
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 88
POLYCHLORINATED DIBENSO- PARA-DIOXINS AND DIBENZOFURANS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR POLYCHLORINATED
DIBENZO-PARA-DIOXINS AND DIBENZOFURANS
1. SUMMARY AND RECOMMENDATIONS
1.1. Summary
1.1.1. Sources
1.1.2. Ambient levels and routes of exposure
1.1.3. Toxicokinetics, biotransformation, and
biological monitoring
1.1.4. Health effects
1.1.4.1 Animals
1.1.4.2 Humans
1.1.5. Conclusion
1.2. Recommendations
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES,
ANALYTICAL METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Analytical methods
2.3.1. General aspects
2.3.2. Sampling strategy and sampling methods
2.3.3. Extraction procedures
2.3.4. Sample clean-up
2.3.5. Isomer identification
2.3.6. Quantification
2.3.7. Confirmation
2.3.8. Other analytical methods
3. SOURCES OF ENVIRONMENTAL POLLUTION
3.1. Production, synthesis, and use
3.2. Industrial processes
3.3. Contamination of commercial products
3.3.1. Chlorophenoxyacetic acid herbicides
3.3.2. Hexachlorophene
3.3.3. Chlorophenols
3.3.4. Polychlorinated biphenyls (PCBs)
3.3.5. Chlorodiphenyl ether herbicides
3.3.6. Hexachlorobenzene
3.3.7. Rice oil
3.4. Sources of heavy environmental pollution
3.4.1. Industrial accidents
3.4.2. Improper disposal of industrial waste
3.4.3. Heavy use of chemicals
3.5. Other sources of PCDDs and PCDFs in the
environment
3.5.1. Thermal degradation of technical
products
3.5.2. Incineration of municipal waste
3.5.3. Incineration of sewage sludge
3.5.4. Incineration of hospital waste
3.5.5. Incineration of hazardous waste
3.5.6. Metal industry and metal treatment
industry
3.5.7. Wire reclamation
3.5.8. Traffic
3.5.9. Fires and accidents in PCB-filled
electrical equipment
3.5.10. Pulp and paper industry
3.5.11. Incineration of coal, peat, and wood
3.5.12. Inorganic chlorine precursors
3.5.13. Photochemical processes
3.6. Comparison of isomeric pattern and congener
profiles from various sources
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND
TRANSFORMATIONS
4.1. Environmental transport
4.1.1. Air
4.1.2. Water
4.1.3. Soil and sediments
4.2. Environmental transformation
4.2.1. Abiotic transformation
4.2.2. Biotransformation and biodegradation
4.3. Bioaccumulation
4.4. Levels in biota
4.4.1. Vegetation
4.4.2. Aquatic organisms
4.4.3. Terrestrial animals
4.4.4. Human data
4.4.4.1 Adipose tissue
4.4.4.2 Blood plasma
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Air
5.2. Water and leachate
5.3. Soil and sediment
5.4. Food
5.4.1. Meat and bovine milk
5.4.2. Human milk
5.4.3. Rice
5.5. Yusho and Yu-cheng episodes
6. KINETICS AND METABOLISM OF 2,3,7,8-TETRACHLORODIBENZO-
P-DIOXIN (TCDD) AND OTHER PCDDs
6.1. Uptake, distribution, and excretion
6.1.1. Studies on rats
6.1.2. Studies on mice
6.1.3. Studies on guinea-pigs
6.1.4. Studies on hamsters
6.1.5. Studies on monkeys
6.1.6. Studies on dogs
6.1.7. Studies on cows
6.1.8. In vitro studies
6.2. Metabolic transformation
6.2.1. Studies on mammals
6.2.1.1 In vivo studies
6.2.1.2 In vitro studies
6.3. Transfer via placenta and/or milk
6.4. Matrix effects on the uptake
("bio-availability")
7. EFFECTS OF TCDD AND OTHER PCDDs ON EXPERIMENTAL
ANIMALS AND IN VITRO TEST SYSTEMS
7.1. Acute toxicity
7.1.1. In vivo studies on mammals
7.1.2. In vitro studies on mammalian cells
7.1.3. Studies on birds
7.1.4. Toxicity of metabolites
7.1.5. Modulation of the acute toxicity
7.2. Short-term toxicity
7.2.1. Studies on rats
7.2.2. Studies on mice
7.2.3. Studies on guinea-pigs
7.2.4. Studies on hamsters
7.2.5. Studies on monkeys
7.3. Long-term toxicity
7.3.1. Studies on rats
7.3.2. Studies on mice
7.3.3. Studies on monkeys
7.4. Effects detected by special studies
7.4.1. Wasting syndrome
7.4.2. Hepatotoxicity
7.4.2.1 Morphological alterations
7.4.2.2 Hepatic plasma membrane
function
7.4.2.3 Biliary excretion
7.4.3. Porphyria
7.4.4. Epidermal effects
7.4.4.1 In vivo studies
7.4.4.2 In vitro studies
7.4.5. Effects on the immune system
7.4.5.1 Histopathology
7.4.5.2 Humoral-mediated immunity
7.4.5.3 Cell-mediated immunity
7.4.5.4 Macrophage function
7.4.6. Myelotoxicity
7.4.7. Effects on the intermediary
metabolism
7.4.8. Enzyme induction
7.4.8.1 Studies on rats
7.4.8.2 Studies on mice
7.4.8.3 Studies on guinea-pigs
7.4.8.4 Studies on rabbits
7.4.8.5 Studies on hamsters
7.4.8.6 Studies on cows
7.4.8.7 Studies on chick embryos
7.4.8.8 Studies on cell cultures
7.4.9. Endocrine effects
7.4.10. Vitamin A storage
7.5. Embryotoxicity and reproductive effects
7.5.1. Studies on rats
7.5.2. Studies on mice
7.5.3. Studies on rabbits
7.5.4. Studies on monkeys
7.5.5. Studies on chickens
7.6. Mutagenicity and related end-points
7.6.1. Mutagenicity
7.6.1.1 Studies on bacteria
7.6.1.2 Studies on eukaryotic cells
7.6.1.3 In vivo studies
7.6.2. Interaction with nucleic acids
7.6.3. Cytogenetic effects
7.6.4. Cell transformation
7.7. Carcinogenicity
7.7.1. Long-term animal studies on single
compounds
7.7.2. Long-term animal studies with mixed
compounds
7.7.3. Short-term and interaction studies
7.8. Mechanisms of action
7.8.1. Receptor-mediated effects
7.8.2. Toxicokinetics
7.8.3. Impairment of normal cellular regulatory
systems
7.8.3.1 Endocrine imbalance
7.8.3.2 Body weight regulation
7.8.3.3 Plasma membrane function
7.8.3.4 Impaired vitamin A storage
7.8.4. Lipid peroxidation
8. EFFECTS OF PCDDs ON HUMAN BEINGS - EPIDEMIOLOGICAL
AND CASE STUDIES
8.1. Occupational studies - historical perspective
8.2. General population studies
8.2.1. Missouri, USA
8.2.2. Seveso, Italy
8.2.3. Viet Nam
8.3. Signs and symptoms in humans associated with
TCDD exposure
8.3.1. Skin manifestations
8.3.2. Systemic effects
8.3.3. Neurological effects
8.3.4. Psychiatric effects
8.4. Epidemiological studies
8.5. Human experimental studies
9. TOXICOKINETICS OF PCDFs
9.1. Uptake, distribution, and excretion
9.1.1. Studies with 2,3,7,8-tetrachlorodibenzo-
furan (2,3,7,8-TCDF)
9.1.2. Studies with other PCDFs
9.2. Metabolic transformation
9.3. Transfer via placenta and/or milk
10. EFFECTS OF PCDFs ON ANIMALS
10.1. Acute toxicity
10.1.1. Studies on rats
10.1.2. Studies on mice
10.1.3. Studies on guinea-pigs
10.1.4. Studies on rabbits
10.1.5. Studies on monkeys
10.2. Short-term toxicity
10.2.1. Studies on rats
10.2.2. Studies on mice
10.2.3. Studies on guinea-pigs
10.2.4. Studies on rabbits
10.2.5. Studies on hamsters
10.2.6. Studies on monkeys
10.2.7. Studies on chickens
10.3. Chronic toxicity
10.3.1. Studies on monkeys
10.4. Effects detected by special studies
10.4.1. Immunobiological effects
10.4.1.1 Histopathology
10.4.1.2 Humoral-mediated immunity
10.4.1.3 Cell-mediated immunity
10.4.2. Enzyme induction
10.4.2.1 Studies on rats
10.4.2.2 Studies on mice
10.4.2.3 Studies on chickens
10.4.2.4 Studies on cell cultures
10.4.3. Receptor binding
10.5. Embryotoxicity and reproductive effects
10.6. Mutagenicity
10.7. Carcinogenicity
11. EFFECTS OF PCDFs ON HUMAN BEINGS
11.1. Yusho and Yu-cheng
12. EVALUATION OF HEALTH RISKS FROM THE EXPOSURE TO
CHLORINATED DIBENZO-P-DIOXINS (PCDDs) AND
DIBENZOFURANS (PCDFs)
12.1. Introduction
12.2. Exposure assessment
12.2.1. Sources of contamination
12.2.2. Ambient levels
12.2.3. Routes of exposure
12.2.4. Bioavailability
12.3. Animal data
12.3.1. Toxicokinetics of 2,3,7,8-TCDD
12.3.2. Toxicokinetics of PCDDs and PCDFs,
other than TCDD
12.3.3. Toxic effects 2,3,7,8-TCDD
12.3.4. Toxic effects of PCDDs and PCDFs,
other than TCDD
12.3.5. Review of species differences
12.4. Human health effects
12.4.1. PCDDs
12.4.2. PCDFs
12.4.3. Human body burden and kinetics
12.5. General conclusions
13. RECOMMENDATIONS
14. EVALUATIONS BY INTERNATIONAL BODIES AND THE CONCEPT
OF TCDD EQUIVALENTS
14.1. International evaluations
14.2. Methodologies used in assessment of
risk from PCDDs and PCDFs
14.2.1. Individual congeners
14.2.2. Mixtures of PCDD and PCDF congeners and
isomers - concept of TCDD toxic
equivalents
REFERENCES
FRENCH TRANSLATION OF SUMMARY, EVALUATION, AND
RECOMMENDATIONS
WHO TASK GROUP ON CHLORINATED DIBENZO-p-DIOXINS AND
DIBENZOFURANS
Members
Dr U.G. Ahlborg, Unit of Toxicology, National Institute of
Environmental Medicine, Stockholm, Sweden
Dr J.S. Bellin, Office of Toxic Substances, US Environmental
Protection Agency, Washington, DC, USA
Dr B. Birmingham, Ministry of the Environment, Hazardous Contaminants
Section, Toronto, Ontario, Canada
Professor A.D. Dayan, Department of Health and Social Security,
St Bartholomew's Hospital Medical College, London, United
Kingdom (Chairman)
Dr A. di Domenica, Instituto Superiore di Sanita, Rome, Italy
Dr M. Greenberg, Department of Health and Social Security,
Division of Toxicology and Environmental Protection, London,
United Kingdom
Dr R.D. Kimbrough, United States Department of Health and Human
Services, Center for Disease Control, Atlanta, Georgia, USA
(Now at the US Environmental Protection Agency Washington,
DC, USA)
Dr R. Koch, Department of Toxicology, Institute of Hygiene,
Gera, DDR
Professor C. Rappe, Department of Chemistry, University of
Umea, Umea, Sweden
Dr S. Safe, Texas A and M University, College Station, Texas,
USA
Dr H. Spielmann, Max von Pettenkofer Institute, Bundesgesundheitsamt,
Berlin (West)
Dr J. Vos, National Institute of Public Health and Environmental
Hygiene, Bilthoven, Netherlands
Representatives
Dr A. Berlin, Health and Safety Directorate, Commission of the
European Communities, Luxembourg
Mrs E. Cox, Department of the Environment, London, United
Kingdom
Miss F.D. Pollitt, Department of the Environment, London,
United Kingdom
Secretariat
Dr G.C. Becking, International Programme on Chemical Safety,
World Health Organization, Research Triangle Park, North
Carolina, USA (Secretary)
Secretariat (contd)
Dr H. Hakensson, Unit of Toxicology, National Institute of
Environmental Medicine, Stockholm, Sweden (Temporary
Adviser) (Rapporteur)
Dr E. Johnson, International Agency for Research on Cancer,
World Health Organization, Lyons, France
Dr S. Tarkowski, Regional Office for Europe, World Health
Organization, Copenhagen, Denmark
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the criteria
documents as accurately as possible without unduly delaying their
publication. In the interest of all users of the environmental health
criteria documents, readers are kindly requested to communicate any
errors that may have occurred to the Manager of the International
Programme on Chemical Safety, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda, which
will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Palais des
Nations, 1211 Geneva 10, Switzerland (Telephone No. 7988400 -
7985850).
ENVIRONMENTAL HEALTH CRITERIA FOR POLYCHLORINATED DIBENZO-PARA-
DIOXINS AND DIBENZOFURANS
A WHO Task Group on Environmental Health Criteria for
Polychlorinated Dibenzo-para-dioxins and Dibenzofurans met at the
Monitoring and Assessment Research Centre, London, United Kingdom,
from 9 to 13 February, 1987. Dr M. Berlin opened the meeting and
welcomed the members on behalf of the host Institute and on behalf of
the United Kingdom Department of Health and Social Security, who
sponsored the meeting. Dr G.C. Becking addressed the meeting on behalf
of the three cooperating organizations of the IPCS (UNEP, ILO, and
WHO). The Task Group reviewed and revised the draft criteria document
and made an evaluation of the risks for human health and for the
environment from exposure to polychlorinated dibenzo-p-dioxins and
dibenzofurans.
The drafts of this document were prepared by Dr U.G. Ahlborg, Dr
H. Hakensson, and Dr B. Holmstedt, all of the National Institute of
Environmental Medicine, Stockholm, Sweden, and by Professor C. Rappe
of the University of Umea, Umea, Sweden.
The efforts of all who helped in the preparation and finalization
of the document are gratefully acknowledged.
* * *
Partial financial support for the publication of this criteria
document was kindly provided by the United States Department of Health
and Human Services, through a contract from the National Institute of
Environmental Health Sciences, Research Triangle Park, North Carolina,
USA - a WHO Collaborating Centre for Environmental Health Effects. The
United Kingdom Department of Health and Social Security generously
supported the cost of printing.
ABBREVIATIONS
AHH aryl hydrocarbon hydroxylase
ALA aminolevulinic acid
BGG bovine gammaglobulin
BHA butylated hydroxyanisole
BP benzo(a)-pyrene
CMI cell-mediated immunity
DEN diethylnitrosamine
diCDD dichlorinated dibenzo-p-dioxin
diCDF dichlorinated dibenzofuran
DMBA dimethylbenzathraline
ECOD 7-ethoxycoumarin-o-deethylase
EGF epidermal growth factor
EH epoxide hydratase
EI electron impact
EROD 7-ethoxyresurofin-o-deethylase
ETG epidermal transglutaminase
fg femtogram (10-15g)
GC gas chromatography
heptaCDD heptachlorinated dibenzo-p-dioxin
heptaCDF heptachlorinated dibenzofuran
hexaCDD hexachlorinated dibenzo-p-dioxin
hexaCDF hexachlorinated dibenzofuran
HMI humoral-mediated immunity
HPLC high pressure liquid chromatography
IARC International Agency for Research on Cancer
ip intraperitoneal
IR infrared
LOEL lowest-observed-effect level
MCPA 4-chloro-o-tolyloxyacetic acid
MFO mixed-function oxidase
MS mass spectrometry
MSW municipal solid waste
ng nanogram (10-9g)
NMR nuclear magnetic resonance
NOEL no-observed-effect level
octaCDD octachlorinated dibenzo-p-dioxin
octaCDF octachlorinated dibenzofuran
PAH polyaromatic hydrocarbons
PCB polychlorinated biphenyl
PCDD polychlorinated dibenzo-p-dioxin
PCDF polychlorinated dibenzofuran
PCDPE polychlorinated diphenylether
PCPY polychlorinated pyrene
PCQ polychlorinated quaterphenyl
pentaCDD pentachlorinated dibenzo-p-dioxin
pentaCDF pentachlorinated dibenzofuran
pg picogram (10-12g)
SC subcutaneous
SCE sister chromatid exchange
SD standard deviation
SEM standard error of the mean
SIM selected ion monitoring
TCDD 2,3,7,8-tetrachlorinated dibenzo-p-dioxin
TCDF 2,3,7,8-tetrachlorinated dibenzofuran
TCP trichlorophenol
tetraCDD tetrachlorinated dibenzofuran
tetraCDF tetrachlorinated dibenzofuran
TPA 12-o-tetradecanoylphorbol-13-acetate
triCDD trichlorinated dibenzo-p-dioxin
triCDF trichlorinated dibenzofuran
t3 triiodothyronine
t4 thyroxine
UDPGT UDP-glucuronosyltransferase
UV ultraviolet
2,4-D 2,4-dichlorophenoxyacetic acid
2,4,5-T 2,4,5-trichlorophenoxyacetic acid
3-MC 3-methylcholanthrene
1. SUMMARY AND RECOMMENDATIONS
1.1 Summary
1.1.1 Sources
Polychlorinated dibenzo-p-dioxins (PCDDs) and polychlorinated
dibenzofurans (PCDFs) are two series of tricyclic aromatic compounds
with similar chemical and physical properties; they are ubiquitous in
the environment. They do not occur naturally, nor are they
intentionally produced. There are 75 positional isomers of PCDDs and
135 isomers of PCDFs.
The most important sources of contamination with PCDDs and PCDFs
include:
- contaminated commercial chemical products, such as
chlorinated phenols and their derivatives, and PCBs;
- incineration of municipal, hazardous, and hospital
wastes, and of sewage sludges;
- automobile operation;
- fossil fuel combustion;
- overheating and emissions from fires involving PCBs;
- disposal of industrial wastes resulting from
processes such as the production of chlorophenols and
their derivatives, chlorophenol wood treatment, use
of PCB fluids in electrical equipment, and wastes
from pulp and paper processing.
1.1.2 Ambient levels and routes of exposure
The limited data available indicate that ambient levels of these
compounds are very low in air, soil, and sediment, i.e. fg/m3 in
air, ng/kg in soil and sediment. Levels of PCDDs and PCDFs up to 50
ng/kg have been found in aquatic organisms in the general environment.
Data on contamination of drinking water and commercial food are very
limited.
Exposure to these compounds in the general population probably
occurs mainly through the food-chain.
Some workers engaged in the production, use, and destruction of
materials containing PCDDs and PCDFs and their precursors may receive
high exposure. For these persons, inhalation and dermal contact are
the primary exposure routes of concern.
1.1.3 Toxicokinetics, biotransformation, and biological monitoring
The bioavailability of PCDDs and PCDFs depends on the matrix they
are in and the route of exposure. Data on bioavailability through
inhalation are not available for any species.
The quantity absorbed by humans after any route of exposure is
not known.
Studies on rodents given single or repeated oral doses of
2,3,7,8-TCDD have shown that about half of the administered dose is
absorbed from the gastrointestinal tract. The reported half-lives for
elimination were between 12 and 94 days for rodents. The half-life of
2,3,7,8-TCDD in adipose tissue of the rhesus monkey is about 1 year.
Animal data on the toxicokinetics of PCDDs other than
2,3,7,8-TCDD are limited. The half-life for 2,3,7,8-TCDD has been
reported to be in the range of 2 and 8 days for rats, mice, and
monkeys and more than 20 days for guinea-pigs. Studies on rats have
shown that 2,3,4,7,8-pentaCDF is more highly retained than is
2,3,7,8-TCDD.
Data on the retention of PCDDs and PCDFs in tissues of various
species, exposed to synthetic mixtures or to environmental samples
containing PCDDs and PCDFs, show a high variability in retention time
between congeners with or without chlorine substitution in the 2,3,7,
and 8 positions.
Limited human data indicate half-lives for some 2,3,7,8-
substituted PCDDs and PCDFs in the range of 2-6 years.
The PCDDs and PCDFs are predominately stored in fat, but they are
also excreted in milk and pass through the placenta. They also appear
in the blood and vital organs at lower concentrations.
The tissue distribution in humans is not clear at present,
although it has been suggested that the ratio between fatty tissue and
liver is higher in humans than in rodents.
In human fat, background levels of TCDD up to 20 ng/kg have been
found in the general population, with no known specific exposure, but
higher levels have been reported in some cases without evidence of
disease. None of these populations were randomly sampled. The more
highly chlorinated PCDDs and PCDFs, particularly octaCDD, are also
present in these samples. Average tissue levels of TCCD tend to
increase with age.
1.1.4 Health effects
1.1.4.1 Animals
The toxic and biological effects resulting from exposure to
2,3,7,8-TCDD are dependent on a number of factors, which include the
species, strain, age, and sex of the animals used. The toxic responses
observed in several animal species include body weight loss,
hepatotoxicity, porphyria, dermal toxicity, gastric lesions, thymus
atrophy and immunotoxicity, teratogenicity, reproductive effects, and
carcinogenicity. TCDD induces a wide spectrum of biological effects
including enzyme induction and vitamin A depletion. Not all of these
effects are observed in any single animal species. The most
characteristic toxic effects observed in all laboratory animals are
body weight loss, thymus atrophy, and immunotoxicity. Chloracne and
related dermal lesions are the most frequently noted signs of
2,3,7,8-TCDD toxicosis in humans; dermal lesions are also observed in
rhesus monkeys, hairless mice, and rabbits. In contrast, most rodents
do not develop chloracne and related dermal toxic lesions after
exposure to 2,3,7,8-TCDD. Many of the toxic lesions are noted
primarily in epithelial tissues.
Reproductive effects have been reported in rhesus monkeys and
rats. The lowest-observed-effect levels have been reported to be
approximately 1-2 ng/kg body weight per day. In two cancer studies in
rats, hepatocellular carcinomas were produced at approximate dose
levels of 0.1 µg/kg body weight per day and 0.01 µg/kg body weight per
day. Doses of 0.001 µg/kg body weight resulted in foci or areas of
hepatocellular alteration. The incidence of certain hormone-dependent
tumours was lower than in the control animals.
TCDD does not appear to have mutagenic properties, and is
therefore not likely to be genotoxic. Thus, it is assumed to be
carcinogenic through an indirect mechanism.
Several other PCDDs and PCDFs cause signs and symptoms similar to
those of 2,3,7,8-TCDD, but there is a wide variation with regard to
potency. There are 12 isomers that display higher toxicity, i.e., the
tetra-, penta-, hexa-, and heptaCDDs and CDFs with four chlorine atoms
in the symmetrical lateral positions 2,3,7, and 8. A mixture of two
hexachlorodibenzo-p-dioxins (1,2,3,7,8,9- and 1,2,3,6,7,8-hexaCDD)
has been demonstrated to possess carcinogenic properties in long-term
animal studies, but at higher doses than those used in the study of
TCDD. Dibenzo-p-dioxin and 2,7-diCDD failed to demonstrate
carcinogenic properties. The relative toxic and biological potencies
of PCDDs and PCDFs have been estimated using short-term studies in
rats and mammalian cell cultures.
There are marked species differences in the susceptibility of
animals to the biological and toxic effects elicited by
2,3,7,8-substituted PCDDs and PCDFs. For example, the oral LD50 values
range from 0.6 µg/kg body weight in guinea-pigs, to 5051 µg/kg body
weight in Golden Syrian hamsters for 2,3,7,8-TCDD. The tremendous
variation in species and strain sensitivity to 2,3,7,8-TCDD and
related compounds cannot be explained by the observed toxicokinetic
differences. The toxicity and toxicokinetics of TCDD in monkeys most
closely resemble the effects observed in humans. There is evidence in
inbred mice that the cellular levels of the Ah receptor correlate, in
part, with susceptibility to the biological and toxic effects of these
compounds. The receptor has also been identified in other species
including man. However, interspecies comparison of cellular Ah
receptor levels do not explain fully the differences in sensitivity.
1.1.4.2 Humans
For occupational and accidental exposures to PCDDs and PCDFs, in
spite of many clinical and follow-up studies, no clear-cut persistent
systemic effects have been delineated except for chloracne. Other
effects have been noted, but, apart from chloracne and perhaps minor
functional disorders, none has been persistent.
In some epidemiological studies of people exposed to a mixture of
dioxins, furans, and other chemicals, an increased incidence of cancer
at different sites has been claimed, but a number of factors limits
confidence in the findings.
In the Seveso accident, the only clear-cut adverse health effect
recorded has been chloracne. Chloracne (193 cases) occurred in 1976
and 1977, and 20 of those individuals still had active chloracne in
1984. Many studies have been performed to find possible links between
exposure to Agent Orange and health effects in civilians or military
personnel in Viet Nam. However, the information available to date does
not allow definite conclusions to be drawn with regard to effects on
human reproduction or any other significant health effects.
In the Missouri incident, children who showed acute illness when
the contamination occurred in 1971 are now reportedly in good health.
Furthermore, epidemiological studies in Missouri on populations
exposed to lower concentrations of dioxins over longer periods of time
have so far not revealed any significant health effects. Although no
clinical symptoms were observed, there were indications of an effect
on the cell-mediated immune system.
The only documented intoxications with PCDFs in humans are the
two instances of contamination of rice oil with PCDFs, PCBs, and PCQs,
i.e., Yusho in Japan, 1968, and Yu-cheng in Taiwan, 1979. In total,
several thousand people were acutely intoxicated. From the data it
appears most likely that the causative agent was the PCDFs. The
general symptomatology was similar to that seen in intoxications with
TCDD, with the differences reflecting the intensity of exposure and
the ages and sex of those exposed.
The average daily intake of 2,3,7,8-substituted PCDFs by Yusho
patients was estimated to be 0.1-0.2 µg/kg body weight for a period of
several months, while the lowest dose causing disease was estimated to
be 0.05-0.1 µg/kg body weight per day over a period of 30 days.
1.1.5 Conclusion
PCDDs and PCDFs occur throughout the environment and we all
probably carry a body burden of them. They have sometimes produced
complex toxic effects following occupational and accidental exposure.
Based on the Yusho disease and experiments in sensitive species
of monkeys, and making assumptions about the relative potencies of
PCDDs and PCDFs, man and certain monkeys may have comparable
sensitivity to these compounds. However, the uncertainties related to
the real dose received by humans and the difficulties of assessing
toxic effects other than chloracne in humans prevents a firm
conclusion as to the relative resistance of humans to the toxic
effects of these compounds. Exposure should be reduced to levels as
low as reasonably practicable.
1.2 Recommendations
1. Analytical interlaboratory validation and "round-robin" studies
using standardized quality assurance and quality control procedures
are needed to improve analytical methodology.
Sampling strategy and analytical procedures and data
interpretation should be optimized and standardized before undertaking
surveys.
2. Further information is required about the origins and
environmental distributon and fate of PCDDs and PCDFs.
Further monitoring data, including time trends and determinations
of isomer patterns, are required for environmental levels of PCDDs and
PCDFs, especially for food, ambient air, and sediments.
3. Data should be obtained about the effects of PCDDs and PCDFs on
environmental biota.
4. More information is required on the bioavailability of PCDDs and
PCDFs from different matrices in the environment and from the diet.
Exposure from these sources should be correlated with agricultural and
industrial practices.
5. Simpler and less expensive chemical and biological methods
suitable for screening for the presence of PCDDs and PCDFs should be
developed and validated.
6. Studies to determine the mechanisms of toxicity of PCDDs and
PCDFs are needed to support an evaluation of the differences in
effects between species and to support an extrapolation to man.
7. Further investigation of immunotoxicity is important, including
cytotoxic T-lymphocyte function. Studies of the effects of perinatal
exposure and of the duration of actions on the immune system are
important.
8. Long-term toxicity studies should be carried out, including
multigeneration reproductive studies in different species with three
of the most widespread PCDDs and PCDFs, namely 2,3,4,7,8-pentaCDF,
1,2,3,7,8-pentaCDD, and octaCDD.
9. Because humans are exposed to complex mixtures of PCDDs and
PCDFs, test systems, including human cell culture systems, should be
developed further and validated for evaluating the toxic potency of
these compounds and other mixtures. These systems can be used to study
mechanisms of action, structure activity relationships, and
interactive effects.
10. Investigations to examine the body burden and to correlate it
with clinical effects and laboratory findings are indicated. Follow-up
studies of previously exposed groups are important.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1 Identity
The polychlorinated dibenzo-para-dioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs) are two series of almost planar
tricyclic aromatic compounds with very similar chemical properties.
The general formulae are given in Fig. 1.
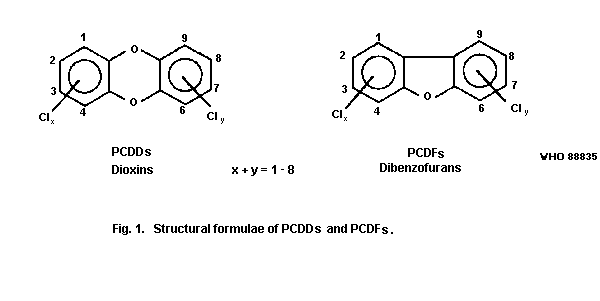 The number of chlorine atoms can vary between 1 and 8. The term
isomers refers to comparisons between compounds with the same
empirical formulae. The term congeners refers to comparisons between
compounds within the same series but with a different number of
chlorine atoms. The number of positional isomers is quite large; in
all there are 75 PCDDs and 135 PCDFs and the number of isomers for a
certain number of chlorine atoms is given in Table 1.
The nomenclature used in this document is based on the system
used by Chemical Abstracts. The Chemical Abstracts System Registry
Numbers (CAS RN) for a few PCDDs and PCDFs that have been cited in the
literature are provided in Table 2.
2.2 Physical and Chemical Properties
A large number of the individual PCDDs have been synthesized by
various methods and characterized, mainly by gas chromatography-mass
spectrometry (GC/MS) (Buser & Rappe, 1980, 1984; Taylor et al., 1985;
Rappe et al., 1985a) but also by using nuclear magnetic resonance
(NMR) or ultraviolet (UV), infrared (IR), (Pohland & Yang, 1972; Kende
et al., 1974), or X-ray analyses (Boer et al., 1973; Slonecker et al.,
1983).
Table 1. Number of PCDD and PCDF isomers
Number Number Number
of chlorine atoms of PCDD isomers of PCDF isomers
1 2 4
2 10 16
3 14 28
4 22 38
5 14 28
6 10 16
7 2 4
8 1 1
75 135
Table 2. CAS RN for some PCDDs and PCDFs
PCDD congener CAS RN PCDF congener CAS RN
2,3,7,8-TetraCDD 1746-01-6 2,3,7,8-TetraCDF 51207-31-9
1,2,3,7,8-PentaCDD 40321-76-4 1,2,3,7,8-PentaCDF 57117-41-6
1,2,3,6,7,8-HexaCDD 57653-85-7 2,3,4,7,8-PentaCDF 57117-31-4
1,2,3,7,8,9-HexaCDD 19408-74-3 1,2,3,4,7,8-HexaCDF 70648-26-9
1,2,3,4,6,7,8-HeptaCDD 35822-46-9 1,2,3,6,7,8-HexaCDF 57117-44-9
1,2,3,4,7,8,9-HeptaCDD 58200-70-7 1,2,3,7,8,9-HexaCDF 72918-38-8
OctaCDD 3268-87-9 2,3,4,6,7,8-HexaCDF 60851-34-5
Pyrolysis of chlorinated phenols yields small amounts of one or
more PCDD isomers. Using this technique all the 22 tetraCDDs have been
prepared (Nestrick et al., 1979; Buser & Rappe, 1980) as well as the
14 pentaCDDs (Buser & Rappe, 1984) and 10 hexaCDDs (Lamparski &
Nestrick, 1981; Buser & Rappe, 1984).
Taylor et al. (1985) have synthesized, separated, and isolated
all the 22 tetraCDD isomers. In Table 3 are listed some other isomers
that have been synthesized and isolated.
The most toxic and most extensively studied representative of the
chlorinated dioxins (PCDDs) is
2,3,7,8-tetrachlorodibenzo-para-dioxin (2,3,7,8-tetraCDD) (Fig. 2).
It is commercially available, as are more than 10 other PCDD
congeners.
The empirical formulae, molecular weights, and some physical
properties of a few PCDDs are given in Table 4.
Table 3. Synthetic method and melting point for some PCDDs
PCDD Synthetic Melting point Reference
Isomer methoda °C
1-Chloro- 1 80-90 Pohland & Yang, 1972
2-Chloro- 1 88-89 Pohland & Yang, 1972
1,3-Dichloro- 1 113.5-114.5 Kende et al., 1974
2,3-Dichloro- 1 163-164 Pohland & Yang, 1972
2,7-Dichloro- 2 209-210 Pohland & Yang, 1972
2,8-Dichloro- 3 150.5-151 Pohland & Yang, 1972
1,2,4-Trichloro- 4 128-129 Pohland & Yang, 1972
2,3,7-Trichloro- 1 157-158 Kende et al., 1974
2,3,7,8-Tetrachloro- 2 305-306 Pohland & Yang, 1972
2,3,7,8-Tetrachloro- 5 305-307 Kende et al., 1974
1,2,3,4-Tetrachloro- 4 188-190 Pohland & Yang, 1972
1,3,7,8-Tetrachloro- 1 193.5-195 Kende et al., 1974
1,3,6,8-Tetrachloro- 2 219-219.5 Pohland & Yang, 1972
1,2,3,4,7-Pentachloro- 5 195-196 Kende et al., 1974
1,2,3,4,7,8-Hexachloro- 5 275 Pohland & Yang, 1972
1,2,4,6,7,9-Hexachloro- 2 238-240 Pohland & Yang, 1972
Octachloro 2 330 Pohland & Yang, 1972
a Synthetic methods as follows:
1 = Catechol + chlorobenzene
2 = Pyrolysis of chlorphenols
3 = Cyclization of chlorophenoxyphenol
4 = Catechol + chloronitrobenzene
5 = Chlorination of chlorodibenzodioxin
The number of chlorine atoms can vary between 1 and 8. The term
isomers refers to comparisons between compounds with the same
empirical formulae. The term congeners refers to comparisons between
compounds within the same series but with a different number of
chlorine atoms. The number of positional isomers is quite large; in
all there are 75 PCDDs and 135 PCDFs and the number of isomers for a
certain number of chlorine atoms is given in Table 1.
The nomenclature used in this document is based on the system
used by Chemical Abstracts. The Chemical Abstracts System Registry
Numbers (CAS RN) for a few PCDDs and PCDFs that have been cited in the
literature are provided in Table 2.
2.2 Physical and Chemical Properties
A large number of the individual PCDDs have been synthesized by
various methods and characterized, mainly by gas chromatography-mass
spectrometry (GC/MS) (Buser & Rappe, 1980, 1984; Taylor et al., 1985;
Rappe et al., 1985a) but also by using nuclear magnetic resonance
(NMR) or ultraviolet (UV), infrared (IR), (Pohland & Yang, 1972; Kende
et al., 1974), or X-ray analyses (Boer et al., 1973; Slonecker et al.,
1983).
Table 1. Number of PCDD and PCDF isomers
Number Number Number
of chlorine atoms of PCDD isomers of PCDF isomers
1 2 4
2 10 16
3 14 28
4 22 38
5 14 28
6 10 16
7 2 4
8 1 1
75 135
Table 2. CAS RN for some PCDDs and PCDFs
PCDD congener CAS RN PCDF congener CAS RN
2,3,7,8-TetraCDD 1746-01-6 2,3,7,8-TetraCDF 51207-31-9
1,2,3,7,8-PentaCDD 40321-76-4 1,2,3,7,8-PentaCDF 57117-41-6
1,2,3,6,7,8-HexaCDD 57653-85-7 2,3,4,7,8-PentaCDF 57117-31-4
1,2,3,7,8,9-HexaCDD 19408-74-3 1,2,3,4,7,8-HexaCDF 70648-26-9
1,2,3,4,6,7,8-HeptaCDD 35822-46-9 1,2,3,6,7,8-HexaCDF 57117-44-9
1,2,3,4,7,8,9-HeptaCDD 58200-70-7 1,2,3,7,8,9-HexaCDF 72918-38-8
OctaCDD 3268-87-9 2,3,4,6,7,8-HexaCDF 60851-34-5
Pyrolysis of chlorinated phenols yields small amounts of one or
more PCDD isomers. Using this technique all the 22 tetraCDDs have been
prepared (Nestrick et al., 1979; Buser & Rappe, 1980) as well as the
14 pentaCDDs (Buser & Rappe, 1984) and 10 hexaCDDs (Lamparski &
Nestrick, 1981; Buser & Rappe, 1984).
Taylor et al. (1985) have synthesized, separated, and isolated
all the 22 tetraCDD isomers. In Table 3 are listed some other isomers
that have been synthesized and isolated.
The most toxic and most extensively studied representative of the
chlorinated dioxins (PCDDs) is
2,3,7,8-tetrachlorodibenzo-para-dioxin (2,3,7,8-tetraCDD) (Fig. 2).
It is commercially available, as are more than 10 other PCDD
congeners.
The empirical formulae, molecular weights, and some physical
properties of a few PCDDs are given in Table 4.
Table 3. Synthetic method and melting point for some PCDDs
PCDD Synthetic Melting point Reference
Isomer methoda °C
1-Chloro- 1 80-90 Pohland & Yang, 1972
2-Chloro- 1 88-89 Pohland & Yang, 1972
1,3-Dichloro- 1 113.5-114.5 Kende et al., 1974
2,3-Dichloro- 1 163-164 Pohland & Yang, 1972
2,7-Dichloro- 2 209-210 Pohland & Yang, 1972
2,8-Dichloro- 3 150.5-151 Pohland & Yang, 1972
1,2,4-Trichloro- 4 128-129 Pohland & Yang, 1972
2,3,7-Trichloro- 1 157-158 Kende et al., 1974
2,3,7,8-Tetrachloro- 2 305-306 Pohland & Yang, 1972
2,3,7,8-Tetrachloro- 5 305-307 Kende et al., 1974
1,2,3,4-Tetrachloro- 4 188-190 Pohland & Yang, 1972
1,3,7,8-Tetrachloro- 1 193.5-195 Kende et al., 1974
1,3,6,8-Tetrachloro- 2 219-219.5 Pohland & Yang, 1972
1,2,3,4,7-Pentachloro- 5 195-196 Kende et al., 1974
1,2,3,4,7,8-Hexachloro- 5 275 Pohland & Yang, 1972
1,2,4,6,7,9-Hexachloro- 2 238-240 Pohland & Yang, 1972
Octachloro 2 330 Pohland & Yang, 1972
a Synthetic methods as follows:
1 = Catechol + chlorobenzene
2 = Pyrolysis of chlorphenols
3 = Cyclization of chlorophenoxyphenol
4 = Catechol + chloronitrobenzene
5 = Chlorination of chlorodibenzodioxin
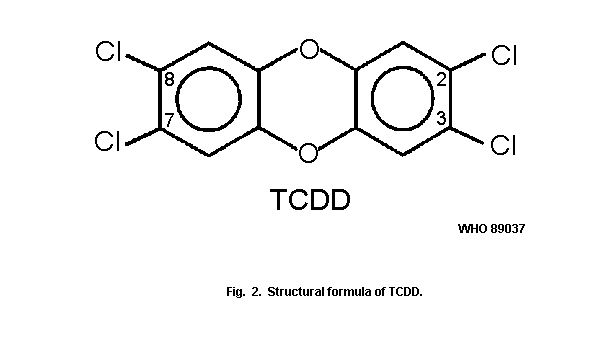 Table 4. Physical properties of some PCDDs
Molecular Molecular Absorption
Compound formulae weight maximum Reference
(chloroform)
(nm)
2,3,7,8-TCDD C12H4Cl4O2 321.9 310 Pohland &
Yang (1972)
1,2,3,7,8-PentaCDD C12H3Cl5O2 356.5 308 Gray et al.
(1976)
1,2,3,6,7,8-HexaCDD C12H2Cl6O2 390.9 316 Gray et al.
(1975)
1,2,3,7,8,9-HexaCDD C12H2Cl6O2 390.9 317 Gray et al.
(1975)
Although tetraCDD is lipophilic, it is only slightly soluble in
most solvents and very slightly soluble in water (Table 5).
Table 5. Solubility of 2,3,7,8-tetraCDD in various solventsa
Solvent Solubility at 25 °C
g/litre g/kg
O-Dichlorobenzene 1.8 1.4
Chlorobenzene 0.8 0.72
Perchloroethylene 0.68 0.48
Chloroform 0.55 0.37
Benzene 0.47 0.57
Acetone 0.09 0.11
Dimethylsulfoxideb < 0.1 < 0.1
Methanol 0.01 0.01
Water 2 x 10-7 2 x 10-7
a From: Crummett & Stehl (1973).
b DMSO caused detector fouling and a better value could not be obtained.
Table 6. Water solubility of PCDDsa
Compound Water solubility (g/litre)
20.0 °C 40.0 °C
1,3,6,8-TetraCDD (3.2±0.2) x 10-7 (3.9±0.4) x 10-7
1,2,3,7-TetraCDD (4.3±0.1) x 10-7 (12.7±0.8) x 10-7
1,2,3,4,7-PentaCDD (1.2±0.1) x 10-7 (4.6±0.1) x 10-7
1,2,3,4,7,8-HexaCDD (4.4±0.1) x 10-9 (19.0±0.1) x 10-9
1,2,3,4,6,7,8-HeptaCDD (2.4±0.3) x 10-9 (6.3±0.2) x 10-9
OctaCDD (0.4±0.1) x 10-9 (2.0±0.2) x 10-9
a From: Friesen et al. (1985).
Marple et al. (1986a) have reanalysed the water solubility of
2,3,7,8-TCDD and found it to be considerably less (12.5-19.2
ng/litre). The log water-octanol partition coefficient (Kow) has
been determined as 6.64 by Marple et al. (1986b).
Friesen et al. (1985) have determined the water solubility for
some PCDDs other than the 2,3,7,8-TCDD compound and these are given in
Table 6.
Similarly Webster et al. (1985) have determined the log
octanol-water partition coefficients for a number of PCDDs (Table 7).
2,3,7,8-TetraCDD is considered to be a stable compound, but due
to its extreme toxicity its chemistry has not been fully evaluated.
However, it undergoes substitution reactions (Baughman, 1974) as well
as photochemical dechlorination (Crosby et al., 1971; Crosby & Wong,
1977; Gebefugi et al., 1977). Thermally it is very stable and rapid
decomposition of 2,3,7,8-tetraCDD occurs only at temperatures above
750 °C (Stehl et al., 1973). The other PCDDs have been much less
studied; however, octaCDD is completely destroyed by treatment with
hot alkali (Albro & Corbett, 1977).
The first synthesis of 2,3,7,8-tetraCDD was reported by
Sandermann et al. (1957), who used catalytic chlorination of the
unchlorinated dioxin. It has also been prepared in good yields by the
dimerization of 2,4,5-trichlorophenol salts (Buu-Hoi et al., 1971b;
Langer et al., 1973).
In the PCDF series, Mazer et al. (1983) synthesized all the 38
positional tetraCDF isomers. The products were mixtures of isomers,
and each of these isomers could be identified. Later Bell & Gari,
(1985) isolated and characterized all the 38 tetraCDFs, 28 pentaCDFs,
and 16 hexaCDFs.
Table 7. Values for log Kow for some PCDDs from linear and quadratic plots
log Kow (linear) log Kow (quadratic)
Waters Waters Waters Waters
Bondapak Bondapak Bondapak Bondapak
Compound (Woodburn data) (Woodburn data)
Dibenzo-p-dioxin 4.26 4.01 4.34 4.17
1-MonoCDD 4.81 4.52 4.91 4.75
2-MonoCDD 5.33 5.00 5.45 5.29
2,7-DiCDD 6.27 5.86 6.39 6.17
1,2,4-TriCDD 7.36 6.86 7.45 7.11
1,2,3,7-TetraCDD 8.15 7.58 8.19 7.72
1,2,3,4-TetraCDD 8.63 8.02 8.64 8.07
1,3,6,8-TetraCDD 8.70 8.08 8.70 8.12
1,2,3,4,7-PentaCDD 9.48 8.80 9.40 8.64
1,2,3,4,7,8-HexaCDD 10.40 9.65 10.22 9.19
1,2,3,4,6,7,8-HeptaCDD 11.38 10.55 11.05 9.69
OctaCDD 12.26 11.35 11.76 10.07
a From: Webster et al. (1985).
Kuroki et al. (1984) have synthesized 51 congeners of PCDFs by a
structure specific method from chlorophenols and chloronitrobenzenes
or chlorophenols and chlorodiphenyls iodonium salts. The structures
were confirmed by MS and NMR.
Safe & Safe (1984) described the synthesis of 22 PCDF congeners
resulting in quantities of 10-320 mg of purified product. They also
reported NMR data on the compounds synthesized.
Sarna et al. (1984) and Burkhard & Kuehl, (1986) have documented
the octanol/water partition coefficients for some PCDFs (Table 8). The
disagreement for OCDF arises because of uncertainties in the Kow
values of reference compounds of high Kow. The partitioning of
organic chemicals between lipid and water is an important determinant
of the bioconcentration potential of a toxicant and has sometimes been
effectively used as an indicator of the preferred degradative in in
vivo pathways.
Table 8. The logarithm of the octanol/water partition coefficients (Kow) of some PCDFs using HPLC methods
PCDF log Kow Reference
2,8-dichloro- 5.95 Sarna et al. 1984a
5.30b Burkhard & Kuehl, 1986c
2,3,7,8-tetrachloro- 5.82±0.02 Burkhard & Kuehl, 1986c
octachloro- 13.37 Sarna et al. 1984a
8.78 Burkhard & Kuehl, 1986c
a Quadratic equation treatment: Biorad Biosil (10 mm) data.
b Quadratic equation treatment: unspecified "microbore" HPLC column.
c Sarna et al. (1984) data recalculated from experimental data.
2.3. Analytical Methods
2.3.1 General aspects
The earliest reported method used to detect 2,3,7,8-tetraCDD was
a rabbit skin test (Adams et al., 1941). Test samples were applied to
the inner surface of the ear and to the shaven belly of albino
rabbits, and inflammatory responses were observed. Subsequently, Jones
& Krizek (1962) developed a test based on the recovery and weight of
the keratin formed on the rabbit ear after application of a sample.
These biological methods were non-specific as to isomers and not
sufficiently sensitive to detect low levels of contamination.
In the late 1960s and early 1970s, gas chromatographic methods
were used for the quantification mainly of 2,3,7,8-tetraCDD in
commercial 2,4,5-T formations. The detection level was normally in the
range of µg/g. These analyses were not isomer-specific and the results
could not be confirmed. Ryhage (1964) solved the problem of combining
a gas chromatograph with a mass spectrometer. During the 1970s and
1980s, various types of mass spectrometer and gas chromatograph/mass
spectrometer combinations were used in analytical work. Use of these
more sophisticated instruments allowed for the development of
isomer-specific and validated analyses for the tetraCDDs in the very
late 1970s and for the other PCDDs and PCDFs in the early 1980s.
A number of spectroscopic methods are available for the
laboratory identification of 2,3,7,8-tetraCDD, but their use is highly
restricted, with the exception of mass spectroscopy (MS). Data on
X-ray, infra-red (IR), ultra-violet (UV), nuclear magnetic resonance
(NMR), electron spin resonance (ESR), and mass spectra were obtained
by Pohland & Yang (1972), Baughman (1974), and Slonecker et al.
(1983).
Because of the large number of isomers and congeners, and due to
the extreme toxicity of some PCDD and PCDF isomers, highly sensitive
and specific analytical techniques are required for the measurements.
Detection limits for the analysis of environmental and human samples
should be orders of magnitude lower than the usual detection levels
required for pesticide analysis. A detection level of 1 pg or less
might be required to measure 2,3,7,8-tetraCDD and the other toxic
isomers in a 1-g environmental sample. Analyses at such low levels are
complicated by the presence of a multitude of other interfering
compounds and clean-up procedures are required.
The mono-, di-, and trichloro congeners are not usually included
in these analyses. Such compounds are considered to be much less toxic
than the higher chlorinated congeners and are also much more volatile
and losses may occur during clean-up.
It should be mentioned that the level of sophistication needed in
the analyses for PCDD and PCDFs will depend upon the objectives
thereof. In cases where the objectives were primarily to screen
samples to identify groups of PCDDs and/or PCDFs (in a qualitative or
semiquantitative manner), routine assays and bioassays were adequate.
In other instances, where the objective of the analysis was to
quantify accurately specific PCDD and/or PCDF isomers in the samples,
sophisticated analytical procedures were required. Clearly, both types
of analyses can be useful, depending on the purpose for which the
analytical results are to be used.
Many analytical methods have been developed in recent years for
the analysis of trace amounts of PCDDs and PCDFs in environmental
samples, especially for 2,3,7,8-tetraCDD. The most specific of these
methods are based on MS. There are many requirements to be met by such
an analytical method, including representative sampling and
appropriate storage, efficient extraction, high selectivity in the
clean-up, high specificity in the gas chromatography, high sensitivity
in the detection, safe and reliable quantification, good
reproducibility, useful confirmatory information.
Several review articles discussing methods of analyzing PCDDs and
PCDFs have appeared (McKinney, 1978; Esposito et al., 1980; Rappe &
Buser, 1980; Harless & Lewis, 1982; Karasek & Anuska, 1982; Tiernan,
1983; Crummett et al., 1985). Most of the older methods have been
critically reviewed by a panel of experts assembled by the National
Research Council of Canada (1981).
2.3.2 Sampling strategy and sampling methods
The quality and utility of analytical data depend on the validity
of the sample and the adequacy of the sampling program. The purpose of
sampling is to obtain specimens that represent the situation being
studied. Sampling plans may require that systematic samples be
obtained at specified times and places, or simple random sampling may
be necessary. Generally, the sample should be an unbiased
representation of the environmental situation.
All aspects of a sampling programme should be planned and
documented in detail, and the expected relationship of the sampling
protocol to the analytical result should be defined. A sampling
programme should include reasons for choosing sampling sites, the
number and type of samples, the timing of sample acquisition and the
sampling equipment used. A detailed sampling procedure should include
a description of the sampling situation, the sampling methodology,
labelling of samples, field blank preparation, pretreatment
procedures, and transportation and storage procedures.
The quality assurance programme should include means to
demonstrate that containers or storage procedures do not alter the
qualitative or quantitative composition of the sample. Special
transportation and storage procedures (refrigeration or exclusion of
light) should be described, if they are required.
Because environmental samples are typically heterogeneous, a
sufficiently large number of samples (ten or more) must normally be
analyzed to obtain meaningful data on chemical composition. The number
of individual samples that should be analyzed will depend on the kind
of information required by the investigation. If an average
compositional value is required, a number of randomly selected
individual samples may be obtained, combined, and blended to provide
a homogeneous composite sample from which a sufficient number of
subsamples could be analyzed. If composition profiles, time trends, or
the variability of the sample population are of interest, many samples
need to be collected and analyzed individually.
If field blanks are not available, efforts should be made to
obtain blank samples that best simulate a sample that does not contain
the specific chemical. In addition, measurements should be made to
ascertain whether, and to what extent, any reagent or solvent used may
contribute to or interfere with the analytical results (laboratory and
solvent blanks).
The recovery tests are frequently used and necessary to evaluate
the analytical methodology. Uncontaminated samples from control sites
that have been spiked with the chemical of interest provide the best
information because they simulate any matrix effect. When feasible,
isotopically labelled (13C, 37Cl) chemicals spiked into the sample
provide the greatest accuracy since they are subjected to the same
matrix effects. The 13C- and 37Cl-labelled compounds can be used
to validate:
(a) sampling (sampling surrogate),
(b) analytical pretreatment (clean-up surrogate),
(c) quantification (internal standard).
Very few laboratories in the world have access to and experience
in working with these complicated analyses.
In order to be able to compare data generated in different
laboratories, the same quantitative standard compounds should be used.
Interlaboratory calibrations or "round-robin" studies have been
performed in very few cases.
2.3.3 Extraction procedures
In this step, the sample is homogenized or digested and extracted
with a suitable solvent or solvent mixture to remove the bulk of the
sample matrix and transfer the PCDD and PCDF residue into the solvent.
Both the selection of the proper solvent and the method of extraction
can be critical in obtaining a satisfactory recovery of PCDDs and
PCDFs from the sample matrix.
Many different procedures for the extraction of PCDDs/PCDFs from
various samples are described. In some cases this involves digestion
or destruction of the matrix. Some of these methods have been
evaluated in the report from the National Research Council of Canada
(1982), while other methods are discussed by Tiernan (1983).
An interlaboratory "round-robin" study involving 13 laboratories
was carried out to evaluate the reliability of data on
2,3,7,8-tetraCDD in fish samples. No significant differences were
found from methods differing in the digestion or extraction procedures
(Ryan et al., 1983b).
In a study described by Albro et al. (1985), eight different
approaches were applied in eight laboratories to quantify four PCDDs
(2,3,7,8-tetraCDD; 1,2,3,7,8-pentaCDD; 1,2,3,4,7,8-hexaCDD; and
octaCDD) and three PCDFs (2,3,7,8-tetraCDF; 2,3,4,7,8-pentaCDF; and
1,2,3,7,8,9-hexaCDF) in spiked samples of an extract from human
adipose tissue. Levels of fortification, unknown to the participating
laboratories, were in the 5-50 ng/kg range, except for octaCDD (up to
500 ng/kg). The results indicated that most of the procedures tested
gave a high degree of qualitative reliability. However, other methods
were not so accurate, a large portion of the reported data consisting
of false positives or false negatives.
Lustenhouwer et al. (1980) studied the extraction of PCDDs and
PCDFs from a fly ash sample. A dramatic difference was found between
different solvents.
2.3.4 Sample clean-up
In the sample clean-up, the PCDDs and PCDFs present in the sample
should be separated from a multitude of other co-extracted and
possibly interfering compounds. The clean-up methods, normally three
steps or more, can vary for different sample matrices. Two different
procedural trends can be recognized:
(a) all PCDD and PCDF isomers can be analyzed in one
single fraction by the containment enrichment
procedure (Norstrom et al., 1982; Stalling et al.,
1983; Tiernan, 1983; Rappe, 1984),
(b) specific isomers are analyzed in different fractions
mainly after normal-phase and reverse-phase high
pressure liquid chromatography (HPLC) separation
(Lamparski et al., 1979; Niemann et al., 1983; Tosine
et al., 1983).
This latter method allows the identification of only a few PCDD
isomers in each fraction, and is mainly used to monitor TCDD and a few
other congeners. For a monitoring program of all PCDDs and PCDFs a
more general method might be preferred.
The method described by Stalling et al. (1983) was originally
designed for the analyses of fish samples. In a "round-robin" study of
fish samples it gave good results (Ryan et al., 1983b). This method
has now been used for the clean-up of other biological samples like
bird muscle, seal fat, turtle fat, and human adipose tissue - blood,
liver, kidney, and milk (Rappe et al., 1983c; Nygren et al., 1986;
Rappe et al., 1986b).
2.3.5 Isomer identification
The purified extracts are used directly for the final analyses
with the aid of a gas-chromatograph/mass spectrometer (GC/MS) equipped
with a glass capillary or a fused-silica column. The column leads
directly into the ion source of the mass spectrometer, which operates
either in the electron impact (EI) or the negative ion-chemical
ionization (NCI) mode. In view of the large variation in toxicological
and biological effects of the PCDD and PCDF isomers, it is imperative
that the isomers, particularly those having high toxicity, be
identified. For an unambiguous isomer identification it is necessary
to have access to all analytical standards within a specific group of
isomers, e.g. all the 22 tetraCDDs and all the 38 tetraCDFs. All the
22 tetraCDDs have been prepared and, using a Silar 10c glass capillary
column, the 2,3,7,8-tetraCDD can be separated from all the other 21
tetra isomers (Buser & Rappe, 1980). Recently all the 14 pentaCDDs and
the 10 hexaCDDs have been prepared. Using the Silar 10c column all the
2,3,7,8- substituted isomers can be separated from all the other
isomers (Buser & Rappe, 1984). The SP 2330 fused silica column can
also be used for this separation (Rappe, 1984).
In the PCDF series, Mazer et al. (1983) have synthesized all the
38 positional tetraCDF isomers. The products were mixtures of isomers,
and each of these isomers could be identified using both an SP 2330
and an SE 54 capillary column. Later, Bell & Gara (1985) isolated and
characterized all tetra-, penta- and hexaCDFs. The SP 2330 column can
separate most of these isomers (Rappe, 1984). The 1,2,3,7,8-pentaCDF
co-elutes with the 1,2,3,4,8-isomer and the 1,2,3,4,7,8- hexaCDF with
the 1,2,3,4,7,9-isomer, but they can be separated on less polar
columns like OV-17 and DB-5.
A very limited number of investigations has been performed using
these complete sets of synthetic standards.
2.3.6 Quantification
Mass selective detection (mass fragmentography) has been used to
quantify trace amounts of PCDDs and PCDFs in the samples by
selectively monitoring M, M + 2, and/or M + 4 ions (SIM). The
quantification is based on peak area measurements and a comparison of
these areas using either isotopically labelled internal standards
(13C or 37Cl) or calibration curves of external standards. As a
first approach, it has been generally assumed that with the MS
quantification technique, all isomers of a particular congener of PCDD
or PCDF (e.g. the tetrachloro-isomers) have the same response factors.
However, an investigation of 13 well-defined tetraCDF isomers has
shown a three-fold variation in response factors with the EI mode and
up to a 20-fold variation with the negative ion-chemical ionization
mode. For the higher chlorinated homologues (penta, hexa) the
variation was found to be less (Rappe et al., 1983b).
Fung et al. (1985) have studied the mass spectra of 26 PCDF
congeners. They found that the EI spectra are not particularly isomer
specific, while positive ion-chemical ionization spectra show a
greater degree of isomer distinction.
2.3.7 Confirmation
Quality control and quality assurance programs help to assure
that positive data reported actually refer to specific PCDDs and PCDFs
(Kloepfer et al., 1983). To provide reliable data:
(a) isomer specificity must be demonstrated initially and verified
daily,
(b) the retention time must equal (within 3 seconds) the retention
time for the isotopically labelled congener,
(c) the signal to noise ratio must be 2.5:1 or higher,
(d) the chlorine cluster must be within ± 10% of the theoretical
values, given in Table 9,
(e) correct fragments, e.g., M+-COCl ions, must be with correct
chlorine clusters.
For confirmation, mass spectroscopy is the best technique now
available. The EI mass spectral properties of PCDFs and PCDD have been
described (Buser, 1975). The molecular (M+) and fragment ions of
PCDDs and PCDFs show the typical, expected clustering due to the
chlorine isotopes (Table 9). The typical fragmentation is M-COCl+,
which is a useful fragment to study.
Buser & Rappe (1978) have shown that observation of low mass ions
can be used for the identification of the substitution pattern of
PCDDs, which can be defined as the number of chlorine atoms on each
carbon ring of the dioxin molecular; the 2,3,7,8-isomer has a 2:2
pattern while 1,2,3,4-tetraCDD has a 4:0 pattern. However, these low
mass ions may not be observed in spectra from environmental or
biological samples.
In the negative ion-chemical ionization mode, the PCDFs have the
base peak due to M-, and the fragmentation produces the unusual
M--34 ions (uptake of H and loss of Cl). Fragmentation of PCDDs in
this mode is more conventional via loss of Cl yielding M--35 ions
(Buser et al., 1985).
Using EI technique and a quadropole instrument, the detection
limits are 1-10 pg for the tetrachloro compounds and up to 10-50 pg
for the octachloro compounds using selected ion monitoring or multiple
ion detection (SIM or MID). Full mass spectra require 0.1-1 ng of
compound (Buser et al., 1985). High resolution instruments can improve
the sensitivity by one order of magnitude.
The negative ion-chemical ionization mode, using methane gas as
reagent, gas provides extremely good sensitivity for all PCDFs (tetra-
to octachloro- compounds) and for the higher chlorinated PCDDs (penta-
to octaCDD). The detection limits are in the 10-100 fg (10-15g)
range using SIR or MID, which is 1 to 2 orders of magnitude better
than EI (Buser et al., 1985). However, the negative ion-chemical
ionization mode has very poor sensitivity for 2,3,7,8- tetraCDD under
these conditions.
Using low resolution MS instruments, a series of interfering
compounds has been identified (Table 10). Some of this interference
can be eliminated using high resolution MS instruments operating at
8000 - 10 000 daltons. However, compounds with the same empirical
formulae cannot be separated by MS technique; they are normally
eliminated during the clean-up or separated by the gas chromatography
step.
2.3.8 Other analytical methods
Paasivirta et al. (1977) have shown that 2,3,7,8-tetraCDD can be
detected down to the pg level using a glass capillary column and a
63Ni electron-capture detector. Combined with efficient clean-up
procedures, this method has shown to be useful down to a level of 9
ppt (Niemann et al., 1983), although positive samples need
confirmation by mass spectroscopy (MID, SIM).
Other techniques, such as enzyme induction and radioimmunoassay
have been described and discussed by Firestone (1978) and McKinney
(1978). McKinney et al. (1982) have used the radioimmunoassay method
for determining 2,3,7,8-tetraCDD in human fat, and found the reliable
sensitivity at 95% confidence interval to be 100 pg per sample.
An analytical method based on the keratonization response of
epithelial cells in an in vitro system has been described by
Gierthy & Crane (1985b). This method can be an assay for dioxin-like
activity in environmental and biological samples. A positive response
was found for 2,3,7,8-tetraCDD at a concentration of 10-11 mol/litre.
Table 9. Isotopic abundance ratio ("cluster") of polychlorinated dioxins and dibenzofurans
Number of
chlorine M M + 2 M + 4 M + 6 M + 8 M + 10 M + 12 M + 14
atoms
1 100.0 33.7
2 100.0 66.1 11.3
3 100.0 98.4 32.7 3.8
4 76.4 100.0 49.4 11.0 1.0
5 61.2 100.0 65.5 21.6 3.6 0.3
6 51.1 100.0 81.7 35.8 8.9 1.2 0.1
7 43.8 100.0 97.9 53.4 17.6 3.5 0.4
8 33.7 87.6 100.0 65.3 26.8 7.0 1.2 0.1
Table 10. List of molecular ions of polychlorinated compounds present in some human and environmental samples
and possibly interfering in the mass spectral analysis of PCDFs and PCDDsa
Molecular ions (m/z,m+,m-) (chlorination)
Compounds mono- di- tri- tetra- penta- hexa- hepta- octa- nona- deca-
PCDDs 320 354 388 422 456 - -
PCDFs 304 338 372 406 440 - -
PCBs 290 324 358 392 426 460 494
PCNs 264 298 332 366 400 - -
PCTs 298 332 366 400 434 468 502 536 570
PCDPEsb 238 272 306 340 374 408 442 476 510
PCPYsc 36 270 304 338 372 406
a From: Buser et al. (1985).
b PCDPEs: Polychlorinated diphenylethers.
c PCPYs: Polychlorinated pyrenes.
3. SOURCES OF ENVIRONMENTAL POLLUTION
3.1 Production, Synthesis, and Use
PCDDs and PCDFs are not produced commercially. These compounds
are in fact formed as trace amounts of undesired impurities in the
manufacture of other chemicals such as chlorinated phenols and their
derivatives, chlorinated diphenyl ethers, and polychlorinated
biphenyls (PCBs). There is no known technical use for the PCDDs and
PCDFs.
The amount of total PCDDs entering the Canadian environment/year
has been estimated to be about 1500 kg, and 75% of this amount has
been estimated to be due to octaCDD alone (National Research Council
of Canada, 1981). There is no estimation of the amount of PCDFs
entering the environment anywhere in the world.
Although the polychlorinated dioxins and dibenzofurans are not
commercially produced, most of these compounds have been synthesized
for research purposes in small quantities according to the reactions
discussed in section 2.
3.2 Industrial Processes
In addition to the synthetic methods mentioned in section 2,
2,3,7,8-tetraCDD may be formed during the industrial preparation of
2,4,5-trichlorophenol from 1,2,4,5-tetra-chlorobenzene. This
substitution reaction takes place at about 180 °C, and when the
solvent is methanol, the pressure rises to about 7 KPa. The formation
of TCDD is an unwanted side reaction which takes place when the
reaction mixture is heated to 230-260 °C (Milnes, 1971). This reaction
is exothermic, so that even higher temperatures may be attained
resulting in uncontrolled conditions.
In some factories ethylene glycol is used as a solvent in order
to avoid the high pressure. As already pointed out by Milnes (1971),
however, use of this solvent requires special precautions because of
the occurrence of a base-promoted polymerization of ethylene glycol
and decomposition reactions that produce ethylene oxide. These
reactions are also exothermic; they may start spontaneously at
temperatures above 180 °C and proceed rapidly and uncontrollably to
result in the formation of relatively large amounts of TCDD.
After most of the solvent has been removed, the reaction mixture
is acidified; the 2,4,5-trichlorophenol can be separated from
2,3,7,8-tetraCDD by one or two distillations, with the result that
2,3,7,8-tetraCDD is concentrated in the still-bottom residues. Up to
1 mg/g of 2,3,7,8-tetraCDD in such residues has been reported
(Kimbrough et al., 1984). Improper disposal of such residues is
discussed in sections 4.4.2 and 9.
Most of the 2,4,5-trichlorophenol produced is used for the
preparation of herbicides such as 2,4,5-T (including various esters
and salts, and the bactericide hexachlorophene).
PCDDs and PCDFs are both formed as by-products during the
manufacture of chlorinated phenols (2,4-dichloro-, 2,4,6-trichloro-,
2,3,4,6-tetrachloro- and pentachlorophenol). The commercial
chlorophenols are produced by two processes, i.e., by chlorination of
the phenol using various catalysts and by the alkaline hydrolysis of
an appropriate chlorobenzene. Apparently both reactions can lead to
the formation of PCDDs as well as PCDFs, and the level of
contamination is normally much higher here than in the production of
2,4,5-trichloro-phenol (see section 3.3).
PCDDs and PCDFs are also formed during the preparation of
chlorinated diphenyl ether herbicides (Yamagishi et al., 1981) and
hexachlorobenzene (Villeneuve et al., 1974). A series of PCDFs are
formed during the production of PCBs (see section 3.3).
Production equipment is often used for the production of several
different chemicals. In the manufacture of chemicals on such equipment
previously contaminated by PCDDs and PCDFs, both the products and
waste generated can be contaminated. Thus, manufactured
2,4-dichlorophenoxyacetic esters (2,4-D), which otherwise should not
be contaminated by 2,3,7,8-tetraCDD, did indeed contain this dioxin
because the equipment used had been employed previously to produce
2,4,5-T and had not been cleaned properly (Federal Register, 1980).
It should be pointed out that the primary occurrence of TCDD in
the environment is possibly related to the synthesis of
2,4,5-trichlorophenol, the use of products prepared from this compound
(Table 11), and to incinerations reactions. The occurrence of the
other PCDDs and PCDFs is related to the synthesis and use of a variety
of other products (Table 12), some of which are quite common.
The other PCDDs and PCDFs are also formed in a variety of
incineration reactions (see section 4.5).
3.3 Contamination of Commercial Products
3.3.1 Chlorophenoxyacetic acid herbicides
Depending on the temperature control and purification efficiency,
the levels of 2,3,7,8-tetraCDD in commercial products may vary
greatly. For example, the levels of 2,3,7,8-tetraCDD in drums of the
herbicide Agent Orange placed in storage in the USA and in the Pacific
before 1970 were between 0.02 and 47 mg/g. More than 450 samples were
analyzed in this study, and the mean value was 1.98 mg/g (Young et
al., 1983). Since Agent Orange was formulated as a 1:1 mixture of the
butyl esters of 2,4,5-T and 2,4-D, the levels of 2,3,7,8-tetraCDD in
individual 2,4,5-T preparations manufactured and used in the 1960s
could have been as high as 100 mg/g.
In analyses using high-resolution GC-MS, Rappe et al. (1978a)
have reported that in other samples of Agent Orange (as well as in
European and the USA 2,4,5-T formulations from the 1950s and 1960s),
2,3,7,8-tetraCDD was the dominating compound of this group of
contaminants. Only minor amounts of other PCDDs and PCDFs could be
found, primarily lower chlorinated PCDDs, in samples of Agent Orange.
As a result of governmental regulations, efforts were made during
the 1970s to minimize the formation of 2,3,7,8-tetraCDD during 2,4,5-T
production, and now all producers claim that their products contain
less than 0.1 µg 2,3,7,8-tetraCDD/g of product (Rappe et al., 1978a).
At present, the chloro-phenoxy herbicides are not the major source of
PCDDs and PCDFs in the environment.
Sixteen samples of 2,4-D esters and amine salts from Canada were
analyzed for the presence of PCDDs. Eight out of nine esters and four
out of seven amine salts were found to be contaminated, with the
esters showing significantly higher levels (210-1752 ng/g) than the
salts (20-278 ng/g). The tetraCDD observed was the 1,3,6,8-isomer, as
verified by a synthetically prepared authentic standard (Cochrane et
al., 1981). In other studies, it has been found that no tetraCDD other
than the 1,3,6,8-isomer elutes in this window. Hagenmaier et al.
(1986) has reported that, unexpectedly, a German 2,4-D formulation
contained 6.8 ng of 2,3,7,8-tetraCDD/g.
Table 11. Some commercial products that may be contaminated with
2,3,7,8-tetraCDD, depending on the method of preparation
Common name Chemical name
2,4,5-Ta 2,4,5-Trichlorophenoxyacetic acid
2,4,5-T estersa n-butyl-, butoxy ethyl-, and
iso-octyl-esters of 2,4,5-
trichlorophenoxyacetic acid
2,4,5-T saltsa dimethylamine salts of 2,4,5-
trichlorophenoxyacetic acid
Fenoprop esters of 2-(2,4,5-trichlorophenoxy)-
propanoic acid
Erbon ethyl ester of 2-(2,4,5-trichloro-
phenoxy)-2,2-dichloropropanoic acid
2,4,5-Trichlorophenol 2,4,5-Trichlorophenol
Fenochlorphos O,O-Dimethyl O-2,4,5-trichlorophenyl
phosphonothioate
Trichloronate O-Ethyl 0-2,4,5-trichlorophenyl
ethylphosphonothioate
Hexachlorophene/isobac 20 2,2'-Methylene-bis (3,4,6-trichloro-
phenol)
a There are numerous trade names for this product.
Table 12. Some commercial products which may be contaminated with PCDDs
other than 2,3,7,8-tetraCDD, and with PCDFs, depending on the method of
preparation
Common name Chemical name
Bifenox Methyl-5-2,4-dichlorophenoxy-2-nitrobenzoate
Chloranil 2,3,5,6-Tetrachloro-2,
5-cyclo-hexadiene-1,4-dione.
2,4-D (esters and salts) 2,4-Dichlorophenoxyacetic acid
and esters and salts
2,4-DB and salts 2,4-Dichlorophenoxybutyric acid and
salts
Dicamba 3,6-Dichloro-2-methoxybenzoic acid
Dicamba, dimethylamine salt 3,6-Dichloro-2-methoxybenzoic acid,
dimethylamine salt
Dicapthon Phosphorothioic acid
o-(2-chloro-4-nitrophenyl)
o,o-dimethyl ester
Dichlofenthion Phosphorothioic acid
o-2,4-dichloro-phenyl
o,o-dialkyl ester
Disul sodium (sesone) 2,4-Dichlorophenoxyethyl sulfate,
sodium salt
2,4-DP 2- 2,4-Dichlorophenoxy propionic acid
HCB Hexachlorobenzene
Nitrofen 2,4-Dichlorophenyl-p-nitrophenyl
ether
PCP and salts Pentachlorophenol and salts
PCB Polychlorinated biphenyls
2,4,6-TCP 2,4,6-Trichlorophenol and salts
2,3,4,6-Tetrachlorophenol and salts
Common name Chemical name
CNP 1,3,5-Trichloro-2-(4-nitrophenoxy)
benzene
NIP 2,4-Dichloro-1-(4-nitrophenoxy)
benzene
X-52 2,4-Dichloro-1-(3-methoxy-4-nitro-
phenoxy) benzene
3.3.2 Hexachlorophene
The bactericide hexachlorophene is prepared from
2,4,5-trichlorophenol, also the key intermediate in the production of
2,4,5-T. Due to additional purification, the level of 2,3,7,8-tetraCDD
in this product is usually < 0.03 mg/kg (Baughman, 1974). Ligon & May
(1986) reported 0.0047 mg/kg of TCDD in one hexachlorophene sample.
However, hexachlorophene also contains about 100 mg/kg of a
hexachloroxanthene, the 1,2,4,6,8,9-substituted isomer (Göthe &
Wachtmeister, 1972).
3.3.3 Chlorophenols
Chlorophenols have been used extensively since the 1950s as
insecticides, fungicides, mold inhibitors, antiseptics, and
disinfectants. In 1978 the annual world production was estimated to be
approximately 200 000 tons. The most important use of 2,4,6-tri-,
2,3,4,6-tetra-, and pentachlorophenol, and their salts, is for wood
preservation. Pentachlorophenol is also used as a fungicide for slime
control in the manufacture of paper pulp and for a variety of other
purposes such as in cutting oils and fluids, for tanning leather, and
in paint, glues, and outdoor textiles. 2,4-Di- and
2,4,5-trichloro-phenol are used for the production of 2,4-D and
2,4,5-T herbicides (phenoxy acids), and 2,4,5-trichlorophenol for the
production of hexachlorophene.
Chlorophenols are produced industrially either by direct
chlorination of phenol or by hydrolysis of chlorobenzenes, the actual
process used depending on the isomer desired. Chlorination of phenol
yields 2,4-di-, 2,4,6-tri-, 2,3,4,6-tetra-, or pentachlorophenol,
while hydrolysis of chlorobenzenes is mainly used for the production
of 2,4,5-tri- and pentachlorophenol (Nilsson et al., 1978).
Chlorophenols may contain a variety of by-products and contaminants,
such as other chlorophenols, polychlorinated phenoxyphenols, and
neutral compounds like polychlorinated benzene and diphenyl ethers
(PCDPEs), PCDDs, and PCDFs. Some of these contaminants may also occur
in chlorophenol derivatives like phenoxy acids, other pesticides, and
hexachlorophene. The possible presence of PCDDs and PCDFs in
commercial products is of special significance because of their
extraordinary persistence and toxicological properties (see sections
7-9). A scientific criteria document for chlorophenols and their
impurities in the Canadian environment has been prepared by Jones
(1981, 1984). Chlorophenols were estimated to be the major chemical
sources of PCDDs and PCDFs in the Canadian environment (Sheffield,
1985).
Buser & Bosshardt (1976) reported on the results of a survey of
the PCDD and PCDF contents of pentachlorophenol (PCP) and PCP-Na from
commercial sources in Switzerland. From the results, a grouping of the
samples into two series can be observed: a first series with generally
low levels (hexaCDD <1 µg/g) and a second series with much higher
levels (hexaCDD >1 µg/g) of PCDDs and PCDFs. Samples with high PCDD
values had also high PCDF values. For most samples, the contents of
the PCDF contaminants were in the order:
tetra- = penta- < hexa- < hepta- < octaCDD/CDF.
The ranges of the combined levels of PCDDs and PCDFs were 2-16 and
1-26 µg/g, respectively, for the first series of samples, and 120-500
and 85-570 µg/g, respectively, for the second series of samples. The
levels of octaCDD and octaCDF were as high as 370 and 300 µg/g,
respectively.
Some PCP-Na samples analyzed showed the unexpected presence of a
tetraCDD (0.05-0.25 µg/g), which was later identified by Buser & Rappe
(1978) as the unusual 1,2,3,4-substituted isomer. Table 13 collects a
number of relevant analyses of these chlorophenol formulations. The
levels of PCDDs and PCDFs are higher than for the phenoxy-acetic acid
herbicides.
It has also been reported that several positional isomers of
PCDDs and PCDFs are present in the chlorophenols. However,
isomer-specific methods have not been used in most of these
investigations, and more research is necessary to identify all the
isomers present for a risk evaluation of these products.
Miles et al. (1985) have analyzed PCP samples for hexaCDDs from
five different manufacturers using an isomer-specific analytical
method. The study included both free PCPs as well as the sodium salts.
Total hexaCDDs in PCPs ranged from 0.66 to 38.5 mg/kg, while in the
sodium salts levels of hexaCDDs between 1.55 and 16.3 mg/kg were
found. The most abundant hexaCDD isomer found in the free PCPs was the
1,2,3,6,7,8 isomer; however, in the sodium salts the 1,2,3,6,7,9- and
1,2,3,6,8,9-hexaCDD pair was the most abundant.
Table 13. Levels of PCDDs and PCDFs in commercial chlorophenols (µg/g)a
2,4,6- 2,3,4,6- PCP PCP
Trichlorophenol Tetrachlorophenol Sample A Sample B
TetraCDDs < 0.1 < 0.1 < 0.1 < 0.1
PentaCDDs < 0.1 < 0.1 < 0.1 < 0.1
HexaCDDs < 1 < 1 < 1 2.5
HeptaCDDs < 1 10 0.5 175
OctaCDD < 1 2 4.3 500
TetraCDFs 1.5 0.5 < 0.1 < 0.1
PentaCDFs 17.5 10 < 0.1 < 0.1
HexaCDFs 36 70 0.03 < 0.3
HeptaCDFs 4.8 70 0.5 19
OctaCDF < 1 10 1.1 25
a From: Rappe et al. (1979).
Hagenmaier & Brunner (1987) has reported that 2,3,7,8-tetraCDD
can be found in commercial pentachlorphenol formulation at levels of
0.21-0.56 ng/g, while Hagenmeyer & Brunner (1986) report that
1,2,3,7,8-pentaCDD was found in pentachlorophenol and
Na-pentachlorophenates in concentrations of 0.9-18 ng/g.
3.3.4 Polychlorinated biphenyls (PCBs)
Vos et al. (1970) were able to identify PCDFs (tetra- and
pentaCDFs) in samples of European PCBs (Phenoclor DP-6 and Clophen A
60) but not in a sample of Aroclor 1260. The toxic effects of these
PCB products were found to parallel the levels of PCDFs present. Bowes
et al. (1975) examined a series of Aroclors, as well as the samples of
Aroclor 1260, Phenoclor DP-6, and Clophen A-60 previously analyzed by
Vos et al. (1970). They used packed columns and very few standard
compounds, and reported that the most abundant PCDFs had the same
retention time as 2,3,7,8-tetraCDF and 2,3,4,7,8-pentaCDF. Using a
complete set of PCDF standards and an isomer-specific analytical
method, Rappe et al. (1985d) determined the levels of
2,3,7,8-substituted PCDFs in commercial PCB products (see Table 14).
3.3.5 Chlorodiphenyl ether herbicides
In 1981, Yamagishi et al. reported on the occurrence of PCDDs and
PCDFs in the commercial diphenyl ether herbicides
1,3,5-trichloro-2-(4-nitrophenoxy) benzene (CNP),
2,4-di-chloro-1-(4-nitrophenoxy)benzene (NIP), and
2,4-dichloro-1-(3-methoxy-4-nitrophenoxy)benzene (X-52). The total
tetraCDD found was 14.0 mg/kg in CNP, 0.38 mg/kg in NIP, and 0.03 in
X-52. Very few synthetic standards were used, but the major tetraCDDs
were identified as the 1,3,6,8- and 1,3,7,9-isomers, the expected
impurities in the starting material 2,4,6-trichlorophenol. No
2,3,7,8-tetraCDD could be detected in these samples. In all three
herbicides, total tetraCDF was between 0.3 and 0.4 mg/kg.
3.3.6 Hexachlorobenzene
Hexachlorobenzene was used for the control of wheat bunt and
fungi. Villeneuve et al. (1974), analyzing three commercial
hexachlorobenzene preparations, identified octaCDD and hepta- and
octaCDFs. The levels and identity of the heptaCDF isomers were not
given. Great variation in levels of octaCDDs between the three samples
(0.05-211.9 mg/kg) was noted, as well as in the level of octaCDF
(0.35-58.3 mg/kg).
3.3.7 Rice oil
In 1968 more than 1500 people in southwest Japan were intoxicated
by the consumption of a commercial rice oil accidentally contaminated
by PCBs, PCDFs, and polychlorinated quarterphenyls (Masuda &
Yoshimura, 1982; Masuda et al., 1985). In 1979 a similar episode
occurred in central Taiwan, the number of people involved here
approaching 2000 (Chen et al., 1980, 1981). Both these accidents have
been referred to as Yusho episodes, but now the Taiwan episode has
been renamed Yu-cheng (see section 5.4.4.4).
The total level of PCDFs in the Japanese rice oil was reported to
be 5 µg/g (Nagayama et al., 1976) and 5.6 µg/g (Buser et al., 1978d).
For the rice oil from Taiwan, Chen et al. (1985) reported the PCDFs
levels to be in the range 0.18-1.68 µg/g.
Buser et al. (1978) analyzed the Japanese rice oil using glass
capillary columns. They found about 50-60 PCDF congeners and also
reported that the 2,3,7,8-tetraCDF was the major isomer among the
tetraCDFs. However, it was later shown that in this column system the
2,3,4,8-tetraCDF co-elutes with the 2,3,7,8-isomer, and in fact the
2,3,4,8-isomer was the main constituent in this peak (Chen & Hites,
1983; Masuda et al., 1985). The 2,3,7,8-substituted congeners were
estimated to account for 10-15% of the total amount of PCDFs (Buser et
al., 1978).
Table 14. PCDFs in commercial PCBs (ng/g)a
TRI- TETRA- PENTA- HEXA- HEPTA-
Total 2378 Total 12348 23478 Total 123479 123678 123789 234678 Total Total
PCB-type 12378 123478
Pyralene 700 53 630 10 T 35 ND ND ND ND ND ND
A1254 63 19 1400 690 490 4000 2500 2100 190 130 10 000 960
A1260 10 13 110 48 56 260 500 120 190 27 1500 1300
A30 500 35 573 14 28 160 50 59 ND ND 220 T
A40 1300 180 2600 96 8 1700 79 68 ND T 310 ND
A50 7400 3300 20 000 760 1100 8000 700 360 18 98 3100 75
A60 770 840 6900 1100 990 8100 1600 330 170 330 6800 2000
T64 47 23 360 97 122 840 520 390 58 41 2600 220
Clophen C 710 54 1200 34 30 270 ND T ND ND T ND
a From: Rappe et al. (1985d).
T = traces.
ND = not detected.
3.4 Sources of Heavy Environmental Pollution
3.4.1 Industrial accidents
Several industrial accidents occurring during the production of
2,4,5-trichlorophenol have been described in the literature. In most
of these accidents the pollution of 2,3,7,8-tetraCDD has been to
factories with circumscribed occupational exposure (section 9).
However, on 10 July, 1976, a runaway reaction in a factory at Meda
near Seveso in Northern Italy resulted in the escape of a chemical
cloud of trichlorophenol/phenate containing 2,3,7,8-tetraCDD.
The cloud initially covered an area outside the factory 5 km long
and 700 m wide. On the basis of the TCDD levels found in the
contaminated soil samples, it has been estimated that 2-3 kg of TCDD
was released in this accident. About 80% of this amount was deposited
in an area of 15 ha, within a distance of about 500 m from the plant.
The levels of soil contamination in three zones are given in Table 15
(Pocchiari, 1978).
3.4.2 Improper disposal of industrial waste
In 1973, three horse arenas in Missouri, USA, were found to be
contaminated by high levels of 2,3,7,8-tetraCDD; the highest value
reported was about 30 µg/g of soil (Kimbrough et al., 1977). This
contamination resulted from the application, in 1971, of contaminated
waste oil to control dust at these locations. The TCDD had originated
at a hexachlorophene-producing factory in Verona, Missouri. Additional
tri- and tetraCDDs were also found, but the major component was
1,2,4,6,8,9-hexachloroxanthene, a compound which apparently can serve
as a marker for this type of contamination. The xanthene is a normal
by-product of hexachlorophene production and has never been associated
with the production of 2,4,5-tri-chlorophenol or 2,4,5-T derivatives
(Viswanathan & Kloepfer, 1986).
In 1982, numerous sites of potential 2,3,7,8-tetraCDD
contamination were discovered in eastern Missouri. The contamination
originated from the same waste oil from the factory in Verona. The
streets of the entire town of Times Beach, Missouri, had been sprayed.
More than 10 000 soil samples from Missouri were analyzed. In this
state more than 40 hazardous waste sites containing 2,3,7,8-tetraCDD
were identified. Most of these contaminated sites resulted from the
disposal of waste from the same factory in Verona. The highest level
reported in these soil samples was 9648 mg TCDD/g (Viswanathan &
Kloepfer, 1986).
Another location of great concern is Love Canal, Niagara Falls,
USA. Here, Smith et al. (1983) found high levels of 2,3,7,8-tetraCDD
in storm sewer sediments taken from around the Love Canal waste
disposal site. The highest value was 312 ng/g sediment.
Table 15. Distribution of TCDD contamination in the Seveso area on the basis of soil sample analysesa
Range Number of soil samples
(µg/m2)
Zone A Zone B Surrounding monitored area
< 0.750 32 25 249
0.750 - 4.99 32 53 128
5.0 - 14.99 6 19 2
15.0 - 49.99 18 6 0
50.0 - 499.99 31 0 0
500.0 - 4999.99 18 0 0
> 5000 3 0 0
a From: Pocchiari (1978).
Zone A: high-level contamination, about 115 ha.
Zone B: low level contamination, about 255 ha.
Surrounding area: about 1400 ha.
3.4.3 Heavy use of chemicals
The Eglin Air Force Base in Northwest Florida, USA, has been used
for the development and testing of aerial spraying equipment for
military defoliation operations. During the period 1962-1970, a
3-km2 test area was sprayed with 73 tons of 2,4,5-T. Analyses of
archived samples of the formulations indicated that approximately 2.8
kg of 2,3,7,8-tetraCDD had been applied as a contaminant of the
herbicide. However, one 37-ha test grid received 2.6 kg of this TCDD
from 1962 to 1964. Levels of 10-1500 ng/kg were found in 22 soil
samples (the top 15 cm) collected and analyzed 14 years after the last
application of herbicide to this site (Young, 1983).
3.5 Other Sources of PCDDs and PCDFs in the Environment
3.5.1 Thermal degradation of technical products
The formation of 2,3,7,8-tetraCDD as a result of thermal
reactions of 2,4,5-T and 2,4,5-T derivatives has been the subject of
controversy. Heating 2,4,5-T salts at 400-450 °C for 30 minutes or
longer yielded approximately 1 g of 2,3,7,8-tetraCDD per kg of 2,4,5-T
salt, while no TCDD was identified from the same treatment of 2,4,5-T
acid or esters (Langer et al., 1973; Baughman, 1974). Using a more
sensitive analytical method, Ahling et al. (1977) reported that 0.2-3
mg of 2,3,7,8-tetraCDD was formed per kg of 2,4,5-T esters during
combustion at 500-850 °C. Two reports (Stehl & Lamparski, 1977;
Andersson et al., 1978) have shown that 2,3,7,8-tetraCDD could not be
found after burning samples of spiked or sprayed vegetation at 600 °C.
The combustion gases, soot, particles, and ashes were analyzed and the
detection limit was 4 mg of TCDD/kg 2,4,5-T burned.
Rappe (1978b) have studied the burning of material impregnated
with various salts of chlorophenols. Very carefully purified
2,4,6-tri- and pentachlorophenate were studied, in addition to a
commercial formulation of 2,3,4,6-tetra-chlorophenate. The analytical
method used in this study was not isomer specific, but the following
conclusions can be drawn concerning the formation of PCDDs by thermal
reactions:
(a) the expected dimerization products and the products formed
in the "Smiles rearrangement" are the major PCDDs;
(b) no other thermal isomerization of the PCDDs formed can be
observed;
(c) no formation of higher chlorinated PCDDs can be observed;
(d) octaCDD and other higher chlorinated PCDDs yield lower
chlorinated dioxins in a nonspecific dechlorination
reaction;
(e) a series of PCDFs was also observed.
It has been found that PCBs can be converted to PCDFs under
pyrolytic conditions. The pyrolysis of commercial PCBs in sealed
quartz ampoules in the presence of air yielded about 30 major, and
more than 30 minor, PCDFs. The optimal yield of PCDFs was about 10%,
calculated on the amount of PCB decomposed. Thus, uncontrolled burning
of PCBs can be an important environmental source of hazardous PCDFs.
Therefore, it was recommended (Buser et al., 1978a, 1978d) that all
destruction of PCB-contaminated waste using incinerators must be
carefully controlled. In the temperature range 300-400 °C, the
conversion yield seems to be in the part-per-million range (Morita et
al., 1978).
Buser & Rappe (1979) studied the pyrolysis of 15 individual
synthetic PCB congeners and showed that the formation of PCDFs can
follow several competing reaction pathways. In another study where a
series of chlorobenzenes were pyrolyzed in the same way, Buser (1979)
found that significant amounts (> 1%) of PCDDs and PCDFs were formed.
A complex mixture of isomers of PCDDs and PCDFs was found, suggesting
several reaction routes. Using the same technique as above, Lindahl et
al. (1980) studied the thermal decomposition of polychlorinated
diphenyl ethers. Both PCDDs and PCDFs were formed, involving several
pathways. The temperature range was 500-600 °C and the yields varied
from 0.1 to 4.5%.
Bergman et al. (1984) studied the thermal degradation of two
polychlorinated alkanes containing 59% and 70% chlorine, respectively,
and also a commercial chlorinated paraffin containing 70% chlorine.
Their studies indicated the presence of at least mono- and diCDFs.
Ahling et al. (1978) reported that chlorinated benzenes can be
found in the pyrolysis of PVC.
Direct evidence for the conversion of PVC to PCDDs and PCDFs has
recently been reported by Marklund et al. (1986). They found that
laboratory pyrolysis of PVC resulted in the formation of PCDDs and
PCDFs, mainly hexa- and heptaCDDs, and tetra- to heptaCDFs. In some
cases, the pattern of isomers seemed to be similar to those found in
municipal and hazardous waste incinerators, e.g. the pentaCDFs (Rappe
et al., 1987).
The data discussed in this section are summarized in Table 16.
3.5.2 Incineration of municipal waste
For some time, emissions from municipal incinerators, heating
facilities, and thermal power plants have been the subject of concern.
Whereas previously the emission of dust, smoke, toxic metals, and
noxious gases were of prime concern, the presence of potentially
hazardous organic compounds from these emissions has been recognized
only recently. Lahaniatis et al. (1977) reported the presence of
chlorinated organic compounds (chlorinated aliphatics, benzenes, PCBs,
and pesticides) in fly ash from a municipal incinerator.
Olie et al. (1977) reported the occurrence of PCDDs and PCDFs in
fly ash from three municipal incinerators in the Netherlands. Their
results indicated the presence of up to 17 PCDD peaks, but isomer
identification and quantification was not possible due to the lack of
synthetic standards. Buser & Bosshardt (1978) studied fly ash from a
municipal incinerator and an industrial heating facility, both in
Switzerland. In the former, the level of PCDDs was 0.2 µg/g and of
PCDFs 0.1 µg/g. In the industrial incinerator, the levels were 0.6
µg/g and 0.3 µg/g, respectively.
During the period 1978-1982 a series of papers, reports, and
reviews were published confirming the original findings of Olie et al.
(1977) and Buser & Bosshardt (1978) regarding fly ash. Less data have
been published on the levels of PCDDs and PCDFs in other incineration
by-products, e.g., particulates and flue gas condensate, and in total
flue gas, which are the true emissions (Marklund et al., 1986).
A risk evaluation should be based on the emission levels of PCDD
and PCDF isomers found in isomer-specific analyses using validated
sampling and clean-up methods. However, in many studies non-validated
sampling and analytical methods are used and the results are given in
terms of total levels of tetra-, penta-, hexa-, hepta-, and octaCDDs
and CDFs. The value of such studies is limited, particularly in this
situation where the number of isomers is quite large. More than 30
PCDDs and 60 PCDFs have been found in fly ash samples (Buser et al.,
1978b, 1978c).
In March 1986, a working group of experts convened by the World
Health Organization Regional Office for Europe reviewed the available
data on emissions of PCDDs and PCDFs from municipal solid-waste (MSW)
incinerators. It was found that the origin of these compounds was not
completely understood, but they appear to result from complex thermal
reactions occurring during periods of poor combustion. Because of
their high thermal stability, the PCDDs and PCDFs can be destroyed
only after adequate residence times at temperatures above 800 °C
(WHO/EURO, 1987).
Available data on total emissions of PCDDs and PCDFs from tests
on MSW incinerators range between a few and several thousand ng/Nm3
dry gas at 10% carbon dioxide (CO2). The working group prepared a
table giving a range of estimated isomer specific emissions for those
isomers of major concern with respect to MSW incinerators operating
under various conditions (Table 17).
The emissions tabulated in column 1 are those which the working
group considered to be achievable in the most modern, highly
controlled, and carefully operated plants in use at the present time.
Such results do not represent what is considered to be achievable by
the use of acid gas cleaning equipment; use of such equipment should
result in much lower values (probably at least one order of
magnitude). The results given in column 1 are not representative of
emissions that might be expected from such plants during start-up or
during occasional abnormal conditions. Emission levels listed in
column 2 were considered by the working group to be indicative of the
higher limit of emissions from modern MSW incinerators. These plants
might experience such emissions during start-up or during occasional
upset conditions. Consequently, the majority of the available
concentration data falls between columns 1 and 2. Some of the data
reviewed has shown that the figures in column 2 should not be
considered an absolute maximum. However, most existing plants, if
carefully operated, will have PCDD and PCDF emisions in the range
between columns 1 and 2.
The highest values for MSW incinerators (column 3) were obtained
by multiplying the values in column 2 by a factor of 5. Column 3
includes emission data that were reported to the working group from
all tests and under all circumstances. Generally, these emission
levels are associated with irregular or unstable operating conditions,
high moisture content of the MSW, low combustion or afterburner
temperatures, less than adequate technologies, etc.
Table 16. Formation of PCDDs and PCDFs by thermal processes
Precursor Conditions Products
2,4,5-T salt Pyrolysis 2,3,7,8-tetraCDD
2,4,5-T (vegetation) Pyrolysis No TCDD
Burning No TCDD
Cl-phenate Burning PCDDsa + PCDFs
PCBs Pyrolysis PCDFsb
PCBzc Pyrolysis PCDFs + PCDDsd
Cl-Diphenyl ethers Pyrolysis PCDFs + PCDDs
Cl-Alkanes (Paraffins) Pyrolysis PCDFs
PVC Pyrolysis PCDDs + PCDFs
a = PCDDs formed by dimerization and a non-specific dechlorination.
b = other products: hexa- and pentaCBs.
c = polychlorinated benzenes.
d = other products: PCBs, polychlorinated naphthalenes.
The working group was aware of both lower and higher emission
levels than those included in Table 17. However, it was felt that the
values included in Table 17 were likely to be representative of
emissions from current facilities (WHO/EURO, 1987).
Of special importance is the observation that the emission of
1,2,3,7,8-pentaCDD normally exceeds the emission of 2,3,7,8-tetraCDD
by a factor of three to ten.
3.5.3 Incineration of sewage sludge
Sludge from municipal waste water treatment plants may be
incinerated after being dewatered. The WHO working group (see 3.5.2)
reviewed the available data from municipal sewage sludge (MSS)
incinerators, and found that PCDD and PCDF emissions from this type of
plant were generally lower than emissions from MSW incinerators (see
Table 17, column 4) (WHO, 1986).
3.5.4 Incineration of hospital waste
Doyle et al. (1985) claimed that the incomplete combustion of
certain hospital waste containing halogenated organics could produce
high levels of PCDDs and PCDFs. They found the mean values of total
PCDDs to be 69 ng/m3 and total PCDFs to be 156 ng/m3. No
isomer-specific data seems to be available. Hagenmaier et al. (1986)
reported the analyses of stack gas from 10 hospital waste incineration
plants. The mean value of 2,3,7,8-tetraCCD emitted was 0.28 ng/m3,
the mean of all TCDDs being 20 ng/m3. The mean value for total PCDDs
was 118 ng/m3 and for total PCDFs 434 ng/m3.
Table 17. Estimated range of emissions from municipal solid waste (MSW) and municipal sewage sludge (MSS) incineratorsa
Emissions from MSW combustion Emissions
from MSS
combustion
1 2 3 4
Congeners Achievable with Maximum High Most
modern plants from emissions likely
with no acid average highest
gas cleaning operation emissions
(ng/Nm3, dry, at 10% CO2)
2,3,7,8-TetraCDD 0.1 1.5 7.5 0.1
1,2,3,7,8-PentaCDD 0.3 14 70 0.3
1,2,3,4,7,8-HexaCDD 0.2 31 155 0.2
1,2,3,6,7,8-HexaCDD 0.6 56 280 0.6
1,2,3,7,8,9-HexaCDD 0.4 20 100 0.4
2,3,7,8-TetraCDF 0.9 10 50 0.9
1,2,3,7,8-/1,2,3,4,8-PentaCDF 2.3 52 260 2.3
2,3,4,7,8-PentaCDF 2.0 40 200 2.0
1,2,3,4,7,8/1,2,3,4,7,9-HexaCDF 1.1 48 240 1.1
1,2,3,6,7,8-HexaCDF 1.3 40 200 1.3
1,2,3,7,8,9-HexaCDF 0.06 52 260 0.06
2,3,4,6,7,8-HexaCDF 2.0 36 180 2.0
a From: WHO/EURO (1987).
3.5.5 Incineration of hazardous waste
Analyses from a test burn of pentachlorophenol waste have been
reported by Rappe et al. (1983c). PCP is a well known precursor to
octaCDD (section 2). Samples of baghouse ash and bottom ash were
analyzed. In the baghouse ash the total level of octaCDD was only 0.2
µg/g. The major constituents were lower chlorinated PCDDs such as
hepta-, hexa-, penta-, and tetraCDDs. The isomeric distribution was
reported to be very similar to a "normal" fly ash. In both cases
2,3,7,8-tetraCDD was a very minor constituent. The level of PCDD in
the bottom ash was 0.31 µg/g. The baghouse ash was also reported to
contain PCDFs at a total level of 2.5 µg/g. For the tetra- and penta-
chlorinated compounds, equal amounts of PCDDs and PCDFs were reported.
Oberg & Bergstrém (1986) reported on test data from a Swedish
hazardous waste incinerator equipped with a rotary kiln, an
afterburner, and a dry scrubbing unit. Combustion tests were performed
with PCB (Aroclor 1242) as a fluid, and as a contaminant in solid
waste (Aroclor 1016 in capacitors). The results of these tests
indicated no correlation between the amount of PCB incinerated and the
amount of PCDDs and PCDFs found in the emissions.
3.5.6 Metal industry and metal treatment industry
It has been reported by Marklund et al. (1986) that industrial
high-temperature processes like copper smelters and electric arc
furnaces in steel mills have been identified as sources of
environmental contamination by PCDDs and PCDFs. The results are
reported in "TCDD equivalents" according to US EPA (1987). The
emission from the copper smelter contained 11 ng of TCDD
equivalents/Nm3 dry gas and 10% CO2, while the dust from the steel
mill contained 0.8 ng TCDD equivalents/g dust. Marklund et al. (1986)
also considered the emissions from industrial incinerators to be of
the same magnitude, or even higher, than the emissions from MSW
incinerators.
Southerland et al. (1987) analyzed emissions from various
incinerators within Tier 4 of the USA. The highest levels were found
in a secondary copper smelter, which contained 170 ng of
2,3,7,8-tetraCDD/Nm3 and 3% oxygen. This was by far the highest
level found within the US EPA National Dioxin Strategy.
3.5.7 Wire reclamation
Hryhorczuk et al. (1981) studied a wire reclamation incinerator
in the USA. Using a non-isomer-specific analytical method, they
determined total levels of tetraCDDs and tetraCDFs. Two samples were
analyzed, one from the furnace and one from the stack. The furnace
sample contained 58 ng/kg of total TCDDs and 730 ng/kg of total TCDFs,
whereas the stack sample contained 410 ng/kg of total TCDDs and 11 600
ng/kg of total TCDFs.
3.5.8 Traffic
Marklund et al. (1987) reported a study where automobile exhaust
emissions were analyzed for PCDDs and PCDFs. Two groups of test cars
were utilized: (1) cars equipped with a catalytic converter using
unleaded gasoline with no halogenated scavengers; (2) cars with no
catalytic converter using leaded gasoline (0.15 g/litre) and a
dichloroethane scavenger (0.1 g/litre). Before the test runs, the
motor oil was changed in all cars. No PCDDs and PCDFs could be
identifed in the cars using the unleaded gasoline, while the average
emission from the cars running on leaded gasoline was found to be
30-540 pg/kg of TCDD equivalents. It was assumed that the chlorinated
scavenger (dichloroethane) was the precursor of the PCDDs and PCDFs
formed. It was estimated that the total amount of PCDDs and PCDFs from
cars in Sweden using leaded gasoline with halogenated scavengers is in
the range of 10-100 g TCDD equivalents/year.
3.5.9 Fires and accidents in PCB-filled electrical equipment
In February 1981 a fire in the State Office Building in
Binghamton, New York, USA, caused a transformer to rupture, releasing
soot throughout the building. The dielectric fluid in the transformer
consisted of a mixture of PCB (65%) and chlorinated benzenes (35%).
The soot was found to be highly contaminated with PCDFs (total PCDFs
> 2000 µg/g). The most toxic isomers (2,3,7,8-tetraCDF; 1,2,3,7,8-
and 2,3,4,7,8-pentaCDF; and 1,2,3,4,7,8- and 1,2,3,6,7,8-hexaCDF) were
found to be the major constituents within each group of congeners.
Levels reported were 12 mg/g of 2,3,7,8-tetra CDF, 670 mg/g of total
penta-CDFs, and 965 mg/g of total hexa-CDFs, 46 mg/g of total
hepta-CDFs, and 460 mg/g of octa-CDFs (Rappe, 1984; Rappe et al.,
1985b). In addition, a series of PCDDs were identified, including the
highly toxic 2,3,7,8-tetraCDD, and 1,2,3,7,8-pentaCDD (Rappe et al.,
1983a; Buser & Rappe, 1984). It is assumed that the chlorinated
benzenes were the dioxin precursors.
Between 1981 and 1985, a series of transformer accidents (7 in
all) similar to the one in Binghamton were reported in the USA and
Canada (Rappe et al., 1986a). In January 1985, an explosion followed
by a fire ruptured a transformer in the basement of a residential
complex in Rheims, France. The transformer was filled with PCB (60%)
and trichlorobenzene (40%). Total levels of PCDFs were as high as 2570
µg/m2 before clean-up. Only traces of hepta- and octaCDD were found
(Rappe et al., 1985a).
In Europe, between 1981 and 1985, 19 accidents involving indoor
capacitor fires and explosions were reported from Scandinavian
countries (Rappe et al., 1986a). All capacitors were mineral-oil
filled, and contamination of the sites averaged 1-5 µg total
PCDFs/m2.
3.5.10 Pulp and paper industry
Large amounts of chlorine or chlorine compounds are used in the
pulp and paper industry for the bleaching of the pulp. Three black
liquor boilers from the craft paper process were included in the US
EPA study of combustion sources. No 2,3,7,8-tetraCDD was found in
these emissions, but low levels of other PCDDs and PCDFs were found in
one of the three incinerators and a yearly emission of 0.25 g was
calculated (Southerland et al., 1987).
Rappe et al. (1987) recently identified both 2,3,7,8-tetraCDD
(170 ng/kg) and 2,3,7,8-tetraCDF (890 ng/kg) in a sample taken in a
sedimentation lagoon at a Swedish paper mill. A series of other PCDDs
and PCDFs was also identified but at lower levels. The isomeric
pattern in this sample differed markedly from other sediment samples,
indicating pulping processes to be a source of environmental pollution
by 2,3,7,8-tetraCDD and 2,3,7,8-tetraCDF (Rappe et al., 1987).
3.5.11 Incineration of coal, peat, and wood
The emissions of PCDDs and PCDFs from coal-fired power plants
(Kimble & Gross 1980), wood stoves (Clement et al., 1985), and peat
burning (Marklund et al., 1986) seem to be very low when calculated
per m3. However, the very high flow rates and the large number of
units could make a significant total contribution. The occurrence of
pentaCDDs and all PCDFs was not discussed in this report.
3.5.12 Inorganic chlorine precursors
It is well known that certain organochlorine compounds are
efficient precursors to PCDDs and PCDFs during pyrolysis. However, it
was proposed by scientists from Dow Chemical Company that PCDDs, and
especially 2,3,7,8-tetraCDD, are ubiquitous and formed as trace level
by-products of any normal combustion (Bumb et al., 1980).
Consequently, dioxins should have been present in the environment
since the advent of fire. This suggests that inorganic chloride can
serve as a useful precursor to the formation of PCDDs and PCDFs. A
recent survey of PCDD levels, in particular from residential wood
combustion units, has been quoted in support of the above. The survey
showed PCDD levels in the ng/kg range (see also section 3.5.11).
However, this hypothesis has been criticized. One of the main
arguments against such a hypothesis is that 2,3,7,8-tetraCDD does not
appear to be formed in coal-fired power plants (Kimble & Gross, 1980;
Junk & Richard, 1981). Another argument is that the Dow studies lack
data on levels of dioxin precursors in the material being burned,
including the air in the flames (Rappe, 1984).
The analyses of historical samples gives additional support to
the theory that organochlorine compounds are more important as
precursors than inorganic chloride. When Czuczwa & Hites (1985)
analyzed sediment core samples from Lake Huron, N. America, the first
indication of PCDDs and PCDFs was found in sediments from 1940. There
was also a good correlation between the trend in the levels of PCDDs
and PCDFs in these sediments and the trend in the production of
chlorinated aromatic compounds (section 5.4).
Schecter et al. (1986a) were unable to detect PCDDs and PCDFs in
human liver and lung tissue from two female Eskimos frozen over one
hundred years ago (see also section 4.4.4).
3.5.13 Photochemical processes
Sundström et al. (1979) studied the formation of 2,3,7,8-tetraCDD
in six re-forestation areas that were sprayed with 2,4,5-T esters.
Leaf samples from the areas were analyzed for 2,4,5-T esters and TCDD.
TCDD was found in one leaf sample only, at levels lower than expected
from the level of dioxin contamination of the herbicide formulations
used.
The photochemical formation of PCDDs and PCDFs has also been
studied in laboratory experiments.
The photochemical dimerization of chlorophenols to PCDDs was
studied by Crosby & Wong (1976). The only PCDD formed in this study
was the octaCDD. Other PCDDs can be formed by photochemical
cyclization of chlorinated o-phenoxyphenols, also called pre-dioxins
(Nilsson et al., 1974). These pre-dioxins are very common impurities
(1-5%) in commercial chlorophenols (Nilsson et al., 1978), but the
cyclization is only a minor reaction pathway; the main reaction being
the dechlorination of the pre-dioxin.
Akermark (1978) studied the formation of 2,3,7,8-tetraCDD from
the appropriate pre-dioxins. He could identify the product, but
claimed the reaction to be very inefficient.
Another photochemical process of potential environmental
importance is dechlorination of the higher chlorinated PCDDs and
PCDFs, e.g., octaCDD and octaCDF. The products formed by photolysis of
octaCDD in organic solvent have now been identified (Buser & Rappe,
1978). By comparison with authentic standards, it was found that the
main tetrachloro isomer was the 1,4,6,9-tetraCDD; the major
pentachloro compound was expected to be the 1,2,4,6,9-isomer, and the
main hexa- and heptachloro compounds were the 1,2,4,6,7,9- (or
1,2,4,6,8,9-) and the 1,2,3,4,6,7,9-isomers, respectively. The
reaction scheme deduced from this data indicates that the chlorine
atoms are removed preferentially from the lateral positions on the
carbon rings. Consequently, the most toxic PCDD isomers, such as
2,3,7,8-tetraCDD, are not likely to be formed from the photolysis of
the higher PCDDs in solution.
Crosby et al. (1973) studied the photolysis of a series of PCBs
dispersed in water. For two isomers, the 2,5-dichloro- and
2,2',5,5'-tetrachlorobiphenyls, small amounts (0.2%) of 2-mono-CDF
could be found among the products (for photo-chemical transformations,
see section 4.2.1).
3.6 Comparison of Isomeric Pattern and Congener Profiles From Various Sources
There is a pronounced difference between technical products and
incineration emissions in both isomeric patterns and congener profiles
of PCDDs and PCDFs. In technical products the number of isomers
present is limited, whereas in incineration emissions most isomers
seem to be present. Rappe (1987) has pointed out the large similarity
qualitatively in isomeric patterns between different incineration
sources.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATIONS
4.1 Environmental Transport
4.1.1 Air
The PCDDs and PCDFs are believed to be transported in the
atmosphere. The transport of these compounds from stacks and other
stationary point sources, as well as from waste disposal sites and
other area sources, can be predicted from dispersion modelling (SAI,
1980). In the case of the accidental release of a toxic cloud
containing 2,3,7,8-tetraCDD at Seveso, Italy, Cavallaro et al. (1982)
determined the transport pattern and the ground deposition. They
determined that the deposition of 2,3,7-8-tetraCDD from air to soil
should follow an exponential decay pattern in the
Gaussian-distribution along the cross-section of the downwind
direction. Thibodeaux (1983) studied the air transport of
2,3,7,8-tetraCDD at a herbicide production facility in Jacksonville,
Arkansas, USA.
The dispersion modelling has limitations. If possible, the
modelling calculations should be combined with true air measurements.
4.1.2 Water
The solubility of 2,3,7,8-tetraCDD in water has been extensively
studied (see section 2), but much less data are available for the
other PCDDs and PCDFs. However, data from microbiological experiments
indicate that 2,3,7,8-tetraCDD is highly adsorbed to sediments and
biota. Matsumura et al. (1983) suggested that more than 90% of the
2,3,7,8-tetraCDD in an aquatic medium could be present in the adsorbed
state. Rappe et al. (1985c) studied a suspension of soot/dust in the
wash water from a PCB fire. The suspension contained 100 ng/ml of
various PCDFs, but when the soot was settled the water contained no
detectable levels of PCDFs (detection level: 0.1 ng/ml of each
isomer). Most of the PCDDs and PCDFs, if present in waterways, should
be in the sediments or attached to suspended particles.
Thibodeaux (1983) has calculated the amount of 2,3,7,8-tetraCDD
transported by a creek within the contaminated herbicide factory in
Jacksonville, Arkansas, USA. The value was 0.89 g/year as an average
rate, and a maximum of 2.1 g/year.
4.1.3 Soil and sediments
The mobility of 2,3,7,8-tetraCDD and of a dichlorodioxin in soils
has been studied by Helling et al. (1973). Both were found to be
immobile in all soils and, therefore, would not be leached out by
rainfall or irrigation, though lateral transport during surface
erosion of the soil could occur.
The US Air Force conducted studies in an area of north-west
Florida that had been heavily sprayed with the herbicide Agent Orange
between 1962 and 1964 (Young et al., 1975). This herbicide mixture was
contaminated with TCDD (section 3.2). A 7.8-ha test grid received a
total of 40 metric tons of 2,4,5-T between 1962 and 1964. When 15-cm
soil core samples were taken in 1974, they showed TCDD concentrations
ranging from 10 to 710 ng/kg. This study illustrates that significant
levels of TCDD residues remained 10 years after the last herbicide
application. Similar TCDD concentrations were obtained from areas that
had been sprayed between 1962 and 1969 (Bartelson et al., 1975).
In another study Young (1983) measured the concentration of
2,3,7,8-tetraCDD in a soil profile. The samples were collected in 1974
and the data suggested that most of the 2,3,7,8-tetraCDD would be
found in the top 15 cm of the soil profile (Table 18).
The probable media and modes of transport of PCDDs from soils are
the following: (1) to air via contaminated airborne dust particles;
(2) to surface water via eroded soil transported by water; (3) to
groundwater via leaching; (4) to air via volatilization. Movement of
particulate matter containing adsorbed PCDDs and PCDFs has been
considered to be a much more important transport mechanism than
leaching and volatilization because of the low water solubility and
volatility of these compounds (Josephson, 1983). However, the
monitoring of Seveso soil one year after the accident showed that the
highest 2,3,7,8-tetraCDD levels were not present in the topmost soil
layer (0.5 cm), but very often in the second (0.5-1.0 cm) or third
(1.0-1.5 cm) layers. This disappearance of at least a part of the
2,3,7,8-tetraCDD from the topmost soil layer was speculated to be due
to volatilization or vertical movement through the soil (DiDomenico et
al., 1980). Therefore, it appears that volatilization from soil and
leaching to groundwater can be responsible for the transport of PCDDs
and PCDFs from soils under certain conditions, namely, heavy rainfall
on sandy soils. Studies by Young (1983) indicate that the half-life
for 2,3,7,8-tetraCDD in soil is 10-12 years.
Thibodeaux (1983) has calculated the vaporization of
2,3,7,8-tetraCDD from a herbicide plant in Jacksonville, Arkansas,
USA. The vaporization can take place from soil surfaces, from landfill
cells, and from the surface of a pond. In Table 19 a summary of yearly
emission rates from these sources is presented.
It was found that vaporization from the soil surface in the
highly contaminated blow out area was the major contributing source of
emissions from this plant.
Table 18. Concentration of 2,3,7,8-tetraCDD in a soil profilea, b
Depth (cm) 2,3,7,8-tetraCDD (ng/kg)
0 - 2.5 150
2.5 - 5.0 160
5.0 - 10 700
10 - 15 44
15 - 90 NDc
a From: Young (1983).
b The area received 1,069 kg/ha of 2,4,5-T Agent Orange during
1962-1964. The soil samples were collected and analyzed in 1974.
c None detected (minimum detection limit: 10 ng/kg).
Table 19. Surface source areas and emission rates of 2,3,7,8-tetraCDDa
Source Area (m2) Emission rate (g/year)
Blow-out area, volatilization 753 120-1200
Blow-out area, entrainment 753 28-37
Rocky Branch Creek, dissolved 0.89-2.1
Reasor-Hill dump 1129 0.1-1.0
Rocky Branch Creek, sediment 0.094-0.22
Cooling water pond 15050 0.015-0.016
Total 150-1240
a From: Thibodeaux (1983).
Freeman et al. (1986) have developed a model to describe the
vaporization and diffusion through a column of soil of low volatility
organic chemicals like PCDDs and PCDFs. This model has been used to
make predictions on the transport of 2,3,7,8-tetraCDD at a site in
Times Beach, Missouri, USA. The model predicted that the 1983 levels
in this soil would be only 10% of the original loading. The model also
predicted that 57% of the initial amount of 2,3,7,8-tetraCDD was
vaporized through the soil column to the surface in the first year
after the spraying and that most transport of the vapour occurred
during the summer months. The same results were also obtained in
studies reported by Facchetti et al. (1986) and Palausky et al.
(1986).
4.2 Environmental Transformation
4.2.1 Abiotic transformation
Like other PCDDs and PCDFs, 2,3,7,8-tetraCDD is chemically quite
stable, and is not likely to be degraded at a significant rate by
hydrolytic reactions under environmental conditions. Under these
conditions, TCDD seems also to be rather stable to photochemical
degradation (Crosby et al., 1971). The half-life of TCDD of about
10-12 years, as found by Young et al. (1983) for soil, is in agreement
with this observation.
However, three reports on rapid photochemical degradation of
2,3,7,8-tetraCDD under experimental conditions make the situation more
complicated. In a methanol solution, TCDD is fairly easily degraded by
photolysis in the laboratory (Crosby et al., 1971). Other studies
using 2,4,5-T ester formulations with known amounts of TCDD and
exposed to natural sunlight on leaves, soil, or glass plates showed
that most of the TCDD was lost during a single day (Crosby & Wong,
1977). In these two studies, a "hydrogen donor", such as methanol or
2,4,5-T ester, enhanced the photochemical dechlorination (Akermark,
1978); they do not therefore truly reflect environmental conditions,
where the 2,4,5-T ester would be rapidly hydrolyzed on the surface of
the leaves. At Seveso the TCDD was released together with salts of
2,4,5-trichlorophenol, ethylene glycol, and inorganic constituents
(Rappe, 1978b). Like water, none of these is a potent hydrogen donor.
According to Bertoni et al. (1978), the addition of a solution of
ethyl oleate in xylene enhances the breakdown of TCDD in soil by UV
light. Similarly, a cationic surfactant, 1-hexadecylpyridinium
chloride, was also reported to enhance photodecomposition (Botre et
al., 1978).
Another experiment, which might be a good model for the
degradation of TCDD bound to dust particles in the air, has shown that
TCDD adsorbed on silica gel undergoes rapid photo-chemical degradation
(Gebefugi et al., 1977).
In order to explain the longer half-life of 2,3,7,8-tetraCDD in
a model laboratory ecosystem than in an outdoor pond, Matsumura et al.
(1983) speculated that photolysis was the most likely cause. In the
outdoor environment, algae-mediated photosensitization of
2,3,7,8-tetra-CDD may have caused some photodecomposition of this
compound.
An increase in chlorine substitution is expected to decrease the
rate of photodegradation. For example, Crosby et al. (1971) showed
that although complete decompostion of 2,3,7,8-tetraCDD in methanol
occurred in 24 h under UV irradiation, > 80% octaCDD in methanol
remained unreacted during the same period under similar irradiation
conditions.
Although the degree of photolysis may be related to the extent of
chlorination, different chlorine substitution patterns also play a
critical part. In higher chlorinated PCDDs, there appears to be
preferential loss of chlorine from the 2,3,7, and 8 positions (Buser
& Rappe, 1978). Thus, PCDDs with chlorine substitutions in positions
2,3,7, and 8 are likely to be photochemically degraded faster than
compounds not having these positions substituted. For example, the
photolysis half-life of 1,2,3,7,8-pentaCDD has been estimated to be
7.8 h in n-hexadecane solution under sunlamp irradiation (Nestrick
et al., 1980). Similarly, the photolytic half-lives of
1,2,3,7,8-pentaCDD, 1,2,3,6,7,9-, and 1,2,4,6,7,9-hexaCDD in hexane
solutions under sunlight irradiation have been determined to be 5.4,
17, and 47 hours, respectively (Dobles & Grant, 1979). Nestrick et al.
(1980) reported a half-life value of 6.8 h for 1,2,3,6,7,8-hexaCDD in
n-hexadecane under sunlamp irradiation. The primary intermediates of
the photo-degradation of higher chlorinated PCDDs are probably lower
chlorinated dioxins (Buser & Rappe, 1978), but the pathways of
degradation are not known with certainty (National Research Council of
Canada, 1981).
From these discussions of the photolysis of PCDDs in the presence
of organic hydrogen-donating substrates, it is difficult to predict
the photolytic fate of these compounds in natural aquatic media, where
hydrogen donors may or may not be available. The situation is
complicated further by the fact that a predominant amount of PCDDs in
surface water may be adsorbed or suspended on particles and sediments,
rather than in solution. Moreover, since the penetration of UV light
into natural water may be very limited, photolytic degradation of
PCDDs in water is not likely to be of environmental importance.
Hutzinger et al. (1973) have studied the photochemical
degradation of 2,8-diCDF and octaCDF. They found that a reductive
dechlorination takes place, especially in methanolic solution. The
reaction was much slower when a thin film was exposed to sunlight.
Thermally, 2,3,7,8-tetraCDD is quite stable, rapid decomposition
occurring only at temperatures above 750 °C (Stehl et al., 1973).
4.2.2 Biotransformation and biodegradation
The 2,3,7,8-tetraCDD isomer is very resistant to biodegradation.
Only 5 of about 100 microbial strains with the ability to degrade
persistent pesticides were able to degrade 2,3,7,8-tetraCDD (Matsumura
& Benezet, 1973). Ward & Matsumura (1978) studied the biodegradation
of 14C-labelled 2,3,7,8-tetraCDD in lake waters and sediments from
Wisconsin, USA, and observed a half-life of 2,3,7,8-tetraCDD in lake
waters containing sediment of 550-590 days. In lake water alone, about
70% of the 2,3,7,8-tetraCDD remained after 589 days. Using an outdoor
pond as a model aquatic ecosystem, and dosing it with 14C-labelled
2,3,7,8-tetraCDD, Matsumura et al. (1983) estimated the half-life of
2,3,7,8-tetraCDD to be approximately 1 year. Although biodegradation
may have been responsible for part of the degradation, it is almost
impossible to estimate the biodegradation half-life of
2,3,7,8-tetraCDD in aquatic systems from this experiment.
Philippi et al. (1982) detected a polar metabolite of
2,3,7,8-tetraCDD in several microbiological cultures after long-term
incubation. They reported chromatographic and MS data that supported
the conclusion that the metabolite was 1-hydroxy-2,3,7,8-tetraCDD,
although a synthetic standard compound was not available.
Tulp & Hutzinger (1978) reported that in rats, dibenzo-p-dioxin,
1-monoCDD, 2-monoCDD, 2,3-diCDD, 2,7-diCDD, 1,2,4-triCDD, and
1,2,3,4-tetraCDD are metabolized to mono- and di-hydroxy derivatives.
In the case of dibenzo-p-dioxin and both of the two monochloro
isomers, sulfur-containing metabolites were also excreted. Primary
hydroxylation exclusively took place at the 2, 3, 7, or 8 positions in
the molecule. In these studies, no metabolites resulting from fission
of the C-O bonds (ortho, ortho'-dihydroxychlorodiphenyl ethers,
chloro-catechols), or hydroxylated derivatives thereof, were detected.
No metabolites were found from octaCDD.
4.3 Bioaccumulation
The bioaccumulation of 2,3,7,8-tetraCDD has been investigated in
several studies, using several aquatic species and different model
ecosystems. In the experiments in which 14C-TCDD was introduced into
the model ecosystem in the form of residues on sand, particularly high
values were found in the mosquito (Aedes egypti) larvae, the level
exceeding that found in water by more than 9000 times. Under similar
conditions, the level in brine shrimp (Artemia salina) was 1570 times
higher than that found in water (Matsumura & Benezet, 1973). In the
second study (Isensee & Jones, 1975; Isensee, 1978), 14C-TCDD was
absorbed, at a broad range of levels, into soil and placed at the
bottom of an aquarium. Five species of organisms were added (though
not simultaneously) 1-30 days after flooding, and exposed for 3-32
days. The correlation between the TCDD level in the water and in the
organisms of each species was highly significant (correlation
coefficient of 0.94 or higher).
Bioaccumulation factors for 2,3,7,8-tetraCDD are given in Table
20 (US EPA, 1985).
4.4 Levels in Biota
4.4.1 Vegetation
When 14C-labelled 2,3,7,8-tetraCDD was added to soil, both oats
and soya beans accumulated small quantities of TCDD, at all stages of
growth. TCDD was also detected in control plants housed with the
experimental plants after treatment (Isensee & Jones, 1971). A maximum
of 0.15% of the TCDD present in the soil was translocated to the
aerial portion of the oats and the soya beans, but neither the grain
nor the beans harvested at maturity showed any detectable level of
14C-labelled TCDD. When TCDD was applied to the central leaflet of
3-week-old soya bean plants and 12-day-old oat plants, very little
TCDD was lost from the soya bean leaves in 21 days, but there was a
gradual loss (38% in 21 days) from the oat leaves.
Analyses of vegetation from Seveso, Italy, after the industrial
accident, gave values of up to 50 mg TCDD/kg (Firestone, 1978). In the
following years, when there was no direct contact of the newly grown
vegetation with the aerosol cloud, the levels of dioxin in plants
decreased by several orders of magnitude (Wipf & Schmid, 1983). In
1977 (one year after the accident in Seveso), no traces of TCDD were
found in the flesh of apples, pears, and peaches, or in corn cobs or
kernels, grown near the factory (the detection limit for the analyses
was 1 ng/kg). At the same time about 100 ng TCDD/kg was detected in
the fruit peels. This strongly suggests that the contamination was due
to dust and not from plant uptake. The TCDD level in the soil was
found to be in the order of 10 ng/g, which corresponds to about 1000
µg/m2 (Wipf et al., 1982).
Facchetti et al. (1986) studied plants grown in soil spiked with
2,3,7,8-tetraCDD in the range 1-752 ng TCDD/kg. At the end of
cultivation, root samples were collected, carefully washed, and
analyzed. The levels of TCDD in the roots were found to be higher than
the levels found in the soil in which the plants were grown. On the
parts above ground, Facchetti et al. (1986) could not find any
significant increase in the levels of TCDD. However, the TCDD
concentration was found to vary with the location, being higher if the
plants were grown in the vicinity of other pots containing
contaminated soil. The conclusion was drawn that evaporation is the
predominant process for the contamination of the aerial parts of
plants. However, studies by Sacchi et al. (1986) indicated that maize
and bean plants grown in soil contaminated by 3H-2,3,7,8-tetraCDD
accumulated radioactivity in the aerial parts progressively with time
and with soil contamination (Sacci et al., 1986). It was suggested
that the distribution of the TCDD into the leaves occurred via the
transpiration stream.
Very few analyses of sprayed vegetation have been reported. A
rough estimate of 20-1000 ng/kg for 2,3,7,8-tetraCDD contamination can
be made on the basis of the level of 2,4,5-T found in newly sprayed
vegetation and the level of 2,3,7,8-tetraCDD in the spray formulation
used. Higher values could be obtained for Agent Orange. Sundström et
al. (1979) reported data in agreement with this estimate. However, the
analytical technique used in their study was not isomer specific.
Vegetation was sprayed with 2,4,5-T ester contaminated by only 0.06 mg
2,3,7,8-tetraCDD/g. A sample of leaves collected 42-45 days after the
spraying was found to have 170 ng TCDD/kg, somewhat lower than the
expected value 600 ng TCDD/kg, indicating a slow photochemical
breakdown.
Table 20. Measured bioaccumulation factor for 2,3,7,8-TCDD in freshwater aquatic organismsa
Species Tissue Duration Bioconcentration Reference
(days) factor
Alga 33 3094b Isensee (1978)
(Oedogonium cardiacum)
Alga 32 2075c Isensee (1978)
(Oedogonium cardiacum) Yockim et al. (1978)
Snail whole body 33 5471b Isensee (1978)
(Physa sp.)
Snail whole body 32 3095c Isensee (1978)
(Physa sp.) 3731 Yockim et al. (1978)
Cladoceran whole body 32 3895b Isensee (1978)
(Daphnia magna)
Cladoceran whole body 30 7070c Isensee (1978)
(Daphnia magna) 7125 Yockim et al. (1978)
Catfish whole body 28 4875 Yockim et al. (1978)
(Italurus punctatus)
Mosquitofish whole body 14 4850c Isensee (1978)
(Gambusia affinis) 4875 Yockim et al. (1978)
a From: US EPA (1985).
b Arithmetic mean of several values reported.
c Tissue concentrations at equilibrium.
Table 21. Levels of TCDDs in fish and shellfisha
Sample Tissue Concentration of
number typeb 2,3,7,8-TCDD (ng/kg)c
1 Fish (edible flesh) 480
2 Catfish 40
3 Buffalo fish ND(13)
4 Fish (predator) 230
5 Fish (bottom feeder) 77
6 Catfish 50
7 Buffalo fish ND(7)
8 Catfish ND(7)
a From: Mitchum et al. (1980).
b All samples were obtained from the Arkansas River, USA, or
a tributary, the Bayou Meto.
c These are averages of samples that had detectable levels of
TCDD.
ND = none detected; the number in parenthesis is the measured
detection limited for that sample.
4.4.2 Aquatic organisms
Fish and shellfish taken from areas in South Viet Nam that were
heavily exposed to Agent Orange during military defoliation operations
in the 1960s have been reported to contain 18-810 ng TCDD/kg (Baughman
& Meselson, 1973). The analytical technique of direct-inlet high
resolution MS used in this study is not considered isomer specific and
did not include any GC separation at all.
In two streams associated with the US Air Force test area in
north-west Florida (section 4.1.3), which had been heavily sprayed
with Agent Orange between 1962 and 1964, the silt contained, 10 years
later, 10 and 35 ng TCDD/kg where eroded soil entered the water.
Concentrations of 12 ng TCDD/kg were found in two species of fish from
this stream, the sailfin shiner (Notropis hypselopterus) and the
mosquito fish (Gambusia affinis). The spotted sunfish (Lepomis
punctatus) contained 4 ng TCDD/kg in skin and muscle, 18 ng/kg in the
gonads, and 85 ng/kg in the gut (Young et al., 1976).
Table 22. Analytical results for 2,3,7,8-tetraCDD residues in fish from Saginaw Bay Region, Michigan, USAa
Species Number of Number of TCDD
samplesb positive detected (ng/kg)c
samples low high mean
Channel
catfish 8 8 28 695 157 (13)
Carp 14 10 20 153 55 (7)
Yellow
perch 6 3 10 20 13 (5)
Smallmouth
bass 2 2 7 8 8 (6)
Sucker 4 3 4 21 10 (4)
Lake trout 2 0 0 0 0 (5)
a From: Harless et al. 1982).
b Mean % recovery for 2.5-10 ng 37Cl4-TCDD added to 5 or 10
g of tissue prior to sample preparation was between 78 and
100%.
c Corrected for losses in efficiency of sample preparation for
particular species. The numbers in parenthesis indicate the
limit of detection for TCDD.
The levels of TCDD in fish from the Atlantic or from ponds in the
USA in areas sprayed with 2,4,5-T were below the detection levels (1-2
ng/kg) (Baughman, 1974; Shadoff et al., 1977).
Mitchum et al. (1980) reported levels of 400 ng
2,3,7,8-tetraCDD/kg in fish samples collected in Bayou Meto/Arkansas
River, USA, a waterway associated with industrial plants for the
production of 2,4,5-T (Thibodeux, 1983) (see Table 21).
Levels ranging from 4-695 ng TCDD/kg were found in the edible
portion of channel catfish, carp, yellow perch, small-mouth bass, and
suckers from Saginaw Bay, Michigan, USA, near facilities used for the
production of 2,4,5-T herbicides. The highest concentrations were
detected in bottom-feeding catfish and carp, while the lowest
concentrations were detected in bass, perch, and suckers (see Table
22) (Harless et al., 1982).
Rappe et al. (1981) identified a series of tetra- to octaCDFs in
fat samples of a snapping turtle from the Hudson River and of gray
seal from the Baltic Sea. The total levels of PCDFs in these samples
were 3 ng/g and 40 ng/kg, respectively. In both samples the major
PCDFs consisted of the most toxic isomers (2,3,7,8-tetra-;
2,3,4,7,8-penta-; and 1,2,3,4,7,8- and 1,2,3,6,7,8-hexaCDFs).
Norstrom et al. (1982) have analyzed pooled samples of herring
gull eggs collected in 1982 from various parts of the Great Lakes, N.
America. In all samples, 2,3,7,8-tetraCDD was found in levels ranging
from 9 to 90 ng/kg. The identity of the 2,3,7,8-isomer was confirmed
by retention times on three capillary columns. In another study
Stalling et al. (1983) were not able to detect measurable levels of
tetraCDDs and other PCDDs in fish samples from Lake Superior,
N.America (the detection level was 2-5 pg/g). The difference could be
explained by the migration of the herring gulls during the winter. On
the other hand, a series of PCDFs could be identified in the Lake
Superior fish samples, indicating more widespread background levels
for the PCDFs than for the PCDDs. Stalling et al. (1983) found the
total levels of PCDFs in fish samples from Lakes Michigan, Huron, and
Ontario, N. America, to be 12-290 ng/kg. The toxic 2,3,7,8-substituted
PCDDs and PCDFs were present in all samples, the highest levels being
found in samples from Lake Huron, Lake Ontario, and the Tittabawasee
River, which flows into Saginaw Bay. The residue pattern found in the
fish and locally high levels suggest a strong influence by local point
source discharges (Stalling et al., 1983). The data of O'Keefe et al.
(1983) are also in agreement with this theory.
Norstrom et al. (1986) have studied the long-term trends of
2,3,7,8-substituted PCDDs and PCDFs in herring gull eggs in the Great
Lakes. The levels of 2,3,7,8-tetraCDD were found to decline
exponentially in Lake Ontario, with a half-life of 3-4 years, from a
high of 2000-5000 ng/kg in the early 1970s to a level of 80-100 ng/kg
in 1984/1985. The levels of TCDD in Lake Michigan were 249 ng/kg in
1971, 70 ng/kg in 1972, and 10-20 ng/kg in 1984/1985. These levels
have not changed significantly since 1979. This suggests that an
equilibrium between input and removal mechanisms has been established
in this water system for most PCDDs and PCDFs. The same trend is
reported for various fish species in Lake Ontario (Ontario, 1986).
Ryan et al. (1983a) analyzed a series of commercial and sport
fish from the Great Lakes and from the Pacific coast of Canada for
2,3,7,8-tetraCDD (Table 23). The highest levels were found in Lake
Huron and Lake Ontario. In a preliminary study they also reported
finding levels of 2,3,7,8-tetraCDFs and other unidentified tetraCDFs
of 3-200 ng/kg of fish.
The Baltic Sea is an area of interest because this region is
without any known point sources of dioxins. Rappe et al. (1987)
reported on the analyses of two samples of homing salmon and two
samples of pooled herring; one herring sample from the Baltic Sea
(Karlskrona) and the other from the northern part of the Gulf of
Bothnia (Lulea) (Table 24). As expected, the levels in the salmon
muscle were much higher than the levels found in the herrings, but,
unexpectedly, the levels in the herring sample from the Gulf of
Bothnia (Lulea) were somewhat higher than levels found in the sample
from the Baltic Sea (Karlskrona).
An interesting observation is that in the majority of the aquatic
samples only the 2,3,7,8-substituted PCDD and PCDF congeners were
found. However, crustaceans seemed to be an exception from this
general trend. Norström et al. (1988) reported that crab
hepatopancreas from the Canadian Pacific Coast contain other
congeners, e.g. 1,2,4,7,8-pentaCDD and
1,2,3,6,7,9-/1,2,3,6,8,9-hexaCDD. Rappe et al. (1987) collected and
analyzed crab hepatopancreas from three different locations along the
west coast of Sweden. The crab samples from the locations Grebbestad
and Idefjord should represent background levels, while Väröfjord has
a potential point source of dioxins from a pulp mill using chlorine
for bleaching. The results are given in Table 25.
Low background levels of series PCDDs and PCDFs were found in all
samples. In addition, the sample from the Väröfjord also contained
much higher levels of some congeners, especially 2,3,7,8-tetraCDF and
2,3,7,8-tetraCDD. This is another indication that pulp bleaching could
be a potential source of 2,3,7,8-tetraCDD and 2,3,7,8-tetraCDF (see
section 3.5.10).
4.4.3 Terrestrial animals
In a heavily sprayed test area in north-west Florida (Young et
al., 1976), a total of 106 adult and 67 fetuses of beach mice
(Peromysous polionotus) were collected in 1973 and 1974 and
examined (method not specified). Livers from the beach mice contained
from 540-1300 ng TCDD/kg and the pelts 130-140 ng/kg. The visceral
mass of race runners (Cnemidophorus sexlineatus) which were caught
in that area contained 360 ng TCDD/kg and the trunk of the reptiles
contained 370 ng/kg.
At the time of the accident in Seveso, Italy, more than 81 000
animals were inhabiting the contaminated zones. Most were rabbits (25
000), poultry, and other small animals (55 500), with 349 cattle, 233
pigs, 49 horses, 21 sheep, and 49 goats also in the zones. Many of
these animals died and others were killed. A large number of these
animals were analyzed for 2,3,7,8-tetraCDD by a method with a
detection level of 250 ng/kg (Pocchiari et al., 1983). The results are
summarized in Tables 26 and 27.
Table 23. Levels of 2,3,7,8-tetraCDD and PCB in Great Lakes Canadian sport
fish (1980) and smelt (1979)a
Species Origin TCDD PCB
(ng/kg) (µg/g)
Lake troutb Lake Ontario 58 7.28
Lake Huron 37 5.03
Rainbow troutb Lake Ontario 33 1.77
Coho salmon Lake Ontario 28c 7.39
Pacific Coast NDd (4) 0.03
Smelt Lake Ontario 11
16
11
Lake Erie NDd (2)
a From: Ryan et al. (1983a).
b Whole fish.
c Also contained 36 ng hexaCDD/kg (three isomers) and 93 ng
octaCDD/kg.
d ND = not detected at bracketed detection limit.
Table 24. Levels of PCDDs and PCDFs in fish samples from the Baltic Sea
(pg/g) a,b
Salmon Salmon Herring Herring
Ume River Ume River Karlskrona Lulea
1985 1985 1983 1983
2,3,7,8-TetraCDF 29 12 5.5 3.0
2,3,7,8-TetraCDD 1.9 1.3 < 0.3 < 0.6
1,2,3,7,8-/1,2,3,4,8-PentaCDF 6.9 3.3 1.4 0.9
2,3,4,7,8-PentaCDF 49.0 23.0 6.8 8.8
1,2,3,7,8-PentaCDD 8.8 4.3 1.1 4.7
1,2,3,4,7,8-/1,2,3,4,7,9-HexaCDF 1.1 0.7 0.4 0.3
1,2,3,6,7,8-HexaCDF 1.3 0.8 0.4 0.3
1,2,3,7,8,9-HexaCDF ND ND 0.4 0.2
2,3,4,6,7,8-HexaCDF 1.1 0.6 0.4 0.2
1,2,3,4,7,8-HexaCDD ND 0.4 0.2 ND
1,2,3,6,7,8-HexaCDD 4.6 2.3 ND 8.1
1,2,3,7,8,9-HexaCDD ND ND ND ND
Total HeptaCDFs ND 2.7 0.8 ND
Total HeptaCDDs ND ND ND ND
OctaCDF ND 1.0 ND ND
OctaCDD ND ND ND ND
Table 24.(cont'd) Levels of 2,3,7,8-tetraCDD and PCB in Great
Lakes Canadian sport fish (1980) and smelt (1979)a
a From: Rappe et al. (1987).
b ND indicates a level < 0.1 pg/g.
Harless et al. (1983) reported a study in which 2,4,5-T
containing less than 0.1 mg of 2,3,7,8-tetraCDD/kg was applied at a
rate of 3.4 kg/ha to approximately 3 ha of an enclosed plot (4.5 ha).
Twelve deer were placed in the enclosure prior to the application of
2,4,5-T. One deer died two days later of unknown causes. The remaining
deer were sacrificed prior to, and at specific intervals during, the
course of the 30-day study. The analytical results are summarized in
Table 28.
In another study (Hryhorczuk et al., 1981), samples from a horse
grazing close to a wire reclamation incinerator were analyzed and
found to contain unspecified tetraCDFs (165 ng/kg in the fat, 57 ng/kg
in the liver) and unspecified tetraCDDs (45 ng/kg in the fat and less
than 6 ng/kg in the liver) (compare section 3.5.7).
In order to identify PCDD and PCDF levels in the general
terrestrial background, Nygren et al. (1986) analyzed bovine samples
- fat, liver, and milk - and identified the same 2,3,7,8-substituted
PCDDs and PCDFs as were found in the aquatic samples. However, the
levels were lower and normally close to the detection limit.
4.4.4 Human data
Occupational exposure to 2,3,7,8-tetraCDD can occur during the
production of 2,4,5-trichlorophenol and the subsequent production and
use of 2,4,5-T acid and esters. The first commercial production of
2,4,5-T in the United States was in 1944, and the use of 2,4,5-T
herbicides increased in the 1940s and 1950s. However, the problem of
dioxin contamination in 2,4,5-T was not recognized until 1957 (Kimmig
& Schulz, 1957a, b).
During the normal production of 2,4,5-T, the heaviest exposure to
TCDD is during purification steps. The residues are far more
contaminated than the purified products. Only limited information is
available on the levels of TCDD contamination of products prepared
prior to the 1970s, and absolutely no information is available on the
dioxin levels in the corresponding residues. Consequently it is a
difficult task to estimate the levels of occupational and general
population exposures during the period prior to 1970.
Table 25. Levels of PCDDs and PCDFs in samples of crab hepatopancreas
from the west coast of Swedena
Crab Hepatopancreas
Idefjorden Grebbestad Väröfjord
(pg/g) (pg/g) (pg/g)
2,3,7,8-tetraCDF 31 47 590
Total tetraCDFs 90 114 800
2,3,7,8-tetraCDD 17 17 170
Total tetraCDDs 17 17 170
1,2,3,7,8-pentaCDFb 6 7.6 45
2,3,4,7,8-pentaCDF 44 50 130
Total pentaCDFs 130 150 490
1,2,3,7,8-pentaCDD 13 11 28
Total pentaCDDs 86 76 270
1,2,3,4,7,8-hexaCDFc 12 16 50
1,2,3,6,7,8-hexaCDF 3 5 10
1,2,3,7,8,9-hexaCDF 3 3 11
2,3,4,6,7,8-hexaCDF 16 18 63
Total hexaCDFs 70 88 280
1,2,3,4,7,8-hexaCDD 8 5 14
1,2,3,6,7,8-hexaCDD 26 18 71
1,2,3,7,8,9-hexaCDD 3 4 7
Total hexaCDDs 154 170 465
Total heptaCDFs 23 28 90
Total heptaCDDs 32 30 85
octaCDF < 1 < 1 < 2
octaCDD < 1 < 1 < 2
a From: Rappe et al. (1987).
b Not separated from 1,2,3,4,8-pentaCDF.
c Not separated from 1,2,3,4,7,9-hexaCDF.
Table 26. TCDD content of the livers of farm animals from Seveso
contaminated zones and surrounding areas (1976-1979)a
Animal Number TCDD-containing TCDD maximum
of samples samples level (ng/g)
Rabbitsb 698 433 633
Poultry 83 35 24
Cattle 43 21 94
Horses 12 2 88
Pigs 13 0 -
Goats 25 17 1
Cats 1 0 -
a From: Pocchiari et al. (1983).
b Figures include rabbits kept in the special test plots on
contaminated ground for experimental purposes.
Table 27. CDD analyses of wildlife from Seveso contaminated zones and
surrounding areas (1976-1979)a
Animal Tested organs Number of Maximum level
and number of samples TCD-containing of TCDD
samples (ng/g)
Rabbits 6 (liver) 4 13
Field mice 14 (whole body) 14 49
Rats 1 (pool-4 livers) 28
Earthworms 2 (pool) 12
Frogs 1 (liver) 0.2
Snakes 1 (liver) 3
a From: Pocchiari et al. (1983).
Table 28. Analytical results for 2,3,7,8-tetraCDD residuesa
Sections of 11 No. of deer No. of Concentration range Limit of
deer in study samples positive of TCDD detected detection
analyzed samples (ng/kg)b range
(ng/kg)b
Muscle 11 3 12 - 27 0.5 - 5
Adipose tissue 10 8 3 - 12 1 - 3
Table 28. (cont'd) Analytical results for 2,3,7,8-tetraCDD residuesa
Sections of 11 No. of deer No. of Concentration range Limit of
deer in study samples positive of TCDD detected detection
analyzed samples (ng/kg)b range
(ng/kg)b
Liver 11 4 2 - 5 0.4 - 4
Bone marrow 5 0 ND 1 - 3
a From: Harless et al. (1983).
b Results corrected for efficiency of sample preparation.
ND = not detected.
4.4.4.1 Adipose tissue
Gross et al. (1984) reported a study in which 30 coded samples of
adipose tissue from Viet Nam veterans were analyzed for TCDD. The TCDD
levels found for two of the three heavily exposed men were 99 pg/g and
63 pg/g, which is higher than for the other Viet Nam veterans or for
the controls (all well below 15 pg/g). Only one single isomer of
tetraCDDs was found, and it was assumed that this was the
2,3,7,8-isomer. The data in this study has also been discussed by
Young et al. (1983). These authors concluded that the levels do not
correlate well with known exposure data or with health status.
Rappe et al. (1984) reported the presence of 2,3,7,8-substituted
PCDDs and PCDFs in samples of human adipose tissue from Northern
Sweden. A series of reports presented during the period 1984-1986
confirms these observations and it has been clearly shown that there
is a background of 2,3,7,8-substituted PCDDs and PCDFs in the general
population in the industrialized part of the world. Most of these
reports are lacking data on how the sampled people were selected and
possible exposure to PCDDs and PCDFs. Consequently, these studies
might not be representative. A series of earlier studies failed to
detect these background levels due to insufficiently low detection
levels. The Swedish study included 31 people, of which 18 were exposed
to phenoxy esters and 13 were nonexposed. The group included 17 cancer
patients and 14 non-cancer patients. The different groups were matched
against each other. No difference in the levels, isomer patterns, or
ranges could be found between these subgroups (Nygren et al., 1986).
The mean values for these 31 people are given in Table 29.
Schecter et al. (1986a) reported the mean levels of PCDDs and
PCDFs in 46 samples of adipose tissue collected in Canada and in 8
samples from the USA (see Table 29). The Canadian samples were taken
from people who had died in 1976 from car accidents, drownings,
trauma, and suicide. The samples included all ages and both sexes and
came from all over the country. The USA samples, (1983-1984), were
taken from biopsies from New York State residents during the course of
normal medical procedures, and also from autopsies. Table 29 also
includes the PCDD and PCDF levels in adipose tissue samples from Viet
Nam (Schecter et al., 1986b) and from cancer patients in Japan (Ono et
al., 1986).
It is interesting to note the similarity between isomers present,
levels of isomers, isomeric patterns, and congener profiles in samples
collected from the general population in industrialized countries on
three continents. The profile of the PCDD isomers shows increasing
levels with an increasing number of chlorine atoms; the level of OCDD
is 230-900 pg/g. On the other hand, the profile of PCDFs shows a
maximum for 2,3,4,7,8-penta- or 1,2,3,6,7,8-hexaCDF. The difference in
levels found between samples from South and North Viet Nam may be
explained by spraying during the war in the 1960s and by the
difference in industrial activities between the two parts of the
country.
Four samples of adipose tissue taken from German workers exposed
to TCDD in the early 1950s have also been analyzed (Rappe et al.,
1987). In spite of the fact that these workers were exposed more than
30 years before collecting the samples, enhanced levels of
2,3,7,8-tetraCDD could be identified, but the levels of the other
PCDDs and PCDFs seem to be in the normal range (Table 29, last
column).
Patterson et al. (1986) studied the levels of 2,3,7,8-tetraCDD in
the adipose tissue of 39 exposed people and 57 controls in Missouri,
USA. The exposed group had subgroups of recreational, residential, and
occupational exposure. All persons in both the exposed and control
groups had detectable levels of 2,3,7,8-tetraCDD in their adipose
tissue. Nineteen of the 39 exposed people had measurements higher than
the highest level in the control group and six of the exposed people
had levels greater than 100 ng/kg, which was five times higher than
the highest control (Table 30).
Ryan et al. (1986) analyzed autopsy tissue samples that were
collected from three subjects who died in New York State, USA, from
natural causes. The tissue types were: fat (both abdominal and
subcutaneous), adrenal, bone marrow, liver, muscle, spleen, kidney,
and lungs. As far as could be ascertained, no subjects had known
abnormal exposure to PCDDs or PCDFs, yet these chemicals were found in
all tissues analyzed. The highest concentrations of all PCDDs and
PCDFs were found in adipose tissue. In individual tissues of the three
subjects, the levels of individual congeners detected were within a
narrow range, with much higher levels of the higher chlorinated PCDDs
and PCDFs (e.g. hepta- and octa-CDD). In adipose tissue, the level of
2,3,7,8-tetraCDD was 3.7-8.4 ng/kg and that of 2,3,4,7,8-pentaCDF
5.2-13 ng/kg, while octaCDD ranged between 430 and 700 ng/kg. No major
differences were seen between abdominal and subcutaneous fat samples
or between these two types and perirenal fat when the lower lipid
content of the latter was considered. Smaller concentrations of PCDDs
and PCDFs were measured (on a wet weight basis) in decreasing order:
adrenal, bone marrow, liver, muscle, spleen, kidney, and lungs.
The high levels found in exposed Viet Nam veterans and German
workers indicate a very slow excretion rate or metabolism of TCDD in
humans. This indicates a dramatic difference between man and rodents;
in the latter the half-life of TCDD is reported to be in the range of
a few weeks.
4.4.4.2 Blood plasma
Analysis of blood plasma has been used to evaluate occupational
exposure to PCDDs and PCDFs, which can occur during the production or
use of 2,4,6-tri-, 2,3,4,6-tetra-, and pentachlorophenol. Rappe et al.
(1983) investigated such exposure through the analysis of blood plasma
of exposed workers and unexposed controls. Good correlations were
found between the plasma levels and:
(a) the nature of exposure - dermal contact with liquids resulted in
higher levels than inhalation of contaminated dust;
(b) the duration of exposure - higher levels for longer exposure
times.
The isomers present in the formulations used could also be found in
the blood plasma.
Kochman et al. (1986) studied a group of people exposed to PCBs
and PCDFs after a transformer fire in Rheims, France in 1985. Low
levels (1-25 pg/g) of 2,3,7,8-substituted penta-, hexa-, hepta-, and
octaCDFs were found in the blood plasma of these people, and there was
a slight variation in the T-lymphocytes cells.
Kahn et al. (1986) measured the levels of PCDDs and PCDFs in the
blood plasma from 10 heavily exposed Viet Nam veterans and their 17
controls. The levels of TCDD was much higher in the exposed men than
in their controls.
Table 29. Levels of PCDDs and PCDFs in human adipose tissue (ng/kg wet weight)
Sweden USA/NYf Canadaf Japanf N Viet Namff FRG
Isomer n=31a n=8b n=46b n=13c n=9d n=15d n=4e
2,3,7,8-tetraCDD 3 7.2 6.4 (25) 9 (12) < 2 28 (12) 150
1,2,3,7,8-pentaCDD 10 11.1 10 (46) 15 (13) < 2 15 (14) 19.2
1,2,3,6,7,8-hexaCDD 15 96 81 (46) 70 (12) 11 (6) 100 (15) 77
1,2,3,7,8,9-hexaCDD 4 NA NA 12 (10) NA NA 9.4
1,2,3,4,6,7,8-heptaCDD 97 164 135 (46) 77 (12) 28 (6) 178 (15) 56
octaCDD 414 707 830 (46) 230 (12) 104 (8) 1256 (15) 267
2,3,7,8-tetraCDF 3.9 NA NA 9 (13) NA NA 0.9
2,3,4,7,8-pentaCDF 54 14.3 15 (46) 25 (13) 13 (7) 21 (15) 44
1,2,3,4,7,8-hexaCDF 6 NA NA 15 (11) NA NA 10.0
1,2,3,6,7,8-hexaCDF 5 31.3 16 (34) 14 (11) 13 (7) 58 (15) 6.7
2,3,4,6,7,8-hexaCDF 2 NA NA 8 (3) NA NA 3.8
1,2,3,4,6,7,8-heptaCDF 11 16.5 30 (44) NA 7 (3) 29 (15) 19.5
octaCDF 4 NA NA NA NA NA 1
n = number of tissue samples.
NA = not analyzed.
a Nygren et al. (1986).
b Schecter et al. (1986a).
c Ono et al. (1986).
d Schecter et al. (1986b).
e Refers to occupationally exposed workers. Rappe et al. (1987).
f Mean values of positives. Number of positives within brackets.
Levels below the detection level (< 1.0 ng/kg) not included.
Table 30. Comparison of Levels of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (ng/kg) in Adipose Tissue of Exposed and Control
Groupsa
Exposed
Variable Controls Total Recreational Residential Occupational
Number of subjects 57 39 8 16 15
Arithmetic mean 7.4 79.7 90.8 21.1 136.2
Median 6.4 17.0 23.5 14.5 24.7
Range 1.4-20.2 2.8-750 5.0-577 2.8-59.1 3.5-750
Geometric mean 6.4 21.8 24.8 15.3 29.8
Mean age, years (SD) 52.6 (15.7) 44.3 (13.7) 42.1 (14.7) 39.7 (14.9) 50.3 (9.8)
% Men 35.1 61.5 37.5 43.8 93.3
a From: Patterson et al. (1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Air
Owing to sampling and analytical problems, very few data are
available on the levels of 2,3,7,8-TCDD and other PCDDs and PCDFs in
normal urban air.
Rappe & Kjeller (1987) reported the levels of PCDDs and PCDFs in
total air samples collected in Hamburg, FRG, and samples of air
particulates collected in Sweden (Table 31). Sample 1 was collected on
the outskirts of Hamburg (representing an urban area), sample 2 in a
traffic tunnel, sample 3 downwind from a MSW incinerator, and sample
4 in the vicinity of a dumpsite and metal refinery. PCDDs and PCDFs
were found in all samples. Lower levels of PCDDs and PCDFs were found
in air particulates than in total air samples (Table 31). Sample 5 was
taken when "clean air" was blowing into a street in Gothenburg,
Sweden, and sample 6 was taken in the same street during an inversion
situation. Samples 7 and 8 were taken at a rural research station
outside Gothenburg when the air was blowing from the sea (sample 7) or
from Gothenburg (sample 8). The isomeric pattern found in these
samples were very similar (Rappe & Kjeller, 1987).
Airborne dust was monitored in 1977 in the Seveso area to
evaluate the possibility that 2,3,7,8-tetraCDD-contaminated particles
might have drifted outside the contaminated areas. A high-volume
sampling technique was used. When pooled particulate samples were
analyzed, levels of 0.17-0.50 pg TCDD/m3 were reported (Wipf et al.,
1982).
The atmospheric concentrations of 2,3,7,8-tetraCDD near two
hazardous waste sites have been monitored. In one study, US EPA (1982)
failed to detect (detection limit: 1-20 pg/m3) any 2,3,7,8-TCDD in the
atmosphere at the Love Canal (New York, USA) area. In another study of
a waste disposal site (near Jacksonville, Arkansas, USA), Thibodeaux
(1983) reported an average concentration of 1100 pg of 2,3,7,8-TCDD/g
in two air particulate samples collected near the disposal site.
Rappe et al. (1985c) analyzed indoor air samples for PCDFs
resulting from fires and explosions in PCB-filled electrical
equipment, and in an industrial situation (locomotive shop) (Table
32).
O'Keefe et al. (1985) analyzed air samples collected in an office
building in Binghamton, New York, USA, after a transformer accident in
the basement in February 1981. The samples were collected after a
primary clean-up and the values are given in Table 33.
Table 31. Levels of PCDDs and PCDFs in samples of total air and air
particulatesa
Total air samples Air particulates
1 2 3 4 5 6 7 8
pg/m3 pg/m3 pg/m3 pg/m3 fg/m3 fg/m3 fg/m3 fg/m3
2,3,7,8-tetraCDFb 0.04 0.72 0.38 0.18 30 240 5 62
Total tetraCDFs 0.36 6.2 4.9 3.3 320 2000 54 490
2,3,7,8-tetraCDD 0.02 0.06 0.02 0.08 3 9 < 1 5
Total tetraCDDs 0.10 0.22 0.21 1.5 150 350 9 130
1,2,3,7,8-pentaCDFc 0.04 0.36 0.42 1.0 39 190 7 58
2,3,4,7,8-pentaCDF 0.04 Inte 0.43 1.2 51 240 6 69
Total pentaCDFs 0.51 4.1 5.0 10 470 2500 85 610
1,2,3,7,8-pentaCDD < 0.02 0.28 0.22 0.6 17 66 5 35
Total pentaCDDs 0.07 1.3 2.4 5.0 200 840 31 280
1,2,3,4,7,8-hexaCDFd 0.03 0.13 0.27 1.1 23 100 8 38
1,2,3,6,7,8-hexaCDF 0.03 0.15 0.24 1.4 20 78 8 33
1,2,3,7,8,9-hexaCDF < 0.01 < 0.05 < 0.02 0.33 4 17 3 14
2,3,4,6,7,8-hexaCDF < 0.01 < 0.05 0.12 0.80 10 84 7 32
Total hexaCDFs 0.18 1.1 2.2 9.5 180 800 70 310
1,2,3,4,7,8-hexaCDD < 0.08 < 0.17 0.19 1.0 3 19 < 1 7
1,2,3,6,7,8-hexaCDD 0.23 0.66 0.71 2.2 11 46 4 14
1,2,3,7,8,9-hexaCDD < 0.08 < 0.17 0.36 5.2 6 92 5 32
Total hexaCDDs 0.74 2.7 5.3 24. 100 520 32 190
Total heptaCDFs 0.10 1.2 2.0 5. 200 1100 120 500
Total heptaCDDs 0.60 3.4 5.3 15. 380 2900 140 1000
OctaCDF < 0.11 < 1.0 0.78 7.0 150 480 100 440
OctaCDD 0.37 6.4 7.4 40.0 290 1900 64 540
a From: Rappe & Kjeller (1987).
b Not separated from 2,3,4,8-tetraCDF.
c Not separated from 1,2,3,4,8-pentaCDF.
d Not separated from 1,2,3,4,7,9-hexaCDF.
e Int = Interferences.
5.2 Water and Leachate
Shadoff et al. (1977) failed to identify 2,3,7,8-tetraCDD in
water from areas in the USA where 2,4,5-T herbicides had been used.
In the 2,4,5-T plant in Jacksonville, Arkansas, USA, Thibodeaux
(1983) could not detect any 2,3,7,8-tetraCDD in the creek water (no
limit of detection levels given).
Since August 1976, a number of tests have been periodically
conducted in Seveso on streams running through the affected area, as
far south as the River Lambro, with consistently negative results.
During the same period sediment samples were taken from Torrents,
Certesa, and Seveso. Positive results of the order of 1 pg/g were
obtained within the first few kilometers downstream from their
confluence, but further downstream results were negative. The
intensive rainfalls after the accident caused the Seveso to repeatedly
overflow its embankments at the point of entry into Milan, thus
depositing silt on adjacent areas. Tests conducted to determine TCDD
in these silts yielded negative findings for the first four floods
while the fifth flood yielded positive findings (pg/g). Since August
1976, the monthly determinations conducted on pipeline and ground
waters have consistently yielded negative results, even when the
analytical detection threshold was as low as 1 pg/litre (parts per
quadrillion) (Pocchiari, 1983).
During 1983 and 1984, the Dow Chemical Company conducted a study
to determine the 2,3,7,8-tetraCDD contamination at its plant in
Midland, Michigan, USA. It was estimated that 0.6 g of
2,3,7,8-tetraCDD was being emitted per year in 2.5x107 m3 of
wastewater effluent (Lamparski et al., 1986).
The Ontario Ministry of Environment has included PCDDs and PCDFs
in its Drinking Water Surveillance Program for the St Clair/Detroit
River area (Ontario, 1986). No 2,3,7,8-tetraCDD has been found in any
sample of raw or treated water. Unspecified congeners of PCDDs and
PCDFs have been found mainly in raw water, octaCDD being the most
frequent congener found. The highest value reported was 1.1 pg
octaCDD/litre raw water in Amherstburg. The octaCDD level in the
treated water was below the detection level of 0.01 pg/litre.
Götz (1986) reported on levels of PCDDs and PCDFs in the oily
leachate from a sanitary landfill in Georgswerder, Hamburg, FRG (table
34).
5.3 Soil and Sediment
The levels of 2,3,7,8-tetraCDD in point source after improper
disposal of industrial waste are discussed in section 3.4.2.
Table 32. Analyses of PCDFs in air samples (pg/m3)a
total
Sample tetra- 2,3,7,8- penta- hexa- hepta- octa-
CDF tetraCDF CDF CDF CDF CDF
Surahammar < 20 < 2 < 10 < 10 < 10 < 10
(during cleaning)
Surahammar < 10 < 2.5 < 10 < 10 < 10 < 10
(after cleaning)
Railway locomotive 500 50 50 30 20 20
(during cleaning
operations)
a From: Rappe et al. (1985c).
Table 33. Concentrations of PCDFs in air samples collected on various
floors of a Binghamton, New York (USA) office after primary cleanup
Analytical results (pg/m3)
Floor/sample typea 2,3,7,8- Total Penta- Hexa-
tetraCDF tetraCDFs CDFs CDFs
3 16 151 43
5 11 126 30 2.0
5 (NE) 20 195 60 8.7
7 11 121 36
9 volatiles 14 140 42
9 particulates 1.8 4.8 4.7
9 (SE) volatiles 13 146 31 3.7
9 (SE) particulates 0.8 3.9 3.2
11 23 76 16
11 (SE + NW) 16 133 19
14 11 92 21
14 (NE) 14 185 13
16 16 118 21
17 volatiles 12 79 24
17 particulates 0.8 3.9 NDb
17 volatiles 9 59 6.6
17 particulates 0.9 NDb 2.9
Table 33. cont'd
a Abbreviations in parentheses designate sampling location on the floor,
e.g., SE = south-east corner. Unless otherwise specified samples were
collected in the north-west corner and analyzed as combined particulates
and volatiles.
b ND = not detected.
Table 34. Levels (ng/g) of the 2,3,7,8-substituted PCDDs and PCDFs in
leachate from a sanitary landfilla
Isomer Concentration Isomer Concentration
2,3,7,8-tetraCDD 60 2,3,7,8-tetraCDF 9
1,2,3,7,8-pentaCDD 28 1,2,3,7,8-pentaCDFb 322
1,2,3,4,7,8-hexaCDD 476 2,3,4,7,8-pentaCDF 261
1,2,3,6,7,8-hexaCDD 1440 1,2,3,4,7,8-hexaCDF 748
1,2,3,7,8,9-hexaCDD 310 1,2,3,6,7,8-hexaCDF 336
1,2,3,7,8,9-hexaCDF 558
2,3,4,6,7,8-hexaCDF 114
a From: Götz (1986).
b Overlapping isomer: 1,2,3,4,8-pentaCDF.
Analytical results of the 1976-1977 survey of Zones B and R in
Seveso were discussed by Pocchiari (1983). TCDD levels in Zones B and
R were, in general, considerably lower than those in Zone A. In fact,
most TCDD levels were lower than 50 µg/m2 in Zone B and 5 µg/m2 in
Zone R. In 1980, a large part of Zone R was remonitored to evaluate
the persistence of TCDD in the soil. This zone had been ploughed and
worked since 1978. A comparison, as well as a statistical evaluation,
of the relevant data indicated a significant decrease (40%) in the
geometric mean level of TCDD in the soil of Zone R.
In 1980 and 1981, soil samples from ten sites in Zone R and five
sites outside Zone R were analyzed using a high resolution GS-MS
system to establish whether other isomers of 2,3,7,8-tetraCDD were
also present. A significant percentage decrease in tetraCDDs could be
accounted for by two isomers (1,3,6,8-tetraCDD and 1,3,7,9-tetraCDD)
present in the majority of the samples tested. These two isomers were
not related to the chemical accident at the factory.
Table 35. PCDD contamination in soil from Zone R in Seveso (1981)
(values in ng/kg)a
Sample 2,3,7,8b TCDDc Penta- Hexa- Hepta- Octa- Total
tetraCDD CDDs CDDs CDDs CDD PCDDs
S1 0.8 0.3 0.4 6.0 1.4 1.7 10.6
S2 3.4 1.0 0.7 9.5 2.1 2.2 18.9
S3 4.0 1.9 1.2 8.2 8.6 27.0 50.9
S4 2.3 0.8 0.6 10.2 2.1 2.0 18.0
S5 < 0.1 < 0.3 0.5 9.5 1.9 1.4 13.7
S6 6.3 1.5 1.1 10.4 2.6 1.3 24.8
S7 1.7 1.0 0.8 12.4 1.9 1.8 19.6
S8 2.2 0.4 0.8 8.8 1.8 0.8 14.8
S9 1.0 2.8 2.3 21.2 9.6 13.5 50.4
a From: Wipf & Schmid (1983).
b Probably related to the accident.
c Total levels of isomers other than 2,3,7,8-tetraCDD,
probably not related to accident.
Wipf & Schmid (1983) reported the presence of PCDDs other than
2,3,7,8-tetraCDD in the soil from Zone R (Table 35). They suggest that
a municipal incinerator and the burning of wood shavings treated with
chlorinated phenols could be the source of the other PCDDs.
Nestrick et al. (1986) reported the levels of 2,3,7,8-tetraCDD in
soil samples collected from industrialized areas of US cities. They
observed a widespread occurrence of 2,3,7,8-tetraCDD in urban soils,
with levels of 1-10 ng/kg, and suggested that local combustion
sources, including MSW and industrial incinerators, were the probable
origin.
McLaughlin & Pearson (1984) measured soil concentrations of PCDDs
and PCDFs in the vicinity of a municipal refuse incinerator in
Ontario, Canada. Urban and rural control locations were also sampled.
All soil samples (14) had detectable quantities of at least one of the
five PCDD congener classes (tetraCDD to octaCCD) tested for, whereas
eight samples contained detectable levels of one or more of the five
PCDF congener groups (tetraCDF to octaCDF). The levels ranged from
non-detectable (0.003 - 0.008 ng/g) to 3.5 ng/g (octaCDD); only one
site had a measurable quantity (0.007 ng/g) of tetraCDD in the soil.
The most abundant PCDD or PCDF congener was octaCDD, which had similar
levels whether samples were taken close to or remote from the
incinerator. Similarly, no concentration gradients, relative to
distance from the incinerator, were apparent for any of the other
PCDDs or PCDFs.
Soil samples near a chemical waste incinerator in Scotland have
also been analyzed for PCDDs and PCDFs, together with samples from
control locations (Edulgee et al., 1986). Detectable levels of all
PCDFs or PCDFs that were examined were found in each of the soil
samples (13). Levels found ranged from 1.2 ng/kg (2,3,7,8-tetraCDD) to
1900 ng/kg (total hexaCDF). No consistent pattern was observed to
differentiate levels found in control samples from levels in soil near
the incinerator.
These studies from widely separate areas of the world support the
suggestion that diffuse combustion sources are the major source of
PCDDs and PCDFs in the soil.
Rappe & Kjeller (1987) analyzed soil samples from various parts
of Europe (Table 36). They represent rural areas (samples 1, 2, and 3)
as well as more industrialized areas (samples 4 and 5). PCDDs and
PCDFs could be identified in all samples. The 2,3,7,8-tetraCDD
concentration was below the detection level in the soil samples from
the non-industrialized areas. Trapped sediments from the archipelago
of Stockholm, Sweden, were also analyzed. The samples were collected
in the inner (sample 6), middle (sample 7), and outer archipelago
(sample 8). Levels decreased with increasing distance from the city of
Stockholm. The isomeric patterns for tetra- and pentaCDF isomers are
very similar to those found for samples of total air and air
particulates (section 6.1). A sediment sample from the mouth of River
Viskan, Sweden, was also analyzed (sample 9). A slight difference in
congener profile was found between this sample and the sediments from
the archipelago of Stockholm.
Czuczwa & Hites (1985) found PCDDs and PCDFs in sediment samples
from several locations in Saginaw River and Bay, and southern Lake
Huron, levels ranging from 100 ng/g in urban areas to 100 ng/kg at
remote sites. Although no isomers were identified, the analytical
profiles in the sediments followed closely those found in combustion
samples, suggesting that combustion is the major source of PCDDs and
PCDFs found in the sediments. Analyses of sediment cores showed a
dramatic increase in the PCDD and PCDF concentrations at a depth
corresponding to approximately the year 1940, and levels remained high
up to the present. There is no good correlation between the trend in
these levels and the trend for coal burning in the United States.
However, the levels in the sediments correlate with the production and
use of chlorinated aromatic compounds within this area of the Great
Lakes.
5.4 Food
5.4.1 Meat and bovine milk
The levels of PCDDs and PCDFs in fish and other seafood are
discussed in section 4.4.2.
Table 36. Levels (pg/g) of PCDDs and PCDFs in samples of sediments and soila
Soil samples Sediments
1 2 3 4 5 6 7 8 9
2,3,7,8-tetraCDFb 2.9 1.6 1.1 34 38 30 17 14 1.6
Total tetraCDFs 9.3 7.7 11 320 370 290 150 120 24
2,3,7,8-tetraCDD < 2.0 < 2.1 < 0.2 2.4 0.8 2.4 2.0 < 2.0 0.2
Total tetraCDDs - - 3.2 55.5 11.2 69 21 23 6.4
1,2,3,7,8-pentaCDFc 2.5 1.6 0.5 17 31 16 8.6 8.5 1.3
2,3,4,7,8-pentaCDF 0.8 1.0 0.6 23 65 20 14 16 1.7
Total pentaCDFs 14 13 6.7 200 450 260 140 130 30
1,2,3,7,8-pentaCDD < 2.0 < 2.0 < 0.1 18 34 7.6 5.2 5.5 0.9
Total pentaCDDs - - 4.6 220 270 230 99 86 13
1,2,3,4,7,8-hexaCDFd 3.8 2.2 0.9 30 45 16 10 8.8 1.9
1,2,3,6,7,8-hexaCDF 1.8 1.5 0.4 11 25 12 7.1 5.9 1.2
1,2,3,7,8,9-hexaCDF 1.0 0.9 4.3 110 1100 5 < 1 < 1 2.0
2,3,4,6,7,8-hexaCDF 1.9 1.0 0.7 26 57 16 31 23 1.6
Total hexaCDFs 16 12 11 270 1900 250 220 92 44
1,2,3,4,7,8-hexaCDD < 2 < 2 < 0.1 13 28 1.6 0.8 1.0 1.6
1,2,3,6,7,8-hexaCDD < 2 < 2 < 0.1 19 64 48 2.0 2.0 10
1,2,3,7,8,9-hexaCDD < 2 < 2 < 0.1 6.2 19 2.5 0.9 1.0 4.3
Total hexaCDDs - - 4.7 200 330 49 16 19 64
Total heptaCDFs 22 14 18 260 4500 1300 1500 190 300
Total heptaCDDs < 10 < 10 17 370 1600 5700 1200 880 190
OctaCDF - - 5.7 68 71 39 < 20 < 20 330
OctaCDD - - 14 140 180 3100 510 260 900
a From: Rappe & Kjeller (1987).
b Not separated from 2,3,4,8-tetraCDF, except in the case of sample 9.
c Not separated from 1,2,3,4,8-pentaCDF.
d Not separated from 1,2,3,4,7,9-hexaCDF.
The US Environmental Protection Agency (US EPA) initiated a
2,3,7,8-tetraCDD monitoring programme of beef fat samples taken from
cattle that had grazed on rangelands known to have been treated with
2,4,5-T. The analytical collaborators in this programme were Dow
Chemical Co. (USA), Wright State University, and Harvard University.
All the laboratories used mass spectroscopic techniques for
quantification. Two different extraction techniques were used. All
three laboratories analyzed control samples taken from cattle that had
grazed on non-treated areas. Some of these control samples were spiked
with known amounts of TCDD. All the controls were prepared by the US
EPA (Firestone, 1978). Good agreement was found between the amounts of
TCDD spiked into the control beef fat samples and the reported levels
found, even down to TCDD levels of 10 ng/kg. The average reported TCDD
level was 10 ng/kg; the amount actually added by the EPA was 9 ng/kg.
Of a total of 34 analyses of controls to which no TCDD was added, in
only one case was there a false positive report of TCDD (O'Keefe et
al., 1977). Of 52 samples of beef fat from 2,4,5-T-treated rangeland,
19 (37%) were reported by one or more laboratories to have TCDD. The
average range of levels reported was 5-66 ng/kg, and the overall
average was 7 ng/kg. If one considers only the 40 beef fat samples
from areas receiving at least 1.1 kg of 2,4,5-T/ha, all 19 positive
samples (48%) belong to this group, and the average reported TCDD
level would be 9 ng/kg. The results indicated a consistent trend,
relating the average reported TCDD level in beef fat to the intensity
of the 2,4,5-T application to rangeland (O'Keffe et al., 1978;
McKinney, 1978).
None of the three collaborating laboratories used the most
selective and sensitive analytical method now known (capillary glass
column gas chromatography - high resolution mass fragmentography).
Kocher et al. (1978) also analyzed specimens of fat taken from
steers that had grazed on rangeland previously treated with 2,4,5-T
herbicides. The limit of detection of TCDD (2.5 times peak to peak
noise) was found to be in the 30-60 pg range (3-6 pg/g in beef fat
using 10 gram samples). None of the sixteen samples analyzed in two of
three studies revealed TCDD. In the third study, the animals were
confined to a fenced pasture sprayed in its entirety with 2,4,5-T
herbicides. The samples from three of the seven animals gave a
positive response at the extremely low level of 3 to 4 ng TCDD/kg,
which is at the detection limit; the highest reported value (without
interfering components) was 13 pg/g. The level of 2,3,7,8-tetraCDD in
the 2,4,5-T used was, however, unknown. Beck et al. (1986) analyzed
seven randomly collected samples of cow's milk from different areas of
the Federal Republic of Germany and one sample from the German
Democratic Republic. The cow's milk was taken from road transport
tankers. In all samples, 2,3,7,8-substituted PCDDs and PCDFs were
found at low levels of pg/g on a fat weight basis. Detection limits
were in the range 0.1-0.3 ng/kg. The levels of PCDDs and PCDFs found
in the milk samples were lower than those measured by Rappe et al.
(1987b) in cow's milk from Switzerland (Table 38). In addition, there
was no evidence of high levels of the higher chlorinated congeners
(e.g. hepta and octa- ) (see Table 37). Levels were much lower than
those in human milk samples (see section 5.4.2).
Rappe et al. (1987b) analyzed PCDDs and PCDFs in six samples of
bovine milk from various locations in Switzerland. In all samples,
2,3,7,8-substituted PCDDs and PCDFs were found at levels of pg/kg in
whole milk (Table 38). However, the levels were lower in commercial
milk samples than in samples collected directly from cows grazing in
the vicinity of incinerators.
When Ryan et al. (1985) analyzed PCDDs and PCDFs in chicken and
pork samples in Canada, the incidence of positives for hexa-, hepta-,
and octaCDD in selected samples of chicken fat was 50, 62, and 46%,
with averages of 27, 52, and 90 ng/kg, respectively. Similar levels of
hexa- and heptaCDFs were also found in some of these samples, but
tetra- and pentaCDDs and tetra-, penta-, and octaCDFs were not
detected. A comparison between the tissue analyses and those of the
wood (treated with pentachlorophenol) used to house the animals showed
a marked similarity (see Table 38), indicating that pentachlorophenol
was the probable source of contamination of the food samples.
Firestone et al. (1986) reported the analyses of various food
items collected in the period beginning in 1979. Low levels (< 300
ng/kg) of 1,2,3,4,6,7,8- and 1,2,3,4,6,7,9-heptaCDD were found in some
samples of chicken, bacon, pork chops, and beef liver. HexaCDD was not
found in any of the foods. Several beef livers had high levels of OCDD
residues, the highest reported value being 3830 ng/kg. No PCDDs (at a
detection limit of 10-40 ng/kg) were found in ground beef.
5.4.2 Human milk
Rappe et al. (1984) reported low levels of 2,3,7,8-substituted
PCDDs and PCDFs in five samples of human milk from the Federal
Republic of Germany (FRG) (Table 39).
Table 39 also indicates the results of analyses of four samples
of human milk from the Umea region of northern Sweden (Rappe, 1985),
92 samples from Rheinland-Westfalia in the FRG (Furst et al., 1987),
30 other samples from the FRG (Beck et al., 1987), and five samples
from the Netherlands or Yugoslavia (Rappe et al., 1987).
Van der Berg et al. (1986) also reported the levels of PCDDs and
PCDFs in human milk samples from the Netherlands, but these levels
were reported on milk basis, not on fat basis, and are therefore not
included in Table 39.
A comparison between the isomers, levels, isomeric pattern, and
congener profiles found in human milk (Table 39) and in adipose tissue
(Tables 29 and 30) shows a remarkable degree of similarity.
5.4.3 Rice
Rice from fields in Arkansas, Louisiana, and Texas, USA, treated
at a maximum rate to give 2.52 kg 2,4,5-T/ha, were analyzed for
possible 2,3,7,8-tetraCDD residues. A specification of 1 µg
2,3,7,8-tetraCDD/g 2,4,5-T was given in the report, but no analytical
data were given for the herbicide. No 2,3,7,8-TCDD was detected in the
rice (detection limit = 2-7 µg/kg) and no 2,3,7,8-TCDD residues
(detection limit = 2-10 µg/kg) were found in 30 samples of rice
purchased in retail stores throughout the USA (Jensen et al., 1983).
5.5 Yusho and Yu-cheng Episodes
In 1968, more than 1500 people in south-west Japan were
intoxicated through consuming commercial rice oil accidentally
contaminated by PCBs, PCDFs, and polychlorinated quarterphenyls
(Masuda & Yoshimura, 1982; Masuda et al., 1985). In 1979, a similar
episode occurred in central Taiwan, the number of persons involved
here approaching 2000 (Chen et al., 1980; Chen et al., 1985). In the
past, both accidents were referred to as Yusho episodes, but now the
Taiwan episode has been renamed Yu-cheng.
The Japanese rice oil contained more than 40 PCDF isomers (tri-
to hexaCDFs) (Buser et al., 1978d), whereas the number of isomers in
the Taiwanese oil seems to have been less (Chen & Hites, 1983). The
toxic 2,3,7,8-substituted PCDFs were middle or minor constituents,
about 10-15% of the total amount of PCDFs (Buser et al., 1978d; Masuda
et al., 1985; Chen & Hites, 1983). The mean total consumption of PCDFs
of the Yusho and Yu-cheng patients has been estimated to be 3.3-3.8
mg/person (Hayabuchi et al., 1979; Chen et al., 1985), or a daily
intake of total PCDFs of 0.9 µg/kg body weight (Hayabuchi et al.,
1979). The average intake of 2,3,7,8-substituted PCDFs was 90-135
ng/kg body weight per day. The smallest amount of total PCDFs causing
chloracne has been estimated to be 0.16 µg/kg body weight per day
(Hayabuchi et al., 1979) or 20-30 ng/kg per day of the
2,3,7,8-substituted congeners.
Analysis of liver samples taken from the Yusho patients about 18
months after the exposure showed a dramatic decrease in the number of
PCDF isomers. Apparently most of the PCDF isomers were metabolized or
excreted during the period between exposure and sampling (Rappe et
al., 1979). A comparison between the PCDF isomers found in the Yusho
oil and the liver samples revealed an interesting relationship. Most
of the isomers retained had lateral positions (2, 3, 7, and 8)
substituted with chlorine; these isomers have the highest toxicity
(Rappe et al., 1979).
Table 37. PCDD and PCDF levels in samples of cow's milk (ng/kg on a fat weight basis)a
SAMPLE
1 2 3 4 5 6 7 8
2,3,7,8-tetraCDF < 0.1 0.29 0.28 1.1 1.0 1.4 1.4 0.27
2,3,7,8-tetraCDD 0.33 < 0.2 < 0.2 < 0.2 ND < 0.2 ND ND
1,2,3,7,8-pentaCDF ND 0.4 ND 0.26 0.24 ND 0.39 < 0.2
2,3,4,7,8-pentaCDF 1.3 0.91 1.1 1.6 1.5 1.3 2.9 0.8
1,2,3,7,8-pentaCDD 1.0 0.72 ND 0.81 0.6 0.78 1.2 < 0.5
1,2,3,4,7,8-hexaCDF 0.93 0.67 0.70 0.85 < 0.3 0.84 1.9 0.57
1,2,3,6,7,8-hexaCDF 0.73 0.58 0.57 0.85 < 0.3 0.73 2.1 0.41
2,3,4,6,7,8-hexaCDF 0.65 0.48 0.53 0.68 ND 0.64 1.8 0.37
1,2,3,4,7,8-hexaCDD 0.33 0.34 < 0.3 < 0.3 < 0.3 0.36 0.33 < 0.3
1,2,3,6,7,8-hexaCDD 1.3 1.2 1.0 0.82 0.32 1.7 1.9 0.80
1,2,3,7,8,9-hexaCDD < 0.3 0.34 0.36 0.39 ND 0.55 0.48 < 0.3
1,2,3,4,6,7,8-heptaCDFb < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5
1,2,3,4,6,7,8-heptaCDDb < 2 < 2 < 2 < 2 < 2 < 2 < 2 < 2
octaCDDb <10 <10 <10 <10 <10 <10 <10 <10
octaCDFb < 1 < 1 < 1 < 1 < 1 < 1 < 1 < 1
a From: Beck et al. (1987).
b Not significantly higher than blanks.
ND = not detectable.
Table 38. PCDF and PCDD content of bovine milk from Switzerland. Results in ng/kg (ppt), whole milk basisa
Compound Commercial Incinerators
1 2 3 4 5 6
2,3,7,8-tetraCDF < 0.028 < 0.035 < 0.021 < 0.022 < 0.032 < 0.028
1,2,3,7,8-pentaCDF < 0.020 < 0.022 < 0.021 < 0.020 < 0.036 < 0.032
2,3,4,7,8-pentaCDF 0.084 0.066 0.069 0.43 0.22 0.23
1,2,3,4,7,8-hexaCDF < 0.020 < 0.026 < 0.017 0.13 0.06 0.084
1,2,3,6,7,8-hexaCDF 0.028 < 0.018 < 0.021 0.19 0.095 0.059
2,3,4,6,7,8-hexaCDF < 0.020 < 0.018 ND 0.28 0.12 0.049
(< 0.02)
1,2,3,4,6,7,8-heptaCDF < 0.12 ND ND 0.49 0.28 < 0.18
(< 0.13) (< 0.08)
octaCDF < 0.20 ND ND ND ND < 0.52
(< 0.13) (< 0.09) (< 0.16) (< 0.21)
2,3,7,8-tetraCDD ND ND ND 0.049 0.038 0.021
(< 0.012) (< 0.013) (< 0.013)
1,2,3,7,8-pentaCDD ND ND ND 0.25 < 0.086 ND
(< 0.04) (< 0.08) (< 0.06) (< 0.1)
1,2,3,4,7,8-hexaCDD < 0.068 ND ND 0.23 0.14 < 0.14
(< 0.1) (< 0.06)
1,2,3,6,7,8-hexaCDD < 0.068 ND ND 0.29 0.16 < 0.21
(< 0.1) (< 0.06)
1,2,3,7,8,9-hexaCDD < 0.068 ND ND 0.17 < 0.080 < 0.11
(< 0.1) (< 0.06)
1,2,3,4,6,7,8-heptaCDD < 0.064 < 0.066 < 0.064 0.26 < 0.095 0.42
octaCDD < 0.16 < 0.26 < 0.12 0.28 < 0.16 0.59
a From: Rappe et al. (1987b).
ND = not detectable.
Table 39. Levels of PCDDs and PCDFs found in human milk (ng/kg of fat weight).
Sweden FRG FRG FRG Netherlands Yugoslavia
n=4a n=5b n=92c n=30d n=3e n=2e
2,3,7,8-tetraCDD 0.6 1.9 < 5 3.4 9.7 < 1.0
1,2,3,7,8-pentaCDD 6.5 12.9 10.7 15 44 5.5
1,2,3,4,7,8-hexaCDD 2.5 4.6 8.1 12 25 3.5
1,2,3,6,7,8-hexaCDD 19 17.3 32.7 59 251 15
1,2,3,7,8,9-hexaCDD 6.3 1.6 6.4 11 23 ND
1,2,3,4,6,7,8-heptaCDD 59.5 72.8 49.9 61 130 106
octaCDD 302 434 181 530 744 106
2,3,7,8-tetraCDF 4.2 5.4 2.6 2.5 2.8 < 1.0
1,2,3,7,8-pentaCDF < 1 < 1 1.8 < 1 ND ND
2,3,4,7,8-pentaCDF 21.3 36.4 22.9 20 79 25
1,2,3,4,7,8-hexaCDF 4.7 11.4 8.2 8.5 8.9 3.7
1,2,3,6,7,8-hexaCDF 3.4 10.2 6.6 7.8 10.3 3.6
2,3,4,6,7,8-hexaCDF 1.4 4.3 3.3 3.0 6.4 1.3
1,2,3,4,6,7,8-heptaCDF 7.4 9.2 6.4 8.5 39 ND
octaCDF 3.2 2.4 22.8 < 3 ND ND
ND = not detected; NA = not analyzed; n = number of samples.
a Rappe (1985).
b Rappe et al. (1984).
c Furst et al. (1987).
d Beck et al. (1987).
e Rappe et al. (1987) (pooled samples)
Kunita et al. (1984) studied the blood levels of PCDFs in people
involved in the Japanese and Taiwanese episodes. They used a
non-isomer-specific analytical method, and reported higher blood
levels in persons with severe dermal symptoms than in persons with
light symptoms. The levels 2 years after exposure were lower than 0.5
year after exposure; for the six persons studied the levels of total
PCDFs decreased on average by 57% ± 12% (Kunita et al., 1984).
The rate of excretion of these toxic PCDF isomers is very low.
Rappe & Nygren (1984) could detect 2,3,4,7,8-pentaCDF in blood plasma
from Yusho patients when the samples were collected 11 years after
exposure. However, higher levels were found in blood from Yu-cheng
patients one year after exposure; these analyses also showed a 15-20%
reduction in levels in one year (Rappe, 1984).
6. KINETICS AND METABOLISM OF 2,3,7,8-TETRACHLORO-
DIBENZO-P-DIOXIN (TCDD) AND OTHER PCDDs
6.1 Uptake, Distribution, and Excretion
Most of the available toxicokinetic data arise from studies of
gastrointestinal exposure and oral or intraperitoneal administration.
There has been one dermal study on rats, but no studies on exposure
via the respiratory tract. Data on the gastrointestinal absorption,
distribution, and elimination of TCDD in various species are
summarized in Tables 40 and 41. Principle organ depots in all species
studied are the liver and adipose tissue. In addition skin and muscle
have been found to be organ depots in monkeys, and skin in
guinea-pigs. In all species studied clearance of TCDD from the body
follows apparent first-order kinetics.
6.1.1 Studies on rats
Male Sprague Dawley rats were dosed by gavage with 14C TCDD in
acetone: corn oil (1:9), at a concentration of 50 µg/kg body weight
(Piper et al., 1973). Rats lost weight and their physical condition
was generally poor, although no deaths occurred. Approximately 30% of
the administered dose of radioactivity was eliminated in the faeces
during the first 48 h, this being most probably unabsorbed TCDD. The
total faecal TCDD content was equivalent to 53.2% of the dose over a
21-day period. During this time 13.2% was eliminated in the urine and
3.2% in the air. From these results the half-time for the clearance of
TCDD was calculated as 17.4 (± 5.6) days. In another study, groups of
two to three rats were killed at different time intervals after
administration of the same dose of 14C-TCDD. The liver contained
3.2, 4.5, and 1.3% of the administered dose per gram of tissue 3, 7,
and 21 days, respectively, after dosing. The concentrations in adipose
tissue at the same time intervals were 2.6, 3.2, and 0.4% of the dose
per gram of tissue. Other tissues showed lower concentrations.
In a study by Allen et al. (1975), male Sprague Dawley rats were
given 14C-TCDD in a single dose of 50 µg/kg body weight by stomach
tube. Groups of five rats were sacrificed 1, 3, 5, 7, 14, and 21 days
after dosing. The dose given resulted in marked liver hypertrophy,
thymic regression, weight loss, and death in 50% of the animals within
25 days. Twenty-five percent of the dose was eliminated within the
first 3 days through the faeces. During the next 18 days, 1 to 2% of
the dose was found in the faeces daily. The total amount in the faeces
during the 21 days following administration was about 52%.
Table 40. Gastrointestinal absorption of TCDD
Species/Strain Vehicle Dose % Absorption Reference
(µg/kg body weight) (mean ± SD)
Rats
Sprague Dawley acetone:corn oil (1:9) 50a 70 Piper et al. (1973)
Sprague Dawley corn oil 50a > 75 Allen et al. (1975)
Sprague Dawley diet 7 or 20; for 42 days 50-60 Fries & Marrow (1975)
Sprague Dawley acetone:corn oil (1:24) 1a 84±11 Rose et al. (1976)
Sprague Dawley acetone:corn oil (1:24) 0.1 or 1.0; 5 days/week 86±12 Rose et al. (1976)
for 7 weeks
Mice
ICR/Ha Swiss ethanol:Tween 80:saline 135a 27-28 Koshakji et al. (1984)
(1:10:89)
Hamsters
Golden Syrian olive oil 650a 73.5±22.8 Olson et al. (1980a)
a Single dose.
Table 41. Elimination of TCDD in different species
Species/Strain Route/Vehicle Dosea Duration Half-life Eliminated radioactivity Reference
(µg/kg of for elimination (% of administered dose)
body study (days)
weight) (days) Faeces Urine
Rats
Sprague Dawley oral/acetone: 50 21 17.4 ±5.6 53.2 13.2 Piper et al. (1973)
corn oil (1:9)
Sprague Dawley oral/corn oil 50 21 21.3 ±2.9 53.3 4.5 Allen et al. (1975)
Sprague Dawley oral/acetone: 1 22 31 ± 6 NR ND Rose et al. (1976)
corn oil (1:24)
Sprague Dawley oral/acetone: 1b 49 23.7 NR 3.1 ±0.2 (m) Rose et al. (1976)
corn oil (1:24) 12.5 ±5.1 (f)
Sprague Dawley ip/corn oil 400 7 NR 4.96 ±0.3 0.51 ±0.05 van Miller et al.
(1976)
Mice
C57BL/6 ip/olive oil 10 30 11 59.3 20.5 Gasiewicz et al.
(1983b)
DBA/2 ip/olive oil 10 30 24.4 39.8 16.8 Gasiewicz et al.
(1983b)
B6D2F1 ip/olive oil 10 30 12.6 56.3 20.5 Gasiewicz et al.
(1983b)
ICR/Ha Swiss oral/ethanol: 135 11 20 78 4 Koshakji et al.
tween 80: (1984)
saline (1:10:89)
C57BL/6 Ahb/Ahd ip/emulphor: 0.5 42 9.6 61.8 19.9 Birnbaum (1986)
ethanol:H2O
(1:1:18)
C57BL/6 Ahd/Ahd ip/emulphor: 0.5 42 9.6 55.9 26.8 Birnbaum (1986)
ethanol:H2O
(1:1:18)
DBA/2 Ahb/Ahd ip/emulphor: 0.5 42 10.8 71.5 13.6 Birnbaum (1986)
ethanol:H2O
(1:1:18)
Table 41 (contd).
Species/Strain Route/Vehicle Dosea Duration Half-life Eliminated radioactivity Reference
(µg/kg of for elimination (% of administered dose)
body study (days)
weight) (days) Faeces Urine
Mice (contd)
DBA/2 Ahd/Ahd ip/emulphor: 0.5 42 10.8 76.1 11.1 Birnbaum (1986)
ethanol:H2O
(1:1:18)
Guinea-pigs
Hartley ip/olive oil 2 23 33 2 Gasiewicz &
Neal (1979)
NR oral/NR NR 22 22-43 ND ND Nolan et al. (1979)
Hartley ip/olive oil 0.56 45 93.7 ± 15.5 26.2 ±3.6 3.1 ±1.2 Olson (1986)
Hamsters
Golden Syrian oral/olive oil 650 35 15.0 ±2.5 NR NR Olson et al.
(1980a)
Golden Syrian ip/olive oil 650 35 12.0 ±2.0 50.0±2.7 34.6±5.4 Olson et al.
(1980a)
Monkeys
Rhesus (adult) ip/corn oil 400 7 NR 3.75 1.06 van Miller et al.
(1976)
Rhesus (infant) ip/corn oil 400 7 NR 1.26 2 van Miller et al.
(1976)
a Single dose, unless otherwise stated.
b 5 days/week for 7 weeks.
ND = not detectable, NR = not reported, m = males, f = females, ip = intraperitoneal.
The authors concluded that the faecal content during the first 3
days represented mainly unabsorbed TCDD and that apparently more than
75% of the administered dose had been absorbed from the
gastrointestinal tract. The radioactivity excreted daily through the
urine ranged between 0.1 and 0.2% of the administered dose during the
initial 12 days. Thereafter, the daily urinary radioactivity excretion
increased from 0.25% of the administered dose to 0.43% by day 21. The
total amount of 14C excreted through the urine over a 21-day period
was about 4.5% of the dose. On the basis of the daily faecal and
urinary excretion, the authors calculated a half-time of 21.3 (± 2.9)
days. In the rats killed on days 1, 3, 5, 7, 14, and 21 after
administration, the total liver content was 56 (± 5), 54 (± 14), 54 (±
6), 54 (± 8), 45 (± 5), and 24 (± 4)% of the administered dose,
respectively. At all intervals, the levels found in the liver exceeded
those found in other organs. On days 5, 7, and 14 about 90% of the
total radioactivity in the liver was present in the microsomal
fraction.
Fries & Marrow (1975) fed Sprague Dawley male and female rats a
diet containing 7 or 20 µg 14C-TCDD/kg diet for 42 days. Thereafter
all rats received the control diet for another 30 days. Two animals of
each sex and TCDD dietary level were sacrificed at 14-day intervals.
This treatment resulted in decreased food consumption, decreased
weight gain, and increased relative liver weight among both males and
females. The concentration of TCDD-derived radioactivity in the liver,
which was the principal tissue depot of both males and females, was
directly proportional to the dietary intake of 14C-TCDD. At the end
of the 42-day feeding period, the 14C levels in male livers
indicated TCDD contents of 5.8 and 15.9 µg/kg of tissue for the lower
and higher feed concentrations, respectively. The concentrations in
the liver of female rats were similar. Analysis of the liver of rats
killed 14 and 30 days after discontinuing TCDD exposure indicated a
gradual decrease in TCDD liver concentration in both sexes. The
steady-state body burden for TCDD was estimated to be 10-11 times the
daily intake in both sexes. The whole-body and liver half-lives were
calculated to be 12 and 11 days in males and 15 and 13 days,
respectively, in females.
Rose et al. (1976) estimated the absorption of a single non-toxic
dose of 1 µg 14C-TCDD/kg body weight in male and female Sprague
Dawley rats to be 84 ± 11% of the administered dose. The elimination
of TCDD was followed for 22 days after administration and the faecal
excretion accounted for most if not all of the elimination of TCDD
and/or its metabolites. No radioactivity was detectable in urine and
expired air. Twenty-two days after dosing, mean values of 1.26% and
1.25% of the dose/g liver and adipose tissue, respectively, were
found. Much lower 14C-activities were found in the thymus, kidney,
and spleen, namely 0.09, 0.06, and 0.02% of the dose/g, respectively.
The whole-body half-life was estimated to be 31 days. Rose et al.
(1976) also followed the fate of 5 daily doses per week of 0.01, 0.1,
and 1.0 mg 14C-TCDD/kg body weight given for 1, 3, and 7 weeks to
male and female Sprague Dawley rats (Table 42).
According to Kociba et al. (1976), a dose level of 0.01 µg/kg per
day, 5 days/week for 13 weeks, produced no overt toxic effects,
whereas 0.1 or 1.0 µg/kg per day produced adverse effects, including
some deaths in the high dose group. In the study by Rose et al.
(1976), the dose level of 0.01 µg/kg per day resulted in no detectable
14C, except in liver, fat, and excreta. Thus no kinetic calculations
on a truly non-toxic dose could be performed. Pooling the results for
all rats receiving 0.1 or 1.0 µg/kg per day, the absorbed dose
corresponded to 86 ± 12% of the administered dose, with an individual
variation of 66 to 93%. The overall rate constant for elimination
corresponded to a half-time of 23.7 days, with an individual variation
of 16-37 days. The radioactivity was eliminated primarily in the
faeces, but the percentage of the dose excreted in the urine compared
to that eliminated in the faeces tended to increase with time. At the
dose level of 1.0 µg/kg per day, males excreted in the urine 3.1 (±
0.2)% and females 12.5 (± 5.1)% of the cumulative dose over 7 weeks of
exposure. The one female rat that died during the 7th week excreted
17.8% of the cumulative dose in the urine. Exhaled air was not
examined in these studies. After 7 weeks of exposure, the average body
burdens were 47.7 (± 8.8) and 37.1 (± 7.5)%, respectively, of the
administered dose for the rats given 0.1 and 1.0 mg TCDD/kg per day.
The main tissue depots of 14C were the liver and adipose tissue.
Radioactivity was also detected in thymus, kidney, and spleen at
levels between 1 and 2% of that in liver. Direct chemical
determination of liver samples confirmed the TCDD-concentrations
calculated by radioactivity measurements. TCDD and/or its metabolites
approached a steady-state body burden calculated to be 21.3 Do for
rats given a daily dose (Do, µg/kg body weight) on 5 consecutive
days per week for an infinite number of weeks, within 13 weeks over
the dose range 0.01 to 1.0 µg/kg per day. It was thought unlikely that
the dietary intake of extremely low levels of TCDD would result in the
accumulation of toxic amounts in the rat.
The excretion and distribution of a toxic dose of 400 µg
3H-TCDD/kg body weight was followed in male Sprague Dawley rats (Van
Miller et al., 1976). Within 7 days, about 5.0% of the dose had been
excreted in faeces and 0.5% in urine. The principal tissue depots for
radioactivity were liver, muscle, and skin, which contained 43.0, 4.6,
and 4.4% of the administered dose, respectively.
Table 42. Tissue distribution of TCDD-derived radioactivity in rats given
oral doses of 14C-TCDDa
Tissue content of 14C (µg equivalents of TCDD/kg tissue)b
Tissue 1 week 3 weeks 7 weeks
Exposure level: 1.0 µg/kg body weight per day
liver 49.5±3.6 110.2±37.1 204.0±52.2
adipose tissue 10.0±3.0 23.5±7.5 61.4±36.7
thymus 0.9±0.2 7.3±4.5 6.9±2.5
kidney 0.9±0.2 1.9±0.9 5.5±5.1
spleen 0.4±0.1 1.6±0.8 1.9±1.1
Exposure level: 0.1 µg/kg body weight per day
liver 3.9±1.1 11.8±2.2 19.8±3.1
adipose tissue 0.9±0.6 2.7±0.8 4.5±0.7
thymus ND 1.0±0.6 0.6±0.2
spleen ND 0.6±0.4 0.3±0.1
kidney ND ND ND
Exposure level: 0.01 µg/kg body weight per day
liver ND 0.8±0.1 1.6±0.5
adipose tissue ND 0.3 (2)c 0.3±0.1 (4)c
thymus ND 0.6±0.1 ND
spleen ND 0.6±0.3 ND
kidney ND ND ND
a From: Rose et al. (1976).
b The mean ± standard deviation of 3 male and 3 female rats.
c Indicates the number of animals with detectable levels of
14C-activity.
ND = not detected.
Kociba et al. (1976) gave Sprague Dawley rats TCDD in daily doses
of 0.01, 0.1, and 1 µg/kg body weight 5 days per week for 13 weeks by
gavage. The liver contained TCDD at a level of 324 (± 53) µg/kg wet
weight in males and 284 (± 21) µg/kg wet weight in females given
repeated TCDD doses of 1 µg/kg body weight per day. For the dose level
of 0.1 µg/kg body weight per day, the liver TCDD levels were 36 (± 4)
and 35 (± 4) µg/kg wet weight for males and females, respectively. A
dose of 0.01 µg/kg body weight per day resulted in TCDD liver levels
of 2.6 ( 0.6) µg/kg wet weight in males and 3.7 (± 0.4) µg/kg wet
weight in females.
After feeding diets with TCDD levels corresponding to daily
dietary intakes of 0.001, 0.01, or 0.1 µg/kg body weight for 2 years,
the average concentrations of TCDD found in the liver of female
Sprague Dawley rats were 0.54, 5.1, and 24.0 µg/kg wet weight,
respectively (Kociba et al., 1978). The corresponding levels in
adipose tissue were 0.54, 1.7, and 8.1 µg/kg wet weight. A comparison
of liver TCDD levels found in rats given comparable daily doses of
TCDD for 13 weeks (Kociba et al., 1976) or 2 years (Kociba et al.,
1978) indicates that with prolonged exposure the liver TCDD content
reaches a plateau. This finding agrees well with the prediction
obtained by mathematical analysis of the data resulting from
experiments involving exposure of a few weeks (Rose et al., 1976).
The proportion of a single oral dose of 3H-TCDD found in the
liver of female Sprague Dawley rats was dependent both on the dose
level and on the vehicle used (Poiger & Schlatter, 1980). Maximal
retention occurred within 48 h after dosing. Increasing retention was
observed up to a dose of 280 ng TCDD/rat. Hepatic retention 24 h after
dosing was higher (36.7% of the dose) if TCDD was given in 50% ethanol
than if it was given as an aqueous suspension of soil (37%, w/w),
where it was 16-24.1% of the dose, or activated carbon (25%, w/w),
where it was < 0.07% of the dose (see section 7.4).
Biliary excretion of radioactivity originating from 3H-labelled
TCDD occurred at a more or less constant rate of 0.5 to 1% of the
administered dose per day for 12 days following the administration of
100 µg TCDD/kg body weight in a female Sprague Dawley rat (Poiger &
Buser, 1983). No severe toxic effects were observed within that time
period.
McConnell et al. (1984) dosed female Sprague Dawley rats orally
with either pure TCDD in corn oil or amounts of contaminated soil
giving similar doses of TCDD. In general TCDD in soil was as potent an
inducer of aryl hydrocarbon hydroxylase (AHH) as pure TCDD in corn
oil. The hepatic concentration of TCDD was 40.8 µg/kg in the corn oil
group, receiving 5 µg TCDD/kg body weight, and 20.3 µg/kg in the soil
group, receiving 5.5 µg TCDD/kg body weight (see section 7.4).
Poiger & Schlatter (1980) studied the dermal absorption of
3H-TCDD in hairless rats of the Naked ex Back-Cross and Holzman
strain (200-250 g). Using the amount of TCDD-derived radioactivity
found in the liver as an indicator of its absorption, they reported
that the permeation of TCDD across the epidermis was highly dependent
on the formulation used. The highest radioactivity in the liver, 14.8%
of the administered dose, was detected when TCDD was applied as a
methanolic solution. The hepatic recovery of the administered dose
observed when TCDD was applied in polyethylene glycol 1500 with and
without 15% water was 14.1 and 1.4%, respectively. Dermal application
of TCDD in vaseline or adsorbed onto soil or activated carbon
decreased the percentage of the dose recovered in the liver to 1.4,
1.7-2.2, and < 0.05%, respectively.
In studies by van den Berg et al. (1983), fly ash and crude or
purified toluene extracts of PCDD- and PCDF-containing fly ash from a
municipal incinerator were mixed with ordinary laboratory diet for
rats. Small portions (2 g) of these diets were fed to male Wistar rats
(300 g) every 24 h for 19 days, at which time the animals were
sacrified. Tetra-, penta-, and hexa-chlorinated PCDDs and PCDFs in the
liver and adipose tissues of these rats were determined. Rats fed the
fly ash containing diet stored PCDDs and PCDFs in their livers at
concentrations that were at least 3 to 5 times lower than in the case
of rats fed comparable amounts of fly ash extracts (for the pentaCDD,
hexaCDF, and hexaCDD isomers, the concentrations were approximately
10-20 times lower). Generally PCDFs showed a higher retention in rat
liver than did the corresponding PCDDs. In the adipose tissue of rats
fed with fly ash extracts, retention was higher for penta- and
hexaCDDs than for the corresponding PCDFs.
In further fly ash studies, male Wistar rats (275 g) were fed for
up to 99 days a diet that included 2.5% HC1-pretreated fly ash
(containing PCDDs and PCDFs) from a municipal incinerator (van den
Berg et al., 1986a). A control group received standard diet. All
congeners retained in the liver of the rats had a 2,3,7,8-chlorine
substitution pattern. With the exception of 2,3,4,7,8-pentaCDF and
2,3,4,6,7,8-hexaCDF, liver retention for each congener was below 10%
of the group dose. The retention percentages of the various congeners
in the liver were almost equal at the time-points studied (34, 59, and
99 days), thus indicating a long half-life of these congeners in rat
liver.
Male Wistar rats fed 22.7 (± 1) µg or 120.7 (± 2.8) µg octaCDD
over a two-week period were found to retain about 1-2% of the given
dose in the liver (Williams et al., 1972). The heart, kidneys, spleen,
lung, skeletal muscle, testes, and urine contained no detectable
levels of octaCDD, but minor amounts were found in the adipose tissue
of the high-dose group. Faeces contained 61% and 37%, respectively, of
the low and high dose given. The presence of a large quantity of
octaCDD in the faeces compared to that in the bile 24-72 h after a
single oral dose of 58 mg octaCDD to bile-cannulated male Wistar rats
(400 g) indicated that the dioxin present in the faeces was mainly
unabsorbed octaCDD (Williams et al., 1972).
After 21 daily doses of 100 mg octaCDD containing 12.6 pg
35S-thio-heptaCDD to male Sprague Dawley rats, the radioactivity was
mainly recovered in the faeces and urine, the percentages of the
ingested radioactive dose being 93 (± 6) and 5.2 (± 0.8)% respectively
(Norback et al., 1975). The high faecal excretion suggests poor
absorption. Of the radio-active body burden, 50% was contained in the
liver. The microsomal fraction contained 96.3 (± 8.2)% of the hepatic
radioactivity.
6.1.2 Studies on mice
In studies by Vinopal & Casida (1973), male white mice (20 g)
were given tritium-labelled TCDD intraperitoneally in a single dose of
130 µg/kg body weight. Three days after TCDD administration to one
mouse, 13% of the administered tritium was recovered in the faeces and
0.3% in the urine, while 32% was found in the liver and 0.3% in the
kidneys. In another study, groups of two to six mice were killed at
various time intervals after similar treatment with the same dose of
3H-TCDD. One and 4 days after dosing, the liver contained about 15%
of the dose, on the 8th day, 26%, on the 11th day, 22%, and on the
15th and 20th days, about 10%. The highest amount of 3H-activity was
found in the microsomal fraction of the liver. Somewhat lower activity
was detected in the mitochondrial fraction and the nuclei. The
supernatant fraction was practically devoid of any radioactivity. On
day 8 when the highest levels were observed, the whole liver
homogenate contained 26.7 (± 4.8)% of the administered dose, the
microsomes 12.6 (± 3.8)%, the nuclei 7.8 (± 1.2)%, the mitochondria
6.2 (± 1.3)%, and the supernatant fraction 0.1 (± 0.0)% of the
administered dose.
Coccia et al. (1981) described the effect of adding different
substances to food on the persistence of TCDD in the liver of male
C57Bl/6 mice. In one of the experiments, the test diet was given
immediately after the administration of a single oral dose of 7.6 µg
3H-TCDD/kg body weight. The hepatic radioactivity 14 days after
dosing was 17.3, 6.3, 13.1, and 14.5% of the administered dose in
animals fed standard chow containing 5% vegetable charcoal, 0.5%
cholic acid, and 4% cholestyramine, respectively. When feeding of the
test diet started 3 days after dosing, the hepatic retention of
3H-TCDD was decreased to a similar extent.
Faecal and urinary excretion (Table 41), along with the formation
of faecal, biliary, and urinary metabolites (see 7.2.1.1), of
3H-TCDD was studied in male C57BL/6, DBA/2, and B6D2F1 mice after a
single intraperitoneal dose of 10 µg 3H-TCDD/kg body weight
(Gasiewicz et al., 1983b). The principal tissue depot in C57BL/6 and
B6D2F1 mice was the liver, followed by the adipose tissue. Most other
tissues examined contained less than 1% of the administered dose. In
DBA/2 mice, the adipose tissue contained more radioactivity than the
liver. This difference may be due to the fact that these three strains
of mice differ in their adipose tissue content, being 5.9, 11.5, and
5.0% of the body weight in C57BL/6, DBA/2, and B6D2F1 mice,
respectively. The estimated half-lives of clearance of 3H-TCDD from
the liver of C57BL/6, DBA/2, and B6D2F1 mice were 17, 27, and 13 days,
respectively, and the corresponding figures for the half-life in the
adipose tissue were 11, 42, and 11 days. The cumulative faecal
elimination 30 days after dosing was 59.3, 39.8, and 56.3% of the
administered dose in C57Bl/6, DBA/2, and B6D2F1 mice, respectively,
and the corresponding figures for urinary elimination were 20.5, 16.8,
and 20.5%.
Birnbaum (1986) studied in mice the distribution and excretion of
a single intraperitoneal dose of 500 ng (45 µCi)3H-TCDD/kg body
weight for up to 42 days after treatment. Two sets of congenic strains
of mice were used, i.e., male C57Bl/6 and female DBA/2 mice, where
within each congenic pair the mice differed only at the Ah locus (or
at a limited number of genes closely linked to the Ah locus). The mice
were bred and phenotyped by zoxazolamine paralysis time. The results,
some of them summarized in Tables 47 and 48, suggested that, at the
dose level studied, the distribution and excretion of TCDD were
primarily governed by the total genetic background rather than by the
allele present at the Ah locus.
When male ICR/Ha Swiss mice (27 - 35 g) were given a single oral
dose of 135 µg 14C-TCDD/kg body weight, about 71% and 1-2%,
respectively, of the administered dose was eliminated via faeces and
urine within the first 24 h (Koshakji et al., 1984). During the
following 10 days, an additional 7% and 2% of the administered
radioactivity were recovered in faeces and urine, respectively. Based
on the estimated body burden of radioactivity, a whole-body half-life
of 20 days was calculated.
The distribution of 3H-TCDD in the skin of hairless (SKH:HR-1)
mice after a single intraperitoneal dose of 6.3 µg TCDD/kg body weight
was examined for up to 14 days (Puhvel et al., 1986). Most of the
3H-TCDD in skin was localized in the dermis, although the
concentration of 3H-TCDD was consistently higher in the epidermis.
6.1.3 Studies on guinea-pigs
The retention of a single intraperitoneal dose of 2 µg
14C-TCDD/kg body weight in various tissues of male Hartley
guinea-pigs was determined 1, 3, 5, 7, 11, and 15 days after exposure
(Table 43) (Gasiewicz & Neal, 1979). Three animals died and all
animals lost 24 to 35% of the body weight during the study. The
highest amount of radioactivity per tissue was found in the liver and
skin. The radioactivity in the liver increased with time, concomitant
to the depletion of adipose tissues. Radioactivity in the skin
decreased also with time. No signs of toxicity were seen when the
cumulative excretion of a single i.p dose of 0.5 µg 3H-TCDD/kg body
weight in male Hartley guinea-pigs was studied (Gasiewicz & Neal,
1979). The faecal and urinary excretion of radioactivity was linear
throughout the 23-day study. Approximately 1.4% of the administered
dose was excreted daily during that period, and the faeces contained
94% of the excreted radioactivity.
The microsomal fraction of the liver in male Hartley guinea-pigs
contained 40.7 to 47.4% of the hepatic radioactivity 1 day after a
single ip dose of 0.3, 2.0, or 7.0 µg of 3H- or 14C-TCDD/kg body
weight (Gasiewicz & Neal, 1979). Corresponding values for the crude
nuclear fraction, the mitochondrial fraction, and the soluble fraction
were 20.1-35.6%, 9.5-12.9%, and 7.6-26.4%, respectively. The
subcellular distribution was similar 1 and 6 days after the low dose
but, following the high dose, more radioactivity was present in the
microsomal fraction and less was recovered in the crude nuclear and
soluble fractions on day 6 after exposure.
Olson (1986) followed the distribution, elimination and
metabolism (see section 6.2.1.1) of a single ip dose of 0.56 µg
3H-TCDD/kg body weight in adult (335 to 625 g) male Hartley
guinea-pigs for 45 days. One of seven animals died on day 27, but the
remaining animals gained weight and exhibited no gross signs of
toxicity. At termination the body composition was normal, and 61% of
the administered radioactivity was recovered in the twelve
investigated tissues at the end of the study. The adipose tissue
contained 36% of the dose; liver, pelt, and skeletal muscle plus
carcass contained each 7% of the dose; the gastrointestinal tract
contained about 2% of the dose, and remaining tissues contained less
than 0.5% of the dose. Urinary and faecal elimination followed
apparent first-order kinetics, with half-lives of 82.5 (± 22.4) and
94.4 (± 14.7) days, respectively.
Table 43. Tissue content of TCDD-derived 14C (% of dose/g tissue)a in guinea-pigs following a single
dose of 14C-TCDDd
Days after exposure
Tissue 1 3 5 7 11 15
Perirenal adipose 3.2±1.0 4.1±0.4 2.1±0.4 1.3±0.2 2.1±0.2
Epididymal adipose 1.5±0.8 3.8±0.5 3.4±0.7 3.2±0.1 3.9b 2.5±1.1
Adrenal 1.4±0.3 1.4±0.2 0.9±0.1 1.2±0.3 2.1±0.9 1.7±0.2
Liver 1.1±0.4 1.5±0.4 1.3±0.2 1.1±0.2 2.2±0.2 3.2±0.3
Liverc 11.4±3.3 15.5±3.3 14.0±2.3 12.0±1.9 21.2±2.3 29.6±2.7
Spleen 0.7±0.3 0.5±0.3 0.2±0.1 0.4±0.2 0.4±0.2 0.5±0.1
Duodenum 0.4±0.2 0.2±0.1 0.2±0.1 0.2±0.1 0.2±0.1 0.3±0.1
Pancreas - - 0.2±0.1 0.5±0.3 0.4±0.3 0.3±0.1
Stomach 0.2±0.1 0.3±0.1 0.1±0.1 0.2±0.1 0.3±0.1 0.3±0.1
Testes 0.2±0.1 0.3±0.1 0.2±0.1 0.3±0.1 0.3±0.1 0.2±0.1
Kidneys 0.3±0.1 0.3±0.1 0.2±0.1 0.4±0.1 0.8±0.4 0.7±0.1
Bone marrow 0.3±0.1 0.5±0.1 0.2±0.1 0.4±0.1 0.4b 0.2±0.1
Lungs 0.3±0.1 0.2±0.1 0.2±0.1 0.4±0.1 0.5±0.2 0.6±0.1
Skinc 13.8±0.7 16.3±0.3 15.8±2.4 6.5±0.8 6.5±0.7 6.7±0.6
Brain, heart,
skeletal muscle <0.25
a Mean ± standard error for three animals, unless indicated otherwise.
b Mean of two animals.
c Percentage of dose/tissue.
d From: Gasiewicz & Neal (1979).
6.1.4 Studies on hamsters
In studies by Olson et al. (1980a), Golden Syrian hamsters
absorbed about 73.5% of a single oral dose of 650 µg 3H-TCDD/kg body
weight, a dose that produced thymic atrophy and body weight loss in
several of the animals. The distribution of radioactitivity in various
tissues 1, 3, 10, and 20 days after administration is given in Table
44. The principal depots were the liver and adipose tissue. A similar
pattern of distribution was obtained when the same dose was given
intraperitoneally. The elimination of radioactivity in faeces and
urine was followed for 35 days after a single intraperitoneal (ip) or
oral dose of 650 µg/kg body weight (Olson et al., 1980a). The
half-life for elimination was 12.0 (± 2.0) days and 15.0 (± 2.5) days
for the ip and oral routes, respectively. Of the excreted
radioactivity 41% occurred in urine and 59% in faeces.
The hepatic retention of PCDDs and PCDFs from dietary intake of
HC1-pretreated fly ash from a municipal incinerator was studied in
male Golden syrian hamsters (van den Berg et al., 1986b). The livers
were analysed for tetra-, penta-, and hexaCDDs and PCDFs after feeding
the diet, which contained 25% fly ash, for 34, 58, and 95 days. No
detectable hepatic retention was observed after 34 days. The highest
retention after 95 days was 8.4% for 2,3,4,7,8-pentaCDF, but the
retention was generally below 5% of the total dose. With the exception
of 2,3,4,6,7-pentaCDF, only 2,3,7,8-substituted PCDDs and PCDFs were
retained. Constant relative concentrations were found for the
2,3,7,8-substituted PCDDs and PCDFs at the time points studied.
6.1.5 Studies on monkeys
Van Miller et al. (1976) gave three adult female rhesus monkeys
and four male infant rhesus monkeys a single intraperitoneal dose of
400 µg 3H-TCDD/kg body weight in corn oil. This dose resulted in a
loss of body weight: 10.8% for adults and 20.7% for infants, and light
microscopic changes in the liver. Over a 7-day period the adult
monkeys excreted 1.06% of the dose in the urine and 3.75% in the
faeces. During the same period the infant monkeys excreted
approximately 2% of the administered dose in urine and about 1.26% in
the faeces. The authors questioned the accuracy of these figures owing
to the difficulty of separating the two types of excreta from infant
monkeys. The total tissue concentrations of radioactivity 7 days after
dosing are given in Table 45. The principal tissue depots of
radioactivity in adult monkeys were adipose tissue, skin, liver, and
muscle; they contained 16.2, 13.1, 10.4, and 8.6% of the administered
dose, respectively. The distribution in infant monkeys was 35.6% in
muscle, 22.7% in skin, and only 4.5% of the dose in the liver.
Table 44. Tissue distribution of TCDD-derived radioactivity in Golden
Syrian hamsters at 1, 3, 10, and 20 days following a single oral
dose of 650 µg 3H-TCDD/kg body weighta
Tissue content of 3H (% of dose/g tissue)b
Tissue Day 1 Day 3 Day 10 Day 20
Liver 4.03±1.00 5.32±.82 3.19±.93 0.86±0.09
Liverb 12.74±3.21 20.44±3.45 9.69±0.99 3.70±0.29
Perirenal adipose 2.93±0.87 3.48±0.56 1.38±0.28 0.32±0.03
Adrenals 1.56±0.52 1.12±0.14 0.47±0.08 0.10±0.01
Pancreas 0.39±0.20 0.61±0.13 0.62±0.26 0.21±0.04
Kidneys 0.60±0.16 0.64±0.11 0.60±0.32 0.12±0.03
Spleen 0.30±0.08 0.24±0.05 0.43±0.26 0.07±0.02
Thymus 0.49±0.14 0.34±0.11 - 0.05±0.02
Skin 0.84±0.26 0.31±0.07 0.56±0.18 0.03±0.01
Stomach 0.34±0.07 0.55±0.09 0.65±0.39 0.16±0.06
Duodenum 0.51±0.13 0.47±0.09 0.55±0.28 0.07±0.02
Jejunum 0.59±0.15 0.71±0.20 0.39±0.16 0.08±0.02
Ileum 0.41±0.12 0.35±0.05 0.37±0.21 0.06±0.02
Colon 0.92±0.27 0.60±0.14 0.34±0.07 0.06±0.01
Caecum 0.39±0.11 0.41±0.10 0.28±0.12 0.05±0.01
Lungs 0.38±0.09 0.37±0.05 0.41±0.25 0.07±0.03
Skeletal muscle 0.20±0.07 0.15±0.05 0.15±0.03 0.04±0.02
Heart 0.14±0.03 0.13±0.02 0.15±0.08 0.03±0.01
Testes 0.10±0.04 0.32±0.13 0.13±0.04 0.03±0.01
Blood 0.12±0.02 0.14±0.03 0.12±0.06 0.02±0.01
Brain 0.03±0.01 0.05±0.01 0.06±0.02 0.01
a From: Olson et al. (1980a).
b All values are the mean (± standard error) of four hamsters.
c Percentage of dose/liver.
Table 45. Tissue distribution of TCDD-derived radioactivity in adult
and infant rhesus monkeys 7 days following a single intraperitoneal
dose of 400 µg 3H-TCDD/kg body weighta
(% of dose/tissue)
Tissue content of 3Hb
Tissue Adult Infant
Liver 10.4±6.9 4.51±1.60
Brain 0.58±0.34 1.41±1.40
Spleen 0.028±0.013 0.026±0.004
Small intestine 0.87±0.39 1.47±0.64
Large intestine 1.29±0.12 0.64±0.24
Musclec 8.62±2.39 35.6±14.4
Skin 13.1±4.9 22.7±8.8
Adipose tissued 16.2±5.8
a Fom: Van Miller et al. (1976).
b Mean (± standard deviation) of three adults or four infants.
c Total muscle was taken as 40% of body weight.
d Quantities in infant monkey were insignificant. For adult
monkeys, the calculation was based on an estimate of 300 g
mesenteric fat.
The concentration of TCDD in samples of faeces, urine, and fat
were measured by GC-MS at intervals after dosing up to 715 days in an
adult female rhesus monkey (Macaca mulatta) given a single oral
dose of 1 µg TCDD/kg body weight (McNulty et al., 1982b). During the
3 months after dosing the monkey lost 50% of its body weight but then
began to gain weight again. The level of TCDD in faeces was high for
4 days and then fell to very low or undetectable levels. TCDD in urine
was very low at all time points. The apparent half-life of TCDD in
adipose tissue was about 1 year.
There were no significant time-dependent changes in TCDD-derived
radioactivity found in the tissues investigated from marmosets during
the 3-week-period following subcutaneous treatment with 5 µg
14C-TCDD/kg body weight, except for a minor decrease in levels found
in the adipose tissue (Krowke, 1986). The data thus indicate a long
half-life for TCDD in marmosets.
6.1.6 Studies on dogs
A one-year-old male beagle dog received a total dose of 5.4 µg of
TCDD enterally by direct introduction into the duodenal lumen in four
portions of 1-2 µg, with intervals of 2-7 days between treatments
(Poiger et al., 1982). Severe toxic symtoms preceeded the death of the
animal 17 days after the first dose. The excretion of radioactivity in
the bile reached a maximum on day 1 or day 2 following administration.
Significantly more biliary radioactivity was found after
administration of doses 3 and 4 than after the administration of doses
1 and 2, suggesting that TCDD-administration stimulated its own
metabolism. It was later demonstrated in a 18-months-old male boxer
that TCDD-pretreatment (10 µg/kg body weight) stimulated the biliary
excretion of 3H-labelled TCDD (32.8 ng/kg body weight), whereas
phenobarbital-pretreatment had no effect when compared to the biliary
excretion without pretreatment (Poiger & Schlatter, 1985). All the
experiments were carried out in the same animal, which had enough time
between treatments for the radioactivity to return almost to the
background level.
6.1.7 Studies on cows
The major routes for elimination of 3H-TCDD in Holstein cows
(500-650 kg) after oral doses of 0.05µg (two cows) or 7.5µg (one cow)
TCDD/kg body weight were faeces > milk > urine (Jones et al., 1987).
Fifty percent of the administered dose was eliminated in faeces, the
major part in the first few days after treatment. Three lactating
Holstein cows received commercial technical grade pentachlorophenol
orally by gelatine capsule at a dose rate of 10 mg/kg body weight
twice daily for 10 days and once daily for the following 60 days
(Firestone et al., 1979). One cow served as a control and received
gelatine capsules containing only ground corn. The pentachlorophenol
composite used contained ten PCDD congeners (0.1 to 690 mg/kg) and
eight PCDF congeners (0.9 to 130 mg/kg). Faeces collected on day 28 of
the treatment period contained three hexaCDDs (0.05 to 0.63 µg/kg),
two heptaCDDs (21.3 to 33.1 µg/kg), and octaCDD (290 to 429 µg/kg).
Faeces also contained hexa-, hepta-, and octaCDF. Milk, body fat, and
blood contained only three of the PCDD congeners present in the
pentachlorophenol composite, namely 1,2,3,6,7,8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, and octaCDD. Milk samples also contained
hexa-, hepta-, and octaCDF. The average concentrations of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, octaCDD, and octaCDF in
the composite milk fat at the end of the treatment period were 20, 40,
25, and 2 mg/kg respectively. Similar concentrations were found in
body (shoulder) fat at the end of the treatment period (13, 24, and 32
mg/kg, respectively, of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD,
and octaCDD). Levels of dioxins in the blood were approximately 1000
times below the values in milk or body fat. The average daily
excretion of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD
in the milk during days 40-70 was about 20, 40, and 23 mg,
corresponding to 33, 3, and 0.6% of the daily intake of PCDDs. One
hundred days after the cessation of treatment, the average values for
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in shoulder
fat and milk fat were 2.5, 6.6, 5.6 mg/kg and 4.3, 6.9, 3.0 mg/kg,
respectively.
6.1.8 In vitro studies
The uptake of 3H-TCDD in human fibroblasts was less efficient
when the TCDD was associated with the high density lipoprotein (HDL)
than with the low density lipoprotein (LDL), and even less when
associated with serum (Shireman & Wei, 1986). From studies in mutant
human fibroblasts lacking the normal LDL cellular receptor, the
authors concluded that the LDL receptor pathway was involved in the
cellular uptake of TCDD. The uptake from LDL was time, temperature,
and concentration dependent.
6.2 Metabolic Transformation
6.2.1 Studies on mammals
6.2.1.1 In vivo studies
The possible existence of a urinary metabolite of TCDD was first
suggested by the finding of Allen et al. (1975) that the radioactivity
in urine of 14C-TCDD treated rats was highest from week 2 to 3. Later
both Poiger & Schlatter (1979) and Ramsey et al. (1982) presented
evidence for the in vivo biotransformation of TCDD in the rat.
Male Sprague Dawley rats were given two, four, or six daily oral doses
of approximately 15 µg 14C-TCDD/kg body weight, and the bile was
collected for 24 h following the last dose (Ramsey et al., 1982).
Using high pressure liquid chromatography, at least eight metabolites
of TCDD were found in the bile from these rats. Incubation of the bile
with ß-glucuronidase indicated the presence of glucuronide conjugates
among the metabolites. Poiger & Schlatter (1979) incubated similarly
the bile or the dialysate with glucuronidase/arylsulfatase, and the
dichloromethane-extractable radioactivity increased from 1.5% to 75%.
Their results indicate the elimination of TCDD-metabolites in the form
of water-soluble sulfate and glucuronide conjugates. 3H-TCDD
metabolites extracted from the bile of one-year-old beagle dogs with
ethanol (Poiger et al., 1982) were given to bile-duct-cannulated
female Sprague Dawley rats (250 g) as single oral doses of 7.8-20.8 µg
3H-TCDD-metabolites/kg body weight (Weber et al., 1982b). The mean
24-h elimination of radioactivity was 86.7 (± 6.7)% of the dose, 8.2%
occurring in urine, 31.3% in bile, and 46.9% in faeces. A delay in the
excretion of radioactivity in rats whose bile ducts were not
cannulated suggested an enterohepatic circulation in the rat of the
3H-TCDD-metabolites from the dog. The radioactive material in the
rat bile seemed to be conjugated forms of the metabolites from the
dogs. A metabolic breakdown scheme of TCDD in the rat and dog (Fig. 3)
was proposed by Poiger & Buser (1983). The major metabolite seems to
be formed via cleavage of an ether bond.
The metabolic fate of a single ip dose of 10 µg, 500 µCi TCDD/kg
body weight in male C57Bl/6, DBA/2, and B6D2F1 mice was studied by
Gasiewicz et al. (1983b). Samples of urine, bile, and faeces,
collected on days 5 to 8, 8, and 7 after treatment, respectively, were
extracted and analyzed for metabolites of TCDD by HPLC. Unmetabolized
TCDD was detected in faeces but not in urine or in bile. More than 85%
of the total radioactivity eliminated was present as metabolites of
TCDD in all three mouse strains. Metabolites in the bile appear to be
less polar than in urine. Qualitatively the elution profiles for
urine, bile, and faeces from all three strains appeared to be quite
similar.
A dose of 3H-TCDD (0.56 µg/kg body weight) that produced no
gross toxicity was given ip in olive oil to six adult male Hartley
guinea-pigs (Olson, 1986). Metabolites of TCDD were found in organic
extracts of the liver, kidney, perirenal adipose tissue, and skeletal
muscle in amounts corresponding to 13, 4, 8, and 28%, respectively, of
the recovered radioactivity in these organs 45 days after dosing.
These figures suggest that TCDD-metabolites are not efficiently
eliminated from tissues in guinea-pigs. All radioactivity in urine and
bile represented metabolites of TCDD, whereas in faeces most (70-90%)
contained unchanged TCDD. Of the radioactivity administered, 73.4% was
eliminated as unchanged TCDD in faeces and 25.7% as metabolites of
TCDD in urine and faeces. The presence of TCDD in faeces and its
absence in bile suggest that direct elimination of TCDD from the blood
to the intestinal lumen may occur. The HPLC elution profiles for
metabolites were similar, although not identical, for bile, urine, and
tissues. Taken together, the data by Olson (1986) indicate that
metabolism does not appear to have a major role in the ultimate
elimination of TCDD in the guinea-pig.
Olson et al. (1980a) collected bile and urine from Golden Syrian
hamsters that had been treated with a single ip dose of 650 µg
14C-TCDD/kg body weight 7 days earlier. By means of HPLC, one major
and several minor metabolites of 14C-TCDD were demonstrated both in
bile and urine. No metabolites of 14C-TCDD were detectable in liver
and adipose tissue, thus suggesting a rapid clearance of
biotransformed products of TCDD.
A one-year-old beagle dog was cholecystectomized and a Thomas
cannula was implanted about 3 months before the first dose of TCDD
(Poiger et al., 1982, Poiger & Buser, 1983). A total dose of 5.4 mg
was administered enterally in four portions of 1.8, 1.08, 1.08, and
1.44 mg on days 0, 2, 7, and 13. Five phenolic metabolites of TCDD
excreted in dog bile were identified by combined gas
chromatography-mass spectrometry. Severe toxic symptoms preceded the
death of the dog 17 days after the first dose. A metabolic breakdown
scheme of TCDD in the dog (Fig. 3) was proposed by Poiger & Buser
(1983), lateral hydroxylation of TCDD seeming to be the major route of
metabolism.
Table 4. Physical properties of some PCDDs
Molecular Molecular Absorption
Compound formulae weight maximum Reference
(chloroform)
(nm)
2,3,7,8-TCDD C12H4Cl4O2 321.9 310 Pohland &
Yang (1972)
1,2,3,7,8-PentaCDD C12H3Cl5O2 356.5 308 Gray et al.
(1976)
1,2,3,6,7,8-HexaCDD C12H2Cl6O2 390.9 316 Gray et al.
(1975)
1,2,3,7,8,9-HexaCDD C12H2Cl6O2 390.9 317 Gray et al.
(1975)
Although tetraCDD is lipophilic, it is only slightly soluble in
most solvents and very slightly soluble in water (Table 5).
Table 5. Solubility of 2,3,7,8-tetraCDD in various solventsa
Solvent Solubility at 25 °C
g/litre g/kg
O-Dichlorobenzene 1.8 1.4
Chlorobenzene 0.8 0.72
Perchloroethylene 0.68 0.48
Chloroform 0.55 0.37
Benzene 0.47 0.57
Acetone 0.09 0.11
Dimethylsulfoxideb < 0.1 < 0.1
Methanol 0.01 0.01
Water 2 x 10-7 2 x 10-7
a From: Crummett & Stehl (1973).
b DMSO caused detector fouling and a better value could not be obtained.
Table 6. Water solubility of PCDDsa
Compound Water solubility (g/litre)
20.0 °C 40.0 °C
1,3,6,8-TetraCDD (3.2±0.2) x 10-7 (3.9±0.4) x 10-7
1,2,3,7-TetraCDD (4.3±0.1) x 10-7 (12.7±0.8) x 10-7
1,2,3,4,7-PentaCDD (1.2±0.1) x 10-7 (4.6±0.1) x 10-7
1,2,3,4,7,8-HexaCDD (4.4±0.1) x 10-9 (19.0±0.1) x 10-9
1,2,3,4,6,7,8-HeptaCDD (2.4±0.3) x 10-9 (6.3±0.2) x 10-9
OctaCDD (0.4±0.1) x 10-9 (2.0±0.2) x 10-9
a From: Friesen et al. (1985).
Marple et al. (1986a) have reanalysed the water solubility of
2,3,7,8-TCDD and found it to be considerably less (12.5-19.2
ng/litre). The log water-octanol partition coefficient (Kow) has
been determined as 6.64 by Marple et al. (1986b).
Friesen et al. (1985) have determined the water solubility for
some PCDDs other than the 2,3,7,8-TCDD compound and these are given in
Table 6.
Similarly Webster et al. (1985) have determined the log
octanol-water partition coefficients for a number of PCDDs (Table 7).
2,3,7,8-TetraCDD is considered to be a stable compound, but due
to its extreme toxicity its chemistry has not been fully evaluated.
However, it undergoes substitution reactions (Baughman, 1974) as well
as photochemical dechlorination (Crosby et al., 1971; Crosby & Wong,
1977; Gebefugi et al., 1977). Thermally it is very stable and rapid
decomposition of 2,3,7,8-tetraCDD occurs only at temperatures above
750 °C (Stehl et al., 1973). The other PCDDs have been much less
studied; however, octaCDD is completely destroyed by treatment with
hot alkali (Albro & Corbett, 1977).
The first synthesis of 2,3,7,8-tetraCDD was reported by
Sandermann et al. (1957), who used catalytic chlorination of the
unchlorinated dioxin. It has also been prepared in good yields by the
dimerization of 2,4,5-trichlorophenol salts (Buu-Hoi et al., 1971b;
Langer et al., 1973).
In the PCDF series, Mazer et al. (1983) synthesized all the 38
positional tetraCDF isomers. The products were mixtures of isomers,
and each of these isomers could be identified. Later Bell & Gari,
(1985) isolated and characterized all the 38 tetraCDFs, 28 pentaCDFs,
and 16 hexaCDFs.
Table 7. Values for log Kow for some PCDDs from linear and quadratic plots
log Kow (linear) log Kow (quadratic)
Waters Waters Waters Waters
Bondapak Bondapak Bondapak Bondapak
Compound (Woodburn data) (Woodburn data)
Dibenzo-p-dioxin 4.26 4.01 4.34 4.17
1-MonoCDD 4.81 4.52 4.91 4.75
2-MonoCDD 5.33 5.00 5.45 5.29
2,7-DiCDD 6.27 5.86 6.39 6.17
1,2,4-TriCDD 7.36 6.86 7.45 7.11
1,2,3,7-TetraCDD 8.15 7.58 8.19 7.72
1,2,3,4-TetraCDD 8.63 8.02 8.64 8.07
1,3,6,8-TetraCDD 8.70 8.08 8.70 8.12
1,2,3,4,7-PentaCDD 9.48 8.80 9.40 8.64
1,2,3,4,7,8-HexaCDD 10.40 9.65 10.22 9.19
1,2,3,4,6,7,8-HeptaCDD 11.38 10.55 11.05 9.69
OctaCDD 12.26 11.35 11.76 10.07
a From: Webster et al. (1985).
Kuroki et al. (1984) have synthesized 51 congeners of PCDFs by a
structure specific method from chlorophenols and chloronitrobenzenes
or chlorophenols and chlorodiphenyls iodonium salts. The structures
were confirmed by MS and NMR.
Safe & Safe (1984) described the synthesis of 22 PCDF congeners
resulting in quantities of 10-320 mg of purified product. They also
reported NMR data on the compounds synthesized.
Sarna et al. (1984) and Burkhard & Kuehl, (1986) have documented
the octanol/water partition coefficients for some PCDFs (Table 8). The
disagreement for OCDF arises because of uncertainties in the Kow
values of reference compounds of high Kow. The partitioning of
organic chemicals between lipid and water is an important determinant
of the bioconcentration potential of a toxicant and has sometimes been
effectively used as an indicator of the preferred degradative in in
vivo pathways.
Table 8. The logarithm of the octanol/water partition coefficients (Kow) of some PCDFs using HPLC methods
PCDF log Kow Reference
2,8-dichloro- 5.95 Sarna et al. 1984a
5.30b Burkhard & Kuehl, 1986c
2,3,7,8-tetrachloro- 5.82±0.02 Burkhard & Kuehl, 1986c
octachloro- 13.37 Sarna et al. 1984a
8.78 Burkhard & Kuehl, 1986c
a Quadratic equation treatment: Biorad Biosil (10 mm) data.
b Quadratic equation treatment: unspecified "microbore" HPLC column.
c Sarna et al. (1984) data recalculated from experimental data.
2.3. Analytical Methods
2.3.1 General aspects
The earliest reported method used to detect 2,3,7,8-tetraCDD was
a rabbit skin test (Adams et al., 1941). Test samples were applied to
the inner surface of the ear and to the shaven belly of albino
rabbits, and inflammatory responses were observed. Subsequently, Jones
& Krizek (1962) developed a test based on the recovery and weight of
the keratin formed on the rabbit ear after application of a sample.
These biological methods were non-specific as to isomers and not
sufficiently sensitive to detect low levels of contamination.
In the late 1960s and early 1970s, gas chromatographic methods
were used for the quantification mainly of 2,3,7,8-tetraCDD in
commercial 2,4,5-T formations. The detection level was normally in the
range of µg/g. These analyses were not isomer-specific and the results
could not be confirmed. Ryhage (1964) solved the problem of combining
a gas chromatograph with a mass spectrometer. During the 1970s and
1980s, various types of mass spectrometer and gas chromatograph/mass
spectrometer combinations were used in analytical work. Use of these
more sophisticated instruments allowed for the development of
isomer-specific and validated analyses for the tetraCDDs in the very
late 1970s and for the other PCDDs and PCDFs in the early 1980s.
A number of spectroscopic methods are available for the
laboratory identification of 2,3,7,8-tetraCDD, but their use is highly
restricted, with the exception of mass spectroscopy (MS). Data on
X-ray, infra-red (IR), ultra-violet (UV), nuclear magnetic resonance
(NMR), electron spin resonance (ESR), and mass spectra were obtained
by Pohland & Yang (1972), Baughman (1974), and Slonecker et al.
(1983).
Because of the large number of isomers and congeners, and due to
the extreme toxicity of some PCDD and PCDF isomers, highly sensitive
and specific analytical techniques are required for the measurements.
Detection limits for the analysis of environmental and human samples
should be orders of magnitude lower than the usual detection levels
required for pesticide analysis. A detection level of 1 pg or less
might be required to measure 2,3,7,8-tetraCDD and the other toxic
isomers in a 1-g environmental sample. Analyses at such low levels are
complicated by the presence of a multitude of other interfering
compounds and clean-up procedures are required.
The mono-, di-, and trichloro congeners are not usually included
in these analyses. Such compounds are considered to be much less toxic
than the higher chlorinated congeners and are also much more volatile
and losses may occur during clean-up.
It should be mentioned that the level of sophistication needed in
the analyses for PCDD and PCDFs will depend upon the objectives
thereof. In cases where the objectives were primarily to screen
samples to identify groups of PCDDs and/or PCDFs (in a qualitative or
semiquantitative manner), routine assays and bioassays were adequate.
In other instances, where the objective of the analysis was to
quantify accurately specific PCDD and/or PCDF isomers in the samples,
sophisticated analytical procedures were required. Clearly, both types
of analyses can be useful, depending on the purpose for which the
analytical results are to be used.
Many analytical methods have been developed in recent years for
the analysis of trace amounts of PCDDs and PCDFs in environmental
samples, especially for 2,3,7,8-tetraCDD. The most specific of these
methods are based on MS. There are many requirements to be met by such
an analytical method, including representative sampling and
appropriate storage, efficient extraction, high selectivity in the
clean-up, high specificity in the gas chromatography, high sensitivity
in the detection, safe and reliable quantification, good
reproducibility, useful confirmatory information.
Several review articles discussing methods of analyzing PCDDs and
PCDFs have appeared (McKinney, 1978; Esposito et al., 1980; Rappe &
Buser, 1980; Harless & Lewis, 1982; Karasek & Anuska, 1982; Tiernan,
1983; Crummett et al., 1985). Most of the older methods have been
critically reviewed by a panel of experts assembled by the National
Research Council of Canada (1981).
2.3.2 Sampling strategy and sampling methods
The quality and utility of analytical data depend on the validity
of the sample and the adequacy of the sampling program. The purpose of
sampling is to obtain specimens that represent the situation being
studied. Sampling plans may require that systematic samples be
obtained at specified times and places, or simple random sampling may
be necessary. Generally, the sample should be an unbiased
representation of the environmental situation.
All aspects of a sampling programme should be planned and
documented in detail, and the expected relationship of the sampling
protocol to the analytical result should be defined. A sampling
programme should include reasons for choosing sampling sites, the
number and type of samples, the timing of sample acquisition and the
sampling equipment used. A detailed sampling procedure should include
a description of the sampling situation, the sampling methodology,
labelling of samples, field blank preparation, pretreatment
procedures, and transportation and storage procedures.
The quality assurance programme should include means to
demonstrate that containers or storage procedures do not alter the
qualitative or quantitative composition of the sample. Special
transportation and storage procedures (refrigeration or exclusion of
light) should be described, if they are required.
Because environmental samples are typically heterogeneous, a
sufficiently large number of samples (ten or more) must normally be
analyzed to obtain meaningful data on chemical composition. The number
of individual samples that should be analyzed will depend on the kind
of information required by the investigation. If an average
compositional value is required, a number of randomly selected
individual samples may be obtained, combined, and blended to provide
a homogeneous composite sample from which a sufficient number of
subsamples could be analyzed. If composition profiles, time trends, or
the variability of the sample population are of interest, many samples
need to be collected and analyzed individually.
If field blanks are not available, efforts should be made to
obtain blank samples that best simulate a sample that does not contain
the specific chemical. In addition, measurements should be made to
ascertain whether, and to what extent, any reagent or solvent used may
contribute to or interfere with the analytical results (laboratory and
solvent blanks).
The recovery tests are frequently used and necessary to evaluate
the analytical methodology. Uncontaminated samples from control sites
that have been spiked with the chemical of interest provide the best
information because they simulate any matrix effect. When feasible,
isotopically labelled (13C, 37Cl) chemicals spiked into the sample
provide the greatest accuracy since they are subjected to the same
matrix effects. The 13C- and 37Cl-labelled compounds can be used
to validate:
(a) sampling (sampling surrogate),
(b) analytical pretreatment (clean-up surrogate),
(c) quantification (internal standard).
Very few laboratories in the world have access to and experience
in working with these complicated analyses.
In order to be able to compare data generated in different
laboratories, the same quantitative standard compounds should be used.
Interlaboratory calibrations or "round-robin" studies have been
performed in very few cases.
2.3.3 Extraction procedures
In this step, the sample is homogenized or digested and extracted
with a suitable solvent or solvent mixture to remove the bulk of the
sample matrix and transfer the PCDD and PCDF residue into the solvent.
Both the selection of the proper solvent and the method of extraction
can be critical in obtaining a satisfactory recovery of PCDDs and
PCDFs from the sample matrix.
Many different procedures for the extraction of PCDDs/PCDFs from
various samples are described. In some cases this involves digestion
or destruction of the matrix. Some of these methods have been
evaluated in the report from the National Research Council of Canada
(1982), while other methods are discussed by Tiernan (1983).
An interlaboratory "round-robin" study involving 13 laboratories
was carried out to evaluate the reliability of data on
2,3,7,8-tetraCDD in fish samples. No significant differences were
found from methods differing in the digestion or extraction procedures
(Ryan et al., 1983b).
In a study described by Albro et al. (1985), eight different
approaches were applied in eight laboratories to quantify four PCDDs
(2,3,7,8-tetraCDD; 1,2,3,7,8-pentaCDD; 1,2,3,4,7,8-hexaCDD; and
octaCDD) and three PCDFs (2,3,7,8-tetraCDF; 2,3,4,7,8-pentaCDF; and
1,2,3,7,8,9-hexaCDF) in spiked samples of an extract from human
adipose tissue. Levels of fortification, unknown to the participating
laboratories, were in the 5-50 ng/kg range, except for octaCDD (up to
500 ng/kg). The results indicated that most of the procedures tested
gave a high degree of qualitative reliability. However, other methods
were not so accurate, a large portion of the reported data consisting
of false positives or false negatives.
Lustenhouwer et al. (1980) studied the extraction of PCDDs and
PCDFs from a fly ash sample. A dramatic difference was found between
different solvents.
2.3.4 Sample clean-up
In the sample clean-up, the PCDDs and PCDFs present in the sample
should be separated from a multitude of other co-extracted and
possibly interfering compounds. The clean-up methods, normally three
steps or more, can vary for different sample matrices. Two different
procedural trends can be recognized:
(a) all PCDD and PCDF isomers can be analyzed in one
single fraction by the containment enrichment
procedure (Norstrom et al., 1982; Stalling et al.,
1983; Tiernan, 1983; Rappe, 1984),
(b) specific isomers are analyzed in different fractions
mainly after normal-phase and reverse-phase high
pressure liquid chromatography (HPLC) separation
(Lamparski et al., 1979; Niemann et al., 1983; Tosine
et al., 1983).
This latter method allows the identification of only a few PCDD
isomers in each fraction, and is mainly used to monitor TCDD and a few
other congeners. For a monitoring program of all PCDDs and PCDFs a
more general method might be preferred.
The method described by Stalling et al. (1983) was originally
designed for the analyses of fish samples. In a "round-robin" study of
fish samples it gave good results (Ryan et al., 1983b). This method
has now been used for the clean-up of other biological samples like
bird muscle, seal fat, turtle fat, and human adipose tissue - blood,
liver, kidney, and milk (Rappe et al., 1983c; Nygren et al., 1986;
Rappe et al., 1986b).
2.3.5 Isomer identification
The purified extracts are used directly for the final analyses
with the aid of a gas-chromatograph/mass spectrometer (GC/MS) equipped
with a glass capillary or a fused-silica column. The column leads
directly into the ion source of the mass spectrometer, which operates
either in the electron impact (EI) or the negative ion-chemical
ionization (NCI) mode. In view of the large variation in toxicological
and biological effects of the PCDD and PCDF isomers, it is imperative
that the isomers, particularly those having high toxicity, be
identified. For an unambiguous isomer identification it is necessary
to have access to all analytical standards within a specific group of
isomers, e.g. all the 22 tetraCDDs and all the 38 tetraCDFs. All the
22 tetraCDDs have been prepared and, using a Silar 10c glass capillary
column, the 2,3,7,8-tetraCDD can be separated from all the other 21
tetra isomers (Buser & Rappe, 1980). Recently all the 14 pentaCDDs and
the 10 hexaCDDs have been prepared. Using the Silar 10c column all the
2,3,7,8- substituted isomers can be separated from all the other
isomers (Buser & Rappe, 1984). The SP 2330 fused silica column can
also be used for this separation (Rappe, 1984).
In the PCDF series, Mazer et al. (1983) have synthesized all the
38 positional tetraCDF isomers. The products were mixtures of isomers,
and each of these isomers could be identified using both an SP 2330
and an SE 54 capillary column. Later, Bell & Gara (1985) isolated and
characterized all tetra-, penta- and hexaCDFs. The SP 2330 column can
separate most of these isomers (Rappe, 1984). The 1,2,3,7,8-pentaCDF
co-elutes with the 1,2,3,4,8-isomer and the 1,2,3,4,7,8- hexaCDF with
the 1,2,3,4,7,9-isomer, but they can be separated on less polar
columns like OV-17 and DB-5.
A very limited number of investigations has been performed using
these complete sets of synthetic standards.
2.3.6 Quantification
Mass selective detection (mass fragmentography) has been used to
quantify trace amounts of PCDDs and PCDFs in the samples by
selectively monitoring M, M + 2, and/or M + 4 ions (SIM). The
quantification is based on peak area measurements and a comparison of
these areas using either isotopically labelled internal standards
(13C or 37Cl) or calibration curves of external standards. As a
first approach, it has been generally assumed that with the MS
quantification technique, all isomers of a particular congener of PCDD
or PCDF (e.g. the tetrachloro-isomers) have the same response factors.
However, an investigation of 13 well-defined tetraCDF isomers has
shown a three-fold variation in response factors with the EI mode and
up to a 20-fold variation with the negative ion-chemical ionization
mode. For the higher chlorinated homologues (penta, hexa) the
variation was found to be less (Rappe et al., 1983b).
Fung et al. (1985) have studied the mass spectra of 26 PCDF
congeners. They found that the EI spectra are not particularly isomer
specific, while positive ion-chemical ionization spectra show a
greater degree of isomer distinction.
2.3.7 Confirmation
Quality control and quality assurance programs help to assure
that positive data reported actually refer to specific PCDDs and PCDFs
(Kloepfer et al., 1983). To provide reliable data:
(a) isomer specificity must be demonstrated initially and verified
daily,
(b) the retention time must equal (within 3 seconds) the retention
time for the isotopically labelled congener,
(c) the signal to noise ratio must be 2.5:1 or higher,
(d) the chlorine cluster must be within ± 10% of the theoretical
values, given in Table 9,
(e) correct fragments, e.g., M+-COCl ions, must be with correct
chlorine clusters.
For confirmation, mass spectroscopy is the best technique now
available. The EI mass spectral properties of PCDFs and PCDD have been
described (Buser, 1975). The molecular (M+) and fragment ions of
PCDDs and PCDFs show the typical, expected clustering due to the
chlorine isotopes (Table 9). The typical fragmentation is M-COCl+,
which is a useful fragment to study.
Buser & Rappe (1978) have shown that observation of low mass ions
can be used for the identification of the substitution pattern of
PCDDs, which can be defined as the number of chlorine atoms on each
carbon ring of the dioxin molecular; the 2,3,7,8-isomer has a 2:2
pattern while 1,2,3,4-tetraCDD has a 4:0 pattern. However, these low
mass ions may not be observed in spectra from environmental or
biological samples.
In the negative ion-chemical ionization mode, the PCDFs have the
base peak due to M-, and the fragmentation produces the unusual
M--34 ions (uptake of H and loss of Cl). Fragmentation of PCDDs in
this mode is more conventional via loss of Cl yielding M--35 ions
(Buser et al., 1985).
Using EI technique and a quadropole instrument, the detection
limits are 1-10 pg for the tetrachloro compounds and up to 10-50 pg
for the octachloro compounds using selected ion monitoring or multiple
ion detection (SIM or MID). Full mass spectra require 0.1-1 ng of
compound (Buser et al., 1985). High resolution instruments can improve
the sensitivity by one order of magnitude.
The negative ion-chemical ionization mode, using methane gas as
reagent, gas provides extremely good sensitivity for all PCDFs (tetra-
to octachloro- compounds) and for the higher chlorinated PCDDs (penta-
to octaCDD). The detection limits are in the 10-100 fg (10-15g)
range using SIR or MID, which is 1 to 2 orders of magnitude better
than EI (Buser et al., 1985). However, the negative ion-chemical
ionization mode has very poor sensitivity for 2,3,7,8- tetraCDD under
these conditions.
Using low resolution MS instruments, a series of interfering
compounds has been identified (Table 10). Some of this interference
can be eliminated using high resolution MS instruments operating at
8000 - 10 000 daltons. However, compounds with the same empirical
formulae cannot be separated by MS technique; they are normally
eliminated during the clean-up or separated by the gas chromatography
step.
2.3.8 Other analytical methods
Paasivirta et al. (1977) have shown that 2,3,7,8-tetraCDD can be
detected down to the pg level using a glass capillary column and a
63Ni electron-capture detector. Combined with efficient clean-up
procedures, this method has shown to be useful down to a level of 9
ppt (Niemann et al., 1983), although positive samples need
confirmation by mass spectroscopy (MID, SIM).
Other techniques, such as enzyme induction and radioimmunoassay
have been described and discussed by Firestone (1978) and McKinney
(1978). McKinney et al. (1982) have used the radioimmunoassay method
for determining 2,3,7,8-tetraCDD in human fat, and found the reliable
sensitivity at 95% confidence interval to be 100 pg per sample.
An analytical method based on the keratonization response of
epithelial cells in an in vitro system has been described by
Gierthy & Crane (1985b). This method can be an assay for dioxin-like
activity in environmental and biological samples. A positive response
was found for 2,3,7,8-tetraCDD at a concentration of 10-11 mol/litre.
Table 9. Isotopic abundance ratio ("cluster") of polychlorinated dioxins and dibenzofurans
Number of
chlorine M M + 2 M + 4 M + 6 M + 8 M + 10 M + 12 M + 14
atoms
1 100.0 33.7
2 100.0 66.1 11.3
3 100.0 98.4 32.7 3.8
4 76.4 100.0 49.4 11.0 1.0
5 61.2 100.0 65.5 21.6 3.6 0.3
6 51.1 100.0 81.7 35.8 8.9 1.2 0.1
7 43.8 100.0 97.9 53.4 17.6 3.5 0.4
8 33.7 87.6 100.0 65.3 26.8 7.0 1.2 0.1
Table 10. List of molecular ions of polychlorinated compounds present in some human and environmental samples
and possibly interfering in the mass spectral analysis of PCDFs and PCDDsa
Molecular ions (m/z,m+,m-) (chlorination)
Compounds mono- di- tri- tetra- penta- hexa- hepta- octa- nona- deca-
PCDDs 320 354 388 422 456 - -
PCDFs 304 338 372 406 440 - -
PCBs 290 324 358 392 426 460 494
PCNs 264 298 332 366 400 - -
PCTs 298 332 366 400 434 468 502 536 570
PCDPEsb 238 272 306 340 374 408 442 476 510
PCPYsc 36 270 304 338 372 406
a From: Buser et al. (1985).
b PCDPEs: Polychlorinated diphenylethers.
c PCPYs: Polychlorinated pyrenes.
3. SOURCES OF ENVIRONMENTAL POLLUTION
3.1 Production, Synthesis, and Use
PCDDs and PCDFs are not produced commercially. These compounds
are in fact formed as trace amounts of undesired impurities in the
manufacture of other chemicals such as chlorinated phenols and their
derivatives, chlorinated diphenyl ethers, and polychlorinated
biphenyls (PCBs). There is no known technical use for the PCDDs and
PCDFs.
The amount of total PCDDs entering the Canadian environment/year
has been estimated to be about 1500 kg, and 75% of this amount has
been estimated to be due to octaCDD alone (National Research Council
of Canada, 1981). There is no estimation of the amount of PCDFs
entering the environment anywhere in the world.
Although the polychlorinated dioxins and dibenzofurans are not
commercially produced, most of these compounds have been synthesized
for research purposes in small quantities according to the reactions
discussed in section 2.
3.2 Industrial Processes
In addition to the synthetic methods mentioned in section 2,
2,3,7,8-tetraCDD may be formed during the industrial preparation of
2,4,5-trichlorophenol from 1,2,4,5-tetra-chlorobenzene. This
substitution reaction takes place at about 180 °C, and when the
solvent is methanol, the pressure rises to about 7 KPa. The formation
of TCDD is an unwanted side reaction which takes place when the
reaction mixture is heated to 230-260 °C (Milnes, 1971). This reaction
is exothermic, so that even higher temperatures may be attained
resulting in uncontrolled conditions.
In some factories ethylene glycol is used as a solvent in order
to avoid the high pressure. As already pointed out by Milnes (1971),
however, use of this solvent requires special precautions because of
the occurrence of a base-promoted polymerization of ethylene glycol
and decomposition reactions that produce ethylene oxide. These
reactions are also exothermic; they may start spontaneously at
temperatures above 180 °C and proceed rapidly and uncontrollably to
result in the formation of relatively large amounts of TCDD.
After most of the solvent has been removed, the reaction mixture
is acidified; the 2,4,5-trichlorophenol can be separated from
2,3,7,8-tetraCDD by one or two distillations, with the result that
2,3,7,8-tetraCDD is concentrated in the still-bottom residues. Up to
1 mg/g of 2,3,7,8-tetraCDD in such residues has been reported
(Kimbrough et al., 1984). Improper disposal of such residues is
discussed in sections 4.4.2 and 9.
Most of the 2,4,5-trichlorophenol produced is used for the
preparation of herbicides such as 2,4,5-T (including various esters
and salts, and the bactericide hexachlorophene).
PCDDs and PCDFs are both formed as by-products during the
manufacture of chlorinated phenols (2,4-dichloro-, 2,4,6-trichloro-,
2,3,4,6-tetrachloro- and pentachlorophenol). The commercial
chlorophenols are produced by two processes, i.e., by chlorination of
the phenol using various catalysts and by the alkaline hydrolysis of
an appropriate chlorobenzene. Apparently both reactions can lead to
the formation of PCDDs as well as PCDFs, and the level of
contamination is normally much higher here than in the production of
2,4,5-trichloro-phenol (see section 3.3).
PCDDs and PCDFs are also formed during the preparation of
chlorinated diphenyl ether herbicides (Yamagishi et al., 1981) and
hexachlorobenzene (Villeneuve et al., 1974). A series of PCDFs are
formed during the production of PCBs (see section 3.3).
Production equipment is often used for the production of several
different chemicals. In the manufacture of chemicals on such equipment
previously contaminated by PCDDs and PCDFs, both the products and
waste generated can be contaminated. Thus, manufactured
2,4-dichlorophenoxyacetic esters (2,4-D), which otherwise should not
be contaminated by 2,3,7,8-tetraCDD, did indeed contain this dioxin
because the equipment used had been employed previously to produce
2,4,5-T and had not been cleaned properly (Federal Register, 1980).
It should be pointed out that the primary occurrence of TCDD in
the environment is possibly related to the synthesis of
2,4,5-trichlorophenol, the use of products prepared from this compound
(Table 11), and to incinerations reactions. The occurrence of the
other PCDDs and PCDFs is related to the synthesis and use of a variety
of other products (Table 12), some of which are quite common.
The other PCDDs and PCDFs are also formed in a variety of
incineration reactions (see section 4.5).
3.3 Contamination of Commercial Products
3.3.1 Chlorophenoxyacetic acid herbicides
Depending on the temperature control and purification efficiency,
the levels of 2,3,7,8-tetraCDD in commercial products may vary
greatly. For example, the levels of 2,3,7,8-tetraCDD in drums of the
herbicide Agent Orange placed in storage in the USA and in the Pacific
before 1970 were between 0.02 and 47 mg/g. More than 450 samples were
analyzed in this study, and the mean value was 1.98 mg/g (Young et
al., 1983). Since Agent Orange was formulated as a 1:1 mixture of the
butyl esters of 2,4,5-T and 2,4-D, the levels of 2,3,7,8-tetraCDD in
individual 2,4,5-T preparations manufactured and used in the 1960s
could have been as high as 100 mg/g.
In analyses using high-resolution GC-MS, Rappe et al. (1978a)
have reported that in other samples of Agent Orange (as well as in
European and the USA 2,4,5-T formulations from the 1950s and 1960s),
2,3,7,8-tetraCDD was the dominating compound of this group of
contaminants. Only minor amounts of other PCDDs and PCDFs could be
found, primarily lower chlorinated PCDDs, in samples of Agent Orange.
As a result of governmental regulations, efforts were made during
the 1970s to minimize the formation of 2,3,7,8-tetraCDD during 2,4,5-T
production, and now all producers claim that their products contain
less than 0.1 µg 2,3,7,8-tetraCDD/g of product (Rappe et al., 1978a).
At present, the chloro-phenoxy herbicides are not the major source of
PCDDs and PCDFs in the environment.
Sixteen samples of 2,4-D esters and amine salts from Canada were
analyzed for the presence of PCDDs. Eight out of nine esters and four
out of seven amine salts were found to be contaminated, with the
esters showing significantly higher levels (210-1752 ng/g) than the
salts (20-278 ng/g). The tetraCDD observed was the 1,3,6,8-isomer, as
verified by a synthetically prepared authentic standard (Cochrane et
al., 1981). In other studies, it has been found that no tetraCDD other
than the 1,3,6,8-isomer elutes in this window. Hagenmaier et al.
(1986) has reported that, unexpectedly, a German 2,4-D formulation
contained 6.8 ng of 2,3,7,8-tetraCDD/g.
Table 11. Some commercial products that may be contaminated with
2,3,7,8-tetraCDD, depending on the method of preparation
Common name Chemical name
2,4,5-Ta 2,4,5-Trichlorophenoxyacetic acid
2,4,5-T estersa n-butyl-, butoxy ethyl-, and
iso-octyl-esters of 2,4,5-
trichlorophenoxyacetic acid
2,4,5-T saltsa dimethylamine salts of 2,4,5-
trichlorophenoxyacetic acid
Fenoprop esters of 2-(2,4,5-trichlorophenoxy)-
propanoic acid
Erbon ethyl ester of 2-(2,4,5-trichloro-
phenoxy)-2,2-dichloropropanoic acid
2,4,5-Trichlorophenol 2,4,5-Trichlorophenol
Fenochlorphos O,O-Dimethyl O-2,4,5-trichlorophenyl
phosphonothioate
Trichloronate O-Ethyl 0-2,4,5-trichlorophenyl
ethylphosphonothioate
Hexachlorophene/isobac 20 2,2'-Methylene-bis (3,4,6-trichloro-
phenol)
a There are numerous trade names for this product.
Table 12. Some commercial products which may be contaminated with PCDDs
other than 2,3,7,8-tetraCDD, and with PCDFs, depending on the method of
preparation
Common name Chemical name
Bifenox Methyl-5-2,4-dichlorophenoxy-2-nitrobenzoate
Chloranil 2,3,5,6-Tetrachloro-2,
5-cyclo-hexadiene-1,4-dione.
2,4-D (esters and salts) 2,4-Dichlorophenoxyacetic acid
and esters and salts
2,4-DB and salts 2,4-Dichlorophenoxybutyric acid and
salts
Dicamba 3,6-Dichloro-2-methoxybenzoic acid
Dicamba, dimethylamine salt 3,6-Dichloro-2-methoxybenzoic acid,
dimethylamine salt
Dicapthon Phosphorothioic acid
o-(2-chloro-4-nitrophenyl)
o,o-dimethyl ester
Dichlofenthion Phosphorothioic acid
o-2,4-dichloro-phenyl
o,o-dialkyl ester
Disul sodium (sesone) 2,4-Dichlorophenoxyethyl sulfate,
sodium salt
2,4-DP 2- 2,4-Dichlorophenoxy propionic acid
HCB Hexachlorobenzene
Nitrofen 2,4-Dichlorophenyl-p-nitrophenyl
ether
PCP and salts Pentachlorophenol and salts
PCB Polychlorinated biphenyls
2,4,6-TCP 2,4,6-Trichlorophenol and salts
2,3,4,6-Tetrachlorophenol and salts
Common name Chemical name
CNP 1,3,5-Trichloro-2-(4-nitrophenoxy)
benzene
NIP 2,4-Dichloro-1-(4-nitrophenoxy)
benzene
X-52 2,4-Dichloro-1-(3-methoxy-4-nitro-
phenoxy) benzene
3.3.2 Hexachlorophene
The bactericide hexachlorophene is prepared from
2,4,5-trichlorophenol, also the key intermediate in the production of
2,4,5-T. Due to additional purification, the level of 2,3,7,8-tetraCDD
in this product is usually < 0.03 mg/kg (Baughman, 1974). Ligon & May
(1986) reported 0.0047 mg/kg of TCDD in one hexachlorophene sample.
However, hexachlorophene also contains about 100 mg/kg of a
hexachloroxanthene, the 1,2,4,6,8,9-substituted isomer (Göthe &
Wachtmeister, 1972).
3.3.3 Chlorophenols
Chlorophenols have been used extensively since the 1950s as
insecticides, fungicides, mold inhibitors, antiseptics, and
disinfectants. In 1978 the annual world production was estimated to be
approximately 200 000 tons. The most important use of 2,4,6-tri-,
2,3,4,6-tetra-, and pentachlorophenol, and their salts, is for wood
preservation. Pentachlorophenol is also used as a fungicide for slime
control in the manufacture of paper pulp and for a variety of other
purposes such as in cutting oils and fluids, for tanning leather, and
in paint, glues, and outdoor textiles. 2,4-Di- and
2,4,5-trichloro-phenol are used for the production of 2,4-D and
2,4,5-T herbicides (phenoxy acids), and 2,4,5-trichlorophenol for the
production of hexachlorophene.
Chlorophenols are produced industrially either by direct
chlorination of phenol or by hydrolysis of chlorobenzenes, the actual
process used depending on the isomer desired. Chlorination of phenol
yields 2,4-di-, 2,4,6-tri-, 2,3,4,6-tetra-, or pentachlorophenol,
while hydrolysis of chlorobenzenes is mainly used for the production
of 2,4,5-tri- and pentachlorophenol (Nilsson et al., 1978).
Chlorophenols may contain a variety of by-products and contaminants,
such as other chlorophenols, polychlorinated phenoxyphenols, and
neutral compounds like polychlorinated benzene and diphenyl ethers
(PCDPEs), PCDDs, and PCDFs. Some of these contaminants may also occur
in chlorophenol derivatives like phenoxy acids, other pesticides, and
hexachlorophene. The possible presence of PCDDs and PCDFs in
commercial products is of special significance because of their
extraordinary persistence and toxicological properties (see sections
7-9). A scientific criteria document for chlorophenols and their
impurities in the Canadian environment has been prepared by Jones
(1981, 1984). Chlorophenols were estimated to be the major chemical
sources of PCDDs and PCDFs in the Canadian environment (Sheffield,
1985).
Buser & Bosshardt (1976) reported on the results of a survey of
the PCDD and PCDF contents of pentachlorophenol (PCP) and PCP-Na from
commercial sources in Switzerland. From the results, a grouping of the
samples into two series can be observed: a first series with generally
low levels (hexaCDD <1 µg/g) and a second series with much higher
levels (hexaCDD >1 µg/g) of PCDDs and PCDFs. Samples with high PCDD
values had also high PCDF values. For most samples, the contents of
the PCDF contaminants were in the order:
tetra- = penta- < hexa- < hepta- < octaCDD/CDF.
The ranges of the combined levels of PCDDs and PCDFs were 2-16 and
1-26 µg/g, respectively, for the first series of samples, and 120-500
and 85-570 µg/g, respectively, for the second series of samples. The
levels of octaCDD and octaCDF were as high as 370 and 300 µg/g,
respectively.
Some PCP-Na samples analyzed showed the unexpected presence of a
tetraCDD (0.05-0.25 µg/g), which was later identified by Buser & Rappe
(1978) as the unusual 1,2,3,4-substituted isomer. Table 13 collects a
number of relevant analyses of these chlorophenol formulations. The
levels of PCDDs and PCDFs are higher than for the phenoxy-acetic acid
herbicides.
It has also been reported that several positional isomers of
PCDDs and PCDFs are present in the chlorophenols. However,
isomer-specific methods have not been used in most of these
investigations, and more research is necessary to identify all the
isomers present for a risk evaluation of these products.
Miles et al. (1985) have analyzed PCP samples for hexaCDDs from
five different manufacturers using an isomer-specific analytical
method. The study included both free PCPs as well as the sodium salts.
Total hexaCDDs in PCPs ranged from 0.66 to 38.5 mg/kg, while in the
sodium salts levels of hexaCDDs between 1.55 and 16.3 mg/kg were
found. The most abundant hexaCDD isomer found in the free PCPs was the
1,2,3,6,7,8 isomer; however, in the sodium salts the 1,2,3,6,7,9- and
1,2,3,6,8,9-hexaCDD pair was the most abundant.
Table 13. Levels of PCDDs and PCDFs in commercial chlorophenols (µg/g)a
2,4,6- 2,3,4,6- PCP PCP
Trichlorophenol Tetrachlorophenol Sample A Sample B
TetraCDDs < 0.1 < 0.1 < 0.1 < 0.1
PentaCDDs < 0.1 < 0.1 < 0.1 < 0.1
HexaCDDs < 1 < 1 < 1 2.5
HeptaCDDs < 1 10 0.5 175
OctaCDD < 1 2 4.3 500
TetraCDFs 1.5 0.5 < 0.1 < 0.1
PentaCDFs 17.5 10 < 0.1 < 0.1
HexaCDFs 36 70 0.03 < 0.3
HeptaCDFs 4.8 70 0.5 19
OctaCDF < 1 10 1.1 25
a From: Rappe et al. (1979).
Hagenmaier & Brunner (1987) has reported that 2,3,7,8-tetraCDD
can be found in commercial pentachlorphenol formulation at levels of
0.21-0.56 ng/g, while Hagenmeyer & Brunner (1986) report that
1,2,3,7,8-pentaCDD was found in pentachlorophenol and
Na-pentachlorophenates in concentrations of 0.9-18 ng/g.
3.3.4 Polychlorinated biphenyls (PCBs)
Vos et al. (1970) were able to identify PCDFs (tetra- and
pentaCDFs) in samples of European PCBs (Phenoclor DP-6 and Clophen A
60) but not in a sample of Aroclor 1260. The toxic effects of these
PCB products were found to parallel the levels of PCDFs present. Bowes
et al. (1975) examined a series of Aroclors, as well as the samples of
Aroclor 1260, Phenoclor DP-6, and Clophen A-60 previously analyzed by
Vos et al. (1970). They used packed columns and very few standard
compounds, and reported that the most abundant PCDFs had the same
retention time as 2,3,7,8-tetraCDF and 2,3,4,7,8-pentaCDF. Using a
complete set of PCDF standards and an isomer-specific analytical
method, Rappe et al. (1985d) determined the levels of
2,3,7,8-substituted PCDFs in commercial PCB products (see Table 14).
3.3.5 Chlorodiphenyl ether herbicides
In 1981, Yamagishi et al. reported on the occurrence of PCDDs and
PCDFs in the commercial diphenyl ether herbicides
1,3,5-trichloro-2-(4-nitrophenoxy) benzene (CNP),
2,4-di-chloro-1-(4-nitrophenoxy)benzene (NIP), and
2,4-dichloro-1-(3-methoxy-4-nitrophenoxy)benzene (X-52). The total
tetraCDD found was 14.0 mg/kg in CNP, 0.38 mg/kg in NIP, and 0.03 in
X-52. Very few synthetic standards were used, but the major tetraCDDs
were identified as the 1,3,6,8- and 1,3,7,9-isomers, the expected
impurities in the starting material 2,4,6-trichlorophenol. No
2,3,7,8-tetraCDD could be detected in these samples. In all three
herbicides, total tetraCDF was between 0.3 and 0.4 mg/kg.
3.3.6 Hexachlorobenzene
Hexachlorobenzene was used for the control of wheat bunt and
fungi. Villeneuve et al. (1974), analyzing three commercial
hexachlorobenzene preparations, identified octaCDD and hepta- and
octaCDFs. The levels and identity of the heptaCDF isomers were not
given. Great variation in levels of octaCDDs between the three samples
(0.05-211.9 mg/kg) was noted, as well as in the level of octaCDF
(0.35-58.3 mg/kg).
3.3.7 Rice oil
In 1968 more than 1500 people in southwest Japan were intoxicated
by the consumption of a commercial rice oil accidentally contaminated
by PCBs, PCDFs, and polychlorinated quarterphenyls (Masuda &
Yoshimura, 1982; Masuda et al., 1985). In 1979 a similar episode
occurred in central Taiwan, the number of people involved here
approaching 2000 (Chen et al., 1980, 1981). Both these accidents have
been referred to as Yusho episodes, but now the Taiwan episode has
been renamed Yu-cheng (see section 5.4.4.4).
The total level of PCDFs in the Japanese rice oil was reported to
be 5 µg/g (Nagayama et al., 1976) and 5.6 µg/g (Buser et al., 1978d).
For the rice oil from Taiwan, Chen et al. (1985) reported the PCDFs
levels to be in the range 0.18-1.68 µg/g.
Buser et al. (1978) analyzed the Japanese rice oil using glass
capillary columns. They found about 50-60 PCDF congeners and also
reported that the 2,3,7,8-tetraCDF was the major isomer among the
tetraCDFs. However, it was later shown that in this column system the
2,3,4,8-tetraCDF co-elutes with the 2,3,7,8-isomer, and in fact the
2,3,4,8-isomer was the main constituent in this peak (Chen & Hites,
1983; Masuda et al., 1985). The 2,3,7,8-substituted congeners were
estimated to account for 10-15% of the total amount of PCDFs (Buser et
al., 1978).
Table 14. PCDFs in commercial PCBs (ng/g)a
TRI- TETRA- PENTA- HEXA- HEPTA-
Total 2378 Total 12348 23478 Total 123479 123678 123789 234678 Total Total
PCB-type 12378 123478
Pyralene 700 53 630 10 T 35 ND ND ND ND ND ND
A1254 63 19 1400 690 490 4000 2500 2100 190 130 10 000 960
A1260 10 13 110 48 56 260 500 120 190 27 1500 1300
A30 500 35 573 14 28 160 50 59 ND ND 220 T
A40 1300 180 2600 96 8 1700 79 68 ND T 310 ND
A50 7400 3300 20 000 760 1100 8000 700 360 18 98 3100 75
A60 770 840 6900 1100 990 8100 1600 330 170 330 6800 2000
T64 47 23 360 97 122 840 520 390 58 41 2600 220
Clophen C 710 54 1200 34 30 270 ND T ND ND T ND
a From: Rappe et al. (1985d).
T = traces.
ND = not detected.
3.4 Sources of Heavy Environmental Pollution
3.4.1 Industrial accidents
Several industrial accidents occurring during the production of
2,4,5-trichlorophenol have been described in the literature. In most
of these accidents the pollution of 2,3,7,8-tetraCDD has been to
factories with circumscribed occupational exposure (section 9).
However, on 10 July, 1976, a runaway reaction in a factory at Meda
near Seveso in Northern Italy resulted in the escape of a chemical
cloud of trichlorophenol/phenate containing 2,3,7,8-tetraCDD.
The cloud initially covered an area outside the factory 5 km long
and 700 m wide. On the basis of the TCDD levels found in the
contaminated soil samples, it has been estimated that 2-3 kg of TCDD
was released in this accident. About 80% of this amount was deposited
in an area of 15 ha, within a distance of about 500 m from the plant.
The levels of soil contamination in three zones are given in Table 15
(Pocchiari, 1978).
3.4.2 Improper disposal of industrial waste
In 1973, three horse arenas in Missouri, USA, were found to be
contaminated by high levels of 2,3,7,8-tetraCDD; the highest value
reported was about 30 µg/g of soil (Kimbrough et al., 1977). This
contamination resulted from the application, in 1971, of contaminated
waste oil to control dust at these locations. The TCDD had originated
at a hexachlorophene-producing factory in Verona, Missouri. Additional
tri- and tetraCDDs were also found, but the major component was
1,2,4,6,8,9-hexachloroxanthene, a compound which apparently can serve
as a marker for this type of contamination. The xanthene is a normal
by-product of hexachlorophene production and has never been associated
with the production of 2,4,5-tri-chlorophenol or 2,4,5-T derivatives
(Viswanathan & Kloepfer, 1986).
In 1982, numerous sites of potential 2,3,7,8-tetraCDD
contamination were discovered in eastern Missouri. The contamination
originated from the same waste oil from the factory in Verona. The
streets of the entire town of Times Beach, Missouri, had been sprayed.
More than 10 000 soil samples from Missouri were analyzed. In this
state more than 40 hazardous waste sites containing 2,3,7,8-tetraCDD
were identified. Most of these contaminated sites resulted from the
disposal of waste from the same factory in Verona. The highest level
reported in these soil samples was 9648 mg TCDD/g (Viswanathan &
Kloepfer, 1986).
Another location of great concern is Love Canal, Niagara Falls,
USA. Here, Smith et al. (1983) found high levels of 2,3,7,8-tetraCDD
in storm sewer sediments taken from around the Love Canal waste
disposal site. The highest value was 312 ng/g sediment.
Table 15. Distribution of TCDD contamination in the Seveso area on the basis of soil sample analysesa
Range Number of soil samples
(µg/m2)
Zone A Zone B Surrounding monitored area
< 0.750 32 25 249
0.750 - 4.99 32 53 128
5.0 - 14.99 6 19 2
15.0 - 49.99 18 6 0
50.0 - 499.99 31 0 0
500.0 - 4999.99 18 0 0
> 5000 3 0 0
a From: Pocchiari (1978).
Zone A: high-level contamination, about 115 ha.
Zone B: low level contamination, about 255 ha.
Surrounding area: about 1400 ha.
3.4.3 Heavy use of chemicals
The Eglin Air Force Base in Northwest Florida, USA, has been used
for the development and testing of aerial spraying equipment for
military defoliation operations. During the period 1962-1970, a
3-km2 test area was sprayed with 73 tons of 2,4,5-T. Analyses of
archived samples of the formulations indicated that approximately 2.8
kg of 2,3,7,8-tetraCDD had been applied as a contaminant of the
herbicide. However, one 37-ha test grid received 2.6 kg of this TCDD
from 1962 to 1964. Levels of 10-1500 ng/kg were found in 22 soil
samples (the top 15 cm) collected and analyzed 14 years after the last
application of herbicide to this site (Young, 1983).
3.5 Other Sources of PCDDs and PCDFs in the Environment
3.5.1 Thermal degradation of technical products
The formation of 2,3,7,8-tetraCDD as a result of thermal
reactions of 2,4,5-T and 2,4,5-T derivatives has been the subject of
controversy. Heating 2,4,5-T salts at 400-450 °C for 30 minutes or
longer yielded approximately 1 g of 2,3,7,8-tetraCDD per kg of 2,4,5-T
salt, while no TCDD was identified from the same treatment of 2,4,5-T
acid or esters (Langer et al., 1973; Baughman, 1974). Using a more
sensitive analytical method, Ahling et al. (1977) reported that 0.2-3
mg of 2,3,7,8-tetraCDD was formed per kg of 2,4,5-T esters during
combustion at 500-850 °C. Two reports (Stehl & Lamparski, 1977;
Andersson et al., 1978) have shown that 2,3,7,8-tetraCDD could not be
found after burning samples of spiked or sprayed vegetation at 600 °C.
The combustion gases, soot, particles, and ashes were analyzed and the
detection limit was 4 mg of TCDD/kg 2,4,5-T burned.
Rappe (1978b) have studied the burning of material impregnated
with various salts of chlorophenols. Very carefully purified
2,4,6-tri- and pentachlorophenate were studied, in addition to a
commercial formulation of 2,3,4,6-tetra-chlorophenate. The analytical
method used in this study was not isomer specific, but the following
conclusions can be drawn concerning the formation of PCDDs by thermal
reactions:
(a) the expected dimerization products and the products formed
in the "Smiles rearrangement" are the major PCDDs;
(b) no other thermal isomerization of the PCDDs formed can be
observed;
(c) no formation of higher chlorinated PCDDs can be observed;
(d) octaCDD and other higher chlorinated PCDDs yield lower
chlorinated dioxins in a nonspecific dechlorination
reaction;
(e) a series of PCDFs was also observed.
It has been found that PCBs can be converted to PCDFs under
pyrolytic conditions. The pyrolysis of commercial PCBs in sealed
quartz ampoules in the presence of air yielded about 30 major, and
more than 30 minor, PCDFs. The optimal yield of PCDFs was about 10%,
calculated on the amount of PCB decomposed. Thus, uncontrolled burning
of PCBs can be an important environmental source of hazardous PCDFs.
Therefore, it was recommended (Buser et al., 1978a, 1978d) that all
destruction of PCB-contaminated waste using incinerators must be
carefully controlled. In the temperature range 300-400 °C, the
conversion yield seems to be in the part-per-million range (Morita et
al., 1978).
Buser & Rappe (1979) studied the pyrolysis of 15 individual
synthetic PCB congeners and showed that the formation of PCDFs can
follow several competing reaction pathways. In another study where a
series of chlorobenzenes were pyrolyzed in the same way, Buser (1979)
found that significant amounts (> 1%) of PCDDs and PCDFs were formed.
A complex mixture of isomers of PCDDs and PCDFs was found, suggesting
several reaction routes. Using the same technique as above, Lindahl et
al. (1980) studied the thermal decomposition of polychlorinated
diphenyl ethers. Both PCDDs and PCDFs were formed, involving several
pathways. The temperature range was 500-600 °C and the yields varied
from 0.1 to 4.5%.
Bergman et al. (1984) studied the thermal degradation of two
polychlorinated alkanes containing 59% and 70% chlorine, respectively,
and also a commercial chlorinated paraffin containing 70% chlorine.
Their studies indicated the presence of at least mono- and diCDFs.
Ahling et al. (1978) reported that chlorinated benzenes can be
found in the pyrolysis of PVC.
Direct evidence for the conversion of PVC to PCDDs and PCDFs has
recently been reported by Marklund et al. (1986). They found that
laboratory pyrolysis of PVC resulted in the formation of PCDDs and
PCDFs, mainly hexa- and heptaCDDs, and tetra- to heptaCDFs. In some
cases, the pattern of isomers seemed to be similar to those found in
municipal and hazardous waste incinerators, e.g. the pentaCDFs (Rappe
et al., 1987).
The data discussed in this section are summarized in Table 16.
3.5.2 Incineration of municipal waste
For some time, emissions from municipal incinerators, heating
facilities, and thermal power plants have been the subject of concern.
Whereas previously the emission of dust, smoke, toxic metals, and
noxious gases were of prime concern, the presence of potentially
hazardous organic compounds from these emissions has been recognized
only recently. Lahaniatis et al. (1977) reported the presence of
chlorinated organic compounds (chlorinated aliphatics, benzenes, PCBs,
and pesticides) in fly ash from a municipal incinerator.
Olie et al. (1977) reported the occurrence of PCDDs and PCDFs in
fly ash from three municipal incinerators in the Netherlands. Their
results indicated the presence of up to 17 PCDD peaks, but isomer
identification and quantification was not possible due to the lack of
synthetic standards. Buser & Bosshardt (1978) studied fly ash from a
municipal incinerator and an industrial heating facility, both in
Switzerland. In the former, the level of PCDDs was 0.2 µg/g and of
PCDFs 0.1 µg/g. In the industrial incinerator, the levels were 0.6
µg/g and 0.3 µg/g, respectively.
During the period 1978-1982 a series of papers, reports, and
reviews were published confirming the original findings of Olie et al.
(1977) and Buser & Bosshardt (1978) regarding fly ash. Less data have
been published on the levels of PCDDs and PCDFs in other incineration
by-products, e.g., particulates and flue gas condensate, and in total
flue gas, which are the true emissions (Marklund et al., 1986).
A risk evaluation should be based on the emission levels of PCDD
and PCDF isomers found in isomer-specific analyses using validated
sampling and clean-up methods. However, in many studies non-validated
sampling and analytical methods are used and the results are given in
terms of total levels of tetra-, penta-, hexa-, hepta-, and octaCDDs
and CDFs. The value of such studies is limited, particularly in this
situation where the number of isomers is quite large. More than 30
PCDDs and 60 PCDFs have been found in fly ash samples (Buser et al.,
1978b, 1978c).
In March 1986, a working group of experts convened by the World
Health Organization Regional Office for Europe reviewed the available
data on emissions of PCDDs and PCDFs from municipal solid-waste (MSW)
incinerators. It was found that the origin of these compounds was not
completely understood, but they appear to result from complex thermal
reactions occurring during periods of poor combustion. Because of
their high thermal stability, the PCDDs and PCDFs can be destroyed
only after adequate residence times at temperatures above 800 °C
(WHO/EURO, 1987).
Available data on total emissions of PCDDs and PCDFs from tests
on MSW incinerators range between a few and several thousand ng/Nm3
dry gas at 10% carbon dioxide (CO2). The working group prepared a
table giving a range of estimated isomer specific emissions for those
isomers of major concern with respect to MSW incinerators operating
under various conditions (Table 17).
The emissions tabulated in column 1 are those which the working
group considered to be achievable in the most modern, highly
controlled, and carefully operated plants in use at the present time.
Such results do not represent what is considered to be achievable by
the use of acid gas cleaning equipment; use of such equipment should
result in much lower values (probably at least one order of
magnitude). The results given in column 1 are not representative of
emissions that might be expected from such plants during start-up or
during occasional abnormal conditions. Emission levels listed in
column 2 were considered by the working group to be indicative of the
higher limit of emissions from modern MSW incinerators. These plants
might experience such emissions during start-up or during occasional
upset conditions. Consequently, the majority of the available
concentration data falls between columns 1 and 2. Some of the data
reviewed has shown that the figures in column 2 should not be
considered an absolute maximum. However, most existing plants, if
carefully operated, will have PCDD and PCDF emisions in the range
between columns 1 and 2.
The highest values for MSW incinerators (column 3) were obtained
by multiplying the values in column 2 by a factor of 5. Column 3
includes emission data that were reported to the working group from
all tests and under all circumstances. Generally, these emission
levels are associated with irregular or unstable operating conditions,
high moisture content of the MSW, low combustion or afterburner
temperatures, less than adequate technologies, etc.
Table 16. Formation of PCDDs and PCDFs by thermal processes
Precursor Conditions Products
2,4,5-T salt Pyrolysis 2,3,7,8-tetraCDD
2,4,5-T (vegetation) Pyrolysis No TCDD
Burning No TCDD
Cl-phenate Burning PCDDsa + PCDFs
PCBs Pyrolysis PCDFsb
PCBzc Pyrolysis PCDFs + PCDDsd
Cl-Diphenyl ethers Pyrolysis PCDFs + PCDDs
Cl-Alkanes (Paraffins) Pyrolysis PCDFs
PVC Pyrolysis PCDDs + PCDFs
a = PCDDs formed by dimerization and a non-specific dechlorination.
b = other products: hexa- and pentaCBs.
c = polychlorinated benzenes.
d = other products: PCBs, polychlorinated naphthalenes.
The working group was aware of both lower and higher emission
levels than those included in Table 17. However, it was felt that the
values included in Table 17 were likely to be representative of
emissions from current facilities (WHO/EURO, 1987).
Of special importance is the observation that the emission of
1,2,3,7,8-pentaCDD normally exceeds the emission of 2,3,7,8-tetraCDD
by a factor of three to ten.
3.5.3 Incineration of sewage sludge
Sludge from municipal waste water treatment plants may be
incinerated after being dewatered. The WHO working group (see 3.5.2)
reviewed the available data from municipal sewage sludge (MSS)
incinerators, and found that PCDD and PCDF emissions from this type of
plant were generally lower than emissions from MSW incinerators (see
Table 17, column 4) (WHO, 1986).
3.5.4 Incineration of hospital waste
Doyle et al. (1985) claimed that the incomplete combustion of
certain hospital waste containing halogenated organics could produce
high levels of PCDDs and PCDFs. They found the mean values of total
PCDDs to be 69 ng/m3 and total PCDFs to be 156 ng/m3. No
isomer-specific data seems to be available. Hagenmaier et al. (1986)
reported the analyses of stack gas from 10 hospital waste incineration
plants. The mean value of 2,3,7,8-tetraCCD emitted was 0.28 ng/m3,
the mean of all TCDDs being 20 ng/m3. The mean value for total PCDDs
was 118 ng/m3 and for total PCDFs 434 ng/m3.
Table 17. Estimated range of emissions from municipal solid waste (MSW) and municipal sewage sludge (MSS) incineratorsa
Emissions from MSW combustion Emissions
from MSS
combustion
1 2 3 4
Congeners Achievable with Maximum High Most
modern plants from emissions likely
with no acid average highest
gas cleaning operation emissions
(ng/Nm3, dry, at 10% CO2)
2,3,7,8-TetraCDD 0.1 1.5 7.5 0.1
1,2,3,7,8-PentaCDD 0.3 14 70 0.3
1,2,3,4,7,8-HexaCDD 0.2 31 155 0.2
1,2,3,6,7,8-HexaCDD 0.6 56 280 0.6
1,2,3,7,8,9-HexaCDD 0.4 20 100 0.4
2,3,7,8-TetraCDF 0.9 10 50 0.9
1,2,3,7,8-/1,2,3,4,8-PentaCDF 2.3 52 260 2.3
2,3,4,7,8-PentaCDF 2.0 40 200 2.0
1,2,3,4,7,8/1,2,3,4,7,9-HexaCDF 1.1 48 240 1.1
1,2,3,6,7,8-HexaCDF 1.3 40 200 1.3
1,2,3,7,8,9-HexaCDF 0.06 52 260 0.06
2,3,4,6,7,8-HexaCDF 2.0 36 180 2.0
a From: WHO/EURO (1987).
3.5.5 Incineration of hazardous waste
Analyses from a test burn of pentachlorophenol waste have been
reported by Rappe et al. (1983c). PCP is a well known precursor to
octaCDD (section 2). Samples of baghouse ash and bottom ash were
analyzed. In the baghouse ash the total level of octaCDD was only 0.2
µg/g. The major constituents were lower chlorinated PCDDs such as
hepta-, hexa-, penta-, and tetraCDDs. The isomeric distribution was
reported to be very similar to a "normal" fly ash. In both cases
2,3,7,8-tetraCDD was a very minor constituent. The level of PCDD in
the bottom ash was 0.31 µg/g. The baghouse ash was also reported to
contain PCDFs at a total level of 2.5 µg/g. For the tetra- and penta-
chlorinated compounds, equal amounts of PCDDs and PCDFs were reported.
Oberg & Bergstrém (1986) reported on test data from a Swedish
hazardous waste incinerator equipped with a rotary kiln, an
afterburner, and a dry scrubbing unit. Combustion tests were performed
with PCB (Aroclor 1242) as a fluid, and as a contaminant in solid
waste (Aroclor 1016 in capacitors). The results of these tests
indicated no correlation between the amount of PCB incinerated and the
amount of PCDDs and PCDFs found in the emissions.
3.5.6 Metal industry and metal treatment industry
It has been reported by Marklund et al. (1986) that industrial
high-temperature processes like copper smelters and electric arc
furnaces in steel mills have been identified as sources of
environmental contamination by PCDDs and PCDFs. The results are
reported in "TCDD equivalents" according to US EPA (1987). The
emission from the copper smelter contained 11 ng of TCDD
equivalents/Nm3 dry gas and 10% CO2, while the dust from the steel
mill contained 0.8 ng TCDD equivalents/g dust. Marklund et al. (1986)
also considered the emissions from industrial incinerators to be of
the same magnitude, or even higher, than the emissions from MSW
incinerators.
Southerland et al. (1987) analyzed emissions from various
incinerators within Tier 4 of the USA. The highest levels were found
in a secondary copper smelter, which contained 170 ng of
2,3,7,8-tetraCDD/Nm3 and 3% oxygen. This was by far the highest
level found within the US EPA National Dioxin Strategy.
3.5.7 Wire reclamation
Hryhorczuk et al. (1981) studied a wire reclamation incinerator
in the USA. Using a non-isomer-specific analytical method, they
determined total levels of tetraCDDs and tetraCDFs. Two samples were
analyzed, one from the furnace and one from the stack. The furnace
sample contained 58 ng/kg of total TCDDs and 730 ng/kg of total TCDFs,
whereas the stack sample contained 410 ng/kg of total TCDDs and 11 600
ng/kg of total TCDFs.
3.5.8 Traffic
Marklund et al. (1987) reported a study where automobile exhaust
emissions were analyzed for PCDDs and PCDFs. Two groups of test cars
were utilized: (1) cars equipped with a catalytic converter using
unleaded gasoline with no halogenated scavengers; (2) cars with no
catalytic converter using leaded gasoline (0.15 g/litre) and a
dichloroethane scavenger (0.1 g/litre). Before the test runs, the
motor oil was changed in all cars. No PCDDs and PCDFs could be
identifed in the cars using the unleaded gasoline, while the average
emission from the cars running on leaded gasoline was found to be
30-540 pg/kg of TCDD equivalents. It was assumed that the chlorinated
scavenger (dichloroethane) was the precursor of the PCDDs and PCDFs
formed. It was estimated that the total amount of PCDDs and PCDFs from
cars in Sweden using leaded gasoline with halogenated scavengers is in
the range of 10-100 g TCDD equivalents/year.
3.5.9 Fires and accidents in PCB-filled electrical equipment
In February 1981 a fire in the State Office Building in
Binghamton, New York, USA, caused a transformer to rupture, releasing
soot throughout the building. The dielectric fluid in the transformer
consisted of a mixture of PCB (65%) and chlorinated benzenes (35%).
The soot was found to be highly contaminated with PCDFs (total PCDFs
> 2000 µg/g). The most toxic isomers (2,3,7,8-tetraCDF; 1,2,3,7,8-
and 2,3,4,7,8-pentaCDF; and 1,2,3,4,7,8- and 1,2,3,6,7,8-hexaCDF) were
found to be the major constituents within each group of congeners.
Levels reported were 12 mg/g of 2,3,7,8-tetra CDF, 670 mg/g of total
penta-CDFs, and 965 mg/g of total hexa-CDFs, 46 mg/g of total
hepta-CDFs, and 460 mg/g of octa-CDFs (Rappe, 1984; Rappe et al.,
1985b). In addition, a series of PCDDs were identified, including the
highly toxic 2,3,7,8-tetraCDD, and 1,2,3,7,8-pentaCDD (Rappe et al.,
1983a; Buser & Rappe, 1984). It is assumed that the chlorinated
benzenes were the dioxin precursors.
Between 1981 and 1985, a series of transformer accidents (7 in
all) similar to the one in Binghamton were reported in the USA and
Canada (Rappe et al., 1986a). In January 1985, an explosion followed
by a fire ruptured a transformer in the basement of a residential
complex in Rheims, France. The transformer was filled with PCB (60%)
and trichlorobenzene (40%). Total levels of PCDFs were as high as 2570
µg/m2 before clean-up. Only traces of hepta- and octaCDD were found
(Rappe et al., 1985a).
In Europe, between 1981 and 1985, 19 accidents involving indoor
capacitor fires and explosions were reported from Scandinavian
countries (Rappe et al., 1986a). All capacitors were mineral-oil
filled, and contamination of the sites averaged 1-5 µg total
PCDFs/m2.
3.5.10 Pulp and paper industry
Large amounts of chlorine or chlorine compounds are used in the
pulp and paper industry for the bleaching of the pulp. Three black
liquor boilers from the craft paper process were included in the US
EPA study of combustion sources. No 2,3,7,8-tetraCDD was found in
these emissions, but low levels of other PCDDs and PCDFs were found in
one of the three incinerators and a yearly emission of 0.25 g was
calculated (Southerland et al., 1987).
Rappe et al. (1987) recently identified both 2,3,7,8-tetraCDD
(170 ng/kg) and 2,3,7,8-tetraCDF (890 ng/kg) in a sample taken in a
sedimentation lagoon at a Swedish paper mill. A series of other PCDDs
and PCDFs was also identified but at lower levels. The isomeric
pattern in this sample differed markedly from other sediment samples,
indicating pulping processes to be a source of environmental pollution
by 2,3,7,8-tetraCDD and 2,3,7,8-tetraCDF (Rappe et al., 1987).
3.5.11 Incineration of coal, peat, and wood
The emissions of PCDDs and PCDFs from coal-fired power plants
(Kimble & Gross 1980), wood stoves (Clement et al., 1985), and peat
burning (Marklund et al., 1986) seem to be very low when calculated
per m3. However, the very high flow rates and the large number of
units could make a significant total contribution. The occurrence of
pentaCDDs and all PCDFs was not discussed in this report.
3.5.12 Inorganic chlorine precursors
It is well known that certain organochlorine compounds are
efficient precursors to PCDDs and PCDFs during pyrolysis. However, it
was proposed by scientists from Dow Chemical Company that PCDDs, and
especially 2,3,7,8-tetraCDD, are ubiquitous and formed as trace level
by-products of any normal combustion (Bumb et al., 1980).
Consequently, dioxins should have been present in the environment
since the advent of fire. This suggests that inorganic chloride can
serve as a useful precursor to the formation of PCDDs and PCDFs. A
recent survey of PCDD levels, in particular from residential wood
combustion units, has been quoted in support of the above. The survey
showed PCDD levels in the ng/kg range (see also section 3.5.11).
However, this hypothesis has been criticized. One of the main
arguments against such a hypothesis is that 2,3,7,8-tetraCDD does not
appear to be formed in coal-fired power plants (Kimble & Gross, 1980;
Junk & Richard, 1981). Another argument is that the Dow studies lack
data on levels of dioxin precursors in the material being burned,
including the air in the flames (Rappe, 1984).
The analyses of historical samples gives additional support to
the theory that organochlorine compounds are more important as
precursors than inorganic chloride. When Czuczwa & Hites (1985)
analyzed sediment core samples from Lake Huron, N. America, the first
indication of PCDDs and PCDFs was found in sediments from 1940. There
was also a good correlation between the trend in the levels of PCDDs
and PCDFs in these sediments and the trend in the production of
chlorinated aromatic compounds (section 5.4).
Schecter et al. (1986a) were unable to detect PCDDs and PCDFs in
human liver and lung tissue from two female Eskimos frozen over one
hundred years ago (see also section 4.4.4).
3.5.13 Photochemical processes
Sundström et al. (1979) studied the formation of 2,3,7,8-tetraCDD
in six re-forestation areas that were sprayed with 2,4,5-T esters.
Leaf samples from the areas were analyzed for 2,4,5-T esters and TCDD.
TCDD was found in one leaf sample only, at levels lower than expected
from the level of dioxin contamination of the herbicide formulations
used.
The photochemical formation of PCDDs and PCDFs has also been
studied in laboratory experiments.
The photochemical dimerization of chlorophenols to PCDDs was
studied by Crosby & Wong (1976). The only PCDD formed in this study
was the octaCDD. Other PCDDs can be formed by photochemical
cyclization of chlorinated o-phenoxyphenols, also called pre-dioxins
(Nilsson et al., 1974). These pre-dioxins are very common impurities
(1-5%) in commercial chlorophenols (Nilsson et al., 1978), but the
cyclization is only a minor reaction pathway; the main reaction being
the dechlorination of the pre-dioxin.
Akermark (1978) studied the formation of 2,3,7,8-tetraCDD from
the appropriate pre-dioxins. He could identify the product, but
claimed the reaction to be very inefficient.
Another photochemical process of potential environmental
importance is dechlorination of the higher chlorinated PCDDs and
PCDFs, e.g., octaCDD and octaCDF. The products formed by photolysis of
octaCDD in organic solvent have now been identified (Buser & Rappe,
1978). By comparison with authentic standards, it was found that the
main tetrachloro isomer was the 1,4,6,9-tetraCDD; the major
pentachloro compound was expected to be the 1,2,4,6,9-isomer, and the
main hexa- and heptachloro compounds were the 1,2,4,6,7,9- (or
1,2,4,6,8,9-) and the 1,2,3,4,6,7,9-isomers, respectively. The
reaction scheme deduced from this data indicates that the chlorine
atoms are removed preferentially from the lateral positions on the
carbon rings. Consequently, the most toxic PCDD isomers, such as
2,3,7,8-tetraCDD, are not likely to be formed from the photolysis of
the higher PCDDs in solution.
Crosby et al. (1973) studied the photolysis of a series of PCBs
dispersed in water. For two isomers, the 2,5-dichloro- and
2,2',5,5'-tetrachlorobiphenyls, small amounts (0.2%) of 2-mono-CDF
could be found among the products (for photo-chemical transformations,
see section 4.2.1).
3.6 Comparison of Isomeric Pattern and Congener Profiles From Various Sources
There is a pronounced difference between technical products and
incineration emissions in both isomeric patterns and congener profiles
of PCDDs and PCDFs. In technical products the number of isomers
present is limited, whereas in incineration emissions most isomers
seem to be present. Rappe (1987) has pointed out the large similarity
qualitatively in isomeric patterns between different incineration
sources.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATIONS
4.1 Environmental Transport
4.1.1 Air
The PCDDs and PCDFs are believed to be transported in the
atmosphere. The transport of these compounds from stacks and other
stationary point sources, as well as from waste disposal sites and
other area sources, can be predicted from dispersion modelling (SAI,
1980). In the case of the accidental release of a toxic cloud
containing 2,3,7,8-tetraCDD at Seveso, Italy, Cavallaro et al. (1982)
determined the transport pattern and the ground deposition. They
determined that the deposition of 2,3,7-8-tetraCDD from air to soil
should follow an exponential decay pattern in the
Gaussian-distribution along the cross-section of the downwind
direction. Thibodeaux (1983) studied the air transport of
2,3,7,8-tetraCDD at a herbicide production facility in Jacksonville,
Arkansas, USA.
The dispersion modelling has limitations. If possible, the
modelling calculations should be combined with true air measurements.
4.1.2 Water
The solubility of 2,3,7,8-tetraCDD in water has been extensively
studied (see section 2), but much less data are available for the
other PCDDs and PCDFs. However, data from microbiological experiments
indicate that 2,3,7,8-tetraCDD is highly adsorbed to sediments and
biota. Matsumura et al. (1983) suggested that more than 90% of the
2,3,7,8-tetraCDD in an aquatic medium could be present in the adsorbed
state. Rappe et al. (1985c) studied a suspension of soot/dust in the
wash water from a PCB fire. The suspension contained 100 ng/ml of
various PCDFs, but when the soot was settled the water contained no
detectable levels of PCDFs (detection level: 0.1 ng/ml of each
isomer). Most of the PCDDs and PCDFs, if present in waterways, should
be in the sediments or attached to suspended particles.
Thibodeaux (1983) has calculated the amount of 2,3,7,8-tetraCDD
transported by a creek within the contaminated herbicide factory in
Jacksonville, Arkansas, USA. The value was 0.89 g/year as an average
rate, and a maximum of 2.1 g/year.
4.1.3 Soil and sediments
The mobility of 2,3,7,8-tetraCDD and of a dichlorodioxin in soils
has been studied by Helling et al. (1973). Both were found to be
immobile in all soils and, therefore, would not be leached out by
rainfall or irrigation, though lateral transport during surface
erosion of the soil could occur.
The US Air Force conducted studies in an area of north-west
Florida that had been heavily sprayed with the herbicide Agent Orange
between 1962 and 1964 (Young et al., 1975). This herbicide mixture was
contaminated with TCDD (section 3.2). A 7.8-ha test grid received a
total of 40 metric tons of 2,4,5-T between 1962 and 1964. When 15-cm
soil core samples were taken in 1974, they showed TCDD concentrations
ranging from 10 to 710 ng/kg. This study illustrates that significant
levels of TCDD residues remained 10 years after the last herbicide
application. Similar TCDD concentrations were obtained from areas that
had been sprayed between 1962 and 1969 (Bartelson et al., 1975).
In another study Young (1983) measured the concentration of
2,3,7,8-tetraCDD in a soil profile. The samples were collected in 1974
and the data suggested that most of the 2,3,7,8-tetraCDD would be
found in the top 15 cm of the soil profile (Table 18).
The probable media and modes of transport of PCDDs from soils are
the following: (1) to air via contaminated airborne dust particles;
(2) to surface water via eroded soil transported by water; (3) to
groundwater via leaching; (4) to air via volatilization. Movement of
particulate matter containing adsorbed PCDDs and PCDFs has been
considered to be a much more important transport mechanism than
leaching and volatilization because of the low water solubility and
volatility of these compounds (Josephson, 1983). However, the
monitoring of Seveso soil one year after the accident showed that the
highest 2,3,7,8-tetraCDD levels were not present in the topmost soil
layer (0.5 cm), but very often in the second (0.5-1.0 cm) or third
(1.0-1.5 cm) layers. This disappearance of at least a part of the
2,3,7,8-tetraCDD from the topmost soil layer was speculated to be due
to volatilization or vertical movement through the soil (DiDomenico et
al., 1980). Therefore, it appears that volatilization from soil and
leaching to groundwater can be responsible for the transport of PCDDs
and PCDFs from soils under certain conditions, namely, heavy rainfall
on sandy soils. Studies by Young (1983) indicate that the half-life
for 2,3,7,8-tetraCDD in soil is 10-12 years.
Thibodeaux (1983) has calculated the vaporization of
2,3,7,8-tetraCDD from a herbicide plant in Jacksonville, Arkansas,
USA. The vaporization can take place from soil surfaces, from landfill
cells, and from the surface of a pond. In Table 19 a summary of yearly
emission rates from these sources is presented.
It was found that vaporization from the soil surface in the
highly contaminated blow out area was the major contributing source of
emissions from this plant.
Table 18. Concentration of 2,3,7,8-tetraCDD in a soil profilea, b
Depth (cm) 2,3,7,8-tetraCDD (ng/kg)
0 - 2.5 150
2.5 - 5.0 160
5.0 - 10 700
10 - 15 44
15 - 90 NDc
a From: Young (1983).
b The area received 1,069 kg/ha of 2,4,5-T Agent Orange during
1962-1964. The soil samples were collected and analyzed in 1974.
c None detected (minimum detection limit: 10 ng/kg).
Table 19. Surface source areas and emission rates of 2,3,7,8-tetraCDDa
Source Area (m2) Emission rate (g/year)
Blow-out area, volatilization 753 120-1200
Blow-out area, entrainment 753 28-37
Rocky Branch Creek, dissolved 0.89-2.1
Reasor-Hill dump 1129 0.1-1.0
Rocky Branch Creek, sediment 0.094-0.22
Cooling water pond 15050 0.015-0.016
Total 150-1240
a From: Thibodeaux (1983).
Freeman et al. (1986) have developed a model to describe the
vaporization and diffusion through a column of soil of low volatility
organic chemicals like PCDDs and PCDFs. This model has been used to
make predictions on the transport of 2,3,7,8-tetraCDD at a site in
Times Beach, Missouri, USA. The model predicted that the 1983 levels
in this soil would be only 10% of the original loading. The model also
predicted that 57% of the initial amount of 2,3,7,8-tetraCDD was
vaporized through the soil column to the surface in the first year
after the spraying and that most transport of the vapour occurred
during the summer months. The same results were also obtained in
studies reported by Facchetti et al. (1986) and Palausky et al.
(1986).
4.2 Environmental Transformation
4.2.1 Abiotic transformation
Like other PCDDs and PCDFs, 2,3,7,8-tetraCDD is chemically quite
stable, and is not likely to be degraded at a significant rate by
hydrolytic reactions under environmental conditions. Under these
conditions, TCDD seems also to be rather stable to photochemical
degradation (Crosby et al., 1971). The half-life of TCDD of about
10-12 years, as found by Young et al. (1983) for soil, is in agreement
with this observation.
However, three reports on rapid photochemical degradation of
2,3,7,8-tetraCDD under experimental conditions make the situation more
complicated. In a methanol solution, TCDD is fairly easily degraded by
photolysis in the laboratory (Crosby et al., 1971). Other studies
using 2,4,5-T ester formulations with known amounts of TCDD and
exposed to natural sunlight on leaves, soil, or glass plates showed
that most of the TCDD was lost during a single day (Crosby & Wong,
1977). In these two studies, a "hydrogen donor", such as methanol or
2,4,5-T ester, enhanced the photochemical dechlorination (Akermark,
1978); they do not therefore truly reflect environmental conditions,
where the 2,4,5-T ester would be rapidly hydrolyzed on the surface of
the leaves. At Seveso the TCDD was released together with salts of
2,4,5-trichlorophenol, ethylene glycol, and inorganic constituents
(Rappe, 1978b). Like water, none of these is a potent hydrogen donor.
According to Bertoni et al. (1978), the addition of a solution of
ethyl oleate in xylene enhances the breakdown of TCDD in soil by UV
light. Similarly, a cationic surfactant, 1-hexadecylpyridinium
chloride, was also reported to enhance photodecomposition (Botre et
al., 1978).
Another experiment, which might be a good model for the
degradation of TCDD bound to dust particles in the air, has shown that
TCDD adsorbed on silica gel undergoes rapid photo-chemical degradation
(Gebefugi et al., 1977).
In order to explain the longer half-life of 2,3,7,8-tetraCDD in
a model laboratory ecosystem than in an outdoor pond, Matsumura et al.
(1983) speculated that photolysis was the most likely cause. In the
outdoor environment, algae-mediated photosensitization of
2,3,7,8-tetra-CDD may have caused some photodecomposition of this
compound.
An increase in chlorine substitution is expected to decrease the
rate of photodegradation. For example, Crosby et al. (1971) showed
that although complete decompostion of 2,3,7,8-tetraCDD in methanol
occurred in 24 h under UV irradiation, > 80% octaCDD in methanol
remained unreacted during the same period under similar irradiation
conditions.
Although the degree of photolysis may be related to the extent of
chlorination, different chlorine substitution patterns also play a
critical part. In higher chlorinated PCDDs, there appears to be
preferential loss of chlorine from the 2,3,7, and 8 positions (Buser
& Rappe, 1978). Thus, PCDDs with chlorine substitutions in positions
2,3,7, and 8 are likely to be photochemically degraded faster than
compounds not having these positions substituted. For example, the
photolysis half-life of 1,2,3,7,8-pentaCDD has been estimated to be
7.8 h in n-hexadecane solution under sunlamp irradiation (Nestrick
et al., 1980). Similarly, the photolytic half-lives of
1,2,3,7,8-pentaCDD, 1,2,3,6,7,9-, and 1,2,4,6,7,9-hexaCDD in hexane
solutions under sunlight irradiation have been determined to be 5.4,
17, and 47 hours, respectively (Dobles & Grant, 1979). Nestrick et al.
(1980) reported a half-life value of 6.8 h for 1,2,3,6,7,8-hexaCDD in
n-hexadecane under sunlamp irradiation. The primary intermediates of
the photo-degradation of higher chlorinated PCDDs are probably lower
chlorinated dioxins (Buser & Rappe, 1978), but the pathways of
degradation are not known with certainty (National Research Council of
Canada, 1981).
From these discussions of the photolysis of PCDDs in the presence
of organic hydrogen-donating substrates, it is difficult to predict
the photolytic fate of these compounds in natural aquatic media, where
hydrogen donors may or may not be available. The situation is
complicated further by the fact that a predominant amount of PCDDs in
surface water may be adsorbed or suspended on particles and sediments,
rather than in solution. Moreover, since the penetration of UV light
into natural water may be very limited, photolytic degradation of
PCDDs in water is not likely to be of environmental importance.
Hutzinger et al. (1973) have studied the photochemical
degradation of 2,8-diCDF and octaCDF. They found that a reductive
dechlorination takes place, especially in methanolic solution. The
reaction was much slower when a thin film was exposed to sunlight.
Thermally, 2,3,7,8-tetraCDD is quite stable, rapid decomposition
occurring only at temperatures above 750 °C (Stehl et al., 1973).
4.2.2 Biotransformation and biodegradation
The 2,3,7,8-tetraCDD isomer is very resistant to biodegradation.
Only 5 of about 100 microbial strains with the ability to degrade
persistent pesticides were able to degrade 2,3,7,8-tetraCDD (Matsumura
& Benezet, 1973). Ward & Matsumura (1978) studied the biodegradation
of 14C-labelled 2,3,7,8-tetraCDD in lake waters and sediments from
Wisconsin, USA, and observed a half-life of 2,3,7,8-tetraCDD in lake
waters containing sediment of 550-590 days. In lake water alone, about
70% of the 2,3,7,8-tetraCDD remained after 589 days. Using an outdoor
pond as a model aquatic ecosystem, and dosing it with 14C-labelled
2,3,7,8-tetraCDD, Matsumura et al. (1983) estimated the half-life of
2,3,7,8-tetraCDD to be approximately 1 year. Although biodegradation
may have been responsible for part of the degradation, it is almost
impossible to estimate the biodegradation half-life of
2,3,7,8-tetraCDD in aquatic systems from this experiment.
Philippi et al. (1982) detected a polar metabolite of
2,3,7,8-tetraCDD in several microbiological cultures after long-term
incubation. They reported chromatographic and MS data that supported
the conclusion that the metabolite was 1-hydroxy-2,3,7,8-tetraCDD,
although a synthetic standard compound was not available.
Tulp & Hutzinger (1978) reported that in rats, dibenzo-p-dioxin,
1-monoCDD, 2-monoCDD, 2,3-diCDD, 2,7-diCDD, 1,2,4-triCDD, and
1,2,3,4-tetraCDD are metabolized to mono- and di-hydroxy derivatives.
In the case of dibenzo-p-dioxin and both of the two monochloro
isomers, sulfur-containing metabolites were also excreted. Primary
hydroxylation exclusively took place at the 2, 3, 7, or 8 positions in
the molecule. In these studies, no metabolites resulting from fission
of the C-O bonds (ortho, ortho'-dihydroxychlorodiphenyl ethers,
chloro-catechols), or hydroxylated derivatives thereof, were detected.
No metabolites were found from octaCDD.
4.3 Bioaccumulation
The bioaccumulation of 2,3,7,8-tetraCDD has been investigated in
several studies, using several aquatic species and different model
ecosystems. In the experiments in which 14C-TCDD was introduced into
the model ecosystem in the form of residues on sand, particularly high
values were found in the mosquito (Aedes egypti) larvae, the level
exceeding that found in water by more than 9000 times. Under similar
conditions, the level in brine shrimp (Artemia salina) was 1570 times
higher than that found in water (Matsumura & Benezet, 1973). In the
second study (Isensee & Jones, 1975; Isensee, 1978), 14C-TCDD was
absorbed, at a broad range of levels, into soil and placed at the
bottom of an aquarium. Five species of organisms were added (though
not simultaneously) 1-30 days after flooding, and exposed for 3-32
days. The correlation between the TCDD level in the water and in the
organisms of each species was highly significant (correlation
coefficient of 0.94 or higher).
Bioaccumulation factors for 2,3,7,8-tetraCDD are given in Table
20 (US EPA, 1985).
4.4 Levels in Biota
4.4.1 Vegetation
When 14C-labelled 2,3,7,8-tetraCDD was added to soil, both oats
and soya beans accumulated small quantities of TCDD, at all stages of
growth. TCDD was also detected in control plants housed with the
experimental plants after treatment (Isensee & Jones, 1971). A maximum
of 0.15% of the TCDD present in the soil was translocated to the
aerial portion of the oats and the soya beans, but neither the grain
nor the beans harvested at maturity showed any detectable level of
14C-labelled TCDD. When TCDD was applied to the central leaflet of
3-week-old soya bean plants and 12-day-old oat plants, very little
TCDD was lost from the soya bean leaves in 21 days, but there was a
gradual loss (38% in 21 days) from the oat leaves.
Analyses of vegetation from Seveso, Italy, after the industrial
accident, gave values of up to 50 mg TCDD/kg (Firestone, 1978). In the
following years, when there was no direct contact of the newly grown
vegetation with the aerosol cloud, the levels of dioxin in plants
decreased by several orders of magnitude (Wipf & Schmid, 1983). In
1977 (one year after the accident in Seveso), no traces of TCDD were
found in the flesh of apples, pears, and peaches, or in corn cobs or
kernels, grown near the factory (the detection limit for the analyses
was 1 ng/kg). At the same time about 100 ng TCDD/kg was detected in
the fruit peels. This strongly suggests that the contamination was due
to dust and not from plant uptake. The TCDD level in the soil was
found to be in the order of 10 ng/g, which corresponds to about 1000
µg/m2 (Wipf et al., 1982).
Facchetti et al. (1986) studied plants grown in soil spiked with
2,3,7,8-tetraCDD in the range 1-752 ng TCDD/kg. At the end of
cultivation, root samples were collected, carefully washed, and
analyzed. The levels of TCDD in the roots were found to be higher than
the levels found in the soil in which the plants were grown. On the
parts above ground, Facchetti et al. (1986) could not find any
significant increase in the levels of TCDD. However, the TCDD
concentration was found to vary with the location, being higher if the
plants were grown in the vicinity of other pots containing
contaminated soil. The conclusion was drawn that evaporation is the
predominant process for the contamination of the aerial parts of
plants. However, studies by Sacchi et al. (1986) indicated that maize
and bean plants grown in soil contaminated by 3H-2,3,7,8-tetraCDD
accumulated radioactivity in the aerial parts progressively with time
and with soil contamination (Sacci et al., 1986). It was suggested
that the distribution of the TCDD into the leaves occurred via the
transpiration stream.
Very few analyses of sprayed vegetation have been reported. A
rough estimate of 20-1000 ng/kg for 2,3,7,8-tetraCDD contamination can
be made on the basis of the level of 2,4,5-T found in newly sprayed
vegetation and the level of 2,3,7,8-tetraCDD in the spray formulation
used. Higher values could be obtained for Agent Orange. Sundström et
al. (1979) reported data in agreement with this estimate. However, the
analytical technique used in their study was not isomer specific.
Vegetation was sprayed with 2,4,5-T ester contaminated by only 0.06 mg
2,3,7,8-tetraCDD/g. A sample of leaves collected 42-45 days after the
spraying was found to have 170 ng TCDD/kg, somewhat lower than the
expected value 600 ng TCDD/kg, indicating a slow photochemical
breakdown.
Table 20. Measured bioaccumulation factor for 2,3,7,8-TCDD in freshwater aquatic organismsa
Species Tissue Duration Bioconcentration Reference
(days) factor
Alga 33 3094b Isensee (1978)
(Oedogonium cardiacum)
Alga 32 2075c Isensee (1978)
(Oedogonium cardiacum) Yockim et al. (1978)
Snail whole body 33 5471b Isensee (1978)
(Physa sp.)
Snail whole body 32 3095c Isensee (1978)
(Physa sp.) 3731 Yockim et al. (1978)
Cladoceran whole body 32 3895b Isensee (1978)
(Daphnia magna)
Cladoceran whole body 30 7070c Isensee (1978)
(Daphnia magna) 7125 Yockim et al. (1978)
Catfish whole body 28 4875 Yockim et al. (1978)
(Italurus punctatus)
Mosquitofish whole body 14 4850c Isensee (1978)
(Gambusia affinis) 4875 Yockim et al. (1978)
a From: US EPA (1985).
b Arithmetic mean of several values reported.
c Tissue concentrations at equilibrium.
Table 21. Levels of TCDDs in fish and shellfisha
Sample Tissue Concentration of
number typeb 2,3,7,8-TCDD (ng/kg)c
1 Fish (edible flesh) 480
2 Catfish 40
3 Buffalo fish ND(13)
4 Fish (predator) 230
5 Fish (bottom feeder) 77
6 Catfish 50
7 Buffalo fish ND(7)
8 Catfish ND(7)
a From: Mitchum et al. (1980).
b All samples were obtained from the Arkansas River, USA, or
a tributary, the Bayou Meto.
c These are averages of samples that had detectable levels of
TCDD.
ND = none detected; the number in parenthesis is the measured
detection limited for that sample.
4.4.2 Aquatic organisms
Fish and shellfish taken from areas in South Viet Nam that were
heavily exposed to Agent Orange during military defoliation operations
in the 1960s have been reported to contain 18-810 ng TCDD/kg (Baughman
& Meselson, 1973). The analytical technique of direct-inlet high
resolution MS used in this study is not considered isomer specific and
did not include any GC separation at all.
In two streams associated with the US Air Force test area in
north-west Florida (section 4.1.3), which had been heavily sprayed
with Agent Orange between 1962 and 1964, the silt contained, 10 years
later, 10 and 35 ng TCDD/kg where eroded soil entered the water.
Concentrations of 12 ng TCDD/kg were found in two species of fish from
this stream, the sailfin shiner (Notropis hypselopterus) and the
mosquito fish (Gambusia affinis). The spotted sunfish (Lepomis
punctatus) contained 4 ng TCDD/kg in skin and muscle, 18 ng/kg in the
gonads, and 85 ng/kg in the gut (Young et al., 1976).
Table 22. Analytical results for 2,3,7,8-tetraCDD residues in fish from Saginaw Bay Region, Michigan, USAa
Species Number of Number of TCDD
samplesb positive detected (ng/kg)c
samples low high mean
Channel
catfish 8 8 28 695 157 (13)
Carp 14 10 20 153 55 (7)
Yellow
perch 6 3 10 20 13 (5)
Smallmouth
bass 2 2 7 8 8 (6)
Sucker 4 3 4 21 10 (4)
Lake trout 2 0 0 0 0 (5)
a From: Harless et al. 1982).
b Mean % recovery for 2.5-10 ng 37Cl4-TCDD added to 5 or 10
g of tissue prior to sample preparation was between 78 and
100%.
c Corrected for losses in efficiency of sample preparation for
particular species. The numbers in parenthesis indicate the
limit of detection for TCDD.
The levels of TCDD in fish from the Atlantic or from ponds in the
USA in areas sprayed with 2,4,5-T were below the detection levels (1-2
ng/kg) (Baughman, 1974; Shadoff et al., 1977).
Mitchum et al. (1980) reported levels of 400 ng
2,3,7,8-tetraCDD/kg in fish samples collected in Bayou Meto/Arkansas
River, USA, a waterway associated with industrial plants for the
production of 2,4,5-T (Thibodeux, 1983) (see Table 21).
Levels ranging from 4-695 ng TCDD/kg were found in the edible
portion of channel catfish, carp, yellow perch, small-mouth bass, and
suckers from Saginaw Bay, Michigan, USA, near facilities used for the
production of 2,4,5-T herbicides. The highest concentrations were
detected in bottom-feeding catfish and carp, while the lowest
concentrations were detected in bass, perch, and suckers (see Table
22) (Harless et al., 1982).
Rappe et al. (1981) identified a series of tetra- to octaCDFs in
fat samples of a snapping turtle from the Hudson River and of gray
seal from the Baltic Sea. The total levels of PCDFs in these samples
were 3 ng/g and 40 ng/kg, respectively. In both samples the major
PCDFs consisted of the most toxic isomers (2,3,7,8-tetra-;
2,3,4,7,8-penta-; and 1,2,3,4,7,8- and 1,2,3,6,7,8-hexaCDFs).
Norstrom et al. (1982) have analyzed pooled samples of herring
gull eggs collected in 1982 from various parts of the Great Lakes, N.
America. In all samples, 2,3,7,8-tetraCDD was found in levels ranging
from 9 to 90 ng/kg. The identity of the 2,3,7,8-isomer was confirmed
by retention times on three capillary columns. In another study
Stalling et al. (1983) were not able to detect measurable levels of
tetraCDDs and other PCDDs in fish samples from Lake Superior,
N.America (the detection level was 2-5 pg/g). The difference could be
explained by the migration of the herring gulls during the winter. On
the other hand, a series of PCDFs could be identified in the Lake
Superior fish samples, indicating more widespread background levels
for the PCDFs than for the PCDDs. Stalling et al. (1983) found the
total levels of PCDFs in fish samples from Lakes Michigan, Huron, and
Ontario, N. America, to be 12-290 ng/kg. The toxic 2,3,7,8-substituted
PCDDs and PCDFs were present in all samples, the highest levels being
found in samples from Lake Huron, Lake Ontario, and the Tittabawasee
River, which flows into Saginaw Bay. The residue pattern found in the
fish and locally high levels suggest a strong influence by local point
source discharges (Stalling et al., 1983). The data of O'Keefe et al.
(1983) are also in agreement with this theory.
Norstrom et al. (1986) have studied the long-term trends of
2,3,7,8-substituted PCDDs and PCDFs in herring gull eggs in the Great
Lakes. The levels of 2,3,7,8-tetraCDD were found to decline
exponentially in Lake Ontario, with a half-life of 3-4 years, from a
high of 2000-5000 ng/kg in the early 1970s to a level of 80-100 ng/kg
in 1984/1985. The levels of TCDD in Lake Michigan were 249 ng/kg in
1971, 70 ng/kg in 1972, and 10-20 ng/kg in 1984/1985. These levels
have not changed significantly since 1979. This suggests that an
equilibrium between input and removal mechanisms has been established
in this water system for most PCDDs and PCDFs. The same trend is
reported for various fish species in Lake Ontario (Ontario, 1986).
Ryan et al. (1983a) analyzed a series of commercial and sport
fish from the Great Lakes and from the Pacific coast of Canada for
2,3,7,8-tetraCDD (Table 23). The highest levels were found in Lake
Huron and Lake Ontario. In a preliminary study they also reported
finding levels of 2,3,7,8-tetraCDFs and other unidentified tetraCDFs
of 3-200 ng/kg of fish.
The Baltic Sea is an area of interest because this region is
without any known point sources of dioxins. Rappe et al. (1987)
reported on the analyses of two samples of homing salmon and two
samples of pooled herring; one herring sample from the Baltic Sea
(Karlskrona) and the other from the northern part of the Gulf of
Bothnia (Lulea) (Table 24). As expected, the levels in the salmon
muscle were much higher than the levels found in the herrings, but,
unexpectedly, the levels in the herring sample from the Gulf of
Bothnia (Lulea) were somewhat higher than levels found in the sample
from the Baltic Sea (Karlskrona).
An interesting observation is that in the majority of the aquatic
samples only the 2,3,7,8-substituted PCDD and PCDF congeners were
found. However, crustaceans seemed to be an exception from this
general trend. Norström et al. (1988) reported that crab
hepatopancreas from the Canadian Pacific Coast contain other
congeners, e.g. 1,2,4,7,8-pentaCDD and
1,2,3,6,7,9-/1,2,3,6,8,9-hexaCDD. Rappe et al. (1987) collected and
analyzed crab hepatopancreas from three different locations along the
west coast of Sweden. The crab samples from the locations Grebbestad
and Idefjord should represent background levels, while Väröfjord has
a potential point source of dioxins from a pulp mill using chlorine
for bleaching. The results are given in Table 25.
Low background levels of series PCDDs and PCDFs were found in all
samples. In addition, the sample from the Väröfjord also contained
much higher levels of some congeners, especially 2,3,7,8-tetraCDF and
2,3,7,8-tetraCDD. This is another indication that pulp bleaching could
be a potential source of 2,3,7,8-tetraCDD and 2,3,7,8-tetraCDF (see
section 3.5.10).
4.4.3 Terrestrial animals
In a heavily sprayed test area in north-west Florida (Young et
al., 1976), a total of 106 adult and 67 fetuses of beach mice
(Peromysous polionotus) were collected in 1973 and 1974 and
examined (method not specified). Livers from the beach mice contained
from 540-1300 ng TCDD/kg and the pelts 130-140 ng/kg. The visceral
mass of race runners (Cnemidophorus sexlineatus) which were caught
in that area contained 360 ng TCDD/kg and the trunk of the reptiles
contained 370 ng/kg.
At the time of the accident in Seveso, Italy, more than 81 000
animals were inhabiting the contaminated zones. Most were rabbits (25
000), poultry, and other small animals (55 500), with 349 cattle, 233
pigs, 49 horses, 21 sheep, and 49 goats also in the zones. Many of
these animals died and others were killed. A large number of these
animals were analyzed for 2,3,7,8-tetraCDD by a method with a
detection level of 250 ng/kg (Pocchiari et al., 1983). The results are
summarized in Tables 26 and 27.
Table 23. Levels of 2,3,7,8-tetraCDD and PCB in Great Lakes Canadian sport
fish (1980) and smelt (1979)a
Species Origin TCDD PCB
(ng/kg) (µg/g)
Lake troutb Lake Ontario 58 7.28
Lake Huron 37 5.03
Rainbow troutb Lake Ontario 33 1.77
Coho salmon Lake Ontario 28c 7.39
Pacific Coast NDd (4) 0.03
Smelt Lake Ontario 11
16
11
Lake Erie NDd (2)
a From: Ryan et al. (1983a).
b Whole fish.
c Also contained 36 ng hexaCDD/kg (three isomers) and 93 ng
octaCDD/kg.
d ND = not detected at bracketed detection limit.
Table 24. Levels of PCDDs and PCDFs in fish samples from the Baltic Sea
(pg/g) a,b
Salmon Salmon Herring Herring
Ume River Ume River Karlskrona Lulea
1985 1985 1983 1983
2,3,7,8-TetraCDF 29 12 5.5 3.0
2,3,7,8-TetraCDD 1.9 1.3 < 0.3 < 0.6
1,2,3,7,8-/1,2,3,4,8-PentaCDF 6.9 3.3 1.4 0.9
2,3,4,7,8-PentaCDF 49.0 23.0 6.8 8.8
1,2,3,7,8-PentaCDD 8.8 4.3 1.1 4.7
1,2,3,4,7,8-/1,2,3,4,7,9-HexaCDF 1.1 0.7 0.4 0.3
1,2,3,6,7,8-HexaCDF 1.3 0.8 0.4 0.3
1,2,3,7,8,9-HexaCDF ND ND 0.4 0.2
2,3,4,6,7,8-HexaCDF 1.1 0.6 0.4 0.2
1,2,3,4,7,8-HexaCDD ND 0.4 0.2 ND
1,2,3,6,7,8-HexaCDD 4.6 2.3 ND 8.1
1,2,3,7,8,9-HexaCDD ND ND ND ND
Total HeptaCDFs ND 2.7 0.8 ND
Total HeptaCDDs ND ND ND ND
OctaCDF ND 1.0 ND ND
OctaCDD ND ND ND ND
Table 24.(cont'd) Levels of 2,3,7,8-tetraCDD and PCB in Great
Lakes Canadian sport fish (1980) and smelt (1979)a
a From: Rappe et al. (1987).
b ND indicates a level < 0.1 pg/g.
Harless et al. (1983) reported a study in which 2,4,5-T
containing less than 0.1 mg of 2,3,7,8-tetraCDD/kg was applied at a
rate of 3.4 kg/ha to approximately 3 ha of an enclosed plot (4.5 ha).
Twelve deer were placed in the enclosure prior to the application of
2,4,5-T. One deer died two days later of unknown causes. The remaining
deer were sacrificed prior to, and at specific intervals during, the
course of the 30-day study. The analytical results are summarized in
Table 28.
In another study (Hryhorczuk et al., 1981), samples from a horse
grazing close to a wire reclamation incinerator were analyzed and
found to contain unspecified tetraCDFs (165 ng/kg in the fat, 57 ng/kg
in the liver) and unspecified tetraCDDs (45 ng/kg in the fat and less
than 6 ng/kg in the liver) (compare section 3.5.7).
In order to identify PCDD and PCDF levels in the general
terrestrial background, Nygren et al. (1986) analyzed bovine samples
- fat, liver, and milk - and identified the same 2,3,7,8-substituted
PCDDs and PCDFs as were found in the aquatic samples. However, the
levels were lower and normally close to the detection limit.
4.4.4 Human data
Occupational exposure to 2,3,7,8-tetraCDD can occur during the
production of 2,4,5-trichlorophenol and the subsequent production and
use of 2,4,5-T acid and esters. The first commercial production of
2,4,5-T in the United States was in 1944, and the use of 2,4,5-T
herbicides increased in the 1940s and 1950s. However, the problem of
dioxin contamination in 2,4,5-T was not recognized until 1957 (Kimmig
& Schulz, 1957a, b).
During the normal production of 2,4,5-T, the heaviest exposure to
TCDD is during purification steps. The residues are far more
contaminated than the purified products. Only limited information is
available on the levels of TCDD contamination of products prepared
prior to the 1970s, and absolutely no information is available on the
dioxin levels in the corresponding residues. Consequently it is a
difficult task to estimate the levels of occupational and general
population exposures during the period prior to 1970.
Table 25. Levels of PCDDs and PCDFs in samples of crab hepatopancreas
from the west coast of Swedena
Crab Hepatopancreas
Idefjorden Grebbestad Väröfjord
(pg/g) (pg/g) (pg/g)
2,3,7,8-tetraCDF 31 47 590
Total tetraCDFs 90 114 800
2,3,7,8-tetraCDD 17 17 170
Total tetraCDDs 17 17 170
1,2,3,7,8-pentaCDFb 6 7.6 45
2,3,4,7,8-pentaCDF 44 50 130
Total pentaCDFs 130 150 490
1,2,3,7,8-pentaCDD 13 11 28
Total pentaCDDs 86 76 270
1,2,3,4,7,8-hexaCDFc 12 16 50
1,2,3,6,7,8-hexaCDF 3 5 10
1,2,3,7,8,9-hexaCDF 3 3 11
2,3,4,6,7,8-hexaCDF 16 18 63
Total hexaCDFs 70 88 280
1,2,3,4,7,8-hexaCDD 8 5 14
1,2,3,6,7,8-hexaCDD 26 18 71
1,2,3,7,8,9-hexaCDD 3 4 7
Total hexaCDDs 154 170 465
Total heptaCDFs 23 28 90
Total heptaCDDs 32 30 85
octaCDF < 1 < 1 < 2
octaCDD < 1 < 1 < 2
a From: Rappe et al. (1987).
b Not separated from 1,2,3,4,8-pentaCDF.
c Not separated from 1,2,3,4,7,9-hexaCDF.
Table 26. TCDD content of the livers of farm animals from Seveso
contaminated zones and surrounding areas (1976-1979)a
Animal Number TCDD-containing TCDD maximum
of samples samples level (ng/g)
Rabbitsb 698 433 633
Poultry 83 35 24
Cattle 43 21 94
Horses 12 2 88
Pigs 13 0 -
Goats 25 17 1
Cats 1 0 -
a From: Pocchiari et al. (1983).
b Figures include rabbits kept in the special test plots on
contaminated ground for experimental purposes.
Table 27. CDD analyses of wildlife from Seveso contaminated zones and
surrounding areas (1976-1979)a
Animal Tested organs Number of Maximum level
and number of samples TCD-containing of TCDD
samples (ng/g)
Rabbits 6 (liver) 4 13
Field mice 14 (whole body) 14 49
Rats 1 (pool-4 livers) 28
Earthworms 2 (pool) 12
Frogs 1 (liver) 0.2
Snakes 1 (liver) 3
a From: Pocchiari et al. (1983).
Table 28. Analytical results for 2,3,7,8-tetraCDD residuesa
Sections of 11 No. of deer No. of Concentration range Limit of
deer in study samples positive of TCDD detected detection
analyzed samples (ng/kg)b range
(ng/kg)b
Muscle 11 3 12 - 27 0.5 - 5
Adipose tissue 10 8 3 - 12 1 - 3
Table 28. (cont'd) Analytical results for 2,3,7,8-tetraCDD residuesa
Sections of 11 No. of deer No. of Concentration range Limit of
deer in study samples positive of TCDD detected detection
analyzed samples (ng/kg)b range
(ng/kg)b
Liver 11 4 2 - 5 0.4 - 4
Bone marrow 5 0 ND 1 - 3
a From: Harless et al. (1983).
b Results corrected for efficiency of sample preparation.
ND = not detected.
4.4.4.1 Adipose tissue
Gross et al. (1984) reported a study in which 30 coded samples of
adipose tissue from Viet Nam veterans were analyzed for TCDD. The TCDD
levels found for two of the three heavily exposed men were 99 pg/g and
63 pg/g, which is higher than for the other Viet Nam veterans or for
the controls (all well below 15 pg/g). Only one single isomer of
tetraCDDs was found, and it was assumed that this was the
2,3,7,8-isomer. The data in this study has also been discussed by
Young et al. (1983). These authors concluded that the levels do not
correlate well with known exposure data or with health status.
Rappe et al. (1984) reported the presence of 2,3,7,8-substituted
PCDDs and PCDFs in samples of human adipose tissue from Northern
Sweden. A series of reports presented during the period 1984-1986
confirms these observations and it has been clearly shown that there
is a background of 2,3,7,8-substituted PCDDs and PCDFs in the general
population in the industrialized part of the world. Most of these
reports are lacking data on how the sampled people were selected and
possible exposure to PCDDs and PCDFs. Consequently, these studies
might not be representative. A series of earlier studies failed to
detect these background levels due to insufficiently low detection
levels. The Swedish study included 31 people, of which 18 were exposed
to phenoxy esters and 13 were nonexposed. The group included 17 cancer
patients and 14 non-cancer patients. The different groups were matched
against each other. No difference in the levels, isomer patterns, or
ranges could be found between these subgroups (Nygren et al., 1986).
The mean values for these 31 people are given in Table 29.
Schecter et al. (1986a) reported the mean levels of PCDDs and
PCDFs in 46 samples of adipose tissue collected in Canada and in 8
samples from the USA (see Table 29). The Canadian samples were taken
from people who had died in 1976 from car accidents, drownings,
trauma, and suicide. The samples included all ages and both sexes and
came from all over the country. The USA samples, (1983-1984), were
taken from biopsies from New York State residents during the course of
normal medical procedures, and also from autopsies. Table 29 also
includes the PCDD and PCDF levels in adipose tissue samples from Viet
Nam (Schecter et al., 1986b) and from cancer patients in Japan (Ono et
al., 1986).
It is interesting to note the similarity between isomers present,
levels of isomers, isomeric patterns, and congener profiles in samples
collected from the general population in industrialized countries on
three continents. The profile of the PCDD isomers shows increasing
levels with an increasing number of chlorine atoms; the level of OCDD
is 230-900 pg/g. On the other hand, the profile of PCDFs shows a
maximum for 2,3,4,7,8-penta- or 1,2,3,6,7,8-hexaCDF. The difference in
levels found between samples from South and North Viet Nam may be
explained by spraying during the war in the 1960s and by the
difference in industrial activities between the two parts of the
country.
Four samples of adipose tissue taken from German workers exposed
to TCDD in the early 1950s have also been analyzed (Rappe et al.,
1987). In spite of the fact that these workers were exposed more than
30 years before collecting the samples, enhanced levels of
2,3,7,8-tetraCDD could be identified, but the levels of the other
PCDDs and PCDFs seem to be in the normal range (Table 29, last
column).
Patterson et al. (1986) studied the levels of 2,3,7,8-tetraCDD in
the adipose tissue of 39 exposed people and 57 controls in Missouri,
USA. The exposed group had subgroups of recreational, residential, and
occupational exposure. All persons in both the exposed and control
groups had detectable levels of 2,3,7,8-tetraCDD in their adipose
tissue. Nineteen of the 39 exposed people had measurements higher than
the highest level in the control group and six of the exposed people
had levels greater than 100 ng/kg, which was five times higher than
the highest control (Table 30).
Ryan et al. (1986) analyzed autopsy tissue samples that were
collected from three subjects who died in New York State, USA, from
natural causes. The tissue types were: fat (both abdominal and
subcutaneous), adrenal, bone marrow, liver, muscle, spleen, kidney,
and lungs. As far as could be ascertained, no subjects had known
abnormal exposure to PCDDs or PCDFs, yet these chemicals were found in
all tissues analyzed. The highest concentrations of all PCDDs and
PCDFs were found in adipose tissue. In individual tissues of the three
subjects, the levels of individual congeners detected were within a
narrow range, with much higher levels of the higher chlorinated PCDDs
and PCDFs (e.g. hepta- and octa-CDD). In adipose tissue, the level of
2,3,7,8-tetraCDD was 3.7-8.4 ng/kg and that of 2,3,4,7,8-pentaCDF
5.2-13 ng/kg, while octaCDD ranged between 430 and 700 ng/kg. No major
differences were seen between abdominal and subcutaneous fat samples
or between these two types and perirenal fat when the lower lipid
content of the latter was considered. Smaller concentrations of PCDDs
and PCDFs were measured (on a wet weight basis) in decreasing order:
adrenal, bone marrow, liver, muscle, spleen, kidney, and lungs.
The high levels found in exposed Viet Nam veterans and German
workers indicate a very slow excretion rate or metabolism of TCDD in
humans. This indicates a dramatic difference between man and rodents;
in the latter the half-life of TCDD is reported to be in the range of
a few weeks.
4.4.4.2 Blood plasma
Analysis of blood plasma has been used to evaluate occupational
exposure to PCDDs and PCDFs, which can occur during the production or
use of 2,4,6-tri-, 2,3,4,6-tetra-, and pentachlorophenol. Rappe et al.
(1983) investigated such exposure through the analysis of blood plasma
of exposed workers and unexposed controls. Good correlations were
found between the plasma levels and:
(a) the nature of exposure - dermal contact with liquids resulted in
higher levels than inhalation of contaminated dust;
(b) the duration of exposure - higher levels for longer exposure
times.
The isomers present in the formulations used could also be found in
the blood plasma.
Kochman et al. (1986) studied a group of people exposed to PCBs
and PCDFs after a transformer fire in Rheims, France in 1985. Low
levels (1-25 pg/g) of 2,3,7,8-substituted penta-, hexa-, hepta-, and
octaCDFs were found in the blood plasma of these people, and there was
a slight variation in the T-lymphocytes cells.
Kahn et al. (1986) measured the levels of PCDDs and PCDFs in the
blood plasma from 10 heavily exposed Viet Nam veterans and their 17
controls. The levels of TCDD was much higher in the exposed men than
in their controls.
Table 29. Levels of PCDDs and PCDFs in human adipose tissue (ng/kg wet weight)
Sweden USA/NYf Canadaf Japanf N Viet Namff FRG
Isomer n=31a n=8b n=46b n=13c n=9d n=15d n=4e
2,3,7,8-tetraCDD 3 7.2 6.4 (25) 9 (12) < 2 28 (12) 150
1,2,3,7,8-pentaCDD 10 11.1 10 (46) 15 (13) < 2 15 (14) 19.2
1,2,3,6,7,8-hexaCDD 15 96 81 (46) 70 (12) 11 (6) 100 (15) 77
1,2,3,7,8,9-hexaCDD 4 NA NA 12 (10) NA NA 9.4
1,2,3,4,6,7,8-heptaCDD 97 164 135 (46) 77 (12) 28 (6) 178 (15) 56
octaCDD 414 707 830 (46) 230 (12) 104 (8) 1256 (15) 267
2,3,7,8-tetraCDF 3.9 NA NA 9 (13) NA NA 0.9
2,3,4,7,8-pentaCDF 54 14.3 15 (46) 25 (13) 13 (7) 21 (15) 44
1,2,3,4,7,8-hexaCDF 6 NA NA 15 (11) NA NA 10.0
1,2,3,6,7,8-hexaCDF 5 31.3 16 (34) 14 (11) 13 (7) 58 (15) 6.7
2,3,4,6,7,8-hexaCDF 2 NA NA 8 (3) NA NA 3.8
1,2,3,4,6,7,8-heptaCDF 11 16.5 30 (44) NA 7 (3) 29 (15) 19.5
octaCDF 4 NA NA NA NA NA 1
n = number of tissue samples.
NA = not analyzed.
a Nygren et al. (1986).
b Schecter et al. (1986a).
c Ono et al. (1986).
d Schecter et al. (1986b).
e Refers to occupationally exposed workers. Rappe et al. (1987).
f Mean values of positives. Number of positives within brackets.
Levels below the detection level (< 1.0 ng/kg) not included.
Table 30. Comparison of Levels of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (ng/kg) in Adipose Tissue of Exposed and Control
Groupsa
Exposed
Variable Controls Total Recreational Residential Occupational
Number of subjects 57 39 8 16 15
Arithmetic mean 7.4 79.7 90.8 21.1 136.2
Median 6.4 17.0 23.5 14.5 24.7
Range 1.4-20.2 2.8-750 5.0-577 2.8-59.1 3.5-750
Geometric mean 6.4 21.8 24.8 15.3 29.8
Mean age, years (SD) 52.6 (15.7) 44.3 (13.7) 42.1 (14.7) 39.7 (14.9) 50.3 (9.8)
% Men 35.1 61.5 37.5 43.8 93.3
a From: Patterson et al. (1986).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Air
Owing to sampling and analytical problems, very few data are
available on the levels of 2,3,7,8-TCDD and other PCDDs and PCDFs in
normal urban air.
Rappe & Kjeller (1987) reported the levels of PCDDs and PCDFs in
total air samples collected in Hamburg, FRG, and samples of air
particulates collected in Sweden (Table 31). Sample 1 was collected on
the outskirts of Hamburg (representing an urban area), sample 2 in a
traffic tunnel, sample 3 downwind from a MSW incinerator, and sample
4 in the vicinity of a dumpsite and metal refinery. PCDDs and PCDFs
were found in all samples. Lower levels of PCDDs and PCDFs were found
in air particulates than in total air samples (Table 31). Sample 5 was
taken when "clean air" was blowing into a street in Gothenburg,
Sweden, and sample 6 was taken in the same street during an inversion
situation. Samples 7 and 8 were taken at a rural research station
outside Gothenburg when the air was blowing from the sea (sample 7) or
from Gothenburg (sample 8). The isomeric pattern found in these
samples were very similar (Rappe & Kjeller, 1987).
Airborne dust was monitored in 1977 in the Seveso area to
evaluate the possibility that 2,3,7,8-tetraCDD-contaminated particles
might have drifted outside the contaminated areas. A high-volume
sampling technique was used. When pooled particulate samples were
analyzed, levels of 0.17-0.50 pg TCDD/m3 were reported (Wipf et al.,
1982).
The atmospheric concentrations of 2,3,7,8-tetraCDD near two
hazardous waste sites have been monitored. In one study, US EPA (1982)
failed to detect (detection limit: 1-20 pg/m3) any 2,3,7,8-TCDD in the
atmosphere at the Love Canal (New York, USA) area. In another study of
a waste disposal site (near Jacksonville, Arkansas, USA), Thibodeaux
(1983) reported an average concentration of 1100 pg of 2,3,7,8-TCDD/g
in two air particulate samples collected near the disposal site.
Rappe et al. (1985c) analyzed indoor air samples for PCDFs
resulting from fires and explosions in PCB-filled electrical
equipment, and in an industrial situation (locomotive shop) (Table
32).
O'Keefe et al. (1985) analyzed air samples collected in an office
building in Binghamton, New York, USA, after a transformer accident in
the basement in February 1981. The samples were collected after a
primary clean-up and the values are given in Table 33.
Table 31. Levels of PCDDs and PCDFs in samples of total air and air
particulatesa
Total air samples Air particulates
1 2 3 4 5 6 7 8
pg/m3 pg/m3 pg/m3 pg/m3 fg/m3 fg/m3 fg/m3 fg/m3
2,3,7,8-tetraCDFb 0.04 0.72 0.38 0.18 30 240 5 62
Total tetraCDFs 0.36 6.2 4.9 3.3 320 2000 54 490
2,3,7,8-tetraCDD 0.02 0.06 0.02 0.08 3 9 < 1 5
Total tetraCDDs 0.10 0.22 0.21 1.5 150 350 9 130
1,2,3,7,8-pentaCDFc 0.04 0.36 0.42 1.0 39 190 7 58
2,3,4,7,8-pentaCDF 0.04 Inte 0.43 1.2 51 240 6 69
Total pentaCDFs 0.51 4.1 5.0 10 470 2500 85 610
1,2,3,7,8-pentaCDD < 0.02 0.28 0.22 0.6 17 66 5 35
Total pentaCDDs 0.07 1.3 2.4 5.0 200 840 31 280
1,2,3,4,7,8-hexaCDFd 0.03 0.13 0.27 1.1 23 100 8 38
1,2,3,6,7,8-hexaCDF 0.03 0.15 0.24 1.4 20 78 8 33
1,2,3,7,8,9-hexaCDF < 0.01 < 0.05 < 0.02 0.33 4 17 3 14
2,3,4,6,7,8-hexaCDF < 0.01 < 0.05 0.12 0.80 10 84 7 32
Total hexaCDFs 0.18 1.1 2.2 9.5 180 800 70 310
1,2,3,4,7,8-hexaCDD < 0.08 < 0.17 0.19 1.0 3 19 < 1 7
1,2,3,6,7,8-hexaCDD 0.23 0.66 0.71 2.2 11 46 4 14
1,2,3,7,8,9-hexaCDD < 0.08 < 0.17 0.36 5.2 6 92 5 32
Total hexaCDDs 0.74 2.7 5.3 24. 100 520 32 190
Total heptaCDFs 0.10 1.2 2.0 5. 200 1100 120 500
Total heptaCDDs 0.60 3.4 5.3 15. 380 2900 140 1000
OctaCDF < 0.11 < 1.0 0.78 7.0 150 480 100 440
OctaCDD 0.37 6.4 7.4 40.0 290 1900 64 540
a From: Rappe & Kjeller (1987).
b Not separated from 2,3,4,8-tetraCDF.
c Not separated from 1,2,3,4,8-pentaCDF.
d Not separated from 1,2,3,4,7,9-hexaCDF.
e Int = Interferences.
5.2 Water and Leachate
Shadoff et al. (1977) failed to identify 2,3,7,8-tetraCDD in
water from areas in the USA where 2,4,5-T herbicides had been used.
In the 2,4,5-T plant in Jacksonville, Arkansas, USA, Thibodeaux
(1983) could not detect any 2,3,7,8-tetraCDD in the creek water (no
limit of detection levels given).
Since August 1976, a number of tests have been periodically
conducted in Seveso on streams running through the affected area, as
far south as the River Lambro, with consistently negative results.
During the same period sediment samples were taken from Torrents,
Certesa, and Seveso. Positive results of the order of 1 pg/g were
obtained within the first few kilometers downstream from their
confluence, but further downstream results were negative. The
intensive rainfalls after the accident caused the Seveso to repeatedly
overflow its embankments at the point of entry into Milan, thus
depositing silt on adjacent areas. Tests conducted to determine TCDD
in these silts yielded negative findings for the first four floods
while the fifth flood yielded positive findings (pg/g). Since August
1976, the monthly determinations conducted on pipeline and ground
waters have consistently yielded negative results, even when the
analytical detection threshold was as low as 1 pg/litre (parts per
quadrillion) (Pocchiari, 1983).
During 1983 and 1984, the Dow Chemical Company conducted a study
to determine the 2,3,7,8-tetraCDD contamination at its plant in
Midland, Michigan, USA. It was estimated that 0.6 g of
2,3,7,8-tetraCDD was being emitted per year in 2.5x107 m3 of
wastewater effluent (Lamparski et al., 1986).
The Ontario Ministry of Environment has included PCDDs and PCDFs
in its Drinking Water Surveillance Program for the St Clair/Detroit
River area (Ontario, 1986). No 2,3,7,8-tetraCDD has been found in any
sample of raw or treated water. Unspecified congeners of PCDDs and
PCDFs have been found mainly in raw water, octaCDD being the most
frequent congener found. The highest value reported was 1.1 pg
octaCDD/litre raw water in Amherstburg. The octaCDD level in the
treated water was below the detection level of 0.01 pg/litre.
Götz (1986) reported on levels of PCDDs and PCDFs in the oily
leachate from a sanitary landfill in Georgswerder, Hamburg, FRG (table
34).
5.3 Soil and Sediment
The levels of 2,3,7,8-tetraCDD in point source after improper
disposal of industrial waste are discussed in section 3.4.2.
Table 32. Analyses of PCDFs in air samples (pg/m3)a
total
Sample tetra- 2,3,7,8- penta- hexa- hepta- octa-
CDF tetraCDF CDF CDF CDF CDF
Surahammar < 20 < 2 < 10 < 10 < 10 < 10
(during cleaning)
Surahammar < 10 < 2.5 < 10 < 10 < 10 < 10
(after cleaning)
Railway locomotive 500 50 50 30 20 20
(during cleaning
operations)
a From: Rappe et al. (1985c).
Table 33. Concentrations of PCDFs in air samples collected on various
floors of a Binghamton, New York (USA) office after primary cleanup
Analytical results (pg/m3)
Floor/sample typea 2,3,7,8- Total Penta- Hexa-
tetraCDF tetraCDFs CDFs CDFs
3 16 151 43
5 11 126 30 2.0
5 (NE) 20 195 60 8.7
7 11 121 36
9 volatiles 14 140 42
9 particulates 1.8 4.8 4.7
9 (SE) volatiles 13 146 31 3.7
9 (SE) particulates 0.8 3.9 3.2
11 23 76 16
11 (SE + NW) 16 133 19
14 11 92 21
14 (NE) 14 185 13
16 16 118 21
17 volatiles 12 79 24
17 particulates 0.8 3.9 NDb
17 volatiles 9 59 6.6
17 particulates 0.9 NDb 2.9
Table 33. cont'd
a Abbreviations in parentheses designate sampling location on the floor,
e.g., SE = south-east corner. Unless otherwise specified samples were
collected in the north-west corner and analyzed as combined particulates
and volatiles.
b ND = not detected.
Table 34. Levels (ng/g) of the 2,3,7,8-substituted PCDDs and PCDFs in
leachate from a sanitary landfilla
Isomer Concentration Isomer Concentration
2,3,7,8-tetraCDD 60 2,3,7,8-tetraCDF 9
1,2,3,7,8-pentaCDD 28 1,2,3,7,8-pentaCDFb 322
1,2,3,4,7,8-hexaCDD 476 2,3,4,7,8-pentaCDF 261
1,2,3,6,7,8-hexaCDD 1440 1,2,3,4,7,8-hexaCDF 748
1,2,3,7,8,9-hexaCDD 310 1,2,3,6,7,8-hexaCDF 336
1,2,3,7,8,9-hexaCDF 558
2,3,4,6,7,8-hexaCDF 114
a From: Götz (1986).
b Overlapping isomer: 1,2,3,4,8-pentaCDF.
Analytical results of the 1976-1977 survey of Zones B and R in
Seveso were discussed by Pocchiari (1983). TCDD levels in Zones B and
R were, in general, considerably lower than those in Zone A. In fact,
most TCDD levels were lower than 50 µg/m2 in Zone B and 5 µg/m2 in
Zone R. In 1980, a large part of Zone R was remonitored to evaluate
the persistence of TCDD in the soil. This zone had been ploughed and
worked since 1978. A comparison, as well as a statistical evaluation,
of the relevant data indicated a significant decrease (40%) in the
geometric mean level of TCDD in the soil of Zone R.
In 1980 and 1981, soil samples from ten sites in Zone R and five
sites outside Zone R were analyzed using a high resolution GS-MS
system to establish whether other isomers of 2,3,7,8-tetraCDD were
also present. A significant percentage decrease in tetraCDDs could be
accounted for by two isomers (1,3,6,8-tetraCDD and 1,3,7,9-tetraCDD)
present in the majority of the samples tested. These two isomers were
not related to the chemical accident at the factory.
Table 35. PCDD contamination in soil from Zone R in Seveso (1981)
(values in ng/kg)a
Sample 2,3,7,8b TCDDc Penta- Hexa- Hepta- Octa- Total
tetraCDD CDDs CDDs CDDs CDD PCDDs
S1 0.8 0.3 0.4 6.0 1.4 1.7 10.6
S2 3.4 1.0 0.7 9.5 2.1 2.2 18.9
S3 4.0 1.9 1.2 8.2 8.6 27.0 50.9
S4 2.3 0.8 0.6 10.2 2.1 2.0 18.0
S5 < 0.1 < 0.3 0.5 9.5 1.9 1.4 13.7
S6 6.3 1.5 1.1 10.4 2.6 1.3 24.8
S7 1.7 1.0 0.8 12.4 1.9 1.8 19.6
S8 2.2 0.4 0.8 8.8 1.8 0.8 14.8
S9 1.0 2.8 2.3 21.2 9.6 13.5 50.4
a From: Wipf & Schmid (1983).
b Probably related to the accident.
c Total levels of isomers other than 2,3,7,8-tetraCDD,
probably not related to accident.
Wipf & Schmid (1983) reported the presence of PCDDs other than
2,3,7,8-tetraCDD in the soil from Zone R (Table 35). They suggest that
a municipal incinerator and the burning of wood shavings treated with
chlorinated phenols could be the source of the other PCDDs.
Nestrick et al. (1986) reported the levels of 2,3,7,8-tetraCDD in
soil samples collected from industrialized areas of US cities. They
observed a widespread occurrence of 2,3,7,8-tetraCDD in urban soils,
with levels of 1-10 ng/kg, and suggested that local combustion
sources, including MSW and industrial incinerators, were the probable
origin.
McLaughlin & Pearson (1984) measured soil concentrations of PCDDs
and PCDFs in the vicinity of a municipal refuse incinerator in
Ontario, Canada. Urban and rural control locations were also sampled.
All soil samples (14) had detectable quantities of at least one of the
five PCDD congener classes (tetraCDD to octaCCD) tested for, whereas
eight samples contained detectable levels of one or more of the five
PCDF congener groups (tetraCDF to octaCDF). The levels ranged from
non-detectable (0.003 - 0.008 ng/g) to 3.5 ng/g (octaCDD); only one
site had a measurable quantity (0.007 ng/g) of tetraCDD in the soil.
The most abundant PCDD or PCDF congener was octaCDD, which had similar
levels whether samples were taken close to or remote from the
incinerator. Similarly, no concentration gradients, relative to
distance from the incinerator, were apparent for any of the other
PCDDs or PCDFs.
Soil samples near a chemical waste incinerator in Scotland have
also been analyzed for PCDDs and PCDFs, together with samples from
control locations (Edulgee et al., 1986). Detectable levels of all
PCDFs or PCDFs that were examined were found in each of the soil
samples (13). Levels found ranged from 1.2 ng/kg (2,3,7,8-tetraCDD) to
1900 ng/kg (total hexaCDF). No consistent pattern was observed to
differentiate levels found in control samples from levels in soil near
the incinerator.
These studies from widely separate areas of the world support the
suggestion that diffuse combustion sources are the major source of
PCDDs and PCDFs in the soil.
Rappe & Kjeller (1987) analyzed soil samples from various parts
of Europe (Table 36). They represent rural areas (samples 1, 2, and 3)
as well as more industrialized areas (samples 4 and 5). PCDDs and
PCDFs could be identified in all samples. The 2,3,7,8-tetraCDD
concentration was below the detection level in the soil samples from
the non-industrialized areas. Trapped sediments from the archipelago
of Stockholm, Sweden, were also analyzed. The samples were collected
in the inner (sample 6), middle (sample 7), and outer archipelago
(sample 8). Levels decreased with increasing distance from the city of
Stockholm. The isomeric patterns for tetra- and pentaCDF isomers are
very similar to those found for samples of total air and air
particulates (section 6.1). A sediment sample from the mouth of River
Viskan, Sweden, was also analyzed (sample 9). A slight difference in
congener profile was found between this sample and the sediments from
the archipelago of Stockholm.
Czuczwa & Hites (1985) found PCDDs and PCDFs in sediment samples
from several locations in Saginaw River and Bay, and southern Lake
Huron, levels ranging from 100 ng/g in urban areas to 100 ng/kg at
remote sites. Although no isomers were identified, the analytical
profiles in the sediments followed closely those found in combustion
samples, suggesting that combustion is the major source of PCDDs and
PCDFs found in the sediments. Analyses of sediment cores showed a
dramatic increase in the PCDD and PCDF concentrations at a depth
corresponding to approximately the year 1940, and levels remained high
up to the present. There is no good correlation between the trend in
these levels and the trend for coal burning in the United States.
However, the levels in the sediments correlate with the production and
use of chlorinated aromatic compounds within this area of the Great
Lakes.
5.4 Food
5.4.1 Meat and bovine milk
The levels of PCDDs and PCDFs in fish and other seafood are
discussed in section 4.4.2.
Table 36. Levels (pg/g) of PCDDs and PCDFs in samples of sediments and soila
Soil samples Sediments
1 2 3 4 5 6 7 8 9
2,3,7,8-tetraCDFb 2.9 1.6 1.1 34 38 30 17 14 1.6
Total tetraCDFs 9.3 7.7 11 320 370 290 150 120 24
2,3,7,8-tetraCDD < 2.0 < 2.1 < 0.2 2.4 0.8 2.4 2.0 < 2.0 0.2
Total tetraCDDs - - 3.2 55.5 11.2 69 21 23 6.4
1,2,3,7,8-pentaCDFc 2.5 1.6 0.5 17 31 16 8.6 8.5 1.3
2,3,4,7,8-pentaCDF 0.8 1.0 0.6 23 65 20 14 16 1.7
Total pentaCDFs 14 13 6.7 200 450 260 140 130 30
1,2,3,7,8-pentaCDD < 2.0 < 2.0 < 0.1 18 34 7.6 5.2 5.5 0.9
Total pentaCDDs - - 4.6 220 270 230 99 86 13
1,2,3,4,7,8-hexaCDFd 3.8 2.2 0.9 30 45 16 10 8.8 1.9
1,2,3,6,7,8-hexaCDF 1.8 1.5 0.4 11 25 12 7.1 5.9 1.2
1,2,3,7,8,9-hexaCDF 1.0 0.9 4.3 110 1100 5 < 1 < 1 2.0
2,3,4,6,7,8-hexaCDF 1.9 1.0 0.7 26 57 16 31 23 1.6
Total hexaCDFs 16 12 11 270 1900 250 220 92 44
1,2,3,4,7,8-hexaCDD < 2 < 2 < 0.1 13 28 1.6 0.8 1.0 1.6
1,2,3,6,7,8-hexaCDD < 2 < 2 < 0.1 19 64 48 2.0 2.0 10
1,2,3,7,8,9-hexaCDD < 2 < 2 < 0.1 6.2 19 2.5 0.9 1.0 4.3
Total hexaCDDs - - 4.7 200 330 49 16 19 64
Total heptaCDFs 22 14 18 260 4500 1300 1500 190 300
Total heptaCDDs < 10 < 10 17 370 1600 5700 1200 880 190
OctaCDF - - 5.7 68 71 39 < 20 < 20 330
OctaCDD - - 14 140 180 3100 510 260 900
a From: Rappe & Kjeller (1987).
b Not separated from 2,3,4,8-tetraCDF, except in the case of sample 9.
c Not separated from 1,2,3,4,8-pentaCDF.
d Not separated from 1,2,3,4,7,9-hexaCDF.
The US Environmental Protection Agency (US EPA) initiated a
2,3,7,8-tetraCDD monitoring programme of beef fat samples taken from
cattle that had grazed on rangelands known to have been treated with
2,4,5-T. The analytical collaborators in this programme were Dow
Chemical Co. (USA), Wright State University, and Harvard University.
All the laboratories used mass spectroscopic techniques for
quantification. Two different extraction techniques were used. All
three laboratories analyzed control samples taken from cattle that had
grazed on non-treated areas. Some of these control samples were spiked
with known amounts of TCDD. All the controls were prepared by the US
EPA (Firestone, 1978). Good agreement was found between the amounts of
TCDD spiked into the control beef fat samples and the reported levels
found, even down to TCDD levels of 10 ng/kg. The average reported TCDD
level was 10 ng/kg; the amount actually added by the EPA was 9 ng/kg.
Of a total of 34 analyses of controls to which no TCDD was added, in
only one case was there a false positive report of TCDD (O'Keefe et
al., 1977). Of 52 samples of beef fat from 2,4,5-T-treated rangeland,
19 (37%) were reported by one or more laboratories to have TCDD. The
average range of levels reported was 5-66 ng/kg, and the overall
average was 7 ng/kg. If one considers only the 40 beef fat samples
from areas receiving at least 1.1 kg of 2,4,5-T/ha, all 19 positive
samples (48%) belong to this group, and the average reported TCDD
level would be 9 ng/kg. The results indicated a consistent trend,
relating the average reported TCDD level in beef fat to the intensity
of the 2,4,5-T application to rangeland (O'Keffe et al., 1978;
McKinney, 1978).
None of the three collaborating laboratories used the most
selective and sensitive analytical method now known (capillary glass
column gas chromatography - high resolution mass fragmentography).
Kocher et al. (1978) also analyzed specimens of fat taken from
steers that had grazed on rangeland previously treated with 2,4,5-T
herbicides. The limit of detection of TCDD (2.5 times peak to peak
noise) was found to be in the 30-60 pg range (3-6 pg/g in beef fat
using 10 gram samples). None of the sixteen samples analyzed in two of
three studies revealed TCDD. In the third study, the animals were
confined to a fenced pasture sprayed in its entirety with 2,4,5-T
herbicides. The samples from three of the seven animals gave a
positive response at the extremely low level of 3 to 4 ng TCDD/kg,
which is at the detection limit; the highest reported value (without
interfering components) was 13 pg/g. The level of 2,3,7,8-tetraCDD in
the 2,4,5-T used was, however, unknown. Beck et al. (1986) analyzed
seven randomly collected samples of cow's milk from different areas of
the Federal Republic of Germany and one sample from the German
Democratic Republic. The cow's milk was taken from road transport
tankers. In all samples, 2,3,7,8-substituted PCDDs and PCDFs were
found at low levels of pg/g on a fat weight basis. Detection limits
were in the range 0.1-0.3 ng/kg. The levels of PCDDs and PCDFs found
in the milk samples were lower than those measured by Rappe et al.
(1987b) in cow's milk from Switzerland (Table 38). In addition, there
was no evidence of high levels of the higher chlorinated congeners
(e.g. hepta and octa- ) (see Table 37). Levels were much lower than
those in human milk samples (see section 5.4.2).
Rappe et al. (1987b) analyzed PCDDs and PCDFs in six samples of
bovine milk from various locations in Switzerland. In all samples,
2,3,7,8-substituted PCDDs and PCDFs were found at levels of pg/kg in
whole milk (Table 38). However, the levels were lower in commercial
milk samples than in samples collected directly from cows grazing in
the vicinity of incinerators.
When Ryan et al. (1985) analyzed PCDDs and PCDFs in chicken and
pork samples in Canada, the incidence of positives for hexa-, hepta-,
and octaCDD in selected samples of chicken fat was 50, 62, and 46%,
with averages of 27, 52, and 90 ng/kg, respectively. Similar levels of
hexa- and heptaCDFs were also found in some of these samples, but
tetra- and pentaCDDs and tetra-, penta-, and octaCDFs were not
detected. A comparison between the tissue analyses and those of the
wood (treated with pentachlorophenol) used to house the animals showed
a marked similarity (see Table 38), indicating that pentachlorophenol
was the probable source of contamination of the food samples.
Firestone et al. (1986) reported the analyses of various food
items collected in the period beginning in 1979. Low levels (< 300
ng/kg) of 1,2,3,4,6,7,8- and 1,2,3,4,6,7,9-heptaCDD were found in some
samples of chicken, bacon, pork chops, and beef liver. HexaCDD was not
found in any of the foods. Several beef livers had high levels of OCDD
residues, the highest reported value being 3830 ng/kg. No PCDDs (at a
detection limit of 10-40 ng/kg) were found in ground beef.
5.4.2 Human milk
Rappe et al. (1984) reported low levels of 2,3,7,8-substituted
PCDDs and PCDFs in five samples of human milk from the Federal
Republic of Germany (FRG) (Table 39).
Table 39 also indicates the results of analyses of four samples
of human milk from the Umea region of northern Sweden (Rappe, 1985),
92 samples from Rheinland-Westfalia in the FRG (Furst et al., 1987),
30 other samples from the FRG (Beck et al., 1987), and five samples
from the Netherlands or Yugoslavia (Rappe et al., 1987).
Van der Berg et al. (1986) also reported the levels of PCDDs and
PCDFs in human milk samples from the Netherlands, but these levels
were reported on milk basis, not on fat basis, and are therefore not
included in Table 39.
A comparison between the isomers, levels, isomeric pattern, and
congener profiles found in human milk (Table 39) and in adipose tissue
(Tables 29 and 30) shows a remarkable degree of similarity.
5.4.3 Rice
Rice from fields in Arkansas, Louisiana, and Texas, USA, treated
at a maximum rate to give 2.52 kg 2,4,5-T/ha, were analyzed for
possible 2,3,7,8-tetraCDD residues. A specification of 1 µg
2,3,7,8-tetraCDD/g 2,4,5-T was given in the report, but no analytical
data were given for the herbicide. No 2,3,7,8-TCDD was detected in the
rice (detection limit = 2-7 µg/kg) and no 2,3,7,8-TCDD residues
(detection limit = 2-10 µg/kg) were found in 30 samples of rice
purchased in retail stores throughout the USA (Jensen et al., 1983).
5.5 Yusho and Yu-cheng Episodes
In 1968, more than 1500 people in south-west Japan were
intoxicated through consuming commercial rice oil accidentally
contaminated by PCBs, PCDFs, and polychlorinated quarterphenyls
(Masuda & Yoshimura, 1982; Masuda et al., 1985). In 1979, a similar
episode occurred in central Taiwan, the number of persons involved
here approaching 2000 (Chen et al., 1980; Chen et al., 1985). In the
past, both accidents were referred to as Yusho episodes, but now the
Taiwan episode has been renamed Yu-cheng.
The Japanese rice oil contained more than 40 PCDF isomers (tri-
to hexaCDFs) (Buser et al., 1978d), whereas the number of isomers in
the Taiwanese oil seems to have been less (Chen & Hites, 1983). The
toxic 2,3,7,8-substituted PCDFs were middle or minor constituents,
about 10-15% of the total amount of PCDFs (Buser et al., 1978d; Masuda
et al., 1985; Chen & Hites, 1983). The mean total consumption of PCDFs
of the Yusho and Yu-cheng patients has been estimated to be 3.3-3.8
mg/person (Hayabuchi et al., 1979; Chen et al., 1985), or a daily
intake of total PCDFs of 0.9 µg/kg body weight (Hayabuchi et al.,
1979). The average intake of 2,3,7,8-substituted PCDFs was 90-135
ng/kg body weight per day. The smallest amount of total PCDFs causing
chloracne has been estimated to be 0.16 µg/kg body weight per day
(Hayabuchi et al., 1979) or 20-30 ng/kg per day of the
2,3,7,8-substituted congeners.
Analysis of liver samples taken from the Yusho patients about 18
months after the exposure showed a dramatic decrease in the number of
PCDF isomers. Apparently most of the PCDF isomers were metabolized or
excreted during the period between exposure and sampling (Rappe et
al., 1979). A comparison between the PCDF isomers found in the Yusho
oil and the liver samples revealed an interesting relationship. Most
of the isomers retained had lateral positions (2, 3, 7, and 8)
substituted with chlorine; these isomers have the highest toxicity
(Rappe et al., 1979).
Table 37. PCDD and PCDF levels in samples of cow's milk (ng/kg on a fat weight basis)a
SAMPLE
1 2 3 4 5 6 7 8
2,3,7,8-tetraCDF < 0.1 0.29 0.28 1.1 1.0 1.4 1.4 0.27
2,3,7,8-tetraCDD 0.33 < 0.2 < 0.2 < 0.2 ND < 0.2 ND ND
1,2,3,7,8-pentaCDF ND 0.4 ND 0.26 0.24 ND 0.39 < 0.2
2,3,4,7,8-pentaCDF 1.3 0.91 1.1 1.6 1.5 1.3 2.9 0.8
1,2,3,7,8-pentaCDD 1.0 0.72 ND 0.81 0.6 0.78 1.2 < 0.5
1,2,3,4,7,8-hexaCDF 0.93 0.67 0.70 0.85 < 0.3 0.84 1.9 0.57
1,2,3,6,7,8-hexaCDF 0.73 0.58 0.57 0.85 < 0.3 0.73 2.1 0.41
2,3,4,6,7,8-hexaCDF 0.65 0.48 0.53 0.68 ND 0.64 1.8 0.37
1,2,3,4,7,8-hexaCDD 0.33 0.34 < 0.3 < 0.3 < 0.3 0.36 0.33 < 0.3
1,2,3,6,7,8-hexaCDD 1.3 1.2 1.0 0.82 0.32 1.7 1.9 0.80
1,2,3,7,8,9-hexaCDD < 0.3 0.34 0.36 0.39 ND 0.55 0.48 < 0.3
1,2,3,4,6,7,8-heptaCDFb < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5 < 0.5
1,2,3,4,6,7,8-heptaCDDb < 2 < 2 < 2 < 2 < 2 < 2 < 2 < 2
octaCDDb <10 <10 <10 <10 <10 <10 <10 <10
octaCDFb < 1 < 1 < 1 < 1 < 1 < 1 < 1 < 1
a From: Beck et al. (1987).
b Not significantly higher than blanks.
ND = not detectable.
Table 38. PCDF and PCDD content of bovine milk from Switzerland. Results in ng/kg (ppt), whole milk basisa
Compound Commercial Incinerators
1 2 3 4 5 6
2,3,7,8-tetraCDF < 0.028 < 0.035 < 0.021 < 0.022 < 0.032 < 0.028
1,2,3,7,8-pentaCDF < 0.020 < 0.022 < 0.021 < 0.020 < 0.036 < 0.032
2,3,4,7,8-pentaCDF 0.084 0.066 0.069 0.43 0.22 0.23
1,2,3,4,7,8-hexaCDF < 0.020 < 0.026 < 0.017 0.13 0.06 0.084
1,2,3,6,7,8-hexaCDF 0.028 < 0.018 < 0.021 0.19 0.095 0.059
2,3,4,6,7,8-hexaCDF < 0.020 < 0.018 ND 0.28 0.12 0.049
(< 0.02)
1,2,3,4,6,7,8-heptaCDF < 0.12 ND ND 0.49 0.28 < 0.18
(< 0.13) (< 0.08)
octaCDF < 0.20 ND ND ND ND < 0.52
(< 0.13) (< 0.09) (< 0.16) (< 0.21)
2,3,7,8-tetraCDD ND ND ND 0.049 0.038 0.021
(< 0.012) (< 0.013) (< 0.013)
1,2,3,7,8-pentaCDD ND ND ND 0.25 < 0.086 ND
(< 0.04) (< 0.08) (< 0.06) (< 0.1)
1,2,3,4,7,8-hexaCDD < 0.068 ND ND 0.23 0.14 < 0.14
(< 0.1) (< 0.06)
1,2,3,6,7,8-hexaCDD < 0.068 ND ND 0.29 0.16 < 0.21
(< 0.1) (< 0.06)
1,2,3,7,8,9-hexaCDD < 0.068 ND ND 0.17 < 0.080 < 0.11
(< 0.1) (< 0.06)
1,2,3,4,6,7,8-heptaCDD < 0.064 < 0.066 < 0.064 0.26 < 0.095 0.42
octaCDD < 0.16 < 0.26 < 0.12 0.28 < 0.16 0.59
a From: Rappe et al. (1987b).
ND = not detectable.
Table 39. Levels of PCDDs and PCDFs found in human milk (ng/kg of fat weight).
Sweden FRG FRG FRG Netherlands Yugoslavia
n=4a n=5b n=92c n=30d n=3e n=2e
2,3,7,8-tetraCDD 0.6 1.9 < 5 3.4 9.7 < 1.0
1,2,3,7,8-pentaCDD 6.5 12.9 10.7 15 44 5.5
1,2,3,4,7,8-hexaCDD 2.5 4.6 8.1 12 25 3.5
1,2,3,6,7,8-hexaCDD 19 17.3 32.7 59 251 15
1,2,3,7,8,9-hexaCDD 6.3 1.6 6.4 11 23 ND
1,2,3,4,6,7,8-heptaCDD 59.5 72.8 49.9 61 130 106
octaCDD 302 434 181 530 744 106
2,3,7,8-tetraCDF 4.2 5.4 2.6 2.5 2.8 < 1.0
1,2,3,7,8-pentaCDF < 1 < 1 1.8 < 1 ND ND
2,3,4,7,8-pentaCDF 21.3 36.4 22.9 20 79 25
1,2,3,4,7,8-hexaCDF 4.7 11.4 8.2 8.5 8.9 3.7
1,2,3,6,7,8-hexaCDF 3.4 10.2 6.6 7.8 10.3 3.6
2,3,4,6,7,8-hexaCDF 1.4 4.3 3.3 3.0 6.4 1.3
1,2,3,4,6,7,8-heptaCDF 7.4 9.2 6.4 8.5 39 ND
octaCDF 3.2 2.4 22.8 < 3 ND ND
ND = not detected; NA = not analyzed; n = number of samples.
a Rappe (1985).
b Rappe et al. (1984).
c Furst et al. (1987).
d Beck et al. (1987).
e Rappe et al. (1987) (pooled samples)
Kunita et al. (1984) studied the blood levels of PCDFs in people
involved in the Japanese and Taiwanese episodes. They used a
non-isomer-specific analytical method, and reported higher blood
levels in persons with severe dermal symptoms than in persons with
light symptoms. The levels 2 years after exposure were lower than 0.5
year after exposure; for the six persons studied the levels of total
PCDFs decreased on average by 57% ± 12% (Kunita et al., 1984).
The rate of excretion of these toxic PCDF isomers is very low.
Rappe & Nygren (1984) could detect 2,3,4,7,8-pentaCDF in blood plasma
from Yusho patients when the samples were collected 11 years after
exposure. However, higher levels were found in blood from Yu-cheng
patients one year after exposure; these analyses also showed a 15-20%
reduction in levels in one year (Rappe, 1984).
6. KINETICS AND METABOLISM OF 2,3,7,8-TETRACHLORO-
DIBENZO-P-DIOXIN (TCDD) AND OTHER PCDDs
6.1 Uptake, Distribution, and Excretion
Most of the available toxicokinetic data arise from studies of
gastrointestinal exposure and oral or intraperitoneal administration.
There has been one dermal study on rats, but no studies on exposure
via the respiratory tract. Data on the gastrointestinal absorption,
distribution, and elimination of TCDD in various species are
summarized in Tables 40 and 41. Principle organ depots in all species
studied are the liver and adipose tissue. In addition skin and muscle
have been found to be organ depots in monkeys, and skin in
guinea-pigs. In all species studied clearance of TCDD from the body
follows apparent first-order kinetics.
6.1.1 Studies on rats
Male Sprague Dawley rats were dosed by gavage with 14C TCDD in
acetone: corn oil (1:9), at a concentration of 50 µg/kg body weight
(Piper et al., 1973). Rats lost weight and their physical condition
was generally poor, although no deaths occurred. Approximately 30% of
the administered dose of radioactivity was eliminated in the faeces
during the first 48 h, this being most probably unabsorbed TCDD. The
total faecal TCDD content was equivalent to 53.2% of the dose over a
21-day period. During this time 13.2% was eliminated in the urine and
3.2% in the air. From these results the half-time for the clearance of
TCDD was calculated as 17.4 (± 5.6) days. In another study, groups of
two to three rats were killed at different time intervals after
administration of the same dose of 14C-TCDD. The liver contained
3.2, 4.5, and 1.3% of the administered dose per gram of tissue 3, 7,
and 21 days, respectively, after dosing. The concentrations in adipose
tissue at the same time intervals were 2.6, 3.2, and 0.4% of the dose
per gram of tissue. Other tissues showed lower concentrations.
In a study by Allen et al. (1975), male Sprague Dawley rats were
given 14C-TCDD in a single dose of 50 µg/kg body weight by stomach
tube. Groups of five rats were sacrificed 1, 3, 5, 7, 14, and 21 days
after dosing. The dose given resulted in marked liver hypertrophy,
thymic regression, weight loss, and death in 50% of the animals within
25 days. Twenty-five percent of the dose was eliminated within the
first 3 days through the faeces. During the next 18 days, 1 to 2% of
the dose was found in the faeces daily. The total amount in the faeces
during the 21 days following administration was about 52%.
Table 40. Gastrointestinal absorption of TCDD
Species/Strain Vehicle Dose % Absorption Reference
(µg/kg body weight) (mean ± SD)
Rats
Sprague Dawley acetone:corn oil (1:9) 50a 70 Piper et al. (1973)
Sprague Dawley corn oil 50a > 75 Allen et al. (1975)
Sprague Dawley diet 7 or 20; for 42 days 50-60 Fries & Marrow (1975)
Sprague Dawley acetone:corn oil (1:24) 1a 84±11 Rose et al. (1976)
Sprague Dawley acetone:corn oil (1:24) 0.1 or 1.0; 5 days/week 86±12 Rose et al. (1976)
for 7 weeks
Mice
ICR/Ha Swiss ethanol:Tween 80:saline 135a 27-28 Koshakji et al. (1984)
(1:10:89)
Hamsters
Golden Syrian olive oil 650a 73.5±22.8 Olson et al. (1980a)
a Single dose.
Table 41. Elimination of TCDD in different species
Species/Strain Route/Vehicle Dosea Duration Half-life Eliminated radioactivity Reference
(µg/kg of for elimination (% of administered dose)
body study (days)
weight) (days) Faeces Urine
Rats
Sprague Dawley oral/acetone: 50 21 17.4 ±5.6 53.2 13.2 Piper et al. (1973)
corn oil (1:9)
Sprague Dawley oral/corn oil 50 21 21.3 ±2.9 53.3 4.5 Allen et al. (1975)
Sprague Dawley oral/acetone: 1 22 31 ± 6 NR ND Rose et al. (1976)
corn oil (1:24)
Sprague Dawley oral/acetone: 1b 49 23.7 NR 3.1 ±0.2 (m) Rose et al. (1976)
corn oil (1:24) 12.5 ±5.1 (f)
Sprague Dawley ip/corn oil 400 7 NR 4.96 ±0.3 0.51 ±0.05 van Miller et al.
(1976)
Mice
C57BL/6 ip/olive oil 10 30 11 59.3 20.5 Gasiewicz et al.
(1983b)
DBA/2 ip/olive oil 10 30 24.4 39.8 16.8 Gasiewicz et al.
(1983b)
B6D2F1 ip/olive oil 10 30 12.6 56.3 20.5 Gasiewicz et al.
(1983b)
ICR/Ha Swiss oral/ethanol: 135 11 20 78 4 Koshakji et al.
tween 80: (1984)
saline (1:10:89)
C57BL/6 Ahb/Ahd ip/emulphor: 0.5 42 9.6 61.8 19.9 Birnbaum (1986)
ethanol:H2O
(1:1:18)
C57BL/6 Ahd/Ahd ip/emulphor: 0.5 42 9.6 55.9 26.8 Birnbaum (1986)
ethanol:H2O
(1:1:18)
DBA/2 Ahb/Ahd ip/emulphor: 0.5 42 10.8 71.5 13.6 Birnbaum (1986)
ethanol:H2O
(1:1:18)
Table 41 (contd).
Species/Strain Route/Vehicle Dosea Duration Half-life Eliminated radioactivity Reference
(µg/kg of for elimination (% of administered dose)
body study (days)
weight) (days) Faeces Urine
Mice (contd)
DBA/2 Ahd/Ahd ip/emulphor: 0.5 42 10.8 76.1 11.1 Birnbaum (1986)
ethanol:H2O
(1:1:18)
Guinea-pigs
Hartley ip/olive oil 2 23 33 2 Gasiewicz &
Neal (1979)
NR oral/NR NR 22 22-43 ND ND Nolan et al. (1979)
Hartley ip/olive oil 0.56 45 93.7 ± 15.5 26.2 ±3.6 3.1 ±1.2 Olson (1986)
Hamsters
Golden Syrian oral/olive oil 650 35 15.0 ±2.5 NR NR Olson et al.
(1980a)
Golden Syrian ip/olive oil 650 35 12.0 ±2.0 50.0±2.7 34.6±5.4 Olson et al.
(1980a)
Monkeys
Rhesus (adult) ip/corn oil 400 7 NR 3.75 1.06 van Miller et al.
(1976)
Rhesus (infant) ip/corn oil 400 7 NR 1.26 2 van Miller et al.
(1976)
a Single dose, unless otherwise stated.
b 5 days/week for 7 weeks.
ND = not detectable, NR = not reported, m = males, f = females, ip = intraperitoneal.
The authors concluded that the faecal content during the first 3
days represented mainly unabsorbed TCDD and that apparently more than
75% of the administered dose had been absorbed from the
gastrointestinal tract. The radioactivity excreted daily through the
urine ranged between 0.1 and 0.2% of the administered dose during the
initial 12 days. Thereafter, the daily urinary radioactivity excretion
increased from 0.25% of the administered dose to 0.43% by day 21. The
total amount of 14C excreted through the urine over a 21-day period
was about 4.5% of the dose. On the basis of the daily faecal and
urinary excretion, the authors calculated a half-time of 21.3 (± 2.9)
days. In the rats killed on days 1, 3, 5, 7, 14, and 21 after
administration, the total liver content was 56 (± 5), 54 (± 14), 54 (±
6), 54 (± 8), 45 (± 5), and 24 (± 4)% of the administered dose,
respectively. At all intervals, the levels found in the liver exceeded
those found in other organs. On days 5, 7, and 14 about 90% of the
total radioactivity in the liver was present in the microsomal
fraction.
Fries & Marrow (1975) fed Sprague Dawley male and female rats a
diet containing 7 or 20 µg 14C-TCDD/kg diet for 42 days. Thereafter
all rats received the control diet for another 30 days. Two animals of
each sex and TCDD dietary level were sacrificed at 14-day intervals.
This treatment resulted in decreased food consumption, decreased
weight gain, and increased relative liver weight among both males and
females. The concentration of TCDD-derived radioactivity in the liver,
which was the principal tissue depot of both males and females, was
directly proportional to the dietary intake of 14C-TCDD. At the end
of the 42-day feeding period, the 14C levels in male livers
indicated TCDD contents of 5.8 and 15.9 µg/kg of tissue for the lower
and higher feed concentrations, respectively. The concentrations in
the liver of female rats were similar. Analysis of the liver of rats
killed 14 and 30 days after discontinuing TCDD exposure indicated a
gradual decrease in TCDD liver concentration in both sexes. The
steady-state body burden for TCDD was estimated to be 10-11 times the
daily intake in both sexes. The whole-body and liver half-lives were
calculated to be 12 and 11 days in males and 15 and 13 days,
respectively, in females.
Rose et al. (1976) estimated the absorption of a single non-toxic
dose of 1 µg 14C-TCDD/kg body weight in male and female Sprague
Dawley rats to be 84 ± 11% of the administered dose. The elimination
of TCDD was followed for 22 days after administration and the faecal
excretion accounted for most if not all of the elimination of TCDD
and/or its metabolites. No radioactivity was detectable in urine and
expired air. Twenty-two days after dosing, mean values of 1.26% and
1.25% of the dose/g liver and adipose tissue, respectively, were
found. Much lower 14C-activities were found in the thymus, kidney,
and spleen, namely 0.09, 0.06, and 0.02% of the dose/g, respectively.
The whole-body half-life was estimated to be 31 days. Rose et al.
(1976) also followed the fate of 5 daily doses per week of 0.01, 0.1,
and 1.0 mg 14C-TCDD/kg body weight given for 1, 3, and 7 weeks to
male and female Sprague Dawley rats (Table 42).
According to Kociba et al. (1976), a dose level of 0.01 µg/kg per
day, 5 days/week for 13 weeks, produced no overt toxic effects,
whereas 0.1 or 1.0 µg/kg per day produced adverse effects, including
some deaths in the high dose group. In the study by Rose et al.
(1976), the dose level of 0.01 µg/kg per day resulted in no detectable
14C, except in liver, fat, and excreta. Thus no kinetic calculations
on a truly non-toxic dose could be performed. Pooling the results for
all rats receiving 0.1 or 1.0 µg/kg per day, the absorbed dose
corresponded to 86 ± 12% of the administered dose, with an individual
variation of 66 to 93%. The overall rate constant for elimination
corresponded to a half-time of 23.7 days, with an individual variation
of 16-37 days. The radioactivity was eliminated primarily in the
faeces, but the percentage of the dose excreted in the urine compared
to that eliminated in the faeces tended to increase with time. At the
dose level of 1.0 µg/kg per day, males excreted in the urine 3.1 (±
0.2)% and females 12.5 (± 5.1)% of the cumulative dose over 7 weeks of
exposure. The one female rat that died during the 7th week excreted
17.8% of the cumulative dose in the urine. Exhaled air was not
examined in these studies. After 7 weeks of exposure, the average body
burdens were 47.7 (± 8.8) and 37.1 (± 7.5)%, respectively, of the
administered dose for the rats given 0.1 and 1.0 mg TCDD/kg per day.
The main tissue depots of 14C were the liver and adipose tissue.
Radioactivity was also detected in thymus, kidney, and spleen at
levels between 1 and 2% of that in liver. Direct chemical
determination of liver samples confirmed the TCDD-concentrations
calculated by radioactivity measurements. TCDD and/or its metabolites
approached a steady-state body burden calculated to be 21.3 Do for
rats given a daily dose (Do, µg/kg body weight) on 5 consecutive
days per week for an infinite number of weeks, within 13 weeks over
the dose range 0.01 to 1.0 µg/kg per day. It was thought unlikely that
the dietary intake of extremely low levels of TCDD would result in the
accumulation of toxic amounts in the rat.
The excretion and distribution of a toxic dose of 400 µg
3H-TCDD/kg body weight was followed in male Sprague Dawley rats (Van
Miller et al., 1976). Within 7 days, about 5.0% of the dose had been
excreted in faeces and 0.5% in urine. The principal tissue depots for
radioactivity were liver, muscle, and skin, which contained 43.0, 4.6,
and 4.4% of the administered dose, respectively.
Table 42. Tissue distribution of TCDD-derived radioactivity in rats given
oral doses of 14C-TCDDa
Tissue content of 14C (µg equivalents of TCDD/kg tissue)b
Tissue 1 week 3 weeks 7 weeks
Exposure level: 1.0 µg/kg body weight per day
liver 49.5±3.6 110.2±37.1 204.0±52.2
adipose tissue 10.0±3.0 23.5±7.5 61.4±36.7
thymus 0.9±0.2 7.3±4.5 6.9±2.5
kidney 0.9±0.2 1.9±0.9 5.5±5.1
spleen 0.4±0.1 1.6±0.8 1.9±1.1
Exposure level: 0.1 µg/kg body weight per day
liver 3.9±1.1 11.8±2.2 19.8±3.1
adipose tissue 0.9±0.6 2.7±0.8 4.5±0.7
thymus ND 1.0±0.6 0.6±0.2
spleen ND 0.6±0.4 0.3±0.1
kidney ND ND ND
Exposure level: 0.01 µg/kg body weight per day
liver ND 0.8±0.1 1.6±0.5
adipose tissue ND 0.3 (2)c 0.3±0.1 (4)c
thymus ND 0.6±0.1 ND
spleen ND 0.6±0.3 ND
kidney ND ND ND
a From: Rose et al. (1976).
b The mean ± standard deviation of 3 male and 3 female rats.
c Indicates the number of animals with detectable levels of
14C-activity.
ND = not detected.
Kociba et al. (1976) gave Sprague Dawley rats TCDD in daily doses
of 0.01, 0.1, and 1 µg/kg body weight 5 days per week for 13 weeks by
gavage. The liver contained TCDD at a level of 324 (± 53) µg/kg wet
weight in males and 284 (± 21) µg/kg wet weight in females given
repeated TCDD doses of 1 µg/kg body weight per day. For the dose level
of 0.1 µg/kg body weight per day, the liver TCDD levels were 36 (± 4)
and 35 (± 4) µg/kg wet weight for males and females, respectively. A
dose of 0.01 µg/kg body weight per day resulted in TCDD liver levels
of 2.6 ( 0.6) µg/kg wet weight in males and 3.7 (± 0.4) µg/kg wet
weight in females.
After feeding diets with TCDD levels corresponding to daily
dietary intakes of 0.001, 0.01, or 0.1 µg/kg body weight for 2 years,
the average concentrations of TCDD found in the liver of female
Sprague Dawley rats were 0.54, 5.1, and 24.0 µg/kg wet weight,
respectively (Kociba et al., 1978). The corresponding levels in
adipose tissue were 0.54, 1.7, and 8.1 µg/kg wet weight. A comparison
of liver TCDD levels found in rats given comparable daily doses of
TCDD for 13 weeks (Kociba et al., 1976) or 2 years (Kociba et al.,
1978) indicates that with prolonged exposure the liver TCDD content
reaches a plateau. This finding agrees well with the prediction
obtained by mathematical analysis of the data resulting from
experiments involving exposure of a few weeks (Rose et al., 1976).
The proportion of a single oral dose of 3H-TCDD found in the
liver of female Sprague Dawley rats was dependent both on the dose
level and on the vehicle used (Poiger & Schlatter, 1980). Maximal
retention occurred within 48 h after dosing. Increasing retention was
observed up to a dose of 280 ng TCDD/rat. Hepatic retention 24 h after
dosing was higher (36.7% of the dose) if TCDD was given in 50% ethanol
than if it was given as an aqueous suspension of soil (37%, w/w),
where it was 16-24.1% of the dose, or activated carbon (25%, w/w),
where it was < 0.07% of the dose (see section 7.4).
Biliary excretion of radioactivity originating from 3H-labelled
TCDD occurred at a more or less constant rate of 0.5 to 1% of the
administered dose per day for 12 days following the administration of
100 µg TCDD/kg body weight in a female Sprague Dawley rat (Poiger &
Buser, 1983). No severe toxic effects were observed within that time
period.
McConnell et al. (1984) dosed female Sprague Dawley rats orally
with either pure TCDD in corn oil or amounts of contaminated soil
giving similar doses of TCDD. In general TCDD in soil was as potent an
inducer of aryl hydrocarbon hydroxylase (AHH) as pure TCDD in corn
oil. The hepatic concentration of TCDD was 40.8 µg/kg in the corn oil
group, receiving 5 µg TCDD/kg body weight, and 20.3 µg/kg in the soil
group, receiving 5.5 µg TCDD/kg body weight (see section 7.4).
Poiger & Schlatter (1980) studied the dermal absorption of
3H-TCDD in hairless rats of the Naked ex Back-Cross and Holzman
strain (200-250 g). Using the amount of TCDD-derived radioactivity
found in the liver as an indicator of its absorption, they reported
that the permeation of TCDD across the epidermis was highly dependent
on the formulation used. The highest radioactivity in the liver, 14.8%
of the administered dose, was detected when TCDD was applied as a
methanolic solution. The hepatic recovery of the administered dose
observed when TCDD was applied in polyethylene glycol 1500 with and
without 15% water was 14.1 and 1.4%, respectively. Dermal application
of TCDD in vaseline or adsorbed onto soil or activated carbon
decreased the percentage of the dose recovered in the liver to 1.4,
1.7-2.2, and < 0.05%, respectively.
In studies by van den Berg et al. (1983), fly ash and crude or
purified toluene extracts of PCDD- and PCDF-containing fly ash from a
municipal incinerator were mixed with ordinary laboratory diet for
rats. Small portions (2 g) of these diets were fed to male Wistar rats
(300 g) every 24 h for 19 days, at which time the animals were
sacrified. Tetra-, penta-, and hexa-chlorinated PCDDs and PCDFs in the
liver and adipose tissues of these rats were determined. Rats fed the
fly ash containing diet stored PCDDs and PCDFs in their livers at
concentrations that were at least 3 to 5 times lower than in the case
of rats fed comparable amounts of fly ash extracts (for the pentaCDD,
hexaCDF, and hexaCDD isomers, the concentrations were approximately
10-20 times lower). Generally PCDFs showed a higher retention in rat
liver than did the corresponding PCDDs. In the adipose tissue of rats
fed with fly ash extracts, retention was higher for penta- and
hexaCDDs than for the corresponding PCDFs.
In further fly ash studies, male Wistar rats (275 g) were fed for
up to 99 days a diet that included 2.5% HC1-pretreated fly ash
(containing PCDDs and PCDFs) from a municipal incinerator (van den
Berg et al., 1986a). A control group received standard diet. All
congeners retained in the liver of the rats had a 2,3,7,8-chlorine
substitution pattern. With the exception of 2,3,4,7,8-pentaCDF and
2,3,4,6,7,8-hexaCDF, liver retention for each congener was below 10%
of the group dose. The retention percentages of the various congeners
in the liver were almost equal at the time-points studied (34, 59, and
99 days), thus indicating a long half-life of these congeners in rat
liver.
Male Wistar rats fed 22.7 (± 1) µg or 120.7 (± 2.8) µg octaCDD
over a two-week period were found to retain about 1-2% of the given
dose in the liver (Williams et al., 1972). The heart, kidneys, spleen,
lung, skeletal muscle, testes, and urine contained no detectable
levels of octaCDD, but minor amounts were found in the adipose tissue
of the high-dose group. Faeces contained 61% and 37%, respectively, of
the low and high dose given. The presence of a large quantity of
octaCDD in the faeces compared to that in the bile 24-72 h after a
single oral dose of 58 mg octaCDD to bile-cannulated male Wistar rats
(400 g) indicated that the dioxin present in the faeces was mainly
unabsorbed octaCDD (Williams et al., 1972).
After 21 daily doses of 100 mg octaCDD containing 12.6 pg
35S-thio-heptaCDD to male Sprague Dawley rats, the radioactivity was
mainly recovered in the faeces and urine, the percentages of the
ingested radioactive dose being 93 (± 6) and 5.2 (± 0.8)% respectively
(Norback et al., 1975). The high faecal excretion suggests poor
absorption. Of the radio-active body burden, 50% was contained in the
liver. The microsomal fraction contained 96.3 (± 8.2)% of the hepatic
radioactivity.
6.1.2 Studies on mice
In studies by Vinopal & Casida (1973), male white mice (20 g)
were given tritium-labelled TCDD intraperitoneally in a single dose of
130 µg/kg body weight. Three days after TCDD administration to one
mouse, 13% of the administered tritium was recovered in the faeces and
0.3% in the urine, while 32% was found in the liver and 0.3% in the
kidneys. In another study, groups of two to six mice were killed at
various time intervals after similar treatment with the same dose of
3H-TCDD. One and 4 days after dosing, the liver contained about 15%
of the dose, on the 8th day, 26%, on the 11th day, 22%, and on the
15th and 20th days, about 10%. The highest amount of 3H-activity was
found in the microsomal fraction of the liver. Somewhat lower activity
was detected in the mitochondrial fraction and the nuclei. The
supernatant fraction was practically devoid of any radioactivity. On
day 8 when the highest levels were observed, the whole liver
homogenate contained 26.7 (± 4.8)% of the administered dose, the
microsomes 12.6 (± 3.8)%, the nuclei 7.8 (± 1.2)%, the mitochondria
6.2 (± 1.3)%, and the supernatant fraction 0.1 (± 0.0)% of the
administered dose.
Coccia et al. (1981) described the effect of adding different
substances to food on the persistence of TCDD in the liver of male
C57Bl/6 mice. In one of the experiments, the test diet was given
immediately after the administration of a single oral dose of 7.6 µg
3H-TCDD/kg body weight. The hepatic radioactivity 14 days after
dosing was 17.3, 6.3, 13.1, and 14.5% of the administered dose in
animals fed standard chow containing 5% vegetable charcoal, 0.5%
cholic acid, and 4% cholestyramine, respectively. When feeding of the
test diet started 3 days after dosing, the hepatic retention of
3H-TCDD was decreased to a similar extent.
Faecal and urinary excretion (Table 41), along with the formation
of faecal, biliary, and urinary metabolites (see 7.2.1.1), of
3H-TCDD was studied in male C57BL/6, DBA/2, and B6D2F1 mice after a
single intraperitoneal dose of 10 µg 3H-TCDD/kg body weight
(Gasiewicz et al., 1983b). The principal tissue depot in C57BL/6 and
B6D2F1 mice was the liver, followed by the adipose tissue. Most other
tissues examined contained less than 1% of the administered dose. In
DBA/2 mice, the adipose tissue contained more radioactivity than the
liver. This difference may be due to the fact that these three strains
of mice differ in their adipose tissue content, being 5.9, 11.5, and
5.0% of the body weight in C57BL/6, DBA/2, and B6D2F1 mice,
respectively. The estimated half-lives of clearance of 3H-TCDD from
the liver of C57BL/6, DBA/2, and B6D2F1 mice were 17, 27, and 13 days,
respectively, and the corresponding figures for the half-life in the
adipose tissue were 11, 42, and 11 days. The cumulative faecal
elimination 30 days after dosing was 59.3, 39.8, and 56.3% of the
administered dose in C57Bl/6, DBA/2, and B6D2F1 mice, respectively,
and the corresponding figures for urinary elimination were 20.5, 16.8,
and 20.5%.
Birnbaum (1986) studied in mice the distribution and excretion of
a single intraperitoneal dose of 500 ng (45 µCi)3H-TCDD/kg body
weight for up to 42 days after treatment. Two sets of congenic strains
of mice were used, i.e., male C57Bl/6 and female DBA/2 mice, where
within each congenic pair the mice differed only at the Ah locus (or
at a limited number of genes closely linked to the Ah locus). The mice
were bred and phenotyped by zoxazolamine paralysis time. The results,
some of them summarized in Tables 47 and 48, suggested that, at the
dose level studied, the distribution and excretion of TCDD were
primarily governed by the total genetic background rather than by the
allele present at the Ah locus.
When male ICR/Ha Swiss mice (27 - 35 g) were given a single oral
dose of 135 µg 14C-TCDD/kg body weight, about 71% and 1-2%,
respectively, of the administered dose was eliminated via faeces and
urine within the first 24 h (Koshakji et al., 1984). During the
following 10 days, an additional 7% and 2% of the administered
radioactivity were recovered in faeces and urine, respectively. Based
on the estimated body burden of radioactivity, a whole-body half-life
of 20 days was calculated.
The distribution of 3H-TCDD in the skin of hairless (SKH:HR-1)
mice after a single intraperitoneal dose of 6.3 µg TCDD/kg body weight
was examined for up to 14 days (Puhvel et al., 1986). Most of the
3H-TCDD in skin was localized in the dermis, although the
concentration of 3H-TCDD was consistently higher in the epidermis.
6.1.3 Studies on guinea-pigs
The retention of a single intraperitoneal dose of 2 µg
14C-TCDD/kg body weight in various tissues of male Hartley
guinea-pigs was determined 1, 3, 5, 7, 11, and 15 days after exposure
(Table 43) (Gasiewicz & Neal, 1979). Three animals died and all
animals lost 24 to 35% of the body weight during the study. The
highest amount of radioactivity per tissue was found in the liver and
skin. The radioactivity in the liver increased with time, concomitant
to the depletion of adipose tissues. Radioactivity in the skin
decreased also with time. No signs of toxicity were seen when the
cumulative excretion of a single i.p dose of 0.5 µg 3H-TCDD/kg body
weight in male Hartley guinea-pigs was studied (Gasiewicz & Neal,
1979). The faecal and urinary excretion of radioactivity was linear
throughout the 23-day study. Approximately 1.4% of the administered
dose was excreted daily during that period, and the faeces contained
94% of the excreted radioactivity.
The microsomal fraction of the liver in male Hartley guinea-pigs
contained 40.7 to 47.4% of the hepatic radioactivity 1 day after a
single ip dose of 0.3, 2.0, or 7.0 µg of 3H- or 14C-TCDD/kg body
weight (Gasiewicz & Neal, 1979). Corresponding values for the crude
nuclear fraction, the mitochondrial fraction, and the soluble fraction
were 20.1-35.6%, 9.5-12.9%, and 7.6-26.4%, respectively. The
subcellular distribution was similar 1 and 6 days after the low dose
but, following the high dose, more radioactivity was present in the
microsomal fraction and less was recovered in the crude nuclear and
soluble fractions on day 6 after exposure.
Olson (1986) followed the distribution, elimination and
metabolism (see section 6.2.1.1) of a single ip dose of 0.56 µg
3H-TCDD/kg body weight in adult (335 to 625 g) male Hartley
guinea-pigs for 45 days. One of seven animals died on day 27, but the
remaining animals gained weight and exhibited no gross signs of
toxicity. At termination the body composition was normal, and 61% of
the administered radioactivity was recovered in the twelve
investigated tissues at the end of the study. The adipose tissue
contained 36% of the dose; liver, pelt, and skeletal muscle plus
carcass contained each 7% of the dose; the gastrointestinal tract
contained about 2% of the dose, and remaining tissues contained less
than 0.5% of the dose. Urinary and faecal elimination followed
apparent first-order kinetics, with half-lives of 82.5 (± 22.4) and
94.4 (± 14.7) days, respectively.
Table 43. Tissue content of TCDD-derived 14C (% of dose/g tissue)a in guinea-pigs following a single
dose of 14C-TCDDd
Days after exposure
Tissue 1 3 5 7 11 15
Perirenal adipose 3.2±1.0 4.1±0.4 2.1±0.4 1.3±0.2 2.1±0.2
Epididymal adipose 1.5±0.8 3.8±0.5 3.4±0.7 3.2±0.1 3.9b 2.5±1.1
Adrenal 1.4±0.3 1.4±0.2 0.9±0.1 1.2±0.3 2.1±0.9 1.7±0.2
Liver 1.1±0.4 1.5±0.4 1.3±0.2 1.1±0.2 2.2±0.2 3.2±0.3
Liverc 11.4±3.3 15.5±3.3 14.0±2.3 12.0±1.9 21.2±2.3 29.6±2.7
Spleen 0.7±0.3 0.5±0.3 0.2±0.1 0.4±0.2 0.4±0.2 0.5±0.1
Duodenum 0.4±0.2 0.2±0.1 0.2±0.1 0.2±0.1 0.2±0.1 0.3±0.1
Pancreas - - 0.2±0.1 0.5±0.3 0.4±0.3 0.3±0.1
Stomach 0.2±0.1 0.3±0.1 0.1±0.1 0.2±0.1 0.3±0.1 0.3±0.1
Testes 0.2±0.1 0.3±0.1 0.2±0.1 0.3±0.1 0.3±0.1 0.2±0.1
Kidneys 0.3±0.1 0.3±0.1 0.2±0.1 0.4±0.1 0.8±0.4 0.7±0.1
Bone marrow 0.3±0.1 0.5±0.1 0.2±0.1 0.4±0.1 0.4b 0.2±0.1
Lungs 0.3±0.1 0.2±0.1 0.2±0.1 0.4±0.1 0.5±0.2 0.6±0.1
Skinc 13.8±0.7 16.3±0.3 15.8±2.4 6.5±0.8 6.5±0.7 6.7±0.6
Brain, heart,
skeletal muscle <0.25
a Mean ± standard error for three animals, unless indicated otherwise.
b Mean of two animals.
c Percentage of dose/tissue.
d From: Gasiewicz & Neal (1979).
6.1.4 Studies on hamsters
In studies by Olson et al. (1980a), Golden Syrian hamsters
absorbed about 73.5% of a single oral dose of 650 µg 3H-TCDD/kg body
weight, a dose that produced thymic atrophy and body weight loss in
several of the animals. The distribution of radioactitivity in various
tissues 1, 3, 10, and 20 days after administration is given in Table
44. The principal depots were the liver and adipose tissue. A similar
pattern of distribution was obtained when the same dose was given
intraperitoneally. The elimination of radioactivity in faeces and
urine was followed for 35 days after a single intraperitoneal (ip) or
oral dose of 650 µg/kg body weight (Olson et al., 1980a). The
half-life for elimination was 12.0 (± 2.0) days and 15.0 (± 2.5) days
for the ip and oral routes, respectively. Of the excreted
radioactivity 41% occurred in urine and 59% in faeces.
The hepatic retention of PCDDs and PCDFs from dietary intake of
HC1-pretreated fly ash from a municipal incinerator was studied in
male Golden syrian hamsters (van den Berg et al., 1986b). The livers
were analysed for tetra-, penta-, and hexaCDDs and PCDFs after feeding
the diet, which contained 25% fly ash, for 34, 58, and 95 days. No
detectable hepatic retention was observed after 34 days. The highest
retention after 95 days was 8.4% for 2,3,4,7,8-pentaCDF, but the
retention was generally below 5% of the total dose. With the exception
of 2,3,4,6,7-pentaCDF, only 2,3,7,8-substituted PCDDs and PCDFs were
retained. Constant relative concentrations were found for the
2,3,7,8-substituted PCDDs and PCDFs at the time points studied.
6.1.5 Studies on monkeys
Van Miller et al. (1976) gave three adult female rhesus monkeys
and four male infant rhesus monkeys a single intraperitoneal dose of
400 µg 3H-TCDD/kg body weight in corn oil. This dose resulted in a
loss of body weight: 10.8% for adults and 20.7% for infants, and light
microscopic changes in the liver. Over a 7-day period the adult
monkeys excreted 1.06% of the dose in the urine and 3.75% in the
faeces. During the same period the infant monkeys excreted
approximately 2% of the administered dose in urine and about 1.26% in
the faeces. The authors questioned the accuracy of these figures owing
to the difficulty of separating the two types of excreta from infant
monkeys. The total tissue concentrations of radioactivity 7 days after
dosing are given in Table 45. The principal tissue depots of
radioactivity in adult monkeys were adipose tissue, skin, liver, and
muscle; they contained 16.2, 13.1, 10.4, and 8.6% of the administered
dose, respectively. The distribution in infant monkeys was 35.6% in
muscle, 22.7% in skin, and only 4.5% of the dose in the liver.
Table 44. Tissue distribution of TCDD-derived radioactivity in Golden
Syrian hamsters at 1, 3, 10, and 20 days following a single oral
dose of 650 µg 3H-TCDD/kg body weighta
Tissue content of 3H (% of dose/g tissue)b
Tissue Day 1 Day 3 Day 10 Day 20
Liver 4.03±1.00 5.32±.82 3.19±.93 0.86±0.09
Liverb 12.74±3.21 20.44±3.45 9.69±0.99 3.70±0.29
Perirenal adipose 2.93±0.87 3.48±0.56 1.38±0.28 0.32±0.03
Adrenals 1.56±0.52 1.12±0.14 0.47±0.08 0.10±0.01
Pancreas 0.39±0.20 0.61±0.13 0.62±0.26 0.21±0.04
Kidneys 0.60±0.16 0.64±0.11 0.60±0.32 0.12±0.03
Spleen 0.30±0.08 0.24±0.05 0.43±0.26 0.07±0.02
Thymus 0.49±0.14 0.34±0.11 - 0.05±0.02
Skin 0.84±0.26 0.31±0.07 0.56±0.18 0.03±0.01
Stomach 0.34±0.07 0.55±0.09 0.65±0.39 0.16±0.06
Duodenum 0.51±0.13 0.47±0.09 0.55±0.28 0.07±0.02
Jejunum 0.59±0.15 0.71±0.20 0.39±0.16 0.08±0.02
Ileum 0.41±0.12 0.35±0.05 0.37±0.21 0.06±0.02
Colon 0.92±0.27 0.60±0.14 0.34±0.07 0.06±0.01
Caecum 0.39±0.11 0.41±0.10 0.28±0.12 0.05±0.01
Lungs 0.38±0.09 0.37±0.05 0.41±0.25 0.07±0.03
Skeletal muscle 0.20±0.07 0.15±0.05 0.15±0.03 0.04±0.02
Heart 0.14±0.03 0.13±0.02 0.15±0.08 0.03±0.01
Testes 0.10±0.04 0.32±0.13 0.13±0.04 0.03±0.01
Blood 0.12±0.02 0.14±0.03 0.12±0.06 0.02±0.01
Brain 0.03±0.01 0.05±0.01 0.06±0.02 0.01
a From: Olson et al. (1980a).
b All values are the mean (± standard error) of four hamsters.
c Percentage of dose/liver.
Table 45. Tissue distribution of TCDD-derived radioactivity in adult
and infant rhesus monkeys 7 days following a single intraperitoneal
dose of 400 µg 3H-TCDD/kg body weighta
(% of dose/tissue)
Tissue content of 3Hb
Tissue Adult Infant
Liver 10.4±6.9 4.51±1.60
Brain 0.58±0.34 1.41±1.40
Spleen 0.028±0.013 0.026±0.004
Small intestine 0.87±0.39 1.47±0.64
Large intestine 1.29±0.12 0.64±0.24
Musclec 8.62±2.39 35.6±14.4
Skin 13.1±4.9 22.7±8.8
Adipose tissued 16.2±5.8
a Fom: Van Miller et al. (1976).
b Mean (± standard deviation) of three adults or four infants.
c Total muscle was taken as 40% of body weight.
d Quantities in infant monkey were insignificant. For adult
monkeys, the calculation was based on an estimate of 300 g
mesenteric fat.
The concentration of TCDD in samples of faeces, urine, and fat
were measured by GC-MS at intervals after dosing up to 715 days in an
adult female rhesus monkey (Macaca mulatta) given a single oral
dose of 1 µg TCDD/kg body weight (McNulty et al., 1982b). During the
3 months after dosing the monkey lost 50% of its body weight but then
began to gain weight again. The level of TCDD in faeces was high for
4 days and then fell to very low or undetectable levels. TCDD in urine
was very low at all time points. The apparent half-life of TCDD in
adipose tissue was about 1 year.
There were no significant time-dependent changes in TCDD-derived
radioactivity found in the tissues investigated from marmosets during
the 3-week-period following subcutaneous treatment with 5 µg
14C-TCDD/kg body weight, except for a minor decrease in levels found
in the adipose tissue (Krowke, 1986). The data thus indicate a long
half-life for TCDD in marmosets.
6.1.6 Studies on dogs
A one-year-old male beagle dog received a total dose of 5.4 µg of
TCDD enterally by direct introduction into the duodenal lumen in four
portions of 1-2 µg, with intervals of 2-7 days between treatments
(Poiger et al., 1982). Severe toxic symtoms preceeded the death of the
animal 17 days after the first dose. The excretion of radioactivity in
the bile reached a maximum on day 1 or day 2 following administration.
Significantly more biliary radioactivity was found after
administration of doses 3 and 4 than after the administration of doses
1 and 2, suggesting that TCDD-administration stimulated its own
metabolism. It was later demonstrated in a 18-months-old male boxer
that TCDD-pretreatment (10 µg/kg body weight) stimulated the biliary
excretion of 3H-labelled TCDD (32.8 ng/kg body weight), whereas
phenobarbital-pretreatment had no effect when compared to the biliary
excretion without pretreatment (Poiger & Schlatter, 1985). All the
experiments were carried out in the same animal, which had enough time
between treatments for the radioactivity to return almost to the
background level.
6.1.7 Studies on cows
The major routes for elimination of 3H-TCDD in Holstein cows
(500-650 kg) after oral doses of 0.05µg (two cows) or 7.5µg (one cow)
TCDD/kg body weight were faeces > milk > urine (Jones et al., 1987).
Fifty percent of the administered dose was eliminated in faeces, the
major part in the first few days after treatment. Three lactating
Holstein cows received commercial technical grade pentachlorophenol
orally by gelatine capsule at a dose rate of 10 mg/kg body weight
twice daily for 10 days and once daily for the following 60 days
(Firestone et al., 1979). One cow served as a control and received
gelatine capsules containing only ground corn. The pentachlorophenol
composite used contained ten PCDD congeners (0.1 to 690 mg/kg) and
eight PCDF congeners (0.9 to 130 mg/kg). Faeces collected on day 28 of
the treatment period contained three hexaCDDs (0.05 to 0.63 µg/kg),
two heptaCDDs (21.3 to 33.1 µg/kg), and octaCDD (290 to 429 µg/kg).
Faeces also contained hexa-, hepta-, and octaCDF. Milk, body fat, and
blood contained only three of the PCDD congeners present in the
pentachlorophenol composite, namely 1,2,3,6,7,8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, and octaCDD. Milk samples also contained
hexa-, hepta-, and octaCDF. The average concentrations of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, octaCDD, and octaCDF in
the composite milk fat at the end of the treatment period were 20, 40,
25, and 2 mg/kg respectively. Similar concentrations were found in
body (shoulder) fat at the end of the treatment period (13, 24, and 32
mg/kg, respectively, of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD,
and octaCDD). Levels of dioxins in the blood were approximately 1000
times below the values in milk or body fat. The average daily
excretion of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD
in the milk during days 40-70 was about 20, 40, and 23 mg,
corresponding to 33, 3, and 0.6% of the daily intake of PCDDs. One
hundred days after the cessation of treatment, the average values for
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in shoulder
fat and milk fat were 2.5, 6.6, 5.6 mg/kg and 4.3, 6.9, 3.0 mg/kg,
respectively.
6.1.8 In vitro studies
The uptake of 3H-TCDD in human fibroblasts was less efficient
when the TCDD was associated with the high density lipoprotein (HDL)
than with the low density lipoprotein (LDL), and even less when
associated with serum (Shireman & Wei, 1986). From studies in mutant
human fibroblasts lacking the normal LDL cellular receptor, the
authors concluded that the LDL receptor pathway was involved in the
cellular uptake of TCDD. The uptake from LDL was time, temperature,
and concentration dependent.
6.2 Metabolic Transformation
6.2.1 Studies on mammals
6.2.1.1 In vivo studies
The possible existence of a urinary metabolite of TCDD was first
suggested by the finding of Allen et al. (1975) that the radioactivity
in urine of 14C-TCDD treated rats was highest from week 2 to 3. Later
both Poiger & Schlatter (1979) and Ramsey et al. (1982) presented
evidence for the in vivo biotransformation of TCDD in the rat.
Male Sprague Dawley rats were given two, four, or six daily oral doses
of approximately 15 µg 14C-TCDD/kg body weight, and the bile was
collected for 24 h following the last dose (Ramsey et al., 1982).
Using high pressure liquid chromatography, at least eight metabolites
of TCDD were found in the bile from these rats. Incubation of the bile
with ß-glucuronidase indicated the presence of glucuronide conjugates
among the metabolites. Poiger & Schlatter (1979) incubated similarly
the bile or the dialysate with glucuronidase/arylsulfatase, and the
dichloromethane-extractable radioactivity increased from 1.5% to 75%.
Their results indicate the elimination of TCDD-metabolites in the form
of water-soluble sulfate and glucuronide conjugates. 3H-TCDD
metabolites extracted from the bile of one-year-old beagle dogs with
ethanol (Poiger et al., 1982) were given to bile-duct-cannulated
female Sprague Dawley rats (250 g) as single oral doses of 7.8-20.8 µg
3H-TCDD-metabolites/kg body weight (Weber et al., 1982b). The mean
24-h elimination of radioactivity was 86.7 (± 6.7)% of the dose, 8.2%
occurring in urine, 31.3% in bile, and 46.9% in faeces. A delay in the
excretion of radioactivity in rats whose bile ducts were not
cannulated suggested an enterohepatic circulation in the rat of the
3H-TCDD-metabolites from the dog. The radioactive material in the
rat bile seemed to be conjugated forms of the metabolites from the
dogs. A metabolic breakdown scheme of TCDD in the rat and dog (Fig. 3)
was proposed by Poiger & Buser (1983). The major metabolite seems to
be formed via cleavage of an ether bond.
The metabolic fate of a single ip dose of 10 µg, 500 µCi TCDD/kg
body weight in male C57Bl/6, DBA/2, and B6D2F1 mice was studied by
Gasiewicz et al. (1983b). Samples of urine, bile, and faeces,
collected on days 5 to 8, 8, and 7 after treatment, respectively, were
extracted and analyzed for metabolites of TCDD by HPLC. Unmetabolized
TCDD was detected in faeces but not in urine or in bile. More than 85%
of the total radioactivity eliminated was present as metabolites of
TCDD in all three mouse strains. Metabolites in the bile appear to be
less polar than in urine. Qualitatively the elution profiles for
urine, bile, and faeces from all three strains appeared to be quite
similar.
A dose of 3H-TCDD (0.56 µg/kg body weight) that produced no
gross toxicity was given ip in olive oil to six adult male Hartley
guinea-pigs (Olson, 1986). Metabolites of TCDD were found in organic
extracts of the liver, kidney, perirenal adipose tissue, and skeletal
muscle in amounts corresponding to 13, 4, 8, and 28%, respectively, of
the recovered radioactivity in these organs 45 days after dosing.
These figures suggest that TCDD-metabolites are not efficiently
eliminated from tissues in guinea-pigs. All radioactivity in urine and
bile represented metabolites of TCDD, whereas in faeces most (70-90%)
contained unchanged TCDD. Of the radioactivity administered, 73.4% was
eliminated as unchanged TCDD in faeces and 25.7% as metabolites of
TCDD in urine and faeces. The presence of TCDD in faeces and its
absence in bile suggest that direct elimination of TCDD from the blood
to the intestinal lumen may occur. The HPLC elution profiles for
metabolites were similar, although not identical, for bile, urine, and
tissues. Taken together, the data by Olson (1986) indicate that
metabolism does not appear to have a major role in the ultimate
elimination of TCDD in the guinea-pig.
Olson et al. (1980a) collected bile and urine from Golden Syrian
hamsters that had been treated with a single ip dose of 650 µg
14C-TCDD/kg body weight 7 days earlier. By means of HPLC, one major
and several minor metabolites of 14C-TCDD were demonstrated both in
bile and urine. No metabolites of 14C-TCDD were detectable in liver
and adipose tissue, thus suggesting a rapid clearance of
biotransformed products of TCDD.
A one-year-old beagle dog was cholecystectomized and a Thomas
cannula was implanted about 3 months before the first dose of TCDD
(Poiger et al., 1982, Poiger & Buser, 1983). A total dose of 5.4 mg
was administered enterally in four portions of 1.8, 1.08, 1.08, and
1.44 mg on days 0, 2, 7, and 13. Five phenolic metabolites of TCDD
excreted in dog bile were identified by combined gas
chromatography-mass spectrometry. Severe toxic symptoms preceded the
death of the dog 17 days after the first dose. A metabolic breakdown
scheme of TCDD in the dog (Fig. 3) was proposed by Poiger & Buser
(1983), lateral hydroxylation of TCDD seeming to be the major route of
metabolism.
 6.2.1.2 In vitro studies
Although there is evidence of different metabolites of TCDD and
the possibility of its metabolic transformation was suggested as early
as 1975 (Allen et al., 1975; Rose et al., 1976), it was only in 1982
that specific metabolites were identified by Sawahata et al. (1982).
They incubated TCDD with isolated rat hepatocytes at 37 ©C for 8 h,
and the resulting incubation mixture was subjected to HPLC. The major
peak of radioactivity not corresponding to TCDD was incubated with
ß-glucuronidase in order to split the possible glucuronide
conjugate(s) of TCDD or its metabolite(s). They found that
4,5-dichlorocatechol and 4,5-dichloroguaiacol are potential
metabolites of TCDD, but due to the limited amount of material the
identity of these metabolites was not confirmed by gas
chromatography-mass spectrometry. Two other metabolites of TCDD,
namely 1-hydroxy-2,3,7,8-tetrachloro-dibenzo-p-dioxin and
8-hydroxy-2,3,7-trichlorodibenzo-p-dioxin, were isolated by means of
HPLC, and were identified by mass spectrometry.
Primary hepatocytes from Sprague Dawley rats and Hartley
guinea-pigs have been used to study the metabolism of 14C-TCDD
(Wroblewski & Olson, 1985). The overall metabolism was 2.8 times
greater in rats than in guinea-pigs. The metabolism of 14C-TCDD was
increased 3.2-fold in rats pretreated with TCDD (5 µg TCDD/kg body
weight ip 72 h prior to isolation of hepatocytes), but no effect was
found in similarly TCDD-pretreated guinea-pigs, or in phenobarbital-
pretreated rats (80 mg/kg body weight ip for 3 days, beginning 4 days
prior to isolation of hepatocytes). Hepatocytes from TCDD-pretreated
rats metabolized TCDD 9 times more rapidly than similarly pretreated
guinea-pig hepatocytes. TCDD may be metabolized by an inducible form
of cytochrome P448 which is expressed in rats but not guinea-pigs.
These differences in metabolism may play a major role in explaining
the differences in species susceptibility to the acute effects of
TCDD.
6.3 Transfer Via Placenta and/or Milk
The transplacental passage of 14C-TCDD has been studied by
Khera & Ruddick (1973). Pregnant Wistar rats were given 14C-TCDD in
a single oral dose of 200 µg/kg body weight on gestation days 16, 17,
or 18 and were killed 6 h after dosing. 14C-activity was detected in
maternal tissues and also in the fetuses and the placenta. Assuming
that all the 14C-activity found in the samples was present as
14C-TCDD, the following levels (ng/gram tissue) were found for
gestation days 16, 17, and 18 respectively: maternal liver 339 (± 15),
339 (± 19), and 275 (± 20); maternal blood 25 (± 11), 19 (± 9), and 10
(± 3); placenta 25 (± 6), 38 (± 4), and 41 (± 3); and fetus 11 (± 3),
15 (± 1), and 16 (± 1). Studies by Moore et al. (1973) indicated that
the passage of TCDD or its metabolites into milk could be of
importance, as TCDD-related effects were observed in sucklings,
nourished by lactating mothers given, after delivery, a single oral
dose of 1 or 3 µg/kg body weight.
The TCDD concentration in livers of pregnant NMRI mice, at a
given dose, was significantly lower than in livers of non-pregnant
mice (Krowke 1986). The concentration of TCDD in the liver of
non-pregnant mice was about 5 times higher than in pregnant mice 7
days after a s.c. dose.
Nau & Bass (1981) studied the transfer of 14C-TCDD to embryos
and fetuses in NMRI mice. The animals were given a single dose of 5,
12.5, or 25 µg TCDD/kg body weight by gavage or by s.c. or ip
injection at day 16 of gestation to study the transfer of TCDD to the
fetus. The animals were killed two days later and various tissues were
analyzed for radioactivity. No evidence was found to indicate a major
first pass effect following oral administration. Maternal livers
contained the highest levels of TCDD, 4.1 to 10.5% of the radioactive
dose administered, which was about one order of magnitude higher than
in extrahepatic maternal tissues, including placenta. Fetal liver and
extrahepatic tissues contained low levels of radioactivity
corresponding to 0.09 to 1.41% and 0.05 to 0.14%, respectively, of the
dose administered to the dams. More radioactivity was recovered in the
placenta and fetus when TCDD was given either as a single ip dose of
25 µg/kg body weight on day 10 of gestation or as 5 daily ip doses of
5 µg/kg body weight on days 7 to 11, when compared to a single i.p.
dose of 25 µg/kg body weight on gestation day 7. Oral dosing of 30 µg
14C-TCDD (0.332 µCi/ µg)/kg body weight to pregnant C57Bl/6 mice on
gestation day 11 resulted in 0 to 14% embryomortality on gestation
days 12 to 14 (Weber & Birnbaum, 1985). About 0.03% of the radioactive
dose was contained in the embryo and in the placenta on days 12 to 14
of gestation. The maternal liver contained 67.4, 67.9, and 50.6% of
the administered radioactive dose on gestation days 12, 13, and 14,
during which days the cumulative elimination of radioactivity in urine
and faeces was 2.4 and 53.3% of the dose, respectively.
The transfer of 14C-TCDD via placenta and milk and the
distribution of the transformed TCDD between various embryonic and
fetal tissues were studied in NMRI mice (Nau et al., 1986). Dams were
given a single dose of 25 µg 14C-TCDD (45 mCi/mmol)/kg body weight
either orally, subcutaneously, or intraperitoneally. To differentiate
between postnatal and in utero exposure, the experimental design
included cross fostering. Depending on the route of administration,
from 0.02 to 0.07% of the administered radioactivity was found in the
liver of the fetus at birth. The highest levels were noted after ip
administration and the lowest after oral intubation. The corresponding
values one week after birth were 0.05 to 0.20%. The hepatic
radioactivity in the neonate reached a peak 1 week after birth and
then decreased slowly throughout and after lactation. The levels of
TCDD-derived radioactivity in extra-hepatic tissues of the offspring
were approximately one order of magnitude lower than the hepatic
levels. Very little radioactivity was found in the stomach filled with
milk, indicating that TCDD ingested from milk was rapidly absorbed in
the stomach.
Arstila et al. (1981) studied the excretion of TCDD in goat milk
after a subchronic administration of 200 ng TCDD per day for 2 months
in the first experiment and of 400 ng TCDD per day for one month in
the second experiment. The minimal detectable concentration in this
study was declared to be below 5 ng/litre. The maximum concentration
of TCDD in milk in the first experiment was 20.8 (± 6.6) ng/litre and
in the second experiment 19.3 (± 6.6) ng/litre. After 18 weeks feeding
with TCDD the levels had dropped to 4.2 and 3.6 ng/litre,
respectively.
The secretion of TCDD in milk and cream has also been studied in
lactating dairy cows kept on a diet containing 10, 30, 100, 300, or
1000 mg/litre 2,4,5-T, corresponding to TCDD levels of 5, 15, 50, 150,
and 500 ng/litre (Jensen & Hummel, 1982). This resulted in levels of
TCDD in the excreted milk of below detection limit, 3, 10, 16-22, and
42-89 ng/litre, respectively, indicating that about 10-20% of the dose
given was eliminated in the milk. The levels in cream were about ten
times higher than those in milk.
One lactating Holstein cow, receiving commercial technical-grade
pentachlorophenol containing several PCDDs and PCDFs orally in
gelatine capsules at a dose rate of 10 mg/kg body weight twice daily
for 10 days and once daily for the following 60 days, calved 151 days
after treatment was stopped (Firestone et al., 1979). The PCDD content
of blood, body fat (shoulder from the cow and hind quarter of the
calf), and milk fat was determined 14 days later. The detected
congeners were 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and
octaCDD and their levels were 12, 13, 20 ng/litre and 27, 14, 6
ng/litre in the blood of the cow and calf, respectively. Corresponding
values for body fat were 4.8, 11.1, and 6.1 µg/kg in the cow and 2.3,
1.9, and 0.5 µg/kg in the calf. The milk fat from the cow contained
2.2, 4.4, and 3.3 µg/kg of the respective congeners.
6.4 Matrix Effects on the Uptake ("Bio-availability")
Uptake of TCDD and other PCDD congeners is highly dependent upon
the formulation in which it is applied. Although conflicting results
have been obtained from studies where TCDD in soil was administered
(McConnell et al., 1984; Umbreit et al., 1985, 1986a, 1986b), most
available data support the idea that mixing TCDD with soil or
activated carbon results in the adsorption of TCDD to the soil
particles, thus reducing the availability of TCDD. Contact time of
TCDD with soil seem to influence the availability, probably because
the binding of TCDD to soil particles becomes strengthened (Poiger &
Schlatter, 1980). Studies elucidating the matrix effects of various
soils or activated carbon on the TCDD-responses in several animal
species are summarized in Table 46.
Poiger & Schlatter (1980) showed decreasing hepatic recovery of
TCDD in rats 1 day after dosing using suspensions of ethanol, soil, or
activated carbon as vehicles. About 50% lower hepatic retention of
TCDD was obtained in the rat, 6 days after dosing, when Minker stout
site soil was the vehicle as compared to corn oil (Lucier et al.,
1986). However, in this study hepatic enzyme-induction (AHH and UDPGT)
was similar in the two vehicle groups.
The bioavailability of TCDD from environmentally contaminated
soil samples has been studied in young guinea-pigs after intragastric
administration (McConnell et al., 1984). Groups of six animals each
were given soil samples corresponding to doses of approximately 1, 3,
or 10 µg TCDD per kg body weight. The doses were based on analyses of
soil siftings (60-gauge mesh) from the Times Beach and Minker Stout
sites, which indicated concentrations of 770 and 880 µg TCDD/kg,
respectively. Controls received soil samples in which no TCDD, PCBs,
or PCDFs were detected. For comparison, pure TCDD in corn oil was
given at either 0, 1, or 3 µg/kg. The observation time was 30 days.
LD50 values calculated in this study were 1.75 µg/kg for TCDD in
corn oil, 7.15 µg/kg for Times Beach soil, and 5.50 µg/kg for the
Minker Stout site soil. An exact percentage for bioavailability was
not calculated in this study, but the TCDD content of the livers of
exposed guinea-pigs indicated a highly efficient absorption of TCDD
from soil.
TCDD in contaminated soil from a 2,4,5-T manufacturing plant and
from a metal salvage yard (Newark, New Jersey, USA) had a low
bioavailability (0.5% and 21.3%, respectively) in guinea-pigs (Umbreit
et al., 1985, 1986a). The soils were given as single oral doses in a
10% aqueous suspension in 5% gum acacia. The bioavailability was
judged by the hepatic concentration of TCDD 60 days after dosing in
guinea-pigs receiving site soils, decontaminated soil, and
TCDD-recontaminated soil.
The difference in bioavailability of TCDD from
2,4,5-T-manufacturing site soil in Newark (New Jersey) and from Times
Beach Site soil (Missouri) when given orally to guinea-pigs was
confirmed by Umbreit et al. (1986b).
Bonaccorsi et al. (1984) gave to rabbits seven daily doses of
TCDD in corn oil, TCDD-contaminated Seveso soil, or recontaminated
soil. Taking the hepatic recovery of TCDD in the corn oil group as
100% "bioavailability", the decrease in "bioavailability" of TCDD from
Seveso soil was 68%. The decrease in "bioavailability" of
recontaminated soil varied from 0 to 44% with the doses used.
Table 46. Matrix effects of various soils on TCDD-responses, as related to the estimated bioavailability of TCDD given
orally to different species.
Species/ Vehicle/matrix Dose of Lethality Hepatic Estimated Reference
Length TCDDc recovery of "bio-
of study TCDDe availability"
(days)
Rat 50% ethanol 14.7 ng 36.7f Poigner &
1 day recontaminated soila 12.7, 22.9 ng 24.1f Schlatter
recontaminated soilb 21.2, 22.7 ng 16.0f (1980)
activated carbon 14.7 ng < 0.07f
Rat corn oil 1.0, 5.0 7.6 and 40.8 Lucier et al.
6 days Minker Stout site soil 1.1, 5.5 1.8 and 20.3 50% (1986)
Guinea- corn oil 1, 3 1/6, 6/6 1.6, 13.3 McConnell et
pig Times Beach site soil 1.3, 3.8, 12.8 0/6, 1/5, 5/5 < 1.0-34.3 Efficient al. (1984)
30 days Minker Stout site soil 1.1, 3.3, 11.0 0/6, 2/6, 6/6 < 1.0-25.7 absorption from
recontaminated soil 10.0 6/6 45.4 soil (85%)
Guinea- corn oil 6 5/8 Umbreit et al.
pig 2,4,5-T-manufacturing 3, 6, 12 0/8, 0/7, 0/7 0.09 (high dose) 0.5% (1985, 1986a)
60 days site soil (Newark,
New Jersey, USA).
Metal salvage yard soil 0.32 0.23 21.3%
(Newark)
recontaminated soil 6 6/7 18
Table 46 (contd).
Species/ Vehicle/matrix Dose of Lethality Hepatic Estimated Reference
Length TCDDc recovery of "bio-
of study TCDDe availability"
(days)
Guinea- 2,4,5-T-Manufacturing 3, 5, 10 0/18, 1/20, 1/18 Umbreit et al.
pig site soil, (Newark) (1986b)
60 days Times Beach site soil 1, 3, 10 2/19, 2/20, 8/14
recontaminated soil 6 19/20
Rabbit corn oil 20, 40, 80d 0.26-2.7 Bonaccorsi et
7 days Seveso soil 80, 160d 0.88-2.2 > 32% al. (1984)
recontaminated soil 20, 40, 80d 0.26-1.5 > 66-100%
a Contact time = 10-15 h.
b Contact time = 8 days.
c µg/kg body weight, unless otherwise stated.
d ng/kg body weight per day.
e µg/kg liver, unless otherwise stated.
f % of dose.
Considerably lower hepatic levels of PCDDs and PCDFs were
observed in rats fed a diet containing fly ash from municipal
incinerators compared with those fed a diet containing extracts from
the same fly ash (van den Berg et al., 1983).
Dietary intake of soot-containing TCDD produced 60% mortality in
male and female guinea-pigs on days 46 and 60, respectively, at which
time the estimated TCDD-consumption was 1.3 µg/kg body weight for
males and 1.9 µg/kg body weight for females (DeCaprio et al., 1983,
see also section 8.2.3). These data thus suggest a high uptake of TCDD
from the soot matrix.
7. EFFECTS OF TCDD AND OTHER PCDDs ON EXPERIMENTAL ANIMALS AND
IN VITRO TEST SYSTEMS
7.1 Acute Toxicity
7.1.1 In vivo studies on mammals
The range of doses required to cause death varies considerably
between species, as well as between strains of species, and with sex,
age, and route of administration within a single strain (Table 47).
More than an 8000-fold difference exists between the dose of TCDD
reported to cause 50% lethality to male Hartley guinea-pigs, the most
sensitive species tested (Schwetz et al., 1973), and the corresponding
dose for male Golden Syrian hamsters (Henck et al., 1981). The rat
seems to be the second most sensitive species, although there is a
more than 200-fold variability in LD50 values between different
strains. The oral LD50 value was 22 µg TCDD/kg body weight for male
Sherman rats (Schwetz et al., 1973), whereas Walden & Schiller (1985)
found LD50 values ranging from 164 to 340 µg TCDD/kg body weight
when male Fisher 334 N rats from three different suppliers were
tested. The Han/ Wistar-strain of rat has been demonstrated to be
particularly resistant to TCDD-exposure (Pohjanvirta & Tuomisto,
1986). Among the five rats per dose group (0, 1500, 2000, 2500, or
3000 mg TCDD/kg body weight) only one animal died within the 39-40
days observation period.
Monkeys (McConnell et al., 1978a), New Zealand rabbits (Schwetz
et al., 1973), C57Bl/6 mice (Chapman & Schiller, 1985; Jones & Greig,
1975; McConnell et al., 1978b; Smith et al., 1981; Vos et al., 1974),
DBA/2-mice (Chapman & Schiller, 1985), and B6D2F1-mice (Chapman &
Schiller, 1985) gave oral LD50 values of 70, 115, 114, 2570, and 296
mg TCDD/kg body weight, respectively. The difference in sensitivity
towards TCDD among various strains of mice has been claimed to depend
on a genetic variability in the Ah and/or hr-locus (see section
7.8.1).
Male Sherman rats were found to be more sensitive to TCDD than
were females (Schwetz et al., 1973), whereas Beatty et al. (1978)
reported that male Sprague Dawley rats were more resistant to TCDD
than were females. Smith et al. (1981) found adult female C57BL/10
mice to be more resistant to TCDD than adult males of the same strain.
No differences in sensitivity to TCDD between sexes were recorded for
guinea-pigs (McConnell et al., 1978b; Silkworth et al., 1982) or
hamsters (Olson et al., 1980b). Thus data on sex differences in
sensitivity to lethal effects of TCDD are conflicting.
Data on the effect of age at exposure to TCDD on the sensitivity
of acute response are scarce, and comparisons are hampered by the
absence of information, or incomplete information, on the age and/or
body weight of the tested animals. However, Beatty et al. (1978) found
that weanling male Sprague Dawley rats were more sensitive to TCDD
than were adult males. A dose of 25 µg TCDD/kg body weight caused,
after 35 days, a cumulative lethality of 62% in weanling Sprague
Dawley rats and 25% in young adults (Christian et al., 1986a). When
weanling and mature adults were exposed to a similar toxic dose of
TCDD (LD62 and LD60, respectively), onset of death occurred 9 days
later in the adults (Christian et al., 1986a).
Schwetz et al. (1973) found LD50 values in rabbits of 115 µg
TCDD/kg body weight after oral exposure, as compared to 275 µg TCDD/kg
body weight after dermal exposure. C57Bl/6-mice seem to be more
sensitive to ip administration of TCDD (Gasiewicz et al., 1983b) than
to oral administration (McConnell et al., 1978b). The LD50 value in
guinea-pigs was increased from 2.5 µg/kg body weight to 19 µg/kg body
weight when the vehicle for the oral administration of TCDD was
changed from corn oil to methyl cellulose (Silkworth et al., 1982).
Umbreit et al. (1985) compared mortality, and time to death, among
guinea-pigs given single oral doses of TCDD in corn oil, TCDD in a
suspension of cleaned soil, from an industrial site, or
TCDD-contaminated soil from the same industrial site. Animals treated
with corn oil, cleaned soil and contaminated site soil survived the
60-day study without any sign of TCDD intoxication. Among animals that
received 6 µg TCDD/kg body weight either in corn oil or mixed with
cleaned soil, only 3/8 and 1/7 survived, respectively. Deaths occurred
between days 9 to 31 and 15 to 25, respectively. These results are
different from those reported by McConnell et al. (1984) where
TCDD-contaminated soils from Times Beach and Minker Stout were highly
toxic to guinea-pigs. LD50 values calculated in this study were 1.75
µg/kg for TCDD in corn oil, 7.15 µg/kg for TCDD in Times Beach soil,
and 5.50 µg/kg for TCDD in Minker Stout site soil. The Minker Stout
site soil was also potent in inducing AHH-activity in female Sprague
Dawley rats. The different results may be due to the vehicle used
and/or to the presence of other substances in the soils that may
potentiate or retard the TCDD-induced toxicity (Umbreit et al.,
1986a). In section 6.4, the results from McConnell et al. (1984) and
Umbreit et al. (1985, 1986a) are further discussed from the viewpoint
of the "bioavailability" of TCDD in soils (Table 46).
Despite similar routes and vehicles for the administration of
TCDD to Golden Syrian hamsters, LD50 values varied between 1157
µg/kg body weight (Olson et al., 1980b) and 5051 µg/kg body weight
(Henck et al., 1981). A possible explanation for this difference could
be the spontaneous occurrence of ileitis observed in the former study,
which might have increased the susceptibility of those hamsters to
TCDD toxicity.
Table 47. Single lethal dose values for TCDDa
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats
Porton F/5-12 8-9 weeks/ oral/DMSO 0 90 days NR 40
170-200 g 30
(Greig et al., 48
1973) 75
120
190
300
Porton F/6 9-10 weeks/ oral/arachis 0 90 days NR 40
170-200 g oil 126
(Greig et al., 199
1973) 315
500
Sherman M/5-10 NR oral/corn oil 8 2-8 weeks 22 9-27
acetone (9:1) 16
(Schwetz et al., 32
1973) 63
Sherman F/NR NR oral/corn oil NR 2-8 weeks 45 13-43
acetone(9:1)
(Schwetz et al.,
1973)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats (contd)
Sprague M/6 adult/NR ip/olive oil 4 doses 20 60 NR
Dawley 20-80 60
(Beatty et al.,
1978)
Sprague F/6 adult/NR ip/olive oil 4 doses 20 60 NR
Dawley 10-60
(Beatty et al.,
1978)
Sprague M/6 25 days/NR ip/olive oil 4 doses 20 25 NR
Dawley 5-50
(Beatty et al.,
1978)
Fisher 334N M/7 11-12 weeks/ oral/corn oil 0 30 days 340b 28b
230-280 g 75 303c 26c
(Walden & 150 164d 25d
Shiller, 1985) 225
275
325
375
CD M/7 10-11 weeks/ oral/corn oil 0 30 days 297d 25d
350-370 g 75
(Walden & 150
Shiller, 1985) 225
275
325
375
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats (contd)
Han/Wistar M/5 NR/300-350 oral/corn oil 1500 39-40 days > 3000 NR
2000
(Pohjanvirta 2500
& Tuomisto, 3000
1986)
Mice
C57BL/6 M/14 3 months/ oral/corn oil 0 2 months 114 15-30
23.6-30.8 g acetone (6:1) 100
(Vos et al., 1974) 150
200
C57BL/6 M/NR 7-15 weeks/ oral/arachis NR 35 days 126 21±1.6
14-30 g oil
(Jones & Greig,
1975)
C57BL/6 M/8 9 weeks/ oral/corn oil NR 30 days 284 22-25
21-25 g
(McConnell et al.,
1978b)
C57BL/6J M/NR NR ip/olive oil NR 30 days 132 NR
(Gasiewicz et al.,
1983bd)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Mice (contd)
C57BL/6J M/10-15 10-12 weeks/ oral/corn oil 95 30 days 182 24
22-32 g 145
(Chapman & 190
Schiller, 1985) 285
C57BL/10 M/5 42-121 days/NR oral/arachis 85 45 days 146 22-38
oil 107
(Smith et al., 135
1981) 170
213
C57BL/10 F/5 42-121 days/NR oral/arachis 85 45 days > 450 22-38
oil 107
(Smith et al., 135
1981) 170
213
269
338
426
536
DBA/2J M/NR NR ip/olive oil NR 30 days 620 NR
(Gasiewicz et al.,
1983be)
DBA/2J M/10-15 10-12 weeks/ oral/corn oil 1370 30 days 2570 21
22-32 g 1870
(Chapman & 2610
Schiller, 1985) 3500
4470
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Mice (contd)
B6D2F1/J M/NR NR ip/olive oil NR 30 days 300 NR
(Gasiewicz et al.,
1983e
B6D2F1J M/10-15 10-12 weeks/ oral/corn oil 170 30 days 296 25
22-32 g 220
(Chapman & 265
Schiller, 1985) 325
425
450
Guinea-pigs
Hartley M/NR NR oral/corn oil NR 2-8 weeks 0.6 5-34
(Schwetz et al.,
1973)
Hartley M/NR NR oral/corn oil NR 2-8 weeks 2.1 9-42
acetone (9:1)
(Schwetz et al.,
1973)
Hartley M/6 3-4 weeks/ oral/corn oil NR 30 days 2 17-20
200-250 g
(McConnell et al.,
1978b)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Guinea-pigs (contd)
Hartley F/6 NR/500-600 g oral/corn oil 0.1 42 days 2.5 32-42
0.5
(Silkworth et al., 2.5
1982) 12.5
20.0
Hartley F/6 NR/500-600 g oral/methyl- 0.1 42 days 19 12-42
cellulose 0.5
(Silkworth et al., 2.5
1982) 12.5
20.0
Rabbits
New Zealand M,F/NR NR oral/corn oil NR 2-8 weeks 115 6-39
acetone (9:1)
(Schwetz et al.,
1973)
New Zealand M,F/NR NR dermal/acetone 31.6 3 weeks 275 12-22
63
(Schwetz et al., 126
1973) 252
500
New Zealand M,F/5 NR ip/corn oil 31.6 4 weeks NR 6-23
63
(Schwetz et al., 126
1973) 252
500
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Hamsters
Golden Syrian M/5-6 NR/50-80 g ip/olive oil 0 50 days > 3000
500
(Olson et al., 1000
1980b) 2000
3000
Golden Syrian F/5 NR/50-80 g ip/olive oil 0 50 days > 3000 14-32
500
(Olson et al., 1000
1980b) 2000
3000
Golden Syrian M/5 NR/50-80 g oral/olive oil 500 50 days 1157 2-47
1000
(Olson et al., 2000
1980b) 3000
Golden Syrian M/6 NR/70-120 g oral/corn oil 0 55 days 5051 9-43
acetone (9:1) 300
(Henck et al., 600
1981) 1000
3000
6000
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Monkeys
Macaca F/3 juvenile/ oral/corn oil 0 47 days < 70 14-34
mulatta 2.1-2.6 kg 70
350
(McConnell et al.,
1978a)
Dogs
Beagle M/2 NR oral/corn oil 300 2-8 weeks NA 9-15
acetone (9:1) 3000
(Schwetz et al.,
1973)
Beagle F/2 NR oral/corn oil 30 2-8 weeks NA all
acetone (9:1) 100 animals
(Schwetz et al., sur-
1973) vived
Chickens
Leghorn NR 4-6 weeks/NR oral/NR NR NR 25-50 12-21
(Grieg et al.,
1973)
a M = male, F = female, NR = not reported, NA = not applicable, ip = intraperitoneal, DMSO = dimethyl sulfoxide.
b supplied by Harlan.
c supplied by Frederick.
d supplied by Charles River.
e based on unpublished studies by Gasiewicz et al., 1983.
TCDD affects a variety of organ systems in different species. The
organ primarily affected in rodents and rabbits is the liver. In
guinea-pigs atrophy of the thymus and lymphatic tissues seems to be
the main effect, while dermal effects are prominent signs in non-human
primates. Generally it is not possible to specify a single organ whose
dysfunction is responsible for death. Overall, TCDD seems to have a
predilection for causing pathological changes in epithelial tissues,
both cutaneous and internal. This is particularly apparent in
non-human primates (Macaca mulatta), and is note-worthy that the
lesions mimic to some degree the effects in human beings. The
histopathological alterations in tissues include hyperplastic and/or
metaplastic alterations as well as hypoplastic responses. The toxic
responses of various species to TCDD are summarized in Table 48,
adapted from Poland & Knutson (1982). In all animal species studied,
death occurred after a time lapse ranging from several days to more
than one month after exposure. The delay was dependent on dose but not
on species (Table 48). Progressive loss of body weight was a
characteristic sign observed in animals given a lethal dose of TCDD.
The weight loss became manifest usually within a few days after
exposure and resulted in a substantial reduction of the adipose tissue
observed at autopsy. At sublethal doses of TCDD a dose-dependent
decrease in body weight gain occurred. This TCDD-induced wasting
syndrome has been thoroughly investigated in several studies discussed
more fully in section 7.4.1.
The greatest difference between species at necropsy, both in
gross and histological effects, concerns pathological alterations in
the liver. As discussed in detail in section 7.4.2, a dose of TCDD
lethal to guinea-pigs did not result in liver damage comparable to the
liver lesions described in rabbits and rats or to liver changes
observed in mice dying after doses higher than those needed to cause
death in these species. In the hamster, frank liver lesions do not
occur even after fatal doses.
Chloracne-like lesions can be induced by topical application
and/or systemic administration of TCDD in rabbits, non-human primates,
and hairless mice. These lesions are further discussed in section
7.4.4.
Severe thymus atrophy was also found at autopsy in all animal
species given lethal doses of TCDD. Histological examinations revealed
lymphoid cell depletion in thymus cortex, spleen, and lymph nodes.
These consistent findings in TCDD poisoning will be discussed in
detail in section 7.4.5 together with the other lymphoid
tissue-related effects.
Table 48. Specific differences in toxic responses following exposure to 2,3,7,8-TCDDa, b
Monkey Guinea- Cowc Rat Mouse Rabbitc Chickenc Hamster
pig
Hyperplasia and/or metaplasia
Gastric mucosa ++ 0 + 0 0 0
Intestinal mucosa + ++
Urinary tract ++ ++ ++ 0 0
Bile duct and/or gall bladder ++ 0 + ++ 0
Lung: focal alveolar ++
Skin ++ 0 +d 0 0 ++ 0
Hypoplasia, atrophy, or necrosis
Thymus + + + + + + +
Bone marrow + + ± +
Testicle + + + +
Other responses
Liver lesions + + ++ + ++ + ±
Porphyria 0 0 + ++ + 0
Oedema + 0 0 + ++ +
a References: monkey (Norback & Allen, 1973; Allen et al., 1977; McConnell et al., 1978a), guinea-pig (McConnell
et al., 1978b; Moore et al., 1979; McConnell, 1980; Turner & Collins, 1983), cow (McConnell, 1980), rat (Kociba et
al., 1978; Kociba et al., 1979a; McConnell, 1980), mouse (Vos et al., 1973; Schwetz et al., 1973; McConnell et al.,
1978b), rabbit (Vos & Beems, 1971; Schwetz et al., 1973), chicken (Allen & Lalich, 1962; Vos & Koeman, 1970;
Norback & Allen, 1973; Schwetz et al., 1973), hamster (Olson et al., 1980b; Henck et al., 1981).
b Symbols: 0 = lesion not observed, + = lesion observed (number of "+" denote severity), ± = lesion observed to
a very limited extent, blank = no evidence reported in literature.
c Responses followed exposure to 2,3,7,8-TCDD or structurally related chlorinated aromatic hydrocarbons.
d Skin lesions in cattle have been observed, but they differ from the skin lesions observed in other species.
There are also substantial interspecies differences in the
effects observed in other organs of animals given lethal doses of
TCDD. Icterus was reported in rats (Buu-Hoi et al., 1972a; Gupta et
al., 1973), hepatic porphyria occurred in mice and rats (see section
7.4.3), and ascites with subcutaneous oedema and hydrothorax appeared
in mice (Jones & Greig, 1975; Vos et al., 1974) and monkeys (Allen et
al., 1977). The accumulation of serous fluid in the pericardial sac
occurred in chickens after a single lethal dose of TCDD (see section
7.1.4). Haemorrhages were frequently observed in many organs following
lethal doses in monkeys (Allen et al., 1977), rats, and guinea-pigs
(Gupta et al., 1973). In mice, death was frequently attributed to
terminal haemorrhages (Vos et al., 1974).
When administered in doses sufficient to cause overt toxicity,
TCDD causes testicular atrophy and degeneration characterized by
reduced spermatogenic activity in mice (McConnell et al., 1978b), rats
(Kociba et al., 1976; Van Miller et al., 1977), and guinea-pigs
(McConnell et al., 1978b). The same symptoms were present in monkeys
fed dioxin-containing toxic fat (Allen & Carstens, 1967; Norback &
Allen, 1973). The decreases in male sex organ weights (seminal
vesicles, ventral prostate, testes, and caput epididymis) were
dose-dependent. ED50 values were around 15 mg TCDD/kg body weight in
Sprague Dawley rats 7 days after TCDD-treatment when compared to
pair-fed control rats (Moore et al., 1985). Shrunken, hyperchromatic
nuclei in the two layers of seminiferous tubules closest to the
basement membrane were observed in testes of young Sprague Dawley rats
90 h after a single ip dose of 5 mg TCDD/kg body weight (Mittler et
al., 1984). Epididymal lesions (Khera & Ruddick, 1973) and decreased
amounts of secretory material within the accessory sex glands (Kociba
et al., 1976) have been reported in TCDD-treated rats. Reduced
prostate weights, both absolute and relative, were found in Han/Wistar
rats at non-lethal doses of TCDD (Pohjanvirta & Tuomisto, 1986).
Reduced relative uterine weight, accompanied by decreased mucosa,
stroma, and glands, occurred in young C57Bl/6 mice dosed with 6 µg
TCDD/kg body weight three times a week for 1 month, but there was no
effect on the ovaries (Gallo et al., 1986). TCDD had no effect on the
uterine weight in 25 day-old Long Evans rats 2 - 10 days after
treatment with single ip doses of 20 or 80 µg/kg body weight (Romkes
et al., 1987). However, the increase in uterine weight induced by
estradiol treatment was counteracted by simultaneous TCDD-treatment.
Proliferative lesions of the gastrointestinal tract have been
found primarily in non-human primates (Allen et al., 1977; McConnell
et al., 1978a) whereas proliferative changes of the transitional
epithelium in the urinary tract have been found in both guinea-pigs
(Gupta et al., 1973; McConnell et al., 1978b) and monkeys (Norback &
Allen, 1973).
Reduced or unaffected spleen weight and slight to moderate loss
of lymphocytes from spleen germinal centers have been common findings
in laboratory animals exposed to sublethal to lethal doses of TCDD
(Gasiewicz et al., 1980; Greig et al., 1973; Kociba et al., 1978;
McConnell et al., 1978a; Olson et al., 1980b; Vos et al., 1973, 1974).
Spleen cellularity in C57BL/6 mice was decreased 14 and 21 days after
treatment with 30 µg TCDD/kg body weight (Chastain & Pazdernik, 1985),
but not in B6C3F1 or DBA/2 mice 7 days after oral doses of up to 10 µg
TCDD/kg body weight (Luster et al., 1984).
The thyroid weight, both absolute and relative to body weight, of
the rat has been found to be increased by TCDD-treatment (Bastomsky,
1977; Potter et al., 1983, 1986b). Degenerative changes in the
epithelial cells of the thyroid gland were observed in rats after 31
daily oral doses of 1 µg TCDD/kg body weight (Gupta et al., 1973) and
7 days after a single ip dose of 150 µg TCDD/kg body weight (Rozman et
al., 1986). However, Potter et al., (1986b) found no consistent
changes in follicle size or colloid content, variation in follicle
size, height of follicular epithelium, or resorption of colloid at the
periphery of follicles in rats one week after an oral dose of TCDD in
the range 6.25 to 100 µg/kg body weight.
Histological changes in the pancreas and in the interscapular
brown adipose tissue have been found in Sprague Dawley rats exposed to
single ip doses of 150 µg TCDD/kg body weight (Rozman et al., 1986).
Lesions of the adrenal glands have been observed in mice and
guinea-pigs treated with (Gupta et al., 1973; McConnell et al.,
1978b).
Acute exposure to lethal doses of TCDD has produced minor
haematological alterations in all species studied. Anaemia was not
observed in mice or guinea-pigs (Zinkl et al., 1973), but a moderate
anaemia and leukocytosis occurred in rats (Buu-Hoi et al., 1972a) and
monkeys (McConnell et al., 1978a). On the contrary, haemoconcentration
was observed in rats after exposure to a somewhat lower but still
lethal dose of TCDD (Greig et al., 1973; Zinkl et al., 1973).
Thrombocytopenia and clotting abnormalities were observed in rats
after acute exposure to lethal levels of TCDD (Weissberg & Zinkl,
1973). Hypocellularity of the bone marrow was found in guinea-pigs
(McConnell et al., 1978b), rhesus monkeys (Allen et al., 1977;
McConnell et al., 1978a), and mice (McConnell et al., 1978b). However,
in these studies decreased bone marrow cellularity, as judged by
histological examination, appeared only at doses high enough to cause
severe toxicity in the experimental animals. More recent studies
(Chastain & Pazdernik, 1985; Luster et al., 1980, 1985) have
demonstrated that collection and enumeration of bone marrow cells in
suspension provides a more sensitive and quantitative method than
histological preparation for assessing bone marrow cellularity.
Decreased bone marrow cellularity was found in adult male C57Bl/6 mice
3 days after exposure to 120 µg TCDD/kg body weight (Chastain &
Pazdernik, 1985), and in female B6C3F1 mice 5 days after exposure to
10 µg TCDD/kg body weight (Luster et al., 1985), but not in female
DBA/2 mice, even at a dose of 50 µg TCDD/kg body weight (Luster et
al., 1985). The myelotoxic potential of TCDD is described in section
7.4.6.
Changes in clinical chemistry, including serum enzyme activities,
serum protein concentrations, and lipid levels, observed in animals
after acute exposure to TCDD, primarily reflect damage to other organ
systems, mainly the liver (McConnell et al., 1978a; McConnell & Moore,
1979; Olson et al., 1980b). Liver-related enzyme activities in serum
are affected in those animal species where liver damage is a prominent
sign of TCDD toxicity. In those animal species where hepatotoxicity is
not as apparent, such as monkeys and guinea-pigs, these enzyme
activities are essentially normal. Also the TCDD-related decrease in
serum albumin seems to be secondary to the hepatotoxic effect, since
the decrease is less evident or non-existent in those animals that
show little liver damage. Generally serum triglycerides and free fatty
acid levels are increased after TCDD exposure, while that of serum
cholesterol is decreased. However, marked species differences exist
and again these effects seem to be secondary to liver damage. For
further details on hyperlipidaemia, see section 7.4.7.
Indicators of renal damage such as blood urea nitrogen,
creatinine, and blood electrolytes are usually within normal limits
after TCDD exposure.
In view of the interspecies differences in the organ distribution
of TCDD and the variability in the effects on various organs in the
different species, the effects of TCDD in non-human primates are of
particular interest. Nine female juvenile rhesus monkeys (Macaca
mulatta) were divided into three groups of three and given TCDD in
a single oral dose of 0, 70, or 350 µg per kg/body weight (McConnell
et al., 1978a). The first indication of a toxic effect of TCDD, at day
3, was weight loss, followed by periorbital oedema, conjunctivitis,
and thickening of the Meibomian glands on day 12. Subsequently, the
eye lashes, facial hair, and toe and finger nails were lost. Monkeys
given the highest dose showed a moderate absolute lymphopenia and
thrombocytopenia. Serum cholesterol levels dropped, while the serum
triglyceride levels increased. Alkaline phosphatase and total
bilirubin were normal, but glutamic oxalic transaminase and aldolase
were increased. A decrease in the albumin fraction of the total serum
protein was noticed. The three monkeys given TCDD in a dose of 350
µg/kg body weight died or became moribund between days 28 and 34 after
administration. One of the monkeys given TCDD in a dose of 70 µg/kg
body weight died 14 days after administration. At autopsy body fat was
almost completely absent in all treated monkeys. Ascites was noticed
in two animals and all monkeys had markedly distended and thickened
bile ducts and gall-bladders. Small focal ulcerated areas in the
fundus of the stomach were observed in two monkeys. Microscopic
examination showed that the Meibomian glands were dilated and filled
with keratinaceous debris. Squamous metaplasia of the glandular
portion, with atrophy of sebaceous cells, was present. Occasional
scattered necrotic hepatocytes were noted in the liver on microscopic
examination. The gastric ulcers that were found extended into the
lamina propria.
Schwetz et al. (1973) reported that no signs of toxicity were
observed in four male mice and two female rats given single oral doses
of respectively 2 and 1 g 2,7-diCDD/kg body weight (purity
99.6-99.8%). For octaCDD (purity 98.86%), oral doses of 1 g/kg body
weight to five female rats and 4 g/kg body weight to four male mice
did not cause any toxic symptoms. A sample containing two different
isomers of hexaCDD (purity > 99%, 89:11) killed 1 of 2 and 0 of 2
male rats when given as single oral doses of 100 and 10 mg,
respectively. The only toxic sign observed among these rats was loss
of body weight. These studies lasted for 2 to 8 weeks.
The toxicities of single oral doses of nine PCDDs, including
TCDD, in C57BL/6J mice and Hartley guinea-pigs were compared by
McConnell et al. (1978b) (Table 49). The purity of the various isomers
tested exceeded 97%. Groups of eight male mice and of six male
guinea-pigs were used for each dose of any of the tested compounds,
and the animals were followed for 30 days after administration. The
toxic effects observed after administration of the different PCDDs
were similar, the only difference among the congeners being the amount
needed to produce a given effect. Progressive decrease of body weight,
more pronounced in guinea-pigs than in mice, was observed after a
lethal dose of any of the congeners tested. Marked reduction in
deposited adipose tissue deposits was a constant finding in animals
given a lethal dose of any of the PCDDs. Reduction of muscle mass and
severe dehydration were observed in guinea-pigs, and ascites,
subcutaneous oedema, and hydrothorax in some of the treated mice.
Decrease of thymus weight was a constant finding in both species,
being more pronounced in mice and in the guinea-pigs that died.
Histological examination of the thymus of the guinea-pigs that died as
early as 5 days after administration of PCDDs revealed scattered
necrosis of lymphocytes throughout the cortex, with concomitant
phagocytosis by macrophages. This was even more apparent in animals
that died 14 days after administration, in which a noticeable decrease
in the thickness of the cortex was observed. In the animals that had
died by day 20 it was difficult to differentiate the cortex from the
medulla, but little evidence of necrosis was present.
Table 49. Estimated single oral LD50 values for some PCDDsa
Chlorination Guinea-pigs Mice
of PCDDs (mg/kg)b (mg/kg)b
2,8 > 300 000 NR
2,3,7 29 444 > 3000
2,3,7,8 2 284
1,2,3,7,8 3 338
1,2,4,7,8 1125 > 5000
1,2,3,4,7,8 73 825
1,2,3,6,7,8 70-100c 1250
1,2,3,7,8,9 60-100c > 440
1,2,3,4,6,7,8 > 600 NR
a From: McConnell et al. (1978b).
b Spearman-Karber method.
c Estimated range due to variability in replicates.
NR = not reported.
In guinea-pigs that survived 30 days after a lethal dose of any
of the congeners, the thymus was reduced to one-fourth of its size in
controls. However, thymus histology at this time was often
comparatively normal. A reduction of the lymphoid follicles in the
spleen and of the Peyer's patches in the intestine was observed with
less conspicuous necrosis, which again was not evident in the 30-day
survivors. Striking hypocellularity was found in the sternal bone
marrow in the guinea-pigs that died, but was less obvious in
survivors. Similar thymic and splenic changes were found in mice.
However, bone marrow atrophy occurred only rarely in this species and
then it was less pronounced than in guinea-pigs. In the guinea-pigs
that died, and occasionally in survivors, a marked hyperplasia of the
renal pelvis was observed, invariably extending into the ureter and at
times involving the urinary bladder mucosa. Gastrointestinal
haemorrhages and occasional microscopic dilatation of crypts in the
glandular portion of the gastric mucosa were observed in dead
PCDD-treated animals of both species. Retro-orbital haemorrhages with
exophthalmus and haemorrhages with detachment of the retina were seen
in mice that died after a lethal dose of any of the congeners tested.
Adrenal haemorrhages and moderate atrophy of the zona glomerulosa were
seen in guinea-pigs that died. These changes were not observed in mice
or in surviving guinea-pigs. In guinea-pigs, primarily in animals that
died during the observation period, changes in the spermatogenic
epithelium were observed, with testicular tubules containing only
spermatogonia and Sertoli cells in severely affected animals. Reduced
spermatogenesis, necrotic spermatocytes, and spermatozoa within the
lumen of the testicular tubules and in the epididymis, and
multi-nucleated giant cells within the seminiferous tubules, were
found in the mice that died but not in those that survived. On the
other hand, liver lesions were observed with the same frequency and
degree of involvement in all the mice given the same dose, whether
they died or survived. Minimal liver changes were detected in
guinea-pigs, changes largely confined to central congestion with
occasional degeneration of hepatocytes in dead animals. Fluorescence,
as an indication of porphyria, was found only in mice, particularly in
the liver but also in the incisors, cranial bones, costochondral
junction, and stifle joint. It was dose related and detectable at
doses several times less than the LD50.
Haemolysis and hyperproteinaemia were found in dying animals of
both species. In mice surviving 30 days, but not in guinea-pigs, the
blood a-globulin level was decreased, with a resultant increase in the
albumin/globulin ratio.
With all PCDDs for which it was possible to establish an LD50
values were in the range 10 to 100 times lower in guinea-pigs than in
mice. To produce the greatest toxicity the lateral positions 2,3,7,
and 8 must be fully chlorinated. With 2,3,7-triCDD or 2,8-diCDD the
LD50 values were in the range 1000 to 100 000 times higher than with
TCDD. The addition of a chlorine atom at an ortho-position, e.g.,
1,2,3,7,8-pentaCDD, resulted in only a minor reduction of toxicity. An
additional chlorine atom further reduced the toxic potency but the
LD50 values of 1,2,3,4,7,8-hexaCDD and 1,2,3,6,7,8-hexaCDD remained
comparatively low. The toxicity of 1,2,3,4,6,7,8-heptaCDD was further
reduced. A reduction in the rate of weight gain was observable in
guinea-pigs given doses of this compound exceeding 200 µg/kg body
weight. However, no deaths were observed during the experiment even at
doses as high as 600 µg/kg body weight.
The acute oral toxicities of soot and benzene extracts of soot,
containing PCDDs and PCDFs from a fire in a PCB-containing transformer
(Binghamton, New York, USA) were determined to be 410 mg/kg body
weight and 327 mg equivalents/kg body weight, respectively, in female
Hartley guinea-pigs (Silkworth et al., 1982). The observation period
was 42 days. The test substances were given in 0.75% aqueous methyl
cellulose and the doses used were 250, 500, 750, 1000, and 1250 mg
soot/kg body weight and benzene extracts corresponding to 4, 20, 100,
500, and 1000 mg soot/kg body weight. An extensive investigation,
including pathology, haematology, and serum chemistry alterations, in
groups of six male and female Hartley guinea-pigs, 42 days after
single oral doses of 1, 10, 100, or 500 mg Binghamton soot/kg body
weight in 0.75% methyl cellulose, was reported by Silkworth et al.
(1982). Control animals received 500 mg activated carbon/kg body
weight in the same vehicle. No treatment-related differences were
observed at 1 and 10 mg soot/kg body weight. Decreased body weight
gain occurred in both sexes at 100 and 500 mg/kg, and decreased thymus
weight occurred at 500 mg/kg for males and at 100 and 500 mg/kg for
females. The kidney weight was decreased only in males at 100 and 500
mg/kg. There were no treatment-related alterations in haematological
values. Male guinea-pigs had significantly increased serum
triglyceride levels at 100 and 500 mg/kg and females at 500 mg/kg
only. Elevated aspartate amino transferase (at 100 and 500 mg/kg) and
decreased Y-glutamyltransferase (at 500 mg/kg) were observed in the
serum of female guinea-pigs. The only clearly dose-related
microscopical findings in soot-exposed guinea-pigs were metaplasia of
salivary gland interlobular duct epithelium and goblet cell
hyperplasia of pancreatic interlobular ducts. These lesions occurred
only in males at doses of 100 or 500 mg soot/kg body weight.
Microscopic lesions, which tended to be more frequent and/or severe in
treatment groups than in controls, included bile duct hyperplasia,
hepatocellular cytoplasmic inclusions, vacuolation of the adrenal
cortex, and focal lacrimal adenitis.
7.1.2 In vitro studies on mammalian cells
Over 30 cell types, including primary cultures and cells from
established and transformed cell lines derived from various tissues of
at least six animal species, have been examined for their response to
TCDD (Beatty et al., 1975; Knutson & Poland, 1980a; Niwa et al., 1975;
Yang et al., 1983). The effects studied were viability, growth rate,
and morphological alterations. No toxic effects were observed except
in one rat hepatoma cell line in which Niwa et al. (1975) reported
decreased viability after exposure for 72 h to TCDD at a concentration
of 300 nmol/litre. This concentration is high when compared to the
LD50 in rats and mice. No effect on these cells was observable at 30
nmol/litre.
The biochemical responses found in primary hepatocytes from rats
exposed to 10 µg TCDD/kg body weight in vivo was not present when
the hepatocytes were exposed in vitro to TCDD at 50, 100, or 200
nmol/litre for 48 h (Yang et al., 1983a).
7.1.3 Studies on birds
Chick oedema disease first gained attention in the United States
in 1957. An extensive outbreak among chickens occurred in that year in
Georgia (Firestone, 1973; Sanger et al., 1958; Simpson et al., 1959).
The cause of the disease was traced to the presence in the feed of
toxic components later identified as chlorinated dibenzo-p-dioxins.
TCDD was identified as one of the isomers in the mixture of
chlorinated dibenzo-p-dioxins capable of producing chick oedema
(Flick et al., 1973).
Clinical signs of chick oedema disease consist of dyspnea,
reduced body weight gain, stunted growth, subcutaneous oedema, pallor,
and sudden death. In young chickens gasping was the first noticeable
sign, followed by a waddling gait. Gross inspection of the birds at
autopsy revealed an increased amount of fluid in the pericardial sac
and pale livers with a mottled and irregular granular surface. In
advanced stages the chickens developed a distended abdomen filled with
fluid. Endotheliosis of the vascular system was observed at
microscopic examination, as well as pronounced proliferation of the
endothelium of the glomerular capillaries and necrosis of the liver
cells. Diseased chickens developed pulmonary oedema and perivascular
lymphocyte infiltration, as well as oedema of the cardiac muscle with
interstitial lymphocytic infiltration (Allen, 1964; Allen & Lalich,
1962, McCune et al., 1962; Simpson et al., 1959).
Experimentally, chick oedema has been produced with a single dose
of 25 to 50 µg TCDD/kg body weight (Greig et al., 1973). When mixtures
of tri- and tetraCDDs were fed at dietary concentrations of 0.01
µg/kg, the chickens developed oedema and 83% of them died (Flick et
al., 1972).
Potency for inducing chick oedema was compared for three
different PCDDs: TCDD (purity 91% and > 99%), hexaCDD (purity > 99%,
two isomers), and octaCDD (purity 98.86%) (Schwetz et al., 1973). In
the experiments, 3-day-old white Leghorn cockerels were exposed for 20
to 21 days to one of these congeners at several dose levels. Chick
oedema occurred in birds given oral doses of 1 or 10 µg TCDD/kg per
day, or of 10 or 100 µg hexaCDD/kg per day. Chick oedema was not
observed in chicks maintained on a diet containing 0.1 or 0.5%
octaCDD. The weight of the Bursa of Fabricius was significantly
decreased in 2-week-old white Leghorn cockerels decapitated 2 days
after three daily ip doses of 10 µg TCDD/kg body weight (Sawyer et
al., 1986).
7.1.4 Toxicity of metabolites
Not only has identification of several in vivo metabolites been
achieved, but the acute oral toxicity of TCDD metabolites excreted in
the bile of dogs has been studied in the guinea-pig (Weber et al.,
1982a). 3H-labelled TCDD was administered directly into the duodenal
lumen of two 1-year-old Beagle dogs in four portions of 1 to 2 mg at
time intervals of 2 to 7 days. Excretion of radioactive material from
pooled bile samples collected daily for 4 or 7 days, and thereafter in
pooled samples of 2 or 3 days, was performed with a method yielding
about 50% of the total radioactivity of the bile. The extracts
containing the metabolites were concentrated and dissolved in
1,3-propanediol for administration. Male Pirbright guinea-pigs were
used in a 5-week toxicity study. Five animals in each of four
dose-groups were given a single oral dose via gastric intubation. The
amount of TCDD metabolites was calculated by means of radioactivity
measurements to be 0.6, 6.0, 30.0, or 60.0 µg/kg body weight. Three
animals, one control and two in the high-dose group, died within 48 h
after administration. One death in the high-dose group was due to
gastric perforation during dosing. The other animals which died
exhibited histological lesions which, according to the authors, were
due to material coextracted from bile together with the metabolites.
The light microscopic examination of the liver, spleen, pancreas,
thymus, kidneys, lungs, and adrenals revealed no histological changes,
and no other toxic effects due to the metabolites of TCDD could be
recorded in this study. The authors concluded that TCDD metabolites
from the dog are at least 100 times less toxic to male guinea-pigs
than TCDD itself.
When the metabolites 2-hydroxy-3,7,8-triCDD and
2-hydroxy-1,3,7,8-tetra CDD were given as single ip injections of 100,
1000, or 5000 µg/kg body weight to young male Wistar rats, no decrease
in body weight gain was noted and thymic atrophy was not seen 14 days
later (Mason & Safe, 1986). When compared to TCDD,
1-hydroxy-3,7,8-triCDD was at least 3 orders of magnitude less
effective in inducing hepatic AHH and EROD activities, whereas
2-hydroxy-1,3,7,8-tetra CDD was inactive at all dose levels tested.
7.1.5 Modulation of the acute toxicity
Several studies have been performed which attempt to modify the
acute toxicity of TCDD. Manara et al. (1984) studied the effect of
activated charcoal or cholic acids in the diet on the mortality after
60 days and mean time to death in mice, rats, and guinea-pigs exposed
to TCDD. Dietary levels of charcoal (2.5%), cholic acid (0.25%), and
dehydrocholic acid (0.5%) decreased the mortality induced in C57Bl/6
mice by a single sc dose of 110 mg TCDD/kg body weight from 93% to
53, 21, and 53%, respectively. The mean time to death was prolonged
from 29 days with a normal chow diet to 35-48 days with the above
dietary additives. In CD rats the addition of charcoal (2.5%) and
cholic acid (0.15%) decreased mortality from 80% to 50 and 70%
respectively. The mean time to death was not affected. Charcoal (5%)
added to guinea-pig chow decreased TCDD induced mortality from 64% to
29% and reduced the mean time to death from 29 to 14 days. These
additions to the diet did not prevent body weight loss or liver
enlargement in the mice or guinea-pigs (Manara et al., 1984), but 5%
charcoal in the diet protected against TCDD-induced thymus athrophy in
C57Bl/6 mice 14 days after a single oral dose of 10 mg TCDD/kg body
weight (Manara et al., 1982). The addition of 5% n-hexadecane in the
diet increased the TCDD induced mortality from 60% to 100% and the
mean time to death, within the 50 day study, from 19.8 to 28.8 days in
Sprague Dawley rats treated with a single ip dose of 60 mg TCDD/kg
body weight (Rozman, 1984). N-hexadecane itself did not affect
animal viability.
Increased survival times have been demonstrated in mice receiving
daily injections of triiodothyronine (T3) after TCDD treatment (Neal
et al., 1979) and in rats thyroidectomized before TCDD treatment
(Rozman et al., 1984). Although thyroidectomy increases the mean time
to death, it does not prevent TCDD-related mortality in rats (Rozman
et al., 1985a). Thyroidectomy has been demonstrated to counteract
TCDD-induced decreases in thymus weight and to reduce the spleen
plaque-forming cell response (Pazdernik & Rozman, 1985). A number of
hepatic enzyme activities were induced by TCDD to an equal magnitude
in thyroidectomized and normal rats (Rozman et al., 1985b). It has
been suggested that TCDD-treatment of rats leads to hypothyroidism, a
possible protective mechanism against TCDD toxicity (Pazdernik &
Rozman, 1985). However, available data on changes in T3, thyroxine
(T4), and thyroid-stimulating hormone (TSH) levels in TCDD-treated
rats are not sufficient to state whether the animals are functionally
hypothyroid, euthyroid, or hyperthyroid. Results presented by Potter
et al. (1986b) (see section 8.4.9) suggest that TCDD-treated rats
remain essentially euthyroid and that the altered thyroid status is
neither a major contributor to TCDD toxicity nor a key response to
TCDD exposure.
Daily injections of butylated hydroxyanisole protected against
TCDD-induced lethality in female Sprague Dawley rats, whereas vitamins
E and A, two other antioxidants, did not (Hassan et al., 1985a,b).
Dietary selenium, if given in an optimal dose, provides partial
protection from the lethal effects of TCDD in female Sprague Dawley
rats (Hassan et al., 1985c). None of these treatments could counteract
the TCDD-induced body weight loss (Hassan et al., 1985a,b,c).
7.2 Short-term Toxicity
7.2.1 Studies on rats
Data from the earliest subchronic laboratory study, in which rats
were exposed to daily and/or weekly doses of TCDD, were reported in
four separate papers (published simultaneously) covering general
effects (Harris et al., 1973), pathology (Gupta et al., 1973),
haematological and clinical chemistry changes (Zinkl et al., 1973),
and immuno-biological effects (Vos et al., 1973). Female CD rats were
given TCDD by gavage in daily doses of 0.1, 1, or 10 µg/kg body
weight for 31 days. The body weight of animals exposed to the highest
dose started to decrease within the first week of exposure and 15/16
animals died or became moribund 17 to 31 days after administration of
the compound began. Pathological changes were comparable to those
observed in rats given a single lethal dose, and included severe
thymic atrophy and liver damage, icterus, haemorrhages in various
organs, and the depletion of lymphoid organs. Weight gain was also
reduced at the daily dose level of 1 µg/kg body weight. However, there
were no deaths and in the animals that were killed moderate thymic
atrophy, slight to moderate liver damage, and, in some of the
animals, degenerative changes in the kidneys and in the thyroid gland
were reported. The weight gain was not affected and significant
histopathological changes were not found in rats that received 0.1
µg/kg body weight per day. A decrease in thymic weight that was
significant on day 24, was observed at the lowest exposure level
(Gupta et al., 1973; Harris et al., 1973). Blood samples were
collected 3, 6, 10, 13, 17, 24, and 31 days after administration of
TCDD began. Serum enzyme activities related to liver damage began to
increase after 10 days of exposure and remained high until death
occurred in the 10 mg TCDD/kg per day group. This group of animals
also exhibited increased serum bilirubin levels commencing on day 13.
These parameters were only slightly affected in rats receiving 1 µg
TCDD/kg per day. Thrombocytopenia occurred at all dose levels. After
3 days of treatment with either 1 or 10 µg/kg per day, animals had
depressed platelets counts that remained low throughout the study. In
the low-dose rats, platelets were significantly decreased by day 17.
No significant leukocytopenia or lymphocytopenia occurred in rats at
any dose level. These results are in good agreement with the results
from a more detailed haematological study on female CD rats given
daily oral doses of 10 µg TCDD/kg body weight for 10 and 14 days
(Weissberg & Zinkl, 1973).
When oral doses of 0.02, 1.0, or 5.0 mg TCDD/kg body weight were
given weekly to groups of 10 female CD rats for 6 weeks, all the
animals survived (Harris et al., 1973). However, body weight gain
decreased in the 5.0 µg/kg group during the exposure period, and at
the end of this period the thymus/body weight ratio was approximately
50% of the ratio found in the controls. Liver damage was reported as
slight at this dose level. No effect on body or thymic weight and no
significant histopathological changes were observed in rats given
1 µg/kg body weight or less.
Adult male and female Sprague Dawley rats, in groups of 12, were
given 0, 0.001, 0.01, 0.1, and 1.0 µg TCDD/kg body weight by gavage,
5 days per week for 13 weeks (Kociba et al., 1976). At the end of the
treatment period, five rats of each sex were killed for
histopathological examination, while the remaining animals were
retained for post-exposure observation. Doses of 1 µg TCDD/kg body
weight per day caused five deaths in females, with three occurring
during treatment and two after treatment, and two deaths in males,
both occurring in the post treatment period. Decreased body weights
and food consumption were found at the two highest dose levels both in
males and females. Decreased relative thymus weight and increased
relative liver weight occurred only in the males given the highest
dose but in both the 0.1 and 1.0 µg/kg female groups. Male rats had
significantly depressed haematological values (packed cell volume, red
blood cell count, and haemoglobin) in the two high-dose groups, while
these values were normal in all female rats. Gross, as well as
histological, examination revealed treatment-related effects only in
the high-dose groups with some minor findings in the 0.1 µg/kg group.
Subcutaneous oedema, decreased sizes of testes and uteri, and a
decreased number of corpora lutea were found at necropsy. Histological
findings were limited to lymphoid tissues, liver, and epithelial
linings. The lymphoid tissues, including thymus, were depleted of
lymphocytes. The liver of both male and female rats showed pleomorphic
and multinucleated hepatocytes. Foci of necrosis, with focal
reticuloendothelial aggregations in the areas of parenchymal cell
necrosis, were observed. Hyperplasia of Kupffer cells and an increased
amount of a golden-brown pigment were noted. The hepatic changes were
more pronounced near the periphery of the lobules. Slight hyperplasia
of bile ducts and ductular epithelium was present. The uterus was
lined by cuboidal epithelium in the female rats. The rats given 0.01
mg TCDD/kg did not differ from the controls in any of these
parameters, except for a slight increase in the mean liver to body
weight ratio.
Goldstein et al. (1982) exposed groups of eight female Sprague
Dawley rats to 16 weekly oral doses of 0, 0.01, 0.1, 1.0, and 10.0 µg
TCDD/kg body weight in a study (further discussed in section 8.4.3)
primarily aimed at investigating TCDD-induced porphyria. All animals
given the highest dose died, or were moribund after eight to twelve
doses and were killed. A decrease in body weight gain was seen in this
group within one week of treatment. Decreased body weight gain was
observed also in the 1.0 µg TCDD/kg per week group, but not until
several weeks after the start of treatment. Hepatic porphyria was
found in 7 out of 8 animals receiving weekly doses of 1.0 µg TCDD/kg
body weight, in 1 out of 8 receiving 0.1 µg/kg per week, and in none
of the animals receiving 0.01 µg/kg per week or the lethal dose of
10.0 µg/kg per week. Porphyria was not reversed after six months
recovery from a 16-week exposure to 1.0 µg/kg body weight per week.
Feeding male Wistar rats (110 g) 0, 0.2, or 1.0 µg octaCDD/kg
diet for two weeks, resulting in a total intake of 0, 22.7 (± 1.0) or
120.7 (± 2.8) mg octaCDD, had no effect on body weight gain, feed
consumption, or tissue weights (liver, thymus, testes, heart, and
kidney) (Williams et al., 1972). Congestion of the liver occurred in
the high-dose group.
Daily doses of 100 mg octaCDD for 21 days produced no effects on
appearance, activity, or eating habits in male Sprague Dawley rats,
but slightly increased relative liver weight. A moderate increase in
the hepatic smooth endoplasmic reticulum was noted (Norback et al.,
1975).
7.2.2 Studies on mice
Oral doses of 0.2, 1, 5, or 25 µg TCDD/kg body weight were given
in corn oil to male C57Bl/6 mice weekly for four weeks (Harris et al.,
1973; Vos et al., 1973). One animal of the 25 µg/kg group died after
24 days. Significant weight loss was observed only in the high-dose
group. Thymic atrophy, characterized by nearly complete loss of
cortex, occurred in the 5 and 25 µg TCDD/kg body weight groups.
7.2.3 Studies on guinea-pigs
All 10 female Hartley guinea-pigs that received weekly oral doses
of 1 µg TCDD/kg body weight died, or were killed when moribund,
between days 24 and 32 after the first dose (Gupta et al., 1973;
Harris et al., 1973; Vos et al., 1973). Light microscopic findings of
moribund or dead animals revealed severe atrophy of the cortex of the
thymus with destruction of lymphocytes. There was lymphoid cell
depletion in spleen and lymph nodes. Haemorrhages, mitotic figures,
and loss of lipid vacuoles were observed in the adrenals. Liver
effects were restricted to diffuse single-cell necrosis, predominantly
in the periportal area. Haemorrhages were found in the urinary bladder
and gastrointestinal tract. The lymphocyte count was decreased at all
doses, whereas total leukocyte values were decreased at doses > 0.04
µg/kg. Animals that received eight weekly doses of 0.008, 0.04, or 0.2
µg TCDD/kg body weight all survived. At the 0.2 µg/kg dose level,
decreased body weight gain and decreased relative thymus weight were
observed.
A 90-day feeding study of TCDD in male (250-370 g) and female
(230-340 g) Hartley guinea-pigs was performed by DeCaprio et al.
(1986) and included extensive pathology, haematology, and serum
chemistry on surviving animals. The diets contained 0, 2, 10, 76, or
430 ng TCDD/kg. Animals that received the highest dose exhibited
severe body weight loss, decreased feed consumption, and mortality.
When 60% mortality was reached, on day 46 for males and day 60 for
females, the remaining animals in these groups were sacrificed. The
estimated total TCDD consumption at that time was 1.3 and 1.9 µg
TCDD/kg body weight for males and females, respectively. No
treatment-related mortality was observed at the 76 ng/kg dose level,
which corresponded to a total estimated intake of 0.44 µg TCDD/kg body
weight over the 90 days. Decreased body weight gain and increased
relative liver weight were seen in both sexes, whereas reduced
relative thymus weight occurred in males only. At doses of 2 and 10
ng/kg diet, no dose-related alterations were observed. The only
treatment-related effect on haematology and serum chemistry parameters
was the elevation of serum triglycerides for male guinea-pigs at the
76 ng/kg dose level. The presence of hepatocellular cytoplasmic
inclusion bodies in female guinea-pigs was the only significant
microscopical finding, except for thymus atrophy. Based on this
study, a no-observed-effect level of 0.6 ng TCDD/kg body weight per
day in guinea-pigs was estimated.
DeCaprio et al. (1986) also followed the body weight changes and
mortality during and after feeding male Hartley guinea-pigs (250-360
g) a diet containing 430 ng TCDD/kg. The diet was fed for 11, 21, or
35 days and was then withdrawn during a 79, 69, or 55-day recovery
period. The rate of change of weight per day was the same as that of
animals on a control diet, after an initial weight loss of
approximately 10% during the first 5 days. When animals were fed the
TCDD-diet for 21 and 35 days, a significant mortality, 10% and 70%,
respectively, was apparent. Both body weight gain and absolute body
weight were severely depressed in surviving animals throughout the
study. Animals destined to die generally lost more than 20% of the
original weight, whereas less pronounced weight losses were usually
followed by increases in body weight during the recovery period.
A PCB-containing transformer fire at the State office building,
Binghamton, New York, USA, resulted in contamination of the building
with soot-like material containing various PCDDs and PCDFs (see
section 4.5.10). This soot, mixed with diet, was fed to groups of 10
male (250-350 g) and female (200-350 g) Hartley guinea-pigs for 90
days (0, 0.2, 1.9, 9.3, and 46.3 mg soot/kg diet) or 32 days (231.5 mg
soot/kg diet) (DeCaprio et al., 1983). The total intake of soot during
the study corresponded to approximately 0.3, 3, 13, 67, and 100% of
the LD50 dose of Binghamton soot in guinea-pigs (Silkworth et al.,
1982) (see section 8.1.1.1). A dose-related decrease in body weight
gain occurred at 9.3 and 46.3 mg soot/kg diet. Body weight loss and
decreased feed intake was evident in the animals given 231.5 mg
soot/kg diet. Three male and three female guinea-pigs given 46.3 mg
soot/kg died. Seven animals in the highest-dose group died within 28
to 31 days and the remaining moribund animals were killed on day 32.
Gross pathology revealed no effects at 0.2 mg soot/kg diet. Thymic
atrophy occurred only in males at 1.9 mg soot/kg diet, but in both
sexes at higher doses. The relative spleen weight was significantly
increased at 46.3 mg TCDD/kg in both sexes. Treatment-related
microscopical findings included metaplasia of salivary gland epithelia
(> 1.9 mg soot/kg diet), increased number of goblet cells in
pancreatic ducts (> 46.3 mg soot/kg diet for males), focal lacrimal
gland adenitis (> 9.3 mg soot/kg diet for males and > 46.3 mg
soot/kg diet for females), depletion of haematopoietic cells from the
bone marrow (> 46.3 mg soot/kg diet for females, > 231.5 mg soot/kg
diet for males), and hepatocellular cytoplasmic inclusion bodies (9.3
and 46.3 mg soot/kg diet in both sexes). Fatty infiltration of the
liver, reduced thickness of thymic cortex, and degenerative changes of
the stomach and intestine were observed only in high-dose animals.
Haematological alterations were observed only in animals at the 46.3
mg soot/kg diet dose level, whereas alterations in serum chemistry
values were found also at lower levels. Toxic effects of feeding
Binghamton soot for 90 days were similar to the effects occurring
after acute exposure (Silkworth et al., 1982) (see section 8.1.1.1),
but the effects were seen at a lower total dose after subchronic
exposure than after acute dosing. The effect seen at the lowest level
in this study was thymic atrophy at 1.9 mg soot/kg diet, which was
equivalent to 7.8 ng TCDD/kg body weight per day. A comparison between
the effects of feeding pure TCDD and TCDD-contaminated soot to
guinea-pigs (DeCaprio et al., 1986) demonstrated that pure TCDD
produced less variability of alterations and gave a steeper
dose-response relationship for many effects. Exposure to Binghamton
soot characteristically resulted in salivary gland duct metaplasia and
decreased serum sodium and potassium levels (DeCaprio et al., 1983).
When male guinea-pigs (200 g) were fed a diet containing 2.5%
HCl-pretreated fly ash from a municipal incinerator (Zaanstad, The
Netherlands) for up to 95 days, the animals exhibited progressive
weight loss, hair loss, and increased relative liver weight (van den
Berg et al., 1986b). One animal died on day 76.
Table 50. Studies on long-term exposure (excluding cancer studies) to TCDD in laboratory animals
Species/Strain Sex/number/ Doses tested Treatment Parameters
groupd schedule monitored
Rats
Sprague Dawleya M/10 0 ng/kg in diet survival
1 ng/kg continuously
5 ng/kg for 65 weeks
50 ng/kg
500 ng/kg
1000 ng/kg
5000 ng/kg
50 000 ng/kg
500 000 ng/kg
1 000 000 ng/kg
Sprague Dawleyb M and F/10 0.001 µg/kg per day in diet extensive
0.01 µg/kg per day continuously histopathology,
0.1 µg/kg per day for 2 years haematology and
clinical chemistry
Mice
Swiss M/38-44 0 µg/kg/week by gavage weekly histopathology
0.007 µg/kg per week for 1 year
0.7 µg/kg per week
7.0 µg/kg per week
Monkeys
Macaca mulattac F/8 500 ng/kg continuous in extensive
the diet for histopathology,
9 months haematology and
clinical chemistry
a Van Miller et al. (1977). b Kociba et al. (1978, 1979a,b).
c Allen et al. (1977). d M = male; F = female.
7.2.4 Studies on hamsters
No toxic effects were reported in male Golden Syrian hamsters
(50-70 g) given a diet containing 2.5% HCl-pretreated fly ash from a
municipal incinerator in Zaanstad, The Netherlands for up to 95 days
(van den Berg et al., 1986b).
7.2.5 Studies on monkeys
A cumulative dose of 0.2 µg TCDD/kg body weight, divided into
nine oral doses given three times per week, produced no clinical
toxicity in female rhesus monkeys (Macaca mulatta) (McNulty,
1984). However, clearly toxic signs did occur in those monkeys that
received cumulative doses of 1.0 and 5.0 µg TCDD/kg body weight over
the same time period. The first signs were thickening and reddening of
the eyelids, followed by weight loss, dryness and granularity of the
skin, and loss of hair, and in some cases anaemia, purpura, and
bleeding from the nose and mouth. Animals that died showed squamous
metaplasia of the sebaceous glands, mucous metaplasia of the gastric
mucosa, hyperplasia of biliary ductal epithelium, gingivitis, and
hypoplasia of the bone marrow. The times to death after dose were 65
and 116 days at 5 µg/kg and 130 to 211 days at 1 µg/kg.
7.3 Long-term Toxicity
Chronic toxicity studies performed on laboratory animals exposed
to TCDD are summarized in Table 50. Studies on carcinogenicity are
presented in section 7.7.
7.3.1 Studies on rats
In studies by Van Miller et al. (1977), male Sprague Dawley rats
were maintained in groups of 10 on diets containing 0, 1, 5, 50, 500,
1000, 5000, 50 000, 500 000, and 1 000 000 ng TCDD/kg for 65 weeks and
survival was monitored. At the five highest dose levels, all animals
died before the study was completed. The first deaths in these treated
groups occurred by weeks 31, 31, 3, 2, and 2 of treatment,
respectively. Groups receiving 50, 500, or 1000 ng TCDD/kg in the diet
died from acute toxic effects including severe liver necrosis, bile
duct hyperplasia and oedema, atrophy of spleen and thymus, and
gastrointestinal haemorrhages.
Groups of 50 male and 50 female Sprague Dawley rats were fed
diets providing daily doses of 0.001, 0.01, and 0.1 µg TCDD/kg body
weight for 2 years (Kociba et al., 1978, 1979a,b). Control rats, 86
males and 86 females, received diets to which the vehicle acetone had
been added. The dose levels corresponded to a dietary content of 22,
208, and 2193 ng TCDD/kg feed. Increased mortality was observed in
females given 0.1 µg/kg per day, while no increased mortality was
observed in male rats at this dose or in animals receiving doses of
0.01 or 0.001 µg/kg per day. From month 6 to the end of the study the
mean body weights of males and females decreased at the highest dose
and to a lesser degree in females given 0.01 µg/kg per day. During the
course of the study, subnormal body weights were occasionally also
recorded in the low-dose group, although during the last quarter of
the study the body weights were similar to those of the controls.
Increased urinary coproporphyrin and uroporphyrin were noted in
females, but not in males, given TCDD at a dose rate of 0.01 and 0.1
µg/kg per day. Analyses of blood serum collected at terminal necropsy
revealed increased enzyme activities related to impaired liver
function in female rats given 0.1 µg TCDD/kg per day. Necropsy
examination of the rats surviving TCDD exposure to the end of the
study revealed that liver effects constituted the most consistent
alteration in both males and females. Histopathological examination
revealed multiple degenerative inflammatory and necrotic changes in
the liver that were more extensive in females. Multinucleated
hepatocytes and bile duct hyperplasia were also noted. Liver damage
was dose-related and no effect was observable at the lowest dose
studied.
7.3.2 Studies on mice
Weekly oral doses of 0, 0.007, 0.7, and 7.0 µg TCDD/kg body
weight for 1 year resulted in amyloidosis and dermatitis in male Swiss
mice (Toth et al., 1979). The incidence of these lesions was 0/38,
5/44, 10/44, and 17/43 in the control, low-, medium-, and high-dose
groups, respectively.
7.3.3 Studies on monkeys
In a study by Allen & Carstens (1967) groups of four to five
rhesus monkeys were fed diets containing 0, 0.125, 0.25, 0.5, 1.0, and
10.0% of fat (which had been shown to be toxic to chickens) until
death. The "toxic fat" was later demonstrated to contain various
PCDDs, of which 65% by mass of the total PCDDs present was TCDD
(Norback & Allen, 1973). The survival time became shorter with
increasing doses of "toxic fat". Mean time to death was 445 days for
the low dose and 91 days for the high dose. Decreased food consumption
and progressive body weight loss, compared with controls, were noted.
Both clinical and histological changes near the time of death appeared
similar regardless of dose. The monkeys developed subcutaneous oedema,
progressing from the eyelids and face, ascites, and hydropericardium.
Characteristic skin changes were observed as well as anaemia,
leukopenia, and hypoproteinaemia. The bone marrow was hypoplastic.
Centrilobular necrosis, bile duct hyperplasia, and multinucleated
hepatocytes were found in the liver. In more than half of the
animals, there was marked hypertrophy of the gastric mucosa, with
crypts and mucin-containing cysts penetrating into the submucosa.
Ulcerations in the fundic and pyloric regions were observed.
Allen et al. (1977) fed eight adult female rhesus monkeys a diet
containing 500 ng TCDD/kg for 9 months. There after, surviving animals
were removed from the TCDD diet and were observed for another 4
months. No control animals were included, and so data were compared
with pre-exposure values where possible. During the first 3 months of
exposure animals developed periorbital oedema, acne, and loss of
facial hair and eyelashes. By 6 months these changes were quite
prominent in six out of eight monkeys, and a decrease in haemoglobin
haematocrit was noticed. The animals lost weight even though their
food intake was unaltered. Two animals died within the 9-month
exposure period and three monkeys continued to develop toxic symptoms
and died after 3 months on a TCDD-free diet. The three surviving
animals continued to experience periorbital oedema and loss of hair.
The total intake of TCDD over the 9-month period was calculated to be
2-3 µg/kg body weight. Death was preceded by severe anaemia, a
decreased white blood cell count, and severe thrombocytopenia. Autopsy
findings included haemorrhage into a variety of organs, ascites, and
subcutaneous oedema. Hypertrophy, dilatation, oedema, and hydropic
degeneration of the myocardium were noted in all animals. The biliary
ducts showed marked dilatation. Moderate hyperkeratosis of the skin,
with cystic keratosis of the hair follicles, was noted, and
hypocellularity of the lymphoid tissue and the bone marrow were
observed. The hyperplastic mucous-secreting cells of the gastric
epithelium had invaded the submucosa, and ulceration and mucinous
cysts were common in the modified gastric mucosa. Hypertrophy and
hyperplasia of the epithelial lining of the biliary system were
present. The bronchial epithelium, salivary glands, bile ducts, and
pancreatic ducts showed metaplastic changes. Death was attributed to
complications from the severe pancytopenia. The same pattern of
morphological changes were reported to occur in a similar study
performed by Barsotti et al. (1979).
Similar, though less severe, effects were observed in four adult
female rhesus monkeys fed a diet of 50 ng TCDD/kg for 20 months
(Schantz et al., 1978).
7.4 Effects Detected By Special Studies
7.4.1 Wasting syndrome
TCDD causes a starvation-like or wasting syndrome in several
animal species. In young animals, or following a sublethal dose in
adults, this response is manifested as a cessation of weight gain.
Early studies suggested that acute or chronic treatment with TCDD
decreased food consumption, but insufficiently to account for the
weight loss (Allen et al., 1975, 1977; Greig et al., 1973; Harris et
al., 1973; Kociba et al., 1976; McConnell et al., 1978a,b). To
elucidate whether malabsorption could explain the wasting syndrome,
the transfer of a number of nutrients has been studied with everted
intestinal sacs from TCDD-treated rats. A transient increase in the
serosal transfer of 59Fe in Sprague Dawley rats was reported by
Manis & Kim (1979). Absorption of glucose (Ball & Chabra, 1981; Madge,
1977) and lipids (Shoaf & Shiller, 1981) was decreased by TCDD
treatment. The absorption of cobalt, galactose, and proline (Manis &
Kim, 1977) as well as of D-galactose, L-arginine, L-histidine (Madge,
1977), and penicillin (Manis & Apap, 1979) was reported to be
unaffected by TCDD treatment. Leucine transport was depressed in
Sprague Dawley rats 4 h after a single oral dose of 100 µg TCDD/kg
body weight (Ball & Chabra, 1981), whereas no effect was observed in
Fisher rats 7 days after exposure to 80 µg TCDD/kg body weight
(Schiller et al., 1982). Neal et al. (1979) found normal absorption
and intermediary metabolism of glucose, L-alanine, and oleate in
guinea-pigs treated with a single oral dose of 2 µg TCDD/kg body
weight. Apparently there was no generalized impairment of intestinal
absorption. The effects reported may well be secondary to decreased
food consumption which by itself causes structural changes in the
intestine (Steiner et al., 1968) as well as impaired absorption of
nutrients (Esposito et al., 1967).
The connection between the wasting syndrome and the lethal effect
of TCDD has been investigated in pair-feeding and forced nutrition
studies. Courtney et al. (1978) fed TCDD- treated female Wistar rats
a normal pelleted diet ad libitum. Supplementation with water,
electrolyte solution, or liquid diet, administered by gavage, could
not reverse or change the pattern or extent of TCDD-induced weight
loss or mortality.
To bypass gastrointestinal absorption, Gasiewicz et al. (1980)
fed rats intravenously with total parenteral nutrition (TPN). Rats
that had received a single ip dose of 100 µg TCDD/kg body weight
gained weight similarly to their TPN-fed controls, yet still died at
days 13 to 17 following treatment. TCDD-treated rats fed a chow diet
ad libitum lost weight progressively (as compared to pair-fed
controls that maintained their starting weight) and died at days 11 to
20. In TPN-fed TCDD-treated rats, liver damage was more severe and fat
depots were increased as compared to chow-fed TCDD-treated animals.
Similar results were obtained with TPN-fed male Hartley guinea-pigs
treated with a single ip dose of 2 µg TCDD/kg body weight in olive
oil, when compared to TPN-fed control guinea-pigs (Lu et al., 1986).
Similar signs of toxicity were present in TPN- and ad libitum-fed
TCDD-treated guinea-pigs. In contrast to TPN-fed rats (Gasiewicz et
al., 1980), TCDD- treatment in TPN-fed guinea-pigs only produced mild
hepatic changes, including increased liver lipid and cytochrome P 450
content, but no morphological changes (Lu et al., 1986).
Seefeld et al. (1984a) suggested that TPN-fed TCDD- treated rats
might have suffered from overnutrition and, secondary to that,
enhanced hepatotoxicity, as compared to chow-fed, TCDD-treated rats.
These same investigators have presented a heuristic model for the
TCDD-induced wasting syndrome based on the assumption that body weight
in rats is regulated around an internal standard or set point (Keesey
et al., 1976). Prevailing weight at a given age is constantly being
compared to this set point value and if differences occur, feed
consumption is adjusted so as to raise or lower body weight to match
the set point level. If TCDD lowers this setpoint, reduction in food
consumption would result, as the rat attempts to reduce its weight to
a new lower level of regulation determined by the dose of TCDD
administered. This hypothesis has been tested in several experiments
under carefully controlled feeding procedures.
Repeated studies have demonstrated that reduction of feed intake,
due to increased food spillage, is sufficient to account for the loss
of body weight in TCDD-treated Sprague- Dawley rats (Seefeld &
Peterson, 1983, 1984; Seefeld et al., 1984a,b). TCDD-treated rats
maintain and defend their reduced weight level with the same precision
as control rats (fed ad libitum) defend their normal weight level
(Seefeld et al., 1984b). The percentage of the daily feed intake that
is absorbed by the gastrointestinal tract of TCDD-treated and control
rats is similar (Potter et al., 1986a; Seefeld & Peterson, 1984).
Water intake, resting and total oxygen consumption, carbon dioxide
production, respiratory quotient, and spontaneous motoractivity were
decreased in a dose-dependent manner by TCDD-treatment (Potter et al.,
1986a; Seefeld & Peterson, 1983; Seefeld et al., 1984a). Urine output
was unaffected by TCDD-treatment, despite decreased water intake,
whereas urinary excretion of energy and urea were decreased and
urinary ammonia was increased (Potter et al., 1986a).
Hypophagia was the major cause of the loss of adipose and lean
tissue in male Fisher F-344 rats, C57Bl/6 mice, and albino guinea-pigs
when exposed to a calculated LD80 dose of TCDD (Kelling et al., 1985).
Body weight loss followed a similar time course in TCDD-treated and
pair-fed control animals of all three species. Lethalities for
TCDD-exposed rats, mice, and guinea-pigs were 95%, 69%, and 81%,
respectively, compared to lethalities in the appropriate pair-fed
controls of 48, 14, and 64%, respectively (Kelling et al., 1985).
Lethality and body weight loss followed almost identical
time-course-curves in Sprague Dawley rats that received a single oral
dose of 75 µg TCDD/kg body weight and in pair-fed controls (Christian
et al., 1986a). Thus the contribution to lethality made by body weight
loss seems to depend on the species and strain. Weight loss appears to
play a greater role in causing death in Sprague Dawley rats and
guinea-pigs than in Fisher F-344 rats and C57Bl/6 mice. Christian et
al. (1986a) demonstrated differences in organ weights and
histopathology in TCDD-treated and pair-fed animals, despite similar
time-courses and magnitude of body weight loss and lethality. Pair-fed
animals exhibited lesions in the gastro-intestinal tract, which were
absent in TCDD-treated rats, that may have contributed to death. The
hepatic carbohydrate, protein, and lipid metabolism was affected
differently in TCDD-treated and pair-fed Sprague Dawley rats
(Christian et al., 1986b). To distinguish direct effects of TCDD from
effects secondary to hypophagia, the studies by Christian et al.
(1986a,b) and Potter et al. (1986a) were performed with schedule-fed
animals. The reason for this was the finding that the 24-h feeding
pattern for TCDD-treated rats was different from the feeding pattern
for pair-fed controls, although it was similar to that of control rats
fed ad libitum. Decreased feed consumption did not contribute to
weight loss in C57BL/6 mice exposed to TCDD until the animals were
moribund (Chapman & Schiller, 1985).
Besides being typical signs of TCDD toxicity, loss of body weight
and appetite are also prominent signs of thyroid dysfunction (see
section 8.4.9). Serum glucose levels were also decreased by TCDD
independently of hypophagia, whereas the decrease in serum insulin
appeared to result from hypophagia, since it was seen in both
TCDD-treated and pair-fed controls. These results indicate that the
effect of TCDD on thyroid hormones cannot explain the TCDD-induced
decrease in body weight gain.
An interesting biochemical effect in TCDD-induced wasting is the
ability of TCDD to decrease hepatic vitamin A storage in rats (see
section 8.4.10). It has long been known that vitamin A is necessary
for growth and that vitamin A deficiency will result in depressed body
weight gain as well as in reduced food intake. However, the animal
continues to eat and grow though body weight gain is less than normal
(Brown & Morgan, 1948; Coward, 1947; Hayes, 1971; Orr & Richards,
1934; Patterson et al., 1942).
The effect of chemical structure on the ability of several PCDDs
to cause body weight loss in rats has been investigated (Mason et al.,
1986). Of the congeners studied, 2,3,7,8-TCDD was the most active.
Those congeners fully substituted in the 2,3,7, and 8 positions but
containing additional chlorosubstituents in the non-lateral 1,4,6, and
9 positions were less active.
7.4.2 Hepatotoxicity
TCDD produces hepatomegaly, due to hyperplasia and hypertrophy of
parenchymal cells, in all species that have been investigated, even at
sublethal doses. However, there is considerable variation between
species in the extent of this lesion. Other liver lesions are more
species specific.
Liver lesions alone cannot explain lethality following TCDD
administration, though it may be a contributing factor at least in the
rat and rabbit.
The morphological changes in the liver are accompanied by
impaired liver function, characterized by liver enzyme leakage,
increased microsomal monooxygenase activities, porphyria, impaired
plasma membrane function, hyperlipidaemia, and increased regenerative
DNA synthesis.
7.4.2.1 Morphological alterations
In Charles River rats given single oral sublethal doses of TCDD
(5 or 25 µg/kg body weight) a dose-related increase was observed 3
days after dosing in the amount of hepatic smooth endoplasmatic
reticulum (SER) around the periphery of cells, particularly in the
areas around bile canaliculi. The effect progressed by days 6 and 9,
when an increased amount of rough endoplasmatic reticulum (RER) was
also present. By day 28 these changes had returned essentially to
normal levels (Fowler et al., 1973).
The livers of CD rats given high sublethal doses of TCDD (5.0
mg/kg body weight per week) for six weeks showed transient
degenerative changes, followed by megalocytosis, regeneration, and the
occurrence of multinucleated giant hepatocytes (Gupta et al., 1973).
They also showed that the hepatotoxic reaction in rats given lethal
doses of TCDD (10 µg/kg body weight per day for 16-31 days) was
characterized by degenerative and necrotic changes, with the
appearance of mononuclear cell infiltration, multinucleated giant
hepatocytes, increased numbers of mitotic figures, and pleomorphism of
cord cells. These lesions were considered severe enough to be a
contributing factor to death.
Parenchymal cell necrosis was observed by Greig et al. (1973) in
Porton rats 3 weeks after exposure to an LD50 dose of TCDD. The
necrosis, which was located in the centrilobular zone close to the
central vein, became more severe with time.
Jones & Butler (1974) further investigated the time course of the
TCDD-induced liver lesions appearing in the centrilobular zone. They
confirmed the transient degenerative and inflammatory lesions
previously reported (Greig et al., 1973; Gupta et al., 1973). At the
ultrastructural level, consistent changes occurred in the cytoplasm
whereas normal nuclear morphology and division were found throughout
the study. Two weeks after a single oral dose of 200 µg TCDD/kg body
weight to Porton rats, extensive fusion of parenchymal cell plasma
membranes in the centrilobular zone was replaced by a diffuse zone
with islands of normal membrane occurring at intervals. Normal tight
and gap junctions were present in control animals and in periportal
areas of the test animals. These findings suggest that the
multinucleated cells occurring in TCDD-treated rats might form by
coalescence of parenchymal cells. The effect of TCDD on plasma
membranes demonstrates a specific subcellular site of action, which
might be involved in the toxic action of TCDD. This lesion was,
however, not observed until 2 weeks after treatment and thus could not
explain the immediate effects on, for instance, food intake, body
weight gain, and general health.
The time course for liver lesions in male Sprague Dawley rats
(200 g) given a single ip dose of 20 µg TCDD/kg body weight was
followed for up to 32 weeks by Weber et al. (1983). The lesions became
progressively worse up to the 16th week after injection, and
thereafter appeared to regress slowly. The lesions were almost
identical to those reported previously (Fowler et al., 1973; Jones &
Butler, 1974). The histological findings were accompanied by
hyperbilirubinaemia, hypercholesterolaemia, hyperproteinaemia, and
increased serum glutamicoxaloacetic transaminase and serum
glutamic-pyruvic transaminase activities, further indicating decreased
liver function (Greig et al., 1973; Zinkl et al., 1973).
Fewer studies of liver lesions produced by TCDD have been made in
other species than in the rat.
In C57BL/6 mice given single oral doses of 100, 150, or 200 µg
TCDD/kg body weight (Vos et al., 1974) and 250 µg TCDD/kg body weight
(Jones & Greig, 1975), centrilobular degenerative and necrotic changes
were present but multinucleated parenchymal cells were not seen.
Proliferation of the bile ducts and bile duct epithelial cells, as
well as lipid accumulation, have been observed, with a substantial
increase in the hepatic levels of esterified fatty acids and
cholesterol. Only slight damage, hepatocellular swelling, was reported
in CD-1 mice 21 days after a single dose of 50 µg TCDD/kg body weight,
and no histological changes were detected 7 or 35 days after
administration (Gupta et al., 1973).
The guinea-pig, while being very sensitive to TCDD, as indicated
by LD50 data, shows less severe morphological alterations in the
liver than do other species. No manifest liver lesions at the light
microscopical or ultrastructural levels has been found (Gupta et al.,
1973; McConnell et al., 1978b; Moore et al., 1979; Turner & Collins,
1983).
The hamster is very resistant to TCDD toxicity (Table 47) and
exhibits no manifest liver damage even after a fatal dose (Henck et
al., 1981; Olson et al., 1980b). However, Gasiewicz et al. (1986)
found bile duct hyperplasia, numerous inflammatory cells, and
increased number of multinucleated cells in the livers of male Golden
Syrian hamsters 35 days after they received a single ip dose of 500 µg
TCDD/kg body weight in olive oil.
7.4.2.2 Hepatic plasma membrane function
The morphological impairment of hepatic plasma membranes in the
centrilobular parenchymal cells of TCDD-treated Porton rats was
demonstrated histochemically to be preceeded by a loss of ATPase
activity (Jones, 1975). The effect occurred 3 days after treatment in
an area five to six cells deep around the central vein along the
canalicular borders, and became more severe with time. At the end of
the study (day 42 posttreatment) the ATPase activity was completely
abolished around the central vein, including the mid-zonal region, and
encroached on the periportal area in moribund animals. The loss of
ATPase activity was related to the clinical state of the animal. Thus
animals displaying minimal signs of intoxication retained the normal
distribution of ATPase in the periportal zone. In animals killed 9
months after treatment, partial restoration of the normal liver
architecture and the ATPase activity were evident.
Biochemical studies of isolated heptatic plasma membranes from
Holtzman rats treated with 10 or 25 µg TCDD/kg body weight revealed
depressed ATPase activities (Peterson et al., 1979a). The activity of
Na/K-ATPase was depressed to the same extent for both doses from day
2 to 40 after treatment, while a similar depression of Mg-ATPase
activity was observed only in the high-dose group. The Mg-ATPase
activity tended to recover by day 40, whereas Na/K-ATPase activities
did not. A pair-feeding experiment demonstrated that these effects
were independent of the TCDD-induced decrease in food consumption. In
vitro incubation of plasma membranes indicated that ATPase inhibition
did not occur by direct interaction with TCDD. Greig & Osborne (1981)
demonstrated a decrease in K/Mg-ATPase activity, but not in the
Na/K-ATPase or 5'-nucleotidase activities of hepatic plasma membranes
prepared from female Porton rats 6 and 11 days after a dose of 200 µg
TCDD/kg body weight.
Many physiological homeostatic mechanisms are dependent on proper
plasma membrane function and composition. Matsumura et al. (1984)
reported that a single ip dose of 25 µg TCDD/kg body weight to male
Sprague Dawley rats had reduced hepatic plasma membrane
ATPase-activities by 40% 10 days after treatment. The marker enzyme
for putative preneoplastic hepatocytes, glutamyl transpeptidase, was
reduced, while protein kinase (both c-AMP-stimulated and
c-AMP-nonstimulated) was increased (Matsumura et al., 1984). Both
c-AMP-dependent and c-AMP-independent protein kinase, in hepatic
plasma membranes from male Sprague Dawley rats treated with an ip dose
of 25 µg TCDD/kg body weight, were maximally increased on day 20 after
treatment (4.5- and 12-fold, respectively). The induction was
measurable within 2 days after the administration and was still
persistent after 40 days (Bombick et al 1985). Protein kinase C was
significantly increased in hepatic plasma membranes from Sprague
Dawley rats but not from guinea-pigs 10 days after single ip doses of
25 and 1 µg TCDD/kg body weight, respectively (Bombick et al., 1985).
TCDD treatment in vivo also affected the in vitro binding of
concanavalin A, epidermal growth factor, and insulin to their cell
surface membrane receptors.
The binding of glucagon and prostaglandin E were not affected by
a dose of 25 µg TCDD/kg body weight (Matsumura et al., 1984). Studies
with Sprague Dawley rats demonstrated that the TCDD-induced decrease
in epidermal growth factor binding (EGF) was observable within 2 days
after dosing, reached its maximum by day 20, and was still significant
40 days after dosing. The decrease was observable after a single ip
dose of 0.1 µg TCDD/kg body weight (Madhukar et al., 1984). The
relative doses of TCDD needed to suppress EGF binding to 50% of the
control level were 1, 14, and 32 µg/kg body weight for the guinea-pig,
the Sprague Dawley rat, and the Golden Syrian hamster, respectively
(Madhukar et al., 1984). A single ip dose of 115 µg TCDD/kg body
weight had decreased the EGF binding 10 days after treatment by 93.1,
97.8, and 46.0% in C57Bl/6, CBA, and AKR mice, respectively (Madhukar
et al., 1984). The effect of daily sc injections of 2 ng EGF/kg body
weight to newborn Balb/c-mice was compared with the effect of a single
ip dose of 10 mg TCDD/kg body weight given to the dam of newborn
Balb/c-mice within 3 h of delivery (Madhukar et al., 1984). The
parameters studied, all well known in vivo effects of EGF, were:
time for eyelid opening and tooth eruption; hair length and diameter
on day 14; and body weight and thymus weight on day 22. All parameters
were significantly reduced both by TCDD treatment and EGF treatment,
as compared to controls.
Bombick et al. (1984) found that, 10 days after treatment, in
vitro binding of 125I-low density lipoprotein (LDL) to its receptor
on hepatic plasma membranes was decreased by 73% in TCDD-treated
guinea-pigs (1 µg TCDD/kg body weight) as compared to pair-fed
controls. Primary hepatocytes from guinea-pigs treated in the same way
had a reduced ability to internalize 125I-LDL. The reduction of LDL
receptors on hepatic plasma membranes might be responsible for the
increase in plasma of very low density lipoproteins (VLDL) and LDL
noted in TCDD-treated animals.
Quantitative changes in the protein composition of plasma
membranes following TCDD treatment have been reported (Brewster et
al., 1982; Matsumara et al., 1984). The membranes were isolated from
male Sprague Dawley rats 10 days after an ip injection of 25 µg
TCDD/kg body weight and analyzed by SDS-polyacrylamide gel
electrophoresis. Some small proteins (14 000-30 000 daltons) were
completely abolished by TCDD treatment. The effect was most pronounced
10 to 20 days after treatment.
7.4.2.3 Biliary excretion
The early proliferation of liver cells around bile canaliculi
seen after TCDD treatment (Fowler et al., 1973) was suggestive of an
effect on biliary excretion.
The cumulative biliary excretion of indocyanine green (ICG), an
organic anion, was decreased in a dose-dependent manner by treatment
with 5 or 25 µg TCDD/kg body weight in CD rats (Hwang, 1973). On the
contrary, biliary excretion of the organic anions sulfobromophthalein
and phenol-3,6-dibromophthalein was unaffected by treatment with 10 or
25 µg TCDD/kg body weight in Holtzman rats (Yang et al., 1977).
Biliary excretion of ouabain, a model compound for neutral
non-metabolized substrates such as estradiol, progesterone, and
cortisol, was depressed in male Holtzman rats in a dose-related manner
by a single oral dose of 10 or 25 µg TCDD/kg body weight (Yang et al.,
1977). The effect was detectable two days after treatment, reached a
peak between 10 and 20 days, and recovery was only slight by day 40.
Increased plasma concentration and decreased bilary excretion of
ouabain was also measured in male Sprague Dawley rats 10 days after a
single oral dose (25 µg/kg body weight) of TCDD, or
1,2,3,7,8,9-hexaCDD (Yang et al., 1983b). Two other congeners,
1,2,4,6,7,9-hexaCDD and 1,3,6,8-tetraCDD had no effect on these
parameters.
When hepatocytes from TCDD-treated (10 µg/kg body weight) male
Sprague Dawley rats were incubated with labelled ouabain or procaine
amide ethobromide (PAEB) 10 days post-treatment, both the rate of
uptake and the steady-state concentration of ouabain were decreased,
whereas the uptake of PAEB was unaffected by TCDD (Eaton & Klaassen,
1979). The dose of TCDD was very small relative to ouabain
(approximately 150 µmol/litre), so it is not likely that TCDD exerted
its effect by competing with the drug for transport into bile. These
data suggest that the hepatic membrane transport process for ouabain
may be selectively damaged by TCDD.
Peterson et al. (1979a) observed a positive correlation between
the levels of hepatic plasma membrane ATPase activities, biliary
excretion of ouabain, and bile flow in vivo after TCDD treatment.
However, in a further experiment, using perfused rat liver, Peterson
et al. (1979b) demonstrated that biliary excretion of ouabain and
liver membrane ATPase activities could be decreased independently.
Therefore, ATPase activities cannot be directly responsible for the
reduced ouabain excretion.
7.4.3 Porphyria
Chronic sublethal exposure to TCDD produces an accumulation of
porphyrins in the liver and an increase in urinary porphyrin
excretion. In stages of manifest porphyria, accumulation of porphyrins
occurs not only in the liver but also in the kidney and spleen
(Goldstein et al., 1982). It was demonstrated that mice respond to
four weekly doses of 25 µg TCDD/kg body weight with hepatic porphyria,
accompanied by an increase in aminolevulinic acid (ALA) synthetase
activity, and liver lesions (Goldstein et al., 1973), whereas a single
dose of 5, 25, or 100 µg TCDD/kg body weight did not induce porphyria
nor ALA synthetase activity in the rat (Woods, 1973). The suggested
species difference was later ruled out by Cantoni et al. (1981) and
Goldstein et al. (1976, 1982). Chronic administration of 1 µg TCDD/kg
body weight per week to rats for 16 weeks resulted in hepatic
porphyria (Goldstein, 1976a,b, 1982). In contrast, single oral doses
as high as 30 µg TCDD/kg did not produce porphyria either acutely or
16 weeks later. A 6-month recovery period following the final dose was
not long enough to reverse the porphyria. Urinary porphyrins and
hepatic ALA synthetase activity remained maximally elevated, while
hepatic porphyrin levels decreased during this period. Failure to
demonstrate porphyria in rats after chronic administration of TCDD for
13 weeks (Kociba et al., 1976) or 2 years (Kociba et al., 1978) was
suggested to be the result of unsatisfactory porphyrin analysis
(Goldstein et al., 1982).
To further characterize TCDD-induced porphyria, Cantoni et al.
(1981) performed a 45-week study to follow the pattern of porphyrin
excretion in rats exposed orally to 0.01, 0.1, or 1.0 µg TCDD/kg body
weight/week. They found an increase in the coproporphyrin level in the
initial phase of exposure, which remained the only sign of exposure in
the lowest-dose group. A marked porphyric state appeared only in the
1.0 µg/kg dose group, commencing 8 months after dosing started. At
that time urinary porphyrin excretion was 70 times higher than in
control rats. The excretion pattern was characterized by increased
levels of carboxylated porphyrins.
In attempts to elucidate the mechanism of TCDD-induced porphyria,
the effects of TCDD on the enzymes involved in the synthesis and
catabolism of porphyrins have been studied. TCDD was found to be a
potent inducer of ALA synthetase, the initial and rate-limiting enzyme
in haeme synthesis in the liver of chicken embryos (Poland & Glover,
1973b). Elevated ALA synthetase activity has since been demonstrated
also in mice and rats (Goldstein et al., 1973, 1982; Kociba et al.,
1976, 1978). However, the TCDD-induced increase does not appear after
acute exposure and only after several weeks of chronic exposure to
TCDD. Jones & Sweeny (1980) failed to demonstrate increased ALA
activity in mice exposed to 25 µg TCDD/kg body weight per week for 11
weeks, although porphyria was evident. Thus, induction of ALA
synthetase does not seem to be the primary event in TCDD-induced
porphyria. Elder et al. (1976, 1978) suggested that decreased hepatic
porphyrinogen decarboxylase is the primary event in porphyria induced
by halogenated aromatics. TCDD depresses this enzyme activity in
vivo in the liver of mice (Jones & Sweeny, 1980; Elder & Sheppard,
1982; Cantoni et al., 1984a,b) but not in vitro (Cantoni et al.,
1984b).
A decrease in porphyrinogen decarboxylase activity was present
one week after a single dose of 75 µg/kg body weight and continued to
decrease with time, thus preceding the increase in hepatic porphyrins,
which started to rise during the first 2 weeks after treatment (Smith
et al., 1981). TCDD- induced depression of hepatic uroporphyrinogen
decarboxylase activity occurs also in the newly regenerating liver, as
demonstrated by Smith et al. (1985) in C57Bl/10 mice 10 days after
partial (2/3) hepatectomy. The mice were treated with 75 µg TCDD/kg
body weight orally 4 weeks before hepatectomy. Greig et al. (1984)
demonstrated that pretreatment of five different strains of mice with
12.5 mg Fe2+ one week before the administration of 75 µg TCDD/kg
body weight had a synergistic effect on porphyria assessed 5 weeks
after dosing, expressed as increased hepatic porphyrin and decreased
porphyrinogen decarboxylase activity. Iron alone did not increase
hepatic porphyrin levels, nor did it affect hepatic porphyrinogen
decarboxylase activity.
7.4.4 Epidermal effects
Chloracne and associated pathological changes in the skin are
among the most sensitive and widespread responses to TCDD in humans.
Similar skin toxicity is expressed only in a limited number of animal
species, namely rabbits, monkeys, and hairless mice. To characterize
the epidermal response and to elucidate the mechanism(s) of toxicity
to epidermal cells, numerous studies have been performed both in
vivo and in vitro.
7.4.4.1 In vivo studies
The acnegenic activity of TCDD and related compounds has been
tested in the rabbit ear bioassay, first developed by Adams et al.
(1941) for industrial applications. The test substance is applied to
the inner surface of one of the ears, while pure vehicle is applied to
the other. Responses indicative of acnegenic activity include comedo
formation, increased ear thickness, and hyperkeratosis. Mild
irritation, increased ear thickness, slight enlargement of follicular
aperture, slight exfoliation and slight crust formation alone are not
considered indicative of acnegenic activity. Microscopically there is
conversion of sebaceous cells in the hair follicles into
keratin-forming cells. A dose-dependent, positive response was found
in this assay when a total dose of 1, 3, or 10 µg TCDD was applied on
three successive days (Jones & Krizek, 1962). Also Schwetz et al.
(1973) found a dose-dependent acnegenic response in the rabbit-ear
bioassay after repeated applications of 4-40 µg TCDD/ear, five days
per week for four weeks, corresponding to total doses of 80-800 µg. No
response was obtained when the total application was 8 ng. Poiger &
Schlatter (1980) applied a single dose of TCDD in various vehicles on
the inner surface of the rabbit ear and followed the appearance of
inflammation, hyperkeratosis, and chloracne. The minimum dose that
induced skin lesions was around 1 µg TCDD/ear when the vehicle was
acetone, vaseline, or polyethylene glycol 1500 with 15% water. When
TCDD was mixed with soil-water (2:1), or activated carbon-water (1:8)
before application, 2-3 and 160 µg TCDD/ear, respectively, were needed
to induce lesions. Even 160 µg TCDD/ear produced very small changes on
the skin surface when applied as an activated carbon-water paste.
Hairless mice constitute another in vivo model for studies of
epidermal effects of TCDD. Puhvel et al. (1982) studied cutaneous
changes induced by topical application of 0.1 µg TCDD, three times per
week for four weeks, in two strains of hairless mice (Skh:HR-1 and
HRS/J). Epidermal hyperplasia, hyperkeratinization, loss of sebaceous
glands and follicles, and keratin build-up in the dermal cysts were
noted in both strains of mice. Follicular keratosis, considered the
pathognomonic lesion in human chloracne, did not appear within 4 weeks
of application. In the same study, follicular keratosis did develop
after topical application of 2 mg 3,4,3',4'- tetrachlorobiphenyl, five
times per week for 8 weeks, suggesting that follicular keratosis is an
extension of the epidermal response and, thus, not related to
metabolic changes in sebaceous glands. The authors considered hairless
mice a less sensitive model for the chloracnegenic response than the
rabbit ear bioassay. Similar findings were obtained when HRS/J mice
were exposed to TCDD topically applied two to three times per week for
four weeks (Knutson & Poland, 1982; Poland & Knutson, 1982). They
found, with a total applied dose of 1.2 µg TCDD, a moderate to severe
response, including hyperplasia, hyperkeratosis of the interfollicular
epidermis, squamous metaplasia of the sebaceous glands, and
hyperkeratosis within the dermal cysts, but no keratosis in the
sebaceous follicles.
Soot or a benzene extract of soot, containing PCDDs and PCDFs
from a PCB-containing transformer fire (Binghamton, New York, USA),
were applied to 64.5 cm2 of the shaved, unabraded dorsal surface of
(3 + 3) and (1 + 1) male and female New Zealand white rabbits (3.5
kg), respectively (Silkworth et al., 1982). The dose applied was 500
mg soot or benzene extract corresponding to 500 mg soot/kg body
weight. Controls, one male plus one female, were exposed to activated
carbon or benzene in corresponding amounts. Exposure lasted for 24 h
and the observation period was 67 days. The soot produced no overt
toxicity, no weight loss, and no histological findings in thymus,
kidney, or skin, but hepatic centrilobular hypertrophy was found in
both sexes. The soot extract gave rise to a reversible skin
inflammation and hepatic centrilobular hypertrophy in the female only.
No weight loss was recorded and the kidney, thymus, and skin were
histologically normal at necropsy.
7.4.4.2 In vitro studies
Keratinocytes, the principal cell type of epidermis, form an in
vitro model for studies of TCDD-induced hyperkeratosis both in human
and animal-derived cell cultures. The response is analogous to
hyperkeratinization in vivo. Newly confluent epidermal cell cultures
exhibit proliferative properties, while the number of basal cells
tends to decrease with increasing time of post-confluency growth.
Thus, with the appropriate selection of culture medium and time of
treatment, different aspects of TCDD toxicity in vivo can be
modelled in vitro.
A TCDD-induced response of in vitro keratinization was first
demonstrated in XB cell cultures, an established keratinocyte cell
line derived from a mouse teratoma, plated at high density to avoid
spontaneous keratinization (Knutson & Poland, 1980b). Keratinization
was dose-related and histologically similar to that which occurs
spontaneously when XB cells are plated at low density. The epidermal
proliferation in XB cells produced by TCDD could not be biochemically
related to the response produced by cholera toxin, epidermal growth
factor, or 12-O-tetradecanoylphorbol-13-acetate, other compounds
known to affect cell proliferation in XB cells (Knutson & Poland,
1984). Late passage XB cells, i.e. XBF cells, show increased cell
density at saturation and a fusiform morphology at high density.
Additionally, they have lost their ability to respond with
keratinization upon TCDD treatment. Exposing XBF cells to TCDD
concentrations in the range 10 to 11 X 10-8 mol/litre resulted in
normal growth until confluency was reached by day 7. Thereafter
TCDD-treated cultures showed a persistent decrease in cell growth and
cell proliferation as well as changed morphology, whereas viability
was unaffected (Gierthy & Crane, 1984). Reseeding these quiescent XBF
cells, previously exposed to 10-9 mol/litre TCDD for 14 days,
resulted in normal growth and proliferation until confluency. These
TCDD-pretreated cells maintained their susceptibility to TCDD-induced
changes in cell growth and morphology. Both XB cells (keratinization
assay) and XBF cells (flat-cell-assay) have proved to be useful in
vitro bioassays to measure "dioxin-like" activity of both
environmental samples and of pure isomers (Gierthy et al., 1984;
Gierthy & Crane, 1985a,b). Although XBF cells, a highly transformed
variant of XB cells, seem to be less appropriate as a model for TCDD
action on normal mammalian epithelial cell proliferation and
differentiation, they seem to be more stable and easier to maintain
than XB cells. Several continuous lines of human keratinocytes derived
from neonatal foreskin (Milstone & Lavigne, 1984; Osborne et al.,
1984) or squamous cell carcinomas (SCC) (Rice & Cline, 1984; Willey et
al., 1984; Hudson et al., 1985, 1986) have been shown to respond to
TCDD in nanomolar concentrations with a variety of signs that indicate
alterations in the normal differentiation process. Stimulation of
3H-thymidine incorporation was seen in post-confluent human
epidermal cells derived from neonatal foreskin after exposure to TCDD
(Milstone & Lavigne, 1984). Newly confluent human epidermal cells,
derived from foreskin, responded to exposure for 4 days to 10 nmol
TCDD/litre with decreased DNA synthesis, a decrease in the number of
proliferating basal cells, decreased binding of epidermal growth
factor (EGF), an increase in the number of differentiated cells, and
increased envelope formation, i.e., a decrease in the proliferative
capacity and an increase in the state of differentiation (Osborne &
Greenlee, 1985). The decreases in small (basal) cell number and EGF
binding were dose dependent, with EC50 values for TCDD of 2 and 1
nmol/litre, respectively. The responses were also obtained with TCDF
but not with 2,4-diCDD.
The proliferation and differentiation of epidermal cells is
normally regulated by several growth factors and hormones, e.g., EGF,
vitamin A and hydrocortisone. Mouse hepatoma cells exposed to TCDD for
24 h showed 20% inhibition of EGF binding (Karenlampi et al., 1983).
Hudson et al. (1985, 1986) demonstrated that TCDD decreases, in
a dose-dependent manner, the specific binding as well as the cellular
uptake of EGF in cultures of human epidermal cells. The EC50 dose
for inhibition was 1.8 nmol/litre. A similar inhibitory effect was
obtained by TCDF, while 2,7-diCDD was inactive even at doses 100-fold
greater. Maximal inhibition, almost 60%, of EGF binding in confluent
SCC-12F cells exposed to 100 nmol TCDD/litre was obtained after a
pretreatment period of 72 h. No effect was obtained when TCDD was
added at the same time as EGF; thus TCDD did not compete for
EGF-binding sites, neither did TCDD affect the process of
internalizing the EGF. Further studies of the SCC-12F cell line
revealed data suggesting that TCDD specifically reduces the high
affinity EGF-binding sites in the basal cell population of this cell
line (Hudson et al., 1986).
The addition of 10-6 mol hydrocortisone/litre to the medium
antagonized the growth inhibition of SCC cells grown in 10-10 mol
TCDD/litre (Rice & Cline, 1984). Hydrocortisone stimulated several
aspects of keratinocyte differentiation. These stimulatory effects
were abolished in the presence of 10-8 mol TCDD/litre, although TCDD
alone had no effect on these parameters. The hydrocortisone level in
the medium was unaffected by TCDD. TCDD, and even more so
hydrocortisone, were able to stimulate stratification in SCC cultures
held at confluence for extended periods. This effect was opposed by
vitamin A (Rice & Cline, 1984).
Like TCDD, vitamin A suppressed the stimulation of keratinocyte
differentiation by hydrocortisone. However, vitamin A had no effect on
TCDD-induced growth inhibition or its reversal by hydrocortisone (Rice
et al., 1983; Rice & Cline, 1984).
Epidermal transglutaminase (ETG) activity, the marker enzyme for
terminal differentiation, was increased by treatment of basal
keratinocyte cultures from neonatal BALB mice with 10-9 mol
TCDD/litre for 5 to 12 days, although morphologically no signs of
terminal differentiation were present. A parallel increase in ETG
activity was present when these cells were grown in medium rich in
Ca2+, although these cells did stratify and differentiate (Puhvel et
al., 1984).
7.4.5 Effects on the immune system
TCDD produces a pronounced atrophy of the thymus, spleen, and, to
a lesser, extent the peripheral lymph nodes of most experimental
animals. Since Buu-Hoi et al. (1972a) reported on TCDD-induced thymic
atrophy, many studies in rats, mice, guinea-pigs, and monkeys have
shown that the thymus is one of the organs most severely affected by
TCDD. Lesions in the thymus appear at exposure levels well below those
inducing lesions in other organs. Although there is species variation
in the degree and severity of other organ effects, the effects of TCDD
on lymphoid tissues is consistent in all species. Further
investigations of the effect of TCDD on the immune system, which is a
rapidly proliferating and differentiating organ system containing many
cellular components in a highly organized and regulated network, have
revealed that TCDD affects both the humoral-mediated (section 7.4.5.2)
and the cell-mediated immune response (section 7.4.5.3). Also the
complement system, a key component of the innate immunity, is affected
by TCDD treatment (White et al., 1986). Damage to the thymus and to
the cell-mediated immune system seems to be rather specific in that it
occurs at doses considerably lower than those affecting other immune
functions (Faith & Luster, 1979). Thymic involution is believed to be
a direct effect on the gland, and not secondary to factors such as
undernutrition (van Logten et al., 1980), altered levels of hormones
including corticosteroids (Vos et al., 1973; van Logten et al., 1980),
pituitary hormones (Vos et al., 1973), and thymosin (Vos et al.,
1978a,b), or zinc deficiency (Vos et al., 1978a,b), or to a direct
cytotoxic effect on lymphocytes (Vos et al., 1978a,b). A direct effect
of TCDD on mouse fetal thymus organ cells grown in vitro was
demonstrated at concentrations as low as 10-10 mol/litre (Dencker et
al., 1985). Within the thymus, lymphocytes in the cortex, i.e., the
immature T-cells, are more severely affected in TCDD-treated animals
than are lymphocytes in the medulla, i.e., the mature T-cells. Thus,
it seems that TCDD impairs the differentiation of thymocytes into
immunocompetent T cells. Greenlee et al., (1985) obtained results
demonstrating a direct effect of TCDD on thymus epithelial (TE) cells.
High levels of Ah receptors (section 7.8.1) have been found in
the thymus (Carlstedt-Duke, 1979; Mason & Okey, 1982; Gasiewicz &
Rucci, 1984). Studies with C57Bl/6 mice (responsive to TCDD), DBA/2
mice (less responsive to TCDD), and B6D2F1 mice, hybrid mice from
crosses between these strains, suggest that TCDD-induced thymic
involution, as well as immunosuppression, segregates with the Ah locus
in these strains of mice (Poland & Glover, 1980; Clark et al., 1983;
Vecchi et al., 1983; Nagarkatti et al., 1984; Dencker et al., 1985).
The most profound and persistent effect of TCDD on the immune system
is found when TCDD is administered during pre- and/or immediate
postnatal life. Contrary to other experimental animals investigated,
rainbow trout (Salmo gairdneri) are relatively resistant to the
immunosuppressive effects of TCDD (Spitzbergen et al., 1986). No
humoral-mediated effects, and only minor cell-mediated effects, were
present at doses which caused clinical toxicity.
7.4.5.1 Histopathology
Lymphoid organs, primarily thymus but also spleen and lymph
nodes, have been found to be affected by TCDD over a wide spectrum of
dose ranges in adult rats, guinea-pigs, and mice (Gupta et al., 1973;
Vos et al., 1973; Vos and Moore, 1974; McConnell et al., 1978b). The
marked reduction in the size of thymus has been referred to as atrophy
(Gupta et al., 1973; Vos & Moore, 1974), regression (Allen et al.,
1975), or involution (Kociba et al., 1976), though these terms do not
clearly represent the pathogenesis of this lesion but are more a
description of the final event. Toxic effects on thymus appeared in
adult guinea-pigs, rats, and mice exposed to eight weekly doses of 0.2
µg TCDD/kg body weight, six weekly doses of 5 µg TCDD/kg body weight
and four weekly doses of 5 µg TCDD/kg body weight respectively (Vos et
al., 1973). The thymus from moribund animals, or from animals that
died from TCDD exposure, showed a dose-dependent decrease in the
number of cortical lymphocytes, markedly smaller thymic lobules, and
loss of demarcation between the cortex and medulla. Guinea-pigs, the
species most severely affected by TCDD, showed large cystic Hassall
bodies, filled with polymorphonuclear leukocytes (Gupta et al., 1973;
Vos et al., 1973). Guinea-pigs that received lethal doses of TCDD
showed scattered necrosis of lymphocytes in the cortical region with
concomitant phagocytosis by macrophages as early as 5 days
post-exposure (McConnell et al., 1978b). The effect was more apparent
at day 14, and by day 20 it was difficult to differentiate the cortex
from the medulla. At day 20 little evidence of necrosis remained,
though karyorrhectic debris and prominent phagocytosis indicated that
this had occurred. Since the thymus from guinea-pigs surviving the
TCDD dose for 30 days was usually normal microscopically (Vos et al.,
1973; McConnell et al., 1978b), it seems that thymic necrosis must be
an early event in the course of the toxic syndrome. Furthermore, in
animals which survive, thymic regeneration seemed to be rapid.
Decreased weight of thymus, loss of cortical cells, and cell necrosis
have also been found in hamsters exposed to TCDD (Gasiewicz et al.,
1986).
Depletion of lymphoid cells in the spleen, intestinal tract, and
various lymph nodes observed in guinea-pigs, rats, and mice (Gupta et
al., 1973; McConnell et al., 1978b) was less extensive than in the
thymus. The major effect in the spleen of rats is the loss of the
T-cell-dependent areas, namely the periarterial lymphoid sheet and the
paracortical areas (Vos & Moore, 1974).
Depressed immunoglobulin levels were reported for 1- and
4-month-old C57Bl/6 mice exposed to four and six weekly doses,
respectively, of 25 µg TCDD/kg body weight (Vos & Moore, 1974).
Feeding 10, 20, 50, or 100 µg TCDD/kg in the diet depressed
dose-dependently the Y-globulin level in 7-week-old Swiss-Webster mice
(Hinsdill et al., 1980).
Vos & Moore (1974) demonstrated a dose-related lymphocyte
depletion of thymus cortex, spleen, and intestinal lymph nodes in
maternally exposed pups of rats and mice. However, no effect on
immunoglobulin levels was observed in 25-day-old rats maternally
exposed (5 µg TCDD/kg body weight) on days 0, 7, and 14. The
developing lymphoid tissues were found to be more sensitive to TCDD
than were the lymphoid tissues of adults or young.
7.4.5.2 Humoral-mediated immunity
The humoral-mediated immunity (Tables 51 and 52) operates through
antibody-producing cells and is transferable by serum. This system
includes classical antibody-mediated protective immunity and immediate
hypersensitivity reactions. Vos et al. (1974) reported a significant
decrease in the alpha-, ß-, and gamma-globulin levels in C57BL/6 mice
given non-toxic doses of TCDD. The effects of TCDD on specific humoral
immunity responses in adult animals are summarized in Table 51.
Feeding levels of 10 µg TCDD/kg body weight or more reduced the
primary and secondary antibody response to both sheep red blood cells
(sRBC) and tetanus toxin in male Swiss-Webster mice (Hinsdill et al.,
1980). Weekly oral doses of 1 or 10 µg TCDD/kg body weight reduced
both the primary and secondary serum antitetanus titres in male New
Zealand rabbits (Sharma et al., 1984). The secondary, but not the
primary, serum tetanus antitoxin level was decreased in Hartley
guinea-pigs given eight weekly doses of 0.2 µg TCDD/kg body weight
(Vos et al., 1973). Results presented by Vecchi et al. (1980, 1983)
show that single doses as low as 1.2 µg TCDD/kg body weight to C57BL/6
mice decreased the number of plaque-forming spleen cells in response
to an injection of the thymus-dependent antigen sRBC. The response was
dose dependent and lasted for at least 42 days. Luster et al. (1985)
found a decrease in anti-sRBC plaque-forming spleen cell production in
B6C3F1 and DBA/2 mice 5 days after single oral doses of 2 and 10 µg
TCDD/kg body weight, respectively. A dose of 30 µg TCDD/kg body weight
was needed to produce a significant antibody response to the
thymus-independent antigen type III pneumococcal polysaccharide (sIII)
in C57Bl/6 mice (Vecchi et al., 1980). With sRBC and the
thymus-independent antigen trinitrophenylated Brucella abortus,
Clark et al. (1981) found a depressed number of spleen plaque-forming
cells in C57BL/6 mice only with a total dose of 40 µg/kg body weight
given as four equal weekly doses. Chastain & Pazdernik (1985)
demonstrated decreased numbers of plaque-forming spleen and bone
marrow cells in response to the thymus-independent antigen
trinitrophenylated lipopolysaccharide (LPS). Spleen and bone marrow
cells were collected from male C57BL/6 mice 7 days after a single ip
dose of 30, 60, 90, or 120 µg TCDD/kg body weight. The decrease
occurred at lower doses in the spleen cell assay but was more
pronounced at higher doses in the bone marrow cell assay. The number
of antibody-producing cells, measured asplaque-forming cells,
following immunization with either sRBC or LPS was reduced in B6C3F1
mice treated with 1 and 5 µg TCDD/kg body weight (Tucker et al.,
1986).
Table 51. Effects of TCDD on humoral-mediated immune responses in adult animals
Species/strain Sex/age/weightf TCDD exposure Parameter measurede
(reference) Frequency/route/dose
Mice
Swiss-Webster F/4-7 weeks/NR fed 10, 20, 50, 100, or primary and secondary sRBCa antibody
500 µg/kg continuously level,
(Hinsdill et al., in the diet for 5 weeks primary and secondary serum tetanus
1980) antitoxin level
C57BL/6J M/6-8 weeks/NR single ip dose of anti-sRBCa plaque-forming spleen cells
1.2, 6, or 30 µg/kg body anti-SIIIb plaque-forming spleen cells
(Vecchi et al., weight
1980)
C57BL/6J M/6-8 weeks/NR four weekly ip doses of anti-sRBCa plaque-forming spleen cells
0.1, 1, or 10 µg/kg body anti-TNP-BAc plaque-forming spleen cells
(Clark et al., weight
1981)
C57BL/6 M/8-10 weeks/NR single ip dose of anti-sRBCa plaque-forming spleen cells
C3H/HeN 1.2, 6, or 30 µg/kg body
DBA/2 weight
AKR
B6D2F1
(Vecchi et al.,
1983)
C57BL/6 M/6-8 weeks/NR single ip dose of 30, 60, anti-TNP-LPSd plaque-forming spleen cells
90, or 120 µg/kg body weight anti-TNP-LPSd plaque-forming bone marrow
(Chastain & cells
Pazdernik, 1985)
Table 51. (contd - 2)
Species/strain Sex/age/weightf TCDD exposure Parameter measurede
(reference) Frequency/route/dose
Mice (contd).
DBA/2N F/6-8 weeks/ single oral doses of 5, anti-sRBCa plaque-forming spleen cells
18-21 g 10, or 50 µg/kg body weight
(Luster et al.,
1985)
B6C3F1 F/6-8 weeks/ single oral doses of 0.2, anti-sRBCa plaque-forming spleen cells
18-21 g 1, 2, 5, or 10 µg/kg body
(Luster et al., weight
1985)
Guinea-pigs
Hartley F/NR/256 g eight weekly oral doses of primary serum tetanus antitoxin level
0.008, 0.04, 0.2, or secondary serum tetanus antitoxin level
(Vos et al., 1973) 1.0 µg/kg body weight
Rabbit
New Zealand M/Adult/3 kg eight weekly oral doses of primary and secondary serum tetanus
0, 0.01, 0.1, 1.0, or 10.0 antitoxin level
(Sharma et al., µg/kg body weight
1984)
a sRBC = sheep red blood cell.
b SIII = type III pneumoccal polysaccharide.
c TNP-BA = trinitrophenylated Brucella abortus.
d TNP-LPS = trinitrophenylated lipolysaccharide.
e = all parameters measured were decreased except for primary serum tetanus antitoxin level in guinea-pigs
(Vosal., 1973).
f M = male; F = female; NR = not reported.
Table 52. Effects of TCDD on humoral-mediated immune responses in maternally exposed animals
Species/strain Time of TCDD exposure Route/dose Parameter measured-response
(reference)
Rats
Fisher-344 N Prenatal day 18 and/or oral/5 µg/kg body weight primary and secondary BGGa
postnatal days 0, 7, and 14 antibody level - no effect
(Faith & Moore,
1977)
Fisher/Wistar Prenatal day 18 and/or oral/5 µg/kg body weight primary and secondary BGGa
postnatal days 0, 7, and 14 antibody level - no effect
(Faith & Luster,
1979)
Mice
Swiss-Webster four weeks before mating, in the diet/ primary and secondary sRBCb
throughout gestation and 1, 2.5, 5, 10, or 20 µ/kg antibody level - no effect;
(Thomas & lactation primary anti sRBCb plaque
Hinsdill, 1979) forming spleen cells - decreased
a = BGG - bovine gamma-globuline.
b = sRBC - sheep red blood cell.
Only minor effects on antibody responses have been reported in
rodents maternally exposed to TCDD (Table 52). Single oral doses of 5
µg TCDD to pregnant Fisher-344 N (poor immunological responder) and
Fisher-Wistar rats (good immunological responder) on gestation day 18
and/or postnatal days 0, 7, and 14 did not affect the antibody reponse
to bovine gammaglobulin (BGG) in the offspring (Faith & Moore 1977;
Faith & Luster, 1979).
Dietary exposure of female Swiss-Webster mice to 2.5 or 5 µg
TCDD/kg for 4 weeks before mating and throughout gestation and
lactation resulted in normal antibody production in the offspring but
in a decrease in anti-sRBC plaque-forming spleen cells.
In vitro exposure of B6C3F1 spleen cells to 10-9 mol
TCDD/litre decreased the production of anti-sRBC plaqueforming cells
(Luster et al., 1984). The ED50 for this effect was found to be 7
nmol/litre when TCDD was present from the first day of culture (Tucker
et al., 1986). Similar activity to that of TCDD was found with
2,3,7-triCDD and 1,2,3,7,8-pentaCDD, whereas 2,8-diCDD and octaCDD
were without effect at concentrations up to 5 x 10-8 mol/litre
(Tucker et al., 1986).
The doses producing 50% suppression of splenic IgM response to
sRBC in C57Bl/6 mice were 7.1 and 85 µg/kg body weight, respectively,
for 1,2,3,6,7,8-hexaCDD and 1,2,3,4,6,7,8-heptaCDD given as single
oral doses 2 days prior to sRBC challenge (Kerkvliet et al., 1985).
OctaCDD had no effect even at doses of 100 and 500 µg/kg body weight.
Humoral immune responses of the rainbow trout (Salmo
gairdneri) were not significantly impaired even at doses of TCDD
that caused clinical toxicity (Spitsbergen et al., 1986).
7.4.5.3 Cell-mediated immunity
Cell-mediated immunity (CMI) operates through specifically
sensitized lymphocytes and is transferred by these cells. Processes
included in this system are classical cell-mediated protective
immunity (which protects against fungi, bacteria, and viruses),
delayed type hypersensitivity, rejection of tumors and foreign tissues
such as transplants, and graft versus host response. Many assays, both
in vivo and in vitro, have been developed to test CMI functions.
Besides a reduction in the number of immunologically competent cells
after TCDD exposure (Gupta et al., 1973; Vos et al., 1973; Zinkl et
al., 1973) TCDD has been demonstrated to induce a decreased CMI
response in adults (Table 53) and even more in maternally exposed
animals (Table 54). Delayed hypersensitivity response, correlatingwith
decreased host resistance to infectious agents in man, was depressed
in rodents exposed to low levels of TCDD (Vos et al., 1973; Faith &
Moore, 1977; Sharma et al., 1978; Faith & Luster, 1979; Thomas &
Hinsdill, 1979; Hinsdill et al., 1980; Clark et al., 1981).
TCDD-exposure also adversely affects host susceptibility to bacteria,
viruses, tumor cells and endotoxins (Thigpen et al., 1975; Thomas &
Hinsdill, 1979; Hinsdill et al., 1980; Luster et al., 1980; Clark et
al., 1983).
Depressed graft versus host response was reported in 2-month-old
C57BL/6 mice exposed to 4 weekly oral doses of 5 µg TCDD/kg body
weight (Vos et al., 1973), whereas no effect was seen in a subsequent
study on 1- and 4-months old C57BL/6Sch mice (Vos & Moore, 1974). In
the same study, Fisher-344 rats maternally exposed to TCDD showed
decreased graft versus host response and prolonged allograft-rejection
time. The latter effect was also demonstrated in maternally
TCDD-exposed C57BL/6Sch mice (Vos et al., 1973). Proliferative
responses of spleen and/or thymus lymphocytes, stimulated by mitogens
specific for the generation of B-lymphocytes and/or T-lymphocytes from
TCDD-exposed animals, were depressed both in adults (Vos & Moore,
1974; Sharma et al., 1978) and maternally exposed rodents (Vos &
Moore, 1974; Faith & Moore, 1977; Vos et al., 1978; Faith & Luster,
1979; Luster et al., 1980). However, Thomas & Hinsdill (1979) found no
effect on the lymphoproliferative response in offspring from
Swiss-Webster mice fed with up to 5 µg TCDD/kg diet 4 weeks before
mating and throughout gestation and lactation. Depressed
lymphoproliferative response is regarded as an extremly sensitive
indicator of immunotoxicity, rather than as a predictor of immune
dysfunction. Cytotoxic T-cell generation in response to allogeneic
antigens has been demonstrated in male DBA/2, C57Bl/6, and B6D2F1 mice
given four weekly ip injections of 1 ng/kg body weight (Clark et al.,
1981, 1983). At this dose no effects were seen on delayed
hypersensitivity, antibody response, thymus cellularity, or enzyme
induction. The adverse effect of TCDD on CMI function seems to be an
age-related phenomenon in rodents. In order to obtain a complete and
persistent immune suppression, TCDD exposure must occur during
ontogenesis of the immune system. In the initial experiments on the
developing immune system, Vos & Moore (1974) exposed Fisher-344 rats
to 1 mg TCDD/kg body weight on gestation days 11 and 18 and on
postnatal days 4, 11, and 18, or to 5 µg TCDD/kg body weight on
postnatal days 0, 7, and 14. CMI functions adversely affected included
in vitro immune competence of spleen and thymus lymphoid cells,
delayed hypersensitivity reaction, prolonged allograft-rejection times
and reduced graft versus host activity. The immune suppression
demonstrated persisted throughout the study, i.e., for 145 days. The
depression of T-cell-dependent immune functions appeared to occur
without helper-cell function being affected (Faith & Moore 1977).
Attempts to study direct effects of TCDD on lymphocytes in
vitro were previously hampered by the low solubility of TCDD in
physiological buffers (Matsumura & Benezet, 1973). Vos & Moore
(1974)obtained no lymphoproliferative response in unstimulated or PHA-
or concanavalin-A-stimulated rat thymus organ cells and mouse spleen
cells when cultured in the presence of up to 20 ng TCDD/ml. Dencker et
al. (1985) demonstrated that mouse fetal thymus cells cultured in
vitro in the presence of TCDD gave a similar response similarly to
that occurring in vivo, i.e., with a dose-dependent inhibition of
the time-dependent increase in the number of lymphoid cells
(EC50=10-10 mol TCDD/litre). It could not be determined with
certainty whether the decreased cell number caused by TCDD was due to
reduced cell proliferation or to increased cell death. The
TCDD-induced suppression of mitogen-stimulated lymphoproliferation has
recently been demonstrated to be mediated by thymus epithelial cells
(Greenlee et al., 1985).
7.4.5.4 Macrophage function
The primary pathway of endotoxin detoxification is thought to be
macrophage-dependent. Thus the increased sensitivity to endotoxin
following TCDD treatment (Vos et al., 1978; Thomas & Hinsdill, 1979)
was suggestive of macrophage dysfunction. However, the number of
peritoneal macrophages, as well as their capacity to mediate cytolytic
and cytostatic effects, was not adversely affected by single ip doses
of 1.2, 6 or 30 µg TCDD/kg body weight to male C57Bl/6J mice
(Mantovani et al., 1980), nor was the ability of macrophages to reduce
nitroblue tetrazolium affected by four to five weekly oral doses of 50
µg TCDD/kg body weight in Swiss-Webster mice (Vos et al., 1978a).
Macrophage function does not appear to be altered by TCDD.
7.4.6 Myelotoxicity
TCDD treatment inhibits the bone marrow haematopoiesis in mice,
both in vivo and in vitro, by directly altering colony growth of
stem cells (Luster et al., 1980, 1985; Chastain & Pazdernik, 1985).
The bone marrow granulocyte-macrophage progenitor cell (CFU-GM)
production was reduced in B6C3F1 mice, receiving 1 µg TCDD/kg body
weight, but was unaffected in DBA/2 mice, even at a dose of 50 µg
TCDD/kg body weight (Luster et al., 1985). Chastain & Pazdernik (1985)
used the B-lymphocyte colony-forming unit assay to demonstrate
reductions in spleen and bone marrow B-cell function of C57BL/6 mice
exposed to single ip doses of TCDD in the range 30 to 120 µg TCDD/kg
body weight. Bone marrow B-cells tended to be more suseptible to TCDD
than spleen B-cells. A maternal oral dose of 5 µg TCDD/kg body weight
on the 14th day of gestation and postnatal days 1, 7, and 14 resulted
in a weak normocytic anaemia indicative of depressed erythrogenesis,
decreased bone marrow cellularity, and decreased stem cell
proliferation in B6C3F1 offspring, assessed 7 days after weaning
(Luster et al., 1980). In vitro exposure of B6C3F1 bone marrow cells
to 10-9 mol TCDD/litre resulted in decreased CFU-GM development and
decreased number of erythrocyte colony-forming units. The decrease in
CFU-GM occurred 1 day post-treatment and remained below control values
until 10 days post-treatment (Luster et al., 1985).
Table 53. Effects of TCDD on cell-mediated immunity responses in adult animals
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Rats
CD F/NR/185 g six weekly oral doses of delayed hypersensitivity to tuberculin - NE
0.2, 1.0, or 5.0 µg/kg
(Vos et al., 1973) body weight
Mice
C57BL/6 M/6-8 weeks/NR four weekly ip doses of delayed hypersensitivity to sRBCa - Dec;
0.1, 1.0, or 10.0 µg/kg delayed hypersensitivity to oxazalone - Dec;
(Clark et al., body weight generation of alloantigen-specific cytotoxic
1981) T-cells - Dec
C57BL/6 M/NR/NR four weekly ip doses of resistance to Herpes virus challenge - Dec;
0.001, 0.01, 1.0, or generation of alloantigen-specific cytotoxic
(Clark et al., 10.0 µg/kg body weight T-cells - Dec
1983)
DBA/2 M/NR/NR four weekly ip doses of generation of alloantigen-specific cytotoxic
0.001, 0.01, 0.1, 1.0, or T-cells - Dec
(Clark et al., 10.0 µg/kg body weight
1983)
Swiss-Webster F/4-7 weeks/NR five weeks feeding of diets resistance to Salmonella typhimurium challenge - Dec;
containing 10, 50, or resistance to Listeria monocytogenes challenge - Dec;
(Hinsdill et al., 100 µg/kg body weight contact sensitivity to 2,4-dinitro-1-fluoro-
1980) benzene - Dec
Table 53 (contd - 2)
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Mice (continued)
C57BL/6J M/6-8 weeks/NR single ip doses of 1.2, number of peritoneal macrophages - Dec;
6 or 30 µg/kg body weight number of splenic natural killer cells - Dec;
(Mantovani macrophage mediated cytolysis - NE;
et al., 1980) macrophage mediated cytostasis - NE
C57BL/6 M/NR/NR four weekly ip doses of generation of allospecific cytotoxic T-cells - Dec
DBA 0.001 µg/kg body weight - NE
B6D2F1 - Dec
(Nagarkatti
et al., 1984)
CD-1 M/NR/28 two, four, or eight weekly lymphoproliferative response of PHAb- and PWMc-
doses of 0.01, 0.1, 1.0, or stimulated splenic cells - Dec
(Sharma & 10.0 µg/kg body weight
Gehring, 1979)
C57BL/6Jfh M/4 weeks/NR four weekly oral doses of resistance to Salmonella bern challenge - Dec;
0.5, 1.0, 5.0, 10.0, or resistance to Herpes virus challenge - NE
(Thigpen et al., 20.0 µg/kg body weight
1975)
C57BL/6 M/2 months/ four weekly oral doses of graft versus host activity - Dec
24.4 g 0.2, 1.0, 5.0, or 25.0
(Vos et al., µg/kg body weight
1973)
C57BL/6Sch M/1 month/NR four weekly oral doses of lymphoproliferative response of PHAb-stimulated
1.0, 5.0, or 25.0 µg/kg spleen cells - Dec;
(Vos & Moore, body weight graft versus host activity - NE
1974)
Table 53 (contd - 3)
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Mice (continued)
C57BL/6Sch M/4 months/NR six weekly oral doses of lymphoproliferative response PHA-stimulated
1.0, 5.0, or 25.0 µg/kg spleen cells - NE
(Vos & Moore, body weight graft versus host activity - NE
1974
Swiss M/3-4 weeks/NR four or five weekly oral resistance of Listeria monocytogenes - NE;
doses of 50 µg/kg body number of peritoneal macrophages - NE;
(Vos et al., weight macrophage reduction of nitroblue tetrazodium - NE
1978a)
Guinea-pigs
Hartley F/NR/256 g eight weekly oral doses delayed hypersensitivity to tuberculin - Dec
of 0.008, 0.04, 0.2, or
(Vos et al, 1973) 1.0 µg/kg body weight
Rabbits
New Zealand M/adult/3 kg eight weekly oral doses delayed hypersensitivity to tuberculin - Dec
of 0.01, 0.1, 1.0, or
(Sharma et al., 10.0 µg/kg body weight
1984)
a sRBC = sheep red blood cells.
b PHA = phytohaemagglutinin.
c PWM = poke weed.
d NR = not reported; NE = no effect; Dec = decreased; M = male; F = female.
Table 54. Effects of TCDD on cell-mediated immunity responses in maternally exposed animals
Species/strain TCDD exposure Age when Parameter measured - responsef
(reference) Frequency/route/dose testedg
Rats
Fisher/Wistar 5 µg/kg body weight on gestation day 18 25 days lymphoproliferative response of
and on postnatal days, 0, 7, and 14, or PHAa- and ConAb-stimulated spleen
(Faith & Luster, 5 µg/kg body weight on postnatal days and thymus cells - Dec;
1979) 0, 7, and 14 delayed hypersensitivity to
tuberculin - Dec
Fisher-344 5 µg/kg body weight on gestation day 18 25 days lymphoproliferative response of
and on postnatal days 0, 7, and 14, or PHAa- and Conb-stimulated spleen
(Faith & Moore, 5 µg/kg body weight postnatal days 0, 7, and thymus cells - Dec;
1977) and 14 delayed hypersensitivity to
oxazolone - Dec
Fisher 344 1 µg/kg body weight on gestation days 11 25 days lymphoproliferative response of
and 18 and on postnatal days 4, 11 and 18 or PHAa-stimulated spleen cells and of
(Vos & Moore, 5 µg/kg body weight on postnatal days 0, PHAa- and ConAb-stimulated thymus-
1974) 7 and 14 cells - Dec
Mice
B6C3F1 c 1, 5, or 15 µg/kg body weight on NR lymphoproliferative response of
gestation day 14 and on postnatal days 1, mitogen-stimulated spleen cells:
(Luster et al., 7, and 14 PHAa - Dec;
1980) ConAb - Dec;
LPSd - NE;
Macrophage proliferation - NE;
Phagocytizing ability - NE;
Resistance to Listeria monocyto-
genes - Dec;
Resistance to PYB6 tumor cells - Dec
Table 54 (contd).
Species/strain TCDD exposure Age when Parameter measured - responsef
(reference) Frequency/route/dose testedg
Mice (continued)
Swiss-Webster Feeding 1, 2.5, or 5 µg TCDD/kg 5-6 weeks Contact sensitivity to 2,4-dinitro-
4 weeks before mating, throughout 1-fluorobenzene - Dec;
(Thomas & gestation, and 3 weeks postnatally lymphoproliferative response of
Hinsdill, 1979) PHAa- and ConAb-stimulated spleen
and thymus cells - NE;
Resistance to Salmonella typhimurium
endotoxin - Dec; Resistance
to Listeria monocytogenes - NE
C57BL/6Sch 2 or 5 µg/kg body weight on gestation days 23 days Skin graft assay - prolonged skin
14 and 17 and on postnatal days 1, 8, and 15 graft rejection time
(Vos et al., 1974)
Swiss 10 µg/kg body weight on postnatal days 22 days lymphoproliferative response of
1, 4, 8, 11, 15, and 18 PHAa-, ConAb- &
(Vos et al., PWMe-stimulated thymus cells - Dec
1978a)
a PHA = phytohaemagglutinin.
b ConA = concanavalin A.
c B6C3F1 = progeny to female C57BL/6N and male C3H mice.
d LPS = lipopolysaccharide.
e PWM = poke weed.
f Dec = decreased, NE = no effect.
g NR = not recorded.
7.4.7 Effects on the intermediary metabolism
Changes in intermediary metabolism have been demonstrated in
TCDD-treated experimental animals. The circulating concentration of
glucose in TCDD-treated rats was decreased relative to ad
libitum-fed (Zinkl et al., 1973; Schiller et al., 1985) and pair-fed
(Gasiewicz et al., 1980; Potter et al., 1983) control rats.
TCDD-treated and pair-fed controls (both groups were schedule fed) had
similar concentrations of serum glucose (Christian et al., 1986b). The
TCDD-induced hypoglycaemia was not caused by altered pancreatic
function, as judged by insulin and glucagon levels (Potter et al.,
1983).
Reduced hepatic glycogen content, compared to the value in
control rats, was reported in Sprague Dawley rats 16 days after a
single ip dose of 20 µg TCDD/kg body weight (Weber et al., 1983).
However, in studies by Christian et al. (1986b), compared to pair-and
schedule-fed control rats, TCDD-treated (75 µg/kg body weight daily)
Sprague Dawley rats had significantly increased hepatic glycogen
levels but unaffected cardiac and muscle (gastrocnemius) levels of
glycogen 2-8 days post-treatment.
TCDD-treated rats maintained a normal overall nitrogen balance,
as judged by urinary urea, creatinine, and ammonia levels, but
exhibited changes in certain plasma protein levels (Christian et al.,
1986b).
Elevated circulating cholesterol levels were found in
TCDD-treated rats (Albro et al., 1978; Poli et al., 1980; Schiller et
al., 1985), whereas circulating free fatty acids and triacylglycerols
were decreased in TCDD-treated rats, when compared to pair-and
schedule-fed control rats (Christian et al., 1986b).
A marked accumulation of hepatic lipid has been found in rats
after single doses of TCDD (Cunningham & Williams, 1972; Gupta et al.,
1973; Albro et al., 1978; Schiller et al., 1985). At a sublethal dose
of TCDD, hepatic triglyceride and free fatty acid levels were elevated
already one day after dosing. Abnormal lipid deposition patterns
persisted for at least 2 months (Albro et al., 1978). The increased
level of triglycerides in the liver (Schiller et al., 1985; Christian
et al., 1986b) of TCDD-treated rats was not accompanied by increases
in cardiac or muscle (gastrocnemius) triacylglycerol levels (Christian
et al., 1986b). Hepatic lipid synthesis in Wistar rats, measured as
the 1-h incorporation of 3H-acetate, was not affected by TCDD
treatment when studied 7 days after exposure to 10 µg TCDD/kg body
weight (Cunningham & Williams, 1972).
Mice (both C57BL/6 and DBA/2 strains) responded to TCDD treatment
with dose-dependent decreases in serum levels of glucose, cholesterol,
and triglycerides, and increases in hepatic triglycerides, whereas
serum glycerol and free fatty acid levels were unaffected (Chapman &
Schiller, 1985).
Hartley guinea-pigs given a lethal ip dose of 2 µg TCDD/kg body
weight had significantly increased circulating levels of cholesterol
esters, triglycerides, and phospholipids but a normal free fatty acid
level 7 days post-treatment. The increase in serum lipids was
accompanied by a pronounced increase in low density lipoproteins,
particularly the very low density lipoprotein fraction (Swift et al.,
1981). The plasma cholesterol and triglyceride levels were elevated
also when TCDD-treated guinea-pigs were compared to pair-fed controls
(Gasiewicz & Neal, 1979).
A TCDD-induced increase in plasma triglyceride levels was also
noted in New Zealand rabbits, fed 20 µg TCDD/kg body weight, whereas
plasma cholesterol was unaffected 12 weeks after dosing (Lovati et
al., 1984). TCDD treatment did not alter the liver lipid levels (free
and esterified cholesterol, triglycerides, and phospholipids), but the
triglyceride level was significantly increased. These results were
valid both for rabbits fed normal chow and those fed a
cholesterol-rich (0.5%) diet. Golden Syrian hamsters exposed to 1000
µg TCDD/kg body weight, orally or ip, had elevated plasma cholesterol
levels until 20 days after exposure but normal levels on day 50,
whereas serum triglyceride levels were normal until day 20 and then
became significantly lower than those of controls (Olson et al.,
1980b).
7.4.8 Enzyme induction
Primarily, TCDD has been found to increase enzyme activities
although observations on enzyme inhibition have also been made. Since
the first reports of enzyme systems as targets for TCDD (Buu-Hoi et
al., 1971a, 1972b; Greig, 1972; Poland & Glover, 1973b,c), enzyme
induction has become the most extensively studied biochemical response
produced by TCDD. The mixed function oxidase system (MFO), capable of
metabolizing both endogenous and foreign lipophilic compounds to more
polar products, has been the most thoroughly investigated one, and
arylhydrocarbon hydroxylase (AHH) and 7-ethoxy-resorufin 0-deethylase
(EROD) are the most frequently assayed enzyme activities in this
system. TCDD has also been reported to affect UDP-glucuronosyl
transferases (UDPGT) (Thunberg et al., 1980, 1984) and
glutathione-S-transferases (GT) (Manis & Apap, 1979), which are
multifunctional enzyme systems involved in conjugating a wide variety
of compounds.
Most studies have been performed with microsomal enzymes, but
TCDD has also been found to have effects on enzymes in the cytosolic
fraction. It seems that TCDD produces organ-specific effects, and
although, quantitatively, hepatic enzyme induction is of more concern
than extrahepatic enzyme effects, the latter may qualitatively be as
important. Studies in different species have revealed that enzyme
induction due to TCDD exposure also is a species-specific phenomenon.
Time course studies have shown that maximal increases in enzyme
activities are reached within 3 to 4 days post-treatment. After a lag
period of about 2 to 3 weeks, enzyme activities begin to return to
normal levels (Hook et al., 1975a,b; Lee & Suzuki, 1980; Lucier et
al., 1973; Poland & Glover, 1973a).
According to Kitchin & Woods (1979), TCDD-induced AHH activity
did not reach the normal level until 6 months after rats were exposed
to a single daily dose of 2 µg/kg body weight.
Hook et al. (1975a) found no apparent dependence on age when
studying AHH induction in CD rats which were 10 to 335 days old at the
time of exposure to 25 µg TCDD/kg body weight.
Several investigators have studied the relative potency of various
PCDDs and PCDFs to induce AHH and/or EROD activities (Bradlaw et al.,
1980; Poland et al., 1976; Bandiera et al., 1984a,b; Sawyer & Safe,
1985; Mason et al., 1986). They found an apparent structure-activity
relationship between the location of the halogen atoms on the
dibenzo-p-dioxin molecule and the ability to induce AHH activity
both in vivo and in vitro. Isomers with halogens at the four
lateral ring positions produced a greater biological response than
those with halogens at three lateral ring positions, while two
lateral halogen atoms seemed to be insufficient to produce a
biological response. TCDD was the most potent enzyme inducer of the
compounds tested.
On a molecular basis TCDD is the most potent MFO-inducing
compound known and MFO induction seems to be the most sensitive
biochemical response produced by this chemical. According to Kitchin
& Woods (1979), induction in the rat takes place after a single daily
dose of only 0.002 µg TCDD/kg body weight. In the guinea-pig (the
animal most sensitive to TCDD toxicity), MFO induction has been
observed, but the induced activities were low even at lethal doses
(Hook et al., 1975a). Neither is there a correlation in cell cultures
between induction of MFO and toxicity. Furthermore, it is known that
metabolites of TCDD are less toxic and more readily excreted than the
parent compound (see section 8.1.5). Thus, TCDD-induced MFO activities
represent a detoxification process rather than one leading to toxic
effects.
However, induction of MFO activities might potentiate the
toxicity of other foreign compounds requiring metabolic transformation
by the MFO system before they can exert their toxic effect. A number
of studies have shown that induction of MFO activities alters the
metabolism of the model xenobiotic, benzo(a)pyrene by increasing the
rate of microsomal metabolism, changing the metabolic profile to more
toxic metabolites, and increasing the extent of covalent binding to
liver microsomes (Berry et al., 1976, 1977; Uotila et al., 1978).
TCDD, applied topically or subcutaneously increased the
carcinogenicity of 3-methyl cholanthrene (MC) in DBA/2 mice (Kuori,
1978), but decreased the carcinogenicity of
7,12-dimethylbenz(a)anthracene (DMBA) in CD-1 mice (DiGiovanni et al.,
1979a). These authors suggested that TCDD induces the MFO system and
thus increases activation of MC to the ultimate carcinogen, as well as
inactivation of DMBA, which would explain these effects.
Furthermore, increased MFO activities might adversely affect
important metabolic pathways of endogenous compounds. The effects of
TCDD on enzyme activities, both MFO and others, involved in such
biological pathways as keratinization, steroid metabolism, lipid
metabolism, plasma membrane function and porphyrin metabolism, are
discussed under separate sections. The minute quantities of TCDD
required for maximal enzyme induction or suppression, the long
duration of the effect, and the stereospecific requirements suggest a
specific interaction of TCDD with a cellular species, possibly at the
gene level. Accordingly considerable research has been directed toward
the study of the genetic regulation of AHH induction by TCDD. A
hepatic cytosolic species that bound TCDD has been suggested as the
receptor for the hepatic AHH activity. Numerous studies of this
cytosolic receptor in several species and tissues have been performed
and it seems that there is a structural gene, the Ah locus, for this
receptor, which is responsible for the expression of various enzyme
activities (see sections 7.8 and 7.8.1).
7.4.8.1 Studies on rats
The effect of TCDD on enzyme activities has been most extensively
investigated in the rat. In the liver, TCDD has been shown to increase
both the content of cytochrome P-450 (Lucier et al., 1973, 1986;
Poland & Glover, 1974a,b; Hook et al., 1975a; Aitio & Parkki, 1978;
Kitchin & Woods, 1979; Madhukar & Matsumura, 1981; Goldstein & Linko,
1984) and cytochrome b5 (Lucier et al., 1973; Hook et al., 1975a), as
well as the microsomal enzyme activities involved in the oxidative
transformation and conjugation of xenobiotics, e.g., aniline
hydroxylase, arylhydrocarbon hydroxylase (AHH), biphenyl hydroxylase,
7-ethoxycoumarin-0-deethylase (ECOD), EROD, and UDPGT. These enzyme
activities have been investigated in a vast number of studies, some of
them quoted in Table 55. Goldstein & Linko (1984) demonstrated that
TCDD induced two isozymes of cytochrome P-450 (P-448) in the liver but
only one of these in extrahepatic tissues of young Sprague Dawley rats
2 days after a single oral dose of 25 µg/kg body weight.
Table 55. Studies demonstrating in vivo induction of mixed function
oxidases and UDP-glucuronosyltransferases in TCDD-exposed strains of rats
Rat
Enzyme activity strain Reference
Aniline hydroxylase SD Beatty et al. (1978)
CD Lucier et al. (1973); Hook et al. (1975a)
Aryl hydrocarbon SD Poland & Glover (1973a, 1974a); Beatty et
hydroxylase al. (1978); Manis & Apap (1979);
Haaparanta et al. (1983); Thunberg et al.
(1984); Lucier et al. (1986); Ahlborg et
al. (1987)
CD Lucier et al. (1973); Hook et al.
(1975a); Kitchin & Woods (1979)
Wistar Nagayama et al. (1983); Keys et al.
(1985); Bannister et al. (1986); Farrell
& Safe (1986); Mason et al. (1986);
Tsyrlov et al. (1986)
Biphenyl hydroxylase CD Hook et al. (1975a,b,); Kitchin & Woods
(1979)
7-Ethoxycoumarin-O- Wistar Aitio & Parkki (1978)
deethylase
7-Ethoxyresurofin-O- SD Haaparanta et al. (1983)
deethylase
CD Kitchin & Woods (1979)
Wistar Keys et al. (1985); Bannister et al.
(1986); Farrell & Safe (1986); Mason et
al. (1986)
UDP-glucuronosyl-
transferase:
p-nitrophenol SD Thunberg et al. (1980, 1984); Ahlborg et
al. (1987)
CD Lucier et al. (1973, 1975a, 1986); Hook
et al. (1975a)
Table 55 (Cont.) Studies demonstrating in vivo induction of mixed
function oxidases and UDP-glucuronosyltransferases in TCDD-exposed
strains of rats
Rat
Enzyme activity strain Reference
UDP-glucuronosyl-
transferase:
p-nitrophenol Wistar Aitio et al. (1979); Thunberg & Híkansson
(cont'd.) (1983)
o-aminophenol Wistar Aitio et al. (1979)
4-methylumbelli-
ferone Wistar Aitio & Parkki (1978)
Microsomal glutathion-s-transferase (GT) did not respond to
TCDD (Aitio & Parkki, 1978; Mukitari et al., 1981; Baars et al.,
1982), but cytosolic GT was induced both by a single dose of 17 µg
TCDD/kg body weight 2 days post-treatment (Manis & Apap, 1979), and by
near lethal or lethal doses 1 and 6 days after dosing (Mukitari et
al., 1981; Baars et al., 1982; Hassan et al., 1983). Glutathione
reductase was also increased, while glutathione peroxidase, both total
and Se-dependent, and the content of reduced glutathione were reduced
by TCDD treatment (Hassan et al., 1983, 1985a,b,c).
The following hepatic enzyme activities involved in drug
metabolism have been reported to be unaffected by TCDD treatment in
the rat: N-and O-demethylation (Lucier et al., 1973; Poland &
Glover, 1973a; Hook et al., 1975a; Beatty et al., 1978; Kitchin &
Woods, 1979; Madhukar & Matsumura, 1981), epoxide hydratase (EH)
(Aitio & Parkki, 1978), ß-glucuronidase (Lucier et al., 1973, 1975)
and NADPH cytochrome c reductase (Poland & Glover, 1974; Aitio &
Parkki, 1978; Kitchin & Woods, 1979; Madhukar & Matsumura, 1981). The
glucuronide conjugation of bilirubin (Aitio et al., 1979), estrone,
and testosterone (Lucier et al., 1975a) by liver microsomes from
TCDD-treated rats was not different when compared to control rats.
Some hepatic enzyme activities not belonging to the MFO system,
which are affected by TCDD treatment include aldehyde dehydrogenase
(Deitrich et al., 1978; Lindahl et al., 1978), delta-aminolevulinic
acid synthetase (see section 7.4.3), DT-diaphorase (Beatty & Neal,
1977), transglutaminase (see section 7.4.4), ornithine decarboxylase
(Nebert et al., 1980; Potter et al., 1982; Farrell & Safe, 1986),
plasma membrane ATPases (see section 7.4.2.2), porphyrinogen
carboxylase (see section 8.4.3), prostaglandin synthetase (see section
7.4.9), enzymes involved in testosterone metabolism (see section
7.4.9), and RNA polymerase (Kurl et al., 1982).
Prenatal and postnatal exposure via milk to TCDD, at doses of 3
µg/kg body weight to pregnant rats on days 5, 10 and 16 of gestation,
induced hepatic AHH and UDPGT activities in the offspring. The effect
was seen 8 days post-partum and persisted for at least 2 weeks. The
inductive effect was due both to exposure to TCDD via milk and to the
activation of an inducing mechanism after birth. Fetal liver AHH was
slightly increased during late gestation, although the UDPGT activity
and the cytochrome P-450 content were not (Lucier et al., 1975b).
Administration of 2.5 µg TCDD/kg body weight to pregnant rats on
day 17 of gestation increased the AHH- and N-hydroxylation activities
and cytochrome P-450 content in the fetal liver on day 20 of gestation
(Berry et al., 1976).
AHH induction due to TCDD has been reported to occur also in the
brain (Hook et al., 1975a), kidney (Poland & Glover, 1973a; Hook et
al., 1975a; Aitio & Parkki, 1978; Potter et al., 1982; Nagayama et
al., 1983), lung (Poland & Glover, 1973a; Hook et al., 1975a; Aitio &
Parkki, 1978; Nagayama et al., 1983), prostate (Lee & Suzuki, 1980;
Haaparanta et al., 1983; Nagayama et al., 1983), thymus (Nagayama et
al., 1983), and intestine (Poland & Glover, 1973a; Hook et al.,
1975a), but intestinal AHH activity was found to be unaffected by 17
and 20 µg TCDD/kg body weight (Aitio & Parkki, 1978; Manis & Apap,
1979). Testicular (Poland & Glover, 1973a; Hook et al., 1975a; Aitio
& Parkki, 1978) and adrenal (Guenthner et al., 1979) AHH activities
were not induced by sublethal doses of TCDD. The O-deethylation
activity in kidney, lung, and prostate was increased, but no effect
was seen on the activity in testes or intestine (Aitio & Parkki, 1978;
Haaparanta et al., 1983). UDPGT activities in kidney, lung, intestine,
and brain were increased, while no effect was seen on testicular UDPGT
(Hook et al., 1975a). Similar results were reported by Aitio & Parkki
(1978), though in their study intestinal UDPGT was not affected.
Renal biphenyl hydroxylation activity has been found to increase
after TCDD treatment, but no effect on this enzyme activity was seen
in lung, intestine, brain, or testes (Hook et al., 1975a). Elevated
levels of cytochrome P-450 were found in prostate (Lee & Suzuki, 1980)
and mammary gland (Rikans et al., 1979), but not in adrenals
(Guenthner et al., 1979). Less testicular cytochrome P-450 was found
after a single dose of 25 µg TCDD/kg body weight (Tofilon & Piper,
1982). The GSH tranferase activity was increased in the lung but not
in kidney, intestine, testes (Aitio & Parkki, 1978), or prostate (Lee
& Suzuki, 1980).
Neither EH nor NADPH cytochrome c reductase were inducible by
TCDD in kidney, lung, intestine, testes (Aitio & Parkki, 1978),
mammary gland (Rikans et al, 1979), or prostate (Lee & Suzuki, 1980).
The ED50 values for hepatic AHH and EROD induction were determined
in immature male Wistar rats 13 days after a single ip dose of
2,3,7-triCDD, TCDD, 1,3,7,8-tetraCDD, 1,2,3,7,8-pentaCDD,
1,2,4,7,8-pentaCDD or 1,2,3,4,7,8-hexaCDD (Mason et al., 1986). The
order of enzyme-inducing capacity was TCDD > 1,2,3,7,8-pentaCDD >
1,2,3,4,7,8-hexaCDD > 1,2,4,7,8-pentaCDD > 2,3,7-triCDD >
1,3,7,8-tetraCDD (Table 56).
7.4.8.2 Studies on mice
Enzyme induction studies in mice have been performed mainly with
two strains genetically separated at the Ah locus, thus making them
responsive (C57Bl/6 (B6)) or non-responsive (DBA/2 (D2)) to induction
of hepatic cytochrome P-450-related enzyme activities by aromatic
hydrocarbons, e.g., 3-methyl-cholanthrene (3-MC). However, the
extraordinary potency of TCDD for enzyme induction revealed increased
hepatic cytochrome P-450 content as well as AHH and O-deethylase
activities both in B6 and D2 mice after sublethal exposure to TCDD
(Poland & Glover, 1974a,b,c; Jones & Sweeney, 1977; Greenlee & Poland,
1978). Studies of MFO induction in five responsive and five
non-responsive strains of mice by Poland & Glover (1974a,b,c) revealed
that there were no consistent differences between the strains when
considering the extent to which TCDD induced AHH, O-deethylase,
N-demethylase and O-demethylase activities in the liver, kidney,
lung, skin, or bowel. The ED50 for hepatic AHH induction was
determined to be 10-9 mol/kg body weight in the responsive strain
and > 10-8 mol/kg body weight in the non-responsive strain (Poland &
Glover, 1975). Fully induced hepatic AHH activity was obtained both in
responsive (C57Bl/6 and AKR/Qdj) and non-responsive (DBA/2 and DDD)
strains of mice 3 days after an ip dose of 30 µg TCDD/kg body weight
(Nagayama et al., 1985a). Both AHH and EROD were induced in C57Bl/6
mice 7 days after an ip dose of 0.32 µg/kg body weight (Bannister et
al., 1986). Both hepatic AHH and ornithine decarboxylase activities
were similarly induced in C57Bl/6 and DBA/2 mice after a single ip
dose of 100 µg TCDD/kg body weight, but at 2 µg TCDD/kg body weight
these enzymes were induced only in the C57Bl/6 strain (Nebert et al.,
1980). Two daily doses of 0.1 µg TCDD, topically applied, increased
the epidermal AHH activity in two strains of hairless mice (Puhvel et
al., 1982). Contrary to observations in rats, TCDD induces testicular
AHH activity both in B6 and D2 mice 40h after an ip dose of 50 µg/kg
body weight (Mattison & Thorgeirsson, 1978).
Table 56. Structure activity relationships for some PCDDs
PCDD In vitro EC50 values (M)a In vivo ED50 values (mmol/kg)bLD50 c
Congener
Receptor AHH EROD AHH EROD Body Thymic Guinea-pig
binding weight atrophy µg/kg
body
weight
1- > 1.0x10-4 > 1.0x10-4 > 1.0x10-4
2,8- 3.2x10-6 > 1.0x10-4 > 1.0x10-4 > 300 000
1,2,4- 1.3x10-5 4.8x10-5 2.2x10-6
2,3,6- 2.2x10-7
2,3,7- 7.1x10-8 3.6x10-7 1.4x10-7 19.6 19.6 98.1 29 400
1,2,3,4- 1.3x10-6 3.7x10-6 2.4x10-6
1,2,3,8- 6.1x10-7
1,3,7,8- 7.9x10-7 5.9x10-7 3.2x10-7 31.2 77.6 132 100
2,3,6,7- 1.6x10-7 6.1x10-8 1.1x10-8
2,3,7,8- 1.0x10-8 7.2x10-11 1.9x10-10 0.004 0.003 0.05 0.09 2
1,2,3,4,7- 6.4x10-6 6.6x10-7 8.2x10-7
1,2,3,7,8- 7.9x10-8 1.1x10-8 1.7x10-8 0.031 0.056 0.62 0.17 3.1
1,2,4,7,8- 1.1x10-6 2.1x10-8 1.1x10-8 2.82 0.56 34 11.2 1125
1,2,3,4,7,8- 2.8x10-7 2.1x10-9 4.1x10-9 0.03 0.130 1.63 1.07 72.5
1,2,3,6,7,8-c 5.7x10-10 3.1x10-8 70-100
1,2,3,7,8,9-c 1.4x10-9 4.6x10-8 60-100
1,2,3,4,6,7,8c 1.3x10-7 > 600
1,2,3,4,6,7,9-c 3.7x10-6
1,2,3,4,6,7,8,9- > 1.0x10-5 > 1.0x10-4 > 1.0x10-4
a Estimated concentrations needed to displace 50% of 3H-TCDD bound to liver cytosol receptor from Wistar
rats and to produce 50% maximum enzyme induction in the rat hepatoma 11-4-II E cell line (Bradlaw & Casterline,
1979; Mason et al., 1986).
b Studies in immature male Wistar rats (Mason et al., 1986).
c McConnell et al., 1978b.
7.4.8.3 Studies on guinea-pigs
The guinea-pig, the species most sensitive to the toxic effects
of TCDD, does not respond with liver toxicity or with extensive
enzyme induction.
Hook et al. (1975a) investigated the effect of a single oral dose
of 0.175 µg TCDD/kg body weight on Hartley guinea-pig MFO and UDPGT
activities in liver, kidney, and lung. AHH induction was found only in
the kidney. In none of the tissues was there an effect on UDPGT
activity. The biphenyl 4-hydroxylase activity was increased in all
tissues, whereas hepatic biphenyl 2-hydroxylase was decreased. With
three daily doses of 1 µg TCDD/kg body weight, Hassan et al. (1983)
found an increase in hepatic AHH 6 days post-treatment. They also
found slightly increased in vitro lipid peroxidation but no effect
on glutathione content or on the enzyme activities facilitating
peroxidation, reduction, or transfer of glutathione. The DT-diaphorase
activity was not affected by a single oral dose of 0.6, 3.0, or 6.0 mg
TCDD/kg body weight (Beatty & Neal, 1977). The maximal increases were
4.4 and 22 times for AHH and EROD activities, respectively. The
testicular cytochrome P-450 content in Hartley guinea-pigs was
decreased by 50% one day after a single dose of 1 mg TCDD/kg body
weight. The effect persisted for at least 9 days (Tofilon, 1980). No
effect was seen on microsomal haeme content or on the activities of
NADPH-cytochrome C reductase and sorbitol dehydrogenase, the marker
enzyme for testicular protein synthesis. Thus, the decrease in
cytochrome P-450 induced by TCDD does not seem to be a nonspecific
inhibition of protein synthesis.
7.4.8.4 Studies on rabbits
Studies on enzymes in rabbits have been performed in the New
Zealand albino strain exposed to single doses of 10 to 30 mg TCDD/kg
body weight for 1 to 5 days. Both in adults (Johnson &
Muller-Eberhard, 1977a,b) and in neonates exposed in utero (Norman
et al., 1978; Kohli & Goldstein, 1981), TCDD increased the content of
cytochrome P-450. It also induced the formation of immunologically
distinct cytochromes P-450 in adult and neonatal liver (Norman et al.,
1978). Increased cytochrome P-450 was observed in the kidney but not
in the lung (Liem et al., 1980; Kohli & Goldstein, 1981). Renal and
pulmonary cytochrome P-450 reductase, investigated by Liem et al.
(1980), were not affected by TCDD treatment. Data on MFO induction and
suppression are conflicting. Liem et al. (1980) reported increased AHH
and O-deethylase activities in lung and kidney, whereas Hook et al.
(1975a) saw no effect on the AHH activity in the lung and reported a
decrease in hepatic AHH activity after a single oral dose of 0.5 µg
TCDD/kg body weight. Biphenyl 4-hydroxylase induction was seen in the
liver by Johnson et al. (1979). Hook et al. (1975a) detected such
induction in lung, but no effect in the liver and kidney (Hook et al.
1975a). Furthermore, Hook et al. (1975a) reported no effects on
biphenyl-2-hydroxylation and UDPGT activities in liver, kidney, or
lung. A decrease in hepatic, but not in renal or pulmonary
N-demethylation, was found by these authors.
7.4.8.5 Studies on hamsters
Golden Syrian hamsters are among the animals most resistant to
acute lethal effects induced by TCDD. Although the liver is a target
tissue, hepatic enzyme induction has barely been studied in this
species. When given an oral dose of 200 µg/TCDD/kg body weight for a
period of 3 days, increased hepatic glutathione-S-transferase and
glutathione reductase activities were found, but no effects were seen
on AHH or glutathione peroxidase activities. Neither the hepatic level
of glutathione nor the in vitro lipid peroxidation were affected
(Hassan et al., 1983). The ED50 values for induction of hepatic ECOD
and reduced NAD(P), menadione oxidoreductase activities, and
cytochrome P-450 content in male Golden Syrian hamsters were 1.0, 2.0,
and 0.5 µg TCDD/kg body weight, i.e. extremely low doses as compared
to doses that produce tissue damage and lethality in this species
(Gasiewicz et al., 1986).
7.4.8.6 Studies on cows
Three dairy Holstein cows (500-600 kg) received a single oral
dose of 0.05 (two cows) or 7.5 µg TCDD/kg body weight (Jones et al.,
1986). The cow receiving the high dose was killed on day 7 and those
receiving the low dose on day 14. AHH and EROD activities were
markedly induced in the high-dose but not in the low-dose animals.
7.4.8.7 Studies on chick embryos
AHH and delta-aminolevulinic acid synthetase in the chick embryo
have been reported to be extremely sensitive to the inductive effects
of TCDD (Poland & Glover, 1973b,c). Maximal induction occurred with
155 pmol TCDD/egg. The induction was relatively long lasting, with 70%
of the maximum induced activity present 5 days following a single dose
of TCDD. Structure-activity studies demonstrated a good correspondence
between the toxicity and induction potency of a series of
dibenzo-p-dioxin congeners (Poland & Glover, 1973c).
ED50 values for the induction of hepatic microsomal EROD, AHH,
and 4-dimethylaminoantipyrine-N-demethylase in 2-week-old white
Leghorn cockerels on day 5 after TCDD exposure were 778, 302, and 561
ng/kg body weight, respectively, and aldrin epoxidase was inhibited by
TCDD treatment (Sawyer et al., 1986). Hepatic and cardiac EROD
activities were increased in white Leghorn chicken embryos exposed to
TCDD in ovo at doses between 1000 and 10 000 pmol/egg (Quilley &
Rifkind, 1986).
7.4.8.8 Studies on cell cultures
TCDD has a very low toxicity in cell cultures, yet it is a very
potent inducer of AHH activity in these systems, including lymphocytes
and primary hepatocytes, as well as established and transformed cell
lines.
The inducibility of lymphocyte AHH has been investigated in
mitogen-stimulated human lymphocytes from the venous blood of healthy
volunteers. Kouri et al. (1974) found a dose-dependent increase in AHH
activity (0, 0.1, 1.0, 10, or 100 ng TCDD/ml medium for 24 h). The
optimal dose was about 10 ng/ml, and the maximal induction was by a
factor of 2 to 3. On the contrary, Gurtoo et al. (1979) found no
dose-response correlation, in the dose range 1.7 to 20 ng TCDD/ml,
when measuring lymphocyte AHH induction. To circumvent the limitation
of prior mitogen activation when studying AHH induction in
lymphocytes, Freedman et al. (1979) used the human B-lymphocyte
RPMI-1788 cell line, which does not require prior activation for the
induction of AHH activity. The optimal concentration to stimulate AHH
activity was determined to be 10 ng/ml medium. Highly variable
induction of AHH (between 3- and 28- fold) was obtained by Nagayama et
al. (1985b) in human lympho-blastoid cell lines derived from the
peripheral blood of healthy volunteers of both sexes and of variable
ages. The cells were exposed to 7.5 ng TCDD/ml medium for 48 h.
In a study by Niwa et al. (1975), the estimated ED50 values for
AHH induction by TCDD in 11 established cell lines, in fetal primary
cultures from five animal species and cultured human lymphocytes,
ranged from 0.04 ng/ml medium in C57Bl/6 mouse cultures and 0.08 ng/ml
in the rat hepatoma H-4-IIE cell line to more than 66 ng/ml in the HTC
rat hepatoma cell line. TCDD was demonstrated to be the most potent
AHH inducer out of 24 chlorinated dibenzo-p-dioxin analogues
(Bradlaw et al., 1980) tested in a rat hepatoma cell culture extremely
sensitive to AHH induction, the ED50 being about 0.5 pg/106 cells.
A 165-fold increase in AHH-activity and a 54-fold increase in EROD
activity were obtained in rat hepatoma H-4-IIE cells when exposed to
2 x 10-10 mol TCDD/litre for 3 days (Keys et al., 1986). In this
system co-exposure of TCDD with 1,3,6,8-tetraCDF and 2,4,6,8-tetraCDF
reduced the TCDD-induced enzyme induction, whereas co-exposure of TCDD
and TCDF resulted in an additive effect on enzyme induction (Keys et
al., 1986). The EC50 values for AHH and EROD induction in the same
cell system varied over 7 orders of magnitude for 14 different PCDDs
(Table 56), the most potent being TCDD and the least potent being
2,3,6-triCDD (Mason et al., 1986). A 2-to 650-fold AHH induction was
observable in 8 of 22 different cell cultures exposed to 10-9 mol
TCDD/litre for 24 h (Knutson & Poland, 1980a). The cells were derived
from tissues and/or species susceptible to TCDD toxicity in vivo.
Nanomolar concentrations of TCDD induced AHH activity in keratinocyte
cultures of human (Willey et al., 1984) and animal origin (Knutson &
Poland, 1980a).
Five human squamous carcinoma cell lines derived from tumours of
the epidermis and tongue responded to TCDD with increased
O-deethylase activity, the EC50 being 10-10 to 10-9 mol/litre
(Hudson et al., 1983a).
Steward & Byard (1981) treated primary hepatocytes isolated from
Sprague Dawley rats, for 48 h with various concentrations of TCDD.
They found a 2-fold induction of the AHH activity with 3 pg TCDD/106
cells. Maximal induction occurred with 2.4 ng TCDD/106 cells. Primary
hepatocytes, isolated from adult male Wistar rats, exhibited a linear
increase (from 2-to 4-fold) in AHH activity when exposed to TCDD in
the range 10-11 to 10-8 mol/litre for 72 h (Jansing & Shain,
1985). Primary hepatocytes from TCDD-treated (5, 10, or 25 µg TCDD/kg
body weight) rats, isolated 2 to 30 days post-treatment, showed
decreased ouabain and alpha-aminoisobutyric acid uptake as well as
tyrosine aminotransferase activity (Yang et al., 1983a). Treatment of
rats with 25 mg 1,3,6,8-tetraCDD/kg body weight did not affect these
parameters. Neither could these effects be demonstrated in primary
hepatocytes from control rats that were treated with TCDD (50, 100, or
200 nmol/litre medium) in vitro for 48 h.
The induction of AHH and EROD activities of a complex PCDD/PCDF
mixture from a fly ash extract has been reported (Safe et al., 1987).
7.4.9 Endocrine effects
Human exposure to TCDD has resulted in hirsutism and chloracne
(Table 64), symptoms that suggest an alteration in endocrine
regulation. Furthermore, chronic exposure to TCDD impaired
reproduction in experimental animals, possibly by interfering with the
estrous cycle (Kociba et al., 1976; Allen et al., 1977; Barsotti et
al., 1979; Murray et al., 1979). The ability of TCDD to mimic natural
steroids with steroid-like actions has prompted studies on the binding
of TCDD to steroid hormone receptors.
Over-production of glucocorticoids mimics some of the symptoms of
TCDD toxicity, e.g., involution of lymphoid tissues, oedema, and
mobilization of fatty acids from adipose tissues. Thus TCDD might
increase glucocorticoid activity by binding to glucocorticoid
receptors. However, TCDD was unable to displace 3H-dexamethasone, a
potent synthetic glucocorticoid, from the normal rat cytosol
glucocorticoid receptor even when present in 200-fold molar excess
(Neal et al., 1979). Poland et al. (1976) demonstrated that cortisol
and synthetic glucocorticoids did not bind to the TCDD receptor. An
increase in the plasma level of corticosterone was found in male
Sprague Dawley rats 7 and 14 days after a single oral dose of 50 µg
TCDD/kg body weight (Neal et al., 1979). With the method used, a
variety of fluorescent adrenocortical steroid hormone derivatives was
measured. In contrast, Balk & Piper (1984), using a competitive
binding radioassay for corticosterone, reported decreased blood levels
(29 and 26% of controls) of corticosterone in male Sprague Dawley rats
on days 14 and 21, respectively, after a single oral dose of 25 µg
TCDD/kg body weight. Accumulation of 11-ß-hydroxy-progesterone in the
blood of TCDD-treated rats was noticed on day 14 (Balk & Piper, 1984).
Neal et al. (1979) reported 100% mortality within 6 days in
adrenalectomized rats given 10, 20, 40, or 80 µg TCDD/kg body weight.
Adrenalectomy and hypophysectomy could not prevent liver lesions,
reduced growth rate, or thymic involution in female Fisher-344 rats
given a single oral dose of 10 or 20 µg TCDD/kg body weight (van
Logten et al., 1980). Thymic effects of TCDD became even more severe
after hypophysectomy. Daily sc injections of 0.25 mg growth hormone
had a positive influence on body weight gain but did not protect
against thymic involution in hypophysectomized rats.
Single oral or interperitoneal doses of TCDD between 7 and 100
µg/kg body weight decreased the serum thyroxine (T4) level in the rat
(Bastomsky, 1977; Potter et al., 1983, 1986b; McKinney et al., 1985;
Pazdernik & Rozman, 1985; Rozman et al., 1985b), but not in the
guinea-pig, after a single oral dose of 2 µg/kg body weight (McKinney
et al., 1985). The serum triiodothyronine (T3) level in TCDD-treated
rats was reported to be increased (Bastomsky, 1977; Potter et al.,
1986b), unaffected (Potter et al., 1983), or decreased (Pazdernik &
Rozman, 1985; Rozman et al., 1985b). Increased serum thyrotropin (TSH)
(Bastomsky, 1977; Potter et al., 1986b), increased thyroid
131I-uptake and increased biliary excretion of T4, but not of T3
(Bastomsky, 1977) have been reported in TCDD-treated rats.
Rats treated with TCDD at doses up to about 30 mg TCDD/kg body
weight and pair-fed controls had similar serum T4, T3, and TSH levels
when compared to ad libitum fed control rats (Potter et al., 1983,
1986b). Thus, it is unlikely that hypophagia is responsible for the
TCDD-induced changes in serum thyroid hormone levels. When mature
TCDD-treated rats were compared to pair-fed controls, there were no
functional alterations in thyroid status or thermogenesis, including
increased serum levels of T3, T4, and TSH after acute cold
challenge, increased total oxygen consumption after moderate cold
exposure or decreased basal metabolic rate as compared to ad
libitum fed control rats (Potter et al., 1986b). A significant
hypothermia was, however, observed in young TCDD-exposed rats
receiving 45 µg/kg body weight as a single ip dose (Potter et al.,
1983).
Available data on serum T4, T3, and TSH levels are not
sufficient to state whether TCDD-treated rats are functionally
hypothyroid, euthyroid, or hyperthyroid.
In ovo exposure of white Leghorn chicken embryos to TCDD in
the dose range 1 to 10 000 pmol/egg increased the cardiac release of
prostaglandins (Quilley & Rifkind, 1986). Potter et al. (1983)
reported decreased levels of insulin in serum and pancreas and of
somatostatin in the gastric antrum of Sprague Dawley rats 7 days after
a single ip dose of 45 µg TCDD/kg body weight, when compared to
pair-fed control rats. The somatostatin levels in serum, liver, and
pancreas were not affected, neither was the serum glucagon level.
The finding that steroids are an endogenous substrate for the
hepatic MFO system (Kuntzman et al., 1965) suggests that compounds,
such as TCDD, that influence the activity of this enzyme system may
alter steroid metabolism in vivo, and consequently also the
magnitude of steroid-mediated functions. As demonstrated by Gustafsson
& Ingelman-Sundberg (1979), the metabolic profiles of 4-androstene-3,
17-dione, 5 alpha-androstane-3 alpha, 17 ß-diol and 4-pregnene-3,
20-dione in hepatic microsomes from SD rats, treated with 20 µg
TCDD/kg body weight for 4 consecutive days, were changed when compared
to control rats 1 day post-treatment. The changes were most pronounced
in female rats. When five daily doses of 1 µg TCDD/kg body weight were
given to pregnant rats for 12 or 13 days during gestation, hepatic
microsomes showed decreased ability to form catechol estrogens and to
hydroxylate testosterone. However, this decrease did not relate to
altered circulating estradiol levels (Shiverick & Muther, 1983). TCDD
treatment did not affect the glucuronidation of testosterone and
estrogen (Lucier et al., 1975a) or prostaglandin synthesis (Kohli &
Goldstein, 1981). TCDD decreased the hepatic and uterine estrogen
receptor levels in 25-day-old Long-Evans rats 2 to 10 days after
treatment with single ip doses of 20 or 80 µg/kg body weight (Romkes
et al., 1987). Decreased estrogen receptor levels in the liver and
uterus also occurred 2 days after treatment with 1,2,7,8-tetraCDD,
1,2,3,7,8- pentaCDD, and 1,2,4,7,8-pentaCDD, but dose-response
relationships were present only for TCDD and 1,2,3,7,8-pentaCDD. The
estradiol-induced increases in hepatic and uterine estrogen receptor
levels were counteracted by simultaneous TCDD treatment, although the
effect of TCDD was not dose dependent. Hepatic hydroxylation of
testosterone in 2 ß-and 16 alpha- positions was not affected in male
Sprague Dawley rats (190-200 g) 3 days prior to a single oral dose of
15 µg TCDD/kg body weight (Hook et al., 1975b). Somewhat younger male
Wistar rats (100 g) treated with a single dose of 0.06 mmol TCDD/kg
body weight exhibited increased levels of 7 alpha-hydroxytestosterone
and decreased levels of 3alpha-, 16alpha- and 16 ß-hydroxylated
testosterone, as well as of androstenedione, in hepatic microsomes
when compared to control rats (Keys et al., 1985). Decreased
cytochrome P-450 content was found in guinea-pig (Tofilon, 1980) and
rat (Tofilon & Piper, 1982) testes for at least one week after oral
TCDD treatment of 1 µg/kg and 25 µg/kg body weight, respectively.
Testicular AHH activity was induced by TCDD in mice (Mattison &
Thorgeirsson, 1978), but not in rats after a single oral dose of 20 µg
TCDD/kg body weight (Aitio & Parkki, 1978). Mittler et al. (1984)
studied the effect of single ip doses of 0.2, 1, or 5 µg TCDD/kg body
weight on testicular 16-alpha-testosterone hydroxylase (16-TH),
6-ß-hydroxytestosterone (6-HT), and 7-alpha-hydroxy testosterone
(7-HT) activities in young Sprague Dawley rats 90 h after exposure.
Seminiferus 16-TH activity was increased from a non-detectable level
to 0.07-0.14 pg/mg protein and interstitial 6-HT activity was
increased 4-fold in TCDD-treated animals. The 7-HT activity was not
affected by TCDD, neither in the seminiferous tubules nor in the
interstitial fraction. Serum testosterone and dihydrotestosterone were
depressed dose-dependently by TCDD treatment, the ED50s being about
15 µg/kg body weight in male Sprague Dawley rats, when compared to
pair-fed and ad libitum fed controls (Moore et al., 1985). The
plasma clearance and biliary excretion of 3H-testosterone was not
affected in Sprague Dawley rats treated with 100 µg TCDD/kg body
weight before an i.v. injection of 3H-testosterone, neither did
castrated rats with implanted testosterone-leaking capsules respond to
a dose of 15 or 100 µg TCDD/kg body weight with decreased accessory
sex organ weights (Moore & Peterson, 1985). Increased AHH and
O-deethylase activities and cytochrome P-450 content have been
reported in rat prostate after single ip and oral doses, respectively,
of 10 µg/TCDD/kg body weight (Haaparanta et al., 1983; Lee & Suzuki,
1980).
7.4.10 Vitamin A storage
Decreased hepatic vitamin A storage has been reported in animals
exposed to various chlorinated aromatic compounds (Table 57). Compared
to other chlorinated hydrocarbons for which this effect has been
evaluated, TCDD is more potent in its ability to reduce the vitamin A
content of the liver.
A single oral dose of 10 µg TCDD/kg body weight decreased both
the total amount and the concentration of vitamin A in the liver of
adult male Sprague Dawley rats (Thunberg et al., 1979). The decrease
was evident 4 days after dosing and progressed with time. After 8
weeks the treated animals had a total liver vitamin A content
corresponding to 33% of that of controls. Decreased dietary intake of
vitamin A could not account for this difference. In a four-week study
TCDD was given as a single oral dose of 0, 0.1, 1.0, or 10 µg per kg
body weight to adult male Sprague Dawley rats fed ad libitum with
pelleted diets containing 1.2 (low), 3.0 (normal), or 6.0 (high) mg
vitamin A/kg diet (Thunberg et al, 1980). Both the concentration and
the total amount of vitamin A were decreased in a dose-dependent
manner in the animals receiving the high vitamin A diet. In the
animals on the normal and low vitamin A diets, significant differences
were seen only at doses of 1.0 and 10 µg TCDD/kg body weight. A
significant increase in the UDPGT activity was observed in all dietary
groups treated with 1.0 and 10 µg TCDD per kg body weight, suggestive
of an increased excretion of vitamin A conjugated with glucuronic acid
(Thunberg & Hakansson, 1983). However, no correlation between the
UDPGT activity and the reduction of hepatic vitamin A levels was seen
when homozygous Gunn rats lacking inducible UDPGT were treated with a
single oral dose of 20 µg/kg body weight (Aitio et al., 1979) nor in
heterozygous Gunn rats with inducible UDPGT after a single oral dose
of 10 µg/kg body weight (Thunberg & Hakansson, 1983).
Table 57. The potency of various chlorinated cyclic hydrocarbons to reduce hepatic vitamin A content in the rat
Compound Strain/sex/age Dose and route of Duration of % Reduction
(Reference) administration the study of hepatic
vitamin A
Arochlor 1242 Rattus norvegicus/M,Ff/21 days 100 mg/kg in dieta 2 months 49
(Cecil et al., 1973)
p,p-DDT Rattus norvegicus/M,F/21 days 100 mg/kg in dieta 2 months 38
(Cecil et al., 1973)
Methoxychlor Sprague Dawley/NRf/23 days 10 mg/kg in dietb 16 weeks 7
(Davison & Cox, 1976) 100 mg/kg in diet 12
1000 mg/kg in diet 37
10 000 mg/kg in diet 68
PCB Sprague Dawley/M/21 days 100 mg/kg in dietc 8 weeks 82
(Innami et al., 1976)
TCDD Sprague Dawley/M/NR 10 µg/kg bwf 7 days 29
(Thunberg et al., 1979) (single oral dose) 14 days 39
28 days 59
56 days 67
Table 57 (contd).
Compound Strain/sex/age Dose and route of Duration of % Reduction
(Reference) administration the study of hepatic
vitamin A
TCDD Sprague Dawley/M/NR 0.1 µg/kg bwg 28 days 2
(Thunberg et al., 1.0 µg/kg bwg 27
1980) 10.0 µg/kg bwg 65
TCDD Sprague Dawley/M/2 months 15 µg/kg bwg 44 days 59d 88e
(Hakansson, 1988) 30 µg/kg bwg 78d 98e
60 µg/kg bwg 81d 97e
120 µg/kg bwg 90d 99e
Toxaphene Sprague Dawley/M/NR 20 mg/kg bwh 4 weeks 0
(Thunberg et al.,
1984)
a 9-12 mg vitamin A/kg diet ad libitum.
b 33 000 IU vitamin A/kg diet ad libitum.
c 3000 IU vitamin A/kg diet ad libitum.
d 21 000 IU vitamin A/kg diet ad libitum.
e 8000 IU vitamin A/kg diet ad libitum.
f M = male; F = female; NR = not reported; bw = body weight.
g Single oral doses.
h Orally twice weekly.
Male Sprague Dawley rats received a single oral dose of 10 µg
TCDD/kg body weight 4 days prior to the oral administration of a
single physiological dose of labelled vitamin A,
(11,12-3H)retinylacetate (RA) (Hakansson & Ahlborg, 1985a). The
distribution and elimination of the radiolabel, and the vitamin A
content in various tissues were determined 1, 6, 12, 24, 72, and 192
h after the administration of 3H-vitamin A. The body burden of
radioactivity remained around 40% of the administered dose in control
rats throughout the study, whereas in TCDD-pretreated rats the body
burden decreased continuously from 21% at 12 h after administration to
11% at the end of the study. More of the radiolabel was recovered in
the kidney, testes, epididymis, and serum of TCDD-treated animals than
in controls, when calculated as percentage of body burden, whereas
less was recovered in the liver. Forty percent of the dose was
eliminated via faeces and urine in TCDD-treated rats, compared to 23%
in controls. More radioactivity was eliminated in faeces than in urine
both in control and TCDD-pretreated animals, although urinary
elimination was more pronounced in TCDD-pretreated than in control
rats. It was concluded that TCDD-treated rats handled the newly
administered dose of vitamin A in a similar way to rats deficient in
Vitamin A (Huque, 1981; Blomhoff et al., 1982). This finding is
remarkable since the TCDD-treated animals in this study still had
considerable stores of hepatic vitamin A, and did not show decreased
levels of serum vitamin A, i.e., they were not deficient in vitamin A.
In a similarly designed study the effect of a single oral dose of 10
µg TCDD/kg body weight on the endogenous pool of vitamin A
(radiolabelled 15-3H-retinol given 5 to 7 days prior to TCDD
treatment) was studied in Sprague Dawley rats with stores of low liver
vitamin A (Hakansson et al., 1986). It was demonstrated that
endogenously stored vitamin A was rapidly depleted from the liver of
TCDD-treated rats and was eliminated both in faeces and in urine. An
increased distribution of the vitamin A stored in the liver to
extrahepatic tissues was also seen in the treated rats.
To elucidate whether dietary vitamin A would reduce TCDD
toxicity, Hakansson (1988) fed male Sprague Dawley rats ad libitum
from weaning throughout the experiment with diets containing 2000 (I),
5000 (II), 8000 (III), or 21 000 (IV) IU of vitamin A/kg. A single
oral dose of TCDD (15, 30, 60, or 120 µg TCDD/kg body weight) was
given when the rats were 8 weeks old and the animals were killed 44
days post-treatment. With diet IV, TCDD reduced in a dose-dependent
manner hepatic vitamin A by 59 to 90%. With diets II and III,
reduction of hepatic vitamin A was more than 95% after dosing with 15
and 30 µg TCDD/kg, respectively. In control animals fed diet I, total
hepatic vitamin A was less than 1 µg and TCDD had no further effect at
any dose. Serum vitamin A was dose-dependently decreased by TCDD
treatment in dietary groups I, II, and III, whereas in dietary group
IV TCDD increased serum vitamin A. Only with the highest TCDD dose was
there a counteraction by dietary vitamin A on all of the above
parameters.
A single dose of 10 µg TCDD/kg body weight to female Sprague
Dawley rats on the day of delivery affected the vitamin A content in
the liver and kidney of the offspring (Hakansson et al., 1987). The
effect became clearly visible after weaning, i.e., when the dietary
intake of vitamin A was high enough to allow for storage. At the end
of the study (postnatal day 32), the vitamin A content in the liver
was 225 µg in control pups and 102 µg in TCDD-exposed pups. The
corresponding values for the kidney vitamin A content were 1.4 and 8.4
µg, respectively. The TCDD-induced effects on vitamin A levels in the
liver and kidneys followed a similar time-course as the growth
reduction, i.e., a minor effect throughout the lactation period, which
became more pronounced post-weaning. This was in contrast to the liver
enlargement and thymus involution in TCDD-exposed pups, which were
most pronounced throughout lactation and tended to diminish post-
weaning.
Single oral doses of 0, 1, 5 and 10 µg TCDD/kg body weight or a
mixture of PCDDs and PCDFs, reconstituting the levels found in human
milk, were given to male Sprague Dawley rats (80 g) in corn oil
(Ahlborg et al., 1987). The mixture was given at three dose levels
resulting in 1, 5, or 10 µg TCDD/kg body weight. Four weeks after
dosing there was a dose-dependent decrease in hepatic vitamin A in
TCDD-treated rats; no further effect was seen in the animals treated
with PCDD/PCDF mixtures. In contrast, mixture treatment had an
additive effect, as compared to TCDD alone, on the increases in renal
vitamin A and hepatic cytochrome P-450 contents, AHH activity, and
UDPGT activity.
Taken together these data indicate that TCDD interferes with the
storage mechanism for vitamin A. In the liver this mechanism has been
thoroughly investigated (Hirosawa & Yamada, 1973; Blomhoff et al.,
1982; Olson & Gunning, 1983). As dietary vitamin A seems unable to
counteract all toxic effects, this would imply either that the effect
on vitamin A storage is secondary to TCDD toxicity or that the
cellular utilization of vitamin A is affected by TCDD.
7.5 Embryotoxicity and Reproductive Effects
The teratogenic potential of TCDD was first demonstrated in rats
and mice by Courtney & Moore (1971). This followed the finding that
2,4,5-trichlorophenoxyacetic acid contaminated with 30 mg TCDD/kg was
teratogenic in two strains of mice and one strain of rat (Courtney et
al., 1970), leading to an increased incidence of cleft palate and
cystic kidney in both strains of mice and cystic kidney in rats.
Further studies have revealed that TCDD is fetotoxic rather than
teratogenic in the rat, producing subcutaneous oedema, haemorrhages,
and slight kidney anomalies (see section 7.5.1). In contrast, TCDD
produces a specific teratogenic response, consisting of cleft palate
and kidney malformations, in several strains of mice (see section
7.5.2 and Table 58). Extra ribs, minor abnormalities in the palate,
and cardiovascular malformations have been demonstrated in rabbits,
monkeys, and chickens, respectively, after exposure to TCDD in utero
(see sections 8.5.3-8.5.6). Impaired reproductive performance due to
TCDD exposure has been demonstrated in rats and monkeys (see sections
8.5.1 and 8.5.4).
7.5.1 Studies on rats
The initial studies on the embryotoxic effects of TCDD in the rat
were performed with Charles River CD (Courtney & Moore, 1971), Sprague
Dawley (Sparschu et al., 1971), and Wistar rats (Khera & Ruddick,
1973). In these studies maternal toxicity was noted at doses > 0.5
(Sparschu et al., 1971) or 1 µg/kg body weight per day (Khera &
Ruddick, 1973). Decreases in gestational survival, fetal weight, and
postnatal survival were reported at dose levels in the range 0.125-0.5
µg TCDD/kg body weight per day on gestation days 6-15 or 5-14.
Haemorrhages, mainly intestinal, and subcutaneous oedema were common
findings at similar doses. The teratogenic findings were limited to
kidney anomalies, described as unilocular cystinephrotic kidney or as
hydronephrosis, in the CD rat at or above 0.5 µg TCDD/kg per day
(Courtney & Moore, 1971). Slightly dilated renal pelvis was reported
in the F1 generation at the 0.01 µg/kg per day dose level in a
three-generation reproductive study of TCDD in Sprague Dawley rats
(Murray et al., 1979). Giavini et al. (1983) found an increased
incidence of renal anomalies in Charles River CD offspring exposed to
2 µg TCDD/kg body weight on gestation days 0 to 2, but not in Sprague
Dawley offspring when the dam was given daily oral doses of 0, 0.125,
0.5, and 2 µg TCDD/kg body weight for 2 weeks before mating (Giavini
et al., 1982a).
Sparschu et al. (1971) found an increased number of resorption
sites in Sprague Dawley rats at doses > 0.5 g TCDD/kg per day, but
no effect on ovulation rate and preimplantation loss was reported even
at 2 µg/kg per day. TCDD exposure (0.1, 0.5, or 2.0 µg/kg per day) on
gestation days 0 to 2 had no effect on the reproductive performance of
Sprague Dawley rats (Giavini et al., 1983). In contrast, impaired
reproductive performance was found in Charles River CD rats after
receiving TCDD for two weeks before mating (Giavini et al., 1982a).
The number of resorption sites was elevated at doses of 0.5 µg TCDD/kg
per day or more, whereas the ovulation rate and preimplantation loss
was affected only at 2 µg/kg per day.
The male reproductive ability was affected by TCDD treatment only
at toxic doses, as judged by studies of Khera & Ruddick (1973) and
Murray et al. (1979). Nevertheless, decreased mating frequency was
noted in the groups, of the F0 generation that received 0.1 µg
TCDD/kg per day in the diet (Murray et al., 1979). No effect on the
mating frequency was found by Giavini et al. (1982a, 1983).
In a three-generation reproduction study, Murray et al. (1979)
maintained Sprague Dawley rats on diets providing doses of 0, 0.001,
0.01, or 0.1 µg TCDD/kg body weight per day. The F0 generation
received the diet for 90 days before mating. No toxic effects were
observed in the F0 generation but decreased body weight and reduced
food consumption were noted in the F1 and F2 generations at 0.01
µg/kg per day. Fertility was greatly reduced at 0.1 µg/kg per day in
the F0 generation. This group was discontinued because of the low
number of offspring. In the F1 and F2 generations fertility was
significantly reduced at 0.001 and 0.01 µg/kg per day, respectively.
At 0.01 µg/kg per day, litter sizes were reduced, and fetal and
neonatal survival were decreased as well as postnatal growth. Murray
et al. (1979) concluded that doses of 0.1 and 0.01 µg/kg per day
impaired reproduction in rats. A dose of 0.001 µg/kg per day had no
effect on fertility, litter size, postnatal body weight, or neonatal
survival and was therefore suggested to be a no-effect dose for
reproductive lesions.
Reevaluation of these data by Murray et al. (1979) using another
statistical model, including pooling of the data from the four
generations, led to the conclusion that the 0.001 µg/kg per day dose
level did affect reproduction and thus was not a no-effect level, but
a low-effect level (Nisbet & Paxton, 1982). Kimbrough et al. (1984)
considered that the data by Murray et al. (1979) could not be used for
risk assessment calculations due to the great variation in fertility
index both in controls and exposed rats.
No embryotoxic and/or reproductive effects were found when female
Wistar rats were exposed to 1,2,3,4-tetraCDD (50, 100, 200, 400, or
800 µg/kg body weight per day), 2,7-diCDD (250, 500, 1000 or 2000
µg/kg body weight per day); 2,3-diCDD (1000 or 2000 µg/kg body weight
per day), or 2-monoCDD (1000 or 2000 µg/kg body weight per day) on
gestation days 6-15 (Khera & Ruddick, 1973). The maturation process of
the lung was not affected in Sherman rats exposed to 2,7-diCDD (40
µg/kg per day on gestation days 7-15) (Kimbrough et al., 1974).
7.5.2 Studies on mice
TCDD-induced embryo mortality in NMRI mice was significantly
increased at doses of 4.5 and 9 µg TCDD/kg body weight per day when
given on gestation days 6-15, but no embryotoxic effect was observed
when daily doses of 9 µg/kg body weight were given on gestation days
9-13 (Neubert & Dillman, 1982). The number of resorptions on day 13 of
gestation in NMRI mice was increased, as compared to controls, when 25
µg TCDD/kg body weight was given, divided into five daily doses on
days 7-11 of gestation, but no effect was observed when the same dose
was given as single ip injections on days 7 or 10 of gestation (Nau &
Bass, 1981). A single ip dose of 30 µg TCDD/kg body weight on
gestation day 11 had no effect on the fetal mortality in C57Bl/6 mice
on days 12, 13, or 14 of gestation (Weber & Birnbaum, 1985). Decreased
pre- and postnatal survival rates and retarded postnatal development
were observed in NMRI mice given four oral doses of 12.5 µg TCDD/kg
body weight on gestation days 14-17 (Nau et al., 1986). The cumulative
mortality was 45%, 68.5%, and 75% in exposed offspring on postnatal
days 1, 14, and 22, respectively, as compared to 6% in controls on day
22.
Table 58 summarizes the early studies on the teratogenic effects
of TCDD in various strains of mice. These studies (Courtney & Moore,
1971; Neubert & Dillman, 1972; Courtney, 1976; Smith et al., 1976),
revealed that TCDD is a specific teratogen in mice causing increased
frequencies of kidney anomalies and cleft palate at doses well below
those which result in fetal mortality and maternal toxicity.
The TCDD-induced kidney anomaly is morphologically described as
a progressive hydronephrosis, preferentially occurring in the right
kidney, and never accompanied by hydroureter or abnormal nephron
development (Courtney & Moore, 1971; Moore et al., 1973; Birnbaum et
al., 1985; Weber et al., 1985).
The fetal kidney seems to be more susceptible to TCDD exposure
than the developing palate (Courtney & Moore, 1971; Moore et al.,
1973; Birnbaum et al., 1986; Weber et al.,1984). The incidence of
kidney anomalies after a single dose of 1 µg TCDD/kg body weight on
gestation day 10 in C57Bl/6 mice was 34.3%. If the same dose was
divided and given on gestation days 10-13, the incidence of kidney
anomalies was 58.9%. The incidences of cleft palate in the same
studies were 0 and 1.9%, respectively (Moore et al., 1973). In
contrast, in the CF-1 strain of mice, cleft palate was a more
sensitive parameter than hydronephrosis, occurring at 1 and 3 µg
TCDD/kg body weight per day on gestation days 6 to 15, respectively
(Smith et al., 1976). It would appear that 3 µg TCDD/kg per day on
gestation days 10-13 is close to a threshold dose for cleft palate
induction in C57Bl/6 mice (Moore et al., 1973; Birnbaum et al., 1986).
The maximum increase in cleft palate incidence is produced if TCDD is
administered on any individual day from gestation day 8-10 in C57Bl/6
mice (Pratt et al., 1984) or on gestation day 10-11 in NMRI mice (Nau
& Bass, 1981; Neubert & Dillman, 1982; Krowke, 1986). The cleft palate
incidence in C57Bl/6 mice was more pronounced (36%) when TCDD was
given as a single oral dose of 12 µg/kg body weight on gestation day
11 than if 3 µg/kg body weight per day was given on gestation days
10-13 (Birnbaum et al., 1985). No difference was seen in the incidence
of kidney anomalies with the same treatment. TCDD is not a potent
inducer of cleft palate when given on day 13 of gestation or later
(Neubert & Dillman, 1982; Pratt et al., 1984). Examination of cryostat
sections taken from C57Bl/6 embryos during the time of palatal
elevation and fusion demonstrated that TCDD does not interfere with
growth, elevation, or initial contact of the palatal shelves but does
interfere with the firm adhesion and/or degeneration of the medial
epithelial cells, i.e., programmed epithelial cell death does not
appear to occur in the medial epithelium in embryos exposed to TCDD
(Pratt et al., 1984). The cleft palates that were observed were
complete clefts of the entire hard and soft palate and no clefts of
the primary palate were observed (Pratt et al., 1984).
Table 58. Early studies on embryotoxic effects of TCDD in mice
Strain/sexb Route/vehicle/dose Treatment/ Parental toxicity Embryotoxic effects
(reference) (µg/kg body weight observation
day) (days)a
CD-1/F subcutaneous/ 6-15 / 18 No effect Increased incidence of kidney
(Courtney & DMSO/1.0, 3.0 anomalies at doses > 1 µg/kg
Moore, 1971) per day.
BDA/2J subcutaneous/ 6-15 / 17 Increased relative Increased incidence of cleft
(Courtney & DMSO/3.0 liver weight palate and kidney anomalies.
Moore, 1971)
C56BL/6J subcutaneous/ 6-15 / 17 Increased relative Increased incidence of cleft
(Courtney & DMSO/3.0 liver weight palate and kidney anomalies.
Moore, 1971)
NMRI/F oral/rape seed 6-15 / 18 None reported Increased number of resorptions
(Neubert & oil/0.3, 3.0, 4.5, at 9 µg/kg per day. Increased
Dillman, 1972) 9.0 incidence of cleft palate at
doses > 3 µg/kg per day.
C56BL/6J oral/corn oil:acetone 10-13 / 18 None reported Increased incidence of cleft
(Neubert & (9:1)/1.0, 3.0 palate at doses > 3 µg/kg
Dillman, 1972) per day increased incidence
of kidney anomalies at doses
> 1 µg/kg per day.
Table 58 (contd).
Strain/sexb Route/vehicle/dose Treatment/ Parental toxicity Embryotoxic effects
(reference) (µg/kg body weight observation
day) (days)a
CD-1/F oral/corn oil:anisole 7-16 / 18 Increased maternal Increased fetal mortality at all
(Courtney, 1976) (95:5)/25, 50, 100, relative liver weight doses. Increased incidence of
200, 400 at 25 and 50 µg/kg per kidney anomalies and cleft palate
day. Marked oedema and at doses > 25 µg/kg per day
vaginal bleeding at and 50 µg/kg per day,
doses > 200 µg/kg per respectively. Increased incidence
day. of club foot was found in the
high-dose group. Hydrocephalus and
open eyes were seen occasionally.
CF-1 oral/corn oil:acetone 6-15 / 18 Increased maternal Increased number of resorptions
(Smith et al., (98:2)/0.001, 0.01, relative liver weight at 1 µg/kg per day. Increased
1976) 0.1, 1.0, 3.0 at 3 µg/kg per day. incidence of cleft palate at
doses > 1 µg/kg per day.
Dilated renal pelvis occurred at
3 µg/kg per day.
a First day of gestation designated day zero.
b F = female.
Poland & Glover (1980) reported that in nine out of ten inbred
strains of mice, the susceptibility to cleft palate formation produced
by TCDD (30 µg TCDD/kg body weight sc on day 10 of pregnancy) followed
the distribution of the Ah locus within that strain. The five strains
with a low affinity TCDD receptor in the liver (DBA/2, RI, AKR, SWR,
and 129) developed cleft palate at an incidence of 0 to 3% while four
of the five strains with a high affinity TCDD receptor in the liver
(C57Bl/6, A, BALB/cBy, and SEC) developed cleft palate at an incidence
of 54 to 95%. The only strain with a high affinity hepatic TCDD
receptor that did not develop cleft palate was CBA. Mid-gestational
mice embryos from C57Bl/6 exhibit high levels of the TCDD receptor in
the maxillary processes and secondary palatal shelves whereas no
specific binding of TCDD could be demonstrated in the AKR strain
(Dencker & Pratt, 1981). Evidence that TCDD interferes in embryonic
development by directly interacting with embryonic cells rather than
being secondary to maternal effects was presented by D'Argy et al.,
(1984) in a study where mouse blastocysts were transplanted between
NMRI (sensitive) and DBA (non-sensitive) dams on gestation day 3. TCDD
treatment (30 µg/kg body weight) on gestation day 10 resulted in 75 to
100% incidence of cleft palate among NMRI fetuses whether they
remained in their own dams or as aliens in DBA dams. Also, none of the
DBA fetuses developed cleft palate whether or not they remained in
their own dams or as aliens in NMRI dams.
Attempts to modify and further characterize the TCDD-induced
cleft palate formation have been performed with several interacting
substances. The non-teratogenic ß-naphthoflavone (ßN) enhanced the
TCDD-induced fetal mortality and the increase in the incidence of
cleft palate in C57Bl/6 and NMRI mice when ßN was administered
simultaneously or 8 h before TCDD, but not 24 h before or after
(Hassoun & Dencker, 1982). TCDD was given as a single ip dose of 25 or
16 mg/kg body weight to C57Bl/6 and NMRI mice, respectively, on
gestation days 10, 11, 12, or 13. TCDD-induced cleft palate incidence
was enhanced by 2,3,4,5,3',4'-hexachlorobiphenyl, but not
2,4,5,2',4',5'-hexachlorobiphenyl, in C57Bl/6 mice, although none of
the isomers alone were teratogenic at the doses used (Birnbaum et al.,
1985). Neither of the hexachlorobiphenyls used affected the incidence
of renal anomalies, as compared to TCDD alone.
A dose-related enhancement of the TCDD-induced incidence of cleft
palate was found in C57Bl/6 mice exposed to either triiodothyronine or
thyroxine, as compared to TCDD alone (Lamb et al., 1986). The
hydrocortisone-induced cleft palate response was enhanced by
simultaneous administration of TCDD in C57Bl/6 mice (Birnbaum et al.,
1986). Co-administration of TCDD and TCDF to C57Bl/6 mice on gestation
day 10 resulted in a cleft palate incidence compatible with an
additive toxicity model in which TCDF contributes to the toxicity of
TCDD in a weight ratio of 1:30 (Weber et al., 1985). Also 1,2,3,7,8-
pentaCDD and 1,2,3,4,7,8-hexaCDD had additive effects on the
TCDD-induced cleft palate incidence in NMRI mice (Krowke, 1986). No
clearly dose-related teratogenic effects were observed in C57Bl/6 mice
exposed to oral doses of 1,2,3,4- tetraCDD (100, 250, 500, or 1000
mg/kg per day), octaCDD (5 or 20 mg/kg per day) or to a mixture of 40%
2,7-diCDD and 60% 2,3,7-triCDD (100 or 200 mg/kg per day) on gestation
days 7-16 (Courtney, 1976).
7.5.3 Studies on rabbits
New Zealand rabbits were administered TCDD by gavage in doses of
0, 0.1, 0.25, 0.5, and 1 µg/kg per day on days 6-15 of gestation and
the fetuses were examined on day 28 of gestation (Giavani et al.,
1982b). Above 0.25 µg/kg per day, decreased maternal weight gain and
unspecified signs of maternal toxicity were reported. At doses of 0.5
and 1 µg/kg per day, there were 2/15 and 4/10 maternal deaths,
respectively. An increase in abortion and resorption rates occurred at
doses above 0.25 µg/kg per day, with no live fetuses detected in the
1 µg per kg per day dose group. There was a significant increase in
extra ribs, compared to a level of 33.3% in the controls to 82, 66.6,
and 82%. In the 0.1, 0.25, and 0.5 µg/kg per day dose groups, 82,
66.6, and 82% extra ribs were noted. There were no increases in
specific soft tissue anomalies.
7.5.4 Studies on monkeys
The effect of exposure to TCDD in the diet, before mating and
throughout gestation, on the reproductive performance and production
of progeny of healthy, fertile rhesus monkeys (Macaca mulatta) was
studied by Allen and co-workers (Allen et al., 1979a,b; Barsotti et
al., 1979; Schantz et al., 1979). At a level of 500 ng TCDD/kg diet
(11 ng/kg body weight per day), there were no effects on the length,
intensity, or duration of the menstrual cycle, but decreases in serum
estradiol and progesterone levels were observed (Barsotti et al.,
1979). Mating with control males at the end of the sixth month
resulted in three pregnancies, out of which two resulted in abortion,
and three animals failed to conceive. The remaining TCDD-treated
female was not bred due to toxic symptoms. The two females that
survived the study were returned to a control diet and later gave
birth to well developed infants. After 7 months on the 50 ng TCDD/kg
diet (1.5 ng/kg body weight per day), the reproductive outcome was:
four abortions, one stillbirth, two failures to conceive and two
normal births (Schantz et al., 1979).
All controls conceived and gave birth to normal infants (Barsotti
et al., 1979; Schantz et al., 1979).
McNulty (1984) demonstrated that rhesus monkeys (Macaca mulatta)
receiving 1 µg TCDD/kg body weight either as a single dose on
gestation days 25, 30, 35, or 40, or divided into nine doses between
gestation days 20-40, failed to give birth normally. Of 16 pregnancies
13 resulted in abortions. Two of the three live fetuses, obtained by
Caesarean section on day 145 of gestation, showed minor abnormalities
in the palate. Maternal toxicity, manifest in 8 out of 16 females,
appeared only after a period of 44-111 days after abortion. From the
results obtained it was not possible to conclude whether fetal death
was a direct effect of TCDD on the fetus or placenta or an indirect
effect through maternal toxicity.
7.5.5 Studies on chickens
Treatment of fertile White Leghorn chicken eggs, on day 0 of
development, with single doses of TCDD, ranging from 0.009 to 77.5
pmol/egg, resulted in a dose-related increase in the following types
of cardiovascular malformations: ventricular septal defects, aortic
arch anomalies, aortic arch anomaly plus ventricular septal defect,
and conotruncal malformation (Cheung et al., 1981). The dose producing
cardiovascular malformations in 50% of the embryos was about 1 pmole
TCDD/egg.
A dose-dependent decrease in hatchability and increased
incidences of beak, brain, and leg malformations were found in the
embryos when fertile White Leghorn eggs were injected with toxic fat
material containing 0.9, 1.8, or 4.5 ng of a mixture of dioxins (14%
diCDDs, 1% triCDDs, 38 to 45% tetraCDDs, 13% pentaCDDs, 14% hexaCDDs,
12% heptaCDDs, 8% octaCDD) (Flick et al., 1973).
7.6 Mutagenicity and Related End-Points
7.6.1 Mutagenicity
7.6.1.1 Studies on bacteria
Results from bacterial mutagenicity tests with TCDD are
conflicting.
In studies by Hussain et al. (1972) and Seiler (1973), a positive
response was reported in the Salmonella typhimurium strain TA 1532
without metabolic activation in a plate test after preincubation of
bacteria in medium containing TCDD and also in a spot test. A
mutagenic response was also obtained with Escherichia coli Sd-4,
measuring reversion to streptomycin independence (Hussain et al.,
1972). OctaCDD was not mutagenic to various strains of Salmonella
typhimurium (Seiler, 1973) without metabolic activation. More recent
publications, however, do not report any mutagenic effect of TCDD in
the Ames' Salmonella plate incorporation assay using strains TA
1530, TA 1532, TA 1535, TA 1537, TA 1538, TA 98, or TA 100 in the
presence or absence of metabolic activation systems from rat and
Syrian hamster liver (Gilbert et al., 1980; Geiger & Neal, 1981;
Mortelmans, 1984). In these studies the earlier reported positive
tester strain TA 1532 was either replaced by strain TA 1537 (Geiger &
Neal, 1981; Mortelmans, 1984) or was tested in addition to TA 1537
(Gilbert et al., 1980). Strain TA 1537 has been derived from TA 1532
and is more sensitive, due to its improved uptake of large molecules.
TCDD was tested in the dose range 0.2-2000 µg/plate. Due to the
limited solubility of TCDD, a maximal dose in the Salmonella system
was reported to be 20 µg TCDD/plate (Geiger & Neal, 1981).
7.6.1.2 Studies on eukaryotic cells
Bronzetti et al. (1983) reported a mutagenic response of TCDD in
yeast in an in vitro suspension test and a host mediated assay. In
both assays Saccharomyces cerevisiae strain D7 was used. Positive
responses were obtained in vitro in the presence of metabolic
activation at doses of TCDD up to 10 µg TCDD/ml, and in the
host-mediated assay after treatment of mice with a single dose of TCDD
(25 µg/kg body weight).
In L5178Y mouse lymphoma cells, TCDD induced mutations in a
dose-dependent manner at doses of 0.05-0.5 µg TCDD/ml; survival of
cells at the highest concentration was at least 75% (Rogers et al.,
1982).
7.6.1.3 In vivo studies
A dominant lethal test on male Wistar rats has been reported by
Khera & Ruddick (1973). The rats were given 4, 8, or 12 µg TCDD/kg
body weight per day orally for seven consecutive days, after which
seven sequential mating trials, 5 days at a time, were conducted in
the surviving males. Nine days after separation of females from the
males, the females were killed. The highest dose was lethal for all
males exposed (20/20); 8 µg/kg per day killed 11/20 males, and 4 µg/kg
per day was lethal for 2/20. All animals in the control group
survived.
The results did not indicate a dominant lethal effect during the
35 days post-treatment period, corresponding to the postmeiotic stages
of spermatogenesis.
7.6.2 Interaction with nucleic acids
Poland & Glover (1979) found that very little TCDD bound to rat
liver nucleic acids after treatment with 3H-TCDD in vivo; the
maximum covalently bound TCDD was calculated to be 6 and 12 pmol per
mol of nucleotide residues from DNA and RNA, respectively. After iv
injection of 3H-TCDD in rats, the radioactivity taken up by the
liver cytosol decreased at the same rate as the radioactivity in the
nuclear fraction increased. The radioactivity in the nuclei was at a
maximum 2 h after injection (Carlstedt-Duke et al., 1982). Guenthner
et al. (1979) demonstrated in vitro metabolism of TCDD to reactive
intermediates that bound covalently to cellular macromolecules,
principally to proteins. However, no isomer-specific methods were used
for analysis of metabolites in this study.
Liver slices from Sprague Dawley rats treated with 5 µg TCDD
incorporated twice the level of thymidine into nuclear DNA 10 days
post-treatment than did controls (Conaway & Matsumura, 1975).
Christian & Peterson (1983a) found no effect on the in vivo
incorporation of thymidine into liver DNA 35 to 36 h after the
administration of 10 µg TCDD/kg body weight to Sprague Dawley rats.
DNA synthesis in Porton rats, stimulated by 70% hepatectomy and
measured as the 1 h in vivo incorporation of thymidine, was not
affected by treatment with 10 or 200 µg TCDD/kg body weight 0, 24, or
72 h before the hepatectomy was performed (Greig et al., 1974). In
contrast, Dickins et al. (1981) found an 8-to 10-fold increase in DNA
synthesis in response to the proliferation caused by a 1/3 hepatectomy
in Sprague Dawley rats given 5 µg TCDD/kg body weight 5 days prior to
the hepatectomy. In this study thymidine incorporation into DNA was
measured in vitro at various times after hepatectomy. The
TCDD-induced increase was most pronounced 24 to 32 h after the
hepatectomy. The somewhat conflicting results may be due to
differences in the in vivo and in vitro incorporation of
thymidine, as well as to differences in the time points studied.
According to Dickins et al. (1981), the discrepancy in proliferative
DNA synthesis could be due to the degree of hepatectomy. Thus 70%
hepatectomy would by itself enhance DNA synthesis to near maximum
level, making it difficult to measure any effect of TCDD under the
experimental conditions. This suggestion was confirmed by Christian &
Peterson (1983a), who compared the effect of TCDD on proliferative DNA
synthesis after 1/3 and 2/3 hepatectomy. This study also revealed that
the effect could be seen only when a certain amount of time, namely 5
to 10 days, elapsed between TCDD administration and hepatectomy.
The transfectivity of bacteriophage Qß/RNA was evaluated after
treatment in vitro with TCDD. No effect was noticed in the tested
dose range (0.2-4 µg TCDD/ml) (Kondorosi et al., 1973).
The effect of TCDD on the repair of DNA damage induced by
2-aminofluorene (AF) and 2-acetylaminofluorene (AAF) in primary
hepatocytes from B6 and D2 mice has been investigated (Moller et al.,
1984). Pretreatment in vivo with TCDD (50 mmol/litre) resulted in
a slight increase in DNA damage (measured by the alkaline elution
technique) following incubation with either AF or AAF for 60 min,
suggesting induction of aromatic amine activating enzymes.
7.6.3 Cytogenetic effects
Green & Mooreland (1975) and Loprieno et al. (1982a) did not
observe any induction of chromosomal aberrations in rats administered
TCDD intraperitoneally or by gavage (5 to 20 µg/kg body weight). Later
Green et al. (1977) showed a significant increase in the induction of
chromosomal abnormalities in bone marrow cells of male rats at doses
of 2 and 4 µg TCDD/kg body weight and in females at 4 µg/kg body
weight. In a study by Meyne et al. (1985) C57Bl/6 or DBA/2 mice (with
high- and low-affinity TCDD receptors, respectively) were ip injected
at doses 0, 50, 100, and 150 µg/kg body weight. There was no increase
in the frequency of chromosomal aberrations in bone marrow cells of
the TCDD-treated mice of either strain 8, 16, or 24 h after treatment.
All doses were high enough to induce hepatotoxic damage in C57Bl/6
mice. In male and female CD-1 mice, however, a weak but significant
increase in chromosomal aberrations was obtained 96 h post-treatment
(ip 10 µg TCDD/kg body weight) (Loprieno et al., 1982b).
Lamb et al. (1981) evaluated the frequency of sister chromatid
exchange (SCE) in bone marrow cells of C57Bl/6 mice given single ip
injections of mixtures of chlorinated phenoxy acids that contained
0.16, 1.2, or 2.4 µg TCDD/kg body weight. The mean SCE frequency in
treated and in control mice did not differ significantly. Mice fed
diets containing the same daily doses as above for 4 to 8 weeks did
not show any increase in SCE frequencies.
At doses (50, 100, and 150 µg per kg body weight) hepatotoxic to
C57Bl/6 mice, there was no significant increase of SCEs in C57Bl/6 or
DBA/2 mice 18 h after ip injection of TCDD (Meyne et al., 1985). Meyne
et al. (1985) also performed a micronucleus test under the same
conditions as above. Mice, killed 24 or 48 h after treatment, did not
show any increase of micronuclei in polychromatic erythrocytes of bone
marrow in either strain.
7.6.4 Cell transformation
Single treatments (11 concentrations in the range 0 to 5
µmol/litre) of mouse embryo fibroblast (C3H/10T1/2) tissue cultures
with TCDD did not transform or initiate the process of transformation
in cultures subsequently exposed to
12-O-tetradecanoylphorbol-13-acetate (Abernethy et al., 1985).
Continued treatment of these cells with low concentrations (> 4
pmol/litre) of TCDD enhanced the production of foci in cultures
pretreated with N-methyl-N'-nitro-N-nitrosoguanidine. Maximal
enhancement occurred at 40 pmol/litre. Higher doses, 120 to 4000
pmol/litre, did not further increase the incidence of foci production.
Promotion of transformation is thus the predominant effect of TCDD in
the C3H/10T1/2 cell-transformation system.
7.7 Carcinogenicity
7.7.1 Long-term animal studies on single compounds
Several studies on the carcinogenicity of TCDD and related
compounds have been performed.
The data from studies using oral exposure are summarized in Table
59. Van Miller et al. (1977) exposed male Sprague Dawley rats to
various dietary levels of TCDD ranging from 0.001 µg/kg and 1 µg/kg to
1 µg/kg for 78 weeks. Pronounced mortality was observed at higher
doses. Neoplastic changes in different organs were noted in a number
of rats that died. At 95 weeks, the small number of surviving animals
were killed. At dietary levels of 5, 50, and 500 ppt TCDD (ng/kg
feed), a variety of tumours were noted, but no particular trend
emerged. However, at a level of 5 µg/kg feed, four squamous cell
tumours of the lung, four neoplastic nodules (hyperplastic nodules),
and two cholangiocarcinomas of the liver were found in seven rats.
Kociba et al. (1978) fed groups of 50 male and female Sprague
Dawley rats 0.1, 0.01, and 0.001 µg TCDD/kg body weight for 2 years.
86 male and 86 female control rats received the vehicle only. The
doses corresponded to 2193, 208, and 22 ng TCDD/kg diet. A variety of
tumours were found in the control and experimental groups. Tumours
caused by the ingestion of TCDD were confined to the liver, lungs,
hard palate/nasal turbinates, and tongue. In the female rats that had
received doses of 0.1 and 0.01 µg/kg body weight, a statistically
significant increase of neoplastic nodules (hyperplastic nodules,
hepatomas) of the liver was noted, and in the rats that had received
0.1 mg TCDD/kg body weight there was a statistically significant
increase of hepatocellular carcinomas. Epithelial tumours along the
respiratory tract, tongue, and hard palate consisted of well
differentiated squamous cell carcinomas. There was an increased
incidence, compared with the controls, of squamous cell carcinomas of
the hard palate and nasal turbinate in both male and female rats
receiving 0.1 µg TCDD/kg body weight, while the incidence of squamous
cell carcinoma of the lungs at this dose showed an increase only in
the females. The authors also noted a decreased incidence of tumours
of the pituitary gland, uterus, mammary glands, pancreas, and adrenal
glands in the treated groups, possibly secondary to an effect on the
hormonal functions of different glands. This decrease was in some
instances statistically significant.
Two further studies on the carcinogenicity of TCDD are available
(NIH, 1982a; NIH, 1982b). The TCDD used in these studies was reported
to be 99.4% pure, based on gas chromatographic analysis.
In two gavage studies both Osborne-Mendel rats and B6C3F1 mice
were used (NIH, 1982a). All animals were about 6 weeks old. Dosages,
duration, and outcome are summarized in Table 59. The statistical
analysis performed was similar to that in the dermal study (NIH,
1982b). Mean body weights of the high-dose groups of rats were lower
than those of the corresponding controls after week 55 and 45 for
males and females, respectively, but no other clinical signs were
observed. No such dose-related depression in mean body weight gain was
observed in mice when compared to the vehicle-control groups.
Table 59. Carcinogenicity bioassays of PCDDs after oral administration
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
2,3,7,8-TCDD Oral (diet), 0.0, 0.001, 0.005, Rat/Sprague Dawley/M all tumours: 0/10 at 0.0,
(Van Miller et al., 0.05, 0.5, 1.0, 5.0 µg/kg. 0/10 at 0.001, 5/10 at 0.005,
1977) 78/95 weeks 3/10 at 0.05, 4/10 at 0.5, 4/10
at 1.0, and 7/10 at 5.0 µg/kg
2,3,7,8-TCDD Oral (diet), 0.0, 0.001, 0.01, Rat/Sprague Dawley/M squamous cell carcinoma hard
(Kociba et al., 1978) 0.1 µg/kg body weight per day. palate: 4/50 at 0.1 µg/kg per day;
105/105 weeks squamous cell carcinoma tongue:
1/50 at 0.001 and 0.01, 3/50 at
0.1 µg/kg per day; adenoma of
adrenal cortex: 2/5 at 0.01 and
5/50 at 0.1 mg/kg per day
Rat/Sprague Dawley/F hepatocellular carcinoma: 0/86
at 0.0, 0/50 at 0.001, 2/50 at
0.01, and 11/49 at 0.1 µg/kg per
day; squamous cell carcinoma of
tongue: 1/50 at 0.01 and 4/49 at
0.1 µg/kg per day, squamous cell
carcinoma of lung: 7/49 at 0.1
µg/kg per day.
2,3,7,8-TCDD Oral (gavage corn oil:acetone, Rats/Osborne-Mendel/M follicular cell adenomas or
(NIH, 1982a) 9:1), 0.0, 0.1, 0.05, 0.5 carcinoma of thyroid: 1/69 at
µg/kg body weight per week. 0.0, 5/48 at 0.10, 8/50 at 0.05,
104/105-107 weeks and 11/50 at 0.5 µg/kg per week
Table 59 (contd - 2).
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
Rats/Osborne-Mendel/F follicular cell adenomas or carcinoma
of thyroid: 3/73 at 0.0, 2/45 at 0.1,
1/49 at 0.05, and 6/47 at 0.5 µg/kg
per week; neoplastic nodules or
hepatocellular carcinoma: 5/75 at
0.0, 1/49 at 0.1, 3/50 at 0.05, and
14/49 at 0.5 µg/kg per week
2,3,7,8-TCDD Oral (gavage corn oil:acetone, Mice/B6C3F18/M hepatocellular carcinoma: 8/73
(NIH, 1982a) 9:1), Males 0.0, 0.01, 0.05, at 0.0, 9/49 at 0.01, 8/49 at
0.5 µg/kg body weight per 0.05, and 17/50 at 0.5 µg/kg per
week. Females 0.0, 0.04, 0.2, week
2.0 µg/kg body weight per
week, 104/105 weeks Mice/B6C3F18/F hepatocellular carcinoma: 1/73
at 0.0, 2/50 at 0.04, 2/48 at
0.2, and 6/47 at 2.0 µg/kg per
week; follicular cell adenomas
of thyroid: 0/69 at 0.0, 3/50
at 0.04, 1/47 at 0.2, and 5/46
at 2.0 µg/kg per week
1,2,3,6,7,8/ Oral (gavage corn oil:acetone, Rats/Osborne-Mendel/M liver neoplastic nodules or
1,2,3,7,8,9- 9:1), 0.0, 1.25, 2.5, 5.0 µg/kg hepatocellular carcinoma: 0/74
hexaCDD (1:2) body weight per week, at 0.0, 0/49 at 1.25, 1/50 at
(NIH, 1980b) 104/105 weeks 2.5, and 4/48 at 5.0 µg/kg per
week
Rats/Osborne-Mendel/F liver neoplastic nodules or
hepatocellular carcinoma: 5/75
at 0.0, 10/50 at 1.25, 12/50 at
2.5, and 30/50 at 5.0 µg/kg per
week
Table 59 (contd - 3).
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
1,2,3,6,7,8-/ Oral (gavage corn oil:acetone, Mice(B6C3F1)/M hepatocellular adenomas or carcinomas:
1,2,3,7,8,9- 9:1), Males 0.0, 1.25, 2.5, 15/73 at 0.0, 14/50 at 1.25, 14.49 at
hexaCDD (1.2) 5.0 µg/kg body weight per week 2.5, and 24/48 at 5.0 µg/kg per
(NIH, 1980b) Females 0.0, 2.5, 5.0, 10.0 µg/ week
kg body weight per week,
104/105-108 weeks Mice(B6C3F1)/F hepatocellular adenomas or carcinomas:
3/73 at 0.0, 4/48 at 2.5, 6.47 at 5.0,
and 10/47 at 10.0 µg/kg per week
Dibenzo-p-dioxin Oral (diet), 0, 5000, 10 000 Mice/B6C3F1/M hepatocellular carcinoma: 4/49 at 0,
(NCI, 1977) µg/kg diet, 87-90/91-97 weeks 7/50 at 5000, and 3/48 at 10 000
µg/kg diet; hepatocellular adenomas:
4/49 at 0, 1/50 at 5000, and 2/48 at
10,000 mg/kg diet; malignant
tumours: 5/49 at 0, 11/50 at 5000,
and 8/50 at 10 000 mg/kg diet
Mice/B6C3F1/F malignant tumours: 8/50 at 0,
9/49 at 5000, and 3/39 at 10,000
mg/kg diet; hepatocellular
carcinoma: 1/47 at 5000 mg/kg
diet
2,7-diCDD Oral (diet), 0, 5000, 10 000 Rats/Osborne-Mendel/M malignant tumours: 5/33 at 0,
(NCI, 1979) mg/kg diet, 110/110-117 weeks. 7/34 at 5000, and 4/33 at 10 000
mg/kg diet; hepatocellular
adenoma: 1/33 at 0; hepatocellular
carcinoma: 1/33 at 10,000 mg/kg diet
Rats/Osborne-Mendel/F malignant tumours: 5/31 at 0;
4/33 at 5000, and 5/30 at
10 000 mg/kg diet
In the male rats, increased incidences of follicular cell
adenomas or carcinomas in the thyroid were dose related and were
significantly higher (P < 0.001) in the high-dose group than in the
vehicle controls (1%, 10%, 16%, and 22%). In the female rats, an
increase (though not statistically significant) was seen only in the
high-dose group (4%, 4%, 2%, and 13%). The incidence of neoplastic
nodules of the liver in the high-dose group of female rats was
significantly (P < 0.006) higher than that in the vehicle-control
group (7%, 2%, 6%, and 28%).
In male and female mice, incidences of hepatocellular carcinomas
were dose related and, in the high-dose groups, were significantly (P
< 0.002 and 0.014, respectively) higher than those in the
corresponding vehicle-control groups (males: 11%, 18%, 16%, and 34%;
females: 1%, 4%, 4%, and 13%).
Follicular cell adenomas in the thyroid occurred at dose-related
incidences in female mice, and were significantly (P < 0.009) higher
in the high-dose groups than those in the vehicle controls (0%, 6%,
2%, and 11%). In conclusion, under the conditions of this bioassay,
TCDD was carcinogenic for Osborne-Mendel rats, inducing follicular
cell thyroid adenomas in males and neoplastic nodules of the liver in
females. TCDD was also carcinogenic for B6C3F1 mice, inducing
hepatocellular carcinomas in males and females and follicular cell
thyroid adenomas in females.
Toth et al. (1979) administered TCDD orally by gavage to groups
of 45 male Swiss/H/Riop mice at doses of 0, 0.007, 0.7, and 7 µg/kg
body weight once a week for one year, and the animals were followed
for their lifetime. Liver tumours were found at 18%, 29%, 48%, and
30%, respectively. The tumour incidence at 0.7 µg/kg was significantly
higher when compared to controls (P < 0.01), while the increase at
the highest dose level (7 µg/kg) was not statistically significant (P
= 0.11). The latter finding may be due to a much reduced average
survival in comparison with the control group (average life span 424
and 588 days, respectively).
In a study using dermal application of TCDD (NIH, 1982b), male
and female Swiss-Webster mice were about 6 weeks old at the beginning
of the bioassay. The one-tailed Fisher exact test was used to compare
the tumour incidence of a control group with that of a group of dosed
animals. Mean body weights of dosed animals were essentially the same
as those of the corresponding vehicle-control groups, but less than
those of the untreated controls, for males throughout the study and
for females during the first 80 weeks. The incidence of fibrosarcoma
in the integumentary system of female mice treated with TCDD or TCDD
and dimethylbenzathraline (DMBA) was significantly higher than that of
the controls (P < 0.007 and P < 0.010, respectively). An increase in
the same tumour type, although not statistically significant (P =
0.084), was also observed in the male mice (7% and 21% for the control
and TCDD-treated groups, respectively). In conclusion, under the
conditions of this bioassay, TCDD was carcinogenic for female
Swiss-Webster mice, causing fibrosarcomas in the integumentary system.
However, the study has been criticized in several areas, namely, a
maximal tolerated dose (MTD) was not achieved, especially in male
mice, only one dose per sex was used, and the number of mice (30) in
the TCDD-exposed groups was considered less than optimal.
7.7.2 Long-term animal studies with mixed compounds
Toth et al. (1979) studied groups of 100 male and 100 female,
10-week-old random-bred Swiss H/Riop mice that were given weekly oral
doses of 2,4,5-trichlorophenoxyethanol (TCPE) at 67-70 mg/kg body
weight, together with 0.112 mg TCDD/kg body weight or 0.007 mg TCDD/kg
body weight in 0.5% carboxymethyl cellulose by gastric intubation for
12 months. The incidences of liver tumours in males after 2 years were
reported to be 48% and 58% in the two treated groups, compared with
26-33% in the untreated male mice of the colony that survived up to 3
years. Three additional groups of mice were given 7 µg TCPE/kg body
weight with 0.0007 µg TCDD/kg body weight, 0.7 µg TCPE/kg body weight
with 0.00007 µg TCDD/kg body weight, or 7 µg TCPE/kg body weight with
0.7 µg TCDD/kg body weight. There was no increased incidence of liver
tumours in any of the treatment groups.
A 1:2 mixture of 1,2,3,6,7,8- and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxins (HxCDDs) has been tested for
carcinogenicity by dermal application to mice and by gavage in rats
and mice (NIH, 1980a,). The following impurities were detected in the
mixture: pentaCDD 0.04%, TCDD 0.09%, triCDD 0.004%, and bromopentaCDD
< 0.004%. The specific isomers of these impurities were not
identified. The doses used and duration of the gavage studies (NIH,
1980b) are given in Table 59. In both species and either sex, only
tumours of the liver occurred at a significantly greater incidence
than controls. In male rats and male and female mice, the liver tumour
incidence was significantly increased over control values only in the
high dose groups (5 µg/kg per week), while in female rats the
incidence was significantly greater at both medium- and high-dose
levels (2.5-5 µg/kg per week). In the dermal study, no
treatment-related tumours were recorded in either the carcinogenicity
bioassay or the tumour promotion assay using DMBA as an initiator
(NIH, 1980a). It was concluded that the mixture of hexaCDDs tested was
carcinogenic to rats and mice following administration by gavage.
However, there was no tumorigenic activity when hexaCDD was applied to
mouse skin.
When added to the diet in concentrations up to 10 000 µg/kg,
2,7-dichlorodibenzo-p-dioxin and dibenzo-p-dioxin were found to be
non-carcinogenic in chronic feeding studies in mice and rats of either
sex (NCI, 1977; NCI, 1979).
7.7.3 Short-term and interaction studies
Poland & Glover (1979) estimated the maximum covalent binding of
TCDD in vivo to rat liver protein, ribosomal RNA (rRNA), and DNA
after 3H-TCDD (39 Ci/mmol) was administered to immature male and
female Sprague Dawley rats (105-135 g) as a single ip injection of 7.5
µg/kg. The rats were killed 12 h, 24 h, 48 h, or 7 days after dosing
with TCDD. The level of radioactivity in the liver varied from 18 to
64% of the administered dose, and only a small fraction was associated
with the purified macromolecular fractions. The radioactivity
associated with rRNA and DNA was very low and essentially all the
unextracted radioactivity was associated with protein (0.03 to 0.1% of
the total radioactivity in the liver). The maximum amount of 3H-TCDD
that could have been covalently bound to DNA was estimated as 1.8 x
10-17 mol TCDD per mg DNA, or 6.2 nmol TCDD per mol DNA nucleotide,
which means binding of about 1 molecule TCDD to the DNA in 35 cells.
Phenobarbital treatment, or prior administration of TCDD did not
significantly alter the amount of unextractable 3H-TCDD associated
with any macromolecular fraction. Similarly, there were no differences
in the levels of 3H-TCDD associated with protein, rRNA, or DNA in
male or female rats pretreated with TCDD.
TCDD was found to be a carcinogen in chronic feeding studies in
rats and mice. Most carcinogens bind covalently, either directly or
after a conversion to electrophilic intermediates, to protein, rRNA,
and DNA to the extent of 10-4 to 10-6 mol of carcinogen per mol of
amino acid or nucleotide residue. The maximum binding of TCDD is 4-6
orders of magnitude lower than that of most chemical carcinogens and
is of questionable biological significance. The results obtained in
the study of Poland & Glover (1979) thus indicate that it is unlikely
that the mechanism of TCDD-induced carcinogenesis would include the
covalent binding of TCDD.
In female Charles River CD-1 mice, TCDD was found to be a weak
initiator when given alone in a single dose of 2 µg/ mouse by dermal
application (Di Giovanni et al., 1977). In these studies
12-O-tetradecanoylphorbol-13-acetate (TPA) was used as a promoter.
When TCDD and 7,12-dimethylbenzanthracene were given together, a
slight additive effect was found. As mentioned earlier, in a study on
a hexaCDD mixture, no treatment-related tumours were found in a tumour
promotion test on mice using DMBA as an initiator (NIH, 1980a).
The possible role of TCDD as a promoter in
diethylnitrosamine-induced hepatocarcinogenesis was studied by Pitot
et al. (1980) in female Charles River rats (200-250 g). A single oral
dose (10 mg/kg) of diethylnitrosamine (DEN) was given 24 h after a
70% hepatectomy, and treatment with TCDD (0.14 or 1.4 mg/kg sc once
every 2 weeks for 7 months) was started one week after the
hepatectomy. The promoting effect of TCDD in this 2-stage model of
liver cancer was also compared with the effect of a known promoting
agent, phenobarbital (0.05% in the diet for 7 months). Enzyme-altered
foci, which are thought to be precursors of hepatocellular carcinomas,
were greatly increased in number, total volume, and phenotypic
heterogeneity by the administration of TCDD. A significant incidence
of hepatocellular carcinomas (5 out of 7) was observed in the
DEN-treated rats that were given the high dose of TCDD, but no
carcinomas were seen in the rats treated with DEN only (0 out of 4).
The results indicated that TCDD was a potent promoting agent for
hepatocarcinogenesis, and the authors suggested that all the tumours
associated with the chronic administration of TCDD arise from its
promoting activity of cells previously initiated by exposure to
carcinogens in the environment.
Studies utilizing a two-stage system of mouse skin tumorigenesis
(Berry et al., 1979), which allows separate evaluation of the
initiation and promotion phases of carcinogenesis, have demonstrated
that TCDD does not promote the development of skin tumours at a dose
of 0.1 µg given twice weekly, whereas in the animals pretreated with
1.0 µg TCDD for 1, 3, or 5 days prior to initiation with DMBA, TCDD
was shown to act as a potent inhibitor of PAH-induced skin tumour
initiation. Almost complete inhibition (96%) was achieved with a
single non-toxic topical dose of 0.1 µg, and 3 days pretreatment with
0.01 µg TCDD gave over 80% inhibition. The authors suggested that this
potent anticarcinogenic effect of TCDD may be related to its ability
to induce epidermal enzyme pathways involved in detoxifying PAH
carcinogens in the skin. According to Kimbrough (1979), TCDD and other
compounds of this type, which are potent enzyme inducers, may prevent
or enhance the tumour-inducing ability of other chemicals by enhancing
the metabolism of these xenobiotics.
Poland et al. (1982) studied the promoting effects of TCDD in the
mouse skin two-stage tumorigenesis model. The effects of TCDD and TPA
were compared in DMBA-initiated HRS/S mice that were either
heterozygous or homozygous for the recessive "hairless" trait. TCDD
was found to have a tumour-promoting effect only in the homozygous
mice. The data suggested to the authors that TCDD might act as a
promoter by a mechanism different from that of TPA.
The interaction of TCDD with 3-methylcholanthrene (3-MC) was
studied by Kouri et al. (1978), who found that TCDD was a
co-carcinogen with 3-MC when administered by subcutaneous injection.
Both sexes of two inbred strains of mice (C57Bl/6C and DBA/2),
responsive and non-responsive to the induction of AHH by 3-MC,
respectively, were used. TCDD at a concentration of 1 or 100 µg/kg
body weight was administered as a single dose alone or in combination
with 3-MC (150 µg/kg). The duration of the study was 36 weeks. The
number of animals in each group at the start of the experiment was not
stated, but seems to have been between 30 and 100. No subcutaneous
tumours were observed in controls or in mice treated with TCDD alone.
In responsive mice no enhancement occurred, while in non-responsive
mice the simultaneous administration of TCDD and 3-MC enhanced the
carcinogenic response of TCDD at 100 mg/kg. At 1 mg TCDD/kg, a
reduction in latency time to tumour was noted.
An anticarcinogenic effect of TCDD has been reported by Cohen et
al. (1979) and by Di Giovanni et al. (1979a). When TCDD was topically
applied to Sencar or CD-1 mice 72 h prior to the administration of
either DMBA (10 nmol) or benzo(a)-pyrene (BP) (100 nmol), it markedly
decreased the skin tumour initiation by both DMBA and BP. This
inhibition of tumorigenesis correlated with the decreased in vivo
binding of DMBA to DNA after TCDD administration, but not with the
total binding of BP to DNA. However, the
hydrocarbon-deoxyribonucleoside adducts from the DNA of
TCDD-pretreated mice showed a striking absence of
BP-7,8-dihyrodiol-9,10-epoxide adduct bound to guanine. It is
suggested, accordingly, that the formation of this adduct may be a
critical step in BP-induced skin carcinogenesis in mice. In further
studies of the tumour-inhibitory effect of TCDD (Di Giovanni et al.
1980), it was demonstrated that exposure of CD-1 mice to TCDD 3 days
before initiation with BP or 3-MC resulted in a decreased tumour
yield, compared to acetone-pretreated animals, while treatment with
TCDD 5 min before and 1 day after initiation failed to affect the
tumour yield. However, when TCDD was administered 3 days or 5 min
before or 1 day after initiation with BP-diol epoxide, there was a
decreased tumour yield in all cases. The authors concluded that the
ability of TCDD to inhibit tumour yield when administered after the
BP-diol epoxide, indicated the possible existence of more than one
mechanism involved in the anticarcinogenic effect of TCDD.
7.8 Mechanisms of Action
The toxicity of TCDD apparently depends on the fact that the four
lateral positions of the molecule are occupied by chlorine (see
section 7.8.1) Toxicity decreases with decreasing lateral substitution
and increasing total chlorine substitution. As has been outlined in
sections 7.1-7.7, TCDD toxicity involves many different types of
symptoms and these symptoms vary from species to species and from
tissue to tissue, both quantitatively and qualitatively. Furthermore,
age- and sex-related differences in sensitivity to TCDD have been
reported. Characteristic for TCDD toxicity is also the delay in
expression of toxicity, from 2 weeks to 2 months, seen in all species.
It has been suggested that the initial event in TCDD-induced toxicity
is the binding of TCDD to the so-called Ah receptor. This complex,
whether of cytosolic or nuclear origin, exerts its action in the
nucleus by triggering a pleiotropic response including the induction
of mixed function oxidases. Present knowledge, however, rules out
enzyme induction per se as being the cause of toxicity and death
(see section 7.8.1). Although the toxicokinetics of TCDD vary between
species, these differences are not sufficient to explain the
variabilities in sensitivity to TCDD toxicity (see section 7.8.2).
Available data indicate an involvement of TCDD in processes regulating
cellular differentiation and/or division. Alterations in the
regulation of such processes, which are not equally active in all
cells throughout the organism, would be expected to result in effects
that vary among tissues as well as among species (see section 7.8.3).
7.8.1 Receptor-mediated effects
The binding of TCDD to the Ah receptor has been postulated to be
the necessary first step in the induction of cytochrome P-450
synthesis and of related enzyme activities, as well as in the
mechanism of toxicity (Poland et al., 1976; Okey et al., 1979; Poland
& Glover, 1979; Poland & Knutson, 1982). So far, no conclusive data
exist for the direct involvement of the Ah receptor in the
TCDD-induced toxicity.
Knowledge of the mechanism involved in the Ah locus
enzyme-induction response has grown rapidly since the initial indings
that binding of TCDD to the receptor resulted in increased levels of
cytochrome P-450 mRNA in genetic variants of mice (Tukey et al., 1982)
and mouse hepatoma cell lines (Israel & Whitlock, 1983). These
findings have been confirmed and further expanded in studies using
mice genetics and recombinant DNA techniques (Tukey et al., 1982;
Miller et al., 1983; Gonzales et al., 1984; Israel & Whitlock, 1984;
Jones et al., 1984, 1985, 1986; Okino et al., 1985; Tuteja et al.,
1985; Kimura et al., 1986), thus providing more data to the
understanding of the mechanism for the Ah locus enzyme induction.
In early experiments (Poland et al., 1976; Carlstedt-Duke, 1979;
Okey et al., 1979), Ah receptors appeared to be localized in the
cytosol when in its unoccupied state and was translocated into nuclei
only when occupied by a ligand (Greenlee & Poland, 1979; Okey et al,
1980; Poellinger et al., 1982; Gasiewicz & Rucci, 1984). However,
Whitlock & Galeazzi (1984) concluded that unoccupied Ah receptor in
the intact cell was primarily located in the nucleus and that apparent
cytosolic Ah receptor was a redistribution artifact. Following the
distribution of Ah receptor and three cytosolic marker enzymes between
the nuclear and cytosolic fractions during fractionation (Denison et
al., 1986a,c), it was again concluded that unoccupied Ah receptor is
primarily cytosolic or that this receptor protein is in equilibrium
between the cytoplasm and nucleus.
However, it is generally agreed that the ultimate biological
regulation by the Ah receptor is due to specific interaction of
ligand-receptor complexes with chromatin sites (Greenlee & Poland,
1979; Okey et al., 1979, 1980; Mason and Okey, 1982; Poellinger et
al., 1982; Poland and Knutson, 1982; Tukey et al., 1982a; Gonzales et
al., 1984; Israel & Whitlock, 1984).
Table 60. Physicochemical data for the hepatic Ah receptor in Sprague Dawley rats
Physicochemical data Denison et al. (1986a) Poellinger et al. (1983)
Stokes radius (nm) 5.2 ± 0.2 6.1 ± 0.2
Sedimentation coefficient (S) 5.6 ± 0.6 4.4
Relative molecular mass 121 000 111 000
Several investigators have estimated the molecular size and other
physicochemical properties for the cytosolic hepatic Ah receptor (Okey
et al., 1979, 1980, 1982; Tukey et al., 1982b; Poellinger et al.,
1983; Gasiewicz et al., 1983a,b; Denison et al., 1986a; Hannah et al.,
1986). To obtain reliable results in the isolation and
characterization of the Ah receptor, it is necessary to use perfused
liver, in order to reduce the contribution from blood proteins, and to
use a radioactive ligand of high purity and specific activity quality
(Poellinger et al., 1983; Denison et al., 1986a). Further more, the
ionic strength of the medium during isolation has a marked effect upon
the apparent molecular weight of the receptor (Denison et al., 1986a).
The physicochemical data for the Ah receptor presented in Table 60
were obtained from two studies (Denison et al., 1986a,c; Poellinger et
al., 1983) in which the receptor was isolated from perfused liver of
Sprague Dawley rats under conditions of high ionic strength.
The receptor protein has been found also in extrahepatic tissues
(Carlstedt-Duke, 1979; Carlstedt-Duke et al., 1979, 1981; Johansson et
al., 1982; Mason & Okey, 1982; Gasiewicz & Rucci, 1984; Gasiewicz et
al., 1984; Furuhashi et al., 1986; Kurl et al., 1985; Söderkvist et
al., 1986). Different mammalian species possess Ah receptors with
similar, though not identical, properties (Gasiewicz & Rucci, 1984;
Denison et al., 1986a,b; Kurl et al., 1985). Jaiswal et al. (1985a,b)
have shown species differences in the TCDD-inducible P-450 gene
subfamily. Humans appear to only have the P1-450. The function of
human P1-450 may be equivalent to a combination of P1-450 and P3-450
in the mouse. A complete lack of measurable cytosolic and almost
total absence of inducer-receptor complexes in the nucleus of human
MCF-1 cells (cells derived from an adenocarcinoma of the breast) were
reported. This absence was out of proportion to the ability of TCDD to
induce AHH and acetamide-4-hydroxylase activities in these cells.
Further studies in different cell lines are thus needed to
characterize the level of receptor in humans. The only non-mammalian
species demonstrated to have significant Ah receptor is the chick
embryo (Denison et al., 1986b). However, no detectable level of the
receptor was found in 2-week-old White Leghorn chickens (Sawyer et
al., 1986). Based on certain similarities in the biochemical behaviour
between the Ah receptor and steroid hormone receptors, it has been
proposed that there is a natural ligand for the Ah receptor (Neal et
al., 1979; Poland et al., 1976). So far such a ligand has not been
identified, either among steroid hormones (Poland et al., 1976;
Carlstedt-Duke et al., 1979; Romkes et al., 1987) or among certain
dietary factors (Johansson et al., 1982), although lumichrome, a
metabolite of riboflavin, was suggested as an endogenous ligand for
the receptor (Kurl & Villee, 1985). TCDD does not bind to the
glucocorticoid, estrogen or progesterone receptors (Neal et al.,
1979; Romkes et al., 1987). Monoclonal anti-glucocorticoid
receptor-IgG antibodies did not react with the TCDD receptor
(Poellinger et al., 1983) and the hydrophobic properties of the Ah
receptor were more pronounced than those of the steroid hormone
receptors (Poellinger & Gullberg, 1985).
Convincing data for the importance of the receptor in
TCDD-induced toxicity could be based on structure activity
relationships, i.e., that the binding affinities of TCDD and other
PCDDs or PCDFs to the receptor correlate with their biological
potencies. The binding affinities of PCDDs and PCDFs have been
demonstrated to correlate with their biological potencies,
particularly the induction of enzyme activities as well as the
production of acute toxic effects (Tables 56 and 61) (Poland & Kende,
1976; Poland et al., 1976; Knutson & Poland, 1982).
Furthermore, the structure-activity relationships observed for
enzyme induction, thymic atrophy, body weight loss, and LD50 values
were comparable to the structure-activity relationships observed for
receptor binding (Tables 56, 61) (Bandiera et al., 1984a,b; Mason et
al., 1985, 1986; Sawyer & Safe, 1985; Safe et al., 1986). Interactive
studies, i.e., studies where PCDD and PCDF congeners have been given
both separately and as mixtures, have also been used to investigate
the role of the Ah receptor in the mechanism of action of TCDD.
Depending on the mechanism of action the biological responses may be
synergistic potentiated, additive, unaffected, or antagonistic. Such
studies have been performed for enzyme induction (Sawyer & Safe, 1985;
Keys et al., 1986; Ahlborg et al., 1987), vitamin A reduction
(Hakansson et al., 1987), teratogenicity (Birnbaum et al., 1985; Weber
et al., 1985), thymic atrophy (Bannister & Safe, 1987), and immune
suppression (Rizzardini et al., 1983). So far this kind of data is
scattered and difficult to interpret.
Table 61. Structure-activity relationships for some PCDFs
In vitro EC50 values (mol/litre)a,b In vivo ED50 values (µmol/kg) LD50 c
PCDF Receptor AHH EROD AHH weight Thymic Guinea-pig
congener binding loss atrophy µg/kg body weight
Dibenzofuran < 10-3 ND ND
2- 2.8 x 10-4 ND ND
3- 4.2 x 10-5 ND ND
4- < 10-3 1.0 x 10-5 1.71 x 10-5
2,3- 4.72 x 10-6 2.19 x 10-6 4.84 x 10-6
2,6- 2.46 x 10-4 6.17 x 10-5 6.31 x 10-5
2,8- 2.57 x 10-4 3.95 x 10-5 4.0 x 10-5
1,3,6- 4.40 x 10-6 2.53 x 10-6 3.37 x 10-6
1,3,8- 8.50 x 10-5 1.94 x 10-5 3.02 x 10-5
2,3,4- 1.9 x 10-5 1.51 x 10-7 2.48 x 10-7
2,3,8- 1.0 x 10-6 2.49 x 10-6 1.56 x 10-6
2,6,7- 4.5 x 10-7 2.80 x 10-6 3.13 x 10-6
2,3,4,6- 3.5 x 10-7 1.32 x 10-6 1.13 x 10-6
2,3,4,7- 2.51 x 10-8 1.79 x 10-8 1.48 x 10-8 46 34 7.8
2,3,4,8- 2.0 x 10-7 4.14 x 10-8 3.76 x 10-8 ND 130 > 150
2,3,6,8- 2.2 x 10-7 1.04 x 10-6 7.79 x 10-7
2,3,7,8- 4.1 x 10-8 3.91 x 10-9 2.02 x 10-9 0.65 3.2 3.6 5-10
1,2,3,6- 3.54 x 10-7 > 10-4 > 10-4 > 160 > 250 > 250
1,2,3,7- 1.12 x 10-7 2.7 x 10-5 6.3 x 10-5 110 87 110
1,2,4,8- > 10-5 1.20 x 10-5 9.26 x 10-5
1,2,4,6,7- 6.77 x 10-8 3.25 x 10-7 3.48 x 10-7
1,2,4,7,9- 2.0 x 10-5 3.77 x 10-8 3.84 x 10-8
1,2,3,4,8- 1.2 x 10-7 2.09 x 10-7 1.63 x 10-7
1,2,3,7,8- 7.45 x 10-8 2.54 x 10-9 3.06 x 10-9 1.5 2.6 1.8
1,2,3,7,9- 3.98 x 10-7 8.6 x 10-8 8.6 x 10-8 15 49 23
1,2,4,6,8- 3.09 x 10-6 1.0 x 10-5 1.2 x 10-5 > 150 7150 > 150
Table 61 (contd).
In vitro EC50 values (mol/litre)a,b In vivo ED50 values (µmol/kg) LD50 c
PCDF Receptor AHH EROD AHH weight Thymic Guinea-pig
congener binding loss atrophy mg/kg body weight
1,2,4,7,8- 1.3 x 10-6 1.06 x 10-7 1.48 x 10-7 7.8 49 46
1,3,4,7,8- 2.0 x 10-7 1.60 x 10-9 1.40 x 10-9 3.5 26 0.70
2,3,4,7,8- 1.5 x 10-8 2.56 x 10-10 1.34 x 10-10 0.037 1.0 0.26
2,3,4,7,9- 2.0 x 10-7 7.9 x 10-9 5.8 x 10-9 7.0 22 5.5
1,2,3,4,7,8- 2.3 x 10-7 3.56 x 10-10 3.79 x 10-10 0.29 1.3 0.56
1,2,3,6,7,8- 2.7 x 10-7 1.47 x 10-9 1.24 x 10-9 0.35 3.2 0.90
1,2,4,6,7,8- 8.3 x 10-6 4.24 x 10-8 2.93 x 10-8
2,3,4,6,7,8- 4.7 x 10-8 6.87 x 10-10 5.75 x 10-10 0.27 2.8 0.90
a Estimated concentration needed to displace 50% of 3H-TCDD bound to liver cytosol receptor from
Wistar rats and to produce 50% maximum enzyme induction in the rat hepatoma H-4-IIE cell line (Bandiera
et al., 1984b).
b Studies in immature male Wistar rats (Mason et al., 1985).
c Moore et al. (1979).
Polymorphism in the Ah locus, which is suggested to be structural
gene for the cytosolic receptor, seems to determine the sensitivity of
genetically different strains of mice to TCDD and congeners. Ah
responsive strains of mice, e.g., C57Bl/6, are characterized by (a)
high hepatic levels of the TCDD receptor protein, (b) highly elevated
levels of hepatic cytochrome P-448 and associated enzyme activities in
response to treatment with 3-MC, and (c) sensitivity to the ulcerative
action of DMBA on the skin. Ah-non-responsive mice, e.g., DBA/2, lack
these attributes (Nebert et al., 1975). Based on these findings
several genetic studies have been performed to elucidate the role of
the receptor in TCDD toxicity. Contrary to 3-MC, TCDD induces AHH
activity and several toxic effects both in Ah-responsive and
Ah-non-responsive strains of mice. However, the dose required to
produce the effect in an Ah-non-responsive strain is approximately
10-fold greater than that needed for a responsive strain, thus
demonstrating that the Ah-non-responsive strain also contains the
TCDD-receptor but that this receptor is defective (Okey & Vella,
1982).
Crosses and backcrosses of C57BL/6 and DBA/2 mice have shown that
sensitivity to TCDD-induced thymic atrophy immune system disturbances
(section 7.4.5) and teratogenic effects (section 7.5.2) segregate with
the Ah locus. Furthermore, data from studies of DBA/2 mice given
either single or multiple doses of TCDD (Jones & Sweeney, 1980; Smith
et al., 1981) suggest that the LD50 in this strain of mice is at
least 5-fold greater than the values recorded for the C57Bl/6 and
C57Bl/10 strains (Vos et al., 1974; Jones & Greig, 1975; Smith et al.,
1981). TCDD-induced hepatic porphyria has also been shown to segregate
with the Ah locus in mice (Jones & Sweeney, 1980). However, Greig et
al. (1984) found that additional genetic loci must be involved in this
lesion. The correlative differences between the C57Bl/6 and DBA/2
strains of mice, in terms of altered specific binding of TCDD and
sensitivity to this compound, may be unique and may not be applicable
to other species (Gasiewicz & Rucci, 1984).
Less convincing data for the model of receptor-mediated toxicity
of TCDD arise from studies of toxicity, receptor levels, and/or enzyme
induction of TCDD in various species, tissues, and cell cultures.
Despite enormous variability in recorded LD50 values for guinea-pig,
rat, mouse, rabbit, and hamster (Table 47), the amounts and physical
properties of the hepatic as well as the extrahepatic receptors, do
not vary extensively in these species (Poland & Knutson, 1982;
Gasiewicz & Rucci, 1984). Furthermore, although recorded LD50 values
for TCDD vary more than 100 times in chick embryos, C3H/HeN mice, and
Sprague Dawley rats, the ED50 doses for AHH induction in these
species are comparable (Poland & Glover, 1974b). In the guinea-pig,
the most TCDD-susceptible species, enzyme induction is several times
lower even at lethal doses. A number of cell types, including primary
cultures and established and transformed cell lines from several
species and tissues, are inducible for AHH activity, indicating the
presence of the receptor, yet toxicity is not expressed in these
systems (Knutson & Poland, 1980a).
Available data thus suggest that the receptor for TCDD may be a
prerequisite, but is not sufficient in itself for the expression of
TCDD toxicity.
7.8.2 Toxicokinetics
The interspecies variation in sensitivity to TCDD may be
attributable, at least in part, to different rates at which various
species distribute, metabolize, and excrete the compound. Table 62
summarizes some of the data on the elimination, toxicity, and
metabolism of TCDD in different species, some previously discussed in
section 6. Distribution data (section 6.1) has been obtained mainly
from animals exposed to toxic doses of TCDD. Interspecies comparisons
based on these data are difficult to perform, since studies in
different species have been performed with non-comparable relative
toxic doses and collection of data has occurred at variable time
points. However, the available data suggest that tissue levels alone
cannot explain the interspecies differences in pathology and acute
toxicity of TCDD. For example, the molar concentration of TCDD in the
hamster may be orders of magnitude greater than in the rat and mice,
without development of hepatotoxicity, yet definite liver damage
occurs both in rats and mice.
Based on the findings that the toxicity of TCDD is lower in rats
after the stimulation of hepatic mixed function oxidases (Beatty et
al., 1978), and that the metabolites of TCDD are less toxic than the
parent compound (Weber et al., 1982a; Mason & Safe, 1986) (see section
7.1.5), the metabolism of TCDD has been considered as a detoxification
mechanism.
Following the administration of TCDD, most of it appears to be
eliminated through a first-order process in most species (see section
7.1). In all species investigated TCDD is largely eliminated in the
faeces (Table 48). Only in hamsters and certain strains of mice is
urinary elimination a major route of excretion (Table 41). Available
data demonstrate that TCDD is converted to more polar metabolites
prior to elimination in the urine and bile (Table 62). Unchanged TCDD
does not appear in the bile or urine of any species, but it is the
major excretory product in the faeces of mice, guinea-pigs, and
hamsters (Table 62). As the urine and bile appear to be free of
unmetabolized TCDD, the existence of unchanged TCDD in faeces
indicates that a significant amount of unchanged TCDD may be excreted
into the intestinal lumen by some route other than bile.
Table 62. Rates of elimination, toxicity, and metabolic transformation of TCDD in different species
Species/strain Elimination LD50 value TCDD-derived radioactivity occurring in:
half-life (µg/kg
(days) body weight) Urine Bile Faeces Tissues
Ratsa,b,c,d
Sprague Dawley 17-31 25-60 5 polar 4-8 polar unchanged TCDD
metabolites metabolites;
little, if any,
unchanged TCDD.
Micee,f,g
C57BL/6, DBA/2, 10-24 114-2570 4-7 polar 4-6 polar 3-4 polar unchanged TCDD
B6D2F1 metabolites metabolites metabolites,
unchanged TCDD.
Guinea-pigh
Hartley 22-94 0.6-2.5 4 polar 5 polar mainly unchanged TCDD,
metabolites metabolites unchanged polar metabolites
TCDD
Hamstersa,i
Golden Syrian 12-15 1157-5051 4 polar 5-6 polar metabolites, unchanged TCDD
metabolites metabolites unchanged
TCDD
Dogsj
Beagle not reported not reported metabolites
a Gasiewicz et al., 1983a. b Poiger & Schlatter, 1979. c Ramsey et al., 1982. d Rose et al., 1976.
e Gasiewicz et al., 1983b. f Koshakji et al., 1984. g Vinopal & Cassida, 1973. h Olson, 1986.
i Olson et al., 1980a. j Poiger et al., 1982.
The metabolic profiles of TCDD in excreta differs between
species, but generally urinary metabolites are more polar than biliary
or faecal metabolites. Furthermore, several metabolites, both in urine
and bile, are glucuronide conjugates.
The apparent absence of TCDD metabolites in the tissues of all
species, except for the guinea-pig (Table 62), suggests that, once
formed, the metabolites of TCDD are readily excreted. Another factor,
besides metabolism, that may influence the total rate of elimination
of TCDD is the amount of adipose tissue stores, which may vary between
species.
At present there is no clear relationship between the ability of
a given species to excrete TCDD and/or its metabolites and the acute
toxicity of TCDD in that species. However, the somewhat greater rate
of elimination of TCDD in the hamster and the lower rate of
elimination in the guinea-pig (Table 62) may contribute to their
relative resistance and sensitivity, respectively, to the acute toxic
effects of TCDD.
The rate of metabolism and excretion of PCDDs and PCDFs varies
with molecular structure. In most species PCDFs are much more readily
eliminated than their PCDD counterparts. Less halogenated congeners
are usually metabolised and excreted more rapidly than the more
halogenated ones, especially the 2,3,7,8-substituted congeners (see
sections 6 and 9).
7.8.3 Impairment of normal cellular regulatory systems
When considered together, the diverse pattern of toxic effects,
the species and tissue-specific responses, and the time-course for
effects, as well as the non-toxic action of TCDD on most cell cultures
in vitro, seem to indicate that TCDD-induced toxicity occurs as a
result of an impairment of a normal cellular regulatory system. Such
a system might be present in all cells throughout the organism, though
the activity may vary with cell type, tissue, age, sex, and strain,
and/or species.
7.8.3.1 Endocrine imbalance
In many aspects, TCDD toxicity mimics endocrine imbalance,
although no evidence exists to indicate a direct involvement of
hormones in the toxic action of TCDD (section 7.4.9).
7.8.3.2 Body weight regulation
The most reliable and consistent symptom of TCDD toxicity among
all experimental animals is weight loss. The cause of the body weight
loss seems to be reduced food intake, apparently occurring secondarily
to a physiological adjustment that reduces the body weight to a
maintenance level lower than normal. The physiological trigger for
controlling this body weight set-point might be a target for TCDD
action (section 7.4.1).
7.8.3.3 Plasma membrane function
The changes in the surface characteristics of the plasma
membranes induced by TCDD in vivo (7.4.2.2) resemble changes
occurring in precancerous and transformed cells (Pitot & Sirica,
1980). Such changes, including reduction of gap junctions and surface
glyco-proteins, would be expected to curtail cell-cell communication
and to reduce intercellular recognition and attachment events
implicated in the process of tumour promotion.
It has been shown by Pitot et al. (1980) that TCDD promotes
diethylnitrosamine (DEN)-induced hepatocarcinoma in rats. In this
study, canalicular ATPase was used as a marker in detecting
enzyme-altered foci, whose number increased when TCDD was given to
DEN-treated partially hepatectomized rats. The foci exhibited
decreased ATPase activity in agreement with previous observations that
TCDD in vivo reduces the ATPase level in canaliculi-rich plasma
membranes.
TCDD, unlike other well known promoters, requires a prolonged
treatment period in vivo to exert its effect. The lack of effect
of TCDD in vitro would imply that the promoter activity is
mediated through some in vivo process and not by its direct
interaction with plasma membranes.
7.8.3.4 Impaired vitamin A storage
The histomorphological appearance of chloracne, the most
characteristic and prominent sign of TCDD-induced toxicity in humans,
resembles in some respects effects seen in the skin of patients
suffering from vitamin A deficiency (Kimbrough, 1974). Many of the
effects of TCDD poisoning observed in animal studies, including
failure of normal growth, keratosis, epithelial lesions,
immunosuppression, and reproductive and teratological effects, are
similar to the effects of dietary vitamin A deficiency (Thunberg et
al., 1980). The most intriguing similarities between symptoms due to
vitamin A deficiency and TCDD toxicity concern effects on epithelial
tissues, particularly the process of keratinization. TCDD induces
terminal differentiation of epithelial tissues both in vivo and
in vitro. However, lack of epithelial degeneration (programmed
cell death) of the medial epithelial cells of palatal shelves has been
reported in mice exposed to TCDD in utero (Pratt et al., 1984).
Vitamin A is essential for normal differentiation. It diminishes
the expression of differentiation in stratified squamous epithelia and
accentuates the expression of differentiation in secretory epithelia.
Vitamin A deficiency can convert secretory epithelia to squamous
epithelia, while excess of the vitamin can convert stratified squamous
epithelia to secretory epithelia (Wolf, 1980). With the use of
cultured human keratinocytes, it has been demonstrated that vitamin A
at the cellular level affects cell motility, cell-cell interaction,
and epithelial morphogenesis. At the molecular level, vitamin A
determines, by controlling the level of the corresponding mRNA, the
nature of keratins synthesized (Fuchs & Green, 1981). Keratins
constitute a cytoskeleton in epithelial cells, and the keratin pattern
may be used as a marker for epithelial differentiation (Sun et al.,
1979, 1983a,b). Removal of vitamin A from the medium of cultivated
human keratinocytes of various origin led to increased synthesis of
large keratins and reduced synthesis of lower molecular weight
keratins (Fuchs & Green, 1981). This pattern was reversed by the
addition of vitamin A to the medium. Each tissue and cell type
controlled its synthesis of keratins differently, depending on the
vitamin A concentration in the medium. The ability of TCDD to impair
vitamin A storage (section 7.4.10) may be responsible for some of the
toxic effects produced by TCDD.
7.8.4 Lipid peroxidation
Based on indirect lines of evidence, Sweeney & Jones (1983)
proposed that increased in vivo lipid peroxidation, resulting in
the formation of free radicals, might be a mechanism of TCDD toxicity.
Firstly, lipofuscin pigments, considered to be by-products of lipid
peroxidation, accumulate in the heart muscle cells of TCDD-treated
rats (Albro et al., 1978). Secondly, iron deficiency inhibits in
vitro lipid peroxidation (Bus & Gibson, 1979; Sweeney et al., 1979)
and has been demonstrated to reduce hepatic TCDD toxicity in vivo
in rats (Sweeney et al., 1979). Thirdly, 0.25% butylated
hydroxyanisole (BHA) in the diet has been shown to provide protection
from TCDD-induced porphyria and lipid accumulation in mice. In
contrast, 0.01% vitamin E, another antioxidant, in the diet had no
protective effect (Hassan et al., 1985a,b). Stohs et al. (1983)
demonstrated increased in vivo (conjugated diene method) and in
vitro (microsomal malondialdehyde formation) hepatic lipid
peroxidation in female Sprague Dawley rats given a total of 70 µg/kg
body weight in three daily oral doses of 10, 20, and 40 µg TCDD/kg
body weight, or a single oral dose of 80 µg TCDD/kg body weight. Lipid
peroxidation was determined at days 1, 6, and 11 after the last
treatment. The maximal increase of lipid peroxidation in vivo was
2-fold one day post-treatment, whereas the 5- to 6-fold increase in
in vitro lipid peroxidation reached its maximum at 6 days
post-treatment. The TCDD-induced in vitro lipid peroxidation could
be inhibited by repeated treatment with BHA, glutathione, vitamin E,
and vitamin A (Hassan et al., 1985a,b). Dietary selenium had no
inhibitory effect on TCDD-induced lipid peroxidation (Hassan et al.,
1985c).
Robertson et al. (1985) found no evidence for TCDD-induced in
vivo lipid peroxidation, as judged by levels of exhaled endogenous
ethane, and metabolic clearance of both externally and internally
applied exogenous ethane in male Sprague Dawley rats after a single ip
dose of 60 µg TCDD/kg body weight. Neither was there a correlation
between TCDD-induced lipid peroxidation in vitro and sensitivity
towards the lethal effect of TCDD in Sprague Dawley rats, Golden
Syrian hamsters or guinea-pigs (Hassan et al., 1983). Hepatic
microsomes from Sprague Dawley rats and Golden Syrian hamsters exposed
to TCDD in vitro responded with increased lipid peroxidation only
in the presence of Fe3+-ADP in the incubation mixture (Albro et al.,
1986).
From all these data on TCDD and lipid peroxidation, it was
concluded by Albro et al. (1986) that it was premature to attempt to
define a relationship between lipid peroxidation and TCDD-induced
lethality.
8. EFFECT OF PCDDs ON HUMAN BEINGS - EPIDEMIOLOGICAL AND
CASE STUDIES
8.1 Occupational Studies - Historical Perspective
The illness most frequently observed in workers engaged in the
manufacture of trichlorophenol, 2,4,5-T, and related products is a
skin disease called chloracne. This skin disease has also been called
"Pernakrankheit" (perchlorinated naphthalene illness or halogen wax
acne) and was described by Herxheimer (1899). In addition to the
halogenated phenols, chloracne is caused by a number of chlorinated
compounds such as the chlorinated biphenyls and chlorinated
naphthalenes (Muller, 1937; Braun, 1955; Crow, 1970; Kimbrough, 1974).
Although chloracne is well known to those engaged in the
treatment of occupational diseases, many outbreaks that have occurred
over the years, particularly in the USA, have not been reported in the
scientific literature. In the Federal Republic of Germany, chloracne
is now considered an occupational disease for which compensation is
mandatory (Braun, 1970).
Herxheimer (1899) also described general toxic signs and symptoms
in his patients, such as lack of appetite, weight loss, headache, and
vertigo. After his original observations and publication, several
other reports followed. The technique of obtaining chlorine gas
consisted of an electrolytic procedure where a mixture of potassium,
sodium, and magnesium chlorides was subjected to a current with a
central carbon electrode where the chlorine was obtained and piped
off. The workers who took care of the chlorine gas never developed
chloracne thus refuting the original hypothesis by Herxheimer. By
contrast, those who handled the electrodes and cleaned the reaction
vessels were those afflicted with chloracne. Already at this time
chlorinated phenolic compounds were considered as possible noxious
agents (Fraenkel, 1902). This however could never be proven and even
at present, when satisfactory analytical techniques are now available,
no analysis of the so-called "tuffy tar" has been carried out.
Another class of chlorinated organic compounds causing skin
damage appeared during the First World War (1914-1918). At this time
perchlorinated naphthalenes had come into use as insulation materials,
e.g., in the radio and electronic industry. The first description of
Pernakrankheit was that by Wauer (1918). The use of the unspecified
technical mixture of chlorinated naphthalenes spread all over the
world and caused numerous intoxications notably among workers in
manufacture. The perna disease has been summarized by von Wedel et al.
(1943) and described in particular detailed by Braun (1955). Apart
from chloracne the systemic effects of the same compounds have been
dealt with by Drinker et al. (1937) and Greenburg et al. (1939).
Both in man and experimental animals, serious liver damage
occurred after exposure to chlorinated naphthalenes, consisting of
liver necrosis and toxic jaundice (acute yellow liver atrophy). Among
several hundred cases of chloracne due to these compounds, Braun
(1955) tabulated 24 deaths due to toxic jaundice and 14 recoveries. It
should be pointed out that a fulminant liver disease with jaundice of
this kind is an extremely rare condition. For comparison, it has never
occurred after exposure to trichlorophenol (TCP) and TCDD as described
below. Note should also be taken of the fact that not only were the
perchlorinated naphthalenes an ill identified mixture of chemical
species, but exposure frequently occurred at the same time to mixtures
of chlorinated biphenyls, the latter now known to be contaminated with
chlorinated dibenzofurans. The potentiation of toxicity by these
mixtures and other chlorinated compounds were discussed by Drinker et
al. (1937), Greenburg et al. (1939), von Wedel et al. (1943), and
Risse-Sunderman (1959).
Several accidental ingestions of chloracnegenic compounds have
occurred. They are of particular importance in relation to discussions
on whether chloracne is a systemic or local disease. The so-called
Yusho disease is discussed in section 11.
Herzberg (1947) described several cases of chloracne, in which
other toxic signs and symptoms were seen, due to consumption of
"chlorinated paraffin" used as a substitute for butter during cooking
in postwar Berlin. Among general signs and symptoms observed were
gastrointestinal disturbances with abdominal pain, headache, pain in
joints, neuropathy, depression, and lack of appetite. The
dermatological symptoms were erythema, exanthema, comedones, and
retention cysts in sebaceous glands. It was noted as remarkable that
the skin signs had a follicular predilection, as in seborrhoea (face,
head, bosom, and back). The slow development of chloracne, and
particularily the fact that the sebaceous glands were affected, led
the author to conclude that it was a secretory disease
(Ausscheidungstoxikose). With regard to the chloracnegenic component,
it is unlikely that paraffin itself was active. Herzberg speculated
that something else, possibly a pyrolysis product that arose during
cooking, could have caused the disease.
Accidents in chemical plants involved in the manufacture of
chlorinated phenolic compounds are listed in Table 63. It should be
stressed that all these intoxications are due to mixtures, e.g., TCP
and TCDD and other compounds. Summaries of the industrial accidents
are to be found in Holmstedt (1980) and only some of them will be
dealt with here.
Table 63. Summary of accidents in chemical plants involving the manufacture of chlorinated phenolic compounds
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
Germany 1910 CP Explosion + 5 Same year Teleky (1913), Wahle (1914),
occupational Dohmeier & Janson (1983)
United States 1949 TCP Explosion + 228 30 Ashe & Suskind (1949, 1950),
Occupational Suskind et al. (1953),
Suskind (1978), Huff et al.
(1980), Zack & Suskind (1980)
Zack & Gaffey (1983), Moses
et al. (1984), Suskind &
Hertzberg (1984)
Federal Republic TCP Occupational 17 1 Baader et al. (1951)
of Germany 1949 (PCP)
Federal Republic TCP Occupational 60 Bauer et al. (1961)
of Germany 1952
Federal Republic TCP Occupational 37 Hay (1977)
of Germany 1952-1953
Federal Republic TCP Explosion + 75 29 Hoffman (1957), Goldmann (1972,
of Germany 1953 Occupational 1973), Huff et al. (1980), Thiess
et al. (1982)
France 1953 TCP Explosion + 17 2 Dugois & Colomb (1956, 1957),
Occupational Dugois et al. (1958)
Federal Republic TCP, Occupational 31 24 Schultz (1957a,b), Bauer et al.
of Germany 1954 2,4,5-T (1961), Kimmig & Schultz (1957a,b)
Kleu & Göltz (1971), von Krause &
Brassow (1978)
Table 63 (contd - 2).
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
Federal Republic TCP Occupational 24 4 Risse-Sundermann (1959)
of Germany 1954 2,4,5-T
United States 1956 2,4,5-T Occupational 48 6 Bleiberg et al. (1964), Poland
2,4,5-T et al. (1971)
United States 1956 TCP Occupational Many Hay (1977)
Italy 1959 TCP Explosion + 5 2 Hofman et al. (1962)
Occupational
United States 1959 TCP Occupational Hay (1977)
United States 1960 TCP Occupational Many Hay (1977)
Netherlands 1963 TCP Explosion 106 11 Dalderup, (1974a,b), Berlin
2,4,5-T et al. (1976), Huff et al. (1980)
USSR 1964 2,4,5-T Occupational 128 Telegina et al. (1970), IARC
(1977)
United States 1964 TCP Occupational 61 6 Vahrenholt (1977), Cook et al.
(1980), Ott et al. (1980)
Czechoslovakia 1964-1969 TCP Occupational 80 6 Jirásek et al. (1973, 1976),
Pazderova et al. (1974, 1980,
1981)
United Kingdom 1968 TCP Explosion 90 14 May (1973, 1982), Huff et al.
(1980)
Japan 1970 2,4,5-T Occupational 25 3 Mivra et al. (1974)
Table 63 (contd - 3).
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
USSR 1972 TCP Occupational 1 1 Zelikov & Danilov
(1974)
Austria 1972-1973 2,4,5-T Occupational 50 Forth (1977), Hay
(1977)
Federal Republic 2,4,5-T Occupational 5 Forth (1977), Hay
of Germany 1974 (1977)
Italy 1976 TCP Explosion 193 8 Reggiani (1977, 1978, 1983a),
Vahrenholt (1977), Filippini
et al. (1981), Abate et al.
(1982), Ideo et al. (1982)
a Products: TCP = trichlorophenols; CP = chlorophenols; PCP = pentachlorophenol;
2,4,5-T = 2,4,5-trichlorophenoxyacetic acid.
The first reported intoxication with a mixture probably
containing TCDD, although the chemical structure was not given,
occurred in February 1910. Five people were said to have been
contaminated after a reactor explosion and two of these were described
in some detail in a dermatological thesis (Teleky, 1913; Wahle 1914).
Wahle (1914), however, in his thesis emphazised that this intoxication
was not due to any of the chlorinated naphthalene derivatives that
were well known by then.
An industrial poisoning was reported in 1949, due to the
formation of TCDD in uncontrolled exothermic reactions occurring
during the manufacture of TCP at a 2,4,5-T-producing factory in Nitro,
West Virginia, USA. The temperature in one of the reactors containing
tetrachlorobenzene, methanol, and sodium hydroxide increased, a relief
valve opened, and the contents of the vessel were discharged into the
interior of the building and over a wide area outside of the building.
A total of 228 people were affected.
Symptoms included chloracne, nausea, vomiting, headaches, severe
muscular aches and pains, fatigue, emotional instability, and
intolerance to cold. Laboratory findings showed an increase in total
serum lipids and an initially prolonged prothrombin time. Among those
affected were not only workmen, but also laboratory personnel, medical
personnel, and even the Safety Director who visited the area of
exposure. Several wives who had never visited the plant also developed
acne, usually at the same time as their husbands working at the plant.
A man from the nearby town who purchased a truck that was parked in
the vicinity of the accident at the time it occurred, and his child,
also developed chloracne. The disabling symptoms, which kept men from
their jobs for as long as 2 years, were severe aches and pains and
fatigability, the manifestations of peripheral neuropathy. Liver tests
4 years later were normal, but mild cases of acne were common. TCDD
was still an unknown chemical. The follow-up to this accident will be
discussed in section 8.4.
In 1953, at the Badische Anilin and Soda Fabrik, during the
alkaline hydrolysis of 1,2,4,5-tetrachlorobenzene to
2,4,5-trichlorophenol, the temperature and pressure in an autoclave
increased rapidly and resulted in an exothermic reaction releasing a
great deal of steam through a safety valve of the reaction vessel.
This steam covered the walls, windows, doors, and machinery in the
rooms of four floors, and finally precipitated in solid form on
everything in these rooms. Forty-two workers involved in the clean-up
operations developed chloracne, and even after the extensive clean-up
operations occasional workers still developed chloracne. Thereafter
the autoclaves were used for 2 years without incident but in 1958 a
mechanic who conducted repair work on an autoclave subsequently
developed chloracne (Hofmann, 1957; Goldmann, 1972). In 1968 and 1969
the building containing the autoclaves was dismantled. Goldmann (1972,
1973) conducted a study of the 42 workers exposed in this accident. In
21 cases, the chloracne was preceded by a non-specific dermatitis and
in two cases very persistent chronic conjunctivitis and blepharitis
were observed; 14 cases also showed involvement of other organs. In
four instances the liver was affected, and microscopic examination of
the liver again showed a very characteristic grey pigment that did not
stain positive for iron. A transient involvement of the myocardium was
also noted. In five instances the upper respiratory tract was involved
with tracheitis and bronchitis. There was one instance of haemorrhagic
pleuritis and one instance of afebrile gingivitis and stomatitis. In
a number of cases a high susceptibility to infection was noted,
sometimes accompanied by a decrease in gamma-globulin. One worker died
of pancreatitis, in seven cases the central nervous system was
affected, three instances of toxic polyneuritis were recorded, and in
two instances hearing, sense of smell, and taste were impaired. The
child of one of these workers also developed chloracne, and in most of
the workers active chloracne persisted for many years - in one
instance for 18 years. Follow-up studies are described in section 9.4.
Of particular interest is a study by Risse-Sundermann (1959).
According to oral reports by the treating physician, all 24 members of
a team working in a trichlorophenol operation became ill after the
production process was switched to the pressurized phenol process in
the spring and summer of 1954. Slightly different acneiform skin
conditions appeared as symptoms of the toxic exposure. In addition,
the patients suffered from dizziness, nausea, vomiting, lacrimation,
burning of the eyes, difficulty in hearing, gastrointestinal spasms,
intolerance to fatty foods, diarrhoea, jaundice, hepatitis (which was
fatal in one case), and paresthesias and hyperesthesias, as well as
extreme irritability. One patient became psychotic and committed
suicide. In addition, some of the patients complained of impotence.
Ten workers at this chemical factory were followed for five years
by Risse-Sundermann (1959). In addition to the signs and symptoms
mentioned above, she noticed swollen lymph glands and a considerable
decrease in body weight. The patients underwent neurological
examination, with no objective signs being observable. Of particular
interest in this well documented study is the fact that in three
patients the general symptoms (e.g., tiredness, depression, lack of
appetite, stomach pains, sexual dysfunction) preceded that of the skin
manifestations .
Bauer et al. (1961) reported a study of workers affected by three
different outbreaks of chloracne. In this study more than 100 workers
were examined. Of these, 31 Hamburg workers had been exposed 5 years
ealier. Nine were examined in detail and their symptoms tabulated.
Initially, there was dermatitis and irritation of the face, sometimes
accompanied by conjunctivitis, and followed by the gradual development
of chloracne and patchy pigmentation of the skin. In some cases
irritations of the mucous membranes of the face and upper respiratory
tract, together with a persistent blepharoconjunctivitis, were also
noted. In the follow-up study, a number of cases of liver injury were
observed and, at liver biopsy, a typical grey pigment was observed in
liver sections, which did not stain positive for iron. Viral hepatitis
was suspected. In a few cases, chronic bronchitis and occasional
myocardial damage were also observed. In all cases, fatigue was the
main complaint and muscle weakness and muscle pain were described by
the workers, particularly in the proximal muscles of the lower
extremities. All nine also reported decreased libido. In a few
instances, paresthesia and hyperesthesia or pronounced sensory
neuropathy were observed, and minor circumscribed pareses were found.
A psychovegetative syndrome occurred in most of the workers. Other
signs recorded were: inability to concentrate, memory deficits, sleep
disturbances, particularly increased somnolence, decreased drive, and
alcohol intolerance. Psychological tests also showed abnormalities.
Following the malfunction of a reaction vessel in northern Italy,
in which 2,4,5-trichlorophenol was produced, the temperature in the
vessel increased rapidly and an intense black vapour filled the
work-room covering everything with a black deposit. Five workers
engaged in clean-up operations developed chloracne (Hofmann &
Meneghini, 1962). None of those involved in the clean-up exhibited any
involvement of general systemic toxicity (even after 16 months) that
could be related to exposure to the tar and soot. However, one
15-year-old worker developed folliculitis and superficial nodular
elements on the face a few days after initial exposure. A slow but
progressive generalization of the dermatosis developed on the trunk,
scalp, and lower extremities. An examination several months later
revealed no damage to the renal or liver parenchyma. However, this
worker was found to still suffer from outbursts of chloracne in 1980
(Holmstedt, 1980).
Duverne et al. (1964) reported a case that occurred at a plant at
Lyon, France, where products that used 2,4,5-tri-chlorophenol as a
starting material were manufactured. This worker developed chloracne
as well as serofibrinous pleuritis.
Ten workers also developed chloracne at a plant near Grenoble,
France, which produced 2,4,5-trichlorophenol that served as the
starting material for phenoxy pesticides and germicides for cosmetics.
These workers showed symptoms of systemic poisoning similar to those
reported by Goldmann (1972), and hepatic insufficiency with lipaemia
and elevated serum cholesterol levels (Dugois & Colomb, 1956). Another
accident resulting in TCDD exposure of workers occurred in the same
factory in 1966 (Dugois et al., 1967).
An exothermic reaction resulted in an explosion at a plant in
Chesterfield, England, in 1968. The company made 2,4,5-trichlorophenol
from tetrachlorobenzene and the explosion occurred during the process
involving ethylene glycol and caustic soda under atmospheric pressure
(Milnes, 1971). In this incident, 79 workers developed chloracne but
there was no evidence of systemic illness (May, 1973). In 1971, 3
years after the explosion, two workers who had not been involved in
the explosion or its aftermath were employed as pipe-fitters at a new
installation, away from the site of the explosion, to refit one of the
cleaned tanks. They both developed severe chloracne, and the son of
one of these workers and the wife of the other also developed this
condition (Jensen & Walker, 1972). May (1973) cited two incidents
involving explosions in a similar process. In the first incident,
fatal injuries were recorded; in the second incident, all 50 exposed
persons fell ill after 10 days and had liver injury.
In the USA, an outbreak of chloracne occurred among workers
manufacturing 2,4-dichlorophenoxyacetic acid and
2,4,5-trichlorophenoxyacetic acid (Bleiberg et al., 1964); 29 workers
developed chloracne and 11 of these had elevated urinary uroporphyrins
and exhibited varying degrees of acquired porphyria cutanea tarda. At
least one of these workers had abnormal liver-function tests and
microscopic examination of a liver biopsy specimen showed parenchymal
cell regeneration and haemofuscin pigment. Many of the workers with
chloracne showed hyperpigmentation of the skin. A second study of the
workers at this plant was conducted in 1969 by Poland et al. (1971).
A total of 73 male employees were examined, and moderate to severe
chloracne was found in 13 workers (18%), mild chloracne in 35 (48%),
hyperpigmentation in 30, and uroporphyrinuria in 1. No definite
systemic illness could be documented in these workers. Of those
studied, 33 had been employed at the plant for 0-4 years, 10 for 4-8
years, and 30 for more than 9 years. The mean duration of employment
was 8.3 ± 7.6 years (mean ± 1 SD). The trichlorophenol manufactured in
this plant contained 10-25 mg TCDD/kg. Twenty-six of the workers seen
by Bleiberg et al. (1964) were also seen in a follow-up study. Six
months prior to the second survey (Poland et al., 1971), the
manufacturing process was altered so that the 2,4,5-T produced
contained less than 1 mg TCDD/kg.
In 1964, in the USSR, many workers developed chloracne while
engaged in producing 2,4,5-T. Production was then discontinued
(Telegina & Bikbulatova, 1970).
On 10 July 1976, an explosion occurred at the ICMESA plant at
Meda, near Seveso, Italy, when 12 workers were present. All 176
workers of the plant were examined 3 or 4 weeks after the accident.
Chloracne was suspected in 1 of them; the others showed minor symptoms
that could not be correlated with exposure. Alkaline phosphatase and
delta-glutamyltransferase seemed slightly increased in 32 and 37
cases, respectively, while five workers showed a reduction in their
delta-aminolevulinic acid dehydratase blood levels, and three showed
moderately increased urinary gamma-aminolevulinic acid (Zedda et al.,
1976). Similar findings were reported by Fara et al. (1976) and
Reggiani (1978). The follow-up of the general population is described
in section 8.2.
8.2 General Population Studies
8.2.1 Missouri, USA
Environmental exposures have occurred in a small area of
Missouri, USA (Carter et al., 1975; Kimbrough et al., 1977; Kimbrough,
1984). In the summer of 1971 many birds, rodents, cats, dogs, insects,
and horses died after exposure in a horse arena in eastern Missouri.
The incident followed the spraying of "waste oil" on the horse arena
for dust control. Within 3 weeks of the spraying of this arena, two
other arenas were sprayed. In all, 57 adult horses died, 26 abortions
occurred among the horses at the most heavily exposed farm and many
foals died soon after birth. At the time, the nature of the chemical
that had caused the problem was unknown. The arenas were excavated and
the contaminated dirt dumped at other sites. After many fruitless
attempts to identify the cause of this outbreak, it was discovered in
1973-1974 that the original soil from one of the arenas contained
5600-6500 mg trichlorophenol/kg, 31.8-33.0 mg TCDD/kg, and 1350-1590
mg polychlorinated biphenyls/kg. Because of this finding, the episode
was reinvestigated. It was found that the salvage oil company that
sprayed the three arenas routinely collected discarded motor oil and
lubricants from over 2000 service stations in eastern Missouri and
southwestern Illinois. It also collected, from various sources, a
limited amount of used organic solvents such as transformer oils and
other compounds. A company in southwestern Missouri was finally
identified as a source of TCDD. This company had manufactured
trichlorophenol as an intermediate for hexachlorophene. The production
of 2,4,5-tri-chlorophenol had generated a distillate residue which was
emptied once a week into a residue storage tank. Initially this
chemical waste was collected and incinerated but, in 1971 when the
trichlorophenol producer experienced a financial crisis, he arranged
for the chemical wastes to be disposed of by a chemical supplier. The
chemical supplier subcontracted the chemical waste disposal to the
salvage oil dealer. The salvage oil dealer added the toxic chemical
waste to his salvage oil storage tank, having collected a total of 18
000 gallons. This material, mixed with salvage oil and other
chemicals, was sprayed on the riding arenas and some of it was taken
to re-refining companies. Soil samples from arenas where contaminated
dirt had been dumped in 1974 contained trichlorophenol levels that
ranged from 1.5-32.6 mg/kg, TCDD levels that ranged from 0.22-0.85
mg/kg, and polychlorinated biphenyl levels that ranged from 10-25
mg/kg.
A 6-year-old girl, who had used one of the arenas for
sandbox-like play in 1971, developed epistaxis, headache, diarrhoea
and lethargy, haemorrhagic cystitis, and signs of pyelonephritis. She
had an uneventful recovery. Three other females exposed to the same
arena had recurring headaches, skin lesions, and polyarthralgias. Two
3-year-old boys in another arena developed chloracne on the exposed
skin surfaces which lasted for more than a year. Evaluation of the
three female patients 5.4 years after exposure to TCDD-containing oil
showed them to be in good health (Beale et al., 1977).
A comprehensive medical examination of 154 residents exposed to
TCDD, and 155 unexposed residents in similar type housing in eastern
Missouri, revealed no consistent differences between the two groups.
The examination included a medical history, physical examination,
serum and urinary chemistries, and immunological and neurological
tests. The findings may suggest that long-term TCDD exposure is
associated with depressed cell-mediated immunity (decreased
delayed-type hypersensitivity skin reactions to standard antigens)
(Hoffman et al., 1986; Stehr et al., 1986). Urinary concentrations of
glucaric acid were not significantly different between persons
identified as being at high or low risk (Steinberg et al., 1985).
8.2.2 Seveso, Italy
The scientific follow-up on the population of Seveso, N. Italy,
which had been accidentally exposed to TCDD in 1976 (see sections
4.1.1 and 8.1), was guided by an international steering group headed
by Professor M.A. Klingberg. The group completed its work in February
1984 and concluded that "it is obvious that no clear-cut adverse
health effects attributable to TCDD, besides chloracne, have been
observed" (Regione Lombardia, 1984). A total of 193 people had
displayed symptoms of chloracne, but at the beginning of 1984 only 20
presented active symptoms. After 15-20 days exposure to TCDD soil
levels of 270-1200 µg/m2, there was a marked incidence of chloracne.
No disturbance of biochemical functions were seen when the exposure
had been limited to soil with TCDD levels at or below 30-70 µg/m2.
Later evaluations failed to confirm earlier findings of a decrease in
motor nerve conduction velocity in some individuals. A significant
increase in urinary glucaric acid levels, indicating increased
microsomal enzyme activity, was found 3 years after exposure in 67
exposed children, as compared to 86 non-exposed children (Ideo et al.,
1982, 1985). The steering group found the data difficult to evaluate
as analytical and individual biological variabilities were not
explained (Regione Lombardia, 1984).
Studies performed on the rate of spontaneous abortions and birth
defects in the Seveso area do not allow any conclusions to be drawn
(Tognoni & Bonaccorsi, 1982). The hypothesis that low exposure might
cause pre-pregnancy or pregnancy effects that adversely affect the
outcome was tested using several exposure models. The only finding was
a slightly higher rate of haemangioma among newborns in the exposed
group. However, this showed up only with one of the exposure models.
It was considered doubtful that this was due to TCDD exposure (Regione
Lombardia, 1984).
Lymphocytes from inhabitants of Seveso were examined for
chromosomal aberrations by Regianni (1980a,b) and Mottura et al.
(1981). In 17 TCDD-exposed individuals examined within two weeks of
the accident, no increase in chromosomal aberrations was observed
(Regianni, 1980). In the abstract by Mottura et al. (1981),
chromosomal aberration analysis was performed on subjects distributed
into three classes: acute exposure, chronic exposure, and a control
group of non-exposed subjects. No significant difference in the
frequency of chromosomal aberrations in the three exposure categories
was reported. Data on number of subjects, chromosomal aberrations, and
exposure level and time were not given.
Tenchini et al. (1983) published a comparative cytogenetic study
on induced abortions from women exposed to TCDD after the Seveso
accident, and in non-exposed subjects. Chromosome analysis was
performed on maternal peripheral blood, placental and umbilical cord
tissues, and fetal tissues. No significant differences were found in
the level of chromosomal aberrations in the blood of placenta and
umbilical cord from TCDD-exposed and non-exposed women. The exception
was fetal samples from non-exposed women, in which a significant
increase in chromosomal aberrations was obtained, possibly an artefact
due to experimental techniques. The effect of TCDD on fetal
chromosomes is therefore still unclear.
Several epidemiological follow-up studies are continuing in and
around Seveso.
8.2.3 Viet Nam
From 1960 to 1969 a mixture of 2,4-dichlorophenoxyacetic acid and
2,4,5-trichlorophenoxyacetic acid (Agent Orange), which was
contaminated with TCDD (concentrations ranging from 0.5 to 47 mg/kg)
(Kearny et al., 1972), was sprayed over areas of Viet Nam as a
defoliant. The spraying from 1960 to 1965 was minimal; in 1966 it
covered slightly more than 800 000 acres, in 1967 almost 1.7 million
acres, in 1968 over 1.3 million acres, and in 1969 1.2 million acres.
Studies have been carried out since the early 1970s to ascertain
whether the exposure of the general population in Viet Nam to this
herbicide could have resulted in an increased incidence of birth
defects. However, the results of such investigations have not been
published in readily available peer-reviewed journals, making it
difficult to assess the scientific significance of the findings. Such
studies have been reviewed by Westing (1984) and Constable & Hatch
(1985). Those studies reviewed indicate a range of effects including
spontaneous abortions, infertility, and birth defects. However, there
are marked deficiencies in experimental design in most, if not all,
studies, including potential bias in the selection of populations,
poor record-keeping of populations and biological effects, such as
congenital malformations, and a lack of control over possible
confounding factors. These deficiencies make it difficult, if not
impossible, to use this body of data in assessing the human health
risks from exposure to phenoxyherbicides contaminated with TCDD and
other PCDDs.
Tung (1973) reported an increased incidence of liver tumours in
Viet Nam. From 1955 to 1961 there were 159 cases of liver cancer out
of a total of 5492 cancer cases, and from 1962 to 1968, 791 out of a
total of 7911 cancer cases. Van (1984) continued Tung's investigation.
Previous exposure to herbicides of 21 male cases of primary liver
cancer and 42 controls was ascertained. Six of the 21 cases and three
of the controls had lived or worked in areas sprayed with herbicides
or had moved there shortly after spraying ceased. Residence time
varied from 8 to 77 months. There is a lack of information on
confounding factors and there was a chance for bias in these studies.
In general, the possibility of exposure to multiple chemicals and the
short latency period noted make the study by Van (1984) of little
value for assessing risk (IARC, 1986).
In 1979, the United States Air Force (USAF) initiated an
epidemiological study into the possible health effects from chemical
exposure of Air Force personnel who conducted aerial dissemination of
herbicide in Viet Nam (Operation Ranch Hand) (Lathrop et al., 1984).
The purpose of this investigation was to determine whether long-term
health effects exist and can be attributed to occupational exposure to
herbicides. This study used a matched cohort design in a
non-concurrent prospective setting, incorporating mortality,
morbidity, and follow-up studies. The report presented the results of
health information on 2706 Ranch Handers and comparison individuals
obtained by questionnaire and 2269 Ranch Handers and comparison
individuals undergoing an extensive physical examination. It was
concluded that there was insufficient evidence to support a cause and
effect relationship between herbicide exposure and adverse health in
the Ranch Hand group at this time. The study disclosed numerous
medical findings, mostly of a minor or undetermined nature, that
require detailed follow-up.
In a study of 15 soldiers in Australia exposed to Agent Orange,
no increases in structural chromosomal aberrations or sister chromatid
exchanges were noticed, compared to a control group of 8 subjects
(Mulcahy, 1980). In 1980 the Australian Commonwealth Institute of
Health agreed to conduct a series of scientific investigations into
the health of Viet Nam veterans and their families. After
considerating the most appropriate study programme, it was decided in
1981 to conduct, as part of that programme, a case-control study of
congenital anomalies and Viet Nam service (Donovan et al., 1984). The
report is largely negative, as is that of Erickson et al. (1984) which
reported a similar study of American veterans.
Table 64. Signs and symptoms reported in association with human exposure
to TCDD or mixtures containing TCDD
A. Skin Manifestations C. Neurological Effects
1. Chloracne 1. Sexual dysfunction
2. Hyperkeratosis 2. Headache
3. Hyperpigmentation 3. Neuropathy
4. Hirsutism 4. Sight disturbance
5. Elastosis 5. Loss of hearing, taste,
and smell
B. Systemic Effects D. Psychiatric Effects
1. Mild fibrosis of liver 1. Sleep disturbance
2. Raised transaminase values 2. Depression
in blood 3. Loss of energy and drive
3. Hypercholesterolaemia 4. Uncharacteristic bouts of anger
4. Hypertriglyceridaemia
5. Loss of appetite and weight loss
6. Digestive disorders (intolerance to
alcohol or fatty food, flatulence,
nausea, vomiting, diarrhoea)
7. Muscular aches and pains, joint
pain, lower extremity weakness
8. Swollen lymph glands
9. Cardiovascular, urinary tract,
respiratory, and pancreatic disorders
8.3 Signs and Symptoms in Humans Associated With TCDD Exposure
Many signs and symptoms have been reported in studies of human
exposures to PCDDs, both occupationally and from the general
environment. These have been compiled from the various studies and are
shown in Table 64.
8.3.1 Skin manifestations
Chloracne is a sign of exposure to several chlorinated cyclic
organic compounds, the most potent being TCDD. Chloracne thus may
serve as a marker of such exposure. The most distinctive lesion in
chloracne is the so-called cyst, a skin-coloured elevation that may
measure from 1 mm to 1 cm in diameter, with a central opening that may
be difficult to detect. Comedones that contain black or
black-appearing material in their openings are also present. There may
be a secondary inflammatory reaction, melanosis, and hyperkeratosis,
and these skin changes may be preceded by a "cable rash" or "cable
itch". These skin lesions resemble photosensitivity reactions and the
bearers may suffer severe pruritus. Microscopic examination of the
skin lesions shows marked dilatation of the hair follicles which are
filled with keratinous material, the sebaceous glands may be partly or
completely atrophied and, occasionally, hyperplasia of these glands
has also been reported. Hyperkeratosis and acanthosis of the
surrounding epidermis usually accompany these lesions. Atrophy of the
epithelium and thinning of the epithelial walls surrounding these
keratinous cysts are observed at a later stage of the disease. If the
follicular cysts rupture, foreign body granulomata may also be
observed. Healing of these skin lesions usually results in deeply
pitted scars. The distribution of chloracne is predominantly facial,
affecting in particular the malar areas, the jaws, and the regions
behind the ears. At times it may involve the ear canal and, with
increasing severity, also the rest of the face and neck. In more
extensive cases, the outer upper arms, neck, back, abdomen, outer
thighs, and genitalia may also be involved (Crow, 1970).
While the absence of chloracne does not absolutely rule out
exposure to TCDD, it usually indicates that there has been no exposure
to a toxic dose of the substance. "Toxic" is used here to indicate
both systemic and local effects. Where there has been exposure to TCDD
and chloracne has resulted, it is the only known clinical sign that
persists for a long period of time, even for the remainder of the
exposed person's life time. In a large group of people exposed to
mixtures containing TCDD, the absence of chloracne usually indicates
that exposure to a toxic dose was unlikely and also makes it unlikely
that severe, persistent systemic disorders will result.
Hyperkeratosis is a fairly common phenomenon whereas
hyperpigmentation and hirsutism are rare. It should be noted that
hyperkeratosis is prominent in the exposed Seveso children who have no
affected sebaceous glands. These glands develop only at puberty.
Elastosis of the skin has been noted as a long-term effect of
TCDD exposure.
8.3.2 Systemic effects
Liver effects following exposure to PCDDs have been diagnosed
even by histological examination, and account for temporarily raised
transaminases in blood, hypercholesteraemia, and
hypertriglyceridaemia. Bauer et al. (1961) and Risse-Sundermann (1959)
do not however exclude viral hepatitis as a cause of such findings in
their patients exposed to TCDD. Loss of appetite, weight loss, and
digestive disorders are common complaints from humans exposed to
either TCDD itself, or to technical mixtures containing TCDD. Muscular
aches and pain and weakness in extremities have been reported,
particularly after exposure to technical mixtures containing TCDD.
Swollen lymph nodes have also been reported, both after exposure to
"pure" TCDD and to mixtures. The cardiovascular, urinary tract,
respiratory, and pancreatic disorders reported are of doubtful
significance with regard to a causal relationship to TCDD exposure.
Porphyria cutanea tarda has been reported in two cases of
occupational exposure where chlorinated organic compounds were
manufactured in addition to trichlorophenol. These were the incidents
at the factory of Diamond Alkali, Newark, New Jersey, USA, in 1956
(Poland et al., 1971) and at Spolana, Czechoslovakia, between 1964 and
1969 (Pazerova-Vejlupkova et al., 1981). The porphyria cutanea tarda
observed in these cases was very unlikely to have been induced by
exposure to TCDD but rather by exposure to other chlorinated organic
compounds manufactured in these plants (Jones & Chelsky, 1986).
8.3.3 Neurological effects
Sexual dysfunction (lack of libido and impotence) has been
reported after acute exposure to both "pure" TCDD and technical
mixtures (Schulz, 1968). The frequency of its occurrence may have been
underestimated to date. Headache is a frequent symptom after exposures
to technical mixtures containing TCDD.
Sensory neuropathy has been noted in many instances. Usually
workers in the initial stages of exposure will complain of pains in
their joints after they have very acute severe chloracne; however,
there are usually no abnormal physical findings in the joints, but the
complaints may continue. In early studies of workers affected by TCDD,
no attempts were made to objectively measure the effects on the
sensory nervous system. Tests have now been developed that evaluate
sensory nerves and that can be used in future field studies. The nerve
conduction tests, which primarily have been used so far, are actually
not very useful to measure neuropathy. Differences in nerve conduction
were shown among residents from Seveso, Italy, who had chloracne and
those who did not (Richert, von, 1962; Fillipini et al., 1981).
Sight disturbance may be related to alkaline exposure or to
conjunctivitis related to effects on the glands of Meibom. Loss of
hearing, taste, and smell have been reported in a few cases, but a
causal relationship to TCDD exposure is doubtful.
8.3.4 Psychiatric effects
The symptoms have been listed in Table 64 in what is believed to
be their order of frequency and degree of severity.
8.4 Epidemiological Studies
Signs and symptoms related to accidental exposure to TCDD are
given in Table 64. However, it should be observed that all the
accidents and occupational contamination concern exposure to a mixture
of compounds where TCDD was only one component. In all cases, its
concentration in the mixtures was unknown. Only two cases of
intoxication with "pure" TCDD have been reported.
The story of the discovery of TCDD is by now well documented
(Holmstedt, 1980; Sandermann, 1984a,b). TCDD was synthetized in 1955.
Four people were intoxicated, one co-worker severely so while drying
crystals. In all cases, decreased libido was the first symptom,
followed by other symptoms such as moderate to severe chloracne,
sleeping difficulties, inability to concentrate, depression, and, in
at least one case, swelling of the lymph nodes. In all cases, the
signs and symptoms disappeared within a couple of years, with the
exception of the chloracne in the most heavily exposed man.
The second occasion of exposure to what one must assume to be
pure TCDD is the one reported by Oliver (1975). The toxic effects on
three young scientists who suffered "transient minimal exposure to
TCDD" were described. Two of them suffered from typical chloracne.
Delayed symptoms about two years after initial exposure occurred in
two of the scientists. These symptoms were said to have included
personality changes, other neurological disturbances, and hirsutism.
All three scientists were found to have raised serum cholesterol
levels, but no other biochemical disturbances and no porphyrinuria or
liver damage were demonstrated. Whether the unusually delayed
physiological effects were in fact due to the initial dioxin exposure
is a question that was discussed by the author. Although conclusive
evidence is lacking, it seems likely that these delayed effects were
in fact due to dioxin intoxication. The conditions of exposure remain
unexplained.
Of the many cases of exposure reported in Table 63, only two (at
Monsanto in 1949 and at BASF in 1953) have been adequately followed up
epidemiologically with matched control groups.
The workers of Monsanto, USA, have been investigated several
times between 1949 and 1984. Immediately after the accident, Ashe &
Suskind (1949) hospitalized and studied four cases of severe poisoning
among the workers. These four workers were diagnosed as having
chloracne, but by the time of examination these men had recovered from
earlier symptoms of peripheral neuropathy. In 1950, a further
examination of these four workers and two additional men revealed
continued irritability, nervousness, and insomnia (Ashe & Suskind,
1950). A consistent loss of libido and some impotence was reported.
Further clinical examination revealed hepatomegaly, altered
prothrombin times, and disturbed lipid metabolism.
A further study of 36 workers from this plant was undertaken in
1953 (Suskind et al., 1953). It was noted that even those who
developed, to a moderate or severe degree, the skin eruptions, pains
in back, dyspnoea, fatigue, nervousness, and decreased libido
generally improved. Even those suffering the most severe cutaneous
eruptions initially had only a few or no lesions in 1953.
More recent studies on these workers are those of Zack & Suskind
(1980) and Zack & Gaffey (1983). Zack and Gaffey reported on a
121-member study cohort, with a presumptive high-peak exposure to TCDD
base on chloracne occurrence, which was followed for mortality until
1978. The entire cohort was traced; there were 32 deaths; 89 people
were still alive. There was no excess in total mortality or in deaths
from malignant neoplasms. The proportional mortality analysis of
decedents according to exposure by 2,4,5-trichlorophenoxyacetic acid
(2,4,5-T) indicated no unusual patterns of mortality. The proportional
mortality ratio (PMR) for malignant neoplasms was low (PMR = 82) in
the exposed group. Lung cancer was the only site among the malignant
neoplasms for which the value was somewhat higher in the exposed
group.
The Monsanto workers were again examined in 1984 (Suskind &
Herzberg, 1984). A clinical epidemiological study was conducted to
determine the long-term health effects of workplace exposures during
the process of manufacturing the herbicide 2,4,5-T, including
contaminants such as TCDD. The population consisted of two cohorts,
204 clearly exposed and 163 not exposed (controls). Among the exposed
workers, clinical evidence of chloracne persisted in 55.7%. None of
the controls experienced chloracne development. An association was
found between the persistence of chloracne and the presence and
severity of actinic elastosis of the skin. There was an association
between exposure and the history of ulcers of the gastrointestinal
tract. Pulmonary function values among those who were exposed and who
currently smoked were lower than those who were not exposed and who
currently smoked. No disturbances of sexual functions were found at
this time after age adjustment. The data assembled in the study
indicated no evidence of increased risk for cardio-vascular disease,
hepatic disease, renal damage, or central or peripheral nervous system
problems.
Another selection of the population from the same plant has been
examined by another group of epidemiologists (Moses et al., 1984).
Since the degree of exposure was unknown to these investigators and
since chloracne is generally considered a quite reliable indicator of
heavy dioxin exposure, it was decided to use chloracne as a
"surrogate" for exposure and to classify the study population by its
presence or absence. It was recognized that those without chloracne,
but with appropriate work-exposure history, might also have had TCDD
exposure and were not therefore used as "unexposed controls".
Chloracne was found in 52% of 226 workers in a 1979 cross-sectional
survey at the plant where 2,4,5-T had been manufactured from 1948 to
1969. Mean duration of residual chloracne was 26 years, and in 29
subjects it had been present for 30 years. A significantly increased
prevalence of abnormal gamma-glutamyl transpeptidase (GGT) and higher
mean GGT were found in those with chloracne compared to those without.
Although mean triglyceride values were higher in those with chloracne,
the difference was not statistically significant. Neurological
examination showed a statistically significant higher prevalence of
abnormal sensory findings in those with chloracne. Increased
prevalence of angina and reported myocardial infarction in those with
chloracne was not significant when age-adjusted. Increased prevalence
of reported sexual dysfunction and decreased libido in those with
chloracne, compared to those without, was statistically significant
after age adjustment. No differences were found between those with and
without chloracne in serum cholesterol, total urinary porphyrins, or
in reproductive outcomes. Exposure to TCDD in 2,4,5-T production may
thus result in apparently permanent changes in the skin. Sensory
changes in peripheral nerves and possible changes in liver metabolism
in those with current or past chloracne are also suggested by these
data. Based on worker histories, even severe acute toxicological
effects of TCDD are reversible, or improve markedly over time. While
the cross-sectional nature of this study, the low participation rate,
and the highly select nature of the population limit the conclusions
that can be drawn, it is unlikely that permanent, severe, and
debilitating toxicological sequelae are inevitable after exposure to
TCDD sufficient to produce chloracne. It must be noted, however, that
individual susceptibility may make certain workers with heavy
exposures more vulnerable.
The exposure of workers at BASF in 1953 has been the subject of
several reviews, the latest one being that of Thiess et al. (1982).
Twenty-seven years after the accident that occurred in the BASF
Ludwigshafen plant, a mortality study was undertaken of people exposed
in the uncontrolled reaction which occurred during the trichlorophenol
process. The follow-up was 100% successful and involved 74 people.
Overall mortality (21 deaths) did not differ in this group from the
rate expected in three external reference populations, or from that
observed in two internal comparison groups, where 18-20 deaths were
observed. Of the 21 deceased people, 7 had had cancer, compared with
4.1 expected. In addition, two other cases of cancer (one bronchial
carcinoma and one carcinoma of the prostate) were still alive at the
time of writing. Three deaths due to stomach cancer at ages 64, 66,
and 69 years, were found, compared with 0.6 expected from regional
mortality data. One stomach cancer occurred among 148 individuals in
the two comparison cohorts. The incidence of cancer in these workers
was considerably greater than expected and cannot be explained only as
a chance event. Of 74 people, 66 had severe chloracne or severe
dermatitis. There is a possibility that some members of the BASF
cohort were exposed to other unknown occupational hazards before or
after the accident. However, the use of two internal comparison groups
composed of matched controls from the same factory was designed to
control for, as far as possible, other occupational exposures that
could be important etiological or confounding factors. Because of the
small size of the cohort and the small absolute number of deaths from
any particular cause, the results of this study do not permit any
definite conclusions concerning the carcinogenic effect of exposure.
In comparison with the above-mentioned, well conducted long-term
epidemiological studies, a host of other follow-up studies have been
published, none of which used adequate controls. They are, therefore,
of less value but will be briefly summarized here.
Jirasek et al. (1973, 1976) and Pazderova et al. (1974, 1980,
1981) examined 55 of a total of 80 workers who suffered intoxication
during the manufacture of sodium pentachlorophenate and the sodium
salt and butyl ester of 2,4,5-T. One worker died from severe acute
intoxication at an early stage (Jirasek et al., 1976), and 76 workers
developed chloracne. The following additional symptoms were found:
porphyria cutanea tarda, disorders of the metabolism of lipids,
porphyrins, and carbohydrates, and alteration of plasma proteins.
Hepatic lesions were also present. Neurological and electromyographic
(EMG) examinations revealed peripheral nerve changes in 17 people,
first detected in 8 people during the second year of the study. A
neurasthenic syndrome was also observed. The patients with porphyria
cutanea tarda showed hyperpigmentation, hypertrichosis, and bullosis
actinica mechanica. Porphyrin excretion in urine ranged from 172 to
2230 µg/24 h. Polyneuropathies, confirmed by EMG examination, were
noted, predominantly in the lower extremities. In this outbreak, the
disease was progressive during the first 2 years; subsequently the
dermatological symptoms as well as the porphyric disease and the
neurological disorders improved. The impaired lipid metabolism
improved only very slowly.
In this plant, the toxic substances were led off through the
breathing zone of the workers. The concentrations of the chlorinated
hydrocarbons in the air were never measured. Due to insufficient data,
the real hygienic conditions at the work place could not be accurately
reconstructed. The manufacturing of 2,4,5-T was halted permanently in
1968 so that it was impossible to obtain the necessary information in
an adequate manner. From 1959 to 1964, according to information from
the plant, only sodium pentachlorophenate was manufactured. Not until
1965 was the manufacture of sodium 2,4,5-T commenced on a pilot scale,
and later the butyl ester of 2,4,5-T was also manufactured. After each
year of production, something was always changed or modified in the
process and technology so that actually there was never full-scale
production in the true sense of the word. Many of the herbicides
manufactured could not be found from the documentation (Pazderova et
al., 1974). The uncertain mixture of compounds involved in the Spolana
episode makes interpretation of signs and symptoms almost impossible.
In all likelihood the porphyria observed was due to the
hexachlorobenzene stated to be produced at this factory.
Signs of disturbance in the porphyrin metabolism in workers
manufacturing 2,4,5-T was also described by Poland et al. (1971).
Chloracne was not correlated significantly with job location within
the plant, duration of employment, or coproporphyrin excretion.
Although 11 subjects with uroporphyria and at least three with overt
porphyria cutanea tarda had been found in a study of the same plant
six years earlier (Bleiberg et al., 1964), no clinical porphyria could
be documented at the time of the second investigation, and only one
worker had persistent uroporphyrinuria. Evidence of toxicity in other
organ systems was markedly less than that reported in previous studies
and in most instances there was no difference from normal populations.
In all likelihood the porphyria cutanea tarda in this case, as in the
study from Czechoslovakia, was due to a compound other than TCDD. This
is corroborated by a recent re-evaluation of the literature (Jones &
Chelsky, 1986).
A study from northern Germany was published by Bauer et al.
(1961). It is not clear where the cases orginated and only nine
patients were studied in depth. A summary of the findings from these
patients, and another person suffering from chloracne after
occupational exposure to trichlorphenol, was reported by Kleu & Göltz
(1971). These patients were followed for 15 years after exposure. The
severity and types of symptoms varied in a dose-related manner. Major
complaints were decreased sexual activity, muscular weakness, easy
fatigability, irritability, and loss of appetite and memory. The
authors concluded that a permanent defect had occurred, the late form
of which resembled a cerebral involutionary syndrome, combined with
mental depression and neurasthenia.
A follow-up study of 11 of the 24 employees at Boehringer
exhibiting skin lesions in 1955 was published by von Krause & Brassow
(1978). Many continued to suffer from their earlier complaints. In
seven of the eleven, nausea and intolerance to heavy fatty food was
still common, and six men complained of alcohol intolerance. Although
conjunctivitis had disappeared, chloracne was still clearly visible in
most of the 11 subjects. Neurological problems were still severe in
six of the workers.
Ten years after the incident at Coalite in 1968 when 79 workers
developed chloracne due to exposure to a chlorophenol-TCDD mixture, a
study was undertaken to establish the state of health of the affected
employees (46) remaining in the company's employment (May, 1982).
Forty-one of the 46 employees participated. The opportunity was used
to examine effects on mortality, morbidity, carcinogenesis,
reproduction, teratogenicity, fetotoxicity, biochemistry, immunology,
and genetic changes. Concurrently, two control groups were
established, one with no dioxin exposure and the other with possible
dioxin exposure. These groups were selected from within the works and
matched the study group with respect to sex and age, but it was not
possible to match them for occupation and social status. Half the
affected subjects still had minor chloracne. Other than this finding,
the authors concluded that the subjects had not been had been
adversely affected in any way.
Data on the mortality of workers at the Dow Chemical company have
been provided in two papers (Cook et al., 1980; Ott et al., 1980). The
first of these studies describes the mortality of a cohort of 61 males
involved in the preparation of trichlorophenol. Forty-nine of these
workers developed chloracne, presumably as a result of skin absorption
of the process contaminant TCDD. Within the limitations posed by
cohort size and length of follow-up, the exposure to chlorophenol-TCDD
mixtures did not appear to have adversely affected mortality
experience. Overall, four deaths occurred and 7.8 were expected. Of
these, one death was due to cardiovascular disease (3.8 expected) and
three deaths were attributed to cancer (1.6 expected). None of the
findings was statistically significant. The second paper examined the
mortality experience of 204 people exposed to 2,4,5-T during its
manufacture from 1950 to 1971. Length of employment within the 2,4,5-T
process area ranged from less than one year to a maximum of
approximately ten years. Efforts to minimize TCDD contamination of the
product resulted in non-detectable concentrations (less than 1 mg/kg)
near the end of this period. Within the scope of this mortality
survey, no adverse effects were observed with respect to occupational
exposure to 2,4,5-T or to its feedstock, 2,4,5-trichlorophenol.
Hardell and his co-workers in Sweden have conducted a series of
case-control studies and reported an increased risk of soft-tissue
sarcomas in men who were exposed to phenoxy herbicides and/or
chlorophenols (Hardell & Sandsstrom, 1979; Hardell, 1981; Hardell et
al., 1981; Hardell & Ericksson, 1981). These authors also reported a
case-control study that suggested that phenoxyacetic acids and
chlorophenols may predispose to Hodgkin's lymphoma (Hardell et al.,
1981). The relative risk was higher for a group exposed to phenoxy
herbicides including 2,4,5-T and chlorophenols, i.e., pesticides that
may be contaminated with PCDDs and PCDFs. However, an increased risk
was still found in a group exposed mainly to phenoxy herbicides such
as MCPA, 2,4-D, mecoprop and dichloroprop, i.e., pesticides with low
or no contamination with PCDDs and PCDFs.
Analysis of fat levels of PCDDs and PCDFs in patients with soft
tissue sarcomas and in controls failed to reveal any differences
between the two groups (Nygren et al., 1986) (section 4.4.4.1).
A cohort study on Swedish farmers and gardeners has been carried
out recently (Wiklund & Holm, 1986). Despite the greatly increased use
of phenoxyacetic acid herbicides from 1947 to 1970, no time-related
increase in the relative risk of soft-tissue sarcoma was found in the
cohort or in any of the subcohorts. The same was found by Hoar et al.
(1986) although the latter study points to an increase in non-Hodgkin
lymphoma. It should be noted that in all these studies the majority of
the herbicides used did not contain TCDD.
In follow-up studies of workers exposed to 2,4,5-T and its
precursor 2,4,5-trichlorophenol (and therefore, presumably, also to
TCDD), no excessive deaths due to any cause were registered (Cook et
al., 1980; Ott et al., 1980; Zack & Suskind, 1980; Zack & Gaffey,
1983).
Honchar & Halperin (1981) merged the above four cohorts and found
that three (2.9%) of the total 105 deaths were reported to be from
soft-tissue sarcoma. Based on national statistics only 0.07% was
expected to be due to this cause. Fingerhut et al. (1984) reviewed the
employment records, medical and pathological reports, tissue
specimens, and death certificates for these three cases and four
additional cases of deaths from soft-tissue sarcomas in these and
related cohorts reported by Cook (1981), Moses & Selikoff (1981), and
Johnson et al. (1981). Three out of the seven cases had a record of
chloracne and one of dermatitis. After review of the tissue specimens,
five of the seven cases were diagnosed as soft-tissue sarcoma. The
remaining two (which were among the three cases in the merged cohort
of Honchar & Halperin (1981)) were found to be carcinoma. For three of
the cases with confirmed soft-tissue sarcoma the exposure was not well
documented, although an undocumented contact with 2,4,5-T,
2,4,5-trichlorophenol, or TCDD could not be excluded.
8.5 Human Experimental Studies
Poiger & Schlatter (1986) studied a human volunteer after
ingestion of a single dose of 1.14 µg 3H-TCDD/kg body weight. The
absorption from the intestine was > 87% and adipose tissue levels
were 3.09 (± 0.05) and 2.85 (± 0.28) ng/kg after 13 and 69 days,
respectively. The estimated half-life of TCDD was 2120 days.
Gorski et al. (1984) calculated the half-lives of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD and octaCDF to be about
3.5, 3.6 and 2 years, respectively. The estimation was based on the
analysis of fat tissue biopsies collected with an interval of 28
months from one 14-year-old girl who for a period of about 2-3 years
had been exposed to technical pentachlorophenol. Analysis was
performed by gas chromatography with electron capture detection, and
the isomers were confirmed by the use of several different packed and
capillary columns.
9. TOXICOKINETICS OF PCDFs
9.1 Uptake, Distribution, and Excretion
Toxicokinetic data for TCDF and other PCDFs arise from iv
injections or gastrointestinal exposure. There are no studies on
exposure via the respiratory tract or via dermal application.
9.1.1 Studies with 2,3,7,8-tetrachlorodibenzofuran (2,3,7,8-TCDF)
Table 65 summarizes the distribution of radioactivity to the
major tissue depots at various time-points after an iv injection of
14C-TCDF into rats and mice. Similar studies in guinea-pigs and
monkeys are summarized in Table 66. Tables 67 and 68 give the tissue
distribution of 14C-TCDF in more detail for rats and mice,
respectively.
TCDF has been used for kinetic studies in the rat (Birnbaum et
al., 1980), mouse (Decad et al., 1981a), guinea-pig (Decad et al.,
1981b), and monkey (Birnbaum et al., 1981). A single iv dose of 30.6
µg 14C-TCDF/kg body weight was given to rats, mice, and monkeys,
while the guinea-pigs received an iv dose of 6 µg/kg body weight. The
distribution of the radio-label was followed in tissues and excreta
for 3 weeks in rats and monkeys, for 10 days in mice, and for 9 days
in guinea-pigs. The distribution of radioactivity in the main tissues
and excreta of the different species at some of the intervals studied
is presented in Tables 65 and 66 along with the respective half-lives
and LD50 values for TCDF. Radioactivity recovered from the tissues
represented the parent compound, while radioactivity in faeces and
urine represented metabolites of TCDF. In the faeces of guinea-pigs
only the parent substance was present. Analysis by thin-layer
chromatography revealed Rf values of 0.5 and 0.1 for metabolites of
TCDF in faeces and urine as compared to an Rf of 0.8 for the parent
compound.
TCDF has a short half-life (2-4 days) and is quickly eliminated
from the liver, both in the rat and the mouse (Birnbaum et al., 1980;
Decad et al., 1981a). Elimination occurs rapidly also from the skin
and muscle, whereas retention is longer in adipose tissues. The
difference in the retention of TCDF in adipose tissues between C57Bl/6
and DBA/2 mice may be explained by the fact that DBA/2 mice have
substantially more adipose tissues than C57Bl/6 mice.
The distribution of TCDF in the guinea-pig was different from
that in the rat or mouse (Decad et al., 1981b). The maximum uptake in
the liver occurred within one hour after dosing; thereafter the
radioactivity was distributed in the fat and skin during the
succeeding hours. After one day, as a result of loss of body fat, the
radioactivity in adipose tissues was redistributed to the liver.
Within 3 days after dosing there was no elimination of radioactivity
from the liver and adipose tissues, whereas in the skin radioactivity
decreased only slightly. The estimated half-life for TCDF in the
guinea-pig was more than 20 days.
Table 65. LD50, whole-body half-life, and distribution of radioactivity (percentage of administered dose) at
various intervals after an iv dose of 30.6 µg 14C-TCDF/kg to ratsa and miceb
Fisher 344 Rats C57BL/6 Mice DBA/2 Mice
3 hr 3 days 10 days 3 h 3 days 10 days 3 h 3 days 10 days
Liver 41.4±3.6 5.9±0.3 1.3 51.0±13.4 22.7±1.8 1.1±0.3 39.4±0.6 16.8±1.4 5.6±2.0
Fat 10.0±1.0 11.1±2.3 1.8 6.0±1.6 2.9±2.1 ND 9.6±3.9 22.3±2.9 7.2±0.9
Skin 6.6±0.3 1.2±0.3 0.5 3.6±0.7 3.0±1.1 ND 5.5±4.3 3.3±0.9 ND
Muscle 5.9±0.4 0.3 < 0.3 7.5±2.8 1.5±0.9 ND 10.8±3.4 5.4±1.9 1.8±0.4
Faeces 63.1±0.6 > 85 43.1 81.9±13.0 27.7 55.8±4.8
Urine 2.0±0.4 < 6 7.7 12.6±0.1 9.2 19.9±4.6
Half-life < 2b 2b 4b
(days)
LD50 > 1000c > 6000c,d
(µg/kg)
a Birnbaum et al. (1980).
b Decad et al. (1981b).
c Moore et al. (1976).
d Moore et al. (1979).
ND = not detectable.
Table 66. LD50, whole-body half-life, and distribution of
radioactivity (percentage of administered dose) at various intervals
after an iv dose of 14C-TCDF in guinea-pigsa and monkeysb
Hartley guinea-pigs Rhesus monkeys
3 h 3 days 9 days 21 days
Liver 23.6±3.8 29.3±0.6 54.2±14.5 1.02±0.80
Fat 31.4±0.7 56.9±7.6 21.8±11.6 3.66±2.83
Skin 22.5±0.1 17.1+±0.6 15.2±3.1 2.44±1.60
Muscle 15.6±4.5 8.8±3.0 1.55±0.14
Faeces 4.7±1.3 6.6 42.9
Urine 2.3±0.4 6.6 7.9
Half-life > 20c 8a
(days)
LD50 > 5c < 10d 1000d
(µg/kg)
a 6 µg/kg (Decad et al., 1981a).
b 30.6 µg/kg (Birnbaum et al., 1981).
c Moore et al. (1976).
d Moore et al. (1979).
Table 67. Tissue distribution of TCDF-derived radioactivity in
Fisher 344 rats at 15 min, 3 h, and 24 h following a single iv dose
of 30.6 µg 14C-TCDF/kg b
Tissue Tissue content of 14C (% of dose/g tissue)a
15 min 3 h 24 h
Blood 0.12±0.04 0.04±0.01 0.03±0.01
Liver 4.4 ±0.2 5.1 ±0.4 2.2 ±0.4
Fat 0.20±0.03 0.44±0.07 0.64±0.11
Muscle 0.25±0.01 0.06±0.00 0.30±0.02
Skin 0.17±0.02 0.20±0.01 0.07±0.01
Kidneys 0.67±0.04 0.17±0.03 0.08±0.01
Adrenals 7.4 ±6.9 4.7 ±1.3 0.34±0.14
Thymus 0.52±0.12 0.54±0.13 0.07±0.03
Spleen 0.37±0.07 0.08±0.02 0.02±0.00
Testes 0.09±0.01 0.09±0.01 0.06±0.02
Brain 0.25±0.01 0.15±0.03 0.02±0.00
Lungs 1.08±0.08 0.24±0.02 0.07±0.02
Heart 0.66±0.03 0.11±0.00 0.02±0.02
a Mean ± SD for three animals.
b From: Birnbaum et al. (1980).
Based on data from three monkeys, the half-life for TCDF was
calculated to be 8 days (Birnbaum et al., 1981). At the end of the
study, more radioactivity remained in adipose tissues and skin than in
the liver. The retention of TCDF in the liver of monkeys 21 days after
dosing was comparable to that in the liver of the rat and C57Bl/6
mouse 10 days after injection. Urinary elimination of radioactivity
was a minor route when compared to faecal elimination both in the rat,
mouse, and monkey, whereas in the guinea-pig these routes were of
comparable importance (Birnbaum et al., 1980, 1981; Decad et al.,
1981a,b). The cumulated excretion of radioactivity 3 days
post-treatment amounted to approximately 64, 51, 11, and 7% in the
rat, C57Bl/6-mouse, monkey, and guinea-pig, respectively.
Against this background of data on tissue distributions,
half-lives, and LD50 values of TCDF in the rat, guinea-pig, and
monkey, Birnbaum et al. (1980, 1981) concluded that TCDF, measured as
excreted radioactivity, is metabolized to less toxic compounds and
that animal species with a high capacity to metabolize TCDF are more
resistant to its acute toxicity. This conclusion was considered
applicable also to the mouse (Decad et al., 1981a). Based on the same
data King et al. (1983) produced a pharmacokinetic model for TCDF in
rats, mice, and monkeys. However, there are objections to this
comprehensive conclusion. First, the kinetic studies on guinea-pigs
(Decad et al., 1981b) were (for analytical reasons) carried out with
such a high dose of TCDF that all of the animals showed marked signs
of toxicity, even within 3 days. After 9 days all the animals were
killed due to toxic symptoms. It is not advisable to draw any
conclusions on normal kinetic behaviour from data obtained on dying
animals with their abnormal metabolism and physiology. As far as the
kinetic data from the monkey are concerned, the conclusions were based
on a single time-point, and the number of animals in that study was
also very limited (Birnbaum et al., 1981).
Table 68. Tissue distributiona of TCDF-derived radioactivity in C57Bl/6 and DBA/2 mice at 15 min, 3 h, and 24 h after
a single iv dose of 30.6 µg 14C-TCDF/kg b
Tissue content of 14C (% of dose/g tissue)a
15 min 3 h 24 h
Tissue C57Bl/6 DBA/2 C57Bl/6 DBA/2 C57Bl/6 DBA/2
Blood 1.1 ND 0.6±0.3 0.22±0.04 ND 0.2±0.0
Liver 28.0±4.2 30.7±2.8 39.3±2.8 38.0 ±3.8 25.3±4.2 19.2±1.9
Adipose 2.6±0.4 2.7±1.0 3.7±1.4 4.9 ±0.1 6.1±1.2 6.1±0.4
Muscle 1.3±0.1 1.6±0.2 0.7±0.2 0.9 ±0.3 0.3±0.1 0.8±0.1
Skin 2.2±0.1 2.3±0.7 1.4±0.2 2.7 ±0.6 1.3±0.7 2.7±0.6
Kidneys 3.4±0.3 4.1±0.5 1.1±0.3 1.3 ±0.3 0.6±0.1 0.7±0.2
Adrenals 18.8±4.9 ND 6.9±5.4 9.2 6.5 ND
Thymus 3.9±2.4 3.0±1.9 2.0±0.7 4.7 ±3.2 0.3±0.2 2.5±0.6
Spleen 1.4±0.1 1.7±0.3 0.8±0.1 0.5 ±0.1 0.2±0.1 0.27
Testes 0.4±0.1 0.6±0.1 0.5±0.2 1.0 ±0.2 0.2±0.2 0.4±0.3
Brain 1.7±0.5 2.4±0.3 0.8±0.1 1.3 ±0.3 0.2±0.3 2.7±2.7
Lungs 6.7±0.5 8.2±0.7 2.4±0.9 3.1 ±1.5 0.4±0.2 0.8±0.4
Heart 2.1±1.3 3.4±2.2 0.6±0.3 0.8 ±0.4 0.2±0.1 0.3±0.1
a Mean ± SD for three animals.
b From: Decad et al. (1981b).
ND = below limit for accurate detection.
Ioannou et al. (1983) calculated a whole-body half-life of
approximately 40 days for a non-toxic dose of TCDF in young male
Hartley guinea-pigs. Their estimation was based on the distribution of
TCDF-derived radioactivity in liver, adipose tissue, skin, and muscle
in three animals 36 days after a single oral dose of 4 mg TCDF/kg body
weight and on certain approximations obtained from a previous study
(Decad et al., 1981b). Failure to demonstrate a correlation between
degree of bioaccumulation and lethality of TCDF in this study may be
due partly to the calculations of body burden based on the uncertain
estimate of the 40 days half-life for TCDF, which may not be valid for
both toxic and non-toxic doses of TCDF.
9.1.2 Studies with other PCDFs
Young male Wistar rats absorbed approximately 68% of a single
oral dose of 1.0 mg 2,3,4,7,8-pentaCDF/kg body weight given in salad
oil (Yoshimura et al., 1986). The daily faecal excretion was about
0.1% of the administered dose/day, whereas no 2,3,4,7,8-pentaCDF was
detected in urine. Four weeks after dosing the retention of
2,3,4,7,8-pentaCDF in the liver was 48.8% of the dose. The addition of
5% of activated charcoal beads to the diet, one week after dosing and
throughout the study, increased the faecal elimination of
2,3,4,7,8-penta CDF about 3-fold, but had no effect on urinary
elimination. Both the liver and extrahepatic tissues, except the
kidney, from rats on basal diet supplemented with activated charcoal
beads had lower levels of 2,3,4,7,8-pentaCDF than rats on basal diet
only.
Yoshihara et al. (1981) administered single ip injections of 13
individual PCDF congeners (at 1, 5, or 10 µg/kg) to young male Wistar
rats, and retention of the respective isomers in the liver was
determined 5 days later. The great variation observed in the hepatic
accumulation of the various isomers seemed to depend on the position
as well as the number of chlorine atoms substituted. Isomers having
vicinal hydrogens were accumulated to a lesser degree, although three
of the six isomers having no vicinal hydrogens (1,3,6,8-tetraCDF,
TCDF, and 1,2,4,6,8-pentaCDF) also showed low accumulation. The isomer
most highly accumulated was 2,3,4,7,8-pentaCDF, more than 65% of the
dose being retained, whereas only 3.8% of TCDF, which is equally
potent biologically, was retained. These results would imply that
there is no relationship between hepatic distribution of PCDFs and
their potential for acute toxicity. All animals in this study showed
toxic symptoms and liver microsomal AHH activity was strongly induced,
except in the cases of the following isomers: 2,8-diCDF,
1,2,7,8-tetraCDF, 1,3,6,7-tetraCDF, 1,3,6,8-tetraCDF,
1,2,4,6,8-pentaCDF. It is important to take this into consideration
when judging the kinetic data. A mixture of 14% 1,2,7,8-tetraCDF, 35%
TCDF, 1% 1,2,4,7,8-pentaCDF, 49% 1,2,3,7,8-pentaCDF, 1%
2,3,4,7,8-pentaCDF, and 1% hexaCDF was administered as a single ip
dose of 10 mg PCDF/kg body weight to young male Wistar rats (Kuroki et
al., 1980). The retention of the isomers in the liver, 5 days
post-treatment, showed good agreement with the results of Yoshihara et
al. (1981).
Based on the purification of three isoenzymes of cytochrome P-450
and the recovery of 14C-radioactivity from the hepatic microsomes of
Wistar rats treated with 14C-2,3,4,7,8-pentaCDF (single ip dose of
1 mg/kg body weight, 5 days previously), Kuroki et al. (1986)
suggested that one of these isoenzymes, P-448 H, functions as the
storage site of 2,3,4,7,8-pentaCDF in the rat liver.
Fly ash and crude or purified toluene extracts of PCDD- and
PCDF-containing fly ash from a municipal incinerator (Zaanstad, The
Netherlands) were mixed with ordinary laboratory diet for rats (van
den Berg et al., 1983). Small portions (2 g) of these diets were fed
to male Wistar rats (300 g) every 24 h for 19 days, at which time the
animals were sacrificed. The levels of tetra-. penta-, and
hexa-chlorinated PCDDs and PCDFs in samples of liver and adipose
tissue from these rats were determined. Rats fed the fly
ash-containing diet stored PCDDs and PCDFs in their livers at
concentrations which were at least 3 to 5 times lower than those of
rats fed with comparable amounts of fly ash extracts. For the
pentaCDD, hexaCDF, and hexaCDD isomers these concentrations were
approximately 10-20 times lower. Generally PCDFs had a higher
retention in the liver of rats than the corresponding PCDDs. In the
adipose tissue of rats fed with fly ash extracts, retention was higher
for penta- and hexaCDDs than for the corresponding PCDFs.
In a later study, male Wistar rats (275 g) were fed a diet
containing the same fly ash, pretreated with 2.5% HCl (van den Berg et
al., 1986a). A control group received standard diet. All congeners
retained in the livers of the rats had a 2,3,7,8-chlorine substitution
pattern. With the exception of 2,3,4,7,8-pentaCDF and
2,3,4,6,7,8-hexaCDF, the retention for each congener was below 10% of
the dose. The retention percentages of the various congeners in the
liver were almost equal at all time-points studied (34, 59, and 99
days), thus indicating a long half-life of these congeners in the
liver of the rat.
A mixture of two tetraCDFs, four pentaCDFs, and four hexaCDFs,
was given as a single ip injection of 500 µg to male ICR mice (Morita
& Oishi, 1977). The distribution patterns of the isomers in various
tissues were followed for up to 8 weeks. Analyses were performed with
GC (with electron capture detection) and isomers were identified by
peak number only. PCDFs were mainly located in the liver, spleen, and
fat tissues, but low to minimal amounts were found also in the kidney,
testes, lungs, heart, and brain. The GC patterns of liver samples
changed markedly with time, in contrast to those of the other tissues,
including fat, where the GC patterns remained similar throughout the
study. Most isomers with shorter retention times were readily absorbed
and then rapidly disappeared from the liver. Isomers with longer
retention times were slowly absorbed, and thus appeared later and
persisted longer in the liver. If the mixture had been administered
orally, those isomers with long retention times might have passed the
gastrointestinal tract with very low absorption.
The hepatic retention of PCDDs and PCDFs after dietary intake of
the above-mentioned HCl-pretreated fly ash was studied in male Golden
Syrian hamsters (van den Berg et al., 1986b). The livers were analyzed
for tetra-, penta-, and hexaCDDs and -CDFs after feeding the diet,
which contained 25% fly ash. No detectable hepatic retention was
observed after 34 days. The highest retention after 95 days was 8.4%
for 2,3,4,7,8-pentaCDF, but the retention was generally below 5% of
the total dose. With the exception of 2,3,4,6,7- pentaCDF, only
2,3,7,8-substituted PCDDs and PCDFs were retained. Constant relative
concentrations were found for the 2,3,7,8-substituted PCDDs and PCDFs
at the time-points studied.
In studies by Firestone et al. (1979), three lactating Holstein
cows received commercial grade pentachlorophenol orally by gelatine
capsule at a dose rate of 10 mg/kg body weight twice daily for 10 days
and once daily for the following 60 days. One cow served as a control
and received gelatine capsules containing only ground corn. The
pentachlorophenol composite used contained ten PCDD congeners (0.1-
690 mg/kg) and eight PCDF congeners (0.9-130 mg/kg). Faeces collected
on day 28 of the treatment period contained three hexaCDDs (0.05-0.63
µg/kg), two heptaCDDs (21.3-33.1 µg/kg), and octaCDD (290-429 µg/kg).
Faeces also contained hexa-, hepta-, and octaCDF. Milk, body fat, and
blood contained only three of the PCDD congeners present in the
pentachlorophenol composite, namely 1,2,3,6,7,8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, and octaCDD. Milk samples also contained
hexa-, hepta-, and octaCDF. The average concentrations of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, octaCDD, and octaCDF in
the composite milk fat at the end of the treatment period were 20, 40,
25, and 2 mg/kg, respectively. Similar concentrations were found in
body (shoulder) fat at the end of the treatment period (13, 24, and 32
mg/kg, respectively, for 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD,
and octaCDD). Levels of dioxins in the blood were about 1000 times
below the values in milk or body fat. The average daily excretion of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in the milk
during days 40 to 70 of treatment was about 20, 40, and 23 mg
(corresponding, respectively, to 33, 3, and 0.6% of the daily intake
of PCDDs). One hundred days after cessation of treatment the average
levels of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in
shoulder fat and milk fat were 2.5, 6.6, 5.6 mg/kg and 4.3, 6.9, 3.0
mg/kg, respectively.
Table 69. Levels of PCDFs in the liver of dams and in fetuses and offspring after oral administration of PCDFs to
mice for 18 days during pregnancyf
PCDF Total intake % of PCDF intake in:a Total intake % of PCDF intake in:b
congener of PCDFs by of PCDFs by
dams killed dams killed
on day 18 of liver 2 weeks after liver offspring
pregnancy (µg)d of dam fetus delivery (µg) of dam (week)
1 2
tetraCDFc 1.4±0.06d 5.4 ND 1.6±0.10 ND ND ND
tetraCDFc 8.1±0.32 ND ND 9.0±0.57 ND ND ND
2,3,7,8-tetraCDF 11.4±9.46 5.5 0.007 12.5±0.79 0.03 0.05 Te
pentaCDFc 2.9±0.12 5.7 ND 3.1±0.20 4.0 0.10 0.27
pentaCDFc 13.2±0.53 3.9 0.004 14.5±0.91 0.1 0.03 Te
2,3,4,7,8-pentaCDF 4.9±0.20 14.5 ND 5.4±0.34 10.0 0.29 0.89
hexaCDFc 1.4±0.06 10.7 ND 1.6±0.10 6.4 0.28 0.76
Total 43.2±1.73 5.2 0.003 47.7±3.0 1.6 0.07 0.14
a Nine dams in group killed on day 18 of pregnancy.
b Ten dams in group killed on day 14 after delivery.
c Specific isomer not determined.
d Mean ± SEM.
e T = 0.01-0.1 µg/kg total congener.
f From: Nagayama et al. (1980).
ND = not detected.
9.2 Metabolic Transformation
Thirteen chlorinated compounds were detected in bile collected
for 48 h from female Sprague Dawley rats given a single oral dose of
678 µg TCDF (79.4% pure)/kg body weight (Poiger et al., 1984). The
four major metabolites considered to originate from TCDF were
trichloromethoxy-dibenzofuran, two trichlorodimethoxy-dibenzofurans,
and tetrachloromethoxy-dibenzofuran. The remaining nine metabolites,
detected in minute amounts, originated most likely from contaminating
PCDFs (1% triCDF, 8.4% tetraCDFs, 11.2% pentaCDFs).
Metabolites of 2-monoCDF, 2,8-diCDF, 2,3,8-triCDF, and octaCDF
were determined in the urine, faeces, fat, and liver of male Wistar
rats given single oral doses of 250 mg/kg body weight of the
respective isomers (Veerkamp et al., 1981). Analyses were performed
with GC-MS. No metabolites in any samples were found in rats given
octaCDF. Monohydroxy and dihydroxy derivatives were obtained with all
other isomers, whereas sulfur-containing metabolites were detected
only with the monoCDF and diCDF. Metabolites from 2-monoCDF and 2,3,8-
triCDF were found in urine and faeces only, but with 2,8-diCDF
metabolites appeared also in the tissues.
9.3 Transfer Via Placenta and/or Milk
Nagayama et al. (1980) studied the transport of a mixture of
PCDFs into the placenta and milk in the mouse (Table 69). A diet
containing 0.6 mg PCDFs/kg (48% tetraCDFs, 49% pentaCDFs, and 3%
hexaCDFs) was given for 18 days after mating. Nine dams were killed on
day 18 of pregnancy and 10 on day 14 after delivery. After giving
birth, the mothers were fed a diet free of PCDFs. The placental
transport, calculated from the amount of PCDFs in the neonates, was
about 0.003% of the administered dose. The isomers that remained in
the tissues were TCDF and 2,3,4,7,8-pentaCDF. While the levels of
PCDFs in the mothers dropped from 5.2% to 1.6% of the total intake,
the whole-body levels in the sucklings increased from 0.003% at the
time of birth to 0.07% after one week and to 0.14% of the total intake
after 2 weeks. TCDF and 2,3,4,7,8- pentaCDF were the dominant species
in both the mothers and the pups. To study the transport through milk
only, the same PCDF-containing diet was given to pregnant rats for 14
days from day 18 after mating, including the lactation period
(Nagayama et al., 1980) (Table 70). After 14 days 5.1% of the total
intake was found in the liver of the mother. The offspring contained
0.3% and 1.2% of the dam's intake after 1 and 2 weeks, respectively.
The dominant isomers recovered in the offspring were TCDF,
2,3,4,7,8-pentaCDF, and one unidentified pentaCDF, i.e., the same
isomers found in the largest amounts in the mother's liver. The data
demonstrated that the amounts of PCDFs transferred through milk were
much larger than the amounts transferred across the placenta.
Table 70. Levels of PCDFs in the liver of mouse dams and in offspring after oral
administration of PCDFs to dams for 14 days following deliverye
PCDF Total % of PCDF intake in:a
congener intake
of PCDF liver of dam offspring (week)
(µg)c 1 2
tetraCDFb 2.3±0.13 6.9 ND ND
tetraCDFb 12.9±0.71 ND ND ND
2,3,7,8-tetraCDF 17.9±3.10 5.6 0.5 1.4
pentaCDFb 4.4±0.24 6.7 Td 0.6
pentaCDFb 21.0±1.14 3.7 0.4 1.6
2,3,4,7,8-pentaCDF 7.7±0.42 13.8 0.4 2.2
hexaCDFb 2.3±0.13 7.9 Td 1.5
Total 68.3±3.75 5.1 0.3 1.2
a Ten dams in the group.
b Specific isomer not known.
c Mean ± SEM.
d T = 0.01-0.1 µg/kg total congener.
e From: Nagayama et al. (1980).
ND = not detected.
Weber & Birnbaum (1985) studied the distribution and placental
transfer of a single oral dose of 800 µg 14C- TCDF (0.0485
µCi/µg)/kg body weight to pregnant C57Bl/6 mice on gestation day 11.
Embryo mortality on gestation days 12 to 14 was in the range 7.9 to
17.8%. No detectable radioactivity was found in the embryos whereas
about 0.01% of the radioactive dose was contained in the placenta. The
hepatic radioactivity in the dams decreased rapidly from 30.0% of the
dose on gestation day 12 to 12.1% of the dose on gestation day 14. The
cumulative urinary and faecal excretion from gestation day 12 to 14
were 5.4 and 80.1% of the administered dose, respectively.
10. EFFECTS OF PCDFs ON ANIMALS
10.1 Acute Toxicity
Single oral LD50 values for TCDF in three species are listed in
Table 71.
10.1.1 Studies on rats
No histological changes associated with TCDF toxicity could be
observed in rats at oral doses up to 1000 µg TCDF/kg body weight
(Moore et al., 1976). These preliminary results, which also mentioned
that only mild toxicological changes occurred in rats at 6000 µg
TCDF/kg body weight, have not been presented in a final report.
Intravenous administration of 30.6 µg TCDF/kg body weight to male
Fisher rats has been shown to cause listlessness, excessive hair loss,
and decreased weight gain 2 days post-treatment. These adverse effects
were reversible and 3 weeks after dosing the animals appeared healthy
with normal body weight. There were no signs of thymic or splenic
atrophy or of liver hypertrophy (Birnbaum et al., 1980).
Single ip injection of 1 or 10 mg/kg body weight of nine
individual PCDF isomers, with at least three chlorines in the lateral
positions, to male Wistar rats produced thymus atrophy and liver
hypertrophy 5 days post-treatment. Five other congeners having no more
than two chlorine atoms in the lateral positions did not cause any
effects on the thymus or liver within the same dose range (Yoshihara
et al., 1981).
The ability of 15 individual PCDFs to affect body weight gain and
thymic atrophy in immature male Wistar rats was investigated 14 days
after a single ip injection (Ganon et al., 1985). The ED50 values
for both effects were estimated for each congener (Table 61).
10.1.2 Studies on mice
Moore et al. (1976) failed to establish a lethal dose for TCDF in
C57Bl/6 mice after giving single oral doses of up to 6000 µg/kg body
weight with an observation period of 30 days. However, there was a
transient depression in body weight gain, thymic involution, and mild
hepatotoxicity when 6000 µg TCDF/kg was given subcutaneously. Poland
& Glover (1980) found TCDF-induced thymus atrophy in C57Bl/6 mice 5
days after a single ip dose of 3 x 10-7 mol/kg body weight.
Single doses of 100 to 1000 µg TCDF/kg body weight to pregnant
C57Bl/6 mice on gestation days 10 to 13 produced no toxic effects on
the dams within the time studied (Hassoun et al., 1984a; Weber et al.,
1984).
A mixture of two tetraCDFs, four pentaCDFs and four hexaCDFs
given as a single ip dose of 500 mg to ICR mice produced no
deathswithin 8 weeks (Morita & Oishi, 1977). CF-1 mice given a PCDF
mixture containing 42% tetraCDFs, 54% pentaCDFs, and 4% hexaCDFs as a
single oral (10 to 1000 mg/kg body weight), sc (10 to 200 mg/kg), or
ip (10 to 100 mg/kg) dose developed no toxic signs during the first
week, although a modest weight loss was noted (Nishizumi, 1978). The
first deaths occurred 8 days after an oral dose of 1000 mg/kg, 5 weeks
after a sc dose of 200 mg/kg, and 11 days after an ip dose of 100
mg/kg. The oral LD50 (30 day) was 184 mg/kg for males and 414 mg/kg
for females. Hepatomegaly and thymus atrophy were consistent findings
in mice that died. In surviving mice on high dosages, the liver
exhibited small necrotic foci accompanied by cellular infiltrates. The
hepatic lesions occurred in the centrilobular area, and enlarged
hepatocytes containing foamy cytoplasm, increased numbers of lipid
droplets, and proliferation of smooth endoplasmatic reticulum were
also seen.
10.1.3 Studies on guinea-pigs
In studies by Moore et al. (1979), the patterns of toxicity were
similar for TCDF, 2,3,4,7,8-pentaCDF, and 2,3,7,8-
tetrabromodibenzofuran when given to young Hartley guinea-pigs. The
single oral LD50 was 5-10 µg/kg body weight for all three isomers,
and the time to death ranged from 8 to 26 days. Overt signs at lethal
doses were immediate and progressive weight loss, rough soiled hair
coat, listlessness, and dehydration. Similar symptoms appeared 3 days
after an iv injection of 6 µg TCDF/kg body weight (Decad et al.,
1981b). At necropsy lack of body fat and reduced body mass and thymus
weight were found. Histological findings were primarily associated
with the depletion of lymphoid cells in the thymic cortex, but
hypocellularity of bone marrow and hyperplasia in epithelial cells of
the renal pelvis, ureter, and urinary bladder were also observed.
Liver lesions were not observed. Surviving animals showed mild thymic
lymphoid hypoplasia only. Sublethal doses resulted in decreased body
weight gain.
In a study by Ioannou et al. (1983), all three adult male Hartley
guinea-pigs survived a single oral dose of 6 µg TCDF/kg body weight
for at least 17 days. At 10 and 15 µg TCDF/kg body weight, deaths
occurred on days 15-39 (two animals were sacrificed on day 17) and
13-20, respectively. The acute oral toxicity of soot (and of benzene
extracts of the soot) containing PCDDs and PCDFs from a PCB-containing
transformer fire (Binghamton, New York, USA) were studied in female
Hartley guinea-pigs (Silkworth et al., 1982). As discussed in section
8.1.1.1, toxicities were noted at 100 and 500 mg/kg body weight of
soot, but not when 1 and 10 mg/kg were administered.
Table 71. Single lethal dose values for TCDFa
Species/strain Sex/No Age or Dose Duration LD50 Time to
weight tested of study (µg/kg death
(µg/kgc (days) body weight) (days)
body weight)
Mice
(C57Bl/6) M/8 6 weeks 0 30 > 6000 not
400 reported
600
800
1200
1500
2500
4000
6000
Guinea-pigs
(Hartley) M/6 3-4 weeks 0 30 5-10 9-20
1
5
10
15
Monkeys
(Macaca F/2 2.0-3.7 kg 0 60 1000 14-31
mulatta) 300
1000
1500
a From: Moore et al. (1979).
b M = male; F = female.
c Doses were given orally in corn oil.
10.1.4 Studies on rabbits
When the above-mentioned soot (or benzene extracts thereof) was
applied dermally to New Zealand white rabbits (Silkworth et al., 1982)
(see section 7.4.4.1), it produced no overt toxicity, weight loss, or
histological changes in thymus, kidney, or skin, but centrilobular
hypertrophy was found in both sexes. The soot extract gave rise to a
reversible skin inflammation and hepatic centrilobular hypertrophy in
females only. Histological examination showed no changes in kidney,
thymus, and skin.
10.1.5 Studies on monkeys
The single oral LD50 value for TCDF in the young female rhesus
monkey (Macaca mulatta) was found to be 1000 µg/kg in a 60-days
study within the dose range 0, 500, 1000, and 1500 µg/kg and with two
or four animals at each dose level (Moore et al., 1979). The two
monkeys that received the sublethal dose developed skin lesions and
had decreased body weight gain. With lethal doses the following overt
signs occurred after 7 to 10 days: progressive weight loss, loss of
body fat, facial oedema, loss of facial hair, loss of finger and toe
nails, and thickening of skin. Death occurred within 2 to 4 weeks.
Major histological findings included hyperkeratosis of the skin,
thymic atrophy with lymphoid hypoplasia, and adverse effects on
epithelial linings. No structural liver lesions were observed though
the liver weight was increased. Increases in serum albumin and
cholesterol were also recorded.
Three male rhesus monkeys given a single dose of 30.6 µg TCDF/kg
body weight did not gain weight during the three following weeks, and
they developed facial skin lesions, mainly of sebaceous glands
(Birnbaum et al., 1981).
10.2 Short-Term Toxicity
10.2.1 Studies on rats
Male Sprague Dawley rats were fed 1 or 10 µg/kg of a PCDF
mixture, containing two tetraCDFs, four pentaCDFs, and four hexaCDFs
for 4 weeks (Oishi et al., 1978). Both diets gave rise to decreases in
growth rate, food consumption, haemoglobin and haematocrit values,
erythrocyte counts, serum levels of triglyceride, testosterone,
glutamic pyruvic transaminase, and leucine aminopeptidase activities,
as well as increases in serum cholesterol, cholinesterase, and
glutamic oxaloacetic transaminase activities. Rats fed the 10 µg/kg
diet developed chloracne-like lesions on the ears within 3 weeks.
Furthermore, this diet decreased the relative weights of thymus,
prostate, and seminal vesicles and increased the relative weights of
liver, testes, spleen, adrenals, lung, heart, and brain. In another
similar study, no effects were seen on total serum proteins or
leukocyte counts (Oishi, 1977).
In studies by Hori et al. (1986), male Sprague Dawley rats (aged
5 weeks) were for 21 days given daily oral doses of a mixture of PCBs,
PCQs, and PCDFs having a similar composition and isomeric ratio to
those found in the contaminated rice oil causing "Yusho" (section
11.1). The toxicities noted included thymic atrophy, suppression of
weight gain, hepatic enlargement, and an increased serum cholesterol
level and a decrease in serum glutamic pyruvic transaminase activity.
The mixture caused an induction of the AAH drug-metabolizing enzyme
similar to that caused by PCDFs alone. These results support the
hypothesis that the predominant etiology of "Yusho" involves PCDFs
contained in the PCB-contaminated toxic rice oil.
10.2.2 Studies on mice
Mice (C57Bl/6), given TCDF orally 5 times per week for 30 days
did not develop clinical signs of toxicity at doses of 30, 100, or 300
µg/kg body weight. However, thymus atrophy, liver hypertrophy,
decreased leukocyte count, and slightly elevated total serum protein
did occur in the high dose group at the end of the study (Moore et
al., 1979). Daily doses of 10, 30, or 50 µg TCDF/kg body weight on
gestation days 10-13 produced a dose-related increase in maternal
liver weight in C57Bl/6 mice (Weber et al., 1984). Decreased thymus
weight was recorded in ICR/JCL mice exposed to four weekly doses of a
mixture (at 100 µg/kg) of 12% tetraCDF and 88% pentaCDF (Oishi &
Hiraga, 1980). When PCDFs of unknown composition were given in the
diet at 0.6 mg/kg to mice for 10 weeks, severe dermal lesions,
hyperkeratosis, and dilated hair follicles filled with keratinous
material occurred in 7 of 12 mice. Furthermore, hepatocytes had
enlarged nuclei and vacuolations in the cytoplasm (Nagayama et al.,
1979). Feeding female ddN mice 0.6 mg PCDFs/kg diet (48% tetraCDFs,
49% pentaCDFs, and 3% hexaCDFs) for 18 days after mating, or for 14
days after delivery, produced no overt toxic effects in dams or in
offspring (Nagayama et al., 1980).
10.2.3 Studies on guinea-pigs
In studies by Luster et al. (1979b), oral administration of 0.05,
0.17, 0.5, or 1.0 µg 2,3,7,8-TCDF/kg body weight once weekly for 6
weeks to young female Hartley guinea-pigs produced 30% mortality in
the high-dose group. The thymus weight was decreased in the 0.5 and
1.0 µg/kg dose groups, though histologically only a slight decrease in
the density of thymic cortex was observable. Reduction in spleen
weight or alterations in splenic morphology did not occur, neither was
there a consistent decrease in body weight.
Four adult male Hartley guinea-pigs were given six or seven
weekly doses of 1 µg TCDF/kg body weight (Decad et al., 1981a). The
animals started to lose weight rapidly after the fifth or sixth dose,
the cumulative dose then being comparable to the oral LD50 value for
young guinea-pigs. At this time all animals were moribund and by day
44 the first animal died. Neither hepatomegaly nor thymic atrophy was
observed in this study. Thus multiple sublethal doses of TCDF appear
to have a cumulative effect, and may lead to a critical body burden
that will result in irreversible and progressive weight loss
eventually followed by death.
Weekly oral doses of 1 µg TCDF/kg body weight or biweekly doses
of 2 µg TCDF/kg body weight (interrupted by a 4-week period of no
dosing after the fourth dose) to young male Hartley guinea-pigs in
groups of four resulted in deaths on days 47, 51, 84; 31, 38, 88; and
32, 70, 85, respectively (Ioannou et al., 1983). At each dosing
schedule one animal was sacrificed at 101 days after exposure.
Repeated small doses, with various intervals in between, resulted in
a similar lethality but a less dramatic weight loss than with a high
acute dose.
10.2.4 Studies on rabbits
The 25% ether-hexane extracts from two commercial polychlorinated
biphenyl (PCB) preparations containing tetraCDFs and pentaCDFs
produced hyperplasia and hyperkeratosis of the follicular epithelium
of the rabbit ear skin when applied dermally weekly for 3 weeks in a
dose corresponding to 200 mg PCB. Liver lesions or decreased weight
gain were not observed. No dermal effects could be found when an
ether-hexane extract from a PCB preparation lacking PCDF impurities
was applied in the same manner (Vos & Beems, 1971).
A mixture of tetraCDFs and pentaCDFs was much less potent than
was TCDD in producing hyperkeratosis when applied to the inside of
depilated rabbit ears for 3 consecutive days (Nishizumi et al., 1975).
10.2.5 Studies on hamsters
No toxic effects were reported in male Golden Syrian hamsters
(50-70 g) given a diet containing 2.5% HCl-pretreated fly ash from a
municipal incinerator (Zaanstad, The Netherlands) for up to 95 days
(van den Berg et al., 1986b).
10.2.6 Studies on monkeys
A two-month study with three young male rhesus monkeys (Macaca
mulatta), serving as their own controls and fed a diet with 50 µg
TCDF/kg, resulted in one case of illness after 1 month and one death
after 2 months when the cumulative dose was calculated to be 300 µg/kg
(McNulty et al., 1981, 1982a). Toxic changes observed after 1 month
included periorbital oedema, reddening and thickening of the eyelids,
enlargement of facial hair follicles, and decreased number and size of
sebaceous glands in the skin. After 2 months these changes had become
more severe and were accompanied by decreased physical activity and
elevated (or eventual loss of) toe and finger nails. There were no
changes in haematology or serum chemical values. The diseased and the
surviving monkeys recovered rapidly when they were returned to
uncontaminated food. Within 3 months, behaviour, clinical appearance,
and histological structure of the skin were normal. The monkey that
died had lost 23% of its initial weight and most of its body hair.
Sebaceous glands were replaced by small squamous cysts. Severe lesions
were confined to the skin, thymus, and the stomach epithelium, whereas
liver lesions were modest. Decreased bone marrow cellularity was a
postmortem finding which was not reflected in the peripheral blood
count taken before death.
10.2.7 Studies on chickens
Mortality in one-day-old White Leghorn chickens given 1 or 5 µg
TCDF/kg body weight orally for 3 weeks was 16% and 100%, respectively,
with an average time to death of 19 and 11.5 days (McKinney et al.,
1976). Body weight gain and food consumption were decreased during the
third week post-treatment. Dose-related subcutaneous oedema, ascites,
and hydropericardium, as well as thymus atrophy, occurred. Depletion
of lymphatic cells was evident both in the spleen and thymus. Mild
liver lesions were found only in the high-dose group. Total serum
protein and serum albumin were reduced.
The significant difference in toxicity in chickens between three
commercial PCB preparations (Vos & Koeman, 1970) was later
demonstrated to be caused by the presence of tetra- and pentaCDFs in
two of the three preparations (Vos et al., 1970). The 25% ether-hexane
extracts from these two PCB preparations were highly toxic in the
chick embryo assay (Vos et al., 1970), whereas no effect could be
produced by the extract from the PCB preparation lacking PCDF
impurities.
10.3 Chronic Toxicity
10.3.1 Studies on monkeys
In studies by McNulty et al. (1981, 1982a), three young male
rhesus monkeys (Macaca mulatta) were exposed for 6 months to 5 µg
TCDF/kg diet, and one animal served as a control. One animal was
killed after 6 weeks when moribund. Overt toxic signs in the two
remaining animals started to appear after 3 months and the symptoms
remained for the following 3 months. One of these animals died
suddenly after 6 months. The remaining monkey was returned to normal
food and rapidly recovered. Clinically and pathologically, chronic
intake of small amounts of TCDF caused symptoms similar to those
following a single large dose of TCDF (section 9.1.2.4) or acute or
chronic ingestion of TCDD (see sections 7.1.1 and 7.3). The major
histopathological changes in all cases were seen in the thymus,
sebaceous glands, nail beds, bone marrow, and mucosa of the stomach
and bile ducts. The toxic potency of TCDF when ingested chronically
was approximately equal to that of TCDD. This contrasts with the acute
toxic effect of TCDF, which is approximately 20 times less than that
of its TCDD counterpart. The reason for death in the TCDF-poisoned
monkeys was obscure; it was preceded by weight loss, anorexia, and
depression. Only modest thymic and epithelial changes were present,
and there was no evidence for liver damage. The quick recovery of
animals returned to normal diet contrasted with the course of TCDD
poisoning in which illness progressed to death, or recovery was much
delayed, even after exposure had ended.
10.4 Effects Detected by Special Studies
10.4.1 Immunobiological effects
To date no studies have been performed on the effects of PCDFs on
the developing immune system.
Comparative studies on humoral immune responses in mice have
revealed that TCDF produces a pattern of responses similar to that
found for TCDD but only at 30-fold higher doses. Furthermore the
immunosuppressive effect of TCDD is much more persistent (Vecchi et
al., 1983b).
10.4.1.1 Histopathology
During toxicity studies with pure isomers of PCDFs or with
mixtures of PCDFs, thymus atrophy has been noted as a consistent
effect in the mouse (Nishizumi et al., 1978; Moore et al., 1979), rat
(Oishi, 1977; Oishi et al., 1978), guinea-pig (Moore et al., 1979),
and monkey (Moore et al., 1979; McNulty et al., 1981). Studies aimed
at investigating immunobiological effects revealed decreased thymic
weights (Luster et al., 1979b; Vecchi et al., 1983). The histological
findings are similar to those occurring after TCDD exposure, i.e.,
loss of lymphoid cells in the thymic cortex. A reduced number of
spleen cells was obtained from mice treated with a single ip dose of
180 µg TCDF/kg body weight (Vecchi et al., 1983), but no splenic
pathology was reported in mice given four weekly oral doses of a PCDF
mixture (at 100 µg/kg) containing 12% tetraCDFs and 88% pentaCDFs
(Oishi & Hiraga, 1980). Peritoneal cell and macrophage counts in mice
were not modified by an ip dose of TCDF (180 µg/kg body weight)
(Vecchi et al., 1983).
10.4.1.2 Humoral-mediated immunity
Adult female Hartley guinea-pigs exposed orally to 0.05, 0.17,
and 0.5 µg TCDF/kg body weight once weekly for 6 weeks showed somewhat
depressed serum IgG concentrations. A dose-related depression in
splenic lymphocyte proliferation was seen in TCDF-treated animals
after stimulation with the B-lymphocyte mitogen Escherichia coli
0127 lipopolysaccharide at 50 µg/ml medium. There were no effects on
any of the major serum proteins, neither was there an effect on the
antibody response towards bovine gamma globulin (BGG) (Luster et al.,
1979b).
The antibody response to sheep red blood cells given 7 days after
a single ip injection of 180 µg TCDF/kg body weight was inhibited by
85% and 35% in C57Bl/6 and DBA/2 mice, respectively (Vecchi et al.,
1983), whereas a single ip dose of 10 µg TCDF/kg body weight to
C57Bl/6 mice had no effect (Rizzardini et al., 1983). The suppression
noted by Vecchi et al. (1983) was dose dependent as well as time
dependent; by day 42 post-treatment a near-normal antibody response
was obtained.
10.4.1.3 Cell-mediated immunity
Oral intubation of 10 or 100 mg PCDF (12% tetraCDFs and 88%
pentaCDFs) per kg body weight once weekly for four weeks increased the
sensitivity to endotoxin of ICR/JCL mice. Following an ip injection of
50, 250, or 500 µg endotoxin per mouse a dose-dependent increased
mortality was noted two days after the final treatment with PCDF
(Oishi & Hiraga, 1980). Only at high dose levels were there any
effects on cell-mediated immunity functions in female Hartley
guinea-pigs given 0.05, 0.17, 0.5, or 1.0 mg TCDF/kg body weight
orally once weekly for six weeks (Luster et al., 1979b).
Both the depression in delayed hypersensitivity response to
purified protein derivative and the ability of BGG-sensitized
lymphocytes to release the macrophage inhibition factor were related
to the dose of TCDF. Splenic lymphocytes from TCDF-treated animals,
stimulated with the T-lymphocyte mitogen phyto-haemagglutinin (PHA),
showed a decreased proliferation. On the other hand proliferation of
splenic lymphocytes stimulated with concanavalin A (Con A), another
T-lymphocyte mitogen, showed no TCDF-related effect. The increased
proliferative response to Con A and PHA in thymocytes co-cultivated
with thymus epithelial (TE) cells or cultivated in TE-conditioned
medium was inhibited if the TE cells were pretreated with TCDF for 48
h, thus suggesting a direct effect on TE cells (Osborne et al., 1984).
10.4.2 Enzyme induction
Studies discussed below show that PCDFs are potent enzyme
inducers, the enzyme-inducing potencies varying greatly depending on
the position as well as on the number of chlorine atoms substituted.
The structure-activity relationships of the PCDFs with regard to
enzyme induction are similar to those for PCDDs, with TCDF and
2,3,4,7,8-pentaCDF being the most potent (Tables 56 and 61).
10.4.2.1 Studies on rats
Intraperitoneal doses of 2.5 mg TCDF/kg body weight given once
daily for three days to female CD rats induced 38- and 3-fold
increases, respectively, in AHH and UDPGT activities 24 h after the
final dose. The cytochrome P-450 content was doubled but no effect was
found on the aminopyrine N-demethylase activity (Goldstein et al.,
1978).
Increased AHH and EROD activities were found in the hepatic
microsomal fraction from immature male Wistar rats 5 days after ip
injection of single doses of TCDF (1.7 µmol/kg body weight) or
2,3,4,7,8-pentaCDF (0.3, 1.5, 3.0 µmol/kg body weight) (Keys et al.,
1985). This study also detected an alteration by TCDF in the hepatic
metabolism of testosterone in these rats. Yoshihara et al. (1981) gave
a single ip dose of 1, 5, or 10 mg PCDF/kg body weight of 13
individual PCDFs to young male Wistar rats five days prior to the
determination of hepatic enzyme activities. Congeners having at least
three chlorine atoms in the lateral positions typically showed
increased AHH and DT-diaphorase activities, while those congeners
having no more than two chlorine atoms in these positions were not
inductive. The cytochrome P-448 content was increased by 5 of the 13
congeners whereas the benz-phetamine-N-demethylase activity was
depressed by 7 of the 13. The most potent isomers, TCDF and
2,3,4,7,8-pentaCDF, were effective at a single dose of 1 µg/kg body
weight. The ranking of the potency for enzyme-inducing abilities did
not coincide with the hepatic distribution of the test substances.
Hepatic AHH activity in male Wistar rats was significantly enhanced
only by TCDF and 2,3,4,7,8-pentaCDF among the 15 individual PCDF
isomers tested, the dose administered intraperitoneally being 5 µg
PCDF/kg body weight (Nagayama et al., 1983).
Eight of the 15 PCDF isomers tested increased the pulmonary AHH
activity from 5-fold to 30-fold. In this study no PCDF-related AHH
induction was present in the kidney, prostate, thymus, or spleen.
Bandiera et al. (1984b) investigated the effect of three tetraCDFs and
three pentaCDFs at doses of 500 and 1000 µg/kg body weight,
respectively, on hepatic AHH, aminopyrine N-demethylase,
4-chlorobiphenyl hydroxylase, and EROD activities in male Wistar rats.
The most active compounds, TCDF and 2,3,4,7,8-pentaCDF, were potent
inducers of the cytochrome P-448-dependent monooxygenases. Some
induction of microsomal AHH, EROD, and 4-chlorobiphenyl hydroxylase
was observed also for the TCDF and 1,2,4,7,9-pentaCDF.
The ED50 values for hepatic AHH (Table 61) and 4-chlorobiphenyl
hydroxylase induction were established for 15 individual PCDFs in
immature male Wistar rats 14 days after a single ip injection (Mason
et al., 1985).
Significant induction of hepatic AHH activity in male Sprague
Dawley rats was given only by 3 out of 25 individual PCDFs given as
single oral doses of 40 µg/kg body weight (Doyle & Fries, 1986). The
active congeners were 2,7-diCDF, TCDF, and 2,3,4,7,8-pentaCDF.
A mixture of PCDFs, reconstituting the approximate composition
found in the liver of Yusho victims (7.4% tetraCDF, 6.1%
1,2,4,7,8-pentaCDF, 19.0% 1,2,3,7,8-pentaCDF, 29.4% 2,3,4,7,8-pentaCDF
and 39.1% 1,2,3,4,7,8-hexaCDF by weight) was given as a single ip
injection to male Wistar rats 14 days before measuring the induction
of cytochrome P-448-related enzyme activities (Bandiera et al.,
1984a). A dose-related enhancement of AHH and EROD activities was
found within the range 10 to 400 µg PCDF mixture/kg body weight.
10.4.2.2 Studies on mice
No induction of cytochrome P-448 content, or of ECOD activities,
was found 12 days after a single ip injection of 10 µg TCDF/kg body
weight to male C57Bl/6J mice (Rizzardini et al., 1983).
Nagayama et al. (1985) investigated the AHH-inducing potency of
TCDF, 2,3,6,7-tetraCDF, 1,2,3,6,7-pentaCDF, 1,2,3,7,8-pentaCDF,
2,3,4,6,7-pentaCDF, 2,3,4,7,8-pentaCDF, 1,2,3,4,6,7-hexaCDF, and
1,2,3,4,7,8-hexaCDF in two strains of responsive (C57Bl/6 and AKR/Qdj)
and two strains of non-responsive (DBA/2 and DDD) mice. All congeners
were given as single ip doses of 30 µg/kg body weight in olive oil. No
single congener induced the AHH activity above the control level in
the non-responsive mice. Significantly increased AHH activity was
found in both responsive strains exposed to TCDF, 1,2,3,7,8-pentaCDF,
and 2,3,4,7,8-pentaCDF. Mice (C57BL/6) treated with 2,3,4,6,7-pentaCDF
and 1,2,3,4,7,8-hexaCDF also responded with increased AHH activity.
10.4.2.3 Studies on chickens
Hepatic AHH activity in chick embryos was inducible by TCDF,
2,3,4,7,8-pentaCDF, and 1,2,3,7,8-pentaCDF, with ED50 values of
0.015, 0.014, and 0.071 nmol/egg, respectively (Poland et al., 1976).
No induction was produced by unchlorinated dibenzofuran, 2,8-diCDF,
2,4-diCDF, 2,4,8-triCDF or 1,3,6,7-tetraCDF at the doses tested. There
were no effects on ALA synthetase, p-nitrophenol-UDPGT and
testosterone-UDPGT activities. However, a modest increase in
cytochrome P-450 content was present in one-day-old White Leghorn
chickens 3 weeks after treatment with a single oral dose of 1 µg TCDF
(Goldstein et al., 1976).
10.4.2.4 Studies on cell cultures
Exposure of primary hepatocytes isolated from adult male Wistar
rats to TCDF for 72 h resulted in a 2-fold increase in AHH induction
at 10-9 mol/litre and half-maximal induction at 3 x 10-10
mol/litre. However, no AHH induction was observed with 2,7-diCDF in
the range 10-11 to 10-8 mol/litre in the same system (Jansing &
Shain, 1985). A 59-fold increase in AHH activity and a 40-fold
increase in EROD activity were obtained in rat hepatoma H-4-II E cells
when exposed to 5 x 10-10 mol TCDF/litre for 3 days (Keys et al.,
1986). In this same study it was demonstrated that TCDF had an
additive effect, whereas 1,3,6,8-tetraCDF and 2,4,6,8-tetraCDF had
counteracting effects on TCDD-induced enzyme induction.
The EC50 values for AHH and EROD induction (Table 61) have been
established for 35 individual PCDFs in the rat hepatoma H-4-II E cell
line (Bandiera et al., 1984b; Mason et al., 1985). AHH and EROD
activities were determined after exposing the cells to optimal doses
of PCDFs for 5 days. Unchlorinated dibenzofuran, or 2- and
3-chlorodibenzofuran did not induce these enzyme activities. EC50
values for all the remaining congeners varied between 10-4 and 1.3 x
10-10 mol/litre, the most active inducer being 2,3,4,7,8-PCDF.
Human lymphoblastoid cell lines, derived from the peripheral
blood of healthy male and female volunteers of various ages, were
exposed to eight individual PCDF isomers for 48 h (Nagayama et al.,
1985b). The AHH inducibility was highly variable between individuals
but less variable between isomers. In this system TCDF was about half
as potent as 2,3,4,7,8-pentaCDF, 1,2,3,4,6,7-hexaCDF, or
1,2,3,4,7,8-hexaCDF, which were equally as potent as TCDD in inducing
AHH.
A mixture of PCDFs, reconstituted on the basis of PCDF residues
in the liver samples from Yusho victims (see section 10.4.2.1), had
EC50 values for induction of AHH and EROD activities of 1.02 x
10-10 and 3.23 x 10-10 mol/litre, respectively, in the rat
hepatoma H-4-II E assay (Sawyer & Safe, 1985). The calculated EC50
values based on the relative isomer content of the mixture were 3.07
x 10-10 and 4.43 x 10-10 mol/litre, respectively.
10.4.3 Receptor binding
The competitive binding of PCDFs to the TCDD receptor protein has
been studied in vitro both in the hepatic cytosol (Poland et al.,
1976; Bandiera et al., 1984b) and in the nucleus (Poellinger et al.,
1982). Poland et al. (1974) investigated the ability of seven PCDF
congeners to compete with TCDD in binding to the hepatic cytosol
receptor from C57Bl/6J mice. They found the relative binding
affinities for TCDF, 2,3,4,7,8-pentaCDF, and 1,2,3,7,8-pentaCDF to be
37%, 34%, and 38%, respectively, of the binding affinity between TCDD
and the receptor. The EC50 values for the competitive binding of 33
individual PCDFs to the receptor from rat hepatoma H-4-II E cell
cultures varied from less than 10-3 mol/litre for
4-chlorodibenzofuran to 1.5 x 10-8 mol/litre for the most active
competitor, 2,3,4,7,8-pentaCDF, which had an EC50 value comparable
to that for TCDD, i.e., 1.0 x 10-8 mol/litre (Table 61) (Bandiera et
al., 1984b; Mason et al., 1985). Of the TCDD bound to the nuclear
receptor in vitro, 58% was displaced by a 100-fold molar excess of
TCDF. These nuclei were isolated from the liver of Sprague Dawley rats
pretreated intravenously with 1 µg TCDF 2 h prior to the incubation
(Poellinger et al., 1982).
10.5 Embryotoxicity and Reproductive Effects
TCDF has been found to be a potent teratogen in mice at doses
that produce no overt toxic effects in dams. Malformations observed
include cleft palate and kidney malformation similar to
hydronephrosis. Dose-related increases in fetal mortality occur with
single high doses. The teratogenic pattern of TCDF thus is strikingly
similar to that of TCDD (see section 7.5).
Single doses of 100 to 1000 µg TCDF/kg body weight to pregnant
C57Bl/6 mice on gestation days 10 to 13 produced dose-related
increases in the number of cleft palates and kidney malformations;
both the number of litters and the number of fetuses were affected
(Hassoun et al., 1984a; Weber et al., 1984). No other
treatment-related malformations were reported. A cleft palate
incidence of 40% was obtained in NMRI mice offspring after sc
treatment of the dams on gestation days 9 to 11 with 200 nmol TCDF/kg
body weight (Krowke, 1986).
Palatal closure in mice occurs late on day 14 of gestation, and
so it is somewhat peculiar that the peak sensitivity for cleft palate
occurs on day 12 (Hassoun et al., 1984a). The peak sensitivity for
kidney malformation in mice occurs on day 11 of gestation (Hassoun et
al., 1984a). The quantitative data on this malformation somewhat
conflict in the two studies. Weber et al. (1984) reported that 95.5%
of the fetuses had kidney malformations after a dose of 500 µg/kg body
weight on day 10 of gestation. However, only 17% of the fetuses per
dam had this malformation after a single dose of 400 µg/kg body weight
on the same day in the study of Hassoun et al. (1984a). The difference
might be due to unequal judging of the malformation. Preliminary
results (Weber et al., 1984), suggested that TCDF-induced kidney
malformations, up to a certain degree, represent a reversible defect
since no hydronephrotic kidneys were found in neonates, whereas in
identically treated dams examined on day 18 of gestation over 80% of
the fetuses/litter were affected. Fetal mortality increased in a
dose-related manner with high single doses administered on days 10 to
12. Peak sensitivity occurred on day 10 (Hassoun et al., 1984a).
Multiple low dosing on gestation days 10 to 13 was more effective in
producing fetal malformations, but less effective in producing fetal
deaths, than single high dosing on day 10 (Weber et al., 1984). No
effect on fetal mortality (days 12, 13, and 14) was observed in
C57BL/6N mice given a single oral dose of 800 mg TCDF/kg body weight
on day 11 of gestation (Weber & Birnbaum, 1985).
Recombinant inbred strains of C57Bl/6 and DBA/2 mice segregating
at the Ah locus respond differently to the teratogenic effect of TCDF
(Hassoun et al., 1984b). Fetuses of Ah-responsive strains responded
with a high frequency of cleft palates and kidney malformations after
a single ip dose of 600 µg TCDF/kg body weight on day 12 of gestation.
However, no cleft palates and only modestly increased numbers of
kidney malformations in a few strains were found with the same
treatment in Ah-nonresponsive strains.
A diet containing 0.6 mg PCDFs/kg (48% tetraCDFs, 49% pentaCDFs,
and 3% hexaCDFs), fed to mice for 18 days after mating, had no effect
on the number or body weight gain of the offspring, neither were there
any malformations related to the diet (Nagayama et al., 1980).
Three PCDFs, namely 1,2,3,7,8-pentaCDF, 2,3,4,7,8-pentaCDF, and
1,2,3,4,7,8-hexa CDF, are teratogenic to C57BL/6N mice when
administered orally by gavage on gestation days 10-13. A significant
increase in hydronephrosis and cleft palate was found, with
2,3,4,7,8-PCDF being the most potent PCDF studied, having an ED50 of
36 µg/kg body weight for cleft palate and 7 µg/kg for hydronephrosis.
For all three PCDFs, hydronephrosis occurred at a lower dose than did
cleft palate (Birnbaum et al., 1987).
It has been pointed out by McNulty (1985) that chlorinated
compounds such as 2,3,7,8-TCDD produce cystic periodontal lesions and
squamous metaplasia of the ameloblasts surrounding unerupted teeth in
rhesus monkeys. These findings are similar to those on the teeth
development seen in Yusho patients (section 11.1).
10.6 Mutagenicity
When tested in Salmonella typhimurium strains KTA98 and TA100
with and without metabolic activation, no mutagenic activity was found
for 2,9-diCDF, 3,6-diCDF, TCDF, or octaCDF (Schoeny, 1982). TCDF was
also studied in Saccharomyces cerevisiae strain MP-1 and was found to
be negative for forward mutation, mitotic crossing over, and mitotic
gene conversion at concentrations up to 1000 mg/litre. Stationary
phase cells were tested in the absence of exogenous activation (Fahrig
et al., 1978).
10.7 Carcinogenicity
The hepatic tumour-promoting activity of a commercial
polychlorinated biphenyl mixture, Aroclor 1254, with (Ar 1254) or
without (Ar 1254-PCDF) PCDF impurities, was studied in Sprague Dawley
rats pretreated with 66 µg diethylnitrosamine/ml drinking water for 5
weeks (Preston et al., 1981). Thereafter the rats were fed a diet
supplemented with 100 µg Ar 1254/kg (> 3 mg PCDF/kg) or Ar
1254-PCDF (<0.1 mg PCDF/kg) for 18 weeks. Examination of liver
lesions by light microscopy demonstrated that Ar 1254 promotes
formation of hepatocellular carcinomas in rats. The promoting
incidence of 64% remained essentially unchanged when PCDF was removed
from Ar 1254 by adsorption chromatography. Due to the high incidence
of hepatocellular carcinomas produced by Ar 1254-PCDF itself, an
additional effect of PCDF might have been difficult to measure in this
study.
11. EFFECTS OF PCDFs ON HUMAN BEINGS
Braun (1955) was the first to report chloracne due to chlorinated
dibenzofurans, subsequently experimentally proven by Bauer et al.
(1961). Vos et al. (1970) identified by mass spectrometry the presence
of chlorinated dibenzofurans in commercial PCB mixtures, accounting
for their acnegenic properties.
11.1 Yusho and Yu-cheng
A mass outbreak of food poisoning occurred in western Japan in
1968 following ingestion of a commercial brand of rice oil
contaminated with polychlorinated biphenyls (PCBs) and related
hydrocarbons. The poisoning was named "Yusho" (oil disease).
Epidemiological proof of the cause of the epidemic depended on the
demonstration of a dose-response relationship between the consumption
of the toxic rice oil and the incidence of the poisoning or between
the oil consumption and the clinical severity of the reaction.
Approximately 2000 cases were recognized. In 1969, Japanese scientists
first reported that the toxic rice oil which caused Yusho was
contaminated with polychlorinated biphenyls (Tsukamoto et al., 1969).
A few years later, the oil was found also to be contaminated with a
smaller quantity of PCDFs (Nagayama et al., 1976) and a relatively
large amount of polychlorinated quaterphenyls (PCQs) (Masuda &
Yoshimura, 1982).
In March 1979, an epidemic of a peculiar skin disease broke out
in Taichung and Changhwa in Central Taiwan. The cause of the disease
was later identified to be the ingestion of rice-bran oil contaminated
with polychlorinated biphenyls (Chen et al., 1980, 1981; Hsu et al.,
1984; Masuda et al., 1986). By the end of 1980, the total number of
reported cases was about 2000. The local name for the disease was
Yu-cheng. The mean consumption of total PCDFs of the Yusho and
Yu-cheng patients has been estimated to be 3.3-3.8 mg/person or
400-500 mg of toxic 2,3,7,8-substituted PCDFs per person. (Hayabuchi
et al., 1979). Hayabuchi et al. (1979) estimated the daily intake of
total PCDFs in the Yusho intoxication to have been 0.9 µg/kg body
weight. Analyses of liver samples taken from the Yusho patients about
18 months after the exposure showed a dramatic decrease in the number
of PCDF isomers. Apparently most of the PCDF isomers were metabolized
or excreted during the period between exposure and sampling (Rappe et
al., 1979). A comparison between the PCDF isomers found in the Yusho
oil and the liver samples revealed an interesting relationship. Most
of the isomers retained had all lateral positions (2-, 3-, 7-, and 8-)
substituted with chlorine (Rappe et al., 1979).
Table 72. Clinical symptomatology of Yusho 1969-1972a
1. Skin (82-87%).
Acneiform eruptions, districtive hair follicles, red plaques on limbs,
dark brown pigmentation of nail, skin, and mucous membranes, itching,
sweating of palms.
2. Ocular manifestations (83-88%).
Increased eye discharge, swelling of the upper eyelids, hyperaemia of
conjunctiva, transient visual disturbance.
3. Jaundice (10%).
No abnormalities of liver function in the majority of cases.
4. Numbness of the limbs, feeling of weakness, muscular spasms (32-39%).
Reduced sensory and motor nerve conduction velocity in a few cases
(9%).
5. Hearing difficulties (18%).
6. Headaches, vomiting, diarrhoea (17-39%).
7. Chronic bronchitis (40%).
Low serum IgA and IgM, PCB in the sputum.
8. Irregular menstrual cycles (60%).
9. Dark brown skin pigmentation (which gradually fades) of newborn,
retarded growth, abnormal teeth number and shape.
a Numbers in brackets refer to per cent of patients exhibiting the
symptoms.
Table 73. Changes in the clinical symptomatology of Yusho
in the years 1968-1978
1. Skin lesions.
All skin symptoms diminished gradually, subcutaneous cyst formation
still present in some of the most severe cases.
2. Ocular manifestations.
Eye discharge, oedema of the eyelids, pigmentation of eyelids and
conjunctiva, and cyst formation of tarsal gland still present in some
of the cases.
Table 73.(cont'd) Changes in the clinical symptomatology of Yusho
in the years 1968-1978
3. Stomatological alterations.
Pigmentation of oral mucosa decreased gradually; anomalies in number
of teeth and shape of the root still present.
4. Chronic bronchitis correlated in severity with concentration of PCBs
in sputum and blood.
5. Serum triglycerides.
The hyperglycidaemia observed in 1968-1970 returned to a normal level
by 1973 in females and by 1975 in males.
6. Mortality.
Of 737 cases in the Fukuoka region, 51 (6.92%) died between 1968 and
1978; there were 11 cancer deaths (3 stomach cancer, 2 lung cancer,
1 breast cancer, 1 liver cancer, 2 malignant lymphoma).
The rate of excretion of these toxic PCDF isomers is very slow.
Rappe et al., (1983c) could detect 2,3,4,7,8-pentaCDF in blood plasma
from Yusho patients when the samples were collected 11 years after
exposure. Higher levels were found in blood from Yu-cheng patients one
year after exposure and these analyses also showed a 15-20% reduction
in one year (Rappe, 1984). PCDFs are selectively retained in the
liver, with levels corresponding to the fatty level of the tissue.
They are not found in unexposed controls or in PCB-exposed workers.
PCB levels in Yusho patients were only about two times higher than
those of normal people several years after the outbreak. PCB-exposed
workers had more than 10 times greater PCB blood levels than Yusho
patients, whereas the PCQ levels of these two groups were similar.
Generally a correlation between degree or severity of clinical
signs and the amount of PCDFs retained in the blood exists, whereas
there is no correspondence between the severity of disease and PCB
concentrations in blood.
Mild dermal lesions seen in workers exposed to PCB disappeared
quickly after discontinuation of PCB handling, in contrast to the
persistence of Yusho and Yu-cheng symptoms. Everything thus suggests
that the PCDF contaminant is the causative agent.
Immunological evaluation of patients exposed in 1979 in Taiwan
(Yu-cheng) has been reported by Chang et al. (1981, 1982a, 1982b) and
Wu et al. (1984). Serum immunoglobulin concentrations and lymphocyte
subpopulations were determined in the peripheral blood of 30 patients
exposed to PCBs and 23 healthy individuals. The groups were age and
sex matched. In the patients, serum concentrations of IgA and IgM, but
not of IgG, were significantly decreased. Also the percentages of
total T lymphocytes and T-helper lymphocytes were significantly
reduced, whereas the percentages of T-suppressor cells and B
lymphocytes were not affected (Chang et al., 1981).
In a later report (Chang et al., 1982a), monocyte and
polymorphonuclear lymphocyte (PMN) complement and Fc receptors were
evaluated in peripheral blood from 30 Yu-cheng patients and 23 normal
human subjects. Monocytes and PMNs from patients had significantly
lower percentages of cells bearing immunoglobulin Fc and complement
receptors. The immune system was further investigated by determining
the delayed-type hypersensitivity skin response to streptokinase and
streptodornase, as parameters of cellular immune function. The
response was studied in 30 PCB-poisoned patients and 50 healthy
volunteers. Results of the study showed that 80% of the controls had
positive hypersensitivity skin tests, compared to only 43% of the
patients. The significant suppression of cellular immunity correlated
with the severity of the dermal lesions; the size of the
hypersensitivity skin reaction was negatively correlated with the
dermal lesions and also with PCB concentrations in whole blood (Chang
et al., 1982b). More recently, the delayed-type hypersensitivity
response to tuberculin was reported in 83 PCB-poisoned Yu-cheng
patients and in 30 age-and sex-matched healthy controls. Compared to
a positive response rate of 74% in the control group, the patients had
a significantly lower skin response of 48% (40 out of 83 patients). In
contrast to the delayed-type hypersensitivity response, the in
vitro proliferation responses of peripheral blood lymphocytes
treated with phytohaemagglutinin and pokeweed mitogens, as well as
tuberculin, were significantly enhanced (Wu et al., 1984).
There are also indications of immunosuppression in the Yusho
poisoning. Serum IgA and IgM levels decreased considerably within 2
years after the onset of the disease. Respiratory involvement included
bronchiolitis, and respiratory distress was often exacerbated by viral
or bacterial infection (Shigematsu et al., 1978).
The symptomatology of Yusho has been summarized by Reggiani
(1983b) and is to be found in Tables 72 and 73. It is similar to that
of Yu-cheng, but there are differences such as the frequency of
transient visual disturbances, hearing difficulties, and a persistent
bronchitis.
12. EVALUATION OF HEALTH RISKS FROM THE EXPOSURE TO CHLORINATED
DIBENZO-P-DIOXINS (PCDDs) AND DIBENZOFURANS (PCDFs)
12.1 Introduction
In order to evaluate the human health risk of PCDDs and PCDFs, it
is necessary to know both the levels of human exposure and the
corresponding human health effects.
Human exposure assessment is complex. Several approaches may be
taken to estimate it, such as the following.
(a) Use of standard physiological models of inhalation,
ingestion, and dermal absorption. Data requirements
include detailed information on ambient levels in
environmental media and food, and on bioavailability.
(b) Intake estimates based on simple pharmacokinetic models and
known levels in human tissue for accurate assessment of
exposure. Detailed knowledge is also required of the uptake,
distribution, metabolism, and elimination of PCDDs and PCDFs
in humans.
12.2 Exposure Assessment
12.2.1 Sources of contamination
The main sources of PCDDs and PCDFs that have so far been
identified are contaminated commercial chemicals (see section 3.3),
emissions from combustion sources (see section 3.5), and disposal of
industrial wastes containing PCDDs and PCDFs (sections 3.4, 3.5.9, and
3.5.10).
In some cases estimates of the relative contribution of these
sources can be generated on a local basis. However, the data and
methods available today do not allow firm conclusions with regard to
the relative quantitative importance of these sources on a nation-wide
or world-wide basis.
12.2.2 Ambient levels
The limited data available indicate very low (fg/m3) background
levels in ambient air of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs. (If hepta- and octachlorinated
congeners are included, pg/m3 levels are noted).
The few data available indicate that the 2,3,7,8-tetra-, penta-,
and hexachlorinated PCDDs and PCDFs are unlikely to occur in finished
drinking-water, even at a level of 1 pg/litre (see section 5.2). In
all soil and sediment samples analyzed (both from industrialized and
non-industrialized areas), PCDDs and PCDFs were identified at levels
ranging from a few ng/kg to several hundred ng/kg (the latter in
sediments and in urban soil).
There are no available data on background levels of PCDDs and
PCDFs in vegetation in the general environment.
Levels of up to 50 ng/kg of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs (principally the tetra- and
pentachlorinated congeners) have been found in fish from the general
environment. For the most part these have been detected in fatty or
bottom-feeding fish (see section 4.4.2).
Data from terrestrial organisms are inadequate for estimation of
background levels (see section 4.4.3). Three samples of pooled cow's
milk showed a maximum of 100 pg/kg of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs in whole milk (see section 5.4). Data
on contamination of other commercial foods are also very limited.
Analyses of several samples of chicken and pork have shown
contamination with highly chlorinated congeners at about 5-30 ng/kg.
The congener profile differs from that noted in aquatic organisms (see
section 5.4).
The data available are not sufficient for assessing the total
exposure of general populations. They are sufficient to perform a
limited evaluation of exposure for local populations. Based on the
environmental levels discussed above and the usual assumptions
regarding intakes of foodstuffs, air, and water, food is more likely
to be a significant source of PCDD/PCDF exposure than air, while
drinking-water is likely to be of much less concern.
12.2.3 Routes of exposure
Human adipose tissue contains 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs. This contamination is presumably due
to exposure at the ambient concentrations noted in the preceding
sections.
In addition, infants may be exposed through breast milk, and
small children may also be exposed through ingestion of contaminated
soil. However, this latter route of exposure, in most instances, is
likely to be of concern only in heavily contaminated areas.
Some populations have been at special risk through exposure in
industrial accidents (and their clean-up) that have occurred during
the normal production and use of chlorphenols and phenoxy herbicides
and PCBs. In these situations inhalation and dermal contact are the
exposure routes of greatest concern. However, quantitative information
on the nature and concentration of contaminants is not available.
Based on the environmental levels discussed above and the usual
assumptions regarding physiological and intake parameters, ingestion
is likely to be the exposure route of greatest concern. Inhalation of
ambient air is not likely to be a problem, although inhalation of
heavily contaminated air may make a significant contribution to
exposure. In general, it is not possible, at present, to estimate the
relative contribution of dermal exposure.
12.2.4 Bioavailability
No bioavailability data from studies in humans are available.
From animal studies, it is clear that bioavailability of PCDDs and
PCDFs following ingestion depends on the matrix ingested. Table 46
summarizes the data available on oral intake.
Studies on hairless rats indicate that dermal exposure through
intact skin from contact with contaminated soil is about 1 to 2%. No
data are available for inhalation exposure.
12.3 Animal Data
12.3.1 Toxicokinetics of 2,3,7,8-TCDD
Studies on rodents given single or repeated oral doses of
2,3,7,8-TCDD have shown that 50% or more of the administered amount is
absorbed from the gastrointestinal tract in rats, guinea-pigs, and
hamsters, but less than 30% in mice (Table 40). The reported
half-lives for elimination were in the ranges 12-31 days for rats,
mice, and hamsters and 22-94 days for guinea-pigs (Table 41). However,
most of these studies have been performed at toxic doses. The
half-life of 2,3,7,8-TCDD in primates has not been well established,
but available data for the rhesus monkey suggest an apparent half-life
in the adipose tissue of about 1 year.
2,3,7,8-TCDD does accumulate in animal tissues. In rodents
accumulation occurs predominantly in the liver and adipose tissue
(Tables 42 to 44). In rhesus monkeys (Table 45), high levels of
2,3,7,8-TCDD are recovered from the adipose tissues, liver, skin, and
muscles.
At a daily dose of 1 ng 2,3,7,8-TCDD/kg body weight for 2 years,
rats accumulated 540 ng 2,3,7,8-TCDD/kg body weight in the liver where
some morphological changes were also observed. Similar levels were
found in beach mice that had been exposed to 2,3,7,8-TCDD soil levels
ranging from 10-710 ng/kg. Total body exposure of animals to
2,3,7,8-TCDD in soil at such concentrations may thus result in tissue
levels that have been demonstrated to cause effects in experimental
animals.
TCDD is largely eliminated in the faeces, although some urinary
excretion occurs. The hamster has a higher urinary elimination than
other species studied.
Transformation of 2,3,7,8-TCDD to more polar metabolites occurs
in all animal species investigated (see section 6.2 and Table 62).
Elimination of metabolites from tissues into faeces and urine occurs
rapidly in all of these species except in the case of the guinea-pig.
Known metabolites are much less toxic than the parent compound.
12.3.2 Toxicokinetics of PCDDs and PCDFs, other than TCDD
Animal data on the toxicokinetics of pure PCDDs other than
2,3,7,8-TCDD are limited. PCDFs have been more extensively studied in
this respect. The half-life for 2,3,7,8-TCDF has been reported to be
in the range of 2-8 days for rats, mice, and rhesus monkeys and more
than 20 days for guinea-pigs (Table 65). Studies on rats have shown
that 2,3,4,7,8-pentaCDF is more highly retained than is 2,3,7,8-TCDF
(65% and 3.8%, respectively, after 5 days).
Tissue retention data of PCDDs and PCDFs in various species
exposed to synthetic mixtures or to environmental samples containing
PCDDs and PCDFs show a high variability in retention time between
congeners with or without chlorine substitution in all the positions
2,3,7, and 8.
12.3.3 Toxic effects of 2,3,7,8-TCDD
The toxic and biological effects resulting from exposure to
2,3,7,8-TCDD are dependent on a number of factors, including the
species, strain, age, and sex of the animals used. The toxic responses
observed in several animal species include body weight loss,
hepatotoxicity, porphyria, dermal toxicity, gastric lesions, thymus
atrophy and immunotoxicity, teratogenicity, reproductive effects, and
carcinogenicity. TCDD induces a wide spectrum of biological effects
including enzyme induction and vitamin A depletion. The complete
spectrum of toxic and biological effects is not usually observed in
any single animal species. The two most characteristic toxic effects
observed in all laboratory animals are body weight loss and thymus
atrophy and immunotoxicity. Chloracne and related dermal lesions are
the most frequently noted signs of 2,3,7,8-TCDD toxicosis in humans;
dermal lesions are also observed in rhesus monkeys, hairless mice, and
rabbits. In contrast, rats, most strains of mice, guinea-pigs, and
hamsters do not develop chloracne and related dermal toxic lesions
after exposure to 2,3,7,8-TCDD. Many of the observed toxic lesions are
either hyperplastic/metaplastic or hypoplastic, and primarily affect
epithelial tissues.
Reproductive toxicity has been reported in rhesus monkeys: the
lowest-observed-effect level (LOEL) was calculated to be 1 to 2 ng/kg
body weight per day. A no-observed-effect level (NOEL), or possibly a
LOEL, of 1 ng/kg body weight per day for reproductive effects in rats
has been discussed (Murray et al., 1979; Nisbet & Paxton, 1982).
If the cancer studies in rats conducted by Kociba et al. (1978)
and by the NIH (1982a,b) are compared, it is evident that the liver
tumours, including hepatocellular carcinomas, are produced at similar
dose levels. Although an increased incidence of tumours in other
organs was observed by the NTP, and by Kociba et al. (1978), the other
target organs varied in the two studies. This may be caused, in part,
by differences in dosing (gavage versus exposure in ground feed) and
by differences in strains. In the Kociba study, doses of 10 ng/kg body
weight caused an increased incidence of neoplastic (hyperplastic)
nodules in females, and doses of 1 ng/kg body weight resulted in foci
or areas of hepatocellular alteration (swollen hepatocytes). At these
dose rates in experimental groups, the incidence of certain
hormone-dependent tumours was lower than in the control animals,
suggesting endocrine changes induced by 2,3,7,8-TCDD. Based on these
animal studies and on available human data IARC (1982 suppl. 4)
concluded that TCDD showed sufficient evidence for carcinogenicity in
animals, but inadequate evidence for carcinogenicity in humans.
TCDD does not appear to have mutagenic properties, and is,
therefore, not likely to be genotoxic. Thus, it is assumed to be
carcinogenic through an indirect (epigenetic) mechanism.
12.3.4 Toxic effects of PCDDs and PCDFs, other than TCDD
Several other PCDDs and PCDFs cause signs and symptoms similar to
those of 2,3,7,8-TCDD, but there is a wide variation with regard to
potency (Tables 56, 62). In summary, there are 12 isomers that display
high toxicity, i.e., the tetra-, penta-, hexa-, and heptaCDDs and CDFs
with four chlorine atoms in the symmetrical lateral positions 2,3,7,
and 8. A mixture of two hexaCDDs (1,2,3,7,8,9- and
1,2,3,6,7,8-hexaCDD) has been demonstrated to possess carcinogenic
properties in long-term animal studies, but at higher doses than those
used in the study of TCDD. Unsubstituted dioxin and 2,7-diCDD failed
to demonstrate carcinogenic properties.
The relative toxic and biological potencies of PCDDs and PCDFs
have been estimated using short-term studies in rats and mammalian
cell cultures. Endpoints used include inhibition of body weight gain,
thymic atrophy, enzyme induction, teratogenicity, acnegenic response,
and keratinization. In the absence of long-term toxicity data, results
obtained from such short-term tests are at present the only source for
ranking the toxicity for human risk assessment.
When investigated, mixtures of these compounds have shown
additive or less than additive responses.
12.3.5 Review of species differences
There are marked species differences in the susceptibility to the
biological and toxic effects elicited by 2,3,7,8-TCDD. For example,
the oral LD50 values range from 0.6 µg/kg body weight in
guinea-pigs, to 5051 µg/kg body weight in Golden Syrian hamsters
(Table 47); moreover, pronounced differences in LD50 values have
also been reported in different strains of the same species (e.g.,
rats and mice). The toxicity and toxicokinetics of TCDD in monkeys
most closely resemble the effects observed in human beings. However,
the tremendous variation in species and strain sensitivity to
2,3,7,8-TCDD and related compounds cannot be explained by the observed
toxicokinetic differences. There is evidence in inbred mice, that the
cellular levels of the Ah receptor correlate, in part, with
susceptibility to the biological and toxic effects of these compounds.
The receptor has also been identified in other species, including
human beings. However, interspecies comparison of cellular Ah receptor
levels do not explain their differences in sensitivity to
2,3,7,8-TCDD; this is consistent with complex as yet unknown
mechanisms of toxicity that involve multiple factors in addition to
the Ah receptor.
12.4 Human Health Effects
12.4.1 PCDDs
Exposure of the general population is to small amounts of PCDDs
and PCDFs in complex mixtures and these have not been associated with
disease. In a few incidents workers and others have been exposed to
larger amounts of a limited number of these compounds, e.g., Seveso
and in Yusho disease.
For occupational and accidental exposure the most prominent
clinical effect has been chloracne. Other effects (Table 64) have been
noted, but, apart from chloracne and perhaps minor functional
disorders, none has been persistent.
In some, but not all mortality studies, an increased incidence of
cancer at different sites has been claimed, but the small numbers of
cases limit confidence in the findings.
The overall impression from the follow-up studies is that even
severe acute systemic effects of TCDD are usually reversible, except
for chloracne, or markedly improved over time following cessation of
exposure. In Seveso, the only clear-cut adverse health effect recorded
has been chloracne. 193 cases of chloracne occurred in 1976 and 1977,
and 20 of those still presented active symptoms in 1984. Many studies
have been performed to find possible links between exposure and health
effects in civilians or military personnel exposed to Agent Orange in
Viet Nam. However, the information available to date does not allow
definite conclusions to be drawn with regard to effects on human
reproduction or any other significant health effects (see section
9.2).
In a number of studies, exposed populations and various control
groups have been compared by measuring serum lipids, liver function
tests, and other variables. Although certain statistically significant
differences have been reported there, and also in isolated case
reports, lack of uniformity, various technical shortcomings, and the
inability to exclude confounding factors means that the results have
been inconclusive.
In the Missouri (USA) incident, children who showed acute illness
when the contamination occurred in 1971 are now reportedly in good
health. Epidemiological studies in Missouri on populations exposed to
lower concentration over longer periods of time have so far not
revealed any significant health effects. Although no clinical illness
was observed, there were indications of an effect on the cell-mediated
immune system.
The ranges of health effects produced by TCDD in human beings
have yet to be defined. It can be concluded that the data from human
exposure and effects, when taken together, do not allow any
determinations of dose/effect or dose/response relationships in human
beings.
In spite of many clinical and follow-up studies, no clearcut
persistent systemic effects have been delineated, except for
chloracne. In the light of present information, it seems unlikely that
permanent, severe, and debilitating toxicological sequelae are
inevitable after exposure to TCDD.
12.4.2 PCDFs
The only well documented intoxications with PCDFs in human beings
are the two instances of contamination of rice oil with PCDFs, PCBs,
and PCQs, i.e., Yusho in Japan (1968) and Yu-cheng in Taiwan (1979)
(see section 11.1). In total, several thousand people were acutely
intoxicated. The summarized data makes it most likely that the
causative agent was PCDFs. The general symptomatology was similar to
that found in intoxications with TCDD. The differences may reflect
intensity in exposure and the ages and sex of the exposed human
beings. Attempts to estimate the average daily intake of PCDFs over
several months in Yusho patients indicated a figure of 0.9 µg/kg body
weight of total PCDFs, 0.1-0.2 µg/kg of 2,3,7,8-substituted tetra-,
penta-, and hexaPCDFs, together with 157 µg PCBs and 148 µg PCQs/kg
body weight (Hayabuchi et al., 1979). The lowest dose causing disease
was estimated to be 0.6 mg total PCDFs per person over 30 days,
corresponding to a daily dose of 0.05-0.1 µg/kg body weight of
2,3,7,8-substituted PCDFs. However, the data available are not
sufficient to permit any conclusions as to what dose might be safe for
human intake.
12.4.3 Human body burden and kinetics
In human fat, background levels of TCDD up to 20 ng/kg have been
found in the general population with no known specific exposure, but
higher levels have been reported in some cases without evidence of
disease. None of these populations have been randomly sampled. The
more highly chlorinated other PCDDs and PCDFs, especially octaDD, also
occur in these samples (see Tables 29, 30). Averages values seem to
increase with age.
In special situations, higher levels (in the low µg/kg range)
have been found that have not been associated with disease.
In the Yusho and Yu-cheng incidents, symptoms were noticed at
higher levels of PCDFs, e.g., 2,3,4,7,8-pentaCDF was found at 6.9
µg/kg fat tissue one year after the exposure to contaminated rice oil.
Based on the very limited data available, the levels of, for
instance, 2,3,4,7,8-pentaCDF in the general population, seem to be two
orders of magnitude lower than the levels associated with the Yusho
disease.
No such comparisons can be made for PCDDs.
Limited data indicate that those isomers chlorinated at the
2,3,7, and 8 positions are selectively retained, except for TCDF.
A half-life for TCDD in human beings of 5 years has been
indicated by one experimental study. In another study half-lives in
the range of 2-6 years were estimated for 1,2,3,6,7, 8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, octaCDD, 1,2,3,4,6,7,8-heptaCDF, and octaCDF.
These data need to be expanded since they are based on studies in only
two subjects and since the toxicokinetics of these types of compounds
may not simply be controlled by first-order kinetics. However, even if
there are limitations in the present data, it is apparent that the
half-lives of these compounds are in the range of one or more years.
These reported half-lives for human beings are very different
from those reported in rodents. However, animal experiments have
usually been performed with toxic doses. Furthermore, animals with a
short life span have a higher metabolic rate, thus shorter half-lives
could be expected.
The PCDDs and PCDFs are predominantly stored in fat, but they are
also excreted in milk (Table 39) and pass the placenta. They also
appear in the blood and vital organs at lower concentrations. The
distribution between different tissues in human beings is not at
present clear, although it has been suggested that the ratio between
fatty tissue and liver is higher in human beings than that in rodents.
However, this conclusion is based on very limited data from autopsy
specimens. Whether this is relevant for the general population remains
to be seen.
The intake route for human beings is at present not very well
delineated, but it has been assumed that intake from food is the main
route. However, the human infant represents a special case; because of
transplacental transfer of these compounds, the neonate might be
expected to be exposed in utero. Levels measured so far in human
milk suggest that this food might be an important source of these
compounds.
No data are available regarding what dose of PCDDs is toxic to
human beings. However, in the Yusho and Yu-cheng episodes, total
intakes of total PCDFs in the range 3.3-3.8 mg/person and total
intakes of 2,3,7,8-substituted PCDFs in the range 400-500 µg/person
were associated with the disease. No good data are available as to
what intakes occurred without causing disease.
12.5 General Conclusions
PCDDs and PCDFs occur throughout the environment and we all
probably carry a body burden of them. They have sometimes produced
complex toxic effects following occupational and accidental exposure.
Based on the Yusho disease and experiments in sensitive species
of monkeys, and making assumptions about the relative potencies of
PCDDs and PCDFs, human beings and certain monkey species may have
comparable sensitivity to these compounds. However, the uncertainties
related to the real dose received by human beings and the difficulties
of assessing toxic effects other than chloracne in our species prevent
a firm conclusion as to the relative resistance of human beings to the
toxic effects of these compounds. Exposure should be reduced to levels
as low as are reasonably practicable.
13. RECOMMENDATIONS
1. Analytical interlaboratory validation and round-robin studies
using standardized quality assurance and quality control
procedures are needed to improve analytical methodology.
Sampling strategy and analytical procedures and data
interpretation should be optimized and standardized before
undertaking surveys.
2. Further information is required about the origins and
environmental distribution and fate of PCDDs and PCDFs. Further
monitoring data, including time trends and determinations of
isomer patterns, are required for environmental levels of PCDDs
and PCDFs, especially for food, ambient air, and sediments.
3. Data should be obtained about the effects of PCDDs and PCDFs on
environmental biota.
4. More information is required about the bioavailability of PCDDs
and PCDFs from different matrices in the environment and from the
diet. Exposure from these sources should be correlated with
agricultural and industrial practices.
5. Simpler and less expensive methods suitable for screening should
be developed and validated.
6. Studies to determine the mechanisms of toxicity of PCDDs and
PCDFs are needed to support an evaluation of the differences in
effects between species and to allow extrapolation to human
beings.
7. Further investigation of immunotoxicity is important, including
cytotoxic T lymphocyte function. Studies of the effects of
perinatal exposure and of the duration of actions on the immune
system are important.
8. Long-term toxicity studies, including multigeneration
reproductive studies, in different species with three of the most
widespread PCDDs and PCDFs, namely 2,3,4,7,8-pentaCDF,
1,2,3,7,8-pentaCDD, and octaCDD, should be carried out.
9. Because humans are exposed to complex mixtures of PCDDs and
PCDFs, test systems, including techniques applicable to
evaluating human tissues, should be further developed and
validated for the toxic potency of these compounds and other
mixtures. These systems can be used to study mechanisms of
action, structure activity relationships, and interactive
effects.
10. Investigations to examine the body burden and to correlate it
with clinical effects and laboratory findings are indicated.
Follow-up studies of previously exposed groups are important.
14. EVALUATIONS BY INTERNATIONAL BODIES AND THE CONCEPT OF TCDD
EQUIVALENTS
14.1 International Evaluations
IARC evaluated the carcinogenic risk of TCDD to man (IARC, 1977,
1982) and concluded that there was sufficient evidence that it was
carcinogenic to animals, but that the data for carcinogenicity to
human beings was inadequate. None of the other PCDDs or PCDFs have
been evaluated by IARC.
Regulatory standards for TCDD and mixtures containing TCDD,
established by national bodies in different countries and the European
Economic Community, are summarized in the Legal File of the
International Register of Potentially Toxic Chemicals (IRPTC, 1987).
14.2 Methodologies Used in Assessment of Risk from PCDDs and PCDFs
14.2.1 Individual Congeners
As shown in this monograph, sufficient high quality data for
assessing human health risks exist only for TCDD. For the other
congeners and isomers where data do exist, they are generally derived
from studies using acute exposures in experimental animals and/or from
in vitro tests.
The several risk evaluations on TCDD from various countries have
utilized the long-term ocogenic rat studies (Kociba et al., 1978) or
the reproduction studies on rats (Murray et al., 1979) or monkeys
(Schantz et al., 1979). Mathematical models have been applied to the
cancer data and "virtually safe doses" between 0.006 and 0.028 pg/kg
body weight per day have been calculated (Kimbrough, 1984). The
biological relevance of such models has been questioned, since TCDD
has not been shown to be genotoxic, and has been found to be a strong
promoter of liver tumours in a two-stage precarcinogenesis study
(Pitot et al., 1980) To avoid the use of mathematical models, several
evaluations have used safety factors in the range of 100 to 1000
applied to the assumed no-effect, or lowest-observed-effect levels in
the cancer study of Kociba et al. (1978) or the reproduction studies
of Murray et al. (1979) or Schantz et al. (1978). Using this
methodology "tolerable daily intakes" have been calculated for human
beings in the range of 1-10 pg/kg body weight (Denmark, 1984; Ontario,
1985; US EPA, 1985; Ahlborg & Victorin, 1987).
14.2.2 Mixtures of PCDD and PCDF congeners and isomers -
concept of TCDD toxic equivalents.
The results of recent isomer-specific analyses of such diverse
environmental samples as emissions from the combustion of hazardous
industrial and municipal wastes, soil, industrial process wastes,
human adipose tissue, and milk indicate that the majority of the 75
CDD and 135 CDF isomers can be detected. Humans are therefore exposed
to complex mixtures of these environmental contaminants (sometimes
2,3,7,8-tetraCDD is only a minor component), and the level of risk
from such exposures must be assessed.
In the absence of long-term whole animal tests on complex
mixtures of PCDDs and PCDFs, as well as similar studies on individual
isomers and/or congeners, several models have been proposed to relate
the toxicity of environmental mixtures to the well studied isomer
2,3,7,8-TCDD. The results from these models are presented as "TCDD
Toxic Equivalents". A summary of some models which have been published
is given in Table 74. The toxic potencies used, relative to TCDD, are
shown. The scientific basis for deriving these relative toxicities is
somewhat different for each model. The Swiss (Switzerland, 1982) and
Danish (Denmark, 1984) models are based essentially on the relative
potency for AHH induction, whereas the German (Germany, 1985) and
Canadian models (Ontario, 1985) are based on a weighting of all
available quantitative data. The USA model (US EPA, 1987) utilizes
primarily the relative carcinogenic potencies of TCDD and hexaCDD,
with consideration given to other relevant quantitative data. A
discussion of the structure-activity relationships and relative
biological activities of many PCDD and PCDF isomers is given in
sections 7 and 10.
Data requirements, and assumptions made in the application of
these models to various environmental mixtures have been reviewed by
US EPA (1987) and Ontario (1985). Suter-Hofmann & Schlatter (1985) fed
toluene extracts of acid-washed particulates from a municipal waste
incinerator to Sprague Dawley rats at levels in the diet corresponding
to 4, 12, and 24% of particulates. The calculated daily TCDD intakes
at these doses were 4.8, 14.4, and 28.8 ng/kg body weight. Depending
upon which model was used from Table 74, the estimated intake of PCDDs
and PCDFs would be 48-300 (4% particulates), 144-900 (12%
particulates), and 288-1800 ng TCDD-equivalents/kg body weight per day
(24% particulates). No mortality was noted in the groups fed 24%
particulates, but at this dose body weight gain was depressed in
females and depressed thymus and increased liver weights were noted in
both sexes. No adverse affects were seen at the 4% and 12% dose
levels. From the known toxicity of TCDD, more severe effects would
have been expected from the calculated dose of between 288 and 1800 ng
TCDD-equivalents/kg body weight per day. These data support the
conclusion that the application of TCDD-equivalence models may
over-estimate the inherent risk from exposures to dioxin-containing
environmental mixtures, depending largely upon the assumptions used in
deriving the model. However, a definitive conclusion on this point
awaits further research (US EPA, 1987) (see section 13).
Table 74. Examples of TCDD-equivalence models
Relative PCDD and PCDF toxicities in model
Olie et al. Switzerland Germany Denmark Ontario United States
Compound (1983) (1982) (1985) (1984) (1985) (US EPA 1987)
MonoCDD 0 0.001 0
DiCDD 0 0.001 0
TriCDD 0 0.01 0
TetraCDD-2,3,7,8 1.0 1.0 1.0 1.0 1.0 1.0
-non 2,3,7,8 1.0 0.01 0.01 0.01 0.01
PentaCDD-1,2,3,7,8 0.1 0.01
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.5
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.005
HexaCDD-1,2,3,4,7,8 0.1
-1,2,3,6,7,8 0.01
-1,2,3,7,8,9 0.01
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.04
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.0004
HeptaCDD-1,2,3,4,6,7,8 0.01
-all 2,3,7,8 0.1 0.1 0.01 0.01 0.001
-non 2,3,7,8 0.1 0.1 0.001 0.01 0.00001
OctaCDD 0 0.001 0.0001 0
MonoCDF 0.0001
DiCDF 0.0001
TriCDF 0.01
Table 74 (contd).
Relative PCDD and PCDF toxicities in model
Olie et al. Switzerland Germany Denmark Ontario United States
Compound (1983) (1982) (1985) (1984) (1985) (US EPA 1987)
TetraCDF-2,3,7,8 0.1 0.1 0.1 0.1 0.5 0.1
-non 2,3,7,8 0.1 0.1 0.01 0.5 0.001
PentaCDF-1,2,3,7,8 0.2
-2,3,4,7,8 0.2
-all 2,3,7,8 0.1 0.1 0.1 0.5 0.1
-non 2,3,7,8 0.1 0.1 0.01 0.5 0.001
HexaCDF-1,2,3,4,7,8 0.2
-1,2,3,6,7,8 0.05
-1,2,3,7,8,9
-2,3,4,6,7,8 0.1
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.01
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.0001
HeptaCDF-1,2,3,4,6,7,8 0.01
-1,2,3,4,7,8,9
-all 2,3,7,8 0.1 0.1 0.01 0.01 0.00001
-non 2,3,7,8 0.1 0.001 0.01 0.00001
OctaCDF 0 0.001 0.0001 0
It must be recognized that an approach such as the use of
"TCDD-equivalents" must be regarded as an interim procedure for the
measurement of the toxicity of environmental samples in the absence of
long-term toxicity data on specific PCDD and PCDF isomers and mixtures
of these compounds. At present it is an imprecise evaluation
methodology with many data gaps in the supporting data base.
The TCDD-equivalent models have two major sources of uncertainty,
i.e., firstly, the unanswered scientific questions related to the
toxicity of TCDD itself, and secondly, the lack of data on the other
PCDD and PCDF congeners and isomers that would permit a more accurate
determination of the potency of these chemicals relative to TCDD. The
research recommendations in this document address some of these
concerns. As such data become available, TCDD-equivalent models must
be continuously updated and risk assessments based on the present
models (Table 74) considered only as interim evaluations.
REFERENCES
ABATE, L., BASSO, P., BELLONI, A., BISANTI, L., BORGNA, C., BRUZZI,
P., DORIGOTTI, G., FALLIVA, L., FANUZZI, A., FORMIGARO, M., MAGGIORE,
G., MARNI, E., MEAZZA, L., MERLO, F., PUNTONI, R., ROSA, A., STAGNARO,
E., & VERCELLI, M. (1982) Mortality and birth defects from 1976 to
1979 in the population living in the TCDD polluted area of Seveso. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
571-598.
ABERNETHY, D.J., GREENLEE, W.F., HUBAND, J.C., & BOREIKO, C.J. (1985)
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) promotes the transformation
of C3H/10T1/2 cells. Carcinogenesis, 6: 651-653.
ADAMS, E.M., IRISH, D.D., SPENCER, H.C., & ROWE, V.K. (1941) The
response of rabbit skin to compounds reported to have caused acneform
dermatits. Ind. Med., 2: 1-4.
AHLBORG, U.G. & VICTORIN, K. (1987) Impact on health and environment
from trace organic emissions. Waste Manage. Res., 5: 203-224.
AHLBORG, U.G., WAERN, F., & HAKANSSON, H. (1987) Interactive effects
of PCDDs and PCDFs occurring in human mother's milk. Chemosphere,
16(8/9): 1701-1706.
AHLING, B., BJORSETH, A., & LUNE, G. (1978) Formation of chlorinated
hydro- carbons during combustion of poly(vinyl chloride).
Chemosphere, 8(10): 799-806.
AHLING, B., LINDSKOG, A., JANSSON, B., & SUNDSTROM, G. (1977)
Formation of polychlorinated dibenzo-p-dioxins and dibenzo-furans
during combustion of a 2,4,5-T formulation. Chemosphere, 6(8):
461-468.
AITIO, A. & PARKKI, M.G. (1978) Organ specific induction of drug
metabolizing enzymes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the
rat. Toxicol. appl. Pharmacol., 44: 107-114.
AITIO, A., PARKKI, M.G., & MARNIEMI, J. (1979) Different effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on glucuronide conjugation of
various aglycones. Studies in Wistar and Gunn rats. Toxicol. appl.
Pharmacol., 47: 55-60.
AKERMARK, B. (1978) Photochemical reactions of phenoxy acids and
dioxins. Chlorinated phenoxy acids and their dioxins. Ecol. Bull.,
21: 75-81.
ALBRO, P.W. & CORBETT, B.J. (1977) Extraction and clean-up of animal
tissues for subsequent determination of mixtures of chlorinated
dibenzo-p-dioxins and dibenzofurans. Chemosphere, 7: 381-385.
ALBRO, P.W., CORBETT, J.T., HARRIS, M., & LAWSON, L.D. (1978) Effects
of 2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid profiles in tissue of
the Fisher rat. Chem.-biol. Interact., 23: 315-330.
ALBRO, P.W., CRUMMETT, W.B., DUPUY., A.E., Jr, GROSS, M.L., HANSON,
M., HARLESS, R.L., HILEMAN, F.D., HILKER, D., JASON, C., JOHNSON,
J.L., LAMPARSKI, L.L., LAU, B.P.Y., MCDANIEL, D.D., MEEHAN, J.L.,
NESTRICK, T.J., NYGREN, M., O'KEEFE, P., PETERS, T.L., RAPPE, C.,
RYAN, J.J., SMITH, L.M., STALLING, D.L., WEERASINGHE, N.C.A., &
WENDLING, J.M. (1985) Methods for the quantitative determination of
multiple, specific poly-chlorinated dibenzo-p-dioxin and dibenzofuran
isomers in human adipose tissue in the parts-per-trillion range. An
inter-laboratory study. Anal. Chem., 57: 2717-2725.
ALBRO, P.W., CORBETT, J.T., & SCHROEDER, J.L. (1986) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid peroxidation in
microsomal systems in vitro. Chem.-biol. Interact., 57: 301-313.
ALLEN, J.R. (1964) The role of "toxic fat" in the production of
hydro-pericardium and ascites in chickens. Am. J. vet. Res., 25:
1210-1219.
ALLEN, J.R. & CARSTENS, L.A. (1967) Light and electron microscopic
observations in Macaca mulatta monkeys fed toxic fat. Am. J. vet.
Res., 28: 1513-1526.
ALLEN, J.R. & LALICH, J.J. (1962) Response of chickens to prolonged
feeding of crude "toxic fat". Proc. Soc. Exp. Biol. Med., 109:
48-51.
ALLEN, J.R., VAN MILLER, J.P., & NORBACK, D.H. (1975) Tissue
distribution, excretion and biological effects of (14C)
tetrachlorodibenzo-p-dioxin in rats. Food Cosmet. Toxicol., 13:
501-505.
ALLEN, J.R., BARSOTTI, D.A., VAN MILLER, J.P., ABRAHAMSON, L.J., &
LALICH, J.J. (1977) Morphological changes in monkeys consuming a diet
containing low levels of 2,3,7,8-tetrachloro-dibenzo-p-dioxin. Food
Cosmet. Toxicol., 15(5): 401-410.
ALLEN, J.R., HARGRAVES, W.A., HSIA, M.T.S., & LIN, F.S.D. (1979a)
Comparative toxicology of chlorinated compounds on mammalian species.
Pharmacol. Ther., 7: 513-547.
ALLEN, J.R., BARSOTTI, D.A., LAMBRECHT, L.K., & VAN MILLER, J.P.
(1979b) Reproductive effects of halogenated aromatic hydrocarbons on
nonhuman primates. Ann. N.Y. Acad. Sci., 320: 19-27.
ANDERSSON, K., BOSSHARDT, H.P., BUSER, H.R., MARKLUND, S., & RAPPE, C.
(1978) Chlorinated phenoxy acids and their dioxins: Chemistry-summary.
Ecol. Bull., 27: 19-27.
ARSTILA, A.U., REGGIANI, G., SORVARI, T.E., RAISANEN, S., & WIPF, H.K.
(1981) Elimination of 2,3,7,8-tetrachlorodibenzo-p-dioxin in goat
milk. Toxicol. Lett., 9: 215-219.
ASHE, W.F. & SUSKIND, R.R. (1949) Clinical Report on four patients
from Monsanto Chemical Company in Nitro, West Virginia, USA, 14 pp.
(Submitted December 1949).
ASHE, W.F. & SUSKIND, R.R. (1950) Clinical report on six patients
from Monsanto Chemical Company in Nitro, West Virginia, USA, 16 pp.
(Submitted April 1950).
BAADER, E.W. & BAUER, H.J. (1951) Industrial intoxication due to
pentachlorophenol. Ind. Med. Surg., 20(6): 286-290.
BAARS, A.J., MUKHTAR, H., JANSEN, M., & BREIMER, D.D. (1982)
Induction of rat hepatic glutathione-s-tranferase activities by
2,3,7,8-tetrachlorodibenzo-p-dioxin, Oxford, New York, Pergamon
Press, pp. 393-401 (Pergamon Series on Environmental Science, Vol. 5).
BALK, J.L. & PIPER, W.N. (1984) Altered blood levels of
corticosteroids in the rat after exposure to
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Biochem. Pharmacol., 33(15):
2531-2534.
BALL, L.M. & CHABRA, R.S. (1981) Intestinal absorption of nutrients in
rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J.
Toxicol. environ. Health, 8: 629-638.
BANDIERA, S., FARRELL, K., MASON, G., KELLEY, M., ROMKES, M.,
BANNISTER, R., & SAFE, S. (1984a) Comparative toxicities of the
polychlorinated dibenzofuran (PCDF) and biphenyl (PCB) mixtures which
persist in yusho victims. Chemosphere, 13(4): 507-512.
BANDIERA, S., SAWYER, T., ROMKES, M., ZMUDZKA, B., SAFE, L., MASON,
G., KEYS, B., & SAFE, S. (1984b) Polychlorinated dibenzofurans
(PCDFs): Effects of structure on binding to the 2,3,7,8-TCDD cytosolic
receptor protein, AHH induction and toxicity. Toxicology, 32:
131-144.
BANNISTER, R. & SAFE, S. (1987) Aroclor 1254 as a
2,3,7,8-tetrachlorodibenzo-p-dioxin antagonist in C57BL/6J and DBA/2J
mice - effects of enzyme induction. Toxicologist, 7: 160.
BANNISTER, R., MASON, G., KELLEY, M., & SAFE, S. (1986) The effects of
cytosolic receptor modulation on the AHH-inducing activity of
2,3,7,8-TCDD. Chemosphere, 15: 1909-1911.
BARSOTTI, D.A., ABRAHAMSON, L.J., & ALLEN, J.R. (1979) Hormonal
alterations in female rhesus monkeys fed a diet containing
2,3,7,8-tetrachlorodibenzo-p-dioxin. Bull. environ. Contam.
Toxicol., 21: 463-469.
BARTELSON, F.D., HARRISON, D.D., & MORGAN, J.D. (1975) Field
studies of wildlife exposed to TCDD contaminated soil, Florida,
Airforce Armament Laboratory, Eglin Air Force Base (AFATL-TR-75-49).
BASTOMSKY, C.H. (1977) Enhanced thyroxine metabolism and high uptake
goiters in rats after a single dose of
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Endocrinology, 101(1):
292-296.
BAUER, H., SCHULZ, K.H., & SPIEGELBERG, U. (1961) [Occupational
poisoning in the manufacture of chlorophenol compounds.] Arch.
Gewerbepathol. Gewerbehyg., 18: 538-555 (in German).
BAUGHMAN, R.W. (1974) Tetrachlorodibenzo-p-dioxins in the
environment. High resolution mass spectrometry of the picogram
level, Cambridge, Massachussets, Harvard University (Thesis).
BAUGHMAN, R.W. & MESELSON, M. (1973) An analytical method for
detecting TCDD (dioxin): Levels of TCDD in samples from Vietnam.
Environ. Health Perspect., 5: 27-35.
BEALE, M.G., SHEARER, W.T., KARL, M.M., & ROBSON, A.M. (1977)
Long-term effects of dioxin exposure. Lancet, 1: 748.
BEATTY, P.W. & NEAL, R.A. (1977) Factors affecting the induction of
DT-diaphorase by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem.
Pharmacol., 27: 505-510.
BEATTY, P.W., LEMBACH, K.J., HOLSCHER, M.A., & NEAL, R.A. (1975)
Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on mammalian
cells in tissue cultures. Toxicol. appl. Pharmacol., 31: 309-312.
BEATTY, P.W., VAUGHN, W.K., & NEAL, R.A. (1978) Effect of alteration
of rat hepatic mixed-function oxidase (MFO) activity on the toxicity
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. appl.
Pharmacol., 45: 513-519.
BECK, H., ECKART, K., KELLERT, M., MATHAR, W., RUHL, CH.-S., &
WITTOWSKI, R. (1987) Levels of PCDF and PCDD in samples of human
origin and food in the Federal Republic of Germany. Chemosphere,
16: 1977-1982.
BELL, R.A. & GARA, A. (1985) Synthesis and characterization of the
isomers of polychlorinated dibenzofurans, tetra-through octa-. In:
Keith, L.H., Rappe, C., & Choudhary, C.G., ed. Chlorinated dioxins
and dibenzofurans in the total environment, Boston, Butterworth
Publishers, pp. 3-16.
BERGMAN, A., HAGMAN, A., JACOBSSON, S., JANSSON, B., & AHLMAN, M.
(1984) Thermal degradation of polychlorinated alkanes. Chemosphere,
13: 237-250.
BERLIN, A., BURRATTA, A., & VAN DER VENNE, M.-T. (1967) Proceedings
of the Expert Meeting on the Problems raised by TCDD-Pollution,
Milan, Italy, Luxembourg, Commission of the European Communities, p.
179.
BERRY, D.L., ZACHARIAH, P.K., NAMKUNG, M.J., & JUCHAU, M.R. (1976)
Transplacental induction of carcinogen-hydroxylating systems with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol., 36:
569-548.
BERRY, D.L., SLAGA, T.J., WILSON, N.M., ZACHARIAH, P.K., NAMKUNG,
M.J., BRACKEN, W.M., & JUCHAU, M.R. (1977) Tranplacental induction of
mixed-function oxygenases in extra-hepatic tissues by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 26:
1383-1388.
BERRY, D.L., SLAGA, T.J., DIGIOVANNI, J., & JUCHAU, M.R. (1979)
Studies with chlorinated dibenzo-p-dioxins, polybrominated biphenyls,
and poly chlorinated biphenyls in a two-stage system of mouse skin
tumorigenesis: potent anticarcinogenic effects. Ann. N.Y. Acad. Sci.,
320: 405-414.
BERTONI, G., BROCCO, D., DI PALO, V., LIBERTI, A., POSSANZINI, M., &
BRUNER, F. (1978) Gas chromatographic determination of 2,3,7,8-tetra
chloro-dibenzodioxin in the experimental decon-tamination of Seveso
soil by ultraviolet radiation. Anal. Chem., 50: 732-735.
BIRNBAUM, L.S. (1986) Distribution and excretion of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in congenic strains of mice which
differ at the Ah locus. Drug Metab. Disp., 14(1): 34-40.
BIRNBAUM, L.S., DECAD, G.M., & MATTHEWS, H.B. (1980) Disposition and
excretion of 2,3,7,8-tetrachlorodibenzofuran in the rat. Toxicol.
appl. Pharmacol., 55: 342-352.
BIRNBAUM, L.S., DECAD, G.M., MATTHEWS, H.B., & MCCONNELL, E.E. (1981)
Fate of 2,3,7,8-tetrachlorodibenzofuran in the monkey. Toxicol. appl.
Pharmacol., 57: 189-196.
BIRNBAUM, L.S., WEBER, H., HARRIS, M.W., LAMB, J.C., IV, & MCKINNEY,
J.D. (1985) Toxic interaction of specific polychlorinated biphenyls
and 2,3,7,8-tetrachlorodibenzo-p-dioxin: Increased incidence of cleft
palate in mice. Toxicol. appl. Pharmacol., 77: 292-302.
BIRNBAUM, L.S., HARRIS, M.W., MILLER, C.P., PRATT, R.M., & LAMB, J.C.
(1986) Synergistic interaction of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
and hydro-cortisone in the induction of cleft palate in mice.
Teratology, 33: 29-35.
BIRNBAUM, L.S., HARRIS, M.W., BARNHART, E.R., & MORRISSEY, R.E. (1987)
Teratogenicity of three polychlorinated dibenzo-furans in C57BL/6N
mice. Toxicol. appl. Pharmacol., 90: 206-216.
BLEIBERG, J., WALLEN, M., BRODKIN, R., & APPLEBAUM, I.L. (1964)
Industrially acquired porphyria. Arch. Dermatol., 89: 793-797.
BLOMHOFF, R., HELGERUD, P., RASMUSSEN, M., BERG, T., & NORUM, K.R.
(1982) In vivo uptake of chylomicron (3H)retinyl ester by rat liver:
Evidence for retinol transfer from parenchymal to nonparenchymal
cells. Proc. Natl Acad. Sci. (USA), 79: 7326-7330.
BOER, F.P., NEUMAN, M.A., VAN REMOORTERE, F.P., NORTH, P.P., & RINN,
H.W., (1973) X-Ray diffraction studies of chlorinated
dibenzo-p-dioxins. Chlorodioxins - origin and fate. Adv. Chem.
Ser., 120, 14-25.
BOMBICK, D.W., MATSUMURA, F., & MADHUKAR, B.V. (1984) TCDD
(2,3,7,8-Tetrachlorodibenzo-p-dioxin) causes reduction in the low
density lipoprotein (LDL) receptor activities in the hepatic plasma
membrane of the guinea pig and rat. Biochem. biophys. Res.
Commun., 118: 548-554.
BOMBICK, D.W., MADHUKAR, B.V., BREWSTER, D.W., & MATSUMURA, F. (1985)
TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) causes increases in protein
kinases particularly protein kinase C in the hepatic plasma membrane
of the rat and the guinea pig. Biochem. biophys. Res. Commun.,
127(1): 296-302.
BONACCORSI, A., DI DOMENICO, A., FANELLI, R., MERLI, F., MOTTA, R.,
VANZATI, R., & ZAPPONI, G.A. (1984) The influence of soil particle
adsorption on 2,3,7,8-tetrachlorodibenzo-p-dioxin biological uptake in
the rabbit. Arch. Toxicol., 7: 431-434.
BOTRE, C., MEMOLI, A., & ALHAIQUE, F. (1978) TCDD solubilization and
photodecomposition in aqueous solutions. Environ. Sci. Technol.,
12: 335-336.
BOWES, G.W., MULVIHILL, M.J., DECAMP, M.R., & KENDE, A.S. (1975) Gas
chromatographic characteristics of authentic chlorinated
dibenzofurans; identification of two isomers in American and Japanese
polychlorinated biphenyls. J. agric. food Chem., 23(6): 1222-1223.
BRADLAW, J.A. & CASTERLINE, J.L., Jr (1979) Induction of enzyme
activity in cell culture: a rapid screen for detection of planar
polychlorinated organic compounds. J. Assoc. Off. Anal. Chem.,
62(4): 904-916.
BRADLAW, J.A., GARTHOFF, L.H., HURLEY, N.E., & FIRESTONE, D. (1980)
Comparative induction of aryl hydrocarbon hydroxylase activity in
vitro by analogues of dibenzo-p-dioxin. Food Cosmet. Toxicol., 18:
627-635.
BRAUN, W. (1955) [Chloracne. Monographs with the Journal
Berufsdermatosen,] Aulendorf i. Wurtt., Editio Cantor, Vol. 1. (in
German).
BREWSTER, D.W., MADHUKAR, B.V., & MATSUMURA, F. (1982) Influence of
2,3,7,8-TCDD on the protein composition of the plasma membrane of
hepatic cells from the rat. Biochem. biophys. Res. Commun., 107:
68-74.
BRONZETTI, G., BAUER, C., CORSI, C., DEL CARRATORE, R., NIERI, R., &
PAOLINI, M. (1983) Mutagenicity study of TCDD and ashes from urban
incinerator "in vitro" and "in vivo" using yeast D7 strain.
Chemosphere, 12(4/5): 549-553.
BROWN, E.F. & MORGAN, A.F. (1948) The effect of vitamin A deficiency
upon the nitrogen metabolism of the rat. J. Nutr., 35: 425-438.
BUMB, R.R., CRUMMETT, W.B., CUTIE, S.S., GLADHILL, R.H., VAGEL, R.O.,
LAMPAVSKI, L.L., LUOMA, E.V., MILLER, D.L., NESTRIDE, T.J., SHADOFF,
L.A., STEHL, R.H., & WOODS, J.S., (1980) Trace chemistries of fire: A
source of chlorinated dioxins. Science, 210: 385-390.
BURKHARD, L.P. & KUEHL, D.W. (1986) N-Octanol/water partition
coefficients by reverse phase liquid chromatography/mass spectrometry
for eight tetrachlorinated planar molecules. Chemosphere, 15:
163-167.
BUS, J.S. & GIBSON, J.E. (1979) Lipid peroxidation and its role in
toxicology. Rev. Biochem. Toxicol., 1: 125-149.
BUSER, H.R. (1975) Polychlorinated dibenzo-p-dioxins. Separation and
identification of isomers by gas chromotagrophy mass spectrometry. J.
Chromatogr., 114: 95-108.
BUSER, H.R. (1979) Formation of polychlorinated dibenzofurans (PCDFs)
and dibenzo-p-dioxins (PCDDs) from the pyrolysis of chlorobenzenes.
Chemosphere, 6: 415-424.
BUSER, H.R. & BOSSHARDT, H.P. (1976) Determination of polychlorinated
dibenzo-p-dioxins and dibenzofurans in commercial pentachlorophenols
by combined gas chromatography - mass spectrometry. J. Assoc. Off.
Anal. Chem., 59: 562-569.
BUSER, H.R. & BOSSHARDT, H.P. (1978) [Polychlorinated
dibenzo-p-dioxin, dibenzofuran and benzene in the ashes from municipal
and industrial incinerators.] Mitt. Geb. Lebensm. Hyg., 69:
191-199 (in German).
BUSER, H.R. & RAPPE, C. (1978) Identification of substitution patterns
in polychlorinated dibenzo-p-dioxins (PCDDs) by mass spectrometry.
Chemosphere, 7: 199-211.
BUSER, H.R. & RAPPE, C. (1979) Formation of polychlorinated
dibenzofurans (PCDFs) from the pyrolysis of individual PCB isomers.
Chemosphere, 3: 157-174.
BUSER, H.R. & RAPPE, C. (1980) High-resolution gas chromato-graphy of
the 22 tetrachlorodibenzo-p-dioxin isomers. Anal. Chem., 52:
2257-2262.
BUSER, H.R. & RAPPE, C. (1984) Isomer-specific separation of
2,3,7,8-substituted polychlorinated dibenzo-p-dioxins by
high-resolution gas chromatography/mass spectrometry. Anal. Chem.,
56: 442-448.
BUSER, H.R., BOSSHARDT, H.P., & RAPPE, C. (1978a) Formation of
polychlorinated dibenzofurans (PCDFs) from the pyrolysis of PCBs.
Chemosphere, 1: 109-119.
BUSER, H.R., BOSSHARDT, H.P., & RAPPE, C. (1978b) Identification of
polychlorinated dibenzo-p-dioxin isomers found in fly ash.
Chemosphere, 7(2): 165-172.
BUSER, H.R., BOSSHARDT, H.P., RAPPE, C., & LINDAHL, R. (1978c)
Identification of polychlorinated dibenzofuran isomers in fly ash and
PCB pyrolysis. Chemosphere, 5: 419-429.
BUSER, H.R., RAPPE, C., & GARA, A. (1978d) Polychlorinated
dibenzofurans (PCDFs) found in Yosho oil and in used Japanese PCB.
Chemosphere, 5: 439-449.
BUSER, H.R., RAPPE, C., & BERGQVIST, P.-A. (1985) Analysis of
polychlorinated dibenzofurans, dioxins, and related compounds in
environmental samples. Environ. Health Perspect., 60: 293-302.
BUU-HOI, N.P., HIEN, D.P., SAINT-RUF, G., & SERVOIN-SIDOINE, J.
(1971a) Propriétés cancéromimétiques de la tétrachloro-2,3,7,8
dibenzo-p-dioxin. C. R. Acad. Sci. Paris, 272: 1447-1450.
BUU-HOI, N.P., SAINT-RUF, G., BIGOT, P., & MANGANE, M. (1971b)
Préparation, propriétés et identification de la "dioxine"
(tétrachloro-2,3,7,8-dibenzo-p-dioxine) dans les pyrolysats de
défoliants à base d'acide trichloro-2,4,5 phénoxyacétique et de ses
esters et des végétaux contaminés. C. R. Acad. Sci. Paris, 273:
708-711.
BUU-HOI, N.P., CHANH, P.H., SESQUE, G., AZUM-GELADE, M.C., &
SAINT-RUF, G. (1972a) Organs as targets of "dioxin"
(2,3,7,8-tetrachlorodibenzo-p-dioxin) intoxication.
Natur-wissenschaften, 59: 174-175.
BUU-HOI, N.P., CHANH, P.H., SESQUE, G., AZUM-GELADE, M.C., &
SAINT-RUF, G. (1972b) Enzymatic functions as targets of the toxicity
of "dioxin" (2,3,7,8-tetrachlorodibnzo-p-dioxin.
Naturwissenschaften, 59: 173-174.
CANTONI, L., SALMONA, M., & RIZZARDINI, M. (1981) Porphyrogenic effect
of chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin in
female rats. Dose-effect relationship following urinary excretion of
porphyrins. Toxicol. appl. Pharmacol., 57: 156-163.
CANTONI, L., DAL FIUME, D., FERRAROLI, A., SALMONA, M., & RUGGIERI, R.
(1984a) Different susceptibility of mouse tissues to porphyrogenic
effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Lett., 20:
201-210.
CANTONI, L., DAL FIUME, D., RIZZARDINI, M., & RUGGIERI, R. (1984b) In
vitro inhibitory effect on porphyrinogen carboxylase of liver extracts
from TCDD treated mice. Toxicol. Lett., 20: 211-217.
CARLSTEDT-DUKE, J.M.B. (1979) Tissue distribution of the receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Cancer Res., 39:
3172-3176.
CARLSTEDT-DUKE, J.M.B, ELFSTROEM, G., HOEGBERG, B., & GUSTAFSSON,
J.-A. (1979) Oncogeny of the rat hepatic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin and its endocrine independence.
Cancer Res., 39: 4653-4656.
CARLSTEDT-DUKE, J.M.B., HARNEMO, U.-B., HOEGBERG, B., & GUSTAFSSON,
J.-A. (1981) Interaction of the hepatic receptor protein for
2,3,7,8-tetrachlorodibenzo-p-dioxin with DNA. Biochim. Biophys.
Acta, 672: 131-141.
CARLSTEDT-DUKE, J.M.B., KURL, R., POELLINGER, L., GILLNER, M.,
HANSSON, L.-A., TOFTGARD, R., HOEGBERG, B., & GUSTAFSSON, J.-A. (1982)
The detection and function of the cytosolic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related cocarcinogens.
In: Hutzinger, O., ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
355-365
CARTER, C.D., KIMBROUGH, R.D., LIDDLE, J.A., CLINE, R.E., ZACH, M.M.,
Jr, BARTHEL, W.F., KOEHLER, R.E., & PHILLIPS, P.E. (1975)
Tetrachlorodibenzo-dioxin: An accidental poisoning episode in horse
arenas. Science, 188: 738-740.
CAVALLARO, A., BANDI, G., INVERNIZZI, G., LUCIANI, L., MONGINI, E., &
GORNI, G. (1982) Negative ion chemical ionization ms as a structure
tool in the determination of small amounts of PCDD and PCDF. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds,
Oxford, New York, Pergamon Press, Vol. 5, pp. 55-65.
CECIL, H.C., HARRIS, S.J., BITMAN, J., & FRIES, G.F. (1973)
Polychlorinated biphenyl-induced decrease in liver vitamin A in
Japanese Quail and rats. Bull. environ. Contam. Toxicol., 9:
179-185.
CHANG, K.J., HSIEH, K.H., LEE, T.P., TANG, S.Y., & TUNG, T.C. (1981)
Immunologic evaluation of patients with polychlorinated biphenyl
poisoning: determination of lymphocyte subpopulations. Toxicol.
appl. Pharmacol., 61: 58-63.
CHANG, K.J., HSIEH, K.H., LEE, T.P., & TUNG, T.C. (1982a) Immunologic
evaluation of patients with polychlorinated biphenyl poisoning:
determination of phagocyte Fc and complement receptors. Environ. Res.,
28: 329-334.
CHANG, K.J., HSIEH, K.H., TANG, S.Y., TUNG, T.C., & LEE, T.P. (1982b)
Immunologic evaluation of patients with polychlorinated biphenyl
poisoning: evaluation of delayed-type skin hypersensitive response and
its relation to clinical studies. J. Toxicol. environ. Health,
9: 217-223.
CHAPMAN, D.E. & SCHILLER, C.M. (1985) Dose-related effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57BL/6J and DBA/2J
mice. Toxicol. appl. Pharmacol., 78: 147-157.
CHASTAIN, J.E. & PAZDERNIK, T.L. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD)-induced immunotoxicity.
Int. J. Immunopharmacol., 7(6): 849-856.
CHEN, P.H. & HITES, R.A. (1983) Polychlorinated biphenyls and
dibenzofurans retained in the tissues of a deceased patient with
Yu-Cheng in Taiwan. Chemosphere, 12: 1507-1516.
CHEN, P.H., GAW, J.M., WONG, C.K., & CHEN, C.J. (1980) Levels and gas
chromatographic patterns of polychlorinated biphenyls in the blood of
patients after PCB poisoning in Taiwan. Bull. environ. Contam.
Toxicol., 25: 325-329.
CHEN, P.H., CHANG, K.T., & LU, Y.D. (1981) Polychlorinated biphenyls
and polychlorinated dibenzofurans in the toxic rice-bran oil that
caused PCB poisoning in Taichung. Bull. environ. Contam. Toxicol.,
26, 489-495.
CHEN, P.H., WONG, C.K., RAPPE, C., & NYGREN, M. (1985) Polychlorinated
biphenyls, dibenzofurans and quaterphenyls in toxic rice-bran oil and
in the blood and tissues of patients with PCB poisoning (Yu-Cheng) in
Taiwan. Environ. Health Perspect., 59: 59-65.
CHEUNG, M.O., GILBERT, E.F., & PETERSON, R.E. (1981) Cardiovascular
teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the chick
embryo. Toxicol. appl. Pharmacol., 61: 197-204.
CHRISTIAN, B.J., INHORN, S.L., & PETERSON, R.E. (1986a) Relationship
of the wasting syndrome to lethality in rats treated with
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
82: 239-255.
CHRISTIAN, B.J., MENAHAN, L.A., & PETERSON, R.E. (1986b) Intermediary
metabolism of the mature rat following
2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Toxicol. appl.
Pharmacol., 83: 360-378.
CLARK, D.A., GAULDIE, J., SZEWCZUK, M.R., & SWEENEY, G. (1981)
Enhanced suppressor cell activity as a mechanism of immunosuppression
by 2,3,7,8-tetrachlorodibenzo-p-dioxin (41275). Proc. Soc. Exp. Biol.
Med., 168: 290-299.
CLARK, D.A., SWEENEY, G., SAFE, S., HANCOCK, E., KILBURN, D.G., &
GAULDIE, J. (1983) Cellular and genetic basis for suppression of
cytotoxic T-cell generation by haloaromatic hydrocarbons.
Immunopharmacology, 6: 143-153.
CLEMENT, R.E., TOSINE, H.M., & ALI, B. (1985) Levels of
polychlorinated dibenzo-p-dioxins and dibenzofurans in wood burning
stoves and fireplaces. Chemosphere, 14: 815-819.
COCCIA, P., CROCI, T., & MANARA, L. (1981) Less TCDD persists in liver
2 weeks after a single dose to mice fed chow with added charcoal or
cholic acid. Br. J. Pharmacol., 72: 181-182.
COCHRANE, W.P., SINGH, J., MILES, W., WAKEFORD, B., & SCOTT, J. (1981)
Analysis of technical and formulated products of 2,4-dichlorophenoxy
acid for the presence of chlorinated dibenzo-p-dioxins. In: Hutzinger,
O., Frei, R.W., Merian, E., & Pocchiari, F., ed. Impact of
chlorinated dioxins and related compounds on the environment,
Oxford, New York, Pergamon Press, pp. 209-213.
COHEN, G.M., BRACKEN, W.M., IYER, R.P., BERRY, D.L., SELKIRK, J.K., &
SLAGA, T.J. (1979) Anticarcinogenic effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on benzo(a)pyrene and
7,12-dimethylbenz(a)anthracene tumor initiation and its relationship
to DNA binding. Cancer Res., 39: 4027-4033.
CONAWAY, C.C. & MATSUMURA, F. (1975) Alteration of cellular
utilization of thymidine by TCDD (2,3,7,8-tetrachlorodibenzo-p
-dioxin). Bull. environ. Contam. Toxicol., 12: 52-56.
CONSTABLE, J.D. & HATCH, M.C. (1985) Reproductive effects of herbicide
exposure in Vietnam: Recent studies by the Vietnamese and others.
Teratog. Carcinog. Mutagen., 5: 231-250.
COOK, R.R. (1981) Dioxin, chloracne, and soft tissue sarcoma.
Lancet, 1: 618-619.
COOK, R.R., TOWNSEND, J.C., OTT, M.G., & SILVERSTEIN, L.G. (1980)
Mortality experience of employees exposed to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. occup. Med., 22:
530-532.
COURTNEY, K.D. (1976) Mouse teratology studies with
chlorodibenzo-p-dioxins. Bull. environ. Contam. Toxicol., 16:
674-681.
COURTNEY, K.D. & MOORE, J.A. (1971) Teratology studies with
2,4,5-trichlorophenoxy-acetic acid and
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
20: 396-403.
COURTNEY, K.D., GAYLOR, D.W., HOGAN, M.D., FALK, H.L., BATES, R.R., &
MITCHEL, I. (1970) Teratogenic evaluation of 2,4,5-T. Science,
168: 864-866.
COURTNEY, K.D., PUTNAM, J.P., & ANDREWS, J.E. (1978) Metabolic studies
with TCDD (Dioxin) treated rats. Arch. environ. Contam. Toxicol.,
7: 385-396.
COWARD, K.H. (1947) The determination of vitamin A. In: The
biological standardisation of the vitamins, London, Baillière,
Tindall and Cox, pp. 23-58.
CROSBY, D.G. & WONG, A.S. (1976) Photochemical generation of
chlorinated dioxins. Chemosphere, 5: 327-332.
CROSBY, D.G. & WONG, A.S. (1977) Environmental degradation of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Science, 195:
1337-1338.
CROSBY, D.G., WONG, A.S., PLIMMER, J.R., & WOOLSON, E.A. (1971)
Photodecomposition of chlorinated dibenzo-p-dioxins. Science, 173:
748-749.
CROSBY, D.G., MOILANEN, K.W., & WONG, A.S. (1973) Environmental
generation and degradation of dibenzodioxins and dibenzofurans.
Environ. Health Perspect., 5: 259-266.
CROW, K.D. (1970) Chloracne. A critical review including a comparison
of two series of cases of acne from chlornaphtalene and pitch fumes.
Trans. St. John's Hosp. Dermatol. Soc., 56: 79-99.
CRUMMETT, W.B. & STEHL, R.H. (1973) Determination of chlorinated
dibenzo-p-dioxins and dibenzofurans in various materials. Environ.
Health Perspect., 5: 15-25.
CRUMMETT, W.B., NESTRICK, T.J., & LAMPARSKI, L.L., (1985) Analytical
methodology for the determination of PCDDs in environmental samples:
an overview and critique. In: Kamrin, M.A. & Rodgers, P.W., ed.
Dioxins in the environment, Washington, DC, Hemisphere Publishing,
pp. 57-83.
CUNNINGHAM, H.M. & WILLIAMS, D.T. (1972) Effect of
tetrachlorodibenzo-p-dioxin on growth rate and the synthesis of lipids
and proteins in rats. Bull. environ. Contam. Toxicol., 7: 45-51.
CZUCZWA, J.M. & HITES, R.A. (1985) Historical record of
polychlorinated dioxins and furans in Lake Huron sediments. In: Keith,
L.H., Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment, Stoneham, Maine, Butterworth
Publishers, pp. 59-63.
DALDERUP, L.M. (1974a) Safety measures for taking down buildings
contaminated with toxic material I. T. Soc. Geneeskd., 52: 582-588.
DALDERUP, L.M. ((1974b) Safety measures for taking down buildings
contaminated with toxic material II. T. Soc. Geneeskd., 52:
616-623.
D'ARGY, R., HASSOUN, E., & DENCKER, L. (1984) Teratogenicity of TCDD
and the congener 3,3',4,4'-tetrachloroazoxybenzene in sensitive and
nonsensitive mouse strains after reciprocal blastocyst transfer.
Toxicol. Lett., 21: 197-202.
DAVISON, K.L. & COX, J.H. (1976) Methoxychlor effects on hepatic
storage of vitamin A in rats. Bull. environ. Contam. Toxicol., 16:
145-148.
DECAD, G.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1981a)
2,3,7,8-tetrachlorodibenzofuran tissue distribution and excretion in
guinea pigs. Toxicol. appl. Pharmacol., 57: 231-240.
DECAD, G.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1981b) Distribution and
excretion of 2,3,7,8-tetrachlorodibenzofuran in C57BL/6J and DBA/2J
mice. Toxicol. appl. Pharmacol., 59: 564-573.
DECAPRIO, A.P., MCMARTIN, D.N., SILKWORTH, J.B., REJ, R., PAUSE, R.,
& KAMINSKY, L.S. (1983) Subchronic oral toxicity in guinea pigs of
soot from a polychlorinated biphenyl-containing transformer fire.
Toxicol. appl. Pharmacol., 68: 308-322.
DECAPRIO, A.P., MCMARTIN, D.N., O'KEEFE, P.W., REJ, R., SILKWORTH,
J.B., & KAMINSKY, L.S. (1986) Subchronic oral toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the guinea pig: comparisons
with a PCB-containing transformer fluid pyrolysate. Fundam. appl.
Toxicol., 6: 454-463.
DEITRICH, R.A., BLUDEAU, P., ROPER, M., & SCHMUCK, J. (1978) Induction
of aldehyde dehydrogenases. Biochem. Pharmacol., 27: 2343-2347.
DENCKER, L. & PRATT, R.M. (1981) Association between the presence of
the Ah receptor in embryonic murine tissues and sensitivity to
TCDD-induced cleft palate. Teratog. Carcinog. Mutagen., 1:
399-406.
DENCKER, L., HASSOUN, E., D'ARGY, R., & ALM, G. (1985) Fetal thymus
organ culture as an in vitro model for the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin and its congeners. Mol.
Pharmacol., 27: 133-140.
DENISON, M.S., VELLA, L.M., & OKEY, A.B. (1986a) Structure and
function of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Species difference in molecular properties of the receptors from mouse
and rat hepatic cytosols. J. biol. Chem., 261: 3987-3995.
DENISON, M.S., WILKINSON, C.F., & OKEY, A.B. (1986b) Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin: comparative studies in mammalian
and nonmammalian species. Chemosphere, 15: 1665-1672.
DENISON, M.S., HARPER, P.A., & OKEY, A.B. (1986c) Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin. Codistribution of unoccupied
receptor with cytosolic marker enzymes during fractionation of mouse
liver, rat liver and cultured Hepa-1c1 cells. Eur. J. Biochem.,
155: 223-229.
DENMARK (1984) [Formation and emission of dioxins especially in
connection with waste incineration: supplement,] Copenhagen,
Miljöstyrelsen (in Danish).
DICKINS, M., SEEFELD, M.D., & PETERSON, R.E. (1981) Enhanced liver DNA
synthesis in partially hepatectomized rats pre-treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
58: 389-398.
DIDOMENICO, A., SILANO, V., VIVIANO, G., & ZAPPONI, G. (1980)
Accidental release of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) at
Seveso, Italy. II. TCDD distribution in the soil surface layer.
Ecotoxicol. environ. Saf., 4: 298-320.
DIGIOVANNI, J., VIAJE, A., BERRY, D.L., SLAGA, T.J., & JUCHAU, M.R.
(1977) Tumor-initiating ability of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) and arochlor 1254 in the
two-stage system of mouse skin carcinogenesis. Bull. environ.
Contam. Toxicol., 18: 552-557.
DIGIOVANNI, J., BERRY, D.L., JUCHAU, M.R., & SLAGA, T.J. (1979)
2,3,7,8-Tetra-chlorodibenzo-p-dioxin: potent anti-carcinogenic
activity in CD-1 mice. Biochem. biophys. Res. Commun., 86:
577-584.
DIGIOVANNI, J., BERRY, D.L., GLEASON, G.L., KISHORE, G.S., & SLAGA,
T.J. (1980) Time dependent inhibition by
2,3,7,8-tetra-chlorodibenzo-p-dioxin of skin tumorogenesis with
polycyclic hydrocarbons. Cancer Res., 40: 1580-1587.
DOBLES, A.J. & GRANT, C. (1979) Photolysis of highly chlorinated
dibenzo-p-dioxins by sunlight. Nature (Lond.), 278: 162-165.
DOHMEIER, H.J. & JANSON, E. (1983) [Killing flies and people:
Dioxin - the poison of Seveso and Viet Nam and how we come into
daily contact with it.] Reinbek, Aktuell Rororo, p. 25.
DONOVAN, J.W., MACLENNAN, R., & ADENA, N. (1984) Vietnam service and
the risk of congenital anomalies: a case-control study. Med. J.
Aust., 140: 394-397.
DOYLE, B.W., DRUM, D.A., & LAUDER, J.D. (1985) The smoldering question
of hospital wastes. Pollut. Eng., 17: 35-39.
DOYLE, E.A. & FRIES, G.F. (1986) Induction of aryl hydrocarbon
hydroxylase by chlorinated dibenzofurans in rats. Chemosphere, 15:
1745-1748.
DRINKER, C.K., FIELD WARREN, M., & GRANVILLE, A. (1937) The problem of
possible systemic effects from certain chlorinated hydrocarbons. J.
ind. Hyg. Toxicol., 19: 283-311.
DUGOIS, P. & COLOMB, L. (1956) Acné chlorique au 2-4-5
trichlorophénol. J. Méd. Lyon, 88: 446-447.
DUGOIS, P. & COLOMB, L. (1957) Remarques sur l'acné chlorique. A
propos d'une éclosion de cas provoqués par la préparation du
2,4,5-trichlorophénol. J. Méd. Lyon, 38: 899-903.
DUGOIS, P., MARECHAL, J., & COLOMB, L. (1958) Acné chlorique au
2,4,5-tri-chlorophénol. Arch. Mal. prof. Hyg. Toxicol. ind., 19:
626-627.
DUGOIS, P., AMBLARD, P., AIMARD, M., & DESHORS, G. (1967) Acné
chlorique collective et accidentelle d'un type nouveau. Bull. Soc.
Dermatol., 75: 260-261.
DUVERNE, J., THIVOLET, J., & BERARD, J. (1964) Acné chlorique profuse
avec épanchement pleural récidivant chez un sujet à réactions
tuberculiniques négatives. Bull. Soc. Fr. Dermatol. Syphiligr.,
71: 649-652.
EATON, D.L. & KLAASSEN, C.D. (1979) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin, kepone, and polybrominated
biphenyls on transport systems in isolated rat hepatocytes. Toxicol.
appl. Pharmacol., 51: 137-144.
EDULJEE, G.H., ATKINS, D.H.F., & EGGLETON, A.E. (1986) Observations
and assessment relating to incineration of chlorinated chemical
wastes. Chemosphere, 15: 1577-1584.
ELDER, D.G., LEE, G.B., & TOVEY, J.A. (1978) Decreased activity of
hepatic uroporphyrinogen decarboxylase in sporadic porphyria cutanea
tarda. New Engl. J. Med., 299: 274-278.
ELDER, G.H. & SHEPPARD, D.M. (1982) Immunoreactive uroporphyrinogen
decarboxylase is unchanged in porphyria caused by TCDD and
hexachlorobenzene. Biochem. biophys. Res. Commun., 109: 113-120.
ELDER, G.H., EVANS, J.O., & MATLIN, S.A. (1976) The effect of
porhyrogenic compound, hexachlorobenzene, on the activity of hepatic
uroporphyrinogen decarboxylase in the rat. Clin. Sci. mol. Med.,
51: 71-80.
ERICKSON, J.D., MULINARE, J., MCCLAIN, P.W., FITCH, T.G., JAMES, L.M.,
MCCLEARN, A.B., & ADAMS, M.J. (1984) Vietnam veterans' risks for
fathering babies with birth defects. J. Am. Med. Assoc., 252:
903-937.
ESPOSITO, G., FAELLI, A., & CAPRARO, V. (1967) Metabolism and
transport phenomena in isolated intestine of normal and semi-starved
rats. Arch. int. Physiol. Biochim., 75: 601-608.
ESPOSITO, M.P., TIERNAN, T.O., & DRYDEN, F.E. (1980) Dioxins,
Cincinnati, Ohio, US Environmental Protection Agency, Office of
Research and Development (EPA-600/2-80-197).
FACCHETTI, S., BALASSO, A., FICHTNER, C., FRARE, G., LEONI, A., MAURI,
C., & VASCONI, M. (1986) Studies on the absorption of TCDD by plant
species. In: Rappe, C., Choudhary, G., & Keith, L.H., ed. Chlorinated
dioxins and dibenzofurans in perspective, Chelsea, Michigan, Lewis
Publishers, pp. 225-239.
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic activity of
chlorophenols and chlorophenol impurities. Environ. Sci. Res., 12:
325-338.
FAITH, R.E. & LUSTER, M.I. (1979) Investigations on the effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on parameters of various
immune functions. Ann. N.Y. Acad. Sci., 320: 564-571.
FAITH, R.E. & MOORE, J.A. (1977) Impairment of thymus-dependent immune
functions by exposure of the developing immune system to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. Toxicol. environ.
Health, 3: 451-464.
FARA, G.M., DEL CORNO, G., BONETTI, F., CARAMASHI, F., DARDANONI, L.,
FAVARETTI, C., GIAMBELLUCA, S.E., MARNI, E., MOCARELLI, P.,
MONTESARCHIO, E., PUCCINELLI, V., & VOLPATO, C. (1976) Chloracne after
release of TCDD at Seveso, Italy. In: Hutzinger, O., Frei, R., Merian,
E., & Pocchiari, F. ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
545-559.
FARRELL, K. & SAFE, S. (1986) 2,3,7,8-tetrachlorodibenzo-p-dioxin:
Relationship between toxicity and the induction of aryl hydrocarbon
hydroxylase and ornithine decarboxylase. Chemosphere, 15:
1971-1976.
FEDERAL REGISTER (1980) Storage and disposal of waste material:
Prohibition of disposal of tetrachlorodibenzo-p-dioxin. Fed. Reg.,
45(98): 32676.
FILIPPINI, G., BORDO, B., CRENNA, P., MASSETTO, N., MUSICCO, M., &
BOERI, R. (1981) Relationship between clinical and
electrophysiological findings and indicators of heavy exposure to
2,3,7,8-tetrachlorodibenzo-dioxin. Scand. J. Work Environ. Health,
7: 257-262.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B., GROTH,
D.H., & RUSSELL, W.O. (1984) Review of exposure and pathology data for
seven cases reported as soft tissue sarcoma among persons
occupationally exposed to dioxin-contaminated herbicides. In:
Lowrance, W.W., ed. Public health risks of the dioxins. Proceedings
of a Symposium held at the Rockefeller University, New York, 19-20
October, 1983, Los Altos, California, William Kaufmann, pp. 187-203.
FIRESTONE, D. (1973) Etiology of chick edema disease. Environ.
Health Perspect., 5: 59-66.
FIRESTONE, D., (1978) The 2,3,7,8-tetrachlorodibenzo-para-dioxin
problem: A review. Proceedings of a Conference on Chlorinated
Phenoxyacids and their Dioxins, Stockholm, 1977. Ecol. Bull., 27:
39-52.
FIRESTONE, D., CLOWER, M., Jr, BORSETTI, A.P., TESKE, R.H., & LONG,
P.E. (1979) Polychlorodibenzo-p-dioxin and pentachloro-phenol residues
in milk and blood of cows fed technical pentachlorophenol. J. agric.
food Chem., 27: 1171-1177.
FIRESTONE, D., NIEMANN, R.A., SCHNIDER, L.F., GRIDLEY, J.R., & BROWN,
D.E., (1986) Dioxin residues in fish and other food. In: Rappe, C.,
Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers.
FLICK, D.F., FIRESTONE, D., & HIGGINBOTHAM, G.R. (1972) Studies of the
chick edema disease. 9. Response of chicks fed or singly administered
synthetic edema-producing compounds. Poult. Sci., 51: 2026-2034.
FLICK, D.F., FIRESTONE, D., RESS, J., & ALLEN, J.R. (1973) Studies of
the chick edema disease. 10. Toxicity of chick edema factors in the
chick, chick embryo, and monkey. Poult. Sci., 52: 1637-1641.
FORTH, W. (1977) [The Seveso Disaster.] Dtsch. Arztebl., 44:
2617-2626 (in German).
FOWLER, B.A., LUCIER, G.W., BROWN, H.W., & MCDANIEL, O.S. (1973)
Ultrastructural changes in rat liver cells following a single oral
dose of TCDD. Environ. Health Perspect., 5: 141-148.
FRAENKEL, C. (1902) [Reports of associations and congresses. Medical
Association of Hall: Meeting of 23 October.] Münchener Med.
Wochenschr., October: 39-41.
FREEDMAN, H.J., PARKER, N.B., MARINELLO, A.J., GURTOO, H.L., &
MINOWADA, J. (1979) Induction, inhibition and biological properties of
aryl hydrocarbon hydroxylase in a stable human B-lymphocyte cell line,
RPMI-1788. Cancer Res., 39: 4612-4619.
FREEMAN, R.A., SCHROY, J.M., HILEMAN, F.D., & NOBLE, R.W. (1986)
Environmental mobility of 2,3,7,8-TCDD and companion chemicals in a
roadway soil matrix. In: Rappe, C., Choudhary, G., & Keith, L., ed.
Chlorinated dioxins and dibenzofurans in perspective, Chelsea,
Michigan, Lewis Publishers, pp. 171-183.
FRIES, G.F. & MARROW, G.S. (1975) Retention and excretion of
2,3,7,8-tetrachlorodibenzo-p-dioxin by rats. J. agric. food Chem.,
23: 265-269.
FRIESEN, K.J., SARNA, L.P., & WEBSTER, G.R.B. (1985) Aqueous
solubility of polychlorinated dibenzo-p-dioxins determined by high
pressure liquid chromatography. Chemosphere, 14: 1267-1274.
FUCHS, E. & GREEN, H. (1981) Regulation of terminal differentiation of
cultured human keratinocytes by vitamin A. Cell, 25: 617-625.
FUNG, D., BOYD, R.K., SAFE, S., & CHITTIM, B.G. (1985) Gas
chromatographic/mass spectrometric analysis of specific isomers of
polychlorodibenzofurans. Biomed. mass Spectrom., 12: 247-253.
FURST, P., MEEMKEN, H.-A. KRUGER, Chr., & GROEBEL, W. (1987)
Polychlorinated dibenzodioxins and dibenzofurans in human milk samples
from Western Germany. Chemosphere, 16: 1983-1988.
FURUHASHI, N., KURL, R.N., WONG, J., & VILLEE, C.A. (1986) A cytosolic
binding protein for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the
uterus and deciduoma of rats. Pharmacology, 33: 110-120.
GALLO, M.A., HESSE, E.J., MACDONALD, G.J., & UMBREIT, T.H. (1986)
Interactive effects of estradiol and
2,3,7-8-tetra-chlorodibenzo-p-dioxin on hepatic cytochrome P-450 and
mouse uterus. Toxicol. Lett., 32: 123-132.
GASIEWICZ, T.A. & NEAL, R.A. (1979)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin tissue distribution, excretion,
and effects on clinical chemical parameters in guinea pigs. Toxicol.
appl. Pharmacol., 51: 329-339.
GASIEWICZ, T.A. & RUCCI, G. (1984) Cytosolic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin. Evidence for a homologous nature
among various mammalian species. Mol. Pharmacol., 26: 90-98.
GASIEWICZ, T.A., HOLSCHER, M.A., & NEAL, R.A. (1980) The effect of
total parenteral nutrition on the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Toxicol. appl.
Pharmacol., 54: 469-488.
GASIEWCICZ, T.A., OLSON, J.R., GEIGER, L.H., & NEAL, R.A. (1983a)
Absorption, distribution and metabolism of
2,3,7,8-tetrachlorodibenzodioxin (TCDD) in experimental animals.
Environ. Sci. Res., 26: 495-525.
GASIEWICZ, T.A., GEIGER, L.E., RUCCI, G., & NEAL, R.A. (1983b)
Distribution, excretion, and metabolism of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in C57Bl/6J, DBA/2J, and B602F,/J
mice. Drug Metab. Disp., 11: 397-403.
GASIEWICZ, T.A., NESS, W.C., & RUCCI, G. (1984) Ontogeny of the
cytosolic receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat
liver, lung, and thymus. Biochem. biophys. Res. Commun., 118:
183-190.
GASIEWICZ, T.A., RUCCI, G., HENRY, E.C., & BAGGS, R.B. (1986) Changes
in hamster hepatic cytochrome P-450, ethoxycoumarin o-deethylase, and
reduced NAD(P): Menadione oxidoreductase following treatment with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 35:
2737-2742.
GEBEFUGI, I., BAUMANN, R., & KORTE, F. (1977) [Photochemical breakdown
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) under stimulated
environmental conditions.] Naturwissenschaften, 64: 486-487 (in
German).
GEIGER, L.E. & NEAL, R.A. (1981) Mutagenicity testing of
2,3,7,8-tetrachlorodibenzo-p-dioxin in histidine auxotrophs of
Salmonella typhimurium. Toxicol. appl. Pharmacol., 59: 125-129.
GERMANY (1985) [Review of dioxins], Berlin, Erich Schmidt Verlag, pp.
257-266 (Federal Office for the Environment, Report 5/85, November
1984) (in German).
GIAVINI, E., PRATI, M., & VISMARA, C. (1982a) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin administered to pregnant rats
during the pre-implantation period. Environ. Res., 29: 185-189.
GIAVINI, E., PRATI, M., & VISMARA, C. (1982b) Rabbit teratology study
with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Res., 27:
74-78.
GIAVINI, E., PRATI, M., & VISMARA, C. (1983) Embryotoxic effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin administered to female rats before
mating. Environ. Res., 31: 105-110.
GIERTHY, J.F. & CRANE, D. (1984) Reversible inhibition of in vitro
epithelial cell proliferation by 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Toxicol. appl. Pharmacol., 74: 91-98.
GIERTHY, J.F. & CRANE, D. (1985a) In vitro bioassay for dioxin-like
activity based on alterations in epithelial cell proliferation and
morphology. Fundam. appl. Toxicol., 5: 754-759.
GIERTHY, J.F. & CRANE, D. (1985b) Development of in vitro
bioassays for chlorinated dioxins and dibenzofurans. In: Keith, L.H.,
Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment II, Stoneham, Maine,
Butterworth Publishers, pp. 267-284.
GIERTHY, J.F., CRANE, D., & FRENKEL, G.D. (1984) Application of an
in vitro keratinization assay to extracts of soot from a fire in
a polychlorinated biphenyl-containing transformer. Fundam. appl.
Toxicol., 4: 1036-1041.
GILBERT, P., SAINT-RUF, G., PONCELET, F., & MERCIER, M. (1980) Genetic
effects of chlorinated anilines and azobenzenes on salmonella
typhimurium. Arch. environ. Contam. Toxicol., 9: 533-541.
GOLDMANN, P.J. (1972) [Very severe acute chloracne caused by
trichlorophenol decomposition products.] Arbeitsmed. Sozialmed.
Arbeitshyg., 7: 12-18 (in German).
GOLDMANN, P.J. (1973) [Very severe acute chloracne: mass poisoning by
2,3,6,7-tetrachlorodibenzodioxin.] Hautarzt, 24: 149-152 (in
German).
GOLDSTEIN, J.A. & LINKO, P. (1984) Differential induction of two
2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible forms of cytochrome
P-450 in extrahepatic versus hepatic tissues. Mol. Pharmacol., 25:
185-191.
GOLDSTEIN, J.A., HICKMAN, P., BERGMAN, H., & VOS, J.G. (1973) Hepatic
porphyria induced by 2,3,7,8-tetrachloro-dibenzo-p-dioxin in the
mouse. Res. Commun. chem. Pathol. Pharmacol., 6: 919-928.
GOLDSTEIN, J.A., MCKINNEY, J.D., LUCIER, G.W., HICKMAN, P., BERGMAN,
H., & MOORE, J.A. (1976) Toxicological assessment of
hexachlorobiphenyl isomers and 2,3,7,8-tetrachlorodibenzofuran in
chicks. II. Effects on drug metabolism and porphyrin accumulation.
Toxicol. appl. Pharmacol., 36: 81-92.
GOLDSTEIN, J.A., HASS, J.R., LINKO, P., & HARVAN, D.J. (1978)
2,3,7,8-Tetrachlorodibenzofuran in a commercially available 99% pure
polychlorinated biphenyl isomer identified as the inducer of hepatic
cytochrome P-448 and aryl hydrocarbon hydroxylase in the rat. Drug
Metab. Disp., 6: 258-264.
GOLDSTEIN, J.A., LINKO, P., & BERGMAN, H. (1982) Induction of
porphyria in the rat by chronic versus acute exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 31:
1607-1613.
GONZALES, F.J., TUKEY, R.H., & NEBERT, D.W. (1984) Structural gene
products of the Ah locus. Transcriptional regulation of cytochrome
P1-450 and P3-450mRNA levels by 3-methylchol-anthrene. Mol.
Pharmacol., 26: 117-121.
GORSKI, T., KONOPKA, L., & BRODZKI, M. (1984) Persistence of some
polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans of
pentachlorophenol in human adipose tissue. Rocz. Pzh, XXXV(4):
297-301.
GOTHE, R. & WACHTMEISTER, C.A. (1972) Synthesis of
1,2,4,5,7,8-hexa-chloroxanthene. Acta chem. Scand., 26: 2523-2576.
GOTZ, R. (1986) Chlorinated dioxins and dibenzofurans in leachate and
sediments of the sanitary landfill in Hamburg-Georswerder.
Chemosphere, 15: 1981-1984.
GRAY, A.P., STEVEN, P.C., & CANTRELL, J.S. (1975) Intervention of the
Smiles rearrangement in synthesis of dibenzo-p-dioxins:
1,2,3,6,7,8-and 1,2,3,7,8,9-hexachlorodibenzo-dioxin. Tetrahydron
Lett., 33: 2873-2876.
GRAY, A.P., STEVEN, P.C., SOLOMON, I.J., & ANILINE, O. (1976)
Synthesis of specific polychlorinated dibenzo-p-dioxins. J. org.
Chem., 41: 2435-2437.
GREEN, S. & MORELAND, F.S. (1975) Cytogenetic evaluation of several
dioxins in the rat. Toxicol. appl. Pharmacol., 33: 161.
GREEN, S., MORELAND, F., & SHEU, C. (1977) Cytogenetic effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on rat bone marrow cells. FDA
By-lines, 6: 292-294.
GREENBURG, L., MAYERS, M.R., & SMITH, A.R. (1939) The systemic effects
resulting from exposure to certain chlorinated hydro-carbons. J. ind.
Hyg. Toxicol., 21: 29-38.
GREENLEE, W.F. & POLAND, A. (1978) An improved assay of
7-ethoxycoumarin O-deethylase activity: induction of hepatic enzyme
activity in C57BL/6J and DBA/2J mice by phenobarbital,
3-methylcholanthrene and 2,3,7,8-tetrachlorodibenzo-p-dioxin. J.
Pharmacol. exp. Ther., 205: 596-605.
GREENLEE, W.F. & POLAND, A. (1979) Nuclear uptake of
2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J and DBA/2J mice. Role
of the hepatic cytosol receptor protein. J. biol. Chem., 254:
9814-9821.
GREENLEE, W.F., DOLD, K.M., IRONS, R.D., & OSBORNE, R. (1985) Evidence
for direct action of 2,3,7,8-tetrachlorodidibenzo-p-dioxin (TCDD) on
thymic epithelium. Toxicol. appl. Pharmacol., 79: 112-120.
GREIG, J.B. (1972) Effect of 2,3,7,8-tetrachlorodibenzo-1, 4-dioxin on
drug metabolism in the rat. Biochem. Pharmacol., 21: 3196-3198.
GREIG, J.B. & OSBORNE, G. (1981) Biochemical and morphological changes
induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat liver cell
plasma membrane. J. appl. Toxicol., 1(6): 334-338.
GREIG, J.B., JONES, G., BUTLER, W.H., & BARNES, J.M. (1973) Toxic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Food Cosmet.
Toxicol., 11: 585-595.
GREIG, J.B., TAYLOR, D.M., & JONES, J.D. (1974) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on stimulated DNA synthesis in the
liver and kidney of the rat. Chem.-biol. Interact., 8: 31-39.
GREIG, J.B., FRANCIS, J.E., KAY, S.J.E., LOVELL, D.P., & SMITH, A.G.
(1984) Incomplete correlation of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
hepatotoxicity with Ah phenotype in mice. Toxicol. appl. Pharmacol.,
74: 17-25.
GROSS, M.L., LAY, J.O., Jr, LYON, P.A., LIPPSTREU, D., KANGAS, N.,
HARLESS, R.L., TAYLOR, S.E., & DUPUY, A.E. Jr (1984)
2,3,7,8-Tetrachlorodibenzo-p-dioxin levels in adipose tissue of
Vietnam veterans. Environ. Res., 33: 261-268.
GUENTHNER, T.M., FYSH, J.M., & NEBERT, D.W. (1979)
2,3,7,8-Tetrachlorodibenzo-p-dioxin: covalent binding of reactive
metabolic intermediates principally to protein in vitro.
Pharmacology, 19: 12-22.
GUPTA, B.N., VOS, J.G., MOORE, J.A., ZINKL, J.G., & BULLOCK, B.C.
(1973) Pathologic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in
laboratory animals. Environ. Health Perspect., 5: 125-140.
GURTOO, H.L., PARKER, N.B., PAIGEN, B., HAVENS, M.B., MINOWADA, J., &
FREEDMAN, H.J. (1979) Induction, inhibition and some enzymological
properties of aryl hydrocarbon hydroxylase in fresh mitogen-activated
human lymphocytes. Cancer Res., 39: 4620-4629.
GUSTAFSSON, J.-A. & INGELMAN-SUNDBERG, M. (1979) Changes in steroid
hormone metabolism in rat liver microsomes following administration of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Biochem. Pharmacol.,
28: 497-499.
HAAPARANTA, T., GLAUMANN, H., & GUSTAFSSON, J.-A. (1983) Induction of
cytochrome P-450 dependent reactions in the rat ventral prostate by
beta-naphthoflavone and 2,3,7,8-tetra-chlorodibenzo-p-dioxin.
Toxicology, 29: 61-75.
HAGENMAIER, H. & BRUNNER, H. (1987) Isomer specific analysis of PCDD
and PCDF in pentachlorophenol and sodium pentachloro- phenate samples
in the sub-PPB range. Chemosphere, 16: 1759-1764.
HAGENMAIER, H., DRAFT, M., JAGER, W., MAYER, U., LUETZKE, K., &
SIEGAL, D. (1986) Comparison of various sampling methods for PCDDs and
PCDFs in stack gas. Chemosphere, 15(9-12): 1187-1192.
HAKANSSON, H. (1988) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin
of the fate of vitamin A in rodents, Stockholm, Department of
Toxicology and Institute of Environmental Medicine, Karolinska
Institute (Ph.D. Thesis).
HAKANSSON, H. & AHLBORG, U.G. (1985) The effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the uptake, distribution
and excretion of a single oral dose of 11,12, 3H-retinylacetate and on
the vitamin A status in the rat. J. Nutr., 115: 759-771.
HAKANSSON, H., AHLBORG, U.G., & GOTTLING, L. (1986) The effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the distribution and
excretion of the endogenous pool of vitamin A in rats with low liver
vitamin A stores. Chemosphere, 15: 1715-1723.
HAKANSSON, H., WAERN, F., & AHLBORG, U.G. (1987) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the lactating rat on
maternal and neonatal vitamin A status. J. Nutr., 117: 580-586.
HANNAH, R.R., LUND, J., POELLINGER, L., GILLNER, M., & GUSTAFSSON,
J.-A. (1986) Characterization of the DNA-binding properties of the
receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Eur. J.
Biochem., 156: 237-242.
HARDELL, L. (1981) Relation of soft-tissue sarcoma, malignant lymphoma
and colon cancer to phenoxy acids, chlorophenols and other agents.
Scand. J. Work Environ. Health, 7: 119-130.
HARDELL, L. & ERIKSSON, M. (1981) Soft-tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 2: 250.
HARDELL, L. & SANDSTROM, A. (1979) Case-control study: soft-tissue
sarcomas and exposure to phenoxyacetic acids or chlorophenols. Br.
J. Cancer, 39: 711-717.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981) Malignant
lymphoma and exposure to chemicals, especially organic solvents,
chlorophenols and phenoxy acids: a case-control study. Br. J.
Cancer, 43: 169-176.
HARLESS, R.L. & LEWIS, R.G. (1982) Quantitative determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin residues by gas
chromatography/mass spectrometry. In: Hutzinger, O., ed. Chlorinated
dioxins and related compounds, Oxford, London, Pergamon Press, pp.
25-36.
HARLESS, R.L., OSWALD, E.O., LEWIS, R.G., DUPUY, A.E., MCDANIEL, D.D.,
& TAI, H. (1982) Determination of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in fresh water fish. Chemosphere, 11, 193-198.
HARLESS, R.L., LEWIS, R.G., DUPUY, A.E., & MCDANIEL, D.D. (1983)
Analyses for 2,3,7,8-tetrachlorodibenzo-p-dioxin residues in
environmental samples. In: Tucker, R.E., ed. Human and
environmental risks of chlorinated dioxins and related compounds,
New York, London, Plenum Press, pp. 161-172.
HARRIS, M.W., MOORE, J.A., VOS, J.G., & GUPTA, B.N. (1973) General
biological effects of TCDD in laboratory animals. Environ. Health
Perspect., 5: 101-109.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1983) Comparative ability
of TCDD to induce lipid peroxidation in rats, guinea pigs and syrian
golden hamsters. Bull. environ. Contam. Toxicol., 31: 649-657.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1985a) Inhibition of
TCDD-induced lipid peroxidation, glutathione peroxidase activity and
toxicity by BHA and glutathione. Bull. environ. Contam. Toxicol.,
34: 787-796.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1985b) Effects of vitamins
E and A on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced lipid
peroxidation and other biochemical changes in the rat. Arch. environ.
Contam. Toxicol., 14: 437-442.
HASSAN, M.Q., STOHS, S.J., MURRAY, W.J., & BIRT,D.F. (1985c) Dietary
selenium, glutathione peroxidase activity, and toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Toxicol. environ. Health,
15: 405-415.
HASSOUN, E.M. & DENCKER, L. (1982) TCDD embryotoxicity in the mouse
may be enhanced by beta-naphtoflavone, another ligand of the
Ah-receptor. Toxicol. Lett., 12: 191-198.
HASSOUN, E.M., D'ARGY, R., & DENCKER, L. (1984a) Teratogenicity of
2,3,7,8-tetrachlorodibenzofuran in the mouse. J. Toxicol. environ.
Health, 14: 337-351.
HASSOUN, E.M., D'ARGY, R., DENCKER, L., LUNDING, L.G., & BORWELL, P.
(1984b) Teratogenicity of 2,3,7,8-tetrachloro-dibenzofuran in BXD
recombinant inbred strains. Toxicol. Lett., 23: 37-42.
HAY, A. (1984) Experimental toxicology and cytogenetics: an overview.
In: Westing, A.N., ed. Herbicides in war, the long-term ecological
and human consequences, London, Taylor & Francis, pp. 161-166.
HAY, A.W.M. (1977) Tetrachlorodibenzo-p-dioxin release at Seveso.
Disasters, 1: 289-308.
HAYABUCHI, H., YOSHIMURA, T., & KURATSUNE, M. (1979) Consumption of
toxic rice oil by Yusho patients and its relation to the clinical
response and latent period. Food Cosmet. Toxicol., 17: 455-461.
HAYES, K.C. (1971) On the pathophysiology of vitamin a deficiency.
Nutr. Rev., 29: 3-6.
HELLING, C.S., ISENSEE, A.R., WOOLSON, E.A., ENZOR, P.D.J., JONES,
G.E., PLIMMER, J.R., & KEARNEY, P.C. (1973) Chlorodioxins in
pesticides, soils and plants. J. environ. Qual., 2: 171-178.
HENCK, J.M., NEW, M.A., KOBICA, R.J., & RAO, K.S. (1981)
2,3,7,8-tetrachlorodibenzo-p-dioxin: acute oral toxicity in hamsters.
Toxicol. appl. Pharmacol., 59: 405-407.
HERXHEIMER, K. (1899) [Chloracne.] Münchenere med. Wochenschr.,
46: 278 (in German).
HERZBERG, J.J. (1947) [Chloracne after ingestion of chlorinated
paraffin.] Dermatol. Wochenschr., 7: 425-433 (in German).
HINSDILL, R.D., COUCH, D.L., & SPEIRS, R.S. (1980) Immunosuppression
in mice induced by dioxin (TCDD) in feed. J. environ. Pathol.
Toxicol., 3: 401-425.
HIROSAWA, K. & YAMADA, E. (1973). The localization of the vitamin A in
the mouse liver as revealed by electron microscope radioautography.
J. electron. Microsc., 22: 337-346.
HOFFMAN, R.E., STEHR-GREEN, P.A., WEBB, K.B., EVANS, R.G., KNUTSEN,
A.P., SCHRAMM, W.F., STAAKE, J.L., GIBSON, B.B., & STEINBERG, K.K.
(1986) Health effects of long-term exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Am. Med. Assoc., 255:
2031-2038.
HOFMANN, H.Th. (1957) [Recent findings with highly toxic chlorinated
hydrocarbons.] Arch. exp. Pathol. Pharmakol., 232: 228-230 (in
German).
HOFMANN, M.F. & MENEGHINI, C.L. (1962) [Folliculosis from
chlorine-substituted hydrocarbons (chloracne).] G. Ital. Dermatol.,
103: 427-450 (in Italian).
HOLMSTEDT, B. (1980) Prolegomena to Seveso. Arch. Toxicol., 44:
211-230.
HONCHAR, P.A. & HALPERIN, W.E. (1981) 2,4,5-trichlorophenol and
soft-tissue sarcoma. Lancet, 21: 268-269.
HOOK, G.E.R., HASEMAN, J.K., & LUCIER, G.W. (1975a) Induction and
suppression of hepatic and extrahepatic microsomal
foreign-compound-metabolizing enzyme systems by
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Chem.-biol. Interact., 10:
199-214.
HOOK, G.E.R., ORTON, T.C., MOORE, J.A., & LUCIER, G.W. (1975b)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced changes in the
hydroxylation of biphenyl by rat liver microsomes. Biochem.
Pharmacol., 24: 335-340.
HORI, S., OBANA, H., TANAKA, R., & KASHIMOTO, T. (1986) Comparative
toxicity in rats of polychlorinated biphenyls (PCBs), polychlorinated
quaterphenyls (PCQs) and poly-chlorinated dibenzofurans (PCDFs)
present in rice oil causing "Yusho". Eisei Kagaku, 32: 13-21.
HRYHORCZUK, D.O., WITHROW, W.A., HESSE, C.S., & BEASLEY, V.R. (1981)
A wire reclamation incinerator as a source of environmental
contamination with tetrachlorodibenzo-p-dioxins and
tetrachlorodibenzofurans. Arch. environ. Health, 36: 228-234.
HSU, S., MA, C., HSU, S.K., WU, S., HSU, N.H., & YEH, C. (1984)
Discovery and epidemiology of PCB poisoning in Taiwan. Am. J. ind.
Med., 5: 71-79.
HUDSON, L.G., SHAIKH, R., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1983)
Induction of 7-ethoxycoumarin o-deethylase activity in cultured human
epithelial cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD):
Evidence for TCDD receptor. Biochem. biophys. Res. Commun., 115:
611-617.
HUDSON, L.G., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1985) Regulation of
epidermal growth factor binding in a human keratinocyte cell line by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
77: 251-259.
HUDSON, L.G., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1986)
2,3,7,8-Tetrachorodibenzo-p-dioxin (TCDD) modulates epidermal growth
factor (EGF) binding to basal cells from a human keratinocyte cell
line. Toxicol. appl. Pharmacol., 82: 481-492.
HUFF, J.E., MOORE, J.A., SARACCI, R., & TOMATIS, L. (1980) Long-term
hazards of polychlorinated dibenzodioxins and polychlorinated
dibenzofurans. Environ. Health Perspect., 36: 221-240.
HUQUE, T. (1981) Excretion of radioactive metabolites of retinal as an
index of vitamin A status in rats. Nutr. Rep. Int., 24: 171-179.
HUSSAIN, S., EHRENBERG, L., LOFROTH, G., & GEJVALL, T. (1972)
Mutagenic effects of TCDD on bacterial systems. Ambio, 1: 32-33.
HUTZINGER, O., SAFE, S., WENTZELL, B.R., & ZITKO, V. (1973)
Photochemical degradation of di-and octachlorodibenzofuran. Environ
Health Perspect., 5: 267-271.
HWANG, S.W. (1973) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on
the biliary excretion of indocyanine Green in rat. Environ. Health
Perspect., 5: 227-231.
IARC (1977) Some fumigants, the herbicides 2,4-D and 2,4,5-T,
chlorinated dibenzodioxins and miscellaneous industrial chemicals,
Lyon, France, International Agency for Research on Cancer, pp. 41-102
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Vol. 15).
IARC (1982) Chemicals, industrial processes and industries
associated with cancer in humans, Lyon, France, International Agency
for Research on Cancer (IARC Monograph on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Suppl. No. 4).
IARC (1986) Some halogenated hydrocarbons and pesticide exposures,
Lyon, International Agency for Research on Cancer (IARC Monographs on
the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol.
41).
IDEO, G., BELLATI, G., BELLOBUONO, A., MOCARELLI, P., MAROCCHI, A., &
BRAMBILLA, P. (1982) Increased urinary D-glucaric acid excretion by
children living in an area polluted with
tetrachlorodibenzo-para-dioxin (TCDD). Clin. Chim. Acta, 120:
273-283.
IDEO, G., BELLATI, G., BELLOBUONO, A., & BISSANTI, L. (1985) Urinary
d-glucaric acid excretion in the Seveso area, polluted by
tetrachlorodibenzo-p-dioxin (TCDD): Five years of experience.
Environ. Health Perspect., 60: 151-157.
INNAMI, S.I., NAKAMURA, A., MIYAZAKI, M., NAGAYAMA, S., & NISHIDE, E.
(1976) Further studies on the reduction of vitamin A content in the
liver of rats given polychlorinated biphenyls. J. nutr. Sci.
Vitaminol., 22: 409-418.
IOANNOU, Y.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1983) Toxicity and
distribution of 2,3,7,8-tetrachlorodibenzofuran in male guinea pigs.
J. Toxicol. environ. Health, 12: 541-553.
IRPTC (1987) IRPTC Legal file 1986, Geneva, International Register
of Potentially Toxic Chemicals, United Nations Environment Programme,
Geneva, Switzerland, Vol. I (Data Profile Series No. 7).
ISENSEE, A.R. (1978) Bioaccumulation of
2,3,7,8-tetrachloro-dibenzo-para-dioxin. Ecol. Bull. (Stockholm),
27: 255-262.
ISENSEE, A.R. & JONES, G.E. (1971) Absorption and trans-location of
root and foliage applied 2,4-dichlorophenol,
2,7-dichlorodibenzo-p-dioxin and 2,3,7,8-tetrachlorodibenzo-p-dioxin.
J. agric. food. Chem., 19: 1210-1214.
ISENSEE, A.R. & JONES, G.E. (1975) Distribution of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in aquatic model ecosystem.
Environ. Sci. Technol., 9: 668-672.
ISRAEL, D.I. & WHITLOCK, J.P., Jr (1983) Induction of mRNA specific
for cytochrome P1-450 in wild type and variant mouse hepatoma cells.
J. biol. Chem., 258: 10390-10394.
ISRAEL, D.I. & WHITLOCK, J.P., Jr (1984) Regulation of cytochrome
P1-450 gene transcription by 2,3,7,8-tetrachloro-dibenzo-p-dioxin in
wild type and variant mouse hepatoma cells. J. biol. Chem., 259:
5400-5402.
JANSING, R.L. & SHAIN, W. (1985) Aryl hydrocarbon hydroxylase
induction in adult rat hepatocytes in primary culture by several
chlorinated aromatic hydrocarbons including
2,3,7,8-tetrachloridibenzo-p-dioxin. Fundam. appl. Toxicol., 5:
713-720.
JENSEN, D.J. & HUMMEL, R.A. (1982) Secretion of TCDD in milk and cream
following the feeding of TCDD to lactating dairy cows. Bull.
environ. Contam. Toxicol., 29: 440-446.
JENSEN, D.J., GETZENDANER, M.E., HUMMEL, R.A., & TURLEY, J. (1983)
Residue studies for (2,4,5-trichlorophenoxy)acetic acid and
2,3,7,8-tetrachlorodibenzo-p-dioxin in grass and rice. J. agric.
food Chem., 31: 118-122.
JENSEN, N.E. & WALKER, A.E. (1972) Chloracne: Three cases. Proc. R.
Soc. Med., 65: 687-688.
JIRASEK, L., KALENSKY, J., & KUBEC, K. (1973) [Acne chlorine and
porphyria-cutanea tarda by the production of herbicides.] Cs.
dermatol., 48: 306-317 (in Czech).
JIRASEK, L., KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS, E.
(1976) [Chloracne, porphyria-cutanea tarda and other intoxication by
herbicides.] Hautarzt, 27: 328-333 (in German).
JOHANSSON, G., GILLNER, M., HOGBERG, B., & GUSTAFSSON, J.-A. (1982)
The TCDD receptor in rat intestinal mucosa and its possible dietary
ligands. Nutr. Cancer, 3: 134-143.
JOHNSON, E.F. & MULLER-EBERHARD, U. (1977a) Purification of the major
cytochrome P-450 of liver microsomes from rabbits treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Biochem. biophys. Res.
Commun., 76: 652-659.
JOHNSON, E.F. & MULLER-EBERHARD, U. (1977b) Resolution of two forms of
cytochrome P-450 from liver microsomes of rabbits treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. biol. Chem., 252:
2839-2845.
JOHNSON, E.F., SCHWAB, G.E., & MULLER-EBERHARD, U. (1979) Multiple
forms of cytochrome P-450: Catalytic differences exhibited by two
homogeneous forms of rabbit cytochrome P-450. Mol. Pharmacol., 15:
708-718.
JOHNSON, F.E., KUGLER, N.A., & BROWN, S.M. (1981) Soft tissue sarcomas
and chlorinated phenols. Lancet, 1: 40.
JONES, D., SAFE, S., MORCOM, E., HOLCOMB, C., COPPOCK, C., & IVIE, W.
(1987) Bioavailability of tritiates
2,3,7,8-tetra-chlorodibenzo-p-dioxin administered to Holstein dairy
cows. Chemosphere, 16: 1743-1748.
JONES, E.L. & KRIZEK, H. (1962) A technic for testing acnegenic
potency in rabbits, applied to the potent acnegen,
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. invest. Dermatol., 39:
511-517.
JONES, G. (1975) A histochemical study of the liver lesion induced by
2,3,7,8-tetrachlorodibenzo-p-dioxin (dioxin) in rats. J. Pathol.,
116: 101-105.
JONES, G. & BUTLER, W.H. (1974) A morphological study of the liver
lesion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. J.
Pathol., 112: 93-97.
JONES, G. & GREIG, J.B. (1975) Pathological changes in the liver of
mice given 2,3,7,8-tetrachlorodibenzo-p-dioxin. Experientia (Basel),
31: 1315-1317.
JONES, K.G. & SWEENEY, G.D. (1977) Association between induction of
aryl hydrocarbon hydroxylase and depression of uroporphyrinogen
decarboxylase activity. Res. Commun. chem. Pathol. Pharmacol., 17:
631-637.
JONES, K.G. & SWEENEY, G.D. (1980) Dependence of the porphyrogenic
effect of 2,3,7,8-tetrachlorodibenzo(p)dioxin upon inheritance of aryl
hydrocarbon hydroxylase responsiveness. Toxicol. appl. Pharmacol.,
53: 42-49.
JONES, P.A. (1981) Chlorophenols and their impurities in the
Canadian environment, Ottawa, Environment Canada (EPS 3-EC-81-2).
JONES, P.A. (1984) Chlorophenols and their impurities in the
Canadian environment: 1983 supplement, Ottawa, Environment Canada
(EPS 3-EP-84-3).
JONES, P.B.C., MILLER, A.G., ISRAEL, D.I., GALEAZZI, D.R., & WHITLOCK,
J.P., Jr (1984) Biochemical and genetic analysis of variant mouse
hepatoma cells which overtranscribe the cytochrome P1-450 gene in
response to 2,3,7,8-tetrachloro-dibenzo-p-dioxin. J. biol. Chem.,
259: 12357-12363.
JONES, P.B.C., GALEAZZI, D.R., & WHITLOCK, J.P., Jr (1985) Control of
cytochrome P1-450 gene expression by dioxin. Science, 227:
1499-1502.
JONES, P.B.C., DURRIN, L.K., GALEAZZI, D.R., & WHITLOCK, J.P., Jr
(1986) Control of cytochrome P1-450 gene expression: Analysis of a
dioxin-responsive enhancer system. Proc. Natl Acad. Sci. (USA),
83: 2802-2806.
JONES, R.E. & CHELSKY, M. (1986) Further discussion concerning
porphyria cutanea tarda and TCDD exposure. Arch. environ. Health,
41: 100-103.
JOSEPHSON, J. (1983) Chlorinated dioxins and furans in the
environment. Environ. Sci. Technol., 17: 124A-128A.
JUNK, G.A. & RICHARD, J. (1981) Dioxins not detected in effluents from
coal/refuse combustion. Chemosphere, 10: 1237-1241.
KARASEK, F.W. & ONUSKA, F.I. (1982) Trace analysis of the dioxins.
Anal. Chem., 54: 309A-324A.
KARENLAMPI, S.O., EISEN, H.J., HANKINSON, O., & NEBER, D.W. (1983)
Effects of cytochrome Pl-450 inducers on the cell-surface receptors
for epidermal growth factor, phorbol 12,13-dibutyrate, or insulin of
cultured mouse hepatoma cells. J. biol. Chem., 17: 10378-10383.
KEARNEY, P.C., WOOLSON, E.A., & ELLINGTON, C.P., Jr (1972) Persistence
and metabolism of chlorodioxins in soils. Environ. Sci. Technol.,
6: 1017-1019.
KEESEY, R.E., BOYLE, P.C., KEMNITZ, J.W., & MITCHEL, J.S. (1976) The
role of the lateral hypothalamus in determining the body weight set
point. In: Novin, D., Wyrwicks, W., & Bray, G., ed. Hunger: Basic
mechanisms and clinical implications, New York, Raven Press, pp.
243-255.
KELLING, C.K., CHRISTIAN, B.J., INHORN, S.L., & PETERSON, R.E. (1985)
Hypophagia-induced weight loss in mice, rats, and guinea pigs treated
with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. appl. Toxicol.,
5: 700-712.
KENDE, A.S., WADE, J.J., RIDGE, D., & POLAND, A. (1974) Synthesis and
fourier transform carbon - 13 nuclear magnetic resonance spectroscopy
of new toxic polyhalodibenzo-p-dioxins. J. org. Chem., 39:
931-937.
KERKVLIET, N.I., BRAUNER, J.A., & MATLOCK, J.P. (1985) Humoral
immunotoxicity of polychlorinated diphenyl ethers, phenoxy-phenols,
dioxins and furans present as contaminants of technical grade
pentachlorophenol. Toxicology, 36: 307-324.
KEYS, B., HLAVINKA, M., MASON, G., & SAFE, S. (1985) Modulation of rat
hepatic microsomal testosterone hydroxylases by
2,3,7,8-tetrachlorodibenzo-p-dioxin and related toxic isostereomers.
Can. J. Pharmacol., 63: 1537-1542.
KEYS, B., PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1986)
Polychlorinated dibenzofurans as 2,3,7,8-TCDD antagonists: in
vitro inhibition of monooxygenase enzyme induction. Toxicol.
Lett., 31: 151-158.
KHERA, K.S. & RUDDICK, J.A. (1973) Polychlorodibenzo-p-dioxins:
Perinatal effects and the dominant lethal test in Wistar rats. Adv.
Chem. Ser., 120: 70-84.
KIMBLE, B.J. & GROSS, M.L. (1980) Tetrachlorodibenzo-p-dioxin
quantitation in stack-collected coal fly ash. Science, 207: 59-61.
KIMBROUGH, R.D. (1974) The toxicity of polychlorinated polycyclic
compounds and related chemicals. CRC Crit. Rev. Toxicol., 2:
445-498.
KIMBROUGH, R.D. (1979) The carcinogenic and other chronic effects of
persistent halogenated organic compounds. Ann. N.Y. Acad. Sci.,
320: 415-418.
KIMBROUGH, R.D. (1984) The epidemiology and toxicology of TCDD. Bull.
environ. Contam. Toxicol., 33: 636-647.
KIMBROUGH, R.D., GAINES, T.B., & LINDER, R.E. (1974)
2,4-Dichlorophenyl-p-nitrophenyl ether (TOK). Effects on the lung
maturation of rat fetus. Arch. environ. Health, 28: 316-319.
KIMBROUGH, R.D., CARTER, C.D., LIDDLE, J.A., CLINE, R.E., & PHILLIPS,
P.E. (1977) Epidemiology and pathology of a tetrachlorodibenzodioxin
poisoning episode. Arch. environ. Health, 32: 77-86.
KIMBROUGH, R.D., FALK, H., & STEHR, P. (1984) Health implications of
2,3,7,8-tetrachlorodibenzodioxin (TCDD) contamination of residential
soil. J. Toxicol. environ. Health, 14: 47-93.
KIMMIG, J. & SCHULZ, K.H. (1957a) [Occupational acne (so called
chloracne) caused by chlorinated aromatic cyclic ethers.]
Dermatologica, 115: 540-546 (in German).
KIMMIG, J. & SCHULZ, K.H. (1957b) [Chlorinated aromatic cyclic ethers
as cause of chloracne.] Naturwissenschaften, 11: 337-338 (in
German).
KIMURA, S., GONZALEZ, F.J., & NEBERT, D.W. (1986) Tissue-specific
expression of the mouse dioxin-inducible P1450 and P3450 genes:
Differential transcriptional activation and mRNA stability in liver
and extrahepatic tissues. Mol. cell. Biol., 6: 1471-1477.
KING, F.G., DEDRICK, R.L., & COLLINS, J.M. (1983) Physiological model
for the pharmacokinetics of 2,3,7,8-tetrachloro-dibenzofuran in
several species. Toxicol. appl. Pharmacol., 67: 390-400.
KITCHIN, K.T. & WOODS, J.S. (1979) 2,3,7,8-Tetrachlorodibenzo-p-dioxin
(TCDD) effects on hepatic microsomal cytochrome P-448-mediated enzyme
activities. Toxicol. appl. Pharmacol., 47: 537-546.
KLEU, G. & GOLTZ, R. (1971) [Late and lasting damage resulting from
the chronic occupational action of chlorophenol compounds.] Med.
Klin., 66: 53-58 (in German).
KNUTSON, J.C. & POLAND, A. (1980a)
2,3,7,8-Tetrachlorodibenzo-p-dioxin: failure to demonstrate toxicity
in twenty-three cultured cell types. Toxicol. appl. Pharmacol.,
54: 377-383.
KNUTSON, J.C. & POLAND, A. (1980b) Keratinization of mouse teratoma
cell line XB produced by 2,3,7,8-tetrachlorodibenzo-p-dioxin: an in
vitro model of toxicity. Cell, 22: 27-36.
KNUTSON, J.C. & POLAND, A. (1982) Response of murine epidermis to
2,3,7,8-tetrachlorodibenzo-p-dioxin: Interaction of the Ah and hr
Loci. Cell, 30: 225-234.
KNUTSON, J.C. & POLAND, A. (1984) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Examination of biochemical effects involved in the proliferation and
differentiation of XB cells. J. cell. Physiol., 121: 143-151.
KOCHER, C.W., MAHLE, N.H., HUMMEL, R.A., & SHADOFF, L.A. (1978) A
search for the presence of 2,3,7,8-tetrachloro-dibenzo-p-dioxin in
beef fat. Bull. environ. Contam. Toxicol., 19: 229-236.
KOCHMAN, S., BERNARD, J., CAZABAT, A., LAVAUD, F., LORTON, C., &
RAPPE, C. (1986) Phenotypical dissection of immunoregulatory T cell
subsets in human after furan exposure. Chemosphere, 15: 1799-1804.
KOCIBA, R.J., KEELER, P.A., PARK, C.N., & GEHRING, P.J. (1976)
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD): Results of a 13-week oral
toxicity study in rats. Toxicol. appl. Pharmacol., 35: 553-574.
KOCIBA, R.J., KEYES, D.G., BEYER, J.E., CARREON, R.M., WADE, C.E.,
DITTENBER, D., KALNINS, R., FRAUSON, L., PARK, C.N., BARNARD, S.,
HUMMEL, R., & HUMISTON, C.G. (1978) Results of a two year chronic
toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin
(TCDD) in rats. Toxicol. appl. Pharmacol., 46: 279-303.
KOCIBA, R.J., KEYES, D.G., LISOWE, R.W., KALNINS, R.P., DITTENBER,
D.D., WADE, C.E., GORZINSKI, S.J., HAHLE, N.H., & SCHWETZ, B.A.
(1979a) Results of a two-year chronic toxicity and encogenic study of
rats ingesting diets containing 2,4,5-trichlorophenoxyacetic acid
(2,4,5-T). Food Cosmet. Toxicol., 17: 205-221.
KOCIBA, R.J., KEYES, D.G., BEYER, J.E., CARREON, R.M., & GEHRING, P.J.
(1979b) Long-term toxicologic studies of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in laboratory animals.
Ann. N.Y. Acad. Sci., 320: 397-404.
KOHLI, K.K. & GOLDSTEIN, J.A. (1981) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin on hepatic and renal
prostaglandin synthetase. Life Sci., 29: 299-305.
KONDOROSI, A., FEDORCSAK, I., SOLYMOSY, F., EHRENBERG, L., &
OSTERMAN-GOLKAR, S. (1973) Inactivation of Q-beta RNA by
electrophiles. Mutat. Res., 17: 149-161.
KOSHAKJI, R.P., HARBISON, R.D., & BUSH, M.T. (1984) Studies on the
metabolic fate of (14C)2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in
the mouse. Toxicol. appl. Pharmacol., 73: 69-77.
KOURI, R.E., RATRIE, H., III, ATLAS, C.A., NIWA, A., & NEBERT, D.W.
(1974) Aryl hydrocarbon hydroxylase induction in human lymphocyte
cultures by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Life Sci., 15:
1585-1595.
KOURI, R.E., RUDE, T.H., & JOGLEKAR, R. (1978)
2,3,7,8-Tetra-chlorodibenzo-p-dioxin as cocarcinogen causing
3-methylchol-antrene-initiated subcutaneous tumors in mice genetically
"non responsive" at Ah locus. Cancer Res., 38: 2777-2783.
KRAUSE, L., VON & BRASSOW, H. (1978) [Contamnestic study of chloracne
cases from the year 1954/55.] Arbeitsmed. Socialmed.
Präventivmed., 13: 19-21 (in German).
KROWKE, R. (1986) Studies on distribution and embryotoxicity of
different PCDD and PCDF in mice and marmosets. Chemosphere, 15:
2011-2022.
KUNITA, N., KASHIMATO, T., MIYATA, H., FUKUSHIMA, S., HARI, S., &
OBANA, H. (1984) Causal agents of Yusho. Am. J. ind. Med., 5:
45-58.
KUNTZMAN, D., LAWRENCE, D., & CONNEY, A.H. (1965) Michaelis constants
for the hydroxylation of steroid hormones and drugs by rat liver
microsomes. Mol. Pharmacol., 1: 163-167.
KURL, R.N. & VILLEE, C.A. (1985) A metabolite of riboflavin binds to
the 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) receptor.
Pharmacology, 30: 241-244.
KURL, R.N., LUND, J., POELLINGER, L., & GUSTAFSSON, J-A. (1982)
Differential effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on nuclear
RNA polymerase activity in the rat liver and thymus. Biochem.
Pharmacol., 31: 2459-2462.
KURL, R.N., LORING, J.M., & VILLEE, C.A. (1985) Control of
2,3,7,8-tetrachlorodibenzo-p-dioxin binding protein(s) in the hamster
kidney. Pharmacology, 30: 245-254.
KUROKI, H., MASUDA, Y., YOSHIHARA, S., & YOSHIMURA, H. (1980)
Accumulation of polychlorinated dibenzofurans in the livers of monkeys
and rats. Food Cosmet. Toxicol., 18: 387-392.
KUROKI, H., HARAGUCHI, K., & MASUDA, Y. (1984) Synthesis of
polychlorinated dibenzofuran isomers and their gas chromato-graphic
profiles. Chemosphere, 13: 561-573.
KUROKI, J., KOGA, N., & YOSHIMURA, H. (1986) High affinity of
2,3,4,7,8-pentachloridibenzofuran to cytochrome P-450 in the hepatic
microsomes of rats. Chemosphere, 15: 731-738.
LAHANIATIS, E.S., PARLAR, H., & KORTE, F. (1977) [Contributions to
ecological chemistry CXXXII. On the occurrence of chlorinated
hydrocarbons in the flue dust of refuse incinerators.] Chemosphere,
7: 11-16 (in German).
LAMB, J.C., MARKS, T.A., GLADEN, B.C., ALLEN, J.W., & MOORE, J.A.
(1981) Male fertility, sister chromatid exchange, and germ cell
toxicity following exposure to mixtures of chlorinated phenoxy acids
containing 2,3,7,8-tetrachloro-dibenzo-p-dioxin. J. Toxicol.
environ. Health, 8: 825-834.
LAMB, J.C., IV, HARRIS, M.W., MCKINNEY, J.D., & BIRNBAUM, L.S. (1986)
Effects of thyroid hormones on the induction of cleft palate by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57Bl/6N mice. Toxicol.
appl. Pharmacol., 84: 115-124.
LAMPARSKI, L.L. & NESTRICK, T.J. (1981) Synthesis and identification
of the 10 hexachlorodibenzo-p-dioxin isomers by high performance
liquid and packed column gas chromatography. Chemosphere, 10:
3-18.
LAMPARSKI, L.L., NESTRICK, T.J., & STEHL, R.H. (1979) Determination of
part-per-trillion concentration of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in fish. Anal. Chem., 51: 1453-1458.
LAMPARSKI, L.L., NESTRICK, T.J., FRAWLEY, N.N., HUMMEL, R.A., KOCKER,
C.W., MAHLE, N.H., MCCOY, J.W., MILLER, D.L., PETERS, T.L., PILLIPICH,
J.L., SMITH, W.E., & TOBEY, S.W. (1986) Perspectives of a large scale
environmental survey for chlorinated dioxins: Water analyses.
Chemosphere, 15: 1445-1452.
LANGER, H.G., BRADEY, T.P., & BRIGGS, P.R. (1973) Formation of
dibenzodioxins and other condensation products from chlorinated
phenols and derivatives. Environ. Health Perspect., 5: 3-7.
LATHROP, G.D., WOLFE, W.H., ALBANESE, R.A., & MOYANAHAN, P.M. (1984)
An epidemiologic investigation of health effects in air force
personnel following exposure to herbicides. Baseline morbidity
study results, San Antonio, Texas, USAF School of Aerospace Medicine
(EK), Aerospace Medical Division, Brooks Air Force Base.
LEE, P. & SUZUKI, K. (1980) Induction of aryl hydrocarbon hydroxylase
activity in the rat prostate glands by
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Pharmacol. exp. Ther.,
215: 601-605.
LIEM, H.H., MULLER-EBERHARD, U., & JOHNSON, E.F. (1980) Differential
induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin of multiple forms of
rabbit microsomal cytochrome P-450: Evidence for tissue specificity.
Mol. Pharmacol., 18: 565-570.
LIGON, W.V., Jr & MAY, R.J. (1986) Determination of selected
chlorodibenzofurans and chlorodibenzodioxins using two-dimensional gas
chromatography/mass spectrometry. Anal. Chem., 58: 558-561.
LINDAHL, R., RAPPE, C., & BUSER, H.R. (1980) Formation of
polychlorinated dibenzofurans (PCDFs) and polychlorinated
dibenzo-p-dioxins (PCDDs) from the pyrolysis of poly-chlorinated
diphenyl ethers. Chemosphere, 9: 351-361.
LINDAHL, R., ROPER, M., & DIETRICH, R.A. (1978) Rat liver aldehyde
dehydrogenase-immunochemical identity of
2,3,7,8-tetrachlorodibenzo-p-dioxin inducible normal liver and
2-acetylaminofluorene inducible hepatoma isozymes. Biochem.
Pharmacol., 27: 2463-2465.
LOPRIENO, N., SBRANA, I., RUSCIANO, D., LASCIALFARI, D., & LARI, T.
(1982a) In vivo cytogenetic studies on mice and rats exposed to
2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Hutzinger, O., Frei, R.W.,
Merian, E., & Pocchiari, P., ed. Chlorinated dioxins and related
compounds. Impact on the environment, Oxford, New York, Pergamon
Press, pp. 419-428.
LOPRIENO, N., SBRANA, I., RUSCIANO, D., LASCIALFARI, D., LARI, T.,
STRETTI, G., & FREZZA, D. (1982b) In vitro and in vivo genotoxicity
studies on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). In: Report
of the 5th Meeting of the Seveso International Scientific Advisory
Committee, Milan, Regione Lombardia (Report No. 32).
LOVATI, M.R., GALBUSSERA, M., FRANCESCHINI, G., WEBER, G., RESI, L.,
TANGANELLI, P., & SIRTORI, C.R. (1984) Increased plasma and aortic
triglycerides in rabbits after acute administration of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
75: 91-97.
LU, C.-J.H., BAGGS, R.B., REDMOND, D., HENRY, E.C., SCHECTER, A., &
GASIEWICZ, T.A. (1986) Toxicity and evidence for meta-bolic
alterations in 2,3,7,8-tetrachlorodibenzo-p-treated guinea pigs fed by
total parenteral nutrition. Toxicol. appl. Pharmacol., 84:
439-453.
LUCIER, G.W., MCDANIEL, O.S., HOOK, G.E.R., FOWLER, B.A., SONAWANE,
B.R., & FAEDER, E. (1973) TCDD-induced changes in rat liver microsomal
enzymes. Environ. Health Perspect., 5: 199-209.
LUCIER, G.W., MCDANIEL, O.S., & HOOK, G.E.R. (1975a) Nature of the
enhancement of hepatic uridine diphosphate gluburonyl-transferase
activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Biochem.
Pharmacol., 24: 325-334.
LUCIER, G.W., SONAWANE, B.R., MCDANIEL, O.S., & HOOK, G.E.R. (1975b)
Postnatal stimulation of hepatic microsomal enzymes following
administration of TCDD to pregnant rats. Chem.-biol. Interact.,
11: 15-26.
LUCIER, G.W., RUMBAUGH, R.C., MCCOY, Z., HASS, R., HARVAN, D., &
ALBRO, P. (1986) Ingestion of soil contaminated with
2,3,7,8-tetrachlorodibenzop-dioxin (TCDD) alters hepatic enzyme
activities in rats. Fundam. appl. Toxicol., 6: 364-371.
LUSTENHOUWER, J.W.A., OLIE, K., & HUTZINGER, O. (1980) Chlorinated
dibenzo-p-dioxins and related compounds in incinerator effluents: A
review of measurements and mechanisms of formation. Chemosphere,
9: 501-522.
LUSTER, M.I., CLARK, G., LAWSON, L.D., & FAITH, R.E. (1979a) Effects
of brief in vitro exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxin
(TCDD) on mouse lymphocytes. J. environ. Pathol. Toxicol., 2:
965-977.
LUSTER, M.I., FAITH, R.E., & LAWSON, L.D. (1979b) Effects of
2,3,7,8-tetrachlorodibenzofuran (TCDF) on the immune system in guinea
pigs. Drug chem. Toxicol., 2: 49-60.
LUSTER, M.I., BOORMAN, G.A., DEAN, J.H., HARRIS, M.W., LUEBKE, R.W.,
PADARATHSINGH, M.L., & MOORE, J.A. (1980) Examination of bone marrow,
immunologic parameters and host susceptibility following pre- and
postnatal exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD).
Int. J. Immunopharmacol., 2: 301-310.
LUSTER, M.I., TUCKER, A.N., HONG, L., BOORMAN, G.A., & PATTERSON, R.
(1984) In vivo and in vitro effects of TCDD on stem cell and
B cell differentiation. In: Biological mechanisms of dioxin action,
Cold Spring Harbor, New York, Cold Spring Harbor Laboratory, pp.
411-417 (Banbury Report No. 18).
LUSTER, M.I., HONG, L.H., BOORMAN, G.A., CLARK, G., HAYES, H.T.,
GREENLEE, W.F., DOLD, K., & TUCKER, A.N. (1985) Acute myelotoxic
responses in mice exposed to 2,3,7,8-tetrachloro-dibenzo-p-dioxin
(TCDD). Toxicol. appl. Pharmacol., 81: 156-165.
MCCONNELL, E.E. (1980) Acute and chronic toxicity, carcinogenesis,
reproduction, teratogenesis and mutagenesis in animals. In: Kimbrough,
R.D., ed. Halogenated biphenyls, perphenyls, naphthalenes,
dibenzodioxins and related products, Amsterdam, Oxford, New York,
Elsevier Science Publishers, pp. 109-150.
MCCONNELL, E.E. & MOORE, J.A. (1979) Toxicopathology characteristis of
the halogenated aromatics. Ann. N.Y. Acad. Sci., 320: 138-150.
MCCONNELL, E.E., MOORE, J.A., & DALGARD, D.W. (1978a) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in Rhesus monkeys (Macaca
mulatta) following a single oral dose. Toxicol. appl. Pharmacol.,
43: 175-187.
MCCONNELL, E.E., MOORE, J.A., HASEMAN, J.K., & HARRIS, M.W. (1978b)
The comparative toxicity of chlorinated dibenzo-p-dioxins in mice and
guinea pigs. Toxicol. appl. Pharmacol., 44: 335-356.
MCCONNELL, E.E., LUCIER, G.W., RUMBAUGH, R.C., ALBRO, P.W., HARVAN,
D.J., HASS, J.R., & HARRIS, M.W. (1984) Dioxin in soil:
bioavailability after ingestion by rats and guinea pigs. Science,
223: 1077-1079.
MCCUNE, E.L., SAVAGE, J.E., & O'DELL, B.L. (1962) Hydro-pericardium
and ascites in chicks fed a chlorinated hydro-carbon. Poult. Sci.,
41: 295-299.
MCKINNEY, J., ALBRO, P., LUSTER, M., CORBETT, B., SCHROEDER, J., &
LAWSON, L. (1982) Development and reliability of a radioimmunoassay
for 2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Hutzinger, O., ed.
Chlorinated dioxins and related compounds: Impact on the
environment, Oxford, New York, Pergammon Press, pp. 67-77.
MCKINNEY, J.D. (1978) Analysis of
2,3,7,8-tetrachlorodibenzo-para-dioxin in environmental samples. Ecol.
Bull. (Stockholm), 27: 53-66.
MCKINNEY, J.D., CHAE, K., GUPTA, B.N., MOORE, J.A., & GOLDSTEIN, J.A.
(1976) Toxicological assessment of hexachlorobiphenyl isomers and
2,3,7,8-tetrachlorodibenzo- furan in chicks. I. Relationship of
chemical parameters. Toxicol. appl. Pharmacol., 36: 65-80.
MCKINNEY, J.D., FAWKES, J., JORDAN, S., CHAE, K., OATLEY, S., COLEMAN,
R.E., & BRINER, W. (1985) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)
as a potent and persistent thyroxine agonist: a mechanistic model for
toxicity based on molecular reactivity. Environ. Health Perspect.,
61: 41-53.
MCLAUGHLIN, D.L. & PEARSON, R.G. (1984) Concentrations of PCDD and
PCDF in soil from the vicinity of the SWARU incinerator, Stoney
Creek, July, 1983, Toronto, Ontario, Air Resources Branch, Ontario
Ministry of the Environment, 22 pp.
MCNULTY, W.P. (1984) Fetotoxicity of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) for rhesus macaques
(Macaca mulatta). Am. J. Primatol., 6: 41-47.
MCNULTY, W.P. (1985) Toxicity and fetotoxicity of TCDD, TCDF and PCB
isomers in rhesus macaques (Macaca mulatta). Environ. Health
Perspect., 60: 77-78.
MCNULTY, W.P., POMERANTZ, I., & FARRELL, T. (1981) Chronic toxicity of
2,3,7,8-tetrachlorodibenzofuran for rhesus macaques. Food Cosmet.
Toxicol., 19: 57-65.
MCNULTY, W.P., POMERANTZ, L.H., & FARRELL, T.J. (1982a) Chronic
toxicity of 2,3,7,8-tetrachlorodibenzofuran for rhesus macaques. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds:
Impact on the environment, Oxford, New York, Pergamon Press, pp.
411-418.
MCNULTY, W.P., NIELSEN-SMITH, K.A., & LAY, J.O., Jr (1982b)
Persistence of TCDD in monkey adipose tissue. Food Cosmet.
Toxicol., 20: 985-987.
MADGE, D.S. (1977) Effects of trichlorophenoxyacetic acid and
chlorodioxins on small intestinal function. Gen. Pharmacol., 8:
319-324.
MADHUKAR, B.V. & MATSUMURA, F. (1981) Differences in the nature of
induction of mixed-function oxidase systems of the rat liver among
phenobarbital, DDT, 3-methylcholanthrene, and TCDD. Toxicol. appl.
Pharmacol., 61: 109-118.
MADHUKAR, B.V., BREWSTER, D.W., & MATSUMURA, F. (1984) Effects of in
vivo-administered 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor
binding of epidermal growth factor in the hepatic plasma membrane of
rat, guinea pig, mouse, and hamster. Proc. Natl Acad. Sci. (USA),
81: 7407-7411.
MANARA, L., COCCIA, P., & CROCI, T. (1982) Persistent tissue levels of
TCDD in the mouse and their reduction as related to prevention of
toxicity. Drug Metab. Rev., 13(3): 423-446.
MANARA, L., COCCIA, P., & CROCI, T. (1984) Prevention of TCDD toxicity
in laboratory rodents by addition of charcoal or cholic acids to chow.
Food chem. Toxicol., 22(10): 815-818.
MANIS, J. & APAP, R. (1979) Intestinal organic anion transport,
glutathione transferase and aryl hydrocarbon hydroxylase activity:
effect of dioxin. Life Sci., 24: 1373-1380.
MANIS, J. & KIM, G. (1977) Induction of intestinal iron transport by
2,3,7,8-tetrachlorodibenzo-p-dioxin on environmental pollutant and
potent inducer of aryl hydrocarbon hydroxylase. Clin. Res., 25:
468A.
MANIS, J. & KIM, G. (1979) Introduction of iron transport by a potent
inducer of aryl hydrocarbon hydroxylase,
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Arch. environ. Health,
34(3): 141-145.
MANTOVANI, A., VECCHI, A., LUINI, W., SIRONI, M., CANDIANI, G.P.,
SPREAFICO, F., & GARATTINI, S. (1980) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on macrophage and natural killer
cell-mediated cytotoxicity in mice. Biomedicine, 32: 200-204.
MARKLUND, S., KJELLER, L.-O., HANSSON, M., TYSKLIND, M., RAPPE, C.,
RYAN, C., COLLAZO, H., & DOUGHERTY, R. (1986) Determination of PCDDs
and PCDFs in incineration samples and pyrolytic products. In: Rappe,
C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 79-92.
MARKLUND, S., RAPPE, C., TYSKLIND, M., & EGEBACK, K. (1987)
Identification of polychlorinated dibenzofurans and dioxins in
exhausts from cars run on unleaded gasoline. Chemosphere, 16:
29-36.
MARPLE, L., BRUNCK, R., & THROOP, L. (1986a) Water solubility of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Sci. Technol., 20:
180-182.
MARPLE, L., BERRIDGE, B., & THROOP, L. (1986b) Measurement of the
water-octanol partition coefficient of
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Environ. Sci. Technol., 20:
397-399.
MASON, G. & SAFE, S. (1986) Synthesis, biologic and toxic effects of
the major 2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites in the rat.
Toxicology, 41: 153-159.
MASON, G., SAWYER, T., KEYS, B., BANDIERA, S., ROMKES, M.,
PISKORSKA-PLISZCZYNSKA, J., ZMUDZKA, B., & SAFE, S. (1985)
Polychlorinated dibenzofurans (PCDFs): correlation between in vivo
and in vitro structure-activity relationships. Toxicology, 37:
1-12.
MASON, G., FARRELL, K., KEYS, B., PISKORSKA-PLISZCZYNSKA, J., SAFE,
L., & SAFE, S. (1986) Polychlorinated dibenzo-p-dioxins: Quantitative
in vitro and in vivo structure-activity relationships.
Toxicology, 41: 21-31.
MASON, M.E. & OKEY, A.B. (1982) Cytosolic and nuclear binding of
2,3,7,8-tetrachlorodibenzo-p-dioxin to the Ah receptor in
extra-hepatic tissues of rats and mice. Eur. J. Biochem., 123:
209-215.
MASUDA, Y., KUROKI, H., HARAGUCHI, K., & NAGAYAMA, J. (1985) PCB and
PCDF congeners in the blood and tissues of Yusho and Yu-Cheng
patients. Environ. Health Perspect., 59: 53-58.
MASUDA, Y., KUROKI, H., HARAGUCHI, K., & NAGAYAMA, J. (1986) PCDFs and
related compounds in humans from Yusho and Yu-Cheng incidents.
Chemosphere, 15: 1621-1628.
MATSUMURA, F. & BENEZET, H.J. (1973) Studies on the bioaccumu-lation
and microbial degradation of 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Environ. Health Perspect., 5: 253-258.
MATSUMURA, F., QUENSEN, J., & TSUSHIMOTO, G. (1983) Microbial
degradation of TCDD in a model ecosystem. In: Tucker, R.E., Young,
A.L., & Gray, A.P., ed. Human and environmental risks of
chlorinated dioxins and related compounds, New York, London, Plenum
Press, pp. 191-220.
MATSUMURA, F., BREWSTER, D.W., MADHUKAR, B.V., & BOMBICK, D.W. (1984)
Alteration of rat hepatic plasma membrane functions by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Arch. environ. Contam.
Toxicol., 13: 509-515.
MATTISON, D.R. & THORGEIRSSON, S.S. (1978) Gonadal aryl hydrocarbon
hydroxylase in rats and mice. Cancer Res., 38: 1368-1373.
MAY, G. (1973) Chloracne from the accidental production of
tetrachlorodibenzodioxin. Br. J. ind. Med., 30: 276-283.
MAY, G. (1982) Tetrachlorodibenzodioxin: a survey of subjects ten
years after exposure. Br. J. ind. Med., 39: 128-135.
MAZER, T., HILEMAN, F.D., NOBLE, R.W., & BROOKS, J.J. (1983) Synthesis
of the 38 tetrachlorodibenzofuran isomers and identification by
capillary column gas chromatography/mass spectrometry. Anal. Chem.,
55: 104-110.
MEYNE, J., ALLISON, D.C., BOSE, K., JORDAN, S.W., RIDOLPHO, P.F., &
SMITH, J. (1985) Hepatotoxic doses of dioxin do not damage mouse bone
marrow chromosomes. Mutat. Res., 157: 63-69.
MILES, W.F., SINGH, J., GURPRASAD, N.P., & MALIS, G.P. (1985) Isomer
specific determination of hexachlorodioxins in technical
pentachlorophenol (PCP) and its sodium salt. Chemosphere, 14(6/7):
807-810.
MILLER, A.G., ISRAEL, D., & WHITLOCK, J.P., Jr (1983) Bio-chemical and
genetic analysis of variant mouse hepatoma cells defective in the
induction of benzo(a)pyrene-metabolizing enzyme. J. biol. Chem.,
258: 3523-3527.
MILNES, M.H. (1971) Formation of 2,3,7,8-tetrachlorodibenzo-dioxin by
thermal decomposition of sodium 2,4,5-trichloro-phentane. Nature
(Lond.), 232: 395-396.
MILSTONE, L.M. & LAVIGNE, J.F. (1984)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin induces hyperplasia in confluent
cultures of human keratinocytes. J. invest. Dermatol., 82: 532-534.
MITCHUM, R.K., MOLER, G.F., & KORFMACHER, W.A. (1980) Combined
capillary gas chromatography/atmospheric pressure negative chemical
ionization/mass spectrometry for the determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin in tissue. Anal. Chem., 52:
2278-2282.
MITTLER, J.C., ERTEL, N.H., PENG, R.X., YANG, C.S., & KIERNAN, T.
(1984) Changes in testosterone hydroxylase activity in rat testis
following administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Ann.
N.Y. Acad. Sci., 438: 645-648.
MIVRA, H., OMORI, A., & SHIBUE, M. (1974) The effect of chlorophenols
on the excretion of porphyrins in urine. Jpn. J. ind. Health,
16: 575-577.
MOLLER, M.E., GLOWINSKI, I.B., & THORGEIRSSON, S.S. (1984) The
genotoxicity of aromatic amines in primary hepatocytes iso-lated from
C57BL/6 and DBA/2 mice. Carcinogenesis, 5: 797-804.
MOORE, J.A., GUPTA, B.N., ZINKL, J.G., & VOS, J.G. (1973) Postnatal
effects of maternal exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxins
(TCDD). Environ. Health Perspect., 5: 81-85.
MOORE, J.A., GUPTA, B.N., & VOS, J.G. (1976) Toxicity of
2,3,7,8-tetrachloro-dibenzofuran - preliminary results. In:
Proceedings from the National Conference on Polychlorinated
Biphenyls, Chicago, Illinois, 19-21 November, 1975, Research
Triangle Park, North Carolina, Research Triangle Park Institute.
MOORE, J.A., MCCONNELL, E.E., DALGARD, D.W., & HARRIS, M.W. (1979)
Comparative toxicity of three halogenated dibenzofurans in guinea
pigs, mice and rhesus monkeys. Ann. N.Y. Acad. Sci., 320: 151-163.
MOORE, R.W. & PETERSON, R.E. (1985) Enhanced catabolism and
elimination of androgens do not cause the androgenic de-ficiency in
2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Fed. Proc., 44:
518.
MOORE, R.W., POTTER, C.L., THEOBALD, H.M., ROBINSON, J.A., & PETERSON,
R.E. (1985) Androgenic deficiency in male rats treated with
2,3,7,8-tetra-chlorodbenzo-p-dioxin. Toxicol. appl. Pharmacol.,
79: 99-111.
MORITA, M. & OISHI, S. (1977) Clearance and tissue distribution of
polychlorinated dibenzofurans in mice. Bull. environ. Contam.
Toxicol., 18: 61-66.
MORITA, M., NAKAGAWA, J., & RAPPE, C. (1978) Polychlorinated
dibenzofuran (PCDF) formation from PCB mixture by heat and oxygen.
Bull. environ. Contam. Toxicol., 19: 665-670.
MORTELMANS, K., HAWORTH, S., SPECK, W., & ZEIGER, E. (1984)
Mutagenicity testing of agent orange components and related chemicals.
Toxicol. appl. Pharmarcol., 75: 137-146.
MOSES, M. & SELIKOFF, I.J. (1981) Soft tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 1: 1370.
MOSES, M., LILIS, R., CROW, K.D., THORNTON, J., FISCHBEIN, A.,
ANDERSON, H.A., & SELIKOFF, I.J. (1984) Health status of workers with
past exposure to 2,3,7,8-tetrachloridibenzo-p-dioxin in manufacture of
2,4,5-trichlorophenoxyacetic acid: Comparison of findings with and
without chloracne. Am. J. ind. Med., 5: 161-182
MOTTURA, A., ZEI, G., NUZZO, F., CRIMAUDO, C., GIORGI, R., VENERONI,
P., PAGGINI, P., MACARELLI, P., FRACCARO, M., NICOLETTI, B., &
DECARLI, L. (1981) Evaluation of results of chromosome analyses on
lymphocytes of TCDD exposed subjects after the Seveso accident.
Mutat. Res., 85: 238-239.
MUKITARI, H., BAARS, A.J., & BREIMER, D.D. (1981) Differences in
inducibility of particulate and cytosolic rat liver glutathione
S-transferase activities. Xenobiotica, 11: 367-371.
MULCAHY, M.T. (1980) Correspondence. Chromosome aberrations and "agent
orange". Med. J. Aust., 2: 573-574.
MULLER, E. (1937) [Chloracne (caused by chlorinated benzenes).
Inaugural Dissertation,] Speyer am Rhein, Pilger-Druckerei (Thesis,
Friedrich-Wilhelm University, Breslau).
MURRAY, F.J., SMITH, F.A., NITSCHKE, K.D., HUMISTON, C.G., KOCIBA,
R.J., & SCHWETZ, B.A. (1979) Three-generation reproduction study of
rats given 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the diet.
Toxicol. appl. Pharmacol., 50: 241-252.
NAGARKATTI, P.S., SWEENEY, G.D., GAULDIE, J., & CLARK, D.A. (1984)
Sensitivity to suppression of cytotoxic T cells generation by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in dependent on the Ah
genotype of the murine host. Toxicol. appl. Pharmacol., 72:
169-176.
NAGAYAMA, J., KURATSUNE, M., & MASUDA, Y. (1976) Determination of
chlorinated dibenzofurans in kanechlors and "Yusho oil". Bull.
environ. Contam. Toxicol., 15: 9-13.
NAGAYAMA, J., NISHIZUMI, M., & MASUDA, Y. (1979) [Subacute toxicity of
polychlorinated dibenzofurans in mice.] Fukuoka Igakuzassh, 70:
109-113 (in Japanese).
NAGAYAMA, J., TOKUDOME, S., & KURATSUNE, M. (1980) Transfer of
polychlorinated dibenzofurans to the foetuses and offspring of mice.
Food Cosmet. Toxicol., 18: 153-157.
NAGAYAMA, J., KUROKI, H., MASUDA, Y., & KURATSUME, M. (1983) A
comparative study of polychlorinated dibenzofurans, poly-chlorinated
biphenyls and 2,3,7,8-tetrachlorodibenzo-p-dioxin on aryl hydrocarbon
hydroxylase inducing potency in rats. Arch. Toxicol., 53: 177-184.
NAGAYAMA, J., KUROKI, H., MASUDA, Y., HANDA, S., & KURATSUNE, M.
(1985a) Genetically mediated induction of aryl hydrocarbon hydroxylase
activity in mice by polychlorinated dibenzofuran isomers and
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Toxicol., 56: 226-229.
NAGAYAMA, J., KIYOHARA, C., KURATSUNE, M., & MASUDA, Y. (1985b)
Induction of aryl hydrocarbon hydroxylase activity in human
lymphoblastoid cells by chlorinated dibenzofuran isomers and
2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Keith, L.H., Rappe, C., &
Choudhary, G., ed. Chlorinated dioxins and dibenzofurans in the
total environment II, Stoneham, Maine, Butterworth Publishers, pp.
285-295.
NAU, H. & BASS, R. (1981) Transfer of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) to the mouse embryo and
fetus. Toxicology, 20: 299-308.
NAU, H., BASS, R., & NEUBERT, D. (1986) Transfer of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) via placenta and milk, and
postnatal toxicity in the mouse. Arch. Toxicol., 59: 36-40.
NCI (1977) Bioassay of dibenzo-p-dioxin for possible
carcinogenicity, Washington, DC, National Cancer Institute, 104 pp
(Technical Report No. 122).
NCI (1979) Bioassay of 2,7-dichlorodibenzo-p-dioxin (DCDD) for
possible carcinogenicity, Washington, DC, National Cancer Institute,
103 pp (Technical Report No. 123).
NEAL, R.A., BEATTY, P.W., & GASIEWICZ, T.A. (1979) Studies of the
mechanisms of toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
Ann. N.Y. Acad. Sci., 320: 204-213.
NEBERT, D.W., ROBINSON, J.R., NIWA, A., KUMAKI, K., & POLAND, A.P.
(1975) Genetic expression of aryl hydrocarbon hydroxylase activity in
the mouse. J. cell. Physiol., 85: 393-414.
NEBERT, D.W., JENSEN, N.M., PERRY, J.W., & OKA, T. (1980) Association
between ornithine decarboxylase induction and the Ah locus in mice
treated with polycyclic aromatic compounds. J. biol. Chem., 255:
6836-6842.
NESTRICK, T.J., LAMPARSKI, L.L., & STEHL, R.H. (1979) Synthesis and
identification of the 22 tetrachlorodienzo-p-dioxin isomers by high
performance liquid chromatography and gas chromatography. Anal.
Chem., 51: 2273-2281.
NESTRICK, T.J., LAMPARSKI, L.L., & TOWNSEND, D.I. (1980)
Identification of tetrachlorodibenzo-p-dioxin isomers at the 1 ng
level by photolytic degradation and patter recognition techniques.
Anal. Chem., 52: 1865-1874.
NESTRICK, T.J., LAMPARSKI, L.L., FRAWLEY, N.N., HUMMEL, R.A., KOCHER,
C.W., MAHLE, N.H., MCCOY, J.W., MILLER, D.L., PETERS, T.L., PILLEPICH,
J.L., SMITH, W.E., & TOBEY, S.W. (1986) Perspectives of a large scale
environmental survey for chlorinated dioxins: Overview and soil data.
Chemosphere, 15: 1453-1460.
NEUBERT, D. & DILLMANN, I. (1972) Embryotoxic effects in mice treated
with 2,4,5-trichlorophenoxyacetic acid and
2,3,7,8-tetrachlorodibenzo-p-dioxin. Naunyn-Schmiedeberg's Arch.
Pharmacol., 272: 243-264.
NIEMANN, R.A., BRUMLEY, W.C., FIRESTONE, D., & SPHON, J.A. (1983)
Analysis of fish for 2,3,7,8-tetrachlorodibenzo-p-dioxin by electron
capture capillary gas chromatography. Anal. Chem., 55: 1497-1504.
NIH (1980a) Bioassay of 1,2,3,6,7,8-and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxin for possible
carcinogenicity (dermal study), Bethesda, Maryland, US Department of
Health and Human Services, National Institutes of Health (DHSS
Publication No. (NIH) 80-1758).
NIH (1980b) Bioassay of 1,2,3,6,7,8-and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxin for possible
carcinogenicity (gavage), Bethseda, Maryland, US Department of
Health and Human Services, National Institutes of Health (DHHS
Publication No. (NIH) 80-1754).
NIH (1982a) Carcinogenesis bioassay of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (CAS No 1746-01-6) in
Osborne-Mendel rats and B6C3F1 mice (gavage study), Research
Triangle Park, North Carolina, US National Institutes of Health,
National Toxicology Program (NTP Technical Report Series No. 209).
NIH (1982b) Carcinogenesis bioassay of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (CAS No 1746-01-6) in
Swiss-Webster mice (dermal study), Research Triangle Park, North
Carolina, US National Institutes of Health, National Toxicology
Program (NTP Technical Report Series No. 201).
NILSSON, C.A., ANDERSSON, K., RAPPE, C., & WESTERMARK, S.O. (1974)
Chromatographic evidence for the formation of chloro-dioxins from
chloro-2-phenoxyphenols. J. Chromatogr., 96: 137-147.
NILSSON, C.A., NORSTROM, A., ANDERSSON, K., & RAPPE, C. (1978)
Impurities in commercial products related to pentachlorophenol. In:
Rao, K.R., ed. Pentachlorophenol: Chemistry, pharmacology and
environmental toxicology, New York, London, Plenum Press, pp.
313-324.
NISBET, I.C.T. & PAXTON, M.B. (1982) Statistical aspects of
three-generation studies of the reproductive toxicity of TCDD and
2,4,5,-T. Am. Stat., 36(3): 290-298.
NISHIZUMI, M. (1978) Acute toxicity of polychlorinated dibenzofurans
in CF-1 mice. Toxicol. appl. Pharmacol., 45: 209-212.
NISHIZUMI, M., KURATSUNE, M., & MASSUDA, Y. (1975) [Comparison of
hyperkeratosis induced by PCBs, PCDF and PCDD application.
Polychlorinated biphenyls, polychlorinated dibenzofuran and
polychlorinated dibenzodioxin.] Fukuoka Asa Med., 66: 600-604 (in
Japanese).
NIWA, A., KUMAKI, K., & NEBERT, D.W. (1975) Induction of aryl
hydrocarbon - hydroxylase activity in various cell cultures by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol. Pharmacol., 11: 399-408.
NOLAN, R.J., SMITH, F.A., & HEFNER, J.G. (1979) Elimination and tissue
distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in female
guinea pigs following a single oral dose. Tox. appl. Pharmacol.,
48: 167 (abstract).
NORBACK, D.H. & ALLEN, J.R. (1973) Biological responses of the
nonhuman primate, chicken, and rat to chlorinated dibenzo-p-dioxin
ingestion. Environ. Health Perspect., 5: 233-240.
NORBACK, D.H., ENGBLOM, J.F., & ALLEN, J.R. (1975) Tissue distribution
and excretion of octachlorodibenzo-p-dioxin in the rat. Toxicol.
appl. Pharmacol., 32: 330-338.
NORMAN, L.R., JOHNSON, E.F., & MULLER-EBERHARD, U. (1978)
Identification of the major cytochrome P-450 form transplacentally
induced in neonatal rabbits by 2,3,7,8-tetra-chlorodibenzo-p-dioxin.
J. biol. Chem., 253: 8640-8647.
NORSTROM, R.J., HALLETT, D.J., SIMON, M., & MULVIHILL, M.J. (1982)
Analysis of Great Lakes Herring Gull eggs for
tetra-chlorodibenzo-p-dioxins. In: Hutzinger, O., ed. Chlorinated
dioxins and related compounds. Impact on the environment, Oxford,
New York, Pergammon Press, pp. 173-182.
NORSTROM, R.J., SIMON, M., & WESELOH, D.V. (1986) Long-term trends
of PCDD and PCDF contamination in the Great Lakes. Presented at
Dioxin 86, 6th International Symposium on Chlorinated Dioxins and
Related Compounds, Fukuoka, Japan, 16-19 September, 1986.
NORSTROM, R.J., SIMON, M., WHITEHEAD, P.E., KUSSAT, R., & GARRET, C.
(1988) Level of polychlorinated dibenzo-p-dioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs) in biota and sediments near
potential sources of contamination in British Columbia, 1987, West
Vancouver, British Columbia, Environment Canada (Analytical Report
CRD-88-5).
NRC CANADA (1981) Polychlorinated dibenzo-p-dioxins. Limitation to
the current analytical technique, Ottawa, National Research Council
of Canada (Publication No. NRCC/NRC 18576).
NYGREN, M., RAPPE, C., LINDSTROM, G., HANSSON, M., BERGQVIST, P.-A.,
MARKLUND, S., DOMELLOF, L., HARDELL, L., & OLSSON, M. (1986)
Identification of 2,3,7,8-substituted polychlorinated dioxins and
dibenzofurans in environmental and human samples. In: Rappe, C.,
Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 17-34.
OBERG, T. & BERGSTROM, J. (1986) Combustion test data from a Swedish
hazardous waste incinerator. Chemosphere, 15: 2045-2048.
OISHI, S. (1977) Influence of polychlorinated dibenzofurans (PCDFs)
and polychlorinated biphenyls (PCBs) to serum protein components in
rats. Bull. environ. Contam. Toxicol., 18: 773-777.
OISHI, S. & HIRAGA, K. (1980) Effect of polychlorinated biphenyl,
dibenzofuran and dibenzo-p-dioxin on the susceptibility of male mice
to endotoxin. J. environ. Sci. Health, B15: 77-85.
OISHI, S., MORITA, M., & FUKUDA, H. (1978) Comparative toxicity of
polychlorinated biphenyls and dibenzofurans in rats. Toxicol. appl.
Pharmacol., 43: 13-22.
O'KEEFE, P., MESELSON, M.S., & BAUGHMAN, R. (1977) Neutral clean-up
procedure for 2,3,7,8-tetrachlorodibenzo-p-dioxin residues in bovine
fat and milk. J. Assoc. Off. Anal. Chem., 61: 621-626.
O'KEEFE, P., MEYER, C., HILKER, D., ALDOVES, K., JELUS-TYROR, B.,
DILLON, K., DONNOLLY, R., HORN, E., & SLOAN, R. (1983) Analysis of
2,3,7,8-tetrachlorodibenzo-p-dioxin in Great Lakes fish. Chemosphere,
12: 325-332.
O'KEEFE, P., SILKWORTH, J.B., BIERTHY, J.F., SMITH, R.M., DECAPRIO,
A.P., TURNER, J.N., EADON, G., HILKER, D.R., ALDOUS, K.M., KAMINSKY,
L.S., & COLLINS, D.N. (1985) Chemical and biological investigations of
a transformer accident at Binghamton, NY. Environ. Health
Perspect., 60: 201-209.
OKEY, A.B. & VELLA, L.M. (1982) Binding of 3-methylchlo-anthrene and
2,3,7,8-tetrachlorodibenzo-p-dioxin to a common Ah receptor site in
mouse and rat hepatic cytosols. Eur. J. Biochem., 127: 39-47.
OKEY, A.B., BONDY, G.P., MASON, M.E., KAHL, G.F., EISEN, H.J.,
GUENTHNER, T.M., & NEBERT, D.W. (1979) Regulatory gene product of the
Ah locus. J. biol. Chem., 254: 11636-11648.
OKEY, A.B., BONDY, G.P., MASON, M.E., NEBERT, D.W., FORSTER-GIBSON,
C.J., MUNCHAN, J., & DUFRESNE, M.J. (1980) Temperature-dependent
cytosol-to-nucleus translocation of the Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin in continuous cell culture lines.
J. biol. Chem., 225: 11415-11422.
OKINO, S.T., QUATTROCHI, L.C., BARNES, H.J., OSANTO, S., GRIFFIN,
K.J., JOHNSON, E.F., & TUKEY, R.H. (1985) Cloning and characterization
of cDNAs encoding 2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible rabbit
mRNAs for cytochrome P-450 isozymes 4 and 6. Proc. Natl Acad. Sci.
(USA), 82: 5310-5314.
OLIE, K., VERMEULEN, P.L., & HUTZINGER, O. (1977)
Chlorodibenzo-p-dioxins and chlorodibenzofurans are trace components
of fly ash and flue gas of some municipal incinerators in the
Netherlands. Chemosphere, 8: 455-459.
OLIE, K., BERG, M.V.D., & HUTZINGER, O. (1983) Formation and fate of
PCDD and PCDF from combustion processes. Chemosphere, 12: 627-636.
OLIVER, R.M. (1975) Toxic effects of
2,3,7,8-tetrachloro-dibenzo-1,4-dioxin in laboratory workers. Br. J.
ind. Med., 32: 49-53.
OLSON, J.A. & GUNNING, D. (1983) The storage form of vitamin A in rat
liver cells. J. Nutr., 113: 2184-2191.
OLSON, J.R. (1986) Metabolism and disposition of
2,3,7,8-tetrachlorodibenzo-p-dioxin in guinea pigs. Toxicol. appl.
Pharmacol., 85: 263-273.
OLSON, J.R., GASIEWICZ, T.A., & NEAL, R.A. (1980a) Tissue
distribution, excretion, and metabolism of
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) in the golden Syrian
hamster. Toxicol. appl. Pharmacol., 56: 78-85.
OLSON, J.R., HOLSCHER, M.A., & NEAL, R.A. (1980b) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the golden syrian hamster.
Toxicol. appl. Pharmacol., 55: 67-78.
ONO, M., WAKIMOTO, T., TATSUKAWA, R., & MASUDA, Y. (1986)
Polychlorinated dibenzo-p-dioxins and dibenzofurans in human adipose
tissues of Japan. Chemosphere, 15: 1629-1634.
ONTARIO (1985) Scientific criteria document for standard
development. Polychlorinated dibenzo-p-dioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs), Toronto, Ontario Ministry of
the Environment (Report No. 4-84).
ONTARIO (1986) Drinking water survey St. Clair river - Detroit river
area, Toronto, Ontario Ministry of the Environment.
ORR, J.B. & RICHARDS, M.B. (1934) Growth and vitamin A deficiency.
Biochem. J., 28: 1259-1273.
OSBORNE, R. & GREENLEE, W.F. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) enhances terminal
differentiation of cultured human epidermal cells. Toxicol. appl.
Pharmacol., 77: 434-443.
OSBORNE, R., DOLD, K.M., & GREENLEE, W.F. (1984) Cell culture models
to study mechanisms of toxicity of chlorinated aromatic compounds to
skin and thymus. Chem. Ind. Inst. Toxicol., 4: 2-7.
OTT, M.G., HOLDER, B.B., & OLSON, R.D. (1980) A mortality analysis of
employees engaged in the manufacture of 2,4,5-trichlorophenoxyacetic
acid. J. occup. Med., 22: 47-50.
PAASIVIRTA, J., ENQVIST, J., RAISANEN, S., & PAASIVUO, P. (1977) On
the limit of detection of TCDD in gas chromatography. Chemosphere,
6: 355.
PALAUSKY, J., HARWOOD, J.J., CLEVENGER, T.E., KAPILA, S., & YANDERS,
A.F. (1986) Disposition of tetrachlorodibenzo-p-dioxin in soil. In:
Rappe, C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 211-223.
PATTERSON, D.G., Jr, HOFFMAN, R.E., NEEDHAM, L.L., ROBERTS, D.W.,
BAGBY, J.R., PIRKLE, J.L., FALK, H., SAMPSON, E.J., & HOUK, V.N.
(1986) 2,3,7,8-Tetrachlorodibenzo-p-dioxin levels in adipose tissue of
exposed and control persons in Missouri. J. Am. Med. Assoc., 256:
2683-2686.
PATTERSON, J.M., MCHENRY, E.W., & CRANDALL, W.A. (1942) The
physiological properties of vitamin A. 1. A specific effect upon body
weight and body composition in the Albino rat. Biochem. J., 36:
792-794.
PAZDERNIK, T.L. & ROZMAN, K.K. (1985) Effect of thyroidectomy and
thyroxine on 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced
immunotoxicity. Life Sci., 36: 695-703.
PAZDEROVA-VEJLUPKOVA, J., LUKAS, E., NEMCOVA, M., SPACILOVA, M.,
JIRASEK, L., KALENSKY, J., JOHN, J., JIRASEK, A., & PICKOVA, J. (1974)
[Chronic intoxication by chlorinated hydrocarbons formed during the
production of sodium 2,4,5-trichlorophenoxyacetate.] Pracov. Lek.,
26: 332-339 (in Czech).
PAZDEROVA-VEJLUPKOVA, J., LUKAS, E., NEMCOVA, M., PICKOVA, J., &
JIRASEK, L. (1980) [Chronic poisoning by
2,3,7,8-tetra-chlorodibenzo-p-dioxin.] Pracov. Lek., 32: 204-209
(in Czech).
PAZDEROVA-VEJLUPKOVA, J., NEMCOVA, M., PICKOVA, J., JIRASEK, L., &
LUKAS, E. (1981) The development and prognosis of chronic intoxication
by tetrachlorodibenzo-p-dioxin in men. Arch. environ. Health, 36:
5-11.
PETERSON, R.E., MADHUKAR, B.V., YANG, K.H., & MATSUMURA, F. (1979a)
Depression of adenosine triphosphatase activities in isolated liver
surface membranes of 2,3,7,8-tetrahlorodibenzo-p-dioxin-treated rats:
correlation with effects on ouabain biliary excretion and bile flow.
J. Pharmacol. exp. Ther., 210: 275-210.
PETERSON, R.E., HAMADA, N., YANG, K.H., & MADHUKAR, B.V. (1979b)
Reversal of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced depression of
ouabain biliary excretion by pregnenolone-16-beta-carbonitrile and
spironolactone in isolated perfused rat livers. Toxicol. appl.
Pharmacol., 50: 407-416.
PHILIPPI, M., SCHMID, J., WIPF, H.K., & HUTTER, R. (1982) A microbial
metabolite of TCDD. Experientia (Basel), 38: 659-661.
PIPER, W.N., ROSE, J.Q., & GEHRING, P.J. (1973) Excretion and tissue
distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Adv.
Chem. Ser., 120: 85-91.
PITOT, H.C. & SIRICA, A.E. (1980) The stages of initiation and
promotion in hepatocarcinogenesis. Biochim. Biophys. Acta, 605:
191-215.
PITOT, H.C., GOLDSWORTHY, T., CAMPBELL, H.A., & POLAND, A. (1980)
Quantitative evaluation of the promotion by
2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from
diethylnitrosamine. Cancer Res., 40: 3616-3620.
POCCHIARI, F. (1978) 2,3,7,8-Tetrachlorodibenzo-para-dioxin
decontamination. Ecol. Bull. (Stockholm), 27: 67-70.
POCCHIARI, F., DIDOMENICO, A., SILANO, V., & ZAPPONI, G. (1983)
Environmental impact of the accidental release of
tetrachlorodibenzo-p-dioxn (TCDD) at Seveso (Italy). In: Coulston, F.
& Pocchiari, F., ed. Accidental exposure to dioxins. human health
aspects, New York, London, Academic Press, pp. 5-35.
POELLINGER, L. & GULLBERG, D. (1985) Characterization of the
hydrophobic properties of the receptor for
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Mol. Pharmacol., 27:
271-276.
POELLINGER, L., KURL, R.N., LUND, J., GILLNER, M., CARLSTEDT-DUKE, J.,
HOGBERG, B., & GUSTAFSSON. J.-A. (1982) High-affinity binding of
2,3,7,8-tetrachlorodibenzo-p-dioxin in cell nuclei from rat liver.
Biochem. Biophys. Acta, 714: 516-523.
POELLINGER, L., LUND, J., HANSSON, L.-A., & GUSTAFSSON, J.-A. (1983)
Physicochemical characterization of specific and non-specific
polyaromatic hydrocarbon binders in rat and mouse liver cytosol. J.
biol. Chem., 258: 13535-13542.
POHJANVIRTA, R. & TUOMISTO, J. (1986) Han/Wistar rats are
exceptionally resistant to TCDD. II. Arch. Toxicol., 11 (Suppl.):
344-347.
POHLAND, A.E. & YANG, G.C. (1972) Preparation and characterization of
chlorinated dibenzo-p-dioxins. J. agric. food Chem., 20(6):
1093-1099.
POIGER, H. & BUSER, H.R. (1983) Structure elucidation of mammalian
TCDD-metabolites. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 483-492.
POIGER, H. & SCHLATTER, Ch. (1979) Biological degradation of TCDD in
rats. Nature (London), 281: 706-707.
POIGER, H. & SCHLATTER, Ch. (1980) Influence of solvents and
adsorbents on dermal and intestinal absorption of TCDD. Food Cosmet.
Toxicol., 18: 477-481.
POIGER, H. & SCHLATTER, Ch. (1985) Influence of phenobarbital and TCDD
on the hepatic metabolism of TCDD in the dog. Experientia (Basel),
41: 376-378.
POIGER, H. & SCHLATTER, Ch. (1986) Pharmacokinetics of 2,3,7,8-TCDD in
man. Chemosphere, 15: 1489-1494.
POIGER, H., BUSER, H.R., WEBER, H., ZWEIFEL, U., & SCHLATTER, Ch.
(1982) Structure elucidation of mammalian TCDD-metabolites.
Experientia (Basel), 38: 484-486.
POIGER, H., BUSER, H.R., & SCHLATTER, Ch. (1984) The metabolism of
2,3,7,8-tetrachlorodibenzofuran in the rat. Chemosphere, 13:
351-357.
POLAND, A. & GLOVER, E. (1973a) Studies on the mechanism of toxicity
of the chlorinated dibenzo-p-dioxins. Environ. Health Perspect.,
5: 245-251.
POLAND, A. & GLOVER, E. (1973b) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
A potent inducer of aminolevulinic acid synthetase. Science, 179:
476-477.
POLAND, A. & GLOVER, E. (1973c) Chlorinated dibenzo-p-dioxins: Potent
inducers of aminolevulinic acid synthetase and aryl hydrocarbon
hydroxylase. II. A study of the structure-activity relationship.
Mol. Pharmacol., 9: 736-747.
POLAND, A. & GLOVER, E. (1974a) The induction of aryl hydrocarbon
hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a
receptor mutation in genetically non-responsive mice. Pharmacologist,
16: 240 (Abstract 282).
POLAND, A. & GLOVER, E. (1974b) Comparison of
2,3,7,8-tetra-chlorodibenzo-p-dioxin, a potent inducer of aryl
hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol. Pharmacol.,
10: 349-359.
POLAND, A. & GLOVER, E. (1974c) Genetic expression of aryl hydrocarbon
hydroxylase activity. Induction of monooxygenase activities and
cytochrome P1-450 formation by 2,3,7,8-tetra-chlorodibenzo-p-dioxin in
mice genetically "nonresponsive" to other aromatic hydrocarbons. J.
biol. Chem., 249: 5599-5606.
POLAND, A. & GLOVER, E. (1975) Genetic expression of arylhydrocarbon
hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a
receptor mutation in genetically non-responsive mice. Mol.
Pharmacol., 11: 389-398.
POLAND, A. & GLOVER, E. (1979) An estimate of the maximum in vivo
covalent binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to rat liver
protein, ribosomal RNA, and DNA. Cancer Res., 39: 3341-3344.
POLAND, A. & GLOVER, E. (1980) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Segregation of toxicity with the Ah-locus. Mol. Pharmacol., 17:
86-94.
POLAND, A. & KENDE, A. (1976) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Environmental contaminant and molecular probe. Fed. Proc., 35:
2404-2411.
POLAND, A. & KNUTSON, J.C. (1982) 2,3,7,8-Tetrachlorodibenzo-p-dioxin
and related halogenated aromatic hydrocarbons: Examination of the
mechanisms of toxicity. Annu. Rev. Pharmacol. Toxicol., 22:
517-554.
POLAND, A., SMITH, D., METTER, G., & POSSICK, P. (1971) A health
survey of workers in a 2,4-D and 2,4,5-T plant. Arch. environ.
Health, 22: 316-327.
POLAND, A., GLOVER, E., & KENDE, A.S. (1976) Stereospecific, high
affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic
cytosol. Evidence that the binding species is receptor for induction
of aryl hydrocarbon hydroxylase. J. biol. Chem., 251: 4936-4946.
POLAND, A., PALEN, D., & GLOVER, E. (1982) Tumor promotion by TCDD in
HRS/J mice. Nature (Lond.), 300(5889): 271-273.
POLI, A., FRANCESCHINI, G., PUGLISI, L., & SIRTORI, C.R. (1980)
Increased total and high density lipoprotein cholesterol with
apoprotein changes resembling streptozotocin diabetes in
tetrachlorodibenzodioxin (TCDD) treated rats. Biochem. Pharmacol.,
29: 835-838.
POTTER, C.L., SIPES, I.G., & RUSSELL, D.H. (1982) Inhibition of
ornithine decarboxylase activity by
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Biochem. Pharmacol., 31:
3367-3371.
POTTER, C.L., SIPES, I.G., & RUSSELL, D.H. (1983) Hypothyroxinemia and
hypothermia in rats in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin
administration. Toxicol. appl. Pharmacol., 69: 89-95.
POTTER, C.L., MENAHAN, L.A., & PETERSON, R.E. (1986a) Relationship of
alterations in energy metabolism to hypophagia in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. appl. Toxicol., 6:
89-97.
POTTER, C.L., MOORE, R.W., INHORN, S.L., HAGEN, T.C., & PETERSON, R.E.
(1986b) Thyroid status and thermogenesis in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
84: 45-55.
PRATT, R.M., DENCKER, L., & DIEWERT, V.M. (1984)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced cleft palate in the mouse:
Evidence for alterations in palatal shelf fusion. Teratog.
Carcinogen. Mutagen., 4: 427-436.
PRESTON, B.D., VAN MILLER, P.J., MOORE, R.W., & ALLEN, J.R. (1981)
Promoting effects of polychlorinated biphenyls (Aroclor 1254) and
polychlorinated dibenzofuran-free Aroclor 1254 on
diethylnitrosamine-induced tumorigenesis in the rat. J. Natl Cancer
Inst., 66: 509-515.
PUHVEL, S.M., SAKAMOTO, M., ERTL, D.C., & REISNER, R.M. (1982)
Hairless mice as models for chloracne: A study of cutaneous changes
induced by topical application of established chloracnegens.
Toxicol. appl. Pharmacol., 64: 492-503.
PUHVEL, S.M., ERTL, D.C., & LYNBERG, C.A. (1984) Increased epidermal
transglutaminase activity following
2,3,7,8-tetra-chlorodibenzo-p-dioxin: In vivo and in vitro studies
with mouse skin. Toxicol. appl. Pharmacol., 73: 42-47.
PUHVEL, S.M., SAKAMOTO, M., & REISNER, R.M. (1986) Localization of
TCDD in hairless mouse skin. Chemosphere, 15: 2065-2067.
QUILLEY, C.P. & RIFKIND, A.B. (1986) Prostaglandin release by the
chick embryo heart is increased by
2,3,7,8-tetrachloro-dibenzo-p-dioxin and by other cytochrome P-448
inducers. Biochem. biophys. Res. Commun., 136(2): 582-589.
RAMSEY, J.C., HEFNER, J.G., KARBOWSKI, R.J., BRAUN, W.H., & GEHRING,
P.J. (1982) The in vivo biotransformation of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat. Toxicol.
appl. Pharmacol., 65: 180-184.
RAPPE, C. (1978a) Chemical background of the phenoxy acids and
dioxins. Ecol. Bull. (Stockholm), 27: 28-30.
RAPPE, C. (1978b) Decontamination of products formed during the
industrial preparation of 2,4,5-trichlorophenol. In: Cattabeni, F.,
Cavallero, A., & Galli, G., ed. Dioxin: toxicological and chemical
aspects, New York, SP Medical and Scientific Books, pp. 179-183.
RAPPE, C. (1984) Analysis of polychlorinated dioxins and furans.
Environ. Sci. Technol., 18: 78A-90A.
RAPPE C. (1985) Problems in analysis of PCDDs and PCDFs and presence
of these compounds in human milk, Copenhagen, WHO Regional Office for
Europe (ICP/CEH/501/05/7).
RAPPE, C. (1987) Global distribution of polychlorinated dioxins
(PCDDs) and dibenzofurans (PCDFs). In: Solving hazardous waste
problems: Learning from dioxins, New York, American Chemical
Society, pp. 20-23 (ACS Symposium Series No. 338).
RAPPE, C. & BUSER, H.R. (1980) Chemical properties and analytical
methods. In: Kimbrough, R.D., ed. Halogenated biphenyls, terphenyls,
naphthalenes, dibenzodioxins and related products, Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp. 41-80.
RAPPE, C. & KJELLER, L.-O. (1987) PCDDs and PCDFs in environ-mental
samples air, particulates, sediments and soil. Chemosphere, 16:
1775-1780.
RAPPE, C. & NYGREN, M. (1984) Chemical analysis of human samples.
Identification and quantification of polychlorinated dioxins and
dibenzofurans. In: de Serres, F.J. & Pero, R.W., ed. Individual
susceptibility to genotoxic agents in the human population, New
York, London, Plenum Press, pp. 305-314.
RAPPE, C., BUSER, H.R., & BOSHARDT, H.P. (1978) Identification and
quantification of polychlorinated dibenzo-p-dioxins (PCDDs) and
dibenzofurans (PCDFs) in 2,4,5-T-ester formulations and herbicide
orange. Chemosphere, 7: 431-438.
RAPPE, C., BUSER, H.R., KUROKI, H., & MASUDA, Y. (1979) Identification
of polychlorinated dibenzofurans (PCDFs) retained in patients with
Yusho. Chemosphere, 4: 259-266.
RAPPE, C., BUSER, H.R., STALLING, D.L., SMITH, L.M., & DOUGHERTY, R.C.
(1981) Identification of polychlorinated dibenzofurans in
environmental samples. Nature (Lond.), 292: 524-526.
RAPPE, C., MARKLUND, S., BERGQVIST, P.-A., & HANSSON, M. (1983a)
Polychlorinated dibenzo-p-dioxins, dibenzofurans and other polynuclear
aromatics formed during incineration and polychlorinated biphenyl
fires. In: Choudhary, G., Keith, L.H., & Rappe, C., ed. Chlorinated
dioxins and dibenzofurans in the total environment, Boston,
Butterworth Publishers, pp. 99-124.
RAPPE, C., MARKLUND, S., NYGREN, M., & GARA, A. (1983b) Parameters for
identification and confirmation in trace analyses of polychlorinated
dibenzo-p-dioxins and dibenzo-furans. In: Choudhary, G., Keith, L.H.,
& Rappe, C., ed., Chlorinated dioxins and dibenzofurans in the total
environment, Boston, Butterworth Publishers, pp. 240-259.
RAPPE, C., NYGREN, M., BUSER, H.R., MASUDA, Y., KUROKI, H., & CHEN,
P.H. (1983c) Identification of polychlorinated dioxins (PCDDs) and
dibenzofurans (PCDFs) in human samples, occupational exposure and
Yusho patients. In: Tucker, R.E., Young, A.L., & Gray, A.P., eds.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 241-254.
RAPPE, C., BERGQVIST, P.-A., & MARKLUND, S. (1985a) Analysis of
polychlorinated dibenzofurans and dioxins in ecological samples. In:
Keith, L.H., Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment II, Boston, Butterworth
Publishers, pp. 135-138.
RAPPE, C., MARKLUND, S., KJELLER, L-O., BERGQVIST, P.-A., & HANSSON,
M. (1985b) Composition of polychlorinated dibenzofurans (PCDF) formed
in PCB fires. In: Keith, L.H., Rappe, C., & Choudhary, G., ed.
Chlorinated dioxins and dibenzofurans in the total environment II,
Boston, Butterworth Publishers, p. 401-424.
RAPPE, C., NYGREN, M., MARKLUND, S., KJELLER, L.-O., BERGQVIST, P.A.,
& HANSON, M. (1985c) Assessment of human exposure to polychlorinated
dibenzofurans and dioxins. Environ. Health Perspect., 60: 303-304.
RAPPE, C., KJELLER, L.-O., & MARKLUND, S. (1985d) PCDF isomers and
isomer levels found in PCBs. In: Komai, R.Y. & Addis, G., ed.
Proceedings of a Workshop on PCB By-Product Formation, Palo Alto,
California, 4-6 December, 1984, Palo Alto, California, Electric
Power Research Institute, pp. 20-23.
RAPPE, C., KJELLER, L.-O., MARKLUND, S., & NYGREN, M. (1986a)
Electrical PCB accident, an update. Chemosphere, 15: 1291-1295.
RAPPE, C., NYGREN, M., HANSSON, M., & KAHN, P.C. (1986b) Analysis of
adipose tissue and blood samples from Vietnam veterans. Clean-up,
analysis and quality control, P 41, In: Dioxin 86, 6th International
Symposium on Chlorinated Dioxins and Related Compounds, Fukuoka,
Japan, 16-19 September, 1986.
RAPPE, C., ANDERSSON, R., BERGQVIST, P.-A., BROHEDE, C., HANSSON, M.,
KJELLER, L.-O., LINDSTROM, G., MARKLUND, S., NYGREN, M., SWANSON,
S.E., TYSKLIND, M., & WIBERG, K. (1987) Overview on environmental fate
of chlorinated dioxins and dibenzofurans, sources, levels and isomeric
pattern in various matrices. Chemosphere, 16: 1603-1618.
RAPPE, C., NYGREN, M., LINDSTROM, G., BUSER, H.R., BLASER, O., &
WUTHRICH, C. (1987) Polychlorinated dibenzofurans and
dibenzo-p-dioxins and other chlorinated contaminants in cow milk from
various locations in Switzerland. Environ. Sci, Technol., 21:
964-970.
REGGIANI, G. (1978) Medical problems raised by the TCDD contamination
in Seveso, Italy. Arch. Toxicol., 40: 161-188.
REGGIANI, G. (1980a) Acute human exposure to TCDD in Seveso, Italy.
J. Toxicol. environ. Health, 6: 27-43.
REGGIANI, G. (1980b) Toxicology of TCDD and related compounds:
observation in man. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds. Impact
on the environment, Oxford, New York, Pergamon Press, pp. 463-493.
REGGIANI, G. (1983a) Anatomy of TCDD spill: The Seveso accident.
Curr. Dev., 2: 269-341.
REGGIANI, G. (1983b) An overview on the health effects of halogenated
dioxins and related compounds - the Yusho and Taiwan episodes. In:
Coulston, F. & Pocchiari, F., ed. Accidental exposure to dioxins.
Human health aspects, New York, London, Academic Press, pp. 39-67.
REGIONE LOMBARDIA (1984) Final report and recommendations of the 6th
International Steering Committee Meeting, Milan, 19-21 February,
1984, Milan, Regione Lombardia, pp. 1-17.
RICE, R.H. & CLINE, P.R. (1984) Opposing effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin and hydrocortisone on growth and
differentiation of cultured malignant human keratinocytes.
Carcinogenesis, 5(3): 367-371.
RICE, R.H., CLINE, P.R., & COE, E.L. (1983) Mutually antagonistic
effects of hydrocortisone and retinylacetate on envelope competence in
cultured malignant human keratinocytes. J. invest. Dermatol., 81:
176s-178s.
RICHERT, J., VON (1962) [On neurological complications in a case of
chlorinated hydrocarbon poisoning accompanied by chloracne.]
Nervenarzt, 33(4): 180-184 (in German).
RIKANS, L.E., GIBSON, D.D., & MCCAY, P.B. (1979) Evidence for the
presence of cytochrome P-450 in rat mammary gland. Biochem.
Pharmacol., 28: 3039-3042.
RISSE-SUNDERMAN, A. (1959) [Intoxication by chloro-aromatics,]
Cologne-Lindenburg, University of Cologne, Department of Dermatology
(Thesis) (in German).
RIZZARDINI, M., ROMANO, M., TURSI, F., SALMONA, F., VECCHI, A.,
SIRONI, M., GIZZI, F., BENFENATI, E., GARATTINI, S., & FANELLI, R.
(1983) Toxicological evaluation of urban waste incinerator emissions.
Chemosphere, 12: 559-564.
ROBERTSON, L.W., REGEL, U., FILSER, J.G., & OESCH, F. (1985) Absence
of lipid peroxidation as determined by ethane exha-lation in rats
treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Arch.
Toxicol., 57: 13-16.
ROGERS, A.M., ANDERSEN, M.E., & BACK, K.C. (1982) Mutagenicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin and perfluoro-n-decanoic acid in
L5178Y mouse-lymphoma cells. Mutat. Res., 105: 445-449.
ROMKES, K., PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1987) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on hepatic and uterine estrogen
receptor levels in rats. Toxicol. appl. Pharmacol., 87: 306-314.
ROSE, J.Q., RAMSEY, J.C., WENTZLER, T.H., HUMMEL, R.A., & GEHRING,
P.J. (1976) The fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin following
single and repeated oral doses to the rat. Toxicol. appl.
Pharmacol., 36: 209-226.
ROZMAN, K. (1984) Hexadecane increases the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): Is brown adipose tissue
the primary target in TCDD-induced wasting syndrome? Biochem. biophys.
Res. Commun., 125(3): 996-1004.
ROZMAN, K., ROZMAN, T., & GREIM, H. (1984) Effect of thyroidectomy and
thyroxine on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induced
toxicity. Toxicol. appl. Pharmacol., 72: 372-376.
ROZMAN, K., ROZMAN, T., SCHEUFLER, E., PAZDERNIK, T., & GREIM, H.
(1985a) Thyroid hormones modulate the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. Toxicol. environ.
Health, 16: 481-491.
ROZMAN, K., HAZELTON, G.A., KLAASSEN, C.D., ARLOTTO, M.P., &
PARKINSON, A. (1985b) Effect of thyroid hormones on liver microsomal
enzyme induction in rats exposed to
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Toxicology, 37: 51-63.
ROZMAN, K., PEREIRA, D., & IATROPOULOS, M.J. (1986) Histo-pathology of
interscapular brown adipose tissue, thyroid, and pancreas in
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicol.
appl. Pharmacol., 82: 551-559.
RYAN, J.J., LAU, P.-Y., PILON, J.C., & LEWIS, D. (1983a)
2,3,7,8-Tetrachlorodibenzo-p-dioxin and
2,3,7,8-tetrachloro-dibenzofuran residues in great lakes commercial
and sport fish. In: Choudhary, G., Keith, L.H., & Rappe, C., ed.
Chlorinated dioxins and dibenzofurans in the total environment,
Boston, Buttersworth Publishers, pp. 87-97.
RYAN, J.J., PILON, J.C., CONACHER, H.B.S., & FIRESTONE, D. (1983b)
Inter-laboratory study on determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin in fish. J. Assoc. Off. Anal.
Chem., 66: 700-707.
RYAN, J.J., LIZOTTE, R., SAKUMA, T., & MORI, B. (1985) Chlorinated
dibenzo-p-dioxins, chlorinated dibenzofurans and pentachlorophenol in
Canadian chicken and pork samples. J. agric. food Chem., 33:
1021-1026.
RYAN, J.J., SCHECTER, A., SUN, W.-F., & LIZOTTE, R. (1986)
Distribution of chlorinated dibenzo-p-dioxins and chlorinated
dibenzofurans in human tissues from the general population. In: Rappe,
C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 3-16.
RYHAGE, R. (1964) Use of mass spectrometer as a detector and analyzer
for effluents emerging from high temperature gas liquid chromatography
columns. Anal. Chem., 36: 759-764.
SACCHI, G.A., VIGANO, P., FORTUNATI, G., & COCUCCI, S.M. (1986)
Accumulation of 2,3,7,8-tetrachlorodibenzo-p-dioxin from soil and
nutriculture solution by bean and maize plants. Experientia (Basel),
42: 586-588.
SAFE, S.H. & SAFE, L.M (1984) Synthesis and characterization of
twenty-two purified polychlorinated dibenzofuran congeners. J.
agric. food Chem., 32: 68-71.
SAFE, S.H., MASON, G., KEYS, B., FARRELL, K., ZMUDZKA, B., SAWYER, T.,
PISKORSKA-PLISZEZYNSKA, J., SAFE, L.M., ROMKES, M., & BANDIERA, S.
(1986) Polychlorinated dibenzo-p-dioxins and dibenzofurans:
correlation between in vitro and in vivo structure-activity
relationships (SARs). Chemosphere, 15: 1725-173.
SAFE, S.H., MASON, G., FARRELL, K., KEYS, B., PISKORSKA-PLISZCZYNSKA,
J., MADGE, J.A., & CHITTIM, B. (1987) Validation of in vitro bioassays
for 2,3,7,8-TCDD equivalents. Chemosphere, 16: 1723-1728.
SAI (SYSTEMS APPLICATIONS, INC.) (1980) Human exposure to
atmospheric concentration of selected chemicals, Springfield,
Virginia, National Technical Information Service, Vol. 1 (Report
prepared for US Environmental Protection Agency, Research Triangle
Park, North Carolina) (PB 81-193252).
SANDERMANN, W. (1984a) [Dioxin. The history of the discovery of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin, Seveso poison.]
Naturwiss. Rundsch., 37(5): 173-178 (in German).
SANDERMANN, W. (1984b) [Breakdown of dioxin by beta-radiation. The
history of the discovery of 2,3,7,8-tetrachlorodibenzo-p-dioxin
(Second communication).] Naturwiss. Rundsch., 37(11): 445-446 (in
German).
SANDERMANN, W., STOCKMANN, H., & GASTEN, R. (1957) [On the pyrolysis
of pentachlorphenol.] Chem. Ber., 90: 690-692 (in German).
SANGER, V.L., SCOTT, L., HAMDY, A., GALE, C., & POUNDEN, W.D. (1958)
Alimentary toxemia in chickens. J. Am. Vet. Med. Assoc., 133:
172-176.
SARNA, L.P., HODGE, P.E., & WEBSTER, G.R.B. (1984) Octanol-water
partition coefficients of chlorinated dioxins and dibenzofurans by
reversed-phase HPLC using several C18 columns. Chemosphere, 13:
975-983.
SAWAHATA, T., OLSON, J.R., & NEAL, R.A. (1982) Identification of
metabolites of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) formed on
incubation with isolated rat hepatocytes. Biochem. biophys. Res.
Commun., 105: 341-346.
SAWYER, T.W. & SAFE, S. (1985) In vitro AHH induction by
polychlorinated biphenyl and dibenzofuran mixtures: Additive effects.
Chemosphere, 14: 79-84.
SAWYER, T.W., JONES, D., ROSANOFF, K., MASON, G.,
PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1986) The biologic and toxic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in chickens.
Toxicology, 39: 197-206.
SCHANTZ, S.L., BARSOTTI, D.A., & ALLEN, J.R. (1978) Toxicological
effects produced in nonhuman primates chronically exposed to fifty
parts per trillion 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
Toxicol. appl. Pharmacol., 48(1): A180.
SCHECTER, A., WEERASINGHE, W.C.A., GROSS, M., & DEKIN, A. (1986a)
Human tissue levels of PCDDs and PCDFs in over one hundred year old
frozen Eskimo tissue, meat, and California redwood charcoal and their
relation to the trace chemistry theory of dioxin origin. In: Dioxin
86. 6th International Symposium on Chlorinated Dioxins and Related
Compounds, Fukuoka, Japan, 16-19 September, 1986, p.135.
SCHECTER, A.J., RYAN, J.J., & CONSTABLE, J.D. (1986b) Chlorinated
dibenzo-p-dioxin and dibenzofuran levels in human adipose tissue and
milk samples from the north and south of Vietnam. Chemosphere, 15:
1613-1620.
SCHILLER, C.M., WALDEN, R., & SHOAF, C.R. (1982) Studies on the
mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity: Nutrient
assimilation. Fed. Proc., 41: 1426 (Abstract).
SCHILLER, C.M., ADCOCK, C.M., MOORE, R.A., & WALDEN, R. (1985) Effect
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and fasting on body
weight and lipid parameters in rats. Toxicol. appl. Pharmacol.,
81: 356-361.
SCHILLER, C.M., ADCOCK, C.M., SGIAF, C.R., & WALDEN, R. (1986) Effects
of adenine and its isomer 4-aminopyrazolo-(3,4-d)-pyrimidine on
2,3,7,8-tetrachlorodibenzo-p-dioxin-induced mortality in rats.
Toxicol. appl. Pharmacol., 84: 369-378.
SCHOENY, R. (1982) Mutagenicity testing of chlorinated biphenyls and
chlorinated dibenzofurans. Mutat. Res., 101: 45-56.
SCHULZ, K.H. (1968) [On the symptoms and etiology of chloracne.]
Arbeitsmed. Sozialmed. Arbeitshyg., 3: 25-29 (in German).
SCHWETZ, B.A., NORRIS, J.M., SPARSCHU, G.L., ROWE, V.K., GEHRING,
P.J., EMERSON, J.L., & GEHRING, C.G. (1973) Toxicology of chlorinated
dibenzo-p-dioxins. Environ. Health Perspect., 5: 87-99.
SEEFELD, M.D. & PETERSON, R.E. (1983)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin-induced weight loss: A proposed
mechanism. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed. Human
and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 405-412
(Environmental Science Research Series).
SEEFELD, M.D. & PETERSON, R.E. (1984) Digestible energy and efficiency
of feed utilization in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol. 74:
214-222.
SEEFELD, M.D., CORBETT, S.W., KEESEY, R.E., & PETERSON, R.E. (1984a)
Characterization of the wasting syndrome in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
73: 311-322.
SEEFELD, M.D., KEESEY, R.E., & PETERSON, R.E. (1984b) Body weight
regulation in rats treated with 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Toxicol. appl. Pharmacol., 76: 526-536.
SEILER, J.P. (1973) A survey on the mutagenicity of various
pesticides. Experientia (Basel), 29: 622-623.
SHADOFF, L.A., HUMMEL, R.A., & LAMPARSKI, L. (1977) A research for
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in an environment exposed
annually to 2,4,5-trichlorophenoxyacetic acid ester (2,3,5-T)
herbicides. Bull. environ. Contam. Toxicol., 18: 478-485.
SHARMA, R.P. & GEHRING, P.J. (1979) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) on splenic lymphocyte
transform-ation in mice after single and repeated exposures. Ann.
N.Y. Acad. Sci., 320: 487-497.
SHARMA, R.P., KOCIBA, R.J., & GEHRING, P.J. (1984) Immuno-toxicologic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rabbits. J.
environ. Pathol. Toxicol. Oncogen., 5(4/5): 321-328.
SHEFFIELD, A. (1985) Sources and releases of PCDDs and PCDFs to the
Canadian environment. Chemosphere, 14: 811-814.
SHIGEMATSU, N., ISHIMARU, S., SAITO, R., IDEDA, T., MATSUBA, K.,
SUGIYAMA, K., & MASUDA, Y. (1978) Respiratory involvement in
polychlorinated biphenyls poisoning. Environ. Res., 16: 92-100.
SHIREMAN, R.B. & WEI, CHENG-I. (1986) Uptake of
2,3,7,8-tetra-chlorodibenzo-p-dioxin from plasma lipoproteins by
cultured human fibroblasts. Chem.-biol. Interact., 58: 1-12.
SHIVERICK, K.T. & MUTHER, T.F. (1983)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) effects on hepatic
microsomal steroid metabolism and serum estradiol of pregnant rats.
Biochem. Pharmacol., 32: 991-995.
SHOAF, C.R. & SCHILLER, C.M. (1981) Studies on the mechanism of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity-lipid
assimilation. II. Pharmacologist, 23: 176 (Abstract).
SILKWORTH, J., MCMARTIN, D., DECAPRIO, A., REJ, R., O'KEEFE, P., &
KAMINSKY, L. (1982) Acute toxicity in guinea pigs and rabbits of soot
from a polychlorinated biphenyl-containing transformer fire.
Toxicol. appl. Pharmacol., 65: 425-439.
SIMPSON, C.F., PRITCHARD, W.R., & HARMS, R.H. (1959) An endotheliosis
in chickens and turkeys caused by an unidentified dietary factor. J.
Am. Vet. Med. Assoc., 134: 410-416.
SLONECKER, P.J., PYLE, J.R., & CANTRELL, J.S. (1983) Identifi-cation
of polychlorinated dibenzo-p-dioxin isomers by powder X-ray
diffraction with electron capture gas chromatography. Anal. Chem.,
55: 1543-1547.
SMITH, A.G., FRANCIS, J.E., KAY, S.J.E., & GREIG, J.B. (1981) Hepatic
toxicity and uroporphyrinogen decarboxylase activity following a
single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin to mice. Biochem.
Pharmacol., 30: 2825-2830.
SMITH, A.G., FRANCIS, J.E., & GREIG, J.B. (1985) Continued depression
of hepatic uroporphyrinogen decarboxylase activity caused by
hexachlorobenzene 2,3,7,8-tetrachlorodibenzo-p-dioxin despite
regeneration after partial hepatectomy. Biochem. Pharmacol., 34:
1817-1820.
SMITH, A.H., FISHER, D.O., GILES, H.J., & PEARCE, N. (1983) The New
Zealand soft tissue sarcoma case-control study: Interview findings
concerning phenoxyacetic acid exposure. Chemosphere, 12: 565-571.
SMITH, F.A., SCHWETZ, B.A., & NITSCHKE, K.D. (1976) Terato-genicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in CF-1 mice. Toxicol. appl.
Pharmacol., 38: 517-523.
SMITH, R.M., O'KEEFE, P.W., ALDOUS, K.M., HILKER, D.R., & O'BRIEN,
J.E. (1983) 2,3,7,8-Tetrachlorodibenzo-p-dioxin in sediment samples
from love canal storm sewers and creeks. Environ. Sci. Technol., 17:
6-10.
SODERKVIST, P., POELLINGER, L., & GUSTAFSSON, J.-A. (1986)
Carcinogen-binding proteins in the rat ventral prostate: Specific and
nonspecific high-affinity binging sites for benso(a)pyrene,
3-methylcholanthrene, and 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Cancer Res., 46: 651-657.
SOUTHERLAND, J.H., KUYKENDAL, W.B., LAMASON, W.H. II, & OBERACKER,
D.A. (1987) Assessment of combustion sources as emitters of
chlorinated dioxin compounds: A report on the result of tier 4 of the
national dioxin strategy. Chemosphere, 16: 2161-2168.
SPARSCHU, G.L., DUNN, F.L., & ROWE, V.K. (1971) Study of the
teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Food
Cosmet. Toxicol., 9: 405-412.
SPITSBERGEN, J.M., SCHAT, K.A., KLEEMAN, J.M., & PETERSON, R.E. (1986)
Interactions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with immune
responses of rainbow trout. Vet. Immunol. Immunopathol., 12:
263-280.
STALLING, D.L., SMITH, L.M., PETTY, J.D., HOGAN, J.W., JOHNSON, J.L.,
RAPPE, C., & BUSER, H.R. (1983) Residues of polychlorinated
dibenzo-p-dioxins and dibenzofurans in laurentian great lakes fish.
In: Tucker, R.E., Young, A.L., & Gray, A.P., ed. Human and
environmental riks of chlorinated dioxins and related compounds, New
York, London, Plenum Press, pp. 221-240.
STEHL, R.H. & LAMPARSKI, L.L. (1977) Combustion of several
2,4,5-trichloro-phenoxy compounds: formation of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Science, 197: 1008-1009.
STEHL, R.H., PAPENFUSS, R.R., BREDEWEG, R.A., & ROBERTS, R.W. (1973)
The stability of pentachlorophenol and chlorinated dioxins to
sunlight, heat, and combustion. Adv. Chem. Ser., 120: 119-125.
STEHR, P.A., STEIN, G., FALK, H., SAMPSON, E., SMITH, S.M., STEINBERG,
K., WEBB, K., AYRES, S., SCHRAMM, W., DONNELL, H.D., & GEDNEY, W.B.
(1986) A pilot epidemiologic study of possible health effects
associated with 2,3,7,8-tetrachloro-dibenzo-p-dioxin contaminations in
Missouri. Arch. environ. Health, 41: 16-21.
STEINBERG, K.K., MACNEIL, M.L., KARON, J.M., STEHR, P.A., NEESE, J.W.,
& NEEDHAM, L.L. (1985) Assessment of
2,3,7,8-tetrachlorodibenzo-p-dioxin exposure using a modified
d-glucaric acid assay. J. Toxicol. environ. Health, 16: 743-752.
STEINER, M., BORGES, H., FREEDMAN, L., & GRAY, S.J. (1968) Effects of
starvation on the tissue composition of the small intestine in the
rat. Am. J. Physiol., 215: 75-77.
STEWARD, A.R. & BYARD, J.L. (1981) Induction of benzo(a)pyrene
metabolism by 2,3,7,8-tetrachlorodibenzo-p-dioxin in primary cultures
of adult rat hepatocytes. Toxicol. appl. Pharmacol., 59: 603-616.
STOHS, S.J., HASSAN, M.Q., & MURRAY, W.J. (1983) Lipid peroxidation as
a possible cause of TCDD toxicity. Biochem. biophys. Res. Commun.,
111: 854-859.
SUN, T.-T., SHIH, C., & GREEN, H. (1979) Keratin cytoskeletons in
epithelial cells of internal organs. Proc. Natl Acad. Sci. (USA),
76: 2813-2817.
SUN, T.-T., EICHNER, R., NELSON, W.G., TSENG, S.C.G., WEISS, R.A.,
JARVINEN, M., & WOODCOCK-MITCHELL, J. (1983a) Keratin classes:
Molecular markers for different types of epithelial differentiation.
J. invest. Dermatol., 81: 109-115.
SUN, T.-T., EICHNER, R., NELSON, W.G., VIDRICH, A., &
WOODCOCK-MITCHELL, J. (1983b) Keratin expression during normal
epidermal differentiation. In: Seji, M. & Bernstein, I.A., ed.
Current problems in dermatology. Normal and abnormal epidermal
differentiation, New York, Karger, pp. 277-291.
SUNDSTROM, G., JENSEN, S., JANSSON, B., & ERNE, K. (1979) Chlorinated
phenoxyacetic acid derivatives and tetrachloro-dibenzo-p-dioxin in
foliage after application of 2,4,5-trichlorophenoxyacetic acid esters.
Arch. environ. Contam. Toxicol., 8: 441-448.
SUSKIND, R.R. & HERTZBERG, V.S. (1984) Human health effects of 2,4,5-T
and its toxic contaminants. J. Am. Med. Assoc., 251: 2372-2380.
SUSKIND, R.R., CLEVELAND, F., KEENAN, C., AKIN, R., DAVIS, A., &
KEHOE, R.A. (1953) Report on a clinical and environmental survey,
Nitro, West Virginia, Monsanto Chemical Co..
SUTER-HOFMANN, M. & SCHLATTER, Ch. (1985) Toxicity of particulate
emissions from a municipal incinerator: critique of the concept of
TCDD-equivalents. Chemosphere, 15: 1733-1743.
SWEENEY, G.D. & JONES, K.G. (1983) Studies of the mechanism of action
of hepatotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and
related compounds. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 415-422.
SWEENEY, G.D., JONES, K.G., COLE, F.M., BASFORD, D., & KRESTYNSKI, F.
(1979) Iron deficiency prevents liver toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Science, 204: 332-335.
SWIFT, L.L., GASIEWICZ, T.A., DUNN, G.D., SOULE, P.D., & NEAL, R.A.
(1981) Characterization of the hyperlipidemia in guinea pigs induced
by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
59: 489-499.
SWITZERLAND (1982) Environmental pollution due to dioxins and furans
from chemical rubbish incineration plants, Bern, Ministry of
Environment, Federal Swiss Government.
TAYLOR, M.L., TIERNAN, T.O., RAMALINGAM, B., WAGEL, D.J., GARRETT,
J.H., SOLCH, J.G., & FERGUSON, G.L. (1985) Synthesis, isolation, and
characterization of the tetrachlorinated dibenzo-p-dioxins and other
related compound. In: Keith, L., Rappe, C., & Choudhary, G., ed.
Chlorinated dioxins and dibenzofurans in the total environment. II,
Boston, Butterworth Publishers, pp. 17-35.
TELEGINA, K.A. & BIKBULATOVA, L.I. (1970) [Affection of the follicular
apparatus of the skin in workers occupied in production of butyl ether
of 2,3,4-trichlorphenoxyacetic acid.] Vestn. Dermat. Venerol
(Moscow), 44: 35-39 (in Russian).
TELEKY, L. (1913) [Chlorine and hydrochloric acid. German Empire.]
Wiener Arb. Geb. Soz. Med., 4: 55-56 (in German).
TENCHINI, M.L., CRIMAUDO, C., PACCHETTI, G., MOTTURA, A., AGOSTI, S.,
& DE CARLI, L. (1983) A comparative cytogenetic study on cases of
induced abortions in TCDD-exposed and non-exposed women. Environ.
Mutagen., 5: 73-85.
THIBODEAUX, L.J. (1983) Offsite transport of
2,3,7,8-tetra-chlorodibenzo-p-dioxin from a production disposal
facility. In: Choudhary, G., Keith, L.H., & Rappe, C., ed.
Chlorinated dioxins and dibenzofurans in the total environment,
Boston, Butterworth Publishers, pp. 75-85.
THIESS, A.M., FRENTZEL-BEYME, R., & LINK, R. (1982) Mortality study of
persons exposed to dioxin in a trichlorophenol-pro-cess accident that
occurred in the BASF AG on November 17, 1953. Am. J. ind. Med., 3:
179-189.
THIGPEN, J.E., FAITH, R.E., MCCONNELL, E.E., & MOORE, J.A. (1975)
Increases susceptibility to bacterial infection as a sequela of
exposure to an environmental contaminant
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Infect. Immun., 12:
1319-1324.
THOMAS, P.T., & HINSDILL, R.D. (1979) The effect of perinatal exposure
to tetrachlorodibenzo-p-dioxin on the immune response of young mice.
Drug chem. Toxicol., 2: 77-98
THUNBERG, T. & HAKANSSON, H. (1983) Vitamin A (retinol) status in the
Gunn rat: the effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch.
Toxicol., 53: 225-233.
THUNBERG, T., AHLBORG, U.G., & JOHNSSON, H. (1979) Vitamin A (retinol)
status in the rat after a single oral dose of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Toxicol., 42: 265-274.
THUNBERG, T., AHLBORG, U.G., HAKANSSON, H., KRANTZ, C., & MONIER, M.
(1980) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the hepatic
storage of retinol in rats with different dietary supplies of vitamin
A (retinol). Arch. Toxicol., 45: 273-285.
THUNBERG, T., AHLBORG, U.G., & WAHLSTROM, B. (1984) Comparison between
the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and six other
compounds on the vitamin A storage, the UDP-glucuronosyltransferase
and the aryl hydrocarbon hydroxylase activity in the rat liver.
Arch. Toxicol., 55: 16-19.
TIERNAN, T.O. (1983) Analytical chemistry of polychlorinated
dibenzo-p-dioxins and dibenzofurans: A review of the current status.
In: Choudhary, G., Keith, L., & Rappe, C., ed. Chlorinated dioxins
and dibenzofurans in the total environment, Boston, Butterworth
Publishers, pp. 211-237.
TOFILON, P.J. (1980) Depressed guinea pig testicular microsomal
cytochrome P-450 content by 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Life Sci., 27: 871-876.
TOFILON, P.J. & PIPER, W.N. (1982)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated depression of rat
testicular heme synthesis and microsomal cytochrome P-450. Biochem.
Pharmacol., 31: 3663-3666.
TOGNONI, G. & BONACCORSI, A. (1982) Epidemiological problems with
TCDD. Drug Metab. Rev., 13: 447-469.
TOSINE, H., SMILLIE, D., & REES, G.A.V. (1983) Comparative monitoring
and analytical methodology for 2,3,7,8-TCDD in fish. In: Tucker, R.E.,
Young, A.L., & Gray, A.P., ed. Human and environmental risks of
chlorinated dioxins and related compounds, New York, London, Plenum
Press, pp. 127-142.
TOTH, K., SOMFAI-RELLE, S., SUGAR, J., & BENCE, J. (1979)
Carcinogenicity testing of herbicide 2,4,5-trichlorophenoxy-ethanol
containing dioxin and of pure dioxin in Swiss mice. Nature (Lond.),
278: 548-549.
TSUKAMOTO, H., MAKISUMI, S., HIROSE, H., KOJIMA, T., FUKUMOTO, H.,
FUKOMOTO, K., KURATSUNE, M., NISHIZUMI, M., SHIBATA, M., NAGAI, J.,
YAE, Y., SAWADA, K., FURUKAWA, M., YOSHIMURA, H., TATSUMI, K., OGURI,
K., SHIMENO, H., KENO, K., KOBAYASHI, H., YANO, T., ITO, A., OKADA,
T., INAGAMI, K., KOGA, T., TOMITA, Y., KOGA, T., YAMADA, Y.,
MIYAGUCHI, M., SUGANO, M., HORI, K., TAKESHITA, K., MANAKO, K.,
NAKAMURA, Y., & SHIGEMORI, N. (1969) [The chemical studies on detecton
of toxic compounds in rice bran oils used by the patients of Yusho.]
Fukuoka Acta med., 6: 496-512 (in Japanese).
TSYRLOV, I.B., CHASOVNIKOVA, O.B., GRISHANOVA, A.YU., & LYAKHOVICH,
V.V. (1986) Reappraisal of the liver benzpyrene hydroxylase
synthesized de novo after treatment of rats with
2,3,7,8-tetrachlorodibenzo-p-dioxin and 3-methylcholanthrene. FEBS
Lett., 198: 225-228.
TUCKER, A.N., VORE, S.J., & LUSTER, M.I. (1986) Supppression of B cell
differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol.
Pharmacol., 29: 372-377.
TUKEY, R.H., HANNAH, R.R., NEGISHI, M., NEBERT, D.W., & EISEN, H.J.
(1982a) The Ah locus: Correlation of intranuclear appearance of
inducer-receptor complex with induction of cytochrome P1-450 mRNA.
Cell, 31: 275-284.
TUKEY, R.H., NEGISHI, M., & NEBERT, D.W. (1982b) Quantitation of
hepatic cytochrome P1-450 mRNA with the use of a cloned DNA probe.
Effects of various P-450 inducers in C57BL/6N and DBA/2N mice. Mol.
Pharmacol., 22: 779-786.
TULP, M. & HUTZINGER, O. (1978) Rat metabolism of polychlori-nated
dibenzo-p-dioxins. Chemosphere, 9: 761-768.
TUNG, T.T. (1973) Le cancer primaire du foie au Vietnam. Chirurgie,
99: 427-436.
TURNER, J.N. & COLLINS, D.N. (1983) Liver morphology in guinea pigs
administered either pyrolysis products of a polychlorinated biphenyl
trans former fluid or 2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol.
appl. Pharmacol., 67: 417-429.
TUTEJA, N., GONZALEZ, F.J., & NEBERT, D.W. (1985) Developmental and
tissue-specific differential regulation of the mouse dioxin-inducible
P1-450 and P3-450 genes. Dev. Biol., 112: 177-184.
UMBREIT, T.H., PATEL, D., & GALLO, M.A. (1985) Acute toxicity of TCDD
contaminated soil from an industrial site. Chemosphere, 14(6/7):
945-947.
UMBREIT, T.H., HESSE, E.J., & GALLO, M.A. (1986a) Bioavailability of
dioxin in soil from a 2,4,5-T manufacturing site. Science, 232:
497-499.
UMBREIT, T.H., HESSE, E.J., & GALLO, M.A. (1986b) Comparative toxicity
of TCDD contaminated soil from Times Beach, Missouri, and Newark, New
Jersey. Chemosphere, 15: 2121-2124.
UOTILA, P., PARKKI, M.G., & AITO, A. (1978) Quantitative and
qualitative changes in the metabolism of benzo(a)pyrene in rat tissues
after intragastric administration of TCDD. Toxicol. appl.
Pharmacol., 46: 671-683.
US EPA (1982) Environmental monitoring at Love Canal, Washington,
DC, US Environmental Protection Agency, Office of Research and
Development, Vol. I (EPA/600/4-82-030a).
US EPA (1985) Health assessment document for polychlorinated
dibenzo-p-dioxins, Washington, DC, US Environmental Protection
Agency, Office of Health and Environmental Assessment
(EPA/600/8-84/0146).
US EPA (1987) Interim procedures for estimating risk associated with
exposures to mixtures of chlorinated dibenzo-p-dioxins and
-dibenzofurans (CDDs and CDFs). Risk assessment forum, Washington,
DC, US Environmental Protection Agency, 48 pp (EPA/625/3-87/012).
VAHRENHOLT, F. (1977) [Seveso - An unparalleled catastrophe.]
Umwelt, 1: 59, 60, 62, 64 (in German).
VAN, D.D. (1984) Herbicides as a possible cause of liver cancer. In:
Westing, A.H., ed. Herbicides in war, the long-term ecological and
human consequences, London, Taylor and Francis, pp. 119-121.
VAN DEN BERG, M., OLIE, K., & HUTZINGER, O. (1983) Uptake and
selective retention in rats of orally administered chlorinated dioxins
and dibenzofurans from fly-ash and fly-ash extract. Chemosphere,
12(4/5): 537-544.
VAN DEN BERG, M., VAN GREEVENBROEK, M., & OLIE, K. (1986a)
Bioavailability of PCDDs and PCDFs on fly ash after semi-chronic oral
ingestion by the rat. Chemosphere, 15(4): 509-518.
VAN DEN BERG, M., DE VROOM, E., & OLIE, K. (1986b) Bioavailability of
PCDDs and PCDFs on fly ash after semi-chronic oral ingestion by guinea
pig and syrian golden hamster. Chemosphere, 15(4): 519-533.
VAN DEN BERG, M., VAN DER WIELEN, F.W.M., & OLIE, K. (1986) The
presence of PCDDs and PCDFs in human breast milk from the Netherlands.
Chemosphere, 15: 693-706.
VAN LOGTEN, M.J., GUPTA, B.N., MCCONNELL, E.E., & MOORE, J.A. (1980)
Role of the endocrine system in the action of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the thymus.
Toxicology, 15: 135-144.
VAN MILLER, J.P., MARLAR, R.J., & ALLEN, J.R. (1976) Tissue
distribution and excretion of tritiated tetrachlorodibenzo-p-dioxin in
non-human primates and rates. Food Cosmet. Toxicol., 14: 31-34.
VAN MILLER, J.P., LALICH, J.J., & ALLEN, J.R. (1977) Increased
incidence of neoplasms in rats exposed to low levels of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere, 61: 625-632.
VECCHI, A., MANTOVANI, A., SIRONI, M., LUINI, W., CAIRO, M., &
GARATTINI, S. (1980) Effect of acute exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin on humoral antibody production in
mice. Chem.-biol. Interact., 30: 337-342.
VECCHI, A., SIRONI, M., CANEGRATI, M.A., RECCHIA, M., & GARATTINI, S.
(1983) Immunosuppressive effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in strains of mice with different susceptibility to induction of aryl
hydrocarbon hydroxylase. Toxicol. appl. Pharmacol., 68: 434-441.
VEERKAMP, W., WEVER, J., & HUTZINGER, O. (1981) The metabolism of some
chlorinated dibenzofurans by rats. Chemosphere, 10: 397-403.
VILLENUEVE, E.C., JENNINGS, R.W., BURSE, V.M., & KIMBROUGH, R.D.
(1974) Evidence of chlorodibenzo-p-dioxin and chlorodibenzofuran in
hexachlorobenzene. J. agric. food Chem., 22: 916-917.
VINOPAL, J.H. & CASIDA, J.E. (1973) Metabolic stability of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in mammalian liver microsomal
systems and in living mice. Arch. environ. Contam. Toxicol., 1:
122-132.
VISWANATHAN, T.S. & KLEOPFER, R.D. (1986) The presence of
hexachloroxanthene at Missouri dioxin sites, In: Rappe, C., Choudhary,
G., & Keith, L., ed. Chlorinated dioxins and dibenzofurans in
perspective, Chelsea, Michigan, Lewis Publishers, pp. 201-210.
VOS, J.G. & BEEMS, R.B. (1971) Dermal toxicity studies of technical
polychlorinated biphenyls and fractions thereof in rabbits. Toxicol.
appl. Pharmacol., 19: 617-633.
VOS, J.G. & KOEMAN, J.H. (1970) Comparative toxicologic study with
polychlorinated biphenyls in chickens with special reference to
porphyria, edema formation, liver necrosis, and tissue residues.
Toxicol. appl. Pharmacol., 17: 656-668.
VOS, J.G. & MOORE, J.A. (1974) Suppression of cellular immunity in
rats and mice by maternal treatment with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Int. Arch. Allergy, 47:
777-794.
VOS, J.G., KOEMAN, J.H., VAN DER MAAS, H.L., TEN NOEVER DE BRAUW,
M.C., & DE VOS, R.H. (1970) Identification and toxicological
evaluation of chlorinated dibenzofuran and chlorinated napthalene in
two commercial polychlorinated biphenyls. Food Cosmet. Toxicol.,
8: 625-633.
VOS, J.G., MOORE, J.A., & ZINKL, J.G. (1973) Effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on the immune system of laboratory
animals. Environ. Health Perspect., 5: 149-162.
VOS, J.G., MOORE, J.A., & ZINKL, J.G. (1974) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57 B1/6 mice.
Toxicol. appl. Pharmacol., 29: 229-241.
VOS, J.G., KREEFTENBERG, J.G., ENGEL, H.W.B., MINDERHOUD, A., & VAN
NOORLE JANSEN, L.M. (1978a) Studies on
2,3,7,8-tetra-chlorodibenzo-p-dioxin-induced immune suppression and
decreased resistance to infection: Endotoxin hypersensitivity, serum
zinc concentrations and effect of thymosin treatment. Toxicology,
9: 75-86.
VOS, J.G., KREEFTENBERG, J.G., & KATER, L. (1978b) Immune suppression
by TCDD. In: Cattabeni, F., Cavallaro, A., & Galli, ed. Dioxin
(TCDD), New York, John Wiley & Sons, Halsted Press Division, pp.
163-175.
WAHLE, P. (1914) [On two cases of chloracne.] Inaugural dissertation
for the Doctorate in Medicine, Surgery and Obstetrics of the Medical
Faculty, University of Leipzig (in German).
WALDEN, R. & SCHILLER, C.M. (1985) Short communications. Comparative
toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in four
(sub)strains of adult male rats. Toxicol. appl. Pharmacol., 77:
490-495.
WARD, C.T. & MATSUMURA, F. (1978) Fate of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) in a model aquatic
environment. Arch. environ. Contam. Toxicol., 7: 349-357.
WAUER, (1918) [Occupational illness caused by chlorinated hydrocarbons
(perna disease).] Zentralbl. Gewerbehyg., 6: 100-101 (in German).
WEBER, H., & BIRNBAUM, L. S. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) and
2,3,7,8-tetrachlorodibenzofuran (TCDF) in pregnant C57Bl/6N mice:
Distribution to the embryo and excretion. Arch. Toxicol., 57:
159-162.
WEBER, H., POIGER, H., & SCHLATTER, Ch. (1982a) Acute oral toxicity of
TCDD-metabolites in male guinea pigs. Toxicol. Lett., 14: 117-122.
WEBER, H., POIGER, H., & SCHLATTER, Ch. (1982b) Fate of
2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites from dogs in rats.
Xenobiotica, 12: 353-357.
WEBER, H., LUZI, P., RESI, L., TANGANELLI, P., LOVATI, M.R., & POLI,
A. (1983) Natural history of TCDD-induced liver lesions in rats as
observed by transmission electron microscopy during a 32-week period
after a single intraperitoneal injection. J. Toxicol. environ.
Health, 12: 533-540.
WEBER, H., LAMB, J.C., HARRIS, M.W., & MOORE, J.A. (1984)
Teratogenicity of 2,3,7,8-tetrachlorodibenzofuran (TCDF) in mice.
Toxicol. Lett., 20: 183-188.
WEBER, H., HARRIS, M.W., HASEMAN, J.K., & BIRNBAUM, L.S. (1985)
Teratogenic potency of TCDD, TCDF and TCDD-TCDF combinations in
C57Bl/6N mice. Toxicol. Lett., 26: 159-167.
WEBSTER, G.R.B., FRIESEN, K.J., SARNA, L.P., & MUIR, D.C.G. (1985)
Environmental fate modelling of chlorodioxins: Determination of
physical constants. Chemosphere, 14: 609-622.
WEDEL, J., VON, HOLLA, W.A., & DENTON, J. (1943) Observations on the
toxic effects resulting from exposure to chlorinated naphtalene and
chlorinated phenyls with suggestions for prevention. Rubber Age,
53: 419-426.
WEISSBERG, J.B. & ZINKL, J.G. (1973) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin upon hemostasis and hematologic
function in the rat. Environ. Health Perspect., 5: 119-123.
WESTING, A.H., (1984) Herbicides in war: past and present. In:
Westing, A.H., ed. Herbicides in war, the long-term ecological and
human consquences, London, Taylor and Francis.
WHITE, K.L., Jr, LYSY, H.H., MCCAY, J.A., & ANDERSON, A.C. (1986)
Modulation of serum complement levels following exposure to
polychlorinated dibenzo-p-dioxins. Toxicol. appl. Pharmacol., 84:
209-219.
WHITLOCK, J.P. & GALEAZZI, D.R. (1984)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin receptors in wild type and
variant mouse hepatoma cells. J. biol. Chem., 259: 980-985.
WHO/EURO (1987) Dioxins and furans from municipal incinerators,
Copenhagen, WHO Regional Office for Europe (Environmental Health
Series 17).
WIKLUND, K. & HOLM, L.-E. (1986) Soft tissue sarcoma risk in Swedish
agricultural and forestry workers. J. Natl Cancer Inst., 76:
229-234.
WILLEY, J.C., SALADINO, A.J., OZANNE, C., LECHNER, J.F., & HARRIS,
C.C. (1984) Acute effects of 12-O-tetradecanoyl-phorbol-13-acetate,
teleocidin B, or 2,3,7,8-tetrachloro-dibenzo-p-dioxin on cultured
normal human bronchial epithelial cells. Carcinogenesis, 5:
209-215.
WILLIAMS, D.T., CUNNINGHAM, H.M., & BLANCHFIELD, B.J. (1972)
Distribution and excretion studies of octachlorodibenzo-p-dioxin in
the rat. Bull. Environ. Contam. Toxicol., 7: 57-62.
WIPF, H.K. & SCHMID, J. (1983) Seveso - An environmental assessment.
In: Tucker, R.E., Young, A.L., & Gray, R., ed. Human and
environmental risks of chlorinated dioxins and related compounds,
New York, London, Plenum Press, pp. 255-276.
WIPF, H.K., HOMBERGER, E., NEUNER, N., RANALDER, U.B., VETTER, W., &
VUILLEUMIER, J.P. (1982) TCDD-levels in soil and plant samples from
the Seveso area. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds. Impact
on the environment, Oxford, New York, Pergamon Press, pp. 115-127.
WOLF, G. (1980) Vitamin A. In: Alfin-Slater, R.B. & Kritchevsky, D.,
ed. Human nutrition, a comprehensive treatise. Part B. Nutrition and
the adult, New York, London, Plenum Press, Vol. 3, pp. 97-203.
WOODS, J.S. (1973) Studies of the effects of 2,3,7,8-tetra-
chlorodibenzo-p-dioxin on mammalian hepatic delta-amino- levulinic
acid synthetase. Environ. Health Perspect., 5: 221-225.
WROBLEWSKI, V.J. & OLSON, J.R. (1985) Hepatic metabolism of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat and guinea pig.
Toxicol. appl. Pharmacol., 81: 231-240.
WU, Y.C., LO, Y.C., KAO, H.Y., PAN, C.C., & LIN, R.Y. (1984)
Cell-mediated immunity in patients with polychlorinated bi-phenyl
poisoning. J. Formoson Med. Assoc., 83: 419-429.
YAMAGISHI, T., MIYAZAKI, T., AKIYAMA, K., MORITA, M., NAKAGAWA, J.,
HORII, S., & KANEKO, S. (1981) Polychlorinated dibenzo-p-dioxins and
dibenzofurans in commercial diphenyl ether herbicides, and in fresh
water fish collected from the application area. Chemosphere, 10:
1137-1144.
YANG, K.H., CROFT, W.A., & PETERSON, R.E. (1977) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on plasma disappearance and
biliary excretion of foreign compounds in rats. Toxicol. appl.
Pharmacol., 40: 485-496.
YANG, K.H., CHOI, E.J., & CHOE, S.Y. (1983a) Cytotoxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin on primary cultures of adult rat
hepatocytes. Arch. environ. Contam. Toxicol., 12: 183-188.
YANG, K.H., YOO, B.S., & CHOE, S.Y. (1983b) Effects of halogenated
dibenzo-p-dioxins on plasma disappearance and biliary excretion of
ouabain in rats. Toxicol. Lett., 15: 259-264.
YOCKIM, R.S., ISENSEE, A.R., & JONES, G.E. (1978) Distribution and
toxicity of TCDD and 2,4,5-T in an aquatic model ecosystem.
Chemosphere, 7: 215-220.
YOSHIHARA, S., NAGATA, K., YOSHIMURA, H., KUROKI, H., & MASUDA, Y.
(1981) Inductive effect on hepatic enzymes and acute toxicity of
individual polychlorinated dibenzofuran congeners in rats. Toxicol.
appl. Pharmacol., 59: 580-588.
YOSHIMURA, H., KAMIMURA, H., OGURI, K., HONDA, Y., & NAKANO, M. (1986)
Stimulating effect of activated charcoal beads on fecal excretion.
Chemosphere, 15(3): 219-227.
YOUNG, A.L. (1983) Long-term studies on the persistence and movement
of TCDD in a natural ecosystem. In: Tucker, R.E., Young, A.L., & Gray,
P., ed. Human and environmental risks of chlorinated dioxins and
related compounds, New York, London, Plenum Press, pp. 173-190.
YOUNG, A.L., KANG, H.K., & SHEPARD, B.M. (1983) Clorinated dioxins as
herbicide contaminants. Environ. Sci. Technol., 17: 530A-532A.
YOUNG, A.L., THALKEN, C.E., & WARD, W.E. (1975) Studies of the
ecological impact of repetitive aerial applications of herbicides on
the ecosystem of test area C-52A, Eglin Air Force Base, Florida,
Washington, DC, US Department of Defence, 127 pp. (Final Report
October 1975).
YOUNG, A.L., THALKEN, C.E., ARNOLD, E.L., CUPELLO, J.M., & COCKERHAM,
L.G. (1976) Fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in
the environment: Summary and decontamination recommendations, US Air
Force Academy, Colorado, Washington, DC, US Department of Defence
(Document 19 USAFA TR-76-8).
ZACK, J.A. & GAFFEY, W.R. (1983) A mortality study of workers employed
at the Monsanto Company Plant in Nitro, West Virginia. Environ. Sci.
Res., 26: 575-591.
ZACK, J.A. & SUSKIND, R.R. (1980) The mortality experience of workers
exposed to tetrachlorodibenzodioxin in a trichloro-phenol process
accident. J. occup. Med., 22: 11-14.
ZEDDA, S., CIRLA, A.M., & SALA, C. (1976) [Accidental
tetrachlorodibenzoparadioxin contamination. Considerations on the
I.C.M.E.S.A. episode.] Med. Lav., 67: 371-378 (in Italian).
ZELIKOV, A.K. & DANILOV, L.N. (1974) Occupational dermatoses (acnes)
in workers engaged in production of 2,4,5-trichloro-phenol. Sov.
Med., 7: 145-146.
ZINKL, J.G., VOS, J.G., MOORE, J.A., & GUPTA, B.N. (1973) Hematologic
and clinical chemistry effects of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
in laboratory animals. Environ. Health Perspect., 5: 111-118.
RESUME ET RECOMMANDATIONS
1. Résumé
1.1 Sources
Les dibenzo-para-dioxines polychlorées (PCDD) et les
dibenzofurannes polychlorés (PCDF) constituent deux séries
d'hydrocarbures aromatiques tricycliques halogénés de propriétés
physico-chimiques très voisines, présentes un peu partout dans
l'environnement. Leur origine n'est pas naturelle et ils ne sont pas
produits intentionnellement. On connait 75 isomères (isomérie de
position) pour les PCDD et 135 pour les PCDF.
Les sources les plus importantes de contamination par ces deux
séries de composés sont les suivantes:
- contamination de produits chimiques du commerce, par exemple
chlorophénols et dérivés, PCB;
- incinération de déchets municipaux, de déchets dangereux et
de déchets d'hôpitaux, ainsi que de boues provenant du
traitement des effluents;
- fonctionnement des véhicules automobiles;
- utilisation de combustibles fossiles;
- surchauffe et émissions de foyers faisant intervenir des
béniphyles polychlorés (PCB);
- élimination de déchets industriels provenant, par exemple,
de la production de chlorophénols et de leurs dérivés, du
traitement du bois par le chlorophénol, de l'emploi de PCB
liquides dans l'appareillage électrique et du traitement du
papier et de la pâte à papier.
1.2 Concentrations dans l'environnement et voies d'exposition
D'après les quelques données dont on dispose, la concentration de
ces composés serait très faible dans l'air, le sol et les sédiments,
de l'ordre du femtogramme (fg) par mètre cube dans l'air et du
nanogramme (ng) par kilogramme dans le sol et les sédiments. On a
observé des concentrations allant jusqu'à 50 mg/kg, aussi bien pour
les PCDD que pour les PCDF, dans certains organismes aquatiques à
l'état naturel. On dispose de très peu de données sur la contamination
de l'eau de boisson et des denrées alimentaires vendues dans le
commerce.
Pour ce qui est de la population en général, l'exposition à ces
composés semble principalement associée à la chaîne alimentaire.
Certains travailleurs peuvent être très exposés lorsqu'ils sont
affectés à la production, la mise en oeuvre ou à la destruction de
matériaux contenant des PCDD ou des PCDF, ainsi que leurs précurseurs.
Pour ce personnel, l'inhalation et les contacts cutanés constituent
les principales voies d'exposition à prendre en compte.
1.3 Toxicocinétique, biotransformation et surveillance
biologique
La biodisponibilité des PCDD et des PCDF dépend de la matrice au
sein de laquelle ils sont incorporés et de la voie d'exposition. On ne
dispose pour aucune des espèces en cause de données sur la
biodisponibilité par inhalation.
On ignore la quantité absorbée par l'homme en cas d'exposition,
par quelque voie que ce soit.
Les études sur des rongeurs à qui l'on a administré une ou
plusieurs doses de 2,3,7,8-TCDD par voie orale montrent qu'environ la
moitié de la dose administrée est absorbée dans les voies digestives.
La demi-vie d'élimination se situe, d'après la littérature, entre 12
et 94 jours chez les rongeurs. Chez le singe rhésus, la demi-vie de
la 2,3,7,8-TCDD est d'environ 1 an dans le tissu adipeux.
Les observations concernant la toxicocinétique des PCDD chez les
animaux sont limitées, en dehors du cas de la 2,3,7,8-TCDD. Pour ce
dérivé, on a observé une demi-vie de l'ordre de 2 à 8 jours chez le
rat, la souris et le singe et de plus de 20 jours chez le cobaye.
Chez le rat, on a constaté que la pentachlore-2,3,4,7,8 dibenzofuranne
s'élimine moins rapidement que la 2,3,7,8-TCDD.
Les études sur la rétention tissulaire des PCDD et des PCDF chez
diverses espèces exposées à des mélanges synthétiques ou à des
échantillons prélevés dans l'environnement et contenant des PCDD ou
des PCDF montrent que la durée de rétention des divers analogues est
très variable selon qu'ils sont ou non substitués en 2, 3, 7, ou 8 par
un atome de chlore.
D'après quelques observations effectuées sur l'homme, la demi-vie
serait de 2 à 6 ans pour certains PCDD et PCDF substitués en 2, 3, 7
et 8.
Les PCDD et les PCDF s'accumulent principalement dans les
graisses; ils sont également excrétés dans le lait et traversent la
barrière placentaire. On en trouve également à faible concentration,
dans le sang et les organes vitaux.
La distribution tissulaire chez l'homme est encore mal élucidée,
encore qu'il semble que le rapport des concentrations dans les tissus
adipeux et dans le foie soit plus élevé que chez les rongeurs.
Dans l'ensemble de la population, on observe une concentration
résiduelle de PCDD dans le tissu adipeux qui atteint 20 mg/kg, en
l'absence de toute exposition particulière connue; mais des chiffres
plus élevés ont été signalés quelquefois, sans aucun signe de maladie.
Dans les deux cas, les groupes de population étudiés n'avaient pas été
constitués par sondage aléatoire. Dans ces échantillons, on a observé
les PCDD et les PCDF les plus substitués par du chlore, en particulier
des octachlorodibenzo-p-dioxines. La concentration tissulaire des TCDD
avait tendance à augmenter avec l'âge.
1.4 Effets sur la santé
1.4.1 Animaux
Les effets toxiques et biologiques découlant de l'exposition aux
2,3,7,8-TCDD dépendent de divers facteurs, dont l'espèce, la souche,
l'âge et le sexe des animaux d'expérience. Entre autres signes de
toxicité observés chez plusieurs espèces animales, on note une perte
de poids, une hépatoxicité, une porphyrie, une atteinte cutanée, des
lésions gastriques, une atrophie du thymus, une atteinte du système
immunitaire, des effets tératogènes, des effets sur la reproduction et
des effets cancérogènes. Les TCDD exercent des effets biologiques
extrêmement divers, notamment l'induction de certaines enzymes et une
carence en vitamine A. Ces effets ne sont pas tous observés chez la
même espèce animale. Les effets toxiques les plus caractéristiques
qu'on retrouve chez tous les animaux de laboratoire sont la perte de
poids, l'atrophie du thymus et l'immunotoxicité. Chez l'homme, une
chloracné et des lésions cutanées de ce type constituent les signes
les plus fréquents d'une intoxication par les 2,3,7,8-TCDD; on observe
également des lésions cutanées chez le rhésus, la souris glabre et le
lapin. En revanche, l'exposition au 2,3,7,8-TCDD ne provoque ni
chloracné ni lésions cutanées de ce type chez la plupart des rongeurs.
De nombreuses lésions toxiques s'observent principalement au niveau
des tissus épithéliaux.
Des effets ont été observés sur la reproduction chez le rhésus et
chez le rat. La plus faible dose exerçant un effet serait de l'ordre
de 1-2 ng/kg de poids corporel par jour. Dans deux études de
cancérogénicité chez le rat, on a obtenu un épithélioma
hépatocellulaire avec des doses respectives d'environ 0,1 et 0,01
µg/kg de poids corporel par jour. Une dose réduite à 0,001 µg/kg de
poids corporel se traduit par une modification des hépatocytes au
niveau de certains foyers ou territoires. L'incidence de certaines
tumeurs à déterminisme hormonal était plus faible que chez les animaux
témoins.
Apparemment, les TCDD n'ont pas de propriétés mutagènes et ne
devraient donc pas être génotoxiques. Il faut donc admettre que les
effets cancérogènes s'expliquent par un mécanisme indirect.
Plusieurs autres PCDD et PCDF déterminent des manifestations
analogues à celles que provoquent les 2,3,7,8-TCDD, mais leur activité
est très inégale. On connaît 12 isomères qui présentent une toxicité
plus élevée, à savoir les tétra-, penta-, hexa-et heptaCDD ou CDF qui
possèdent quatre atomes de chlore symétriques deux à deux, en
poisition 2,3,7 et 8. Des études prolongées sur l'animal ont révélé
l'action cancérogène d'un mélange de deux
hexachlorodibenzo-p-dioxines (1,2,3,7,8,9-et 1,2,3,6,7,8-hexaCDD),
mais en présence de doses plus élevées que celles qu'on avait
utilisées dans l'étude des TCDD. La dibenzo-p-dioxine et la
dichloro-2,7 dibenzo-p-dioxine ne se sont pas révélées cancérogènes.
Les activités relatives, toxiques et biologiques, des PCDD et des PCDF
ont été estimées au moyen d'études de courte durée chez le rat et en
culture de cellules mammaliennes.
Selon les espèces, les animaux présentent une sensibilité très
inégale aux effets biologiques et toxiques des PCDD et PCDF substitués
en 2,3,7,8. Par exemple, la DL50 par voie orale varie, pour les
2,3,7,8-TCDD, de 0,6 µg/kg de poids corporel chez le cobaye à 5051
µg/kg chez le hamster doré syrien. Les différences considérables de
sensibilité vis-à-vis des 2,3,7,8-TCDD et des composés apparentés
qu'on observe ainsi chez les diverses espèces et souches est
impossible à expliquer par les différences qu'on constate dans la
toxicocinétique. La toxicité et la toxicocinétique des TCDD chez les
singes sont à peu près les mêmes que chez l'homme. Chez des souris de
lignée pure, on a des raisons de penser que la concentration
cellulaire du récepteur aux Ah est partiellement corrélée avec la
sensibilité à l'action biologique et toxique de ces composés. Le
récepteur a été retrouvé chez d'autres espèces, notamment chez
l'homme. Cependant, l'abondance relative du récepteur aux Ah selon les
espèces n'explique pas entièrement les différences de sensibilité.
1.4.2 Homme
Malgré de nombreuses études cliniques et études de suivi
post-thérapeutiques, aucun effet général persistant bien net n'a été
mis en évidence, si ce n'est une chloracné, à la suite d'expositions
professionnelles ou accidentelles aux PCDD et aux PCDF. D'autres
effets ont été notés mais aucun n'est persistant, en dehors de la
chloracné et, peut-être, de troubles fonctionnels minimes.
Dans quelques études épidémiologiques consacrées à des cas
d'exposition à un mélange de dioxines, de furannes et d'autres
produits chimiques, on a fait état d'une augmentation de l'incidence
des cancers de localisations diverses, mais plusieurs facteurs ne
permettent pas de faire entièrement foi à ces observations.
Lors de l'accident de Seveso, le seul effet nocif indiscutable a
été la chloracné. Cette affection (193 cas) s'est déclarée en 1976 et
1977 et elle était encore active en 1984 chez 20 des victimes. De
nombreuses études ont été consacrées à la recherche de liens possibles
entre l'exposition à l'Agent orange et des effets nocifs chez les
civils ou les militaires, au Viet Nam. Cependant, les données dont on
dispose à l'heure actuelle ne permettent pas de conclure
catégoriquement à l'existence d'effets sur la reproduction ou d'autres
effets biologiques notables.
Lors de l'incident survenu au Missouri, les enfants chez qui l'on
avait observé des signes aigus d'intoxication lors de la
contamination, en 1971, sont, semble-t-il, aujourd'hui en bonne santé.
En outre, des études épidémiologiques poursuivies dans ce même Etat au
sein de populations plus longuement exposées à des concentrations plus
faibles de dioxine n'ont jusqu'ici révélé aucun effet appréciable sur
la santé. Mais si aucun symptôme clinique n'a été observé, on a
constaté un effet sur l'immunité à médiation cellulaire.
Les seuls cas d'intoxication humaine par les PCDF pour lesquels
on dispose d'observations circonstanciées concernent la contamination
d'huile de riz par des PCDF, des PCB et des PCQ, observée à Yusho au
Japon, en 1968, et Yu-cheng à Taiwan, en 1979. Au total, plusieurs
milliers de personnes ont été gravement intoxiquées. Il semble très
probable que les PCDF sont à incriminer dans ces intoxications. La
symptomatologie générale était analogue à celle qui accompagne les
intoxi-cations par les TCDD, compte tenu des différences traduisant
l'intensité de l'exposition et la structure d'âge et de sexe des
groupes exposés.
A Yusho, l'apport quotidien moyen de PCDF substitués en 2,3,7,8
a été estimé à 0,1-0,2 µg/kg de poids corporel sur plusieurs mois,
alors que la dose pathogène minimale a été estimée à 0,05-0,1 µg/kg de
poids corporel par jour, sur 30 jours.
1.5 Conclusion
Les PCDD et les PCDF se rencontrent dans tout l'environnement et
il est probable que chacun en est porteur. Ces composés déterminent
parfois des effets toxiques complexes en cas d'exposition
professionnelle ou accidentelle.
D'après les observations faites à Yusho, les expériences
conduites chez des espèces sensibles de singes, et compte tenu de
certaines hypothèses concernant l'activité relative des PCDD et des
PCDF, il se peut que l'homme et certains singes présentent une
sensibilité comparable à l'égard de ces composés. Cependant, les
incertitudes concernant la dose effectivement reçue et les difficultés
de l'évaluation des effets toxiques autres que la chloracné
interdisent, dans le cas de l'homme, une conclusion catégorique quant
à la résistance relative de l'homme aux effets toxiques de ces
composés. Il convient de limiter l'exposition aux plus faibles niveaux
compatibles avec les impératifs pratiques.
2. Recommandations
1. Des études interlaboratoires de validation, selon une sorte de
"poule" avec protocoles normalisés de contrôle et d'assurance de
qualité, sont indispensables pour améliorer les méthodes d'analyse.
La méthodologie de l'échantillonnage, les méthodes d'analyse et
l'interprétation des données doivent être optimalisées et normalisées
avant le démarrage des enquêtes.
2. Des données complémentaires sont nécessaires sur l'origine des
PCDD et des PCDF, sur leur répartition dans l'environnement et sur
leur destinée.
D'autres observations sont indispensables au sujet de la présence
des PCDD ou des PCDF dans l'environnement, notamment sur l'évolution
des concentrations dans le temps et sur la détermination des profils
isomériques, notamment dans les produits alimentaires, dans l'air
ambiant et dans les sédiments.
3. Il serait bon de faire des observations sur les effets
qu'exercent les PCDD et les PCDF sur la biocénose.
4. Des données plus complètes sont nécessaires en ce qui concerne la
biodisponibilité des PCDD et des PCDF provenant de diverses matrices,
dans l'environnement et dans la ration alimentaire. L'exposition à ces
sources devrait être mise en corrélation avec les pratiques agricoles
et industrielles.
5. Il convient de mettre au point et de valider des méthodes
chimiques et biologiques plus simples et moins coûteuses pour
rechercher la présence de PCDD et de PCDF.
6. Des études sur les mécanismes toxiques des PCDD et des PCDF sont
nécessaires pour élucider les différences observées dans les effets
produits chez les diverses espèces et permettre une extrapolation à
l'homme.
7. Il importe de poursuivre l'étude de l'immunotoxicité, notamment
en ce qui concerne la fonction des lymphocytes T cytotoxiques. En
particulier, il est capital d'étudier la nature et la durée des effets
sur le système immunitaire d'une exposition pendant la période
périnatale.
8. Des études de toxicité à long terme devraient être effectuées,
notamment des études de reproduction sur plusieurs générations chez
diverses espèces, au moyen de trois des PCDD et des PCDF les plus
répandus, à savoir le 2,3,4,7,8-pentaCDF, le 1,2,3,7,8-pentaCDD et
l'octaCDD.
9. Etant donné que l'homme est exposé à des mélanges complexes de
PCDD et de PCDF, il faut poursuivre la mise au point et la validation
de divers systèmes d'épreuve, notamment des cultures de cellules
humaines en vue d'apprécier l'activité toxique de ces composés et de
leurs mélanges. Ces systèmes permettront d'étudier les mécanismes
d'action, les relations structure/activité et les effets interactifs.
10. Il serait bon d'étudier la charge de l'organisme en PCDD et PCDF
et d'établir une corrélation entre cette charge, les effets cliniques
constatés et les résultats des examens de laboratoire. Le suivi de
groupes exposés antérieurement est important à cet égard.
EVALUATION DES DANGERS POUR LA SANTE DE L'HOMME
DE L'EXPOSITION AUX DIBENZO-P-DIOXINES POLYCHLORES (PCDD)
ET AUX DIBENZOFURANNES POLYCHLORES (PCDF)
1. Introduction
Pour évaluer les dangers que représentent les PCDD et les PCDF
pour la santé humaine, il faut connaître à la fois l'intensité de
l'exposition et les effets biologiques correspondants.
Il est fort difficile d'évaluer l'exposition humaine. Plusieurs
méthodes sont possibles, par exemple:
a) Utilisation de modèles physiologiques classiques de
l'inhalation, de l'ingestion et de l'absorption cutanée. Il
faut connaître de façon précise la concentration des
produits en cause dans les différents milieux et dans les
denrées alimentaires, ainsi que leur biodisponibilité.
b) Estimations de l'apport sur la base de modèles
pharmacocinétiques simples et des teneurs observées dans les
divers tissus humains en vue d'une évaluation précise de
l'exposition. Il faut en outre connaître en détail l'apport,
la distribution, le métabolisme et l'élimination des PCDD et
des PCDF chez l'homme.
2. Evaluation de l'exposition
2.1 Sources de contamination
Les principales sources de PCDD et de PCDF identifiées jusqu'ici
sont les contaminants contenus dans les produits chimiques vendus dans
le commerce (voir section 3.3), les émissions par suite de combustion
(voir section 3.5) et l'élimination de déchets industriels contenant
des PCDD ou des PCDF (sections 3.4, 3.5.9 et 3.5.10).
Dans certains cas, on peut estimer au niveau local la
contribution relative de ces diverses sources. Mais, les méthodes et
les données dont on dispose aujourd'hui ne permettent pas de
conclusions définitives quant à leur importance relative au niveau
d'un pays ou du monde entier.
2.2 Concentrations dans le milieu ambiant
Les données restreintes dont on dispose montrent que la
concentration atmosphérique résiduelle est très faible (fg/m3) pour
les 2,3,7,8-tétrachloro-PCDD ou PCDF ainsi que pour les dérivés
pentachlorés ou hexachlorés. (Si l'on tient compte des analogues
heptachlorés ou octachlorés, on observe des concentrations de l'ordre
du picogramme par mètre cube).
D'après les rares données disponibles, il est peu probable qu'on
trouve des tétrachloro-2,3,7,8-PCDD ou PCDF ni des dérivés
pentachlorés ou hexachlorés dans une eau de boisson traitée, même à la
concentration de 1 pg/litre (voir section 5.2). Dans tous les
échantillons de sols et de sédiments analysés (provenant de régions
industrialisées ou non), la concentration de PCDD et de PCDF allait de
quelques nanogrammes à plusieurs centaines de nanogrammes par
kilogramme (la concentration la plus élevée concernant les sédiments
et le sol en zone urbaine).
On ne connaît pas la concentration résiduelle des PCDD et des
PCDF dans les végétaux en général.
Des concentrations atteignant 50 ng/kg ont été observées chez des
poissons capturés dans la nature pour les mêmes dérivés
(principalement les dérivés tétrachlorés et pentachlorés). Le plus
souvent, ces composés ont été observés dans des poissons gras ou des
poissons qui se nourrissent sur le fond (voir section 4.4.2).
Les observations concernant les organismes terrestres ne
permettent pas d'estimer les concentrations résiduelles (voir section
4.4.3). Dans trois échantillons d'un mélange de différents laits de
vache, on a trouvé, pour le lait entier, une concentration maximale de
100 pg/kg (voir section 5.4). La contamination des autres denrées
alimentaires vendues dans le commerce est également très mal connue.
L'analyse de plusieurs échantillons de poulet et de porc montre que
ces produits contiennent environ 5-30 ng/kg d'analogues fortement
chlorés. Le profil de ces analogues n'est pas le même que chez les
organismes aquatiques (voir section 5.4).
Les données disponibles ne permettent pas d'évaluer l'exposition
globale de la population dans son ensemble. Elles suffisent pour une
évaluation limitée de l'exposition dans le cas des populations
locales. Compte tenu des concentrations environnementales examinées
ci-dessus et des hypothèses habituelles sur l'apport d'aliments, d'air
et d'eau, les denrées alimentaires risquent davantage de contribuer
notablement à l'exposition aux PCDD/PCDF que l'air, tandis que l'eau
de boisson est sans doute beaucoup moins en cause.
2.3 Modalités d'exposition
Chez l'homme, le tissu adipeux contient des tétrachloro-2,3,7,8
PCDD/PCDF ainsi que des dérivés pentachlorés et hexachlorés. Cette
contamination s'explique sans doute par une exposition aux produits
présents dans l'environnement, aux concentrations indiquées plus haut.
En outre, les nourrissons peuvent être exposés par
l'intermédiaire du lait maternel et les jeunes enfants par l'ingestion
de terre contaminée. Mais, dans la plupart des cas, ce dernier mode
d'exposition ne devrait être pris en compte que dans les régions
fortement polluées.
Certaines populations sont particulièrement exposées à l'occasion
d'accidents survenus dans l'industrie (et des opérations de nettoyage
ultérieures) lors de la production et de l'utilisation normales de
chlorophénols, d'herbicides phénoxylés et de PCB. En pareil cas, ce
sont l'inhalation et le contact cutané qui constituent les modes
d'exposition les plus importants. Cependant, on ne dispose pas de
données quantitatives sur la nature et la concentration des
contaminants.
Sur la base des valeurs indiquées plus haut pour les
concentrations dans le milieu ambiant et des hypothèses habituelles
sur les paramètres physiologiques et les valeurs de l'apport, il est
probable que l'ingestion constitue le mode d'exposition essentiel.
L'inhalation de l'air ambiant ne devrait pas poser de problème encore
qu'un air fortement contaminé puisse contribuer sensiblement à
l'exposition. En général, il n'est pas possible actuellement d'estimer
l'importance relative de l'exposition par voie cutanée.
2.4 Biodisponibilité
On ne dispose d'aucune donnée sur la biodisponibilité provenant
d'études sur l'homme. L'expérimentation animale montre que la
biodisponibilité des PCDD ou des PCDF après ingestion dépend de la
matrice du produit ingéré. Les données disponibles au sujet de
l'apport par voie orale sont résumées au tableau 46.
Des études sur des rats glabres montrent que l'exposition cutanée
par contact de la peau intacte avec un sol contaminé représente
environ 1-2%. On ne dispose d'aucune donnée pour l'exposition par
inhalation.
3. Résultats de l'exposition animale
3.1 Toxicocinétique des 2,3,7,8-TCDD
Des études sur des rongeurs à qui l'on administrait du
2,3,7,8-TCDD par voie orale, en une ou plusieurs prises, ont montré
que l'absorption dans les voies digestives était d'au moins 50% chez
le rat, le cobaye et le hamster, mais de moins de 30% chez la souris
(tableau 40). Les valeurs indiquées pour la demi-vie d'élimination
étaient de l'ordre de 12-31 jours pour le rat, la souris et le hamster
et de 22-94 jours pour le cobaye (tableau 41). Cependant, la plupart
de ces études ont été effectuées en utilisant des doses toxiques. La
demi-vie des 2,3,7,8-TCDD chez les primates est mal connue mais il
semble, d'après les données disponibles pour le rhésus, que la
demi-vie apparente soit de l'ordre de 1 an dans les tissus adipeux.
Les 2,3,7,8-TCDD ne s'accumulent pas dans les tissus animaux.
Chez les rongeurs, l'accumulation intervient principalement au niveau
du foie et du tissu adipeux (tableaux 42 à 44). Chez des rhésus
(tableau 45), on a observé une concentration élevée de ces mêmes
produits dans le tissu adipeux, le foie, la peau et les muscles.
Chez des rats ayant reçu quotidiennement pendant 2 ans des
2,3,7,8-TCDD à raison de 1 ng/kg de poids corporel, on a observé une
accumulation de 540 ng/kg de poids corporel dans le foie où l'on a
également noté certaines altérations morphologiques. Des quantités du
même ordre ont été notées chez le péromysque après exposition à un sol
contenant des 2,3,7,8-TCDD à la concentration de 10-710 ng/kg
(Cockherham et al., 1980). L'exposition des animaux, au niveau de
l'ensemble de leur corps, à un sol présentant de telles teneurs peut
donc se traduire par une concentration tissulaire qui a des effets
observables chez les animaux d'expérience.
Les TCDD sont en grande partie éliminés dans les déjections,
encore qu'il existe une certaine excrétion urinaire. Elle est plus
importante chez le hamster que chez les autres espèces étudiées.
La transformation des 2,3,7,8-TCDD en métabolites à caractère
polaire plus prononcé s'observe chez toutes les espèces animales
étudiées (voir section 6.2 et tableau 62). L'élimination fécale et
urinaire des métabolites est rapide chez toutes ces espèces, sauf chez
le cobaye. Les métabolites connus sont beaucoup moins toxiques que les
composés dont ils dérivent.
3.2 Toxicocinétique des PCDD et des PCDF autres que les TCDD
En dehors du cas des dérivés tétrachlorés, les observations sur
la toxicocinétique des PCDD chez les animaux sont limitées. De ce
point de vue, les PCDF ont été davantage étudiés. La demi-vie des
2,3,7,8-TCDF serait de l'ordre de 2 à 8 jours chez le rat, la souris
et le rhésus et de plus de 20 jours chez le cobaye (tableau 65). Les
études sur le rat montrent que les 2,3,7,8-pentaCDF se fixent
davantage dans les tissus que les 2,3,7,8-TCDF (65% et 3,8%
respectivement, au bout de 5 jours).
La rétention tissulaire des PCDD et des PCDF chez diverses
espèces exposées à des mélanges de synthèse ou à des échantillons
prélevés dans l'environnement et contenant des PCDD et des PCDF se
révèle très inégale selon que les produits sont ou non substitués par
des atomes de chlore dans toutes les positions 2,3,7 et 8.
3.3 Effets toxiques des 2,3,7,8-TCDD
Les effets toxiques et biologiques découlant de l'exposition aux
2,3,7,8-TCDD dépendent de divers facteurs, dont l'espèce, la souche,
l'âge et le sexe des animaux d'expérience. Entre autres signes de
toxicité observés chez plusieurs espèces animales, on note une perte
de poids, une hépatoxicité, une porphyrie, une atteinte cutanée, des
lésions gastriques, une atrophie du thymus, une atteinte du système
immunitaire, des effets tératogènes, des effets sur la reproduction et
des effets cancérogènes. Les TCDD exercent des effets biologiques
extrêmement divers, notamment l'induction de certaines enzymes et une
carence en vitamine A. Ces effets ne sont pas tous observés chez la
même espèce animale. Les effets toxiques les plus caractéristiques
qu'on retrouve chez tous les animaux de laboratoire sont la perte de
poids, l'atrophie du thymus et l'immunotoxicité. Chez l'homme, une
chloracné et des lésions cutanées apparentées constituent les signes
les plus fréquents d'une intoxication par les 2,3,7,8-TCDD; on observe
également des lésions cutanées chez le rhésus, la souris glabre et le
lapin. En revanche, l'exposition au 2,3,7,8-TCDD ne provoque ni
chloracné ni lésions cutanées de ce type chez le rat, la plupart des
souris, le cobaye et le hamster. Les lésions toxiques observées sont
fréquemment soit des lésions hyperplasiques/ métaplasiques, soit des
lésions hypoplasiques et elles intéressent principalement les tissus
épithéliaux.
Des effets toxiques sur la reproduction ont été observés chez les
singes rhésus: la plus faible dose sans effets observables serait,
d'après les calculs, de 1 à 2 ng/kg de poids corporel par jour. Il se
peut qu'il existe une dose sans effets observables de 1 ng/kg de poids
corporel par jour chez le rat, s'agissant des effets sur la
reproduction (Murray et al., 1979; Nisbet & Paxton, 1982).
Lorsqu'on compare les études consacrées à la production de
cancers chez le rat par Kociba et al. (1978) et par le NTP (1982a, b),
il apparaît clairement que les tumeurs hépatiques, notamment les
épithéliomas hépatocellulaires, s'obtiennent à des doses similaires.
Bien que le NTP ainsi que Kociba et al. (1978) aient observé une
augmentation de l'incidence des tumeurs au niveau d'autres organes,
les résultats ne sont pas analogues dans les deux études. Cela tient
peut-être, en partie, à des différences dans les doses administrées
(gavage ou exposition par incorporation à des aliments broyés) et dans
les souches. Dans l'étude de Kociba, l'administration d'une dose de 10
ng/kg de poids corporel a provoqué une augmentation de l'incidence des
nodules néoplasiques (hyperplasiques) chez les femelles, tandis qu'une
dose de 1 ng/kg de poids corporel se traduisait par un gonflement des
hépatocytes en foyers ou par places. A ces doses, l'incidence de
certaines tumeurs à déterminisme hormonal était plus faible chez les
animaux d'expérience que chez les témoins, ce qui donne à penser que
les 2,3,7,8-TCDD déterminent des altérations au niveau des glandes
endocrines. Compte tenu de ces résultats de l'expérimentation animale
et des données dont on dispose au sujet de l'homme, le CIRC (1982,
suppl. 4) est arrivé à la conclusion que les preuves de
cancérogénicité des TCDD étaient suffisantes chez les animaux, mais
insuffisantes chez l'homme.
Apparemment, les TCDD n'ont pas de propriétés mutagènes et ne
devraient donc pas être génotoxiques. Leur action cancérogène est
donc attribuée à un mécanisme indirect (épigénétique).
3.4 Effets toxiques des PCDD et des PCDF autres que les TCDD
Plusieurs autres PCDD et PCDF provoquent les mêmes symptômes que
les 2,3,7,8-TCDD, mais leur activité est très inégale (tableaux 56,
62). Pour résumer, il existe 12 isomères qui présentent une toxicité
élevée, à savoir les tétra-, penta-, hexa- et heptaCDD et CDF
contenant quatre atomes de chlore symétriques deux à deux en position
2,3,7 et 8. On a montré, lors d'études prolongées sur l'animal, qu'un
mélange de deux hexaCDD (1,2,3,7,8-9 et 1,2,3,6,7,8-haxaCDD) était
cancérogène, mais à des doses plus élevées que celles utilisées dans
l'étude des TCDD. La dioxine non substituée et la dichloro-2,7 dioxine
n'ont manifesté aucun pouvoir cancérogène.
L'activité relative, toxique et biologique, des PCDD et des PCDF
a été estimée au moyen d'études de courte durée chez le rat et
d'études en culture de cellules mammaliennes. Comme critères, on a
retenu l'inhibition de la prise de poids, l'atrophie du thymus,
l'induction d'enzymes, la térato-génicité, la production d'acné et la
kératinisation. En l'absence d'observations sur la toxicité à long
terme, les résultats fournis par ces épreuves de durée limitée
constituent actuellement la seule source qui permette un classement
par toxicité en vue de l'évaluation des dangers pour la santé de
l'homme.
Les études consacrées à des mélanges de ces composés ont mis en
évidence une synergie additive tout au plus.
3.5 Etude des différences interspécifiques
La sensibilité aux effets biologiques et toxiques des
2,3,7,8-TCDD est très inégale selon les espèces. Par exemple, la
DL50 par voie orale varie de 0,6 µg/kg de poids corporel chez le
cobaye à 5051 µg/kg de poids corporel chez le hamster doré syrien
(tableau 47); en outre, on a signalé des différences prononcées de
DL50 chez différentes souches de la même espèce (par exemple le rat
et la souris). La toxicité et la toxicocinétique des TCDD sont très
analogues chez les singes et chez l'homme. Cependant, il est
impossible d'expliquer les différences considérables de sensibilité,
selon le sexe et la souche, aux 2,3,7,8-TCDD et aux composés analogues
par les différences qu'on observe sur le plan toxicocinétique. Les
observations montrent que, chez des souris de lignée pure, l'abondance
cellulaire du récepteur Ah est partiellement corrélée avec la
sensibilité aux effets biologiques et toxiques de ces composés. On a
également repéré ce récepteur chez d'autres espèces, notamment chez
l'homme. Cependant, l'abondance relative de ce récepteur selon les
espèces n'explique pas les différences de sensibilité aux
2,3,7,8-TCDD; cette constatation n'a rien d'étonnant quand on sait
qu'il existe des mécanismes de toxicité complexes et encore inconnus
qui font intervenir de multiples facteurs en plus du récepteur aux Ah.
4. Effets sur la santé de l'homme
4.1 PCDD
La population générale est exposée à de faibles quantités de PCDD
et de PCDF présentes dans des mélanges complexes qui ne provoquent
aucun cas de maladie. Lors de quelques incidents, par exemple à Seveso
et à Yusho, des ouvriers et d'autres personnes sont tombés malades à
la suite d'une exposition à des quantités plus élevées d'un nombre
restreint de ces composés.
Dans le cas de l'exposition professionnelle ou accidentelle,
l'effet clinique le plus net réside dans une chloracné. D'autres
effets (tableau 64) ont été observés mais, à part la chloracné et
peut-être des troubles fonctionnels mineurs, aucun n'a été durable.
Dans certaines des études de mortalité réalisées, on a fait état
d'une augmentation de l'incidence des cancers de diverses
localisations, mais le trop petit nombre de cas incite à la prudence.
L'impression générale qui se dégage des études de suivi est que
les effets généraux aigus des TCDD, même lorsqu'ils sont graves, sont
habituellement réversibles, à l'exception de la chloracné, ou
s'améliorent peu à peu après l'arrêt de l'exposition. A Seveso, le
seul effet nocif sur la santé qui soit indiscutable a été la
chloracné: sur les 193 cas qui se sont déclarés en 1976 et 1977, 20
présentaient encore des symptômes actifs en 1984. De nombreuses études
ont été consacrées à la recherche de liens possibles entre
l'exposition à l'Agent orange, au Viet Nam, et divers effets sur la
santé, tant chez les civils que chez les militaires. Cependant, les
données dont on dispose à ce jour ne permettent pas de conclure
catégoriquement à l'existence d'effets biologiques notables, en
particulier sur la reproduction (voir section 9.2).
Dans un certain nombre d'études, on a comparé les populations
exposées à divers groupes témoins sur la base du dosage des lipides
sériques, d'épreuves de la fonction hépatique et de divers autres
paramètres. Bien que dans ces études ainsi que dans des études de cas
isolés, on ait signalé des différences statistiquement significatives,
il est impossible de formuler des conclusions définitives par suite du
manque d'uniformité, de diverses insuffisances sur le plan technique
et de l'impossibilité d'exclure les facteurs de confusion.
Lors de la contamination accidentelle survenue en 1971 au
Missouri (Etats-Unis d'Amérique), des enfants ont manifesté des signes
d'intoxication aiguë mais ils sont aujourd'hui en bonne santé,
semble-t-il. Les études épidémiologiques conduites dans le même Etat
sur des populations durablement exposées à des concentrations plus
faibles n'ont jusqu'ici révélé aucun effet biologique notable.
Cependant, malgré l'absence de manifestations cliniques, certains
signes témoignent d'une action sur le système immunitaire à médiation
cellulaire.
La gamme des effets des TCDD sur la santé humaine reste à
préciser. On peut conclure que, dans l'ensemble, les observations
concernant l'exposition et les effets sur la santé ne permettent pas
d'établir pour l'homme des relations dose/effet ou dose/réponse.
En dépit de nombreuses études cliniques et études de suivi, aucun
effet général persistant bien net n'a été observé, à l'exception de la
chloracné. Compte tenu de ce qu'on sait actuellement, il semble peu
probable que l'exposition aux TCDD ait des séquelles toxicologiques
permanentes, graves et invalidantes.
4.2 PCDF
Les seuls cas d'intoxication humaine par les PCDF pour lesquels
on dispose d'informations circonstanciées concernent les incidents
survenus à Yusho, au Japon (1968) et à Yu-cheng, à Taiwan (1979), à la
suite d'une contamination d'huile de riz par des PCDF, des PCD et des
PCQ (voir section 11.1). Au total, plusieurs milliers de personnes ont
été gravement intoxiquées. La récapitulation des données montre que,
selon toute vraisemblance, il faut incriminer les PCDF. La
symptomatologie générale était sensiblement la même que lors d'une
intoxication par les PCDD. Les différences reflètent peut-être
l'intensité de l'exposition et la structure, par âge et sexe, de la
population exposée. Dans le cas des malades de Yusho, les estimations
de l'apport quotidien moyen de PCDF pendant plusieurs mois ont abouti
à une dose de 0,9 µg/kg de poids corporel pour les PCDF totaux, de
0,1-0,2 µg/kg pour les tétrachloro-2,3,7,8 dibenzofurannes ainsi que
les penta- et hexaPCDF, à quoi il faut ajouter 150 µg pour les PCB et
148 µg pour les PCQ par kg de poids corporel (Hayabuchi et al., 1979).
La dose pathogène minimale a été estimée à 0,6 mg de PCDF totaux par
personne sur 30 jours, ce qui correspond à une dose quotidienne de
0,05-0,1 µg/kg de poids corporel pour les PCDF substitués en 2,3,7 et
8. Quoi qu'il en soit, les données disponibles sont insuffisantes pour
qu'on puisse se prononcer sur la valeur de la dose qui serait sans
danger pour l'homme.
4.3 Charge de l'organisme humain et cinétique
Chez l'homme, la concentration résiduelle des TCDD dans le tissu
adipeux s'élève jusqu'à 20 mg/kg dans la population générale, en
dehors de toute exposition précise connue, mais elle peut atteindre
une valeur plus élevée dans certains cas sans entraîner de
manifestations pathologiques. Aucune des populations ainsi étudiées
n'avait été constituée par sondage aléatoire. Les autres PCDD et PCDF
contenant un plus grand nombre d'atomes de chlore, en particulier les
octaDD, se rencontrent également dans ces échantillons (voir tableaux
29, 30). Les concentrations moyennes semblent augmenter avec l'âge.
Dans certaines situations, on a observé des concen-trations plus
élevées (de l'ordre de quelques microgrammes par kilogramme) qui ne
s'accompagnaient d'aucune manifestation pathologique.
Lors des accidents de Yusho et de Yu-cheng, les symptômes
observés étaient associés à des concentrations de PCDF plus élevées;
par exemple, on a trouvé, un an après exposition à de l'huile de riz
contaminée, une concentration de 6,9 µg/kg de tissu adipeux pour les
pentachloro-2,3,4,7,8 dibenzofurannes.
D'après les données très limitées dont on dispose, la
concentration dans la population générale du 2,3,4,7,8- pentaCDF, par
exemple, semble cent fois plus faible que la concentration observée
chez les victimes de l'accident de Yusho.
Aucune comparaison de ce type n'est possible pour les PCDD.
Des données limitées montrent que, à l'exception des TCDF, les
isomères chlorés en position 2,3,7 et 8 font l'objet d'une rétention
sélective.
D'après une étude expérimentale unique, la demi-vie des TCDD
serait de 5 ans chez l'homme. Une autre étude a permis d'évaluer cette
demi-vie à 2-6 ans pour les 1,2,3,6,7,8- hexaCDD, les
1,2,3,4,6,7,8-heptaCDD, les octaCDD, les 1,2,3,4,6,7,8-heptaCDF et les
octaCDF. Ces observations devront être complétées car elles
proviennent d'études qui portaient sur deux sujets seulement, sans
compter que la toxicinétique de ces types de composés ne se ramène
peut-être pas simplement à une cinétique du premier ordre. Cependant,
même si les données actuelles ont des limites, il est clair que la
demi-vie de ces composés est de l'ordre d'une ou plusieurs années.
Les valeurs indiquées pour la demi-vie sont très différentes chez
l'homme et chez les rongeurs. Mais, les expériences conduites sur les
animaux ont généralement été exécutées au moyen de doses toxiques. En
outre, les animaux qui ont une moindre longévité ont un métabolisme
plus important de sorte qu'il est normal que la demi-vie soit plus
courte.
Les PCDD et les PCDF sont essentiellement retenus dans les
graisses, mais ces produits sont aussi excrétés dans le lait (tableau
39) et traversent la barrière placentaire. On les trouve aussi, à plus
faibles concentrations, dans le sang et les organes vitaux. La
distribution entre les différents tissus humains est mal connue mais
selon certains, le rapport des concentrations dans le tissu adipeux et
dans le foie est plus élevé chez l'homme que chez les rongeurs.
Cependant, cette conclusion repose sur des données fort restreintes
fondées sur des pièces d'autopsie. Il reste donc à voir si elle vaut
pour la population dans son ensemble.
Les voies de pénétration dans l'organisme humain sont encore mal
connues mais on admet que l'apport alimentaire représente l'essentiel.
Cependant, le nourrisson représente un cas particulier puisque, par
suite du passage trans-placentaire, il peut y avoir une exposition
in utero. D'après les dosages effectués jusqu'ici dans le lait
maternel, ce dernier pourrait être une source importante de
contamination.
Il n'existe aucune donnée concernant la dose des PCDD qui est
toxique pour l'homme. Toutefois, lors des accidents de Yusho et de
Yu-cheng, les victimes avaient absorbé une dose totale de l'ordre de
3,3-3,8 mg pour les PCDF totaux et de 400-500 µg pour les PCDF
substitués en 2,3,7 et 8. Il n'existe aucune donnée fiable sur les
doses sans effet nocif.
5. Conclusions générales
Les PCDD et les PCDF se rencontrent dans tout l'environnement et
il est probable que chacun en est porteur. Parfois, ces produits
déterminent des effets toxiques complexes à la suite d'une activité
professionnelle ou accidentelle.
D'après les observations faites à Yusho, les expériences
conduites chez des espèces sensibles de singes et compte tenu de
certaines hypothèses concernant l'activité relative des PCDD et des
PCDF, il se peut que l'homme et certains singes présentent une
sensibilité comparable à l'égard de ces composés. Cependant, les
incertitudes concernant la dose effectivement reçue et les difficultés
de l'évaluation des effets toxiques autres que la chloracné
interdisent, dans le cas de l'homme, une conclusion catégorique quant
à la résistance relative de l'homme aux effets toxiques de ces
composés. Il convient de limiter l'exposition aux plus faibles niveaux
compatibles avec les impératifs pratiques.
RECOMMENDATIONS
1. Des études interlaboratoires de validation, selon une sorte de
"poule" et l'utilisation de protocoles normalisés de contrôle et
d'assurance de qualité, sont indispensables pour améliorer les
méthodes d'analyse.
La méthodologie de l'échantillonnage, les méthodes d'analyse et
l'interprétation des données doivent être optimalisées et
normalisées avant le démarrage des enquêtes.
2. Des données complémentaires sont nécessaires sur l'origine des
PCDD et des PCDF, sur leur répartition dans l'environnement et
sur leur destinée.
D'autres observations sont indispensables au sujet de la présence
des PCDD ou des PCDF dans l'environnement, notamment sur
l'évolution des concentrations dans le temps et sur la
détermination des profils isomériques, notamment dans les
produits alimentaires, dans l'air ambiant et dans les sédiments.
3. Il serait bon de faire des observations sur les effets
qu'exercent les PCDD et les PCDF sur la biocénose.
4. Des données plus complètes sont nécessaires en ce qui concerne la
biodisponibilité des PCDD et des PCDF provenant de diverses
matrices, dans l'environnement et dans la ration alimentaire.
L'exposition à ces sources devrait être mise en corrélation avec
les pratiques agricoles et industrielles.
5. Il convient de mettre au point et de valider des méthodes
chimiques et biologiques plus simples et moins coûteuses pour
rechercher la présence de PCDD et de PCDF.
6. Des études sur les mécanismes toxiques des PCDD et des PCDF sont
nécessaires pour élucider les différences observées dans les
effets produits chez les diverses espèces et permettre une
extrapolation à l'homme.
7. Il importe de poursuivre l'étude de l'immunotoxicité, notamment
en ce qui concerne la fonction des lymphocytes T cytotoxiques. En
particulier, il est capital d'étudier les effets sur le système
immunitaire d'une exposition à la période périnatale et de la
durée de cette action.
8. Des études de toxicité à long terme devraient être effectuées,
notamment des études de reproduction sur plusieurs générations
chez diverses espèces, au moyen de trois des PCDD et des PCDF les
plus répandus, à savoir le 2,3,4,7,8-pentaCDF, le
1,2,3,7,8-pentaCDD et l'octaCDD.
9. Etant donné que l'homme est exposé à des mélanges complexes de
PCDD et de PCDF, il faut poursuivre la mise au point et la
validation de systèmes d'épreuve, permmettant notamment l'étude
des réactions des tissus humains en vue d'apprécier l'activité
toxique de ces composés et de leurs mélanges. Ces systèmes
permettront d'étudier les mécanismes d'action, les relations
structure/activité et les effets interactifs.
10. Il serait bon d'étudier la charge de l'organisme en PCDD et PCDF
et d'établir une corrélation entre cette charge, les effets
cliniques constatés et les résultats des examens de laboratoire.
Le suivi des groupes exposés antérieurement est important à cet
égard.
6.2.1.2 In vitro studies
Although there is evidence of different metabolites of TCDD and
the possibility of its metabolic transformation was suggested as early
as 1975 (Allen et al., 1975; Rose et al., 1976), it was only in 1982
that specific metabolites were identified by Sawahata et al. (1982).
They incubated TCDD with isolated rat hepatocytes at 37 ©C for 8 h,
and the resulting incubation mixture was subjected to HPLC. The major
peak of radioactivity not corresponding to TCDD was incubated with
ß-glucuronidase in order to split the possible glucuronide
conjugate(s) of TCDD or its metabolite(s). They found that
4,5-dichlorocatechol and 4,5-dichloroguaiacol are potential
metabolites of TCDD, but due to the limited amount of material the
identity of these metabolites was not confirmed by gas
chromatography-mass spectrometry. Two other metabolites of TCDD,
namely 1-hydroxy-2,3,7,8-tetrachloro-dibenzo-p-dioxin and
8-hydroxy-2,3,7-trichlorodibenzo-p-dioxin, were isolated by means of
HPLC, and were identified by mass spectrometry.
Primary hepatocytes from Sprague Dawley rats and Hartley
guinea-pigs have been used to study the metabolism of 14C-TCDD
(Wroblewski & Olson, 1985). The overall metabolism was 2.8 times
greater in rats than in guinea-pigs. The metabolism of 14C-TCDD was
increased 3.2-fold in rats pretreated with TCDD (5 µg TCDD/kg body
weight ip 72 h prior to isolation of hepatocytes), but no effect was
found in similarly TCDD-pretreated guinea-pigs, or in phenobarbital-
pretreated rats (80 mg/kg body weight ip for 3 days, beginning 4 days
prior to isolation of hepatocytes). Hepatocytes from TCDD-pretreated
rats metabolized TCDD 9 times more rapidly than similarly pretreated
guinea-pig hepatocytes. TCDD may be metabolized by an inducible form
of cytochrome P448 which is expressed in rats but not guinea-pigs.
These differences in metabolism may play a major role in explaining
the differences in species susceptibility to the acute effects of
TCDD.
6.3 Transfer Via Placenta and/or Milk
The transplacental passage of 14C-TCDD has been studied by
Khera & Ruddick (1973). Pregnant Wistar rats were given 14C-TCDD in
a single oral dose of 200 µg/kg body weight on gestation days 16, 17,
or 18 and were killed 6 h after dosing. 14C-activity was detected in
maternal tissues and also in the fetuses and the placenta. Assuming
that all the 14C-activity found in the samples was present as
14C-TCDD, the following levels (ng/gram tissue) were found for
gestation days 16, 17, and 18 respectively: maternal liver 339 (± 15),
339 (± 19), and 275 (± 20); maternal blood 25 (± 11), 19 (± 9), and 10
(± 3); placenta 25 (± 6), 38 (± 4), and 41 (± 3); and fetus 11 (± 3),
15 (± 1), and 16 (± 1). Studies by Moore et al. (1973) indicated that
the passage of TCDD or its metabolites into milk could be of
importance, as TCDD-related effects were observed in sucklings,
nourished by lactating mothers given, after delivery, a single oral
dose of 1 or 3 µg/kg body weight.
The TCDD concentration in livers of pregnant NMRI mice, at a
given dose, was significantly lower than in livers of non-pregnant
mice (Krowke 1986). The concentration of TCDD in the liver of
non-pregnant mice was about 5 times higher than in pregnant mice 7
days after a s.c. dose.
Nau & Bass (1981) studied the transfer of 14C-TCDD to embryos
and fetuses in NMRI mice. The animals were given a single dose of 5,
12.5, or 25 µg TCDD/kg body weight by gavage or by s.c. or ip
injection at day 16 of gestation to study the transfer of TCDD to the
fetus. The animals were killed two days later and various tissues were
analyzed for radioactivity. No evidence was found to indicate a major
first pass effect following oral administration. Maternal livers
contained the highest levels of TCDD, 4.1 to 10.5% of the radioactive
dose administered, which was about one order of magnitude higher than
in extrahepatic maternal tissues, including placenta. Fetal liver and
extrahepatic tissues contained low levels of radioactivity
corresponding to 0.09 to 1.41% and 0.05 to 0.14%, respectively, of the
dose administered to the dams. More radioactivity was recovered in the
placenta and fetus when TCDD was given either as a single ip dose of
25 µg/kg body weight on day 10 of gestation or as 5 daily ip doses of
5 µg/kg body weight on days 7 to 11, when compared to a single i.p.
dose of 25 µg/kg body weight on gestation day 7. Oral dosing of 30 µg
14C-TCDD (0.332 µCi/ µg)/kg body weight to pregnant C57Bl/6 mice on
gestation day 11 resulted in 0 to 14% embryomortality on gestation
days 12 to 14 (Weber & Birnbaum, 1985). About 0.03% of the radioactive
dose was contained in the embryo and in the placenta on days 12 to 14
of gestation. The maternal liver contained 67.4, 67.9, and 50.6% of
the administered radioactive dose on gestation days 12, 13, and 14,
during which days the cumulative elimination of radioactivity in urine
and faeces was 2.4 and 53.3% of the dose, respectively.
The transfer of 14C-TCDD via placenta and milk and the
distribution of the transformed TCDD between various embryonic and
fetal tissues were studied in NMRI mice (Nau et al., 1986). Dams were
given a single dose of 25 µg 14C-TCDD (45 mCi/mmol)/kg body weight
either orally, subcutaneously, or intraperitoneally. To differentiate
between postnatal and in utero exposure, the experimental design
included cross fostering. Depending on the route of administration,
from 0.02 to 0.07% of the administered radioactivity was found in the
liver of the fetus at birth. The highest levels were noted after ip
administration and the lowest after oral intubation. The corresponding
values one week after birth were 0.05 to 0.20%. The hepatic
radioactivity in the neonate reached a peak 1 week after birth and
then decreased slowly throughout and after lactation. The levels of
TCDD-derived radioactivity in extra-hepatic tissues of the offspring
were approximately one order of magnitude lower than the hepatic
levels. Very little radioactivity was found in the stomach filled with
milk, indicating that TCDD ingested from milk was rapidly absorbed in
the stomach.
Arstila et al. (1981) studied the excretion of TCDD in goat milk
after a subchronic administration of 200 ng TCDD per day for 2 months
in the first experiment and of 400 ng TCDD per day for one month in
the second experiment. The minimal detectable concentration in this
study was declared to be below 5 ng/litre. The maximum concentration
of TCDD in milk in the first experiment was 20.8 (± 6.6) ng/litre and
in the second experiment 19.3 (± 6.6) ng/litre. After 18 weeks feeding
with TCDD the levels had dropped to 4.2 and 3.6 ng/litre,
respectively.
The secretion of TCDD in milk and cream has also been studied in
lactating dairy cows kept on a diet containing 10, 30, 100, 300, or
1000 mg/litre 2,4,5-T, corresponding to TCDD levels of 5, 15, 50, 150,
and 500 ng/litre (Jensen & Hummel, 1982). This resulted in levels of
TCDD in the excreted milk of below detection limit, 3, 10, 16-22, and
42-89 ng/litre, respectively, indicating that about 10-20% of the dose
given was eliminated in the milk. The levels in cream were about ten
times higher than those in milk.
One lactating Holstein cow, receiving commercial technical-grade
pentachlorophenol containing several PCDDs and PCDFs orally in
gelatine capsules at a dose rate of 10 mg/kg body weight twice daily
for 10 days and once daily for the following 60 days, calved 151 days
after treatment was stopped (Firestone et al., 1979). The PCDD content
of blood, body fat (shoulder from the cow and hind quarter of the
calf), and milk fat was determined 14 days later. The detected
congeners were 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and
octaCDD and their levels were 12, 13, 20 ng/litre and 27, 14, 6
ng/litre in the blood of the cow and calf, respectively. Corresponding
values for body fat were 4.8, 11.1, and 6.1 µg/kg in the cow and 2.3,
1.9, and 0.5 µg/kg in the calf. The milk fat from the cow contained
2.2, 4.4, and 3.3 µg/kg of the respective congeners.
6.4 Matrix Effects on the Uptake ("Bio-availability")
Uptake of TCDD and other PCDD congeners is highly dependent upon
the formulation in which it is applied. Although conflicting results
have been obtained from studies where TCDD in soil was administered
(McConnell et al., 1984; Umbreit et al., 1985, 1986a, 1986b), most
available data support the idea that mixing TCDD with soil or
activated carbon results in the adsorption of TCDD to the soil
particles, thus reducing the availability of TCDD. Contact time of
TCDD with soil seem to influence the availability, probably because
the binding of TCDD to soil particles becomes strengthened (Poiger &
Schlatter, 1980). Studies elucidating the matrix effects of various
soils or activated carbon on the TCDD-responses in several animal
species are summarized in Table 46.
Poiger & Schlatter (1980) showed decreasing hepatic recovery of
TCDD in rats 1 day after dosing using suspensions of ethanol, soil, or
activated carbon as vehicles. About 50% lower hepatic retention of
TCDD was obtained in the rat, 6 days after dosing, when Minker stout
site soil was the vehicle as compared to corn oil (Lucier et al.,
1986). However, in this study hepatic enzyme-induction (AHH and UDPGT)
was similar in the two vehicle groups.
The bioavailability of TCDD from environmentally contaminated
soil samples has been studied in young guinea-pigs after intragastric
administration (McConnell et al., 1984). Groups of six animals each
were given soil samples corresponding to doses of approximately 1, 3,
or 10 µg TCDD per kg body weight. The doses were based on analyses of
soil siftings (60-gauge mesh) from the Times Beach and Minker Stout
sites, which indicated concentrations of 770 and 880 µg TCDD/kg,
respectively. Controls received soil samples in which no TCDD, PCBs,
or PCDFs were detected. For comparison, pure TCDD in corn oil was
given at either 0, 1, or 3 µg/kg. The observation time was 30 days.
LD50 values calculated in this study were 1.75 µg/kg for TCDD in
corn oil, 7.15 µg/kg for Times Beach soil, and 5.50 µg/kg for the
Minker Stout site soil. An exact percentage for bioavailability was
not calculated in this study, but the TCDD content of the livers of
exposed guinea-pigs indicated a highly efficient absorption of TCDD
from soil.
TCDD in contaminated soil from a 2,4,5-T manufacturing plant and
from a metal salvage yard (Newark, New Jersey, USA) had a low
bioavailability (0.5% and 21.3%, respectively) in guinea-pigs (Umbreit
et al., 1985, 1986a). The soils were given as single oral doses in a
10% aqueous suspension in 5% gum acacia. The bioavailability was
judged by the hepatic concentration of TCDD 60 days after dosing in
guinea-pigs receiving site soils, decontaminated soil, and
TCDD-recontaminated soil.
The difference in bioavailability of TCDD from
2,4,5-T-manufacturing site soil in Newark (New Jersey) and from Times
Beach Site soil (Missouri) when given orally to guinea-pigs was
confirmed by Umbreit et al. (1986b).
Bonaccorsi et al. (1984) gave to rabbits seven daily doses of
TCDD in corn oil, TCDD-contaminated Seveso soil, or recontaminated
soil. Taking the hepatic recovery of TCDD in the corn oil group as
100% "bioavailability", the decrease in "bioavailability" of TCDD from
Seveso soil was 68%. The decrease in "bioavailability" of
recontaminated soil varied from 0 to 44% with the doses used.
Table 46. Matrix effects of various soils on TCDD-responses, as related to the estimated bioavailability of TCDD given
orally to different species.
Species/ Vehicle/matrix Dose of Lethality Hepatic Estimated Reference
Length TCDDc recovery of "bio-
of study TCDDe availability"
(days)
Rat 50% ethanol 14.7 ng 36.7f Poigner &
1 day recontaminated soila 12.7, 22.9 ng 24.1f Schlatter
recontaminated soilb 21.2, 22.7 ng 16.0f (1980)
activated carbon 14.7 ng < 0.07f
Rat corn oil 1.0, 5.0 7.6 and 40.8 Lucier et al.
6 days Minker Stout site soil 1.1, 5.5 1.8 and 20.3 50% (1986)
Guinea- corn oil 1, 3 1/6, 6/6 1.6, 13.3 McConnell et
pig Times Beach site soil 1.3, 3.8, 12.8 0/6, 1/5, 5/5 < 1.0-34.3 Efficient al. (1984)
30 days Minker Stout site soil 1.1, 3.3, 11.0 0/6, 2/6, 6/6 < 1.0-25.7 absorption from
recontaminated soil 10.0 6/6 45.4 soil (85%)
Guinea- corn oil 6 5/8 Umbreit et al.
pig 2,4,5-T-manufacturing 3, 6, 12 0/8, 0/7, 0/7 0.09 (high dose) 0.5% (1985, 1986a)
60 days site soil (Newark,
New Jersey, USA).
Metal salvage yard soil 0.32 0.23 21.3%
(Newark)
recontaminated soil 6 6/7 18
Table 46 (contd).
Species/ Vehicle/matrix Dose of Lethality Hepatic Estimated Reference
Length TCDDc recovery of "bio-
of study TCDDe availability"
(days)
Guinea- 2,4,5-T-Manufacturing 3, 5, 10 0/18, 1/20, 1/18 Umbreit et al.
pig site soil, (Newark) (1986b)
60 days Times Beach site soil 1, 3, 10 2/19, 2/20, 8/14
recontaminated soil 6 19/20
Rabbit corn oil 20, 40, 80d 0.26-2.7 Bonaccorsi et
7 days Seveso soil 80, 160d 0.88-2.2 > 32% al. (1984)
recontaminated soil 20, 40, 80d 0.26-1.5 > 66-100%
a Contact time = 10-15 h.
b Contact time = 8 days.
c µg/kg body weight, unless otherwise stated.
d ng/kg body weight per day.
e µg/kg liver, unless otherwise stated.
f % of dose.
Considerably lower hepatic levels of PCDDs and PCDFs were
observed in rats fed a diet containing fly ash from municipal
incinerators compared with those fed a diet containing extracts from
the same fly ash (van den Berg et al., 1983).
Dietary intake of soot-containing TCDD produced 60% mortality in
male and female guinea-pigs on days 46 and 60, respectively, at which
time the estimated TCDD-consumption was 1.3 µg/kg body weight for
males and 1.9 µg/kg body weight for females (DeCaprio et al., 1983,
see also section 8.2.3). These data thus suggest a high uptake of TCDD
from the soot matrix.
7. EFFECTS OF TCDD AND OTHER PCDDs ON EXPERIMENTAL ANIMALS AND
IN VITRO TEST SYSTEMS
7.1 Acute Toxicity
7.1.1 In vivo studies on mammals
The range of doses required to cause death varies considerably
between species, as well as between strains of species, and with sex,
age, and route of administration within a single strain (Table 47).
More than an 8000-fold difference exists between the dose of TCDD
reported to cause 50% lethality to male Hartley guinea-pigs, the most
sensitive species tested (Schwetz et al., 1973), and the corresponding
dose for male Golden Syrian hamsters (Henck et al., 1981). The rat
seems to be the second most sensitive species, although there is a
more than 200-fold variability in LD50 values between different
strains. The oral LD50 value was 22 µg TCDD/kg body weight for male
Sherman rats (Schwetz et al., 1973), whereas Walden & Schiller (1985)
found LD50 values ranging from 164 to 340 µg TCDD/kg body weight
when male Fisher 334 N rats from three different suppliers were
tested. The Han/ Wistar-strain of rat has been demonstrated to be
particularly resistant to TCDD-exposure (Pohjanvirta & Tuomisto,
1986). Among the five rats per dose group (0, 1500, 2000, 2500, or
3000 mg TCDD/kg body weight) only one animal died within the 39-40
days observation period.
Monkeys (McConnell et al., 1978a), New Zealand rabbits (Schwetz
et al., 1973), C57Bl/6 mice (Chapman & Schiller, 1985; Jones & Greig,
1975; McConnell et al., 1978b; Smith et al., 1981; Vos et al., 1974),
DBA/2-mice (Chapman & Schiller, 1985), and B6D2F1-mice (Chapman &
Schiller, 1985) gave oral LD50 values of 70, 115, 114, 2570, and 296
mg TCDD/kg body weight, respectively. The difference in sensitivity
towards TCDD among various strains of mice has been claimed to depend
on a genetic variability in the Ah and/or hr-locus (see section
7.8.1).
Male Sherman rats were found to be more sensitive to TCDD than
were females (Schwetz et al., 1973), whereas Beatty et al. (1978)
reported that male Sprague Dawley rats were more resistant to TCDD
than were females. Smith et al. (1981) found adult female C57BL/10
mice to be more resistant to TCDD than adult males of the same strain.
No differences in sensitivity to TCDD between sexes were recorded for
guinea-pigs (McConnell et al., 1978b; Silkworth et al., 1982) or
hamsters (Olson et al., 1980b). Thus data on sex differences in
sensitivity to lethal effects of TCDD are conflicting.
Data on the effect of age at exposure to TCDD on the sensitivity
of acute response are scarce, and comparisons are hampered by the
absence of information, or incomplete information, on the age and/or
body weight of the tested animals. However, Beatty et al. (1978) found
that weanling male Sprague Dawley rats were more sensitive to TCDD
than were adult males. A dose of 25 µg TCDD/kg body weight caused,
after 35 days, a cumulative lethality of 62% in weanling Sprague
Dawley rats and 25% in young adults (Christian et al., 1986a). When
weanling and mature adults were exposed to a similar toxic dose of
TCDD (LD62 and LD60, respectively), onset of death occurred 9 days
later in the adults (Christian et al., 1986a).
Schwetz et al. (1973) found LD50 values in rabbits of 115 µg
TCDD/kg body weight after oral exposure, as compared to 275 µg TCDD/kg
body weight after dermal exposure. C57Bl/6-mice seem to be more
sensitive to ip administration of TCDD (Gasiewicz et al., 1983b) than
to oral administration (McConnell et al., 1978b). The LD50 value in
guinea-pigs was increased from 2.5 µg/kg body weight to 19 µg/kg body
weight when the vehicle for the oral administration of TCDD was
changed from corn oil to methyl cellulose (Silkworth et al., 1982).
Umbreit et al. (1985) compared mortality, and time to death, among
guinea-pigs given single oral doses of TCDD in corn oil, TCDD in a
suspension of cleaned soil, from an industrial site, or
TCDD-contaminated soil from the same industrial site. Animals treated
with corn oil, cleaned soil and contaminated site soil survived the
60-day study without any sign of TCDD intoxication. Among animals that
received 6 µg TCDD/kg body weight either in corn oil or mixed with
cleaned soil, only 3/8 and 1/7 survived, respectively. Deaths occurred
between days 9 to 31 and 15 to 25, respectively. These results are
different from those reported by McConnell et al. (1984) where
TCDD-contaminated soils from Times Beach and Minker Stout were highly
toxic to guinea-pigs. LD50 values calculated in this study were 1.75
µg/kg for TCDD in corn oil, 7.15 µg/kg for TCDD in Times Beach soil,
and 5.50 µg/kg for TCDD in Minker Stout site soil. The Minker Stout
site soil was also potent in inducing AHH-activity in female Sprague
Dawley rats. The different results may be due to the vehicle used
and/or to the presence of other substances in the soils that may
potentiate or retard the TCDD-induced toxicity (Umbreit et al.,
1986a). In section 6.4, the results from McConnell et al. (1984) and
Umbreit et al. (1985, 1986a) are further discussed from the viewpoint
of the "bioavailability" of TCDD in soils (Table 46).
Despite similar routes and vehicles for the administration of
TCDD to Golden Syrian hamsters, LD50 values varied between 1157
µg/kg body weight (Olson et al., 1980b) and 5051 µg/kg body weight
(Henck et al., 1981). A possible explanation for this difference could
be the spontaneous occurrence of ileitis observed in the former study,
which might have increased the susceptibility of those hamsters to
TCDD toxicity.
Table 47. Single lethal dose values for TCDDa
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats
Porton F/5-12 8-9 weeks/ oral/DMSO 0 90 days NR 40
170-200 g 30
(Greig et al., 48
1973) 75
120
190
300
Porton F/6 9-10 weeks/ oral/arachis 0 90 days NR 40
170-200 g oil 126
(Greig et al., 199
1973) 315
500
Sherman M/5-10 NR oral/corn oil 8 2-8 weeks 22 9-27
acetone (9:1) 16
(Schwetz et al., 32
1973) 63
Sherman F/NR NR oral/corn oil NR 2-8 weeks 45 13-43
acetone(9:1)
(Schwetz et al.,
1973)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats (contd)
Sprague M/6 adult/NR ip/olive oil 4 doses 20 60 NR
Dawley 20-80 60
(Beatty et al.,
1978)
Sprague F/6 adult/NR ip/olive oil 4 doses 20 60 NR
Dawley 10-60
(Beatty et al.,
1978)
Sprague M/6 25 days/NR ip/olive oil 4 doses 20 25 NR
Dawley 5-50
(Beatty et al.,
1978)
Fisher 334N M/7 11-12 weeks/ oral/corn oil 0 30 days 340b 28b
230-280 g 75 303c 26c
(Walden & 150 164d 25d
Shiller, 1985) 225
275
325
375
CD M/7 10-11 weeks/ oral/corn oil 0 30 days 297d 25d
350-370 g 75
(Walden & 150
Shiller, 1985) 225
275
325
375
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Rats (contd)
Han/Wistar M/5 NR/300-350 oral/corn oil 1500 39-40 days > 3000 NR
2000
(Pohjanvirta 2500
& Tuomisto, 3000
1986)
Mice
C57BL/6 M/14 3 months/ oral/corn oil 0 2 months 114 15-30
23.6-30.8 g acetone (6:1) 100
(Vos et al., 1974) 150
200
C57BL/6 M/NR 7-15 weeks/ oral/arachis NR 35 days 126 21±1.6
14-30 g oil
(Jones & Greig,
1975)
C57BL/6 M/8 9 weeks/ oral/corn oil NR 30 days 284 22-25
21-25 g
(McConnell et al.,
1978b)
C57BL/6J M/NR NR ip/olive oil NR 30 days 132 NR
(Gasiewicz et al.,
1983bd)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Mice (contd)
C57BL/6J M/10-15 10-12 weeks/ oral/corn oil 95 30 days 182 24
22-32 g 145
(Chapman & 190
Schiller, 1985) 285
C57BL/10 M/5 42-121 days/NR oral/arachis 85 45 days 146 22-38
oil 107
(Smith et al., 135
1981) 170
213
C57BL/10 F/5 42-121 days/NR oral/arachis 85 45 days > 450 22-38
oil 107
(Smith et al., 135
1981) 170
213
269
338
426
536
DBA/2J M/NR NR ip/olive oil NR 30 days 620 NR
(Gasiewicz et al.,
1983be)
DBA/2J M/10-15 10-12 weeks/ oral/corn oil 1370 30 days 2570 21
22-32 g 1870
(Chapman & 2610
Schiller, 1985) 3500
4470
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Mice (contd)
B6D2F1/J M/NR NR ip/olive oil NR 30 days 300 NR
(Gasiewicz et al.,
1983e
B6D2F1J M/10-15 10-12 weeks/ oral/corn oil 170 30 days 296 25
22-32 g 220
(Chapman & 265
Schiller, 1985) 325
425
450
Guinea-pigs
Hartley M/NR NR oral/corn oil NR 2-8 weeks 0.6 5-34
(Schwetz et al.,
1973)
Hartley M/NR NR oral/corn oil NR 2-8 weeks 2.1 9-42
acetone (9:1)
(Schwetz et al.,
1973)
Hartley M/6 3-4 weeks/ oral/corn oil NR 30 days 2 17-20
200-250 g
(McConnell et al.,
1978b)
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Guinea-pigs (contd)
Hartley F/6 NR/500-600 g oral/corn oil 0.1 42 days 2.5 32-42
0.5
(Silkworth et al., 2.5
1982) 12.5
20.0
Hartley F/6 NR/500-600 g oral/methyl- 0.1 42 days 19 12-42
cellulose 0.5
(Silkworth et al., 2.5
1982) 12.5
20.0
Rabbits
New Zealand M,F/NR NR oral/corn oil NR 2-8 weeks 115 6-39
acetone (9:1)
(Schwetz et al.,
1973)
New Zealand M,F/NR NR dermal/acetone 31.6 3 weeks 275 12-22
63
(Schwetz et al., 126
1973) 252
500
New Zealand M,F/5 NR ip/corn oil 31.6 4 weeks NR 6-23
63
(Schwetz et al., 126
1973) 252
500
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Hamsters
Golden Syrian M/5-6 NR/50-80 g ip/olive oil 0 50 days > 3000
500
(Olson et al., 1000
1980b) 2000
3000
Golden Syrian F/5 NR/50-80 g ip/olive oil 0 50 days > 3000 14-32
500
(Olson et al., 1000
1980b) 2000
3000
Golden Syrian M/5 NR/50-80 g oral/olive oil 500 50 days 1157 2-47
1000
(Olson et al., 2000
1980b) 3000
Golden Syrian M/6 NR/70-120 g oral/corn oil 0 55 days 5051 9-43
acetone (9:1) 300
(Henck et al., 600
1981) 1000
3000
6000
Table 47 (contd).
Species/strain Sex/No/ Age/weight Route/vehicle Dose Duration of LD50 Time to
(Reference) group tested observation (µg/kg) death
(µg/kg) (days)
Monkeys
Macaca F/3 juvenile/ oral/corn oil 0 47 days < 70 14-34
mulatta 2.1-2.6 kg 70
350
(McConnell et al.,
1978a)
Dogs
Beagle M/2 NR oral/corn oil 300 2-8 weeks NA 9-15
acetone (9:1) 3000
(Schwetz et al.,
1973)
Beagle F/2 NR oral/corn oil 30 2-8 weeks NA all
acetone (9:1) 100 animals
(Schwetz et al., sur-
1973) vived
Chickens
Leghorn NR 4-6 weeks/NR oral/NR NR NR 25-50 12-21
(Grieg et al.,
1973)
a M = male, F = female, NR = not reported, NA = not applicable, ip = intraperitoneal, DMSO = dimethyl sulfoxide.
b supplied by Harlan.
c supplied by Frederick.
d supplied by Charles River.
e based on unpublished studies by Gasiewicz et al., 1983.
TCDD affects a variety of organ systems in different species. The
organ primarily affected in rodents and rabbits is the liver. In
guinea-pigs atrophy of the thymus and lymphatic tissues seems to be
the main effect, while dermal effects are prominent signs in non-human
primates. Generally it is not possible to specify a single organ whose
dysfunction is responsible for death. Overall, TCDD seems to have a
predilection for causing pathological changes in epithelial tissues,
both cutaneous and internal. This is particularly apparent in
non-human primates (Macaca mulatta), and is note-worthy that the
lesions mimic to some degree the effects in human beings. The
histopathological alterations in tissues include hyperplastic and/or
metaplastic alterations as well as hypoplastic responses. The toxic
responses of various species to TCDD are summarized in Table 48,
adapted from Poland & Knutson (1982). In all animal species studied,
death occurred after a time lapse ranging from several days to more
than one month after exposure. The delay was dependent on dose but not
on species (Table 48). Progressive loss of body weight was a
characteristic sign observed in animals given a lethal dose of TCDD.
The weight loss became manifest usually within a few days after
exposure and resulted in a substantial reduction of the adipose tissue
observed at autopsy. At sublethal doses of TCDD a dose-dependent
decrease in body weight gain occurred. This TCDD-induced wasting
syndrome has been thoroughly investigated in several studies discussed
more fully in section 7.4.1.
The greatest difference between species at necropsy, both in
gross and histological effects, concerns pathological alterations in
the liver. As discussed in detail in section 7.4.2, a dose of TCDD
lethal to guinea-pigs did not result in liver damage comparable to the
liver lesions described in rabbits and rats or to liver changes
observed in mice dying after doses higher than those needed to cause
death in these species. In the hamster, frank liver lesions do not
occur even after fatal doses.
Chloracne-like lesions can be induced by topical application
and/or systemic administration of TCDD in rabbits, non-human primates,
and hairless mice. These lesions are further discussed in section
7.4.4.
Severe thymus atrophy was also found at autopsy in all animal
species given lethal doses of TCDD. Histological examinations revealed
lymphoid cell depletion in thymus cortex, spleen, and lymph nodes.
These consistent findings in TCDD poisoning will be discussed in
detail in section 7.4.5 together with the other lymphoid
tissue-related effects.
Table 48. Specific differences in toxic responses following exposure to 2,3,7,8-TCDDa, b
Monkey Guinea- Cowc Rat Mouse Rabbitc Chickenc Hamster
pig
Hyperplasia and/or metaplasia
Gastric mucosa ++ 0 + 0 0 0
Intestinal mucosa + ++
Urinary tract ++ ++ ++ 0 0
Bile duct and/or gall bladder ++ 0 + ++ 0
Lung: focal alveolar ++
Skin ++ 0 +d 0 0 ++ 0
Hypoplasia, atrophy, or necrosis
Thymus + + + + + + +
Bone marrow + + ± +
Testicle + + + +
Other responses
Liver lesions + + ++ + ++ + ±
Porphyria 0 0 + ++ + 0
Oedema + 0 0 + ++ +
a References: monkey (Norback & Allen, 1973; Allen et al., 1977; McConnell et al., 1978a), guinea-pig (McConnell
et al., 1978b; Moore et al., 1979; McConnell, 1980; Turner & Collins, 1983), cow (McConnell, 1980), rat (Kociba et
al., 1978; Kociba et al., 1979a; McConnell, 1980), mouse (Vos et al., 1973; Schwetz et al., 1973; McConnell et al.,
1978b), rabbit (Vos & Beems, 1971; Schwetz et al., 1973), chicken (Allen & Lalich, 1962; Vos & Koeman, 1970;
Norback & Allen, 1973; Schwetz et al., 1973), hamster (Olson et al., 1980b; Henck et al., 1981).
b Symbols: 0 = lesion not observed, + = lesion observed (number of "+" denote severity), ± = lesion observed to
a very limited extent, blank = no evidence reported in literature.
c Responses followed exposure to 2,3,7,8-TCDD or structurally related chlorinated aromatic hydrocarbons.
d Skin lesions in cattle have been observed, but they differ from the skin lesions observed in other species.
There are also substantial interspecies differences in the
effects observed in other organs of animals given lethal doses of
TCDD. Icterus was reported in rats (Buu-Hoi et al., 1972a; Gupta et
al., 1973), hepatic porphyria occurred in mice and rats (see section
7.4.3), and ascites with subcutaneous oedema and hydrothorax appeared
in mice (Jones & Greig, 1975; Vos et al., 1974) and monkeys (Allen et
al., 1977). The accumulation of serous fluid in the pericardial sac
occurred in chickens after a single lethal dose of TCDD (see section
7.1.4). Haemorrhages were frequently observed in many organs following
lethal doses in monkeys (Allen et al., 1977), rats, and guinea-pigs
(Gupta et al., 1973). In mice, death was frequently attributed to
terminal haemorrhages (Vos et al., 1974).
When administered in doses sufficient to cause overt toxicity,
TCDD causes testicular atrophy and degeneration characterized by
reduced spermatogenic activity in mice (McConnell et al., 1978b), rats
(Kociba et al., 1976; Van Miller et al., 1977), and guinea-pigs
(McConnell et al., 1978b). The same symptoms were present in monkeys
fed dioxin-containing toxic fat (Allen & Carstens, 1967; Norback &
Allen, 1973). The decreases in male sex organ weights (seminal
vesicles, ventral prostate, testes, and caput epididymis) were
dose-dependent. ED50 values were around 15 mg TCDD/kg body weight in
Sprague Dawley rats 7 days after TCDD-treatment when compared to
pair-fed control rats (Moore et al., 1985). Shrunken, hyperchromatic
nuclei in the two layers of seminiferous tubules closest to the
basement membrane were observed in testes of young Sprague Dawley rats
90 h after a single ip dose of 5 mg TCDD/kg body weight (Mittler et
al., 1984). Epididymal lesions (Khera & Ruddick, 1973) and decreased
amounts of secretory material within the accessory sex glands (Kociba
et al., 1976) have been reported in TCDD-treated rats. Reduced
prostate weights, both absolute and relative, were found in Han/Wistar
rats at non-lethal doses of TCDD (Pohjanvirta & Tuomisto, 1986).
Reduced relative uterine weight, accompanied by decreased mucosa,
stroma, and glands, occurred in young C57Bl/6 mice dosed with 6 µg
TCDD/kg body weight three times a week for 1 month, but there was no
effect on the ovaries (Gallo et al., 1986). TCDD had no effect on the
uterine weight in 25 day-old Long Evans rats 2 - 10 days after
treatment with single ip doses of 20 or 80 µg/kg body weight (Romkes
et al., 1987). However, the increase in uterine weight induced by
estradiol treatment was counteracted by simultaneous TCDD-treatment.
Proliferative lesions of the gastrointestinal tract have been
found primarily in non-human primates (Allen et al., 1977; McConnell
et al., 1978a) whereas proliferative changes of the transitional
epithelium in the urinary tract have been found in both guinea-pigs
(Gupta et al., 1973; McConnell et al., 1978b) and monkeys (Norback &
Allen, 1973).
Reduced or unaffected spleen weight and slight to moderate loss
of lymphocytes from spleen germinal centers have been common findings
in laboratory animals exposed to sublethal to lethal doses of TCDD
(Gasiewicz et al., 1980; Greig et al., 1973; Kociba et al., 1978;
McConnell et al., 1978a; Olson et al., 1980b; Vos et al., 1973, 1974).
Spleen cellularity in C57BL/6 mice was decreased 14 and 21 days after
treatment with 30 µg TCDD/kg body weight (Chastain & Pazdernik, 1985),
but not in B6C3F1 or DBA/2 mice 7 days after oral doses of up to 10 µg
TCDD/kg body weight (Luster et al., 1984).
The thyroid weight, both absolute and relative to body weight, of
the rat has been found to be increased by TCDD-treatment (Bastomsky,
1977; Potter et al., 1983, 1986b). Degenerative changes in the
epithelial cells of the thyroid gland were observed in rats after 31
daily oral doses of 1 µg TCDD/kg body weight (Gupta et al., 1973) and
7 days after a single ip dose of 150 µg TCDD/kg body weight (Rozman et
al., 1986). However, Potter et al., (1986b) found no consistent
changes in follicle size or colloid content, variation in follicle
size, height of follicular epithelium, or resorption of colloid at the
periphery of follicles in rats one week after an oral dose of TCDD in
the range 6.25 to 100 µg/kg body weight.
Histological changes in the pancreas and in the interscapular
brown adipose tissue have been found in Sprague Dawley rats exposed to
single ip doses of 150 µg TCDD/kg body weight (Rozman et al., 1986).
Lesions of the adrenal glands have been observed in mice and
guinea-pigs treated with (Gupta et al., 1973; McConnell et al.,
1978b).
Acute exposure to lethal doses of TCDD has produced minor
haematological alterations in all species studied. Anaemia was not
observed in mice or guinea-pigs (Zinkl et al., 1973), but a moderate
anaemia and leukocytosis occurred in rats (Buu-Hoi et al., 1972a) and
monkeys (McConnell et al., 1978a). On the contrary, haemoconcentration
was observed in rats after exposure to a somewhat lower but still
lethal dose of TCDD (Greig et al., 1973; Zinkl et al., 1973).
Thrombocytopenia and clotting abnormalities were observed in rats
after acute exposure to lethal levels of TCDD (Weissberg & Zinkl,
1973). Hypocellularity of the bone marrow was found in guinea-pigs
(McConnell et al., 1978b), rhesus monkeys (Allen et al., 1977;
McConnell et al., 1978a), and mice (McConnell et al., 1978b). However,
in these studies decreased bone marrow cellularity, as judged by
histological examination, appeared only at doses high enough to cause
severe toxicity in the experimental animals. More recent studies
(Chastain & Pazdernik, 1985; Luster et al., 1980, 1985) have
demonstrated that collection and enumeration of bone marrow cells in
suspension provides a more sensitive and quantitative method than
histological preparation for assessing bone marrow cellularity.
Decreased bone marrow cellularity was found in adult male C57Bl/6 mice
3 days after exposure to 120 µg TCDD/kg body weight (Chastain &
Pazdernik, 1985), and in female B6C3F1 mice 5 days after exposure to
10 µg TCDD/kg body weight (Luster et al., 1985), but not in female
DBA/2 mice, even at a dose of 50 µg TCDD/kg body weight (Luster et
al., 1985). The myelotoxic potential of TCDD is described in section
7.4.6.
Changes in clinical chemistry, including serum enzyme activities,
serum protein concentrations, and lipid levels, observed in animals
after acute exposure to TCDD, primarily reflect damage to other organ
systems, mainly the liver (McConnell et al., 1978a; McConnell & Moore,
1979; Olson et al., 1980b). Liver-related enzyme activities in serum
are affected in those animal species where liver damage is a prominent
sign of TCDD toxicity. In those animal species where hepatotoxicity is
not as apparent, such as monkeys and guinea-pigs, these enzyme
activities are essentially normal. Also the TCDD-related decrease in
serum albumin seems to be secondary to the hepatotoxic effect, since
the decrease is less evident or non-existent in those animals that
show little liver damage. Generally serum triglycerides and free fatty
acid levels are increased after TCDD exposure, while that of serum
cholesterol is decreased. However, marked species differences exist
and again these effects seem to be secondary to liver damage. For
further details on hyperlipidaemia, see section 7.4.7.
Indicators of renal damage such as blood urea nitrogen,
creatinine, and blood electrolytes are usually within normal limits
after TCDD exposure.
In view of the interspecies differences in the organ distribution
of TCDD and the variability in the effects on various organs in the
different species, the effects of TCDD in non-human primates are of
particular interest. Nine female juvenile rhesus monkeys (Macaca
mulatta) were divided into three groups of three and given TCDD in
a single oral dose of 0, 70, or 350 µg per kg/body weight (McConnell
et al., 1978a). The first indication of a toxic effect of TCDD, at day
3, was weight loss, followed by periorbital oedema, conjunctivitis,
and thickening of the Meibomian glands on day 12. Subsequently, the
eye lashes, facial hair, and toe and finger nails were lost. Monkeys
given the highest dose showed a moderate absolute lymphopenia and
thrombocytopenia. Serum cholesterol levels dropped, while the serum
triglyceride levels increased. Alkaline phosphatase and total
bilirubin were normal, but glutamic oxalic transaminase and aldolase
were increased. A decrease in the albumin fraction of the total serum
protein was noticed. The three monkeys given TCDD in a dose of 350
µg/kg body weight died or became moribund between days 28 and 34 after
administration. One of the monkeys given TCDD in a dose of 70 µg/kg
body weight died 14 days after administration. At autopsy body fat was
almost completely absent in all treated monkeys. Ascites was noticed
in two animals and all monkeys had markedly distended and thickened
bile ducts and gall-bladders. Small focal ulcerated areas in the
fundus of the stomach were observed in two monkeys. Microscopic
examination showed that the Meibomian glands were dilated and filled
with keratinaceous debris. Squamous metaplasia of the glandular
portion, with atrophy of sebaceous cells, was present. Occasional
scattered necrotic hepatocytes were noted in the liver on microscopic
examination. The gastric ulcers that were found extended into the
lamina propria.
Schwetz et al. (1973) reported that no signs of toxicity were
observed in four male mice and two female rats given single oral doses
of respectively 2 and 1 g 2,7-diCDD/kg body weight (purity
99.6-99.8%). For octaCDD (purity 98.86%), oral doses of 1 g/kg body
weight to five female rats and 4 g/kg body weight to four male mice
did not cause any toxic symptoms. A sample containing two different
isomers of hexaCDD (purity > 99%, 89:11) killed 1 of 2 and 0 of 2
male rats when given as single oral doses of 100 and 10 mg,
respectively. The only toxic sign observed among these rats was loss
of body weight. These studies lasted for 2 to 8 weeks.
The toxicities of single oral doses of nine PCDDs, including
TCDD, in C57BL/6J mice and Hartley guinea-pigs were compared by
McConnell et al. (1978b) (Table 49). The purity of the various isomers
tested exceeded 97%. Groups of eight male mice and of six male
guinea-pigs were used for each dose of any of the tested compounds,
and the animals were followed for 30 days after administration. The
toxic effects observed after administration of the different PCDDs
were similar, the only difference among the congeners being the amount
needed to produce a given effect. Progressive decrease of body weight,
more pronounced in guinea-pigs than in mice, was observed after a
lethal dose of any of the congeners tested. Marked reduction in
deposited adipose tissue deposits was a constant finding in animals
given a lethal dose of any of the PCDDs. Reduction of muscle mass and
severe dehydration were observed in guinea-pigs, and ascites,
subcutaneous oedema, and hydrothorax in some of the treated mice.
Decrease of thymus weight was a constant finding in both species,
being more pronounced in mice and in the guinea-pigs that died.
Histological examination of the thymus of the guinea-pigs that died as
early as 5 days after administration of PCDDs revealed scattered
necrosis of lymphocytes throughout the cortex, with concomitant
phagocytosis by macrophages. This was even more apparent in animals
that died 14 days after administration, in which a noticeable decrease
in the thickness of the cortex was observed. In the animals that had
died by day 20 it was difficult to differentiate the cortex from the
medulla, but little evidence of necrosis was present.
Table 49. Estimated single oral LD50 values for some PCDDsa
Chlorination Guinea-pigs Mice
of PCDDs (mg/kg)b (mg/kg)b
2,8 > 300 000 NR
2,3,7 29 444 > 3000
2,3,7,8 2 284
1,2,3,7,8 3 338
1,2,4,7,8 1125 > 5000
1,2,3,4,7,8 73 825
1,2,3,6,7,8 70-100c 1250
1,2,3,7,8,9 60-100c > 440
1,2,3,4,6,7,8 > 600 NR
a From: McConnell et al. (1978b).
b Spearman-Karber method.
c Estimated range due to variability in replicates.
NR = not reported.
In guinea-pigs that survived 30 days after a lethal dose of any
of the congeners, the thymus was reduced to one-fourth of its size in
controls. However, thymus histology at this time was often
comparatively normal. A reduction of the lymphoid follicles in the
spleen and of the Peyer's patches in the intestine was observed with
less conspicuous necrosis, which again was not evident in the 30-day
survivors. Striking hypocellularity was found in the sternal bone
marrow in the guinea-pigs that died, but was less obvious in
survivors. Similar thymic and splenic changes were found in mice.
However, bone marrow atrophy occurred only rarely in this species and
then it was less pronounced than in guinea-pigs. In the guinea-pigs
that died, and occasionally in survivors, a marked hyperplasia of the
renal pelvis was observed, invariably extending into the ureter and at
times involving the urinary bladder mucosa. Gastrointestinal
haemorrhages and occasional microscopic dilatation of crypts in the
glandular portion of the gastric mucosa were observed in dead
PCDD-treated animals of both species. Retro-orbital haemorrhages with
exophthalmus and haemorrhages with detachment of the retina were seen
in mice that died after a lethal dose of any of the congeners tested.
Adrenal haemorrhages and moderate atrophy of the zona glomerulosa were
seen in guinea-pigs that died. These changes were not observed in mice
or in surviving guinea-pigs. In guinea-pigs, primarily in animals that
died during the observation period, changes in the spermatogenic
epithelium were observed, with testicular tubules containing only
spermatogonia and Sertoli cells in severely affected animals. Reduced
spermatogenesis, necrotic spermatocytes, and spermatozoa within the
lumen of the testicular tubules and in the epididymis, and
multi-nucleated giant cells within the seminiferous tubules, were
found in the mice that died but not in those that survived. On the
other hand, liver lesions were observed with the same frequency and
degree of involvement in all the mice given the same dose, whether
they died or survived. Minimal liver changes were detected in
guinea-pigs, changes largely confined to central congestion with
occasional degeneration of hepatocytes in dead animals. Fluorescence,
as an indication of porphyria, was found only in mice, particularly in
the liver but also in the incisors, cranial bones, costochondral
junction, and stifle joint. It was dose related and detectable at
doses several times less than the LD50.
Haemolysis and hyperproteinaemia were found in dying animals of
both species. In mice surviving 30 days, but not in guinea-pigs, the
blood a-globulin level was decreased, with a resultant increase in the
albumin/globulin ratio.
With all PCDDs for which it was possible to establish an LD50
values were in the range 10 to 100 times lower in guinea-pigs than in
mice. To produce the greatest toxicity the lateral positions 2,3,7,
and 8 must be fully chlorinated. With 2,3,7-triCDD or 2,8-diCDD the
LD50 values were in the range 1000 to 100 000 times higher than with
TCDD. The addition of a chlorine atom at an ortho-position, e.g.,
1,2,3,7,8-pentaCDD, resulted in only a minor reduction of toxicity. An
additional chlorine atom further reduced the toxic potency but the
LD50 values of 1,2,3,4,7,8-hexaCDD and 1,2,3,6,7,8-hexaCDD remained
comparatively low. The toxicity of 1,2,3,4,6,7,8-heptaCDD was further
reduced. A reduction in the rate of weight gain was observable in
guinea-pigs given doses of this compound exceeding 200 µg/kg body
weight. However, no deaths were observed during the experiment even at
doses as high as 600 µg/kg body weight.
The acute oral toxicities of soot and benzene extracts of soot,
containing PCDDs and PCDFs from a fire in a PCB-containing transformer
(Binghamton, New York, USA) were determined to be 410 mg/kg body
weight and 327 mg equivalents/kg body weight, respectively, in female
Hartley guinea-pigs (Silkworth et al., 1982). The observation period
was 42 days. The test substances were given in 0.75% aqueous methyl
cellulose and the doses used were 250, 500, 750, 1000, and 1250 mg
soot/kg body weight and benzene extracts corresponding to 4, 20, 100,
500, and 1000 mg soot/kg body weight. An extensive investigation,
including pathology, haematology, and serum chemistry alterations, in
groups of six male and female Hartley guinea-pigs, 42 days after
single oral doses of 1, 10, 100, or 500 mg Binghamton soot/kg body
weight in 0.75% methyl cellulose, was reported by Silkworth et al.
(1982). Control animals received 500 mg activated carbon/kg body
weight in the same vehicle. No treatment-related differences were
observed at 1 and 10 mg soot/kg body weight. Decreased body weight
gain occurred in both sexes at 100 and 500 mg/kg, and decreased thymus
weight occurred at 500 mg/kg for males and at 100 and 500 mg/kg for
females. The kidney weight was decreased only in males at 100 and 500
mg/kg. There were no treatment-related alterations in haematological
values. Male guinea-pigs had significantly increased serum
triglyceride levels at 100 and 500 mg/kg and females at 500 mg/kg
only. Elevated aspartate amino transferase (at 100 and 500 mg/kg) and
decreased Y-glutamyltransferase (at 500 mg/kg) were observed in the
serum of female guinea-pigs. The only clearly dose-related
microscopical findings in soot-exposed guinea-pigs were metaplasia of
salivary gland interlobular duct epithelium and goblet cell
hyperplasia of pancreatic interlobular ducts. These lesions occurred
only in males at doses of 100 or 500 mg soot/kg body weight.
Microscopic lesions, which tended to be more frequent and/or severe in
treatment groups than in controls, included bile duct hyperplasia,
hepatocellular cytoplasmic inclusions, vacuolation of the adrenal
cortex, and focal lacrimal adenitis.
7.1.2 In vitro studies on mammalian cells
Over 30 cell types, including primary cultures and cells from
established and transformed cell lines derived from various tissues of
at least six animal species, have been examined for their response to
TCDD (Beatty et al., 1975; Knutson & Poland, 1980a; Niwa et al., 1975;
Yang et al., 1983). The effects studied were viability, growth rate,
and morphological alterations. No toxic effects were observed except
in one rat hepatoma cell line in which Niwa et al. (1975) reported
decreased viability after exposure for 72 h to TCDD at a concentration
of 300 nmol/litre. This concentration is high when compared to the
LD50 in rats and mice. No effect on these cells was observable at 30
nmol/litre.
The biochemical responses found in primary hepatocytes from rats
exposed to 10 µg TCDD/kg body weight in vivo was not present when
the hepatocytes were exposed in vitro to TCDD at 50, 100, or 200
nmol/litre for 48 h (Yang et al., 1983a).
7.1.3 Studies on birds
Chick oedema disease first gained attention in the United States
in 1957. An extensive outbreak among chickens occurred in that year in
Georgia (Firestone, 1973; Sanger et al., 1958; Simpson et al., 1959).
The cause of the disease was traced to the presence in the feed of
toxic components later identified as chlorinated dibenzo-p-dioxins.
TCDD was identified as one of the isomers in the mixture of
chlorinated dibenzo-p-dioxins capable of producing chick oedema
(Flick et al., 1973).
Clinical signs of chick oedema disease consist of dyspnea,
reduced body weight gain, stunted growth, subcutaneous oedema, pallor,
and sudden death. In young chickens gasping was the first noticeable
sign, followed by a waddling gait. Gross inspection of the birds at
autopsy revealed an increased amount of fluid in the pericardial sac
and pale livers with a mottled and irregular granular surface. In
advanced stages the chickens developed a distended abdomen filled with
fluid. Endotheliosis of the vascular system was observed at
microscopic examination, as well as pronounced proliferation of the
endothelium of the glomerular capillaries and necrosis of the liver
cells. Diseased chickens developed pulmonary oedema and perivascular
lymphocyte infiltration, as well as oedema of the cardiac muscle with
interstitial lymphocytic infiltration (Allen, 1964; Allen & Lalich,
1962, McCune et al., 1962; Simpson et al., 1959).
Experimentally, chick oedema has been produced with a single dose
of 25 to 50 µg TCDD/kg body weight (Greig et al., 1973). When mixtures
of tri- and tetraCDDs were fed at dietary concentrations of 0.01
µg/kg, the chickens developed oedema and 83% of them died (Flick et
al., 1972).
Potency for inducing chick oedema was compared for three
different PCDDs: TCDD (purity 91% and > 99%), hexaCDD (purity > 99%,
two isomers), and octaCDD (purity 98.86%) (Schwetz et al., 1973). In
the experiments, 3-day-old white Leghorn cockerels were exposed for 20
to 21 days to one of these congeners at several dose levels. Chick
oedema occurred in birds given oral doses of 1 or 10 µg TCDD/kg per
day, or of 10 or 100 µg hexaCDD/kg per day. Chick oedema was not
observed in chicks maintained on a diet containing 0.1 or 0.5%
octaCDD. The weight of the Bursa of Fabricius was significantly
decreased in 2-week-old white Leghorn cockerels decapitated 2 days
after three daily ip doses of 10 µg TCDD/kg body weight (Sawyer et
al., 1986).
7.1.4 Toxicity of metabolites
Not only has identification of several in vivo metabolites been
achieved, but the acute oral toxicity of TCDD metabolites excreted in
the bile of dogs has been studied in the guinea-pig (Weber et al.,
1982a). 3H-labelled TCDD was administered directly into the duodenal
lumen of two 1-year-old Beagle dogs in four portions of 1 to 2 mg at
time intervals of 2 to 7 days. Excretion of radioactive material from
pooled bile samples collected daily for 4 or 7 days, and thereafter in
pooled samples of 2 or 3 days, was performed with a method yielding
about 50% of the total radioactivity of the bile. The extracts
containing the metabolites were concentrated and dissolved in
1,3-propanediol for administration. Male Pirbright guinea-pigs were
used in a 5-week toxicity study. Five animals in each of four
dose-groups were given a single oral dose via gastric intubation. The
amount of TCDD metabolites was calculated by means of radioactivity
measurements to be 0.6, 6.0, 30.0, or 60.0 µg/kg body weight. Three
animals, one control and two in the high-dose group, died within 48 h
after administration. One death in the high-dose group was due to
gastric perforation during dosing. The other animals which died
exhibited histological lesions which, according to the authors, were
due to material coextracted from bile together with the metabolites.
The light microscopic examination of the liver, spleen, pancreas,
thymus, kidneys, lungs, and adrenals revealed no histological changes,
and no other toxic effects due to the metabolites of TCDD could be
recorded in this study. The authors concluded that TCDD metabolites
from the dog are at least 100 times less toxic to male guinea-pigs
than TCDD itself.
When the metabolites 2-hydroxy-3,7,8-triCDD and
2-hydroxy-1,3,7,8-tetra CDD were given as single ip injections of 100,
1000, or 5000 µg/kg body weight to young male Wistar rats, no decrease
in body weight gain was noted and thymic atrophy was not seen 14 days
later (Mason & Safe, 1986). When compared to TCDD,
1-hydroxy-3,7,8-triCDD was at least 3 orders of magnitude less
effective in inducing hepatic AHH and EROD activities, whereas
2-hydroxy-1,3,7,8-tetra CDD was inactive at all dose levels tested.
7.1.5 Modulation of the acute toxicity
Several studies have been performed which attempt to modify the
acute toxicity of TCDD. Manara et al. (1984) studied the effect of
activated charcoal or cholic acids in the diet on the mortality after
60 days and mean time to death in mice, rats, and guinea-pigs exposed
to TCDD. Dietary levels of charcoal (2.5%), cholic acid (0.25%), and
dehydrocholic acid (0.5%) decreased the mortality induced in C57Bl/6
mice by a single sc dose of 110 mg TCDD/kg body weight from 93% to
53, 21, and 53%, respectively. The mean time to death was prolonged
from 29 days with a normal chow diet to 35-48 days with the above
dietary additives. In CD rats the addition of charcoal (2.5%) and
cholic acid (0.15%) decreased mortality from 80% to 50 and 70%
respectively. The mean time to death was not affected. Charcoal (5%)
added to guinea-pig chow decreased TCDD induced mortality from 64% to
29% and reduced the mean time to death from 29 to 14 days. These
additions to the diet did not prevent body weight loss or liver
enlargement in the mice or guinea-pigs (Manara et al., 1984), but 5%
charcoal in the diet protected against TCDD-induced thymus athrophy in
C57Bl/6 mice 14 days after a single oral dose of 10 mg TCDD/kg body
weight (Manara et al., 1982). The addition of 5% n-hexadecane in the
diet increased the TCDD induced mortality from 60% to 100% and the
mean time to death, within the 50 day study, from 19.8 to 28.8 days in
Sprague Dawley rats treated with a single ip dose of 60 mg TCDD/kg
body weight (Rozman, 1984). N-hexadecane itself did not affect
animal viability.
Increased survival times have been demonstrated in mice receiving
daily injections of triiodothyronine (T3) after TCDD treatment (Neal
et al., 1979) and in rats thyroidectomized before TCDD treatment
(Rozman et al., 1984). Although thyroidectomy increases the mean time
to death, it does not prevent TCDD-related mortality in rats (Rozman
et al., 1985a). Thyroidectomy has been demonstrated to counteract
TCDD-induced decreases in thymus weight and to reduce the spleen
plaque-forming cell response (Pazdernik & Rozman, 1985). A number of
hepatic enzyme activities were induced by TCDD to an equal magnitude
in thyroidectomized and normal rats (Rozman et al., 1985b). It has
been suggested that TCDD-treatment of rats leads to hypothyroidism, a
possible protective mechanism against TCDD toxicity (Pazdernik &
Rozman, 1985). However, available data on changes in T3, thyroxine
(T4), and thyroid-stimulating hormone (TSH) levels in TCDD-treated
rats are not sufficient to state whether the animals are functionally
hypothyroid, euthyroid, or hyperthyroid. Results presented by Potter
et al. (1986b) (see section 8.4.9) suggest that TCDD-treated rats
remain essentially euthyroid and that the altered thyroid status is
neither a major contributor to TCDD toxicity nor a key response to
TCDD exposure.
Daily injections of butylated hydroxyanisole protected against
TCDD-induced lethality in female Sprague Dawley rats, whereas vitamins
E and A, two other antioxidants, did not (Hassan et al., 1985a,b).
Dietary selenium, if given in an optimal dose, provides partial
protection from the lethal effects of TCDD in female Sprague Dawley
rats (Hassan et al., 1985c). None of these treatments could counteract
the TCDD-induced body weight loss (Hassan et al., 1985a,b,c).
7.2 Short-term Toxicity
7.2.1 Studies on rats
Data from the earliest subchronic laboratory study, in which rats
were exposed to daily and/or weekly doses of TCDD, were reported in
four separate papers (published simultaneously) covering general
effects (Harris et al., 1973), pathology (Gupta et al., 1973),
haematological and clinical chemistry changes (Zinkl et al., 1973),
and immuno-biological effects (Vos et al., 1973). Female CD rats were
given TCDD by gavage in daily doses of 0.1, 1, or 10 µg/kg body
weight for 31 days. The body weight of animals exposed to the highest
dose started to decrease within the first week of exposure and 15/16
animals died or became moribund 17 to 31 days after administration of
the compound began. Pathological changes were comparable to those
observed in rats given a single lethal dose, and included severe
thymic atrophy and liver damage, icterus, haemorrhages in various
organs, and the depletion of lymphoid organs. Weight gain was also
reduced at the daily dose level of 1 µg/kg body weight. However, there
were no deaths and in the animals that were killed moderate thymic
atrophy, slight to moderate liver damage, and, in some of the
animals, degenerative changes in the kidneys and in the thyroid gland
were reported. The weight gain was not affected and significant
histopathological changes were not found in rats that received 0.1
µg/kg body weight per day. A decrease in thymic weight that was
significant on day 24, was observed at the lowest exposure level
(Gupta et al., 1973; Harris et al., 1973). Blood samples were
collected 3, 6, 10, 13, 17, 24, and 31 days after administration of
TCDD began. Serum enzyme activities related to liver damage began to
increase after 10 days of exposure and remained high until death
occurred in the 10 mg TCDD/kg per day group. This group of animals
also exhibited increased serum bilirubin levels commencing on day 13.
These parameters were only slightly affected in rats receiving 1 µg
TCDD/kg per day. Thrombocytopenia occurred at all dose levels. After
3 days of treatment with either 1 or 10 µg/kg per day, animals had
depressed platelets counts that remained low throughout the study. In
the low-dose rats, platelets were significantly decreased by day 17.
No significant leukocytopenia or lymphocytopenia occurred in rats at
any dose level. These results are in good agreement with the results
from a more detailed haematological study on female CD rats given
daily oral doses of 10 µg TCDD/kg body weight for 10 and 14 days
(Weissberg & Zinkl, 1973).
When oral doses of 0.02, 1.0, or 5.0 mg TCDD/kg body weight were
given weekly to groups of 10 female CD rats for 6 weeks, all the
animals survived (Harris et al., 1973). However, body weight gain
decreased in the 5.0 µg/kg group during the exposure period, and at
the end of this period the thymus/body weight ratio was approximately
50% of the ratio found in the controls. Liver damage was reported as
slight at this dose level. No effect on body or thymic weight and no
significant histopathological changes were observed in rats given
1 µg/kg body weight or less.
Adult male and female Sprague Dawley rats, in groups of 12, were
given 0, 0.001, 0.01, 0.1, and 1.0 µg TCDD/kg body weight by gavage,
5 days per week for 13 weeks (Kociba et al., 1976). At the end of the
treatment period, five rats of each sex were killed for
histopathological examination, while the remaining animals were
retained for post-exposure observation. Doses of 1 µg TCDD/kg body
weight per day caused five deaths in females, with three occurring
during treatment and two after treatment, and two deaths in males,
both occurring in the post treatment period. Decreased body weights
and food consumption were found at the two highest dose levels both in
males and females. Decreased relative thymus weight and increased
relative liver weight occurred only in the males given the highest
dose but in both the 0.1 and 1.0 µg/kg female groups. Male rats had
significantly depressed haematological values (packed cell volume, red
blood cell count, and haemoglobin) in the two high-dose groups, while
these values were normal in all female rats. Gross, as well as
histological, examination revealed treatment-related effects only in
the high-dose groups with some minor findings in the 0.1 µg/kg group.
Subcutaneous oedema, decreased sizes of testes and uteri, and a
decreased number of corpora lutea were found at necropsy. Histological
findings were limited to lymphoid tissues, liver, and epithelial
linings. The lymphoid tissues, including thymus, were depleted of
lymphocytes. The liver of both male and female rats showed pleomorphic
and multinucleated hepatocytes. Foci of necrosis, with focal
reticuloendothelial aggregations in the areas of parenchymal cell
necrosis, were observed. Hyperplasia of Kupffer cells and an increased
amount of a golden-brown pigment were noted. The hepatic changes were
more pronounced near the periphery of the lobules. Slight hyperplasia
of bile ducts and ductular epithelium was present. The uterus was
lined by cuboidal epithelium in the female rats. The rats given 0.01
mg TCDD/kg did not differ from the controls in any of these
parameters, except for a slight increase in the mean liver to body
weight ratio.
Goldstein et al. (1982) exposed groups of eight female Sprague
Dawley rats to 16 weekly oral doses of 0, 0.01, 0.1, 1.0, and 10.0 µg
TCDD/kg body weight in a study (further discussed in section 8.4.3)
primarily aimed at investigating TCDD-induced porphyria. All animals
given the highest dose died, or were moribund after eight to twelve
doses and were killed. A decrease in body weight gain was seen in this
group within one week of treatment. Decreased body weight gain was
observed also in the 1.0 µg TCDD/kg per week group, but not until
several weeks after the start of treatment. Hepatic porphyria was
found in 7 out of 8 animals receiving weekly doses of 1.0 µg TCDD/kg
body weight, in 1 out of 8 receiving 0.1 µg/kg per week, and in none
of the animals receiving 0.01 µg/kg per week or the lethal dose of
10.0 µg/kg per week. Porphyria was not reversed after six months
recovery from a 16-week exposure to 1.0 µg/kg body weight per week.
Feeding male Wistar rats (110 g) 0, 0.2, or 1.0 µg octaCDD/kg
diet for two weeks, resulting in a total intake of 0, 22.7 (± 1.0) or
120.7 (± 2.8) mg octaCDD, had no effect on body weight gain, feed
consumption, or tissue weights (liver, thymus, testes, heart, and
kidney) (Williams et al., 1972). Congestion of the liver occurred in
the high-dose group.
Daily doses of 100 mg octaCDD for 21 days produced no effects on
appearance, activity, or eating habits in male Sprague Dawley rats,
but slightly increased relative liver weight. A moderate increase in
the hepatic smooth endoplasmic reticulum was noted (Norback et al.,
1975).
7.2.2 Studies on mice
Oral doses of 0.2, 1, 5, or 25 µg TCDD/kg body weight were given
in corn oil to male C57Bl/6 mice weekly for four weeks (Harris et al.,
1973; Vos et al., 1973). One animal of the 25 µg/kg group died after
24 days. Significant weight loss was observed only in the high-dose
group. Thymic atrophy, characterized by nearly complete loss of
cortex, occurred in the 5 and 25 µg TCDD/kg body weight groups.
7.2.3 Studies on guinea-pigs
All 10 female Hartley guinea-pigs that received weekly oral doses
of 1 µg TCDD/kg body weight died, or were killed when moribund,
between days 24 and 32 after the first dose (Gupta et al., 1973;
Harris et al., 1973; Vos et al., 1973). Light microscopic findings of
moribund or dead animals revealed severe atrophy of the cortex of the
thymus with destruction of lymphocytes. There was lymphoid cell
depletion in spleen and lymph nodes. Haemorrhages, mitotic figures,
and loss of lipid vacuoles were observed in the adrenals. Liver
effects were restricted to diffuse single-cell necrosis, predominantly
in the periportal area. Haemorrhages were found in the urinary bladder
and gastrointestinal tract. The lymphocyte count was decreased at all
doses, whereas total leukocyte values were decreased at doses > 0.04
µg/kg. Animals that received eight weekly doses of 0.008, 0.04, or 0.2
µg TCDD/kg body weight all survived. At the 0.2 µg/kg dose level,
decreased body weight gain and decreased relative thymus weight were
observed.
A 90-day feeding study of TCDD in male (250-370 g) and female
(230-340 g) Hartley guinea-pigs was performed by DeCaprio et al.
(1986) and included extensive pathology, haematology, and serum
chemistry on surviving animals. The diets contained 0, 2, 10, 76, or
430 ng TCDD/kg. Animals that received the highest dose exhibited
severe body weight loss, decreased feed consumption, and mortality.
When 60% mortality was reached, on day 46 for males and day 60 for
females, the remaining animals in these groups were sacrificed. The
estimated total TCDD consumption at that time was 1.3 and 1.9 µg
TCDD/kg body weight for males and females, respectively. No
treatment-related mortality was observed at the 76 ng/kg dose level,
which corresponded to a total estimated intake of 0.44 µg TCDD/kg body
weight over the 90 days. Decreased body weight gain and increased
relative liver weight were seen in both sexes, whereas reduced
relative thymus weight occurred in males only. At doses of 2 and 10
ng/kg diet, no dose-related alterations were observed. The only
treatment-related effect on haematology and serum chemistry parameters
was the elevation of serum triglycerides for male guinea-pigs at the
76 ng/kg dose level. The presence of hepatocellular cytoplasmic
inclusion bodies in female guinea-pigs was the only significant
microscopical finding, except for thymus atrophy. Based on this
study, a no-observed-effect level of 0.6 ng TCDD/kg body weight per
day in guinea-pigs was estimated.
DeCaprio et al. (1986) also followed the body weight changes and
mortality during and after feeding male Hartley guinea-pigs (250-360
g) a diet containing 430 ng TCDD/kg. The diet was fed for 11, 21, or
35 days and was then withdrawn during a 79, 69, or 55-day recovery
period. The rate of change of weight per day was the same as that of
animals on a control diet, after an initial weight loss of
approximately 10% during the first 5 days. When animals were fed the
TCDD-diet for 21 and 35 days, a significant mortality, 10% and 70%,
respectively, was apparent. Both body weight gain and absolute body
weight were severely depressed in surviving animals throughout the
study. Animals destined to die generally lost more than 20% of the
original weight, whereas less pronounced weight losses were usually
followed by increases in body weight during the recovery period.
A PCB-containing transformer fire at the State office building,
Binghamton, New York, USA, resulted in contamination of the building
with soot-like material containing various PCDDs and PCDFs (see
section 4.5.10). This soot, mixed with diet, was fed to groups of 10
male (250-350 g) and female (200-350 g) Hartley guinea-pigs for 90
days (0, 0.2, 1.9, 9.3, and 46.3 mg soot/kg diet) or 32 days (231.5 mg
soot/kg diet) (DeCaprio et al., 1983). The total intake of soot during
the study corresponded to approximately 0.3, 3, 13, 67, and 100% of
the LD50 dose of Binghamton soot in guinea-pigs (Silkworth et al.,
1982) (see section 8.1.1.1). A dose-related decrease in body weight
gain occurred at 9.3 and 46.3 mg soot/kg diet. Body weight loss and
decreased feed intake was evident in the animals given 231.5 mg
soot/kg diet. Three male and three female guinea-pigs given 46.3 mg
soot/kg died. Seven animals in the highest-dose group died within 28
to 31 days and the remaining moribund animals were killed on day 32.
Gross pathology revealed no effects at 0.2 mg soot/kg diet. Thymic
atrophy occurred only in males at 1.9 mg soot/kg diet, but in both
sexes at higher doses. The relative spleen weight was significantly
increased at 46.3 mg TCDD/kg in both sexes. Treatment-related
microscopical findings included metaplasia of salivary gland epithelia
(> 1.9 mg soot/kg diet), increased number of goblet cells in
pancreatic ducts (> 46.3 mg soot/kg diet for males), focal lacrimal
gland adenitis (> 9.3 mg soot/kg diet for males and > 46.3 mg
soot/kg diet for females), depletion of haematopoietic cells from the
bone marrow (> 46.3 mg soot/kg diet for females, > 231.5 mg soot/kg
diet for males), and hepatocellular cytoplasmic inclusion bodies (9.3
and 46.3 mg soot/kg diet in both sexes). Fatty infiltration of the
liver, reduced thickness of thymic cortex, and degenerative changes of
the stomach and intestine were observed only in high-dose animals.
Haematological alterations were observed only in animals at the 46.3
mg soot/kg diet dose level, whereas alterations in serum chemistry
values were found also at lower levels. Toxic effects of feeding
Binghamton soot for 90 days were similar to the effects occurring
after acute exposure (Silkworth et al., 1982) (see section 8.1.1.1),
but the effects were seen at a lower total dose after subchronic
exposure than after acute dosing. The effect seen at the lowest level
in this study was thymic atrophy at 1.9 mg soot/kg diet, which was
equivalent to 7.8 ng TCDD/kg body weight per day. A comparison between
the effects of feeding pure TCDD and TCDD-contaminated soot to
guinea-pigs (DeCaprio et al., 1986) demonstrated that pure TCDD
produced less variability of alterations and gave a steeper
dose-response relationship for many effects. Exposure to Binghamton
soot characteristically resulted in salivary gland duct metaplasia and
decreased serum sodium and potassium levels (DeCaprio et al., 1983).
When male guinea-pigs (200 g) were fed a diet containing 2.5%
HCl-pretreated fly ash from a municipal incinerator (Zaanstad, The
Netherlands) for up to 95 days, the animals exhibited progressive
weight loss, hair loss, and increased relative liver weight (van den
Berg et al., 1986b). One animal died on day 76.
Table 50. Studies on long-term exposure (excluding cancer studies) to TCDD in laboratory animals
Species/Strain Sex/number/ Doses tested Treatment Parameters
groupd schedule monitored
Rats
Sprague Dawleya M/10 0 ng/kg in diet survival
1 ng/kg continuously
5 ng/kg for 65 weeks
50 ng/kg
500 ng/kg
1000 ng/kg
5000 ng/kg
50 000 ng/kg
500 000 ng/kg
1 000 000 ng/kg
Sprague Dawleyb M and F/10 0.001 µg/kg per day in diet extensive
0.01 µg/kg per day continuously histopathology,
0.1 µg/kg per day for 2 years haematology and
clinical chemistry
Mice
Swiss M/38-44 0 µg/kg/week by gavage weekly histopathology
0.007 µg/kg per week for 1 year
0.7 µg/kg per week
7.0 µg/kg per week
Monkeys
Macaca mulattac F/8 500 ng/kg continuous in extensive
the diet for histopathology,
9 months haematology and
clinical chemistry
a Van Miller et al. (1977). b Kociba et al. (1978, 1979a,b).
c Allen et al. (1977). d M = male; F = female.
7.2.4 Studies on hamsters
No toxic effects were reported in male Golden Syrian hamsters
(50-70 g) given a diet containing 2.5% HCl-pretreated fly ash from a
municipal incinerator in Zaanstad, The Netherlands for up to 95 days
(van den Berg et al., 1986b).
7.2.5 Studies on monkeys
A cumulative dose of 0.2 µg TCDD/kg body weight, divided into
nine oral doses given three times per week, produced no clinical
toxicity in female rhesus monkeys (Macaca mulatta) (McNulty,
1984). However, clearly toxic signs did occur in those monkeys that
received cumulative doses of 1.0 and 5.0 µg TCDD/kg body weight over
the same time period. The first signs were thickening and reddening of
the eyelids, followed by weight loss, dryness and granularity of the
skin, and loss of hair, and in some cases anaemia, purpura, and
bleeding from the nose and mouth. Animals that died showed squamous
metaplasia of the sebaceous glands, mucous metaplasia of the gastric
mucosa, hyperplasia of biliary ductal epithelium, gingivitis, and
hypoplasia of the bone marrow. The times to death after dose were 65
and 116 days at 5 µg/kg and 130 to 211 days at 1 µg/kg.
7.3 Long-term Toxicity
Chronic toxicity studies performed on laboratory animals exposed
to TCDD are summarized in Table 50. Studies on carcinogenicity are
presented in section 7.7.
7.3.1 Studies on rats
In studies by Van Miller et al. (1977), male Sprague Dawley rats
were maintained in groups of 10 on diets containing 0, 1, 5, 50, 500,
1000, 5000, 50 000, 500 000, and 1 000 000 ng TCDD/kg for 65 weeks and
survival was monitored. At the five highest dose levels, all animals
died before the study was completed. The first deaths in these treated
groups occurred by weeks 31, 31, 3, 2, and 2 of treatment,
respectively. Groups receiving 50, 500, or 1000 ng TCDD/kg in the diet
died from acute toxic effects including severe liver necrosis, bile
duct hyperplasia and oedema, atrophy of spleen and thymus, and
gastrointestinal haemorrhages.
Groups of 50 male and 50 female Sprague Dawley rats were fed
diets providing daily doses of 0.001, 0.01, and 0.1 µg TCDD/kg body
weight for 2 years (Kociba et al., 1978, 1979a,b). Control rats, 86
males and 86 females, received diets to which the vehicle acetone had
been added. The dose levels corresponded to a dietary content of 22,
208, and 2193 ng TCDD/kg feed. Increased mortality was observed in
females given 0.1 µg/kg per day, while no increased mortality was
observed in male rats at this dose or in animals receiving doses of
0.01 or 0.001 µg/kg per day. From month 6 to the end of the study the
mean body weights of males and females decreased at the highest dose
and to a lesser degree in females given 0.01 µg/kg per day. During the
course of the study, subnormal body weights were occasionally also
recorded in the low-dose group, although during the last quarter of
the study the body weights were similar to those of the controls.
Increased urinary coproporphyrin and uroporphyrin were noted in
females, but not in males, given TCDD at a dose rate of 0.01 and 0.1
µg/kg per day. Analyses of blood serum collected at terminal necropsy
revealed increased enzyme activities related to impaired liver
function in female rats given 0.1 µg TCDD/kg per day. Necropsy
examination of the rats surviving TCDD exposure to the end of the
study revealed that liver effects constituted the most consistent
alteration in both males and females. Histopathological examination
revealed multiple degenerative inflammatory and necrotic changes in
the liver that were more extensive in females. Multinucleated
hepatocytes and bile duct hyperplasia were also noted. Liver damage
was dose-related and no effect was observable at the lowest dose
studied.
7.3.2 Studies on mice
Weekly oral doses of 0, 0.007, 0.7, and 7.0 µg TCDD/kg body
weight for 1 year resulted in amyloidosis and dermatitis in male Swiss
mice (Toth et al., 1979). The incidence of these lesions was 0/38,
5/44, 10/44, and 17/43 in the control, low-, medium-, and high-dose
groups, respectively.
7.3.3 Studies on monkeys
In a study by Allen & Carstens (1967) groups of four to five
rhesus monkeys were fed diets containing 0, 0.125, 0.25, 0.5, 1.0, and
10.0% of fat (which had been shown to be toxic to chickens) until
death. The "toxic fat" was later demonstrated to contain various
PCDDs, of which 65% by mass of the total PCDDs present was TCDD
(Norback & Allen, 1973). The survival time became shorter with
increasing doses of "toxic fat". Mean time to death was 445 days for
the low dose and 91 days for the high dose. Decreased food consumption
and progressive body weight loss, compared with controls, were noted.
Both clinical and histological changes near the time of death appeared
similar regardless of dose. The monkeys developed subcutaneous oedema,
progressing from the eyelids and face, ascites, and hydropericardium.
Characteristic skin changes were observed as well as anaemia,
leukopenia, and hypoproteinaemia. The bone marrow was hypoplastic.
Centrilobular necrosis, bile duct hyperplasia, and multinucleated
hepatocytes were found in the liver. In more than half of the
animals, there was marked hypertrophy of the gastric mucosa, with
crypts and mucin-containing cysts penetrating into the submucosa.
Ulcerations in the fundic and pyloric regions were observed.
Allen et al. (1977) fed eight adult female rhesus monkeys a diet
containing 500 ng TCDD/kg for 9 months. There after, surviving animals
were removed from the TCDD diet and were observed for another 4
months. No control animals were included, and so data were compared
with pre-exposure values where possible. During the first 3 months of
exposure animals developed periorbital oedema, acne, and loss of
facial hair and eyelashes. By 6 months these changes were quite
prominent in six out of eight monkeys, and a decrease in haemoglobin
haematocrit was noticed. The animals lost weight even though their
food intake was unaltered. Two animals died within the 9-month
exposure period and three monkeys continued to develop toxic symptoms
and died after 3 months on a TCDD-free diet. The three surviving
animals continued to experience periorbital oedema and loss of hair.
The total intake of TCDD over the 9-month period was calculated to be
2-3 µg/kg body weight. Death was preceded by severe anaemia, a
decreased white blood cell count, and severe thrombocytopenia. Autopsy
findings included haemorrhage into a variety of organs, ascites, and
subcutaneous oedema. Hypertrophy, dilatation, oedema, and hydropic
degeneration of the myocardium were noted in all animals. The biliary
ducts showed marked dilatation. Moderate hyperkeratosis of the skin,
with cystic keratosis of the hair follicles, was noted, and
hypocellularity of the lymphoid tissue and the bone marrow were
observed. The hyperplastic mucous-secreting cells of the gastric
epithelium had invaded the submucosa, and ulceration and mucinous
cysts were common in the modified gastric mucosa. Hypertrophy and
hyperplasia of the epithelial lining of the biliary system were
present. The bronchial epithelium, salivary glands, bile ducts, and
pancreatic ducts showed metaplastic changes. Death was attributed to
complications from the severe pancytopenia. The same pattern of
morphological changes were reported to occur in a similar study
performed by Barsotti et al. (1979).
Similar, though less severe, effects were observed in four adult
female rhesus monkeys fed a diet of 50 ng TCDD/kg for 20 months
(Schantz et al., 1978).
7.4 Effects Detected By Special Studies
7.4.1 Wasting syndrome
TCDD causes a starvation-like or wasting syndrome in several
animal species. In young animals, or following a sublethal dose in
adults, this response is manifested as a cessation of weight gain.
Early studies suggested that acute or chronic treatment with TCDD
decreased food consumption, but insufficiently to account for the
weight loss (Allen et al., 1975, 1977; Greig et al., 1973; Harris et
al., 1973; Kociba et al., 1976; McConnell et al., 1978a,b). To
elucidate whether malabsorption could explain the wasting syndrome,
the transfer of a number of nutrients has been studied with everted
intestinal sacs from TCDD-treated rats. A transient increase in the
serosal transfer of 59Fe in Sprague Dawley rats was reported by
Manis & Kim (1979). Absorption of glucose (Ball & Chabra, 1981; Madge,
1977) and lipids (Shoaf & Shiller, 1981) was decreased by TCDD
treatment. The absorption of cobalt, galactose, and proline (Manis &
Kim, 1977) as well as of D-galactose, L-arginine, L-histidine (Madge,
1977), and penicillin (Manis & Apap, 1979) was reported to be
unaffected by TCDD treatment. Leucine transport was depressed in
Sprague Dawley rats 4 h after a single oral dose of 100 µg TCDD/kg
body weight (Ball & Chabra, 1981), whereas no effect was observed in
Fisher rats 7 days after exposure to 80 µg TCDD/kg body weight
(Schiller et al., 1982). Neal et al. (1979) found normal absorption
and intermediary metabolism of glucose, L-alanine, and oleate in
guinea-pigs treated with a single oral dose of 2 µg TCDD/kg body
weight. Apparently there was no generalized impairment of intestinal
absorption. The effects reported may well be secondary to decreased
food consumption which by itself causes structural changes in the
intestine (Steiner et al., 1968) as well as impaired absorption of
nutrients (Esposito et al., 1967).
The connection between the wasting syndrome and the lethal effect
of TCDD has been investigated in pair-feeding and forced nutrition
studies. Courtney et al. (1978) fed TCDD- treated female Wistar rats
a normal pelleted diet ad libitum. Supplementation with water,
electrolyte solution, or liquid diet, administered by gavage, could
not reverse or change the pattern or extent of TCDD-induced weight
loss or mortality.
To bypass gastrointestinal absorption, Gasiewicz et al. (1980)
fed rats intravenously with total parenteral nutrition (TPN). Rats
that had received a single ip dose of 100 µg TCDD/kg body weight
gained weight similarly to their TPN-fed controls, yet still died at
days 13 to 17 following treatment. TCDD-treated rats fed a chow diet
ad libitum lost weight progressively (as compared to pair-fed
controls that maintained their starting weight) and died at days 11 to
20. In TPN-fed TCDD-treated rats, liver damage was more severe and fat
depots were increased as compared to chow-fed TCDD-treated animals.
Similar results were obtained with TPN-fed male Hartley guinea-pigs
treated with a single ip dose of 2 µg TCDD/kg body weight in olive
oil, when compared to TPN-fed control guinea-pigs (Lu et al., 1986).
Similar signs of toxicity were present in TPN- and ad libitum-fed
TCDD-treated guinea-pigs. In contrast to TPN-fed rats (Gasiewicz et
al., 1980), TCDD- treatment in TPN-fed guinea-pigs only produced mild
hepatic changes, including increased liver lipid and cytochrome P 450
content, but no morphological changes (Lu et al., 1986).
Seefeld et al. (1984a) suggested that TPN-fed TCDD- treated rats
might have suffered from overnutrition and, secondary to that,
enhanced hepatotoxicity, as compared to chow-fed, TCDD-treated rats.
These same investigators have presented a heuristic model for the
TCDD-induced wasting syndrome based on the assumption that body weight
in rats is regulated around an internal standard or set point (Keesey
et al., 1976). Prevailing weight at a given age is constantly being
compared to this set point value and if differences occur, feed
consumption is adjusted so as to raise or lower body weight to match
the set point level. If TCDD lowers this setpoint, reduction in food
consumption would result, as the rat attempts to reduce its weight to
a new lower level of regulation determined by the dose of TCDD
administered. This hypothesis has been tested in several experiments
under carefully controlled feeding procedures.
Repeated studies have demonstrated that reduction of feed intake,
due to increased food spillage, is sufficient to account for the loss
of body weight in TCDD-treated Sprague- Dawley rats (Seefeld &
Peterson, 1983, 1984; Seefeld et al., 1984a,b). TCDD-treated rats
maintain and defend their reduced weight level with the same precision
as control rats (fed ad libitum) defend their normal weight level
(Seefeld et al., 1984b). The percentage of the daily feed intake that
is absorbed by the gastrointestinal tract of TCDD-treated and control
rats is similar (Potter et al., 1986a; Seefeld & Peterson, 1984).
Water intake, resting and total oxygen consumption, carbon dioxide
production, respiratory quotient, and spontaneous motoractivity were
decreased in a dose-dependent manner by TCDD-treatment (Potter et al.,
1986a; Seefeld & Peterson, 1983; Seefeld et al., 1984a). Urine output
was unaffected by TCDD-treatment, despite decreased water intake,
whereas urinary excretion of energy and urea were decreased and
urinary ammonia was increased (Potter et al., 1986a).
Hypophagia was the major cause of the loss of adipose and lean
tissue in male Fisher F-344 rats, C57Bl/6 mice, and albino guinea-pigs
when exposed to a calculated LD80 dose of TCDD (Kelling et al., 1985).
Body weight loss followed a similar time course in TCDD-treated and
pair-fed control animals of all three species. Lethalities for
TCDD-exposed rats, mice, and guinea-pigs were 95%, 69%, and 81%,
respectively, compared to lethalities in the appropriate pair-fed
controls of 48, 14, and 64%, respectively (Kelling et al., 1985).
Lethality and body weight loss followed almost identical
time-course-curves in Sprague Dawley rats that received a single oral
dose of 75 µg TCDD/kg body weight and in pair-fed controls (Christian
et al., 1986a). Thus the contribution to lethality made by body weight
loss seems to depend on the species and strain. Weight loss appears to
play a greater role in causing death in Sprague Dawley rats and
guinea-pigs than in Fisher F-344 rats and C57Bl/6 mice. Christian et
al. (1986a) demonstrated differences in organ weights and
histopathology in TCDD-treated and pair-fed animals, despite similar
time-courses and magnitude of body weight loss and lethality. Pair-fed
animals exhibited lesions in the gastro-intestinal tract, which were
absent in TCDD-treated rats, that may have contributed to death. The
hepatic carbohydrate, protein, and lipid metabolism was affected
differently in TCDD-treated and pair-fed Sprague Dawley rats
(Christian et al., 1986b). To distinguish direct effects of TCDD from
effects secondary to hypophagia, the studies by Christian et al.
(1986a,b) and Potter et al. (1986a) were performed with schedule-fed
animals. The reason for this was the finding that the 24-h feeding
pattern for TCDD-treated rats was different from the feeding pattern
for pair-fed controls, although it was similar to that of control rats
fed ad libitum. Decreased feed consumption did not contribute to
weight loss in C57BL/6 mice exposed to TCDD until the animals were
moribund (Chapman & Schiller, 1985).
Besides being typical signs of TCDD toxicity, loss of body weight
and appetite are also prominent signs of thyroid dysfunction (see
section 8.4.9). Serum glucose levels were also decreased by TCDD
independently of hypophagia, whereas the decrease in serum insulin
appeared to result from hypophagia, since it was seen in both
TCDD-treated and pair-fed controls. These results indicate that the
effect of TCDD on thyroid hormones cannot explain the TCDD-induced
decrease in body weight gain.
An interesting biochemical effect in TCDD-induced wasting is the
ability of TCDD to decrease hepatic vitamin A storage in rats (see
section 8.4.10). It has long been known that vitamin A is necessary
for growth and that vitamin A deficiency will result in depressed body
weight gain as well as in reduced food intake. However, the animal
continues to eat and grow though body weight gain is less than normal
(Brown & Morgan, 1948; Coward, 1947; Hayes, 1971; Orr & Richards,
1934; Patterson et al., 1942).
The effect of chemical structure on the ability of several PCDDs
to cause body weight loss in rats has been investigated (Mason et al.,
1986). Of the congeners studied, 2,3,7,8-TCDD was the most active.
Those congeners fully substituted in the 2,3,7, and 8 positions but
containing additional chlorosubstituents in the non-lateral 1,4,6, and
9 positions were less active.
7.4.2 Hepatotoxicity
TCDD produces hepatomegaly, due to hyperplasia and hypertrophy of
parenchymal cells, in all species that have been investigated, even at
sublethal doses. However, there is considerable variation between
species in the extent of this lesion. Other liver lesions are more
species specific.
Liver lesions alone cannot explain lethality following TCDD
administration, though it may be a contributing factor at least in the
rat and rabbit.
The morphological changes in the liver are accompanied by
impaired liver function, characterized by liver enzyme leakage,
increased microsomal monooxygenase activities, porphyria, impaired
plasma membrane function, hyperlipidaemia, and increased regenerative
DNA synthesis.
7.4.2.1 Morphological alterations
In Charles River rats given single oral sublethal doses of TCDD
(5 or 25 µg/kg body weight) a dose-related increase was observed 3
days after dosing in the amount of hepatic smooth endoplasmatic
reticulum (SER) around the periphery of cells, particularly in the
areas around bile canaliculi. The effect progressed by days 6 and 9,
when an increased amount of rough endoplasmatic reticulum (RER) was
also present. By day 28 these changes had returned essentially to
normal levels (Fowler et al., 1973).
The livers of CD rats given high sublethal doses of TCDD (5.0
mg/kg body weight per week) for six weeks showed transient
degenerative changes, followed by megalocytosis, regeneration, and the
occurrence of multinucleated giant hepatocytes (Gupta et al., 1973).
They also showed that the hepatotoxic reaction in rats given lethal
doses of TCDD (10 µg/kg body weight per day for 16-31 days) was
characterized by degenerative and necrotic changes, with the
appearance of mononuclear cell infiltration, multinucleated giant
hepatocytes, increased numbers of mitotic figures, and pleomorphism of
cord cells. These lesions were considered severe enough to be a
contributing factor to death.
Parenchymal cell necrosis was observed by Greig et al. (1973) in
Porton rats 3 weeks after exposure to an LD50 dose of TCDD. The
necrosis, which was located in the centrilobular zone close to the
central vein, became more severe with time.
Jones & Butler (1974) further investigated the time course of the
TCDD-induced liver lesions appearing in the centrilobular zone. They
confirmed the transient degenerative and inflammatory lesions
previously reported (Greig et al., 1973; Gupta et al., 1973). At the
ultrastructural level, consistent changes occurred in the cytoplasm
whereas normal nuclear morphology and division were found throughout
the study. Two weeks after a single oral dose of 200 µg TCDD/kg body
weight to Porton rats, extensive fusion of parenchymal cell plasma
membranes in the centrilobular zone was replaced by a diffuse zone
with islands of normal membrane occurring at intervals. Normal tight
and gap junctions were present in control animals and in periportal
areas of the test animals. These findings suggest that the
multinucleated cells occurring in TCDD-treated rats might form by
coalescence of parenchymal cells. The effect of TCDD on plasma
membranes demonstrates a specific subcellular site of action, which
might be involved in the toxic action of TCDD. This lesion was,
however, not observed until 2 weeks after treatment and thus could not
explain the immediate effects on, for instance, food intake, body
weight gain, and general health.
The time course for liver lesions in male Sprague Dawley rats
(200 g) given a single ip dose of 20 µg TCDD/kg body weight was
followed for up to 32 weeks by Weber et al. (1983). The lesions became
progressively worse up to the 16th week after injection, and
thereafter appeared to regress slowly. The lesions were almost
identical to those reported previously (Fowler et al., 1973; Jones &
Butler, 1974). The histological findings were accompanied by
hyperbilirubinaemia, hypercholesterolaemia, hyperproteinaemia, and
increased serum glutamicoxaloacetic transaminase and serum
glutamic-pyruvic transaminase activities, further indicating decreased
liver function (Greig et al., 1973; Zinkl et al., 1973).
Fewer studies of liver lesions produced by TCDD have been made in
other species than in the rat.
In C57BL/6 mice given single oral doses of 100, 150, or 200 µg
TCDD/kg body weight (Vos et al., 1974) and 250 µg TCDD/kg body weight
(Jones & Greig, 1975), centrilobular degenerative and necrotic changes
were present but multinucleated parenchymal cells were not seen.
Proliferation of the bile ducts and bile duct epithelial cells, as
well as lipid accumulation, have been observed, with a substantial
increase in the hepatic levels of esterified fatty acids and
cholesterol. Only slight damage, hepatocellular swelling, was reported
in CD-1 mice 21 days after a single dose of 50 µg TCDD/kg body weight,
and no histological changes were detected 7 or 35 days after
administration (Gupta et al., 1973).
The guinea-pig, while being very sensitive to TCDD, as indicated
by LD50 data, shows less severe morphological alterations in the
liver than do other species. No manifest liver lesions at the light
microscopical or ultrastructural levels has been found (Gupta et al.,
1973; McConnell et al., 1978b; Moore et al., 1979; Turner & Collins,
1983).
The hamster is very resistant to TCDD toxicity (Table 47) and
exhibits no manifest liver damage even after a fatal dose (Henck et
al., 1981; Olson et al., 1980b). However, Gasiewicz et al. (1986)
found bile duct hyperplasia, numerous inflammatory cells, and
increased number of multinucleated cells in the livers of male Golden
Syrian hamsters 35 days after they received a single ip dose of 500 µg
TCDD/kg body weight in olive oil.
7.4.2.2 Hepatic plasma membrane function
The morphological impairment of hepatic plasma membranes in the
centrilobular parenchymal cells of TCDD-treated Porton rats was
demonstrated histochemically to be preceeded by a loss of ATPase
activity (Jones, 1975). The effect occurred 3 days after treatment in
an area five to six cells deep around the central vein along the
canalicular borders, and became more severe with time. At the end of
the study (day 42 posttreatment) the ATPase activity was completely
abolished around the central vein, including the mid-zonal region, and
encroached on the periportal area in moribund animals. The loss of
ATPase activity was related to the clinical state of the animal. Thus
animals displaying minimal signs of intoxication retained the normal
distribution of ATPase in the periportal zone. In animals killed 9
months after treatment, partial restoration of the normal liver
architecture and the ATPase activity were evident.
Biochemical studies of isolated heptatic plasma membranes from
Holtzman rats treated with 10 or 25 µg TCDD/kg body weight revealed
depressed ATPase activities (Peterson et al., 1979a). The activity of
Na/K-ATPase was depressed to the same extent for both doses from day
2 to 40 after treatment, while a similar depression of Mg-ATPase
activity was observed only in the high-dose group. The Mg-ATPase
activity tended to recover by day 40, whereas Na/K-ATPase activities
did not. A pair-feeding experiment demonstrated that these effects
were independent of the TCDD-induced decrease in food consumption. In
vitro incubation of plasma membranes indicated that ATPase inhibition
did not occur by direct interaction with TCDD. Greig & Osborne (1981)
demonstrated a decrease in K/Mg-ATPase activity, but not in the
Na/K-ATPase or 5'-nucleotidase activities of hepatic plasma membranes
prepared from female Porton rats 6 and 11 days after a dose of 200 µg
TCDD/kg body weight.
Many physiological homeostatic mechanisms are dependent on proper
plasma membrane function and composition. Matsumura et al. (1984)
reported that a single ip dose of 25 µg TCDD/kg body weight to male
Sprague Dawley rats had reduced hepatic plasma membrane
ATPase-activities by 40% 10 days after treatment. The marker enzyme
for putative preneoplastic hepatocytes, glutamyl transpeptidase, was
reduced, while protein kinase (both c-AMP-stimulated and
c-AMP-nonstimulated) was increased (Matsumura et al., 1984). Both
c-AMP-dependent and c-AMP-independent protein kinase, in hepatic
plasma membranes from male Sprague Dawley rats treated with an ip dose
of 25 µg TCDD/kg body weight, were maximally increased on day 20 after
treatment (4.5- and 12-fold, respectively). The induction was
measurable within 2 days after the administration and was still
persistent after 40 days (Bombick et al 1985). Protein kinase C was
significantly increased in hepatic plasma membranes from Sprague
Dawley rats but not from guinea-pigs 10 days after single ip doses of
25 and 1 µg TCDD/kg body weight, respectively (Bombick et al., 1985).
TCDD treatment in vivo also affected the in vitro binding of
concanavalin A, epidermal growth factor, and insulin to their cell
surface membrane receptors.
The binding of glucagon and prostaglandin E were not affected by
a dose of 25 µg TCDD/kg body weight (Matsumura et al., 1984). Studies
with Sprague Dawley rats demonstrated that the TCDD-induced decrease
in epidermal growth factor binding (EGF) was observable within 2 days
after dosing, reached its maximum by day 20, and was still significant
40 days after dosing. The decrease was observable after a single ip
dose of 0.1 µg TCDD/kg body weight (Madhukar et al., 1984). The
relative doses of TCDD needed to suppress EGF binding to 50% of the
control level were 1, 14, and 32 µg/kg body weight for the guinea-pig,
the Sprague Dawley rat, and the Golden Syrian hamster, respectively
(Madhukar et al., 1984). A single ip dose of 115 µg TCDD/kg body
weight had decreased the EGF binding 10 days after treatment by 93.1,
97.8, and 46.0% in C57Bl/6, CBA, and AKR mice, respectively (Madhukar
et al., 1984). The effect of daily sc injections of 2 ng EGF/kg body
weight to newborn Balb/c-mice was compared with the effect of a single
ip dose of 10 mg TCDD/kg body weight given to the dam of newborn
Balb/c-mice within 3 h of delivery (Madhukar et al., 1984). The
parameters studied, all well known in vivo effects of EGF, were:
time for eyelid opening and tooth eruption; hair length and diameter
on day 14; and body weight and thymus weight on day 22. All parameters
were significantly reduced both by TCDD treatment and EGF treatment,
as compared to controls.
Bombick et al. (1984) found that, 10 days after treatment, in
vitro binding of 125I-low density lipoprotein (LDL) to its receptor
on hepatic plasma membranes was decreased by 73% in TCDD-treated
guinea-pigs (1 µg TCDD/kg body weight) as compared to pair-fed
controls. Primary hepatocytes from guinea-pigs treated in the same way
had a reduced ability to internalize 125I-LDL. The reduction of LDL
receptors on hepatic plasma membranes might be responsible for the
increase in plasma of very low density lipoproteins (VLDL) and LDL
noted in TCDD-treated animals.
Quantitative changes in the protein composition of plasma
membranes following TCDD treatment have been reported (Brewster et
al., 1982; Matsumara et al., 1984). The membranes were isolated from
male Sprague Dawley rats 10 days after an ip injection of 25 µg
TCDD/kg body weight and analyzed by SDS-polyacrylamide gel
electrophoresis. Some small proteins (14 000-30 000 daltons) were
completely abolished by TCDD treatment. The effect was most pronounced
10 to 20 days after treatment.
7.4.2.3 Biliary excretion
The early proliferation of liver cells around bile canaliculi
seen after TCDD treatment (Fowler et al., 1973) was suggestive of an
effect on biliary excretion.
The cumulative biliary excretion of indocyanine green (ICG), an
organic anion, was decreased in a dose-dependent manner by treatment
with 5 or 25 µg TCDD/kg body weight in CD rats (Hwang, 1973). On the
contrary, biliary excretion of the organic anions sulfobromophthalein
and phenol-3,6-dibromophthalein was unaffected by treatment with 10 or
25 µg TCDD/kg body weight in Holtzman rats (Yang et al., 1977).
Biliary excretion of ouabain, a model compound for neutral
non-metabolized substrates such as estradiol, progesterone, and
cortisol, was depressed in male Holtzman rats in a dose-related manner
by a single oral dose of 10 or 25 µg TCDD/kg body weight (Yang et al.,
1977). The effect was detectable two days after treatment, reached a
peak between 10 and 20 days, and recovery was only slight by day 40.
Increased plasma concentration and decreased bilary excretion of
ouabain was also measured in male Sprague Dawley rats 10 days after a
single oral dose (25 µg/kg body weight) of TCDD, or
1,2,3,7,8,9-hexaCDD (Yang et al., 1983b). Two other congeners,
1,2,4,6,7,9-hexaCDD and 1,3,6,8-tetraCDD had no effect on these
parameters.
When hepatocytes from TCDD-treated (10 µg/kg body weight) male
Sprague Dawley rats were incubated with labelled ouabain or procaine
amide ethobromide (PAEB) 10 days post-treatment, both the rate of
uptake and the steady-state concentration of ouabain were decreased,
whereas the uptake of PAEB was unaffected by TCDD (Eaton & Klaassen,
1979). The dose of TCDD was very small relative to ouabain
(approximately 150 µmol/litre), so it is not likely that TCDD exerted
its effect by competing with the drug for transport into bile. These
data suggest that the hepatic membrane transport process for ouabain
may be selectively damaged by TCDD.
Peterson et al. (1979a) observed a positive correlation between
the levels of hepatic plasma membrane ATPase activities, biliary
excretion of ouabain, and bile flow in vivo after TCDD treatment.
However, in a further experiment, using perfused rat liver, Peterson
et al. (1979b) demonstrated that biliary excretion of ouabain and
liver membrane ATPase activities could be decreased independently.
Therefore, ATPase activities cannot be directly responsible for the
reduced ouabain excretion.
7.4.3 Porphyria
Chronic sublethal exposure to TCDD produces an accumulation of
porphyrins in the liver and an increase in urinary porphyrin
excretion. In stages of manifest porphyria, accumulation of porphyrins
occurs not only in the liver but also in the kidney and spleen
(Goldstein et al., 1982). It was demonstrated that mice respond to
four weekly doses of 25 µg TCDD/kg body weight with hepatic porphyria,
accompanied by an increase in aminolevulinic acid (ALA) synthetase
activity, and liver lesions (Goldstein et al., 1973), whereas a single
dose of 5, 25, or 100 µg TCDD/kg body weight did not induce porphyria
nor ALA synthetase activity in the rat (Woods, 1973). The suggested
species difference was later ruled out by Cantoni et al. (1981) and
Goldstein et al. (1976, 1982). Chronic administration of 1 µg TCDD/kg
body weight per week to rats for 16 weeks resulted in hepatic
porphyria (Goldstein, 1976a,b, 1982). In contrast, single oral doses
as high as 30 µg TCDD/kg did not produce porphyria either acutely or
16 weeks later. A 6-month recovery period following the final dose was
not long enough to reverse the porphyria. Urinary porphyrins and
hepatic ALA synthetase activity remained maximally elevated, while
hepatic porphyrin levels decreased during this period. Failure to
demonstrate porphyria in rats after chronic administration of TCDD for
13 weeks (Kociba et al., 1976) or 2 years (Kociba et al., 1978) was
suggested to be the result of unsatisfactory porphyrin analysis
(Goldstein et al., 1982).
To further characterize TCDD-induced porphyria, Cantoni et al.
(1981) performed a 45-week study to follow the pattern of porphyrin
excretion in rats exposed orally to 0.01, 0.1, or 1.0 µg TCDD/kg body
weight/week. They found an increase in the coproporphyrin level in the
initial phase of exposure, which remained the only sign of exposure in
the lowest-dose group. A marked porphyric state appeared only in the
1.0 µg/kg dose group, commencing 8 months after dosing started. At
that time urinary porphyrin excretion was 70 times higher than in
control rats. The excretion pattern was characterized by increased
levels of carboxylated porphyrins.
In attempts to elucidate the mechanism of TCDD-induced porphyria,
the effects of TCDD on the enzymes involved in the synthesis and
catabolism of porphyrins have been studied. TCDD was found to be a
potent inducer of ALA synthetase, the initial and rate-limiting enzyme
in haeme synthesis in the liver of chicken embryos (Poland & Glover,
1973b). Elevated ALA synthetase activity has since been demonstrated
also in mice and rats (Goldstein et al., 1973, 1982; Kociba et al.,
1976, 1978). However, the TCDD-induced increase does not appear after
acute exposure and only after several weeks of chronic exposure to
TCDD. Jones & Sweeny (1980) failed to demonstrate increased ALA
activity in mice exposed to 25 µg TCDD/kg body weight per week for 11
weeks, although porphyria was evident. Thus, induction of ALA
synthetase does not seem to be the primary event in TCDD-induced
porphyria. Elder et al. (1976, 1978) suggested that decreased hepatic
porphyrinogen decarboxylase is the primary event in porphyria induced
by halogenated aromatics. TCDD depresses this enzyme activity in
vivo in the liver of mice (Jones & Sweeny, 1980; Elder & Sheppard,
1982; Cantoni et al., 1984a,b) but not in vitro (Cantoni et al.,
1984b).
A decrease in porphyrinogen decarboxylase activity was present
one week after a single dose of 75 µg/kg body weight and continued to
decrease with time, thus preceding the increase in hepatic porphyrins,
which started to rise during the first 2 weeks after treatment (Smith
et al., 1981). TCDD- induced depression of hepatic uroporphyrinogen
decarboxylase activity occurs also in the newly regenerating liver, as
demonstrated by Smith et al. (1985) in C57Bl/10 mice 10 days after
partial (2/3) hepatectomy. The mice were treated with 75 µg TCDD/kg
body weight orally 4 weeks before hepatectomy. Greig et al. (1984)
demonstrated that pretreatment of five different strains of mice with
12.5 mg Fe2+ one week before the administration of 75 µg TCDD/kg
body weight had a synergistic effect on porphyria assessed 5 weeks
after dosing, expressed as increased hepatic porphyrin and decreased
porphyrinogen decarboxylase activity. Iron alone did not increase
hepatic porphyrin levels, nor did it affect hepatic porphyrinogen
decarboxylase activity.
7.4.4 Epidermal effects
Chloracne and associated pathological changes in the skin are
among the most sensitive and widespread responses to TCDD in humans.
Similar skin toxicity is expressed only in a limited number of animal
species, namely rabbits, monkeys, and hairless mice. To characterize
the epidermal response and to elucidate the mechanism(s) of toxicity
to epidermal cells, numerous studies have been performed both in
vivo and in vitro.
7.4.4.1 In vivo studies
The acnegenic activity of TCDD and related compounds has been
tested in the rabbit ear bioassay, first developed by Adams et al.
(1941) for industrial applications. The test substance is applied to
the inner surface of one of the ears, while pure vehicle is applied to
the other. Responses indicative of acnegenic activity include comedo
formation, increased ear thickness, and hyperkeratosis. Mild
irritation, increased ear thickness, slight enlargement of follicular
aperture, slight exfoliation and slight crust formation alone are not
considered indicative of acnegenic activity. Microscopically there is
conversion of sebaceous cells in the hair follicles into
keratin-forming cells. A dose-dependent, positive response was found
in this assay when a total dose of 1, 3, or 10 µg TCDD was applied on
three successive days (Jones & Krizek, 1962). Also Schwetz et al.
(1973) found a dose-dependent acnegenic response in the rabbit-ear
bioassay after repeated applications of 4-40 µg TCDD/ear, five days
per week for four weeks, corresponding to total doses of 80-800 µg. No
response was obtained when the total application was 8 ng. Poiger &
Schlatter (1980) applied a single dose of TCDD in various vehicles on
the inner surface of the rabbit ear and followed the appearance of
inflammation, hyperkeratosis, and chloracne. The minimum dose that
induced skin lesions was around 1 µg TCDD/ear when the vehicle was
acetone, vaseline, or polyethylene glycol 1500 with 15% water. When
TCDD was mixed with soil-water (2:1), or activated carbon-water (1:8)
before application, 2-3 and 160 µg TCDD/ear, respectively, were needed
to induce lesions. Even 160 µg TCDD/ear produced very small changes on
the skin surface when applied as an activated carbon-water paste.
Hairless mice constitute another in vivo model for studies of
epidermal effects of TCDD. Puhvel et al. (1982) studied cutaneous
changes induced by topical application of 0.1 µg TCDD, three times per
week for four weeks, in two strains of hairless mice (Skh:HR-1 and
HRS/J). Epidermal hyperplasia, hyperkeratinization, loss of sebaceous
glands and follicles, and keratin build-up in the dermal cysts were
noted in both strains of mice. Follicular keratosis, considered the
pathognomonic lesion in human chloracne, did not appear within 4 weeks
of application. In the same study, follicular keratosis did develop
after topical application of 2 mg 3,4,3',4'- tetrachlorobiphenyl, five
times per week for 8 weeks, suggesting that follicular keratosis is an
extension of the epidermal response and, thus, not related to
metabolic changes in sebaceous glands. The authors considered hairless
mice a less sensitive model for the chloracnegenic response than the
rabbit ear bioassay. Similar findings were obtained when HRS/J mice
were exposed to TCDD topically applied two to three times per week for
four weeks (Knutson & Poland, 1982; Poland & Knutson, 1982). They
found, with a total applied dose of 1.2 µg TCDD, a moderate to severe
response, including hyperplasia, hyperkeratosis of the interfollicular
epidermis, squamous metaplasia of the sebaceous glands, and
hyperkeratosis within the dermal cysts, but no keratosis in the
sebaceous follicles.
Soot or a benzene extract of soot, containing PCDDs and PCDFs
from a PCB-containing transformer fire (Binghamton, New York, USA),
were applied to 64.5 cm2 of the shaved, unabraded dorsal surface of
(3 + 3) and (1 + 1) male and female New Zealand white rabbits (3.5
kg), respectively (Silkworth et al., 1982). The dose applied was 500
mg soot or benzene extract corresponding to 500 mg soot/kg body
weight. Controls, one male plus one female, were exposed to activated
carbon or benzene in corresponding amounts. Exposure lasted for 24 h
and the observation period was 67 days. The soot produced no overt
toxicity, no weight loss, and no histological findings in thymus,
kidney, or skin, but hepatic centrilobular hypertrophy was found in
both sexes. The soot extract gave rise to a reversible skin
inflammation and hepatic centrilobular hypertrophy in the female only.
No weight loss was recorded and the kidney, thymus, and skin were
histologically normal at necropsy.
7.4.4.2 In vitro studies
Keratinocytes, the principal cell type of epidermis, form an in
vitro model for studies of TCDD-induced hyperkeratosis both in human
and animal-derived cell cultures. The response is analogous to
hyperkeratinization in vivo. Newly confluent epidermal cell cultures
exhibit proliferative properties, while the number of basal cells
tends to decrease with increasing time of post-confluency growth.
Thus, with the appropriate selection of culture medium and time of
treatment, different aspects of TCDD toxicity in vivo can be
modelled in vitro.
A TCDD-induced response of in vitro keratinization was first
demonstrated in XB cell cultures, an established keratinocyte cell
line derived from a mouse teratoma, plated at high density to avoid
spontaneous keratinization (Knutson & Poland, 1980b). Keratinization
was dose-related and histologically similar to that which occurs
spontaneously when XB cells are plated at low density. The epidermal
proliferation in XB cells produced by TCDD could not be biochemically
related to the response produced by cholera toxin, epidermal growth
factor, or 12-O-tetradecanoylphorbol-13-acetate, other compounds
known to affect cell proliferation in XB cells (Knutson & Poland,
1984). Late passage XB cells, i.e. XBF cells, show increased cell
density at saturation and a fusiform morphology at high density.
Additionally, they have lost their ability to respond with
keratinization upon TCDD treatment. Exposing XBF cells to TCDD
concentrations in the range 10 to 11 X 10-8 mol/litre resulted in
normal growth until confluency was reached by day 7. Thereafter
TCDD-treated cultures showed a persistent decrease in cell growth and
cell proliferation as well as changed morphology, whereas viability
was unaffected (Gierthy & Crane, 1984). Reseeding these quiescent XBF
cells, previously exposed to 10-9 mol/litre TCDD for 14 days,
resulted in normal growth and proliferation until confluency. These
TCDD-pretreated cells maintained their susceptibility to TCDD-induced
changes in cell growth and morphology. Both XB cells (keratinization
assay) and XBF cells (flat-cell-assay) have proved to be useful in
vitro bioassays to measure "dioxin-like" activity of both
environmental samples and of pure isomers (Gierthy et al., 1984;
Gierthy & Crane, 1985a,b). Although XBF cells, a highly transformed
variant of XB cells, seem to be less appropriate as a model for TCDD
action on normal mammalian epithelial cell proliferation and
differentiation, they seem to be more stable and easier to maintain
than XB cells. Several continuous lines of human keratinocytes derived
from neonatal foreskin (Milstone & Lavigne, 1984; Osborne et al.,
1984) or squamous cell carcinomas (SCC) (Rice & Cline, 1984; Willey et
al., 1984; Hudson et al., 1985, 1986) have been shown to respond to
TCDD in nanomolar concentrations with a variety of signs that indicate
alterations in the normal differentiation process. Stimulation of
3H-thymidine incorporation was seen in post-confluent human
epidermal cells derived from neonatal foreskin after exposure to TCDD
(Milstone & Lavigne, 1984). Newly confluent human epidermal cells,
derived from foreskin, responded to exposure for 4 days to 10 nmol
TCDD/litre with decreased DNA synthesis, a decrease in the number of
proliferating basal cells, decreased binding of epidermal growth
factor (EGF), an increase in the number of differentiated cells, and
increased envelope formation, i.e., a decrease in the proliferative
capacity and an increase in the state of differentiation (Osborne &
Greenlee, 1985). The decreases in small (basal) cell number and EGF
binding were dose dependent, with EC50 values for TCDD of 2 and 1
nmol/litre, respectively. The responses were also obtained with TCDF
but not with 2,4-diCDD.
The proliferation and differentiation of epidermal cells is
normally regulated by several growth factors and hormones, e.g., EGF,
vitamin A and hydrocortisone. Mouse hepatoma cells exposed to TCDD for
24 h showed 20% inhibition of EGF binding (Karenlampi et al., 1983).
Hudson et al. (1985, 1986) demonstrated that TCDD decreases, in
a dose-dependent manner, the specific binding as well as the cellular
uptake of EGF in cultures of human epidermal cells. The EC50 dose
for inhibition was 1.8 nmol/litre. A similar inhibitory effect was
obtained by TCDF, while 2,7-diCDD was inactive even at doses 100-fold
greater. Maximal inhibition, almost 60%, of EGF binding in confluent
SCC-12F cells exposed to 100 nmol TCDD/litre was obtained after a
pretreatment period of 72 h. No effect was obtained when TCDD was
added at the same time as EGF; thus TCDD did not compete for
EGF-binding sites, neither did TCDD affect the process of
internalizing the EGF. Further studies of the SCC-12F cell line
revealed data suggesting that TCDD specifically reduces the high
affinity EGF-binding sites in the basal cell population of this cell
line (Hudson et al., 1986).
The addition of 10-6 mol hydrocortisone/litre to the medium
antagonized the growth inhibition of SCC cells grown in 10-10 mol
TCDD/litre (Rice & Cline, 1984). Hydrocortisone stimulated several
aspects of keratinocyte differentiation. These stimulatory effects
were abolished in the presence of 10-8 mol TCDD/litre, although TCDD
alone had no effect on these parameters. The hydrocortisone level in
the medium was unaffected by TCDD. TCDD, and even more so
hydrocortisone, were able to stimulate stratification in SCC cultures
held at confluence for extended periods. This effect was opposed by
vitamin A (Rice & Cline, 1984).
Like TCDD, vitamin A suppressed the stimulation of keratinocyte
differentiation by hydrocortisone. However, vitamin A had no effect on
TCDD-induced growth inhibition or its reversal by hydrocortisone (Rice
et al., 1983; Rice & Cline, 1984).
Epidermal transglutaminase (ETG) activity, the marker enzyme for
terminal differentiation, was increased by treatment of basal
keratinocyte cultures from neonatal BALB mice with 10-9 mol
TCDD/litre for 5 to 12 days, although morphologically no signs of
terminal differentiation were present. A parallel increase in ETG
activity was present when these cells were grown in medium rich in
Ca2+, although these cells did stratify and differentiate (Puhvel et
al., 1984).
7.4.5 Effects on the immune system
TCDD produces a pronounced atrophy of the thymus, spleen, and, to
a lesser, extent the peripheral lymph nodes of most experimental
animals. Since Buu-Hoi et al. (1972a) reported on TCDD-induced thymic
atrophy, many studies in rats, mice, guinea-pigs, and monkeys have
shown that the thymus is one of the organs most severely affected by
TCDD. Lesions in the thymus appear at exposure levels well below those
inducing lesions in other organs. Although there is species variation
in the degree and severity of other organ effects, the effects of TCDD
on lymphoid tissues is consistent in all species. Further
investigations of the effect of TCDD on the immune system, which is a
rapidly proliferating and differentiating organ system containing many
cellular components in a highly organized and regulated network, have
revealed that TCDD affects both the humoral-mediated (section 7.4.5.2)
and the cell-mediated immune response (section 7.4.5.3). Also the
complement system, a key component of the innate immunity, is affected
by TCDD treatment (White et al., 1986). Damage to the thymus and to
the cell-mediated immune system seems to be rather specific in that it
occurs at doses considerably lower than those affecting other immune
functions (Faith & Luster, 1979). Thymic involution is believed to be
a direct effect on the gland, and not secondary to factors such as
undernutrition (van Logten et al., 1980), altered levels of hormones
including corticosteroids (Vos et al., 1973; van Logten et al., 1980),
pituitary hormones (Vos et al., 1973), and thymosin (Vos et al.,
1978a,b), or zinc deficiency (Vos et al., 1978a,b), or to a direct
cytotoxic effect on lymphocytes (Vos et al., 1978a,b). A direct effect
of TCDD on mouse fetal thymus organ cells grown in vitro was
demonstrated at concentrations as low as 10-10 mol/litre (Dencker et
al., 1985). Within the thymus, lymphocytes in the cortex, i.e., the
immature T-cells, are more severely affected in TCDD-treated animals
than are lymphocytes in the medulla, i.e., the mature T-cells. Thus,
it seems that TCDD impairs the differentiation of thymocytes into
immunocompetent T cells. Greenlee et al., (1985) obtained results
demonstrating a direct effect of TCDD on thymus epithelial (TE) cells.
High levels of Ah receptors (section 7.8.1) have been found in
the thymus (Carlstedt-Duke, 1979; Mason & Okey, 1982; Gasiewicz &
Rucci, 1984). Studies with C57Bl/6 mice (responsive to TCDD), DBA/2
mice (less responsive to TCDD), and B6D2F1 mice, hybrid mice from
crosses between these strains, suggest that TCDD-induced thymic
involution, as well as immunosuppression, segregates with the Ah locus
in these strains of mice (Poland & Glover, 1980; Clark et al., 1983;
Vecchi et al., 1983; Nagarkatti et al., 1984; Dencker et al., 1985).
The most profound and persistent effect of TCDD on the immune system
is found when TCDD is administered during pre- and/or immediate
postnatal life. Contrary to other experimental animals investigated,
rainbow trout (Salmo gairdneri) are relatively resistant to the
immunosuppressive effects of TCDD (Spitzbergen et al., 1986). No
humoral-mediated effects, and only minor cell-mediated effects, were
present at doses which caused clinical toxicity.
7.4.5.1 Histopathology
Lymphoid organs, primarily thymus but also spleen and lymph
nodes, have been found to be affected by TCDD over a wide spectrum of
dose ranges in adult rats, guinea-pigs, and mice (Gupta et al., 1973;
Vos et al., 1973; Vos and Moore, 1974; McConnell et al., 1978b). The
marked reduction in the size of thymus has been referred to as atrophy
(Gupta et al., 1973; Vos & Moore, 1974), regression (Allen et al.,
1975), or involution (Kociba et al., 1976), though these terms do not
clearly represent the pathogenesis of this lesion but are more a
description of the final event. Toxic effects on thymus appeared in
adult guinea-pigs, rats, and mice exposed to eight weekly doses of 0.2
µg TCDD/kg body weight, six weekly doses of 5 µg TCDD/kg body weight
and four weekly doses of 5 µg TCDD/kg body weight respectively (Vos et
al., 1973). The thymus from moribund animals, or from animals that
died from TCDD exposure, showed a dose-dependent decrease in the
number of cortical lymphocytes, markedly smaller thymic lobules, and
loss of demarcation between the cortex and medulla. Guinea-pigs, the
species most severely affected by TCDD, showed large cystic Hassall
bodies, filled with polymorphonuclear leukocytes (Gupta et al., 1973;
Vos et al., 1973). Guinea-pigs that received lethal doses of TCDD
showed scattered necrosis of lymphocytes in the cortical region with
concomitant phagocytosis by macrophages as early as 5 days
post-exposure (McConnell et al., 1978b). The effect was more apparent
at day 14, and by day 20 it was difficult to differentiate the cortex
from the medulla. At day 20 little evidence of necrosis remained,
though karyorrhectic debris and prominent phagocytosis indicated that
this had occurred. Since the thymus from guinea-pigs surviving the
TCDD dose for 30 days was usually normal microscopically (Vos et al.,
1973; McConnell et al., 1978b), it seems that thymic necrosis must be
an early event in the course of the toxic syndrome. Furthermore, in
animals which survive, thymic regeneration seemed to be rapid.
Decreased weight of thymus, loss of cortical cells, and cell necrosis
have also been found in hamsters exposed to TCDD (Gasiewicz et al.,
1986).
Depletion of lymphoid cells in the spleen, intestinal tract, and
various lymph nodes observed in guinea-pigs, rats, and mice (Gupta et
al., 1973; McConnell et al., 1978b) was less extensive than in the
thymus. The major effect in the spleen of rats is the loss of the
T-cell-dependent areas, namely the periarterial lymphoid sheet and the
paracortical areas (Vos & Moore, 1974).
Depressed immunoglobulin levels were reported for 1- and
4-month-old C57Bl/6 mice exposed to four and six weekly doses,
respectively, of 25 µg TCDD/kg body weight (Vos & Moore, 1974).
Feeding 10, 20, 50, or 100 µg TCDD/kg in the diet depressed
dose-dependently the Y-globulin level in 7-week-old Swiss-Webster mice
(Hinsdill et al., 1980).
Vos & Moore (1974) demonstrated a dose-related lymphocyte
depletion of thymus cortex, spleen, and intestinal lymph nodes in
maternally exposed pups of rats and mice. However, no effect on
immunoglobulin levels was observed in 25-day-old rats maternally
exposed (5 µg TCDD/kg body weight) on days 0, 7, and 14. The
developing lymphoid tissues were found to be more sensitive to TCDD
than were the lymphoid tissues of adults or young.
7.4.5.2 Humoral-mediated immunity
The humoral-mediated immunity (Tables 51 and 52) operates through
antibody-producing cells and is transferable by serum. This system
includes classical antibody-mediated protective immunity and immediate
hypersensitivity reactions. Vos et al. (1974) reported a significant
decrease in the alpha-, ß-, and gamma-globulin levels in C57BL/6 mice
given non-toxic doses of TCDD. The effects of TCDD on specific humoral
immunity responses in adult animals are summarized in Table 51.
Feeding levels of 10 µg TCDD/kg body weight or more reduced the
primary and secondary antibody response to both sheep red blood cells
(sRBC) and tetanus toxin in male Swiss-Webster mice (Hinsdill et al.,
1980). Weekly oral doses of 1 or 10 µg TCDD/kg body weight reduced
both the primary and secondary serum antitetanus titres in male New
Zealand rabbits (Sharma et al., 1984). The secondary, but not the
primary, serum tetanus antitoxin level was decreased in Hartley
guinea-pigs given eight weekly doses of 0.2 µg TCDD/kg body weight
(Vos et al., 1973). Results presented by Vecchi et al. (1980, 1983)
show that single doses as low as 1.2 µg TCDD/kg body weight to C57BL/6
mice decreased the number of plaque-forming spleen cells in response
to an injection of the thymus-dependent antigen sRBC. The response was
dose dependent and lasted for at least 42 days. Luster et al. (1985)
found a decrease in anti-sRBC plaque-forming spleen cell production in
B6C3F1 and DBA/2 mice 5 days after single oral doses of 2 and 10 µg
TCDD/kg body weight, respectively. A dose of 30 µg TCDD/kg body weight
was needed to produce a significant antibody response to the
thymus-independent antigen type III pneumococcal polysaccharide (sIII)
in C57Bl/6 mice (Vecchi et al., 1980). With sRBC and the
thymus-independent antigen trinitrophenylated Brucella abortus,
Clark et al. (1981) found a depressed number of spleen plaque-forming
cells in C57BL/6 mice only with a total dose of 40 µg/kg body weight
given as four equal weekly doses. Chastain & Pazdernik (1985)
demonstrated decreased numbers of plaque-forming spleen and bone
marrow cells in response to the thymus-independent antigen
trinitrophenylated lipopolysaccharide (LPS). Spleen and bone marrow
cells were collected from male C57BL/6 mice 7 days after a single ip
dose of 30, 60, 90, or 120 µg TCDD/kg body weight. The decrease
occurred at lower doses in the spleen cell assay but was more
pronounced at higher doses in the bone marrow cell assay. The number
of antibody-producing cells, measured asplaque-forming cells,
following immunization with either sRBC or LPS was reduced in B6C3F1
mice treated with 1 and 5 µg TCDD/kg body weight (Tucker et al.,
1986).
Table 51. Effects of TCDD on humoral-mediated immune responses in adult animals
Species/strain Sex/age/weightf TCDD exposure Parameter measurede
(reference) Frequency/route/dose
Mice
Swiss-Webster F/4-7 weeks/NR fed 10, 20, 50, 100, or primary and secondary sRBCa antibody
500 µg/kg continuously level,
(Hinsdill et al., in the diet for 5 weeks primary and secondary serum tetanus
1980) antitoxin level
C57BL/6J M/6-8 weeks/NR single ip dose of anti-sRBCa plaque-forming spleen cells
1.2, 6, or 30 µg/kg body anti-SIIIb plaque-forming spleen cells
(Vecchi et al., weight
1980)
C57BL/6J M/6-8 weeks/NR four weekly ip doses of anti-sRBCa plaque-forming spleen cells
0.1, 1, or 10 µg/kg body anti-TNP-BAc plaque-forming spleen cells
(Clark et al., weight
1981)
C57BL/6 M/8-10 weeks/NR single ip dose of anti-sRBCa plaque-forming spleen cells
C3H/HeN 1.2, 6, or 30 µg/kg body
DBA/2 weight
AKR
B6D2F1
(Vecchi et al.,
1983)
C57BL/6 M/6-8 weeks/NR single ip dose of 30, 60, anti-TNP-LPSd plaque-forming spleen cells
90, or 120 µg/kg body weight anti-TNP-LPSd plaque-forming bone marrow
(Chastain & cells
Pazdernik, 1985)
Table 51. (contd - 2)
Species/strain Sex/age/weightf TCDD exposure Parameter measurede
(reference) Frequency/route/dose
Mice (contd).
DBA/2N F/6-8 weeks/ single oral doses of 5, anti-sRBCa plaque-forming spleen cells
18-21 g 10, or 50 µg/kg body weight
(Luster et al.,
1985)
B6C3F1 F/6-8 weeks/ single oral doses of 0.2, anti-sRBCa plaque-forming spleen cells
18-21 g 1, 2, 5, or 10 µg/kg body
(Luster et al., weight
1985)
Guinea-pigs
Hartley F/NR/256 g eight weekly oral doses of primary serum tetanus antitoxin level
0.008, 0.04, 0.2, or secondary serum tetanus antitoxin level
(Vos et al., 1973) 1.0 µg/kg body weight
Rabbit
New Zealand M/Adult/3 kg eight weekly oral doses of primary and secondary serum tetanus
0, 0.01, 0.1, 1.0, or 10.0 antitoxin level
(Sharma et al., µg/kg body weight
1984)
a sRBC = sheep red blood cell.
b SIII = type III pneumoccal polysaccharide.
c TNP-BA = trinitrophenylated Brucella abortus.
d TNP-LPS = trinitrophenylated lipolysaccharide.
e = all parameters measured were decreased except for primary serum tetanus antitoxin level in guinea-pigs
(Vosal., 1973).
f M = male; F = female; NR = not reported.
Table 52. Effects of TCDD on humoral-mediated immune responses in maternally exposed animals
Species/strain Time of TCDD exposure Route/dose Parameter measured-response
(reference)
Rats
Fisher-344 N Prenatal day 18 and/or oral/5 µg/kg body weight primary and secondary BGGa
postnatal days 0, 7, and 14 antibody level - no effect
(Faith & Moore,
1977)
Fisher/Wistar Prenatal day 18 and/or oral/5 µg/kg body weight primary and secondary BGGa
postnatal days 0, 7, and 14 antibody level - no effect
(Faith & Luster,
1979)
Mice
Swiss-Webster four weeks before mating, in the diet/ primary and secondary sRBCb
throughout gestation and 1, 2.5, 5, 10, or 20 µ/kg antibody level - no effect;
(Thomas & lactation primary anti sRBCb plaque
Hinsdill, 1979) forming spleen cells - decreased
a = BGG - bovine gamma-globuline.
b = sRBC - sheep red blood cell.
Only minor effects on antibody responses have been reported in
rodents maternally exposed to TCDD (Table 52). Single oral doses of 5
µg TCDD to pregnant Fisher-344 N (poor immunological responder) and
Fisher-Wistar rats (good immunological responder) on gestation day 18
and/or postnatal days 0, 7, and 14 did not affect the antibody reponse
to bovine gammaglobulin (BGG) in the offspring (Faith & Moore 1977;
Faith & Luster, 1979).
Dietary exposure of female Swiss-Webster mice to 2.5 or 5 µg
TCDD/kg for 4 weeks before mating and throughout gestation and
lactation resulted in normal antibody production in the offspring but
in a decrease in anti-sRBC plaque-forming spleen cells.
In vitro exposure of B6C3F1 spleen cells to 10-9 mol
TCDD/litre decreased the production of anti-sRBC plaqueforming cells
(Luster et al., 1984). The ED50 for this effect was found to be 7
nmol/litre when TCDD was present from the first day of culture (Tucker
et al., 1986). Similar activity to that of TCDD was found with
2,3,7-triCDD and 1,2,3,7,8-pentaCDD, whereas 2,8-diCDD and octaCDD
were without effect at concentrations up to 5 x 10-8 mol/litre
(Tucker et al., 1986).
The doses producing 50% suppression of splenic IgM response to
sRBC in C57Bl/6 mice were 7.1 and 85 µg/kg body weight, respectively,
for 1,2,3,6,7,8-hexaCDD and 1,2,3,4,6,7,8-heptaCDD given as single
oral doses 2 days prior to sRBC challenge (Kerkvliet et al., 1985).
OctaCDD had no effect even at doses of 100 and 500 µg/kg body weight.
Humoral immune responses of the rainbow trout (Salmo
gairdneri) were not significantly impaired even at doses of TCDD
that caused clinical toxicity (Spitsbergen et al., 1986).
7.4.5.3 Cell-mediated immunity
Cell-mediated immunity (CMI) operates through specifically
sensitized lymphocytes and is transferred by these cells. Processes
included in this system are classical cell-mediated protective
immunity (which protects against fungi, bacteria, and viruses),
delayed type hypersensitivity, rejection of tumors and foreign tissues
such as transplants, and graft versus host response. Many assays, both
in vivo and in vitro, have been developed to test CMI functions.
Besides a reduction in the number of immunologically competent cells
after TCDD exposure (Gupta et al., 1973; Vos et al., 1973; Zinkl et
al., 1973) TCDD has been demonstrated to induce a decreased CMI
response in adults (Table 53) and even more in maternally exposed
animals (Table 54). Delayed hypersensitivity response, correlatingwith
decreased host resistance to infectious agents in man, was depressed
in rodents exposed to low levels of TCDD (Vos et al., 1973; Faith &
Moore, 1977; Sharma et al., 1978; Faith & Luster, 1979; Thomas &
Hinsdill, 1979; Hinsdill et al., 1980; Clark et al., 1981).
TCDD-exposure also adversely affects host susceptibility to bacteria,
viruses, tumor cells and endotoxins (Thigpen et al., 1975; Thomas &
Hinsdill, 1979; Hinsdill et al., 1980; Luster et al., 1980; Clark et
al., 1983).
Depressed graft versus host response was reported in 2-month-old
C57BL/6 mice exposed to 4 weekly oral doses of 5 µg TCDD/kg body
weight (Vos et al., 1973), whereas no effect was seen in a subsequent
study on 1- and 4-months old C57BL/6Sch mice (Vos & Moore, 1974). In
the same study, Fisher-344 rats maternally exposed to TCDD showed
decreased graft versus host response and prolonged allograft-rejection
time. The latter effect was also demonstrated in maternally
TCDD-exposed C57BL/6Sch mice (Vos et al., 1973). Proliferative
responses of spleen and/or thymus lymphocytes, stimulated by mitogens
specific for the generation of B-lymphocytes and/or T-lymphocytes from
TCDD-exposed animals, were depressed both in adults (Vos & Moore,
1974; Sharma et al., 1978) and maternally exposed rodents (Vos &
Moore, 1974; Faith & Moore, 1977; Vos et al., 1978; Faith & Luster,
1979; Luster et al., 1980). However, Thomas & Hinsdill (1979) found no
effect on the lymphoproliferative response in offspring from
Swiss-Webster mice fed with up to 5 µg TCDD/kg diet 4 weeks before
mating and throughout gestation and lactation. Depressed
lymphoproliferative response is regarded as an extremly sensitive
indicator of immunotoxicity, rather than as a predictor of immune
dysfunction. Cytotoxic T-cell generation in response to allogeneic
antigens has been demonstrated in male DBA/2, C57Bl/6, and B6D2F1 mice
given four weekly ip injections of 1 ng/kg body weight (Clark et al.,
1981, 1983). At this dose no effects were seen on delayed
hypersensitivity, antibody response, thymus cellularity, or enzyme
induction. The adverse effect of TCDD on CMI function seems to be an
age-related phenomenon in rodents. In order to obtain a complete and
persistent immune suppression, TCDD exposure must occur during
ontogenesis of the immune system. In the initial experiments on the
developing immune system, Vos & Moore (1974) exposed Fisher-344 rats
to 1 mg TCDD/kg body weight on gestation days 11 and 18 and on
postnatal days 4, 11, and 18, or to 5 µg TCDD/kg body weight on
postnatal days 0, 7, and 14. CMI functions adversely affected included
in vitro immune competence of spleen and thymus lymphoid cells,
delayed hypersensitivity reaction, prolonged allograft-rejection times
and reduced graft versus host activity. The immune suppression
demonstrated persisted throughout the study, i.e., for 145 days. The
depression of T-cell-dependent immune functions appeared to occur
without helper-cell function being affected (Faith & Moore 1977).
Attempts to study direct effects of TCDD on lymphocytes in
vitro were previously hampered by the low solubility of TCDD in
physiological buffers (Matsumura & Benezet, 1973). Vos & Moore
(1974)obtained no lymphoproliferative response in unstimulated or PHA-
or concanavalin-A-stimulated rat thymus organ cells and mouse spleen
cells when cultured in the presence of up to 20 ng TCDD/ml. Dencker et
al. (1985) demonstrated that mouse fetal thymus cells cultured in
vitro in the presence of TCDD gave a similar response similarly to
that occurring in vivo, i.e., with a dose-dependent inhibition of
the time-dependent increase in the number of lymphoid cells
(EC50=10-10 mol TCDD/litre). It could not be determined with
certainty whether the decreased cell number caused by TCDD was due to
reduced cell proliferation or to increased cell death. The
TCDD-induced suppression of mitogen-stimulated lymphoproliferation has
recently been demonstrated to be mediated by thymus epithelial cells
(Greenlee et al., 1985).
7.4.5.4 Macrophage function
The primary pathway of endotoxin detoxification is thought to be
macrophage-dependent. Thus the increased sensitivity to endotoxin
following TCDD treatment (Vos et al., 1978; Thomas & Hinsdill, 1979)
was suggestive of macrophage dysfunction. However, the number of
peritoneal macrophages, as well as their capacity to mediate cytolytic
and cytostatic effects, was not adversely affected by single ip doses
of 1.2, 6 or 30 µg TCDD/kg body weight to male C57Bl/6J mice
(Mantovani et al., 1980), nor was the ability of macrophages to reduce
nitroblue tetrazolium affected by four to five weekly oral doses of 50
µg TCDD/kg body weight in Swiss-Webster mice (Vos et al., 1978a).
Macrophage function does not appear to be altered by TCDD.
7.4.6 Myelotoxicity
TCDD treatment inhibits the bone marrow haematopoiesis in mice,
both in vivo and in vitro, by directly altering colony growth of
stem cells (Luster et al., 1980, 1985; Chastain & Pazdernik, 1985).
The bone marrow granulocyte-macrophage progenitor cell (CFU-GM)
production was reduced in B6C3F1 mice, receiving 1 µg TCDD/kg body
weight, but was unaffected in DBA/2 mice, even at a dose of 50 µg
TCDD/kg body weight (Luster et al., 1985). Chastain & Pazdernik (1985)
used the B-lymphocyte colony-forming unit assay to demonstrate
reductions in spleen and bone marrow B-cell function of C57BL/6 mice
exposed to single ip doses of TCDD in the range 30 to 120 µg TCDD/kg
body weight. Bone marrow B-cells tended to be more suseptible to TCDD
than spleen B-cells. A maternal oral dose of 5 µg TCDD/kg body weight
on the 14th day of gestation and postnatal days 1, 7, and 14 resulted
in a weak normocytic anaemia indicative of depressed erythrogenesis,
decreased bone marrow cellularity, and decreased stem cell
proliferation in B6C3F1 offspring, assessed 7 days after weaning
(Luster et al., 1980). In vitro exposure of B6C3F1 bone marrow cells
to 10-9 mol TCDD/litre resulted in decreased CFU-GM development and
decreased number of erythrocyte colony-forming units. The decrease in
CFU-GM occurred 1 day post-treatment and remained below control values
until 10 days post-treatment (Luster et al., 1985).
Table 53. Effects of TCDD on cell-mediated immunity responses in adult animals
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Rats
CD F/NR/185 g six weekly oral doses of delayed hypersensitivity to tuberculin - NE
0.2, 1.0, or 5.0 µg/kg
(Vos et al., 1973) body weight
Mice
C57BL/6 M/6-8 weeks/NR four weekly ip doses of delayed hypersensitivity to sRBCa - Dec;
0.1, 1.0, or 10.0 µg/kg delayed hypersensitivity to oxazalone - Dec;
(Clark et al., body weight generation of alloantigen-specific cytotoxic
1981) T-cells - Dec
C57BL/6 M/NR/NR four weekly ip doses of resistance to Herpes virus challenge - Dec;
0.001, 0.01, 1.0, or generation of alloantigen-specific cytotoxic
(Clark et al., 10.0 µg/kg body weight T-cells - Dec
1983)
DBA/2 M/NR/NR four weekly ip doses of generation of alloantigen-specific cytotoxic
0.001, 0.01, 0.1, 1.0, or T-cells - Dec
(Clark et al., 10.0 µg/kg body weight
1983)
Swiss-Webster F/4-7 weeks/NR five weeks feeding of diets resistance to Salmonella typhimurium challenge - Dec;
containing 10, 50, or resistance to Listeria monocytogenes challenge - Dec;
(Hinsdill et al., 100 µg/kg body weight contact sensitivity to 2,4-dinitro-1-fluoro-
1980) benzene - Dec
Table 53 (contd - 2)
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Mice (continued)
C57BL/6J M/6-8 weeks/NR single ip doses of 1.2, number of peritoneal macrophages - Dec;
6 or 30 µg/kg body weight number of splenic natural killer cells - Dec;
(Mantovani macrophage mediated cytolysis - NE;
et al., 1980) macrophage mediated cytostasis - NE
C57BL/6 M/NR/NR four weekly ip doses of generation of allospecific cytotoxic T-cells - Dec
DBA 0.001 µg/kg body weight - NE
B6D2F1 - Dec
(Nagarkatti
et al., 1984)
CD-1 M/NR/28 two, four, or eight weekly lymphoproliferative response of PHAb- and PWMc-
doses of 0.01, 0.1, 1.0, or stimulated splenic cells - Dec
(Sharma & 10.0 µg/kg body weight
Gehring, 1979)
C57BL/6Jfh M/4 weeks/NR four weekly oral doses of resistance to Salmonella bern challenge - Dec;
0.5, 1.0, 5.0, 10.0, or resistance to Herpes virus challenge - NE
(Thigpen et al., 20.0 µg/kg body weight
1975)
C57BL/6 M/2 months/ four weekly oral doses of graft versus host activity - Dec
24.4 g 0.2, 1.0, 5.0, or 25.0
(Vos et al., µg/kg body weight
1973)
C57BL/6Sch M/1 month/NR four weekly oral doses of lymphoproliferative response of PHAb-stimulated
1.0, 5.0, or 25.0 µg/kg spleen cells - Dec;
(Vos & Moore, body weight graft versus host activity - NE
1974)
Table 53 (contd - 3)
Species/strain Sex/age/weightd TCDD exposure Parameter measured-responsed
(reference) Frequency/route/dose
Mice (continued)
C57BL/6Sch M/4 months/NR six weekly oral doses of lymphoproliferative response PHA-stimulated
1.0, 5.0, or 25.0 µg/kg spleen cells - NE
(Vos & Moore, body weight graft versus host activity - NE
1974
Swiss M/3-4 weeks/NR four or five weekly oral resistance of Listeria monocytogenes - NE;
doses of 50 µg/kg body number of peritoneal macrophages - NE;
(Vos et al., weight macrophage reduction of nitroblue tetrazodium - NE
1978a)
Guinea-pigs
Hartley F/NR/256 g eight weekly oral doses delayed hypersensitivity to tuberculin - Dec
of 0.008, 0.04, 0.2, or
(Vos et al, 1973) 1.0 µg/kg body weight
Rabbits
New Zealand M/adult/3 kg eight weekly oral doses delayed hypersensitivity to tuberculin - Dec
of 0.01, 0.1, 1.0, or
(Sharma et al., 10.0 µg/kg body weight
1984)
a sRBC = sheep red blood cells.
b PHA = phytohaemagglutinin.
c PWM = poke weed.
d NR = not reported; NE = no effect; Dec = decreased; M = male; F = female.
Table 54. Effects of TCDD on cell-mediated immunity responses in maternally exposed animals
Species/strain TCDD exposure Age when Parameter measured - responsef
(reference) Frequency/route/dose testedg
Rats
Fisher/Wistar 5 µg/kg body weight on gestation day 18 25 days lymphoproliferative response of
and on postnatal days, 0, 7, and 14, or PHAa- and ConAb-stimulated spleen
(Faith & Luster, 5 µg/kg body weight on postnatal days and thymus cells - Dec;
1979) 0, 7, and 14 delayed hypersensitivity to
tuberculin - Dec
Fisher-344 5 µg/kg body weight on gestation day 18 25 days lymphoproliferative response of
and on postnatal days 0, 7, and 14, or PHAa- and Conb-stimulated spleen
(Faith & Moore, 5 µg/kg body weight postnatal days 0, 7, and thymus cells - Dec;
1977) and 14 delayed hypersensitivity to
oxazolone - Dec
Fisher 344 1 µg/kg body weight on gestation days 11 25 days lymphoproliferative response of
and 18 and on postnatal days 4, 11 and 18 or PHAa-stimulated spleen cells and of
(Vos & Moore, 5 µg/kg body weight on postnatal days 0, PHAa- and ConAb-stimulated thymus-
1974) 7 and 14 cells - Dec
Mice
B6C3F1 c 1, 5, or 15 µg/kg body weight on NR lymphoproliferative response of
gestation day 14 and on postnatal days 1, mitogen-stimulated spleen cells:
(Luster et al., 7, and 14 PHAa - Dec;
1980) ConAb - Dec;
LPSd - NE;
Macrophage proliferation - NE;
Phagocytizing ability - NE;
Resistance to Listeria monocyto-
genes - Dec;
Resistance to PYB6 tumor cells - Dec
Table 54 (contd).
Species/strain TCDD exposure Age when Parameter measured - responsef
(reference) Frequency/route/dose testedg
Mice (continued)
Swiss-Webster Feeding 1, 2.5, or 5 µg TCDD/kg 5-6 weeks Contact sensitivity to 2,4-dinitro-
4 weeks before mating, throughout 1-fluorobenzene - Dec;
(Thomas & gestation, and 3 weeks postnatally lymphoproliferative response of
Hinsdill, 1979) PHAa- and ConAb-stimulated spleen
and thymus cells - NE;
Resistance to Salmonella typhimurium
endotoxin - Dec; Resistance
to Listeria monocytogenes - NE
C57BL/6Sch 2 or 5 µg/kg body weight on gestation days 23 days Skin graft assay - prolonged skin
14 and 17 and on postnatal days 1, 8, and 15 graft rejection time
(Vos et al., 1974)
Swiss 10 µg/kg body weight on postnatal days 22 days lymphoproliferative response of
1, 4, 8, 11, 15, and 18 PHAa-, ConAb- &
(Vos et al., PWMe-stimulated thymus cells - Dec
1978a)
a PHA = phytohaemagglutinin.
b ConA = concanavalin A.
c B6C3F1 = progeny to female C57BL/6N and male C3H mice.
d LPS = lipopolysaccharide.
e PWM = poke weed.
f Dec = decreased, NE = no effect.
g NR = not recorded.
7.4.7 Effects on the intermediary metabolism
Changes in intermediary metabolism have been demonstrated in
TCDD-treated experimental animals. The circulating concentration of
glucose in TCDD-treated rats was decreased relative to ad
libitum-fed (Zinkl et al., 1973; Schiller et al., 1985) and pair-fed
(Gasiewicz et al., 1980; Potter et al., 1983) control rats.
TCDD-treated and pair-fed controls (both groups were schedule fed) had
similar concentrations of serum glucose (Christian et al., 1986b). The
TCDD-induced hypoglycaemia was not caused by altered pancreatic
function, as judged by insulin and glucagon levels (Potter et al.,
1983).
Reduced hepatic glycogen content, compared to the value in
control rats, was reported in Sprague Dawley rats 16 days after a
single ip dose of 20 µg TCDD/kg body weight (Weber et al., 1983).
However, in studies by Christian et al. (1986b), compared to pair-and
schedule-fed control rats, TCDD-treated (75 µg/kg body weight daily)
Sprague Dawley rats had significantly increased hepatic glycogen
levels but unaffected cardiac and muscle (gastrocnemius) levels of
glycogen 2-8 days post-treatment.
TCDD-treated rats maintained a normal overall nitrogen balance,
as judged by urinary urea, creatinine, and ammonia levels, but
exhibited changes in certain plasma protein levels (Christian et al.,
1986b).
Elevated circulating cholesterol levels were found in
TCDD-treated rats (Albro et al., 1978; Poli et al., 1980; Schiller et
al., 1985), whereas circulating free fatty acids and triacylglycerols
were decreased in TCDD-treated rats, when compared to pair-and
schedule-fed control rats (Christian et al., 1986b).
A marked accumulation of hepatic lipid has been found in rats
after single doses of TCDD (Cunningham & Williams, 1972; Gupta et al.,
1973; Albro et al., 1978; Schiller et al., 1985). At a sublethal dose
of TCDD, hepatic triglyceride and free fatty acid levels were elevated
already one day after dosing. Abnormal lipid deposition patterns
persisted for at least 2 months (Albro et al., 1978). The increased
level of triglycerides in the liver (Schiller et al., 1985; Christian
et al., 1986b) of TCDD-treated rats was not accompanied by increases
in cardiac or muscle (gastrocnemius) triacylglycerol levels (Christian
et al., 1986b). Hepatic lipid synthesis in Wistar rats, measured as
the 1-h incorporation of 3H-acetate, was not affected by TCDD
treatment when studied 7 days after exposure to 10 µg TCDD/kg body
weight (Cunningham & Williams, 1972).
Mice (both C57BL/6 and DBA/2 strains) responded to TCDD treatment
with dose-dependent decreases in serum levels of glucose, cholesterol,
and triglycerides, and increases in hepatic triglycerides, whereas
serum glycerol and free fatty acid levels were unaffected (Chapman &
Schiller, 1985).
Hartley guinea-pigs given a lethal ip dose of 2 µg TCDD/kg body
weight had significantly increased circulating levels of cholesterol
esters, triglycerides, and phospholipids but a normal free fatty acid
level 7 days post-treatment. The increase in serum lipids was
accompanied by a pronounced increase in low density lipoproteins,
particularly the very low density lipoprotein fraction (Swift et al.,
1981). The plasma cholesterol and triglyceride levels were elevated
also when TCDD-treated guinea-pigs were compared to pair-fed controls
(Gasiewicz & Neal, 1979).
A TCDD-induced increase in plasma triglyceride levels was also
noted in New Zealand rabbits, fed 20 µg TCDD/kg body weight, whereas
plasma cholesterol was unaffected 12 weeks after dosing (Lovati et
al., 1984). TCDD treatment did not alter the liver lipid levels (free
and esterified cholesterol, triglycerides, and phospholipids), but the
triglyceride level was significantly increased. These results were
valid both for rabbits fed normal chow and those fed a
cholesterol-rich (0.5%) diet. Golden Syrian hamsters exposed to 1000
µg TCDD/kg body weight, orally or ip, had elevated plasma cholesterol
levels until 20 days after exposure but normal levels on day 50,
whereas serum triglyceride levels were normal until day 20 and then
became significantly lower than those of controls (Olson et al.,
1980b).
7.4.8 Enzyme induction
Primarily, TCDD has been found to increase enzyme activities
although observations on enzyme inhibition have also been made. Since
the first reports of enzyme systems as targets for TCDD (Buu-Hoi et
al., 1971a, 1972b; Greig, 1972; Poland & Glover, 1973b,c), enzyme
induction has become the most extensively studied biochemical response
produced by TCDD. The mixed function oxidase system (MFO), capable of
metabolizing both endogenous and foreign lipophilic compounds to more
polar products, has been the most thoroughly investigated one, and
arylhydrocarbon hydroxylase (AHH) and 7-ethoxy-resorufin 0-deethylase
(EROD) are the most frequently assayed enzyme activities in this
system. TCDD has also been reported to affect UDP-glucuronosyl
transferases (UDPGT) (Thunberg et al., 1980, 1984) and
glutathione-S-transferases (GT) (Manis & Apap, 1979), which are
multifunctional enzyme systems involved in conjugating a wide variety
of compounds.
Most studies have been performed with microsomal enzymes, but
TCDD has also been found to have effects on enzymes in the cytosolic
fraction. It seems that TCDD produces organ-specific effects, and
although, quantitatively, hepatic enzyme induction is of more concern
than extrahepatic enzyme effects, the latter may qualitatively be as
important. Studies in different species have revealed that enzyme
induction due to TCDD exposure also is a species-specific phenomenon.
Time course studies have shown that maximal increases in enzyme
activities are reached within 3 to 4 days post-treatment. After a lag
period of about 2 to 3 weeks, enzyme activities begin to return to
normal levels (Hook et al., 1975a,b; Lee & Suzuki, 1980; Lucier et
al., 1973; Poland & Glover, 1973a).
According to Kitchin & Woods (1979), TCDD-induced AHH activity
did not reach the normal level until 6 months after rats were exposed
to a single daily dose of 2 µg/kg body weight.
Hook et al. (1975a) found no apparent dependence on age when
studying AHH induction in CD rats which were 10 to 335 days old at the
time of exposure to 25 µg TCDD/kg body weight.
Several investigators have studied the relative potency of various
PCDDs and PCDFs to induce AHH and/or EROD activities (Bradlaw et al.,
1980; Poland et al., 1976; Bandiera et al., 1984a,b; Sawyer & Safe,
1985; Mason et al., 1986). They found an apparent structure-activity
relationship between the location of the halogen atoms on the
dibenzo-p-dioxin molecule and the ability to induce AHH activity
both in vivo and in vitro. Isomers with halogens at the four
lateral ring positions produced a greater biological response than
those with halogens at three lateral ring positions, while two
lateral halogen atoms seemed to be insufficient to produce a
biological response. TCDD was the most potent enzyme inducer of the
compounds tested.
On a molecular basis TCDD is the most potent MFO-inducing
compound known and MFO induction seems to be the most sensitive
biochemical response produced by this chemical. According to Kitchin
& Woods (1979), induction in the rat takes place after a single daily
dose of only 0.002 µg TCDD/kg body weight. In the guinea-pig (the
animal most sensitive to TCDD toxicity), MFO induction has been
observed, but the induced activities were low even at lethal doses
(Hook et al., 1975a). Neither is there a correlation in cell cultures
between induction of MFO and toxicity. Furthermore, it is known that
metabolites of TCDD are less toxic and more readily excreted than the
parent compound (see section 8.1.5). Thus, TCDD-induced MFO activities
represent a detoxification process rather than one leading to toxic
effects.
However, induction of MFO activities might potentiate the
toxicity of other foreign compounds requiring metabolic transformation
by the MFO system before they can exert their toxic effect. A number
of studies have shown that induction of MFO activities alters the
metabolism of the model xenobiotic, benzo(a)pyrene by increasing the
rate of microsomal metabolism, changing the metabolic profile to more
toxic metabolites, and increasing the extent of covalent binding to
liver microsomes (Berry et al., 1976, 1977; Uotila et al., 1978).
TCDD, applied topically or subcutaneously increased the
carcinogenicity of 3-methyl cholanthrene (MC) in DBA/2 mice (Kuori,
1978), but decreased the carcinogenicity of
7,12-dimethylbenz(a)anthracene (DMBA) in CD-1 mice (DiGiovanni et al.,
1979a). These authors suggested that TCDD induces the MFO system and
thus increases activation of MC to the ultimate carcinogen, as well as
inactivation of DMBA, which would explain these effects.
Furthermore, increased MFO activities might adversely affect
important metabolic pathways of endogenous compounds. The effects of
TCDD on enzyme activities, both MFO and others, involved in such
biological pathways as keratinization, steroid metabolism, lipid
metabolism, plasma membrane function and porphyrin metabolism, are
discussed under separate sections. The minute quantities of TCDD
required for maximal enzyme induction or suppression, the long
duration of the effect, and the stereospecific requirements suggest a
specific interaction of TCDD with a cellular species, possibly at the
gene level. Accordingly considerable research has been directed toward
the study of the genetic regulation of AHH induction by TCDD. A
hepatic cytosolic species that bound TCDD has been suggested as the
receptor for the hepatic AHH activity. Numerous studies of this
cytosolic receptor in several species and tissues have been performed
and it seems that there is a structural gene, the Ah locus, for this
receptor, which is responsible for the expression of various enzyme
activities (see sections 7.8 and 7.8.1).
7.4.8.1 Studies on rats
The effect of TCDD on enzyme activities has been most extensively
investigated in the rat. In the liver, TCDD has been shown to increase
both the content of cytochrome P-450 (Lucier et al., 1973, 1986;
Poland & Glover, 1974a,b; Hook et al., 1975a; Aitio & Parkki, 1978;
Kitchin & Woods, 1979; Madhukar & Matsumura, 1981; Goldstein & Linko,
1984) and cytochrome b5 (Lucier et al., 1973; Hook et al., 1975a), as
well as the microsomal enzyme activities involved in the oxidative
transformation and conjugation of xenobiotics, e.g., aniline
hydroxylase, arylhydrocarbon hydroxylase (AHH), biphenyl hydroxylase,
7-ethoxycoumarin-0-deethylase (ECOD), EROD, and UDPGT. These enzyme
activities have been investigated in a vast number of studies, some of
them quoted in Table 55. Goldstein & Linko (1984) demonstrated that
TCDD induced two isozymes of cytochrome P-450 (P-448) in the liver but
only one of these in extrahepatic tissues of young Sprague Dawley rats
2 days after a single oral dose of 25 µg/kg body weight.
Table 55. Studies demonstrating in vivo induction of mixed function
oxidases and UDP-glucuronosyltransferases in TCDD-exposed strains of rats
Rat
Enzyme activity strain Reference
Aniline hydroxylase SD Beatty et al. (1978)
CD Lucier et al. (1973); Hook et al. (1975a)
Aryl hydrocarbon SD Poland & Glover (1973a, 1974a); Beatty et
hydroxylase al. (1978); Manis & Apap (1979);
Haaparanta et al. (1983); Thunberg et al.
(1984); Lucier et al. (1986); Ahlborg et
al. (1987)
CD Lucier et al. (1973); Hook et al.
(1975a); Kitchin & Woods (1979)
Wistar Nagayama et al. (1983); Keys et al.
(1985); Bannister et al. (1986); Farrell
& Safe (1986); Mason et al. (1986);
Tsyrlov et al. (1986)
Biphenyl hydroxylase CD Hook et al. (1975a,b,); Kitchin & Woods
(1979)
7-Ethoxycoumarin-O- Wistar Aitio & Parkki (1978)
deethylase
7-Ethoxyresurofin-O- SD Haaparanta et al. (1983)
deethylase
CD Kitchin & Woods (1979)
Wistar Keys et al. (1985); Bannister et al.
(1986); Farrell & Safe (1986); Mason et
al. (1986)
UDP-glucuronosyl-
transferase:
p-nitrophenol SD Thunberg et al. (1980, 1984); Ahlborg et
al. (1987)
CD Lucier et al. (1973, 1975a, 1986); Hook
et al. (1975a)
Table 55 (Cont.) Studies demonstrating in vivo induction of mixed
function oxidases and UDP-glucuronosyltransferases in TCDD-exposed
strains of rats
Rat
Enzyme activity strain Reference
UDP-glucuronosyl-
transferase:
p-nitrophenol Wistar Aitio et al. (1979); Thunberg & Híkansson
(cont'd.) (1983)
o-aminophenol Wistar Aitio et al. (1979)
4-methylumbelli-
ferone Wistar Aitio & Parkki (1978)
Microsomal glutathion-s-transferase (GT) did not respond to
TCDD (Aitio & Parkki, 1978; Mukitari et al., 1981; Baars et al.,
1982), but cytosolic GT was induced both by a single dose of 17 µg
TCDD/kg body weight 2 days post-treatment (Manis & Apap, 1979), and by
near lethal or lethal doses 1 and 6 days after dosing (Mukitari et
al., 1981; Baars et al., 1982; Hassan et al., 1983). Glutathione
reductase was also increased, while glutathione peroxidase, both total
and Se-dependent, and the content of reduced glutathione were reduced
by TCDD treatment (Hassan et al., 1983, 1985a,b,c).
The following hepatic enzyme activities involved in drug
metabolism have been reported to be unaffected by TCDD treatment in
the rat: N-and O-demethylation (Lucier et al., 1973; Poland &
Glover, 1973a; Hook et al., 1975a; Beatty et al., 1978; Kitchin &
Woods, 1979; Madhukar & Matsumura, 1981), epoxide hydratase (EH)
(Aitio & Parkki, 1978), ß-glucuronidase (Lucier et al., 1973, 1975)
and NADPH cytochrome c reductase (Poland & Glover, 1974; Aitio &
Parkki, 1978; Kitchin & Woods, 1979; Madhukar & Matsumura, 1981). The
glucuronide conjugation of bilirubin (Aitio et al., 1979), estrone,
and testosterone (Lucier et al., 1975a) by liver microsomes from
TCDD-treated rats was not different when compared to control rats.
Some hepatic enzyme activities not belonging to the MFO system,
which are affected by TCDD treatment include aldehyde dehydrogenase
(Deitrich et al., 1978; Lindahl et al., 1978), delta-aminolevulinic
acid synthetase (see section 7.4.3), DT-diaphorase (Beatty & Neal,
1977), transglutaminase (see section 7.4.4), ornithine decarboxylase
(Nebert et al., 1980; Potter et al., 1982; Farrell & Safe, 1986),
plasma membrane ATPases (see section 7.4.2.2), porphyrinogen
carboxylase (see section 8.4.3), prostaglandin synthetase (see section
7.4.9), enzymes involved in testosterone metabolism (see section
7.4.9), and RNA polymerase (Kurl et al., 1982).
Prenatal and postnatal exposure via milk to TCDD, at doses of 3
µg/kg body weight to pregnant rats on days 5, 10 and 16 of gestation,
induced hepatic AHH and UDPGT activities in the offspring. The effect
was seen 8 days post-partum and persisted for at least 2 weeks. The
inductive effect was due both to exposure to TCDD via milk and to the
activation of an inducing mechanism after birth. Fetal liver AHH was
slightly increased during late gestation, although the UDPGT activity
and the cytochrome P-450 content were not (Lucier et al., 1975b).
Administration of 2.5 µg TCDD/kg body weight to pregnant rats on
day 17 of gestation increased the AHH- and N-hydroxylation activities
and cytochrome P-450 content in the fetal liver on day 20 of gestation
(Berry et al., 1976).
AHH induction due to TCDD has been reported to occur also in the
brain (Hook et al., 1975a), kidney (Poland & Glover, 1973a; Hook et
al., 1975a; Aitio & Parkki, 1978; Potter et al., 1982; Nagayama et
al., 1983), lung (Poland & Glover, 1973a; Hook et al., 1975a; Aitio &
Parkki, 1978; Nagayama et al., 1983), prostate (Lee & Suzuki, 1980;
Haaparanta et al., 1983; Nagayama et al., 1983), thymus (Nagayama et
al., 1983), and intestine (Poland & Glover, 1973a; Hook et al.,
1975a), but intestinal AHH activity was found to be unaffected by 17
and 20 µg TCDD/kg body weight (Aitio & Parkki, 1978; Manis & Apap,
1979). Testicular (Poland & Glover, 1973a; Hook et al., 1975a; Aitio
& Parkki, 1978) and adrenal (Guenthner et al., 1979) AHH activities
were not induced by sublethal doses of TCDD. The O-deethylation
activity in kidney, lung, and prostate was increased, but no effect
was seen on the activity in testes or intestine (Aitio & Parkki, 1978;
Haaparanta et al., 1983). UDPGT activities in kidney, lung, intestine,
and brain were increased, while no effect was seen on testicular UDPGT
(Hook et al., 1975a). Similar results were reported by Aitio & Parkki
(1978), though in their study intestinal UDPGT was not affected.
Renal biphenyl hydroxylation activity has been found to increase
after TCDD treatment, but no effect on this enzyme activity was seen
in lung, intestine, brain, or testes (Hook et al., 1975a). Elevated
levels of cytochrome P-450 were found in prostate (Lee & Suzuki, 1980)
and mammary gland (Rikans et al., 1979), but not in adrenals
(Guenthner et al., 1979). Less testicular cytochrome P-450 was found
after a single dose of 25 µg TCDD/kg body weight (Tofilon & Piper,
1982). The GSH tranferase activity was increased in the lung but not
in kidney, intestine, testes (Aitio & Parkki, 1978), or prostate (Lee
& Suzuki, 1980).
Neither EH nor NADPH cytochrome c reductase were inducible by
TCDD in kidney, lung, intestine, testes (Aitio & Parkki, 1978),
mammary gland (Rikans et al, 1979), or prostate (Lee & Suzuki, 1980).
The ED50 values for hepatic AHH and EROD induction were determined
in immature male Wistar rats 13 days after a single ip dose of
2,3,7-triCDD, TCDD, 1,3,7,8-tetraCDD, 1,2,3,7,8-pentaCDD,
1,2,4,7,8-pentaCDD or 1,2,3,4,7,8-hexaCDD (Mason et al., 1986). The
order of enzyme-inducing capacity was TCDD > 1,2,3,7,8-pentaCDD >
1,2,3,4,7,8-hexaCDD > 1,2,4,7,8-pentaCDD > 2,3,7-triCDD >
1,3,7,8-tetraCDD (Table 56).
7.4.8.2 Studies on mice
Enzyme induction studies in mice have been performed mainly with
two strains genetically separated at the Ah locus, thus making them
responsive (C57Bl/6 (B6)) or non-responsive (DBA/2 (D2)) to induction
of hepatic cytochrome P-450-related enzyme activities by aromatic
hydrocarbons, e.g., 3-methyl-cholanthrene (3-MC). However, the
extraordinary potency of TCDD for enzyme induction revealed increased
hepatic cytochrome P-450 content as well as AHH and O-deethylase
activities both in B6 and D2 mice after sublethal exposure to TCDD
(Poland & Glover, 1974a,b,c; Jones & Sweeney, 1977; Greenlee & Poland,
1978). Studies of MFO induction in five responsive and five
non-responsive strains of mice by Poland & Glover (1974a,b,c) revealed
that there were no consistent differences between the strains when
considering the extent to which TCDD induced AHH, O-deethylase,
N-demethylase and O-demethylase activities in the liver, kidney,
lung, skin, or bowel. The ED50 for hepatic AHH induction was
determined to be 10-9 mol/kg body weight in the responsive strain
and > 10-8 mol/kg body weight in the non-responsive strain (Poland &
Glover, 1975). Fully induced hepatic AHH activity was obtained both in
responsive (C57Bl/6 and AKR/Qdj) and non-responsive (DBA/2 and DDD)
strains of mice 3 days after an ip dose of 30 µg TCDD/kg body weight
(Nagayama et al., 1985a). Both AHH and EROD were induced in C57Bl/6
mice 7 days after an ip dose of 0.32 µg/kg body weight (Bannister et
al., 1986). Both hepatic AHH and ornithine decarboxylase activities
were similarly induced in C57Bl/6 and DBA/2 mice after a single ip
dose of 100 µg TCDD/kg body weight, but at 2 µg TCDD/kg body weight
these enzymes were induced only in the C57Bl/6 strain (Nebert et al.,
1980). Two daily doses of 0.1 µg TCDD, topically applied, increased
the epidermal AHH activity in two strains of hairless mice (Puhvel et
al., 1982). Contrary to observations in rats, TCDD induces testicular
AHH activity both in B6 and D2 mice 40h after an ip dose of 50 µg/kg
body weight (Mattison & Thorgeirsson, 1978).
Table 56. Structure activity relationships for some PCDDs
PCDD In vitro EC50 values (M)a In vivo ED50 values (mmol/kg)bLD50 c
Congener
Receptor AHH EROD AHH EROD Body Thymic Guinea-pig
binding weight atrophy µg/kg
body
weight
1- > 1.0x10-4 > 1.0x10-4 > 1.0x10-4
2,8- 3.2x10-6 > 1.0x10-4 > 1.0x10-4 > 300 000
1,2,4- 1.3x10-5 4.8x10-5 2.2x10-6
2,3,6- 2.2x10-7
2,3,7- 7.1x10-8 3.6x10-7 1.4x10-7 19.6 19.6 98.1 29 400
1,2,3,4- 1.3x10-6 3.7x10-6 2.4x10-6
1,2,3,8- 6.1x10-7
1,3,7,8- 7.9x10-7 5.9x10-7 3.2x10-7 31.2 77.6 132 100
2,3,6,7- 1.6x10-7 6.1x10-8 1.1x10-8
2,3,7,8- 1.0x10-8 7.2x10-11 1.9x10-10 0.004 0.003 0.05 0.09 2
1,2,3,4,7- 6.4x10-6 6.6x10-7 8.2x10-7
1,2,3,7,8- 7.9x10-8 1.1x10-8 1.7x10-8 0.031 0.056 0.62 0.17 3.1
1,2,4,7,8- 1.1x10-6 2.1x10-8 1.1x10-8 2.82 0.56 34 11.2 1125
1,2,3,4,7,8- 2.8x10-7 2.1x10-9 4.1x10-9 0.03 0.130 1.63 1.07 72.5
1,2,3,6,7,8-c 5.7x10-10 3.1x10-8 70-100
1,2,3,7,8,9-c 1.4x10-9 4.6x10-8 60-100
1,2,3,4,6,7,8c 1.3x10-7 > 600
1,2,3,4,6,7,9-c 3.7x10-6
1,2,3,4,6,7,8,9- > 1.0x10-5 > 1.0x10-4 > 1.0x10-4
a Estimated concentrations needed to displace 50% of 3H-TCDD bound to liver cytosol receptor from Wistar
rats and to produce 50% maximum enzyme induction in the rat hepatoma 11-4-II E cell line (Bradlaw & Casterline,
1979; Mason et al., 1986).
b Studies in immature male Wistar rats (Mason et al., 1986).
c McConnell et al., 1978b.
7.4.8.3 Studies on guinea-pigs
The guinea-pig, the species most sensitive to the toxic effects
of TCDD, does not respond with liver toxicity or with extensive
enzyme induction.
Hook et al. (1975a) investigated the effect of a single oral dose
of 0.175 µg TCDD/kg body weight on Hartley guinea-pig MFO and UDPGT
activities in liver, kidney, and lung. AHH induction was found only in
the kidney. In none of the tissues was there an effect on UDPGT
activity. The biphenyl 4-hydroxylase activity was increased in all
tissues, whereas hepatic biphenyl 2-hydroxylase was decreased. With
three daily doses of 1 µg TCDD/kg body weight, Hassan et al. (1983)
found an increase in hepatic AHH 6 days post-treatment. They also
found slightly increased in vitro lipid peroxidation but no effect
on glutathione content or on the enzyme activities facilitating
peroxidation, reduction, or transfer of glutathione. The DT-diaphorase
activity was not affected by a single oral dose of 0.6, 3.0, or 6.0 mg
TCDD/kg body weight (Beatty & Neal, 1977). The maximal increases were
4.4 and 22 times for AHH and EROD activities, respectively. The
testicular cytochrome P-450 content in Hartley guinea-pigs was
decreased by 50% one day after a single dose of 1 mg TCDD/kg body
weight. The effect persisted for at least 9 days (Tofilon, 1980). No
effect was seen on microsomal haeme content or on the activities of
NADPH-cytochrome C reductase and sorbitol dehydrogenase, the marker
enzyme for testicular protein synthesis. Thus, the decrease in
cytochrome P-450 induced by TCDD does not seem to be a nonspecific
inhibition of protein synthesis.
7.4.8.4 Studies on rabbits
Studies on enzymes in rabbits have been performed in the New
Zealand albino strain exposed to single doses of 10 to 30 mg TCDD/kg
body weight for 1 to 5 days. Both in adults (Johnson &
Muller-Eberhard, 1977a,b) and in neonates exposed in utero (Norman
et al., 1978; Kohli & Goldstein, 1981), TCDD increased the content of
cytochrome P-450. It also induced the formation of immunologically
distinct cytochromes P-450 in adult and neonatal liver (Norman et al.,
1978). Increased cytochrome P-450 was observed in the kidney but not
in the lung (Liem et al., 1980; Kohli & Goldstein, 1981). Renal and
pulmonary cytochrome P-450 reductase, investigated by Liem et al.
(1980), were not affected by TCDD treatment. Data on MFO induction and
suppression are conflicting. Liem et al. (1980) reported increased AHH
and O-deethylase activities in lung and kidney, whereas Hook et al.
(1975a) saw no effect on the AHH activity in the lung and reported a
decrease in hepatic AHH activity after a single oral dose of 0.5 µg
TCDD/kg body weight. Biphenyl 4-hydroxylase induction was seen in the
liver by Johnson et al. (1979). Hook et al. (1975a) detected such
induction in lung, but no effect in the liver and kidney (Hook et al.
1975a). Furthermore, Hook et al. (1975a) reported no effects on
biphenyl-2-hydroxylation and UDPGT activities in liver, kidney, or
lung. A decrease in hepatic, but not in renal or pulmonary
N-demethylation, was found by these authors.
7.4.8.5 Studies on hamsters
Golden Syrian hamsters are among the animals most resistant to
acute lethal effects induced by TCDD. Although the liver is a target
tissue, hepatic enzyme induction has barely been studied in this
species. When given an oral dose of 200 µg/TCDD/kg body weight for a
period of 3 days, increased hepatic glutathione-S-transferase and
glutathione reductase activities were found, but no effects were seen
on AHH or glutathione peroxidase activities. Neither the hepatic level
of glutathione nor the in vitro lipid peroxidation were affected
(Hassan et al., 1983). The ED50 values for induction of hepatic ECOD
and reduced NAD(P), menadione oxidoreductase activities, and
cytochrome P-450 content in male Golden Syrian hamsters were 1.0, 2.0,
and 0.5 µg TCDD/kg body weight, i.e. extremely low doses as compared
to doses that produce tissue damage and lethality in this species
(Gasiewicz et al., 1986).
7.4.8.6 Studies on cows
Three dairy Holstein cows (500-600 kg) received a single oral
dose of 0.05 (two cows) or 7.5 µg TCDD/kg body weight (Jones et al.,
1986). The cow receiving the high dose was killed on day 7 and those
receiving the low dose on day 14. AHH and EROD activities were
markedly induced in the high-dose but not in the low-dose animals.
7.4.8.7 Studies on chick embryos
AHH and delta-aminolevulinic acid synthetase in the chick embryo
have been reported to be extremely sensitive to the inductive effects
of TCDD (Poland & Glover, 1973b,c). Maximal induction occurred with
155 pmol TCDD/egg. The induction was relatively long lasting, with 70%
of the maximum induced activity present 5 days following a single dose
of TCDD. Structure-activity studies demonstrated a good correspondence
between the toxicity and induction potency of a series of
dibenzo-p-dioxin congeners (Poland & Glover, 1973c).
ED50 values for the induction of hepatic microsomal EROD, AHH,
and 4-dimethylaminoantipyrine-N-demethylase in 2-week-old white
Leghorn cockerels on day 5 after TCDD exposure were 778, 302, and 561
ng/kg body weight, respectively, and aldrin epoxidase was inhibited by
TCDD treatment (Sawyer et al., 1986). Hepatic and cardiac EROD
activities were increased in white Leghorn chicken embryos exposed to
TCDD in ovo at doses between 1000 and 10 000 pmol/egg (Quilley &
Rifkind, 1986).
7.4.8.8 Studies on cell cultures
TCDD has a very low toxicity in cell cultures, yet it is a very
potent inducer of AHH activity in these systems, including lymphocytes
and primary hepatocytes, as well as established and transformed cell
lines.
The inducibility of lymphocyte AHH has been investigated in
mitogen-stimulated human lymphocytes from the venous blood of healthy
volunteers. Kouri et al. (1974) found a dose-dependent increase in AHH
activity (0, 0.1, 1.0, 10, or 100 ng TCDD/ml medium for 24 h). The
optimal dose was about 10 ng/ml, and the maximal induction was by a
factor of 2 to 3. On the contrary, Gurtoo et al. (1979) found no
dose-response correlation, in the dose range 1.7 to 20 ng TCDD/ml,
when measuring lymphocyte AHH induction. To circumvent the limitation
of prior mitogen activation when studying AHH induction in
lymphocytes, Freedman et al. (1979) used the human B-lymphocyte
RPMI-1788 cell line, which does not require prior activation for the
induction of AHH activity. The optimal concentration to stimulate AHH
activity was determined to be 10 ng/ml medium. Highly variable
induction of AHH (between 3- and 28- fold) was obtained by Nagayama et
al. (1985b) in human lympho-blastoid cell lines derived from the
peripheral blood of healthy volunteers of both sexes and of variable
ages. The cells were exposed to 7.5 ng TCDD/ml medium for 48 h.
In a study by Niwa et al. (1975), the estimated ED50 values for
AHH induction by TCDD in 11 established cell lines, in fetal primary
cultures from five animal species and cultured human lymphocytes,
ranged from 0.04 ng/ml medium in C57Bl/6 mouse cultures and 0.08 ng/ml
in the rat hepatoma H-4-IIE cell line to more than 66 ng/ml in the HTC
rat hepatoma cell line. TCDD was demonstrated to be the most potent
AHH inducer out of 24 chlorinated dibenzo-p-dioxin analogues
(Bradlaw et al., 1980) tested in a rat hepatoma cell culture extremely
sensitive to AHH induction, the ED50 being about 0.5 pg/106 cells.
A 165-fold increase in AHH-activity and a 54-fold increase in EROD
activity were obtained in rat hepatoma H-4-IIE cells when exposed to
2 x 10-10 mol TCDD/litre for 3 days (Keys et al., 1986). In this
system co-exposure of TCDD with 1,3,6,8-tetraCDF and 2,4,6,8-tetraCDF
reduced the TCDD-induced enzyme induction, whereas co-exposure of TCDD
and TCDF resulted in an additive effect on enzyme induction (Keys et
al., 1986). The EC50 values for AHH and EROD induction in the same
cell system varied over 7 orders of magnitude for 14 different PCDDs
(Table 56), the most potent being TCDD and the least potent being
2,3,6-triCDD (Mason et al., 1986). A 2-to 650-fold AHH induction was
observable in 8 of 22 different cell cultures exposed to 10-9 mol
TCDD/litre for 24 h (Knutson & Poland, 1980a). The cells were derived
from tissues and/or species susceptible to TCDD toxicity in vivo.
Nanomolar concentrations of TCDD induced AHH activity in keratinocyte
cultures of human (Willey et al., 1984) and animal origin (Knutson &
Poland, 1980a).
Five human squamous carcinoma cell lines derived from tumours of
the epidermis and tongue responded to TCDD with increased
O-deethylase activity, the EC50 being 10-10 to 10-9 mol/litre
(Hudson et al., 1983a).
Steward & Byard (1981) treated primary hepatocytes isolated from
Sprague Dawley rats, for 48 h with various concentrations of TCDD.
They found a 2-fold induction of the AHH activity with 3 pg TCDD/106
cells. Maximal induction occurred with 2.4 ng TCDD/106 cells. Primary
hepatocytes, isolated from adult male Wistar rats, exhibited a linear
increase (from 2-to 4-fold) in AHH activity when exposed to TCDD in
the range 10-11 to 10-8 mol/litre for 72 h (Jansing & Shain,
1985). Primary hepatocytes from TCDD-treated (5, 10, or 25 µg TCDD/kg
body weight) rats, isolated 2 to 30 days post-treatment, showed
decreased ouabain and alpha-aminoisobutyric acid uptake as well as
tyrosine aminotransferase activity (Yang et al., 1983a). Treatment of
rats with 25 mg 1,3,6,8-tetraCDD/kg body weight did not affect these
parameters. Neither could these effects be demonstrated in primary
hepatocytes from control rats that were treated with TCDD (50, 100, or
200 nmol/litre medium) in vitro for 48 h.
The induction of AHH and EROD activities of a complex PCDD/PCDF
mixture from a fly ash extract has been reported (Safe et al., 1987).
7.4.9 Endocrine effects
Human exposure to TCDD has resulted in hirsutism and chloracne
(Table 64), symptoms that suggest an alteration in endocrine
regulation. Furthermore, chronic exposure to TCDD impaired
reproduction in experimental animals, possibly by interfering with the
estrous cycle (Kociba et al., 1976; Allen et al., 1977; Barsotti et
al., 1979; Murray et al., 1979). The ability of TCDD to mimic natural
steroids with steroid-like actions has prompted studies on the binding
of TCDD to steroid hormone receptors.
Over-production of glucocorticoids mimics some of the symptoms of
TCDD toxicity, e.g., involution of lymphoid tissues, oedema, and
mobilization of fatty acids from adipose tissues. Thus TCDD might
increase glucocorticoid activity by binding to glucocorticoid
receptors. However, TCDD was unable to displace 3H-dexamethasone, a
potent synthetic glucocorticoid, from the normal rat cytosol
glucocorticoid receptor even when present in 200-fold molar excess
(Neal et al., 1979). Poland et al. (1976) demonstrated that cortisol
and synthetic glucocorticoids did not bind to the TCDD receptor. An
increase in the plasma level of corticosterone was found in male
Sprague Dawley rats 7 and 14 days after a single oral dose of 50 µg
TCDD/kg body weight (Neal et al., 1979). With the method used, a
variety of fluorescent adrenocortical steroid hormone derivatives was
measured. In contrast, Balk & Piper (1984), using a competitive
binding radioassay for corticosterone, reported decreased blood levels
(29 and 26% of controls) of corticosterone in male Sprague Dawley rats
on days 14 and 21, respectively, after a single oral dose of 25 µg
TCDD/kg body weight. Accumulation of 11-ß-hydroxy-progesterone in the
blood of TCDD-treated rats was noticed on day 14 (Balk & Piper, 1984).
Neal et al. (1979) reported 100% mortality within 6 days in
adrenalectomized rats given 10, 20, 40, or 80 µg TCDD/kg body weight.
Adrenalectomy and hypophysectomy could not prevent liver lesions,
reduced growth rate, or thymic involution in female Fisher-344 rats
given a single oral dose of 10 or 20 µg TCDD/kg body weight (van
Logten et al., 1980). Thymic effects of TCDD became even more severe
after hypophysectomy. Daily sc injections of 0.25 mg growth hormone
had a positive influence on body weight gain but did not protect
against thymic involution in hypophysectomized rats.
Single oral or interperitoneal doses of TCDD between 7 and 100
µg/kg body weight decreased the serum thyroxine (T4) level in the rat
(Bastomsky, 1977; Potter et al., 1983, 1986b; McKinney et al., 1985;
Pazdernik & Rozman, 1985; Rozman et al., 1985b), but not in the
guinea-pig, after a single oral dose of 2 µg/kg body weight (McKinney
et al., 1985). The serum triiodothyronine (T3) level in TCDD-treated
rats was reported to be increased (Bastomsky, 1977; Potter et al.,
1986b), unaffected (Potter et al., 1983), or decreased (Pazdernik &
Rozman, 1985; Rozman et al., 1985b). Increased serum thyrotropin (TSH)
(Bastomsky, 1977; Potter et al., 1986b), increased thyroid
131I-uptake and increased biliary excretion of T4, but not of T3
(Bastomsky, 1977) have been reported in TCDD-treated rats.
Rats treated with TCDD at doses up to about 30 mg TCDD/kg body
weight and pair-fed controls had similar serum T4, T3, and TSH levels
when compared to ad libitum fed control rats (Potter et al., 1983,
1986b). Thus, it is unlikely that hypophagia is responsible for the
TCDD-induced changes in serum thyroid hormone levels. When mature
TCDD-treated rats were compared to pair-fed controls, there were no
functional alterations in thyroid status or thermogenesis, including
increased serum levels of T3, T4, and TSH after acute cold
challenge, increased total oxygen consumption after moderate cold
exposure or decreased basal metabolic rate as compared to ad
libitum fed control rats (Potter et al., 1986b). A significant
hypothermia was, however, observed in young TCDD-exposed rats
receiving 45 µg/kg body weight as a single ip dose (Potter et al.,
1983).
Available data on serum T4, T3, and TSH levels are not
sufficient to state whether TCDD-treated rats are functionally
hypothyroid, euthyroid, or hyperthyroid.
In ovo exposure of white Leghorn chicken embryos to TCDD in
the dose range 1 to 10 000 pmol/egg increased the cardiac release of
prostaglandins (Quilley & Rifkind, 1986). Potter et al. (1983)
reported decreased levels of insulin in serum and pancreas and of
somatostatin in the gastric antrum of Sprague Dawley rats 7 days after
a single ip dose of 45 µg TCDD/kg body weight, when compared to
pair-fed control rats. The somatostatin levels in serum, liver, and
pancreas were not affected, neither was the serum glucagon level.
The finding that steroids are an endogenous substrate for the
hepatic MFO system (Kuntzman et al., 1965) suggests that compounds,
such as TCDD, that influence the activity of this enzyme system may
alter steroid metabolism in vivo, and consequently also the
magnitude of steroid-mediated functions. As demonstrated by Gustafsson
& Ingelman-Sundberg (1979), the metabolic profiles of 4-androstene-3,
17-dione, 5 alpha-androstane-3 alpha, 17 ß-diol and 4-pregnene-3,
20-dione in hepatic microsomes from SD rats, treated with 20 µg
TCDD/kg body weight for 4 consecutive days, were changed when compared
to control rats 1 day post-treatment. The changes were most pronounced
in female rats. When five daily doses of 1 µg TCDD/kg body weight were
given to pregnant rats for 12 or 13 days during gestation, hepatic
microsomes showed decreased ability to form catechol estrogens and to
hydroxylate testosterone. However, this decrease did not relate to
altered circulating estradiol levels (Shiverick & Muther, 1983). TCDD
treatment did not affect the glucuronidation of testosterone and
estrogen (Lucier et al., 1975a) or prostaglandin synthesis (Kohli &
Goldstein, 1981). TCDD decreased the hepatic and uterine estrogen
receptor levels in 25-day-old Long-Evans rats 2 to 10 days after
treatment with single ip doses of 20 or 80 µg/kg body weight (Romkes
et al., 1987). Decreased estrogen receptor levels in the liver and
uterus also occurred 2 days after treatment with 1,2,7,8-tetraCDD,
1,2,3,7,8- pentaCDD, and 1,2,4,7,8-pentaCDD, but dose-response
relationships were present only for TCDD and 1,2,3,7,8-pentaCDD. The
estradiol-induced increases in hepatic and uterine estrogen receptor
levels were counteracted by simultaneous TCDD treatment, although the
effect of TCDD was not dose dependent. Hepatic hydroxylation of
testosterone in 2 ß-and 16 alpha- positions was not affected in male
Sprague Dawley rats (190-200 g) 3 days prior to a single oral dose of
15 µg TCDD/kg body weight (Hook et al., 1975b). Somewhat younger male
Wistar rats (100 g) treated with a single dose of 0.06 mmol TCDD/kg
body weight exhibited increased levels of 7 alpha-hydroxytestosterone
and decreased levels of 3alpha-, 16alpha- and 16 ß-hydroxylated
testosterone, as well as of androstenedione, in hepatic microsomes
when compared to control rats (Keys et al., 1985). Decreased
cytochrome P-450 content was found in guinea-pig (Tofilon, 1980) and
rat (Tofilon & Piper, 1982) testes for at least one week after oral
TCDD treatment of 1 µg/kg and 25 µg/kg body weight, respectively.
Testicular AHH activity was induced by TCDD in mice (Mattison &
Thorgeirsson, 1978), but not in rats after a single oral dose of 20 µg
TCDD/kg body weight (Aitio & Parkki, 1978). Mittler et al. (1984)
studied the effect of single ip doses of 0.2, 1, or 5 µg TCDD/kg body
weight on testicular 16-alpha-testosterone hydroxylase (16-TH),
6-ß-hydroxytestosterone (6-HT), and 7-alpha-hydroxy testosterone
(7-HT) activities in young Sprague Dawley rats 90 h after exposure.
Seminiferus 16-TH activity was increased from a non-detectable level
to 0.07-0.14 pg/mg protein and interstitial 6-HT activity was
increased 4-fold in TCDD-treated animals. The 7-HT activity was not
affected by TCDD, neither in the seminiferous tubules nor in the
interstitial fraction. Serum testosterone and dihydrotestosterone were
depressed dose-dependently by TCDD treatment, the ED50s being about
15 µg/kg body weight in male Sprague Dawley rats, when compared to
pair-fed and ad libitum fed controls (Moore et al., 1985). The
plasma clearance and biliary excretion of 3H-testosterone was not
affected in Sprague Dawley rats treated with 100 µg TCDD/kg body
weight before an i.v. injection of 3H-testosterone, neither did
castrated rats with implanted testosterone-leaking capsules respond to
a dose of 15 or 100 µg TCDD/kg body weight with decreased accessory
sex organ weights (Moore & Peterson, 1985). Increased AHH and
O-deethylase activities and cytochrome P-450 content have been
reported in rat prostate after single ip and oral doses, respectively,
of 10 µg/TCDD/kg body weight (Haaparanta et al., 1983; Lee & Suzuki,
1980).
7.4.10 Vitamin A storage
Decreased hepatic vitamin A storage has been reported in animals
exposed to various chlorinated aromatic compounds (Table 57). Compared
to other chlorinated hydrocarbons for which this effect has been
evaluated, TCDD is more potent in its ability to reduce the vitamin A
content of the liver.
A single oral dose of 10 µg TCDD/kg body weight decreased both
the total amount and the concentration of vitamin A in the liver of
adult male Sprague Dawley rats (Thunberg et al., 1979). The decrease
was evident 4 days after dosing and progressed with time. After 8
weeks the treated animals had a total liver vitamin A content
corresponding to 33% of that of controls. Decreased dietary intake of
vitamin A could not account for this difference. In a four-week study
TCDD was given as a single oral dose of 0, 0.1, 1.0, or 10 µg per kg
body weight to adult male Sprague Dawley rats fed ad libitum with
pelleted diets containing 1.2 (low), 3.0 (normal), or 6.0 (high) mg
vitamin A/kg diet (Thunberg et al, 1980). Both the concentration and
the total amount of vitamin A were decreased in a dose-dependent
manner in the animals receiving the high vitamin A diet. In the
animals on the normal and low vitamin A diets, significant differences
were seen only at doses of 1.0 and 10 µg TCDD/kg body weight. A
significant increase in the UDPGT activity was observed in all dietary
groups treated with 1.0 and 10 µg TCDD per kg body weight, suggestive
of an increased excretion of vitamin A conjugated with glucuronic acid
(Thunberg & Hakansson, 1983). However, no correlation between the
UDPGT activity and the reduction of hepatic vitamin A levels was seen
when homozygous Gunn rats lacking inducible UDPGT were treated with a
single oral dose of 20 µg/kg body weight (Aitio et al., 1979) nor in
heterozygous Gunn rats with inducible UDPGT after a single oral dose
of 10 µg/kg body weight (Thunberg & Hakansson, 1983).
Table 57. The potency of various chlorinated cyclic hydrocarbons to reduce hepatic vitamin A content in the rat
Compound Strain/sex/age Dose and route of Duration of % Reduction
(Reference) administration the study of hepatic
vitamin A
Arochlor 1242 Rattus norvegicus/M,Ff/21 days 100 mg/kg in dieta 2 months 49
(Cecil et al., 1973)
p,p-DDT Rattus norvegicus/M,F/21 days 100 mg/kg in dieta 2 months 38
(Cecil et al., 1973)
Methoxychlor Sprague Dawley/NRf/23 days 10 mg/kg in dietb 16 weeks 7
(Davison & Cox, 1976) 100 mg/kg in diet 12
1000 mg/kg in diet 37
10 000 mg/kg in diet 68
PCB Sprague Dawley/M/21 days 100 mg/kg in dietc 8 weeks 82
(Innami et al., 1976)
TCDD Sprague Dawley/M/NR 10 µg/kg bwf 7 days 29
(Thunberg et al., 1979) (single oral dose) 14 days 39
28 days 59
56 days 67
Table 57 (contd).
Compound Strain/sex/age Dose and route of Duration of % Reduction
(Reference) administration the study of hepatic
vitamin A
TCDD Sprague Dawley/M/NR 0.1 µg/kg bwg 28 days 2
(Thunberg et al., 1.0 µg/kg bwg 27
1980) 10.0 µg/kg bwg 65
TCDD Sprague Dawley/M/2 months 15 µg/kg bwg 44 days 59d 88e
(Hakansson, 1988) 30 µg/kg bwg 78d 98e
60 µg/kg bwg 81d 97e
120 µg/kg bwg 90d 99e
Toxaphene Sprague Dawley/M/NR 20 mg/kg bwh 4 weeks 0
(Thunberg et al.,
1984)
a 9-12 mg vitamin A/kg diet ad libitum.
b 33 000 IU vitamin A/kg diet ad libitum.
c 3000 IU vitamin A/kg diet ad libitum.
d 21 000 IU vitamin A/kg diet ad libitum.
e 8000 IU vitamin A/kg diet ad libitum.
f M = male; F = female; NR = not reported; bw = body weight.
g Single oral doses.
h Orally twice weekly.
Male Sprague Dawley rats received a single oral dose of 10 µg
TCDD/kg body weight 4 days prior to the oral administration of a
single physiological dose of labelled vitamin A,
(11,12-3H)retinylacetate (RA) (Hakansson & Ahlborg, 1985a). The
distribution and elimination of the radiolabel, and the vitamin A
content in various tissues were determined 1, 6, 12, 24, 72, and 192
h after the administration of 3H-vitamin A. The body burden of
radioactivity remained around 40% of the administered dose in control
rats throughout the study, whereas in TCDD-pretreated rats the body
burden decreased continuously from 21% at 12 h after administration to
11% at the end of the study. More of the radiolabel was recovered in
the kidney, testes, epididymis, and serum of TCDD-treated animals than
in controls, when calculated as percentage of body burden, whereas
less was recovered in the liver. Forty percent of the dose was
eliminated via faeces and urine in TCDD-treated rats, compared to 23%
in controls. More radioactivity was eliminated in faeces than in urine
both in control and TCDD-pretreated animals, although urinary
elimination was more pronounced in TCDD-pretreated than in control
rats. It was concluded that TCDD-treated rats handled the newly
administered dose of vitamin A in a similar way to rats deficient in
Vitamin A (Huque, 1981; Blomhoff et al., 1982). This finding is
remarkable since the TCDD-treated animals in this study still had
considerable stores of hepatic vitamin A, and did not show decreased
levels of serum vitamin A, i.e., they were not deficient in vitamin A.
In a similarly designed study the effect of a single oral dose of 10
µg TCDD/kg body weight on the endogenous pool of vitamin A
(radiolabelled 15-3H-retinol given 5 to 7 days prior to TCDD
treatment) was studied in Sprague Dawley rats with stores of low liver
vitamin A (Hakansson et al., 1986). It was demonstrated that
endogenously stored vitamin A was rapidly depleted from the liver of
TCDD-treated rats and was eliminated both in faeces and in urine. An
increased distribution of the vitamin A stored in the liver to
extrahepatic tissues was also seen in the treated rats.
To elucidate whether dietary vitamin A would reduce TCDD
toxicity, Hakansson (1988) fed male Sprague Dawley rats ad libitum
from weaning throughout the experiment with diets containing 2000 (I),
5000 (II), 8000 (III), or 21 000 (IV) IU of vitamin A/kg. A single
oral dose of TCDD (15, 30, 60, or 120 µg TCDD/kg body weight) was
given when the rats were 8 weeks old and the animals were killed 44
days post-treatment. With diet IV, TCDD reduced in a dose-dependent
manner hepatic vitamin A by 59 to 90%. With diets II and III,
reduction of hepatic vitamin A was more than 95% after dosing with 15
and 30 µg TCDD/kg, respectively. In control animals fed diet I, total
hepatic vitamin A was less than 1 µg and TCDD had no further effect at
any dose. Serum vitamin A was dose-dependently decreased by TCDD
treatment in dietary groups I, II, and III, whereas in dietary group
IV TCDD increased serum vitamin A. Only with the highest TCDD dose was
there a counteraction by dietary vitamin A on all of the above
parameters.
A single dose of 10 µg TCDD/kg body weight to female Sprague
Dawley rats on the day of delivery affected the vitamin A content in
the liver and kidney of the offspring (Hakansson et al., 1987). The
effect became clearly visible after weaning, i.e., when the dietary
intake of vitamin A was high enough to allow for storage. At the end
of the study (postnatal day 32), the vitamin A content in the liver
was 225 µg in control pups and 102 µg in TCDD-exposed pups. The
corresponding values for the kidney vitamin A content were 1.4 and 8.4
µg, respectively. The TCDD-induced effects on vitamin A levels in the
liver and kidneys followed a similar time-course as the growth
reduction, i.e., a minor effect throughout the lactation period, which
became more pronounced post-weaning. This was in contrast to the liver
enlargement and thymus involution in TCDD-exposed pups, which were
most pronounced throughout lactation and tended to diminish post-
weaning.
Single oral doses of 0, 1, 5 and 10 µg TCDD/kg body weight or a
mixture of PCDDs and PCDFs, reconstituting the levels found in human
milk, were given to male Sprague Dawley rats (80 g) in corn oil
(Ahlborg et al., 1987). The mixture was given at three dose levels
resulting in 1, 5, or 10 µg TCDD/kg body weight. Four weeks after
dosing there was a dose-dependent decrease in hepatic vitamin A in
TCDD-treated rats; no further effect was seen in the animals treated
with PCDD/PCDF mixtures. In contrast, mixture treatment had an
additive effect, as compared to TCDD alone, on the increases in renal
vitamin A and hepatic cytochrome P-450 contents, AHH activity, and
UDPGT activity.
Taken together these data indicate that TCDD interferes with the
storage mechanism for vitamin A. In the liver this mechanism has been
thoroughly investigated (Hirosawa & Yamada, 1973; Blomhoff et al.,
1982; Olson & Gunning, 1983). As dietary vitamin A seems unable to
counteract all toxic effects, this would imply either that the effect
on vitamin A storage is secondary to TCDD toxicity or that the
cellular utilization of vitamin A is affected by TCDD.
7.5 Embryotoxicity and Reproductive Effects
The teratogenic potential of TCDD was first demonstrated in rats
and mice by Courtney & Moore (1971). This followed the finding that
2,4,5-trichlorophenoxyacetic acid contaminated with 30 mg TCDD/kg was
teratogenic in two strains of mice and one strain of rat (Courtney et
al., 1970), leading to an increased incidence of cleft palate and
cystic kidney in both strains of mice and cystic kidney in rats.
Further studies have revealed that TCDD is fetotoxic rather than
teratogenic in the rat, producing subcutaneous oedema, haemorrhages,
and slight kidney anomalies (see section 7.5.1). In contrast, TCDD
produces a specific teratogenic response, consisting of cleft palate
and kidney malformations, in several strains of mice (see section
7.5.2 and Table 58). Extra ribs, minor abnormalities in the palate,
and cardiovascular malformations have been demonstrated in rabbits,
monkeys, and chickens, respectively, after exposure to TCDD in utero
(see sections 8.5.3-8.5.6). Impaired reproductive performance due to
TCDD exposure has been demonstrated in rats and monkeys (see sections
8.5.1 and 8.5.4).
7.5.1 Studies on rats
The initial studies on the embryotoxic effects of TCDD in the rat
were performed with Charles River CD (Courtney & Moore, 1971), Sprague
Dawley (Sparschu et al., 1971), and Wistar rats (Khera & Ruddick,
1973). In these studies maternal toxicity was noted at doses > 0.5
(Sparschu et al., 1971) or 1 µg/kg body weight per day (Khera &
Ruddick, 1973). Decreases in gestational survival, fetal weight, and
postnatal survival were reported at dose levels in the range 0.125-0.5
µg TCDD/kg body weight per day on gestation days 6-15 or 5-14.
Haemorrhages, mainly intestinal, and subcutaneous oedema were common
findings at similar doses. The teratogenic findings were limited to
kidney anomalies, described as unilocular cystinephrotic kidney or as
hydronephrosis, in the CD rat at or above 0.5 µg TCDD/kg per day
(Courtney & Moore, 1971). Slightly dilated renal pelvis was reported
in the F1 generation at the 0.01 µg/kg per day dose level in a
three-generation reproductive study of TCDD in Sprague Dawley rats
(Murray et al., 1979). Giavini et al. (1983) found an increased
incidence of renal anomalies in Charles River CD offspring exposed to
2 µg TCDD/kg body weight on gestation days 0 to 2, but not in Sprague
Dawley offspring when the dam was given daily oral doses of 0, 0.125,
0.5, and 2 µg TCDD/kg body weight for 2 weeks before mating (Giavini
et al., 1982a).
Sparschu et al. (1971) found an increased number of resorption
sites in Sprague Dawley rats at doses > 0.5 g TCDD/kg per day, but
no effect on ovulation rate and preimplantation loss was reported even
at 2 µg/kg per day. TCDD exposure (0.1, 0.5, or 2.0 µg/kg per day) on
gestation days 0 to 2 had no effect on the reproductive performance of
Sprague Dawley rats (Giavini et al., 1983). In contrast, impaired
reproductive performance was found in Charles River CD rats after
receiving TCDD for two weeks before mating (Giavini et al., 1982a).
The number of resorption sites was elevated at doses of 0.5 µg TCDD/kg
per day or more, whereas the ovulation rate and preimplantation loss
was affected only at 2 µg/kg per day.
The male reproductive ability was affected by TCDD treatment only
at toxic doses, as judged by studies of Khera & Ruddick (1973) and
Murray et al. (1979). Nevertheless, decreased mating frequency was
noted in the groups, of the F0 generation that received 0.1 µg
TCDD/kg per day in the diet (Murray et al., 1979). No effect on the
mating frequency was found by Giavini et al. (1982a, 1983).
In a three-generation reproduction study, Murray et al. (1979)
maintained Sprague Dawley rats on diets providing doses of 0, 0.001,
0.01, or 0.1 µg TCDD/kg body weight per day. The F0 generation
received the diet for 90 days before mating. No toxic effects were
observed in the F0 generation but decreased body weight and reduced
food consumption were noted in the F1 and F2 generations at 0.01
µg/kg per day. Fertility was greatly reduced at 0.1 µg/kg per day in
the F0 generation. This group was discontinued because of the low
number of offspring. In the F1 and F2 generations fertility was
significantly reduced at 0.001 and 0.01 µg/kg per day, respectively.
At 0.01 µg/kg per day, litter sizes were reduced, and fetal and
neonatal survival were decreased as well as postnatal growth. Murray
et al. (1979) concluded that doses of 0.1 and 0.01 µg/kg per day
impaired reproduction in rats. A dose of 0.001 µg/kg per day had no
effect on fertility, litter size, postnatal body weight, or neonatal
survival and was therefore suggested to be a no-effect dose for
reproductive lesions.
Reevaluation of these data by Murray et al. (1979) using another
statistical model, including pooling of the data from the four
generations, led to the conclusion that the 0.001 µg/kg per day dose
level did affect reproduction and thus was not a no-effect level, but
a low-effect level (Nisbet & Paxton, 1982). Kimbrough et al. (1984)
considered that the data by Murray et al. (1979) could not be used for
risk assessment calculations due to the great variation in fertility
index both in controls and exposed rats.
No embryotoxic and/or reproductive effects were found when female
Wistar rats were exposed to 1,2,3,4-tetraCDD (50, 100, 200, 400, or
800 µg/kg body weight per day), 2,7-diCDD (250, 500, 1000 or 2000
µg/kg body weight per day); 2,3-diCDD (1000 or 2000 µg/kg body weight
per day), or 2-monoCDD (1000 or 2000 µg/kg body weight per day) on
gestation days 6-15 (Khera & Ruddick, 1973). The maturation process of
the lung was not affected in Sherman rats exposed to 2,7-diCDD (40
µg/kg per day on gestation days 7-15) (Kimbrough et al., 1974).
7.5.2 Studies on mice
TCDD-induced embryo mortality in NMRI mice was significantly
increased at doses of 4.5 and 9 µg TCDD/kg body weight per day when
given on gestation days 6-15, but no embryotoxic effect was observed
when daily doses of 9 µg/kg body weight were given on gestation days
9-13 (Neubert & Dillman, 1982). The number of resorptions on day 13 of
gestation in NMRI mice was increased, as compared to controls, when 25
µg TCDD/kg body weight was given, divided into five daily doses on
days 7-11 of gestation, but no effect was observed when the same dose
was given as single ip injections on days 7 or 10 of gestation (Nau &
Bass, 1981). A single ip dose of 30 µg TCDD/kg body weight on
gestation day 11 had no effect on the fetal mortality in C57Bl/6 mice
on days 12, 13, or 14 of gestation (Weber & Birnbaum, 1985). Decreased
pre- and postnatal survival rates and retarded postnatal development
were observed in NMRI mice given four oral doses of 12.5 µg TCDD/kg
body weight on gestation days 14-17 (Nau et al., 1986). The cumulative
mortality was 45%, 68.5%, and 75% in exposed offspring on postnatal
days 1, 14, and 22, respectively, as compared to 6% in controls on day
22.
Table 58 summarizes the early studies on the teratogenic effects
of TCDD in various strains of mice. These studies (Courtney & Moore,
1971; Neubert & Dillman, 1972; Courtney, 1976; Smith et al., 1976),
revealed that TCDD is a specific teratogen in mice causing increased
frequencies of kidney anomalies and cleft palate at doses well below
those which result in fetal mortality and maternal toxicity.
The TCDD-induced kidney anomaly is morphologically described as
a progressive hydronephrosis, preferentially occurring in the right
kidney, and never accompanied by hydroureter or abnormal nephron
development (Courtney & Moore, 1971; Moore et al., 1973; Birnbaum et
al., 1985; Weber et al., 1985).
The fetal kidney seems to be more susceptible to TCDD exposure
than the developing palate (Courtney & Moore, 1971; Moore et al.,
1973; Birnbaum et al., 1986; Weber et al.,1984). The incidence of
kidney anomalies after a single dose of 1 µg TCDD/kg body weight on
gestation day 10 in C57Bl/6 mice was 34.3%. If the same dose was
divided and given on gestation days 10-13, the incidence of kidney
anomalies was 58.9%. The incidences of cleft palate in the same
studies were 0 and 1.9%, respectively (Moore et al., 1973). In
contrast, in the CF-1 strain of mice, cleft palate was a more
sensitive parameter than hydronephrosis, occurring at 1 and 3 µg
TCDD/kg body weight per day on gestation days 6 to 15, respectively
(Smith et al., 1976). It would appear that 3 µg TCDD/kg per day on
gestation days 10-13 is close to a threshold dose for cleft palate
induction in C57Bl/6 mice (Moore et al., 1973; Birnbaum et al., 1986).
The maximum increase in cleft palate incidence is produced if TCDD is
administered on any individual day from gestation day 8-10 in C57Bl/6
mice (Pratt et al., 1984) or on gestation day 10-11 in NMRI mice (Nau
& Bass, 1981; Neubert & Dillman, 1982; Krowke, 1986). The cleft palate
incidence in C57Bl/6 mice was more pronounced (36%) when TCDD was
given as a single oral dose of 12 µg/kg body weight on gestation day
11 than if 3 µg/kg body weight per day was given on gestation days
10-13 (Birnbaum et al., 1985). No difference was seen in the incidence
of kidney anomalies with the same treatment. TCDD is not a potent
inducer of cleft palate when given on day 13 of gestation or later
(Neubert & Dillman, 1982; Pratt et al., 1984). Examination of cryostat
sections taken from C57Bl/6 embryos during the time of palatal
elevation and fusion demonstrated that TCDD does not interfere with
growth, elevation, or initial contact of the palatal shelves but does
interfere with the firm adhesion and/or degeneration of the medial
epithelial cells, i.e., programmed epithelial cell death does not
appear to occur in the medial epithelium in embryos exposed to TCDD
(Pratt et al., 1984). The cleft palates that were observed were
complete clefts of the entire hard and soft palate and no clefts of
the primary palate were observed (Pratt et al., 1984).
Table 58. Early studies on embryotoxic effects of TCDD in mice
Strain/sexb Route/vehicle/dose Treatment/ Parental toxicity Embryotoxic effects
(reference) (µg/kg body weight observation
day) (days)a
CD-1/F subcutaneous/ 6-15 / 18 No effect Increased incidence of kidney
(Courtney & DMSO/1.0, 3.0 anomalies at doses > 1 µg/kg
Moore, 1971) per day.
BDA/2J subcutaneous/ 6-15 / 17 Increased relative Increased incidence of cleft
(Courtney & DMSO/3.0 liver weight palate and kidney anomalies.
Moore, 1971)
C56BL/6J subcutaneous/ 6-15 / 17 Increased relative Increased incidence of cleft
(Courtney & DMSO/3.0 liver weight palate and kidney anomalies.
Moore, 1971)
NMRI/F oral/rape seed 6-15 / 18 None reported Increased number of resorptions
(Neubert & oil/0.3, 3.0, 4.5, at 9 µg/kg per day. Increased
Dillman, 1972) 9.0 incidence of cleft palate at
doses > 3 µg/kg per day.
C56BL/6J oral/corn oil:acetone 10-13 / 18 None reported Increased incidence of cleft
(Neubert & (9:1)/1.0, 3.0 palate at doses > 3 µg/kg
Dillman, 1972) per day increased incidence
of kidney anomalies at doses
> 1 µg/kg per day.
Table 58 (contd).
Strain/sexb Route/vehicle/dose Treatment/ Parental toxicity Embryotoxic effects
(reference) (µg/kg body weight observation
day) (days)a
CD-1/F oral/corn oil:anisole 7-16 / 18 Increased maternal Increased fetal mortality at all
(Courtney, 1976) (95:5)/25, 50, 100, relative liver weight doses. Increased incidence of
200, 400 at 25 and 50 µg/kg per kidney anomalies and cleft palate
day. Marked oedema and at doses > 25 µg/kg per day
vaginal bleeding at and 50 µg/kg per day,
doses > 200 µg/kg per respectively. Increased incidence
day. of club foot was found in the
high-dose group. Hydrocephalus and
open eyes were seen occasionally.
CF-1 oral/corn oil:acetone 6-15 / 18 Increased maternal Increased number of resorptions
(Smith et al., (98:2)/0.001, 0.01, relative liver weight at 1 µg/kg per day. Increased
1976) 0.1, 1.0, 3.0 at 3 µg/kg per day. incidence of cleft palate at
doses > 1 µg/kg per day.
Dilated renal pelvis occurred at
3 µg/kg per day.
a First day of gestation designated day zero.
b F = female.
Poland & Glover (1980) reported that in nine out of ten inbred
strains of mice, the susceptibility to cleft palate formation produced
by TCDD (30 µg TCDD/kg body weight sc on day 10 of pregnancy) followed
the distribution of the Ah locus within that strain. The five strains
with a low affinity TCDD receptor in the liver (DBA/2, RI, AKR, SWR,
and 129) developed cleft palate at an incidence of 0 to 3% while four
of the five strains with a high affinity TCDD receptor in the liver
(C57Bl/6, A, BALB/cBy, and SEC) developed cleft palate at an incidence
of 54 to 95%. The only strain with a high affinity hepatic TCDD
receptor that did not develop cleft palate was CBA. Mid-gestational
mice embryos from C57Bl/6 exhibit high levels of the TCDD receptor in
the maxillary processes and secondary palatal shelves whereas no
specific binding of TCDD could be demonstrated in the AKR strain
(Dencker & Pratt, 1981). Evidence that TCDD interferes in embryonic
development by directly interacting with embryonic cells rather than
being secondary to maternal effects was presented by D'Argy et al.,
(1984) in a study where mouse blastocysts were transplanted between
NMRI (sensitive) and DBA (non-sensitive) dams on gestation day 3. TCDD
treatment (30 µg/kg body weight) on gestation day 10 resulted in 75 to
100% incidence of cleft palate among NMRI fetuses whether they
remained in their own dams or as aliens in DBA dams. Also, none of the
DBA fetuses developed cleft palate whether or not they remained in
their own dams or as aliens in NMRI dams.
Attempts to modify and further characterize the TCDD-induced
cleft palate formation have been performed with several interacting
substances. The non-teratogenic ß-naphthoflavone (ßN) enhanced the
TCDD-induced fetal mortality and the increase in the incidence of
cleft palate in C57Bl/6 and NMRI mice when ßN was administered
simultaneously or 8 h before TCDD, but not 24 h before or after
(Hassoun & Dencker, 1982). TCDD was given as a single ip dose of 25 or
16 mg/kg body weight to C57Bl/6 and NMRI mice, respectively, on
gestation days 10, 11, 12, or 13. TCDD-induced cleft palate incidence
was enhanced by 2,3,4,5,3',4'-hexachlorobiphenyl, but not
2,4,5,2',4',5'-hexachlorobiphenyl, in C57Bl/6 mice, although none of
the isomers alone were teratogenic at the doses used (Birnbaum et al.,
1985). Neither of the hexachlorobiphenyls used affected the incidence
of renal anomalies, as compared to TCDD alone.
A dose-related enhancement of the TCDD-induced incidence of cleft
palate was found in C57Bl/6 mice exposed to either triiodothyronine or
thyroxine, as compared to TCDD alone (Lamb et al., 1986). The
hydrocortisone-induced cleft palate response was enhanced by
simultaneous administration of TCDD in C57Bl/6 mice (Birnbaum et al.,
1986). Co-administration of TCDD and TCDF to C57Bl/6 mice on gestation
day 10 resulted in a cleft palate incidence compatible with an
additive toxicity model in which TCDF contributes to the toxicity of
TCDD in a weight ratio of 1:30 (Weber et al., 1985). Also 1,2,3,7,8-
pentaCDD and 1,2,3,4,7,8-hexaCDD had additive effects on the
TCDD-induced cleft palate incidence in NMRI mice (Krowke, 1986). No
clearly dose-related teratogenic effects were observed in C57Bl/6 mice
exposed to oral doses of 1,2,3,4- tetraCDD (100, 250, 500, or 1000
mg/kg per day), octaCDD (5 or 20 mg/kg per day) or to a mixture of 40%
2,7-diCDD and 60% 2,3,7-triCDD (100 or 200 mg/kg per day) on gestation
days 7-16 (Courtney, 1976).
7.5.3 Studies on rabbits
New Zealand rabbits were administered TCDD by gavage in doses of
0, 0.1, 0.25, 0.5, and 1 µg/kg per day on days 6-15 of gestation and
the fetuses were examined on day 28 of gestation (Giavani et al.,
1982b). Above 0.25 µg/kg per day, decreased maternal weight gain and
unspecified signs of maternal toxicity were reported. At doses of 0.5
and 1 µg/kg per day, there were 2/15 and 4/10 maternal deaths,
respectively. An increase in abortion and resorption rates occurred at
doses above 0.25 µg/kg per day, with no live fetuses detected in the
1 µg per kg per day dose group. There was a significant increase in
extra ribs, compared to a level of 33.3% in the controls to 82, 66.6,
and 82%. In the 0.1, 0.25, and 0.5 µg/kg per day dose groups, 82,
66.6, and 82% extra ribs were noted. There were no increases in
specific soft tissue anomalies.
7.5.4 Studies on monkeys
The effect of exposure to TCDD in the diet, before mating and
throughout gestation, on the reproductive performance and production
of progeny of healthy, fertile rhesus monkeys (Macaca mulatta) was
studied by Allen and co-workers (Allen et al., 1979a,b; Barsotti et
al., 1979; Schantz et al., 1979). At a level of 500 ng TCDD/kg diet
(11 ng/kg body weight per day), there were no effects on the length,
intensity, or duration of the menstrual cycle, but decreases in serum
estradiol and progesterone levels were observed (Barsotti et al.,
1979). Mating with control males at the end of the sixth month
resulted in three pregnancies, out of which two resulted in abortion,
and three animals failed to conceive. The remaining TCDD-treated
female was not bred due to toxic symptoms. The two females that
survived the study were returned to a control diet and later gave
birth to well developed infants. After 7 months on the 50 ng TCDD/kg
diet (1.5 ng/kg body weight per day), the reproductive outcome was:
four abortions, one stillbirth, two failures to conceive and two
normal births (Schantz et al., 1979).
All controls conceived and gave birth to normal infants (Barsotti
et al., 1979; Schantz et al., 1979).
McNulty (1984) demonstrated that rhesus monkeys (Macaca mulatta)
receiving 1 µg TCDD/kg body weight either as a single dose on
gestation days 25, 30, 35, or 40, or divided into nine doses between
gestation days 20-40, failed to give birth normally. Of 16 pregnancies
13 resulted in abortions. Two of the three live fetuses, obtained by
Caesarean section on day 145 of gestation, showed minor abnormalities
in the palate. Maternal toxicity, manifest in 8 out of 16 females,
appeared only after a period of 44-111 days after abortion. From the
results obtained it was not possible to conclude whether fetal death
was a direct effect of TCDD on the fetus or placenta or an indirect
effect through maternal toxicity.
7.5.5 Studies on chickens
Treatment of fertile White Leghorn chicken eggs, on day 0 of
development, with single doses of TCDD, ranging from 0.009 to 77.5
pmol/egg, resulted in a dose-related increase in the following types
of cardiovascular malformations: ventricular septal defects, aortic
arch anomalies, aortic arch anomaly plus ventricular septal defect,
and conotruncal malformation (Cheung et al., 1981). The dose producing
cardiovascular malformations in 50% of the embryos was about 1 pmole
TCDD/egg.
A dose-dependent decrease in hatchability and increased
incidences of beak, brain, and leg malformations were found in the
embryos when fertile White Leghorn eggs were injected with toxic fat
material containing 0.9, 1.8, or 4.5 ng of a mixture of dioxins (14%
diCDDs, 1% triCDDs, 38 to 45% tetraCDDs, 13% pentaCDDs, 14% hexaCDDs,
12% heptaCDDs, 8% octaCDD) (Flick et al., 1973).
7.6 Mutagenicity and Related End-Points
7.6.1 Mutagenicity
7.6.1.1 Studies on bacteria
Results from bacterial mutagenicity tests with TCDD are
conflicting.
In studies by Hussain et al. (1972) and Seiler (1973), a positive
response was reported in the Salmonella typhimurium strain TA 1532
without metabolic activation in a plate test after preincubation of
bacteria in medium containing TCDD and also in a spot test. A
mutagenic response was also obtained with Escherichia coli Sd-4,
measuring reversion to streptomycin independence (Hussain et al.,
1972). OctaCDD was not mutagenic to various strains of Salmonella
typhimurium (Seiler, 1973) without metabolic activation. More recent
publications, however, do not report any mutagenic effect of TCDD in
the Ames' Salmonella plate incorporation assay using strains TA
1530, TA 1532, TA 1535, TA 1537, TA 1538, TA 98, or TA 100 in the
presence or absence of metabolic activation systems from rat and
Syrian hamster liver (Gilbert et al., 1980; Geiger & Neal, 1981;
Mortelmans, 1984). In these studies the earlier reported positive
tester strain TA 1532 was either replaced by strain TA 1537 (Geiger &
Neal, 1981; Mortelmans, 1984) or was tested in addition to TA 1537
(Gilbert et al., 1980). Strain TA 1537 has been derived from TA 1532
and is more sensitive, due to its improved uptake of large molecules.
TCDD was tested in the dose range 0.2-2000 µg/plate. Due to the
limited solubility of TCDD, a maximal dose in the Salmonella system
was reported to be 20 µg TCDD/plate (Geiger & Neal, 1981).
7.6.1.2 Studies on eukaryotic cells
Bronzetti et al. (1983) reported a mutagenic response of TCDD in
yeast in an in vitro suspension test and a host mediated assay. In
both assays Saccharomyces cerevisiae strain D7 was used. Positive
responses were obtained in vitro in the presence of metabolic
activation at doses of TCDD up to 10 µg TCDD/ml, and in the
host-mediated assay after treatment of mice with a single dose of TCDD
(25 µg/kg body weight).
In L5178Y mouse lymphoma cells, TCDD induced mutations in a
dose-dependent manner at doses of 0.05-0.5 µg TCDD/ml; survival of
cells at the highest concentration was at least 75% (Rogers et al.,
1982).
7.6.1.3 In vivo studies
A dominant lethal test on male Wistar rats has been reported by
Khera & Ruddick (1973). The rats were given 4, 8, or 12 µg TCDD/kg
body weight per day orally for seven consecutive days, after which
seven sequential mating trials, 5 days at a time, were conducted in
the surviving males. Nine days after separation of females from the
males, the females were killed. The highest dose was lethal for all
males exposed (20/20); 8 µg/kg per day killed 11/20 males, and 4 µg/kg
per day was lethal for 2/20. All animals in the control group
survived.
The results did not indicate a dominant lethal effect during the
35 days post-treatment period, corresponding to the postmeiotic stages
of spermatogenesis.
7.6.2 Interaction with nucleic acids
Poland & Glover (1979) found that very little TCDD bound to rat
liver nucleic acids after treatment with 3H-TCDD in vivo; the
maximum covalently bound TCDD was calculated to be 6 and 12 pmol per
mol of nucleotide residues from DNA and RNA, respectively. After iv
injection of 3H-TCDD in rats, the radioactivity taken up by the
liver cytosol decreased at the same rate as the radioactivity in the
nuclear fraction increased. The radioactivity in the nuclei was at a
maximum 2 h after injection (Carlstedt-Duke et al., 1982). Guenthner
et al. (1979) demonstrated in vitro metabolism of TCDD to reactive
intermediates that bound covalently to cellular macromolecules,
principally to proteins. However, no isomer-specific methods were used
for analysis of metabolites in this study.
Liver slices from Sprague Dawley rats treated with 5 µg TCDD
incorporated twice the level of thymidine into nuclear DNA 10 days
post-treatment than did controls (Conaway & Matsumura, 1975).
Christian & Peterson (1983a) found no effect on the in vivo
incorporation of thymidine into liver DNA 35 to 36 h after the
administration of 10 µg TCDD/kg body weight to Sprague Dawley rats.
DNA synthesis in Porton rats, stimulated by 70% hepatectomy and
measured as the 1 h in vivo incorporation of thymidine, was not
affected by treatment with 10 or 200 µg TCDD/kg body weight 0, 24, or
72 h before the hepatectomy was performed (Greig et al., 1974). In
contrast, Dickins et al. (1981) found an 8-to 10-fold increase in DNA
synthesis in response to the proliferation caused by a 1/3 hepatectomy
in Sprague Dawley rats given 5 µg TCDD/kg body weight 5 days prior to
the hepatectomy. In this study thymidine incorporation into DNA was
measured in vitro at various times after hepatectomy. The
TCDD-induced increase was most pronounced 24 to 32 h after the
hepatectomy. The somewhat conflicting results may be due to
differences in the in vivo and in vitro incorporation of
thymidine, as well as to differences in the time points studied.
According to Dickins et al. (1981), the discrepancy in proliferative
DNA synthesis could be due to the degree of hepatectomy. Thus 70%
hepatectomy would by itself enhance DNA synthesis to near maximum
level, making it difficult to measure any effect of TCDD under the
experimental conditions. This suggestion was confirmed by Christian &
Peterson (1983a), who compared the effect of TCDD on proliferative DNA
synthesis after 1/3 and 2/3 hepatectomy. This study also revealed that
the effect could be seen only when a certain amount of time, namely 5
to 10 days, elapsed between TCDD administration and hepatectomy.
The transfectivity of bacteriophage Qß/RNA was evaluated after
treatment in vitro with TCDD. No effect was noticed in the tested
dose range (0.2-4 µg TCDD/ml) (Kondorosi et al., 1973).
The effect of TCDD on the repair of DNA damage induced by
2-aminofluorene (AF) and 2-acetylaminofluorene (AAF) in primary
hepatocytes from B6 and D2 mice has been investigated (Moller et al.,
1984). Pretreatment in vivo with TCDD (50 mmol/litre) resulted in
a slight increase in DNA damage (measured by the alkaline elution
technique) following incubation with either AF or AAF for 60 min,
suggesting induction of aromatic amine activating enzymes.
7.6.3 Cytogenetic effects
Green & Mooreland (1975) and Loprieno et al. (1982a) did not
observe any induction of chromosomal aberrations in rats administered
TCDD intraperitoneally or by gavage (5 to 20 µg/kg body weight). Later
Green et al. (1977) showed a significant increase in the induction of
chromosomal abnormalities in bone marrow cells of male rats at doses
of 2 and 4 µg TCDD/kg body weight and in females at 4 µg/kg body
weight. In a study by Meyne et al. (1985) C57Bl/6 or DBA/2 mice (with
high- and low-affinity TCDD receptors, respectively) were ip injected
at doses 0, 50, 100, and 150 µg/kg body weight. There was no increase
in the frequency of chromosomal aberrations in bone marrow cells of
the TCDD-treated mice of either strain 8, 16, or 24 h after treatment.
All doses were high enough to induce hepatotoxic damage in C57Bl/6
mice. In male and female CD-1 mice, however, a weak but significant
increase in chromosomal aberrations was obtained 96 h post-treatment
(ip 10 µg TCDD/kg body weight) (Loprieno et al., 1982b).
Lamb et al. (1981) evaluated the frequency of sister chromatid
exchange (SCE) in bone marrow cells of C57Bl/6 mice given single ip
injections of mixtures of chlorinated phenoxy acids that contained
0.16, 1.2, or 2.4 µg TCDD/kg body weight. The mean SCE frequency in
treated and in control mice did not differ significantly. Mice fed
diets containing the same daily doses as above for 4 to 8 weeks did
not show any increase in SCE frequencies.
At doses (50, 100, and 150 µg per kg body weight) hepatotoxic to
C57Bl/6 mice, there was no significant increase of SCEs in C57Bl/6 or
DBA/2 mice 18 h after ip injection of TCDD (Meyne et al., 1985). Meyne
et al. (1985) also performed a micronucleus test under the same
conditions as above. Mice, killed 24 or 48 h after treatment, did not
show any increase of micronuclei in polychromatic erythrocytes of bone
marrow in either strain.
7.6.4 Cell transformation
Single treatments (11 concentrations in the range 0 to 5
µmol/litre) of mouse embryo fibroblast (C3H/10T1/2) tissue cultures
with TCDD did not transform or initiate the process of transformation
in cultures subsequently exposed to
12-O-tetradecanoylphorbol-13-acetate (Abernethy et al., 1985).
Continued treatment of these cells with low concentrations (> 4
pmol/litre) of TCDD enhanced the production of foci in cultures
pretreated with N-methyl-N'-nitro-N-nitrosoguanidine. Maximal
enhancement occurred at 40 pmol/litre. Higher doses, 120 to 4000
pmol/litre, did not further increase the incidence of foci production.
Promotion of transformation is thus the predominant effect of TCDD in
the C3H/10T1/2 cell-transformation system.
7.7 Carcinogenicity
7.7.1 Long-term animal studies on single compounds
Several studies on the carcinogenicity of TCDD and related
compounds have been performed.
The data from studies using oral exposure are summarized in Table
59. Van Miller et al. (1977) exposed male Sprague Dawley rats to
various dietary levels of TCDD ranging from 0.001 µg/kg and 1 µg/kg to
1 µg/kg for 78 weeks. Pronounced mortality was observed at higher
doses. Neoplastic changes in different organs were noted in a number
of rats that died. At 95 weeks, the small number of surviving animals
were killed. At dietary levels of 5, 50, and 500 ppt TCDD (ng/kg
feed), a variety of tumours were noted, but no particular trend
emerged. However, at a level of 5 µg/kg feed, four squamous cell
tumours of the lung, four neoplastic nodules (hyperplastic nodules),
and two cholangiocarcinomas of the liver were found in seven rats.
Kociba et al. (1978) fed groups of 50 male and female Sprague
Dawley rats 0.1, 0.01, and 0.001 µg TCDD/kg body weight for 2 years.
86 male and 86 female control rats received the vehicle only. The
doses corresponded to 2193, 208, and 22 ng TCDD/kg diet. A variety of
tumours were found in the control and experimental groups. Tumours
caused by the ingestion of TCDD were confined to the liver, lungs,
hard palate/nasal turbinates, and tongue. In the female rats that had
received doses of 0.1 and 0.01 µg/kg body weight, a statistically
significant increase of neoplastic nodules (hyperplastic nodules,
hepatomas) of the liver was noted, and in the rats that had received
0.1 mg TCDD/kg body weight there was a statistically significant
increase of hepatocellular carcinomas. Epithelial tumours along the
respiratory tract, tongue, and hard palate consisted of well
differentiated squamous cell carcinomas. There was an increased
incidence, compared with the controls, of squamous cell carcinomas of
the hard palate and nasal turbinate in both male and female rats
receiving 0.1 µg TCDD/kg body weight, while the incidence of squamous
cell carcinoma of the lungs at this dose showed an increase only in
the females. The authors also noted a decreased incidence of tumours
of the pituitary gland, uterus, mammary glands, pancreas, and adrenal
glands in the treated groups, possibly secondary to an effect on the
hormonal functions of different glands. This decrease was in some
instances statistically significant.
Two further studies on the carcinogenicity of TCDD are available
(NIH, 1982a; NIH, 1982b). The TCDD used in these studies was reported
to be 99.4% pure, based on gas chromatographic analysis.
In two gavage studies both Osborne-Mendel rats and B6C3F1 mice
were used (NIH, 1982a). All animals were about 6 weeks old. Dosages,
duration, and outcome are summarized in Table 59. The statistical
analysis performed was similar to that in the dermal study (NIH,
1982b). Mean body weights of the high-dose groups of rats were lower
than those of the corresponding controls after week 55 and 45 for
males and females, respectively, but no other clinical signs were
observed. No such dose-related depression in mean body weight gain was
observed in mice when compared to the vehicle-control groups.
Table 59. Carcinogenicity bioassays of PCDDs after oral administration
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
2,3,7,8-TCDD Oral (diet), 0.0, 0.001, 0.005, Rat/Sprague Dawley/M all tumours: 0/10 at 0.0,
(Van Miller et al., 0.05, 0.5, 1.0, 5.0 µg/kg. 0/10 at 0.001, 5/10 at 0.005,
1977) 78/95 weeks 3/10 at 0.05, 4/10 at 0.5, 4/10
at 1.0, and 7/10 at 5.0 µg/kg
2,3,7,8-TCDD Oral (diet), 0.0, 0.001, 0.01, Rat/Sprague Dawley/M squamous cell carcinoma hard
(Kociba et al., 1978) 0.1 µg/kg body weight per day. palate: 4/50 at 0.1 µg/kg per day;
105/105 weeks squamous cell carcinoma tongue:
1/50 at 0.001 and 0.01, 3/50 at
0.1 µg/kg per day; adenoma of
adrenal cortex: 2/5 at 0.01 and
5/50 at 0.1 mg/kg per day
Rat/Sprague Dawley/F hepatocellular carcinoma: 0/86
at 0.0, 0/50 at 0.001, 2/50 at
0.01, and 11/49 at 0.1 µg/kg per
day; squamous cell carcinoma of
tongue: 1/50 at 0.01 and 4/49 at
0.1 µg/kg per day, squamous cell
carcinoma of lung: 7/49 at 0.1
µg/kg per day.
2,3,7,8-TCDD Oral (gavage corn oil:acetone, Rats/Osborne-Mendel/M follicular cell adenomas or
(NIH, 1982a) 9:1), 0.0, 0.1, 0.05, 0.5 carcinoma of thyroid: 1/69 at
µg/kg body weight per week. 0.0, 5/48 at 0.10, 8/50 at 0.05,
104/105-107 weeks and 11/50 at 0.5 µg/kg per week
Table 59 (contd - 2).
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
Rats/Osborne-Mendel/F follicular cell adenomas or carcinoma
of thyroid: 3/73 at 0.0, 2/45 at 0.1,
1/49 at 0.05, and 6/47 at 0.5 µg/kg
per week; neoplastic nodules or
hepatocellular carcinoma: 5/75 at
0.0, 1/49 at 0.1, 3/50 at 0.05, and
14/49 at 0.5 µg/kg per week
2,3,7,8-TCDD Oral (gavage corn oil:acetone, Mice/B6C3F18/M hepatocellular carcinoma: 8/73
(NIH, 1982a) 9:1), Males 0.0, 0.01, 0.05, at 0.0, 9/49 at 0.01, 8/49 at
0.5 µg/kg body weight per 0.05, and 17/50 at 0.5 µg/kg per
week. Females 0.0, 0.04, 0.2, week
2.0 µg/kg body weight per
week, 104/105 weeks Mice/B6C3F18/F hepatocellular carcinoma: 1/73
at 0.0, 2/50 at 0.04, 2/48 at
0.2, and 6/47 at 2.0 µg/kg per
week; follicular cell adenomas
of thyroid: 0/69 at 0.0, 3/50
at 0.04, 1/47 at 0.2, and 5/46
at 2.0 µg/kg per week
1,2,3,6,7,8/ Oral (gavage corn oil:acetone, Rats/Osborne-Mendel/M liver neoplastic nodules or
1,2,3,7,8,9- 9:1), 0.0, 1.25, 2.5, 5.0 µg/kg hepatocellular carcinoma: 0/74
hexaCDD (1:2) body weight per week, at 0.0, 0/49 at 1.25, 1/50 at
(NIH, 1980b) 104/105 weeks 2.5, and 4/48 at 5.0 µg/kg per
week
Rats/Osborne-Mendel/F liver neoplastic nodules or
hepatocellular carcinoma: 5/75
at 0.0, 10/50 at 1.25, 12/50 at
2.5, and 30/50 at 5.0 µg/kg per
week
Table 59 (contd - 3).
Compound Exposure: route, dose, frequency Species/strain/sex Tumour type and incidence
(Reference) and duration-treatment/test
1,2,3,6,7,8-/ Oral (gavage corn oil:acetone, Mice(B6C3F1)/M hepatocellular adenomas or carcinomas:
1,2,3,7,8,9- 9:1), Males 0.0, 1.25, 2.5, 15/73 at 0.0, 14/50 at 1.25, 14.49 at
hexaCDD (1.2) 5.0 µg/kg body weight per week 2.5, and 24/48 at 5.0 µg/kg per
(NIH, 1980b) Females 0.0, 2.5, 5.0, 10.0 µg/ week
kg body weight per week,
104/105-108 weeks Mice(B6C3F1)/F hepatocellular adenomas or carcinomas:
3/73 at 0.0, 4/48 at 2.5, 6.47 at 5.0,
and 10/47 at 10.0 µg/kg per week
Dibenzo-p-dioxin Oral (diet), 0, 5000, 10 000 Mice/B6C3F1/M hepatocellular carcinoma: 4/49 at 0,
(NCI, 1977) µg/kg diet, 87-90/91-97 weeks 7/50 at 5000, and 3/48 at 10 000
µg/kg diet; hepatocellular adenomas:
4/49 at 0, 1/50 at 5000, and 2/48 at
10,000 mg/kg diet; malignant
tumours: 5/49 at 0, 11/50 at 5000,
and 8/50 at 10 000 mg/kg diet
Mice/B6C3F1/F malignant tumours: 8/50 at 0,
9/49 at 5000, and 3/39 at 10,000
mg/kg diet; hepatocellular
carcinoma: 1/47 at 5000 mg/kg
diet
2,7-diCDD Oral (diet), 0, 5000, 10 000 Rats/Osborne-Mendel/M malignant tumours: 5/33 at 0,
(NCI, 1979) mg/kg diet, 110/110-117 weeks. 7/34 at 5000, and 4/33 at 10 000
mg/kg diet; hepatocellular
adenoma: 1/33 at 0; hepatocellular
carcinoma: 1/33 at 10,000 mg/kg diet
Rats/Osborne-Mendel/F malignant tumours: 5/31 at 0;
4/33 at 5000, and 5/30 at
10 000 mg/kg diet
In the male rats, increased incidences of follicular cell
adenomas or carcinomas in the thyroid were dose related and were
significantly higher (P < 0.001) in the high-dose group than in the
vehicle controls (1%, 10%, 16%, and 22%). In the female rats, an
increase (though not statistically significant) was seen only in the
high-dose group (4%, 4%, 2%, and 13%). The incidence of neoplastic
nodules of the liver in the high-dose group of female rats was
significantly (P < 0.006) higher than that in the vehicle-control
group (7%, 2%, 6%, and 28%).
In male and female mice, incidences of hepatocellular carcinomas
were dose related and, in the high-dose groups, were significantly (P
< 0.002 and 0.014, respectively) higher than those in the
corresponding vehicle-control groups (males: 11%, 18%, 16%, and 34%;
females: 1%, 4%, 4%, and 13%).
Follicular cell adenomas in the thyroid occurred at dose-related
incidences in female mice, and were significantly (P < 0.009) higher
in the high-dose groups than those in the vehicle controls (0%, 6%,
2%, and 11%). In conclusion, under the conditions of this bioassay,
TCDD was carcinogenic for Osborne-Mendel rats, inducing follicular
cell thyroid adenomas in males and neoplastic nodules of the liver in
females. TCDD was also carcinogenic for B6C3F1 mice, inducing
hepatocellular carcinomas in males and females and follicular cell
thyroid adenomas in females.
Toth et al. (1979) administered TCDD orally by gavage to groups
of 45 male Swiss/H/Riop mice at doses of 0, 0.007, 0.7, and 7 µg/kg
body weight once a week for one year, and the animals were followed
for their lifetime. Liver tumours were found at 18%, 29%, 48%, and
30%, respectively. The tumour incidence at 0.7 µg/kg was significantly
higher when compared to controls (P < 0.01), while the increase at
the highest dose level (7 µg/kg) was not statistically significant (P
= 0.11). The latter finding may be due to a much reduced average
survival in comparison with the control group (average life span 424
and 588 days, respectively).
In a study using dermal application of TCDD (NIH, 1982b), male
and female Swiss-Webster mice were about 6 weeks old at the beginning
of the bioassay. The one-tailed Fisher exact test was used to compare
the tumour incidence of a control group with that of a group of dosed
animals. Mean body weights of dosed animals were essentially the same
as those of the corresponding vehicle-control groups, but less than
those of the untreated controls, for males throughout the study and
for females during the first 80 weeks. The incidence of fibrosarcoma
in the integumentary system of female mice treated with TCDD or TCDD
and dimethylbenzathraline (DMBA) was significantly higher than that of
the controls (P < 0.007 and P < 0.010, respectively). An increase in
the same tumour type, although not statistically significant (P =
0.084), was also observed in the male mice (7% and 21% for the control
and TCDD-treated groups, respectively). In conclusion, under the
conditions of this bioassay, TCDD was carcinogenic for female
Swiss-Webster mice, causing fibrosarcomas in the integumentary system.
However, the study has been criticized in several areas, namely, a
maximal tolerated dose (MTD) was not achieved, especially in male
mice, only one dose per sex was used, and the number of mice (30) in
the TCDD-exposed groups was considered less than optimal.
7.7.2 Long-term animal studies with mixed compounds
Toth et al. (1979) studied groups of 100 male and 100 female,
10-week-old random-bred Swiss H/Riop mice that were given weekly oral
doses of 2,4,5-trichlorophenoxyethanol (TCPE) at 67-70 mg/kg body
weight, together with 0.112 mg TCDD/kg body weight or 0.007 mg TCDD/kg
body weight in 0.5% carboxymethyl cellulose by gastric intubation for
12 months. The incidences of liver tumours in males after 2 years were
reported to be 48% and 58% in the two treated groups, compared with
26-33% in the untreated male mice of the colony that survived up to 3
years. Three additional groups of mice were given 7 µg TCPE/kg body
weight with 0.0007 µg TCDD/kg body weight, 0.7 µg TCPE/kg body weight
with 0.00007 µg TCDD/kg body weight, or 7 µg TCPE/kg body weight with
0.7 µg TCDD/kg body weight. There was no increased incidence of liver
tumours in any of the treatment groups.
A 1:2 mixture of 1,2,3,6,7,8- and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxins (HxCDDs) has been tested for
carcinogenicity by dermal application to mice and by gavage in rats
and mice (NIH, 1980a,). The following impurities were detected in the
mixture: pentaCDD 0.04%, TCDD 0.09%, triCDD 0.004%, and bromopentaCDD
< 0.004%. The specific isomers of these impurities were not
identified. The doses used and duration of the gavage studies (NIH,
1980b) are given in Table 59. In both species and either sex, only
tumours of the liver occurred at a significantly greater incidence
than controls. In male rats and male and female mice, the liver tumour
incidence was significantly increased over control values only in the
high dose groups (5 µg/kg per week), while in female rats the
incidence was significantly greater at both medium- and high-dose
levels (2.5-5 µg/kg per week). In the dermal study, no
treatment-related tumours were recorded in either the carcinogenicity
bioassay or the tumour promotion assay using DMBA as an initiator
(NIH, 1980a). It was concluded that the mixture of hexaCDDs tested was
carcinogenic to rats and mice following administration by gavage.
However, there was no tumorigenic activity when hexaCDD was applied to
mouse skin.
When added to the diet in concentrations up to 10 000 µg/kg,
2,7-dichlorodibenzo-p-dioxin and dibenzo-p-dioxin were found to be
non-carcinogenic in chronic feeding studies in mice and rats of either
sex (NCI, 1977; NCI, 1979).
7.7.3 Short-term and interaction studies
Poland & Glover (1979) estimated the maximum covalent binding of
TCDD in vivo to rat liver protein, ribosomal RNA (rRNA), and DNA
after 3H-TCDD (39 Ci/mmol) was administered to immature male and
female Sprague Dawley rats (105-135 g) as a single ip injection of 7.5
µg/kg. The rats were killed 12 h, 24 h, 48 h, or 7 days after dosing
with TCDD. The level of radioactivity in the liver varied from 18 to
64% of the administered dose, and only a small fraction was associated
with the purified macromolecular fractions. The radioactivity
associated with rRNA and DNA was very low and essentially all the
unextracted radioactivity was associated with protein (0.03 to 0.1% of
the total radioactivity in the liver). The maximum amount of 3H-TCDD
that could have been covalently bound to DNA was estimated as 1.8 x
10-17 mol TCDD per mg DNA, or 6.2 nmol TCDD per mol DNA nucleotide,
which means binding of about 1 molecule TCDD to the DNA in 35 cells.
Phenobarbital treatment, or prior administration of TCDD did not
significantly alter the amount of unextractable 3H-TCDD associated
with any macromolecular fraction. Similarly, there were no differences
in the levels of 3H-TCDD associated with protein, rRNA, or DNA in
male or female rats pretreated with TCDD.
TCDD was found to be a carcinogen in chronic feeding studies in
rats and mice. Most carcinogens bind covalently, either directly or
after a conversion to electrophilic intermediates, to protein, rRNA,
and DNA to the extent of 10-4 to 10-6 mol of carcinogen per mol of
amino acid or nucleotide residue. The maximum binding of TCDD is 4-6
orders of magnitude lower than that of most chemical carcinogens and
is of questionable biological significance. The results obtained in
the study of Poland & Glover (1979) thus indicate that it is unlikely
that the mechanism of TCDD-induced carcinogenesis would include the
covalent binding of TCDD.
In female Charles River CD-1 mice, TCDD was found to be a weak
initiator when given alone in a single dose of 2 µg/ mouse by dermal
application (Di Giovanni et al., 1977). In these studies
12-O-tetradecanoylphorbol-13-acetate (TPA) was used as a promoter.
When TCDD and 7,12-dimethylbenzanthracene were given together, a
slight additive effect was found. As mentioned earlier, in a study on
a hexaCDD mixture, no treatment-related tumours were found in a tumour
promotion test on mice using DMBA as an initiator (NIH, 1980a).
The possible role of TCDD as a promoter in
diethylnitrosamine-induced hepatocarcinogenesis was studied by Pitot
et al. (1980) in female Charles River rats (200-250 g). A single oral
dose (10 mg/kg) of diethylnitrosamine (DEN) was given 24 h after a
70% hepatectomy, and treatment with TCDD (0.14 or 1.4 mg/kg sc once
every 2 weeks for 7 months) was started one week after the
hepatectomy. The promoting effect of TCDD in this 2-stage model of
liver cancer was also compared with the effect of a known promoting
agent, phenobarbital (0.05% in the diet for 7 months). Enzyme-altered
foci, which are thought to be precursors of hepatocellular carcinomas,
were greatly increased in number, total volume, and phenotypic
heterogeneity by the administration of TCDD. A significant incidence
of hepatocellular carcinomas (5 out of 7) was observed in the
DEN-treated rats that were given the high dose of TCDD, but no
carcinomas were seen in the rats treated with DEN only (0 out of 4).
The results indicated that TCDD was a potent promoting agent for
hepatocarcinogenesis, and the authors suggested that all the tumours
associated with the chronic administration of TCDD arise from its
promoting activity of cells previously initiated by exposure to
carcinogens in the environment.
Studies utilizing a two-stage system of mouse skin tumorigenesis
(Berry et al., 1979), which allows separate evaluation of the
initiation and promotion phases of carcinogenesis, have demonstrated
that TCDD does not promote the development of skin tumours at a dose
of 0.1 µg given twice weekly, whereas in the animals pretreated with
1.0 µg TCDD for 1, 3, or 5 days prior to initiation with DMBA, TCDD
was shown to act as a potent inhibitor of PAH-induced skin tumour
initiation. Almost complete inhibition (96%) was achieved with a
single non-toxic topical dose of 0.1 µg, and 3 days pretreatment with
0.01 µg TCDD gave over 80% inhibition. The authors suggested that this
potent anticarcinogenic effect of TCDD may be related to its ability
to induce epidermal enzyme pathways involved in detoxifying PAH
carcinogens in the skin. According to Kimbrough (1979), TCDD and other
compounds of this type, which are potent enzyme inducers, may prevent
or enhance the tumour-inducing ability of other chemicals by enhancing
the metabolism of these xenobiotics.
Poland et al. (1982) studied the promoting effects of TCDD in the
mouse skin two-stage tumorigenesis model. The effects of TCDD and TPA
were compared in DMBA-initiated HRS/S mice that were either
heterozygous or homozygous for the recessive "hairless" trait. TCDD
was found to have a tumour-promoting effect only in the homozygous
mice. The data suggested to the authors that TCDD might act as a
promoter by a mechanism different from that of TPA.
The interaction of TCDD with 3-methylcholanthrene (3-MC) was
studied by Kouri et al. (1978), who found that TCDD was a
co-carcinogen with 3-MC when administered by subcutaneous injection.
Both sexes of two inbred strains of mice (C57Bl/6C and DBA/2),
responsive and non-responsive to the induction of AHH by 3-MC,
respectively, were used. TCDD at a concentration of 1 or 100 µg/kg
body weight was administered as a single dose alone or in combination
with 3-MC (150 µg/kg). The duration of the study was 36 weeks. The
number of animals in each group at the start of the experiment was not
stated, but seems to have been between 30 and 100. No subcutaneous
tumours were observed in controls or in mice treated with TCDD alone.
In responsive mice no enhancement occurred, while in non-responsive
mice the simultaneous administration of TCDD and 3-MC enhanced the
carcinogenic response of TCDD at 100 mg/kg. At 1 mg TCDD/kg, a
reduction in latency time to tumour was noted.
An anticarcinogenic effect of TCDD has been reported by Cohen et
al. (1979) and by Di Giovanni et al. (1979a). When TCDD was topically
applied to Sencar or CD-1 mice 72 h prior to the administration of
either DMBA (10 nmol) or benzo(a)-pyrene (BP) (100 nmol), it markedly
decreased the skin tumour initiation by both DMBA and BP. This
inhibition of tumorigenesis correlated with the decreased in vivo
binding of DMBA to DNA after TCDD administration, but not with the
total binding of BP to DNA. However, the
hydrocarbon-deoxyribonucleoside adducts from the DNA of
TCDD-pretreated mice showed a striking absence of
BP-7,8-dihyrodiol-9,10-epoxide adduct bound to guanine. It is
suggested, accordingly, that the formation of this adduct may be a
critical step in BP-induced skin carcinogenesis in mice. In further
studies of the tumour-inhibitory effect of TCDD (Di Giovanni et al.
1980), it was demonstrated that exposure of CD-1 mice to TCDD 3 days
before initiation with BP or 3-MC resulted in a decreased tumour
yield, compared to acetone-pretreated animals, while treatment with
TCDD 5 min before and 1 day after initiation failed to affect the
tumour yield. However, when TCDD was administered 3 days or 5 min
before or 1 day after initiation with BP-diol epoxide, there was a
decreased tumour yield in all cases. The authors concluded that the
ability of TCDD to inhibit tumour yield when administered after the
BP-diol epoxide, indicated the possible existence of more than one
mechanism involved in the anticarcinogenic effect of TCDD.
7.8 Mechanisms of Action
The toxicity of TCDD apparently depends on the fact that the four
lateral positions of the molecule are occupied by chlorine (see
section 7.8.1) Toxicity decreases with decreasing lateral substitution
and increasing total chlorine substitution. As has been outlined in
sections 7.1-7.7, TCDD toxicity involves many different types of
symptoms and these symptoms vary from species to species and from
tissue to tissue, both quantitatively and qualitatively. Furthermore,
age- and sex-related differences in sensitivity to TCDD have been
reported. Characteristic for TCDD toxicity is also the delay in
expression of toxicity, from 2 weeks to 2 months, seen in all species.
It has been suggested that the initial event in TCDD-induced toxicity
is the binding of TCDD to the so-called Ah receptor. This complex,
whether of cytosolic or nuclear origin, exerts its action in the
nucleus by triggering a pleiotropic response including the induction
of mixed function oxidases. Present knowledge, however, rules out
enzyme induction per se as being the cause of toxicity and death
(see section 7.8.1). Although the toxicokinetics of TCDD vary between
species, these differences are not sufficient to explain the
variabilities in sensitivity to TCDD toxicity (see section 7.8.2).
Available data indicate an involvement of TCDD in processes regulating
cellular differentiation and/or division. Alterations in the
regulation of such processes, which are not equally active in all
cells throughout the organism, would be expected to result in effects
that vary among tissues as well as among species (see section 7.8.3).
7.8.1 Receptor-mediated effects
The binding of TCDD to the Ah receptor has been postulated to be
the necessary first step in the induction of cytochrome P-450
synthesis and of related enzyme activities, as well as in the
mechanism of toxicity (Poland et al., 1976; Okey et al., 1979; Poland
& Glover, 1979; Poland & Knutson, 1982). So far, no conclusive data
exist for the direct involvement of the Ah receptor in the
TCDD-induced toxicity.
Knowledge of the mechanism involved in the Ah locus
enzyme-induction response has grown rapidly since the initial indings
that binding of TCDD to the receptor resulted in increased levels of
cytochrome P-450 mRNA in genetic variants of mice (Tukey et al., 1982)
and mouse hepatoma cell lines (Israel & Whitlock, 1983). These
findings have been confirmed and further expanded in studies using
mice genetics and recombinant DNA techniques (Tukey et al., 1982;
Miller et al., 1983; Gonzales et al., 1984; Israel & Whitlock, 1984;
Jones et al., 1984, 1985, 1986; Okino et al., 1985; Tuteja et al.,
1985; Kimura et al., 1986), thus providing more data to the
understanding of the mechanism for the Ah locus enzyme induction.
In early experiments (Poland et al., 1976; Carlstedt-Duke, 1979;
Okey et al., 1979), Ah receptors appeared to be localized in the
cytosol when in its unoccupied state and was translocated into nuclei
only when occupied by a ligand (Greenlee & Poland, 1979; Okey et al,
1980; Poellinger et al., 1982; Gasiewicz & Rucci, 1984). However,
Whitlock & Galeazzi (1984) concluded that unoccupied Ah receptor in
the intact cell was primarily located in the nucleus and that apparent
cytosolic Ah receptor was a redistribution artifact. Following the
distribution of Ah receptor and three cytosolic marker enzymes between
the nuclear and cytosolic fractions during fractionation (Denison et
al., 1986a,c), it was again concluded that unoccupied Ah receptor is
primarily cytosolic or that this receptor protein is in equilibrium
between the cytoplasm and nucleus.
However, it is generally agreed that the ultimate biological
regulation by the Ah receptor is due to specific interaction of
ligand-receptor complexes with chromatin sites (Greenlee & Poland,
1979; Okey et al., 1979, 1980; Mason and Okey, 1982; Poellinger et
al., 1982; Poland and Knutson, 1982; Tukey et al., 1982a; Gonzales et
al., 1984; Israel & Whitlock, 1984).
Table 60. Physicochemical data for the hepatic Ah receptor in Sprague Dawley rats
Physicochemical data Denison et al. (1986a) Poellinger et al. (1983)
Stokes radius (nm) 5.2 ± 0.2 6.1 ± 0.2
Sedimentation coefficient (S) 5.6 ± 0.6 4.4
Relative molecular mass 121 000 111 000
Several investigators have estimated the molecular size and other
physicochemical properties for the cytosolic hepatic Ah receptor (Okey
et al., 1979, 1980, 1982; Tukey et al., 1982b; Poellinger et al.,
1983; Gasiewicz et al., 1983a,b; Denison et al., 1986a; Hannah et al.,
1986). To obtain reliable results in the isolation and
characterization of the Ah receptor, it is necessary to use perfused
liver, in order to reduce the contribution from blood proteins, and to
use a radioactive ligand of high purity and specific activity quality
(Poellinger et al., 1983; Denison et al., 1986a). Further more, the
ionic strength of the medium during isolation has a marked effect upon
the apparent molecular weight of the receptor (Denison et al., 1986a).
The physicochemical data for the Ah receptor presented in Table 60
were obtained from two studies (Denison et al., 1986a,c; Poellinger et
al., 1983) in which the receptor was isolated from perfused liver of
Sprague Dawley rats under conditions of high ionic strength.
The receptor protein has been found also in extrahepatic tissues
(Carlstedt-Duke, 1979; Carlstedt-Duke et al., 1979, 1981; Johansson et
al., 1982; Mason & Okey, 1982; Gasiewicz & Rucci, 1984; Gasiewicz et
al., 1984; Furuhashi et al., 1986; Kurl et al., 1985; Söderkvist et
al., 1986). Different mammalian species possess Ah receptors with
similar, though not identical, properties (Gasiewicz & Rucci, 1984;
Denison et al., 1986a,b; Kurl et al., 1985). Jaiswal et al. (1985a,b)
have shown species differences in the TCDD-inducible P-450 gene
subfamily. Humans appear to only have the P1-450. The function of
human P1-450 may be equivalent to a combination of P1-450 and P3-450
in the mouse. A complete lack of measurable cytosolic and almost
total absence of inducer-receptor complexes in the nucleus of human
MCF-1 cells (cells derived from an adenocarcinoma of the breast) were
reported. This absence was out of proportion to the ability of TCDD to
induce AHH and acetamide-4-hydroxylase activities in these cells.
Further studies in different cell lines are thus needed to
characterize the level of receptor in humans. The only non-mammalian
species demonstrated to have significant Ah receptor is the chick
embryo (Denison et al., 1986b). However, no detectable level of the
receptor was found in 2-week-old White Leghorn chickens (Sawyer et
al., 1986). Based on certain similarities in the biochemical behaviour
between the Ah receptor and steroid hormone receptors, it has been
proposed that there is a natural ligand for the Ah receptor (Neal et
al., 1979; Poland et al., 1976). So far such a ligand has not been
identified, either among steroid hormones (Poland et al., 1976;
Carlstedt-Duke et al., 1979; Romkes et al., 1987) or among certain
dietary factors (Johansson et al., 1982), although lumichrome, a
metabolite of riboflavin, was suggested as an endogenous ligand for
the receptor (Kurl & Villee, 1985). TCDD does not bind to the
glucocorticoid, estrogen or progesterone receptors (Neal et al.,
1979; Romkes et al., 1987). Monoclonal anti-glucocorticoid
receptor-IgG antibodies did not react with the TCDD receptor
(Poellinger et al., 1983) and the hydrophobic properties of the Ah
receptor were more pronounced than those of the steroid hormone
receptors (Poellinger & Gullberg, 1985).
Convincing data for the importance of the receptor in
TCDD-induced toxicity could be based on structure activity
relationships, i.e., that the binding affinities of TCDD and other
PCDDs or PCDFs to the receptor correlate with their biological
potencies. The binding affinities of PCDDs and PCDFs have been
demonstrated to correlate with their biological potencies,
particularly the induction of enzyme activities as well as the
production of acute toxic effects (Tables 56 and 61) (Poland & Kende,
1976; Poland et al., 1976; Knutson & Poland, 1982).
Furthermore, the structure-activity relationships observed for
enzyme induction, thymic atrophy, body weight loss, and LD50 values
were comparable to the structure-activity relationships observed for
receptor binding (Tables 56, 61) (Bandiera et al., 1984a,b; Mason et
al., 1985, 1986; Sawyer & Safe, 1985; Safe et al., 1986). Interactive
studies, i.e., studies where PCDD and PCDF congeners have been given
both separately and as mixtures, have also been used to investigate
the role of the Ah receptor in the mechanism of action of TCDD.
Depending on the mechanism of action the biological responses may be
synergistic potentiated, additive, unaffected, or antagonistic. Such
studies have been performed for enzyme induction (Sawyer & Safe, 1985;
Keys et al., 1986; Ahlborg et al., 1987), vitamin A reduction
(Hakansson et al., 1987), teratogenicity (Birnbaum et al., 1985; Weber
et al., 1985), thymic atrophy (Bannister & Safe, 1987), and immune
suppression (Rizzardini et al., 1983). So far this kind of data is
scattered and difficult to interpret.
Table 61. Structure-activity relationships for some PCDFs
In vitro EC50 values (mol/litre)a,b In vivo ED50 values (µmol/kg) LD50 c
PCDF Receptor AHH EROD AHH weight Thymic Guinea-pig
congener binding loss atrophy µg/kg body weight
Dibenzofuran < 10-3 ND ND
2- 2.8 x 10-4 ND ND
3- 4.2 x 10-5 ND ND
4- < 10-3 1.0 x 10-5 1.71 x 10-5
2,3- 4.72 x 10-6 2.19 x 10-6 4.84 x 10-6
2,6- 2.46 x 10-4 6.17 x 10-5 6.31 x 10-5
2,8- 2.57 x 10-4 3.95 x 10-5 4.0 x 10-5
1,3,6- 4.40 x 10-6 2.53 x 10-6 3.37 x 10-6
1,3,8- 8.50 x 10-5 1.94 x 10-5 3.02 x 10-5
2,3,4- 1.9 x 10-5 1.51 x 10-7 2.48 x 10-7
2,3,8- 1.0 x 10-6 2.49 x 10-6 1.56 x 10-6
2,6,7- 4.5 x 10-7 2.80 x 10-6 3.13 x 10-6
2,3,4,6- 3.5 x 10-7 1.32 x 10-6 1.13 x 10-6
2,3,4,7- 2.51 x 10-8 1.79 x 10-8 1.48 x 10-8 46 34 7.8
2,3,4,8- 2.0 x 10-7 4.14 x 10-8 3.76 x 10-8 ND 130 > 150
2,3,6,8- 2.2 x 10-7 1.04 x 10-6 7.79 x 10-7
2,3,7,8- 4.1 x 10-8 3.91 x 10-9 2.02 x 10-9 0.65 3.2 3.6 5-10
1,2,3,6- 3.54 x 10-7 > 10-4 > 10-4 > 160 > 250 > 250
1,2,3,7- 1.12 x 10-7 2.7 x 10-5 6.3 x 10-5 110 87 110
1,2,4,8- > 10-5 1.20 x 10-5 9.26 x 10-5
1,2,4,6,7- 6.77 x 10-8 3.25 x 10-7 3.48 x 10-7
1,2,4,7,9- 2.0 x 10-5 3.77 x 10-8 3.84 x 10-8
1,2,3,4,8- 1.2 x 10-7 2.09 x 10-7 1.63 x 10-7
1,2,3,7,8- 7.45 x 10-8 2.54 x 10-9 3.06 x 10-9 1.5 2.6 1.8
1,2,3,7,9- 3.98 x 10-7 8.6 x 10-8 8.6 x 10-8 15 49 23
1,2,4,6,8- 3.09 x 10-6 1.0 x 10-5 1.2 x 10-5 > 150 7150 > 150
Table 61 (contd).
In vitro EC50 values (mol/litre)a,b In vivo ED50 values (µmol/kg) LD50 c
PCDF Receptor AHH EROD AHH weight Thymic Guinea-pig
congener binding loss atrophy mg/kg body weight
1,2,4,7,8- 1.3 x 10-6 1.06 x 10-7 1.48 x 10-7 7.8 49 46
1,3,4,7,8- 2.0 x 10-7 1.60 x 10-9 1.40 x 10-9 3.5 26 0.70
2,3,4,7,8- 1.5 x 10-8 2.56 x 10-10 1.34 x 10-10 0.037 1.0 0.26
2,3,4,7,9- 2.0 x 10-7 7.9 x 10-9 5.8 x 10-9 7.0 22 5.5
1,2,3,4,7,8- 2.3 x 10-7 3.56 x 10-10 3.79 x 10-10 0.29 1.3 0.56
1,2,3,6,7,8- 2.7 x 10-7 1.47 x 10-9 1.24 x 10-9 0.35 3.2 0.90
1,2,4,6,7,8- 8.3 x 10-6 4.24 x 10-8 2.93 x 10-8
2,3,4,6,7,8- 4.7 x 10-8 6.87 x 10-10 5.75 x 10-10 0.27 2.8 0.90
a Estimated concentration needed to displace 50% of 3H-TCDD bound to liver cytosol receptor from
Wistar rats and to produce 50% maximum enzyme induction in the rat hepatoma H-4-IIE cell line (Bandiera
et al., 1984b).
b Studies in immature male Wistar rats (Mason et al., 1985).
c Moore et al. (1979).
Polymorphism in the Ah locus, which is suggested to be structural
gene for the cytosolic receptor, seems to determine the sensitivity of
genetically different strains of mice to TCDD and congeners. Ah
responsive strains of mice, e.g., C57Bl/6, are characterized by (a)
high hepatic levels of the TCDD receptor protein, (b) highly elevated
levels of hepatic cytochrome P-448 and associated enzyme activities in
response to treatment with 3-MC, and (c) sensitivity to the ulcerative
action of DMBA on the skin. Ah-non-responsive mice, e.g., DBA/2, lack
these attributes (Nebert et al., 1975). Based on these findings
several genetic studies have been performed to elucidate the role of
the receptor in TCDD toxicity. Contrary to 3-MC, TCDD induces AHH
activity and several toxic effects both in Ah-responsive and
Ah-non-responsive strains of mice. However, the dose required to
produce the effect in an Ah-non-responsive strain is approximately
10-fold greater than that needed for a responsive strain, thus
demonstrating that the Ah-non-responsive strain also contains the
TCDD-receptor but that this receptor is defective (Okey & Vella,
1982).
Crosses and backcrosses of C57BL/6 and DBA/2 mice have shown that
sensitivity to TCDD-induced thymic atrophy immune system disturbances
(section 7.4.5) and teratogenic effects (section 7.5.2) segregate with
the Ah locus. Furthermore, data from studies of DBA/2 mice given
either single or multiple doses of TCDD (Jones & Sweeney, 1980; Smith
et al., 1981) suggest that the LD50 in this strain of mice is at
least 5-fold greater than the values recorded for the C57Bl/6 and
C57Bl/10 strains (Vos et al., 1974; Jones & Greig, 1975; Smith et al.,
1981). TCDD-induced hepatic porphyria has also been shown to segregate
with the Ah locus in mice (Jones & Sweeney, 1980). However, Greig et
al. (1984) found that additional genetic loci must be involved in this
lesion. The correlative differences between the C57Bl/6 and DBA/2
strains of mice, in terms of altered specific binding of TCDD and
sensitivity to this compound, may be unique and may not be applicable
to other species (Gasiewicz & Rucci, 1984).
Less convincing data for the model of receptor-mediated toxicity
of TCDD arise from studies of toxicity, receptor levels, and/or enzyme
induction of TCDD in various species, tissues, and cell cultures.
Despite enormous variability in recorded LD50 values for guinea-pig,
rat, mouse, rabbit, and hamster (Table 47), the amounts and physical
properties of the hepatic as well as the extrahepatic receptors, do
not vary extensively in these species (Poland & Knutson, 1982;
Gasiewicz & Rucci, 1984). Furthermore, although recorded LD50 values
for TCDD vary more than 100 times in chick embryos, C3H/HeN mice, and
Sprague Dawley rats, the ED50 doses for AHH induction in these
species are comparable (Poland & Glover, 1974b). In the guinea-pig,
the most TCDD-susceptible species, enzyme induction is several times
lower even at lethal doses. A number of cell types, including primary
cultures and established and transformed cell lines from several
species and tissues, are inducible for AHH activity, indicating the
presence of the receptor, yet toxicity is not expressed in these
systems (Knutson & Poland, 1980a).
Available data thus suggest that the receptor for TCDD may be a
prerequisite, but is not sufficient in itself for the expression of
TCDD toxicity.
7.8.2 Toxicokinetics
The interspecies variation in sensitivity to TCDD may be
attributable, at least in part, to different rates at which various
species distribute, metabolize, and excrete the compound. Table 62
summarizes some of the data on the elimination, toxicity, and
metabolism of TCDD in different species, some previously discussed in
section 6. Distribution data (section 6.1) has been obtained mainly
from animals exposed to toxic doses of TCDD. Interspecies comparisons
based on these data are difficult to perform, since studies in
different species have been performed with non-comparable relative
toxic doses and collection of data has occurred at variable time
points. However, the available data suggest that tissue levels alone
cannot explain the interspecies differences in pathology and acute
toxicity of TCDD. For example, the molar concentration of TCDD in the
hamster may be orders of magnitude greater than in the rat and mice,
without development of hepatotoxicity, yet definite liver damage
occurs both in rats and mice.
Based on the findings that the toxicity of TCDD is lower in rats
after the stimulation of hepatic mixed function oxidases (Beatty et
al., 1978), and that the metabolites of TCDD are less toxic than the
parent compound (Weber et al., 1982a; Mason & Safe, 1986) (see section
7.1.5), the metabolism of TCDD has been considered as a detoxification
mechanism.
Following the administration of TCDD, most of it appears to be
eliminated through a first-order process in most species (see section
7.1). In all species investigated TCDD is largely eliminated in the
faeces (Table 48). Only in hamsters and certain strains of mice is
urinary elimination a major route of excretion (Table 41). Available
data demonstrate that TCDD is converted to more polar metabolites
prior to elimination in the urine and bile (Table 62). Unchanged TCDD
does not appear in the bile or urine of any species, but it is the
major excretory product in the faeces of mice, guinea-pigs, and
hamsters (Table 62). As the urine and bile appear to be free of
unmetabolized TCDD, the existence of unchanged TCDD in faeces
indicates that a significant amount of unchanged TCDD may be excreted
into the intestinal lumen by some route other than bile.
Table 62. Rates of elimination, toxicity, and metabolic transformation of TCDD in different species
Species/strain Elimination LD50 value TCDD-derived radioactivity occurring in:
half-life (µg/kg
(days) body weight) Urine Bile Faeces Tissues
Ratsa,b,c,d
Sprague Dawley 17-31 25-60 5 polar 4-8 polar unchanged TCDD
metabolites metabolites;
little, if any,
unchanged TCDD.
Micee,f,g
C57BL/6, DBA/2, 10-24 114-2570 4-7 polar 4-6 polar 3-4 polar unchanged TCDD
B6D2F1 metabolites metabolites metabolites,
unchanged TCDD.
Guinea-pigh
Hartley 22-94 0.6-2.5 4 polar 5 polar mainly unchanged TCDD,
metabolites metabolites unchanged polar metabolites
TCDD
Hamstersa,i
Golden Syrian 12-15 1157-5051 4 polar 5-6 polar metabolites, unchanged TCDD
metabolites metabolites unchanged
TCDD
Dogsj
Beagle not reported not reported metabolites
a Gasiewicz et al., 1983a. b Poiger & Schlatter, 1979. c Ramsey et al., 1982. d Rose et al., 1976.
e Gasiewicz et al., 1983b. f Koshakji et al., 1984. g Vinopal & Cassida, 1973. h Olson, 1986.
i Olson et al., 1980a. j Poiger et al., 1982.
The metabolic profiles of TCDD in excreta differs between
species, but generally urinary metabolites are more polar than biliary
or faecal metabolites. Furthermore, several metabolites, both in urine
and bile, are glucuronide conjugates.
The apparent absence of TCDD metabolites in the tissues of all
species, except for the guinea-pig (Table 62), suggests that, once
formed, the metabolites of TCDD are readily excreted. Another factor,
besides metabolism, that may influence the total rate of elimination
of TCDD is the amount of adipose tissue stores, which may vary between
species.
At present there is no clear relationship between the ability of
a given species to excrete TCDD and/or its metabolites and the acute
toxicity of TCDD in that species. However, the somewhat greater rate
of elimination of TCDD in the hamster and the lower rate of
elimination in the guinea-pig (Table 62) may contribute to their
relative resistance and sensitivity, respectively, to the acute toxic
effects of TCDD.
The rate of metabolism and excretion of PCDDs and PCDFs varies
with molecular structure. In most species PCDFs are much more readily
eliminated than their PCDD counterparts. Less halogenated congeners
are usually metabolised and excreted more rapidly than the more
halogenated ones, especially the 2,3,7,8-substituted congeners (see
sections 6 and 9).
7.8.3 Impairment of normal cellular regulatory systems
When considered together, the diverse pattern of toxic effects,
the species and tissue-specific responses, and the time-course for
effects, as well as the non-toxic action of TCDD on most cell cultures
in vitro, seem to indicate that TCDD-induced toxicity occurs as a
result of an impairment of a normal cellular regulatory system. Such
a system might be present in all cells throughout the organism, though
the activity may vary with cell type, tissue, age, sex, and strain,
and/or species.
7.8.3.1 Endocrine imbalance
In many aspects, TCDD toxicity mimics endocrine imbalance,
although no evidence exists to indicate a direct involvement of
hormones in the toxic action of TCDD (section 7.4.9).
7.8.3.2 Body weight regulation
The most reliable and consistent symptom of TCDD toxicity among
all experimental animals is weight loss. The cause of the body weight
loss seems to be reduced food intake, apparently occurring secondarily
to a physiological adjustment that reduces the body weight to a
maintenance level lower than normal. The physiological trigger for
controlling this body weight set-point might be a target for TCDD
action (section 7.4.1).
7.8.3.3 Plasma membrane function
The changes in the surface characteristics of the plasma
membranes induced by TCDD in vivo (7.4.2.2) resemble changes
occurring in precancerous and transformed cells (Pitot & Sirica,
1980). Such changes, including reduction of gap junctions and surface
glyco-proteins, would be expected to curtail cell-cell communication
and to reduce intercellular recognition and attachment events
implicated in the process of tumour promotion.
It has been shown by Pitot et al. (1980) that TCDD promotes
diethylnitrosamine (DEN)-induced hepatocarcinoma in rats. In this
study, canalicular ATPase was used as a marker in detecting
enzyme-altered foci, whose number increased when TCDD was given to
DEN-treated partially hepatectomized rats. The foci exhibited
decreased ATPase activity in agreement with previous observations that
TCDD in vivo reduces the ATPase level in canaliculi-rich plasma
membranes.
TCDD, unlike other well known promoters, requires a prolonged
treatment period in vivo to exert its effect. The lack of effect
of TCDD in vitro would imply that the promoter activity is
mediated through some in vivo process and not by its direct
interaction with plasma membranes.
7.8.3.4 Impaired vitamin A storage
The histomorphological appearance of chloracne, the most
characteristic and prominent sign of TCDD-induced toxicity in humans,
resembles in some respects effects seen in the skin of patients
suffering from vitamin A deficiency (Kimbrough, 1974). Many of the
effects of TCDD poisoning observed in animal studies, including
failure of normal growth, keratosis, epithelial lesions,
immunosuppression, and reproductive and teratological effects, are
similar to the effects of dietary vitamin A deficiency (Thunberg et
al., 1980). The most intriguing similarities between symptoms due to
vitamin A deficiency and TCDD toxicity concern effects on epithelial
tissues, particularly the process of keratinization. TCDD induces
terminal differentiation of epithelial tissues both in vivo and
in vitro. However, lack of epithelial degeneration (programmed
cell death) of the medial epithelial cells of palatal shelves has been
reported in mice exposed to TCDD in utero (Pratt et al., 1984).
Vitamin A is essential for normal differentiation. It diminishes
the expression of differentiation in stratified squamous epithelia and
accentuates the expression of differentiation in secretory epithelia.
Vitamin A deficiency can convert secretory epithelia to squamous
epithelia, while excess of the vitamin can convert stratified squamous
epithelia to secretory epithelia (Wolf, 1980). With the use of
cultured human keratinocytes, it has been demonstrated that vitamin A
at the cellular level affects cell motility, cell-cell interaction,
and epithelial morphogenesis. At the molecular level, vitamin A
determines, by controlling the level of the corresponding mRNA, the
nature of keratins synthesized (Fuchs & Green, 1981). Keratins
constitute a cytoskeleton in epithelial cells, and the keratin pattern
may be used as a marker for epithelial differentiation (Sun et al.,
1979, 1983a,b). Removal of vitamin A from the medium of cultivated
human keratinocytes of various origin led to increased synthesis of
large keratins and reduced synthesis of lower molecular weight
keratins (Fuchs & Green, 1981). This pattern was reversed by the
addition of vitamin A to the medium. Each tissue and cell type
controlled its synthesis of keratins differently, depending on the
vitamin A concentration in the medium. The ability of TCDD to impair
vitamin A storage (section 7.4.10) may be responsible for some of the
toxic effects produced by TCDD.
7.8.4 Lipid peroxidation
Based on indirect lines of evidence, Sweeney & Jones (1983)
proposed that increased in vivo lipid peroxidation, resulting in
the formation of free radicals, might be a mechanism of TCDD toxicity.
Firstly, lipofuscin pigments, considered to be by-products of lipid
peroxidation, accumulate in the heart muscle cells of TCDD-treated
rats (Albro et al., 1978). Secondly, iron deficiency inhibits in
vitro lipid peroxidation (Bus & Gibson, 1979; Sweeney et al., 1979)
and has been demonstrated to reduce hepatic TCDD toxicity in vivo
in rats (Sweeney et al., 1979). Thirdly, 0.25% butylated
hydroxyanisole (BHA) in the diet has been shown to provide protection
from TCDD-induced porphyria and lipid accumulation in mice. In
contrast, 0.01% vitamin E, another antioxidant, in the diet had no
protective effect (Hassan et al., 1985a,b). Stohs et al. (1983)
demonstrated increased in vivo (conjugated diene method) and in
vitro (microsomal malondialdehyde formation) hepatic lipid
peroxidation in female Sprague Dawley rats given a total of 70 µg/kg
body weight in three daily oral doses of 10, 20, and 40 µg TCDD/kg
body weight, or a single oral dose of 80 µg TCDD/kg body weight. Lipid
peroxidation was determined at days 1, 6, and 11 after the last
treatment. The maximal increase of lipid peroxidation in vivo was
2-fold one day post-treatment, whereas the 5- to 6-fold increase in
in vitro lipid peroxidation reached its maximum at 6 days
post-treatment. The TCDD-induced in vitro lipid peroxidation could
be inhibited by repeated treatment with BHA, glutathione, vitamin E,
and vitamin A (Hassan et al., 1985a,b). Dietary selenium had no
inhibitory effect on TCDD-induced lipid peroxidation (Hassan et al.,
1985c).
Robertson et al. (1985) found no evidence for TCDD-induced in
vivo lipid peroxidation, as judged by levels of exhaled endogenous
ethane, and metabolic clearance of both externally and internally
applied exogenous ethane in male Sprague Dawley rats after a single ip
dose of 60 µg TCDD/kg body weight. Neither was there a correlation
between TCDD-induced lipid peroxidation in vitro and sensitivity
towards the lethal effect of TCDD in Sprague Dawley rats, Golden
Syrian hamsters or guinea-pigs (Hassan et al., 1983). Hepatic
microsomes from Sprague Dawley rats and Golden Syrian hamsters exposed
to TCDD in vitro responded with increased lipid peroxidation only
in the presence of Fe3+-ADP in the incubation mixture (Albro et al.,
1986).
From all these data on TCDD and lipid peroxidation, it was
concluded by Albro et al. (1986) that it was premature to attempt to
define a relationship between lipid peroxidation and TCDD-induced
lethality.
8. EFFECT OF PCDDs ON HUMAN BEINGS - EPIDEMIOLOGICAL AND
CASE STUDIES
8.1 Occupational Studies - Historical Perspective
The illness most frequently observed in workers engaged in the
manufacture of trichlorophenol, 2,4,5-T, and related products is a
skin disease called chloracne. This skin disease has also been called
"Pernakrankheit" (perchlorinated naphthalene illness or halogen wax
acne) and was described by Herxheimer (1899). In addition to the
halogenated phenols, chloracne is caused by a number of chlorinated
compounds such as the chlorinated biphenyls and chlorinated
naphthalenes (Muller, 1937; Braun, 1955; Crow, 1970; Kimbrough, 1974).
Although chloracne is well known to those engaged in the
treatment of occupational diseases, many outbreaks that have occurred
over the years, particularly in the USA, have not been reported in the
scientific literature. In the Federal Republic of Germany, chloracne
is now considered an occupational disease for which compensation is
mandatory (Braun, 1970).
Herxheimer (1899) also described general toxic signs and symptoms
in his patients, such as lack of appetite, weight loss, headache, and
vertigo. After his original observations and publication, several
other reports followed. The technique of obtaining chlorine gas
consisted of an electrolytic procedure where a mixture of potassium,
sodium, and magnesium chlorides was subjected to a current with a
central carbon electrode where the chlorine was obtained and piped
off. The workers who took care of the chlorine gas never developed
chloracne thus refuting the original hypothesis by Herxheimer. By
contrast, those who handled the electrodes and cleaned the reaction
vessels were those afflicted with chloracne. Already at this time
chlorinated phenolic compounds were considered as possible noxious
agents (Fraenkel, 1902). This however could never be proven and even
at present, when satisfactory analytical techniques are now available,
no analysis of the so-called "tuffy tar" has been carried out.
Another class of chlorinated organic compounds causing skin
damage appeared during the First World War (1914-1918). At this time
perchlorinated naphthalenes had come into use as insulation materials,
e.g., in the radio and electronic industry. The first description of
Pernakrankheit was that by Wauer (1918). The use of the unspecified
technical mixture of chlorinated naphthalenes spread all over the
world and caused numerous intoxications notably among workers in
manufacture. The perna disease has been summarized by von Wedel et al.
(1943) and described in particular detailed by Braun (1955). Apart
from chloracne the systemic effects of the same compounds have been
dealt with by Drinker et al. (1937) and Greenburg et al. (1939).
Both in man and experimental animals, serious liver damage
occurred after exposure to chlorinated naphthalenes, consisting of
liver necrosis and toxic jaundice (acute yellow liver atrophy). Among
several hundred cases of chloracne due to these compounds, Braun
(1955) tabulated 24 deaths due to toxic jaundice and 14 recoveries. It
should be pointed out that a fulminant liver disease with jaundice of
this kind is an extremely rare condition. For comparison, it has never
occurred after exposure to trichlorophenol (TCP) and TCDD as described
below. Note should also be taken of the fact that not only were the
perchlorinated naphthalenes an ill identified mixture of chemical
species, but exposure frequently occurred at the same time to mixtures
of chlorinated biphenyls, the latter now known to be contaminated with
chlorinated dibenzofurans. The potentiation of toxicity by these
mixtures and other chlorinated compounds were discussed by Drinker et
al. (1937), Greenburg et al. (1939), von Wedel et al. (1943), and
Risse-Sunderman (1959).
Several accidental ingestions of chloracnegenic compounds have
occurred. They are of particular importance in relation to discussions
on whether chloracne is a systemic or local disease. The so-called
Yusho disease is discussed in section 11.
Herzberg (1947) described several cases of chloracne, in which
other toxic signs and symptoms were seen, due to consumption of
"chlorinated paraffin" used as a substitute for butter during cooking
in postwar Berlin. Among general signs and symptoms observed were
gastrointestinal disturbances with abdominal pain, headache, pain in
joints, neuropathy, depression, and lack of appetite. The
dermatological symptoms were erythema, exanthema, comedones, and
retention cysts in sebaceous glands. It was noted as remarkable that
the skin signs had a follicular predilection, as in seborrhoea (face,
head, bosom, and back). The slow development of chloracne, and
particularily the fact that the sebaceous glands were affected, led
the author to conclude that it was a secretory disease
(Ausscheidungstoxikose). With regard to the chloracnegenic component,
it is unlikely that paraffin itself was active. Herzberg speculated
that something else, possibly a pyrolysis product that arose during
cooking, could have caused the disease.
Accidents in chemical plants involved in the manufacture of
chlorinated phenolic compounds are listed in Table 63. It should be
stressed that all these intoxications are due to mixtures, e.g., TCP
and TCDD and other compounds. Summaries of the industrial accidents
are to be found in Holmstedt (1980) and only some of them will be
dealt with here.
Table 63. Summary of accidents in chemical plants involving the manufacture of chlorinated phenolic compounds
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
Germany 1910 CP Explosion + 5 Same year Teleky (1913), Wahle (1914),
occupational Dohmeier & Janson (1983)
United States 1949 TCP Explosion + 228 30 Ashe & Suskind (1949, 1950),
Occupational Suskind et al. (1953),
Suskind (1978), Huff et al.
(1980), Zack & Suskind (1980)
Zack & Gaffey (1983), Moses
et al. (1984), Suskind &
Hertzberg (1984)
Federal Republic TCP Occupational 17 1 Baader et al. (1951)
of Germany 1949 (PCP)
Federal Republic TCP Occupational 60 Bauer et al. (1961)
of Germany 1952
Federal Republic TCP Occupational 37 Hay (1977)
of Germany 1952-1953
Federal Republic TCP Explosion + 75 29 Hoffman (1957), Goldmann (1972,
of Germany 1953 Occupational 1973), Huff et al. (1980), Thiess
et al. (1982)
France 1953 TCP Explosion + 17 2 Dugois & Colomb (1956, 1957),
Occupational Dugois et al. (1958)
Federal Republic TCP, Occupational 31 24 Schultz (1957a,b), Bauer et al.
of Germany 1954 2,4,5-T (1961), Kimmig & Schultz (1957a,b)
Kleu & Göltz (1971), von Krause &
Brassow (1978)
Table 63 (contd - 2).
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
Federal Republic TCP Occupational 24 4 Risse-Sundermann (1959)
of Germany 1954 2,4,5-T
United States 1956 2,4,5-T Occupational 48 6 Bleiberg et al. (1964), Poland
2,4,5-T et al. (1971)
United States 1956 TCP Occupational Many Hay (1977)
Italy 1959 TCP Explosion + 5 2 Hofman et al. (1962)
Occupational
United States 1959 TCP Occupational Hay (1977)
United States 1960 TCP Occupational Many Hay (1977)
Netherlands 1963 TCP Explosion 106 11 Dalderup, (1974a,b), Berlin
2,4,5-T et al. (1976), Huff et al. (1980)
USSR 1964 2,4,5-T Occupational 128 Telegina et al. (1970), IARC
(1977)
United States 1964 TCP Occupational 61 6 Vahrenholt (1977), Cook et al.
(1980), Ott et al. (1980)
Czechoslovakia 1964-1969 TCP Occupational 80 6 Jirásek et al. (1973, 1976),
Pazderova et al. (1974, 1980,
1981)
United Kingdom 1968 TCP Explosion 90 14 May (1973, 1982), Huff et al.
(1980)
Japan 1970 2,4,5-T Occupational 25 3 Mivra et al. (1974)
Table 63 (contd - 3).
Years from
Cause of Personnel incident to
Country and date Producta exposure affected last observation References
USSR 1972 TCP Occupational 1 1 Zelikov & Danilov
(1974)
Austria 1972-1973 2,4,5-T Occupational 50 Forth (1977), Hay
(1977)
Federal Republic 2,4,5-T Occupational 5 Forth (1977), Hay
of Germany 1974 (1977)
Italy 1976 TCP Explosion 193 8 Reggiani (1977, 1978, 1983a),
Vahrenholt (1977), Filippini
et al. (1981), Abate et al.
(1982), Ideo et al. (1982)
a Products: TCP = trichlorophenols; CP = chlorophenols; PCP = pentachlorophenol;
2,4,5-T = 2,4,5-trichlorophenoxyacetic acid.
The first reported intoxication with a mixture probably
containing TCDD, although the chemical structure was not given,
occurred in February 1910. Five people were said to have been
contaminated after a reactor explosion and two of these were described
in some detail in a dermatological thesis (Teleky, 1913; Wahle 1914).
Wahle (1914), however, in his thesis emphazised that this intoxication
was not due to any of the chlorinated naphthalene derivatives that
were well known by then.
An industrial poisoning was reported in 1949, due to the
formation of TCDD in uncontrolled exothermic reactions occurring
during the manufacture of TCP at a 2,4,5-T-producing factory in Nitro,
West Virginia, USA. The temperature in one of the reactors containing
tetrachlorobenzene, methanol, and sodium hydroxide increased, a relief
valve opened, and the contents of the vessel were discharged into the
interior of the building and over a wide area outside of the building.
A total of 228 people were affected.
Symptoms included chloracne, nausea, vomiting, headaches, severe
muscular aches and pains, fatigue, emotional instability, and
intolerance to cold. Laboratory findings showed an increase in total
serum lipids and an initially prolonged prothrombin time. Among those
affected were not only workmen, but also laboratory personnel, medical
personnel, and even the Safety Director who visited the area of
exposure. Several wives who had never visited the plant also developed
acne, usually at the same time as their husbands working at the plant.
A man from the nearby town who purchased a truck that was parked in
the vicinity of the accident at the time it occurred, and his child,
also developed chloracne. The disabling symptoms, which kept men from
their jobs for as long as 2 years, were severe aches and pains and
fatigability, the manifestations of peripheral neuropathy. Liver tests
4 years later were normal, but mild cases of acne were common. TCDD
was still an unknown chemical. The follow-up to this accident will be
discussed in section 8.4.
In 1953, at the Badische Anilin and Soda Fabrik, during the
alkaline hydrolysis of 1,2,4,5-tetrachlorobenzene to
2,4,5-trichlorophenol, the temperature and pressure in an autoclave
increased rapidly and resulted in an exothermic reaction releasing a
great deal of steam through a safety valve of the reaction vessel.
This steam covered the walls, windows, doors, and machinery in the
rooms of four floors, and finally precipitated in solid form on
everything in these rooms. Forty-two workers involved in the clean-up
operations developed chloracne, and even after the extensive clean-up
operations occasional workers still developed chloracne. Thereafter
the autoclaves were used for 2 years without incident but in 1958 a
mechanic who conducted repair work on an autoclave subsequently
developed chloracne (Hofmann, 1957; Goldmann, 1972). In 1968 and 1969
the building containing the autoclaves was dismantled. Goldmann (1972,
1973) conducted a study of the 42 workers exposed in this accident. In
21 cases, the chloracne was preceded by a non-specific dermatitis and
in two cases very persistent chronic conjunctivitis and blepharitis
were observed; 14 cases also showed involvement of other organs. In
four instances the liver was affected, and microscopic examination of
the liver again showed a very characteristic grey pigment that did not
stain positive for iron. A transient involvement of the myocardium was
also noted. In five instances the upper respiratory tract was involved
with tracheitis and bronchitis. There was one instance of haemorrhagic
pleuritis and one instance of afebrile gingivitis and stomatitis. In
a number of cases a high susceptibility to infection was noted,
sometimes accompanied by a decrease in gamma-globulin. One worker died
of pancreatitis, in seven cases the central nervous system was
affected, three instances of toxic polyneuritis were recorded, and in
two instances hearing, sense of smell, and taste were impaired. The
child of one of these workers also developed chloracne, and in most of
the workers active chloracne persisted for many years - in one
instance for 18 years. Follow-up studies are described in section 9.4.
Of particular interest is a study by Risse-Sundermann (1959).
According to oral reports by the treating physician, all 24 members of
a team working in a trichlorophenol operation became ill after the
production process was switched to the pressurized phenol process in
the spring and summer of 1954. Slightly different acneiform skin
conditions appeared as symptoms of the toxic exposure. In addition,
the patients suffered from dizziness, nausea, vomiting, lacrimation,
burning of the eyes, difficulty in hearing, gastrointestinal spasms,
intolerance to fatty foods, diarrhoea, jaundice, hepatitis (which was
fatal in one case), and paresthesias and hyperesthesias, as well as
extreme irritability. One patient became psychotic and committed
suicide. In addition, some of the patients complained of impotence.
Ten workers at this chemical factory were followed for five years
by Risse-Sundermann (1959). In addition to the signs and symptoms
mentioned above, she noticed swollen lymph glands and a considerable
decrease in body weight. The patients underwent neurological
examination, with no objective signs being observable. Of particular
interest in this well documented study is the fact that in three
patients the general symptoms (e.g., tiredness, depression, lack of
appetite, stomach pains, sexual dysfunction) preceded that of the skin
manifestations .
Bauer et al. (1961) reported a study of workers affected by three
different outbreaks of chloracne. In this study more than 100 workers
were examined. Of these, 31 Hamburg workers had been exposed 5 years
ealier. Nine were examined in detail and their symptoms tabulated.
Initially, there was dermatitis and irritation of the face, sometimes
accompanied by conjunctivitis, and followed by the gradual development
of chloracne and patchy pigmentation of the skin. In some cases
irritations of the mucous membranes of the face and upper respiratory
tract, together with a persistent blepharoconjunctivitis, were also
noted. In the follow-up study, a number of cases of liver injury were
observed and, at liver biopsy, a typical grey pigment was observed in
liver sections, which did not stain positive for iron. Viral hepatitis
was suspected. In a few cases, chronic bronchitis and occasional
myocardial damage were also observed. In all cases, fatigue was the
main complaint and muscle weakness and muscle pain were described by
the workers, particularly in the proximal muscles of the lower
extremities. All nine also reported decreased libido. In a few
instances, paresthesia and hyperesthesia or pronounced sensory
neuropathy were observed, and minor circumscribed pareses were found.
A psychovegetative syndrome occurred in most of the workers. Other
signs recorded were: inability to concentrate, memory deficits, sleep
disturbances, particularly increased somnolence, decreased drive, and
alcohol intolerance. Psychological tests also showed abnormalities.
Following the malfunction of a reaction vessel in northern Italy,
in which 2,4,5-trichlorophenol was produced, the temperature in the
vessel increased rapidly and an intense black vapour filled the
work-room covering everything with a black deposit. Five workers
engaged in clean-up operations developed chloracne (Hofmann &
Meneghini, 1962). None of those involved in the clean-up exhibited any
involvement of general systemic toxicity (even after 16 months) that
could be related to exposure to the tar and soot. However, one
15-year-old worker developed folliculitis and superficial nodular
elements on the face a few days after initial exposure. A slow but
progressive generalization of the dermatosis developed on the trunk,
scalp, and lower extremities. An examination several months later
revealed no damage to the renal or liver parenchyma. However, this
worker was found to still suffer from outbursts of chloracne in 1980
(Holmstedt, 1980).
Duverne et al. (1964) reported a case that occurred at a plant at
Lyon, France, where products that used 2,4,5-tri-chlorophenol as a
starting material were manufactured. This worker developed chloracne
as well as serofibrinous pleuritis.
Ten workers also developed chloracne at a plant near Grenoble,
France, which produced 2,4,5-trichlorophenol that served as the
starting material for phenoxy pesticides and germicides for cosmetics.
These workers showed symptoms of systemic poisoning similar to those
reported by Goldmann (1972), and hepatic insufficiency with lipaemia
and elevated serum cholesterol levels (Dugois & Colomb, 1956). Another
accident resulting in TCDD exposure of workers occurred in the same
factory in 1966 (Dugois et al., 1967).
An exothermic reaction resulted in an explosion at a plant in
Chesterfield, England, in 1968. The company made 2,4,5-trichlorophenol
from tetrachlorobenzene and the explosion occurred during the process
involving ethylene glycol and caustic soda under atmospheric pressure
(Milnes, 1971). In this incident, 79 workers developed chloracne but
there was no evidence of systemic illness (May, 1973). In 1971, 3
years after the explosion, two workers who had not been involved in
the explosion or its aftermath were employed as pipe-fitters at a new
installation, away from the site of the explosion, to refit one of the
cleaned tanks. They both developed severe chloracne, and the son of
one of these workers and the wife of the other also developed this
condition (Jensen & Walker, 1972). May (1973) cited two incidents
involving explosions in a similar process. In the first incident,
fatal injuries were recorded; in the second incident, all 50 exposed
persons fell ill after 10 days and had liver injury.
In the USA, an outbreak of chloracne occurred among workers
manufacturing 2,4-dichlorophenoxyacetic acid and
2,4,5-trichlorophenoxyacetic acid (Bleiberg et al., 1964); 29 workers
developed chloracne and 11 of these had elevated urinary uroporphyrins
and exhibited varying degrees of acquired porphyria cutanea tarda. At
least one of these workers had abnormal liver-function tests and
microscopic examination of a liver biopsy specimen showed parenchymal
cell regeneration and haemofuscin pigment. Many of the workers with
chloracne showed hyperpigmentation of the skin. A second study of the
workers at this plant was conducted in 1969 by Poland et al. (1971).
A total of 73 male employees were examined, and moderate to severe
chloracne was found in 13 workers (18%), mild chloracne in 35 (48%),
hyperpigmentation in 30, and uroporphyrinuria in 1. No definite
systemic illness could be documented in these workers. Of those
studied, 33 had been employed at the plant for 0-4 years, 10 for 4-8
years, and 30 for more than 9 years. The mean duration of employment
was 8.3 ± 7.6 years (mean ± 1 SD). The trichlorophenol manufactured in
this plant contained 10-25 mg TCDD/kg. Twenty-six of the workers seen
by Bleiberg et al. (1964) were also seen in a follow-up study. Six
months prior to the second survey (Poland et al., 1971), the
manufacturing process was altered so that the 2,4,5-T produced
contained less than 1 mg TCDD/kg.
In 1964, in the USSR, many workers developed chloracne while
engaged in producing 2,4,5-T. Production was then discontinued
(Telegina & Bikbulatova, 1970).
On 10 July 1976, an explosion occurred at the ICMESA plant at
Meda, near Seveso, Italy, when 12 workers were present. All 176
workers of the plant were examined 3 or 4 weeks after the accident.
Chloracne was suspected in 1 of them; the others showed minor symptoms
that could not be correlated with exposure. Alkaline phosphatase and
delta-glutamyltransferase seemed slightly increased in 32 and 37
cases, respectively, while five workers showed a reduction in their
delta-aminolevulinic acid dehydratase blood levels, and three showed
moderately increased urinary gamma-aminolevulinic acid (Zedda et al.,
1976). Similar findings were reported by Fara et al. (1976) and
Reggiani (1978). The follow-up of the general population is described
in section 8.2.
8.2 General Population Studies
8.2.1 Missouri, USA
Environmental exposures have occurred in a small area of
Missouri, USA (Carter et al., 1975; Kimbrough et al., 1977; Kimbrough,
1984). In the summer of 1971 many birds, rodents, cats, dogs, insects,
and horses died after exposure in a horse arena in eastern Missouri.
The incident followed the spraying of "waste oil" on the horse arena
for dust control. Within 3 weeks of the spraying of this arena, two
other arenas were sprayed. In all, 57 adult horses died, 26 abortions
occurred among the horses at the most heavily exposed farm and many
foals died soon after birth. At the time, the nature of the chemical
that had caused the problem was unknown. The arenas were excavated and
the contaminated dirt dumped at other sites. After many fruitless
attempts to identify the cause of this outbreak, it was discovered in
1973-1974 that the original soil from one of the arenas contained
5600-6500 mg trichlorophenol/kg, 31.8-33.0 mg TCDD/kg, and 1350-1590
mg polychlorinated biphenyls/kg. Because of this finding, the episode
was reinvestigated. It was found that the salvage oil company that
sprayed the three arenas routinely collected discarded motor oil and
lubricants from over 2000 service stations in eastern Missouri and
southwestern Illinois. It also collected, from various sources, a
limited amount of used organic solvents such as transformer oils and
other compounds. A company in southwestern Missouri was finally
identified as a source of TCDD. This company had manufactured
trichlorophenol as an intermediate for hexachlorophene. The production
of 2,4,5-tri-chlorophenol had generated a distillate residue which was
emptied once a week into a residue storage tank. Initially this
chemical waste was collected and incinerated but, in 1971 when the
trichlorophenol producer experienced a financial crisis, he arranged
for the chemical wastes to be disposed of by a chemical supplier. The
chemical supplier subcontracted the chemical waste disposal to the
salvage oil dealer. The salvage oil dealer added the toxic chemical
waste to his salvage oil storage tank, having collected a total of 18
000 gallons. This material, mixed with salvage oil and other
chemicals, was sprayed on the riding arenas and some of it was taken
to re-refining companies. Soil samples from arenas where contaminated
dirt had been dumped in 1974 contained trichlorophenol levels that
ranged from 1.5-32.6 mg/kg, TCDD levels that ranged from 0.22-0.85
mg/kg, and polychlorinated biphenyl levels that ranged from 10-25
mg/kg.
A 6-year-old girl, who had used one of the arenas for
sandbox-like play in 1971, developed epistaxis, headache, diarrhoea
and lethargy, haemorrhagic cystitis, and signs of pyelonephritis. She
had an uneventful recovery. Three other females exposed to the same
arena had recurring headaches, skin lesions, and polyarthralgias. Two
3-year-old boys in another arena developed chloracne on the exposed
skin surfaces which lasted for more than a year. Evaluation of the
three female patients 5.4 years after exposure to TCDD-containing oil
showed them to be in good health (Beale et al., 1977).
A comprehensive medical examination of 154 residents exposed to
TCDD, and 155 unexposed residents in similar type housing in eastern
Missouri, revealed no consistent differences between the two groups.
The examination included a medical history, physical examination,
serum and urinary chemistries, and immunological and neurological
tests. The findings may suggest that long-term TCDD exposure is
associated with depressed cell-mediated immunity (decreased
delayed-type hypersensitivity skin reactions to standard antigens)
(Hoffman et al., 1986; Stehr et al., 1986). Urinary concentrations of
glucaric acid were not significantly different between persons
identified as being at high or low risk (Steinberg et al., 1985).
8.2.2 Seveso, Italy
The scientific follow-up on the population of Seveso, N. Italy,
which had been accidentally exposed to TCDD in 1976 (see sections
4.1.1 and 8.1), was guided by an international steering group headed
by Professor M.A. Klingberg. The group completed its work in February
1984 and concluded that "it is obvious that no clear-cut adverse
health effects attributable to TCDD, besides chloracne, have been
observed" (Regione Lombardia, 1984). A total of 193 people had
displayed symptoms of chloracne, but at the beginning of 1984 only 20
presented active symptoms. After 15-20 days exposure to TCDD soil
levels of 270-1200 µg/m2, there was a marked incidence of chloracne.
No disturbance of biochemical functions were seen when the exposure
had been limited to soil with TCDD levels at or below 30-70 µg/m2.
Later evaluations failed to confirm earlier findings of a decrease in
motor nerve conduction velocity in some individuals. A significant
increase in urinary glucaric acid levels, indicating increased
microsomal enzyme activity, was found 3 years after exposure in 67
exposed children, as compared to 86 non-exposed children (Ideo et al.,
1982, 1985). The steering group found the data difficult to evaluate
as analytical and individual biological variabilities were not
explained (Regione Lombardia, 1984).
Studies performed on the rate of spontaneous abortions and birth
defects in the Seveso area do not allow any conclusions to be drawn
(Tognoni & Bonaccorsi, 1982). The hypothesis that low exposure might
cause pre-pregnancy or pregnancy effects that adversely affect the
outcome was tested using several exposure models. The only finding was
a slightly higher rate of haemangioma among newborns in the exposed
group. However, this showed up only with one of the exposure models.
It was considered doubtful that this was due to TCDD exposure (Regione
Lombardia, 1984).
Lymphocytes from inhabitants of Seveso were examined for
chromosomal aberrations by Regianni (1980a,b) and Mottura et al.
(1981). In 17 TCDD-exposed individuals examined within two weeks of
the accident, no increase in chromosomal aberrations was observed
(Regianni, 1980). In the abstract by Mottura et al. (1981),
chromosomal aberration analysis was performed on subjects distributed
into three classes: acute exposure, chronic exposure, and a control
group of non-exposed subjects. No significant difference in the
frequency of chromosomal aberrations in the three exposure categories
was reported. Data on number of subjects, chromosomal aberrations, and
exposure level and time were not given.
Tenchini et al. (1983) published a comparative cytogenetic study
on induced abortions from women exposed to TCDD after the Seveso
accident, and in non-exposed subjects. Chromosome analysis was
performed on maternal peripheral blood, placental and umbilical cord
tissues, and fetal tissues. No significant differences were found in
the level of chromosomal aberrations in the blood of placenta and
umbilical cord from TCDD-exposed and non-exposed women. The exception
was fetal samples from non-exposed women, in which a significant
increase in chromosomal aberrations was obtained, possibly an artefact
due to experimental techniques. The effect of TCDD on fetal
chromosomes is therefore still unclear.
Several epidemiological follow-up studies are continuing in and
around Seveso.
8.2.3 Viet Nam
From 1960 to 1969 a mixture of 2,4-dichlorophenoxyacetic acid and
2,4,5-trichlorophenoxyacetic acid (Agent Orange), which was
contaminated with TCDD (concentrations ranging from 0.5 to 47 mg/kg)
(Kearny et al., 1972), was sprayed over areas of Viet Nam as a
defoliant. The spraying from 1960 to 1965 was minimal; in 1966 it
covered slightly more than 800 000 acres, in 1967 almost 1.7 million
acres, in 1968 over 1.3 million acres, and in 1969 1.2 million acres.
Studies have been carried out since the early 1970s to ascertain
whether the exposure of the general population in Viet Nam to this
herbicide could have resulted in an increased incidence of birth
defects. However, the results of such investigations have not been
published in readily available peer-reviewed journals, making it
difficult to assess the scientific significance of the findings. Such
studies have been reviewed by Westing (1984) and Constable & Hatch
(1985). Those studies reviewed indicate a range of effects including
spontaneous abortions, infertility, and birth defects. However, there
are marked deficiencies in experimental design in most, if not all,
studies, including potential bias in the selection of populations,
poor record-keeping of populations and biological effects, such as
congenital malformations, and a lack of control over possible
confounding factors. These deficiencies make it difficult, if not
impossible, to use this body of data in assessing the human health
risks from exposure to phenoxyherbicides contaminated with TCDD and
other PCDDs.
Tung (1973) reported an increased incidence of liver tumours in
Viet Nam. From 1955 to 1961 there were 159 cases of liver cancer out
of a total of 5492 cancer cases, and from 1962 to 1968, 791 out of a
total of 7911 cancer cases. Van (1984) continued Tung's investigation.
Previous exposure to herbicides of 21 male cases of primary liver
cancer and 42 controls was ascertained. Six of the 21 cases and three
of the controls had lived or worked in areas sprayed with herbicides
or had moved there shortly after spraying ceased. Residence time
varied from 8 to 77 months. There is a lack of information on
confounding factors and there was a chance for bias in these studies.
In general, the possibility of exposure to multiple chemicals and the
short latency period noted make the study by Van (1984) of little
value for assessing risk (IARC, 1986).
In 1979, the United States Air Force (USAF) initiated an
epidemiological study into the possible health effects from chemical
exposure of Air Force personnel who conducted aerial dissemination of
herbicide in Viet Nam (Operation Ranch Hand) (Lathrop et al., 1984).
The purpose of this investigation was to determine whether long-term
health effects exist and can be attributed to occupational exposure to
herbicides. This study used a matched cohort design in a
non-concurrent prospective setting, incorporating mortality,
morbidity, and follow-up studies. The report presented the results of
health information on 2706 Ranch Handers and comparison individuals
obtained by questionnaire and 2269 Ranch Handers and comparison
individuals undergoing an extensive physical examination. It was
concluded that there was insufficient evidence to support a cause and
effect relationship between herbicide exposure and adverse health in
the Ranch Hand group at this time. The study disclosed numerous
medical findings, mostly of a minor or undetermined nature, that
require detailed follow-up.
In a study of 15 soldiers in Australia exposed to Agent Orange,
no increases in structural chromosomal aberrations or sister chromatid
exchanges were noticed, compared to a control group of 8 subjects
(Mulcahy, 1980). In 1980 the Australian Commonwealth Institute of
Health agreed to conduct a series of scientific investigations into
the health of Viet Nam veterans and their families. After
considerating the most appropriate study programme, it was decided in
1981 to conduct, as part of that programme, a case-control study of
congenital anomalies and Viet Nam service (Donovan et al., 1984). The
report is largely negative, as is that of Erickson et al. (1984) which
reported a similar study of American veterans.
Table 64. Signs and symptoms reported in association with human exposure
to TCDD or mixtures containing TCDD
A. Skin Manifestations C. Neurological Effects
1. Chloracne 1. Sexual dysfunction
2. Hyperkeratosis 2. Headache
3. Hyperpigmentation 3. Neuropathy
4. Hirsutism 4. Sight disturbance
5. Elastosis 5. Loss of hearing, taste,
and smell
B. Systemic Effects D. Psychiatric Effects
1. Mild fibrosis of liver 1. Sleep disturbance
2. Raised transaminase values 2. Depression
in blood 3. Loss of energy and drive
3. Hypercholesterolaemia 4. Uncharacteristic bouts of anger
4. Hypertriglyceridaemia
5. Loss of appetite and weight loss
6. Digestive disorders (intolerance to
alcohol or fatty food, flatulence,
nausea, vomiting, diarrhoea)
7. Muscular aches and pains, joint
pain, lower extremity weakness
8. Swollen lymph glands
9. Cardiovascular, urinary tract,
respiratory, and pancreatic disorders
8.3 Signs and Symptoms in Humans Associated With TCDD Exposure
Many signs and symptoms have been reported in studies of human
exposures to PCDDs, both occupationally and from the general
environment. These have been compiled from the various studies and are
shown in Table 64.
8.3.1 Skin manifestations
Chloracne is a sign of exposure to several chlorinated cyclic
organic compounds, the most potent being TCDD. Chloracne thus may
serve as a marker of such exposure. The most distinctive lesion in
chloracne is the so-called cyst, a skin-coloured elevation that may
measure from 1 mm to 1 cm in diameter, with a central opening that may
be difficult to detect. Comedones that contain black or
black-appearing material in their openings are also present. There may
be a secondary inflammatory reaction, melanosis, and hyperkeratosis,
and these skin changes may be preceded by a "cable rash" or "cable
itch". These skin lesions resemble photosensitivity reactions and the
bearers may suffer severe pruritus. Microscopic examination of the
skin lesions shows marked dilatation of the hair follicles which are
filled with keratinous material, the sebaceous glands may be partly or
completely atrophied and, occasionally, hyperplasia of these glands
has also been reported. Hyperkeratosis and acanthosis of the
surrounding epidermis usually accompany these lesions. Atrophy of the
epithelium and thinning of the epithelial walls surrounding these
keratinous cysts are observed at a later stage of the disease. If the
follicular cysts rupture, foreign body granulomata may also be
observed. Healing of these skin lesions usually results in deeply
pitted scars. The distribution of chloracne is predominantly facial,
affecting in particular the malar areas, the jaws, and the regions
behind the ears. At times it may involve the ear canal and, with
increasing severity, also the rest of the face and neck. In more
extensive cases, the outer upper arms, neck, back, abdomen, outer
thighs, and genitalia may also be involved (Crow, 1970).
While the absence of chloracne does not absolutely rule out
exposure to TCDD, it usually indicates that there has been no exposure
to a toxic dose of the substance. "Toxic" is used here to indicate
both systemic and local effects. Where there has been exposure to TCDD
and chloracne has resulted, it is the only known clinical sign that
persists for a long period of time, even for the remainder of the
exposed person's life time. In a large group of people exposed to
mixtures containing TCDD, the absence of chloracne usually indicates
that exposure to a toxic dose was unlikely and also makes it unlikely
that severe, persistent systemic disorders will result.
Hyperkeratosis is a fairly common phenomenon whereas
hyperpigmentation and hirsutism are rare. It should be noted that
hyperkeratosis is prominent in the exposed Seveso children who have no
affected sebaceous glands. These glands develop only at puberty.
Elastosis of the skin has been noted as a long-term effect of
TCDD exposure.
8.3.2 Systemic effects
Liver effects following exposure to PCDDs have been diagnosed
even by histological examination, and account for temporarily raised
transaminases in blood, hypercholesteraemia, and
hypertriglyceridaemia. Bauer et al. (1961) and Risse-Sundermann (1959)
do not however exclude viral hepatitis as a cause of such findings in
their patients exposed to TCDD. Loss of appetite, weight loss, and
digestive disorders are common complaints from humans exposed to
either TCDD itself, or to technical mixtures containing TCDD. Muscular
aches and pain and weakness in extremities have been reported,
particularly after exposure to technical mixtures containing TCDD.
Swollen lymph nodes have also been reported, both after exposure to
"pure" TCDD and to mixtures. The cardiovascular, urinary tract,
respiratory, and pancreatic disorders reported are of doubtful
significance with regard to a causal relationship to TCDD exposure.
Porphyria cutanea tarda has been reported in two cases of
occupational exposure where chlorinated organic compounds were
manufactured in addition to trichlorophenol. These were the incidents
at the factory of Diamond Alkali, Newark, New Jersey, USA, in 1956
(Poland et al., 1971) and at Spolana, Czechoslovakia, between 1964 and
1969 (Pazerova-Vejlupkova et al., 1981). The porphyria cutanea tarda
observed in these cases was very unlikely to have been induced by
exposure to TCDD but rather by exposure to other chlorinated organic
compounds manufactured in these plants (Jones & Chelsky, 1986).
8.3.3 Neurological effects
Sexual dysfunction (lack of libido and impotence) has been
reported after acute exposure to both "pure" TCDD and technical
mixtures (Schulz, 1968). The frequency of its occurrence may have been
underestimated to date. Headache is a frequent symptom after exposures
to technical mixtures containing TCDD.
Sensory neuropathy has been noted in many instances. Usually
workers in the initial stages of exposure will complain of pains in
their joints after they have very acute severe chloracne; however,
there are usually no abnormal physical findings in the joints, but the
complaints may continue. In early studies of workers affected by TCDD,
no attempts were made to objectively measure the effects on the
sensory nervous system. Tests have now been developed that evaluate
sensory nerves and that can be used in future field studies. The nerve
conduction tests, which primarily have been used so far, are actually
not very useful to measure neuropathy. Differences in nerve conduction
were shown among residents from Seveso, Italy, who had chloracne and
those who did not (Richert, von, 1962; Fillipini et al., 1981).
Sight disturbance may be related to alkaline exposure or to
conjunctivitis related to effects on the glands of Meibom. Loss of
hearing, taste, and smell have been reported in a few cases, but a
causal relationship to TCDD exposure is doubtful.
8.3.4 Psychiatric effects
The symptoms have been listed in Table 64 in what is believed to
be their order of frequency and degree of severity.
8.4 Epidemiological Studies
Signs and symptoms related to accidental exposure to TCDD are
given in Table 64. However, it should be observed that all the
accidents and occupational contamination concern exposure to a mixture
of compounds where TCDD was only one component. In all cases, its
concentration in the mixtures was unknown. Only two cases of
intoxication with "pure" TCDD have been reported.
The story of the discovery of TCDD is by now well documented
(Holmstedt, 1980; Sandermann, 1984a,b). TCDD was synthetized in 1955.
Four people were intoxicated, one co-worker severely so while drying
crystals. In all cases, decreased libido was the first symptom,
followed by other symptoms such as moderate to severe chloracne,
sleeping difficulties, inability to concentrate, depression, and, in
at least one case, swelling of the lymph nodes. In all cases, the
signs and symptoms disappeared within a couple of years, with the
exception of the chloracne in the most heavily exposed man.
The second occasion of exposure to what one must assume to be
pure TCDD is the one reported by Oliver (1975). The toxic effects on
three young scientists who suffered "transient minimal exposure to
TCDD" were described. Two of them suffered from typical chloracne.
Delayed symptoms about two years after initial exposure occurred in
two of the scientists. These symptoms were said to have included
personality changes, other neurological disturbances, and hirsutism.
All three scientists were found to have raised serum cholesterol
levels, but no other biochemical disturbances and no porphyrinuria or
liver damage were demonstrated. Whether the unusually delayed
physiological effects were in fact due to the initial dioxin exposure
is a question that was discussed by the author. Although conclusive
evidence is lacking, it seems likely that these delayed effects were
in fact due to dioxin intoxication. The conditions of exposure remain
unexplained.
Of the many cases of exposure reported in Table 63, only two (at
Monsanto in 1949 and at BASF in 1953) have been adequately followed up
epidemiologically with matched control groups.
The workers of Monsanto, USA, have been investigated several
times between 1949 and 1984. Immediately after the accident, Ashe &
Suskind (1949) hospitalized and studied four cases of severe poisoning
among the workers. These four workers were diagnosed as having
chloracne, but by the time of examination these men had recovered from
earlier symptoms of peripheral neuropathy. In 1950, a further
examination of these four workers and two additional men revealed
continued irritability, nervousness, and insomnia (Ashe & Suskind,
1950). A consistent loss of libido and some impotence was reported.
Further clinical examination revealed hepatomegaly, altered
prothrombin times, and disturbed lipid metabolism.
A further study of 36 workers from this plant was undertaken in
1953 (Suskind et al., 1953). It was noted that even those who
developed, to a moderate or severe degree, the skin eruptions, pains
in back, dyspnoea, fatigue, nervousness, and decreased libido
generally improved. Even those suffering the most severe cutaneous
eruptions initially had only a few or no lesions in 1953.
More recent studies on these workers are those of Zack & Suskind
(1980) and Zack & Gaffey (1983). Zack and Gaffey reported on a
121-member study cohort, with a presumptive high-peak exposure to TCDD
base on chloracne occurrence, which was followed for mortality until
1978. The entire cohort was traced; there were 32 deaths; 89 people
were still alive. There was no excess in total mortality or in deaths
from malignant neoplasms. The proportional mortality analysis of
decedents according to exposure by 2,4,5-trichlorophenoxyacetic acid
(2,4,5-T) indicated no unusual patterns of mortality. The proportional
mortality ratio (PMR) for malignant neoplasms was low (PMR = 82) in
the exposed group. Lung cancer was the only site among the malignant
neoplasms for which the value was somewhat higher in the exposed
group.
The Monsanto workers were again examined in 1984 (Suskind &
Herzberg, 1984). A clinical epidemiological study was conducted to
determine the long-term health effects of workplace exposures during
the process of manufacturing the herbicide 2,4,5-T, including
contaminants such as TCDD. The population consisted of two cohorts,
204 clearly exposed and 163 not exposed (controls). Among the exposed
workers, clinical evidence of chloracne persisted in 55.7%. None of
the controls experienced chloracne development. An association was
found between the persistence of chloracne and the presence and
severity of actinic elastosis of the skin. There was an association
between exposure and the history of ulcers of the gastrointestinal
tract. Pulmonary function values among those who were exposed and who
currently smoked were lower than those who were not exposed and who
currently smoked. No disturbances of sexual functions were found at
this time after age adjustment. The data assembled in the study
indicated no evidence of increased risk for cardio-vascular disease,
hepatic disease, renal damage, or central or peripheral nervous system
problems.
Another selection of the population from the same plant has been
examined by another group of epidemiologists (Moses et al., 1984).
Since the degree of exposure was unknown to these investigators and
since chloracne is generally considered a quite reliable indicator of
heavy dioxin exposure, it was decided to use chloracne as a
"surrogate" for exposure and to classify the study population by its
presence or absence. It was recognized that those without chloracne,
but with appropriate work-exposure history, might also have had TCDD
exposure and were not therefore used as "unexposed controls".
Chloracne was found in 52% of 226 workers in a 1979 cross-sectional
survey at the plant where 2,4,5-T had been manufactured from 1948 to
1969. Mean duration of residual chloracne was 26 years, and in 29
subjects it had been present for 30 years. A significantly increased
prevalence of abnormal gamma-glutamyl transpeptidase (GGT) and higher
mean GGT were found in those with chloracne compared to those without.
Although mean triglyceride values were higher in those with chloracne,
the difference was not statistically significant. Neurological
examination showed a statistically significant higher prevalence of
abnormal sensory findings in those with chloracne. Increased
prevalence of angina and reported myocardial infarction in those with
chloracne was not significant when age-adjusted. Increased prevalence
of reported sexual dysfunction and decreased libido in those with
chloracne, compared to those without, was statistically significant
after age adjustment. No differences were found between those with and
without chloracne in serum cholesterol, total urinary porphyrins, or
in reproductive outcomes. Exposure to TCDD in 2,4,5-T production may
thus result in apparently permanent changes in the skin. Sensory
changes in peripheral nerves and possible changes in liver metabolism
in those with current or past chloracne are also suggested by these
data. Based on worker histories, even severe acute toxicological
effects of TCDD are reversible, or improve markedly over time. While
the cross-sectional nature of this study, the low participation rate,
and the highly select nature of the population limit the conclusions
that can be drawn, it is unlikely that permanent, severe, and
debilitating toxicological sequelae are inevitable after exposure to
TCDD sufficient to produce chloracne. It must be noted, however, that
individual susceptibility may make certain workers with heavy
exposures more vulnerable.
The exposure of workers at BASF in 1953 has been the subject of
several reviews, the latest one being that of Thiess et al. (1982).
Twenty-seven years after the accident that occurred in the BASF
Ludwigshafen plant, a mortality study was undertaken of people exposed
in the uncontrolled reaction which occurred during the trichlorophenol
process. The follow-up was 100% successful and involved 74 people.
Overall mortality (21 deaths) did not differ in this group from the
rate expected in three external reference populations, or from that
observed in two internal comparison groups, where 18-20 deaths were
observed. Of the 21 deceased people, 7 had had cancer, compared with
4.1 expected. In addition, two other cases of cancer (one bronchial
carcinoma and one carcinoma of the prostate) were still alive at the
time of writing. Three deaths due to stomach cancer at ages 64, 66,
and 69 years, were found, compared with 0.6 expected from regional
mortality data. One stomach cancer occurred among 148 individuals in
the two comparison cohorts. The incidence of cancer in these workers
was considerably greater than expected and cannot be explained only as
a chance event. Of 74 people, 66 had severe chloracne or severe
dermatitis. There is a possibility that some members of the BASF
cohort were exposed to other unknown occupational hazards before or
after the accident. However, the use of two internal comparison groups
composed of matched controls from the same factory was designed to
control for, as far as possible, other occupational exposures that
could be important etiological or confounding factors. Because of the
small size of the cohort and the small absolute number of deaths from
any particular cause, the results of this study do not permit any
definite conclusions concerning the carcinogenic effect of exposure.
In comparison with the above-mentioned, well conducted long-term
epidemiological studies, a host of other follow-up studies have been
published, none of which used adequate controls. They are, therefore,
of less value but will be briefly summarized here.
Jirasek et al. (1973, 1976) and Pazderova et al. (1974, 1980,
1981) examined 55 of a total of 80 workers who suffered intoxication
during the manufacture of sodium pentachlorophenate and the sodium
salt and butyl ester of 2,4,5-T. One worker died from severe acute
intoxication at an early stage (Jirasek et al., 1976), and 76 workers
developed chloracne. The following additional symptoms were found:
porphyria cutanea tarda, disorders of the metabolism of lipids,
porphyrins, and carbohydrates, and alteration of plasma proteins.
Hepatic lesions were also present. Neurological and electromyographic
(EMG) examinations revealed peripheral nerve changes in 17 people,
first detected in 8 people during the second year of the study. A
neurasthenic syndrome was also observed. The patients with porphyria
cutanea tarda showed hyperpigmentation, hypertrichosis, and bullosis
actinica mechanica. Porphyrin excretion in urine ranged from 172 to
2230 µg/24 h. Polyneuropathies, confirmed by EMG examination, were
noted, predominantly in the lower extremities. In this outbreak, the
disease was progressive during the first 2 years; subsequently the
dermatological symptoms as well as the porphyric disease and the
neurological disorders improved. The impaired lipid metabolism
improved only very slowly.
In this plant, the toxic substances were led off through the
breathing zone of the workers. The concentrations of the chlorinated
hydrocarbons in the air were never measured. Due to insufficient data,
the real hygienic conditions at the work place could not be accurately
reconstructed. The manufacturing of 2,4,5-T was halted permanently in
1968 so that it was impossible to obtain the necessary information in
an adequate manner. From 1959 to 1964, according to information from
the plant, only sodium pentachlorophenate was manufactured. Not until
1965 was the manufacture of sodium 2,4,5-T commenced on a pilot scale,
and later the butyl ester of 2,4,5-T was also manufactured. After each
year of production, something was always changed or modified in the
process and technology so that actually there was never full-scale
production in the true sense of the word. Many of the herbicides
manufactured could not be found from the documentation (Pazderova et
al., 1974). The uncertain mixture of compounds involved in the Spolana
episode makes interpretation of signs and symptoms almost impossible.
In all likelihood the porphyria observed was due to the
hexachlorobenzene stated to be produced at this factory.
Signs of disturbance in the porphyrin metabolism in workers
manufacturing 2,4,5-T was also described by Poland et al. (1971).
Chloracne was not correlated significantly with job location within
the plant, duration of employment, or coproporphyrin excretion.
Although 11 subjects with uroporphyria and at least three with overt
porphyria cutanea tarda had been found in a study of the same plant
six years earlier (Bleiberg et al., 1964), no clinical porphyria could
be documented at the time of the second investigation, and only one
worker had persistent uroporphyrinuria. Evidence of toxicity in other
organ systems was markedly less than that reported in previous studies
and in most instances there was no difference from normal populations.
In all likelihood the porphyria cutanea tarda in this case, as in the
study from Czechoslovakia, was due to a compound other than TCDD. This
is corroborated by a recent re-evaluation of the literature (Jones &
Chelsky, 1986).
A study from northern Germany was published by Bauer et al.
(1961). It is not clear where the cases orginated and only nine
patients were studied in depth. A summary of the findings from these
patients, and another person suffering from chloracne after
occupational exposure to trichlorphenol, was reported by Kleu & Göltz
(1971). These patients were followed for 15 years after exposure. The
severity and types of symptoms varied in a dose-related manner. Major
complaints were decreased sexual activity, muscular weakness, easy
fatigability, irritability, and loss of appetite and memory. The
authors concluded that a permanent defect had occurred, the late form
of which resembled a cerebral involutionary syndrome, combined with
mental depression and neurasthenia.
A follow-up study of 11 of the 24 employees at Boehringer
exhibiting skin lesions in 1955 was published by von Krause & Brassow
(1978). Many continued to suffer from their earlier complaints. In
seven of the eleven, nausea and intolerance to heavy fatty food was
still common, and six men complained of alcohol intolerance. Although
conjunctivitis had disappeared, chloracne was still clearly visible in
most of the 11 subjects. Neurological problems were still severe in
six of the workers.
Ten years after the incident at Coalite in 1968 when 79 workers
developed chloracne due to exposure to a chlorophenol-TCDD mixture, a
study was undertaken to establish the state of health of the affected
employees (46) remaining in the company's employment (May, 1982).
Forty-one of the 46 employees participated. The opportunity was used
to examine effects on mortality, morbidity, carcinogenesis,
reproduction, teratogenicity, fetotoxicity, biochemistry, immunology,
and genetic changes. Concurrently, two control groups were
established, one with no dioxin exposure and the other with possible
dioxin exposure. These groups were selected from within the works and
matched the study group with respect to sex and age, but it was not
possible to match them for occupation and social status. Half the
affected subjects still had minor chloracne. Other than this finding,
the authors concluded that the subjects had not been had been
adversely affected in any way.
Data on the mortality of workers at the Dow Chemical company have
been provided in two papers (Cook et al., 1980; Ott et al., 1980). The
first of these studies describes the mortality of a cohort of 61 males
involved in the preparation of trichlorophenol. Forty-nine of these
workers developed chloracne, presumably as a result of skin absorption
of the process contaminant TCDD. Within the limitations posed by
cohort size and length of follow-up, the exposure to chlorophenol-TCDD
mixtures did not appear to have adversely affected mortality
experience. Overall, four deaths occurred and 7.8 were expected. Of
these, one death was due to cardiovascular disease (3.8 expected) and
three deaths were attributed to cancer (1.6 expected). None of the
findings was statistically significant. The second paper examined the
mortality experience of 204 people exposed to 2,4,5-T during its
manufacture from 1950 to 1971. Length of employment within the 2,4,5-T
process area ranged from less than one year to a maximum of
approximately ten years. Efforts to minimize TCDD contamination of the
product resulted in non-detectable concentrations (less than 1 mg/kg)
near the end of this period. Within the scope of this mortality
survey, no adverse effects were observed with respect to occupational
exposure to 2,4,5-T or to its feedstock, 2,4,5-trichlorophenol.
Hardell and his co-workers in Sweden have conducted a series of
case-control studies and reported an increased risk of soft-tissue
sarcomas in men who were exposed to phenoxy herbicides and/or
chlorophenols (Hardell & Sandsstrom, 1979; Hardell, 1981; Hardell et
al., 1981; Hardell & Ericksson, 1981). These authors also reported a
case-control study that suggested that phenoxyacetic acids and
chlorophenols may predispose to Hodgkin's lymphoma (Hardell et al.,
1981). The relative risk was higher for a group exposed to phenoxy
herbicides including 2,4,5-T and chlorophenols, i.e., pesticides that
may be contaminated with PCDDs and PCDFs. However, an increased risk
was still found in a group exposed mainly to phenoxy herbicides such
as MCPA, 2,4-D, mecoprop and dichloroprop, i.e., pesticides with low
or no contamination with PCDDs and PCDFs.
Analysis of fat levels of PCDDs and PCDFs in patients with soft
tissue sarcomas and in controls failed to reveal any differences
between the two groups (Nygren et al., 1986) (section 4.4.4.1).
A cohort study on Swedish farmers and gardeners has been carried
out recently (Wiklund & Holm, 1986). Despite the greatly increased use
of phenoxyacetic acid herbicides from 1947 to 1970, no time-related
increase in the relative risk of soft-tissue sarcoma was found in the
cohort or in any of the subcohorts. The same was found by Hoar et al.
(1986) although the latter study points to an increase in non-Hodgkin
lymphoma. It should be noted that in all these studies the majority of
the herbicides used did not contain TCDD.
In follow-up studies of workers exposed to 2,4,5-T and its
precursor 2,4,5-trichlorophenol (and therefore, presumably, also to
TCDD), no excessive deaths due to any cause were registered (Cook et
al., 1980; Ott et al., 1980; Zack & Suskind, 1980; Zack & Gaffey,
1983).
Honchar & Halperin (1981) merged the above four cohorts and found
that three (2.9%) of the total 105 deaths were reported to be from
soft-tissue sarcoma. Based on national statistics only 0.07% was
expected to be due to this cause. Fingerhut et al. (1984) reviewed the
employment records, medical and pathological reports, tissue
specimens, and death certificates for these three cases and four
additional cases of deaths from soft-tissue sarcomas in these and
related cohorts reported by Cook (1981), Moses & Selikoff (1981), and
Johnson et al. (1981). Three out of the seven cases had a record of
chloracne and one of dermatitis. After review of the tissue specimens,
five of the seven cases were diagnosed as soft-tissue sarcoma. The
remaining two (which were among the three cases in the merged cohort
of Honchar & Halperin (1981)) were found to be carcinoma. For three of
the cases with confirmed soft-tissue sarcoma the exposure was not well
documented, although an undocumented contact with 2,4,5-T,
2,4,5-trichlorophenol, or TCDD could not be excluded.
8.5 Human Experimental Studies
Poiger & Schlatter (1986) studied a human volunteer after
ingestion of a single dose of 1.14 µg 3H-TCDD/kg body weight. The
absorption from the intestine was > 87% and adipose tissue levels
were 3.09 (± 0.05) and 2.85 (± 0.28) ng/kg after 13 and 69 days,
respectively. The estimated half-life of TCDD was 2120 days.
Gorski et al. (1984) calculated the half-lives of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD and octaCDF to be about
3.5, 3.6 and 2 years, respectively. The estimation was based on the
analysis of fat tissue biopsies collected with an interval of 28
months from one 14-year-old girl who for a period of about 2-3 years
had been exposed to technical pentachlorophenol. Analysis was
performed by gas chromatography with electron capture detection, and
the isomers were confirmed by the use of several different packed and
capillary columns.
9. TOXICOKINETICS OF PCDFs
9.1 Uptake, Distribution, and Excretion
Toxicokinetic data for TCDF and other PCDFs arise from iv
injections or gastrointestinal exposure. There are no studies on
exposure via the respiratory tract or via dermal application.
9.1.1 Studies with 2,3,7,8-tetrachlorodibenzofuran (2,3,7,8-TCDF)
Table 65 summarizes the distribution of radioactivity to the
major tissue depots at various time-points after an iv injection of
14C-TCDF into rats and mice. Similar studies in guinea-pigs and
monkeys are summarized in Table 66. Tables 67 and 68 give the tissue
distribution of 14C-TCDF in more detail for rats and mice,
respectively.
TCDF has been used for kinetic studies in the rat (Birnbaum et
al., 1980), mouse (Decad et al., 1981a), guinea-pig (Decad et al.,
1981b), and monkey (Birnbaum et al., 1981). A single iv dose of 30.6
µg 14C-TCDF/kg body weight was given to rats, mice, and monkeys,
while the guinea-pigs received an iv dose of 6 µg/kg body weight. The
distribution of the radio-label was followed in tissues and excreta
for 3 weeks in rats and monkeys, for 10 days in mice, and for 9 days
in guinea-pigs. The distribution of radioactivity in the main tissues
and excreta of the different species at some of the intervals studied
is presented in Tables 65 and 66 along with the respective half-lives
and LD50 values for TCDF. Radioactivity recovered from the tissues
represented the parent compound, while radioactivity in faeces and
urine represented metabolites of TCDF. In the faeces of guinea-pigs
only the parent substance was present. Analysis by thin-layer
chromatography revealed Rf values of 0.5 and 0.1 for metabolites of
TCDF in faeces and urine as compared to an Rf of 0.8 for the parent
compound.
TCDF has a short half-life (2-4 days) and is quickly eliminated
from the liver, both in the rat and the mouse (Birnbaum et al., 1980;
Decad et al., 1981a). Elimination occurs rapidly also from the skin
and muscle, whereas retention is longer in adipose tissues. The
difference in the retention of TCDF in adipose tissues between C57Bl/6
and DBA/2 mice may be explained by the fact that DBA/2 mice have
substantially more adipose tissues than C57Bl/6 mice.
The distribution of TCDF in the guinea-pig was different from
that in the rat or mouse (Decad et al., 1981b). The maximum uptake in
the liver occurred within one hour after dosing; thereafter the
radioactivity was distributed in the fat and skin during the
succeeding hours. After one day, as a result of loss of body fat, the
radioactivity in adipose tissues was redistributed to the liver.
Within 3 days after dosing there was no elimination of radioactivity
from the liver and adipose tissues, whereas in the skin radioactivity
decreased only slightly. The estimated half-life for TCDF in the
guinea-pig was more than 20 days.
Table 65. LD50, whole-body half-life, and distribution of radioactivity (percentage of administered dose) at
various intervals after an iv dose of 30.6 µg 14C-TCDF/kg to ratsa and miceb
Fisher 344 Rats C57BL/6 Mice DBA/2 Mice
3 hr 3 days 10 days 3 h 3 days 10 days 3 h 3 days 10 days
Liver 41.4±3.6 5.9±0.3 1.3 51.0±13.4 22.7±1.8 1.1±0.3 39.4±0.6 16.8±1.4 5.6±2.0
Fat 10.0±1.0 11.1±2.3 1.8 6.0±1.6 2.9±2.1 ND 9.6±3.9 22.3±2.9 7.2±0.9
Skin 6.6±0.3 1.2±0.3 0.5 3.6±0.7 3.0±1.1 ND 5.5±4.3 3.3±0.9 ND
Muscle 5.9±0.4 0.3 < 0.3 7.5±2.8 1.5±0.9 ND 10.8±3.4 5.4±1.9 1.8±0.4
Faeces 63.1±0.6 > 85 43.1 81.9±13.0 27.7 55.8±4.8
Urine 2.0±0.4 < 6 7.7 12.6±0.1 9.2 19.9±4.6
Half-life < 2b 2b 4b
(days)
LD50 > 1000c > 6000c,d
(µg/kg)
a Birnbaum et al. (1980).
b Decad et al. (1981b).
c Moore et al. (1976).
d Moore et al. (1979).
ND = not detectable.
Table 66. LD50, whole-body half-life, and distribution of
radioactivity (percentage of administered dose) at various intervals
after an iv dose of 14C-TCDF in guinea-pigsa and monkeysb
Hartley guinea-pigs Rhesus monkeys
3 h 3 days 9 days 21 days
Liver 23.6±3.8 29.3±0.6 54.2±14.5 1.02±0.80
Fat 31.4±0.7 56.9±7.6 21.8±11.6 3.66±2.83
Skin 22.5±0.1 17.1+±0.6 15.2±3.1 2.44±1.60
Muscle 15.6±4.5 8.8±3.0 1.55±0.14
Faeces 4.7±1.3 6.6 42.9
Urine 2.3±0.4 6.6 7.9
Half-life > 20c 8a
(days)
LD50 > 5c < 10d 1000d
(µg/kg)
a 6 µg/kg (Decad et al., 1981a).
b 30.6 µg/kg (Birnbaum et al., 1981).
c Moore et al. (1976).
d Moore et al. (1979).
Table 67. Tissue distribution of TCDF-derived radioactivity in
Fisher 344 rats at 15 min, 3 h, and 24 h following a single iv dose
of 30.6 µg 14C-TCDF/kg b
Tissue Tissue content of 14C (% of dose/g tissue)a
15 min 3 h 24 h
Blood 0.12±0.04 0.04±0.01 0.03±0.01
Liver 4.4 ±0.2 5.1 ±0.4 2.2 ±0.4
Fat 0.20±0.03 0.44±0.07 0.64±0.11
Muscle 0.25±0.01 0.06±0.00 0.30±0.02
Skin 0.17±0.02 0.20±0.01 0.07±0.01
Kidneys 0.67±0.04 0.17±0.03 0.08±0.01
Adrenals 7.4 ±6.9 4.7 ±1.3 0.34±0.14
Thymus 0.52±0.12 0.54±0.13 0.07±0.03
Spleen 0.37±0.07 0.08±0.02 0.02±0.00
Testes 0.09±0.01 0.09±0.01 0.06±0.02
Brain 0.25±0.01 0.15±0.03 0.02±0.00
Lungs 1.08±0.08 0.24±0.02 0.07±0.02
Heart 0.66±0.03 0.11±0.00 0.02±0.02
a Mean ± SD for three animals.
b From: Birnbaum et al. (1980).
Based on data from three monkeys, the half-life for TCDF was
calculated to be 8 days (Birnbaum et al., 1981). At the end of the
study, more radioactivity remained in adipose tissues and skin than in
the liver. The retention of TCDF in the liver of monkeys 21 days after
dosing was comparable to that in the liver of the rat and C57Bl/6
mouse 10 days after injection. Urinary elimination of radioactivity
was a minor route when compared to faecal elimination both in the rat,
mouse, and monkey, whereas in the guinea-pig these routes were of
comparable importance (Birnbaum et al., 1980, 1981; Decad et al.,
1981a,b). The cumulated excretion of radioactivity 3 days
post-treatment amounted to approximately 64, 51, 11, and 7% in the
rat, C57Bl/6-mouse, monkey, and guinea-pig, respectively.
Against this background of data on tissue distributions,
half-lives, and LD50 values of TCDF in the rat, guinea-pig, and
monkey, Birnbaum et al. (1980, 1981) concluded that TCDF, measured as
excreted radioactivity, is metabolized to less toxic compounds and
that animal species with a high capacity to metabolize TCDF are more
resistant to its acute toxicity. This conclusion was considered
applicable also to the mouse (Decad et al., 1981a). Based on the same
data King et al. (1983) produced a pharmacokinetic model for TCDF in
rats, mice, and monkeys. However, there are objections to this
comprehensive conclusion. First, the kinetic studies on guinea-pigs
(Decad et al., 1981b) were (for analytical reasons) carried out with
such a high dose of TCDF that all of the animals showed marked signs
of toxicity, even within 3 days. After 9 days all the animals were
killed due to toxic symptoms. It is not advisable to draw any
conclusions on normal kinetic behaviour from data obtained on dying
animals with their abnormal metabolism and physiology. As far as the
kinetic data from the monkey are concerned, the conclusions were based
on a single time-point, and the number of animals in that study was
also very limited (Birnbaum et al., 1981).
Table 68. Tissue distributiona of TCDF-derived radioactivity in C57Bl/6 and DBA/2 mice at 15 min, 3 h, and 24 h after
a single iv dose of 30.6 µg 14C-TCDF/kg b
Tissue content of 14C (% of dose/g tissue)a
15 min 3 h 24 h
Tissue C57Bl/6 DBA/2 C57Bl/6 DBA/2 C57Bl/6 DBA/2
Blood 1.1 ND 0.6±0.3 0.22±0.04 ND 0.2±0.0
Liver 28.0±4.2 30.7±2.8 39.3±2.8 38.0 ±3.8 25.3±4.2 19.2±1.9
Adipose 2.6±0.4 2.7±1.0 3.7±1.4 4.9 ±0.1 6.1±1.2 6.1±0.4
Muscle 1.3±0.1 1.6±0.2 0.7±0.2 0.9 ±0.3 0.3±0.1 0.8±0.1
Skin 2.2±0.1 2.3±0.7 1.4±0.2 2.7 ±0.6 1.3±0.7 2.7±0.6
Kidneys 3.4±0.3 4.1±0.5 1.1±0.3 1.3 ±0.3 0.6±0.1 0.7±0.2
Adrenals 18.8±4.9 ND 6.9±5.4 9.2 6.5 ND
Thymus 3.9±2.4 3.0±1.9 2.0±0.7 4.7 ±3.2 0.3±0.2 2.5±0.6
Spleen 1.4±0.1 1.7±0.3 0.8±0.1 0.5 ±0.1 0.2±0.1 0.27
Testes 0.4±0.1 0.6±0.1 0.5±0.2 1.0 ±0.2 0.2±0.2 0.4±0.3
Brain 1.7±0.5 2.4±0.3 0.8±0.1 1.3 ±0.3 0.2±0.3 2.7±2.7
Lungs 6.7±0.5 8.2±0.7 2.4±0.9 3.1 ±1.5 0.4±0.2 0.8±0.4
Heart 2.1±1.3 3.4±2.2 0.6±0.3 0.8 ±0.4 0.2±0.1 0.3±0.1
a Mean ± SD for three animals.
b From: Decad et al. (1981b).
ND = below limit for accurate detection.
Ioannou et al. (1983) calculated a whole-body half-life of
approximately 40 days for a non-toxic dose of TCDF in young male
Hartley guinea-pigs. Their estimation was based on the distribution of
TCDF-derived radioactivity in liver, adipose tissue, skin, and muscle
in three animals 36 days after a single oral dose of 4 mg TCDF/kg body
weight and on certain approximations obtained from a previous study
(Decad et al., 1981b). Failure to demonstrate a correlation between
degree of bioaccumulation and lethality of TCDF in this study may be
due partly to the calculations of body burden based on the uncertain
estimate of the 40 days half-life for TCDF, which may not be valid for
both toxic and non-toxic doses of TCDF.
9.1.2 Studies with other PCDFs
Young male Wistar rats absorbed approximately 68% of a single
oral dose of 1.0 mg 2,3,4,7,8-pentaCDF/kg body weight given in salad
oil (Yoshimura et al., 1986). The daily faecal excretion was about
0.1% of the administered dose/day, whereas no 2,3,4,7,8-pentaCDF was
detected in urine. Four weeks after dosing the retention of
2,3,4,7,8-pentaCDF in the liver was 48.8% of the dose. The addition of
5% of activated charcoal beads to the diet, one week after dosing and
throughout the study, increased the faecal elimination of
2,3,4,7,8-penta CDF about 3-fold, but had no effect on urinary
elimination. Both the liver and extrahepatic tissues, except the
kidney, from rats on basal diet supplemented with activated charcoal
beads had lower levels of 2,3,4,7,8-pentaCDF than rats on basal diet
only.
Yoshihara et al. (1981) administered single ip injections of 13
individual PCDF congeners (at 1, 5, or 10 µg/kg) to young male Wistar
rats, and retention of the respective isomers in the liver was
determined 5 days later. The great variation observed in the hepatic
accumulation of the various isomers seemed to depend on the position
as well as the number of chlorine atoms substituted. Isomers having
vicinal hydrogens were accumulated to a lesser degree, although three
of the six isomers having no vicinal hydrogens (1,3,6,8-tetraCDF,
TCDF, and 1,2,4,6,8-pentaCDF) also showed low accumulation. The isomer
most highly accumulated was 2,3,4,7,8-pentaCDF, more than 65% of the
dose being retained, whereas only 3.8% of TCDF, which is equally
potent biologically, was retained. These results would imply that
there is no relationship between hepatic distribution of PCDFs and
their potential for acute toxicity. All animals in this study showed
toxic symptoms and liver microsomal AHH activity was strongly induced,
except in the cases of the following isomers: 2,8-diCDF,
1,2,7,8-tetraCDF, 1,3,6,7-tetraCDF, 1,3,6,8-tetraCDF,
1,2,4,6,8-pentaCDF. It is important to take this into consideration
when judging the kinetic data. A mixture of 14% 1,2,7,8-tetraCDF, 35%
TCDF, 1% 1,2,4,7,8-pentaCDF, 49% 1,2,3,7,8-pentaCDF, 1%
2,3,4,7,8-pentaCDF, and 1% hexaCDF was administered as a single ip
dose of 10 mg PCDF/kg body weight to young male Wistar rats (Kuroki et
al., 1980). The retention of the isomers in the liver, 5 days
post-treatment, showed good agreement with the results of Yoshihara et
al. (1981).
Based on the purification of three isoenzymes of cytochrome P-450
and the recovery of 14C-radioactivity from the hepatic microsomes of
Wistar rats treated with 14C-2,3,4,7,8-pentaCDF (single ip dose of
1 mg/kg body weight, 5 days previously), Kuroki et al. (1986)
suggested that one of these isoenzymes, P-448 H, functions as the
storage site of 2,3,4,7,8-pentaCDF in the rat liver.
Fly ash and crude or purified toluene extracts of PCDD- and
PCDF-containing fly ash from a municipal incinerator (Zaanstad, The
Netherlands) were mixed with ordinary laboratory diet for rats (van
den Berg et al., 1983). Small portions (2 g) of these diets were fed
to male Wistar rats (300 g) every 24 h for 19 days, at which time the
animals were sacrificed. The levels of tetra-. penta-, and
hexa-chlorinated PCDDs and PCDFs in samples of liver and adipose
tissue from these rats were determined. Rats fed the fly
ash-containing diet stored PCDDs and PCDFs in their livers at
concentrations which were at least 3 to 5 times lower than those of
rats fed with comparable amounts of fly ash extracts. For the
pentaCDD, hexaCDF, and hexaCDD isomers these concentrations were
approximately 10-20 times lower. Generally PCDFs had a higher
retention in the liver of rats than the corresponding PCDDs. In the
adipose tissue of rats fed with fly ash extracts, retention was higher
for penta- and hexaCDDs than for the corresponding PCDFs.
In a later study, male Wistar rats (275 g) were fed a diet
containing the same fly ash, pretreated with 2.5% HCl (van den Berg et
al., 1986a). A control group received standard diet. All congeners
retained in the livers of the rats had a 2,3,7,8-chlorine substitution
pattern. With the exception of 2,3,4,7,8-pentaCDF and
2,3,4,6,7,8-hexaCDF, the retention for each congener was below 10% of
the dose. The retention percentages of the various congeners in the
liver were almost equal at all time-points studied (34, 59, and 99
days), thus indicating a long half-life of these congeners in the
liver of the rat.
A mixture of two tetraCDFs, four pentaCDFs, and four hexaCDFs,
was given as a single ip injection of 500 µg to male ICR mice (Morita
& Oishi, 1977). The distribution patterns of the isomers in various
tissues were followed for up to 8 weeks. Analyses were performed with
GC (with electron capture detection) and isomers were identified by
peak number only. PCDFs were mainly located in the liver, spleen, and
fat tissues, but low to minimal amounts were found also in the kidney,
testes, lungs, heart, and brain. The GC patterns of liver samples
changed markedly with time, in contrast to those of the other tissues,
including fat, where the GC patterns remained similar throughout the
study. Most isomers with shorter retention times were readily absorbed
and then rapidly disappeared from the liver. Isomers with longer
retention times were slowly absorbed, and thus appeared later and
persisted longer in the liver. If the mixture had been administered
orally, those isomers with long retention times might have passed the
gastrointestinal tract with very low absorption.
The hepatic retention of PCDDs and PCDFs after dietary intake of
the above-mentioned HCl-pretreated fly ash was studied in male Golden
Syrian hamsters (van den Berg et al., 1986b). The livers were analyzed
for tetra-, penta-, and hexaCDDs and -CDFs after feeding the diet,
which contained 25% fly ash. No detectable hepatic retention was
observed after 34 days. The highest retention after 95 days was 8.4%
for 2,3,4,7,8-pentaCDF, but the retention was generally below 5% of
the total dose. With the exception of 2,3,4,6,7- pentaCDF, only
2,3,7,8-substituted PCDDs and PCDFs were retained. Constant relative
concentrations were found for the 2,3,7,8-substituted PCDDs and PCDFs
at the time-points studied.
In studies by Firestone et al. (1979), three lactating Holstein
cows received commercial grade pentachlorophenol orally by gelatine
capsule at a dose rate of 10 mg/kg body weight twice daily for 10 days
and once daily for the following 60 days. One cow served as a control
and received gelatine capsules containing only ground corn. The
pentachlorophenol composite used contained ten PCDD congeners (0.1-
690 mg/kg) and eight PCDF congeners (0.9-130 mg/kg). Faeces collected
on day 28 of the treatment period contained three hexaCDDs (0.05-0.63
µg/kg), two heptaCDDs (21.3-33.1 µg/kg), and octaCDD (290-429 µg/kg).
Faeces also contained hexa-, hepta-, and octaCDF. Milk, body fat, and
blood contained only three of the PCDD congeners present in the
pentachlorophenol composite, namely 1,2,3,6,7,8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, and octaCDD. Milk samples also contained
hexa-, hepta-, and octaCDF. The average concentrations of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, octaCDD, and octaCDF in
the composite milk fat at the end of the treatment period were 20, 40,
25, and 2 mg/kg, respectively. Similar concentrations were found in
body (shoulder) fat at the end of the treatment period (13, 24, and 32
mg/kg, respectively, for 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD,
and octaCDD). Levels of dioxins in the blood were about 1000 times
below the values in milk or body fat. The average daily excretion of
1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in the milk
during days 40 to 70 of treatment was about 20, 40, and 23 mg
(corresponding, respectively, to 33, 3, and 0.6% of the daily intake
of PCDDs). One hundred days after cessation of treatment the average
levels of 1,2,3,6,7,8-hexaCDD, 1,2,3,4,6,7,8-heptaCDD, and octaCDD in
shoulder fat and milk fat were 2.5, 6.6, 5.6 mg/kg and 4.3, 6.9, 3.0
mg/kg, respectively.
Table 69. Levels of PCDFs in the liver of dams and in fetuses and offspring after oral administration of PCDFs to
mice for 18 days during pregnancyf
PCDF Total intake % of PCDF intake in:a Total intake % of PCDF intake in:b
congener of PCDFs by of PCDFs by
dams killed dams killed
on day 18 of liver 2 weeks after liver offspring
pregnancy (µg)d of dam fetus delivery (µg) of dam (week)
1 2
tetraCDFc 1.4±0.06d 5.4 ND 1.6±0.10 ND ND ND
tetraCDFc 8.1±0.32 ND ND 9.0±0.57 ND ND ND
2,3,7,8-tetraCDF 11.4±9.46 5.5 0.007 12.5±0.79 0.03 0.05 Te
pentaCDFc 2.9±0.12 5.7 ND 3.1±0.20 4.0 0.10 0.27
pentaCDFc 13.2±0.53 3.9 0.004 14.5±0.91 0.1 0.03 Te
2,3,4,7,8-pentaCDF 4.9±0.20 14.5 ND 5.4±0.34 10.0 0.29 0.89
hexaCDFc 1.4±0.06 10.7 ND 1.6±0.10 6.4 0.28 0.76
Total 43.2±1.73 5.2 0.003 47.7±3.0 1.6 0.07 0.14
a Nine dams in group killed on day 18 of pregnancy.
b Ten dams in group killed on day 14 after delivery.
c Specific isomer not determined.
d Mean ± SEM.
e T = 0.01-0.1 µg/kg total congener.
f From: Nagayama et al. (1980).
ND = not detected.
9.2 Metabolic Transformation
Thirteen chlorinated compounds were detected in bile collected
for 48 h from female Sprague Dawley rats given a single oral dose of
678 µg TCDF (79.4% pure)/kg body weight (Poiger et al., 1984). The
four major metabolites considered to originate from TCDF were
trichloromethoxy-dibenzofuran, two trichlorodimethoxy-dibenzofurans,
and tetrachloromethoxy-dibenzofuran. The remaining nine metabolites,
detected in minute amounts, originated most likely from contaminating
PCDFs (1% triCDF, 8.4% tetraCDFs, 11.2% pentaCDFs).
Metabolites of 2-monoCDF, 2,8-diCDF, 2,3,8-triCDF, and octaCDF
were determined in the urine, faeces, fat, and liver of male Wistar
rats given single oral doses of 250 mg/kg body weight of the
respective isomers (Veerkamp et al., 1981). Analyses were performed
with GC-MS. No metabolites in any samples were found in rats given
octaCDF. Monohydroxy and dihydroxy derivatives were obtained with all
other isomers, whereas sulfur-containing metabolites were detected
only with the monoCDF and diCDF. Metabolites from 2-monoCDF and 2,3,8-
triCDF were found in urine and faeces only, but with 2,8-diCDF
metabolites appeared also in the tissues.
9.3 Transfer Via Placenta and/or Milk
Nagayama et al. (1980) studied the transport of a mixture of
PCDFs into the placenta and milk in the mouse (Table 69). A diet
containing 0.6 mg PCDFs/kg (48% tetraCDFs, 49% pentaCDFs, and 3%
hexaCDFs) was given for 18 days after mating. Nine dams were killed on
day 18 of pregnancy and 10 on day 14 after delivery. After giving
birth, the mothers were fed a diet free of PCDFs. The placental
transport, calculated from the amount of PCDFs in the neonates, was
about 0.003% of the administered dose. The isomers that remained in
the tissues were TCDF and 2,3,4,7,8-pentaCDF. While the levels of
PCDFs in the mothers dropped from 5.2% to 1.6% of the total intake,
the whole-body levels in the sucklings increased from 0.003% at the
time of birth to 0.07% after one week and to 0.14% of the total intake
after 2 weeks. TCDF and 2,3,4,7,8- pentaCDF were the dominant species
in both the mothers and the pups. To study the transport through milk
only, the same PCDF-containing diet was given to pregnant rats for 14
days from day 18 after mating, including the lactation period
(Nagayama et al., 1980) (Table 70). After 14 days 5.1% of the total
intake was found in the liver of the mother. The offspring contained
0.3% and 1.2% of the dam's intake after 1 and 2 weeks, respectively.
The dominant isomers recovered in the offspring were TCDF,
2,3,4,7,8-pentaCDF, and one unidentified pentaCDF, i.e., the same
isomers found in the largest amounts in the mother's liver. The data
demonstrated that the amounts of PCDFs transferred through milk were
much larger than the amounts transferred across the placenta.
Table 70. Levels of PCDFs in the liver of mouse dams and in offspring after oral
administration of PCDFs to dams for 14 days following deliverye
PCDF Total % of PCDF intake in:a
congener intake
of PCDF liver of dam offspring (week)
(µg)c 1 2
tetraCDFb 2.3±0.13 6.9 ND ND
tetraCDFb 12.9±0.71 ND ND ND
2,3,7,8-tetraCDF 17.9±3.10 5.6 0.5 1.4
pentaCDFb 4.4±0.24 6.7 Td 0.6
pentaCDFb 21.0±1.14 3.7 0.4 1.6
2,3,4,7,8-pentaCDF 7.7±0.42 13.8 0.4 2.2
hexaCDFb 2.3±0.13 7.9 Td 1.5
Total 68.3±3.75 5.1 0.3 1.2
a Ten dams in the group.
b Specific isomer not known.
c Mean ± SEM.
d T = 0.01-0.1 µg/kg total congener.
e From: Nagayama et al. (1980).
ND = not detected.
Weber & Birnbaum (1985) studied the distribution and placental
transfer of a single oral dose of 800 µg 14C- TCDF (0.0485
µCi/µg)/kg body weight to pregnant C57Bl/6 mice on gestation day 11.
Embryo mortality on gestation days 12 to 14 was in the range 7.9 to
17.8%. No detectable radioactivity was found in the embryos whereas
about 0.01% of the radioactive dose was contained in the placenta. The
hepatic radioactivity in the dams decreased rapidly from 30.0% of the
dose on gestation day 12 to 12.1% of the dose on gestation day 14. The
cumulative urinary and faecal excretion from gestation day 12 to 14
were 5.4 and 80.1% of the administered dose, respectively.
10. EFFECTS OF PCDFs ON ANIMALS
10.1 Acute Toxicity
Single oral LD50 values for TCDF in three species are listed in
Table 71.
10.1.1 Studies on rats
No histological changes associated with TCDF toxicity could be
observed in rats at oral doses up to 1000 µg TCDF/kg body weight
(Moore et al., 1976). These preliminary results, which also mentioned
that only mild toxicological changes occurred in rats at 6000 µg
TCDF/kg body weight, have not been presented in a final report.
Intravenous administration of 30.6 µg TCDF/kg body weight to male
Fisher rats has been shown to cause listlessness, excessive hair loss,
and decreased weight gain 2 days post-treatment. These adverse effects
were reversible and 3 weeks after dosing the animals appeared healthy
with normal body weight. There were no signs of thymic or splenic
atrophy or of liver hypertrophy (Birnbaum et al., 1980).
Single ip injection of 1 or 10 mg/kg body weight of nine
individual PCDF isomers, with at least three chlorines in the lateral
positions, to male Wistar rats produced thymus atrophy and liver
hypertrophy 5 days post-treatment. Five other congeners having no more
than two chlorine atoms in the lateral positions did not cause any
effects on the thymus or liver within the same dose range (Yoshihara
et al., 1981).
The ability of 15 individual PCDFs to affect body weight gain and
thymic atrophy in immature male Wistar rats was investigated 14 days
after a single ip injection (Ganon et al., 1985). The ED50 values
for both effects were estimated for each congener (Table 61).
10.1.2 Studies on mice
Moore et al. (1976) failed to establish a lethal dose for TCDF in
C57Bl/6 mice after giving single oral doses of up to 6000 µg/kg body
weight with an observation period of 30 days. However, there was a
transient depression in body weight gain, thymic involution, and mild
hepatotoxicity when 6000 µg TCDF/kg was given subcutaneously. Poland
& Glover (1980) found TCDF-induced thymus atrophy in C57Bl/6 mice 5
days after a single ip dose of 3 x 10-7 mol/kg body weight.
Single doses of 100 to 1000 µg TCDF/kg body weight to pregnant
C57Bl/6 mice on gestation days 10 to 13 produced no toxic effects on
the dams within the time studied (Hassoun et al., 1984a; Weber et al.,
1984).
A mixture of two tetraCDFs, four pentaCDFs and four hexaCDFs
given as a single ip dose of 500 mg to ICR mice produced no
deathswithin 8 weeks (Morita & Oishi, 1977). CF-1 mice given a PCDF
mixture containing 42% tetraCDFs, 54% pentaCDFs, and 4% hexaCDFs as a
single oral (10 to 1000 mg/kg body weight), sc (10 to 200 mg/kg), or
ip (10 to 100 mg/kg) dose developed no toxic signs during the first
week, although a modest weight loss was noted (Nishizumi, 1978). The
first deaths occurred 8 days after an oral dose of 1000 mg/kg, 5 weeks
after a sc dose of 200 mg/kg, and 11 days after an ip dose of 100
mg/kg. The oral LD50 (30 day) was 184 mg/kg for males and 414 mg/kg
for females. Hepatomegaly and thymus atrophy were consistent findings
in mice that died. In surviving mice on high dosages, the liver
exhibited small necrotic foci accompanied by cellular infiltrates. The
hepatic lesions occurred in the centrilobular area, and enlarged
hepatocytes containing foamy cytoplasm, increased numbers of lipid
droplets, and proliferation of smooth endoplasmatic reticulum were
also seen.
10.1.3 Studies on guinea-pigs
In studies by Moore et al. (1979), the patterns of toxicity were
similar for TCDF, 2,3,4,7,8-pentaCDF, and 2,3,7,8-
tetrabromodibenzofuran when given to young Hartley guinea-pigs. The
single oral LD50 was 5-10 µg/kg body weight for all three isomers,
and the time to death ranged from 8 to 26 days. Overt signs at lethal
doses were immediate and progressive weight loss, rough soiled hair
coat, listlessness, and dehydration. Similar symptoms appeared 3 days
after an iv injection of 6 µg TCDF/kg body weight (Decad et al.,
1981b). At necropsy lack of body fat and reduced body mass and thymus
weight were found. Histological findings were primarily associated
with the depletion of lymphoid cells in the thymic cortex, but
hypocellularity of bone marrow and hyperplasia in epithelial cells of
the renal pelvis, ureter, and urinary bladder were also observed.
Liver lesions were not observed. Surviving animals showed mild thymic
lymphoid hypoplasia only. Sublethal doses resulted in decreased body
weight gain.
In a study by Ioannou et al. (1983), all three adult male Hartley
guinea-pigs survived a single oral dose of 6 µg TCDF/kg body weight
for at least 17 days. At 10 and 15 µg TCDF/kg body weight, deaths
occurred on days 15-39 (two animals were sacrificed on day 17) and
13-20, respectively. The acute oral toxicity of soot (and of benzene
extracts of the soot) containing PCDDs and PCDFs from a PCB-containing
transformer fire (Binghamton, New York, USA) were studied in female
Hartley guinea-pigs (Silkworth et al., 1982). As discussed in section
8.1.1.1, toxicities were noted at 100 and 500 mg/kg body weight of
soot, but not when 1 and 10 mg/kg were administered.
Table 71. Single lethal dose values for TCDFa
Species/strain Sex/No Age or Dose Duration LD50 Time to
weight tested of study (µg/kg death
(µg/kgc (days) body weight) (days)
body weight)
Mice
(C57Bl/6) M/8 6 weeks 0 30 > 6000 not
400 reported
600
800
1200
1500
2500
4000
6000
Guinea-pigs
(Hartley) M/6 3-4 weeks 0 30 5-10 9-20
1
5
10
15
Monkeys
(Macaca F/2 2.0-3.7 kg 0 60 1000 14-31
mulatta) 300
1000
1500
a From: Moore et al. (1979).
b M = male; F = female.
c Doses were given orally in corn oil.
10.1.4 Studies on rabbits
When the above-mentioned soot (or benzene extracts thereof) was
applied dermally to New Zealand white rabbits (Silkworth et al., 1982)
(see section 7.4.4.1), it produced no overt toxicity, weight loss, or
histological changes in thymus, kidney, or skin, but centrilobular
hypertrophy was found in both sexes. The soot extract gave rise to a
reversible skin inflammation and hepatic centrilobular hypertrophy in
females only. Histological examination showed no changes in kidney,
thymus, and skin.
10.1.5 Studies on monkeys
The single oral LD50 value for TCDF in the young female rhesus
monkey (Macaca mulatta) was found to be 1000 µg/kg in a 60-days
study within the dose range 0, 500, 1000, and 1500 µg/kg and with two
or four animals at each dose level (Moore et al., 1979). The two
monkeys that received the sublethal dose developed skin lesions and
had decreased body weight gain. With lethal doses the following overt
signs occurred after 7 to 10 days: progressive weight loss, loss of
body fat, facial oedema, loss of facial hair, loss of finger and toe
nails, and thickening of skin. Death occurred within 2 to 4 weeks.
Major histological findings included hyperkeratosis of the skin,
thymic atrophy with lymphoid hypoplasia, and adverse effects on
epithelial linings. No structural liver lesions were observed though
the liver weight was increased. Increases in serum albumin and
cholesterol were also recorded.
Three male rhesus monkeys given a single dose of 30.6 µg TCDF/kg
body weight did not gain weight during the three following weeks, and
they developed facial skin lesions, mainly of sebaceous glands
(Birnbaum et al., 1981).
10.2 Short-Term Toxicity
10.2.1 Studies on rats
Male Sprague Dawley rats were fed 1 or 10 µg/kg of a PCDF
mixture, containing two tetraCDFs, four pentaCDFs, and four hexaCDFs
for 4 weeks (Oishi et al., 1978). Both diets gave rise to decreases in
growth rate, food consumption, haemoglobin and haematocrit values,
erythrocyte counts, serum levels of triglyceride, testosterone,
glutamic pyruvic transaminase, and leucine aminopeptidase activities,
as well as increases in serum cholesterol, cholinesterase, and
glutamic oxaloacetic transaminase activities. Rats fed the 10 µg/kg
diet developed chloracne-like lesions on the ears within 3 weeks.
Furthermore, this diet decreased the relative weights of thymus,
prostate, and seminal vesicles and increased the relative weights of
liver, testes, spleen, adrenals, lung, heart, and brain. In another
similar study, no effects were seen on total serum proteins or
leukocyte counts (Oishi, 1977).
In studies by Hori et al. (1986), male Sprague Dawley rats (aged
5 weeks) were for 21 days given daily oral doses of a mixture of PCBs,
PCQs, and PCDFs having a similar composition and isomeric ratio to
those found in the contaminated rice oil causing "Yusho" (section
11.1). The toxicities noted included thymic atrophy, suppression of
weight gain, hepatic enlargement, and an increased serum cholesterol
level and a decrease in serum glutamic pyruvic transaminase activity.
The mixture caused an induction of the AAH drug-metabolizing enzyme
similar to that caused by PCDFs alone. These results support the
hypothesis that the predominant etiology of "Yusho" involves PCDFs
contained in the PCB-contaminated toxic rice oil.
10.2.2 Studies on mice
Mice (C57Bl/6), given TCDF orally 5 times per week for 30 days
did not develop clinical signs of toxicity at doses of 30, 100, or 300
µg/kg body weight. However, thymus atrophy, liver hypertrophy,
decreased leukocyte count, and slightly elevated total serum protein
did occur in the high dose group at the end of the study (Moore et
al., 1979). Daily doses of 10, 30, or 50 µg TCDF/kg body weight on
gestation days 10-13 produced a dose-related increase in maternal
liver weight in C57Bl/6 mice (Weber et al., 1984). Decreased thymus
weight was recorded in ICR/JCL mice exposed to four weekly doses of a
mixture (at 100 µg/kg) of 12% tetraCDF and 88% pentaCDF (Oishi &
Hiraga, 1980). When PCDFs of unknown composition were given in the
diet at 0.6 mg/kg to mice for 10 weeks, severe dermal lesions,
hyperkeratosis, and dilated hair follicles filled with keratinous
material occurred in 7 of 12 mice. Furthermore, hepatocytes had
enlarged nuclei and vacuolations in the cytoplasm (Nagayama et al.,
1979). Feeding female ddN mice 0.6 mg PCDFs/kg diet (48% tetraCDFs,
49% pentaCDFs, and 3% hexaCDFs) for 18 days after mating, or for 14
days after delivery, produced no overt toxic effects in dams or in
offspring (Nagayama et al., 1980).
10.2.3 Studies on guinea-pigs
In studies by Luster et al. (1979b), oral administration of 0.05,
0.17, 0.5, or 1.0 µg 2,3,7,8-TCDF/kg body weight once weekly for 6
weeks to young female Hartley guinea-pigs produced 30% mortality in
the high-dose group. The thymus weight was decreased in the 0.5 and
1.0 µg/kg dose groups, though histologically only a slight decrease in
the density of thymic cortex was observable. Reduction in spleen
weight or alterations in splenic morphology did not occur, neither was
there a consistent decrease in body weight.
Four adult male Hartley guinea-pigs were given six or seven
weekly doses of 1 µg TCDF/kg body weight (Decad et al., 1981a). The
animals started to lose weight rapidly after the fifth or sixth dose,
the cumulative dose then being comparable to the oral LD50 value for
young guinea-pigs. At this time all animals were moribund and by day
44 the first animal died. Neither hepatomegaly nor thymic atrophy was
observed in this study. Thus multiple sublethal doses of TCDF appear
to have a cumulative effect, and may lead to a critical body burden
that will result in irreversible and progressive weight loss
eventually followed by death.
Weekly oral doses of 1 µg TCDF/kg body weight or biweekly doses
of 2 µg TCDF/kg body weight (interrupted by a 4-week period of no
dosing after the fourth dose) to young male Hartley guinea-pigs in
groups of four resulted in deaths on days 47, 51, 84; 31, 38, 88; and
32, 70, 85, respectively (Ioannou et al., 1983). At each dosing
schedule one animal was sacrificed at 101 days after exposure.
Repeated small doses, with various intervals in between, resulted in
a similar lethality but a less dramatic weight loss than with a high
acute dose.
10.2.4 Studies on rabbits
The 25% ether-hexane extracts from two commercial polychlorinated
biphenyl (PCB) preparations containing tetraCDFs and pentaCDFs
produced hyperplasia and hyperkeratosis of the follicular epithelium
of the rabbit ear skin when applied dermally weekly for 3 weeks in a
dose corresponding to 200 mg PCB. Liver lesions or decreased weight
gain were not observed. No dermal effects could be found when an
ether-hexane extract from a PCB preparation lacking PCDF impurities
was applied in the same manner (Vos & Beems, 1971).
A mixture of tetraCDFs and pentaCDFs was much less potent than
was TCDD in producing hyperkeratosis when applied to the inside of
depilated rabbit ears for 3 consecutive days (Nishizumi et al., 1975).
10.2.5 Studies on hamsters
No toxic effects were reported in male Golden Syrian hamsters
(50-70 g) given a diet containing 2.5% HCl-pretreated fly ash from a
municipal incinerator (Zaanstad, The Netherlands) for up to 95 days
(van den Berg et al., 1986b).
10.2.6 Studies on monkeys
A two-month study with three young male rhesus monkeys (Macaca
mulatta), serving as their own controls and fed a diet with 50 µg
TCDF/kg, resulted in one case of illness after 1 month and one death
after 2 months when the cumulative dose was calculated to be 300 µg/kg
(McNulty et al., 1981, 1982a). Toxic changes observed after 1 month
included periorbital oedema, reddening and thickening of the eyelids,
enlargement of facial hair follicles, and decreased number and size of
sebaceous glands in the skin. After 2 months these changes had become
more severe and were accompanied by decreased physical activity and
elevated (or eventual loss of) toe and finger nails. There were no
changes in haematology or serum chemical values. The diseased and the
surviving monkeys recovered rapidly when they were returned to
uncontaminated food. Within 3 months, behaviour, clinical appearance,
and histological structure of the skin were normal. The monkey that
died had lost 23% of its initial weight and most of its body hair.
Sebaceous glands were replaced by small squamous cysts. Severe lesions
were confined to the skin, thymus, and the stomach epithelium, whereas
liver lesions were modest. Decreased bone marrow cellularity was a
postmortem finding which was not reflected in the peripheral blood
count taken before death.
10.2.7 Studies on chickens
Mortality in one-day-old White Leghorn chickens given 1 or 5 µg
TCDF/kg body weight orally for 3 weeks was 16% and 100%, respectively,
with an average time to death of 19 and 11.5 days (McKinney et al.,
1976). Body weight gain and food consumption were decreased during the
third week post-treatment. Dose-related subcutaneous oedema, ascites,
and hydropericardium, as well as thymus atrophy, occurred. Depletion
of lymphatic cells was evident both in the spleen and thymus. Mild
liver lesions were found only in the high-dose group. Total serum
protein and serum albumin were reduced.
The significant difference in toxicity in chickens between three
commercial PCB preparations (Vos & Koeman, 1970) was later
demonstrated to be caused by the presence of tetra- and pentaCDFs in
two of the three preparations (Vos et al., 1970). The 25% ether-hexane
extracts from these two PCB preparations were highly toxic in the
chick embryo assay (Vos et al., 1970), whereas no effect could be
produced by the extract from the PCB preparation lacking PCDF
impurities.
10.3 Chronic Toxicity
10.3.1 Studies on monkeys
In studies by McNulty et al. (1981, 1982a), three young male
rhesus monkeys (Macaca mulatta) were exposed for 6 months to 5 µg
TCDF/kg diet, and one animal served as a control. One animal was
killed after 6 weeks when moribund. Overt toxic signs in the two
remaining animals started to appear after 3 months and the symptoms
remained for the following 3 months. One of these animals died
suddenly after 6 months. The remaining monkey was returned to normal
food and rapidly recovered. Clinically and pathologically, chronic
intake of small amounts of TCDF caused symptoms similar to those
following a single large dose of TCDF (section 9.1.2.4) or acute or
chronic ingestion of TCDD (see sections 7.1.1 and 7.3). The major
histopathological changes in all cases were seen in the thymus,
sebaceous glands, nail beds, bone marrow, and mucosa of the stomach
and bile ducts. The toxic potency of TCDF when ingested chronically
was approximately equal to that of TCDD. This contrasts with the acute
toxic effect of TCDF, which is approximately 20 times less than that
of its TCDD counterpart. The reason for death in the TCDF-poisoned
monkeys was obscure; it was preceded by weight loss, anorexia, and
depression. Only modest thymic and epithelial changes were present,
and there was no evidence for liver damage. The quick recovery of
animals returned to normal diet contrasted with the course of TCDD
poisoning in which illness progressed to death, or recovery was much
delayed, even after exposure had ended.
10.4 Effects Detected by Special Studies
10.4.1 Immunobiological effects
To date no studies have been performed on the effects of PCDFs on
the developing immune system.
Comparative studies on humoral immune responses in mice have
revealed that TCDF produces a pattern of responses similar to that
found for TCDD but only at 30-fold higher doses. Furthermore the
immunosuppressive effect of TCDD is much more persistent (Vecchi et
al., 1983b).
10.4.1.1 Histopathology
During toxicity studies with pure isomers of PCDFs or with
mixtures of PCDFs, thymus atrophy has been noted as a consistent
effect in the mouse (Nishizumi et al., 1978; Moore et al., 1979), rat
(Oishi, 1977; Oishi et al., 1978), guinea-pig (Moore et al., 1979),
and monkey (Moore et al., 1979; McNulty et al., 1981). Studies aimed
at investigating immunobiological effects revealed decreased thymic
weights (Luster et al., 1979b; Vecchi et al., 1983). The histological
findings are similar to those occurring after TCDD exposure, i.e.,
loss of lymphoid cells in the thymic cortex. A reduced number of
spleen cells was obtained from mice treated with a single ip dose of
180 µg TCDF/kg body weight (Vecchi et al., 1983), but no splenic
pathology was reported in mice given four weekly oral doses of a PCDF
mixture (at 100 µg/kg) containing 12% tetraCDFs and 88% pentaCDFs
(Oishi & Hiraga, 1980). Peritoneal cell and macrophage counts in mice
were not modified by an ip dose of TCDF (180 µg/kg body weight)
(Vecchi et al., 1983).
10.4.1.2 Humoral-mediated immunity
Adult female Hartley guinea-pigs exposed orally to 0.05, 0.17,
and 0.5 µg TCDF/kg body weight once weekly for 6 weeks showed somewhat
depressed serum IgG concentrations. A dose-related depression in
splenic lymphocyte proliferation was seen in TCDF-treated animals
after stimulation with the B-lymphocyte mitogen Escherichia coli
0127 lipopolysaccharide at 50 µg/ml medium. There were no effects on
any of the major serum proteins, neither was there an effect on the
antibody response towards bovine gamma globulin (BGG) (Luster et al.,
1979b).
The antibody response to sheep red blood cells given 7 days after
a single ip injection of 180 µg TCDF/kg body weight was inhibited by
85% and 35% in C57Bl/6 and DBA/2 mice, respectively (Vecchi et al.,
1983), whereas a single ip dose of 10 µg TCDF/kg body weight to
C57Bl/6 mice had no effect (Rizzardini et al., 1983). The suppression
noted by Vecchi et al. (1983) was dose dependent as well as time
dependent; by day 42 post-treatment a near-normal antibody response
was obtained.
10.4.1.3 Cell-mediated immunity
Oral intubation of 10 or 100 mg PCDF (12% tetraCDFs and 88%
pentaCDFs) per kg body weight once weekly for four weeks increased the
sensitivity to endotoxin of ICR/JCL mice. Following an ip injection of
50, 250, or 500 µg endotoxin per mouse a dose-dependent increased
mortality was noted two days after the final treatment with PCDF
(Oishi & Hiraga, 1980). Only at high dose levels were there any
effects on cell-mediated immunity functions in female Hartley
guinea-pigs given 0.05, 0.17, 0.5, or 1.0 mg TCDF/kg body weight
orally once weekly for six weeks (Luster et al., 1979b).
Both the depression in delayed hypersensitivity response to
purified protein derivative and the ability of BGG-sensitized
lymphocytes to release the macrophage inhibition factor were related
to the dose of TCDF. Splenic lymphocytes from TCDF-treated animals,
stimulated with the T-lymphocyte mitogen phyto-haemagglutinin (PHA),
showed a decreased proliferation. On the other hand proliferation of
splenic lymphocytes stimulated with concanavalin A (Con A), another
T-lymphocyte mitogen, showed no TCDF-related effect. The increased
proliferative response to Con A and PHA in thymocytes co-cultivated
with thymus epithelial (TE) cells or cultivated in TE-conditioned
medium was inhibited if the TE cells were pretreated with TCDF for 48
h, thus suggesting a direct effect on TE cells (Osborne et al., 1984).
10.4.2 Enzyme induction
Studies discussed below show that PCDFs are potent enzyme
inducers, the enzyme-inducing potencies varying greatly depending on
the position as well as on the number of chlorine atoms substituted.
The structure-activity relationships of the PCDFs with regard to
enzyme induction are similar to those for PCDDs, with TCDF and
2,3,4,7,8-pentaCDF being the most potent (Tables 56 and 61).
10.4.2.1 Studies on rats
Intraperitoneal doses of 2.5 mg TCDF/kg body weight given once
daily for three days to female CD rats induced 38- and 3-fold
increases, respectively, in AHH and UDPGT activities 24 h after the
final dose. The cytochrome P-450 content was doubled but no effect was
found on the aminopyrine N-demethylase activity (Goldstein et al.,
1978).
Increased AHH and EROD activities were found in the hepatic
microsomal fraction from immature male Wistar rats 5 days after ip
injection of single doses of TCDF (1.7 µmol/kg body weight) or
2,3,4,7,8-pentaCDF (0.3, 1.5, 3.0 µmol/kg body weight) (Keys et al.,
1985). This study also detected an alteration by TCDF in the hepatic
metabolism of testosterone in these rats. Yoshihara et al. (1981) gave
a single ip dose of 1, 5, or 10 mg PCDF/kg body weight of 13
individual PCDFs to young male Wistar rats five days prior to the
determination of hepatic enzyme activities. Congeners having at least
three chlorine atoms in the lateral positions typically showed
increased AHH and DT-diaphorase activities, while those congeners
having no more than two chlorine atoms in these positions were not
inductive. The cytochrome P-448 content was increased by 5 of the 13
congeners whereas the benz-phetamine-N-demethylase activity was
depressed by 7 of the 13. The most potent isomers, TCDF and
2,3,4,7,8-pentaCDF, were effective at a single dose of 1 µg/kg body
weight. The ranking of the potency for enzyme-inducing abilities did
not coincide with the hepatic distribution of the test substances.
Hepatic AHH activity in male Wistar rats was significantly enhanced
only by TCDF and 2,3,4,7,8-pentaCDF among the 15 individual PCDF
isomers tested, the dose administered intraperitoneally being 5 µg
PCDF/kg body weight (Nagayama et al., 1983).
Eight of the 15 PCDF isomers tested increased the pulmonary AHH
activity from 5-fold to 30-fold. In this study no PCDF-related AHH
induction was present in the kidney, prostate, thymus, or spleen.
Bandiera et al. (1984b) investigated the effect of three tetraCDFs and
three pentaCDFs at doses of 500 and 1000 µg/kg body weight,
respectively, on hepatic AHH, aminopyrine N-demethylase,
4-chlorobiphenyl hydroxylase, and EROD activities in male Wistar rats.
The most active compounds, TCDF and 2,3,4,7,8-pentaCDF, were potent
inducers of the cytochrome P-448-dependent monooxygenases. Some
induction of microsomal AHH, EROD, and 4-chlorobiphenyl hydroxylase
was observed also for the TCDF and 1,2,4,7,9-pentaCDF.
The ED50 values for hepatic AHH (Table 61) and 4-chlorobiphenyl
hydroxylase induction were established for 15 individual PCDFs in
immature male Wistar rats 14 days after a single ip injection (Mason
et al., 1985).
Significant induction of hepatic AHH activity in male Sprague
Dawley rats was given only by 3 out of 25 individual PCDFs given as
single oral doses of 40 µg/kg body weight (Doyle & Fries, 1986). The
active congeners were 2,7-diCDF, TCDF, and 2,3,4,7,8-pentaCDF.
A mixture of PCDFs, reconstituting the approximate composition
found in the liver of Yusho victims (7.4% tetraCDF, 6.1%
1,2,4,7,8-pentaCDF, 19.0% 1,2,3,7,8-pentaCDF, 29.4% 2,3,4,7,8-pentaCDF
and 39.1% 1,2,3,4,7,8-hexaCDF by weight) was given as a single ip
injection to male Wistar rats 14 days before measuring the induction
of cytochrome P-448-related enzyme activities (Bandiera et al.,
1984a). A dose-related enhancement of AHH and EROD activities was
found within the range 10 to 400 µg PCDF mixture/kg body weight.
10.4.2.2 Studies on mice
No induction of cytochrome P-448 content, or of ECOD activities,
was found 12 days after a single ip injection of 10 µg TCDF/kg body
weight to male C57Bl/6J mice (Rizzardini et al., 1983).
Nagayama et al. (1985) investigated the AHH-inducing potency of
TCDF, 2,3,6,7-tetraCDF, 1,2,3,6,7-pentaCDF, 1,2,3,7,8-pentaCDF,
2,3,4,6,7-pentaCDF, 2,3,4,7,8-pentaCDF, 1,2,3,4,6,7-hexaCDF, and
1,2,3,4,7,8-hexaCDF in two strains of responsive (C57Bl/6 and AKR/Qdj)
and two strains of non-responsive (DBA/2 and DDD) mice. All congeners
were given as single ip doses of 30 µg/kg body weight in olive oil. No
single congener induced the AHH activity above the control level in
the non-responsive mice. Significantly increased AHH activity was
found in both responsive strains exposed to TCDF, 1,2,3,7,8-pentaCDF,
and 2,3,4,7,8-pentaCDF. Mice (C57BL/6) treated with 2,3,4,6,7-pentaCDF
and 1,2,3,4,7,8-hexaCDF also responded with increased AHH activity.
10.4.2.3 Studies on chickens
Hepatic AHH activity in chick embryos was inducible by TCDF,
2,3,4,7,8-pentaCDF, and 1,2,3,7,8-pentaCDF, with ED50 values of
0.015, 0.014, and 0.071 nmol/egg, respectively (Poland et al., 1976).
No induction was produced by unchlorinated dibenzofuran, 2,8-diCDF,
2,4-diCDF, 2,4,8-triCDF or 1,3,6,7-tetraCDF at the doses tested. There
were no effects on ALA synthetase, p-nitrophenol-UDPGT and
testosterone-UDPGT activities. However, a modest increase in
cytochrome P-450 content was present in one-day-old White Leghorn
chickens 3 weeks after treatment with a single oral dose of 1 µg TCDF
(Goldstein et al., 1976).
10.4.2.4 Studies on cell cultures
Exposure of primary hepatocytes isolated from adult male Wistar
rats to TCDF for 72 h resulted in a 2-fold increase in AHH induction
at 10-9 mol/litre and half-maximal induction at 3 x 10-10
mol/litre. However, no AHH induction was observed with 2,7-diCDF in
the range 10-11 to 10-8 mol/litre in the same system (Jansing &
Shain, 1985). A 59-fold increase in AHH activity and a 40-fold
increase in EROD activity were obtained in rat hepatoma H-4-II E cells
when exposed to 5 x 10-10 mol TCDF/litre for 3 days (Keys et al.,
1986). In this same study it was demonstrated that TCDF had an
additive effect, whereas 1,3,6,8-tetraCDF and 2,4,6,8-tetraCDF had
counteracting effects on TCDD-induced enzyme induction.
The EC50 values for AHH and EROD induction (Table 61) have been
established for 35 individual PCDFs in the rat hepatoma H-4-II E cell
line (Bandiera et al., 1984b; Mason et al., 1985). AHH and EROD
activities were determined after exposing the cells to optimal doses
of PCDFs for 5 days. Unchlorinated dibenzofuran, or 2- and
3-chlorodibenzofuran did not induce these enzyme activities. EC50
values for all the remaining congeners varied between 10-4 and 1.3 x
10-10 mol/litre, the most active inducer being 2,3,4,7,8-PCDF.
Human lymphoblastoid cell lines, derived from the peripheral
blood of healthy male and female volunteers of various ages, were
exposed to eight individual PCDF isomers for 48 h (Nagayama et al.,
1985b). The AHH inducibility was highly variable between individuals
but less variable between isomers. In this system TCDF was about half
as potent as 2,3,4,7,8-pentaCDF, 1,2,3,4,6,7-hexaCDF, or
1,2,3,4,7,8-hexaCDF, which were equally as potent as TCDD in inducing
AHH.
A mixture of PCDFs, reconstituted on the basis of PCDF residues
in the liver samples from Yusho victims (see section 10.4.2.1), had
EC50 values for induction of AHH and EROD activities of 1.02 x
10-10 and 3.23 x 10-10 mol/litre, respectively, in the rat
hepatoma H-4-II E assay (Sawyer & Safe, 1985). The calculated EC50
values based on the relative isomer content of the mixture were 3.07
x 10-10 and 4.43 x 10-10 mol/litre, respectively.
10.4.3 Receptor binding
The competitive binding of PCDFs to the TCDD receptor protein has
been studied in vitro both in the hepatic cytosol (Poland et al.,
1976; Bandiera et al., 1984b) and in the nucleus (Poellinger et al.,
1982). Poland et al. (1974) investigated the ability of seven PCDF
congeners to compete with TCDD in binding to the hepatic cytosol
receptor from C57Bl/6J mice. They found the relative binding
affinities for TCDF, 2,3,4,7,8-pentaCDF, and 1,2,3,7,8-pentaCDF to be
37%, 34%, and 38%, respectively, of the binding affinity between TCDD
and the receptor. The EC50 values for the competitive binding of 33
individual PCDFs to the receptor from rat hepatoma H-4-II E cell
cultures varied from less than 10-3 mol/litre for
4-chlorodibenzofuran to 1.5 x 10-8 mol/litre for the most active
competitor, 2,3,4,7,8-pentaCDF, which had an EC50 value comparable
to that for TCDD, i.e., 1.0 x 10-8 mol/litre (Table 61) (Bandiera et
al., 1984b; Mason et al., 1985). Of the TCDD bound to the nuclear
receptor in vitro, 58% was displaced by a 100-fold molar excess of
TCDF. These nuclei were isolated from the liver of Sprague Dawley rats
pretreated intravenously with 1 µg TCDF 2 h prior to the incubation
(Poellinger et al., 1982).
10.5 Embryotoxicity and Reproductive Effects
TCDF has been found to be a potent teratogen in mice at doses
that produce no overt toxic effects in dams. Malformations observed
include cleft palate and kidney malformation similar to
hydronephrosis. Dose-related increases in fetal mortality occur with
single high doses. The teratogenic pattern of TCDF thus is strikingly
similar to that of TCDD (see section 7.5).
Single doses of 100 to 1000 µg TCDF/kg body weight to pregnant
C57Bl/6 mice on gestation days 10 to 13 produced dose-related
increases in the number of cleft palates and kidney malformations;
both the number of litters and the number of fetuses were affected
(Hassoun et al., 1984a; Weber et al., 1984). No other
treatment-related malformations were reported. A cleft palate
incidence of 40% was obtained in NMRI mice offspring after sc
treatment of the dams on gestation days 9 to 11 with 200 nmol TCDF/kg
body weight (Krowke, 1986).
Palatal closure in mice occurs late on day 14 of gestation, and
so it is somewhat peculiar that the peak sensitivity for cleft palate
occurs on day 12 (Hassoun et al., 1984a). The peak sensitivity for
kidney malformation in mice occurs on day 11 of gestation (Hassoun et
al., 1984a). The quantitative data on this malformation somewhat
conflict in the two studies. Weber et al. (1984) reported that 95.5%
of the fetuses had kidney malformations after a dose of 500 µg/kg body
weight on day 10 of gestation. However, only 17% of the fetuses per
dam had this malformation after a single dose of 400 µg/kg body weight
on the same day in the study of Hassoun et al. (1984a). The difference
might be due to unequal judging of the malformation. Preliminary
results (Weber et al., 1984), suggested that TCDF-induced kidney
malformations, up to a certain degree, represent a reversible defect
since no hydronephrotic kidneys were found in neonates, whereas in
identically treated dams examined on day 18 of gestation over 80% of
the fetuses/litter were affected. Fetal mortality increased in a
dose-related manner with high single doses administered on days 10 to
12. Peak sensitivity occurred on day 10 (Hassoun et al., 1984a).
Multiple low dosing on gestation days 10 to 13 was more effective in
producing fetal malformations, but less effective in producing fetal
deaths, than single high dosing on day 10 (Weber et al., 1984). No
effect on fetal mortality (days 12, 13, and 14) was observed in
C57BL/6N mice given a single oral dose of 800 mg TCDF/kg body weight
on day 11 of gestation (Weber & Birnbaum, 1985).
Recombinant inbred strains of C57Bl/6 and DBA/2 mice segregating
at the Ah locus respond differently to the teratogenic effect of TCDF
(Hassoun et al., 1984b). Fetuses of Ah-responsive strains responded
with a high frequency of cleft palates and kidney malformations after
a single ip dose of 600 µg TCDF/kg body weight on day 12 of gestation.
However, no cleft palates and only modestly increased numbers of
kidney malformations in a few strains were found with the same
treatment in Ah-nonresponsive strains.
A diet containing 0.6 mg PCDFs/kg (48% tetraCDFs, 49% pentaCDFs,
and 3% hexaCDFs), fed to mice for 18 days after mating, had no effect
on the number or body weight gain of the offspring, neither were there
any malformations related to the diet (Nagayama et al., 1980).
Three PCDFs, namely 1,2,3,7,8-pentaCDF, 2,3,4,7,8-pentaCDF, and
1,2,3,4,7,8-hexa CDF, are teratogenic to C57BL/6N mice when
administered orally by gavage on gestation days 10-13. A significant
increase in hydronephrosis and cleft palate was found, with
2,3,4,7,8-PCDF being the most potent PCDF studied, having an ED50 of
36 µg/kg body weight for cleft palate and 7 µg/kg for hydronephrosis.
For all three PCDFs, hydronephrosis occurred at a lower dose than did
cleft palate (Birnbaum et al., 1987).
It has been pointed out by McNulty (1985) that chlorinated
compounds such as 2,3,7,8-TCDD produce cystic periodontal lesions and
squamous metaplasia of the ameloblasts surrounding unerupted teeth in
rhesus monkeys. These findings are similar to those on the teeth
development seen in Yusho patients (section 11.1).
10.6 Mutagenicity
When tested in Salmonella typhimurium strains KTA98 and TA100
with and without metabolic activation, no mutagenic activity was found
for 2,9-diCDF, 3,6-diCDF, TCDF, or octaCDF (Schoeny, 1982). TCDF was
also studied in Saccharomyces cerevisiae strain MP-1 and was found to
be negative for forward mutation, mitotic crossing over, and mitotic
gene conversion at concentrations up to 1000 mg/litre. Stationary
phase cells were tested in the absence of exogenous activation (Fahrig
et al., 1978).
10.7 Carcinogenicity
The hepatic tumour-promoting activity of a commercial
polychlorinated biphenyl mixture, Aroclor 1254, with (Ar 1254) or
without (Ar 1254-PCDF) PCDF impurities, was studied in Sprague Dawley
rats pretreated with 66 µg diethylnitrosamine/ml drinking water for 5
weeks (Preston et al., 1981). Thereafter the rats were fed a diet
supplemented with 100 µg Ar 1254/kg (> 3 mg PCDF/kg) or Ar
1254-PCDF (<0.1 mg PCDF/kg) for 18 weeks. Examination of liver
lesions by light microscopy demonstrated that Ar 1254 promotes
formation of hepatocellular carcinomas in rats. The promoting
incidence of 64% remained essentially unchanged when PCDF was removed
from Ar 1254 by adsorption chromatography. Due to the high incidence
of hepatocellular carcinomas produced by Ar 1254-PCDF itself, an
additional effect of PCDF might have been difficult to measure in this
study.
11. EFFECTS OF PCDFs ON HUMAN BEINGS
Braun (1955) was the first to report chloracne due to chlorinated
dibenzofurans, subsequently experimentally proven by Bauer et al.
(1961). Vos et al. (1970) identified by mass spectrometry the presence
of chlorinated dibenzofurans in commercial PCB mixtures, accounting
for their acnegenic properties.
11.1 Yusho and Yu-cheng
A mass outbreak of food poisoning occurred in western Japan in
1968 following ingestion of a commercial brand of rice oil
contaminated with polychlorinated biphenyls (PCBs) and related
hydrocarbons. The poisoning was named "Yusho" (oil disease).
Epidemiological proof of the cause of the epidemic depended on the
demonstration of a dose-response relationship between the consumption
of the toxic rice oil and the incidence of the poisoning or between
the oil consumption and the clinical severity of the reaction.
Approximately 2000 cases were recognized. In 1969, Japanese scientists
first reported that the toxic rice oil which caused Yusho was
contaminated with polychlorinated biphenyls (Tsukamoto et al., 1969).
A few years later, the oil was found also to be contaminated with a
smaller quantity of PCDFs (Nagayama et al., 1976) and a relatively
large amount of polychlorinated quaterphenyls (PCQs) (Masuda &
Yoshimura, 1982).
In March 1979, an epidemic of a peculiar skin disease broke out
in Taichung and Changhwa in Central Taiwan. The cause of the disease
was later identified to be the ingestion of rice-bran oil contaminated
with polychlorinated biphenyls (Chen et al., 1980, 1981; Hsu et al.,
1984; Masuda et al., 1986). By the end of 1980, the total number of
reported cases was about 2000. The local name for the disease was
Yu-cheng. The mean consumption of total PCDFs of the Yusho and
Yu-cheng patients has been estimated to be 3.3-3.8 mg/person or
400-500 mg of toxic 2,3,7,8-substituted PCDFs per person. (Hayabuchi
et al., 1979). Hayabuchi et al. (1979) estimated the daily intake of
total PCDFs in the Yusho intoxication to have been 0.9 µg/kg body
weight. Analyses of liver samples taken from the Yusho patients about
18 months after the exposure showed a dramatic decrease in the number
of PCDF isomers. Apparently most of the PCDF isomers were metabolized
or excreted during the period between exposure and sampling (Rappe et
al., 1979). A comparison between the PCDF isomers found in the Yusho
oil and the liver samples revealed an interesting relationship. Most
of the isomers retained had all lateral positions (2-, 3-, 7-, and 8-)
substituted with chlorine (Rappe et al., 1979).
Table 72. Clinical symptomatology of Yusho 1969-1972a
1. Skin (82-87%).
Acneiform eruptions, districtive hair follicles, red plaques on limbs,
dark brown pigmentation of nail, skin, and mucous membranes, itching,
sweating of palms.
2. Ocular manifestations (83-88%).
Increased eye discharge, swelling of the upper eyelids, hyperaemia of
conjunctiva, transient visual disturbance.
3. Jaundice (10%).
No abnormalities of liver function in the majority of cases.
4. Numbness of the limbs, feeling of weakness, muscular spasms (32-39%).
Reduced sensory and motor nerve conduction velocity in a few cases
(9%).
5. Hearing difficulties (18%).
6. Headaches, vomiting, diarrhoea (17-39%).
7. Chronic bronchitis (40%).
Low serum IgA and IgM, PCB in the sputum.
8. Irregular menstrual cycles (60%).
9. Dark brown skin pigmentation (which gradually fades) of newborn,
retarded growth, abnormal teeth number and shape.
a Numbers in brackets refer to per cent of patients exhibiting the
symptoms.
Table 73. Changes in the clinical symptomatology of Yusho
in the years 1968-1978
1. Skin lesions.
All skin symptoms diminished gradually, subcutaneous cyst formation
still present in some of the most severe cases.
2. Ocular manifestations.
Eye discharge, oedema of the eyelids, pigmentation of eyelids and
conjunctiva, and cyst formation of tarsal gland still present in some
of the cases.
Table 73.(cont'd) Changes in the clinical symptomatology of Yusho
in the years 1968-1978
3. Stomatological alterations.
Pigmentation of oral mucosa decreased gradually; anomalies in number
of teeth and shape of the root still present.
4. Chronic bronchitis correlated in severity with concentration of PCBs
in sputum and blood.
5. Serum triglycerides.
The hyperglycidaemia observed in 1968-1970 returned to a normal level
by 1973 in females and by 1975 in males.
6. Mortality.
Of 737 cases in the Fukuoka region, 51 (6.92%) died between 1968 and
1978; there were 11 cancer deaths (3 stomach cancer, 2 lung cancer,
1 breast cancer, 1 liver cancer, 2 malignant lymphoma).
The rate of excretion of these toxic PCDF isomers is very slow.
Rappe et al., (1983c) could detect 2,3,4,7,8-pentaCDF in blood plasma
from Yusho patients when the samples were collected 11 years after
exposure. Higher levels were found in blood from Yu-cheng patients one
year after exposure and these analyses also showed a 15-20% reduction
in one year (Rappe, 1984). PCDFs are selectively retained in the
liver, with levels corresponding to the fatty level of the tissue.
They are not found in unexposed controls or in PCB-exposed workers.
PCB levels in Yusho patients were only about two times higher than
those of normal people several years after the outbreak. PCB-exposed
workers had more than 10 times greater PCB blood levels than Yusho
patients, whereas the PCQ levels of these two groups were similar.
Generally a correlation between degree or severity of clinical
signs and the amount of PCDFs retained in the blood exists, whereas
there is no correspondence between the severity of disease and PCB
concentrations in blood.
Mild dermal lesions seen in workers exposed to PCB disappeared
quickly after discontinuation of PCB handling, in contrast to the
persistence of Yusho and Yu-cheng symptoms. Everything thus suggests
that the PCDF contaminant is the causative agent.
Immunological evaluation of patients exposed in 1979 in Taiwan
(Yu-cheng) has been reported by Chang et al. (1981, 1982a, 1982b) and
Wu et al. (1984). Serum immunoglobulin concentrations and lymphocyte
subpopulations were determined in the peripheral blood of 30 patients
exposed to PCBs and 23 healthy individuals. The groups were age and
sex matched. In the patients, serum concentrations of IgA and IgM, but
not of IgG, were significantly decreased. Also the percentages of
total T lymphocytes and T-helper lymphocytes were significantly
reduced, whereas the percentages of T-suppressor cells and B
lymphocytes were not affected (Chang et al., 1981).
In a later report (Chang et al., 1982a), monocyte and
polymorphonuclear lymphocyte (PMN) complement and Fc receptors were
evaluated in peripheral blood from 30 Yu-cheng patients and 23 normal
human subjects. Monocytes and PMNs from patients had significantly
lower percentages of cells bearing immunoglobulin Fc and complement
receptors. The immune system was further investigated by determining
the delayed-type hypersensitivity skin response to streptokinase and
streptodornase, as parameters of cellular immune function. The
response was studied in 30 PCB-poisoned patients and 50 healthy
volunteers. Results of the study showed that 80% of the controls had
positive hypersensitivity skin tests, compared to only 43% of the
patients. The significant suppression of cellular immunity correlated
with the severity of the dermal lesions; the size of the
hypersensitivity skin reaction was negatively correlated with the
dermal lesions and also with PCB concentrations in whole blood (Chang
et al., 1982b). More recently, the delayed-type hypersensitivity
response to tuberculin was reported in 83 PCB-poisoned Yu-cheng
patients and in 30 age-and sex-matched healthy controls. Compared to
a positive response rate of 74% in the control group, the patients had
a significantly lower skin response of 48% (40 out of 83 patients). In
contrast to the delayed-type hypersensitivity response, the in
vitro proliferation responses of peripheral blood lymphocytes
treated with phytohaemagglutinin and pokeweed mitogens, as well as
tuberculin, were significantly enhanced (Wu et al., 1984).
There are also indications of immunosuppression in the Yusho
poisoning. Serum IgA and IgM levels decreased considerably within 2
years after the onset of the disease. Respiratory involvement included
bronchiolitis, and respiratory distress was often exacerbated by viral
or bacterial infection (Shigematsu et al., 1978).
The symptomatology of Yusho has been summarized by Reggiani
(1983b) and is to be found in Tables 72 and 73. It is similar to that
of Yu-cheng, but there are differences such as the frequency of
transient visual disturbances, hearing difficulties, and a persistent
bronchitis.
12. EVALUATION OF HEALTH RISKS FROM THE EXPOSURE TO CHLORINATED
DIBENZO-P-DIOXINS (PCDDs) AND DIBENZOFURANS (PCDFs)
12.1 Introduction
In order to evaluate the human health risk of PCDDs and PCDFs, it
is necessary to know both the levels of human exposure and the
corresponding human health effects.
Human exposure assessment is complex. Several approaches may be
taken to estimate it, such as the following.
(a) Use of standard physiological models of inhalation,
ingestion, and dermal absorption. Data requirements
include detailed information on ambient levels in
environmental media and food, and on bioavailability.
(b) Intake estimates based on simple pharmacokinetic models and
known levels in human tissue for accurate assessment of
exposure. Detailed knowledge is also required of the uptake,
distribution, metabolism, and elimination of PCDDs and PCDFs
in humans.
12.2 Exposure Assessment
12.2.1 Sources of contamination
The main sources of PCDDs and PCDFs that have so far been
identified are contaminated commercial chemicals (see section 3.3),
emissions from combustion sources (see section 3.5), and disposal of
industrial wastes containing PCDDs and PCDFs (sections 3.4, 3.5.9, and
3.5.10).
In some cases estimates of the relative contribution of these
sources can be generated on a local basis. However, the data and
methods available today do not allow firm conclusions with regard to
the relative quantitative importance of these sources on a nation-wide
or world-wide basis.
12.2.2 Ambient levels
The limited data available indicate very low (fg/m3) background
levels in ambient air of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs. (If hepta- and octachlorinated
congeners are included, pg/m3 levels are noted).
The few data available indicate that the 2,3,7,8-tetra-, penta-,
and hexachlorinated PCDDs and PCDFs are unlikely to occur in finished
drinking-water, even at a level of 1 pg/litre (see section 5.2). In
all soil and sediment samples analyzed (both from industrialized and
non-industrialized areas), PCDDs and PCDFs were identified at levels
ranging from a few ng/kg to several hundred ng/kg (the latter in
sediments and in urban soil).
There are no available data on background levels of PCDDs and
PCDFs in vegetation in the general environment.
Levels of up to 50 ng/kg of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs (principally the tetra- and
pentachlorinated congeners) have been found in fish from the general
environment. For the most part these have been detected in fatty or
bottom-feeding fish (see section 4.4.2).
Data from terrestrial organisms are inadequate for estimation of
background levels (see section 4.4.3). Three samples of pooled cow's
milk showed a maximum of 100 pg/kg of the 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs in whole milk (see section 5.4). Data
on contamination of other commercial foods are also very limited.
Analyses of several samples of chicken and pork have shown
contamination with highly chlorinated congeners at about 5-30 ng/kg.
The congener profile differs from that noted in aquatic organisms (see
section 5.4).
The data available are not sufficient for assessing the total
exposure of general populations. They are sufficient to perform a
limited evaluation of exposure for local populations. Based on the
environmental levels discussed above and the usual assumptions
regarding intakes of foodstuffs, air, and water, food is more likely
to be a significant source of PCDD/PCDF exposure than air, while
drinking-water is likely to be of much less concern.
12.2.3 Routes of exposure
Human adipose tissue contains 2,3,7,8-tetra-, penta-, and
hexachlorinated PCDDs and PCDFs. This contamination is presumably due
to exposure at the ambient concentrations noted in the preceding
sections.
In addition, infants may be exposed through breast milk, and
small children may also be exposed through ingestion of contaminated
soil. However, this latter route of exposure, in most instances, is
likely to be of concern only in heavily contaminated areas.
Some populations have been at special risk through exposure in
industrial accidents (and their clean-up) that have occurred during
the normal production and use of chlorphenols and phenoxy herbicides
and PCBs. In these situations inhalation and dermal contact are the
exposure routes of greatest concern. However, quantitative information
on the nature and concentration of contaminants is not available.
Based on the environmental levels discussed above and the usual
assumptions regarding physiological and intake parameters, ingestion
is likely to be the exposure route of greatest concern. Inhalation of
ambient air is not likely to be a problem, although inhalation of
heavily contaminated air may make a significant contribution to
exposure. In general, it is not possible, at present, to estimate the
relative contribution of dermal exposure.
12.2.4 Bioavailability
No bioavailability data from studies in humans are available.
From animal studies, it is clear that bioavailability of PCDDs and
PCDFs following ingestion depends on the matrix ingested. Table 46
summarizes the data available on oral intake.
Studies on hairless rats indicate that dermal exposure through
intact skin from contact with contaminated soil is about 1 to 2%. No
data are available for inhalation exposure.
12.3 Animal Data
12.3.1 Toxicokinetics of 2,3,7,8-TCDD
Studies on rodents given single or repeated oral doses of
2,3,7,8-TCDD have shown that 50% or more of the administered amount is
absorbed from the gastrointestinal tract in rats, guinea-pigs, and
hamsters, but less than 30% in mice (Table 40). The reported
half-lives for elimination were in the ranges 12-31 days for rats,
mice, and hamsters and 22-94 days for guinea-pigs (Table 41). However,
most of these studies have been performed at toxic doses. The
half-life of 2,3,7,8-TCDD in primates has not been well established,
but available data for the rhesus monkey suggest an apparent half-life
in the adipose tissue of about 1 year.
2,3,7,8-TCDD does accumulate in animal tissues. In rodents
accumulation occurs predominantly in the liver and adipose tissue
(Tables 42 to 44). In rhesus monkeys (Table 45), high levels of
2,3,7,8-TCDD are recovered from the adipose tissues, liver, skin, and
muscles.
At a daily dose of 1 ng 2,3,7,8-TCDD/kg body weight for 2 years,
rats accumulated 540 ng 2,3,7,8-TCDD/kg body weight in the liver where
some morphological changes were also observed. Similar levels were
found in beach mice that had been exposed to 2,3,7,8-TCDD soil levels
ranging from 10-710 ng/kg. Total body exposure of animals to
2,3,7,8-TCDD in soil at such concentrations may thus result in tissue
levels that have been demonstrated to cause effects in experimental
animals.
TCDD is largely eliminated in the faeces, although some urinary
excretion occurs. The hamster has a higher urinary elimination than
other species studied.
Transformation of 2,3,7,8-TCDD to more polar metabolites occurs
in all animal species investigated (see section 6.2 and Table 62).
Elimination of metabolites from tissues into faeces and urine occurs
rapidly in all of these species except in the case of the guinea-pig.
Known metabolites are much less toxic than the parent compound.
12.3.2 Toxicokinetics of PCDDs and PCDFs, other than TCDD
Animal data on the toxicokinetics of pure PCDDs other than
2,3,7,8-TCDD are limited. PCDFs have been more extensively studied in
this respect. The half-life for 2,3,7,8-TCDF has been reported to be
in the range of 2-8 days for rats, mice, and rhesus monkeys and more
than 20 days for guinea-pigs (Table 65). Studies on rats have shown
that 2,3,4,7,8-pentaCDF is more highly retained than is 2,3,7,8-TCDF
(65% and 3.8%, respectively, after 5 days).
Tissue retention data of PCDDs and PCDFs in various species
exposed to synthetic mixtures or to environmental samples containing
PCDDs and PCDFs show a high variability in retention time between
congeners with or without chlorine substitution in all the positions
2,3,7, and 8.
12.3.3 Toxic effects of 2,3,7,8-TCDD
The toxic and biological effects resulting from exposure to
2,3,7,8-TCDD are dependent on a number of factors, including the
species, strain, age, and sex of the animals used. The toxic responses
observed in several animal species include body weight loss,
hepatotoxicity, porphyria, dermal toxicity, gastric lesions, thymus
atrophy and immunotoxicity, teratogenicity, reproductive effects, and
carcinogenicity. TCDD induces a wide spectrum of biological effects
including enzyme induction and vitamin A depletion. The complete
spectrum of toxic and biological effects is not usually observed in
any single animal species. The two most characteristic toxic effects
observed in all laboratory animals are body weight loss and thymus
atrophy and immunotoxicity. Chloracne and related dermal lesions are
the most frequently noted signs of 2,3,7,8-TCDD toxicosis in humans;
dermal lesions are also observed in rhesus monkeys, hairless mice, and
rabbits. In contrast, rats, most strains of mice, guinea-pigs, and
hamsters do not develop chloracne and related dermal toxic lesions
after exposure to 2,3,7,8-TCDD. Many of the observed toxic lesions are
either hyperplastic/metaplastic or hypoplastic, and primarily affect
epithelial tissues.
Reproductive toxicity has been reported in rhesus monkeys: the
lowest-observed-effect level (LOEL) was calculated to be 1 to 2 ng/kg
body weight per day. A no-observed-effect level (NOEL), or possibly a
LOEL, of 1 ng/kg body weight per day for reproductive effects in rats
has been discussed (Murray et al., 1979; Nisbet & Paxton, 1982).
If the cancer studies in rats conducted by Kociba et al. (1978)
and by the NIH (1982a,b) are compared, it is evident that the liver
tumours, including hepatocellular carcinomas, are produced at similar
dose levels. Although an increased incidence of tumours in other
organs was observed by the NTP, and by Kociba et al. (1978), the other
target organs varied in the two studies. This may be caused, in part,
by differences in dosing (gavage versus exposure in ground feed) and
by differences in strains. In the Kociba study, doses of 10 ng/kg body
weight caused an increased incidence of neoplastic (hyperplastic)
nodules in females, and doses of 1 ng/kg body weight resulted in foci
or areas of hepatocellular alteration (swollen hepatocytes). At these
dose rates in experimental groups, the incidence of certain
hormone-dependent tumours was lower than in the control animals,
suggesting endocrine changes induced by 2,3,7,8-TCDD. Based on these
animal studies and on available human data IARC (1982 suppl. 4)
concluded that TCDD showed sufficient evidence for carcinogenicity in
animals, but inadequate evidence for carcinogenicity in humans.
TCDD does not appear to have mutagenic properties, and is,
therefore, not likely to be genotoxic. Thus, it is assumed to be
carcinogenic through an indirect (epigenetic) mechanism.
12.3.4 Toxic effects of PCDDs and PCDFs, other than TCDD
Several other PCDDs and PCDFs cause signs and symptoms similar to
those of 2,3,7,8-TCDD, but there is a wide variation with regard to
potency (Tables 56, 62). In summary, there are 12 isomers that display
high toxicity, i.e., the tetra-, penta-, hexa-, and heptaCDDs and CDFs
with four chlorine atoms in the symmetrical lateral positions 2,3,7,
and 8. A mixture of two hexaCDDs (1,2,3,7,8,9- and
1,2,3,6,7,8-hexaCDD) has been demonstrated to possess carcinogenic
properties in long-term animal studies, but at higher doses than those
used in the study of TCDD. Unsubstituted dioxin and 2,7-diCDD failed
to demonstrate carcinogenic properties.
The relative toxic and biological potencies of PCDDs and PCDFs
have been estimated using short-term studies in rats and mammalian
cell cultures. Endpoints used include inhibition of body weight gain,
thymic atrophy, enzyme induction, teratogenicity, acnegenic response,
and keratinization. In the absence of long-term toxicity data, results
obtained from such short-term tests are at present the only source for
ranking the toxicity for human risk assessment.
When investigated, mixtures of these compounds have shown
additive or less than additive responses.
12.3.5 Review of species differences
There are marked species differences in the susceptibility to the
biological and toxic effects elicited by 2,3,7,8-TCDD. For example,
the oral LD50 values range from 0.6 µg/kg body weight in
guinea-pigs, to 5051 µg/kg body weight in Golden Syrian hamsters
(Table 47); moreover, pronounced differences in LD50 values have
also been reported in different strains of the same species (e.g.,
rats and mice). The toxicity and toxicokinetics of TCDD in monkeys
most closely resemble the effects observed in human beings. However,
the tremendous variation in species and strain sensitivity to
2,3,7,8-TCDD and related compounds cannot be explained by the observed
toxicokinetic differences. There is evidence in inbred mice, that the
cellular levels of the Ah receptor correlate, in part, with
susceptibility to the biological and toxic effects of these compounds.
The receptor has also been identified in other species, including
human beings. However, interspecies comparison of cellular Ah receptor
levels do not explain their differences in sensitivity to
2,3,7,8-TCDD; this is consistent with complex as yet unknown
mechanisms of toxicity that involve multiple factors in addition to
the Ah receptor.
12.4 Human Health Effects
12.4.1 PCDDs
Exposure of the general population is to small amounts of PCDDs
and PCDFs in complex mixtures and these have not been associated with
disease. In a few incidents workers and others have been exposed to
larger amounts of a limited number of these compounds, e.g., Seveso
and in Yusho disease.
For occupational and accidental exposure the most prominent
clinical effect has been chloracne. Other effects (Table 64) have been
noted, but, apart from chloracne and perhaps minor functional
disorders, none has been persistent.
In some, but not all mortality studies, an increased incidence of
cancer at different sites has been claimed, but the small numbers of
cases limit confidence in the findings.
The overall impression from the follow-up studies is that even
severe acute systemic effects of TCDD are usually reversible, except
for chloracne, or markedly improved over time following cessation of
exposure. In Seveso, the only clear-cut adverse health effect recorded
has been chloracne. 193 cases of chloracne occurred in 1976 and 1977,
and 20 of those still presented active symptoms in 1984. Many studies
have been performed to find possible links between exposure and health
effects in civilians or military personnel exposed to Agent Orange in
Viet Nam. However, the information available to date does not allow
definite conclusions to be drawn with regard to effects on human
reproduction or any other significant health effects (see section
9.2).
In a number of studies, exposed populations and various control
groups have been compared by measuring serum lipids, liver function
tests, and other variables. Although certain statistically significant
differences have been reported there, and also in isolated case
reports, lack of uniformity, various technical shortcomings, and the
inability to exclude confounding factors means that the results have
been inconclusive.
In the Missouri (USA) incident, children who showed acute illness
when the contamination occurred in 1971 are now reportedly in good
health. Epidemiological studies in Missouri on populations exposed to
lower concentration over longer periods of time have so far not
revealed any significant health effects. Although no clinical illness
was observed, there were indications of an effect on the cell-mediated
immune system.
The ranges of health effects produced by TCDD in human beings
have yet to be defined. It can be concluded that the data from human
exposure and effects, when taken together, do not allow any
determinations of dose/effect or dose/response relationships in human
beings.
In spite of many clinical and follow-up studies, no clearcut
persistent systemic effects have been delineated, except for
chloracne. In the light of present information, it seems unlikely that
permanent, severe, and debilitating toxicological sequelae are
inevitable after exposure to TCDD.
12.4.2 PCDFs
The only well documented intoxications with PCDFs in human beings
are the two instances of contamination of rice oil with PCDFs, PCBs,
and PCQs, i.e., Yusho in Japan (1968) and Yu-cheng in Taiwan (1979)
(see section 11.1). In total, several thousand people were acutely
intoxicated. The summarized data makes it most likely that the
causative agent was PCDFs. The general symptomatology was similar to
that found in intoxications with TCDD. The differences may reflect
intensity in exposure and the ages and sex of the exposed human
beings. Attempts to estimate the average daily intake of PCDFs over
several months in Yusho patients indicated a figure of 0.9 µg/kg body
weight of total PCDFs, 0.1-0.2 µg/kg of 2,3,7,8-substituted tetra-,
penta-, and hexaPCDFs, together with 157 µg PCBs and 148 µg PCQs/kg
body weight (Hayabuchi et al., 1979). The lowest dose causing disease
was estimated to be 0.6 mg total PCDFs per person over 30 days,
corresponding to a daily dose of 0.05-0.1 µg/kg body weight of
2,3,7,8-substituted PCDFs. However, the data available are not
sufficient to permit any conclusions as to what dose might be safe for
human intake.
12.4.3 Human body burden and kinetics
In human fat, background levels of TCDD up to 20 ng/kg have been
found in the general population with no known specific exposure, but
higher levels have been reported in some cases without evidence of
disease. None of these populations have been randomly sampled. The
more highly chlorinated other PCDDs and PCDFs, especially octaDD, also
occur in these samples (see Tables 29, 30). Averages values seem to
increase with age.
In special situations, higher levels (in the low µg/kg range)
have been found that have not been associated with disease.
In the Yusho and Yu-cheng incidents, symptoms were noticed at
higher levels of PCDFs, e.g., 2,3,4,7,8-pentaCDF was found at 6.9
µg/kg fat tissue one year after the exposure to contaminated rice oil.
Based on the very limited data available, the levels of, for
instance, 2,3,4,7,8-pentaCDF in the general population, seem to be two
orders of magnitude lower than the levels associated with the Yusho
disease.
No such comparisons can be made for PCDDs.
Limited data indicate that those isomers chlorinated at the
2,3,7, and 8 positions are selectively retained, except for TCDF.
A half-life for TCDD in human beings of 5 years has been
indicated by one experimental study. In another study half-lives in
the range of 2-6 years were estimated for 1,2,3,6,7, 8-hexaCDD,
1,2,3,4,6,7,8-heptaCDD, octaCDD, 1,2,3,4,6,7,8-heptaCDF, and octaCDF.
These data need to be expanded since they are based on studies in only
two subjects and since the toxicokinetics of these types of compounds
may not simply be controlled by first-order kinetics. However, even if
there are limitations in the present data, it is apparent that the
half-lives of these compounds are in the range of one or more years.
These reported half-lives for human beings are very different
from those reported in rodents. However, animal experiments have
usually been performed with toxic doses. Furthermore, animals with a
short life span have a higher metabolic rate, thus shorter half-lives
could be expected.
The PCDDs and PCDFs are predominantly stored in fat, but they are
also excreted in milk (Table 39) and pass the placenta. They also
appear in the blood and vital organs at lower concentrations. The
distribution between different tissues in human beings is not at
present clear, although it has been suggested that the ratio between
fatty tissue and liver is higher in human beings than that in rodents.
However, this conclusion is based on very limited data from autopsy
specimens. Whether this is relevant for the general population remains
to be seen.
The intake route for human beings is at present not very well
delineated, but it has been assumed that intake from food is the main
route. However, the human infant represents a special case; because of
transplacental transfer of these compounds, the neonate might be
expected to be exposed in utero. Levels measured so far in human
milk suggest that this food might be an important source of these
compounds.
No data are available regarding what dose of PCDDs is toxic to
human beings. However, in the Yusho and Yu-cheng episodes, total
intakes of total PCDFs in the range 3.3-3.8 mg/person and total
intakes of 2,3,7,8-substituted PCDFs in the range 400-500 µg/person
were associated with the disease. No good data are available as to
what intakes occurred without causing disease.
12.5 General Conclusions
PCDDs and PCDFs occur throughout the environment and we all
probably carry a body burden of them. They have sometimes produced
complex toxic effects following occupational and accidental exposure.
Based on the Yusho disease and experiments in sensitive species
of monkeys, and making assumptions about the relative potencies of
PCDDs and PCDFs, human beings and certain monkey species may have
comparable sensitivity to these compounds. However, the uncertainties
related to the real dose received by human beings and the difficulties
of assessing toxic effects other than chloracne in our species prevent
a firm conclusion as to the relative resistance of human beings to the
toxic effects of these compounds. Exposure should be reduced to levels
as low as are reasonably practicable.
13. RECOMMENDATIONS
1. Analytical interlaboratory validation and round-robin studies
using standardized quality assurance and quality control
procedures are needed to improve analytical methodology.
Sampling strategy and analytical procedures and data
interpretation should be optimized and standardized before
undertaking surveys.
2. Further information is required about the origins and
environmental distribution and fate of PCDDs and PCDFs. Further
monitoring data, including time trends and determinations of
isomer patterns, are required for environmental levels of PCDDs
and PCDFs, especially for food, ambient air, and sediments.
3. Data should be obtained about the effects of PCDDs and PCDFs on
environmental biota.
4. More information is required about the bioavailability of PCDDs
and PCDFs from different matrices in the environment and from the
diet. Exposure from these sources should be correlated with
agricultural and industrial practices.
5. Simpler and less expensive methods suitable for screening should
be developed and validated.
6. Studies to determine the mechanisms of toxicity of PCDDs and
PCDFs are needed to support an evaluation of the differences in
effects between species and to allow extrapolation to human
beings.
7. Further investigation of immunotoxicity is important, including
cytotoxic T lymphocyte function. Studies of the effects of
perinatal exposure and of the duration of actions on the immune
system are important.
8. Long-term toxicity studies, including multigeneration
reproductive studies, in different species with three of the most
widespread PCDDs and PCDFs, namely 2,3,4,7,8-pentaCDF,
1,2,3,7,8-pentaCDD, and octaCDD, should be carried out.
9. Because humans are exposed to complex mixtures of PCDDs and
PCDFs, test systems, including techniques applicable to
evaluating human tissues, should be further developed and
validated for the toxic potency of these compounds and other
mixtures. These systems can be used to study mechanisms of
action, structure activity relationships, and interactive
effects.
10. Investigations to examine the body burden and to correlate it
with clinical effects and laboratory findings are indicated.
Follow-up studies of previously exposed groups are important.
14. EVALUATIONS BY INTERNATIONAL BODIES AND THE CONCEPT OF TCDD
EQUIVALENTS
14.1 International Evaluations
IARC evaluated the carcinogenic risk of TCDD to man (IARC, 1977,
1982) and concluded that there was sufficient evidence that it was
carcinogenic to animals, but that the data for carcinogenicity to
human beings was inadequate. None of the other PCDDs or PCDFs have
been evaluated by IARC.
Regulatory standards for TCDD and mixtures containing TCDD,
established by national bodies in different countries and the European
Economic Community, are summarized in the Legal File of the
International Register of Potentially Toxic Chemicals (IRPTC, 1987).
14.2 Methodologies Used in Assessment of Risk from PCDDs and PCDFs
14.2.1 Individual Congeners
As shown in this monograph, sufficient high quality data for
assessing human health risks exist only for TCDD. For the other
congeners and isomers where data do exist, they are generally derived
from studies using acute exposures in experimental animals and/or from
in vitro tests.
The several risk evaluations on TCDD from various countries have
utilized the long-term ocogenic rat studies (Kociba et al., 1978) or
the reproduction studies on rats (Murray et al., 1979) or monkeys
(Schantz et al., 1979). Mathematical models have been applied to the
cancer data and "virtually safe doses" between 0.006 and 0.028 pg/kg
body weight per day have been calculated (Kimbrough, 1984). The
biological relevance of such models has been questioned, since TCDD
has not been shown to be genotoxic, and has been found to be a strong
promoter of liver tumours in a two-stage precarcinogenesis study
(Pitot et al., 1980) To avoid the use of mathematical models, several
evaluations have used safety factors in the range of 100 to 1000
applied to the assumed no-effect, or lowest-observed-effect levels in
the cancer study of Kociba et al. (1978) or the reproduction studies
of Murray et al. (1979) or Schantz et al. (1978). Using this
methodology "tolerable daily intakes" have been calculated for human
beings in the range of 1-10 pg/kg body weight (Denmark, 1984; Ontario,
1985; US EPA, 1985; Ahlborg & Victorin, 1987).
14.2.2 Mixtures of PCDD and PCDF congeners and isomers -
concept of TCDD toxic equivalents.
The results of recent isomer-specific analyses of such diverse
environmental samples as emissions from the combustion of hazardous
industrial and municipal wastes, soil, industrial process wastes,
human adipose tissue, and milk indicate that the majority of the 75
CDD and 135 CDF isomers can be detected. Humans are therefore exposed
to complex mixtures of these environmental contaminants (sometimes
2,3,7,8-tetraCDD is only a minor component), and the level of risk
from such exposures must be assessed.
In the absence of long-term whole animal tests on complex
mixtures of PCDDs and PCDFs, as well as similar studies on individual
isomers and/or congeners, several models have been proposed to relate
the toxicity of environmental mixtures to the well studied isomer
2,3,7,8-TCDD. The results from these models are presented as "TCDD
Toxic Equivalents". A summary of some models which have been published
is given in Table 74. The toxic potencies used, relative to TCDD, are
shown. The scientific basis for deriving these relative toxicities is
somewhat different for each model. The Swiss (Switzerland, 1982) and
Danish (Denmark, 1984) models are based essentially on the relative
potency for AHH induction, whereas the German (Germany, 1985) and
Canadian models (Ontario, 1985) are based on a weighting of all
available quantitative data. The USA model (US EPA, 1987) utilizes
primarily the relative carcinogenic potencies of TCDD and hexaCDD,
with consideration given to other relevant quantitative data. A
discussion of the structure-activity relationships and relative
biological activities of many PCDD and PCDF isomers is given in
sections 7 and 10.
Data requirements, and assumptions made in the application of
these models to various environmental mixtures have been reviewed by
US EPA (1987) and Ontario (1985). Suter-Hofmann & Schlatter (1985) fed
toluene extracts of acid-washed particulates from a municipal waste
incinerator to Sprague Dawley rats at levels in the diet corresponding
to 4, 12, and 24% of particulates. The calculated daily TCDD intakes
at these doses were 4.8, 14.4, and 28.8 ng/kg body weight. Depending
upon which model was used from Table 74, the estimated intake of PCDDs
and PCDFs would be 48-300 (4% particulates), 144-900 (12%
particulates), and 288-1800 ng TCDD-equivalents/kg body weight per day
(24% particulates). No mortality was noted in the groups fed 24%
particulates, but at this dose body weight gain was depressed in
females and depressed thymus and increased liver weights were noted in
both sexes. No adverse affects were seen at the 4% and 12% dose
levels. From the known toxicity of TCDD, more severe effects would
have been expected from the calculated dose of between 288 and 1800 ng
TCDD-equivalents/kg body weight per day. These data support the
conclusion that the application of TCDD-equivalence models may
over-estimate the inherent risk from exposures to dioxin-containing
environmental mixtures, depending largely upon the assumptions used in
deriving the model. However, a definitive conclusion on this point
awaits further research (US EPA, 1987) (see section 13).
Table 74. Examples of TCDD-equivalence models
Relative PCDD and PCDF toxicities in model
Olie et al. Switzerland Germany Denmark Ontario United States
Compound (1983) (1982) (1985) (1984) (1985) (US EPA 1987)
MonoCDD 0 0.001 0
DiCDD 0 0.001 0
TriCDD 0 0.01 0
TetraCDD-2,3,7,8 1.0 1.0 1.0 1.0 1.0 1.0
-non 2,3,7,8 1.0 0.01 0.01 0.01 0.01
PentaCDD-1,2,3,7,8 0.1 0.01
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.5
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.005
HexaCDD-1,2,3,4,7,8 0.1
-1,2,3,6,7,8 0.01
-1,2,3,7,8,9 0.01
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.04
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.0004
HeptaCDD-1,2,3,4,6,7,8 0.01
-all 2,3,7,8 0.1 0.1 0.01 0.01 0.001
-non 2,3,7,8 0.1 0.1 0.001 0.01 0.00001
OctaCDD 0 0.001 0.0001 0
MonoCDF 0.0001
DiCDF 0.0001
TriCDF 0.01
Table 74 (contd).
Relative PCDD and PCDF toxicities in model
Olie et al. Switzerland Germany Denmark Ontario United States
Compound (1983) (1982) (1985) (1984) (1985) (US EPA 1987)
TetraCDF-2,3,7,8 0.1 0.1 0.1 0.1 0.5 0.1
-non 2,3,7,8 0.1 0.1 0.01 0.5 0.001
PentaCDF-1,2,3,7,8 0.2
-2,3,4,7,8 0.2
-all 2,3,7,8 0.1 0.1 0.1 0.5 0.1
-non 2,3,7,8 0.1 0.1 0.01 0.5 0.001
HexaCDF-1,2,3,4,7,8 0.2
-1,2,3,6,7,8 0.05
-1,2,3,7,8,9
-2,3,4,6,7,8 0.1
-all 2,3,7,8 0.1 0.1 0.1 0.1 0.01
-non 2,3,7,8 0.1 0.1 0.01 0.1 0.0001
HeptaCDF-1,2,3,4,6,7,8 0.01
-1,2,3,4,7,8,9
-all 2,3,7,8 0.1 0.1 0.01 0.01 0.00001
-non 2,3,7,8 0.1 0.001 0.01 0.00001
OctaCDF 0 0.001 0.0001 0
It must be recognized that an approach such as the use of
"TCDD-equivalents" must be regarded as an interim procedure for the
measurement of the toxicity of environmental samples in the absence of
long-term toxicity data on specific PCDD and PCDF isomers and mixtures
of these compounds. At present it is an imprecise evaluation
methodology with many data gaps in the supporting data base.
The TCDD-equivalent models have two major sources of uncertainty,
i.e., firstly, the unanswered scientific questions related to the
toxicity of TCDD itself, and secondly, the lack of data on the other
PCDD and PCDF congeners and isomers that would permit a more accurate
determination of the potency of these chemicals relative to TCDD. The
research recommendations in this document address some of these
concerns. As such data become available, TCDD-equivalent models must
be continuously updated and risk assessments based on the present
models (Table 74) considered only as interim evaluations.
REFERENCES
ABATE, L., BASSO, P., BELLONI, A., BISANTI, L., BORGNA, C., BRUZZI,
P., DORIGOTTI, G., FALLIVA, L., FANUZZI, A., FORMIGARO, M., MAGGIORE,
G., MARNI, E., MEAZZA, L., MERLO, F., PUNTONI, R., ROSA, A., STAGNARO,
E., & VERCELLI, M. (1982) Mortality and birth defects from 1976 to
1979 in the population living in the TCDD polluted area of Seveso. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
571-598.
ABERNETHY, D.J., GREENLEE, W.F., HUBAND, J.C., & BOREIKO, C.J. (1985)
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) promotes the transformation
of C3H/10T1/2 cells. Carcinogenesis, 6: 651-653.
ADAMS, E.M., IRISH, D.D., SPENCER, H.C., & ROWE, V.K. (1941) The
response of rabbit skin to compounds reported to have caused acneform
dermatits. Ind. Med., 2: 1-4.
AHLBORG, U.G. & VICTORIN, K. (1987) Impact on health and environment
from trace organic emissions. Waste Manage. Res., 5: 203-224.
AHLBORG, U.G., WAERN, F., & HAKANSSON, H. (1987) Interactive effects
of PCDDs and PCDFs occurring in human mother's milk. Chemosphere,
16(8/9): 1701-1706.
AHLING, B., BJORSETH, A., & LUNE, G. (1978) Formation of chlorinated
hydro- carbons during combustion of poly(vinyl chloride).
Chemosphere, 8(10): 799-806.
AHLING, B., LINDSKOG, A., JANSSON, B., & SUNDSTROM, G. (1977)
Formation of polychlorinated dibenzo-p-dioxins and dibenzo-furans
during combustion of a 2,4,5-T formulation. Chemosphere, 6(8):
461-468.
AITIO, A. & PARKKI, M.G. (1978) Organ specific induction of drug
metabolizing enzymes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the
rat. Toxicol. appl. Pharmacol., 44: 107-114.
AITIO, A., PARKKI, M.G., & MARNIEMI, J. (1979) Different effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on glucuronide conjugation of
various aglycones. Studies in Wistar and Gunn rats. Toxicol. appl.
Pharmacol., 47: 55-60.
AKERMARK, B. (1978) Photochemical reactions of phenoxy acids and
dioxins. Chlorinated phenoxy acids and their dioxins. Ecol. Bull.,
21: 75-81.
ALBRO, P.W. & CORBETT, B.J. (1977) Extraction and clean-up of animal
tissues for subsequent determination of mixtures of chlorinated
dibenzo-p-dioxins and dibenzofurans. Chemosphere, 7: 381-385.
ALBRO, P.W., CORBETT, J.T., HARRIS, M., & LAWSON, L.D. (1978) Effects
of 2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid profiles in tissue of
the Fisher rat. Chem.-biol. Interact., 23: 315-330.
ALBRO, P.W., CRUMMETT, W.B., DUPUY., A.E., Jr, GROSS, M.L., HANSON,
M., HARLESS, R.L., HILEMAN, F.D., HILKER, D., JASON, C., JOHNSON,
J.L., LAMPARSKI, L.L., LAU, B.P.Y., MCDANIEL, D.D., MEEHAN, J.L.,
NESTRICK, T.J., NYGREN, M., O'KEEFE, P., PETERS, T.L., RAPPE, C.,
RYAN, J.J., SMITH, L.M., STALLING, D.L., WEERASINGHE, N.C.A., &
WENDLING, J.M. (1985) Methods for the quantitative determination of
multiple, specific poly-chlorinated dibenzo-p-dioxin and dibenzofuran
isomers in human adipose tissue in the parts-per-trillion range. An
inter-laboratory study. Anal. Chem., 57: 2717-2725.
ALBRO, P.W., CORBETT, J.T., & SCHROEDER, J.L. (1986) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid peroxidation in
microsomal systems in vitro. Chem.-biol. Interact., 57: 301-313.
ALLEN, J.R. (1964) The role of "toxic fat" in the production of
hydro-pericardium and ascites in chickens. Am. J. vet. Res., 25:
1210-1219.
ALLEN, J.R. & CARSTENS, L.A. (1967) Light and electron microscopic
observations in Macaca mulatta monkeys fed toxic fat. Am. J. vet.
Res., 28: 1513-1526.
ALLEN, J.R. & LALICH, J.J. (1962) Response of chickens to prolonged
feeding of crude "toxic fat". Proc. Soc. Exp. Biol. Med., 109:
48-51.
ALLEN, J.R., VAN MILLER, J.P., & NORBACK, D.H. (1975) Tissue
distribution, excretion and biological effects of (14C)
tetrachlorodibenzo-p-dioxin in rats. Food Cosmet. Toxicol., 13:
501-505.
ALLEN, J.R., BARSOTTI, D.A., VAN MILLER, J.P., ABRAHAMSON, L.J., &
LALICH, J.J. (1977) Morphological changes in monkeys consuming a diet
containing low levels of 2,3,7,8-tetrachloro-dibenzo-p-dioxin. Food
Cosmet. Toxicol., 15(5): 401-410.
ALLEN, J.R., HARGRAVES, W.A., HSIA, M.T.S., & LIN, F.S.D. (1979a)
Comparative toxicology of chlorinated compounds on mammalian species.
Pharmacol. Ther., 7: 513-547.
ALLEN, J.R., BARSOTTI, D.A., LAMBRECHT, L.K., & VAN MILLER, J.P.
(1979b) Reproductive effects of halogenated aromatic hydrocarbons on
nonhuman primates. Ann. N.Y. Acad. Sci., 320: 19-27.
ANDERSSON, K., BOSSHARDT, H.P., BUSER, H.R., MARKLUND, S., & RAPPE, C.
(1978) Chlorinated phenoxy acids and their dioxins: Chemistry-summary.
Ecol. Bull., 27: 19-27.
ARSTILA, A.U., REGGIANI, G., SORVARI, T.E., RAISANEN, S., & WIPF, H.K.
(1981) Elimination of 2,3,7,8-tetrachlorodibenzo-p-dioxin in goat
milk. Toxicol. Lett., 9: 215-219.
ASHE, W.F. & SUSKIND, R.R. (1949) Clinical Report on four patients
from Monsanto Chemical Company in Nitro, West Virginia, USA, 14 pp.
(Submitted December 1949).
ASHE, W.F. & SUSKIND, R.R. (1950) Clinical report on six patients
from Monsanto Chemical Company in Nitro, West Virginia, USA, 16 pp.
(Submitted April 1950).
BAADER, E.W. & BAUER, H.J. (1951) Industrial intoxication due to
pentachlorophenol. Ind. Med. Surg., 20(6): 286-290.
BAARS, A.J., MUKHTAR, H., JANSEN, M., & BREIMER, D.D. (1982)
Induction of rat hepatic glutathione-s-tranferase activities by
2,3,7,8-tetrachlorodibenzo-p-dioxin, Oxford, New York, Pergamon
Press, pp. 393-401 (Pergamon Series on Environmental Science, Vol. 5).
BALK, J.L. & PIPER, W.N. (1984) Altered blood levels of
corticosteroids in the rat after exposure to
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Biochem. Pharmacol., 33(15):
2531-2534.
BALL, L.M. & CHABRA, R.S. (1981) Intestinal absorption of nutrients in
rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J.
Toxicol. environ. Health, 8: 629-638.
BANDIERA, S., FARRELL, K., MASON, G., KELLEY, M., ROMKES, M.,
BANNISTER, R., & SAFE, S. (1984a) Comparative toxicities of the
polychlorinated dibenzofuran (PCDF) and biphenyl (PCB) mixtures which
persist in yusho victims. Chemosphere, 13(4): 507-512.
BANDIERA, S., SAWYER, T., ROMKES, M., ZMUDZKA, B., SAFE, L., MASON,
G., KEYS, B., & SAFE, S. (1984b) Polychlorinated dibenzofurans
(PCDFs): Effects of structure on binding to the 2,3,7,8-TCDD cytosolic
receptor protein, AHH induction and toxicity. Toxicology, 32:
131-144.
BANNISTER, R. & SAFE, S. (1987) Aroclor 1254 as a
2,3,7,8-tetrachlorodibenzo-p-dioxin antagonist in C57BL/6J and DBA/2J
mice - effects of enzyme induction. Toxicologist, 7: 160.
BANNISTER, R., MASON, G., KELLEY, M., & SAFE, S. (1986) The effects of
cytosolic receptor modulation on the AHH-inducing activity of
2,3,7,8-TCDD. Chemosphere, 15: 1909-1911.
BARSOTTI, D.A., ABRAHAMSON, L.J., & ALLEN, J.R. (1979) Hormonal
alterations in female rhesus monkeys fed a diet containing
2,3,7,8-tetrachlorodibenzo-p-dioxin. Bull. environ. Contam.
Toxicol., 21: 463-469.
BARTELSON, F.D., HARRISON, D.D., & MORGAN, J.D. (1975) Field
studies of wildlife exposed to TCDD contaminated soil, Florida,
Airforce Armament Laboratory, Eglin Air Force Base (AFATL-TR-75-49).
BASTOMSKY, C.H. (1977) Enhanced thyroxine metabolism and high uptake
goiters in rats after a single dose of
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Endocrinology, 101(1):
292-296.
BAUER, H., SCHULZ, K.H., & SPIEGELBERG, U. (1961) [Occupational
poisoning in the manufacture of chlorophenol compounds.] Arch.
Gewerbepathol. Gewerbehyg., 18: 538-555 (in German).
BAUGHMAN, R.W. (1974) Tetrachlorodibenzo-p-dioxins in the
environment. High resolution mass spectrometry of the picogram
level, Cambridge, Massachussets, Harvard University (Thesis).
BAUGHMAN, R.W. & MESELSON, M. (1973) An analytical method for
detecting TCDD (dioxin): Levels of TCDD in samples from Vietnam.
Environ. Health Perspect., 5: 27-35.
BEALE, M.G., SHEARER, W.T., KARL, M.M., & ROBSON, A.M. (1977)
Long-term effects of dioxin exposure. Lancet, 1: 748.
BEATTY, P.W. & NEAL, R.A. (1977) Factors affecting the induction of
DT-diaphorase by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem.
Pharmacol., 27: 505-510.
BEATTY, P.W., LEMBACH, K.J., HOLSCHER, M.A., & NEAL, R.A. (1975)
Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on mammalian
cells in tissue cultures. Toxicol. appl. Pharmacol., 31: 309-312.
BEATTY, P.W., VAUGHN, W.K., & NEAL, R.A. (1978) Effect of alteration
of rat hepatic mixed-function oxidase (MFO) activity on the toxicity
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. appl.
Pharmacol., 45: 513-519.
BECK, H., ECKART, K., KELLERT, M., MATHAR, W., RUHL, CH.-S., &
WITTOWSKI, R. (1987) Levels of PCDF and PCDD in samples of human
origin and food in the Federal Republic of Germany. Chemosphere,
16: 1977-1982.
BELL, R.A. & GARA, A. (1985) Synthesis and characterization of the
isomers of polychlorinated dibenzofurans, tetra-through octa-. In:
Keith, L.H., Rappe, C., & Choudhary, C.G., ed. Chlorinated dioxins
and dibenzofurans in the total environment, Boston, Butterworth
Publishers, pp. 3-16.
BERGMAN, A., HAGMAN, A., JACOBSSON, S., JANSSON, B., & AHLMAN, M.
(1984) Thermal degradation of polychlorinated alkanes. Chemosphere,
13: 237-250.
BERLIN, A., BURRATTA, A., & VAN DER VENNE, M.-T. (1967) Proceedings
of the Expert Meeting on the Problems raised by TCDD-Pollution,
Milan, Italy, Luxembourg, Commission of the European Communities, p.
179.
BERRY, D.L., ZACHARIAH, P.K., NAMKUNG, M.J., & JUCHAU, M.R. (1976)
Transplacental induction of carcinogen-hydroxylating systems with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol., 36:
569-548.
BERRY, D.L., SLAGA, T.J., WILSON, N.M., ZACHARIAH, P.K., NAMKUNG,
M.J., BRACKEN, W.M., & JUCHAU, M.R. (1977) Tranplacental induction of
mixed-function oxygenases in extra-hepatic tissues by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 26:
1383-1388.
BERRY, D.L., SLAGA, T.J., DIGIOVANNI, J., & JUCHAU, M.R. (1979)
Studies with chlorinated dibenzo-p-dioxins, polybrominated biphenyls,
and poly chlorinated biphenyls in a two-stage system of mouse skin
tumorigenesis: potent anticarcinogenic effects. Ann. N.Y. Acad. Sci.,
320: 405-414.
BERTONI, G., BROCCO, D., DI PALO, V., LIBERTI, A., POSSANZINI, M., &
BRUNER, F. (1978) Gas chromatographic determination of 2,3,7,8-tetra
chloro-dibenzodioxin in the experimental decon-tamination of Seveso
soil by ultraviolet radiation. Anal. Chem., 50: 732-735.
BIRNBAUM, L.S. (1986) Distribution and excretion of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in congenic strains of mice which
differ at the Ah locus. Drug Metab. Disp., 14(1): 34-40.
BIRNBAUM, L.S., DECAD, G.M., & MATTHEWS, H.B. (1980) Disposition and
excretion of 2,3,7,8-tetrachlorodibenzofuran in the rat. Toxicol.
appl. Pharmacol., 55: 342-352.
BIRNBAUM, L.S., DECAD, G.M., MATTHEWS, H.B., & MCCONNELL, E.E. (1981)
Fate of 2,3,7,8-tetrachlorodibenzofuran in the monkey. Toxicol. appl.
Pharmacol., 57: 189-196.
BIRNBAUM, L.S., WEBER, H., HARRIS, M.W., LAMB, J.C., IV, & MCKINNEY,
J.D. (1985) Toxic interaction of specific polychlorinated biphenyls
and 2,3,7,8-tetrachlorodibenzo-p-dioxin: Increased incidence of cleft
palate in mice. Toxicol. appl. Pharmacol., 77: 292-302.
BIRNBAUM, L.S., HARRIS, M.W., MILLER, C.P., PRATT, R.M., & LAMB, J.C.
(1986) Synergistic interaction of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
and hydro-cortisone in the induction of cleft palate in mice.
Teratology, 33: 29-35.
BIRNBAUM, L.S., HARRIS, M.W., BARNHART, E.R., & MORRISSEY, R.E. (1987)
Teratogenicity of three polychlorinated dibenzo-furans in C57BL/6N
mice. Toxicol. appl. Pharmacol., 90: 206-216.
BLEIBERG, J., WALLEN, M., BRODKIN, R., & APPLEBAUM, I.L. (1964)
Industrially acquired porphyria. Arch. Dermatol., 89: 793-797.
BLOMHOFF, R., HELGERUD, P., RASMUSSEN, M., BERG, T., & NORUM, K.R.
(1982) In vivo uptake of chylomicron (3H)retinyl ester by rat liver:
Evidence for retinol transfer from parenchymal to nonparenchymal
cells. Proc. Natl Acad. Sci. (USA), 79: 7326-7330.
BOER, F.P., NEUMAN, M.A., VAN REMOORTERE, F.P., NORTH, P.P., & RINN,
H.W., (1973) X-Ray diffraction studies of chlorinated
dibenzo-p-dioxins. Chlorodioxins - origin and fate. Adv. Chem.
Ser., 120, 14-25.
BOMBICK, D.W., MATSUMURA, F., & MADHUKAR, B.V. (1984) TCDD
(2,3,7,8-Tetrachlorodibenzo-p-dioxin) causes reduction in the low
density lipoprotein (LDL) receptor activities in the hepatic plasma
membrane of the guinea pig and rat. Biochem. biophys. Res.
Commun., 118: 548-554.
BOMBICK, D.W., MADHUKAR, B.V., BREWSTER, D.W., & MATSUMURA, F. (1985)
TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) causes increases in protein
kinases particularly protein kinase C in the hepatic plasma membrane
of the rat and the guinea pig. Biochem. biophys. Res. Commun.,
127(1): 296-302.
BONACCORSI, A., DI DOMENICO, A., FANELLI, R., MERLI, F., MOTTA, R.,
VANZATI, R., & ZAPPONI, G.A. (1984) The influence of soil particle
adsorption on 2,3,7,8-tetrachlorodibenzo-p-dioxin biological uptake in
the rabbit. Arch. Toxicol., 7: 431-434.
BOTRE, C., MEMOLI, A., & ALHAIQUE, F. (1978) TCDD solubilization and
photodecomposition in aqueous solutions. Environ. Sci. Technol.,
12: 335-336.
BOWES, G.W., MULVIHILL, M.J., DECAMP, M.R., & KENDE, A.S. (1975) Gas
chromatographic characteristics of authentic chlorinated
dibenzofurans; identification of two isomers in American and Japanese
polychlorinated biphenyls. J. agric. food Chem., 23(6): 1222-1223.
BRADLAW, J.A. & CASTERLINE, J.L., Jr (1979) Induction of enzyme
activity in cell culture: a rapid screen for detection of planar
polychlorinated organic compounds. J. Assoc. Off. Anal. Chem.,
62(4): 904-916.
BRADLAW, J.A., GARTHOFF, L.H., HURLEY, N.E., & FIRESTONE, D. (1980)
Comparative induction of aryl hydrocarbon hydroxylase activity in
vitro by analogues of dibenzo-p-dioxin. Food Cosmet. Toxicol., 18:
627-635.
BRAUN, W. (1955) [Chloracne. Monographs with the Journal
Berufsdermatosen,] Aulendorf i. Wurtt., Editio Cantor, Vol. 1. (in
German).
BREWSTER, D.W., MADHUKAR, B.V., & MATSUMURA, F. (1982) Influence of
2,3,7,8-TCDD on the protein composition of the plasma membrane of
hepatic cells from the rat. Biochem. biophys. Res. Commun., 107:
68-74.
BRONZETTI, G., BAUER, C., CORSI, C., DEL CARRATORE, R., NIERI, R., &
PAOLINI, M. (1983) Mutagenicity study of TCDD and ashes from urban
incinerator "in vitro" and "in vivo" using yeast D7 strain.
Chemosphere, 12(4/5): 549-553.
BROWN, E.F. & MORGAN, A.F. (1948) The effect of vitamin A deficiency
upon the nitrogen metabolism of the rat. J. Nutr., 35: 425-438.
BUMB, R.R., CRUMMETT, W.B., CUTIE, S.S., GLADHILL, R.H., VAGEL, R.O.,
LAMPAVSKI, L.L., LUOMA, E.V., MILLER, D.L., NESTRIDE, T.J., SHADOFF,
L.A., STEHL, R.H., & WOODS, J.S., (1980) Trace chemistries of fire: A
source of chlorinated dioxins. Science, 210: 385-390.
BURKHARD, L.P. & KUEHL, D.W. (1986) N-Octanol/water partition
coefficients by reverse phase liquid chromatography/mass spectrometry
for eight tetrachlorinated planar molecules. Chemosphere, 15:
163-167.
BUS, J.S. & GIBSON, J.E. (1979) Lipid peroxidation and its role in
toxicology. Rev. Biochem. Toxicol., 1: 125-149.
BUSER, H.R. (1975) Polychlorinated dibenzo-p-dioxins. Separation and
identification of isomers by gas chromotagrophy mass spectrometry. J.
Chromatogr., 114: 95-108.
BUSER, H.R. (1979) Formation of polychlorinated dibenzofurans (PCDFs)
and dibenzo-p-dioxins (PCDDs) from the pyrolysis of chlorobenzenes.
Chemosphere, 6: 415-424.
BUSER, H.R. & BOSSHARDT, H.P. (1976) Determination of polychlorinated
dibenzo-p-dioxins and dibenzofurans in commercial pentachlorophenols
by combined gas chromatography - mass spectrometry. J. Assoc. Off.
Anal. Chem., 59: 562-569.
BUSER, H.R. & BOSSHARDT, H.P. (1978) [Polychlorinated
dibenzo-p-dioxin, dibenzofuran and benzene in the ashes from municipal
and industrial incinerators.] Mitt. Geb. Lebensm. Hyg., 69:
191-199 (in German).
BUSER, H.R. & RAPPE, C. (1978) Identification of substitution patterns
in polychlorinated dibenzo-p-dioxins (PCDDs) by mass spectrometry.
Chemosphere, 7: 199-211.
BUSER, H.R. & RAPPE, C. (1979) Formation of polychlorinated
dibenzofurans (PCDFs) from the pyrolysis of individual PCB isomers.
Chemosphere, 3: 157-174.
BUSER, H.R. & RAPPE, C. (1980) High-resolution gas chromato-graphy of
the 22 tetrachlorodibenzo-p-dioxin isomers. Anal. Chem., 52:
2257-2262.
BUSER, H.R. & RAPPE, C. (1984) Isomer-specific separation of
2,3,7,8-substituted polychlorinated dibenzo-p-dioxins by
high-resolution gas chromatography/mass spectrometry. Anal. Chem.,
56: 442-448.
BUSER, H.R., BOSSHARDT, H.P., & RAPPE, C. (1978a) Formation of
polychlorinated dibenzofurans (PCDFs) from the pyrolysis of PCBs.
Chemosphere, 1: 109-119.
BUSER, H.R., BOSSHARDT, H.P., & RAPPE, C. (1978b) Identification of
polychlorinated dibenzo-p-dioxin isomers found in fly ash.
Chemosphere, 7(2): 165-172.
BUSER, H.R., BOSSHARDT, H.P., RAPPE, C., & LINDAHL, R. (1978c)
Identification of polychlorinated dibenzofuran isomers in fly ash and
PCB pyrolysis. Chemosphere, 5: 419-429.
BUSER, H.R., RAPPE, C., & GARA, A. (1978d) Polychlorinated
dibenzofurans (PCDFs) found in Yosho oil and in used Japanese PCB.
Chemosphere, 5: 439-449.
BUSER, H.R., RAPPE, C., & BERGQVIST, P.-A. (1985) Analysis of
polychlorinated dibenzofurans, dioxins, and related compounds in
environmental samples. Environ. Health Perspect., 60: 293-302.
BUU-HOI, N.P., HIEN, D.P., SAINT-RUF, G., & SERVOIN-SIDOINE, J.
(1971a) Propriétés cancéromimétiques de la tétrachloro-2,3,7,8
dibenzo-p-dioxin. C. R. Acad. Sci. Paris, 272: 1447-1450.
BUU-HOI, N.P., SAINT-RUF, G., BIGOT, P., & MANGANE, M. (1971b)
Préparation, propriétés et identification de la "dioxine"
(tétrachloro-2,3,7,8-dibenzo-p-dioxine) dans les pyrolysats de
défoliants à base d'acide trichloro-2,4,5 phénoxyacétique et de ses
esters et des végétaux contaminés. C. R. Acad. Sci. Paris, 273:
708-711.
BUU-HOI, N.P., CHANH, P.H., SESQUE, G., AZUM-GELADE, M.C., &
SAINT-RUF, G. (1972a) Organs as targets of "dioxin"
(2,3,7,8-tetrachlorodibenzo-p-dioxin) intoxication.
Natur-wissenschaften, 59: 174-175.
BUU-HOI, N.P., CHANH, P.H., SESQUE, G., AZUM-GELADE, M.C., &
SAINT-RUF, G. (1972b) Enzymatic functions as targets of the toxicity
of "dioxin" (2,3,7,8-tetrachlorodibnzo-p-dioxin.
Naturwissenschaften, 59: 173-174.
CANTONI, L., SALMONA, M., & RIZZARDINI, M. (1981) Porphyrogenic effect
of chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin in
female rats. Dose-effect relationship following urinary excretion of
porphyrins. Toxicol. appl. Pharmacol., 57: 156-163.
CANTONI, L., DAL FIUME, D., FERRAROLI, A., SALMONA, M., & RUGGIERI, R.
(1984a) Different susceptibility of mouse tissues to porphyrogenic
effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Lett., 20:
201-210.
CANTONI, L., DAL FIUME, D., RIZZARDINI, M., & RUGGIERI, R. (1984b) In
vitro inhibitory effect on porphyrinogen carboxylase of liver extracts
from TCDD treated mice. Toxicol. Lett., 20: 211-217.
CARLSTEDT-DUKE, J.M.B. (1979) Tissue distribution of the receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Cancer Res., 39:
3172-3176.
CARLSTEDT-DUKE, J.M.B, ELFSTROEM, G., HOEGBERG, B., & GUSTAFSSON,
J.-A. (1979) Oncogeny of the rat hepatic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin and its endocrine independence.
Cancer Res., 39: 4653-4656.
CARLSTEDT-DUKE, J.M.B., HARNEMO, U.-B., HOEGBERG, B., & GUSTAFSSON,
J.-A. (1981) Interaction of the hepatic receptor protein for
2,3,7,8-tetrachlorodibenzo-p-dioxin with DNA. Biochim. Biophys.
Acta, 672: 131-141.
CARLSTEDT-DUKE, J.M.B., KURL, R., POELLINGER, L., GILLNER, M.,
HANSSON, L.-A., TOFTGARD, R., HOEGBERG, B., & GUSTAFSSON, J.-A. (1982)
The detection and function of the cytosolic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related cocarcinogens.
In: Hutzinger, O., ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
355-365
CARTER, C.D., KIMBROUGH, R.D., LIDDLE, J.A., CLINE, R.E., ZACH, M.M.,
Jr, BARTHEL, W.F., KOEHLER, R.E., & PHILLIPS, P.E. (1975)
Tetrachlorodibenzo-dioxin: An accidental poisoning episode in horse
arenas. Science, 188: 738-740.
CAVALLARO, A., BANDI, G., INVERNIZZI, G., LUCIANI, L., MONGINI, E., &
GORNI, G. (1982) Negative ion chemical ionization ms as a structure
tool in the determination of small amounts of PCDD and PCDF. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds,
Oxford, New York, Pergamon Press, Vol. 5, pp. 55-65.
CECIL, H.C., HARRIS, S.J., BITMAN, J., & FRIES, G.F. (1973)
Polychlorinated biphenyl-induced decrease in liver vitamin A in
Japanese Quail and rats. Bull. environ. Contam. Toxicol., 9:
179-185.
CHANG, K.J., HSIEH, K.H., LEE, T.P., TANG, S.Y., & TUNG, T.C. (1981)
Immunologic evaluation of patients with polychlorinated biphenyl
poisoning: determination of lymphocyte subpopulations. Toxicol.
appl. Pharmacol., 61: 58-63.
CHANG, K.J., HSIEH, K.H., LEE, T.P., & TUNG, T.C. (1982a) Immunologic
evaluation of patients with polychlorinated biphenyl poisoning:
determination of phagocyte Fc and complement receptors. Environ. Res.,
28: 329-334.
CHANG, K.J., HSIEH, K.H., TANG, S.Y., TUNG, T.C., & LEE, T.P. (1982b)
Immunologic evaluation of patients with polychlorinated biphenyl
poisoning: evaluation of delayed-type skin hypersensitive response and
its relation to clinical studies. J. Toxicol. environ. Health,
9: 217-223.
CHAPMAN, D.E. & SCHILLER, C.M. (1985) Dose-related effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57BL/6J and DBA/2J
mice. Toxicol. appl. Pharmacol., 78: 147-157.
CHASTAIN, J.E. & PAZDERNIK, T.L. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD)-induced immunotoxicity.
Int. J. Immunopharmacol., 7(6): 849-856.
CHEN, P.H. & HITES, R.A. (1983) Polychlorinated biphenyls and
dibenzofurans retained in the tissues of a deceased patient with
Yu-Cheng in Taiwan. Chemosphere, 12: 1507-1516.
CHEN, P.H., GAW, J.M., WONG, C.K., & CHEN, C.J. (1980) Levels and gas
chromatographic patterns of polychlorinated biphenyls in the blood of
patients after PCB poisoning in Taiwan. Bull. environ. Contam.
Toxicol., 25: 325-329.
CHEN, P.H., CHANG, K.T., & LU, Y.D. (1981) Polychlorinated biphenyls
and polychlorinated dibenzofurans in the toxic rice-bran oil that
caused PCB poisoning in Taichung. Bull. environ. Contam. Toxicol.,
26, 489-495.
CHEN, P.H., WONG, C.K., RAPPE, C., & NYGREN, M. (1985) Polychlorinated
biphenyls, dibenzofurans and quaterphenyls in toxic rice-bran oil and
in the blood and tissues of patients with PCB poisoning (Yu-Cheng) in
Taiwan. Environ. Health Perspect., 59: 59-65.
CHEUNG, M.O., GILBERT, E.F., & PETERSON, R.E. (1981) Cardiovascular
teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the chick
embryo. Toxicol. appl. Pharmacol., 61: 197-204.
CHRISTIAN, B.J., INHORN, S.L., & PETERSON, R.E. (1986a) Relationship
of the wasting syndrome to lethality in rats treated with
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
82: 239-255.
CHRISTIAN, B.J., MENAHAN, L.A., & PETERSON, R.E. (1986b) Intermediary
metabolism of the mature rat following
2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Toxicol. appl.
Pharmacol., 83: 360-378.
CLARK, D.A., GAULDIE, J., SZEWCZUK, M.R., & SWEENEY, G. (1981)
Enhanced suppressor cell activity as a mechanism of immunosuppression
by 2,3,7,8-tetrachlorodibenzo-p-dioxin (41275). Proc. Soc. Exp. Biol.
Med., 168: 290-299.
CLARK, D.A., SWEENEY, G., SAFE, S., HANCOCK, E., KILBURN, D.G., &
GAULDIE, J. (1983) Cellular and genetic basis for suppression of
cytotoxic T-cell generation by haloaromatic hydrocarbons.
Immunopharmacology, 6: 143-153.
CLEMENT, R.E., TOSINE, H.M., & ALI, B. (1985) Levels of
polychlorinated dibenzo-p-dioxins and dibenzofurans in wood burning
stoves and fireplaces. Chemosphere, 14: 815-819.
COCCIA, P., CROCI, T., & MANARA, L. (1981) Less TCDD persists in liver
2 weeks after a single dose to mice fed chow with added charcoal or
cholic acid. Br. J. Pharmacol., 72: 181-182.
COCHRANE, W.P., SINGH, J., MILES, W., WAKEFORD, B., & SCOTT, J. (1981)
Analysis of technical and formulated products of 2,4-dichlorophenoxy
acid for the presence of chlorinated dibenzo-p-dioxins. In: Hutzinger,
O., Frei, R.W., Merian, E., & Pocchiari, F., ed. Impact of
chlorinated dioxins and related compounds on the environment,
Oxford, New York, Pergamon Press, pp. 209-213.
COHEN, G.M., BRACKEN, W.M., IYER, R.P., BERRY, D.L., SELKIRK, J.K., &
SLAGA, T.J. (1979) Anticarcinogenic effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on benzo(a)pyrene and
7,12-dimethylbenz(a)anthracene tumor initiation and its relationship
to DNA binding. Cancer Res., 39: 4027-4033.
CONAWAY, C.C. & MATSUMURA, F. (1975) Alteration of cellular
utilization of thymidine by TCDD (2,3,7,8-tetrachlorodibenzo-p
-dioxin). Bull. environ. Contam. Toxicol., 12: 52-56.
CONSTABLE, J.D. & HATCH, M.C. (1985) Reproductive effects of herbicide
exposure in Vietnam: Recent studies by the Vietnamese and others.
Teratog. Carcinog. Mutagen., 5: 231-250.
COOK, R.R. (1981) Dioxin, chloracne, and soft tissue sarcoma.
Lancet, 1: 618-619.
COOK, R.R., TOWNSEND, J.C., OTT, M.G., & SILVERSTEIN, L.G. (1980)
Mortality experience of employees exposed to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. occup. Med., 22:
530-532.
COURTNEY, K.D. (1976) Mouse teratology studies with
chlorodibenzo-p-dioxins. Bull. environ. Contam. Toxicol., 16:
674-681.
COURTNEY, K.D. & MOORE, J.A. (1971) Teratology studies with
2,4,5-trichlorophenoxy-acetic acid and
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
20: 396-403.
COURTNEY, K.D., GAYLOR, D.W., HOGAN, M.D., FALK, H.L., BATES, R.R., &
MITCHEL, I. (1970) Teratogenic evaluation of 2,4,5-T. Science,
168: 864-866.
COURTNEY, K.D., PUTNAM, J.P., & ANDREWS, J.E. (1978) Metabolic studies
with TCDD (Dioxin) treated rats. Arch. environ. Contam. Toxicol.,
7: 385-396.
COWARD, K.H. (1947) The determination of vitamin A. In: The
biological standardisation of the vitamins, London, Baillière,
Tindall and Cox, pp. 23-58.
CROSBY, D.G. & WONG, A.S. (1976) Photochemical generation of
chlorinated dioxins. Chemosphere, 5: 327-332.
CROSBY, D.G. & WONG, A.S. (1977) Environmental degradation of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Science, 195:
1337-1338.
CROSBY, D.G., WONG, A.S., PLIMMER, J.R., & WOOLSON, E.A. (1971)
Photodecomposition of chlorinated dibenzo-p-dioxins. Science, 173:
748-749.
CROSBY, D.G., MOILANEN, K.W., & WONG, A.S. (1973) Environmental
generation and degradation of dibenzodioxins and dibenzofurans.
Environ. Health Perspect., 5: 259-266.
CROW, K.D. (1970) Chloracne. A critical review including a comparison
of two series of cases of acne from chlornaphtalene and pitch fumes.
Trans. St. John's Hosp. Dermatol. Soc., 56: 79-99.
CRUMMETT, W.B. & STEHL, R.H. (1973) Determination of chlorinated
dibenzo-p-dioxins and dibenzofurans in various materials. Environ.
Health Perspect., 5: 15-25.
CRUMMETT, W.B., NESTRICK, T.J., & LAMPARSKI, L.L., (1985) Analytical
methodology for the determination of PCDDs in environmental samples:
an overview and critique. In: Kamrin, M.A. & Rodgers, P.W., ed.
Dioxins in the environment, Washington, DC, Hemisphere Publishing,
pp. 57-83.
CUNNINGHAM, H.M. & WILLIAMS, D.T. (1972) Effect of
tetrachlorodibenzo-p-dioxin on growth rate and the synthesis of lipids
and proteins in rats. Bull. environ. Contam. Toxicol., 7: 45-51.
CZUCZWA, J.M. & HITES, R.A. (1985) Historical record of
polychlorinated dioxins and furans in Lake Huron sediments. In: Keith,
L.H., Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment, Stoneham, Maine, Butterworth
Publishers, pp. 59-63.
DALDERUP, L.M. (1974a) Safety measures for taking down buildings
contaminated with toxic material I. T. Soc. Geneeskd., 52: 582-588.
DALDERUP, L.M. ((1974b) Safety measures for taking down buildings
contaminated with toxic material II. T. Soc. Geneeskd., 52:
616-623.
D'ARGY, R., HASSOUN, E., & DENCKER, L. (1984) Teratogenicity of TCDD
and the congener 3,3',4,4'-tetrachloroazoxybenzene in sensitive and
nonsensitive mouse strains after reciprocal blastocyst transfer.
Toxicol. Lett., 21: 197-202.
DAVISON, K.L. & COX, J.H. (1976) Methoxychlor effects on hepatic
storage of vitamin A in rats. Bull. environ. Contam. Toxicol., 16:
145-148.
DECAD, G.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1981a)
2,3,7,8-tetrachlorodibenzofuran tissue distribution and excretion in
guinea pigs. Toxicol. appl. Pharmacol., 57: 231-240.
DECAD, G.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1981b) Distribution and
excretion of 2,3,7,8-tetrachlorodibenzofuran in C57BL/6J and DBA/2J
mice. Toxicol. appl. Pharmacol., 59: 564-573.
DECAPRIO, A.P., MCMARTIN, D.N., SILKWORTH, J.B., REJ, R., PAUSE, R.,
& KAMINSKY, L.S. (1983) Subchronic oral toxicity in guinea pigs of
soot from a polychlorinated biphenyl-containing transformer fire.
Toxicol. appl. Pharmacol., 68: 308-322.
DECAPRIO, A.P., MCMARTIN, D.N., O'KEEFE, P.W., REJ, R., SILKWORTH,
J.B., & KAMINSKY, L.S. (1986) Subchronic oral toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the guinea pig: comparisons
with a PCB-containing transformer fluid pyrolysate. Fundam. appl.
Toxicol., 6: 454-463.
DEITRICH, R.A., BLUDEAU, P., ROPER, M., & SCHMUCK, J. (1978) Induction
of aldehyde dehydrogenases. Biochem. Pharmacol., 27: 2343-2347.
DENCKER, L. & PRATT, R.M. (1981) Association between the presence of
the Ah receptor in embryonic murine tissues and sensitivity to
TCDD-induced cleft palate. Teratog. Carcinog. Mutagen., 1:
399-406.
DENCKER, L., HASSOUN, E., D'ARGY, R., & ALM, G. (1985) Fetal thymus
organ culture as an in vitro model for the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin and its congeners. Mol.
Pharmacol., 27: 133-140.
DENISON, M.S., VELLA, L.M., & OKEY, A.B. (1986a) Structure and
function of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Species difference in molecular properties of the receptors from mouse
and rat hepatic cytosols. J. biol. Chem., 261: 3987-3995.
DENISON, M.S., WILKINSON, C.F., & OKEY, A.B. (1986b) Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin: comparative studies in mammalian
and nonmammalian species. Chemosphere, 15: 1665-1672.
DENISON, M.S., HARPER, P.A., & OKEY, A.B. (1986c) Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin. Codistribution of unoccupied
receptor with cytosolic marker enzymes during fractionation of mouse
liver, rat liver and cultured Hepa-1c1 cells. Eur. J. Biochem.,
155: 223-229.
DENMARK (1984) [Formation and emission of dioxins especially in
connection with waste incineration: supplement,] Copenhagen,
Miljöstyrelsen (in Danish).
DICKINS, M., SEEFELD, M.D., & PETERSON, R.E. (1981) Enhanced liver DNA
synthesis in partially hepatectomized rats pre-treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
58: 389-398.
DIDOMENICO, A., SILANO, V., VIVIANO, G., & ZAPPONI, G. (1980)
Accidental release of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) at
Seveso, Italy. II. TCDD distribution in the soil surface layer.
Ecotoxicol. environ. Saf., 4: 298-320.
DIGIOVANNI, J., VIAJE, A., BERRY, D.L., SLAGA, T.J., & JUCHAU, M.R.
(1977) Tumor-initiating ability of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) and arochlor 1254 in the
two-stage system of mouse skin carcinogenesis. Bull. environ.
Contam. Toxicol., 18: 552-557.
DIGIOVANNI, J., BERRY, D.L., JUCHAU, M.R., & SLAGA, T.J. (1979)
2,3,7,8-Tetra-chlorodibenzo-p-dioxin: potent anti-carcinogenic
activity in CD-1 mice. Biochem. biophys. Res. Commun., 86:
577-584.
DIGIOVANNI, J., BERRY, D.L., GLEASON, G.L., KISHORE, G.S., & SLAGA,
T.J. (1980) Time dependent inhibition by
2,3,7,8-tetra-chlorodibenzo-p-dioxin of skin tumorogenesis with
polycyclic hydrocarbons. Cancer Res., 40: 1580-1587.
DOBLES, A.J. & GRANT, C. (1979) Photolysis of highly chlorinated
dibenzo-p-dioxins by sunlight. Nature (Lond.), 278: 162-165.
DOHMEIER, H.J. & JANSON, E. (1983) [Killing flies and people:
Dioxin - the poison of Seveso and Viet Nam and how we come into
daily contact with it.] Reinbek, Aktuell Rororo, p. 25.
DONOVAN, J.W., MACLENNAN, R., & ADENA, N. (1984) Vietnam service and
the risk of congenital anomalies: a case-control study. Med. J.
Aust., 140: 394-397.
DOYLE, B.W., DRUM, D.A., & LAUDER, J.D. (1985) The smoldering question
of hospital wastes. Pollut. Eng., 17: 35-39.
DOYLE, E.A. & FRIES, G.F. (1986) Induction of aryl hydrocarbon
hydroxylase by chlorinated dibenzofurans in rats. Chemosphere, 15:
1745-1748.
DRINKER, C.K., FIELD WARREN, M., & GRANVILLE, A. (1937) The problem of
possible systemic effects from certain chlorinated hydrocarbons. J.
ind. Hyg. Toxicol., 19: 283-311.
DUGOIS, P. & COLOMB, L. (1956) Acné chlorique au 2-4-5
trichlorophénol. J. Méd. Lyon, 88: 446-447.
DUGOIS, P. & COLOMB, L. (1957) Remarques sur l'acné chlorique. A
propos d'une éclosion de cas provoqués par la préparation du
2,4,5-trichlorophénol. J. Méd. Lyon, 38: 899-903.
DUGOIS, P., MARECHAL, J., & COLOMB, L. (1958) Acné chlorique au
2,4,5-tri-chlorophénol. Arch. Mal. prof. Hyg. Toxicol. ind., 19:
626-627.
DUGOIS, P., AMBLARD, P., AIMARD, M., & DESHORS, G. (1967) Acné
chlorique collective et accidentelle d'un type nouveau. Bull. Soc.
Dermatol., 75: 260-261.
DUVERNE, J., THIVOLET, J., & BERARD, J. (1964) Acné chlorique profuse
avec épanchement pleural récidivant chez un sujet à réactions
tuberculiniques négatives. Bull. Soc. Fr. Dermatol. Syphiligr.,
71: 649-652.
EATON, D.L. & KLAASSEN, C.D. (1979) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin, kepone, and polybrominated
biphenyls on transport systems in isolated rat hepatocytes. Toxicol.
appl. Pharmacol., 51: 137-144.
EDULJEE, G.H., ATKINS, D.H.F., & EGGLETON, A.E. (1986) Observations
and assessment relating to incineration of chlorinated chemical
wastes. Chemosphere, 15: 1577-1584.
ELDER, D.G., LEE, G.B., & TOVEY, J.A. (1978) Decreased activity of
hepatic uroporphyrinogen decarboxylase in sporadic porphyria cutanea
tarda. New Engl. J. Med., 299: 274-278.
ELDER, G.H. & SHEPPARD, D.M. (1982) Immunoreactive uroporphyrinogen
decarboxylase is unchanged in porphyria caused by TCDD and
hexachlorobenzene. Biochem. biophys. Res. Commun., 109: 113-120.
ELDER, G.H., EVANS, J.O., & MATLIN, S.A. (1976) The effect of
porhyrogenic compound, hexachlorobenzene, on the activity of hepatic
uroporphyrinogen decarboxylase in the rat. Clin. Sci. mol. Med.,
51: 71-80.
ERICKSON, J.D., MULINARE, J., MCCLAIN, P.W., FITCH, T.G., JAMES, L.M.,
MCCLEARN, A.B., & ADAMS, M.J. (1984) Vietnam veterans' risks for
fathering babies with birth defects. J. Am. Med. Assoc., 252:
903-937.
ESPOSITO, G., FAELLI, A., & CAPRARO, V. (1967) Metabolism and
transport phenomena in isolated intestine of normal and semi-starved
rats. Arch. int. Physiol. Biochim., 75: 601-608.
ESPOSITO, M.P., TIERNAN, T.O., & DRYDEN, F.E. (1980) Dioxins,
Cincinnati, Ohio, US Environmental Protection Agency, Office of
Research and Development (EPA-600/2-80-197).
FACCHETTI, S., BALASSO, A., FICHTNER, C., FRARE, G., LEONI, A., MAURI,
C., & VASCONI, M. (1986) Studies on the absorption of TCDD by plant
species. In: Rappe, C., Choudhary, G., & Keith, L.H., ed. Chlorinated
dioxins and dibenzofurans in perspective, Chelsea, Michigan, Lewis
Publishers, pp. 225-239.
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic activity of
chlorophenols and chlorophenol impurities. Environ. Sci. Res., 12:
325-338.
FAITH, R.E. & LUSTER, M.I. (1979) Investigations on the effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on parameters of various
immune functions. Ann. N.Y. Acad. Sci., 320: 564-571.
FAITH, R.E. & MOORE, J.A. (1977) Impairment of thymus-dependent immune
functions by exposure of the developing immune system to
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. Toxicol. environ.
Health, 3: 451-464.
FARA, G.M., DEL CORNO, G., BONETTI, F., CARAMASHI, F., DARDANONI, L.,
FAVARETTI, C., GIAMBELLUCA, S.E., MARNI, E., MOCARELLI, P.,
MONTESARCHIO, E., PUCCINELLI, V., & VOLPATO, C. (1976) Chloracne after
release of TCDD at Seveso, Italy. In: Hutzinger, O., Frei, R., Merian,
E., & Pocchiari, F. ed. Chlorinated dioxins and related compounds.
Impact on the environment, Oxford, New York, Pergamon Press, pp.
545-559.
FARRELL, K. & SAFE, S. (1986) 2,3,7,8-tetrachlorodibenzo-p-dioxin:
Relationship between toxicity and the induction of aryl hydrocarbon
hydroxylase and ornithine decarboxylase. Chemosphere, 15:
1971-1976.
FEDERAL REGISTER (1980) Storage and disposal of waste material:
Prohibition of disposal of tetrachlorodibenzo-p-dioxin. Fed. Reg.,
45(98): 32676.
FILIPPINI, G., BORDO, B., CRENNA, P., MASSETTO, N., MUSICCO, M., &
BOERI, R. (1981) Relationship between clinical and
electrophysiological findings and indicators of heavy exposure to
2,3,7,8-tetrachlorodibenzo-dioxin. Scand. J. Work Environ. Health,
7: 257-262.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B., GROTH,
D.H., & RUSSELL, W.O. (1984) Review of exposure and pathology data for
seven cases reported as soft tissue sarcoma among persons
occupationally exposed to dioxin-contaminated herbicides. In:
Lowrance, W.W., ed. Public health risks of the dioxins. Proceedings
of a Symposium held at the Rockefeller University, New York, 19-20
October, 1983, Los Altos, California, William Kaufmann, pp. 187-203.
FIRESTONE, D. (1973) Etiology of chick edema disease. Environ.
Health Perspect., 5: 59-66.
FIRESTONE, D., (1978) The 2,3,7,8-tetrachlorodibenzo-para-dioxin
problem: A review. Proceedings of a Conference on Chlorinated
Phenoxyacids and their Dioxins, Stockholm, 1977. Ecol. Bull., 27:
39-52.
FIRESTONE, D., CLOWER, M., Jr, BORSETTI, A.P., TESKE, R.H., & LONG,
P.E. (1979) Polychlorodibenzo-p-dioxin and pentachloro-phenol residues
in milk and blood of cows fed technical pentachlorophenol. J. agric.
food Chem., 27: 1171-1177.
FIRESTONE, D., NIEMANN, R.A., SCHNIDER, L.F., GRIDLEY, J.R., & BROWN,
D.E., (1986) Dioxin residues in fish and other food. In: Rappe, C.,
Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers.
FLICK, D.F., FIRESTONE, D., & HIGGINBOTHAM, G.R. (1972) Studies of the
chick edema disease. 9. Response of chicks fed or singly administered
synthetic edema-producing compounds. Poult. Sci., 51: 2026-2034.
FLICK, D.F., FIRESTONE, D., RESS, J., & ALLEN, J.R. (1973) Studies of
the chick edema disease. 10. Toxicity of chick edema factors in the
chick, chick embryo, and monkey. Poult. Sci., 52: 1637-1641.
FORTH, W. (1977) [The Seveso Disaster.] Dtsch. Arztebl., 44:
2617-2626 (in German).
FOWLER, B.A., LUCIER, G.W., BROWN, H.W., & MCDANIEL, O.S. (1973)
Ultrastructural changes in rat liver cells following a single oral
dose of TCDD. Environ. Health Perspect., 5: 141-148.
FRAENKEL, C. (1902) [Reports of associations and congresses. Medical
Association of Hall: Meeting of 23 October.] Münchener Med.
Wochenschr., October: 39-41.
FREEDMAN, H.J., PARKER, N.B., MARINELLO, A.J., GURTOO, H.L., &
MINOWADA, J. (1979) Induction, inhibition and biological properties of
aryl hydrocarbon hydroxylase in a stable human B-lymphocyte cell line,
RPMI-1788. Cancer Res., 39: 4612-4619.
FREEMAN, R.A., SCHROY, J.M., HILEMAN, F.D., & NOBLE, R.W. (1986)
Environmental mobility of 2,3,7,8-TCDD and companion chemicals in a
roadway soil matrix. In: Rappe, C., Choudhary, G., & Keith, L., ed.
Chlorinated dioxins and dibenzofurans in perspective, Chelsea,
Michigan, Lewis Publishers, pp. 171-183.
FRIES, G.F. & MARROW, G.S. (1975) Retention and excretion of
2,3,7,8-tetrachlorodibenzo-p-dioxin by rats. J. agric. food Chem.,
23: 265-269.
FRIESEN, K.J., SARNA, L.P., & WEBSTER, G.R.B. (1985) Aqueous
solubility of polychlorinated dibenzo-p-dioxins determined by high
pressure liquid chromatography. Chemosphere, 14: 1267-1274.
FUCHS, E. & GREEN, H. (1981) Regulation of terminal differentiation of
cultured human keratinocytes by vitamin A. Cell, 25: 617-625.
FUNG, D., BOYD, R.K., SAFE, S., & CHITTIM, B.G. (1985) Gas
chromatographic/mass spectrometric analysis of specific isomers of
polychlorodibenzofurans. Biomed. mass Spectrom., 12: 247-253.
FURST, P., MEEMKEN, H.-A. KRUGER, Chr., & GROEBEL, W. (1987)
Polychlorinated dibenzodioxins and dibenzofurans in human milk samples
from Western Germany. Chemosphere, 16: 1983-1988.
FURUHASHI, N., KURL, R.N., WONG, J., & VILLEE, C.A. (1986) A cytosolic
binding protein for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the
uterus and deciduoma of rats. Pharmacology, 33: 110-120.
GALLO, M.A., HESSE, E.J., MACDONALD, G.J., & UMBREIT, T.H. (1986)
Interactive effects of estradiol and
2,3,7-8-tetra-chlorodibenzo-p-dioxin on hepatic cytochrome P-450 and
mouse uterus. Toxicol. Lett., 32: 123-132.
GASIEWICZ, T.A. & NEAL, R.A. (1979)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin tissue distribution, excretion,
and effects on clinical chemical parameters in guinea pigs. Toxicol.
appl. Pharmacol., 51: 329-339.
GASIEWICZ, T.A. & RUCCI, G. (1984) Cytosolic receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin. Evidence for a homologous nature
among various mammalian species. Mol. Pharmacol., 26: 90-98.
GASIEWICZ, T.A., HOLSCHER, M.A., & NEAL, R.A. (1980) The effect of
total parenteral nutrition on the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Toxicol. appl.
Pharmacol., 54: 469-488.
GASIEWCICZ, T.A., OLSON, J.R., GEIGER, L.H., & NEAL, R.A. (1983a)
Absorption, distribution and metabolism of
2,3,7,8-tetrachlorodibenzodioxin (TCDD) in experimental animals.
Environ. Sci. Res., 26: 495-525.
GASIEWICZ, T.A., GEIGER, L.E., RUCCI, G., & NEAL, R.A. (1983b)
Distribution, excretion, and metabolism of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in C57Bl/6J, DBA/2J, and B602F,/J
mice. Drug Metab. Disp., 11: 397-403.
GASIEWICZ, T.A., NESS, W.C., & RUCCI, G. (1984) Ontogeny of the
cytosolic receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat
liver, lung, and thymus. Biochem. biophys. Res. Commun., 118:
183-190.
GASIEWICZ, T.A., RUCCI, G., HENRY, E.C., & BAGGS, R.B. (1986) Changes
in hamster hepatic cytochrome P-450, ethoxycoumarin o-deethylase, and
reduced NAD(P): Menadione oxidoreductase following treatment with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 35:
2737-2742.
GEBEFUGI, I., BAUMANN, R., & KORTE, F. (1977) [Photochemical breakdown
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) under stimulated
environmental conditions.] Naturwissenschaften, 64: 486-487 (in
German).
GEIGER, L.E. & NEAL, R.A. (1981) Mutagenicity testing of
2,3,7,8-tetrachlorodibenzo-p-dioxin in histidine auxotrophs of
Salmonella typhimurium. Toxicol. appl. Pharmacol., 59: 125-129.
GERMANY (1985) [Review of dioxins], Berlin, Erich Schmidt Verlag, pp.
257-266 (Federal Office for the Environment, Report 5/85, November
1984) (in German).
GIAVINI, E., PRATI, M., & VISMARA, C. (1982a) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin administered to pregnant rats
during the pre-implantation period. Environ. Res., 29: 185-189.
GIAVINI, E., PRATI, M., & VISMARA, C. (1982b) Rabbit teratology study
with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Res., 27:
74-78.
GIAVINI, E., PRATI, M., & VISMARA, C. (1983) Embryotoxic effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin administered to female rats before
mating. Environ. Res., 31: 105-110.
GIERTHY, J.F. & CRANE, D. (1984) Reversible inhibition of in vitro
epithelial cell proliferation by 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Toxicol. appl. Pharmacol., 74: 91-98.
GIERTHY, J.F. & CRANE, D. (1985a) In vitro bioassay for dioxin-like
activity based on alterations in epithelial cell proliferation and
morphology. Fundam. appl. Toxicol., 5: 754-759.
GIERTHY, J.F. & CRANE, D. (1985b) Development of in vitro
bioassays for chlorinated dioxins and dibenzofurans. In: Keith, L.H.,
Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment II, Stoneham, Maine,
Butterworth Publishers, pp. 267-284.
GIERTHY, J.F., CRANE, D., & FRENKEL, G.D. (1984) Application of an
in vitro keratinization assay to extracts of soot from a fire in
a polychlorinated biphenyl-containing transformer. Fundam. appl.
Toxicol., 4: 1036-1041.
GILBERT, P., SAINT-RUF, G., PONCELET, F., & MERCIER, M. (1980) Genetic
effects of chlorinated anilines and azobenzenes on salmonella
typhimurium. Arch. environ. Contam. Toxicol., 9: 533-541.
GOLDMANN, P.J. (1972) [Very severe acute chloracne caused by
trichlorophenol decomposition products.] Arbeitsmed. Sozialmed.
Arbeitshyg., 7: 12-18 (in German).
GOLDMANN, P.J. (1973) [Very severe acute chloracne: mass poisoning by
2,3,6,7-tetrachlorodibenzodioxin.] Hautarzt, 24: 149-152 (in
German).
GOLDSTEIN, J.A. & LINKO, P. (1984) Differential induction of two
2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible forms of cytochrome
P-450 in extrahepatic versus hepatic tissues. Mol. Pharmacol., 25:
185-191.
GOLDSTEIN, J.A., HICKMAN, P., BERGMAN, H., & VOS, J.G. (1973) Hepatic
porphyria induced by 2,3,7,8-tetrachloro-dibenzo-p-dioxin in the
mouse. Res. Commun. chem. Pathol. Pharmacol., 6: 919-928.
GOLDSTEIN, J.A., MCKINNEY, J.D., LUCIER, G.W., HICKMAN, P., BERGMAN,
H., & MOORE, J.A. (1976) Toxicological assessment of
hexachlorobiphenyl isomers and 2,3,7,8-tetrachlorodibenzofuran in
chicks. II. Effects on drug metabolism and porphyrin accumulation.
Toxicol. appl. Pharmacol., 36: 81-92.
GOLDSTEIN, J.A., HASS, J.R., LINKO, P., & HARVAN, D.J. (1978)
2,3,7,8-Tetrachlorodibenzofuran in a commercially available 99% pure
polychlorinated biphenyl isomer identified as the inducer of hepatic
cytochrome P-448 and aryl hydrocarbon hydroxylase in the rat. Drug
Metab. Disp., 6: 258-264.
GOLDSTEIN, J.A., LINKO, P., & BERGMAN, H. (1982) Induction of
porphyria in the rat by chronic versus acute exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol., 31:
1607-1613.
GONZALES, F.J., TUKEY, R.H., & NEBERT, D.W. (1984) Structural gene
products of the Ah locus. Transcriptional regulation of cytochrome
P1-450 and P3-450mRNA levels by 3-methylchol-anthrene. Mol.
Pharmacol., 26: 117-121.
GORSKI, T., KONOPKA, L., & BRODZKI, M. (1984) Persistence of some
polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans of
pentachlorophenol in human adipose tissue. Rocz. Pzh, XXXV(4):
297-301.
GOTHE, R. & WACHTMEISTER, C.A. (1972) Synthesis of
1,2,4,5,7,8-hexa-chloroxanthene. Acta chem. Scand., 26: 2523-2576.
GOTZ, R. (1986) Chlorinated dioxins and dibenzofurans in leachate and
sediments of the sanitary landfill in Hamburg-Georswerder.
Chemosphere, 15: 1981-1984.
GRAY, A.P., STEVEN, P.C., & CANTRELL, J.S. (1975) Intervention of the
Smiles rearrangement in synthesis of dibenzo-p-dioxins:
1,2,3,6,7,8-and 1,2,3,7,8,9-hexachlorodibenzo-dioxin. Tetrahydron
Lett., 33: 2873-2876.
GRAY, A.P., STEVEN, P.C., SOLOMON, I.J., & ANILINE, O. (1976)
Synthesis of specific polychlorinated dibenzo-p-dioxins. J. org.
Chem., 41: 2435-2437.
GREEN, S. & MORELAND, F.S. (1975) Cytogenetic evaluation of several
dioxins in the rat. Toxicol. appl. Pharmacol., 33: 161.
GREEN, S., MORELAND, F., & SHEU, C. (1977) Cytogenetic effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on rat bone marrow cells. FDA
By-lines, 6: 292-294.
GREENBURG, L., MAYERS, M.R., & SMITH, A.R. (1939) The systemic effects
resulting from exposure to certain chlorinated hydro-carbons. J. ind.
Hyg. Toxicol., 21: 29-38.
GREENLEE, W.F. & POLAND, A. (1978) An improved assay of
7-ethoxycoumarin O-deethylase activity: induction of hepatic enzyme
activity in C57BL/6J and DBA/2J mice by phenobarbital,
3-methylcholanthrene and 2,3,7,8-tetrachlorodibenzo-p-dioxin. J.
Pharmacol. exp. Ther., 205: 596-605.
GREENLEE, W.F. & POLAND, A. (1979) Nuclear uptake of
2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J and DBA/2J mice. Role
of the hepatic cytosol receptor protein. J. biol. Chem., 254:
9814-9821.
GREENLEE, W.F., DOLD, K.M., IRONS, R.D., & OSBORNE, R. (1985) Evidence
for direct action of 2,3,7,8-tetrachlorodidibenzo-p-dioxin (TCDD) on
thymic epithelium. Toxicol. appl. Pharmacol., 79: 112-120.
GREIG, J.B. (1972) Effect of 2,3,7,8-tetrachlorodibenzo-1, 4-dioxin on
drug metabolism in the rat. Biochem. Pharmacol., 21: 3196-3198.
GREIG, J.B. & OSBORNE, G. (1981) Biochemical and morphological changes
induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat liver cell
plasma membrane. J. appl. Toxicol., 1(6): 334-338.
GREIG, J.B., JONES, G., BUTLER, W.H., & BARNES, J.M. (1973) Toxic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Food Cosmet.
Toxicol., 11: 585-595.
GREIG, J.B., TAYLOR, D.M., & JONES, J.D. (1974) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on stimulated DNA synthesis in the
liver and kidney of the rat. Chem.-biol. Interact., 8: 31-39.
GREIG, J.B., FRANCIS, J.E., KAY, S.J.E., LOVELL, D.P., & SMITH, A.G.
(1984) Incomplete correlation of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
hepatotoxicity with Ah phenotype in mice. Toxicol. appl. Pharmacol.,
74: 17-25.
GROSS, M.L., LAY, J.O., Jr, LYON, P.A., LIPPSTREU, D., KANGAS, N.,
HARLESS, R.L., TAYLOR, S.E., & DUPUY, A.E. Jr (1984)
2,3,7,8-Tetrachlorodibenzo-p-dioxin levels in adipose tissue of
Vietnam veterans. Environ. Res., 33: 261-268.
GUENTHNER, T.M., FYSH, J.M., & NEBERT, D.W. (1979)
2,3,7,8-Tetrachlorodibenzo-p-dioxin: covalent binding of reactive
metabolic intermediates principally to protein in vitro.
Pharmacology, 19: 12-22.
GUPTA, B.N., VOS, J.G., MOORE, J.A., ZINKL, J.G., & BULLOCK, B.C.
(1973) Pathologic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in
laboratory animals. Environ. Health Perspect., 5: 125-140.
GURTOO, H.L., PARKER, N.B., PAIGEN, B., HAVENS, M.B., MINOWADA, J., &
FREEDMAN, H.J. (1979) Induction, inhibition and some enzymological
properties of aryl hydrocarbon hydroxylase in fresh mitogen-activated
human lymphocytes. Cancer Res., 39: 4620-4629.
GUSTAFSSON, J.-A. & INGELMAN-SUNDBERG, M. (1979) Changes in steroid
hormone metabolism in rat liver microsomes following administration of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Biochem. Pharmacol.,
28: 497-499.
HAAPARANTA, T., GLAUMANN, H., & GUSTAFSSON, J.-A. (1983) Induction of
cytochrome P-450 dependent reactions in the rat ventral prostate by
beta-naphthoflavone and 2,3,7,8-tetra-chlorodibenzo-p-dioxin.
Toxicology, 29: 61-75.
HAGENMAIER, H. & BRUNNER, H. (1987) Isomer specific analysis of PCDD
and PCDF in pentachlorophenol and sodium pentachloro- phenate samples
in the sub-PPB range. Chemosphere, 16: 1759-1764.
HAGENMAIER, H., DRAFT, M., JAGER, W., MAYER, U., LUETZKE, K., &
SIEGAL, D. (1986) Comparison of various sampling methods for PCDDs and
PCDFs in stack gas. Chemosphere, 15(9-12): 1187-1192.
HAKANSSON, H. (1988) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin
of the fate of vitamin A in rodents, Stockholm, Department of
Toxicology and Institute of Environmental Medicine, Karolinska
Institute (Ph.D. Thesis).
HAKANSSON, H. & AHLBORG, U.G. (1985) The effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the uptake, distribution
and excretion of a single oral dose of 11,12, 3H-retinylacetate and on
the vitamin A status in the rat. J. Nutr., 115: 759-771.
HAKANSSON, H., AHLBORG, U.G., & GOTTLING, L. (1986) The effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the distribution and
excretion of the endogenous pool of vitamin A in rats with low liver
vitamin A stores. Chemosphere, 15: 1715-1723.
HAKANSSON, H., WAERN, F., & AHLBORG, U.G. (1987) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the lactating rat on
maternal and neonatal vitamin A status. J. Nutr., 117: 580-586.
HANNAH, R.R., LUND, J., POELLINGER, L., GILLNER, M., & GUSTAFSSON,
J.-A. (1986) Characterization of the DNA-binding properties of the
receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Eur. J.
Biochem., 156: 237-242.
HARDELL, L. (1981) Relation of soft-tissue sarcoma, malignant lymphoma
and colon cancer to phenoxy acids, chlorophenols and other agents.
Scand. J. Work Environ. Health, 7: 119-130.
HARDELL, L. & ERIKSSON, M. (1981) Soft-tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 2: 250.
HARDELL, L. & SANDSTROM, A. (1979) Case-control study: soft-tissue
sarcomas and exposure to phenoxyacetic acids or chlorophenols. Br.
J. Cancer, 39: 711-717.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981) Malignant
lymphoma and exposure to chemicals, especially organic solvents,
chlorophenols and phenoxy acids: a case-control study. Br. J.
Cancer, 43: 169-176.
HARLESS, R.L. & LEWIS, R.G. (1982) Quantitative determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin residues by gas
chromatography/mass spectrometry. In: Hutzinger, O., ed. Chlorinated
dioxins and related compounds, Oxford, London, Pergamon Press, pp.
25-36.
HARLESS, R.L., OSWALD, E.O., LEWIS, R.G., DUPUY, A.E., MCDANIEL, D.D.,
& TAI, H. (1982) Determination of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in fresh water fish. Chemosphere, 11, 193-198.
HARLESS, R.L., LEWIS, R.G., DUPUY, A.E., & MCDANIEL, D.D. (1983)
Analyses for 2,3,7,8-tetrachlorodibenzo-p-dioxin residues in
environmental samples. In: Tucker, R.E., ed. Human and
environmental risks of chlorinated dioxins and related compounds,
New York, London, Plenum Press, pp. 161-172.
HARRIS, M.W., MOORE, J.A., VOS, J.G., & GUPTA, B.N. (1973) General
biological effects of TCDD in laboratory animals. Environ. Health
Perspect., 5: 101-109.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1983) Comparative ability
of TCDD to induce lipid peroxidation in rats, guinea pigs and syrian
golden hamsters. Bull. environ. Contam. Toxicol., 31: 649-657.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1985a) Inhibition of
TCDD-induced lipid peroxidation, glutathione peroxidase activity and
toxicity by BHA and glutathione. Bull. environ. Contam. Toxicol.,
34: 787-796.
HASSAN, M.Q., STOHS, S.J., & MURRAY, W.J. (1985b) Effects of vitamins
E and A on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced lipid
peroxidation and other biochemical changes in the rat. Arch. environ.
Contam. Toxicol., 14: 437-442.
HASSAN, M.Q., STOHS, S.J., MURRAY, W.J., & BIRT,D.F. (1985c) Dietary
selenium, glutathione peroxidase activity, and toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Toxicol. environ. Health,
15: 405-415.
HASSOUN, E.M. & DENCKER, L. (1982) TCDD embryotoxicity in the mouse
may be enhanced by beta-naphtoflavone, another ligand of the
Ah-receptor. Toxicol. Lett., 12: 191-198.
HASSOUN, E.M., D'ARGY, R., & DENCKER, L. (1984a) Teratogenicity of
2,3,7,8-tetrachlorodibenzofuran in the mouse. J. Toxicol. environ.
Health, 14: 337-351.
HASSOUN, E.M., D'ARGY, R., DENCKER, L., LUNDING, L.G., & BORWELL, P.
(1984b) Teratogenicity of 2,3,7,8-tetrachloro-dibenzofuran in BXD
recombinant inbred strains. Toxicol. Lett., 23: 37-42.
HAY, A. (1984) Experimental toxicology and cytogenetics: an overview.
In: Westing, A.N., ed. Herbicides in war, the long-term ecological
and human consequences, London, Taylor & Francis, pp. 161-166.
HAY, A.W.M. (1977) Tetrachlorodibenzo-p-dioxin release at Seveso.
Disasters, 1: 289-308.
HAYABUCHI, H., YOSHIMURA, T., & KURATSUNE, M. (1979) Consumption of
toxic rice oil by Yusho patients and its relation to the clinical
response and latent period. Food Cosmet. Toxicol., 17: 455-461.
HAYES, K.C. (1971) On the pathophysiology of vitamin a deficiency.
Nutr. Rev., 29: 3-6.
HELLING, C.S., ISENSEE, A.R., WOOLSON, E.A., ENZOR, P.D.J., JONES,
G.E., PLIMMER, J.R., & KEARNEY, P.C. (1973) Chlorodioxins in
pesticides, soils and plants. J. environ. Qual., 2: 171-178.
HENCK, J.M., NEW, M.A., KOBICA, R.J., & RAO, K.S. (1981)
2,3,7,8-tetrachlorodibenzo-p-dioxin: acute oral toxicity in hamsters.
Toxicol. appl. Pharmacol., 59: 405-407.
HERXHEIMER, K. (1899) [Chloracne.] Münchenere med. Wochenschr.,
46: 278 (in German).
HERZBERG, J.J. (1947) [Chloracne after ingestion of chlorinated
paraffin.] Dermatol. Wochenschr., 7: 425-433 (in German).
HINSDILL, R.D., COUCH, D.L., & SPEIRS, R.S. (1980) Immunosuppression
in mice induced by dioxin (TCDD) in feed. J. environ. Pathol.
Toxicol., 3: 401-425.
HIROSAWA, K. & YAMADA, E. (1973). The localization of the vitamin A in
the mouse liver as revealed by electron microscope radioautography.
J. electron. Microsc., 22: 337-346.
HOFFMAN, R.E., STEHR-GREEN, P.A., WEBB, K.B., EVANS, R.G., KNUTSEN,
A.P., SCHRAMM, W.F., STAAKE, J.L., GIBSON, B.B., & STEINBERG, K.K.
(1986) Health effects of long-term exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Am. Med. Assoc., 255:
2031-2038.
HOFMANN, H.Th. (1957) [Recent findings with highly toxic chlorinated
hydrocarbons.] Arch. exp. Pathol. Pharmakol., 232: 228-230 (in
German).
HOFMANN, M.F. & MENEGHINI, C.L. (1962) [Folliculosis from
chlorine-substituted hydrocarbons (chloracne).] G. Ital. Dermatol.,
103: 427-450 (in Italian).
HOLMSTEDT, B. (1980) Prolegomena to Seveso. Arch. Toxicol., 44:
211-230.
HONCHAR, P.A. & HALPERIN, W.E. (1981) 2,4,5-trichlorophenol and
soft-tissue sarcoma. Lancet, 21: 268-269.
HOOK, G.E.R., HASEMAN, J.K., & LUCIER, G.W. (1975a) Induction and
suppression of hepatic and extrahepatic microsomal
foreign-compound-metabolizing enzyme systems by
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Chem.-biol. Interact., 10:
199-214.
HOOK, G.E.R., ORTON, T.C., MOORE, J.A., & LUCIER, G.W. (1975b)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced changes in the
hydroxylation of biphenyl by rat liver microsomes. Biochem.
Pharmacol., 24: 335-340.
HORI, S., OBANA, H., TANAKA, R., & KASHIMOTO, T. (1986) Comparative
toxicity in rats of polychlorinated biphenyls (PCBs), polychlorinated
quaterphenyls (PCQs) and poly-chlorinated dibenzofurans (PCDFs)
present in rice oil causing "Yusho". Eisei Kagaku, 32: 13-21.
HRYHORCZUK, D.O., WITHROW, W.A., HESSE, C.S., & BEASLEY, V.R. (1981)
A wire reclamation incinerator as a source of environmental
contamination with tetrachlorodibenzo-p-dioxins and
tetrachlorodibenzofurans. Arch. environ. Health, 36: 228-234.
HSU, S., MA, C., HSU, S.K., WU, S., HSU, N.H., & YEH, C. (1984)
Discovery and epidemiology of PCB poisoning in Taiwan. Am. J. ind.
Med., 5: 71-79.
HUDSON, L.G., SHAIKH, R., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1983)
Induction of 7-ethoxycoumarin o-deethylase activity in cultured human
epithelial cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD):
Evidence for TCDD receptor. Biochem. biophys. Res. Commun., 115:
611-617.
HUDSON, L.G., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1985) Regulation of
epidermal growth factor binding in a human keratinocyte cell line by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
77: 251-259.
HUDSON, L.G., TOSCANO, W.A., Jr, & GREENLEE, W.F. (1986)
2,3,7,8-Tetrachorodibenzo-p-dioxin (TCDD) modulates epidermal growth
factor (EGF) binding to basal cells from a human keratinocyte cell
line. Toxicol. appl. Pharmacol., 82: 481-492.
HUFF, J.E., MOORE, J.A., SARACCI, R., & TOMATIS, L. (1980) Long-term
hazards of polychlorinated dibenzodioxins and polychlorinated
dibenzofurans. Environ. Health Perspect., 36: 221-240.
HUQUE, T. (1981) Excretion of radioactive metabolites of retinal as an
index of vitamin A status in rats. Nutr. Rep. Int., 24: 171-179.
HUSSAIN, S., EHRENBERG, L., LOFROTH, G., & GEJVALL, T. (1972)
Mutagenic effects of TCDD on bacterial systems. Ambio, 1: 32-33.
HUTZINGER, O., SAFE, S., WENTZELL, B.R., & ZITKO, V. (1973)
Photochemical degradation of di-and octachlorodibenzofuran. Environ
Health Perspect., 5: 267-271.
HWANG, S.W. (1973) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on
the biliary excretion of indocyanine Green in rat. Environ. Health
Perspect., 5: 227-231.
IARC (1977) Some fumigants, the herbicides 2,4-D and 2,4,5-T,
chlorinated dibenzodioxins and miscellaneous industrial chemicals,
Lyon, France, International Agency for Research on Cancer, pp. 41-102
(IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Vol. 15).
IARC (1982) Chemicals, industrial processes and industries
associated with cancer in humans, Lyon, France, International Agency
for Research on Cancer (IARC Monograph on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Suppl. No. 4).
IARC (1986) Some halogenated hydrocarbons and pesticide exposures,
Lyon, International Agency for Research on Cancer (IARC Monographs on
the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol.
41).
IDEO, G., BELLATI, G., BELLOBUONO, A., MOCARELLI, P., MAROCCHI, A., &
BRAMBILLA, P. (1982) Increased urinary D-glucaric acid excretion by
children living in an area polluted with
tetrachlorodibenzo-para-dioxin (TCDD). Clin. Chim. Acta, 120:
273-283.
IDEO, G., BELLATI, G., BELLOBUONO, A., & BISSANTI, L. (1985) Urinary
d-glucaric acid excretion in the Seveso area, polluted by
tetrachlorodibenzo-p-dioxin (TCDD): Five years of experience.
Environ. Health Perspect., 60: 151-157.
INNAMI, S.I., NAKAMURA, A., MIYAZAKI, M., NAGAYAMA, S., & NISHIDE, E.
(1976) Further studies on the reduction of vitamin A content in the
liver of rats given polychlorinated biphenyls. J. nutr. Sci.
Vitaminol., 22: 409-418.
IOANNOU, Y.M., BIRNBAUM, L.S., & MATTHEWS, H.B. (1983) Toxicity and
distribution of 2,3,7,8-tetrachlorodibenzofuran in male guinea pigs.
J. Toxicol. environ. Health, 12: 541-553.
IRPTC (1987) IRPTC Legal file 1986, Geneva, International Register
of Potentially Toxic Chemicals, United Nations Environment Programme,
Geneva, Switzerland, Vol. I (Data Profile Series No. 7).
ISENSEE, A.R. (1978) Bioaccumulation of
2,3,7,8-tetrachloro-dibenzo-para-dioxin. Ecol. Bull. (Stockholm),
27: 255-262.
ISENSEE, A.R. & JONES, G.E. (1971) Absorption and trans-location of
root and foliage applied 2,4-dichlorophenol,
2,7-dichlorodibenzo-p-dioxin and 2,3,7,8-tetrachlorodibenzo-p-dioxin.
J. agric. food. Chem., 19: 1210-1214.
ISENSEE, A.R. & JONES, G.E. (1975) Distribution of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in aquatic model ecosystem.
Environ. Sci. Technol., 9: 668-672.
ISRAEL, D.I. & WHITLOCK, J.P., Jr (1983) Induction of mRNA specific
for cytochrome P1-450 in wild type and variant mouse hepatoma cells.
J. biol. Chem., 258: 10390-10394.
ISRAEL, D.I. & WHITLOCK, J.P., Jr (1984) Regulation of cytochrome
P1-450 gene transcription by 2,3,7,8-tetrachloro-dibenzo-p-dioxin in
wild type and variant mouse hepatoma cells. J. biol. Chem., 259:
5400-5402.
JANSING, R.L. & SHAIN, W. (1985) Aryl hydrocarbon hydroxylase
induction in adult rat hepatocytes in primary culture by several
chlorinated aromatic hydrocarbons including
2,3,7,8-tetrachloridibenzo-p-dioxin. Fundam. appl. Toxicol., 5:
713-720.
JENSEN, D.J. & HUMMEL, R.A. (1982) Secretion of TCDD in milk and cream
following the feeding of TCDD to lactating dairy cows. Bull.
environ. Contam. Toxicol., 29: 440-446.
JENSEN, D.J., GETZENDANER, M.E., HUMMEL, R.A., & TURLEY, J. (1983)
Residue studies for (2,4,5-trichlorophenoxy)acetic acid and
2,3,7,8-tetrachlorodibenzo-p-dioxin in grass and rice. J. agric.
food Chem., 31: 118-122.
JENSEN, N.E. & WALKER, A.E. (1972) Chloracne: Three cases. Proc. R.
Soc. Med., 65: 687-688.
JIRASEK, L., KALENSKY, J., & KUBEC, K. (1973) [Acne chlorine and
porphyria-cutanea tarda by the production of herbicides.] Cs.
dermatol., 48: 306-317 (in Czech).
JIRASEK, L., KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS, E.
(1976) [Chloracne, porphyria-cutanea tarda and other intoxication by
herbicides.] Hautarzt, 27: 328-333 (in German).
JOHANSSON, G., GILLNER, M., HOGBERG, B., & GUSTAFSSON, J.-A. (1982)
The TCDD receptor in rat intestinal mucosa and its possible dietary
ligands. Nutr. Cancer, 3: 134-143.
JOHNSON, E.F. & MULLER-EBERHARD, U. (1977a) Purification of the major
cytochrome P-450 of liver microsomes from rabbits treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Biochem. biophys. Res.
Commun., 76: 652-659.
JOHNSON, E.F. & MULLER-EBERHARD, U. (1977b) Resolution of two forms of
cytochrome P-450 from liver microsomes of rabbits treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. biol. Chem., 252:
2839-2845.
JOHNSON, E.F., SCHWAB, G.E., & MULLER-EBERHARD, U. (1979) Multiple
forms of cytochrome P-450: Catalytic differences exhibited by two
homogeneous forms of rabbit cytochrome P-450. Mol. Pharmacol., 15:
708-718.
JOHNSON, F.E., KUGLER, N.A., & BROWN, S.M. (1981) Soft tissue sarcomas
and chlorinated phenols. Lancet, 1: 40.
JONES, D., SAFE, S., MORCOM, E., HOLCOMB, C., COPPOCK, C., & IVIE, W.
(1987) Bioavailability of tritiates
2,3,7,8-tetra-chlorodibenzo-p-dioxin administered to Holstein dairy
cows. Chemosphere, 16: 1743-1748.
JONES, E.L. & KRIZEK, H. (1962) A technic for testing acnegenic
potency in rabbits, applied to the potent acnegen,
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. invest. Dermatol., 39:
511-517.
JONES, G. (1975) A histochemical study of the liver lesion induced by
2,3,7,8-tetrachlorodibenzo-p-dioxin (dioxin) in rats. J. Pathol.,
116: 101-105.
JONES, G. & BUTLER, W.H. (1974) A morphological study of the liver
lesion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. J.
Pathol., 112: 93-97.
JONES, G. & GREIG, J.B. (1975) Pathological changes in the liver of
mice given 2,3,7,8-tetrachlorodibenzo-p-dioxin. Experientia (Basel),
31: 1315-1317.
JONES, K.G. & SWEENEY, G.D. (1977) Association between induction of
aryl hydrocarbon hydroxylase and depression of uroporphyrinogen
decarboxylase activity. Res. Commun. chem. Pathol. Pharmacol., 17:
631-637.
JONES, K.G. & SWEENEY, G.D. (1980) Dependence of the porphyrogenic
effect of 2,3,7,8-tetrachlorodibenzo(p)dioxin upon inheritance of aryl
hydrocarbon hydroxylase responsiveness. Toxicol. appl. Pharmacol.,
53: 42-49.
JONES, P.A. (1981) Chlorophenols and their impurities in the
Canadian environment, Ottawa, Environment Canada (EPS 3-EC-81-2).
JONES, P.A. (1984) Chlorophenols and their impurities in the
Canadian environment: 1983 supplement, Ottawa, Environment Canada
(EPS 3-EP-84-3).
JONES, P.B.C., MILLER, A.G., ISRAEL, D.I., GALEAZZI, D.R., & WHITLOCK,
J.P., Jr (1984) Biochemical and genetic analysis of variant mouse
hepatoma cells which overtranscribe the cytochrome P1-450 gene in
response to 2,3,7,8-tetrachloro-dibenzo-p-dioxin. J. biol. Chem.,
259: 12357-12363.
JONES, P.B.C., GALEAZZI, D.R., & WHITLOCK, J.P., Jr (1985) Control of
cytochrome P1-450 gene expression by dioxin. Science, 227:
1499-1502.
JONES, P.B.C., DURRIN, L.K., GALEAZZI, D.R., & WHITLOCK, J.P., Jr
(1986) Control of cytochrome P1-450 gene expression: Analysis of a
dioxin-responsive enhancer system. Proc. Natl Acad. Sci. (USA),
83: 2802-2806.
JONES, R.E. & CHELSKY, M. (1986) Further discussion concerning
porphyria cutanea tarda and TCDD exposure. Arch. environ. Health,
41: 100-103.
JOSEPHSON, J. (1983) Chlorinated dioxins and furans in the
environment. Environ. Sci. Technol., 17: 124A-128A.
JUNK, G.A. & RICHARD, J. (1981) Dioxins not detected in effluents from
coal/refuse combustion. Chemosphere, 10: 1237-1241.
KARASEK, F.W. & ONUSKA, F.I. (1982) Trace analysis of the dioxins.
Anal. Chem., 54: 309A-324A.
KARENLAMPI, S.O., EISEN, H.J., HANKINSON, O., & NEBER, D.W. (1983)
Effects of cytochrome Pl-450 inducers on the cell-surface receptors
for epidermal growth factor, phorbol 12,13-dibutyrate, or insulin of
cultured mouse hepatoma cells. J. biol. Chem., 17: 10378-10383.
KEARNEY, P.C., WOOLSON, E.A., & ELLINGTON, C.P., Jr (1972) Persistence
and metabolism of chlorodioxins in soils. Environ. Sci. Technol.,
6: 1017-1019.
KEESEY, R.E., BOYLE, P.C., KEMNITZ, J.W., & MITCHEL, J.S. (1976) The
role of the lateral hypothalamus in determining the body weight set
point. In: Novin, D., Wyrwicks, W., & Bray, G., ed. Hunger: Basic
mechanisms and clinical implications, New York, Raven Press, pp.
243-255.
KELLING, C.K., CHRISTIAN, B.J., INHORN, S.L., & PETERSON, R.E. (1985)
Hypophagia-induced weight loss in mice, rats, and guinea pigs treated
with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. appl. Toxicol.,
5: 700-712.
KENDE, A.S., WADE, J.J., RIDGE, D., & POLAND, A. (1974) Synthesis and
fourier transform carbon - 13 nuclear magnetic resonance spectroscopy
of new toxic polyhalodibenzo-p-dioxins. J. org. Chem., 39:
931-937.
KERKVLIET, N.I., BRAUNER, J.A., & MATLOCK, J.P. (1985) Humoral
immunotoxicity of polychlorinated diphenyl ethers, phenoxy-phenols,
dioxins and furans present as contaminants of technical grade
pentachlorophenol. Toxicology, 36: 307-324.
KEYS, B., HLAVINKA, M., MASON, G., & SAFE, S. (1985) Modulation of rat
hepatic microsomal testosterone hydroxylases by
2,3,7,8-tetrachlorodibenzo-p-dioxin and related toxic isostereomers.
Can. J. Pharmacol., 63: 1537-1542.
KEYS, B., PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1986)
Polychlorinated dibenzofurans as 2,3,7,8-TCDD antagonists: in
vitro inhibition of monooxygenase enzyme induction. Toxicol.
Lett., 31: 151-158.
KHERA, K.S. & RUDDICK, J.A. (1973) Polychlorodibenzo-p-dioxins:
Perinatal effects and the dominant lethal test in Wistar rats. Adv.
Chem. Ser., 120: 70-84.
KIMBLE, B.J. & GROSS, M.L. (1980) Tetrachlorodibenzo-p-dioxin
quantitation in stack-collected coal fly ash. Science, 207: 59-61.
KIMBROUGH, R.D. (1974) The toxicity of polychlorinated polycyclic
compounds and related chemicals. CRC Crit. Rev. Toxicol., 2:
445-498.
KIMBROUGH, R.D. (1979) The carcinogenic and other chronic effects of
persistent halogenated organic compounds. Ann. N.Y. Acad. Sci.,
320: 415-418.
KIMBROUGH, R.D. (1984) The epidemiology and toxicology of TCDD. Bull.
environ. Contam. Toxicol., 33: 636-647.
KIMBROUGH, R.D., GAINES, T.B., & LINDER, R.E. (1974)
2,4-Dichlorophenyl-p-nitrophenyl ether (TOK). Effects on the lung
maturation of rat fetus. Arch. environ. Health, 28: 316-319.
KIMBROUGH, R.D., CARTER, C.D., LIDDLE, J.A., CLINE, R.E., & PHILLIPS,
P.E. (1977) Epidemiology and pathology of a tetrachlorodibenzodioxin
poisoning episode. Arch. environ. Health, 32: 77-86.
KIMBROUGH, R.D., FALK, H., & STEHR, P. (1984) Health implications of
2,3,7,8-tetrachlorodibenzodioxin (TCDD) contamination of residential
soil. J. Toxicol. environ. Health, 14: 47-93.
KIMMIG, J. & SCHULZ, K.H. (1957a) [Occupational acne (so called
chloracne) caused by chlorinated aromatic cyclic ethers.]
Dermatologica, 115: 540-546 (in German).
KIMMIG, J. & SCHULZ, K.H. (1957b) [Chlorinated aromatic cyclic ethers
as cause of chloracne.] Naturwissenschaften, 11: 337-338 (in
German).
KIMURA, S., GONZALEZ, F.J., & NEBERT, D.W. (1986) Tissue-specific
expression of the mouse dioxin-inducible P1450 and P3450 genes:
Differential transcriptional activation and mRNA stability in liver
and extrahepatic tissues. Mol. cell. Biol., 6: 1471-1477.
KING, F.G., DEDRICK, R.L., & COLLINS, J.M. (1983) Physiological model
for the pharmacokinetics of 2,3,7,8-tetrachloro-dibenzofuran in
several species. Toxicol. appl. Pharmacol., 67: 390-400.
KITCHIN, K.T. & WOODS, J.S. (1979) 2,3,7,8-Tetrachlorodibenzo-p-dioxin
(TCDD) effects on hepatic microsomal cytochrome P-448-mediated enzyme
activities. Toxicol. appl. Pharmacol., 47: 537-546.
KLEU, G. & GOLTZ, R. (1971) [Late and lasting damage resulting from
the chronic occupational action of chlorophenol compounds.] Med.
Klin., 66: 53-58 (in German).
KNUTSON, J.C. & POLAND, A. (1980a)
2,3,7,8-Tetrachlorodibenzo-p-dioxin: failure to demonstrate toxicity
in twenty-three cultured cell types. Toxicol. appl. Pharmacol.,
54: 377-383.
KNUTSON, J.C. & POLAND, A. (1980b) Keratinization of mouse teratoma
cell line XB produced by 2,3,7,8-tetrachlorodibenzo-p-dioxin: an in
vitro model of toxicity. Cell, 22: 27-36.
KNUTSON, J.C. & POLAND, A. (1982) Response of murine epidermis to
2,3,7,8-tetrachlorodibenzo-p-dioxin: Interaction of the Ah and hr
Loci. Cell, 30: 225-234.
KNUTSON, J.C. & POLAND, A. (1984) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Examination of biochemical effects involved in the proliferation and
differentiation of XB cells. J. cell. Physiol., 121: 143-151.
KOCHER, C.W., MAHLE, N.H., HUMMEL, R.A., & SHADOFF, L.A. (1978) A
search for the presence of 2,3,7,8-tetrachloro-dibenzo-p-dioxin in
beef fat. Bull. environ. Contam. Toxicol., 19: 229-236.
KOCHMAN, S., BERNARD, J., CAZABAT, A., LAVAUD, F., LORTON, C., &
RAPPE, C. (1986) Phenotypical dissection of immunoregulatory T cell
subsets in human after furan exposure. Chemosphere, 15: 1799-1804.
KOCIBA, R.J., KEELER, P.A., PARK, C.N., & GEHRING, P.J. (1976)
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD): Results of a 13-week oral
toxicity study in rats. Toxicol. appl. Pharmacol., 35: 553-574.
KOCIBA, R.J., KEYES, D.G., BEYER, J.E., CARREON, R.M., WADE, C.E.,
DITTENBER, D., KALNINS, R., FRAUSON, L., PARK, C.N., BARNARD, S.,
HUMMEL, R., & HUMISTON, C.G. (1978) Results of a two year chronic
toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin
(TCDD) in rats. Toxicol. appl. Pharmacol., 46: 279-303.
KOCIBA, R.J., KEYES, D.G., LISOWE, R.W., KALNINS, R.P., DITTENBER,
D.D., WADE, C.E., GORZINSKI, S.J., HAHLE, N.H., & SCHWETZ, B.A.
(1979a) Results of a two-year chronic toxicity and encogenic study of
rats ingesting diets containing 2,4,5-trichlorophenoxyacetic acid
(2,4,5-T). Food Cosmet. Toxicol., 17: 205-221.
KOCIBA, R.J., KEYES, D.G., BEYER, J.E., CARREON, R.M., & GEHRING, P.J.
(1979b) Long-term toxicologic studies of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in laboratory animals.
Ann. N.Y. Acad. Sci., 320: 397-404.
KOHLI, K.K. & GOLDSTEIN, J.A. (1981) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin on hepatic and renal
prostaglandin synthetase. Life Sci., 29: 299-305.
KONDOROSI, A., FEDORCSAK, I., SOLYMOSY, F., EHRENBERG, L., &
OSTERMAN-GOLKAR, S. (1973) Inactivation of Q-beta RNA by
electrophiles. Mutat. Res., 17: 149-161.
KOSHAKJI, R.P., HARBISON, R.D., & BUSH, M.T. (1984) Studies on the
metabolic fate of (14C)2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in
the mouse. Toxicol. appl. Pharmacol., 73: 69-77.
KOURI, R.E., RATRIE, H., III, ATLAS, C.A., NIWA, A., & NEBERT, D.W.
(1974) Aryl hydrocarbon hydroxylase induction in human lymphocyte
cultures by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Life Sci., 15:
1585-1595.
KOURI, R.E., RUDE, T.H., & JOGLEKAR, R. (1978)
2,3,7,8-Tetra-chlorodibenzo-p-dioxin as cocarcinogen causing
3-methylchol-antrene-initiated subcutaneous tumors in mice genetically
"non responsive" at Ah locus. Cancer Res., 38: 2777-2783.
KRAUSE, L., VON & BRASSOW, H. (1978) [Contamnestic study of chloracne
cases from the year 1954/55.] Arbeitsmed. Socialmed.
Präventivmed., 13: 19-21 (in German).
KROWKE, R. (1986) Studies on distribution and embryotoxicity of
different PCDD and PCDF in mice and marmosets. Chemosphere, 15:
2011-2022.
KUNITA, N., KASHIMATO, T., MIYATA, H., FUKUSHIMA, S., HARI, S., &
OBANA, H. (1984) Causal agents of Yusho. Am. J. ind. Med., 5:
45-58.
KUNTZMAN, D., LAWRENCE, D., & CONNEY, A.H. (1965) Michaelis constants
for the hydroxylation of steroid hormones and drugs by rat liver
microsomes. Mol. Pharmacol., 1: 163-167.
KURL, R.N. & VILLEE, C.A. (1985) A metabolite of riboflavin binds to
the 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) receptor.
Pharmacology, 30: 241-244.
KURL, R.N., LUND, J., POELLINGER, L., & GUSTAFSSON, J-A. (1982)
Differential effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on nuclear
RNA polymerase activity in the rat liver and thymus. Biochem.
Pharmacol., 31: 2459-2462.
KURL, R.N., LORING, J.M., & VILLEE, C.A. (1985) Control of
2,3,7,8-tetrachlorodibenzo-p-dioxin binding protein(s) in the hamster
kidney. Pharmacology, 30: 245-254.
KUROKI, H., MASUDA, Y., YOSHIHARA, S., & YOSHIMURA, H. (1980)
Accumulation of polychlorinated dibenzofurans in the livers of monkeys
and rats. Food Cosmet. Toxicol., 18: 387-392.
KUROKI, H., HARAGUCHI, K., & MASUDA, Y. (1984) Synthesis of
polychlorinated dibenzofuran isomers and their gas chromato-graphic
profiles. Chemosphere, 13: 561-573.
KUROKI, J., KOGA, N., & YOSHIMURA, H. (1986) High affinity of
2,3,4,7,8-pentachloridibenzofuran to cytochrome P-450 in the hepatic
microsomes of rats. Chemosphere, 15: 731-738.
LAHANIATIS, E.S., PARLAR, H., & KORTE, F. (1977) [Contributions to
ecological chemistry CXXXII. On the occurrence of chlorinated
hydrocarbons in the flue dust of refuse incinerators.] Chemosphere,
7: 11-16 (in German).
LAMB, J.C., MARKS, T.A., GLADEN, B.C., ALLEN, J.W., & MOORE, J.A.
(1981) Male fertility, sister chromatid exchange, and germ cell
toxicity following exposure to mixtures of chlorinated phenoxy acids
containing 2,3,7,8-tetrachloro-dibenzo-p-dioxin. J. Toxicol.
environ. Health, 8: 825-834.
LAMB, J.C., IV, HARRIS, M.W., MCKINNEY, J.D., & BIRNBAUM, L.S. (1986)
Effects of thyroid hormones on the induction of cleft palate by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57Bl/6N mice. Toxicol.
appl. Pharmacol., 84: 115-124.
LAMPARSKI, L.L. & NESTRICK, T.J. (1981) Synthesis and identification
of the 10 hexachlorodibenzo-p-dioxin isomers by high performance
liquid and packed column gas chromatography. Chemosphere, 10:
3-18.
LAMPARSKI, L.L., NESTRICK, T.J., & STEHL, R.H. (1979) Determination of
part-per-trillion concentration of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in fish. Anal. Chem., 51: 1453-1458.
LAMPARSKI, L.L., NESTRICK, T.J., FRAWLEY, N.N., HUMMEL, R.A., KOCKER,
C.W., MAHLE, N.H., MCCOY, J.W., MILLER, D.L., PETERS, T.L., PILLIPICH,
J.L., SMITH, W.E., & TOBEY, S.W. (1986) Perspectives of a large scale
environmental survey for chlorinated dioxins: Water analyses.
Chemosphere, 15: 1445-1452.
LANGER, H.G., BRADEY, T.P., & BRIGGS, P.R. (1973) Formation of
dibenzodioxins and other condensation products from chlorinated
phenols and derivatives. Environ. Health Perspect., 5: 3-7.
LATHROP, G.D., WOLFE, W.H., ALBANESE, R.A., & MOYANAHAN, P.M. (1984)
An epidemiologic investigation of health effects in air force
personnel following exposure to herbicides. Baseline morbidity
study results, San Antonio, Texas, USAF School of Aerospace Medicine
(EK), Aerospace Medical Division, Brooks Air Force Base.
LEE, P. & SUZUKI, K. (1980) Induction of aryl hydrocarbon hydroxylase
activity in the rat prostate glands by
2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Pharmacol. exp. Ther.,
215: 601-605.
LIEM, H.H., MULLER-EBERHARD, U., & JOHNSON, E.F. (1980) Differential
induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin of multiple forms of
rabbit microsomal cytochrome P-450: Evidence for tissue specificity.
Mol. Pharmacol., 18: 565-570.
LIGON, W.V., Jr & MAY, R.J. (1986) Determination of selected
chlorodibenzofurans and chlorodibenzodioxins using two-dimensional gas
chromatography/mass spectrometry. Anal. Chem., 58: 558-561.
LINDAHL, R., RAPPE, C., & BUSER, H.R. (1980) Formation of
polychlorinated dibenzofurans (PCDFs) and polychlorinated
dibenzo-p-dioxins (PCDDs) from the pyrolysis of poly-chlorinated
diphenyl ethers. Chemosphere, 9: 351-361.
LINDAHL, R., ROPER, M., & DIETRICH, R.A. (1978) Rat liver aldehyde
dehydrogenase-immunochemical identity of
2,3,7,8-tetrachlorodibenzo-p-dioxin inducible normal liver and
2-acetylaminofluorene inducible hepatoma isozymes. Biochem.
Pharmacol., 27: 2463-2465.
LOPRIENO, N., SBRANA, I., RUSCIANO, D., LASCIALFARI, D., & LARI, T.
(1982a) In vivo cytogenetic studies on mice and rats exposed to
2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Hutzinger, O., Frei, R.W.,
Merian, E., & Pocchiari, P., ed. Chlorinated dioxins and related
compounds. Impact on the environment, Oxford, New York, Pergamon
Press, pp. 419-428.
LOPRIENO, N., SBRANA, I., RUSCIANO, D., LASCIALFARI, D., LARI, T.,
STRETTI, G., & FREZZA, D. (1982b) In vitro and in vivo genotoxicity
studies on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). In: Report
of the 5th Meeting of the Seveso International Scientific Advisory
Committee, Milan, Regione Lombardia (Report No. 32).
LOVATI, M.R., GALBUSSERA, M., FRANCESCHINI, G., WEBER, G., RESI, L.,
TANGANELLI, P., & SIRTORI, C.R. (1984) Increased plasma and aortic
triglycerides in rabbits after acute administration of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
75: 91-97.
LU, C.-J.H., BAGGS, R.B., REDMOND, D., HENRY, E.C., SCHECTER, A., &
GASIEWICZ, T.A. (1986) Toxicity and evidence for meta-bolic
alterations in 2,3,7,8-tetrachlorodibenzo-p-treated guinea pigs fed by
total parenteral nutrition. Toxicol. appl. Pharmacol., 84:
439-453.
LUCIER, G.W., MCDANIEL, O.S., HOOK, G.E.R., FOWLER, B.A., SONAWANE,
B.R., & FAEDER, E. (1973) TCDD-induced changes in rat liver microsomal
enzymes. Environ. Health Perspect., 5: 199-209.
LUCIER, G.W., MCDANIEL, O.S., & HOOK, G.E.R. (1975a) Nature of the
enhancement of hepatic uridine diphosphate gluburonyl-transferase
activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Biochem.
Pharmacol., 24: 325-334.
LUCIER, G.W., SONAWANE, B.R., MCDANIEL, O.S., & HOOK, G.E.R. (1975b)
Postnatal stimulation of hepatic microsomal enzymes following
administration of TCDD to pregnant rats. Chem.-biol. Interact.,
11: 15-26.
LUCIER, G.W., RUMBAUGH, R.C., MCCOY, Z., HASS, R., HARVAN, D., &
ALBRO, P. (1986) Ingestion of soil contaminated with
2,3,7,8-tetrachlorodibenzop-dioxin (TCDD) alters hepatic enzyme
activities in rats. Fundam. appl. Toxicol., 6: 364-371.
LUSTENHOUWER, J.W.A., OLIE, K., & HUTZINGER, O. (1980) Chlorinated
dibenzo-p-dioxins and related compounds in incinerator effluents: A
review of measurements and mechanisms of formation. Chemosphere,
9: 501-522.
LUSTER, M.I., CLARK, G., LAWSON, L.D., & FAITH, R.E. (1979a) Effects
of brief in vitro exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxin
(TCDD) on mouse lymphocytes. J. environ. Pathol. Toxicol., 2:
965-977.
LUSTER, M.I., FAITH, R.E., & LAWSON, L.D. (1979b) Effects of
2,3,7,8-tetrachlorodibenzofuran (TCDF) on the immune system in guinea
pigs. Drug chem. Toxicol., 2: 49-60.
LUSTER, M.I., BOORMAN, G.A., DEAN, J.H., HARRIS, M.W., LUEBKE, R.W.,
PADARATHSINGH, M.L., & MOORE, J.A. (1980) Examination of bone marrow,
immunologic parameters and host susceptibility following pre- and
postnatal exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD).
Int. J. Immunopharmacol., 2: 301-310.
LUSTER, M.I., TUCKER, A.N., HONG, L., BOORMAN, G.A., & PATTERSON, R.
(1984) In vivo and in vitro effects of TCDD on stem cell and
B cell differentiation. In: Biological mechanisms of dioxin action,
Cold Spring Harbor, New York, Cold Spring Harbor Laboratory, pp.
411-417 (Banbury Report No. 18).
LUSTER, M.I., HONG, L.H., BOORMAN, G.A., CLARK, G., HAYES, H.T.,
GREENLEE, W.F., DOLD, K., & TUCKER, A.N. (1985) Acute myelotoxic
responses in mice exposed to 2,3,7,8-tetrachloro-dibenzo-p-dioxin
(TCDD). Toxicol. appl. Pharmacol., 81: 156-165.
MCCONNELL, E.E. (1980) Acute and chronic toxicity, carcinogenesis,
reproduction, teratogenesis and mutagenesis in animals. In: Kimbrough,
R.D., ed. Halogenated biphenyls, perphenyls, naphthalenes,
dibenzodioxins and related products, Amsterdam, Oxford, New York,
Elsevier Science Publishers, pp. 109-150.
MCCONNELL, E.E. & MOORE, J.A. (1979) Toxicopathology characteristis of
the halogenated aromatics. Ann. N.Y. Acad. Sci., 320: 138-150.
MCCONNELL, E.E., MOORE, J.A., & DALGARD, D.W. (1978a) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in Rhesus monkeys (Macaca
mulatta) following a single oral dose. Toxicol. appl. Pharmacol.,
43: 175-187.
MCCONNELL, E.E., MOORE, J.A., HASEMAN, J.K., & HARRIS, M.W. (1978b)
The comparative toxicity of chlorinated dibenzo-p-dioxins in mice and
guinea pigs. Toxicol. appl. Pharmacol., 44: 335-356.
MCCONNELL, E.E., LUCIER, G.W., RUMBAUGH, R.C., ALBRO, P.W., HARVAN,
D.J., HASS, J.R., & HARRIS, M.W. (1984) Dioxin in soil:
bioavailability after ingestion by rats and guinea pigs. Science,
223: 1077-1079.
MCCUNE, E.L., SAVAGE, J.E., & O'DELL, B.L. (1962) Hydro-pericardium
and ascites in chicks fed a chlorinated hydro-carbon. Poult. Sci.,
41: 295-299.
MCKINNEY, J., ALBRO, P., LUSTER, M., CORBETT, B., SCHROEDER, J., &
LAWSON, L. (1982) Development and reliability of a radioimmunoassay
for 2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Hutzinger, O., ed.
Chlorinated dioxins and related compounds: Impact on the
environment, Oxford, New York, Pergammon Press, pp. 67-77.
MCKINNEY, J.D. (1978) Analysis of
2,3,7,8-tetrachlorodibenzo-para-dioxin in environmental samples. Ecol.
Bull. (Stockholm), 27: 53-66.
MCKINNEY, J.D., CHAE, K., GUPTA, B.N., MOORE, J.A., & GOLDSTEIN, J.A.
(1976) Toxicological assessment of hexachlorobiphenyl isomers and
2,3,7,8-tetrachlorodibenzo- furan in chicks. I. Relationship of
chemical parameters. Toxicol. appl. Pharmacol., 36: 65-80.
MCKINNEY, J.D., FAWKES, J., JORDAN, S., CHAE, K., OATLEY, S., COLEMAN,
R.E., & BRINER, W. (1985) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)
as a potent and persistent thyroxine agonist: a mechanistic model for
toxicity based on molecular reactivity. Environ. Health Perspect.,
61: 41-53.
MCLAUGHLIN, D.L. & PEARSON, R.G. (1984) Concentrations of PCDD and
PCDF in soil from the vicinity of the SWARU incinerator, Stoney
Creek, July, 1983, Toronto, Ontario, Air Resources Branch, Ontario
Ministry of the Environment, 22 pp.
MCNULTY, W.P. (1984) Fetotoxicity of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) for rhesus macaques
(Macaca mulatta). Am. J. Primatol., 6: 41-47.
MCNULTY, W.P. (1985) Toxicity and fetotoxicity of TCDD, TCDF and PCB
isomers in rhesus macaques (Macaca mulatta). Environ. Health
Perspect., 60: 77-78.
MCNULTY, W.P., POMERANTZ, I., & FARRELL, T. (1981) Chronic toxicity of
2,3,7,8-tetrachlorodibenzofuran for rhesus macaques. Food Cosmet.
Toxicol., 19: 57-65.
MCNULTY, W.P., POMERANTZ, L.H., & FARRELL, T.J. (1982a) Chronic
toxicity of 2,3,7,8-tetrachlorodibenzofuran for rhesus macaques. In:
Hutzinger, O., ed. Chlorinated dioxins and related compounds:
Impact on the environment, Oxford, New York, Pergamon Press, pp.
411-418.
MCNULTY, W.P., NIELSEN-SMITH, K.A., & LAY, J.O., Jr (1982b)
Persistence of TCDD in monkey adipose tissue. Food Cosmet.
Toxicol., 20: 985-987.
MADGE, D.S. (1977) Effects of trichlorophenoxyacetic acid and
chlorodioxins on small intestinal function. Gen. Pharmacol., 8:
319-324.
MADHUKAR, B.V. & MATSUMURA, F. (1981) Differences in the nature of
induction of mixed-function oxidase systems of the rat liver among
phenobarbital, DDT, 3-methylcholanthrene, and TCDD. Toxicol. appl.
Pharmacol., 61: 109-118.
MADHUKAR, B.V., BREWSTER, D.W., & MATSUMURA, F. (1984) Effects of in
vivo-administered 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor
binding of epidermal growth factor in the hepatic plasma membrane of
rat, guinea pig, mouse, and hamster. Proc. Natl Acad. Sci. (USA),
81: 7407-7411.
MANARA, L., COCCIA, P., & CROCI, T. (1982) Persistent tissue levels of
TCDD in the mouse and their reduction as related to prevention of
toxicity. Drug Metab. Rev., 13(3): 423-446.
MANARA, L., COCCIA, P., & CROCI, T. (1984) Prevention of TCDD toxicity
in laboratory rodents by addition of charcoal or cholic acids to chow.
Food chem. Toxicol., 22(10): 815-818.
MANIS, J. & APAP, R. (1979) Intestinal organic anion transport,
glutathione transferase and aryl hydrocarbon hydroxylase activity:
effect of dioxin. Life Sci., 24: 1373-1380.
MANIS, J. & KIM, G. (1977) Induction of intestinal iron transport by
2,3,7,8-tetrachlorodibenzo-p-dioxin on environmental pollutant and
potent inducer of aryl hydrocarbon hydroxylase. Clin. Res., 25:
468A.
MANIS, J. & KIM, G. (1979) Introduction of iron transport by a potent
inducer of aryl hydrocarbon hydroxylase,
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Arch. environ. Health,
34(3): 141-145.
MANTOVANI, A., VECCHI, A., LUINI, W., SIRONI, M., CANDIANI, G.P.,
SPREAFICO, F., & GARATTINI, S. (1980) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on macrophage and natural killer
cell-mediated cytotoxicity in mice. Biomedicine, 32: 200-204.
MARKLUND, S., KJELLER, L.-O., HANSSON, M., TYSKLIND, M., RAPPE, C.,
RYAN, C., COLLAZO, H., & DOUGHERTY, R. (1986) Determination of PCDDs
and PCDFs in incineration samples and pyrolytic products. In: Rappe,
C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 79-92.
MARKLUND, S., RAPPE, C., TYSKLIND, M., & EGEBACK, K. (1987)
Identification of polychlorinated dibenzofurans and dioxins in
exhausts from cars run on unleaded gasoline. Chemosphere, 16:
29-36.
MARPLE, L., BRUNCK, R., & THROOP, L. (1986a) Water solubility of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Sci. Technol., 20:
180-182.
MARPLE, L., BERRIDGE, B., & THROOP, L. (1986b) Measurement of the
water-octanol partition coefficient of
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Environ. Sci. Technol., 20:
397-399.
MASON, G. & SAFE, S. (1986) Synthesis, biologic and toxic effects of
the major 2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites in the rat.
Toxicology, 41: 153-159.
MASON, G., SAWYER, T., KEYS, B., BANDIERA, S., ROMKES, M.,
PISKORSKA-PLISZCZYNSKA, J., ZMUDZKA, B., & SAFE, S. (1985)
Polychlorinated dibenzofurans (PCDFs): correlation between in vivo
and in vitro structure-activity relationships. Toxicology, 37:
1-12.
MASON, G., FARRELL, K., KEYS, B., PISKORSKA-PLISZCZYNSKA, J., SAFE,
L., & SAFE, S. (1986) Polychlorinated dibenzo-p-dioxins: Quantitative
in vitro and in vivo structure-activity relationships.
Toxicology, 41: 21-31.
MASON, M.E. & OKEY, A.B. (1982) Cytosolic and nuclear binding of
2,3,7,8-tetrachlorodibenzo-p-dioxin to the Ah receptor in
extra-hepatic tissues of rats and mice. Eur. J. Biochem., 123:
209-215.
MASUDA, Y., KUROKI, H., HARAGUCHI, K., & NAGAYAMA, J. (1985) PCB and
PCDF congeners in the blood and tissues of Yusho and Yu-Cheng
patients. Environ. Health Perspect., 59: 53-58.
MASUDA, Y., KUROKI, H., HARAGUCHI, K., & NAGAYAMA, J. (1986) PCDFs and
related compounds in humans from Yusho and Yu-Cheng incidents.
Chemosphere, 15: 1621-1628.
MATSUMURA, F. & BENEZET, H.J. (1973) Studies on the bioaccumu-lation
and microbial degradation of 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Environ. Health Perspect., 5: 253-258.
MATSUMURA, F., QUENSEN, J., & TSUSHIMOTO, G. (1983) Microbial
degradation of TCDD in a model ecosystem. In: Tucker, R.E., Young,
A.L., & Gray, A.P., ed. Human and environmental risks of
chlorinated dioxins and related compounds, New York, London, Plenum
Press, pp. 191-220.
MATSUMURA, F., BREWSTER, D.W., MADHUKAR, B.V., & BOMBICK, D.W. (1984)
Alteration of rat hepatic plasma membrane functions by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Arch. environ. Contam.
Toxicol., 13: 509-515.
MATTISON, D.R. & THORGEIRSSON, S.S. (1978) Gonadal aryl hydrocarbon
hydroxylase in rats and mice. Cancer Res., 38: 1368-1373.
MAY, G. (1973) Chloracne from the accidental production of
tetrachlorodibenzodioxin. Br. J. ind. Med., 30: 276-283.
MAY, G. (1982) Tetrachlorodibenzodioxin: a survey of subjects ten
years after exposure. Br. J. ind. Med., 39: 128-135.
MAZER, T., HILEMAN, F.D., NOBLE, R.W., & BROOKS, J.J. (1983) Synthesis
of the 38 tetrachlorodibenzofuran isomers and identification by
capillary column gas chromatography/mass spectrometry. Anal. Chem.,
55: 104-110.
MEYNE, J., ALLISON, D.C., BOSE, K., JORDAN, S.W., RIDOLPHO, P.F., &
SMITH, J. (1985) Hepatotoxic doses of dioxin do not damage mouse bone
marrow chromosomes. Mutat. Res., 157: 63-69.
MILES, W.F., SINGH, J., GURPRASAD, N.P., & MALIS, G.P. (1985) Isomer
specific determination of hexachlorodioxins in technical
pentachlorophenol (PCP) and its sodium salt. Chemosphere, 14(6/7):
807-810.
MILLER, A.G., ISRAEL, D., & WHITLOCK, J.P., Jr (1983) Bio-chemical and
genetic analysis of variant mouse hepatoma cells defective in the
induction of benzo(a)pyrene-metabolizing enzyme. J. biol. Chem.,
258: 3523-3527.
MILNES, M.H. (1971) Formation of 2,3,7,8-tetrachlorodibenzo-dioxin by
thermal decomposition of sodium 2,4,5-trichloro-phentane. Nature
(Lond.), 232: 395-396.
MILSTONE, L.M. & LAVIGNE, J.F. (1984)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin induces hyperplasia in confluent
cultures of human keratinocytes. J. invest. Dermatol., 82: 532-534.
MITCHUM, R.K., MOLER, G.F., & KORFMACHER, W.A. (1980) Combined
capillary gas chromatography/atmospheric pressure negative chemical
ionization/mass spectrometry for the determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin in tissue. Anal. Chem., 52:
2278-2282.
MITTLER, J.C., ERTEL, N.H., PENG, R.X., YANG, C.S., & KIERNAN, T.
(1984) Changes in testosterone hydroxylase activity in rat testis
following administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Ann.
N.Y. Acad. Sci., 438: 645-648.
MIVRA, H., OMORI, A., & SHIBUE, M. (1974) The effect of chlorophenols
on the excretion of porphyrins in urine. Jpn. J. ind. Health,
16: 575-577.
MOLLER, M.E., GLOWINSKI, I.B., & THORGEIRSSON, S.S. (1984) The
genotoxicity of aromatic amines in primary hepatocytes iso-lated from
C57BL/6 and DBA/2 mice. Carcinogenesis, 5: 797-804.
MOORE, J.A., GUPTA, B.N., ZINKL, J.G., & VOS, J.G. (1973) Postnatal
effects of maternal exposure to 2,3,7,8-tetrachloro-dibenzo-p-dioxins
(TCDD). Environ. Health Perspect., 5: 81-85.
MOORE, J.A., GUPTA, B.N., & VOS, J.G. (1976) Toxicity of
2,3,7,8-tetrachloro-dibenzofuran - preliminary results. In:
Proceedings from the National Conference on Polychlorinated
Biphenyls, Chicago, Illinois, 19-21 November, 1975, Research
Triangle Park, North Carolina, Research Triangle Park Institute.
MOORE, J.A., MCCONNELL, E.E., DALGARD, D.W., & HARRIS, M.W. (1979)
Comparative toxicity of three halogenated dibenzofurans in guinea
pigs, mice and rhesus monkeys. Ann. N.Y. Acad. Sci., 320: 151-163.
MOORE, R.W. & PETERSON, R.E. (1985) Enhanced catabolism and
elimination of androgens do not cause the androgenic de-ficiency in
2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Fed. Proc., 44:
518.
MOORE, R.W., POTTER, C.L., THEOBALD, H.M., ROBINSON, J.A., & PETERSON,
R.E. (1985) Androgenic deficiency in male rats treated with
2,3,7,8-tetra-chlorodbenzo-p-dioxin. Toxicol. appl. Pharmacol.,
79: 99-111.
MORITA, M. & OISHI, S. (1977) Clearance and tissue distribution of
polychlorinated dibenzofurans in mice. Bull. environ. Contam.
Toxicol., 18: 61-66.
MORITA, M., NAKAGAWA, J., & RAPPE, C. (1978) Polychlorinated
dibenzofuran (PCDF) formation from PCB mixture by heat and oxygen.
Bull. environ. Contam. Toxicol., 19: 665-670.
MORTELMANS, K., HAWORTH, S., SPECK, W., & ZEIGER, E. (1984)
Mutagenicity testing of agent orange components and related chemicals.
Toxicol. appl. Pharmarcol., 75: 137-146.
MOSES, M. & SELIKOFF, I.J. (1981) Soft tissue sarcomas, phenoxy
herbicides, and chlorinated phenols. Lancet, 1: 1370.
MOSES, M., LILIS, R., CROW, K.D., THORNTON, J., FISCHBEIN, A.,
ANDERSON, H.A., & SELIKOFF, I.J. (1984) Health status of workers with
past exposure to 2,3,7,8-tetrachloridibenzo-p-dioxin in manufacture of
2,4,5-trichlorophenoxyacetic acid: Comparison of findings with and
without chloracne. Am. J. ind. Med., 5: 161-182
MOTTURA, A., ZEI, G., NUZZO, F., CRIMAUDO, C., GIORGI, R., VENERONI,
P., PAGGINI, P., MACARELLI, P., FRACCARO, M., NICOLETTI, B., &
DECARLI, L. (1981) Evaluation of results of chromosome analyses on
lymphocytes of TCDD exposed subjects after the Seveso accident.
Mutat. Res., 85: 238-239.
MUKITARI, H., BAARS, A.J., & BREIMER, D.D. (1981) Differences in
inducibility of particulate and cytosolic rat liver glutathione
S-transferase activities. Xenobiotica, 11: 367-371.
MULCAHY, M.T. (1980) Correspondence. Chromosome aberrations and "agent
orange". Med. J. Aust., 2: 573-574.
MULLER, E. (1937) [Chloracne (caused by chlorinated benzenes).
Inaugural Dissertation,] Speyer am Rhein, Pilger-Druckerei (Thesis,
Friedrich-Wilhelm University, Breslau).
MURRAY, F.J., SMITH, F.A., NITSCHKE, K.D., HUMISTON, C.G., KOCIBA,
R.J., & SCHWETZ, B.A. (1979) Three-generation reproduction study of
rats given 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the diet.
Toxicol. appl. Pharmacol., 50: 241-252.
NAGARKATTI, P.S., SWEENEY, G.D., GAULDIE, J., & CLARK, D.A. (1984)
Sensitivity to suppression of cytotoxic T cells generation by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in dependent on the Ah
genotype of the murine host. Toxicol. appl. Pharmacol., 72:
169-176.
NAGAYAMA, J., KURATSUNE, M., & MASUDA, Y. (1976) Determination of
chlorinated dibenzofurans in kanechlors and "Yusho oil". Bull.
environ. Contam. Toxicol., 15: 9-13.
NAGAYAMA, J., NISHIZUMI, M., & MASUDA, Y. (1979) [Subacute toxicity of
polychlorinated dibenzofurans in mice.] Fukuoka Igakuzassh, 70:
109-113 (in Japanese).
NAGAYAMA, J., TOKUDOME, S., & KURATSUNE, M. (1980) Transfer of
polychlorinated dibenzofurans to the foetuses and offspring of mice.
Food Cosmet. Toxicol., 18: 153-157.
NAGAYAMA, J., KUROKI, H., MASUDA, Y., & KURATSUME, M. (1983) A
comparative study of polychlorinated dibenzofurans, poly-chlorinated
biphenyls and 2,3,7,8-tetrachlorodibenzo-p-dioxin on aryl hydrocarbon
hydroxylase inducing potency in rats. Arch. Toxicol., 53: 177-184.
NAGAYAMA, J., KUROKI, H., MASUDA, Y., HANDA, S., & KURATSUNE, M.
(1985a) Genetically mediated induction of aryl hydrocarbon hydroxylase
activity in mice by polychlorinated dibenzofuran isomers and
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Toxicol., 56: 226-229.
NAGAYAMA, J., KIYOHARA, C., KURATSUNE, M., & MASUDA, Y. (1985b)
Induction of aryl hydrocarbon hydroxylase activity in human
lymphoblastoid cells by chlorinated dibenzofuran isomers and
2,3,7,8-tetrachlorodibenzo-p-dioxin. In: Keith, L.H., Rappe, C., &
Choudhary, G., ed. Chlorinated dioxins and dibenzofurans in the
total environment II, Stoneham, Maine, Butterworth Publishers, pp.
285-295.
NAU, H. & BASS, R. (1981) Transfer of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) to the mouse embryo and
fetus. Toxicology, 20: 299-308.
NAU, H., BASS, R., & NEUBERT, D. (1986) Transfer of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) via placenta and milk, and
postnatal toxicity in the mouse. Arch. Toxicol., 59: 36-40.
NCI (1977) Bioassay of dibenzo-p-dioxin for possible
carcinogenicity, Washington, DC, National Cancer Institute, 104 pp
(Technical Report No. 122).
NCI (1979) Bioassay of 2,7-dichlorodibenzo-p-dioxin (DCDD) for
possible carcinogenicity, Washington, DC, National Cancer Institute,
103 pp (Technical Report No. 123).
NEAL, R.A., BEATTY, P.W., & GASIEWICZ, T.A. (1979) Studies of the
mechanisms of toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
Ann. N.Y. Acad. Sci., 320: 204-213.
NEBERT, D.W., ROBINSON, J.R., NIWA, A., KUMAKI, K., & POLAND, A.P.
(1975) Genetic expression of aryl hydrocarbon hydroxylase activity in
the mouse. J. cell. Physiol., 85: 393-414.
NEBERT, D.W., JENSEN, N.M., PERRY, J.W., & OKA, T. (1980) Association
between ornithine decarboxylase induction and the Ah locus in mice
treated with polycyclic aromatic compounds. J. biol. Chem., 255:
6836-6842.
NESTRICK, T.J., LAMPARSKI, L.L., & STEHL, R.H. (1979) Synthesis and
identification of the 22 tetrachlorodienzo-p-dioxin isomers by high
performance liquid chromatography and gas chromatography. Anal.
Chem., 51: 2273-2281.
NESTRICK, T.J., LAMPARSKI, L.L., & TOWNSEND, D.I. (1980)
Identification of tetrachlorodibenzo-p-dioxin isomers at the 1 ng
level by photolytic degradation and patter recognition techniques.
Anal. Chem., 52: 1865-1874.
NESTRICK, T.J., LAMPARSKI, L.L., FRAWLEY, N.N., HUMMEL, R.A., KOCHER,
C.W., MAHLE, N.H., MCCOY, J.W., MILLER, D.L., PETERS, T.L., PILLEPICH,
J.L., SMITH, W.E., & TOBEY, S.W. (1986) Perspectives of a large scale
environmental survey for chlorinated dioxins: Overview and soil data.
Chemosphere, 15: 1453-1460.
NEUBERT, D. & DILLMANN, I. (1972) Embryotoxic effects in mice treated
with 2,4,5-trichlorophenoxyacetic acid and
2,3,7,8-tetrachlorodibenzo-p-dioxin. Naunyn-Schmiedeberg's Arch.
Pharmacol., 272: 243-264.
NIEMANN, R.A., BRUMLEY, W.C., FIRESTONE, D., & SPHON, J.A. (1983)
Analysis of fish for 2,3,7,8-tetrachlorodibenzo-p-dioxin by electron
capture capillary gas chromatography. Anal. Chem., 55: 1497-1504.
NIH (1980a) Bioassay of 1,2,3,6,7,8-and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxin for possible
carcinogenicity (dermal study), Bethesda, Maryland, US Department of
Health and Human Services, National Institutes of Health (DHSS
Publication No. (NIH) 80-1758).
NIH (1980b) Bioassay of 1,2,3,6,7,8-and
1,2,3,7,8,9-hexa-chlorodibenzo-p-dioxin for possible
carcinogenicity (gavage), Bethseda, Maryland, US Department of
Health and Human Services, National Institutes of Health (DHHS
Publication No. (NIH) 80-1754).
NIH (1982a) Carcinogenesis bioassay of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (CAS No 1746-01-6) in
Osborne-Mendel rats and B6C3F1 mice (gavage study), Research
Triangle Park, North Carolina, US National Institutes of Health,
National Toxicology Program (NTP Technical Report Series No. 209).
NIH (1982b) Carcinogenesis bioassay of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (CAS No 1746-01-6) in
Swiss-Webster mice (dermal study), Research Triangle Park, North
Carolina, US National Institutes of Health, National Toxicology
Program (NTP Technical Report Series No. 201).
NILSSON, C.A., ANDERSSON, K., RAPPE, C., & WESTERMARK, S.O. (1974)
Chromatographic evidence for the formation of chloro-dioxins from
chloro-2-phenoxyphenols. J. Chromatogr., 96: 137-147.
NILSSON, C.A., NORSTROM, A., ANDERSSON, K., & RAPPE, C. (1978)
Impurities in commercial products related to pentachlorophenol. In:
Rao, K.R., ed. Pentachlorophenol: Chemistry, pharmacology and
environmental toxicology, New York, London, Plenum Press, pp.
313-324.
NISBET, I.C.T. & PAXTON, M.B. (1982) Statistical aspects of
three-generation studies of the reproductive toxicity of TCDD and
2,4,5,-T. Am. Stat., 36(3): 290-298.
NISHIZUMI, M. (1978) Acute toxicity of polychlorinated dibenzofurans
in CF-1 mice. Toxicol. appl. Pharmacol., 45: 209-212.
NISHIZUMI, M., KURATSUNE, M., & MASSUDA, Y. (1975) [Comparison of
hyperkeratosis induced by PCBs, PCDF and PCDD application.
Polychlorinated biphenyls, polychlorinated dibenzofuran and
polychlorinated dibenzodioxin.] Fukuoka Asa Med., 66: 600-604 (in
Japanese).
NIWA, A., KUMAKI, K., & NEBERT, D.W. (1975) Induction of aryl
hydrocarbon - hydroxylase activity in various cell cultures by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol. Pharmacol., 11: 399-408.
NOLAN, R.J., SMITH, F.A., & HEFNER, J.G. (1979) Elimination and tissue
distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in female
guinea pigs following a single oral dose. Tox. appl. Pharmacol.,
48: 167 (abstract).
NORBACK, D.H. & ALLEN, J.R. (1973) Biological responses of the
nonhuman primate, chicken, and rat to chlorinated dibenzo-p-dioxin
ingestion. Environ. Health Perspect., 5: 233-240.
NORBACK, D.H., ENGBLOM, J.F., & ALLEN, J.R. (1975) Tissue distribution
and excretion of octachlorodibenzo-p-dioxin in the rat. Toxicol.
appl. Pharmacol., 32: 330-338.
NORMAN, L.R., JOHNSON, E.F., & MULLER-EBERHARD, U. (1978)
Identification of the major cytochrome P-450 form transplacentally
induced in neonatal rabbits by 2,3,7,8-tetra-chlorodibenzo-p-dioxin.
J. biol. Chem., 253: 8640-8647.
NORSTROM, R.J., HALLETT, D.J., SIMON, M., & MULVIHILL, M.J. (1982)
Analysis of Great Lakes Herring Gull eggs for
tetra-chlorodibenzo-p-dioxins. In: Hutzinger, O., ed. Chlorinated
dioxins and related compounds. Impact on the environment, Oxford,
New York, Pergammon Press, pp. 173-182.
NORSTROM, R.J., SIMON, M., & WESELOH, D.V. (1986) Long-term trends
of PCDD and PCDF contamination in the Great Lakes. Presented at
Dioxin 86, 6th International Symposium on Chlorinated Dioxins and
Related Compounds, Fukuoka, Japan, 16-19 September, 1986.
NORSTROM, R.J., SIMON, M., WHITEHEAD, P.E., KUSSAT, R., & GARRET, C.
(1988) Level of polychlorinated dibenzo-p-dioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs) in biota and sediments near
potential sources of contamination in British Columbia, 1987, West
Vancouver, British Columbia, Environment Canada (Analytical Report
CRD-88-5).
NRC CANADA (1981) Polychlorinated dibenzo-p-dioxins. Limitation to
the current analytical technique, Ottawa, National Research Council
of Canada (Publication No. NRCC/NRC 18576).
NYGREN, M., RAPPE, C., LINDSTROM, G., HANSSON, M., BERGQVIST, P.-A.,
MARKLUND, S., DOMELLOF, L., HARDELL, L., & OLSSON, M. (1986)
Identification of 2,3,7,8-substituted polychlorinated dioxins and
dibenzofurans in environmental and human samples. In: Rappe, C.,
Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 17-34.
OBERG, T. & BERGSTROM, J. (1986) Combustion test data from a Swedish
hazardous waste incinerator. Chemosphere, 15: 2045-2048.
OISHI, S. (1977) Influence of polychlorinated dibenzofurans (PCDFs)
and polychlorinated biphenyls (PCBs) to serum protein components in
rats. Bull. environ. Contam. Toxicol., 18: 773-777.
OISHI, S. & HIRAGA, K. (1980) Effect of polychlorinated biphenyl,
dibenzofuran and dibenzo-p-dioxin on the susceptibility of male mice
to endotoxin. J. environ. Sci. Health, B15: 77-85.
OISHI, S., MORITA, M., & FUKUDA, H. (1978) Comparative toxicity of
polychlorinated biphenyls and dibenzofurans in rats. Toxicol. appl.
Pharmacol., 43: 13-22.
O'KEEFE, P., MESELSON, M.S., & BAUGHMAN, R. (1977) Neutral clean-up
procedure for 2,3,7,8-tetrachlorodibenzo-p-dioxin residues in bovine
fat and milk. J. Assoc. Off. Anal. Chem., 61: 621-626.
O'KEEFE, P., MEYER, C., HILKER, D., ALDOVES, K., JELUS-TYROR, B.,
DILLON, K., DONNOLLY, R., HORN, E., & SLOAN, R. (1983) Analysis of
2,3,7,8-tetrachlorodibenzo-p-dioxin in Great Lakes fish. Chemosphere,
12: 325-332.
O'KEEFE, P., SILKWORTH, J.B., BIERTHY, J.F., SMITH, R.M., DECAPRIO,
A.P., TURNER, J.N., EADON, G., HILKER, D.R., ALDOUS, K.M., KAMINSKY,
L.S., & COLLINS, D.N. (1985) Chemical and biological investigations of
a transformer accident at Binghamton, NY. Environ. Health
Perspect., 60: 201-209.
OKEY, A.B. & VELLA, L.M. (1982) Binding of 3-methylchlo-anthrene and
2,3,7,8-tetrachlorodibenzo-p-dioxin to a common Ah receptor site in
mouse and rat hepatic cytosols. Eur. J. Biochem., 127: 39-47.
OKEY, A.B., BONDY, G.P., MASON, M.E., KAHL, G.F., EISEN, H.J.,
GUENTHNER, T.M., & NEBERT, D.W. (1979) Regulatory gene product of the
Ah locus. J. biol. Chem., 254: 11636-11648.
OKEY, A.B., BONDY, G.P., MASON, M.E., NEBERT, D.W., FORSTER-GIBSON,
C.J., MUNCHAN, J., & DUFRESNE, M.J. (1980) Temperature-dependent
cytosol-to-nucleus translocation of the Ah receptor for
2,3,7,8-tetrachlorodibenzo-p-dioxin in continuous cell culture lines.
J. biol. Chem., 225: 11415-11422.
OKINO, S.T., QUATTROCHI, L.C., BARNES, H.J., OSANTO, S., GRIFFIN,
K.J., JOHNSON, E.F., & TUKEY, R.H. (1985) Cloning and characterization
of cDNAs encoding 2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible rabbit
mRNAs for cytochrome P-450 isozymes 4 and 6. Proc. Natl Acad. Sci.
(USA), 82: 5310-5314.
OLIE, K., VERMEULEN, P.L., & HUTZINGER, O. (1977)
Chlorodibenzo-p-dioxins and chlorodibenzofurans are trace components
of fly ash and flue gas of some municipal incinerators in the
Netherlands. Chemosphere, 8: 455-459.
OLIE, K., BERG, M.V.D., & HUTZINGER, O. (1983) Formation and fate of
PCDD and PCDF from combustion processes. Chemosphere, 12: 627-636.
OLIVER, R.M. (1975) Toxic effects of
2,3,7,8-tetrachloro-dibenzo-1,4-dioxin in laboratory workers. Br. J.
ind. Med., 32: 49-53.
OLSON, J.A. & GUNNING, D. (1983) The storage form of vitamin A in rat
liver cells. J. Nutr., 113: 2184-2191.
OLSON, J.R. (1986) Metabolism and disposition of
2,3,7,8-tetrachlorodibenzo-p-dioxin in guinea pigs. Toxicol. appl.
Pharmacol., 85: 263-273.
OLSON, J.R., GASIEWICZ, T.A., & NEAL, R.A. (1980a) Tissue
distribution, excretion, and metabolism of
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) in the golden Syrian
hamster. Toxicol. appl. Pharmacol., 56: 78-85.
OLSON, J.R., HOLSCHER, M.A., & NEAL, R.A. (1980b) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in the golden syrian hamster.
Toxicol. appl. Pharmacol., 55: 67-78.
ONO, M., WAKIMOTO, T., TATSUKAWA, R., & MASUDA, Y. (1986)
Polychlorinated dibenzo-p-dioxins and dibenzofurans in human adipose
tissues of Japan. Chemosphere, 15: 1629-1634.
ONTARIO (1985) Scientific criteria document for standard
development. Polychlorinated dibenzo-p-dioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs), Toronto, Ontario Ministry of
the Environment (Report No. 4-84).
ONTARIO (1986) Drinking water survey St. Clair river - Detroit river
area, Toronto, Ontario Ministry of the Environment.
ORR, J.B. & RICHARDS, M.B. (1934) Growth and vitamin A deficiency.
Biochem. J., 28: 1259-1273.
OSBORNE, R. & GREENLEE, W.F. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) enhances terminal
differentiation of cultured human epidermal cells. Toxicol. appl.
Pharmacol., 77: 434-443.
OSBORNE, R., DOLD, K.M., & GREENLEE, W.F. (1984) Cell culture models
to study mechanisms of toxicity of chlorinated aromatic compounds to
skin and thymus. Chem. Ind. Inst. Toxicol., 4: 2-7.
OTT, M.G., HOLDER, B.B., & OLSON, R.D. (1980) A mortality analysis of
employees engaged in the manufacture of 2,4,5-trichlorophenoxyacetic
acid. J. occup. Med., 22: 47-50.
PAASIVIRTA, J., ENQVIST, J., RAISANEN, S., & PAASIVUO, P. (1977) On
the limit of detection of TCDD in gas chromatography. Chemosphere,
6: 355.
PALAUSKY, J., HARWOOD, J.J., CLEVENGER, T.E., KAPILA, S., & YANDERS,
A.F. (1986) Disposition of tetrachlorodibenzo-p-dioxin in soil. In:
Rappe, C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 211-223.
PATTERSON, D.G., Jr, HOFFMAN, R.E., NEEDHAM, L.L., ROBERTS, D.W.,
BAGBY, J.R., PIRKLE, J.L., FALK, H., SAMPSON, E.J., & HOUK, V.N.
(1986) 2,3,7,8-Tetrachlorodibenzo-p-dioxin levels in adipose tissue of
exposed and control persons in Missouri. J. Am. Med. Assoc., 256:
2683-2686.
PATTERSON, J.M., MCHENRY, E.W., & CRANDALL, W.A. (1942) The
physiological properties of vitamin A. 1. A specific effect upon body
weight and body composition in the Albino rat. Biochem. J., 36:
792-794.
PAZDERNIK, T.L. & ROZMAN, K.K. (1985) Effect of thyroidectomy and
thyroxine on 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced
immunotoxicity. Life Sci., 36: 695-703.
PAZDEROVA-VEJLUPKOVA, J., LUKAS, E., NEMCOVA, M., SPACILOVA, M.,
JIRASEK, L., KALENSKY, J., JOHN, J., JIRASEK, A., & PICKOVA, J. (1974)
[Chronic intoxication by chlorinated hydrocarbons formed during the
production of sodium 2,4,5-trichlorophenoxyacetate.] Pracov. Lek.,
26: 332-339 (in Czech).
PAZDEROVA-VEJLUPKOVA, J., LUKAS, E., NEMCOVA, M., PICKOVA, J., &
JIRASEK, L. (1980) [Chronic poisoning by
2,3,7,8-tetra-chlorodibenzo-p-dioxin.] Pracov. Lek., 32: 204-209
(in Czech).
PAZDEROVA-VEJLUPKOVA, J., NEMCOVA, M., PICKOVA, J., JIRASEK, L., &
LUKAS, E. (1981) The development and prognosis of chronic intoxication
by tetrachlorodibenzo-p-dioxin in men. Arch. environ. Health, 36:
5-11.
PETERSON, R.E., MADHUKAR, B.V., YANG, K.H., & MATSUMURA, F. (1979a)
Depression of adenosine triphosphatase activities in isolated liver
surface membranes of 2,3,7,8-tetrahlorodibenzo-p-dioxin-treated rats:
correlation with effects on ouabain biliary excretion and bile flow.
J. Pharmacol. exp. Ther., 210: 275-210.
PETERSON, R.E., HAMADA, N., YANG, K.H., & MADHUKAR, B.V. (1979b)
Reversal of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced depression of
ouabain biliary excretion by pregnenolone-16-beta-carbonitrile and
spironolactone in isolated perfused rat livers. Toxicol. appl.
Pharmacol., 50: 407-416.
PHILIPPI, M., SCHMID, J., WIPF, H.K., & HUTTER, R. (1982) A microbial
metabolite of TCDD. Experientia (Basel), 38: 659-661.
PIPER, W.N., ROSE, J.Q., & GEHRING, P.J. (1973) Excretion and tissue
distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Adv.
Chem. Ser., 120: 85-91.
PITOT, H.C. & SIRICA, A.E. (1980) The stages of initiation and
promotion in hepatocarcinogenesis. Biochim. Biophys. Acta, 605:
191-215.
PITOT, H.C., GOLDSWORTHY, T., CAMPBELL, H.A., & POLAND, A. (1980)
Quantitative evaluation of the promotion by
2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from
diethylnitrosamine. Cancer Res., 40: 3616-3620.
POCCHIARI, F. (1978) 2,3,7,8-Tetrachlorodibenzo-para-dioxin
decontamination. Ecol. Bull. (Stockholm), 27: 67-70.
POCCHIARI, F., DIDOMENICO, A., SILANO, V., & ZAPPONI, G. (1983)
Environmental impact of the accidental release of
tetrachlorodibenzo-p-dioxn (TCDD) at Seveso (Italy). In: Coulston, F.
& Pocchiari, F., ed. Accidental exposure to dioxins. human health
aspects, New York, London, Academic Press, pp. 5-35.
POELLINGER, L. & GULLBERG, D. (1985) Characterization of the
hydrophobic properties of the receptor for
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Mol. Pharmacol., 27:
271-276.
POELLINGER, L., KURL, R.N., LUND, J., GILLNER, M., CARLSTEDT-DUKE, J.,
HOGBERG, B., & GUSTAFSSON. J.-A. (1982) High-affinity binding of
2,3,7,8-tetrachlorodibenzo-p-dioxin in cell nuclei from rat liver.
Biochem. Biophys. Acta, 714: 516-523.
POELLINGER, L., LUND, J., HANSSON, L.-A., & GUSTAFSSON, J.-A. (1983)
Physicochemical characterization of specific and non-specific
polyaromatic hydrocarbon binders in rat and mouse liver cytosol. J.
biol. Chem., 258: 13535-13542.
POHJANVIRTA, R. & TUOMISTO, J. (1986) Han/Wistar rats are
exceptionally resistant to TCDD. II. Arch. Toxicol., 11 (Suppl.):
344-347.
POHLAND, A.E. & YANG, G.C. (1972) Preparation and characterization of
chlorinated dibenzo-p-dioxins. J. agric. food Chem., 20(6):
1093-1099.
POIGER, H. & BUSER, H.R. (1983) Structure elucidation of mammalian
TCDD-metabolites. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 483-492.
POIGER, H. & SCHLATTER, Ch. (1979) Biological degradation of TCDD in
rats. Nature (London), 281: 706-707.
POIGER, H. & SCHLATTER, Ch. (1980) Influence of solvents and
adsorbents on dermal and intestinal absorption of TCDD. Food Cosmet.
Toxicol., 18: 477-481.
POIGER, H. & SCHLATTER, Ch. (1985) Influence of phenobarbital and TCDD
on the hepatic metabolism of TCDD in the dog. Experientia (Basel),
41: 376-378.
POIGER, H. & SCHLATTER, Ch. (1986) Pharmacokinetics of 2,3,7,8-TCDD in
man. Chemosphere, 15: 1489-1494.
POIGER, H., BUSER, H.R., WEBER, H., ZWEIFEL, U., & SCHLATTER, Ch.
(1982) Structure elucidation of mammalian TCDD-metabolites.
Experientia (Basel), 38: 484-486.
POIGER, H., BUSER, H.R., & SCHLATTER, Ch. (1984) The metabolism of
2,3,7,8-tetrachlorodibenzofuran in the rat. Chemosphere, 13:
351-357.
POLAND, A. & GLOVER, E. (1973a) Studies on the mechanism of toxicity
of the chlorinated dibenzo-p-dioxins. Environ. Health Perspect.,
5: 245-251.
POLAND, A. & GLOVER, E. (1973b) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
A potent inducer of aminolevulinic acid synthetase. Science, 179:
476-477.
POLAND, A. & GLOVER, E. (1973c) Chlorinated dibenzo-p-dioxins: Potent
inducers of aminolevulinic acid synthetase and aryl hydrocarbon
hydroxylase. II. A study of the structure-activity relationship.
Mol. Pharmacol., 9: 736-747.
POLAND, A. & GLOVER, E. (1974a) The induction of aryl hydrocarbon
hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a
receptor mutation in genetically non-responsive mice. Pharmacologist,
16: 240 (Abstract 282).
POLAND, A. & GLOVER, E. (1974b) Comparison of
2,3,7,8-tetra-chlorodibenzo-p-dioxin, a potent inducer of aryl
hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol. Pharmacol.,
10: 349-359.
POLAND, A. & GLOVER, E. (1974c) Genetic expression of aryl hydrocarbon
hydroxylase activity. Induction of monooxygenase activities and
cytochrome P1-450 formation by 2,3,7,8-tetra-chlorodibenzo-p-dioxin in
mice genetically "nonresponsive" to other aromatic hydrocarbons. J.
biol. Chem., 249: 5599-5606.
POLAND, A. & GLOVER, E. (1975) Genetic expression of arylhydrocarbon
hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a
receptor mutation in genetically non-responsive mice. Mol.
Pharmacol., 11: 389-398.
POLAND, A. & GLOVER, E. (1979) An estimate of the maximum in vivo
covalent binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to rat liver
protein, ribosomal RNA, and DNA. Cancer Res., 39: 3341-3344.
POLAND, A. & GLOVER, E. (1980) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Segregation of toxicity with the Ah-locus. Mol. Pharmacol., 17:
86-94.
POLAND, A. & KENDE, A. (1976) 2,3,7,8-Tetrachlorodibenzo-p-dioxin:
Environmental contaminant and molecular probe. Fed. Proc., 35:
2404-2411.
POLAND, A. & KNUTSON, J.C. (1982) 2,3,7,8-Tetrachlorodibenzo-p-dioxin
and related halogenated aromatic hydrocarbons: Examination of the
mechanisms of toxicity. Annu. Rev. Pharmacol. Toxicol., 22:
517-554.
POLAND, A., SMITH, D., METTER, G., & POSSICK, P. (1971) A health
survey of workers in a 2,4-D and 2,4,5-T plant. Arch. environ.
Health, 22: 316-327.
POLAND, A., GLOVER, E., & KENDE, A.S. (1976) Stereospecific, high
affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic
cytosol. Evidence that the binding species is receptor for induction
of aryl hydrocarbon hydroxylase. J. biol. Chem., 251: 4936-4946.
POLAND, A., PALEN, D., & GLOVER, E. (1982) Tumor promotion by TCDD in
HRS/J mice. Nature (Lond.), 300(5889): 271-273.
POLI, A., FRANCESCHINI, G., PUGLISI, L., & SIRTORI, C.R. (1980)
Increased total and high density lipoprotein cholesterol with
apoprotein changes resembling streptozotocin diabetes in
tetrachlorodibenzodioxin (TCDD) treated rats. Biochem. Pharmacol.,
29: 835-838.
POTTER, C.L., SIPES, I.G., & RUSSELL, D.H. (1982) Inhibition of
ornithine decarboxylase activity by
2,3,7,8-tetrachloro-dibenzo-p-dioxin. Biochem. Pharmacol., 31:
3367-3371.
POTTER, C.L., SIPES, I.G., & RUSSELL, D.H. (1983) Hypothyroxinemia and
hypothermia in rats in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin
administration. Toxicol. appl. Pharmacol., 69: 89-95.
POTTER, C.L., MENAHAN, L.A., & PETERSON, R.E. (1986a) Relationship of
alterations in energy metabolism to hypophagia in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. appl. Toxicol., 6:
89-97.
POTTER, C.L., MOORE, R.W., INHORN, S.L., HAGEN, T.C., & PETERSON, R.E.
(1986b) Thyroid status and thermogenesis in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
84: 45-55.
PRATT, R.M., DENCKER, L., & DIEWERT, V.M. (1984)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced cleft palate in the mouse:
Evidence for alterations in palatal shelf fusion. Teratog.
Carcinogen. Mutagen., 4: 427-436.
PRESTON, B.D., VAN MILLER, P.J., MOORE, R.W., & ALLEN, J.R. (1981)
Promoting effects of polychlorinated biphenyls (Aroclor 1254) and
polychlorinated dibenzofuran-free Aroclor 1254 on
diethylnitrosamine-induced tumorigenesis in the rat. J. Natl Cancer
Inst., 66: 509-515.
PUHVEL, S.M., SAKAMOTO, M., ERTL, D.C., & REISNER, R.M. (1982)
Hairless mice as models for chloracne: A study of cutaneous changes
induced by topical application of established chloracnegens.
Toxicol. appl. Pharmacol., 64: 492-503.
PUHVEL, S.M., ERTL, D.C., & LYNBERG, C.A. (1984) Increased epidermal
transglutaminase activity following
2,3,7,8-tetra-chlorodibenzo-p-dioxin: In vivo and in vitro studies
with mouse skin. Toxicol. appl. Pharmacol., 73: 42-47.
PUHVEL, S.M., SAKAMOTO, M., & REISNER, R.M. (1986) Localization of
TCDD in hairless mouse skin. Chemosphere, 15: 2065-2067.
QUILLEY, C.P. & RIFKIND, A.B. (1986) Prostaglandin release by the
chick embryo heart is increased by
2,3,7,8-tetrachloro-dibenzo-p-dioxin and by other cytochrome P-448
inducers. Biochem. biophys. Res. Commun., 136(2): 582-589.
RAMSEY, J.C., HEFNER, J.G., KARBOWSKI, R.J., BRAUN, W.H., & GEHRING,
P.J. (1982) The in vivo biotransformation of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat. Toxicol.
appl. Pharmacol., 65: 180-184.
RAPPE, C. (1978a) Chemical background of the phenoxy acids and
dioxins. Ecol. Bull. (Stockholm), 27: 28-30.
RAPPE, C. (1978b) Decontamination of products formed during the
industrial preparation of 2,4,5-trichlorophenol. In: Cattabeni, F.,
Cavallero, A., & Galli, G., ed. Dioxin: toxicological and chemical
aspects, New York, SP Medical and Scientific Books, pp. 179-183.
RAPPE, C. (1984) Analysis of polychlorinated dioxins and furans.
Environ. Sci. Technol., 18: 78A-90A.
RAPPE C. (1985) Problems in analysis of PCDDs and PCDFs and presence
of these compounds in human milk, Copenhagen, WHO Regional Office for
Europe (ICP/CEH/501/05/7).
RAPPE, C. (1987) Global distribution of polychlorinated dioxins
(PCDDs) and dibenzofurans (PCDFs). In: Solving hazardous waste
problems: Learning from dioxins, New York, American Chemical
Society, pp. 20-23 (ACS Symposium Series No. 338).
RAPPE, C. & BUSER, H.R. (1980) Chemical properties and analytical
methods. In: Kimbrough, R.D., ed. Halogenated biphenyls, terphenyls,
naphthalenes, dibenzodioxins and related products, Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp. 41-80.
RAPPE, C. & KJELLER, L.-O. (1987) PCDDs and PCDFs in environ-mental
samples air, particulates, sediments and soil. Chemosphere, 16:
1775-1780.
RAPPE, C. & NYGREN, M. (1984) Chemical analysis of human samples.
Identification and quantification of polychlorinated dioxins and
dibenzofurans. In: de Serres, F.J. & Pero, R.W., ed. Individual
susceptibility to genotoxic agents in the human population, New
York, London, Plenum Press, pp. 305-314.
RAPPE, C., BUSER, H.R., & BOSHARDT, H.P. (1978) Identification and
quantification of polychlorinated dibenzo-p-dioxins (PCDDs) and
dibenzofurans (PCDFs) in 2,4,5-T-ester formulations and herbicide
orange. Chemosphere, 7: 431-438.
RAPPE, C., BUSER, H.R., KUROKI, H., & MASUDA, Y. (1979) Identification
of polychlorinated dibenzofurans (PCDFs) retained in patients with
Yusho. Chemosphere, 4: 259-266.
RAPPE, C., BUSER, H.R., STALLING, D.L., SMITH, L.M., & DOUGHERTY, R.C.
(1981) Identification of polychlorinated dibenzofurans in
environmental samples. Nature (Lond.), 292: 524-526.
RAPPE, C., MARKLUND, S., BERGQVIST, P.-A., & HANSSON, M. (1983a)
Polychlorinated dibenzo-p-dioxins, dibenzofurans and other polynuclear
aromatics formed during incineration and polychlorinated biphenyl
fires. In: Choudhary, G., Keith, L.H., & Rappe, C., ed. Chlorinated
dioxins and dibenzofurans in the total environment, Boston,
Butterworth Publishers, pp. 99-124.
RAPPE, C., MARKLUND, S., NYGREN, M., & GARA, A. (1983b) Parameters for
identification and confirmation in trace analyses of polychlorinated
dibenzo-p-dioxins and dibenzo-furans. In: Choudhary, G., Keith, L.H.,
& Rappe, C., ed., Chlorinated dioxins and dibenzofurans in the total
environment, Boston, Butterworth Publishers, pp. 240-259.
RAPPE, C., NYGREN, M., BUSER, H.R., MASUDA, Y., KUROKI, H., & CHEN,
P.H. (1983c) Identification of polychlorinated dioxins (PCDDs) and
dibenzofurans (PCDFs) in human samples, occupational exposure and
Yusho patients. In: Tucker, R.E., Young, A.L., & Gray, A.P., eds.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 241-254.
RAPPE, C., BERGQVIST, P.-A., & MARKLUND, S. (1985a) Analysis of
polychlorinated dibenzofurans and dioxins in ecological samples. In:
Keith, L.H., Rappe, C., & Choudhary, G., ed. Chlorinated dioxins and
dibenzofurans in the total environment II, Boston, Butterworth
Publishers, pp. 135-138.
RAPPE, C., MARKLUND, S., KJELLER, L-O., BERGQVIST, P.-A., & HANSSON,
M. (1985b) Composition of polychlorinated dibenzofurans (PCDF) formed
in PCB fires. In: Keith, L.H., Rappe, C., & Choudhary, G., ed.
Chlorinated dioxins and dibenzofurans in the total environment II,
Boston, Butterworth Publishers, p. 401-424.
RAPPE, C., NYGREN, M., MARKLUND, S., KJELLER, L.-O., BERGQVIST, P.A.,
& HANSON, M. (1985c) Assessment of human exposure to polychlorinated
dibenzofurans and dioxins. Environ. Health Perspect., 60: 303-304.
RAPPE, C., KJELLER, L.-O., & MARKLUND, S. (1985d) PCDF isomers and
isomer levels found in PCBs. In: Komai, R.Y. & Addis, G., ed.
Proceedings of a Workshop on PCB By-Product Formation, Palo Alto,
California, 4-6 December, 1984, Palo Alto, California, Electric
Power Research Institute, pp. 20-23.
RAPPE, C., KJELLER, L.-O., MARKLUND, S., & NYGREN, M. (1986a)
Electrical PCB accident, an update. Chemosphere, 15: 1291-1295.
RAPPE, C., NYGREN, M., HANSSON, M., & KAHN, P.C. (1986b) Analysis of
adipose tissue and blood samples from Vietnam veterans. Clean-up,
analysis and quality control, P 41, In: Dioxin 86, 6th International
Symposium on Chlorinated Dioxins and Related Compounds, Fukuoka,
Japan, 16-19 September, 1986.
RAPPE, C., ANDERSSON, R., BERGQVIST, P.-A., BROHEDE, C., HANSSON, M.,
KJELLER, L.-O., LINDSTROM, G., MARKLUND, S., NYGREN, M., SWANSON,
S.E., TYSKLIND, M., & WIBERG, K. (1987) Overview on environmental fate
of chlorinated dioxins and dibenzofurans, sources, levels and isomeric
pattern in various matrices. Chemosphere, 16: 1603-1618.
RAPPE, C., NYGREN, M., LINDSTROM, G., BUSER, H.R., BLASER, O., &
WUTHRICH, C. (1987) Polychlorinated dibenzofurans and
dibenzo-p-dioxins and other chlorinated contaminants in cow milk from
various locations in Switzerland. Environ. Sci, Technol., 21:
964-970.
REGGIANI, G. (1978) Medical problems raised by the TCDD contamination
in Seveso, Italy. Arch. Toxicol., 40: 161-188.
REGGIANI, G. (1980a) Acute human exposure to TCDD in Seveso, Italy.
J. Toxicol. environ. Health, 6: 27-43.
REGGIANI, G. (1980b) Toxicology of TCDD and related compounds:
observation in man. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds. Impact
on the environment, Oxford, New York, Pergamon Press, pp. 463-493.
REGGIANI, G. (1983a) Anatomy of TCDD spill: The Seveso accident.
Curr. Dev., 2: 269-341.
REGGIANI, G. (1983b) An overview on the health effects of halogenated
dioxins and related compounds - the Yusho and Taiwan episodes. In:
Coulston, F. & Pocchiari, F., ed. Accidental exposure to dioxins.
Human health aspects, New York, London, Academic Press, pp. 39-67.
REGIONE LOMBARDIA (1984) Final report and recommendations of the 6th
International Steering Committee Meeting, Milan, 19-21 February,
1984, Milan, Regione Lombardia, pp. 1-17.
RICE, R.H. & CLINE, P.R. (1984) Opposing effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin and hydrocortisone on growth and
differentiation of cultured malignant human keratinocytes.
Carcinogenesis, 5(3): 367-371.
RICE, R.H., CLINE, P.R., & COE, E.L. (1983) Mutually antagonistic
effects of hydrocortisone and retinylacetate on envelope competence in
cultured malignant human keratinocytes. J. invest. Dermatol., 81:
176s-178s.
RICHERT, J., VON (1962) [On neurological complications in a case of
chlorinated hydrocarbon poisoning accompanied by chloracne.]
Nervenarzt, 33(4): 180-184 (in German).
RIKANS, L.E., GIBSON, D.D., & MCCAY, P.B. (1979) Evidence for the
presence of cytochrome P-450 in rat mammary gland. Biochem.
Pharmacol., 28: 3039-3042.
RISSE-SUNDERMAN, A. (1959) [Intoxication by chloro-aromatics,]
Cologne-Lindenburg, University of Cologne, Department of Dermatology
(Thesis) (in German).
RIZZARDINI, M., ROMANO, M., TURSI, F., SALMONA, F., VECCHI, A.,
SIRONI, M., GIZZI, F., BENFENATI, E., GARATTINI, S., & FANELLI, R.
(1983) Toxicological evaluation of urban waste incinerator emissions.
Chemosphere, 12: 559-564.
ROBERTSON, L.W., REGEL, U., FILSER, J.G., & OESCH, F. (1985) Absence
of lipid peroxidation as determined by ethane exha-lation in rats
treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Arch.
Toxicol., 57: 13-16.
ROGERS, A.M., ANDERSEN, M.E., & BACK, K.C. (1982) Mutagenicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin and perfluoro-n-decanoic acid in
L5178Y mouse-lymphoma cells. Mutat. Res., 105: 445-449.
ROMKES, K., PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1987) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on hepatic and uterine estrogen
receptor levels in rats. Toxicol. appl. Pharmacol., 87: 306-314.
ROSE, J.Q., RAMSEY, J.C., WENTZLER, T.H., HUMMEL, R.A., & GEHRING,
P.J. (1976) The fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin following
single and repeated oral doses to the rat. Toxicol. appl.
Pharmacol., 36: 209-226.
ROZMAN, K. (1984) Hexadecane increases the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): Is brown adipose tissue
the primary target in TCDD-induced wasting syndrome? Biochem. biophys.
Res. Commun., 125(3): 996-1004.
ROZMAN, K., ROZMAN, T., & GREIM, H. (1984) Effect of thyroidectomy and
thyroxine on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induced
toxicity. Toxicol. appl. Pharmacol., 72: 372-376.
ROZMAN, K., ROZMAN, T., SCHEUFLER, E., PAZDERNIK, T., & GREIM, H.
(1985a) Thyroid hormones modulate the toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). J. Toxicol. environ.
Health, 16: 481-491.
ROZMAN, K., HAZELTON, G.A., KLAASSEN, C.D., ARLOTTO, M.P., &
PARKINSON, A. (1985b) Effect of thyroid hormones on liver microsomal
enzyme induction in rats exposed to
2,3,7,8-tetra-chlorodibenzo-p-dioxin. Toxicology, 37: 51-63.
ROZMAN, K., PEREIRA, D., & IATROPOULOS, M.J. (1986) Histo-pathology of
interscapular brown adipose tissue, thyroid, and pancreas in
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicol.
appl. Pharmacol., 82: 551-559.
RYAN, J.J., LAU, P.-Y., PILON, J.C., & LEWIS, D. (1983a)
2,3,7,8-Tetrachlorodibenzo-p-dioxin and
2,3,7,8-tetrachloro-dibenzofuran residues in great lakes commercial
and sport fish. In: Choudhary, G., Keith, L.H., & Rappe, C., ed.
Chlorinated dioxins and dibenzofurans in the total environment,
Boston, Buttersworth Publishers, pp. 87-97.
RYAN, J.J., PILON, J.C., CONACHER, H.B.S., & FIRESTONE, D. (1983b)
Inter-laboratory study on determination of
2,3,7,8-tetrachlorodibenzo-p-dioxin in fish. J. Assoc. Off. Anal.
Chem., 66: 700-707.
RYAN, J.J., LIZOTTE, R., SAKUMA, T., & MORI, B. (1985) Chlorinated
dibenzo-p-dioxins, chlorinated dibenzofurans and pentachlorophenol in
Canadian chicken and pork samples. J. agric. food Chem., 33:
1021-1026.
RYAN, J.J., SCHECTER, A., SUN, W.-F., & LIZOTTE, R. (1986)
Distribution of chlorinated dibenzo-p-dioxins and chlorinated
dibenzofurans in human tissues from the general population. In: Rappe,
C., Choudhary, G., & Keith, L., ed. Chlorinated dioxins and
dibenzofurans in perspective, Chelsea, Michigan, Lewis Publishers,
pp. 3-16.
RYHAGE, R. (1964) Use of mass spectrometer as a detector and analyzer
for effluents emerging from high temperature gas liquid chromatography
columns. Anal. Chem., 36: 759-764.
SACCHI, G.A., VIGANO, P., FORTUNATI, G., & COCUCCI, S.M. (1986)
Accumulation of 2,3,7,8-tetrachlorodibenzo-p-dioxin from soil and
nutriculture solution by bean and maize plants. Experientia (Basel),
42: 586-588.
SAFE, S.H. & SAFE, L.M (1984) Synthesis and characterization of
twenty-two purified polychlorinated dibenzofuran congeners. J.
agric. food Chem., 32: 68-71.
SAFE, S.H., MASON, G., KEYS, B., FARRELL, K., ZMUDZKA, B., SAWYER, T.,
PISKORSKA-PLISZEZYNSKA, J., SAFE, L.M., ROMKES, M., & BANDIERA, S.
(1986) Polychlorinated dibenzo-p-dioxins and dibenzofurans:
correlation between in vitro and in vivo structure-activity
relationships (SARs). Chemosphere, 15: 1725-173.
SAFE, S.H., MASON, G., FARRELL, K., KEYS, B., PISKORSKA-PLISZCZYNSKA,
J., MADGE, J.A., & CHITTIM, B. (1987) Validation of in vitro bioassays
for 2,3,7,8-TCDD equivalents. Chemosphere, 16: 1723-1728.
SAI (SYSTEMS APPLICATIONS, INC.) (1980) Human exposure to
atmospheric concentration of selected chemicals, Springfield,
Virginia, National Technical Information Service, Vol. 1 (Report
prepared for US Environmental Protection Agency, Research Triangle
Park, North Carolina) (PB 81-193252).
SANDERMANN, W. (1984a) [Dioxin. The history of the discovery of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin, Seveso poison.]
Naturwiss. Rundsch., 37(5): 173-178 (in German).
SANDERMANN, W. (1984b) [Breakdown of dioxin by beta-radiation. The
history of the discovery of 2,3,7,8-tetrachlorodibenzo-p-dioxin
(Second communication).] Naturwiss. Rundsch., 37(11): 445-446 (in
German).
SANDERMANN, W., STOCKMANN, H., & GASTEN, R. (1957) [On the pyrolysis
of pentachlorphenol.] Chem. Ber., 90: 690-692 (in German).
SANGER, V.L., SCOTT, L., HAMDY, A., GALE, C., & POUNDEN, W.D. (1958)
Alimentary toxemia in chickens. J. Am. Vet. Med. Assoc., 133:
172-176.
SARNA, L.P., HODGE, P.E., & WEBSTER, G.R.B. (1984) Octanol-water
partition coefficients of chlorinated dioxins and dibenzofurans by
reversed-phase HPLC using several C18 columns. Chemosphere, 13:
975-983.
SAWAHATA, T., OLSON, J.R., & NEAL, R.A. (1982) Identification of
metabolites of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) formed on
incubation with isolated rat hepatocytes. Biochem. biophys. Res.
Commun., 105: 341-346.
SAWYER, T.W. & SAFE, S. (1985) In vitro AHH induction by
polychlorinated biphenyl and dibenzofuran mixtures: Additive effects.
Chemosphere, 14: 79-84.
SAWYER, T.W., JONES, D., ROSANOFF, K., MASON, G.,
PISKORSKA-PLISZCZYNSKA, J., & SAFE, S. (1986) The biologic and toxic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in chickens.
Toxicology, 39: 197-206.
SCHANTZ, S.L., BARSOTTI, D.A., & ALLEN, J.R. (1978) Toxicological
effects produced in nonhuman primates chronically exposed to fifty
parts per trillion 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
Toxicol. appl. Pharmacol., 48(1): A180.
SCHECTER, A., WEERASINGHE, W.C.A., GROSS, M., & DEKIN, A. (1986a)
Human tissue levels of PCDDs and PCDFs in over one hundred year old
frozen Eskimo tissue, meat, and California redwood charcoal and their
relation to the trace chemistry theory of dioxin origin. In: Dioxin
86. 6th International Symposium on Chlorinated Dioxins and Related
Compounds, Fukuoka, Japan, 16-19 September, 1986, p.135.
SCHECTER, A.J., RYAN, J.J., & CONSTABLE, J.D. (1986b) Chlorinated
dibenzo-p-dioxin and dibenzofuran levels in human adipose tissue and
milk samples from the north and south of Vietnam. Chemosphere, 15:
1613-1620.
SCHILLER, C.M., WALDEN, R., & SHOAF, C.R. (1982) Studies on the
mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity: Nutrient
assimilation. Fed. Proc., 41: 1426 (Abstract).
SCHILLER, C.M., ADCOCK, C.M., MOORE, R.A., & WALDEN, R. (1985) Effect
of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and fasting on body
weight and lipid parameters in rats. Toxicol. appl. Pharmacol.,
81: 356-361.
SCHILLER, C.M., ADCOCK, C.M., SGIAF, C.R., & WALDEN, R. (1986) Effects
of adenine and its isomer 4-aminopyrazolo-(3,4-d)-pyrimidine on
2,3,7,8-tetrachlorodibenzo-p-dioxin-induced mortality in rats.
Toxicol. appl. Pharmacol., 84: 369-378.
SCHOENY, R. (1982) Mutagenicity testing of chlorinated biphenyls and
chlorinated dibenzofurans. Mutat. Res., 101: 45-56.
SCHULZ, K.H. (1968) [On the symptoms and etiology of chloracne.]
Arbeitsmed. Sozialmed. Arbeitshyg., 3: 25-29 (in German).
SCHWETZ, B.A., NORRIS, J.M., SPARSCHU, G.L., ROWE, V.K., GEHRING,
P.J., EMERSON, J.L., & GEHRING, C.G. (1973) Toxicology of chlorinated
dibenzo-p-dioxins. Environ. Health Perspect., 5: 87-99.
SEEFELD, M.D. & PETERSON, R.E. (1983)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin-induced weight loss: A proposed
mechanism. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed. Human
and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 405-412
(Environmental Science Research Series).
SEEFELD, M.D. & PETERSON, R.E. (1984) Digestible energy and efficiency
of feed utilization in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol. 74:
214-222.
SEEFELD, M.D., CORBETT, S.W., KEESEY, R.E., & PETERSON, R.E. (1984a)
Characterization of the wasting syndrome in rats treated with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
73: 311-322.
SEEFELD, M.D., KEESEY, R.E., & PETERSON, R.E. (1984b) Body weight
regulation in rats treated with 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Toxicol. appl. Pharmacol., 76: 526-536.
SEILER, J.P. (1973) A survey on the mutagenicity of various
pesticides. Experientia (Basel), 29: 622-623.
SHADOFF, L.A., HUMMEL, R.A., & LAMPARSKI, L. (1977) A research for
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in an environment exposed
annually to 2,4,5-trichlorophenoxyacetic acid ester (2,3,5-T)
herbicides. Bull. environ. Contam. Toxicol., 18: 478-485.
SHARMA, R.P. & GEHRING, P.J. (1979) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) on splenic lymphocyte
transform-ation in mice after single and repeated exposures. Ann.
N.Y. Acad. Sci., 320: 487-497.
SHARMA, R.P., KOCIBA, R.J., & GEHRING, P.J. (1984) Immuno-toxicologic
effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rabbits. J.
environ. Pathol. Toxicol. Oncogen., 5(4/5): 321-328.
SHEFFIELD, A. (1985) Sources and releases of PCDDs and PCDFs to the
Canadian environment. Chemosphere, 14: 811-814.
SHIGEMATSU, N., ISHIMARU, S., SAITO, R., IDEDA, T., MATSUBA, K.,
SUGIYAMA, K., & MASUDA, Y. (1978) Respiratory involvement in
polychlorinated biphenyls poisoning. Environ. Res., 16: 92-100.
SHIREMAN, R.B. & WEI, CHENG-I. (1986) Uptake of
2,3,7,8-tetra-chlorodibenzo-p-dioxin from plasma lipoproteins by
cultured human fibroblasts. Chem.-biol. Interact., 58: 1-12.
SHIVERICK, K.T. & MUTHER, T.F. (1983)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) effects on hepatic
microsomal steroid metabolism and serum estradiol of pregnant rats.
Biochem. Pharmacol., 32: 991-995.
SHOAF, C.R. & SCHILLER, C.M. (1981) Studies on the mechanism of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity-lipid
assimilation. II. Pharmacologist, 23: 176 (Abstract).
SILKWORTH, J., MCMARTIN, D., DECAPRIO, A., REJ, R., O'KEEFE, P., &
KAMINSKY, L. (1982) Acute toxicity in guinea pigs and rabbits of soot
from a polychlorinated biphenyl-containing transformer fire.
Toxicol. appl. Pharmacol., 65: 425-439.
SIMPSON, C.F., PRITCHARD, W.R., & HARMS, R.H. (1959) An endotheliosis
in chickens and turkeys caused by an unidentified dietary factor. J.
Am. Vet. Med. Assoc., 134: 410-416.
SLONECKER, P.J., PYLE, J.R., & CANTRELL, J.S. (1983) Identifi-cation
of polychlorinated dibenzo-p-dioxin isomers by powder X-ray
diffraction with electron capture gas chromatography. Anal. Chem.,
55: 1543-1547.
SMITH, A.G., FRANCIS, J.E., KAY, S.J.E., & GREIG, J.B. (1981) Hepatic
toxicity and uroporphyrinogen decarboxylase activity following a
single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin to mice. Biochem.
Pharmacol., 30: 2825-2830.
SMITH, A.G., FRANCIS, J.E., & GREIG, J.B. (1985) Continued depression
of hepatic uroporphyrinogen decarboxylase activity caused by
hexachlorobenzene 2,3,7,8-tetrachlorodibenzo-p-dioxin despite
regeneration after partial hepatectomy. Biochem. Pharmacol., 34:
1817-1820.
SMITH, A.H., FISHER, D.O., GILES, H.J., & PEARCE, N. (1983) The New
Zealand soft tissue sarcoma case-control study: Interview findings
concerning phenoxyacetic acid exposure. Chemosphere, 12: 565-571.
SMITH, F.A., SCHWETZ, B.A., & NITSCHKE, K.D. (1976) Terato-genicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin in CF-1 mice. Toxicol. appl.
Pharmacol., 38: 517-523.
SMITH, R.M., O'KEEFE, P.W., ALDOUS, K.M., HILKER, D.R., & O'BRIEN,
J.E. (1983) 2,3,7,8-Tetrachlorodibenzo-p-dioxin in sediment samples
from love canal storm sewers and creeks. Environ. Sci. Technol., 17:
6-10.
SODERKVIST, P., POELLINGER, L., & GUSTAFSSON, J.-A. (1986)
Carcinogen-binding proteins in the rat ventral prostate: Specific and
nonspecific high-affinity binging sites for benso(a)pyrene,
3-methylcholanthrene, and 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Cancer Res., 46: 651-657.
SOUTHERLAND, J.H., KUYKENDAL, W.B., LAMASON, W.H. II, & OBERACKER,
D.A. (1987) Assessment of combustion sources as emitters of
chlorinated dioxin compounds: A report on the result of tier 4 of the
national dioxin strategy. Chemosphere, 16: 2161-2168.
SPARSCHU, G.L., DUNN, F.L., & ROWE, V.K. (1971) Study of the
teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Food
Cosmet. Toxicol., 9: 405-412.
SPITSBERGEN, J.M., SCHAT, K.A., KLEEMAN, J.M., & PETERSON, R.E. (1986)
Interactions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with immune
responses of rainbow trout. Vet. Immunol. Immunopathol., 12:
263-280.
STALLING, D.L., SMITH, L.M., PETTY, J.D., HOGAN, J.W., JOHNSON, J.L.,
RAPPE, C., & BUSER, H.R. (1983) Residues of polychlorinated
dibenzo-p-dioxins and dibenzofurans in laurentian great lakes fish.
In: Tucker, R.E., Young, A.L., & Gray, A.P., ed. Human and
environmental riks of chlorinated dioxins and related compounds, New
York, London, Plenum Press, pp. 221-240.
STEHL, R.H. & LAMPARSKI, L.L. (1977) Combustion of several
2,4,5-trichloro-phenoxy compounds: formation of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Science, 197: 1008-1009.
STEHL, R.H., PAPENFUSS, R.R., BREDEWEG, R.A., & ROBERTS, R.W. (1973)
The stability of pentachlorophenol and chlorinated dioxins to
sunlight, heat, and combustion. Adv. Chem. Ser., 120: 119-125.
STEHR, P.A., STEIN, G., FALK, H., SAMPSON, E., SMITH, S.M., STEINBERG,
K., WEBB, K., AYRES, S., SCHRAMM, W., DONNELL, H.D., & GEDNEY, W.B.
(1986) A pilot epidemiologic study of possible health effects
associated with 2,3,7,8-tetrachloro-dibenzo-p-dioxin contaminations in
Missouri. Arch. environ. Health, 41: 16-21.
STEINBERG, K.K., MACNEIL, M.L., KARON, J.M., STEHR, P.A., NEESE, J.W.,
& NEEDHAM, L.L. (1985) Assessment of
2,3,7,8-tetrachlorodibenzo-p-dioxin exposure using a modified
d-glucaric acid assay. J. Toxicol. environ. Health, 16: 743-752.
STEINER, M., BORGES, H., FREEDMAN, L., & GRAY, S.J. (1968) Effects of
starvation on the tissue composition of the small intestine in the
rat. Am. J. Physiol., 215: 75-77.
STEWARD, A.R. & BYARD, J.L. (1981) Induction of benzo(a)pyrene
metabolism by 2,3,7,8-tetrachlorodibenzo-p-dioxin in primary cultures
of adult rat hepatocytes. Toxicol. appl. Pharmacol., 59: 603-616.
STOHS, S.J., HASSAN, M.Q., & MURRAY, W.J. (1983) Lipid peroxidation as
a possible cause of TCDD toxicity. Biochem. biophys. Res. Commun.,
111: 854-859.
SUN, T.-T., SHIH, C., & GREEN, H. (1979) Keratin cytoskeletons in
epithelial cells of internal organs. Proc. Natl Acad. Sci. (USA),
76: 2813-2817.
SUN, T.-T., EICHNER, R., NELSON, W.G., TSENG, S.C.G., WEISS, R.A.,
JARVINEN, M., & WOODCOCK-MITCHELL, J. (1983a) Keratin classes:
Molecular markers for different types of epithelial differentiation.
J. invest. Dermatol., 81: 109-115.
SUN, T.-T., EICHNER, R., NELSON, W.G., VIDRICH, A., &
WOODCOCK-MITCHELL, J. (1983b) Keratin expression during normal
epidermal differentiation. In: Seji, M. & Bernstein, I.A., ed.
Current problems in dermatology. Normal and abnormal epidermal
differentiation, New York, Karger, pp. 277-291.
SUNDSTROM, G., JENSEN, S., JANSSON, B., & ERNE, K. (1979) Chlorinated
phenoxyacetic acid derivatives and tetrachloro-dibenzo-p-dioxin in
foliage after application of 2,4,5-trichlorophenoxyacetic acid esters.
Arch. environ. Contam. Toxicol., 8: 441-448.
SUSKIND, R.R. & HERTZBERG, V.S. (1984) Human health effects of 2,4,5-T
and its toxic contaminants. J. Am. Med. Assoc., 251: 2372-2380.
SUSKIND, R.R., CLEVELAND, F., KEENAN, C., AKIN, R., DAVIS, A., &
KEHOE, R.A. (1953) Report on a clinical and environmental survey,
Nitro, West Virginia, Monsanto Chemical Co..
SUTER-HOFMANN, M. & SCHLATTER, Ch. (1985) Toxicity of particulate
emissions from a municipal incinerator: critique of the concept of
TCDD-equivalents. Chemosphere, 15: 1733-1743.
SWEENEY, G.D. & JONES, K.G. (1983) Studies of the mechanism of action
of hepatotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and
related compounds. In: Tucker, R.E., Young, A.L., & Gray, A.P., ed.
Human and environmental risks of chlorinated dioxins and related
compounds, New York, London, Plenum Press, pp. 415-422.
SWEENEY, G.D., JONES, K.G., COLE, F.M., BASFORD, D., & KRESTYNSKI, F.
(1979) Iron deficiency prevents liver toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Science, 204: 332-335.
SWIFT, L.L., GASIEWICZ, T.A., DUNN, G.D., SOULE, P.D., & NEAL, R.A.
(1981) Characterization of the hyperlipidemia in guinea pigs induced
by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. appl. Pharmacol.,
59: 489-499.
SWITZERLAND (1982) Environmental pollution due to dioxins and furans
from chemical rubbish incineration plants, Bern, Ministry of
Environment, Federal Swiss Government.
TAYLOR, M.L., TIERNAN, T.O., RAMALINGAM, B., WAGEL, D.J., GARRETT,
J.H., SOLCH, J.G., & FERGUSON, G.L. (1985) Synthesis, isolation, and
characterization of the tetrachlorinated dibenzo-p-dioxins and other
related compound. In: Keith, L., Rappe, C., & Choudhary, G., ed.
Chlorinated dioxins and dibenzofurans in the total environment. II,
Boston, Butterworth Publishers, pp. 17-35.
TELEGINA, K.A. & BIKBULATOVA, L.I. (1970) [Affection of the follicular
apparatus of the skin in workers occupied in production of butyl ether
of 2,3,4-trichlorphenoxyacetic acid.] Vestn. Dermat. Venerol
(Moscow), 44: 35-39 (in Russian).
TELEKY, L. (1913) [Chlorine and hydrochloric acid. German Empire.]
Wiener Arb. Geb. Soz. Med., 4: 55-56 (in German).
TENCHINI, M.L., CRIMAUDO, C., PACCHETTI, G., MOTTURA, A., AGOSTI, S.,
& DE CARLI, L. (1983) A comparative cytogenetic study on cases of
induced abortions in TCDD-exposed and non-exposed women. Environ.
Mutagen., 5: 73-85.
THIBODEAUX, L.J. (1983) Offsite transport of
2,3,7,8-tetra-chlorodibenzo-p-dioxin from a production disposal
facility. In: Choudhary, G., Keith, L.H., & Rappe, C., ed.
Chlorinated dioxins and dibenzofurans in the total environment,
Boston, Butterworth Publishers, pp. 75-85.
THIESS, A.M., FRENTZEL-BEYME, R., & LINK, R. (1982) Mortality study of
persons exposed to dioxin in a trichlorophenol-pro-cess accident that
occurred in the BASF AG on November 17, 1953. Am. J. ind. Med., 3:
179-189.
THIGPEN, J.E., FAITH, R.E., MCCONNELL, E.E., & MOORE, J.A. (1975)
Increases susceptibility to bacterial infection as a sequela of
exposure to an environmental contaminant
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Infect. Immun., 12:
1319-1324.
THOMAS, P.T., & HINSDILL, R.D. (1979) The effect of perinatal exposure
to tetrachlorodibenzo-p-dioxin on the immune response of young mice.
Drug chem. Toxicol., 2: 77-98
THUNBERG, T. & HAKANSSON, H. (1983) Vitamin A (retinol) status in the
Gunn rat: the effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch.
Toxicol., 53: 225-233.
THUNBERG, T., AHLBORG, U.G., & JOHNSSON, H. (1979) Vitamin A (retinol)
status in the rat after a single oral dose of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Toxicol., 42: 265-274.
THUNBERG, T., AHLBORG, U.G., HAKANSSON, H., KRANTZ, C., & MONIER, M.
(1980) Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the hepatic
storage of retinol in rats with different dietary supplies of vitamin
A (retinol). Arch. Toxicol., 45: 273-285.
THUNBERG, T., AHLBORG, U.G., & WAHLSTROM, B. (1984) Comparison between
the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and six other
compounds on the vitamin A storage, the UDP-glucuronosyltransferase
and the aryl hydrocarbon hydroxylase activity in the rat liver.
Arch. Toxicol., 55: 16-19.
TIERNAN, T.O. (1983) Analytical chemistry of polychlorinated
dibenzo-p-dioxins and dibenzofurans: A review of the current status.
In: Choudhary, G., Keith, L., & Rappe, C., ed. Chlorinated dioxins
and dibenzofurans in the total environment, Boston, Butterworth
Publishers, pp. 211-237.
TOFILON, P.J. (1980) Depressed guinea pig testicular microsomal
cytochrome P-450 content by 2,3,7,8-tetrachloro-dibenzo-p-dioxin.
Life Sci., 27: 871-876.
TOFILON, P.J. & PIPER, W.N. (1982)
2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated depression of rat
testicular heme synthesis and microsomal cytochrome P-450. Biochem.
Pharmacol., 31: 3663-3666.
TOGNONI, G. & BONACCORSI, A. (1982) Epidemiological problems with
TCDD. Drug Metab. Rev., 13: 447-469.
TOSINE, H., SMILLIE, D., & REES, G.A.V. (1983) Comparative monitoring
and analytical methodology for 2,3,7,8-TCDD in fish. In: Tucker, R.E.,
Young, A.L., & Gray, A.P., ed. Human and environmental risks of
chlorinated dioxins and related compounds, New York, London, Plenum
Press, pp. 127-142.
TOTH, K., SOMFAI-RELLE, S., SUGAR, J., & BENCE, J. (1979)
Carcinogenicity testing of herbicide 2,4,5-trichlorophenoxy-ethanol
containing dioxin and of pure dioxin in Swiss mice. Nature (Lond.),
278: 548-549.
TSUKAMOTO, H., MAKISUMI, S., HIROSE, H., KOJIMA, T., FUKUMOTO, H.,
FUKOMOTO, K., KURATSUNE, M., NISHIZUMI, M., SHIBATA, M., NAGAI, J.,
YAE, Y., SAWADA, K., FURUKAWA, M., YOSHIMURA, H., TATSUMI, K., OGURI,
K., SHIMENO, H., KENO, K., KOBAYASHI, H., YANO, T., ITO, A., OKADA,
T., INAGAMI, K., KOGA, T., TOMITA, Y., KOGA, T., YAMADA, Y.,
MIYAGUCHI, M., SUGANO, M., HORI, K., TAKESHITA, K., MANAKO, K.,
NAKAMURA, Y., & SHIGEMORI, N. (1969) [The chemical studies on detecton
of toxic compounds in rice bran oils used by the patients of Yusho.]
Fukuoka Acta med., 6: 496-512 (in Japanese).
TSYRLOV, I.B., CHASOVNIKOVA, O.B., GRISHANOVA, A.YU., & LYAKHOVICH,
V.V. (1986) Reappraisal of the liver benzpyrene hydroxylase
synthesized de novo after treatment of rats with
2,3,7,8-tetrachlorodibenzo-p-dioxin and 3-methylcholanthrene. FEBS
Lett., 198: 225-228.
TUCKER, A.N., VORE, S.J., & LUSTER, M.I. (1986) Supppression of B cell
differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol.
Pharmacol., 29: 372-377.
TUKEY, R.H., HANNAH, R.R., NEGISHI, M., NEBERT, D.W., & EISEN, H.J.
(1982a) The Ah locus: Correlation of intranuclear appearance of
inducer-receptor complex with induction of cytochrome P1-450 mRNA.
Cell, 31: 275-284.
TUKEY, R.H., NEGISHI, M., & NEBERT, D.W. (1982b) Quantitation of
hepatic cytochrome P1-450 mRNA with the use of a cloned DNA probe.
Effects of various P-450 inducers in C57BL/6N and DBA/2N mice. Mol.
Pharmacol., 22: 779-786.
TULP, M. & HUTZINGER, O. (1978) Rat metabolism of polychlori-nated
dibenzo-p-dioxins. Chemosphere, 9: 761-768.
TUNG, T.T. (1973) Le cancer primaire du foie au Vietnam. Chirurgie,
99: 427-436.
TURNER, J.N. & COLLINS, D.N. (1983) Liver morphology in guinea pigs
administered either pyrolysis products of a polychlorinated biphenyl
trans former fluid or 2,3,7,8-tetrachloro-dibenzo-p-dioxin. Toxicol.
appl. Pharmacol., 67: 417-429.
TUTEJA, N., GONZALEZ, F.J., & NEBERT, D.W. (1985) Developmental and
tissue-specific differential regulation of the mouse dioxin-inducible
P1-450 and P3-450 genes. Dev. Biol., 112: 177-184.
UMBREIT, T.H., PATEL, D., & GALLO, M.A. (1985) Acute toxicity of TCDD
contaminated soil from an industrial site. Chemosphere, 14(6/7):
945-947.
UMBREIT, T.H., HESSE, E.J., & GALLO, M.A. (1986a) Bioavailability of
dioxin in soil from a 2,4,5-T manufacturing site. Science, 232:
497-499.
UMBREIT, T.H., HESSE, E.J., & GALLO, M.A. (1986b) Comparative toxicity
of TCDD contaminated soil from Times Beach, Missouri, and Newark, New
Jersey. Chemosphere, 15: 2121-2124.
UOTILA, P., PARKKI, M.G., & AITO, A. (1978) Quantitative and
qualitative changes in the metabolism of benzo(a)pyrene in rat tissues
after intragastric administration of TCDD. Toxicol. appl.
Pharmacol., 46: 671-683.
US EPA (1982) Environmental monitoring at Love Canal, Washington,
DC, US Environmental Protection Agency, Office of Research and
Development, Vol. I (EPA/600/4-82-030a).
US EPA (1985) Health assessment document for polychlorinated
dibenzo-p-dioxins, Washington, DC, US Environmental Protection
Agency, Office of Health and Environmental Assessment
(EPA/600/8-84/0146).
US EPA (1987) Interim procedures for estimating risk associated with
exposures to mixtures of chlorinated dibenzo-p-dioxins and
-dibenzofurans (CDDs and CDFs). Risk assessment forum, Washington,
DC, US Environmental Protection Agency, 48 pp (EPA/625/3-87/012).
VAHRENHOLT, F. (1977) [Seveso - An unparalleled catastrophe.]
Umwelt, 1: 59, 60, 62, 64 (in German).
VAN, D.D. (1984) Herbicides as a possible cause of liver cancer. In:
Westing, A.H., ed. Herbicides in war, the long-term ecological and
human consequences, London, Taylor and Francis, pp. 119-121.
VAN DEN BERG, M., OLIE, K., & HUTZINGER, O. (1983) Uptake and
selective retention in rats of orally administered chlorinated dioxins
and dibenzofurans from fly-ash and fly-ash extract. Chemosphere,
12(4/5): 537-544.
VAN DEN BERG, M., VAN GREEVENBROEK, M., & OLIE, K. (1986a)
Bioavailability of PCDDs and PCDFs on fly ash after semi-chronic oral
ingestion by the rat. Chemosphere, 15(4): 509-518.
VAN DEN BERG, M., DE VROOM, E., & OLIE, K. (1986b) Bioavailability of
PCDDs and PCDFs on fly ash after semi-chronic oral ingestion by guinea
pig and syrian golden hamster. Chemosphere, 15(4): 519-533.
VAN DEN BERG, M., VAN DER WIELEN, F.W.M., & OLIE, K. (1986) The
presence of PCDDs and PCDFs in human breast milk from the Netherlands.
Chemosphere, 15: 693-706.
VAN LOGTEN, M.J., GUPTA, B.N., MCCONNELL, E.E., & MOORE, J.A. (1980)
Role of the endocrine system in the action of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the thymus.
Toxicology, 15: 135-144.
VAN MILLER, J.P., MARLAR, R.J., & ALLEN, J.R. (1976) Tissue
distribution and excretion of tritiated tetrachlorodibenzo-p-dioxin in
non-human primates and rates. Food Cosmet. Toxicol., 14: 31-34.
VAN MILLER, J.P., LALICH, J.J., & ALLEN, J.R. (1977) Increased
incidence of neoplasms in rats exposed to low levels of
2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere, 61: 625-632.
VECCHI, A., MANTOVANI, A., SIRONI, M., LUINI, W., CAIRO, M., &
GARATTINI, S. (1980) Effect of acute exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin on humoral antibody production in
mice. Chem.-biol. Interact., 30: 337-342.
VECCHI, A., SIRONI, M., CANEGRATI, M.A., RECCHIA, M., & GARATTINI, S.
(1983) Immunosuppressive effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in strains of mice with different susceptibility to induction of aryl
hydrocarbon hydroxylase. Toxicol. appl. Pharmacol., 68: 434-441.
VEERKAMP, W., WEVER, J., & HUTZINGER, O. (1981) The metabolism of some
chlorinated dibenzofurans by rats. Chemosphere, 10: 397-403.
VILLENUEVE, E.C., JENNINGS, R.W., BURSE, V.M., & KIMBROUGH, R.D.
(1974) Evidence of chlorodibenzo-p-dioxin and chlorodibenzofuran in
hexachlorobenzene. J. agric. food Chem., 22: 916-917.
VINOPAL, J.H. & CASIDA, J.E. (1973) Metabolic stability of
2,3,7,8-tetra-chlorodibenzo-p-dioxin in mammalian liver microsomal
systems and in living mice. Arch. environ. Contam. Toxicol., 1:
122-132.
VISWANATHAN, T.S. & KLEOPFER, R.D. (1986) The presence of
hexachloroxanthene at Missouri dioxin sites, In: Rappe, C., Choudhary,
G., & Keith, L., ed. Chlorinated dioxins and dibenzofurans in
perspective, Chelsea, Michigan, Lewis Publishers, pp. 201-210.
VOS, J.G. & BEEMS, R.B. (1971) Dermal toxicity studies of technical
polychlorinated biphenyls and fractions thereof in rabbits. Toxicol.
appl. Pharmacol., 19: 617-633.
VOS, J.G. & KOEMAN, J.H. (1970) Comparative toxicologic study with
polychlorinated biphenyls in chickens with special reference to
porphyria, edema formation, liver necrosis, and tissue residues.
Toxicol. appl. Pharmacol., 17: 656-668.
VOS, J.G. & MOORE, J.A. (1974) Suppression of cellular immunity in
rats and mice by maternal treatment with
2,3,7,8-tetrachlorodibenzo-p-dioxin. Int. Arch. Allergy, 47:
777-794.
VOS, J.G., KOEMAN, J.H., VAN DER MAAS, H.L., TEN NOEVER DE BRAUW,
M.C., & DE VOS, R.H. (1970) Identification and toxicological
evaluation of chlorinated dibenzofuran and chlorinated napthalene in
two commercial polychlorinated biphenyls. Food Cosmet. Toxicol.,
8: 625-633.
VOS, J.G., MOORE, J.A., & ZINKL, J.G. (1973) Effect of
2,3,7,8-tetrachlorodibenzo-p-dioxin on the immune system of laboratory
animals. Environ. Health Perspect., 5: 149-162.
VOS, J.G., MOORE, J.A., & ZINKL, J.G. (1974) Toxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57 B1/6 mice.
Toxicol. appl. Pharmacol., 29: 229-241.
VOS, J.G., KREEFTENBERG, J.G., ENGEL, H.W.B., MINDERHOUD, A., & VAN
NOORLE JANSEN, L.M. (1978a) Studies on
2,3,7,8-tetra-chlorodibenzo-p-dioxin-induced immune suppression and
decreased resistance to infection: Endotoxin hypersensitivity, serum
zinc concentrations and effect of thymosin treatment. Toxicology,
9: 75-86.
VOS, J.G., KREEFTENBERG, J.G., & KATER, L. (1978b) Immune suppression
by TCDD. In: Cattabeni, F., Cavallaro, A., & Galli, ed. Dioxin
(TCDD), New York, John Wiley & Sons, Halsted Press Division, pp.
163-175.
WAHLE, P. (1914) [On two cases of chloracne.] Inaugural dissertation
for the Doctorate in Medicine, Surgery and Obstetrics of the Medical
Faculty, University of Leipzig (in German).
WALDEN, R. & SCHILLER, C.M. (1985) Short communications. Comparative
toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in four
(sub)strains of adult male rats. Toxicol. appl. Pharmacol., 77:
490-495.
WARD, C.T. & MATSUMURA, F. (1978) Fate of
2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD) in a model aquatic
environment. Arch. environ. Contam. Toxicol., 7: 349-357.
WAUER, (1918) [Occupational illness caused by chlorinated hydrocarbons
(perna disease).] Zentralbl. Gewerbehyg., 6: 100-101 (in German).
WEBER, H., & BIRNBAUM, L. S. (1985)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin (TCDD) and
2,3,7,8-tetrachlorodibenzofuran (TCDF) in pregnant C57Bl/6N mice:
Distribution to the embryo and excretion. Arch. Toxicol., 57:
159-162.
WEBER, H., POIGER, H., & SCHLATTER, Ch. (1982a) Acute oral toxicity of
TCDD-metabolites in male guinea pigs. Toxicol. Lett., 14: 117-122.
WEBER, H., POIGER, H., & SCHLATTER, Ch. (1982b) Fate of
2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites from dogs in rats.
Xenobiotica, 12: 353-357.
WEBER, H., LUZI, P., RESI, L., TANGANELLI, P., LOVATI, M.R., & POLI,
A. (1983) Natural history of TCDD-induced liver lesions in rats as
observed by transmission electron microscopy during a 32-week period
after a single intraperitoneal injection. J. Toxicol. environ.
Health, 12: 533-540.
WEBER, H., LAMB, J.C., HARRIS, M.W., & MOORE, J.A. (1984)
Teratogenicity of 2,3,7,8-tetrachlorodibenzofuran (TCDF) in mice.
Toxicol. Lett., 20: 183-188.
WEBER, H., HARRIS, M.W., HASEMAN, J.K., & BIRNBAUM, L.S. (1985)
Teratogenic potency of TCDD, TCDF and TCDD-TCDF combinations in
C57Bl/6N mice. Toxicol. Lett., 26: 159-167.
WEBSTER, G.R.B., FRIESEN, K.J., SARNA, L.P., & MUIR, D.C.G. (1985)
Environmental fate modelling of chlorodioxins: Determination of
physical constants. Chemosphere, 14: 609-622.
WEDEL, J., VON, HOLLA, W.A., & DENTON, J. (1943) Observations on the
toxic effects resulting from exposure to chlorinated naphtalene and
chlorinated phenyls with suggestions for prevention. Rubber Age,
53: 419-426.
WEISSBERG, J.B. & ZINKL, J.G. (1973) Effects of
2,3,7,8-tetra-chlorodibenzo-p-dioxin upon hemostasis and hematologic
function in the rat. Environ. Health Perspect., 5: 119-123.
WESTING, A.H., (1984) Herbicides in war: past and present. In:
Westing, A.H., ed. Herbicides in war, the long-term ecological and
human consquences, London, Taylor and Francis.
WHITE, K.L., Jr, LYSY, H.H., MCCAY, J.A., & ANDERSON, A.C. (1986)
Modulation of serum complement levels following exposure to
polychlorinated dibenzo-p-dioxins. Toxicol. appl. Pharmacol., 84:
209-219.
WHITLOCK, J.P. & GALEAZZI, D.R. (1984)
2,3,7,8-Tetrachloro-dibenzo-p-dioxin receptors in wild type and
variant mouse hepatoma cells. J. biol. Chem., 259: 980-985.
WHO/EURO (1987) Dioxins and furans from municipal incinerators,
Copenhagen, WHO Regional Office for Europe (Environmental Health
Series 17).
WIKLUND, K. & HOLM, L.-E. (1986) Soft tissue sarcoma risk in Swedish
agricultural and forestry workers. J. Natl Cancer Inst., 76:
229-234.
WILLEY, J.C., SALADINO, A.J., OZANNE, C., LECHNER, J.F., & HARRIS,
C.C. (1984) Acute effects of 12-O-tetradecanoyl-phorbol-13-acetate,
teleocidin B, or 2,3,7,8-tetrachloro-dibenzo-p-dioxin on cultured
normal human bronchial epithelial cells. Carcinogenesis, 5:
209-215.
WILLIAMS, D.T., CUNNINGHAM, H.M., & BLANCHFIELD, B.J. (1972)
Distribution and excretion studies of octachlorodibenzo-p-dioxin in
the rat. Bull. Environ. Contam. Toxicol., 7: 57-62.
WIPF, H.K. & SCHMID, J. (1983) Seveso - An environmental assessment.
In: Tucker, R.E., Young, A.L., & Gray, R., ed. Human and
environmental risks of chlorinated dioxins and related compounds,
New York, London, Plenum Press, pp. 255-276.
WIPF, H.K., HOMBERGER, E., NEUNER, N., RANALDER, U.B., VETTER, W., &
VUILLEUMIER, J.P. (1982) TCDD-levels in soil and plant samples from
the Seveso area. In: Hutzinger, O., Frei, R.W., Merian, E., &
Pocchiari, F., ed. Chlorinated dioxins and related compounds. Impact
on the environment, Oxford, New York, Pergamon Press, pp. 115-127.
WOLF, G. (1980) Vitamin A. In: Alfin-Slater, R.B. & Kritchevsky, D.,
ed. Human nutrition, a comprehensive treatise. Part B. Nutrition and
the adult, New York, London, Plenum Press, Vol. 3, pp. 97-203.
WOODS, J.S. (1973) Studies of the effects of 2,3,7,8-tetra-
chlorodibenzo-p-dioxin on mammalian hepatic delta-amino- levulinic
acid synthetase. Environ. Health Perspect., 5: 221-225.
WROBLEWSKI, V.J. & OLSON, J.R. (1985) Hepatic metabolism of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat and guinea pig.
Toxicol. appl. Pharmacol., 81: 231-240.
WU, Y.C., LO, Y.C., KAO, H.Y., PAN, C.C., & LIN, R.Y. (1984)
Cell-mediated immunity in patients with polychlorinated bi-phenyl
poisoning. J. Formoson Med. Assoc., 83: 419-429.
YAMAGISHI, T., MIYAZAKI, T., AKIYAMA, K., MORITA, M., NAKAGAWA, J.,
HORII, S., & KANEKO, S. (1981) Polychlorinated dibenzo-p-dioxins and
dibenzofurans in commercial diphenyl ether herbicides, and in fresh
water fish collected from the application area. Chemosphere, 10:
1137-1144.
YANG, K.H., CROFT, W.A., & PETERSON, R.E. (1977) Effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin on plasma disappearance and
biliary excretion of foreign compounds in rats. Toxicol. appl.
Pharmacol., 40: 485-496.
YANG, K.H., CHOI, E.J., & CHOE, S.Y. (1983a) Cytotoxicity of
2,3,7,8-tetrachlorodibenzo-p-dioxin on primary cultures of adult rat
hepatocytes. Arch. environ. Contam. Toxicol., 12: 183-188.
YANG, K.H., YOO, B.S., & CHOE, S.Y. (1983b) Effects of halogenated
dibenzo-p-dioxins on plasma disappearance and biliary excretion of
ouabain in rats. Toxicol. Lett., 15: 259-264.
YOCKIM, R.S., ISENSEE, A.R., & JONES, G.E. (1978) Distribution and
toxicity of TCDD and 2,4,5-T in an aquatic model ecosystem.
Chemosphere, 7: 215-220.
YOSHIHARA, S., NAGATA, K., YOSHIMURA, H., KUROKI, H., & MASUDA, Y.
(1981) Inductive effect on hepatic enzymes and acute toxicity of
individual polychlorinated dibenzofuran congeners in rats. Toxicol.
appl. Pharmacol., 59: 580-588.
YOSHIMURA, H., KAMIMURA, H., OGURI, K., HONDA, Y., & NAKANO, M. (1986)
Stimulating effect of activated charcoal beads on fecal excretion.
Chemosphere, 15(3): 219-227.
YOUNG, A.L. (1983) Long-term studies on the persistence and movement
of TCDD in a natural ecosystem. In: Tucker, R.E., Young, A.L., & Gray,
P., ed. Human and environmental risks of chlorinated dioxins and
related compounds, New York, London, Plenum Press, pp. 173-190.
YOUNG, A.L., KANG, H.K., & SHEPARD, B.M. (1983) Clorinated dioxins as
herbicide contaminants. Environ. Sci. Technol., 17: 530A-532A.
YOUNG, A.L., THALKEN, C.E., & WARD, W.E. (1975) Studies of the
ecological impact of repetitive aerial applications of herbicides on
the ecosystem of test area C-52A, Eglin Air Force Base, Florida,
Washington, DC, US Department of Defence, 127 pp. (Final Report
October 1975).
YOUNG, A.L., THALKEN, C.E., ARNOLD, E.L., CUPELLO, J.M., & COCKERHAM,
L.G. (1976) Fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in
the environment: Summary and decontamination recommendations, US Air
Force Academy, Colorado, Washington, DC, US Department of Defence
(Document 19 USAFA TR-76-8).
ZACK, J.A. & GAFFEY, W.R. (1983) A mortality study of workers employed
at the Monsanto Company Plant in Nitro, West Virginia. Environ. Sci.
Res., 26: 575-591.
ZACK, J.A. & SUSKIND, R.R. (1980) The mortality experience of workers
exposed to tetrachlorodibenzodioxin in a trichloro-phenol process
accident. J. occup. Med., 22: 11-14.
ZEDDA, S., CIRLA, A.M., & SALA, C. (1976) [Accidental
tetrachlorodibenzoparadioxin contamination. Considerations on the
I.C.M.E.S.A. episode.] Med. Lav., 67: 371-378 (in Italian).
ZELIKOV, A.K. & DANILOV, L.N. (1974) Occupational dermatoses (acnes)
in workers engaged in production of 2,4,5-trichloro-phenol. Sov.
Med., 7: 145-146.
ZINKL, J.G., VOS, J.G., MOORE, J.A., & GUPTA, B.N. (1973) Hematologic
and clinical chemistry effects of 2,3,7,8-tetra-chlorodibenzo-p-dioxin
in laboratory animals. Environ. Health Perspect., 5: 111-118.
RESUME ET RECOMMANDATIONS
1. Résumé
1.1 Sources
Les dibenzo-para-dioxines polychlorées (PCDD) et les
dibenzofurannes polychlorés (PCDF) constituent deux séries
d'hydrocarbures aromatiques tricycliques halogénés de propriétés
physico-chimiques très voisines, présentes un peu partout dans
l'environnement. Leur origine n'est pas naturelle et ils ne sont pas
produits intentionnellement. On connait 75 isomères (isomérie de
position) pour les PCDD et 135 pour les PCDF.
Les sources les plus importantes de contamination par ces deux
séries de composés sont les suivantes:
- contamination de produits chimiques du commerce, par exemple
chlorophénols et dérivés, PCB;
- incinération de déchets municipaux, de déchets dangereux et
de déchets d'hôpitaux, ainsi que de boues provenant du
traitement des effluents;
- fonctionnement des véhicules automobiles;
- utilisation de combustibles fossiles;
- surchauffe et émissions de foyers faisant intervenir des
béniphyles polychlorés (PCB);
- élimination de déchets industriels provenant, par exemple,
de la production de chlorophénols et de leurs dérivés, du
traitement du bois par le chlorophénol, de l'emploi de PCB
liquides dans l'appareillage électrique et du traitement du
papier et de la pâte à papier.
1.2 Concentrations dans l'environnement et voies d'exposition
D'après les quelques données dont on dispose, la concentration de
ces composés serait très faible dans l'air, le sol et les sédiments,
de l'ordre du femtogramme (fg) par mètre cube dans l'air et du
nanogramme (ng) par kilogramme dans le sol et les sédiments. On a
observé des concentrations allant jusqu'à 50 mg/kg, aussi bien pour
les PCDD que pour les PCDF, dans certains organismes aquatiques à
l'état naturel. On dispose de très peu de données sur la contamination
de l'eau de boisson et des denrées alimentaires vendues dans le
commerce.
Pour ce qui est de la population en général, l'exposition à ces
composés semble principalement associée à la chaîne alimentaire.
Certains travailleurs peuvent être très exposés lorsqu'ils sont
affectés à la production, la mise en oeuvre ou à la destruction de
matériaux contenant des PCDD ou des PCDF, ainsi que leurs précurseurs.
Pour ce personnel, l'inhalation et les contacts cutanés constituent
les principales voies d'exposition à prendre en compte.
1.3 Toxicocinétique, biotransformation et surveillance
biologique
La biodisponibilité des PCDD et des PCDF dépend de la matrice au
sein de laquelle ils sont incorporés et de la voie d'exposition. On ne
dispose pour aucune des espèces en cause de données sur la
biodisponibilité par inhalation.
On ignore la quantité absorbée par l'homme en cas d'exposition,
par quelque voie que ce soit.
Les études sur des rongeurs à qui l'on a administré une ou
plusieurs doses de 2,3,7,8-TCDD par voie orale montrent qu'environ la
moitié de la dose administrée est absorbée dans les voies digestives.
La demi-vie d'élimination se situe, d'après la littérature, entre 12
et 94 jours chez les rongeurs. Chez le singe rhésus, la demi-vie de
la 2,3,7,8-TCDD est d'environ 1 an dans le tissu adipeux.
Les observations concernant la toxicocinétique des PCDD chez les
animaux sont limitées, en dehors du cas de la 2,3,7,8-TCDD. Pour ce
dérivé, on a observé une demi-vie de l'ordre de 2 à 8 jours chez le
rat, la souris et le singe et de plus de 20 jours chez le cobaye.
Chez le rat, on a constaté que la pentachlore-2,3,4,7,8 dibenzofuranne
s'élimine moins rapidement que la 2,3,7,8-TCDD.
Les études sur la rétention tissulaire des PCDD et des PCDF chez
diverses espèces exposées à des mélanges synthétiques ou à des
échantillons prélevés dans l'environnement et contenant des PCDD ou
des PCDF montrent que la durée de rétention des divers analogues est
très variable selon qu'ils sont ou non substitués en 2, 3, 7, ou 8 par
un atome de chlore.
D'après quelques observations effectuées sur l'homme, la demi-vie
serait de 2 à 6 ans pour certains PCDD et PCDF substitués en 2, 3, 7
et 8.
Les PCDD et les PCDF s'accumulent principalement dans les
graisses; ils sont également excrétés dans le lait et traversent la
barrière placentaire. On en trouve également à faible concentration,
dans le sang et les organes vitaux.
La distribution tissulaire chez l'homme est encore mal élucidée,
encore qu'il semble que le rapport des concentrations dans les tissus
adipeux et dans le foie soit plus élevé que chez les rongeurs.
Dans l'ensemble de la population, on observe une concentration
résiduelle de PCDD dans le tissu adipeux qui atteint 20 mg/kg, en
l'absence de toute exposition particulière connue; mais des chiffres
plus élevés ont été signalés quelquefois, sans aucun signe de maladie.
Dans les deux cas, les groupes de population étudiés n'avaient pas été
constitués par sondage aléatoire. Dans ces échantillons, on a observé
les PCDD et les PCDF les plus substitués par du chlore, en particulier
des octachlorodibenzo-p-dioxines. La concentration tissulaire des TCDD
avait tendance à augmenter avec l'âge.
1.4 Effets sur la santé
1.4.1 Animaux
Les effets toxiques et biologiques découlant de l'exposition aux
2,3,7,8-TCDD dépendent de divers facteurs, dont l'espèce, la souche,
l'âge et le sexe des animaux d'expérience. Entre autres signes de
toxicité observés chez plusieurs espèces animales, on note une perte
de poids, une hépatoxicité, une porphyrie, une atteinte cutanée, des
lésions gastriques, une atrophie du thymus, une atteinte du système
immunitaire, des effets tératogènes, des effets sur la reproduction et
des effets cancérogènes. Les TCDD exercent des effets biologiques
extrêmement divers, notamment l'induction de certaines enzymes et une
carence en vitamine A. Ces effets ne sont pas tous observés chez la
même espèce animale. Les effets toxiques les plus caractéristiques
qu'on retrouve chez tous les animaux de laboratoire sont la perte de
poids, l'atrophie du thymus et l'immunotoxicité. Chez l'homme, une
chloracné et des lésions cutanées de ce type constituent les signes
les plus fréquents d'une intoxication par les 2,3,7,8-TCDD; on observe
également des lésions cutanées chez le rhésus, la souris glabre et le
lapin. En revanche, l'exposition au 2,3,7,8-TCDD ne provoque ni
chloracné ni lésions cutanées de ce type chez la plupart des rongeurs.
De nombreuses lésions toxiques s'observent principalement au niveau
des tissus épithéliaux.
Des effets ont été observés sur la reproduction chez le rhésus et
chez le rat. La plus faible dose exerçant un effet serait de l'ordre
de 1-2 ng/kg de poids corporel par jour. Dans deux études de
cancérogénicité chez le rat, on a obtenu un épithélioma
hépatocellulaire avec des doses respectives d'environ 0,1 et 0,01
µg/kg de poids corporel par jour. Une dose réduite à 0,001 µg/kg de
poids corporel se traduit par une modification des hépatocytes au
niveau de certains foyers ou territoires. L'incidence de certaines
tumeurs à déterminisme hormonal était plus faible que chez les animaux
témoins.
Apparemment, les TCDD n'ont pas de propriétés mutagènes et ne
devraient donc pas être génotoxiques. Il faut donc admettre que les
effets cancérogènes s'expliquent par un mécanisme indirect.
Plusieurs autres PCDD et PCDF déterminent des manifestations
analogues à celles que provoquent les 2,3,7,8-TCDD, mais leur activité
est très inégale. On connaît 12 isomères qui présentent une toxicité
plus élevée, à savoir les tétra-, penta-, hexa-et heptaCDD ou CDF qui
possèdent quatre atomes de chlore symétriques deux à deux, en
poisition 2,3,7 et 8. Des études prolongées sur l'animal ont révélé
l'action cancérogène d'un mélange de deux
hexachlorodibenzo-p-dioxines (1,2,3,7,8,9-et 1,2,3,6,7,8-hexaCDD),
mais en présence de doses plus élevées que celles qu'on avait
utilisées dans l'étude des TCDD. La dibenzo-p-dioxine et la
dichloro-2,7 dibenzo-p-dioxine ne se sont pas révélées cancérogènes.
Les activités relatives, toxiques et biologiques, des PCDD et des PCDF
ont été estimées au moyen d'études de courte durée chez le rat et en
culture de cellules mammaliennes.
Selon les espèces, les animaux présentent une sensibilité très
inégale aux effets biologiques et toxiques des PCDD et PCDF substitués
en 2,3,7,8. Par exemple, la DL50 par voie orale varie, pour les
2,3,7,8-TCDD, de 0,6 µg/kg de poids corporel chez le cobaye à 5051
µg/kg chez le hamster doré syrien. Les différences considérables de
sensibilité vis-à-vis des 2,3,7,8-TCDD et des composés apparentés
qu'on observe ainsi chez les diverses espèces et souches est
impossible à expliquer par les différences qu'on constate dans la
toxicocinétique. La toxicité et la toxicocinétique des TCDD chez les
singes sont à peu près les mêmes que chez l'homme. Chez des souris de
lignée pure, on a des raisons de penser que la concentration
cellulaire du récepteur aux Ah est partiellement corrélée avec la
sensibilité à l'action biologique et toxique de ces composés. Le
récepteur a été retrouvé chez d'autres espèces, notamment chez
l'homme. Cependant, l'abondance relative du récepteur aux Ah selon les
espèces n'explique pas entièrement les différences de sensibilité.
1.4.2 Homme
Malgré de nombreuses études cliniques et études de suivi
post-thérapeutiques, aucun effet général persistant bien net n'a été
mis en évidence, si ce n'est une chloracné, à la suite d'expositions
professionnelles ou accidentelles aux PCDD et aux PCDF. D'autres
effets ont été notés mais aucun n'est persistant, en dehors de la
chloracné et, peut-être, de troubles fonctionnels minimes.
Dans quelques études épidémiologiques consacrées à des cas
d'exposition à un mélange de dioxines, de furannes et d'autres
produits chimiques, on a fait état d'une augmentation de l'incidence
des cancers de localisations diverses, mais plusieurs facteurs ne
permettent pas de faire entièrement foi à ces observations.
Lors de l'accident de Seveso, le seul effet nocif indiscutable a
été la chloracné. Cette affection (193 cas) s'est déclarée en 1976 et
1977 et elle était encore active en 1984 chez 20 des victimes. De
nombreuses études ont été consacrées à la recherche de liens possibles
entre l'exposition à l'Agent orange et des effets nocifs chez les
civils ou les militaires, au Viet Nam. Cependant, les données dont on
dispose à l'heure actuelle ne permettent pas de conclure
catégoriquement à l'existence d'effets sur la reproduction ou d'autres
effets biologiques notables.
Lors de l'incident survenu au Missouri, les enfants chez qui l'on
avait observé des signes aigus d'intoxication lors de la
contamination, en 1971, sont, semble-t-il, aujourd'hui en bonne santé.
En outre, des études épidémiologiques poursuivies dans ce même Etat au
sein de populations plus longuement exposées à des concentrations plus
faibles de dioxine n'ont jusqu'ici révélé aucun effet appréciable sur
la santé. Mais si aucun symptôme clinique n'a été observé, on a
constaté un effet sur l'immunité à médiation cellulaire.
Les seuls cas d'intoxication humaine par les PCDF pour lesquels
on dispose d'observations circonstanciées concernent la contamination
d'huile de riz par des PCDF, des PCB et des PCQ, observée à Yusho au
Japon, en 1968, et Yu-cheng à Taiwan, en 1979. Au total, plusieurs
milliers de personnes ont été gravement intoxiquées. Il semble très
probable que les PCDF sont à incriminer dans ces intoxications. La
symptomatologie générale était analogue à celle qui accompagne les
intoxi-cations par les TCDD, compte tenu des différences traduisant
l'intensité de l'exposition et la structure d'âge et de sexe des
groupes exposés.
A Yusho, l'apport quotidien moyen de PCDF substitués en 2,3,7,8
a été estimé à 0,1-0,2 µg/kg de poids corporel sur plusieurs mois,
alors que la dose pathogène minimale a été estimée à 0,05-0,1 µg/kg de
poids corporel par jour, sur 30 jours.
1.5 Conclusion
Les PCDD et les PCDF se rencontrent dans tout l'environnement et
il est probable que chacun en est porteur. Ces composés déterminent
parfois des effets toxiques complexes en cas d'exposition
professionnelle ou accidentelle.
D'après les observations faites à Yusho, les expériences
conduites chez des espèces sensibles de singes, et compte tenu de
certaines hypothèses concernant l'activité relative des PCDD et des
PCDF, il se peut que l'homme et certains singes présentent une
sensibilité comparable à l'égard de ces composés. Cependant, les
incertitudes concernant la dose effectivement reçue et les difficultés
de l'évaluation des effets toxiques autres que la chloracné
interdisent, dans le cas de l'homme, une conclusion catégorique quant
à la résistance relative de l'homme aux effets toxiques de ces
composés. Il convient de limiter l'exposition aux plus faibles niveaux
compatibles avec les impératifs pratiques.
2. Recommandations
1. Des études interlaboratoires de validation, selon une sorte de
"poule" avec protocoles normalisés de contrôle et d'assurance de
qualité, sont indispensables pour améliorer les méthodes d'analyse.
La méthodologie de l'échantillonnage, les méthodes d'analyse et
l'interprétation des données doivent être optimalisées et normalisées
avant le démarrage des enquêtes.
2. Des données complémentaires sont nécessaires sur l'origine des
PCDD et des PCDF, sur leur répartition dans l'environnement et sur
leur destinée.
D'autres observations sont indispensables au sujet de la présence
des PCDD ou des PCDF dans l'environnement, notamment sur l'évolution
des concentrations dans le temps et sur la détermination des profils
isomériques, notamment dans les produits alimentaires, dans l'air
ambiant et dans les sédiments.
3. Il serait bon de faire des observations sur les effets
qu'exercent les PCDD et les PCDF sur la biocénose.
4. Des données plus complètes sont nécessaires en ce qui concerne la
biodisponibilité des PCDD et des PCDF provenant de diverses matrices,
dans l'environnement et dans la ration alimentaire. L'exposition à ces
sources devrait être mise en corrélation avec les pratiques agricoles
et industrielles.
5. Il convient de mettre au point et de valider des méthodes
chimiques et biologiques plus simples et moins coûteuses pour
rechercher la présence de PCDD et de PCDF.
6. Des études sur les mécanismes toxiques des PCDD et des PCDF sont
nécessaires pour élucider les différences observées dans les effets
produits chez les diverses espèces et permettre une extrapolation à
l'homme.
7. Il importe de poursuivre l'étude de l'immunotoxicité, notamment
en ce qui concerne la fonction des lymphocytes T cytotoxiques. En
particulier, il est capital d'étudier la nature et la durée des effets
sur le système immunitaire d'une exposition pendant la période
périnatale.
8. Des études de toxicité à long terme devraient être effectuées,
notamment des études de reproduction sur plusieurs générations chez
diverses espèces, au moyen de trois des PCDD et des PCDF les plus
répandus, à savoir le 2,3,4,7,8-pentaCDF, le 1,2,3,7,8-pentaCDD et
l'octaCDD.
9. Etant donné que l'homme est exposé à des mélanges complexes de
PCDD et de PCDF, il faut poursuivre la mise au point et la validation
de divers systèmes d'épreuve, notamment des cultures de cellules
humaines en vue d'apprécier l'activité toxique de ces composés et de
leurs mélanges. Ces systèmes permettront d'étudier les mécanismes
d'action, les relations structure/activité et les effets interactifs.
10. Il serait bon d'étudier la charge de l'organisme en PCDD et PCDF
et d'établir une corrélation entre cette charge, les effets cliniques
constatés et les résultats des examens de laboratoire. Le suivi de
groupes exposés antérieurement est important à cet égard.
EVALUATION DES DANGERS POUR LA SANTE DE L'HOMME
DE L'EXPOSITION AUX DIBENZO-P-DIOXINES POLYCHLORES (PCDD)
ET AUX DIBENZOFURANNES POLYCHLORES (PCDF)
1. Introduction
Pour évaluer les dangers que représentent les PCDD et les PCDF
pour la santé humaine, il faut connaître à la fois l'intensité de
l'exposition et les effets biologiques correspondants.
Il est fort difficile d'évaluer l'exposition humaine. Plusieurs
méthodes sont possibles, par exemple:
a) Utilisation de modèles physiologiques classiques de
l'inhalation, de l'ingestion et de l'absorption cutanée. Il
faut connaître de façon précise la concentration des
produits en cause dans les différents milieux et dans les
denrées alimentaires, ainsi que leur biodisponibilité.
b) Estimations de l'apport sur la base de modèles
pharmacocinétiques simples et des teneurs observées dans les
divers tissus humains en vue d'une évaluation précise de
l'exposition. Il faut en outre connaître en détail l'apport,
la distribution, le métabolisme et l'élimination des PCDD et
des PCDF chez l'homme.
2. Evaluation de l'exposition
2.1 Sources de contamination
Les principales sources de PCDD et de PCDF identifiées jusqu'ici
sont les contaminants contenus dans les produits chimiques vendus dans
le commerce (voir section 3.3), les émissions par suite de combustion
(voir section 3.5) et l'élimination de déchets industriels contenant
des PCDD ou des PCDF (sections 3.4, 3.5.9 et 3.5.10).
Dans certains cas, on peut estimer au niveau local la
contribution relative de ces diverses sources. Mais, les méthodes et
les données dont on dispose aujourd'hui ne permettent pas de
conclusions définitives quant à leur importance relative au niveau
d'un pays ou du monde entier.
2.2 Concentrations dans le milieu ambiant
Les données restreintes dont on dispose montrent que la
concentration atmosphérique résiduelle est très faible (fg/m3) pour
les 2,3,7,8-tétrachloro-PCDD ou PCDF ainsi que pour les dérivés
pentachlorés ou hexachlorés. (Si l'on tient compte des analogues
heptachlorés ou octachlorés, on observe des concentrations de l'ordre
du picogramme par mètre cube).
D'après les rares données disponibles, il est peu probable qu'on
trouve des tétrachloro-2,3,7,8-PCDD ou PCDF ni des dérivés
pentachlorés ou hexachlorés dans une eau de boisson traitée, même à la
concentration de 1 pg/litre (voir section 5.2). Dans tous les
échantillons de sols et de sédiments analysés (provenant de régions
industrialisées ou non), la concentration de PCDD et de PCDF allait de
quelques nanogrammes à plusieurs centaines de nanogrammes par
kilogramme (la concentration la plus élevée concernant les sédiments
et le sol en zone urbaine).
On ne connaît pas la concentration résiduelle des PCDD et des
PCDF dans les végétaux en général.
Des concentrations atteignant 50 ng/kg ont été observées chez des
poissons capturés dans la nature pour les mêmes dérivés
(principalement les dérivés tétrachlorés et pentachlorés). Le plus
souvent, ces composés ont été observés dans des poissons gras ou des
poissons qui se nourrissent sur le fond (voir section 4.4.2).
Les observations concernant les organismes terrestres ne
permettent pas d'estimer les concentrations résiduelles (voir section
4.4.3). Dans trois échantillons d'un mélange de différents laits de
vache, on a trouvé, pour le lait entier, une concentration maximale de
100 pg/kg (voir section 5.4). La contamination des autres denrées
alimentaires vendues dans le commerce est également très mal connue.
L'analyse de plusieurs échantillons de poulet et de porc montre que
ces produits contiennent environ 5-30 ng/kg d'analogues fortement
chlorés. Le profil de ces analogues n'est pas le même que chez les
organismes aquatiques (voir section 5.4).
Les données disponibles ne permettent pas d'évaluer l'exposition
globale de la population dans son ensemble. Elles suffisent pour une
évaluation limitée de l'exposition dans le cas des populations
locales. Compte tenu des concentrations environnementales examinées
ci-dessus et des hypothèses habituelles sur l'apport d'aliments, d'air
et d'eau, les denrées alimentaires risquent davantage de contribuer
notablement à l'exposition aux PCDD/PCDF que l'air, tandis que l'eau
de boisson est sans doute beaucoup moins en cause.
2.3 Modalités d'exposition
Chez l'homme, le tissu adipeux contient des tétrachloro-2,3,7,8
PCDD/PCDF ainsi que des dérivés pentachlorés et hexachlorés. Cette
contamination s'explique sans doute par une exposition aux produits
présents dans l'environnement, aux concentrations indiquées plus haut.
En outre, les nourrissons peuvent être exposés par
l'intermédiaire du lait maternel et les jeunes enfants par l'ingestion
de terre contaminée. Mais, dans la plupart des cas, ce dernier mode
d'exposition ne devrait être pris en compte que dans les régions
fortement polluées.
Certaines populations sont particulièrement exposées à l'occasion
d'accidents survenus dans l'industrie (et des opérations de nettoyage
ultérieures) lors de la production et de l'utilisation normales de
chlorophénols, d'herbicides phénoxylés et de PCB. En pareil cas, ce
sont l'inhalation et le contact cutané qui constituent les modes
d'exposition les plus importants. Cependant, on ne dispose pas de
données quantitatives sur la nature et la concentration des
contaminants.
Sur la base des valeurs indiquées plus haut pour les
concentrations dans le milieu ambiant et des hypothèses habituelles
sur les paramètres physiologiques et les valeurs de l'apport, il est
probable que l'ingestion constitue le mode d'exposition essentiel.
L'inhalation de l'air ambiant ne devrait pas poser de problème encore
qu'un air fortement contaminé puisse contribuer sensiblement à
l'exposition. En général, il n'est pas possible actuellement d'estimer
l'importance relative de l'exposition par voie cutanée.
2.4 Biodisponibilité
On ne dispose d'aucune donnée sur la biodisponibilité provenant
d'études sur l'homme. L'expérimentation animale montre que la
biodisponibilité des PCDD ou des PCDF après ingestion dépend de la
matrice du produit ingéré. Les données disponibles au sujet de
l'apport par voie orale sont résumées au tableau 46.
Des études sur des rats glabres montrent que l'exposition cutanée
par contact de la peau intacte avec un sol contaminé représente
environ 1-2%. On ne dispose d'aucune donnée pour l'exposition par
inhalation.
3. Résultats de l'exposition animale
3.1 Toxicocinétique des 2,3,7,8-TCDD
Des études sur des rongeurs à qui l'on administrait du
2,3,7,8-TCDD par voie orale, en une ou plusieurs prises, ont montré
que l'absorption dans les voies digestives était d'au moins 50% chez
le rat, le cobaye et le hamster, mais de moins de 30% chez la souris
(tableau 40). Les valeurs indiquées pour la demi-vie d'élimination
étaient de l'ordre de 12-31 jours pour le rat, la souris et le hamster
et de 22-94 jours pour le cobaye (tableau 41). Cependant, la plupart
de ces études ont été effectuées en utilisant des doses toxiques. La
demi-vie des 2,3,7,8-TCDD chez les primates est mal connue mais il
semble, d'après les données disponibles pour le rhésus, que la
demi-vie apparente soit de l'ordre de 1 an dans les tissus adipeux.
Les 2,3,7,8-TCDD ne s'accumulent pas dans les tissus animaux.
Chez les rongeurs, l'accumulation intervient principalement au niveau
du foie et du tissu adipeux (tableaux 42 à 44). Chez des rhésus
(tableau 45), on a observé une concentration élevée de ces mêmes
produits dans le tissu adipeux, le foie, la peau et les muscles.
Chez des rats ayant reçu quotidiennement pendant 2 ans des
2,3,7,8-TCDD à raison de 1 ng/kg de poids corporel, on a observé une
accumulation de 540 ng/kg de poids corporel dans le foie où l'on a
également noté certaines altérations morphologiques. Des quantités du
même ordre ont été notées chez le péromysque après exposition à un sol
contenant des 2,3,7,8-TCDD à la concentration de 10-710 ng/kg
(Cockherham et al., 1980). L'exposition des animaux, au niveau de
l'ensemble de leur corps, à un sol présentant de telles teneurs peut
donc se traduire par une concentration tissulaire qui a des effets
observables chez les animaux d'expérience.
Les TCDD sont en grande partie éliminés dans les déjections,
encore qu'il existe une certaine excrétion urinaire. Elle est plus
importante chez le hamster que chez les autres espèces étudiées.
La transformation des 2,3,7,8-TCDD en métabolites à caractère
polaire plus prononcé s'observe chez toutes les espèces animales
étudiées (voir section 6.2 et tableau 62). L'élimination fécale et
urinaire des métabolites est rapide chez toutes ces espèces, sauf chez
le cobaye. Les métabolites connus sont beaucoup moins toxiques que les
composés dont ils dérivent.
3.2 Toxicocinétique des PCDD et des PCDF autres que les TCDD
En dehors du cas des dérivés tétrachlorés, les observations sur
la toxicocinétique des PCDD chez les animaux sont limitées. De ce
point de vue, les PCDF ont été davantage étudiés. La demi-vie des
2,3,7,8-TCDF serait de l'ordre de 2 à 8 jours chez le rat, la souris
et le rhésus et de plus de 20 jours chez le cobaye (tableau 65). Les
études sur le rat montrent que les 2,3,7,8-pentaCDF se fixent
davantage dans les tissus que les 2,3,7,8-TCDF (65% et 3,8%
respectivement, au bout de 5 jours).
La rétention tissulaire des PCDD et des PCDF chez diverses
espèces exposées à des mélanges de synthèse ou à des échantillons
prélevés dans l'environnement et contenant des PCDD et des PCDF se
révèle très inégale selon que les produits sont ou non substitués par
des atomes de chlore dans toutes les positions 2,3,7 et 8.
3.3 Effets toxiques des 2,3,7,8-TCDD
Les effets toxiques et biologiques découlant de l'exposition aux
2,3,7,8-TCDD dépendent de divers facteurs, dont l'espèce, la souche,
l'âge et le sexe des animaux d'expérience. Entre autres signes de
toxicité observés chez plusieurs espèces animales, on note une perte
de poids, une hépatoxicité, une porphyrie, une atteinte cutanée, des
lésions gastriques, une atrophie du thymus, une atteinte du système
immunitaire, des effets tératogènes, des effets sur la reproduction et
des effets cancérogènes. Les TCDD exercent des effets biologiques
extrêmement divers, notamment l'induction de certaines enzymes et une
carence en vitamine A. Ces effets ne sont pas tous observés chez la
même espèce animale. Les effets toxiques les plus caractéristiques
qu'on retrouve chez tous les animaux de laboratoire sont la perte de
poids, l'atrophie du thymus et l'immunotoxicité. Chez l'homme, une
chloracné et des lésions cutanées apparentées constituent les signes
les plus fréquents d'une intoxication par les 2,3,7,8-TCDD; on observe
également des lésions cutanées chez le rhésus, la souris glabre et le
lapin. En revanche, l'exposition au 2,3,7,8-TCDD ne provoque ni
chloracné ni lésions cutanées de ce type chez le rat, la plupart des
souris, le cobaye et le hamster. Les lésions toxiques observées sont
fréquemment soit des lésions hyperplasiques/ métaplasiques, soit des
lésions hypoplasiques et elles intéressent principalement les tissus
épithéliaux.
Des effets toxiques sur la reproduction ont été observés chez les
singes rhésus: la plus faible dose sans effets observables serait,
d'après les calculs, de 1 à 2 ng/kg de poids corporel par jour. Il se
peut qu'il existe une dose sans effets observables de 1 ng/kg de poids
corporel par jour chez le rat, s'agissant des effets sur la
reproduction (Murray et al., 1979; Nisbet & Paxton, 1982).
Lorsqu'on compare les études consacrées à la production de
cancers chez le rat par Kociba et al. (1978) et par le NTP (1982a, b),
il apparaît clairement que les tumeurs hépatiques, notamment les
épithéliomas hépatocellulaires, s'obtiennent à des doses similaires.
Bien que le NTP ainsi que Kociba et al. (1978) aient observé une
augmentation de l'incidence des tumeurs au niveau d'autres organes,
les résultats ne sont pas analogues dans les deux études. Cela tient
peut-être, en partie, à des différences dans les doses administrées
(gavage ou exposition par incorporation à des aliments broyés) et dans
les souches. Dans l'étude de Kociba, l'administration d'une dose de 10
ng/kg de poids corporel a provoqué une augmentation de l'incidence des
nodules néoplasiques (hyperplasiques) chez les femelles, tandis qu'une
dose de 1 ng/kg de poids corporel se traduisait par un gonflement des
hépatocytes en foyers ou par places. A ces doses, l'incidence de
certaines tumeurs à déterminisme hormonal était plus faible chez les
animaux d'expérience que chez les témoins, ce qui donne à penser que
les 2,3,7,8-TCDD déterminent des altérations au niveau des glandes
endocrines. Compte tenu de ces résultats de l'expérimentation animale
et des données dont on dispose au sujet de l'homme, le CIRC (1982,
suppl. 4) est arrivé à la conclusion que les preuves de
cancérogénicité des TCDD étaient suffisantes chez les animaux, mais
insuffisantes chez l'homme.
Apparemment, les TCDD n'ont pas de propriétés mutagènes et ne
devraient donc pas être génotoxiques. Leur action cancérogène est
donc attribuée à un mécanisme indirect (épigénétique).
3.4 Effets toxiques des PCDD et des PCDF autres que les TCDD
Plusieurs autres PCDD et PCDF provoquent les mêmes symptômes que
les 2,3,7,8-TCDD, mais leur activité est très inégale (tableaux 56,
62). Pour résumer, il existe 12 isomères qui présentent une toxicité
élevée, à savoir les tétra-, penta-, hexa- et heptaCDD et CDF
contenant quatre atomes de chlore symétriques deux à deux en position
2,3,7 et 8. On a montré, lors d'études prolongées sur l'animal, qu'un
mélange de deux hexaCDD (1,2,3,7,8-9 et 1,2,3,6,7,8-haxaCDD) était
cancérogène, mais à des doses plus élevées que celles utilisées dans
l'étude des TCDD. La dioxine non substituée et la dichloro-2,7 dioxine
n'ont manifesté aucun pouvoir cancérogène.
L'activité relative, toxique et biologique, des PCDD et des PCDF
a été estimée au moyen d'études de courte durée chez le rat et
d'études en culture de cellules mammaliennes. Comme critères, on a
retenu l'inhibition de la prise de poids, l'atrophie du thymus,
l'induction d'enzymes, la térato-génicité, la production d'acné et la
kératinisation. En l'absence d'observations sur la toxicité à long
terme, les résultats fournis par ces épreuves de durée limitée
constituent actuellement la seule source qui permette un classement
par toxicité en vue de l'évaluation des dangers pour la santé de
l'homme.
Les études consacrées à des mélanges de ces composés ont mis en
évidence une synergie additive tout au plus.
3.5 Etude des différences interspécifiques
La sensibilité aux effets biologiques et toxiques des
2,3,7,8-TCDD est très inégale selon les espèces. Par exemple, la
DL50 par voie orale varie de 0,6 µg/kg de poids corporel chez le
cobaye à 5051 µg/kg de poids corporel chez le hamster doré syrien
(tableau 47); en outre, on a signalé des différences prononcées de
DL50 chez différentes souches de la même espèce (par exemple le rat
et la souris). La toxicité et la toxicocinétique des TCDD sont très
analogues chez les singes et chez l'homme. Cependant, il est
impossible d'expliquer les différences considérables de sensibilité,
selon le sexe et la souche, aux 2,3,7,8-TCDD et aux composés analogues
par les différences qu'on observe sur le plan toxicocinétique. Les
observations montrent que, chez des souris de lignée pure, l'abondance
cellulaire du récepteur Ah est partiellement corrélée avec la
sensibilité aux effets biologiques et toxiques de ces composés. On a
également repéré ce récepteur chez d'autres espèces, notamment chez
l'homme. Cependant, l'abondance relative de ce récepteur selon les
espèces n'explique pas les différences de sensibilité aux
2,3,7,8-TCDD; cette constatation n'a rien d'étonnant quand on sait
qu'il existe des mécanismes de toxicité complexes et encore inconnus
qui font intervenir de multiples facteurs en plus du récepteur aux Ah.
4. Effets sur la santé de l'homme
4.1 PCDD
La population générale est exposée à de faibles quantités de PCDD
et de PCDF présentes dans des mélanges complexes qui ne provoquent
aucun cas de maladie. Lors de quelques incidents, par exemple à Seveso
et à Yusho, des ouvriers et d'autres personnes sont tombés malades à
la suite d'une exposition à des quantités plus élevées d'un nombre
restreint de ces composés.
Dans le cas de l'exposition professionnelle ou accidentelle,
l'effet clinique le plus net réside dans une chloracné. D'autres
effets (tableau 64) ont été observés mais, à part la chloracné et
peut-être des troubles fonctionnels mineurs, aucun n'a été durable.
Dans certaines des études de mortalité réalisées, on a fait état
d'une augmentation de l'incidence des cancers de diverses
localisations, mais le trop petit nombre de cas incite à la prudence.
L'impression générale qui se dégage des études de suivi est que
les effets généraux aigus des TCDD, même lorsqu'ils sont graves, sont
habituellement réversibles, à l'exception de la chloracné, ou
s'améliorent peu à peu après l'arrêt de l'exposition. A Seveso, le
seul effet nocif sur la santé qui soit indiscutable a été la
chloracné: sur les 193 cas qui se sont déclarés en 1976 et 1977, 20
présentaient encore des symptômes actifs en 1984. De nombreuses études
ont été consacrées à la recherche de liens possibles entre
l'exposition à l'Agent orange, au Viet Nam, et divers effets sur la
santé, tant chez les civils que chez les militaires. Cependant, les
données dont on dispose à ce jour ne permettent pas de conclure
catégoriquement à l'existence d'effets biologiques notables, en
particulier sur la reproduction (voir section 9.2).
Dans un certain nombre d'études, on a comparé les populations
exposées à divers groupes témoins sur la base du dosage des lipides
sériques, d'épreuves de la fonction hépatique et de divers autres
paramètres. Bien que dans ces études ainsi que dans des études de cas
isolés, on ait signalé des différences statistiquement significatives,
il est impossible de formuler des conclusions définitives par suite du
manque d'uniformité, de diverses insuffisances sur le plan technique
et de l'impossibilité d'exclure les facteurs de confusion.
Lors de la contamination accidentelle survenue en 1971 au
Missouri (Etats-Unis d'Amérique), des enfants ont manifesté des signes
d'intoxication aiguë mais ils sont aujourd'hui en bonne santé,
semble-t-il. Les études épidémiologiques conduites dans le même Etat
sur des populations durablement exposées à des concentrations plus
faibles n'ont jusqu'ici révélé aucun effet biologique notable.
Cependant, malgré l'absence de manifestations cliniques, certains
signes témoignent d'une action sur le système immunitaire à médiation
cellulaire.
La gamme des effets des TCDD sur la santé humaine reste à
préciser. On peut conclure que, dans l'ensemble, les observations
concernant l'exposition et les effets sur la santé ne permettent pas
d'établir pour l'homme des relations dose/effet ou dose/réponse.
En dépit de nombreuses études cliniques et études de suivi, aucun
effet général persistant bien net n'a été observé, à l'exception de la
chloracné. Compte tenu de ce qu'on sait actuellement, il semble peu
probable que l'exposition aux TCDD ait des séquelles toxicologiques
permanentes, graves et invalidantes.
4.2 PCDF
Les seuls cas d'intoxication humaine par les PCDF pour lesquels
on dispose d'informations circonstanciées concernent les incidents
survenus à Yusho, au Japon (1968) et à Yu-cheng, à Taiwan (1979), à la
suite d'une contamination d'huile de riz par des PCDF, des PCD et des
PCQ (voir section 11.1). Au total, plusieurs milliers de personnes ont
été gravement intoxiquées. La récapitulation des données montre que,
selon toute vraisemblance, il faut incriminer les PCDF. La
symptomatologie générale était sensiblement la même que lors d'une
intoxication par les PCDD. Les différences reflètent peut-être
l'intensité de l'exposition et la structure, par âge et sexe, de la
population exposée. Dans le cas des malades de Yusho, les estimations
de l'apport quotidien moyen de PCDF pendant plusieurs mois ont abouti
à une dose de 0,9 µg/kg de poids corporel pour les PCDF totaux, de
0,1-0,2 µg/kg pour les tétrachloro-2,3,7,8 dibenzofurannes ainsi que
les penta- et hexaPCDF, à quoi il faut ajouter 150 µg pour les PCB et
148 µg pour les PCQ par kg de poids corporel (Hayabuchi et al., 1979).
La dose pathogène minimale a été estimée à 0,6 mg de PCDF totaux par
personne sur 30 jours, ce qui correspond à une dose quotidienne de
0,05-0,1 µg/kg de poids corporel pour les PCDF substitués en 2,3,7 et
8. Quoi qu'il en soit, les données disponibles sont insuffisantes pour
qu'on puisse se prononcer sur la valeur de la dose qui serait sans
danger pour l'homme.
4.3 Charge de l'organisme humain et cinétique
Chez l'homme, la concentration résiduelle des TCDD dans le tissu
adipeux s'élève jusqu'à 20 mg/kg dans la population générale, en
dehors de toute exposition précise connue, mais elle peut atteindre
une valeur plus élevée dans certains cas sans entraîner de
manifestations pathologiques. Aucune des populations ainsi étudiées
n'avait été constituée par sondage aléatoire. Les autres PCDD et PCDF
contenant un plus grand nombre d'atomes de chlore, en particulier les
octaDD, se rencontrent également dans ces échantillons (voir tableaux
29, 30). Les concentrations moyennes semblent augmenter avec l'âge.
Dans certaines situations, on a observé des concen-trations plus
élevées (de l'ordre de quelques microgrammes par kilogramme) qui ne
s'accompagnaient d'aucune manifestation pathologique.
Lors des accidents de Yusho et de Yu-cheng, les symptômes
observés étaient associés à des concentrations de PCDF plus élevées;
par exemple, on a trouvé, un an après exposition à de l'huile de riz
contaminée, une concentration de 6,9 µg/kg de tissu adipeux pour les
pentachloro-2,3,4,7,8 dibenzofurannes.
D'après les données très limitées dont on dispose, la
concentration dans la population générale du 2,3,4,7,8- pentaCDF, par
exemple, semble cent fois plus faible que la concentration observée
chez les victimes de l'accident de Yusho.
Aucune comparaison de ce type n'est possible pour les PCDD.
Des données limitées montrent que, à l'exception des TCDF, les
isomères chlorés en position 2,3,7 et 8 font l'objet d'une rétention
sélective.
D'après une étude expérimentale unique, la demi-vie des TCDD
serait de 5 ans chez l'homme. Une autre étude a permis d'évaluer cette
demi-vie à 2-6 ans pour les 1,2,3,6,7,8- hexaCDD, les
1,2,3,4,6,7,8-heptaCDD, les octaCDD, les 1,2,3,4,6,7,8-heptaCDF et les
octaCDF. Ces observations devront être complétées car elles
proviennent d'études qui portaient sur deux sujets seulement, sans
compter que la toxicinétique de ces types de composés ne se ramène
peut-être pas simplement à une cinétique du premier ordre. Cependant,
même si les données actuelles ont des limites, il est clair que la
demi-vie de ces composés est de l'ordre d'une ou plusieurs années.
Les valeurs indiquées pour la demi-vie sont très différentes chez
l'homme et chez les rongeurs. Mais, les expériences conduites sur les
animaux ont généralement été exécutées au moyen de doses toxiques. En
outre, les animaux qui ont une moindre longévité ont un métabolisme
plus important de sorte qu'il est normal que la demi-vie soit plus
courte.
Les PCDD et les PCDF sont essentiellement retenus dans les
graisses, mais ces produits sont aussi excrétés dans le lait (tableau
39) et traversent la barrière placentaire. On les trouve aussi, à plus
faibles concentrations, dans le sang et les organes vitaux. La
distribution entre les différents tissus humains est mal connue mais
selon certains, le rapport des concentrations dans le tissu adipeux et
dans le foie est plus élevé chez l'homme que chez les rongeurs.
Cependant, cette conclusion repose sur des données fort restreintes
fondées sur des pièces d'autopsie. Il reste donc à voir si elle vaut
pour la population dans son ensemble.
Les voies de pénétration dans l'organisme humain sont encore mal
connues mais on admet que l'apport alimentaire représente l'essentiel.
Cependant, le nourrisson représente un cas particulier puisque, par
suite du passage trans-placentaire, il peut y avoir une exposition
in utero. D'après les dosages effectués jusqu'ici dans le lait
maternel, ce dernier pourrait être une source importante de
contamination.
Il n'existe aucune donnée concernant la dose des PCDD qui est
toxique pour l'homme. Toutefois, lors des accidents de Yusho et de
Yu-cheng, les victimes avaient absorbé une dose totale de l'ordre de
3,3-3,8 mg pour les PCDF totaux et de 400-500 µg pour les PCDF
substitués en 2,3,7 et 8. Il n'existe aucune donnée fiable sur les
doses sans effet nocif.
5. Conclusions générales
Les PCDD et les PCDF se rencontrent dans tout l'environnement et
il est probable que chacun en est porteur. Parfois, ces produits
déterminent des effets toxiques complexes à la suite d'une activité
professionnelle ou accidentelle.
D'après les observations faites à Yusho, les expériences
conduites chez des espèces sensibles de singes et compte tenu de
certaines hypothèses concernant l'activité relative des PCDD et des
PCDF, il se peut que l'homme et certains singes présentent une
sensibilité comparable à l'égard de ces composés. Cependant, les
incertitudes concernant la dose effectivement reçue et les difficultés
de l'évaluation des effets toxiques autres que la chloracné
interdisent, dans le cas de l'homme, une conclusion catégorique quant
à la résistance relative de l'homme aux effets toxiques de ces
composés. Il convient de limiter l'exposition aux plus faibles niveaux
compatibles avec les impératifs pratiques.
RECOMMENDATIONS
1. Des études interlaboratoires de validation, selon une sorte de
"poule" et l'utilisation de protocoles normalisés de contrôle et
d'assurance de qualité, sont indispensables pour améliorer les
méthodes d'analyse.
La méthodologie de l'échantillonnage, les méthodes d'analyse et
l'interprétation des données doivent être optimalisées et
normalisées avant le démarrage des enquêtes.
2. Des données complémentaires sont nécessaires sur l'origine des
PCDD et des PCDF, sur leur répartition dans l'environnement et
sur leur destinée.
D'autres observations sont indispensables au sujet de la présence
des PCDD ou des PCDF dans l'environnement, notamment sur
l'évolution des concentrations dans le temps et sur la
détermination des profils isomériques, notamment dans les
produits alimentaires, dans l'air ambiant et dans les sédiments.
3. Il serait bon de faire des observations sur les effets
qu'exercent les PCDD et les PCDF sur la biocénose.
4. Des données plus complètes sont nécessaires en ce qui concerne la
biodisponibilité des PCDD et des PCDF provenant de diverses
matrices, dans l'environnement et dans la ration alimentaire.
L'exposition à ces sources devrait être mise en corrélation avec
les pratiques agricoles et industrielles.
5. Il convient de mettre au point et de valider des méthodes
chimiques et biologiques plus simples et moins coûteuses pour
rechercher la présence de PCDD et de PCDF.
6. Des études sur les mécanismes toxiques des PCDD et des PCDF sont
nécessaires pour élucider les différences observées dans les
effets produits chez les diverses espèces et permettre une
extrapolation à l'homme.
7. Il importe de poursuivre l'étude de l'immunotoxicité, notamment
en ce qui concerne la fonction des lymphocytes T cytotoxiques. En
particulier, il est capital d'étudier les effets sur le système
immunitaire d'une exposition à la période périnatale et de la
durée de cette action.
8. Des études de toxicité à long terme devraient être effectuées,
notamment des études de reproduction sur plusieurs générations
chez diverses espèces, au moyen de trois des PCDD et des PCDF les
plus répandus, à savoir le 2,3,4,7,8-pentaCDF, le
1,2,3,7,8-pentaCDD et l'octaCDD.
9. Etant donné que l'homme est exposé à des mélanges complexes de
PCDD et de PCDF, il faut poursuivre la mise au point et la
validation de systèmes d'épreuve, permmettant notamment l'étude
des réactions des tissus humains en vue d'apprécier l'activité
toxique de ces composés et de leurs mélanges. Ces systèmes
permettront d'étudier les mécanismes d'action, les relations
structure/activité et les effets interactifs.
10. Il serait bon d'étudier la charge de l'organisme en PCDD et PCDF
et d'établir une corrélation entre cette charge, les effets
cliniques constatés et les résultats des examens de laboratoire.
Le suivi des groupes exposés antérieurement est important à cet
égard.