evaluate exposure but with the currently available methods
remain relatively short-term exposure markers; biomarkers are of much
less use in predicting long-term or lifetime human exposure. As such,
they reflect poorly the natural pattern of exposure to aflatoxin
(i.e., seasonality, manual sorting of foodstuffs, age at exposure,
etc.); therefore, it is not surprising that studies conducted using
aflatoxin biomarkers as markers of exposure show conflicting results.
At present, it is not fully understood how the functional status
of the liver or the coexistence of other risk factors for liver cancer
may affect the different biomarkers that are being proposed for
epidemiological studies. Few studies have described the natural
history of these markers in patients with chronic liver disease
including chronic hepatitis and liver cirrhosis, conditions that
usually precede liver cancer by months or years (Wild et al., 1993;
Wang et al., 1996a). In this circumstance, the interpretation of the
findings is complicated since the aflatoxin biomarker may be
confounded by the presence of some risk factors, (e.g., HBV, HCV,
alcohol), the presence of some protective factors (e.g., retinol in
the diet) or by the presence of liver disease (i.e. chronic active
hepatitis B infection).
The mutations in the p53 gene claimed to be specific markers of
exposure to aflatoxin are being actively investigated to confirm the
strength and the specificity of the association in human populations
(Harris, 1995). Recent data have been summarized earlier in this
paper.
The best evidence of an interaction between HBV and aflatoxin in
the causation of human liver cancer is the cohort study in Shanghai
(Ross et al., 1992, Qian et al., 1994; Yuan et al., 1995). This
is an ongoing prospective study of 18 244 middle-aged men in Shanghai,
China. Assays for urinary AFB1, its metabolites AFP1 and AFM1 and DNA
adducts have been performed to assess the relationship between
aflatoxin exposure and liver cancer. After 35 299 person-years of
follow-up, 22 cases of liver cancer had been identified. For each
case, 5 or 10 controls were randomly selected from cohort members
without liver cancer on the date the disorder was diagnosed in the
case and matched to within 1 year of age, within 1 month for sample
collection, and for neighbourhood of residence. Each subject provided
a blood sample and a urine sample. A positive result was defined as
the presence of at least 1 ng of an individual aflatoxin compound in
the sample. Hepatitis B surface antigen was measured by a standard
radioimmunoassay method.
Subjects with liver cancer were significantly more likely than
were controls to have detectable concentrations of any of the
aflatoxin compounds; the strongest association was for AFP1.
Positivity for HBsAg was strongly associated with risk of liver
cancer. The authors concluded that their results are based on too few
cases to give a reliable estimate of attributable risk, but they
estimated that up to 50% of cases of liver cancer in Shanghai may be
due to aflatoxin exposure.
In further follow-up of the Shanghai study, Qian et al. (1994)
reported on 70 000 person-years of follow-up and 55 cases of HCC.
Levels of urinary AFB1 and the oxidative metabolites, including the
major aflatoxin nucleic acid adduct, aflatoxin-N7-guanine, were
determined for 50 of the 55 identified cases of HCC; 267 controls were
matched against the 50 cases as above. A nested case-control analysis
showed highly significant association between the presence of at least
one of the four urinary aflatoxin metabolites, serum HBsAg positivity
and HCC risk. Risk was especially elevated in individuals who were
positive for both of these biomarkers. However, the number of liver
cancer cases in which the interaction was explored is small (i.e., 13
cases of AFB1 positive and HBsAg negative), and there is room for
misclassification of cases in relation to their viral exposure. Thus
risk estimates become unstable and the barely significant increase in
the ORs for the AFB1 exposed may be easily lost if only 2/3 of cases
now considered HBV negative turn out to be HBV (or HCV) positive. On
the other hand, a cohort analysis using all 55 cases of HCC revealed
no statistically significant association between HCC risk and dietary
aflatoxin consumption, as determined from the in-person food frequency
interview combined with the survey of market foods in the study
region, adding additional uncertainty of the value of the biomarkers
used. HCV prevalence was low in this cohort (Yuan et al., 1995).
An exchange of correspondence on this Shanghai study has occurred.
Campbell (1994) has pointed out that the HCC risk putatively
attributed to aflatoxin in the study of Ross et al. (1992) appears
to be accounted for mostly by the urinary AFB-N7-guanine adduct.
Instead Campbell postulates that this risk could also be caused by
factors that enhance enzymatic activation of AFB1 by the hepatic P450
enzyme system to produce more AFB-N7-guanine. These enzyme-inducing
factors could readily be nutritional, especially those that also are
associated with elevated plasma cholesterol. This interpretation would
be in accord with the ecological study by Youngman et al. (1992) in
48 survey counties in China showing that the most significant and
robust determinants of HCC risk were elevated cholesterol levels and
HBsAg positivity, not aflatoxin. This finding is given further
plausibility by animal experiments (Preston et al., 1976; Hu
et al., 1994). These experiments have shown that in vivo
activation of AFB1 to form hepatic DNA adducts could be markedly
enhanced by a modest elevation in the intake of animal protein. This
same modest animal protein intake that markedly elevates AFB1
activation also markedly increased the over-expression of a hepatitis
B virus transcript in mice.
Ross et al. have responded to the comments of Campbell (1994).
There is an overwhelming amount of experimental data across species
and in experimental models demonstrating the potency of AFB1 as a
carcinogen and mutagen (IARC, 1993). There is also evidence that
humans have the metabolic capacity to activate AFB1 to the same
DNA-damaging products that occur in animal models. A well-established
major risk factor for liver cancer is hepatitis B virus; however,
there is at least a 5-8 fold variation in liver cancer incidence
across regions of the world where the prevalence of hepatitis B viral
markers is comparable. Ross et al. emphasize that their Shanghai
study using aflatoxin-specific biomarkers and HBV markers has provided
the first direct evidence in human studies that aflatoxins are major
risk factors for HCC and that a synergistic interaction between HBV
and aflatoxin exposure occurs. Ross et al. criticize the Chinese
study of Campbell (1994) as an ecological study, "well known to be
highly limited in their ability to address cause and effect
relationships". In addition, a questionnaire administered to the
Shanghai subjects showed no difference in daily intake of animal
protein between liver cancer cases and their matched controls, or
between Hbsag positive cases and controls.
Groopman et al. (1993) stated that, based on urinary measures of
AFB-N7-guanine and dose-response characteristics of people living in
China and The Gambia, that 1) levels of daily urinary excretion of
total aflatoxin metabolites are unrelated to risk of aflatoxin-induced
disease; 2) the AFB-N7-guanine adduct in urine is a good,
non-invasive, short-term biomarker for determining both aflatoxin
exposure and risk of genetic damage in target organs.
However, the Shanghai study is clearly limited for purposes of
quantitative risk assessment of the risk to humans of aflatoxin
exposure. By the authors' own admission (Qian et al., 1994), no
dose-dependent association between the dietary aflatoxin index and
either liver cancer risk or biomarker status was found. This is due at
least in part to the facts that urinary levels of aflatoxin accurately
reflect intake levels of the past 24 hours and dietary assessment is
inadequate to reflect lifetime aflatoxin exposure. An unexplained
observation was the rather marked decline in the prevalence of
unmetabolized AFB1 with longer follow-up. Urinary adducts were
measured with inadequate precision; a patient was scored "positive" or
"negative" if an adduct was detected. Levels of adducts were not
considered, nor was the fact that one measurement represents one
"snapshot" out of a lifetime. One might question whether or not the
increased excretion of aflatoxin-DNA adducts represents the activity
of a diseased liver rather than a causal relationship. Exposure to
AFB1 may be expected to fluctuate greatly on a day-to-day basis (as a
result of varying behaviour and AFB1 concentrations); exposure for
each individual was evaluated at a single time point. The authors are
correct in their call for pharmacokinetic investigations of ingested
aflatoxins in humans. Until this work is done, AFB adducts cannot be
considered to be a true indicator of aflatoxin exposure, especially
over the probable lengthy timeframe required for human cancer
induction.
This study strongly suggests that aflatoxin exposure in the
presence of a persistent HBV infection increases the risk of liver
cancer. It is less convincing support for the conclusion that AFB1 is
capable of independently inducing liver cancer and provides limited
quantitative data on aflatoxin's relationship to liver cancer.
Follow-up of this cohort is awaited with great interest.
In a study by Wild et al. (1993), blood samples were collected
over a one-month period from 117 children aged 3 to 4 years residing
in Kuntair or Kerr Cherno in the Upper Niumi District of The Gambia.
Samples were analysed for aflatoxin-albumin (AF-alb) adducts, markers
of HBV infection, liver enzymes (serum alanine aminotransferase (ALT))
as markers of liver damage, and glutathione-S-transferase (GST) M1
genotype. All but two children showed detectable serum AF-alb with
levels ranging from 2.2 to 250 pg AFB1-lysine equivalent/mg albumin.
There was a statistically significant positive correlation between
AF-alb and ALT. HBV carriers showed moderately higher levels of AF-alb
than non-carriers, but the difference was not statistically
significant and the association between AF-alb and ALT was unchanged
when the HBV carriers were excluded from the analysis, suggesting that
factors other than HBV infection contributed to the association. The
null GSTM1 genotype was infrequent in this population and was not
associated with any difference in AF-alb adduct levels compared to
GSTM1-positive subjects. However, the percentage of individuals with
the null genotype varied significantly between ethnic groups. The
association between AF-alb and ALT could be a result of the
hepatotoxicity of aflatoxin, but the data are also consistent with the
hypothesis that liver damage resulting from HBV and/other factors can
alter aflatoxin metabolism resulting in an increased binding to
cellular macromolecules including DNA. The authors recommended more
study of this hypothesis.
Srivatanakul et al. (1991) conducted a case control study on
hepatocellular carcinoma in Thailand using the aflatoxin-albumin
adduct as a marker of recent exposure to aflatoxin. HBV exposure and
anti-HCV were assessed using standard methods. HBV was the predominant
risk factor; neither aflatoxin-albumin nor HCV were associated with
liver cancer. The Committee concluded that the study lacked sufficient
statistical power to detect aflatoxin or HCV as independent risk
factors. In addition, aflatoxin-albumin samples were taken from liver
cancer cases; levels or kinds of aflatoxin metabolites might have been
affected by illness.
Hatch et al. (1993) conducted a survey in eight areas in Taiwan
with a gradient in the estimates of exposure to aflatoxin and in the
incidence of PLC. Exposure to aflatoxin was assessed using urinary
tests, and a regression model was used to predict aflatoxin urinary
metabolites using mortality due to PLC as a predictor (as well as five
other variables). The conclusion of the study was that aflatoxin
played an independent role in PLC in Taiwan.
Monoclonal antibodies recognizing the stable imidazole ring-opened
form of the major N7-guanine aflatoxin B1-DNA adduct have been used in
competitive enzyme-liked immunosorbent assays (ELISA) and indirect
immunofluorescence assays to quantify adduct levels in liver tissue.
Santella et al. (1993) developed methods in AFB1-treated animals,
then applied these to paired tumour and non-tumour liver tissues of
hepatocellular carcinoma patients from Taiwan. An avidin-biotin
complex staining method was also used for the detection of HBsAg and
HBxAg antigens in liver sections. In all, 8 (30%) HCC samples and 7
(26%) adjacent non-tumour liver tissue samples from Taiwan were
positive for AFB1-DNA adducts. For HBsAg, 10 (37%) HCC samples and 22
(81%) adjacent non-tumorous liver samples were positive, and 9 (33%)
HCC samples and 11 (41%) adjacent non-tumour liver samples were HBsAg
positive. No association with AFB1-DNA adducts was observed for HBsAg
and HBxAg. The authors concluded that these results were compatible
with the conclusion that HBV and AFB1 do not act synergistically in
the genesis of HCC, but called for further investigation to define the
relationship between HBV and AFB1.
Wang et al. (1996a) conducted studies in Qidong, China, where
liver cancer accounts for 10% of all adult deaths and both HBV and
AFB1 exposures are common. Serum samples were collected during a
longitudinal study designed to measure aflatoxin molecular biomarkers
in residents of Daxin Township, Qidong City, China. In this study, the
temporal modulation of aflatoxin adduct formation with albumin over
multiple lifetimes of serum albumin was examined in both HBV-positive
and HBV-negative people in two periods: September-December 1993 (wave
1) and June-September 1994 (wave 2). During the 12-week monitoring
period of wave 1, 120 persons (balanced by gender and HBV status)
provided a total of 792 blood samples. AFB1-albumin adducts were
detected in all but one of the serum samples. During wave 2, 103
individuals from wave 1 provided 396 blood samples collected monthly
over wave 2. Using linear regression models, the mean
aflatoxin-albumin adduct levels increased during the 12 weeks of wave
1 and decreased over the 4 months of wave 2.
Neither HBV status nor gender modified either the baseline mean or
the temporal trend. High-performance liquid chromatography
confirmation was done on a subset of serum samples, and the results
showed an excellent association between the immunoassay data and
high-performance liquid chromatography. The investigators concluded
that AFB1-albumin is a sensitive and specific biomarker for assessing
exposure to AFB1 in the Qidong population.
The authors noted that aflatoxin-albumin binding is a longer term
biomarker of aflatoxin exposure than any of the urinary markers; the
rate of turnover of these adducts is similar to that of the blood
protein. The half-life of albumin in normal people is about 14-20
days, but there is some information to indicate that people with
serious liver disease have a much more variable turnover time. In this
study, an 8-fold range of adduct formation existed among individuals
within a cycle. Such factors as liver disease may account for the lack
of tracking shown in this study between HBV status and adducts. The
authors stressed the need to follow-up on this investigation with
studies that have more frequent and longer sampling intervals for
albumin adducts.
The authors discussed the data supporting the hypothesis that HBV
enhances aflatoxin metabolism to genotoxic derivatives. This study did
not seem to support this hypothesis nor did previous results in adult
populations in West Africa and Taiwan. HBV may affect the metabolism
of aflatoxin in children at a time when hepatocytes are maximally
dividing; more data are needed in this regard.
Wang et al. (1996b) investigated the carcinogenic effect of
aflatoxin exposure in Taiwan. Fifty-six cases of HCC diagnosed between
1991 and 1995 were identified and individually matched by age, sex,
residence and date of recruitment to 220 healthy controls from the
same large cohort in Taiwan. Blood samples were analysed for hepatitis
B and C virus markers and for aflatoxin-albumin adducts. Urine was
tested for aflatoxin metabolites. Information was obtained about
sociodemographic characteristics, habitual alcohol drinking, cigarette
smoking and diet in a structured interview.
HBsAg carriers had a significantly increased risk for HCC. After
adjustment for HBsAg serostatus, the matched odds ratio (ORm) was
significantly elevated for subjects with high levels of urinary
aflatoxin metabolites. When stratified into tertiles, a dose-response
relationship with HCC was observed. The ORm for detectable
aflatoxin-albumin adducts was not significant after adjustment for
HBsAg serostatus. HBsAg-seropositive subjects with high aflatoxin
exposure had a higher risk than subjects with high aflatoxin exposure
only or HBsAg seropositivity only. The OR for developing HCC was found
to increase in the presence of anti-HCV alone, HBsAg alone and both
anti-HCV and HBsAg. There was a poor correlation between
aflatoxin-albumin adducts and urinary metabolites in the same
controls, although both were related to HCC risk. The investigators
suggested that environmental aflatoxin exposure may enhance the
hepatic carcinogenic potential of hepatitis B virus, expressed concern
about the small sample size in their study, and urged the mounting of
a large-scale study to evaluate the effect of aflatoxin exposure on
HBsAg non-carriers.
Olubuyide et al. (1993) screened for the presence of HBsAg and
aflatoxin in the sera of 100 non-hospitalized individuals from the
rural population of Igobo-Ora and 89 non-hospitalized individuals from
the urban population of Ibadan, Nigeria. Controls were 31 healthy
British Caucasians who had not travelled to the tropics or subtropics
in the six months before the venipuncture. Forty-nine per cent of
rural subjects and 47% of urban subjects were consistently and
reproducibly seropositive for HBsAg (as determined by the ELISA test).
Two of the former subjects and five of the latter were positive for
both HBV DNA (as measured by spot hybridization) and HBsAg. Total
aflatoxin levels were less than 17 pg/ml in the British controls;
serum levels of aflatoxins greater than this were detected in 8% of
rural subjects and in 9% of urban subjects. The types and amounts of
aflatoxins and the amounts of aflatoxins found were so widely
dispersed that it was not possible to draw any conclusions about
differences in types and amounts of aflatoxins between rural and urban
populations. The authors intend to follow their subjects to determine
their propensity to develop HCC.
Groopman et al. (1994) have reviewed the subject of molecular
biomarkers for aflatoxins and their application to human cancer
prevention. Cancer prevention trials that use biological markers as
intermediate end-points provide the ability to assess the efficacy of
promising chemoprotective agents in an efficient manner by reducing
sample size requirements, as well as reducing the time required to
conduct the studies, compared to trials that have cancer incidence or
mortality as end-points. The key issue in trials that use biomarkers
as the outcome of interest is to use a marker that is directly
associated with the evolution or development of neoplasia. The authors
briefly discussed the possible impact of a short-term intervention
with oltipraz (a substituted dithioethione, which is a potent
inhibitor of AFB1-induced tumorigenesis and carcinogenesis in rats) on
levels of two aflatoxin biomarkers in individuals exposed to
aflatoxin-contaminated foods.
(i) Oltipraz chemoprevention trial
In 1995, 234 adults from Qidong, Jiangsu Province, China, where
hepato-cellular carcinoma is the leading cause of cancer death and
exposure to dietary aflatoxins is widespread, were enrolled and
followed in a Phase II chemo-prevention trial (Jacobson et al.,
1997). The goals of the study were to define a dose and schedule of
oltipraz for reducing levels of validated aflatoxin biomarkers and to
characterize dose-limiting toxicities. Healthy eligible individuals,
including those infected with hepatitis B virus, were randomized to
receive either 125 mg of oltipraz daily, 500 mg of oltipraz weekly, or
placebo. Blood and urine specimens were collected to monitor
toxicities and evaluate biomarkers over the 8-week intervention period
and subsequent 8-week follow-up period. The authors reported excellent
compliance (>70%); 21% of subjects reported clinical adverse events.
The oltipraz arms did not differ in symptom type or severity, most
commonly an extremity syndrome. There were no indications of
exacerbated drug intolerance among the few participants infected with
hepatitis B virus. The authors concluded that chemoprevention trials
with biomarker end-points are feasible in such populations.
(j) Genetic susceptibility
AFB1 is metabolized via the phase I and II detoxification pathway;
hence, genetic variation at those loci may predict susceptibility to
the effects of AFB1. To test this hypothesis, McGlynn et al. (1995)
contrasted genetic variation in two AFB1 detoxification genes, epoxide
hydrolase (EPHX) and GSTM1 with the presence of serum AFB1-albumin
adducts, the presence of hepatocellular carcinoma (HCC), and with p53
codon 249 mutations. Subjects were 40 unrelated Ghanaian males
(healthy gold miners employed by the Ashanti Goldfields Corp. in
Obuasi, Ghana) and 52 patients with HCC and 116 healthy controls from
the Zhong Shan Hospital in Shanghai, China.
Mutant alleles at both loci were significantly over represented in
individuals with serum AFB1-albumin adducts in a cross-sectional
study. Mutant alleles of EPHX were significantly over-represented in
subjects with HCC and also in a case-control study. The relationship
of EPHX to HCC varied by hepatitis B surface antigen status and
indicated that a synergistic effect may exist. p53 codon 249 mutations
were observed only among HCC patients with one or both high risk
genotypes. These results indicate that individuals with mutant
genotypes at EPHX and GSTM1 may be at greater risk of developing
AFB1-adducts, p53 mutations and HCC when exposed to AFB1. Hepatitis B
carriers with the high-risk genotypes may be an even greater risk than
carriers with low-risk genotypes. The authors concluded that these
findings support the existence of genetic susceptibility in humans to
the environmental carcinogen AFB1 and indicate that there is a
synergistic increase in risk of HCC with the combination of hepatitis
B virus infection and susceptible genotype. However, it has been
pointed out that the control and experimental samples came from
different populations, which may weaken the case for genetic
susceptibility (J. Groopman, personal communication).
The Committee concluded that the currently available studies
utilizing aflatoxin biomarkers do not provide a reliable quantitative
measure of aflatoxin exposure in humans, especially over the
long-term. Biomarker levels in relation to cancer risk in the Wang
et al. and Qian et al. studies have been used for modelling data;
however, the interpretation is limited.
At present, it seems reasonable to subscribe to the 1993
conclusion of the IARC in qualitative terms that AFB1 is carcinogenic
(group 1), and as such to recommend reducing exposure of human
populations as much as possible. There is still some uncertainty
concerning the independent status of aflatoxin as a human carcinogen
and concerning the relationship between aflatoxin dose and liver
cancer incidence.
(k) Conclusions from epidemiology studies
The potential carcinogenicity in humans of the aflatoxins (either
total or AFB1) has been examined in a large number of epidemiology
studies, generally carried out in Africa and Asia, where substantial
quantities of aflatoxin occur in basic foodstuffs. Exposure to
aflatoxins appears to present an additional risk which is enhanced by
simultaneous exposure to hepatitis B virus, and possibly hepatitis C
virus. This relationship, which may affect not only carcinogenic
potency but also the metabolism, biochemistry and pharmacology of the
aflatoxins, and other multiple etiological agents for primary liver
cancer makes it difficult to interpret the epidemiological studies in
the context of the risk of primary liver cancer from aflatoxins.
Perhaps, further development of biochemical and pharmacological
markers will help to clarify exposure, although these can cause other
problems.
Further clarification of the relative roles of hepatitis and
aflatoxin in liver cancer awaits studies that comply with the
following requirements: 1) the studies should be cohort studies with
long-term follow-up; 2) the studies should be conducted in countries
with variability in the exposure to aflatoxins; 3) the studies should
provide for storage and analyses of biological specimens from repeated
sampling, preferably with concurrent sampling of aflatoxin in the
diet; 4) the studies should provide evidence that the aflatoxin
biomarker used is not affected by the presence of chronic liver
disease; this will be difficult to achieve; the different measures of
aflatoxin exposure, i.e., biomarker vs. dietary analysis, should
correlate with liver cancer; 5) a large number of liver cancer cases
should be included, preferably confirmed by biopsy; 6) liver cancer
cases, if shown to contain the p53 specific aflatoxin mutation, would
strengthen the case.
The Committee identified some on-going studies that comply with
some of these requirements and may produce relevant results in the
near future.
i) A study in Qidong, China, screened 45 000 males (ages 30-59) of
which 20% were HBsAg positive. Questionnaires, urine and serum were
collected at different intervals; 260 cases of liver cancer have been
identified although the number of biopsies is small. Laboratory tests
and statistical analyses are required; funds are needed.
ii) A cohort study in Thailand collected questionnaires, blood and
urine specimens at different intervals in a cohort of HBsAg carriers
who were regularly screened for AFP, ALT and ultrasound. Field work is
completed at this point and lab results are pending.
iii) The Shanghai study described in the text may be of value if
sampling and follow-up continue.
iv) Finally, the on-going HBV vaccination trials and campaigns in
China, Taiwan and The Gambia may provide evidence in the future for
the occurrence of AFB1-induced liver cancer cases in individuals
vaccinated against HBV.
2.2.11 Summary of information on other aflatoxins
2.2.11.1 Aflatoxin B2
Aflatoxin B2 (AFB2) has not been studied extensively, and most
data are derived from single reports. AFB2 becomes bound to DNA of
rats treated in vivo, after its metabolic conversion to AFB1. In
rodent cells, AFB2 induced DNA damage, sister chromatid exchange and
cell transformation, but not gene mutation. AFB2 produces gene
mutation in bacteria. IARC concluded in 1993 that there is limited
evidence for carcinogenicity of AFB2 in experimental animals.
No additional toxicological information on AFB2 has appeared in
the literature since IARC (1993).
2.2.11.2 Aflatoxin G1
Aflatoxin G1 (AFG1) binds to DNA and produces chromosomal
aberrations in rodents treated in vivo. In cultured human and animal
cells, it induces DNA damage, and also induces chromosomal anomalies
in single studies. AFG1 induces gene mutation in bacteria. IARC
concluded in 1993 that there was sufficient evidence in experimental
animals for the carcinogenicity of AFG1.
No additional toxicological information on AFG1 has appeared in
the literature since IARC (1993).
2.2.11.3 Aflatoxin G2
Aflatoxin G2 (AFG2) has been the subject of very little research.
IARC concluded in 1993 that there was inadequate evidence for the
carcinogenicity of AFG2.
No additional toxicological information on AFG1 has appeared in
the literature since IARC (1993).
2.2.11.4 Aflatoxin M1
Aflatoxin M1 (AFM1) is a metabolic hydroxylation product of AFB1,
and can occur in the absence of the other aflatoxins. Human exposure
occurs primarily via milk and milk products from animals that have
consumed contaminated feed. IARC concluded in 1993 that there was
sufficient evidence in experimental animals for the carcinogenicity of
AFM1 and inadequate evidence for the carcinogenicity of AFM1 in
humans. Although AFM1 has been tested less extensively, it appears to
be toxicologically similar to AFB1. AFM1 is considered to be a
genotoxic agent, based on its activity in vitro and its structural
similarity with AFB1. It is a less potent liver carcinogen, with a
probable carcinogenic potency in laboratory animals within a factor of
10 of AFB1 (Cullen et al., 1987).
No additional toxicological information on AFM1 has appeared in
the literature since IARC (1993).
3. ESTIMATING CARCINOGENIC RISKS FROM THE INTAKE OF AFLATOXINS
Risks from specific exposures to aflatoxins are difficult to
estimate and predict, despite extensive information available from
epidemiological studies, mutagenicity tests, animal bioassays,
in vitro and in vivo metabolic studies, and p53 mutation studies.
Many questions remain regarding the independence of aflatoxin as a
human carcinogen, the extent to which hepatitis B, hepatitis C and
other factors modify the effect of aflatoxin, how findings from
countries with high liver cancer rates and high prevalence of
hepatitis B may be compared to those from countries with low rates,
how to deal with the wide range of susceptibility to aflatoxin
carcinogenesis among experimental animals, and how to describe the
dose-response curve over the wide range of aflatoxin exposure found
worldwide.
3.1 Information from various scientific disciplines and its
contribution to aflatoxin carcinogenic risk
3.1.1 Laboratory animal, mutagenicity and metabolic studies
The liver is the primary target organ in most species, but tumours
of other organs have also been observed in aflatoxin-treated animals.
The effective dose of AFB1 for induction of liver tumours varied over
a wide range in different animals species when the carcinogen was
administered by continuous feeding, generally for the lifetime of the
animal. Effective doses were 10-30 µg/kg in the diet in fish and
birds. Rats responded according to strain at levels of 15-1000 µg/kg,
while some strains of mice showed no response at doses up to 150 000
µg/kg. Tree shrews responded to 2000 µg/kg. In subhuman primate
species, AFB1 potency in induction of liver tumours differed widely,
squirrel monkeys developing liver tumours when fed AFB1 at 2000 µg/kg
for 13 months, and rhesus, African green and cynomolgus monkeys
developing a low (7-20%) incidence of liver tumours when fed average
doses of 99-1225 mg/animal over 28-179 months (Wogan, 1992).
The aflatoxins are among the most potent mutagenic and
carcinogenic substances known. Much of the information available
regarding mutagenesis has been performed in bacterial systems, but
also to a lesser extent in eukaryotes. Aflatoxin falls in the category
of bulky mutagens, including the polycyclic aromatic hydrocarbons and
the aromatic amines. A large body of literature suggests that a
chemical causes a cell to become tumorigenic by reacting readily with
DNA to give DNA adducts and these adducts or their breakdown products
must then cause mutations efficiently (Loechler, 1994).
Much of the recent aflatoxin metabolic data has been discussed in
more detail in section 2.1.3. In brief, it has been demonstrated that
many isoforms of P450 are able to biotransform AFB1 to
DNA-binding/mutagenic species. Differences in P450 isoform activities,
due either to genetic polymorphisms or to environmental alteration in
expression, may be important contributors to human susceptibility to
AFB1 (Massey et al., 1995). For example, there is some evidence that
AFB1 is strongly metabolized to DNA-binding species in areas of
damaged liver or in individual cells where CYP2A5 activity is high
(Camus-Randon et al., 1996). It is well known that a host of other
risk factors affecting metabolism exist, including infection with
hepatitis B and C, parasites such as liver flukes, alcohol
consumption, cigarette smoking, long-term use of oral contraceptives,
and nutritional status.
There is increasing evidence that AFB1 can be activated by lipid
hydroperoxide-dependent mechanisms, involving microsomal prostaglandin
H synthase and lipoxygenases. Although the maximum activity of this
co-oxidation is low relative to P450, these processes may contribute
significantly to bioactivation of AFB1 in vivo in humans, who are
generally exposed to low levels of AFB1. Co-oxidation may be
particularly important for AFB1 carcinogenicity in extrahepatic
tissues, in view of relatively low cytochrome P450 activity in these
organs. In the search for target cell types in the human lung, a
thorough analysis of the cellular distribution of potential AFB1
metabolizing systems will be necessary (Massey et al., 1995).
Detoxification (mediated by cytochrome P450 as well as conjugation
of the epoxide with glutathione) must also be considered. Animal
studies suggest that GST-catalysed detoxification is the crucial
factor in susceptibility to AFB1, and humans appear to lack
significant GST-mediated protection against AFB1 (Massey et al.,
1995). There is suggestive evidence that human GSTs in the alpha,
mu and theta families may all have roles in the detoxification of
the epoxide. It is not yet known with certainty whether there is a
role for epoxide hydrolase.
A possibly important factor in the assessment of aflatoxin risk is
the variation of human susceptibility due to individual differences in
human metabolism, such as polymorphisms in cytochrome P450s, GSTs, and
epoxide hydrolase. Data are beginning to become available; it is
unclear as yet what impact gene polymorphisms may have on human
activating as well as detoxifying enzymes, and therefore on aflatoxin
risk (Cardis et al., in press). With all the enzymes, it is
necessary to consider the stereochemistry of the aflatoxin epoxide,
which is critical in genotoxicity (Guengerich et al., 1996).
Most studies comparing AFB1 metabolism in different species have
been conducted in vitro, using subcellular fractions such as
microsomes or cytosol, or purified components of either fraction. The
exclusion of enzymes and cofactors for competing metabolic pathways
restricts quantitative comparisons of metabolism between different
species; therefore, conclusions based on data from in vitro
experiments are limited to qualitative comparisons of individual
pathways of AFB1 metabolism. Although specific metabolism pathways may
be associated with increasing sensitivity to AFB1 carcinogenicity,
quantification of sensitivity requires a supply of metabolic factors
for competing reactions found by either reconstitution of total
cellular fractions, use of primary cell cultures with representative
metabolic capacities, or whole animal studies (Gorelick, 1990).
The P450s both activate and detoxify AFB1 and the effect of
inducing individual P450s is not easily predicted. Also, the small
intestine, site of absorption of orally ingested AFB1, expresses P450
3A4. Activation of AFB1 and DNA alkylation in the small intestine may
be considered also to be a detoxification process since the cells are
sloughed rapidly and cancers of the small intestine are very rare
(Guengerich et al., 1996).
After reviewing the available data from the metabolic,
mutagenicity and laboratory animal studies, the Committee concluded
that there is at the present time insufficient quantitative
information available about competing aspects of metabolic activation
and detoxification of AFB1 in vivo in various species to describe
quantitatively a species-dependent effect of metabolism on AFB1
carcinogenicity (Gorelick, 1990; Massey et al., 1995; Guengerich
et al., 1996; Wild et al., 1996). It is, however, probable, that
differential sensitivity to AFB1-induced tumours between species can
be partially attributed to differences in metabolism.
3.1.2 Studies on the p53 gene
Studies on p53 mutations have been extensively discussed earlier
in this paper. The Committee concluded that there is currently
insufficient straightforward information available on the specificity
of the aflatoxin/p53 association to assess and quantify the
independence of aflatoxin as a cause of human liver cancer.
3.1.3 Epidemiological studies
The relevant epidemiological studies have been discussed earlier;
only the conclusions are presented here. Most of the epidemiological
studies show a correlation between exposure to aflatoxins and liver
cancer; some studies suggest that aflatoxin exposure poses no
detectable independent risk and other studies suggest that it poses a
risk only in the presence of other risk factors such as HBV infection.
Several ongoing studies are likely to improve further the estimates of
human risks from aflatoxin exposures; most notable among these are
cohort studies in Shanghai, Thailand and Qidong, China, and the HBV
vaccination trials in The Gambia, Taiwan and Qidong. When these
studies are complete, JECFA may want to re-evaluate the risks of
aflatoxins in humans.
A number of factors influence the risk of primary liver cancer,
most notably carriage of HBV; the potency of aflatoxins appears to be
significantly enhanced in individuals with simultaneous HBV infection.
Most of the epidemiological data are from geographical areas where
both the prevalence of HBsAg+ individuals and aflatoxins are high; the
relationship between these risk factors in areas of low aflatoxin
contamination and low HBV prevalence is unknown. This interaction
makes it difficult to interpret the epidemiological studies in the
context of aflatoxin as an independent risk. The Committee therefore
has made decisions contingent upon the dynamics of HBV infection in a
human population for which aflatoxin potency is to be determined.
The identification of HCV is a major breakthrough in understanding
the etiology of liver cancer. Two studies have investigated
interactions between HCV infection, aflatoxins and liver cancer; the
evidence so far is inconclusive. As shown in Table 1, it has been
estimated that 50 to 100% of liver cancer cases are associated with
persistent infection with HBV and/or HCV.
In Latin America both liver cancer and HBV infection are rare, yet
aflatoxin exposure is relatively high. Unfortunately, few studies are
available on the occurrence of liver cancer in Latin America; much
could be learned about aflatoxin as a risk factor in liver cancer by
conducting appropriately designed epidemiology studies in Latin
America.
3.1.4 Aflatoxin biomarker studies
The Committee concluded that the currently available studies
utilizing aflatoxin biomarkers do not provide a quantitative measure
of aflatoxin exposure in humans especially over the long term.
Biomarker levels in relation to cancer risk in the studies of Wang
et al. (1996a) and Qian et al. (1994) studies have been used for
modelling data; however, the interpretation is limited.
3.2 General modelling issues
Quantitative risk assessment for food contaminants involves four
basic issues: 1) choice of data; 2) measure of exposure; 3) measure of
response; and 4) choice of a mathematical relationship between dose
and response for a given data set. General comments can be made for
each of these areas, as well as specific comments concerning what has
been done regarding estimating risk from exposure to aflatoxin.
3.2.1 Choice of data
In general, the best data set to use for dose-response analysis
would be a human study in which dose is accurately measured, response
is determined without error and there are no confounding factors which
are unexplained. It is rare to find an epidemiological study without
one of these factors causing difficulty in the interpretation and
utility of the data. In contrast to the human data, test species data
are generally devoid of confounders. There is a clear and accurate
measure of response, and dose is an integral part of the design of the
study. Numerous dose-response assessments have been conducted by
modelling animal data and extrapolating the results to humans.
However, the extrapolation may be problematical, given outstanding
questions concerning the overall relevance of the animal data. For
aflatoxin, several epidemiological studies are capable of providing a
dose-response assessment. However, the study of Yeh et al. (1989)
has some limitations as described in section 2.2.10.3(e). The cohort
study in Shanghai (Ross et al., 1992; Qian et al., 1994)
considered both biomarker information and dietary questionnaires as
sources of aflatoxin exposure information. The study conducted by Wang
et al. (1996b) in Taiwan considered HCV, but results were
inconclusive.
3.2.2 Measure of exposure
In all of the risk assessments performed for aflatoxin, dose has
been expressed as lifetime average exposure in ng/kg per day. Note
that if peak exposure or early lifetime exposure has an impact on the
risk other than through the increase in the lifetime average ng/kg per
day, this choice of exposure measure could bias the risk estimates.
3.2.3 Measure of response
The major toxicological impact of aflatoxin on humans and animals
is an increase in primary liver cancer; that is the focus of this risk
assessment and all others performed to date.
3.2.4 Choice of mathematical model
Two basic risk models are routinely used in cancer epidemiology to
describe the relationship between dose of a contaminant and the risk
of disease or death. These models are of the form:
rM(t,E) = rO(t) × fM(E) multiplicative model
and
rA(t,E) = rO(t) + fA(E) additive model
where rM(t,E) and rA(t,E) are functions that describe disease
incidence as a function of age (t) and exposure (E). Exposure is
used here generically to include factors other than age that could
affect on the incidence rate. For unexposed individuals the incidence
rate is rO(t), and fM(E) and fA(E) are functions describing
the effect of exposure on the background. Typically, the forms for
fM(E) and fA(E) are assumed to be either linear or log-linear
(exponential). For example,
fM(E) = 1 + a1E1 + a2E2 + a3E3E2 or log[FM(E)] = a1E1
+ a2E2 + a3E1E2, where a1, a2 and a3 are parameters to be
estimated. The multiplicative model with log-linear effect of exposure
is commonly known as the Cox model and is related to logistic
regression. Tests of significance for any one effect (say E1) are
performed by testing whether its associated parameter (a1 for E1)
differs significantly from 0. The term a3E1E2 describes an
interaction between the two factors E1 and E2 and allows one to test
for such an interaction amongst the exposures. Choice of an additive
or multiplicative model can have a substantial impact upon resulting
risk estimates, particularly when extrapolating to a different
population. In the case of the additive model, differences in
background incidence have no impact on predictions of additional risk
(i.e., r(t,E) - r(t,0) = fA(E) and does not include rO(t)). On
the other hand, in the multiplicative model, predictions of additional
risk depend on the background incidence rate (i.e., r(t,E) - r(t,0)
= r0(t)(fM(t,E)-1), which is proportional to r0(t)).
Other plausible models not of the additive or the multiplicative
form include "mechanistically-based" models such as the two-stage
model for cancer. Although such models are not additive or
multiplicative per se, dose effects on the parameters that drive the
background cancer rate are usually modelled as linear or exponential,
as described earlier, and the choice of the relationship can affect
the risk estimates.
3.3 Potency estimates
3.3.1 Potency estimates based upon epidemiological data
In analysing any epidemiological study, there are many plausible
alternatives as to the form of the mathematical relationship between
exposure and response. For aflatoxin, the range of potencies derived
by using different models provides an indication of the uncertainty in
risk when one extrapolates from human data based upon studying areas
with relatively high background incidence of liver cancers and with
relatively high prevalence of HBV. In the following sections, selected
risk analyses will be reviewed briefly and the resulting potency
estimates presented and compared. In all of the analyses cited, it
should be noted that the potential effect of misspecification of the
dose that went into the derivation of the potency has not been
quantitatively addressed. As for all retrospective constructions of
exposure, use of recent levels of aflatoxin exposure to describe
current incidence rates assumes that current exposures are comparable
to past exposures. Owing to the long latency period predicted for most
cancers, uncertainty in the lifetime dose is an additional source of
variability that could lower (if the historical exposures were
actually higher than reported) or raise (if the historical exposures
were lower than actually reported) the resulting potency.
3.3.2 Potency estimates not accounting for HBV infection
Table 2 summarizes potency estimates based on analyses of
epidemio-logical studies in which regional cancer rates were compared
to estimates of aflatoxin intake without regard to differences in HBV
infection rates. As a reality check, the values in Table 2 can be
applied to the average aflatoxin exposure in the USA to obtain a
prediction of added incidence for the population. Using the largest
potency value in Table 2 (0.375) and assuming an average aflatoxin
intake of 0.26 ng/kg per day (Henry et al., 1997) for the USA
population, the added incidence is calculated to be approximately
0.375 × 0.26 or 0.0975 per 100 000 per year. Since this calculation is
based on the highest predicted potency, none of the potency estimates
in Table 2 are overtly inconsistent with the current USA rate of
approximately 3.4 per 100 000 and estimates of current levels of
aflatoxin intake (assumed to reflect past exposure levels). However,
as already indicated, these potency estimates are largely based on
studies in Africa and Southeast Asia where HBV infection rates are
much higher than in the USA or other Western countries.
Table 2. Potency estimates of the risk of liver cancer in humans based upon epidemiological
data with no correction for HBV status assuming an exposure of 1 ng/kg per day
Author Incidence/year per 100 0001
Peers & Linsell (1977) 0.11
Stoloff & Friedman (1976) 0
Carlborg (1979) 0.21
Bruce (1990)
based upon Stoloff (1983) 0
based upon van Rensburg et al. (1985),
Shank et al. (1972a,b), Peers et al. (1976, 1987) 0.10
Croy & Crouch (1991)
based on Peers et al. (1976) 0.15 (0.09, 0.23)
based on Yeh et al. (1989) 0.14 (0.08, 0.21)
Calif. Dept. Health Serv. (1990)
based on Peers et al. (1976) 0.38 (0.15, 0.60)
based on van Rensburg et al. (1985) 0.14 (0.10, 0.17)
based on Peers et al. (1987) 0.17 (NA, 0.3)
based on Yeh et al. (1989) 0.18 (NA)
1 Numbers in parentheses represent (lower, upper) 95% confidence limits on the predicted
risk when available from the authors.
3.3.3 Potency estimates accounting for HBV infection
The epidemiology study by Yeh et al. (1989) has been the focus
of several recent quantitative risk assessments and is described in
section 2.2.10.3(e). This study took place in Guangxi Province in
southern China and was a prospective cohort study of 7917 men.
In the analysis of their study, Yeh et al. (1989) adjusted
mortality rates for each region based on the age distribution of the
composite study cohort as an internal standard. Wu-Williams et al.
(1992) calculated that the age-adjusted PLC rate for the total cohort
was 121.5 per 100 000 when standardized to the age distribution of the
world population versus 226.3 per 100 000 when standardized to the age
distribution of the study cohort. The ratio of these rates (0.54) was
then used to adjust the regional PLC mortality rates reported by Yeh
et al. (1989) to obtain expected incidence rates for a
(hypothetical) cohort with age-distribution similar to the world
population. Adjusted person-years of observation (APY) were calculated
in each region as the number of PLC deaths observed in that region
divided by the adjusted mortality rate. Adjusted person-years of
observation were assumed to be distributed among HBsAg+ and
HBsAg-carriers according to the regional prevalence of hepatitis B.
The data are summarized in Table 3.
Table 3. Epidemiological data from Yeh et al. (1989)
Dose aflatoxin PLC cases APY1
(ng/kg per day) HBsAg- HBsAg+ HBsAg- HBsAg+
12 0 12 9932 2727
90 1 7 6114 2017
705 4 12 7733 2537
2028 2 23 5803 1743
-2 7 54 29582 9034
1 Adjusted person-years (see text)
2 No data are available for this group
Croy & Crouch (1991) separately analysed the HBV negative and HBV
positive cancer mortality rates in the Yeh et al. (1989) study using
additive linear models. They estimated potencies of 0.036 cancers per
100 000 per year for every ng/kg per day exposure for the HBV
negatives and 0.50 cancers per 100 000 per year for every ng/kg per
day exposure for the HBV positives.
Their analysis did not look at the combined data under a single
model and has been criticized for the use of only the small numbers of
cancers in the HBV negatives. Hoseyni (1992) analysed the Yeh et
al. (1989) data using regression techniques applied to several
different models including multiplicative and additive background
combined with linear and linear-exponential (multiplicative only)
models. He compared these various models based upon goodness-of-fit as
well as rejection by a likelihood ratio test and concluded that the
multiplicative model with a linear-exponential effect on mortality
rates by aflatoxin and HBV status (an added constant in the model if
HBsAg was positive) best fit these data. He did not explicitly include
an interaction term in this model (although the multiplicative model
implies a specific type of interaction) nor did he include an
interaction term in the additive linear model (there is no implicit
interaction in this model). The potency of the preferred
multiplicative model changes as a function of the background (in the
absence of aflatoxin and HBV) so that potencies can only be given with
respect to an explicit population liver cancer rate. Focusing on risk
prediction for the USA population, Hoseyni (1992) chose a background
cancer rate of 3.4 per 100 000 in deriving potency estimates. The
resulting estimates were 0.0018 cancers per 100 000 per year for every
ng/kg per day exposure to aflatoxin in HBV negative individuals and
0.046 cancers per 100 000 per year for every ng/kg per day exposure in
HBV positives.
Wu-Williams et al. (1992) examined the fit of a variety of
multiplicative and additive models that incorporated interaction
terms. These models were fit to the adjusted person-years data as
discussed above. Two models were found to fit the data adequately and
equally; an additive-linear model that includes an interaction term
and a multiplicative-linear model (very similar to that of Hoseyni)
with no interaction term. Under the additive-linear model, the potency
estimates were 0.031 and 0.43 for HBV-negative and HBV-positive
populations, respectively. Under the multiplicative-linear model, the
same risks for a USA population with a background cancer risk of 2.8
per 100 000 were 0.0037 and 0.094, respectively.
Finally, Bowers et al. (1993) applied an approximation to the
two-stage model of carcinogenesis (discussed in Kopp-Schneider &
Portier, 1989) to the adjusted person-years data. This model is
similar to a mixed additive model suggested by Bowers (1993), but the
parameters are tied to the biological concepts of induction of
mutations and growth of mutated cells (Thorslund et al., 1987). In
their analysis, it was assumed that aflatoxin had a linear effect on
the formation of mutations while HBV had no effect on the mutation
rate. For the growth of mutated cells, Bowers et al. (1993) assumed
a linear effect of HBV (presence or absence) and an interaction effect
of HBV and AFB1. The resulting potencies for the HBV-negative and
HBV-positive populations were 0.013 and 0.328 cancers per 100 000 per
year for every ng/kg per day exposure, respectively.
Potencies for all these studies are summarized in Table 4. Also
summarized in Table 4 are new analyses performed for the Committee
that analysed aflatoxin biomarker data from Qian et al. (1994) and
Wang et al. (1996b).
3.3.4 Potency estimates based on biomarker studies
Recent studies in Shanghai (Qian et al., 1994) and Taiwan (Wang
et al., 1996b) have measured biomarkers of aflatoxin exposure on the
individual level. The Committee has calculated potency estimates based
on these studies for comparison to estimates determined on the basis
of the data of Yeh et al. (1989). Concerning these studies, an
additional difficulty is that the internal markers of exposure were
frequently below the level of analytical quantification and
consequently individual determinations were necessarily classified on
an ordinal scale. Estimating potency requires estimating quantitative
mean levels of internal biomarkers and daily aflatoxin intake
corresponding to these classifications.
Table 4. Potency estimates of the risk of liver cancer in humans
based upon epidemiological data with correction for HBV status
assuming an exposure of 1 ng/kg per day
Study HbsAg status Incidence per 100 0001
Croy & Crouch (1991) - 0.036 (0.079)
+ 0.50 (0.77)
Wu-Williams et al.(1992)
multiplicative-linear - 0.0037 (0.006)
+ 0.094 (0.19)
additive-linear - 0.031 (0.06)
+ 0.43 (0.64)
Hosenyi (1992)
(background=3.4/100 000) - 0.0018 (0.0032)
+ 0.046 (0.08)
Bowers et al. (1993) - 0.013
+ 0.328
Qian et al. (1994)
(background=3.4/100 000) - 0.011
+ 0.11
Wang et al. (1996b)
(background=3.4/100 000) - 0.0082
+ 0.37
1 Numbers in parentheses represent upper 95% confidence limits on
the predicted risk when available from the authors.
In the study of Qian et al. (1994), 18 out of 50 (36%) cases and
31 out of 267 (12%) controls had quantified levels of urinary AFB1-
N7-Gua above the detection limit of 0.07 ng/ml. The overall range of
quantified levels was 0.3-1.81 ng/ml but the ranges for cases and
controls were not reported separately and the individual
determinations are no longer readily available (J.D. Groopman,
personal communication). The HBV-adjusted relative risk associated
with detectable levels of AFB1-N7-Gua was 9.1 (95% CI = 2.1, 29.2).
Though not reported, the HBV-adjusted relative risk associated with
detectable versus non-detectable levels of AFB1-N7-Gua was 4.6 (95%
CI = 1.8, 11.3) with a corresponding relative risk of 10.2 (95% CI =
4.9, 21.2) for HBV positivity.
Assuming an exponential distribution, estimates of mean levels of
AFB1-N7-Gua were obtained by fitting the cumulative probability
below the limit of detection to the proportion of non-detectables. The
estimated conditional mean values corresponding to non-detectable and
detectable classifications are 0.031 and 0.18 ng/ml for cases and 0.02
and 0.095 ng/ml for controls. For the logistic regression
(multiplicative linear-exponential) model, potency is the product of a
regression coefficient for AFB effect (on a ratio scale) and the
background incidence rate. Assuming that the distribution of
AFB1-N7-Gua in the general population is similar to controls and
adjusting the regression coefficient by dividing by the difference in
estimated mean levels corresponding to detectable versus
non-detectable levels gives:
J = log(4.6)/(0.095-0.02) = 20 (ng/ml)-1.
Adjusting for the relative molecular mass of AFB1-N7-Gua versus
AFB1, and assuming an average body weight of 70 kg, a daily urine
volume of 1400 ml (ICRP, 1975) and that 0.2% of daily AFB1 intake is
excreted as AFB1-N7-Gua (Groopman et al., 1992), the regression
coefficient is equivalently expressed as 0.0031 (ng AFB1/kg per
day)-1. For an annual background cancer rate of 3.4 per 100 000, the
corresponding potency estimate is 0.011 cancers per 100 000 per year
for every ng/kg per day exposure in HBV negative individuals and
10-fold higher for HBV-positive individuals.
In the study of Wang et al. (1996b), urinary metabolites were
fully quantified for all individual samples analysed and AFB1-albumin
adduct levels were quantified for only 93 of 232 (40%) blood samples
tested with a detection limit of 0.01 fm adduct/µg albumin. Estimated
HBV-adjusted relative risks were 1.6 (95% CI = 0.4, 5.5) for
detectable versus non-detectable AFB1-albumin adducts and 3.8 (95% CI
= 1.1, 12.8) for high versus low levels of urinary metabolites.
Although the urinary biomarker was fully quantified, levels of
AFB1-albumin adducts better reflect average AFB1 intake. Mean levels
corresponding to detectable and non-detectable classifications of
AFB1-albumin were calculated by fitting a log-normal distribution to
the quantified levels (Santella, personal communication) by log-probit
analysis (Travis & Land, 1990). For controls, the estimated mean
values corresponding to non-detectable and detectable classifications
were 0.0048 and 0.035 fm adduct/µg albumin.
Assuming that the distribution of AFB1-albumin adduct levels in
the general population is similar to controls, the regression
coefficient adjusted with respect to quantified levels of
AFB1-albumin is:
J = log(1.6)/(0.035-0.0048) = 16 (fm/µg albumin)-1
Based on data from China (Gan et al., 1988), 1.05 ng AFB1-albumin
adduct per g albumin corresponds to 1 µg AFB1 intake per day.
Correcting for the relative molecular mass of the AFB1 adduct, the
conversion factor is 0.00015 fm adduct/µg albumin per 1 ng/kg per day
AFB1 intake and the regression coefficient is equivalently expressed
as 0.0024 (ng/kg per day)-1. For a population with an annual
background cancer rate of 3.4 per 100 000, the corresponding potency
estimate is 0.0082 cancers per 100 000 per year for every ng/kg per
day exposure in HBV-negative individuals. The estimated relative risk
of 45.5 for HBV reported in the study of Wang et al. (1996b)
suggests a potency of about 0.37 cancers per 100 000 per year for
every ng/kg per day exposure for HBV-positive individuals.
The similarity of the estimates suggests that the magnitude of the
associations detected in Shanghai and Taiwan are relatively consistent
with that observed in Guangxi. However, the calculations presented
here are subject to a number of reservations. First, the estimates of
mean levels corresponding to detectable and non-detectable
classifications of AFB1-N7-Gua or AFB1-albumin are based on very
limited data. Furthermore, the conversion factors relating internal
exposure (AFB1-N7-Gua or AFB1-albumin) to dietary AFB1 intake are
based on studies in human populations that may have different genetic
characteristics than the study populations to which the conversion
factor is applied. For the Taiwan study, there is the additional
consideration of how the case series was obtained. About half of the
identified cases were prevalent cases diagnosed at the onset of the
study. Consequently, the AFB1 exposure determinations for these cases
may reflect alterations in metabolism directly related to the presence
of PLC per se.
3.3.5 Potency estimates from test species
Several investigators have studied the carcinogenic potential of
aflatoxins in vivo using laboratory animals (Wieder et al., 1968;
Butler et al., 1969; Epstein et al., 1969; Merkow et al., 1973;
Newberne & Rogers, 1973; Wogan et al., 1974; Vesselinovitch et
al., 1972; Ward et al., 1975; Reddy & Svoboda, 1976; Sieber et
al., 1979; Stoner et al., 1986; Angsubhakorn et al., 1981a,b;
Butler & Hempsall, 1981; Nixon et al., 1981; Moore et al., 1982;
Cullen et al., 1987). In most of these studies, hepatocarcinogenesis
was the main focus although other cancers have been noted such as
colon, kidney, lung and lymphoreticular system. The majority of these
studies focused on aflatoxin B1 with one study of aflatoxin M1
(Cullen et al., 1987), one study comparing aflatoxins B1, G1 and
B2 (Butler et al., 1969) and another study considering the
aflatoxin metabolite aflatoxicol (Nixon et al., 1981). All of these
laboratory results are amenable to quantitative estimation of risks;
however, some only contain one experimental dose group, have little
indication of dose-response due to 100% response in all dosed animals
or include the use of other agents (e.g., vitamin A) in their
protocols. Cardis et al. (1997) summarized the calculated potencies
from aflatoxin exposure in these test species. With regard to
quantitative estimations and prediction of risks for aflatoxin B1,
the study by Epstein et al. (1969) contains the most experimental
dose-groups and the most complete data for fitting a model. Using a
simple multistage model of carcinogenesis (Cardis et al., 1997),
these data predict an added incidence of 0.97 cancers per 100 000 per
year for an exposure of 1 ng/kg per day of aflatoxin B1 (scaled from
the animal data to human risk estimates using body weight raised to
the 0.75 power). Other potency estimates (extrapolated to humans)
ranged from as low as 0.05 per 100 000 per year for the Syrian golden
hamster (Moore et al., 1982) to as high as 37 per 100 000 per year
for the Fischer 344 rat (Cullen et al., 1987), with median estimate
across all experiments of 1.4 per 100 000 per year.
The human potency estimates for aflatoxin B1 alone (Table 4 shows
that these are in the range of 0.002-0.036 per 100 000 per year for
exposure to 1 ng/kg per day of aflatoxin) fall well below this range,
suggesting that humans are considerably less sensitive than other
species. There are several possible explanations for this. First, it
is possible that humans are in fact less sensitive than species tested
in laboratory experiments. Considering the proposed mechanism by which
aflatoxin induces liver tumours at low levels of exposure (DNA damage)
and the efficiency by which humans repair DNA damage, it is plausible
that humans are less sensitive. For this to be a reasonable
explanation, the rate per day of DNA repair in the human system for
the critical aflatoxin B1 lesions would have to be approximately 5
times more efficient than that of other species on a surface area
basis. A second possibility is that estimated exposures in the Yeh et
al. (1989) study were larger than the true exposures. While exposure
estimates in epidemiological studies are generally a point of concern
for any dose-response assessment, it is unlikely that the estimates
obtained in the Yeh et al. (1989) study are biased to this degree. A
third possibility is misclassification of the HBV cases, with a large
proportion of the HBV negatives actually being classified as positives
(the misclassification would need to be about 50% for the human
potency estimates to move into the range of the test species
estimates). This level of misclassification is highly unlikely; in
fact it is more likely that HBV positives have been incorrectly
classified as negatives. A fourth possibility is that many of the HBV-
positive individuals with liver tumours were actually infected late in
life and that, consequently, there was not sufficient time for HBV to
contribute to development of liver cancer in these individuals. If
true, this would suggest that the risk of liver cancer due to exposure
to aflatoxin in the absence of HBV could be as high as the risks seen
in Table 2 (approximately 0.15 cancers per 100 000 per year for an
exposure of 1 ng/kg per day). However, once again, this would seem to
be an unlikely explanation, and still falls well below the animal-
based estimates.
Finally, it is possible that the usual conversion factor for
converting potency estimates in experimental species to potency
estimates in humans is inappropriate for these data. For the case of
malignant hepatomas in Wistar rats (Epstein et al., 1969), as
mentioned above, conversion based upon surface area scaling using body
weight raised to the 0.75 power yielded a potency of 0.97 per 100 000
per year for an exposure of 1 ng/kg per day. If no conversion had been
made, the potency estimate in the Wistar rat would have been 0.23 per
100 000 per year. Considering the different sizes of the test species
involved, the potencies range from 0.014 per 100 000 per year (Syrian
golden hamster) to 1 per 100 000 per year (Fischer rat), with most of
the larger mammals being on the low range of the potency scale (0.029
per 100 000 per year for the tree shrew; 0.057 per 100 000 per year
for the rhesus and cynomolgus monkeys). In this case, although the
human estimates are still at the lowest end of the potency scale, they
would be comparable to the values estimated for other primates.
Figure 2 presents potencies estimated from animal studies and
epidemi-ological studies. Epidemiological data for which HBV infection
status was unknown and for which potencies were estimated gave
potencies in between the range predicted by the HBV
infected/non-infected numbers. Potencies given in Figure 2 do not
generally apply to aflatoxin M1, since exposure estimates given in
many of the epidemiological studies ignored the contributions to total
aflatoxins exposure from milk and milk products. From one comparative
toxicology study in rats, it has been possible to estimate that
aflatoxin M1 has a potency approximately one order of magnitude less
than that of aflatoxin B1 in that species (Cullen et al., 1987).
2.2 Toxicological studies
2.2.1 Acute toxicity
No additional acute toxicity studies have been reported in the
literature since the review by Eaton & Groopman (1994).
2.2.2 Special studies on reproductive toxicity
Ankrah et al. (1993) exposed ddy mice to AFB1 and AFG1 via
their feed (4.8 ng AFG1, 0.8 ng AFB1 (or both) per kg bw per day while
in utero. Levels of aflatoxin used were realistic relative to the
level of human exposure currently seen in Ghanaian foods. Offspring of
these animals (control and aflatoxin-fed) were continued on the
respective diets received by the parent stock until sacrifice at six
months of age. Blood obtained by cardiac puncture was used to
determine haema-tological indices and the sera were used to determine
glucose, triglyceride, total protein and albumin. AFG1 caused
significant accumulation of only neutral fat in the liver, a slight
rise in serum triglyceride and intensified hepatorenal inflammation,
necrosis and bile duct proliferation. AFB1 caused the accumulation of
both neutral fat and fatty acids in the liver, and was cytotoxic to
the liver and kidney. Iron storage in the liver, haematological
indices, serum total protein and albumin levels were not affected by
the aflatoxins. At the level used, AFG1 was six times in excess of
AFB1, but the latter was more severe in the observed hepatorenal
effects.
The authors pointed out that the mouse liver has been shown to
metabolize aflatoxin in a manner similar in some ways to the human
liver, although not all investigators would agree on this point. Hence
they postulated that the action of aflatoxin on mouse organs may shed
light on aflatoxin cytotoxicity in humans; results of this study are
of particular relevance to population groups that ingest foods known
to contain mainly AFG1 and to some extent AFB1.
2.2.3 Special studies on genotoxicity
AFB1 covalently binds to DNA and efficiently induces G to T
trans-versions; codon 249, one site in p53, is a striking hot spot for
AFB1 mutagenesis (Sengstag & Wurgler, 1994). Often, such mutations are
followed by the loss of the second functional alleles of tumour
suppressor genes, a phenomenon called loss of heterozygosity. To test
whether mitotic recombination leading to loss of heterozygosity is
induced by certain carcinogens, the authors genetically engineered a
Saccharomyces cerevisiae tester strain so that it metabolized two
important classes of carcinogens including AFB1. Human cDNAs coding
for the cytochrome P450 (CYP) enzymes CYP1A1 or CYP1A2 in combination
with NADPH-CYP oxidoreductase in a strain heterozygous for two
mutations in the trp5 gene were inserted. AFB1, when activated
intracellularly in microsomes isolated from the yeast strains
containing either human CYP enzyme, significantly induced mitotic
recombination. The authors concluded that activated AFB1 is a potent
inducer of DNA recombination in S. cerevisiae strains harbouring
various heterologous xenobiotic-metabolizing systems.
Young weanling Swiss albino mice were orally administered crude
AFB1 in a dose mimicking human exposure, i.e., at 0.05 µg/kg bw per
day for 14 weeks (Sinha & Dharmshila, 1994). Vitamin A (retinol) was
orally administered along with the toxin at double (132 IU/kg bw per
day) the human equivalent therapeutic dose. The authors concluded that
vitamin A minimized the frequency of toxin-induced clastogeny in both
mitotic and meiotic chromosomes. The decreases in sperm count, as well
as increases in abnormality in the gross morphology of the sperm head,
as observed upon toxin treatment, was ameliorated by the vitamin A.
Marquez-Marquez et al. (1993) evaluated the effects of an AFB1
inactivating system with ammonia on the genotoxicity of AFB1 measuring
micronucleus (MN) and sister chromatid exchange (SCE) analyses. Four
groups of CD1 male mice were fed for 8 weeks with a special diet
mainly composed of maize: 1) uncontaminated; 2) uncontaminated/
inactivated; 3) contaminated/ inactivated; and 4) contaminated. The
inactivating treatment was performed with ammonium hydroxide by
homogeneously impregnating the grain and leaving it for 20 days in
hermetically closed plastic bags and then heating in an oven for 24
hours to eliminate the residual ammonia. AFB1 was quantified before
and after inactivation. MN was evaluated at weekly intervals in
peripheral blood; SCE was quantified in bone marrow cells at weeks 4
and 8. The results showed that mice fed with AFB1 contaminated/
inactivated maize had a 45% lower level of induced cytogenetic damage
than those animals fed with AFB1 contaminated (but not inactivated)
maize. A residual amount of AFB1 remaining after the inactivating
treatment and the reconversion back to AFB1 in the organism may
account for the remaining increased levels of SCE and MN.
Marquez-Marquez et al. (1995) evaluated the efficiency of the
AFB1 inactivating system with ammonia, as described above, and using
mice (male CD-1) and micronucleus (MN) and sister chromatid exchange
(SCE) analysis. Apparently this study was the same as that published
earlier.
Occupational exposures to respirable dusts contaminated with the
mycotoxin AFB1 have been associated with an increased incidence of
upper airway tumours. To investigate this possible etiology Ball
et al. (1995) compared the abilities of tracheal and lung S9 from
rabbit (male, New Zealand white), hamster (male Syrian Golden) and rat
(male Sprague-Dawley) to activate AFB1 to mutagens in Salmonella
typhimurium TA98. These species differ in airway morphology with
respect to numbers of metabolically active non-ciliated tracheal
epithelial cells. Tracheas from hamster and rabbit and lung from
rabbit were active in converting AFB1 to bacterial mutagens. Tracheas
from hamster were more efficient in activating AFB1 to mutagens than
lung, while rabbit lung was over 4 times more active in converting
AFB1 to mutagens than that from trachea. In all cases, AFB1 was more
mutagenic than B[a]P. The relative capabilities of trachea to activate
AFB1 were in agreement with the ability of cultured tracheas from
these species to form AFB1-DNA adducts. These results demonstrate that
AFB1 is activated more efficiently than B[a]P in the lung, and that
the metabolic capabilities of airway epithelium to activate AFB1 are
not predictable by airway morphology.
A study by Shi et al. (1995a) examined the effect of two
selenium compounds, namely, sodium selenite and selenium-enriched
yeast extract (SeY) on the cytotoxicity, DNA binding, and mutagenicity
of AFB1 in cultured Chinese hamster ovary (CHO) cells. CHO cells,
after treatment with 2 µg/ml selenite or 80 µg/ml SeY, exhibited
increased resistance to AFB1-induced cell killing. At a concentration
of 50 µg/ml AFB1, cell survival, measured by the clonogenicity assay,
was increased by 21- and 10-fold in selenite- and SeY-treated cells,
respectively. However, selenium treatment did not appear to affect
AFB1-DNA binding. Similarly, no effect was observed on AFB1
mutagenicity, as determined by the hypoxanthine-guanine phosphoribosyl
transferase (HPRT) gene mutation assay. The results showed that
selenium could effectively protect cells from AFB1 cytotoxicity in
cultured cells, but had no effect on AFB1-DNA adduct formation or
mutagenesis. The authors suggested that there are multiple pathways of
AFB1 toxicity and that selenium can modulate AFB1-induced cell killing
independent of its genotoxicity.
Rats and mice differ markedly in sensitivity to AFB1
hepatocarcinogenicity, the former being sensitive and the latter
resistant. The purpose of this study was to determine whether the
formation of AFB1-albumin (AF-alb) adducts was related to the
induction of cytogenetic changes in vivo as a step to understanding
whether such markers of exposure may be informative with respect to
genetic alterations important in the carcinogenic process (Anwar
et al., 1994). The comparison was made at two levels: between
species and between individuals within a species. Animals (male Helwan
Wistar albino rats and Swiss albino mice) were treated with single
doses of different concentrations of AFB1 between 0.01 and 1.0 µg
AFB1/g bw. The frequency of chromosomal aberrations and micronuclei in
the bone marrow was measured and compared to the level of AFB1 bound
covalently to albumin in the peripheral blood. Both chromosomal
aberrations and micronuclei were significantly increased in treated
rats compared to the control group at doses above 0.1 µg/g. In
contrast, in mice, a slight increase in chromosome aberrations was
seen in the highest dose group (1.0 µg/g), but no increase in
micronuclei was observed at any of the doses. The level of chromosomal
aberrations was about 10 times higher in rats than in mice at the
highest dose of AFB1. AFB1-albumin increased linearly with dose of
AFB1, and there were strong statistically significant correlations at
the individual rat level with both chromosomal aberrations.
The level of AF-alb adducts was higher for a given dose in rats
than mice, as has been seen for the level of liver DNA adducts in the
two species. The metabolic basis of these differences has been
investigated and has been shown to be associated with the expression
of a specific glutathione-S-transferase isoenzyme in mice, which
efficiently conjugates the AFB1-epoxide to glutathione.
In rats, the level of AF-alb adducts was strongly correlated with
the frequency of both micronuclei and chromosomal aberrations in the
bone marrow. An increase in adduct levels was seen with exposures as
low as a few ng AFB1/g bw, whereas the genetic alterations were only
increased above control levels at doses around 0.1 µg/g.
The authors suggested two considerations for interpretation of
the present studies. First, the cells in which the micronuclei and
chromosomal aberrations were examined are not the target cells for
AFB1 hepatocarcinogenesis, and second, that this type of genetic
marker is relatively non-specific. Thus, the genetic alterations being
measured are not directly relevant to the carcinogenic process; this
limitation may be overcome as sensitive molecular techniques are
developed to measure mutation induced by aflatoxin in specific gene
sequences in somatic cells (See Aguilar et al., 1993). Recent
studies by these authors (Wild et al., in preparation) suggest that
AF-alb adducts reflect the differing species sensitivity to AFB1
carcinogenesis. This peripheral blood marker could be an indicator of
risk of liver cancer development in addition to being a marker of
exposure, the authors suggest, as has been further supported by the
study of Ross et al. (1992), in which the level of AFB1-N7 guanine
adduct in the urine was related to the subsequent risk of developing
hepatocellular carcinoma in a Chinese cohort. This study will be
discussed in section 2.2.10.
2.2.4 Special studies on immunosuppression
The immunosuppressive potential of AFB1 was evaluated in growing
rats (Raisuddin et al., 1993). The weanling rats (species
unspecified) were sub-chronically exposed to 60, 300 or 600 µg AFB1/kg
bw for four weeks on alternate days by oral feeding. Various
parameters of cell-mediated immunity (CMI) and humoral immunity were
assessed in control and treated animals. CMI was evaluated by
measuring delayed type of hypersensitivity (DTH) response and humoral
immunity was measured by plaque forming (PFC) assay. The
lympho-proliferative response assay for T- and B-cells was also
performed. It was observed that AFB1 selectively suppressed
cell-mediated immunity in growing rats. AFB1 suppressed CMI at the 300
and 600 µg dose levels only as measured by DTH response assay. The
authors concluded that continuous low level exposure of aflatoxin to
the growing host may enhance its susceptibility to infection and
tumorigenesis.
Jakab et al. (1994) conducted experiments to demonstrate the
immunosuppressive effects of AFB1 ingestion, in this case respiratory
tract exposure to AFB1. Rats (male Fischer 344) and mice (female
Swiss) were exposed either by aerosol inhalation or intratrachael
instillation to AFB1. Nose-only inhalation exposure of rats to AFB1
aerosols suppressed alveolar macrophage (AM) phagocytosis at an
estimated dose of 16.8 µg/kg with the effect of persisting for
approximately 2 weeks. To determine whether another mode of
respiratory tract exposure, intratrachael instillation, reflected
inhalation exposure, animals were treated with increasing
concentrations of AFB1, which also suppressed AM phagocytosis in a
dose-related manner, albeit at doses at least an order of magnitude
more than that obtained by aerosol inhalation. Intratrachael
administration of AFB1 also suppressed the release of tumour necrosis
factor-alpha from AMs and impaired systemic innate and acquired immune
defences as shown, respectively, by suppression of peritoneal
macrophage phagocytosis and the primary splenic antibody response. The
authors concluded that experimental respiratory tract exposure to AFB1
suppressed pulmonary and systemic host defences; they indicated that
inhalation exposure to AFB1 is an occupational hazard where exposure
to AFB1-laden dust is common, such as in grain dust.
2.2.5 Factors modifying carcinogenicity of aflatoxins
Young adult male Fischer rats maintained on a reduced calory diet
(60% of ad libitum food consumption) for 6 weeks showed a decrease
in the binding of AFB1 to hepatic or renal nuclear DNA and a reduction
of AFB-induced hepatocellular damage (Chou et al., 1993). Repeated
dosing of rats with AFB1 resulted in the inhibition of hepatic and
renal DNA synthesis as measured by [3H]thymidine incorporation.
However, the rate of DNA synthesis was greater in ad libitum (AL)
rats than in calorically restricted (CR) animals. Three days after
AFB1 dosing, the rate of DNA synthesis had recovered to the control
level. Cell cycle analyses measured by a flow cytometric method on
kidney cells of both AL and CR rats showed that there were no
significant changes in cell populations in the S phase between these
two groups of rats. AFB1 inhibited the cell proliferation by 33% (on
average). The restoration of the cell proliferation in kidney cells
was found on the third day after AFB1 dosing. The rate of regenerative
cell proliferation was found to be slightly greater in AL rats than in
CR animals. The AFB1-induced regenerative DNA synthesis in both liver
and kidney was retarded by CR.
Youngman & Campbell (1992) demonstrated that with young Fischer
344 rats the post-initiation development of AFB1-induced
gamma-glutamyltranspeptidase-positive (GGT+) hepatic foci was
markedly inhibited by low protein feeding, even though the energy
intake was greater. These investigators also studied this dietary
effect upon the development of hepatic tumours and the correlation of
foci development with tumour development. Following AFB1 dosing (15
daily doses of 0.3 mg/kg each), animals were fed diets containing 6,
14 or 22% casein (5.2, 12.2 or 19.1% protein) for 6, 12, 40, 58 or 100
weeks. Foci at 12 weeks and tumours at 40, 58 and 100 weeks developed
dose-dependently to protein intake. Foci development, tumour
incidence, tumour size and the number of tumours per animal were
markedly reduced, while the time to tumour emergence was increased
with low-protein feeding. Non-hepatic tumour incidence also was lower
in the animals fed the lowest protein diet. Foci development indices
(foci number, per cent liver volume occupied) were highly correlated
with tumour incidence at 58 and 100 weeks (r = 0.90-1.00). Tumour and
foci inhibition occurred in spite of the greater energy intake.
Previous results from a large ecological study in 65 rural
counties in China suggested that primary liver cancer in humans
primarily is associated with chronic HBV infection, coupled with
nutritional factors (e.g., animal protein) that elevate plasma
cholesterol level and encourage cancer growth (Campbell et al.,
1990). To test this hypothesis, the authors investigated the effect of
dietary animal protein on tumour development in HBV transgenic mice.
Male F2 offspring of 50-4 HBV transgenic mice were randomly assigned
to 6, 14 and 22% dietary casein. Serum was collected from the retro-
orbital vein and was analysed for the level of hepatitis B virus
antigen (HBsAg), the products of the S-transgene. The increases from
baseline in S-gene product observed for the normal protein animal
(22%) at 3 months was inhibited in the mid- and low-protein animals by
42% and 72%, respectively, with a highly significant dose-response
relationship (P<0.001). Serum glutamic-pyruvic transaminase activity
was not affected by diet treatment. The authors concluded that their
results strongly suggest that dietary casein controls, in a
dose-response manner, S-transgene expression in these experimental
animals.
Hasler et al. (1994) fed Fischer 344 rats a low-fat high
carbohydrate (HC) diet, an isocaloric fat-containing (IC) diet, a
hypercaloric fat-containing (HF) diet or a commercial rodent chow. The
effects of these diets were studied on the binding of AFB1 to
exogenous DNA and on the activities of hepatic glutathione
transferases (GSTs), cytochromes 2B1 and 1A1. Microsome-mediated
binding of [3H]AFB1 to exogenous DNA was significantly lower in the
HC rats than in the chow- and IC-fed rats. No significant differences
were noted between HF and either HC or IC rats. There was no
significant difference in hepatic GST activity of rats fed the
different diets. The authors suggested that high carbohydrate/low fat
diets may reduce microsome-mediated epoxidation of AFB1 to a larger
extent than high-fat diets. In general, high-fat diets increased
cytochrome 1A1 and 2B1 activities relative to chow and
high-carbohydrate diet. This suggested greater detoxification of AFB1,
thus reducing the amount of AFB1 available for hepatic macromolecular
binding, the authors concluded.
An excellent review by Massey et al. (1995) covers the
biochemical and molecular aspects of mammalian susceptibility to AFB1
carcinogenicity. Important considerations include: 1) different
mechanisms for bioactivation of AFB1 to its ultimate carcinogenic
epoxide metabolite; 2) the balance between bioactivation to and
detoxification of the epoxide; 3) the interaction of AFB1 epoxide with
DNA and the mutational events leading to neoplastic transformation; 4)
the role of cyto-toxicity in AFB1 carcinogenesis; 5) the significance
of non-epoxide metabolites in toxicity; and 6) the contribution of
mycotoxin-unrelated disease processes.
2.2.6 Special studies on covalent binding of aflatoxin residues with
nucleic acids and proteins
Shi et al. (1994) studied the effect of selenium on AFB1-DNA
binding and adduct formation. Male Fischer 344 rats, fed with up to 8
mg/litre sodium selenite in drinking-water for 8 weeks, were given a
single i.p. dose of AFB1. The rats were killed 24 hours later and the
amount of AFB1 bound to hepatic DNA and the amount of DNA adducts
formed were determined. Selenium pretreatment resulted in a
dose-dependent inhibition of AFB1-DNA binding as well as adduct
formation. This was accompanied by an increase of reduced glutathione
(GSH) in the liver of selenium-treated animals. These results
suggested that selenium could effectively inhibit AFB1-induced DNA
damage, which may be partially responsible for its anticarcinogenic
effect against AFB1.
Choy (1993) has reviewed the dose-response induction of DNA
adducts by AFB1 and its implication to quantitative cancer risk
assessment. Dose-response curves of DNA adduct formation after
ingestion or injection treatments in the rat were reviewed; a linear
dose-response relationship was observed in both injection and
ingestion studies at low doses. The author concluded that this
observation is consistent with the assumption of the linear
dose-response risk assessment model for genotoxic agents and justifies
the use of this model for quantitative cancer risk assessment for
aflatoxins. The author also concluded that although AFB1-DNA adducts
generated in rats, mice and humans reflect the "molecular dose" and
DNA damage in the target organ, bypassing the need for interspecies
pharmacokinetic dose adjustments, it is not possible to extrapolate
from rodents to humans at this time because human DNA adduct data are
incomplete.
2.2.7 Special studies on glucose tolerance
Glyoxalase-1 activity plays an important role in glucose
metabolism and has been reported to be depressed in mice fed low
levels of AFB1 (Ankrah, 1995). In the present study, glyoxalase-1
activity, glucose tolerance and pancreatic beta cell sensitivity were
examined in mice (male and female ddy) fed 0.045 ng AFB1 plus 0.450 ng
ABG1/g feed prenatally and for 6 months after birth. After glucose
challenge, the ratios between 0- and 2-hour serum glucose levels were
significantly higher than controls, indicating an increase in
tolerance of glucose in the aflatoxin-fed mice with lower glyoxalase-1
activity. Pancreatic beta cell sensitivity to stimulation by
tolbutamide was similar in both groups. However, liver malonic
dialdehyde was significantly higher in the aflatoxin-fed mice,
suggesting that the altered tolerance for glucose in the aflatoxin-fed
mice might be a consequence of aflatoxin-mediated peroxidative action
in the liver, the authors suggested.
2.2.8 Special studies on effect of ammoniation of AFB1 in
contaminated cottonseed
The effectiveness of ammonia in inactivating aflatoxin in
contaminated cottonseed was investigated (Bailey et al., 1994). Two
aflatoxin-contaminated cottonseed lots were treated separately using
atmospheric pressure, ambient temperature ammoniation procedure (APAT)
or a high pressure, high temperature ammoniation procedure (HPHT), and
incorporated into dairy cow rations. Isocalorific diets containing 25%
defatted, dried milk from cows fed aflatoxin-contaminated cottonseed
without or with APAT or HPHT treatment, or an aflatoxin-free human
grade commercial milk powder, were then fed for 12 months to rainbow
trout (Oncorhynchus mykiss). AFM1 concentrations in milk powders
without and with seed treatment were: APAT, 85 and <0.05 µg/kg; HPHT,
32 and <0.05 µg/kg. In the APAT experiment, trout consuming the diet
containing milk from cows fed the aflatoxin-contaminated cottonseed
had a 42% incidence of hepatic tumours; APAT cottonseed treatment
reduced this to 2.5%. Positive controls were included to demonstrate
trout responsiveness. AFB1 fed continuously for 12 months at 4 µg/kg
resulted in a 34% tumour incidence, whereas positive controls fed 20
µg AFB1/kg, 80 µg AFM1/kg, or 800 µg AFM1/kg for 2 weeks and killed 9
months later had a 37, 5.7 and 50% incidence of tumours, respectively.
The authors concluded that APAT ammonia treatment of
aflatoxin-contaminated dairy cattle cottonseed feedstock abolished the
detectable transfer of AFM1 or AFB1 into milk powder, and greatly
reduced the carcinogenic risk posed by any carry-over of aflatoxins or
their derivatives into milk.
In addition, the results confirm AFM1 to be a lower level
hepatocarcinogen in comparison with AFB1 in the trout carcinogenicity
assay. In the separate HPHT experiment, no tumours were observed in
the livers of trout fed diets containing milk from either the
ammonia-treated or untreated source, or the control diet containing 8
µg AFM1/kg. Positive controls fed 64 µg AFB1/kg for 2 weeks exhibited
a 29% tumour incidence 12 months later. Thus in this experiment,
neither AFM1 at 8 µg/kg nor any HPHT-derived aflatoxin derivatives
that might have been carried over into milk represented a detectably
carcinogenic hazard to trout, the authors conclude.
2.2.9 Special studies on aflatoxin and hepatitis B virus infection in
woodchucks, ducks, ground squirrels and tree shrews
Interactive hepadnavirus and chemical hepatocarcinogenesis has
been studied in woodchucks inoculated as newborns with woodchuck
hepatitis virus (WHV), which is closely related to the human hepatitis
B virus (Bannasch et al., 1995). When the woodchucks reached 12
months of age, AFB1 was administered in the diet at dose levels of 40
µg/kg bw per day for 4 months and subsequently 20 µg/kg bw per day
(5 days/week) for a lifetime. WHV DNA was demonstrated by Southern
blot hybridization in the serum and by PCR in the serum and/or liver
tissue. The histomorphology and cytomorphology of the liver were
investigated by light and electron microscopy. WHV carriers with and
without AFB1 treatment developed a high incidence of preneoplastic
foci or altered hepatocytes, hepatocellular adenoma and hepatocellular
carcinomas that appeared 6-26 months after the beginning of the
combination experiment. Administration of AFB1 to WHV carriers
resulted in a significantly earlier appearance of hepatocellular
neoplasms and a higher incidence of hepatocellular carcinomas compared
to WHV carriers not treated with AFB1. Neither hepatocellular adenomas
nor carcinomas (but preneoplastic foci of altered hepatocytes) were
detected in woodchucks receiving AFB1 alone, and no preneoplastic or
neoplastic lesions were found in untreated controls.
The authors pointed out that these results provide conclusive
evidence of a synergistic hepatocarcinogenic effect of hepadnaviral
infection and dietary AFB1. The striking similarities in altered
cellular phenotypes of preneoplastic hepatic foci similarities in
altered cellular phenotypes of preneoplastic hepatic foci emerging
after both hepadnaviral infection and exposure to AFB1 suggested
closely related underlying molecular mechanisms that may be mainly
responsible for the synergistic hepatocarcinogenic effect of these
oncogenic agents.
In addition, the authors observed that the decisive role of the
chronic WHV infection for hepatocarcinogenesis became particularly
evident in those animals that seroconverted after 1 year and showed
neither a chronic active hepatitis nor hepatocellular neoplasms, no
matter when AFB1 was given. From this observation, the authors
concluded that chronic hepatitis is not an absolutely necessary
condition for the development of HCC in WHV carriers.
To determine whether p53 mutations are common to HCCs of hosts
infected with related hepadnaviruses with and without treatment with
aflatoxin, Rivkina et al. (1994) studied the occurrence of mutations
in the p53 gene in HCCs of ground squirrels and woodchucks with a
history of infection with ground squirrel hepatitis virus (GSHV) and
woodchuck hepatitis virus (WHV). Sequencing of wild type p53 genes
from ground squirrels and woodchucks revealed remarkable homology
between the two species; using direct polymerase chain reaction
sequencing, the investigators analysed the state of the p53 gene in 20
HCCs from ground squirrels (2 uninfected, 7 with past and 11 with
ongoing infection with GSHV) and in 11 HCCs from woodchucks
persistently infected with WHV. Five GSHV carrier and two uninfected
ground squirrels received i.p. administration of AFB1. Only one
mutation - located in codon 176 of exon 5 - in the p53 gene of the
tested animals was detected and that in a GSHV-positive ground
squirrel treated with AFB1. The investigators suggested that in view
of the considerably lower apparent rate of mutations in comparison to
human HCCs, other etiological factors may be of greater significance
in the development of HCC in ground squirrels and woodchucks.
The unique mutation from G to T at the third base in codon 249
observed in human hepatocellular carcinoma has been suggested to be
linked to aflatoxin exposure. Imazeki et al. (1995) studied six
ducks with HCC, three of which were infected with duck hepatitis B
virus and five of which were fed a diet containing AFB1 for 1-2 years.
Liver tissues were analysed for the presence of point mutations at
this codon of the p53 gene by polymerase chain reaction and direct
nucleotide sequencing. None of the six ducks with HCC showed the
change at this codon regardless of duck hepatitis B virus infection.
Integration of duck hepatitis B virus DNA into the host genome was not
observed in two ducks that were chronically infected with the virus
and treated with AFB1. A third duck from Qitong Province in China,
where HBV and AFB1 are risk factors for HCC in humans, did show viral
integration. This suggested, in the opinion of the authors, that AFB1
itself might not be involved in the unique mutation at codon 249 in
hepatocarcinogenesis, or that other factors coincident with aflatoxin
may be responsible for this unique mutation.
Cova et al. (1996) used a Pekin duck model to examine the
effect of congenital duck hepatitis B virus (DHBV) infection and AFB1
exposure in the induction and development of liver cancer. The study
of the two major risk factors in the development of HCC, i.e.,
persistent hepatitis virus infection and exposure to dietary
aflatoxins, has been hampered by lack of an animal model, and these
experiments were undertaken to this end. AFB1 was administered to
groups of 13 DHBV infected or non-infected ducks at two doses (0.08
and 0.02 mg/kg) by i.p. injection once a week from the third month
posthatch until they were sacrificed 2.3 years later. Two control
groups of ducks not treated with AFB1 (one of which was infected with
DHBV) were observed for the same period. Higher mortality was observed
in ducks infected with DHBV and treated with AFB1 compared to
non-infected ducks treated with AFB1 and other control ducks. In the
groups of non-infected ducks treated with high and low doses of AFB1,
liver tumours developed in 3 out of 10 and 2 of 10 ducks,
respectively. In infected ducks treated with the high dose, 3 of 6
showed liver tumours; there were none with the low dose of AFB1. No
liver tumours were observed in the two control groups. Ducks infected
with DHBV and treated with AFB1 showed more pronounced periportal
inflammatory change, fibrosis and focal necrosis compared to other
groups. All DHBV carrier ducks showed persistent viraemia throughout
the observation period. An increase of viral DNA titres in livers and
sera of AFB1-treated animals compared to infected controls was
frequently observed.
No DHBV DNA integration into the host genome was observed,
although in one hepatocellular carcinoma from an AFB1-treated duck, an
accumulation of viral multimer DNA forms was detected. Unlike the
situation observed for woodchuck and ground squirrel, HCC has rarely
been associated with DHBV infection or integration of viral DNA in the
duck. HCC has to date been reported only in Chinese ducks from
Chi-tung County, not always associated with detectable virus, and with
only a single reported case of integrated DHBV. Colonies of
DHBV-infected ducks from other parts of the world do not develop HCC.
Prevalence of liver tumours observed in Chi-tung County ducks
reportedly correlated with the AFB1 food contamination and with the
incidence of primary liver cancer in these areas.
The authors observed a lower level of AFB1 binding to liver DNA
and plasma protein in the DHBV-infected ducks compared to non-infected
ducks after a single dose of AFB1; this finding appeared inconsistent
with the hypothesis that DHBV infection could increase the metabolic
activation of AFB1, as has been observed in woodchucks and in some
human data. The investigators noted that their observations were made
at a single dose at a single exposure and using one specific age of
ducks; all of these factors could have influenced the AFB1-DNA adduct
level.
Yan et al. (1996) reported the successful establishment of an
animal model in tree shrews (Tupaia belangeri chinensis) captured
from the wild and experimentally infected with human hepatitis B
virus. In animals exposed to AFB1 and infected with HBV, the incidence
of HCC was significantly higher than in the animals solely infected
with HBV or exposed to AFB1. AFB1-exposed animals received a total
dosage of 15-16 mg/animal. No HCC or precancerous lesions were found
in the controls that were neither HBV-infected nor AFB1-exposed. HBV
DNA and the protein it encodes were detected in the cancer cells
and/or the surrounding hepatocytes. Integration of HBV DNA into the
host liver genome was found during hepatocarcinogenesis among the
animals infected by HBV.
The investigators pointed out that the cumulative dose of AFB
used in their experiment was much lower than those (24-66 mg/animal)
used in previous experiments on tree shrews where HCC was seen. This
suggested that HBV infection might increase the hepatocarcinogenic
effect of AFB1. The occurrence of precancerous GT foci in the tree
shrews exposed only to AFB1 was much more frequent than in those
infected by HBV alone. Among the animals exposed to the same dose of
AFB1, the gamma-glutamyltranspeptidase (GGT) foci were more numerous
and larger in HBV-infected than in uninfected animals during the late
state (after the 83rd week), but not at the early state. This suggests
that, although both AFB1 and HBV may induce GGT foci and have a
synergistic effect, the effect of HBV is weaker and slower than that
of AFB1.
2.2.10 Observations in humans
2.2.10.1 Biomarkers of aflatoxin exposure
A key issue in the use of aflatoxin biomarkers is whether the
ratio of AFB1-albumin adduct to DNA adduct suggested in rodent
experiments is the same in humans (Wild et al., 1996). Direct
evidence for this is not available due to the limitation of measuring
DNA adducts in human liver. The human populations where AFB1 intake
vs. AFB1-albumin adduct relationship has been examined are populations
in which aflatoxin intake is relatively high. Examination of the
relationship in low-exposure populations would be important to test
whether the linear dose-response relationships seen in rats at
exposures as low as 1 ng AFB1/kg bw are also observed in man. There
are some data discussed in Wild et al. (1996) indicating that the
amounts of AFB1 intake bound to albumin are similar for rats and
humans; assuming that the majority of AFB1-DNA adducts are formed in
liver, then the initial ratio between the serum albumin and liver DNA
adducts would be expected also to be similar in humans and Fischer
rats. However, the capacity of human intestine to metabolize AFB1 must
be further explored to clarify this point.
It would appear that the AFB1-albumin adduct in peripheral blood
is a reliable marker of AFB1-DNA adducts in the liver in rodents (Wild
et al., 1996). Both of these parameters are at least qualitatively
associated with species susceptibility to AFB1 hepatocarcinogenesis.
Cross-species extrapolation to man suggests that the amount of
AFB1-albumin formed for a given exposure more closely approximates
that in the sensitive species rather than the resistant, and indicates
that the Fischer rat may be a more appropriate model than the mouse
for molecular dosimetry studies of AFB1 when, for example, validating
approaches for chemoprevention studies.
However, carcinogenesis is a multistep process; as pointed out in
Wild et al. (1996) AFB1-albumin adduct is acting as a surrogate
marker only for one critical step, the formation of AFB1-DNA adducts
in the target cell. The relationship between this marker and the
genetic consequences of exposure as well as the quantitative
association with HCC risk in man remain to be determined. In addition,
HBV and possibly HCC infection, are major risk factors. The
availability of more reliable markers of biologically effective dose
of AFB1 should contribute to improving attempts to understand the
mechanism of interaction between these two and other risk factors.
2.2.10.2 Mutations in p53 tumour-suppressor gene in human
hepatocellular carcinoma
Molecular epidemiological studies have found that a G to T
mis-sense mutation at the third base of codon 249 of the p53 gene,
effecting an arginine to serine substitution, occurs in high frequency
(up to 67%) in human liver tumours in regions with high risk of
aflatoxin exposure, but not in regions of low aflatoxin exposure
(Ozturk, 1991). Hsieh & Atkinson (1995) performed experiments to
confirm this, using liver tissue from liver cancer patients in Taiwan
and Japan. This was analysed for the presence of aflatoxin-DNA adducts
(ADA) as a marker for aflatoxin exposure and an AGG to AGT
transversion at codon 249 of the p53 gene. Ten per cent of samples
containing ADA, indicating definite exposure of the subjects to
aflatoxin, were found to harbour the codon 249 mutation, whereas 18%
of the samples with no detectable adducts also contained the mutation.
Since the presence of ADA in the liver tissue samples is an indication
of definite recent exposure of the liver cancer patients to aflatoxin,
these data indicated that the codon 249 mutation is not a high
frequency event associated with recent aflatoxin exposure. If recent
exposure to aflatoxin is indeed involved in the late stage
hepatocarcinogenesis, these data suggested that it is through some
mechanism other than codon 249 mutation. If either mutation at codon
249 of the p53 gene or exposure to aflatoxin is involved in earlier
stages of hepatocarcinogenesis, whether codon 249 of the p53 gene is a
"hot spot" for aflatoxin attack could be shown by the present
experiment, the authors concluded.
The tumour suppressor p53 exerts important protective functions
towards DNA-damaging agents (Gerbes & Caselmann, 1993). Its
inactivation by allelic deletions or point mutations within the p53
gene as well as complex formation of wildtype p53 with cellular or
viral proteins is a common and crucial event in carcinogenesis.
Mutations increase the half-life of the p53 protein allowing the
immunohistochemical detection and anti-p53 antibody formation.
Distinct G to T mutations in codon 249 leading to a substitution
of the basic amino acid arginine by the neutral amino acid serine are
responsible for the altered functionality of the mutation gene product
and were originally identified in 8 of 16 Chinese and 5 of 10 African
HCC patients, both groups living in regions with traditionally high
exposure to mycotoxins. None of these mutations was detectable in 20
patients with HCCs recently studied in the United Kingdom; only two of
13 HCC DNAs from Germany displayed a C to T and a T to A transversion,
respectively, in codons 257 or 273, but not in codon 249. An average
p53 gene nutation rate of 25% is currently assumed for high-AFB1
exposure regions; this is double the rate observed in low-AFB1
exposure countries. The authors concluded that although many HCC
patients displaying P53 mutations also suffer from HBV infection,
which itself can lead to rearrangement of P53 coding regions or induce
the synthesis of viral proteins possibly interacting with p53, the
specific G to T transversion within codon 249 of the P53 gene seems to
directly reflect the extent of AFB1 exposure and is not pathognomonic
for all HCCs.
Yap et al. (1993) analysed 24 HCC liver biopsy samples from
patients in Durban, South Africa, for p53 mutations and HBV infection.
One patient was negative for HBV (Hbsag, anti-HBcAb, anti-HBsAb) and
possessed the p53 249 mutation (which results in an arginine to serine
substitution). The authors suggested that HBV infection or integration
increases the likelihood of, but is not essential for, this p53
"hot-spot" mutation in HCC. AFB1 or other as yet unidentified
environmental carcinogens and cofactors are implicated; the mechanisms
by which cells exposed to these agents acquire such a specific
mutation and then expand clonally remains to be elucidated.
The subject of the mutation at codon 249 of the p53 tumour
suppressor gene has continued to be the subject of much research.
Fifty-eight per cent of HCCs from Quidong, China, contain this
mutation which is rarely seen in HCCs from Western countries (Aguilar
et al., 1994). The population of Qidong is exposed to high levels of
AFB1 and this toxin has been shown to induce the same mutation in
cultured human HCC cells. To investigate the role of AFB1 and of these
p53 mutations in hepatocarcinogenesis, normal liver samples from the
USA (5), Thailand (3), and Qidong (14) (where AFB1 exposures are
negligible, low, and high, respectively), were examined for p53
mutations. The frequency of the AGG to AGT mutation at codon 249
paralleled the level of AFB1 exposure, which supports the hypothesis
that this toxin has a causative - and probably early - role in
hepatocarcinogenesis. However, a role for other carcinogens cannot be
ruled out, the authors point out; bulky heterocyclic amines in cooked
foods and oxidants released by inflammatory leukocytes possess the
same specificity for G to T transversion and HBV infection is
associated with inflammation. All of the liver samples from Qidong and
Thailand were from HBV-infected individuals.
The presence of elevated frequencies of codon 249 AGT mutations
in the non-malignant tissue of HCC patents from Qidong suggested that
the mutagenic event occurred early in hepatocarcinogenesis. In
contrast, p53 mutations in HCCs from geographic areas with low
exposure to AFB1 could be late events. For example, p53 mutations have
been observed more frequently in large tumours and in advanced grades
of malignancy in HCCs from Japan. In other organs, such as the colon
and the bladder, p53 mutations are thought to occur late in
tumorigenesis. However, the methods used in previous work may not have
been sensitive enough to detect mutations at early stages of
tumorigenesis.
Fujimoto et al. (1994) tested the hypothesis that exposure to
AFB1 alone or coincident with other environmental carcinogens might be
associated with allelic losses occurring during development of human
hepatocarcinogenesis (HCC) in China. The HCCs were obtained from two
different areas in China: Qidong, where exposure to HBV and AFB1 is
high; and Beijing, where exposure to HBV is high, but that to AFB1 is
low. Tumours were analysed for mutations in the p53 gene and loss of
heterozygosity for the p53, Rb and APC genes and at marker loci on
chromosomes 4, 13 and 16. The data indicated that mutation and/or loss
of heterozygosity in the p53 gene, independent of the 249 mutation,
played a critical role in the development of HBV-associated HCCs in
China. The authors postulated that different mechanisms appeared to be
responsible for the development of HCC in Beijing and may have
resulted from exposure to unknown environmental carcinogens or a
different subtype of HBV. Also, the results demonstrated that multiple
alterations in DNA located on different chromosomes may be involved in
the development of HCC.
Additional support for the etiological role of AFB1 in
hepatocarcinogenesis in regions of the world with AFB1-contaminated
food has come from the studies of Aguilar et al. (1993). These
investigators studied the mutagenesis of codons 247-250 of p53 by rat
liver microsome-activated AFB1 in human HCC cells HepG2 by restriction
fragment length polymorphism/polymerase chain reaction genotypic
analysis. AFB1 preferentially induced the transversion of G to T in
the third position of codon 249, and also induced G to T and C to A
transversions into adjacent codons, albeit at lower frequencies. Since
the latter mutations are not observed in HCC, the investigators
concluded that both mutability on the DNA level and altered function
of the mutant serine 249 p53 protein are responsible for the observed
mutational hot spot in p53 HCC from AFB1-contaminated areas. The fact
that this mutation is only rarely found in HCC from low AFB1 regions
indicates that it is not a prerequisite for hepatocarcinogenesis;
perhaps HBV and the mutant serine 240 p53 protein play a synergistic
role.
In a later study, Aguilar et al. (1995) examined normal liver
samples from the USA, Thailand and Qidong, where AFB1 exposures are
negligible, low and high, respectively, for p53 mutations. The
frequency of the AGG to AGT mutation at codon 249 paralleled the level
of AFB1 exposure, which, according to the authors, provides additional
support for the hypothesis that this toxin has a causative and
probably early role in hepatocarcinogenesis.
Hulla et al. (1993) analysed the p53 gene at the site
corresponding to codon 249 of the human gene in AFB-induced
preneoplastic hepatic nodules from rats. No mutations were detected in
the tissues examined. Thus, at least in the rat, the authors suggested
that AFB exposure alone may not be sufficient for the specificity of
p53 mutations observed in HCC. The selective mutations have been
identified only in populations at risk for hepatitis B; it is possible
that both AFB1 and chronic hepatitis are essential for mutation at
codon 249 in the human p53 gene.
In another study in rats, Liu et al. (1996) looked at the
effects of AFB1 on the p53 locus at the preneoplastic stage of rat
liver oncogenesis. Male Wistar rats received a single dose of 1.5 mg
AFB1/kg bw by a gastric tube. Liver biopsies over a period of one year
were examined for aberrations of the p53 gene together with the
expression of placental GST, a marker for preneoplasia.
Immunohistochemistry, Western blot, polymerase chain
reaction-single-strand conformation polymorphism and DNA sequencing
techniques were used. AFB1 induction resulted in GST overexpression,
forming GST-positive multi-foci and nodules of hepatocytes but no
aberrations in the p53 expression and the microstructure of exons 5-8
of the p53 gene. Thus, the authors concluded that p53 mutations might
not occur at this early stage of AFB1-induced hepatocarcinogenesis.
Shi et al. (1995b) characterized p53 mutations in 44
hepatocellular carcinomas from Chinese patients residing in a
high-incidence area. In contrast to HCCs from other high HCC incidence
areas with endemic aflatoxin exposures, in which codon 249 is a
mutational hotspot, no mutations were observed at codon 249. The
authors concluded that risk factors other that dietary exposure to
aflatoxin may contribute to the high HCC incidence in Singapore.
Liang (1995) recently reviewed the relationship of p53 proteins
and AFB1. He pointed out firstly that the murine mutant p53 gene
p53Ser249 appears to have a hepatocyte-specific phenotype, which
suggests that this gene may interact with cellular factors(s) in a
liver-specific manner to alter the growth property of hepatocytes. It
is not known if the human form of p53Ser249 exhibits the same
properties. Secondly, cooperative interaction of this p53 mutation and
viral-induced cellular changes are probably involved in the
transformation of hepatocytes in situations where aflatoxin exposure
and hepatitis viral infection are evident. Recent studies of
non-aflatoxin-associated HCC showed that p53 mutations are not as
common as other human malignancies. This difference could be explained
by the relatively low proliferation rate of hepatocytes as compared
with other epithelial cells, such as colonic mucosa and mammary gland.
Because p53 plays a critical role in "damage control" of proliferating
cells and in regulation of abnormal proliferation, it is reasonable to
speculate that p53 mutations may play a lesser role in
hepatocarcinogenesis. However, dysregulated p53 function may still be
an important step in this process, in view of the recent observation
that HBX protein encoded by HBV appears to interact with p53 and
inhibit its function.
Thirdly, it has not been possible to induce the same p53 mutation
with aflatoxin exposure in a murine model, which casts a shadow of
doubt on the applicability of studies in the murine model to human
hepatocarcinogenesis. Liang (1995) recommended using human p53 genes
to perform parallel experiments.
Harris (1995) has also reviewed this subject. He reiterated that
in high-incidence liver cancer areas such as China and Mozambique, the
high frequency of G:C to T:A transversions in human hepatocellular
carcinomas in this region could be due to the high mutability of the
third base of codon 249 by AFB1 or a selective growth advantage of
hepatocyte clones carrying this specific p53 mutant in liver
chronically infected with HBV. The third base of codon 240 in a human
liver cell line exposed to AFB1 has been shown to be preferentially
mutated, and transfected 240Serp53 mutant enhances the growth rate of
the p53 null hepatocellular carcinoma cell line Hep3B.
The hypothesis that some of the mutations observed in the p53
tumour-suppressor gene may be specific markers of exposure to
aflatoxin may represent a real breakthrough in the field of liver
cancer epidemiology. In particular, the confirmation of the
specificity of the p53/aflatoxin association could be useful in
assessing and quantifying the responsibility of aflatoxin as an
independent cause of liver cancer and in evaluating the likely
interactions with the hepatitis viruses in humans. A word of caution
should be raised regarding the interpretation of the early studies
because of: 1) the small sample size and limited methodology as to the
criteria of specimen inclusion; 2) inadequate adjustment of the
correlations for exposures to other viral and non-viral risk factors
at the individual level; 3) limited information on the sensitivity and
specificity of the proposed genetic markers; in particular, some
animal data and cell system data are inconsistent in showing a
specific association between p53 codon 249 mutations and previous
exposure to aflatoxin; and 4) insufficient knowledge of the additional
genetic changes in p53 and other genes (i.e., N-ras, C-myc, c-fos,
alpha-TGF) associated with liver cancer development.
2.2.10.3 Epidemiology of primary liver cancer
(a) Descriptive epidemiology.
Liver cancer is a disease prevalent in some of the developing
parts of the world. It is frequent in China, South East Asia and
subsaharan Africa. In some of these regions, like the Qidong area in
Southern China, liver cancer is the major cause of death to cancer
among men. It is relatively common in Japan and in the countries in
the Mediterranean basin and it is rare in the Americas and Northern
Europe. Pockets of high risk populations have been described in the
Amazonian basin, among Eskimos and in special populations like the
renal transplant patients. The incidence of liver cancer is
consistently higher in men than in women with a sex ratio ranging from
2 to 3 in most countries. Within countries, further variation in
incidence rates is observed across cancer registries, men showing
greater variation than women. Worldwide, the incidence of liver cancer
in men and women shows a strong correlation.
Migration from high risk areas to lower risk areas tends to
reduce the risk to the levels of the host country, and this is
observable within first and second generations.
(b) Etiology
The etiology of primary liver cancer is nowadays largely
understood. Table 1 summarizes the range and the point estimates of
the attributable fractions in two different settings, the low-risk
areas in Europe and the USA and the high-risk areas in Africa and
Asia.
In both scenarios, viral infections to hepatitis B or C virus are
associated with liver cancer in a range from 65% to 100% of cases. In
low-risk countries HBV predominates and the other relevant factors are
alcohol, tobacco and oral contraceptives. In high-risk areas HBV
predominates and aflatoxins play a role, although quantification has
been difficult.
The evidence points to a synergistic interaction between HBV and
AF in the etiology of liver cancer and some debate exists as to the
independency of AF as an etiologic agent in humans.
It is noteworthy that the large majority of the available
epidemiological studies including data on aflatoxin exposure are based
on high-risk countries where both HBV and AF are highly prevalent.
Since the nature of the interaction at low levels of exposure is
unknown, extrapolation of results from available studies to other
settings is questionable.
In addition to these established factors, studies have identified
other factors that may modulate the incidence of the disease. Risk
factors identified are the use of contaminated drinking-water, liver
flukes and severe malnourishment. Protection from liver cancer has
Table 1. Causal factors of liver cancer and estimates of the attributable fractions
Factor Low-risk countries High-risk countries
Japan Europe and the USA Africa and Asia
Estimate Range Estimate Range Estimate Range
Hepatitis B <15% 4-50% 20% 18-44% 60% 40-90%
Hepatitis C3 60% 12-64% 50% 40-80% <10% NE
Aflatoxin limited exposure limited exposure important exposure1
Alcohol <15%4 <20% 11-30%5 NE
Tobacco <12%4 40% 38-51%5 NE
Oral contraceptive 10-50%2 NE NE
Other <5% <5%
1 Attributable risk not quantified. One study suggested attributable fraction close
to 50%.
2 Restricted to liver cancer in women. Likely to increase in future generations.
Uncertain if hepatitis infections (notably HCV) are necessary co-factors.
3 Not including double infections with HBV and HCV. Very few studies available using
second-generation assays.
4 Estimates for the USA
5 Estimates from three studies of LC in men
NE non evaluated.
Note: attributable fractions do not necessarily add to 100% due to multiple exposures and
possible interactions between risk factors.
Adapted from CDC, 1989; Bosch & Muñoz, 1991; Thomas, 1991; Tanaka et al., 1993; IARC,
1994; Bosch, 1995
been reported in the case of diets rich in retinol and protein.
Associations have been reported between liver cancer and blood
testosterone levels, HLA types, and predisposition due to
polimorphisms in some of the SGT and CYT metabolic regulatory genes.
(c) Vaccination against HBV as a preventive measure against liver
cancer
In 1983, the World Health Organization proposed as a medium-term
objective trials of immunization against hepatitis B to prevent liver
cancer. Since then more than 70 countries have introduced HBV
vaccination into their routine vaccination schemes. A recently
published study in Taiwan (Chang et al., 1997) has described the
rigorous application of universal immunization against hepatitis B and
the prevention of the carrier state in children; these data provide
further evidence of a direct causal relationship between HBV and liver
cancer.
The immunization programme against hepatitis B in Taiwan, an area
of hyperendemic infection and moderate to high aflatoxin exposure,
reduced the rate of HBV carriage in six-year-old children from about
10% in the period from 1981 to 1986 to between 0.9 and 0.8% in the
period from 1990 to 1994. The drop in the rate of carriage occurred as
the proportion of infants immunized against hepatitis B increased from
15% of children born to mothers at high risk during the earlier period
to 84-94% of all newborn infants during the later period. This
significant reduction in the prevalence of hepatitis B surface antigen
was accompanied by a decline in the average annual incidence of
hepatocellular carcinoma in children 6 to 14 years of age, from 0.7
per 100 000 between 1981 and 1986 to 0.57 between 1986 and 1989 and
0.36 between 1990 and 1994. The incidence of hepatocellular carcinoma
in children 6 to 19 years old fell even more dramatically, from 0.52
among those born between 1974 and 1984 to 0.13 among those born
between 1984 and 1986. As the investigators pointed out, since the
incidence of hepatocellular carcinoma in Taiwan peaks in the sixth
decade of life, it may take 40 years or longer to see an overall
decrease in the rate of hepatocellular carcinoma as a result of the
vaccination programme.
The Committee noted that studies like this one need to be observed
carefully in coming years for the light they may shed on the
relationship between aflatoxin, HBV and liver cancer.
(d) Effects of exposure to aflatoxins
Ahmed et al.(1995) undertook two prospective studies to
determine a possible relationship between perinatal aflatoxin exposure
and neonatal jaundice. First, cord blood samples from 37 neonates who
subsequently developed jaundice and from 40 non-jaundiced (control)
babies were analysed for six major aflatoxins and aflatoxicol.
Peripheral blood samples of both groups were also analysed postnatally
for aflatoxins. In a second study, serum aflatoxin levels of 64
jaundiced neonates admitted from outside the hospital were compared
with levels in 60 non-jaundiced control babies. Aflatoxins were
detected in 14 (38%) cord blood samples of jaundiced neonates and in
nine (23%) of the controls. The mean cord aflatoxin concentration was
highest in jaundiced neonates with septicaemia, but the difference was
not statistically significant. The frequency of detection of
aflatoxins in peripheral blood was not significantly different in
jaundiced and non-jaundiced babies. Aflatoxins were detected in the
blood of over 50% of neonates with jaundice of unknown etiology. There
was no correlation between severity of hyperbilirubinaemia and serum
aflatoxin levels. Further studies are needed to determine the extent
of pre-and postnatal exposure to aflatoxin in Nigerian infants and the
effects of such exposure on fetal and neonatal health, the authors
concluded.
In October 1988, 13 Chinese children died of acute hepatic
encephalopathy in the northwestern state of Perak in peninsular
Malaysia (Lye et al., 1995). Symptoms included vomiting,
haematemesis, seizures, diarrhoea, fever and abdominal pain. All had
liver dysfunction with increased aspartate aminotransferase and
alanine aminotransferase levels greater than 100 IU/litre. The
acuteness of the illness differed from previously reported outbreaks
described in Kenya, India and Thailand; median incubation period for
this outbreak was 8 hours, whereas the exposure was over a period of
days to weeks of consumption of highly contaminated food such as maize
in outbreaks in Kenya and India. Epidemiological investigations
determined that the children had eaten a Chinese noodle, loh see
fun, hours before they died. The attack rates among those who had
eaten the noodles were significantly higher than those who had not
(P < 0.0001). The cases were geographically scattered in six towns in
two districts along the route of distribution of the noodle supplied
by one factory in Kampar town. Aflatoxins were confirmed in the
postmortem samples from patients, but the noodles or their ingredients
were not analysed for aflatoxins. The authors questioned the etiology
of the outbreak.
Ibeh et al. (1994) examined the relationship between aflatoxin
levels in serum of infertile men in comparison with random controls
from the community. The subjects were 100 adult males, yielding 50
semen samples, from men attending infertility clinics at a university
teaching hospital and 50 normal men in the same community. The staple
foods of the men were assayed for aflatoxin content. Aflatoxin was
found in 20 semen samples from the infertile group (40 %) with a mean
concentration of 1.7 µg/ml and four samples from the fertile group
(8%) with a mean concentration of 1.0 µg/ml. Infertile men showed a
higher percentage of spermatozoa abnormality (50%) than the fertile
men (10-15 %).
In a parallel experiment, adult male rats were given an aflatoxin-
contaminated diet (8.5 µg purified AFB1/g of feed) for 14 days while
10 control age-matched rats were fed a normal aflatoxin-free diet
during the same period. Seven days after the withdrawal of aflatoxin
from the diet of test rats, two rats were randomly selected from the
test and control groups, killed and their semen harvested from the
epididymis and vasa deferentia and analysed. The process was repeated
at weekly intervals until four rats were left in the test and control
groups. Thereafter, four fertile adult female rats were introduced to
mate with test and control rats, and rats were observed for 90 days
with an adequate, non-aflatoxin-contaminated diet. Results
showed that rats exposed to dietary aflatoxin experienced changes in
spermatozoal profile which differed in a statistically significant
manner from the control rats; test rats showed depression in the
motility, viability and number of sperm cells which resembled features
seen in semen of infertile men exposed to aflatoxin. The four test
rats who were mated in the conclusion of the study were unable to
effect conception of fertile female rats, while the four control rats
were able to do so.
The authors hypothesized that aflatoxin may affect the
reproductive system by its toxic effect on the liver, leading to the
desquamation of the membranes of hepatocytes, the mitochondria, the
cytosol and the endoplasmic reticulum. This cellular damage could
include inhibition of enzyme synthesis and/or enzyme activities or
inhibition of lipid metabolism or fatty acid synthesis, which may
derail the capacity of the hepatocytes to handle the conversion of
intermediate biomolecules, such as precursor molecules for hormones,
e.g., testosterone and progesterone. Depression or absence of normal
hormone levels could cause a wide range of degenerative changes in
sexual organs. Aflatoxin may also affect the male reproductive system
by causing lysis of sperm cells as a result of constant reversible
reaction with the mycotoxin, binding of the toxin to free and/or bound
amino acids in the seminal fluid, depressing the motility of
spermatozoa and the formation of aflatoxin adducts with nucleic acids,
giving a risk of mutations of the spermatogonia.
El-Nazami et al. (1995) examined the exposure of infants to
aflatoxin M1 (AFM1) and of lactating mothers to AFB1, using AFM1 in
breast milk as a biomarker for exposure to AFB1. Prevalence of AFM1 in
breast milk samples from 73 women from Victoria, Australia
(low-exposure area) and 11 women from Thailand (high-exposure area)
was also compared. Assays were done by both HPLC and by ELISA. AFM1
was detected in 11 samples from Victoria and five samples from
Thailand at median concentrations of 0.071 mg/ml (range 0.028 - 1.031)
and 0.664 ng/ml (range 0.039 - 1.736). Levels of AFM1 were
significantly higher in milk samples from Thailand than in milk
samples from Victoria.
Ankrah et al. (1994) attempted to ascertain if the presumed
intake of dietary aflatoxins (AFB1 and AFG1) has adverse effect on the
liver; aflatoxins were measured in serum, urine and faecal specimens
obtained from a group of 40 apparently healthy adults (11 females and
29 males) from the Greater Accra region of Ghana. Liver status of the
subjects was monitored with serum alpha-fetoprotein (AFP),
alpha-1-antitrypsin (AAT) and direct: total bilirubin ratio. Aliquots
of serum were tested for HBsAg. AFG1, AFB1, AFQ1, and AFM1 were
detected in one or more of the body specimens in 35% of the subjects
(AFB1+ group). Sixty-five per cent of the subjects had only AFG1 in
their body specimens (AFB-group). Serum levels of AFP (greater than 20
ng/ml), AAT (greater than 170 ng/dl) and direct: total bilirubin ratio
(greater than 0.5), which indicated absence of predisposition to liver
cancer in all the subjects but were suggestive of liver inflammation,
were noted in both the AFB+ and AFB1-subjects. None of the subjects
had malaria or hepatitis B virus infection. The authors suggested that
the pattern of distribution of the aflatoxins in the subjects
indicates that the suspected liver inflammation may involve other
factors and may not be only due to present intake levels of
aflatoxins.
(e) Epidemiological studies on dietary aflatoxins and liver cancer
A number of important epidemiological studies have been published
since the Committee's last review of aflatoxins at its thirty-first
meeting (Annex 1, reference 77).
Yeh et al. (1989) examined the roles of the hepatitis B virus
and AFB1 in the development of primary hepatocellular carcinoma (PHC)
in a cohort of 7917 men aged 25 to 64 years old in southern Guangxi,
China, where the incidence of PHC is among the highest in the world.
After accumulating 30 188 person-years of observation, 149 deaths were
observed, 76 (51%) of which were due to PHC. Ninety-one per cent (69
of 76) were HBsAg+ at enrollment into the study in contrast to 23% of
all members of the cohort. Three of the four patients who died of
liver cirrhosis were also HBsAg+ at enrollment. There was no
association between HBsAg positivity and other causes of death. Within
the cohort, there was a 3.5-fold difference in PHC mortality by place
of residence.
To estimate AFB1 exposure, between 1978 and 1984, the Fusui Liver
Cancer Institute regularly sampled and tested staple foods consumed in
the counties of southern Guangxi for contamination by AFB1. Twice a
year, samples of raw foods were collected from all over the region and
analysed for AFB1 content by TLC. An estimated mean level was computed
for each commune as follows. The yearly amount consumed of a given raw
foodstuff was multiplied by the average AFB1 content as determined
from tested samples of raw foodstuffs. These cross-product terms were
then summed over all staple foods, and the resultant figure was
divided by the total population to obtain an estimated intake per
person per year. These population-based levels of AFB1 were correlated
with mortality rates of PHC among members of the cohort by the
communes from which the subjects were derived.
When estimated AFB1 levels in the subpopulations were plotted
against the corresponding mortality rates of PHC, a positive and
almost perfectly linear relationship was observed. On the other hand
the prevalence of HBsAg was very high and homogeneous across the study
areas (range 21.6%-24.7%) and therefore, no significant association
was observed when the prevalence of HBsAg positivity in the
subpopulations was compared with their corresponding rates of PHC
mortality. The authors conclude that despite the "crudeness" of their
exposure estimate, (i.e., population-based instead of personal
exposure assessments), it is reasonable to conclude that AFB1 seems to
play a role in the unusually high rates of PHC in southern Guangxi.
The population prevalence of HBsAg is extraordinarily high in this
study population, almost one in every four adult men being a
positive carrier of HBV. Primary infection occurs very early in this
high-risk population, possibly through vertical transmission from
carrier mothers to infants during the perinatal period, based on a
survey of serum HBsAg in children ages 1 to 9 years in a county
adjacent to Fusui. Even though most cases of liver cancer in this
study did not have histopathological confirmation, the authors
indicate that probably all were PHCs.
The Yeh et al. (1989) report is an early important study showing
that, in a region where HBV is highly prevalent and PLC is common, the
HBV carriers are at very high risk. It further indicates that in an
area of high AFB1 exposure, the PLC mortality rates are higher than in
areas of lower AFB1 exposure. This study provides the basic
information for most potency estimates. Most of the early correlation
studies (with or without HBV consideration) are consistent with the
basic conclusion of the study of Yeh et al. (1989), but other
studies are not (Campbell et al., 1990; Hsing et al., 1991).
However, the study has the general limitations of correlation studies
in which: i) exposure to AFB1 is estimated from raw foodstuffs
available to populations and attributed to individuals; ii) the
correlation between PLC and AFB1 was not adjusted for any of the
possible confounders such as HCV, alcohol, tobacco or nutritional
status as shown in Taiwan by Yu et al. (1995); iii) HBV exposure may
have been underestimated due to lack of use of PCR methodology; iv)
HBsAg prevalence was measured in a 25% sample of the cohort and
attributed to the region.
Campbell et al. (1990) conducted a comprehensive cross-sectional
survey in the People's Republic of China of possible risk factors for
primary liver cancer (PLC) to include 48 survey sites, an
approximately 600-fold aflatoxin exposure range, a 39-fold range of
HCC mortality rates, a 28-fold range of hepatitis B virus surface
antigen (HBsAg+) carrier prevalence, and estimation of exposures for a
large number of other nutritional, dietary and life-style features
(Campbell et al., 1990). PLC mortality was unrelated to aflatoxin
intake, but was positively correlated with HBsAg+ prevalence, plasma
cholesterol, frequency of liquor consumption, and mean daily intake of
cadmium from foods of plant origin. Multiple regression analysis for
various combinations of risk factors showed that aflatoxin exposure
consistently remained unassociated with PLC mortality regardless of
variable adjustment. In contrast, associations of PLC mortality with
HBsAg+, plasma cholesterol, and cadmium intake remained, regardless of
model specifications, while the association with liquor consumption
was markedly attenuated (nonsignificant) with adjustment for plasma
cholesterol.
The authors commented on the lack of an association between
aflatoxin exposure and PLC mortality in this study, in view of the
findings of most previous investigations. The absence of an
aflatoxin-PLC association was consistent with a similar lack of
association of PLC mortality with the consumption of the two foods
most commonly contaminated with aflatoxin i.e. maize and mouldy
groundnuts. In contrast to the lack of an association with aflatoxin,
PLC mortality was highly correlated with HBsAg+ prevalence and not
with past HBV infection, as assessed by the prevalence of antibody to
the HBV core protein. In this study, the association of plasma
cholesterol with PLC mortality was even more consistent than the
association of PLC mortality with HBsAg+ prevalence. Mortalities from
colon cancer, rectal cancer, lung cancer, leukemia, brain cancer and
total aggregate cancer are also known to be associated with plasma
cholesterol. This association was even more surprising in China where
plasma cholesterol in this cohort ranged up to about 190 mg/dl, which
is near the low end of the range for comparable Western subjects (Chen
et al., 1990).
The authors offered several explanations for the lack of an
association between aflatoxin intake and PLC mortality, which
contrasts with the finding of other previous studies. First, Chinese
people might respond differently to aflatoxin, perhaps because of
unique genetic or environmental characteristics. This is unlikely
given the previously shown positive association between aflatoxin
intake and PLC mortality in Chinese subjects (Yeh et al., 1985; Yeh
et al., 1989) and because major ethnic differences in risk for other
cancers are greatly reduced or eliminated after migration to new
environments.
A second argument could be that the lack of an effect in this
study may have been because measurement of aflatoxin exposure during
the survey period was not representative of past intakes when the
cancers were forming. However, a similar limitation existed for all
other Chinese studies; this study is more reliable, in the opinion of
the authors, because it is based on urinary aflatoxin metabolite
excretion which directly represents and integrates over a day or so
actual consumption. In addition, aflatoxin contamination rates in a
county in the Guangxi Autonomous Region were relatively stable during
the years 1972-1983.
A third line of reasoning suggests that aflatoxin may not be a
significant human carcinogen, in the opinion of the authors. The
present study has greater statistical power and more comprehensive
range, diversity and inclusiveness of risk factors than other previous
studies. Humans may also be resistant to aflatoxin carcinogenesis, a
finding which is supported by in vitro aflatoxin studies on species
of varying resistance (Booth et al., 1981). Humans may also be more
refractory when consuming lower protein diets; whereas acute toxicity
of aflatoxin is increased in protein-malnourished children (Hendrickse
et al., 1982).
The authors pointed out that data from animal studies have shown
that when animals were fed either lower levels of animal protein (5-
or the same level (20%) of plant protein after completion of aflatoxin
dosing, development or preneoplastic lesions and tumours was markedly
inhibited (Appleton & Campbell, 1983; Schulsinger et al., 1989).
Protein in the Chinese diet is primarily of plant or fish origin, as
compared to protein in the USA diet which is primarily of animal
origin (Food and Nutrition Board, 1989; Chen et al., 1990).
The authors continued with a critique of previous aflatoxin
epidemiology studies and offered the following model to explain the
etiology of PLC. The vast majority of individuals who are susceptible
to PLC are those who are persistently infected with HBV. Within this
HBsAg+ population, additional risk is contributed chiefly by
nutritional and dietary practices that enhance liver cell
proliferation, such as diets containing significant amounts of animal
protein. Aflatoxin may act as a carcinogenic initiator, but
contributes only a very small proportion of the initiating activity
routinely exposing the liver. Therefore, HBsAg+ is a necessary but
insufficient cause of PLC, aflatoxin is an unnecessary and
insufficient cause, and sustained nourishment causing liver cell
proliferation (and elevated plasma cholesterol) is a necessary and
insufficient cause for HBsAg negative carriers, but a necessary and
sufficient cause for HBsAg positive carriers. Why is PLC so much more
common in undernourished and impoverished societies? The authors
concluded that PLC is more common because HBsAg+ carriers are more
common.
In evaluating the significance of this study by Campbell et
al. (1990), a number of issues, both statistical and
non-statistical, should be considered. For example, PLC rates were
determined for the years 1973-1975 and the biochemical analyses
(covariate ascertainment) was conducted in 1983. With regard to the
statistical analysis presented in the paper, there is some indication
that the sample data do not adequately satisfy the normality
assumptions upon which the univariate correlation and multiple
regression analyses are based. Finally, the urinary aflatoxin
measurements were of total aflatoxin metabolites, which have been
shown not to correlate well with levels of AFB1 consumed (Wild
et al., 1992; Groopman et al., 1993).
(f) What can we learn from epidemiological studies that considered
HBV, HCV and AFB in relation to liver cancer?
Viral hepatitis is a major worldwide public health problem. It is
estimated that over 300 million individuals are chronically infected
with HBV and perhaps 100 million with HCV. Chronic infection with
either virus has been linked to cirrhosis and liver cancer. HBV is
prevalent in the developing parts of the world, and HCV is emerging as
a major cause of hepatocellular cancer in Japan and western societies
(Table 1).
Tests to detect HBV markers have increased in sensitivity, largely
due to the use of the polymerase chain reaction (PCR) to amplify HBV
DNA in serum and liver tissues. HBV infection has been shown to
persist in the serum (49.7%) or in the liver cancer tissue (24.9%) in
a number of patients with liver cancer that are at the same time HBSAg
negative (Bosch & Muñoz, 1991). This pattern has been documented in
cases from areas at low and high risk for HBV infection.
Although the significance of detecting low levels of HBV DNA in a
patient with liver cancer is not fully understood, from an
epidemiological viewpoint, these subjects could easily be classified
as persistently exposed to HBV and grouped with the HBsAg carriers in
computing risk estimates. Although data are sparse on the prevalence
of equivalent markers in the general population, it is likely that
among controls the prevalence of PCR-detected HBV DNA in the absence
of any other HBV marker is extremely low. If this is the case, the
case control studies that use PCR would increase the Risk Ratio
estimates for HBV as well as the estimates of the Attributable
Fraction. There are no case control studies that have used PCR methods
(in serum or liver tissue) to detect HBV exposure.
Ramesh & Panda (1993) have questioned the hypothesis that HBV
causes chronic liver disease and liver cell carcinoma in
HBsAg-positive individuals only. The presence of HBV in patients with
HCC who are seropositive for the envelope antigen (HBsAg) is well
established. Epidemiological studies have shown a small percentage of
patients with HCC with past HBV infection, positive for anti-HBsAb or
HBcAb. However, the role of HBV in HCC cases who seroconverted from
HBsAg to HBsAb is unclear. The authors described a study of 36 HCC
cases where four cases negative for HBsAg and with underlying
cirrhosis were found. Biopsy tissue was investigated by polymerase
chain reaction; all four samples tested were positive for a portion of
the surface region (nucleotide position: 636-735), but were negative
for the "X" and the "C" regions of HBV genome. Since hepatitis C virus
(HCV) has been associated with HCC, the authors tested serum samples
of the four cases for anti-HCV; one out of four was positive for
anti-HCV. The authors concluded that these observations indicate that
parts of the HBV genome can persist in liver cells of individuals who
have recovered from clinical illness and seroconverted to HBsAg
positive. However, the significance of these sub-genomic fragments in
the development of HCC is not clear.
The identification of HCV in the last decade has been a major step
forward in the understanding of the origins of liver cancer and in the
quantification of the proportion of cases related directly to viral
infections (IARC, 1994). Epidemiological studies are largely
consistent in showing a strong association between carriers of
anti-HCV and liver cancer. The specific potential to induce PLC by
each of the HVC types and variants of types as well as the impact of
other factors from the host and the environment still requires further
research. Likewise, few studies are available exploring the role of
aflatoxin in the presence of HBV and HCV. High estimates of the
Relative Risk for carriers of anti-HCV have also been reported in
areas of HBV endemicity. The risk linked to HCV is independent of HBV
and persons who are carriers of both HBsAg and anti-HCV are at a very
high risk of developing liver cancer. HCV is likely to be the major
cause of liver cancer in countries at low/intermediate risk like the
USA and Europe.
(g) Epidemiological studies including aflatoxins in countries where
the risk of liver cancer is low
In countries where liver cancer is rare and aflatoxin exposure is
low, most etiological studies on liver cancer have not considered
aflatoxins as a risk factor. The populations with higher exposures are
the workers occupationally exposed to grain dust in the animal feed
processing plants. In studies conducted in the Nordic countries in
Europe, Sweden, Denmark, the Netherlands and in the USA, aflatoxins
have been isolated from dust samples and an excess of mortality of
several such cohorts has been documented for liver cancer (risk, 2.4
times the expected rates) liver and biliary tract cancer (risk, 2.5
times the expected rates), lung cancer and lymphomas (risk, 1.5-3
times the expected rates (Hayes et al., 1984; Alavanja et al.,
1987; Olsen et al., 1988). It should be noted that some of these
studies did not evaluate other relevant exposures such as hepatitis
infections and alcohol.
It is of interest that few studies are available on liver cancer
in Latin America. In this extensive region of the world, agricultural
products are prone to mould growth, and consumption of maize is part
of the staple food in many countries. Yet liver cancer is rare in
these populations as is HBV infection. If that is the case, Latin
America would be potentially a very informative field to investigate
the occurrence of liver cancer in populations exposed to AF as the
central risk factor.
(h) Epidemiological studies that used biomarkers of exposure to
aflatoxins including studies on genetic susceptibility to aflatoxins
Biomarkers have been developed and are being introduced in
epidemiological studies with the purpose of increasing the accuracy of
the assessment of exposure to aflatoxins. Various biomarkers have been
developed, including urinary total aflatoxins, aflatoxin adducts in
urine, aflatoxin albumin adduct in serum, aflatoxin adducts in liver
cancer tissue and more recently p53 specific mutations in liver cancer
specimens. Other studies are investigating genetic polymorphisms in
some key genes involved in the metabolism of aflatoxin that may
introduce some variability in the response to aflatoxin. (Groopman
et al., 1994; Wild et al., 1996; IARC, 1997).
The importance of the major aflatoxin-nucleic acid adduct,
AFB-N7-guanine, in urine as a biomarker was enhanced by the finding
that this metabolite is excreted exclusively in urine of exposed rats,
thus simplifying pharmacokinetic considerations. The aflatoxin-albumin
adduct in serum has also been examined as a biomarker of exposure;
because of the longer half-life in vivo of albumin compared to the
urinary AFB-N7-guanine, the serum albumin adduct can integrate
exposures over longer time periods. Data from human exposure studies
have shown that the excretion of the urinary aflatoxin nucleic acid
adduct and formation of the serum albumin adduct are highly
correlated. In the rat, validation studies for the dose-dependent
excretion of urinary aflatoxin biomarkers were conducted in rats
following a single exposure to AFB1; excellent linear correspondence
between oral AFB1 dose and excretion of AFB-N7-guanine in urine was
shown (Scholl et al., 1995).
Aflatoxin metabolites in urine or adducts in serum can be a useful
tool to evaluate exposure but with the currently available methods
remain relatively short-term exposure markers; biomarkers are of much
less use in predicting long-term or lifetime human exposure. As such,
they reflect poorly the natural pattern of exposure to aflatoxin
(i.e., seasonality, manual sorting of foodstuffs, age at exposure,
etc.); therefore, it is not surprising that studies conducted using
aflatoxin biomarkers as markers of exposure show conflicting results.
At present, it is not fully understood how the functional status
of the liver or the coexistence of other risk factors for liver cancer
may affect the different biomarkers that are being proposed for
epidemiological studies. Few studies have described the natural
history of these markers in patients with chronic liver disease
including chronic hepatitis and liver cirrhosis, conditions that
usually precede liver cancer by months or years (Wild et al., 1993;
Wang et al., 1996a). In this circumstance, the interpretation of the
findings is complicated since the aflatoxin biomarker may be
confounded by the presence of some risk factors, (e.g., HBV, HCV,
alcohol), the presence of some protective factors (e.g., retinol in
the diet) or by the presence of liver disease (i.e. chronic active
hepatitis B infection).
The mutations in the p53 gene claimed to be specific markers of
exposure to aflatoxin are being actively investigated to confirm the
strength and the specificity of the association in human populations
(Harris, 1995). Recent data have been summarized earlier in this
paper.
The best evidence of an interaction between HBV and aflatoxin in
the causation of human liver cancer is the cohort study in Shanghai
(Ross et al., 1992, Qian et al., 1994; Yuan et al., 1995). This
is an ongoing prospective study of 18 244 middle-aged men in Shanghai,
China. Assays for urinary AFB1, its metabolites AFP1 and AFM1 and DNA
adducts have been performed to assess the relationship between
aflatoxin exposure and liver cancer. After 35 299 person-years of
follow-up, 22 cases of liver cancer had been identified. For each
case, 5 or 10 controls were randomly selected from cohort members
without liver cancer on the date the disorder was diagnosed in the
case and matched to within 1 year of age, within 1 month for sample
collection, and for neighbourhood of residence. Each subject provided
a blood sample and a urine sample. A positive result was defined as
the presence of at least 1 ng of an individual aflatoxin compound in
the sample. Hepatitis B surface antigen was measured by a standard
radioimmunoassay method.
Subjects with liver cancer were significantly more likely than
were controls to have detectable concentrations of any of the
aflatoxin compounds; the strongest association was for AFP1.
Positivity for HBsAg was strongly associated with risk of liver
cancer. The authors concluded that their results are based on too few
cases to give a reliable estimate of attributable risk, but they
estimated that up to 50% of cases of liver cancer in Shanghai may be
due to aflatoxin exposure.
In further follow-up of the Shanghai study, Qian et al. (1994)
reported on 70 000 person-years of follow-up and 55 cases of HCC.
Levels of urinary AFB1 and the oxidative metabolites, including the
major aflatoxin nucleic acid adduct, aflatoxin-N7-guanine, were
determined for 50 of the 55 identified cases of HCC; 267 controls were
matched against the 50 cases as above. A nested case-control analysis
showed highly significant association between the presence of at least
one of the four urinary aflatoxin metabolites, serum HBsAg positivity
and HCC risk. Risk was especially elevated in individuals who were
positive for both of these biomarkers. However, the number of liver
cancer cases in which the interaction was explored is small (i.e., 13
cases of AFB1 positive and HBsAg negative), and there is room for
misclassification of cases in relation to their viral exposure. Thus
risk estimates become unstable and the barely significant increase in
the ORs for the AFB1 exposed may be easily lost if only 2/3 of cases
now considered HBV negative turn out to be HBV (or HCV) positive. On
the other hand, a cohort analysis using all 55 cases of HCC revealed
no statistically significant association between HCC risk and dietary
aflatoxin consumption, as determined from the in-person food frequency
interview combined with the survey of market foods in the study
region, adding additional uncertainty of the value of the biomarkers
used. HCV prevalence was low in this cohort (Yuan et al., 1995).
An exchange of correspondence on this Shanghai study has occurred.
Campbell (1994) has pointed out that the HCC risk putatively
attributed to aflatoxin in the study of Ross et al. (1992) appears
to be accounted for mostly by the urinary AFB-N7-guanine adduct.
Instead Campbell postulates that this risk could also be caused by
factors that enhance enzymatic activation of AFB1 by the hepatic P450
enzyme system to produce more AFB-N7-guanine. These enzyme-inducing
factors could readily be nutritional, especially those that also are
associated with elevated plasma cholesterol. This interpretation would
be in accord with the ecological study by Youngman et al. (1992) in
48 survey counties in China showing that the most significant and
robust determinants of HCC risk were elevated cholesterol levels and
HBsAg positivity, not aflatoxin. This finding is given further
plausibility by animal experiments (Preston et al., 1976; Hu
et al., 1994). These experiments have shown that in vivo
activation of AFB1 to form hepatic DNA adducts could be markedly
enhanced by a modest elevation in the intake of animal protein. This
same modest animal protein intake that markedly elevates AFB1
activation also markedly increased the over-expression of a hepatitis
B virus transcript in mice.
Ross et al. have responded to the comments of Campbell (1994).
There is an overwhelming amount of experimental data across species
and in experimental models demonstrating the potency of AFB1 as a
carcinogen and mutagen (IARC, 1993). There is also evidence that
humans have the metabolic capacity to activate AFB1 to the same
DNA-damaging products that occur in animal models. A well-established
major risk factor for liver cancer is hepatitis B virus; however,
there is at least a 5-8 fold variation in liver cancer incidence
across regions of the world where the prevalence of hepatitis B viral
markers is comparable. Ross et al. emphasize that their Shanghai
study using aflatoxin-specific biomarkers and HBV markers has provided
the first direct evidence in human studies that aflatoxins are major
risk factors for HCC and that a synergistic interaction between HBV
and aflatoxin exposure occurs. Ross et al. criticize the Chinese
study of Campbell (1994) as an ecological study, "well known to be
highly limited in their ability to address cause and effect
relationships". In addition, a questionnaire administered to the
Shanghai subjects showed no difference in daily intake of animal
protein between liver cancer cases and their matched controls, or
between Hbsag positive cases and controls.
Groopman et al. (1993) stated that, based on urinary measures of
AFB-N7-guanine and dose-response characteristics of people living in
China and The Gambia, that 1) levels of daily urinary excretion of
total aflatoxin metabolites are unrelated to risk of aflatoxin-induced
disease; 2) the AFB-N7-guanine adduct in urine is a good,
non-invasive, short-term biomarker for determining both aflatoxin
exposure and risk of genetic damage in target organs.
However, the Shanghai study is clearly limited for purposes of
quantitative risk assessment of the risk to humans of aflatoxin
exposure. By the authors' own admission (Qian et al., 1994), no
dose-dependent association between the dietary aflatoxin index and
either liver cancer risk or biomarker status was found. This is due at
least in part to the facts that urinary levels of aflatoxin accurately
reflect intake levels of the past 24 hours and dietary assessment is
inadequate to reflect lifetime aflatoxin exposure. An unexplained
observation was the rather marked decline in the prevalence of
unmetabolized AFB1 with longer follow-up. Urinary adducts were
measured with inadequate precision; a patient was scored "positive" or
"negative" if an adduct was detected. Levels of adducts were not
considered, nor was the fact that one measurement represents one
"snapshot" out of a lifetime. One might question whether or not the
increased excretion of aflatoxin-DNA adducts represents the activity
of a diseased liver rather than a causal relationship. Exposure to
AFB1 may be expected to fluctuate greatly on a day-to-day basis (as a
result of varying behaviour and AFB1 concentrations); exposure for
each individual was evaluated at a single time point. The authors are
correct in their call for pharmacokinetic investigations of ingested
aflatoxins in humans. Until this work is done, AFB adducts cannot be
considered to be a true indicator of aflatoxin exposure, especially
over the probable lengthy timeframe required for human cancer
induction.
This study strongly suggests that aflatoxin exposure in the
presence of a persistent HBV infection increases the risk of liver
cancer. It is less convincing support for the conclusion that AFB1 is
capable of independently inducing liver cancer and provides limited
quantitative data on aflatoxin's relationship to liver cancer.
Follow-up of this cohort is awaited with great interest.
In a study by Wild et al. (1993), blood samples were collected
over a one-month period from 117 children aged 3 to 4 years residing
in Kuntair or Kerr Cherno in the Upper Niumi District of The Gambia.
Samples were analysed for aflatoxin-albumin (AF-alb) adducts, markers
of HBV infection, liver enzymes (serum alanine aminotransferase (ALT))
as markers of liver damage, and glutathione-S-transferase (GST) M1
genotype. All but two children showed detectable serum AF-alb with
levels ranging from 2.2 to 250 pg AFB1-lysine equivalent/mg albumin.
There was a statistically significant positive correlation between
AF-alb and ALT. HBV carriers showed moderately higher levels of AF-alb
than non-carriers, but the difference was not statistically
significant and the association between AF-alb and ALT was unchanged
when the HBV carriers were excluded from the analysis, suggesting that
factors other than HBV infection contributed to the association. The
null GSTM1 genotype was infrequent in this population and was not
associated with any difference in AF-alb adduct levels compared to
GSTM1-positive subjects. However, the percentage of individuals with
the null genotype varied significantly between ethnic groups. The
association between AF-alb and ALT could be a result of the
hepatotoxicity of aflatoxin, but the data are also consistent with the
hypothesis that liver damage resulting from HBV and/other factors can
alter aflatoxin metabolism resulting in an increased binding to
cellular macromolecules including DNA. The authors recommended more
study of this hypothesis.
Srivatanakul et al. (1991) conducted a case control study on
hepatocellular carcinoma in Thailand using the aflatoxin-albumin
adduct as a marker of recent exposure to aflatoxin. HBV exposure and
anti-HCV were assessed using standard methods. HBV was the predominant
risk factor; neither aflatoxin-albumin nor HCV were associated with
liver cancer. The Committee concluded that the study lacked sufficient
statistical power to detect aflatoxin or HCV as independent risk
factors. In addition, aflatoxin-albumin samples were taken from liver
cancer cases; levels or kinds of aflatoxin metabolites might have been
affected by illness.
Hatch et al. (1993) conducted a survey in eight areas in Taiwan
with a gradient in the estimates of exposure to aflatoxin and in the
incidence of PLC. Exposure to aflatoxin was assessed using urinary
tests, and a regression model was used to predict aflatoxin urinary
metabolites using mortality due to PLC as a predictor (as well as five
other variables). The conclusion of the study was that aflatoxin
played an independent role in PLC in Taiwan.
Monoclonal antibodies recognizing the stable imidazole ring-opened
form of the major N7-guanine aflatoxin B1-DNA adduct have been used in
competitive enzyme-liked immunosorbent assays (ELISA) and indirect
immunofluorescence assays to quantify adduct levels in liver tissue.
Santella et al. (1993) developed methods in AFB1-treated animals,
then applied these to paired tumour and non-tumour liver tissues of
hepatocellular carcinoma patients from Taiwan. An avidin-biotin
complex staining method was also used for the detection of HBsAg and
HBxAg antigens in liver sections. In all, 8 (30%) HCC samples and 7
(26%) adjacent non-tumour liver tissue samples from Taiwan were
positive for AFB1-DNA adducts. For HBsAg, 10 (37%) HCC samples and 22
(81%) adjacent non-tumorous liver samples were positive, and 9 (33%)
HCC samples and 11 (41%) adjacent non-tumour liver samples were HBsAg
positive. No association with AFB1-DNA adducts was observed for HBsAg
and HBxAg. The authors concluded that these results were compatible
with the conclusion that HBV and AFB1 do not act synergistically in
the genesis of HCC, but called for further investigation to define the
relationship between HBV and AFB1.
Wang et al. (1996a) conducted studies in Qidong, China, where
liver cancer accounts for 10% of all adult deaths and both HBV and
AFB1 exposures are common. Serum samples were collected during a
longitudinal study designed to measure aflatoxin molecular biomarkers
in residents of Daxin Township, Qidong City, China. In this study, the
temporal modulation of aflatoxin adduct formation with albumin over
multiple lifetimes of serum albumin was examined in both HBV-positive
and HBV-negative people in two periods: September-December 1993 (wave
1) and June-September 1994 (wave 2). During the 12-week monitoring
period of wave 1, 120 persons (balanced by gender and HBV status)
provided a total of 792 blood samples. AFB1-albumin adducts were
detected in all but one of the serum samples. During wave 2, 103
individuals from wave 1 provided 396 blood samples collected monthly
over wave 2. Using linear regression models, the mean
aflatoxin-albumin adduct levels increased during the 12 weeks of wave
1 and decreased over the 4 months of wave 2.
Neither HBV status nor gender modified either the baseline mean or
the temporal trend. High-performance liquid chromatography
confirmation was done on a subset of serum samples, and the results
showed an excellent association between the immunoassay data and
high-performance liquid chromatography. The investigators concluded
that AFB1-albumin is a sensitive and specific biomarker for assessing
exposure to AFB1 in the Qidong population.
The authors noted that aflatoxin-albumin binding is a longer term
biomarker of aflatoxin exposure than any of the urinary markers; the
rate of turnover of these adducts is similar to that of the blood
protein. The half-life of albumin in normal people is about 14-20
days, but there is some information to indicate that people with
serious liver disease have a much more variable turnover time. In this
study, an 8-fold range of adduct formation existed among individuals
within a cycle. Such factors as liver disease may account for the lack
of tracking shown in this study between HBV status and adducts. The
authors stressed the need to follow-up on this investigation with
studies that have more frequent and longer sampling intervals for
albumin adducts.
The authors discussed the data supporting the hypothesis that HBV
enhances aflatoxin metabolism to genotoxic derivatives. This study did
not seem to support this hypothesis nor did previous results in adult
populations in West Africa and Taiwan. HBV may affect the metabolism
of aflatoxin in children at a time when hepatocytes are maximally
dividing; more data are needed in this regard.
Wang et al. (1996b) investigated the carcinogenic effect of
aflatoxin exposure in Taiwan. Fifty-six cases of HCC diagnosed between
1991 and 1995 were identified and individually matched by age, sex,
residence and date of recruitment to 220 healthy controls from the
same large cohort in Taiwan. Blood samples were analysed for hepatitis
B and C virus markers and for aflatoxin-albumin adducts. Urine was
tested for aflatoxin metabolites. Information was obtained about
sociodemographic characteristics, habitual alcohol drinking, cigarette
smoking and diet in a structured interview.
HBsAg carriers had a significantly increased risk for HCC. After
adjustment for HBsAg serostatus, the matched odds ratio (ORm) was
significantly elevated for subjects with high levels of urinary
aflatoxin metabolites. When stratified into tertiles, a dose-response
relationship with HCC was observed. The ORm for detectable
aflatoxin-albumin adducts was not significant after adjustment for
HBsAg serostatus. HBsAg-seropositive subjects with high aflatoxin
exposure had a higher risk than subjects with high aflatoxin exposure
only or HBsAg seropositivity only. The OR for developing HCC was found
to increase in the presence of anti-HCV alone, HBsAg alone and both
anti-HCV and HBsAg. There was a poor correlation between
aflatoxin-albumin adducts and urinary metabolites in the same
controls, although both were related to HCC risk. The investigators
suggested that environmental aflatoxin exposure may enhance the
hepatic carcinogenic potential of hepatitis B virus, expressed concern
about the small sample size in their study, and urged the mounting of
a large-scale study to evaluate the effect of aflatoxin exposure on
HBsAg non-carriers.
Olubuyide et al. (1993) screened for the presence of HBsAg and
aflatoxin in the sera of 100 non-hospitalized individuals from the
rural population of Igobo-Ora and 89 non-hospitalized individuals from
the urban population of Ibadan, Nigeria. Controls were 31 healthy
British Caucasians who had not travelled to the tropics or subtropics
in the six months before the venipuncture. Forty-nine per cent of
rural subjects and 47% of urban subjects were consistently and
reproducibly seropositive for HBsAg (as determined by the ELISA test).
Two of the former subjects and five of the latter were positive for
both HBV DNA (as measured by spot hybridization) and HBsAg. Total
aflatoxin levels were less than 17 pg/ml in the British controls;
serum levels of aflatoxins greater than this were detected in 8% of
rural subjects and in 9% of urban subjects. The types and amounts of
aflatoxins and the amounts of aflatoxins found were so widely
dispersed that it was not possible to draw any conclusions about
differences in types and amounts of aflatoxins between rural and urban
populations. The authors intend to follow their subjects to determine
their propensity to develop HCC.
Groopman et al. (1994) have reviewed the subject of molecular
biomarkers for aflatoxins and their application to human cancer
prevention. Cancer prevention trials that use biological markers as
intermediate end-points provide the ability to assess the efficacy of
promising chemoprotective agents in an efficient manner by reducing
sample size requirements, as well as reducing the time required to
conduct the studies, compared to trials that have cancer incidence or
mortality as end-points. The key issue in trials that use biomarkers
as the outcome of interest is to use a marker that is directly
associated with the evolution or development of neoplasia. The authors
briefly discussed the possible impact of a short-term intervention
with oltipraz (a substituted dithioethione, which is a potent
inhibitor of AFB1-induced tumorigenesis and carcinogenesis in rats) on
levels of two aflatoxin biomarkers in individuals exposed to
aflatoxin-contaminated foods.
(i) Oltipraz chemoprevention trial
In 1995, 234 adults from Qidong, Jiangsu Province, China, where
hepato-cellular carcinoma is the leading cause of cancer death and
exposure to dietary aflatoxins is widespread, were enrolled and
followed in a Phase II chemo-prevention trial (Jacobson et al.,
1997). The goals of the study were to define a dose and schedule of
oltipraz for reducing levels of validated aflatoxin biomarkers and to
characterize dose-limiting toxicities. Healthy eligible individuals,
including those infected with hepatitis B virus, were randomized to
receive either 125 mg of oltipraz daily, 500 mg of oltipraz weekly, or
placebo. Blood and urine specimens were collected to monitor
toxicities and evaluate biomarkers over the 8-week intervention period
and subsequent 8-week follow-up period. The authors reported excellent
compliance (>70%); 21% of subjects reported clinical adverse events.
The oltipraz arms did not differ in symptom type or severity, most
commonly an extremity syndrome. There were no indications of
exacerbated drug intolerance among the few participants infected with
hepatitis B virus. The authors concluded that chemoprevention trials
with biomarker end-points are feasible in such populations.
(j) Genetic susceptibility
AFB1 is metabolized via the phase I and II detoxification pathway;
hence, genetic variation at those loci may predict susceptibility to
the effects of AFB1. To test this hypothesis, McGlynn et al. (1995)
contrasted genetic variation in two AFB1 detoxification genes, epoxide
hydrolase (EPHX) and GSTM1 with the presence of serum AFB1-albumin
adducts, the presence of hepatocellular carcinoma (HCC), and with p53
codon 249 mutations. Subjects were 40 unrelated Ghanaian males
(healthy gold miners employed by the Ashanti Goldfields Corp. in
Obuasi, Ghana) and 52 patients with HCC and 116 healthy controls from
the Zhong Shan Hospital in Shanghai, China.
Mutant alleles at both loci were significantly over represented in
individuals with serum AFB1-albumin adducts in a cross-sectional
study. Mutant alleles of EPHX were significantly over-represented in
subjects with HCC and also in a case-control study. The relationship
of EPHX to HCC varied by hepatitis B surface antigen status and
indicated that a synergistic effect may exist. p53 codon 249 mutations
were observed only among HCC patients with one or both high risk
genotypes. These results indicate that individuals with mutant
genotypes at EPHX and GSTM1 may be at greater risk of developing
AFB1-adducts, p53 mutations and HCC when exposed to AFB1. Hepatitis B
carriers with the high-risk genotypes may be an even greater risk than
carriers with low-risk genotypes. The authors concluded that these
findings support the existence of genetic susceptibility in humans to
the environmental carcinogen AFB1 and indicate that there is a
synergistic increase in risk of HCC with the combination of hepatitis
B virus infection and susceptible genotype. However, it has been
pointed out that the control and experimental samples came from
different populations, which may weaken the case for genetic
susceptibility (J. Groopman, personal communication).
The Committee concluded that the currently available studies
utilizing aflatoxin biomarkers do not provide a reliable quantitative
measure of aflatoxin exposure in humans, especially over the
long-term. Biomarker levels in relation to cancer risk in the Wang
et al. and Qian et al. studies have been used for modelling data;
however, the interpretation is limited.
At present, it seems reasonable to subscribe to the 1993
conclusion of the IARC in qualitative terms that AFB1 is carcinogenic
(group 1), and as such to recommend reducing exposure of human
populations as much as possible. There is still some uncertainty
concerning the independent status of aflatoxin as a human carcinogen
and concerning the relationship between aflatoxin dose and liver
cancer incidence.
(k) Conclusions from epidemiology studies
The potential carcinogenicity in humans of the aflatoxins (either
total or AFB1) has been examined in a large number of epidemiology
studies, generally carried out in Africa and Asia, where substantial
quantities of aflatoxin occur in basic foodstuffs. Exposure to
aflatoxins appears to present an additional risk which is enhanced by
simultaneous exposure to hepatitis B virus, and possibly hepatitis C
virus. This relationship, which may affect not only carcinogenic
potency but also the metabolism, biochemistry and pharmacology of the
aflatoxins, and other multiple etiological agents for primary liver
cancer makes it difficult to interpret the epidemiological studies in
the context of the risk of primary liver cancer from aflatoxins.
Perhaps, further development of biochemical and pharmacological
markers will help to clarify exposure, although these can cause other
problems.
Further clarification of the relative roles of hepatitis and
aflatoxin in liver cancer awaits studies that comply with the
following requirements: 1) the studies should be cohort studies with
long-term follow-up; 2) the studies should be conducted in countries
with variability in the exposure to aflatoxins; 3) the studies should
provide for storage and analyses of biological specimens from repeated
sampling, preferably with concurrent sampling of aflatoxin in the
diet; 4) the studies should provide evidence that the aflatoxin
biomarker used is not affected by the presence of chronic liver
disease; this will be difficult to achieve; the different measures of
aflatoxin exposure, i.e., biomarker vs. dietary analysis, should
correlate with liver cancer; 5) a large number of liver cancer cases
should be included, preferably confirmed by biopsy; 6) liver cancer
cases, if shown to contain the p53 specific aflatoxin mutation, would
strengthen the case.
The Committee identified some on-going studies that comply with
some of these requirements and may produce relevant results in the
near future.
i) A study in Qidong, China, screened 45 000 males (ages 30-59) of
which 20% were HBsAg positive. Questionnaires, urine and serum were
collected at different intervals; 260 cases of liver cancer have been
identified although the number of biopsies is small. Laboratory tests
and statistical analyses are required; funds are needed.
ii) A cohort study in Thailand collected questionnaires, blood and
urine specimens at different intervals in a cohort of HBsAg carriers
who were regularly screened for AFP, ALT and ultrasound. Field work is
completed at this point and lab results are pending.
iii) The Shanghai study described in the text may be of value if
sampling and follow-up continue.
iv) Finally, the on-going HBV vaccination trials and campaigns in
China, Taiwan and The Gambia may provide evidence in the future for
the occurrence of AFB1-induced liver cancer cases in individuals
vaccinated against HBV.
2.2.11 Summary of information on other aflatoxins
2.2.11.1 Aflatoxin B2
Aflatoxin B2 (AFB2) has not been studied extensively, and most
data are derived from single reports. AFB2 becomes bound to DNA of
rats treated in vivo, after its metabolic conversion to AFB1. In
rodent cells, AFB2 induced DNA damage, sister chromatid exchange and
cell transformation, but not gene mutation. AFB2 produces gene
mutation in bacteria. IARC concluded in 1993 that there is limited
evidence for carcinogenicity of AFB2 in experimental animals.
No additional toxicological information on AFB2 has appeared in
the literature since IARC (1993).
2.2.11.2 Aflatoxin G1
Aflatoxin G1 (AFG1) binds to DNA and produces chromosomal
aberrations in rodents treated in vivo. In cultured human and animal
cells, it induces DNA damage, and also induces chromosomal anomalies
in single studies. AFG1 induces gene mutation in bacteria. IARC
concluded in 1993 that there was sufficient evidence in experimental
animals for the carcinogenicity of AFG1.
No additional toxicological information on AFG1 has appeared in
the literature since IARC (1993).
2.2.11.3 Aflatoxin G2
Aflatoxin G2 (AFG2) has been the subject of very little research.
IARC concluded in 1993 that there was inadequate evidence for the
carcinogenicity of AFG2.
No additional toxicological information on AFG1 has appeared in
the literature since IARC (1993).
2.2.11.4 Aflatoxin M1
Aflatoxin M1 (AFM1) is a metabolic hydroxylation product of AFB1,
and can occur in the absence of the other aflatoxins. Human exposure
occurs primarily via milk and milk products from animals that have
consumed contaminated feed. IARC concluded in 1993 that there was
sufficient evidence in experimental animals for the carcinogenicity of
AFM1 and inadequate evidence for the carcinogenicity of AFM1 in
humans. Although AFM1 has been tested less extensively, it appears to
be toxicologically similar to AFB1. AFM1 is considered to be a
genotoxic agent, based on its activity in vitro and its structural
similarity with AFB1. It is a less potent liver carcinogen, with a
probable carcinogenic potency in laboratory animals within a factor of
10 of AFB1 (Cullen et al., 1987).
No additional toxicological information on AFM1 has appeared in
the literature since IARC (1993).
3. ESTIMATING CARCINOGENIC RISKS FROM THE INTAKE OF AFLATOXINS
Risks from specific exposures to aflatoxins are difficult to
estimate and predict, despite extensive information available from
epidemiological studies, mutagenicity tests, animal bioassays,
in vitro and in vivo metabolic studies, and p53 mutation studies.
Many questions remain regarding the independence of aflatoxin as a
human carcinogen, the extent to which hepatitis B, hepatitis C and
other factors modify the effect of aflatoxin, how findings from
countries with high liver cancer rates and high prevalence of
hepatitis B may be compared to those from countries with low rates,
how to deal with the wide range of susceptibility to aflatoxin
carcinogenesis among experimental animals, and how to describe the
dose-response curve over the wide range of aflatoxin exposure found
worldwide.
3.1 Information from various scientific disciplines and its
contribution to aflatoxin carcinogenic risk
3.1.1 Laboratory animal, mutagenicity and metabolic studies
The liver is the primary target organ in most species, but tumours
of other organs have also been observed in aflatoxin-treated animals.
The effective dose of AFB1 for induction of liver tumours varied over
a wide range in different animals species when the carcinogen was
administered by continuous feeding, generally for the lifetime of the
animal. Effective doses were 10-30 µg/kg in the diet in fish and
birds. Rats responded according to strain at levels of 15-1000 µg/kg,
while some strains of mice showed no response at doses up to 150 000
µg/kg. Tree shrews responded to 2000 µg/kg. In subhuman primate
species, AFB1 potency in induction of liver tumours differed widely,
squirrel monkeys developing liver tumours when fed AFB1 at 2000 µg/kg
for 13 months, and rhesus, African green and cynomolgus monkeys
developing a low (7-20%) incidence of liver tumours when fed average
doses of 99-1225 mg/animal over 28-179 months (Wogan, 1992).
The aflatoxins are among the most potent mutagenic and
carcinogenic substances known. Much of the information available
regarding mutagenesis has been performed in bacterial systems, but
also to a lesser extent in eukaryotes. Aflatoxin falls in the category
of bulky mutagens, including the polycyclic aromatic hydrocarbons and
the aromatic amines. A large body of literature suggests that a
chemical causes a cell to become tumorigenic by reacting readily with
DNA to give DNA adducts and these adducts or their breakdown products
must then cause mutations efficiently (Loechler, 1994).
Much of the recent aflatoxin metabolic data has been discussed in
more detail in section 2.1.3. In brief, it has been demonstrated that
many isoforms of P450 are able to biotransform AFB1 to
DNA-binding/mutagenic species. Differences in P450 isoform activities,
due either to genetic polymorphisms or to environmental alteration in
expression, may be important contributors to human susceptibility to
AFB1 (Massey et al., 1995). For example, there is some evidence that
AFB1 is strongly metabolized to DNA-binding species in areas of
damaged liver or in individual cells where CYP2A5 activity is high
(Camus-Randon et al., 1996). It is well known that a host of other
risk factors affecting metabolism exist, including infection with
hepatitis B and C, parasites such as liver flukes, alcohol
consumption, cigarette smoking, long-term use of oral contraceptives,
and nutritional status.
There is increasing evidence that AFB1 can be activated by lipid
hydroperoxide-dependent mechanisms, involving microsomal prostaglandin
H synthase and lipoxygenases. Although the maximum activity of this
co-oxidation is low relative to P450, these processes may contribute
significantly to bioactivation of AFB1 in vivo in humans, who are
generally exposed to low levels of AFB1. Co-oxidation may be
particularly important for AFB1 carcinogenicity in extrahepatic
tissues, in view of relatively low cytochrome P450 activity in these
organs. In the search for target cell types in the human lung, a
thorough analysis of the cellular distribution of potential AFB1
metabolizing systems will be necessary (Massey et al., 1995).
Detoxification (mediated by cytochrome P450 as well as conjugation
of the epoxide with glutathione) must also be considered. Animal
studies suggest that GST-catalysed detoxification is the crucial
factor in susceptibility to AFB1, and humans appear to lack
significant GST-mediated protection against AFB1 (Massey et al.,
1995). There is suggestive evidence that human GSTs in the alpha,
mu and theta families may all have roles in the detoxification of
the epoxide. It is not yet known with certainty whether there is a
role for epoxide hydrolase.
A possibly important factor in the assessment of aflatoxin risk is
the variation of human susceptibility due to individual differences in
human metabolism, such as polymorphisms in cytochrome P450s, GSTs, and
epoxide hydrolase. Data are beginning to become available; it is
unclear as yet what impact gene polymorphisms may have on human
activating as well as detoxifying enzymes, and therefore on aflatoxin
risk (Cardis et al., in press). With all the enzymes, it is
necessary to consider the stereochemistry of the aflatoxin epoxide,
which is critical in genotoxicity (Guengerich et al., 1996).
Most studies comparing AFB1 metabolism in different species have
been conducted in vitro, using subcellular fractions such as
microsomes or cytosol, or purified components of either fraction. The
exclusion of enzymes and cofactors for competing metabolic pathways
restricts quantitative comparisons of metabolism between different
species; therefore, conclusions based on data from in vitro
experiments are limited to qualitative comparisons of individual
pathways of AFB1 metabolism. Although specific metabolism pathways may
be associated with increasing sensitivity to AFB1 carcinogenicity,
quantification of sensitivity requires a supply of metabolic factors
for competing reactions found by either reconstitution of total
cellular fractions, use of primary cell cultures with representative
metabolic capacities, or whole animal studies (Gorelick, 1990).
The P450s both activate and detoxify AFB1 and the effect of
inducing individual P450s is not easily predicted. Also, the small
intestine, site of absorption of orally ingested AFB1, expresses P450
3A4. Activation of AFB1 and DNA alkylation in the small intestine may
be considered also to be a detoxification process since the cells are
sloughed rapidly and cancers of the small intestine are very rare
(Guengerich et al., 1996).
After reviewing the available data from the metabolic,
mutagenicity and laboratory animal studies, the Committee concluded
that there is at the present time insufficient quantitative
information available about competing aspects of metabolic activation
and detoxification of AFB1 in vivo in various species to describe
quantitatively a species-dependent effect of metabolism on AFB1
carcinogenicity (Gorelick, 1990; Massey et al., 1995; Guengerich
et al., 1996; Wild et al., 1996). It is, however, probable, that
differential sensitivity to AFB1-induced tumours between species can
be partially attributed to differences in metabolism.
3.1.2 Studies on the p53 gene
Studies on p53 mutations have been extensively discussed earlier
in this paper. The Committee concluded that there is currently
insufficient straightforward information available on the specificity
of the aflatoxin/p53 association to assess and quantify the
independence of aflatoxin as a cause of human liver cancer.
3.1.3 Epidemiological studies
The relevant epidemiological studies have been discussed earlier;
only the conclusions are presented here. Most of the epidemiological
studies show a correlation between exposure to aflatoxins and liver
cancer; some studies suggest that aflatoxin exposure poses no
detectable independent risk and other studies suggest that it poses a
risk only in the presence of other risk factors such as HBV infection.
Several ongoing studies are likely to improve further the estimates of
human risks from aflatoxin exposures; most notable among these are
cohort studies in Shanghai, Thailand and Qidong, China, and the HBV
vaccination trials in The Gambia, Taiwan and Qidong. When these
studies are complete, JECFA may want to re-evaluate the risks of
aflatoxins in humans.
A number of factors influence the risk of primary liver cancer,
most notably carriage of HBV; the potency of aflatoxins appears to be
significantly enhanced in individuals with simultaneous HBV infection.
Most of the epidemiological data are from geographical areas where
both the prevalence of HBsAg+ individuals and aflatoxins are high; the
relationship between these risk factors in areas of low aflatoxin
contamination and low HBV prevalence is unknown. This interaction
makes it difficult to interpret the epidemiological studies in the
context of aflatoxin as an independent risk. The Committee therefore
has made decisions contingent upon the dynamics of HBV infection in a
human population for which aflatoxin potency is to be determined.
The identification of HCV is a major breakthrough in understanding
the etiology of liver cancer. Two studies have investigated
interactions between HCV infection, aflatoxins and liver cancer; the
evidence so far is inconclusive. As shown in Table 1, it has been
estimated that 50 to 100% of liver cancer cases are associated with
persistent infection with HBV and/or HCV.
In Latin America both liver cancer and HBV infection are rare, yet
aflatoxin exposure is relatively high. Unfortunately, few studies are
available on the occurrence of liver cancer in Latin America; much
could be learned about aflatoxin as a risk factor in liver cancer by
conducting appropriately designed epidemiology studies in Latin
America.
3.1.4 Aflatoxin biomarker studies
The Committee concluded that the currently available studies
utilizing aflatoxin biomarkers do not provide a quantitative measure
of aflatoxin exposure in humans especially over the long term.
Biomarker levels in relation to cancer risk in the studies of Wang
et al. (1996a) and Qian et al. (1994) studies have been used for
modelling data; however, the interpretation is limited.
3.2 General modelling issues
Quantitative risk assessment for food contaminants involves four
basic issues: 1) choice of data; 2) measure of exposure; 3) measure of
response; and 4) choice of a mathematical relationship between dose
and response for a given data set. General comments can be made for
each of these areas, as well as specific comments concerning what has
been done regarding estimating risk from exposure to aflatoxin.
3.2.1 Choice of data
In general, the best data set to use for dose-response analysis
would be a human study in which dose is accurately measured, response
is determined without error and there are no confounding factors which
are unexplained. It is rare to find an epidemiological study without
one of these factors causing difficulty in the interpretation and
utility of the data. In contrast to the human data, test species data
are generally devoid of confounders. There is a clear and accurate
measure of response, and dose is an integral part of the design of the
study. Numerous dose-response assessments have been conducted by
modelling animal data and extrapolating the results to humans.
However, the extrapolation may be problematical, given outstanding
questions concerning the overall relevance of the animal data. For
aflatoxin, several epidemiological studies are capable of providing a
dose-response assessment. However, the study of Yeh et al. (1989)
has some limitations as described in section 2.2.10.3(e). The cohort
study in Shanghai (Ross et al., 1992; Qian et al., 1994)
considered both biomarker information and dietary questionnaires as
sources of aflatoxin exposure information. The study conducted by Wang
et al. (1996b) in Taiwan considered HCV, but results were
inconclusive.
3.2.2 Measure of exposure
In all of the risk assessments performed for aflatoxin, dose has
been expressed as lifetime average exposure in ng/kg per day. Note
that if peak exposure or early lifetime exposure has an impact on the
risk other than through the increase in the lifetime average ng/kg per
day, this choice of exposure measure could bias the risk estimates.
3.2.3 Measure of response
The major toxicological impact of aflatoxin on humans and animals
is an increase in primary liver cancer; that is the focus of this risk
assessment and all others performed to date.
3.2.4 Choice of mathematical model
Two basic risk models are routinely used in cancer epidemiology to
describe the relationship between dose of a contaminant and the risk
of disease or death. These models are of the form:
rM(t,E) = rO(t) × fM(E) multiplicative model
and
rA(t,E) = rO(t) + fA(E) additive model
where rM(t,E) and rA(t,E) are functions that describe disease
incidence as a function of age (t) and exposure (E). Exposure is
used here generically to include factors other than age that could
affect on the incidence rate. For unexposed individuals the incidence
rate is rO(t), and fM(E) and fA(E) are functions describing
the effect of exposure on the background. Typically, the forms for
fM(E) and fA(E) are assumed to be either linear or log-linear
(exponential). For example,
fM(E) = 1 + a1E1 + a2E2 + a3E3E2 or log[FM(E)] = a1E1
+ a2E2 + a3E1E2, where a1, a2 and a3 are parameters to be
estimated. The multiplicative model with log-linear effect of exposure
is commonly known as the Cox model and is related to logistic
regression. Tests of significance for any one effect (say E1) are
performed by testing whether its associated parameter (a1 for E1)
differs significantly from 0. The term a3E1E2 describes an
interaction between the two factors E1 and E2 and allows one to test
for such an interaction amongst the exposures. Choice of an additive
or multiplicative model can have a substantial impact upon resulting
risk estimates, particularly when extrapolating to a different
population. In the case of the additive model, differences in
background incidence have no impact on predictions of additional risk
(i.e., r(t,E) - r(t,0) = fA(E) and does not include rO(t)). On
the other hand, in the multiplicative model, predictions of additional
risk depend on the background incidence rate (i.e., r(t,E) - r(t,0)
= r0(t)(fM(t,E)-1), which is proportional to r0(t)).
Other plausible models not of the additive or the multiplicative
form include "mechanistically-based" models such as the two-stage
model for cancer. Although such models are not additive or
multiplicative per se, dose effects on the parameters that drive the
background cancer rate are usually modelled as linear or exponential,
as described earlier, and the choice of the relationship can affect
the risk estimates.
3.3 Potency estimates
3.3.1 Potency estimates based upon epidemiological data
In analysing any epidemiological study, there are many plausible
alternatives as to the form of the mathematical relationship between
exposure and response. For aflatoxin, the range of potencies derived
by using different models provides an indication of the uncertainty in
risk when one extrapolates from human data based upon studying areas
with relatively high background incidence of liver cancers and with
relatively high prevalence of HBV. In the following sections, selected
risk analyses will be reviewed briefly and the resulting potency
estimates presented and compared. In all of the analyses cited, it
should be noted that the potential effect of misspecification of the
dose that went into the derivation of the potency has not been
quantitatively addressed. As for all retrospective constructions of
exposure, use of recent levels of aflatoxin exposure to describe
current incidence rates assumes that current exposures are comparable
to past exposures. Owing to the long latency period predicted for most
cancers, uncertainty in the lifetime dose is an additional source of
variability that could lower (if the historical exposures were
actually higher than reported) or raise (if the historical exposures
were lower than actually reported) the resulting potency.
3.3.2 Potency estimates not accounting for HBV infection
Table 2 summarizes potency estimates based on analyses of
epidemio-logical studies in which regional cancer rates were compared
to estimates of aflatoxin intake without regard to differences in HBV
infection rates. As a reality check, the values in Table 2 can be
applied to the average aflatoxin exposure in the USA to obtain a
prediction of added incidence for the population. Using the largest
potency value in Table 2 (0.375) and assuming an average aflatoxin
intake of 0.26 ng/kg per day (Henry et al., 1997) for the USA
population, the added incidence is calculated to be approximately
0.375 × 0.26 or 0.0975 per 100 000 per year. Since this calculation is
based on the highest predicted potency, none of the potency estimates
in Table 2 are overtly inconsistent with the current USA rate of
approximately 3.4 per 100 000 and estimates of current levels of
aflatoxin intake (assumed to reflect past exposure levels). However,
as already indicated, these potency estimates are largely based on
studies in Africa and Southeast Asia where HBV infection rates are
much higher than in the USA or other Western countries.
Table 2. Potency estimates of the risk of liver cancer in humans based upon epidemiological
data with no correction for HBV status assuming an exposure of 1 ng/kg per day
Author Incidence/year per 100 0001
Peers & Linsell (1977) 0.11
Stoloff & Friedman (1976) 0
Carlborg (1979) 0.21
Bruce (1990)
based upon Stoloff (1983) 0
based upon van Rensburg et al. (1985),
Shank et al. (1972a,b), Peers et al. (1976, 1987) 0.10
Croy & Crouch (1991)
based on Peers et al. (1976) 0.15 (0.09, 0.23)
based on Yeh et al. (1989) 0.14 (0.08, 0.21)
Calif. Dept. Health Serv. (1990)
based on Peers et al. (1976) 0.38 (0.15, 0.60)
based on van Rensburg et al. (1985) 0.14 (0.10, 0.17)
based on Peers et al. (1987) 0.17 (NA, 0.3)
based on Yeh et al. (1989) 0.18 (NA)
1 Numbers in parentheses represent (lower, upper) 95% confidence limits on the predicted
risk when available from the authors.
3.3.3 Potency estimates accounting for HBV infection
The epidemiology study by Yeh et al. (1989) has been the focus
of several recent quantitative risk assessments and is described in
section 2.2.10.3(e). This study took place in Guangxi Province in
southern China and was a prospective cohort study of 7917 men.
In the analysis of their study, Yeh et al. (1989) adjusted
mortality rates for each region based on the age distribution of the
composite study cohort as an internal standard. Wu-Williams et al.
(1992) calculated that the age-adjusted PLC rate for the total cohort
was 121.5 per 100 000 when standardized to the age distribution of the
world population versus 226.3 per 100 000 when standardized to the age
distribution of the study cohort. The ratio of these rates (0.54) was
then used to adjust the regional PLC mortality rates reported by Yeh
et al. (1989) to obtain expected incidence rates for a
(hypothetical) cohort with age-distribution similar to the world
population. Adjusted person-years of observation (APY) were calculated
in each region as the number of PLC deaths observed in that region
divided by the adjusted mortality rate. Adjusted person-years of
observation were assumed to be distributed among HBsAg+ and
HBsAg-carriers according to the regional prevalence of hepatitis B.
The data are summarized in Table 3.
Table 3. Epidemiological data from Yeh et al. (1989)
Dose aflatoxin PLC cases APY1
(ng/kg per day) HBsAg- HBsAg+ HBsAg- HBsAg+
12 0 12 9932 2727
90 1 7 6114 2017
705 4 12 7733 2537
2028 2 23 5803 1743
-2 7 54 29582 9034
1 Adjusted person-years (see text)
2 No data are available for this group
Croy & Crouch (1991) separately analysed the HBV negative and HBV
positive cancer mortality rates in the Yeh et al. (1989) study using
additive linear models. They estimated potencies of 0.036 cancers per
100 000 per year for every ng/kg per day exposure for the HBV
negatives and 0.50 cancers per 100 000 per year for every ng/kg per
day exposure for the HBV positives.
Their analysis did not look at the combined data under a single
model and has been criticized for the use of only the small numbers of
cancers in the HBV negatives. Hoseyni (1992) analysed the Yeh et
al. (1989) data using regression techniques applied to several
different models including multiplicative and additive background
combined with linear and linear-exponential (multiplicative only)
models. He compared these various models based upon goodness-of-fit as
well as rejection by a likelihood ratio test and concluded that the
multiplicative model with a linear-exponential effect on mortality
rates by aflatoxin and HBV status (an added constant in the model if
HBsAg was positive) best fit these data. He did not explicitly include
an interaction term in this model (although the multiplicative model
implies a specific type of interaction) nor did he include an
interaction term in the additive linear model (there is no implicit
interaction in this model). The potency of the preferred
multiplicative model changes as a function of the background (in the
absence of aflatoxin and HBV) so that potencies can only be given with
respect to an explicit population liver cancer rate. Focusing on risk
prediction for the USA population, Hoseyni (1992) chose a background
cancer rate of 3.4 per 100 000 in deriving potency estimates. The
resulting estimates were 0.0018 cancers per 100 000 per year for every
ng/kg per day exposure to aflatoxin in HBV negative individuals and
0.046 cancers per 100 000 per year for every ng/kg per day exposure in
HBV positives.
Wu-Williams et al. (1992) examined the fit of a variety of
multiplicative and additive models that incorporated interaction
terms. These models were fit to the adjusted person-years data as
discussed above. Two models were found to fit the data adequately and
equally; an additive-linear model that includes an interaction term
and a multiplicative-linear model (very similar to that of Hoseyni)
with no interaction term. Under the additive-linear model, the potency
estimates were 0.031 and 0.43 for HBV-negative and HBV-positive
populations, respectively. Under the multiplicative-linear model, the
same risks for a USA population with a background cancer risk of 2.8
per 100 000 were 0.0037 and 0.094, respectively.
Finally, Bowers et al. (1993) applied an approximation to the
two-stage model of carcinogenesis (discussed in Kopp-Schneider &
Portier, 1989) to the adjusted person-years data. This model is
similar to a mixed additive model suggested by Bowers (1993), but the
parameters are tied to the biological concepts of induction of
mutations and growth of mutated cells (Thorslund et al., 1987). In
their analysis, it was assumed that aflatoxin had a linear effect on
the formation of mutations while HBV had no effect on the mutation
rate. For the growth of mutated cells, Bowers et al. (1993) assumed
a linear effect of HBV (presence or absence) and an interaction effect
of HBV and AFB1. The resulting potencies for the HBV-negative and
HBV-positive populations were 0.013 and 0.328 cancers per 100 000 per
year for every ng/kg per day exposure, respectively.
Potencies for all these studies are summarized in Table 4. Also
summarized in Table 4 are new analyses performed for the Committee
that analysed aflatoxin biomarker data from Qian et al. (1994) and
Wang et al. (1996b).
3.3.4 Potency estimates based on biomarker studies
Recent studies in Shanghai (Qian et al., 1994) and Taiwan (Wang
et al., 1996b) have measured biomarkers of aflatoxin exposure on the
individual level. The Committee has calculated potency estimates based
on these studies for comparison to estimates determined on the basis
of the data of Yeh et al. (1989). Concerning these studies, an
additional difficulty is that the internal markers of exposure were
frequently below the level of analytical quantification and
consequently individual determinations were necessarily classified on
an ordinal scale. Estimating potency requires estimating quantitative
mean levels of internal biomarkers and daily aflatoxin intake
corresponding to these classifications.
Table 4. Potency estimates of the risk of liver cancer in humans
based upon epidemiological data with correction for HBV status
assuming an exposure of 1 ng/kg per day
Study HbsAg status Incidence per 100 0001
Croy & Crouch (1991) - 0.036 (0.079)
+ 0.50 (0.77)
Wu-Williams et al.(1992)
multiplicative-linear - 0.0037 (0.006)
+ 0.094 (0.19)
additive-linear - 0.031 (0.06)
+ 0.43 (0.64)
Hosenyi (1992)
(background=3.4/100 000) - 0.0018 (0.0032)
+ 0.046 (0.08)
Bowers et al. (1993) - 0.013
+ 0.328
Qian et al. (1994)
(background=3.4/100 000) - 0.011
+ 0.11
Wang et al. (1996b)
(background=3.4/100 000) - 0.0082
+ 0.37
1 Numbers in parentheses represent upper 95% confidence limits on
the predicted risk when available from the authors.
In the study of Qian et al. (1994), 18 out of 50 (36%) cases and
31 out of 267 (12%) controls had quantified levels of urinary AFB1-
N7-Gua above the detection limit of 0.07 ng/ml. The overall range of
quantified levels was 0.3-1.81 ng/ml but the ranges for cases and
controls were not reported separately and the individual
determinations are no longer readily available (J.D. Groopman,
personal communication). The HBV-adjusted relative risk associated
with detectable levels of AFB1-N7-Gua was 9.1 (95% CI = 2.1, 29.2).
Though not reported, the HBV-adjusted relative risk associated with
detectable versus non-detectable levels of AFB1-N7-Gua was 4.6 (95%
CI = 1.8, 11.3) with a corresponding relative risk of 10.2 (95% CI =
4.9, 21.2) for HBV positivity.
Assuming an exponential distribution, estimates of mean levels of
AFB1-N7-Gua were obtained by fitting the cumulative probability
below the limit of detection to the proportion of non-detectables. The
estimated conditional mean values corresponding to non-detectable and
detectable classifications are 0.031 and 0.18 ng/ml for cases and 0.02
and 0.095 ng/ml for controls. For the logistic regression
(multiplicative linear-exponential) model, potency is the product of a
regression coefficient for AFB effect (on a ratio scale) and the
background incidence rate. Assuming that the distribution of
AFB1-N7-Gua in the general population is similar to controls and
adjusting the regression coefficient by dividing by the difference in
estimated mean levels corresponding to detectable versus
non-detectable levels gives:
J = log(4.6)/(0.095-0.02) = 20 (ng/ml)-1.
Adjusting for the relative molecular mass of AFB1-N7-Gua versus
AFB1, and assuming an average body weight of 70 kg, a daily urine
volume of 1400 ml (ICRP, 1975) and that 0.2% of daily AFB1 intake is
excreted as AFB1-N7-Gua (Groopman et al., 1992), the regression
coefficient is equivalently expressed as 0.0031 (ng AFB1/kg per
day)-1. For an annual background cancer rate of 3.4 per 100 000, the
corresponding potency estimate is 0.011 cancers per 100 000 per year
for every ng/kg per day exposure in HBV negative individuals and
10-fold higher for HBV-positive individuals.
In the study of Wang et al. (1996b), urinary metabolites were
fully quantified for all individual samples analysed and AFB1-albumin
adduct levels were quantified for only 93 of 232 (40%) blood samples
tested with a detection limit of 0.01 fm adduct/µg albumin. Estimated
HBV-adjusted relative risks were 1.6 (95% CI = 0.4, 5.5) for
detectable versus non-detectable AFB1-albumin adducts and 3.8 (95% CI
= 1.1, 12.8) for high versus low levels of urinary metabolites.
Although the urinary biomarker was fully quantified, levels of
AFB1-albumin adducts better reflect average AFB1 intake. Mean levels
corresponding to detectable and non-detectable classifications of
AFB1-albumin were calculated by fitting a log-normal distribution to
the quantified levels (Santella, personal communication) by log-probit
analysis (Travis & Land, 1990). For controls, the estimated mean
values corresponding to non-detectable and detectable classifications
were 0.0048 and 0.035 fm adduct/µg albumin.
Assuming that the distribution of AFB1-albumin adduct levels in
the general population is similar to controls, the regression
coefficient adjusted with respect to quantified levels of
AFB1-albumin is:
J = log(1.6)/(0.035-0.0048) = 16 (fm/µg albumin)-1
Based on data from China (Gan et al., 1988), 1.05 ng AFB1-albumin
adduct per g albumin corresponds to 1 µg AFB1 intake per day.
Correcting for the relative molecular mass of the AFB1 adduct, the
conversion factor is 0.00015 fm adduct/µg albumin per 1 ng/kg per day
AFB1 intake and the regression coefficient is equivalently expressed
as 0.0024 (ng/kg per day)-1. For a population with an annual
background cancer rate of 3.4 per 100 000, the corresponding potency
estimate is 0.0082 cancers per 100 000 per year for every ng/kg per
day exposure in HBV-negative individuals. The estimated relative risk
of 45.5 for HBV reported in the study of Wang et al. (1996b)
suggests a potency of about 0.37 cancers per 100 000 per year for
every ng/kg per day exposure for HBV-positive individuals.
The similarity of the estimates suggests that the magnitude of the
associations detected in Shanghai and Taiwan are relatively consistent
with that observed in Guangxi. However, the calculations presented
here are subject to a number of reservations. First, the estimates of
mean levels corresponding to detectable and non-detectable
classifications of AFB1-N7-Gua or AFB1-albumin are based on very
limited data. Furthermore, the conversion factors relating internal
exposure (AFB1-N7-Gua or AFB1-albumin) to dietary AFB1 intake are
based on studies in human populations that may have different genetic
characteristics than the study populations to which the conversion
factor is applied. For the Taiwan study, there is the additional
consideration of how the case series was obtained. About half of the
identified cases were prevalent cases diagnosed at the onset of the
study. Consequently, the AFB1 exposure determinations for these cases
may reflect alterations in metabolism directly related to the presence
of PLC per se.
3.3.5 Potency estimates from test species
Several investigators have studied the carcinogenic potential of
aflatoxins in vivo using laboratory animals (Wieder et al., 1968;
Butler et al., 1969; Epstein et al., 1969; Merkow et al., 1973;
Newberne & Rogers, 1973; Wogan et al., 1974; Vesselinovitch et
al., 1972; Ward et al., 1975; Reddy & Svoboda, 1976; Sieber et
al., 1979; Stoner et al., 1986; Angsubhakorn et al., 1981a,b;
Butler & Hempsall, 1981; Nixon et al., 1981; Moore et al., 1982;
Cullen et al., 1987). In most of these studies, hepatocarcinogenesis
was the main focus although other cancers have been noted such as
colon, kidney, lung and lymphoreticular system. The majority of these
studies focused on aflatoxin B1 with one study of aflatoxin M1
(Cullen et al., 1987), one study comparing aflatoxins B1, G1 and
B2 (Butler et al., 1969) and another study considering the
aflatoxin metabolite aflatoxicol (Nixon et al., 1981). All of these
laboratory results are amenable to quantitative estimation of risks;
however, some only contain one experimental dose group, have little
indication of dose-response due to 100% response in all dosed animals
or include the use of other agents (e.g., vitamin A) in their
protocols. Cardis et al. (1997) summarized the calculated potencies
from aflatoxin exposure in these test species. With regard to
quantitative estimations and prediction of risks for aflatoxin B1,
the study by Epstein et al. (1969) contains the most experimental
dose-groups and the most complete data for fitting a model. Using a
simple multistage model of carcinogenesis (Cardis et al., 1997),
these data predict an added incidence of 0.97 cancers per 100 000 per
year for an exposure of 1 ng/kg per day of aflatoxin B1 (scaled from
the animal data to human risk estimates using body weight raised to
the 0.75 power). Other potency estimates (extrapolated to humans)
ranged from as low as 0.05 per 100 000 per year for the Syrian golden
hamster (Moore et al., 1982) to as high as 37 per 100 000 per year
for the Fischer 344 rat (Cullen et al., 1987), with median estimate
across all experiments of 1.4 per 100 000 per year.
The human potency estimates for aflatoxin B1 alone (Table 4 shows
that these are in the range of 0.002-0.036 per 100 000 per year for
exposure to 1 ng/kg per day of aflatoxin) fall well below this range,
suggesting that humans are considerably less sensitive than other
species. There are several possible explanations for this. First, it
is possible that humans are in fact less sensitive than species tested
in laboratory experiments. Considering the proposed mechanism by which
aflatoxin induces liver tumours at low levels of exposure (DNA damage)
and the efficiency by which humans repair DNA damage, it is plausible
that humans are less sensitive. For this to be a reasonable
explanation, the rate per day of DNA repair in the human system for
the critical aflatoxin B1 lesions would have to be approximately 5
times more efficient than that of other species on a surface area
basis. A second possibility is that estimated exposures in the Yeh et
al. (1989) study were larger than the true exposures. While exposure
estimates in epidemiological studies are generally a point of concern
for any dose-response assessment, it is unlikely that the estimates
obtained in the Yeh et al. (1989) study are biased to this degree. A
third possibility is misclassification of the HBV cases, with a large
proportion of the HBV negatives actually being classified as positives
(the misclassification would need to be about 50% for the human
potency estimates to move into the range of the test species
estimates). This level of misclassification is highly unlikely; in
fact it is more likely that HBV positives have been incorrectly
classified as negatives. A fourth possibility is that many of the HBV-
positive individuals with liver tumours were actually infected late in
life and that, consequently, there was not sufficient time for HBV to
contribute to development of liver cancer in these individuals. If
true, this would suggest that the risk of liver cancer due to exposure
to aflatoxin in the absence of HBV could be as high as the risks seen
in Table 2 (approximately 0.15 cancers per 100 000 per year for an
exposure of 1 ng/kg per day). However, once again, this would seem to
be an unlikely explanation, and still falls well below the animal-
based estimates.
Finally, it is possible that the usual conversion factor for
converting potency estimates in experimental species to potency
estimates in humans is inappropriate for these data. For the case of
malignant hepatomas in Wistar rats (Epstein et al., 1969), as
mentioned above, conversion based upon surface area scaling using body
weight raised to the 0.75 power yielded a potency of 0.97 per 100 000
per year for an exposure of 1 ng/kg per day. If no conversion had been
made, the potency estimate in the Wistar rat would have been 0.23 per
100 000 per year. Considering the different sizes of the test species
involved, the potencies range from 0.014 per 100 000 per year (Syrian
golden hamster) to 1 per 100 000 per year (Fischer rat), with most of
the larger mammals being on the low range of the potency scale (0.029
per 100 000 per year for the tree shrew; 0.057 per 100 000 per year
for the rhesus and cynomolgus monkeys). In this case, although the
human estimates are still at the lowest end of the potency scale, they
would be comparable to the values estimated for other primates.
Figure 2 presents potencies estimated from animal studies and
epidemi-ological studies. Epidemiological data for which HBV infection
status was unknown and for which potencies were estimated gave
potencies in between the range predicted by the HBV
infected/non-infected numbers. Potencies given in Figure 2 do not
generally apply to aflatoxin M1, since exposure estimates given in
many of the epidemiological studies ignored the contributions to total
aflatoxins exposure from milk and milk products. From one comparative
toxicology study in rats, it has been possible to estimate that
aflatoxin M1 has a potency approximately one order of magnitude less
than that of aflatoxin B1 in that species (Cullen et al., 1987).
 4. AFLATOXIN DIETARY INTAKE ESTIMATES
4.1 Introduction
This report summarizes the results of monitoring and available
national estimates of intake of aflatoxins in order to provide a
framework for the task of estimating increments in intake of
aflatoxins. Estimates are based on the results of available monitoring
data. Total aflatoxin intake based on the GEMS/FOODS regional diets
are used to evaluate the impact of four different scenarios: no limit,
and limits set at 20, 15 and 10 µg/kg. This evaluation was conducted
for ground-nuts and for maize for total aflatoxins and for aflatoxin
B1 alone. Generally they are not considered by the submitters to be
representative because sampling has focused on those lots that are
more likely to contain the highest levels of aflatoxin. However, this
analysis provides useful qualitative comparisons between regulator
options.
4.2 Background
Aflatoxins are found as contaminants in human and animal food as a
result of fungal contamination both pre- and post-harvest, with the
rate and degree of contamination being dependent on temperature,
humidity, soil and storage conditions. Though a wide range of foods
may be contaminated with aflatoxins, they have been most commonly
associated with groundnuts (groundnuts and groundnut products), dried
fruit, tree nuts, spices, figs, crude vegetable oils, cocoa beans,
maize (maize), rice, cottonseed and copra. There are practices that
reduce but do not completely eliminate aflatoxins in grains,
groundnuts, figs and other crops. However, even in the most tropical
of climates, many lots of these crops do not contain detectable levels
of aflatoxins.
4.3 Methods
4.3.1 Period of intake of relevance
Chronic (lifetime) intake is assumed to be the period of
relevance. Therefore, the average concentrations in the diet are of
primary interest.
4.3.2 Estimated levels of aflatoxins in foodstuff
Data were available for this analysis from at least one country on
every continent. Typically the data were uniformly judged by the
submitters not to be representative. In most instances, the data were
thought to be biased towards the upper end of intake. Nonetheless,
caution must be exercised in using the data to generate intake
estimates and in interpreting the results of such analyses. However,
the data did provide a framework to explore the relative impact of
regulatory activities. The data are summarized in Table 5.
For some of the analyses used in this paper individual data points
were required in order to generate distributions and to evaluate the
impact of imposing upper limits on aflatoxins in foodstuffs.
Specifically, a series of analyses was conducted to determine the
impact of truncating the distribution at 10, 15 and 20 µg/kg,
respectively, in order to simulate the potential impact of proposed
limits. For these analyses, data reported by the USA, China and Europe
were evaluated because the raw data were available. These analyses
were conducted for maize and groundnuts for total aflatoxins and for
aflatoxin B1.
4.3.3 Estimated intakes
Four pieces of information are required to estimate the potential
intakes due to aflatoxins in crops that are imported: (1) the levels
of aflatoxin in imported crops; (2) the amount of each imported crops
consumed; (3) the impact of any subsequent processing on aflatoxins
levels; and (4) methods for combining the first 3 to estimate intake.
Table 5. Summary of aflatoxin monitoring data submitted for consideration by the Committee
Commodity Country/Region Number of samples Results Comments Reference
NUTS
Groundnuts
(groundnuts and
groundnut products) Australia 9 samples Mean = 2 µg/kg; Australia Market
Max = 10 µg/kg Basket (1992)
913 lots 83.1% <5 µg/kg presorted Read (1997)
(b1) 14.2% 5-10 µg/kg
2.7% 10-15 µg/kg
Brazil 199 samples Mean = 14 µg/kg Sabino (1997)
(b1) Max = 181 µg/kg
51%> LOD
China 174 groundnuts 3 >30 µg/kg selected to be worst Chen (1997)
40 groundnut meal 1 >251 µg/kg case for research
(b1) (see Table 3) purposes
Cuba 1114 samples 49% >LOD Regueiro
(b1) (no date)
European Union data from 12 Mean B1 35-64 µg/kg data submitters SCOOP (1996)
countries (country means) emphatically state that
Max = 789 µg/kg DATA ARE NOT
(see Tables 3 and 4) REPRESENTATIVE
Japan 22 789 samples >97% >LOD imported from >25 Japanese Ministry
(1972-1989) 238 >10 µg/kg (B1) countries of Health (1995)
Max = 8070 µg/kg
Mexico 107 samples 104 <20 µg/kg Mexico (1996)
(1992-1995) 3 >20 µg/kg
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Nicaragua 9 samples (1996) 1 >15 µg/kg (B1) Nicaragua (1996)
USA >600 000 lots >90% <15 µg/kg Wood (1995)
(1975-1992) (total); see Table 6
Zimbabwe 286 samples 39% <5 µg (1995) analysed B1 and total Zimbabwe Government
(1995-1996) 54% <10 µg (1995) Analyst Laboratory
46% >10 µg (1995) (1995-1996)
85% <5 µg (Jan-Jun 96)
92% <10 µg/kg
8% >D12 10 µg/kg+D24
Brazil nuts European Union not available Max = 15 µg/kg (B1) see comment under SCOOP (1996)
Max = 35 µg/g (total) groundnuts
Japan 74 samples 70 <LOD Japanese Ministry
2 >10 µg/kg of Health (1995)
2 >10 µg/kg
Max = 123 µg/kg
Pistachio nuts European Union 11 countries Mean = 2-23 µg/kg
provided data (B1; country means)
Mean = 44-27 µg/kg
(total; country means)
Max = 450 µg/kg (B1)
Max = 813 µg/kg (total)
Japan 2422 samples 2339 <LOD (B1) Japanese Ministry
(1972-1989) 48 >10 µg/kg of Health (1995)
35 <10 µg/kg
Max = 8030 µg/kg
Mexico 244 samples 5 samples >20 µg/kg Mexico (1996)
(1993-1996)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Sunflower seeds Argentina 20 samples no detects Argentina (1996)
Almonds Australia 9 samples no detects Australian Market
Basket Survey (1992)
Japan 93 749 samples >98% <LOD reported all Japanese Ministry
Max = 128 µg/kg miscellaneous nuts of Health (1995)
in one statistics;
covered years 1972-
1989; all were imported
Cashews Japan 1227 samples >98% <LOD Japanese Ministry
of Health (1995)
Walnuts Japan 321 samples >98% <LOD Japanese Ministry
of Health (1995)
Macadamia nuts Japan 149 samples >98% <LOD Japanese Ministry
of Health (1995)
Hazel nuts Japan 103 samples >98% <LOD Japanese Ministry
of Health (1995)
FIG PRODUCTS
European Union Mean = 0.5-26 µg/kg see comment for SCOOP (1996)
(country mean B1) groundnuts
CEREAL & CEREAL PRODUCTS
rice, wheat, maize
Bolivia number unknown 2 rice 25-168 µg/kg (B1)
(1992-1995) 1 wheat 10 µg/kg (B1)
1 wheat 18 µg/kg (B1 + g1)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
2460 samples
(1986-1997) 1273 >LOD
Mean = 34 µg/kg
(range = 7-144 µg/kg)
Maize Brazil 321 samples 179 <LOD Sabino (1997)
27 <30 µg/kg
116 >30 µg/kg
(B1 or total not reported)
Max = 2440 µg/kg
Maize Brazil 2546 samples Mean = 35 µg/kg
(51% >LOD)
Rice Brazil 401 samples Mean = 2 µg/kg
(10% >LOD)
Wheat Brazil 237 samples Mean = 2 µg/kg
(19% >LOD)
Malt Brazil 30 samples Mean = 30 µg/kg
Popcorn Brazil 32% >LOD
Sorghum Brazil 59 samples Mean = 3 µg/kg
(33% >LOD)
Maize China 486 samples Mean 5-251 µg/kg
(117 >LOD) Chen (1997)
Wheat China 597 samples Max = < 31 µg/kg
(9 >LOD) see also Table 7 Chen (1997)
Sorghum China 58 samples 1 >LOD Chen (1997)
Rice China 747 samples 7 >LOD Chen (1997)
Sorghum Columbia 45 samples 11 >LOD (B1) Diaz (1996)
(1995-1996) (1.4-43 µg/kg)
Mean = 11 µg/kg
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Maize Columbia 33 samples 4 >LOD (B1) Diaz (1996)
(4-66 µg/kg)
Mean = 21 µg/kg
Soybeans Columbia 25 samples 0 >LOD (B1) Diaz (1996)
Rice Columbia 22 samples 8 >LOD (B1) Diaz (1996)
(1-53 µg/kg)
Mean = 21 µg/kg
Cotton Columbia 17 samples
(1995-1996) 15 >LOD (B1) Diaz (1996)
(2-11 µg/kg)
Mean = 5 µg/kg
Maize Costa Rica 49 samples 1 >15 µg/kg (B1) Pacin (no date)
48 <10 µg/kg
Maize Cuba 4620 samples 20% >LOD (B1) Regueiro (no date)
Rice Cuba 340 samples 16% >LOD Regueiro (no date)
Sorghum Cuba 12% >LOD Regueiro (no date)
Wheat Cuba 1% >LOD Regueiro (no date)
CEREAL PRODUCTS
European Union 0.1-7.6 µg/kg see comment for SCOOP (1996)
(country means B1) groundnuts
0.25-5.9 µg/kg
(country means total)
(see Tables 6 and 7)
Maize Japan 371 samples 16 >LOD Japanese Ministry
Max B1 = 1.5 µg/kg of Health (1995)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Maize Mexico 1710 samples 265 >20 µg/kg Mexico (1996)
(1992-1996) 35 tortilla samples
>20 µg/kg
Maize Thailand 18 samples <LOD - 606 µg/kg Aflatoxin was visible Yoshizawa et al.,
1996
PULSES
Soybeans Brazil 143 samples 17% >LOD
Mean = 1 µg/kg
Soybeans China 388 samples 11 >LOD Jiangu Province Chen (1977)
(51-100 µg/kg)
Soy sauce China 308 samples 4.6% >LOD Jiangu Province Chen (1977)
Bean paste China 1 sample <100 µg/kg Jiangu Province Chen (1977)
3.3% > LOD
Max = >251 µg/kg
Soy oil China 379 samples 10 samples >LOD Jiangu Province Chen (1977)
Max = <251 µg/kg
Soybean meal China 6.7% >LOD Jiangu Province Chen (1977)
Max = <50 µg/kg
Frijoles (beans) Cuba 413 samples 13% >LOD (B1) Regueiro (no date)
Pulses Japan >2000 samples 2 samples >LOD (B1) Japanese Ministry
Max = <10 µg/kg of Health (1995)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
SPICES
European Union Max B1 = 323 µg/kg reported in at SCOOP (1996)
(0.5-17 µg country least one sample
mean levels) of every type of
Max total = 72 µg/kg spice analysed- see
comment re: data
under groundnuts
Japan 1804 >LOD in chilli, nutmeg, none found in 25 Japanese Ministry
(1979-1988) white pepper, paprika, other spices of Health (1995)
turmeric, cardamon,
pimento, ginger, celery
seed spices
CONFECTIONARY PRODUCTS
few samples 0.05 = - 1.1 µg/kg SCOOP (1996)
(country means B1)
FATS, OILS, OILSEEDS
France 56 samples Mean 4.5 µg/kg (France)
Max = <25 µg/kg
MILK AND MILK PRODUCTS
see text
a) National intakes
Available national intake studies were reviewed to provide
estimates of intake levels. This information was obtained from reports
submitted by members of the EU (SCOOP, 1996), China (Chen, 1997), USA
(1992), Brazil, Australia, Costa Rica, Argentina, Mexico, Nicaragua,
Colombia and Thailand. Thirteen European countries provided data for
the EU SCOOP project.
b) International intakes
The available data provide a general idea of the range of
aflatoxin levels in foodstuffs and the frequency of detection.
Information on the proportion of the crop imported was not available
for most countries, nor was it possible to determine the distribution
of aflatoxin levels in imported crops versus domestic crops.
Therefore, the data from individual countries were reviewed to obtain
a general overview of the likely levels in foods and to determine the
impact on average intakes if extreme levels of aflatoxins could be
removed from foodstuffs.
Impact on dietary intake if upper concentrations of aflatoxin
in foodstuffs are successfully limited to 10, 15 or 20 µg/kg
foodstuff versus no limit
It was assumed that methods are available to ensure that aflatoxin
levels above the specified limit are excluded from the food supply and
that the same proportion of the commodity would be imported (versus
domestic) regardless of the limit.
For these analyses all of the food consumed in the country was
assumed to contain the average residue concentration under that
scenario. Each analysis was repeated twice: (1) using European
monitoring data (b1 and total aflatoxin; and (2) using either Chinese
monitoring data (aflatoxin b1) or USA monitoring data (total
aflatoxin). Using these assumptions, the GEMS/FOODS regional diets
were used to evaluate the difference in dietary intake to aflatoxin
from groundnuts and maize under different limits.
c) Amounts consumed
The GEMS/FOODS regional diets were used as estimates of the
consumption of each of the commodities (Tables 4 and 5).
4.4 Results
4.4.1 Aflatoxin levels in foods: general
The 1995 FAO compendium, Worldwide regulations for mycotoxins
(FAO, 1995), summarized reports from 48 countries. The data submitted
by 33 countries for aflatoxin B1 and total aflatoxins (B1,B2,G1,G2)
were used to estimate median levels of 4 and 8 µg/kg, respectively, in
foodstuffs. The range of levels reported for B1 was from 0 to 30 µg/kg
and for total from 0 to 50 µg/kg. Seventeen countries provided
information on aflatoxin M1 in milk with a median of 0.05 µg/kg and a
range of 0-1 µg/kg. The FAO report did not provide additional details
about sampling, treatment of non-detects, imported versus domestic
foodstuffs, etc.
Reports from individual countries that were submitted to FAO and
to JECFA have been used to provide additional detail. The data are
summarized in Table 5 by commodity and by country.
The participants in the European Union Scientific Co-operation
Assessment of aflatoxin (SCOOP) reviewed data submitted by member
countries and by Norway. The participants concluded that the results
were unlikely to be representative and should not be used to estimate
total aflatoxin intake for individual countries or for Europe.
However, the studies did provide some insight into issues surrounding
aflatoxin intake assessments. Based on the data and subsequent
discussions, SCOOP concluded: (1) aflatoxins are found in a broader
range of foods than had been previously assumed; (2) most of the
samples did not contain detectable aflatoxin; (3) sampling methods are
important in accurately estimating aflatoxin levels; and (4) different
methods of collecting food consumption data may make a difference in
estimating aflatoxin intakes.
4.4.2 Aflatoxin levels in foodstuffs: Occurrence data by commodity
Most countries provided data for aflatoxin B1 in selected crops.
Some countries also provided an estimate of the level of total
aflatoxins. Total aflatoxin estimates typically included aflatoxin B1,
B2 and G. M1 was the common aflatoxin reported for milk and milk
products. The data are summarized in Table 1 for crops.
The data for aflatoxin M1 are summarized below:
Australia: No aflatoxins were detected in 34 whole milk samples.
European Union: Ten countries reported results of sampling for M1
in milk. The maximum level of 0.37 µg/kg M1 was reported by France.
The United Kingdom reported the highest level (0.22 µg/kg) in cheese.
Brazil: 204 samples of milk, cheese and yoghurt were analysed. Of
these only four samples of pasteurized milk contained detectable
levels of aflatoxin M1 (de Sylos, 1996). The levels ranged from 73-370
ng/litre.
Spain: Aflatoxin M1 was estimated in 19 total diet studies in 1990
and 1991. All but one sample was below the limit of detection. The one
sample contained 0.025 µg/kg.
4.4.2.1 Amount of commodity imported
The proportion of any commodity that is imported varies from 0 to
100%. For example, in the case of groundnuts all groundnuts, are
imported into the Nordic countries while virtually no groundnuts are
imported into China and the USA. If import/export data were available,
aflatoxin intakes could be more accurately estimated. However, this
information was not available for most countries.
4.4.2.2 Accounting for the change in aflatoxin levels during
processing
a) Maize
Processing of maize causes reduction in aflatoxin levels. Wet
milling reduces the concentration of aflatoxin in maize starch to 1%
of the levels found in the raw grain (Yahl, 1971). Similarly dry
milling reduces aflatoxin in food products (grits, low-fat meal and
low-fat flour) to 6-10% of the original concentrations (Brekke, 1975).
b) Groundnuts
The roasting of groundnuts reduces aflatoxin levels by 50-80%
(Waltking, 1971; Read, 1989; Billy, 1996).
c) Milk
Aflatoxin M1 is a metabolite of aflatoxin B1 and is found
associated with the casein in milk. Pasteurization does not affect the
level of aflatoxin M1 in milk or yogurt (Wiseman, 1983). Aflatoxin M1
has been reported to concentrate 3-6 fold during cheese making (Van
Egmond, 1983). MAFF (1994-5, No. 22) reported that aflatoxin was not
destroyed under domestic cooking conditions (microwave or heading in
gas oven).
The effects of processing were considered in many of the national
estimates. Processing factors have not been included in any of the
distributions generated for the estimates of aflatoxin intake.
However, it would be appropriate to adjust further the international
estimates of intake to reflect the impact of processing where the
commodity is always processed/cooked prior to consumption.
4.4.3 National estimates of aflatoxin intake
4.4.3.1 Australia
Australia conducts market basket surveys and estimates intake for
average and extreme consumers. The average diet was estimated to
contain 0.15 ng aflatoxin/kg bw per day and the upper 95th percentile
diet to contain approximately twice that level. Children's diets were
estimated to be somewhat higher - up to approximately 0.45 ng/kg bw
per day for the 95th percentile 2-year-old (Australia Market Basket
Survey, 1992).
4.4.3.2 China
A series of intake and market basket studies have been conducted
since 1980 to estimate the aflatoxin B1 intake. The reported intakes
have ranged from 0 to 91 µg aflatoxin B1/kg bw per day (Chen, 1997).
4.4.3.3 European Union
Estimates of aflatoxin intake were provided to the EU SCOOP
project by 9 countries. However, it should be noted that in every
instance, it was clearly stated that these estimates were not
representative. They were regarded as being no more than indicators of
intake of aflatoxin and as clearly not useful for predictive purposes.
These indicators of intake ranged from 2 to 77 ng/person per day for
aflatoxin B1 and from 0.4 to 6 ng/person per day for aflatoxin M1.
Although these may be useful to guide understanding of potential
intake, they are not to be used as an estimate of intake either for a
particular country or for Europe.
Of the countries that computed intakes of M1, the computed intakes
ranged from 0.04 µg/kg bw per day M1 (France, Germany) to 0.19 ng/kg
bw per day (Netherlands). The value reported by the Netherlands was
from baby food. In addition, the Netherlands also estimate 0.04 ng/kg
bw per day M1 from milk.
France provided additional information on intakes from cereals,
nuts, spices and milk. Two estimates of aflatoxin intake were
computed. The first was the product of the maximum level of aflatoxin
reported and the maximum consumption for each category of food. The
second estimate was the product of the average aflatoxin concentration
reported and the maximum consumption. The resulting estimates are
given in Table 6.
Table 6. Estimated aflatoxin intake in France (µg per day)
Maximum Mean
Cereal 13.33 2.42
Nuts 7.13 0.04
Spices 0.68 0.01
Milk 0.12 0.06
4.4.3.4 USA
The US FDA estimated intakes using data from the National
Compliance program for maize, groundnut and milk products using Monte
Carlo simulation procedures. The data were from the 1980s. The
eaters-only mean lifetime intake of total aflatoxin was 18 ng/person
per day and intake for the 90th percentile individuals was 40
ng/person per day. Mean aflatoxin M1 intake was 44 ng/person per day
and for the 90th percentile individuals 87 ng/person per day. The
authors noted that many assumptions were made that bias these
estimates upwards. The same analysis was repeated in 1992 with only
slightly different results (DiNovi, 1992).
4.4.3.5 Zimbabwe
The theoretical maximum intake of aflatoxin B1 was estimated for a
child's diet containing 150 grams maize with 5 µg/kg aflatoxin B1 and
30 grams ground-nuts with 10 µg/kg aflatoxin B1. The total aflatoxin
intake per day would be 1.05 µg per day if all maize consumed
contained 5 µg/kg and all groundnuts contained 10 µg/kg. If all maize
were to contain 15 µg per day the intake would be 2.55 µg per day.
4.4.4 Relative impact of establishing maximum limits on estimate of
intake
4.4.4.1. Average aflatoxin concentrations using four possible
scenarios
Data from the EU, China and the USA were used to assess the
potential impact of successfully eliminating aflatoxin levels above 20
µg/kg versus 15 µg/kg versus 10 µg/kg versus no limit for maize and
groundnuts. For each commodity, two sets of analyses were conducted,
(1) total aflatoxins and (2) aflatoxin B1, in order to determine
whether different conclusions would be reached. For each analysis the
data were evaluated as reported. In all cases in which samples
contained no detectable aflatoxins it was assumed that aflatoxins were
present at the LOD - obviously an overestimation. No impact of
processing was included, which again is an overestimation since
processing is known to result in 50-90% reduction in concentrations.
Table 6 summarizes the impact of successfully limiting aflatoxin
concentrations to less than 10, 15 or 20 µg/kg foodstuff on the mean
total aflatoxin concentrations in maize and groundnuts.
Table 6. Anticipated mean residues of total aflatoxins in maize and groundnuts under four assumptions for
acceptable residue levels in samples:
Scenario 1: no samples excluded
Scenario 2: samples > 10 µg/kg excluded
Scenario 3: samples > 15 µg/kg excluded
Scenario 4: samples > 20 µg/kg excluded
Scenario Samples USA maize European cereals European groundnuts USA groundnuts
excluded
µg total aflatoxin/kg commodity
1 No limit Mean 4.7 0.2 13.3 14.3
SD 30.7 0.4 97.2 58.8
2 Limit = 10 µg/kg Mean 0.6 NA 0.9 0.4
SD 1.4 1.4 0.9
3 Limit = 15 µg/kg Mean 0.7 NA 0.9 0.5
SD 2.0 1.6 1.4
4 Limit = 20 µg/kg Mean 0.9 NA 1.0 0.6
SD 2.6 2.0 1.7
Note: Samples were taken in either USA or Europe but the crop may have come from other geographic locations
No aflatoxins levels above 5 µg/kg have been reported on cereals
in Europe, so there would be no impact of imposing a regulatory
programme that would reduce aflatoxin levels1. In contrast, for
groundnuts and for groundnuts and maize in the USA, the largest
difference in mean aflatoxins levels is between no limit and a maximum
aflatoxin level of 20 µg/kg. In the USA, the greatest impact is
achieved by establishing a maximum level of 20 µg/kg for both maize
and ground-nuts. For example, using data for USA maize, the average
total aflatoxin levels would drop from 4.7 µg/kg to 0.9 µg/kg if all
maize with aflatoxin levels above 20 µg/kg were rejected (Table 6).
Small additional declines in average aflatoxin levels would be found
if the acceptable limit were 15 or 10 µg/kg. The average aflatoxin
levels would be 0.7 and 0.6 µg/kg if 15 and 10 µg/kg limits were
established (Table 7) for aflatoxin B1 in maize.
The same analysis was conducted for groundnuts. The reported total
afla-toxin levels in groundnut and groundnut products sampled in
Europe and USA are presented separately Table 6. If all groundnuts are
included, the average aflatoxin concentration would be 14 µg/kg. The
average aflatoxin concentration would be 0.6 µg/kg if all samples with
levels above 20 µg/kg were excluded and 0.5 and 0.4 µg/kg if all
samples with levels above 15 and 10 µg/kg, respectively, were
excluded.
The distribution of total aflatoxins in crops sampled in the USA
and Europe is provided in Table 8 and the distribution of aflatoxin B1
in crops in Europe and China is provided in Table 9.
1 The major part of cereals for human consumption in Europe are
domestically grown. If the source of cereals were to change this might
no longer be the situation.
Table 7. Anticipated mean residues of aflatoxin B1 in maize and groundnuts under four
assumptions for acceptable residue levels in samples:
Scenario 1: no samples excluded
Scenario 2: samples > 10 µg/kg excluded
Scenario 3: samples > 15 µg/kg excluded
Scenario 4: samples > 20 µg/kg excluded
Scenario Samples European Chinese European Chinese
excluded cereals maize groundnuts groundnuts
µg total aflatoxin B1/kg commodity
1 No limit Mean 1.5 11.8 6.9 8.3
SD 2.3 52.3 72.2 33.1
2 Limit = 10 µg/kg Mean 1.5 2.8 0.6 2.7
SD 2.3 1.9 0.9 1.6
3 Limit = 15 µg/kg Mean 1.5 3.1 0.6 2.7
SD 2.3 2.5 1.2 1.8
4 Limit = 20 µg/kg Mean 1.5 3.5 0.7 2.9
SD 2.3 3.3 1.7 2.3
Note: Samples were taken in either China or Europe but the crop may have come from
other geographic locations
Table 8. Distribution of total aflatoxins in maize and groundnuts
sampled in the USA and Europe1
Percentile USA maize European European USA
cereals groundnuts groundnuts
µg total aflatoxin/kg commodity
10.0% 0.1 0.0 0.1 0.1
20.0% 0.1 0.0 0.2 0.1
30.0% 0.2 0.0 0.3 0.2
40.0% 0.2 0.0 0.5 0.2
50.0% 0.3 0.1 0.6 0.3
60.0% 0.3 0.1 0.7 0.3
70.0% 0.4 0.1 0.8 0.4
80.0% 0.5 0.1 1.0 0.4
90.0% 4.0 0.1 3.3 1.9
95.0% 15.1 1.1 11.5 106
97.5% 38.1 1.5 55.3 267
99.0% 93.8 2.1 379 304
99.5% 149 2.6 767 314
99.8% 247 3.2 1110 323
99.9% 482 3.4 1320 327
1 European cereals include other crops in addition to maize.
Table 9. Distribution of aflatoxin B1 in maize and groundnuts sampled
in Europe1 and China
Percentile European Chinese European Chinese
cereals maize groundnuts groundnuts
µg aflatoxin B1/kg commodity
10.0% 0.1 0.6 0.0 0.6
20.0% 0.1 1.2 0.1 1.2
30.0% 0.3 1.8 0.1 1.7
40.0% 0.5 2.5 0.2 2.3
50.0% 0.7 3.0 0.4 3.0
60.0% 0.9 3.6 0.6 3.5
70.0% 1.2 4.3 0.7 4.0
80.0% 1.9 4.9 0.8 4.6
90.0% 5.4 16.1 1.2 7.8
95.0% 7.6 43.5 4.2 43.8
97.5% 8.8 75.9 16.9 69.9
99.0% 9.4 169 61.4 90.8
99.5% 9.7 453 367 97.3
99.8% 9.9 630 809 150
99.9% 10.0 720 1220 488
1 European cereals include other crops in addition to maize
From: Chen (1997) and SCOOP (1996)
The proportion of samples that would be excluded under each
scenario is identified in Table 10 for total aflatoxins. For example,
if all maize that contains total aflatoxin levels > 20 µg/kg is to be
eliminated, 4% of USA maize would be rejected. If the limit is 10
µg/kg, 6% of USA maize would be rejected. No European maize would be
rejected under either limit. Six per cent of USA and 4% of European
groundnuts would be rejected with a limit of 20 µg/kg and 7% and 5%,
respectively, with a limit of 10 µg/kg. The same information is
provided in Table 11 for aflatoxin B1. If all maize that contains
aflatoxin B1 levels > 20 µg/kg is eliminated, 8% of Chinese maize
would be rejected. If the limit is 10 µg/kg, 13% of Chinese maize
would be rejected. No European maize would be rejected under either
limit. A limit of 20 µg/kg would result in rejection of 2% of European
groundnuts and 8% of Chinese groundnuts. A limit of 10 µg/kg, would
rejected 9% of Chinese groundnuts and 3% of European groundnuts.
4.4.4.2 Intake of total aflatoxins using four scenarios
The four scenarios are:
Scenario 1: no change in current aflatoxin levels;
Scenario 2: exclude groundnuts and maize with > 10 µg/kg total
aflatoxin;
Scenario 3: exclude groundnuts and maize with > 15 µg/kg total
aflatoxin;
Scenario 4: exclude groundnuts and maize with > 20 µg/kg total
aflatoxin).
This analysis was conducted using the average aflatoxin levels
that would result under each scenario and was repeated using only the
data sampled in the USA and only the European data (for total
aflatoxins and only the data in Europe and China for aflatoxin B1).
The range of estimated intakes of total aflatoxin from maize is
shown in Table 12. The estimated range of intakes is 2-501 ng total
aflatoxin/person perday. These data should not be used as true
estimates of likely intake but rather as a measure of the relative
impact of establishing limits. Thus with an upper limit of 20 µg/kg
aflatoxin, the estimated intake using the European WHO regional diet
and European monitoring data would be 2 ng/person per day. There would
be no impact in establishing a limit since there are no European maize
data with levels > 5 µg/kg. Using the USA monitoring data and the
European WHO diet (which includes North America) the intake would be
47 ng/person per day with no limit, 9 ng/person with a 20 µg/kg limit
and 7 ng/person per day with a 15 µg/kg limit. A limit of 10 ng/person
per day would lower the estimated intake to 6 ng/person per day (using
the USA monitoring data). This exercise is repeated using the other
four WHO regional diets and either the European or USA maize/cereal
monitoring data.
Table 10. Distribution of estimated concentrations of total
aflatoxin in maize and groundnuts sampled in the USA and Europe1
Aflatoxin USA maize European European USA
(µg/kg) cereals groundnuts groundnuts
percentiles
0.5 87.4% 91.5% 44.2% 89.1%
1 88.0% 94.7% 81.8% 89.1%
2.5 88.9% 99.2% 88.8% 90.3%
5 90.6% NA 91.7% 92.0%
7.5 92.1% NA 93.8% 92.6%
10 93.8% NA 94.9% 93.0%
12.5 94.2% NA 95.1% 93.4%
15 95.0% NA 95.3% 93.7%
17.5 95.7% NA 95.5% 94.0%
20 96.1% NA 95.8% 94.1%
30 96.8% NA 96.4% 94.6%
40 97.6% NA 96.8% 94.7%
50 98.0% NA 97.4% 94.8%
1 European cereals include other crops in addition to maize
From: Chen (1997) and Wood (1995)
Table 11. Distribution of estimated concentrations of aflatoxins B1
in maize and groundnuts sampled in Europe1 and China
Aflatoxin B1 European Chinese European Chinese
levels (µg/kg) cereals maize groundnuts groundnuts
0.5 41.6% 7.5% 56.9% 8.3%
1 66.2% 16.3% 89.8% 17.3%
2.5 83.4% 40.4% 93.1% 43.0%
5 89.3% 80.9% 96.4% 88.0%
7.5 94.8% 83.5% 96.7% 89.8%
10 NA 86.5% 97.1% 90.6%
12.5 NA 88.2% 97.2% 90.9%
15 NA 89.5% 97.4% 91.3%
17.5 NA 90.7% 97.6% 91.8%
20 NA 91.7% 97.9% 92.1%
30 NA 92.7% 98.3% 93.4%
40 NA 94.4% 98.5% 94.7%
50 NA 96.0% 98.9% 95.8%
1 European cereals include other crops in addition to maize.
The range of estimated intakes of total aflatoxin from groundnuts
was 2-162 ng/person per day (Table 12). However, these data should not
be used as true estimates of likely intake. Rather they should be used
as a measure of the relative impact of establishing limits. Thus, for
example, establishing a programme that successfully limits aflatoxin
levels to 20 µg/kg would reduce the estimated intake of aflatoxin from
groundnuts to 5 ng per day from 66 ng/person per day (European diet).
Likewise, reducing aflatoxin levels to no more than 15 µg/kg limit
would maintain the estimated intake at 5 ng/person per day (Table 12).
Similarly, the estimated intake using the European monitoring data and
a 10 µg/kg limit would be 4.4 ng/person per day. Similar comparisons
are shown in Table 12 for each of the regional diets combined with
either the European or USA aflatoxin monitoring results.
4.4.4.3 Intake of aflatoxin B1 within four scenarios
These analyses were repeated for aflatoxin B1 using the results of
the European and Chinese monitoring results. The results are presented
in Table 13.
Table 12. Estimated intake of total aflatoxin under 4 different scenarios
Scenario 1: no samples excluded
Scenario 2: samples >10 µg/kg excluded
Scenario 3: samples >15 µg/kg excluded
Scenario 4: samples >20 µg/kg excluded
I. FOOD CONSUMPTION ESTIMATES (GEMS/Foods, World Health Organization) + 431
Middle Eastern Far Eastern African Latin American European
(g/person per day) (g/person per day) (g/person per day) (g/person per day) (g/person per day)
Maize 50 31 106 42 10
Groundnuts 0.3 6 11.3 2 5
II. MEAN TOTAL AFLATOXIN RESIDUE CONCENTRATIONS UNDER EACH SCENARIO (from Table 6)
A. MAIZE/CEREALS Mean residues
European monitoring data USA monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 0.2 4.7
Scenario 2: samples >10 µg/kg excluded 0.2 0.6
Scenario 3: samples >15 µg/kg excluded 0.2 0.7
Scenario 4: samples >20 µg/kg excluded 0.2 0.9
B. GROUNDNUTS Mean residues
European monitoring data USA monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 13 14
Scenario 2: samples >10 µg/kg excluded 0.9 0.4
Scenario 3: samples >15 µg/kg excluded 0.9 0.5
Scenario 4: samples >20 µg/kg excluded 1.0 0.6
Table 12. Continued...
III. ESTIMATED DIETARY INTAKE OF AFLATOXIN (TOTAL) UNDER THE FOUR SCENARIOS AND DIFFERENT RESIDUE DATA SETS
A. TOTAL AFLATOXIN INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 10 6 21 8.4 2
Scenario 2: samples >10 µg/kg excluded 10 6 21 8.4 2
Scenario 3: samples >15 µg/kg excluded 10 6 21 8.4 2
Scenario 4: samples >20 µg/kg excluded 10 6 21 8.4 2
B. TOTAL AFLATOXIN INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN THE USA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 240 150 500 200 47
Scenario 2: samples >10 µg/kg excluded 30 19 64 25 6
Scenario 3: samples >15 µg/kg excluded 35 22 74 29 7
Scenario 4: samples >20 µg/kg excluded 45 28 95 38 9
GROUNDNUTS
A. TOTAL AFLATOXIN INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 39 78 150 26 65
Scenario 2: samples >10 µg/kg excluded 0.3 3.6 10 1.8 4.5
Scenario 3: samples >15 µg/kg excluded 0.3 3.6 10 1.8 4.5
Scenario 4: samples >20 µg/kg excluded 0.3 6.0 11 2.0 5.0
(ng total
Table 12. Continued...
B. TOTAL AFLATOXIN INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN THE USA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 4.2 84 160 28 70
Scenario 2: samples >10 µg/kg excluded 0.12 2.4 4.5 0.8 2.0
Scenario 3: samples >15 µg/kg excluded 0.15 3.0 5.7 1.0 2.5
Scenario 4: samples >20 µg/kg excluded 0.18 3.6 6.8 1.2 3.0
Table 13. Estimated intake of aflatoxin B1 under 4 different scenarios with 2 residue datasets
Scenario 1: no samples excluded
Scenario 2: samples >10 µg/kg excluded
Scenario 3: samples >15 µg/kg excluded
Scenario 4: samples >20 µg/kg excluded
I. FOOD CONSUMPTION ESTIMATES (GEMS/Foods, World Health Organization)
Middle Eastern Far Eastern African Latin American European
(g/person per day) (g/person per day) (g/person per day) (g/person per day) (g/person per day)
Maize 50 31 106 42 10
Groundnuts 0.3 6 11.3 2 5
II. MEAN AFLATOXIN B1 RESIDUE CONCENTRATIONS UNDER EACH SCENARIO (from Table 7)
A. MAIZE/CEREALS Mean residues
European monitoring data Chinese monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 1.6 12
Scenario 2: samples >10 µg/kg excluded 1.6 2.8
Scenario 3: samples >15 µg/kg excluded 1.6 3.1
Scenario 4: samples >20 µg/kg excluded 1.6 3.5
B. GROUNDNUTS Mean residues
European monitoring data Chinese monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 6.9 8.3
Scenario 2: samples >10 µg/kg excluded 0.6 2.7
Scenario 3: samples >15 µg/kg excluded 0.6 2.7
Scenario 4: samples >20 µg/kg excluded 0.7 2.9
Table 13. Continued...
III. ESTIMATED DIETARY INTAKE OF AFLATOXIN (B1) UNDER THE FOUR SCENARIOS AND DIFFERENT RESIDUE DATASETS
A. AFLATOXIN B1 INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 75 46 160 63 15
Scenario 2: samples >10 µg/kg excluded 75 46 160 63 15
Scenario 3: samples >15 µg/kg excluded 75 46 160 63 15
Scenario 4: samples >20 µg/kg excluded 75 46 160 63 15
B. AFLATOXIN B1 INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN CHINA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 600 370 1270 500 120
Scenario 2: samples >10 µg/kg excluded 140 87 300 120 28
Scenario 3: samples >15 µg/kg excluded 160 96 330 130 31
Scenario 4: samples >20 µg/kg excluded 180 108 370 150 35
GROUNDNUTS
A. AFLATOXIN B1 INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 2.1 41 78 14 34
Scenario 2: samples >10 µg/kg excluded 0.2 3.6 6.8 1.2 3.0
Scenario 3: samples >15 µg/kg excluded 0.2 3.6 6.8 1.2 3.0
Scenario 4: samples >20 µg/kg excluded 0.2 4.2 7.9 1.4 3.5
Table 13. Continued...
B. AFLATOXIN B1 INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN CHINA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 2.5 50 94 17 42
Scenario 2: samples >10 µg/kg excluded 0.8 16 30 5.4 14
Scenario 3: samples >15 µg/kg excluded 0.8 16 30 5.4 14
Scenario 4: samples >20 µg/kg excluded 0.9 17 33 5.8 15
4.4.5 Summary
The values for aflatoxin levels presented above are not considered
to be representative of the food supply in any country nor of the
commodities moving in international trade. Quantitative estimates of
intake of aflatoxin at the international level are severely limited by
the lack of representative data. Although intake estimates are
available at the national level for many countries, the submitters of
all of these studies are emphatic in stating that the results are not
truly "representative." In general, the results appear to be biased
upwards because monitoring studies focus on lots of commodity that are
thought to be contaminated.
However, the data do provide sufficient information to evaluate
the likely impact of limiting aflatoxin levels in foodstuffs. Of the
scenarios considered, the greatest relative impact on estimated
average aflatoxin levels is achieved by establishing a programme that
would limit aflatoxin contamination to less than 20 µg/kg. This
assumes that the controls would successfully exclude all samples
containing aflatoxin above that limit. Depending upon assumptions
regarding the distribution of residues, some small incremental
reductions can be achieved by limiting aflatoxin levels to no more
than 15 or 10 µg/kg, respectively.
Additional data that would better simulate the actual aflatoxin
levels in foods moving in international trade would provide much more
accurate estimates of intake. Most likely these estimates would result
in lower estimates of the average intake since the available data
appear to be biased upwards. The incorporation of data on the effects
of food processing on aflatoxin levels would also improve the accuracy
of estimates of intake since aflatoxin is removed during many
procedures.
5. COMMENTS AND EVALUATION
The aflatoxins are among the most potent mutagenic and
carcinogenic substances known. Extensive experimental evidence in test
species shows that aflatoxins are capable of inducing liver cancer in
most species studied. In addition, most epidemiological studies show a
correlation between exposure to aflatoxin B1 and increased incidence
of liver cancer. Aflatoxins are metabolized in humans and test species
to an epoxide, which usually is considered to be the ultimate reactive
intermediate. There is some evidence suggesting that humans are at
substantially lower risk to aflatoxins than test species. The
Committee was aware of epidemiological studies suggesting that intake
of aflatoxin poses no detectable independent risk and studies that
suggest it poses risks only in the presence of other risk factors such
as hepatitis B infection. Several ongoing studies are likely to
improve further the estimates of human risks from the intake of
aflatoxin, most notably cohort studies in Shanghai, Thailand and
Qidong and hepatitis B vaccination trials in The Gambia, Taiwan and
Qidong. When these studies are complete, the Committee may want to
reevaluate the risks of aflatoxins in humans.
A number of factors influence the risk of primary liver cancer,
most notably carriage of hepatitis B virus as determined by the
presence in serum of the hepatitis B surface antigen (presence denoted
HBsAg+ and absence denoted HbsAg-). The potency of aflatoxins
appears to be significantly enhanced in individuals with simultaneous
hepatitis B infection. This interaction makes it difficult to
interpret the epidemiological studies in the context of aflatoxin as
an independent risk factor. The conclusions of the Committee regarding
aflatoxin potency therefore are contingent upon the dynamics of
hepatitis B infection in a human population.
The identification of hepatitis C virus is an important recent
advance in understanding the etiology of liver cancer. Two studies
have investigated interactions between hepatitis C infection,
aflatoxins and liver cancer; the evidence so far is inconclusive. It
is estimated that 50 to 100% of liver cancer cases are associated with
persistent infection with hepatitis B and/or hepatitis C.
The Committee considered that the weight of scientific evidence,
which includes epidemiological data, laboratory animal studies and
in vivo and in vitro metabolism studies, supports a conclusion
that aflatoxins should be treated as carcinogenic food contaminants,
the intake of which should be reduced to levels as low as reasonably
achievable.
5.1 Aflatoxin potencies
The Committee reviewed dose-response analyses that have been
performed on aflatoxins. All of these analyses suffer limitations,
three of which predominate. First, all of the epidemiological data
from which a dose-response relationship can be developed are
confounded by concurrent hepatitis B infection. The epidemiological
data are from geographical areas where both the prevalence of HBsAg+
individuals and aflatoxins are high; the relationship between these
risk factors in areas of low aflatoxin contamination and low hepatitis
B prevalence is unknown. Second, the reliability and precision of the
estimates of aflatoxin exposure in the relevant study populations are
unknown. For example, aflatoxin biomarkers in humans do not reflect
long-term aflatoxin intake; analysis of crops for aflatoxins do not
reflect levels of aflatoxins consumed in foods after selection and
processing. Finally, the shape of the dose-response relationship is
unknown, which introduces an additional element of uncertainty when
choosing mathematical models for interpolation.
Observations concerning the interaction of hepatitis B and
aflatoxins suggest two separate aflatoxin potencies in populations in
which chronic hepatitis infections are common versus populations in
which chronic hepatitis infections are rare. In analyses based on
toxicological and epidemiological data, potency estimates for
aflatoxin were divided into two basic groups, potencies applicable to
individuals without hepatitis B infection and those applicable to
individuals with chronic hepatitis B infection. The Committee found
these estimates useful even though, through the use of differing
mathematical models, they covered a broad range of possible values
(Figure 2). Epidemiological data for which hepatitis B infection
status was unknown and for which potencies were calculated were also
reviewed and found to be in the range of potencies for hepatitis B
infected/non-infected individuals. The review also considered the
extrapolation of animal data to estimate potency in humans; these also
generally fell within the range of the potency estimates derived from
the epidemiological data.
Some discussion is warranted on the potential biases in the
potencies depicted in Figure 2: (i) only studies showing a positive
association between aflatoxins and liver cancer were used, as opposed
to considering all studies (positive as well as negative), leading to
overestimation of the aflatoxin potency; (ii) by relating current
levels of intake (i.e. using biomarkers or dietary surveys) to current
levels of liver cancer (presumably with a long induction period),
historical levels of intake are ignored; they are likely to have been
higher, in which case aflatoxin potency will be overestimated; (iii)
the earliest studies systematically underestimated hepatitis B
prevalence in cases of liver cancer by a factor as high as 20-30%,
owing to limitations in the methodology used to detect hepatitis B,
which also leads to an overestimate of the relative potency of any
other factor, including aflatoxins; (iv) histological confirmation of
the liver cancer cases is limited in most epidemiological studies,
allowing the possibility that non-primary liver cancer cases have been
included, which could lead to an underestimation or overestimation of
the aflatoxin potency. Considering these biases, the values in Figure
2 should be viewed as overestimates of the potency of the aflatoxins,
leading to the hypothesis that it is possible that humans are in fact
less sensitive to aflatoxins than the species tested in laboratory
experiments.
The Committee reviewed the extensive data available on the
metabolism of aflatoxins in various species. It was agreed that
differential potency to aflatoxins between species can be partially
attributed to differences in metabolism. However, there is at the
present time insufficient quantitative information available about
competing aspects of metabolic activation and detoxification of
aflatoxin B1 in various species to identify an adequate animal model
for humans and to explain the apparent species differences in potency.
Intake assessments used in many of the epidemiological studies
ignored the contributions to total aflatoxin intake through milk and
milk products. Thus, the potencies shown in Figure 2 do not generally
apply to aflatoxin M1. From one comparative toxicity study in rats,
it is possible to estimate that aflatoxin M1 has a potency
approximately one order of magnitude less than that of aflatoxin B1
in this species.
The Committee reviewed the potencies estimated from the positive
epidemiological studies and chose separate central tendency estimated
potencies and ranges for HBsAg+ and for HBsAg- individuals. Potency
values of 0.3 cancers/year per 100 000 population per ng aflatoxin/kg
bw per day with an uncertainty range of 0.05 to 0.5 in HBsAg+
individuals and of 0.01 cancers/year per 100 000 population per ng
aflatoxin/kg bw per day with an uncertainty range of 0.002 to 0.03 in
HBsAg- individuals were chosen.
5.2 Population risks
The fraction of the incidence of liver cancer in a population
attributable to intake of aflatoxins is derived by combining aflatoxin
potency estimates (risk per unit dose) and estimates of aflatoxin
intake (dose per person). The Committee reviewed the frequency and
amount of aflatoxin contamination in a variety of products (e.g.,
groundnuts, cereals and maize) in numerous countries (e.g., China,
Denmark, Italy and the USA). Many of the data on contamination levels
were derived from non-random samples, which appeared to be biased
upwards because monitoring studies focus on lots of commodities that
are thought to be contaminated. Some of the data on contaminant levels
are unlikely to be based on current Codex sampling recommendations for
aflatoxins. These contamination levels can only be used with caution
to infer patterns of importance in setting standards and not to
provide exact contamination estimates.
Through the use of hypothetical standards, it was noted that the
magnitude of the difference between two hypothetical standards is
substantially larger than the magnitude of the difference in the mean
contamination levels resulting from the separate standards. This point
is illustrated in Figure 3 in which the derived distribution of
aflatoxin contamination in maize in the USA is shown. Application of a
hypothetical 20 µg/kg standard would result in rejection of 4% of the
maize crop and a mean aflatoxin level in maize of 0.9 µg/kg. Imposing
the stricter hypothetical standard of 10 µg/kg would result in
rejection of 6.2% of the samples to achieve a drop in the mean
aflatoxin contamination level by 0.3 µg/kg to 0.6 µg/kg. Similar
results were obtained when examining aflatoxin B1 levels in maize and
also for total aflatoxins or B1 alone in groundnuts.
Using the Global Environment Monitoring System - Food
Contamination Monitoring and Assessment Programme (GEMS/Food) regional
diets combined with contamination levels, the Committee was able to
provide relative estimates of mean dietary intake of aflatoxins for
various regions under differing standard dietary choices. Linking
these intakes to the potencies shown in Figure 2 allows for the
calculation of overall population risks based upon the prevalence of
hepatitis B infection in various regions.
4. AFLATOXIN DIETARY INTAKE ESTIMATES
4.1 Introduction
This report summarizes the results of monitoring and available
national estimates of intake of aflatoxins in order to provide a
framework for the task of estimating increments in intake of
aflatoxins. Estimates are based on the results of available monitoring
data. Total aflatoxin intake based on the GEMS/FOODS regional diets
are used to evaluate the impact of four different scenarios: no limit,
and limits set at 20, 15 and 10 µg/kg. This evaluation was conducted
for ground-nuts and for maize for total aflatoxins and for aflatoxin
B1 alone. Generally they are not considered by the submitters to be
representative because sampling has focused on those lots that are
more likely to contain the highest levels of aflatoxin. However, this
analysis provides useful qualitative comparisons between regulator
options.
4.2 Background
Aflatoxins are found as contaminants in human and animal food as a
result of fungal contamination both pre- and post-harvest, with the
rate and degree of contamination being dependent on temperature,
humidity, soil and storage conditions. Though a wide range of foods
may be contaminated with aflatoxins, they have been most commonly
associated with groundnuts (groundnuts and groundnut products), dried
fruit, tree nuts, spices, figs, crude vegetable oils, cocoa beans,
maize (maize), rice, cottonseed and copra. There are practices that
reduce but do not completely eliminate aflatoxins in grains,
groundnuts, figs and other crops. However, even in the most tropical
of climates, many lots of these crops do not contain detectable levels
of aflatoxins.
4.3 Methods
4.3.1 Period of intake of relevance
Chronic (lifetime) intake is assumed to be the period of
relevance. Therefore, the average concentrations in the diet are of
primary interest.
4.3.2 Estimated levels of aflatoxins in foodstuff
Data were available for this analysis from at least one country on
every continent. Typically the data were uniformly judged by the
submitters not to be representative. In most instances, the data were
thought to be biased towards the upper end of intake. Nonetheless,
caution must be exercised in using the data to generate intake
estimates and in interpreting the results of such analyses. However,
the data did provide a framework to explore the relative impact of
regulatory activities. The data are summarized in Table 5.
For some of the analyses used in this paper individual data points
were required in order to generate distributions and to evaluate the
impact of imposing upper limits on aflatoxins in foodstuffs.
Specifically, a series of analyses was conducted to determine the
impact of truncating the distribution at 10, 15 and 20 µg/kg,
respectively, in order to simulate the potential impact of proposed
limits. For these analyses, data reported by the USA, China and Europe
were evaluated because the raw data were available. These analyses
were conducted for maize and groundnuts for total aflatoxins and for
aflatoxin B1.
4.3.3 Estimated intakes
Four pieces of information are required to estimate the potential
intakes due to aflatoxins in crops that are imported: (1) the levels
of aflatoxin in imported crops; (2) the amount of each imported crops
consumed; (3) the impact of any subsequent processing on aflatoxins
levels; and (4) methods for combining the first 3 to estimate intake.
Table 5. Summary of aflatoxin monitoring data submitted for consideration by the Committee
Commodity Country/Region Number of samples Results Comments Reference
NUTS
Groundnuts
(groundnuts and
groundnut products) Australia 9 samples Mean = 2 µg/kg; Australia Market
Max = 10 µg/kg Basket (1992)
913 lots 83.1% <5 µg/kg presorted Read (1997)
(b1) 14.2% 5-10 µg/kg
2.7% 10-15 µg/kg
Brazil 199 samples Mean = 14 µg/kg Sabino (1997)
(b1) Max = 181 µg/kg
51%> LOD
China 174 groundnuts 3 >30 µg/kg selected to be worst Chen (1997)
40 groundnut meal 1 >251 µg/kg case for research
(b1) (see Table 3) purposes
Cuba 1114 samples 49% >LOD Regueiro
(b1) (no date)
European Union data from 12 Mean B1 35-64 µg/kg data submitters SCOOP (1996)
countries (country means) emphatically state that
Max = 789 µg/kg DATA ARE NOT
(see Tables 3 and 4) REPRESENTATIVE
Japan 22 789 samples >97% >LOD imported from >25 Japanese Ministry
(1972-1989) 238 >10 µg/kg (B1) countries of Health (1995)
Max = 8070 µg/kg
Mexico 107 samples 104 <20 µg/kg Mexico (1996)
(1992-1995) 3 >20 µg/kg
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Nicaragua 9 samples (1996) 1 >15 µg/kg (B1) Nicaragua (1996)
USA >600 000 lots >90% <15 µg/kg Wood (1995)
(1975-1992) (total); see Table 6
Zimbabwe 286 samples 39% <5 µg (1995) analysed B1 and total Zimbabwe Government
(1995-1996) 54% <10 µg (1995) Analyst Laboratory
46% >10 µg (1995) (1995-1996)
85% <5 µg (Jan-Jun 96)
92% <10 µg/kg
8% >D12 10 µg/kg+D24
Brazil nuts European Union not available Max = 15 µg/kg (B1) see comment under SCOOP (1996)
Max = 35 µg/g (total) groundnuts
Japan 74 samples 70 <LOD Japanese Ministry
2 >10 µg/kg of Health (1995)
2 >10 µg/kg
Max = 123 µg/kg
Pistachio nuts European Union 11 countries Mean = 2-23 µg/kg
provided data (B1; country means)
Mean = 44-27 µg/kg
(total; country means)
Max = 450 µg/kg (B1)
Max = 813 µg/kg (total)
Japan 2422 samples 2339 <LOD (B1) Japanese Ministry
(1972-1989) 48 >10 µg/kg of Health (1995)
35 <10 µg/kg
Max = 8030 µg/kg
Mexico 244 samples 5 samples >20 µg/kg Mexico (1996)
(1993-1996)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Sunflower seeds Argentina 20 samples no detects Argentina (1996)
Almonds Australia 9 samples no detects Australian Market
Basket Survey (1992)
Japan 93 749 samples >98% <LOD reported all Japanese Ministry
Max = 128 µg/kg miscellaneous nuts of Health (1995)
in one statistics;
covered years 1972-
1989; all were imported
Cashews Japan 1227 samples >98% <LOD Japanese Ministry
of Health (1995)
Walnuts Japan 321 samples >98% <LOD Japanese Ministry
of Health (1995)
Macadamia nuts Japan 149 samples >98% <LOD Japanese Ministry
of Health (1995)
Hazel nuts Japan 103 samples >98% <LOD Japanese Ministry
of Health (1995)
FIG PRODUCTS
European Union Mean = 0.5-26 µg/kg see comment for SCOOP (1996)
(country mean B1) groundnuts
CEREAL & CEREAL PRODUCTS
rice, wheat, maize
Bolivia number unknown 2 rice 25-168 µg/kg (B1)
(1992-1995) 1 wheat 10 µg/kg (B1)
1 wheat 18 µg/kg (B1 + g1)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
2460 samples
(1986-1997) 1273 >LOD
Mean = 34 µg/kg
(range = 7-144 µg/kg)
Maize Brazil 321 samples 179 <LOD Sabino (1997)
27 <30 µg/kg
116 >30 µg/kg
(B1 or total not reported)
Max = 2440 µg/kg
Maize Brazil 2546 samples Mean = 35 µg/kg
(51% >LOD)
Rice Brazil 401 samples Mean = 2 µg/kg
(10% >LOD)
Wheat Brazil 237 samples Mean = 2 µg/kg
(19% >LOD)
Malt Brazil 30 samples Mean = 30 µg/kg
Popcorn Brazil 32% >LOD
Sorghum Brazil 59 samples Mean = 3 µg/kg
(33% >LOD)
Maize China 486 samples Mean 5-251 µg/kg
(117 >LOD) Chen (1997)
Wheat China 597 samples Max = < 31 µg/kg
(9 >LOD) see also Table 7 Chen (1997)
Sorghum China 58 samples 1 >LOD Chen (1997)
Rice China 747 samples 7 >LOD Chen (1997)
Sorghum Columbia 45 samples 11 >LOD (B1) Diaz (1996)
(1995-1996) (1.4-43 µg/kg)
Mean = 11 µg/kg
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Maize Columbia 33 samples 4 >LOD (B1) Diaz (1996)
(4-66 µg/kg)
Mean = 21 µg/kg
Soybeans Columbia 25 samples 0 >LOD (B1) Diaz (1996)
Rice Columbia 22 samples 8 >LOD (B1) Diaz (1996)
(1-53 µg/kg)
Mean = 21 µg/kg
Cotton Columbia 17 samples
(1995-1996) 15 >LOD (B1) Diaz (1996)
(2-11 µg/kg)
Mean = 5 µg/kg
Maize Costa Rica 49 samples 1 >15 µg/kg (B1) Pacin (no date)
48 <10 µg/kg
Maize Cuba 4620 samples 20% >LOD (B1) Regueiro (no date)
Rice Cuba 340 samples 16% >LOD Regueiro (no date)
Sorghum Cuba 12% >LOD Regueiro (no date)
Wheat Cuba 1% >LOD Regueiro (no date)
CEREAL PRODUCTS
European Union 0.1-7.6 µg/kg see comment for SCOOP (1996)
(country means B1) groundnuts
0.25-5.9 µg/kg
(country means total)
(see Tables 6 and 7)
Maize Japan 371 samples 16 >LOD Japanese Ministry
Max B1 = 1.5 µg/kg of Health (1995)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
Maize Mexico 1710 samples 265 >20 µg/kg Mexico (1996)
(1992-1996) 35 tortilla samples
>20 µg/kg
Maize Thailand 18 samples <LOD - 606 µg/kg Aflatoxin was visible Yoshizawa et al.,
1996
PULSES
Soybeans Brazil 143 samples 17% >LOD
Mean = 1 µg/kg
Soybeans China 388 samples 11 >LOD Jiangu Province Chen (1977)
(51-100 µg/kg)
Soy sauce China 308 samples 4.6% >LOD Jiangu Province Chen (1977)
Bean paste China 1 sample <100 µg/kg Jiangu Province Chen (1977)
3.3% > LOD
Max = >251 µg/kg
Soy oil China 379 samples 10 samples >LOD Jiangu Province Chen (1977)
Max = <251 µg/kg
Soybean meal China 6.7% >LOD Jiangu Province Chen (1977)
Max = <50 µg/kg
Frijoles (beans) Cuba 413 samples 13% >LOD (B1) Regueiro (no date)
Pulses Japan >2000 samples 2 samples >LOD (B1) Japanese Ministry
Max = <10 µg/kg of Health (1995)
Table 5. Continued...
Commodity Country/Region Number of samples Results Comments Reference
SPICES
European Union Max B1 = 323 µg/kg reported in at SCOOP (1996)
(0.5-17 µg country least one sample
mean levels) of every type of
Max total = 72 µg/kg spice analysed- see
comment re: data
under groundnuts
Japan 1804 >LOD in chilli, nutmeg, none found in 25 Japanese Ministry
(1979-1988) white pepper, paprika, other spices of Health (1995)
turmeric, cardamon,
pimento, ginger, celery
seed spices
CONFECTIONARY PRODUCTS
few samples 0.05 = - 1.1 µg/kg SCOOP (1996)
(country means B1)
FATS, OILS, OILSEEDS
France 56 samples Mean 4.5 µg/kg (France)
Max = <25 µg/kg
MILK AND MILK PRODUCTS
see text
a) National intakes
Available national intake studies were reviewed to provide
estimates of intake levels. This information was obtained from reports
submitted by members of the EU (SCOOP, 1996), China (Chen, 1997), USA
(1992), Brazil, Australia, Costa Rica, Argentina, Mexico, Nicaragua,
Colombia and Thailand. Thirteen European countries provided data for
the EU SCOOP project.
b) International intakes
The available data provide a general idea of the range of
aflatoxin levels in foodstuffs and the frequency of detection.
Information on the proportion of the crop imported was not available
for most countries, nor was it possible to determine the distribution
of aflatoxin levels in imported crops versus domestic crops.
Therefore, the data from individual countries were reviewed to obtain
a general overview of the likely levels in foods and to determine the
impact on average intakes if extreme levels of aflatoxins could be
removed from foodstuffs.
Impact on dietary intake if upper concentrations of aflatoxin
in foodstuffs are successfully limited to 10, 15 or 20 µg/kg
foodstuff versus no limit
It was assumed that methods are available to ensure that aflatoxin
levels above the specified limit are excluded from the food supply and
that the same proportion of the commodity would be imported (versus
domestic) regardless of the limit.
For these analyses all of the food consumed in the country was
assumed to contain the average residue concentration under that
scenario. Each analysis was repeated twice: (1) using European
monitoring data (b1 and total aflatoxin; and (2) using either Chinese
monitoring data (aflatoxin b1) or USA monitoring data (total
aflatoxin). Using these assumptions, the GEMS/FOODS regional diets
were used to evaluate the difference in dietary intake to aflatoxin
from groundnuts and maize under different limits.
c) Amounts consumed
The GEMS/FOODS regional diets were used as estimates of the
consumption of each of the commodities (Tables 4 and 5).
4.4 Results
4.4.1 Aflatoxin levels in foods: general
The 1995 FAO compendium, Worldwide regulations for mycotoxins
(FAO, 1995), summarized reports from 48 countries. The data submitted
by 33 countries for aflatoxin B1 and total aflatoxins (B1,B2,G1,G2)
were used to estimate median levels of 4 and 8 µg/kg, respectively, in
foodstuffs. The range of levels reported for B1 was from 0 to 30 µg/kg
and for total from 0 to 50 µg/kg. Seventeen countries provided
information on aflatoxin M1 in milk with a median of 0.05 µg/kg and a
range of 0-1 µg/kg. The FAO report did not provide additional details
about sampling, treatment of non-detects, imported versus domestic
foodstuffs, etc.
Reports from individual countries that were submitted to FAO and
to JECFA have been used to provide additional detail. The data are
summarized in Table 5 by commodity and by country.
The participants in the European Union Scientific Co-operation
Assessment of aflatoxin (SCOOP) reviewed data submitted by member
countries and by Norway. The participants concluded that the results
were unlikely to be representative and should not be used to estimate
total aflatoxin intake for individual countries or for Europe.
However, the studies did provide some insight into issues surrounding
aflatoxin intake assessments. Based on the data and subsequent
discussions, SCOOP concluded: (1) aflatoxins are found in a broader
range of foods than had been previously assumed; (2) most of the
samples did not contain detectable aflatoxin; (3) sampling methods are
important in accurately estimating aflatoxin levels; and (4) different
methods of collecting food consumption data may make a difference in
estimating aflatoxin intakes.
4.4.2 Aflatoxin levels in foodstuffs: Occurrence data by commodity
Most countries provided data for aflatoxin B1 in selected crops.
Some countries also provided an estimate of the level of total
aflatoxins. Total aflatoxin estimates typically included aflatoxin B1,
B2 and G. M1 was the common aflatoxin reported for milk and milk
products. The data are summarized in Table 1 for crops.
The data for aflatoxin M1 are summarized below:
Australia: No aflatoxins were detected in 34 whole milk samples.
European Union: Ten countries reported results of sampling for M1
in milk. The maximum level of 0.37 µg/kg M1 was reported by France.
The United Kingdom reported the highest level (0.22 µg/kg) in cheese.
Brazil: 204 samples of milk, cheese and yoghurt were analysed. Of
these only four samples of pasteurized milk contained detectable
levels of aflatoxin M1 (de Sylos, 1996). The levels ranged from 73-370
ng/litre.
Spain: Aflatoxin M1 was estimated in 19 total diet studies in 1990
and 1991. All but one sample was below the limit of detection. The one
sample contained 0.025 µg/kg.
4.4.2.1 Amount of commodity imported
The proportion of any commodity that is imported varies from 0 to
100%. For example, in the case of groundnuts all groundnuts, are
imported into the Nordic countries while virtually no groundnuts are
imported into China and the USA. If import/export data were available,
aflatoxin intakes could be more accurately estimated. However, this
information was not available for most countries.
4.4.2.2 Accounting for the change in aflatoxin levels during
processing
a) Maize
Processing of maize causes reduction in aflatoxin levels. Wet
milling reduces the concentration of aflatoxin in maize starch to 1%
of the levels found in the raw grain (Yahl, 1971). Similarly dry
milling reduces aflatoxin in food products (grits, low-fat meal and
low-fat flour) to 6-10% of the original concentrations (Brekke, 1975).
b) Groundnuts
The roasting of groundnuts reduces aflatoxin levels by 50-80%
(Waltking, 1971; Read, 1989; Billy, 1996).
c) Milk
Aflatoxin M1 is a metabolite of aflatoxin B1 and is found
associated with the casein in milk. Pasteurization does not affect the
level of aflatoxin M1 in milk or yogurt (Wiseman, 1983). Aflatoxin M1
has been reported to concentrate 3-6 fold during cheese making (Van
Egmond, 1983). MAFF (1994-5, No. 22) reported that aflatoxin was not
destroyed under domestic cooking conditions (microwave or heading in
gas oven).
The effects of processing were considered in many of the national
estimates. Processing factors have not been included in any of the
distributions generated for the estimates of aflatoxin intake.
However, it would be appropriate to adjust further the international
estimates of intake to reflect the impact of processing where the
commodity is always processed/cooked prior to consumption.
4.4.3 National estimates of aflatoxin intake
4.4.3.1 Australia
Australia conducts market basket surveys and estimates intake for
average and extreme consumers. The average diet was estimated to
contain 0.15 ng aflatoxin/kg bw per day and the upper 95th percentile
diet to contain approximately twice that level. Children's diets were
estimated to be somewhat higher - up to approximately 0.45 ng/kg bw
per day for the 95th percentile 2-year-old (Australia Market Basket
Survey, 1992).
4.4.3.2 China
A series of intake and market basket studies have been conducted
since 1980 to estimate the aflatoxin B1 intake. The reported intakes
have ranged from 0 to 91 µg aflatoxin B1/kg bw per day (Chen, 1997).
4.4.3.3 European Union
Estimates of aflatoxin intake were provided to the EU SCOOP
project by 9 countries. However, it should be noted that in every
instance, it was clearly stated that these estimates were not
representative. They were regarded as being no more than indicators of
intake of aflatoxin and as clearly not useful for predictive purposes.
These indicators of intake ranged from 2 to 77 ng/person per day for
aflatoxin B1 and from 0.4 to 6 ng/person per day for aflatoxin M1.
Although these may be useful to guide understanding of potential
intake, they are not to be used as an estimate of intake either for a
particular country or for Europe.
Of the countries that computed intakes of M1, the computed intakes
ranged from 0.04 µg/kg bw per day M1 (France, Germany) to 0.19 ng/kg
bw per day (Netherlands). The value reported by the Netherlands was
from baby food. In addition, the Netherlands also estimate 0.04 ng/kg
bw per day M1 from milk.
France provided additional information on intakes from cereals,
nuts, spices and milk. Two estimates of aflatoxin intake were
computed. The first was the product of the maximum level of aflatoxin
reported and the maximum consumption for each category of food. The
second estimate was the product of the average aflatoxin concentration
reported and the maximum consumption. The resulting estimates are
given in Table 6.
Table 6. Estimated aflatoxin intake in France (µg per day)
Maximum Mean
Cereal 13.33 2.42
Nuts 7.13 0.04
Spices 0.68 0.01
Milk 0.12 0.06
4.4.3.4 USA
The US FDA estimated intakes using data from the National
Compliance program for maize, groundnut and milk products using Monte
Carlo simulation procedures. The data were from the 1980s. The
eaters-only mean lifetime intake of total aflatoxin was 18 ng/person
per day and intake for the 90th percentile individuals was 40
ng/person per day. Mean aflatoxin M1 intake was 44 ng/person per day
and for the 90th percentile individuals 87 ng/person per day. The
authors noted that many assumptions were made that bias these
estimates upwards. The same analysis was repeated in 1992 with only
slightly different results (DiNovi, 1992).
4.4.3.5 Zimbabwe
The theoretical maximum intake of aflatoxin B1 was estimated for a
child's diet containing 150 grams maize with 5 µg/kg aflatoxin B1 and
30 grams ground-nuts with 10 µg/kg aflatoxin B1. The total aflatoxin
intake per day would be 1.05 µg per day if all maize consumed
contained 5 µg/kg and all groundnuts contained 10 µg/kg. If all maize
were to contain 15 µg per day the intake would be 2.55 µg per day.
4.4.4 Relative impact of establishing maximum limits on estimate of
intake
4.4.4.1. Average aflatoxin concentrations using four possible
scenarios
Data from the EU, China and the USA were used to assess the
potential impact of successfully eliminating aflatoxin levels above 20
µg/kg versus 15 µg/kg versus 10 µg/kg versus no limit for maize and
groundnuts. For each commodity, two sets of analyses were conducted,
(1) total aflatoxins and (2) aflatoxin B1, in order to determine
whether different conclusions would be reached. For each analysis the
data were evaluated as reported. In all cases in which samples
contained no detectable aflatoxins it was assumed that aflatoxins were
present at the LOD - obviously an overestimation. No impact of
processing was included, which again is an overestimation since
processing is known to result in 50-90% reduction in concentrations.
Table 6 summarizes the impact of successfully limiting aflatoxin
concentrations to less than 10, 15 or 20 µg/kg foodstuff on the mean
total aflatoxin concentrations in maize and groundnuts.
Table 6. Anticipated mean residues of total aflatoxins in maize and groundnuts under four assumptions for
acceptable residue levels in samples:
Scenario 1: no samples excluded
Scenario 2: samples > 10 µg/kg excluded
Scenario 3: samples > 15 µg/kg excluded
Scenario 4: samples > 20 µg/kg excluded
Scenario Samples USA maize European cereals European groundnuts USA groundnuts
excluded
µg total aflatoxin/kg commodity
1 No limit Mean 4.7 0.2 13.3 14.3
SD 30.7 0.4 97.2 58.8
2 Limit = 10 µg/kg Mean 0.6 NA 0.9 0.4
SD 1.4 1.4 0.9
3 Limit = 15 µg/kg Mean 0.7 NA 0.9 0.5
SD 2.0 1.6 1.4
4 Limit = 20 µg/kg Mean 0.9 NA 1.0 0.6
SD 2.6 2.0 1.7
Note: Samples were taken in either USA or Europe but the crop may have come from other geographic locations
No aflatoxins levels above 5 µg/kg have been reported on cereals
in Europe, so there would be no impact of imposing a regulatory
programme that would reduce aflatoxin levels1. In contrast, for
groundnuts and for groundnuts and maize in the USA, the largest
difference in mean aflatoxins levels is between no limit and a maximum
aflatoxin level of 20 µg/kg. In the USA, the greatest impact is
achieved by establishing a maximum level of 20 µg/kg for both maize
and ground-nuts. For example, using data for USA maize, the average
total aflatoxin levels would drop from 4.7 µg/kg to 0.9 µg/kg if all
maize with aflatoxin levels above 20 µg/kg were rejected (Table 6).
Small additional declines in average aflatoxin levels would be found
if the acceptable limit were 15 or 10 µg/kg. The average aflatoxin
levels would be 0.7 and 0.6 µg/kg if 15 and 10 µg/kg limits were
established (Table 7) for aflatoxin B1 in maize.
The same analysis was conducted for groundnuts. The reported total
afla-toxin levels in groundnut and groundnut products sampled in
Europe and USA are presented separately Table 6. If all groundnuts are
included, the average aflatoxin concentration would be 14 µg/kg. The
average aflatoxin concentration would be 0.6 µg/kg if all samples with
levels above 20 µg/kg were excluded and 0.5 and 0.4 µg/kg if all
samples with levels above 15 and 10 µg/kg, respectively, were
excluded.
The distribution of total aflatoxins in crops sampled in the USA
and Europe is provided in Table 8 and the distribution of aflatoxin B1
in crops in Europe and China is provided in Table 9.
1 The major part of cereals for human consumption in Europe are
domestically grown. If the source of cereals were to change this might
no longer be the situation.
Table 7. Anticipated mean residues of aflatoxin B1 in maize and groundnuts under four
assumptions for acceptable residue levels in samples:
Scenario 1: no samples excluded
Scenario 2: samples > 10 µg/kg excluded
Scenario 3: samples > 15 µg/kg excluded
Scenario 4: samples > 20 µg/kg excluded
Scenario Samples European Chinese European Chinese
excluded cereals maize groundnuts groundnuts
µg total aflatoxin B1/kg commodity
1 No limit Mean 1.5 11.8 6.9 8.3
SD 2.3 52.3 72.2 33.1
2 Limit = 10 µg/kg Mean 1.5 2.8 0.6 2.7
SD 2.3 1.9 0.9 1.6
3 Limit = 15 µg/kg Mean 1.5 3.1 0.6 2.7
SD 2.3 2.5 1.2 1.8
4 Limit = 20 µg/kg Mean 1.5 3.5 0.7 2.9
SD 2.3 3.3 1.7 2.3
Note: Samples were taken in either China or Europe but the crop may have come from
other geographic locations
Table 8. Distribution of total aflatoxins in maize and groundnuts
sampled in the USA and Europe1
Percentile USA maize European European USA
cereals groundnuts groundnuts
µg total aflatoxin/kg commodity
10.0% 0.1 0.0 0.1 0.1
20.0% 0.1 0.0 0.2 0.1
30.0% 0.2 0.0 0.3 0.2
40.0% 0.2 0.0 0.5 0.2
50.0% 0.3 0.1 0.6 0.3
60.0% 0.3 0.1 0.7 0.3
70.0% 0.4 0.1 0.8 0.4
80.0% 0.5 0.1 1.0 0.4
90.0% 4.0 0.1 3.3 1.9
95.0% 15.1 1.1 11.5 106
97.5% 38.1 1.5 55.3 267
99.0% 93.8 2.1 379 304
99.5% 149 2.6 767 314
99.8% 247 3.2 1110 323
99.9% 482 3.4 1320 327
1 European cereals include other crops in addition to maize.
Table 9. Distribution of aflatoxin B1 in maize and groundnuts sampled
in Europe1 and China
Percentile European Chinese European Chinese
cereals maize groundnuts groundnuts
µg aflatoxin B1/kg commodity
10.0% 0.1 0.6 0.0 0.6
20.0% 0.1 1.2 0.1 1.2
30.0% 0.3 1.8 0.1 1.7
40.0% 0.5 2.5 0.2 2.3
50.0% 0.7 3.0 0.4 3.0
60.0% 0.9 3.6 0.6 3.5
70.0% 1.2 4.3 0.7 4.0
80.0% 1.9 4.9 0.8 4.6
90.0% 5.4 16.1 1.2 7.8
95.0% 7.6 43.5 4.2 43.8
97.5% 8.8 75.9 16.9 69.9
99.0% 9.4 169 61.4 90.8
99.5% 9.7 453 367 97.3
99.8% 9.9 630 809 150
99.9% 10.0 720 1220 488
1 European cereals include other crops in addition to maize
From: Chen (1997) and SCOOP (1996)
The proportion of samples that would be excluded under each
scenario is identified in Table 10 for total aflatoxins. For example,
if all maize that contains total aflatoxin levels > 20 µg/kg is to be
eliminated, 4% of USA maize would be rejected. If the limit is 10
µg/kg, 6% of USA maize would be rejected. No European maize would be
rejected under either limit. Six per cent of USA and 4% of European
groundnuts would be rejected with a limit of 20 µg/kg and 7% and 5%,
respectively, with a limit of 10 µg/kg. The same information is
provided in Table 11 for aflatoxin B1. If all maize that contains
aflatoxin B1 levels > 20 µg/kg is eliminated, 8% of Chinese maize
would be rejected. If the limit is 10 µg/kg, 13% of Chinese maize
would be rejected. No European maize would be rejected under either
limit. A limit of 20 µg/kg would result in rejection of 2% of European
groundnuts and 8% of Chinese groundnuts. A limit of 10 µg/kg, would
rejected 9% of Chinese groundnuts and 3% of European groundnuts.
4.4.4.2 Intake of total aflatoxins using four scenarios
The four scenarios are:
Scenario 1: no change in current aflatoxin levels;
Scenario 2: exclude groundnuts and maize with > 10 µg/kg total
aflatoxin;
Scenario 3: exclude groundnuts and maize with > 15 µg/kg total
aflatoxin;
Scenario 4: exclude groundnuts and maize with > 20 µg/kg total
aflatoxin).
This analysis was conducted using the average aflatoxin levels
that would result under each scenario and was repeated using only the
data sampled in the USA and only the European data (for total
aflatoxins and only the data in Europe and China for aflatoxin B1).
The range of estimated intakes of total aflatoxin from maize is
shown in Table 12. The estimated range of intakes is 2-501 ng total
aflatoxin/person perday. These data should not be used as true
estimates of likely intake but rather as a measure of the relative
impact of establishing limits. Thus with an upper limit of 20 µg/kg
aflatoxin, the estimated intake using the European WHO regional diet
and European monitoring data would be 2 ng/person per day. There would
be no impact in establishing a limit since there are no European maize
data with levels > 5 µg/kg. Using the USA monitoring data and the
European WHO diet (which includes North America) the intake would be
47 ng/person per day with no limit, 9 ng/person with a 20 µg/kg limit
and 7 ng/person per day with a 15 µg/kg limit. A limit of 10 ng/person
per day would lower the estimated intake to 6 ng/person per day (using
the USA monitoring data). This exercise is repeated using the other
four WHO regional diets and either the European or USA maize/cereal
monitoring data.
Table 10. Distribution of estimated concentrations of total
aflatoxin in maize and groundnuts sampled in the USA and Europe1
Aflatoxin USA maize European European USA
(µg/kg) cereals groundnuts groundnuts
percentiles
0.5 87.4% 91.5% 44.2% 89.1%
1 88.0% 94.7% 81.8% 89.1%
2.5 88.9% 99.2% 88.8% 90.3%
5 90.6% NA 91.7% 92.0%
7.5 92.1% NA 93.8% 92.6%
10 93.8% NA 94.9% 93.0%
12.5 94.2% NA 95.1% 93.4%
15 95.0% NA 95.3% 93.7%
17.5 95.7% NA 95.5% 94.0%
20 96.1% NA 95.8% 94.1%
30 96.8% NA 96.4% 94.6%
40 97.6% NA 96.8% 94.7%
50 98.0% NA 97.4% 94.8%
1 European cereals include other crops in addition to maize
From: Chen (1997) and Wood (1995)
Table 11. Distribution of estimated concentrations of aflatoxins B1
in maize and groundnuts sampled in Europe1 and China
Aflatoxin B1 European Chinese European Chinese
levels (µg/kg) cereals maize groundnuts groundnuts
0.5 41.6% 7.5% 56.9% 8.3%
1 66.2% 16.3% 89.8% 17.3%
2.5 83.4% 40.4% 93.1% 43.0%
5 89.3% 80.9% 96.4% 88.0%
7.5 94.8% 83.5% 96.7% 89.8%
10 NA 86.5% 97.1% 90.6%
12.5 NA 88.2% 97.2% 90.9%
15 NA 89.5% 97.4% 91.3%
17.5 NA 90.7% 97.6% 91.8%
20 NA 91.7% 97.9% 92.1%
30 NA 92.7% 98.3% 93.4%
40 NA 94.4% 98.5% 94.7%
50 NA 96.0% 98.9% 95.8%
1 European cereals include other crops in addition to maize.
The range of estimated intakes of total aflatoxin from groundnuts
was 2-162 ng/person per day (Table 12). However, these data should not
be used as true estimates of likely intake. Rather they should be used
as a measure of the relative impact of establishing limits. Thus, for
example, establishing a programme that successfully limits aflatoxin
levels to 20 µg/kg would reduce the estimated intake of aflatoxin from
groundnuts to 5 ng per day from 66 ng/person per day (European diet).
Likewise, reducing aflatoxin levels to no more than 15 µg/kg limit
would maintain the estimated intake at 5 ng/person per day (Table 12).
Similarly, the estimated intake using the European monitoring data and
a 10 µg/kg limit would be 4.4 ng/person per day. Similar comparisons
are shown in Table 12 for each of the regional diets combined with
either the European or USA aflatoxin monitoring results.
4.4.4.3 Intake of aflatoxin B1 within four scenarios
These analyses were repeated for aflatoxin B1 using the results of
the European and Chinese monitoring results. The results are presented
in Table 13.
Table 12. Estimated intake of total aflatoxin under 4 different scenarios
Scenario 1: no samples excluded
Scenario 2: samples >10 µg/kg excluded
Scenario 3: samples >15 µg/kg excluded
Scenario 4: samples >20 µg/kg excluded
I. FOOD CONSUMPTION ESTIMATES (GEMS/Foods, World Health Organization) + 431
Middle Eastern Far Eastern African Latin American European
(g/person per day) (g/person per day) (g/person per day) (g/person per day) (g/person per day)
Maize 50 31 106 42 10
Groundnuts 0.3 6 11.3 2 5
II. MEAN TOTAL AFLATOXIN RESIDUE CONCENTRATIONS UNDER EACH SCENARIO (from Table 6)
A. MAIZE/CEREALS Mean residues
European monitoring data USA monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 0.2 4.7
Scenario 2: samples >10 µg/kg excluded 0.2 0.6
Scenario 3: samples >15 µg/kg excluded 0.2 0.7
Scenario 4: samples >20 µg/kg excluded 0.2 0.9
B. GROUNDNUTS Mean residues
European monitoring data USA monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 13 14
Scenario 2: samples >10 µg/kg excluded 0.9 0.4
Scenario 3: samples >15 µg/kg excluded 0.9 0.5
Scenario 4: samples >20 µg/kg excluded 1.0 0.6
Table 12. Continued...
III. ESTIMATED DIETARY INTAKE OF AFLATOXIN (TOTAL) UNDER THE FOUR SCENARIOS AND DIFFERENT RESIDUE DATA SETS
A. TOTAL AFLATOXIN INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 10 6 21 8.4 2
Scenario 2: samples >10 µg/kg excluded 10 6 21 8.4 2
Scenario 3: samples >15 µg/kg excluded 10 6 21 8.4 2
Scenario 4: samples >20 µg/kg excluded 10 6 21 8.4 2
B. TOTAL AFLATOXIN INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN THE USA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 240 150 500 200 47
Scenario 2: samples >10 µg/kg excluded 30 19 64 25 6
Scenario 3: samples >15 µg/kg excluded 35 22 74 29 7
Scenario 4: samples >20 µg/kg excluded 45 28 95 38 9
GROUNDNUTS
A. TOTAL AFLATOXIN INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 39 78 150 26 65
Scenario 2: samples >10 µg/kg excluded 0.3 3.6 10 1.8 4.5
Scenario 3: samples >15 µg/kg excluded 0.3 3.6 10 1.8 4.5
Scenario 4: samples >20 µg/kg excluded 0.3 6.0 11 2.0 5.0
(ng total
Table 12. Continued...
B. TOTAL AFLATOXIN INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN THE USA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 4.2 84 160 28 70
Scenario 2: samples >10 µg/kg excluded 0.12 2.4 4.5 0.8 2.0
Scenario 3: samples >15 µg/kg excluded 0.15 3.0 5.7 1.0 2.5
Scenario 4: samples >20 µg/kg excluded 0.18 3.6 6.8 1.2 3.0
Table 13. Estimated intake of aflatoxin B1 under 4 different scenarios with 2 residue datasets
Scenario 1: no samples excluded
Scenario 2: samples >10 µg/kg excluded
Scenario 3: samples >15 µg/kg excluded
Scenario 4: samples >20 µg/kg excluded
I. FOOD CONSUMPTION ESTIMATES (GEMS/Foods, World Health Organization)
Middle Eastern Far Eastern African Latin American European
(g/person per day) (g/person per day) (g/person per day) (g/person per day) (g/person per day)
Maize 50 31 106 42 10
Groundnuts 0.3 6 11.3 2 5
II. MEAN AFLATOXIN B1 RESIDUE CONCENTRATIONS UNDER EACH SCENARIO (from Table 7)
A. MAIZE/CEREALS Mean residues
European monitoring data Chinese monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 1.6 12
Scenario 2: samples >10 µg/kg excluded 1.6 2.8
Scenario 3: samples >15 µg/kg excluded 1.6 3.1
Scenario 4: samples >20 µg/kg excluded 1.6 3.5
B. GROUNDNUTS Mean residues
European monitoring data Chinese monitoring data
(µg/kg) (µg/kg)
Scenario 1: no samples excluded 6.9 8.3
Scenario 2: samples >10 µg/kg excluded 0.6 2.7
Scenario 3: samples >15 µg/kg excluded 0.6 2.7
Scenario 4: samples >20 µg/kg excluded 0.7 2.9
Table 13. Continued...
III. ESTIMATED DIETARY INTAKE OF AFLATOXIN (B1) UNDER THE FOUR SCENARIOS AND DIFFERENT RESIDUE DATASETS
A. AFLATOXIN B1 INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 75 46 160 63 15
Scenario 2: samples >10 µg/kg excluded 75 46 160 63 15
Scenario 3: samples >15 µg/kg excluded 75 46 160 63 15
Scenario 4: samples >20 µg/kg excluded 75 46 160 63 15
B. AFLATOXIN B1 INTAKE FROM MAIZE IF RESIDUES ARE THOSE REPORTED IN CHINA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 600 370 1270 500 120
Scenario 2: samples >10 µg/kg excluded 140 87 300 120 28
Scenario 3: samples >15 µg/kg excluded 160 96 330 130 31
Scenario 4: samples >20 µg/kg excluded 180 108 370 150 35
GROUNDNUTS
A. AFLATOXIN B1 INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN EUROPE
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 2.1 41 78 14 34
Scenario 2: samples >10 µg/kg excluded 0.2 3.6 6.8 1.2 3.0
Scenario 3: samples >15 µg/kg excluded 0.2 3.6 6.8 1.2 3.0
Scenario 4: samples >20 µg/kg excluded 0.2 4.2 7.9 1.4 3.5
Table 13. Continued...
B. AFLATOXIN B1 INTAKE FROM GROUNDNUTS IF RESIDUES ARE THOSE REPORTED IN CHINA
Middle Eastern Far Eastern African Latin American European
(ng total aflatoxin/person per day)
Scenario 1: no samples excluded 2.5 50 94 17 42
Scenario 2: samples >10 µg/kg excluded 0.8 16 30 5.4 14
Scenario 3: samples >15 µg/kg excluded 0.8 16 30 5.4 14
Scenario 4: samples >20 µg/kg excluded 0.9 17 33 5.8 15
4.4.5 Summary
The values for aflatoxin levels presented above are not considered
to be representative of the food supply in any country nor of the
commodities moving in international trade. Quantitative estimates of
intake of aflatoxin at the international level are severely limited by
the lack of representative data. Although intake estimates are
available at the national level for many countries, the submitters of
all of these studies are emphatic in stating that the results are not
truly "representative." In general, the results appear to be biased
upwards because monitoring studies focus on lots of commodity that are
thought to be contaminated.
However, the data do provide sufficient information to evaluate
the likely impact of limiting aflatoxin levels in foodstuffs. Of the
scenarios considered, the greatest relative impact on estimated
average aflatoxin levels is achieved by establishing a programme that
would limit aflatoxin contamination to less than 20 µg/kg. This
assumes that the controls would successfully exclude all samples
containing aflatoxin above that limit. Depending upon assumptions
regarding the distribution of residues, some small incremental
reductions can be achieved by limiting aflatoxin levels to no more
than 15 or 10 µg/kg, respectively.
Additional data that would better simulate the actual aflatoxin
levels in foods moving in international trade would provide much more
accurate estimates of intake. Most likely these estimates would result
in lower estimates of the average intake since the available data
appear to be biased upwards. The incorporation of data on the effects
of food processing on aflatoxin levels would also improve the accuracy
of estimates of intake since aflatoxin is removed during many
procedures.
5. COMMENTS AND EVALUATION
The aflatoxins are among the most potent mutagenic and
carcinogenic substances known. Extensive experimental evidence in test
species shows that aflatoxins are capable of inducing liver cancer in
most species studied. In addition, most epidemiological studies show a
correlation between exposure to aflatoxin B1 and increased incidence
of liver cancer. Aflatoxins are metabolized in humans and test species
to an epoxide, which usually is considered to be the ultimate reactive
intermediate. There is some evidence suggesting that humans are at
substantially lower risk to aflatoxins than test species. The
Committee was aware of epidemiological studies suggesting that intake
of aflatoxin poses no detectable independent risk and studies that
suggest it poses risks only in the presence of other risk factors such
as hepatitis B infection. Several ongoing studies are likely to
improve further the estimates of human risks from the intake of
aflatoxin, most notably cohort studies in Shanghai, Thailand and
Qidong and hepatitis B vaccination trials in The Gambia, Taiwan and
Qidong. When these studies are complete, the Committee may want to
reevaluate the risks of aflatoxins in humans.
A number of factors influence the risk of primary liver cancer,
most notably carriage of hepatitis B virus as determined by the
presence in serum of the hepatitis B surface antigen (presence denoted
HBsAg+ and absence denoted HbsAg-). The potency of aflatoxins
appears to be significantly enhanced in individuals with simultaneous
hepatitis B infection. This interaction makes it difficult to
interpret the epidemiological studies in the context of aflatoxin as
an independent risk factor. The conclusions of the Committee regarding
aflatoxin potency therefore are contingent upon the dynamics of
hepatitis B infection in a human population.
The identification of hepatitis C virus is an important recent
advance in understanding the etiology of liver cancer. Two studies
have investigated interactions between hepatitis C infection,
aflatoxins and liver cancer; the evidence so far is inconclusive. It
is estimated that 50 to 100% of liver cancer cases are associated with
persistent infection with hepatitis B and/or hepatitis C.
The Committee considered that the weight of scientific evidence,
which includes epidemiological data, laboratory animal studies and
in vivo and in vitro metabolism studies, supports a conclusion
that aflatoxins should be treated as carcinogenic food contaminants,
the intake of which should be reduced to levels as low as reasonably
achievable.
5.1 Aflatoxin potencies
The Committee reviewed dose-response analyses that have been
performed on aflatoxins. All of these analyses suffer limitations,
three of which predominate. First, all of the epidemiological data
from which a dose-response relationship can be developed are
confounded by concurrent hepatitis B infection. The epidemiological
data are from geographical areas where both the prevalence of HBsAg+
individuals and aflatoxins are high; the relationship between these
risk factors in areas of low aflatoxin contamination and low hepatitis
B prevalence is unknown. Second, the reliability and precision of the
estimates of aflatoxin exposure in the relevant study populations are
unknown. For example, aflatoxin biomarkers in humans do not reflect
long-term aflatoxin intake; analysis of crops for aflatoxins do not
reflect levels of aflatoxins consumed in foods after selection and
processing. Finally, the shape of the dose-response relationship is
unknown, which introduces an additional element of uncertainty when
choosing mathematical models for interpolation.
Observations concerning the interaction of hepatitis B and
aflatoxins suggest two separate aflatoxin potencies in populations in
which chronic hepatitis infections are common versus populations in
which chronic hepatitis infections are rare. In analyses based on
toxicological and epidemiological data, potency estimates for
aflatoxin were divided into two basic groups, potencies applicable to
individuals without hepatitis B infection and those applicable to
individuals with chronic hepatitis B infection. The Committee found
these estimates useful even though, through the use of differing
mathematical models, they covered a broad range of possible values
(Figure 2). Epidemiological data for which hepatitis B infection
status was unknown and for which potencies were calculated were also
reviewed and found to be in the range of potencies for hepatitis B
infected/non-infected individuals. The review also considered the
extrapolation of animal data to estimate potency in humans; these also
generally fell within the range of the potency estimates derived from
the epidemiological data.
Some discussion is warranted on the potential biases in the
potencies depicted in Figure 2: (i) only studies showing a positive
association between aflatoxins and liver cancer were used, as opposed
to considering all studies (positive as well as negative), leading to
overestimation of the aflatoxin potency; (ii) by relating current
levels of intake (i.e. using biomarkers or dietary surveys) to current
levels of liver cancer (presumably with a long induction period),
historical levels of intake are ignored; they are likely to have been
higher, in which case aflatoxin potency will be overestimated; (iii)
the earliest studies systematically underestimated hepatitis B
prevalence in cases of liver cancer by a factor as high as 20-30%,
owing to limitations in the methodology used to detect hepatitis B,
which also leads to an overestimate of the relative potency of any
other factor, including aflatoxins; (iv) histological confirmation of
the liver cancer cases is limited in most epidemiological studies,
allowing the possibility that non-primary liver cancer cases have been
included, which could lead to an underestimation or overestimation of
the aflatoxin potency. Considering these biases, the values in Figure
2 should be viewed as overestimates of the potency of the aflatoxins,
leading to the hypothesis that it is possible that humans are in fact
less sensitive to aflatoxins than the species tested in laboratory
experiments.
The Committee reviewed the extensive data available on the
metabolism of aflatoxins in various species. It was agreed that
differential potency to aflatoxins between species can be partially
attributed to differences in metabolism. However, there is at the
present time insufficient quantitative information available about
competing aspects of metabolic activation and detoxification of
aflatoxin B1 in various species to identify an adequate animal model
for humans and to explain the apparent species differences in potency.
Intake assessments used in many of the epidemiological studies
ignored the contributions to total aflatoxin intake through milk and
milk products. Thus, the potencies shown in Figure 2 do not generally
apply to aflatoxin M1. From one comparative toxicity study in rats,
it is possible to estimate that aflatoxin M1 has a potency
approximately one order of magnitude less than that of aflatoxin B1
in this species.
The Committee reviewed the potencies estimated from the positive
epidemiological studies and chose separate central tendency estimated
potencies and ranges for HBsAg+ and for HBsAg- individuals. Potency
values of 0.3 cancers/year per 100 000 population per ng aflatoxin/kg
bw per day with an uncertainty range of 0.05 to 0.5 in HBsAg+
individuals and of 0.01 cancers/year per 100 000 population per ng
aflatoxin/kg bw per day with an uncertainty range of 0.002 to 0.03 in
HBsAg- individuals were chosen.
5.2 Population risks
The fraction of the incidence of liver cancer in a population
attributable to intake of aflatoxins is derived by combining aflatoxin
potency estimates (risk per unit dose) and estimates of aflatoxin
intake (dose per person). The Committee reviewed the frequency and
amount of aflatoxin contamination in a variety of products (e.g.,
groundnuts, cereals and maize) in numerous countries (e.g., China,
Denmark, Italy and the USA). Many of the data on contamination levels
were derived from non-random samples, which appeared to be biased
upwards because monitoring studies focus on lots of commodities that
are thought to be contaminated. Some of the data on contaminant levels
are unlikely to be based on current Codex sampling recommendations for
aflatoxins. These contamination levels can only be used with caution
to infer patterns of importance in setting standards and not to
provide exact contamination estimates.
Through the use of hypothetical standards, it was noted that the
magnitude of the difference between two hypothetical standards is
substantially larger than the magnitude of the difference in the mean
contamination levels resulting from the separate standards. This point
is illustrated in Figure 3 in which the derived distribution of
aflatoxin contamination in maize in the USA is shown. Application of a
hypothetical 20 µg/kg standard would result in rejection of 4% of the
maize crop and a mean aflatoxin level in maize of 0.9 µg/kg. Imposing
the stricter hypothetical standard of 10 µg/kg would result in
rejection of 6.2% of the samples to achieve a drop in the mean
aflatoxin contamination level by 0.3 µg/kg to 0.6 µg/kg. Similar
results were obtained when examining aflatoxin B1 levels in maize and
also for total aflatoxins or B1 alone in groundnuts.
Using the Global Environment Monitoring System - Food
Contamination Monitoring and Assessment Programme (GEMS/Food) regional
diets combined with contamination levels, the Committee was able to
provide relative estimates of mean dietary intake of aflatoxins for
various regions under differing standard dietary choices. Linking
these intakes to the potencies shown in Figure 2 allows for the
calculation of overall population risks based upon the prevalence of
hepatitis B infection in various regions.
 From its analysis the Committee noted that the application of a
hypothetical standard removes from human consumption the samples most
highly contaminated, thus greatly reducing average estimated intakes.
Use of standards by all countries should be encouraged. Assuming a
standard is in place, the Committee considered the effect of modifying
that standard through the use of several hypothetical calculations.
Two illustrations are given below.
The first example pertains to areas with low contamination of food
by aflatoxins and with a population having a small prevalence of
carriers of hepatitis B. Aflatoxin levels based on European monitoring
of aflatoxin B1 in groundnuts, maize and products derived from
groundnuts and maize1 were used. In this example a population with
1% carriers of hepatitis B was assumed. From the potencies given
earlier, this yields an estimated population potency of 0.01 × 99% +
0.3 × 1% = 0.013 cancers/year per 100 000 population per ng
aflatoxin/kg bw per day with a range of 0.002 to 0.035. Based on
European monitoring, if all samples with contamination above 20 µg/kg
are removed and it is assumed that these foods are ingested according
to the "European diet", the mean estimated intake of aflatoxin is 19
ng/person per day. Assuming an adult human weight of 60 kg, the
estimated population risk (potency × intake) is 0.0041 cancers/year
per 100 000 people with a range of 0.0006 to 0.01. In contrast, using
the same assumptions but applying a 10 µg/kg hypothetical standard,
the average aflatoxin intake is 18 ng/person per day, resulting in an
estimated population risk of 0.0039 cancers/year per 100 000 people
with a range of 0.0006 to 0.01. Thus, reducing the hypothetical
standard from 20 µg/kg to 10 µg/kg yields a drop in the estimated
population risk of approximately 2 additional cancers/year per 109
people.
The second example pertains to areas with higher contamination
(for these purposes, Chinese monitoring data of aflatoxin B1 in
groundnuts, maize and their products were used) and areas with a
larger population fraction as carriers of hepatitis B (in this case,
25% hepatitis B carriers was assumed). The estimated potency for this
population is 0.01 × 75% + 0.3 × 25% = 0.083, with a range of 0.014 to
0.15. Using a 20 µg/kg hypothetical standard and the "Far Eastern
diet", the average estimated intake is 125 ng/person per day yielding
an average population risk of 0.17 cancers/year per 100 000 people
with a range of 0.03 to 0.3. Using a 10 µg/kg hypothetical standard,
the average estimated intake drops to 103 ng per person, yielding an
estimated population risk of 0.14 cancers/year per 100 000 people with
a range of 0.02 to 0.3. Thus, reducing the hypothetical standard for
this population from 20 µg/kg to 10 µg/kg yields a drop in the
estimated population risk of 300 cancers/year per 109 people.
1 The Committee noted that aflatoxin data for Europe was for "all
cereals". However, in these calculations, it was assumed that the
aflatoxin level for "all cereals" applied to maize consumption only.
5.3 Conclusions
1. Aflatoxins are considered to be human liver carcinogens. Aflatoxin
B1 is the most potent carcinogen of the aflatoxins; most of the
toxicological data available are related to aflatoxin B1. Aflatoxin
M1, the hydroxylated metabolite of B1, has a potency approximately
one order of magnitude less than that of B1.
2. The potency of aflatoxins in HBsAg+ individuals is substantially
higher than the potency in HBsAg- individuals. Thus, reduction of the
intake of aflatoxins in populations with a high prevalence of HBsAg+
individuals will have greater impact on reducing liver cancer rates
than reductions in populations with a low prevalence of HBsAg+
individuals.
3. Vaccination against hepatitis B will reduce the prevalence of
carriers. The present analysis suggests that this would reduce the
potency of the aflatoxins in vaccinated populations and consequently
reduce liver cancer risks.
4. Analyses of the application of hypothetical standards (10 mg/kg or
20 µg/kg aflatoxin in food) to model populations indicate that: (i)
populations with a low prevalence of HBsAg+ individuals and/or with
a low mean intake (less than 1 ng/kg bw per day) are unlikely to
exhibit detectable1 differences in population risks for standards in
the range of the hypothetical cases; and (ii) populations with a high
prevalence of HBsAg+ individuals and high mean intake of aflatoxins
would benefit from reductions in aflatoxin intake.
5. The Committee has previously noted that reductions can be achieved
through avoidance measures such as improved farming and proper storage
practices and/or through enforcing standards for food or feed within
countries and across borders (Annex 1, reference 77).
6. In considering two competing standards, if the fraction of the
samples excluded under the two standards is similar, the higher
standard will yield almost the same liver cancer risks as the lower
standard. When a substantial fraction of the current food supply is
heavily contaminated, reducing the aflatoxin contamination levels may
detectably lower liver cancer rates. Conversely, when only a small
fraction of the current food supply is heavily contaminated, reducing
the standard by an apparently substantial amount may have little
appreciable effect on public health.
1 In the context of this statement "detectable" refers to an
aflatoxin-induced change in liver cancer rates that exceeds the year-
to-year variability around the current incidence and mortality rates.
Hence "detectable" refers to our ability to observe a significant
effect in the occurrence of liver cancer following intervention and
will depend upon the quality of the data available on historical
trends in incidence and mortality.
6. REFERENCES
Aguilar, F., Hussain, S.P., & Cerutti, P. (1993) Aflatoxin B1 induces
the transversion of G to T in codon 249 of the p53 tumor suppressor
gene in human hepatocytes. PNAS, 90: 8586-8590.
Aguilar, F., Harris, C.C., Sun, T., Hollstein, M., & Cerutti, P.
(1994) Geographic variation of p53 mutational profile in nonmalignant
human liver. Science, 264(5163): 1317-1319.
Aguilar, F., Harris, C.C., Sun, T., Hollstein, M., & Cerutti, P.
(1995) p53 mutations in nonmalignant human liver: fingerprints of
aflatoxins? Hepatology, 21(2): 600-601.
Ahmed, H., Hendrickse, R.G., Maxwell, S.M., & Yakabu, A.M. (1995)
Neonatal jaundice with reference to aflatoxins: an aetiological study
in Zaria, northern Nigeria. Ann. Trop. Paediatr., 15: 11-20.
Alavanja M.J., Rush, G.A., Stewart, P., & Blair, A. (1987)
Proportionate mortality study of workers in the grain industry.
J. Natl Cancer Inst., 78: 247-252.
Angsubhakorn, S., Bharmarapravati, N., Romruen, K., Sahaphong, S.,
Thamnavit, W., & Miyamoto, M. (1981a) Further study of alpha benzene
hexachloride inhibition of aflatoxin Bl hepatocarcinogenesis in rats.
Br. J. Cancer, 43: 881-883.
Angsubhakorn, S., Bharmarapravati, N., Romruen, K., & Sahaphong, S.
(1981b) Enhancing effects of dimethylnitrosamine on aflatoxin Bl
hepatocarcinogenesis in rats. Int. J. Cancer, 28: 621-626.
Ankrah, N.-Y. (1995) Alteration of glucose tolerance in mice fed low
levels of aflatoxins and with depressed glyoxalase-1 activity.
Vet. Hum. Toxicol., 37(1): 59-61.
Ankrah, N-A., Addo, P.G.A., Abrahmas, C.A., Ekuban, F.A., & Addae,
M.M. (1993) Comparative effects of aflatoxin G1 and B1 at levels
within human exposure limits on mouse liver and kidney. West Afr.
J. Med., 12(2): 105-109.
Ankrah, N.-A., Rikimaru, T., & Ekuban, F.A. (1994) Observations on
aflatoxins and the liver status of Ghanaian subjects. East. Afr.
Med. J., 71(11): 739-741.
Anwar, W.A., Khalil, M.M., & Wild, C.P. (1994) Micronuclei,
chromosomal aberrations and aflatoxin-albumin adducts in experimental
animals after exposure to aflatoxin B1. Mutat. Res., 322: 61-67.
Appleton, B.S. & Campbell, T.C. (1983) Dietary protein intervention
during the post-dosing phase of aflatoxin B1-induced hepatic
preneoplastic lesion development. J. Natl Cancer Inst., 70: 547-549.
Argentina (1996) Results of studies conducted on the presence of
mycotoxins during the period 1992-1996 (in Spanish).
Australia Market Basket Survey (1992). National Food Authority
(Information submitted to WHO by Australia).
Bailey, G.S., Price, R.L., Park, D.L., & Hendricks, J.D. (1994) Effect
of ammoniation of aflatoxin B1-contaminated cottonseed feedstock on
the aflatoxin M1 content of cows' milk and hepatocarcinogenicity in
the trout bioassay. Food Chem. Toxicicol., 32(8): 707-715.
Ball, R.W., Huyie, J.M., & Coulombe, R.A. Jr. (1995) Comparative
activation of aflatoxin B1 by mammalian pulmonary tissues.
Toxicol. Lett., 75: 119-125.
Bannasch, P., Khoshkhou, N.I., Hacker, H.J., Radaeva, S., Mrozek, M.,
Zillman, U., Kopp-Schneider, A., Haberkorn, U., Elgas, M., Tolle, T.,
Toggendork, M., & Toshkov, I. (1995) Synergistic hepatocarcinogenic
effect of hepadnaviral infection and dietary aflatoxin B1 in
woodchucks. Cancer Res., 55: 3318-3350.
Billy, T.J. (1996) Consideration of Government comments on the Draft
Guideline Level and Sampling Plans for Aflatoxin in Groundnuts.
Personal communication to H. Van der Kooi.
Booth, S.C., Bosenberg, H., Garner, R.C., Herzog, P.J., & Norpoth, K.
(1981) The activation of aflatoxin B1 in liver slices and in bacterial
mutagenicity assays using livers from different species including man.
Carcinogenesis, 2: 1063-1068.
Bosch, F.X. (1995) HCV and liver cancer: the epidemiological evidence.
In: Kobayashi, K., Purcell, R., Shimotohno, K., & Tabor, E. ed.
Hepatitis C virus and its involvement in the development of
hepatocellular carcinoma. Princeton Scientific Publishers, N.J., pp.
15-25.
Bosch, F.X. & Munoz, N. (1991) Hepatocellular carcinoma in the world:
Epidemiologic questions. In: Tabor, E., Di Bisceglie, A.M., & Purcell,
R.H. ed. Etiology, pathology, and treatment of hepatocellular
carcinoma in North America. Gulf Publishing Company, Houston, London,
Paris, Zurich, Tokyo, pp. 35-54 (Advances in Applied Biotechnology
Series, Volume 13).
Bowers, J.C. (1993) Relative versus absolute risk modeling of
aflatoxin. Risk Anal., 13: 9-10.
Bowers, J.C., Brown, B., Springer, J., Tollefson, L., Lorentzen, R., &
Henry, S. (1993) Risk assessment for aflatoxin: an evaluation based on
the multistage model. Risk Anal., 13: 637-642.
Brekke, O.L., et al (1975) Cereal Chem., 52: 205-211.
Bruce, R.D. (1990) Risk assessment for aflatoxin: II. Implications of
human epidemiology data. Risk Anal., 10: 561-569.
Butler, W.H. & Hempsall, V. (1981) Histochemical studies of
hepatocellular carcinomas in the rat induced by aflatoxin. J.
Pathol., 134: 157-170.
Butler, W.H., Greenblut, M., & Lijinsky, W. (1969) Carcinogenesis in
rats by aflatoxins B1, G1 and B2. Cancer Res., 29: 2206-2211.
Campbell, T.C. (1994) Correspondence re: Qian, et al., A follow-up
study of urinary markers of aflatoxin exposure and liver cancer risk
in Shanghai, People's Republic of China. Cancer Epidemiol.
Biomarkers Prev., 3: 3-10, 1994, and C.C. Harris, Solving the
viral-chemical puzzle of human liver carcinogenesis. Cancer
Epidemiol. Biomarkers Prev., 3: 1-2. Cancer Epidemiol. Biomarkers
Prev., 3: 519-521.
Campbell, T.C., Chen, J., Liu, C., Li, J., & Parpia, B. (1990)
Nonassociation of aflatoxin with primary liver cancer in a
cross-sectional ecological survey in the People's Republic of China.
Cancer Res., 50: 6882-6893.
Camus-Randon, A.-M., Raffalli, F., Bereziat, J.-C., McGregor, D.,
Konstandi, M., & Lang, M.A. (1996) Liver injury and expression of
cytochrome P450: evidence that regulation of CYP2A5 is different from
that of other major xenobiotic metabolizing CYP enzymes. Toxicol.
Appl. Pharmacol., 138: 140-148.
Cardis, E., Wild, C.P., Moolgavkar, S., & Zeise, L. (in press)
Aflatoxin and liver cancer. International Agency for Research on
Cancer, Lyon, Chapter 8 (IARC Scientific Publications).
Carlborg, F.W. (1979) Cancer, mathematical models, and aflatoxin.
Food Cosmet. Toxicol., 17: 169-166.
CDHS (California Department of Health Services) (1990). DRAFT - Risk
specific intake levels for aflatoxin (March). Presented to the
Proposition 65 Scientific Advisory Panel, Sacramento,University of
California at Davis.
Chang, M.-H., Chen, C.-J., Lai, M.-S., Hsu, H.-M., Wu, T.-C., Kong,
M.-S., Lian, D.-C., Shau, W.-Y., & Chen, D.-S. (1997) Universal
hepatitis B vaccination in Taiwan and the incidence of hepatocellular
carcinoma in children. New Engl. J. Med., 336(26): 1855-1859.
Chen, J. (1997) Dietary aflatoxin intake levels in China: data
compilation (Unpublished information submitted to WHO).
Chen, J., Campbell, T.C., Li, J., & Peto, R. (1990) Diet, lifestyle
and mortality in China: A study of the characteristics of 65 Chinese
counties. Joint publication of Oxford University Press (Oxford,
England), Cornell University Press (Ithaca, NY), and China People's
Medical Publishing House (Beijing, China).
Chen, C.J., Wang, L.Y., Lu, S.N., Wu, M.H., You, S.L., Zhang, Y.J.,
Wang, L.W., & Santella, R.M. (1996) Elevated aflatoxin exposure and
increased risk of hepatocellular carcinoma. Hepatology, 24: 38-42.
Chou, M.L., Lu, M.H., Pegram, R.A., Gao, P., Cao, S., Kong, J., &
Hart, R.W. (1993) Effect of caloric restriction on aflatoxin
B1-induced DNA synthesis, AFB1-DNA binding and cell proliferation in
Fischer 344 rats. Mech. Ageing Dev., 70: 23-33.
Choy, W.N. (1993) A review of the dose-response induction of DNA
adducts by aflatoxin B1 and its implication to quantitative
cancer-risk assessment. Mutat. Res., 296: 181-198.
Cova, L., Wild, C.P., Mehrotra, R., Turusov, V., Shirai, T., Lambert,
V., Jacquet, C., Tomatis, L., Trepo, C., & Montesano, R. (1996)
Contribution of aflatoxin B1 and hepatitis B virus infection in the
induction of liver tumors in ducks. Cancer Res., 50: 2156-2163.
Croy, R.G. & Crouch, E.A.C. (1991) Interaction of aflatoxin and
hepatitis B virus as carcinogens in human populations. In: Bray, G.A.
& Ryan, D.H. ed. Mycotoxins, cancer and health. Louisiana State
University Press, Baton Rouge, LA, pp. 87-100 (Pennington Nutrition
Series, Vol. 1).
Cullen, J.M. & Newberne, P.M. (1994) Acute hepatotoxicity of
aflatoxins. In: Eaton, D.L. & Groopman, J.D. ed. The toxicology of
aflatoxins: Human health, veterinary, and agricultural significance.
Academic Press, San Diego, CA, pp 1-26.
Cullen, J.M., Ruebner, B.H., Hsieh, L.S., Hyde, D.M., & Hsieh, D.P.
(1987) Carcinogenicity of dietary aflatoxin M1 in male Fischer rats
compared to aflatoxin B1. Cancer Res., 47: 1913-1917.
De Sylos, C.M., et al. (1996) Occurrence of aflatoxin M1 in milk and
dairy products commercialized in Campinas, Brazil (Unpublished
information submitted to WHO).
Diaz, P.V. (1996) Natural occurrence of aflatoxin B1 in raw material
and foodstuffs for birds and pigs in Columbia (1995-1996).
Dragan, Y.P. & Pitot, H.C. (1994) Aflatoxin carcinogenesis in the
context of the multistage nature of cancer. In: Eaton, D.L. &
Groopman, J.D. ed. The toxicology of aflatoxins: Human health,
veterinary, and agricultural significance. Academic Press, San Diego,
CA, pp. 179-198.
Eaton, D.L. & Gallagher, E.P. (1994) Mechanisms of aflatoxin
carcinogenesis. Annu. Rev. Pharmacol. Toxicol., 34: 135-172.
Eaton, D.L. & Groopman, J.D. (1994) The toxicology of aflatoxins:
Human health, veterinary, and agricultural significance. Academic
Press, San Diego, CA.
El-Nazami, H.S., Nicoletti, G., Neal, G.E., Donohue, D.C., & Ahokas,
J.T. (1995) Aflatoxin M1 in human breast milk samples from Victoria,
Australia and Thailand. Food Chem. Toxicol., 33(3): 173-179.
Epstein, S.M., Bartus, B., & Farber, E. (1969) Renal epithelial
neoplasms induced in male Wistar rats by oral aflatoxin B1. Cancer
Res., 29: 1045-1050.
FAO (1993) Sampling plans for aflatoxin analysis in peanuts and corn.
Food and Agriculture Organization of the United Nations, Rome (FAO
Food and Nutrition Paper 55).
FAO (1995) Worldwide regulations for mycotoxins: A compendium. Food
and Agriculture Organization of the United Nations, Rome (FAO Food
and Nutrition Paper 64).
Food and Nutrition Board (1989) Recommended dietary allowances, 6th
ed. National Research Council, National Academy of Sciences,
Washington, D.C., Chapter 6, pp. 52-77.
Fujimoto, Y., Hampton, L.L., Wirth, P.J., Wang, N.J., Xie, J.P., &
Thorgeirsson, S.S. (1994) Alterations of tumor suppressor genes and
allelic losses in human hepatocellular carcinomas in China. Cancer
Res., 54: 281-286.
Gallagher, E.P. & Eaton, D.L. (1995) In vitro biotransformation of
aflatoxin (AFB1) in channel catfish liver. Toxicol. Appl.
Pharmacol., 132: 82-90.
Gallagher, E.P., Wienkers, L.C., Stapleton, P.L., Kunze, K.L., &
Eaton, E.L. (1994) Role of human microsomal and human complementary
DNA-expressed cytochromes P4501A2 and P4503A4 in the bioactivation of
aflatoxin B1. Cancer Res., 54: 101-108.
Gallagher, E.P., Kunze, K.L., Stapleton, P.L., & Eaton, D.L. (1996)
The kinetics of aflatoxin B1 oxidation by human cDNA-expressed and
human liver microsomal cytochromes P450 1A2 and 3A4. Toxicol. Appl.
Pharmacol., 141: 595-606.
Gan, L.S., Skipper, P.L., Peng, X., Groopman, J.D., Chen, J.S, Wogan,
G.N., &Tannenbaum, S.R. (1988) Serum albumin adducts in the molecular
epidemiology of aflatoxin carcinogenesis: correlation with aflatoxin
B1 intake and urinary excretion of aflatoxin M1. Carcinogenesis, 9:
1323-1325.
Gaylor, D.W. & Kodell, R.L. (1980) Linear interpolation algorithm for
low dose risk assessment of toxic substances. J. Environ. Pathol.
Toxicol., 4: 305-315.
Gerbes, A.L. & Caselmann, W.H. (1993) Point mutations of the P53
gene, human hepatocellular carcinoma and aflatoxins. J. Hepatol.,
19: 312-315.
Gopalan, P., Tsuji, K., Lehmann, K., Kimura, M., Shinozuka, H., Sato,
K., & Lotlikar, P.D. (1993) Modulation of aflatoxin B1-induced
glutathione S-transferase placental form positive hepatic foci by
pre-treatment of rats with phenobarbital and buthioninesufoximine.
Carcinogenesis, 14(7): 1469-1470.
Gorelick, N.J. (1990) Risk assessment for aflatoxin: I. Metabolism of
aflatoxin B1 by different species. Risk Anal., 10(4): 539-559.
Griciute, L. (1980) Investigation on the combined action of
N-nitrosodiethylamine with other carcinogens. In: Walker, E.A.,
Griciute, L., Castegnaro, M., & Borzsonyi, M. ed. N-nitroso compounds:
Analysis, formation and occurrence. International Agency for Research
on Cancer, Lyon, pp 813-822 (IARC Scientific Publications No. 31).
Groopman, J.D., Zhu, J., Donahue, P.R., Pikul, A., Zhang, L., Chen,
J.S., & Wogan, G.N. (1992) Molecular dosimetry of urinary
aflatoxin-DNA adducts in people living in Guangxi Autonomous Region,
People's Republic of China. Cancer Res., 52: 45-52.
Groopman, J.D., Wild, C.P., Hasler, J., Chen, J., Wogan, G.N., &
Kansler, T.W. (1993) Molecular epidemiology of aflatoxin exposures:
Validation of aflatoxin-N-7-guanine levels in urine as a biomarker in
experimental rat models and humans. Environ. Health Perspect., 99:
107-113.
Groopman, J.D., Wogan, G.N., Roebuck, B.D., & Kensler, T.W. (1994)
Molecular biomarkers for aflatoxins and their application to human
cancer prevention. Cancer Res., 54(Suppl.): 1907s-1911s.
Guengerich, F.P., Johnson, W.W., Ueng, Y.-F., & Shimade, T. (1996)
Involvement of cytochrome P450, glutathione S-transferase, and epoxide
hydrolase in the metabolism of aflatoxin B1 and relevance to risk of
human liver cancer. Environ. Health Perspect., 104(3): 557-562.
Hall, A.J. & Wild, C.P. (1994) Epidemiology of aflatoxin-related
disease. In: Eaton, D.L. & Groopman, J.D. ed. The toxicology of
aflatoxins: Human health, veterinary, and agricultural significance.
Academic Press, San Diego, CA, pp. 233-258.
Harris, C.C. (1994) Solving the viral-chemical puzzle of human liver
carcinogenesis. Cancer Epidemiol. Biomarkers Prev., 3: 1-2.
Harris, C.C. (1995 At the crossroads of molecular carcinogenesis and
risk assessment. Chem. Ind. Inst. Toxicol. Act., 15(5): 1-6.
Hasler, J.A., Dube, N., Nyathi, C.B., Fuhrmann, H., & Sallmann, H.-P.
(1994) The influence of dietary fat on hepatic bioactivation of
aflatoxin B1 in rats. Res. Commun. Chem. Pathol. Pharmacol., 83(3):
279-287.
Hatch, M.C., Chen, C.J., Levin, B., Ji, B.T., Yang, G.Y., Hsu, S.W.,
Wang, L.W., & Santella, R.M. (1993) Urinary aflatoxin levels,
hepatitis B virus infection and hepatocellular carcinoma in Taiwan.
Int. J. Cancer, 54: 931-934.
Hayes, R.B., van Niewenhuize, J.P., Raatgever, J.W., & ten Kate,
F.J.W. (1984) Aflatoxin exposures in the industrial setting: an
epidemiological study of mortality. Food Chem. Toxicol., 22: 39-43.
Hayes, J.D., Nguyen, T., Judah, D.J., Petersson, D.G., & Neal, G.E.
(1994) Cloning of CDNAS from fetal rat liver encoding glutathione
S-transferase Yc polypeptides. J. Biol. Chem., 269(32): 20707-20717.
Heinonen, J.T., Fisher, R., Brendel, K., & Eaton, D.L.(1996)
Determination of aflatoxin B1 biotransformation and binding to hepatic
macromolecules in human precision liver slices. Toxicol. Appl.
Pharmacol., 136: 1-7.
Hendrickse, R.G., Coulter, J.B.S., Lamplugh, S.M., MacFarlane, S.B.J.,
Williams, T.E., Omer, M.J.A., & Suliman, G.I. (1982) Aflatoxins and
kwashiorkor: a study in Sudanese children. Br. Med. J., 285:
843-846.
Henry, S.H., DiNovi, M.J., Bowers, J.C., & Bolger, P.M. (1997) Risk
assessment for aflatoxin in corn and peanuts in the United States.
Poster presented at the Society of Toxicology meeting, Cincinnati,
Ohio, March 9-13.
Hoseyni, M.S. (1992) Risk assessment for aflatoxin: III. Modeling the
relative risk of hepatocellular carcinoma. Risk Anal., 12: 123-126.
Hsieh, D.P.H. & Atkinson, D.N. (1995) Recent aflatoxin exposure and
mutation at codon 249 of the human p53 gene: lack of association.
Food Addit. Contam., 12(3): 421-424.
Hsing, A.W., Guo, W., Chen, J., Li, J-Y., Stone, B.J., Blot, W.J., &
Fraumeni, J.F. Jr. (1991) Correlates of liver cancer mortality in
China. Int. J. Epidemiol., 20(1): 54
Hu, J-F., Chisari, F.V., & Campbell, T.C. (1994) Modulating effect of
dietary protein on transgene expression in hepatitis B virus (HBV)
transgenic mice. Proc. Am Assoc. Cancer Res., 35: 104.
Hulla, J.E., Chen, Z.Y., & Eaton, D.L. (1993) Aflatoxin B1-induced rat
hepatic hyperplastic nodules do not exhibit a site-specific mutation
within the p53 gene. Cancer Res., 53: 9-11.
IARC (1993) Some naturally occurring substances: food items and
constituents, heterocyclic aromatic amines and mycotoxin.
International Agency for Research on Cancer, Lyon, pp 245-395 (IARC
Monographs on the Evaluation of Carcinogenic Risks of Chemicals to
Humans, Volume 56).
IARC (1994) Hepatitis viruses. International Agency for Research on
Cancer, Lyon (IARC Monographs on the Evaluation of Carcinogenic Risks
of Chemicals to Humans, Volume 59).
IARC (1997) In: Toniolo, P., Boffetta, P., Shuker, D., Rothman, N.,
Hulka, B. & Pearce, H. ed. Application of biomarkers in cancer
epidemiology. International Agency for Research on Cancer, Lyon, pp.
251-264 (IARC Scientific Publications No. 142).
Ibeh, I.N., Uraih, N., & Ogonar, J.I. (1994) Dietary exposure of
aflatoxin in human male infertility in Benin City, Nigeria. Int. J.
Fertil., 39(4): 208-214.
Imazeki, F., Yokosuka, O., Ohto, M., & Omata, M. (1995) Aflatoxin and
p53 abnormality in duck hepatocellular carcinoma. J. Gastroenterol.
Hepatol., 10: 646-649.
International Commission on Radiological Protection (1975) Report of
the Task Group on Reference Man. Pergamon Press, New York (ICRP
Publication No. 23).
Itoh, S., Tagawa, S., Sawada, M., & Kamataki, T. (1993) High
susceptibility to aflatoxin B1 and benzo[ a]pyrene of BALB3T3
A31-1-1 cells expressing monkey CYP1A1. J. Toxicol. Sci., 18:
207-210.
Jacobson, L.P., Zhang, B.C., Zhu, Y.R., Wang, J.B., Wu, Y., Zang,
Q.N., Yu, L.Y., Qian, G.S., Kuang, S.Y., Li, Y.F., Fang, X., Xarba,
A., Chen, B., Enger, C., Davidson, N.E., Gorman, M.B., Gordon, G.B.,
Prochaska, H.J., Egner, P.A., Groopman, J.D., Munoz, A., Helzlsouer,
K.J., & Kensler, T.W. (1997) Oltipraz chemoprevention trial in Qidong,
People's Republic of China: study design and clinical outcomes.
Cancer Epidemiol. Biomarkers Prev., 6(4): 257-265.
Jakab, G.J., Hmielski, R.R., Zarba, A., Hemenway, D.R., & Groopman,
J.D. (1994) Respiratory aflatoxicosis: suppression of pulmonary and
systemic host defenses in rats and mice. Toxicol. Appl. Pharmacol.,
125: 198-205.
Japanese Ministry of Health, Food Sanitation Division (1995) The
situation of aflatoxin contamination in Japan, 1972-1989 and related
information prepared for the 47th JECFA..
Jennings, G.S., Heck, R., Oesch, F., & Steinberg, P. (1994) Metabolism
and cytotoxicity of AFB1 in cultured rat hepatocytes and
nonparenchymal cells: implications for tumorigenesis. Toxicol.
Appl. Pharmacol., 129: 86-94.
Judah, D.J., Hayes, J.D., Yang, J.-C., Lian, L.-Y., Roberts, G.C.K.,
Farmer, P.B., Lamb, J.H., & Neal, G.E. (1993) A novel aldehyde
reductase with activity towards a metabolite of aflatoxin B1 is
expressed in rat liver during carcinogenesis and following the
administration of an anti-oxidant. Biochem. J., 292: 13-18.
Kirby, G.M., Chemin, I., Montesano, R., Chisari, F.V., Lang, M.A., &
Wild, C.P. (1994) Induction of specific cytochrome P450s involved in
aflatoxin B1 metabolism in hepatitis B virus transgenic mice. Mol.
Carcinog., 11: 74-80.
Kirby, G.M., Wolf, C.R., Neal, G.E., Judah, D.J., Henderson, C.J.,
Srivatanakul, P., & Wild, C.P. (1993) In vitro metabolism of
aflatoxin B1 by normal and tumorous liver tissue from Thailand.
Carcinogensis, 14(12): 2613-2620.
Kopp-Schneider, A. & Portier, C.J. (1989) A note on approximating the
cumulative distribution function of the time to tumor onset in
multistage models. Biometrics, 45: 1259-1264.
Langouet, S., Coles, B., Morel., F., Becquemont, L., Beaune, P.,
Guengerich, F.P., Ketterer, B., & Guillouzo, A. (1995) Inhibition of
CYP1A2 and CYP3A4 by oltipraz results in reduction of aflatoxin B1
metabolism in human hepatocytes in primary culture. Cancer Res., 55:
5574-5579.
Liang, T.J. (1995) p53 proteins and aflatoxin B1: the good, the bad,
and the ugly. Hepatology, 22(4): 1330-1332.
Liu, Y.H., Taylor, J., Linko, P., Lucier, G.W., & Thompson, C.L.
(1991) Glutathione S-transferase mu in human lymphocyte and liver:
role in modulating formation of carcinogen-derived DNA adducts.
Carcinogenesis, 12(12): 2269-2275
Loechler, E.L. (1994) Mechanisms by which aflatoxins and other bulky
carcinogens induce mutations. In: Eaton, D.L. & Groopman, J.D. ed. The
toxicology of aflatoxins: Human health, veterinary, and agricultural
significance. Academic Press, San Diego, CA, pp. 149-178.
Lye, M.S., Ghazali, A.A., Mohan, J., Alwin, N., & Nair, R.C. (1995) An
outbreak of acute hepatic encephalopathy due to severe aflatoxicosis
in Malaysia. Am. J. Trop. Med. Hyg., 53(1): 68-72.
McGlynn, K.A., Rosvold, E.A., Lustbader, E.D., Hu, Y., Clapper, M.L.,
Zhou, T., Wild, C.P., Xia, X.-L., Baffoe-Bonnie, A., Ofori-Adjei, D.,
Chen, G.-C., London, W.T., Shen, F.-M., & Buetow, D.H.(1995)
Susceptibility to hepatocellular carcinoma is associated with genetic
variation in the enzymatic detoxification of aflatoxin B1. Proc.
Natl Acad. Sci., 92: 2384-2387.
McLean, M & Dutton, M.F. (1995) Cellular interactions and metabolism
of aflatoxin: an update. Pharmacol. Ther., 65: 163-192.
Marquez-Marquez, R., Badrigal-Bujaidar, E., & Hernandez, I.T. (1993)
Genotoxic evaluation of ammonium inactivated aflatoxin B1 in mice fed
with contaminated corn. Mutat. Res., 299: 1-8.
Marquez-Marquez, R., Hernandez, I.T., & Bujaidar, E.M. (1995)
Genotoxicity of aflatoxin B1 and its ammonium derivatives. Food
Addit. Contam., 12(3): 425-429.
Massey, T.E., Stewart, R.K., Daniels, J.M., & Liu, L. (1995)
Biochemical and molecular aspects of mammalian susceptibility to
aflatoxin B1 carcinogenicity. Proc. Soc. Exp. Biol. Med., 208:
213-227.
Merkow, L.P., Epstein, S.M., Slifkin, M., & Pardo, M. (1973) The
ultrastructure of renal neoplasms induced by aflatoxin B1. Cancer
Res., 33: 1608-1614.
Mexico D.F. (1996) Monitoring results 1992-1996: Document presented at
the Course on Mycotoxins (in Spanish).
Moore, M.R., Pitot, H.C., Miller, E.C., & Miller, J.A. (1982)
Cholangiocellular carcinoma incidence in Syrian hamsters administered
aflatoxin B1 in large doses. J. Natl Cancer Inst., 68: 271-278.
Newberne, P.M. & Rogers, A.W. (1973) Rat colon carcinomas associated
with aflatoxin and marginal vitamin A. J. Natl Cancer Inst., 50:
439-444.
Nicaragua (1996) Detection of aflatoxin in samples from the
Directorate of Food and Zoonosis Control, analyzed at the Department
of Environmental Toxicology, National Reference and Diagnosis Centre
(in Spanish).
Nixon, J.E., Hendricks, J.D., Pawlowski, N.E., Loveland, P.M., &
Sinnhuber, R.O. (1981) Carcinogenicity of aflatoxin in Fischer 344
rats. J. Natl Cancer Inst., 66: 1159-1163.
Olsen, J.H., Dragsted, L., & Autrup, H. (1988) Cancer risk and
occupational exposure to aflatoxins in Denmark. Br. J. Cancer, 58:
392-396.
Olubuyide, I.O., Maxwell, S.M., Akinyinka, O.O., Hart, C.A., Neal,
G.E., & Hendrickse, R.G. (1993) Hbsag and aflatoxins in sera of rural
(Igbo-Ora) and urban (Ibadan) populations in Nigeria. Afr. J. Med.
Med. Sci., 22: 77-80.
Ozturk, M. (1991) p53 mutation in hepatocellular carcinoma after
aflatoxin exposure. Lancet, 338: 1356-1359.
Pacin, A. (no date) Personal communication to B.J. Petersen.
Peers, F.G. & Linsell, C.A. (1977) Dietary aflatoxins and human
primary liver cancer. Ann. Nutr. Alim., 31: 1005-1018.
Peers, F.G., Gilman, G.A., & Linsell C.A. (1976) Dietary aflatoxins
and human liver cancer: A study in Swaziland. Int. J. Cancer, 17:
167-176.
Peers, F., Bosch, X., Kaldo, J., Linsell, A., & Pluijem, M. (1987)
Aflatoxin exposure, hepatitis B virus infection and liver cancer in
Swaziland. Int. J. Cancer, 39: 545-553.
Preston, R.S., Hayes, J.R., & Campbell, T.C. (1976) The effect of
protein deficiency on the in vivo binding of aflatoxin B1 to rat
liver macromolecules. Life Sci., 19: 1191-1198.
Primiano, T., Egner, P.A., Sutter, T.R., Kelloff, G.J., Roebuck, B.D.,
& Kensler, T.W. (1995) Intermittent dosing with oltipraz: relationship
bewteen chemoprevention of aflatoxin-induced tumorigenesis and
induction of glutathione S-transferase. Cancer Res., 55: 4319-4324.
Qian, G.-S., Ross, R.K., Yu, M.C., Yuan, J.-M., Gao, Y.-T., Henderson,
B.E., Wogan, G.N., & Groopman, J.D. (1994) A follow-up study of
urinary markers of aflatoxin exposure and liver cancer risk in
Shanghai, People`s Republic of China. Cancer Epidemiol. Biomarkers
Prev., 3: 3-10.
Raisuddin, S., Singh, K.P., Zaidi, B.N., Paul, B.N., & Ray, P.K.
(1993) Immunosuppressive effects of aflatoxin in growing rats.
Mycopathologia, 124: 189-194.
Ramesh, R. & Panda, S.K. (1993) Hepatitis B virus (HBV) genome in
antibody positive hepatocellular carcinoma (HCC). Letters to the
Editor. J. Hepatol., 19: 319.
Read, M. (1989) Removal of aflatoxin contamination from the Australian
groundnut crop. Proceedings of the ICRISAT International Workshop,
6-9 October 1987. International Crops Research Institute for the
Semi-Arid Tropics.
Read, M. (1997) Letter to J. Baines providing estimates of aflatoxins
in peanuts.
Reddy, J.K., Svoboda, D.J., & Rao, M.S. (1976) Induction of liver
tumors by aflatoxin B1 in the tree shrew (Tupaia glis), a nonhuman
primate. Cancer Res., 36: 151-160.
Regueiro, O.S. (no date) Report on criteria for aflatoxins B1, G1 and
M presented for review at the XLVI meeting of the FAO/WHO Expert
Committee on Food Additives (in Spanish).
Rivkina, M.B., Cullen, J.M., Robinson, W.S., & Marion, P.L. (1994)
State of the p53 gene in hepatocellular carcinomas of ground squirrels
and woodchucks with past and ongoing infection with hepadnaviruses.
Cancer Res., 54: 5430-5437.
Ross, R.K., Yuan, J.-M., Yu, M.C., Wogan, G.N., Qian, G.-S., Tu, J.T.,
Groopman, J.D., Gao, Y.-T., & Henderson, B.E. (1992) Urinary aflatoxin
biomarkers and risk of hepatocellular carcinoma. Lancet, 339(8799):
943-946.
Sabino, M. (1997) Personal communication to B.J. Petersen from A.
Pacin dated 4.8.1997: Maize monitoring results for 321 samples.
Santella, R.M., Zhang, Y.-J., Chen, C.-J., Lee, C.-S., Haghighi, B.,
Hang, G.-Y., Wang, L.-W., & Feitelson, M. (1993) Immunohistochemical
detection of aflatoxin-B1-DNA adducts and hepatitis B virus antigens
in hepatocellular carcinoma and nontumorous liver tissue. Environ.
Health Perspect., 99: 199-202.
Sawada, M., Kitamura, R., Norose, T., Kitada, M., Itahashi, K., &
Kamataki, T. (1993) Metabolic activation of aflatoxin B1 by human
placental microsomes. J. Toxicol. Sci., 18: 129-132.
Scholl, P., Musser, S.M., Kensler, T.W., & Groopman, J.D. (1995)
Molecular biomarkers for aflatoxin and their application to human
liver cancer. Pharmacogenetics, 5: S171-S176.
Schulsinger, D.A., Root, M.M., & Campbell, T.C. (1989) Effect of
dietary protein quality on development of aflatoxin B1-induced hepatic
preneoplastic lesions. J. Natl Cancer Inst., 81: 1241-1245.
SCOOP (1996) Scientific co-operation on questions relating to food:
Working document in support of a SCF risk assessment of aflatoxin:
Task 3.2.1 (SCOOP/CNTM/1). Task Co-ordinator, UK.
Sengstag, C. & Wurgler, F.E. (1994) DNA recombination induced by
aflatoxin B1 activated by cytochrome P450 1A enzymes. Mol.
Carcinog., 11: 227-235.
Shank, R.C., Gordon, J.E., Wogan, G.N., Nondasuta, A., & Subhamani, B.
(1972a) Dietary aflatoxins and human liver cancer. III. Field survey
of rural Thai families for ingested aflatoxins. Food Cosmet.
Toxicol., 10: 71-84.
Shank, R.C., Bhamarapravati, N., Gordon, J.E., & Wogan, G.N. (1972b)
Dietary aflatoxins and human liver cancer. IV. Incidence of primary
liver cancer in two municipal populations of Thailand. Food
Cosmet. Toxicol., 10: 171-179.
Shen, H.-M., Ong, D.N., & Shi, C.-Y. (1995) Involvement of reactive
oxygen species in aflatoxin B1-induced cell injury in cultured rat
hepatocytes. Toxicology, 99: 115-123.
Shi, C.-Y., Chua, S.-C., Lee, H.-P., & Ong, C.-N. (1994) Inhibition of
aflatoxin B1-DNA binding and adduct formation by selenium in rats.
Cancer Lett., 82: 203-208.
Shi, C.-Y, Hew, Y.-C., & Ong, C.-N. (1995a) Inhibition of aflatoxin
B1-induced cell injury by selenium: an in vitro study. Hum. Exp.
Toxicol., 14(1): 55-60.
Shi, C.-Y., Phang, T.W., Lin, Y., Wee, A., Li, B., Lee, H.P., & Ong,
C.N. (1995b) Codon 249 mutation of the p53 gene is a rare event in
hepatocellular carcinomas from ethnic Chinese in Singapore. Br. J.
Cancer, 72: 146-149.
Sieber, S.M., Correa, P., Dalgard, D.W., & Adamson, R.H. (1979)
Induction of osteogenic sarcomas and tumors of the hepatobiliary
system in human primates with aflatoxin B1. Cancer Res., 39:
4545-4554.
Sinha, S.P. & Dharmshila, K. (1994) Vitamin A ameliorates the
genotoxicity in mice of aflatoxin B1-containing Aspergillus
flavus infested food. Cytobios, 79: 85-95.
Srivatanakul, P., Parkin, D.M., Khlat, M., Chenvidhya, D., Chotiwan,
P., Insiripong, S.L., Abbé, K.A., & Wild, C.P. (1991) Liver cancer in
Thailand: II. A case-control study of hepatocellular carcinoma.
Int. J. Cancer, 48: 329-332.
Stoner, G.D., Conran, P.B., Greisiger, E.A., Stober, J., Morgan, M., &
Pereira, M.A. (1986) Comparison of two routes of chemical
administration on the lung adenoma response in strain A/J mice.
Toxicol. Appl. Pharmacol., 82: 19-31.
Stoloff, L. (1983) Aflatoxin as a cause of primary liver-cell cancer
in the United States: a probability study. Nutr. Cancer, 5:
165-186.
Stoloff, L. & Friedman, L. (1976) Information bearing on the
evaluation of the hazard to man from aflatoxin ingestion. PAG Bull.,
6: 21-32.
Stresser, D.M., Bailey, G.S., & Williams, D.E. (1994a)
Indole-3-carbinol and beta-naphthoflavone induction of aflatoxin
B12 metabolism and cytochromes P-450 associated with bioactivation
and detoxication of aflatoxin B1 in the rat. Drug Metab. Dispos.,
22(3): 383-391.
Stresser, D.M., Williams, D.E., McLellan, L.I., Harris, T.M., &
Bailey, G.S. (1994b) Indole-3-carbinol induces a rat liver glutathione
transferase subunit (Yc2) with high activity toward aflatoxin B1
exo-epoxide. Drug Metab. Dispos., 22(3): 392-399.
Tanaka, N., Chiba, T., Matsuzaki, Y., Osuga, T., Aikawa, T., &
Mitamura, K. (1993) High prevalence of hepatitis B and C viral markers
in Japanese patients with hepatocellular carcinoma. Gastroenterol.
Jpn., 28(4): 547-553.
Thomas, D.B. (1991) Exogenous steroid hormones and hepatocellular
carcinoma. In: Tabor, E., Di Bisceglie, A.M., & Purcell, R. ed.
Etiology, pathology and treatment of hepatocellular carcinoma.
PPC-Gulf Publishing Company, Houston, TX, Chapter 6, pp. 77-91.
Thorslund, T.W., Brown, C.C., & Charnley, G. (1987) Biologically
motivated cancer risk models. Risk Anal., 7: 109-119.
Travis C.C. & Land, M.L. (1990) Estimating the mean of data sets with
nondetectable values. Environ Sci. Technol., 24: 961-962.
Ueng, Y-F., Shimada, T., Yamazaki, H., & Guengerich, F.P. (1995)
Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome
P450 enzymes. Chem. Res. Toxicol., 8: 218-225.
Van Egmond, H.P. (1983) Food Chem., 11: 289-307.
van Rensburg, S.J., Cook-Mozaffari, P., van Schalkwyk, D.J., van der
Watt, J.J., Vincent, T.J., & Purchase, I.F. (1985) Hepatocellular
carcinoma and dietary aflatoxin in Mozambique and Transkei. Br. J.
Cancer, 51: 713-726.
Venable, W.N. & Ripley, D.B. (1994) Modern applied statistics with
S-Plus - Chapter 13: Tree based methods. Springer-Verlag, Basel,
Heidelberg.
Vesselinovitch, S.D., Mihailovich, N., Wogan, G.N., Lombard, L.S., &
Rao, K.V.N. (1972) Aflatoxin B1, a hepatocarcinogen in the infant
mouse. Cancer Res., 32: 2289-2291.
Waltking, A.E. (1971) Fate of aflatoxin during roasting and storage of
contaminated peanut products. J. Assoc. Off. Anal. Chem., 54:
533-539.
Wang, J-S., Qian, G.-S., Zarba, A., He, X., Zhu, Y.-R., Zhang, B.-C.,
Jacobson, L., Gange, S.J., Munoz, A., Kensler, T.W., & Groopman, J.D.
(1996a) Temporal patterns of aflatoxin-albumin adducts in hepatitis B
surface antigen-positive and antigen-negative residents of Daxin,
Qidong County, People's Republic of China. Cancer Epidemiol.
Biomarkers Prev., 5: 253-261.
Wang, L.-Y., Hatch, M., Chen, C.-J., Levin, B., You, S.-L., Lu, S.-N.,
Wu, M.-H., Wu, W.-P., Wang, L.-W., Wang, Q., Huang, G.-T., Yang,
P.-M., Lee, H-.S., & Santella, R.M. (1996b) Aflatoxin exposure and
risk of hepatocellular carcinoma in Taiwan. Int. J. Cancer, 67:
620-625.
Ward, J.M., Sontag, J.M., Weisburger, E.K., & Brown, C.A. (1975)
Effect of lifetime exposure to aflatoxin B1 in rats. J. Natl
Cancer Inst., 55: 107-110.
Wieder, R., Wogan, G.N., & Shimkin, M.A. (1968) Pulmonary tumors in
strain A mice given injections of aflatoxin B1. J. Natl Cancer
Inst., 40: 1195-1197.
Wild, C.P., Hudson, G.J., Sabbioni, G., Chapot, B., Hall, A.J., Wogan,
G.N., Whittle, H., Montesano, R., & Groopman, J.D. (1992) Dietary
intake of aflatoxins and the level of albumin-bound aflatoxin in
peripheral blood in the Gambia, West Africa. Cancer Epidemiol.
Biomarkers Prev., 1: 229-234.
Wild, C.P., Fortuin, M., Donato, F., Whittle, H.C., Hall, A.J., Wolf,
C.R., & Montesano, R. (1993) Aflatoxin, liver enzymes and hepatitis B
virus infection in Gambian children. Cancer Epidemiol. Biomarkers
Prev., 2: 555-561.
Wild, C.P., Hasegawa, R., Barraud, L., Chutimataewin, S., Chapot, B.,
Ito, N., & Montesano, R. (1996) Aflatoxin-albumin adducts: a basis for
comparative carcinogenesis between animals and humans. Cancer
Epidemiol. Biomarkers. Prev., 5: 179-189.
Wiseman, W.W., et al (1983) J. Food Prot., 46: 530-532.
Wogan, G.N., Palianlunga, S., & Newberne, P.M. (1974) Carcinogenic
effects of low dietary levels of aflatoxin Bl in rats. Food Cosmet.
Toxicol., 12: 681-685.
Wogan, G.N. (1992) Aflatoxins as risk factors for hepatocellular
carcinoma in humans. Cancer Res., 52(Suppl.): 2114-2118s.
Wood, G.E. (1995) Communication from Dr G.E. Wood (US FDA) to Dr J.
Paakkanen (FAO) dated 21.12.1995.
Wu-Williams, A.H., Zeise, L., & Thomas, D. (1992) Risk assessment for
aflatoxin B1: A modeling approach. Risk Anal., 12: 559-567.
Yahl, K.R., et al. (1971) Laboratory wet-milling of corn containing
high-levels of aflatoxin and a survey of commercial wet-milling
products. Cereal Chem., 48: 385-391.
Yan, R.Q., Su, J.J., Huang, D.R., Gan, Y.C., Yang, C., & Huang, G.H.
(1996) Human hepatitis B virus and hepatocellular carcinoma II.
Experimental induction of hepatocellular carcinoma in tree shrews
exposed to hepatitis B virus and aflatoxin B1.
Yap, E.P.H., Cooper, K., Maharaj, B., & McGee, J.O'D. (1993) p53 codon
249ser hot-spot mutation in HBV-negative hepatocellular carcinoma.
Lancet, 341(8839): 251.
Yeh, F.-S., Mo, C.-C.,& Yen, R.-C. (1985) Risk factors for
hepatocellular carcinoma in Guangxi, People's Republic of China.
Natl Cancer Inst. Monogr., 69: 47-48.
Yeh, F.-S., Yu, M.C., Mo, C.-C., Luo, S., Tong, M.J., & Henderson,
B.E. (1989) Hepatitis B virus, aflatoxin, and hepatocellular carcinoma
in Southern Guangxi, China. Cancer Res., 49: 2506-25509.
Yoshizawa, T., Yamashita, A., & Chokethaworn, N. (1996) Occurrence of
fumonisins and aflatoxins in maize from Thailand. Food Addit.
Contam., 13: 163-168.
Youngman, L.D. & Campbell, T.C. (1992) Inhibition of aflatoxin
B1-induced gamma-glutamyltranspeptidase positive (GGT+) hepatic
preneoplastic foci and tumors by low protein diets: evidence that
altered GGT+ foci indicate neoplastic potential. Carcinogenesis,
13(9): 1607-1613.
Yuan, J.M., Ross, R.K., Stanczyk, F.Z., Govindaragan, S., & Gao, Y.T.
(1995) A cohort study of serum testosterone and hepatocellular
carcinoma in Shanghai, China. Int. J. Cancer, 63: 491-493.
Zimbawbe Government Analyst Laboratory. Aflatoxin surveillance:
Monitoring results 1995-1996.
From its analysis the Committee noted that the application of a
hypothetical standard removes from human consumption the samples most
highly contaminated, thus greatly reducing average estimated intakes.
Use of standards by all countries should be encouraged. Assuming a
standard is in place, the Committee considered the effect of modifying
that standard through the use of several hypothetical calculations.
Two illustrations are given below.
The first example pertains to areas with low contamination of food
by aflatoxins and with a population having a small prevalence of
carriers of hepatitis B. Aflatoxin levels based on European monitoring
of aflatoxin B1 in groundnuts, maize and products derived from
groundnuts and maize1 were used. In this example a population with
1% carriers of hepatitis B was assumed. From the potencies given
earlier, this yields an estimated population potency of 0.01 × 99% +
0.3 × 1% = 0.013 cancers/year per 100 000 population per ng
aflatoxin/kg bw per day with a range of 0.002 to 0.035. Based on
European monitoring, if all samples with contamination above 20 µg/kg
are removed and it is assumed that these foods are ingested according
to the "European diet", the mean estimated intake of aflatoxin is 19
ng/person per day. Assuming an adult human weight of 60 kg, the
estimated population risk (potency × intake) is 0.0041 cancers/year
per 100 000 people with a range of 0.0006 to 0.01. In contrast, using
the same assumptions but applying a 10 µg/kg hypothetical standard,
the average aflatoxin intake is 18 ng/person per day, resulting in an
estimated population risk of 0.0039 cancers/year per 100 000 people
with a range of 0.0006 to 0.01. Thus, reducing the hypothetical
standard from 20 µg/kg to 10 µg/kg yields a drop in the estimated
population risk of approximately 2 additional cancers/year per 109
people.
The second example pertains to areas with higher contamination
(for these purposes, Chinese monitoring data of aflatoxin B1 in
groundnuts, maize and their products were used) and areas with a
larger population fraction as carriers of hepatitis B (in this case,
25% hepatitis B carriers was assumed). The estimated potency for this
population is 0.01 × 75% + 0.3 × 25% = 0.083, with a range of 0.014 to
0.15. Using a 20 µg/kg hypothetical standard and the "Far Eastern
diet", the average estimated intake is 125 ng/person per day yielding
an average population risk of 0.17 cancers/year per 100 000 people
with a range of 0.03 to 0.3. Using a 10 µg/kg hypothetical standard,
the average estimated intake drops to 103 ng per person, yielding an
estimated population risk of 0.14 cancers/year per 100 000 people with
a range of 0.02 to 0.3. Thus, reducing the hypothetical standard for
this population from 20 µg/kg to 10 µg/kg yields a drop in the
estimated population risk of 300 cancers/year per 109 people.
1 The Committee noted that aflatoxin data for Europe was for "all
cereals". However, in these calculations, it was assumed that the
aflatoxin level for "all cereals" applied to maize consumption only.
5.3 Conclusions
1. Aflatoxins are considered to be human liver carcinogens. Aflatoxin
B1 is the most potent carcinogen of the aflatoxins; most of the
toxicological data available are related to aflatoxin B1. Aflatoxin
M1, the hydroxylated metabolite of B1, has a potency approximately
one order of magnitude less than that of B1.
2. The potency of aflatoxins in HBsAg+ individuals is substantially
higher than the potency in HBsAg- individuals. Thus, reduction of the
intake of aflatoxins in populations with a high prevalence of HBsAg+
individuals will have greater impact on reducing liver cancer rates
than reductions in populations with a low prevalence of HBsAg+
individuals.
3. Vaccination against hepatitis B will reduce the prevalence of
carriers. The present analysis suggests that this would reduce the
potency of the aflatoxins in vaccinated populations and consequently
reduce liver cancer risks.
4. Analyses of the application of hypothetical standards (10 mg/kg or
20 µg/kg aflatoxin in food) to model populations indicate that: (i)
populations with a low prevalence of HBsAg+ individuals and/or with
a low mean intake (less than 1 ng/kg bw per day) are unlikely to
exhibit detectable1 differences in population risks for standards in
the range of the hypothetical cases; and (ii) populations with a high
prevalence of HBsAg+ individuals and high mean intake of aflatoxins
would benefit from reductions in aflatoxin intake.
5. The Committee has previously noted that reductions can be achieved
through avoidance measures such as improved farming and proper storage
practices and/or through enforcing standards for food or feed within
countries and across borders (Annex 1, reference 77).
6. In considering two competing standards, if the fraction of the
samples excluded under the two standards is similar, the higher
standard will yield almost the same liver cancer risks as the lower
standard. When a substantial fraction of the current food supply is
heavily contaminated, reducing the aflatoxin contamination levels may
detectably lower liver cancer rates. Conversely, when only a small
fraction of the current food supply is heavily contaminated, reducing
the standard by an apparently substantial amount may have little
appreciable effect on public health.
1 In the context of this statement "detectable" refers to an
aflatoxin-induced change in liver cancer rates that exceeds the year-
to-year variability around the current incidence and mortality rates.
Hence "detectable" refers to our ability to observe a significant
effect in the occurrence of liver cancer following intervention and
will depend upon the quality of the data available on historical
trends in incidence and mortality.
6. REFERENCES
Aguilar, F., Hussain, S.P., & Cerutti, P. (1993) Aflatoxin B1 induces
the transversion of G to T in codon 249 of the p53 tumor suppressor
gene in human hepatocytes. PNAS, 90: 8586-8590.
Aguilar, F., Harris, C.C., Sun, T., Hollstein, M., & Cerutti, P.
(1994) Geographic variation of p53 mutational profile in nonmalignant
human liver. Science, 264(5163): 1317-1319.
Aguilar, F., Harris, C.C., Sun, T., Hollstein, M., & Cerutti, P.
(1995) p53 mutations in nonmalignant human liver: fingerprints of
aflatoxins? Hepatology, 21(2): 600-601.
Ahmed, H., Hendrickse, R.G., Maxwell, S.M., & Yakabu, A.M. (1995)
Neonatal jaundice with reference to aflatoxins: an aetiological study
in Zaria, northern Nigeria. Ann. Trop. Paediatr., 15: 11-20.
Alavanja M.J., Rush, G.A., Stewart, P., & Blair, A. (1987)
Proportionate mortality study of workers in the grain industry.
J. Natl Cancer Inst., 78: 247-252.
Angsubhakorn, S., Bharmarapravati, N., Romruen, K., Sahaphong, S.,
Thamnavit, W., & Miyamoto, M. (1981a) Further study of alpha benzene
hexachloride inhibition of aflatoxin Bl hepatocarcinogenesis in rats.
Br. J. Cancer, 43: 881-883.
Angsubhakorn, S., Bharmarapravati, N., Romruen, K., & Sahaphong, S.
(1981b) Enhancing effects of dimethylnitrosamine on aflatoxin Bl
hepatocarcinogenesis in rats. Int. J. Cancer, 28: 621-626.
Ankrah, N.-Y. (1995) Alteration of glucose tolerance in mice fed low
levels of aflatoxins and with depressed glyoxalase-1 activity.
Vet. Hum. Toxicol., 37(1): 59-61.
Ankrah, N-A., Addo, P.G.A., Abrahmas, C.A., Ekuban, F.A., & Addae,
M.M. (1993) Comparative effects of aflatoxin G1 and B1 at levels
within human exposure limits on mouse liver and kidney. West Afr.
J. Med., 12(2): 105-109.
Ankrah, N.-A., Rikimaru, T., & Ekuban, F.A. (1994) Observations on
aflatoxins and the liver status of Ghanaian subjects. East. Afr.
Med. J., 71(11): 739-741.
Anwar, W.A., Khalil, M.M., & Wild, C.P. (1994) Micronuclei,
chromosomal aberrations and aflatoxin-albumin adducts in experimental
animals after exposure to aflatoxin B1. Mutat. Res., 322: 61-67.
Appleton, B.S. & Campbell, T.C. (1983) Dietary protein intervention
during the post-dosing phase of aflatoxin B1-induced hepatic
preneoplastic lesion development. J. Natl Cancer Inst., 70: 547-549.
Argentina (1996) Results of studies conducted on the presence of
mycotoxins during the period 1992-1996 (in Spanish).
Australia Market Basket Survey (1992). National Food Authority
(Information submitted to WHO by Australia).
Bailey, G.S., Price, R.L., Park, D.L., & Hendricks, J.D. (1994) Effect
of ammoniation of aflatoxin B1-contaminated cottonseed feedstock on
the aflatoxin M1 content of cows' milk and hepatocarcinogenicity in
the trout bioassay. Food Chem. Toxicicol., 32(8): 707-715.
Ball, R.W., Huyie, J.M., & Coulombe, R.A. Jr. (1995) Comparative
activation of aflatoxin B1 by mammalian pulmonary tissues.
Toxicol. Lett., 75: 119-125.
Bannasch, P., Khoshkhou, N.I., Hacker, H.J., Radaeva, S., Mrozek, M.,
Zillman, U., Kopp-Schneider, A., Haberkorn, U., Elgas, M., Tolle, T.,
Toggendork, M., & Toshkov, I. (1995) Synergistic hepatocarcinogenic
effect of hepadnaviral infection and dietary aflatoxin B1 in
woodchucks. Cancer Res., 55: 3318-3350.
Billy, T.J. (1996) Consideration of Government comments on the Draft
Guideline Level and Sampling Plans for Aflatoxin in Groundnuts.
Personal communication to H. Van der Kooi.
Booth, S.C., Bosenberg, H., Garner, R.C., Herzog, P.J., & Norpoth, K.
(1981) The activation of aflatoxin B1 in liver slices and in bacterial
mutagenicity assays using livers from different species including man.
Carcinogenesis, 2: 1063-1068.
Bosch, F.X. (1995) HCV and liver cancer: the epidemiological evidence.
In: Kobayashi, K., Purcell, R., Shimotohno, K., & Tabor, E. ed.
Hepatitis C virus and its involvement in the development of
hepatocellular carcinoma. Princeton Scientific Publishers, N.J., pp.
15-25.
Bosch, F.X. & Munoz, N. (1991) Hepatocellular carcinoma in the world:
Epidemiologic questions. In: Tabor, E., Di Bisceglie, A.M., & Purcell,
R.H. ed. Etiology, pathology, and treatment of hepatocellular
carcinoma in North America. Gulf Publishing Company, Houston, London,
Paris, Zurich, Tokyo, pp. 35-54 (Advances in Applied Biotechnology
Series, Volume 13).
Bowers, J.C. (1993) Relative versus absolute risk modeling of
aflatoxin. Risk Anal., 13: 9-10.
Bowers, J.C., Brown, B., Springer, J., Tollefson, L., Lorentzen, R., &
Henry, S. (1993) Risk assessment for aflatoxin: an evaluation based on
the multistage model. Risk Anal., 13: 637-642.
Brekke, O.L., et al (1975) Cereal Chem., 52: 205-211.
Bruce, R.D. (1990) Risk assessment for aflatoxin: II. Implications of
human epidemiology data. Risk Anal., 10: 561-569.
Butler, W.H. & Hempsall, V. (1981) Histochemical studies of
hepatocellular carcinomas in the rat induced by aflatoxin. J.
Pathol., 134: 157-170.
Butler, W.H., Greenblut, M., & Lijinsky, W. (1969) Carcinogenesis in
rats by aflatoxins B1, G1 and B2. Cancer Res., 29: 2206-2211.
Campbell, T.C. (1994) Correspondence re: Qian, et al., A follow-up
study of urinary markers of aflatoxin exposure and liver cancer risk
in Shanghai, People's Republic of China. Cancer Epidemiol.
Biomarkers Prev., 3: 3-10, 1994, and C.C. Harris, Solving the
viral-chemical puzzle of human liver carcinogenesis. Cancer
Epidemiol. Biomarkers Prev., 3: 1-2. Cancer Epidemiol. Biomarkers
Prev., 3: 519-521.
Campbell, T.C., Chen, J., Liu, C., Li, J., & Parpia, B. (1990)
Nonassociation of aflatoxin with primary liver cancer in a
cross-sectional ecological survey in the People's Republic of China.
Cancer Res., 50: 6882-6893.
Camus-Randon, A.-M., Raffalli, F., Bereziat, J.-C., McGregor, D.,
Konstandi, M., & Lang, M.A. (1996) Liver injury and expression of
cytochrome P450: evidence that regulation of CYP2A5 is different from
that of other major xenobiotic metabolizing CYP enzymes. Toxicol.
Appl. Pharmacol., 138: 140-148.
Cardis, E., Wild, C.P., Moolgavkar, S., & Zeise, L. (in press)
Aflatoxin and liver cancer. International Agency for Research on
Cancer, Lyon, Chapter 8 (IARC Scientific Publications).
Carlborg, F.W. (1979) Cancer, mathematical models, and aflatoxin.
Food Cosmet. Toxicol., 17: 169-166.
CDHS (California Department of Health Services) (1990). DRAFT - Risk
specific intake levels for aflatoxin (March). Presented to the
Proposition 65 Scientific Advisory Panel, Sacramento,University of
California at Davis.
Chang, M.-H., Chen, C.-J., Lai, M.-S., Hsu, H.-M., Wu, T.-C., Kong,
M.-S., Lian, D.-C., Shau, W.-Y., & Chen, D.-S. (1997) Universal
hepatitis B vaccination in Taiwan and the incidence of hepatocellular
carcinoma in children. New Engl. J. Med., 336(26): 1855-1859.
Chen, J. (1997) Dietary aflatoxin intake levels in China: data
compilation (Unpublished information submitted to WHO).
Chen, J., Campbell, T.C., Li, J., & Peto, R. (1990) Diet, lifestyle
and mortality in China: A study of the characteristics of 65 Chinese
counties. Joint publication of Oxford University Press (Oxford,
England), Cornell University Press (Ithaca, NY), and China People's
Medical Publishing House (Beijing, China).
Chen, C.J., Wang, L.Y., Lu, S.N., Wu, M.H., You, S.L., Zhang, Y.J.,
Wang, L.W., & Santella, R.M. (1996) Elevated aflatoxin exposure and
increased risk of hepatocellular carcinoma. Hepatology, 24: 38-42.
Chou, M.L., Lu, M.H., Pegram, R.A., Gao, P., Cao, S., Kong, J., &
Hart, R.W. (1993) Effect of caloric restriction on aflatoxin
B1-induced DNA synthesis, AFB1-DNA binding and cell proliferation in
Fischer 344 rats. Mech. Ageing Dev., 70: 23-33.
Choy, W.N. (1993) A review of the dose-response induction of DNA
adducts by aflatoxin B1 and its implication to quantitative
cancer-risk assessment. Mutat. Res., 296: 181-198.
Cova, L., Wild, C.P., Mehrotra, R., Turusov, V., Shirai, T., Lambert,
V., Jacquet, C., Tomatis, L., Trepo, C., & Montesano, R. (1996)
Contribution of aflatoxin B1 and hepatitis B virus infection in the
induction of liver tumors in ducks. Cancer Res., 50: 2156-2163.
Croy, R.G. & Crouch, E.A.C. (1991) Interaction of aflatoxin and
hepatitis B virus as carcinogens in human populations. In: Bray, G.A.
& Ryan, D.H. ed. Mycotoxins, cancer and health. Louisiana State
University Press, Baton Rouge, LA, pp. 87-100 (Pennington Nutrition
Series, Vol. 1).
Cullen, J.M. & Newberne, P.M. (1994) Acute hepatotoxicity of
aflatoxins. In: Eaton, D.L. & Groopman, J.D. ed. The toxicology of
aflatoxins: Human health, veterinary, and agricultural significance.
Academic Press, San Diego, CA, pp 1-26.
Cullen, J.M., Ruebner, B.H., Hsieh, L.S., Hyde, D.M., & Hsieh, D.P.
(1987) Carcinogenicity of dietary aflatoxin M1 in male Fischer rats
compared to aflatoxin B1. Cancer Res., 47: 1913-1917.
De Sylos, C.M., et al. (1996) Occurrence of aflatoxin M1 in milk and
dairy products commercialized in Campinas, Brazil (Unpublished
information submitted to WHO).
Diaz, P.V. (1996) Natural occurrence of aflatoxin B1 in raw material
and foodstuffs for birds and pigs in Columbia (1995-1996).
Dragan, Y.P. & Pitot, H.C. (1994) Aflatoxin carcinogenesis in the
context of the multistage nature of cancer. In: Eaton, D.L. &
Groopman, J.D. ed. The toxicology of aflatoxins: Human health,
veterinary, and agricultural significance. Academic Press, San Diego,
CA, pp. 179-198.
Eaton, D.L. & Gallagher, E.P. (1994) Mechanisms of aflatoxin
carcinogenesis. Annu. Rev. Pharmacol. Toxicol., 34: 135-172.
Eaton, D.L. & Groopman, J.D. (1994) The toxicology of aflatoxins:
Human health, veterinary, and agricultural significance. Academic
Press, San Diego, CA.
El-Nazami, H.S., Nicoletti, G., Neal, G.E., Donohue, D.C., & Ahokas,
J.T. (1995) Aflatoxin M1 in human breast milk samples from Victoria,
Australia and Thailand. Food Chem. Toxicol., 33(3): 173-179.
Epstein, S.M., Bartus, B., & Farber, E. (1969) Renal epithelial
neoplasms induced in male Wistar rats by oral aflatoxin B1. Cancer
Res., 29: 1045-1050.
FAO (1993) Sampling plans for aflatoxin analysis in peanuts and corn.
Food and Agriculture Organization of the United Nations, Rome (FAO
Food and Nutrition Paper 55).
FAO (1995) Worldwide regulations for mycotoxins: A compendium. Food
and Agriculture Organization of the United Nations, Rome (FAO Food
and Nutrition Paper 64).
Food and Nutrition Board (1989) Recommended dietary allowances, 6th
ed. National Research Council, National Academy of Sciences,
Washington, D.C., Chapter 6, pp. 52-77.
Fujimoto, Y., Hampton, L.L., Wirth, P.J., Wang, N.J., Xie, J.P., &
Thorgeirsson, S.S. (1994) Alterations of tumor suppressor genes and
allelic losses in human hepatocellular carcinomas in China. Cancer
Res., 54: 281-286.
Gallagher, E.P. & Eaton, D.L. (1995) In vitro biotransformation of
aflatoxin (AFB1) in channel catfish liver. Toxicol. Appl.
Pharmacol., 132: 82-90.
Gallagher, E.P., Wienkers, L.C., Stapleton, P.L., Kunze, K.L., &
Eaton, E.L. (1994) Role of human microsomal and human complementary
DNA-expressed cytochromes P4501A2 and P4503A4 in the bioactivation of
aflatoxin B1. Cancer Res., 54: 101-108.
Gallagher, E.P., Kunze, K.L., Stapleton, P.L., & Eaton, D.L. (1996)
The kinetics of aflatoxin B1 oxidation by human cDNA-expressed and
human liver microsomal cytochromes P450 1A2 and 3A4. Toxicol. Appl.
Pharmacol., 141: 595-606.
Gan, L.S., Skipper, P.L., Peng, X., Groopman, J.D., Chen, J.S, Wogan,
G.N., &Tannenbaum, S.R. (1988) Serum albumin adducts in the molecular
epidemiology of aflatoxin carcinogenesis: correlation with aflatoxin
B1 intake and urinary excretion of aflatoxin M1. Carcinogenesis, 9:
1323-1325.
Gaylor, D.W. & Kodell, R.L. (1980) Linear interpolation algorithm for
low dose risk assessment of toxic substances. J. Environ. Pathol.
Toxicol., 4: 305-315.
Gerbes, A.L. & Caselmann, W.H. (1993) Point mutations of the P53
gene, human hepatocellular carcinoma and aflatoxins. J. Hepatol.,
19: 312-315.
Gopalan, P., Tsuji, K., Lehmann, K., Kimura, M., Shinozuka, H., Sato,
K., & Lotlikar, P.D. (1993) Modulation of aflatoxin B1-induced
glutathione S-transferase placental form positive hepatic foci by
pre-treatment of rats with phenobarbital and buthioninesufoximine.
Carcinogenesis, 14(7): 1469-1470.
Gorelick, N.J. (1990) Risk assessment for aflatoxin: I. Metabolism of
aflatoxin B1 by different species. Risk Anal., 10(4): 539-559.
Griciute, L. (1980) Investigation on the combined action of
N-nitrosodiethylamine with other carcinogens. In: Walker, E.A.,
Griciute, L., Castegnaro, M., & Borzsonyi, M. ed. N-nitroso compounds:
Analysis, formation and occurrence. International Agency for Research
on Cancer, Lyon, pp 813-822 (IARC Scientific Publications No. 31).
Groopman, J.D., Zhu, J., Donahue, P.R., Pikul, A., Zhang, L., Chen,
J.S., & Wogan, G.N. (1992) Molecular dosimetry of urinary
aflatoxin-DNA adducts in people living in Guangxi Autonomous Region,
People's Republic of China. Cancer Res., 52: 45-52.
Groopman, J.D., Wild, C.P., Hasler, J., Chen, J., Wogan, G.N., &
Kansler, T.W. (1993) Molecular epidemiology of aflatoxin exposures:
Validation of aflatoxin-N-7-guanine levels in urine as a biomarker in
experimental rat models and humans. Environ. Health Perspect., 99:
107-113.
Groopman, J.D., Wogan, G.N., Roebuck, B.D., & Kensler, T.W. (1994)
Molecular biomarkers for aflatoxins and their application to human
cancer prevention. Cancer Res., 54(Suppl.): 1907s-1911s.
Guengerich, F.P., Johnson, W.W., Ueng, Y.-F., & Shimade, T. (1996)
Involvement of cytochrome P450, glutathione S-transferase, and epoxide
hydrolase in the metabolism of aflatoxin B1 and relevance to risk of
human liver cancer. Environ. Health Perspect., 104(3): 557-562.
Hall, A.J. & Wild, C.P. (1994) Epidemiology of aflatoxin-related
disease. In: Eaton, D.L. & Groopman, J.D. ed. The toxicology of
aflatoxins: Human health, veterinary, and agricultural significance.
Academic Press, San Diego, CA, pp. 233-258.
Harris, C.C. (1994) Solving the viral-chemical puzzle of human liver
carcinogenesis. Cancer Epidemiol. Biomarkers Prev., 3: 1-2.
Harris, C.C. (1995 At the crossroads of molecular carcinogenesis and
risk assessment. Chem. Ind. Inst. Toxicol. Act., 15(5): 1-6.
Hasler, J.A., Dube, N., Nyathi, C.B., Fuhrmann, H., & Sallmann, H.-P.
(1994) The influence of dietary fat on hepatic bioactivation of
aflatoxin B1 in rats. Res. Commun. Chem. Pathol. Pharmacol., 83(3):
279-287.
Hatch, M.C., Chen, C.J., Levin, B., Ji, B.T., Yang, G.Y., Hsu, S.W.,
Wang, L.W., & Santella, R.M. (1993) Urinary aflatoxin levels,
hepatitis B virus infection and hepatocellular carcinoma in Taiwan.
Int. J. Cancer, 54: 931-934.
Hayes, R.B., van Niewenhuize, J.P., Raatgever, J.W., & ten Kate,
F.J.W. (1984) Aflatoxin exposures in the industrial setting: an
epidemiological study of mortality. Food Chem. Toxicol., 22: 39-43.
Hayes, J.D., Nguyen, T., Judah, D.J., Petersson, D.G., & Neal, G.E.
(1994) Cloning of CDNAS from fetal rat liver encoding glutathione
S-transferase Yc polypeptides. J. Biol. Chem., 269(32): 20707-20717.
Heinonen, J.T., Fisher, R., Brendel, K., & Eaton, D.L.(1996)
Determination of aflatoxin B1 biotransformation and binding to hepatic
macromolecules in human precision liver slices. Toxicol. Appl.
Pharmacol., 136: 1-7.
Hendrickse, R.G., Coulter, J.B.S., Lamplugh, S.M., MacFarlane, S.B.J.,
Williams, T.E., Omer, M.J.A., & Suliman, G.I. (1982) Aflatoxins and
kwashiorkor: a study in Sudanese children. Br. Med. J., 285:
843-846.
Henry, S.H., DiNovi, M.J., Bowers, J.C., & Bolger, P.M. (1997) Risk
assessment for aflatoxin in corn and peanuts in the United States.
Poster presented at the Society of Toxicology meeting, Cincinnati,
Ohio, March 9-13.
Hoseyni, M.S. (1992) Risk assessment for aflatoxin: III. Modeling the
relative risk of hepatocellular carcinoma. Risk Anal., 12: 123-126.
Hsieh, D.P.H. & Atkinson, D.N. (1995) Recent aflatoxin exposure and
mutation at codon 249 of the human p53 gene: lack of association.
Food Addit. Contam., 12(3): 421-424.
Hsing, A.W., Guo, W., Chen, J., Li, J-Y., Stone, B.J., Blot, W.J., &
Fraumeni, J.F. Jr. (1991) Correlates of liver cancer mortality in
China. Int. J. Epidemiol., 20(1): 54
Hu, J-F., Chisari, F.V., & Campbell, T.C. (1994) Modulating effect of
dietary protein on transgene expression in hepatitis B virus (HBV)
transgenic mice. Proc. Am Assoc. Cancer Res., 35: 104.
Hulla, J.E., Chen, Z.Y., & Eaton, D.L. (1993) Aflatoxin B1-induced rat
hepatic hyperplastic nodules do not exhibit a site-specific mutation
within the p53 gene. Cancer Res., 53: 9-11.
IARC (1993) Some naturally occurring substances: food items and
constituents, heterocyclic aromatic amines and mycotoxin.
International Agency for Research on Cancer, Lyon, pp 245-395 (IARC
Monographs on the Evaluation of Carcinogenic Risks of Chemicals to
Humans, Volume 56).
IARC (1994) Hepatitis viruses. International Agency for Research on
Cancer, Lyon (IARC Monographs on the Evaluation of Carcinogenic Risks
of Chemicals to Humans, Volume 59).
IARC (1997) In: Toniolo, P., Boffetta, P., Shuker, D., Rothman, N.,
Hulka, B. & Pearce, H. ed. Application of biomarkers in cancer
epidemiology. International Agency for Research on Cancer, Lyon, pp.
251-264 (IARC Scientific Publications No. 142).
Ibeh, I.N., Uraih, N., & Ogonar, J.I. (1994) Dietary exposure of
aflatoxin in human male infertility in Benin City, Nigeria. Int. J.
Fertil., 39(4): 208-214.
Imazeki, F., Yokosuka, O., Ohto, M., & Omata, M. (1995) Aflatoxin and
p53 abnormality in duck hepatocellular carcinoma. J. Gastroenterol.
Hepatol., 10: 646-649.
International Commission on Radiological Protection (1975) Report of
the Task Group on Reference Man. Pergamon Press, New York (ICRP
Publication No. 23).
Itoh, S., Tagawa, S., Sawada, M., & Kamataki, T. (1993) High
susceptibility to aflatoxin B1 and benzo[ a]pyrene of BALB3T3
A31-1-1 cells expressing monkey CYP1A1. J. Toxicol. Sci., 18:
207-210.
Jacobson, L.P., Zhang, B.C., Zhu, Y.R., Wang, J.B., Wu, Y., Zang,
Q.N., Yu, L.Y., Qian, G.S., Kuang, S.Y., Li, Y.F., Fang, X., Xarba,
A., Chen, B., Enger, C., Davidson, N.E., Gorman, M.B., Gordon, G.B.,
Prochaska, H.J., Egner, P.A., Groopman, J.D., Munoz, A., Helzlsouer,
K.J., & Kensler, T.W. (1997) Oltipraz chemoprevention trial in Qidong,
People's Republic of China: study design and clinical outcomes.
Cancer Epidemiol. Biomarkers Prev., 6(4): 257-265.
Jakab, G.J., Hmielski, R.R., Zarba, A., Hemenway, D.R., & Groopman,
J.D. (1994) Respiratory aflatoxicosis: suppression of pulmonary and
systemic host defenses in rats and mice. Toxicol. Appl. Pharmacol.,
125: 198-205.
Japanese Ministry of Health, Food Sanitation Division (1995) The
situation of aflatoxin contamination in Japan, 1972-1989 and related
information prepared for the 47th JECFA..
Jennings, G.S., Heck, R., Oesch, F., & Steinberg, P. (1994) Metabolism
and cytotoxicity of AFB1 in cultured rat hepatocytes and
nonparenchymal cells: implications for tumorigenesis. Toxicol.
Appl. Pharmacol., 129: 86-94.
Judah, D.J., Hayes, J.D., Yang, J.-C., Lian, L.-Y., Roberts, G.C.K.,
Farmer, P.B., Lamb, J.H., & Neal, G.E. (1993) A novel aldehyde
reductase with activity towards a metabolite of aflatoxin B1 is
expressed in rat liver during carcinogenesis and following the
administration of an anti-oxidant. Biochem. J., 292: 13-18.
Kirby, G.M., Chemin, I., Montesano, R., Chisari, F.V., Lang, M.A., &
Wild, C.P. (1994) Induction of specific cytochrome P450s involved in
aflatoxin B1 metabolism in hepatitis B virus transgenic mice. Mol.
Carcinog., 11: 74-80.
Kirby, G.M., Wolf, C.R., Neal, G.E., Judah, D.J., Henderson, C.J.,
Srivatanakul, P., & Wild, C.P. (1993) In vitro metabolism of
aflatoxin B1 by normal and tumorous liver tissue from Thailand.
Carcinogensis, 14(12): 2613-2620.
Kopp-Schneider, A. & Portier, C.J. (1989) A note on approximating the
cumulative distribution function of the time to tumor onset in
multistage models. Biometrics, 45: 1259-1264.
Langouet, S., Coles, B., Morel., F., Becquemont, L., Beaune, P.,
Guengerich, F.P., Ketterer, B., & Guillouzo, A. (1995) Inhibition of
CYP1A2 and CYP3A4 by oltipraz results in reduction of aflatoxin B1
metabolism in human hepatocytes in primary culture. Cancer Res., 55:
5574-5579.
Liang, T.J. (1995) p53 proteins and aflatoxin B1: the good, the bad,
and the ugly. Hepatology, 22(4): 1330-1332.
Liu, Y.H., Taylor, J., Linko, P., Lucier, G.W., & Thompson, C.L.
(1991) Glutathione S-transferase mu in human lymphocyte and liver:
role in modulating formation of carcinogen-derived DNA adducts.
Carcinogenesis, 12(12): 2269-2275
Loechler, E.L. (1994) Mechanisms by which aflatoxins and other bulky
carcinogens induce mutations. In: Eaton, D.L. & Groopman, J.D. ed. The
toxicology of aflatoxins: Human health, veterinary, and agricultural
significance. Academic Press, San Diego, CA, pp. 149-178.
Lye, M.S., Ghazali, A.A., Mohan, J., Alwin, N., & Nair, R.C. (1995) An
outbreak of acute hepatic encephalopathy due to severe aflatoxicosis
in Malaysia. Am. J. Trop. Med. Hyg., 53(1): 68-72.
McGlynn, K.A., Rosvold, E.A., Lustbader, E.D., Hu, Y., Clapper, M.L.,
Zhou, T., Wild, C.P., Xia, X.-L., Baffoe-Bonnie, A., Ofori-Adjei, D.,
Chen, G.-C., London, W.T., Shen, F.-M., & Buetow, D.H.(1995)
Susceptibility to hepatocellular carcinoma is associated with genetic
variation in the enzymatic detoxification of aflatoxin B1. Proc.
Natl Acad. Sci., 92: 2384-2387.
McLean, M & Dutton, M.F. (1995) Cellular interactions and metabolism
of aflatoxin: an update. Pharmacol. Ther., 65: 163-192.
Marquez-Marquez, R., Badrigal-Bujaidar, E., & Hernandez, I.T. (1993)
Genotoxic evaluation of ammonium inactivated aflatoxin B1 in mice fed
with contaminated corn. Mutat. Res., 299: 1-8.
Marquez-Marquez, R., Hernandez, I.T., & Bujaidar, E.M. (1995)
Genotoxicity of aflatoxin B1 and its ammonium derivatives. Food
Addit. Contam., 12(3): 425-429.
Massey, T.E., Stewart, R.K., Daniels, J.M., & Liu, L. (1995)
Biochemical and molecular aspects of mammalian susceptibility to
aflatoxin B1 carcinogenicity. Proc. Soc. Exp. Biol. Med., 208:
213-227.
Merkow, L.P., Epstein, S.M., Slifkin, M., & Pardo, M. (1973) The
ultrastructure of renal neoplasms induced by aflatoxin B1. Cancer
Res., 33: 1608-1614.
Mexico D.F. (1996) Monitoring results 1992-1996: Document presented at
the Course on Mycotoxins (in Spanish).
Moore, M.R., Pitot, H.C., Miller, E.C., & Miller, J.A. (1982)
Cholangiocellular carcinoma incidence in Syrian hamsters administered
aflatoxin B1 in large doses. J. Natl Cancer Inst., 68: 271-278.
Newberne, P.M. & Rogers, A.W. (1973) Rat colon carcinomas associated
with aflatoxin and marginal vitamin A. J. Natl Cancer Inst., 50:
439-444.
Nicaragua (1996) Detection of aflatoxin in samples from the
Directorate of Food and Zoonosis Control, analyzed at the Department
of Environmental Toxicology, National Reference and Diagnosis Centre
(in Spanish).
Nixon, J.E., Hendricks, J.D., Pawlowski, N.E., Loveland, P.M., &
Sinnhuber, R.O. (1981) Carcinogenicity of aflatoxin in Fischer 344
rats. J. Natl Cancer Inst., 66: 1159-1163.
Olsen, J.H., Dragsted, L., & Autrup, H. (1988) Cancer risk and
occupational exposure to aflatoxins in Denmark. Br. J. Cancer, 58:
392-396.
Olubuyide, I.O., Maxwell, S.M., Akinyinka, O.O., Hart, C.A., Neal,
G.E., & Hendrickse, R.G. (1993) Hbsag and aflatoxins in sera of rural
(Igbo-Ora) and urban (Ibadan) populations in Nigeria. Afr. J. Med.
Med. Sci., 22: 77-80.
Ozturk, M. (1991) p53 mutation in hepatocellular carcinoma after
aflatoxin exposure. Lancet, 338: 1356-1359.
Pacin, A. (no date) Personal communication to B.J. Petersen.
Peers, F.G. & Linsell, C.A. (1977) Dietary aflatoxins and human
primary liver cancer. Ann. Nutr. Alim., 31: 1005-1018.
Peers, F.G., Gilman, G.A., & Linsell C.A. (1976) Dietary aflatoxins
and human liver cancer: A study in Swaziland. Int. J. Cancer, 17:
167-176.
Peers, F., Bosch, X., Kaldo, J., Linsell, A., & Pluijem, M. (1987)
Aflatoxin exposure, hepatitis B virus infection and liver cancer in
Swaziland. Int. J. Cancer, 39: 545-553.
Preston, R.S., Hayes, J.R., & Campbell, T.C. (1976) The effect of
protein deficiency on the in vivo binding of aflatoxin B1 to rat
liver macromolecules. Life Sci., 19: 1191-1198.
Primiano, T., Egner, P.A., Sutter, T.R., Kelloff, G.J., Roebuck, B.D.,
& Kensler, T.W. (1995) Intermittent dosing with oltipraz: relationship
bewteen chemoprevention of aflatoxin-induced tumorigenesis and
induction of glutathione S-transferase. Cancer Res., 55: 4319-4324.
Qian, G.-S., Ross, R.K., Yu, M.C., Yuan, J.-M., Gao, Y.-T., Henderson,
B.E., Wogan, G.N., & Groopman, J.D. (1994) A follow-up study of
urinary markers of aflatoxin exposure and liver cancer risk in
Shanghai, People`s Republic of China. Cancer Epidemiol. Biomarkers
Prev., 3: 3-10.
Raisuddin, S., Singh, K.P., Zaidi, B.N., Paul, B.N., & Ray, P.K.
(1993) Immunosuppressive effects of aflatoxin in growing rats.
Mycopathologia, 124: 189-194.
Ramesh, R. & Panda, S.K. (1993) Hepatitis B virus (HBV) genome in
antibody positive hepatocellular carcinoma (HCC). Letters to the
Editor. J. Hepatol., 19: 319.
Read, M. (1989) Removal of aflatoxin contamination from the Australian
groundnut crop. Proceedings of the ICRISAT International Workshop,
6-9 October 1987. International Crops Research Institute for the
Semi-Arid Tropics.
Read, M. (1997) Letter to J. Baines providing estimates of aflatoxins
in peanuts.
Reddy, J.K., Svoboda, D.J., & Rao, M.S. (1976) Induction of liver
tumors by aflatoxin B1 in the tree shrew (Tupaia glis), a nonhuman
primate. Cancer Res., 36: 151-160.
Regueiro, O.S. (no date) Report on criteria for aflatoxins B1, G1 and
M presented for review at the XLVI meeting of the FAO/WHO Expert
Committee on Food Additives (in Spanish).
Rivkina, M.B., Cullen, J.M., Robinson, W.S., & Marion, P.L. (1994)
State of the p53 gene in hepatocellular carcinomas of ground squirrels
and woodchucks with past and ongoing infection with hepadnaviruses.
Cancer Res., 54: 5430-5437.
Ross, R.K., Yuan, J.-M., Yu, M.C., Wogan, G.N., Qian, G.-S., Tu, J.T.,
Groopman, J.D., Gao, Y.-T., & Henderson, B.E. (1992) Urinary aflatoxin
biomarkers and risk of hepatocellular carcinoma. Lancet, 339(8799):
943-946.
Sabino, M. (1997) Personal communication to B.J. Petersen from A.
Pacin dated 4.8.1997: Maize monitoring results for 321 samples.
Santella, R.M., Zhang, Y.-J., Chen, C.-J., Lee, C.-S., Haghighi, B.,
Hang, G.-Y., Wang, L.-W., & Feitelson, M. (1993) Immunohistochemical
detection of aflatoxin-B1-DNA adducts and hepatitis B virus antigens
in hepatocellular carcinoma and nontumorous liver tissue. Environ.
Health Perspect., 99: 199-202.
Sawada, M., Kitamura, R., Norose, T., Kitada, M., Itahashi, K., &
Kamataki, T. (1993) Metabolic activation of aflatoxin B1 by human
placental microsomes. J. Toxicol. Sci., 18: 129-132.
Scholl, P., Musser, S.M., Kensler, T.W., & Groopman, J.D. (1995)
Molecular biomarkers for aflatoxin and their application to human
liver cancer. Pharmacogenetics, 5: S171-S176.
Schulsinger, D.A., Root, M.M., & Campbell, T.C. (1989) Effect of
dietary protein quality on development of aflatoxin B1-induced hepatic
preneoplastic lesions. J. Natl Cancer Inst., 81: 1241-1245.
SCOOP (1996) Scientific co-operation on questions relating to food:
Working document in support of a SCF risk assessment of aflatoxin:
Task 3.2.1 (SCOOP/CNTM/1). Task Co-ordinator, UK.
Sengstag, C. & Wurgler, F.E. (1994) DNA recombination induced by
aflatoxin B1 activated by cytochrome P450 1A enzymes. Mol.
Carcinog., 11: 227-235.
Shank, R.C., Gordon, J.E., Wogan, G.N., Nondasuta, A., & Subhamani, B.
(1972a) Dietary aflatoxins and human liver cancer. III. Field survey
of rural Thai families for ingested aflatoxins. Food Cosmet.
Toxicol., 10: 71-84.
Shank, R.C., Bhamarapravati, N., Gordon, J.E., & Wogan, G.N. (1972b)
Dietary aflatoxins and human liver cancer. IV. Incidence of primary
liver cancer in two municipal populations of Thailand. Food
Cosmet. Toxicol., 10: 171-179.
Shen, H.-M., Ong, D.N., & Shi, C.-Y. (1995) Involvement of reactive
oxygen species in aflatoxin B1-induced cell injury in cultured rat
hepatocytes. Toxicology, 99: 115-123.
Shi, C.-Y., Chua, S.-C., Lee, H.-P., & Ong, C.-N. (1994) Inhibition of
aflatoxin B1-DNA binding and adduct formation by selenium in rats.
Cancer Lett., 82: 203-208.
Shi, C.-Y, Hew, Y.-C., & Ong, C.-N. (1995a) Inhibition of aflatoxin
B1-induced cell injury by selenium: an in vitro study. Hum. Exp.
Toxicol., 14(1): 55-60.
Shi, C.-Y., Phang, T.W., Lin, Y., Wee, A., Li, B., Lee, H.P., & Ong,
C.N. (1995b) Codon 249 mutation of the p53 gene is a rare event in
hepatocellular carcinomas from ethnic Chinese in Singapore. Br. J.
Cancer, 72: 146-149.
Sieber, S.M., Correa, P., Dalgard, D.W., & Adamson, R.H. (1979)
Induction of osteogenic sarcomas and tumors of the hepatobiliary
system in human primates with aflatoxin B1. Cancer Res., 39:
4545-4554.
Sinha, S.P. & Dharmshila, K. (1994) Vitamin A ameliorates the
genotoxicity in mice of aflatoxin B1-containing Aspergillus
flavus infested food. Cytobios, 79: 85-95.
Srivatanakul, P., Parkin, D.M., Khlat, M., Chenvidhya, D., Chotiwan,
P., Insiripong, S.L., Abbé, K.A., & Wild, C.P. (1991) Liver cancer in
Thailand: II. A case-control study of hepatocellular carcinoma.
Int. J. Cancer, 48: 329-332.
Stoner, G.D., Conran, P.B., Greisiger, E.A., Stober, J., Morgan, M., &
Pereira, M.A. (1986) Comparison of two routes of chemical
administration on the lung adenoma response in strain A/J mice.
Toxicol. Appl. Pharmacol., 82: 19-31.
Stoloff, L. (1983) Aflatoxin as a cause of primary liver-cell cancer
in the United States: a probability study. Nutr. Cancer, 5:
165-186.
Stoloff, L. & Friedman, L. (1976) Information bearing on the
evaluation of the hazard to man from aflatoxin ingestion. PAG Bull.,
6: 21-32.
Stresser, D.M., Bailey, G.S., & Williams, D.E. (1994a)
Indole-3-carbinol and beta-naphthoflavone induction of aflatoxin
B12 metabolism and cytochromes P-450 associated with bioactivation
and detoxication of aflatoxin B1 in the rat. Drug Metab. Dispos.,
22(3): 383-391.
Stresser, D.M., Williams, D.E., McLellan, L.I., Harris, T.M., &
Bailey, G.S. (1994b) Indole-3-carbinol induces a rat liver glutathione
transferase subunit (Yc2) with high activity toward aflatoxin B1
exo-epoxide. Drug Metab. Dispos., 22(3): 392-399.
Tanaka, N., Chiba, T., Matsuzaki, Y., Osuga, T., Aikawa, T., &
Mitamura, K. (1993) High prevalence of hepatitis B and C viral markers
in Japanese patients with hepatocellular carcinoma. Gastroenterol.
Jpn., 28(4): 547-553.
Thomas, D.B. (1991) Exogenous steroid hormones and hepatocellular
carcinoma. In: Tabor, E., Di Bisceglie, A.M., & Purcell, R. ed.
Etiology, pathology and treatment of hepatocellular carcinoma.
PPC-Gulf Publishing Company, Houston, TX, Chapter 6, pp. 77-91.
Thorslund, T.W., Brown, C.C., & Charnley, G. (1987) Biologically
motivated cancer risk models. Risk Anal., 7: 109-119.
Travis C.C. & Land, M.L. (1990) Estimating the mean of data sets with
nondetectable values. Environ Sci. Technol., 24: 961-962.
Ueng, Y-F., Shimada, T., Yamazaki, H., & Guengerich, F.P. (1995)
Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome
P450 enzymes. Chem. Res. Toxicol., 8: 218-225.
Van Egmond, H.P. (1983) Food Chem., 11: 289-307.
van Rensburg, S.J., Cook-Mozaffari, P., van Schalkwyk, D.J., van der
Watt, J.J., Vincent, T.J., & Purchase, I.F. (1985) Hepatocellular
carcinoma and dietary aflatoxin in Mozambique and Transkei. Br. J.
Cancer, 51: 713-726.
Venable, W.N. & Ripley, D.B. (1994) Modern applied statistics with
S-Plus - Chapter 13: Tree based methods. Springer-Verlag, Basel,
Heidelberg.
Vesselinovitch, S.D., Mihailovich, N., Wogan, G.N., Lombard, L.S., &
Rao, K.V.N. (1972) Aflatoxin B1, a hepatocarcinogen in the infant
mouse. Cancer Res., 32: 2289-2291.
Waltking, A.E. (1971) Fate of aflatoxin during roasting and storage of
contaminated peanut products. J. Assoc. Off. Anal. Chem., 54:
533-539.
Wang, J-S., Qian, G.-S., Zarba, A., He, X., Zhu, Y.-R., Zhang, B.-C.,
Jacobson, L., Gange, S.J., Munoz, A., Kensler, T.W., & Groopman, J.D.
(1996a) Temporal patterns of aflatoxin-albumin adducts in hepatitis B
surface antigen-positive and antigen-negative residents of Daxin,
Qidong County, People's Republic of China. Cancer Epidemiol.
Biomarkers Prev., 5: 253-261.
Wang, L.-Y., Hatch, M., Chen, C.-J., Levin, B., You, S.-L., Lu, S.-N.,
Wu, M.-H., Wu, W.-P., Wang, L.-W., Wang, Q., Huang, G.-T., Yang,
P.-M., Lee, H-.S., & Santella, R.M. (1996b) Aflatoxin exposure and
risk of hepatocellular carcinoma in Taiwan. Int. J. Cancer, 67:
620-625.
Ward, J.M., Sontag, J.M., Weisburger, E.K., & Brown, C.A. (1975)
Effect of lifetime exposure to aflatoxin B1 in rats. J. Natl
Cancer Inst., 55: 107-110.
Wieder, R., Wogan, G.N., & Shimkin, M.A. (1968) Pulmonary tumors in
strain A mice given injections of aflatoxin B1. J. Natl Cancer
Inst., 40: 1195-1197.
Wild, C.P., Hudson, G.J., Sabbioni, G., Chapot, B., Hall, A.J., Wogan,
G.N., Whittle, H., Montesano, R., & Groopman, J.D. (1992) Dietary
intake of aflatoxins and the level of albumin-bound aflatoxin in
peripheral blood in the Gambia, West Africa. Cancer Epidemiol.
Biomarkers Prev., 1: 229-234.
Wild, C.P., Fortuin, M., Donato, F., Whittle, H.C., Hall, A.J., Wolf,
C.R., & Montesano, R. (1993) Aflatoxin, liver enzymes and hepatitis B
virus infection in Gambian children. Cancer Epidemiol. Biomarkers
Prev., 2: 555-561.
Wild, C.P., Hasegawa, R., Barraud, L., Chutimataewin, S., Chapot, B.,
Ito, N., & Montesano, R. (1996) Aflatoxin-albumin adducts: a basis for
comparative carcinogenesis between animals and humans. Cancer
Epidemiol. Biomarkers. Prev., 5: 179-189.
Wiseman, W.W., et al (1983) J. Food Prot., 46: 530-532.
Wogan, G.N., Palianlunga, S., & Newberne, P.M. (1974) Carcinogenic
effects of low dietary levels of aflatoxin Bl in rats. Food Cosmet.
Toxicol., 12: 681-685.
Wogan, G.N. (1992) Aflatoxins as risk factors for hepatocellular
carcinoma in humans. Cancer Res., 52(Suppl.): 2114-2118s.
Wood, G.E. (1995) Communication from Dr G.E. Wood (US FDA) to Dr J.
Paakkanen (FAO) dated 21.12.1995.
Wu-Williams, A.H., Zeise, L., & Thomas, D. (1992) Risk assessment for
aflatoxin B1: A modeling approach. Risk Anal., 12: 559-567.
Yahl, K.R., et al. (1971) Laboratory wet-milling of corn containing
high-levels of aflatoxin and a survey of commercial wet-milling
products. Cereal Chem., 48: 385-391.
Yan, R.Q., Su, J.J., Huang, D.R., Gan, Y.C., Yang, C., & Huang, G.H.
(1996) Human hepatitis B virus and hepatocellular carcinoma II.
Experimental induction of hepatocellular carcinoma in tree shrews
exposed to hepatitis B virus and aflatoxin B1.
Yap, E.P.H., Cooper, K., Maharaj, B., & McGee, J.O'D. (1993) p53 codon
249ser hot-spot mutation in HBV-negative hepatocellular carcinoma.
Lancet, 341(8839): 251.
Yeh, F.-S., Mo, C.-C.,& Yen, R.-C. (1985) Risk factors for
hepatocellular carcinoma in Guangxi, People's Republic of China.
Natl Cancer Inst. Monogr., 69: 47-48.
Yeh, F.-S., Yu, M.C., Mo, C.-C., Luo, S., Tong, M.J., & Henderson,
B.E. (1989) Hepatitis B virus, aflatoxin, and hepatocellular carcinoma
in Southern Guangxi, China. Cancer Res., 49: 2506-25509.
Yoshizawa, T., Yamashita, A., & Chokethaworn, N. (1996) Occurrence of
fumonisins and aflatoxins in maize from Thailand. Food Addit.
Contam., 13: 163-168.
Youngman, L.D. & Campbell, T.C. (1992) Inhibition of aflatoxin
B1-induced gamma-glutamyltranspeptidase positive (GGT+) hepatic
preneoplastic foci and tumors by low protein diets: evidence that
altered GGT+ foci indicate neoplastic potential. Carcinogenesis,
13(9): 1607-1613.
Yuan, J.M., Ross, R.K., Stanczyk, F.Z., Govindaragan, S., & Gao, Y.T.
(1995) A cohort study of serum testosterone and hepatocellular
carcinoma in Shanghai, China. Int. J. Cancer, 63: 491-493.
Zimbawbe Government Analyst Laboratory. Aflatoxin surveillance:
Monitoring results 1995-1996.