
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 71
PENTACHLOROPHENOL
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1987
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
ISBN 92 4 154271 3
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1987
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR PENTACHLOROPHENOL
1. SUMMARY
1.1. Identity, physical and chemical properties, analytical
methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution, and transformation
1.4. Environmental levels and human exposure
1.5. Effects on organisms in the environment
1.6. Kinetics and metabolism
1.7. Effects on experimental animals and in vitro test systems
1.8. Effects on man
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
2.1. Identity
2.1.1. Pentachlorophenol (PCP)
2.1.2. Sodium pentachlorphenate (Na-PCP)
2.1.3. Pentachlorophenyl laurate
2.2. Impurities in pentachlorophenol
2.2.1. Formation of PCDDs and PCDFs during thermal
decomposition
2.3. Physical, chemical, and organoleptic properties
2.4. Conversion factors
2.5. Analytical methods
2.5.1. Sampling methods
2.5.2. Analytical methods
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Man-made sources
3.2.1. Industrial production
3.2.1.1 Manufacturing processes
3.2.1.2 Emissions during production
3.2.1.3 Disposal of production wastes
3.2.1.4 Production levels
3.3. Uses
3.3.1. Commercial use
3.3.2. Agricultural use
3.3.3. Domestic use
3.3.4. Use for control of vectors
3.3.5. Formulations
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Volatilization
4.1.2. Adsorption
4.1.3. Leaching
4.2. Biotransformation
4.2.1. Abiotic degradation
4.2.2. Microbial degradation
4.2.2.1 Aquatic degradation
4.2.2.2 Degradation in soil
4.3. Degradation by plants
4.4. Ultimate fate following use
4.4.1. General aspects
4.4.2. Disposal of waste water
4.4.3. Incineration of wastes
5. ENVIRONMENTAL LEVELS ANS HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water and sediments
5.1.3. Soil
5.1.4. Aquatic and terrestrial organisms
5.1.4.1 Aquatic organisms
5.1.4.2 Terrestrial organisms
5.1.5. Drinking-water and food
5.1.6. Consumer products
5.1.7. Treated wood
5.2. Occupational exposure
5.3. General population exposure
5.4. Human monitoring data
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Animal studies
6.1.2. Human studies
6.2. Distribution
6.2.1. Animal studies
6.2.2. Human studies
6.3. Metabolic transformation
6.3.1. Animal studies
6.3.2. Human studies
6.4. Elimination and excretion
6.4.1. Animal studies
6.4.2. Human studies
6.5. Retention and turnover
6.5.1. Animal studies
6.5.2. Human studies
6.6. Reaction with body components
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
7.1. Microorganisms
7.2. Aquatic organisms
7.2.1. Plants
7.2.2. Invertebrates
7.2.3. Vertebrates
7.3. Terrestrial organisms
7.3.1. Plants
7.3.2. Animals
7.4. Population and ecosystem effects
7.5. Biotransformation, bioaccumulation, and
biomagnification
7.5.1. Aquatic organisms
7.5.2. Terrestrial organisms
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
8.1. Acute toxicity
8.2. Short-term toxicity
8.2.1. Pure or purified PCP
8.2.2. Technical grade PCP
8.2.3. Comparative studies
8.3. Long-term toxicity
8.4. Effects on reproduction and fetal development
8.5. Mutagenicity
8.6. Carcinogenicity
8.7. Other studies
8.8. Contaminants affecting toxicity
8.8.1. Octachlorodibenzodioxin (OCDD)
8.8.2. Heptachlorodibenzodioxin (H7CDD)
8.8.3. Hexachlorodibenzodioxin (H6CDD)
8.8.4. Polychlorinated dibenzofurans (PCDFs)
8.8.5. Polychlorodiphenyl ethers (PCDPEs)
8.8.6. Other microcontaminants
8.9. Mechanism of toxicity
9. EFFECTS ON MAN
9.1. Acute toxicity - poisoning incidents
9.2. Effects of short- and long-term exposures
9.2.1. Occupational exposure
9.2.1.1 Skin and mucous membranes
9.2.1.2 Liver and kidney
9.2.1.3 Blood and haemopoetic system
9.2.1.4 Nervous system
9.2.1.5 Immunological system
9.2.1.6 Reproduction
9.2.1.7 Cytogenetic effects
9.2.1.8 Carcinogenicity
9.2.1.9 Other systems
9.2.2. General population exposure
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of human health risks
10.1.1. Occupational exposure
10.1.1.1 Exposure levels and routes
10.1.1.2 Toxic effects
10.1.1.3 Risk evaluation
10.1.2. Non-occupational exposure
10.1.2.1 Exposure levels and routes
10.1.2.2 Risk evaluation
10.1.3. General population exposure
10.1.3.1 Exposure levels and routes
10.1.3.2 Risk evaluation
10.2. Evaluation of effects on the environment
10.3. Conclusions
11. RECOMMENDATIONS
11.1. Environmental contamination and human exposure
11.2. Future research
11.2.1. Human exposure and effects
11.2.2. Effects on experimental animals and in vitro test
systems
11.2.3. Effects on the ecosystem
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
WHO TASK GROUP ON PENTACHLOROPHENOL
Members
Dr U.G. Ahlborg, Unit of Toxicology, National Institute of
Environmental Medicine, Stockholm, Sweden
Dr R.C. Dougherty, Department of Chemistry, Florida State
University, Tallahassee, Florida, USA
Dr H.H. Dieter, Federal Health Office, Institute for Water, Soil,
and Air Hygiene, Berlin (West)
Dr A.H. El Sabae, Pesticide Division, Facultry of Agriculture,
University of Alexandria, Alexandria, Egypta
Dr A. Furtado Rahde, Ministry of Public Health, Porto Alegre,
Brazil
Dr S. Gupta, Department of Zoology, Faculty of Basic Sciences,
Punjab Agricultural University, Ludhiana, Punjab, Indiaa
Dr L.V. Martson, All Union Scientific Research Institute of the
Hygiene and Toxicology of Pesticides, Polymers, and
Plastics, Kiev, USSRa
Dr U.G. Oleru, Department of Community Health, College of Medicine,
University of Lagos, Lagos, Nigeria
Dr Shou-Zheng Xue, Toxicology Programme, School of Public Health,
Shanghai Medical University, Shanghai, China
Observers
Dr F.F. Hertel, Fraunhofer Institute for Toxicology and
Aerosol Research, Hanover, Federal Republic of Germany
Dr E. Kramer (European Chemical Industry Ecology and Toxicology
Centre), Dynamit Nobel A.G., Cologne, Federal Republic of
Germany
Dr D. Streelman (International Group of National Associations of
Agrochemical Manufacturers), Agricultural Chemicals Registration
and Regulatory Affairs, Philadelphia, Pennsylvania, USA
Mr G. Ozanne (European Chemical Industry Ecology and Toxicology
Centre), Rhone Poulenc DSE/TOX, Neuilly-sur-Seine, France
Mr V. Quarg, Federal Ministry for Environment, Nature Conservation
and Nuclear Safety, Bonn, Federal Republic of Germany
---------------------------------------------------------------------------
a Invited but unable to attend.
Observers (contd)
Dr U. Schlottmann, Chemical Safety, Federal Ministry for
Environment, Nature Conservation and Nuclear Safety, Bonn,
Federal Republic of Germany
Dr M. Sonneborn, Federal Health Office, Berlin (West)
Dr W. Stober, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Federal Republic of Germany
Secretariat
Dr K.W. Jager, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
Mrs B. Bender, International Register for Potentially Toxic
Chemicals, Geneva, Switzerland
Dr A. Gilman, Industrial Chemicals and Product Safety Section,
Health Protection Branch, Department of National Health and
Welfare, Tunney's Pasture, Ottawa, Ontario, Canada (Temporary
Adviser) (Rapporteur)
Dr L. Ivanova-Chemishankska, Institute of Hygiene and Occupational
Health, Medical Academy, Sofia, Bulgaria (Temporary Adviser)
Dr E. Johnson, Unit of Analytical Epidemiology, International
Agency for Research on Cancer, Lyons, France
Dr G. Rosner, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Federal Republic of Germany (Rapporteur)
Dr G.J. Van Esch, Bilthoven, Netherlands (Temporary Adviser)
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as accurately as possible without unduly
delaying their publication. In the interest of all users of the
environmental health criteria documents, readers are kindly
requested to communicate any errors that may have occurred to the
Manager of the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland, in order that they may be
included in corrigenda, which will appear in subsequent volumes.
* * *
A detailed data profile and a legal file can be obtained from
the International Register of Potentially Toxic Chemicals, Palais
des Nations, 1211 Geneva 10, Switzerland (Telephone no. 988400 -
985850).
ENVIRONMENTAL HEALTH CRITERIA FOR PENTACHLOROPHENOL
A WHO Task Group on Environmental Health Criteria for
Pentachlorophenol met at the Fraunhofer Institute for Toxicology
and Aerosol Research, Hanover, Federal Republic of Germany from 20
to 24 October, 1986. Dr W. Stöber opened the meeting and welcomed
the members on behalf of the host Institute, and Dr U. Schlottmann
spoke on behalf of the Federal Government, who sponsored the
meeting. Dr K.W. Jager addressed the meeting on behalf of the
three co-operating organizations of the IPCS (UNEP/ILO/WHO). The
Task Group reviewed and revised the draft criteria document and
made an evaluation of the risks for human health and the
environment from exposure to pentachlorophenol.
The drafts of this document were prepared by DR G. ROSNER of
the Fraunhofer Institute for Toxicology and Aerosol Research,
Hanover, Federal Republic of Germany, and DR A. GILMAN of the
Health Protection Branch, Ottawa, Canada.
The efforts of all who helped in the preparation and
finalization of the document are gratefully acknowledged.
* * *
Partial financial support for the publication of this criteria
document was kindly provided by the United States Department of
Health and Human Services, through a contract from the National
Institute of Environmental Health Sciences, Research Triangle Park,
North Carolina, USA - a WHO Collaborating Centre for Environmental
Health Effects. The United Kingdom Department of Health and Social
Security generously supported the costs of printing.
1. SUMMARY
1.1. Identity, Physical and Chemical Properties, Analytical Methods
Pure pentachlorophenol (PCP) consists of light tan to white,
needlelike crystals and is relatively volatile. It is soluble in
most organic solvents, but practically insoluble in water at the
slightly acidic pH generated by its dissociation (pKa 4.7).
However, its salts, such as sodium pentachlorophenate (Na-PCP),
are readily soluble in water. At the approximately neutral pH of
most natural waters, PCP is more than 99% ionized.
Apart from other chlorophenols, unpurified technical PCP
contains several microcontaminants, particularly polychlorinated
dibenzo- p-dioxins (PCDDs) and polychlorinated dibenzofurans
(PCDFs), of which H6CDD is the most relevant congener
toxicologically. 2,3,7,8-T4CDD has only once been confirmed in
commercial PCP samples (0.25 - 1.1 µg/kg). Depending on the
thermolytic conditions, thermal decomposition of PCP or Na-PCP may
yield significant amounts of PCDDs and PCDFs. The use and the
uncontrolled incineration of technical grade PCP is one of the most
important sources of PCDDs and PCDFs in the environment.
Most of the analytical methods used today involve acidification
of the sample to convert PCP to its non-ionized form, extraction
into an organic solvent, possible cleaning by back-extraction into
a basic solution, and determination by gas chromatography with
electron-capture detector (GC-EC) or other chromatographic methods
as ester or ether derivatives (e.g., acetyl-PCP). Depending on
sampling procedures and matrices, detection limits as low as
0.05 µg/m3 in air or 0.01 µg/litre in water can be achieved.
1.2. Sources of Human and Environmental Exposure
PCP is mainly produced by the stepwise chlorination of phenols
in the presence of catalysts. Until 1984, Na-PCP was partly
synthesized by means of the alkaline hydrolysis of
hexachlorobenzene, but it is now produced by dissolving PCP flakes
in sodium hydroxide solution.
World production of PCP is estimated to be of the order of
30 000 tonnes per year. Because of their broad pesticidal efficiency
spectrum and low cost, PCP and its salts have been used as
algicides, bactericides, fungicides, herbicides, insecticides, and
molluscicides with a variety of applications in the industrial,
agricultural, and domestic fields. However, in recent years, most
developed countries have restricted the use of PCP, especially for
agricultural and domestic applications.
PCP is mainly used as a wood preservative, particularly on a
commercial scale. The domestic use of PCP is of minor importance
in the overall PCP market, but has been of particular concern
because of possible health hazards associated with the indoor
application of wood preservatives containing PCP.
1.3. Environmental Transport, Distribution, and Transformation
The relatively high volatility of PCP and the water solubility
of its ionized form have led to widespread contamination of the
environment with this compound. Depending on the solvent,
temperature, pH, and type of wood, up to 80% of PCP may evaporate
from treated wood within 12 months.
The adsorption and leaching behaviour of PCP varies from soil
to soil. Adsorption of PCP decreases with rising pH and so PCP is
most mobile in mineral soils, and least mobile in acidic clay and
sandy soils.
Solid or water-dissolved PCP can be photolysed by sunlight
within a few days, yielding aromatic (lower chlorinated phenols,
etc.) and nonaromatic fragments, as well as hydrogen chloride (HCl)
and carbon dioxide (CO2). Traces of PCDDs, mainly OCDD are formed
photochemically on irradiation of Na-PCP in aqueous solution.
PCP degrading microorganisms have been isolated from waters and
soils. High organic matter and moisture content, median
temperatures, and high pH enhance microbial breakdown in soil
(half-life = 7 - 14 days). Low oxygen conditions are generally
unfavourable for the biodegradation of PCP, allowing it to persist
in soil (half-life = 10 - 70 days under flooded conditions), water
(half-life = 80 - 192 days in anaerobic water), and sediments (10%
decomposition within 5 weeks to almost no degradation). Several
studies have proved that PCP can be degraded by activated sludge.
However, in full-scale treatment plants the treatment efficiency is
often reduced.
Numerous metabolites have been identified resulting from the
methylation, acetylation, dechlorination, or hydroxylation of PCP.
Of the possible metabolites, at least tetrachlorocatechol seems to
be relatively persistent. However, there is a lack of data
concerning the fate of the intermediate products of both the
abiotic and biotic degradation of PCP.
1.4. Environmental Levels and Human Exposure
The ubiquitous occurrence of PCP is indicated by its detection,
even in ambient air of mountain rural areas (0.25 - 0.93 ng/m3). In
urban areas, PCP levels of 5.7 - 7.8 ng/m3 have been detected.
While elevated PCP concentrations can be found in groundwater
(3 - 23 µg/litre) and surface water (0.07 - 31.9 µg/litre) within
wood-treatment areas, the PCP level of surface waters is usually in
the range of 0.1 - 1.0 µg/litre, with maximum values of up to 11
µg/litre. PCP concentrations in the mg/litre range can be
encountered near industrial discharges.
Sediments of water bodies generally contain much higher levels
of PCP than the overlying waters. Soil samples from PCP or
pesticide plants contain around 100 µg PCP/kg (dry weight); heavily
contaminated soil (up to 45.6 mg PCP/kg) can be found in the
vicinity of wood-treatment areas.
Residues of PCP in the aquatic invertebrate and vertebrate
fauna are in the low µg/kg range (wet weight). Very high levels
(up to 6400 µg/kg) are found in fish from waters that are
contaminated with wood preservatives, while sediment-dwelling
organisms, such as clams, show PCP levels of up to 133 000 µg/kg.
Fish kills result in PCP residues in fish of between 10 and
30 mg/kg.
After agricultural PCP application, birds can be highly
contaminated (47 mg/kg wet weight in liver). Exposure of farm
animals to PCP-treated wood shavings used as litter causes a musty
taint of the flesh as a result of contamination with
pentachloroanisole, a metabolite of PCP biodecomposition. PCP
levels ranging from not detectable to 8571 µg/kg have been found in
the muscle tissue of wild birds.
The general population is exposed to PCP through the ingestion
of drinking-water (0.01 - 0.1 µg/litre) and food (up to 40 µg/kg in
composite food samples). Apart from the daily dietary intake (0.1
- 6 µg/person per day) resulting from direct food contamination
with PCP, continuous exposure to hexachlorobenzene and related
compounds in food, which are biotransformed to PCP, may be another
important source.
In addition, because of its widespread use, the general
population can be exposed to PCP in treated items such as textiles,
leather, and paper products, and above all, through inhalation of
indoor air contaminated with PCP. Generally, PCP concentrations of
up to about 30 µg/m3 can be expected, for up to the first month,
after indoor treatment of large surfaces; considerably higher
levels (up to 160 µg/m3) cannot be excluded under unfavourable
conditions. In the long term, values of between 1 and 10 µg/m3 are
typical PCP concentrations after extensive treatments, though
higher levels, up to 25 µg/m3, have been found in rooms treated one
to several years earlier. For comparison, PCP indoor air levels in
untreated houses are generally below 0.1 µg/m3.
According to the usage pattern, the main sources of
occupational exposure to PCP are the treatment of lumber in
sawmills and treatment plants, and exposure to treated wood during
carpentry and other wood-working activities. Most of the reported
air concentrations at the work-place are below the TWA MAC value of
500 µg/m3 that has been established by several countries.
Occupational exposure to PCP mainly occurs via inhalation and
dermal exposure.
Since the PCP concentrations in the sources (air, food) do not
directly indicate the actual PCP intake by the different routes,
extrapolation from urine residue data has been used to estimate
human total body exposure. Mean or median urine-PCP levels range
around 10 µg/litre for the general population without known
exposure, around 40 µg/litre for non-occupationally exposed
persons, and around 1000 µg/litre for occupationally exposed
people.
The ranges of urine levels observed in exposed and unexposed
persons overlap considerably. This overlap probably occurs because
occupational exposure does not necessarily involve high loading,
while non-occupationally exposed people may, in some instances, be
exposed to PCP at levels encountered at the work-place.
1.5. Effects on Organisms in the Environment
As a result of its biocidal properties, PCP negatively affects
non-target organisms in soil and water at relatively low
concentrations. Algae appear to be the most sensitive aquatic
organisms; as little as 1 µg/litre can cause significant inhibition
of the most sensitive algal species. Less sensitive species show
EC50 values of around 1 mg/litre.
Most aquatic invertebrates (annelids, molluscs, crustacea) and
vertebrates (fish) are affected by PCP concentrations below 1
mg/litre in acute toxicity tests. Generally, reproductive and
juvenile stages are the most sensitive, with LC50 values as low as
0.01 mg/litre for fish larvae. Low levels of dissolved oxygen, low
pH, and high temperature increase the toxic effects of PCP.
Concentrations causing sublethal effects on fish are in the low
µg/litre range. As PCP contamination in many surface waters is in
this range, population and community effects cannot be ruled out.
This is also indicated by the substantial alterations in the
community structure of model ecosystems that are induced by PCP.
PCP is accumulated by aquatic organisms. Fresh-water fish show
bioconcentration factors of up to 1000 compared to < 100 in marine
fish. The portion of PCP taken up, either through the surrounding
water or along the food chain, is probably species specific.
PCP taken up by terrestrial plants remains in the roots and is
partly metabolized.
1.6. Kinetics and Metabolism
PCP is readily absorbed through the intact skin and respiratory
and gastrointestinal tracts, and distributed in the tissues.
Highest levels are observed in liver and kidney, and lower levels
are found in body fat, brain, and muscle tissue. There is only a
slight tendency to bioaccumulate, and so relatively low PCP
concentrations are found in tissues. In rodent species,
detoxication occurs through the oxidative conversion of PCP to
tetrachlorohydroquinone, to a small extent also to
trichlorohydroquinone, as well as through conjugation with
glucuronic acid. In rhesus monkeys, no specific metabolites have
been detected. In man, metabolism of PCP to
tetrachlorohydroquinone seems to occur only to a small extent.
Rats, mice, and monkeys excrete PCP and their metabolites,
either free or conjugated with glucuronic acid, mainly in urine
(rodents, 62 - 83%; monkeys, 45 - 75%) and to a lesser extent with
the faeces (rodents, 4 - 34%; monkeys, 4 - 17%). The
pharmacokinetic profile following single doses depends on the
species and possibly on the sex of the test animals. Rats and mice
eliminate PCP rapidly, with a half-life of 6 - 27 h. The kinetics
in rats follow a biphasic elimination scheme with a comparatively
slow second elimination phase (half-life, 33 - 374 h), perhaps
because extensive enterohepatic circulation retains PCP in the
liver. Retention may also be the result of plasma-protein binding
of PCP, which seems to become stronger at lower PCP concentrations.
In rats, 90% of an applied single oral dose is excreted by day
3 with small amounts still remaining in the liver (0.3%) and kidney
(0.05%) after 9 days. On the other hand, monkeys show a much
slower elimination rate (half-life, 41 - 92 h), apparently because
they do not metabolize PCP; even 15 days after oral application of
a single dose (10 mg/kg bodyweight), about 11% of the total dose
remained in the body, particularly in the intestines and liver.
The elimination kinetics of PCP in human beings are a
controversial subject. A study on 4 male volunteers ingesting a
single oral dose of water-soluble Na-PCP at 0.1 mg/kg body weight
showed a rapid elimination of PCP both in urine (half-life, 33 h)
and plasma (30 h). Within 168 h, 74% of the dose was excreted in
urine as free PCP and 12% as its glucuronide, while about 4% was
eliminated in the faeces. In contrast to this study, the
application of oral doses of between 0.016 and 0.31 mg PCP/kg body
weight in 40% ethanol revealed a substantially slower PCP
excretion rate, with elimination half-lives of 16 days (plasma) and
18 - 20 days (urine). These low elimination rates have been
ascribed to the high protein binding tendency of PCP.
Some animal data indicate that there may be long-term
accumulation and storage of small amounts of PCP in human beings.
The fact that urine- or blood-PCP levels do not completely
disappear in some occupationally exposed people, even after a long
absence of exposure, seems to confirm this, though the
biotransformation of hexachlorobenzene and related compounds
provides an alternative explanation of this phenomenon. However,
there is a lack of data concerning the long-term fate of low PCP
levels in animals as well as in man. Furthermore, no data are
available on the accumulation and effects of microcontaminants
taken up by people together with PCP.
1.7. Effects on Experimental Animals and In Vitro Test Systems
In the main, mammalian studies have been relatively consistent
in their demonstration of the effects of exposure to PCP. In rats,
lethal doses induce an increased respiratory rate, a marked rise in
temperature, tremors, and a loss of righting reflex. Asphyxial
spasms and cessation of breathing occur soon before cardiac arrest,
which is in turn followed by a rapid, intense rigor mortis.
PCP is highly toxic, regardless of the route, length, and
frequency of exposure. Oral LD50 values for a variety of species
range between 27 and 205 mg/kg body weight according to the
different solvent vehicles and grades of PCP. There is limited
evidence that the most dangerous route of exposure to PCP is
through the air.
PCP is also an irritant for exposed epithelial tissue,
especially the mucosal tissues of the eyes, nose, and throat. Other
localized acute effects include swelling, skin damage, and hair
loss, as well as flushed skin areas where PCP affects surface blood
vessels. Exposure to technical formulations of PCP may produce
chloracne. Comparative studies indicate that this is a response to
microcontaminants, principally PCDDs, present in the commercial
product. The parent molecule appears responsible for immediate
acute effects, including irritation and the uncoupling of oxidative
phosphorylation with a resultant elevated temperature.
Short- and long-term studies indicate that purified PCP has a
fairly limited range of effects in test organisms, primarily rats.
Exposure to fairly high concentrations of PCP may reduce growth
rates and serum-thyroid hormone levels, and increase liver weights
and/or the activity of some liver enzymes. In contrast, technical
formulations of PCP usually at much lower concentrations can
decrease growth rates, increase the weights of liver, lungs,
kidneys, and adrenals, increase the activity of a number of liver
enzymes, interfere with porphyrin metabolism, alter haematological
and biochemical parameters and interfere with renal function.
Apparently microcontaminants are the principal active moities in
the nonacute toxicity of commercial PCP.
PCP is fetotoxic, delaying the development of rat embryos and
reducing litter size, neonatal body weight, neonatal survival, and
the growth of weanlings. The no-observed-adverse-effect-level
(NOAEL) for technical PCP is a maternal dose of 5 mg/kg body weight
per day during organogenesis. The NOAEL for purified PCP is lower.
In one study, it was reported that purified PCP was slightly more
embryo/fetotoxic than technical PCP, presumably because
contaminants induced enzymes that detoxified the parent compound.
PCP is not considered teratogenic, though, in one instance,
birth defects arose as an indirect result of maternal hyperthermia.
The NOAEL in rat reproduction studies is 3 mg/kg body weight per
day. This value is remarkably close to the NOAEL mentioned in the
previous paragraph, but there are no corroborating studies in other
mammalian species.
PCP has also proved immunotoxic to mice, rats, chickens, and
cattle; at least part of this effect is caused by the parent
molecule.
Neurotoxic effects have also been reported, but the possibility
that these are due to microcontaminants has not been excluded.
PCP is not considered carcinogenic for rats. Mutagenicity
studies support this conclusion in as much as pure PCP has not been
found to be highly mutagenic. Its carcinogenicity remains
questionable because of shortcomings in these studies. The
presence of at least one carcinogenic microcontaminant (H6CDD)
suggests that the potential for technical PCP to cause cancer in
laboratory animals cannot be completely ruled out.
Note: Since the publication of this monograph in 1987, however, the
results of adequate carcinogenicity studies with commercial-grade
pentachlorophenol have been published. The conclusions of these studies
are indicated in the addendum to 8.6 Carcinogenicity.
1.8. Effects on Man
The effects of PCP on man are very similar to those reported in
experimental animals. Human data have been obtained primarily from
accidental exposures and from the work-place. Unfortunately, there
are few precise estimates of exposure, hence dose-response
relationships are difficult to establish in human beings.
It is clear that the use of PCP may pose a significant hazard
with regard to specific aspects of the health of workers employed
in the production or use of PCP. Chloracne, skin rashes,
respiratory diseases, neurological changes, headaches, nausea, and
weakness have been documented in workers at numerous production and
manufacturing sites. Similar symptoms have been reported in some
inhabitants of houses treated internally with PCP. Acute
intoxications leading to hyperpyrexia and death have been clearly
associated with exposure to the chlorophenol molecule itself,
whereas chloracne appears to be an effect of the PCDD and PCDF
microcontaminants. Changes in industrial practice have resulted
in fewer high-dosage, acute exposures, but deaths due to
occupational overexposure to PCP are still being reported.
Studies designed to examine biochemical changes in wood-workers
exposed to high levels of PCP for extended periods have failed to
indicate statistically significant effects on major organs, neural
tissues, blood elements, the immune system, or reproductive
capacity. However, many of these studies were based on small
sample sizes; hence, analyses of trends indicating effects on liver
enzymes, kidney function, T-cell suppression, nerve conduction
velocity, etc., have not been statistically significant. Others
have been non-specific in the search for signs of intoxication in
large groups of workers. However, there are mounting indications
that long-term exposure to relatively high levels of PCP leading to
blood-plasma concentrations as high as 4 ppm is likely to cause
borderline effects on some physiological processes. Some of these
effects, especially those involving the liver and the immune
system, may be caused, in whole or in part, by the
microcontaminants of these chlorophenols, especially H6CDD.
Several epidemiological studies from Sweden and the USA have
indicated that occupational exposure to mixtures of chlorophenols
is associated with increased incidences of soft tissue sarcomas,
nasal and nasopharyngeal cancers, and lymphomas. In contrast,
surveys from Finland and New Zealand have not detected such
relationships. The major deficiency in all of these studies
appears to be a lack of specific exposure data.
There are no conclusive reports of increased incidences of
cancers in workers exposed specifically to PCP; however, there have
not been any carefully conducted studies of a suitably exposed
occupational group large enough to provide the necessary
statistical power to identify an increase in cancer mortality.
Furthermore, there are few occupational groups that have been
exposed to a single chemical, such as PCP. Finally, the various
levels of microcontaminants in different formulations make
inferences to PCP in general difficult.
Persons non-occupationally exposed to technical PCP in rooms
complained about relatively unspecific symptoms (headache, fatigue,
hair loss, tonsillitis, etc.); a causative connection with PCP
could not be proved or disproved.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS
Pentachlorophenol (PCP) and its salt, sodium pentachlorophenate
(Na-PCP), are the most important forms of pentachlorophenol in
terms of production and use. Other derivatives such as the
potassium salt, K-PCP, and the lauric acid ester, L-PCP are of
minor importance. Reflecting this minor role, few data on the
physical and chemical properties of K-PCP and L-PCP are reported in
the literature. Hence, this section primarly concerns PCP and its
sodium salt.
2.1. Identity
2.1.1. Pentachlorophenol (PCP)
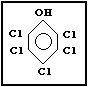 Molecular formula: C6HCl5O
CAS chemical name: Pentachlorophenol
Common synonyms: chlorophen; PCP; penchlorol; penta;
pentachlorofenol; pentachlorofenolo;
pentachlorphenol; 2,3,4,5,6-
pentachlorophenol
Common trade names: Acutox; Chem-Penta; Chem-Tol; Cryptogil
ol; Dowicide 7; Dowicide EC-7; Dow
Pentachlorophenol DP-2 Antimicrobial;
Durotox; EP 30; Fungifen; Fungol; Glazd
Penta; Grundier Arbezol; Lauxtol; Lauxtol
A; Liroprem; Moosuran; NCI-C 54933; NCI-C
55378; NCI-C 56655; Pentacon; Penta-Kil;
Pentasol; Penwar; Peratox; Permacide;
Permagard; Permasan; Permatox; Priltox;
Permite; Santophen; Santophen 20;
Sinituho; Term-i-Trol; Thompson's Wood
Fix; Weedone; Witophen P
CAS registry number: 87-86-5
2.1.2. Sodium pentachlorphenate (Na-PCP)
Molecular formula: C6HCl5O
CAS chemical name: Pentachlorophenol
Common synonyms: chlorophen; PCP; penchlorol; penta;
pentachlorofenol; pentachlorofenolo;
pentachlorphenol; 2,3,4,5,6-
pentachlorophenol
Common trade names: Acutox; Chem-Penta; Chem-Tol; Cryptogil
ol; Dowicide 7; Dowicide EC-7; Dow
Pentachlorophenol DP-2 Antimicrobial;
Durotox; EP 30; Fungifen; Fungol; Glazd
Penta; Grundier Arbezol; Lauxtol; Lauxtol
A; Liroprem; Moosuran; NCI-C 54933; NCI-C
55378; NCI-C 56655; Pentacon; Penta-Kil;
Pentasol; Penwar; Peratox; Permacide;
Permagard; Permasan; Permatox; Priltox;
Permite; Santophen; Santophen 20;
Sinituho; Term-i-Trol; Thompson's Wood
Fix; Weedone; Witophen P
CAS registry number: 87-86-5
2.1.2. Sodium pentachlorphenate (Na-PCP)
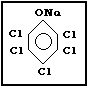 Molecular formula: C6Cl5ONa
C6Cl5ONa x H2O (as monohydrate)
Common synonyms: penta-ate; pentachlorophenate sodium;
pentachlorophenol, sodium salt;
pentachlorophenoxy sodium; pentaphenate;
phenol, pentachloro-, sodium derivative
monohydrate; sodium PCP; sodium
pentachlorophenate; sodium
pentachlorophenolate; sodium
pentachlorophenoxide
Common trade names: Albapin; Cryptogil Na; Dow Dormant
Fungicide; Dowicide G-St; Dowicide G;
Napclor-G; Santobrite; Weed-beads;
Xylophene Na; Witophen N
CAS registry number: 131-52-2 (Na-PCP);
27735-64-4 (Na-PCP monohydrate)
2.1.3. Pentachlorophenyl laurate
The molecular formula of pentachlorophenyl laurate is
C6Cl5OCOR; R is the fatty acid moiety, which consists of a mixture
of fatty acids ranging in carbon chain length from C6 to C20, the
predominant fatty acid being lauric acid (C12) (Cirelli, 1978b).
2.2. Impurities in Pentachlorophenol
Technical PCP has been shown to contain a large number of
impurities, depending on the manufacturing method (section 3.2.1).
These consist of other chlorophenols, particularly isomeric
tetrachlorophenols, and several microcontaminants, mainly
polychlorodibenzodioxins (PCDDs), polychlorodibenzofurans (PCDFs),
polychlorodiphenyl ethers, polychlorophenoxyphenols, chlorinated
cyclohexenons and cyclohexadienons, hexachlorobenzene, and
polychlorinated biphenyls (PCBs). Table 1 presents analyses of PCP
formulations taken from several publications. According to Crosby
et al. (1981), the quality of PCP is depends on the source and date
of manufacture. Furthermore, analytical results may be extremely
variable, particularly with regard to earlier results, which should
be considered with caution. Jensen & Renberg (1972) detected
chlorinated 2-hydroxydiphenyl ethers, which obviously may transform
to dioxins during gas chromatography, thus giving a false
indication of a higher level of PCDDs. Unlike these "predioxins",
other isomers are not direct precursors of dioxins, and are
labelled "isopredioxins".
Table 1. Impurities (mg/kg PCP) in different technical PCP products
---------------------------------------------------------------------------------------------------
Component Specification, producer, PCP content (%)
Tech- Tech- Tech- Techni- Techni- Techni- Technicalf
nicala nicalb nicalb,e calc,g,h cald,i cale
Monsanto Dow Dow Dow Dow Dyn. Nobel Rhône-Poulenc
(84.6%) (88.4%) (98%) (90.4%) (ns)j (87%) (86%)
---------------------------------------------------------------------------------------------------
Phenols
Tetrachloro- 30 000 44 000 2700 10 4000 ns 50 000 70 000
Trichloro- ns < 1000 500 < 1000 ns 20 ns
Higher chlorinated ns 62 000 5000 ns ns ns 70 000
phenoxyphenols
Dibenzo-p-dioxins
Tetrachloro- < 0.1 < 0.05 < 0.05 < 0.05 < 0.2k < 0.001 < 0.01
Pentachloro- < 0.1 ns ns ns < 0.2 ns ns
Hexachloro- 8 4 < 0.5 1 9 3.5 5
Heptachloro- 520 125 < 0.5 6.5 235 130 150
Octachloro- 1380 2500 < 1.0 15 250 600 600
Dibenzofurans
Tetrachloro- < 4 ns ns ns < 0.2 ns ns
Pentachloro- 40 ns ns ns < 0.2 0.2 ns
Hexachloro- 90 30 < 0.5 3.4 39 10 ns
Heptachloro- 400 80 < 0.5 1.8 280 60 ns
Octachloro- 260 80 < 0.5 < 1.0 230 150 ns
Hexachlorobenzene ns ns ns 400 ns ns ns
---------------------------------------------------------------------------------------------------
a From: Goldstein et al. (1977).
b From: Schwetz et al. (1974).
c From: Schwetz et al. (1978).
d From: Buser (1975).
e From: Umweltbundesamt (1985).
f From: Anon (1983b).
g Purified.
h Dowicide EC-7.
i Dowicide 7.
j ns = not specified.
k < = below detection limit.
In Fig. 1, the structures and numbering system for the
polychlorinated dibenzodioxins (PCDDs) and dibenzofurans (PCDFs)
are illustrated.
Molecular formula: C6Cl5ONa
C6Cl5ONa x H2O (as monohydrate)
Common synonyms: penta-ate; pentachlorophenate sodium;
pentachlorophenol, sodium salt;
pentachlorophenoxy sodium; pentaphenate;
phenol, pentachloro-, sodium derivative
monohydrate; sodium PCP; sodium
pentachlorophenate; sodium
pentachlorophenolate; sodium
pentachlorophenoxide
Common trade names: Albapin; Cryptogil Na; Dow Dormant
Fungicide; Dowicide G-St; Dowicide G;
Napclor-G; Santobrite; Weed-beads;
Xylophene Na; Witophen N
CAS registry number: 131-52-2 (Na-PCP);
27735-64-4 (Na-PCP monohydrate)
2.1.3. Pentachlorophenyl laurate
The molecular formula of pentachlorophenyl laurate is
C6Cl5OCOR; R is the fatty acid moiety, which consists of a mixture
of fatty acids ranging in carbon chain length from C6 to C20, the
predominant fatty acid being lauric acid (C12) (Cirelli, 1978b).
2.2. Impurities in Pentachlorophenol
Technical PCP has been shown to contain a large number of
impurities, depending on the manufacturing method (section 3.2.1).
These consist of other chlorophenols, particularly isomeric
tetrachlorophenols, and several microcontaminants, mainly
polychlorodibenzodioxins (PCDDs), polychlorodibenzofurans (PCDFs),
polychlorodiphenyl ethers, polychlorophenoxyphenols, chlorinated
cyclohexenons and cyclohexadienons, hexachlorobenzene, and
polychlorinated biphenyls (PCBs). Table 1 presents analyses of PCP
formulations taken from several publications. According to Crosby
et al. (1981), the quality of PCP is depends on the source and date
of manufacture. Furthermore, analytical results may be extremely
variable, particularly with regard to earlier results, which should
be considered with caution. Jensen & Renberg (1972) detected
chlorinated 2-hydroxydiphenyl ethers, which obviously may transform
to dioxins during gas chromatography, thus giving a false
indication of a higher level of PCDDs. Unlike these "predioxins",
other isomers are not direct precursors of dioxins, and are
labelled "isopredioxins".
Table 1. Impurities (mg/kg PCP) in different technical PCP products
---------------------------------------------------------------------------------------------------
Component Specification, producer, PCP content (%)
Tech- Tech- Tech- Techni- Techni- Techni- Technicalf
nicala nicalb nicalb,e calc,g,h cald,i cale
Monsanto Dow Dow Dow Dow Dyn. Nobel Rhône-Poulenc
(84.6%) (88.4%) (98%) (90.4%) (ns)j (87%) (86%)
---------------------------------------------------------------------------------------------------
Phenols
Tetrachloro- 30 000 44 000 2700 10 4000 ns 50 000 70 000
Trichloro- ns < 1000 500 < 1000 ns 20 ns
Higher chlorinated ns 62 000 5000 ns ns ns 70 000
phenoxyphenols
Dibenzo-p-dioxins
Tetrachloro- < 0.1 < 0.05 < 0.05 < 0.05 < 0.2k < 0.001 < 0.01
Pentachloro- < 0.1 ns ns ns < 0.2 ns ns
Hexachloro- 8 4 < 0.5 1 9 3.5 5
Heptachloro- 520 125 < 0.5 6.5 235 130 150
Octachloro- 1380 2500 < 1.0 15 250 600 600
Dibenzofurans
Tetrachloro- < 4 ns ns ns < 0.2 ns ns
Pentachloro- 40 ns ns ns < 0.2 0.2 ns
Hexachloro- 90 30 < 0.5 3.4 39 10 ns
Heptachloro- 400 80 < 0.5 1.8 280 60 ns
Octachloro- 260 80 < 0.5 < 1.0 230 150 ns
Hexachlorobenzene ns ns ns 400 ns ns ns
---------------------------------------------------------------------------------------------------
a From: Goldstein et al. (1977).
b From: Schwetz et al. (1974).
c From: Schwetz et al. (1978).
d From: Buser (1975).
e From: Umweltbundesamt (1985).
f From: Anon (1983b).
g Purified.
h Dowicide EC-7.
i Dowicide 7.
j ns = not specified.
k < = below detection limit.
In Fig. 1, the structures and numbering system for the
polychlorinated dibenzodioxins (PCDDs) and dibenzofurans (PCDFs)
are illustrated.
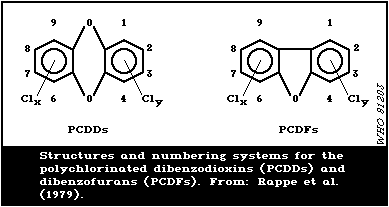 Since the toxicity of PCDDs and PCDFs depends not only on the
number but also on the position of chlorine substituents, a precise
characterisation of PCP impurities is essential. The presence of
highly toxic 2,3,7,8-tetrachlorodibenzo- p-dioxin (2,3,7,8-T4CDD)
has only been confirmed once in commercial PCP samples. In the
course of a collaborative survey, one out of five laboratories
detected 2,3,7,8-T4CDD in technical PCP and Na-PCP samples at
concentrations of 250 - 260 and 890 - 1100 ng/kg, respectively
(Umweltbundesamt, 1985). Buser & Bosshardt (1976) found detectable
amounts of T4CDD (0.05 - 0.23 mg/kg) in some samples of different
technical PCP products, but on re-analysis were unable to confirm
the compound's identity. In other cases, T4CDD has not been
identified at detection limits of 0.2 - 0.001 mg/kg (Table 1).
The higher polychlorinated dibenzodioxins and dibenzofurans
are more characteristic of PCP formulations (Table 1).
Hexachlorodibenzo- p-dioxin (H6CDD), which is also considered
highly toxic and carcinogenic (section 8), was found at levels of
0.03 - 35 mg/kg (Firestone et al., 1972), 9 - 27 mg/kg (Johnson et
al., 1973), and < 0.03 - 10 mg/kg (Buser & Bosshardt, 1976).
According to Fielder et al. (1982), the 1,2,3,6,7,9-, 1,2,3,6,8,9-,
1,2,3,6,7,8-, and 1,2,3,7,8,9-isomers of H6CDD have been detected
in technical PCP. The 1,2,3,6,7,8and 1,2,3,7,8,9-H6CDDs
predominated in commercial samples of technical PCP (Dowicide 7)
and Na-PCP. Octachloro-dibenzo- p-dioxin (OCDD) is present in
relatively high amounts in unpurified technical PCP (Table 1).
Recently, the identification of 2-bromo-3,4,5,6-
tetrachlorophenol as a major contaminant in three commercial PCP
samples (ca. 0.1%) has been reported. This manufacturing by-
product has probably not been detected in other analyses because it
is not resolved from the PCP peak by traditional chromatographic
methods (Timmons et al., 1984).
2.2.1. Formation of PCDDs and PCDFs during thermal decomposition
The thermal decomposition of PCP or Na-PCP yields significant
amounts of PCDDs and PCDFs, depending on the thermolytic
conditions. For pure PCP, dimerization of PCP has been suggested
as an underlying reaction process; in technical PCP, additional
reactions, i.e., dechlorination of higher chlorinated PCDDs and
cyclization of predioxins are involved in forming various and
different PCDD isomers (Rappe et al., 1978b).
Pyrolysis of alkali metal salts of PCP at temperatures above
300 °C results in the condensation of two molecules to produce
OCDD. PCP itself forms traces of OCDD only on prolonged heating in
bulk and at temperatures above 200 °C (Sandermann et al., 1957;
Langer et al., 1973; Stehl et al., 1973).
Although present in original technical PCP products, a number
of PCDDs, other than OCDD, are generated during thermal
decomposition (290 - 310 °C) in the absence of oxygen (Table 2)
(Buser, 1982).
Table 2. PCDDs (mg/kg PCP) in the
pyrolysate of technical PCP and Na-PCPa
--------------------------------------
PCP Na-PCP
--------------------------------------
2,3,7,8-T4CDD -b -c
1,2,3,7,8,9-H6CDD 53 2.1
1,2,3,6,7,8-H6CDD 66 0.95
Total H6CDD 455 10.5
H7CDD 5200 65
OCDD 15 000 200
--------------------------------------
a From: Buser (1982).
b Detection limit (1 mg/kg).
c Detection limit (0.25 mg/kg).
2.3. Physical, Chemical, and Organoleptic Properties
Pure pentachlorophenol consists of light tan to white,
needlelike crystals. It has a pungent odour when heated (Windholz,
1976). Its vapour pressure suggests that it is relatively
volatile, even at ambient temperatures. Since PCP is practically
insoluble in water at the slightly acidic pH generated by its
dissociation, readily water-soluble salts such as Na-PCP are used
as substitutes, where appropriate.
Na-PCP is non-volatile; its sharp PCP odour results from slight
hydrolysis (Crosby et al., 1981). Technical PCP consists of
brownish flakes or brownish oiled, dustless flakes, coated with a
mixture of benzoin polyisopropyl and pine oil. Technical Na-PCP
consists of cream-coloured beads (Anon., 1983a,b). Technically
pure L-PCP consists of a brown oil that is insoluble in water and
alcohols, and soluble in non-polar solvents, oils, fats, waxes, and
plasticizers (Cirelli, 1978b).
PCP is non-inflammable and non-corrosive in its unmixed state,
whereas a solution in oil causes deterioration of rubber (Mercier,
1981).
Because of the electron withdrawal by the ring chlorines, PCP
behaves as an acid, yielding water-soluble salts such as sodium
pentachlorophenate. Due to nucleophilic reactions of the hydroxyl
group, PCP can form esters with organic and inorganic acids and
ethers with alkylating agents, such as methyl iodide and
diazomethane (Crosby et al., 1981). This property has been used
for analytical purposes (section 2.5.2).
PCP may exist in two forms: the anionic phenolate, at neutral
to alkaline pH, and the undissociated phenol at acidic pH. At pH
2.7, PCP is only 1% ionized; at pH 6.7, it is 99% ionized (Crosby
et al., 1981). Other relevant properties of pure PCP and Na-PCP
are shown in Table 3.
PCDDs and PCDFs may also be formed during the combustion of
materials treated with either purified or technical PCP. Smoke from
birch leaves impregnated with purified Na-PCP and burnt on an open
fire showed considerably increased amounts of PCDDs compared with
the original sample (Table 4). The mass fragmentograms revealed 14
of the 22 possible T4CDD isomers with 1,3,6,8- and 1,3,7,9-T4CDD as
the main and 2,3,7,8- T4CDD as minor isomers. The formation of
PCDFs, including small amounts of 2,3,7,8-T4CDF, during either
combustion or micropyrolysis (280 °C, 30 min) was only observed in
technical PCP samples; purified Na-PCP was negative in this respect
(Rappe et al., 1978b).
Table 3. Physical, chemical, and organoleptic properties of PCP
and Na-PCP
------------------------------------------------------------------
PCP Na-PCP
------------------------------------------------------------------
Boiling pointa 310 °C (decomposition)
Relative molecular massb 266.4 288.3
306.3
(monohydrate)
Melting pointa 191 °C
Density (d422 in g/ml)b 1.987 2
Vapour pressure kPa (mmHg)
at 20 °Cc 2 x 10-6 (1.5 x 10-5)
at 19 °Cd 6.7 x 10-7 (5 x 10-6)
Saturation vapour density 220
(µg/m3) (20 °C)c
Steam volatilitye 0.167
(g/100 g water vapour)
(100 °C)
------------------------------------------------------------------
Table 3 (contd.)
------------------------------------------------------------------
PCP Na-PCP
------------------------------------------------------------------
Solubility in fat 213
(g/kg) (37 °C)f
n-Octanol/water partition 4.84 (pH 1.2);
coefficient (log P)g 3.56 (pH 6.5);
3.32 (pH 7.2);
3.86 (pH 13.5)
pKa (25 °C)e 4.7
Solubility in water:
(g/litre)e,h,i
0 °C, pH 5 0.005
20 °C, pH 5 0.014
30 °C, pH 5 0.020
20 °C, pH 7 2
20 °C, pH 8 8
20 °C, pH 10 15 > 200
25 °C 330
Solubility in organic
solvents (g/100 g)
(25 °C)b:
acetone 50 35
benzene 15 insoluble
ethanol (95%) 120 65
ethylene glycol 11 40
isopropanol 85 25
methanol 180 25
Odour threshold 1.6 (in water)
(mg/litre)j:
Olfactory threshold 0.03 (in water)
(mg/litre)j:
------------------------------------------------------------------
a From: IRPTC (1983).
b From: Cirelli (1978b).
c From: Zimmerli (1982).
d From: Dobbs & Grant (1980).
e From: Crosby et al. (1981).
f From: Rippen (1984).
g From: Kaiser & Valdmanis (1982).
h From: Gunther et al. (1968).
i From: Bundesamt für Umweltschutz (1982).
j From: Dietz & Traud (1978a).
Jansson et al. (1978) observed a very wide range of PCDD
concentrations in the smoke from burning wood chips impregnated
with a technical PCP formulation (Table 4). The formation of PCDDs
was favoured by temperatures below 500 °C, oxygen deficit, and
lower gas-retention time. The results given in Table 4 are
corrected for the very low background values obtained by burning
untreated wood chips.
When technical PCP was burnt in a quartz reactor (600 °C, 10
min), Lahaniatis et al. (1985) identified the following thermolytic
products: pentachlorobenzene, hexachlorobenzene, octachlorostyrole,
octachloronaphthaline, decachlorobiphenyl, H6CDF, OCDF, and OCDD.
2,3,7,8-T4CDD was not detected at a detection limit of 1 mg/kg PCP.
Olie et al. (1983) found only slightly higher levels of PCDDs
and PCDFs in the fly ash of burned new wood treated with PCP
compared with painted wood, which was more than 60 years old.
However, because data were missing on PCDD/PCDF levels in the
original samples and on the conditions of burning, meaningful
interpretation of these results is not possible.
2.4. Conversion Factors
1 ppm = 10.9 mg PCP/m3 (25 °C, 101.3 kPa)
1 mg PCP/m3 = 0.09 ppm
Table 4. Amount of PCDDs in the original sample and in
the smoke from combusted materials treated with purified
Na-PCP or technical PCP
----------------------------------------------------------
Birch leavesa Wood chipsb
(mg PCDDs/kg Na-PCP (mg PCDDs/kg PCP
(purified)) (technical))
Original Smokec Original Smokec,d
sample sample
----------------------------------------------------------
T4CDD < 0.02 5.2 nde < 4.7 - 47
P5CDD < 0.03 14 nd < 1.2 - 419
H6CDD < 0.03 56 7 < 9.3 - 93
H7CDD 0.3 172 93 < 4.7 - 279
OCDD 0.9 710 186 < 0.9 - 442
----------------------------------------------------------
a Adapted from: Rappe et al. (1978b).
b Adapted from: Jansson et al. (1978).
c Smoke trapped on charcoal filter.
d Depending on combustion conditions.
e nd = not determined.
2.5. Analytical Methods
A number of methods have been used to determine PCP in a
variety of media. The earlier procedures were reviewed by Bevenue
& Beckman (1967). They were mostly based on colour reactions,
which are not very specific and relatively insensitive. For
several years, more sophisticated devices have been available to
analysts, of which gas chromatography has become the method of
choice (Table 5).
Table 5. Analytical methods for the determination of PCP
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Air
Air- Impinger collection Derivatization with diazo- ECa 0.22 mg/m3 nsc Hoben et al.
aerosol with KOH; hexane methane; purification in (1976a)
extraction chromatoflex Florisil
column; GCd analysis
Air Filter and bubbler HPLCe analysis; column: UV254b 0.27 mg/m3 95.3- NIOSH
collection; ethylene µ Bondapack C18; mobile 100.9% (1978)
glycol extraction phase: methanol/water
Air Bubbler collection; Derivatization with acetyl EC 0.05 µg/m3 ns Dahms &
absorption in K2CO3 chloride; GC analysis Metzner
solution; hexane (1979)
extraction
Air Adsorption on to GC analysis EC 0.5 µg/m3 ns Zimmerli &
filter papers Zimmermann
impregnated with (1979)
adsorbant extraction
concentration in ether
Air Impinger collection; HPLC analysis; column: UV225 0.5 µg/m3 ns Woiwode et
absorption in K2CO3- Lichrosorb C18; mobile al. (1980)
solution; benzene phase: methanol
extraction
Air Air from wood samples GC analysis EC ns 89.7- Warren et
in reactor tube ads- (< 1.5 99.9% al. (1982)
orbed on silica gel; µg/m3)
desorption with benzene
Air Impinger collection; Derivatization with acetic EC ns ns Kauppinen &
absorption in anhydride in presence of Lindroos
toluene pyridine; GC analysis (1985)
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Biological tissues and fluids
Blood Benzene extraction Derivatization with diazo- EC 20 µg/litre 87- Bevenue et
(human) from acidified methane; GC analysis 100% al. (1968)
solution
Blood, Ethyl ether extrac- No derivatization; GC anal- EC-3H 0.1 ng 90- Barthel et
urine, tion from acidified ysis; column: 3% diethylene EC-63Ni 0.02 ng 100% al. (1969)
tissue solution; NaOH ex- glycol succinate + 2% H3PO4;
(human) traction; benzene confirmation by MS and TLC
extraction
Organic Hexane/isopropanol Der. with acetic anhydride EC ns 81-91% Rudling
tissues extraction from in presence of pyridine; (1970)
acidified sample; GC analysis
borax extraction
Urine Ethyl ether extrac- Derivatization with diazo- EC 10 µg/litre 92- Shafik et
(rat) tion from acidified ethane; GC analysis 98% al. (1973)
solution
Adipose NaOH extraction; Derivatization with diazo- EC 5 µg/kg 75% Shafik
tissue diethyl ether methane; GC analysis (1973)
(human) extraction from
acidified solution
Urine, Acidic hydrolysis No derivatization; negative MSf 1 ng 90% Dougherty &
seminal (urine); hexane/2- chemical ionization (NCI) Piotrowska
fluid propanol or hexane/ mass spectrometry; internal (1976a;b)
(human) ether extraction from standard: p-chlorobenzophenone
acidified solution
Tissue, Hexane or benzene Derivatization with diazo- EC 20 µg/kg 91.4- Hoben et al.
plasma, extraction ethane (urine) or diazo- 95.3% (1976a)
urine methane; purification in
(rat) chromatoflex Florisil column;
GC analysis
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Tissues Acetone extraction (a) TLCg analysis on silica Radio- ns ns Glickman et
(fish) gel UV254; mobile phase: active al. (1977)
methylene chloride
Acetone extraction (b) Derivatization with MS ns ns Glickman et
methyl iodide and K2CO3; al. (1977)
GC analysis
Urine Hexane extraction Derivatization with acetyl EC 10 µg/litre ns Dahms &
(human) from acidified chloride in the presence of Metzner
solution pyridine; GC analysis (1979)
Urine Benzene extraction Derivatization with diazo- EC 1 µg/litre 93.2- Edgerton
(human, from acidified methane; separation of 97.2% et al.
rat) solution methylated phenols in acid (1979)
alumina column; GC analysis;
GC-MS confirmation
Plasma Benzene extraction Derivatization with acetic EC 50 µg/litre 91- Eben et
(human) anhydride; GC analysis 102% al. (1981)
Urine Acidic or enzymatic Derivatization with acetic EC 20 µg/litre 77- Eben et
(human) hydrolysis; ethyl anhydride; GC analysis 98% al. (1981)
ether extraction;
benzene extraction
Tissues, Diethyl ether (ethyl Purification on Hypersil- UV216 1 µg/kg 62- Mundy &
serum, acetate-hexane) cartridge; mobile phase: 108% Machin
egg extraction from NaOH methanol; HPLC analysis (1981)
yolk/ (hydrochloric acid)
white solution; concentra-
tion by evaporation
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Urine Acidic hydrolysis; HPLC analysis; column: Sphe- UV313 10 µg/litre 102% Drummond
(human) distillation; methy- risorb-ODS; mobile phase: et al.
lene chloride ex- acetonitrile; internal stan- (1982)
traction from acidi- dard: 3,5-dichloro-2,3,6-
fied solution; tribromophenol (DTP)
evaporation and
redistillation in
acetonitrile
Urine Acidic hydrolysis; LC analysis; column: Spheri- UV254 0.1 83.3± Pekari &
(human) hexane/isopropanol sorb-ODS; mobile phase: µmol/litre 3.7% Antero
extraction; evap- methanol (27 µg/litre) (1982)
oration and redis-
tillation in methanol
Urine Acidic hydrolysis; Derivatization with acetic SIM-MSh 1 nmol/litre ns Harge-
(human) int.standard (4,6- or propionic anhydride; (0.27 sheimer &
dibromo- o-cresol) GC analysis µg/litre) Coutts
added; methylene (1983)
chloride extraction
Food
Milk Benzene extraction; Derivatization with acetic EC 5 µg/litre 80- Erney
(bovine) extraction with anhydride; GC analysis 87.2% (1978)
K2CO3 solution
Milk Sulfuric acid Purification in BioSil EC 10 - 15 80% Lamparski
(bovine) digestion; hexane A-column; derivatization µg/litre et al.
extraction with diazomethane; GC (1978)
analysis
Carrots, Soxhlet extraction Derivatization with diazo- EC 0.2 µg/kg 80- Bruns &
potatoes (carrots) or blending ethane; purification in 108% Currie
with acidified Florisil-column; GC (1980)
acetone analysis
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Canned Methylene chloride Derivatization with diazo- EC ns 92- Heikes &
food and extraction from methane; purification in (< 0.3 103% Griffitt
jar lids acidified solution; Florisil-column; GC analysis µg/kg) (1980)
clean-up by gel perme-
ation chromatography
Edible Alkaline hydrolysis; Direct GC analysis on DEGS/ EC 5 - 10 µg/kg 83- Stijve
gelatins iso-octane extraction phosphoric acid column; con- 108% (1981)
from acidified firmation by GC analysis of
solution acetate derivative
Plant Maceration with acidi- Derivatization with diazo- EC ns 94.2% Fuchs-
mater- fied acetone; methane; purification in bichler
ials chloroform extr.; Florisil-column; GC analysis (1982)
cleanup by automated
gel chromatography
Mush- Steam distillation (a) Derivatization with ace- EC 0.5 µg/kg 92% Schönhaber
rooms from acidified sample; tic anhydride GC analysis fresh weight et al.
dichloromethane (b) HPLC analysis; column: UV220 0.5 µg/kg (1982)
extraction LiChrosorb RP-8; mobile
phase: methanol/ o-phos-
phoric acid
Soil
Soil NaOH extraction; Derivatization with diazo- EC 0.1 - 1 > 97% Renberg
clean-up by ion methane; GC analysis µg/kg (1974)
exchanger
Soil Nielsen-Kryger steam Direct GC analysis on fused EC ns 58- Narang et
distillation from silica SE 54 column 95% al. (1983)
acidified soil;
toluene-methylene
chloride extraction
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Water
Raw and Petroleum ether TLC analysis on Al2O3 Colori- 0.5 µg/litre 75- Zigler &
treated extraction from plates; mobile phase: metric 100% Phillips
water acidified sample; (a) benzene;(b) NaOH/acetone; (1967)
evaporation; chromogenic agent: AgNO3/2-
drying; evaporation phenoxyethanol
Natural, Benzene extraction Derivatization with acetic EC 0.01 84- Chau &
waste and K2CO3-solution anhydride; hexane extrac- µg/litre 93% Coburn
water tion; GC analysis (1974)
River Hexane/ethyl ether Derivatization with boron MS 0.01 ns Matsumoto
water extraction; ethyl trifluoride methanol and µg/litre et al.
acetate extraction trimethylsilyl; GC analysis (1977)
from acidified
solution
Surface Toluene extraction; Derivatization with acetic EC 0.01 85% Wegman &
water extraction with anhydride; petroleum ether µg/litre Hofstee
K2CO3-solution extraction; GC analysis (1979)
Waste Chloroform extraction Direct HPLC analysis of UV254 10 µg/litre ns Ervin &
water from acidified sample; chloroform extract; column: McGinnis
rotary evaporation silica gel; mobile phase: (1980)
cyclohexane-acetic acid and
other solvents
Surface Diethyl ether Direct HPLC analysis UV215 20 µg/litre > 80% Ivanov &
water extraction; evapor- Magee
ation; dissolution in (1980)
methanol-petroleum
Surface Chloroform extraction Direct analysis in double Absorp- 5 - 10 92- Carr et
water from acidified beam UV spectrophotometer tion µg/litre 102% al. (1982)
sample;NaOH extraction (320 nm)
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Surface Concentration by GC analysis (no deriviti- EC 0.01 95 ± Rübelt et
water extraction, ads- zation); column: Carbowax µg/litre 3% al. (1982)
orption, rotary 20 M plus phosphoric acid;
evaporation confirmation by MS
Surface, Addition of Na2HPO4 Extraction and derivitzation EC 1 ng/litre 98- Abrahams-
waste, buffer solution by adding hexane containing 100% son & Xie
drinking-(for acid waste water internal standard (2,6-dibro- (1983)
water pH adjustment to 7 mophenol) and acetic anhy-
with NaOH dride directly to sample;
GC analysis
Water Samples prepared from HPLC analysis with isocratic UV280 21 ng ns Buckman et
stock solutions in elution of various substitu- al. (1984)
acetonitrile ted phenols
Wood
Wood Chloroform extraction TLC analysis on silica gel UV 0.06 µg ns Henshaw et
from wood shavings plates; developing solvent: al. (1975)
cyclohexane-acetone-paraffin
Wood- Soxhlet extraction Derivatization with diazo- EC ns 30-70% Levin &
dust with ether ether; methane; GC analysis Nilsson
evaporation; dissol- (1977)
ution in acetone;
TLC separation;
hexane elution
Lumber Pulverization of wood HPLC analysis; column: Sphe- UV230 0.1 µg/cm2 ns Daniels &
(sur- sample; extraction risorb-ODS; mobile phase: Swan
face with acetonitrile water-acetonitrile-acetic (1979)
treated) containing internal acid
standard; ultrasonic
bath
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Sawdust, Extr. with acetic TLC analysis; different sol- Colour 2 mg/kg ns Ting &
wood- acid-methanol; evap- vent systems; sprayed with reaction Quick
shavings oration; conversion tetrabase reagent (1980)
to chloranil in warm
nitric acid
Various materials
Toy Soxhlet extraction Direct GC analysis; confirm- FIDi 1 - 4 mg 70- von
paints with acetone; concen- ation by TLC analysis /litre 100% Langeveld
tration by evaporation (1975)
PCP Dioxane extraction HPLC analysis; column: Bon- UV254 ns 97% Hayes
formula- dapakC18; mobile phase: (1979)
tions methanol/PIC A (paired ion
chromatography A reagent)
and water/PIC A
Sedi- Homogenization; Derivatization: pyrolytic EC 0.5 - 25 92.8- Butte et
ment, toluene extraction ethylation with triethyl µg/kg 97.6% al.
clams from acidified sample sulfonium-iodide; GC anal- (1983)
containing 2,4,6- ysis; confirmation by MS
tribromophenol as
internal standard
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Tallow Vortex mixing; auto- Derivatization with diazo- EC 1 µg/kg 80- Lee et
mated gel permeation methane; Florisil-column; 107% al.
chromatographic clean- GC analysis; confirmation (1984)
up; rotary evapora- with MS
tion; solution in
hexane
Indus- Hexane extraction Derivatization with penta- 0.1 pg ns Sha &
trial (for starch: after fluorobenzyl bromide; GC EC Duffield
starch, steam distillation) analysis and negative ion (1984)
surface chemical ionization MS MS
water (NICI-MS) analysis
---------------------------------------------------------------------------------------------------------
a EC = electron-capture detector.
b UV = ultraviolet.
c ns = not specified.
d GC = gas chromatography.
e HPLC = high-performance liquid chromatography.
f MS = mass spectrometry.
g TLC = thin-layer chromatography.
h SIM = selected ion monitoring.
i FID = flame ionization detector.
j NICI = negative ion chemical ionization.
The determination of PCP is based on the distinctive properties
of this substance: steam distillation is possible because of its
volatility; its acidic behaviour is used in extracting it into a
base and in ion-exchange chromatography; the electro-positive ring
reinforces selective chromatographic adsorption and the absorption
of ultraviolet radiation; finally, the reactivity of PCP with
certain organic compounds to form esters, ethers, and coloured
derivatives is of great importance for its detection and
measurement (Crosby et al., 1981).
Most of the analytical methods used today involved
acidification of the sample to convert PCP to its non-ionized form,
extraction into an organic solvent, possible cleanup by back-
extraction into basic solution, and analysis by gas chromatography
or other chromatographic methods as ester or ether derivatives. In
the following section, the sampling and analytical methods is
described as reviewed mainly by Bevenue & Beckman (1967), Gebefuegi
et al. (1979), and Crosby et al. (1981). In addition, the more
recently published methods for PCP determination in various
matrices are summarized (Table 5).
2.5.1. Sampling methods
In principle, the sampling techniques summarized by Bevenue &
Beckman (1967) are still the methods of choice; more recent methods
are included in Table 5.
The first step in preparing a sample consisting of a solid
material is a thorough pulverization or homogenization in special
mills or blenders. Maceration of the sample in a blender with an
organic solvent is more rapid than Soxhlet extraction and similar
efficiencies can be achieved with both procedures (Bruns & Currie,
1980).
For cellulose materials, adhesives, agricultural commodities,
biological tissues, and water, an initial extraction with dilute
sodium hydroxide solution at room temperature for several hours,
followed by acidification and steam distillation may be preferable.
For samples that contain components strongly complexed with PCP,
such as soybean oil, treatment with hot concentrated sulfuric acid
is recommended prior to steam distillation. Liquid-liquid
partitioning or distillation of the filtered extract at the boiling
point of water may also be used to isolate PCP (Bevenue & Beckman,
1967).
When alkaline soil extracts are acidified, gel formation can
occur at pH values lower than 6, resulting in interference with the
extraction of PCP. According to Renberg (1974), proper separation
is possible if the acidic substances are bound, under alkaline
conditions, to an anion ion exchanger.
When analysing liquid materials, particularly urine samples,
the sample should first be hydrolysed by heating the acidified
urine to free the PCP moiety of its sulfate and glucuronide
conjugates (Edgerton & Moseman, 1979; Drummond et al., 1982; Butte,
1984). Enzymatic hydrolysis is questionable, because the metabolite
tetrachlorohydroquinone strongly inhibits the enzyme
beta-glucuronidase (Ahlborg et al., 1974).
For determining the PCP content of air several possible
sampling procedures are described by Gebefuegi et al. (1979)
including: absorption in liquids, such as potassium carbonate
(K2CO3) solution or ethylene glycol; adsorption on activated
charcoal or silica gel; freezing and condensing by sucking the air
through cooling traps; or derivatization by phenolate formation in
alkaline solution. By pumping high volumes of air through the
sample-collecting device, PCP is concentrated in the collector,
thus enhancing the detection limit.
Concentration is also required for other matrices with
relatively low PCP contents. For water samples, procedures used
involve the separation of PCP from the water by distillation,
sublimation, freeze-drying, adsorption, and extraction (Rübelt et
al., 1982). The extraction solvents, in turn, are concentrated by
distillation or evaporation.
Only a few investigators have used internal standards, adding
specific substances to the samples to check for completeness of
recovery during the extensive solvent extractions and manipulative
steps required. Drummond et al. (1982) used 3,5-dichloro-2,3,6-
tribromophenol, while Needham et al. (1981) incorporated 2,4,6-
tribromophenol, and Hargesheimer & Coutts (1983) spiked the samples
with 4,6-dibromo- o-cresol.
Most recovery data given in Table 5 were obtained by spiking
samples with known amounts of PCP and carrying them through the
entire analytical procedure. Ernst & Weber (1978a) used 14C-PCP
for this purpose. To check the efficiency of acetylation, Rudling
(1970) compared spiked samples with a pentachlorophenyl acetate
standard. According to NIOSH (1978), an appropriate correction
factor should be used if recovery of PCP in air samples is less
than 95%.
Using the analytical method of Erney (1978) (Table 5), Zimmerli
et al. (1980) found that only about 8% of "endogenous" PCP was
extractable from raw bovine milk, though 82.5% of known amounts of
PCP added had been recovered on average. A complete extraction was
only achieved by acid or alkaline pretreatment of the milk (cf.,
Lamparski et al., 1978) (Table 5), which probably releases the PCP
bound to proteins. Zimmerli et al. (1980) concluded from this
finding that recovery data may indicate values that do not
correspond to the true recovery.
2.5.2. Analytical methods
Earlier methods, which have been thoroughly reviewed by Bevenue
& Beckman (1967), were based on the formation of coloured
derivatives from the reaction of PCP with either nitric acid or 4-
aminoantipyrine. Other reagents commonly used in this respect are
p-nitraniline, sulfanilic acid, and 3-methyl-2-benzenethiazoline-
hydrazine (Koppe et al., 1977). As already mentioned, these
colorimetric or spectrometric methods are not very specific and
comparatively insensitive, and therefore only suitable for pure
solutions or for production and routine controls. They may be of
some importance in determining total phenolics, for example, in the
monitoring of levels of phenolics in surface and waste waters.
However, comparative studies, in which 45 laboratories within the
European Communities participated, revealed that photometric
procedures gave rather different results, depending on specific
laboratory conditions (Sonneborn, 1976; Rübelt et al., 1982).
According to Crosby et al. (1981), colorimetric or
spectrophotometric procedures achieve a sensitivity that is, at
best, in the low ppb-range (1:109).
Gas chromatography, particularly when combined with an
electron-capture detector, substantially lowers the detection
limits to the ppt-range (1:1011 - 1:1012) and is therefore the
preferred method today. Very few investigators have applied direct
gas chromatography after the extraction procedures. To reduce peak
tailing, derivatization of PCP with appropriate compounds prior to
analysis is preferred. Diazomethane is most commonly used to
produce the methyl ether. As shown in Table 5, this method, which
is based on the work of Bevenue et al. (1966), has been used to
determine PCP in a variety of matrices including blood, urine,
fish, soil, and water. According to Crosby et al. (1981), it is
an official method for regulatory analysis in the USA. The
procedure for measuring PCP in blood and urine samples as
recommended by the National Institute for Occupational Safety and
Health (NIOSH), USA, is described by Eller (1984a,b).
Other alkyl ethers have been produced as derivatives of PCP,
including the ethyl, propyl, 1-butyl, isobutyl, amyl, and isoamyl-
PCP (Cranmer & Freal, 1970). Besides the potential health risk
incurred when using hazardous reagents such as diazomethane or
dimethyl sulfate, the alkylation method is subject to interferences
from other compounds with active H-atoms, e.g., carboxy acid
herbicides such as 2,4-dichloro-and 2,4,5-trichlorophenoxyacetic
acid (Chau & Coborn, 1974; Crosby et al., 1981). These drawbacks
are avoided by the acetylation of PCP with acetic anhydride to give
acetyl-PCP as reported by Rudling (1970), Chau & Coborn (1974), and
other research workers (Table 5).
Several techniques, other than gas chromatography, have been
used in connection with electron-capture detection. These include
thermal conductivity and microcoulometric detectors (Bevenue &
Beckman, 1967), thin-layer chromatography (TLC), gas chromatography
in connection with mass spectrometry (MS), and high-performance
liquid chromatography (HPLC) equipped with UV detectors. In
particular, the last two methods have become more and more
prevalent as reflected by Table 5. In many cases, mass
spectrometry has been used to confirm the identity of PCP peaks
determined by EC detectors. Dougherty & Piotrowska (1976a) and
Kuehl & Dougherty (1980) screened environmental and tissue samples
for PCP using negative chemical ionization (NCI) mass spectrometry.
This method provides a sensitivity of detection comparable to GC-
ECD analysis. Moreover, it can be used for compound
identification. Since both of these methods require an extensive
amount of pretreatment, a procedure had to be adopted for PCP
determination by which samples could be measured simply and
precisely, without the tedious extraction and formation of
derivatives needed for the other methods. High-performance liquid
chromatography offers these advantages, as using this method direct
determination of PCP is possible, giving peaks of constant height
and high resolution. Comparative GC-ECD and HPLC analyses of
mushrooms conducted by Schönhaber et al. (1982) resulted in similar
detection limits (Table 5).
Detection limits depend not only on the sensitivity of the
detection systems, but also, to a great extent, on the volume of
the sample. The detection limits given in Table 5 refer to the
smallest amounts of PCP detectable using the procedure and sample
size described by the authors. In many cases, it would be possible
to lower the detection limit by taking larger samples, particularly
in the case of gaseous and fluid matrices.
Analytical interferences may become a problem in PCP analysis
for residues, particularly at low measurement levels. Bevenue &
Ogata (1971) reported errors during the determination of PCP in the
picogram range, because of analytical-grade reagents such as sodium
hydroxide. Arsenault (1976) observed an apparent contamination of
samples with PCP from the general laboratory atmosphere. However,
measuring blank samples as controls and purifying reagents should
exclude false data. For example, Dietz & Traud (1978b) distilled
the extraction solvent diethyl ether to remove the antioxidant BHT
(2,6-di- tert-butyl-4-methyl-phenol). Similarly, the authors
recommended the distillation of dioxan prior to its use as
extraction solvent; otherwise, some volatile impurities could
interfere with the measurement of PCP.
Substances interfering during gas chromatography may cause more
of a problem. These include chloronaphthalenes, polychlorinated
biphenyls (PCBs), pesticides such as diuron, and p-methoxytetra-
chlorophenol. Arsenault (1976) therefore questioned the GC-ECD
method in the µg range. In a thorough study on phenolics in water
(Rübelt et al., 1982), derivatization was omitted because non-
specific reactions might occur in complex mixtures, e.g., in
polluted waters. The working group achieved best results in terms
of separation of chlorophenols with a column of 10% Carbowax 20 M
plus 2% phosphoric acid, the mobile phase being nitrogen enriched
with formic acid. The latter was found to prevent tailing
resulting from adsorption of chlorophenols on the packing material
of the column. For quantitative analysis with an unequivocal
identification, it has been recommended that after gas
chromatographic separation the carrier gas should be split and
conducted to both an electron capture detector (ECD), and a flame
ionization detector (FID), as well as to a mass spectrometer (MS).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural Occurrence
Arsenault (1976) hypothesized the existence of a natural
background level of PCP or analytically similar compounds. He
suggested this on the basis that paramethoxytetrachlorophenol, a
metabolite of a fungus, could interfere with the GC-EC analysis of
PCP, because of its similar molecular size, shape, and retention
time. The hypothesis of a natural background level of PCP has not
been examined further. Unsuccessful attempts to produce higher
chlorophenols by enzymatic conversion (Siuda, 1980) suggest that
sources of environmental PCP are exclusively related to human
activities.
3.2. Man-Made Sources
3.2.1. Industrial production
3.2.1.1 Manufacturing processes
PCP was first synthesized by Merz & Weith (1872) using a
similar preparation method to that currently used during commercial
production (Prager et al., 1923). The use of PCP as a wood
preservative started in the late 1930s (Doedens, 1964).
PCP is produced by one of two methods: direct chlorination of
phenols and hydrolysis of hexachlorobenzene. The direct
chlorination is carried out in two steps. First, liquid phenol,
chlorophenol, or a polychlorophenol is bubbled with chlorine gas at
30 - 40 °C to produce 2,4,6-trichlorophenol, which is then
converted to PCP by further chlorination at progressively higher
temperatures in the presence of catalysts (aluminum, antimony,
their chlorides, and others). The second method involves an
alkaline hydrolysis of hexachlorobenzene (HCB) in methanol and
dihydric alcohols, in water and mixtures of different solvents in
an autoclave at 130 - 170 °C (Melnikov, 1971). In the Federal
Republic of Germany, PCP is synthesized by means of stepwise
chlorination of phenols. Na-PCP was produced until 1984 using
hexachlorobenzene hydrolysis; now, it is produced by dissolving PCP
flakes in sodium hydroxide solution (BUA, 1986). In the USA, the
general reaction used is the chlorination of phenols (Crosby et
al., 1981).
In addition to the formation of PCP, numerous by-products are
generated, as reflected by analytical profiles in Table 1. The
chlorination procedure yields a technical product that usually
contains a considerable amount of tetrachlorophenols (4 - 12%) due
to incomplete chlorination reactions. The formation of
microcontaminants is favoured by elevated temperatures and
pressure. With both manufacturing methods, toxic by-products, such
as chlorinated ethers, dibenzofurans, and dibenzo- p-dioxins, are
formed. In addition, the alkaline HCB hydrolysis method can result
in the presence of hexachlorobenzene in the resulting PCP (Jensen
& Renberg, 1973; Plimmer, 1973; Firestone, 1977; Jones, 1981).
3.2.1.2 Emissions during production
Some data are available concerning the loss of phenolic and
nonphenolic compounds into the environment during the normal
production of PCP or Na-PCP (Umweltbundesamt, 1985). The following
air emission concentrations (mg/m3) and mass flow values (g/h) were
reported: PCP 0.7 mg/m3, 9 g/h; tetrachlorophenols 0.2 mg/m3, 0.8
g/h; trichlorophenols 0.02 mg/m3, 0.04 g/h; hexachlorobenzene 23.9
mg/m3, 12 g/h; pentachlorobenzene 2 mg/m3, 15.5 g/h;
tetrachlorobenzene 2.8 mg/m3, 66.5 g/h; OCDD 0.05 mg/m3, 0.04 g/h;
OCDF 0.02 mg/m3, 0.002 g/h.
The annual air emission values resulting from the production
of approximately 2000 tonnes of PCP or Na-PCP, respectively, per
annum are given in Table 6.
Table 6. Air emissions of phenolic and non-phenolic
compounds during production (maximum values)a
------------------------------------------------------
Annual air emissions (kg/year)
during production of:
2000 tonnes 2000 tonnes
PCP/year Na-PCP/year
------------------------------------------------------
PCP 18 65
Other chlorophenols 9 5
Hexachlorobenzene - 105
Other chlorobenzenes 1 700
OCDD 0.2 0.2
------------------------------------------------------
a Adapted from: BUA (1986).
While no waste water occurs during the production of PCP, the
annual loss of various compounds resulting from Na-PCP production
into the waste water was as follows: PCP, 60 kg; OCDD, 0.34 g;
H7CDDs, 0.1 g; H6CDDs, 0.001 g; OCDF, 0.1 g; H7CDFs, 0.026 g;
H6CDFs, 0.002 g (BUA, 1986).
3.2.1.3 Disposal of production wastes
The volume of contaminated waste water generated during the
production of Na-PCP is small, because manufacturers and regulatory
agencies have emphasized efficient process design (Jones, 1981).
During the production of approximately 2000 tonnes PCP/year,
about 8 tonnes of washing methanol, 4 tonnes of activated charcoal,
and 2 tonnes of other wastes occur. These wastes, as well as the
filtration sludge resulting from Na-PCP production, contain
considerable amounts of hazardous chemicals (Table 7). They are
generally disposed of by either storage in underground disposal
sites (filtration sludge) or incineration at temperatures above
1200 °C (BUA, 1986).
Table 7. Phenolic and non-phenolic compounds in the combined
wastes (PCP production) and filtration sludge (Na-PCP production)
---------------------------------------------------------------
Compound Combined wastes Filtration sludge
(kg/year) (kg/year)
---------------------------------------------------------------
PCP 1350 900
Other chlorophenols 0.7 nsb
Hexachlorobenzene ns 6000
Decachlorobiphenyl ns 3400
Decachlorophenoxybenzene ns 44
OCDD (OCDF) 0.98 0.67 (0.67)
H7CDDs (H7CDFs) 0.13 0.17 (0.045)
H6CDDs (H6CDFs) 0.013 0.092 (0.015)
P5CDDs (P5CDFs) 0.003 x 10-3 0.016 (0.005)
T4CDDs (T4CDFs) 0.002 x 10-3 0.007 (0.001)
2,3,7,8-T4CDD ns 0.001
---------------------------------------------------------------
a Adapted from: BUA (1986).
b ns = not specified.
US EPA (1985) proposed that wastes from the production and
manufacturing use of PCP should be classifed as acutely hazardous
wastes, on the basis of the presence of substantial concentrations
of the carcinogenic congener H6CDD and the chronic toxicity
potential of PCP itself.
3.2.1.4 Production levels
No precise estimates can be made of the total world production
of PCP and Na-PCP. According to the data profile of IRPTC (1983),
90 000 tonnes of PCP per year are produced globally. The Economist
Intelligence Unit (1981) estimated world production to be of the
order of 50 000 - 60 000 tonnes per year, based on the North
American and European Community output. However, the production
figures presented in Table 8 indicate a total production of only
30 000 tonnes per year. The production, foreign trade, and
consumption figures given in this summary table can give only a
rough idea of the true PCP market. Recent restrictions on the use
of PCP (section 3.3), a decline in the forestry industry, and the
increasing use of alternative means of wood preservation have
probably reduced the demand for PCP over the last few years.
The major PCP producers operating in 1980 are shown in Table 9
together with the plant locations and their capacities. Some
additional factories exist in which PCP is mixed or formulated to
yield special end-use products. There are also chemical producers
who sell pure, analytical grade PCP, but do not produce PCP for
technical purposes. The Monsanto Company, which had a capacity of
11.8 kilotonnes in the USA, stopped PCP production in their plant
at Sauget, Illinois, in 1978 (Jones, 1981). Dow Chemical closed
down its manufacturing plant at Midland, Michigan in October 1980
(Jones, 1984). Similarly, the only PCP producing plant in the
United Kingdom, also operated by the Monsanto Company, was closed
down in the same year (Economist Intelligence Unit, 1981), while
Reichhold Chemicals Inc., at Tacoma, Washington, USA ceased PCP
production in 1985. In the Federal Republic of Germany, the
production of PCP and Na-PCP was stopped in 1986.
3.3. Uses
The main advantages of PCP and its derivatives are that they
are effective biocides and soluble in oil (PCP) or water (Na-PCP).
Few pesticides show a similarly broad efficiency spectrum at low
cost. Therefore, PCP and its salts have a variety of applications
in industry, agriculture, and in domestic fields, where they have
been used as algicides, bactericides, fungicides, herbicides,
insecticides, and molluscicides.
3.3.1. Commercial use
In Table 10, the major registered commercial uses of PCP are
broken down for the United Kingdom and the USA. Although PCP and
its derivatives have many uses, by far the major application is
wood preservation. Cirelli (1978a), Hoos (1978), and Jones (1981)
have reported on commercial use patterns in North America. In the
USA, about 80% of PCP is used for commercial wood treatment, 6% is
in use for slime control in pulp and paper production, and 3%
accounts for non-industrial purposes, such as weed control, fence-
post treatment and paint preservation (Crosby et al., 1981);
however, the last two cases imply wood treatment as well. The
remaining 11% is converted to Na-PCP, which in turn is partly used
for wood preservation, mainly sapstain control in waterborne
conditions, e.g., for treating pressboard. Overall, some 95 - 98%
of American PCP production is used directly or indirectly in wood
treatment (Economist Intelligence Unit, 1981).
Table 8. Production, foreign trade, and consumption of PCP and Na-PCP (tonnes per year)
according to data available from government authorities and producers
--------------------------------------------------------------------------------------------------
Belgium/ Francea Germany, Italya Nether- United Canadac USAd
Luxemburga Federal landsa Kingdoma
Republic ofb (year (year (year
(year na)e 1979 1979 1984 na)e na)e na)e 1981 1977
--------------------------------------------------------------------------------------------------
Production
PCP 0 1700 2450 1550 0 0 0 1700 20 349
Na-PCP 0 2800 2100 1750 0 0 0 70
Imports
PCP 150- insigni- 0 0 250 - 30 - na 500 na
Na-PCP 160 ficant 300 0 280 40 na 0 na
Exports
PCP 0 300- 1950 1360 0 0 300 600 approxi-
Na-PCP 0 700 2150 1710 0 0 0 mately
200f
Consumption
PCP approxi- 1000 500 190 250- 30- 500 1536 na
Na-PCP mately 2500 250 40 280 40 32 na
150
--------------------------------------------------------------------------------------------------
a From: Economist Intelligence Unit (1981).
b From: BUA (1986).
c From: Jones (1984).
d From: Jones (1981).
e na = not available.
f Approximately 1% of domestic sales.
Table 9. Pentachlorophenol producers and their capacities in 1980
--------------------------------------------------------------------
Producer Country Plant Capacity
Location (tonnes)
(total PCP)
--------------------------------------------------------------------
Uniroyal Chemical,a Canada Clover Bar, 1800
Division of Uniroyal, Ltd Alta
Rhône-Poulencb France Pont-de-Claix 4500
Dynamit Nobelb Germany, Rheinfelden 4000
Federal
Republic of
Dow Chemical, USAa USA Midland, 13 500
Michigan
--------------------------------------------------------------------
Table 9 (contd.)
--------------------------------------------------------------------
Producer Country Plant Capacity
Location (tonnes)
(total PCP)
--------------------------------------------------------------------
Reichhold Chemicalsa USA Tacoma, 8100
Inc. Washington
Vulcan Materiala USA Wichita, 9000
Company Chemical Division Kansas
--------------------------------------------------------------------
a From: Jones (1981).
b From: Economist Intelligence Unit (1981).
Data from Canada and the Federal Republic of Germany confirm
the main use of PCP as a wood preservative. In Canada, about 95%
of the PCP is used for this purpose (Jones, 1981). Approximately
61% of the volume of PCP used in the Federal Republic of Germany in
1983 was used for wood preservation, while considerable amounts of
PCP were used by the textile (13%), leather (5%), mineral oil (6%),
and glue (6%) industries, respectively (Angerer, 1984). No PCP was
used in the paint or pulp industry whereas, in 1974, as much as 3%
or 7%, respectively, were used in these branches. PCP used on
textiles is usually in the form of the PCP ester rather than PCP or
Na-PCP.
Pentachlorophenyl laurate (L-PCP) was developed especially for
application on fabrics (Hueck & LaBrijn, 1960; Bevenue & Beckman,
1967). The estimates of L-PCP use in the United Kingdom in Table
10 are based on a publication from the year 1974 (HMSO, 1974).
According to an unpublished note submitted to the IPCS by Catomance
Limited, Hertfordshire, the sole manufacturer of pentachlorophenyl
laurate in the United Kingdom, the usage pattern in the United
Kingdom has not changed following the cessation of production of
PCP in 1978. However, most of the PCP ester used there today is
said to be for domestic timber preservation; the use of L-PCP for
textile preservation is supposed to be mainly confined to tropical
or semi-tropical countries.
In the USSR, PCP is used for the preservation of commercial
timber, paints, varnishes, paper, textiles, ropes, and leather
(IRPTC, 1984).
Na-PCP is also used to inhibit algal and fungal growth in
cooling tower waters at electric generating plants (Hoos, 1978); in
1976, about 30% of the Na-PCP used in Canada was for this purpose
(Jones, 1981).
Table 10. Major commercial (non-agricultural) uses of PCP in the
United Kingdom and the USAa
---------------------------------------------------------------------------
Use Active
ingredient
---------------------------------------------------------------------------
United Kingdom
Anti-mildew agent in the wool textile industry L-PCP, Na-PCP
Mothproofing carried out by dyers and cleaners L-PCP
Wood preservation PCP, L-PCP,
Na-PCP
Paint additives PCP
Antimicrobial (slimicide) agents in paper and board PCP
Antifungal agent in textiles other than wool (cotton,
Flax and jute fabric, ropes, cordage and tentage) L-PCP
Cable impregnation L-PCP
Anti-mildew agent in leather nsb
Fungicide in adhesives Na-PCP
Bactericide in drilling fluids nsb
USA
Microbiostat for commercial and industrial water cooling Na-PCP
Microbiocide for leather K-PCP, PCP
Microbiocide for burlap, canvas, cotton, rope, and twine PCP
Microbiocide and insecticide for wood treatment PCP, Na-PCP
Preservative for oil and water-based paint PCP
Slime control for pulp and paper PCP
Microbiocide for petroleum drilling mud and flood water PCP
Fumigant for shipping-van interiors PCP
Preservative for hardboard and particle-board PCP
---------------------------------------------------------------------------
a From: Crosby et al. (1981).
b ns = not specified.
Alterations in the use pattern have taken place during the last
few years as a result of the increased concern about the potential
health hazards from PCP and its impurities. In Japan, the
production of PCP was 14.5 kilotonnes in 1966 and 3.3 kilotonnes in
1971, after which production ceased entirely (IARC, 1979a). In the
Federal Republic of Germany, 3300 tonnes of total PCP were produced
in the year 1984, of which 93% was exported, leaving 230 tonnes for
use in the country (BUA, 1986). Nine years earlier (1974), 4100
tonnes of PCP were produced, 60% of which were exported; in 1979,
84% of the 4503 tonnes produced were sold abroad (Angerer, 1984).
These figures indicate a drastic decrease in the consumption of PCP
in the Federal Republic of Germany during the last few years. In
Canada, the Federal Republic of Germany, and Sweden, where PCP had
been heavily used as a slime control agent in the paper mills, the
use of chlorinated phenols for this purpose was prohibited in
recent years as a consequence of the discharges, which had toxic
effects on the aquatic environment. In Sweden, all use of PCP was
banned in 1977 (Ahlborg & Thunberg, 1980; Jones, 1981). The US
Environmental Protection Agency does not intend to prohibit the use
of PCP in oil-well water or in pulp and paper mills, provided that
impermeable gloves are worn during application and that the H6CDD
content will be reduced to 1 ppm (US EPA, 1984b). Similarly, the
use of PCP (including its salts) for wood protection has not been
cancelled in the USA. However, the US EPA (1984a) intends to
establish certain changes in the terms and conditions of
registration.
3.3.2. Agricultural use
Significant quantities of PCP were previously used in a number
of agricultural applications. These resembled industrial uses in
that most were to prevent wood decay, in farm buildings, fences,
and storage facilities (Jones, 1981). However, PCP or its sodium
salt have also been used as a herbicide and dessicant for forage
seed crops, a herbicide for non-food vegetation control, a biocide
in the post-harvest washing of fruit, and as an insecticide for use
in beehives, seed plots, and greenhouses (Crosby et al., 1981).
PCP was formerly used as a herbicide in paddy and upland rice
fields, particularly in Japan (Kobayashi, 1978; Crosby et al.,
1981). In addition, PCP and Na-PCP have been approved for a number
of applications in the food industry, such as biocides in packaging
materials and glues (Table 11).
In the USSR, PCP is applied as a nonselective herbicide and as
a desiccant on cotton plants. At least 10 days should lapse
following cotton plant treatment (IRPTC, 1984).
Table 11. Other uses of PCP and its salts as a potential
source of food contaminationa
-------------------------------------------------------------
Use Specific compound
-------------------------------------------------------------
Slime control on paper and paperboard K-PCP, Na-PCP
Preservative in can-end cement Na-PCP
Defoaming agents Na-PCP
Paper contacting aqueous and fatty food Na-PCP
Animal glue for containers Na-PCP
Sealing gaskets for containers K-PCP, Na-PCP
Preservative for wood products PCP, Na-PCP
Preservative in coatings Na-PCP
Rubber antioxidant Na-PCP
Preservative for ammonium alginate Na-PCP
-------------------------------------------------------------
a Adapted from: Firestone (1973).
Recently, regulations to limit or even ban some uses of PCP
have been established in a number of countries. The Canadian
government suspended agricultural applications of PCP and Na-PCP in
mushroom culture, above-ground interior woodwork of farm buildings,
and as herbicides and soil sterilants (Jones, 1984). Japan
restricted herbicidal use of PCP because of its high toxicity to
fish (Kobayashi, 1979; Crosby et al., 1981). The paper industry of
the Federal Republic of Germany and Sweden no longer use PCP in
packaging paper (Ahlborg & Thunberg, 1980; Angerer, 1984).
US EPA (1984b) proposed cancelling the registration of
pesticide products containing PCP as the active ingredient for non-
wood preservative uses, i.e., herbicidal and antimicrobial uses.
In addition, PCP-containing wood preservatives for home and farm
use must not be applied where there may be direct contact with
domestic animals or livestock or close contact with food or feed
(US EPA, 1984a).
3.3.3. Domestic use
The largely uncontrolled use of PCP by private individuals is
almost exclusively related to the treatment of wood, both outdoors
and indoors. PCP is the main active ingredient in certain wood
preservatives for home use, and is added to products such as stains
and paints. Although this category of products plays only a minor
role in the overall PCP market, it has been of particular concern,
since cases of apparent PCP intoxication after indoor application
in private homes have been reported (section 5). As a consequence
of such incidents, the use of PCP for the preservation of interior
timber has been banned in Canada (Jones, 1981) and the Netherlands
(Economist Intelligence Unit, 1981). Since 1986, the use of PCP as
a biocide for indoor application has been forbidden in the Federal
Republic of Germany by government regulatory action (FRG, 1986).
Furthermore, there is a gentlemen's agreement between the
government and industry to suspend the use of PCP in wood
preservatives in general (BUA, 1986). The US EPA (1984a) intends
to limit the use of PCP-containing wood preservatives in interiors
to certain support structures. This is also true for the indoor
use of PCP-treated wood. The sale and use of PCP is restricted to
certified applicators. Thus, the domestic use of PCP is not as
significant as it was some years ago.
Other reported applications of PCP include health-care products
and disinfectants for use in the home, farms, and hospitals. PCP
may also be contained in dental-care products (Jones, 1981),
bactericidal soaps, laundry products, and medical products for the
skin (Crosby et al., 1981).
3.3.4. Use for control of vectors
The application of Na-PCP to control vectors of pathogens has
been of some relevance in tropical and subtropical areas. Na-PCP
has been used as a herbicide to control Salvinia sp., a host plant
of Mansonia mosquitos, which transmit the elephantiasis-causing
filarias to man (Chow et al., 1955).
Na-PCP has also been used for control of the intermediate snail
hosts of schistosomiasis (Berry et al., 1950; Toledo et al., 1976).
After World War II, Hunter et al. (1952) proposed Na-PCP as the
molluscicide of choice for use in Japan. In China, Na-PCP is still
in use for this purpose today (Xue, 1986)a.
___________________________________________________________________
a Personal communication to the Task Group on Pentachlorophenol.
3.3.5. Formulations
In the treatment of wood, PCP is usually administered as a 5%
solution in a mineral spirit solvent, such as No. 2 fuel oil or
kerosene (Cirelli, 1978a), or methylene chloride, isopropyl
alcohol, or methanol (Ingram et al., 1981a). Since PCP is not very
soluble in hydrocarbon solvents, and tends to migrate to, and
crystallize on, treated wood surfaces (a phenomenon known as
"blooming"), formulations may also contain co-solvents and anti-
blooming agents (David, 1985a). Most commercial formulations also
contain other chlorophenols, mainly tetrachlorophenol (Nilsson et
al., 1978). The aim of such mixtures is to prevent blooming on
treated wood by lowering the melting point of the chlorophenols.
An aqueous solution of Na-PCP is used for commercial sapstain
control (Konasevich et al., 1983).
Chlorophenols may be combined with other active components such
as methylene bisthiocyanate and copper naphthenate in the
formulation of PCP pesticides (von Rümker et al., 1974).
Conversely, PCP is added to biocides, the primary active ingredient
of which is another compound; for example, sodium fluoride
formulations for wooden poles and posts may contain up to 10%
technical PCP (US EPA, 1973).
---------------------------------------------------------------------------
a Personal communication, Catomance Ltd, Welwyn Garden City,
Hertfordshire, United Kingdom.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and Distribution Between Media
Several physical and chemical parameters affect the transport
and distribution of PCP in soil, water, and air. Volatilization
and adsorption are the major mechanisms; leaching and movements on
surfaces and in air are of minor importance (Jones, 1981).
4.1.1. Volatilization
Volatilization can be an important source of loss of PCP from
water and soil surfaces as well as from PCP-treated materials.
Dip- or brush-treating coniferous wood may lead to a 30 - 80% loss
of PCP, due to evaporation, within 12 months (Morgan & Purslow,
1973; Petrowitz, 1981).
Several factors influence the rate of volatilization of PCP
from wood. Ingram et al. (1981b) showed that certain solvents or
mixtures of solvents may either reduce or increase the
volatilization of PCP. Such solvents, as well as resins, are used
to achieve a timely release of PCP through solubilization and/or
occlusion, e.g., PCP evaporation was decreased by about 20%, when
15% of an alkyd resin was added to a wood preservative containing
5% PCP (Petrowitz, 1981). Temperature appeared to be the external
variable that had the greatest effect on volatilization; a rise in
temperature from 20 to 30 °C caused a 3- to 4-fold increase in
volatilization. Relative humidity and rate of air flow had only a
minor influence on PCP volatilization from wood. The loss of PCP
from pine-wood samples was half as much as that from spruce wood
samples after 21 days, apparently because pine is much more easily
impregnated than spruce. Thus, the depth of penetration of PCP in
wood, the species of wood, and the process of treatment appear to
be other external variables influencing diffusion and
volatilization. As an example, immersion of small blocks of pine
sapwood with a 5% PCP solution leads to a PCP loss of 20% after 6
months compared with only 4%, 9 months following double vacuum
treatment (Morgan & Purslow, 1973).
Evaporation is used for the disposal of PCP in waste water at
some wood-preserving plants (section 4.4). Extensive studies on
the volatilization of PCP from water or soil have not been carried
out, but Klöpffer et al. (1982) studied this process in an aqueous
solution in the absence of other pathways of removal. Temperature
and pH of the solution were the most important factors influencing
the evaporation rate. Since only the un-ionized form of PCP seems
to be volatile, at pH 5.1, when 13.2% of the PCP was present as the
free acid, the 50% residence time was 328 h at 30 °C, whereas, at
pH 6, with more than 98% PCP dissociated to its phenate, a one-half
residence time of 3120 h was measured. These findings suggest that
evaporation of PCP from surface waters with a pH above 6 should be
quite low.
4.1.2. Adsorption
The extent of adsorption of a pesticide governs its
biovailability in soil; hence, both the rate of degradation and the
biocidal activity are likely to be reduced by strong adsorption.
In addition, though possessing a reduced activity, a highly
adsorbed compound would exert a prolonged effect (Su & Lin, 1971;
Choi & Aomine, 1972).
pH seems to be the major factor controlling the magnitude of
PCP adsorption. Choi & Aomine (1974a) investigated PCP retention
in a range of soil types, and determined that adsorption was
maximal in strongly acidic soils, relatively minor in moderately
acidic soils, and absent in weakly acidic or neutral soils. Other
research workers have observed the same relationship between soil
pH and PCP retention (Green & Young, 1970; Kaufmann, 1976).
The organic matter content and surface area of soils exert a
minor, but significant, effect on PCP adsorption. Choi & Aomine
(1974a) found that the magnitude of adsorption decreased in the
following order: humusallophanic, allophanic, montmerillonitic, and
halloysitic soils. This finding confirms the binding of PCP by
organic matter reported by other authors (Su & Lin, 1971; Choi &
Aomine, 1972).
Under the weakly acidic to neutral conditions characterizing
most soils, adsorption is likely to exert a minor effect on PCP
dynamics. In this regard, it is noteworthy that Choi & Aomine
(1974b) studied adsorption using hexane as a solvent, because the
amount of the pesticide sorbed onto soils from aqueous solution was
too small to determine.
4.1.3. Leaching
Leaching of a chemical through the soil is interrelated with
factors such as adsorption, water solubility of the substance, soil
type, moisture, percolation velocity, and pH (Haque & Freed, 1974).
Thus, the leaching behaviour of PCP will vary, depending on the
soil under examination.
Leaching is an important means of transport for PCP, in some
instances. Kuwatsuka (1972) noted that much of the PCP applied to
flooded rice paddies was carried through the soil in solution, and
the Weed Science Society of America Herbicide Handbook (WSSA, 1974)
reported that Na-PCP also leaches readily in soil. This is
consistent with the observation that leaching of PCP occurs more
easily in alkaline soils than in both acidic clay and sandy soils
(Kaufman, 1976).
In addition, substantial quantities of PCP are found in waters
leaching from contaminated sites. For example, 2.05 and 3.35 mg
PCP/litre were detected in groundwater from a wood preservation
plant near Lake Superior (Thompson et al., 1978), and PCP in the
µg/litre range was detected in water seeping from a landfill
(Kotzias et al., 1975).
Some in vitro studies have revealed little or no PCP in soil
leachate (Arsenault, 1976; Weiss et al., 1982b), but these are
difficult to interpret, as residence times are either extremely
long, or not reported. In other percolation tests, PCP did not
leach in the profile of a brown earth-Lessivé within one month, but
was detected in the water seeping from a podzol (1.5 mg PCP/litre)
after two days (Fränzle, 1982).
From the sorption and leaching behaviour of PCP, it can be
concluded that organic matter serves as a reversible storage
compartment, allowing desorption of PCP at elevated soil-water
content and, hence, accumulation of PCP in the soil solution and
eventually, in the groundwater (BUA, 1986).
Stranks (1976) stated that PCP does not leach readily from
treated wood, particularly if applied via an oil carrier.
4.2. Biotransformation
4.2.1. Abiotic degradation
Both PCP and Na-PCP are subject to abiotic (photochemical)
degradation in water, organic solvents, and on solid surfaces. In
the photolysis pathway (Fig. 2) suggested by Wong & Crosby (1978),
three types of degradation products occur: (a) lower chlorinated
phenols, mainly 2,3,4,6- and 2,3,5,6-tetrachlorophenol together
with trichlorophenols; (b) chlorinated dihydroxybenzenes, primarly
tetrachlororesorcinal and tetrachlorocatechol; and (c) non-aromatic
fragments, mostly dichloromaleic acid. Irradiation of the last
compound, in turn, yielded carbon dioxide (CO2) and chloride ions.
Since the toxicity of PCDDs and PCDFs depends not only on the
number but also on the position of chlorine substituents, a precise
characterisation of PCP impurities is essential. The presence of
highly toxic 2,3,7,8-tetrachlorodibenzo- p-dioxin (2,3,7,8-T4CDD)
has only been confirmed once in commercial PCP samples. In the
course of a collaborative survey, one out of five laboratories
detected 2,3,7,8-T4CDD in technical PCP and Na-PCP samples at
concentrations of 250 - 260 and 890 - 1100 ng/kg, respectively
(Umweltbundesamt, 1985). Buser & Bosshardt (1976) found detectable
amounts of T4CDD (0.05 - 0.23 mg/kg) in some samples of different
technical PCP products, but on re-analysis were unable to confirm
the compound's identity. In other cases, T4CDD has not been
identified at detection limits of 0.2 - 0.001 mg/kg (Table 1).
The higher polychlorinated dibenzodioxins and dibenzofurans
are more characteristic of PCP formulations (Table 1).
Hexachlorodibenzo- p-dioxin (H6CDD), which is also considered
highly toxic and carcinogenic (section 8), was found at levels of
0.03 - 35 mg/kg (Firestone et al., 1972), 9 - 27 mg/kg (Johnson et
al., 1973), and < 0.03 - 10 mg/kg (Buser & Bosshardt, 1976).
According to Fielder et al. (1982), the 1,2,3,6,7,9-, 1,2,3,6,8,9-,
1,2,3,6,7,8-, and 1,2,3,7,8,9-isomers of H6CDD have been detected
in technical PCP. The 1,2,3,6,7,8and 1,2,3,7,8,9-H6CDDs
predominated in commercial samples of technical PCP (Dowicide 7)
and Na-PCP. Octachloro-dibenzo- p-dioxin (OCDD) is present in
relatively high amounts in unpurified technical PCP (Table 1).
Recently, the identification of 2-bromo-3,4,5,6-
tetrachlorophenol as a major contaminant in three commercial PCP
samples (ca. 0.1%) has been reported. This manufacturing by-
product has probably not been detected in other analyses because it
is not resolved from the PCP peak by traditional chromatographic
methods (Timmons et al., 1984).
2.2.1. Formation of PCDDs and PCDFs during thermal decomposition
The thermal decomposition of PCP or Na-PCP yields significant
amounts of PCDDs and PCDFs, depending on the thermolytic
conditions. For pure PCP, dimerization of PCP has been suggested
as an underlying reaction process; in technical PCP, additional
reactions, i.e., dechlorination of higher chlorinated PCDDs and
cyclization of predioxins are involved in forming various and
different PCDD isomers (Rappe et al., 1978b).
Pyrolysis of alkali metal salts of PCP at temperatures above
300 °C results in the condensation of two molecules to produce
OCDD. PCP itself forms traces of OCDD only on prolonged heating in
bulk and at temperatures above 200 °C (Sandermann et al., 1957;
Langer et al., 1973; Stehl et al., 1973).
Although present in original technical PCP products, a number
of PCDDs, other than OCDD, are generated during thermal
decomposition (290 - 310 °C) in the absence of oxygen (Table 2)
(Buser, 1982).
Table 2. PCDDs (mg/kg PCP) in the
pyrolysate of technical PCP and Na-PCPa
--------------------------------------
PCP Na-PCP
--------------------------------------
2,3,7,8-T4CDD -b -c
1,2,3,7,8,9-H6CDD 53 2.1
1,2,3,6,7,8-H6CDD 66 0.95
Total H6CDD 455 10.5
H7CDD 5200 65
OCDD 15 000 200
--------------------------------------
a From: Buser (1982).
b Detection limit (1 mg/kg).
c Detection limit (0.25 mg/kg).
2.3. Physical, Chemical, and Organoleptic Properties
Pure pentachlorophenol consists of light tan to white,
needlelike crystals. It has a pungent odour when heated (Windholz,
1976). Its vapour pressure suggests that it is relatively
volatile, even at ambient temperatures. Since PCP is practically
insoluble in water at the slightly acidic pH generated by its
dissociation, readily water-soluble salts such as Na-PCP are used
as substitutes, where appropriate.
Na-PCP is non-volatile; its sharp PCP odour results from slight
hydrolysis (Crosby et al., 1981). Technical PCP consists of
brownish flakes or brownish oiled, dustless flakes, coated with a
mixture of benzoin polyisopropyl and pine oil. Technical Na-PCP
consists of cream-coloured beads (Anon., 1983a,b). Technically
pure L-PCP consists of a brown oil that is insoluble in water and
alcohols, and soluble in non-polar solvents, oils, fats, waxes, and
plasticizers (Cirelli, 1978b).
PCP is non-inflammable and non-corrosive in its unmixed state,
whereas a solution in oil causes deterioration of rubber (Mercier,
1981).
Because of the electron withdrawal by the ring chlorines, PCP
behaves as an acid, yielding water-soluble salts such as sodium
pentachlorophenate. Due to nucleophilic reactions of the hydroxyl
group, PCP can form esters with organic and inorganic acids and
ethers with alkylating agents, such as methyl iodide and
diazomethane (Crosby et al., 1981). This property has been used
for analytical purposes (section 2.5.2).
PCP may exist in two forms: the anionic phenolate, at neutral
to alkaline pH, and the undissociated phenol at acidic pH. At pH
2.7, PCP is only 1% ionized; at pH 6.7, it is 99% ionized (Crosby
et al., 1981). Other relevant properties of pure PCP and Na-PCP
are shown in Table 3.
PCDDs and PCDFs may also be formed during the combustion of
materials treated with either purified or technical PCP. Smoke from
birch leaves impregnated with purified Na-PCP and burnt on an open
fire showed considerably increased amounts of PCDDs compared with
the original sample (Table 4). The mass fragmentograms revealed 14
of the 22 possible T4CDD isomers with 1,3,6,8- and 1,3,7,9-T4CDD as
the main and 2,3,7,8- T4CDD as minor isomers. The formation of
PCDFs, including small amounts of 2,3,7,8-T4CDF, during either
combustion or micropyrolysis (280 °C, 30 min) was only observed in
technical PCP samples; purified Na-PCP was negative in this respect
(Rappe et al., 1978b).
Table 3. Physical, chemical, and organoleptic properties of PCP
and Na-PCP
------------------------------------------------------------------
PCP Na-PCP
------------------------------------------------------------------
Boiling pointa 310 °C (decomposition)
Relative molecular massb 266.4 288.3
306.3
(monohydrate)
Melting pointa 191 °C
Density (d422 in g/ml)b 1.987 2
Vapour pressure kPa (mmHg)
at 20 °Cc 2 x 10-6 (1.5 x 10-5)
at 19 °Cd 6.7 x 10-7 (5 x 10-6)
Saturation vapour density 220
(µg/m3) (20 °C)c
Steam volatilitye 0.167
(g/100 g water vapour)
(100 °C)
------------------------------------------------------------------
Table 3 (contd.)
------------------------------------------------------------------
PCP Na-PCP
------------------------------------------------------------------
Solubility in fat 213
(g/kg) (37 °C)f
n-Octanol/water partition 4.84 (pH 1.2);
coefficient (log P)g 3.56 (pH 6.5);
3.32 (pH 7.2);
3.86 (pH 13.5)
pKa (25 °C)e 4.7
Solubility in water:
(g/litre)e,h,i
0 °C, pH 5 0.005
20 °C, pH 5 0.014
30 °C, pH 5 0.020
20 °C, pH 7 2
20 °C, pH 8 8
20 °C, pH 10 15 > 200
25 °C 330
Solubility in organic
solvents (g/100 g)
(25 °C)b:
acetone 50 35
benzene 15 insoluble
ethanol (95%) 120 65
ethylene glycol 11 40
isopropanol 85 25
methanol 180 25
Odour threshold 1.6 (in water)
(mg/litre)j:
Olfactory threshold 0.03 (in water)
(mg/litre)j:
------------------------------------------------------------------
a From: IRPTC (1983).
b From: Cirelli (1978b).
c From: Zimmerli (1982).
d From: Dobbs & Grant (1980).
e From: Crosby et al. (1981).
f From: Rippen (1984).
g From: Kaiser & Valdmanis (1982).
h From: Gunther et al. (1968).
i From: Bundesamt für Umweltschutz (1982).
j From: Dietz & Traud (1978a).
Jansson et al. (1978) observed a very wide range of PCDD
concentrations in the smoke from burning wood chips impregnated
with a technical PCP formulation (Table 4). The formation of PCDDs
was favoured by temperatures below 500 °C, oxygen deficit, and
lower gas-retention time. The results given in Table 4 are
corrected for the very low background values obtained by burning
untreated wood chips.
When technical PCP was burnt in a quartz reactor (600 °C, 10
min), Lahaniatis et al. (1985) identified the following thermolytic
products: pentachlorobenzene, hexachlorobenzene, octachlorostyrole,
octachloronaphthaline, decachlorobiphenyl, H6CDF, OCDF, and OCDD.
2,3,7,8-T4CDD was not detected at a detection limit of 1 mg/kg PCP.
Olie et al. (1983) found only slightly higher levels of PCDDs
and PCDFs in the fly ash of burned new wood treated with PCP
compared with painted wood, which was more than 60 years old.
However, because data were missing on PCDD/PCDF levels in the
original samples and on the conditions of burning, meaningful
interpretation of these results is not possible.
2.4. Conversion Factors
1 ppm = 10.9 mg PCP/m3 (25 °C, 101.3 kPa)
1 mg PCP/m3 = 0.09 ppm
Table 4. Amount of PCDDs in the original sample and in
the smoke from combusted materials treated with purified
Na-PCP or technical PCP
----------------------------------------------------------
Birch leavesa Wood chipsb
(mg PCDDs/kg Na-PCP (mg PCDDs/kg PCP
(purified)) (technical))
Original Smokec Original Smokec,d
sample sample
----------------------------------------------------------
T4CDD < 0.02 5.2 nde < 4.7 - 47
P5CDD < 0.03 14 nd < 1.2 - 419
H6CDD < 0.03 56 7 < 9.3 - 93
H7CDD 0.3 172 93 < 4.7 - 279
OCDD 0.9 710 186 < 0.9 - 442
----------------------------------------------------------
a Adapted from: Rappe et al. (1978b).
b Adapted from: Jansson et al. (1978).
c Smoke trapped on charcoal filter.
d Depending on combustion conditions.
e nd = not determined.
2.5. Analytical Methods
A number of methods have been used to determine PCP in a
variety of media. The earlier procedures were reviewed by Bevenue
& Beckman (1967). They were mostly based on colour reactions,
which are not very specific and relatively insensitive. For
several years, more sophisticated devices have been available to
analysts, of which gas chromatography has become the method of
choice (Table 5).
Table 5. Analytical methods for the determination of PCP
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Air
Air- Impinger collection Derivatization with diazo- ECa 0.22 mg/m3 nsc Hoben et al.
aerosol with KOH; hexane methane; purification in (1976a)
extraction chromatoflex Florisil
column; GCd analysis
Air Filter and bubbler HPLCe analysis; column: UV254b 0.27 mg/m3 95.3- NIOSH
collection; ethylene µ Bondapack C18; mobile 100.9% (1978)
glycol extraction phase: methanol/water
Air Bubbler collection; Derivatization with acetyl EC 0.05 µg/m3 ns Dahms &
absorption in K2CO3 chloride; GC analysis Metzner
solution; hexane (1979)
extraction
Air Adsorption on to GC analysis EC 0.5 µg/m3 ns Zimmerli &
filter papers Zimmermann
impregnated with (1979)
adsorbant extraction
concentration in ether
Air Impinger collection; HPLC analysis; column: UV225 0.5 µg/m3 ns Woiwode et
absorption in K2CO3- Lichrosorb C18; mobile al. (1980)
solution; benzene phase: methanol
extraction
Air Air from wood samples GC analysis EC ns 89.7- Warren et
in reactor tube ads- (< 1.5 99.9% al. (1982)
orbed on silica gel; µg/m3)
desorption with benzene
Air Impinger collection; Derivatization with acetic EC ns ns Kauppinen &
absorption in anhydride in presence of Lindroos
toluene pyridine; GC analysis (1985)
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Biological tissues and fluids
Blood Benzene extraction Derivatization with diazo- EC 20 µg/litre 87- Bevenue et
(human) from acidified methane; GC analysis 100% al. (1968)
solution
Blood, Ethyl ether extrac- No derivatization; GC anal- EC-3H 0.1 ng 90- Barthel et
urine, tion from acidified ysis; column: 3% diethylene EC-63Ni 0.02 ng 100% al. (1969)
tissue solution; NaOH ex- glycol succinate + 2% H3PO4;
(human) traction; benzene confirmation by MS and TLC
extraction
Organic Hexane/isopropanol Der. with acetic anhydride EC ns 81-91% Rudling
tissues extraction from in presence of pyridine; (1970)
acidified sample; GC analysis
borax extraction
Urine Ethyl ether extrac- Derivatization with diazo- EC 10 µg/litre 92- Shafik et
(rat) tion from acidified ethane; GC analysis 98% al. (1973)
solution
Adipose NaOH extraction; Derivatization with diazo- EC 5 µg/kg 75% Shafik
tissue diethyl ether methane; GC analysis (1973)
(human) extraction from
acidified solution
Urine, Acidic hydrolysis No derivatization; negative MSf 1 ng 90% Dougherty &
seminal (urine); hexane/2- chemical ionization (NCI) Piotrowska
fluid propanol or hexane/ mass spectrometry; internal (1976a;b)
(human) ether extraction from standard: p-chlorobenzophenone
acidified solution
Tissue, Hexane or benzene Derivatization with diazo- EC 20 µg/kg 91.4- Hoben et al.
plasma, extraction ethane (urine) or diazo- 95.3% (1976a)
urine methane; purification in
(rat) chromatoflex Florisil column;
GC analysis
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Tissues Acetone extraction (a) TLCg analysis on silica Radio- ns ns Glickman et
(fish) gel UV254; mobile phase: active al. (1977)
methylene chloride
Acetone extraction (b) Derivatization with MS ns ns Glickman et
methyl iodide and K2CO3; al. (1977)
GC analysis
Urine Hexane extraction Derivatization with acetyl EC 10 µg/litre ns Dahms &
(human) from acidified chloride in the presence of Metzner
solution pyridine; GC analysis (1979)
Urine Benzene extraction Derivatization with diazo- EC 1 µg/litre 93.2- Edgerton
(human, from acidified methane; separation of 97.2% et al.
rat) solution methylated phenols in acid (1979)
alumina column; GC analysis;
GC-MS confirmation
Plasma Benzene extraction Derivatization with acetic EC 50 µg/litre 91- Eben et
(human) anhydride; GC analysis 102% al. (1981)
Urine Acidic or enzymatic Derivatization with acetic EC 20 µg/litre 77- Eben et
(human) hydrolysis; ethyl anhydride; GC analysis 98% al. (1981)
ether extraction;
benzene extraction
Tissues, Diethyl ether (ethyl Purification on Hypersil- UV216 1 µg/kg 62- Mundy &
serum, acetate-hexane) cartridge; mobile phase: 108% Machin
egg extraction from NaOH methanol; HPLC analysis (1981)
yolk/ (hydrochloric acid)
white solution; concentra-
tion by evaporation
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Urine Acidic hydrolysis; HPLC analysis; column: Sphe- UV313 10 µg/litre 102% Drummond
(human) distillation; methy- risorb-ODS; mobile phase: et al.
lene chloride ex- acetonitrile; internal stan- (1982)
traction from acidi- dard: 3,5-dichloro-2,3,6-
fied solution; tribromophenol (DTP)
evaporation and
redistillation in
acetonitrile
Urine Acidic hydrolysis; LC analysis; column: Spheri- UV254 0.1 83.3± Pekari &
(human) hexane/isopropanol sorb-ODS; mobile phase: µmol/litre 3.7% Antero
extraction; evap- methanol (27 µg/litre) (1982)
oration and redis-
tillation in methanol
Urine Acidic hydrolysis; Derivatization with acetic SIM-MSh 1 nmol/litre ns Harge-
(human) int.standard (4,6- or propionic anhydride; (0.27 sheimer &
dibromo- o-cresol) GC analysis µg/litre) Coutts
added; methylene (1983)
chloride extraction
Food
Milk Benzene extraction; Derivatization with acetic EC 5 µg/litre 80- Erney
(bovine) extraction with anhydride; GC analysis 87.2% (1978)
K2CO3 solution
Milk Sulfuric acid Purification in BioSil EC 10 - 15 80% Lamparski
(bovine) digestion; hexane A-column; derivatization µg/litre et al.
extraction with diazomethane; GC (1978)
analysis
Carrots, Soxhlet extraction Derivatization with diazo- EC 0.2 µg/kg 80- Bruns &
potatoes (carrots) or blending ethane; purification in 108% Currie
with acidified Florisil-column; GC (1980)
acetone analysis
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Canned Methylene chloride Derivatization with diazo- EC ns 92- Heikes &
food and extraction from methane; purification in (< 0.3 103% Griffitt
jar lids acidified solution; Florisil-column; GC analysis µg/kg) (1980)
clean-up by gel perme-
ation chromatography
Edible Alkaline hydrolysis; Direct GC analysis on DEGS/ EC 5 - 10 µg/kg 83- Stijve
gelatins iso-octane extraction phosphoric acid column; con- 108% (1981)
from acidified firmation by GC analysis of
solution acetate derivative
Plant Maceration with acidi- Derivatization with diazo- EC ns 94.2% Fuchs-
mater- fied acetone; methane; purification in bichler
ials chloroform extr.; Florisil-column; GC analysis (1982)
cleanup by automated
gel chromatography
Mush- Steam distillation (a) Derivatization with ace- EC 0.5 µg/kg 92% Schönhaber
rooms from acidified sample; tic anhydride GC analysis fresh weight et al.
dichloromethane (b) HPLC analysis; column: UV220 0.5 µg/kg (1982)
extraction LiChrosorb RP-8; mobile
phase: methanol/ o-phos-
phoric acid
Soil
Soil NaOH extraction; Derivatization with diazo- EC 0.1 - 1 > 97% Renberg
clean-up by ion methane; GC analysis µg/kg (1974)
exchanger
Soil Nielsen-Kryger steam Direct GC analysis on fused EC ns 58- Narang et
distillation from silica SE 54 column 95% al. (1983)
acidified soil;
toluene-methylene
chloride extraction
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Water
Raw and Petroleum ether TLC analysis on Al2O3 Colori- 0.5 µg/litre 75- Zigler &
treated extraction from plates; mobile phase: metric 100% Phillips
water acidified sample; (a) benzene;(b) NaOH/acetone; (1967)
evaporation; chromogenic agent: AgNO3/2-
drying; evaporation phenoxyethanol
Natural, Benzene extraction Derivatization with acetic EC 0.01 84- Chau &
waste and K2CO3-solution anhydride; hexane extrac- µg/litre 93% Coburn
water tion; GC analysis (1974)
River Hexane/ethyl ether Derivatization with boron MS 0.01 ns Matsumoto
water extraction; ethyl trifluoride methanol and µg/litre et al.
acetate extraction trimethylsilyl; GC analysis (1977)
from acidified
solution
Surface Toluene extraction; Derivatization with acetic EC 0.01 85% Wegman &
water extraction with anhydride; petroleum ether µg/litre Hofstee
K2CO3-solution extraction; GC analysis (1979)
Waste Chloroform extraction Direct HPLC analysis of UV254 10 µg/litre ns Ervin &
water from acidified sample; chloroform extract; column: McGinnis
rotary evaporation silica gel; mobile phase: (1980)
cyclohexane-acetic acid and
other solvents
Surface Diethyl ether Direct HPLC analysis UV215 20 µg/litre > 80% Ivanov &
water extraction; evapor- Magee
ation; dissolution in (1980)
methanol-petroleum
Surface Chloroform extraction Direct analysis in double Absorp- 5 - 10 92- Carr et
water from acidified beam UV spectrophotometer tion µg/litre 102% al. (1982)
sample;NaOH extraction (320 nm)
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Surface Concentration by GC analysis (no deriviti- EC 0.01 95 ± Rübelt et
water extraction, ads- zation); column: Carbowax µg/litre 3% al. (1982)
orption, rotary 20 M plus phosphoric acid;
evaporation confirmation by MS
Surface, Addition of Na2HPO4 Extraction and derivitzation EC 1 ng/litre 98- Abrahams-
waste, buffer solution by adding hexane containing 100% son & Xie
drinking-(for acid waste water internal standard (2,6-dibro- (1983)
water pH adjustment to 7 mophenol) and acetic anhy-
with NaOH dride directly to sample;
GC analysis
Water Samples prepared from HPLC analysis with isocratic UV280 21 ng ns Buckman et
stock solutions in elution of various substitu- al. (1984)
acetonitrile ted phenols
Wood
Wood Chloroform extraction TLC analysis on silica gel UV 0.06 µg ns Henshaw et
from wood shavings plates; developing solvent: al. (1975)
cyclohexane-acetone-paraffin
Wood- Soxhlet extraction Derivatization with diazo- EC ns 30-70% Levin &
dust with ether ether; methane; GC analysis Nilsson
evaporation; dissol- (1977)
ution in acetone;
TLC separation;
hexane elution
Lumber Pulverization of wood HPLC analysis; column: Sphe- UV230 0.1 µg/cm2 ns Daniels &
(sur- sample; extraction risorb-ODS; mobile phase: Swan
face with acetonitrile water-acetonitrile-acetic (1979)
treated) containing internal acid
standard; ultrasonic
bath
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Sawdust, Extr. with acetic TLC analysis; different sol- Colour 2 mg/kg ns Ting &
wood- acid-methanol; evap- vent systems; sprayed with reaction Quick
shavings oration; conversion tetrabase reagent (1980)
to chloranil in warm
nitric acid
Various materials
Toy Soxhlet extraction Direct GC analysis; confirm- FIDi 1 - 4 mg 70- von
paints with acetone; concen- ation by TLC analysis /litre 100% Langeveld
tration by evaporation (1975)
PCP Dioxane extraction HPLC analysis; column: Bon- UV254 ns 97% Hayes
formula- dapakC18; mobile phase: (1979)
tions methanol/PIC A (paired ion
chromatography A reagent)
and water/PIC A
Sedi- Homogenization; Derivatization: pyrolytic EC 0.5 - 25 92.8- Butte et
ment, toluene extraction ethylation with triethyl µg/kg 97.6% al.
clams from acidified sample sulfonium-iodide; GC anal- (1983)
containing 2,4,6- ysis; confirmation by MS
tribromophenol as
internal standard
---------------------------------------------------------------------------------------------------------
Table 5. (contd.)
---------------------------------------------------------------------------------------------------------
Medium Sampling method Analytical method Detec- Detection Reco- Reference
tion limit very
---------------------------------------------------------------------------------------------------------
Tallow Vortex mixing; auto- Derivatization with diazo- EC 1 µg/kg 80- Lee et
mated gel permeation methane; Florisil-column; 107% al.
chromatographic clean- GC analysis; confirmation (1984)
up; rotary evapora- with MS
tion; solution in
hexane
Indus- Hexane extraction Derivatization with penta- 0.1 pg ns Sha &
trial (for starch: after fluorobenzyl bromide; GC EC Duffield
starch, steam distillation) analysis and negative ion (1984)
surface chemical ionization MS MS
water (NICI-MS) analysis
---------------------------------------------------------------------------------------------------------
a EC = electron-capture detector.
b UV = ultraviolet.
c ns = not specified.
d GC = gas chromatography.
e HPLC = high-performance liquid chromatography.
f MS = mass spectrometry.
g TLC = thin-layer chromatography.
h SIM = selected ion monitoring.
i FID = flame ionization detector.
j NICI = negative ion chemical ionization.
The determination of PCP is based on the distinctive properties
of this substance: steam distillation is possible because of its
volatility; its acidic behaviour is used in extracting it into a
base and in ion-exchange chromatography; the electro-positive ring
reinforces selective chromatographic adsorption and the absorption
of ultraviolet radiation; finally, the reactivity of PCP with
certain organic compounds to form esters, ethers, and coloured
derivatives is of great importance for its detection and
measurement (Crosby et al., 1981).
Most of the analytical methods used today involved
acidification of the sample to convert PCP to its non-ionized form,
extraction into an organic solvent, possible cleanup by back-
extraction into basic solution, and analysis by gas chromatography
or other chromatographic methods as ester or ether derivatives. In
the following section, the sampling and analytical methods is
described as reviewed mainly by Bevenue & Beckman (1967), Gebefuegi
et al. (1979), and Crosby et al. (1981). In addition, the more
recently published methods for PCP determination in various
matrices are summarized (Table 5).
2.5.1. Sampling methods
In principle, the sampling techniques summarized by Bevenue &
Beckman (1967) are still the methods of choice; more recent methods
are included in Table 5.
The first step in preparing a sample consisting of a solid
material is a thorough pulverization or homogenization in special
mills or blenders. Maceration of the sample in a blender with an
organic solvent is more rapid than Soxhlet extraction and similar
efficiencies can be achieved with both procedures (Bruns & Currie,
1980).
For cellulose materials, adhesives, agricultural commodities,
biological tissues, and water, an initial extraction with dilute
sodium hydroxide solution at room temperature for several hours,
followed by acidification and steam distillation may be preferable.
For samples that contain components strongly complexed with PCP,
such as soybean oil, treatment with hot concentrated sulfuric acid
is recommended prior to steam distillation. Liquid-liquid
partitioning or distillation of the filtered extract at the boiling
point of water may also be used to isolate PCP (Bevenue & Beckman,
1967).
When alkaline soil extracts are acidified, gel formation can
occur at pH values lower than 6, resulting in interference with the
extraction of PCP. According to Renberg (1974), proper separation
is possible if the acidic substances are bound, under alkaline
conditions, to an anion ion exchanger.
When analysing liquid materials, particularly urine samples,
the sample should first be hydrolysed by heating the acidified
urine to free the PCP moiety of its sulfate and glucuronide
conjugates (Edgerton & Moseman, 1979; Drummond et al., 1982; Butte,
1984). Enzymatic hydrolysis is questionable, because the metabolite
tetrachlorohydroquinone strongly inhibits the enzyme
beta-glucuronidase (Ahlborg et al., 1974).
For determining the PCP content of air several possible
sampling procedures are described by Gebefuegi et al. (1979)
including: absorption in liquids, such as potassium carbonate
(K2CO3) solution or ethylene glycol; adsorption on activated
charcoal or silica gel; freezing and condensing by sucking the air
through cooling traps; or derivatization by phenolate formation in
alkaline solution. By pumping high volumes of air through the
sample-collecting device, PCP is concentrated in the collector,
thus enhancing the detection limit.
Concentration is also required for other matrices with
relatively low PCP contents. For water samples, procedures used
involve the separation of PCP from the water by distillation,
sublimation, freeze-drying, adsorption, and extraction (Rübelt et
al., 1982). The extraction solvents, in turn, are concentrated by
distillation or evaporation.
Only a few investigators have used internal standards, adding
specific substances to the samples to check for completeness of
recovery during the extensive solvent extractions and manipulative
steps required. Drummond et al. (1982) used 3,5-dichloro-2,3,6-
tribromophenol, while Needham et al. (1981) incorporated 2,4,6-
tribromophenol, and Hargesheimer & Coutts (1983) spiked the samples
with 4,6-dibromo- o-cresol.
Most recovery data given in Table 5 were obtained by spiking
samples with known amounts of PCP and carrying them through the
entire analytical procedure. Ernst & Weber (1978a) used 14C-PCP
for this purpose. To check the efficiency of acetylation, Rudling
(1970) compared spiked samples with a pentachlorophenyl acetate
standard. According to NIOSH (1978), an appropriate correction
factor should be used if recovery of PCP in air samples is less
than 95%.
Using the analytical method of Erney (1978) (Table 5), Zimmerli
et al. (1980) found that only about 8% of "endogenous" PCP was
extractable from raw bovine milk, though 82.5% of known amounts of
PCP added had been recovered on average. A complete extraction was
only achieved by acid or alkaline pretreatment of the milk (cf.,
Lamparski et al., 1978) (Table 5), which probably releases the PCP
bound to proteins. Zimmerli et al. (1980) concluded from this
finding that recovery data may indicate values that do not
correspond to the true recovery.
2.5.2. Analytical methods
Earlier methods, which have been thoroughly reviewed by Bevenue
& Beckman (1967), were based on the formation of coloured
derivatives from the reaction of PCP with either nitric acid or 4-
aminoantipyrine. Other reagents commonly used in this respect are
p-nitraniline, sulfanilic acid, and 3-methyl-2-benzenethiazoline-
hydrazine (Koppe et al., 1977). As already mentioned, these
colorimetric or spectrometric methods are not very specific and
comparatively insensitive, and therefore only suitable for pure
solutions or for production and routine controls. They may be of
some importance in determining total phenolics, for example, in the
monitoring of levels of phenolics in surface and waste waters.
However, comparative studies, in which 45 laboratories within the
European Communities participated, revealed that photometric
procedures gave rather different results, depending on specific
laboratory conditions (Sonneborn, 1976; Rübelt et al., 1982).
According to Crosby et al. (1981), colorimetric or
spectrophotometric procedures achieve a sensitivity that is, at
best, in the low ppb-range (1:109).
Gas chromatography, particularly when combined with an
electron-capture detector, substantially lowers the detection
limits to the ppt-range (1:1011 - 1:1012) and is therefore the
preferred method today. Very few investigators have applied direct
gas chromatography after the extraction procedures. To reduce peak
tailing, derivatization of PCP with appropriate compounds prior to
analysis is preferred. Diazomethane is most commonly used to
produce the methyl ether. As shown in Table 5, this method, which
is based on the work of Bevenue et al. (1966), has been used to
determine PCP in a variety of matrices including blood, urine,
fish, soil, and water. According to Crosby et al. (1981), it is
an official method for regulatory analysis in the USA. The
procedure for measuring PCP in blood and urine samples as
recommended by the National Institute for Occupational Safety and
Health (NIOSH), USA, is described by Eller (1984a,b).
Other alkyl ethers have been produced as derivatives of PCP,
including the ethyl, propyl, 1-butyl, isobutyl, amyl, and isoamyl-
PCP (Cranmer & Freal, 1970). Besides the potential health risk
incurred when using hazardous reagents such as diazomethane or
dimethyl sulfate, the alkylation method is subject to interferences
from other compounds with active H-atoms, e.g., carboxy acid
herbicides such as 2,4-dichloro-and 2,4,5-trichlorophenoxyacetic
acid (Chau & Coborn, 1974; Crosby et al., 1981). These drawbacks
are avoided by the acetylation of PCP with acetic anhydride to give
acetyl-PCP as reported by Rudling (1970), Chau & Coborn (1974), and
other research workers (Table 5).
Several techniques, other than gas chromatography, have been
used in connection with electron-capture detection. These include
thermal conductivity and microcoulometric detectors (Bevenue &
Beckman, 1967), thin-layer chromatography (TLC), gas chromatography
in connection with mass spectrometry (MS), and high-performance
liquid chromatography (HPLC) equipped with UV detectors. In
particular, the last two methods have become more and more
prevalent as reflected by Table 5. In many cases, mass
spectrometry has been used to confirm the identity of PCP peaks
determined by EC detectors. Dougherty & Piotrowska (1976a) and
Kuehl & Dougherty (1980) screened environmental and tissue samples
for PCP using negative chemical ionization (NCI) mass spectrometry.
This method provides a sensitivity of detection comparable to GC-
ECD analysis. Moreover, it can be used for compound
identification. Since both of these methods require an extensive
amount of pretreatment, a procedure had to be adopted for PCP
determination by which samples could be measured simply and
precisely, without the tedious extraction and formation of
derivatives needed for the other methods. High-performance liquid
chromatography offers these advantages, as using this method direct
determination of PCP is possible, giving peaks of constant height
and high resolution. Comparative GC-ECD and HPLC analyses of
mushrooms conducted by Schönhaber et al. (1982) resulted in similar
detection limits (Table 5).
Detection limits depend not only on the sensitivity of the
detection systems, but also, to a great extent, on the volume of
the sample. The detection limits given in Table 5 refer to the
smallest amounts of PCP detectable using the procedure and sample
size described by the authors. In many cases, it would be possible
to lower the detection limit by taking larger samples, particularly
in the case of gaseous and fluid matrices.
Analytical interferences may become a problem in PCP analysis
for residues, particularly at low measurement levels. Bevenue &
Ogata (1971) reported errors during the determination of PCP in the
picogram range, because of analytical-grade reagents such as sodium
hydroxide. Arsenault (1976) observed an apparent contamination of
samples with PCP from the general laboratory atmosphere. However,
measuring blank samples as controls and purifying reagents should
exclude false data. For example, Dietz & Traud (1978b) distilled
the extraction solvent diethyl ether to remove the antioxidant BHT
(2,6-di- tert-butyl-4-methyl-phenol). Similarly, the authors
recommended the distillation of dioxan prior to its use as
extraction solvent; otherwise, some volatile impurities could
interfere with the measurement of PCP.
Substances interfering during gas chromatography may cause more
of a problem. These include chloronaphthalenes, polychlorinated
biphenyls (PCBs), pesticides such as diuron, and p-methoxytetra-
chlorophenol. Arsenault (1976) therefore questioned the GC-ECD
method in the µg range. In a thorough study on phenolics in water
(Rübelt et al., 1982), derivatization was omitted because non-
specific reactions might occur in complex mixtures, e.g., in
polluted waters. The working group achieved best results in terms
of separation of chlorophenols with a column of 10% Carbowax 20 M
plus 2% phosphoric acid, the mobile phase being nitrogen enriched
with formic acid. The latter was found to prevent tailing
resulting from adsorption of chlorophenols on the packing material
of the column. For quantitative analysis with an unequivocal
identification, it has been recommended that after gas
chromatographic separation the carrier gas should be split and
conducted to both an electron capture detector (ECD), and a flame
ionization detector (FID), as well as to a mass spectrometer (MS).
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural Occurrence
Arsenault (1976) hypothesized the existence of a natural
background level of PCP or analytically similar compounds. He
suggested this on the basis that paramethoxytetrachlorophenol, a
metabolite of a fungus, could interfere with the GC-EC analysis of
PCP, because of its similar molecular size, shape, and retention
time. The hypothesis of a natural background level of PCP has not
been examined further. Unsuccessful attempts to produce higher
chlorophenols by enzymatic conversion (Siuda, 1980) suggest that
sources of environmental PCP are exclusively related to human
activities.
3.2. Man-Made Sources
3.2.1. Industrial production
3.2.1.1 Manufacturing processes
PCP was first synthesized by Merz & Weith (1872) using a
similar preparation method to that currently used during commercial
production (Prager et al., 1923). The use of PCP as a wood
preservative started in the late 1930s (Doedens, 1964).
PCP is produced by one of two methods: direct chlorination of
phenols and hydrolysis of hexachlorobenzene. The direct
chlorination is carried out in two steps. First, liquid phenol,
chlorophenol, or a polychlorophenol is bubbled with chlorine gas at
30 - 40 °C to produce 2,4,6-trichlorophenol, which is then
converted to PCP by further chlorination at progressively higher
temperatures in the presence of catalysts (aluminum, antimony,
their chlorides, and others). The second method involves an
alkaline hydrolysis of hexachlorobenzene (HCB) in methanol and
dihydric alcohols, in water and mixtures of different solvents in
an autoclave at 130 - 170 °C (Melnikov, 1971). In the Federal
Republic of Germany, PCP is synthesized by means of stepwise
chlorination of phenols. Na-PCP was produced until 1984 using
hexachlorobenzene hydrolysis; now, it is produced by dissolving PCP
flakes in sodium hydroxide solution (BUA, 1986). In the USA, the
general reaction used is the chlorination of phenols (Crosby et
al., 1981).
In addition to the formation of PCP, numerous by-products are
generated, as reflected by analytical profiles in Table 1. The
chlorination procedure yields a technical product that usually
contains a considerable amount of tetrachlorophenols (4 - 12%) due
to incomplete chlorination reactions. The formation of
microcontaminants is favoured by elevated temperatures and
pressure. With both manufacturing methods, toxic by-products, such
as chlorinated ethers, dibenzofurans, and dibenzo- p-dioxins, are
formed. In addition, the alkaline HCB hydrolysis method can result
in the presence of hexachlorobenzene in the resulting PCP (Jensen
& Renberg, 1973; Plimmer, 1973; Firestone, 1977; Jones, 1981).
3.2.1.2 Emissions during production
Some data are available concerning the loss of phenolic and
nonphenolic compounds into the environment during the normal
production of PCP or Na-PCP (Umweltbundesamt, 1985). The following
air emission concentrations (mg/m3) and mass flow values (g/h) were
reported: PCP 0.7 mg/m3, 9 g/h; tetrachlorophenols 0.2 mg/m3, 0.8
g/h; trichlorophenols 0.02 mg/m3, 0.04 g/h; hexachlorobenzene 23.9
mg/m3, 12 g/h; pentachlorobenzene 2 mg/m3, 15.5 g/h;
tetrachlorobenzene 2.8 mg/m3, 66.5 g/h; OCDD 0.05 mg/m3, 0.04 g/h;
OCDF 0.02 mg/m3, 0.002 g/h.
The annual air emission values resulting from the production
of approximately 2000 tonnes of PCP or Na-PCP, respectively, per
annum are given in Table 6.
Table 6. Air emissions of phenolic and non-phenolic
compounds during production (maximum values)a
------------------------------------------------------
Annual air emissions (kg/year)
during production of:
2000 tonnes 2000 tonnes
PCP/year Na-PCP/year
------------------------------------------------------
PCP 18 65
Other chlorophenols 9 5
Hexachlorobenzene - 105
Other chlorobenzenes 1 700
OCDD 0.2 0.2
------------------------------------------------------
a Adapted from: BUA (1986).
While no waste water occurs during the production of PCP, the
annual loss of various compounds resulting from Na-PCP production
into the waste water was as follows: PCP, 60 kg; OCDD, 0.34 g;
H7CDDs, 0.1 g; H6CDDs, 0.001 g; OCDF, 0.1 g; H7CDFs, 0.026 g;
H6CDFs, 0.002 g (BUA, 1986).
3.2.1.3 Disposal of production wastes
The volume of contaminated waste water generated during the
production of Na-PCP is small, because manufacturers and regulatory
agencies have emphasized efficient process design (Jones, 1981).
During the production of approximately 2000 tonnes PCP/year,
about 8 tonnes of washing methanol, 4 tonnes of activated charcoal,
and 2 tonnes of other wastes occur. These wastes, as well as the
filtration sludge resulting from Na-PCP production, contain
considerable amounts of hazardous chemicals (Table 7). They are
generally disposed of by either storage in underground disposal
sites (filtration sludge) or incineration at temperatures above
1200 °C (BUA, 1986).
Table 7. Phenolic and non-phenolic compounds in the combined
wastes (PCP production) and filtration sludge (Na-PCP production)
---------------------------------------------------------------
Compound Combined wastes Filtration sludge
(kg/year) (kg/year)
---------------------------------------------------------------
PCP 1350 900
Other chlorophenols 0.7 nsb
Hexachlorobenzene ns 6000
Decachlorobiphenyl ns 3400
Decachlorophenoxybenzene ns 44
OCDD (OCDF) 0.98 0.67 (0.67)
H7CDDs (H7CDFs) 0.13 0.17 (0.045)
H6CDDs (H6CDFs) 0.013 0.092 (0.015)
P5CDDs (P5CDFs) 0.003 x 10-3 0.016 (0.005)
T4CDDs (T4CDFs) 0.002 x 10-3 0.007 (0.001)
2,3,7,8-T4CDD ns 0.001
---------------------------------------------------------------
a Adapted from: BUA (1986).
b ns = not specified.
US EPA (1985) proposed that wastes from the production and
manufacturing use of PCP should be classifed as acutely hazardous
wastes, on the basis of the presence of substantial concentrations
of the carcinogenic congener H6CDD and the chronic toxicity
potential of PCP itself.
3.2.1.4 Production levels
No precise estimates can be made of the total world production
of PCP and Na-PCP. According to the data profile of IRPTC (1983),
90 000 tonnes of PCP per year are produced globally. The Economist
Intelligence Unit (1981) estimated world production to be of the
order of 50 000 - 60 000 tonnes per year, based on the North
American and European Community output. However, the production
figures presented in Table 8 indicate a total production of only
30 000 tonnes per year. The production, foreign trade, and
consumption figures given in this summary table can give only a
rough idea of the true PCP market. Recent restrictions on the use
of PCP (section 3.3), a decline in the forestry industry, and the
increasing use of alternative means of wood preservation have
probably reduced the demand for PCP over the last few years.
The major PCP producers operating in 1980 are shown in Table 9
together with the plant locations and their capacities. Some
additional factories exist in which PCP is mixed or formulated to
yield special end-use products. There are also chemical producers
who sell pure, analytical grade PCP, but do not produce PCP for
technical purposes. The Monsanto Company, which had a capacity of
11.8 kilotonnes in the USA, stopped PCP production in their plant
at Sauget, Illinois, in 1978 (Jones, 1981). Dow Chemical closed
down its manufacturing plant at Midland, Michigan in October 1980
(Jones, 1984). Similarly, the only PCP producing plant in the
United Kingdom, also operated by the Monsanto Company, was closed
down in the same year (Economist Intelligence Unit, 1981), while
Reichhold Chemicals Inc., at Tacoma, Washington, USA ceased PCP
production in 1985. In the Federal Republic of Germany, the
production of PCP and Na-PCP was stopped in 1986.
3.3. Uses
The main advantages of PCP and its derivatives are that they
are effective biocides and soluble in oil (PCP) or water (Na-PCP).
Few pesticides show a similarly broad efficiency spectrum at low
cost. Therefore, PCP and its salts have a variety of applications
in industry, agriculture, and in domestic fields, where they have
been used as algicides, bactericides, fungicides, herbicides,
insecticides, and molluscicides.
3.3.1. Commercial use
In Table 10, the major registered commercial uses of PCP are
broken down for the United Kingdom and the USA. Although PCP and
its derivatives have many uses, by far the major application is
wood preservation. Cirelli (1978a), Hoos (1978), and Jones (1981)
have reported on commercial use patterns in North America. In the
USA, about 80% of PCP is used for commercial wood treatment, 6% is
in use for slime control in pulp and paper production, and 3%
accounts for non-industrial purposes, such as weed control, fence-
post treatment and paint preservation (Crosby et al., 1981);
however, the last two cases imply wood treatment as well. The
remaining 11% is converted to Na-PCP, which in turn is partly used
for wood preservation, mainly sapstain control in waterborne
conditions, e.g., for treating pressboard. Overall, some 95 - 98%
of American PCP production is used directly or indirectly in wood
treatment (Economist Intelligence Unit, 1981).
Table 8. Production, foreign trade, and consumption of PCP and Na-PCP (tonnes per year)
according to data available from government authorities and producers
--------------------------------------------------------------------------------------------------
Belgium/ Francea Germany, Italya Nether- United Canadac USAd
Luxemburga Federal landsa Kingdoma
Republic ofb (year (year (year
(year na)e 1979 1979 1984 na)e na)e na)e 1981 1977
--------------------------------------------------------------------------------------------------
Production
PCP 0 1700 2450 1550 0 0 0 1700 20 349
Na-PCP 0 2800 2100 1750 0 0 0 70
Imports
PCP 150- insigni- 0 0 250 - 30 - na 500 na
Na-PCP 160 ficant 300 0 280 40 na 0 na
Exports
PCP 0 300- 1950 1360 0 0 300 600 approxi-
Na-PCP 0 700 2150 1710 0 0 0 mately
200f
Consumption
PCP approxi- 1000 500 190 250- 30- 500 1536 na
Na-PCP mately 2500 250 40 280 40 32 na
150
--------------------------------------------------------------------------------------------------
a From: Economist Intelligence Unit (1981).
b From: BUA (1986).
c From: Jones (1984).
d From: Jones (1981).
e na = not available.
f Approximately 1% of domestic sales.
Table 9. Pentachlorophenol producers and their capacities in 1980
--------------------------------------------------------------------
Producer Country Plant Capacity
Location (tonnes)
(total PCP)
--------------------------------------------------------------------
Uniroyal Chemical,a Canada Clover Bar, 1800
Division of Uniroyal, Ltd Alta
Rhône-Poulencb France Pont-de-Claix 4500
Dynamit Nobelb Germany, Rheinfelden 4000
Federal
Republic of
Dow Chemical, USAa USA Midland, 13 500
Michigan
--------------------------------------------------------------------
Table 9 (contd.)
--------------------------------------------------------------------
Producer Country Plant Capacity
Location (tonnes)
(total PCP)
--------------------------------------------------------------------
Reichhold Chemicalsa USA Tacoma, 8100
Inc. Washington
Vulcan Materiala USA Wichita, 9000
Company Chemical Division Kansas
--------------------------------------------------------------------
a From: Jones (1981).
b From: Economist Intelligence Unit (1981).
Data from Canada and the Federal Republic of Germany confirm
the main use of PCP as a wood preservative. In Canada, about 95%
of the PCP is used for this purpose (Jones, 1981). Approximately
61% of the volume of PCP used in the Federal Republic of Germany in
1983 was used for wood preservation, while considerable amounts of
PCP were used by the textile (13%), leather (5%), mineral oil (6%),
and glue (6%) industries, respectively (Angerer, 1984). No PCP was
used in the paint or pulp industry whereas, in 1974, as much as 3%
or 7%, respectively, were used in these branches. PCP used on
textiles is usually in the form of the PCP ester rather than PCP or
Na-PCP.
Pentachlorophenyl laurate (L-PCP) was developed especially for
application on fabrics (Hueck & LaBrijn, 1960; Bevenue & Beckman,
1967). The estimates of L-PCP use in the United Kingdom in Table
10 are based on a publication from the year 1974 (HMSO, 1974).
According to an unpublished note submitted to the IPCS by Catomance
Limited, Hertfordshire, the sole manufacturer of pentachlorophenyl
laurate in the United Kingdom, the usage pattern in the United
Kingdom has not changed following the cessation of production of
PCP in 1978. However, most of the PCP ester used there today is
said to be for domestic timber preservation; the use of L-PCP for
textile preservation is supposed to be mainly confined to tropical
or semi-tropical countries.
In the USSR, PCP is used for the preservation of commercial
timber, paints, varnishes, paper, textiles, ropes, and leather
(IRPTC, 1984).
Na-PCP is also used to inhibit algal and fungal growth in
cooling tower waters at electric generating plants (Hoos, 1978); in
1976, about 30% of the Na-PCP used in Canada was for this purpose
(Jones, 1981).
Table 10. Major commercial (non-agricultural) uses of PCP in the
United Kingdom and the USAa
---------------------------------------------------------------------------
Use Active
ingredient
---------------------------------------------------------------------------
United Kingdom
Anti-mildew agent in the wool textile industry L-PCP, Na-PCP
Mothproofing carried out by dyers and cleaners L-PCP
Wood preservation PCP, L-PCP,
Na-PCP
Paint additives PCP
Antimicrobial (slimicide) agents in paper and board PCP
Antifungal agent in textiles other than wool (cotton,
Flax and jute fabric, ropes, cordage and tentage) L-PCP
Cable impregnation L-PCP
Anti-mildew agent in leather nsb
Fungicide in adhesives Na-PCP
Bactericide in drilling fluids nsb
USA
Microbiostat for commercial and industrial water cooling Na-PCP
Microbiocide for leather K-PCP, PCP
Microbiocide for burlap, canvas, cotton, rope, and twine PCP
Microbiocide and insecticide for wood treatment PCP, Na-PCP
Preservative for oil and water-based paint PCP
Slime control for pulp and paper PCP
Microbiocide for petroleum drilling mud and flood water PCP
Fumigant for shipping-van interiors PCP
Preservative for hardboard and particle-board PCP
---------------------------------------------------------------------------
a From: Crosby et al. (1981).
b ns = not specified.
Alterations in the use pattern have taken place during the last
few years as a result of the increased concern about the potential
health hazards from PCP and its impurities. In Japan, the
production of PCP was 14.5 kilotonnes in 1966 and 3.3 kilotonnes in
1971, after which production ceased entirely (IARC, 1979a). In the
Federal Republic of Germany, 3300 tonnes of total PCP were produced
in the year 1984, of which 93% was exported, leaving 230 tonnes for
use in the country (BUA, 1986). Nine years earlier (1974), 4100
tonnes of PCP were produced, 60% of which were exported; in 1979,
84% of the 4503 tonnes produced were sold abroad (Angerer, 1984).
These figures indicate a drastic decrease in the consumption of PCP
in the Federal Republic of Germany during the last few years. In
Canada, the Federal Republic of Germany, and Sweden, where PCP had
been heavily used as a slime control agent in the paper mills, the
use of chlorinated phenols for this purpose was prohibited in
recent years as a consequence of the discharges, which had toxic
effects on the aquatic environment. In Sweden, all use of PCP was
banned in 1977 (Ahlborg & Thunberg, 1980; Jones, 1981). The US
Environmental Protection Agency does not intend to prohibit the use
of PCP in oil-well water or in pulp and paper mills, provided that
impermeable gloves are worn during application and that the H6CDD
content will be reduced to 1 ppm (US EPA, 1984b). Similarly, the
use of PCP (including its salts) for wood protection has not been
cancelled in the USA. However, the US EPA (1984a) intends to
establish certain changes in the terms and conditions of
registration.
3.3.2. Agricultural use
Significant quantities of PCP were previously used in a number
of agricultural applications. These resembled industrial uses in
that most were to prevent wood decay, in farm buildings, fences,
and storage facilities (Jones, 1981). However, PCP or its sodium
salt have also been used as a herbicide and dessicant for forage
seed crops, a herbicide for non-food vegetation control, a biocide
in the post-harvest washing of fruit, and as an insecticide for use
in beehives, seed plots, and greenhouses (Crosby et al., 1981).
PCP was formerly used as a herbicide in paddy and upland rice
fields, particularly in Japan (Kobayashi, 1978; Crosby et al.,
1981). In addition, PCP and Na-PCP have been approved for a number
of applications in the food industry, such as biocides in packaging
materials and glues (Table 11).
In the USSR, PCP is applied as a nonselective herbicide and as
a desiccant on cotton plants. At least 10 days should lapse
following cotton plant treatment (IRPTC, 1984).
Table 11. Other uses of PCP and its salts as a potential
source of food contaminationa
-------------------------------------------------------------
Use Specific compound
-------------------------------------------------------------
Slime control on paper and paperboard K-PCP, Na-PCP
Preservative in can-end cement Na-PCP
Defoaming agents Na-PCP
Paper contacting aqueous and fatty food Na-PCP
Animal glue for containers Na-PCP
Sealing gaskets for containers K-PCP, Na-PCP
Preservative for wood products PCP, Na-PCP
Preservative in coatings Na-PCP
Rubber antioxidant Na-PCP
Preservative for ammonium alginate Na-PCP
-------------------------------------------------------------
a Adapted from: Firestone (1973).
Recently, regulations to limit or even ban some uses of PCP
have been established in a number of countries. The Canadian
government suspended agricultural applications of PCP and Na-PCP in
mushroom culture, above-ground interior woodwork of farm buildings,
and as herbicides and soil sterilants (Jones, 1984). Japan
restricted herbicidal use of PCP because of its high toxicity to
fish (Kobayashi, 1979; Crosby et al., 1981). The paper industry of
the Federal Republic of Germany and Sweden no longer use PCP in
packaging paper (Ahlborg & Thunberg, 1980; Angerer, 1984).
US EPA (1984b) proposed cancelling the registration of
pesticide products containing PCP as the active ingredient for non-
wood preservative uses, i.e., herbicidal and antimicrobial uses.
In addition, PCP-containing wood preservatives for home and farm
use must not be applied where there may be direct contact with
domestic animals or livestock or close contact with food or feed
(US EPA, 1984a).
3.3.3. Domestic use
The largely uncontrolled use of PCP by private individuals is
almost exclusively related to the treatment of wood, both outdoors
and indoors. PCP is the main active ingredient in certain wood
preservatives for home use, and is added to products such as stains
and paints. Although this category of products plays only a minor
role in the overall PCP market, it has been of particular concern,
since cases of apparent PCP intoxication after indoor application
in private homes have been reported (section 5). As a consequence
of such incidents, the use of PCP for the preservation of interior
timber has been banned in Canada (Jones, 1981) and the Netherlands
(Economist Intelligence Unit, 1981). Since 1986, the use of PCP as
a biocide for indoor application has been forbidden in the Federal
Republic of Germany by government regulatory action (FRG, 1986).
Furthermore, there is a gentlemen's agreement between the
government and industry to suspend the use of PCP in wood
preservatives in general (BUA, 1986). The US EPA (1984a) intends
to limit the use of PCP-containing wood preservatives in interiors
to certain support structures. This is also true for the indoor
use of PCP-treated wood. The sale and use of PCP is restricted to
certified applicators. Thus, the domestic use of PCP is not as
significant as it was some years ago.
Other reported applications of PCP include health-care products
and disinfectants for use in the home, farms, and hospitals. PCP
may also be contained in dental-care products (Jones, 1981),
bactericidal soaps, laundry products, and medical products for the
skin (Crosby et al., 1981).
3.3.4. Use for control of vectors
The application of Na-PCP to control vectors of pathogens has
been of some relevance in tropical and subtropical areas. Na-PCP
has been used as a herbicide to control Salvinia sp., a host plant
of Mansonia mosquitos, which transmit the elephantiasis-causing
filarias to man (Chow et al., 1955).
Na-PCP has also been used for control of the intermediate snail
hosts of schistosomiasis (Berry et al., 1950; Toledo et al., 1976).
After World War II, Hunter et al. (1952) proposed Na-PCP as the
molluscicide of choice for use in Japan. In China, Na-PCP is still
in use for this purpose today (Xue, 1986)a.
___________________________________________________________________
a Personal communication to the Task Group on Pentachlorophenol.
3.3.5. Formulations
In the treatment of wood, PCP is usually administered as a 5%
solution in a mineral spirit solvent, such as No. 2 fuel oil or
kerosene (Cirelli, 1978a), or methylene chloride, isopropyl
alcohol, or methanol (Ingram et al., 1981a). Since PCP is not very
soluble in hydrocarbon solvents, and tends to migrate to, and
crystallize on, treated wood surfaces (a phenomenon known as
"blooming"), formulations may also contain co-solvents and anti-
blooming agents (David, 1985a). Most commercial formulations also
contain other chlorophenols, mainly tetrachlorophenol (Nilsson et
al., 1978). The aim of such mixtures is to prevent blooming on
treated wood by lowering the melting point of the chlorophenols.
An aqueous solution of Na-PCP is used for commercial sapstain
control (Konasevich et al., 1983).
Chlorophenols may be combined with other active components such
as methylene bisthiocyanate and copper naphthenate in the
formulation of PCP pesticides (von Rümker et al., 1974).
Conversely, PCP is added to biocides, the primary active ingredient
of which is another compound; for example, sodium fluoride
formulations for wooden poles and posts may contain up to 10%
technical PCP (US EPA, 1973).
---------------------------------------------------------------------------
a Personal communication, Catomance Ltd, Welwyn Garden City,
Hertfordshire, United Kingdom.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and Distribution Between Media
Several physical and chemical parameters affect the transport
and distribution of PCP in soil, water, and air. Volatilization
and adsorption are the major mechanisms; leaching and movements on
surfaces and in air are of minor importance (Jones, 1981).
4.1.1. Volatilization
Volatilization can be an important source of loss of PCP from
water and soil surfaces as well as from PCP-treated materials.
Dip- or brush-treating coniferous wood may lead to a 30 - 80% loss
of PCP, due to evaporation, within 12 months (Morgan & Purslow,
1973; Petrowitz, 1981).
Several factors influence the rate of volatilization of PCP
from wood. Ingram et al. (1981b) showed that certain solvents or
mixtures of solvents may either reduce or increase the
volatilization of PCP. Such solvents, as well as resins, are used
to achieve a timely release of PCP through solubilization and/or
occlusion, e.g., PCP evaporation was decreased by about 20%, when
15% of an alkyd resin was added to a wood preservative containing
5% PCP (Petrowitz, 1981). Temperature appeared to be the external
variable that had the greatest effect on volatilization; a rise in
temperature from 20 to 30 °C caused a 3- to 4-fold increase in
volatilization. Relative humidity and rate of air flow had only a
minor influence on PCP volatilization from wood. The loss of PCP
from pine-wood samples was half as much as that from spruce wood
samples after 21 days, apparently because pine is much more easily
impregnated than spruce. Thus, the depth of penetration of PCP in
wood, the species of wood, and the process of treatment appear to
be other external variables influencing diffusion and
volatilization. As an example, immersion of small blocks of pine
sapwood with a 5% PCP solution leads to a PCP loss of 20% after 6
months compared with only 4%, 9 months following double vacuum
treatment (Morgan & Purslow, 1973).
Evaporation is used for the disposal of PCP in waste water at
some wood-preserving plants (section 4.4). Extensive studies on
the volatilization of PCP from water or soil have not been carried
out, but Klöpffer et al. (1982) studied this process in an aqueous
solution in the absence of other pathways of removal. Temperature
and pH of the solution were the most important factors influencing
the evaporation rate. Since only the un-ionized form of PCP seems
to be volatile, at pH 5.1, when 13.2% of the PCP was present as the
free acid, the 50% residence time was 328 h at 30 °C, whereas, at
pH 6, with more than 98% PCP dissociated to its phenate, a one-half
residence time of 3120 h was measured. These findings suggest that
evaporation of PCP from surface waters with a pH above 6 should be
quite low.
4.1.2. Adsorption
The extent of adsorption of a pesticide governs its
biovailability in soil; hence, both the rate of degradation and the
biocidal activity are likely to be reduced by strong adsorption.
In addition, though possessing a reduced activity, a highly
adsorbed compound would exert a prolonged effect (Su & Lin, 1971;
Choi & Aomine, 1972).
pH seems to be the major factor controlling the magnitude of
PCP adsorption. Choi & Aomine (1974a) investigated PCP retention
in a range of soil types, and determined that adsorption was
maximal in strongly acidic soils, relatively minor in moderately
acidic soils, and absent in weakly acidic or neutral soils. Other
research workers have observed the same relationship between soil
pH and PCP retention (Green & Young, 1970; Kaufmann, 1976).
The organic matter content and surface area of soils exert a
minor, but significant, effect on PCP adsorption. Choi & Aomine
(1974a) found that the magnitude of adsorption decreased in the
following order: humusallophanic, allophanic, montmerillonitic, and
halloysitic soils. This finding confirms the binding of PCP by
organic matter reported by other authors (Su & Lin, 1971; Choi &
Aomine, 1972).
Under the weakly acidic to neutral conditions characterizing
most soils, adsorption is likely to exert a minor effect on PCP
dynamics. In this regard, it is noteworthy that Choi & Aomine
(1974b) studied adsorption using hexane as a solvent, because the
amount of the pesticide sorbed onto soils from aqueous solution was
too small to determine.
4.1.3. Leaching
Leaching of a chemical through the soil is interrelated with
factors such as adsorption, water solubility of the substance, soil
type, moisture, percolation velocity, and pH (Haque & Freed, 1974).
Thus, the leaching behaviour of PCP will vary, depending on the
soil under examination.
Leaching is an important means of transport for PCP, in some
instances. Kuwatsuka (1972) noted that much of the PCP applied to
flooded rice paddies was carried through the soil in solution, and
the Weed Science Society of America Herbicide Handbook (WSSA, 1974)
reported that Na-PCP also leaches readily in soil. This is
consistent with the observation that leaching of PCP occurs more
easily in alkaline soils than in both acidic clay and sandy soils
(Kaufman, 1976).
In addition, substantial quantities of PCP are found in waters
leaching from contaminated sites. For example, 2.05 and 3.35 mg
PCP/litre were detected in groundwater from a wood preservation
plant near Lake Superior (Thompson et al., 1978), and PCP in the
µg/litre range was detected in water seeping from a landfill
(Kotzias et al., 1975).
Some in vitro studies have revealed little or no PCP in soil
leachate (Arsenault, 1976; Weiss et al., 1982b), but these are
difficult to interpret, as residence times are either extremely
long, or not reported. In other percolation tests, PCP did not
leach in the profile of a brown earth-Lessivé within one month, but
was detected in the water seeping from a podzol (1.5 mg PCP/litre)
after two days (Fränzle, 1982).
From the sorption and leaching behaviour of PCP, it can be
concluded that organic matter serves as a reversible storage
compartment, allowing desorption of PCP at elevated soil-water
content and, hence, accumulation of PCP in the soil solution and
eventually, in the groundwater (BUA, 1986).
Stranks (1976) stated that PCP does not leach readily from
treated wood, particularly if applied via an oil carrier.
4.2. Biotransformation
4.2.1. Abiotic degradation
Both PCP and Na-PCP are subject to abiotic (photochemical)
degradation in water, organic solvents, and on solid surfaces. In
the photolysis pathway (Fig. 2) suggested by Wong & Crosby (1978),
three types of degradation products occur: (a) lower chlorinated
phenols, mainly 2,3,4,6- and 2,3,5,6-tetrachlorophenol together
with trichlorophenols; (b) chlorinated dihydroxybenzenes, primarly
tetrachlororesorcinal and tetrachlorocatechol; and (c) non-aromatic
fragments, mostly dichloromaleic acid. Irradiation of the last
compound, in turn, yielded carbon dioxide (CO2) and chloride ions.
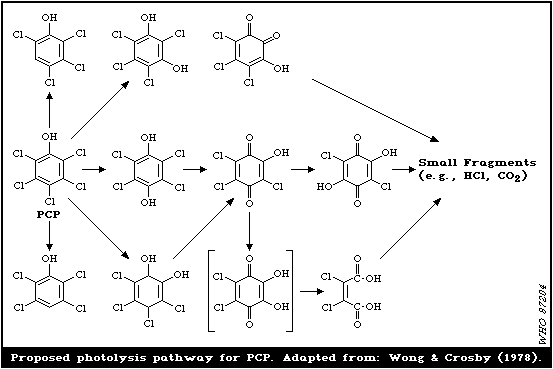 Wong & Crosby (1978, 1981) discovered that photolysis of PCP
(100 mg/litre) in aqueous solutions took place much faster at pH
7.3 than at pH 3.3. The ionized PCP disappeared completely within
20 h (half-life, 3.5 h), whereas the half-life of the un-ionized
form was about 100 h. The increasing rate of photodecomposition
with increasing pH, reported also by Wang (1965), provides evidence
of an ionic mechanism, the initial and rate-limiting reaction being
the photo-nucleophilic replacement of PCP chlorine atoms by
hydroxyl groups (Crosby et al., 1972).
PCP photodegradation proved to be primarily oxidative in the
studies described. However, in natural waters, reduction processes
seem to prevail in an acid, high organic load environment and under
the influence of organic proton-donors. Wong & Crosby (1981)
concluded this from the absence of oxidized substances, such as
dichloromaleic acid, and the detection of reduced products.
Gäb et al. (1975) and Gäb (1981) simulated photochemical
decomposition in, or on, aerosol surfaces, dust particles etc., in
the troposphere, using a mercury high-pressure lamp to match
tropospherical sunlight. Adsorption of PCP on silica gel resulted
in markedly accelerated photodecomposition because of the
bathochromic shift of the maxima of absorption from 210 to 310 nm;
about 14% of 80 mg solid PCP was mineralized to HCl and CO2 within
7 days, while, under otherwise identical conditions, 88% of PCP
deposited on silica gel was decomposed.
Photodecomposition of PCP is even accelerated if catalysed by
semiconductors such as zinc (ZnO) and titanium dioxide (TiO2) in
aqueous suspensions (Barbeni et al., 1985). Under direct summer
sunlight, the half-life of PCP was about 8 min (12 mg PCP/litre
with 2 TiO2/litre, pH 3). Under laboratory conditions, the half-
life was 15 min at 45 - 50 °C, pH 10.5, and lambda > 330 nm;
mineralization of PCP was more than 95%.
Na-PCP, like its parent compound, is readily photolysed. Hiatt
et al. (1960) investigated photodecomposition of Na-PCP as a
possible factor reducing its efficacy as a molluscicide in South
African streamwaters. In vitro exposure to UV radiation (290 - 330
nm) caused chemical degradation of Na-PCP, which was directly
proportional to light intensity, and a corresponding loss of
molluscicidal activity. Similarly, Na-PCP applied to rice paddies
in order to control barnyard grass ( Panicum crusgalli L.) was
readily decomposed by sunlight (Kuwahara et al., 1966a). Aqueous
solutions of Na-PCP exposed to sunlight broke down to form mainly
chloroanilic acid and 3,4,5-trichloro-6-(2'-hydroxy-3',4',5',6'-
tetrachlorophenoxy)- o-benzoquinone (Kuwahara et al., 1966a), and
minor amounts of tetrachlororesorcinol and three benzoquinones
(Kuwahara et al., 1966a,b; Munakata & Kuwahara, 1969).
Field evidence indicates that photolysis is an important means
of PCP loss in situ. In their study of PCP contamination in the Bay
of Quinte, Lake Ontario, Fox & Joshi (1984) found the proportions
of 2,3,4,6- and 2,3,5,6-tetrachlorophenol in environmental samples
to be enriched relative to PCP. Since these compounds are products
of the photodegradation of PCP, they suggested that photolysis was
dominating PCP breakdown. Similarly, Yunker (1981) concluded that
photolysis was the most important pathway for the removal of
pentachlorophenate from enclosed marine pelagic enclosures off the
west coast of Canada.
Crossland & Wolf (1985) found evidence that direct photo-
transformation was mainly responsible for the loss of PCP from
experimental outdoor ponds (50 - 100 µg PCP/litre). They observed
half-lives for the loss of PCP in the range of 2 - 4.7 days (pH 7.3
- 10.3; 10 - 21 °C); decomposition was most rapid in relatively
clear water.
The formation of PCDDs as a result of photochemical reaction
has been described. Crosby & Wong (1976) irradiated Na-PCP in
aqueous solution. Traces of OCDD were found, while 2,3,7,8-T4CDD
was not detectable, presumably because of its rapid photoreduction
(Crosby et al., 1971). The photolysis of OCDD yields a variety of
chlorinated dibenzodioxins with decreasing numbers of chlorine
atoms (Crosby et al., 1971, 1973; Buser, 1976).
This is consistent with the findings of Lamparski et al.
(1980), who observed H6CDD and H7CDD in the course of photolysis
studies with wood samples treated with PCP in methylene chloride.
The OCDD content increased from the initial concentration of 3 mg
OCDD/kg Dowicide EC-7 (containing low initial PCDD concentrations)
or non-detectable amounts of OCDD/kg purified PCP (Aldrich Chemical
Company), respectively, to yield up to about 70 mg OCDD/kg PCP on
the surface of wood in both PCP specifications, while controls
stored in the dark showed no increase in OCDD. Use of a
hydrocarbon oil as a solvent significantly reduced OCDD formation
during irradiation to yield 4.4 mg OCDD/kg Dowicide EC-7 or 2.2 mg
OCDD/kg purified PCP, respectively. Moreover, OCDD concentrations
only slightly increased in wood samples treated with a technical
PCP containing relatively high initial PCDD concentrations (OCDD -
1100 mg/kg).
These results agree with those reported by Cull & Dobbs (1984),
who found no evidence of the formation of OCDD in technical PCP
(solvent - hydrocarbon oil) or Na-PCP (solvent - water). This has
been attributed to the photolytic destruction and volatilisation
of OCDD dominating its formation when the initial OCDD
concentration is relatively high.
4.2.2. Microbial degradation
The microbial degradation of PCP has been studied using natural
and artificial media with mixed or single microbial cultures. Lyr
(1963) demonstrated that fungi were able to attack PCP by means of
phenol oxidase. Cserjesi (1967) observed PCP decomposition by
fungi of the genus Trichoderma in malt extract solution at a
concentration of approximately 10 mg/litre. Similar studies have
also been carried out with a number of other fungal species (Duncan
& Deverall, 1964; Cserjesi, 1972).
Numerous PCP degrading bacterial strains, which are partly
capable of using PCP as a sole source of organic carbon, have been
isolated (Chu & Kirsch, 1972; Kirsch & Etzel, 1973; Watanabe, 1973;
Suzuki, 1977; Reiner et al., 1978; Edgehill, 1982; Stanlake & Finn,
1982; Trevors, 1982a).
Wong & Crosby (1978, 1981) discovered that photolysis of PCP
(100 mg/litre) in aqueous solutions took place much faster at pH
7.3 than at pH 3.3. The ionized PCP disappeared completely within
20 h (half-life, 3.5 h), whereas the half-life of the un-ionized
form was about 100 h. The increasing rate of photodecomposition
with increasing pH, reported also by Wang (1965), provides evidence
of an ionic mechanism, the initial and rate-limiting reaction being
the photo-nucleophilic replacement of PCP chlorine atoms by
hydroxyl groups (Crosby et al., 1972).
PCP photodegradation proved to be primarily oxidative in the
studies described. However, in natural waters, reduction processes
seem to prevail in an acid, high organic load environment and under
the influence of organic proton-donors. Wong & Crosby (1981)
concluded this from the absence of oxidized substances, such as
dichloromaleic acid, and the detection of reduced products.
Gäb et al. (1975) and Gäb (1981) simulated photochemical
decomposition in, or on, aerosol surfaces, dust particles etc., in
the troposphere, using a mercury high-pressure lamp to match
tropospherical sunlight. Adsorption of PCP on silica gel resulted
in markedly accelerated photodecomposition because of the
bathochromic shift of the maxima of absorption from 210 to 310 nm;
about 14% of 80 mg solid PCP was mineralized to HCl and CO2 within
7 days, while, under otherwise identical conditions, 88% of PCP
deposited on silica gel was decomposed.
Photodecomposition of PCP is even accelerated if catalysed by
semiconductors such as zinc (ZnO) and titanium dioxide (TiO2) in
aqueous suspensions (Barbeni et al., 1985). Under direct summer
sunlight, the half-life of PCP was about 8 min (12 mg PCP/litre
with 2 TiO2/litre, pH 3). Under laboratory conditions, the half-
life was 15 min at 45 - 50 °C, pH 10.5, and lambda > 330 nm;
mineralization of PCP was more than 95%.
Na-PCP, like its parent compound, is readily photolysed. Hiatt
et al. (1960) investigated photodecomposition of Na-PCP as a
possible factor reducing its efficacy as a molluscicide in South
African streamwaters. In vitro exposure to UV radiation (290 - 330
nm) caused chemical degradation of Na-PCP, which was directly
proportional to light intensity, and a corresponding loss of
molluscicidal activity. Similarly, Na-PCP applied to rice paddies
in order to control barnyard grass ( Panicum crusgalli L.) was
readily decomposed by sunlight (Kuwahara et al., 1966a). Aqueous
solutions of Na-PCP exposed to sunlight broke down to form mainly
chloroanilic acid and 3,4,5-trichloro-6-(2'-hydroxy-3',4',5',6'-
tetrachlorophenoxy)- o-benzoquinone (Kuwahara et al., 1966a), and
minor amounts of tetrachlororesorcinol and three benzoquinones
(Kuwahara et al., 1966a,b; Munakata & Kuwahara, 1969).
Field evidence indicates that photolysis is an important means
of PCP loss in situ. In their study of PCP contamination in the Bay
of Quinte, Lake Ontario, Fox & Joshi (1984) found the proportions
of 2,3,4,6- and 2,3,5,6-tetrachlorophenol in environmental samples
to be enriched relative to PCP. Since these compounds are products
of the photodegradation of PCP, they suggested that photolysis was
dominating PCP breakdown. Similarly, Yunker (1981) concluded that
photolysis was the most important pathway for the removal of
pentachlorophenate from enclosed marine pelagic enclosures off the
west coast of Canada.
Crossland & Wolf (1985) found evidence that direct photo-
transformation was mainly responsible for the loss of PCP from
experimental outdoor ponds (50 - 100 µg PCP/litre). They observed
half-lives for the loss of PCP in the range of 2 - 4.7 days (pH 7.3
- 10.3; 10 - 21 °C); decomposition was most rapid in relatively
clear water.
The formation of PCDDs as a result of photochemical reaction
has been described. Crosby & Wong (1976) irradiated Na-PCP in
aqueous solution. Traces of OCDD were found, while 2,3,7,8-T4CDD
was not detectable, presumably because of its rapid photoreduction
(Crosby et al., 1971). The photolysis of OCDD yields a variety of
chlorinated dibenzodioxins with decreasing numbers of chlorine
atoms (Crosby et al., 1971, 1973; Buser, 1976).
This is consistent with the findings of Lamparski et al.
(1980), who observed H6CDD and H7CDD in the course of photolysis
studies with wood samples treated with PCP in methylene chloride.
The OCDD content increased from the initial concentration of 3 mg
OCDD/kg Dowicide EC-7 (containing low initial PCDD concentrations)
or non-detectable amounts of OCDD/kg purified PCP (Aldrich Chemical
Company), respectively, to yield up to about 70 mg OCDD/kg PCP on
the surface of wood in both PCP specifications, while controls
stored in the dark showed no increase in OCDD. Use of a
hydrocarbon oil as a solvent significantly reduced OCDD formation
during irradiation to yield 4.4 mg OCDD/kg Dowicide EC-7 or 2.2 mg
OCDD/kg purified PCP, respectively. Moreover, OCDD concentrations
only slightly increased in wood samples treated with a technical
PCP containing relatively high initial PCDD concentrations (OCDD -
1100 mg/kg).
These results agree with those reported by Cull & Dobbs (1984),
who found no evidence of the formation of OCDD in technical PCP
(solvent - hydrocarbon oil) or Na-PCP (solvent - water). This has
been attributed to the photolytic destruction and volatilisation
of OCDD dominating its formation when the initial OCDD
concentration is relatively high.
4.2.2. Microbial degradation
The microbial degradation of PCP has been studied using natural
and artificial media with mixed or single microbial cultures. Lyr
(1963) demonstrated that fungi were able to attack PCP by means of
phenol oxidase. Cserjesi (1967) observed PCP decomposition by
fungi of the genus Trichoderma in malt extract solution at a
concentration of approximately 10 mg/litre. Similar studies have
also been carried out with a number of other fungal species (Duncan
& Deverall, 1964; Cserjesi, 1972).
Numerous PCP degrading bacterial strains, which are partly
capable of using PCP as a sole source of organic carbon, have been
isolated (Chu & Kirsch, 1972; Kirsch & Etzel, 1973; Watanabe, 1973;
Suzuki, 1977; Reiner et al., 1978; Edgehill, 1982; Stanlake & Finn,
1982; Trevors, 1982a).
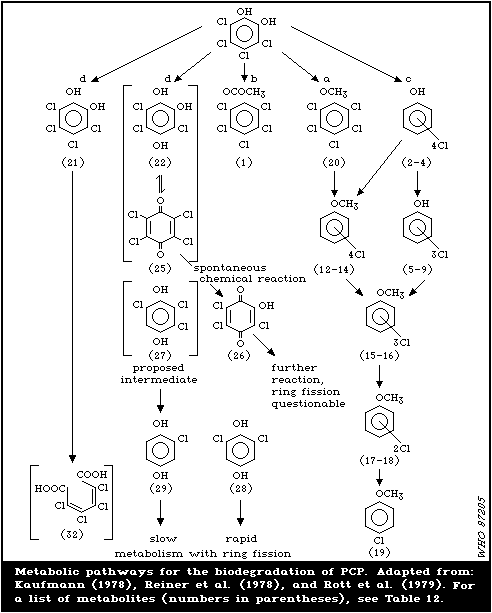 Several pathways of PCP degradation have been suggested.
Because of the tremendous number of microbial strains, numerous
metabolites have been identified as degradation products (Table
12). The possible steps of PCP decomposition as reported by
several authors are summarized in Fig. 3. The major metabolic
processes degrading PCP or its sodium salt are as follows (Suzuki,
1977; Kaufman, 1978; Reiner at al., 1978; Murthy et al., 1979; Rott
et al., 1979):
(a) methylation to yield the methylether of PCP,
pentachloroanisole;
(b) acylation of the hydroxyl group resulting in
pentachlorophenol acetate;
(c) dechlorination to tetrachlorophenols; and
(d) hydroxylation to tetrachlorodihydroxybenzenes.
The metabolites originating from these initial steps are subject to
further transformations as depicted in Fig. 3. Thus, a number of
substances may arise, which accumulate to different extents; the
limiting step is the ring fission to chlorinated aliphatic
compounds such as tetrachloromuconic acid (Lyr, 1962). Further
dechlorination may result in further transformations of the
aliphatic compounds (Janke & Fritsche, 1978) to form low molecular
substances such as acetic acid or succinate, which then enter the
tricarboxylic acid cycle. Suzuki (1983) reported that 34% of the
14C resulting from 14C-PCP microbial decomposition was recovered as
14CO2, the remaining compounds being unidentified 14C-metabolites.
Table 12. Metabolites formed by the microbial transformation of PCPa
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(1) pentachlorophenol acetate Rott et al. (1979)
(2) 2,3,4,5-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975); Murthy et al.
(1979)
(3) 2,3,5,6-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975); Murthy et al.
(1979)
(4) 2,3,4,6-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(5) 2,4,5-trichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(6) 2,3,6-trichlorophenol Kuwatsuka & Igarashi (1975);
Murthy et al. (1979)
(7) 2,3,4-trichlorophenol Kuwatsuka & Igarashi (1975)
(8) 2,3,5-trichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(9) 2,4,6-trichlorophenol Kuwatsuka & Igarashi (1975)
(10) 3,4-dichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(11) 3,5-dichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
---------------------------------------------------------------------------
Table 12. (contd.)
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(12) 2,3,4,5-tetrachloroanisole Ide et al. (1972); Rott et al.
(acetate) (1979)b
(13) 2,3,5,6-tetrachloroanisole Ide et al. (1972); Rott et al.
(acetate) (1979)b
(14) 2,3,4,6-tetrachloroanisole Engel et al. (1966); Ide et al.
(acetate) (1972); Rott et al. (1979)b
(15) 2,3,5-trichloroanisole Ide et al. (1972)
(16) 2,4,5-trichloroanisole Ide et al. (1972)
(17) 3,4-dichloroanisole Ide et al. (1972)
(18) 3,5-dichloroanisole Ide et al. (1972)
(19) 3-chloroanisole Ide et al. (1972)
(20) pentachloroanisole Cserjesi & Johnson (1972); Ide et
al. (1972); Kuwatsuka & Igarashi
(1975); Murthy et al. (1979);
Rott et al. (1979)
(21) tetrachlorocatechol Suzuki (1977); (Rott et al.
(diacetate) (1979)b
(22) tetrachlorohydroquinone Suzuki (1977)
(23) tetrachlororesorcinol Rott et al. (1979)b
(diacetate)
(24) tetrachlorohydroquinone Rott et al. (1979)
dimethylether (diacetate)
(25) tetrachlorobenzoquinone Reiner et al. (1978)
(26) trichlorohydroxybenzoquinone Reiner et al. (1978)
(27) 2,3,6-trichlorohydroquinone Reiner et al. (1978)
(28) 2,6-dichlorohydroquinone Reiner et al. (1978)
(29) 2-chlorohydroquinone Reiner et al. (1978)
(30) 14CO2 Chu & Kirsch (1972); Kirsch &
Etzel (1973); Suzuki (1977)
---------------------------------------------------------------------------
Table 12. (contd.)
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(31) Cl- Watanabe (1973); Suzuki (1977)
(32) tetrachloromuconic acid Lyr (1962)
(33) beta-hydroxytrichloromuconic Lyr (1962)
acid
---------------------------------------------------------------------------
a Adapted from: Kaufman (1978).
b Reference refers to acetate form.
4.2.2.1 Aquatic degradation
Boyle et al. (1980) studied the effects of dissolved oxygen
supply, light, pH, and the presence of hydrosoil on the aquatic
biodegradation of PCP. Light, high pH, and high oxygen
concentrations led to the most rapid and complete breakdown of PCP.
PCP disappeared from the pond water of aquaria at differential
rates, with half-lives ranging from 12.8 to 18.6 days, except for
the anaerobic aquarium without hydrosoil (79.8 days). The absence
of light and mud associated with low pH and anaerobic conditions
favoured the persistence of PCP, indicating that the phenolic form
of PCP is more persistent than the phenate form and that the
oxidative pathway is the major mechanism for PCP degradation in
simulated lake environments. Thus, PCP may be most persistent in
the deoxygenated hypolimnion of stratified lakes.
This effect of oxygen on biotransformation was confirmed by Liu
et al. (1981), who calculated the half-lives of PCP in aerobic and
anaerobic metabolic fermentors to be 0.36 and 192 days,
respectively. These half-lives are much lower than those reported
by Boyle et al. (1980), probably because Liu et al. (1981) used an
acclimatized culture whereas in the other studies natural pond
water was employed. This illustrates how important the
preadaptation of microorganisms is for their capacity to degrade
PCP.
Pignatello et al. (1983) showed that the aquatic micro-flora
can adapt to PCP and can become the most important factor for
clearing contaminated surface water of PCP, particularly in deeper
waters where the photolytic contribution is minimized.
Liu et al. (1981) investigated the impact of co-metabolites
and of different nitrogen sources on PCP biodegradability.
Peptone, glucose, and the sodium salt of 4-chlorophenol suppressed
the degradation rate while yeast extracts stimulated PCP
decomposition; the basis for these various effects is not clear.
In a report by Pierce & Victor (1978), levels of PCP in a man-
made lake in Mississippi increased from background levels (0.3
µg/litre) to 150 - 277 µg/litre, immediately after an accidental
overflow from a pole-treatment plant, decreasing to 5 - 16 µg/litre
within 4 months. Long-term influx of PCP from the contaminated
watershed and persistence in organically rich sediments (4 - 1518
µg/kg dry weight) were thought to provide a source of long-term
pollution of the aquatic ecosystem (sections 5.1.4 and 6.5.1).
Since anaerobic biodegradation of PCP is relatively slow, PCP
persists in sediments considerably longer than in water.
Accordingly, high concentrations of PCP have been measured in river
and lake sediments (section 5.1.2).
The fate of PCP in estuarine sediments was investigated
following contamination with 11 tonnes of PCP after a ship
collision (De Laune et al., 1983). Although the contaminated
sediments had been removed by vacuuming 2 weeks after the accident,
as much as 1.6 mg/kg (dry weight) could be detected 1 1/2 years
later compared with < 5 µg/kg at remote sampling points.
Laboratory studies showed a much faster breakdown under aerobic
conditions (70% decomposition at pH 8 within 5 weeks) compared with
anaerobic conditions (10% decomposition).
The high persistence of PCP in the sediment has been confirmed
by studies on the fate of PCP in a bay of Lake Ontario (Canada),
which has been contaminated by a wood-preserving plant since the
1940s (Fox & Joshi, 1984). The rather constant T4CP:PCP ratio
throughout the core (depth of 15 cm corresponding to the year 1949)
indicates that almost no degradation of these chlorophenols
occurred, once they had been incorporated into sediments.
Of the possible PCP metabolites, tetrachlorocatechol seems to
be among the most persistent. Compared with all other chlorinated
phenols studied, this substance showed the highest levels in the
sediment (3.2 - 348 µg/kg dry weight) or plankton (108 µg/kg wet
weight) of Finnish lakes contaminated with wood preservatives and
chlorobleaching wastes (Paasivirta et al., 1980).
4.2.2.2 Degradation in soil
As in water, the biodegradability of PCP in soil depends on the
type and the physiological state of the microorganisms, but several
environmental factors also influence this process, such as a high
amount of organic matter or high moisture content, which enhance
the biodegradation of PCP (Young & Carrol, 1951; Kuwatsuka, 1972).
Low temperatures (O °C, 4 °C) have proved unfavourable for the
growth of Pseudomonas species and hence their PCP breakdown
activity, while, at 20 °C, PCP (50 mg/litre) was degraded to about
50% within 8 days (Trevors, 1982a). Low pH values also reduced the
microbial breakdown of PCP (Stanlake & Finn, 1982).
The effects of oxygen on biodegradation vary: in some
instances, anaerobic conditions in soils increase the rate of
degradation, apparently as reducing conditions promote reductive
dechlorination (Ide et al., 1972). On the other hand,
biodegradation in terms of formation of the intermediate
pentachloroanisole was significantly greater in aerobic than in
anaerobic soil (Murthy et al., 1979). An average loss of 88% of
100 mg PCP/kg from clay soil was detected under aerobic conditions
(over 160 days at 23 °C) compared with only 7% loss under anaerobic
conditions (Baker & Mayfield, 1980).
The half-life of PCP in moist farm soils (initial
concentration, 100 mg/kg) ranged from 7 to 14 days, depending on
the soil varieties. PCP degradation was inhibited by certain
fungicides and also under submerged conditions (Suzuki, 1983).
Other half-lives reported are 10 - 70 days, under flooded
conditions, and 20 - 120 days under upland conditions (Kaufman,
1978).
Edgehill & Finn (1983) investigated the use of PCP-degrading
bacteria as a prophylactic measure to decontaminate soil after
accidental PCP spills or when PCP-treated poles are set up near
surface waters. Direct inoculation of acclimated Arthrobacter
cells into the soil enhanced the disappearance of PCP at least 10-
fold; the half-life of PCP in the soil incubated at 30 °C in the
laboratory was reduced from 12 - 14 days to about 1 day. However,
outdoor trials demonstrated that the efficacy of this measure was
limited under natural conditions, as thorough mixing of the soil at
the time of inoculation was required to achieve a 85% reduction of
the extractable PCP at 12 days compared with only 15 - 30% without
mixing.
4.3 Degradation by Plants
Degradation of PCP may also take place in plants. Rice plants
were found to absorb about 3% of the radioactivity of 14C-PCP
applied on the soil, of which 50% could be extracted, mainly as
unchanged PCP, 9% as unidentified conjugates, and about 1% as a
tetrachlorophenol-isomer (Haque et al., 1978).
Weiss et al. (1982a) also studied the metabolism of 14C-PCP
(23 kg/ha) in rice plants. Rice roots contained 0.14% of the
applied radioactivity as unchanged PCP, 3.95% as unextractable
residues, and 1.08% as various metabolites. The influence of soil
microorganisms was not excluded, but the authors regarded the
detection of hydroxylated and methoxylated tetrachlorobenzenes as
evidence of plant metabolism, because these substances were not
found in soil (Weiss et al., 1982a,b).
The metabolism of PCP was also investigated under aseptic
conditions using soybean ( Glycine max L.) and wheat ( Triticum
aestivum L.) cell suspension cultures in which the isolated
conversion products could be attributed to the metabolic activity
of the plant cell cultures themselves (Langebartels & Harms, 1984).
The beta-D-glucoside of PCP was identified as the main conjugate
formed by the cell suspensions. Anisoles and lower chlorinated
phenols were not detected; however, some PCP was incorporated into
a non-extractable fraction. Sandermann et al. (1984) isolated
polar conjugates from wheat and soybean cell cultures, and
demonstrated that covalent incorporation of PCP into lignin takes
place.
4.4 Ultimate Fate Following Use
4.4.1 General aspects
The amount of PCP entering the environment and its subsequent
fate can be controlled at point sources where high amounts of PCP
are used, such as preservation plants. However, because of its many
applications, PCP is released into the environment from a number of
diffuse sources and is subject to transport and transformation in
different environmental compartments, as outlined in the previous
sections. The evaporation data cited above (section 4.1.1) suggest
that a significant fraction of the entire production of PCP will
ultimately enter the atmosphere.
4.4.2 Disposal of waste water
As shown in Table 13, municipal sewage discharges contain only
low PCP concentrations, whereas effluents from wood-treating
factories may contain considerable amounts of PCP, depending on the
intensity and efficacy of treatment measures prior to discharge.
One method of handling wood preservation effluents in Canada is to
store waste water on company property and allow it to evaporate,
which obviously contributes to air pollution. The disposal of
waste water is also achieved by incineration (section 4.4.3) and by
secondary treatment before discharge into the receiving water
(Hoos, 1978).
Most small wood-treatment plants handle wastes by incineration
or lagooning, while larger manufacturers treat their wastes.
Primary treatment is often applied when PCP is dissolved in a
carrier oil: gravity separation is used to recover oil and PCP for
recycling or treatment, while some plants remove oil droplets or
wood particles by filtration (Richardson, 1978).
Several laboratory and treatment-plant studies have shown that
PCP can be degraded by activated sludge (Dust & Thompson, 1973;
Kirsch & Etzel, 1973; Etzel & Kirsch, 1974; Moos et al., 1983;
Guthrie et al., 1984; Hickman & Novak, 1984). However, in full-
scale treatment plants, the treatment efficiency is often reduced.
For example, according to a US EPA survey, 8 out of 14 publicly-
owned treatment plants could not remove any of the PCP load. Most
of the removal efficiency of the remaining plants (6 - 87%) was
attributed to adsorption on solids (Feiler, 1980).
Biodegradability strikingly decreases when commercial PCP is
introduced unless the input concentrations are reduced (Reiner et
al., 1978). In the presence of more readily degradable substrates,
PCP degradation is suppressed. Moreover, activated sludge is not
usually protected from shock loads, unless acclimatized to PCP
(Hickman & Novak, 1984).
Some other treatment systems appear to be appropriate for the
treatment of PCP-contaminated waste water, but their suitability
has not as yet been demonstrated on a large scale. Degradation of
PCP in a biofilm reactor only occurred when the microflora was
attached to solid support material, such as soft-wood bark
(Apajalahti & Salkinoja-Salonen, 1984; Salkinoja-Salonen et al.,
1983, 1984). PCP concentrations exceeding 1 mmol (266 mg/litre),
and also some toxic solvents, such as chloroform, inhibited PCP
biodegradation.
Table 13. Levels of PCP in industrial and municipal discharges in
different countries
---------------------------------------------------------------------------
Country Type of waste water PCP Reference
(µg/litre)
---------------------------------------------------------------------------
Canada Effluents from wood 0.6 Environment
preservation industry at 225 Canada (1979)
4 sites in British NDa
Columbia 2760
Canada Industrial and municipal Garrett (1980)
discharges in the Greater
Vancouver Area:
- average from 22 sites 5.1
- range (0.2 - 42.5)
- drainage ditches 6000
2520
1125
Denmark Municipal sewage Folke & Lund
- influents 0.2 - 0.7 (1983)
- effluents 0.1 - 2.4
Germany, Effluents from sewage 20 - 680 Dietz &
Federal treatment plant receiving Traud (1978b)
Republic waste water from paper mill
of
Effluents from various 1 - 130
industries
USA Municipal sewage Buhler et al.
- influents 1.4 - 4.6 (1973)
- effluents 1 - 4.4
USA Samples from wood-treating Ervin &
factories McGinnis
- untreated waste water 17 000 - 32 000 (1980)
- treated waste water 160 - 75 000
USA Effluents from wood- 25 000 - 15 0000 Thompson &
treatment factories Dust (1971)
---------------------------------------------------------------------------
a ND = not detectable.
Hakulinen & Salkinoja-Salonen (1982) reported on the efficiency
of a fluidized bed reactor in removing chlorophenols from pulp and
paper industry bleaching effluents. Chlorophenols including PCP
were completely mineralized in the anaerobic reactor. The aerobic
part of the system served as an after treatment unit to remove the
remaining organic load.
Adsorption on to activated carbon has also been used in
treating contaminated waste waters; removal of PCP approaches 100%
using this method (Richardson, 1978).
4.4.3 Incineration of wastes
As considerably increased amounts of PCDDs can be emitted
during the combustion of PCP-treated material compared with
untreated samples (section 2.2.1), the incineration of PCP-
containing wastes is problematical. Since temperature of burning
and the residence time cannot be controlled in the fire-places of
private homes, the incineration of wood treated with chlorophenols
is a potential source of PCDD/PCDF emission. Moreover, accidental
burning of chlorophenols can lead to considerable emissions of
these compounds: Kauppinen & Lindroos (1985) estimated that the
burning of 100 kg chlorophenol formulation during a saw-mill fire
would result in 20 g of PCDDs.
According to Powers (1976), "the complete and controlled high
temperature oxidation coupled with adequate scrubbing and ash
disposal facilities offers the greatest immediate potential for the
safe disposal of concentrated pentachlorophenol". The destruction
of PCP in treated wood in a controlled air incinerator was achieved
with efficiencies greater than 99.99% at combustion temperatures of
between 916 and 1032 °C (Stretz & Vavruska, 1984). The analytical
results showed no evidence of T4CDD or T4CDF, both in the hot zone
between primary and secondary chambers and in the offgas, at
detection limits of 1 ppb or 5 ppb, respectively.
There are many other sources of PCDDs and PCDFs from
combustion processes. The incineration of municipal waste may be
the largest source of PCDD and PCDF emissions into the environment
(Ballschmiter et al., 1983; Chiu et al., 1983; Tiernan et al.,
1983). The various sources of these compounds are discussed in
more detail in the corresponding reviews on PCDDs and PCDFs, e.g.,
Umweltbundesamt (1985), Karasek & Hutzinger (1986), and in
Boddington et al. (1985).
The thermal conversion of organochlorine compounds, e.g.,
polyvinylchloride and polyvinylidene chloride, can be a source of
atmospheric chlorophenols including PCP (Ahlborg et al., 1986;
Dougherty, 1986a).
Other common methods of waste disposal such as deep-sea or
deep-well disposal, landfill sites, or open pits should not be
considered as a means for disposing of PCP-containing wastes,
because of the mobility of PCP (Powers, 1976; Crosby et al., 1981).
---------------------------------------------------------------------------
a Personal communication to the Task Group on Pentachlorophenol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental Levels
5.1.1. Air
While PCP concentrations in the air at industrial sites and in
rooms contaminated with PCP have been reported (section 5.2), there
is apparently little information on PCP levels in the ambient air.
Cautreels et al. (1977) sampled airborne particulate matter
near La Paz, Bolivia, at an altitude of 5200 m and in a residential
city area of Antwerp, Belgium. At a detection limit of 0.02 ng/m3,
the atmosphere of the Bolivian mountain rural area contained 0.25 -
0.93 ng/m3 and that of the Antwerp urban area 5.7 - 7.8 ng/m3 air,
respectively. More recent analytical results (Bundesamt für
Umweltschutz, 1983) showed PCP air concentrations ranging between
0.9 and 5.1 ng/m3 in Switzerland.
The ubiquitous occurrence of PCP in ambient air can also be
shown from rain water and snow analyses. Rain water collected in
Canada (Jones, 1981), Hawaii (Bevenue et al., 1972) and West Berlin
(Rosskamp, 1982) contained between 0.002 and 0.3 µg PCP/litre.
Water melted from snow in southern Finland revealed PCP
concentrations of 0.15 and 0.98 µg/litre, respectively. PCP
fallout as calculated from Finnish snow samples ranged from 1.49 to
136.0 µg/m2 (Paasivirta et al., 1985).
5.1.2. Water and sediments
Levels of industrial and municipal discharges in different
countries are shown in Table 13 (section 4.4.2). Municipal sewage
discharges contain PCP concentrations at levels comparable with
those in surface waters. However, wood-treating factories may
contribute substantially to the PCP load in surface waters, which
ranges from non detectable to 10 500 µg PCP/litre (Table 14),
depending on the extent of pollution by different sources.
The majority of the water samples analysed for PCP contained
less than 10 µg/litre, most contained less than 1 µ PCP/litre. The
extreme PCP levels of up to 10 500 µg/litre reported by Fountaine
et al. (1976) were found in a highly polluted stream near an
industrial area in the vicinity of Philadelphia, USA.
Ernst & Weber (1978b) calculated the PCP input into the German
Bight via the river Weser to be of the order of 1000 kg per year,
assuming an average PCP level of 0.1 µg PCP/litre and a water flow
of 300 m3/second. Taking an average concentration of 0.5 µg
PCP/litre in the surface waters of the Federal Republic of Germany
(Foquet & Theisen, 1981), the total load in all surface waters of
the Federal Republic of Germany was estimated by Fischer (1983) to
be in the range of 60 tonnes per year, of which 30 - 40 tonnes are
transported by the Rhine river.
Table 14. PCP concentrations in surface waters of different countries
---------------------------------------------------------------------------
Country Surface water PCP (µg/litre) Reference
and location (range, mean)
---------------------------------------------------------------------------
Canada Fresh-water sites in trace - 0.30 Environment,
British Columbia (BC) Canada (1979)
Marine sites in BC NDa - 7.3
Germany, Weser river and estuary 0.05 - 0.5 Ernst & Weber
Federal (1978b)
Republic German Bight < 0.002 - 0.026
of
Ruhr river < 0.1 - 0.2 Dietz & Traud
(0.1) (1978b)
Rhine river, Cologne 0.1 Fischer & Slem-
rova (1978)
Japan Tama river, Tokyo 0.1 - 0.9 Matsumoto et
0.01 - 0.09 al. (1977)
Sumida river, Tokyo 1 - 9
River water, Tokyo 0.18 ± 0.14 Matsumoto
area (1982)
Nether- Rhine river 1976 Max.b 2.4 (0.7) Wegman &
lands Rhine river 1977 Max. 11.0 (1.1) Hofstee (1979)
River Meuse 1976 Max. 1.4 (0.3)
River Meuse 1977 Max. 10.0 (0.8)
South 124 sampling points ND - 0.85 Van Rensburg
Africa (1981)
Sweden River water downstream 9 Rudling (1970)
from pulp mill
Lake receiving 3
discharges
---------------------------------------------------------------------------
Table 14. (contd.)
---------------------------------------------------------------------------
Country Surface water PCP (µg/litre) Reference
and location (range, mean)
---------------------------------------------------------------------------
USA Willamette river 0.1 - 0.7 Buhler et al.
(1973)
Highly polluted stream Fountaine et
near Philadelphia al. (1976)
- factory location 4500 - 10 500
- downstream 49 - 240
Estuary in the ND - 0.01 Murray et al.
Galveston Bay, Texas (1981)
Pond in Mississippi < 1 - 82 Pierce et al.
contaminated by waste (1977)
from pole-treatment
plant
---------------------------------------------------------------------------
a ND = not detectable.
b Max. = maximum values.
Wong & Crosby (1981) reported PCP concentrations ranging from 1
to 800 µg/litre (average, 227 µg/litre) in the surface pond water
near a local wood-treatment factory and of about 20 µg PCP/litre in
agricultural drainage. Elevated PCP concentrations were also found
in groundwater (3.03 - 23.3 µg/litre) and surface water samples
(0.07 - 31.9 µg/litre) within saw-mill areas. Around these sites,
PCP levels ranged between not detectable and 0.6 µg/litre in
groundwater and between 0.01 and 0.07 µg/litre in the water of a
nearby lake (Valo et al., 1984). PCP in the µg/litre range was
detected in the water seeping from a landfill (Kotzias et al.,
1975). A level as high as 3.35 mg/litre was found in groundwater
from a monitoring well near a wood-preservation factory (Thompson
et al., 1978).
A PCP-monitoring study in water was performed by Rahde & Della
Rosa (1984, 1986) in a region of the Amazon jungle (Tucurui,
Brazil). The construction of a dammed reservoir affected a large
area (2430 km2) with sawmills and PCP-treated wood. Water samples
collected from the main river and its affluents before the flooding
in 1984 contained between 5 and 14 µg PCP/litre. In 1984-85, after
the flooding, the area had been covered with about 46 billions m3
of water, PCP was not detectable at a detection limit of 4 µg/litre.
In general, the sediments of a water body contain much higher
levels of PCP than the overlying waters. At several fresh-water
and marine sites in British Columbia, Canada, receiving effluents
from the wood-treatment industry, average PCP levels in the
sediments ranged from not detectable to 590 µg/kg, while the
corresponding range in the overlying waters was from not detectable
to 7.3 µg/litre (Table 14). During a 1978 survey of toxic
substances in the Great Lakes of Canada, sediment samples from the
Thunder Bay, Marathon, and Michipicoten areas of Lake Superior
contained averages of 16 900, 7300, and 2300 µg PCP/kg dry
sediment, respectively. In another study of contamination from a
wood preservation facility on the Bay of Quinte, Lake Ontario, Fox
& Joshi (1984) analysed water and sediment samples for PCP. At a
site distant from the plant discharge, sediment PCP levels ranged
from 1 to 61 µg/kg dry weight, while surface waters contained only
0.015 µg/litre. Sediments from the Mississippi lake monitored by
Pierce & Victor (1978) averaged 364 µg/kg dry sediment, compared
with levels in the lake water of only 0.1 µg/litre. A similar
distribution was observed in surface waters in the Netherlands
(Wegman & van den Broek, 1983); sediment samples from Lake
Ketelmeer, a deposition area for Rhine river sediments, contained a
median PCP concentration of 8.4 µg/kg dry weight, while the
overlying water contained 0.41 µg/litre. PCP concentrations in
sediment samples collected in the vicinity of a paper mill
discharge pipe in a North Sea bight, two years after going out of
use (Butte et al., 1985) and in Finnish lakes contaminated by wood
preservatives (Paasivirta et al., 1980) were of the same order of
magnitude.
These examples indicate that PCP and Na-PCP adsorb on
sediments, which concurs with findings from experimental work.
Strufe (1968) reported a study in which 65% of added Na-PCP
adsorbed on river mud within 20 h.
5.1.3. Soil
Soil samples, taken at 4 sites in the vicinity of a Swiss PCP-
producing facility (Dynamit Nobel), contained between 25 and 140 µg
per kg (dry weight) at a depth of 0 - 10 cm and between 33 and 184
µg/kg at 20 - 30 cm. These levels are higher than the PCP
concentrations of 35 µg/kg (0 - 10 cm) and 26 µg/kg (20 - 30 cm)
determined in soil samples from a "reference site". The
simultaneous presence of some PCDDs and PCDFs (maximum values:
H7CDD, 0.6 µg/kg; OCDD, 7.68 µg/kg; P5CDF, 1 µg/kg at 0 - 10 cm) in
sample sites near the chemical factory compared to only one
positive sample (H7CDF, 0.51 µg/kg) at the remote site confirmed
the contamination (Bundesamt für Umweltschutz, 1983).
The soil surrounding Finnish sawmills was found to be heavily
contaminated with up to 45.6 mg/kg (0 - 5 cm) or 1 mg PCP/kg fresh
weight (80 - 100 cm) near the treatment basin, up to 0.14 mg/kg in
the storage area for preserved wood and 0.012 mg/kg outside the
storage area. The vertical distribution of chlorophenols including
PCP explains the ground-water contamination observed (Valo et al.,
1984).
In Canada, soil samples from the former site of a pesticide
plant contained less than 50 µg PCP/kg (Garrett, 1980). The PCP
levels in the leachate and in soil in the vicinity of 3 waste-
disposal sites were also in the µg/kg range (Kotzias et al., 1975).
Samples of agriculturally used soils in Bavaria (Federal Republic
of Germany) contained about 100 µg PCP/kg (Gebefuegi, 1981).
PCP concentrations in soil samples taken at a distance of 2.5,
30.5, and 152.5 cm from poles treated with PCP were 658, 3.4, and
0.26 mg/kg, respectively. Arsenault (1976) considered the last
value as a "natural background level", which he derived from the
blank of 0.2 - 0.4 ppm found in unexposed soil samples. However,
such a level seems very high for a substance that does not appear
to occur naturally. This high level could be the result of the
contamination of the soil or of the reagents used for analysis.
5.1.4. Aquatic and terrestrial organisms
5.1.4.1 Aquatic organisms
Levels of PCP in aquatic organisms from various collection
sites are listed in Table 15. No data are available on the
background levels of PCP in biota. All sampling sites in Table 15
were more or less contaminated with industrial effluents.
Relatively low contamination is reflected by residues of PCP in
aquatic invertebrate and vertebrate fauna in the low µg/kg-range.
For example, Zitko et al. (1974) found a range of < 0.5 - 4 µg
PCP/kg wet weight in the muscle tissue of different fish species
(Table 15). Higher levels were detected in organisms collected in
surface waters that were thought to be contaminated with wood
preservatives: up to 2100 µg PCP/kg wet weight were found in marine
fish in British Columbia, Canada (Environment Canada, 1979) and up
to 6400 µg/kg in fresh-water fish from Finnish lakes (Paasivirta et
al., 1981) (Table 15). Some sediment-dwelling organisms showed the
highest residues: polychaetes from the Weser estuary contained
between 103 - 339 µg PCP/kg wet weight (Ernst & Weber, 1978a).
Even higher levels (266 - 133 000 µg/kg) were found in clams from a
North Sea bight, near the end of a waste-water pipe from which
about 26 tonnes of PCP were discharged into the mud flats until
1978 (Butte et al., 1985).
Residues of PCP in biota associated with toxic PCP water
concentrations are in the mg/kg range. Following extensive
application of Na-PCP as a molluscicide in rice fields in Surinam,
Vermeer et al. (1974) found 8.1 mg PCP/kg wet weight in dead frogs
(Pseudis paradoxa) and between 31.2 and 59.4 mg/kg in three species
of fish, which were also found dead. Composite samples of snails
(Pomacea glauca) contained, on average, 36.8 mg PCP/kg wet weight.
Whole samples of small fish collected from a river in British
Columbia, Canada, during an accidental fish kill resulting from the
spraying of hydropoles, had levels of 16.3 mg PCP/kg; two large
cutthroat trout (Salmo clarki) contained 10.3 mg/kg (Jones, 1981).
Table 15. PCP residues in aquatic animalsa
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Invertebrates
Jellyfish whole Gulf of Mexico 1979 0.1 - 1 wet Kuehl & Dougherty (1980)
Sponge whole Finnish lakes Summer 1.9 - 13 wet Paasivirta et al. (1980)
contaminated with 1978
wood preservatives
Sagartia whole Weser estuary and 1976-77 2.7 - 7 wet Ernst & Weber (1978a)
troglodytes German Bight
(actinian)
Polychaete whole 1978 103 - 339 wet
(Lanice
conchilega)
Mussel muscle Finnish lakes Summer 1.7 - 5.6 wet Paasivirta et al. (1980)
contaminated with 1978
wood preservatives
Clam muscle Marine sites near Autumn ND - 12 wet Environment Canada
( Macoma sp.) wood-preservation 1978 (1979)
factories in British
Columbia, Canada
Clam whole Wadden sediment of 1980-81 266 - 133 000 dry Butte et al. (1985)
(Mya (without Jadebusen, bight of (median: 800)
arenaria) shells) the North Sea, PCP
discharged area until
1978
Reference site 266 - 532
Crayfish muscle Fresh-water sites Autumn ND - trace wet Environment Canada
( Pacifas- near wood-preserv- 1978 (1979)
tacus sp.) ation factories in
British Columbia,
Canada
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Crab muscle Marine sites near Autumn ND - 20 wet Environment Canada
(Cancer wood-preservation 1978 (1979)
magister) factories in
British Columbia,
Canada
Crab muscle Marine sites near Autumn trace - 7 wet
(Cancer wood-preservation 1978
productus) factories in
British Columbia,
Canada
Brown shrimp whole Estuary of the 1980 4 - 17 wet Murray et al. (1981)
(Penaeus Galveston Bay area
aztecus) of Texas
Blue crab soft 1.9 - 4.1 wet
(Calinectes tissues
sapidus)
Dwarf squid whole 1.4 - 4.3 wet
(Lollingnuula
brevis)
Vertebrates
Sculpin muscle Marine sites near Autumn trace - 84 wet Environment Canada
(marine) liver wood-preservation 1978 trace - 2100 wet (1979)
(Leptocottus factories in
armatus) British Columbia,
Canada
Sculpin muscle Fresh-water sites 5 - 100 wet
(freshwater) liver near wood-preserv- trace - 600 wet
(Cottus ation factories in
asper) British Columbia,
Canada
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Pike muscle Finnish lakes Summer 6.5 - 8 wet Paasivirta et al. (1980)
(Esox contaminated with 1978
lucius) wood preservatives
Roach muscle 0.9 - 12.8 wet
(Rutilus
rutilus)
Pike muscle Finnish lakes May 11.9 - 94.3 wet Paasivirta et al. (1981)
(Esox contaminated with 1980 (maximum 6400)
lucius) wood preservatives
Pike muscle Finnish lakes Spring/ 15.9 - 18.9 wet Paasivirta et al. (1983)
(Esox contaminated with Summer
lucius) wood preservatives 1981
Pike muscle Finnish lakes Summer 8.2 - 17.3 wet Paasivirta et al. (1985)
(Esox contaminated with 1982
lucius) wood preservatives
1983 1.2 wet
Crab muscle 1982 9.4 - 41.5 wet
(Rutilus 1983 0.5 wet
rutilus)
Baltic muscle 1983 1.8 - 4.7 wet
salmon
Winter muscle Estuaries in New Autumn 1.8 - 4 wet Zitko et al. (1974)
flounder Brunswick, Canada 1972
(Pseudopleuro-
nectus)
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Cod muscle 0.8 wet
(Gadus
morhua)
Sea raven muscle < 0.5 wet
(Hemipterus
americanus)
Atlantic whole Estuaries in New Autumn 0.5 - 1.3 wet Zitko et al. (1974)
salmon Brunswick, Canada 1972
(Salmo salar)
White shark liver 10.8 wet
(Carcharodon
carcharias)
Flounder whole Estuary in the 1980 1.6 - 3.5 wet Murray et al. (1981)
Galveston Bay
Longnose whole 4.7 - 5.6 wet
killifish
(Fundulus
similis)
---------------------------------------------------------------------------------------------------------
a ND = not detectable.
5.1.4.2 Terrestrial organisms
As with aquatic plants, almost no data are available on
residues of PCP in terrestrial plants. Grass samples taken in the
vicinity of a PCP producer at Rheinfelden, Switzerland, contained
between 67 - 87 µg PCP/kg dry weight, comparable to the PCP
concentration of 87 µg/kg found in grass from a reference site
(Bundesamt für Umweltschutz, 1983).
Reported residue levels in terrestrial vertebrates are mainly
related to domestic animals exposed to PCP: the tissues and blood
of cows and calves of dairy herds in the USA showed unquantified
PCP contamination (Hoeting, 1977). One herd housed in a PCP-
treated wooden barn had blood-PCP levels of 270 - 570 µg/litre (US
EPA, 1978).
Pentachloroanisole, a metabolite of the PCP biodecomposition,
causes a musty taint in broiler chicken tissues. It appears that
the chloroanisole arises through the microbial methylation of PCP
in wood shavings used as chicken litter (Curtis et al., 1972; Parr
et al., 1974; Harper & Banave, 1975). Wood shavings have been used
as litter not only for broiler chickens, but also for turkeys,
ducks, pigs, and cattle.
Neidert et al. (1984) found low residue levels of PCP in all
1072 chicken liver and 723 fat samples examined (most < 0.01
mg/kg), indicating an overall exposure of poultry to PCP. Only
0.75% of the liver samples contained PCP levels higher than 0.1
mg/kg.
In the field study of Vermeer et al. (1974) mentioned earlier,
PCP was detected in liver samples of birds (0.06 - 0.19 mg/kg wet
weight) residing in the vicinity of PCP-treated rice fields. High
PCP residues were found in the brain (mean, 11.3 mg/kg wet weight),
liver (46.6 mg/kg), and kidney (20.3 mg/kg) of dead snail kites
(Rostrhamus sociabilis), which had probably ingested Na-PCP
contaminated snails.
Only a few data on PCP residues in terrestrial animals,
apparently not exposed to PCP, have been reported: purple martin
fledglings from Alberta, Canada, contained 31 µg PCP/kg (Jones,
1981). The muscle tissue of juvenile starlings, collected from
their nests in South Finland in 1982 and 1983, contained PCP levels
ranging from not detectable to 59 µg/kg wet weight (mean, 5.9
µg/kg) (Paasivirta et al., 1985). The pectoral muscles of white-
tailed eagles, also collected in Finland, contained between 14 and
8571 µg PCP/kg wet weight, while levels in eggs ranged from not
detectable to 25 µg/kg. Eggs of osprey contained between 1 and 803
µg PCP/kg.
5.1.5. Drinking-water and food
PCP concentrations ranging from < 1 to 50 µg/litre were
detected in domestic well water (Oroville, California) (Wong &
Crosby, 1981). Buhler et al. (1973) analysed drinking-water
obtained from the Willamette river (USA). They found 0.06 µg
PCP/litre in the finished water. PCP was found at a level of 0.1
µg/litre in one water sample (Dougherty & Piotrowska, 1976b).
Concentrations of 0.01 - 0.02 µg PCP/litre were detected in
drinking-water in the Ruhr area of the Federal Republic of Germany
(Dietz & Traud, 1978b). PCP levels in Florida drinking-water
supplies ranged from 0.003 to 0.34 µg/litre (Morgade et al., 1980).
Detrick (1977) suggested that the chlorination of phenol in water
supplies might be responsible for the wide occurrence of PCP. The
chlorination of 1 mg phenol/litre by 10 mg chlorine/litre is said
to yield about 0.2 µg PCP/litre, which is comparable with the
levels found in drinking-water. However, the odour threshold for
phenol is in the µg/litre range, thus low levels of phenol can
generally be detected in water.
Most data on PCP residues in food have been collected in the
USA, where a number of pesticides, including PCP, have been
routinely monitored in the FDA Market Basket Survey. In 1973-74,
PCP was found in 10 out of 360 composite food samples, at
concentrations ranging from 10 to 30 µg/kg (Manske & Johnson,
1977). In 1975, 5.4% of a total of 240 samples were contaminated
with PCP at 10 - 40 µg/kg (Johnson & Manske, 1977). Values for the
period 1965-70 are shown in Table 16. PCP concentrations measured
in daily diet samples in the Federal Republic of Germany
(Gebefuegi, 1981) are similar, averaging 16.3 µg/kg (range, 2.6 -
27.5 µg/kg). Krause (1982) found elevated PCP concentrations in
the food-basket samples of persons applying wood preservatives in
private homes. Two-thirds of the samples analysed contained between
2 and 13 µg PCP/kg with a median of about 6 µg PCP/kg, whereas the
control samples fell between less than 0.1 and 5 µg/kg.
Samples of agricultural produce taken by the Alberta Department
of Agriculture (Canada), consisting mainly of potatoes and raw
milk, contained PCP levels of less than 10 µg/kg. In isolated
samples, PCP levels of up to 2700 µg/kg occurred, as a result of
contamination from storage containers made of treated wood (Jones,
1981).
In southern Ontario, Canada, 45 bovine milk samples collected
from bulk transports hauling milk were analysed for chlorophenols
(Frank et al., 1979). PCP was not detected in whole milk at a
detection level of 0.1 µg/litre.
In analysing commercial mushrooms for PCP residues, Meemken et
al. (1982) found that levels in 11 out of 17 fresh mushroom samples
exceeded the recommended limit of 10 µg/kg set in the Federal
Republic of Germany for certain food items. Residues apparently
originated from the treated wooden cases used for culturing
mushrooms, which contained up to 3900 mg PCP/kg.
Table 16. Average incidence of PCP residues
in food composites and daily dietary intake of
PCP in the USAa
------------------------------------------------
Year Number of Percent Daily PCP
composites positive intake
examined composites (µg/person
per day)
------------------------------------------------
1965 216 1.4 -b
1966 312 3.3 6
1967 360 2.2 1
1968 360 1.9 1
1969 360 2.8 2
1970 360 nsc nsc
------------------------------------------------
a From: Duggan & Corneliussen (1972).
b < detection limit (1 µg).
c ns = not specified.
PCP can also enter food during processing, transportation, or
storage. Heikes & Griffitt (1980) demonstrated that canned fruit
and vegetables in Mason jars can be contaminated with PCP due to
PCP residues in sealing gaskets, lids, and enamel. Levels in the
jar lid, sealing gaskets, and enamel were as high as 198 µg/lid,
125 mg/kg, and 4.4 mg/kg, respectively. Six fruits and vegetables
originally free from PCP, contained between 0.29 and 1.1 µg/litre
in the liquid and between 1.4 and 38 µg/kg in the solids, after
being canned and stored for 4 days in contaminated jars.
Kroyer et al. (1982) modelled the transfer of PCP from wooden
storage containers to flour experimentally. Within 24 h,
remarkable quantities of PCP can be adsorbed by flour in contact
with PCP-treated wood: applied wood preservative could be detected
in the foodstuff at levels ranging from 0.2 to 1 mg PCP/kg.
PCP levels in the range of 240 - 1090 µg/kg were found in the
flesh grease from hides treated with PCP. Collagen material
derived from hides, pigskin, and decalcified bones is used for the
manufacture of edible gelatins, and Stijve (1981) detected PCP in
each of 50 samples of commercially available gelatins tested.
Products from western Europe and the USA generally contained less
than 100 µg/kg, while those from tropical countries contained from
1000 to 5000 µg PCP/kg. The US Food and Drug Administration (FDA)
has proposed prohibiting "the use of animal bones, hides, or skins
that have been exposed to pentachlorophenol" (Federal Register,
1977).
A calculated daily dietary intake of PCP for the period 1965-70
is shown in Table 16. The values ranging from 1 to 6 µg/person per
day were based on a market-basket survey of 117 retail food items.
Each market basket represented a 2-week diet, constructed according
to consumer behaviour. The foods were prepared as for eating and
then analysed (Duggan & Corneliussen, 1972). In another survey
(Krause, 1982), in contrast to the above food collection procedure,
samples of complete meals prepared and eaten by different families
were collected over a period of 3 - 7 days and pooled. This
sampling procedure is more realistic, as it takes into
consideration the fact that food may be contaminated during storage
in PCP-contaminated rooms. In fact, in households where PCP-
containing wood preservatives had been applied, meals averaged 6 µg
PCP/kg (2/3 range, 2 - 13 µg/kg), while the meals of a control
group were less contaminated (< 0.1 - 5 µg/kg). According to
Fischer (1983), the daily dietary intake on the basis of these data
is 0.1 - 1 µg/person per day for people without known exposure and
6 µg/person per day for persons exposed to PCP-treated interiors.
5.1.6. Consumer products
On the basis of the diverse applications of PCP, it would be
expected that a large number of consumer products contain this
compound. Furthermore, PCP from the indoor atmosphere can
contaminate a number of household items. However, there are few
data on PCP levels in consumer products. In one example, van
Langeveld (1975) analysed 65 commercial samples of paints used on
children's toys and found that 14% contained PCP in the range of
100 - 2700 mg/kg.
The Swiss Federal Office of Health (Siegwart, 1983) found PCP
in various clothes including socks, pantyhose, and insoles, with
PCP concentrations between 0.015 and 0.96 mg/kg; one insole
contained 13.2 mg PCP/kg (Siegwart, personal communication, 1986).
5.1.7. Treated wood
Obviously, PCP-treated wood contains substantial quantities of
the compound itself. The Ontario Ministry of Agriculture detected
tetrachlorophenol and PCP in samples of wood shavings used as
livestock litter in southern Ontario (Jones, 1981); PCP levels were
as high as 628 mg/kg in fresh litter, but fell off sharply, after
56 days use to 96 mg/kg or less. In the United Kingdom, Parr et
al. (1974) found an average PCP concentration of 12 mg/kg (range,
1 - 83 mg/kg) in fresh broiler house litter, while spent litter
contained an average of 0.3 mg/kg. Curtis et al. (1972) found as
much as 40 mg PCP/kg in samples of shavings and sawdust. Levin &
Nilsson (1977) assayed for tetrachlorophenol, PCP, and several
contaminants in wood dust from a Swedish sawmill. PCP levels in
dust from wood treated with 2% Na-2,3,4,6-tetrachlorophenol ranged
from 30 to 100 mg/kg.
Analysis of timber samples taken from homes in the Federal
Republic of Germany, several years after it had been recom-mended
to avoid the indoor use of PCP, revealed PCP levels ranging from
0.1 to 615 mg/kg (mean, 35 mg/kg) (Ruh et al., 1984). This means
that the timber had either been treated with PCP or that it was
contaminated. In this context, it is noteworthy that Ruh &
Gebefuegi (1984) also analysed wooden material which, according to
consumer information, was supposed to be untreated. Only 30% of
the samples were PCP-free, 40% contained from 0.05 to 0.8 mg
PCP/kg, and 31% contained from 1 to 20 mg/kg.
Untreated wood samples from furniture in a living room in
which panelling had been painted with a wood preservative (6% PCP)
according to instructions, were analysed by Gebefuegi et al.
(1979). PCP concentrations ranged from 15.5 to 26 mg/kg in the top
layer (0 - 1.5 mm) and from 2.5 to 7 mg/kg at 3 - 8 mm. For
comparison, treated wood samples contained between 1570 and 2754
mg/kg in the top layer, 612 - 1800 mg/kg in the middle layer (1.5 -
3 mm), and 117 - 340 mg/kg in the bottom layer (3 - 8 mm). Freshly
treated wood surfaces showed PCP concentrations of between 4000 and
6000 mg/kg.
5.2. Occupational Exposure
A list of potential sources of occupational exposure to PCP is
presented in Table 17. However, the actual PCP concentrations that
workers are exposed to during such industrial or commercial
activities are rarely measured.
Since PCP is extensively used for wood protection and
preservation, most studies of occupational exposure have been
conducted in this field of industry. If the pressure treating
method is used, respiratory exposure to PCP occurs mainly when the
door of the pressure vessel is opened and PCP can escape into the
breathing zone of the worker. With non-pressure treatment,
continuous evaporation of PCP into the air takes place, since the
tanks or vats are open. With both processes, dermal exposure of
workers is possible during the handling of the treated wood
(Williams, 1982). The polychlorinated impurities in PCP may be
enriched relative to freshly prepared PCP solution during the
recirculation of the preservatives (Levin et al., 1976; Lamberton
et al., 1979). Hence, during the periodic tank and cylinder
cleaning processes, much higher exposures to PCP impurities may
occur than expected theoretically.
In Table 20, a number of PCP air concentrations as measured in
the course of human monitoring studies is shown. In addition, air
samples taken at the breathing zone in 11 wood-treating factories
in the USA ranged from 10 to 510 µg/m3 (Todd & Timbie, 1983). Most
of the data in Table 20 are below the TWA or MAC value of 500 µg
PCP/m3 (IRPTC, 1983), which has been established by several
countries. However, this value is derived "by analogy with other
compounds of similar action and toxicity in addition to the
specific available information" (ACGIH, 1980). With lumber
treatment, exposure to airborne PCP is generally below 100 µg/m3;
concentrations higher than the TWA value commonly occur during PCP
or Na-PCP production.
Table 17. Potential sources of occupational exposure to
PCP or its sodium salta
--------------------------------------------------------
Manufacture and shipping of industrial chlorophenols
Sawmills
Wood-treatment plants
Carpentry and other timber and wood-working
Termite control
Agricultural pesticide application
Greenhouses
Industrial cooling towers and evaporative condensers
Treatment and handling of wool
Treatment and handling of burlap, canvas, rope, leather
Paper manufacture
Petroleum and other drilling
Paint and adhesive manufacture and use
Telephone and electrical line work
Dyeing and cleaning of garments
--------------------------------------------------------
a Adapted from: Crosby et al. (1981).
Extremely high occupational exposures have been reported as a
result of the agricultural use of PCP. Following PCP application
on cotton fields over a 2-year period, Demidenko (1969) reported
that unusually high concentrations of up to 38 000 µg PCP/m3 air
were found where the sprayers were working and that the workers
exhibited typical symptoms of PCP intoxication (eye and nasal
irritation, headaches, fatigue). In the breathing zone of the
workers formulating the spray, air-PCP levels of 320 µg/m3 were
measured. Pilots in spraying aircraft were exposed to an average
of 880 µg PCP/m3. Air levels, at a distance of 10 - 50 m from the
treated field, varied from 960 to 4400 µg/m3.
The magnitude of PCP exposure depends particularly on the
methods used in handling the chemical, and on measures to minimize
PCP levels in the work-place. According to Wood et al. (1983),
automated processes and closed systems have greatly reduced the
exposure level in large-scale manufacturing and wood-treatment
factories. However, in small-scale operations, overexposure may
occur through inadequate control measures. In addition to
measurements of ambient PCP levels during industrial or
agricultural use, several attempts have been made to estimate
exposure on the basis of urine-PCP levels in workers involved in
the direct and controlled application of PCP (section 5.4).
5.3. General Population Exposure
Because of the widespread use of PCP products, the general
population also comes into contact with this substance. PCP has
been detected at the µg/litre level in the urine of people, not
occupationally exposed, in widely different groups and locations
(section 5.4). It is likely that this wide-spread occurrence of
PCP in human populations results from PCP intake or from the
metabolism of other chlorinated compounds rather than from the
natural occurrence of PCP.
Some possible sources of non-occupational exposure to PCP in
the home, work, and outdoor environments are listed in Table 18.
In such cases, the general population can be incidentally exposed
to PCP-treated items such as textiles, leather, and paper products
(Jones, 1981, 1984). In addition, a variety of consumer products
including food may contain PCP, though no direct PCP application
has been involved, if they are stored in PCP-treated wooden
containers or exposed to PCP in the atmosphere of rooms where
woodwork has been treated with PCP. This is consistent with the
laboratory studies of Morgan & Purslow (1973), Ingram et al.
(1981a,b), and Petrowitz (1981) who observed considerable losses of
PCP from wood samples (section 4.1.1).
Table 18. Some possible sources of non-occupational
exposure to PCP or its sodium salta
--------------------------------------------------------
Use of retail trade pesticide products containing PCP
(for wood preservation, termite control, etc.)
Use of PCP-treated lumber for construction of dwellings
Smoke from sawmills and burning scrap lumber
Sawdust (fuel, floor covering, particle-board, etc.)
Burlap, canvas, and rope
Wool and other textiles
Leather products
Paper products
Contact with adhesives, paint, and painted surfaces
Used telephone poles and railroad ties
Ornamental wood-chips
Fat trimmed from treated hides (used as feed additive)
Water treated for mollusc control
Contaminated food
--------------------------------------------------------
a Adapted from: Crosby et al. (1981).
Wood preservatives containing PCP or its sodium salt have been
widely applied in the domestic field both indoors and outdoors. In
some countries, the indoor use of PCP-containing wood preservatives
has been regulated by government authorities (section 3.3.3).
Cases of PCP intoxication in persons residing in treated houses
have roused public concern as to whether the general population
might be endangered by domestic applications of PCP and have led to
investigations of PCP levels in rooms and the evaluation of
potential health effects.
In one instance, volatilization of PCP within a room from
treated interior wood led to PCP deposition on untreated wood,
furniture, and curtains at levels of between 23 and 26 mg/kg and to
residues (mg/kg) on other household items such as carpets, books,
oil paintings, and cassette tapes. The air levels of PCP varied
between 50 and 100 µg/m3 in the living room of the house, which had
been under examination since 1974, because of reported PCP
intoxications (Gebefuegi et al., 1979).
Krause & Englert (1980), Aurand et al. (1981), and Krause
(1982) reported the results of a survey in the Federal Republic of
Germany, which included the analysis of indoor air samples in 104
homes and of the urine of more than 1000 persons. Depending on the
intensity of, and the time elapsed since, indoor PCP application,
PCP concentrations in the air ranged from not detectable to about
25 µg/m3, frequently from 2 to 10 µg/m3. The median value of 5 µg
PCP/m3 is 1000 times higher than the outdoor air levels found in
residential areas (section 5.1.1). For comparison, indoor air
levels of PCP in houses of a control group without known exposure
were generally below the detection limit of 0.1 µg PCP/m3.
Air concentrations of PCP of between 1 and 10 µg/m3 have been
reported in the living- and bedrooms of Swiss homes (Zimmerli et
al., 1979). Similar levels (1 - 25 µg/m3) were found in rooms
treated one to several years earlier; levels of 25 and 30 µg/m3
were found in rooms a few weeks after treatment (Dahms & Metzner,
1979).
In an unpublished study carried out in the United Kingdom in
1980 and cited by Dobbs & Williams (1983), PCP air concentrations
were monitored as a function of time. During the first week after
treatment of the roof void of a house, PCP levels were as follows:
16 - 67 µg/m3 in the treated roof void, 3.9 - 15 µg/m3 in the
landing, and 1.6 - 2.8 µg/m3 in the bedroom; 5 - 10 weeks after
treatment, PCP concentrations in the treated roof void ranged from
1.7 to 6.7 µg/m3 compared with 0.6 - 5 µg/m3 in the landing and
1.6 - 2.8 µg/m3 in the bedroom. Although the PCP air concentration
in or near the treated room rapidly decreased, levels in the
untreated bedroom remained stable, perhaps as a result of adsorption
and desorption processes.
To follow the volatilization of PCP from treated wood in
enclosed environments, studies were conducted in a building with an
enclosed swimming pool, the walls and ceiling of which had been
treated with PCP (Gebefuegi, 1981; Gebefuegi et al., 1983). Within
a fortnight, an estimated 178.5 mg PCP had diffused from the
wooden panels (210 m2), based on detection of 1 µg/litre in the
water of the swimming pool, 60 µg/litre in the water condensed from
the heat pump, and 4 µg/m3 in the air of the hall housing the
swimming pool. Air levels of PCP inside this building ranged from
1 to 160 µg/m3, one year after the application of the wood
preservative. Two years later, PCP concentrations in the indoor
air averaged 4 µg/m3, and, even after 7 - 8 years, PCP was still
present at about 0.4 µg/m3, though, in the interim, the wood panels
had been painted with another wood preservative, which undoubtedly
reduced the rate of PCP diffusion.
In a study conducted in the USA, PCP vapour levels were
measured in the air of treated wooden structures. The highest
level detected was 38 µg/m3 in the basement of the building where
there was the highest ratio of treated wood surface area to room
volume, and no ventilation. PCP levels were higher in the main
floor of this house (8.8 µg/m3) and in a warehouse (3.52 µg/m3)
than in 11 other rooms of different buildings, which ranged from
0.09 to 1 µg/m3. Variability in PCP concentrations was mainly
attributable to ventilation differences (Saur et al., 1982).
Compared with the aforementioned studies, the values (0.06 -
1.6 µg/m3) reported by Dobbs & Williams (1983) from houses in the
United Kingdom represent rather low PCP levels. Generally, PCP
concentrations of up to about 30 µg/m3 can be expected during the
first month following treatment; considerably higher levels (up to
160 µg/m3) may not be excluded under unfavourable conditions. In
the long term, values of between 1 and 10 µg/m3 are typical PCP
concentrations after extensive treatments.
Krause (1982) considered household dust to be very suitable for
screening purposes, because it accumulates PCP and can be easily
collected. Household dust collected in the houses of residents
using PCP contained about 1000 times more PCP (mean, 15 mg/kg; 2/3
range, 6 - 20 mg/kg) than that of control households (mean, 0.008
mg/kg; 2/3 range, 0.003 - 0.013 mg/kg).
In similar, more recent studies, relatively high PCP levels
(range, 0.2 - 217.3 mg/kg; mean, 28.5 mg/kg) were found in
household dust samples, indicating indoor air contamination
resulting from PCP contaminated surfaces (Ruh et al., 1984).
The concentration of PCDDs or PCDFs in PCP-contaminated
interiors has not yet been determined. However, it may be assumed
that the ratio of their air concentrations will be proportional to
their rates of volatilization. Because of their lower vapour
pressure (e.g., H6CDD, 8.8 x 10-5 Pa), volatilization of PCDDs will
be lower than that of PCP. According to Cull et al. (1983), the
following PCDD concentrations can be predicted in houses where PCP
air concentrations range from 1 to 10 µg/m3: OCDD, 0.2 - 20 ng/m3;
H6CDD, 0.007 - 0.7 ng/m3; H7CDD, 0.04 - 4 ng/m3.
First reports of indoor air analyses of PCDDs and PCDFs in a
house in the Federal Republic of Germany (Eckrich, 1986), which had
been treated with PCP-containing wood preservatives several years
before, seem to confirm these estimates; however, the data
reported are still insufficient for a conclusion to be drawn.
Further studies are under way that could be used to characterize
the PCDD and PCDF levels in indoor air of PCP-treated homes.
Residues of PCP in the general population may arise not only
from oral, dermal, or inhalation uptake of PCP, but also from the
metabolic transformation of other chlorinated compounds (Table 19).
In studying the biotransformation of hexa- and pentachlorobenzene
in the rat, mouse, guinea-pig, laying hen, and rainbow trout, Koss
& Koransky (1978) found PCP and other metabolites in both the
excreta and tissues of the animals. Considering the substantial
residues of hexachlorobenzene in the human body and in human milk,
PCP intake arises via this route in both adults and new-born
children. Similarly, the continuous low-level PCP excretion from
people, who apparently are not exposed to PCP, might be partly due
to continuous exposure to hexachlorobenzene and related compounds.
Table 19. Chlorinated compounds metabolized to PCP
----------------------------------------------------------------
Source Reference
----------------------------------------------------------------
hexachlorobenzene Mehendale et al. (1975); Engst et
al. (1976); Rozman et al. (1977);
Sanborn et al. (1977); Koss &
Koransky (1978); van Ommen et al.
(1985)
pentachloronitrobenzene Murthy & Kaufman (1978); Koegel et
al. (1979)
BHC isomers (i.e., lindane) Balba & Saha (1974); Engst et al.
(1978)
----------------------------------------------------------------
5.4. Human Monitoring Data
Exposure levels as measured in the indoor air or in consumer
products may provide indirect indications of exposure to PCP.
However, it is not possible to separate oral, respiratory, and
dermal exposures, under non-experimental conditions. Thus, the PCP
concentrations of the sources do not directly indicate the actual
human PCP intake by the different routes.
Several investigations have been carried out to relate the
human body burden to the urinary-PCP level. These are summarized
in Tables 20, 21, and 22) distinguishing between occupationally
exposed workers and the general population. The latter has, in
turn, been divided into individuals exposed non-occupationally,
such as persons exposed to PCP-containing wood preservatives
applied to the interior of private homes, offices, public
facilities etc., and people without known exposure.
In many cases, values for populations without known exposure
overlap those of exposed populations, perhaps because persons
designated as unexposed might unknowingly have been exposed to
obscure sources of PCP. In addition, occupational exposure does
not always involve high loading, for, as pointed out in section
5.2, both very low and very high air levels of PCP have been found
in work-places. Conversely, people exposed non-occupationally,
particularly those who apply PCP-containing wood preservatives
indoors, may be exposed to air levels of PCP encountered at work-
places or even higher levels.
Analytical improvements in the last few years have
substantially lowered the detection limits (section 2.5.2) and,
combined with different methods, this makes the comparability of
analytical data questionable, particularly when urinary-PCP levels
have been determined without hydrolysis, as in the survey of Krause
& Englert (1980), since, under these conditions, PCP levels will
have undoubtedly been underestimated. According to Zimmerli et al.
(1979) and Butte (1984), analysis for total PCP yields
concentrations about 3 times higher than those for only free PCP.
For these reasons, it is not possible to derive exact ranges of
PCP levels in different exposure groups from the data in Tables 20,
21, and 22. However, the mean or median urine-PCP levels are
likely to range around 0.01 mg/litre for the general population
without known exposure, around 0.04 mg/litre for persons exposed
non-occupationally, and around 1 mg/litre for occupationally
exposed people.
Only two particularly detailed studies of domestic PCP exposure
provide comparative data. Comparing the mean urine-PCP levels,
Hernandez & Strassmann-Sundy (1980) (No. 3 in Table 21, No. 5 in
Table 22) observed that residents of log homes treated with PCP had
a body burden that was between 5 and 60 times higher than that of
residents of untreated log homes or conventional homes.
The most thoroughly designed survey (Krause & Englert, 1980;
Aurand et al., 1981; Krause, 1982) included air analyses in the
houses of a control group showing PCP concentrations generally
below the detection limit of 0.1 µg/m3 (No. 6 in Table 22). The
mean urine-PCP level of this group was about 3.5 times lower than
that of the corresponding exposure group (No. 5 in Table 21). Much
greater differences are encountered, if the highest concentrations
measured are compared with the control levels. No significant
correlation was established between the PCP levels in air and in
urine, which again casts doubt on the validity of air-PCP levels as
an exposure parameter. Nevertheless, on subdividing the exposed
persons into 2 groups with PCP exposure either lower and equal to
5 µg PCP/m3 or higher than 5 µg PCP/m3, urinary-PCP levels seem to
be elevated at the higher indoor concentrations. In addition,
younger residents are obviously more exposed to PCP than older
ones, perhaps because, on average, children spend more time at
home.
When comparing PCP concentrations in the urine and blood, the
levels in serum or plasma generally exceed those in urine, the
extent of this difference varying according to the exposed group.
The blood-PCP:urine-PCP ratio in people without known exposure or
in persons exposed non-occupationally is considerably higher than
that in occupationally exposed individuals. For comparison, in most
of the cases of lethal intoxication summarized in Table 24 (section
6.2.2), urine-PCP levels even exceeded the corresponding blood
levels. This pattern may be the result of heterogenous plasma-
protein binding of PCP (section 6.6).
Shafik (1973) detected significant amounts of PCP (mean, 0.025
mg/kg; range, 0.005 - 0.052 mg/kg) in human adipose tissue samples
from the general population. Similar values (mean, 0.0145 mg/kg)
(Table 25, section 6.3.1) were reported by Grimm et al. (1981),
whereas higher PCP contents (mean, 0.14 mg/kg; range, not
detectable - 0.57 mg/kg) were found in adipose tissue samples from
subjects with "no occupational contacts" (Ohe, 1979).
Table 20. Levels of PCP in the air, and in the serum or plasma, and urine of
individuals exposed occupationally
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(1) Lumber, carpentry 1 ns na na 0.024 Cranmer &
(0.022 - 0.025)a Freal (1970)
(2) Lumber, closed 11 1 na na 1.6 (ns) Casarett et
tank procedure al. (1969)
(3) Lumber, dipping 11 1 na na 2.6 (ns) Casarett et
al. (1969)
(4) Lumber, dipping ns ns 19 na 2.83 Arsenault
(3 - 63) (0.12 - 9.68) (1976)
(5) Lumber, dipping 18 - 22 ns na 3.78 0.95 Klemmer et
(0.15 - 17.4) (< 0.01 - 7.80) al. (1980)
(6) Lumber, dipping, 18 ns na 5.14 1.31 Begley et
spraying, or (0.43 - 14) (0.09 - 3.3) al. (1977)
brushing
- 6th day of 18 ns na 4.92 1.36
vacation (0.50 - 13) (0.18 - 3.5)
- 20th day of 18 ns na 2.19 0.59
vacation (0.32 - 5.3) (0.05 - 1.4)
- 51st day of 13 ns na 2.61 0.95
renewed work (0.19 - 8.1) (0.03 - 3.6)
exposure
(7) Lumber, generalb 3 5 1 1.11 0.15 Wyllie et
(2 - 11) (< 1 - 15) (0.35 - 3) (0.044 - 0.47) al. (1975)
(8) Lumber industry 20 ns na ns ns Gossler &
(0.4 - 4.8) (0.07 - 0.57)c Schaller
(1978)
(9) Lumber, officeb 1 10 2 0.65 0.06 Wyllie et
(< 1 - 3) (0.42 - 0.75) (0.04 - 0.11) al. (1975)
--------------------------------------------------------------------------------------------------------------
Table 20. (contd.)
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(10) Lumber, pressure 1 5 6 2.29 0.30 Wyllie et
treatmentb (< 1 - 15) (1.51 - 3.55) (0.09 - 0.76) al. (1975)
(11) Lumber, pressure ns ns 14 na 1.24 Arsenault
treatment (4 - 1000)d (0.17 - 5.57) (1976)
(12) Lumber, pressure 23 - 24 ns na 1.72 0.27 Klemmer et
treatment (0.02 - 7.70) (< 0.01 - 2.40) al. (1980)
(13) Lumber, pressure Embree et
treatment al. (1984)
- Airborne + 10 5 - 10 55.6 0.71 0.11
dermal (± 89) (± 0.38) (± 0.02)
- Airborne 8 5 - 10 66.7 0.24 0.05
exposure (± 100) (± 0.23) (±0.02)
- No known 5 5 - 10 ns 0.06 ns
exposure (± 0.02)
(14) Lumber, spraying 2 ns na na 0.20 Cranmer &
(0.13 - 0.27)a Freal
(1970)
(15) Lumber, spraying ns ns 6 na 0.98 Arsenault
(3 - 69)d (0.13 - 2.58) (1976)
(16) PCP processing 18 12 ns 0.25e 0.112e Triebig et
factory (0.3 - (2.2 - 55.5) (0.02-1.5)f (0.013 - 1.224) al. (1981)g
31)
(17) PCP application 23 3e 2.4 1e ns Zober et
(0.5 - (0.3 - 8) (0.2 - 2.4)f al. (1981)
12)
(18) PCP processing 18 10e 17.5 0.25e ns Zober et
(0.2 - (2 - 50) (0.02 - 1.5)f al. (1981)
31)
--------------------------------------------------------------------------------------------------------------
Table 20. (contd.)
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(19) PCP production 8 ns < 100 - 4.73 ± 3.41 2.38 ± 1.91 Bauchinger
> 500h et al.
Na-PCP production 14 ns < 100 - 2.23 ± 1.51 0.84 ± 0.65 (1982)
> 500i
(20) PCP production 18 ns 270 - 4000 na 0.72 ± 0.55 Ning (1984)
Na-PCP production 50 ns 0 - 50 na 0.35 ± 0.30
(21) PCP synthesis, 9 ns na na 1.2 Siqueira &
full time activity (0.34 - 3.4) Fernicola
- same factory, 12 ns na na 0.15 (1981)
reduced PCP (0.032 - 0.4)
exposure
(22) Pesticide, 130 ns na na 1.80 Bevenue et
spraying (0.003 - 35.7) al. (1967b)
(23) Farmers and pest 210 - ns na 0.25 0.01 Klemmer et
control operators 280 (< 0.01 - 8.4) (< 0.01 - 0.040) al. (1980)
--------------------------------------------------------------------------------------------------------------
a Range of replicate analyses of single urine samples.
b Mean concentrations shown are calculated from sampling data collected over a 5-month period. Mean air
level for workers listed as "lumber, general", is calculated from data provided for all 11 sites over a
5-month period by Wyllie et al. (1975).
c Assuming a daily urine volume of 1.4 litre.
d Mean "average exposure levels" encountered by employees. Air at "maximum exposure" sites, next to sources,
contained 26 µg/m3 (lumber spraying site) and 297 µg/m3 (pressure treatment site).
e Median.
f Plasma.
g Data partly identical with those from Zober et al. (1981).
h From 67 samples, 18 were < 100 and 10 > 500 µg/m3.
i From 55 samples, 7 were < 100 and 8 > 500 µg/m3.
na = Not analysed.
ns = Not specified.
Table 21. Levels of PCP in the air, and in the serum or plasma, and urine of individuals
exposed non-occupationally
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(1) Miscellaneous groups 117 ns na na 0.04 Bevenue et
including house- (nd - al. (1967b)
holds and pesticide 1.84)
users
(2) Indoor application 16 ns ns na ns Zimmerli et
of PCP solutions (1 - 10) (0.030 - al. (1979)
0.150)
(3) Residents of log 5 ns 0.29 1.126 0.084 Hernandez &
homes treated with (0.20 - (0.580 - (0.047 - Strassmann-
PCP solutions 0.38)a 1.750) 0.216) Sundy (1980)
32 ns na 0.330 0.013
(0.116 - (0.002 -
1.084) 0.087)
(4) "No occupational 32 ns na 0.32 0.03 Klemmer et
exposure"; control (0.002 - (< 0.01 - al. (1980)
group for Number 5 7.20) 1)
in Table 20
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(5) Indoor application 989 ns 6.1 na 0.044 Krause &
of an average of (nd - Englert
40 litre PCP (< 9 25)b (1980)
solutions years) 4.9c 0.029c
(2.5 - (0.013 -
9.5) 0.071)
- Subgroups:
1. m < 18 years 16 ns < 5 na 0.047c
(0.017 -
0.107)
2. m > 18 years 39 ns < 5 na 0.023c
(0.011 -
0.052)
3. f < 18 years 22 ns < 5 na 0.033c
(0.016 -
0.066)
4. f > 18 years 39 ns < 5 na 0.026c
(0.015 -
0.059)
5. m < 18 years 23 ns > 5 na 0.079c
(0.014 -
0.125)
6. m > 18 years 31 ns > 5 na 0.043c
(0.011 -
0.146)
7. f < 18 years 25 ns > 5 na 0.059c
(0.011 -
0.103)
8. f > 18 years 43 ns > 5 na 0.039c
(0.021 -
0.125)
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(6) Indoor application Sangster et
of about 70 litres al. (1982)
PCP solution
- before ventilation 6 6 0.60 na 0.0032
(0.14 - (0.0007 -
1.20) 0.0078)
- after ventilation 6 - 0.08 0.080 0.0033
(nd - (0.025 - (0.0018 -
0.24) 0.190)d 0.0080)
Indoor application 2 0.5 0.15 0.033 na
of about 100 litres (nd - (0.031 -
PCP solution 0.40) 0.034)d
about 75 litres PCP
solution
Indoor application 2 1 0.67 0.565 na
of about 100 litres (0.44 - (0.47 -
PCP solution 0.95) 0.66)d
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(7) "Workers non- 27 ns na na 0.009 Siqueira &
occupationally (nd - Fernicola
exposed"; control 0.034) (1981)
group for Number 21
in Table 20
(8) Indoor application 80 ns na (0.0025 - (0.002 - Janssens &
PCP solutions 0.5) 0.075) Schepens
(1984)
(9) Residents in buil- 46 ns na (0.001 - na Ruh et al.
dings with PCP con- 0.110)e (1984)
taminated wood
(10) Residents in homes 204 ns na 0.058c 0.014c Grimm et al.
treated with PCP (234) (1985)
---------------------------------------------------------------------------------------------------
a Air samples taken on the 1st and 2nd floor of a 2-story log house.
A sample of interior surface wood contained 1132 mg PCP/kg (0.11%).
b 104 air indoor samples taken.
c Median (2/3 range).
d Plasma.
e Whole blood.
na = Not analysed.
nd = Not detectable.
ns = Not specified.
f = Female.
m = Male.
Table 22. Levels of PCP in the serum or urine of individuals without known exposure
---------------------------------------------------------------------------------------------------------
Number Comments Sample Indoor Serum Urine (mg/litre) Reference
of size air (mg/litre) mean (range)
study (number) (µg/m3) mean
(range)
---------------------------------------------------------------------------------------------------------
(1) ns 6 na na 0.005 Cranmer & Freal
(0.002 - 0.011)a (1970)
(2) Control group for ns na 0.06b ns Gossler & Schaller
Number 8 in Table 20 (0.03 - (0.001 - 0.057)c (1978)
0.2)
(3) US National Human 418 na na 0.0063 Kutz et al. (1978)
Monitoring Program for (nd - 0.193)
Pesticides
(4) Control group for 12 na na 0.0135 Zimmerli et al.
Number 2 in Table 21 (0.006 - 0.023) (1979)
(5) Control groups for 42 na ns ns Hernandez &
Number 3 in Table 21; (0.004 - (0.0007 - 0.011) Strassmann-Sundy
January 1980 "conven- 0.068) (1980)
tional" homes
March 1980; untreated 2 na 0.051 0.0014
log homes (0.034 - (0.001 - 0.002)
0.075)
March 1980; 11 na 0.048 0.0025
"conventional" (0.015 - (0.001 - 0.007)
homes 0.055)
---------------------------------------------------------------------------------------------------------
Table 22. (contd.)
---------------------------------------------------------------------------------------------------------
Number Comments Sample Indoor Serum Urine (mg/litre) Reference
of size air (mg/litre) mean (range)
study (number) (µg/m3) mean
(range)
---------------------------------------------------------------------------------------------------------
(6) Control group for 207 ndd na 0.0127 Krause &
Number 5 in Table 21 0.0102b Englert (1980)
(0.0038 - 0.0214)
(7) ns 10 na na 0.009 Lores et al. (1981)
(0.003 - 0.016)
(8) Dutch draftees; 99 na 0.128 na Sangster et al.
control group for (< 0.05 - (1982)
Number 6 in Table 21 1.10)
0.088b
(9) Control group for 12 na ns 0.0009 Janssens &
Number 7 in Table 21 (0.0002 - 0.002) Schepens (1984)
(10) Non-specifically 12 na 0.025 0.014 Uhl et al.
exposed persons 30 (0.019 - (0.007 - 0.034)c (1986)
0.036)
---------------------------------------------------------------------------------------------------------
a Range of replicate analysis of single urine samples.
b Median (2/3 range).
c Assuming a daily urine volume of 1.4 litre.
d Below detection limit of 0.1 µg/m3.
na = Not analysed.
nd = Not detectable.
ns = Not specified.
Samples of human milk were found to contain between 0.03 and
2.8 µg PCP/kg (mean, 0.68 ± 0.05 µg/kg), which is considerably less
than the PCP levels usually found in other body fluids or tissues
(Gebefuegi & Korte, 1983).
In investigating the apparent decrease in sperm density in US
males over the last 30 years, Dougherty et al. (1980) and Kuehl &
Dougherty (1980) detected PCP (100 - 200 ppb) in all 50 samples of
human seminal plasma analysed. They also observed that PCP was
selectively concentrated by the cellular material.
Only few monitoring data are available on the human body burden
of microcontaminants as a result of exposure to PCP. Rappe et al.
(1982) analysed urine and blood samples of 9 workers exposed to PCP
or L-PCP. In the case of 5 workers in the textile industry,
urinary levels of PCDDs (total PCDDs, 3 - 365 ng/kg) and PCDFs
(total PCDFs, < 1 - 45 ng/kg) paralleled the urinary-PCP levels
(< 0.01 - 3.12 mg/litre). Similar levels of these impurities were
found in the blood of 4 tannery workers 8 months after last
exposure, but these could not be compared with urinary-PCP levels.
However, in both groups, the concentration pattern of the different
isomers reflected the different proportions of contaminants in
commercial PCP products.
Because of their high fat-solubility and slow metabolic
degradation and elimination, impurities of PCP such as HCB, PCDDs,
and PCDFs are expected to accumulate in body fat. No data are
available concerning the accumulation behaviour of these
microcontaminants as a result of human PCP uptake. However, levels
of dioxins in the milk- or body-fat of cows orally treated with
technical-grade PCP (10 mg/kg body weight per day) were about 1000
times higher than those in blood, indicating a substantial
accumulation (Firestone et al., 1979). In addition, the three
dioxins detected (1,2,3,6,7,8-H6CDD, 1,2,3,4,6,7,8 H7CDD, and OCDD)
declined comparatively slowly from about 20, 40, and 25 µg/kg
composite milk-fat to 4.3, 6.9, and 3 µg/kg, respectively, 100 days
after PCP feeding was stopped. For comparison, the steady-state
PCP level of about 40 mg/kg in blood or 4 mg/kg in composite milk
dropped to basal levels (0.02 - 0.08 mg/kg) within less than 10
days. Firestone et al. (1979) concluded from their results that
"the absence of PCP in milk or biological tissue affords no
guarantee of the absence of biologically active dioxins".
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Animal studies
PCP is readily absorbed through the skin as well as through the
respiratory and gastrointestinal tracts. Braun & Sauerhoff (1976)
administered a single oral dose of 10 mg PCP/kg body weight in corn
oil to 3 male and 3 female rhesus monkeys and calculated the half-
life for absorption to be 3.6 h in males and 1.8 h in females.
After 12 - 24 h, plasma levels peaked in the range of 10 - 30 mg
PCP/litre.
In rats given a single oral dose of 10 mg of 14C-PCP/kg body
weight, the peak plasma concentration (50 mg/litre) was attained
much earlier, after 4 - 6 h. The absorption rate constants were
1.95 and 1.52/h for males and females, respectively (Braun et al.,
1977); assuming first order kinetics, the half-life for absorption
can be calculated to be 0.36 and 0.46 h, respectively.
Rapid absorption of PCP was also observed in rats during 20-min
inhalation of approximately 5.7 mg PCP/kg body weight (Hoben et
al., 1976d) and in mice following intraperitoneal and subcutaneous
injections of 14C-PCP (Jakobson & Yllner, 1971).
The same holds true for fish. Goldfish exposed to PCP medium
(0.4 mg/litre) absorbed PCP rapidly, until a lethal level of
approximately 100 mg/kg body weight was reached after about 5 h
(Kobayashi & Akitake, 1975a). The apparent route of PCP uptake was
via the gills and the skin. Similarly, PCP was rapidly taken up
from the water by rainbow trout and assimilated into various
tissues (Glickman et al., 1977).
6.1.2 Human studies
Braun et al. (1979) studied 4 healthy male volunteers of normal
weight, between 21 and 55 years of age, who ingested a dose of 0.1
mg Na-PCP (> 99% purity)/kg body weight. The observed half-life
for absorption was about 1.3 h. The peak plasma level of 0.245 mg
PCP/litre occurred 4 h after ingestion of PCP. In the study of Uhl
et al. (1986), the PCP level in the plasma of a male volunteer was
approximately 0.185 mg/litre, 2 days after a single oral dose of
0.016 mg 13C-PCP/kg body weight in 40% ethanol. This implies that
absorption of PCP, when dissolved in alcohol, is much greater than
when it is dissolved in water.
For the general population, the uptake of PCP by the oral route
is thought to be more significant than that via other routes of
exposure. For individuals exposed to high airborne concentrations
of PCP in the work-place or in PCP-treated dwellings, the major
routes of exposure are probably via the skin and lungs. No
experimental data are available concerning these routes. However,
the cases of acute intoxication reported were almost exclusively
due to extensive skin contact with PCP or to the inhalation of high
doses of PCP, which subsequently led to high PCP levels in the
human body.
Bevenue et al. (1967a) reported a case of PCP absorption
through the skin. A male individual had skin contact with PCP for
10 min while cleaning a paint brush in a can containing a solution
of 4% PCP. Two days later, a urinary-PCP level of 236 µg/litre was
measured.
One case of oral uptake has been reported (Haley, 1977). A
71-year-old Japanese male had intentionally ingested an amount
estimated at between 113 and 226 g of weed killer containing 12%
PCP. Although the patient was treated with gastric aspiration and
lavage within the next hour, a substantial amount of PCP must have
already been absorbed as indicated by the high serum level of 150
mg PCP/litre, 5 h after the incident.
6.2 Distribution
6.2.1 Animal studies
Available information indicates that usually the highest PCP
levels can be found in the urine immediately after exposure, and
consequently, the PCP concentrations in the tissues account for
only a small fraction of the dose applied. This is reflected by
the data on excretion and recovery of radioactivity from groups of
rats, 9 and 10 days after oral administration of 10 and 100 mg
PCP/kg body weight, respectively (Table 23). It should be noted
that the percentages of the dose recovered are to be considered as
cumulative over the period of 9 days (10 mg/kg dose) or 8 days (100
mg/kg dose) in the case of the excreta, while the PCP contents of
the organs were only analysed 9 days after administration of 10
mg/kg body weight. Thus, PCP is apparently eliminated much more
rapidly from the kidney than from the liver (section 6.5.1).
The results of early studies did not show a uniform
distribution pattern of PCP in experimental animals but indicated
that very high levels of PCP could be found in liver and kidneys
(Truhaut et al., 1952b). However, following long-term exposure,
most PCP was absorbed by the central nervous system.
Several recent studies on the distribution and elimination of
PCP were performed using 14C-labelled PCP, thus making the results
more reliable. Larsen et al. (1972) studied the tissue distribution
in rats after administering oral doses between 31 and 40 mg 14C-
PCP/kg body weight. The body component containing the highest
level appeared to be the liver, followed by the kidney and blood.
Low levels of PCP were found in fat, brain, and muscle tissue.
More than 99% of the total radioactivity in the blood was contained
in the serum, indicating that PCP and/or its metabolites in the
blood are not bound to the cellular constituents.
Table 23. Recovery of radioactivity from rats given a
single oral dose of 10 or 100 mg of 14C-PCP/kg body weighta
------------------------------------------------------------
Percentage of radioactivity (mean ± SD)
dose: 10 mg/kg dose: 100 mg/kg
------------------------------------------------------------
Excreta
urine 79.8 ± 2.9 64.0 ± 14.9
faeces 18.6 ± 3.7 33.6 ± 13.7
expired 14CO2 0.2 ± 0.1 -b
Organsc
liver 0.315 ± 0.137 -b
kidneys 0.045 ± 0.014 -b
Total body 0.437 ± 0.142 -b
Cage rinse 1.4 ± 1.9 -b
Total recovery 99.8 ± 4.4 97.6 ± 2
------------------------------------------------------------
a From: Braun et al. (1977).
b Samples were not analysed.
c Other organs analysed were the stomach, lungs, testes,
ovaries, brain, heart, spleen, and adrenals. Each of
these organs contained 0.005% of the dose or less and
were included with the liver and kidneys in the total
body figure.
Jakobson & Yllner (1971) examined the distribution of PCP in
the mouse after the subcutaneous or intraperitoneal injection of
14C-PCP (15 - 37 mg/kg body weight). Autoradiographic studies
showed that the highest specific activity was in the liver, the
gall bladder and its contents, the wall of the stomach fundus, the
kidney, and the contents of the gastrointestinal tract, while the
lung, heart, and brain contained only negligible amounts of PCP.
PCP concentrations were highest in the plasma of rats
immediately after a 20-min inhalation exposure to an aerosol of Na-
PCP, resulting in a calculated dose of about 5.7 mg/kg body weight
(Hoben et al., 1976d). About 35% of the dose was found in the
plasma, while the liver contained about 25% and the lung a little
less than 2% at time 0. After 24 h, the liver showed the highest
PCP level followed by the plasma and the lung. Other organs were
not examined.
Zenzen (1979) administered a daily dose of 15 mg 14C-PCP/kg
body weight intraperitoneally to rats for 15 days. On day 1, the
highest PCP levels in the organs examined were found in the liver
and kidney of male rats, comprising 14.1 and 14.3 %, respectively,
of the total activity on a dry weight basis. Similar
concentrations were measured in the testicle. However, on a fresh
weight basis, the levels in this organ were similar to those in the
other endocrine organs.
Preliminary data obtained from a single sheep (Wilson et al.,
1982) indicated that PCP is absorbed into the lymphatic system; 47%
of the PCP dose (10 mg/kg in corn oil, given by intraruminal
injection) remained in the digestive tract 36 h after dosing. Of
the absorbed PCP (53% of total dose), approximately 17% was found
in the lymph collected through a thoracic duct canula. The
remainder was probably absorbed from the digestive tract directly
into the blood.
To study the placental transfer of PCP in rats, 60 mg 14C-
PCP/kg body weight was orally administered to pregnant rats on day
15 of gestation. The amount of specific radioactivity in the
maternal blood-serum was greatest at 8 h (about 1.1% of the
administered dose per gram of tissue), but, in the placentas and
fetuses, it never exceeded 0.3% and 0.1%, respectively (Larsen et
al., 1975). Thus, the amount of PCP that crosses the placental
barrier is very low. Contrasting data have been obtained in a
preliminary observation of a single pregnant monkey, but no full
report has been published (Müller, 1981).
Kobayashi (1979) observed an accumulation of 14C in various
organs of goldfish exposed to 0.1 mg 14C-PCP/litre water. The gall
bladder contained the highest 14C level after a 24-h exposure. The
biliary concentration increased, even after fish had been
transferred to clean, running water for 24 h, whereas a decrease
was observed in the levels in all other organs examined (Kobayashi
& Akitake, 1975b). This characteristic accumulation indicates that
conjugated PCP is transferred to the gall bladder and bile due to
an enterohepatic circulation (section 6.5.1).
6.2.2 Human studies
Human data concerning tissue distribution following PCP uptake
are derived mainly from autopsy results on victims of fatal
intoxications (Table 24). No exact conclusions can be drawn from
these data with regard to PCP accumulation, though PCP levels in
the liver, kidney, and lungs are often elevated. The high levels in
the lungs reported in some cases might be related to inhalation
uptake of PCP. Similarly, the stomach of the individual who
committed suicide by ingesting PCP contained 750 mg PCP/kg, which
highly exceeded the concentrations found in the other body parts
examined (Cretney, 1976). An unusually high kidney-PCP level of
639 mg/kg, reported by Wood et al. (1983), might have been due to
kidney malfunction. In general, PCP levels in the various tissues
do not indicate a clear accumulation of PCP, since blood-PCP levels
are often similar to the corresponding tissue concentrations.
Levels in urine can vary depending on the actual urine volume in
the bladder at the time of poisoning and the pH value. From the
data in Table 24, liver and kidney residues associated with acute
lethal intoxications can be estimated to be 10 - 225 mg PCP/kg and
5 - 145 mg PCP/kg, respectively.
Table 24. PCP levels found in human tissues and body fluids
after PCP intoxication resulting in death
---------------------------------------------------------------------------------------------
Reference Case Urine Blood Liver Kidney Lung Brain Supposed
number (mg/ (mg/ (mg/ (mg/kg) (mg/kg) (mg/kg) routes of uptake
litre) litre) kg)
---------------------------------------------------------------------------------------------
Truhaut et al. 1 55 5 10 5 1.5 na dermala
(1952b) 2 96 6 52 21 38 6.5 dermala
Gordon (1956) 1 70 50 65 95 145 20 inhalation, dermala
Menon (1958) 1 160 na na na na na inhalation, dermala
Blair (1961) 1 na na 59 41 na na dermal, orala
2 na na 62 84 76 na inhalationa
3 na na 59 63 na na oralb
Mason et al. 1 na 79 66 na na 10 inhalationa
(1965) 2 365 110 89 86 na 25 inhalationa
Burger (1966) 1 na 39 na na na na orald
Barthel et al. 1 na na na 27.6 na na dermalc
(1969)
Cretney (1976) 1 75 173 225 116 na na oral(?)d
Wood et al. 1 29 16 52 639 116 na inhalation,
(1983) dermala
---------------------------------------------------------------------------------------------
a Occupational exposure.
b Accidentally contaminated food.
c Accidentally contaminated diapers.
d Suicide.
na = Not analysed.
There is a paucity of data on the distribution of PCP in the
tissues and body fluids in the general population. In 2
investigations (Table 25), autopsy samples were collected from
human subjects in Northern Bavaria (Federal Republic of Germany)
with no known history of PCP exposure. Grimm et al. (1981)
concluded from the data that there was only a slight tendency for
PCP to accumulate in both liver and kidney. The relatively high
level observed in the brain samples was attributed to the fact that
most of the persons with high brain-PCP levels had bled to death.
In such cases, a cerebral hypoxia preceding the death might have
led to the breakdown of the blood-brain barrier and accumulation of
PCP in the brain. Löwer (1982) found much lower brain-PCP levels,
in conjunction with liver and kidney levels similar to those
reported by Grimm et al. (1981). There was no correlation between
PCP levels in tissues and the age of the person examined (Löwer,
1982). Moreover, PCP levels in the body fluids were of the same
order of magnitude as those in the tissues (Grimm et al., 1981).
6.3 Metabolic Transformation
6.3.1 Animal studies
The first studies on the fate of PCP in the body were conducted
by Deichmann et al. (1942), who obtained evidence of metabolic
transformation in the rabbit after oral administration and in the
rat after intraperitoneal dosing with Na-PCP. The authors did not
identify any metabolites.
Later studies using more advanced analytical methods have shown
that PCP is metabolized, either to tetrachlorohydroquinone through
oxidation or conjugated to PCP glucuronide. Tetrachlorohydroquinone
was found in its free form in the urine of mice and rats (Jakobson
& Yllner, 1971; Ahlborg et al., 1974); in the rat, it is also
conjugated with glucuronic acid (Ahlborg et al., 1978). Traces of
trichlorohydroquinone formed by the reductive dechlorination of
tetrachlorohydroquinone were found in the urine of rats. The
formation of tetra- and trichlorohydroquinone as well as total
elimination during the first 24 h can be enhanced through
pretreatment with 3-methylcholanthrene or 2,3,7,8-T4CDD.
Phenobarbital only increases the metabolism to tetrachlorohydro-
quinone. These observations were confirmed by in vitro tests on rat
liver microsomes (Ahlborg & Thunberg, 1978). Glucuronidation rates
were not significantly altered by pretreatment with phenobarbital
or 3-methylcholanthrene (Lilienblum, 1985).
Table 25. Medians, means, and ranges of PCP concentrations in tissues
and body fluids at autopsy from people without known exposure
----------------------------------------------------------------------------------------
Number Urine Blood Liver Kidney Brain Body fat Spleen
of (mg/ (mg/ (mg/kg) (mg/kg) (mg/kg) (mg/kg) (mg/kg)
cases litre) litre)
----------------------------------------------------------------------------------------
Median 21a 0.0044 0.0233 0.0670 0.0430 0.0470 0.0127 0.0190
5. Percentile dlb dl 0.0017 0.0240 0.0190 0.0100 0.0070
95. Percentile 0.1603 0.0679 0.1735 0.0950 0.0725 0.0225 0.0325
Mean 0.0297 0.0260 0.0860 0.0641 0.0491 0.0145 0.0208
Median 51c nad na 0.0720 0.0223 0.0180 na na
Lowest value na na 0.0140 nde nd na na
Highest value na na 0.4190 0.1040 0.0560 na na
----------------------------------------------------------------------------------------
a From: Grimm et al. (1981).
b dl = detection limit = 0.001 mg PCP/litre urine; 0.005 mg PCP/litre blood.
c From: Löwer (1982).
d na = not analysed.
e nd = not detected (detection limit = 0.010 mg PCP/kg wet tissue).
In contrast to these rodent species, the rhesus monkey
eliminates PCP unchanged in the urine (Braun & Sauerhoff, 1976).
This is the only recent study that failed to detect any metabolites
of PCP.
Detoxication of PCP has also been observed in fish. While no
dechlorination processes have been reported, conjugation and
subsequent excretion of the PCP conjugates occurs: PCP glucuronide
is formed and excreted into the bile of both goldfish (Kobayashi &
Nakamura, 1979b) and rainbow trout (Glickman et al., 1977), while
pentachlorophenylsulfate is excreted into the surrounding water
through the gills and in the urine of goldfish (Kobayashi &
Nakamura, 1979a,b).
6.3.2 Human studies
Most of the human data available consist of analyses of urine
samples from people exposed to different uncontrolled PCP regimes.
In the studies of Braun et al. (1979) and Uhl et al. (1986) on
male volunteers (section 6.1.2), PCP was eliminated as both the
parent compound and glucuronide. No other metabolites could be
detected. Ahlborg et al. (1974) found tetrachlorohydroquinone in
the urine of 2 occupationally exposed spray operators, who were
also exposed to other chlorophenolic compounds.
Recently, the metabolic transformation of PCP to
tetrachlorohydroquinone was substantiated by Juhl et al. (1985);
human and rat liver homogenates showed similar metabolizing
activities. The rate of PCP metabolism dependedon the PCP
concentration and was 1000 times lower at 1 mmol/litre (266
mg/litre) than at 0.01 mmol/litre (2.66 mg/litre).
6.4 Elimination and Excretion
6.4.1 Animal studies
PCP has been rapidly eliminated by most of the animals
examined. It is cleared from the plasma by its distribution to the
tissues and by excretion via the urine and the faeces; the
metabolites, when produced, are also rapidly excreted.
The uptake of PCP by the tissues accounts for only a small
amount of the total PCP dose taken up by the body (section 6.2.1).
Most of it leaves the body immediately after uptake, mainly through
the urinary excretion of PCP and its metabolites. The proportions
of PCP excreted via the two major excretion routes in the rat,
mouse, and monkey are summarized in Table 26. Although the species
and the test conditions differed, the excretion patterns were very
similar: renal excretion amounted to between 45 and 83% of the
total dose applied, while most of the remaining activity appeared
in the faeces. Thus, excretion of PCP and its metabolites mainly
occurs via the kidneys, and to a lesser extent by the processes of
gastric and biliary secretion.
Table 26. Urinary and faecal excretion of 14C activity as a
percentage of a single dose of 14C-PCP
---------------------------------------------------------------
Species Dose Time % recovery in: Reference
(mg/kg (h) urine faeces
body
weight)
---------------------------------------------------------------
Monkey
male 10a 168 75 12 Braun & Sauerhoff
female 10a 360 70 17 (1976)
Monkey 30a 144 51.7 4.3 Ballhorn et al.
male 50a 144 44.9 11.3 (1981)
Rat 10a 216 80 19 Braun et al. (1977)
100a 192 64 34
Mouse 14.8b 96 83 8 Jakobson & Yllner
18.2b 96 62 5 (1971)
37.2b 96 73 4
35.2b 168 82 10
36.8b 168 80 12
---------------------------------------------------------------
a Oral (corn oil solutions).
b Intraperitoneal.
Only trace amounts of radioisotopes from the metabolism of
labelled compounds are expired. Although this route is of minor
importance, it could indicate other metabolic processes, provided
that analytical errors can be excluded. However, Larsen et al.
(1972) questioned whether expired 14C originated from PCP
metabolism, attributing it, instead, to impurities in the
radiolabelled PCP.
Ahlborg et al. (1974) found that the radioactivity from
labelled PCP excreted in the urine of mice treated intra-
peritoneally (10 mg/kg body weight) consisted of about 41%
unchanged PCP, 13% conjugated PCP, 24% unconjugated, and 22%
conjugated tetrachlorohydroquinone. For rats, the corresponding
values were 60%, 9 - 16%, 7%, and 16 - 22%, respectively (Ahlborg
et al., 1978).
Following an oral dose of 100 mg/kg body weight, the urinary
metabolites of 14C-PCP in rats accounted for 75% unchanged PCP, 9%
PCP glucuronide, and 16% tetrachlorohydroquinone. Levels of the
last compound were below detectable values in the blood-plasma
(Braun et al., 1977). The authors concluded that the rate-limiting
step for the elimination of the metabolites of PCP is the rate of
metabolism rather than that of urinary excretion and therefore, PCP
metabolites were unlikely to accumulate in the body.
The proportions of PCP glucuronide in urine may have been
underestimated to date; this metabolite has recently been shown to
undergo a partial hydrolysis under weakly acidic conditions in
urine (Lilienblum, 1985).
In contrast to the rodents, rhesus monkeys excreted all of the
14C activity in urine as unmetabolized PCP (Braun & Sauerhoff,
1976).
Goldfish, in addition to free PCP, mainly excrete sulfate and
glucuronide conjugates via 3 pathways of elimination: the amounts
of PCP lost by the branchial, renal, and biliary routes were 52,
24, and 22% of the total amount of PCP excreted by the fish in the
24 h following a 24-h exposure to 0.1 mg PCP/litre water. The
excretion of PCP from the body surface was minor. About 30% of the
PCP excreted via the gills was in the unchanged form, whereas
almost all the PCP excreted in both the bile and urine was
conjugated. PCP sulfate was the major conjugate in the branchial
and renal routes, while PCP glucuronide was the primary biliary
conjugate (Kobayashi & Nakamura, 1979b).
6.4.2 Human studies
The PCP concentration in human urine has been widely used as an
indicator of the PCP body burden (section 5.4), based on the fact
that renal excretion of PCP is the major elimination route in man.
In the study of Braun et al. (1979) (section 6.3.2), within 168 h
of ingesting 0.1 mg Na-PCP/kg body weight, volunteers excreted 74%
of the total dose in urine as PCP and 12% as PCP glucuronide.
About 4% of the total dose was eliminated in the faeces; this
amount consisted of equal parts of both free PCP and PCP
glucuronide. The fate of the remaining 10% of the dose was not
discussed.
Uhl et al. (1986) found that about 30% of PCP was excreted as
glucuronide in the urine of a volunteer, up to 4 days after a
single dose of 0.31 mg PCP/kg body weight (section 6.5.2). However,
in contrast to the study of Braun et al. (1979), the percentage of
PCP glucuronide gradually increased to reach about the normal range
determined for people not specifically exposed (65 ± 5% PCP
glucuronide) after about 14 days. The findings of Zimmerli et al.
(1979) and Janssens & Schepens (1984) also indicated that long-term
exposure result in a higher proportion of conjugated PCP than that
reported by Braun et al. (1979). On the average, two thirds of the
PCP detected in the urine samples of non-occupationally exposed
people was conjugated.
6.5 Retention and Turnover
6.5.1 Animal studies
Pharmacokinetic data on the retention and half-life of PCP in
the compartments also indicate that most of the PCP absorbed is
rapidly eliminated from the body.
The dynamics of elimination of PCP and its metabolites depend
on the species and the sex of the test animal. As summarized in
Table 27, the monkey differs from other animal species in showing a
much slower elimination rate as expressed by the half-life for the
clearance from urine and plasma, perhaps because monkeys do not
metabolize PCP.
An extensive enterohepatic circulation of PCP is also suggested
by the slow but steady elimination of 14C activity in the faeces of
the monkeys (Braun & Sauerhoff, 1976). On treating rhesus monkeys
with cholestyramine, this enterohepatic circulation was
interrupted as the cholestyramine bound the PCP and/or its
metabolites and bile acids, thus preventing their reabsorption
(Ballhorn et al., 1981). However, the results were derived from
only 4 monkeys in single studies.
Apart from the monkey studies, pronounced differences in PCP
kinetics that are only in part related to the species or type of
application have been observed in different single-dose studies
(Table 27). Braun & Sauerhoff (1976) and Braun et al. (1977)
observed a monophasic elimination of PCP in monkey, while, in rats,
2 phases could be distinguished (Fig. 4): an alpha-phase with a
rapid elimination rate (elimination half-lives, 13 - 17 h) followed
by a beta-phase (elimination half-lives for male rats, 40 h (10
mg/kg body weight), 121 h (100 mg/kg body weight)) with a
comparatively slow elimination rate. However, the beta-phase, is
not well defined, as it does not remain constant.
Braun et al. (1977) found that, at a higher dose (100 mg/kg
body weight), the female rats followed the monophasic scheme
(elimination half-life, 27 h) (Fig. 4). Hoben et al. (1976d) also
reported a monophasic elimination in rats, while Larsen et al.
(1972) and Zenzen (1979) described a distinct biphasic model. The
accuracy of the unusually slow beta-phase (102 days = 2448 h)
reported by Larsen et al. (1972) has been questioned by Braun et
al. (1977), since this value was obtained by subtracting the
cumulative amount excreted in the urine from the total dose,
without knowing the total recovery. On the other hand, the
monophasic scheme claimed by Braun et al. (1977) for female rats
dosed with 100 mg/kg body weight is not as distinct as it seems to
be; for, on omitting the last sample measured at 192 h, a biphasic
scheme could be fitted to the points as well (Fig. 4).
The amount of the PCP or 14C activity remaining in the body
several days after dosing differs markedly between test species.
According to Braun et al. (1977), in each case, 90% or more of the
radioactivity had been excreted by rats 3 days after dosing;
detectable levels of PCP remained only in liver and kidney, 9 days
after the 10 mg/kg dose (see also Table 23). As a result of a
slower elimination rate, about 11% of the administered 14C activity
remained in the body of rhesus monkeys 15 days after oral
application of 10 mg/kg body weight; approximately 80% of this
remaining activity was identified in the large and small intestines
and liver. Braun & Sauerhoff (1976) calculated that a steady-state
concentration of PCP in plasma would be reached by the 10th
repeated daily dose; the approximate plasma concentration at this
time was estimated to be 50 mg PCP/litre.
Several pathways of PCP degradation have been suggested.
Because of the tremendous number of microbial strains, numerous
metabolites have been identified as degradation products (Table
12). The possible steps of PCP decomposition as reported by
several authors are summarized in Fig. 3. The major metabolic
processes degrading PCP or its sodium salt are as follows (Suzuki,
1977; Kaufman, 1978; Reiner at al., 1978; Murthy et al., 1979; Rott
et al., 1979):
(a) methylation to yield the methylether of PCP,
pentachloroanisole;
(b) acylation of the hydroxyl group resulting in
pentachlorophenol acetate;
(c) dechlorination to tetrachlorophenols; and
(d) hydroxylation to tetrachlorodihydroxybenzenes.
The metabolites originating from these initial steps are subject to
further transformations as depicted in Fig. 3. Thus, a number of
substances may arise, which accumulate to different extents; the
limiting step is the ring fission to chlorinated aliphatic
compounds such as tetrachloromuconic acid (Lyr, 1962). Further
dechlorination may result in further transformations of the
aliphatic compounds (Janke & Fritsche, 1978) to form low molecular
substances such as acetic acid or succinate, which then enter the
tricarboxylic acid cycle. Suzuki (1983) reported that 34% of the
14C resulting from 14C-PCP microbial decomposition was recovered as
14CO2, the remaining compounds being unidentified 14C-metabolites.
Table 12. Metabolites formed by the microbial transformation of PCPa
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(1) pentachlorophenol acetate Rott et al. (1979)
(2) 2,3,4,5-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975); Murthy et al.
(1979)
(3) 2,3,5,6-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975); Murthy et al.
(1979)
(4) 2,3,4,6-tetrachlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(5) 2,4,5-trichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(6) 2,3,6-trichlorophenol Kuwatsuka & Igarashi (1975);
Murthy et al. (1979)
(7) 2,3,4-trichlorophenol Kuwatsuka & Igarashi (1975)
(8) 2,3,5-trichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(9) 2,4,6-trichlorophenol Kuwatsuka & Igarashi (1975)
(10) 3,4-dichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
(11) 3,5-dichlorophenol Ide et al. (1972); Kuwatsuka &
Igarashi (1975)
---------------------------------------------------------------------------
Table 12. (contd.)
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(12) 2,3,4,5-tetrachloroanisole Ide et al. (1972); Rott et al.
(acetate) (1979)b
(13) 2,3,5,6-tetrachloroanisole Ide et al. (1972); Rott et al.
(acetate) (1979)b
(14) 2,3,4,6-tetrachloroanisole Engel et al. (1966); Ide et al.
(acetate) (1972); Rott et al. (1979)b
(15) 2,3,5-trichloroanisole Ide et al. (1972)
(16) 2,4,5-trichloroanisole Ide et al. (1972)
(17) 3,4-dichloroanisole Ide et al. (1972)
(18) 3,5-dichloroanisole Ide et al. (1972)
(19) 3-chloroanisole Ide et al. (1972)
(20) pentachloroanisole Cserjesi & Johnson (1972); Ide et
al. (1972); Kuwatsuka & Igarashi
(1975); Murthy et al. (1979);
Rott et al. (1979)
(21) tetrachlorocatechol Suzuki (1977); (Rott et al.
(diacetate) (1979)b
(22) tetrachlorohydroquinone Suzuki (1977)
(23) tetrachlororesorcinol Rott et al. (1979)b
(diacetate)
(24) tetrachlorohydroquinone Rott et al. (1979)
dimethylether (diacetate)
(25) tetrachlorobenzoquinone Reiner et al. (1978)
(26) trichlorohydroxybenzoquinone Reiner et al. (1978)
(27) 2,3,6-trichlorohydroquinone Reiner et al. (1978)
(28) 2,6-dichlorohydroquinone Reiner et al. (1978)
(29) 2-chlorohydroquinone Reiner et al. (1978)
(30) 14CO2 Chu & Kirsch (1972); Kirsch &
Etzel (1973); Suzuki (1977)
---------------------------------------------------------------------------
Table 12. (contd.)
---------------------------------------------------------------------------
Substance Reference
---------------------------------------------------------------------------
(31) Cl- Watanabe (1973); Suzuki (1977)
(32) tetrachloromuconic acid Lyr (1962)
(33) beta-hydroxytrichloromuconic Lyr (1962)
acid
---------------------------------------------------------------------------
a Adapted from: Kaufman (1978).
b Reference refers to acetate form.
4.2.2.1 Aquatic degradation
Boyle et al. (1980) studied the effects of dissolved oxygen
supply, light, pH, and the presence of hydrosoil on the aquatic
biodegradation of PCP. Light, high pH, and high oxygen
concentrations led to the most rapid and complete breakdown of PCP.
PCP disappeared from the pond water of aquaria at differential
rates, with half-lives ranging from 12.8 to 18.6 days, except for
the anaerobic aquarium without hydrosoil (79.8 days). The absence
of light and mud associated with low pH and anaerobic conditions
favoured the persistence of PCP, indicating that the phenolic form
of PCP is more persistent than the phenate form and that the
oxidative pathway is the major mechanism for PCP degradation in
simulated lake environments. Thus, PCP may be most persistent in
the deoxygenated hypolimnion of stratified lakes.
This effect of oxygen on biotransformation was confirmed by Liu
et al. (1981), who calculated the half-lives of PCP in aerobic and
anaerobic metabolic fermentors to be 0.36 and 192 days,
respectively. These half-lives are much lower than those reported
by Boyle et al. (1980), probably because Liu et al. (1981) used an
acclimatized culture whereas in the other studies natural pond
water was employed. This illustrates how important the
preadaptation of microorganisms is for their capacity to degrade
PCP.
Pignatello et al. (1983) showed that the aquatic micro-flora
can adapt to PCP and can become the most important factor for
clearing contaminated surface water of PCP, particularly in deeper
waters where the photolytic contribution is minimized.
Liu et al. (1981) investigated the impact of co-metabolites
and of different nitrogen sources on PCP biodegradability.
Peptone, glucose, and the sodium salt of 4-chlorophenol suppressed
the degradation rate while yeast extracts stimulated PCP
decomposition; the basis for these various effects is not clear.
In a report by Pierce & Victor (1978), levels of PCP in a man-
made lake in Mississippi increased from background levels (0.3
µg/litre) to 150 - 277 µg/litre, immediately after an accidental
overflow from a pole-treatment plant, decreasing to 5 - 16 µg/litre
within 4 months. Long-term influx of PCP from the contaminated
watershed and persistence in organically rich sediments (4 - 1518
µg/kg dry weight) were thought to provide a source of long-term
pollution of the aquatic ecosystem (sections 5.1.4 and 6.5.1).
Since anaerobic biodegradation of PCP is relatively slow, PCP
persists in sediments considerably longer than in water.
Accordingly, high concentrations of PCP have been measured in river
and lake sediments (section 5.1.2).
The fate of PCP in estuarine sediments was investigated
following contamination with 11 tonnes of PCP after a ship
collision (De Laune et al., 1983). Although the contaminated
sediments had been removed by vacuuming 2 weeks after the accident,
as much as 1.6 mg/kg (dry weight) could be detected 1 1/2 years
later compared with < 5 µg/kg at remote sampling points.
Laboratory studies showed a much faster breakdown under aerobic
conditions (70% decomposition at pH 8 within 5 weeks) compared with
anaerobic conditions (10% decomposition).
The high persistence of PCP in the sediment has been confirmed
by studies on the fate of PCP in a bay of Lake Ontario (Canada),
which has been contaminated by a wood-preserving plant since the
1940s (Fox & Joshi, 1984). The rather constant T4CP:PCP ratio
throughout the core (depth of 15 cm corresponding to the year 1949)
indicates that almost no degradation of these chlorophenols
occurred, once they had been incorporated into sediments.
Of the possible PCP metabolites, tetrachlorocatechol seems to
be among the most persistent. Compared with all other chlorinated
phenols studied, this substance showed the highest levels in the
sediment (3.2 - 348 µg/kg dry weight) or plankton (108 µg/kg wet
weight) of Finnish lakes contaminated with wood preservatives and
chlorobleaching wastes (Paasivirta et al., 1980).
4.2.2.2 Degradation in soil
As in water, the biodegradability of PCP in soil depends on the
type and the physiological state of the microorganisms, but several
environmental factors also influence this process, such as a high
amount of organic matter or high moisture content, which enhance
the biodegradation of PCP (Young & Carrol, 1951; Kuwatsuka, 1972).
Low temperatures (O °C, 4 °C) have proved unfavourable for the
growth of Pseudomonas species and hence their PCP breakdown
activity, while, at 20 °C, PCP (50 mg/litre) was degraded to about
50% within 8 days (Trevors, 1982a). Low pH values also reduced the
microbial breakdown of PCP (Stanlake & Finn, 1982).
The effects of oxygen on biodegradation vary: in some
instances, anaerobic conditions in soils increase the rate of
degradation, apparently as reducing conditions promote reductive
dechlorination (Ide et al., 1972). On the other hand,
biodegradation in terms of formation of the intermediate
pentachloroanisole was significantly greater in aerobic than in
anaerobic soil (Murthy et al., 1979). An average loss of 88% of
100 mg PCP/kg from clay soil was detected under aerobic conditions
(over 160 days at 23 °C) compared with only 7% loss under anaerobic
conditions (Baker & Mayfield, 1980).
The half-life of PCP in moist farm soils (initial
concentration, 100 mg/kg) ranged from 7 to 14 days, depending on
the soil varieties. PCP degradation was inhibited by certain
fungicides and also under submerged conditions (Suzuki, 1983).
Other half-lives reported are 10 - 70 days, under flooded
conditions, and 20 - 120 days under upland conditions (Kaufman,
1978).
Edgehill & Finn (1983) investigated the use of PCP-degrading
bacteria as a prophylactic measure to decontaminate soil after
accidental PCP spills or when PCP-treated poles are set up near
surface waters. Direct inoculation of acclimated Arthrobacter
cells into the soil enhanced the disappearance of PCP at least 10-
fold; the half-life of PCP in the soil incubated at 30 °C in the
laboratory was reduced from 12 - 14 days to about 1 day. However,
outdoor trials demonstrated that the efficacy of this measure was
limited under natural conditions, as thorough mixing of the soil at
the time of inoculation was required to achieve a 85% reduction of
the extractable PCP at 12 days compared with only 15 - 30% without
mixing.
4.3 Degradation by Plants
Degradation of PCP may also take place in plants. Rice plants
were found to absorb about 3% of the radioactivity of 14C-PCP
applied on the soil, of which 50% could be extracted, mainly as
unchanged PCP, 9% as unidentified conjugates, and about 1% as a
tetrachlorophenol-isomer (Haque et al., 1978).
Weiss et al. (1982a) also studied the metabolism of 14C-PCP
(23 kg/ha) in rice plants. Rice roots contained 0.14% of the
applied radioactivity as unchanged PCP, 3.95% as unextractable
residues, and 1.08% as various metabolites. The influence of soil
microorganisms was not excluded, but the authors regarded the
detection of hydroxylated and methoxylated tetrachlorobenzenes as
evidence of plant metabolism, because these substances were not
found in soil (Weiss et al., 1982a,b).
The metabolism of PCP was also investigated under aseptic
conditions using soybean ( Glycine max L.) and wheat ( Triticum
aestivum L.) cell suspension cultures in which the isolated
conversion products could be attributed to the metabolic activity
of the plant cell cultures themselves (Langebartels & Harms, 1984).
The beta-D-glucoside of PCP was identified as the main conjugate
formed by the cell suspensions. Anisoles and lower chlorinated
phenols were not detected; however, some PCP was incorporated into
a non-extractable fraction. Sandermann et al. (1984) isolated
polar conjugates from wheat and soybean cell cultures, and
demonstrated that covalent incorporation of PCP into lignin takes
place.
4.4 Ultimate Fate Following Use
4.4.1 General aspects
The amount of PCP entering the environment and its subsequent
fate can be controlled at point sources where high amounts of PCP
are used, such as preservation plants. However, because of its many
applications, PCP is released into the environment from a number of
diffuse sources and is subject to transport and transformation in
different environmental compartments, as outlined in the previous
sections. The evaporation data cited above (section 4.1.1) suggest
that a significant fraction of the entire production of PCP will
ultimately enter the atmosphere.
4.4.2 Disposal of waste water
As shown in Table 13, municipal sewage discharges contain only
low PCP concentrations, whereas effluents from wood-treating
factories may contain considerable amounts of PCP, depending on the
intensity and efficacy of treatment measures prior to discharge.
One method of handling wood preservation effluents in Canada is to
store waste water on company property and allow it to evaporate,
which obviously contributes to air pollution. The disposal of
waste water is also achieved by incineration (section 4.4.3) and by
secondary treatment before discharge into the receiving water
(Hoos, 1978).
Most small wood-treatment plants handle wastes by incineration
or lagooning, while larger manufacturers treat their wastes.
Primary treatment is often applied when PCP is dissolved in a
carrier oil: gravity separation is used to recover oil and PCP for
recycling or treatment, while some plants remove oil droplets or
wood particles by filtration (Richardson, 1978).
Several laboratory and treatment-plant studies have shown that
PCP can be degraded by activated sludge (Dust & Thompson, 1973;
Kirsch & Etzel, 1973; Etzel & Kirsch, 1974; Moos et al., 1983;
Guthrie et al., 1984; Hickman & Novak, 1984). However, in full-
scale treatment plants, the treatment efficiency is often reduced.
For example, according to a US EPA survey, 8 out of 14 publicly-
owned treatment plants could not remove any of the PCP load. Most
of the removal efficiency of the remaining plants (6 - 87%) was
attributed to adsorption on solids (Feiler, 1980).
Biodegradability strikingly decreases when commercial PCP is
introduced unless the input concentrations are reduced (Reiner et
al., 1978). In the presence of more readily degradable substrates,
PCP degradation is suppressed. Moreover, activated sludge is not
usually protected from shock loads, unless acclimatized to PCP
(Hickman & Novak, 1984).
Some other treatment systems appear to be appropriate for the
treatment of PCP-contaminated waste water, but their suitability
has not as yet been demonstrated on a large scale. Degradation of
PCP in a biofilm reactor only occurred when the microflora was
attached to solid support material, such as soft-wood bark
(Apajalahti & Salkinoja-Salonen, 1984; Salkinoja-Salonen et al.,
1983, 1984). PCP concentrations exceeding 1 mmol (266 mg/litre),
and also some toxic solvents, such as chloroform, inhibited PCP
biodegradation.
Table 13. Levels of PCP in industrial and municipal discharges in
different countries
---------------------------------------------------------------------------
Country Type of waste water PCP Reference
(µg/litre)
---------------------------------------------------------------------------
Canada Effluents from wood 0.6 Environment
preservation industry at 225 Canada (1979)
4 sites in British NDa
Columbia 2760
Canada Industrial and municipal Garrett (1980)
discharges in the Greater
Vancouver Area:
- average from 22 sites 5.1
- range (0.2 - 42.5)
- drainage ditches 6000
2520
1125
Denmark Municipal sewage Folke & Lund
- influents 0.2 - 0.7 (1983)
- effluents 0.1 - 2.4
Germany, Effluents from sewage 20 - 680 Dietz &
Federal treatment plant receiving Traud (1978b)
Republic waste water from paper mill
of
Effluents from various 1 - 130
industries
USA Municipal sewage Buhler et al.
- influents 1.4 - 4.6 (1973)
- effluents 1 - 4.4
USA Samples from wood-treating Ervin &
factories McGinnis
- untreated waste water 17 000 - 32 000 (1980)
- treated waste water 160 - 75 000
USA Effluents from wood- 25 000 - 15 0000 Thompson &
treatment factories Dust (1971)
---------------------------------------------------------------------------
a ND = not detectable.
Hakulinen & Salkinoja-Salonen (1982) reported on the efficiency
of a fluidized bed reactor in removing chlorophenols from pulp and
paper industry bleaching effluents. Chlorophenols including PCP
were completely mineralized in the anaerobic reactor. The aerobic
part of the system served as an after treatment unit to remove the
remaining organic load.
Adsorption on to activated carbon has also been used in
treating contaminated waste waters; removal of PCP approaches 100%
using this method (Richardson, 1978).
4.4.3 Incineration of wastes
As considerably increased amounts of PCDDs can be emitted
during the combustion of PCP-treated material compared with
untreated samples (section 2.2.1), the incineration of PCP-
containing wastes is problematical. Since temperature of burning
and the residence time cannot be controlled in the fire-places of
private homes, the incineration of wood treated with chlorophenols
is a potential source of PCDD/PCDF emission. Moreover, accidental
burning of chlorophenols can lead to considerable emissions of
these compounds: Kauppinen & Lindroos (1985) estimated that the
burning of 100 kg chlorophenol formulation during a saw-mill fire
would result in 20 g of PCDDs.
According to Powers (1976), "the complete and controlled high
temperature oxidation coupled with adequate scrubbing and ash
disposal facilities offers the greatest immediate potential for the
safe disposal of concentrated pentachlorophenol". The destruction
of PCP in treated wood in a controlled air incinerator was achieved
with efficiencies greater than 99.99% at combustion temperatures of
between 916 and 1032 °C (Stretz & Vavruska, 1984). The analytical
results showed no evidence of T4CDD or T4CDF, both in the hot zone
between primary and secondary chambers and in the offgas, at
detection limits of 1 ppb or 5 ppb, respectively.
There are many other sources of PCDDs and PCDFs from
combustion processes. The incineration of municipal waste may be
the largest source of PCDD and PCDF emissions into the environment
(Ballschmiter et al., 1983; Chiu et al., 1983; Tiernan et al.,
1983). The various sources of these compounds are discussed in
more detail in the corresponding reviews on PCDDs and PCDFs, e.g.,
Umweltbundesamt (1985), Karasek & Hutzinger (1986), and in
Boddington et al. (1985).
The thermal conversion of organochlorine compounds, e.g.,
polyvinylchloride and polyvinylidene chloride, can be a source of
atmospheric chlorophenols including PCP (Ahlborg et al., 1986;
Dougherty, 1986a).
Other common methods of waste disposal such as deep-sea or
deep-well disposal, landfill sites, or open pits should not be
considered as a means for disposing of PCP-containing wastes,
because of the mobility of PCP (Powers, 1976; Crosby et al., 1981).
---------------------------------------------------------------------------
a Personal communication to the Task Group on Pentachlorophenol.
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental Levels
5.1.1. Air
While PCP concentrations in the air at industrial sites and in
rooms contaminated with PCP have been reported (section 5.2), there
is apparently little information on PCP levels in the ambient air.
Cautreels et al. (1977) sampled airborne particulate matter
near La Paz, Bolivia, at an altitude of 5200 m and in a residential
city area of Antwerp, Belgium. At a detection limit of 0.02 ng/m3,
the atmosphere of the Bolivian mountain rural area contained 0.25 -
0.93 ng/m3 and that of the Antwerp urban area 5.7 - 7.8 ng/m3 air,
respectively. More recent analytical results (Bundesamt für
Umweltschutz, 1983) showed PCP air concentrations ranging between
0.9 and 5.1 ng/m3 in Switzerland.
The ubiquitous occurrence of PCP in ambient air can also be
shown from rain water and snow analyses. Rain water collected in
Canada (Jones, 1981), Hawaii (Bevenue et al., 1972) and West Berlin
(Rosskamp, 1982) contained between 0.002 and 0.3 µg PCP/litre.
Water melted from snow in southern Finland revealed PCP
concentrations of 0.15 and 0.98 µg/litre, respectively. PCP
fallout as calculated from Finnish snow samples ranged from 1.49 to
136.0 µg/m2 (Paasivirta et al., 1985).
5.1.2. Water and sediments
Levels of industrial and municipal discharges in different
countries are shown in Table 13 (section 4.4.2). Municipal sewage
discharges contain PCP concentrations at levels comparable with
those in surface waters. However, wood-treating factories may
contribute substantially to the PCP load in surface waters, which
ranges from non detectable to 10 500 µg PCP/litre (Table 14),
depending on the extent of pollution by different sources.
The majority of the water samples analysed for PCP contained
less than 10 µg/litre, most contained less than 1 µ PCP/litre. The
extreme PCP levels of up to 10 500 µg/litre reported by Fountaine
et al. (1976) were found in a highly polluted stream near an
industrial area in the vicinity of Philadelphia, USA.
Ernst & Weber (1978b) calculated the PCP input into the German
Bight via the river Weser to be of the order of 1000 kg per year,
assuming an average PCP level of 0.1 µg PCP/litre and a water flow
of 300 m3/second. Taking an average concentration of 0.5 µg
PCP/litre in the surface waters of the Federal Republic of Germany
(Foquet & Theisen, 1981), the total load in all surface waters of
the Federal Republic of Germany was estimated by Fischer (1983) to
be in the range of 60 tonnes per year, of which 30 - 40 tonnes are
transported by the Rhine river.
Table 14. PCP concentrations in surface waters of different countries
---------------------------------------------------------------------------
Country Surface water PCP (µg/litre) Reference
and location (range, mean)
---------------------------------------------------------------------------
Canada Fresh-water sites in trace - 0.30 Environment,
British Columbia (BC) Canada (1979)
Marine sites in BC NDa - 7.3
Germany, Weser river and estuary 0.05 - 0.5 Ernst & Weber
Federal (1978b)
Republic German Bight < 0.002 - 0.026
of
Ruhr river < 0.1 - 0.2 Dietz & Traud
(0.1) (1978b)
Rhine river, Cologne 0.1 Fischer & Slem-
rova (1978)
Japan Tama river, Tokyo 0.1 - 0.9 Matsumoto et
0.01 - 0.09 al. (1977)
Sumida river, Tokyo 1 - 9
River water, Tokyo 0.18 ± 0.14 Matsumoto
area (1982)
Nether- Rhine river 1976 Max.b 2.4 (0.7) Wegman &
lands Rhine river 1977 Max. 11.0 (1.1) Hofstee (1979)
River Meuse 1976 Max. 1.4 (0.3)
River Meuse 1977 Max. 10.0 (0.8)
South 124 sampling points ND - 0.85 Van Rensburg
Africa (1981)
Sweden River water downstream 9 Rudling (1970)
from pulp mill
Lake receiving 3
discharges
---------------------------------------------------------------------------
Table 14. (contd.)
---------------------------------------------------------------------------
Country Surface water PCP (µg/litre) Reference
and location (range, mean)
---------------------------------------------------------------------------
USA Willamette river 0.1 - 0.7 Buhler et al.
(1973)
Highly polluted stream Fountaine et
near Philadelphia al. (1976)
- factory location 4500 - 10 500
- downstream 49 - 240
Estuary in the ND - 0.01 Murray et al.
Galveston Bay, Texas (1981)
Pond in Mississippi < 1 - 82 Pierce et al.
contaminated by waste (1977)
from pole-treatment
plant
---------------------------------------------------------------------------
a ND = not detectable.
b Max. = maximum values.
Wong & Crosby (1981) reported PCP concentrations ranging from 1
to 800 µg/litre (average, 227 µg/litre) in the surface pond water
near a local wood-treatment factory and of about 20 µg PCP/litre in
agricultural drainage. Elevated PCP concentrations were also found
in groundwater (3.03 - 23.3 µg/litre) and surface water samples
(0.07 - 31.9 µg/litre) within saw-mill areas. Around these sites,
PCP levels ranged between not detectable and 0.6 µg/litre in
groundwater and between 0.01 and 0.07 µg/litre in the water of a
nearby lake (Valo et al., 1984). PCP in the µg/litre range was
detected in the water seeping from a landfill (Kotzias et al.,
1975). A level as high as 3.35 mg/litre was found in groundwater
from a monitoring well near a wood-preservation factory (Thompson
et al., 1978).
A PCP-monitoring study in water was performed by Rahde & Della
Rosa (1984, 1986) in a region of the Amazon jungle (Tucurui,
Brazil). The construction of a dammed reservoir affected a large
area (2430 km2) with sawmills and PCP-treated wood. Water samples
collected from the main river and its affluents before the flooding
in 1984 contained between 5 and 14 µg PCP/litre. In 1984-85, after
the flooding, the area had been covered with about 46 billions m3
of water, PCP was not detectable at a detection limit of 4 µg/litre.
In general, the sediments of a water body contain much higher
levels of PCP than the overlying waters. At several fresh-water
and marine sites in British Columbia, Canada, receiving effluents
from the wood-treatment industry, average PCP levels in the
sediments ranged from not detectable to 590 µg/kg, while the
corresponding range in the overlying waters was from not detectable
to 7.3 µg/litre (Table 14). During a 1978 survey of toxic
substances in the Great Lakes of Canada, sediment samples from the
Thunder Bay, Marathon, and Michipicoten areas of Lake Superior
contained averages of 16 900, 7300, and 2300 µg PCP/kg dry
sediment, respectively. In another study of contamination from a
wood preservation facility on the Bay of Quinte, Lake Ontario, Fox
& Joshi (1984) analysed water and sediment samples for PCP. At a
site distant from the plant discharge, sediment PCP levels ranged
from 1 to 61 µg/kg dry weight, while surface waters contained only
0.015 µg/litre. Sediments from the Mississippi lake monitored by
Pierce & Victor (1978) averaged 364 µg/kg dry sediment, compared
with levels in the lake water of only 0.1 µg/litre. A similar
distribution was observed in surface waters in the Netherlands
(Wegman & van den Broek, 1983); sediment samples from Lake
Ketelmeer, a deposition area for Rhine river sediments, contained a
median PCP concentration of 8.4 µg/kg dry weight, while the
overlying water contained 0.41 µg/litre. PCP concentrations in
sediment samples collected in the vicinity of a paper mill
discharge pipe in a North Sea bight, two years after going out of
use (Butte et al., 1985) and in Finnish lakes contaminated by wood
preservatives (Paasivirta et al., 1980) were of the same order of
magnitude.
These examples indicate that PCP and Na-PCP adsorb on
sediments, which concurs with findings from experimental work.
Strufe (1968) reported a study in which 65% of added Na-PCP
adsorbed on river mud within 20 h.
5.1.3. Soil
Soil samples, taken at 4 sites in the vicinity of a Swiss PCP-
producing facility (Dynamit Nobel), contained between 25 and 140 µg
per kg (dry weight) at a depth of 0 - 10 cm and between 33 and 184
µg/kg at 20 - 30 cm. These levels are higher than the PCP
concentrations of 35 µg/kg (0 - 10 cm) and 26 µg/kg (20 - 30 cm)
determined in soil samples from a "reference site". The
simultaneous presence of some PCDDs and PCDFs (maximum values:
H7CDD, 0.6 µg/kg; OCDD, 7.68 µg/kg; P5CDF, 1 µg/kg at 0 - 10 cm) in
sample sites near the chemical factory compared to only one
positive sample (H7CDF, 0.51 µg/kg) at the remote site confirmed
the contamination (Bundesamt für Umweltschutz, 1983).
The soil surrounding Finnish sawmills was found to be heavily
contaminated with up to 45.6 mg/kg (0 - 5 cm) or 1 mg PCP/kg fresh
weight (80 - 100 cm) near the treatment basin, up to 0.14 mg/kg in
the storage area for preserved wood and 0.012 mg/kg outside the
storage area. The vertical distribution of chlorophenols including
PCP explains the ground-water contamination observed (Valo et al.,
1984).
In Canada, soil samples from the former site of a pesticide
plant contained less than 50 µg PCP/kg (Garrett, 1980). The PCP
levels in the leachate and in soil in the vicinity of 3 waste-
disposal sites were also in the µg/kg range (Kotzias et al., 1975).
Samples of agriculturally used soils in Bavaria (Federal Republic
of Germany) contained about 100 µg PCP/kg (Gebefuegi, 1981).
PCP concentrations in soil samples taken at a distance of 2.5,
30.5, and 152.5 cm from poles treated with PCP were 658, 3.4, and
0.26 mg/kg, respectively. Arsenault (1976) considered the last
value as a "natural background level", which he derived from the
blank of 0.2 - 0.4 ppm found in unexposed soil samples. However,
such a level seems very high for a substance that does not appear
to occur naturally. This high level could be the result of the
contamination of the soil or of the reagents used for analysis.
5.1.4. Aquatic and terrestrial organisms
5.1.4.1 Aquatic organisms
Levels of PCP in aquatic organisms from various collection
sites are listed in Table 15. No data are available on the
background levels of PCP in biota. All sampling sites in Table 15
were more or less contaminated with industrial effluents.
Relatively low contamination is reflected by residues of PCP in
aquatic invertebrate and vertebrate fauna in the low µg/kg-range.
For example, Zitko et al. (1974) found a range of < 0.5 - 4 µg
PCP/kg wet weight in the muscle tissue of different fish species
(Table 15). Higher levels were detected in organisms collected in
surface waters that were thought to be contaminated with wood
preservatives: up to 2100 µg PCP/kg wet weight were found in marine
fish in British Columbia, Canada (Environment Canada, 1979) and up
to 6400 µg/kg in fresh-water fish from Finnish lakes (Paasivirta et
al., 1981) (Table 15). Some sediment-dwelling organisms showed the
highest residues: polychaetes from the Weser estuary contained
between 103 - 339 µg PCP/kg wet weight (Ernst & Weber, 1978a).
Even higher levels (266 - 133 000 µg/kg) were found in clams from a
North Sea bight, near the end of a waste-water pipe from which
about 26 tonnes of PCP were discharged into the mud flats until
1978 (Butte et al., 1985).
Residues of PCP in biota associated with toxic PCP water
concentrations are in the mg/kg range. Following extensive
application of Na-PCP as a molluscicide in rice fields in Surinam,
Vermeer et al. (1974) found 8.1 mg PCP/kg wet weight in dead frogs
(Pseudis paradoxa) and between 31.2 and 59.4 mg/kg in three species
of fish, which were also found dead. Composite samples of snails
(Pomacea glauca) contained, on average, 36.8 mg PCP/kg wet weight.
Whole samples of small fish collected from a river in British
Columbia, Canada, during an accidental fish kill resulting from the
spraying of hydropoles, had levels of 16.3 mg PCP/kg; two large
cutthroat trout (Salmo clarki) contained 10.3 mg/kg (Jones, 1981).
Table 15. PCP residues in aquatic animalsa
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Invertebrates
Jellyfish whole Gulf of Mexico 1979 0.1 - 1 wet Kuehl & Dougherty (1980)
Sponge whole Finnish lakes Summer 1.9 - 13 wet Paasivirta et al. (1980)
contaminated with 1978
wood preservatives
Sagartia whole Weser estuary and 1976-77 2.7 - 7 wet Ernst & Weber (1978a)
troglodytes German Bight
(actinian)
Polychaete whole 1978 103 - 339 wet
(Lanice
conchilega)
Mussel muscle Finnish lakes Summer 1.7 - 5.6 wet Paasivirta et al. (1980)
contaminated with 1978
wood preservatives
Clam muscle Marine sites near Autumn ND - 12 wet Environment Canada
( Macoma sp.) wood-preservation 1978 (1979)
factories in British
Columbia, Canada
Clam whole Wadden sediment of 1980-81 266 - 133 000 dry Butte et al. (1985)
(Mya (without Jadebusen, bight of (median: 800)
arenaria) shells) the North Sea, PCP
discharged area until
1978
Reference site 266 - 532
Crayfish muscle Fresh-water sites Autumn ND - trace wet Environment Canada
( Pacifas- near wood-preserv- 1978 (1979)
tacus sp.) ation factories in
British Columbia,
Canada
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Crab muscle Marine sites near Autumn ND - 20 wet Environment Canada
(Cancer wood-preservation 1978 (1979)
magister) factories in
British Columbia,
Canada
Crab muscle Marine sites near Autumn trace - 7 wet
(Cancer wood-preservation 1978
productus) factories in
British Columbia,
Canada
Brown shrimp whole Estuary of the 1980 4 - 17 wet Murray et al. (1981)
(Penaeus Galveston Bay area
aztecus) of Texas
Blue crab soft 1.9 - 4.1 wet
(Calinectes tissues
sapidus)
Dwarf squid whole 1.4 - 4.3 wet
(Lollingnuula
brevis)
Vertebrates
Sculpin muscle Marine sites near Autumn trace - 84 wet Environment Canada
(marine) liver wood-preservation 1978 trace - 2100 wet (1979)
(Leptocottus factories in
armatus) British Columbia,
Canada
Sculpin muscle Fresh-water sites 5 - 100 wet
(freshwater) liver near wood-preserv- trace - 600 wet
(Cottus ation factories in
asper) British Columbia,
Canada
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Pike muscle Finnish lakes Summer 6.5 - 8 wet Paasivirta et al. (1980)
(Esox contaminated with 1978
lucius) wood preservatives
Roach muscle 0.9 - 12.8 wet
(Rutilus
rutilus)
Pike muscle Finnish lakes May 11.9 - 94.3 wet Paasivirta et al. (1981)
(Esox contaminated with 1980 (maximum 6400)
lucius) wood preservatives
Pike muscle Finnish lakes Spring/ 15.9 - 18.9 wet Paasivirta et al. (1983)
(Esox contaminated with Summer
lucius) wood preservatives 1981
Pike muscle Finnish lakes Summer 8.2 - 17.3 wet Paasivirta et al. (1985)
(Esox contaminated with 1982
lucius) wood preservatives
1983 1.2 wet
Crab muscle 1982 9.4 - 41.5 wet
(Rutilus 1983 0.5 wet
rutilus)
Baltic muscle 1983 1.8 - 4.7 wet
salmon
Winter muscle Estuaries in New Autumn 1.8 - 4 wet Zitko et al. (1974)
flounder Brunswick, Canada 1972
(Pseudopleuro-
nectus)
---------------------------------------------------------------------------------------------------------
Table 15. (contd.)
---------------------------------------------------------------------------------------------------------
Organism Type of Location of sample Sample Concentration Basis Reference
sample date (µg/kg)a
---------------------------------------------------------------------------------------------------------
Cod muscle 0.8 wet
(Gadus
morhua)
Sea raven muscle < 0.5 wet
(Hemipterus
americanus)
Atlantic whole Estuaries in New Autumn 0.5 - 1.3 wet Zitko et al. (1974)
salmon Brunswick, Canada 1972
(Salmo salar)
White shark liver 10.8 wet
(Carcharodon
carcharias)
Flounder whole Estuary in the 1980 1.6 - 3.5 wet Murray et al. (1981)
Galveston Bay
Longnose whole 4.7 - 5.6 wet
killifish
(Fundulus
similis)
---------------------------------------------------------------------------------------------------------
a ND = not detectable.
5.1.4.2 Terrestrial organisms
As with aquatic plants, almost no data are available on
residues of PCP in terrestrial plants. Grass samples taken in the
vicinity of a PCP producer at Rheinfelden, Switzerland, contained
between 67 - 87 µg PCP/kg dry weight, comparable to the PCP
concentration of 87 µg/kg found in grass from a reference site
(Bundesamt für Umweltschutz, 1983).
Reported residue levels in terrestrial vertebrates are mainly
related to domestic animals exposed to PCP: the tissues and blood
of cows and calves of dairy herds in the USA showed unquantified
PCP contamination (Hoeting, 1977). One herd housed in a PCP-
treated wooden barn had blood-PCP levels of 270 - 570 µg/litre (US
EPA, 1978).
Pentachloroanisole, a metabolite of the PCP biodecomposition,
causes a musty taint in broiler chicken tissues. It appears that
the chloroanisole arises through the microbial methylation of PCP
in wood shavings used as chicken litter (Curtis et al., 1972; Parr
et al., 1974; Harper & Banave, 1975). Wood shavings have been used
as litter not only for broiler chickens, but also for turkeys,
ducks, pigs, and cattle.
Neidert et al. (1984) found low residue levels of PCP in all
1072 chicken liver and 723 fat samples examined (most < 0.01
mg/kg), indicating an overall exposure of poultry to PCP. Only
0.75% of the liver samples contained PCP levels higher than 0.1
mg/kg.
In the field study of Vermeer et al. (1974) mentioned earlier,
PCP was detected in liver samples of birds (0.06 - 0.19 mg/kg wet
weight) residing in the vicinity of PCP-treated rice fields. High
PCP residues were found in the brain (mean, 11.3 mg/kg wet weight),
liver (46.6 mg/kg), and kidney (20.3 mg/kg) of dead snail kites
(Rostrhamus sociabilis), which had probably ingested Na-PCP
contaminated snails.
Only a few data on PCP residues in terrestrial animals,
apparently not exposed to PCP, have been reported: purple martin
fledglings from Alberta, Canada, contained 31 µg PCP/kg (Jones,
1981). The muscle tissue of juvenile starlings, collected from
their nests in South Finland in 1982 and 1983, contained PCP levels
ranging from not detectable to 59 µg/kg wet weight (mean, 5.9
µg/kg) (Paasivirta et al., 1985). The pectoral muscles of white-
tailed eagles, also collected in Finland, contained between 14 and
8571 µg PCP/kg wet weight, while levels in eggs ranged from not
detectable to 25 µg/kg. Eggs of osprey contained between 1 and 803
µg PCP/kg.
5.1.5. Drinking-water and food
PCP concentrations ranging from < 1 to 50 µg/litre were
detected in domestic well water (Oroville, California) (Wong &
Crosby, 1981). Buhler et al. (1973) analysed drinking-water
obtained from the Willamette river (USA). They found 0.06 µg
PCP/litre in the finished water. PCP was found at a level of 0.1
µg/litre in one water sample (Dougherty & Piotrowska, 1976b).
Concentrations of 0.01 - 0.02 µg PCP/litre were detected in
drinking-water in the Ruhr area of the Federal Republic of Germany
(Dietz & Traud, 1978b). PCP levels in Florida drinking-water
supplies ranged from 0.003 to 0.34 µg/litre (Morgade et al., 1980).
Detrick (1977) suggested that the chlorination of phenol in water
supplies might be responsible for the wide occurrence of PCP. The
chlorination of 1 mg phenol/litre by 10 mg chlorine/litre is said
to yield about 0.2 µg PCP/litre, which is comparable with the
levels found in drinking-water. However, the odour threshold for
phenol is in the µg/litre range, thus low levels of phenol can
generally be detected in water.
Most data on PCP residues in food have been collected in the
USA, where a number of pesticides, including PCP, have been
routinely monitored in the FDA Market Basket Survey. In 1973-74,
PCP was found in 10 out of 360 composite food samples, at
concentrations ranging from 10 to 30 µg/kg (Manske & Johnson,
1977). In 1975, 5.4% of a total of 240 samples were contaminated
with PCP at 10 - 40 µg/kg (Johnson & Manske, 1977). Values for the
period 1965-70 are shown in Table 16. PCP concentrations measured
in daily diet samples in the Federal Republic of Germany
(Gebefuegi, 1981) are similar, averaging 16.3 µg/kg (range, 2.6 -
27.5 µg/kg). Krause (1982) found elevated PCP concentrations in
the food-basket samples of persons applying wood preservatives in
private homes. Two-thirds of the samples analysed contained between
2 and 13 µg PCP/kg with a median of about 6 µg PCP/kg, whereas the
control samples fell between less than 0.1 and 5 µg/kg.
Samples of agricultural produce taken by the Alberta Department
of Agriculture (Canada), consisting mainly of potatoes and raw
milk, contained PCP levels of less than 10 µg/kg. In isolated
samples, PCP levels of up to 2700 µg/kg occurred, as a result of
contamination from storage containers made of treated wood (Jones,
1981).
In southern Ontario, Canada, 45 bovine milk samples collected
from bulk transports hauling milk were analysed for chlorophenols
(Frank et al., 1979). PCP was not detected in whole milk at a
detection level of 0.1 µg/litre.
In analysing commercial mushrooms for PCP residues, Meemken et
al. (1982) found that levels in 11 out of 17 fresh mushroom samples
exceeded the recommended limit of 10 µg/kg set in the Federal
Republic of Germany for certain food items. Residues apparently
originated from the treated wooden cases used for culturing
mushrooms, which contained up to 3900 mg PCP/kg.
Table 16. Average incidence of PCP residues
in food composites and daily dietary intake of
PCP in the USAa
------------------------------------------------
Year Number of Percent Daily PCP
composites positive intake
examined composites (µg/person
per day)
------------------------------------------------
1965 216 1.4 -b
1966 312 3.3 6
1967 360 2.2 1
1968 360 1.9 1
1969 360 2.8 2
1970 360 nsc nsc
------------------------------------------------
a From: Duggan & Corneliussen (1972).
b < detection limit (1 µg).
c ns = not specified.
PCP can also enter food during processing, transportation, or
storage. Heikes & Griffitt (1980) demonstrated that canned fruit
and vegetables in Mason jars can be contaminated with PCP due to
PCP residues in sealing gaskets, lids, and enamel. Levels in the
jar lid, sealing gaskets, and enamel were as high as 198 µg/lid,
125 mg/kg, and 4.4 mg/kg, respectively. Six fruits and vegetables
originally free from PCP, contained between 0.29 and 1.1 µg/litre
in the liquid and between 1.4 and 38 µg/kg in the solids, after
being canned and stored for 4 days in contaminated jars.
Kroyer et al. (1982) modelled the transfer of PCP from wooden
storage containers to flour experimentally. Within 24 h,
remarkable quantities of PCP can be adsorbed by flour in contact
with PCP-treated wood: applied wood preservative could be detected
in the foodstuff at levels ranging from 0.2 to 1 mg PCP/kg.
PCP levels in the range of 240 - 1090 µg/kg were found in the
flesh grease from hides treated with PCP. Collagen material
derived from hides, pigskin, and decalcified bones is used for the
manufacture of edible gelatins, and Stijve (1981) detected PCP in
each of 50 samples of commercially available gelatins tested.
Products from western Europe and the USA generally contained less
than 100 µg/kg, while those from tropical countries contained from
1000 to 5000 µg PCP/kg. The US Food and Drug Administration (FDA)
has proposed prohibiting "the use of animal bones, hides, or skins
that have been exposed to pentachlorophenol" (Federal Register,
1977).
A calculated daily dietary intake of PCP for the period 1965-70
is shown in Table 16. The values ranging from 1 to 6 µg/person per
day were based on a market-basket survey of 117 retail food items.
Each market basket represented a 2-week diet, constructed according
to consumer behaviour. The foods were prepared as for eating and
then analysed (Duggan & Corneliussen, 1972). In another survey
(Krause, 1982), in contrast to the above food collection procedure,
samples of complete meals prepared and eaten by different families
were collected over a period of 3 - 7 days and pooled. This
sampling procedure is more realistic, as it takes into
consideration the fact that food may be contaminated during storage
in PCP-contaminated rooms. In fact, in households where PCP-
containing wood preservatives had been applied, meals averaged 6 µg
PCP/kg (2/3 range, 2 - 13 µg/kg), while the meals of a control
group were less contaminated (< 0.1 - 5 µg/kg). According to
Fischer (1983), the daily dietary intake on the basis of these data
is 0.1 - 1 µg/person per day for people without known exposure and
6 µg/person per day for persons exposed to PCP-treated interiors.
5.1.6. Consumer products
On the basis of the diverse applications of PCP, it would be
expected that a large number of consumer products contain this
compound. Furthermore, PCP from the indoor atmosphere can
contaminate a number of household items. However, there are few
data on PCP levels in consumer products. In one example, van
Langeveld (1975) analysed 65 commercial samples of paints used on
children's toys and found that 14% contained PCP in the range of
100 - 2700 mg/kg.
The Swiss Federal Office of Health (Siegwart, 1983) found PCP
in various clothes including socks, pantyhose, and insoles, with
PCP concentrations between 0.015 and 0.96 mg/kg; one insole
contained 13.2 mg PCP/kg (Siegwart, personal communication, 1986).
5.1.7. Treated wood
Obviously, PCP-treated wood contains substantial quantities of
the compound itself. The Ontario Ministry of Agriculture detected
tetrachlorophenol and PCP in samples of wood shavings used as
livestock litter in southern Ontario (Jones, 1981); PCP levels were
as high as 628 mg/kg in fresh litter, but fell off sharply, after
56 days use to 96 mg/kg or less. In the United Kingdom, Parr et
al. (1974) found an average PCP concentration of 12 mg/kg (range,
1 - 83 mg/kg) in fresh broiler house litter, while spent litter
contained an average of 0.3 mg/kg. Curtis et al. (1972) found as
much as 40 mg PCP/kg in samples of shavings and sawdust. Levin &
Nilsson (1977) assayed for tetrachlorophenol, PCP, and several
contaminants in wood dust from a Swedish sawmill. PCP levels in
dust from wood treated with 2% Na-2,3,4,6-tetrachlorophenol ranged
from 30 to 100 mg/kg.
Analysis of timber samples taken from homes in the Federal
Republic of Germany, several years after it had been recom-mended
to avoid the indoor use of PCP, revealed PCP levels ranging from
0.1 to 615 mg/kg (mean, 35 mg/kg) (Ruh et al., 1984). This means
that the timber had either been treated with PCP or that it was
contaminated. In this context, it is noteworthy that Ruh &
Gebefuegi (1984) also analysed wooden material which, according to
consumer information, was supposed to be untreated. Only 30% of
the samples were PCP-free, 40% contained from 0.05 to 0.8 mg
PCP/kg, and 31% contained from 1 to 20 mg/kg.
Untreated wood samples from furniture in a living room in
which panelling had been painted with a wood preservative (6% PCP)
according to instructions, were analysed by Gebefuegi et al.
(1979). PCP concentrations ranged from 15.5 to 26 mg/kg in the top
layer (0 - 1.5 mm) and from 2.5 to 7 mg/kg at 3 - 8 mm. For
comparison, treated wood samples contained between 1570 and 2754
mg/kg in the top layer, 612 - 1800 mg/kg in the middle layer (1.5 -
3 mm), and 117 - 340 mg/kg in the bottom layer (3 - 8 mm). Freshly
treated wood surfaces showed PCP concentrations of between 4000 and
6000 mg/kg.
5.2. Occupational Exposure
A list of potential sources of occupational exposure to PCP is
presented in Table 17. However, the actual PCP concentrations that
workers are exposed to during such industrial or commercial
activities are rarely measured.
Since PCP is extensively used for wood protection and
preservation, most studies of occupational exposure have been
conducted in this field of industry. If the pressure treating
method is used, respiratory exposure to PCP occurs mainly when the
door of the pressure vessel is opened and PCP can escape into the
breathing zone of the worker. With non-pressure treatment,
continuous evaporation of PCP into the air takes place, since the
tanks or vats are open. With both processes, dermal exposure of
workers is possible during the handling of the treated wood
(Williams, 1982). The polychlorinated impurities in PCP may be
enriched relative to freshly prepared PCP solution during the
recirculation of the preservatives (Levin et al., 1976; Lamberton
et al., 1979). Hence, during the periodic tank and cylinder
cleaning processes, much higher exposures to PCP impurities may
occur than expected theoretically.
In Table 20, a number of PCP air concentrations as measured in
the course of human monitoring studies is shown. In addition, air
samples taken at the breathing zone in 11 wood-treating factories
in the USA ranged from 10 to 510 µg/m3 (Todd & Timbie, 1983). Most
of the data in Table 20 are below the TWA or MAC value of 500 µg
PCP/m3 (IRPTC, 1983), which has been established by several
countries. However, this value is derived "by analogy with other
compounds of similar action and toxicity in addition to the
specific available information" (ACGIH, 1980). With lumber
treatment, exposure to airborne PCP is generally below 100 µg/m3;
concentrations higher than the TWA value commonly occur during PCP
or Na-PCP production.
Table 17. Potential sources of occupational exposure to
PCP or its sodium salta
--------------------------------------------------------
Manufacture and shipping of industrial chlorophenols
Sawmills
Wood-treatment plants
Carpentry and other timber and wood-working
Termite control
Agricultural pesticide application
Greenhouses
Industrial cooling towers and evaporative condensers
Treatment and handling of wool
Treatment and handling of burlap, canvas, rope, leather
Paper manufacture
Petroleum and other drilling
Paint and adhesive manufacture and use
Telephone and electrical line work
Dyeing and cleaning of garments
--------------------------------------------------------
a Adapted from: Crosby et al. (1981).
Extremely high occupational exposures have been reported as a
result of the agricultural use of PCP. Following PCP application
on cotton fields over a 2-year period, Demidenko (1969) reported
that unusually high concentrations of up to 38 000 µg PCP/m3 air
were found where the sprayers were working and that the workers
exhibited typical symptoms of PCP intoxication (eye and nasal
irritation, headaches, fatigue). In the breathing zone of the
workers formulating the spray, air-PCP levels of 320 µg/m3 were
measured. Pilots in spraying aircraft were exposed to an average
of 880 µg PCP/m3. Air levels, at a distance of 10 - 50 m from the
treated field, varied from 960 to 4400 µg/m3.
The magnitude of PCP exposure depends particularly on the
methods used in handling the chemical, and on measures to minimize
PCP levels in the work-place. According to Wood et al. (1983),
automated processes and closed systems have greatly reduced the
exposure level in large-scale manufacturing and wood-treatment
factories. However, in small-scale operations, overexposure may
occur through inadequate control measures. In addition to
measurements of ambient PCP levels during industrial or
agricultural use, several attempts have been made to estimate
exposure on the basis of urine-PCP levels in workers involved in
the direct and controlled application of PCP (section 5.4).
5.3. General Population Exposure
Because of the widespread use of PCP products, the general
population also comes into contact with this substance. PCP has
been detected at the µg/litre level in the urine of people, not
occupationally exposed, in widely different groups and locations
(section 5.4). It is likely that this wide-spread occurrence of
PCP in human populations results from PCP intake or from the
metabolism of other chlorinated compounds rather than from the
natural occurrence of PCP.
Some possible sources of non-occupational exposure to PCP in
the home, work, and outdoor environments are listed in Table 18.
In such cases, the general population can be incidentally exposed
to PCP-treated items such as textiles, leather, and paper products
(Jones, 1981, 1984). In addition, a variety of consumer products
including food may contain PCP, though no direct PCP application
has been involved, if they are stored in PCP-treated wooden
containers or exposed to PCP in the atmosphere of rooms where
woodwork has been treated with PCP. This is consistent with the
laboratory studies of Morgan & Purslow (1973), Ingram et al.
(1981a,b), and Petrowitz (1981) who observed considerable losses of
PCP from wood samples (section 4.1.1).
Table 18. Some possible sources of non-occupational
exposure to PCP or its sodium salta
--------------------------------------------------------
Use of retail trade pesticide products containing PCP
(for wood preservation, termite control, etc.)
Use of PCP-treated lumber for construction of dwellings
Smoke from sawmills and burning scrap lumber
Sawdust (fuel, floor covering, particle-board, etc.)
Burlap, canvas, and rope
Wool and other textiles
Leather products
Paper products
Contact with adhesives, paint, and painted surfaces
Used telephone poles and railroad ties
Ornamental wood-chips
Fat trimmed from treated hides (used as feed additive)
Water treated for mollusc control
Contaminated food
--------------------------------------------------------
a Adapted from: Crosby et al. (1981).
Wood preservatives containing PCP or its sodium salt have been
widely applied in the domestic field both indoors and outdoors. In
some countries, the indoor use of PCP-containing wood preservatives
has been regulated by government authorities (section 3.3.3).
Cases of PCP intoxication in persons residing in treated houses
have roused public concern as to whether the general population
might be endangered by domestic applications of PCP and have led to
investigations of PCP levels in rooms and the evaluation of
potential health effects.
In one instance, volatilization of PCP within a room from
treated interior wood led to PCP deposition on untreated wood,
furniture, and curtains at levels of between 23 and 26 mg/kg and to
residues (mg/kg) on other household items such as carpets, books,
oil paintings, and cassette tapes. The air levels of PCP varied
between 50 and 100 µg/m3 in the living room of the house, which had
been under examination since 1974, because of reported PCP
intoxications (Gebefuegi et al., 1979).
Krause & Englert (1980), Aurand et al. (1981), and Krause
(1982) reported the results of a survey in the Federal Republic of
Germany, which included the analysis of indoor air samples in 104
homes and of the urine of more than 1000 persons. Depending on the
intensity of, and the time elapsed since, indoor PCP application,
PCP concentrations in the air ranged from not detectable to about
25 µg/m3, frequently from 2 to 10 µg/m3. The median value of 5 µg
PCP/m3 is 1000 times higher than the outdoor air levels found in
residential areas (section 5.1.1). For comparison, indoor air
levels of PCP in houses of a control group without known exposure
were generally below the detection limit of 0.1 µg PCP/m3.
Air concentrations of PCP of between 1 and 10 µg/m3 have been
reported in the living- and bedrooms of Swiss homes (Zimmerli et
al., 1979). Similar levels (1 - 25 µg/m3) were found in rooms
treated one to several years earlier; levels of 25 and 30 µg/m3
were found in rooms a few weeks after treatment (Dahms & Metzner,
1979).
In an unpublished study carried out in the United Kingdom in
1980 and cited by Dobbs & Williams (1983), PCP air concentrations
were monitored as a function of time. During the first week after
treatment of the roof void of a house, PCP levels were as follows:
16 - 67 µg/m3 in the treated roof void, 3.9 - 15 µg/m3 in the
landing, and 1.6 - 2.8 µg/m3 in the bedroom; 5 - 10 weeks after
treatment, PCP concentrations in the treated roof void ranged from
1.7 to 6.7 µg/m3 compared with 0.6 - 5 µg/m3 in the landing and
1.6 - 2.8 µg/m3 in the bedroom. Although the PCP air concentration
in or near the treated room rapidly decreased, levels in the
untreated bedroom remained stable, perhaps as a result of adsorption
and desorption processes.
To follow the volatilization of PCP from treated wood in
enclosed environments, studies were conducted in a building with an
enclosed swimming pool, the walls and ceiling of which had been
treated with PCP (Gebefuegi, 1981; Gebefuegi et al., 1983). Within
a fortnight, an estimated 178.5 mg PCP had diffused from the
wooden panels (210 m2), based on detection of 1 µg/litre in the
water of the swimming pool, 60 µg/litre in the water condensed from
the heat pump, and 4 µg/m3 in the air of the hall housing the
swimming pool. Air levels of PCP inside this building ranged from
1 to 160 µg/m3, one year after the application of the wood
preservative. Two years later, PCP concentrations in the indoor
air averaged 4 µg/m3, and, even after 7 - 8 years, PCP was still
present at about 0.4 µg/m3, though, in the interim, the wood panels
had been painted with another wood preservative, which undoubtedly
reduced the rate of PCP diffusion.
In a study conducted in the USA, PCP vapour levels were
measured in the air of treated wooden structures. The highest
level detected was 38 µg/m3 in the basement of the building where
there was the highest ratio of treated wood surface area to room
volume, and no ventilation. PCP levels were higher in the main
floor of this house (8.8 µg/m3) and in a warehouse (3.52 µg/m3)
than in 11 other rooms of different buildings, which ranged from
0.09 to 1 µg/m3. Variability in PCP concentrations was mainly
attributable to ventilation differences (Saur et al., 1982).
Compared with the aforementioned studies, the values (0.06 -
1.6 µg/m3) reported by Dobbs & Williams (1983) from houses in the
United Kingdom represent rather low PCP levels. Generally, PCP
concentrations of up to about 30 µg/m3 can be expected during the
first month following treatment; considerably higher levels (up to
160 µg/m3) may not be excluded under unfavourable conditions. In
the long term, values of between 1 and 10 µg/m3 are typical PCP
concentrations after extensive treatments.
Krause (1982) considered household dust to be very suitable for
screening purposes, because it accumulates PCP and can be easily
collected. Household dust collected in the houses of residents
using PCP contained about 1000 times more PCP (mean, 15 mg/kg; 2/3
range, 6 - 20 mg/kg) than that of control households (mean, 0.008
mg/kg; 2/3 range, 0.003 - 0.013 mg/kg).
In similar, more recent studies, relatively high PCP levels
(range, 0.2 - 217.3 mg/kg; mean, 28.5 mg/kg) were found in
household dust samples, indicating indoor air contamination
resulting from PCP contaminated surfaces (Ruh et al., 1984).
The concentration of PCDDs or PCDFs in PCP-contaminated
interiors has not yet been determined. However, it may be assumed
that the ratio of their air concentrations will be proportional to
their rates of volatilization. Because of their lower vapour
pressure (e.g., H6CDD, 8.8 x 10-5 Pa), volatilization of PCDDs will
be lower than that of PCP. According to Cull et al. (1983), the
following PCDD concentrations can be predicted in houses where PCP
air concentrations range from 1 to 10 µg/m3: OCDD, 0.2 - 20 ng/m3;
H6CDD, 0.007 - 0.7 ng/m3; H7CDD, 0.04 - 4 ng/m3.
First reports of indoor air analyses of PCDDs and PCDFs in a
house in the Federal Republic of Germany (Eckrich, 1986), which had
been treated with PCP-containing wood preservatives several years
before, seem to confirm these estimates; however, the data
reported are still insufficient for a conclusion to be drawn.
Further studies are under way that could be used to characterize
the PCDD and PCDF levels in indoor air of PCP-treated homes.
Residues of PCP in the general population may arise not only
from oral, dermal, or inhalation uptake of PCP, but also from the
metabolic transformation of other chlorinated compounds (Table 19).
In studying the biotransformation of hexa- and pentachlorobenzene
in the rat, mouse, guinea-pig, laying hen, and rainbow trout, Koss
& Koransky (1978) found PCP and other metabolites in both the
excreta and tissues of the animals. Considering the substantial
residues of hexachlorobenzene in the human body and in human milk,
PCP intake arises via this route in both adults and new-born
children. Similarly, the continuous low-level PCP excretion from
people, who apparently are not exposed to PCP, might be partly due
to continuous exposure to hexachlorobenzene and related compounds.
Table 19. Chlorinated compounds metabolized to PCP
----------------------------------------------------------------
Source Reference
----------------------------------------------------------------
hexachlorobenzene Mehendale et al. (1975); Engst et
al. (1976); Rozman et al. (1977);
Sanborn et al. (1977); Koss &
Koransky (1978); van Ommen et al.
(1985)
pentachloronitrobenzene Murthy & Kaufman (1978); Koegel et
al. (1979)
BHC isomers (i.e., lindane) Balba & Saha (1974); Engst et al.
(1978)
----------------------------------------------------------------
5.4. Human Monitoring Data
Exposure levels as measured in the indoor air or in consumer
products may provide indirect indications of exposure to PCP.
However, it is not possible to separate oral, respiratory, and
dermal exposures, under non-experimental conditions. Thus, the PCP
concentrations of the sources do not directly indicate the actual
human PCP intake by the different routes.
Several investigations have been carried out to relate the
human body burden to the urinary-PCP level. These are summarized
in Tables 20, 21, and 22) distinguishing between occupationally
exposed workers and the general population. The latter has, in
turn, been divided into individuals exposed non-occupationally,
such as persons exposed to PCP-containing wood preservatives
applied to the interior of private homes, offices, public
facilities etc., and people without known exposure.
In many cases, values for populations without known exposure
overlap those of exposed populations, perhaps because persons
designated as unexposed might unknowingly have been exposed to
obscure sources of PCP. In addition, occupational exposure does
not always involve high loading, for, as pointed out in section
5.2, both very low and very high air levels of PCP have been found
in work-places. Conversely, people exposed non-occupationally,
particularly those who apply PCP-containing wood preservatives
indoors, may be exposed to air levels of PCP encountered at work-
places or even higher levels.
Analytical improvements in the last few years have
substantially lowered the detection limits (section 2.5.2) and,
combined with different methods, this makes the comparability of
analytical data questionable, particularly when urinary-PCP levels
have been determined without hydrolysis, as in the survey of Krause
& Englert (1980), since, under these conditions, PCP levels will
have undoubtedly been underestimated. According to Zimmerli et al.
(1979) and Butte (1984), analysis for total PCP yields
concentrations about 3 times higher than those for only free PCP.
For these reasons, it is not possible to derive exact ranges of
PCP levels in different exposure groups from the data in Tables 20,
21, and 22. However, the mean or median urine-PCP levels are
likely to range around 0.01 mg/litre for the general population
without known exposure, around 0.04 mg/litre for persons exposed
non-occupationally, and around 1 mg/litre for occupationally
exposed people.
Only two particularly detailed studies of domestic PCP exposure
provide comparative data. Comparing the mean urine-PCP levels,
Hernandez & Strassmann-Sundy (1980) (No. 3 in Table 21, No. 5 in
Table 22) observed that residents of log homes treated with PCP had
a body burden that was between 5 and 60 times higher than that of
residents of untreated log homes or conventional homes.
The most thoroughly designed survey (Krause & Englert, 1980;
Aurand et al., 1981; Krause, 1982) included air analyses in the
houses of a control group showing PCP concentrations generally
below the detection limit of 0.1 µg/m3 (No. 6 in Table 22). The
mean urine-PCP level of this group was about 3.5 times lower than
that of the corresponding exposure group (No. 5 in Table 21). Much
greater differences are encountered, if the highest concentrations
measured are compared with the control levels. No significant
correlation was established between the PCP levels in air and in
urine, which again casts doubt on the validity of air-PCP levels as
an exposure parameter. Nevertheless, on subdividing the exposed
persons into 2 groups with PCP exposure either lower and equal to
5 µg PCP/m3 or higher than 5 µg PCP/m3, urinary-PCP levels seem to
be elevated at the higher indoor concentrations. In addition,
younger residents are obviously more exposed to PCP than older
ones, perhaps because, on average, children spend more time at
home.
When comparing PCP concentrations in the urine and blood, the
levels in serum or plasma generally exceed those in urine, the
extent of this difference varying according to the exposed group.
The blood-PCP:urine-PCP ratio in people without known exposure or
in persons exposed non-occupationally is considerably higher than
that in occupationally exposed individuals. For comparison, in most
of the cases of lethal intoxication summarized in Table 24 (section
6.2.2), urine-PCP levels even exceeded the corresponding blood
levels. This pattern may be the result of heterogenous plasma-
protein binding of PCP (section 6.6).
Shafik (1973) detected significant amounts of PCP (mean, 0.025
mg/kg; range, 0.005 - 0.052 mg/kg) in human adipose tissue samples
from the general population. Similar values (mean, 0.0145 mg/kg)
(Table 25, section 6.3.1) were reported by Grimm et al. (1981),
whereas higher PCP contents (mean, 0.14 mg/kg; range, not
detectable - 0.57 mg/kg) were found in adipose tissue samples from
subjects with "no occupational contacts" (Ohe, 1979).
Table 20. Levels of PCP in the air, and in the serum or plasma, and urine of
individuals exposed occupationally
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(1) Lumber, carpentry 1 ns na na 0.024 Cranmer &
(0.022 - 0.025)a Freal (1970)
(2) Lumber, closed 11 1 na na 1.6 (ns) Casarett et
tank procedure al. (1969)
(3) Lumber, dipping 11 1 na na 2.6 (ns) Casarett et
al. (1969)
(4) Lumber, dipping ns ns 19 na 2.83 Arsenault
(3 - 63) (0.12 - 9.68) (1976)
(5) Lumber, dipping 18 - 22 ns na 3.78 0.95 Klemmer et
(0.15 - 17.4) (< 0.01 - 7.80) al. (1980)
(6) Lumber, dipping, 18 ns na 5.14 1.31 Begley et
spraying, or (0.43 - 14) (0.09 - 3.3) al. (1977)
brushing
- 6th day of 18 ns na 4.92 1.36
vacation (0.50 - 13) (0.18 - 3.5)
- 20th day of 18 ns na 2.19 0.59
vacation (0.32 - 5.3) (0.05 - 1.4)
- 51st day of 13 ns na 2.61 0.95
renewed work (0.19 - 8.1) (0.03 - 3.6)
exposure
(7) Lumber, generalb 3 5 1 1.11 0.15 Wyllie et
(2 - 11) (< 1 - 15) (0.35 - 3) (0.044 - 0.47) al. (1975)
(8) Lumber industry 20 ns na ns ns Gossler &
(0.4 - 4.8) (0.07 - 0.57)c Schaller
(1978)
(9) Lumber, officeb 1 10 2 0.65 0.06 Wyllie et
(< 1 - 3) (0.42 - 0.75) (0.04 - 0.11) al. (1975)
--------------------------------------------------------------------------------------------------------------
Table 20. (contd.)
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(10) Lumber, pressure 1 5 6 2.29 0.30 Wyllie et
treatmentb (< 1 - 15) (1.51 - 3.55) (0.09 - 0.76) al. (1975)
(11) Lumber, pressure ns ns 14 na 1.24 Arsenault
treatment (4 - 1000)d (0.17 - 5.57) (1976)
(12) Lumber, pressure 23 - 24 ns na 1.72 0.27 Klemmer et
treatment (0.02 - 7.70) (< 0.01 - 2.40) al. (1980)
(13) Lumber, pressure Embree et
treatment al. (1984)
- Airborne + 10 5 - 10 55.6 0.71 0.11
dermal (± 89) (± 0.38) (± 0.02)
- Airborne 8 5 - 10 66.7 0.24 0.05
exposure (± 100) (± 0.23) (±0.02)
- No known 5 5 - 10 ns 0.06 ns
exposure (± 0.02)
(14) Lumber, spraying 2 ns na na 0.20 Cranmer &
(0.13 - 0.27)a Freal
(1970)
(15) Lumber, spraying ns ns 6 na 0.98 Arsenault
(3 - 69)d (0.13 - 2.58) (1976)
(16) PCP processing 18 12 ns 0.25e 0.112e Triebig et
factory (0.3 - (2.2 - 55.5) (0.02-1.5)f (0.013 - 1.224) al. (1981)g
31)
(17) PCP application 23 3e 2.4 1e ns Zober et
(0.5 - (0.3 - 8) (0.2 - 2.4)f al. (1981)
12)
(18) PCP processing 18 10e 17.5 0.25e ns Zober et
(0.2 - (2 - 50) (0.02 - 1.5)f al. (1981)
31)
--------------------------------------------------------------------------------------------------------------
Table 20. (contd.)
--------------------------------------------------------------------------------------------------------------
Number Activity Sample Length of Air (µg/m3) Serum (mg/litre) Urine (mg/litre) Reference
of size exposure mean (range) mean (range) mean (range)
study (number) (years)
--------------------------------------------------------------------------------------------------------------
(19) PCP production 8 ns < 100 - 4.73 ± 3.41 2.38 ± 1.91 Bauchinger
> 500h et al.
Na-PCP production 14 ns < 100 - 2.23 ± 1.51 0.84 ± 0.65 (1982)
> 500i
(20) PCP production 18 ns 270 - 4000 na 0.72 ± 0.55 Ning (1984)
Na-PCP production 50 ns 0 - 50 na 0.35 ± 0.30
(21) PCP synthesis, 9 ns na na 1.2 Siqueira &
full time activity (0.34 - 3.4) Fernicola
- same factory, 12 ns na na 0.15 (1981)
reduced PCP (0.032 - 0.4)
exposure
(22) Pesticide, 130 ns na na 1.80 Bevenue et
spraying (0.003 - 35.7) al. (1967b)
(23) Farmers and pest 210 - ns na 0.25 0.01 Klemmer et
control operators 280 (< 0.01 - 8.4) (< 0.01 - 0.040) al. (1980)
--------------------------------------------------------------------------------------------------------------
a Range of replicate analyses of single urine samples.
b Mean concentrations shown are calculated from sampling data collected over a 5-month period. Mean air
level for workers listed as "lumber, general", is calculated from data provided for all 11 sites over a
5-month period by Wyllie et al. (1975).
c Assuming a daily urine volume of 1.4 litre.
d Mean "average exposure levels" encountered by employees. Air at "maximum exposure" sites, next to sources,
contained 26 µg/m3 (lumber spraying site) and 297 µg/m3 (pressure treatment site).
e Median.
f Plasma.
g Data partly identical with those from Zober et al. (1981).
h From 67 samples, 18 were < 100 and 10 > 500 µg/m3.
i From 55 samples, 7 were < 100 and 8 > 500 µg/m3.
na = Not analysed.
ns = Not specified.
Table 21. Levels of PCP in the air, and in the serum or plasma, and urine of individuals
exposed non-occupationally
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(1) Miscellaneous groups 117 ns na na 0.04 Bevenue et
including house- (nd - al. (1967b)
holds and pesticide 1.84)
users
(2) Indoor application 16 ns ns na ns Zimmerli et
of PCP solutions (1 - 10) (0.030 - al. (1979)
0.150)
(3) Residents of log 5 ns 0.29 1.126 0.084 Hernandez &
homes treated with (0.20 - (0.580 - (0.047 - Strassmann-
PCP solutions 0.38)a 1.750) 0.216) Sundy (1980)
32 ns na 0.330 0.013
(0.116 - (0.002 -
1.084) 0.087)
(4) "No occupational 32 ns na 0.32 0.03 Klemmer et
exposure"; control (0.002 - (< 0.01 - al. (1980)
group for Number 5 7.20) 1)
in Table 20
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(5) Indoor application 989 ns 6.1 na 0.044 Krause &
of an average of (nd - Englert
40 litre PCP (< 9 25)b (1980)
solutions years) 4.9c 0.029c
(2.5 - (0.013 -
9.5) 0.071)
- Subgroups:
1. m < 18 years 16 ns < 5 na 0.047c
(0.017 -
0.107)
2. m > 18 years 39 ns < 5 na 0.023c
(0.011 -
0.052)
3. f < 18 years 22 ns < 5 na 0.033c
(0.016 -
0.066)
4. f > 18 years 39 ns < 5 na 0.026c
(0.015 -
0.059)
5. m < 18 years 23 ns > 5 na 0.079c
(0.014 -
0.125)
6. m > 18 years 31 ns > 5 na 0.043c
(0.011 -
0.146)
7. f < 18 years 25 ns > 5 na 0.059c
(0.011 -
0.103)
8. f > 18 years 43 ns > 5 na 0.039c
(0.021 -
0.125)
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(6) Indoor application Sangster et
of about 70 litres al. (1982)
PCP solution
- before ventilation 6 6 0.60 na 0.0032
(0.14 - (0.0007 -
1.20) 0.0078)
- after ventilation 6 - 0.08 0.080 0.0033
(nd - (0.025 - (0.0018 -
0.24) 0.190)d 0.0080)
Indoor application 2 0.5 0.15 0.033 na
of about 100 litres (nd - (0.031 -
PCP solution 0.40) 0.034)d
about 75 litres PCP
solution
Indoor application 2 1 0.67 0.565 na
of about 100 litres (0.44 - (0.47 -
PCP solution 0.95) 0.66)d
---------------------------------------------------------------------------------------------------
Table 21. (contd.)
---------------------------------------------------------------------------------------------------
Number Exposure/ Sample Length Air Serum Urine Reference
of comments size of (µg/m3) (mg/litre) (mg/litre)
study (number) exposure mean mean mean
(months) (range) (range) (range)
---------------------------------------------------------------------------------------------------
(7) "Workers non- 27 ns na na 0.009 Siqueira &
occupationally (nd - Fernicola
exposed"; control 0.034) (1981)
group for Number 21
in Table 20
(8) Indoor application 80 ns na (0.0025 - (0.002 - Janssens &
PCP solutions 0.5) 0.075) Schepens
(1984)
(9) Residents in buil- 46 ns na (0.001 - na Ruh et al.
dings with PCP con- 0.110)e (1984)
taminated wood
(10) Residents in homes 204 ns na 0.058c 0.014c Grimm et al.
treated with PCP (234) (1985)
---------------------------------------------------------------------------------------------------
a Air samples taken on the 1st and 2nd floor of a 2-story log house.
A sample of interior surface wood contained 1132 mg PCP/kg (0.11%).
b 104 air indoor samples taken.
c Median (2/3 range).
d Plasma.
e Whole blood.
na = Not analysed.
nd = Not detectable.
ns = Not specified.
f = Female.
m = Male.
Table 22. Levels of PCP in the serum or urine of individuals without known exposure
---------------------------------------------------------------------------------------------------------
Number Comments Sample Indoor Serum Urine (mg/litre) Reference
of size air (mg/litre) mean (range)
study (number) (µg/m3) mean
(range)
---------------------------------------------------------------------------------------------------------
(1) ns 6 na na 0.005 Cranmer & Freal
(0.002 - 0.011)a (1970)
(2) Control group for ns na 0.06b ns Gossler & Schaller
Number 8 in Table 20 (0.03 - (0.001 - 0.057)c (1978)
0.2)
(3) US National Human 418 na na 0.0063 Kutz et al. (1978)
Monitoring Program for (nd - 0.193)
Pesticides
(4) Control group for 12 na na 0.0135 Zimmerli et al.
Number 2 in Table 21 (0.006 - 0.023) (1979)
(5) Control groups for 42 na ns ns Hernandez &
Number 3 in Table 21; (0.004 - (0.0007 - 0.011) Strassmann-Sundy
January 1980 "conven- 0.068) (1980)
tional" homes
March 1980; untreated 2 na 0.051 0.0014
log homes (0.034 - (0.001 - 0.002)
0.075)
March 1980; 11 na 0.048 0.0025
"conventional" (0.015 - (0.001 - 0.007)
homes 0.055)
---------------------------------------------------------------------------------------------------------
Table 22. (contd.)
---------------------------------------------------------------------------------------------------------
Number Comments Sample Indoor Serum Urine (mg/litre) Reference
of size air (mg/litre) mean (range)
study (number) (µg/m3) mean
(range)
---------------------------------------------------------------------------------------------------------
(6) Control group for 207 ndd na 0.0127 Krause &
Number 5 in Table 21 0.0102b Englert (1980)
(0.0038 - 0.0214)
(7) ns 10 na na 0.009 Lores et al. (1981)
(0.003 - 0.016)
(8) Dutch draftees; 99 na 0.128 na Sangster et al.
control group for (< 0.05 - (1982)
Number 6 in Table 21 1.10)
0.088b
(9) Control group for 12 na ns 0.0009 Janssens &
Number 7 in Table 21 (0.0002 - 0.002) Schepens (1984)
(10) Non-specifically 12 na 0.025 0.014 Uhl et al.
exposed persons 30 (0.019 - (0.007 - 0.034)c (1986)
0.036)
---------------------------------------------------------------------------------------------------------
a Range of replicate analysis of single urine samples.
b Median (2/3 range).
c Assuming a daily urine volume of 1.4 litre.
d Below detection limit of 0.1 µg/m3.
na = Not analysed.
nd = Not detectable.
ns = Not specified.
Samples of human milk were found to contain between 0.03 and
2.8 µg PCP/kg (mean, 0.68 ± 0.05 µg/kg), which is considerably less
than the PCP levels usually found in other body fluids or tissues
(Gebefuegi & Korte, 1983).
In investigating the apparent decrease in sperm density in US
males over the last 30 years, Dougherty et al. (1980) and Kuehl &
Dougherty (1980) detected PCP (100 - 200 ppb) in all 50 samples of
human seminal plasma analysed. They also observed that PCP was
selectively concentrated by the cellular material.
Only few monitoring data are available on the human body burden
of microcontaminants as a result of exposure to PCP. Rappe et al.
(1982) analysed urine and blood samples of 9 workers exposed to PCP
or L-PCP. In the case of 5 workers in the textile industry,
urinary levels of PCDDs (total PCDDs, 3 - 365 ng/kg) and PCDFs
(total PCDFs, < 1 - 45 ng/kg) paralleled the urinary-PCP levels
(< 0.01 - 3.12 mg/litre). Similar levels of these impurities were
found in the blood of 4 tannery workers 8 months after last
exposure, but these could not be compared with urinary-PCP levels.
However, in both groups, the concentration pattern of the different
isomers reflected the different proportions of contaminants in
commercial PCP products.
Because of their high fat-solubility and slow metabolic
degradation and elimination, impurities of PCP such as HCB, PCDDs,
and PCDFs are expected to accumulate in body fat. No data are
available concerning the accumulation behaviour of these
microcontaminants as a result of human PCP uptake. However, levels
of dioxins in the milk- or body-fat of cows orally treated with
technical-grade PCP (10 mg/kg body weight per day) were about 1000
times higher than those in blood, indicating a substantial
accumulation (Firestone et al., 1979). In addition, the three
dioxins detected (1,2,3,6,7,8-H6CDD, 1,2,3,4,6,7,8 H7CDD, and OCDD)
declined comparatively slowly from about 20, 40, and 25 µg/kg
composite milk-fat to 4.3, 6.9, and 3 µg/kg, respectively, 100 days
after PCP feeding was stopped. For comparison, the steady-state
PCP level of about 40 mg/kg in blood or 4 mg/kg in composite milk
dropped to basal levels (0.02 - 0.08 mg/kg) within less than 10
days. Firestone et al. (1979) concluded from their results that
"the absence of PCP in milk or biological tissue affords no
guarantee of the absence of biologically active dioxins".
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Animal studies
PCP is readily absorbed through the skin as well as through the
respiratory and gastrointestinal tracts. Braun & Sauerhoff (1976)
administered a single oral dose of 10 mg PCP/kg body weight in corn
oil to 3 male and 3 female rhesus monkeys and calculated the half-
life for absorption to be 3.6 h in males and 1.8 h in females.
After 12 - 24 h, plasma levels peaked in the range of 10 - 30 mg
PCP/litre.
In rats given a single oral dose of 10 mg of 14C-PCP/kg body
weight, the peak plasma concentration (50 mg/litre) was attained
much earlier, after 4 - 6 h. The absorption rate constants were
1.95 and 1.52/h for males and females, respectively (Braun et al.,
1977); assuming first order kinetics, the half-life for absorption
can be calculated to be 0.36 and 0.46 h, respectively.
Rapid absorption of PCP was also observed in rats during 20-min
inhalation of approximately 5.7 mg PCP/kg body weight (Hoben et
al., 1976d) and in mice following intraperitoneal and subcutaneous
injections of 14C-PCP (Jakobson & Yllner, 1971).
The same holds true for fish. Goldfish exposed to PCP medium
(0.4 mg/litre) absorbed PCP rapidly, until a lethal level of
approximately 100 mg/kg body weight was reached after about 5 h
(Kobayashi & Akitake, 1975a). The apparent route of PCP uptake was
via the gills and the skin. Similarly, PCP was rapidly taken up
from the water by rainbow trout and assimilated into various
tissues (Glickman et al., 1977).
6.1.2 Human studies
Braun et al. (1979) studied 4 healthy male volunteers of normal
weight, between 21 and 55 years of age, who ingested a dose of 0.1
mg Na-PCP (> 99% purity)/kg body weight. The observed half-life
for absorption was about 1.3 h. The peak plasma level of 0.245 mg
PCP/litre occurred 4 h after ingestion of PCP. In the study of Uhl
et al. (1986), the PCP level in the plasma of a male volunteer was
approximately 0.185 mg/litre, 2 days after a single oral dose of
0.016 mg 13C-PCP/kg body weight in 40% ethanol. This implies that
absorption of PCP, when dissolved in alcohol, is much greater than
when it is dissolved in water.
For the general population, the uptake of PCP by the oral route
is thought to be more significant than that via other routes of
exposure. For individuals exposed to high airborne concentrations
of PCP in the work-place or in PCP-treated dwellings, the major
routes of exposure are probably via the skin and lungs. No
experimental data are available concerning these routes. However,
the cases of acute intoxication reported were almost exclusively
due to extensive skin contact with PCP or to the inhalation of high
doses of PCP, which subsequently led to high PCP levels in the
human body.
Bevenue et al. (1967a) reported a case of PCP absorption
through the skin. A male individual had skin contact with PCP for
10 min while cleaning a paint brush in a can containing a solution
of 4% PCP. Two days later, a urinary-PCP level of 236 µg/litre was
measured.
One case of oral uptake has been reported (Haley, 1977). A
71-year-old Japanese male had intentionally ingested an amount
estimated at between 113 and 226 g of weed killer containing 12%
PCP. Although the patient was treated with gastric aspiration and
lavage within the next hour, a substantial amount of PCP must have
already been absorbed as indicated by the high serum level of 150
mg PCP/litre, 5 h after the incident.
6.2 Distribution
6.2.1 Animal studies
Available information indicates that usually the highest PCP
levels can be found in the urine immediately after exposure, and
consequently, the PCP concentrations in the tissues account for
only a small fraction of the dose applied. This is reflected by
the data on excretion and recovery of radioactivity from groups of
rats, 9 and 10 days after oral administration of 10 and 100 mg
PCP/kg body weight, respectively (Table 23). It should be noted
that the percentages of the dose recovered are to be considered as
cumulative over the period of 9 days (10 mg/kg dose) or 8 days (100
mg/kg dose) in the case of the excreta, while the PCP contents of
the organs were only analysed 9 days after administration of 10
mg/kg body weight. Thus, PCP is apparently eliminated much more
rapidly from the kidney than from the liver (section 6.5.1).
The results of early studies did not show a uniform
distribution pattern of PCP in experimental animals but indicated
that very high levels of PCP could be found in liver and kidneys
(Truhaut et al., 1952b). However, following long-term exposure,
most PCP was absorbed by the central nervous system.
Several recent studies on the distribution and elimination of
PCP were performed using 14C-labelled PCP, thus making the results
more reliable. Larsen et al. (1972) studied the tissue distribution
in rats after administering oral doses between 31 and 40 mg 14C-
PCP/kg body weight. The body component containing the highest
level appeared to be the liver, followed by the kidney and blood.
Low levels of PCP were found in fat, brain, and muscle tissue.
More than 99% of the total radioactivity in the blood was contained
in the serum, indicating that PCP and/or its metabolites in the
blood are not bound to the cellular constituents.
Table 23. Recovery of radioactivity from rats given a
single oral dose of 10 or 100 mg of 14C-PCP/kg body weighta
------------------------------------------------------------
Percentage of radioactivity (mean ± SD)
dose: 10 mg/kg dose: 100 mg/kg
------------------------------------------------------------
Excreta
urine 79.8 ± 2.9 64.0 ± 14.9
faeces 18.6 ± 3.7 33.6 ± 13.7
expired 14CO2 0.2 ± 0.1 -b
Organsc
liver 0.315 ± 0.137 -b
kidneys 0.045 ± 0.014 -b
Total body 0.437 ± 0.142 -b
Cage rinse 1.4 ± 1.9 -b
Total recovery 99.8 ± 4.4 97.6 ± 2
------------------------------------------------------------
a From: Braun et al. (1977).
b Samples were not analysed.
c Other organs analysed were the stomach, lungs, testes,
ovaries, brain, heart, spleen, and adrenals. Each of
these organs contained 0.005% of the dose or less and
were included with the liver and kidneys in the total
body figure.
Jakobson & Yllner (1971) examined the distribution of PCP in
the mouse after the subcutaneous or intraperitoneal injection of
14C-PCP (15 - 37 mg/kg body weight). Autoradiographic studies
showed that the highest specific activity was in the liver, the
gall bladder and its contents, the wall of the stomach fundus, the
kidney, and the contents of the gastrointestinal tract, while the
lung, heart, and brain contained only negligible amounts of PCP.
PCP concentrations were highest in the plasma of rats
immediately after a 20-min inhalation exposure to an aerosol of Na-
PCP, resulting in a calculated dose of about 5.7 mg/kg body weight
(Hoben et al., 1976d). About 35% of the dose was found in the
plasma, while the liver contained about 25% and the lung a little
less than 2% at time 0. After 24 h, the liver showed the highest
PCP level followed by the plasma and the lung. Other organs were
not examined.
Zenzen (1979) administered a daily dose of 15 mg 14C-PCP/kg
body weight intraperitoneally to rats for 15 days. On day 1, the
highest PCP levels in the organs examined were found in the liver
and kidney of male rats, comprising 14.1 and 14.3 %, respectively,
of the total activity on a dry weight basis. Similar
concentrations were measured in the testicle. However, on a fresh
weight basis, the levels in this organ were similar to those in the
other endocrine organs.
Preliminary data obtained from a single sheep (Wilson et al.,
1982) indicated that PCP is absorbed into the lymphatic system; 47%
of the PCP dose (10 mg/kg in corn oil, given by intraruminal
injection) remained in the digestive tract 36 h after dosing. Of
the absorbed PCP (53% of total dose), approximately 17% was found
in the lymph collected through a thoracic duct canula. The
remainder was probably absorbed from the digestive tract directly
into the blood.
To study the placental transfer of PCP in rats, 60 mg 14C-
PCP/kg body weight was orally administered to pregnant rats on day
15 of gestation. The amount of specific radioactivity in the
maternal blood-serum was greatest at 8 h (about 1.1% of the
administered dose per gram of tissue), but, in the placentas and
fetuses, it never exceeded 0.3% and 0.1%, respectively (Larsen et
al., 1975). Thus, the amount of PCP that crosses the placental
barrier is very low. Contrasting data have been obtained in a
preliminary observation of a single pregnant monkey, but no full
report has been published (Müller, 1981).
Kobayashi (1979) observed an accumulation of 14C in various
organs of goldfish exposed to 0.1 mg 14C-PCP/litre water. The gall
bladder contained the highest 14C level after a 24-h exposure. The
biliary concentration increased, even after fish had been
transferred to clean, running water for 24 h, whereas a decrease
was observed in the levels in all other organs examined (Kobayashi
& Akitake, 1975b). This characteristic accumulation indicates that
conjugated PCP is transferred to the gall bladder and bile due to
an enterohepatic circulation (section 6.5.1).
6.2.2 Human studies
Human data concerning tissue distribution following PCP uptake
are derived mainly from autopsy results on victims of fatal
intoxications (Table 24). No exact conclusions can be drawn from
these data with regard to PCP accumulation, though PCP levels in
the liver, kidney, and lungs are often elevated. The high levels in
the lungs reported in some cases might be related to inhalation
uptake of PCP. Similarly, the stomach of the individual who
committed suicide by ingesting PCP contained 750 mg PCP/kg, which
highly exceeded the concentrations found in the other body parts
examined (Cretney, 1976). An unusually high kidney-PCP level of
639 mg/kg, reported by Wood et al. (1983), might have been due to
kidney malfunction. In general, PCP levels in the various tissues
do not indicate a clear accumulation of PCP, since blood-PCP levels
are often similar to the corresponding tissue concentrations.
Levels in urine can vary depending on the actual urine volume in
the bladder at the time of poisoning and the pH value. From the
data in Table 24, liver and kidney residues associated with acute
lethal intoxications can be estimated to be 10 - 225 mg PCP/kg and
5 - 145 mg PCP/kg, respectively.
Table 24. PCP levels found in human tissues and body fluids
after PCP intoxication resulting in death
---------------------------------------------------------------------------------------------
Reference Case Urine Blood Liver Kidney Lung Brain Supposed
number (mg/ (mg/ (mg/ (mg/kg) (mg/kg) (mg/kg) routes of uptake
litre) litre) kg)
---------------------------------------------------------------------------------------------
Truhaut et al. 1 55 5 10 5 1.5 na dermala
(1952b) 2 96 6 52 21 38 6.5 dermala
Gordon (1956) 1 70 50 65 95 145 20 inhalation, dermala
Menon (1958) 1 160 na na na na na inhalation, dermala
Blair (1961) 1 na na 59 41 na na dermal, orala
2 na na 62 84 76 na inhalationa
3 na na 59 63 na na oralb
Mason et al. 1 na 79 66 na na 10 inhalationa
(1965) 2 365 110 89 86 na 25 inhalationa
Burger (1966) 1 na 39 na na na na orald
Barthel et al. 1 na na na 27.6 na na dermalc
(1969)
Cretney (1976) 1 75 173 225 116 na na oral(?)d
Wood et al. 1 29 16 52 639 116 na inhalation,
(1983) dermala
---------------------------------------------------------------------------------------------
a Occupational exposure.
b Accidentally contaminated food.
c Accidentally contaminated diapers.
d Suicide.
na = Not analysed.
There is a paucity of data on the distribution of PCP in the
tissues and body fluids in the general population. In 2
investigations (Table 25), autopsy samples were collected from
human subjects in Northern Bavaria (Federal Republic of Germany)
with no known history of PCP exposure. Grimm et al. (1981)
concluded from the data that there was only a slight tendency for
PCP to accumulate in both liver and kidney. The relatively high
level observed in the brain samples was attributed to the fact that
most of the persons with high brain-PCP levels had bled to death.
In such cases, a cerebral hypoxia preceding the death might have
led to the breakdown of the blood-brain barrier and accumulation of
PCP in the brain. Löwer (1982) found much lower brain-PCP levels,
in conjunction with liver and kidney levels similar to those
reported by Grimm et al. (1981). There was no correlation between
PCP levels in tissues and the age of the person examined (Löwer,
1982). Moreover, PCP levels in the body fluids were of the same
order of magnitude as those in the tissues (Grimm et al., 1981).
6.3 Metabolic Transformation
6.3.1 Animal studies
The first studies on the fate of PCP in the body were conducted
by Deichmann et al. (1942), who obtained evidence of metabolic
transformation in the rabbit after oral administration and in the
rat after intraperitoneal dosing with Na-PCP. The authors did not
identify any metabolites.
Later studies using more advanced analytical methods have shown
that PCP is metabolized, either to tetrachlorohydroquinone through
oxidation or conjugated to PCP glucuronide. Tetrachlorohydroquinone
was found in its free form in the urine of mice and rats (Jakobson
& Yllner, 1971; Ahlborg et al., 1974); in the rat, it is also
conjugated with glucuronic acid (Ahlborg et al., 1978). Traces of
trichlorohydroquinone formed by the reductive dechlorination of
tetrachlorohydroquinone were found in the urine of rats. The
formation of tetra- and trichlorohydroquinone as well as total
elimination during the first 24 h can be enhanced through
pretreatment with 3-methylcholanthrene or 2,3,7,8-T4CDD.
Phenobarbital only increases the metabolism to tetrachlorohydro-
quinone. These observations were confirmed by in vitro tests on rat
liver microsomes (Ahlborg & Thunberg, 1978). Glucuronidation rates
were not significantly altered by pretreatment with phenobarbital
or 3-methylcholanthrene (Lilienblum, 1985).
Table 25. Medians, means, and ranges of PCP concentrations in tissues
and body fluids at autopsy from people without known exposure
----------------------------------------------------------------------------------------
Number Urine Blood Liver Kidney Brain Body fat Spleen
of (mg/ (mg/ (mg/kg) (mg/kg) (mg/kg) (mg/kg) (mg/kg)
cases litre) litre)
----------------------------------------------------------------------------------------
Median 21a 0.0044 0.0233 0.0670 0.0430 0.0470 0.0127 0.0190
5. Percentile dlb dl 0.0017 0.0240 0.0190 0.0100 0.0070
95. Percentile 0.1603 0.0679 0.1735 0.0950 0.0725 0.0225 0.0325
Mean 0.0297 0.0260 0.0860 0.0641 0.0491 0.0145 0.0208
Median 51c nad na 0.0720 0.0223 0.0180 na na
Lowest value na na 0.0140 nde nd na na
Highest value na na 0.4190 0.1040 0.0560 na na
----------------------------------------------------------------------------------------
a From: Grimm et al. (1981).
b dl = detection limit = 0.001 mg PCP/litre urine; 0.005 mg PCP/litre blood.
c From: Löwer (1982).
d na = not analysed.
e nd = not detected (detection limit = 0.010 mg PCP/kg wet tissue).
In contrast to these rodent species, the rhesus monkey
eliminates PCP unchanged in the urine (Braun & Sauerhoff, 1976).
This is the only recent study that failed to detect any metabolites
of PCP.
Detoxication of PCP has also been observed in fish. While no
dechlorination processes have been reported, conjugation and
subsequent excretion of the PCP conjugates occurs: PCP glucuronide
is formed and excreted into the bile of both goldfish (Kobayashi &
Nakamura, 1979b) and rainbow trout (Glickman et al., 1977), while
pentachlorophenylsulfate is excreted into the surrounding water
through the gills and in the urine of goldfish (Kobayashi &
Nakamura, 1979a,b).
6.3.2 Human studies
Most of the human data available consist of analyses of urine
samples from people exposed to different uncontrolled PCP regimes.
In the studies of Braun et al. (1979) and Uhl et al. (1986) on
male volunteers (section 6.1.2), PCP was eliminated as both the
parent compound and glucuronide. No other metabolites could be
detected. Ahlborg et al. (1974) found tetrachlorohydroquinone in
the urine of 2 occupationally exposed spray operators, who were
also exposed to other chlorophenolic compounds.
Recently, the metabolic transformation of PCP to
tetrachlorohydroquinone was substantiated by Juhl et al. (1985);
human and rat liver homogenates showed similar metabolizing
activities. The rate of PCP metabolism dependedon the PCP
concentration and was 1000 times lower at 1 mmol/litre (266
mg/litre) than at 0.01 mmol/litre (2.66 mg/litre).
6.4 Elimination and Excretion
6.4.1 Animal studies
PCP has been rapidly eliminated by most of the animals
examined. It is cleared from the plasma by its distribution to the
tissues and by excretion via the urine and the faeces; the
metabolites, when produced, are also rapidly excreted.
The uptake of PCP by the tissues accounts for only a small
amount of the total PCP dose taken up by the body (section 6.2.1).
Most of it leaves the body immediately after uptake, mainly through
the urinary excretion of PCP and its metabolites. The proportions
of PCP excreted via the two major excretion routes in the rat,
mouse, and monkey are summarized in Table 26. Although the species
and the test conditions differed, the excretion patterns were very
similar: renal excretion amounted to between 45 and 83% of the
total dose applied, while most of the remaining activity appeared
in the faeces. Thus, excretion of PCP and its metabolites mainly
occurs via the kidneys, and to a lesser extent by the processes of
gastric and biliary secretion.
Table 26. Urinary and faecal excretion of 14C activity as a
percentage of a single dose of 14C-PCP
---------------------------------------------------------------
Species Dose Time % recovery in: Reference
(mg/kg (h) urine faeces
body
weight)
---------------------------------------------------------------
Monkey
male 10a 168 75 12 Braun & Sauerhoff
female 10a 360 70 17 (1976)
Monkey 30a 144 51.7 4.3 Ballhorn et al.
male 50a 144 44.9 11.3 (1981)
Rat 10a 216 80 19 Braun et al. (1977)
100a 192 64 34
Mouse 14.8b 96 83 8 Jakobson & Yllner
18.2b 96 62 5 (1971)
37.2b 96 73 4
35.2b 168 82 10
36.8b 168 80 12
---------------------------------------------------------------
a Oral (corn oil solutions).
b Intraperitoneal.
Only trace amounts of radioisotopes from the metabolism of
labelled compounds are expired. Although this route is of minor
importance, it could indicate other metabolic processes, provided
that analytical errors can be excluded. However, Larsen et al.
(1972) questioned whether expired 14C originated from PCP
metabolism, attributing it, instead, to impurities in the
radiolabelled PCP.
Ahlborg et al. (1974) found that the radioactivity from
labelled PCP excreted in the urine of mice treated intra-
peritoneally (10 mg/kg body weight) consisted of about 41%
unchanged PCP, 13% conjugated PCP, 24% unconjugated, and 22%
conjugated tetrachlorohydroquinone. For rats, the corresponding
values were 60%, 9 - 16%, 7%, and 16 - 22%, respectively (Ahlborg
et al., 1978).
Following an oral dose of 100 mg/kg body weight, the urinary
metabolites of 14C-PCP in rats accounted for 75% unchanged PCP, 9%
PCP glucuronide, and 16% tetrachlorohydroquinone. Levels of the
last compound were below detectable values in the blood-plasma
(Braun et al., 1977). The authors concluded that the rate-limiting
step for the elimination of the metabolites of PCP is the rate of
metabolism rather than that of urinary excretion and therefore, PCP
metabolites were unlikely to accumulate in the body.
The proportions of PCP glucuronide in urine may have been
underestimated to date; this metabolite has recently been shown to
undergo a partial hydrolysis under weakly acidic conditions in
urine (Lilienblum, 1985).
In contrast to the rodents, rhesus monkeys excreted all of the
14C activity in urine as unmetabolized PCP (Braun & Sauerhoff,
1976).
Goldfish, in addition to free PCP, mainly excrete sulfate and
glucuronide conjugates via 3 pathways of elimination: the amounts
of PCP lost by the branchial, renal, and biliary routes were 52,
24, and 22% of the total amount of PCP excreted by the fish in the
24 h following a 24-h exposure to 0.1 mg PCP/litre water. The
excretion of PCP from the body surface was minor. About 30% of the
PCP excreted via the gills was in the unchanged form, whereas
almost all the PCP excreted in both the bile and urine was
conjugated. PCP sulfate was the major conjugate in the branchial
and renal routes, while PCP glucuronide was the primary biliary
conjugate (Kobayashi & Nakamura, 1979b).
6.4.2 Human studies
The PCP concentration in human urine has been widely used as an
indicator of the PCP body burden (section 5.4), based on the fact
that renal excretion of PCP is the major elimination route in man.
In the study of Braun et al. (1979) (section 6.3.2), within 168 h
of ingesting 0.1 mg Na-PCP/kg body weight, volunteers excreted 74%
of the total dose in urine as PCP and 12% as PCP glucuronide.
About 4% of the total dose was eliminated in the faeces; this
amount consisted of equal parts of both free PCP and PCP
glucuronide. The fate of the remaining 10% of the dose was not
discussed.
Uhl et al. (1986) found that about 30% of PCP was excreted as
glucuronide in the urine of a volunteer, up to 4 days after a
single dose of 0.31 mg PCP/kg body weight (section 6.5.2). However,
in contrast to the study of Braun et al. (1979), the percentage of
PCP glucuronide gradually increased to reach about the normal range
determined for people not specifically exposed (65 ± 5% PCP
glucuronide) after about 14 days. The findings of Zimmerli et al.
(1979) and Janssens & Schepens (1984) also indicated that long-term
exposure result in a higher proportion of conjugated PCP than that
reported by Braun et al. (1979). On the average, two thirds of the
PCP detected in the urine samples of non-occupationally exposed
people was conjugated.
6.5 Retention and Turnover
6.5.1 Animal studies
Pharmacokinetic data on the retention and half-life of PCP in
the compartments also indicate that most of the PCP absorbed is
rapidly eliminated from the body.
The dynamics of elimination of PCP and its metabolites depend
on the species and the sex of the test animal. As summarized in
Table 27, the monkey differs from other animal species in showing a
much slower elimination rate as expressed by the half-life for the
clearance from urine and plasma, perhaps because monkeys do not
metabolize PCP.
An extensive enterohepatic circulation of PCP is also suggested
by the slow but steady elimination of 14C activity in the faeces of
the monkeys (Braun & Sauerhoff, 1976). On treating rhesus monkeys
with cholestyramine, this enterohepatic circulation was
interrupted as the cholestyramine bound the PCP and/or its
metabolites and bile acids, thus preventing their reabsorption
(Ballhorn et al., 1981). However, the results were derived from
only 4 monkeys in single studies.
Apart from the monkey studies, pronounced differences in PCP
kinetics that are only in part related to the species or type of
application have been observed in different single-dose studies
(Table 27). Braun & Sauerhoff (1976) and Braun et al. (1977)
observed a monophasic elimination of PCP in monkey, while, in rats,
2 phases could be distinguished (Fig. 4): an alpha-phase with a
rapid elimination rate (elimination half-lives, 13 - 17 h) followed
by a beta-phase (elimination half-lives for male rats, 40 h (10
mg/kg body weight), 121 h (100 mg/kg body weight)) with a
comparatively slow elimination rate. However, the beta-phase, is
not well defined, as it does not remain constant.
Braun et al. (1977) found that, at a higher dose (100 mg/kg
body weight), the female rats followed the monophasic scheme
(elimination half-life, 27 h) (Fig. 4). Hoben et al. (1976d) also
reported a monophasic elimination in rats, while Larsen et al.
(1972) and Zenzen (1979) described a distinct biphasic model. The
accuracy of the unusually slow beta-phase (102 days = 2448 h)
reported by Larsen et al. (1972) has been questioned by Braun et
al. (1977), since this value was obtained by subtracting the
cumulative amount excreted in the urine from the total dose,
without knowing the total recovery. On the other hand, the
monophasic scheme claimed by Braun et al. (1977) for female rats
dosed with 100 mg/kg body weight is not as distinct as it seems to
be; for, on omitting the last sample measured at 192 h, a biphasic
scheme could be fitted to the points as well (Fig. 4).
The amount of the PCP or 14C activity remaining in the body
several days after dosing differs markedly between test species.
According to Braun et al. (1977), in each case, 90% or more of the
radioactivity had been excreted by rats 3 days after dosing;
detectable levels of PCP remained only in liver and kidney, 9 days
after the 10 mg/kg dose (see also Table 23). As a result of a
slower elimination rate, about 11% of the administered 14C activity
remained in the body of rhesus monkeys 15 days after oral
application of 10 mg/kg body weight; approximately 80% of this
remaining activity was identified in the large and small intestines
and liver. Braun & Sauerhoff (1976) calculated that a steady-state
concentration of PCP in plasma would be reached by the 10th
repeated daily dose; the approximate plasma concentration at this
time was estimated to be 50 mg PCP/litre.
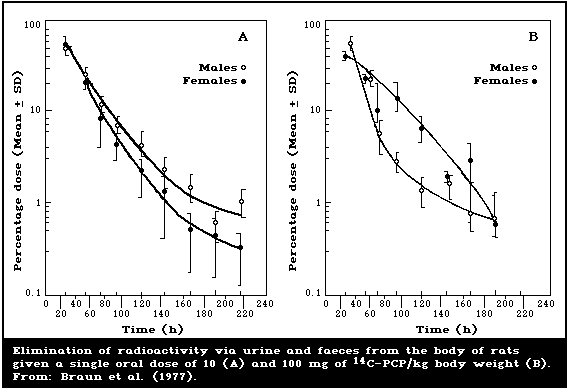 Table 27. Comparison of PCP elimination kinetics in mammals after administration of
single doses
-------------------------------------------------------------------------------------------
Species Dose Elimina- Sex Elimination Kinetics Reference
(mg/kg tion via half-life (h)
body of phase
weight) alpha beta
-------------------------------------------------------------------------------------------
Mousea,b 15 - 37 urine female approxi- - not reported Jakobson &
mately 24 Yllner (1971)
Ratc 37 - 41 urine female approxi- 2448 biphasic Larsen et al.
mately 10 (1972)
Ratd 5.7 urine male approxi- - monophasic Hoben et al.
mately 24 (1976d)
Ratc 10 urine and female 13 33 biphasic Braun et al.
faeces male 17 40 biphasic (1977)
100 urine and female 27 - monophasic
faeces male 13 121 biphasic
Rata 15 all female 6.3 - 9.9 33- biphasic Zenzen (1979)
routes 374
Monkeyc 10 urine female 92.4 - monophasic Braun &
male 40.8 - monophasic Sauerhoff
(1976)
-------------------------------------------------------------------------------------------
a Intraperitoneal. b Subcutaneous.
c Oral. d Inhalation.
When comparing the turnover of PCP in rats after single and
after repeated inhalation exposure, Hoben et al. (1976d) did not
find any evidence of accumulation. The clearance rates for urine,
plasma, liver, and lung were fairly parallel. After repeated
inhalation exposures to about 5.9 mg/kg body weight, the PCP body
burden did not increase as expected from the 24-h half-life
following a single exposure. On the basis of the increased urinary
excretion, the authors concluded that the biotransformation was
accelerated and possibly induced by prior exposure to PCP.
6.5.2 Human studies
Braun et al. (1979) observed a time lag in the urinary
excretion rate following a single oral dose of 0.1 mg Na-PCP/kg
body weight given to 4 volunteers (section 6.1.2). Maximum urinary
excretion was reached 40 h after ingestion and 36 h after the
maximum plasma level of 0.245 mg PCP/litre. The authors ascribed
this delay to a strong enterohepatic circulation similar to that
reported in rats and monkeys. The elimination half-life for PCP in
plasma was about 30 h. The elimination half-life for PCP and PCP
glucuronide in urine was 33 and 13 h, respectively. The
elimination of PCP from plasma in these human subjects followed
linear kinetics and was monophasic, and resembled the elimination
kinetics reported for the monkey. Elimination kinetics in the rat
are biphasic; however, the rate constants and half-lives for the
absorption and elimination of PCP from rat plasma are more similar
to those for men than those for monkeys.
Using the kinetic parameters determined in their single-
exposure studies, Braun et al. (1979) calculated that men ingesting
0.1 mg PCP/kg body weight daily (based on 100% uptake of 0.5 mg
PCP/m3 by a man carrying out light work) would attain a steady-
state plasma concentration of 0.491 mg/litre, after 8.4 days. This
suggests that there is no cumulative effect, even with repeated
daily low-level exposure. This conclusion is based on a simulation
model for the daily ingestion of PCP using data derived from
single-dose studies.
Uhl et al. (1986) studied 3 healthy male volunteers aged 29,
24, and 47 years, exposed to single oral doses of PCP (purity >
99%) in 40% ethanol in 6 different studies. The doses varied from
0.016 to 0.31 mg PCP/kg body weight. Their results are in contrast
to those of Braun et al. (1979). Uhl et al. (1986) determined PCP
elimination half-lives of 16 days (plasma) and 18 - 20 days
(urine), elimination rates being about 13 times slower than those
reported by Braun et al. (1979). Uhl et al. (1986) did not find
any evidence for an enterohepatic accumulation mechanism. They
ascribed the low elimination rate to the high protein-binding
tendency of PCP and the concomitantly low PCP clearance observed
(0.07 ml/min). At normal urinary pH (5 - 6), PCP exists in its
phenolic form and therefore more than 99% of the filtered PCP is
believed to be reabsorbed in the renal tubules. In one study,
alkalinization of the urine by administration of sodium bicarbonate
considerably enhanced the elimination rate of PCP in urine. Over
the pH range 5.4 - 7.8, the rate of PCP elimination varied by a
factor of 8; urinary excretion was approximately 2 µg PCP/h at pH
5.4 and about 16 µg PCP/h at pH 7.4.
The differences in the pharmacokinetic profile established in
the 2 studies may be explained by the different experimental
regimes. Uhl et al. (1986) administered the PCP in ethanol; Braun
et al. (1979) administered the sodium salt of PCP in water. The
role of the dietary status of the volunteers before and after the
PCP ingestion or of any other factors is unknown. According to Uhl
et al. (1986), the time lag necessary to attain a steady state
after a change in the exposure, as calculated from the elimination
half-life, is about 3 months. These authors estimated that the
human body burden of PCP at steady state is 10 - 20 times higher
than that estimated by Braun et al. (1979).
Reported cases of accidental PCP exposure as well as data from
occupationally exposed people seem to confirm the results of Uhl et
al. (1986). In the case of an accidental uptake of PCP through the
skin (section 6.1.2), the urinary-PCP concentration decreased from
236 µg/litre, 2 days after the accident to 17 µg/litre, 51 days
later (Bevenue et al., 1967a). From these data, an elimination
half-life of about 15 days can be derived.
In the case of intentional ingestion of PCP (solvent: 82%
aromatic petroleums) discussed in section 6.1.2, despite forced
diuresis with furosemide and mannitol, the serum-PCP level of the
patient decreased from the highest level on day 2 (155 mg
PCP/litre) to 12 mg PCP/litre on day 37 (Haley, 1977), suggesting
an elimination half-life of approximately 10 days.
Begley et al. (1977) surveyed PCP concentrations in the blood
and urine of 18 workers in a wood-treatment factory before and
during a 20-day vacation. The blood and urine levels of PCP
decreased from an average of 5.14 mg/litre to 2.19 mg/litre and
from 1.31 mg/litre to 0.59 mg/litre, respectively, during vacation.
Elimination half-lives of about 9 days for both urine and plasma
can be derived.
Casarett et al. (1969) calculated an excretion half-life of
about 10 h, based on measurements made one day after a single
inhalation exposure in 2 wood-treatment workers. Despite this rapid
elimination rate, the PCP concentrations in the urine of workers
following a long-term high level exposure (PCP in urine, 1.6 - 2.6
mg/litre) did not decrease by more than 60 - 80% "even after a long
absence from exposure" (Casarett et al., 1969). In a comparable
survey with a group of woodworkers exposed long-term to Permatox
100 (3% PCP, 21% tetrachlorophenol), no clear elimination pattern
could be found during a 16-day vacation (Kalman & Horstman, 1983);
half-times for urinary elimination varied from 4 to 72 days, as
estimated from the urinary-PCP levels at day 0 and day 16. A number
of pre-exposed workers showed an increase in urinary-PCP during the
vacation, either as a result of storage of PCP in tissues (section
6.6), additional exposure to PCP independent of work-place
exposure, or a continuous biotransformation of hexachlorobenzene
and similar compounds to PCP (section 5.2.2). However, the latter
can hardly explain an increase in urinary-PCP levels of 53 - 88
µg/litre, as observed by Kalman & Horstman (1983); urinary-PCP
levels range around 10 µg/litre for the general population and
around 40 µg/litre for people exposed non-occupationally (section
5.2.3).
Since all controlled studies on the metabolism of PCP in
mammals have been performed using pure PCP, a judgement of the
influence of impurities in commercial PCP on the metabolism of PCP
is not possible. In fish, Huckins & Petty (1983) observed a
greater conjugation of PCP to its glucoronide with exposure to
purified PCP compared with exposure to commercial PCP. Conjugation
may be a rate-limiting step in its elimination, in which case the
toxic impurities in industrial PCP formulations could alter the
turnover pattern.
6.6 Reaction With Body Components
Braun et al. (1977) found that 99% of the PCP in rat plasma was
bound to protein. Heterogenous binding has been demonstrated,
indicating that, at very low plasma concentrations of PCP, the
protein binding becomes even stronger, because of preferential
binding to more limited but higher affinity sites. Braun et al.
(1977) and Uhl et al. (1986) suggested that this heterogenous
plasma-protein binding might be the cause of long-term urinary
excretion of PCP. Hoben et al. (1976e) found that human plasma had
a much higher capacity for binding to PCP than rat plasma; this
could explain the longer retention times observed in human beings
compared with that in the rat. The difference in binding capacity
could not be accounted for by albumin fraction.
PCP is conjugated in vitro to palmitic acid in rat liver
incubated with a coenzyme A-fortified microsomal system (Leighty &
Fentiman, 1982). Presumably, the binding of PCP to fatty acids
could contribute to PCP retention in lipid-containing tissues.
Weinbach & Garbus (1965) had already discovered the strong
affinity of PCP for rat liver mitochondrial protein. In more
recent studies of Arrhenius et al. (1977a,b) on the sub-cellular
distribution of PCP, PCP accumulation in microsomes or in the
cytosol was approximately 6 and 3 times, respectively, higher than
in the mitochondria. On this basis, the authors suggested that the
microsomal functions, though 4 times less sensitive than
mitochondrial oxidative phosphorylation, were disturbed by PCP
concentrated in these organelles, possibly increasing the toxic
and carcinogenic action of other xenobiotics.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
The toxic effects of chlorophenols have been studied in a
number of organisms. The degree of oil solubility governs the
toxicity of phenolic compounds by controlling their binding to
lipoid cellular structures, e.g., biological membranes, which
probably are the loci of action of PCP. Blackman et al. (1955a,b)
showed that, with the duckweed Lemna minor and the mould
Trichoderma viride, the lipophilic behaviour and, hence, the
toxicity of chlorophenols increased with the degree of chlorination
of the aromatic ring, PCP being the most effective chlorophenol.
Similar results have been reported from studies on fish (Ingols &
Gaffney, 1965), bacteria (Liu et al., 1982), and algae (Huang &
Gloyna, 1968; Rowe et al., 1982).
Although there have been some studies on the effects of PCP on
ecosystems, most of the toxicity data have been derived from single
species trials. Moreover, most bioassays deal with acute rather
than long-term toxic effects.
7.1 Microorganisms
The microbiocidal effectiveness of PCP has been the basis of
its widespread use as a bactericide and fungicide (section 3.3).
For instance, PCP at a concentration of 0.25% and 0.125% (v/v) was
used to control the sapstain fungi Trichoderma harzianum and
Phialophora sp., respectively (Cserjesi & Roff, 1975). Conkey &
Carlson (1963) screened PCP-containing pesticides against 2
bacterial species and 2 species of fungi that are common in pulp
and paper mill systems. Depending on the PCP formulation and the
microbial species, complete inhibition occurred on agar plates at
4 - 250 ppm.
Ishizawa et al. (1961) found bacterial und fungal growth in
soil to be depressed by PCP at 2 g Na-PCP/kg dry soil. Similarly,
oxidative phosphorylation and ATPase activity in Micrococcus
denitrificans cultures were strongly inhibited by PCP at a
concentration of about 130 mg/litre (Imai et al., 1967).
In anaerobic soil containing 10 mg PCP/kg, Murthy et al. (1979)
observed reduced soil respiration as PCP directly or indirectly
inhibited cellulose degradation. The degradation of PCP itself may
also be affected by the toxicity of this compound for degrading
microorganisms.
Godsy et al. (1986) studied the effects of PCP on the
methanogenic fermentation of phenol in anaerobic laboratory
digestors. With PCP concentrations of 0.1 mg/litre or less, PCP
was dechlorinated to non-toxic levels allowing for complete
bioconversion of phenol (200 mg/litre) and PCP, presumably to
methane and carbon dioxide (CO2). Higher PCP levels inhibited the
methanogenic fermentation; at 5 mg PCP/litre, complete inhibition
occurred.
Tam & Trevors (1981a,b) studied the effects of PCP on
asymbiotic nitrogen fixation in soil. The EC50 values for the
inhibition of nitrogenase activity in non-sterile soil, incubated
aerobically and anaerobically, and in sterilized soil inoculated
with Azotobacter sp. were 49.8 mg Na-PCP/kg, 186.8 mg Na-PCP/kg,
and 660.8 mg Na-PCP/kg, respectively. The inhibition of both CO2
evolution and oxygen uptake by Azotobacter vinelandii was found to
be similar to that of nitrogen fixation activity (Tam & Trevors,
1981a). The high concentrations required for inhibition suggest
that, at normal field application rates, no adverse effects on
nitrogenase activity would be expected.
Na-PCP at 50 and 100 mg/kg had a stimulating effect on soil
microbial electron transport activity, while 200 mg Na-PCP/kg
caused 5.8% inhibition (Trevors, 1982c). Inhibition by Na-PCP was
greater in soil enriched with glucose and yeast extract than in
non-amended soil. Concentrations of 25 - 50 mg PCP/litre of
nutrient broth delayed the growth of the bacterium Pseudomonas
fluorescens, which does not degrade PCP. A 1-h exposure to 75 mg
PCP/litre inhibited growth completely. Higher concentrations of
PCP were required to produce inhibition of respiration. Oxygen
consumption was reduced by 21% at a concentration of 25 mg
PCP/litre, while CO2 evolution was not inhibited (Trevors et al.,
1981a; Trevors, 1982b).
Pre-exposure to PCP lowered the sensitivity of Pseudomonas
fluorescens to PCP (Trevors et al., 1982). In addition, non-toxic
concentrations of the antioxidants butylated hydroxyanisole (BHA)
and butylated hydroxytoluene (BHT), which have also been used as
food additives, enhanced the toxicity of PCP for this bacterial
species (Trevors et al., 1981b).
Izaki et al. (1981) found gram-negative bacteria to be more
resistant to PCP than gram-positive bacteria. Some very resistent
Pseudomonas strains tolerated over 500 mg PCP/litre. Studies on
various mutants, in which the lipopolysaccharide layers were more
or less defective, supported the hypothesis that these layers act
as a barrier for PCP and impart resistance to gram-negative
bacteria.
7.2 Aquatic Organisms
PCP has been used in aquatic environments as a molluscicide
and an algicide. The potential hazard of PCP was recognized early,
leading to a number of toxicological studies on aquatic organisms;
these have been reviewed by US EPA (1978) and Buikema et al.
(1979). In general, PCP is extremely toxic for aquatic organisms.
Apart from very sensitive or resistant species, there is apparently
no significant difference in sensitivity to PCP between the
different taxonomic groups (Adema & Vink, 1981).
7.2.1. Plants
Most toxicity tests on aquatic plants have been performed on
algae, particularly the microscopic, free-floating forms. A
selection of these tests is summarized in Table 28. One of the
more striking features evident from the Table is the extreme
variability in the PCP levels that result in toxic effects. For
example, Adema & Vink (1981) determined that a nominal
concentration of 7 mg/litre inhibited the growth of Chlorella
pyrenoidosa by 50%. In contrast, Huang & Gloyna (1968) found that
as little as 7.5 µg/litre caused complete destruction of chlorophyll
in the same species.
At least part of this variability reflects different
sensitivities to PCP between algal species. Adema & Vink (1981)
found that the green alga Scenedesmus quadricauda had an EC50 for
growth that was 87.5 times lower than that for Chlorella
pyrenoidosa. The most sensitive algae were Ankistrodesmus falcatus
and Microcystis sp., for which as little as 0.001 mg PCP/litre
inhibited photosynthesis by an average of 21%, in semi-continuous
cultures.
However, the tremendous variability in susceptibility to PCP
presented in the Table cannot be accounted for solely by species-
specific differences in sensitivity. They probably also reflect
differences in the experimental conditions under which the tests
were run, such as the ambient pH, which affects PCP toxicity for
invertebrates (section 7.2.2), and presumably algae.
Some aquatic vascular plants have also been used for toxicity
studies. The water hyacinth (Crassipes eichhornia) is relatively
tolerant to Na-PCP; a concentration of 5 mg/litre was required to
affect its appearance, and 80 mg/litre to kill the plant (Hirsch,
1942). Only 7.1 x 10-7 mol PCP/litre (approximately 0.19 mg/litre)
were required to induce 50% chlorosis in the fronds of the duckweed
(Lemna minor) (Blackman et al., 1955a). In contrast, Huber et al.
(1982) noted a 50% decrease in the chlorophyll content of this
aquatic macrophyte on exposure to 3 - 4 mg PCP/litre.
Photosynthesis as well as the activity of glutamate dehydrogenase
and alanine aminotransferase were similarly inhibited, but there
was no pronounced effect on dark respiration.
7.2.2 Invertebrates
As shown in Table 29, the toxicity of PCP or Na-PCP for
invertebrates varies with concentrations ranging from 0.068
mg/litre to 10.39 mg/litre. Most of the reported 50% lethal or
effective concentrations are below 1 mg/litre.
In general, developing embryos and larvae are more affected by
PCP than the adults (Table 29). The most striking difference was
reported by van Dijk et al. (1977), who found that, at the same Na-
PCP concentration, the larvae of the marine decapod Palaemon
elegans were inhibited (in terms of the 96-h LC50) about 130 times
more than the adults.
Table 28. The toxicity of PCP and Na-PCP for algae
--------------------------------------------------------------------------------------------------------
Test species Aquatic Test type Test Concent- Effects Reference
system duration ration
(h) (mg/litre)
--------------------------------------------------------------------------------------------------------
Chlorella freshwater static 72 0.008 complete Huang & Gloyna
pyrenoidosa destruction of (1968)
chlorophyll
Chlorella freshwater static and flow 96 7a EC50, growth Adema & Vink
pyrenoidosa (1981)
Scenedesmus freshwater static and flow 96 0.08a EC50, growth Adema & Vink
quadricauda (1981)
Microcystis freshwater static 96 1 NOEC, growth Sloof & Canton
aeruginosa (1983)
Scenedesmus freshwater static 96 0.1 NOEC, growth Sloof & Canton
pannonicus (1983)
Cylindrospermum freshwater static 72 2 no growth Palmer & Maloney
licheniformeb (1955)
Microcystis freshwater static 72 2 no growth Palmer & Maloney
aeruginosab (1955)
Scenedesmus freshwater static 72 - 168 2 growth inhibition Palmer & Maloney
obliquusb (1955)
Chlorella freshwater static 72 - 504 2 no inhibition Palmer & Maloney
variegatab (1955)
Monochrysis sp. marine static and flow 96 0.2a EC50, growth Adema & Vink
(1981)
--------------------------------------------------------------------------------------------------------
Table 28. (contd.)
--------------------------------------------------------------------------------------------------------
Test species Aquatic Test type Test Concent- Effects Reference
system duration ration
(h) (mg/litre)
--------------------------------------------------------------------------------------------------------
Chlamydomonas marine static and flow 96 1.4a EC50, growth Adema & Vink
sp. (1981)
Phaeodactylum marine static and flow 96 3a EC50, growth Adema & Vink
tricornutum (1981)
Dunaliella sp. marine static and flow 96 3.6a EC50, growth Adema & Vink
(1981)
Chlorella ovalis marine static and flow 96 5.5a EC50, growth Adema & Vink
(1981)
Ankistrodesmus marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
falcatus flow 264 photosynthesis (1982)
Microcystis sp. marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
flow 264 photosynthesis (1982)
Melosira sp. marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
flow 264 photosynthesis (1982)
and growth
Selenastrum marine static 2 0.05 - beginning inhibi- Jayaweera et al.
capricornutum 0.1 tion of carbon (1982)
assimilation rate
Selenastrum marine static 2 2.66 complete inhibi- Jayaweera et al.
capricornutum tion (1982)
--------------------------------------------------------------------------------------------------------
a Average results of static and flow-through tests.
b Na-PCP.
Dragonfly nymphs ( Epicordulia sp.), damsel fly nymphs ( Ischnura
sp.), isopods ( Asellus communis Say), and amphipods ( Hyalella
knickerbockeri Bate) were resistant to Na-PCP compared with other
invertebrate species, and easily survived exposure to 5 mg/litre
(Goodnight, 1942). Turner et al. (1948) found that 0.1 mg Na-
PCP/litre was ineffective against mussels (Mytilus edulis),
anemones, and barnacles, while a concentration of 1 mg/litre
prevented attachment and growth in sea water.
The results of more recent studies, mainly conducted on
annelids, molluscs, and crustaceans, are mostly based on median
lethal or effective concentrations (LC50, EC50, TLm). However, a
comparison of acute toxicity data established within equal test
duration may only be appropriate if the various organisms have
similar life cycles. It is arbitrary to compare, for instance, the
96-h LC50 of copepods, crayfish, and trout. On a basis of the
median life span, a 96-h toxicity test with the cladoceran Daphnia
would correspond to a test with trout or carp lasting as long as
one year.
Very few studies have been carried out to assess chronic or
sublethal effects, or the influence of various environmental
conditions on the toxicity of PCP. Examining the effect of Na-PCP
on the burrowing activity of a lugworm (Arenicola cristata),
Rubinstein (1978) noted a significant adverse effect at
concentrations of 80 and 156 µg Na-PCP/litre. At both
concentrations, no mortality was observed. The reduced lugworm
activity could affect the sediment turnover.
At sublethal concentrations (120 µg PCP/litre), coelomic fluid
glucose in the marine sandworm Neanthes virens increased to about
twice the control levels after 24 h and then gradually decreased
(Carr & Neff, 1981; Thomas et al., 1981). Ascorbic acid levels
became elevated during exposure, indicating sublethal stress.
During the acute lethal exposure (above 365 µg/litre), a
significant hypoglycaemic response and ascorbic acid depletion were
observed.
The acute toxicity of PCP has been found to be pH dependent
(Table 29). The data of Whitley (1968) indicate that the toxicity
of PCP for tubificid worms increased up to almost 5-fold, when the
pH value decreased from 9.5 to 7.5. This is consistent with the
sensitivity pattern of various oligochaete species (Chapman et al.,
1982). Similarly, lowering the pH from 7.5 to 6.5 increased the
toxicity of PCP for the crayfish (Astacus fluviatilis) by a factor
of 5.9 (Kaila & Saarikoski, 1977). The non-ionized form of a
compound penetrates biological membranes much more easily than the
ionized form, which accounts for the effects of pH on PCP toxicity.
Table 29. The toxicity of PCP and Na-PCP for invertebrates
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Tubificid wormsa,b freshwater static pH 7.5 24 0.31 LC50 Whitley (1968)
Tubificid wormsa,b freshwater static pH 8.5 24 0.67 LC50 Whitley (1968)
Tubificid wormsa,b freshwater static pH 9.5 24 1.40 LC50 Whitley (1968)
Water flea freshwater static 24 0.8 EC50 Bringmann &
(Daphnia magna) Kühn (1982)
Fresh-water snail freshwater static 96 0.16 LC50 Gupta & Rao
(Lymnaea acuminata) (1982)
Fresh-water snail freshwater static 96 0.19b LC50 Gupta & Rao
(Lymnaea acuminata) (1982)
Eastern oyster marine static 48 < 0.25 LC50, egg Davis & Hidu
(Crassostrea virginica) development (1969)
Eastern oyster marine static 336 0.07 LC50, Davis & Hidu
(Crassostrea virginica) survival (1969)
of larvae
Pacific oysterb marine static 48 0.027 4.3% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
Pacific oyster marine static 48 0.069 72.4% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
---------------------------------------------------------------------------------------------------------
Table 29. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Pacific oyster marine static 48 0.11 100% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
Eastern oyster marine static 48 0.04 EC50, Borthwick &
(Crassostrea virginica) embryo Schimmel (1978)
development
European brown shrimp adult marine static 96 1.79 LC50 van Dijk et
(Crangon crangon)b al. (1977)
European brown shrimp larvae marine static 96 0.11 LC50 van Dijk et
(Crangon crangon)b al. (1977)
Marine decapod adult marine static 96 10.39 LC50 van Dijk et
(Palaemon elegans)b al. (1977)
Marine decapod larvae marine static 96 0.08 LC50 van Dijk et
(Palaemon elegans)b al. (1977)
Brackish water decapod adult marine static 96 5.09 LC50 van Dijk et
(Palaemonetes varians)b al. (1977)
Brackish water decapod larvae marine static 96 0.36 LC50 van Dijk et
(Palaemonetes varians)b al. (1977)
---------------------------------------------------------------------------------------------------------
Table 29. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Grass shrimp inter- marine static 96 2.63 LC50 Conklin & Rao
(Palaemonetes pugio)b molt (1978)
Grass shrimp early marine static 96 2.74 LC50 Conklin & Rao
(Palaemonetes pugio)b premolt (1978)
Grass shrimp late marine static 96 0.44 LC50 Conklin & Rao
(Palaemonetes pugio)b premolt (1978)
Brown shrimp marine flow 96 > 0.195 LC50 Schimmel et
(Penaeus aztecus)b al. (1978)
Copepod (Pseudodiap- marine static 96 0.068 LC50 Hauch et al.
tomus coronatus)b (1980)
Crayfish marine semi- pH 7.5 192 53 LC50 Kaila &
(Astacus fluviatilis) contin- Saarikoski
uous (1977)
flow
Crayfish marine semi- pH 6.5 192 9 LC50 Kaila &
(Astacus fluviatilis) contin- Saarikoski
uous (1977)
flow
---------------------------------------------------------------------------------------------------------
a Mixed population of Tubifex tubifex and Limnodrilus hoffmeisteri.
b Na-PCP.
7.2.3 Vertebrates
Most studies on vertebrates have been performed with fish. In
short-term studies, the LC50 values for PCP or Na-PCP are generally
less than 1 mg PCP/litre, and, in many cases, even less than 0.1 mg
PCP/litre (Table 30).
The effectiveness of purified PCP and Na-PCP as well as of
commercial products has been investigated under comparable
conditions in only a few cases. Borthwick & Schimmel (1978)
determined that the 96-h LC50 for 14-day-old sheepshead minnow fry
exposed to analytical grade PCP was similar to that of the
commercial formulation Dowicide G (0.392 and 0.516 mg/litre,
respectively, corresponding to 1.47 and 1.41 µmol). Similarly, the
prolarval pinfish was, on a molar basis, about equally affected by
analytical grade Na-PCP (LC50, 0.038 mg/litre = 0.14 µmol) and
Dowicide G (LC50, 0.066 mg/litre = 0.18 µmol).
Differences in the toxicity of PCP and Na-PCP for fish have
been observed under various test conditions. For example, both the
goldfish (Carassius auratus) and the sheepshead minnow (Cyprinodon
variegatus) were more affected by PCP in static than in continuous-
flow bioassays (Table 30). Ruesinck & Smith (1975) noted that the
fathead minnow (Pimephales promelas) was more resistant to Na-PCP
at 25 °C than at 15 °C. In contrast, Crandall & Goodnight (1959)
observed that higher temperatures increased the toxicity of Na-PCP
for the fathead minnow (Pimephales promelas), i.e., the LD50 at
10 °C, 18 °C, and 26 °C was 260, 81, and 46 mg/litre, respectively.
Temperature was also found to control PCP toxicity for rainbow
trout (Salmo gairdneri) (Hodson & Blunt, 1981). Eggs of trout
exposed to PCP at 0.01 - 0.1 mg/litre) showed elevated mortality
between fertilization and hatch and reduced weight at hatch. The
effects on hatch weight were greater at 6 °C than at 10 °C, while
growth rates were reduced more at 20 °C than at 12 °C.
Saarikoski & Viluksela (1981) also demonstrated that the
ambient pH influences the toxicity of PCP for fish. The 96-h LC50
values for the guppy (Poecilia reticulata) were about 0.04 mg/litre
at pH 5, 0.12 mg/litre at pH 6, 0.44 mg/litre at pH 7, and 0.91
mg/litre at pH 8. At pH 5, 33.39% of PCP is in the molecular form,
while, at pH 8, more than 99% exists as phenate ion. Since the
change in toxicity was substantially smaller than it would be if
only the molecular PCP were toxic, the authors concluded that the
phenate ion also contributes to the toxic effect.
Dissolved oxygen also plays an important role in modifying the
toxicity of PCP for fish. Dissolved oxygen levels of 7.8, 6.5, or
5 mg/litre resulted in 96-h LC50 values of 0.107, 0.083, and 0.026
mg PCP/litre, respectively (Gupta et al., 1983a). The increase in
toxicity at low levels of dissolved oxygen may be due to enhanced
absorption of PCP via gills, as the ventilation rate speeds up
under low oxygen regimes.
Table 30. The toxicity of PCP and Na-PCP for various fish species
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effect Reference
stage system type modifier duration tration
(h) (mg/litre)
---------------------------------------------------------------------------------------------------------
Brown trout fresh- static 24 0.2 LC50 Hattula et al.
(Salmo trutta) water (1981)
Bluegill sunfish fresh- static hardness 48 0.03 - LC50 Inglis & Davis
(Lepomis macrochirus) water 0.04 (1972)
Goldfish fresh- static hardness 48 0.08 - LC50 Inglis & Davis
(Carassius auratus) water 0.17 (1972)
Fathead minnow juvenile fresh- flow 48 0.21 LC50 Ruesinck & Smith
(Pimephales promelas) water (1975)
Rainbow trouta fresh- static labora- 96 0.05 - LC50 Davis & Hoos
(Salmo gairdneri) water toryb 0.10 (1975)
Coho salmona fresh- static labora- 96 0.03 - LC50 Davis & Hoos
(Oncorhynchus kisutch) water toryb 0.09 (1975)
Goldfish fresh- flow 96 0.22 LC50 Adelmann & Smith
(Carassius auratus) water (1976)
Fathead minnow juvenile fresh- static 96 0.6 LC50 Mattson et al.
(Pimephales promelas) water (1976)
Common carpa larvae fresh- static 96 0.01 LC50 Verma et al.
(Cyprinus carpio) water (1981b)
---------------------------------------------------------------------------------------------------------
Table 30. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effect Reference
stage system type modifier duration tration
(h) (mg/litre)
---------------------------------------------------------------------------------------------------------
Guppy fresh- static 96 0.97 LC50 Gupta et al.
(Lebistes reticulatus) water (1982)
Sheepshead minnow marine flow 96 0.44 LC50 Parrish et al.
(Cyprinodon variegatus) (1978)
Sheepshead minnow 1-day- marine static 96 0.329 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 14-day- marine static 96 0.392 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 28-day- marine static 96 0.240 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 42-day- marine static 96 0.223 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Pin percha adult marine flow 96 0.053 LC50 Schimmel et al.
(Lagodon rhomboides) (1978)
Pin percha 48-h marine static 96 0.038 LC50 Borthwick &
(Lagodon rhomboides) pro- Schimmel (1978)
larvae
---------------------------------------------------------------------------------------------------------
a Na-PCP.
b Results of an inter-laboratory bioassay standardization exercise.
The size of fresh-water fish may also influence the toxicity of
PCP. The toxicity of PCP for Notopterus notopterus decreased with
increasing fish length up to 14.5 cm, though larger fish were again
more susceptible (Gupta et al., 1982). Similarly, slight
differences in sensitivity were observed when Cyprinodon variegatus
fry of different ages were exposed to PCP (Borthwick & Schimmel,
1978).
As with invertebrates, larvae of fish seem to be more
vulnerable to PCP than adult fish. The lowest value in Table 30 in
terms of LC50 is based on tests with the larvae of the fresh-water
carp (Cyprinus carpio).
Data derived from acute toxicity tests are of limited value in
estimating the long-term effects of PCP on fish. Studies dealing
with sublethal concentrations of PCP may provide more valuable
information, since they involve PCP concentrations approaching
ambient levels. Since responses to sublethal concentrations
require prolonged periods of time, the continuous-flow method is
usually chosen to keep test conditions, particularly the
concentration of the toxicant, constant.
In underyearling sockeye salmon (Oncorhynchus nerka) exposed to
sublethal concentrations of Na-PCP, Webb & Brett (1973) observed
significant reductions in growth rate and food conversion
efficiency. The EC50 for these processes is about 1.80 µg Na-
PCP/litre (approximately 2.8% of the 96-h LC50 of 0.063 mg/litre).
Similarly, low concentrations of PCP (13.6 - 60.2 µg/litre)
were used to induce physiological stress in the fresh-water fish
Notopterus notopterus, as measured by the activity of hepatic acid
and alkaline phosphatases and succinic dehydrogenase (Dalela et
al., 1980a). The activity of these 3 enzymes was reduced after 10,
20, and 30 days of exposure. The greatest effect was observed in
acid phosphatase after 30 days at a concentration of 60.2 µg
PCP/litre caused a 71.09% inhibition of enzyme activity.
In vivo blood variables of Notopterus notopterus (Verma et al.,
1981c) were sensitive to PCP concentrations as low as 1/10th,
1/15th, and 1/20th of the 96-h LC50 of 0.083 mg PCP/litre.
Generally, red and white blood cell counts and packed cell volume
were increased after 30 days of exposure, while clotting time,
erythrocyte sedimentation rate, levels of haemoglobin and mean
corpuscular haemoglobin, and mean cell volume were decreased. In
addition, there were significant increases in the activity of
transaminases in the blood-serum (Verma et al., 1981a) and in the
brain, liver, kidney, and gills (Gupta et al., 1983b). Succinic
dehydrogenase and pyruvic dehydrogenase were inhibited, while the
activity of lactic dehydrogenase was stimulated, thus indicating
the development of anaerobic conditions at the cellular level at
these sublethal concentrations (Verma et al., 1982).
Other biochemical responses were observed in juvenile striped
mullet (Mugil cephalus), a marine fish (Thomas et al., 1981);
environmental stress was indicated by a rapid rise in plasma-
cortisol concentrations at 200 µg PCP/litre, accompanied by a
marked hyperglycaemia and a depletion of hepatic glycogen reserves.
Cleveland et al. (1982) evaluated the chronic toxicities of
commercial PCP, purified PCP, and Dowicide EC-7 in 90-day partial
life-cycle studies with fathead minnows (Pimephales promelas). The
commercial grade PCP used contained relatively large quantities of
hexachlorobenzene, chlorophenoxyphenols, chlorodiphenyloxides,
chlorodibenzodioxins, and chlorodibenzofurans. The purified PCP
included relatively high amounts of chlorinated phenoxyphenols, and
the Dowicide EC-7 contained a broad spectrum of impurities,
generally at lower concentrations than the other 2 formulations.
The commercial composite PCP formulation was the most toxic
preparation; a concentration of 13 µg/litre reduced growth of the
fish, and 27 µg/litre also reduced survival. Growth, but not
survival, was affected by the purified PCP at concentrations equal
to, or greater than, 85 µg/litre. Dowicide EC-7 was the least
toxic of the PCPs tested, and at the maximum level tested (139
µg/litre), did not adversely affect growth or survival of fathead
minnows. Thus, impurities present in PCP were found clearly to
increase toxicity under long-term exposure conditions. Moreover,
degeneration of the fins and opercula, as well as malfunction of
the anterior regions of the skull were also noted in fathead minnows
exposed to the commercial composite of PCP mixture.
7.3 Terrestrial Organisms
7.3.1 Plants
Previously, PCP was widely used as a herbicide, defoliant, and
preharvest desiccant. However, as PCP is not a very specific
herbicide in terms of inhibiting special target species (Kozak et
al., 1979), non-target crop and wildlife species can also be
adversely affected, though no data are available in this respect.
Plants may be damaged by contact with the PCP in treated wooden
material as, for example, when fruit trees in the vicinity of
freshly-treated wooden support posts or stakes suffered bark
lesions and chlorosis (Ferree, 1974). Golden delicious trees
(Malus domesticus) were the most sensitive, some even died.
7.3.2 Animals
Data on the toxicity of PCP for terrestrial animals have been
obtained almost exclusively from laboratory studies. The results
of toxicity tests on experimental animals are presented in section
8. Some fatal cases have been reported in which farm animals were
incidentally exposed to PCP. Blevins (1965), for example, described
a case of acute and lethal poisoning of baby pigs by PCP. The
owners of a newly constructed farrowing house had exceeded the
manufacturer's recommendation in treating the floor with a solution
of PCP in used crankcase oil. All piglets died within one day
after they had been moved into the farrowing house. The sow was
moved outside where she recovered. No information was given
concerning the PCP levels in the air or in the swine.
A recent case, in Canada, of the mortality of young pigs kept
on a PCP-treated wooden floor was reported by Ryan (1983).
Although PCP residues of 310 µg/litre were found in sow's milk
samples, no PCP could be detected in the liver and stomach of the
young pigs. However, µg/kg concentrations of the higher
chlorinated dioxins were found in the skin and liver of the young
pigs, and Ryan (1983) concluded from these findings that these
impurities were responsible for their deaths.
Pesticide poisonings of livestock in the United Kingdom have
been reviewed by Quick (1982) for the period 1977-80. Of 38
suspected PCP poisoning incidents, only 9 were confirmed as PCP
intoxications. High PCP levels found in wood shavings and sawdust,
used as bedding or litter for cats and poultry, apparently caused
the death of animals. Quick (1982) suspected that impurities
present in the commercial PCP products could have been partly
responsible for the deaths.
Hill et al. (1975) reported a study in which the toxicity of
PCP was determined in young birds of 4 species after 5 days of
feeding PCP in the diet; the relatively high LC50 values
(3400 - 5204 mg/kg body weight) do not indicate that PCP is highly
toxic for birds. However, Vermeer et al. (1974) found 50 dead
snail kites after extensive application of Na-PCP as a molluscicide
in the rice fields of Surinam (section 5.1.4).
7.4 Population and Ecosystem Effects
Very few studies have addressed the effects of PCP on aquatic
or terrestrial communities. Field studies on pesticides have
usually been carried out when their accidental release resulted in
visible kills of fish, birds, or other organisms. For instance,
Pierce et al. (1977) and Pierce & Victor (1978) investigated the
fate of PCP in a fresh-water lake after an overflow from a pole
treatment waste pond caused extensive fish kills (section 7.5).
More valuable information about the effects of PCP on
communities can be obtained from model ecosystem studies, in which
the response of portions of the environment placed in a laboratory
was observed. Tagatz et al. (1977) designed a test system
consisting of constant-flow aquaria containing a layer of sand, and
seawater with its plankton as well as animals representing
estuarine macrobenthic communities. The averages and ranges of the
PCP concentrations in the exposed aquaria were 7 µg/litre (3 - 13
µg/litre), 76 µg/litre (47 - 112 µg/litre), and 622 µg/litre (330 -
964 µg/litre). After the 9-week exposure period, a dose-related
decrease was found in the numerically dominant groups. Molluscs in
particular were markedly reduced at 7 µg PCP/litre, and annelids
and arthropods at 76 µg/litre. Almost no animals occurred at 622
µg PCP/litre, while the total numbers of individuals and species
were significantly less in aquaria exposed to 76 µg PCP/litre than
in controls or those exposed to 7 µg PCP/litre. These striking
changes in the relative abundance and diversity of species are
evidence of substantial alterations in the community structure
induced by PCP. In nature, the stability of macrobenthic
communities could be disrupted.
In a second study conducted with Dowicide G-ST (79% Na-PCP),
molluscs were also the most sensitive organisms tested. Levels of
15.8 and 161 µg PCP/litre caused similar reductions in the numbers
of individuals and species (Tagatz et al., 1978).
Using the same test procedure, Cantelmo & Rao (1978) studied
the effects of PCP on meiobenthic communities. Meiofauna comprise
organisms that pass through a 0.5 mm sieve but are retained on a
sieve with mesh widths smaller than 0.1 mm. The Nematoda are
generally the most common taxon in marine sediments (83% in this
study). PCP at 76 µg/litre caused an increase in the biomass and
density of nematodes compared with those in control aquaria, while
higher concentrations of PCP (161 and 622 µg/litre) caused a
decrease. One of the major effects of PCP on nematodes was a shift
from epistrate feeders to deposit feeders at concentrations of 161
and 622 µg PCP/litre. Part of this alteration may have been due to
the reduction of algae serving as a food supply.
Tagatz et al. (1981) reported the effects of PCP on field and
laboratory estuarine benthic communities. In principle, test
apparata were the same as those used in earlier studies, except
that already established communities were exposed to PCP.
Community structure was significantly altered at a PCP
concentration of about 140 µg/litre in both field and laboratory
aquaria; the populations of several invertebrate species were
significantly reduced. There were slight differences in the
effects of PCP on numbers of individuals and species between the
field and laboratory systems.
Cook et al. (1980) examined the effects of PCP on the
microfungal succession of an estuarine benthic microcosm.
Trichoderma sp. was initially the most common fungus isolated from
the sediment. The addition of PCP (140 µg/litre) resulted in an
alteration in the pattern of species; a different species, probably
Penicillium canescens, assumed dominance.
Considerable effects of PCP on the population dynamics of
several species were noted by Schauerte et al. (1982) during an
outdoor study on a natural pond divided into 6 compartments of
about 100 litres each. PCP was applied in the water of 2
compartments at a concentration of 1 mg/litre. Three days after
application, the population of Daphnia pulex pulex was eliminated
from the treated enclosures. The phytoplankton species showed
marked alterations in population dynamics: the autotrophic blue-
green alga Chroococcus limneticus decreased, while the mixotrophic
flagellate Euglena acus significantly increased. This increase was
attributed to the reduced grazing pressure by Daphnia;
interspecific competition was also discussed as possible cause. As
a further secondary effect, the oxygen concentration significantly
decreased, because of the "changed balance between autotrophic and
heterotrophic populations" (Schauerte et al., 1982).
The US Environmental Protection Agency examined the effects of
PCP on a periphyton community in outdoor experimental streams.
Even at the low PCP level corresponding to the water quality
criterion (48 µg/litre) (US EPA, 1980), adverse effects were noted
in terms of community alterations and suppressed community
metabolism (Yount & Richter, 1986). PCP at this concentration also
caused adverse effects on fish growth, larval drift, and larval
yield (Zischke et al., 1985).
7.5 Biotransformation, Bioaccumulation, and Biomagnification
7.5.1. Aquatic organisms
Most research on the bioaccumulation of PCP has been carried
out in aquatic situations. This presents difficulties in that the
bioconcentration factors, which are generally directly related to
the partition coefficients, could vary by several orders of
magnitude, depending on the pH and, at high pH values, on the ionic
strength, the two factors governing the partition coefficient of
PCP (Kaiser & Valdmanis, 1982). In addition, exposure time must
also be taken into account when interpreting PCP residues in
organisms. These influences may, in part, explain the wide range
in bioconcentration factors that has been found.
In general, substances with the solubility properties of PCP
are predominantly taken up by the surrounding water (Niimi &
McFadden, 1982). However, PCP accumulation along the food chains
contributes to its overall bioaccumulation as well.
Table 31 shows the bioconcentration factors derived for several
fish species along with the ambient levels in water. Fresh-water
species seem to accumulate PCP to a much greater extent than marine
fish, possibly because the relevant enzyme systems in marine
species respond faster than those in fresh-water fish (Trujillo et
al., 1982).
Since the ambient concentrations of PCP in the water of natural
aquatic environments are usually less than 1 µg/litre (section
5.1.2), the studies of Niimi & McFadden (1982) are of particular
importance. The authors applied realistic concentrations in
exposing rainbow trout (Salmo gairdneri) to < 10 (control), 35,
and 660 ng Na-PCP/litre, and distinguished between PCP content in
liver and gall bladder, the remaining tissues, and whole fish. As
shown in Table 32, rainbow trout accumulated PCP, even when exposed
to concentrations as low as 35 ng Na-PCP/litre over prolonged
periods. The percentage of PCP stored was highest in the liver and
gall bladder. On the basis of this high bioconcentration at the
low waterborne toxicant concentrations, the authors suggested that
rainbow trout may be less efficient in eliminating PCP than other
species.
On removal from PCP-containing water, fish eliminate previously
accumulated PCP. However, a portion of the PCP incorporated is
more persistent: residues of 0.32 mg/kg (Trujillo et al., 1982) and
0.03 in the muscle to 0.6 mg/kg in the liver (Pruitt et al., 1977)
were still detectable after 18 and 16 days of depuration,
respectively. Elimination half-lives during depuration phases were
4.7 days in killifish (Trujillo et al., 1982) and 6 - 24 days in
rainbow trout (Glickman et al., 1977).
Table 31. Measured bioconcentration factors for PCP for several
fish species
---------------------------------------------------------------------------
Species Time Concentration Bioconcen- Reference
(days) in water tration
(µg/litre) factor
---------------------------------------------------------------------------
Fresh-water fish
Carassius auratus 5 100 1000 Kobayashi &
Akitake (1975a)
Lepomis macro- 1 100 320 Pruitt et al.
chirus (various 4 100 5 - 350 (1977)
tissues) 16 100 4 - 230
Salmo trutta 1 200 100 Hattula et al.
(whole body) (1981)
Salmo gairdneri 20 0.035 200 Niimi &
(whole body) 0.660 130 McFadden (1982)
65 0.035 600
0.660 232
Leuciscus idus 3 42 1050 Freitag et al.
melanotus (whole (1982)
body)
Marine fish
Fundulus similis 4 36 - 306 30 Schimmel et al.
(unspecified (1978)
tissues)
Mugil cephalus 4 26 - 308 38 Schimmel et al.
(unspecified (1978)
tissues)
Mugil cephalus 4 46 6 Faas & Moore
(edible tissues) 4 85 79 (1979)
4 157 56
Fundulus similus 1 57 - 610 8 Trujillo et al.
(whole body) 1 49 (1982)
7 64
7 47
---------------------------------------------------------------------------
In an ecotoxicological profile analysis, Freitag et al. (1982)
determined the bioconcentration of PCP not only in fish (Table 31)
but also in the green alga Chlorella fusca var. vacuolata and in
activated sludge. The bioaccumulation factor in the 24-h algal
test was 1250; in a 5-day activated sludge assay it was 1100 at a
waterborne concentration of 0.05 mg PCP/litre.
Table 32. PCP levels in tissues, organs, and
whole body of rainbow trout (Salmo gairdneri)
exposed to < 10 (control), 35, and 660 ng
Na-PCP/litrea
--------------------------------------------------
Na-PCP PCP concentrations (µg/kg) in:
Days in water liver and remaining whole
(ng/litre) gall bladder tissue body
--------------------------------------------------
0 < 10 2.3 1.1 1.1
20 < 10 1.2 1.5 1.5
35 28 7 7
660 674 77 86
65 < 10 3.9 3.7 3.6
35 135 20 21
660 1984 135 153
115 < 10 3.1 1.9 1.9
35 63 6 7
660 2204 128 160
--------------------------------------------------
a Adapted from: Niimi & McFadden (1982).
Ernst (1979) measured PCP residues in water and benthic
invertebrates at steady state in a static marine system. For the
common mussel (Mytilus edulis) and the polychaete (Lanice
conchilega), bioconcentration factors averaged 390 and 3820 on a
wet weight basis, respectively, at an initial PCP concentration in
sea water of 0.002 - 0.005 mg/litre. The species studied and the
lipid contents of the animals had pronounced effects on the
bioconcentration factor, while temperature and metabolic activity
did not show any remarkable effects on the bioaccumulation. In a
similar study, the polychaete (Neanthes virens) was exposed to 0.1
mg 14C-PCP/litre in sea water (Carr & Neff, 1981). The
bioconcentration factor of 280 was 10 times lower than that
reported by Ernst (1979) for Lanice conchilega.
Clams from the Jadebusen, a North Sea bight, sampled near the
end of a waste-water pipe, accumulated 100 - 1000 times more PCP
than the sediments (Butte et al., 1985).
The studies of Pierce et al. (1977) and Pierce & Victor (1978)
are some of the few field studies concerning the biotransformation
of PCP. The authors followed an accidental discharge into a fresh-
water lake near Hattiesburg, Mississippi, USA. Within two months,
the initially lethal levels of PCP, which had resulted in an
extensive fish kill, decreased to between 6 and 19 µg/litre in the
water and remained near this level throughout the studies,
apparently because of the continuous influx of contaminated water
from other areas. In a control pond with a low background
concentration of 0.5 µg PCP/litre, fish contained only 50 µg
PCP/litre, whereas much higher levels were found in fish in the
contaminated pond. Two months following the spill, levels in fish
averaged 2500 µg PCP/kg dry weight but dropped to 130 µg/kg after
6 months; background levels were achieved within about 10 months.
In a Finnish lake area contaminated through pulp bleaching and
with wood preservative wastes, PCP residues in fish and plankton
did not indicate a strong accumulation via the food chain,
plankton > roach >> pike. However, tetrachlorocatechol, a
possible biodegradation product of PCP, was found to be strongly
absorbed in plankton (Paasivirta et al., 1980).
In littoral microcosms, 14C-PCP was linearly accumulated by
aquatic plants, mainly Potamogeton foliosus and Najos
guadalupensis, during the first 35 days, levelling off to
concentrations over 700 times the initial concentration in water
(41 µg/litre). After 8 weeks, PCP concentration in the macrophytes
rapidly decreased (Knowlton & Huckins, 1983).
In an aquatic model ecosystem, the ecological magnification
value for PCP in fish was 296, the parent compound representing 74%
of the total extractable 14C. The other members had lower
bioconcentration factors: algae, 1.5; mosquito larvae, 16; snail,
121; and Daphnia, 165. In a terrestrial-aquatic model ecosystem,
the bioconcentration factors were: algae, 5; Daphnia, 205; snail,
21; mosquito, 26; and fish, 132 (Lu et al., 1978). These data
indicate that bioaccumulation takes place not only through the
surrounding water but also along the food chains.
7.5.2 Terrestrial organisms
The terrestrial model ecosystem established by Lu et al. (1978)
simulated a crop-soil interaction and included the following
organisms: earthworm (Lumbricus terrestris), slug (Limax maximus),
pillbug (Armadillidium vulgare), saltmarsh caterpillar (Estigmene
acrea), prairie vole (Microtus ochregaster), and corn (Zea mays).
Corn plants grown on the soil of the model system rapidly
accumulated radioactivity. After 14 days, they contained 6.3 mg/kg
of which 16% was intact PCP, 40% were unknown compounds, and 44%
were conjugates. The prairie vole, at the top of the food chain,
consumed virtually all the plant and animal material in the system
within the 5-day exposure time, and then was found to contain 0.5%
of the total dosage applied (25 µg).
Gruttke et al. (1986) investigated the fate of Na-PCP in two
different model food chains representing important groups of
organisms commonly present in soils. In food chain 1, contaminated
bakers-yeast (0.87 µg 14C-Na-PCP/mg dry weight) was fed to
springtails (Folsomia candida), which accumulated up to 0.37 µg
PCP/mg fresh weight after 10 days. Carabid beetles (Nebria
brevicollis) preying on the contaminated springtails showed a body
burden of approximately 4.5 mg PCP/kg fresh weight in the steady
state, from day 4 to 12. Four days after offering uncontaminated
prey, the PCP content in the beetles dropped to 0.4 mg/kg fresh
weight. Similar results were obtained in food chain 2, which
consisted of contaminated leaves of poplar (700 mg Na-PCP/kg dry
weight), with isopods (Oniscus asellus) as primary consumers, and
staphylinid beetles (Ocypus olens) as predators. Because of the
low accumulation tendency, a lasting effect on the predators would
only be expected in case of long-term contamination.
Apart from these model ecosystem studies, little information is
available on the biotransformation of PCP in terrestrial systems.
Miller & Aboul-Ela (1969) observed that cottonseed kernels of bolls
that were closed during spraying accumulated PCP or its metabolites
in quantities of up to 2 mg/kg. No PCP was detected when the bolls
were open during spraying.
PCP, applied in nutrient solution to the roots of growing corn
plants, was taken up by the roots (Schuppener, 1974). At
concentrations of up to 20 mg/litre, roots of sterile hydro-ponic
cultures accumulated as much as 151 mg PCP/kg. Apparently, PCP did
not translocate in the corn plants as it was not detected in the
upper parts of the plants. The root system of sugarcane treated
with 5 mg PCP/litre nutrient solution accumulated 14C-PCP within 4
weeks, retaining over 99% of the total PCP taken up from solution
(Hilton et al., 1970): no measurable translocation into stalks or
leaves occurred. These findings contrast with the translocation
within corn plants described by Lu et al. (1978).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
Toxicology data on both purified and commercial PCP are
provided in this review because both are relevant for a health
assessment. The mode of action of chlorophenol can only be
determined using purified chemicals. Furthermore, any decision to
remove or reduce the levels of microcontaminants in PCP would have
to be based on a clear understanding of the toxicity of these
purified products.
8.1. Acute Toxicity
PCP, regardless of the route of administration, is the most
acutely toxic of the chlorophenols tested in laboratory animal
species. Oral LD50 values range between 27 and 205 mg/kg body
weight for a variety of species, regardless of the vehicle of
administration and the grade of PCP (Table 33). Acute oral exposure
of mice and rats to lethal doses of PCP (Deichmann, 1943;
Farquharson et al., 1958; Borzelleca et al., 1985; Renner et al.,
1986) results in an increase in respiratory rate, a marked rise in
temperature (4 - 4.5 °C), tremors or possibly convulsions, and a
loss of the righting reflex. Asphyxial spasms and cessation of
breathing usually occurs 0.5 - 2 min before cardiac arrest. A
rapid and intense rigor mortis is observed within 3 - 5 min of
death and approximately 45 min sooner than the onset of rigor
mortis in rats given ether. Similar signs are observed with lethal
exposure to PCP and its sodium salt, regardless of the route of
administration.
In addition to its systemic effects, PCP also induces more
localized effects in test organisms. Both dermal and subcutaneous
applications have produced swelling, skin damage, and occasionally
hair loss in a variety of animals (Kehoe et al., 1939; Baader &
Bauer, 1951; Johnson et al., 1973). Localized effects on blood
vessels may result in hyperaemia or erythema (Kehoe et al., 1939;
Baader & Bauer, 1951). Contact with PCP causes irritation of the
eye, skin, or respiratory mucosae in man (section 9). Skin
bioassay techniques have shown that technical PCP, but not purified
PCP, is acnegenic for rabbits, and that the acnegenic effects are
caused by the microcontaminants, particularly H6CDD (Johnson et
al., 1973).
There are far fewer published data on the effects of acute
exposure of animals to PCP and Na-PCP via the dermal and pulmonary
exposure routes compared with oral exposure, which is surprising
considering that the primary exposures in the work-place are via
the skin and lung. Kozak et al. (1979) reported that PCP was
readily absorbed when applied to the skin of experimental animals.
The effects of acute dermal exposure have been examined only in the
rat and rabbit. For rats, but not rabbits, PCP is much more toxic
when given orally than when applied dermally (Table 33). However,
with Na-PCP, toxicity via the 2 routes appears to be similar (Table
34). The only acute inhalation toxicity value reported for Na-PCP
is for rats; Na-PCP is at least 10 times more toxic via inhalation
than by oral ingestion (Hoben et al., 1976c).
Table 33. Acute toxicity of PCP
---------------------------------------------------------------------------------------------------------
Species Para- Sex Doseb Route Purity/carrier Reference
metera
--------------------------------------------------------------------------------------------------------
Mouse LD50 F 74 oral PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 M 36 oral PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 F 150 oral PCP in polypropylene glycol Ahlborg & Larsson (1978)
LD50 M 177 oral PCP in 10% Emulphor Borzelleca et al. (1985)
LD50 F 117 oral PCP in 10% Emulphor Borzelleca et al. (1985)
LD50 M 129 oral PCP in corn oil Renner et al. (1986)
LD50 F 134 oral PCP in corn oil Renner et al. (1986)
LD50 F 32 intraperitoneal PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 M 59 intraperitoneal PCP in polypropylene glycol Ahlborg & Larsson (1978)
LD50 M 59 intraperitoneal PCP in corn oil Renner et al. (1986)
LD50 F 61 intraperitoneal PCP in corn oil Renner et al. (1986)
LD50 M/F 82 subcutaneous Ning et al. (1984)
Rat LD50 150 oral Schwetz et al. (1978)
LD50 F 135 oral commercial PCP Schwetz et al. (1974)
LD50 M 205 oral commercial PCP Schwetz et al. (1974)
LD50 78 oral 1% in olive oil Deichmann et al. (1942)
LD50 65 oral Schwetz et al. (1978)
(3- to
4-day-
old)
LD50 27 oral 0.5% in Stanolex fuel oil Deichmann et al. (1942)
LD50 M/F 83 oral reagent grade Ning et al. (1984)
LD50 M 146 oral peanut oil Gaines (1969)
LD50 F 175 oral peanut oil Gaines (1969)
MLD M/F 160 oral peanut oil Gaines (1969)
LD50 F 149 cutaneous PCP technical 40% w/v Noakes & Sanderson (1969)
solution in glycerol formal
LD50 320 cutaneous xylene Gaines (1969)
LD50 330 cutaneous xylene Gaines (1969)
MLD M/F 300 cutaneous xylene Gaines (1969)
Rat MDLD M 56 intraperitoneal olive oil Farquharson et al. (1958)
LD50 100 subcutaneous 4% in fuel oil Deichmann (1943)
LD50 90 subcutaneous 4% in fuel oil Deichmann & Mergard (1948)
LD50 M/F 40 subcutaneous reagent grade Ning et al. (1984)
--------------------------------------------------------------------------------------------------------
Table 33. (contd.)
--------------------------------------------------------------------------------------------------------
Species Para- Sex Doseb Route Purity/carrier Reference
metera
--------------------------------------------------------------------------------------------------------
Rabbit MLD 100 - oral 11% PCP in olive oil Kehoe et al. (1939)
130
MLD 70 - oral 5% in Stanolex fuel oil No. 1 Deichmann et al. (1942)
90
MLD 40 - cutaneous various carriers Deichmann et al. (1942)
170
MLD 350 cutaneous 11% in olive oil Kehoe et al. (1939)
MLD 39 cutaneous 1.8% in pine oil Kehoe et al. (1939)
MLD 60 cutaneous 5% in Stanoflex fuel oil No.1 Kehoe et al. (1939)
MLD 110 cutaneous 5% in Shell Dione oil Kehoe et al. (1939)
MLD 70 - subcutaneous 5% in Olive Oil Kehoe et al. (1939)
85
Hamster LD50 168 oral Cabral et al. (1979)
LD50 70 - subcutaneous 5% in olive oil Kehoe et al. (1939)
85
Sheep MLD 120 oral aqueous suspension of sawdust Harrison (1959)
treated at 20 lb technical
PCP/cu. ft
Calf MLD 140 oral aqueous suspension of sawdust Harrison (1959)
treated at 20 lb technical
PCP/cu. ft
---------------------------------------------------------------------------------------------------------
a MLD = Minimum lethal dose.
MDLD = Median lethal dose.
LD50 = Estimated dosage capable of causing 50% mortality of a test population.
LC50 = Estimated concentration capable of causing 50% mortality of a test population.
b LD50 or MLD in mg/kg body weight; LC50 in mg/m3 air.
The limited data on other routes of administration (intra-
peritoneal, intravenous, perhaps subcutaneous), which introduce
PCP or Na-PCP directly into the body indicate that these exposures
result in a stronger toxic effect on rats, hamsters, and mice than
either oral or dermal exposure (Tables 33, 34). These differences
probably result from incomplete uptake of the compound via the
oral, dermal, and perhaps subcutaneous routes.
PCP is considerably more toxic than its sodium salt when
administered orally to rats or rabbits, or dermally to rabbits
(Tables 33, 34). However, subcutaneous or intravenous injections
of PCP and Na-PCP are almost equally toxic. These patterns may
reflect differences in the rate of absorption of the parent
compounds when applied to the skin of rats, but this does not
appear to be true of PCP.
One sex is not consistently more strongly affected by PCP than
the other. For a given combination of organism (rats and mice),
vehicle, and exposure route (mostly oral), males are sometimes
more, sometimes equally, and sometimes less sensitive than females
(Table 33). However, technical PCP is more toxic for female rats
than for males (Schwetz et al., 1974) and more toxic for young rats
than for adults (Schwetz et al., 1978). This differential toxicity
between the sexes is apparent in both short- and long-term studies
with technical PCP and with one of its contaminants, hexachloro-
dibenzo- p-dioxin (H6CDD).
Although the influence of exposure route, the form of the PCP
(sodium salt or parent molecule), and the sex of the experimental
animal are evident within a given study, such patterns may be
masked by a number of factors. Unfortunately, the type and extent
of contamination of PCP tested under acute exposure conditions is
rarely described, despite the fact that some of the
microcontaminants, especially some congeners of polychlorinated
dibenzo- p-dioxin (PCDD) and polychlorinated dibenzofuran (PCDF),
are extremely toxic. Furthermore, a variety of solvents is used to
administer chemicals tested for acute toxicity, and some of these
solvents can enhance or decrease absorption of PCP, thus affecting
toxicity. Hence, the variations in the LD50, LDLO and TDLO values
reported in Kozak et al. (1979), Ahlborg & Thunberg (1980), Jones
(1981), NRCC (1981), and NIOSH (1983) for each animal species and
route of exposure may result, in part, from the use of a variety of
purified and commercial products containing several different
solvents, as well as possible differences in animal strains, or
test design (in this regard, note that some of the earlier studies
involved only a small number of animals, sometimes as few as two).
Acute toxic effects, other than chloracne, which are due
exclusively to the presence of PCDDs or PCDFs, are difficult to
identify. PCDDs, in particular, have a delayed toxic effect that is
masked by the rapid onset of signs of acute exposure to the PCP
molecule.
Table 34. Acute toxicity of Na-PCP
---------------------------------------------------------------------------------------------------------
Species Parametera Doseb Route Purity/carrier Reference
---------------------------------------------------------------------------------------------------------
Mouse LD50 83 subcutaneous reagent grade Ning et al. (1984)
Rat LD50 210.6 oral 2% aqueous Deichmann et al. (1942)
LD50 (F) 125 - 200 oral commercial (79%); aqueous Stohlman (1951)
LD50 71 oral reagent grade Ning et al. (1984)
LD50 104 dermal reagent grade Ning et al. (1984)
LD50 66 subcutaneous 2% aqueous Deichmann et al. (1942)
LD50 38 subcutaneous reagent grade Ning et al. (1984)
LC50 294 inhalation reagent grade; 2-h inhalation Ning et al. (1984
LD50c 11.7 inhalation aqueous aerosol Hoben et al. (1976c)
LD50 34 intraperitoneal aqueous Hoben et al. (1976c)
Rabbit MLD 218 oral 1% NaCl Kehoe et al. (1939)
MLD 250 - 300 oral 5% aqueous Deichmann et al. (1942)
MLD 450 - 700 oral aqueous McGavack et al. (1941)
MLD 250 cutaneous 10% aqueous Deichmann et al. (1942)
MLD 257 cutaneous 2% aqueous Kehoe et al. (1939)
MLD 450 - 600 cutaneous aqueous McGavack et al. (1941)
MLD 100 subcutaneous 10% aqueous Deichmann et al. (1942)
MLD 250 - 300 subcutaneous aqueous McGavack et al. (1941)
MLD 50 - 150 intraperitoneal aqueous McGavack et al. (1941)
MLD 22 - 23 intravenous 2% aqueous Deichmann et al. (1942)
MLD 22 intravenous 1% aqueous Kehoe et al. (1939)
Guinea- MLD 266 cutaneous aqueous Kehoe et al. (1939)
pig
Dog MLD 135 subcutaneous aqueous McGavack et al. (1941)
---------------------------------------------------------------------------------------------------------
a MLD = Minimum lethal dose.
LD50 = Estimated dosage capable of causing 50% mortality of a test population.
LC50 = Estimated concentration capable of causing 50% mortality of a test population.
b LD50 or MLD in mg/kg body weight; LC50 in mg/m3 air.
c Inhalation toxicity value expressed by author as LD50, not LC50.
8.2. Short-Term Toxicity
As discussed in the previous section, some of the acute effects
of exposure to commercial PCP are attributable to microcontaminants
present in the technical preparation. In addition, signs of
exposure to some of these microcontaminants, notably PCDDs, may
not appear for weeks. As a consequence, it is particularly
important to consider the possible confounding effects of these
impurities in reviewing the long-term toxicity of chlorophenols.
In this section, studies on the toxicity of purified PCP, technical
grade PCP, and comparative studies are discussed separately.
8.2.1. Pure or purified PCP
Debets et al. (1980) studied the effect of 99% pure PCP fed to
female rats for 5 weeks. The PCP concentration used was 500 mg/kg
feed, which corresponds to an approximate dose of 40 - 50 mg/kg
body weight per day. Of several liver microsomal enzymes tested,
only ethoxyresorufin O-de-ethylase (20-fold) and glucuronyl
transferase (3-fold) increased in activity with exposure. Body
weight gain, urine and liver porphyrin concentrations, and liver
weights were unaffected by PCP. Interestingly, PCP accelerated the
onset of hexachlorobenzene (HCB) porphyria, suggesting that it is
the PCP metabolized from HCB that causes the porphyria.
In 6-week-old pigs, Greichus et al. (1979) found no overt signs
of toxicosis associated with the oral administration of purified
PCP (at 5, 10, and 15 mg/kg body weight per day in capsules) for 30
days. However, at 10 and 15 mg/kg per day, hepatocyte size
increased and enlarged livers were observed.
8.2.2. Technical grade PCP
Knudsen et al. (1974) fed rats diets containing 0, 25, 50 and
200 mg commercial PCP/kg for 12 weeks. The 50 mg/kg exposure
(corresponding to about 2.3 mg/kg body weight per day) increased
liver weights in both sexes, while haemoglobin concentrations,
haematocrit, and glucose concentrations in serum were elevated in
the 2 highest dosage groups. The no-observed-adverse-effect level
of 25 mg/kg corresponded to an ingested dose of about 1.2 mg/kg
body weight per day.
Post-mortem examination of dairy cattle fed 0.2 mg/kg body
weight per day, for 75 - 84 days, and 2 mg/kg body weight per day
for another 56 - 60 days during lactation, revealed enlargement of
the liver, lungs, kidneys, and adrenals, thickening of the urinary
bladder walls, chronic interstitial nephritis, and subacute
urocystitis in exposed animals (Kinzell et al., 1981). In vitro
testing identified a significant loss of renal function associated
with exposure.
8.2.3. Comparative studies
Recent studies of between 2 and 8 months duration are useful in
discerning differences in the effects of purified and technical
grade pentachlorophenol (Table 35). These studies on rats and mice
have been summarized by Fielder et al. (1982) and NRCC (1982).
Rats receiving 500 mg technical PCP (Table 1, section 2.2) per
kg feed for 8 months had slower growth rates, hepatomegaly,
porphyria, and increased hepatic enzyme activities (aryl
hydrocarbon hydroxylase, glucoronyl transferase, and cytochrome
P-450) (Goldstein et al., 1977). When rats were fed purified PCP
under similar conditions, i.e., 500 mg/kg feed for 8 months, only
retardation of growth rate and an increase in liver glucoronyl
transferase activity were observed. Rats fed 20 mg technical
PCP/kg feed for 8 months had elevated liver enzyme activities,
while rats fed purified PCP at the same concentrations did not.
These findings have been confirmed by Kimbrough & Linder (1978)
using rats exposed to 0, 20, 100, and 500 mg/kg of the same
technical and purified PCP and the same protocol as was employed by
Goldstein et al. (1977). Male and female rats exposed to either
purified or technical PCP gained less weight than controls.
Exposure to purified PCP in the diet at 500 mg/kg resulted in
slightly enlarged liver cells and caused occasional cytoplasmic
inclusions. These effects were not observed in rats fed lower
doses. In contrast, exposure of rats to diets containing 500 mg
technical PCP/kg increased liver weights and thickened the walls of
the hepatic central veins in both sexes, and also caused
pleiomorphic hepatocytes with foamy or vacuolar cytoplasm in male
rats. The livers of females exposed to 500 mg/kg were
characterized by vacuolation and degeneration of hepatocytes and
mitotic anomalies. Similar, but less severe, effects were noted in
rats exposed to diets containing 100 mg and 20 mg technical PCP/kg.
Wainstok de Calmanovici & San Martin de Viale (1980) determined
that technical PCP is more toxic for porphyrin metabolism in rats
than the purified compound. Dosing with 45 - 90 mg technical
PCP/kg body weight per day (the amount given by stomach tube for 18
weeks varied) enhanced the excretion of porphyrins and their
precursors, increased the deposition of porphyrins in the spleen,
liver, and kidney, and altered the activity of enzymes involved in
porphyrin metabolism. Larger doses of purified PCP (100 - 195
mg/kg body weight per day) had similar effects.
Table 35. No-observed-adverse-effect-levels (NOAELs) established in rats
exposed orally to pure, technical, and purified technical grades of PCP
---------------------------------------------------------------------------
PCP Sex NOAEL (mg/kg Reference
body weight
per day)
---------------------------------------------------------------------------
Short-term toxicity
Pure 3 Johnson et al. (1973)
Purified 3 Johnson et al. (1973)
technical
Technical < 3 Johnson et al. (1973)
Technical approximately Knudsen et al. (1974)
1.2
Teratogenicity/
fetotoxicitya
Technical male, female 5 Schwetz et al. (1974)
(progeny)
Purified male, female < 5 Schwetz et al. (1974)
technical (progeny)
Reproductive
toxicityb
Purified male, female 3 Schwetz et al. (1978)
technical
Long-term toxicity
Pure approximately Goldstein et al. (1977)
5
Technical < 1 Kimbrough & Linder (1978)
Purified female < 3 Schwetz et al. (1978)
technical male < 10 Schwetz et al. (1978)
---------------------------------------------------------------------------
a Studies involved exposure of females during days 6 - 15 of gestation
and evaluation of progeny.
b Studies involved exposure of both sexes for 62 days before mating, for
15 days during mating, and, subsequently, throughout gestation and
lactation for females.
In a similar comparative study by Johnson et al. (1973), 99%
pure PCP and purified technical PCP (H6CDD reduced to 1 mg/kg) at
10 and 30 mg/kg body weight per day increased liver weight, but did
not affect other variables monitored. In contrast, unpurified PCP
increased liver and kidney weights, enhanced serum-alkaline
phosphatase activity, and reduced serum-albumin, numbers of
erythrocytes, total haemoglobin, and haematocrit.
The toxicity of different grades of PCP has also been studied
in cattle (McConnell et al., 1980; Parker et al., 1980; Hughes et
al., 1985). These studies have confirmed that effects such as
reduced weight gain, anaemia, liver pathology, and a decrease in
thymus weight were induced by microcontaminants in the technical
PCP. Reductions in serum-thyroid hormones (triiodo-thyronine T3 and
thyroxine - T4) observed in cows fed either purified or technical
PCP were probably due to the chlorophenol itself. Significant
levels of octa-, hepta-, and hexachlorodioxins were found in liver,
fat, and milk at the conclusion of 160 days of exposure (Parker et
al., 1980). Firestone et al. (1979) also reported residues of PCP
and related chemicals in cows' milk, body fat, and blood following
short-term exposures. Only 3 out of 7 PCDD congeners identified in
the technical PCP used were found in tissue and body fluids, i.e.,
1,2,3,6,7,8-H6CDD, 1,2,3,4,6,7,8-H6CDD, and OCDD.
Hexachlorobenzene (HCB) and PCP were also detected. Levels of PCP,
HCB, and PCDD in pooled milk fat from 3 cows reached 4 mg/kg, 200
µg/kg, and 85 µg/kg, respectively. PCP levels fell to 100 µg/kg a
few days after the cessation of dosing and levels of HCB and total
dioxin declined by 50% in 50 days.
8.3. Long-Term Toxicity
The 8-month PCP feeding studies on rats by Goldstein et al.
(1977) and Kimbrough & Linder (1978) might be considered by some to
be long-term. The results of these studies (section 8.2) indicate
that the toxicity of technical PCP preparations is primarily
attributable to microcontaminants.
Schwetz et al. (1978) reported a 2-year exposure of rats to
purified PCP (Table 35). In females, no toxic effects were
observed at levels below 3 mg/kg body weight per day; pigment
accumulation was observed at 10 mg/kg body weight per day, and
decreased body weight gain, increased serum-glutamic pyruvic
transaminase (GPT) activity, and pigment accumulation were observed
at 30 mg/kg body weight per day. Fewer changes were observed in
male rats; no effects were observed at levels below 10 mg/kg body
weight per day, and pigment accumulation and increased GPT activity
were reported at 30 mg/kg body weight per day. It is interesting
to note that Schwetz et al. (1978) did not observe any absolute or
relative weight increase in kidney and liver at 30 mg/kg body
weight per day. In contrast, Johnson et al. (1973) reported
increased weights of these tissues in male and female rats after a
90-day exposure to the same purified PCP used by Schwetz et al.
(1978) at 30 mg/kg body weight per day (section 8.2). An earlier
90-day rat study with purified PCP containing < 0.5 mg PCDD or
PCDF/kg also demonstrated changes in liver and kidney weight at the
30 mg/kg body weight per day dose level (Kociba et al., 1971).
Ning et al. (1984) reported test results for laboratory animals
exposed to airborne Na-PCP. Both weanling rats (males) and rabbits
(males and females) were exposed to reagent grade Na-PCP at 21.4
mg/m3 or 3.1 mg/m3 for 4 h per day, 6 days per week, for 4 months.
Rabbits (6 pooled males and females) in the high-dose group showed
a statistically significant increase in serum-gamma-globulin but
not in alpha- or beta-globulin or serum-albumin. Lung weight
increased significantly in the high-dose group and liver weight
increased significantly in both dose groups compared with controls.
In rats, the lung, kidney, liver, and adrenal gland all increased
significantly in weight in the high-dose group compared with the
same organs in control animals. Blood-glucose levels in rats from
the group exposed to 21.4 mg/m3 remained higher than those in
controls, throughout the study.
These results are consistent with previous observations
reported by Demidenko (1969). Rats and rabbits exposed to 28.9 or
2.97 mg PCP/m3 for 4 h per day and 4 months were significantly
adversely affected at the high dose (anaemia, leukocytosis,
eosinophilia, hyperglycaemia, dystrophic processes in the liver).
At the low dose, only minor effects on liver function,
cholinesterase activity, and blood sugar were registered, which
returned to normal one month after completion of exposure.
Although the results of these 2 inhalation toxicity studies are
only preliminary, they give an indication that short-term
inhalation of Na-PCP or PCP at concentrations as low as about 3
mg/m3 can cause biochemical and gross pathological effects in
laboratory mammals. Assessing the data of Demidenko (1969), Kunde
& Böhme (1978) calculated from the 3 mg/m3 concentration a daily
dose of 0.3 mg/kg body weight per day for rats, assuming 100%
pulmonary uptake and absorption. This dose would indicate that
PCP is at least 10 times more toxic with inhalation exposure than
with oral exposure. This finding is corroborated by the results of
studies comparing acute inhalation and acute oral exposure
(section 8.1).
Other long-term studies on animals have been designed
specifically to evaluate the carcinogenic properties of PCP and are
reported in section 8.6.
8.4. Effects on Reproduction and Fetal Development
There is good agreement that PCP is a fetotoxic agent; however,
it does not appear to be teratogenic (Kozak et al., 1979; Ahlborg &
Thunberg, 1980; Fielder et al., 1982; NRCC, 1982). These
conclusions are based primarily on the studies of Schwetz et al.
(1974, 1978) on rats.
Technical grade PCP administered to pregnant female rats from
day 6 to day 15 of gestation did not have any effects on the mother
or fetus at 5 mg/kg body weight per day (Schwetz et al., 1974).
Fetal resorptions and delayed development of fetuses were observed
at 15 mg/kg body weight per day, and signs of maternal toxicity,
based on weight loss, were observed at 35 mg/kg body weight per
day. Reports of delayed ossification of the skull, supernumerary,
fused, or missing vertebrae and lumbar spurs are usually considered
indicative of delayed development rather than teratogenicity, and
are responsible for the differences of opinion between the early
position of US EPA, that PCP is teratogenic (Cirelli, 1978b), and
most other reviews. Purified PCP induced effects similar to those
of technical PCP; however, maternal toxicity and decreased fetal
weights occurred at 30 mg/kg body weight per day, and delayed fetal
development was observed at the 5 mg/kg body weight per day dose
level, which had previously been found to be the no-observed-
adverse-effect-level for technical grade PCP. More limited
fetotoxic effects were observed in rats exposed to PCP by Courtney
et al. (1976).
Na-PCP (> 98% pure) fed to female Wistar rats at 10, 30, or 60
mg/kg body weight during days 8 - 19 of gestation led to
statistically significant reductions in body weight in females,
decreased litter weights, and dramatic increases in fetal
resorption and fetal death in the 2 highest dose groups (Anon,
1981). No birth defects were observed in the control group or in
the group fed 10 mg/kg body weight; however, 3/31 pups examined
from females in the 30 mg/kg body weight group had major
malformations (hare lip, umbilican hernia, exocephalus) and 60%
had spine and rib malformations (supernumerary, fused, bifurcated,
or short ribs). In addition, retardation of ossification and
increased breadth of sagittal fissure were extensive in this group.
No pups were born to females in the 60 mg/kg body weight group.
The authors concluded that 10 mg/kg body weight was the no-
observed-adverse-effect level for teratogenicity, fetotoxicity, and
embryotoxicity in rats administered Na-PCP. Considering the
linearity of the dose-response curve and the fact that 10 mg/kg
body weight was the lowest dose administered, the no-observed-
adverse-effect level reported in this study is not substantially
different from that determined for PCP.
It is probable that the reduced fetotoxicity of technical PCP
relative to the purified material, if not artifactual, is a result
of the presence of microcontaminants (NRCC, 1982). Liver enzymes
that accelerate the rate of PCP metabolism are known to be
activated by phenobarbital, 3-methylcholanthrene, and 2,3,7,8-T4CDD
(Ahlborg & Thunberg, 1978). Other PCDD and PCDF microcontaminants
in technical PCP may also activate microsomes and reduce the amount
of fetal exposure to PCP, causing a concomitant decrease in
toxicity. Indeed, the induction of liver enzymes by technical PCP
has been demonstrated in the rat by Goldstein et al. (1977).
Purified PCP did not increase the activity of liver enzymes.
Technical grade PCP was contaminated with 8 - 1380 mg PCDDs/kg and
4 -1500 mg PCDFs/kg, while purified PCP contained less than 0.1 mg
of these contaminants per kg.
Larsen et al. (1975) reported single instances of dwarfism,
exencephaly, macrophthalmia, and taillessness in fetuses after
pregnant rats were fed single doses of 60 mg purified PCP per kg
body weight per day on days 8, 9, or 10 of gestation. However,
these findings were attributed to maternal hyperthermia, which is
known to cause teratogenic effects in rats (Edwards, 1968).
Hyperthermia is a common outcome of exposure to large, single doses
of chlorophenol.
Exon & Koller (1983a) investigated the effects of PCP on rats
exposed pre- and postnatally. Females were exposed to technical
PCP (95% pure) in the feed from 21 days of age, throughout breeding
(at 90 days), gestation, and up to weaning of the pups, 21 days
after parturition. Exposure doses were 0, 5, 50, and 500 mg/kg
body weight per day. The progeny were exposed to the same levels
of PCP in feed as their mothers for a total exposure period of 12
1/2 months. No significant decreases in mean litter size or
percentage of still-born pups were recorded in the groups given
PCP. There was a significant decrease in survival to weaning in
the group fed 5 mg/kg body weight but not in the 2 higher dose
groups.
A level of purified technical PCP of 30 mg/kg body weight per
day (Schwetz et al., 1978) as well as pure Na-PCP at 26 mg/kg body
weight per day (Kunde & Böhme, 1978) fed to male and female rats
for 62 days before mating, 15 days during mating, and to females
during gestation and lactation, caused reductions in the numbers of
offspring, neonatal body weight, neonatal survival, and growth of
weanlings. The no-observed-adverse-effect level was 3 mg/kg body
weight per day (Table 35). Male fertility did not appear to be
affected in this study.
8.5. Mutagenicity
Williams (1982) reviewed the literature and concluded that,
although there are deficiencies in the data, the Ames Salmonella
typhimurium test (Andersen et al., 1972), a sex-linked lethal test
with Drosophila melanogaster (Vogel & Chandler, 1974), and a host-
mediated assay (Schwetz et al., 1978) all indicate that PCP
probably does not cause point mutations. On the basis of negative
findings in an in vivo mammalian dominant lethal test (Buselmaier
et al., 1973), PCP does not seem to cause chromosomal aberrations.
However, an in vitro study to show primary damage using
Saccharomyces cerevisiae demonstrated an increase in mitotic gene
conversion (Fahrig, 1974; Fahrig et al., 1978). A mammalian spot-
test pointed to weak mutagenic activity (Fahrig et al., 1978). PCP
did not cause single-strand breaks in human fibroblast DNA, while
its metabolite tetrachlorohydroquinone did (Witte et al., 1985).
Lymphocytes taken from workers at a PCP factory showed a small, but
significantly higher, incidence of dicentric and acentric mitoses
(Bauchinger et al., 1982). Sodium pentachlorophenate (20 mmol)
reportedly increased the frequency of both auxotrophic and
morphological variations in the fungus Aspergillus niger strain 350
(Roy et al., 1981). However, because of shortcomings in these
studies, the information available is still insufficient to assess
the mutagenicity of PCP. The host-mediated assay with bacteria
(Schwetz et al., 1978) and the sex-linked recessive lethal assay
(Vogel & Chandler, 1974) have only been carried with single dose
levels. In the Ames test (Andersen et al., 1972), no metabolic
activation system was used. The increase in the incidence of
offspring with coat discoloration in the mouse spot test (Fahrig et
al., 1978) was not statistically significant. The mutagenicity
test of sodium pentachlorophenate on Aspergillus niger 350 (Roy et
al., 1981) did not include a control group.
8.6. Carcinogenicity
The carcinogenicity of chlorophenols in mammals is a
contentious issue. In 1979, the International Agency for Research
on Cancer reviewed the data available on the carcinogenic
properties of PCP (IARC, 1979a) and concluded that the information
was inadequate for a meaningful assessment of carcinogenicity.
Two carcinogenicity bioassays have been carried out using PCP.
Innes et al. (1969) exposed 2 hybrid strains of mice to roughly the
maximum tolerated dose of PCP for a total of 78 weeks, and did not
find any significant increases in tumour incidence in males or
females of either strain. Schwetz et al. (1978) report similar
findings in rats exposed to a maximum of 30 mg technical PCP/kg
body weight per day, for 24 months. Using these data, the
Carcinogenic Assessment Group of the US EPA concluded that PCP was
negative with respect to oncogenic effects (Williams, 1982).
PCP (technical and commercial grade) is currently being tested
by the National Toxicology Program of the US EPA (NRC, 1986),
however, data from this study were not available to the Task Group
for evaluation.
NOTE: The results of these studies have now been published
(US NTP (1989) Toxicology and carcinogenesis studies of two
pentachlorophenol technical-grade mixtures (CAS No. 87-86-5) in
B6C3F1 mice (feed studies), Research Triangle Park,
North Carolina, US National Toxicology Program, Technical Report
No. 349, p. 98). The conclusions were as follows:
"Under the conditions of these 2-year feed studies, there
was clear evidence of carcinogenic activity for male
B6C3F1 mice fed diets containing technical-grade
pentachlorophenol, as shown by increased incidences of adrenal
medullary and hepatocellular neoplasms. There was some
evidence of carcinogenic activity for female B6C3F1
mice exposed to technical-grade pentachlorophenol, as shown by
increased incidences of hemangiosarcomas and hepatocellular
neoplasms. There was clear evidence of carcinogenic
activity for male B6C3F1 mice exposed to
pentachlorophenol, EC-7, as shown by increased incidences of
adrenal medullary and hepatocellular neoplasms. There was
clear evidence of carcinogenic activity for female
B6C3F1 mice exposed to pentachlorophenol, EC-7, as
shown by increased incidences of adrenal medullary and
hepatocellular neoplasms and hemangiosarcomas."
Exon & Koller (1983a) investigated the potential for PCP to act
as a co-carcinogen by administering ethylnitrosourea (ENU) to
female rats exposed pre- and/or postnatally to 0, 5, 50, or 500 mg
PCP/kg body weight per day. ENU was administered as ethylurea in
drinking-water and nitrite in feed during days 14 - 21 of
gestation. The progeny were exposed to the same levels of PCP in
feed as their mothers for a total exposure period of 12.5 months.
High incidences of tumours in progeny exposed to PCP + ENU could
not be separated from those observed in progeny exposed to ENU
alone.
Tumour promotion studies on phenol and chlorophenol using
dimethylbenzanthracene (DMBA) as the initiator on mouse skin
indicated that phenol, 2-MCP, 2,4-DCP, and 2,4,5-T3CP were probable
tumour promoters, but that 2,4,6-T3CP and higher chlorophenols,
including PCP, were not (Boutwell & Bosch, 1959). None of the
chlorophenols tested were tumorigenic, when applied alone.
Carcinogenicity bioassays involving oral exposure have been
conducted on one other chlorophenol, 2,4,6-trichorophenol (NCI,
1979) and on a mixture of 2 hexachlorodibenzo- p-dioxin isomers
(NCI, 1980a). 2,4,6-T3CP caused a significant increase in cancer
in a variety of tissues in both male and mice and in male rats.
A mixture of two H6CDD isomers known to contaminate technical
PCP caused a significant increase in cancer in the livers of female
rats and mice (Table 36).
Thus, animal data indicate that, when PCP is ingested, it does
not cause cancer in mice or rats, and that, when applied to the
skin or administered orally, it does not act as a tumorigen or a
tumour promoter. Nevertheless, there is evidence that one other
chlorophenol is an animal carcinogen, the lower chlorinated phenols
are tumorigens, and that H6CDD found in PCP is carcinogenic, when
ingested by rodents. In addition, Arrhenius et al. (1977b)
suggested that PCP may be able to act as a co-carcinogen, on the
basis of its effects on liver microsomes.
8.7. Other Studies
The immunotoxic effects of PCP have been investigated in a
variety of animal species. Cows fed technical PCP have shown
thymic hypoplasia in one study (McConnell et al., 1980) but no
overt differences in immunological function in another (Forsell et
al., 1981). Mice exposed to technical grade PCP have shown reduced
humoral immunity and an in vitro impairment of T-cell cytolytic
activity (Kerkvliet et al., 1982a,b). Analytical grade PCP did not
have any effect. In the rat, a decrease in humoral immunity and an
increase in cell-mediated immunity was demonstrated following
exposure to technical PCP (97% pure) (Exon & Koller, 1983b).
Hillam & Greichus (1983) reported a suppression of total leukocyte
counts, gamma globulins, and IgG in young pigs exposed to technical
PCP (95% pure) at dose levels of 5 and 10 mg/kg body weight per day
for 30 days. Chickens ingesting 2400 µg/g feed of purified PCP
exhibited significantly reduced humoral responses to injections of
bovine serum-albumin and lymphoproliferative responses to the
mitogens concanavalin and phytohaemaglutinin and lower white blood
cell counts (Prescott et al., 1982). Thus, some immunotoxic effects
are observed when PCP (pure and technical) is administered to
experimental animals. In studies carried out with technical PCP,
it is likely that the non-phenolic contaminants such as PCDD are
responsible for most of the observed immunotoxic effects.
A neurotoxic effect of PCP (grade not specified) has been
reported by Walum & Peterson (1984) on the basis of an in vitro
assay with cultured mice neuroblastoma cells in which an increase
in cell detachments with exposure to PCP was observed. Also, a
transient alteration in brain tissue enzyme activity was found in
rats given 20 mg technical PCP per litre drinking-water over a
period of 3 - 18 weeks (Savolainen & Pekari, 1979). The
significance of these findings is unclear.
8.8. Contaminants Affecting Toxicity
The toxicological evaluation of pentachlorophenol is
complicated by the presence of several impurities in technical
grade formulations of this chemical. Some of these impurities are
extremely toxic in their own right. On the other hand, some
microcontaminants are capable of inducing liver microsomal
enzymes, and in so doing, affect the rates of metabolism and
excretion and the fetotoxicity of PCP (Ahlborg & Thunberg, 1978)
(section 8.4). Thus, meaningful assessments of toxicological
studies on the effects of pentachlorophenol are impossible without
an accurate knowledge of the type and extent of contamination of
the PCP under investigation.
Concern for the toxic effects of microcontaminants has focused
on the dioxins, because of the extreme toxicity of the intensely
studied congener 2,3,7,8-T4CDD. However, it is necessary to
emphasize that this congener has not been found frequently in PCP.
A brief summary of the toxic properties of the microcontaminants of
chlorophenols is provided in section 2.2. Extensive reviews of the
toxicology and residue levels of PCDDs and PCDFs are available in
Hutzinger et al. (1982), Jones (1981), NRCC (1981), Fielder et al.
(1982), Kociba & Schwetz (1982), Umweltbundesamt (1985), and in
several articles published in Boddington et al. (1985).
8.8.1. Octachlorodibenzodioxin (OCDD)
Only one congener exists of this fully substituted isomer. It
was not acutely toxic for rats, when administered orally at 1 mg/kg
body weight or acnegenic for rabbits, when applied to the ear as a
10% solution in chloroform (Fielder et al., 1982). Exposures of
approximately 1 mg/kg body weight per day for 3 weeks did not cause
any toxic signs. Livers were nominally enlarged, but appeared
normal under the light microscope. OCDD does not appear to be
mutagenic in the Ames test and has not undergone a carcinogenicity
bioassay. Studies of the effects of OCDD on reproduction and fetal
development indicate that it is not teratogenic at 500 µg/kg body
weight per day or fetotoxic at 100 µg/kg body weight per day, when
administered to females on days 6 - 15 of gestation.
8.8.2. Heptachlorodibenzodioxin (H7CDD)
Few data available on H7CDD. The LD50 value has not been
determined accurately for either the 1,2,3,4,6,7,8- or
1,2,3,4,6,7,9-isomers.
An in vitro assessment of the induction of aryl hydrocarbon
hydroxylase (AHH) in rat hepatoma cell cultures was used to
calculate the biological potency of both H7CDD isomers relative to
2,3,7,8-T4CDD. The relative potencies of 1,2,3,4,6,7,8-H7CDD and
1,2,3,4,6,7,9-H7CDD were 0.3 - 0.5% and 0.011 - 0.025% respectively
(Bradlaw et al., 1980).
Table 36. Summary of toxicology data for hexachlorodibenzo- p-dioxin (H6CDD)
---------------------------------------------------------------------------------------------------------
Species Toxic for: Vehiclea Isomerb Route Toxicity Observations Reference
(sex) (µg/kg body
body weight
per exposure
period)
---------------------------------------------------------------------------------------------------------
Acute toxicity
Lethality (LD50)
Rat females CO:acetone B/C mix oral 800 Observations in all NCI (1980a)
males 1800 studies included:
Mouse females CO:acetone B/C mix oral 500 weight loss, skin NCI (1980a)
males 750 eruptions, delayed
death; Tissues
females, CO A oral 825 affected: liver, McConnell
males CO B oral 1250 thymus, spleen, et al.
CO C oral 1440 kidney, testes (1978)
Guinea- females, CO A oral 73 McConnell
pig males CO B oral 70 - 100 et al.
CO C oral 60 - 100 (1978)
Short-term toxicity
(NOAEL)
Rat female, CO:acetone B/C mix oral < 2.5 thymic atrophy, splenic NCI (1980a)
male (per week) hypertrophy, and liver
lesions at 10 - 50 µg/kg
body weight per week
Mouse female, CO:acetone B/C mix oral 1.2 only liver damage at NCI (1980a)
male (per week) higher levels (10 - 50
µg/kg body weight
female, acetone dermal << 1.5 per week NCI (1980b)
male (per week)
---------------------------------------------------------------------------------------------------------
Table 36. (contd.)
---------------------------------------------------------------------------------------------------------
Species Toxic for: Vehiclea Isomerb Route Toxicity Observations Reference
(sex) (µg/kg body
body weight
per exposure
period)
---------------------------------------------------------------------------------------------------------
Fetal toxicity
(NOAEL)
Rat females, CO:acetone oral 0.1 (per fetal oedema at 1 µg/kg Schwetz
males day during body weight; resorptions et al.
gestation) at 10 µg/kg body (1973)
weight; teratogenic and
maternal toxicty at
100 µg/kg body weight
Long-term toxicity
Carcinogenicity
bioassay
Rat females CO:acetone B/C mix oral carcinogen Loss of body weight NCI (1980a)
> 1.5 (male and female);
(per week) hepatocellular
carcinomas and
neoplastic nodules
in females only
Mouse females CO:acetone B/C mix oral carcinogen hepatocellular carcin- NCI (1980a)
(incon- > 2.5 omas and adenomas in
clusive (per week) females only;
in males) incidence in males
not significant
incon- acetone B/C mix dermal inconclu- fibrosarcomas observed NCI (1980b)
clusive sive in females; incidence
not significant
---------------------------------------------------------------------------------------------------------
a CO = corn oil; CO:acetone = 9:1 mixture of corn oil and acetone.
b A = 1,2,3,4,7,8-H6CDD; B = 1,2,3,6,7,8-H6CDD; C = 1,2,3,7,8,9-H6CDD.
8.8.3. Hexachlorodibenzodioxin (H6CDD)
Commercial PCP and Na-PCP have been found to contain 4 out of
10 possible H6CDD isomers; however, the 1,2,3,6,8,9- and
1,2,3,6,7,8-isomers predominate (Fielder et al., 1982). Levels have
been in the 5 - 10 mg/kg range in recent commercial samples of PCP
and Na-PCP. The acute oral toxicity (LD50) of 1,2,3,6,7,8-H6CDD in
the mouse is 1250 µg/kg (Table 36). H6CDD is more toxic for female
rats and mice than for males. Signs of toxicity include weight
loss and deterioration of the skin. The onset of mortality is
often delayed for up to 3 weeks. Thymus, liver, spleen, kidney,
and testes are affected by H6CDD. This isomer is also known to be
acnegenic.
Oral exposure of rats to H6CDD isomers indicated that no-
observed-adverse-effect levels for the rat were < 2.5 µg/kg body
weight per week and, for the mouse, < 1.25 µg/kg body weight per
week (NCI, 1980a). Above these levels, weight loss and liver
damage were observed. Dermal exposure of mice to H6CDD also
resulted in liver damage and mortality, even at the lowest dose of
1.5 µg/kg body weight per week (NCI, 1980b). Thus, H6CDD is readily
absorbed through the skin and extremely toxic. A mixture of
1,2,3,6,7,8- and 1,2,3,7,8,9-H6CDD has been shown to be
carcinogenic for female mice and rats; however, males did not
develop hepatocellular carcinomas or adenomas in excess of the
control rate in the same study (NCI, 1980a). Carcinogenicity
assays using dermal exposures were inconclusive (NCI, 1980b).
On the basis of the results of exposure of pregnant rats to an
unidentified mixture of H6CDD isomers, this homologue is considered
fetotoxic and teratogenic (Schwetz et al., 1973). The no-observed-
adverse-effect-level was 0.1 µg/kg body weight. Cleft palate,
vertebrae with split or unfused centra, and split sternebrae were
observed in the fetuses of females fed 100 µg/kg body weight on
days 6 - 15 of gestation. Fetal resorptions were observed at
maternal doses of 10 µg/kg body weight and fetal subcutaneous
oedema at doses of > 1 µg/kg body weight.
8.8.4. Polychlorinated dibenzofurans (PCDFs)
Little is known of the toxicity of the PCDFs, despite their
relatively common occurrence in chlorophenol formulations at
concentrations of 1 - 500 mg/kg. Some have considerable acute oral
toxicity. For example, the LD50 of 2,3,7,8-T4CDF in guinea-pigs
and monkeys is 5 - 10 µg/kg body weight and 1000 µg/kg body weight,
respectively (Jones, 1981). Rappe et al. (1982) indicated that
1,2,3,7,8-P5CDF, 2,3,4,7,8P5CDF, and 2,3,4,6,7,8-H6CDF all had LD50
values in the 1 - 100 µg/kg body weight range for the most
sensitive species tested. As with PCDD, the toxicity of PCDF
isomers appears related to the extent of symmetrical positioning of
chlorine atoms in the 2, 3, 7, and 8 positions of the molecule.
2,3,7,8-T4CDF and 2,3,4,7,8-P5CDF appear to have the longest half-
lives of the PCDFs studied (Masuda & Kuroki, 1982). Furthermore,
these authors have suggested that PCDFs, especially the tetra- and
penta-isomers, may have been largely responsible for the signs of
Yusho disease reported in the Japanese who ingested rice oil
contaminated with PCBs, PCDF, and polychlorinated quarterphenyls
(PCQs). PCDFs have acnegenic properties, but may not be
porphyrinogenic.
8.8.5. Polychlorodiphenyl ethers (PCDPEs)
PCDPEs are common contaminants of chlorophenols and are found
almost exclusively in T4CP and PCP (Jones, 1981). Little is known
of their toxicity.
8.8.6. Other microcontaminants
Less common impurities of chlorophenols include polychlorinated
phenoxyphenols (PCPP) ("predioxins" or "isopredioxins"),
polychlorinated biphenyls (PCBs), and polychlorinated benzenes
(Jones, 1981). Extensive toxicology data exist on the effects of
PCBs (WHO, 1976) and some members of the chlorinated benzene group,
i.e., hexachlorobenzene (IARC, 1979b). Although these 2 groups of
chemicals are not acutely toxic, they can affect reproduction and
are considered carcinogenic. The levels found in technical
formulations of PCP are not likely to increase their toxicity or
carcinogenicity.
8.9. Mechanism of Toxicity
PCP is known to be cytotoxic for mammalian cells (Packham et
al., 1982). All chlorophenols, especially PCP, are uncouplers of
oxidative phosphorylation (Kozak et al., 1979). However, the
molecular basis for the uncoupling action is not clear (NRCC,
1981). PCP binds to mitochondrial protein and inhibits
mitochondrial ATPase activity. PCP may have 2 independent effects
on mitochondria; uncoupling oxidative phosphorylation and also
inhibiting mitochondrial ATPase (Stockdale & Selwyn, 1971a,b).
Thus, both the formation of ATP and the release of energy to the
cell from the breakdown of ATP to ADP are prevented. Electron
transport is not inhibited by PCP, though reactions dependent on
available high-energy bonds, such as oxidative and glycolytic
phosphorylation, are affected. Binding to enzymic protein has
been reported and may lead to the observed inhibition of other
cellular enzymes (Kozak et al., 1979). An increase in cellular
oxygen demand during the uncoupling of oxidative phosphorylation
has also been observed, which gives rise to the initial increase in
respiration rate reported for individuals poisoned by PCP
(Weinbach, 1957; Mitsuda et al., 1963; Wood et al., 1983).
9. EFFECTS ON MAN
There are no studies or case reports of the effects of pure or
purified PCP on human beings. Human exposure is nearly always to
technical grades of PCP or Na-PCP in a variety of formulations.
Reference to "PCP" in this section is to the technical grade.
9.1. Acute Toxicity - Poisoning Incidents
In man, the minimum lethal oral dose (LDLO) of PCP has been
estimated to be 29 mg/kg body weight (Ahlborg & Thunberg, 1980).
Kozak et al. (1979) report that this value depends on the ambient
temperature at the time of exposure, and the general health and
renal competence of the individual. PCP is approximately 5 times
more toxic than phenol (estimated oral LDLO is 140 mg/kg body
weight). The proportional lethality of these 2 chemicals (LDLO
phenol:LDLO pentachlorophenol) in man is almost identical to the
proportional lethality of the LD50 values for these substances in
rats.
Numerous accidental or suicidal poisonings with commercial
chlorinated phenols have been reported (Nomura, 1954; Menon, 1958;
Blair, 1961; Bergner et al., 1965; Mason et al., 1965; Armstrong et
al., 1969; Robson et al., 1969; Watanabe & Watanabe, 1970; Haley,
1977; Stevens & Richardson, 1979; Gjovik et al., 1981; Wood et al.,
1983), and nearly 60% of these acute exposures have resulted in
death. These cases together with the results of animal studies
provide a relatively clear picture of the signs and symptoms of
acute exposure to technical pentachlorophenol in man.
In contrast to the lower chlorinated phenols, PCP does not
cause convulsions. Ataxia, mental and physical fatigue, headaches,
dizziness, disorientation, anorexia, nausea, vomiting, dyspnoea,
hyperpyrexia, tachycardia, and a rise in metabolic rate are common
signs and symptoms of PCP poisoning. Most prominent are extreme
weakness, elevated body temperature, and profuse sweating. Death
is due to cardiac arrest and poison victims usually show a marked
rigor mortis (Truhaut et al., 1952a,b; Nomura, 1954; Mason et al.,
1965; Robson et al., 1969; Watanabe & Watanabe, 1970).
The gross pathology and histological lesions associated with
acute exposures to PCP are generally consistent between laboratory
animals and man. Oral exposures result in gastric and intestinal
inflammation; however, the severity can depend on the carrier
solvent and the presence of other chemicals (Menon, 1958; Stevens &
Richardson, 1979). Pulmonary oedema and congestion have been
reported after inhalation exposure, and occasionally oral exposure,
if aspiration of ingested PCP has occurred. Splenomegaly,
cardiomegaly, renal congestion, hepatomegaly, and hepatic
congestion are also frequently observed at autopsy.
Histologically, fatty degeneration, and necrosis in the
centrilobular region of the liver have been reported, together with
degenerative lesions in renal tubules (Gordon, 1956; Menon, 1958;
Blair, 1961; Bergner et al., 1965; Mason et al., 1965; Robson et
al., 1969).
It is generally agreed that the signs and symptoms of acute
toxicity observed in animals and human beings exposed to
chlorophenols result from the effects of the chlorophenol molecule
itself rather than the microcontaminants, with hyperthermia,
profuse sweating, and the rapid onset of morbidity and early death
associated with acute chlorophenol exposures. These signs are not
observed in animals exposed only to PCDD and PCDF; death is delayed
by up to 3 weeks in acute exposure studies with these
microcontaminants.
9.2. Effects of Short- and Long-Term Exposures
Most data on the effects of non-acute exposures to
chlorophenols in man come from occupational studies. The clinical
outcome of repeated exposure to PCP has been reviewed by Fielder et
al. (1982), Williams (1982), and Exon (1984). The high rate of
employee turnover and variation in the level and duration of
exposure make it difficult to distinguish between subacute, short-
and long-term exposures. For this reason, the following studies
concerning occupational PCP toxicity in man are not separated on
the basis of duration of exposure. Interpretation of these studies
is frequently confounded by factors such as age, alcohol
consumption, tobacco smoking, and other aspects of life style.
9.2.1. Occupational exposure
Clinical studies have identified a number of toxic effects of
short-term PCP exposure in man, some of which are also
characteristic of acute intoxication (section 9.1). Symptoms
include irritation of the skin, mucous membranes, and respiratory
tract, signs of chloracne, neurasthesia, depression, headaches,
porphyria cutanea tarda, and liver and kidney functional changes
(Fielder et al., 1982). These effects are discussed in greater
detail in the following sections. Among workers employed in
pressure-treating wood with PCP, insomnia and vertigo have also
been reported (Arsenault, 1976).
9.2.1.1 Skin and mucous membranes
Workers exposed to airborne concentrations of 1 mg PCP/m3 or
more have reported painful nasal irritation (Deichmann & Keplinger,
1981). Variations in the effect level are associated with the
historical exposure of the individual to inhaled PCP. Workers
accustomed to exposure may have a higher threshold for irritating
effects and may tolerate up to 2.4 mg PCP/m3 air.
As in the case of experimental animals (section 8), persons
exposed to large amounts of technical PCP develop chloracne.
Fielder et al. (1982) summarized published cases of chloracne in
workers at PCP-manufacturing sites in Czechoslovakia, the Federal
Republic of Germany, the United Kingdom, and the USSR. Kozak et
al. (1979) reported other cases in Japan and the USA. The use of
Na-PCP and Na-tetra-chlorophenol has also resulted in chloracne in
woodworkers (Behrbohm, 1959). Baxter (1984) reported chloracne and
minor disturbances of the lipid metabolism among 40 workers from a
PCP-manufacturing plant over a 3-year study period. However, the
author concluded that the abnormalities observed were due to the
PCDD contaminants and could not be attributed to the PCP
preparation.
A survey of sawmill workers in British Columbia, Canada,
carried out using self-administered questionnaires, indicated that
dermatological and respiratory symptoms were significantly higher
in a PCP/T4CP exposed group than in the control group (Sterling et
al., 1982). However, no reliable estimates of exposure were
provided.
A more detailed study carried out in the same geographical area
made use of personal monitors carried by individual workers to
determine exposure levels of PCP and T4CP (Embree et al., 1984).
Blood and urine samples were collected and analysed, and health and
employment histories were recorded by a trained interiewer. The
workers were divided into 3 groups: a high exposure group handling
wet-treated lumber; a medium exposure group with no manual contact
with treated lumber; and a control group with no exposure to PCP,
T4CP, or related chemicals. Exposure concentrations for PCP are
shown in Table 20. The authors reported a correlation between
exposure levels and serum- and urine-chlorophenol concentrations.
However, they were unable to substantiate the findings of Sterling
et al. (1982) of increased incidences of respiratory and
dermatological health problems in workers exposed to PCP/T4CP.
A study on 113 employees at a wood-treatment facility found
that workers were in good health overall, but with a greater than
expected prevalence of skin pustular eruptions (Flickinger &
Lawrence, 1982). Airborne exposures were less than 0.03 mg/m3.
Klemmer et al. (1980) reported the results of a 7-year study on
400 Hawaiians, many of whom had long-term, high-level exposure to
PCP. Concentrations of PCP in blood-serum far exceeded the 1.05
mg/litre reported in Arsenault's (1976) study; workers treating
wood in open-vats had a mean level of 3.78 mg PCP/litre, pressure-
tank workers 1.72 mg/litre, and farmers and controls 0.25 and 0.32,
mg/litre, repectively. After considering data on 189 individuals of
the total of 400, Klemmer et al. (1980) concluded ... "despite high
chronic exposures to PCP, individuals in the wood treatment group
of workers had not undergone any serious health effects from this
exposure. The only evidence of tangible health effects, part of
which could have been caused by exposures to chemicals other than
PCP, were the low-grade infections or inflammations of the skin and
subcutaneous tissue, of the protective membrane of the eye, and of
the mucous membrane of the upper respiratory tract. No specific
long-term effects could be elicited in the exposed group".
9.2.1.2 Liver and kidney
Indications of significant liver damage have not been found.
Elevations in circulating levels of some hepatic enzymes have been
reported; however, they are usually transitory and do not suggest
severe functional impairment (Kozak et al., 1979; Fielder et al.,
1982). These findings are consistent with those reported in
studies on rats with short-term exposure to technical PCP (section
8.2).
Kidney functional changes resulting in reductions in creatinine
clearance and resorption of phosphorus have been reported by Begley
et al. (1977). The spontaneous normalization in kidney function
during a 3-week non-exposure period indicated that this effect on
kidney is largely reversible.
Jirasek et al. (1974) reported the clinical signs exhibited by
workers who suffered intoxication during the manufacture of Na-
2,4,5-T3CP and Na-PCP. These workers displayed abnormal porphyrin
metabolism (increased uroporphyrin and delta-aminolevulinic acid
in urine, UV fluorescence of liver), and indications of
hepatotoxicity (liver enlargement, mild steatosis or fibrosis of
liver tissue, elevated levels/activities of bilirubin, serum-
glutamic-oxaloacetic transaminase and serum-glutamic-pyruvic
transaminase).
Mild dysfunction of the liver has been reported among Soviet
workers engaged in the production of Na-PCP (Vinogradova et al.,
1973) including, for example, a reduced ability to synthesize
protein.
Zober et al. (1981) reported a study on a small group of
woodworkers involved in the application of PCP. The average
concentration of PCP in the air at the time of the study was 2.4
µg/m3, the average exposure period for the cohort was 3 years and
the average levels in urine and serum were 46 µg PCP/g creatinine
and 1 µg/ml, respectively. Elevations in serum-aminotransferases
and alpha-glutamyl transpeptidase were observed; however,
confounding factors of sample size and alcohol consumption
prevented the formation of any conclusions concerning the effects
of PCP on liver function.
As part of a similar study (Embree et al., 1984) (section
9.2.1.1), Enarson et al. (1986) found that serum levels of
creatinine, bilirubin, glutamic oxaloacetic transaminase, and
alkaline phosphatase in sawmill workers exposed to a mixture of Na-
PCP and Na-tetrachloropenate did not differ from those measured in
the controls.
9.2.1.3 Blood and the haematopoietic system
Aplastic anaemia has been associated with PCP use (Roberts,
1981); however, sample sizes were small and exposures were not
quantified. Incidental references to haematological changes in
isolated workers in a German chemical plant manufacturing PCP and
HCB were made by Baader & Bauer (1951). Effects on the
haematopoetic systems of animals have been reported in studies with
PCDD contaminated PCP (Johnson et al., 1973; Knudsen et al., 1974;
McConnell et al., 1980) and 2,3,7,8-T4CDD (Allen & van Miller,
1978). Wood (1980) examined the relationship between anaemias
(with no established cause) in 128 Canadian woodworkers and
exposure to PCP. No significant differences between the
haematology values of exposed and unexposed workers were found.
Wood (1980) concluded that PCP exposure did not appear to have any
significant effects on the prevalence of anaemias in these
woodworkers.
In a companion report to that of Embree et al. (1984) (section
9.2.1.1), Enarson et al. (1986) found few exposure effects in
sawmill workers exposed to Na-PCP and Na-tetra-chlorophenate. Most
blood variables monitored were within normal ranges and did not
differ between exposed and unexposed workers. A significant
decrease in haematocrit and an increase in haematuria were reported
in workers handling treated lumber, but not in workers exposed
solely through inhalation.
Urinary-PCP values (2.2 mg/litre) reported by Shirakawa et al.
(1959) in primarily female workers at several rubber manufacturing
factories indicated that these workers were exposed to high levels
of Na-PCP (presumably technical grade). Increased blood sugar
levels, decreased blood pressure, and dermatoses were reported, but
no worker was reported to have missed work through the effects of
PCP exposure.
9.2.1.4 Nervous system
Investigation of clinical reports of neuropathy did not
indicate any overt significant signs of peripheral neuropathy in a
recent study on PCP workers (Triebig et al., 1981). Sensory nerve
conduction velocity was significantly reduced in exposed workers,
but was not correlated with urinary levels of PCP.
Skin, blood, and neurological disorders have been reported
among workers at a Na-PCP manufacturing factory in the USSR
(Vinogradova et al., 1973). The workers were exposed to air levels
of PCP and Na-PCP ranging between 0.03 and 1 mg/m3. Readings for
21% of the air samples ranged from 0.21 to 1 mg/m3, exceeding the
maximum permissible concentration in the USSR of 0.1 mg/m3.
Washings taken from clothing and exposed skin yielded PCP and Na-
PCP values of 21 - 212 mg/dm2 and 7.6 - 75 mg/dm2, respectively.
However, concentrations of hexachlorobenzene were also 2 - 3 times
higher than the maximum permissible concentration (0.9 mg/m3) set
in the USSR and may have had an influence on the disorders
reported.
9.2.1.5 Immunological system
McGovern (1982) suggested that man may suffer an immunotoxic
response to phenolic compounds, including chlorinated phenolic
compounds. Marked T-cell suppression has been observed in several
patients exposed to phenols. Zober et al. (1981) reported that
some woodworkers exposed to PCP displayed increased concentrations
of immunoglobulins, though this increase was not correlated with
exposure. Ning (1984) reported that workers exposed to PCP showed
significant decreases in IgG and IgA immunoglobulins. The results
of animal studies, while indicating that PCP is not strongly
immunotoxic, confirm that PCP exposure can lead to immunological
changes (section 8.7).
9.2.1.6 Reproduction
There are few published data on the effects on male or female
reproductive capacity of short- or long-term exposure to
chlorophenols. Schrag & Dixon (1985) classified PCP as "agents
with inconclusive effects" on male reproduction. Corddry (1981)
investigated pregnancy outcomes in women married to sawmill workers
in Canada. Analysis of data from 43 women, with a total of 100
pregnancies, did not reveal any significant differences in the
pregnancy outcomes of women living with "exposed" compared with
"unexposed" men. There was a slight trend towards more adverse
pregnancy outcomes in the exposed group, but this trend disappeared
when the alcohol consumption was considered as a confounding
factor. Male fertility was not studied.
9.2.1.7 Cytogenetic effects
There is no evidence to indicate that PCP or other
chlorophenols exert cytogenetic effects on human cells. A study of
circulating lymphocytes in a small group of workers in Idaho, USA,
indicated that individuals exposed to PCP had a slightly higher
rate of chromosome breakage than controls, but the increase was not
statistically significant (Wyllie et al., 1975). Bauchinger et al.
(1982) reported that lymphocytes from 22 workers in a PCP-
manufacturing factory had a significantly elevated number of
chromosomal aberrations (dicentrics and acentrics). These data are
not adequate for assessing the cytogenetic effects of PCP in man.
9.2.1.8 Carcinogenicity
Only 2 reports associating exposure to PCP specifically with
human cancer are available. Greene et al. (1978) suggested that
there was an association between exposure to wood treatment
chemicals (PCP) and the incidence of Hodgkin's disease, on the
basis of a family case history (2 of 4 siblings contracting the
disease were occupationally exposed to PCP) and a relative risk
(RR) of 4.2 (from death certificates, in the USA) for persons
employed in carpentry and lumbering. Bishop & Jones (1981)
reported 2 cases of non-Hodgkin's lymphoma in PCP workers in the
United Kingdom; both cases were associated with chloracne. These
data are not adequate for the identification of a positive and
statistically sound correlation between lymphomas and PCP.
However, there is some epidemiological evidence that exposure
of workers to mixtures of chlorophenols, but not specifically PCP,
increases their risk of developing soft-tissue sarcomas and
lymphomas. Considerable debate has ensued since the initial report
of chlorophenol-related cancer by Hardell (1977) and the subsequent
reports of Hardell and his co-workers in Sweden. Case control
studies of soft-tissue sarcoma patients in Sweden indicated a
relative risk (RR) of 6.6 for those "exposed" to chlorophenols
compared to those who did not appear to have been exposed (Hardell
& Sandström, 1979). Individuals exposed to 2,4,5-T had an RR of
5.8. A follow-up study in another area of Sweden involving 330
subjects tended to confirm the overall risk of soft-tissue sarcomas
in individuals exposed to phenoxyacetic acids and chlorophenols
(Erickson et al., 1981). The authors reported RR values of 6.8 for
all phenoxyacetic acid exposures and 3.3 for chlorophenol
exposures. Exposures to phenoxyacetic acids, assumed by the
authors to be free of PCDD and PCDF impurities resulted in an RR of
4.2. Hence, Erickson et al. (1981) concluded that impurities in
these chlorinated phenols and phenoxyacids were probably not the
sole cause of the elevated cancer rates reported, though they might
have played a role in this apparent carcinogenicity.
The validity of the assumption that 2,4-D is free of PCDD and
PCDF "impurities" is questionable inasmuch as 2,4-D has been found
to be contaminated, in one case, with H6CDD (IARC, 1977) and, in
another, with T4CDF (Norström et al., 1979). However, it is
reasonable to assume that the contamination of the phenoxy-acid
herbicides 2,4-D, MCPA, necoprop, and dichlorprop with PCDDs and
PCDFs is very low.
Hardell et al. (1981) have also applied their case-referent
technique to malignant lymphoma patients (both Hodgkin's disease
and non-Hodgkin's lymphomas) in Sweden. They reported RR values in
individuals exposed to phenoxy acids, chlorophenols, and other
organic solvents to be 4.8, 4.3, and 2.4, respectively. The RR
value for high-level exposure to chlorophenols was as high as 8.4.
A possible explanation for the lymphomas may rest with the
immunological effects (in animals) of the PCDD contaminant,
2,3,7,8-T4CDD. Some immunosuppressive chemicals have been shown to
cause an increase in histiocytic lymphomas in man (Hardell, 1979).
In response to criticisms that recall bias was a significant
factor in his studies, Hardell (1981a) applied his case-control
method to study colon cancer, a disease that correlates positively
with asbestos exposure, but not with chlorophenol exposure. His
findings indicated that recall and observer bias were negligible in
his earlier studies, since colon cancers correlated significantly
only with asbestos exposure, and not with phenoxy acids or
chlorophenols exposure. Hardell et al. (1982) also used their
technique to demonstrate an increased risk of nasal/nasopharyngeal
cancer (RR = 7) among workers exposed to chlorophenols.
Others have not found associations between cancer and human
exposure to chlorophenols. In contrast to Hardell et al. (1982),
Tola et al. (1980) did not find any relationship between nasal
cancer and chlorophenol exposure in Finnish workers. A case-
control study in New Zealand (Smith et al., 1984) did not reveal a
higher incidence of soft-tissue sarcoma in workers exposed to
chlorophenols. Gilbert et al. (1983) conducted a cohort study in
Hawaii with workers employed in the wood-treatment industry, in
which chromated copper-arsenate, tributyl tin oxide, and PCP were
used. They did not find any adverse health effects, but urinary-
PCP levels were higher in the exposed group.
In a recent Swedish study on the risks of soft-tissue sarcoma,
a cohort of 354 620 men employed in agriculture or forestry was
compared to a reference cohort of 1 725 845 men employed in other
industries during the period 1961-79 (Wiklund & Holm, 1986). A
relative risk of 0.9 (95% confidence interval 0.8 - 1) was found.
When the cohort was divided into 6 subgroups, based on assumed
exposure to phenoxy acid herbicides, no significant differences in
relative risks were found. Despite the increased use of phenoxy
acid herbicides in Sweden between 1947 and 1970, no time-related
increase in the relative risk of soft-tissue sarcomas was found.
The authors concluded that their study did not confirm the results
of Hardell (1981b). However, they pointed out that only a small
percentage of their total cohort of agricultural and forestry
workers in Sweden were possibly exposed to phenoxy acid herbicides
(15%) and chlorophenols (2%). Hence, a relative risk of 1.5
observed for sarcomas in these groups, as defined in their study,
would be equivalent to an actual 6-fold risk from exposure to these
compounds. Thus, it is unlikely that their study would have
detected a true increased risk from such exposures, if the risk
were less than 6-fold.
Pearce et al. (1986) studied 82 cases of non-Hodgkin's lymphoma
in New Zealand with 168 cancer controls and 228 general population
controls. They obtained statistically significant odds ratios (OR)
of 2.7 and 2, respectively, for workers in the pelt department of
meat works with potential exposure to 2,4,6-trichlorophenol and for
workers who carried out fencing with potential exposure to both
2,4,6-trichlorophenol and pentachlorophenol. Further examination
of the data revealed that: 2 of the 4 lymphoma cases who worked in
the pelt department were possibly not exposed to TCP; that a
significant proportion of the fencing workers also worked in the
meat works; and that no significant risk was found for exposure to
chlorophenols as a group. Pearce et al. (1986) concluded that the
excess risk observed in these 2 groups of workers might not be due
to chlorophenol exposure. In a second study, Pearce et al. (in
press) added other lymphoma cases to their previous study sample
and found similar relationships. They concluded that, though an
association with chlorophenol exposure was unlikely, it could not
be ruled out. They proposed that alternative hypotheses, such as
exposure to oncogenic zoonotic viruses should be considered to
explain their findings.
While there is some evidence that chlorophenols, and in
particular trichlorophenols, are associated with elevations in the
rates of certain cancers in exposed individuals, there is no clear-
cut dose-effect relationship. "Exposure" has been loosely defined
in most studies and no quantitative assessments have been
published. In addition, it has been suggested that, since other
environmental chemicals such as hexachlorobenzene,
pentachlorobenzene, and pentachloronitrobenzene, are metabolized to
PCP in animals and man, there is no necessary relationship between
PCP concentrations in body fluids and exposure to PCP (Renner &
Mücke, 1986). Other factors that could have a bearing on the
conflicting reports of chlorophenol exposure and cancer incidence
include differences in study methods and the diagnosis of soft-
tissue sarcoma cases, and inadequacies in death-certificate data.
The results of epidemiological studies, currently underway in
several countries, could confirm or refute the association between
chlorophenol exposure and human cancer (Fingerhut et al., 1984).
9.2.1.9 Other systems
It is not unusual to find few or no signs of toxicity in
workers with long-term exposure to low levels of PCP or Na-PCP.
Arsenault (1976) reported a prospective clinical evaluation of 21
PCP workers involved in the pressure-treatment of wood, who had
been exposed for an average of 9 years and had elevated blood-serum
levels of PCP (on average, 1.05 mg/litre, versus 0.1 mg/litre in
controls). The only significant clinical findings in the pressure-
treatment workers were vertigo and insomnia. Arsenault (1976) also
provides information obtained from the health records of 1330
workers in a large wood-processing company. From 1961 to 1971,
only 26 cases of health problems related to PCP use and exposure
were identified; however, it is probable that this is an
underestimate because of under-reporting.
Similarly, in a cohort study comparing 88 wood-treatment
workers with 61 controls (Gilbert et al., 1983), no significant
effects of exposure (by history or physical examination) to wood
preservatives, including PCP, were reported on: skin or mucous
membranes of the eyes or upper respiratory tract; mental status;
cardiovascular, gastrointestinal, genitourinary, or neuromuscular
systems; or reproduction. In the accompanying historical
perspective study, calculations of age-specific deaths rates from
all causes for 125 workers, over 21 years, showed that observed
rates were similar to, or lower than, those expected.
9.2.2. General population exposure
References to non-occupational exposure to chlorophenols, for
example from wood in homes, confirm that pulmonary, and, to a
lesser extent, dermal exposure to PCP can produce symptoms of
poisoning similar to those documented in occupational settings.
These studies (section 5.2) may be of significance in as much as
they identify new sources of exposure; however, they add little to
the toxicology data base for PCP. Concentrations of PCP in the
indoor air of homes and in the urine and serum of their residents
are elevated relative to those in the general population (Table
21). The limited effects of this exposure are considered briefly
here.
In cases where individuals display symptoms of PCP
intoxication, usually as a result of the application of PCP in the
interior of houses, typical acute symptoms are observed, but other
parameters (haematological, biochemical) may be normal. Sangster
et al. (1982) outlined case histories of 3 families in PCP-treated
houses who reported experiencing one or more of the following signs
or symptoms: generalized itching or burning dermatosis, nausea,
vomiting, decreased appetite, headache, dizziness, and fatigue.
Haematological, urinary, and biochemical parameters were unaffected
by exposure. Similarly, a young girl poisoned by bathwater stored
in a PCP-contaminated tank displayed fever, intermittent delirium,
rigors, acidosis, and elevated urine levels of ketones and amino
acids, but her respiratory rate and other clinical symptoms were
normal (Chapman & Robson, 1965). However, longer-term exposure may
have more profound effects. Brandt et al. (1977) reported that
exposure to PCP for several years in the air of a treated wooden
house resulted in liver damage and elevated activities of several
liver enzymes in a German woman (Ahlborg & Thunberg, 1980).
A Sacramento woman lost weight, and complained of weakness and
tightness in the chest after the interior of her house was treated
with PCP (Anon, 1970).
Krause & Englert (1980) examined several medical and laboratory
parameters in 250 persons with elevated PCP exposure (section
5.3). No clear relationship could be found between elevated
concentrations of PCP in the urine and biochemical parameters
related to the liver, kidney, and blood. However, significantly
more complaints of headache, fatigue, tonsillitis, hair loss, and
bronchitis were reported in persons with PCP exposure. Because the
signs and symptoms usually reported in connection with indoor PCP
exposure are relatively non-specific, thye cannot be definitively
ascribed to PCP. However, the observation that many symptoms
disappeared when exposure was reduced (by improving ventilation,
sealing wood surfaces, or leaving the premises) is indicative that
PCP or the substances included in the formulated product might well
be the causative agents. The persistence of some biochemical and
dermatological signs, similar to those reported in the work-place,
is a further indication that PCP may induce subacute effects in
these exposed persons.
In general, however, no adverse effects can be ascribed to the
low ambient concentrations of PCP resulting from the diffuse
sources to which most people are exposed.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of Human Health Risks
In this subsection, PCP and Na-PCP are referred to as PCP.
10.1.1. Occupational exposure
10.1.1.1 Exposure levels and routes
Occupational exposure to technical PCP mainly occurs through
inhalation and dermal contact. Virtually all workers exposed to
airborne concentrations take up PCP through the lungs and skin. In
addition, workers handling treated lumber or maintaining PCP-
contaminated equipment would be exposed dermally to PCP in
solution, and may take up from one-half (based on urinary-PCP
concentrations) to two-thirds (using serum levels) of their total
PCP burden through the skin.
The actual concentrations to which workers have been exposed
are seldom measured but, where they have been monitored, they are
predictably high. Airborne levels at PCP-production and wood-
preservation facilities have ranged from several µg/m3 to more than
500 µg/m3 in some work areas. The outer layer of treated wood can
contain up to several hundred mg/kg, though levels are usually less
than 100 mg/kg.
These exposures result in concentrations of PCP in the serum
and urine that are 1 - 2 orders of magnitude higher than those in
the general population without known exposure. Mean/median urinary-
PCP concentrations of approximately 1 mg/litre are typical for
workers in contact with PCP, compared with urinary concentrations
of approximately 0.01 mg/litre for the general population (section
5.4).
Automated processes and the use of closed systems have greatly
reduced worker exposure in large-scale manufacturing and modern
wood-treatment factories and sawmills. Other improvements in
industrial hygiene can significantly reduce exposure, as measured
by lower urinary-PCP concentrations.
10.1.1.2 Toxic effects
Past use of PCP has affected workers producing or using this
chemical. Chloracne, skin irritation and rashes, respiratory
disorders, neurological changes, headaches, nausea, weakness,
irritability, and drowsiness have been documented in exposed
workers. Work-place exposures are to technical PCP, which usually
contains mg/kg quantities of microcontaminants, particularly H6CDD.
Subacute effects such as chloracne and potential subchronic and
chronic effects such as hepatotoxicity, fetotoxicity, and
immunotoxicity (as reported in animal studies) are probably mainly
caused by microcontaminants. However, the PCP molecule itself
appears to play a role in the pathology of the last 3 effects and
is likely to be wholly responsible for the reports of skin and
mucous membrane irritiation, hyperpyrexia and, in severe cases,
coma and death. The toxicity of pure or purified PCP has not been
evaluated for human beings, because human exposure has usually been
to technical PCP.
Investigations of biochemical changes in woodworkers with long-
term exposure to PCP have failed to detect consistently significant
effects on major organs, nerves, blood, reproduction, or the
immune system. However, the statistical power of these studies has
been limited as a result of the small sample sizes used. Overall,
the body of research suggests that long-term exposure to levels of
PCP encountered in the work-place is likely to cause borderline
effects on some organ systems and biochemical processes.
Some epidemiological studies from Sweden and the USA have
revealed an association between exposure to mixtures of
chlorophenols, especially 2,4,5-T3CP, and the incidences of soft-
tissue sarcomas, lymphomas, and nasal and nasopharyngeal cancers.
Other studies have failed to detect such a relationship. None of
these studies has managed to address the effects of exposure to PCP
itself.
Animal studies designed to assess the carcinogenicity of PCP
and reported to date have been negative. Carcinogenicity bioassays
with one other chlorophenol (2,4,6-T3CP) and a mixture of two H6CDD
congeners found in PCP have been positive. Hence, the carcinogenic
effects of long-term exposure of animals to technical PCP are not
clear.
10.1.1.3 Risk evaluation
It is clear that the levels of PCP found in work-places have
adversely affected some aspects of the health of exposed workers.
Potentially the most deleterious effect of technical PCP is on the
fetus, and pregnant women should avoid exposure, whenever possible.
There is limited evidence that PCP may cause hepatotoxicity,
neurological disorders, and effects on the immune system. No
convincing data for or against a carcinogenic link exists.
Note: Since the publication of this monograph in 1987, however, the
results of adequate carcinogenicity studies with commercial-grade
pentachlorophenol have been published. The conclusions of these studies
are indicated in the addendum to 8.6 Carcinogenicity.
The National Academy of Sciences (1977) calculated an
acceptable daily intake (ADI) for PCP of 3 µg/kg body weight per
day. This ADI is based on data from a feeding study on rats and a
1000-fold safety factor. The results of long-term studies indicate
that the no-observed-adverse-effect level for rats is below 3 mg/kg
body weight per day (section 8.2). A recent human study has shown
that the steady-state body burden is 10 - 20 times higher than the
value extrapolated from rat pharmacokinetic data, suggesting that
caution should be applied when extrapolating directly from the rat
model to man. Furthermore, the US ADI was not based on an
inhalation study, and does not account for the possibly greater
toxicity of PCP via inhalation, as indicated by animal studies
(sections 8.1 and 8.3). Hence, the safety factor of 1000 used to
derive this ADI value is by no means too conservative. The intake
for a 60-kg adult exposed to concentrations of PCP at the ADI level
would be 180 µg/person per day.
A rough estimate of occupational exposure alone can be
calculated, assuming a moderate breathing rate of 1.8 m3/h for a
60-kg worker, 100% uptake of all inhaled PCP (which takes some
account of the often significant dermal uptake), and an 8-h working
shift per day, 5 days per week. Hence, an exposure to 500 µg
PCP/m3 per shift (section 5.2) would result in an average daily PCP
intake of approximately 5000 µg/person per day, averaged over the
entire week. Under these circumstances, the ADI level proposed by
the National Academy of Sciences is significantly exceeded, even
when consideration is given to the effects of intermittant
exposures during the working week and the high health status
assumed for workers.
There is a clear need for a reduction in occupational exposure
to PCP. Emphasis must be placed on reducing airborne
concentrations at production and wood-treatment facilities, as well
as dermal contact with solutions containing PCP. In addition,
reductions in the concentrations of microcontaminants in technical
PCP, particularly PCDDs and PCDFs, would reduce the potential for
expression of several effects and would better protect the health
of workers in these industries.
10.1.2. Non-occupational exposure
10.1.2.1 Exposure levels and routes
Domestic use of products containing technical PCP, especially
the indoor application of wood preservatives and paints based on
PCP, has led to elevated concentrations of PCP in indoor air.
Indoor exposures have been well documented in houses constructed
with PCP-treated wood, or in which interior wood panels or boards
have been treated with PCP. PCP concentrations in indoor air can
be expected to reach 30 µg/m3 during the first month after
treatment. Considerably higher levels, up to 160 µg/m3, have been
reported in houses with concomitant poor indoor ventilation. Even
higher concentrations can be encountered immediately after do-it-
yourself applications of PCP-containing wood preservatives.
In the long term, values of between 1 and 10 µg/m3 are typical,
though higher levels, up to 25 µg/m3, have been found in rooms
treated one to several years earlier. Indoor air concentrations
are influenced by a variety of factors, e.g., intensity of
treatment, solvents and additives involved, species of wood
treated, environmental conditions, and time elapsed since
treatment.
In many cases, levels of PCP in the serum and urine of people
exposed in the home overlap those for occupationally exposed
persons; but, on average, urine-PCP levels are approximately 0.04
mg/litre for non-occupationally exposed persons.
In the long term, exposure to PCP in treated buildings
continuously decreases, because of the high volatility of PCP.
Because of their lower vapour pressure, the volatilization of
PCDDs and PCDFs from the wood surface is much slower than that of
PCP. Hence, these microcontaminants are emitted at a low rate, but
over a longer period of time. Long-term exposure to these
lipophilic contaminants is likely to lead to accumulation of PCDDs
and PCDFs in fatty body tissues.
As a result of regulations restricting the use of PCP, and also
changing use patterns, indoor exposure to PCP is probably declining
in most developed countries.
10.1.2.2 Risk evaluation
Assuming a daily respiratory volume of 20 m3/adult and 100%
uptake of all inhaled PCP (a worst case that takes some account of
dermal uptake), the exposure of persons living in PCP-treated
buildings, shortly after treatment, or, in some cases, after a long
period of time, could be expected to range between 600 and 3200
µg/person per day. Long-term exposure to concentrations of 1 - 25
µg PCP/m3 could result in a daily PCP intake of 20 500 µg/person
per day. The median value of 5 µg/m3 reported from a survey of 104
homes (section 5.3) corresponds to a daily PCP uptake of 100 µg/
person per day. Other potential sources of exposure to PCP
including food, drinking-water, and consumer products contribute
further to PCP uptake (section 10.1.3.1).
The indoor air data suggest that, at least during the first
weeks following indoor treatment, and occasionally for quite
prolonged periods of time, the ADI level of 180 µg/person per day
is significantly exceeded. Under these circumstances, there is a
potential health risk. This conclusion is supported, in part, by
reports of signs and symptoms similar to those in persons
occupationally exposed to PCP (dermatosis, nausea, headache,
dizziness, fatigue). These signs and symptoms are most likely
associated with the effects of the PCP molecule and, in some cases,
the solvents associated with the wood treatment chemicals used.
The long-term significance of exposure to low levels of PCDDs and
PCDFs and their accumulation in human tissues is not entirely
clear; however, at least 2 isomeric groups of the PCDDs family are
carcinogenic for animals. Animal data indicate that low
concentrations of PCP in biological tissues or body fluids do not
signify an absence of biologically active PCDDs and PCDFs.
It is worth noting that exposure in the home is frequently for
longer periods of time than exposures in the work-place and can
affect subpopulations potentially at greater risk than workers, for
example, children, the elderly, pregnant women, or those with an
existing adverse health condition.
10.1.3. General population exposure
10.1.3.1 Exposure levels and routes
Exposure of the general population to low levels of PCP is
common. PCP has been found in air, food, water, and other consumer
products. Biotransformation of some chlorinated hydrocarbons
(e.g., lindane, hexachlorobenzene) to PCP also contribute to the
human body burden.
The ambient air in urban areas typically contains several
ng/m3, while concentrations in less developed areas are roughly an
order of magnitude lower (section 5.1.1).
Drinking-water concentrations of PCP rarely exceed several
µg/litre, even in highly industrialized regions, and most are less
than 1 µg/litre (section 5.1.5).
Fruits, vegetables, and other produce usually contain much less
than 10 µg/kg, but may on occasion exceed this level. Most meats
contain similar concentrations of PCP (< 10 µg/kg) but, a few
samples, particularly liver, can contain over 100 µg/kg. Fish
skeletal muscle typically contains PCP levels of 4 µg/kg or less.
Overall estimates of PCP intake from all foods, based on total diet
samples in the USA and the Federal Republic of Germany, are
remarkably similar, i.e., up to 6 µg/person per day (section
5.1.5).
PCP is also present in a wide variety of consumer products,
including veterinary supplies, disinfectants, photographic
solutions, fabrics, home-care products, and pharmaceutical
products. No calculated estimates of the contribution made by
consumer products to overall exposure to PCP are available.
10.1.3.2 Risk evaluation
On the basis of the PCP levels in the various compartments,
the overall exposure of an average person without known specific
exposure can be estimated to be approximately 6 µg/person per day
from food, 2 µg/person per day from drinking-water, and 2 µg/person
per day from the ambient air. Thus, the total exposure of the
general population could be approximately 10 µg/person per day
(exclusive of exposure to consumer products), which is far below
the intake based on the ADI proposed by the US National Academy of
Science of 180 µg/person per day. On the basis of available data,
this exposure is not likely to constitute a health hazard.
However, the diffuse contamination of the environment with
technical PCP must be considered as an important source of
environmental PCDDs and PCDFs.
10.2. Evaluation of Effects on the Environment
The widespread use of technical PCP and its physical and
chemical properties (water solubility, n-octanol/water partition
coefficient, volatility) lead to ubiquitous contamination of air,
soil, water, sediments, and environmental organisms.
Depending on the soil type, PCP can be very mobile, potentially
leading to contamination of groundwater and hence, of drinking-
water. Because applications in agriculture have been reduced, soil
contamination will, for the most part, be confined to treatment
areas.
Photodecomposition and biodegradation processes may not be
adequate to eliminate PCP from the different compartments.
Unfavourable temperature, pH, and other environmental conditions
may retard degradation of PCP allowing it to persist in the
environment. Biological decomposition may also be limited in
waste-treatment factories resulting in high concentrations in the
final effluents. PCP has also been used in aquatic environments as
a molluscicide and an algicide.
PCP concentrations in surface waters are usually in the range
of 0.1 - 1 µg/litre, though much higher levels can be found near
point sources or after accidental spills (section 5.1.2).
PCP is highly toxic for aquatic organisms. Apart from very
sensitive or resistant species, there is apparently no difference
in the sensitivity to PCP of the different taxonomic groups
(section 7.2). Invertebrates (annelids, molluscs, crustaceans) and
fish are adversely affected by PCP concentrations below 1 mg/litre
in acute toxicity tests. Sublethal concentrations are in the low
µg/litre range.
As little as 1 µg PCP/litre can have adverse effects on very
sensitive algal species. Moreover, low concentrations (µg/litre)
may lead to substantial alterations in community structures, as
seen in model ecosystem studies.
10.3. Conclusions
In this subsection, PCP and Na-PCP are referred to as PCP.
1. Human exposure to PCP is usually from technical products that
contain several toxic microcontaminants, including PCDDs and PCDFs.
2. The acute health effects of exposure to high concentrations of
technical PCP are generally the result of the biological action of
the PCP molecule itself. Sub-chronic effects and the effects of
long-term exposure to technical PCP are most probably largely
related to the biological action of the PCDDs and PCDFs.
3. A dose-effect relationship for the acute or chronic toxicity of
technical PCP for human beings cannot be derived from available
data. Derivation of this relationship is confounded by variations
in individual susceptibility, social and environmental influences,
concomitant exposure to other chemical substances, a lack of
accurate exposure estimates, and inadequate toxicity data.
4. Occupational exposure to technical PCP can lead to adverse
health effects.
5. Non-occupationally exposed persons (using products containing
technical PCP and/or those living in buildings treated with wood
preservatives or paints containing PCP) can be exposed to
concentrations of PCP in air that can have adverse health effects.
6. The exposure of the general population to diffuse sources of
PCP (via food, drinking-water, ambient air, consumer products,
chlorinated compounds that can be metabolized to PCP) is very low
and, on the basis of available data, it is not likely to constitute
a health hazard.
7. Epidemiological investigations and animal studies, conducted to
date, are insufficient for an evaluation of the carcinogenicity of
technical PCP. Uncertainties also exist over the genotoxic and
fetotoxic effects of technical PCP.
8. PCP is rather persistent, quite mobile, and found in all
environmental compartments. At the higher concentrations found in
the surface water near point sources or discharges (mg/litre),
aquatic life is adversely affected. Ambient concentrations of PCP
commonly found in surface waters (0.1 -1 µg/litre) may adversely
affect very sensitive organisms and may lead to alterations in the
ecosystem.
9. Use of technical PCP and its improper disposal (landfill and
low-temperature combustion) can contribute significantly to the
contamination of the environment with PCP, PCDDs, and PCDFs.
11. RECOMMENDATIONS
In this section, PCP and Na-PCP are referred to as PCP.
11.1. Environmental Contamination and Human Exposure
(a) Concentrations of microcontaminants in technical PCP,
especially PCDDs and PCDFs, must be reduced by improving the
quality in production processes.
(b) There is a need for specification of a technical PCP.
(c) Disposal of technical PCP and associated waste should
preferably involve high-temperature combustion or, where this
is not possible, the use of secure land-fill sites.
(d) In order to reduce contamination of surface waters and the
hazards for the aquatic ecosystem, manufacturers and users of
technical PCP should prevent releases into the environment.
(e) Protective measures should be provided for non-target aquatic
organisms in cases where PCP is used as molluscicide or
algicide.
(f) Occupational exposure to technical PCP must be reduced to a
minimum. Reduction in exposure can be achieved by:
- explicit product labelling;
- employee instruction on product handling;
- lowering airborne concentrations; and
- use of effective protective equipment.
(g) Industries handling technical PCP should ensure adequate
routine monitoring and health surveillance of all potentially
exposed employees.
(h) The indoor application of PCP-based wood preservatives and wood
stains and the use of PCP-treated wood products in the interior
of buildings should cease.
(i) The availability and use of consumer products containing PCP
should be reduced and controlled.
(k) The following commercial uses of PCP-based products should be
eliminated, in order to reduce contamination of food and the
environment:
- application as wood preservatives on wooden food
containers, horticultural lumber, wood and tools in
mushroom houses, and above-ground interior wood of
farm buildings;
- application during the curing of hides;
- application as a herbicide or soil sterilant;
- application as a slimicide in wood pulp and paper
operations; and
- application as a molluscicide in surface water if
another control chemical or measure is available that
is less toxic for man and the aquatic ecosystem.
11.2. Future Research
11.2.1. Human exposure and effects
(a) Reliable estimates of human absorption of airborne PCP via the
lung and skin are required.
(b) The importance of the biotransformation of hexachlorobenzene
and related compounds as contributors to human body burdens of
PCP needs to be quantified.
(c) It is necessary to determine the intake and accumulation by
human beings of the lipophilic microcontaminants (especially
the PCDDs and PCDFs) resulting from exposure to technical PCP.
(d) Development of reliable estimates of biochemical and
reproductive no-observed-adverse-effect levels is desirable.
(e) Studies on persons occupationally exposed to technical PCP
should be conducted using a large enough cohort or sufficient
numbers of cases to provide the statistical power necessary to
determine the relationships between exposure to PCP and
morbidity, mortality in general, and cancer. Such studies
should include quantitative estimates of concentrations and
duration of exposure to PCP, wherever possible.
11.2.2. Effects on experimental animals and in vitro test systems
(a) New data on the carcinogenicity of technical and pure PCP in
both sexes of 2 mammalian species are required.
(b) There is a need for a long-term inhalation study on the effects
of both technical and pure PCP.
(c) Studies should be undertaken to clearly determine the
teratogenic effects of pure and technical PCP. The potential
effects of PCP induced maternal hyperthermia on embryological
development and fetal growth warrant investigation.
(d) More research on the genotoxic and mutagenic activity of pure
and technical PCP is required.
11.2.3. Effects on the ecosystem
(a) Studies are needed to clarify the fate of sediment-bound PCP
and its effects on the environment.
(b) Studies of the effects of long-term, low-level exposure on
fresh-water aquatic communities are required to establish no-
observed-adverse-effect levels.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
"The WHO Recommended Classification of Pesticides by Hazard"
(WHO, 1984) distinguishes between the four hazard classes Ia, Ib,
II, and III, based on the toxicity of technical products. In this
report, PCP is classified in class Ib, being highly hazardous.
The WHO manual "Prevention, Diagnosis and Treatment of
Insecticide Poisoning" (Plestina, 1981) provides practical advice
that generally applies to nitro- and chlorophenols.
In the Guidelines for Drinking-Water Quality (WHO, 1985), a
guideline value of 10 µg/litre is recommended for PCP.
No evaluation of the carcinogenicity of PCP was made by the
International Agency for Research on Cancer (IARC, 1979a), because
the available data on the carcinogenic and mutagenic effects of PCP
were considered inadequate for a sound evaluation.
Regulatory standards established by national bodies in
different countries and the EEC are summarized in the data profile
of the International Register of Potentially Toxic Chemicals
(IRPTC, 1983).
IRPTC (1984), in its series "Scientific reviews of Soviet
literature on toxicity and hazard of chemicals", issued a review on
pentachlorophenol.
REFERENCES
ABRAHAMSSON, K. & XIE, T.M. (1983) Direct determination of
trace amounts of chlorophenols in fresh water, waste water and
sea water. J. Chromatogr., 279: 199-208.
ACGIH (1980) Documentation of the threshold limit values,
4th ed., Cincinnati, Ohio, American Conference of Governmental
Industrial Hygienists Inc, p. 323.
ADELMANN, I.R. & SMITH, L.L. (1976) Standard test fish
development. I. Fathead minnows (Pimephales promelas) and
goldfish (Carassius auratus) as standard fish in bioassays and
their reaction to potential reference toxicants, Duluth,
Minnesota, US Environmental Protection Agency, 88 pp (Ecology
Research Series No. EPA 600/3-76-061A).
ADELMANN, I.R., SMITH, L.L., Jr, & SIESENNOP, G.D. (1976)
Acute toxicity of sodium chloride, pentachlorophenol, guthion,
and hexavalent chromium to fathead minnow (Pimephales
promelas) and goldfish (Carassius auratus). J. Fish. Res.
Board Can., 33: 203-208.
ADEMA, D.M.M. & VINK, G.J. (1981) A comparative study of
1,1,2-trichloroethane, dieldrin, pentachlorophenol, and
3,4-dichloroaniline for marine and fresh water organisms.
Chemosphere, 10: 533-554.
AHLBORG, U.G. & LARSSON, K. (1978) Metabolism of tetra-
chlorophenols in the rat. Arch. Toxicol., 40: 63-74.
AHLBORG, U.G. & THUNBERG, T. (1978) Effects of 2,3,7,8-
tetrachlorodibenzo- p-dioxin in the in vivo and in vitro
dechlorination of pentachlorophenol. Arch. Toxicol., 40: 55-61.
AHLBORG, U.G. & THUNBERG, T. M. (1980) Chlorinated phenols:
occurrence, toxicity, metabolism, and environmental impact.
CRC Crit. Rev. Toxicol., 7: 1-35.
AHLBORG, U.G., LINDGREN, J.-E., & MERCIER, M. (1974)
Metabolism of pentachlorophenol. Arch. Toxicol., 32: 271-281.
AHLBORG, U.G., LARSSON, K., & THUNBERG, T. (1978) Metabolism
of pentachlorophenol in vivo and in vitro. Arch. Toxicol., 40:
45-53.
AHLBORG, U.G., VICTORIN, K., CAMNER, P., NORDBERG, G., &
WAERN, F. (1986) [Health effects of waste incineration.] In:
[Energy from waste,] Sweden, National Board for Energy,
Appendix 4 (Report No. 1986:6) (in Swedish).
AHLING, B. & JOHANSSON, L. (1977) Combustion experiments
using pentachlorophenol on a pilot scale and full-scale.
Chemosphere, 6: 425-436.
ALLEN, J.R. & VAN MILLER, J.P. (1978) Health implications of
2,3,7,8-tetrachlorodibenzo- p-dioxin exposure in primates. In:
Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
371-379.
ANDERSEN, K.J., LEIGHTY, E.G., & TAKAHASHI, M.T. (1972)
Evaluation of herbicides for possible mutagenic properties. J.
agric food Chem., 20: 649-656.
ANDO, M., HIRANO, S., & ITOH, Y. (1985) Transfer of hexa-
chlorobenzene (HCB) from mother to new-born baby through
placenta and milk. Arch. Toxicol., 56: 195-200.
ANGERER, C. (1984) [Production and use of pentachlorophenol
in the Federal Republic of Germany.] In: [Pentachlorphenol -
specimen case,] Berlin, Umweltbundesamt, 76 pp (UBA F & E -
Bericht No. 106 04 007) (in German).
ANON (1970) Pentachlorophenol poisoning in the home.
Californian Health, 27: 13.
ANON (1981) [Studies of the teratogenicity of Na-PCP in the
rat.] In: Shanghai Medical College, ed. [Studies on maximal
allowable concentration of hazardous substances in surface
water.] Beijing, China, People's Medical Publication, pp.
215-224 (in Chinese).
ANON (1983a) Cryptogil Na. Technical documentation,
Courbevoie, France, Rhône-Poulenc Specialités chimiques, 3 pp.
ANON (1983b) Cryptogil Ol. Technical documentation,
Courbevoie, France, Rhone-Poulenc Specialités chimiques.
APAJALAHTI, J.H.A. & SALKINOJA-SALONEN, M.S. (1984)
Absorption of pentachlorophenol (PCP) by bark chips and its
role in microbial PCP degradation. Microbiol. Ecol., 10:
359-367.
ARMSTRONG, R.W., EICHNER, E.R., KLEIN, D.E., BARTHEL, W.F.,
BENNETT, J.V., JONSSON, V., BRUCE, P.H.H., & LOVELESS, L.E.
(1969) Pentachlorophenol poisoning in a nursery for newborn
infants. II. Epidemiologic and toxicologic studies. J.
Pediatr., 75(2): 317-325.
ARRHENIUS, E., RENBERG, L., & JOHANSSON, L. (1977a)
Subcellular distribution, a factor in risk evaluation of
pentachlorophenol. Chem.-biol. Interact., 18: 23-34.
ARRHENIUS, E., RENBERG, L., JOHANSSON, L., & ZETTERQVIST,
M.A. (1977b) Disturbance of microsomal detoxication
mechanisms in liver by chlorophenol pesticides. Chem.-biol.
Interact., 18: 35-46.
ARSENAULT, R.D. (1976) Pentachlorophenol and contained
chlorinated dibenzodioxins in the environment. A study of
environmental fate, stability, and significance when used in
wood preservation. Am. Wood Preserv. Assoc., 20: 122-148.
AURAND, K., ENGLERT, N., KRAUSE, CH., ULRICH, D., & WALTER,
R. (1981) [Pentachlorophenol containing wood preservatives
in rooms.] Schr.-Reihe Ver. WaBoLu, 52: 293-313 (in German).
BAADER, E.W. & BAUER, H.J. (1951) Industrial intoxication
due to pentachlorophenol. Ind. Med. Surg., 20: 286-290.
BAKER, M.D. & MAYFIELD, C.I. (1980) Microbial and non-
biological decomposition of chlorophenols and phenol in soil.
Water Air Soil Pollut., 13: 411-424.
BALBA, M.H. & SAHA, J.G. (1974) Metabolism of lindane-14C by
wheat plants grown from treated seed. Environ. Lett., 7:
181-194.
BALLHORN, L., ROZMAN, T. ROZMAN, K., KORTE, F., & GREIM, H.
(1981) Cholestyramine enhances fecal elimination of penta-
chlorophenol in rhesus monkeys. Chemosphere, 10: 877-888.
BALLSCHMITER, K., ZOLLER, W., SCHOLZ, CH., & NOTTRODT, A.
(1983) Occurrence and absence of polychlorodibenzofurans and
polychlorodibenzodioxins in fly ash from municipal
incinerators. Chemosphere, 12: 585-594.
BARBENI, M., PRAMAURO, E., PELIZZENTI, E., BORGARELLO, E., &
SERPONE, N. (1985) Photodegradation of pentachlorophenol
catalyzed by semiconductor particles. Chemosphere, 14: 195-208.
BARTHEL, W.F., CURLEY, A., TRASHER, C.L., SEDLAK, V.A., &
ARMSTRONG, R. (1969) Determination of pentachlorophenol in
blood, urine, tissue, and clothing. J. Assoc. Off. Anal.
Chem., 52: 294-298.
BAUCHINGER, M., DRESP, J., SCHMID, E., & HAUF, R. (1982)
Chromosome changes in lymphocytes after occupational exposure
to pentachlorophenol. Mutat. Res., 102: 83-88.
BAXTER, R.A. (1984) Biochemical study of pentachlorophenol
workers. Ann. occup. Hyg., 28: 429-438.
BEGLEY, J., REICHERT, E.L., RASHAD, M.N., KLEMMER, H.W., &
SIEMSEN, A.W. (1977) Association between renal function
tests and pentachlorophenol. Clin. Toxicol., 11: 97-106.
BEHRBOHM, P. (1959) [On the hazards of exposure to
chlorinated phenols.] Dtsch. Gesundheitswes., 14: 614-619 (in
German).
BERGNER, H., CONSTANTINIDES, P., & MARTIN, J.H. (1965)
Industrial pentachlorophenol poisoning in Winnipeg, Canada.
Can. Med. Assoc. J., 92: 448-451.
BERRY, E.G., NOLAN, M.O., & GONZALES, J.O. (1950) Field
tests of molluscicides against Australorbus glabratus in
endemic areas of schistosomiasis in Puerto Rico. US Public
Health Rep., 65: 939-950.
BETTS, J.J., JAMES, S.P., & THORPE, W.V. (1955) The
metabolism of pentachloronitrobenzene and 2,3,4,6-tetrachloro-
nitrobenzene and the formation of mercapturic acids in the
rabbit. Biochem. J., 61: 611-617.
BEVENUE, A. & BECKMAN, H. (1967) Pentachlorophenol: a
discussion of its properties and its occurrence as a residue
in human and animal tissues. Residue Rev., 19: 83-134.
BEVENUE, A. & OGATA, J.N. (1971) A contributive error from
analytical reagents in the analysis of chlorophenoxy acids and
pentachlorophenol by electron capture gas chromatography. J.
Chromatogr., 61: 147-148.
BEVENUE, A., WILSON, J.R., POTTER, E.F., SONG, M.K., BECKMAN,
H., & MALLETT, G. (1966) A method for the determination of
pentachlorophenol in human urine in picogram quantities. Bull.
environ. Contam. Toxicol., 1: 257-266.
BEVENUE, A., HALEY, T.J., & KLEMMER, H.W. (1967a) A note on
the effect of a temporary exposure of an individual to penta-
chlorophenol. Bull. environ. Contam. Toxicol., 2: 293-296.
BEVENUE, A., WILSON, J.R., CASARETT, L.J., & KLEMMER, H.W.
(1967b) A survey of pentachlorophenol content in human urine.
Bull. environ. Contam. Toxicol., 2: 319-332.
BEVENUE, A., EMERSON, M.L., CASARETT, L.J., & YAUGER, W.L.
(1968) A sensitive gas chromatographic method for the
determination of pentachlorophenol in human blood. J.
Chromatogr., 38: 467-472.
BEVENUE, A., OGATA, J.N., & HYLIN, J.W. (1972) Organo-
chlorine pesticides in rainwater, Oahu, Hawaii, 1971-72. Bull.
environ. Contam. Toxicol., 8: 238-241.
BISHOP, C.M. & JONES, A.H. (1981) Non-Hodgkin's lymphoma of
the scalp in workers exposed to dioxins. Lancet, 2: 369.
BLACKMAN, G.E., PARKE, M.H., & GARTON, G. (1955a) The
physiological activity of substituted phenols. I. Relation-
ships between chemical structure and physiological activity.
Arch. Biochem. Biophys., 54: 45-54.
BLACKMAN, G.E., PARKE, M.H., & GARTON, G. (1955b) The
physiological activity of substituted phenols. II. Relation-
ships between physical properties and physiological activity.
Arch. Biochem. Biophys., 54: 55-71.
BLAIR, D.M. (1961) Dangers in using and handling sodium
pentachlorophenate as a molluscicide. Bull. World Health
Organ., 25: 597-601.
BLEVINS, D. (1965) Pentachlorophenol poisoning in swine.
Vet. Med., 60: 455.
BODDINGTON, M.J., BARRETTE, P., GRANT, D., NORSTROM, R.J.,
RYAN, J.J., & WHITBY, L. (1985) Chlorinated dioxins and
related compounds, 1984. Proceedings of the Fourth Inter-
national Symposium, Ottawa, Canada, 16-18 October 1984.
Chemosphere, 14(6/7): 571-989.
BORTHWICK, P.W. & SCHIMMEL, S.C. (1978) Toxicity of penta-
chlorophenol and related compounds to early life stages of
selected estuarine animals. In: Rao, K.R. ed. Pentachloro-
phenol: chemistry, pharmacology, and environmental toxicology,
New York, London, Plenum Press, pp. 141-146.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr
(1985) Acute toxicity of monochlorophenols, dichlorophenols,
and pentachlorophenol in the mouse. Toxicol. Lett., 29: 39-42.
BOUTWELL, R.K. & BOSCH, D.K. (1959) The tumour-promoting
action of phenol and related compounds for mouse skin. Cancer
Res., 19: 413-424.
BOYLE, T.P., ROBINSON-WILSON, E.F., PETTY, B.D., & WEBER, W.
(1980) Degradation of pentachlorophenol in simulated lentic
environment. Bull. environ. Contam. Toxicol., 24: 177-184.
BRADLAW, J.A., GARTHOFF, L.H., & HURLEY, N.W. (1980)
Comparative induction of aryl hydrocarbon hydroxylase activity
in vitro by analogues of dibenzo- p-dioxin. Food Cosmet.
Toxicol., 18: 627-635.
BRANDT, M., SCHMIDT, E., & SCHMIDT, F.W. (1977) [Chronic liver
disease caused by long term household poisoning with penta-
chlorophenol.] Verh. Dtsch. Ges. Inn. Med., 83: 1609-1611 (in
German).
BRAUN, W.H. & SAUERHOFF, M.W. (1976) The pharmacokinetic
profile of pentachlorophenol in monkeys. Toxicol. appl.
Pharmacol., 38: 525-533.
BRAUN, W.H. & WAECHTER, J.M., Jr (1983) Sources of
uncertainty in pharmacokinetic prediction. J. anim. Sci., 56:
235-243.
BRAUN, W.H., YOUNG, J.D., BLAU, G.E., & GEHRING, P.J. (1977)
The pharmacokinetics and metabolism of pentachlorophenol in
rats. Toxicol. appl. Pharmacol., 41: 395-406.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1979) The
metabolism/pharmacokinetics of pentachlorophenol in man, and a
comparison with the rat and monkey. In: Deichmann, W.B., ed.
Toxicology and occupational medicine, New York, Amsterdam,
Oxford, Elsevier/North-Holland, pp. 289-296.
BRINGMANN, G. & KUHN, R. (1982) [Results of toxic action of
water pollutants on Daphnia magna Straus tested by an improved
standardized procedure.] Z. Wasser Abwasser Forsch., 15: 1-6
(in German).
BRUNS, G.W. & CURRIE, R.A. (1980) Extraction of pentachloro-
phenol and tetrachlorophenol residues from field-contaminated
carrots and potatoes: comparison of several methods. J. Assoc.
Off. Anal. Chem., 63: 56-60.
BUA (1986) In: Ges. Dt. Chemiker, ed. [Pentachlorophenol.
Report of the Advisory Group for existing chemicals which are
relevant from an environmental point of view (BUA),] Weinheim,
Deerfield Beach, VCH Publishers, 183 pp (in German).
BUCKMAN, N.G., HILL, J.O., MAGEE, R.J., & MCCORMICK, M.J.
(1984) Separation of substituted phenols, including eleven
priority pollutants using high-performance liquid
chromatography. J. Chromatogr., 284: 441-446.
BUHLER, D.R., RASMUSSON, M.E., & NAKANE, H.S. (1973)
Occurrence of hexachlorophene and pentachlorophenol in sewage
and water. Environ. Sci. Technol., 7: 929-934.
BUIKEMA, A.L., GINNISS, M.J., & CAIRNS, J. (1979) Phenolics
in aquatic ecosystems: a selected review of recent literature.
Mar. environ. Res., 2: 87-181.
BUNDESAMT FUR UMWELTSCHUTZ (1982) [Impairment of surface
waters by pentachlorophenol.] Mitteilung No. 21, Bern, 12 pp.
(in German).
BUNDESAMT FUR UMWELTSCHUTZ (1983) [Polychlorinated organic
compounds in Rheinfelden (Switzerland). Investigation of dust,
soil, and grass samples: results and evaluation.]
Schriftenreihe Umweltschutz, 18, Bern, 32 pp (in German).
BUNDESGESETZBLATT (1978) [Regulation of agricultural
pesticides in or on foodstuffs of plant origin and tobacco
products (regulation of plant-protective pesticides).] In:
[Bundesgeseztblatt. Part I,] Bonn, Federal Republic of
Germany, Federal Ministry of Youth, Family Affairs and Health,
pp. 718-719, 734 (in German).
BUNDESGESUNDHEITSAMT (1983) [On handling wood preserva-
tives,] Berlin, Bundesgesundheitsamt, 36 pp (Information
Booklet of the Federal Health Office of Federal Republic of
Germany) (in German).
BURGER, E. (1966) Acute fatal poisoning with sodium
pentachlorophenol. Dtsch. Z. Gesamt. Gerichtl. Med., 58:
240-247.
BUSELMAIER, W., ROHRBORN, G., & PROPPING, P. (1973)
Comparative investigations on the mutagenicity of pesticides
in mammalian test systems. Mutat. Res., 21: 25-26.
BUSER, H.R. (1975) Analysis of polychlorinated dibenzo-
p-dioxins and dibenzofurans in chlorinated phenols by mass
fragmentography. J. Chromatogr., 107: 295-310.
BUSER, H.R. (1976) Preparation of qualitative standard
mixtures of polychlorinated dibenzo- p-dioxins and dibenzo-
furans by ultraviolet and gamma-irradiation of the octachloro
compounds. J. Chromatogr., 129: 303-307.
BUSER, H.R. (1982) [Report on the formation of PCDDs during
pyrolysis of PCP and PCP-Na,] Wädenswil, Switzerland, Eidg.
Forschungsanstalt, 2 pp (Unpublished report) (in German).
BUSER, H.R. & BOSSHARDT, H.P. (1976) Determination of
polychlorinated dibenzo- p-dioxins and dibenzofurans in
commercial pentachlorophenols by combined gas chromatography -
mass spectrometry. J. Assoc. Off. Anal. Chem., 59: 562-569.
BUTTE, W. (1984) Determination of free and total penta-
chlorophenol in urine. Fresenius Z. Anal. Chem., 317: 659.
BUTTE, W., KIRSCH, M., & DENKER, J. (1983) The determination
of pentachlorophenol and tetrachlorophenols in Wadden sediment
and clams ( Mya arenaria) using triethylsulfonium hydroxide for
extraction and pyrolytic ethylation. Int. J. environ. anal.
Chem., 13: 141-153.
BUTTE, W., DENKER, J., KIRSCH, M., & HOPNER, TH. (1985)
Pentachlorophenol und tetrachlorophenols in Wadden sediment
and clams Mya arenaria of the Jadebusen after a 14-year period
of waste-water discharge containing pentachlorophenol.
Environ. Pollut. B, 9: 29-39.
CABRAL, J.R.P., RAITANO, F., MOLLNER, T., BRONCZYK, S., &
SHUBIK, P. (1979) Acute toxicity of pesticides in hamster.
Toxicol. appl. Pharmacol., 48: A192.
CANTELMO, A.C. & RAO, K.R. (1978) Effects of pentachloro-
phenol on the meiobenthic nematodes in an experimental system.
In: Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology,
and environmental toxicology, New York, London, Plenum Press,
pp. 165-174.
CARR, R.S. & NEFF, J.M. (1981) Biochemical indices of stress
in the sandworm Neanthes virens (Sars). I. Responses to penta-
chlorophenol. Aquat. Toxicol., 1: 313-327.
CARR, R.S., THOMAS, P., & NEFF, J.M. (1982) A simple
spectrophotometric technique for the determination of penta-
chlorophenol in water. Bull. environ. Contam. Toxicol., 28:
477-479.
CASANOVA, M. & DUBROCA, J. (1972) Residues of pentachloro-
nitrobenzene and its hexachlorobenzene impurity in soils and
lettuce (Fr.). C.R. Séances Acad. Agric. Fr., 58: 990-998.
CASARETT, L.J., BEVENUE, A., YANGER, W.L., & WHALEN, S.A.
(1969) Observations on pentachlorophenol in human blood and
urine. Am. Ind. Hyg. Assoc. J., 30: 360-366.
CASTERLINE, J.L., Jr (1982) Uptake, translocation, and
transformation of pentachlorophenol in soybean and spinach
plants. Diss. Abstr. Int. B 1983, 43(8): 2409.
CAUTREELS, W., VAN COUWENBERGHE, K., & GUZMAN, L.A. (1977)
Comparison between the organic fraction of suspended matter at
a background and an urban station. Sci. total Environ., 8:
79-88.
CHAPMAN, J.B. & ROBSON, P. (1965) Pentachlorophenol
poisoning from bath-water. Lancet, 12 June: 1266-1267.
CHAPMAN, J.C., HIGGINS, V.R., SIMPSON, G.R., & SIYALI, D.S.
(1981) Pentachlorophenol (PCP) exposure: an occupational
health hazard in mushroom growing. In: Mushroom science. XI.
Proceedings of the eleventh International Scientific Congress
on the Cultivation of Edible Fungi, Sydney, Australia, 18 pp.
CHAPMAN, P.M., FARRELL, M.A., & BRINKHURST, R.O. (1982)
Relative tolerances of selected aquatic oligochaetes to
combinations of pollutants and environmental factors. Aquat.
Toxicol., 2: 69-78.
CHAU, A.S.Y. & COBURN, J.A. (1974) Determination of penta-
chlorophenol in natural and waste waters. J. Assoc. Off. Anal.
Chem., 57: 389-393.
CHIU, C., THOMAS, R.S., LOCKWOOD, J., LI, K., HALMAN, R., &
LAO, R.C.C. (1983) Polychlorinated hydrocarbons from power
plants, wood burning and municipal incinerators. Chemosphere,
12: 607-616.
CHOI, J. & AOMINE, S. (1972) Effects of soil on the activity
of pentachlorophenol. Soil Sci. plant Nutr., 18: 255-260.
CHOI, J. & AOMINE, S. (1974a) Adsorption of pentachloro-
phenol by soils. Soil Sci. plant Nutr., 20: 135-144.
CHOI, J. & AOMINE, S. (1974b) Mechanism of pentachlorophenol
adsorption by soils. Soil Sci. plant Nutr., 20: 371-379.
CHOW, C.Y., THEVASAGAYAM, E.S., & WAMBEEK, E.G. (1955) Control
of Salvinia - a host plant of Mansonia mosquitos. Bull. World
Health Organ., 12: 365-369.
CHU, J.P. & KIRSCH, E.J. (1972) Metabolism of pentachloro-
phenol by an axenic bacterial culture. Appl. Microbiol., 23:
1033-1035.
CIRELLI, D.P. (1978a) Patterns of pentachlorophenol usage in
the United States of America: an overview. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology. New York, London, Plenum Press, pp. 13-18.
CIRELLI, D.P. (1978b) Pentachlorophenol: position document
1. Fed. Reg., 43: 48446-48477.
CLEVELAND, L., BUCKLER, D.R., MAYER, F.L., & BRANSON, D.R.
(1982) Toxicity of three preparations of pentachlorophenol to
fathead minnows: a comparative study. Environ. Toxicol. Chem.,
1, 205-212.
CONKEY, J.H. & CARLSON, J.A. (1963) Relative toxicity of
biostatic agents suggested for use in the pulp and paper
industry: 1963 review. Tech. Assoc. Pulp Paper Ind., 46: 23
A-39A.
CONKLIN, P.J. & RAO, K.R. (1978) Toxicity of sodium penta-
chlorophenate (Na-PCP) to the grass shrimp, Palaemonetes
pugio, at different stages of the molt cycle. Bull. environ.
Contam. Toxicol., 20: 275-279.
COOK, W.L., FIEDLER, D., & BOURQUIN, A.W. (1980) Succession
of microfungi in estuarine microcosms perturbed by carbaryl,
methyl parathion, and pentachlorophenol. Bot. Mar., 23:
129-131.
CORDDRY, A.E. (1981) A pregnancy outcome study of the wives
of workers exposed to chlorophenate wood preservatives at a
sawmill, University of Washington, Department of Environmental
Health, 191 pp (Thesis).
COURTNEY, K.D., COPELAND, M.F., & ROBBINS, A. (1976) The
effects of pentachloronitrobenzene, hexachlorobenzene, and
related compounds on fetal development. Toxicol. appl.
Pharmacol., 35: 239-256.
CRANDALL, C.A. & GOODNIGHT, C.J. (1959) The effect of
various factors on the toxicity of sodium pentachlorophenate
to fish. Limnol. Oceanogr., 4: 53-56.
CRANMER, M. & FREAL, J. (1970) Gas chromatographic analysis
of pentachlorophenol in human urine by formation of alkyl
ethers. Life Sci., 9: 121-128.
CRETNEY, M.J. (1976) Pentachlorophenol death. Bull. Int.
Assoc. Forensic Toxicol., 12: 10.
CROSBY, D.G. & WONG, A.S. (1976) Photochemical generation of
chlorinated dioxins. Chemosphere, 5: 327-332.
CROSBY, D.G., WONG, A.S., PLIMMER, J.R., & WOOLSON, E.A.
(1971) Photodecomposition of chlorinated dibenzo- p-dioxins.
Science, 173: 748-749.
CROSBY, D.G., MOILANEN, K.W., NAKAGAWA, M., & WONG, A.S.
(1972) Photonucleophilic reactions of pesticides. In:
Matsumura, F., Boush, G.M., & Misato, T., ed. Environmental
toxicology of pesticides, New York, London, Academic Press,
pp. 423-431.
CROSBY, D.G., MOILANEN, K.W., & WONG, A.S. (1973) Environmental
generation and degradation of dibenzodioxins and
dibenzofurans. Environ. Health Perspect., 5: 259-266.
CROSBY, D.G., BEYNON, K.I., GREVE, P.A., KORTE, F., STILL,
G.G., & VOUK, J.W. (1981) Environmental chemistry of penta-
chlorophenol. Pure appl. Chem., 53: 1051-1080.
CROSSLAND, N.O. & WOLF, C.J.M. (1985) Fate and biological
effects of pentachlorophenol in outdoor ponds. Environ.
Toxicol. Chem., 4: 73-86.
CSERJESI, A.J. (1967) The adaptation of fungi to penta-
chlorophenol and its biodegradation. Can. J. Microbiol., 13:
1243-1249.
CSERJESI, A.J. (1972) Detoxification of chlorinated phenols.
Int. Biodeterioration Bull., 8: 135-138.
CSERJESI, A.J. & JOHNSON, E.L. (1972) Methylation of penta-
chlorophenol by Trichoderma virgatum. Can. J. Microbiol., 18:
45-49.
CSERJESI, A.J. & ROFF, J.W. (1975) Toxicity tests of some
chemicals against certain wood-staining fungi. Int.
Biodeterioration Bull., 11: 90-96.
CULL, M.R. & DOBBS, A.J. (1984) Long-term changes in
polychlorodibenzo- p-dioxin concentrations in wood treated with
technical pentachlorophenol. Chemosphere, 13: 1091-1099.
CULL, M.R., DOBBS, A.J., & WILLIAMS, N. (1983) Polychloro-
dibenzo- p-dioxins (PCDDs) in commercial pentachlorophenol
(PCP) used in wood preservation. Chemosphere, 12: 483-485.
CURTIS, R.F., LAND, D.G., GRIFFITH, N.M., GEE, M., ROBINSON,
D., PEEL, J.L., DENNIS, C., & GEE, J.M. (1972) 2,3,4,6-
tetrachloroanisole association with musty taint in chickens
and microbiological formation. Nature (Lond.), 235: 223.
DAHMS, A. & METZNER, W. (1979) [On the analytics of penta-
chlorophenol and tetrachlorophenol in the air and in urine.]
Holz Roh-Werkst., 37: 341-344 (in German).
DALELA, R.C., RANI, S., & VERMA, S.R. (1980a) Physiological
stress induced by sublethal concentrations of phenol and
pentachlorophenol in Notopterus notopteru: hepatic acid and
alkaline phosphatases and succinic dehydrogenase. Environ.
Pollut., 21: 3-8.
DALELA, R.C., RANI, S., RANI, S., & VERMA, S.R. (1980b)
Influence of pH on the toxicity of phenol and its two
derivatives pentachlorophenol and dinitrophenol to some fresh
water teleosts. Acta hydrochim. hydrobiol., 8: 623-629.
DANIELS, C.R. & SWAN, E.P. (1979) Determination of
chlorinated phenols in surface-treated lumber by HPLC. J.
chromatogr. Sci., 17, 628-630.
DAVIS, H.C. & HIDU, H. (1969) Effects of pesticides on
embryonic development of clams and oysters and on survival and
growth of the larvae. US Fish Wildl. Serv. Fish. Bull., 67:
393-403.
DAVIS, J.C. & HOOS, R.A. (1975) Use of sodium pentachloro-
phenate and dehydroabiotic acid as reference toxicants for
salmonid bioassays. J. Fish. Res. Board Can., 32: 411-416.
DEBETS, F.M.H., STRIK, J.J.T.W.A., & OLIE, K. (1980) Effects
of pentachlorophenol on rat liver changes induced by hexa-
chlorobenzene with special reference to porphyria and
alterations in mixed-function oxygenases. Toxicology, 15:
181-195.
DEICHMANN, W.B. (1943) The toxicity of chlorophenols for
rats. Fed. Proc., 2: 76-77.
DEICHMANN, W.B. & KEPLINGER, M.L. (1981) Phenols and
phenolic compounds. In: Clayton, G.D. & Clayton, F.E., ed.
Patty's industrial hygiene and toxicology 2A, 3rd revised ed.,
New York, John Wiley and Sons, pp. 2567-2627.
DEICHMANN, W.B. & MERGARD, E.G. (1948) Comparative evalua-
tion of methods employed to express the degree of toxicity of
a compound. J. ind. Hyg. Toxicol., 30: 373-378.
DEICHMANN, W.B., MACHLE, W., KITZMILLER, K.V., & THOMAS, G.
(1942) Acute and chronic effects of pentachlorophenol and
sodium pentachlorophenate upon experimental animals. J.
Pharmacol. exp. Ther., 76: 104-117.
DE LAUNE, R.D., GAMBRELL, R.P., & REDDY, K.S. (1983) Fate of
PCP in estuarine sediment. Environ. Pollut. (B), 6: 297-308.
DEMENTI, B.A. (1981) Health hazard alert pentachlorophenol
(news). Am. Ind. Hyg. Assoc. J., 42:: A16, A18.
DEMIDENKO, N.M. (1969) [Materials for establishing the
maximum permissible concentrations in air.] Gig. Tr. Prof.
Zabol., 7: 58-60 (in Russian).
DETRICK, R.S. (1977) Pentachlorophenol: possible sources of
human exposure. For. Prod. J., 27: 13-16.
DIETZ, F. & TRAUD, J. (1978a) [Threshold odour and taste
concentrations of phenolic compounds.] GWF-Wasser/Abwasser,
119: 318-325 (in German).
DIETZ, F. & TRAUD, J. (1978b) [On the analysis of phenols,
particularly chlorophenols, in water by means of gas chromato-
graphy - methods and results.] Vom Wasser, 51: 235-257 (in
German).
DIJK, J.J., VAN, VAN DER MEER, C., & WIJNANS, M. (1977) The
toxicity of sodium pentachlorophenolate for three species of
decapod crustaceans and their larvae. Bull. environ. Contam.
Toxicol., 17: 622-630.
DOBBS, A.J. & GRANT, C. (1980) Pesticide volatilisation
rates: a new measurement of the vapour pressure of penta-
chlorophenol at room temperature. Pestic. Sci., 11: 29-32.
DOBBS, A.J. & WILLIAMS, N. (1983) Indoor air pollution from
pesticides used in wood remedial treatments. Environ. Pollut.
(B), 6: 271-296.
DOEDENS, J.D. (1964) Chlorophenols. In: Kirk, R.E. & Othmer,
D.F., ed. Encyclopedia of chemical technology, 2nd ed., New
York, John Wiley and Sons, Vol. 5, pp. 336-337.
DOUGHERTY, R.C. & PIOTROWSKA, K. (1976a) Multi-residue
screening by negative chemical ionization mass spectrometry of
organic polychlorides. J. Assoc. Off. Anal. Chem., 59:
1023-1027.
DOUGHERTY, R.C. & PIOTROWSKA, K. (1976b) Screening by
negative chemical ionization mass spectrometry for environ-
mental contamination with toxic residues: application to human
urines. Proc. Natl Acad. Sci. (USA), 73: 1777-1781.
DOUGHERTY, R.C., WHITAKER, M.J., SMITH, L.M., STALLING, D.L.,
& KUEHL, D.W. (1980) Negative chemical ionization studies of
human and food chain contamination with xenobiotic chemicals.
Environ. Health Perspect., 36: 103-117.
DRUMMOND, I., VAN ROOSMALEN, P.B., & KORNICKI, M. (1982)
Determination of total pentachlorophenol in the urine of
workers. A method incorporating hydrolysis, an internal
standard and measurement by liquid chromatography. Int. Arch.
occup. environ. Health., 50: 321-327.
DUGGAN, R.E. & CORNELIUSSEN, P.E. (1972) Dietary intake of
pesticide chemicals in the United States. III. June 1968 -
April 1970. Pestic. monit. J., 5: 331-341.
DUNCAN, C.G. & DEVERALL, F.J. (1964) Degradation of wood
preservatives by fungi. Appl. Microbiol., 12: 57-62.
DUST, J.V. & THOMPSON, W.S. (1973) Pollution control in the
wood-preserving industry. Part IV. Biological methods of
treating waste water. For. Prod. J., 23: 59-66.
EBEN, A., SCHALLER, K.H., & KRAUSE, CH. (1981) [Penta-
chlorophenol (PCP): analysis of plasma and urine.] In: German
Science Foundation, Working Group on Analytical Chemistry,
Commission for the Investigation of Health Hazards of Chemical
Compounds in the Work Area, ed. [Analysis of hazardous
substances in biological materials,] Weinheim, VCH Publishers,
Vol. 2, pp. 1-11 (in German).
ECKRICH, W. (1986) [Levels of PCDDs/PCDFs in indoor air and
in human blood.] In: VDI-Kommission Reinhaltung der Luft, ed.
[ Dioxins. Report of the VDI Meeting, Essen, Federal Republic
of Germany, 22-23 April 1986,] Düsseldorf, VDI-Verlag, pp.
70-81 (in German).
ECONOMIST INTELLIGENCE UNIT (1981) Economic implications of
abatement measures of water pollution due to hexachlorobuta-
diene, endosulfan, trichlorophenol, and pentachlorophenol,
Brussels, Commission of the European Communities, 134 pp
(Report prepared for Environment and Consumer Protection
Service).
EDGEHILL, R.U. (1982) Microbial treatment of wastewater and
soil to remove pentachlorophenol. Part 1. Diss. Abstr. Int. B,
42: 4871.
EDGEHILL, R.U. & FINN, R.K. (1983) Treatment of soil to remove
pentachlorophenol. Appl. environ. Microbiol., 45: 1122-1125.
EDGERTON, T.R. & MOSEMAN, R.F. (1979) Determination of
pentachlorophenol in urine: the importance of hydrolysis. J.
agric. food Chem., 27: 197-199.
EDGERTON, T.R., MOSEMAN, R.F., LINDER, R.E., & WRIGHT, L.H.
(1979) Multi-residue method for the determination of
chlorinated phenol metabolites in urine. J. Chromatogr., 170:
331-342.
EDWARDS, M.J. (1968) Congenital malformations in the rat
following induced hyperthermia during gestation. Teratology,
1: 173-178.
ELLER, P.M., ed. (1984a) Pentachlorophenol in blood. In:
NIOSH manual of analytical methods, 3rd ed., Cincinnati, Ohio,
US Department of Health and Human Services, Public Health
Service, Centers for Disease Control, National Institute for
Occupational Safety and Health, pp. 8001-1 - 8001-4 (DHHS
Publication No. 84-100).
ELLER, P.H., ed. (1984b) Pentachlorophenol in urine. In:
NIOSH manual of analytical methods, 3rd ed., Cincinnati, Ohio,
US Department of Health and Human Services, Public Health
Service, Centers for Disease Control, National Institute for
Occupational Safety and Health, pp. 8303-1 - 8303-4 (DHHS
Publication No. 84-100).
EMBREE, V., ENARSON, D.A., CHAN-YEUNG, M., DY BUNCIO, A.,
DENNIS, R., & LEACH J. (1984) Occupatonal exposure to
chlorophenates: toxicology and respiratory effects. Clin.
Toxicol., 22: 317-329.
ENARSON, D.A., CHAN-YEUNG, M., EMBREE, V., WANG, R., &
SCHULZER, M. (1986) Occupational exposure to chlorophenates,
renal, hepatic and other health effects. Scand. J. Work
environ. Health, 12: 144-148.
ENGEL, C., DE GROOT, A.P., & WEURMAN, C. (1966) Tetra-
chloroanisole: a source of musty taste in eggs and broilers.
Science, 154: 270-271
ENGST, R., MACHOLZ, R.M., KUJAWA, M., LEWERENZ, H.J., & PLASS,
R. (1976) The metabolism of lindane and its metabolites
gamma-2,3,4,5,6-pentachlorocyclohexene, pentachlorobenzene,
and pentachlorophenol in rats and the pathways of lindane
metabolism. J. environ. Sci. Health (B), 11: 95-117.
ENGST, R., MACHOLZ, R.M., & KUJAWA, M. (1978) [Metabolites
of hexachlorocyclohexane isomers (HCH) in human blood.]
Pharmazie, 33: 109-111 (in German).
ENVIRONMENT, CANADA (1979) Monitoring environmental
contamination from chlorophenol contaminated wastes generated
in the wood preservation industry, Ottawa, Environmental
Protection Bureau, Environmental Protection Service, Pacific
and Yukon Region, pp. 74 (Regional Program Report No. 79-24)
(Prepared by Can Test Ltd and EVS Consultants Ltd) (DSS File
No. 075B, KE 114-8-1935).
ERIKSON, M., HARDELL, L., BERG, N.O., MOELLER, T., & AXELSSON,
O. (1981) Soft-tissue sarcomas and exposure to chemical
substances: a case reference study. Br. J. ind. Med., 38:
27-33.
ERNEY, D.R. (1978) Gas-liquid chromatographic determination
of pentachlorophenol in milk. J. Assoc. Off. Anal. Chem., 61:
214-216.
ERNST, W. (1979) Factors affecting the evaluation of
chemicals in laboratory experiments using marine organisms.
Ecotoxicol. environ. Saf., 3: 90-98.
ERNST, W. & WEBER, K. (1978a) Chlorinated phenols in selected
estuarine bottom fauna. Chemosphere, 7: 867-822.
ERNST, W. & WEBER, K. (1978b) The fate of pentachlorophenol
in the Weser estuary and the German Bight. Veröff. Inst.
Meeresforsch. Bremerh., 17: 45-53.
ERVIN, H.E. & MCGINNIS, G.D. (1980) Analysis of pentachloro-
phenol in waste-water using high-performance liquid chromato-
graphy. J. Chromatogr., 190: 203-207.
ETZEL, J.E. & KIRSCH, E.J. (1974) Biological treatment of
industrial wastewater containing pentachlorophenol. Dev. ind.
Microbiol., 16: 287-295.
EXON, J.H. (1984) A review of chlorinated phenols. Vet. hum.
Toxicol., 26: 508-520.
EXON, J.H. & KOLLER, L.D. (1982) Effects of transplacental
exposure to chlorinated phenols. Environ. Health Perspect.,
46: 137-140.
EXON, J.H. & KOLLER, L.D. (1983a) Alternation of trans-
placental carcinogenesis by chlorinated phenols. In: Jolley,
R.L., et al., ed. Water chlorination: environmental impact and
health effects, Ann Arbor, Michigan, Ann Arbor Science, Vol.
2, Book 4, pp. 1177-1188.
EXON, J.H. & KOLLER, L.D. (1983b) Effects of chlorinated
phenols on immunity in rats. Int. J. Immunopharmacol., 5:
131-136.
FAAS, L.F. & MOORE, J.C. (1979) Determination of penta-
chlorophenol in marine biota and sea water by gas-liquid
chromatography and high pressure liquid chromatography. J.
agric. food Chem., 27: 554-557.
FAHRIG, R. (1974) Comparative mutagenicity studies with
pesticides, Lyons, International Agency for Research on
Cancer, pp. 161-181 (IARC Scientific Publication Vol. 10).
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic
activity of chlorophenols and chlorophenol impurities. In:
Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
325-338.
FARQUHARSON, H.E., CAGE, J.C., & NORTHOVER, J. (1958) The
biological action of chlorophenols. Br. J. Pharmacol., 13:
20-24.
FARRINGTON, D.S. & MUNDAY, J.W. (1976) Determination of
trace amounts of chlorophenols by gas-liquid chromatography.
Analyst, 101: 639-643.
FEDERAL REGISTER (1977) Gelatin: affirmation of GRAS status
as a direct and indirect human food ingredient. Fed. Reg.,
42(218): 58763-58765.
FEILER, H. (1980) Fate of priority pollutants in publicly
owned treatment works, Washington DC, US Environmental
Protection Agency (EPA-440/1-80-301).
FERREE, D.C. (1974) Influence of freshly treated posts on
growth of newly planted trees. Ohio Agric. Res. Dev. Cent.
Res. Summ., 75: 11-12.
FIELDER, R.J., SORRIE, G.S., BISHOP, C.M., JONES, R.B., & VAN
DEN HEUVEL, M.J. (1982) Pentachlorophenol. In: Toxicity
review, London, Health and Safety Executive, Vol. 5, 20 pp.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B.,
GROTH, D.H., & RUSSELL, W.O. (1984) An evaluation of reports
of dioxin exposure and soft-tissue sarcoma pathology among
chemical workers in the United States. Scand. J. Work environ.
Health, 10: 299-303.
FIRESTONE, D. (1973) Etiology of chick edema disease.
Environ. Health Perspect., 5: 59-66.
FIRESTONE, D. (1977) Chemistry and analysis of pentachloro-
phenol and its contaminants, Washington DC, US Food and Drug
Administration, pp. 57-59 (FDA Bylines No. 2).
FIRESTONE, D., REES, J., BROWN, N.L., BARRON, R.P., & DAMICO,
J.N. (1972) Determination of polychlorodibenzo- p-dioxins and
related compounds in commercial chlorophenols. J. Assoc. Off.
Anal. Chem., 55: 85-92.
FIRESTONE, D., CLOWER, M., Jr, BORSETTI, A.P., TESKE, R.H., &
LONG, P.E. (1979) Polychlorodibenzo- p-dioxin and penta-
chlorophenol residues in milk and blood of cows fed technical
pentachlorophenol. J. agric. food Chem., 27: 1171-1177.
FISCHER, A. & SLEMROVA, J. (1978) [The contamination of the
river Rhine with chlorophenols.] Vom Wasser, 51: 33-46 (in
German).
FISCHER, M. (1983) [Pentachlorophenol as model case of a
xenobiotic compound,] Berlin, Federal Health Office Institute
for Water, Soil and Air Hygiene, 38 pp (Research Report No.
10604007) (in German).
FLEISCHER, M., MEISS, R., ROBENEK, H., THEMANN, H., & ECKHARD,
R. (1980) Ultrastructural morphometric investigations on rat
liver of young and adult rats after treatment with technical
pentachlorophenol. Arch. Toxicol., 44: 243-257.
FLICKINGER, C.W. & LAWRENCE, A.W. (1982) Occupational health
experience in the wood preserving industry. In: Am. Wood
Preserv. Assoc., 78: 11-30.
FOCQUET, J.P. & THEISEN, R. (1981) Détermination de la
réduction de l'hexachlorobutadiène de l'endosulfan, du
pentachlorophenol et du trichlorophénol contenue dans les
effluents en tenant compte des meilleurs moyens techniques
disponsibles, Luxembourg, Commission of the European
Communities (Contract No. ENV/723/74-FR) (unpublished report).
FOLKE, J. & LUND, H. (1983) Occurrence of low- and high-
chlorinated phenols in municipal sewage before and after
passing through biological treatment plants. J. Chromatogr.,
279: 189-198.
FORSELL, J.H., SHULL, L.R., & KATELEY, J.R. (1981)
Subchronic administration of technical pentachlorophenol to
lactating dairy cattle immunotoxicologic evaluation. J.
Toxicol. environ. Health, 8:: 543-558.
FOUNTAINE, J.E., JOSHIPURA, P.B., & KELIHER, P.N. (1976)
Some observations regarding pentachlorophenol levels in
Haverford Township, Pennsylvania. Water Res., 10: 185-188.
FOX, M. & JOSHI, S.R. (1984) The fate of pentachlorophenol
in the Bay of Quinte, Lake Ontario (Canada USA). J. Great
Lakes Res., 10: 190-196.
FRANZLE, O. (1982) [Modelling the transport of environmental
chemicals and their metabolites through the unsaturated zone
of natural soil profiles and sludge in laboratory lysimeters
and in the field.] In: BMI Environmental Research Plan, pp.
XIX + 315 (Research Report No. 10602005/02) (in German).
FRAGIADAKIS, A., KLEIN, W., KORTE, F., MOZA, P.N., SCHEUNERT,
I., VOCKEL, D., & WEISS, U. (1979) [Fate of organochlorine
compounds in plant-soil systems] In: [Organohalogen compounds
in the environment. Project report 1975-78,] Jülich Nuclear
Research Plant (Jül-Spez. 45, July 1979) (BMFT Research Report
037118) (in German).
FRANK. R., BRAUN, H.E., HOLDRINET, M., SIRONS, G.J., SMITH,
E.H., & DIXON, D.W. (1979) Organochlorine insecticides and
industrial pollutants in the milk supply of southern Ontario,
Canada, 1977. J. food Prot., 42: 31-37.
FREITAG, D., GEGER, H., KRAUS, A., VISWANATHAN, R., KOTZIAS,
D., ASSAR, A., KLEIN, W., & KORTE, F. (1982) Ecotoxico-
logical profile analysis. VII. Screening chemicals for their
environmental behaviour by comparative evaluation. Ecotoxicol.
environ. Saf., 6: 60-81.
FRG (1986) [Statuary ordinance by dangerous chemicals.] In:
Gefahrstoffverordnung, Bonn, Ministry of Labour and Social
Affairs (BGBL I), p. 1470 (in German).
FUCHSBICHLER, G. (1982) Automated gel chromatography as
clean-up process for determination of ECD-detectable
substance, chlorinated hydrocarbons, pentachlorophenol,
diphenyl and o-phenylphenol in plant materials. Z.
Lebensm.-Unters. Forsch., 174: 9-12.
GAB, F. & PARLAR, H. (1979) [Transformation of organo-
halogens under abiotic conditions.] In: [Organohalogen
compounds in the environment. Project report 1975-78,] Jülich
Nuclear Research Plant (Jül.-Spez. 45, July 1979) (BMFT
Research Report 037114) (in German).
GAB, S., NITZ, S., PARLAR, H., & KORTE, F. (1975)
Photomineralisation of certain aromatic xenobiotica.
Chemosphere, 4: 251-256.
GAB, S. (1981) [Decomposition of organic chemicals in
adsorbed phase.] In: [Report on the 1980 Symposium of the
Institute for Ecological Chemistry,] Münich, Society for
Radiation and Environmental Research (GSF), pp. 61-75 (in
German).
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol.
appl. Pharmacol., 14: 515-534.
GARRETT, C.L. (1980) Fraser River Estuary study. Water
quality series. Toxic organic contaminants, Ottawa,
Environmental Protection Service, Pacific and Yukon Region,
Environment Canada, pp. 123.
GEBEFUEGI, I. (1981) [Our present knowledge of the
occurrence of pentachlorophenol in the environment.] In:
[Report on the 1980 Symposium of the Institute for Ecological
Chemistry,] Münich, Society for Radiation and Environmental
Research (GSF), pp. 25-33 (in German).
GEBEFUEGI, I. & KORTE, F. (1983) Pentachlorophenol
contamination of human milk samples. Chemosphere, 12:
1055-1060.
GEBEFUEGI, I. & KORTE, F. (1984) Indoor contamination of
household articles through pentachlorophenol and lindane. In:
Berglund, B., Lindvall, T., & Sundell, J., ed. Indoor air. IV.
Chemical characterisation and personal exposure, Stockholm,
Swedish Council for Building Research, pp. 317-322.
GEBEFUEGI, I., PARLAR, H., & KORTE, F. (1979) Occurrence of
pentachlorophenol in closed environments. Ecotoxicol. environ.
Saf., 3: 269.
GEBEFUEGI, I., OXYNOS, K., & KORTE, F. (1983) [Long-term
behaviour of pentachlorophenol in closed rooms.] Chemosphere,
12: 59-63 (in German).
GILBERT, F.I., MINN, C.E., DUNCAN, R.C., ALDRICH, T., LEDERER,
W.H., & WILKINSON, J.E. (1983) Effects of chemical preserva-
tives on the health of wood treating workers in Hawaii, 1981.
Clinical and chemical profiles and historical prospective
study, Honolulu, Hawaii, Pacific Health Research Institute,
233 pp.
GJOVIK, L.R., JOHNSON, D.B., KOZAK, V., WOOLSON, E.A.,
THOMPSON, W.A., MICKLEWRIGHT, J.T., DOST, W.A., & NICHOLAS,
D.D. (1981) The biologic and economic assessment of
pentachlorophenol, inorganic arsenicals, creosote. I. Wood
Preservatives, Washington DC, US Department of Agriculture,
pp. 69-87 (Technical Bulletin No. 1658-1).
GLICKMAN, A.H., STRATHAM, C.N., & LECH, J.J. (1977) Studies
on the uptake, metabolism, and disposition of pentachloro-
phenol and pentachloroanisole in rainbow trout. Toxic. appl.
Pharmacol., 41: 649-658.
GODSY, E.M., GOERLITZ, D.F., & EHRLICH, G.G. (1986) Effects of
pentachlorophenol on methanogenic fermentation of phenol.
Bull. environ. Contam. Toxicol., 36: 271-277.
GOLDSTEIN, J.A., FRIESEN, M., LINDER, R.E., HICKMAN, P., HASS,
J.R., & BERGMAN, H. (1977) Effects of pentachlorophenol on
hepatic drug-metabolizing enzymes and porphyria related to
contamination with chlorinated dibenzo- p-dioxins and dibenzo-
furans. Biochem. Pharmacol., 26: 1549-1557.
GOODNIGHT, C.J. (1942) Toxicity of sodium pentachlorophenate
and pentachlorophenols to fish. Ind. eng. Chem., 34: 868-872.
GORDON, D. (1956) How dangerous is PCP? Med. J. Aust., Sept.
29: 485-488.
GOSSLER, K. & SCHALLER, K.H. (1978) [Quantitative
determination of pentachlorophenol in urine and plasma by gas
chromatography.] Z. anal. Chem., 290: 111-112 (in German).
GOTHAM, I.J. & RHEE, G.Y. (1982) Effects of a hexachloro-
biphenyl and pentachlorophenol on growth and photosynthesis of
phytoplankton. J. Great Lakes Res., 8: 328-335.
GREEN, R.E. & YOUNG, R.H.T. (1970) Herbicide and fertilizer
movement in Hawaiian sugarcane soils in relation to subsurface
water quality. Hawaiian Sugar Technol., 29: 88-96.
GREENE, M.H., BRINTON, L.A., FRAUMENI, J.F., & D'AMICO, R.
(1978) Familial and sporadic Hodgkin's disease associated
with occupational wood exposure. Lancet, 9: 626-627.
GREICHUS, Y.A., LIBAL, G.W., & JOHNSON, D.D. (1979)
Diagnosis and physiologic effects of pentachlorophenols on
young pigs. I. Effects of purified pentachlorophenol. Bull.
environ. Contam. Toxicol., 23: 418-422.
GRIMM, H.G., SCHALLER, K.H., & VALENTIN, H. (1985) [Results
of pentachlorophenol exposure in the workplace and the
environment.] Zentralbl. Arbeitsmed., Arbeitsschutz, Prophyl.
Ergon., 34: 136-142 (in German).
GRIMM, H.G., SCHELLMANN, B., SCHALLER, K.H., & GOSSLER, K.
(1981) [Pentachlorophenol concentrations in tissues and body
fluids of normal persons.] Zbl. Bakt. Hyg., I. Abt. Orig. B,
174: 77-90 (in German).
GRUTTKE, H., KRATZ, W., PAPENHAUSEN, U., WEIGMANN, G., HAQUE,
A., & SCHUPHAN, I. (1986) [Transfer of 14C-Na-PCP in
model-foodchains.] Verhd. Ges. ökol., 14: 451-455 (in German).
GUNTHER, F.A., WESTLAKE, W.E., & JAGLAN, P.S. (1968)
Reported solubilities of 738 pesticide chemicals in water.
Residue Rev., 20: 56-148.
GUPTA, P.K. & RAO, P.S. (1982) Toxicity of phenol, penta-
chlorophenol, and sodium pentachlorophenate to a freshwater
pulmonate snail Lymnaea acuminata. Arch. Hydrobiol., 94:
210-217.
GUPTA, P.K., MUJUMDAR, V.S., RAO, P.S., & DURVE, V.S. (1982)
Toxicity of phenol, pentachlorophenol, and sodium
pentachlorophenolate to a freshwater teleost Lebistes
reticulatus (Peters). Acta hydrochim. hydrobiol., 10: 177-181.
GUPTA, S., VERMA, S.R., & SAXENA, P.K. (1982) Toxicity of
phenolic compounds in relation to the size of a freshwater
fish Notopterus notopterus (Pallas). Ecotoxicol. environ.
Saf., 6: 433-438.
GUPTA, S., DALELA, R.C., & SAXENA, P.K. (1983a) Influence of
dissolved oxygen levels on acute toxicity of phenolic
compounds to fresh water teleost Notopterus notopterus
(Pallas). Water Air Soil Pollut., 19: 223-228.
GUPTA, S., DALELA, R.C., & SAXENA, P.K. (1983b) Effects of
phenolic compounds on in vivo activity of transaminases in
certain tissues of the fish Notopterus notopterus. Environ.
Res., 32: 8-13.
GUTHRIE, M.A., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L.
(1984) Pentachlorophenol biodegradation. II. Anaerobic. Water
Res., 18: 451-461.
HAKULINEN, R. & SALKINOJA-SALONEN, M. (1982) Treatment of
pulp and paper industry waste waters in an anaerobic fluidised
bed reactor. Process Biochem., 3/4: 18-22.
HALAWANI, A. (1951) Endemic diseases control. J. Egypt. Med.
Assoc., 34: 347-358.
HALEY, T.J. (1977) Human poisoning with pentachlorophenol
and its treatment. Ecotoxicol. environ. Saf., 1: 343-347.
HALL, L.H. & KIER, L.B. (1984) Molecular connectivity of
phenols and their toxicity to fish. Bull. environ. Contam.
Toxicol., 32: 354-362.
HAQUE, A., SCHEUNERT, I., & KORTE, F. (1978) Isolation and
identification of a metabolite of pentachlorophenol-14C in
rice plants. Chemosphere, 7: 65-69.
HAQUE, R. & FREED, V.H. (1974) Behaviour of pesticides in
the environment: environmental chemodynamics. Residue Rev.,
52: 89-116.
HARDELL, L. (1977) Soft-tissue sarcoma and exposure to
phenoxy acids: a clinical observation. Läkartidingen, 74:
2853-2854.
HARDELL, L. (1979) Malignant lymphoma of histiocytic type
and exposure to phenoxyacetic acids or chlorophenols. Lancet,
1: 55-56.
HARDELL, L. (1981a) Relation to soft-tissue sarcoma,
malignant lymphoma, and colon cancer to phenoxy acids,
chlorophenols, and other agents. Scand. J. Work environ.
Health, 7: 119-130.
HARDELL, L. (1981b) Epidemiological studies on soft-tissue
sarcoma and malignant lymphoma and their relation to phenoxy
acid or chlorophenol exposure, Umea, Sweden, Umea University
Medical Dissertations (New Series No. 65).
HARDELL, L. & SANDSTROM, A. (1979) Case-control study:
soft-tissue sarcomas and exposure to phenoxyacetic acids or
chlorophenols. Br. J. Cancer, 39: 711-717.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981)
Malignant lymphoma and exposure to chemicals, especially
organic solvents, chlorophenols, and phenoxy acids: a case-
control study. Br. J. Cancer, 43: 169-176.
HARDELL, L., JOHANSSON, B., & AXELSON, O. (1982) Epidemio-
logical study of nasal and nasopharyngeal cancer and their
relation to phenoxy acid or chlorophenol exposure. Am. J. ind.
Med., 3: 247-257.
HARGESHEIMER, E.E. & COUTTS, R.T. (1983) Selected ion mass
spectrometric identification of chlorophenol residues in human
urine. J. Assoc. Off. Anal. Chem., 66: 13-21.
HARPER, D.B. & BANAVE, D. (1975) Chloroanisole residues in
broiler tissues. Pestic. Sci., 6: 159-163.
HARRISON, D.L. (1959) The toxicity of wood preservatives to
stock. Part I. Pentachlorophenol. NZ. Vet. J., 7: 89-98.
HARVEY, W.A. & CRAFTS, A.S. (1952) Toxicity of pentachloro-
phenol and its sodium salts in three Yolo soils. Hilgardia,
21: 487-490.
HATTULA, M.L., WASENIUS, V.M., REUNANEN, H., & ARSTILA, A.U.
(1981) Acute toxicity of some chlorinated phenols, catechols
and cresols to trout. Bull. environ. Contam. Toxicol., 26:
295-298.
HAUCH, R.G., NORRIS, D.R., & PIERCE, R.H. (1980) Acute and
chronic toxicity of sodium pentachlorophenate to the copepod
Pseudodiaptomus coronatus. Bull. environ. Contam. Toxicol.,
25: 562-568.
HAYES, E.H. (1979) High-pressure liquid chromatographic
determination of pentachlorophenol by using paired ion
chromatography reagents. J. Assoc. Off. Anal. Chem., 62:
1004-1006.
HEIKES, D.L. & GRIFFITT, K.R. (1980) Pentachlorophenol in
Mason jar lids and home canned food. J. Assoc. Off. Anal.
Chem., 63: 1125-1128.
HENSHAW, B.G., MORGAN, J.W.W., & WILLIAMS, N. (1975) The
detection of organic solvent preservatives in wood by
thin-layer chromatography. J. Chromatogr., 110: 37-41.
HERNANDEZ, C. & STRASSMAN-SUNDY, S. (1980) Pentachlorophenol
in log homes - Kentucky. Morb. Mortal. Wkly Rep., 29: 431-432,
437.
HERNBERG, S. & PESSI, T. (1964) Peripheral nervous paralysis
due to pentachlorophenate. Int. Arch. Gewerbepathol.
Gewerbehyg., 21: 23-26.
HIATT, C.W., HASKINS, W.T., & OLIVIER, L. (1960) The action
of sunlight on sodium pentachlorophenate. Am. J. trop. Med.
Hyg., 9: 527-531.
HICKMAN, G.T. & NOVAK, J.T. (1984) Acclimation of activated
sludge to pentachlorophenol. J. Water Pollut. Control Fed.,
56: 364-469.
HILL, E.F., HEALTH, R.G., SPANN, J.W., & WILLIAMS, J.D.
(1975) Lethal dietary toxicities of environmental pollutants
to birds, Washington DC, US Fish and Wildlife Service,
Department of the Interior, 61 pp (Special Scientific Report -
Wildlife No. 191).
HILLAM, R.P. & GREICHUS Y.A. (1983) Effects of purified
pentachlorophenol on the serum proteins of young pigs. Bull.
environ. Contam. Toxicol., 31: 599-604.
HILTON, H.W., YUEN, Q.H., & NOMURA, N.S. (1970) Distribution
of residues from atrazine, ametryne, and pentachlorophenol in
sugarcane. J. agric. food Chem., 18: 217-220.
HINKLE, D.K. (1973) Fetotoxic effects of pentachlorophenol
in the golden Syrian hamster. Toxicol. appl. Pharmacol., 25:
455.
HIRSCH, A.A. (1942) Toxicity of sodium pentachlorophenate
and other chemicals on water hyacinth. Bot. Gaz., 103: 620-621.
HMSO (1974) The non-agricultural user of pesticides in Great
Britain, London, Her Majesty's Stationery Office, 65 pp
(Pollution Paper No. 3).
HOBEN, H.J., CHING, S.A., CASARETT, L.J., & YOUNG, R.A.
(1976a) A study of the inhalation of pentachlorophenol by
rats. I. A method for the determination of pentachlorophenol
in rat plasma, urine, and tissue and in aerosol samples. Bull.
environ. Contam. Toxicol., 15: 78-85.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976b) A study
of the inhalation of pentachlorophenol by rats. II. A new
inhalation exposure system for high doses in short exposure
time. Bull. environ. Contam. Toxicol., 15: 86-92.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976c) A study
of inhalation of pentachlorophenol by rats. III. Inhalation
toxicity study. Bull. environ. Contam. Toxicol., 15: 463-465.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976d) A study
of inhalation of pentachlorophenol by rats. IV. Distribution
and excretion of inhaled pentachlorophenol. Bull. environ.
Contam. Toxicol., 15: 466-474.
HOBEN, J.H., CHING, S.A., YOUNG, R.A., & CASARETT, L.J.
(1976e) A study of the inhalation of pentachlorophenol by
rats. V. A protein binding study of pentachlorophenol. Bull.
environ. Contam. Toxicol., 16: 225-232.
HODSON, P.V. & BLUNT, B.R. (1981) Temperature-induced
changes in pentachlorophenol chronic toxicity to early life
stages of rainbow trout (Salmo gairdneri). Aquat. Toxicol., 1:
113-128.
HODSON, P.V., DIXON, D.G., & KAISER, K.L.E. (1984) Measure-
ment of median lethal dose as a rapid indication of contam-
inant toxicity to fish. Environ. Toxicol. Chem., 3: 243-254.
HOETING, A.L. (1977) Penta - another environmental
contaminant. In: Proceedings of the Central States Association
of Food and Drug Officials Spring Meeting, Mason, Ohio, 4-5
May 1977, pp. 65-71.
HOLMBERG, B., JENSEN, S., LARSSON, A., LEWANDER, K., & OLSSON,
M. (1972) Metabolic effects of technical pentachlorophenol
(PCP) on the eel Anguilla anguilla L. Comp. Biochem. Physiol.,
43B: 171-183.
HOOS, R.A.W. (1978) Patterns of pentachlorophenol usage in
Canada - an overview. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 3-11.
HUANG, J.-C. & GLOYNA, E.F. (1968) Effect of organic
compounds on photosynthetic oxygenation. I. Chlorophyll
destruction and suppression of photosynthetic oxygen
production. Water Res., 2: 347-366.
HUBER, W., SCHUBERT, V., & SAUTTER, C. (1982) Effects of
pentachlorophenol on the metabolism of the aquatic macrophyte
Lemna minor. Environ. Pollut. A, 29: 215-224.
HUCKINS, J.N. & PETTY, J.D. (1983) Dynamics of purified and
industrial pentachlorophenol in fathead minnows. Arch.
environ. Contam. Toxicol., 12: 667-672.
HUECK, H.J. & LABRIJN, J. (1960) [Mold protection of cotton
with pentachlorophenol and laurylpentachlorophenol.] Textil-
Rundschau, 15: 467-472 (in German).
HUGHES, B.J., FORSELL, J.H., SLEIGHT, S.D., KUD, S., & SHULL,
L.R. (1985) Assessment of pentachlorophenol toxicity in
newborn calves: clinicopathology and tissue residues. J. anim.
Sci., 61(6): 1587-1603.
HUNTER, G.W., FREYTAG, R.E., RITCHIE, L.S., PAN, C., YOKOGAWA,
M., & POTTS, D.E. (1952) Studies on schistosomiasis. VI.
Control of the snail host of schistosomiasis in Japan with
sodium pentachlorophenate (Santobrite). Am. J. trop. Med.
Hyg., 1: 831-847.
HUTZINGER, O., FREI, R.W., MERIAN, E., & POCCHIARI, F.
(1982) Chlorinated dioxins and related compounds impact on
the environment. In: Proceedings of a Workshop, Rome, 22-24
October, 1980, Oxford, New York, Toronto, Sydney, Paris,
Frankfurt, Pergamon Press, Vol. 5, 658 pp (Pergamon Series on
Environmental Science).
IARC (1977) Some fumigants, the herbicides 2,4-D and 2,4,5-T,
chlorinated dibenzodioxins and miscellaneous industrial
chemicals, Lyons, International Agency for Research on Cancer,
pp. 111-138 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 15).
IARC (1979a) Pentachlorophenol. In: Some halogenated
hydrocarbons, Lyons, International Agency for Research on
Cancer, pp. 303-325 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 20).
IARC (1979b) Hexachlorobenzene. In: Some halogenated
hydrocarbons, Lyons, International Agency for Research on
Cancer, pp. 155-178 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 20).
IDE, A., NIKI, J., SAKAMOTO, F., WATANABE, J., & WATANABE, H.
(1972) Decomposition of pentachlorophenol in paddy soil.
Agric. biol. Chem., 36: 1937-1944.
IFEADI, C.N. (1975) Screening study to develop background
information and determine the significance of air contaminant
emissions from pesticide plants, Washington DC, US EPA Office
of Pesticide Programes (EPA Report No. PB-244734).
IMAI, K., ASANO, A., & SATO, R. (1967) Oxidative phosphory-
lation in Micrococcus denitrificans. I. Preparation and
properties of phosphorylating membrane fragments. Biochim.
Biophys. Acta, 143: 462-476.
INGLIS, A. & DAVIS, E.L. (1972) Effects of water hardness on
the toxicity of several organic and inorganic herbicides to
fish. US Bur. Sport Fish. Wildl. tech. Pap., 67: 1-22.
INGOLS, R.S. & GAFFNEY, P.E. (1965) Biological studies of
halophenols. Proc. Soc. Water Resour. Pollut. Control Conf.,
14: 175-181.
INGOLS, R.S., GAFFNEY, P.E., & STEVENSON, P.C. (1966)
Biological activity of halophenols. J. Water Pollut. Control
Fed., 38: 629-635.
INGRAM, L.L., MCGINNIS, G.D., & GJOVIK, L.R. (1981a) The
relative amount of pentachlorophenol volatilization from
treated wood. In: Proceedings of the American Wood Preservers'
Association Annual Meeting, April 27-29, Hyatt Orlando,
Florida, Florida, Kissimmee, pp. 102-108.
INGRAM, L.L., MCGINNIS, G.D., JASPERSE, G., & GJOVIK, L.R.
(1981b) The effect of solvent systems on the volatilization
of pentachlorophenol from treated wood. In: Proceedings of the
American Wood Preservers' Association Annual Meeting April
27-29, Hyatt Orlando, Florida, Florida, Kissimmee, pp. 105-117.
INNES, J.R.M., ULLAND, B.M., VALERIO, M.G., PETRUCELLI, L.,
FISHBEIN, L., HART, E.R., PALLOTTA, A.J., BATES, R.R., FALK,
H.L., GART, J.J., KLEIN, M., MITCHELL, I., & PETERS, J.
(1969) Bioassay of pesticides and industrial chemicals for
tumourigenicity in mice: a preliminary note. J. Natl Cancer
Inst., 42: 1101-1114.
IRPTC (1983) Data profile on pentachlorophenol, Geneva,
International Register of Potentially Toxic Chemicals, United
Nations Environment Programme.
IRPTC (1984) Scientific review of Soviet literature on
toxicity and hazards of chemicals. Vol. 75. Pentachlorophenol,
Moscow, Centre of International Projects (GKNT, no. 75).
ISHIZAWA, S., TOYODA, H., & MATSUGUCHI, T. (1961) Effects of
DD, EDB, and PCP upon microorganisms and their activities in
soil. Part I. Effects on microflora. Soil Plant Food Tokyo, 6:
145-155.
IVANOV, Z. & MAGEE, R.J., (1980) The determination of trace
amounts of chlorophenols by high-performance liquid chromato-
graphy. Microchem. J., 25: 543-547.
IZAKI, K., TAKAHASHI, M., SATO, Y, SASAGAWA, Y., SATO, K., &
FURUSAKA, C. (1981) Some properties of pentachlorophenol-
resistant gram-negative bacteria. Agric. biol. Chem., 45:
765-767.
JAKOBSON, I. & YLLNER, S. (1971) Metabolism of 14C-
pentachlorophenol in the mouse. Acta pharmacol. toxicol., 29:
513-524.
JANKE, D. & FRITSCHE, W. (1978) [Microbial dechlorination of
pesticides and other xenobiotica.] Z. Allerg. Mikrobiol., 18:
365-382 (in German).
JANSSENS, J.J. & SCHEPENS, P.J.C. (1984) Chronic penta-
chlorophenol intoxication as a result of the usage of wood
protectants. Meded. Fac. Landbouwwet., 49: 1175-1184.
JANSSON, B., SUNDSTROM, G., & AHLING, B. (1978) Formation of
polychlorinated dibenzo- p-dioxins during combustion of chloro-
phenol formulations. Sci. total Environ., 10: 209-217.
JAYAWEERA, R., PETERSEN, R., & SMEJTEK, P. (1982) Induced
hydrogen ion transport in lipid membranes as origin of toxic
effect of pentachlorophenol in an alga. Pestic. Biochem.
Physiol., 18: 197-204.
JENSEN, S. & RENBERG, L. (1972) Contaminants in penta-
chlorophenol: chlorinated dioxins and predioxins (chlorinated
hydroxy-diphenylethers). Ambio, 1: 62-65.
JENSEN, S. & RENBERG, L. (1973) Chlorinated dimers present
in several technical chlorophenols used as fungicides.
Environ. Health Perspect., 5: 37-39.
JIRASEK, L., KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS,
E. (1974) [Acne chlorina, porphyria cutanea tarda, and other
manifestations of general poisoning during the manufacture of
herbicides. II.] Cesk. Dermatol., 49: 145-157 (in Czech with
English summary).
JOHNSON, R.D. & MANSKE, D.D. (1977) Pesticides in food and
feed. Pesticide and other chemical residues in total diet
samples (XI). Pestic. monit. J., 11: 116-131.
JOHNSON, R.L., GEHRING, P.J., KOCIBA, R.J., & SCHWETZ, B.A.
(1973) Chlorinated dibenzodioxins and pentachlorophenol.
Environ. Health. Perspect., 5: 171-175.
JONES, P.A. (1981) Chlorophenols and their impurities in the
Canadian environment, Ottawa, Environment Canada, 434 pp
(Report No. EPS 3-EC-81-2)
JONES, P.A. (1984) Chlorophenols and their impurities in the
Canadian environment: 1983 supplement, Ottawa, Environment
Canada, 93 pp (Report No. EPS 3-Ep-84-3).
JUHL, U., WITTE, I., & BUTTE, W. (1985) Metabolism of
pentachlorophenol to tetrachlorohydroquinone by human liver
homogenate. Bull. environ. Contam. Toxicol., 35: 596-601.
KAILA, K. & SAARIKOSKI, J. (1977) Toxicity of pentachloro-
phenol and 2,3,6-trichlorophenol to the crayfish ( Astacus
fluviatilis L.). Environ. Pollut., 12: 119-123.
KAISER, K.L.E. & VALDMANIS, I. (1982) Apparent octanol/water
partition coefficients of pentachlorophenol as a function of
pH. Can. J. Chem., 60: 2104-2106.
KALMAN, D.A. & HORSTMAN, S.W. (1983) Persistence of tetra-
chlorophenol and pentachlorophenol in exposed woodworkers. J.
Toxicol. clin. Toxicol., 20: 343-352.
KARASEK, F.W. & HUTZINGER, O. (1986) Dioxin danger from
garbage incineration. Anal. Chem., 58: 633A-642A.
KAUFMAN, D.D. (1976) Phenols. In: Kearney, P.C. & Kaufman,
D.D., ed. Chemistry, degradation, and mode of action, New
York, Marcel Dekker, Vol. 2, pp. 665-707.
KAUFMAN, D.D. (1978) Degradation of pentachlorophenol in
soil, and by soil microorganisms. In: Rao, K.R., ed. Penta-
chlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 27-39.
KAUPPINEN, T. & LINDROOS, L. (1985) Chlorophenol exposure in
saw mills. Am. Ind. Hyg. Assoc. J., 46: 34-38.
KEHOE, R.A., DEICHMANN-GRUEBLER, W., & KITZMILLER, K-V.
(1939) Toxic effects upon rabbits of pentachlorophenol and
sodium pentachlorophenate. J. ind. Hyg. Toxicol. 21(5):
160-172.
KENAGA, E.E. (1972) Guidelines for environmental study of
pesticides: determination of bioconcentration potential.
Residue Rev., 44: 73-113.
KERKVLIET, N.I., BAECHER-STEPPAN, L., CLAYCOMB, A.T., CRAIG,
A.M., & SHEGGEBY, G.G. (1982a) Immunotoxicity of technical
pentachlorophenol (PCP-T) depressed humoral immune response
to T-dependent and T-independent antigen stimulation in PCP-T
exposed mice. Fundam. appl. Toxicol., 2:: 90-99.
KERKVLIET, N.I., BAECHER-STEPPAN, L., & SCHMITZ, J.A.
(1982b) Immunotoxicity of pentachlorophenol (PCP): increased
susceptibility to tumour growth in adult mice fed technical
PCP-contaminated diets. Toxicol. appl. Pharmacol., 62: 55-64.
KIMBROUGH, R.D. & LINDER, R.E. (1978) The effect of
technical and purified pentachlorophenol on the rat liver.
Toxicol. appl. Pharmacol., 46: 151-162.
KINZELL, J.H., AMES, N.K., SLEIGHT, S.D., KREHBIEL, J.D., KUO,
C., ZABIK, M.J., & SHULL, L.R. (1981) Subchronic administra-
tion of technical pentachlorophenol to lactating dairy cattle:
performance, general health, and pathologic changes. J. dairy
Sci. 64: 42-51.
KIRSCH, E.J. & ETZEL, J.E. (1973) Microbial decomposition of
pentachlorophenol. J. Water Pollut. Control Fed., 45: 359-364.
KLEMMER, H.W. (1972) Human health and pesticides - community
pesticide studies. Residue Rev., 41: 55-63.
KLEMMER, H.W, WONG, L., SATO, M.M., REICHERT, E.L., KORSAK,
R.J., & RASHAD, M.N. (1980) Clinical findings in workers
exposed to pentachlorophenol. Arch. environ. Contam. Toxicol.,
9(6): 715-725.
KLOPFFER, W., KAUFMANN, G., RIPPEN, G., & POREMSKI, H.-J.
(1982) A laboratory method for testing the volatility from
aqueous solution: first results and comparison with theory.
Ecotoxicol. environ. Saf., 6: 545-559.
KNOWLTON, M.F. & HUCKINS, J.N. (1983) Fate of radiolabelled
sodium pentachlorophenate in littoral microcosms. Bull.
environ. Contam. Toxicol., 30: 206-213.
KNUDSEN, I., VERSCHUUREN, H.G., DEN TONKELAAR, E.M., KROES,
R., & HELLEMAN, P.F.W. (1974) Short-term toxicity of penta-
chlorophenol in rats. Toxicology, 2: 141-152.
KOBAYASHI, K. (1978) Metabolism of pentachlorophenol in
fishes. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 89-105.
KOBAYASHI, K. (1979) Metabolism of pentachlorophenol in
fish. In: Khan, M.A.Q., Lech, J.J., & Menn, J.J., ed.
Pesticide and xenobiotic metabolism in aquatic organisms,
Washington DC, Americal Chemical Society, Vol. 99, pp. 131-143
(ACS Symposium Series).
KOBAYASHI, K. & AKITAKE, H. (1975a) Studies on the meta-
bolism of chlorophenols in fish. I. Absorption and excretion
of PCP by goldfish. Bull. Jpn. Soc. Sci. Fish., 41: 87-92.
KOBAYASHI, K. & AKITAKE, H. (1975b) Studies on the meta-
bolism of chlorophenols in fish. II. Turnover of absorbed PCP
in goldfish. Bull. Jpn. Soc. Sci. Fish., 41: 93-99.
KOBAYASHI, K. & NAKAMURA, N. (1979a) Isolation and
identification of a conjugated PCP excreted in the urine of
goldfish. Bull. Jpn. Soc. Sci. Fish., 45: 1001-1004.
KOBAYASHI, K. & NAKAMURA, N. (1979b) Major detoxification
pathways for pentachlorophenol in goldfish. Bull. Jpn. Soc.
Sci. Fish., 45: 1185-1188.
KOCIBA, R.J. & SCHWETZ, B.A. (1982) A review of the toxicity
of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) with a
comparison of the toxicity of other chlorinated dioxin
isomers. Assoc. Food Drug Off. Q. Bull., 46: 168-188.
KOCIBA, R.J., GEHRING, P.J., MCCOLLISTER, S.B., WADE, C.E.,
LISOWE, R.W., & MEINECKE, B. (1971) Results of 90-day
toxicological study in male rats maintained on diets
containing production grade or "purified" pentachlorophenol,
Midland, Michigan, Dow Chemical Company, 23 pp.
KOEGEL, W., MUELLER, W.F., COULSTON, F., & KORTE, F. (1979)
Biotransformation of pentachloronitrobenzene-14C in rhesus
monkeys after single and chronic oral administration.
Chemosphere, 8: 97-105.
KONASEWICH, D.E., HENNING, F.A., WILE, K.H., & GERENCHER, E.
(1983) Chlorophenate wood protection, Canada, British
Columbia Ministry of Environment (ISBN O-7719-9371-4).
KOPPE, P., DIETZ, F., TRAUD, J., & RUEBELT, C. (1977)
[Detection and photometric determination of 126 phenolic
compounds in water using four group-specific reagents.] Z.
anal. Chem., 285: 1-19 (in German).
KOSS, G. & KORANSKY, W. (1978) Pentachlorophenol in
different species of vertebrates after administration of
hexachlorobenzene and pentachlorobenzene. In: Rao, R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 131-137.
KOTZIAS, D., KLEIN, W., & KORTE, F. (1975) [Occurrence of
xenobiotica in seeping waters of landfills.] Chemosphere, 5:
301-306 (in German).
KOZAK, V.P., SIMSIMAN, G.V., CHESTERS, G. STENSBY, D., &
HARKIN, J. (1979) Reviews of the environmental effects of
pollutants: XI. Chlorophenols, Washington DC, US Environmental
Protection Agency, 492 pp (EPA Report 600/1-79-012).
KRAUSE, CHR. (1982) [Active ingredients of wood preserva-
tives used in homes.] In: Aurand, K., Seifert, B., & Wegner,
J., ed. [Air quality in interiors,] Stuttgart, New York,
Gustav-Fischer-Verlag, pp. 309-316 (in German).
KRAUSE, CHR. & ENGLERT, N. (1980) [Health evaluation of PCP
(pentachlorophenol) containing wood preservatives in rooms.]
Holz Roh Werkst., 38: 429-432 (in German).
KROYER, G., WASHUTTL, J., SCHOERGMEYER, W., STEINER, I., &
WINKER, N. (1982) [Contamination of food with volatile
biocide substances from wood preservatives in model
experiments.] Lebensm.-Wissen. Technol., 15: 111-112 (in
German).
KUEHL, D.W. & DOUGHERTY, R.C. (1980) Pentachlorophenol in
the environment. Evidence for its origin from commercial
pentachlorophenol by negative chemical ionization mass
spectrometry. Environ. Sci. Technol., 14: 447-449.
KUNDE, M. (1982) [Experience in the evaluation of wood
preservatives.] In: Aurand, K., Seifert, B., & Wegner, J., ed.
[Air quality in interiors,] Stuttgart, New York, Gustav-
Fischer-Verlag, pp. 317-325 (in German).
KUNDE, M, & BOHME, C. (1978) [Toxicology of pentachloro-
phenol - an overview]. Bundesgesundheitsblatt. 21: 302-310 (in
German).
KUTZ, F.W., MURPHY, R.S., & STRASSMAN, S.C. (1978) Survey of
pesticide residues and their metabolites in urine from the
general population. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 363-369.
KUWAHARA, M., KATO, N., & MUNAKATA, K. (1966a) The photo-
chemical reaction of pentachlorophenol. Part I. The structure
of the yellow compound. Agric. Biol. Chem. Jpn, 30: 232-238.
KUWAHARA, M., KATO, N., & MUNAKATA K. (1966b) The photo-
chemical reaction of pentachlorophenol. Part II. The chemical
structure of minor products. Agric. biol. Chem. Jpn, 30:
239-245.
KUWATSUKA, S. (1972) Degradation of several herbicides under
different conditions. In: Matsumura, F., Boush, G., & Misato,
T., ed. Environmental toxicology of pesticides, New York,
Academic Press, pp. 385-400.
KUWATSUKA, S. & IGARASHI, M. (1975) Degradation of PCP in
soils. II. The relationship between the degradation of PCP and
the properties of soils, and the identification of the
degradation products of PCP. Soil Sci. Plant Nutr., 21:
405-414.
LAHANIATIS, E.S., CLAUSEN, E., BIENIEK, D., & KORTE, F.
(1985) Formation of 2,3,7,8-tetrachlorodibenzofuran during
thermolysis of selected chlorinated organic compounds.
Chemosphere, 14: 233-238.
LAMBERTON, J., GRIFFIN, B., ARBOGAST, B., INMAN, R., DEINZER,
M., & GRIFFIN, D. (1979) The determination of polychloro-
dibenzo- p-dioxins in pentachlorophenol and wood treatment
solutions. Am. Ind. Hyg. Assoc. J., 40: 816-822.
LAMPARSKI, L.L., MAHLE, N.H., & SHADOFF, L.A. (1978)
Determination of pentachlorophenol, hexachlorodibenzo- p-
dioxin, and octachlorodibenzo- p-dioxin in bovine milk. J.
agric. food Chem., 26: 1113-1116.
LAMPARSKI, L.L., STEHL, R.H., & JOHNSON, R.L. (1980)
Photolysis of pentachlorophenol-treated wood. Chlorinated
dibenzo- p-dioxin formation. Environ. Sci. Technol., 14,
196-200.
LANGEBARTELS, C. & HARMS, H. (1984) Metabolism of penta-
chlorophenol in cell suspension cultures of soybean and wheat:
pentachlorophenol glucoside formation. Z. Pflanzenphysiol.,
113: 201-212.
LANGER, H.G., BRADY, T.P., DALTON, L.A., SHANNON, T.W., &
BRIGGS, P.R. (1973) Thermal chemistry of chlorinated
phenols. In: Blair, E.H., ed. Chlorodioxins: origin and fate,
Washington DC, American Chemical Society, pp. 26-32 (Advances
in Chemistry Series No. 120).
LANGEVELD, VAN, H.E.A.M. (1975) Determination of penta-
chlorophenol in toy paints. J. Assoc. Off. Anal. Chem., 58:
19-22.
LARSEN, R.V., BORN, G.S., KESSLER, W.V., SHAW, S.M., & VAN
SICKLE, D.C. (1975) Placental transfer and teratology of
pentachlorophenol in rats. Environ. Lett., 10: 121-128.
LARSEN, R.V., KIRSCH, L.E., SHAW, S.M., CHRISTIAN, J.E., &
BORN, G.S. (1972) Excretion and tissue distribution of
uniformly labelled 14C-pentachlorophenol in rats. J. Pharm.
Sci., 61: 2004-2006.
LAUWERYS, R. (1970) Biological criteria for selected
industrial toxic chemicals: a review. Scand. J. Work. environ.
Health, 1: 139-172.
LEE, B.E., LACROIX, M.D., DUPONT, G.A.J., & SCOTT, J.A.
(1984) Capillary gas chromatographic determination of
pentachlorophenol residues in tallow following automated gel
permeation clean-up. J. Assoc. Off. Anal. Chem., 67: 546-548.
LEIGHTY, E.G. & FENTIMAN, A.F., Jr (1982) Conjugation of
pentachlorphenol to palmitic acid by liver microsomes. Bull.
environ. Contam. Toxicol., 28(3): 329-333.
LEVIN, J.-O. & NILSSON, C.-A. (1977) Chromatographic
determination of polychlorinated phenols, phenoxyphenols,
dibenzofurans, and dibenzodioxins in wood-dust from worker
environments. Chemosphere, 6: 443-448.
LEVIN, J.-O., RAPPE, C., & NILSSON, C. (1976) Use of
chlorophenols as fungicides in sawmills. Scand. J. Work
environ. Health, 2: 71-81.
LILIENBLUM, W. (1985) Formation of pentachlorophenol
glucuronide in rat and human liver microsomes. Biochem.
Pharmacol., 34: 893-894.
LIU, D., THOMSON, K., & STRACHAN, W.M.J. (1981) Biodegra-
dation of pentachlorophenol in a simulated aquatic environ-
ment. Bull. environ. Contam. Toxicol., 26: 85-90.
LIU, D., THOMSON, K., & KAISER, K.L.E. (1982) Quantitative
structure-toxicity relationship of halogenated phenols on
bacteria. Bull. environ. Contam. Toxicol., 29: 130-136.
LOWER, M. (1982) [Studies on the pentachlorophenol content
of human organs.] Friedrich-Alexander- University Erlangen-
Nürnberg (Thesis) (in German).
LORES, E.M., EDGERTON, T.R., & MOSEMAN, R.F. (1981) Method
for the confirmation of chlorophenols in human urine by LC
with an electrochemical detector. J. chromatogr. Sci., 19,
466-469.
LU, P.Y. & METCALF, R.L. (1975) Environmental fate and
biodegradability of benzene derivatives as studied in a model
aquatic ecosystem. Environ. Health Perspect., 10: 269-284.
LU, P.Y., METCALF, R.L., & COLE, L.K. (1978) The environ-
mental fate of 14C-pentachlorophenol in laboratory model
ecosystems. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 53-63.
LYR, H. (1962) [On the oxidative decomposition of
chlorinated phenols.] Holztechnologie, 3: 201-208 (in German).
LYR, H. (1963) [Enzymatic detoxification of chlorinated
phenols.] Phytopathol. Z., 47: 73-83 (in German).
MCCONNELL, E.E., MOORE, J.A., HASEMANN, J.K., & HARRIS, M.W.
(1978) The comparative toxicity of chlorinated dibenzo- p-
dioxins in mice and guinea-pigs. Toxicol. appl. Pharmacol.,
44: 335-356.
MCCONNELL, E.E., MOORE, J.A., GUPTA, B.N., RAKES, A.H.,
LUSTER, M.I., GOLDSTEIN, J.A., HASEMAN, J.K., & PARKER, C.E.
(1980) The chronic toxicity of technical and analytical
pentachlorophenol in cattle. I. Clinicopathology. Toxicol.
appl. Pharmacol., 52: 468-490.
MCGAVACK, T.H., BOYD, L.J., PICCIONE, F.V., & TERRANOVA, R.
(1941) Acute and chronic intoxications with sodium penta-
chlorophenate in rabbits. J. ind. Hyg. Toxicol., 23(6):
239-251.
MCGOVERN, J.J. (1982) Apparent immunotoxic response to
phenolic compounds. Food Chem. Toxicol., 20: 496.
MANSKE, D.D. & JOHNSON, R.D. (1977) Residues in food and
feed. Pesticide and other chemical residues in total diet
samples. X. Pest. monit. J., 10: 134-148.
MASON, M.F., WALLACE, S.M., FOERSTER, E., & DRUMMOND, W.
(1965) Pentachlorophenol poisoning: report of two cases. J.
Forensic Sci., 10: 136-147.
MASUDA, Y. & KUROKI, H. (1982) Polychlorinated dibenzofurans
and related compounds in patients with "Yusho". In: Hutzinger,
O., Frei, R.W., Merian, E., & Pocchiari, F., ed. Chlorinated
dioxins and related compounds: impact on the environment,
Oxford, Pergamon Press, Vol. 5, pp. 561-570 (Pergamon Series
on Environmental Science).
MATSUMOTO, G. (1982) Comparative study on organic
constituents in polluted and unpolluted inland aquatic
environments. III. Phenols and aromatic acids in polluted and
unpolluted waters. Water Res., 16: 551-557.
MATSUMOTO, G., ISHIWATARI, R., & HANYA, T. (1977) Gas
chromatographic-mass spectrometric identification of phenols
and aromatic acids in river waters. Water Res., 11: 693-698.
MATTSON, V.R., ARTHUR, J.W., & WALBRIDGE, C.T. (1976) Acute
toxicity of selected organic compounds to fathead minnows,
Duluth, Minnesota, US Environmental Protection Agency, 13 pp
(Ecological Research Series No. EPA 600/3-76-097).
MEEMKEN, H.-A., FUERST, P., & HABERSAAT, K. (1982) [Analysis
of pentachlorophenol in cultured champignons.] Dtsch. Lebensm.
Rundsch. 78: 282-287 (in German).
MEHENDALE, H.W., FIELDS, M., & MATTHEWS, H.B. (1975) Meta-
bolism and effects of hexachlorobenzene on hepatic microsomal
enzymes in the rat. J. agric. food Chem., 23: 261-264.
MELNIKOV, N.N. (1971) Chemistry of pesticides. Residue Rev.,
36: 104-107.
MENON, J.A. (1958) Tropical hazards associated with the use
of pentachlorophenol. Br. med. J., 11: 1156-1158.
MERCIER, M. (1981) Criteria (dose/effect relationships) for
organochlorine pesticides, Oxford, Pergamon Press, 381 pp.
MERZ, V. & WEITH, W. (1872) [On the characteristics of
pentachlorophenol.] Ber. Dtsch. Chem. Ges., 5: 458-463 (in
German).
MILLER, C.S. & ABOUL-ELA, M.M. (1969) Fate of penta-
chlorophenol in cotton. J. agric. food Chem., 17: 1244-1246.
MITSUDA, H., MURAKAMI, K., & KAWAI, F. (1963) Effect of
chlorophenol analogues on the oxidative phosphorylation in rat
liver mitochondria. Agric. biol. Chem., 27: 366-372.
MORGADE, C., BARQUET, A., & PFAFFENBERGER, C.D. (1980)
Determination of polyhalogenated phenolic compounds in
drinking-water, human blood-serum, and adipose tissue. Bull.
environ. Contam. Toxicol., 24: 257-264.
MORGAN, J.W.W. & PURSLOW, D.F. (1973) Volatile losses of
wood preservatives. In: Proceedings of the 23rd Annual
Convention of the BWPA, British Wood Preservers Association,
pp. 173-193.
MOOS, L.P., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L.
(1983) Pentachlorophenol biodegradation. I. Aerobic. Water
Res., 17: 1575-1584.
MULLER, W.F. (1981) [Metabolism and pharmacokinetics of
selected chemicals in rhesus monkey and chimpanzee,] Society
for Radiation and Environmental Research (GSF), Münich, pp.
120-128 (Report ö/599) (in German).
MUNAKATA, K. & KUWAHARA, M. (1969) Photochemical degradation
products of pentachlorophenol. Residue Rev., 25: 13-23.
MUNDY, D.E. & MACHIN, A.F. (1981) Determination of penta-
chlorophenol and related compounds in animal materials by
high-performance liquid chromatography and gas chromatography.
J. Chromatogr., 216: 229-238.
MUNRO, I.B., OSTLER, D.C., MACHIN, A.F., & QUICK, M.P. (1977)
Suspected poisoning by pentachlorophenol in sawdust. Vet.
Rec., 101(26/27): 525.
MURRAY, H.E., RAY, L.E., & GIAM, C.S. (1981) Analysis of
marine sediment, water, and biota for selected organic
pollutants. Chemosphere, 10: 1327-1334.
MURTHY, N.B.K. & KAUFMAN, D.D. (1978) Degradation of penta-
chloronitrobenzene (PCNB) in anaerobic soils. J. agric. food
Chem., 26: 1151-1156.
MURTHY, N.B.K., KAUFMAN, D.D., & FRIES, G.F. (1979) Degra-
dation of pentachlorophenol (PCP) in aerobic and anaerobic
soil. J. environ. Sci. Health, B14: 1-14.
NARANG, A.S., VERNOY, C.A., & EADON, G.A. (1983) Evaluation
of Nielsen-Kryger steam distillation technique for recovery of
phenols from soil. J. Assoc. Off. Anal. Chem., 66: 1330-1334.
NCI (1979) Bioassay of 2,4,6-trichlorophenol for possible
carcinogenicity, Bethesda, Maryland, National Cancer
Institute, 115 pp (Technical Report No. PC A06/MF, PB
293-7700).
NCI (1980a) Bioassay of a mixture of 1,2,3,6,7,8-hexachloro-
benzo-p-dioxin and 1,2,3,7,8,9-hexachlorodibenzo-p-dioxin
(gavage) for possible carcinogenicity, Bethesda, Maryland,
National Cancer Institute (Carcinogenesis Technical Report
Series No. 198) (US Department of Health and Human Services
Publication No. (NIH) 80-1754).
NCI (1980b) Bioassay of a mixture of 1,2,3,6,7,8-hexachloro-
dibenzo-p-dioxin and 1,2,3,7,8,9-hexachlorodibenzo-p-dioxin
(dermal) for possible carcinogenicity, Bethesda, Maryland,
National Cancer Institute (Carcinogenesis Technical Report
Series No. 202) (US Department of Health and Human Services
Publication No. (NIH) 80-1758).
NEEDHAM, L.L., CLINE, R.E., HEAD, S.L., & LIDDLE, J.A.
(1981) Determining pentachlorophenol in body fluids by gas
chromatography after acetylation. J. anal. Toxicol., 5:
283-286.
NEIDERT, E., SASCHENBRECKER, P.W., & PATTERSON, J.R. (1984)
Detection and occurrence of pentachlorophenol residues in
chicken liver and fat. J. environ. Sci. Health B, 19: 579-592.
NIIMI, A.J. & MCFADDEN, L.A. (1982) Uptake of sodium penta-
chlorophenate (NaPCP) from water by rainbow trout (Salmo
gairdneri) exposed to concentrations in the ng/litre range.
Bull. environ. Contam. Toxicol., 28(1): 11-19.
NILSSON, C.-A., NORSTROM, A., ANDERSSON, K., & RAPPE, C.
(1978) Impurities in commercial products related to penta-
chlorophenol. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 313-324.
NING, H.S. (1984) [Investigation of the chronic intoxication
of pentachlorophenol.] Chin. J. Ind. Hyg. Occup. Dis., 4:
24-28 (in Chinese).
NING, H.S., ZHEN, H.-Q., LI, L.-S., XU, H., WANG, J.-J., WU,
S.-S., LIANG, T.-Z., JAN, J.-Q., ZHU, B.-H., HUANG, W.-W.,
CHEN, W.-Y., & WANG, G.-F. (1984) [Study of the toxicity of
pentachlorophenol and recommendations of the maximum allowable
concentration in air.] J. Commun. ind. Hyg. (Rail Transp.
Syst.), 4: 7-16 (in Chinese).
NIOSH (1978) Manual of analytical methods, Cincinatti, Ohio,
US Department of Health, Education, and Welfare, Vol. 4, pp.
S297-1-S297-8 (DHEW Publication No. 78-175).
NIOSH (1983) In: Tatken, R.L. & Lewis, R.J., ed. Registry of
toxic effects of chemical substances, Cincinnati, Ohio, US
National Institute for Occupational Safety and Health, pp.
74-75.
NOAKES, D.N. & SANDERSON, D.M. (1969) A method for
determining the dermal toxicity of pesticides. Br. J. ind.
Med., 26: 59-64.
NOMURA, S. (1954) Studies on chlorophenol poisoning. I. A
clinical examination of workers exposed to pentachlorophenol.
Bull. Hyg., 29: 294.
NORSTROM, A., RAPPE, C., LINDAHL, R., & BUSER, H.-R. (1979)
Analysis of some older Scandinavian formulations of 2,4-
dichlorophenoxy acetic acid and 2,4,5-trichlorophenoxy acetic
acid for contents of chlorinated dibenzo- p-dioxins and
dibenzofurans. Scand. J. Work environ. Health, 5: 375-378.
NRC (1986) In: Thomas, R.D., ed. Drinking water and health,
Washington DC, National Research Council, National Academy
Press, Vol. 6, pp. 407-421.
NRCC (1981) Polychlorinated dibenzo-p-dioxins: criteria for
their effects on man and his environment, Ottawa, Ontario,
National Research Council of Canada, Associate Committee on
Scientific Criteria for Environmental Quality (No. 18574),
251 pp.
NRCC (1982) Chlorinated phenols: criteria for environmental
quality, Ottawa, Ontario, National Research Council of Canada,
Associate Committee on Scientific Criteria for Environmental
Quality, (No. 18578) 191 pp.
OHE, T. (1979) Pentachlorophenol residues in human adipose
tissue. Bull. environ. Contam. Toxicol., 22: 287-292.
OLIE, K., BERG, M.V.D., & HUTZINGER, O. (1983) Formation and
fate of PCDD and PCDF combustion processes. Chemosphere, 12:
627-636.
OWEN, J.W. & ROSSO, S.W. (1981) Effects of sublethal
concentrations of pentachlorophenol on the liver of bluegill
sunfish, Lepomis machrochirus. Bull. environ. Contam. Toxicol.,
26: 594-600.
PAASIVIRTA, J., SORKKO, J., LESKIJORVI, T., & ROOS, A.
(1980) Transportation and enrichment of chlorinated phenolic
compounds in different aquatic food chains. Chemosphere, 9:
441-456.
PAASIVIRTA, J., SORKKO, J., AHO, M., SURMA-AHO, K., TARHANEN,
J., & ROOS, A. (1981) Recent trends of biocides in pikes of
the lake Päijänne. Chemosphere, 10: 405-414.
PAASIVIRTA, J., SORKKO, J., SURMA-AHO, K., HUMPPI, T.,
KUOKKANEN, T., & MARTTINEN, M. (1983) Food chain enrichment
of organochlorine compounds and mercury in clean and polluted
lakes of Finland. Chemosphere, 12: 239-252.
PAASIVIRTA, J., HEINOLA, K., HUMPPI, T., KARJALAINEN, A.,
KNUUTINEN, J., MANTYKOSKI, K., PAUKKU, R., PIILOLA, T., SURMA-
AHO, K., TARHANEN, J., WELLING, L., & VIHONEN, H. (1985)
Polychlorinated phenols, guaiacols and catechols in the
environment. Chemosphere, 14: 469-491.
PACKHAM, E.D., DUXBURY, C.L., MAYFIELD, C.I., INNISS, W.E.,
KRUUV, J., & THOMPSON, J.E. (1982) Quantitative analysis of
pollutant-induced lethal and sublethal damage in cultured
mammalian cells. Bull. environ. Contam. Toxicol., 29: 739-746.
PALMER, C.M. & MALONEY, T.E. (1955) Preliminary screening
for potential algicides. Ohio J. Sci., 55: 1-8.
PARKER, C.E., JONES, W.A., MATTHEWS, H.B., MCCONNELL, E.E., &
HASS, J.R. (1980) The chronic toxicity of technical and
analytical pentachlorophenol in cattle. II. Chemical analysis
of tissues. Toxicol. appl. Pharmacol., 55: 359-369.
PARR, L.J., GEE, M.G., LAND, D.G., ROBINSON, D., & CURTIS,
R.F. (1974) Chlorophenols from wood preservatives in broiler
house litter. J. Sci. Food Agric., 25: 835-841.
PARRISH, P.R., DYAR, E.E., ENOS, J.M., & WILSON, W.G. (1978)
Chronic toxicity of chlordane, trifluralin, and pentachloro-
phenol to sheepshead minnows (Cyprinodon variegatus),
Washington DC, US Environmental Protection Agency, 67 pp
(Report No. EPA 600/3-78-010).
PAULI, O. & FRANKE, G. (1972) Behaviour and degradaton of
technical preservatives in the biological purification of
sewage. In: Walters, A.H. & Hueck-Van der Plas, E.H., ed.
Biodegradaton of materials. II, New York, John Wiley and Sons,
pp. 52-60.
PEARCE, N.E., SMITH, A.H., HOWARD, J.K., SHEPPARD, R.A.,
GILES, H.J., & TEAGUE, C.A. (1986) Non-Hodgkin's lymphoma
and exposure to phenoxyherbicides, chlorophenols, fencing
work, and meat works employment: a case control study. Br. J.
ind. Med., 43: 75-83.
PEARCE, N.E., SHEPPARD, R.A., SMITH, A.H., & TEAGUE, C.A. (in
press) Non-Hodgkin's lymphoma and farming: an expanded
case-control study. Int. J. Cancer.
PEKARI, L. & ANTERO, A. (1982) A simple liquid chromato-
graphic method for the analysis of penta- and tetrachloro-
phenols in urine of exposed workers. J. Chromatogr. biomed.
Appl., 232: 129-136.
PETROWITZ, H.J. (1981) [Volatilisation of the active
ingredients of wood preservatives from chemically protected
timber.] In: [Research report of the Expert Group on
Biological Materials Research,] Berlin, Federal Institute for
Materials Research, 32 pp (in German).
PIERCE, R.H., Jr & VICTOR, D.M. (1978) The fate of penta-
chlorophenol in an aquatic ecosystem. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 41-52.
PIERCE, R.H., BRENT, C.R., WILLIAMS, H.P., & REEVES, S.G.
(1977) Pentachlorophenol distribution in a fresh water
ecosystem. Bull. environ. Contam. Toxicol., 18: 251-258.
PIGNATELLO, J.J., MARTINSON, M.M., STEIERT, J.G., CARLSON,
R.E., & CRAWFORD, R.L. (1983) Biodegradation and photolysis
of pentachlorophenol in artificial freshwater streams. Appl.
environ. Microbiol., 46: 1024-1031.
PLESTINA, R. (1984) Prevention, diagnosis and treatment of
insecticide poisoning, Geneva, World Health Organization,
69@pp (unpublished report VBC/84.889).
PLIMMER, J.R. (1973) Technical pentachlorophenol: origin and
analysis of base-insoluble contaminants. Environ. Health
Perspect., 5: 41-48.
POWERS, P.W. (1976) How to dispose of toxic substances and
industrial wastes, Park Ridge, New Jersey, Noyes Data
Corporation, p. 373.
PRAGER, B., JACOBSON, P., SCHMIDT, P., & STERN, D., ed.
(1923) [Beilstein's handbook of organic chemistry.] 4th ed.,
Berlin, Springer-Verlag, Vol. 6, (Syst. No. 522) pp. 194-197
(in German).
PRESCOTT, C.A., WILKIE, B.N., HUNTER, B., & JULIAN, R.J.
(1982) Influence of a purified grade of pentachlorophenol on
the immune-response of chickens. Am. J. vet. Res., 43: 481-487.
PRUITT, G.W., GRANTHAM, B.J., & PIERCE, R.H. (1977)
Accumulation and elimination of pentachlorophenol by the
bluegill Lepomis macrochirus. Am. Fish. Soc., 106: 462-465.
QUICK, M.P. (1982) Pesticide poisoning of livestock: a
review of cases investigated. Vet. Rec., 111: 5-7.
RAHDE, A.F. & DELLA ROSA, H.V. (1984) [Evaluation of
ecotoxicological impact of the hydroelectric dam of Tucurui,
Brazil,] 45 pp (unpublished report presented to Eletronorta
(Eletrobras)) (in Portuguese).
RAHDE, A.F. & DELLA ROSA, H.V. (1986) Pentachlorophenol - an
evaluation of the ecotoxicological impact of the hydroelectric
dam of tucurui, Brazil, 6 pp (unpublished report presented to
Eletronorte (Eletrobras)).
RAPPE, C., GARA, A., & BUSER, H.R. (1978a) Identification of
polychlorinated dibenzofurans (PCDFs) in commercial chloro-
phenol formulations. Chemosphere, 12: 981-991.
RAPPE, C., MARKLUND, S., BUSER, H.R., & BOSSHARDT, H.-P.
(1978b) Formation of polychlorinated dibenzo- p-dioxins
(PCDDs) and dibenzofurans (PCDFs) by burning or heating
chlorophenates. Chemosphere, 3: 269-281.
RAPPE, C., BUSER, H.R., & BOSSHARDT, H.-P. (1979) Dioxins,
dibenzofurans, and other polyhalogenated aromatics: production,
use, formation, and destruction. Ann. NY Acad. Sci.,
320: 1-18
RAPPE, C., NYGREN, M., BUSER, H.R., & KAUPPINEN, T. (1982)
Occupational exposure to polychlorinated dioxins and dibenzo-
furans. In: Hutzinger, O. et al., ed. Chlorinated dioxins and
related compounds: impact on the environment, Oxford, Pergamon
Press, Vol. 5, (Pergamon Series on Environmental Science) pp.
495-514.
REINER, E.A., CHU, J.P., & KIRSCH, E.J. (1978) Microbial
metabolism of pentachlorophenol. In: Rao, K.R., ed. Penta-
chlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 67-81.
RENBERG, L. (1974) Ion exchange technique for the deter-
mination of chlorinated phenols and phenoxy acids in organic
tissue, soil, and water. Anal. Chem., 46: 459-461.
RENNER, G. & MUCKE, W. (1986) Transformations of penta-
chlorophenol. I. Metabolism in animals and man. Toxicol.
environ. Chem., 11: 9-29.
RENNER, G., HOPFER, C., & GOKEL, J.M. (1986) Acute
toxicities of pentachlorophenol, pentachloroanisole, tetra-
chlorohydroquinone, tetrachlorocatechol, tetrachloro-
resorcinol, terachlorodimethoxybenzene diacetates administered
to mice. Toxicol. environ. Chem., 11: 37-50.
RICHARDSON, N.G. (1978) Wood preserving effluents and their
treatment. In: Proceedings of the Technical Transfer Seminar
Timber Processing Industry, March 10-11 1977, Toronto,
Environment Canada, pp. 40-62 (Econ. Technical Review No. EPS
3-WP-78-1).
RIPPEN G. (1984) [Pentachlorophenol.] In: [Handbook of
environmental chemicals: physicochemical and ecotoxicological
data of selected chemicals,] Landsberg/Lech, Ecomed, pp. 1-13
(in German).
ROBERTS, H.J. (1981) Aplastic anemia due to pentachloro-
phenol. New Engl. J. Med., 305: 1650-1651.
ROBERTS, H.J., (1983) Aplastic anemia and red cell aplasia
due to pentachlorophenol. South. Med. J., 76(1): 45-48.
ROBSON, A.L., KISSANE, J.M., ELVICK, N.H., & PUNDAVELA, L.
(1969) Pentachlorophenol poisoning in a nursery for newborn
infants. I. Clinical features and treatment. J. Pediatr.,
75(2): 309-316.
ROSSKAMP, E. (1982) [Pentachlorophenol as a model case of a
xenobiotic compound: PCP in the environment.] Berlin, Federal
Health Office Institute for Water, Soil and Air Hygiene, 22 pp
(Research Report No. 10604007) (in German).
ROTT, B., NITZ, S., & KORTE, F. (1979) Microbial decompo-
sition of sodium pentachlorophenolate. J. agric. food Chem.,
27, 306-310.
ROWE, E.L., ZIOBRO, R.J., WANG, C.J.K., & DENCE, C.W. (1982)
The use of an alga Chlorella pyrenoidosa and a duckweed Lemna
perpusilla as test organisms for toxicity bioassays of spent
bleaching liquors and their components. Environ. Pollut. (Ser.
A), 27: 289-296.
ROY, P., KUNDU, P., & DAS, A. (1981) Sodium pentachloro-
phenate as a mutagenic agent. Biotechnol. Lett., 3: 401-404.
ROZMAN, K., MUELLER, W., COULSTON, F., & KORTE, F. (1977)
Long-term feeding study of hexachlorobenzene in rhesus
monkeys. Chemosphere, 2/3: 81-84.
RUBINSTEIN, N.I. (1978) Effects of sodium pentachlorophenate
on the feeding activity of the lugworm Arenicola cristata
Stimpson. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 175-179.
RUDLING, L. (1970) Determination of pentachlorophenol in
organic tissues and water. Water Res., 4: 533-537.
RUBELT, C., DIETZ, F., KICKUTH, R., KOPPE, P., KUNTE, H.,
PESCHEL, G., & SONNEBORN, M. (1982) [Pollutants in ambient
water. Vol. II. Phenols.] Boppard, Harald Boldt Verlag, 168 pp
(DFG Report) (in German).
RUMKER, VON, R., LAWLESS, E.W., & MEINERS, A.F. (1974)
Production, distribution, use, and environmental impact
potential of selected pesticides. Washington DC, US
Environmental Protection Agency, pp. 308-319 (EPA Production
Report No. EPA 540/1-74-001).
RUSINK, R.G. & SMITH, L.L. (1975) The relationship of the
96-hour LC50 to the lethal threshold concentration of
hexavalent chromium, phenol, and sodium pentachlorophenate for
fathead minnows ( Pimephales promelas Rafinesque). Trans. Am.
Fish. Soc., 104: 567-570.
RUH, C. & GEBEFUEGI, I. (1984) [Sources for indoor space
contamination: presence of pentachlorophenol and lindane in
untreated wood samples.] Chemosphere, 13: 919-925 (in German).
RUH, C., GEBEFUEGI, I., & KORTE, F. (1984) The indoor
biocide pollution: occurrence of pentachlorophenol and lindane
in homes. In: Berglund, B., Lindvall, T., & Sundell, J., ed.
Indoor air. IV. Chemical characterization and personal
exposure, Stockholm, Swedish Council for Building Research,
pp. 309-322.
RYAN, J.J. (1983) Higher chlorinated dioxins implicated in
the mortality of young pigs kept on a pentachlorophenol-
treated wooden floor. Can. vet. J., 24: 72-75.
SAARIKOSKI, I. & VILUKSELA, M. (1981) Influence of pH on the
toxicity of substituted phenols to fish. Arch. environ.
Contam. Toxicol., 10: 747-753.
SALKINOJA-SALONEN, M.S., HAKULINEN, R., VALO, R., &
APAJALAHTI, J. (1983) Biodegradation of recalcitrant
organochlorine compounds in fixed film reactors. Water Sci.
Tech., 15: 309-319.
SALKINOJA-SALONEN, M.S., VALO, R., APAJALAHTI, J., HAKULINEN,
R., SILAKOSKI, L., & JAAKKOLA, T. (1984) Biodegradation of
chlorophenolic compounds in wastes from wood-processing
industry. In: Klug, M. & Reddy, C., ed. Current perspectives
in microbial ecology, Washington DC, American Society for
Microbiology, pp. 668-676.
SANBORN, J.R., CHILDERS, W.F., & HANSEN, L.G. (1977) Uptake
and elimination of 14C-hexachlorobenzene (HCB) by the green
sunfish, Lepomis cyanellus Raf., after feeding contaminated
food. J. agric. food Chem., 25: 551-553.
SANDERMANN, H., Jr, SCHEEL, D., & V.D. TRENCK, T. (1984) Use
of plant cell cultures to study the metabolism of environ-
mental chemicals. Ecotoxicol. environ. Saf., 8: 167-182.
SANDERMANN, H.S., CASTEN, R., & STOCKMANN, H. (1957)
Pyrolysis of pentachlorophenol. Chem. Ber., 90: 690-692.
SANGSTER, B., WEGMAN, R.C.C., & HOFSTEE, A.W.M. (1982)
Non-occupational exposure to pentachlorophenol: clinical
findings and plasma-PCP-concentrations in three families. Hum.
Toxicol., 1: 123-133.
SAUR, J.M., WALCHESKI, P.J., NICHOLAS, D.-D., & GJOVIK, L.R.
(1982) The concentration of airborne pentachlorophenol within
treated wood structures. Proc. Ann. Meet. Am. Wood Preserv.
Assoc., 78: 169-173.
SAVOLAINEN, H. & PEKARI, K. (1979) Neurochemical effects of
peroral administration of technical pentachlorophenol. Res.
Commun. chem. Pathol. Pharmacol., 23(1): 97-105.
SCHAUERTE, W., LAY, J.P., KLEIN, W., & KORTE, F. (1982)
Influence of 2,4,6-trichlorophenol and pentachlorophenol on
the biota of aquatic systems. Outdoor experiments in
compartments of a natural pond. Chemosphere, 11: 71-79.
SCHIMMEL, S.C., PATRICK, J.M., Jr, & FAAS, L.F. (1978)
Effects of sodium pentachlorophenate on several estuarine
animals: toxicity, uptake and depuration. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 147-155.
SCHONHABER, R., SCHWARZ, A., & KRANL, D. (1982) Comparative
studies on the high-pressure liquid chromatographic and gas
chromatographic determination of pentachlorophenol in mush-
rooms. Dtsch. Lebensm. Rundsch., 78: 254-257.
SCHRAG., S.D. & DIXON, R.L. (1985) Occupational exposures
associated with male reproductive dysfunction. Ann Rev.
Pharmacol. Toxicol., 25: 567-592.
SCHUPPENER, H. (1974) [Uptake of pentachlorophenol (PCP) by
corn plants from culture media - a methodological study on
the ecological chemistry of lipophilic toxic compounds,]
University of Göttingen, 105 pp, (Dissertation) (in German).
SCHWETZ, B.A., NORRIS, J.M., SPARSCHU, G.L., ROWE, V.K.,
GEHRING, P.J., EMERSON, J.L., & GERBIG, C.G. (1973)
Toxicology of chlorinated dibenzo- p-dioxins. Environ. Health
Perspect., 5: 87-99.
SCHWETZ, B.A., KEELER, P.A., & GEHRING, P.J. (1974) The
effect of purified and commercial grade pentachlorophenol on
rat embryonal and fetal development. Toxicol. appl.
Pharmacol., 28: 151-161.
SCHWETZ, B.A., QUAST, I.F., KEELER, P.A., HUMISTON, C.G., &
KOCIBA, R.J. (1978) Results of two-year toxicity and
reproduction studies on pentachlorophenol in rats. In: Rao,
K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
301-309.
SHA, S.Z. & DUFFIELD, A.M. (1984) Negative ion chemical
ionization gas chromatography-mass spectrometry of some
derivatives of tri-, tetra-, and pentachlorophenols. J.
Chromatogr., 284: 157-166.
SHAFIK, T.M. (1973) The determination of pentachlorophenol
and hexachlorophene in human adipose tissue. Bull. environ.
Contam. Toxicol., 10: 57-63.
SHAFIK, T.M., SULLIVAN, H.C., & ENOS, H.R. (1973) Multi-
residue procedure for halo- and nitrophenols. Measurement of
exposure to biodegradable pesticides yielding these compounds
as metabolites. J. agric. food Chem., 21: 295-298.
SHEN, S., VILLENEUVE, D.C., CHU, I., KELLY, J., & GILMAN,
A.P. (1983) The acute dermal toxicity of tetrachlorophenols
in the rat. Bull. environ. Contam. Toxicol., 31: 680-685.
SHIRAKAWA, M., NAGATOSHI, H., & HIRAKAWI, M. (1959) Hygienic
investigation on workers dealing with pentachlorophenol (PCP)
in a rubber manufacturing industry. Kurume Med. J., 6(1):
24-35.
SIEGWART, Y. (1983) [Control of foodstuff in Switzerland in
the year 1982.] Mitt. Geb. Lebensm. Hyg., 74: 179-319 (in
German).
SIQUEIRA, M.E.P.B. & FERNICOLA, N.A.C.G. (1981) Determination
of pentachlorophenol in urine. Bull. environ. Contam.
Toxicol., 27: 380-385.
SIUDA, J.F. (1980) Natural production of organohalogens. In:
Jolley, R.L., Brungs, W.A., Cumming, R.B., & Jacobs, V.A., ed.
Water chlorination - environmental impact and health effects,
Ann Arbor, Michigan, Ann Arbor Science Publishers Inc, Vol. 3,
pp. 63-72.
SLOOF, W. & CANTON, J.H. (1983) Comparison of the
susceptibility of 11 freshwater species to 8 chemical
compounds. II. (Semi) chronic toxicity tests. Aquat. Toxicol.,
4: 271-282.
SMITH, A.H., PEARCE, N.F., FISHER, D.O., GILES, H.J., TEAGUE,
C.A., & HOWARD, J.K. (1984) Soft-tissue sarcoma and exposure
to phenoxyherbicides and chlorophenols in New Zealand. J. Natl
Cancer Inst., 73: 1111-1117.
SONNEBORN, M. (1976) A study to determine the comparability
of chemical analyses for drinking-water quality within
European communities, Luxembourg, Commission of European
Communities, 97 pp (EUR 5542e).
STANLAKE, G.J. & FINN, R.K. (1982) Isolation and character-
ization of a pentachlorophenol-degrading bacterium. Appl.
environ. Microbiol., 44: 1421-1427.
STEHL, R.H., PAPENFUSS, R.R., BREDEWEG, R.A., & ROBERTS, R.W.
(1973) The stability of pentachlorophenol and chlorinated
dioxins to sunlight, heat, and combustion. Adv. Chem. Ser.,
120: 119-125.
STERLING, T.D., STOFFMAN, L.D., STERLING, D.A., & MATE, G.
(1982) Health effects of chlorophenol wood preservatives on
sawmill workers. Int. J. Health Serv., 12: 559-571.
STEVENS, H.M. & RICHARDSON, A. (1979) The rapid screening of
body tissues for pentachlorophenol (PCP) with special
reference to poisoning fatality. J. Forensic Sci. Soc., 19:
125-129.
STIJVE, T. (1981) Determination of pentachlorophenol and
2,3,4,6-tetrachlorophenol in edible gelatins. Dtsch. Lebensm.
Rundsch., 77: 249-253.
STOCKDALE, M. & SELWYN, M.J. (1971a) Influence of ring
substituents on the action of phenols on some dehydrogenases,
phosphokinases, and the soluble ATPase from mitochondria. Eur.
J. Biochem., 21: 416-423.
STOCKDALE, M. & SELWYN, M.J. (1971b) Effects of ring
substituents on the activity of phenols as inhibitors and
uncouplers of mitochondrial respiration. Eur. J. Biochem., 21:
565-574.
STOHLMAN, E.F. (1951) The toxicity of some related halo-
genated derivatives of phenols, Bethesda, Maryland, US Public
Health Service, pp. 1303 (Public Health Reports No. 66).
STRANKS, D.W. (1976) Wood preservatives: their depletion as
fungicides and fate in the environment, Ottawa, Environment
Canada, Canadian Forestry Service, pp. 1-35 (Forestry
Technical Report No. 10).
STRETZ, L.A. & VAVRUSKA, J.S. (1984) Controlled air
incineration of pentachlorophenol-treated wood, Washington DC,
US Environmental Protection Agency, 4 pp (EPA-600/S2-84-089).
STRUFE, R. (1968) Problems and results of residue studies
after application of molluscicides. Residue Rev., 24: 80-168.
SU, Y.H. & LIN, H.C. (1971) Influence of soil physico-
chemical characteristics on the efficacy of herbicide penta-
chlorophenol. Chung. Kuo Nung Yeh Hua Hsueh Hui Chih, 8(3-4):
99-104 ( Chem. Abstr. 110707, 74: 301).
SUZUKI, T. (1977) Metabolism of pentachlorophenol by a soil
microbe. J. environ. Sci. Health, B12: 113-127.
SUZUKI, T. (1983) Metabolism of pentachlorophenol (PCP) by
soil microorganisms, Nippon Noyaku Gakkaishi, 8, 385-394.
TAGATZ, M.E., IVEY, I.M., MOORE, I.C., & TOBIA, M. (1977)
Effects of pentachlorophenol on the development of estuarine
communities. J. Toxicol. environ. Health, 3: 501-506.
TAGATZ, M.E., IVEY, I.M., & TOBIA, M. (1978) Effects of
Dowicide(R) G-ST on development of experimental estuarine
macrobenthic communities. In: Rao, K.R., ed. Pentachloro-
phenol: chemistry, pharmacology, and environmental toxicology,
New York, London, Plenum Press, pp. 157-163.
TAGATZ, M.E., IVEY, J.M., GREGORY, N.R., & OGLESBY, J.L.
(1981) Effects of pentachlorophenol on field- and
laboratory-developed estuarine benthic communities. Bull.
environ. Contam. Toxicol., 26: 137-143.
TAM, T.Y. & TREVORS, J.T. (1981a) Toxicity of pentachloro-
phenol to Azotobacter vinelandii. Bull. environ. Contam.
Toxicol., 27: 230-234.
TAM, T.Y. & TREVORS, J.T. (1981b) Effects of pentachloro-
phenol on asymbiotic nitrogen fixation in soil. Water Air Soil
Pollut., 16: 409-414.
THOMAS, P., CARR, R.S., & NEFF, J.M. (1981) Biochemical
stress responses of mullet Mugil cephalus and polychaete worms
Neanthes virens to pentachlorophenol. In: Vernberg, F.J.,
Calabrese, A., Thurnberg, F.P., & Vernberg, W.B., ed.
Biological monitoring of marine pollutants, New York, Academic
Press, pp. 73-103.
THOMPSON, G.E., HUSAIN, H., PARRY, J., WARDROP, W.L., &
GILBRIDE, P.J. (1978) Hydrogeological control and clean-up
of soil and groundwater contaminants at Northern Wood
Preservers, Ltd. In: Proceedings of the Ontario Industrial
Waste Conference, Toronto, June 18-21, Winnipeg, Manitoba,
Engineering Consultants, 18 pp.
THOMPSON, W.S. & DUST, J.V. (1971) Pollution control in the
wood preserving industry. I. Nature and scope of the problem.
For. prod. J., 21: 70-75.
TIERNAN, T.O., TAYLOR, M.L., GARRETT, J.H., VAN NESS, G.F.,
SOLCH, J.G., DEIS, D.A., & WAGEL, D.J. (1983) Chlorodibenzo-
dioxins, chlorodibenzofurans and related compounds in the
effluents from combustion processes. Chemosphere, 12: 595-606.
TIMMONS, L., STEELE, D., CANNON, M., GRESE, R., BROWN, R.,
MURRIL, E., & JAMESON, C.W. (1984) Identification of bromo-
tetrachlorophenol in commercial pentachlorophenol samples. J.
Chromatogr., 314: 476-481.
TING, H.H. & QUICK, M.P. (1980) Simple thin-layer chromato-
graphy method for detection of pentachlorophenol in sawdust
and woodshavings. J. Chromatogr., 195: 441-444.
TODD, A.S. & TIMBIE, C.Y. (1983) Industrial hygiene surveys
of occupational exposure to wood preservative chemicals,
Cincinatti, Ohio, US Department of Health, Education, and
Welfare, National Institute for Occupational Safety and Health.
TOLA, S., HANBERG, S., COLLAN, Y., LINDERBORG, H., & KORKALA,
M.L. (1980) A case-control study of the etiology of nasal
cancer in Finland. Int. Arch. occup. environ. Health, 46:
79-85.
TOLEDO, J.V., MONTEIRO DA SILVA, C.S., BULHOES, M.S., PAES
LEME, L.A., DA SILVA NETTO, J.A., & GILBERT, B. (1976) Snail
control in urban sites in Brazil with slow-release hexabutyl-
distannoxane and pentachlorophenol. Bull. World Health Organ.,
54: 421-425.
TREVORS, J.T. (1982a) Effect of temperature on the
degradation of pentachlorophenol by Pseudomonas spp.
Chemosphere, 11: 471-475.
TREVORS, J.T. (1982b) Differences in the sensitivity of
short-term bioassays. Bull. environ. Contam. Toxicol., 28:
655-659.
TREVORS, J.T. (1982c) Effect of pentachlorophenol on
electron transport system activity in soil. Bull. environ.
Contam. Toxicol., 29: 727-730.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1981a) A
rapid toxicity test using Pseudomonas fluorescens. Bull.
environ. Contam. Toxicol., 26: 433-439.
TREVORS, J.T., MAYFIELD, C.I., INNISS, W.E., & THOMPSON, J.E.
(1981b) Effect of phenolic antioxidants on the toxicity of
pentachlorophenol in short-term bacterial bioassays. Bull.
environ. Contam. Toxicol., 27: 433-439.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1982) Effect
of sequence of exposure to chlorophenols in short-term
bacterial bioassays. Arch. environ. Contam. Toxicol., 11:
203-207.
TRIEBIG, G., KREKELER, H., GOSSLER, K., & VALENTIN, H.
(1981) [Investigations into neurotoxicity of work-related
materials. II. Motor and sensory nerve conduction velocity in
pentachlorophenol-exposed persons.] Int. Arch. occup. environ.
Health, 48: 357-367 (in German).
TRUHAUT, R., VITTE, G., & BOUSSEMART, E. (1952a) Recherches
sur la toxicologie du pentachlorphénol. I. Propriétés.
Caractérisation et dosage dans les milieux biologiques. Arch.
Mal. prof. Méd. Trav. Sécur. soc., 13: 561-567.
TRUHAUT, R., L'EPEE, P., & BOUSSEMART, E. (1952b) Recherches
sur la toxicologie du pentachlorophénol. II. Intoxications
professionelles dans l'industrie du bois. Observations de deux
cas mortels. Arch. Mal. prof. Méd. Trav. Sécur. soc., 13:
567-569.
TRUJILLO, D.A., RAY, L.E., MURRAY, H.E., & GIAM, C.S. (1982)
Bioaccumulation of pentachlorophenol by killifish (Fundulus
similis). Chemosphere, 11: 25-32.
TURNER, H.J., Jr, REYNOLDS, D.M., & REDFIELD, A.C. (1948)
Chlorine and sodium pentachlorophenate as fouling preventives
in sea water conduits. Ind. eng. Chem., 40: 450-453.
UHL, S., SCHMID, P., & SCHLATTER, C. (1986) Pharmacokinetics
of pentachlorophenol in man. Arch. Toxicol., 58: 182-186.
UMWELTBUNDESAMT (1985) [State of the art: dioxins,] Berlin,
Erich Schmidt Verlag, 353 pp (in German).
US EPA (1973) EPA compendium of registered pesticides,
Washington DC, US Environmental Protection Agency, Vol. II.
pp. S-59-001 - S-59-003.
US EPA (1978) Ambient water quality criteria. Pentachloro-
phenol, Washington DC, US Environmental Protection Agency
(PB-292439).
US EPA (1980) Ambient water quality criteria for
pentachlorophenol, Washington DC, Office of Water Regulations
and Standards, US Environmental Protection Agency, 98 pp
(EPA-440/5-80-065).
US EPA (1984a) Creosote, pentachlorophenol, and inorganic
arsenicals; notice of intent to cancel: notice of determina-
tion of availability of position document. Fed. Reg., 49(136):
28666-28689.
US EPA (1984b) Pentachlorophenol: preliminary notice of
determination concluding the rebuttable presumption against
registration of pesticide products containing pentachloro-
phenol for non-wood preservative uses; proposed notice of
intent to cancel such registrations; notice of availability of
position document 2/3. Fed. Reg., 49(240): 48367-48372.
US EPA (1985) Hazardous waste management system: dioxin-
containing wastes. Fed. Reg., 50: 1978-2006.
US NATIONAL ACADEMY OF SCIENCES (1977) Drinking-water and
health, Washington DC, Safe Drinking Water Committee, National
Academy of Sciences, pp. 583.
VALLEJO-FRIERE, A., RIBEIRO, O.F., & RIBEIRO, I.F. (1954)
Quaternary ammonium compounds as molluscicides. Science,
119(3093): 470-472.
VALO, R., KITUNEN, V., SALKINOJA-SALONEN, M., & RAISANEN, S.
(1984) Chlorinated phenols as contaminants of soil and water
in the vicinity of 2 Finnish sawmills. Chemosphere, 13:
835-844.
VAN OMMEN, B., VAN BLADEREN, P.J., TEMMINK, J.H.M., & MUELLER,
F. (1985) Formation of pentachlorophenol as the major
product of microsomal oxidation of hexachlorobenzene. Biochem.
biophys. Res. Commun., 126: 25-32.
VAN RENSBURG, J.F.J. (1981) Health aspects of organic
substances in South African waters - opinions and realities.
Water S.Afr., 7: 139-149.
VERMA, S.R., RANI, S., & DALELA, R.C. (1981a) Responses of
serum transaminases in Notopterus notopterus chronically
exposed to phenolic compounds and their combinations. Environ.
Res., 24: 218-223.
VERMA, S.R., TONK, I.P., & DALELA, R.C. (1981b) Determina-
tion of the maximum acceptable toxicant concentration (MATC)
and the safe concentration for certain aquatic pollutants.
Acta hydrochim. hydrobiol., 9: 247-254.
VERMA, S.R., RANI, S., & DALELA, R.C. (1981c) Effects of
phenolic compounds on in vivo blood parameters of a fish
Notopterus notopterus. J. environ. Sci. Health B, 16: 273-282.
VERMA, S.R., RANI, S., & DALELA, R.C. (1982) Effects of
sodium pentachlorophenate on enzymes of energy metabolism in
tissues of Notopterus notopterus. Toxicol Lett., 10: 297-302.
VERMEER, K., RISEBROUGH, R.W., SPAANS, A.L., & REYNOLDS, L.M.
(1974) Pesticide effects on fish and birds in rice fields of
Surinam, South America. Environ. Pollut., 7: 217-236.
VINOGRADOVA, V.K., KALYAGANOV, P.I., SUDONINA, L.T., &
ELIZAROV, G.P. (1973) [Industrial hygiene and the health
condition of workers engaged in the production of sodium
pentachlorophenolate.] Gig. Tr. Prof. Zabol., 8: 11-13 (in
Russian).
VOGEL, E. & CHANDLER, J.L.R. (1974) Mutagenicity testing of
cyclamate and some pesticides in Drosphila melanogaster.
Experientia (Basel), 30(6): 621-623.
WAINSTOCK DE CALMANOVICI, R. & SAN MARTIN DE VIALE, L.C.
(1980) Effect of chlorophenols on porphyrin metabolism in
rats and chick embryo. Int. J. Biochem., 12: 1039-1044.
WALUM, E. & PETERSON, A. (1984) On the application of
culture neuroblastoma cells in chemical toxicity screening. J.
Toxicol environ. Health, 13: 511-520.
WANG, M. (1965) [Stability and molluscciidal activity of
Na-PCP in the environment.] Chin. J. Hyg., 10: 243-245 (in
Chinese).
WARREN, J.S., LAMPARSKI, L.L., JOHNSON, R.L., & GOOCH, R.M.
(1982) Determination of pentachlorophenol volatilized from
wood via collection on silica gel. Bull. environ. Contam.
Toxicol., 29: 719-726.
WATANABE, I. (1973) Isolation of pentachlorophenol
decomposing bacteria from soil. Soil Sci. plant Nutr., 19:
109-116.
WATANABE, I. (1978) Pentachlorophenol (PCP) decomposing
activity of field soils treated annually with PCP. Soil Biol.
Biochem., 10: 71-75.
WATANABE, S. & WATANABE, S. (1970) [Contact dermatitis due
to herbicide (PCP and MCPB): a fatal case.] Rinsho Hifugaku,
24(10): 945-949 (in Japanese).
WEBB, P.W. & BRETT, J.R. (1973) Effects of sublethal
concentrations of sodium pentachlorophenate on growth rate,
food conversion efficiency, and swimming performance in
underyearling sockeye salmon (Oncorhynchus nerka). J. Fish.
Res. Board Can., 30: 499-507.
WEBER, K. & ERNST, W. (1978) Levels and patterns of
chlorophenols in water of the Weser estuary and the German
bight. Chemosphere, 11: 873.
WEGMAN, R.C.C. & HOFSTEE, A.W.M. (1979) Chlorophenols in
surface waters of the Netherlands. Water Res., 13: 651-657.
WEGMAN, R.C.C. & VAN DEN BROEK, H.H. (1983) Chlorophenol in
river sediment in the Netherlands. Water Res., 17: 227-230.
WEINBACH, E.C. (1957) Biochemical basis for the toxicity of
pentachlorophenol. Proc. Natl Acad. Sci. (USA), 43: 393-397.
WEINBACH, E.C. & GARBUS, J. (1965) The interaction of
uncoupling phenols with mitochondria and with mitochondrial
protein. J. biol. Chem., 240: 1811-1819.
WEISS, U.M., KORTE, F., HAQUE, A.U., MOZA, P., & SCHEUNERT,
I. (1982a) Fate of pentachlorophenol-14C in rice plants
under controlled conditions. J. agric. food Chem., 30:
1186-1190.
WEISS, U.M., SCHEUNERT, I., KLEIN, W., & KORTE, F. (1982b)
Fate of pentachlorophenol-14C in soil under controlled
conditions. J. agric. food Chem., 30: 1191-1194.
WHITLEY, L.S. (1968) The resistance of tubificid worms to
three common pollutants. Hydrobiologia, 32: 193-205.
WHO (1976) EHC 2: Polychlorinated biphenyls and terphenyls,
Geneva, World Health Organization, 85 pp.
WHO (1984) The WHO recommended classification of pesticides
by hazard. Guidelines to classification 1984-1985, Geneva,
World Health Organization (Unpublished report VBC/84.2).
WHO (1985) Guidelines for drinking-water quality: health
criteria and other supporting information, Geneva, World
Health Organization, Vol. 2, pp. 237-239.
WIKLUND, K. & HOLM, L.E. (1986) Soft-tissue sarcoma risk in
Swedish agricultural and forestry workers. J. Natl Cancer
Inst., 76: 229-234.
WILLIAMS, P.L. (1982) Pentachlorophenol: an assessment of
the occupational hazard. Am. Ind. Hyg. Assoc. J., 43: 799-810.
WILSON, R.D., ZIPRIN, R.L., CLARK, D.E., & ELISSALDE, M.H.
(1982) Absorption of pentachlorophenol by the ovine lymphatic
system: a technical note. Vet. hum. Toxicol., 24: 12-14.
WINDHOLZ, M., ed. (1976) The Merck index, 9th ed., Rahway,
New Jersey, Marck & Co., p. 921.
WITTE, I., JUHL, U., & BUTTE, W. (1985) DNA-damaging
properties and cytotoxicity in human fibroblasts of tetra-
chlorohydroquinone, a pentachlorophenol metabolite. Mutat.
Res., 145: 71-75.
WOELKE, C.E. (1972) Development of a receiving water quality
bioassay criterion based on the 48-h Pacific oyster (Crasso-
strea gigas) embryo. Wash. Dep. Fish. Techn. Rep., 9: 93 pp.
WOIWODE, W., WODARZ, R., DRYSCH, K., & WEICHARDT, H. (1980)
Determination of free pentachlorophenol in air and in blood by
efficient chromatographic procedures. Int. Arch. occup.
environ. Health, 45: 153-162.
WONG, A.S. & CROSBY, D.G. (1978) Photolysis of pentachloro-
phenol in water. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 19-25.
WONG, A.S. & CROSBY, D.G. (1981) Photodecomposition of
pentachlorophenol in water. J. agric. food Chem., 29: 125-130.
WOOD, K.M. (1980) Prevalence of unexplained anaemias
associated with occupational exposures to pentachlorophenol
used as a wood preservative, University of Washington,
Department of Environment and Health, 74 pp (Thesis).
WOOD, S., ROM, W.N., WHITE, G.L., & LOGAN, D.C. (1983)
Pentachlorophenol poisoning J. occup. Med., 25(7): 527-530.
WSSA (1974) Herbicide handbook of the Weed Science Society
of America, Champaign, Illinois, Weed Science Society of
America.
WYLLIE, J.A., GABICA, J., BENSON, W.W., & YODER, J. (1975)
Exposure and contamination of the air and employees of a
pentachlorophenol plant, Idaho - 1972. Pestic. Monit. J., 9:
150-153.
YOUNG, H.C. & CARROL, J.C. (1951) The decomposition of
pentachlorophenol when applied as a residual pre-emergence
herbicide. Agron. J., 43: 504-507.
YOUNT, J.D. & RICHTER, J.E. (1986) Effects of pentachloro-
phenol on periphyton communities in outdoor experimental
streams. Arch. environ. Contam. Toxicol., 15: 51-60.
YUNKER, M.B. (1981) A pelagic marine ecosystem study of the
behaviour, pathways, residence time and toxicity of penta-
chlorophenol, Sidney, Institute of Ocean Sciences (DSS file
No. 07sb.fp833-9-09043, contract rpt. Dobrocky Seatch. Ltd).
ZENZEN, C. (1979) [Pentachlorophenol. Studies on the
distribution kinetics after single and repeated dosing,]
University of Düsseldorf, 149 pp (Thesis) (in German).
ZIGLER, G.M. & PHILLIPS, W.F. (1967) Thin-layer chromato-
graphic method for estimation of chlorophenols. Environ. Sci.
Technol., 1: 65.
ZIMMERLI, B. (1982) [Modelling the transfer of pollutants
from painted surfaces in the surrounding air.] In: Aurand, K.,
Seifert, B., & Wegner, J., ed. [Air quality in closed rooms,]
Stuttgart, New York, Gustav Fischer Verlag, pp. 235-267 (in
German).
ZIMMERLI, B. & ZIMMERMANN, H. (1979) [Simple procedure to
estimate the concentrations of toxic chemicals in the indoor
atmosphere.] Mitt. Geb. Lebensmittelunters Hyg., 70: 429-422
(in German).
ZIMMERLI, B., MARSCHALL, T., & MAREK, B. (1979) [Preliminary
studies on occurrence of pentachlorophenol in human urine.]
Mitt. Geb. Lebensmittelunters. Hyg., 70: 443-450 (in German).
ZIMMERLI, B., MARSCHALL, T., & MAREK, B. (1980) [On the
excretion of pentachlorophenol in cow milk.] Mitt. Geb.
Lebensmittelunters. Hyg., 71: 404-414 (in German).
ZISCHKE, J.A., ARTHUR, J.W., HERMANUTZ, R.O., HEDTKE, S.F., &
HELGAN, J.C. (1985) Effects of pentachlorophenol on
invertebrates and fish in outdoor experimental channels.
Aquat. Toxicol., 7: 37-58.
ZITKO, V., HUTZINGER, O., & CHOI, P.M.K. (1974) Determina-
tion of pentachlorophenol and chlorobiphenyls in biological
samples. Bull. environ. Contam. Toxicol., 12: 649-653.
ZOBER, A., GOSSLER, K., MANKE, G., & SCHALLER, K.H. (1979)
[Studies on pentachlorophenol loading in the wood industry.]
Ber. Jahrestag. Dtsch. Ges. Arbeitsmed., 19th, 1979: 447-454
(in German).
ZOBER, A., SCHALLER, K.H., GOSSLER, K., & KREKELER, H.J.
(1981) [Pentachlorophenol and liver function: a pilot study
of occupationally exposed groups.] Int. Arch. occup. environ.
Health, 48: 347-356 (in German).
Table 27. Comparison of PCP elimination kinetics in mammals after administration of
single doses
-------------------------------------------------------------------------------------------
Species Dose Elimina- Sex Elimination Kinetics Reference
(mg/kg tion via half-life (h)
body of phase
weight) alpha beta
-------------------------------------------------------------------------------------------
Mousea,b 15 - 37 urine female approxi- - not reported Jakobson &
mately 24 Yllner (1971)
Ratc 37 - 41 urine female approxi- 2448 biphasic Larsen et al.
mately 10 (1972)
Ratd 5.7 urine male approxi- - monophasic Hoben et al.
mately 24 (1976d)
Ratc 10 urine and female 13 33 biphasic Braun et al.
faeces male 17 40 biphasic (1977)
100 urine and female 27 - monophasic
faeces male 13 121 biphasic
Rata 15 all female 6.3 - 9.9 33- biphasic Zenzen (1979)
routes 374
Monkeyc 10 urine female 92.4 - monophasic Braun &
male 40.8 - monophasic Sauerhoff
(1976)
-------------------------------------------------------------------------------------------
a Intraperitoneal. b Subcutaneous.
c Oral. d Inhalation.
When comparing the turnover of PCP in rats after single and
after repeated inhalation exposure, Hoben et al. (1976d) did not
find any evidence of accumulation. The clearance rates for urine,
plasma, liver, and lung were fairly parallel. After repeated
inhalation exposures to about 5.9 mg/kg body weight, the PCP body
burden did not increase as expected from the 24-h half-life
following a single exposure. On the basis of the increased urinary
excretion, the authors concluded that the biotransformation was
accelerated and possibly induced by prior exposure to PCP.
6.5.2 Human studies
Braun et al. (1979) observed a time lag in the urinary
excretion rate following a single oral dose of 0.1 mg Na-PCP/kg
body weight given to 4 volunteers (section 6.1.2). Maximum urinary
excretion was reached 40 h after ingestion and 36 h after the
maximum plasma level of 0.245 mg PCP/litre. The authors ascribed
this delay to a strong enterohepatic circulation similar to that
reported in rats and monkeys. The elimination half-life for PCP in
plasma was about 30 h. The elimination half-life for PCP and PCP
glucuronide in urine was 33 and 13 h, respectively. The
elimination of PCP from plasma in these human subjects followed
linear kinetics and was monophasic, and resembled the elimination
kinetics reported for the monkey. Elimination kinetics in the rat
are biphasic; however, the rate constants and half-lives for the
absorption and elimination of PCP from rat plasma are more similar
to those for men than those for monkeys.
Using the kinetic parameters determined in their single-
exposure studies, Braun et al. (1979) calculated that men ingesting
0.1 mg PCP/kg body weight daily (based on 100% uptake of 0.5 mg
PCP/m3 by a man carrying out light work) would attain a steady-
state plasma concentration of 0.491 mg/litre, after 8.4 days. This
suggests that there is no cumulative effect, even with repeated
daily low-level exposure. This conclusion is based on a simulation
model for the daily ingestion of PCP using data derived from
single-dose studies.
Uhl et al. (1986) studied 3 healthy male volunteers aged 29,
24, and 47 years, exposed to single oral doses of PCP (purity >
99%) in 40% ethanol in 6 different studies. The doses varied from
0.016 to 0.31 mg PCP/kg body weight. Their results are in contrast
to those of Braun et al. (1979). Uhl et al. (1986) determined PCP
elimination half-lives of 16 days (plasma) and 18 - 20 days
(urine), elimination rates being about 13 times slower than those
reported by Braun et al. (1979). Uhl et al. (1986) did not find
any evidence for an enterohepatic accumulation mechanism. They
ascribed the low elimination rate to the high protein-binding
tendency of PCP and the concomitantly low PCP clearance observed
(0.07 ml/min). At normal urinary pH (5 - 6), PCP exists in its
phenolic form and therefore more than 99% of the filtered PCP is
believed to be reabsorbed in the renal tubules. In one study,
alkalinization of the urine by administration of sodium bicarbonate
considerably enhanced the elimination rate of PCP in urine. Over
the pH range 5.4 - 7.8, the rate of PCP elimination varied by a
factor of 8; urinary excretion was approximately 2 µg PCP/h at pH
5.4 and about 16 µg PCP/h at pH 7.4.
The differences in the pharmacokinetic profile established in
the 2 studies may be explained by the different experimental
regimes. Uhl et al. (1986) administered the PCP in ethanol; Braun
et al. (1979) administered the sodium salt of PCP in water. The
role of the dietary status of the volunteers before and after the
PCP ingestion or of any other factors is unknown. According to Uhl
et al. (1986), the time lag necessary to attain a steady state
after a change in the exposure, as calculated from the elimination
half-life, is about 3 months. These authors estimated that the
human body burden of PCP at steady state is 10 - 20 times higher
than that estimated by Braun et al. (1979).
Reported cases of accidental PCP exposure as well as data from
occupationally exposed people seem to confirm the results of Uhl et
al. (1986). In the case of an accidental uptake of PCP through the
skin (section 6.1.2), the urinary-PCP concentration decreased from
236 µg/litre, 2 days after the accident to 17 µg/litre, 51 days
later (Bevenue et al., 1967a). From these data, an elimination
half-life of about 15 days can be derived.
In the case of intentional ingestion of PCP (solvent: 82%
aromatic petroleums) discussed in section 6.1.2, despite forced
diuresis with furosemide and mannitol, the serum-PCP level of the
patient decreased from the highest level on day 2 (155 mg
PCP/litre) to 12 mg PCP/litre on day 37 (Haley, 1977), suggesting
an elimination half-life of approximately 10 days.
Begley et al. (1977) surveyed PCP concentrations in the blood
and urine of 18 workers in a wood-treatment factory before and
during a 20-day vacation. The blood and urine levels of PCP
decreased from an average of 5.14 mg/litre to 2.19 mg/litre and
from 1.31 mg/litre to 0.59 mg/litre, respectively, during vacation.
Elimination half-lives of about 9 days for both urine and plasma
can be derived.
Casarett et al. (1969) calculated an excretion half-life of
about 10 h, based on measurements made one day after a single
inhalation exposure in 2 wood-treatment workers. Despite this rapid
elimination rate, the PCP concentrations in the urine of workers
following a long-term high level exposure (PCP in urine, 1.6 - 2.6
mg/litre) did not decrease by more than 60 - 80% "even after a long
absence from exposure" (Casarett et al., 1969). In a comparable
survey with a group of woodworkers exposed long-term to Permatox
100 (3% PCP, 21% tetrachlorophenol), no clear elimination pattern
could be found during a 16-day vacation (Kalman & Horstman, 1983);
half-times for urinary elimination varied from 4 to 72 days, as
estimated from the urinary-PCP levels at day 0 and day 16. A number
of pre-exposed workers showed an increase in urinary-PCP during the
vacation, either as a result of storage of PCP in tissues (section
6.6), additional exposure to PCP independent of work-place
exposure, or a continuous biotransformation of hexachlorobenzene
and similar compounds to PCP (section 5.2.2). However, the latter
can hardly explain an increase in urinary-PCP levels of 53 - 88
µg/litre, as observed by Kalman & Horstman (1983); urinary-PCP
levels range around 10 µg/litre for the general population and
around 40 µg/litre for people exposed non-occupationally (section
5.2.3).
Since all controlled studies on the metabolism of PCP in
mammals have been performed using pure PCP, a judgement of the
influence of impurities in commercial PCP on the metabolism of PCP
is not possible. In fish, Huckins & Petty (1983) observed a
greater conjugation of PCP to its glucoronide with exposure to
purified PCP compared with exposure to commercial PCP. Conjugation
may be a rate-limiting step in its elimination, in which case the
toxic impurities in industrial PCP formulations could alter the
turnover pattern.
6.6 Reaction With Body Components
Braun et al. (1977) found that 99% of the PCP in rat plasma was
bound to protein. Heterogenous binding has been demonstrated,
indicating that, at very low plasma concentrations of PCP, the
protein binding becomes even stronger, because of preferential
binding to more limited but higher affinity sites. Braun et al.
(1977) and Uhl et al. (1986) suggested that this heterogenous
plasma-protein binding might be the cause of long-term urinary
excretion of PCP. Hoben et al. (1976e) found that human plasma had
a much higher capacity for binding to PCP than rat plasma; this
could explain the longer retention times observed in human beings
compared with that in the rat. The difference in binding capacity
could not be accounted for by albumin fraction.
PCP is conjugated in vitro to palmitic acid in rat liver
incubated with a coenzyme A-fortified microsomal system (Leighty &
Fentiman, 1982). Presumably, the binding of PCP to fatty acids
could contribute to PCP retention in lipid-containing tissues.
Weinbach & Garbus (1965) had already discovered the strong
affinity of PCP for rat liver mitochondrial protein. In more
recent studies of Arrhenius et al. (1977a,b) on the sub-cellular
distribution of PCP, PCP accumulation in microsomes or in the
cytosol was approximately 6 and 3 times, respectively, higher than
in the mitochondria. On this basis, the authors suggested that the
microsomal functions, though 4 times less sensitive than
mitochondrial oxidative phosphorylation, were disturbed by PCP
concentrated in these organelles, possibly increasing the toxic
and carcinogenic action of other xenobiotics.
7. EFFECTS ON ORGANISMS IN THE ENVIRONMENT
The toxic effects of chlorophenols have been studied in a
number of organisms. The degree of oil solubility governs the
toxicity of phenolic compounds by controlling their binding to
lipoid cellular structures, e.g., biological membranes, which
probably are the loci of action of PCP. Blackman et al. (1955a,b)
showed that, with the duckweed Lemna minor and the mould
Trichoderma viride, the lipophilic behaviour and, hence, the
toxicity of chlorophenols increased with the degree of chlorination
of the aromatic ring, PCP being the most effective chlorophenol.
Similar results have been reported from studies on fish (Ingols &
Gaffney, 1965), bacteria (Liu et al., 1982), and algae (Huang &
Gloyna, 1968; Rowe et al., 1982).
Although there have been some studies on the effects of PCP on
ecosystems, most of the toxicity data have been derived from single
species trials. Moreover, most bioassays deal with acute rather
than long-term toxic effects.
7.1 Microorganisms
The microbiocidal effectiveness of PCP has been the basis of
its widespread use as a bactericide and fungicide (section 3.3).
For instance, PCP at a concentration of 0.25% and 0.125% (v/v) was
used to control the sapstain fungi Trichoderma harzianum and
Phialophora sp., respectively (Cserjesi & Roff, 1975). Conkey &
Carlson (1963) screened PCP-containing pesticides against 2
bacterial species and 2 species of fungi that are common in pulp
and paper mill systems. Depending on the PCP formulation and the
microbial species, complete inhibition occurred on agar plates at
4 - 250 ppm.
Ishizawa et al. (1961) found bacterial und fungal growth in
soil to be depressed by PCP at 2 g Na-PCP/kg dry soil. Similarly,
oxidative phosphorylation and ATPase activity in Micrococcus
denitrificans cultures were strongly inhibited by PCP at a
concentration of about 130 mg/litre (Imai et al., 1967).
In anaerobic soil containing 10 mg PCP/kg, Murthy et al. (1979)
observed reduced soil respiration as PCP directly or indirectly
inhibited cellulose degradation. The degradation of PCP itself may
also be affected by the toxicity of this compound for degrading
microorganisms.
Godsy et al. (1986) studied the effects of PCP on the
methanogenic fermentation of phenol in anaerobic laboratory
digestors. With PCP concentrations of 0.1 mg/litre or less, PCP
was dechlorinated to non-toxic levels allowing for complete
bioconversion of phenol (200 mg/litre) and PCP, presumably to
methane and carbon dioxide (CO2). Higher PCP levels inhibited the
methanogenic fermentation; at 5 mg PCP/litre, complete inhibition
occurred.
Tam & Trevors (1981a,b) studied the effects of PCP on
asymbiotic nitrogen fixation in soil. The EC50 values for the
inhibition of nitrogenase activity in non-sterile soil, incubated
aerobically and anaerobically, and in sterilized soil inoculated
with Azotobacter sp. were 49.8 mg Na-PCP/kg, 186.8 mg Na-PCP/kg,
and 660.8 mg Na-PCP/kg, respectively. The inhibition of both CO2
evolution and oxygen uptake by Azotobacter vinelandii was found to
be similar to that of nitrogen fixation activity (Tam & Trevors,
1981a). The high concentrations required for inhibition suggest
that, at normal field application rates, no adverse effects on
nitrogenase activity would be expected.
Na-PCP at 50 and 100 mg/kg had a stimulating effect on soil
microbial electron transport activity, while 200 mg Na-PCP/kg
caused 5.8% inhibition (Trevors, 1982c). Inhibition by Na-PCP was
greater in soil enriched with glucose and yeast extract than in
non-amended soil. Concentrations of 25 - 50 mg PCP/litre of
nutrient broth delayed the growth of the bacterium Pseudomonas
fluorescens, which does not degrade PCP. A 1-h exposure to 75 mg
PCP/litre inhibited growth completely. Higher concentrations of
PCP were required to produce inhibition of respiration. Oxygen
consumption was reduced by 21% at a concentration of 25 mg
PCP/litre, while CO2 evolution was not inhibited (Trevors et al.,
1981a; Trevors, 1982b).
Pre-exposure to PCP lowered the sensitivity of Pseudomonas
fluorescens to PCP (Trevors et al., 1982). In addition, non-toxic
concentrations of the antioxidants butylated hydroxyanisole (BHA)
and butylated hydroxytoluene (BHT), which have also been used as
food additives, enhanced the toxicity of PCP for this bacterial
species (Trevors et al., 1981b).
Izaki et al. (1981) found gram-negative bacteria to be more
resistant to PCP than gram-positive bacteria. Some very resistent
Pseudomonas strains tolerated over 500 mg PCP/litre. Studies on
various mutants, in which the lipopolysaccharide layers were more
or less defective, supported the hypothesis that these layers act
as a barrier for PCP and impart resistance to gram-negative
bacteria.
7.2 Aquatic Organisms
PCP has been used in aquatic environments as a molluscicide
and an algicide. The potential hazard of PCP was recognized early,
leading to a number of toxicological studies on aquatic organisms;
these have been reviewed by US EPA (1978) and Buikema et al.
(1979). In general, PCP is extremely toxic for aquatic organisms.
Apart from very sensitive or resistant species, there is apparently
no significant difference in sensitivity to PCP between the
different taxonomic groups (Adema & Vink, 1981).
7.2.1. Plants
Most toxicity tests on aquatic plants have been performed on
algae, particularly the microscopic, free-floating forms. A
selection of these tests is summarized in Table 28. One of the
more striking features evident from the Table is the extreme
variability in the PCP levels that result in toxic effects. For
example, Adema & Vink (1981) determined that a nominal
concentration of 7 mg/litre inhibited the growth of Chlorella
pyrenoidosa by 50%. In contrast, Huang & Gloyna (1968) found that
as little as 7.5 µg/litre caused complete destruction of chlorophyll
in the same species.
At least part of this variability reflects different
sensitivities to PCP between algal species. Adema & Vink (1981)
found that the green alga Scenedesmus quadricauda had an EC50 for
growth that was 87.5 times lower than that for Chlorella
pyrenoidosa. The most sensitive algae were Ankistrodesmus falcatus
and Microcystis sp., for which as little as 0.001 mg PCP/litre
inhibited photosynthesis by an average of 21%, in semi-continuous
cultures.
However, the tremendous variability in susceptibility to PCP
presented in the Table cannot be accounted for solely by species-
specific differences in sensitivity. They probably also reflect
differences in the experimental conditions under which the tests
were run, such as the ambient pH, which affects PCP toxicity for
invertebrates (section 7.2.2), and presumably algae.
Some aquatic vascular plants have also been used for toxicity
studies. The water hyacinth (Crassipes eichhornia) is relatively
tolerant to Na-PCP; a concentration of 5 mg/litre was required to
affect its appearance, and 80 mg/litre to kill the plant (Hirsch,
1942). Only 7.1 x 10-7 mol PCP/litre (approximately 0.19 mg/litre)
were required to induce 50% chlorosis in the fronds of the duckweed
(Lemna minor) (Blackman et al., 1955a). In contrast, Huber et al.
(1982) noted a 50% decrease in the chlorophyll content of this
aquatic macrophyte on exposure to 3 - 4 mg PCP/litre.
Photosynthesis as well as the activity of glutamate dehydrogenase
and alanine aminotransferase were similarly inhibited, but there
was no pronounced effect on dark respiration.
7.2.2 Invertebrates
As shown in Table 29, the toxicity of PCP or Na-PCP for
invertebrates varies with concentrations ranging from 0.068
mg/litre to 10.39 mg/litre. Most of the reported 50% lethal or
effective concentrations are below 1 mg/litre.
In general, developing embryos and larvae are more affected by
PCP than the adults (Table 29). The most striking difference was
reported by van Dijk et al. (1977), who found that, at the same Na-
PCP concentration, the larvae of the marine decapod Palaemon
elegans were inhibited (in terms of the 96-h LC50) about 130 times
more than the adults.
Table 28. The toxicity of PCP and Na-PCP for algae
--------------------------------------------------------------------------------------------------------
Test species Aquatic Test type Test Concent- Effects Reference
system duration ration
(h) (mg/litre)
--------------------------------------------------------------------------------------------------------
Chlorella freshwater static 72 0.008 complete Huang & Gloyna
pyrenoidosa destruction of (1968)
chlorophyll
Chlorella freshwater static and flow 96 7a EC50, growth Adema & Vink
pyrenoidosa (1981)
Scenedesmus freshwater static and flow 96 0.08a EC50, growth Adema & Vink
quadricauda (1981)
Microcystis freshwater static 96 1 NOEC, growth Sloof & Canton
aeruginosa (1983)
Scenedesmus freshwater static 96 0.1 NOEC, growth Sloof & Canton
pannonicus (1983)
Cylindrospermum freshwater static 72 2 no growth Palmer & Maloney
licheniformeb (1955)
Microcystis freshwater static 72 2 no growth Palmer & Maloney
aeruginosab (1955)
Scenedesmus freshwater static 72 - 168 2 growth inhibition Palmer & Maloney
obliquusb (1955)
Chlorella freshwater static 72 - 504 2 no inhibition Palmer & Maloney
variegatab (1955)
Monochrysis sp. marine static and flow 96 0.2a EC50, growth Adema & Vink
(1981)
--------------------------------------------------------------------------------------------------------
Table 28. (contd.)
--------------------------------------------------------------------------------------------------------
Test species Aquatic Test type Test Concent- Effects Reference
system duration ration
(h) (mg/litre)
--------------------------------------------------------------------------------------------------------
Chlamydomonas marine static and flow 96 1.4a EC50, growth Adema & Vink
sp. (1981)
Phaeodactylum marine static and flow 96 3a EC50, growth Adema & Vink
tricornutum (1981)
Dunaliella sp. marine static and flow 96 3.6a EC50, growth Adema & Vink
(1981)
Chlorella ovalis marine static and flow 96 5.5a EC50, growth Adema & Vink
(1981)
Ankistrodesmus marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
falcatus flow 264 photosynthesis (1982)
Microcystis sp. marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
flow 264 photosynthesis (1982)
Melosira sp. marine semi-continuous 216 - 0.001 inhibition of Gotham & Rhee
flow 264 photosynthesis (1982)
and growth
Selenastrum marine static 2 0.05 - beginning inhibi- Jayaweera et al.
capricornutum 0.1 tion of carbon (1982)
assimilation rate
Selenastrum marine static 2 2.66 complete inhibi- Jayaweera et al.
capricornutum tion (1982)
--------------------------------------------------------------------------------------------------------
a Average results of static and flow-through tests.
b Na-PCP.
Dragonfly nymphs ( Epicordulia sp.), damsel fly nymphs ( Ischnura
sp.), isopods ( Asellus communis Say), and amphipods ( Hyalella
knickerbockeri Bate) were resistant to Na-PCP compared with other
invertebrate species, and easily survived exposure to 5 mg/litre
(Goodnight, 1942). Turner et al. (1948) found that 0.1 mg Na-
PCP/litre was ineffective against mussels (Mytilus edulis),
anemones, and barnacles, while a concentration of 1 mg/litre
prevented attachment and growth in sea water.
The results of more recent studies, mainly conducted on
annelids, molluscs, and crustaceans, are mostly based on median
lethal or effective concentrations (LC50, EC50, TLm). However, a
comparison of acute toxicity data established within equal test
duration may only be appropriate if the various organisms have
similar life cycles. It is arbitrary to compare, for instance, the
96-h LC50 of copepods, crayfish, and trout. On a basis of the
median life span, a 96-h toxicity test with the cladoceran Daphnia
would correspond to a test with trout or carp lasting as long as
one year.
Very few studies have been carried out to assess chronic or
sublethal effects, or the influence of various environmental
conditions on the toxicity of PCP. Examining the effect of Na-PCP
on the burrowing activity of a lugworm (Arenicola cristata),
Rubinstein (1978) noted a significant adverse effect at
concentrations of 80 and 156 µg Na-PCP/litre. At both
concentrations, no mortality was observed. The reduced lugworm
activity could affect the sediment turnover.
At sublethal concentrations (120 µg PCP/litre), coelomic fluid
glucose in the marine sandworm Neanthes virens increased to about
twice the control levels after 24 h and then gradually decreased
(Carr & Neff, 1981; Thomas et al., 1981). Ascorbic acid levels
became elevated during exposure, indicating sublethal stress.
During the acute lethal exposure (above 365 µg/litre), a
significant hypoglycaemic response and ascorbic acid depletion were
observed.
The acute toxicity of PCP has been found to be pH dependent
(Table 29). The data of Whitley (1968) indicate that the toxicity
of PCP for tubificid worms increased up to almost 5-fold, when the
pH value decreased from 9.5 to 7.5. This is consistent with the
sensitivity pattern of various oligochaete species (Chapman et al.,
1982). Similarly, lowering the pH from 7.5 to 6.5 increased the
toxicity of PCP for the crayfish (Astacus fluviatilis) by a factor
of 5.9 (Kaila & Saarikoski, 1977). The non-ionized form of a
compound penetrates biological membranes much more easily than the
ionized form, which accounts for the effects of pH on PCP toxicity.
Table 29. The toxicity of PCP and Na-PCP for invertebrates
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Tubificid wormsa,b freshwater static pH 7.5 24 0.31 LC50 Whitley (1968)
Tubificid wormsa,b freshwater static pH 8.5 24 0.67 LC50 Whitley (1968)
Tubificid wormsa,b freshwater static pH 9.5 24 1.40 LC50 Whitley (1968)
Water flea freshwater static 24 0.8 EC50 Bringmann &
(Daphnia magna) Kühn (1982)
Fresh-water snail freshwater static 96 0.16 LC50 Gupta & Rao
(Lymnaea acuminata) (1982)
Fresh-water snail freshwater static 96 0.19b LC50 Gupta & Rao
(Lymnaea acuminata) (1982)
Eastern oyster marine static 48 < 0.25 LC50, egg Davis & Hidu
(Crassostrea virginica) development (1969)
Eastern oyster marine static 336 0.07 LC50, Davis & Hidu
(Crassostrea virginica) survival (1969)
of larvae
Pacific oysterb marine static 48 0.027 4.3% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
Pacific oyster marine static 48 0.069 72.4% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
---------------------------------------------------------------------------------------------------------
Table 29. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Pacific oyster marine static 48 0.11 100% Woelke (1972)
(Crassostrea gigas) abnormal
embryos
Eastern oyster marine static 48 0.04 EC50, Borthwick &
(Crassostrea virginica) embryo Schimmel (1978)
development
European brown shrimp adult marine static 96 1.79 LC50 van Dijk et
(Crangon crangon)b al. (1977)
European brown shrimp larvae marine static 96 0.11 LC50 van Dijk et
(Crangon crangon)b al. (1977)
Marine decapod adult marine static 96 10.39 LC50 van Dijk et
(Palaemon elegans)b al. (1977)
Marine decapod larvae marine static 96 0.08 LC50 van Dijk et
(Palaemon elegans)b al. (1977)
Brackish water decapod adult marine static 96 5.09 LC50 van Dijk et
(Palaemonetes varians)b al. (1977)
Brackish water decapod larvae marine static 96 0.36 LC50 van Dijk et
(Palaemonetes varians)b al. (1977)
---------------------------------------------------------------------------------------------------------
Table 29. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effects Reference
stage system type modifier dura- tration
tion (mg/
(h) litre)
---------------------------------------------------------------------------------------------------------
Grass shrimp inter- marine static 96 2.63 LC50 Conklin & Rao
(Palaemonetes pugio)b molt (1978)
Grass shrimp early marine static 96 2.74 LC50 Conklin & Rao
(Palaemonetes pugio)b premolt (1978)
Grass shrimp late marine static 96 0.44 LC50 Conklin & Rao
(Palaemonetes pugio)b premolt (1978)
Brown shrimp marine flow 96 > 0.195 LC50 Schimmel et
(Penaeus aztecus)b al. (1978)
Copepod (Pseudodiap- marine static 96 0.068 LC50 Hauch et al.
tomus coronatus)b (1980)
Crayfish marine semi- pH 7.5 192 53 LC50 Kaila &
(Astacus fluviatilis) contin- Saarikoski
uous (1977)
flow
Crayfish marine semi- pH 6.5 192 9 LC50 Kaila &
(Astacus fluviatilis) contin- Saarikoski
uous (1977)
flow
---------------------------------------------------------------------------------------------------------
a Mixed population of Tubifex tubifex and Limnodrilus hoffmeisteri.
b Na-PCP.
7.2.3 Vertebrates
Most studies on vertebrates have been performed with fish. In
short-term studies, the LC50 values for PCP or Na-PCP are generally
less than 1 mg PCP/litre, and, in many cases, even less than 0.1 mg
PCP/litre (Table 30).
The effectiveness of purified PCP and Na-PCP as well as of
commercial products has been investigated under comparable
conditions in only a few cases. Borthwick & Schimmel (1978)
determined that the 96-h LC50 for 14-day-old sheepshead minnow fry
exposed to analytical grade PCP was similar to that of the
commercial formulation Dowicide G (0.392 and 0.516 mg/litre,
respectively, corresponding to 1.47 and 1.41 µmol). Similarly, the
prolarval pinfish was, on a molar basis, about equally affected by
analytical grade Na-PCP (LC50, 0.038 mg/litre = 0.14 µmol) and
Dowicide G (LC50, 0.066 mg/litre = 0.18 µmol).
Differences in the toxicity of PCP and Na-PCP for fish have
been observed under various test conditions. For example, both the
goldfish (Carassius auratus) and the sheepshead minnow (Cyprinodon
variegatus) were more affected by PCP in static than in continuous-
flow bioassays (Table 30). Ruesinck & Smith (1975) noted that the
fathead minnow (Pimephales promelas) was more resistant to Na-PCP
at 25 °C than at 15 °C. In contrast, Crandall & Goodnight (1959)
observed that higher temperatures increased the toxicity of Na-PCP
for the fathead minnow (Pimephales promelas), i.e., the LD50 at
10 °C, 18 °C, and 26 °C was 260, 81, and 46 mg/litre, respectively.
Temperature was also found to control PCP toxicity for rainbow
trout (Salmo gairdneri) (Hodson & Blunt, 1981). Eggs of trout
exposed to PCP at 0.01 - 0.1 mg/litre) showed elevated mortality
between fertilization and hatch and reduced weight at hatch. The
effects on hatch weight were greater at 6 °C than at 10 °C, while
growth rates were reduced more at 20 °C than at 12 °C.
Saarikoski & Viluksela (1981) also demonstrated that the
ambient pH influences the toxicity of PCP for fish. The 96-h LC50
values for the guppy (Poecilia reticulata) were about 0.04 mg/litre
at pH 5, 0.12 mg/litre at pH 6, 0.44 mg/litre at pH 7, and 0.91
mg/litre at pH 8. At pH 5, 33.39% of PCP is in the molecular form,
while, at pH 8, more than 99% exists as phenate ion. Since the
change in toxicity was substantially smaller than it would be if
only the molecular PCP were toxic, the authors concluded that the
phenate ion also contributes to the toxic effect.
Dissolved oxygen also plays an important role in modifying the
toxicity of PCP for fish. Dissolved oxygen levels of 7.8, 6.5, or
5 mg/litre resulted in 96-h LC50 values of 0.107, 0.083, and 0.026
mg PCP/litre, respectively (Gupta et al., 1983a). The increase in
toxicity at low levels of dissolved oxygen may be due to enhanced
absorption of PCP via gills, as the ventilation rate speeds up
under low oxygen regimes.
Table 30. The toxicity of PCP and Na-PCP for various fish species
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effect Reference
stage system type modifier duration tration
(h) (mg/litre)
---------------------------------------------------------------------------------------------------------
Brown trout fresh- static 24 0.2 LC50 Hattula et al.
(Salmo trutta) water (1981)
Bluegill sunfish fresh- static hardness 48 0.03 - LC50 Inglis & Davis
(Lepomis macrochirus) water 0.04 (1972)
Goldfish fresh- static hardness 48 0.08 - LC50 Inglis & Davis
(Carassius auratus) water 0.17 (1972)
Fathead minnow juvenile fresh- flow 48 0.21 LC50 Ruesinck & Smith
(Pimephales promelas) water (1975)
Rainbow trouta fresh- static labora- 96 0.05 - LC50 Davis & Hoos
(Salmo gairdneri) water toryb 0.10 (1975)
Coho salmona fresh- static labora- 96 0.03 - LC50 Davis & Hoos
(Oncorhynchus kisutch) water toryb 0.09 (1975)
Goldfish fresh- flow 96 0.22 LC50 Adelmann & Smith
(Carassius auratus) water (1976)
Fathead minnow juvenile fresh- static 96 0.6 LC50 Mattson et al.
(Pimephales promelas) water (1976)
Common carpa larvae fresh- static 96 0.01 LC50 Verma et al.
(Cyprinus carpio) water (1981b)
---------------------------------------------------------------------------------------------------------
Table 30. (contd.)
---------------------------------------------------------------------------------------------------------
Test species Life Aquatic Test Test Test Concen- Effect Reference
stage system type modifier duration tration
(h) (mg/litre)
---------------------------------------------------------------------------------------------------------
Guppy fresh- static 96 0.97 LC50 Gupta et al.
(Lebistes reticulatus) water (1982)
Sheepshead minnow marine flow 96 0.44 LC50 Parrish et al.
(Cyprinodon variegatus) (1978)
Sheepshead minnow 1-day- marine static 96 0.329 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 14-day- marine static 96 0.392 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 28-day- marine static 96 0.240 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Sheepshead minnow 42-day- marine static 96 0.223 LC50 Borthwick &
(Cyprinodon variegatus) old fry Schimmel (1978)
Pin percha adult marine flow 96 0.053 LC50 Schimmel et al.
(Lagodon rhomboides) (1978)
Pin percha 48-h marine static 96 0.038 LC50 Borthwick &
(Lagodon rhomboides) pro- Schimmel (1978)
larvae
---------------------------------------------------------------------------------------------------------
a Na-PCP.
b Results of an inter-laboratory bioassay standardization exercise.
The size of fresh-water fish may also influence the toxicity of
PCP. The toxicity of PCP for Notopterus notopterus decreased with
increasing fish length up to 14.5 cm, though larger fish were again
more susceptible (Gupta et al., 1982). Similarly, slight
differences in sensitivity were observed when Cyprinodon variegatus
fry of different ages were exposed to PCP (Borthwick & Schimmel,
1978).
As with invertebrates, larvae of fish seem to be more
vulnerable to PCP than adult fish. The lowest value in Table 30 in
terms of LC50 is based on tests with the larvae of the fresh-water
carp (Cyprinus carpio).
Data derived from acute toxicity tests are of limited value in
estimating the long-term effects of PCP on fish. Studies dealing
with sublethal concentrations of PCP may provide more valuable
information, since they involve PCP concentrations approaching
ambient levels. Since responses to sublethal concentrations
require prolonged periods of time, the continuous-flow method is
usually chosen to keep test conditions, particularly the
concentration of the toxicant, constant.
In underyearling sockeye salmon (Oncorhynchus nerka) exposed to
sublethal concentrations of Na-PCP, Webb & Brett (1973) observed
significant reductions in growth rate and food conversion
efficiency. The EC50 for these processes is about 1.80 µg Na-
PCP/litre (approximately 2.8% of the 96-h LC50 of 0.063 mg/litre).
Similarly, low concentrations of PCP (13.6 - 60.2 µg/litre)
were used to induce physiological stress in the fresh-water fish
Notopterus notopterus, as measured by the activity of hepatic acid
and alkaline phosphatases and succinic dehydrogenase (Dalela et
al., 1980a). The activity of these 3 enzymes was reduced after 10,
20, and 30 days of exposure. The greatest effect was observed in
acid phosphatase after 30 days at a concentration of 60.2 µg
PCP/litre caused a 71.09% inhibition of enzyme activity.
In vivo blood variables of Notopterus notopterus (Verma et al.,
1981c) were sensitive to PCP concentrations as low as 1/10th,
1/15th, and 1/20th of the 96-h LC50 of 0.083 mg PCP/litre.
Generally, red and white blood cell counts and packed cell volume
were increased after 30 days of exposure, while clotting time,
erythrocyte sedimentation rate, levels of haemoglobin and mean
corpuscular haemoglobin, and mean cell volume were decreased. In
addition, there were significant increases in the activity of
transaminases in the blood-serum (Verma et al., 1981a) and in the
brain, liver, kidney, and gills (Gupta et al., 1983b). Succinic
dehydrogenase and pyruvic dehydrogenase were inhibited, while the
activity of lactic dehydrogenase was stimulated, thus indicating
the development of anaerobic conditions at the cellular level at
these sublethal concentrations (Verma et al., 1982).
Other biochemical responses were observed in juvenile striped
mullet (Mugil cephalus), a marine fish (Thomas et al., 1981);
environmental stress was indicated by a rapid rise in plasma-
cortisol concentrations at 200 µg PCP/litre, accompanied by a
marked hyperglycaemia and a depletion of hepatic glycogen reserves.
Cleveland et al. (1982) evaluated the chronic toxicities of
commercial PCP, purified PCP, and Dowicide EC-7 in 90-day partial
life-cycle studies with fathead minnows (Pimephales promelas). The
commercial grade PCP used contained relatively large quantities of
hexachlorobenzene, chlorophenoxyphenols, chlorodiphenyloxides,
chlorodibenzodioxins, and chlorodibenzofurans. The purified PCP
included relatively high amounts of chlorinated phenoxyphenols, and
the Dowicide EC-7 contained a broad spectrum of impurities,
generally at lower concentrations than the other 2 formulations.
The commercial composite PCP formulation was the most toxic
preparation; a concentration of 13 µg/litre reduced growth of the
fish, and 27 µg/litre also reduced survival. Growth, but not
survival, was affected by the purified PCP at concentrations equal
to, or greater than, 85 µg/litre. Dowicide EC-7 was the least
toxic of the PCPs tested, and at the maximum level tested (139
µg/litre), did not adversely affect growth or survival of fathead
minnows. Thus, impurities present in PCP were found clearly to
increase toxicity under long-term exposure conditions. Moreover,
degeneration of the fins and opercula, as well as malfunction of
the anterior regions of the skull were also noted in fathead minnows
exposed to the commercial composite of PCP mixture.
7.3 Terrestrial Organisms
7.3.1 Plants
Previously, PCP was widely used as a herbicide, defoliant, and
preharvest desiccant. However, as PCP is not a very specific
herbicide in terms of inhibiting special target species (Kozak et
al., 1979), non-target crop and wildlife species can also be
adversely affected, though no data are available in this respect.
Plants may be damaged by contact with the PCP in treated wooden
material as, for example, when fruit trees in the vicinity of
freshly-treated wooden support posts or stakes suffered bark
lesions and chlorosis (Ferree, 1974). Golden delicious trees
(Malus domesticus) were the most sensitive, some even died.
7.3.2 Animals
Data on the toxicity of PCP for terrestrial animals have been
obtained almost exclusively from laboratory studies. The results
of toxicity tests on experimental animals are presented in section
8. Some fatal cases have been reported in which farm animals were
incidentally exposed to PCP. Blevins (1965), for example, described
a case of acute and lethal poisoning of baby pigs by PCP. The
owners of a newly constructed farrowing house had exceeded the
manufacturer's recommendation in treating the floor with a solution
of PCP in used crankcase oil. All piglets died within one day
after they had been moved into the farrowing house. The sow was
moved outside where she recovered. No information was given
concerning the PCP levels in the air or in the swine.
A recent case, in Canada, of the mortality of young pigs kept
on a PCP-treated wooden floor was reported by Ryan (1983).
Although PCP residues of 310 µg/litre were found in sow's milk
samples, no PCP could be detected in the liver and stomach of the
young pigs. However, µg/kg concentrations of the higher
chlorinated dioxins were found in the skin and liver of the young
pigs, and Ryan (1983) concluded from these findings that these
impurities were responsible for their deaths.
Pesticide poisonings of livestock in the United Kingdom have
been reviewed by Quick (1982) for the period 1977-80. Of 38
suspected PCP poisoning incidents, only 9 were confirmed as PCP
intoxications. High PCP levels found in wood shavings and sawdust,
used as bedding or litter for cats and poultry, apparently caused
the death of animals. Quick (1982) suspected that impurities
present in the commercial PCP products could have been partly
responsible for the deaths.
Hill et al. (1975) reported a study in which the toxicity of
PCP was determined in young birds of 4 species after 5 days of
feeding PCP in the diet; the relatively high LC50 values
(3400 - 5204 mg/kg body weight) do not indicate that PCP is highly
toxic for birds. However, Vermeer et al. (1974) found 50 dead
snail kites after extensive application of Na-PCP as a molluscicide
in the rice fields of Surinam (section 5.1.4).
7.4 Population and Ecosystem Effects
Very few studies have addressed the effects of PCP on aquatic
or terrestrial communities. Field studies on pesticides have
usually been carried out when their accidental release resulted in
visible kills of fish, birds, or other organisms. For instance,
Pierce et al. (1977) and Pierce & Victor (1978) investigated the
fate of PCP in a fresh-water lake after an overflow from a pole
treatment waste pond caused extensive fish kills (section 7.5).
More valuable information about the effects of PCP on
communities can be obtained from model ecosystem studies, in which
the response of portions of the environment placed in a laboratory
was observed. Tagatz et al. (1977) designed a test system
consisting of constant-flow aquaria containing a layer of sand, and
seawater with its plankton as well as animals representing
estuarine macrobenthic communities. The averages and ranges of the
PCP concentrations in the exposed aquaria were 7 µg/litre (3 - 13
µg/litre), 76 µg/litre (47 - 112 µg/litre), and 622 µg/litre (330 -
964 µg/litre). After the 9-week exposure period, a dose-related
decrease was found in the numerically dominant groups. Molluscs in
particular were markedly reduced at 7 µg PCP/litre, and annelids
and arthropods at 76 µg/litre. Almost no animals occurred at 622
µg PCP/litre, while the total numbers of individuals and species
were significantly less in aquaria exposed to 76 µg PCP/litre than
in controls or those exposed to 7 µg PCP/litre. These striking
changes in the relative abundance and diversity of species are
evidence of substantial alterations in the community structure
induced by PCP. In nature, the stability of macrobenthic
communities could be disrupted.
In a second study conducted with Dowicide G-ST (79% Na-PCP),
molluscs were also the most sensitive organisms tested. Levels of
15.8 and 161 µg PCP/litre caused similar reductions in the numbers
of individuals and species (Tagatz et al., 1978).
Using the same test procedure, Cantelmo & Rao (1978) studied
the effects of PCP on meiobenthic communities. Meiofauna comprise
organisms that pass through a 0.5 mm sieve but are retained on a
sieve with mesh widths smaller than 0.1 mm. The Nematoda are
generally the most common taxon in marine sediments (83% in this
study). PCP at 76 µg/litre caused an increase in the biomass and
density of nematodes compared with those in control aquaria, while
higher concentrations of PCP (161 and 622 µg/litre) caused a
decrease. One of the major effects of PCP on nematodes was a shift
from epistrate feeders to deposit feeders at concentrations of 161
and 622 µg PCP/litre. Part of this alteration may have been due to
the reduction of algae serving as a food supply.
Tagatz et al. (1981) reported the effects of PCP on field and
laboratory estuarine benthic communities. In principle, test
apparata were the same as those used in earlier studies, except
that already established communities were exposed to PCP.
Community structure was significantly altered at a PCP
concentration of about 140 µg/litre in both field and laboratory
aquaria; the populations of several invertebrate species were
significantly reduced. There were slight differences in the
effects of PCP on numbers of individuals and species between the
field and laboratory systems.
Cook et al. (1980) examined the effects of PCP on the
microfungal succession of an estuarine benthic microcosm.
Trichoderma sp. was initially the most common fungus isolated from
the sediment. The addition of PCP (140 µg/litre) resulted in an
alteration in the pattern of species; a different species, probably
Penicillium canescens, assumed dominance.
Considerable effects of PCP on the population dynamics of
several species were noted by Schauerte et al. (1982) during an
outdoor study on a natural pond divided into 6 compartments of
about 100 litres each. PCP was applied in the water of 2
compartments at a concentration of 1 mg/litre. Three days after
application, the population of Daphnia pulex pulex was eliminated
from the treated enclosures. The phytoplankton species showed
marked alterations in population dynamics: the autotrophic blue-
green alga Chroococcus limneticus decreased, while the mixotrophic
flagellate Euglena acus significantly increased. This increase was
attributed to the reduced grazing pressure by Daphnia;
interspecific competition was also discussed as possible cause. As
a further secondary effect, the oxygen concentration significantly
decreased, because of the "changed balance between autotrophic and
heterotrophic populations" (Schauerte et al., 1982).
The US Environmental Protection Agency examined the effects of
PCP on a periphyton community in outdoor experimental streams.
Even at the low PCP level corresponding to the water quality
criterion (48 µg/litre) (US EPA, 1980), adverse effects were noted
in terms of community alterations and suppressed community
metabolism (Yount & Richter, 1986). PCP at this concentration also
caused adverse effects on fish growth, larval drift, and larval
yield (Zischke et al., 1985).
7.5 Biotransformation, Bioaccumulation, and Biomagnification
7.5.1. Aquatic organisms
Most research on the bioaccumulation of PCP has been carried
out in aquatic situations. This presents difficulties in that the
bioconcentration factors, which are generally directly related to
the partition coefficients, could vary by several orders of
magnitude, depending on the pH and, at high pH values, on the ionic
strength, the two factors governing the partition coefficient of
PCP (Kaiser & Valdmanis, 1982). In addition, exposure time must
also be taken into account when interpreting PCP residues in
organisms. These influences may, in part, explain the wide range
in bioconcentration factors that has been found.
In general, substances with the solubility properties of PCP
are predominantly taken up by the surrounding water (Niimi &
McFadden, 1982). However, PCP accumulation along the food chains
contributes to its overall bioaccumulation as well.
Table 31 shows the bioconcentration factors derived for several
fish species along with the ambient levels in water. Fresh-water
species seem to accumulate PCP to a much greater extent than marine
fish, possibly because the relevant enzyme systems in marine
species respond faster than those in fresh-water fish (Trujillo et
al., 1982).
Since the ambient concentrations of PCP in the water of natural
aquatic environments are usually less than 1 µg/litre (section
5.1.2), the studies of Niimi & McFadden (1982) are of particular
importance. The authors applied realistic concentrations in
exposing rainbow trout (Salmo gairdneri) to < 10 (control), 35,
and 660 ng Na-PCP/litre, and distinguished between PCP content in
liver and gall bladder, the remaining tissues, and whole fish. As
shown in Table 32, rainbow trout accumulated PCP, even when exposed
to concentrations as low as 35 ng Na-PCP/litre over prolonged
periods. The percentage of PCP stored was highest in the liver and
gall bladder. On the basis of this high bioconcentration at the
low waterborne toxicant concentrations, the authors suggested that
rainbow trout may be less efficient in eliminating PCP than other
species.
On removal from PCP-containing water, fish eliminate previously
accumulated PCP. However, a portion of the PCP incorporated is
more persistent: residues of 0.32 mg/kg (Trujillo et al., 1982) and
0.03 in the muscle to 0.6 mg/kg in the liver (Pruitt et al., 1977)
were still detectable after 18 and 16 days of depuration,
respectively. Elimination half-lives during depuration phases were
4.7 days in killifish (Trujillo et al., 1982) and 6 - 24 days in
rainbow trout (Glickman et al., 1977).
Table 31. Measured bioconcentration factors for PCP for several
fish species
---------------------------------------------------------------------------
Species Time Concentration Bioconcen- Reference
(days) in water tration
(µg/litre) factor
---------------------------------------------------------------------------
Fresh-water fish
Carassius auratus 5 100 1000 Kobayashi &
Akitake (1975a)
Lepomis macro- 1 100 320 Pruitt et al.
chirus (various 4 100 5 - 350 (1977)
tissues) 16 100 4 - 230
Salmo trutta 1 200 100 Hattula et al.
(whole body) (1981)
Salmo gairdneri 20 0.035 200 Niimi &
(whole body) 0.660 130 McFadden (1982)
65 0.035 600
0.660 232
Leuciscus idus 3 42 1050 Freitag et al.
melanotus (whole (1982)
body)
Marine fish
Fundulus similis 4 36 - 306 30 Schimmel et al.
(unspecified (1978)
tissues)
Mugil cephalus 4 26 - 308 38 Schimmel et al.
(unspecified (1978)
tissues)
Mugil cephalus 4 46 6 Faas & Moore
(edible tissues) 4 85 79 (1979)
4 157 56
Fundulus similus 1 57 - 610 8 Trujillo et al.
(whole body) 1 49 (1982)
7 64
7 47
---------------------------------------------------------------------------
In an ecotoxicological profile analysis, Freitag et al. (1982)
determined the bioconcentration of PCP not only in fish (Table 31)
but also in the green alga Chlorella fusca var. vacuolata and in
activated sludge. The bioaccumulation factor in the 24-h algal
test was 1250; in a 5-day activated sludge assay it was 1100 at a
waterborne concentration of 0.05 mg PCP/litre.
Table 32. PCP levels in tissues, organs, and
whole body of rainbow trout (Salmo gairdneri)
exposed to < 10 (control), 35, and 660 ng
Na-PCP/litrea
--------------------------------------------------
Na-PCP PCP concentrations (µg/kg) in:
Days in water liver and remaining whole
(ng/litre) gall bladder tissue body
--------------------------------------------------
0 < 10 2.3 1.1 1.1
20 < 10 1.2 1.5 1.5
35 28 7 7
660 674 77 86
65 < 10 3.9 3.7 3.6
35 135 20 21
660 1984 135 153
115 < 10 3.1 1.9 1.9
35 63 6 7
660 2204 128 160
--------------------------------------------------
a Adapted from: Niimi & McFadden (1982).
Ernst (1979) measured PCP residues in water and benthic
invertebrates at steady state in a static marine system. For the
common mussel (Mytilus edulis) and the polychaete (Lanice
conchilega), bioconcentration factors averaged 390 and 3820 on a
wet weight basis, respectively, at an initial PCP concentration in
sea water of 0.002 - 0.005 mg/litre. The species studied and the
lipid contents of the animals had pronounced effects on the
bioconcentration factor, while temperature and metabolic activity
did not show any remarkable effects on the bioaccumulation. In a
similar study, the polychaete (Neanthes virens) was exposed to 0.1
mg 14C-PCP/litre in sea water (Carr & Neff, 1981). The
bioconcentration factor of 280 was 10 times lower than that
reported by Ernst (1979) for Lanice conchilega.
Clams from the Jadebusen, a North Sea bight, sampled near the
end of a waste-water pipe, accumulated 100 - 1000 times more PCP
than the sediments (Butte et al., 1985).
The studies of Pierce et al. (1977) and Pierce & Victor (1978)
are some of the few field studies concerning the biotransformation
of PCP. The authors followed an accidental discharge into a fresh-
water lake near Hattiesburg, Mississippi, USA. Within two months,
the initially lethal levels of PCP, which had resulted in an
extensive fish kill, decreased to between 6 and 19 µg/litre in the
water and remained near this level throughout the studies,
apparently because of the continuous influx of contaminated water
from other areas. In a control pond with a low background
concentration of 0.5 µg PCP/litre, fish contained only 50 µg
PCP/litre, whereas much higher levels were found in fish in the
contaminated pond. Two months following the spill, levels in fish
averaged 2500 µg PCP/kg dry weight but dropped to 130 µg/kg after
6 months; background levels were achieved within about 10 months.
In a Finnish lake area contaminated through pulp bleaching and
with wood preservative wastes, PCP residues in fish and plankton
did not indicate a strong accumulation via the food chain,
plankton > roach >> pike. However, tetrachlorocatechol, a
possible biodegradation product of PCP, was found to be strongly
absorbed in plankton (Paasivirta et al., 1980).
In littoral microcosms, 14C-PCP was linearly accumulated by
aquatic plants, mainly Potamogeton foliosus and Najos
guadalupensis, during the first 35 days, levelling off to
concentrations over 700 times the initial concentration in water
(41 µg/litre). After 8 weeks, PCP concentration in the macrophytes
rapidly decreased (Knowlton & Huckins, 1983).
In an aquatic model ecosystem, the ecological magnification
value for PCP in fish was 296, the parent compound representing 74%
of the total extractable 14C. The other members had lower
bioconcentration factors: algae, 1.5; mosquito larvae, 16; snail,
121; and Daphnia, 165. In a terrestrial-aquatic model ecosystem,
the bioconcentration factors were: algae, 5; Daphnia, 205; snail,
21; mosquito, 26; and fish, 132 (Lu et al., 1978). These data
indicate that bioaccumulation takes place not only through the
surrounding water but also along the food chains.
7.5.2 Terrestrial organisms
The terrestrial model ecosystem established by Lu et al. (1978)
simulated a crop-soil interaction and included the following
organisms: earthworm (Lumbricus terrestris), slug (Limax maximus),
pillbug (Armadillidium vulgare), saltmarsh caterpillar (Estigmene
acrea), prairie vole (Microtus ochregaster), and corn (Zea mays).
Corn plants grown on the soil of the model system rapidly
accumulated radioactivity. After 14 days, they contained 6.3 mg/kg
of which 16% was intact PCP, 40% were unknown compounds, and 44%
were conjugates. The prairie vole, at the top of the food chain,
consumed virtually all the plant and animal material in the system
within the 5-day exposure time, and then was found to contain 0.5%
of the total dosage applied (25 µg).
Gruttke et al. (1986) investigated the fate of Na-PCP in two
different model food chains representing important groups of
organisms commonly present in soils. In food chain 1, contaminated
bakers-yeast (0.87 µg 14C-Na-PCP/mg dry weight) was fed to
springtails (Folsomia candida), which accumulated up to 0.37 µg
PCP/mg fresh weight after 10 days. Carabid beetles (Nebria
brevicollis) preying on the contaminated springtails showed a body
burden of approximately 4.5 mg PCP/kg fresh weight in the steady
state, from day 4 to 12. Four days after offering uncontaminated
prey, the PCP content in the beetles dropped to 0.4 mg/kg fresh
weight. Similar results were obtained in food chain 2, which
consisted of contaminated leaves of poplar (700 mg Na-PCP/kg dry
weight), with isopods (Oniscus asellus) as primary consumers, and
staphylinid beetles (Ocypus olens) as predators. Because of the
low accumulation tendency, a lasting effect on the predators would
only be expected in case of long-term contamination.
Apart from these model ecosystem studies, little information is
available on the biotransformation of PCP in terrestrial systems.
Miller & Aboul-Ela (1969) observed that cottonseed kernels of bolls
that were closed during spraying accumulated PCP or its metabolites
in quantities of up to 2 mg/kg. No PCP was detected when the bolls
were open during spraying.
PCP, applied in nutrient solution to the roots of growing corn
plants, was taken up by the roots (Schuppener, 1974). At
concentrations of up to 20 mg/litre, roots of sterile hydro-ponic
cultures accumulated as much as 151 mg PCP/kg. Apparently, PCP did
not translocate in the corn plants as it was not detected in the
upper parts of the plants. The root system of sugarcane treated
with 5 mg PCP/litre nutrient solution accumulated 14C-PCP within 4
weeks, retaining over 99% of the total PCP taken up from solution
(Hilton et al., 1970): no measurable translocation into stalks or
leaves occurred. These findings contrast with the translocation
within corn plants described by Lu et al. (1978).
8. EFFECTS ON EXPERIMENTAL ANIMALS AND IN VITRO TEST SYSTEMS
Toxicology data on both purified and commercial PCP are
provided in this review because both are relevant for a health
assessment. The mode of action of chlorophenol can only be
determined using purified chemicals. Furthermore, any decision to
remove or reduce the levels of microcontaminants in PCP would have
to be based on a clear understanding of the toxicity of these
purified products.
8.1. Acute Toxicity
PCP, regardless of the route of administration, is the most
acutely toxic of the chlorophenols tested in laboratory animal
species. Oral LD50 values range between 27 and 205 mg/kg body
weight for a variety of species, regardless of the vehicle of
administration and the grade of PCP (Table 33). Acute oral exposure
of mice and rats to lethal doses of PCP (Deichmann, 1943;
Farquharson et al., 1958; Borzelleca et al., 1985; Renner et al.,
1986) results in an increase in respiratory rate, a marked rise in
temperature (4 - 4.5 °C), tremors or possibly convulsions, and a
loss of the righting reflex. Asphyxial spasms and cessation of
breathing usually occurs 0.5 - 2 min before cardiac arrest. A
rapid and intense rigor mortis is observed within 3 - 5 min of
death and approximately 45 min sooner than the onset of rigor
mortis in rats given ether. Similar signs are observed with lethal
exposure to PCP and its sodium salt, regardless of the route of
administration.
In addition to its systemic effects, PCP also induces more
localized effects in test organisms. Both dermal and subcutaneous
applications have produced swelling, skin damage, and occasionally
hair loss in a variety of animals (Kehoe et al., 1939; Baader &
Bauer, 1951; Johnson et al., 1973). Localized effects on blood
vessels may result in hyperaemia or erythema (Kehoe et al., 1939;
Baader & Bauer, 1951). Contact with PCP causes irritation of the
eye, skin, or respiratory mucosae in man (section 9). Skin
bioassay techniques have shown that technical PCP, but not purified
PCP, is acnegenic for rabbits, and that the acnegenic effects are
caused by the microcontaminants, particularly H6CDD (Johnson et
al., 1973).
There are far fewer published data on the effects of acute
exposure of animals to PCP and Na-PCP via the dermal and pulmonary
exposure routes compared with oral exposure, which is surprising
considering that the primary exposures in the work-place are via
the skin and lung. Kozak et al. (1979) reported that PCP was
readily absorbed when applied to the skin of experimental animals.
The effects of acute dermal exposure have been examined only in the
rat and rabbit. For rats, but not rabbits, PCP is much more toxic
when given orally than when applied dermally (Table 33). However,
with Na-PCP, toxicity via the 2 routes appears to be similar (Table
34). The only acute inhalation toxicity value reported for Na-PCP
is for rats; Na-PCP is at least 10 times more toxic via inhalation
than by oral ingestion (Hoben et al., 1976c).
Table 33. Acute toxicity of PCP
---------------------------------------------------------------------------------------------------------
Species Para- Sex Doseb Route Purity/carrier Reference
metera
--------------------------------------------------------------------------------------------------------
Mouse LD50 F 74 oral PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 M 36 oral PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 F 150 oral PCP in polypropylene glycol Ahlborg & Larsson (1978)
LD50 M 177 oral PCP in 10% Emulphor Borzelleca et al. (1985)
LD50 F 117 oral PCP in 10% Emulphor Borzelleca et al. (1985)
LD50 M 129 oral PCP in corn oil Renner et al. (1986)
LD50 F 134 oral PCP in corn oil Renner et al. (1986)
LD50 F 32 intraperitoneal PCP in 40% ETOH Ahlborg & Larsson (1978)
LD50 M 59 intraperitoneal PCP in polypropylene glycol Ahlborg & Larsson (1978)
LD50 M 59 intraperitoneal PCP in corn oil Renner et al. (1986)
LD50 F 61 intraperitoneal PCP in corn oil Renner et al. (1986)
LD50 M/F 82 subcutaneous Ning et al. (1984)
Rat LD50 150 oral Schwetz et al. (1978)
LD50 F 135 oral commercial PCP Schwetz et al. (1974)
LD50 M 205 oral commercial PCP Schwetz et al. (1974)
LD50 78 oral 1% in olive oil Deichmann et al. (1942)
LD50 65 oral Schwetz et al. (1978)
(3- to
4-day-
old)
LD50 27 oral 0.5% in Stanolex fuel oil Deichmann et al. (1942)
LD50 M/F 83 oral reagent grade Ning et al. (1984)
LD50 M 146 oral peanut oil Gaines (1969)
LD50 F 175 oral peanut oil Gaines (1969)
MLD M/F 160 oral peanut oil Gaines (1969)
LD50 F 149 cutaneous PCP technical 40% w/v Noakes & Sanderson (1969)
solution in glycerol formal
LD50 320 cutaneous xylene Gaines (1969)
LD50 330 cutaneous xylene Gaines (1969)
MLD M/F 300 cutaneous xylene Gaines (1969)
Rat MDLD M 56 intraperitoneal olive oil Farquharson et al. (1958)
LD50 100 subcutaneous 4% in fuel oil Deichmann (1943)
LD50 90 subcutaneous 4% in fuel oil Deichmann & Mergard (1948)
LD50 M/F 40 subcutaneous reagent grade Ning et al. (1984)
--------------------------------------------------------------------------------------------------------
Table 33. (contd.)
--------------------------------------------------------------------------------------------------------
Species Para- Sex Doseb Route Purity/carrier Reference
metera
--------------------------------------------------------------------------------------------------------
Rabbit MLD 100 - oral 11% PCP in olive oil Kehoe et al. (1939)
130
MLD 70 - oral 5% in Stanolex fuel oil No. 1 Deichmann et al. (1942)
90
MLD 40 - cutaneous various carriers Deichmann et al. (1942)
170
MLD 350 cutaneous 11% in olive oil Kehoe et al. (1939)
MLD 39 cutaneous 1.8% in pine oil Kehoe et al. (1939)
MLD 60 cutaneous 5% in Stanoflex fuel oil No.1 Kehoe et al. (1939)
MLD 110 cutaneous 5% in Shell Dione oil Kehoe et al. (1939)
MLD 70 - subcutaneous 5% in Olive Oil Kehoe et al. (1939)
85
Hamster LD50 168 oral Cabral et al. (1979)
LD50 70 - subcutaneous 5% in olive oil Kehoe et al. (1939)
85
Sheep MLD 120 oral aqueous suspension of sawdust Harrison (1959)
treated at 20 lb technical
PCP/cu. ft
Calf MLD 140 oral aqueous suspension of sawdust Harrison (1959)
treated at 20 lb technical
PCP/cu. ft
---------------------------------------------------------------------------------------------------------
a MLD = Minimum lethal dose.
MDLD = Median lethal dose.
LD50 = Estimated dosage capable of causing 50% mortality of a test population.
LC50 = Estimated concentration capable of causing 50% mortality of a test population.
b LD50 or MLD in mg/kg body weight; LC50 in mg/m3 air.
The limited data on other routes of administration (intra-
peritoneal, intravenous, perhaps subcutaneous), which introduce
PCP or Na-PCP directly into the body indicate that these exposures
result in a stronger toxic effect on rats, hamsters, and mice than
either oral or dermal exposure (Tables 33, 34). These differences
probably result from incomplete uptake of the compound via the
oral, dermal, and perhaps subcutaneous routes.
PCP is considerably more toxic than its sodium salt when
administered orally to rats or rabbits, or dermally to rabbits
(Tables 33, 34). However, subcutaneous or intravenous injections
of PCP and Na-PCP are almost equally toxic. These patterns may
reflect differences in the rate of absorption of the parent
compounds when applied to the skin of rats, but this does not
appear to be true of PCP.
One sex is not consistently more strongly affected by PCP than
the other. For a given combination of organism (rats and mice),
vehicle, and exposure route (mostly oral), males are sometimes
more, sometimes equally, and sometimes less sensitive than females
(Table 33). However, technical PCP is more toxic for female rats
than for males (Schwetz et al., 1974) and more toxic for young rats
than for adults (Schwetz et al., 1978). This differential toxicity
between the sexes is apparent in both short- and long-term studies
with technical PCP and with one of its contaminants, hexachloro-
dibenzo- p-dioxin (H6CDD).
Although the influence of exposure route, the form of the PCP
(sodium salt or parent molecule), and the sex of the experimental
animal are evident within a given study, such patterns may be
masked by a number of factors. Unfortunately, the type and extent
of contamination of PCP tested under acute exposure conditions is
rarely described, despite the fact that some of the
microcontaminants, especially some congeners of polychlorinated
dibenzo- p-dioxin (PCDD) and polychlorinated dibenzofuran (PCDF),
are extremely toxic. Furthermore, a variety of solvents is used to
administer chemicals tested for acute toxicity, and some of these
solvents can enhance or decrease absorption of PCP, thus affecting
toxicity. Hence, the variations in the LD50, LDLO and TDLO values
reported in Kozak et al. (1979), Ahlborg & Thunberg (1980), Jones
(1981), NRCC (1981), and NIOSH (1983) for each animal species and
route of exposure may result, in part, from the use of a variety of
purified and commercial products containing several different
solvents, as well as possible differences in animal strains, or
test design (in this regard, note that some of the earlier studies
involved only a small number of animals, sometimes as few as two).
Acute toxic effects, other than chloracne, which are due
exclusively to the presence of PCDDs or PCDFs, are difficult to
identify. PCDDs, in particular, have a delayed toxic effect that is
masked by the rapid onset of signs of acute exposure to the PCP
molecule.
Table 34. Acute toxicity of Na-PCP
---------------------------------------------------------------------------------------------------------
Species Parametera Doseb Route Purity/carrier Reference
---------------------------------------------------------------------------------------------------------
Mouse LD50 83 subcutaneous reagent grade Ning et al. (1984)
Rat LD50 210.6 oral 2% aqueous Deichmann et al. (1942)
LD50 (F) 125 - 200 oral commercial (79%); aqueous Stohlman (1951)
LD50 71 oral reagent grade Ning et al. (1984)
LD50 104 dermal reagent grade Ning et al. (1984)
LD50 66 subcutaneous 2% aqueous Deichmann et al. (1942)
LD50 38 subcutaneous reagent grade Ning et al. (1984)
LC50 294 inhalation reagent grade; 2-h inhalation Ning et al. (1984
LD50c 11.7 inhalation aqueous aerosol Hoben et al. (1976c)
LD50 34 intraperitoneal aqueous Hoben et al. (1976c)
Rabbit MLD 218 oral 1% NaCl Kehoe et al. (1939)
MLD 250 - 300 oral 5% aqueous Deichmann et al. (1942)
MLD 450 - 700 oral aqueous McGavack et al. (1941)
MLD 250 cutaneous 10% aqueous Deichmann et al. (1942)
MLD 257 cutaneous 2% aqueous Kehoe et al. (1939)
MLD 450 - 600 cutaneous aqueous McGavack et al. (1941)
MLD 100 subcutaneous 10% aqueous Deichmann et al. (1942)
MLD 250 - 300 subcutaneous aqueous McGavack et al. (1941)
MLD 50 - 150 intraperitoneal aqueous McGavack et al. (1941)
MLD 22 - 23 intravenous 2% aqueous Deichmann et al. (1942)
MLD 22 intravenous 1% aqueous Kehoe et al. (1939)
Guinea- MLD 266 cutaneous aqueous Kehoe et al. (1939)
pig
Dog MLD 135 subcutaneous aqueous McGavack et al. (1941)
---------------------------------------------------------------------------------------------------------
a MLD = Minimum lethal dose.
LD50 = Estimated dosage capable of causing 50% mortality of a test population.
LC50 = Estimated concentration capable of causing 50% mortality of a test population.
b LD50 or MLD in mg/kg body weight; LC50 in mg/m3 air.
c Inhalation toxicity value expressed by author as LD50, not LC50.
8.2. Short-Term Toxicity
As discussed in the previous section, some of the acute effects
of exposure to commercial PCP are attributable to microcontaminants
present in the technical preparation. In addition, signs of
exposure to some of these microcontaminants, notably PCDDs, may
not appear for weeks. As a consequence, it is particularly
important to consider the possible confounding effects of these
impurities in reviewing the long-term toxicity of chlorophenols.
In this section, studies on the toxicity of purified PCP, technical
grade PCP, and comparative studies are discussed separately.
8.2.1. Pure or purified PCP
Debets et al. (1980) studied the effect of 99% pure PCP fed to
female rats for 5 weeks. The PCP concentration used was 500 mg/kg
feed, which corresponds to an approximate dose of 40 - 50 mg/kg
body weight per day. Of several liver microsomal enzymes tested,
only ethoxyresorufin O-de-ethylase (20-fold) and glucuronyl
transferase (3-fold) increased in activity with exposure. Body
weight gain, urine and liver porphyrin concentrations, and liver
weights were unaffected by PCP. Interestingly, PCP accelerated the
onset of hexachlorobenzene (HCB) porphyria, suggesting that it is
the PCP metabolized from HCB that causes the porphyria.
In 6-week-old pigs, Greichus et al. (1979) found no overt signs
of toxicosis associated with the oral administration of purified
PCP (at 5, 10, and 15 mg/kg body weight per day in capsules) for 30
days. However, at 10 and 15 mg/kg per day, hepatocyte size
increased and enlarged livers were observed.
8.2.2. Technical grade PCP
Knudsen et al. (1974) fed rats diets containing 0, 25, 50 and
200 mg commercial PCP/kg for 12 weeks. The 50 mg/kg exposure
(corresponding to about 2.3 mg/kg body weight per day) increased
liver weights in both sexes, while haemoglobin concentrations,
haematocrit, and glucose concentrations in serum were elevated in
the 2 highest dosage groups. The no-observed-adverse-effect level
of 25 mg/kg corresponded to an ingested dose of about 1.2 mg/kg
body weight per day.
Post-mortem examination of dairy cattle fed 0.2 mg/kg body
weight per day, for 75 - 84 days, and 2 mg/kg body weight per day
for another 56 - 60 days during lactation, revealed enlargement of
the liver, lungs, kidneys, and adrenals, thickening of the urinary
bladder walls, chronic interstitial nephritis, and subacute
urocystitis in exposed animals (Kinzell et al., 1981). In vitro
testing identified a significant loss of renal function associated
with exposure.
8.2.3. Comparative studies
Recent studies of between 2 and 8 months duration are useful in
discerning differences in the effects of purified and technical
grade pentachlorophenol (Table 35). These studies on rats and mice
have been summarized by Fielder et al. (1982) and NRCC (1982).
Rats receiving 500 mg technical PCP (Table 1, section 2.2) per
kg feed for 8 months had slower growth rates, hepatomegaly,
porphyria, and increased hepatic enzyme activities (aryl
hydrocarbon hydroxylase, glucoronyl transferase, and cytochrome
P-450) (Goldstein et al., 1977). When rats were fed purified PCP
under similar conditions, i.e., 500 mg/kg feed for 8 months, only
retardation of growth rate and an increase in liver glucoronyl
transferase activity were observed. Rats fed 20 mg technical
PCP/kg feed for 8 months had elevated liver enzyme activities,
while rats fed purified PCP at the same concentrations did not.
These findings have been confirmed by Kimbrough & Linder (1978)
using rats exposed to 0, 20, 100, and 500 mg/kg of the same
technical and purified PCP and the same protocol as was employed by
Goldstein et al. (1977). Male and female rats exposed to either
purified or technical PCP gained less weight than controls.
Exposure to purified PCP in the diet at 500 mg/kg resulted in
slightly enlarged liver cells and caused occasional cytoplasmic
inclusions. These effects were not observed in rats fed lower
doses. In contrast, exposure of rats to diets containing 500 mg
technical PCP/kg increased liver weights and thickened the walls of
the hepatic central veins in both sexes, and also caused
pleiomorphic hepatocytes with foamy or vacuolar cytoplasm in male
rats. The livers of females exposed to 500 mg/kg were
characterized by vacuolation and degeneration of hepatocytes and
mitotic anomalies. Similar, but less severe, effects were noted in
rats exposed to diets containing 100 mg and 20 mg technical PCP/kg.
Wainstok de Calmanovici & San Martin de Viale (1980) determined
that technical PCP is more toxic for porphyrin metabolism in rats
than the purified compound. Dosing with 45 - 90 mg technical
PCP/kg body weight per day (the amount given by stomach tube for 18
weeks varied) enhanced the excretion of porphyrins and their
precursors, increased the deposition of porphyrins in the spleen,
liver, and kidney, and altered the activity of enzymes involved in
porphyrin metabolism. Larger doses of purified PCP (100 - 195
mg/kg body weight per day) had similar effects.
Table 35. No-observed-adverse-effect-levels (NOAELs) established in rats
exposed orally to pure, technical, and purified technical grades of PCP
---------------------------------------------------------------------------
PCP Sex NOAEL (mg/kg Reference
body weight
per day)
---------------------------------------------------------------------------
Short-term toxicity
Pure 3 Johnson et al. (1973)
Purified 3 Johnson et al. (1973)
technical
Technical < 3 Johnson et al. (1973)
Technical approximately Knudsen et al. (1974)
1.2
Teratogenicity/
fetotoxicitya
Technical male, female 5 Schwetz et al. (1974)
(progeny)
Purified male, female < 5 Schwetz et al. (1974)
technical (progeny)
Reproductive
toxicityb
Purified male, female 3 Schwetz et al. (1978)
technical
Long-term toxicity
Pure approximately Goldstein et al. (1977)
5
Technical < 1 Kimbrough & Linder (1978)
Purified female < 3 Schwetz et al. (1978)
technical male < 10 Schwetz et al. (1978)
---------------------------------------------------------------------------
a Studies involved exposure of females during days 6 - 15 of gestation
and evaluation of progeny.
b Studies involved exposure of both sexes for 62 days before mating, for
15 days during mating, and, subsequently, throughout gestation and
lactation for females.
In a similar comparative study by Johnson et al. (1973), 99%
pure PCP and purified technical PCP (H6CDD reduced to 1 mg/kg) at
10 and 30 mg/kg body weight per day increased liver weight, but did
not affect other variables monitored. In contrast, unpurified PCP
increased liver and kidney weights, enhanced serum-alkaline
phosphatase activity, and reduced serum-albumin, numbers of
erythrocytes, total haemoglobin, and haematocrit.
The toxicity of different grades of PCP has also been studied
in cattle (McConnell et al., 1980; Parker et al., 1980; Hughes et
al., 1985). These studies have confirmed that effects such as
reduced weight gain, anaemia, liver pathology, and a decrease in
thymus weight were induced by microcontaminants in the technical
PCP. Reductions in serum-thyroid hormones (triiodo-thyronine T3 and
thyroxine - T4) observed in cows fed either purified or technical
PCP were probably due to the chlorophenol itself. Significant
levels of octa-, hepta-, and hexachlorodioxins were found in liver,
fat, and milk at the conclusion of 160 days of exposure (Parker et
al., 1980). Firestone et al. (1979) also reported residues of PCP
and related chemicals in cows' milk, body fat, and blood following
short-term exposures. Only 3 out of 7 PCDD congeners identified in
the technical PCP used were found in tissue and body fluids, i.e.,
1,2,3,6,7,8-H6CDD, 1,2,3,4,6,7,8-H6CDD, and OCDD.
Hexachlorobenzene (HCB) and PCP were also detected. Levels of PCP,
HCB, and PCDD in pooled milk fat from 3 cows reached 4 mg/kg, 200
µg/kg, and 85 µg/kg, respectively. PCP levels fell to 100 µg/kg a
few days after the cessation of dosing and levels of HCB and total
dioxin declined by 50% in 50 days.
8.3. Long-Term Toxicity
The 8-month PCP feeding studies on rats by Goldstein et al.
(1977) and Kimbrough & Linder (1978) might be considered by some to
be long-term. The results of these studies (section 8.2) indicate
that the toxicity of technical PCP preparations is primarily
attributable to microcontaminants.
Schwetz et al. (1978) reported a 2-year exposure of rats to
purified PCP (Table 35). In females, no toxic effects were
observed at levels below 3 mg/kg body weight per day; pigment
accumulation was observed at 10 mg/kg body weight per day, and
decreased body weight gain, increased serum-glutamic pyruvic
transaminase (GPT) activity, and pigment accumulation were observed
at 30 mg/kg body weight per day. Fewer changes were observed in
male rats; no effects were observed at levels below 10 mg/kg body
weight per day, and pigment accumulation and increased GPT activity
were reported at 30 mg/kg body weight per day. It is interesting
to note that Schwetz et al. (1978) did not observe any absolute or
relative weight increase in kidney and liver at 30 mg/kg body
weight per day. In contrast, Johnson et al. (1973) reported
increased weights of these tissues in male and female rats after a
90-day exposure to the same purified PCP used by Schwetz et al.
(1978) at 30 mg/kg body weight per day (section 8.2). An earlier
90-day rat study with purified PCP containing < 0.5 mg PCDD or
PCDF/kg also demonstrated changes in liver and kidney weight at the
30 mg/kg body weight per day dose level (Kociba et al., 1971).
Ning et al. (1984) reported test results for laboratory animals
exposed to airborne Na-PCP. Both weanling rats (males) and rabbits
(males and females) were exposed to reagent grade Na-PCP at 21.4
mg/m3 or 3.1 mg/m3 for 4 h per day, 6 days per week, for 4 months.
Rabbits (6 pooled males and females) in the high-dose group showed
a statistically significant increase in serum-gamma-globulin but
not in alpha- or beta-globulin or serum-albumin. Lung weight
increased significantly in the high-dose group and liver weight
increased significantly in both dose groups compared with controls.
In rats, the lung, kidney, liver, and adrenal gland all increased
significantly in weight in the high-dose group compared with the
same organs in control animals. Blood-glucose levels in rats from
the group exposed to 21.4 mg/m3 remained higher than those in
controls, throughout the study.
These results are consistent with previous observations
reported by Demidenko (1969). Rats and rabbits exposed to 28.9 or
2.97 mg PCP/m3 for 4 h per day and 4 months were significantly
adversely affected at the high dose (anaemia, leukocytosis,
eosinophilia, hyperglycaemia, dystrophic processes in the liver).
At the low dose, only minor effects on liver function,
cholinesterase activity, and blood sugar were registered, which
returned to normal one month after completion of exposure.
Although the results of these 2 inhalation toxicity studies are
only preliminary, they give an indication that short-term
inhalation of Na-PCP or PCP at concentrations as low as about 3
mg/m3 can cause biochemical and gross pathological effects in
laboratory mammals. Assessing the data of Demidenko (1969), Kunde
& Böhme (1978) calculated from the 3 mg/m3 concentration a daily
dose of 0.3 mg/kg body weight per day for rats, assuming 100%
pulmonary uptake and absorption. This dose would indicate that
PCP is at least 10 times more toxic with inhalation exposure than
with oral exposure. This finding is corroborated by the results of
studies comparing acute inhalation and acute oral exposure
(section 8.1).
Other long-term studies on animals have been designed
specifically to evaluate the carcinogenic properties of PCP and are
reported in section 8.6.
8.4. Effects on Reproduction and Fetal Development
There is good agreement that PCP is a fetotoxic agent; however,
it does not appear to be teratogenic (Kozak et al., 1979; Ahlborg &
Thunberg, 1980; Fielder et al., 1982; NRCC, 1982). These
conclusions are based primarily on the studies of Schwetz et al.
(1974, 1978) on rats.
Technical grade PCP administered to pregnant female rats from
day 6 to day 15 of gestation did not have any effects on the mother
or fetus at 5 mg/kg body weight per day (Schwetz et al., 1974).
Fetal resorptions and delayed development of fetuses were observed
at 15 mg/kg body weight per day, and signs of maternal toxicity,
based on weight loss, were observed at 35 mg/kg body weight per
day. Reports of delayed ossification of the skull, supernumerary,
fused, or missing vertebrae and lumbar spurs are usually considered
indicative of delayed development rather than teratogenicity, and
are responsible for the differences of opinion between the early
position of US EPA, that PCP is teratogenic (Cirelli, 1978b), and
most other reviews. Purified PCP induced effects similar to those
of technical PCP; however, maternal toxicity and decreased fetal
weights occurred at 30 mg/kg body weight per day, and delayed fetal
development was observed at the 5 mg/kg body weight per day dose
level, which had previously been found to be the no-observed-
adverse-effect-level for technical grade PCP. More limited
fetotoxic effects were observed in rats exposed to PCP by Courtney
et al. (1976).
Na-PCP (> 98% pure) fed to female Wistar rats at 10, 30, or 60
mg/kg body weight during days 8 - 19 of gestation led to
statistically significant reductions in body weight in females,
decreased litter weights, and dramatic increases in fetal
resorption and fetal death in the 2 highest dose groups (Anon,
1981). No birth defects were observed in the control group or in
the group fed 10 mg/kg body weight; however, 3/31 pups examined
from females in the 30 mg/kg body weight group had major
malformations (hare lip, umbilican hernia, exocephalus) and 60%
had spine and rib malformations (supernumerary, fused, bifurcated,
or short ribs). In addition, retardation of ossification and
increased breadth of sagittal fissure were extensive in this group.
No pups were born to females in the 60 mg/kg body weight group.
The authors concluded that 10 mg/kg body weight was the no-
observed-adverse-effect level for teratogenicity, fetotoxicity, and
embryotoxicity in rats administered Na-PCP. Considering the
linearity of the dose-response curve and the fact that 10 mg/kg
body weight was the lowest dose administered, the no-observed-
adverse-effect level reported in this study is not substantially
different from that determined for PCP.
It is probable that the reduced fetotoxicity of technical PCP
relative to the purified material, if not artifactual, is a result
of the presence of microcontaminants (NRCC, 1982). Liver enzymes
that accelerate the rate of PCP metabolism are known to be
activated by phenobarbital, 3-methylcholanthrene, and 2,3,7,8-T4CDD
(Ahlborg & Thunberg, 1978). Other PCDD and PCDF microcontaminants
in technical PCP may also activate microsomes and reduce the amount
of fetal exposure to PCP, causing a concomitant decrease in
toxicity. Indeed, the induction of liver enzymes by technical PCP
has been demonstrated in the rat by Goldstein et al. (1977).
Purified PCP did not increase the activity of liver enzymes.
Technical grade PCP was contaminated with 8 - 1380 mg PCDDs/kg and
4 -1500 mg PCDFs/kg, while purified PCP contained less than 0.1 mg
of these contaminants per kg.
Larsen et al. (1975) reported single instances of dwarfism,
exencephaly, macrophthalmia, and taillessness in fetuses after
pregnant rats were fed single doses of 60 mg purified PCP per kg
body weight per day on days 8, 9, or 10 of gestation. However,
these findings were attributed to maternal hyperthermia, which is
known to cause teratogenic effects in rats (Edwards, 1968).
Hyperthermia is a common outcome of exposure to large, single doses
of chlorophenol.
Exon & Koller (1983a) investigated the effects of PCP on rats
exposed pre- and postnatally. Females were exposed to technical
PCP (95% pure) in the feed from 21 days of age, throughout breeding
(at 90 days), gestation, and up to weaning of the pups, 21 days
after parturition. Exposure doses were 0, 5, 50, and 500 mg/kg
body weight per day. The progeny were exposed to the same levels
of PCP in feed as their mothers for a total exposure period of 12
1/2 months. No significant decreases in mean litter size or
percentage of still-born pups were recorded in the groups given
PCP. There was a significant decrease in survival to weaning in
the group fed 5 mg/kg body weight but not in the 2 higher dose
groups.
A level of purified technical PCP of 30 mg/kg body weight per
day (Schwetz et al., 1978) as well as pure Na-PCP at 26 mg/kg body
weight per day (Kunde & Böhme, 1978) fed to male and female rats
for 62 days before mating, 15 days during mating, and to females
during gestation and lactation, caused reductions in the numbers of
offspring, neonatal body weight, neonatal survival, and growth of
weanlings. The no-observed-adverse-effect level was 3 mg/kg body
weight per day (Table 35). Male fertility did not appear to be
affected in this study.
8.5. Mutagenicity
Williams (1982) reviewed the literature and concluded that,
although there are deficiencies in the data, the Ames Salmonella
typhimurium test (Andersen et al., 1972), a sex-linked lethal test
with Drosophila melanogaster (Vogel & Chandler, 1974), and a host-
mediated assay (Schwetz et al., 1978) all indicate that PCP
probably does not cause point mutations. On the basis of negative
findings in an in vivo mammalian dominant lethal test (Buselmaier
et al., 1973), PCP does not seem to cause chromosomal aberrations.
However, an in vitro study to show primary damage using
Saccharomyces cerevisiae demonstrated an increase in mitotic gene
conversion (Fahrig, 1974; Fahrig et al., 1978). A mammalian spot-
test pointed to weak mutagenic activity (Fahrig et al., 1978). PCP
did not cause single-strand breaks in human fibroblast DNA, while
its metabolite tetrachlorohydroquinone did (Witte et al., 1985).
Lymphocytes taken from workers at a PCP factory showed a small, but
significantly higher, incidence of dicentric and acentric mitoses
(Bauchinger et al., 1982). Sodium pentachlorophenate (20 mmol)
reportedly increased the frequency of both auxotrophic and
morphological variations in the fungus Aspergillus niger strain 350
(Roy et al., 1981). However, because of shortcomings in these
studies, the information available is still insufficient to assess
the mutagenicity of PCP. The host-mediated assay with bacteria
(Schwetz et al., 1978) and the sex-linked recessive lethal assay
(Vogel & Chandler, 1974) have only been carried with single dose
levels. In the Ames test (Andersen et al., 1972), no metabolic
activation system was used. The increase in the incidence of
offspring with coat discoloration in the mouse spot test (Fahrig et
al., 1978) was not statistically significant. The mutagenicity
test of sodium pentachlorophenate on Aspergillus niger 350 (Roy et
al., 1981) did not include a control group.
8.6. Carcinogenicity
The carcinogenicity of chlorophenols in mammals is a
contentious issue. In 1979, the International Agency for Research
on Cancer reviewed the data available on the carcinogenic
properties of PCP (IARC, 1979a) and concluded that the information
was inadequate for a meaningful assessment of carcinogenicity.
Two carcinogenicity bioassays have been carried out using PCP.
Innes et al. (1969) exposed 2 hybrid strains of mice to roughly the
maximum tolerated dose of PCP for a total of 78 weeks, and did not
find any significant increases in tumour incidence in males or
females of either strain. Schwetz et al. (1978) report similar
findings in rats exposed to a maximum of 30 mg technical PCP/kg
body weight per day, for 24 months. Using these data, the
Carcinogenic Assessment Group of the US EPA concluded that PCP was
negative with respect to oncogenic effects (Williams, 1982).
PCP (technical and commercial grade) is currently being tested
by the National Toxicology Program of the US EPA (NRC, 1986),
however, data from this study were not available to the Task Group
for evaluation.
NOTE: The results of these studies have now been published
(US NTP (1989) Toxicology and carcinogenesis studies of two
pentachlorophenol technical-grade mixtures (CAS No. 87-86-5) in
B6C3F1 mice (feed studies), Research Triangle Park,
North Carolina, US National Toxicology Program, Technical Report
No. 349, p. 98). The conclusions were as follows:
"Under the conditions of these 2-year feed studies, there
was clear evidence of carcinogenic activity for male
B6C3F1 mice fed diets containing technical-grade
pentachlorophenol, as shown by increased incidences of adrenal
medullary and hepatocellular neoplasms. There was some
evidence of carcinogenic activity for female B6C3F1
mice exposed to technical-grade pentachlorophenol, as shown by
increased incidences of hemangiosarcomas and hepatocellular
neoplasms. There was clear evidence of carcinogenic
activity for male B6C3F1 mice exposed to
pentachlorophenol, EC-7, as shown by increased incidences of
adrenal medullary and hepatocellular neoplasms. There was
clear evidence of carcinogenic activity for female
B6C3F1 mice exposed to pentachlorophenol, EC-7, as
shown by increased incidences of adrenal medullary and
hepatocellular neoplasms and hemangiosarcomas."
Exon & Koller (1983a) investigated the potential for PCP to act
as a co-carcinogen by administering ethylnitrosourea (ENU) to
female rats exposed pre- and/or postnatally to 0, 5, 50, or 500 mg
PCP/kg body weight per day. ENU was administered as ethylurea in
drinking-water and nitrite in feed during days 14 - 21 of
gestation. The progeny were exposed to the same levels of PCP in
feed as their mothers for a total exposure period of 12.5 months.
High incidences of tumours in progeny exposed to PCP + ENU could
not be separated from those observed in progeny exposed to ENU
alone.
Tumour promotion studies on phenol and chlorophenol using
dimethylbenzanthracene (DMBA) as the initiator on mouse skin
indicated that phenol, 2-MCP, 2,4-DCP, and 2,4,5-T3CP were probable
tumour promoters, but that 2,4,6-T3CP and higher chlorophenols,
including PCP, were not (Boutwell & Bosch, 1959). None of the
chlorophenols tested were tumorigenic, when applied alone.
Carcinogenicity bioassays involving oral exposure have been
conducted on one other chlorophenol, 2,4,6-trichorophenol (NCI,
1979) and on a mixture of 2 hexachlorodibenzo- p-dioxin isomers
(NCI, 1980a). 2,4,6-T3CP caused a significant increase in cancer
in a variety of tissues in both male and mice and in male rats.
A mixture of two H6CDD isomers known to contaminate technical
PCP caused a significant increase in cancer in the livers of female
rats and mice (Table 36).
Thus, animal data indicate that, when PCP is ingested, it does
not cause cancer in mice or rats, and that, when applied to the
skin or administered orally, it does not act as a tumorigen or a
tumour promoter. Nevertheless, there is evidence that one other
chlorophenol is an animal carcinogen, the lower chlorinated phenols
are tumorigens, and that H6CDD found in PCP is carcinogenic, when
ingested by rodents. In addition, Arrhenius et al. (1977b)
suggested that PCP may be able to act as a co-carcinogen, on the
basis of its effects on liver microsomes.
8.7. Other Studies
The immunotoxic effects of PCP have been investigated in a
variety of animal species. Cows fed technical PCP have shown
thymic hypoplasia in one study (McConnell et al., 1980) but no
overt differences in immunological function in another (Forsell et
al., 1981). Mice exposed to technical grade PCP have shown reduced
humoral immunity and an in vitro impairment of T-cell cytolytic
activity (Kerkvliet et al., 1982a,b). Analytical grade PCP did not
have any effect. In the rat, a decrease in humoral immunity and an
increase in cell-mediated immunity was demonstrated following
exposure to technical PCP (97% pure) (Exon & Koller, 1983b).
Hillam & Greichus (1983) reported a suppression of total leukocyte
counts, gamma globulins, and IgG in young pigs exposed to technical
PCP (95% pure) at dose levels of 5 and 10 mg/kg body weight per day
for 30 days. Chickens ingesting 2400 µg/g feed of purified PCP
exhibited significantly reduced humoral responses to injections of
bovine serum-albumin and lymphoproliferative responses to the
mitogens concanavalin and phytohaemaglutinin and lower white blood
cell counts (Prescott et al., 1982). Thus, some immunotoxic effects
are observed when PCP (pure and technical) is administered to
experimental animals. In studies carried out with technical PCP,
it is likely that the non-phenolic contaminants such as PCDD are
responsible for most of the observed immunotoxic effects.
A neurotoxic effect of PCP (grade not specified) has been
reported by Walum & Peterson (1984) on the basis of an in vitro
assay with cultured mice neuroblastoma cells in which an increase
in cell detachments with exposure to PCP was observed. Also, a
transient alteration in brain tissue enzyme activity was found in
rats given 20 mg technical PCP per litre drinking-water over a
period of 3 - 18 weeks (Savolainen & Pekari, 1979). The
significance of these findings is unclear.
8.8. Contaminants Affecting Toxicity
The toxicological evaluation of pentachlorophenol is
complicated by the presence of several impurities in technical
grade formulations of this chemical. Some of these impurities are
extremely toxic in their own right. On the other hand, some
microcontaminants are capable of inducing liver microsomal
enzymes, and in so doing, affect the rates of metabolism and
excretion and the fetotoxicity of PCP (Ahlborg & Thunberg, 1978)
(section 8.4). Thus, meaningful assessments of toxicological
studies on the effects of pentachlorophenol are impossible without
an accurate knowledge of the type and extent of contamination of
the PCP under investigation.
Concern for the toxic effects of microcontaminants has focused
on the dioxins, because of the extreme toxicity of the intensely
studied congener 2,3,7,8-T4CDD. However, it is necessary to
emphasize that this congener has not been found frequently in PCP.
A brief summary of the toxic properties of the microcontaminants of
chlorophenols is provided in section 2.2. Extensive reviews of the
toxicology and residue levels of PCDDs and PCDFs are available in
Hutzinger et al. (1982), Jones (1981), NRCC (1981), Fielder et al.
(1982), Kociba & Schwetz (1982), Umweltbundesamt (1985), and in
several articles published in Boddington et al. (1985).
8.8.1. Octachlorodibenzodioxin (OCDD)
Only one congener exists of this fully substituted isomer. It
was not acutely toxic for rats, when administered orally at 1 mg/kg
body weight or acnegenic for rabbits, when applied to the ear as a
10% solution in chloroform (Fielder et al., 1982). Exposures of
approximately 1 mg/kg body weight per day for 3 weeks did not cause
any toxic signs. Livers were nominally enlarged, but appeared
normal under the light microscope. OCDD does not appear to be
mutagenic in the Ames test and has not undergone a carcinogenicity
bioassay. Studies of the effects of OCDD on reproduction and fetal
development indicate that it is not teratogenic at 500 µg/kg body
weight per day or fetotoxic at 100 µg/kg body weight per day, when
administered to females on days 6 - 15 of gestation.
8.8.2. Heptachlorodibenzodioxin (H7CDD)
Few data available on H7CDD. The LD50 value has not been
determined accurately for either the 1,2,3,4,6,7,8- or
1,2,3,4,6,7,9-isomers.
An in vitro assessment of the induction of aryl hydrocarbon
hydroxylase (AHH) in rat hepatoma cell cultures was used to
calculate the biological potency of both H7CDD isomers relative to
2,3,7,8-T4CDD. The relative potencies of 1,2,3,4,6,7,8-H7CDD and
1,2,3,4,6,7,9-H7CDD were 0.3 - 0.5% and 0.011 - 0.025% respectively
(Bradlaw et al., 1980).
Table 36. Summary of toxicology data for hexachlorodibenzo- p-dioxin (H6CDD)
---------------------------------------------------------------------------------------------------------
Species Toxic for: Vehiclea Isomerb Route Toxicity Observations Reference
(sex) (µg/kg body
body weight
per exposure
period)
---------------------------------------------------------------------------------------------------------
Acute toxicity
Lethality (LD50)
Rat females CO:acetone B/C mix oral 800 Observations in all NCI (1980a)
males 1800 studies included:
Mouse females CO:acetone B/C mix oral 500 weight loss, skin NCI (1980a)
males 750 eruptions, delayed
death; Tissues
females, CO A oral 825 affected: liver, McConnell
males CO B oral 1250 thymus, spleen, et al.
CO C oral 1440 kidney, testes (1978)
Guinea- females, CO A oral 73 McConnell
pig males CO B oral 70 - 100 et al.
CO C oral 60 - 100 (1978)
Short-term toxicity
(NOAEL)
Rat female, CO:acetone B/C mix oral < 2.5 thymic atrophy, splenic NCI (1980a)
male (per week) hypertrophy, and liver
lesions at 10 - 50 µg/kg
body weight per week
Mouse female, CO:acetone B/C mix oral 1.2 only liver damage at NCI (1980a)
male (per week) higher levels (10 - 50
µg/kg body weight
female, acetone dermal << 1.5 per week NCI (1980b)
male (per week)
---------------------------------------------------------------------------------------------------------
Table 36. (contd.)
---------------------------------------------------------------------------------------------------------
Species Toxic for: Vehiclea Isomerb Route Toxicity Observations Reference
(sex) (µg/kg body
body weight
per exposure
period)
---------------------------------------------------------------------------------------------------------
Fetal toxicity
(NOAEL)
Rat females, CO:acetone oral 0.1 (per fetal oedema at 1 µg/kg Schwetz
males day during body weight; resorptions et al.
gestation) at 10 µg/kg body (1973)
weight; teratogenic and
maternal toxicty at
100 µg/kg body weight
Long-term toxicity
Carcinogenicity
bioassay
Rat females CO:acetone B/C mix oral carcinogen Loss of body weight NCI (1980a)
> 1.5 (male and female);
(per week) hepatocellular
carcinomas and
neoplastic nodules
in females only
Mouse females CO:acetone B/C mix oral carcinogen hepatocellular carcin- NCI (1980a)
(incon- > 2.5 omas and adenomas in
clusive (per week) females only;
in males) incidence in males
not significant
incon- acetone B/C mix dermal inconclu- fibrosarcomas observed NCI (1980b)
clusive sive in females; incidence
not significant
---------------------------------------------------------------------------------------------------------
a CO = corn oil; CO:acetone = 9:1 mixture of corn oil and acetone.
b A = 1,2,3,4,7,8-H6CDD; B = 1,2,3,6,7,8-H6CDD; C = 1,2,3,7,8,9-H6CDD.
8.8.3. Hexachlorodibenzodioxin (H6CDD)
Commercial PCP and Na-PCP have been found to contain 4 out of
10 possible H6CDD isomers; however, the 1,2,3,6,8,9- and
1,2,3,6,7,8-isomers predominate (Fielder et al., 1982). Levels have
been in the 5 - 10 mg/kg range in recent commercial samples of PCP
and Na-PCP. The acute oral toxicity (LD50) of 1,2,3,6,7,8-H6CDD in
the mouse is 1250 µg/kg (Table 36). H6CDD is more toxic for female
rats and mice than for males. Signs of toxicity include weight
loss and deterioration of the skin. The onset of mortality is
often delayed for up to 3 weeks. Thymus, liver, spleen, kidney,
and testes are affected by H6CDD. This isomer is also known to be
acnegenic.
Oral exposure of rats to H6CDD isomers indicated that no-
observed-adverse-effect levels for the rat were < 2.5 µg/kg body
weight per week and, for the mouse, < 1.25 µg/kg body weight per
week (NCI, 1980a). Above these levels, weight loss and liver
damage were observed. Dermal exposure of mice to H6CDD also
resulted in liver damage and mortality, even at the lowest dose of
1.5 µg/kg body weight per week (NCI, 1980b). Thus, H6CDD is readily
absorbed through the skin and extremely toxic. A mixture of
1,2,3,6,7,8- and 1,2,3,7,8,9-H6CDD has been shown to be
carcinogenic for female mice and rats; however, males did not
develop hepatocellular carcinomas or adenomas in excess of the
control rate in the same study (NCI, 1980a). Carcinogenicity
assays using dermal exposures were inconclusive (NCI, 1980b).
On the basis of the results of exposure of pregnant rats to an
unidentified mixture of H6CDD isomers, this homologue is considered
fetotoxic and teratogenic (Schwetz et al., 1973). The no-observed-
adverse-effect-level was 0.1 µg/kg body weight. Cleft palate,
vertebrae with split or unfused centra, and split sternebrae were
observed in the fetuses of females fed 100 µg/kg body weight on
days 6 - 15 of gestation. Fetal resorptions were observed at
maternal doses of 10 µg/kg body weight and fetal subcutaneous
oedema at doses of > 1 µg/kg body weight.
8.8.4. Polychlorinated dibenzofurans (PCDFs)
Little is known of the toxicity of the PCDFs, despite their
relatively common occurrence in chlorophenol formulations at
concentrations of 1 - 500 mg/kg. Some have considerable acute oral
toxicity. For example, the LD50 of 2,3,7,8-T4CDF in guinea-pigs
and monkeys is 5 - 10 µg/kg body weight and 1000 µg/kg body weight,
respectively (Jones, 1981). Rappe et al. (1982) indicated that
1,2,3,7,8-P5CDF, 2,3,4,7,8P5CDF, and 2,3,4,6,7,8-H6CDF all had LD50
values in the 1 - 100 µg/kg body weight range for the most
sensitive species tested. As with PCDD, the toxicity of PCDF
isomers appears related to the extent of symmetrical positioning of
chlorine atoms in the 2, 3, 7, and 8 positions of the molecule.
2,3,7,8-T4CDF and 2,3,4,7,8-P5CDF appear to have the longest half-
lives of the PCDFs studied (Masuda & Kuroki, 1982). Furthermore,
these authors have suggested that PCDFs, especially the tetra- and
penta-isomers, may have been largely responsible for the signs of
Yusho disease reported in the Japanese who ingested rice oil
contaminated with PCBs, PCDF, and polychlorinated quarterphenyls
(PCQs). PCDFs have acnegenic properties, but may not be
porphyrinogenic.
8.8.5. Polychlorodiphenyl ethers (PCDPEs)
PCDPEs are common contaminants of chlorophenols and are found
almost exclusively in T4CP and PCP (Jones, 1981). Little is known
of their toxicity.
8.8.6. Other microcontaminants
Less common impurities of chlorophenols include polychlorinated
phenoxyphenols (PCPP) ("predioxins" or "isopredioxins"),
polychlorinated biphenyls (PCBs), and polychlorinated benzenes
(Jones, 1981). Extensive toxicology data exist on the effects of
PCBs (WHO, 1976) and some members of the chlorinated benzene group,
i.e., hexachlorobenzene (IARC, 1979b). Although these 2 groups of
chemicals are not acutely toxic, they can affect reproduction and
are considered carcinogenic. The levels found in technical
formulations of PCP are not likely to increase their toxicity or
carcinogenicity.
8.9. Mechanism of Toxicity
PCP is known to be cytotoxic for mammalian cells (Packham et
al., 1982). All chlorophenols, especially PCP, are uncouplers of
oxidative phosphorylation (Kozak et al., 1979). However, the
molecular basis for the uncoupling action is not clear (NRCC,
1981). PCP binds to mitochondrial protein and inhibits
mitochondrial ATPase activity. PCP may have 2 independent effects
on mitochondria; uncoupling oxidative phosphorylation and also
inhibiting mitochondrial ATPase (Stockdale & Selwyn, 1971a,b).
Thus, both the formation of ATP and the release of energy to the
cell from the breakdown of ATP to ADP are prevented. Electron
transport is not inhibited by PCP, though reactions dependent on
available high-energy bonds, such as oxidative and glycolytic
phosphorylation, are affected. Binding to enzymic protein has
been reported and may lead to the observed inhibition of other
cellular enzymes (Kozak et al., 1979). An increase in cellular
oxygen demand during the uncoupling of oxidative phosphorylation
has also been observed, which gives rise to the initial increase in
respiration rate reported for individuals poisoned by PCP
(Weinbach, 1957; Mitsuda et al., 1963; Wood et al., 1983).
9. EFFECTS ON MAN
There are no studies or case reports of the effects of pure or
purified PCP on human beings. Human exposure is nearly always to
technical grades of PCP or Na-PCP in a variety of formulations.
Reference to "PCP" in this section is to the technical grade.
9.1. Acute Toxicity - Poisoning Incidents
In man, the minimum lethal oral dose (LDLO) of PCP has been
estimated to be 29 mg/kg body weight (Ahlborg & Thunberg, 1980).
Kozak et al. (1979) report that this value depends on the ambient
temperature at the time of exposure, and the general health and
renal competence of the individual. PCP is approximately 5 times
more toxic than phenol (estimated oral LDLO is 140 mg/kg body
weight). The proportional lethality of these 2 chemicals (LDLO
phenol:LDLO pentachlorophenol) in man is almost identical to the
proportional lethality of the LD50 values for these substances in
rats.
Numerous accidental or suicidal poisonings with commercial
chlorinated phenols have been reported (Nomura, 1954; Menon, 1958;
Blair, 1961; Bergner et al., 1965; Mason et al., 1965; Armstrong et
al., 1969; Robson et al., 1969; Watanabe & Watanabe, 1970; Haley,
1977; Stevens & Richardson, 1979; Gjovik et al., 1981; Wood et al.,
1983), and nearly 60% of these acute exposures have resulted in
death. These cases together with the results of animal studies
provide a relatively clear picture of the signs and symptoms of
acute exposure to technical pentachlorophenol in man.
In contrast to the lower chlorinated phenols, PCP does not
cause convulsions. Ataxia, mental and physical fatigue, headaches,
dizziness, disorientation, anorexia, nausea, vomiting, dyspnoea,
hyperpyrexia, tachycardia, and a rise in metabolic rate are common
signs and symptoms of PCP poisoning. Most prominent are extreme
weakness, elevated body temperature, and profuse sweating. Death
is due to cardiac arrest and poison victims usually show a marked
rigor mortis (Truhaut et al., 1952a,b; Nomura, 1954; Mason et al.,
1965; Robson et al., 1969; Watanabe & Watanabe, 1970).
The gross pathology and histological lesions associated with
acute exposures to PCP are generally consistent between laboratory
animals and man. Oral exposures result in gastric and intestinal
inflammation; however, the severity can depend on the carrier
solvent and the presence of other chemicals (Menon, 1958; Stevens &
Richardson, 1979). Pulmonary oedema and congestion have been
reported after inhalation exposure, and occasionally oral exposure,
if aspiration of ingested PCP has occurred. Splenomegaly,
cardiomegaly, renal congestion, hepatomegaly, and hepatic
congestion are also frequently observed at autopsy.
Histologically, fatty degeneration, and necrosis in the
centrilobular region of the liver have been reported, together with
degenerative lesions in renal tubules (Gordon, 1956; Menon, 1958;
Blair, 1961; Bergner et al., 1965; Mason et al., 1965; Robson et
al., 1969).
It is generally agreed that the signs and symptoms of acute
toxicity observed in animals and human beings exposed to
chlorophenols result from the effects of the chlorophenol molecule
itself rather than the microcontaminants, with hyperthermia,
profuse sweating, and the rapid onset of morbidity and early death
associated with acute chlorophenol exposures. These signs are not
observed in animals exposed only to PCDD and PCDF; death is delayed
by up to 3 weeks in acute exposure studies with these
microcontaminants.
9.2. Effects of Short- and Long-Term Exposures
Most data on the effects of non-acute exposures to
chlorophenols in man come from occupational studies. The clinical
outcome of repeated exposure to PCP has been reviewed by Fielder et
al. (1982), Williams (1982), and Exon (1984). The high rate of
employee turnover and variation in the level and duration of
exposure make it difficult to distinguish between subacute, short-
and long-term exposures. For this reason, the following studies
concerning occupational PCP toxicity in man are not separated on
the basis of duration of exposure. Interpretation of these studies
is frequently confounded by factors such as age, alcohol
consumption, tobacco smoking, and other aspects of life style.
9.2.1. Occupational exposure
Clinical studies have identified a number of toxic effects of
short-term PCP exposure in man, some of which are also
characteristic of acute intoxication (section 9.1). Symptoms
include irritation of the skin, mucous membranes, and respiratory
tract, signs of chloracne, neurasthesia, depression, headaches,
porphyria cutanea tarda, and liver and kidney functional changes
(Fielder et al., 1982). These effects are discussed in greater
detail in the following sections. Among workers employed in
pressure-treating wood with PCP, insomnia and vertigo have also
been reported (Arsenault, 1976).
9.2.1.1 Skin and mucous membranes
Workers exposed to airborne concentrations of 1 mg PCP/m3 or
more have reported painful nasal irritation (Deichmann & Keplinger,
1981). Variations in the effect level are associated with the
historical exposure of the individual to inhaled PCP. Workers
accustomed to exposure may have a higher threshold for irritating
effects and may tolerate up to 2.4 mg PCP/m3 air.
As in the case of experimental animals (section 8), persons
exposed to large amounts of technical PCP develop chloracne.
Fielder et al. (1982) summarized published cases of chloracne in
workers at PCP-manufacturing sites in Czechoslovakia, the Federal
Republic of Germany, the United Kingdom, and the USSR. Kozak et
al. (1979) reported other cases in Japan and the USA. The use of
Na-PCP and Na-tetra-chlorophenol has also resulted in chloracne in
woodworkers (Behrbohm, 1959). Baxter (1984) reported chloracne and
minor disturbances of the lipid metabolism among 40 workers from a
PCP-manufacturing plant over a 3-year study period. However, the
author concluded that the abnormalities observed were due to the
PCDD contaminants and could not be attributed to the PCP
preparation.
A survey of sawmill workers in British Columbia, Canada,
carried out using self-administered questionnaires, indicated that
dermatological and respiratory symptoms were significantly higher
in a PCP/T4CP exposed group than in the control group (Sterling et
al., 1982). However, no reliable estimates of exposure were
provided.
A more detailed study carried out in the same geographical area
made use of personal monitors carried by individual workers to
determine exposure levels of PCP and T4CP (Embree et al., 1984).
Blood and urine samples were collected and analysed, and health and
employment histories were recorded by a trained interiewer. The
workers were divided into 3 groups: a high exposure group handling
wet-treated lumber; a medium exposure group with no manual contact
with treated lumber; and a control group with no exposure to PCP,
T4CP, or related chemicals. Exposure concentrations for PCP are
shown in Table 20. The authors reported a correlation between
exposure levels and serum- and urine-chlorophenol concentrations.
However, they were unable to substantiate the findings of Sterling
et al. (1982) of increased incidences of respiratory and
dermatological health problems in workers exposed to PCP/T4CP.
A study on 113 employees at a wood-treatment facility found
that workers were in good health overall, but with a greater than
expected prevalence of skin pustular eruptions (Flickinger &
Lawrence, 1982). Airborne exposures were less than 0.03 mg/m3.
Klemmer et al. (1980) reported the results of a 7-year study on
400 Hawaiians, many of whom had long-term, high-level exposure to
PCP. Concentrations of PCP in blood-serum far exceeded the 1.05
mg/litre reported in Arsenault's (1976) study; workers treating
wood in open-vats had a mean level of 3.78 mg PCP/litre, pressure-
tank workers 1.72 mg/litre, and farmers and controls 0.25 and 0.32,
mg/litre, repectively. After considering data on 189 individuals of
the total of 400, Klemmer et al. (1980) concluded ... "despite high
chronic exposures to PCP, individuals in the wood treatment group
of workers had not undergone any serious health effects from this
exposure. The only evidence of tangible health effects, part of
which could have been caused by exposures to chemicals other than
PCP, were the low-grade infections or inflammations of the skin and
subcutaneous tissue, of the protective membrane of the eye, and of
the mucous membrane of the upper respiratory tract. No specific
long-term effects could be elicited in the exposed group".
9.2.1.2 Liver and kidney
Indications of significant liver damage have not been found.
Elevations in circulating levels of some hepatic enzymes have been
reported; however, they are usually transitory and do not suggest
severe functional impairment (Kozak et al., 1979; Fielder et al.,
1982). These findings are consistent with those reported in
studies on rats with short-term exposure to technical PCP (section
8.2).
Kidney functional changes resulting in reductions in creatinine
clearance and resorption of phosphorus have been reported by Begley
et al. (1977). The spontaneous normalization in kidney function
during a 3-week non-exposure period indicated that this effect on
kidney is largely reversible.
Jirasek et al. (1974) reported the clinical signs exhibited by
workers who suffered intoxication during the manufacture of Na-
2,4,5-T3CP and Na-PCP. These workers displayed abnormal porphyrin
metabolism (increased uroporphyrin and delta-aminolevulinic acid
in urine, UV fluorescence of liver), and indications of
hepatotoxicity (liver enlargement, mild steatosis or fibrosis of
liver tissue, elevated levels/activities of bilirubin, serum-
glutamic-oxaloacetic transaminase and serum-glutamic-pyruvic
transaminase).
Mild dysfunction of the liver has been reported among Soviet
workers engaged in the production of Na-PCP (Vinogradova et al.,
1973) including, for example, a reduced ability to synthesize
protein.
Zober et al. (1981) reported a study on a small group of
woodworkers involved in the application of PCP. The average
concentration of PCP in the air at the time of the study was 2.4
µg/m3, the average exposure period for the cohort was 3 years and
the average levels in urine and serum were 46 µg PCP/g creatinine
and 1 µg/ml, respectively. Elevations in serum-aminotransferases
and alpha-glutamyl transpeptidase were observed; however,
confounding factors of sample size and alcohol consumption
prevented the formation of any conclusions concerning the effects
of PCP on liver function.
As part of a similar study (Embree et al., 1984) (section
9.2.1.1), Enarson et al. (1986) found that serum levels of
creatinine, bilirubin, glutamic oxaloacetic transaminase, and
alkaline phosphatase in sawmill workers exposed to a mixture of Na-
PCP and Na-tetrachloropenate did not differ from those measured in
the controls.
9.2.1.3 Blood and the haematopoietic system
Aplastic anaemia has been associated with PCP use (Roberts,
1981); however, sample sizes were small and exposures were not
quantified. Incidental references to haematological changes in
isolated workers in a German chemical plant manufacturing PCP and
HCB were made by Baader & Bauer (1951). Effects on the
haematopoetic systems of animals have been reported in studies with
PCDD contaminated PCP (Johnson et al., 1973; Knudsen et al., 1974;
McConnell et al., 1980) and 2,3,7,8-T4CDD (Allen & van Miller,
1978). Wood (1980) examined the relationship between anaemias
(with no established cause) in 128 Canadian woodworkers and
exposure to PCP. No significant differences between the
haematology values of exposed and unexposed workers were found.
Wood (1980) concluded that PCP exposure did not appear to have any
significant effects on the prevalence of anaemias in these
woodworkers.
In a companion report to that of Embree et al. (1984) (section
9.2.1.1), Enarson et al. (1986) found few exposure effects in
sawmill workers exposed to Na-PCP and Na-tetra-chlorophenate. Most
blood variables monitored were within normal ranges and did not
differ between exposed and unexposed workers. A significant
decrease in haematocrit and an increase in haematuria were reported
in workers handling treated lumber, but not in workers exposed
solely through inhalation.
Urinary-PCP values (2.2 mg/litre) reported by Shirakawa et al.
(1959) in primarily female workers at several rubber manufacturing
factories indicated that these workers were exposed to high levels
of Na-PCP (presumably technical grade). Increased blood sugar
levels, decreased blood pressure, and dermatoses were reported, but
no worker was reported to have missed work through the effects of
PCP exposure.
9.2.1.4 Nervous system
Investigation of clinical reports of neuropathy did not
indicate any overt significant signs of peripheral neuropathy in a
recent study on PCP workers (Triebig et al., 1981). Sensory nerve
conduction velocity was significantly reduced in exposed workers,
but was not correlated with urinary levels of PCP.
Skin, blood, and neurological disorders have been reported
among workers at a Na-PCP manufacturing factory in the USSR
(Vinogradova et al., 1973). The workers were exposed to air levels
of PCP and Na-PCP ranging between 0.03 and 1 mg/m3. Readings for
21% of the air samples ranged from 0.21 to 1 mg/m3, exceeding the
maximum permissible concentration in the USSR of 0.1 mg/m3.
Washings taken from clothing and exposed skin yielded PCP and Na-
PCP values of 21 - 212 mg/dm2 and 7.6 - 75 mg/dm2, respectively.
However, concentrations of hexachlorobenzene were also 2 - 3 times
higher than the maximum permissible concentration (0.9 mg/m3) set
in the USSR and may have had an influence on the disorders
reported.
9.2.1.5 Immunological system
McGovern (1982) suggested that man may suffer an immunotoxic
response to phenolic compounds, including chlorinated phenolic
compounds. Marked T-cell suppression has been observed in several
patients exposed to phenols. Zober et al. (1981) reported that
some woodworkers exposed to PCP displayed increased concentrations
of immunoglobulins, though this increase was not correlated with
exposure. Ning (1984) reported that workers exposed to PCP showed
significant decreases in IgG and IgA immunoglobulins. The results
of animal studies, while indicating that PCP is not strongly
immunotoxic, confirm that PCP exposure can lead to immunological
changes (section 8.7).
9.2.1.6 Reproduction
There are few published data on the effects on male or female
reproductive capacity of short- or long-term exposure to
chlorophenols. Schrag & Dixon (1985) classified PCP as "agents
with inconclusive effects" on male reproduction. Corddry (1981)
investigated pregnancy outcomes in women married to sawmill workers
in Canada. Analysis of data from 43 women, with a total of 100
pregnancies, did not reveal any significant differences in the
pregnancy outcomes of women living with "exposed" compared with
"unexposed" men. There was a slight trend towards more adverse
pregnancy outcomes in the exposed group, but this trend disappeared
when the alcohol consumption was considered as a confounding
factor. Male fertility was not studied.
9.2.1.7 Cytogenetic effects
There is no evidence to indicate that PCP or other
chlorophenols exert cytogenetic effects on human cells. A study of
circulating lymphocytes in a small group of workers in Idaho, USA,
indicated that individuals exposed to PCP had a slightly higher
rate of chromosome breakage than controls, but the increase was not
statistically significant (Wyllie et al., 1975). Bauchinger et al.
(1982) reported that lymphocytes from 22 workers in a PCP-
manufacturing factory had a significantly elevated number of
chromosomal aberrations (dicentrics and acentrics). These data are
not adequate for assessing the cytogenetic effects of PCP in man.
9.2.1.8 Carcinogenicity
Only 2 reports associating exposure to PCP specifically with
human cancer are available. Greene et al. (1978) suggested that
there was an association between exposure to wood treatment
chemicals (PCP) and the incidence of Hodgkin's disease, on the
basis of a family case history (2 of 4 siblings contracting the
disease were occupationally exposed to PCP) and a relative risk
(RR) of 4.2 (from death certificates, in the USA) for persons
employed in carpentry and lumbering. Bishop & Jones (1981)
reported 2 cases of non-Hodgkin's lymphoma in PCP workers in the
United Kingdom; both cases were associated with chloracne. These
data are not adequate for the identification of a positive and
statistically sound correlation between lymphomas and PCP.
However, there is some epidemiological evidence that exposure
of workers to mixtures of chlorophenols, but not specifically PCP,
increases their risk of developing soft-tissue sarcomas and
lymphomas. Considerable debate has ensued since the initial report
of chlorophenol-related cancer by Hardell (1977) and the subsequent
reports of Hardell and his co-workers in Sweden. Case control
studies of soft-tissue sarcoma patients in Sweden indicated a
relative risk (RR) of 6.6 for those "exposed" to chlorophenols
compared to those who did not appear to have been exposed (Hardell
& Sandström, 1979). Individuals exposed to 2,4,5-T had an RR of
5.8. A follow-up study in another area of Sweden involving 330
subjects tended to confirm the overall risk of soft-tissue sarcomas
in individuals exposed to phenoxyacetic acids and chlorophenols
(Erickson et al., 1981). The authors reported RR values of 6.8 for
all phenoxyacetic acid exposures and 3.3 for chlorophenol
exposures. Exposures to phenoxyacetic acids, assumed by the
authors to be free of PCDD and PCDF impurities resulted in an RR of
4.2. Hence, Erickson et al. (1981) concluded that impurities in
these chlorinated phenols and phenoxyacids were probably not the
sole cause of the elevated cancer rates reported, though they might
have played a role in this apparent carcinogenicity.
The validity of the assumption that 2,4-D is free of PCDD and
PCDF "impurities" is questionable inasmuch as 2,4-D has been found
to be contaminated, in one case, with H6CDD (IARC, 1977) and, in
another, with T4CDF (Norström et al., 1979). However, it is
reasonable to assume that the contamination of the phenoxy-acid
herbicides 2,4-D, MCPA, necoprop, and dichlorprop with PCDDs and
PCDFs is very low.
Hardell et al. (1981) have also applied their case-referent
technique to malignant lymphoma patients (both Hodgkin's disease
and non-Hodgkin's lymphomas) in Sweden. They reported RR values in
individuals exposed to phenoxy acids, chlorophenols, and other
organic solvents to be 4.8, 4.3, and 2.4, respectively. The RR
value for high-level exposure to chlorophenols was as high as 8.4.
A possible explanation for the lymphomas may rest with the
immunological effects (in animals) of the PCDD contaminant,
2,3,7,8-T4CDD. Some immunosuppressive chemicals have been shown to
cause an increase in histiocytic lymphomas in man (Hardell, 1979).
In response to criticisms that recall bias was a significant
factor in his studies, Hardell (1981a) applied his case-control
method to study colon cancer, a disease that correlates positively
with asbestos exposure, but not with chlorophenol exposure. His
findings indicated that recall and observer bias were negligible in
his earlier studies, since colon cancers correlated significantly
only with asbestos exposure, and not with phenoxy acids or
chlorophenols exposure. Hardell et al. (1982) also used their
technique to demonstrate an increased risk of nasal/nasopharyngeal
cancer (RR = 7) among workers exposed to chlorophenols.
Others have not found associations between cancer and human
exposure to chlorophenols. In contrast to Hardell et al. (1982),
Tola et al. (1980) did not find any relationship between nasal
cancer and chlorophenol exposure in Finnish workers. A case-
control study in New Zealand (Smith et al., 1984) did not reveal a
higher incidence of soft-tissue sarcoma in workers exposed to
chlorophenols. Gilbert et al. (1983) conducted a cohort study in
Hawaii with workers employed in the wood-treatment industry, in
which chromated copper-arsenate, tributyl tin oxide, and PCP were
used. They did not find any adverse health effects, but urinary-
PCP levels were higher in the exposed group.
In a recent Swedish study on the risks of soft-tissue sarcoma,
a cohort of 354 620 men employed in agriculture or forestry was
compared to a reference cohort of 1 725 845 men employed in other
industries during the period 1961-79 (Wiklund & Holm, 1986). A
relative risk of 0.9 (95% confidence interval 0.8 - 1) was found.
When the cohort was divided into 6 subgroups, based on assumed
exposure to phenoxy acid herbicides, no significant differences in
relative risks were found. Despite the increased use of phenoxy
acid herbicides in Sweden between 1947 and 1970, no time-related
increase in the relative risk of soft-tissue sarcomas was found.
The authors concluded that their study did not confirm the results
of Hardell (1981b). However, they pointed out that only a small
percentage of their total cohort of agricultural and forestry
workers in Sweden were possibly exposed to phenoxy acid herbicides
(15%) and chlorophenols (2%). Hence, a relative risk of 1.5
observed for sarcomas in these groups, as defined in their study,
would be equivalent to an actual 6-fold risk from exposure to these
compounds. Thus, it is unlikely that their study would have
detected a true increased risk from such exposures, if the risk
were less than 6-fold.
Pearce et al. (1986) studied 82 cases of non-Hodgkin's lymphoma
in New Zealand with 168 cancer controls and 228 general population
controls. They obtained statistically significant odds ratios (OR)
of 2.7 and 2, respectively, for workers in the pelt department of
meat works with potential exposure to 2,4,6-trichlorophenol and for
workers who carried out fencing with potential exposure to both
2,4,6-trichlorophenol and pentachlorophenol. Further examination
of the data revealed that: 2 of the 4 lymphoma cases who worked in
the pelt department were possibly not exposed to TCP; that a
significant proportion of the fencing workers also worked in the
meat works; and that no significant risk was found for exposure to
chlorophenols as a group. Pearce et al. (1986) concluded that the
excess risk observed in these 2 groups of workers might not be due
to chlorophenol exposure. In a second study, Pearce et al. (in
press) added other lymphoma cases to their previous study sample
and found similar relationships. They concluded that, though an
association with chlorophenol exposure was unlikely, it could not
be ruled out. They proposed that alternative hypotheses, such as
exposure to oncogenic zoonotic viruses should be considered to
explain their findings.
While there is some evidence that chlorophenols, and in
particular trichlorophenols, are associated with elevations in the
rates of certain cancers in exposed individuals, there is no clear-
cut dose-effect relationship. "Exposure" has been loosely defined
in most studies and no quantitative assessments have been
published. In addition, it has been suggested that, since other
environmental chemicals such as hexachlorobenzene,
pentachlorobenzene, and pentachloronitrobenzene, are metabolized to
PCP in animals and man, there is no necessary relationship between
PCP concentrations in body fluids and exposure to PCP (Renner &
Mücke, 1986). Other factors that could have a bearing on the
conflicting reports of chlorophenol exposure and cancer incidence
include differences in study methods and the diagnosis of soft-
tissue sarcoma cases, and inadequacies in death-certificate data.
The results of epidemiological studies, currently underway in
several countries, could confirm or refute the association between
chlorophenol exposure and human cancer (Fingerhut et al., 1984).
9.2.1.9 Other systems
It is not unusual to find few or no signs of toxicity in
workers with long-term exposure to low levels of PCP or Na-PCP.
Arsenault (1976) reported a prospective clinical evaluation of 21
PCP workers involved in the pressure-treatment of wood, who had
been exposed for an average of 9 years and had elevated blood-serum
levels of PCP (on average, 1.05 mg/litre, versus 0.1 mg/litre in
controls). The only significant clinical findings in the pressure-
treatment workers were vertigo and insomnia. Arsenault (1976) also
provides information obtained from the health records of 1330
workers in a large wood-processing company. From 1961 to 1971,
only 26 cases of health problems related to PCP use and exposure
were identified; however, it is probable that this is an
underestimate because of under-reporting.
Similarly, in a cohort study comparing 88 wood-treatment
workers with 61 controls (Gilbert et al., 1983), no significant
effects of exposure (by history or physical examination) to wood
preservatives, including PCP, were reported on: skin or mucous
membranes of the eyes or upper respiratory tract; mental status;
cardiovascular, gastrointestinal, genitourinary, or neuromuscular
systems; or reproduction. In the accompanying historical
perspective study, calculations of age-specific deaths rates from
all causes for 125 workers, over 21 years, showed that observed
rates were similar to, or lower than, those expected.
9.2.2. General population exposure
References to non-occupational exposure to chlorophenols, for
example from wood in homes, confirm that pulmonary, and, to a
lesser extent, dermal exposure to PCP can produce symptoms of
poisoning similar to those documented in occupational settings.
These studies (section 5.2) may be of significance in as much as
they identify new sources of exposure; however, they add little to
the toxicology data base for PCP. Concentrations of PCP in the
indoor air of homes and in the urine and serum of their residents
are elevated relative to those in the general population (Table
21). The limited effects of this exposure are considered briefly
here.
In cases where individuals display symptoms of PCP
intoxication, usually as a result of the application of PCP in the
interior of houses, typical acute symptoms are observed, but other
parameters (haematological, biochemical) may be normal. Sangster
et al. (1982) outlined case histories of 3 families in PCP-treated
houses who reported experiencing one or more of the following signs
or symptoms: generalized itching or burning dermatosis, nausea,
vomiting, decreased appetite, headache, dizziness, and fatigue.
Haematological, urinary, and biochemical parameters were unaffected
by exposure. Similarly, a young girl poisoned by bathwater stored
in a PCP-contaminated tank displayed fever, intermittent delirium,
rigors, acidosis, and elevated urine levels of ketones and amino
acids, but her respiratory rate and other clinical symptoms were
normal (Chapman & Robson, 1965). However, longer-term exposure may
have more profound effects. Brandt et al. (1977) reported that
exposure to PCP for several years in the air of a treated wooden
house resulted in liver damage and elevated activities of several
liver enzymes in a German woman (Ahlborg & Thunberg, 1980).
A Sacramento woman lost weight, and complained of weakness and
tightness in the chest after the interior of her house was treated
with PCP (Anon, 1970).
Krause & Englert (1980) examined several medical and laboratory
parameters in 250 persons with elevated PCP exposure (section
5.3). No clear relationship could be found between elevated
concentrations of PCP in the urine and biochemical parameters
related to the liver, kidney, and blood. However, significantly
more complaints of headache, fatigue, tonsillitis, hair loss, and
bronchitis were reported in persons with PCP exposure. Because the
signs and symptoms usually reported in connection with indoor PCP
exposure are relatively non-specific, thye cannot be definitively
ascribed to PCP. However, the observation that many symptoms
disappeared when exposure was reduced (by improving ventilation,
sealing wood surfaces, or leaving the premises) is indicative that
PCP or the substances included in the formulated product might well
be the causative agents. The persistence of some biochemical and
dermatological signs, similar to those reported in the work-place,
is a further indication that PCP may induce subacute effects in
these exposed persons.
In general, however, no adverse effects can be ascribed to the
low ambient concentrations of PCP resulting from the diffuse
sources to which most people are exposed.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
10.1. Evaluation of Human Health Risks
In this subsection, PCP and Na-PCP are referred to as PCP.
10.1.1. Occupational exposure
10.1.1.1 Exposure levels and routes
Occupational exposure to technical PCP mainly occurs through
inhalation and dermal contact. Virtually all workers exposed to
airborne concentrations take up PCP through the lungs and skin. In
addition, workers handling treated lumber or maintaining PCP-
contaminated equipment would be exposed dermally to PCP in
solution, and may take up from one-half (based on urinary-PCP
concentrations) to two-thirds (using serum levels) of their total
PCP burden through the skin.
The actual concentrations to which workers have been exposed
are seldom measured but, where they have been monitored, they are
predictably high. Airborne levels at PCP-production and wood-
preservation facilities have ranged from several µg/m3 to more than
500 µg/m3 in some work areas. The outer layer of treated wood can
contain up to several hundred mg/kg, though levels are usually less
than 100 mg/kg.
These exposures result in concentrations of PCP in the serum
and urine that are 1 - 2 orders of magnitude higher than those in
the general population without known exposure. Mean/median urinary-
PCP concentrations of approximately 1 mg/litre are typical for
workers in contact with PCP, compared with urinary concentrations
of approximately 0.01 mg/litre for the general population (section
5.4).
Automated processes and the use of closed systems have greatly
reduced worker exposure in large-scale manufacturing and modern
wood-treatment factories and sawmills. Other improvements in
industrial hygiene can significantly reduce exposure, as measured
by lower urinary-PCP concentrations.
10.1.1.2 Toxic effects
Past use of PCP has affected workers producing or using this
chemical. Chloracne, skin irritation and rashes, respiratory
disorders, neurological changes, headaches, nausea, weakness,
irritability, and drowsiness have been documented in exposed
workers. Work-place exposures are to technical PCP, which usually
contains mg/kg quantities of microcontaminants, particularly H6CDD.
Subacute effects such as chloracne and potential subchronic and
chronic effects such as hepatotoxicity, fetotoxicity, and
immunotoxicity (as reported in animal studies) are probably mainly
caused by microcontaminants. However, the PCP molecule itself
appears to play a role in the pathology of the last 3 effects and
is likely to be wholly responsible for the reports of skin and
mucous membrane irritiation, hyperpyrexia and, in severe cases,
coma and death. The toxicity of pure or purified PCP has not been
evaluated for human beings, because human exposure has usually been
to technical PCP.
Investigations of biochemical changes in woodworkers with long-
term exposure to PCP have failed to detect consistently significant
effects on major organs, nerves, blood, reproduction, or the
immune system. However, the statistical power of these studies has
been limited as a result of the small sample sizes used. Overall,
the body of research suggests that long-term exposure to levels of
PCP encountered in the work-place is likely to cause borderline
effects on some organ systems and biochemical processes.
Some epidemiological studies from Sweden and the USA have
revealed an association between exposure to mixtures of
chlorophenols, especially 2,4,5-T3CP, and the incidences of soft-
tissue sarcomas, lymphomas, and nasal and nasopharyngeal cancers.
Other studies have failed to detect such a relationship. None of
these studies has managed to address the effects of exposure to PCP
itself.
Animal studies designed to assess the carcinogenicity of PCP
and reported to date have been negative. Carcinogenicity bioassays
with one other chlorophenol (2,4,6-T3CP) and a mixture of two H6CDD
congeners found in PCP have been positive. Hence, the carcinogenic
effects of long-term exposure of animals to technical PCP are not
clear.
10.1.1.3 Risk evaluation
It is clear that the levels of PCP found in work-places have
adversely affected some aspects of the health of exposed workers.
Potentially the most deleterious effect of technical PCP is on the
fetus, and pregnant women should avoid exposure, whenever possible.
There is limited evidence that PCP may cause hepatotoxicity,
neurological disorders, and effects on the immune system. No
convincing data for or against a carcinogenic link exists.
Note: Since the publication of this monograph in 1987, however, the
results of adequate carcinogenicity studies with commercial-grade
pentachlorophenol have been published. The conclusions of these studies
are indicated in the addendum to 8.6 Carcinogenicity.
The National Academy of Sciences (1977) calculated an
acceptable daily intake (ADI) for PCP of 3 µg/kg body weight per
day. This ADI is based on data from a feeding study on rats and a
1000-fold safety factor. The results of long-term studies indicate
that the no-observed-adverse-effect level for rats is below 3 mg/kg
body weight per day (section 8.2). A recent human study has shown
that the steady-state body burden is 10 - 20 times higher than the
value extrapolated from rat pharmacokinetic data, suggesting that
caution should be applied when extrapolating directly from the rat
model to man. Furthermore, the US ADI was not based on an
inhalation study, and does not account for the possibly greater
toxicity of PCP via inhalation, as indicated by animal studies
(sections 8.1 and 8.3). Hence, the safety factor of 1000 used to
derive this ADI value is by no means too conservative. The intake
for a 60-kg adult exposed to concentrations of PCP at the ADI level
would be 180 µg/person per day.
A rough estimate of occupational exposure alone can be
calculated, assuming a moderate breathing rate of 1.8 m3/h for a
60-kg worker, 100% uptake of all inhaled PCP (which takes some
account of the often significant dermal uptake), and an 8-h working
shift per day, 5 days per week. Hence, an exposure to 500 µg
PCP/m3 per shift (section 5.2) would result in an average daily PCP
intake of approximately 5000 µg/person per day, averaged over the
entire week. Under these circumstances, the ADI level proposed by
the National Academy of Sciences is significantly exceeded, even
when consideration is given to the effects of intermittant
exposures during the working week and the high health status
assumed for workers.
There is a clear need for a reduction in occupational exposure
to PCP. Emphasis must be placed on reducing airborne
concentrations at production and wood-treatment facilities, as well
as dermal contact with solutions containing PCP. In addition,
reductions in the concentrations of microcontaminants in technical
PCP, particularly PCDDs and PCDFs, would reduce the potential for
expression of several effects and would better protect the health
of workers in these industries.
10.1.2. Non-occupational exposure
10.1.2.1 Exposure levels and routes
Domestic use of products containing technical PCP, especially
the indoor application of wood preservatives and paints based on
PCP, has led to elevated concentrations of PCP in indoor air.
Indoor exposures have been well documented in houses constructed
with PCP-treated wood, or in which interior wood panels or boards
have been treated with PCP. PCP concentrations in indoor air can
be expected to reach 30 µg/m3 during the first month after
treatment. Considerably higher levels, up to 160 µg/m3, have been
reported in houses with concomitant poor indoor ventilation. Even
higher concentrations can be encountered immediately after do-it-
yourself applications of PCP-containing wood preservatives.
In the long term, values of between 1 and 10 µg/m3 are typical,
though higher levels, up to 25 µg/m3, have been found in rooms
treated one to several years earlier. Indoor air concentrations
are influenced by a variety of factors, e.g., intensity of
treatment, solvents and additives involved, species of wood
treated, environmental conditions, and time elapsed since
treatment.
In many cases, levels of PCP in the serum and urine of people
exposed in the home overlap those for occupationally exposed
persons; but, on average, urine-PCP levels are approximately 0.04
mg/litre for non-occupationally exposed persons.
In the long term, exposure to PCP in treated buildings
continuously decreases, because of the high volatility of PCP.
Because of their lower vapour pressure, the volatilization of
PCDDs and PCDFs from the wood surface is much slower than that of
PCP. Hence, these microcontaminants are emitted at a low rate, but
over a longer period of time. Long-term exposure to these
lipophilic contaminants is likely to lead to accumulation of PCDDs
and PCDFs in fatty body tissues.
As a result of regulations restricting the use of PCP, and also
changing use patterns, indoor exposure to PCP is probably declining
in most developed countries.
10.1.2.2 Risk evaluation
Assuming a daily respiratory volume of 20 m3/adult and 100%
uptake of all inhaled PCP (a worst case that takes some account of
dermal uptake), the exposure of persons living in PCP-treated
buildings, shortly after treatment, or, in some cases, after a long
period of time, could be expected to range between 600 and 3200
µg/person per day. Long-term exposure to concentrations of 1 - 25
µg PCP/m3 could result in a daily PCP intake of 20 500 µg/person
per day. The median value of 5 µg/m3 reported from a survey of 104
homes (section 5.3) corresponds to a daily PCP uptake of 100 µg/
person per day. Other potential sources of exposure to PCP
including food, drinking-water, and consumer products contribute
further to PCP uptake (section 10.1.3.1).
The indoor air data suggest that, at least during the first
weeks following indoor treatment, and occasionally for quite
prolonged periods of time, the ADI level of 180 µg/person per day
is significantly exceeded. Under these circumstances, there is a
potential health risk. This conclusion is supported, in part, by
reports of signs and symptoms similar to those in persons
occupationally exposed to PCP (dermatosis, nausea, headache,
dizziness, fatigue). These signs and symptoms are most likely
associated with the effects of the PCP molecule and, in some cases,
the solvents associated with the wood treatment chemicals used.
The long-term significance of exposure to low levels of PCDDs and
PCDFs and their accumulation in human tissues is not entirely
clear; however, at least 2 isomeric groups of the PCDDs family are
carcinogenic for animals. Animal data indicate that low
concentrations of PCP in biological tissues or body fluids do not
signify an absence of biologically active PCDDs and PCDFs.
It is worth noting that exposure in the home is frequently for
longer periods of time than exposures in the work-place and can
affect subpopulations potentially at greater risk than workers, for
example, children, the elderly, pregnant women, or those with an
existing adverse health condition.
10.1.3. General population exposure
10.1.3.1 Exposure levels and routes
Exposure of the general population to low levels of PCP is
common. PCP has been found in air, food, water, and other consumer
products. Biotransformation of some chlorinated hydrocarbons
(e.g., lindane, hexachlorobenzene) to PCP also contribute to the
human body burden.
The ambient air in urban areas typically contains several
ng/m3, while concentrations in less developed areas are roughly an
order of magnitude lower (section 5.1.1).
Drinking-water concentrations of PCP rarely exceed several
µg/litre, even in highly industrialized regions, and most are less
than 1 µg/litre (section 5.1.5).
Fruits, vegetables, and other produce usually contain much less
than 10 µg/kg, but may on occasion exceed this level. Most meats
contain similar concentrations of PCP (< 10 µg/kg) but, a few
samples, particularly liver, can contain over 100 µg/kg. Fish
skeletal muscle typically contains PCP levels of 4 µg/kg or less.
Overall estimates of PCP intake from all foods, based on total diet
samples in the USA and the Federal Republic of Germany, are
remarkably similar, i.e., up to 6 µg/person per day (section
5.1.5).
PCP is also present in a wide variety of consumer products,
including veterinary supplies, disinfectants, photographic
solutions, fabrics, home-care products, and pharmaceutical
products. No calculated estimates of the contribution made by
consumer products to overall exposure to PCP are available.
10.1.3.2 Risk evaluation
On the basis of the PCP levels in the various compartments,
the overall exposure of an average person without known specific
exposure can be estimated to be approximately 6 µg/person per day
from food, 2 µg/person per day from drinking-water, and 2 µg/person
per day from the ambient air. Thus, the total exposure of the
general population could be approximately 10 µg/person per day
(exclusive of exposure to consumer products), which is far below
the intake based on the ADI proposed by the US National Academy of
Science of 180 µg/person per day. On the basis of available data,
this exposure is not likely to constitute a health hazard.
However, the diffuse contamination of the environment with
technical PCP must be considered as an important source of
environmental PCDDs and PCDFs.
10.2. Evaluation of Effects on the Environment
The widespread use of technical PCP and its physical and
chemical properties (water solubility, n-octanol/water partition
coefficient, volatility) lead to ubiquitous contamination of air,
soil, water, sediments, and environmental organisms.
Depending on the soil type, PCP can be very mobile, potentially
leading to contamination of groundwater and hence, of drinking-
water. Because applications in agriculture have been reduced, soil
contamination will, for the most part, be confined to treatment
areas.
Photodecomposition and biodegradation processes may not be
adequate to eliminate PCP from the different compartments.
Unfavourable temperature, pH, and other environmental conditions
may retard degradation of PCP allowing it to persist in the
environment. Biological decomposition may also be limited in
waste-treatment factories resulting in high concentrations in the
final effluents. PCP has also been used in aquatic environments as
a molluscicide and an algicide.
PCP concentrations in surface waters are usually in the range
of 0.1 - 1 µg/litre, though much higher levels can be found near
point sources or after accidental spills (section 5.1.2).
PCP is highly toxic for aquatic organisms. Apart from very
sensitive or resistant species, there is apparently no difference
in the sensitivity to PCP of the different taxonomic groups
(section 7.2). Invertebrates (annelids, molluscs, crustaceans) and
fish are adversely affected by PCP concentrations below 1 mg/litre
in acute toxicity tests. Sublethal concentrations are in the low
µg/litre range.
As little as 1 µg PCP/litre can have adverse effects on very
sensitive algal species. Moreover, low concentrations (µg/litre)
may lead to substantial alterations in community structures, as
seen in model ecosystem studies.
10.3. Conclusions
In this subsection, PCP and Na-PCP are referred to as PCP.
1. Human exposure to PCP is usually from technical products that
contain several toxic microcontaminants, including PCDDs and PCDFs.
2. The acute health effects of exposure to high concentrations of
technical PCP are generally the result of the biological action of
the PCP molecule itself. Sub-chronic effects and the effects of
long-term exposure to technical PCP are most probably largely
related to the biological action of the PCDDs and PCDFs.
3. A dose-effect relationship for the acute or chronic toxicity of
technical PCP for human beings cannot be derived from available
data. Derivation of this relationship is confounded by variations
in individual susceptibility, social and environmental influences,
concomitant exposure to other chemical substances, a lack of
accurate exposure estimates, and inadequate toxicity data.
4. Occupational exposure to technical PCP can lead to adverse
health effects.
5. Non-occupationally exposed persons (using products containing
technical PCP and/or those living in buildings treated with wood
preservatives or paints containing PCP) can be exposed to
concentrations of PCP in air that can have adverse health effects.
6. The exposure of the general population to diffuse sources of
PCP (via food, drinking-water, ambient air, consumer products,
chlorinated compounds that can be metabolized to PCP) is very low
and, on the basis of available data, it is not likely to constitute
a health hazard.
7. Epidemiological investigations and animal studies, conducted to
date, are insufficient for an evaluation of the carcinogenicity of
technical PCP. Uncertainties also exist over the genotoxic and
fetotoxic effects of technical PCP.
8. PCP is rather persistent, quite mobile, and found in all
environmental compartments. At the higher concentrations found in
the surface water near point sources or discharges (mg/litre),
aquatic life is adversely affected. Ambient concentrations of PCP
commonly found in surface waters (0.1 -1 µg/litre) may adversely
affect very sensitive organisms and may lead to alterations in the
ecosystem.
9. Use of technical PCP and its improper disposal (landfill and
low-temperature combustion) can contribute significantly to the
contamination of the environment with PCP, PCDDs, and PCDFs.
11. RECOMMENDATIONS
In this section, PCP and Na-PCP are referred to as PCP.
11.1. Environmental Contamination and Human Exposure
(a) Concentrations of microcontaminants in technical PCP,
especially PCDDs and PCDFs, must be reduced by improving the
quality in production processes.
(b) There is a need for specification of a technical PCP.
(c) Disposal of technical PCP and associated waste should
preferably involve high-temperature combustion or, where this
is not possible, the use of secure land-fill sites.
(d) In order to reduce contamination of surface waters and the
hazards for the aquatic ecosystem, manufacturers and users of
technical PCP should prevent releases into the environment.
(e) Protective measures should be provided for non-target aquatic
organisms in cases where PCP is used as molluscicide or
algicide.
(f) Occupational exposure to technical PCP must be reduced to a
minimum. Reduction in exposure can be achieved by:
- explicit product labelling;
- employee instruction on product handling;
- lowering airborne concentrations; and
- use of effective protective equipment.
(g) Industries handling technical PCP should ensure adequate
routine monitoring and health surveillance of all potentially
exposed employees.
(h) The indoor application of PCP-based wood preservatives and wood
stains and the use of PCP-treated wood products in the interior
of buildings should cease.
(i) The availability and use of consumer products containing PCP
should be reduced and controlled.
(k) The following commercial uses of PCP-based products should be
eliminated, in order to reduce contamination of food and the
environment:
- application as wood preservatives on wooden food
containers, horticultural lumber, wood and tools in
mushroom houses, and above-ground interior wood of
farm buildings;
- application during the curing of hides;
- application as a herbicide or soil sterilant;
- application as a slimicide in wood pulp and paper
operations; and
- application as a molluscicide in surface water if
another control chemical or measure is available that
is less toxic for man and the aquatic ecosystem.
11.2. Future Research
11.2.1. Human exposure and effects
(a) Reliable estimates of human absorption of airborne PCP via the
lung and skin are required.
(b) The importance of the biotransformation of hexachlorobenzene
and related compounds as contributors to human body burdens of
PCP needs to be quantified.
(c) It is necessary to determine the intake and accumulation by
human beings of the lipophilic microcontaminants (especially
the PCDDs and PCDFs) resulting from exposure to technical PCP.
(d) Development of reliable estimates of biochemical and
reproductive no-observed-adverse-effect levels is desirable.
(e) Studies on persons occupationally exposed to technical PCP
should be conducted using a large enough cohort or sufficient
numbers of cases to provide the statistical power necessary to
determine the relationships between exposure to PCP and
morbidity, mortality in general, and cancer. Such studies
should include quantitative estimates of concentrations and
duration of exposure to PCP, wherever possible.
11.2.2. Effects on experimental animals and in vitro test systems
(a) New data on the carcinogenicity of technical and pure PCP in
both sexes of 2 mammalian species are required.
(b) There is a need for a long-term inhalation study on the effects
of both technical and pure PCP.
(c) Studies should be undertaken to clearly determine the
teratogenic effects of pure and technical PCP. The potential
effects of PCP induced maternal hyperthermia on embryological
development and fetal growth warrant investigation.
(d) More research on the genotoxic and mutagenic activity of pure
and technical PCP is required.
11.2.3. Effects on the ecosystem
(a) Studies are needed to clarify the fate of sediment-bound PCP
and its effects on the environment.
(b) Studies of the effects of long-term, low-level exposure on
fresh-water aquatic communities are required to establish no-
observed-adverse-effect levels.
12. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
"The WHO Recommended Classification of Pesticides by Hazard"
(WHO, 1984) distinguishes between the four hazard classes Ia, Ib,
II, and III, based on the toxicity of technical products. In this
report, PCP is classified in class Ib, being highly hazardous.
The WHO manual "Prevention, Diagnosis and Treatment of
Insecticide Poisoning" (Plestina, 1981) provides practical advice
that generally applies to nitro- and chlorophenols.
In the Guidelines for Drinking-Water Quality (WHO, 1985), a
guideline value of 10 µg/litre is recommended for PCP.
No evaluation of the carcinogenicity of PCP was made by the
International Agency for Research on Cancer (IARC, 1979a), because
the available data on the carcinogenic and mutagenic effects of PCP
were considered inadequate for a sound evaluation.
Regulatory standards established by national bodies in
different countries and the EEC are summarized in the data profile
of the International Register of Potentially Toxic Chemicals
(IRPTC, 1983).
IRPTC (1984), in its series "Scientific reviews of Soviet
literature on toxicity and hazard of chemicals", issued a review on
pentachlorophenol.
REFERENCES
ABRAHAMSSON, K. & XIE, T.M. (1983) Direct determination of
trace amounts of chlorophenols in fresh water, waste water and
sea water. J. Chromatogr., 279: 199-208.
ACGIH (1980) Documentation of the threshold limit values,
4th ed., Cincinnati, Ohio, American Conference of Governmental
Industrial Hygienists Inc, p. 323.
ADELMANN, I.R. & SMITH, L.L. (1976) Standard test fish
development. I. Fathead minnows (Pimephales promelas) and
goldfish (Carassius auratus) as standard fish in bioassays and
their reaction to potential reference toxicants, Duluth,
Minnesota, US Environmental Protection Agency, 88 pp (Ecology
Research Series No. EPA 600/3-76-061A).
ADELMANN, I.R., SMITH, L.L., Jr, & SIESENNOP, G.D. (1976)
Acute toxicity of sodium chloride, pentachlorophenol, guthion,
and hexavalent chromium to fathead minnow (Pimephales
promelas) and goldfish (Carassius auratus). J. Fish. Res.
Board Can., 33: 203-208.
ADEMA, D.M.M. & VINK, G.J. (1981) A comparative study of
1,1,2-trichloroethane, dieldrin, pentachlorophenol, and
3,4-dichloroaniline for marine and fresh water organisms.
Chemosphere, 10: 533-554.
AHLBORG, U.G. & LARSSON, K. (1978) Metabolism of tetra-
chlorophenols in the rat. Arch. Toxicol., 40: 63-74.
AHLBORG, U.G. & THUNBERG, T. (1978) Effects of 2,3,7,8-
tetrachlorodibenzo- p-dioxin in the in vivo and in vitro
dechlorination of pentachlorophenol. Arch. Toxicol., 40: 55-61.
AHLBORG, U.G. & THUNBERG, T. M. (1980) Chlorinated phenols:
occurrence, toxicity, metabolism, and environmental impact.
CRC Crit. Rev. Toxicol., 7: 1-35.
AHLBORG, U.G., LINDGREN, J.-E., & MERCIER, M. (1974)
Metabolism of pentachlorophenol. Arch. Toxicol., 32: 271-281.
AHLBORG, U.G., LARSSON, K., & THUNBERG, T. (1978) Metabolism
of pentachlorophenol in vivo and in vitro. Arch. Toxicol., 40:
45-53.
AHLBORG, U.G., VICTORIN, K., CAMNER, P., NORDBERG, G., &
WAERN, F. (1986) [Health effects of waste incineration.] In:
[Energy from waste,] Sweden, National Board for Energy,
Appendix 4 (Report No. 1986:6) (in Swedish).
AHLING, B. & JOHANSSON, L. (1977) Combustion experiments
using pentachlorophenol on a pilot scale and full-scale.
Chemosphere, 6: 425-436.
ALLEN, J.R. & VAN MILLER, J.P. (1978) Health implications of
2,3,7,8-tetrachlorodibenzo- p-dioxin exposure in primates. In:
Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
371-379.
ANDERSEN, K.J., LEIGHTY, E.G., & TAKAHASHI, M.T. (1972)
Evaluation of herbicides for possible mutagenic properties. J.
agric food Chem., 20: 649-656.
ANDO, M., HIRANO, S., & ITOH, Y. (1985) Transfer of hexa-
chlorobenzene (HCB) from mother to new-born baby through
placenta and milk. Arch. Toxicol., 56: 195-200.
ANGERER, C. (1984) [Production and use of pentachlorophenol
in the Federal Republic of Germany.] In: [Pentachlorphenol -
specimen case,] Berlin, Umweltbundesamt, 76 pp (UBA F & E -
Bericht No. 106 04 007) (in German).
ANON (1970) Pentachlorophenol poisoning in the home.
Californian Health, 27: 13.
ANON (1981) [Studies of the teratogenicity of Na-PCP in the
rat.] In: Shanghai Medical College, ed. [Studies on maximal
allowable concentration of hazardous substances in surface
water.] Beijing, China, People's Medical Publication, pp.
215-224 (in Chinese).
ANON (1983a) Cryptogil Na. Technical documentation,
Courbevoie, France, Rhône-Poulenc Specialités chimiques, 3 pp.
ANON (1983b) Cryptogil Ol. Technical documentation,
Courbevoie, France, Rhone-Poulenc Specialités chimiques.
APAJALAHTI, J.H.A. & SALKINOJA-SALONEN, M.S. (1984)
Absorption of pentachlorophenol (PCP) by bark chips and its
role in microbial PCP degradation. Microbiol. Ecol., 10:
359-367.
ARMSTRONG, R.W., EICHNER, E.R., KLEIN, D.E., BARTHEL, W.F.,
BENNETT, J.V., JONSSON, V., BRUCE, P.H.H., & LOVELESS, L.E.
(1969) Pentachlorophenol poisoning in a nursery for newborn
infants. II. Epidemiologic and toxicologic studies. J.
Pediatr., 75(2): 317-325.
ARRHENIUS, E., RENBERG, L., & JOHANSSON, L. (1977a)
Subcellular distribution, a factor in risk evaluation of
pentachlorophenol. Chem.-biol. Interact., 18: 23-34.
ARRHENIUS, E., RENBERG, L., JOHANSSON, L., & ZETTERQVIST,
M.A. (1977b) Disturbance of microsomal detoxication
mechanisms in liver by chlorophenol pesticides. Chem.-biol.
Interact., 18: 35-46.
ARSENAULT, R.D. (1976) Pentachlorophenol and contained
chlorinated dibenzodioxins in the environment. A study of
environmental fate, stability, and significance when used in
wood preservation. Am. Wood Preserv. Assoc., 20: 122-148.
AURAND, K., ENGLERT, N., KRAUSE, CH., ULRICH, D., & WALTER,
R. (1981) [Pentachlorophenol containing wood preservatives
in rooms.] Schr.-Reihe Ver. WaBoLu, 52: 293-313 (in German).
BAADER, E.W. & BAUER, H.J. (1951) Industrial intoxication
due to pentachlorophenol. Ind. Med. Surg., 20: 286-290.
BAKER, M.D. & MAYFIELD, C.I. (1980) Microbial and non-
biological decomposition of chlorophenols and phenol in soil.
Water Air Soil Pollut., 13: 411-424.
BALBA, M.H. & SAHA, J.G. (1974) Metabolism of lindane-14C by
wheat plants grown from treated seed. Environ. Lett., 7:
181-194.
BALLHORN, L., ROZMAN, T. ROZMAN, K., KORTE, F., & GREIM, H.
(1981) Cholestyramine enhances fecal elimination of penta-
chlorophenol in rhesus monkeys. Chemosphere, 10: 877-888.
BALLSCHMITER, K., ZOLLER, W., SCHOLZ, CH., & NOTTRODT, A.
(1983) Occurrence and absence of polychlorodibenzofurans and
polychlorodibenzodioxins in fly ash from municipal
incinerators. Chemosphere, 12: 585-594.
BARBENI, M., PRAMAURO, E., PELIZZENTI, E., BORGARELLO, E., &
SERPONE, N. (1985) Photodegradation of pentachlorophenol
catalyzed by semiconductor particles. Chemosphere, 14: 195-208.
BARTHEL, W.F., CURLEY, A., TRASHER, C.L., SEDLAK, V.A., &
ARMSTRONG, R. (1969) Determination of pentachlorophenol in
blood, urine, tissue, and clothing. J. Assoc. Off. Anal.
Chem., 52: 294-298.
BAUCHINGER, M., DRESP, J., SCHMID, E., & HAUF, R. (1982)
Chromosome changes in lymphocytes after occupational exposure
to pentachlorophenol. Mutat. Res., 102: 83-88.
BAXTER, R.A. (1984) Biochemical study of pentachlorophenol
workers. Ann. occup. Hyg., 28: 429-438.
BEGLEY, J., REICHERT, E.L., RASHAD, M.N., KLEMMER, H.W., &
SIEMSEN, A.W. (1977) Association between renal function
tests and pentachlorophenol. Clin. Toxicol., 11: 97-106.
BEHRBOHM, P. (1959) [On the hazards of exposure to
chlorinated phenols.] Dtsch. Gesundheitswes., 14: 614-619 (in
German).
BERGNER, H., CONSTANTINIDES, P., & MARTIN, J.H. (1965)
Industrial pentachlorophenol poisoning in Winnipeg, Canada.
Can. Med. Assoc. J., 92: 448-451.
BERRY, E.G., NOLAN, M.O., & GONZALES, J.O. (1950) Field
tests of molluscicides against Australorbus glabratus in
endemic areas of schistosomiasis in Puerto Rico. US Public
Health Rep., 65: 939-950.
BETTS, J.J., JAMES, S.P., & THORPE, W.V. (1955) The
metabolism of pentachloronitrobenzene and 2,3,4,6-tetrachloro-
nitrobenzene and the formation of mercapturic acids in the
rabbit. Biochem. J., 61: 611-617.
BEVENUE, A. & BECKMAN, H. (1967) Pentachlorophenol: a
discussion of its properties and its occurrence as a residue
in human and animal tissues. Residue Rev., 19: 83-134.
BEVENUE, A. & OGATA, J.N. (1971) A contributive error from
analytical reagents in the analysis of chlorophenoxy acids and
pentachlorophenol by electron capture gas chromatography. J.
Chromatogr., 61: 147-148.
BEVENUE, A., WILSON, J.R., POTTER, E.F., SONG, M.K., BECKMAN,
H., & MALLETT, G. (1966) A method for the determination of
pentachlorophenol in human urine in picogram quantities. Bull.
environ. Contam. Toxicol., 1: 257-266.
BEVENUE, A., HALEY, T.J., & KLEMMER, H.W. (1967a) A note on
the effect of a temporary exposure of an individual to penta-
chlorophenol. Bull. environ. Contam. Toxicol., 2: 293-296.
BEVENUE, A., WILSON, J.R., CASARETT, L.J., & KLEMMER, H.W.
(1967b) A survey of pentachlorophenol content in human urine.
Bull. environ. Contam. Toxicol., 2: 319-332.
BEVENUE, A., EMERSON, M.L., CASARETT, L.J., & YAUGER, W.L.
(1968) A sensitive gas chromatographic method for the
determination of pentachlorophenol in human blood. J.
Chromatogr., 38: 467-472.
BEVENUE, A., OGATA, J.N., & HYLIN, J.W. (1972) Organo-
chlorine pesticides in rainwater, Oahu, Hawaii, 1971-72. Bull.
environ. Contam. Toxicol., 8: 238-241.
BISHOP, C.M. & JONES, A.H. (1981) Non-Hodgkin's lymphoma of
the scalp in workers exposed to dioxins. Lancet, 2: 369.
BLACKMAN, G.E., PARKE, M.H., & GARTON, G. (1955a) The
physiological activity of substituted phenols. I. Relation-
ships between chemical structure and physiological activity.
Arch. Biochem. Biophys., 54: 45-54.
BLACKMAN, G.E., PARKE, M.H., & GARTON, G. (1955b) The
physiological activity of substituted phenols. II. Relation-
ships between physical properties and physiological activity.
Arch. Biochem. Biophys., 54: 55-71.
BLAIR, D.M. (1961) Dangers in using and handling sodium
pentachlorophenate as a molluscicide. Bull. World Health
Organ., 25: 597-601.
BLEVINS, D. (1965) Pentachlorophenol poisoning in swine.
Vet. Med., 60: 455.
BODDINGTON, M.J., BARRETTE, P., GRANT, D., NORSTROM, R.J.,
RYAN, J.J., & WHITBY, L. (1985) Chlorinated dioxins and
related compounds, 1984. Proceedings of the Fourth Inter-
national Symposium, Ottawa, Canada, 16-18 October 1984.
Chemosphere, 14(6/7): 571-989.
BORTHWICK, P.W. & SCHIMMEL, S.C. (1978) Toxicity of penta-
chlorophenol and related compounds to early life stages of
selected estuarine animals. In: Rao, K.R. ed. Pentachloro-
phenol: chemistry, pharmacology, and environmental toxicology,
New York, London, Plenum Press, pp. 141-146.
BORZELLECA, J.F., HAYES, J.R., CONDIE, L.W., & EGLE, J.L., Jr
(1985) Acute toxicity of monochlorophenols, dichlorophenols,
and pentachlorophenol in the mouse. Toxicol. Lett., 29: 39-42.
BOUTWELL, R.K. & BOSCH, D.K. (1959) The tumour-promoting
action of phenol and related compounds for mouse skin. Cancer
Res., 19: 413-424.
BOYLE, T.P., ROBINSON-WILSON, E.F., PETTY, B.D., & WEBER, W.
(1980) Degradation of pentachlorophenol in simulated lentic
environment. Bull. environ. Contam. Toxicol., 24: 177-184.
BRADLAW, J.A., GARTHOFF, L.H., & HURLEY, N.W. (1980)
Comparative induction of aryl hydrocarbon hydroxylase activity
in vitro by analogues of dibenzo- p-dioxin. Food Cosmet.
Toxicol., 18: 627-635.
BRANDT, M., SCHMIDT, E., & SCHMIDT, F.W. (1977) [Chronic liver
disease caused by long term household poisoning with penta-
chlorophenol.] Verh. Dtsch. Ges. Inn. Med., 83: 1609-1611 (in
German).
BRAUN, W.H. & SAUERHOFF, M.W. (1976) The pharmacokinetic
profile of pentachlorophenol in monkeys. Toxicol. appl.
Pharmacol., 38: 525-533.
BRAUN, W.H. & WAECHTER, J.M., Jr (1983) Sources of
uncertainty in pharmacokinetic prediction. J. anim. Sci., 56:
235-243.
BRAUN, W.H., YOUNG, J.D., BLAU, G.E., & GEHRING, P.J. (1977)
The pharmacokinetics and metabolism of pentachlorophenol in
rats. Toxicol. appl. Pharmacol., 41: 395-406.
BRAUN, W.H., BLAU, G.E., & CHENOWETH, M.B. (1979) The
metabolism/pharmacokinetics of pentachlorophenol in man, and a
comparison with the rat and monkey. In: Deichmann, W.B., ed.
Toxicology and occupational medicine, New York, Amsterdam,
Oxford, Elsevier/North-Holland, pp. 289-296.
BRINGMANN, G. & KUHN, R. (1982) [Results of toxic action of
water pollutants on Daphnia magna Straus tested by an improved
standardized procedure.] Z. Wasser Abwasser Forsch., 15: 1-6
(in German).
BRUNS, G.W. & CURRIE, R.A. (1980) Extraction of pentachloro-
phenol and tetrachlorophenol residues from field-contaminated
carrots and potatoes: comparison of several methods. J. Assoc.
Off. Anal. Chem., 63: 56-60.
BUA (1986) In: Ges. Dt. Chemiker, ed. [Pentachlorophenol.
Report of the Advisory Group for existing chemicals which are
relevant from an environmental point of view (BUA),] Weinheim,
Deerfield Beach, VCH Publishers, 183 pp (in German).
BUCKMAN, N.G., HILL, J.O., MAGEE, R.J., & MCCORMICK, M.J.
(1984) Separation of substituted phenols, including eleven
priority pollutants using high-performance liquid
chromatography. J. Chromatogr., 284: 441-446.
BUHLER, D.R., RASMUSSON, M.E., & NAKANE, H.S. (1973)
Occurrence of hexachlorophene and pentachlorophenol in sewage
and water. Environ. Sci. Technol., 7: 929-934.
BUIKEMA, A.L., GINNISS, M.J., & CAIRNS, J. (1979) Phenolics
in aquatic ecosystems: a selected review of recent literature.
Mar. environ. Res., 2: 87-181.
BUNDESAMT FUR UMWELTSCHUTZ (1982) [Impairment of surface
waters by pentachlorophenol.] Mitteilung No. 21, Bern, 12 pp.
(in German).
BUNDESAMT FUR UMWELTSCHUTZ (1983) [Polychlorinated organic
compounds in Rheinfelden (Switzerland). Investigation of dust,
soil, and grass samples: results and evaluation.]
Schriftenreihe Umweltschutz, 18, Bern, 32 pp (in German).
BUNDESGESETZBLATT (1978) [Regulation of agricultural
pesticides in or on foodstuffs of plant origin and tobacco
products (regulation of plant-protective pesticides).] In:
[Bundesgeseztblatt. Part I,] Bonn, Federal Republic of
Germany, Federal Ministry of Youth, Family Affairs and Health,
pp. 718-719, 734 (in German).
BUNDESGESUNDHEITSAMT (1983) [On handling wood preserva-
tives,] Berlin, Bundesgesundheitsamt, 36 pp (Information
Booklet of the Federal Health Office of Federal Republic of
Germany) (in German).
BURGER, E. (1966) Acute fatal poisoning with sodium
pentachlorophenol. Dtsch. Z. Gesamt. Gerichtl. Med., 58:
240-247.
BUSELMAIER, W., ROHRBORN, G., & PROPPING, P. (1973)
Comparative investigations on the mutagenicity of pesticides
in mammalian test systems. Mutat. Res., 21: 25-26.
BUSER, H.R. (1975) Analysis of polychlorinated dibenzo-
p-dioxins and dibenzofurans in chlorinated phenols by mass
fragmentography. J. Chromatogr., 107: 295-310.
BUSER, H.R. (1976) Preparation of qualitative standard
mixtures of polychlorinated dibenzo- p-dioxins and dibenzo-
furans by ultraviolet and gamma-irradiation of the octachloro
compounds. J. Chromatogr., 129: 303-307.
BUSER, H.R. (1982) [Report on the formation of PCDDs during
pyrolysis of PCP and PCP-Na,] Wädenswil, Switzerland, Eidg.
Forschungsanstalt, 2 pp (Unpublished report) (in German).
BUSER, H.R. & BOSSHARDT, H.P. (1976) Determination of
polychlorinated dibenzo- p-dioxins and dibenzofurans in
commercial pentachlorophenols by combined gas chromatography -
mass spectrometry. J. Assoc. Off. Anal. Chem., 59: 562-569.
BUTTE, W. (1984) Determination of free and total penta-
chlorophenol in urine. Fresenius Z. Anal. Chem., 317: 659.
BUTTE, W., KIRSCH, M., & DENKER, J. (1983) The determination
of pentachlorophenol and tetrachlorophenols in Wadden sediment
and clams ( Mya arenaria) using triethylsulfonium hydroxide for
extraction and pyrolytic ethylation. Int. J. environ. anal.
Chem., 13: 141-153.
BUTTE, W., DENKER, J., KIRSCH, M., & HOPNER, TH. (1985)
Pentachlorophenol und tetrachlorophenols in Wadden sediment
and clams Mya arenaria of the Jadebusen after a 14-year period
of waste-water discharge containing pentachlorophenol.
Environ. Pollut. B, 9: 29-39.
CABRAL, J.R.P., RAITANO, F., MOLLNER, T., BRONCZYK, S., &
SHUBIK, P. (1979) Acute toxicity of pesticides in hamster.
Toxicol. appl. Pharmacol., 48: A192.
CANTELMO, A.C. & RAO, K.R. (1978) Effects of pentachloro-
phenol on the meiobenthic nematodes in an experimental system.
In: Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology,
and environmental toxicology, New York, London, Plenum Press,
pp. 165-174.
CARR, R.S. & NEFF, J.M. (1981) Biochemical indices of stress
in the sandworm Neanthes virens (Sars). I. Responses to penta-
chlorophenol. Aquat. Toxicol., 1: 313-327.
CARR, R.S., THOMAS, P., & NEFF, J.M. (1982) A simple
spectrophotometric technique for the determination of penta-
chlorophenol in water. Bull. environ. Contam. Toxicol., 28:
477-479.
CASANOVA, M. & DUBROCA, J. (1972) Residues of pentachloro-
nitrobenzene and its hexachlorobenzene impurity in soils and
lettuce (Fr.). C.R. Séances Acad. Agric. Fr., 58: 990-998.
CASARETT, L.J., BEVENUE, A., YANGER, W.L., & WHALEN, S.A.
(1969) Observations on pentachlorophenol in human blood and
urine. Am. Ind. Hyg. Assoc. J., 30: 360-366.
CASTERLINE, J.L., Jr (1982) Uptake, translocation, and
transformation of pentachlorophenol in soybean and spinach
plants. Diss. Abstr. Int. B 1983, 43(8): 2409.
CAUTREELS, W., VAN COUWENBERGHE, K., & GUZMAN, L.A. (1977)
Comparison between the organic fraction of suspended matter at
a background and an urban station. Sci. total Environ., 8:
79-88.
CHAPMAN, J.B. & ROBSON, P. (1965) Pentachlorophenol
poisoning from bath-water. Lancet, 12 June: 1266-1267.
CHAPMAN, J.C., HIGGINS, V.R., SIMPSON, G.R., & SIYALI, D.S.
(1981) Pentachlorophenol (PCP) exposure: an occupational
health hazard in mushroom growing. In: Mushroom science. XI.
Proceedings of the eleventh International Scientific Congress
on the Cultivation of Edible Fungi, Sydney, Australia, 18 pp.
CHAPMAN, P.M., FARRELL, M.A., & BRINKHURST, R.O. (1982)
Relative tolerances of selected aquatic oligochaetes to
combinations of pollutants and environmental factors. Aquat.
Toxicol., 2: 69-78.
CHAU, A.S.Y. & COBURN, J.A. (1974) Determination of penta-
chlorophenol in natural and waste waters. J. Assoc. Off. Anal.
Chem., 57: 389-393.
CHIU, C., THOMAS, R.S., LOCKWOOD, J., LI, K., HALMAN, R., &
LAO, R.C.C. (1983) Polychlorinated hydrocarbons from power
plants, wood burning and municipal incinerators. Chemosphere,
12: 607-616.
CHOI, J. & AOMINE, S. (1972) Effects of soil on the activity
of pentachlorophenol. Soil Sci. plant Nutr., 18: 255-260.
CHOI, J. & AOMINE, S. (1974a) Adsorption of pentachloro-
phenol by soils. Soil Sci. plant Nutr., 20: 135-144.
CHOI, J. & AOMINE, S. (1974b) Mechanism of pentachlorophenol
adsorption by soils. Soil Sci. plant Nutr., 20: 371-379.
CHOW, C.Y., THEVASAGAYAM, E.S., & WAMBEEK, E.G. (1955) Control
of Salvinia - a host plant of Mansonia mosquitos. Bull. World
Health Organ., 12: 365-369.
CHU, J.P. & KIRSCH, E.J. (1972) Metabolism of pentachloro-
phenol by an axenic bacterial culture. Appl. Microbiol., 23:
1033-1035.
CIRELLI, D.P. (1978a) Patterns of pentachlorophenol usage in
the United States of America: an overview. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology. New York, London, Plenum Press, pp. 13-18.
CIRELLI, D.P. (1978b) Pentachlorophenol: position document
1. Fed. Reg., 43: 48446-48477.
CLEVELAND, L., BUCKLER, D.R., MAYER, F.L., & BRANSON, D.R.
(1982) Toxicity of three preparations of pentachlorophenol to
fathead minnows: a comparative study. Environ. Toxicol. Chem.,
1, 205-212.
CONKEY, J.H. & CARLSON, J.A. (1963) Relative toxicity of
biostatic agents suggested for use in the pulp and paper
industry: 1963 review. Tech. Assoc. Pulp Paper Ind., 46: 23
A-39A.
CONKLIN, P.J. & RAO, K.R. (1978) Toxicity of sodium penta-
chlorophenate (Na-PCP) to the grass shrimp, Palaemonetes
pugio, at different stages of the molt cycle. Bull. environ.
Contam. Toxicol., 20: 275-279.
COOK, W.L., FIEDLER, D., & BOURQUIN, A.W. (1980) Succession
of microfungi in estuarine microcosms perturbed by carbaryl,
methyl parathion, and pentachlorophenol. Bot. Mar., 23:
129-131.
CORDDRY, A.E. (1981) A pregnancy outcome study of the wives
of workers exposed to chlorophenate wood preservatives at a
sawmill, University of Washington, Department of Environmental
Health, 191 pp (Thesis).
COURTNEY, K.D., COPELAND, M.F., & ROBBINS, A. (1976) The
effects of pentachloronitrobenzene, hexachlorobenzene, and
related compounds on fetal development. Toxicol. appl.
Pharmacol., 35: 239-256.
CRANDALL, C.A. & GOODNIGHT, C.J. (1959) The effect of
various factors on the toxicity of sodium pentachlorophenate
to fish. Limnol. Oceanogr., 4: 53-56.
CRANMER, M. & FREAL, J. (1970) Gas chromatographic analysis
of pentachlorophenol in human urine by formation of alkyl
ethers. Life Sci., 9: 121-128.
CRETNEY, M.J. (1976) Pentachlorophenol death. Bull. Int.
Assoc. Forensic Toxicol., 12: 10.
CROSBY, D.G. & WONG, A.S. (1976) Photochemical generation of
chlorinated dioxins. Chemosphere, 5: 327-332.
CROSBY, D.G., WONG, A.S., PLIMMER, J.R., & WOOLSON, E.A.
(1971) Photodecomposition of chlorinated dibenzo- p-dioxins.
Science, 173: 748-749.
CROSBY, D.G., MOILANEN, K.W., NAKAGAWA, M., & WONG, A.S.
(1972) Photonucleophilic reactions of pesticides. In:
Matsumura, F., Boush, G.M., & Misato, T., ed. Environmental
toxicology of pesticides, New York, London, Academic Press,
pp. 423-431.
CROSBY, D.G., MOILANEN, K.W., & WONG, A.S. (1973) Environmental
generation and degradation of dibenzodioxins and
dibenzofurans. Environ. Health Perspect., 5: 259-266.
CROSBY, D.G., BEYNON, K.I., GREVE, P.A., KORTE, F., STILL,
G.G., & VOUK, J.W. (1981) Environmental chemistry of penta-
chlorophenol. Pure appl. Chem., 53: 1051-1080.
CROSSLAND, N.O. & WOLF, C.J.M. (1985) Fate and biological
effects of pentachlorophenol in outdoor ponds. Environ.
Toxicol. Chem., 4: 73-86.
CSERJESI, A.J. (1967) The adaptation of fungi to penta-
chlorophenol and its biodegradation. Can. J. Microbiol., 13:
1243-1249.
CSERJESI, A.J. (1972) Detoxification of chlorinated phenols.
Int. Biodeterioration Bull., 8: 135-138.
CSERJESI, A.J. & JOHNSON, E.L. (1972) Methylation of penta-
chlorophenol by Trichoderma virgatum. Can. J. Microbiol., 18:
45-49.
CSERJESI, A.J. & ROFF, J.W. (1975) Toxicity tests of some
chemicals against certain wood-staining fungi. Int.
Biodeterioration Bull., 11: 90-96.
CULL, M.R. & DOBBS, A.J. (1984) Long-term changes in
polychlorodibenzo- p-dioxin concentrations in wood treated with
technical pentachlorophenol. Chemosphere, 13: 1091-1099.
CULL, M.R., DOBBS, A.J., & WILLIAMS, N. (1983) Polychloro-
dibenzo- p-dioxins (PCDDs) in commercial pentachlorophenol
(PCP) used in wood preservation. Chemosphere, 12: 483-485.
CURTIS, R.F., LAND, D.G., GRIFFITH, N.M., GEE, M., ROBINSON,
D., PEEL, J.L., DENNIS, C., & GEE, J.M. (1972) 2,3,4,6-
tetrachloroanisole association with musty taint in chickens
and microbiological formation. Nature (Lond.), 235: 223.
DAHMS, A. & METZNER, W. (1979) [On the analytics of penta-
chlorophenol and tetrachlorophenol in the air and in urine.]
Holz Roh-Werkst., 37: 341-344 (in German).
DALELA, R.C., RANI, S., & VERMA, S.R. (1980a) Physiological
stress induced by sublethal concentrations of phenol and
pentachlorophenol in Notopterus notopteru: hepatic acid and
alkaline phosphatases and succinic dehydrogenase. Environ.
Pollut., 21: 3-8.
DALELA, R.C., RANI, S., RANI, S., & VERMA, S.R. (1980b)
Influence of pH on the toxicity of phenol and its two
derivatives pentachlorophenol and dinitrophenol to some fresh
water teleosts. Acta hydrochim. hydrobiol., 8: 623-629.
DANIELS, C.R. & SWAN, E.P. (1979) Determination of
chlorinated phenols in surface-treated lumber by HPLC. J.
chromatogr. Sci., 17, 628-630.
DAVIS, H.C. & HIDU, H. (1969) Effects of pesticides on
embryonic development of clams and oysters and on survival and
growth of the larvae. US Fish Wildl. Serv. Fish. Bull., 67:
393-403.
DAVIS, J.C. & HOOS, R.A. (1975) Use of sodium pentachloro-
phenate and dehydroabiotic acid as reference toxicants for
salmonid bioassays. J. Fish. Res. Board Can., 32: 411-416.
DEBETS, F.M.H., STRIK, J.J.T.W.A., & OLIE, K. (1980) Effects
of pentachlorophenol on rat liver changes induced by hexa-
chlorobenzene with special reference to porphyria and
alterations in mixed-function oxygenases. Toxicology, 15:
181-195.
DEICHMANN, W.B. (1943) The toxicity of chlorophenols for
rats. Fed. Proc., 2: 76-77.
DEICHMANN, W.B. & KEPLINGER, M.L. (1981) Phenols and
phenolic compounds. In: Clayton, G.D. & Clayton, F.E., ed.
Patty's industrial hygiene and toxicology 2A, 3rd revised ed.,
New York, John Wiley and Sons, pp. 2567-2627.
DEICHMANN, W.B. & MERGARD, E.G. (1948) Comparative evalua-
tion of methods employed to express the degree of toxicity of
a compound. J. ind. Hyg. Toxicol., 30: 373-378.
DEICHMANN, W.B., MACHLE, W., KITZMILLER, K.V., & THOMAS, G.
(1942) Acute and chronic effects of pentachlorophenol and
sodium pentachlorophenate upon experimental animals. J.
Pharmacol. exp. Ther., 76: 104-117.
DE LAUNE, R.D., GAMBRELL, R.P., & REDDY, K.S. (1983) Fate of
PCP in estuarine sediment. Environ. Pollut. (B), 6: 297-308.
DEMENTI, B.A. (1981) Health hazard alert pentachlorophenol
(news). Am. Ind. Hyg. Assoc. J., 42:: A16, A18.
DEMIDENKO, N.M. (1969) [Materials for establishing the
maximum permissible concentrations in air.] Gig. Tr. Prof.
Zabol., 7: 58-60 (in Russian).
DETRICK, R.S. (1977) Pentachlorophenol: possible sources of
human exposure. For. Prod. J., 27: 13-16.
DIETZ, F. & TRAUD, J. (1978a) [Threshold odour and taste
concentrations of phenolic compounds.] GWF-Wasser/Abwasser,
119: 318-325 (in German).
DIETZ, F. & TRAUD, J. (1978b) [On the analysis of phenols,
particularly chlorophenols, in water by means of gas chromato-
graphy - methods and results.] Vom Wasser, 51: 235-257 (in
German).
DIJK, J.J., VAN, VAN DER MEER, C., & WIJNANS, M. (1977) The
toxicity of sodium pentachlorophenolate for three species of
decapod crustaceans and their larvae. Bull. environ. Contam.
Toxicol., 17: 622-630.
DOBBS, A.J. & GRANT, C. (1980) Pesticide volatilisation
rates: a new measurement of the vapour pressure of penta-
chlorophenol at room temperature. Pestic. Sci., 11: 29-32.
DOBBS, A.J. & WILLIAMS, N. (1983) Indoor air pollution from
pesticides used in wood remedial treatments. Environ. Pollut.
(B), 6: 271-296.
DOEDENS, J.D. (1964) Chlorophenols. In: Kirk, R.E. & Othmer,
D.F., ed. Encyclopedia of chemical technology, 2nd ed., New
York, John Wiley and Sons, Vol. 5, pp. 336-337.
DOUGHERTY, R.C. & PIOTROWSKA, K. (1976a) Multi-residue
screening by negative chemical ionization mass spectrometry of
organic polychlorides. J. Assoc. Off. Anal. Chem., 59:
1023-1027.
DOUGHERTY, R.C. & PIOTROWSKA, K. (1976b) Screening by
negative chemical ionization mass spectrometry for environ-
mental contamination with toxic residues: application to human
urines. Proc. Natl Acad. Sci. (USA), 73: 1777-1781.
DOUGHERTY, R.C., WHITAKER, M.J., SMITH, L.M., STALLING, D.L.,
& KUEHL, D.W. (1980) Negative chemical ionization studies of
human and food chain contamination with xenobiotic chemicals.
Environ. Health Perspect., 36: 103-117.
DRUMMOND, I., VAN ROOSMALEN, P.B., & KORNICKI, M. (1982)
Determination of total pentachlorophenol in the urine of
workers. A method incorporating hydrolysis, an internal
standard and measurement by liquid chromatography. Int. Arch.
occup. environ. Health., 50: 321-327.
DUGGAN, R.E. & CORNELIUSSEN, P.E. (1972) Dietary intake of
pesticide chemicals in the United States. III. June 1968 -
April 1970. Pestic. monit. J., 5: 331-341.
DUNCAN, C.G. & DEVERALL, F.J. (1964) Degradation of wood
preservatives by fungi. Appl. Microbiol., 12: 57-62.
DUST, J.V. & THOMPSON, W.S. (1973) Pollution control in the
wood-preserving industry. Part IV. Biological methods of
treating waste water. For. Prod. J., 23: 59-66.
EBEN, A., SCHALLER, K.H., & KRAUSE, CH. (1981) [Penta-
chlorophenol (PCP): analysis of plasma and urine.] In: German
Science Foundation, Working Group on Analytical Chemistry,
Commission for the Investigation of Health Hazards of Chemical
Compounds in the Work Area, ed. [Analysis of hazardous
substances in biological materials,] Weinheim, VCH Publishers,
Vol. 2, pp. 1-11 (in German).
ECKRICH, W. (1986) [Levels of PCDDs/PCDFs in indoor air and
in human blood.] In: VDI-Kommission Reinhaltung der Luft, ed.
[ Dioxins. Report of the VDI Meeting, Essen, Federal Republic
of Germany, 22-23 April 1986,] Düsseldorf, VDI-Verlag, pp.
70-81 (in German).
ECONOMIST INTELLIGENCE UNIT (1981) Economic implications of
abatement measures of water pollution due to hexachlorobuta-
diene, endosulfan, trichlorophenol, and pentachlorophenol,
Brussels, Commission of the European Communities, 134 pp
(Report prepared for Environment and Consumer Protection
Service).
EDGEHILL, R.U. (1982) Microbial treatment of wastewater and
soil to remove pentachlorophenol. Part 1. Diss. Abstr. Int. B,
42: 4871.
EDGEHILL, R.U. & FINN, R.K. (1983) Treatment of soil to remove
pentachlorophenol. Appl. environ. Microbiol., 45: 1122-1125.
EDGERTON, T.R. & MOSEMAN, R.F. (1979) Determination of
pentachlorophenol in urine: the importance of hydrolysis. J.
agric. food Chem., 27: 197-199.
EDGERTON, T.R., MOSEMAN, R.F., LINDER, R.E., & WRIGHT, L.H.
(1979) Multi-residue method for the determination of
chlorinated phenol metabolites in urine. J. Chromatogr., 170:
331-342.
EDWARDS, M.J. (1968) Congenital malformations in the rat
following induced hyperthermia during gestation. Teratology,
1: 173-178.
ELLER, P.M., ed. (1984a) Pentachlorophenol in blood. In:
NIOSH manual of analytical methods, 3rd ed., Cincinnati, Ohio,
US Department of Health and Human Services, Public Health
Service, Centers for Disease Control, National Institute for
Occupational Safety and Health, pp. 8001-1 - 8001-4 (DHHS
Publication No. 84-100).
ELLER, P.H., ed. (1984b) Pentachlorophenol in urine. In:
NIOSH manual of analytical methods, 3rd ed., Cincinnati, Ohio,
US Department of Health and Human Services, Public Health
Service, Centers for Disease Control, National Institute for
Occupational Safety and Health, pp. 8303-1 - 8303-4 (DHHS
Publication No. 84-100).
EMBREE, V., ENARSON, D.A., CHAN-YEUNG, M., DY BUNCIO, A.,
DENNIS, R., & LEACH J. (1984) Occupatonal exposure to
chlorophenates: toxicology and respiratory effects. Clin.
Toxicol., 22: 317-329.
ENARSON, D.A., CHAN-YEUNG, M., EMBREE, V., WANG, R., &
SCHULZER, M. (1986) Occupational exposure to chlorophenates,
renal, hepatic and other health effects. Scand. J. Work
environ. Health, 12: 144-148.
ENGEL, C., DE GROOT, A.P., & WEURMAN, C. (1966) Tetra-
chloroanisole: a source of musty taste in eggs and broilers.
Science, 154: 270-271
ENGST, R., MACHOLZ, R.M., KUJAWA, M., LEWERENZ, H.J., & PLASS,
R. (1976) The metabolism of lindane and its metabolites
gamma-2,3,4,5,6-pentachlorocyclohexene, pentachlorobenzene,
and pentachlorophenol in rats and the pathways of lindane
metabolism. J. environ. Sci. Health (B), 11: 95-117.
ENGST, R., MACHOLZ, R.M., & KUJAWA, M. (1978) [Metabolites
of hexachlorocyclohexane isomers (HCH) in human blood.]
Pharmazie, 33: 109-111 (in German).
ENVIRONMENT, CANADA (1979) Monitoring environmental
contamination from chlorophenol contaminated wastes generated
in the wood preservation industry, Ottawa, Environmental
Protection Bureau, Environmental Protection Service, Pacific
and Yukon Region, pp. 74 (Regional Program Report No. 79-24)
(Prepared by Can Test Ltd and EVS Consultants Ltd) (DSS File
No. 075B, KE 114-8-1935).
ERIKSON, M., HARDELL, L., BERG, N.O., MOELLER, T., & AXELSSON,
O. (1981) Soft-tissue sarcomas and exposure to chemical
substances: a case reference study. Br. J. ind. Med., 38:
27-33.
ERNEY, D.R. (1978) Gas-liquid chromatographic determination
of pentachlorophenol in milk. J. Assoc. Off. Anal. Chem., 61:
214-216.
ERNST, W. (1979) Factors affecting the evaluation of
chemicals in laboratory experiments using marine organisms.
Ecotoxicol. environ. Saf., 3: 90-98.
ERNST, W. & WEBER, K. (1978a) Chlorinated phenols in selected
estuarine bottom fauna. Chemosphere, 7: 867-822.
ERNST, W. & WEBER, K. (1978b) The fate of pentachlorophenol
in the Weser estuary and the German Bight. Veröff. Inst.
Meeresforsch. Bremerh., 17: 45-53.
ERVIN, H.E. & MCGINNIS, G.D. (1980) Analysis of pentachloro-
phenol in waste-water using high-performance liquid chromato-
graphy. J. Chromatogr., 190: 203-207.
ETZEL, J.E. & KIRSCH, E.J. (1974) Biological treatment of
industrial wastewater containing pentachlorophenol. Dev. ind.
Microbiol., 16: 287-295.
EXON, J.H. (1984) A review of chlorinated phenols. Vet. hum.
Toxicol., 26: 508-520.
EXON, J.H. & KOLLER, L.D. (1982) Effects of transplacental
exposure to chlorinated phenols. Environ. Health Perspect.,
46: 137-140.
EXON, J.H. & KOLLER, L.D. (1983a) Alternation of trans-
placental carcinogenesis by chlorinated phenols. In: Jolley,
R.L., et al., ed. Water chlorination: environmental impact and
health effects, Ann Arbor, Michigan, Ann Arbor Science, Vol.
2, Book 4, pp. 1177-1188.
EXON, J.H. & KOLLER, L.D. (1983b) Effects of chlorinated
phenols on immunity in rats. Int. J. Immunopharmacol., 5:
131-136.
FAAS, L.F. & MOORE, J.C. (1979) Determination of penta-
chlorophenol in marine biota and sea water by gas-liquid
chromatography and high pressure liquid chromatography. J.
agric. food Chem., 27: 554-557.
FAHRIG, R. (1974) Comparative mutagenicity studies with
pesticides, Lyons, International Agency for Research on
Cancer, pp. 161-181 (IARC Scientific Publication Vol. 10).
FAHRIG, R., NILSSON, C.-A., & RAPPE, C. (1978) Genetic
activity of chlorophenols and chlorophenol impurities. In:
Rao, K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
325-338.
FARQUHARSON, H.E., CAGE, J.C., & NORTHOVER, J. (1958) The
biological action of chlorophenols. Br. J. Pharmacol., 13:
20-24.
FARRINGTON, D.S. & MUNDAY, J.W. (1976) Determination of
trace amounts of chlorophenols by gas-liquid chromatography.
Analyst, 101: 639-643.
FEDERAL REGISTER (1977) Gelatin: affirmation of GRAS status
as a direct and indirect human food ingredient. Fed. Reg.,
42(218): 58763-58765.
FEILER, H. (1980) Fate of priority pollutants in publicly
owned treatment works, Washington DC, US Environmental
Protection Agency (EPA-440/1-80-301).
FERREE, D.C. (1974) Influence of freshly treated posts on
growth of newly planted trees. Ohio Agric. Res. Dev. Cent.
Res. Summ., 75: 11-12.
FIELDER, R.J., SORRIE, G.S., BISHOP, C.M., JONES, R.B., & VAN
DEN HEUVEL, M.J. (1982) Pentachlorophenol. In: Toxicity
review, London, Health and Safety Executive, Vol. 5, 20 pp.
FINGERHUT, M.A., HALPERIN, W.E., HONCHAR, P.A., SMITH, A.B.,
GROTH, D.H., & RUSSELL, W.O. (1984) An evaluation of reports
of dioxin exposure and soft-tissue sarcoma pathology among
chemical workers in the United States. Scand. J. Work environ.
Health, 10: 299-303.
FIRESTONE, D. (1973) Etiology of chick edema disease.
Environ. Health Perspect., 5: 59-66.
FIRESTONE, D. (1977) Chemistry and analysis of pentachloro-
phenol and its contaminants, Washington DC, US Food and Drug
Administration, pp. 57-59 (FDA Bylines No. 2).
FIRESTONE, D., REES, J., BROWN, N.L., BARRON, R.P., & DAMICO,
J.N. (1972) Determination of polychlorodibenzo- p-dioxins and
related compounds in commercial chlorophenols. J. Assoc. Off.
Anal. Chem., 55: 85-92.
FIRESTONE, D., CLOWER, M., Jr, BORSETTI, A.P., TESKE, R.H., &
LONG, P.E. (1979) Polychlorodibenzo- p-dioxin and penta-
chlorophenol residues in milk and blood of cows fed technical
pentachlorophenol. J. agric. food Chem., 27: 1171-1177.
FISCHER, A. & SLEMROVA, J. (1978) [The contamination of the
river Rhine with chlorophenols.] Vom Wasser, 51: 33-46 (in
German).
FISCHER, M. (1983) [Pentachlorophenol as model case of a
xenobiotic compound,] Berlin, Federal Health Office Institute
for Water, Soil and Air Hygiene, 38 pp (Research Report No.
10604007) (in German).
FLEISCHER, M., MEISS, R., ROBENEK, H., THEMANN, H., & ECKHARD,
R. (1980) Ultrastructural morphometric investigations on rat
liver of young and adult rats after treatment with technical
pentachlorophenol. Arch. Toxicol., 44: 243-257.
FLICKINGER, C.W. & LAWRENCE, A.W. (1982) Occupational health
experience in the wood preserving industry. In: Am. Wood
Preserv. Assoc., 78: 11-30.
FOCQUET, J.P. & THEISEN, R. (1981) Détermination de la
réduction de l'hexachlorobutadiène de l'endosulfan, du
pentachlorophenol et du trichlorophénol contenue dans les
effluents en tenant compte des meilleurs moyens techniques
disponsibles, Luxembourg, Commission of the European
Communities (Contract No. ENV/723/74-FR) (unpublished report).
FOLKE, J. & LUND, H. (1983) Occurrence of low- and high-
chlorinated phenols in municipal sewage before and after
passing through biological treatment plants. J. Chromatogr.,
279: 189-198.
FORSELL, J.H., SHULL, L.R., & KATELEY, J.R. (1981)
Subchronic administration of technical pentachlorophenol to
lactating dairy cattle immunotoxicologic evaluation. J.
Toxicol. environ. Health, 8:: 543-558.
FOUNTAINE, J.E., JOSHIPURA, P.B., & KELIHER, P.N. (1976)
Some observations regarding pentachlorophenol levels in
Haverford Township, Pennsylvania. Water Res., 10: 185-188.
FOX, M. & JOSHI, S.R. (1984) The fate of pentachlorophenol
in the Bay of Quinte, Lake Ontario (Canada USA). J. Great
Lakes Res., 10: 190-196.
FRANZLE, O. (1982) [Modelling the transport of environmental
chemicals and their metabolites through the unsaturated zone
of natural soil profiles and sludge in laboratory lysimeters
and in the field.] In: BMI Environmental Research Plan, pp.
XIX + 315 (Research Report No. 10602005/02) (in German).
FRAGIADAKIS, A., KLEIN, W., KORTE, F., MOZA, P.N., SCHEUNERT,
I., VOCKEL, D., & WEISS, U. (1979) [Fate of organochlorine
compounds in plant-soil systems] In: [Organohalogen compounds
in the environment. Project report 1975-78,] Jülich Nuclear
Research Plant (Jül-Spez. 45, July 1979) (BMFT Research Report
037118) (in German).
FRANK. R., BRAUN, H.E., HOLDRINET, M., SIRONS, G.J., SMITH,
E.H., & DIXON, D.W. (1979) Organochlorine insecticides and
industrial pollutants in the milk supply of southern Ontario,
Canada, 1977. J. food Prot., 42: 31-37.
FREITAG, D., GEGER, H., KRAUS, A., VISWANATHAN, R., KOTZIAS,
D., ASSAR, A., KLEIN, W., & KORTE, F. (1982) Ecotoxico-
logical profile analysis. VII. Screening chemicals for their
environmental behaviour by comparative evaluation. Ecotoxicol.
environ. Saf., 6: 60-81.
FRG (1986) [Statuary ordinance by dangerous chemicals.] In:
Gefahrstoffverordnung, Bonn, Ministry of Labour and Social
Affairs (BGBL I), p. 1470 (in German).
FUCHSBICHLER, G. (1982) Automated gel chromatography as
clean-up process for determination of ECD-detectable
substance, chlorinated hydrocarbons, pentachlorophenol,
diphenyl and o-phenylphenol in plant materials. Z.
Lebensm.-Unters. Forsch., 174: 9-12.
GAB, F. & PARLAR, H. (1979) [Transformation of organo-
halogens under abiotic conditions.] In: [Organohalogen
compounds in the environment. Project report 1975-78,] Jülich
Nuclear Research Plant (Jül.-Spez. 45, July 1979) (BMFT
Research Report 037114) (in German).
GAB, S., NITZ, S., PARLAR, H., & KORTE, F. (1975)
Photomineralisation of certain aromatic xenobiotica.
Chemosphere, 4: 251-256.
GAB, S. (1981) [Decomposition of organic chemicals in
adsorbed phase.] In: [Report on the 1980 Symposium of the
Institute for Ecological Chemistry,] Münich, Society for
Radiation and Environmental Research (GSF), pp. 61-75 (in
German).
GAINES, T.B. (1969) Acute toxicity of pesticides. Toxicol.
appl. Pharmacol., 14: 515-534.
GARRETT, C.L. (1980) Fraser River Estuary study. Water
quality series. Toxic organic contaminants, Ottawa,
Environmental Protection Service, Pacific and Yukon Region,
Environment Canada, pp. 123.
GEBEFUEGI, I. (1981) [Our present knowledge of the
occurrence of pentachlorophenol in the environment.] In:
[Report on the 1980 Symposium of the Institute for Ecological
Chemistry,] Münich, Society for Radiation and Environmental
Research (GSF), pp. 25-33 (in German).
GEBEFUEGI, I. & KORTE, F. (1983) Pentachlorophenol
contamination of human milk samples. Chemosphere, 12:
1055-1060.
GEBEFUEGI, I. & KORTE, F. (1984) Indoor contamination of
household articles through pentachlorophenol and lindane. In:
Berglund, B., Lindvall, T., & Sundell, J., ed. Indoor air. IV.
Chemical characterisation and personal exposure, Stockholm,
Swedish Council for Building Research, pp. 317-322.
GEBEFUEGI, I., PARLAR, H., & KORTE, F. (1979) Occurrence of
pentachlorophenol in closed environments. Ecotoxicol. environ.
Saf., 3: 269.
GEBEFUEGI, I., OXYNOS, K., & KORTE, F. (1983) [Long-term
behaviour of pentachlorophenol in closed rooms.] Chemosphere,
12: 59-63 (in German).
GILBERT, F.I., MINN, C.E., DUNCAN, R.C., ALDRICH, T., LEDERER,
W.H., & WILKINSON, J.E. (1983) Effects of chemical preserva-
tives on the health of wood treating workers in Hawaii, 1981.
Clinical and chemical profiles and historical prospective
study, Honolulu, Hawaii, Pacific Health Research Institute,
233 pp.
GJOVIK, L.R., JOHNSON, D.B., KOZAK, V., WOOLSON, E.A.,
THOMPSON, W.A., MICKLEWRIGHT, J.T., DOST, W.A., & NICHOLAS,
D.D. (1981) The biologic and economic assessment of
pentachlorophenol, inorganic arsenicals, creosote. I. Wood
Preservatives, Washington DC, US Department of Agriculture,
pp. 69-87 (Technical Bulletin No. 1658-1).
GLICKMAN, A.H., STRATHAM, C.N., & LECH, J.J. (1977) Studies
on the uptake, metabolism, and disposition of pentachloro-
phenol and pentachloroanisole in rainbow trout. Toxic. appl.
Pharmacol., 41: 649-658.
GODSY, E.M., GOERLITZ, D.F., & EHRLICH, G.G. (1986) Effects of
pentachlorophenol on methanogenic fermentation of phenol.
Bull. environ. Contam. Toxicol., 36: 271-277.
GOLDSTEIN, J.A., FRIESEN, M., LINDER, R.E., HICKMAN, P., HASS,
J.R., & BERGMAN, H. (1977) Effects of pentachlorophenol on
hepatic drug-metabolizing enzymes and porphyria related to
contamination with chlorinated dibenzo- p-dioxins and dibenzo-
furans. Biochem. Pharmacol., 26: 1549-1557.
GOODNIGHT, C.J. (1942) Toxicity of sodium pentachlorophenate
and pentachlorophenols to fish. Ind. eng. Chem., 34: 868-872.
GORDON, D. (1956) How dangerous is PCP? Med. J. Aust., Sept.
29: 485-488.
GOSSLER, K. & SCHALLER, K.H. (1978) [Quantitative
determination of pentachlorophenol in urine and plasma by gas
chromatography.] Z. anal. Chem., 290: 111-112 (in German).
GOTHAM, I.J. & RHEE, G.Y. (1982) Effects of a hexachloro-
biphenyl and pentachlorophenol on growth and photosynthesis of
phytoplankton. J. Great Lakes Res., 8: 328-335.
GREEN, R.E. & YOUNG, R.H.T. (1970) Herbicide and fertilizer
movement in Hawaiian sugarcane soils in relation to subsurface
water quality. Hawaiian Sugar Technol., 29: 88-96.
GREENE, M.H., BRINTON, L.A., FRAUMENI, J.F., & D'AMICO, R.
(1978) Familial and sporadic Hodgkin's disease associated
with occupational wood exposure. Lancet, 9: 626-627.
GREICHUS, Y.A., LIBAL, G.W., & JOHNSON, D.D. (1979)
Diagnosis and physiologic effects of pentachlorophenols on
young pigs. I. Effects of purified pentachlorophenol. Bull.
environ. Contam. Toxicol., 23: 418-422.
GRIMM, H.G., SCHALLER, K.H., & VALENTIN, H. (1985) [Results
of pentachlorophenol exposure in the workplace and the
environment.] Zentralbl. Arbeitsmed., Arbeitsschutz, Prophyl.
Ergon., 34: 136-142 (in German).
GRIMM, H.G., SCHELLMANN, B., SCHALLER, K.H., & GOSSLER, K.
(1981) [Pentachlorophenol concentrations in tissues and body
fluids of normal persons.] Zbl. Bakt. Hyg., I. Abt. Orig. B,
174: 77-90 (in German).
GRUTTKE, H., KRATZ, W., PAPENHAUSEN, U., WEIGMANN, G., HAQUE,
A., & SCHUPHAN, I. (1986) [Transfer of 14C-Na-PCP in
model-foodchains.] Verhd. Ges. ökol., 14: 451-455 (in German).
GUNTHER, F.A., WESTLAKE, W.E., & JAGLAN, P.S. (1968)
Reported solubilities of 738 pesticide chemicals in water.
Residue Rev., 20: 56-148.
GUPTA, P.K. & RAO, P.S. (1982) Toxicity of phenol, penta-
chlorophenol, and sodium pentachlorophenate to a freshwater
pulmonate snail Lymnaea acuminata. Arch. Hydrobiol., 94:
210-217.
GUPTA, P.K., MUJUMDAR, V.S., RAO, P.S., & DURVE, V.S. (1982)
Toxicity of phenol, pentachlorophenol, and sodium
pentachlorophenolate to a freshwater teleost Lebistes
reticulatus (Peters). Acta hydrochim. hydrobiol., 10: 177-181.
GUPTA, S., VERMA, S.R., & SAXENA, P.K. (1982) Toxicity of
phenolic compounds in relation to the size of a freshwater
fish Notopterus notopterus (Pallas). Ecotoxicol. environ.
Saf., 6: 433-438.
GUPTA, S., DALELA, R.C., & SAXENA, P.K. (1983a) Influence of
dissolved oxygen levels on acute toxicity of phenolic
compounds to fresh water teleost Notopterus notopterus
(Pallas). Water Air Soil Pollut., 19: 223-228.
GUPTA, S., DALELA, R.C., & SAXENA, P.K. (1983b) Effects of
phenolic compounds on in vivo activity of transaminases in
certain tissues of the fish Notopterus notopterus. Environ.
Res., 32: 8-13.
GUTHRIE, M.A., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L.
(1984) Pentachlorophenol biodegradation. II. Anaerobic. Water
Res., 18: 451-461.
HAKULINEN, R. & SALKINOJA-SALONEN, M. (1982) Treatment of
pulp and paper industry waste waters in an anaerobic fluidised
bed reactor. Process Biochem., 3/4: 18-22.
HALAWANI, A. (1951) Endemic diseases control. J. Egypt. Med.
Assoc., 34: 347-358.
HALEY, T.J. (1977) Human poisoning with pentachlorophenol
and its treatment. Ecotoxicol. environ. Saf., 1: 343-347.
HALL, L.H. & KIER, L.B. (1984) Molecular connectivity of
phenols and their toxicity to fish. Bull. environ. Contam.
Toxicol., 32: 354-362.
HAQUE, A., SCHEUNERT, I., & KORTE, F. (1978) Isolation and
identification of a metabolite of pentachlorophenol-14C in
rice plants. Chemosphere, 7: 65-69.
HAQUE, R. & FREED, V.H. (1974) Behaviour of pesticides in
the environment: environmental chemodynamics. Residue Rev.,
52: 89-116.
HARDELL, L. (1977) Soft-tissue sarcoma and exposure to
phenoxy acids: a clinical observation. Läkartidingen, 74:
2853-2854.
HARDELL, L. (1979) Malignant lymphoma of histiocytic type
and exposure to phenoxyacetic acids or chlorophenols. Lancet,
1: 55-56.
HARDELL, L. (1981a) Relation to soft-tissue sarcoma,
malignant lymphoma, and colon cancer to phenoxy acids,
chlorophenols, and other agents. Scand. J. Work environ.
Health, 7: 119-130.
HARDELL, L. (1981b) Epidemiological studies on soft-tissue
sarcoma and malignant lymphoma and their relation to phenoxy
acid or chlorophenol exposure, Umea, Sweden, Umea University
Medical Dissertations (New Series No. 65).
HARDELL, L. & SANDSTROM, A. (1979) Case-control study:
soft-tissue sarcomas and exposure to phenoxyacetic acids or
chlorophenols. Br. J. Cancer, 39: 711-717.
HARDELL, L., ERIKSSON, M., LENNER, P., & LUNDGREN, E. (1981)
Malignant lymphoma and exposure to chemicals, especially
organic solvents, chlorophenols, and phenoxy acids: a case-
control study. Br. J. Cancer, 43: 169-176.
HARDELL, L., JOHANSSON, B., & AXELSON, O. (1982) Epidemio-
logical study of nasal and nasopharyngeal cancer and their
relation to phenoxy acid or chlorophenol exposure. Am. J. ind.
Med., 3: 247-257.
HARGESHEIMER, E.E. & COUTTS, R.T. (1983) Selected ion mass
spectrometric identification of chlorophenol residues in human
urine. J. Assoc. Off. Anal. Chem., 66: 13-21.
HARPER, D.B. & BANAVE, D. (1975) Chloroanisole residues in
broiler tissues. Pestic. Sci., 6: 159-163.
HARRISON, D.L. (1959) The toxicity of wood preservatives to
stock. Part I. Pentachlorophenol. NZ. Vet. J., 7: 89-98.
HARVEY, W.A. & CRAFTS, A.S. (1952) Toxicity of pentachloro-
phenol and its sodium salts in three Yolo soils. Hilgardia,
21: 487-490.
HATTULA, M.L., WASENIUS, V.M., REUNANEN, H., & ARSTILA, A.U.
(1981) Acute toxicity of some chlorinated phenols, catechols
and cresols to trout. Bull. environ. Contam. Toxicol., 26:
295-298.
HAUCH, R.G., NORRIS, D.R., & PIERCE, R.H. (1980) Acute and
chronic toxicity of sodium pentachlorophenate to the copepod
Pseudodiaptomus coronatus. Bull. environ. Contam. Toxicol.,
25: 562-568.
HAYES, E.H. (1979) High-pressure liquid chromatographic
determination of pentachlorophenol by using paired ion
chromatography reagents. J. Assoc. Off. Anal. Chem., 62:
1004-1006.
HEIKES, D.L. & GRIFFITT, K.R. (1980) Pentachlorophenol in
Mason jar lids and home canned food. J. Assoc. Off. Anal.
Chem., 63: 1125-1128.
HENSHAW, B.G., MORGAN, J.W.W., & WILLIAMS, N. (1975) The
detection of organic solvent preservatives in wood by
thin-layer chromatography. J. Chromatogr., 110: 37-41.
HERNANDEZ, C. & STRASSMAN-SUNDY, S. (1980) Pentachlorophenol
in log homes - Kentucky. Morb. Mortal. Wkly Rep., 29: 431-432,
437.
HERNBERG, S. & PESSI, T. (1964) Peripheral nervous paralysis
due to pentachlorophenate. Int. Arch. Gewerbepathol.
Gewerbehyg., 21: 23-26.
HIATT, C.W., HASKINS, W.T., & OLIVIER, L. (1960) The action
of sunlight on sodium pentachlorophenate. Am. J. trop. Med.
Hyg., 9: 527-531.
HICKMAN, G.T. & NOVAK, J.T. (1984) Acclimation of activated
sludge to pentachlorophenol. J. Water Pollut. Control Fed.,
56: 364-469.
HILL, E.F., HEALTH, R.G., SPANN, J.W., & WILLIAMS, J.D.
(1975) Lethal dietary toxicities of environmental pollutants
to birds, Washington DC, US Fish and Wildlife Service,
Department of the Interior, 61 pp (Special Scientific Report -
Wildlife No. 191).
HILLAM, R.P. & GREICHUS Y.A. (1983) Effects of purified
pentachlorophenol on the serum proteins of young pigs. Bull.
environ. Contam. Toxicol., 31: 599-604.
HILTON, H.W., YUEN, Q.H., & NOMURA, N.S. (1970) Distribution
of residues from atrazine, ametryne, and pentachlorophenol in
sugarcane. J. agric. food Chem., 18: 217-220.
HINKLE, D.K. (1973) Fetotoxic effects of pentachlorophenol
in the golden Syrian hamster. Toxicol. appl. Pharmacol., 25:
455.
HIRSCH, A.A. (1942) Toxicity of sodium pentachlorophenate
and other chemicals on water hyacinth. Bot. Gaz., 103: 620-621.
HMSO (1974) The non-agricultural user of pesticides in Great
Britain, London, Her Majesty's Stationery Office, 65 pp
(Pollution Paper No. 3).
HOBEN, H.J., CHING, S.A., CASARETT, L.J., & YOUNG, R.A.
(1976a) A study of the inhalation of pentachlorophenol by
rats. I. A method for the determination of pentachlorophenol
in rat plasma, urine, and tissue and in aerosol samples. Bull.
environ. Contam. Toxicol., 15: 78-85.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976b) A study
of the inhalation of pentachlorophenol by rats. II. A new
inhalation exposure system for high doses in short exposure
time. Bull. environ. Contam. Toxicol., 15: 86-92.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976c) A study
of inhalation of pentachlorophenol by rats. III. Inhalation
toxicity study. Bull. environ. Contam. Toxicol., 15: 463-465.
HOBEN, H.J., CHING, S.A., & CASARETT, L.J. (1976d) A study
of inhalation of pentachlorophenol by rats. IV. Distribution
and excretion of inhaled pentachlorophenol. Bull. environ.
Contam. Toxicol., 15: 466-474.
HOBEN, J.H., CHING, S.A., YOUNG, R.A., & CASARETT, L.J.
(1976e) A study of the inhalation of pentachlorophenol by
rats. V. A protein binding study of pentachlorophenol. Bull.
environ. Contam. Toxicol., 16: 225-232.
HODSON, P.V. & BLUNT, B.R. (1981) Temperature-induced
changes in pentachlorophenol chronic toxicity to early life
stages of rainbow trout (Salmo gairdneri). Aquat. Toxicol., 1:
113-128.
HODSON, P.V., DIXON, D.G., & KAISER, K.L.E. (1984) Measure-
ment of median lethal dose as a rapid indication of contam-
inant toxicity to fish. Environ. Toxicol. Chem., 3: 243-254.
HOETING, A.L. (1977) Penta - another environmental
contaminant. In: Proceedings of the Central States Association
of Food and Drug Officials Spring Meeting, Mason, Ohio, 4-5
May 1977, pp. 65-71.
HOLMBERG, B., JENSEN, S., LARSSON, A., LEWANDER, K., & OLSSON,
M. (1972) Metabolic effects of technical pentachlorophenol
(PCP) on the eel Anguilla anguilla L. Comp. Biochem. Physiol.,
43B: 171-183.
HOOS, R.A.W. (1978) Patterns of pentachlorophenol usage in
Canada - an overview. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 3-11.
HUANG, J.-C. & GLOYNA, E.F. (1968) Effect of organic
compounds on photosynthetic oxygenation. I. Chlorophyll
destruction and suppression of photosynthetic oxygen
production. Water Res., 2: 347-366.
HUBER, W., SCHUBERT, V., & SAUTTER, C. (1982) Effects of
pentachlorophenol on the metabolism of the aquatic macrophyte
Lemna minor. Environ. Pollut. A, 29: 215-224.
HUCKINS, J.N. & PETTY, J.D. (1983) Dynamics of purified and
industrial pentachlorophenol in fathead minnows. Arch.
environ. Contam. Toxicol., 12: 667-672.
HUECK, H.J. & LABRIJN, J. (1960) [Mold protection of cotton
with pentachlorophenol and laurylpentachlorophenol.] Textil-
Rundschau, 15: 467-472 (in German).
HUGHES, B.J., FORSELL, J.H., SLEIGHT, S.D., KUD, S., & SHULL,
L.R. (1985) Assessment of pentachlorophenol toxicity in
newborn calves: clinicopathology and tissue residues. J. anim.
Sci., 61(6): 1587-1603.
HUNTER, G.W., FREYTAG, R.E., RITCHIE, L.S., PAN, C., YOKOGAWA,
M., & POTTS, D.E. (1952) Studies on schistosomiasis. VI.
Control of the snail host of schistosomiasis in Japan with
sodium pentachlorophenate (Santobrite). Am. J. trop. Med.
Hyg., 1: 831-847.
HUTZINGER, O., FREI, R.W., MERIAN, E., & POCCHIARI, F.
(1982) Chlorinated dioxins and related compounds impact on
the environment. In: Proceedings of a Workshop, Rome, 22-24
October, 1980, Oxford, New York, Toronto, Sydney, Paris,
Frankfurt, Pergamon Press, Vol. 5, 658 pp (Pergamon Series on
Environmental Science).
IARC (1977) Some fumigants, the herbicides 2,4-D and 2,4,5-T,
chlorinated dibenzodioxins and miscellaneous industrial
chemicals, Lyons, International Agency for Research on Cancer,
pp. 111-138 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 15).
IARC (1979a) Pentachlorophenol. In: Some halogenated
hydrocarbons, Lyons, International Agency for Research on
Cancer, pp. 303-325 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 20).
IARC (1979b) Hexachlorobenzene. In: Some halogenated
hydrocarbons, Lyons, International Agency for Research on
Cancer, pp. 155-178 (IARC Monographs on the Evaluation of the
Carcinogenic Risk of Chemicals to Humans, Vol. 20).
IDE, A., NIKI, J., SAKAMOTO, F., WATANABE, J., & WATANABE, H.
(1972) Decomposition of pentachlorophenol in paddy soil.
Agric. biol. Chem., 36: 1937-1944.
IFEADI, C.N. (1975) Screening study to develop background
information and determine the significance of air contaminant
emissions from pesticide plants, Washington DC, US EPA Office
of Pesticide Programes (EPA Report No. PB-244734).
IMAI, K., ASANO, A., & SATO, R. (1967) Oxidative phosphory-
lation in Micrococcus denitrificans. I. Preparation and
properties of phosphorylating membrane fragments. Biochim.
Biophys. Acta, 143: 462-476.
INGLIS, A. & DAVIS, E.L. (1972) Effects of water hardness on
the toxicity of several organic and inorganic herbicides to
fish. US Bur. Sport Fish. Wildl. tech. Pap., 67: 1-22.
INGOLS, R.S. & GAFFNEY, P.E. (1965) Biological studies of
halophenols. Proc. Soc. Water Resour. Pollut. Control Conf.,
14: 175-181.
INGOLS, R.S., GAFFNEY, P.E., & STEVENSON, P.C. (1966)
Biological activity of halophenols. J. Water Pollut. Control
Fed., 38: 629-635.
INGRAM, L.L., MCGINNIS, G.D., & GJOVIK, L.R. (1981a) The
relative amount of pentachlorophenol volatilization from
treated wood. In: Proceedings of the American Wood Preservers'
Association Annual Meeting, April 27-29, Hyatt Orlando,
Florida, Florida, Kissimmee, pp. 102-108.
INGRAM, L.L., MCGINNIS, G.D., JASPERSE, G., & GJOVIK, L.R.
(1981b) The effect of solvent systems on the volatilization
of pentachlorophenol from treated wood. In: Proceedings of the
American Wood Preservers' Association Annual Meeting April
27-29, Hyatt Orlando, Florida, Florida, Kissimmee, pp. 105-117.
INNES, J.R.M., ULLAND, B.M., VALERIO, M.G., PETRUCELLI, L.,
FISHBEIN, L., HART, E.R., PALLOTTA, A.J., BATES, R.R., FALK,
H.L., GART, J.J., KLEIN, M., MITCHELL, I., & PETERS, J.
(1969) Bioassay of pesticides and industrial chemicals for
tumourigenicity in mice: a preliminary note. J. Natl Cancer
Inst., 42: 1101-1114.
IRPTC (1983) Data profile on pentachlorophenol, Geneva,
International Register of Potentially Toxic Chemicals, United
Nations Environment Programme.
IRPTC (1984) Scientific review of Soviet literature on
toxicity and hazards of chemicals. Vol. 75. Pentachlorophenol,
Moscow, Centre of International Projects (GKNT, no. 75).
ISHIZAWA, S., TOYODA, H., & MATSUGUCHI, T. (1961) Effects of
DD, EDB, and PCP upon microorganisms and their activities in
soil. Part I. Effects on microflora. Soil Plant Food Tokyo, 6:
145-155.
IVANOV, Z. & MAGEE, R.J., (1980) The determination of trace
amounts of chlorophenols by high-performance liquid chromato-
graphy. Microchem. J., 25: 543-547.
IZAKI, K., TAKAHASHI, M., SATO, Y, SASAGAWA, Y., SATO, K., &
FURUSAKA, C. (1981) Some properties of pentachlorophenol-
resistant gram-negative bacteria. Agric. biol. Chem., 45:
765-767.
JAKOBSON, I. & YLLNER, S. (1971) Metabolism of 14C-
pentachlorophenol in the mouse. Acta pharmacol. toxicol., 29:
513-524.
JANKE, D. & FRITSCHE, W. (1978) [Microbial dechlorination of
pesticides and other xenobiotica.] Z. Allerg. Mikrobiol., 18:
365-382 (in German).
JANSSENS, J.J. & SCHEPENS, P.J.C. (1984) Chronic penta-
chlorophenol intoxication as a result of the usage of wood
protectants. Meded. Fac. Landbouwwet., 49: 1175-1184.
JANSSON, B., SUNDSTROM, G., & AHLING, B. (1978) Formation of
polychlorinated dibenzo- p-dioxins during combustion of chloro-
phenol formulations. Sci. total Environ., 10: 209-217.
JAYAWEERA, R., PETERSEN, R., & SMEJTEK, P. (1982) Induced
hydrogen ion transport in lipid membranes as origin of toxic
effect of pentachlorophenol in an alga. Pestic. Biochem.
Physiol., 18: 197-204.
JENSEN, S. & RENBERG, L. (1972) Contaminants in penta-
chlorophenol: chlorinated dioxins and predioxins (chlorinated
hydroxy-diphenylethers). Ambio, 1: 62-65.
JENSEN, S. & RENBERG, L. (1973) Chlorinated dimers present
in several technical chlorophenols used as fungicides.
Environ. Health Perspect., 5: 37-39.
JIRASEK, L., KALENSKY, J., KUBEC, K., PAZDEROVA, J., & LUKAS,
E. (1974) [Acne chlorina, porphyria cutanea tarda, and other
manifestations of general poisoning during the manufacture of
herbicides. II.] Cesk. Dermatol., 49: 145-157 (in Czech with
English summary).
JOHNSON, R.D. & MANSKE, D.D. (1977) Pesticides in food and
feed. Pesticide and other chemical residues in total diet
samples (XI). Pestic. monit. J., 11: 116-131.
JOHNSON, R.L., GEHRING, P.J., KOCIBA, R.J., & SCHWETZ, B.A.
(1973) Chlorinated dibenzodioxins and pentachlorophenol.
Environ. Health. Perspect., 5: 171-175.
JONES, P.A. (1981) Chlorophenols and their impurities in the
Canadian environment, Ottawa, Environment Canada, 434 pp
(Report No. EPS 3-EC-81-2)
JONES, P.A. (1984) Chlorophenols and their impurities in the
Canadian environment: 1983 supplement, Ottawa, Environment
Canada, 93 pp (Report No. EPS 3-Ep-84-3).
JUHL, U., WITTE, I., & BUTTE, W. (1985) Metabolism of
pentachlorophenol to tetrachlorohydroquinone by human liver
homogenate. Bull. environ. Contam. Toxicol., 35: 596-601.
KAILA, K. & SAARIKOSKI, J. (1977) Toxicity of pentachloro-
phenol and 2,3,6-trichlorophenol to the crayfish ( Astacus
fluviatilis L.). Environ. Pollut., 12: 119-123.
KAISER, K.L.E. & VALDMANIS, I. (1982) Apparent octanol/water
partition coefficients of pentachlorophenol as a function of
pH. Can. J. Chem., 60: 2104-2106.
KALMAN, D.A. & HORSTMAN, S.W. (1983) Persistence of tetra-
chlorophenol and pentachlorophenol in exposed woodworkers. J.
Toxicol. clin. Toxicol., 20: 343-352.
KARASEK, F.W. & HUTZINGER, O. (1986) Dioxin danger from
garbage incineration. Anal. Chem., 58: 633A-642A.
KAUFMAN, D.D. (1976) Phenols. In: Kearney, P.C. & Kaufman,
D.D., ed. Chemistry, degradation, and mode of action, New
York, Marcel Dekker, Vol. 2, pp. 665-707.
KAUFMAN, D.D. (1978) Degradation of pentachlorophenol in
soil, and by soil microorganisms. In: Rao, K.R., ed. Penta-
chlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 27-39.
KAUPPINEN, T. & LINDROOS, L. (1985) Chlorophenol exposure in
saw mills. Am. Ind. Hyg. Assoc. J., 46: 34-38.
KEHOE, R.A., DEICHMANN-GRUEBLER, W., & KITZMILLER, K-V.
(1939) Toxic effects upon rabbits of pentachlorophenol and
sodium pentachlorophenate. J. ind. Hyg. Toxicol. 21(5):
160-172.
KENAGA, E.E. (1972) Guidelines for environmental study of
pesticides: determination of bioconcentration potential.
Residue Rev., 44: 73-113.
KERKVLIET, N.I., BAECHER-STEPPAN, L., CLAYCOMB, A.T., CRAIG,
A.M., & SHEGGEBY, G.G. (1982a) Immunotoxicity of technical
pentachlorophenol (PCP-T) depressed humoral immune response
to T-dependent and T-independent antigen stimulation in PCP-T
exposed mice. Fundam. appl. Toxicol., 2:: 90-99.
KERKVLIET, N.I., BAECHER-STEPPAN, L., & SCHMITZ, J.A.
(1982b) Immunotoxicity of pentachlorophenol (PCP): increased
susceptibility to tumour growth in adult mice fed technical
PCP-contaminated diets. Toxicol. appl. Pharmacol., 62: 55-64.
KIMBROUGH, R.D. & LINDER, R.E. (1978) The effect of
technical and purified pentachlorophenol on the rat liver.
Toxicol. appl. Pharmacol., 46: 151-162.
KINZELL, J.H., AMES, N.K., SLEIGHT, S.D., KREHBIEL, J.D., KUO,
C., ZABIK, M.J., & SHULL, L.R. (1981) Subchronic administra-
tion of technical pentachlorophenol to lactating dairy cattle:
performance, general health, and pathologic changes. J. dairy
Sci. 64: 42-51.
KIRSCH, E.J. & ETZEL, J.E. (1973) Microbial decomposition of
pentachlorophenol. J. Water Pollut. Control Fed., 45: 359-364.
KLEMMER, H.W. (1972) Human health and pesticides - community
pesticide studies. Residue Rev., 41: 55-63.
KLEMMER, H.W, WONG, L., SATO, M.M., REICHERT, E.L., KORSAK,
R.J., & RASHAD, M.N. (1980) Clinical findings in workers
exposed to pentachlorophenol. Arch. environ. Contam. Toxicol.,
9(6): 715-725.
KLOPFFER, W., KAUFMANN, G., RIPPEN, G., & POREMSKI, H.-J.
(1982) A laboratory method for testing the volatility from
aqueous solution: first results and comparison with theory.
Ecotoxicol. environ. Saf., 6: 545-559.
KNOWLTON, M.F. & HUCKINS, J.N. (1983) Fate of radiolabelled
sodium pentachlorophenate in littoral microcosms. Bull.
environ. Contam. Toxicol., 30: 206-213.
KNUDSEN, I., VERSCHUUREN, H.G., DEN TONKELAAR, E.M., KROES,
R., & HELLEMAN, P.F.W. (1974) Short-term toxicity of penta-
chlorophenol in rats. Toxicology, 2: 141-152.
KOBAYASHI, K. (1978) Metabolism of pentachlorophenol in
fishes. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 89-105.
KOBAYASHI, K. (1979) Metabolism of pentachlorophenol in
fish. In: Khan, M.A.Q., Lech, J.J., & Menn, J.J., ed.
Pesticide and xenobiotic metabolism in aquatic organisms,
Washington DC, Americal Chemical Society, Vol. 99, pp. 131-143
(ACS Symposium Series).
KOBAYASHI, K. & AKITAKE, H. (1975a) Studies on the meta-
bolism of chlorophenols in fish. I. Absorption and excretion
of PCP by goldfish. Bull. Jpn. Soc. Sci. Fish., 41: 87-92.
KOBAYASHI, K. & AKITAKE, H. (1975b) Studies on the meta-
bolism of chlorophenols in fish. II. Turnover of absorbed PCP
in goldfish. Bull. Jpn. Soc. Sci. Fish., 41: 93-99.
KOBAYASHI, K. & NAKAMURA, N. (1979a) Isolation and
identification of a conjugated PCP excreted in the urine of
goldfish. Bull. Jpn. Soc. Sci. Fish., 45: 1001-1004.
KOBAYASHI, K. & NAKAMURA, N. (1979b) Major detoxification
pathways for pentachlorophenol in goldfish. Bull. Jpn. Soc.
Sci. Fish., 45: 1185-1188.
KOCIBA, R.J. & SCHWETZ, B.A. (1982) A review of the toxicity
of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) with a
comparison of the toxicity of other chlorinated dioxin
isomers. Assoc. Food Drug Off. Q. Bull., 46: 168-188.
KOCIBA, R.J., GEHRING, P.J., MCCOLLISTER, S.B., WADE, C.E.,
LISOWE, R.W., & MEINECKE, B. (1971) Results of 90-day
toxicological study in male rats maintained on diets
containing production grade or "purified" pentachlorophenol,
Midland, Michigan, Dow Chemical Company, 23 pp.
KOEGEL, W., MUELLER, W.F., COULSTON, F., & KORTE, F. (1979)
Biotransformation of pentachloronitrobenzene-14C in rhesus
monkeys after single and chronic oral administration.
Chemosphere, 8: 97-105.
KONASEWICH, D.E., HENNING, F.A., WILE, K.H., & GERENCHER, E.
(1983) Chlorophenate wood protection, Canada, British
Columbia Ministry of Environment (ISBN O-7719-9371-4).
KOPPE, P., DIETZ, F., TRAUD, J., & RUEBELT, C. (1977)
[Detection and photometric determination of 126 phenolic
compounds in water using four group-specific reagents.] Z.
anal. Chem., 285: 1-19 (in German).
KOSS, G. & KORANSKY, W. (1978) Pentachlorophenol in
different species of vertebrates after administration of
hexachlorobenzene and pentachlorobenzene. In: Rao, R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 131-137.
KOTZIAS, D., KLEIN, W., & KORTE, F. (1975) [Occurrence of
xenobiotica in seeping waters of landfills.] Chemosphere, 5:
301-306 (in German).
KOZAK, V.P., SIMSIMAN, G.V., CHESTERS, G. STENSBY, D., &
HARKIN, J. (1979) Reviews of the environmental effects of
pollutants: XI. Chlorophenols, Washington DC, US Environmental
Protection Agency, 492 pp (EPA Report 600/1-79-012).
KRAUSE, CHR. (1982) [Active ingredients of wood preserva-
tives used in homes.] In: Aurand, K., Seifert, B., & Wegner,
J., ed. [Air quality in interiors,] Stuttgart, New York,
Gustav-Fischer-Verlag, pp. 309-316 (in German).
KRAUSE, CHR. & ENGLERT, N. (1980) [Health evaluation of PCP
(pentachlorophenol) containing wood preservatives in rooms.]
Holz Roh Werkst., 38: 429-432 (in German).
KROYER, G., WASHUTTL, J., SCHOERGMEYER, W., STEINER, I., &
WINKER, N. (1982) [Contamination of food with volatile
biocide substances from wood preservatives in model
experiments.] Lebensm.-Wissen. Technol., 15: 111-112 (in
German).
KUEHL, D.W. & DOUGHERTY, R.C. (1980) Pentachlorophenol in
the environment. Evidence for its origin from commercial
pentachlorophenol by negative chemical ionization mass
spectrometry. Environ. Sci. Technol., 14: 447-449.
KUNDE, M. (1982) [Experience in the evaluation of wood
preservatives.] In: Aurand, K., Seifert, B., & Wegner, J., ed.
[Air quality in interiors,] Stuttgart, New York, Gustav-
Fischer-Verlag, pp. 317-325 (in German).
KUNDE, M, & BOHME, C. (1978) [Toxicology of pentachloro-
phenol - an overview]. Bundesgesundheitsblatt. 21: 302-310 (in
German).
KUTZ, F.W., MURPHY, R.S., & STRASSMAN, S.C. (1978) Survey of
pesticide residues and their metabolites in urine from the
general population. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 363-369.
KUWAHARA, M., KATO, N., & MUNAKATA, K. (1966a) The photo-
chemical reaction of pentachlorophenol. Part I. The structure
of the yellow compound. Agric. Biol. Chem. Jpn, 30: 232-238.
KUWAHARA, M., KATO, N., & MUNAKATA K. (1966b) The photo-
chemical reaction of pentachlorophenol. Part II. The chemical
structure of minor products. Agric. biol. Chem. Jpn, 30:
239-245.
KUWATSUKA, S. (1972) Degradation of several herbicides under
different conditions. In: Matsumura, F., Boush, G., & Misato,
T., ed. Environmental toxicology of pesticides, New York,
Academic Press, pp. 385-400.
KUWATSUKA, S. & IGARASHI, M. (1975) Degradation of PCP in
soils. II. The relationship between the degradation of PCP and
the properties of soils, and the identification of the
degradation products of PCP. Soil Sci. Plant Nutr., 21:
405-414.
LAHANIATIS, E.S., CLAUSEN, E., BIENIEK, D., & KORTE, F.
(1985) Formation of 2,3,7,8-tetrachlorodibenzofuran during
thermolysis of selected chlorinated organic compounds.
Chemosphere, 14: 233-238.
LAMBERTON, J., GRIFFIN, B., ARBOGAST, B., INMAN, R., DEINZER,
M., & GRIFFIN, D. (1979) The determination of polychloro-
dibenzo- p-dioxins in pentachlorophenol and wood treatment
solutions. Am. Ind. Hyg. Assoc. J., 40: 816-822.
LAMPARSKI, L.L., MAHLE, N.H., & SHADOFF, L.A. (1978)
Determination of pentachlorophenol, hexachlorodibenzo- p-
dioxin, and octachlorodibenzo- p-dioxin in bovine milk. J.
agric. food Chem., 26: 1113-1116.
LAMPARSKI, L.L., STEHL, R.H., & JOHNSON, R.L. (1980)
Photolysis of pentachlorophenol-treated wood. Chlorinated
dibenzo- p-dioxin formation. Environ. Sci. Technol., 14,
196-200.
LANGEBARTELS, C. & HARMS, H. (1984) Metabolism of penta-
chlorophenol in cell suspension cultures of soybean and wheat:
pentachlorophenol glucoside formation. Z. Pflanzenphysiol.,
113: 201-212.
LANGER, H.G., BRADY, T.P., DALTON, L.A., SHANNON, T.W., &
BRIGGS, P.R. (1973) Thermal chemistry of chlorinated
phenols. In: Blair, E.H., ed. Chlorodioxins: origin and fate,
Washington DC, American Chemical Society, pp. 26-32 (Advances
in Chemistry Series No. 120).
LANGEVELD, VAN, H.E.A.M. (1975) Determination of penta-
chlorophenol in toy paints. J. Assoc. Off. Anal. Chem., 58:
19-22.
LARSEN, R.V., BORN, G.S., KESSLER, W.V., SHAW, S.M., & VAN
SICKLE, D.C. (1975) Placental transfer and teratology of
pentachlorophenol in rats. Environ. Lett., 10: 121-128.
LARSEN, R.V., KIRSCH, L.E., SHAW, S.M., CHRISTIAN, J.E., &
BORN, G.S. (1972) Excretion and tissue distribution of
uniformly labelled 14C-pentachlorophenol in rats. J. Pharm.
Sci., 61: 2004-2006.
LAUWERYS, R. (1970) Biological criteria for selected
industrial toxic chemicals: a review. Scand. J. Work. environ.
Health, 1: 139-172.
LEE, B.E., LACROIX, M.D., DUPONT, G.A.J., & SCOTT, J.A.
(1984) Capillary gas chromatographic determination of
pentachlorophenol residues in tallow following automated gel
permeation clean-up. J. Assoc. Off. Anal. Chem., 67: 546-548.
LEIGHTY, E.G. & FENTIMAN, A.F., Jr (1982) Conjugation of
pentachlorphenol to palmitic acid by liver microsomes. Bull.
environ. Contam. Toxicol., 28(3): 329-333.
LEVIN, J.-O. & NILSSON, C.-A. (1977) Chromatographic
determination of polychlorinated phenols, phenoxyphenols,
dibenzofurans, and dibenzodioxins in wood-dust from worker
environments. Chemosphere, 6: 443-448.
LEVIN, J.-O., RAPPE, C., & NILSSON, C. (1976) Use of
chlorophenols as fungicides in sawmills. Scand. J. Work
environ. Health, 2: 71-81.
LILIENBLUM, W. (1985) Formation of pentachlorophenol
glucuronide in rat and human liver microsomes. Biochem.
Pharmacol., 34: 893-894.
LIU, D., THOMSON, K., & STRACHAN, W.M.J. (1981) Biodegra-
dation of pentachlorophenol in a simulated aquatic environ-
ment. Bull. environ. Contam. Toxicol., 26: 85-90.
LIU, D., THOMSON, K., & KAISER, K.L.E. (1982) Quantitative
structure-toxicity relationship of halogenated phenols on
bacteria. Bull. environ. Contam. Toxicol., 29: 130-136.
LOWER, M. (1982) [Studies on the pentachlorophenol content
of human organs.] Friedrich-Alexander- University Erlangen-
Nürnberg (Thesis) (in German).
LORES, E.M., EDGERTON, T.R., & MOSEMAN, R.F. (1981) Method
for the confirmation of chlorophenols in human urine by LC
with an electrochemical detector. J. chromatogr. Sci., 19,
466-469.
LU, P.Y. & METCALF, R.L. (1975) Environmental fate and
biodegradability of benzene derivatives as studied in a model
aquatic ecosystem. Environ. Health Perspect., 10: 269-284.
LU, P.Y., METCALF, R.L., & COLE, L.K. (1978) The environ-
mental fate of 14C-pentachlorophenol in laboratory model
ecosystems. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 53-63.
LYR, H. (1962) [On the oxidative decomposition of
chlorinated phenols.] Holztechnologie, 3: 201-208 (in German).
LYR, H. (1963) [Enzymatic detoxification of chlorinated
phenols.] Phytopathol. Z., 47: 73-83 (in German).
MCCONNELL, E.E., MOORE, J.A., HASEMANN, J.K., & HARRIS, M.W.
(1978) The comparative toxicity of chlorinated dibenzo- p-
dioxins in mice and guinea-pigs. Toxicol. appl. Pharmacol.,
44: 335-356.
MCCONNELL, E.E., MOORE, J.A., GUPTA, B.N., RAKES, A.H.,
LUSTER, M.I., GOLDSTEIN, J.A., HASEMAN, J.K., & PARKER, C.E.
(1980) The chronic toxicity of technical and analytical
pentachlorophenol in cattle. I. Clinicopathology. Toxicol.
appl. Pharmacol., 52: 468-490.
MCGAVACK, T.H., BOYD, L.J., PICCIONE, F.V., & TERRANOVA, R.
(1941) Acute and chronic intoxications with sodium penta-
chlorophenate in rabbits. J. ind. Hyg. Toxicol., 23(6):
239-251.
MCGOVERN, J.J. (1982) Apparent immunotoxic response to
phenolic compounds. Food Chem. Toxicol., 20: 496.
MANSKE, D.D. & JOHNSON, R.D. (1977) Residues in food and
feed. Pesticide and other chemical residues in total diet
samples. X. Pest. monit. J., 10: 134-148.
MASON, M.F., WALLACE, S.M., FOERSTER, E., & DRUMMOND, W.
(1965) Pentachlorophenol poisoning: report of two cases. J.
Forensic Sci., 10: 136-147.
MASUDA, Y. & KUROKI, H. (1982) Polychlorinated dibenzofurans
and related compounds in patients with "Yusho". In: Hutzinger,
O., Frei, R.W., Merian, E., & Pocchiari, F., ed. Chlorinated
dioxins and related compounds: impact on the environment,
Oxford, Pergamon Press, Vol. 5, pp. 561-570 (Pergamon Series
on Environmental Science).
MATSUMOTO, G. (1982) Comparative study on organic
constituents in polluted and unpolluted inland aquatic
environments. III. Phenols and aromatic acids in polluted and
unpolluted waters. Water Res., 16: 551-557.
MATSUMOTO, G., ISHIWATARI, R., & HANYA, T. (1977) Gas
chromatographic-mass spectrometric identification of phenols
and aromatic acids in river waters. Water Res., 11: 693-698.
MATTSON, V.R., ARTHUR, J.W., & WALBRIDGE, C.T. (1976) Acute
toxicity of selected organic compounds to fathead minnows,
Duluth, Minnesota, US Environmental Protection Agency, 13 pp
(Ecological Research Series No. EPA 600/3-76-097).
MEEMKEN, H.-A., FUERST, P., & HABERSAAT, K. (1982) [Analysis
of pentachlorophenol in cultured champignons.] Dtsch. Lebensm.
Rundsch. 78: 282-287 (in German).
MEHENDALE, H.W., FIELDS, M., & MATTHEWS, H.B. (1975) Meta-
bolism and effects of hexachlorobenzene on hepatic microsomal
enzymes in the rat. J. agric. food Chem., 23: 261-264.
MELNIKOV, N.N. (1971) Chemistry of pesticides. Residue Rev.,
36: 104-107.
MENON, J.A. (1958) Tropical hazards associated with the use
of pentachlorophenol. Br. med. J., 11: 1156-1158.
MERCIER, M. (1981) Criteria (dose/effect relationships) for
organochlorine pesticides, Oxford, Pergamon Press, 381 pp.
MERZ, V. & WEITH, W. (1872) [On the characteristics of
pentachlorophenol.] Ber. Dtsch. Chem. Ges., 5: 458-463 (in
German).
MILLER, C.S. & ABOUL-ELA, M.M. (1969) Fate of penta-
chlorophenol in cotton. J. agric. food Chem., 17: 1244-1246.
MITSUDA, H., MURAKAMI, K., & KAWAI, F. (1963) Effect of
chlorophenol analogues on the oxidative phosphorylation in rat
liver mitochondria. Agric. biol. Chem., 27: 366-372.
MORGADE, C., BARQUET, A., & PFAFFENBERGER, C.D. (1980)
Determination of polyhalogenated phenolic compounds in
drinking-water, human blood-serum, and adipose tissue. Bull.
environ. Contam. Toxicol., 24: 257-264.
MORGAN, J.W.W. & PURSLOW, D.F. (1973) Volatile losses of
wood preservatives. In: Proceedings of the 23rd Annual
Convention of the BWPA, British Wood Preservers Association,
pp. 173-193.
MOOS, L.P., KIRSCH, E.J., WUKASCH, R.F., & GRADY, C.P.L.
(1983) Pentachlorophenol biodegradation. I. Aerobic. Water
Res., 17: 1575-1584.
MULLER, W.F. (1981) [Metabolism and pharmacokinetics of
selected chemicals in rhesus monkey and chimpanzee,] Society
for Radiation and Environmental Research (GSF), Münich, pp.
120-128 (Report ö/599) (in German).
MUNAKATA, K. & KUWAHARA, M. (1969) Photochemical degradation
products of pentachlorophenol. Residue Rev., 25: 13-23.
MUNDY, D.E. & MACHIN, A.F. (1981) Determination of penta-
chlorophenol and related compounds in animal materials by
high-performance liquid chromatography and gas chromatography.
J. Chromatogr., 216: 229-238.
MUNRO, I.B., OSTLER, D.C., MACHIN, A.F., & QUICK, M.P. (1977)
Suspected poisoning by pentachlorophenol in sawdust. Vet.
Rec., 101(26/27): 525.
MURRAY, H.E., RAY, L.E., & GIAM, C.S. (1981) Analysis of
marine sediment, water, and biota for selected organic
pollutants. Chemosphere, 10: 1327-1334.
MURTHY, N.B.K. & KAUFMAN, D.D. (1978) Degradation of penta-
chloronitrobenzene (PCNB) in anaerobic soils. J. agric. food
Chem., 26: 1151-1156.
MURTHY, N.B.K., KAUFMAN, D.D., & FRIES, G.F. (1979) Degra-
dation of pentachlorophenol (PCP) in aerobic and anaerobic
soil. J. environ. Sci. Health, B14: 1-14.
NARANG, A.S., VERNOY, C.A., & EADON, G.A. (1983) Evaluation
of Nielsen-Kryger steam distillation technique for recovery of
phenols from soil. J. Assoc. Off. Anal. Chem., 66: 1330-1334.
NCI (1979) Bioassay of 2,4,6-trichlorophenol for possible
carcinogenicity, Bethesda, Maryland, National Cancer
Institute, 115 pp (Technical Report No. PC A06/MF, PB
293-7700).
NCI (1980a) Bioassay of a mixture of 1,2,3,6,7,8-hexachloro-
benzo-p-dioxin and 1,2,3,7,8,9-hexachlorodibenzo-p-dioxin
(gavage) for possible carcinogenicity, Bethesda, Maryland,
National Cancer Institute (Carcinogenesis Technical Report
Series No. 198) (US Department of Health and Human Services
Publication No. (NIH) 80-1754).
NCI (1980b) Bioassay of a mixture of 1,2,3,6,7,8-hexachloro-
dibenzo-p-dioxin and 1,2,3,7,8,9-hexachlorodibenzo-p-dioxin
(dermal) for possible carcinogenicity, Bethesda, Maryland,
National Cancer Institute (Carcinogenesis Technical Report
Series No. 202) (US Department of Health and Human Services
Publication No. (NIH) 80-1758).
NEEDHAM, L.L., CLINE, R.E., HEAD, S.L., & LIDDLE, J.A.
(1981) Determining pentachlorophenol in body fluids by gas
chromatography after acetylation. J. anal. Toxicol., 5:
283-286.
NEIDERT, E., SASCHENBRECKER, P.W., & PATTERSON, J.R. (1984)
Detection and occurrence of pentachlorophenol residues in
chicken liver and fat. J. environ. Sci. Health B, 19: 579-592.
NIIMI, A.J. & MCFADDEN, L.A. (1982) Uptake of sodium penta-
chlorophenate (NaPCP) from water by rainbow trout (Salmo
gairdneri) exposed to concentrations in the ng/litre range.
Bull. environ. Contam. Toxicol., 28(1): 11-19.
NILSSON, C.-A., NORSTROM, A., ANDERSSON, K., & RAPPE, C.
(1978) Impurities in commercial products related to penta-
chlorophenol. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 313-324.
NING, H.S. (1984) [Investigation of the chronic intoxication
of pentachlorophenol.] Chin. J. Ind. Hyg. Occup. Dis., 4:
24-28 (in Chinese).
NING, H.S., ZHEN, H.-Q., LI, L.-S., XU, H., WANG, J.-J., WU,
S.-S., LIANG, T.-Z., JAN, J.-Q., ZHU, B.-H., HUANG, W.-W.,
CHEN, W.-Y., & WANG, G.-F. (1984) [Study of the toxicity of
pentachlorophenol and recommendations of the maximum allowable
concentration in air.] J. Commun. ind. Hyg. (Rail Transp.
Syst.), 4: 7-16 (in Chinese).
NIOSH (1978) Manual of analytical methods, Cincinatti, Ohio,
US Department of Health, Education, and Welfare, Vol. 4, pp.
S297-1-S297-8 (DHEW Publication No. 78-175).
NIOSH (1983) In: Tatken, R.L. & Lewis, R.J., ed. Registry of
toxic effects of chemical substances, Cincinnati, Ohio, US
National Institute for Occupational Safety and Health, pp.
74-75.
NOAKES, D.N. & SANDERSON, D.M. (1969) A method for
determining the dermal toxicity of pesticides. Br. J. ind.
Med., 26: 59-64.
NOMURA, S. (1954) Studies on chlorophenol poisoning. I. A
clinical examination of workers exposed to pentachlorophenol.
Bull. Hyg., 29: 294.
NORSTROM, A., RAPPE, C., LINDAHL, R., & BUSER, H.-R. (1979)
Analysis of some older Scandinavian formulations of 2,4-
dichlorophenoxy acetic acid and 2,4,5-trichlorophenoxy acetic
acid for contents of chlorinated dibenzo- p-dioxins and
dibenzofurans. Scand. J. Work environ. Health, 5: 375-378.
NRC (1986) In: Thomas, R.D., ed. Drinking water and health,
Washington DC, National Research Council, National Academy
Press, Vol. 6, pp. 407-421.
NRCC (1981) Polychlorinated dibenzo-p-dioxins: criteria for
their effects on man and his environment, Ottawa, Ontario,
National Research Council of Canada, Associate Committee on
Scientific Criteria for Environmental Quality (No. 18574),
251 pp.
NRCC (1982) Chlorinated phenols: criteria for environmental
quality, Ottawa, Ontario, National Research Council of Canada,
Associate Committee on Scientific Criteria for Environmental
Quality, (No. 18578) 191 pp.
OHE, T. (1979) Pentachlorophenol residues in human adipose
tissue. Bull. environ. Contam. Toxicol., 22: 287-292.
OLIE, K., BERG, M.V.D., & HUTZINGER, O. (1983) Formation and
fate of PCDD and PCDF combustion processes. Chemosphere, 12:
627-636.
OWEN, J.W. & ROSSO, S.W. (1981) Effects of sublethal
concentrations of pentachlorophenol on the liver of bluegill
sunfish, Lepomis machrochirus. Bull. environ. Contam. Toxicol.,
26: 594-600.
PAASIVIRTA, J., SORKKO, J., LESKIJORVI, T., & ROOS, A.
(1980) Transportation and enrichment of chlorinated phenolic
compounds in different aquatic food chains. Chemosphere, 9:
441-456.
PAASIVIRTA, J., SORKKO, J., AHO, M., SURMA-AHO, K., TARHANEN,
J., & ROOS, A. (1981) Recent trends of biocides in pikes of
the lake Päijänne. Chemosphere, 10: 405-414.
PAASIVIRTA, J., SORKKO, J., SURMA-AHO, K., HUMPPI, T.,
KUOKKANEN, T., & MARTTINEN, M. (1983) Food chain enrichment
of organochlorine compounds and mercury in clean and polluted
lakes of Finland. Chemosphere, 12: 239-252.
PAASIVIRTA, J., HEINOLA, K., HUMPPI, T., KARJALAINEN, A.,
KNUUTINEN, J., MANTYKOSKI, K., PAUKKU, R., PIILOLA, T., SURMA-
AHO, K., TARHANEN, J., WELLING, L., & VIHONEN, H. (1985)
Polychlorinated phenols, guaiacols and catechols in the
environment. Chemosphere, 14: 469-491.
PACKHAM, E.D., DUXBURY, C.L., MAYFIELD, C.I., INNISS, W.E.,
KRUUV, J., & THOMPSON, J.E. (1982) Quantitative analysis of
pollutant-induced lethal and sublethal damage in cultured
mammalian cells. Bull. environ. Contam. Toxicol., 29: 739-746.
PALMER, C.M. & MALONEY, T.E. (1955) Preliminary screening
for potential algicides. Ohio J. Sci., 55: 1-8.
PARKER, C.E., JONES, W.A., MATTHEWS, H.B., MCCONNELL, E.E., &
HASS, J.R. (1980) The chronic toxicity of technical and
analytical pentachlorophenol in cattle. II. Chemical analysis
of tissues. Toxicol. appl. Pharmacol., 55: 359-369.
PARR, L.J., GEE, M.G., LAND, D.G., ROBINSON, D., & CURTIS,
R.F. (1974) Chlorophenols from wood preservatives in broiler
house litter. J. Sci. Food Agric., 25: 835-841.
PARRISH, P.R., DYAR, E.E., ENOS, J.M., & WILSON, W.G. (1978)
Chronic toxicity of chlordane, trifluralin, and pentachloro-
phenol to sheepshead minnows (Cyprinodon variegatus),
Washington DC, US Environmental Protection Agency, 67 pp
(Report No. EPA 600/3-78-010).
PAULI, O. & FRANKE, G. (1972) Behaviour and degradaton of
technical preservatives in the biological purification of
sewage. In: Walters, A.H. & Hueck-Van der Plas, E.H., ed.
Biodegradaton of materials. II, New York, John Wiley and Sons,
pp. 52-60.
PEARCE, N.E., SMITH, A.H., HOWARD, J.K., SHEPPARD, R.A.,
GILES, H.J., & TEAGUE, C.A. (1986) Non-Hodgkin's lymphoma
and exposure to phenoxyherbicides, chlorophenols, fencing
work, and meat works employment: a case control study. Br. J.
ind. Med., 43: 75-83.
PEARCE, N.E., SHEPPARD, R.A., SMITH, A.H., & TEAGUE, C.A. (in
press) Non-Hodgkin's lymphoma and farming: an expanded
case-control study. Int. J. Cancer.
PEKARI, L. & ANTERO, A. (1982) A simple liquid chromato-
graphic method for the analysis of penta- and tetrachloro-
phenols in urine of exposed workers. J. Chromatogr. biomed.
Appl., 232: 129-136.
PETROWITZ, H.J. (1981) [Volatilisation of the active
ingredients of wood preservatives from chemically protected
timber.] In: [Research report of the Expert Group on
Biological Materials Research,] Berlin, Federal Institute for
Materials Research, 32 pp (in German).
PIERCE, R.H., Jr & VICTOR, D.M. (1978) The fate of penta-
chlorophenol in an aquatic ecosystem. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 41-52.
PIERCE, R.H., BRENT, C.R., WILLIAMS, H.P., & REEVES, S.G.
(1977) Pentachlorophenol distribution in a fresh water
ecosystem. Bull. environ. Contam. Toxicol., 18: 251-258.
PIGNATELLO, J.J., MARTINSON, M.M., STEIERT, J.G., CARLSON,
R.E., & CRAWFORD, R.L. (1983) Biodegradation and photolysis
of pentachlorophenol in artificial freshwater streams. Appl.
environ. Microbiol., 46: 1024-1031.
PLESTINA, R. (1984) Prevention, diagnosis and treatment of
insecticide poisoning, Geneva, World Health Organization,
69@pp (unpublished report VBC/84.889).
PLIMMER, J.R. (1973) Technical pentachlorophenol: origin and
analysis of base-insoluble contaminants. Environ. Health
Perspect., 5: 41-48.
POWERS, P.W. (1976) How to dispose of toxic substances and
industrial wastes, Park Ridge, New Jersey, Noyes Data
Corporation, p. 373.
PRAGER, B., JACOBSON, P., SCHMIDT, P., & STERN, D., ed.
(1923) [Beilstein's handbook of organic chemistry.] 4th ed.,
Berlin, Springer-Verlag, Vol. 6, (Syst. No. 522) pp. 194-197
(in German).
PRESCOTT, C.A., WILKIE, B.N., HUNTER, B., & JULIAN, R.J.
(1982) Influence of a purified grade of pentachlorophenol on
the immune-response of chickens. Am. J. vet. Res., 43: 481-487.
PRUITT, G.W., GRANTHAM, B.J., & PIERCE, R.H. (1977)
Accumulation and elimination of pentachlorophenol by the
bluegill Lepomis macrochirus. Am. Fish. Soc., 106: 462-465.
QUICK, M.P. (1982) Pesticide poisoning of livestock: a
review of cases investigated. Vet. Rec., 111: 5-7.
RAHDE, A.F. & DELLA ROSA, H.V. (1984) [Evaluation of
ecotoxicological impact of the hydroelectric dam of Tucurui,
Brazil,] 45 pp (unpublished report presented to Eletronorta
(Eletrobras)) (in Portuguese).
RAHDE, A.F. & DELLA ROSA, H.V. (1986) Pentachlorophenol - an
evaluation of the ecotoxicological impact of the hydroelectric
dam of tucurui, Brazil, 6 pp (unpublished report presented to
Eletronorte (Eletrobras)).
RAPPE, C., GARA, A., & BUSER, H.R. (1978a) Identification of
polychlorinated dibenzofurans (PCDFs) in commercial chloro-
phenol formulations. Chemosphere, 12: 981-991.
RAPPE, C., MARKLUND, S., BUSER, H.R., & BOSSHARDT, H.-P.
(1978b) Formation of polychlorinated dibenzo- p-dioxins
(PCDDs) and dibenzofurans (PCDFs) by burning or heating
chlorophenates. Chemosphere, 3: 269-281.
RAPPE, C., BUSER, H.R., & BOSSHARDT, H.-P. (1979) Dioxins,
dibenzofurans, and other polyhalogenated aromatics: production,
use, formation, and destruction. Ann. NY Acad. Sci.,
320: 1-18
RAPPE, C., NYGREN, M., BUSER, H.R., & KAUPPINEN, T. (1982)
Occupational exposure to polychlorinated dioxins and dibenzo-
furans. In: Hutzinger, O. et al., ed. Chlorinated dioxins and
related compounds: impact on the environment, Oxford, Pergamon
Press, Vol. 5, (Pergamon Series on Environmental Science) pp.
495-514.
REINER, E.A., CHU, J.P., & KIRSCH, E.J. (1978) Microbial
metabolism of pentachlorophenol. In: Rao, K.R., ed. Penta-
chlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 67-81.
RENBERG, L. (1974) Ion exchange technique for the deter-
mination of chlorinated phenols and phenoxy acids in organic
tissue, soil, and water. Anal. Chem., 46: 459-461.
RENNER, G. & MUCKE, W. (1986) Transformations of penta-
chlorophenol. I. Metabolism in animals and man. Toxicol.
environ. Chem., 11: 9-29.
RENNER, G., HOPFER, C., & GOKEL, J.M. (1986) Acute
toxicities of pentachlorophenol, pentachloroanisole, tetra-
chlorohydroquinone, tetrachlorocatechol, tetrachloro-
resorcinol, terachlorodimethoxybenzene diacetates administered
to mice. Toxicol. environ. Chem., 11: 37-50.
RICHARDSON, N.G. (1978) Wood preserving effluents and their
treatment. In: Proceedings of the Technical Transfer Seminar
Timber Processing Industry, March 10-11 1977, Toronto,
Environment Canada, pp. 40-62 (Econ. Technical Review No. EPS
3-WP-78-1).
RIPPEN G. (1984) [Pentachlorophenol.] In: [Handbook of
environmental chemicals: physicochemical and ecotoxicological
data of selected chemicals,] Landsberg/Lech, Ecomed, pp. 1-13
(in German).
ROBERTS, H.J. (1981) Aplastic anemia due to pentachloro-
phenol. New Engl. J. Med., 305: 1650-1651.
ROBERTS, H.J., (1983) Aplastic anemia and red cell aplasia
due to pentachlorophenol. South. Med. J., 76(1): 45-48.
ROBSON, A.L., KISSANE, J.M., ELVICK, N.H., & PUNDAVELA, L.
(1969) Pentachlorophenol poisoning in a nursery for newborn
infants. I. Clinical features and treatment. J. Pediatr.,
75(2): 309-316.
ROSSKAMP, E. (1982) [Pentachlorophenol as a model case of a
xenobiotic compound: PCP in the environment.] Berlin, Federal
Health Office Institute for Water, Soil and Air Hygiene, 22 pp
(Research Report No. 10604007) (in German).
ROTT, B., NITZ, S., & KORTE, F. (1979) Microbial decompo-
sition of sodium pentachlorophenolate. J. agric. food Chem.,
27, 306-310.
ROWE, E.L., ZIOBRO, R.J., WANG, C.J.K., & DENCE, C.W. (1982)
The use of an alga Chlorella pyrenoidosa and a duckweed Lemna
perpusilla as test organisms for toxicity bioassays of spent
bleaching liquors and their components. Environ. Pollut. (Ser.
A), 27: 289-296.
ROY, P., KUNDU, P., & DAS, A. (1981) Sodium pentachloro-
phenate as a mutagenic agent. Biotechnol. Lett., 3: 401-404.
ROZMAN, K., MUELLER, W., COULSTON, F., & KORTE, F. (1977)
Long-term feeding study of hexachlorobenzene in rhesus
monkeys. Chemosphere, 2/3: 81-84.
RUBINSTEIN, N.I. (1978) Effects of sodium pentachlorophenate
on the feeding activity of the lugworm Arenicola cristata
Stimpson. In: Rao, K.R., ed. Pentachlorophenol: chemistry,
pharmacology, and environmental toxicology, New York, London,
Plenum Press, pp. 175-179.
RUDLING, L. (1970) Determination of pentachlorophenol in
organic tissues and water. Water Res., 4: 533-537.
RUBELT, C., DIETZ, F., KICKUTH, R., KOPPE, P., KUNTE, H.,
PESCHEL, G., & SONNEBORN, M. (1982) [Pollutants in ambient
water. Vol. II. Phenols.] Boppard, Harald Boldt Verlag, 168 pp
(DFG Report) (in German).
RUMKER, VON, R., LAWLESS, E.W., & MEINERS, A.F. (1974)
Production, distribution, use, and environmental impact
potential of selected pesticides. Washington DC, US
Environmental Protection Agency, pp. 308-319 (EPA Production
Report No. EPA 540/1-74-001).
RUSINK, R.G. & SMITH, L.L. (1975) The relationship of the
96-hour LC50 to the lethal threshold concentration of
hexavalent chromium, phenol, and sodium pentachlorophenate for
fathead minnows ( Pimephales promelas Rafinesque). Trans. Am.
Fish. Soc., 104: 567-570.
RUH, C. & GEBEFUEGI, I. (1984) [Sources for indoor space
contamination: presence of pentachlorophenol and lindane in
untreated wood samples.] Chemosphere, 13: 919-925 (in German).
RUH, C., GEBEFUEGI, I., & KORTE, F. (1984) The indoor
biocide pollution: occurrence of pentachlorophenol and lindane
in homes. In: Berglund, B., Lindvall, T., & Sundell, J., ed.
Indoor air. IV. Chemical characterization and personal
exposure, Stockholm, Swedish Council for Building Research,
pp. 309-322.
RYAN, J.J. (1983) Higher chlorinated dioxins implicated in
the mortality of young pigs kept on a pentachlorophenol-
treated wooden floor. Can. vet. J., 24: 72-75.
SAARIKOSKI, I. & VILUKSELA, M. (1981) Influence of pH on the
toxicity of substituted phenols to fish. Arch. environ.
Contam. Toxicol., 10: 747-753.
SALKINOJA-SALONEN, M.S., HAKULINEN, R., VALO, R., &
APAJALAHTI, J. (1983) Biodegradation of recalcitrant
organochlorine compounds in fixed film reactors. Water Sci.
Tech., 15: 309-319.
SALKINOJA-SALONEN, M.S., VALO, R., APAJALAHTI, J., HAKULINEN,
R., SILAKOSKI, L., & JAAKKOLA, T. (1984) Biodegradation of
chlorophenolic compounds in wastes from wood-processing
industry. In: Klug, M. & Reddy, C., ed. Current perspectives
in microbial ecology, Washington DC, American Society for
Microbiology, pp. 668-676.
SANBORN, J.R., CHILDERS, W.F., & HANSEN, L.G. (1977) Uptake
and elimination of 14C-hexachlorobenzene (HCB) by the green
sunfish, Lepomis cyanellus Raf., after feeding contaminated
food. J. agric. food Chem., 25: 551-553.
SANDERMANN, H., Jr, SCHEEL, D., & V.D. TRENCK, T. (1984) Use
of plant cell cultures to study the metabolism of environ-
mental chemicals. Ecotoxicol. environ. Saf., 8: 167-182.
SANDERMANN, H.S., CASTEN, R., & STOCKMANN, H. (1957)
Pyrolysis of pentachlorophenol. Chem. Ber., 90: 690-692.
SANGSTER, B., WEGMAN, R.C.C., & HOFSTEE, A.W.M. (1982)
Non-occupational exposure to pentachlorophenol: clinical
findings and plasma-PCP-concentrations in three families. Hum.
Toxicol., 1: 123-133.
SAUR, J.M., WALCHESKI, P.J., NICHOLAS, D.-D., & GJOVIK, L.R.
(1982) The concentration of airborne pentachlorophenol within
treated wood structures. Proc. Ann. Meet. Am. Wood Preserv.
Assoc., 78: 169-173.
SAVOLAINEN, H. & PEKARI, K. (1979) Neurochemical effects of
peroral administration of technical pentachlorophenol. Res.
Commun. chem. Pathol. Pharmacol., 23(1): 97-105.
SCHAUERTE, W., LAY, J.P., KLEIN, W., & KORTE, F. (1982)
Influence of 2,4,6-trichlorophenol and pentachlorophenol on
the biota of aquatic systems. Outdoor experiments in
compartments of a natural pond. Chemosphere, 11: 71-79.
SCHIMMEL, S.C., PATRICK, J.M., Jr, & FAAS, L.F. (1978)
Effects of sodium pentachlorophenate on several estuarine
animals: toxicity, uptake and depuration. In: Rao, K.R., ed.
Pentachlorophenol: chemistry, pharmacology, and environmental
toxicology, New York, London, Plenum Press, pp. 147-155.
SCHONHABER, R., SCHWARZ, A., & KRANL, D. (1982) Comparative
studies on the high-pressure liquid chromatographic and gas
chromatographic determination of pentachlorophenol in mush-
rooms. Dtsch. Lebensm. Rundsch., 78: 254-257.
SCHRAG., S.D. & DIXON, R.L. (1985) Occupational exposures
associated with male reproductive dysfunction. Ann Rev.
Pharmacol. Toxicol., 25: 567-592.
SCHUPPENER, H. (1974) [Uptake of pentachlorophenol (PCP) by
corn plants from culture media - a methodological study on
the ecological chemistry of lipophilic toxic compounds,]
University of Göttingen, 105 pp, (Dissertation) (in German).
SCHWETZ, B.A., NORRIS, J.M., SPARSCHU, G.L., ROWE, V.K.,
GEHRING, P.J., EMERSON, J.L., & GERBIG, C.G. (1973)
Toxicology of chlorinated dibenzo- p-dioxins. Environ. Health
Perspect., 5: 87-99.
SCHWETZ, B.A., KEELER, P.A., & GEHRING, P.J. (1974) The
effect of purified and commercial grade pentachlorophenol on
rat embryonal and fetal development. Toxicol. appl.
Pharmacol., 28: 151-161.
SCHWETZ, B.A., QUAST, I.F., KEELER, P.A., HUMISTON, C.G., &
KOCIBA, R.J. (1978) Results of two-year toxicity and
reproduction studies on pentachlorophenol in rats. In: Rao,
K.R., ed. Pentachlorophenol: chemistry, pharmacology, and
environmental toxicology, New York, London, Plenum Press, pp.
301-309.
SHA, S.Z. & DUFFIELD, A.M. (1984) Negative ion chemical
ionization gas chromatography-mass spectrometry of some
derivatives of tri-, tetra-, and pentachlorophenols. J.
Chromatogr., 284: 157-166.
SHAFIK, T.M. (1973) The determination of pentachlorophenol
and hexachlorophene in human adipose tissue. Bull. environ.
Contam. Toxicol., 10: 57-63.
SHAFIK, T.M., SULLIVAN, H.C., & ENOS, H.R. (1973) Multi-
residue procedure for halo- and nitrophenols. Measurement of
exposure to biodegradable pesticides yielding these compounds
as metabolites. J. agric. food Chem., 21: 295-298.
SHEN, S., VILLENEUVE, D.C., CHU, I., KELLY, J., & GILMAN,
A.P. (1983) The acute dermal toxicity of tetrachlorophenols
in the rat. Bull. environ. Contam. Toxicol., 31: 680-685.
SHIRAKAWA, M., NAGATOSHI, H., & HIRAKAWI, M. (1959) Hygienic
investigation on workers dealing with pentachlorophenol (PCP)
in a rubber manufacturing industry. Kurume Med. J., 6(1):
24-35.
SIEGWART, Y. (1983) [Control of foodstuff in Switzerland in
the year 1982.] Mitt. Geb. Lebensm. Hyg., 74: 179-319 (in
German).
SIQUEIRA, M.E.P.B. & FERNICOLA, N.A.C.G. (1981) Determination
of pentachlorophenol in urine. Bull. environ. Contam.
Toxicol., 27: 380-385.
SIUDA, J.F. (1980) Natural production of organohalogens. In:
Jolley, R.L., Brungs, W.A., Cumming, R.B., & Jacobs, V.A., ed.
Water chlorination - environmental impact and health effects,
Ann Arbor, Michigan, Ann Arbor Science Publishers Inc, Vol. 3,
pp. 63-72.
SLOOF, W. & CANTON, J.H. (1983) Comparison of the
susceptibility of 11 freshwater species to 8 chemical
compounds. II. (Semi) chronic toxicity tests. Aquat. Toxicol.,
4: 271-282.
SMITH, A.H., PEARCE, N.F., FISHER, D.O., GILES, H.J., TEAGUE,
C.A., & HOWARD, J.K. (1984) Soft-tissue sarcoma and exposure
to phenoxyherbicides and chlorophenols in New Zealand. J. Natl
Cancer Inst., 73: 1111-1117.
SONNEBORN, M. (1976) A study to determine the comparability
of chemical analyses for drinking-water quality within
European communities, Luxembourg, Commission of European
Communities, 97 pp (EUR 5542e).
STANLAKE, G.J. & FINN, R.K. (1982) Isolation and character-
ization of a pentachlorophenol-degrading bacterium. Appl.
environ. Microbiol., 44: 1421-1427.
STEHL, R.H., PAPENFUSS, R.R., BREDEWEG, R.A., & ROBERTS, R.W.
(1973) The stability of pentachlorophenol and chlorinated
dioxins to sunlight, heat, and combustion. Adv. Chem. Ser.,
120: 119-125.
STERLING, T.D., STOFFMAN, L.D., STERLING, D.A., & MATE, G.
(1982) Health effects of chlorophenol wood preservatives on
sawmill workers. Int. J. Health Serv., 12: 559-571.
STEVENS, H.M. & RICHARDSON, A. (1979) The rapid screening of
body tissues for pentachlorophenol (PCP) with special
reference to poisoning fatality. J. Forensic Sci. Soc., 19:
125-129.
STIJVE, T. (1981) Determination of pentachlorophenol and
2,3,4,6-tetrachlorophenol in edible gelatins. Dtsch. Lebensm.
Rundsch., 77: 249-253.
STOCKDALE, M. & SELWYN, M.J. (1971a) Influence of ring
substituents on the action of phenols on some dehydrogenases,
phosphokinases, and the soluble ATPase from mitochondria. Eur.
J. Biochem., 21: 416-423.
STOCKDALE, M. & SELWYN, M.J. (1971b) Effects of ring
substituents on the activity of phenols as inhibitors and
uncouplers of mitochondrial respiration. Eur. J. Biochem., 21:
565-574.
STOHLMAN, E.F. (1951) The toxicity of some related halo-
genated derivatives of phenols, Bethesda, Maryland, US Public
Health Service, pp. 1303 (Public Health Reports No. 66).
STRANKS, D.W. (1976) Wood preservatives: their depletion as
fungicides and fate in the environment, Ottawa, Environment
Canada, Canadian Forestry Service, pp. 1-35 (Forestry
Technical Report No. 10).
STRETZ, L.A. & VAVRUSKA, J.S. (1984) Controlled air
incineration of pentachlorophenol-treated wood, Washington DC,
US Environmental Protection Agency, 4 pp (EPA-600/S2-84-089).
STRUFE, R. (1968) Problems and results of residue studies
after application of molluscicides. Residue Rev., 24: 80-168.
SU, Y.H. & LIN, H.C. (1971) Influence of soil physico-
chemical characteristics on the efficacy of herbicide penta-
chlorophenol. Chung. Kuo Nung Yeh Hua Hsueh Hui Chih, 8(3-4):
99-104 ( Chem. Abstr. 110707, 74: 301).
SUZUKI, T. (1977) Metabolism of pentachlorophenol by a soil
microbe. J. environ. Sci. Health, B12: 113-127.
SUZUKI, T. (1983) Metabolism of pentachlorophenol (PCP) by
soil microorganisms, Nippon Noyaku Gakkaishi, 8, 385-394.
TAGATZ, M.E., IVEY, I.M., MOORE, I.C., & TOBIA, M. (1977)
Effects of pentachlorophenol on the development of estuarine
communities. J. Toxicol. environ. Health, 3: 501-506.
TAGATZ, M.E., IVEY, I.M., & TOBIA, M. (1978) Effects of
Dowicide(R) G-ST on development of experimental estuarine
macrobenthic communities. In: Rao, K.R., ed. Pentachloro-
phenol: chemistry, pharmacology, and environmental toxicology,
New York, London, Plenum Press, pp. 157-163.
TAGATZ, M.E., IVEY, J.M., GREGORY, N.R., & OGLESBY, J.L.
(1981) Effects of pentachlorophenol on field- and
laboratory-developed estuarine benthic communities. Bull.
environ. Contam. Toxicol., 26: 137-143.
TAM, T.Y. & TREVORS, J.T. (1981a) Toxicity of pentachloro-
phenol to Azotobacter vinelandii. Bull. environ. Contam.
Toxicol., 27: 230-234.
TAM, T.Y. & TREVORS, J.T. (1981b) Effects of pentachloro-
phenol on asymbiotic nitrogen fixation in soil. Water Air Soil
Pollut., 16: 409-414.
THOMAS, P., CARR, R.S., & NEFF, J.M. (1981) Biochemical
stress responses of mullet Mugil cephalus and polychaete worms
Neanthes virens to pentachlorophenol. In: Vernberg, F.J.,
Calabrese, A., Thurnberg, F.P., & Vernberg, W.B., ed.
Biological monitoring of marine pollutants, New York, Academic
Press, pp. 73-103.
THOMPSON, G.E., HUSAIN, H., PARRY, J., WARDROP, W.L., &
GILBRIDE, P.J. (1978) Hydrogeological control and clean-up
of soil and groundwater contaminants at Northern Wood
Preservers, Ltd. In: Proceedings of the Ontario Industrial
Waste Conference, Toronto, June 18-21, Winnipeg, Manitoba,
Engineering Consultants, 18 pp.
THOMPSON, W.S. & DUST, J.V. (1971) Pollution control in the
wood preserving industry. I. Nature and scope of the problem.
For. prod. J., 21: 70-75.
TIERNAN, T.O., TAYLOR, M.L., GARRETT, J.H., VAN NESS, G.F.,
SOLCH, J.G., DEIS, D.A., & WAGEL, D.J. (1983) Chlorodibenzo-
dioxins, chlorodibenzofurans and related compounds in the
effluents from combustion processes. Chemosphere, 12: 595-606.
TIMMONS, L., STEELE, D., CANNON, M., GRESE, R., BROWN, R.,
MURRIL, E., & JAMESON, C.W. (1984) Identification of bromo-
tetrachlorophenol in commercial pentachlorophenol samples. J.
Chromatogr., 314: 476-481.
TING, H.H. & QUICK, M.P. (1980) Simple thin-layer chromato-
graphy method for detection of pentachlorophenol in sawdust
and woodshavings. J. Chromatogr., 195: 441-444.
TODD, A.S. & TIMBIE, C.Y. (1983) Industrial hygiene surveys
of occupational exposure to wood preservative chemicals,
Cincinatti, Ohio, US Department of Health, Education, and
Welfare, National Institute for Occupational Safety and Health.
TOLA, S., HANBERG, S., COLLAN, Y., LINDERBORG, H., & KORKALA,
M.L. (1980) A case-control study of the etiology of nasal
cancer in Finland. Int. Arch. occup. environ. Health, 46:
79-85.
TOLEDO, J.V., MONTEIRO DA SILVA, C.S., BULHOES, M.S., PAES
LEME, L.A., DA SILVA NETTO, J.A., & GILBERT, B. (1976) Snail
control in urban sites in Brazil with slow-release hexabutyl-
distannoxane and pentachlorophenol. Bull. World Health Organ.,
54: 421-425.
TREVORS, J.T. (1982a) Effect of temperature on the
degradation of pentachlorophenol by Pseudomonas spp.
Chemosphere, 11: 471-475.
TREVORS, J.T. (1982b) Differences in the sensitivity of
short-term bioassays. Bull. environ. Contam. Toxicol., 28:
655-659.
TREVORS, J.T. (1982c) Effect of pentachlorophenol on
electron transport system activity in soil. Bull. environ.
Contam. Toxicol., 29: 727-730.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1981a) A
rapid toxicity test using Pseudomonas fluorescens. Bull.
environ. Contam. Toxicol., 26: 433-439.
TREVORS, J.T., MAYFIELD, C.I., INNISS, W.E., & THOMPSON, J.E.
(1981b) Effect of phenolic antioxidants on the toxicity of
pentachlorophenol in short-term bacterial bioassays. Bull.
environ. Contam. Toxicol., 27: 433-439.
TREVORS, J.T., MAYFIELD, C.I., & INNISS, W.E. (1982) Effect
of sequence of exposure to chlorophenols in short-term
bacterial bioassays. Arch. environ. Contam. Toxicol., 11:
203-207.
TRIEBIG, G., KREKELER, H., GOSSLER, K., & VALENTIN, H.
(1981) [Investigations into neurotoxicity of work-related
materials. II. Motor and sensory nerve conduction velocity in
pentachlorophenol-exposed persons.] Int. Arch. occup. environ.
Health, 48: 357-367 (in German).
TRUHAUT, R., VITTE, G., & BOUSSEMART, E. (1952a) Recherches
sur la toxicologie du pentachlorphénol. I. Propriétés.
Caractérisation et dosage dans les milieux biologiques. Arch.
Mal. prof. Méd. Trav. Sécur. soc., 13: 561-567.
TRUHAUT, R., L'EPEE, P., & BOUSSEMART, E. (1952b) Recherches
sur la toxicologie du pentachlorophénol. II. Intoxications
professionelles dans l'industrie du bois. Observations de deux
cas mortels. Arch. Mal. prof. Méd. Trav. Sécur. soc., 13:
567-569.
TRUJILLO, D.A., RAY, L.E., MURRAY, H.E., & GIAM, C.S. (1982)
Bioaccumulation of pentachlorophenol by killifish (Fundulus
similis). Chemosphere, 11: 25-32.
TURNER, H.J., Jr, REYNOLDS, D.M., & REDFIELD, A.C. (1948)
Chlorine and sodium pentachlorophenate as fouling preventives
in sea water conduits. Ind. eng. Chem., 40: 450-453.
UHL, S., SCHMID, P., & SCHLATTER, C. (1986) Pharmacokinetics
of pentachlorophenol in man. Arch. Toxicol., 58: 182-186.
UMWELTBUNDESAMT (1985) [State of the art: dioxins,] Berlin,
Erich Schmidt Verlag, 353 pp (in German).
US EPA (1973) EPA compendium of registered pesticides,
Washington DC, US Environmental Protection Agency, Vol. II.
pp. S-59-001 - S-59-003.
US EPA (1978) Ambient water quality criteria. Pentachloro-
phenol, Washington DC, US Environmental Protection Agency
(PB-292439).
US EPA (1980) Ambient water quality criteria for
pentachlorophenol, Washington DC, Office of Water Regulations
and Standards, US Environmental Protection Agency, 98 pp
(EPA-440/5-80-065).
US EPA (1984a) Creosote, pentachlorophenol, and inorganic
arsenicals; notice of intent to cancel: notice of determina-
tion of availability of position document. Fed. Reg., 49(136):
28666-28689.
US EPA (1984b) Pentachlorophenol: preliminary notice of
determination concluding the rebuttable presumption against
registration of pesticide products containing pentachloro-
phenol for non-wood preservative uses; proposed notice of
intent to cancel such registrations; notice of availability of
position document 2/3. Fed. Reg., 49(240): 48367-48372.
US EPA (1985) Hazardous waste management system: dioxin-
containing wastes. Fed. Reg., 50: 1978-2006.
US NATIONAL ACADEMY OF SCIENCES (1977) Drinking-water and
health, Washington DC, Safe Drinking Water Committee, National
Academy of Sciences, pp. 583.
VALLEJO-FRIERE, A., RIBEIRO, O.F., & RIBEIRO, I.F. (1954)
Quaternary ammonium compounds as molluscicides. Science,
119(3093): 470-472.
VALO, R., KITUNEN, V., SALKINOJA-SALONEN, M., & RAISANEN, S.
(1984) Chlorinated phenols as contaminants of soil and water
in the vicinity of 2 Finnish sawmills. Chemosphere, 13:
835-844.
VAN OMMEN, B., VAN BLADEREN, P.J., TEMMINK, J.H.M., & MUELLER,
F. (1985) Formation of pentachlorophenol as the major
product of microsomal oxidation of hexachlorobenzene. Biochem.
biophys. Res. Commun., 126: 25-32.
VAN RENSBURG, J.F.J. (1981) Health aspects of organic
substances in South African waters - opinions and realities.
Water S.Afr., 7: 139-149.
VERMA, S.R., RANI, S., & DALELA, R.C. (1981a) Responses of
serum transaminases in Notopterus notopterus chronically
exposed to phenolic compounds and their combinations. Environ.
Res., 24: 218-223.
VERMA, S.R., TONK, I.P., & DALELA, R.C. (1981b) Determina-
tion of the maximum acceptable toxicant concentration (MATC)
and the safe concentration for certain aquatic pollutants.
Acta hydrochim. hydrobiol., 9: 247-254.
VERMA, S.R., RANI, S., & DALELA, R.C. (1981c) Effects of
phenolic compounds on in vivo blood parameters of a fish
Notopterus notopterus. J. environ. Sci. Health B, 16: 273-282.
VERMA, S.R., RANI, S., & DALELA, R.C. (1982) Effects of
sodium pentachlorophenate on enzymes of energy metabolism in
tissues of Notopterus notopterus. Toxicol Lett., 10: 297-302.
VERMEER, K., RISEBROUGH, R.W., SPAANS, A.L., & REYNOLDS, L.M.
(1974) Pesticide effects on fish and birds in rice fields of
Surinam, South America. Environ. Pollut., 7: 217-236.
VINOGRADOVA, V.K., KALYAGANOV, P.I., SUDONINA, L.T., &
ELIZAROV, G.P. (1973) [Industrial hygiene and the health
condition of workers engaged in the production of sodium
pentachlorophenolate.] Gig. Tr. Prof. Zabol., 8: 11-13 (in
Russian).
VOGEL, E. & CHANDLER, J.L.R. (1974) Mutagenicity testing of
cyclamate and some pesticides in Drosphila melanogaster.
Experientia (Basel), 30(6): 621-623.
WAINSTOCK DE CALMANOVICI, R. & SAN MARTIN DE VIALE, L.C.
(1980) Effect of chlorophenols on porphyrin metabolism in
rats and chick embryo. Int. J. Biochem., 12: 1039-1044.
WALUM, E. & PETERSON, A. (1984) On the application of
culture neuroblastoma cells in chemical toxicity screening. J.
Toxicol environ. Health, 13: 511-520.
WANG, M. (1965) [Stability and molluscciidal activity of
Na-PCP in the environment.] Chin. J. Hyg., 10: 243-245 (in
Chinese).
WARREN, J.S., LAMPARSKI, L.L., JOHNSON, R.L., & GOOCH, R.M.
(1982) Determination of pentachlorophenol volatilized from
wood via collection on silica gel. Bull. environ. Contam.
Toxicol., 29: 719-726.
WATANABE, I. (1973) Isolation of pentachlorophenol
decomposing bacteria from soil. Soil Sci. plant Nutr., 19:
109-116.
WATANABE, I. (1978) Pentachlorophenol (PCP) decomposing
activity of field soils treated annually with PCP. Soil Biol.
Biochem., 10: 71-75.
WATANABE, S. & WATANABE, S. (1970) [Contact dermatitis due
to herbicide (PCP and MCPB): a fatal case.] Rinsho Hifugaku,
24(10): 945-949 (in Japanese).
WEBB, P.W. & BRETT, J.R. (1973) Effects of sublethal
concentrations of sodium pentachlorophenate on growth rate,
food conversion efficiency, and swimming performance in
underyearling sockeye salmon (Oncorhynchus nerka). J. Fish.
Res. Board Can., 30: 499-507.
WEBER, K. & ERNST, W. (1978) Levels and patterns of
chlorophenols in water of the Weser estuary and the German
bight. Chemosphere, 11: 873.
WEGMAN, R.C.C. & HOFSTEE, A.W.M. (1979) Chlorophenols in
surface waters of the Netherlands. Water Res., 13: 651-657.
WEGMAN, R.C.C. & VAN DEN BROEK, H.H. (1983) Chlorophenol in
river sediment in the Netherlands. Water Res., 17: 227-230.
WEINBACH, E.C. (1957) Biochemical basis for the toxicity of
pentachlorophenol. Proc. Natl Acad. Sci. (USA), 43: 393-397.
WEINBACH, E.C. & GARBUS, J. (1965) The interaction of
uncoupling phenols with mitochondria and with mitochondrial
protein. J. biol. Chem., 240: 1811-1819.
WEISS, U.M., KORTE, F., HAQUE, A.U., MOZA, P., & SCHEUNERT,
I. (1982a) Fate of pentachlorophenol-14C in rice plants
under controlled conditions. J. agric. food Chem., 30:
1186-1190.
WEISS, U.M., SCHEUNERT, I., KLEIN, W., & KORTE, F. (1982b)
Fate of pentachlorophenol-14C in soil under controlled
conditions. J. agric. food Chem., 30: 1191-1194.
WHITLEY, L.S. (1968) The resistance of tubificid worms to
three common pollutants. Hydrobiologia, 32: 193-205.
WHO (1976) EHC 2: Polychlorinated biphenyls and terphenyls,
Geneva, World Health Organization, 85 pp.
WHO (1984) The WHO recommended classification of pesticides
by hazard. Guidelines to classification 1984-1985, Geneva,
World Health Organization (Unpublished report VBC/84.2).
WHO (1985) Guidelines for drinking-water quality: health
criteria and other supporting information, Geneva, World
Health Organization, Vol. 2, pp. 237-239.
WIKLUND, K. & HOLM, L.E. (1986) Soft-tissue sarcoma risk in
Swedish agricultural and forestry workers. J. Natl Cancer
Inst., 76: 229-234.
WILLIAMS, P.L. (1982) Pentachlorophenol: an assessment of
the occupational hazard. Am. Ind. Hyg. Assoc. J., 43: 799-810.
WILSON, R.D., ZIPRIN, R.L., CLARK, D.E., & ELISSALDE, M.H.
(1982) Absorption of pentachlorophenol by the ovine lymphatic
system: a technical note. Vet. hum. Toxicol., 24: 12-14.
WINDHOLZ, M., ed. (1976) The Merck index, 9th ed., Rahway,
New Jersey, Marck & Co., p. 921.
WITTE, I., JUHL, U., & BUTTE, W. (1985) DNA-damaging
properties and cytotoxicity in human fibroblasts of tetra-
chlorohydroquinone, a pentachlorophenol metabolite. Mutat.
Res., 145: 71-75.
WOELKE, C.E. (1972) Development of a receiving water quality
bioassay criterion based on the 48-h Pacific oyster (Crasso-
strea gigas) embryo. Wash. Dep. Fish. Techn. Rep., 9: 93 pp.
WOIWODE, W., WODARZ, R., DRYSCH, K., & WEICHARDT, H. (1980)
Determination of free pentachlorophenol in air and in blood by
efficient chromatographic procedures. Int. Arch. occup.
environ. Health, 45: 153-162.
WONG, A.S. & CROSBY, D.G. (1978) Photolysis of pentachloro-
phenol in water. In: Rao, K.R., ed. Pentachlorophenol:
chemistry, pharmacology, and environmental toxicology, New
York, London, Plenum Press, pp. 19-25.
WONG, A.S. & CROSBY, D.G. (1981) Photodecomposition of
pentachlorophenol in water. J. agric. food Chem., 29: 125-130.
WOOD, K.M. (1980) Prevalence of unexplained anaemias
associated with occupational exposures to pentachlorophenol
used as a wood preservative, University of Washington,
Department of Environment and Health, 74 pp (Thesis).
WOOD, S., ROM, W.N., WHITE, G.L., & LOGAN, D.C. (1983)
Pentachlorophenol poisoning J. occup. Med., 25(7): 527-530.
WSSA (1974) Herbicide handbook of the Weed Science Society
of America, Champaign, Illinois, Weed Science Society of
America.
WYLLIE, J.A., GABICA, J., BENSON, W.W., & YODER, J. (1975)
Exposure and contamination of the air and employees of a
pentachlorophenol plant, Idaho - 1972. Pestic. Monit. J., 9:
150-153.
YOUNG, H.C. & CARROL, J.C. (1951) The decomposition of
pentachlorophenol when applied as a residual pre-emergence
herbicide. Agron. J., 43: 504-507.
YOUNT, J.D. & RICHTER, J.E. (1986) Effects of pentachloro-
phenol on periphyton communities in outdoor experimental
streams. Arch. environ. Contam. Toxicol., 15: 51-60.
YUNKER, M.B. (1981) A pelagic marine ecosystem study of the
behaviour, pathways, residence time and toxicity of penta-
chlorophenol, Sidney, Institute of Ocean Sciences (DSS file
No. 07sb.fp833-9-09043, contract rpt. Dobrocky Seatch. Ltd).
ZENZEN, C. (1979) [Pentachlorophenol. Studies on the
distribution kinetics after single and repeated dosing,]
University of Düsseldorf, 149 pp (Thesis) (in German).
ZIGLER, G.M. & PHILLIPS, W.F. (1967) Thin-layer chromato-
graphic method for estimation of chlorophenols. Environ. Sci.
Technol., 1: 65.
ZIMMERLI, B. (1982) [Modelling the transfer of pollutants
from painted surfaces in the surrounding air.] In: Aurand, K.,
Seifert, B., & Wegner, J., ed. [Air quality in closed rooms,]
Stuttgart, New York, Gustav Fischer Verlag, pp. 235-267 (in
German).
ZIMMERLI, B. & ZIMMERMANN, H. (1979) [Simple procedure to
estimate the concentrations of toxic chemicals in the indoor
atmosphere.] Mitt. Geb. Lebensmittelunters Hyg., 70: 429-422
(in German).
ZIMMERLI, B., MARSCHALL, T., & MAREK, B. (1979) [Preliminary
studies on occurrence of pentachlorophenol in human urine.]
Mitt. Geb. Lebensmittelunters. Hyg., 70: 443-450 (in German).
ZIMMERLI, B., MARSCHALL, T., & MAREK, B. (1980) [On the
excretion of pentachlorophenol in cow milk.] Mitt. Geb.
Lebensmittelunters. Hyg., 71: 404-414 (in German).
ZISCHKE, J.A., ARTHUR, J.W., HERMANUTZ, R.O., HEDTKE, S.F., &
HELGAN, J.C. (1985) Effects of pentachlorophenol on
invertebrates and fish in outdoor experimental channels.
Aquat. Toxicol., 7: 37-58.
ZITKO, V., HUTZINGER, O., & CHOI, P.M.K. (1974) Determina-
tion of pentachlorophenol and chlorobiphenyls in biological
samples. Bull. environ. Contam. Toxicol., 12: 649-653.
ZOBER, A., GOSSLER, K., MANKE, G., & SCHALLER, K.H. (1979)
[Studies on pentachlorophenol loading in the wood industry.]
Ber. Jahrestag. Dtsch. Ges. Arbeitsmed., 19th, 1979: 447-454
(in German).
ZOBER, A., SCHALLER, K.H., GOSSLER, K., & KREKELER, H.J.
(1981) [Pentachlorophenol and liver function: a pilot study
of occupationally exposed groups.] Int. Arch. occup. environ.
Health, 48: 347-356 (in German).