
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 25
SELECTED RADIONUCLIDES
TRITIUM
CARBON-14
KRYPTON-85
STRONTIUM-90
IODINE
CAESIUM-137
RADON
PLUTONIUM
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1983
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
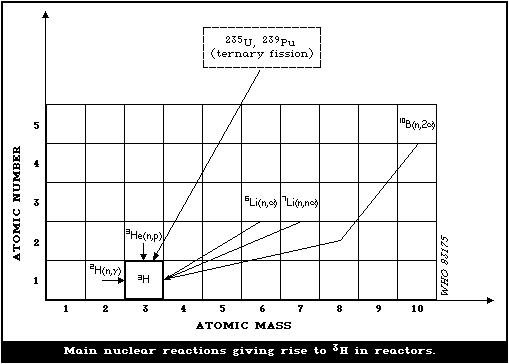
chemicals.
ISBN 92 4 154085 0
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1983
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
Paragraphs
ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED RADIONUCLIDES
PREFACE . . . . . . . . . . . . . . . . . . . . . . 1 - 6
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . 7 - 22
II. TRITIUM . . . . . . . . . . . . . . . . . . . . . . 23 - 77
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 23 - 25
B. SOURCES . . . . . . . . . . . . . . . . . . . . 26 - 57
1. Natural tritium . . . . . . . . . . . . . . 26 - 29
2. Nuclear explosions . . . . . . . . . . . . . 30 - 33
3. Nuclear fuel cycle . . . . . . . . . . . . . 34 - 51
4. Tritium production plants . . . . . . . . . 52 - 54
5. Consumer products . . . . . . . . . . . . . 55 - 56
6. Controlled thermonuclear reactors . . . . . 57
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 58 - 61
1. Natural and fallout tritium . . . . . . . . 58 - 59
2. Industrial releases . . . . . . . . . . . . 60 - 61
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 62 - 63
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 64 - 77
1. Dose per unit intake . . . . . . . . . . . . 64 - 66
2. Dose per unit release . . . . . . . . . . . 67 - 77
F. REFERENCES
III. CARBON-14 . . . . . . . . . . . . . . . . . . . . . 78 - 112
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 78 - 80
B. SOURCES . . . . . . . . . . . . . . . . . . . . 81 - 98
1. Natural carbon-14 . . . . . . . . . . . . . 81
2. Nuclear explosions . . . . . . . . . . . . . 82 - 84
3. Nuclear fuel cycle . . . . . . . . . . . . . 85 - 98
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 99 - 102
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 103 - 105
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 106 - 112
1. Dose per unit intake . . . . . . . . . . . . 106 - 107
2. Dose per unit release . . . . . . . . . . . 108 - 112
F. REFERENCES
IV. KRYPTON-85 . . . . . . . . . . . . . . . . . . . . . 113 - 150
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 113 - 117
B. SOURCES . . . . . . . . . . . . . . . . . . . . 118 - 128
1. Natural krypton-85 . . . . . . . . . . . . . 121
2. Nuclear explosions . . . . . . . . . . . . . 122 - 123
3. Nuclear fuel cycle . . . . . . . . . . . . . 124 - 128
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 129 - 137
1. Dispersion in the atmosphere . . . . . . . . 130 - 133
2. Removal from the atmosphere . . . . . . . . 134 - 137
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 138 - 141
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 142 - 150
1. Dose per unit exposure . . . . . . . . . . . 143 - 144
2. Dose per unit release . . . . . . . . . . . 145 - 150
F. REFERENCES
V. STRONTIUM-90 . . . . . . . . . . . . . . . . . . . . 151 - 211
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 151 - 154
B. SOURCES . . . . . . . . . . . . . . . . . . . . 155 - 165
1. Nuclear explosions . . . . . . . . . . . . . 155 - 156
2. Nuclear fuel cycle . . . . . . . . . . . . . 157 - 165
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 166 - 185
1. Movement in soil . . . . . . . . . . . . . . 166
2. Transfer to plants . . . . . . . . . . . . . 167 - 171
3. Transfer to milk . . . . . . . . . . . . . . 172
4. Transfer to diet . . . . . . . . . . . . . . 173 - 181
5. Aquatic behaviour . . . . . . . . . . . . . 182 - 185
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 189 - 192
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 193 - 211
1. Dose per unit intake . . . . . . . . . . . . 193 - 197
2. Dose per unit release . . . . . . . . . . . 198 - 211
F. REFERENCES
VI. IODINE . . . . . . . . . . . . . . . . . . . . . . . 212 - 269
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 212 - 214
B. SOURCES . . . . . . . . . . . . . . . . . . . . 215 - 234
1. Natural production . . . . . . . . . . . . . 215 - 216
2. Nuclear explosions . . . . . . . . . . . . . 217 - 220
3. Nuclear fuel cycle . . . . . . . . . . . . . 221 - 234
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 235 - 255
1. Nuclear explosions . . . . . . . . . . . . . 235 - 241
2. Industrial releases . . . . . . . . . . . . 242 - 255
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 256 - 259
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 260 - 270
1. Dose per unit intake . . . . . . . . . . . . 260 - 261
2. Dose per unit release . . . . . . . . . . . 262 - 270
F. REFERENCES
VII. CAESIUM-137 . . . . . . . . . . . . . . . . . . . . 271 - 336
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 271 - 274
B. SOURCES . . . . . . . . . . . . . . . . . . . . 275 - 282
1. Nuclear explosions . . . . . . . . . . . . . 275 - 276
2. Nuclear fuel cycle . . . . . . . . . . . . . 277 - 282
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 283 - 309
1. Fixation in soil . . . . . . . . . . . . . . 283 - 286
2. Transfer to plants . . . . . . . . . . . . . 287 - 290
3. Transfer to milk . . . . . . . . . . . . . . 291
4. Transfer to meat . . . . . . . . . . . . . . 292
5. Transfer to diet . . . . . . . . . . . . . . 293 - 301
6. The lichen-caribou-man foodchain . . . . . . 302 - 303
7. Aquatic behaviour . . . . . . . . . . . . . 304 - 309
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 310 - 319
1. Absorption and distribution in tissues . . . 310 - 314
2. Retention half-time . . . . . . . . . . . . 315 - 317
3. Transfer factor . . . . . . . . . . . . . . 318 - 319
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 320 - 336
1. Dose per unit intake . . . . . . . . . . . . 320 - 324
2. Dose per unit release . . . . . . . . . . . 325 - 336
F. REFERENCES
VIII. RADON . . . . . . . . . . . . . . . . . . . . . . . 337 - 395
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 337 - 340
B. SOURCES . . . . . . . . . . . . . . . . . . . . 341 - 351
1. Outdoors . . . . . . . . . . . . . . . . . . 341 - 344
2. Indoors . . . . . . . . . . . . . . . . . . 345 - 351
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 352 - 375
1. Release from soil . . . . . . . . . . . . . 352 - 355
2. Dispersion in air . . . . . . . . . . . . . 356 - 361
3. Indoor behaviour . . . . . . . . . . . . . . 362 - 365
4. Radon daughter concentrations . . . . . . . 366 - 375
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 376 - 380
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 381 - 395
1. Dose per unit exposure . . . . . . . . . . . 381 - 393
2. Dose per unit release . . . . . . . . . . . 394 - 395
F. REFERENCES
IX. PLUTONIUM . . . . . . . . . . . . . . . . . . . . . 396 - 456
A. INTRODUCTION . . . . . . . . . . . . . . . . . . 396 - 401
B. SOURCES . . . . . . . . . . . . . . . . . . . . 402 - 411
1. Nuclear explosions . . . . . . . . . . . . . 402 - 404
2. Nuclear fuel cycle . . . . . . . . . . . . . 405 - 406
3. Other sources . . . . . . . . . . . . . . . 407 - 411
C. BEHAVIOUR IN THE ENVIRONMENT . . . . . . . . . . 412 - 434
1. Movement in soil . . . . . . . . . . . . . . 412 - 416
2. Transfer to plants . . . . . . . . . . . . . 417 - 418
3. Transfer to animals . . . . . . . . . . . . 419 - 420
4. Transfer to diet . . . . . . . . . . . . . . 421 - 425
5. Aquatic behaviour . . . . . . . . . . . . . 426 - 434
D. TRANSFER TO MAN . . . . . . . . . . . . . . . . 435 - 443
E. DOSIMETRY . . . . . . . . . . . . . . . . . . . 444 - 456
1. Dose per unit intake . . . . . . . . . . . . 444 - 448
2. Dose per unit release . . . . . . . . . . . 449 - 456
F. REFERENCES
X. RADIATION EFFECTS . . . . . . . . . . . . . . . . . 457 - 476
A. SOMATIC EFFECTS . . . . . . . . . . . . . . . . 459 - 463
1. Early somatic effects . . . . . . . . . . . 459 - 461
2. Late somatic effects . . . . . . . . . . . . 462 - 463
B. GENETIC EFFECTS . . . . . . . . . . . . . . . . 464 - 465
C. DOSE-RESPONSE RELATIONSHIPS . . . . . . . . . . 466 - 469
D. RISK ESTIMATES . . . . . . . . . . . . . . . . . 470 - 476
XI. CONCLUSIONS . . . . . . . . . . . . . . . . . . . . 477 - 491
A. RADIONUCLIDES AND THE ENVIRONMENT . . . . . . . 477 - 481
B. DOSE ASSESSMENTS . . . . . . . . . . . . . . . . 482 - 487
C. EFFECTS EVALUATION . . . . . . . . . . . . . . . 488 - 491
XII. ANNEX
EXCERPTS FROM "BASIC SAFETY STANDARDS FOR RADIATION
PROTECTION 1982 EDITION"
NOTE TO READERS OF THE CRITERIA DOCUMENTS
While every effort has been made to present information in the
criteria documents as accurately as possible without unduly
delaying their publication, mistakes might have occurred and are
likely to occur in the future. In the interest of all users of the
environmental health criteria documents, readers are kindly
requested to communicate any errors found to the Division of
Environmental Health, World Health Organization, Geneva,
Switzerland, in order that they may be included in corrigenda which
will appear in subsequent volumes.
In addition, experts in any particular field dealt with in the
criteria documents are kindly requested to make available to the
WHO Secretariat any important published information that may have
inadvertently been omitted and which may change the evaluation of
health risks from exposure to the environmental agent under
examination, so that the information may be considered in the event
of updating and re-evaluation of the conclusions contained in the
criteria documents.
ENVIRONMENTAL HEALTH CRITERIA FOR SELECTED RADIONUCLIDES
At the request of the United Nations Environment Programme
(UNEP), the United Nations Scientific Committee on the Effects of
Atomic Radiation (UNSCEAR) prepared a paper on the Environmental
Behaviour and Dosimetry of Radionuclides. In accordance with the
UNEP proposal, the paper, which was prepared during the 27th - 29th
sessions of the Committee and was completed and approved at the
30th session in 1981, is now being published in the WHO/UNEP
Environmental Health Criteria series. The EHC document, which is
entitled "Selected Radionuclides", comprises the integral report
prepared and edited by UNSCEAR, together with an annex consisting
of excerpts taken from "Basic Safety Standards for Radiation
Protection 1982 Edition", Safety Series No 9, a document prepared
jointly by IAEA/ILO/NEA(OECD)/WHO, and published by the
International Atomic Energy Agency, to give guidance to the
appropriate national authorities on the establishment of limits for
radionuclides. The selected radionuclides discussed in the
Environmental Health Criteria document are those of environmental
importance for the general population and radiation workers.
Dr E. Komarov, Environmental Health Division, World Health
Organization, was responsible for the final layout of the
Environmental Health Criteria document.
The assistance of Dr B.G. Bennett (Monitoring and Assessment
Research Centre, MARC) in the scientific editing of the
Environmental Health Criteria document is gratefully acknowledged.
The contents of the 1982 UNSCEAR report to the General Assembly
of the United Nations were taken into account during the
preparation of the paper on the Environmental Behaviour and
Dosimetry of Radionuclides, but the report was not quoted as it had
not been issued at that time.
ENVIRONMENTAL BEHAVIOUR AND DOSIMETRY OF RADIONUCLIDES
1. PREFACE
1. The release of radioactive materials to the environment
potentially exposes populations to ionizing radiation and increases
the risk of incurring deleterious health effects. The associations
of released amounts to effects establish the health criteria for
radionuclides, i.e., the quantitative relationships that would be
required to establish release limits governing the management of
radioactive materials used by man.
2. This report has been prepared by the United Nations Scientific
Committee on the Effects of Atomic Radiation (UNSCEAR) for the
United Nations Environment Programme (UNEP) to provide background
information in establishing such health criteria. In this report
the more general considerations of environmental behaviour of
several radionuclides are discussed, including sources, transport
to man and dosimetry. The radionuclides discussed are those most
frequently released from natural and man-made sources and the
greatest contributors to population radiation exposure under normal
circumstances.
3. The compilation of the relevant information is based largely on
the detailed presentations and evaluations of the sources of
ionizing radiation by UNSCEAR in its reports to the United Nations
General Assembly. The reader is referred to these reports for
general concepts and for assessments of the dose commitments to man
from exposures to sources such as natural radioactivity, fallout
from atmospheric nuclear testing, releases from nuclear power
production, occupational and medical irradiations.
4. Further information to be considered in establishing health
criteria for radionuclides is that on health effects of
irradiations. The relationships between radiation dose and risks
of health effects in man have recently been re-evaluated based on
the available data. This information can be found in the 1977
report of UNSCEAR. Only a brief summary of the general aspects of
radiation effects and of radiation protection considerations is
presented here.
5. The establishment of release limits for radionuclides in
particular situations cannot be accomplished without rather more
detailed considerations of the local and regional environment and
the special pathways of transfer to man. With this in mind, it is
recognized that the material given here can only serve as
background guidance.
6. The following scientists have contributed in the preparation of
this report: Dr. W.J. Bair, Dr. D. Beninson, Dr. B.G. Bennett, Dr.
A. Bouville, Dr. P. Patek, Dr. G. Silini and Dr. J.O. Snihs.
I. INTRODUCTION
7. Radionuclides are a special class of environmental substances.
They are the unstable configurations of chemical elements which
undergo radioactive decay, emitting radiation in the form of alpha
or beta particles and x or gamma rays. The interaction of radiation
with biological materials causes energy to be released to these
materials which may result in a variety of harmful effects.
Radiation is thus a potential hazard to man, although it may also
be used in many beneficial ways, as in medical diagnosis and
treatment, in industrial and consumer products and in the
generation of electricity with nuclear reactors.
8. The realization of the harmful potential of ionizing radiation,
which was dramatically brought to the attention of the public by
the atomic bombing of Hiroshima and Nagasaki in 1945, was the cause
of considerable attention that has been paid throughout the years
to the effects of radiation. As a result of these studies, a great
deal is now known about radionuclide behaviour in the environment
and in man and about the somatic and genetic consequences of
irradiation. This information surpasses by far that relating to
any other class of environmental pollutants.
9. Considerable experience has been gained in environmental
radiation measurements, particularly in tracing the movement of
fallout radionuclides produced in atmospheric testing of nuclear
weapons. Much of this information has in turn contributed to the
general knowledge of atmospheric and oceanic transport processes
and of bio-geochemical cycles of elements. Extensive studies of
radiation effects in animals and numerous epidemiological surveys
of exposed population groups have by now been conducted. They have
considerably enlarged our understanding of the biological effects
of radiation on man and the environment, although uncertainties
still remain, particularly regarding the basic mechanisms of action
and the risk evaluations at low doses and dose rates [U1-U7].
10. A few definitions and general concepts should be introduced
before the detailed presentation of radionuclide assessments. The
basic unit of radioactivity is the becquerel (Bq), corresponding to
one disintegration per second. The previously used unit was the
curie (Ci), one Ci corresponding to 3.7 1010 Bq.
11. The basic measure of radiation interaction in irradiated
materials is the absorbed dose (D). This quantity is also the
basis of health risk estimates, under the assumption of a linear
relationship between dose and risk. The absorbed dose is defined
as the mean energy (joules) imparted to the irradiated material per
unit mass (kg) at the point of interest. The unit of absorbed dose
is ca11ed the gray (Gy) which corresponds to 1 J/kg. The unit of
absorbed dose previously in use, the rad, is one hundred times
smaller than the Gy.
12. Radiations of different types and energies have different
effectiveness for producing effects, depending on the amount of
energy transferred per unit length (LET) along the path of the
charged particles. In order to quantify this differing
effectiveness, use is made of a normalizing quantity called the
quality factor (Q). For general purposes of radiological
protection the assumed values of Q are: 1 for x and gamma rays and
for electrons; 10 for neutrons and protons; 20 for alpha and
multiply charged particles.
13. The product of the absorbed dose, D, and the quality factor,
Q, is termed the dose equivalent (H). The unit of dose equivalent
is the sievert (Sv). The previously used unit was the rem (1 rem =
0.01 Sv). Use of the dose equivalent allows the summation of doses
from all types of radiation of different biological effectiveness.
14. The exposure of an individual to a source of radiation may be
expressed in terms of the absorbed dose or dose equivalent during
the period of exposure. In the natural radiation environment the
exposure is continuous and it is sufficient to give the annual
average dose or dose rate. There are important spatial variations
to be considered, for example, as a function of the altitude in
case of exposure to cosmic radiation or as a function of the
geographical location due to the different radionuclides present in
soil.
15. For specific releases of radioactive materials into the
environment (atmospheric nuclear tests, operation of nuclear
reactors) there are also important temporal variations in the
exposure. In order to account for the exposures which will occur
in the future from specific sources, use is made of the dose
commitment (Dc). This quantity is the infinite time integral of
the average individual dose rate. Dose commitments may not
represent doses to specific individuals. For example, if the
radionuclide released has a very long half-life, the dose
commitment is derived from the doses to successive generations in
the population.
16. The collective harm to a population resulting from the
exposure of all individuals is related to the collective dose in
the population, particularly if the linearity of the relationships
between dose and effects may be assumed for the exposures involved.
The collective dose (S) in a given population is the summation of
products of the average individual doses and the number of
individuals in each range of doses. The summation may become an
integral for continuous variations over the entire range of doses.
The unit of the collective dose is man Gy and the corresponding
unit of collective dose equivalent is man Sv.
17. The measure of the total exposure of a population from a
specified source or release practice is the collective dose
commitment (Sc), defined as the infinite time integral of the
collective dose rate. The relevant units are man Gy, or man Sv in
case of the collective dose equivalent commitment.
18. In radiation exposure assessments, it is often necessary to
account for the different sensitivity of individual organs of the
body with respect to each other or to irradiation of the whole
body, particularly in the case of internally deposited
radionuclides. Weighting factors for the relevant organs may be
derived for this purpose from relative risk estimates. These
factors will be listed in the section on radiation effects with
some additional discussion.
19. The summation of the products of the weighting factors and the
dose equivalents for individual organs gives a single measure to be
used as an index of health detriment, called the effective dose
equivalent (HE). The concepts of collective and committed doses
may also be used with this quantity. Thus a final quantity for
health assessments may be the collective effective dose equivalent
commitment, (ScE) which is a collective dose, weighted for the
effects of doses within the body and dose distributions within the
population.
20. The chain of events leading from the release of radioactive
materials into the environment to the irradiation of human tissues
may be expressed schematically as a series of compartments
connected by transfer pathways. Such models are necessarily
simplifications of the actual transfer pathways. The following
diagram illustrates the transfer stages most usually considered in
assessments by UNSCEAR.
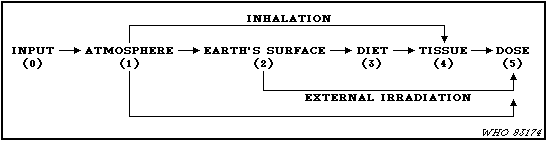 21. The basic task in the assessment process is to evaluate the
transfer factors (Pi,j) which relate the appropriate quantity of
radioactivity amount or dose in step i of the sequence to the
appropriate quantity in the subsequent step j. Since the desired
quantity in the final step is the time integrated dose rate, the
dose commitment from a specific source, the quantities in the other
steps are the time integrated activity concentrations. The
transfer factor is the quotient of time integrated quantities in
successive compartments. The total transfer factor for steps in
series is the product of the transfer factors involved. The total
transfer factor of several parallel pathways is the sum of the
transfer factors of the individual pathways.
22. There are many common features of the behaviour of different
radionuclides in the environment and their transfer to man. For
example, the physical dispersion of radionuclides in the
environment following release from a source is largely the same for
broad classes of material, such as particulates and gases. Several
models used to describe the transfer of radioactive material within
an environmental medium or from one medium to the next have general
applicability. A review of such general behaviour and modelling
procedures can be found in the 1982 report of UNSCEAR [U8].
Therefore, in the presentations which follow only the rather more
specific aspects of environmental behaviour and dosimetry of the
radionuclides are considered.
REFERENCES
U1 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Thirteenth Session,
Supplement No. 17 (A/3838). New York, 1958.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Seventeenth Session,
Supplement No. 16 (A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Nineteenth Session,
Supplement No. 14 (A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Twenty-first Session,
Supplement No. 14 (A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Twenty-fourth Session,
Supplement No. 13 (A/7613). New York, 1969.
U6 United Nations. Ionizing Radiation: Levels and Effects.
A report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication, No. E.72.IX.17
and 18. New York, 1972.
U7 United Nations. Sources and Effects of Ionizing
Radiation. United Nations Scientific Committee on the
Effects of Atomic Radiation 1977 report to the General
Assembly, with annexes. United Nations sales publication
No. E.77.IX.I. New York, 1977.
U8 United Nations. Ionizing Radiation: Sources and
Biological Effects. United Nations Scientific Committee
on the Effects of Atomic Radiation 1982 report to the
General Assembly, with annexes. United Nations sales
publication No. E.82.IX.8. New York, 1982.
II. TRITIUM
A. INTRODUCTION
23. Tritium, 3H, is a radioactive isotope of hydrogen which decays
into the stable nuclide 3He. Tritium is a pure beta-emitter with a
half-life of 12.3 a, a maximum energy of 18 keV and an average
energy of 5.7 keV. Tritium is produced naturally in the
atmosphere, where it results from the interaction of cosmic ray
protons and neutrons with nitrogen, oxygen, and argon. Man-made
tritium, in amounts substantially larger than the natural
inventory, has been injected into the stratosphere by thermonuclear
explosions. In addition, tritium is produced during the operation
of nuclear reactors.
24. There are many applications of tritium in industry. It is
widely used in consumer products, such as radioluminous timepieces
and also as a tracer in biomedical research. Environmental tritium
is mainly found as tritiated water. As such, it follows the
hydrological cycle and penetrates into all components of the
biosphere, including man.
25. This document is mainly based on the 1977 UNSCEAR report [U1],
but makes also extensive use of the contents of recent reviews or
symposia on tritium [I4, J1, M7, N1, N2].
B. SOURCES
1. Natural tritium
26. Natural tritium is produced by nuclear reactions in the
atmosphere and, to a much smaller extent, in the hydrosphere and in
the lithosphere. In addition, some tritium may be created in the
extra-terrestrial environment and enter the atmosphere along with
cosmic rays. Most of the natural tritium is found in the
environment as tritiated water, generally designated as HTO.
27. In the atmosphere, natural tritium is produced by the
interaction of high energy cosmic rays with atmospheric nitrogen
and oxygen. The estimates of the number of atoms of tritium
produced per unit time and per unit area of the earth's surface
range from 0.1 to 1.3 cm-2 s-1 [U1]. In the UNSCEAR 1977 report
[U1], a production rate of 0.25 cm-2 s-1 was adopted; this
corresponds to a production rate of 3.6 1016 Bq a-1 in each
hemisphere and to a global inventory of 1.3 1018 Bq at equilibrium.
28. It has been suggested that tritium might be ejected from the
sun during solar flares [L1] and from stars [F1]. Flamm et al.
[F2] estimated that the solar flares could account for an
additional production rate, averaged over the solar cycle, of 0.1
cm-2 s-1.
29. In the lithosphere and in the hydrosphere, tritium is produced
by interaction of neutrons with 6Li nuclides. The production rates
have been assessed at 10-3 cm-2 s-1 in the lithosphere and at 10-6
cm-2 s-1 in the hydrosphere [F1, K1].
2. Nuclear explosions
30. Nuclear tests have been conducted in the atmosphere since 1945
and have produced tritium in amounts that greatly exceed the global
natural activity. The tritium activity arising from atmospheric
nuclear tests can be estimated from the fission and fusion yields
or from environmental measurements.
31. Bennett [B1] has published an estimate of the total and
fission yields for each reported atmospheric test from 1945 to
1978; according to that compilation, 422 nuclear tests were
conducted in the atmosphere up to 1979, with cumulative yields of
217 Mt and 328 Mt for fission and fusion, respectively. The tritium
activity produced per unit yield depends on the characteristics of
the device, as well as on those of the explosion site, but is in
any case much greater for fusion than for fission [N1]. Miskel
[M1] estimated the yield for fission explosion to be 2.6 1013 Bq
Mt-1 and that for fusion to be typically 7.4 1017 Bq Mt-1. The
total tritium activity produced by atmospheric tests is thus
assessed at
328 Mt (fusion) x 7.4 1017 Bq Mt-1 = 2.4 1020 Bq
Most of this activity was produced during the large yield test
series which took place during 1954-1958 and 1961-1962; the
contribution of the nuclear tests carried out since 1964 is less
than 5% of the total.
32. Almost all the tritium produced by fallout occurs as HTO in
the atmosphere and the hydrosphere, and thus follows the
hydrological cycle. The total activity injected can therefore be
conceivably derived from measured concentrations in water samples.
From the study of Schell et al. [S1] on the tritium concentrations
in precipitation at stations in the IAEA network, it can be
estimated [U1] that the total production was about 1.7 1020 Bq.
Other estimates, using vertical profiles of 3H in the oceans as a
basis, lead to injections of 1.2 1020 Bq (in the oceans only) [O1],
1.3 1020 Bq [B2, U1], and 2.0 1020 Bq [M2].
33. All the estimates presented above are in fairly good
agreement, as they lie in the limited range from 1.2 1020 to 2.4
1020 Bq. In its 1977 report UNSCEAR adopted a value of 1.7 1020 Bq
for the total globally dispersed activity of tritium produced in
atmospheric tests up to 1976 [U1].
3. Nuclear fuel cycle
34. Tritium occurs in nuclear reactors by ternary fission in the
fuel and also by neutron activation reactions with lithium and
boron isotopes dissolved in, or in contact with, the primary
coolant as well as with naturally-occurring deuterium in the
primary coolant (Figure II.I).
21. The basic task in the assessment process is to evaluate the
transfer factors (Pi,j) which relate the appropriate quantity of
radioactivity amount or dose in step i of the sequence to the
appropriate quantity in the subsequent step j. Since the desired
quantity in the final step is the time integrated dose rate, the
dose commitment from a specific source, the quantities in the other
steps are the time integrated activity concentrations. The
transfer factor is the quotient of time integrated quantities in
successive compartments. The total transfer factor for steps in
series is the product of the transfer factors involved. The total
transfer factor of several parallel pathways is the sum of the
transfer factors of the individual pathways.
22. There are many common features of the behaviour of different
radionuclides in the environment and their transfer to man. For
example, the physical dispersion of radionuclides in the
environment following release from a source is largely the same for
broad classes of material, such as particulates and gases. Several
models used to describe the transfer of radioactive material within
an environmental medium or from one medium to the next have general
applicability. A review of such general behaviour and modelling
procedures can be found in the 1982 report of UNSCEAR [U8].
Therefore, in the presentations which follow only the rather more
specific aspects of environmental behaviour and dosimetry of the
radionuclides are considered.
REFERENCES
U1 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Thirteenth Session,
Supplement No. 17 (A/3838). New York, 1958.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Seventeenth Session,
Supplement No. 16 (A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Nineteenth Session,
Supplement No. 14 (A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Twenty-first Session,
Supplement No. 14 (A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Twenty-fourth Session,
Supplement No. 13 (A/7613). New York, 1969.
U6 United Nations. Ionizing Radiation: Levels and Effects.
A report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication, No. E.72.IX.17
and 18. New York, 1972.
U7 United Nations. Sources and Effects of Ionizing
Radiation. United Nations Scientific Committee on the
Effects of Atomic Radiation 1977 report to the General
Assembly, with annexes. United Nations sales publication
No. E.77.IX.I. New York, 1977.
U8 United Nations. Ionizing Radiation: Sources and
Biological Effects. United Nations Scientific Committee
on the Effects of Atomic Radiation 1982 report to the
General Assembly, with annexes. United Nations sales
publication No. E.82.IX.8. New York, 1982.
II. TRITIUM
A. INTRODUCTION
23. Tritium, 3H, is a radioactive isotope of hydrogen which decays
into the stable nuclide 3He. Tritium is a pure beta-emitter with a
half-life of 12.3 a, a maximum energy of 18 keV and an average
energy of 5.7 keV. Tritium is produced naturally in the
atmosphere, where it results from the interaction of cosmic ray
protons and neutrons with nitrogen, oxygen, and argon. Man-made
tritium, in amounts substantially larger than the natural
inventory, has been injected into the stratosphere by thermonuclear
explosions. In addition, tritium is produced during the operation
of nuclear reactors.
24. There are many applications of tritium in industry. It is
widely used in consumer products, such as radioluminous timepieces
and also as a tracer in biomedical research. Environmental tritium
is mainly found as tritiated water. As such, it follows the
hydrological cycle and penetrates into all components of the
biosphere, including man.
25. This document is mainly based on the 1977 UNSCEAR report [U1],
but makes also extensive use of the contents of recent reviews or
symposia on tritium [I4, J1, M7, N1, N2].
B. SOURCES
1. Natural tritium
26. Natural tritium is produced by nuclear reactions in the
atmosphere and, to a much smaller extent, in the hydrosphere and in
the lithosphere. In addition, some tritium may be created in the
extra-terrestrial environment and enter the atmosphere along with
cosmic rays. Most of the natural tritium is found in the
environment as tritiated water, generally designated as HTO.
27. In the atmosphere, natural tritium is produced by the
interaction of high energy cosmic rays with atmospheric nitrogen
and oxygen. The estimates of the number of atoms of tritium
produced per unit time and per unit area of the earth's surface
range from 0.1 to 1.3 cm-2 s-1 [U1]. In the UNSCEAR 1977 report
[U1], a production rate of 0.25 cm-2 s-1 was adopted; this
corresponds to a production rate of 3.6 1016 Bq a-1 in each
hemisphere and to a global inventory of 1.3 1018 Bq at equilibrium.
28. It has been suggested that tritium might be ejected from the
sun during solar flares [L1] and from stars [F1]. Flamm et al.
[F2] estimated that the solar flares could account for an
additional production rate, averaged over the solar cycle, of 0.1
cm-2 s-1.
29. In the lithosphere and in the hydrosphere, tritium is produced
by interaction of neutrons with 6Li nuclides. The production rates
have been assessed at 10-3 cm-2 s-1 in the lithosphere and at 10-6
cm-2 s-1 in the hydrosphere [F1, K1].
2. Nuclear explosions
30. Nuclear tests have been conducted in the atmosphere since 1945
and have produced tritium in amounts that greatly exceed the global
natural activity. The tritium activity arising from atmospheric
nuclear tests can be estimated from the fission and fusion yields
or from environmental measurements.
31. Bennett [B1] has published an estimate of the total and
fission yields for each reported atmospheric test from 1945 to
1978; according to that compilation, 422 nuclear tests were
conducted in the atmosphere up to 1979, with cumulative yields of
217 Mt and 328 Mt for fission and fusion, respectively. The tritium
activity produced per unit yield depends on the characteristics of
the device, as well as on those of the explosion site, but is in
any case much greater for fusion than for fission [N1]. Miskel
[M1] estimated the yield for fission explosion to be 2.6 1013 Bq
Mt-1 and that for fusion to be typically 7.4 1017 Bq Mt-1. The
total tritium activity produced by atmospheric tests is thus
assessed at
328 Mt (fusion) x 7.4 1017 Bq Mt-1 = 2.4 1020 Bq
Most of this activity was produced during the large yield test
series which took place during 1954-1958 and 1961-1962; the
contribution of the nuclear tests carried out since 1964 is less
than 5% of the total.
32. Almost all the tritium produced by fallout occurs as HTO in
the atmosphere and the hydrosphere, and thus follows the
hydrological cycle. The total activity injected can therefore be
conceivably derived from measured concentrations in water samples.
From the study of Schell et al. [S1] on the tritium concentrations
in precipitation at stations in the IAEA network, it can be
estimated [U1] that the total production was about 1.7 1020 Bq.
Other estimates, using vertical profiles of 3H in the oceans as a
basis, lead to injections of 1.2 1020 Bq (in the oceans only) [O1],
1.3 1020 Bq [B2, U1], and 2.0 1020 Bq [M2].
33. All the estimates presented above are in fairly good
agreement, as they lie in the limited range from 1.2 1020 to 2.4
1020 Bq. In its 1977 report UNSCEAR adopted a value of 1.7 1020 Bq
for the total globally dispersed activity of tritium produced in
atmospheric tests up to 1976 [U1].
3. Nuclear fuel cycle
34. Tritium occurs in nuclear reactors by ternary fission in the
fuel and also by neutron activation reactions with lithium and
boron isotopes dissolved in, or in contact with, the primary
coolant as well as with naturally-occurring deuterium in the
primary coolant (Figure II.I).
 35. Most of the fission product tritium produced in the fuel rods
is usually retained within the fuel and is not released into the
environment at the reactor site; it is instead released during fuel
reprocessing, if that practice is carried out. The activity
produced in the coolant is partly or entirely released in the
effluent streams according to the waste management practices at the
plant.
36. Releases into the environment are mainly in the form of HTO in
reactors that use water as primary coolant, as well as in fuel
reprocessing plants.
(a) Nuclear reactors
37. Four types of reactors have been considered (PWR, BWR, HWR,
GCR), the emphasis being on PWRs and BWRs which currently represent
the largest share of nuclear capacity. Estimated generation rates
and appearance of tritium in effluent streams of reactors are
summarized in Table II.1.
38. The annual production of fission product tritium in the fuel
rods of a pressurized water reactor (PWR) is in the range of 6 to 9
1011 Bq per MW(e)a [N1]. A small percentage, 1% or less, is
expected to be released into the coolant through defects in the
cladding, currently made of zirconium alloy. In contrast, the use
of stainless steel cladding in earlier PWRs resulted in the release
to the coolant of most of the tritium produced in the fuel.
39. Tritium generation in the primary coolant (water) of a
PWR is mainly due to reactions with boron (2.6 1010 Bq per
MW(e)a) which is dissolved as boric acid to control
reactivity; in addition, the maintenance of 2 ppm lithium
hydroxide for pH control [L2] results in the formation of
about 7 108 Bq per MW(e)a.
40. Environmental tritium discharges from PWRs depend on waste
management practices as well as on the materials used in the
reactor. Average normalized releases of tritium were shown in the
UNSCEAR 1977 report [U1] to be about 7 1010 Bq per MW(e)a in liquid
effluents and 7 109 Bq per MW(e)a in airborne effluents for the
reactors in operation in 1973-1974. However, large differences
between PWRs are due to the type of fuel cladding. For an old
reactor using stainless steel Kahn et al. [K3] measured 3H releases
of about 4 1011 Bq per MW(e)a in liquid effluents and 4 1010 Bq per
MW(e)a in airborne effluents, whereas the combined releases of 9
PWRs with zirconium alloy clad fuel (current practice) were
reported by NCRP [N1] to be about 3 1010 and 109 Bq per MW(e)a in
liquid and airborne effluents, respectively.
41. In boiling water reactors (BWRs) tritium is produced by
ternary fission in the fuel at about the same rate as in PWRs (6 to
9 1011 Bq per MW(e)a). The generalized use of zirconium alloy
cladding limits the tritium release into the coolant to less than 7
109 Bq per MW(e)a.
42. Tritium can be generated by neutron activation in the coolant
and in the control rods. Prior to 1971, control rods of boron
carbide were used in BWRs [S2]; the production of tritium by
activation of these control rods has been estimated to be about 3
1011 Bq per MW(e)a. However, tritium has not been shown to diffuse
through the boron carbide matrix [T1]. In the coolant itself,
tritium is generated by activation of deuterium at a rate of about
4 108 Bq per MW(e)a.
43. Tritium activities discharged from BWRs into the environment
are lower than those of PWRs because less tritium is produced in or
diffuses into the primary coolant. UNSCEAR [U1] reported the
average discharge rates to be 4 109 and 2 109 Bq MW(e)a in liquid
and airborne effluents, respectively.
44. The amount of tritium generated in fuel of heavy water
reactors (HWR) by ternary fission is approximately the same as in
light water reactors, but it is largely exceeded by the production
in the D2O coolant and moderator by neutron activation, which has
been estimated to be about 2 1013 Bq per MW(e)a [K2].
Table II.1 Estimated rates of generation of tritium and of its release in effluent streams of
different types of reactors (1010 Bq per MW(e)a) [G1, K2, S2, T1, U1]
---------------------------------------------------------------------------------------------------------
PWR BWR HWR GCR
Source ------------------------------------------------------------------------------------------
Generation Effluent Generation Effluent Generation Effluent Generation Effluent
stream stream stream stream
---------------------------------------------------------------------------------------------------------
Fission 75 < 0.7 75 < 0.7 55 < 0.6 75 < 0.7
Activation
Deuterium 0.004 0.004 0.04 0.04 2000 75a/
Lithium 0.07 0.07 2 0.4
Boron 2.6 2.6 30 0
Rounded total 80 3 110 0.5 2000 75 80 1
---------------------------------------------------------------------------------------------------------
a/ Depending on the irradiation time and on the net leakage of heavy water.
45. Environmental discharges depend upon the D2O leakage which is
kept as small as possible for economical and radiological reasons,
and upon the tritium activity in the coolant and moderator, which
builds up with the irradiation time. Annual losses of from 0.5% to
3% are anticipated in HWRs [U1]. For the optimal loss of 0.5% per
year, the normalized tritium release rate ranges from 1011 Bq per
MW(e)a in the first year of operation to about 7 1011 Bq per MW(e)a
in the tenth year. Based on the latter value as representative of
the reactor life, the normalized 3H release rates are estimated to
be 6 1011 and 1.5 1011 Bq per MW(e)a in airborne and liquid
effluents, respectively [G1]. Reported releases roughly agree with
these estimates: they are 6.3 1011 and 2.6 1011 Bq per MW(e)a for
the Pickering A station in Canada, in airborne and liquid
effluents, respectively whereas the Atucha reactor in Argentina
releases about 8 1011 Bq per MW(e)a both in airborne and in liquid
effluents.
46. In gas-cooled reactors (GCR), tritium is produced by ternary
fission (about 7 1011 Bq per MW(e)a) and by activation of lithium
in the graphite moderator. Based on the experience with UK
reactors (mainly Magnox reactors), the tritium release is about 7
109 Bq per MW(e)a in liquid effluents and ranges from 109 to 1010
Bq per MW(e)a in airborne effluents [U1].
(b) Fuel reprocessing plants
47. At the fuel reprocessing stage of the nuclear fuel cycle (if
it is undertaken) the elements uranium and plutonium in the
irradiated nuclear fuel are recovered for reuse in fission
reactors. When the fuel elements are reprocessed, the uranium is
first taken out of its cladding material and then dissolved in
nitric acid. Most of the tritium released from fuel during
dissolution appears in the liquid waste stream while some is
carried out in the dissolver off-gas stream and a portion is
immobilized as a solid zirconium compound in the cladding.
48. In 1980, the only reprocessing plants operating commercially
in the world were at Windscale (U.K.) and La Hague and Marcoule
(France); their combined capacity was much lower than the amount
of fuel discharged from reactors worldwide. Luykx and Fraser [L3]
have expressed the reported releases from the three reprocessing
plants during the 1974-1978 time period in terms of activity
discharged per unit of electricity generated. The average figures
for each plant are given in Table II.2.
Table II.2 Average normalized tritium activities
discharged into the environment by fuel
reprocessing plants (1010 Bq per MW(e)a) [L3]
--------------------------------------------------
Plant Airborne Liquid Total
location effluents effluents
--------------------------------------------------
Windscale 17 55 72
La Hague 0.4 28.5 29
Marcoule 5.2 41 46
--------------------------------------------------
49. As there is no retention system for tritium in the currently
operating reprocessing plants, the activity released corresponds to
that which is contained in the fuel elements (with the exclusion of
cladding) at the time of reprocessing. The production rate of
tritium in reactors being about 75 1010 Bq per MW(e)a (Table II.1),
approximately half of the theoretical fuel content seems to be
unaccounted for at the La Hague and Marcoule plants.
(c) Summary
50. In 1980, the installed nuclear capacity was 1.25 105 MW(e) on
a worldwide scale [I1]. Assuming an average load factor of 0.6,
the energy produced was 7.5 104 MW(e)a. Using the average figures
given previously for production and release in the types of
reactors considered, the global production and release of tritium
at the reactor sites in 1980 are estimated to be about 1.5 1017 Bq
and 4 1015 Bq, respectively. Table II.3 provides a breakdown of
the environmental discharges from reactors according to reactor
type.
Table II.3 Estimated global discharge of tritium from nuclear
power stations in 1980
----------------------------------------------------------------
Estimated tritium discharges
Reactor Number Capacity in 1980 (Bq)
type [MW(e)] Airborne Liquid Total
effluents effluents
----------------------------------------------------------------
PWR 96 64239 3.9 1013 1.2 1015 1.2 1015
BWR 62 35170 4.2 1013 8.4 1013 1.3 1014
HWR 14 5963 5.4 1014 2.1 1015 2.6 1015
GCR 36 7086 1.3 1013 3.0 1013 4.3 1013
Other 33 12527 - - -
----------------------------------------------------------------
Total 241 124985 6.3 1014 3.4 1015 4.0 1015
----------------------------------------------------------------
51. In comparison, the tritium releases reported for the three
currently operating commercial fuel reprocessing plants were about
2 1015 Bq in 1978. All together, the current tritium production
rate in the nuclear fuel cycle is comparable to the natural
production rate, whereas the release rate is about 20 times less.
4. Tritium production plants
52. Artificial production of tritium on an industrial scale is
necessary to provide an essential component of thermonuclear
weapons. In addition, relatively small amounts of tritium are used
for other industrial and scientific applications. The most
economical way to produce tritium is the irradiation of lithium
metal, alloys or salts in a nuclear reactor [J1]. The tritium is
isotopically separated from other hydrogen isotopes and is
processed in tritium-handling plants [C1].
53. Tritium airborne release rates from Savannah River Plant,
which is the primary production source of tritium in the U.S.A.,
have ranged from 1.4 1016 Bq a-1 to 9.9 1016 Bq a-1 from 1974 to
1977 with an average of 4.1 1016 Bq a-1 [M4]. Under normal
operating conditions, the releases are about 20% HT and 80% HTO.
However, accidental airborne releases, which seem to be essentially
in the gaseous HT form, have raised the contribution of HT to the
total activity released to 60% in 1974 and 57% in 1975 [M4]. The
activity of tritium released in the liquid effluents appears to be
about 10% of that in the airborne effluents [N1].
54. Data on releases from other tritium production plants have not
been found in the literature. However, an indirect estimate of 7
1016 Bq for the worldwide release of HT in 1977 has been made by
Mason and Ostlund [M3] on the basis of their measurements of the
atmospheric HT content.
5. Consumer products
55. Tritium has been used extensively in the dial-painting
industry for the illumination of timepieces, the radiation emitted
by 3H being converted into light by a scintillator which is usually
zinc sulfide containing small amounts of copper or silver. In
recent years, this illumination system has been in competition with
the tritium gas-filled glass tubes, coated internally with
phosphor, which are used to illuminate some types of LCD (liquid
crystal display) watches. Exit signs and electronic tubes are
other types of consumer products containing tritium [C2, K4, U1,
W1].
56. In luminous compounds, the fractional release rate of tritium,
in the form of HTO, HT and short-chain organic radicals of the
styrene type, is about 5% annually [K4, K5] while it is negligible
from gas-filled glass tubes. It has been estimated that about 7
1016 Bq was processed in 1978 in the worldwide production of
timepieces and that the activity released is probably under 1014 Bq
a-1 for luminous compounds and 2 1012 Bq a-1 for gas-filled glass
tubes [K5]. Environmental releases due to breakage through accident
or disposal could be more important [C2, W1].
6. Controlled thermonuclear reactors
57. Large-scale use of controlled thermonuclear reactors for heat
or power generation seems quite unlikely in the next 25 years.
However, if thermonuclear reactors come into use, they will contain
substantial inventories of tritium and will pose considerable
tritium management problems [N1]. The production of tritium in a
nominal 1000 MW(e) controlled thermonuclear reactor is anticipated
to be about 5 1017 Bq d-1 and the inventory of the order of 1019 Bq
[C1, C3, H1]. In order to prevent massive releases of tritium into
the environment, an extraordinary degree of control will be
required. However, conceptual designs for fusion power plants show
that the effluent release rate can be limited to 4 1013 Bq a-1 by
applying present-day tritium technology [C3].
C. BEHAVIOUR IN THE ENVIRONMENT
1. Natural and fallout tritium
58. Natural and fallout tritium are mainly produced in the
stratosphere where they are essentially found in the HTO form.
Tritiated water vapour is transferred from the stratosphere to the
troposphere with a half-time of about one year, then from
troposphere to the earth's surface through rainfall and molecular
exchange with a half-time of about ten days. Tritiated water then
follows the hydrological cycle. Water deposited on the ocean
surfaces is diluted in the mixed layer. Part of it evaporates back
to the atmosphere, with a much lower concentration, while a smaller
fraction is transferred to the deep ocean. Tritiated water
deposited on land surfaces is partitioned partly to surface run-off
(leading to a pond, a lake, a stream, or an ocean) and partly to
infiltration in the soil from where it can be absorbed by plants,
evaporate, or move with groundwater to a surface stream or to an
ocean.
59. Part of the tritiated water deposited on soils finds its way
into vegetable and animal products and thus contaminates dietary
foodstuffs. Tritium incorporated into those biological materials,
and in soil and sediments as well, is found to be present in at
least two separable fractions, one easily exchangeable, that is
available by freeze-drying (free water tritium fraction) and one
less easily exchangeable, available by combustion ("organically
bound" fraction) [B6]. The analysis of soil, water, and various
components of the diet in the New York area in 1978 [B6] revealed
that water, soil and diet were in equilibrium with respect to free
water tritium; however, the specific activities (activity
concentration per unit mass of hydrogen) of the "organically bound"
tritium in various foodstuffs were higher by a factor of 2 to 4
than those of the water tritium. It is suggested that tritium was
incorporated uniformly into biological materials during the period
of highest deposition rates in the early 1960s and that differences
in specific activities developed due to longer biological residence
half-time of the "organically bound" fraction compared to the free
water tritium fraction [B6].
2. Industrial releases
60. Industrial releases consist mainly of HTO and HT, and probably
tritiated methane, CH3T [B7]. The residence times of HT and CH3T
in the atmosphere are not known with certainty but the estimates
point to average values of 5 to 10 years [B7]. The main removal
processes are bacterial action and photochemical oxidation for HT
and photochemical oxidation alone for CH3T [B7]. In both cases,
the resulting product is presumably HTO. As HTO is much more
biologically active than HT and CH3T, it is this tritium compound
that is of most concern in the case of industrial releases.
61. Industrial releases may be to the atmosphere or to water
(river or sea). In addition, releases to ground water have taken
place but they are of little consequence as the movement of water
in suitable aquifers is very slow. The environmental behaviour of
HTO released by industry is not different from that from natural or
fallout sources.
D. TRANSFER TO MAN
62. Transfer to man of environmental HTO takes place via
inhalation, diffusion through skin and ingestion of beverages and
foodstuffs; in the case of HT, inhalation is the only meaningful
pathway to man. Exposure to an atmosphere contaminated with
tritiated water vapour results in total absorption of the inhaled
activity through the lungs and absorption of about 50% of that
amount through the intact skin [I2]. Ingested tritiated water is
completely absorbed from the gastro-intestinal tract.
63. Absorbed tritiated water is rapidly distributed throughout the
body via the blood. Tritiated water in blood equilibrates with
extracellular fluid in about 12 minutes. However, in poorly
vascularized tissues, such as bone and fat, equilibrium with plasma
water may take days to weeks [N2, W2]. The biological half-life of
tritium in the body following intake of tritiated water has been
found to range from 2.4 to 18 days among 300 individuals [B3, W3].
The experience from observations of human cases of accidental
tritium exposures with intakes large enough to allow relatively
long-term monitoring shows that the excretion rate can be
represented as the sum of three exponentials with half-times of
residence of the order of 10 days, one month, and one year [L4, M5,
M6, S3]. The first component is believed to reflect the turnover
of body water while the second and the third components are likely
to represent the turnover of tritium incorporated into organic
compounds.
E. DOSIMETRY
1. Dose per unit intake
(a) Tritiated water
64. External irradiation from tritium does not need to be
considered as the range of the electrons emitted by decay (at most
6 µm in soft tissue) is shorter than the depth of the basal cells
in the epidermis. Following a chronic intake of 1 Bq 1-1 of
tritium (as HTO) in air, water and food the equilibrium dose rate
in active wet tissue (the totality of soft tissues with the
exclusion of fat) is 2.6 10-8 Gy a-1. Of that dose, 16% is
calculated to be due to tritium contained in organic pools of the
body. These results were derived by Bennett [B4] based on human
retention data.
65. When all the sources of intake (air, water and food) are
assumed to be contaminated at the same level, use can be made of
the specific activity model which consists in assuming that the
specific activity of tritium (activity concentration per unit mass
of hydrogen) in the body is the same as that in the intake. A
chronic intake of tritium at a concentration of 1 Bq per litre of
water would thus give rise to an absorbed dose averaged over the
whole body of
10-3
1 Bq 1H2O gH2O gH
----- x ----------- x 18 ---- x 0.1 -----
1H2O gH2O gH gbody
MeV s Gy gbody
x 5.7 10-3 ------ x 3.16 107 --- x 1.6 10-10 ----------
Bq s a MeV
= 2.6 10-8 Gy a-1 per Bq 1-1
This result is numerically equal to that of Bennett [B4]. The
doses in individual tissues depend on their hydrogen
concentrations. According to the values adopted for the Reference
Man of ICRP [I2], the hydrogen concentration per unit mass is the
same (10%) in total body and in total soft tissues and is, as a
first approximation, uniform in the soft tissues. Hydrogen content
is lowest in mineral bone (about 4%) and highest in adipose tissue
(12%). Since the range of the beta-particles emitted by tritium
decay is very small, it can be assumed that all the energy emitted
in a given tissue is absorbed in the same tissue. The effective
dose equivalent is therefore numerically equal to the absorbed dose
averaged over the whole body and is 2.6 10-8 Sv a-1 per Bq 1-1.
Assuming a rate of intake of 3 litres of water (in beverages and in
food) per day and a water vapour atmosphere concentration of 8 g m-3,
the effective dose equivalent per unit intake is found to be 2.2
10-11 Sv Bq-1 while the effective dose equivalent rate per unit
atmospheric concentration would be 2.1 10-9 Sv a-1 per Bq m-3.
(b) Tritiated hydrogen
66. The doses from inhalation of HT are much lower than those from
HTO for a given atmospheric concentration of tritium. The dose rate
to the lungs per unit concentration of HT in air is about 10-14 Gy
h-1 per Bq m-3 [I3], while the doses in tissues from the absorbed
gas are 60 to 150 times smaller [I3]. The corresponding effective
dose equivalent rate per unit concentration in air is therefore 1.1
10-11 Sv a-1 per Bq m-3.
2. Dose per unit release
(a) Natural tritium
67. Doses from natural tritium can be estimated from the few
tritium measurements in environmental materials that were carried
out before the contamination with fallout (or that had been
preserved from contamination). Activity concentrations of
continental surface waters were then found to be in the range from
0.2 to 0.9 Bq 1-1 [K1]. The production rate of natural tritium
being constant in time and relatively uniform on the global scale,
the concentrations in all the components of human intake (air,
water and food) of natural tritium are in steady-state equilibrium
with the concentrations in continental surface waters. Using the
specific activity approach, it is assumed that the specific
activity of natural tritium is the same in the continental surface
waters, in all the components of human intake and in the body. The
effective dose equivalent rate is thus found to range from
0.2 Bq 1-1 x 2.6 10-8 Sv a-1 per Bq 1-1 = 5.2 109 Sv a-1 to
0.9 Bq 1-1 x 2.6 10-8 Sv a-1 per Bq 1-1 = 2.3 10-8 Sv a-1, being
therefore of the order of 10-8 Sv a-1. The effective dose equivalent
commitment per unit release would then be
10-8 Sv a-1
--------------- ca. 1.4 10-25 Sv per Bq
7.2 1016 Bq a-1
Taking the world's population to be 4 109 people, the global
collective effective dose equivalent commitment per unit of
activity produced is about 5 10-16 man Sv per Bq.
(b) Nuclear explosions
68. The doses from fallout tritium can be estimated in the same
way as those from natural tritium. On the basis of the variation
with time of the tritium activity concentration in surface waters
[B5] and of the latitudinal distribution of the fallout deposition
[S1], UNSCEAR [U1] estimated the effective dose equivalent
commitments to the populations of the northern and southern
hemispheres to be 2 10-5 and 2 10-6 Sv respectively.
69. The effective dose equivalent commitment from fallout tritium
was also estimated indirectly, using the relationship obtained for
natural tritium between the production rate and the dose rate
W
Hc = gammao -
B
where Hc is the effective dose equivalent commitment (Sv) from
production of fallout tritium in a given hemisphere; gammao is the
effective dose equivalent rate from natural tritium (gammao = 10-8
Sv a-1); W is the activity of tritium released by nuclear
explosions (1.5 1020 Bq in the northern hemisphere and 0.2 1020 Bq
in the southern hemisphere); and B is the natural rate of
production (3.6 1016 Bq a-1 in each hemisphere). The effective
dose equivalent commitments thus derived are 4.2 10-5 Sv for the
population of the northern hemisphere and 5.6 10-6 Sv for the
population of the southern hemisphere. These results are higher
than the direct estimates by a factor of 2 to 3. The global
collective effective dose equivalent commitments per unit activity
released are estimated to be 9 10-16 and 4 10-16 man Sv Bq-1 using
the latter and the former method, respectively. UNSCEAR [U1] used
an intermediate value of 8 10-16 man Sv per Bq.
(c) Nuclear installations
70. While the production of natural and fallout tritium brings
about a relatively uniform contamination of the whole biosphere,
the releases from nuclear installations occur at discrete points on
the earth's surface giving a highly heterogenous spatial
distribution of concentrations.
71. UNSCEAR'S practice is to divide the collective doses into two
components: the local and regional collective doses, which are due
to the first passage of the plume, over distances of 100 to 1000 km
from the point of release, and the global collective doses, which
arise from the mixing of tritium in the whole biosphere. As the
doses per unit concentration of tritium in air are much higher for
HTO than for HT, tritiated water will be the only compound
considered in the estimate of the local and regional collective
doses.
(i) Local and regional collective dose
72. A distinction is made between airborne and liquid effluents.
Tritium present in airborne effluents can contribute to the local
and regional collective doses through inhalation, absorption
through skin and ingestion. As the contribution from the ingestion
pathway is quite variable from site to site owing to differences in
local hydrology and water usage, UNSCEAR [U1] has not taken this
pathway into account in its assessment of the local and regional
collective doses. Assuming an atmospheric dispersion factor of 5
10-7 s m-3 at 1 km from the release and a reduction in
concentration inversely proportional to the 1.5 power of the
distance expressed in kilometres, the local collective dose per
unit activity released can be assessed by integration over the
local area. Integrating from 1 to 100 km for a population density
of 100 km-2, UNSCEAR [U1] estimated the local collective dose from
airborne tritium per unit activity released to be about 5 10-17 man
Sv per Bq.
73. The collective dose commitment from the input of 3H to water
bodies, normalized per unit activity released, can be estimated
[U1], using the expression
c sigmak Nk Ik fk phi
S = -------------------
1 V(lambda + 1/tau)
where V is the volume of the receiving waters, tau is the turnover
time of receiving waters, lambda is the decay constant of 3H, Nk is
the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
74. The quantity V(lambda + 1/tau) is the infinite time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentrations in water and in fish
will be high, but the population which can be served with drinking
water or by fish consumption will limited. For inputs into larger
volumes of water, the concentrations will be smaller, but the
populations involved will be correspondingly larger. It is
reasonable, therefore, to assume as a first approximation that the
quantities Nk/V are relatively constant, independent of V. The
values for these quantities as well as values for the other
parameters of the above expression have been extensively discussed
[U1].
75. A summary of the values used in the assessment, based on
UNSCEAR [U1], and the evaluation of the collective dose commitments
for a release of 1 Bq of 3H in liquid effluents are given in Table
II.4.
(ii) Global collective dose
76. For HTO releases, the global collective effective dose
equivalent commitment established for fallout tritium (8 10-16 man
Sv per Bq) can be applied without change. With respect to HT
releases, if it is assumed that the conversion to HTO takes place
on average 5 years after the discharges, the global collective
effective dose equivalent commitment is estimated to be
-0.693
8 10-16 e x 5/12.3 = 6 10-16 man Sv per Bq.
Table II.4 Collective dose factors for 3H in liquid effluents
---------------------------------------------------------------------------
Fresh water Salt water
---------------------------------------------------------------------------
Activity released, A 1 Bq 1 Bq
Turnover time of receiving water, 10 a 1.0 a
Sediment removal correction factor, s 1.0 1.0
Time integral of activity in water,
As
W = ------------ 6.36 Bq a 0.946 Bq a
1/tau+lambda
Water utilization, V/N 3 107 1/man 3 109 1/man
---------------------------------------------------------------------------
FRESHWATER PATHWAYS
1 Drinking water
Treatment removal factor, f1 1.0
Consumption, I1 438 1 a-1
Collective dose commitment
c NI
S1 = W f1 (--)1D 2 10-15 man Sv
V
Table II.4 (contd.)
---------------------------------------------------------------------------
Fresh water Salt water
---------------------------------------------------------------------------
2. Fish
Concentration factor, f2 1.0
Consumption, I2 1 kg a-1
Collective dose commitment
c NI
S2 = W f2 (--)2D 5 10-18 man Sv
V
SALT WATER PATHWAYS
3. Fish
Concentration factor, f3 1.0
Consumption, I3 6 kg a-1
Collective dose commitment
c NI
S3 = W f3 (--)3D 4 10-20 man Sv
V
4. Shellfish
Concentration factor, f4 1.0
Consumption, I4 1 kg a-1
Collective dose commitment
c NI
S4 = W f4 (--)4D 7 10-21 man Sv
V
---------------------------------------------------------------------------
(iii) Summary of collective dose commitments per unit activity released
77. Table II.5 summarizes the values obtained above for the
collective effective dose equivalent commitments per unit of 3H
activity released. With respect to the local and regional
component due to industrial releases, the largest collective
effective dose equivalent commitment per unit activity released is
obtained for a river discharge and the smallest for a sea discharge
while an intermediate value is found for the airborne discharge.
Table II.5 Summary of collective effective dose
equivalent commitments per unit tritium activity released
(man Sv per Bq)
---------------------------------------------------------
Origin Local and regional Global
component component
---------------------------------------------------------
Natural 5 10-16
Nuclear tests 8 10-16
Industry
Airborne discharge 5 10-17 (HTO) ) 8 10-16 (HTO)
River discharge 2 10-15 )
Sea charge 5 10-20 ) 6 10-16 (HT)
---------------------------------------------------------
F. REFERENCES
B1 Bennett, B.G. Environmental aspects of americium. EML-348
(1978).
B2 Bowen, V.T. and W. Roether. Vertical distributions of
strontium-90, caesium-137 and tritium near 45° north in the
Atlantic. J. Geophys. Res. 78: 6277-6285 (1973).
B3 Butler, H.L. and J.H. LeRoy. Observations of biological half-
life of tritium. Health Phys. 11: 283-285 (1965).
B4 Bennett, B.G. Environmental tritium and the dose to man.
p. 1047-1053 in Proceedings of the Third International
Congress of IRPA. CONF-730907 (1973).
B5 Bennett, B.G. Fallout tritium in the environment and the dose
commitments. HASL-268 (1973).
B6 Bogen, D.C., G.A. Welford and C.G. White. Tritium distribution
in man and his environment. p. 567-574 in Behaviour of Tritium
in the Environment. IAEA, Vienna, 1979.
B7 Burger, L.L. Distribution and reactions of tritiated hydrogen
and methane. p. 47-64 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
C1 Crowson, D.L. Man-made tritium. p. 23-27 in Tritium (A.A.
Moghissi and M.W. Carter, eds.). Messenger Graphics, Las
Vegas, Nevada, 1973.
C2 Comps, F. and R.J. Doda. Large-scale distribution of tritium
in a commercial product. p. 93-99 in Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
C3 Coyle, P.E. Laser fusion. Status, future and tritium control.
p. 139-153 in Behaviour of Tritium in the Environment. IAEA,
Vienna 1979.
F1 Fireman, E.L. Measurement of the (n,H3) cross section in
nitrogen and its relationship to the tritium produced in the
atmosphere. Phys. Rev. 91: 922-926 (1953).
F2 Flamm, E., R.E. Lingenfelter, J. F. MacDonald et al. Tritium
and helium-3 in solar flares and loss of helium from the
earth's atmosphere. Science 138: 48-49 (1962).
G1 Gratwohl, G. Erzeugung und Freisetzung von Tritium durch
Reaktoren und Wiederaufarbeitungsanlagen und die
voraussichtliche radiologische Belastung bis zum Jahr 2000.
Kernforschungszentrum Karlsruhe report KFF-Ext. 4/73-36 (1973).
H1 Häfele, W., J.P. Holdren, G. Kessler et al. Fusion and fast
breeder reactors. IIASA RR-77-8 (1976) (revised July 1977).
I1 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
I2 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP publication 23.
Pergamon Press, 1975.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP publication 30.
Annals of the ICRP 2: 3/4 (1979)
I4 International Atomic Energy Agency. Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
J1 Jacobs, D.G. Sources of tritium and its behaviour upon release
to the environment. AEC Critical Review Series. TID-24635
(1968).
K1 Kaufamn, S. and W.F. Libby. The natural distribution of
tritium. Phys. Rev. 93: 1337-1344 (1954).
K2 Kouts, H. and J. Long. Tritium production in nuclear reactors.
p. 38-45 in Tritium (A.A. Moghissi and M.W. Carter, eds.).
Messenger Graphics, Las Vegas, Nevada, 1973.
K3 Kahn, B., R.L. Blanchard, W.L. Brinck et al. Radiological
surveillance study at the Haddam Neck PWR nuclear power
station. EPA-520/374-007, Washington, 1974.
K4 Krejci, K. and A. Zeller, Jr. Tritium pollution in the Swiss
luminous compound industry. p. 65-77 in Behaviour of Tritium
in the Environment. IAEA, Vienna, 1979.
K5 Krejci, K. Discussion. p. 101 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
L1 Lal, D. and B. Peters. Cosmic ray produced radioactivity
on the earth. p. 551-612 in Encyclopaedia of Physics, Vol.
XLV1/2 on Cosmic Rays (K. Sitte, ed.). Springer-Verlag, New
York, 1967.
L2 Locante, J. and D.D. Malinowski. Tritium in pressurized water
reactors. p. 45-57 in Tritium (A.A. Moghissi and M.W. Carter,
eds.). Messenger Graphics, Las Vegas, Nevada, 1973.
L3 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European community. Discharge data 1974-1978. Radiological
aspects. Commission of the European Communities. V/4116/80
(1980).
L4 Lambert, B.E., H.B.A. Sharpe and K.B. Dawson. Am. Ind. Hyg.
Assoc. J. 32: 682 (1971).
M1 Miskell, J.A. Production of tritium by nuclear weapons. p. 79-
85 in Tritium (A.A. Moghissi and M.W. Carter, eds.).
Messenger Graphics, Phoenix and Las Vegas, 1973.
M2 Michel, L. Tritium inventories of the world oceans and their
implications. Nature 263: 103-106 (1976).
M3 Mason, A.S. and H.G. Ostlund. Atmospheric HT and HTO: V.
Distribution and large-scale circulation. p. 3-16 in Behaviour
of Tritium in the Environment. IAEA, Vienna, 1979.
M4 Murphy, C.E. Jr. and M.M. Pendergast. Environmental transport
and cycling of tritium in the vicinity of atmospheric releases.
p. 361-372 in Behaviour of Tritium in the Environment. IAEA,
Vienna, 1979.
M5 Minder, W. Strahlentherapie. 137: 700 (1969).
M6 Moghissi, A.A., M.W. Carter and E.W. Bretthauer. Further
studies on the long-term evaluation of the biological half-life
of tritium. Health Phys. 23: 805-806 (1972).
M7 Moghissi, A.A. and M.W. Carter, eds. Tritium. Messenger
Graphics, Las Vegas, Nevada, 1973.
N1 National Council on Radiation Protection and Measurements.
Tritium in the environment. NCRP No. 62 (1979).
N2 National Council on Radiation Protection and Measurements.
Tritium and other radionuclide labelled organic compounds
incorporated in genetic material. NCRP No. 63 (1979).
O1 Ostlund, H.G. and R.A. Fine. Oceanic distribution and
transport of tritium. p. 303-314 in Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
S1 Schell, W.R., S. Sauzay and B.R. Payne. World distribution of
environmental tritium. p. 374-385 in Physical Behaviour of
Radioactive Contaminants in the Atmosphere. IAEA, Vienna,
1974.
S2 Smith, J.M. and R.S. Gilbert. Tritium experience in boiling
water reactors. p. 57-68 in Tritium (A.A. Moghissi and M.W.
Carter, eds.). Messenger Graphics, Las Vegas, Nevada, 1973.
S3 Sanders, S.M. Hr. and W. C. Reinig. Assessment of tritium in
man. p. 534-542 in Diagnosis and Treatment of Deposited
Radionuclides (H.A. Kornberg and W.D. Norwood, eds.). Excerpta
Medica Foundation, Amsterdam, 1968.
T1 Trevorrow, L.E., B.J. Kullen, R.L. Jarry et al. Tritium and
noble gas fission products in the nuclear fuel cycle. I.
Reactors. ANL-8102 (1974).
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication no. E.77.IX.I. New York,
1977.
W1 Wehner, G. Discharges of tritium to the environment from
unrestricted use of consumer products containing this
radionuclide. p. 79-91 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
W2 Woodard, H.Q. The biological effects of tritium. United
States Atomic Energy Commission. HASL-229 (1970).
W3 Wylie, K. F., W. A. Bigler and G.R. Grove. Biological half-
life of tritium. Health Phys. 9: 911-914 (1963).
III. CARBON-14
A. INTRODUCTION
78. Carbon-14 has always been present on the earth. It is
produced by cosmic ray interactions in the atmosphere. This
nuclide is a pure beta-emitter, with a half-life of 5730 years, a
maximum energy of 185 keV and an average energy of 49.47 keV [N1].
79. Carbon is one of the elements that are essential to all forms
of life and is involved in most biological and geochemical
processes on the earth. Associated with the stable isotopes of
carbon (12C and about 1.1%13C), there is a very small amount of 14C
formed in the atmosphere and which has subsequently entered in the
carbon cycle. The specific activity of biological carbon, as
measured in wood samples grown in the nineteenth century, was 0.227
± 0.001 Bq per gram of carbon [T1], corresponding to an atmospheric
inventory of 1.4 1017 Bq. During the present century the specific
activity of 14C has decreased due to the diluting effect of
releases into the atmosphere of carbon dioxide from the combustion
of fossil fuels. This effect (the Suess effect) accounts for a
reduction of a few percent.
80. In addition to its natural production, carbon-14 is also
produced by the detonation of nuclear explosives and by the
operation of nuclear reactors. The assessment of the collective
dose commitments from the releases of man-made carbon-14 is
facilitated by knowledge of the production rate of natural
carbon-14.
B. SOURCES
1. Natural carbon-14
81. Carbon-14 is produced by the action of cosmic ray neutrons on
nitrogen atoms, both in the stratosphere and in the upper
troposphere. UNSCEAR [U3] has estimated the natural production
rate to be about 1015 Bq per year, a value which has been derived
from assessments of the natural 14C inventory. The production rate
has also been estimated directly from assessments of cosmic ray
neutrons and the values obtained by different authors range from 1
to 1.4 1015 Bq per year [U3]. Considering the uncertainties
involved in determining both the direct production rate and also
the total 14C inventory of the earth, the estimates are in
reasonable agreement.
2. Nuclear explosions
82. Carbon-14 is formed in nuclear explosions through the capture
of excess neutrons by atmospheric nitrogen. After large
atmospheric nuclear explosions, most of the 14C is transported into
the stratosphere, from where it equilibrates with the troposphere
with a half-time of 1 to 2 years [U3].
83. The inventory of 14C from nuclear explosions has been
estimated from measurements of excess specific activity in the
troposphere and in the surface ocean waters, and models
representing the exchange of 14C between the atmosphere, the
biosphere and the ocean. UNSCEAR [U3] has estimated that nuclear
explosions up to 1972 have injected into the atmosphere 2.1 1017 Bq,
while subsequent injections have increased this amount by about 1%.
84. For the past pattern of atmospheric nuclear explosions, the
production mentioned above corresponds to about 3.7 1014 Bq per
megaton. This value, however, is not representative of any given
nuclear explosion, because the production of 14C will depend on the
type of nuclear device exploded and also on whether the explosion
took place on the surface of the earth or high in the atmosphere.
A "surface" test will produce approximately 50% of the 14C that
would be produced by the same device in an "air" test, because
about one half of the escaping neutrons will be captured in the
soil or water rather than in the atmosphere.
3. Nuclear fuel cycle
85. Carbon-14 is produced in nuclear reactors and is released to
the environment at the reactor itself or at reprocessing plants
where spent fuel is reprocessed. Only recently has attention been
given to the production and release of this radionuclide at nuclear
fuel cycle installations.
(a) Nuclear reactors
86. The production of carbon-14 in nuclear power reactors is due
to several nuclear reactions in the fuel, core construction
materials and moderator. Figure III.I summarizes the relevant
reactions.
35. Most of the fission product tritium produced in the fuel rods
is usually retained within the fuel and is not released into the
environment at the reactor site; it is instead released during fuel
reprocessing, if that practice is carried out. The activity
produced in the coolant is partly or entirely released in the
effluent streams according to the waste management practices at the
plant.
36. Releases into the environment are mainly in the form of HTO in
reactors that use water as primary coolant, as well as in fuel
reprocessing plants.
(a) Nuclear reactors
37. Four types of reactors have been considered (PWR, BWR, HWR,
GCR), the emphasis being on PWRs and BWRs which currently represent
the largest share of nuclear capacity. Estimated generation rates
and appearance of tritium in effluent streams of reactors are
summarized in Table II.1.
38. The annual production of fission product tritium in the fuel
rods of a pressurized water reactor (PWR) is in the range of 6 to 9
1011 Bq per MW(e)a [N1]. A small percentage, 1% or less, is
expected to be released into the coolant through defects in the
cladding, currently made of zirconium alloy. In contrast, the use
of stainless steel cladding in earlier PWRs resulted in the release
to the coolant of most of the tritium produced in the fuel.
39. Tritium generation in the primary coolant (water) of a
PWR is mainly due to reactions with boron (2.6 1010 Bq per
MW(e)a) which is dissolved as boric acid to control
reactivity; in addition, the maintenance of 2 ppm lithium
hydroxide for pH control [L2] results in the formation of
about 7 108 Bq per MW(e)a.
40. Environmental tritium discharges from PWRs depend on waste
management practices as well as on the materials used in the
reactor. Average normalized releases of tritium were shown in the
UNSCEAR 1977 report [U1] to be about 7 1010 Bq per MW(e)a in liquid
effluents and 7 109 Bq per MW(e)a in airborne effluents for the
reactors in operation in 1973-1974. However, large differences
between PWRs are due to the type of fuel cladding. For an old
reactor using stainless steel Kahn et al. [K3] measured 3H releases
of about 4 1011 Bq per MW(e)a in liquid effluents and 4 1010 Bq per
MW(e)a in airborne effluents, whereas the combined releases of 9
PWRs with zirconium alloy clad fuel (current practice) were
reported by NCRP [N1] to be about 3 1010 and 109 Bq per MW(e)a in
liquid and airborne effluents, respectively.
41. In boiling water reactors (BWRs) tritium is produced by
ternary fission in the fuel at about the same rate as in PWRs (6 to
9 1011 Bq per MW(e)a). The generalized use of zirconium alloy
cladding limits the tritium release into the coolant to less than 7
109 Bq per MW(e)a.
42. Tritium can be generated by neutron activation in the coolant
and in the control rods. Prior to 1971, control rods of boron
carbide were used in BWRs [S2]; the production of tritium by
activation of these control rods has been estimated to be about 3
1011 Bq per MW(e)a. However, tritium has not been shown to diffuse
through the boron carbide matrix [T1]. In the coolant itself,
tritium is generated by activation of deuterium at a rate of about
4 108 Bq per MW(e)a.
43. Tritium activities discharged from BWRs into the environment
are lower than those of PWRs because less tritium is produced in or
diffuses into the primary coolant. UNSCEAR [U1] reported the
average discharge rates to be 4 109 and 2 109 Bq MW(e)a in liquid
and airborne effluents, respectively.
44. The amount of tritium generated in fuel of heavy water
reactors (HWR) by ternary fission is approximately the same as in
light water reactors, but it is largely exceeded by the production
in the D2O coolant and moderator by neutron activation, which has
been estimated to be about 2 1013 Bq per MW(e)a [K2].
Table II.1 Estimated rates of generation of tritium and of its release in effluent streams of
different types of reactors (1010 Bq per MW(e)a) [G1, K2, S2, T1, U1]
---------------------------------------------------------------------------------------------------------
PWR BWR HWR GCR
Source ------------------------------------------------------------------------------------------
Generation Effluent Generation Effluent Generation Effluent Generation Effluent
stream stream stream stream
---------------------------------------------------------------------------------------------------------
Fission 75 < 0.7 75 < 0.7 55 < 0.6 75 < 0.7
Activation
Deuterium 0.004 0.004 0.04 0.04 2000 75a/
Lithium 0.07 0.07 2 0.4
Boron 2.6 2.6 30 0
Rounded total 80 3 110 0.5 2000 75 80 1
---------------------------------------------------------------------------------------------------------
a/ Depending on the irradiation time and on the net leakage of heavy water.
45. Environmental discharges depend upon the D2O leakage which is
kept as small as possible for economical and radiological reasons,
and upon the tritium activity in the coolant and moderator, which
builds up with the irradiation time. Annual losses of from 0.5% to
3% are anticipated in HWRs [U1]. For the optimal loss of 0.5% per
year, the normalized tritium release rate ranges from 1011 Bq per
MW(e)a in the first year of operation to about 7 1011 Bq per MW(e)a
in the tenth year. Based on the latter value as representative of
the reactor life, the normalized 3H release rates are estimated to
be 6 1011 and 1.5 1011 Bq per MW(e)a in airborne and liquid
effluents, respectively [G1]. Reported releases roughly agree with
these estimates: they are 6.3 1011 and 2.6 1011 Bq per MW(e)a for
the Pickering A station in Canada, in airborne and liquid
effluents, respectively whereas the Atucha reactor in Argentina
releases about 8 1011 Bq per MW(e)a both in airborne and in liquid
effluents.
46. In gas-cooled reactors (GCR), tritium is produced by ternary
fission (about 7 1011 Bq per MW(e)a) and by activation of lithium
in the graphite moderator. Based on the experience with UK
reactors (mainly Magnox reactors), the tritium release is about 7
109 Bq per MW(e)a in liquid effluents and ranges from 109 to 1010
Bq per MW(e)a in airborne effluents [U1].
(b) Fuel reprocessing plants
47. At the fuel reprocessing stage of the nuclear fuel cycle (if
it is undertaken) the elements uranium and plutonium in the
irradiated nuclear fuel are recovered for reuse in fission
reactors. When the fuel elements are reprocessed, the uranium is
first taken out of its cladding material and then dissolved in
nitric acid. Most of the tritium released from fuel during
dissolution appears in the liquid waste stream while some is
carried out in the dissolver off-gas stream and a portion is
immobilized as a solid zirconium compound in the cladding.
48. In 1980, the only reprocessing plants operating commercially
in the world were at Windscale (U.K.) and La Hague and Marcoule
(France); their combined capacity was much lower than the amount
of fuel discharged from reactors worldwide. Luykx and Fraser [L3]
have expressed the reported releases from the three reprocessing
plants during the 1974-1978 time period in terms of activity
discharged per unit of electricity generated. The average figures
for each plant are given in Table II.2.
Table II.2 Average normalized tritium activities
discharged into the environment by fuel
reprocessing plants (1010 Bq per MW(e)a) [L3]
--------------------------------------------------
Plant Airborne Liquid Total
location effluents effluents
--------------------------------------------------
Windscale 17 55 72
La Hague 0.4 28.5 29
Marcoule 5.2 41 46
--------------------------------------------------
49. As there is no retention system for tritium in the currently
operating reprocessing plants, the activity released corresponds to
that which is contained in the fuel elements (with the exclusion of
cladding) at the time of reprocessing. The production rate of
tritium in reactors being about 75 1010 Bq per MW(e)a (Table II.1),
approximately half of the theoretical fuel content seems to be
unaccounted for at the La Hague and Marcoule plants.
(c) Summary
50. In 1980, the installed nuclear capacity was 1.25 105 MW(e) on
a worldwide scale [I1]. Assuming an average load factor of 0.6,
the energy produced was 7.5 104 MW(e)a. Using the average figures
given previously for production and release in the types of
reactors considered, the global production and release of tritium
at the reactor sites in 1980 are estimated to be about 1.5 1017 Bq
and 4 1015 Bq, respectively. Table II.3 provides a breakdown of
the environmental discharges from reactors according to reactor
type.
Table II.3 Estimated global discharge of tritium from nuclear
power stations in 1980
----------------------------------------------------------------
Estimated tritium discharges
Reactor Number Capacity in 1980 (Bq)
type [MW(e)] Airborne Liquid Total
effluents effluents
----------------------------------------------------------------
PWR 96 64239 3.9 1013 1.2 1015 1.2 1015
BWR 62 35170 4.2 1013 8.4 1013 1.3 1014
HWR 14 5963 5.4 1014 2.1 1015 2.6 1015
GCR 36 7086 1.3 1013 3.0 1013 4.3 1013
Other 33 12527 - - -
----------------------------------------------------------------
Total 241 124985 6.3 1014 3.4 1015 4.0 1015
----------------------------------------------------------------
51. In comparison, the tritium releases reported for the three
currently operating commercial fuel reprocessing plants were about
2 1015 Bq in 1978. All together, the current tritium production
rate in the nuclear fuel cycle is comparable to the natural
production rate, whereas the release rate is about 20 times less.
4. Tritium production plants
52. Artificial production of tritium on an industrial scale is
necessary to provide an essential component of thermonuclear
weapons. In addition, relatively small amounts of tritium are used
for other industrial and scientific applications. The most
economical way to produce tritium is the irradiation of lithium
metal, alloys or salts in a nuclear reactor [J1]. The tritium is
isotopically separated from other hydrogen isotopes and is
processed in tritium-handling plants [C1].
53. Tritium airborne release rates from Savannah River Plant,
which is the primary production source of tritium in the U.S.A.,
have ranged from 1.4 1016 Bq a-1 to 9.9 1016 Bq a-1 from 1974 to
1977 with an average of 4.1 1016 Bq a-1 [M4]. Under normal
operating conditions, the releases are about 20% HT and 80% HTO.
However, accidental airborne releases, which seem to be essentially
in the gaseous HT form, have raised the contribution of HT to the
total activity released to 60% in 1974 and 57% in 1975 [M4]. The
activity of tritium released in the liquid effluents appears to be
about 10% of that in the airborne effluents [N1].
54. Data on releases from other tritium production plants have not
been found in the literature. However, an indirect estimate of 7
1016 Bq for the worldwide release of HT in 1977 has been made by
Mason and Ostlund [M3] on the basis of their measurements of the
atmospheric HT content.
5. Consumer products
55. Tritium has been used extensively in the dial-painting
industry for the illumination of timepieces, the radiation emitted
by 3H being converted into light by a scintillator which is usually
zinc sulfide containing small amounts of copper or silver. In
recent years, this illumination system has been in competition with
the tritium gas-filled glass tubes, coated internally with
phosphor, which are used to illuminate some types of LCD (liquid
crystal display) watches. Exit signs and electronic tubes are
other types of consumer products containing tritium [C2, K4, U1,
W1].
56. In luminous compounds, the fractional release rate of tritium,
in the form of HTO, HT and short-chain organic radicals of the
styrene type, is about 5% annually [K4, K5] while it is negligible
from gas-filled glass tubes. It has been estimated that about 7
1016 Bq was processed in 1978 in the worldwide production of
timepieces and that the activity released is probably under 1014 Bq
a-1 for luminous compounds and 2 1012 Bq a-1 for gas-filled glass
tubes [K5]. Environmental releases due to breakage through accident
or disposal could be more important [C2, W1].
6. Controlled thermonuclear reactors
57. Large-scale use of controlled thermonuclear reactors for heat
or power generation seems quite unlikely in the next 25 years.
However, if thermonuclear reactors come into use, they will contain
substantial inventories of tritium and will pose considerable
tritium management problems [N1]. The production of tritium in a
nominal 1000 MW(e) controlled thermonuclear reactor is anticipated
to be about 5 1017 Bq d-1 and the inventory of the order of 1019 Bq
[C1, C3, H1]. In order to prevent massive releases of tritium into
the environment, an extraordinary degree of control will be
required. However, conceptual designs for fusion power plants show
that the effluent release rate can be limited to 4 1013 Bq a-1 by
applying present-day tritium technology [C3].
C. BEHAVIOUR IN THE ENVIRONMENT
1. Natural and fallout tritium
58. Natural and fallout tritium are mainly produced in the
stratosphere where they are essentially found in the HTO form.
Tritiated water vapour is transferred from the stratosphere to the
troposphere with a half-time of about one year, then from
troposphere to the earth's surface through rainfall and molecular
exchange with a half-time of about ten days. Tritiated water then
follows the hydrological cycle. Water deposited on the ocean
surfaces is diluted in the mixed layer. Part of it evaporates back
to the atmosphere, with a much lower concentration, while a smaller
fraction is transferred to the deep ocean. Tritiated water
deposited on land surfaces is partitioned partly to surface run-off
(leading to a pond, a lake, a stream, or an ocean) and partly to
infiltration in the soil from where it can be absorbed by plants,
evaporate, or move with groundwater to a surface stream or to an
ocean.
59. Part of the tritiated water deposited on soils finds its way
into vegetable and animal products and thus contaminates dietary
foodstuffs. Tritium incorporated into those biological materials,
and in soil and sediments as well, is found to be present in at
least two separable fractions, one easily exchangeable, that is
available by freeze-drying (free water tritium fraction) and one
less easily exchangeable, available by combustion ("organically
bound" fraction) [B6]. The analysis of soil, water, and various
components of the diet in the New York area in 1978 [B6] revealed
that water, soil and diet were in equilibrium with respect to free
water tritium; however, the specific activities (activity
concentration per unit mass of hydrogen) of the "organically bound"
tritium in various foodstuffs were higher by a factor of 2 to 4
than those of the water tritium. It is suggested that tritium was
incorporated uniformly into biological materials during the period
of highest deposition rates in the early 1960s and that differences
in specific activities developed due to longer biological residence
half-time of the "organically bound" fraction compared to the free
water tritium fraction [B6].
2. Industrial releases
60. Industrial releases consist mainly of HTO and HT, and probably
tritiated methane, CH3T [B7]. The residence times of HT and CH3T
in the atmosphere are not known with certainty but the estimates
point to average values of 5 to 10 years [B7]. The main removal
processes are bacterial action and photochemical oxidation for HT
and photochemical oxidation alone for CH3T [B7]. In both cases,
the resulting product is presumably HTO. As HTO is much more
biologically active than HT and CH3T, it is this tritium compound
that is of most concern in the case of industrial releases.
61. Industrial releases may be to the atmosphere or to water
(river or sea). In addition, releases to ground water have taken
place but they are of little consequence as the movement of water
in suitable aquifers is very slow. The environmental behaviour of
HTO released by industry is not different from that from natural or
fallout sources.
D. TRANSFER TO MAN
62. Transfer to man of environmental HTO takes place via
inhalation, diffusion through skin and ingestion of beverages and
foodstuffs; in the case of HT, inhalation is the only meaningful
pathway to man. Exposure to an atmosphere contaminated with
tritiated water vapour results in total absorption of the inhaled
activity through the lungs and absorption of about 50% of that
amount through the intact skin [I2]. Ingested tritiated water is
completely absorbed from the gastro-intestinal tract.
63. Absorbed tritiated water is rapidly distributed throughout the
body via the blood. Tritiated water in blood equilibrates with
extracellular fluid in about 12 minutes. However, in poorly
vascularized tissues, such as bone and fat, equilibrium with plasma
water may take days to weeks [N2, W2]. The biological half-life of
tritium in the body following intake of tritiated water has been
found to range from 2.4 to 18 days among 300 individuals [B3, W3].
The experience from observations of human cases of accidental
tritium exposures with intakes large enough to allow relatively
long-term monitoring shows that the excretion rate can be
represented as the sum of three exponentials with half-times of
residence of the order of 10 days, one month, and one year [L4, M5,
M6, S3]. The first component is believed to reflect the turnover
of body water while the second and the third components are likely
to represent the turnover of tritium incorporated into organic
compounds.
E. DOSIMETRY
1. Dose per unit intake
(a) Tritiated water
64. External irradiation from tritium does not need to be
considered as the range of the electrons emitted by decay (at most
6 µm in soft tissue) is shorter than the depth of the basal cells
in the epidermis. Following a chronic intake of 1 Bq 1-1 of
tritium (as HTO) in air, water and food the equilibrium dose rate
in active wet tissue (the totality of soft tissues with the
exclusion of fat) is 2.6 10-8 Gy a-1. Of that dose, 16% is
calculated to be due to tritium contained in organic pools of the
body. These results were derived by Bennett [B4] based on human
retention data.
65. When all the sources of intake (air, water and food) are
assumed to be contaminated at the same level, use can be made of
the specific activity model which consists in assuming that the
specific activity of tritium (activity concentration per unit mass
of hydrogen) in the body is the same as that in the intake. A
chronic intake of tritium at a concentration of 1 Bq per litre of
water would thus give rise to an absorbed dose averaged over the
whole body of
10-3
1 Bq 1H2O gH2O gH
----- x ----------- x 18 ---- x 0.1 -----
1H2O gH2O gH gbody
MeV s Gy gbody
x 5.7 10-3 ------ x 3.16 107 --- x 1.6 10-10 ----------
Bq s a MeV
= 2.6 10-8 Gy a-1 per Bq 1-1
This result is numerically equal to that of Bennett [B4]. The
doses in individual tissues depend on their hydrogen
concentrations. According to the values adopted for the Reference
Man of ICRP [I2], the hydrogen concentration per unit mass is the
same (10%) in total body and in total soft tissues and is, as a
first approximation, uniform in the soft tissues. Hydrogen content
is lowest in mineral bone (about 4%) and highest in adipose tissue
(12%). Since the range of the beta-particles emitted by tritium
decay is very small, it can be assumed that all the energy emitted
in a given tissue is absorbed in the same tissue. The effective
dose equivalent is therefore numerically equal to the absorbed dose
averaged over the whole body and is 2.6 10-8 Sv a-1 per Bq 1-1.
Assuming a rate of intake of 3 litres of water (in beverages and in
food) per day and a water vapour atmosphere concentration of 8 g m-3,
the effective dose equivalent per unit intake is found to be 2.2
10-11 Sv Bq-1 while the effective dose equivalent rate per unit
atmospheric concentration would be 2.1 10-9 Sv a-1 per Bq m-3.
(b) Tritiated hydrogen
66. The doses from inhalation of HT are much lower than those from
HTO for a given atmospheric concentration of tritium. The dose rate
to the lungs per unit concentration of HT in air is about 10-14 Gy
h-1 per Bq m-3 [I3], while the doses in tissues from the absorbed
gas are 60 to 150 times smaller [I3]. The corresponding effective
dose equivalent rate per unit concentration in air is therefore 1.1
10-11 Sv a-1 per Bq m-3.
2. Dose per unit release
(a) Natural tritium
67. Doses from natural tritium can be estimated from the few
tritium measurements in environmental materials that were carried
out before the contamination with fallout (or that had been
preserved from contamination). Activity concentrations of
continental surface waters were then found to be in the range from
0.2 to 0.9 Bq 1-1 [K1]. The production rate of natural tritium
being constant in time and relatively uniform on the global scale,
the concentrations in all the components of human intake (air,
water and food) of natural tritium are in steady-state equilibrium
with the concentrations in continental surface waters. Using the
specific activity approach, it is assumed that the specific
activity of natural tritium is the same in the continental surface
waters, in all the components of human intake and in the body. The
effective dose equivalent rate is thus found to range from
0.2 Bq 1-1 x 2.6 10-8 Sv a-1 per Bq 1-1 = 5.2 109 Sv a-1 to
0.9 Bq 1-1 x 2.6 10-8 Sv a-1 per Bq 1-1 = 2.3 10-8 Sv a-1, being
therefore of the order of 10-8 Sv a-1. The effective dose equivalent
commitment per unit release would then be
10-8 Sv a-1
--------------- ca. 1.4 10-25 Sv per Bq
7.2 1016 Bq a-1
Taking the world's population to be 4 109 people, the global
collective effective dose equivalent commitment per unit of
activity produced is about 5 10-16 man Sv per Bq.
(b) Nuclear explosions
68. The doses from fallout tritium can be estimated in the same
way as those from natural tritium. On the basis of the variation
with time of the tritium activity concentration in surface waters
[B5] and of the latitudinal distribution of the fallout deposition
[S1], UNSCEAR [U1] estimated the effective dose equivalent
commitments to the populations of the northern and southern
hemispheres to be 2 10-5 and 2 10-6 Sv respectively.
69. The effective dose equivalent commitment from fallout tritium
was also estimated indirectly, using the relationship obtained for
natural tritium between the production rate and the dose rate
W
Hc = gammao -
B
where Hc is the effective dose equivalent commitment (Sv) from
production of fallout tritium in a given hemisphere; gammao is the
effective dose equivalent rate from natural tritium (gammao = 10-8
Sv a-1); W is the activity of tritium released by nuclear
explosions (1.5 1020 Bq in the northern hemisphere and 0.2 1020 Bq
in the southern hemisphere); and B is the natural rate of
production (3.6 1016 Bq a-1 in each hemisphere). The effective
dose equivalent commitments thus derived are 4.2 10-5 Sv for the
population of the northern hemisphere and 5.6 10-6 Sv for the
population of the southern hemisphere. These results are higher
than the direct estimates by a factor of 2 to 3. The global
collective effective dose equivalent commitments per unit activity
released are estimated to be 9 10-16 and 4 10-16 man Sv Bq-1 using
the latter and the former method, respectively. UNSCEAR [U1] used
an intermediate value of 8 10-16 man Sv per Bq.
(c) Nuclear installations
70. While the production of natural and fallout tritium brings
about a relatively uniform contamination of the whole biosphere,
the releases from nuclear installations occur at discrete points on
the earth's surface giving a highly heterogenous spatial
distribution of concentrations.
71. UNSCEAR'S practice is to divide the collective doses into two
components: the local and regional collective doses, which are due
to the first passage of the plume, over distances of 100 to 1000 km
from the point of release, and the global collective doses, which
arise from the mixing of tritium in the whole biosphere. As the
doses per unit concentration of tritium in air are much higher for
HTO than for HT, tritiated water will be the only compound
considered in the estimate of the local and regional collective
doses.
(i) Local and regional collective dose
72. A distinction is made between airborne and liquid effluents.
Tritium present in airborne effluents can contribute to the local
and regional collective doses through inhalation, absorption
through skin and ingestion. As the contribution from the ingestion
pathway is quite variable from site to site owing to differences in
local hydrology and water usage, UNSCEAR [U1] has not taken this
pathway into account in its assessment of the local and regional
collective doses. Assuming an atmospheric dispersion factor of 5
10-7 s m-3 at 1 km from the release and a reduction in
concentration inversely proportional to the 1.5 power of the
distance expressed in kilometres, the local collective dose per
unit activity released can be assessed by integration over the
local area. Integrating from 1 to 100 km for a population density
of 100 km-2, UNSCEAR [U1] estimated the local collective dose from
airborne tritium per unit activity released to be about 5 10-17 man
Sv per Bq.
73. The collective dose commitment from the input of 3H to water
bodies, normalized per unit activity released, can be estimated
[U1], using the expression
c sigmak Nk Ik fk phi
S = -------------------
1 V(lambda + 1/tau)
where V is the volume of the receiving waters, tau is the turnover
time of receiving waters, lambda is the decay constant of 3H, Nk is
the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
74. The quantity V(lambda + 1/tau) is the infinite time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentrations in water and in fish
will be high, but the population which can be served with drinking
water or by fish consumption will limited. For inputs into larger
volumes of water, the concentrations will be smaller, but the
populations involved will be correspondingly larger. It is
reasonable, therefore, to assume as a first approximation that the
quantities Nk/V are relatively constant, independent of V. The
values for these quantities as well as values for the other
parameters of the above expression have been extensively discussed
[U1].
75. A summary of the values used in the assessment, based on
UNSCEAR [U1], and the evaluation of the collective dose commitments
for a release of 1 Bq of 3H in liquid effluents are given in Table
II.4.
(ii) Global collective dose
76. For HTO releases, the global collective effective dose
equivalent commitment established for fallout tritium (8 10-16 man
Sv per Bq) can be applied without change. With respect to HT
releases, if it is assumed that the conversion to HTO takes place
on average 5 years after the discharges, the global collective
effective dose equivalent commitment is estimated to be
-0.693
8 10-16 e x 5/12.3 = 6 10-16 man Sv per Bq.
Table II.4 Collective dose factors for 3H in liquid effluents
---------------------------------------------------------------------------
Fresh water Salt water
---------------------------------------------------------------------------
Activity released, A 1 Bq 1 Bq
Turnover time of receiving water, 10 a 1.0 a
Sediment removal correction factor, s 1.0 1.0
Time integral of activity in water,
As
W = ------------ 6.36 Bq a 0.946 Bq a
1/tau+lambda
Water utilization, V/N 3 107 1/man 3 109 1/man
---------------------------------------------------------------------------
FRESHWATER PATHWAYS
1 Drinking water
Treatment removal factor, f1 1.0
Consumption, I1 438 1 a-1
Collective dose commitment
c NI
S1 = W f1 (--)1D 2 10-15 man Sv
V
Table II.4 (contd.)
---------------------------------------------------------------------------
Fresh water Salt water
---------------------------------------------------------------------------
2. Fish
Concentration factor, f2 1.0
Consumption, I2 1 kg a-1
Collective dose commitment
c NI
S2 = W f2 (--)2D 5 10-18 man Sv
V
SALT WATER PATHWAYS
3. Fish
Concentration factor, f3 1.0
Consumption, I3 6 kg a-1
Collective dose commitment
c NI
S3 = W f3 (--)3D 4 10-20 man Sv
V
4. Shellfish
Concentration factor, f4 1.0
Consumption, I4 1 kg a-1
Collective dose commitment
c NI
S4 = W f4 (--)4D 7 10-21 man Sv
V
---------------------------------------------------------------------------
(iii) Summary of collective dose commitments per unit activity released
77. Table II.5 summarizes the values obtained above for the
collective effective dose equivalent commitments per unit of 3H
activity released. With respect to the local and regional
component due to industrial releases, the largest collective
effective dose equivalent commitment per unit activity released is
obtained for a river discharge and the smallest for a sea discharge
while an intermediate value is found for the airborne discharge.
Table II.5 Summary of collective effective dose
equivalent commitments per unit tritium activity released
(man Sv per Bq)
---------------------------------------------------------
Origin Local and regional Global
component component
---------------------------------------------------------
Natural 5 10-16
Nuclear tests 8 10-16
Industry
Airborne discharge 5 10-17 (HTO) ) 8 10-16 (HTO)
River discharge 2 10-15 )
Sea charge 5 10-20 ) 6 10-16 (HT)
---------------------------------------------------------
F. REFERENCES
B1 Bennett, B.G. Environmental aspects of americium. EML-348
(1978).
B2 Bowen, V.T. and W. Roether. Vertical distributions of
strontium-90, caesium-137 and tritium near 45° north in the
Atlantic. J. Geophys. Res. 78: 6277-6285 (1973).
B3 Butler, H.L. and J.H. LeRoy. Observations of biological half-
life of tritium. Health Phys. 11: 283-285 (1965).
B4 Bennett, B.G. Environmental tritium and the dose to man.
p. 1047-1053 in Proceedings of the Third International
Congress of IRPA. CONF-730907 (1973).
B5 Bennett, B.G. Fallout tritium in the environment and the dose
commitments. HASL-268 (1973).
B6 Bogen, D.C., G.A. Welford and C.G. White. Tritium distribution
in man and his environment. p. 567-574 in Behaviour of Tritium
in the Environment. IAEA, Vienna, 1979.
B7 Burger, L.L. Distribution and reactions of tritiated hydrogen
and methane. p. 47-64 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
C1 Crowson, D.L. Man-made tritium. p. 23-27 in Tritium (A.A.
Moghissi and M.W. Carter, eds.). Messenger Graphics, Las
Vegas, Nevada, 1973.
C2 Comps, F. and R.J. Doda. Large-scale distribution of tritium
in a commercial product. p. 93-99 in Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
C3 Coyle, P.E. Laser fusion. Status, future and tritium control.
p. 139-153 in Behaviour of Tritium in the Environment. IAEA,
Vienna 1979.
F1 Fireman, E.L. Measurement of the (n,H3) cross section in
nitrogen and its relationship to the tritium produced in the
atmosphere. Phys. Rev. 91: 922-926 (1953).
F2 Flamm, E., R.E. Lingenfelter, J. F. MacDonald et al. Tritium
and helium-3 in solar flares and loss of helium from the
earth's atmosphere. Science 138: 48-49 (1962).
G1 Gratwohl, G. Erzeugung und Freisetzung von Tritium durch
Reaktoren und Wiederaufarbeitungsanlagen und die
voraussichtliche radiologische Belastung bis zum Jahr 2000.
Kernforschungszentrum Karlsruhe report KFF-Ext. 4/73-36 (1973).
H1 Häfele, W., J.P. Holdren, G. Kessler et al. Fusion and fast
breeder reactors. IIASA RR-77-8 (1976) (revised July 1977).
I1 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
I2 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP publication 23.
Pergamon Press, 1975.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP publication 30.
Annals of the ICRP 2: 3/4 (1979)
I4 International Atomic Energy Agency. Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
J1 Jacobs, D.G. Sources of tritium and its behaviour upon release
to the environment. AEC Critical Review Series. TID-24635
(1968).
K1 Kaufamn, S. and W.F. Libby. The natural distribution of
tritium. Phys. Rev. 93: 1337-1344 (1954).
K2 Kouts, H. and J. Long. Tritium production in nuclear reactors.
p. 38-45 in Tritium (A.A. Moghissi and M.W. Carter, eds.).
Messenger Graphics, Las Vegas, Nevada, 1973.
K3 Kahn, B., R.L. Blanchard, W.L. Brinck et al. Radiological
surveillance study at the Haddam Neck PWR nuclear power
station. EPA-520/374-007, Washington, 1974.
K4 Krejci, K. and A. Zeller, Jr. Tritium pollution in the Swiss
luminous compound industry. p. 65-77 in Behaviour of Tritium
in the Environment. IAEA, Vienna, 1979.
K5 Krejci, K. Discussion. p. 101 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
L1 Lal, D. and B. Peters. Cosmic ray produced radioactivity
on the earth. p. 551-612 in Encyclopaedia of Physics, Vol.
XLV1/2 on Cosmic Rays (K. Sitte, ed.). Springer-Verlag, New
York, 1967.
L2 Locante, J. and D.D. Malinowski. Tritium in pressurized water
reactors. p. 45-57 in Tritium (A.A. Moghissi and M.W. Carter,
eds.). Messenger Graphics, Las Vegas, Nevada, 1973.
L3 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European community. Discharge data 1974-1978. Radiological
aspects. Commission of the European Communities. V/4116/80
(1980).
L4 Lambert, B.E., H.B.A. Sharpe and K.B. Dawson. Am. Ind. Hyg.
Assoc. J. 32: 682 (1971).
M1 Miskell, J.A. Production of tritium by nuclear weapons. p. 79-
85 in Tritium (A.A. Moghissi and M.W. Carter, eds.).
Messenger Graphics, Phoenix and Las Vegas, 1973.
M2 Michel, L. Tritium inventories of the world oceans and their
implications. Nature 263: 103-106 (1976).
M3 Mason, A.S. and H.G. Ostlund. Atmospheric HT and HTO: V.
Distribution and large-scale circulation. p. 3-16 in Behaviour
of Tritium in the Environment. IAEA, Vienna, 1979.
M4 Murphy, C.E. Jr. and M.M. Pendergast. Environmental transport
and cycling of tritium in the vicinity of atmospheric releases.
p. 361-372 in Behaviour of Tritium in the Environment. IAEA,
Vienna, 1979.
M5 Minder, W. Strahlentherapie. 137: 700 (1969).
M6 Moghissi, A.A., M.W. Carter and E.W. Bretthauer. Further
studies on the long-term evaluation of the biological half-life
of tritium. Health Phys. 23: 805-806 (1972).
M7 Moghissi, A.A. and M.W. Carter, eds. Tritium. Messenger
Graphics, Las Vegas, Nevada, 1973.
N1 National Council on Radiation Protection and Measurements.
Tritium in the environment. NCRP No. 62 (1979).
N2 National Council on Radiation Protection and Measurements.
Tritium and other radionuclide labelled organic compounds
incorporated in genetic material. NCRP No. 63 (1979).
O1 Ostlund, H.G. and R.A. Fine. Oceanic distribution and
transport of tritium. p. 303-314 in Behaviour of Tritium in
the Environment. IAEA, Vienna, 1979.
S1 Schell, W.R., S. Sauzay and B.R. Payne. World distribution of
environmental tritium. p. 374-385 in Physical Behaviour of
Radioactive Contaminants in the Atmosphere. IAEA, Vienna,
1974.
S2 Smith, J.M. and R.S. Gilbert. Tritium experience in boiling
water reactors. p. 57-68 in Tritium (A.A. Moghissi and M.W.
Carter, eds.). Messenger Graphics, Las Vegas, Nevada, 1973.
S3 Sanders, S.M. Hr. and W. C. Reinig. Assessment of tritium in
man. p. 534-542 in Diagnosis and Treatment of Deposited
Radionuclides (H.A. Kornberg and W.D. Norwood, eds.). Excerpta
Medica Foundation, Amsterdam, 1968.
T1 Trevorrow, L.E., B.J. Kullen, R.L. Jarry et al. Tritium and
noble gas fission products in the nuclear fuel cycle. I.
Reactors. ANL-8102 (1974).
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication no. E.77.IX.I. New York,
1977.
W1 Wehner, G. Discharges of tritium to the environment from
unrestricted use of consumer products containing this
radionuclide. p. 79-91 in Behaviour of Tritium in the
Environment. IAEA, Vienna, 1979.
W2 Woodard, H.Q. The biological effects of tritium. United
States Atomic Energy Commission. HASL-229 (1970).
W3 Wylie, K. F., W. A. Bigler and G.R. Grove. Biological half-
life of tritium. Health Phys. 9: 911-914 (1963).
III. CARBON-14
A. INTRODUCTION
78. Carbon-14 has always been present on the earth. It is
produced by cosmic ray interactions in the atmosphere. This
nuclide is a pure beta-emitter, with a half-life of 5730 years, a
maximum energy of 185 keV and an average energy of 49.47 keV [N1].
79. Carbon is one of the elements that are essential to all forms
of life and is involved in most biological and geochemical
processes on the earth. Associated with the stable isotopes of
carbon (12C and about 1.1%13C), there is a very small amount of 14C
formed in the atmosphere and which has subsequently entered in the
carbon cycle. The specific activity of biological carbon, as
measured in wood samples grown in the nineteenth century, was 0.227
± 0.001 Bq per gram of carbon [T1], corresponding to an atmospheric
inventory of 1.4 1017 Bq. During the present century the specific
activity of 14C has decreased due to the diluting effect of
releases into the atmosphere of carbon dioxide from the combustion
of fossil fuels. This effect (the Suess effect) accounts for a
reduction of a few percent.
80. In addition to its natural production, carbon-14 is also
produced by the detonation of nuclear explosives and by the
operation of nuclear reactors. The assessment of the collective
dose commitments from the releases of man-made carbon-14 is
facilitated by knowledge of the production rate of natural
carbon-14.
B. SOURCES
1. Natural carbon-14
81. Carbon-14 is produced by the action of cosmic ray neutrons on
nitrogen atoms, both in the stratosphere and in the upper
troposphere. UNSCEAR [U3] has estimated the natural production
rate to be about 1015 Bq per year, a value which has been derived
from assessments of the natural 14C inventory. The production rate
has also been estimated directly from assessments of cosmic ray
neutrons and the values obtained by different authors range from 1
to 1.4 1015 Bq per year [U3]. Considering the uncertainties
involved in determining both the direct production rate and also
the total 14C inventory of the earth, the estimates are in
reasonable agreement.
2. Nuclear explosions
82. Carbon-14 is formed in nuclear explosions through the capture
of excess neutrons by atmospheric nitrogen. After large
atmospheric nuclear explosions, most of the 14C is transported into
the stratosphere, from where it equilibrates with the troposphere
with a half-time of 1 to 2 years [U3].
83. The inventory of 14C from nuclear explosions has been
estimated from measurements of excess specific activity in the
troposphere and in the surface ocean waters, and models
representing the exchange of 14C between the atmosphere, the
biosphere and the ocean. UNSCEAR [U3] has estimated that nuclear
explosions up to 1972 have injected into the atmosphere 2.1 1017 Bq,
while subsequent injections have increased this amount by about 1%.
84. For the past pattern of atmospheric nuclear explosions, the
production mentioned above corresponds to about 3.7 1014 Bq per
megaton. This value, however, is not representative of any given
nuclear explosion, because the production of 14C will depend on the
type of nuclear device exploded and also on whether the explosion
took place on the surface of the earth or high in the atmosphere.
A "surface" test will produce approximately 50% of the 14C that
would be produced by the same device in an "air" test, because
about one half of the escaping neutrons will be captured in the
soil or water rather than in the atmosphere.
3. Nuclear fuel cycle
85. Carbon-14 is produced in nuclear reactors and is released to
the environment at the reactor itself or at reprocessing plants
where spent fuel is reprocessed. Only recently has attention been
given to the production and release of this radionuclide at nuclear
fuel cycle installations.
(a) Nuclear reactors
86. The production of carbon-14 in nuclear power reactors is due
to several nuclear reactions in the fuel, core construction
materials and moderator. Figure III.I summarizes the relevant
reactions.
 87. Production rates depend upon the neutron flux, the shape of
the respective neutron spectra and the resulting effective cross
sections, on the amount of the target elements present in different
reactor components and on the abundance of the target isotopes in
the target elements. The target elements are uranium, nitrogen,
oxygen, and also carbon in the case of graphite moderated reactors.
Nitrogen is present as an impurity in the fuel, as dissolved gas in
the coolant, as nitrogen compounds sometimes used for pH control in
the coolant, and as an impurity in structural materials. Oxygen is
present in water moderators and coolants, in CO2 coolants, and in
oxide fuels (e.g., UO2).
88. The place of origin of 14C within a nuclear reactor has a
strong influence on the discharge pathway. One can basically
distinguish between three locations of 14C generation, namely, 14C
in the fuel, 14C in structural materials of the core (and solid
moderator, if applicable) and 14C in the reactor coolant (and
liquid moderator, if applicable).
89. The 14C produced in liquid or gaseous coolants will be present
in different chemical compounds (CO2, CO, methane), depending on
the chemical environment. Under the influence of intensive
radiation fields several chemical reactions may occur, influencing
the chemical form of carbon-14. The compounds in the coolant are
released mainly together with off-gas and waste water from the
coolant purification and treatment system. Part of the carbon-14
also leaks from the primary coolant circuit into the plant
ventilation system and is released with ventilation air.
90. Significant reactions for the production of 14C in light water
reactors (LWR) are: (n) reactions with 17O present in the oxide
fuel and in the moderator; (n, p) reactions with 14N present in the
fuel as impurities; and ternary fissions. Ternary fission
production per unit electrical energy generated is practically
independent of reactor design, while the normalized production of
14C by the other reactions depends on the enrichment of the fuel,
the relative masses of the fuel and moderator, the concentration of
nitrogen impurities in the fuel and the temperature of the fuel and
moderator.
91. In boiling water reactors (BWR), the gaseous 14C is
transported with the steam until it arrives at the turbine
condenser. There the gases are continuously withdrawn over a
catalytic recombiner to burn the hydrogen and oxygen produced by
radiolysis of the primary water. Measurements have shown that one
half or more of the total 14C produced in the coolant will be
discharged in the form of CO2 together with the filtered gases from
the turbine condenser. There are other pathways of release of 14C,
mainly caused by leakage from the primary circuit into the reactor
building and the turbine hall. These releases are also mainly in
the form of CO2. A part of the 14C remains dissolved in the
primary water purification and treatment systems, causing smaller
sources of release, for example in the auxiliary building and
finally in the waste water system.
92. The primary circuit water of a pressurized water reactor (PWR)
contains hydrogen in excess to recombine the oxygen produced by
radiolysis. Under such reducing conditions compounds like methane
will be formed. Therefore, contrary to the BWR, a PWR will release
most of the 14C bound in hydrocarbons. The main release pathways
for gaseous compounds of 14C in PWRs are leakages of the primary
water circuit into the containment air and the degasification of
the primary water. The escaping or withdrawn gases may be stored
in decay tanks prior to release, and the gaseous 14C compounds can
be oxidized to CO2 or released through charcoal beds. Leakages may
also arise in the auxiliary building from the primary water
purification and treatment systems by way of degasing. Also, a part
of the 14C compounds stays dissolved in the water and is released
at the different steps of the waste water treatment.
93. The total environmental release of carbon-14 at the reactor,
expressed as a fraction of the production rate, is on the average
about 50% in BWRs and 30% in PWRs, but the value is quite variable,
as has been shown by several recent monitoring programmes [R1, L1].
UNSCEAR summarized the estimates of production in LWRs from several
authors, the values being in the range 0.5 to 1.9 109 Bq per
MW(e)a, and also derived an independent value of about 0.7 109 Bq
per MW(e)a [U3].
94. Carbon-14 is generated in heavy water reactors (HWR) through
reactions similar to those described for LWRs. Owing mainly to the
large moderator mass, the production rate of 14C in HWRs is
expected to be considerably larger than in LWRs [U3]. The
production rate in pressure vessel reactors is estimated to be 1.7
1010 Bq per MW(e)a, with 90% generated in the moderator. The
production of 14C in CANDU reactors is estimated to be 1.6 1010 Bq
per MW(e)a, 95% being produced in the moderator.
95. In gas-cooled graphite-moderated reactors (GCR), the major
source of 14C production is the graphite moderator, due to 13C(n,
gamma)14C reaction and also to the 14N(n, p)14C reaction based
on the incorporated nitrogen impurity. Production rates have been
estimated to be about 0.7 1010 Bq per MW(e)a in Magnox reactors and
1.1 1010 Bq per MW(e)a in advanced gas-cooled reactors (AGR) [U3].
Production of 14C in the carbon dioxide coolant, mainly from
activation of nitrogen impurities and from the 17O(n, alpha)14C
reaction, is a smaller source estimated to be about 108 Bq per
MW(e)a for Magnox reactors and 4 108 Bq per MW(e)a for AGRs.
96. Carbon-14 discharges from Magnox reactors and AGRs result from
coolant leakage and include 14C released to the coolant from
corrosion of the moderator. The fraction released at the reactor
is about 3% in Magnox reactors and about 6% in AGRs, of the total
production rate of 14C in these reactors [U3].
(b) Fuel reprocessing plants
97. While the 14C produced in the reactor coolant and moderator
has a potential for immediate release at the nuclear reactor, the
14C produced in the fuel will be released only later during nuclear
fuel reprocessing. Depending on reprocessing plant operation
characteristics the release may be continuous or discontinuous.
There are few measurements of 14C releases from reprocessing
installations [S1], but it seems reasonable to assume that almost
all the inventory of the fuel elements is released during the
chemical dissolution of the fuel. In the case of the Purex process
the 14C is released in the form of CO2.
(c) Summary
98. A very rough estimate can be made of the total production and
release of 14C from nuclear fuel cycle installations, based on the
average values given above. Installed nuclear capacity worldwide
in 1980 was 1.25 105 GW(e) [I2]. Assuming an average load factor
for reactor operation of 0.6, the energy produced was 7.5 104
GW(e)a. Global production and release of 14C from reactor sites
are thus estimated to be about 1.4 1014 Bq and 6 1013 Bq,
respectively. The estimated discharges by reactor types are given
in Table III.1. There are no estimates of production and release
from other reactor types representing 10% of the total installed
capacity. The difference between production and reactor discharge
estimates will largely represent the release from reprocessing
plants, to the extent that the fuel is eventually reprocessed.
Table III.1 Estimated global discharge of carbon-14
from nuclear power stations in 1980
------------------------------------------------------------------------
Reactor Reactor Capacity Production rate Release Estimated
type number [MW(e)] [Bq per MW(e)] fraction carbon-14
(%) discharge (Bq)
------------------------------------------------------------------------
PWR 96 64239 7 108 30 8 1012
BWR 62 35170 7 108 50 7 1012
HWR 14 5963 1.6 1010 70 4 1013
GCR 36 7086 9 109 5 2 1012
Other 33 12527 - - -
------------------------------------------------------------------------
Total 241 124985 6 1013
------------------------------------------------------------------------
C. BEHAVIOUR IN THE ENVIRONMENT
99. Carbon-14 is present in atmospheric carbon dioxide, in the
biosphere, and in the bicarbonates dissolved in the ocean. The
specific activity of natural 14C in the terrestrial biosphere, as
measured in wood grown in the nineteenth century, was 0.227 ± 0.001
Bq per gram of carbon. The Suess effect, accounting for a few
percent decrease of specific activity at present, could reach a
figure of the order of 20% in the year 2000 [U2], but is of little
importance in the long range, when fossil fuel resources are
exhausted.
100. Leaving aside the Suess effect, it has been suggested,
however, that the present-day inventory does not correspond to the
equilibrium value, but is increasing. In fact, measurements of
wood samples of known age show that only cyclic variations of
atmospheric 14C, amounting to a few percent, have occurred in the
past 6000 years [U2]. Two types of variations have been
recognized: one, with a time scale of the order of 100 years, has
been explained by the solar wind modulation of the cosmic-ray flux
density; the other, with a time constant of more than 1000 years,
may largely be due to a variation of the geomagnetic shielding of
the earth.
101. Contrary to the case of natural carbon-14, the levels of man-
made carbon-14 are not at steady state in the different
compartments of the environment. Due to the very long mean life of
carbon-14, continuing practices are not expected to last long
enough to allow the environmental levels to reach the steady state.
The predictions of the time-evolution of 14C levels in the
atmosphere, biosphere and ocean after a release into the
environment require, therefore, the use of compartment models.
102. Many models describing the dispersion of released 14C, and
the subsequent exchange between the different compartments involved
in the carbon cycle, have been proposed [C1, P1, N2, Y1, N3].
UNSCEAR [U3] also developed a dynamic model for the assessment of
doses from 14C released by nuclear explosions. This model includes
compartments for the atmosphere and short-term biosphere, the
terrestrial biosphere, the surface ocean and the deep ocean, and
represents the thermocline layer in the ocean as a thick diffusion
barrier.
D. TRANSFER TO MAN
103. Carbon-14 released to the environment enters the carbon
cycle, giving rise eventually to increased levels in man. From
measurements of fallout carbon-14, it was noted that the specific
activity in human tissue comes into equilibrium with that of
atmospheric CO2 with a delay time of about 1.4 years [N5].
104. Intake of carbon by man is primarily from diet. Ingestion
intake is of the order of 300 g d-1 with nearly complete
absorption, whereas inhalation intake is about 3 g d-1 with only 1%
retained in the body [U3]. The total carbon content of the body is
1.6 104 [I1]. The quotient of this with the intake rate gives an
estimated mean residence time of carbon in the human body of 53
days.
105. Man comes, therefore, into fairly rapid equilibrium with
carbon-14 in his immediate environment. It is generally sufficient
in carbon-14 dose calculations to adopt a steady-state model which
assumes that the specific activity of carbon in tissues is in
equilibrium with that in air and in the diet.
E. DOSIMETRY
1. Dose per unit intake
106. An intake of carbon-14 at a specific concentration of 0.23 Bq
per gram of carbon, corresponding to the present value for natural
carbon-14, gives rise to the following absorbed dose rate averaged
over the whole body
Bq Gy g 0.049 MeV/Bq s
0.23 -- 1.6 10-10 ---- ----------------
gc MeV 7 104g
3.15 107 s/a 1.6 104 gc = 13 µGy a-1
The dose rates in individual tissues depend on their carbon
concentrations. The carbon content per unit mass averages 23% for
the whole body, but ranges from 9% in gonads and 10% in lungs to
41% in red bone marrow and 67% in adipose tissue [I1]. The annual
absorbed doses are 5 µGy in gonads, 6 µGy in lungs, 20 µGy in bone-
lining cells and 22 µGy in red bone marrow [U3]. The tissue-
weighted annual effective dose equivalent from natural carbon-14 is
12 µSv.
107. This dose is due almost entirely to ingestion intake of
carbon-14. If the carbon intake rate is 300 g d-1 at the specific
activity of 0.23 Bq g-1, the intake rate of 14C is 69 Bq d-1. The
effective dose equivalent per unit ingestion intake of 14C is
12 10-6 Sv/a 1 a
------------ ------- = 5.2 10-10 Gy Bq-1
69 Bq/d 365 d
The dose factor for inhalation intake is less by a factor of 10-2,
since absorption into the body is that much less by this pathway.
2. Dose per unit release
108. The doses given above for natural carbon-14 correspond to
the annual global production of 1015 Bq. This production is
essentially constant in time and uniform over the world. Therefore,
equilibrium has become established. The effective dose equivalent
commitment per unit release is
12 10-6 Sv/a
------------ = 1.2 10-20 Sv Bq-1
1015 Bq/a
The collective dose equivalent rate from natural carbon-14 to the
world population of 4 109 people is 4.8 104 man Sv a-1.
109. The assessment of the dose commitment from a given release of
man-made carbon-14 is carried out by direct analogy with natural
carbon-14. Once the released carbon-14 enters the global carbon
cycle, the effective dose equivalent commitment per unit release is
1.2 10-20 Sv Bq-1.
110. It is difficult to assess with precision the collective dose
commitment per unit release of carbon-14, because the projected
increase in the world population is very uncertain. Assuming that
it will attain an equilibrium value of 1010 persons, in a time
short compared with the mean effective life of 14C [U3], the
collective effective dose equivalent commitment per unit released
is approximately 1.2 10-10 man Sv per Bq.
111. In order to calculate the complete collective dose commitment
[U3] required for assessments of maximum future mean annual doses
from a continuing but finite practice releasing 14C, it is
necessary to use dynamic models predicting the time evolution of
environmental levels. Assuming that power production by nuclear
fission will last for a few hundred years (for example, 500 years),
the incomplete collective dose commitment can be calculated using
the model with diffusion barrier already mentioned. The incomplete
collective dose commitment, integrated over 500 years, is about 2.3
10-11 man Sv per Bq released. This value is somewhat higher than a
value of about 1.4 10-11 man Sv per Bq which can be deduced from a
recent assessment of the environmental significance of 14C [N3],
but in view of the uncertainties involved, the difference is
probably insignificant.
112. The contribution of local and regional exposures to the
collective dose commitment is very small, of the order of a
percent, and can be neglected [N3]. The assessment of individual
doses at some selected locations, however, is necessary for
radiation protection purposes. Its calculations can be carried out
by the use of specific activity methods. One simple model assumes
that the specific activity of 14C in air is equal to that in the
body. A more sophisticated calculation assumes that the specific
activity in the vegetation at the location of interest is equal to
that of air. The dose can then be assessed from knowledge of the
relative proportion of contaminated food in the diet. Both methods
require the use of micrometeorological models to assess
quantitatively the dispersion of 14C from the release point to the
locations of interest. Some publications [U4, N4, C2], present
improvements to the classical formulations describing the local
atmospheric dispersion.
F. REFERENCES
C1 Craig, H. The natural distribution of radiocarbon and the
exchange time of carbon dioxide between atmosphere and sea.
Tellus 9: 1-17 (1957).
C2 Clarke, R. A model for short and medium range dispersion of
radionuclides released to the atmosphere. A first report of a
working group on atmospheric dispersion. NRPB-R91 (1979).
I1 International Commission on Radiological Protection. Report of
the task group on reference man. International Commission on
Radiological Protection publication 23 (1975).
I2 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
L1 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European community: discharge data 1962-76. Radiological
aspects. Commission of the European Communities. V/4604/78-EN
(1978).
N1 National Council on Radiation Protection and Measurements. A
handbook of radioactivity measurements procedures. National
Council on Radiation Protection report No. 58 (1978).
N2 Nydal, R. Further investigation on the transfer of radiocarbon
in nature. J. Geophys. Res. 73: 3617-3635 (1968).
N3 Nuclear Energy Agency, OECD. Radiological significance and
management of H-3, C-14, Kr-85 and I-129 arising from the
nuclear fuel cycle. Report by an NEA group of experts.
OECD/NEA (1980).
N4 NRPB and CEA. Methodology for evaluation of radiological
consequences of radioactive effluents released in normal
operations. Commission of European Communities. V/3865/79
(1979).
N5 Nydal, R., K. Lovseth and O. Syrstad. Bomb 14-C in the human
population. Nature 232: 418-421 (1971).
P1 Plesset, M. and A. Latter. Transient effects in the
distribution of carbon-14 in nature. Proceeding of the
National Academy of Sciences 46: 232-241 (1960).
R1 Riedel, H. and P. Gesewsky. Zweiter Bericht über Messungen zur
Emission von Kohlenstoff-14 mit der Abluft aus Kernkraftwerken
mit Leichtwasserreaktor in der Bundesrepublik Deutschland.
Bundesgesundheitsamt report STH-13/77 (1978).
S1 Schuettelkopf, H. and G. Herrman. 14-CO2 Emissionen aus wer
Wiederaufarbeitungsanlage Karlsruhe. p. 189 in Report for the
Commission of the European Communities. V/2266/78-D (1978).
T1 Telegadas, K. The seasonal atmospheric distribution and
inventories of excess carbon-14 from March 1955 to July 1969.
HASL-243 (1971).
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiaton to the General
Assembly, with annexes. Volume I: Levels, Volume II: Effects.
United Nations sales publication No. E.72.IX.17 and 18. New
York, 1972.
U3 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
U4 U.S. Nuclear Regulatory Commission. Regulatory Guide 1.111
(1977).
Y1 Young, J. and A. Fairhall. Radiocarbon from nuclear weapons
test. J. Geophys. Res. 73: 1185-1200 (1968).
IV. KRYPTON-85
A. INTRODUCTION
113. Krypton is element number 36 in the periodic table. It
belongs to the group of inert gases together with helium, neon,
argon, xenon and radon. It occurs naturally in the atmosphere to
an estimated extent of 1 to 2 10-6 by volume.
114. The naturally occurring stable krypton isotopes and their
atom percentage abundances are: 78Kr (0.35%), 80Kr (2.27%), 82Kr
(11.56%), 83Kr (11.55%), 84Kr (56.9%), 86Kr (17.37%) [N1]. The
radioactive isotopes of krypton include mass numbers of 74-77, 79,
79m, 81, 81m, 85, 85m, 87-95 and 97. Some of these occur naturally
in low trace amounts as a result of cosmic ray induced reactions
with stable krypton isotopes and by spontaneous fission of natural
uranium.
115. The radioactive isotope 85Kr is produced in nuclear fission.
With a half-life of 10.7 years, it can become widely dispersed in
the atmosphere following release. The average fission yields
differ by about a factor of 2 for 239Pu and 235U, being about 0.6
and 1.3 atoms per 100 fissions, respectively (Table IV.1).
Table IV.1 Fission yields of
krypton-85 [C2]
-----------------------------
Fission yield (%)
Nuclide thermal fast
-----------------------------
232Th 4.14
233U 2.28 2.12
235U 1.32 1.33
238U 0.74
239Pu 0.558 0.62
-----------------------------
116. The decay scheme of 85Kr is presented in Figure IV.I. Two
beta particles and a single gamma photon are emitted, along with
several low-energy conversion electrons and x-rays.
117. Being chemically inert, krypton and other inert gases are not
usually involved in biological processes. They are, however,
dissolved in body fluids and tissues following inhalation. Krypton
is characterized by low blood solubility, high lipid solubility and
rapid diffusion in tissue [K1]. The biological involvement of
inert gases has been noted in numerous studies [K1].
87. Production rates depend upon the neutron flux, the shape of
the respective neutron spectra and the resulting effective cross
sections, on the amount of the target elements present in different
reactor components and on the abundance of the target isotopes in
the target elements. The target elements are uranium, nitrogen,
oxygen, and also carbon in the case of graphite moderated reactors.
Nitrogen is present as an impurity in the fuel, as dissolved gas in
the coolant, as nitrogen compounds sometimes used for pH control in
the coolant, and as an impurity in structural materials. Oxygen is
present in water moderators and coolants, in CO2 coolants, and in
oxide fuels (e.g., UO2).
88. The place of origin of 14C within a nuclear reactor has a
strong influence on the discharge pathway. One can basically
distinguish between three locations of 14C generation, namely, 14C
in the fuel, 14C in structural materials of the core (and solid
moderator, if applicable) and 14C in the reactor coolant (and
liquid moderator, if applicable).
89. The 14C produced in liquid or gaseous coolants will be present
in different chemical compounds (CO2, CO, methane), depending on
the chemical environment. Under the influence of intensive
radiation fields several chemical reactions may occur, influencing
the chemical form of carbon-14. The compounds in the coolant are
released mainly together with off-gas and waste water from the
coolant purification and treatment system. Part of the carbon-14
also leaks from the primary coolant circuit into the plant
ventilation system and is released with ventilation air.
90. Significant reactions for the production of 14C in light water
reactors (LWR) are: (n) reactions with 17O present in the oxide
fuel and in the moderator; (n, p) reactions with 14N present in the
fuel as impurities; and ternary fissions. Ternary fission
production per unit electrical energy generated is practically
independent of reactor design, while the normalized production of
14C by the other reactions depends on the enrichment of the fuel,
the relative masses of the fuel and moderator, the concentration of
nitrogen impurities in the fuel and the temperature of the fuel and
moderator.
91. In boiling water reactors (BWR), the gaseous 14C is
transported with the steam until it arrives at the turbine
condenser. There the gases are continuously withdrawn over a
catalytic recombiner to burn the hydrogen and oxygen produced by
radiolysis of the primary water. Measurements have shown that one
half or more of the total 14C produced in the coolant will be
discharged in the form of CO2 together with the filtered gases from
the turbine condenser. There are other pathways of release of 14C,
mainly caused by leakage from the primary circuit into the reactor
building and the turbine hall. These releases are also mainly in
the form of CO2. A part of the 14C remains dissolved in the
primary water purification and treatment systems, causing smaller
sources of release, for example in the auxiliary building and
finally in the waste water system.
92. The primary circuit water of a pressurized water reactor (PWR)
contains hydrogen in excess to recombine the oxygen produced by
radiolysis. Under such reducing conditions compounds like methane
will be formed. Therefore, contrary to the BWR, a PWR will release
most of the 14C bound in hydrocarbons. The main release pathways
for gaseous compounds of 14C in PWRs are leakages of the primary
water circuit into the containment air and the degasification of
the primary water. The escaping or withdrawn gases may be stored
in decay tanks prior to release, and the gaseous 14C compounds can
be oxidized to CO2 or released through charcoal beds. Leakages may
also arise in the auxiliary building from the primary water
purification and treatment systems by way of degasing. Also, a part
of the 14C compounds stays dissolved in the water and is released
at the different steps of the waste water treatment.
93. The total environmental release of carbon-14 at the reactor,
expressed as a fraction of the production rate, is on the average
about 50% in BWRs and 30% in PWRs, but the value is quite variable,
as has been shown by several recent monitoring programmes [R1, L1].
UNSCEAR summarized the estimates of production in LWRs from several
authors, the values being in the range 0.5 to 1.9 109 Bq per
MW(e)a, and also derived an independent value of about 0.7 109 Bq
per MW(e)a [U3].
94. Carbon-14 is generated in heavy water reactors (HWR) through
reactions similar to those described for LWRs. Owing mainly to the
large moderator mass, the production rate of 14C in HWRs is
expected to be considerably larger than in LWRs [U3]. The
production rate in pressure vessel reactors is estimated to be 1.7
1010 Bq per MW(e)a, with 90% generated in the moderator. The
production of 14C in CANDU reactors is estimated to be 1.6 1010 Bq
per MW(e)a, 95% being produced in the moderator.
95. In gas-cooled graphite-moderated reactors (GCR), the major
source of 14C production is the graphite moderator, due to 13C(n,
gamma)14C reaction and also to the 14N(n, p)14C reaction based
on the incorporated nitrogen impurity. Production rates have been
estimated to be about 0.7 1010 Bq per MW(e)a in Magnox reactors and
1.1 1010 Bq per MW(e)a in advanced gas-cooled reactors (AGR) [U3].
Production of 14C in the carbon dioxide coolant, mainly from
activation of nitrogen impurities and from the 17O(n, alpha)14C
reaction, is a smaller source estimated to be about 108 Bq per
MW(e)a for Magnox reactors and 4 108 Bq per MW(e)a for AGRs.
96. Carbon-14 discharges from Magnox reactors and AGRs result from
coolant leakage and include 14C released to the coolant from
corrosion of the moderator. The fraction released at the reactor
is about 3% in Magnox reactors and about 6% in AGRs, of the total
production rate of 14C in these reactors [U3].
(b) Fuel reprocessing plants
97. While the 14C produced in the reactor coolant and moderator
has a potential for immediate release at the nuclear reactor, the
14C produced in the fuel will be released only later during nuclear
fuel reprocessing. Depending on reprocessing plant operation
characteristics the release may be continuous or discontinuous.
There are few measurements of 14C releases from reprocessing
installations [S1], but it seems reasonable to assume that almost
all the inventory of the fuel elements is released during the
chemical dissolution of the fuel. In the case of the Purex process
the 14C is released in the form of CO2.
(c) Summary
98. A very rough estimate can be made of the total production and
release of 14C from nuclear fuel cycle installations, based on the
average values given above. Installed nuclear capacity worldwide
in 1980 was 1.25 105 GW(e) [I2]. Assuming an average load factor
for reactor operation of 0.6, the energy produced was 7.5 104
GW(e)a. Global production and release of 14C from reactor sites
are thus estimated to be about 1.4 1014 Bq and 6 1013 Bq,
respectively. The estimated discharges by reactor types are given
in Table III.1. There are no estimates of production and release
from other reactor types representing 10% of the total installed
capacity. The difference between production and reactor discharge
estimates will largely represent the release from reprocessing
plants, to the extent that the fuel is eventually reprocessed.
Table III.1 Estimated global discharge of carbon-14
from nuclear power stations in 1980
------------------------------------------------------------------------
Reactor Reactor Capacity Production rate Release Estimated
type number [MW(e)] [Bq per MW(e)] fraction carbon-14
(%) discharge (Bq)
------------------------------------------------------------------------
PWR 96 64239 7 108 30 8 1012
BWR 62 35170 7 108 50 7 1012
HWR 14 5963 1.6 1010 70 4 1013
GCR 36 7086 9 109 5 2 1012
Other 33 12527 - - -
------------------------------------------------------------------------
Total 241 124985 6 1013
------------------------------------------------------------------------
C. BEHAVIOUR IN THE ENVIRONMENT
99. Carbon-14 is present in atmospheric carbon dioxide, in the
biosphere, and in the bicarbonates dissolved in the ocean. The
specific activity of natural 14C in the terrestrial biosphere, as
measured in wood grown in the nineteenth century, was 0.227 ± 0.001
Bq per gram of carbon. The Suess effect, accounting for a few
percent decrease of specific activity at present, could reach a
figure of the order of 20% in the year 2000 [U2], but is of little
importance in the long range, when fossil fuel resources are
exhausted.
100. Leaving aside the Suess effect, it has been suggested,
however, that the present-day inventory does not correspond to the
equilibrium value, but is increasing. In fact, measurements of
wood samples of known age show that only cyclic variations of
atmospheric 14C, amounting to a few percent, have occurred in the
past 6000 years [U2]. Two types of variations have been
recognized: one, with a time scale of the order of 100 years, has
been explained by the solar wind modulation of the cosmic-ray flux
density; the other, with a time constant of more than 1000 years,
may largely be due to a variation of the geomagnetic shielding of
the earth.
101. Contrary to the case of natural carbon-14, the levels of man-
made carbon-14 are not at steady state in the different
compartments of the environment. Due to the very long mean life of
carbon-14, continuing practices are not expected to last long
enough to allow the environmental levels to reach the steady state.
The predictions of the time-evolution of 14C levels in the
atmosphere, biosphere and ocean after a release into the
environment require, therefore, the use of compartment models.
102. Many models describing the dispersion of released 14C, and
the subsequent exchange between the different compartments involved
in the carbon cycle, have been proposed [C1, P1, N2, Y1, N3].
UNSCEAR [U3] also developed a dynamic model for the assessment of
doses from 14C released by nuclear explosions. This model includes
compartments for the atmosphere and short-term biosphere, the
terrestrial biosphere, the surface ocean and the deep ocean, and
represents the thermocline layer in the ocean as a thick diffusion
barrier.
D. TRANSFER TO MAN
103. Carbon-14 released to the environment enters the carbon
cycle, giving rise eventually to increased levels in man. From
measurements of fallout carbon-14, it was noted that the specific
activity in human tissue comes into equilibrium with that of
atmospheric CO2 with a delay time of about 1.4 years [N5].
104. Intake of carbon by man is primarily from diet. Ingestion
intake is of the order of 300 g d-1 with nearly complete
absorption, whereas inhalation intake is about 3 g d-1 with only 1%
retained in the body [U3]. The total carbon content of the body is
1.6 104 [I1]. The quotient of this with the intake rate gives an
estimated mean residence time of carbon in the human body of 53
days.
105. Man comes, therefore, into fairly rapid equilibrium with
carbon-14 in his immediate environment. It is generally sufficient
in carbon-14 dose calculations to adopt a steady-state model which
assumes that the specific activity of carbon in tissues is in
equilibrium with that in air and in the diet.
E. DOSIMETRY
1. Dose per unit intake
106. An intake of carbon-14 at a specific concentration of 0.23 Bq
per gram of carbon, corresponding to the present value for natural
carbon-14, gives rise to the following absorbed dose rate averaged
over the whole body
Bq Gy g 0.049 MeV/Bq s
0.23 -- 1.6 10-10 ---- ----------------
gc MeV 7 104g
3.15 107 s/a 1.6 104 gc = 13 µGy a-1
The dose rates in individual tissues depend on their carbon
concentrations. The carbon content per unit mass averages 23% for
the whole body, but ranges from 9% in gonads and 10% in lungs to
41% in red bone marrow and 67% in adipose tissue [I1]. The annual
absorbed doses are 5 µGy in gonads, 6 µGy in lungs, 20 µGy in bone-
lining cells and 22 µGy in red bone marrow [U3]. The tissue-
weighted annual effective dose equivalent from natural carbon-14 is
12 µSv.
107. This dose is due almost entirely to ingestion intake of
carbon-14. If the carbon intake rate is 300 g d-1 at the specific
activity of 0.23 Bq g-1, the intake rate of 14C is 69 Bq d-1. The
effective dose equivalent per unit ingestion intake of 14C is
12 10-6 Sv/a 1 a
------------ ------- = 5.2 10-10 Gy Bq-1
69 Bq/d 365 d
The dose factor for inhalation intake is less by a factor of 10-2,
since absorption into the body is that much less by this pathway.
2. Dose per unit release
108. The doses given above for natural carbon-14 correspond to
the annual global production of 1015 Bq. This production is
essentially constant in time and uniform over the world. Therefore,
equilibrium has become established. The effective dose equivalent
commitment per unit release is
12 10-6 Sv/a
------------ = 1.2 10-20 Sv Bq-1
1015 Bq/a
The collective dose equivalent rate from natural carbon-14 to the
world population of 4 109 people is 4.8 104 man Sv a-1.
109. The assessment of the dose commitment from a given release of
man-made carbon-14 is carried out by direct analogy with natural
carbon-14. Once the released carbon-14 enters the global carbon
cycle, the effective dose equivalent commitment per unit release is
1.2 10-20 Sv Bq-1.
110. It is difficult to assess with precision the collective dose
commitment per unit release of carbon-14, because the projected
increase in the world population is very uncertain. Assuming that
it will attain an equilibrium value of 1010 persons, in a time
short compared with the mean effective life of 14C [U3], the
collective effective dose equivalent commitment per unit released
is approximately 1.2 10-10 man Sv per Bq.
111. In order to calculate the complete collective dose commitment
[U3] required for assessments of maximum future mean annual doses
from a continuing but finite practice releasing 14C, it is
necessary to use dynamic models predicting the time evolution of
environmental levels. Assuming that power production by nuclear
fission will last for a few hundred years (for example, 500 years),
the incomplete collective dose commitment can be calculated using
the model with diffusion barrier already mentioned. The incomplete
collective dose commitment, integrated over 500 years, is about 2.3
10-11 man Sv per Bq released. This value is somewhat higher than a
value of about 1.4 10-11 man Sv per Bq which can be deduced from a
recent assessment of the environmental significance of 14C [N3],
but in view of the uncertainties involved, the difference is
probably insignificant.
112. The contribution of local and regional exposures to the
collective dose commitment is very small, of the order of a
percent, and can be neglected [N3]. The assessment of individual
doses at some selected locations, however, is necessary for
radiation protection purposes. Its calculations can be carried out
by the use of specific activity methods. One simple model assumes
that the specific activity of 14C in air is equal to that in the
body. A more sophisticated calculation assumes that the specific
activity in the vegetation at the location of interest is equal to
that of air. The dose can then be assessed from knowledge of the
relative proportion of contaminated food in the diet. Both methods
require the use of micrometeorological models to assess
quantitatively the dispersion of 14C from the release point to the
locations of interest. Some publications [U4, N4, C2], present
improvements to the classical formulations describing the local
atmospheric dispersion.
F. REFERENCES
C1 Craig, H. The natural distribution of radiocarbon and the
exchange time of carbon dioxide between atmosphere and sea.
Tellus 9: 1-17 (1957).
C2 Clarke, R. A model for short and medium range dispersion of
radionuclides released to the atmosphere. A first report of a
working group on atmospheric dispersion. NRPB-R91 (1979).
I1 International Commission on Radiological Protection. Report of
the task group on reference man. International Commission on
Radiological Protection publication 23 (1975).
I2 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
L1 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European community: discharge data 1962-76. Radiological
aspects. Commission of the European Communities. V/4604/78-EN
(1978).
N1 National Council on Radiation Protection and Measurements. A
handbook of radioactivity measurements procedures. National
Council on Radiation Protection report No. 58 (1978).
N2 Nydal, R. Further investigation on the transfer of radiocarbon
in nature. J. Geophys. Res. 73: 3617-3635 (1968).
N3 Nuclear Energy Agency, OECD. Radiological significance and
management of H-3, C-14, Kr-85 and I-129 arising from the
nuclear fuel cycle. Report by an NEA group of experts.
OECD/NEA (1980).
N4 NRPB and CEA. Methodology for evaluation of radiological
consequences of radioactive effluents released in normal
operations. Commission of European Communities. V/3865/79
(1979).
N5 Nydal, R., K. Lovseth and O. Syrstad. Bomb 14-C in the human
population. Nature 232: 418-421 (1971).
P1 Plesset, M. and A. Latter. Transient effects in the
distribution of carbon-14 in nature. Proceeding of the
National Academy of Sciences 46: 232-241 (1960).
R1 Riedel, H. and P. Gesewsky. Zweiter Bericht über Messungen zur
Emission von Kohlenstoff-14 mit der Abluft aus Kernkraftwerken
mit Leichtwasserreaktor in der Bundesrepublik Deutschland.
Bundesgesundheitsamt report STH-13/77 (1978).
S1 Schuettelkopf, H. and G. Herrman. 14-CO2 Emissionen aus wer
Wiederaufarbeitungsanlage Karlsruhe. p. 189 in Report for the
Commission of the European Communities. V/2266/78-D (1978).
T1 Telegadas, K. The seasonal atmospheric distribution and
inventories of excess carbon-14 from March 1955 to July 1969.
HASL-243 (1971).
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiaton to the General
Assembly, with annexes. Volume I: Levels, Volume II: Effects.
United Nations sales publication No. E.72.IX.17 and 18. New
York, 1972.
U3 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
U4 U.S. Nuclear Regulatory Commission. Regulatory Guide 1.111
(1977).
Y1 Young, J. and A. Fairhall. Radiocarbon from nuclear weapons
test. J. Geophys. Res. 73: 1185-1200 (1968).
IV. KRYPTON-85
A. INTRODUCTION
113. Krypton is element number 36 in the periodic table. It
belongs to the group of inert gases together with helium, neon,
argon, xenon and radon. It occurs naturally in the atmosphere to
an estimated extent of 1 to 2 10-6 by volume.
114. The naturally occurring stable krypton isotopes and their
atom percentage abundances are: 78Kr (0.35%), 80Kr (2.27%), 82Kr
(11.56%), 83Kr (11.55%), 84Kr (56.9%), 86Kr (17.37%) [N1]. The
radioactive isotopes of krypton include mass numbers of 74-77, 79,
79m, 81, 81m, 85, 85m, 87-95 and 97. Some of these occur naturally
in low trace amounts as a result of cosmic ray induced reactions
with stable krypton isotopes and by spontaneous fission of natural
uranium.
115. The radioactive isotope 85Kr is produced in nuclear fission.
With a half-life of 10.7 years, it can become widely dispersed in
the atmosphere following release. The average fission yields
differ by about a factor of 2 for 239Pu and 235U, being about 0.6
and 1.3 atoms per 100 fissions, respectively (Table IV.1).
Table IV.1 Fission yields of
krypton-85 [C2]
-----------------------------
Fission yield (%)
Nuclide thermal fast
-----------------------------
232Th 4.14
233U 2.28 2.12
235U 1.32 1.33
238U 0.74
239Pu 0.558 0.62
-----------------------------
116. The decay scheme of 85Kr is presented in Figure IV.I. Two
beta particles and a single gamma photon are emitted, along with
several low-energy conversion electrons and x-rays.
117. Being chemically inert, krypton and other inert gases are not
usually involved in biological processes. They are, however,
dissolved in body fluids and tissues following inhalation. Krypton
is characterized by low blood solubility, high lipid solubility and
rapid diffusion in tissue [K1]. The biological involvement of
inert gases has been noted in numerous studies [K1].
 B. SOURCES
118. Krypton-85 is produced by cosmic ray interactions in the
atmosphere, in nuclear power reactors, and nuclear explosions. The
main release source is the dissolution step in the reprocessing of
nuclear fuel.
119. Concentrations of 85Kr in the atmosphere increased sharply
after 1955 due to the production and testing of nuclear weapons and
the development of the nuclear power industry. More recently the
input rates of 85Kr into air have decreased [H2]. There have been
reductions in plutonium production for military purposes and in
nuclear fuel reprocessing.
120. A review of 85Kr measurement data for 1950-77 has been
prepared by Rozanski [R1]. The most recent data indicate that
concentrations in air have stablized at about 0.6 Bq/m3 in the
northern hemisphere and 0.4 Bq/m3 in the southern hemisphere [R1].
The major sources are in the northern hemisphere, accounting for
the higher levels in that hemisphere.
1. Natural krypton-85
121. Krypton-85 is present in small amounts in the environment as
a result of spontaneous fission of natural uranium and interactions
of cosmic ray neutrons with atmospheric 84Kr. The steady state
environmental inventories of 85Kr from these sources have been
calculated: 7.4 1010 Bq in the upper 3 m of the total land and
water surface due to spontaneous fission of natural uranium, 3.7
1011 Bq in the atmosphere from cosmic ray production and 3.7 105 Bq
in the oceans from the atmospheric source [D1]. These estimates,
in comparison with the estimates of man-made sources of 85Kr to
follow, are negligible in contributing to the world's total 85Kr
inventory.
2. Nuclear explosions
122. Since 85Kr is produced during fission, it has been generated
by nuclear weapon tests. The total amount of 85Kr produced in
nuclear testing can be calculated from the ratio of 85Kr/90Sr
fission yield of 0.08, giving an activity ratio of 0.22 [C2].
Measurements of 90Sr activity have been reported and discussed in
the reports of UNSCEAR [U1-U7]. There have been 6 1017 Bq of 90Sr
produced in weapon testing through 1976 [U7], corresponding to
about 1.3 1017 Bq of 85Kr.
123. Another source of 85Kr associated with nuclear weapons is in
the production of plutonium in military reactors. The amount of
85Kr released from this source is estimated to be two times higher
than that from the weapon tests [D1]. Naval propulsion reactors
also contribute to the 85Kr inventory with an annual production in
the region of 1.1 to 1.9 1016 Bq [B1]. Including all sources, the
total amount of 85Kr produced in operations for military purposes
is still rather small in comparison to the prospective generation
of 85Kr by the nuclear power industry.
3. Nuclear fuel cycle
124. Krypton-85 is produced by fission in the fuel of nuclear
reactors and in very low trace amounts in the moderator or coolant,
due to contamination with fissile material. The rates of 85Kr
production are related to the type of fuel and degree of burn-up.
Production and emission rates may be conveniently normalized to
unit electrical energy generated (for power reactors) or to the
electrical energy generated by the reactors serviced (for fuel
reprocessing plants).
125. The amounts of 85Kr produced vary according to reactor type.
For thermal reactors, the range of estimated production is about
1.1 to 1.5 1013 Bq/MW(e)a. For FBRs the values are about 25%
smaller [E1, M1], for HTGRs 50% higher [B3]. A production rate of
1.4 1013 Bq/MW(e)a has been correlated with some measurements from
reprocessing plants [U7] and this value can be taken for general
evaluations.
126. An estimate of 85Kr annual generation from reactor operation
can be obtained from the installed capacity of nuclear reactors of
1.25 105 MW(e) worldwide in 1980 [I1], with the assumptions of 60%
utilization and average 85Kr generation rate of 1.4 1013 Bq/MW(e)a:
1.25 105 MW(e)a x 0.6 x 1.4 1013 Bq/MW(e)a = 1 1018 Bq/a
The actual release rate is less, since delays occur before
reprocessing and not all fuel is reprocessed.
127. Reported releases of 85Kr and other fission noble gases were
listed in the 1977 report of UNSCEAR [U7]. There are large
differences in the release values of the various plants. Although
the relevant data are not very extensive, there are indications of
improved retention of 85Kr at reactors in recent years due to the
installation of additional hold-up tanks or adsorption columns.
128. In the reprocessing plant the spent fuel elements are
dismantled and the nuclear material dissolved. Procedures to
separate 85Kr from gaseous effluents and to provide long-term
retention are under study, but current practice is to allow
controlled release to the atmosphere.
C. BEHAVIOUR IN THE ENVIRONMENT
129. Krypton-85 discharged to the environment disperses in the
atmosphere and largely remains there until decay. It can become
washed out by rain and diffuse into surface layers of soil and
oceans, but these processes account for very little transfer of
85Kr from the atmosphere.
1. Dispersion in the atmosphere
130. Materials released to the atmosphere are transported downwind
and dispersed according to atmospheric mixing processes. The
estimation of this dispersion is commonly approached by using a
diffusion-transport equation. Several models have been developed
for this purpose, using a variety of boundary conditions and
simplifying assumptions. Most of them are based on the Gaussian
plume diffusion model [S1, I2], which has been shown to be adequate
in many practical situations. The krypton concentrations in air at
various distances for a release from a 30 m high stack are shown in
Table IV.2 [C5].
Table IV.2 Krypton-85
concentration in air for a
release of 1 Bq/s (stack height
30 m, Pasquill category D)
[C5]
-------------------------------
Distance Concentration
(km) (Bq/m3)
-------------------------------
1 4.8 10-7
10 1.3 10-8
100 4.4 10-10
1000 3.2 10-11
-------------------------------
131. For estimation of dispersion at greater distances, some
shortcomings in the Gaussian model are evident in the assumptions
that the meteorologic conditions and the direction of the wind
remain constant throughout the transit of the plume. To overcome
these difficulties, long-range models have been developed [A1, D3,
M2], which follow the trajectories of masses of air passing over
the release point and take into account the changing meteorologic
conditions with time. A survey of several diffusion models and of
their applications is given in [C5].
132. The global circulation of 85Kr can be approximated by a
simple compartment model, consisting of single compartments
representing the atmosphere in the northern and in the southern
hemispheres. Following a single release, equilibrium
concentrations in the atmosphere are achieved after about two
years. Further decrease in concentrations is due to radioactive
decay. In applying this model, Kelly et al. [K3] determined that
the integral concentration in air would be 5.3 10-18 Bq a m-3 per
Bq released. The atmospheric mass was assumed to be 3.8 1021 g,
equivalent to 3.1 1018 m3 at STP.
133. The dispersion calculations of Machta et al. [M2] are based
on detailed meteorological considerations and allow population-
weighted exposures to be determined. Table IV.3 lists the average
surface air concentrations of 85Kr in latitude bands following
release of 1 Bq in the 30-50° N latitude band. Uniform
concentrations are achieved after two years, after which the
integral concentration until complete decay is
10.73 a
22 10-20 Bq/m3 ------- = 3.4 10-18 Bq a/m3
1n 2
Adding the contributions from the first two years gives
3.9 10-18 Bq a/m3 for the population weighted integral
concentration of 85Kr in air from a release of 1 Bq.
2. Removal from the atmosphere
134. There is very little removal of 85Kr from the atmosphere,
except by radio-active decay. The low solubility of krypton in
water limits the accumulation of 85Kr in rainwater. Adsorption of
85Kr on particulate matter in air and subsequent deposition of the
particles provides a removal means of very low efficiency [N1].
135. The transfer of 85Kr to soil can occur by diffusion
processes; however, estimates of this transfer can account for
only about 0.05% of the total krypton in the atmosphere [N1].
Therefore, soil in general is not an important removal sink for
85Kr.
136. The efficiency of the oceans as a sink for 85Kr can be
determined from the natural krypton content of the atmosphere and
of the mixed layer of the ocean. From estimates of the krypton
concentration in air, the atmospheric volume and the density
krypton (STP), a total mass of about 1.64 1016 g of krypton in the
atmosphere is calculated [N1]. Assuming that the mixed layer of
the ocean extends to 100 m depth and an area of 3.6 1018 cm2, and
using the measured average krypton concentration in this layer of
seawater of 5 10-8 by volume [B4], a total mass of 6.7 1012 g of
krypton in the mixed layer of the ocean is obtained. This
corresponds approximately to 0.04% of the atmospheric mass of
krypton.
Table IV.3 Average surface air concentration of krypton-85
(1 Bq emitted uniformly over one year in 30-50° N latitude band)
[M2]
---------------------------------------------------------------
Krypton-85 concentration Population
Latitude band (10-20 Bq/m3) distribution %
Year 1 Year 2 Year 3
---------------------------------------------------------------
70 - 90° N 23 32 22 -
50 - 70° N 25 31 22 12.6
30 - 50° N 23 30 22 32.0
10 - 30° N 19 27 22 39.0
10° N - 10° S 11 22 22 11.5
10 - 30° S 6.3 22 22 3.4
30 - 50° S 5.1 20 22 1.5
50 - 70° S 4.3 19 22 0.05
70 - 90° S 3.8 19 22 -
Population weighted
integral
concentration
(10-20Bq a/m3) 19.5 27.6 22.0
---------------------------------------------------------------
137. An estimate of the total mass of krypton in the oceans as a
whole is obtained using an average concentration by volume of
krypton in the oceans of 9 10-8 [B4], a total ocean volume of 1.4
1024 cm3, and a krypton density of 3.73 10-3 g/cm3 at STP. This
calculation results in a total ocean inventory of about 4.7 1014 g
of krypton, or approximately 3% of the total atmospheric krypton
[N1]. These figures clearly indicate that the oceans can serve
only as a minor sink for 85Kr discharged into the atmosphere.
D. TRANSFER TO MAN
138. Following release to the atmosphere 85Kr becomes widely
dispersed. Exposure of man occurs by external irradiation from the
passing cloud or the dispersed gas and by internal irradiation
following inhalation of 85Kr and absorption in tissues.
139. After intake, 85Kr is distributed in the body by blood and
lymph fluids and is absorbed in the various tissues. A person
immersed in an atmosphere of 85Kr at low concentration would rather
quickly come into equilibrium with it. The concentrations in body
tissues are determined by multiplying the concentration in air by a
partitioning factor, called the Ostwald's coefficient. The
relevant values reflect the rate at which tissues are perfused with
blood, the solubility of the gas in the several tissues and the
velocity of diffusion of krypton across anatomical boundaries. The
concentration of 85Kr in the body is not uniform, the concentration
in the adipose tissue being nearly 50 times higher than that in
other parts of the body.
140. As a first approximation, one may only account for a
difference in the absorption behaviour of krypton in fat and non-
fat tissues, with values of the Ostwald coefficient of 0.45 for fat
and 0.07 for non-fat tissue [N1]. Other more elaborate models use
weight-related coefficients, where the density of the absorbing
tissue is taken into account [S2].
141. The total body retention of 85Kr has been subjected to
exponential analysis. Several clearance rates have been
recognized. Recent work has suggested a model for krypton in the
body consisting of five compartments [C6]. The fastest component
probably represents the clearance from circulating blood,
particularly blood plasma (T´ = 21.5 ± 5.7 s). The second
component (4.74 ± 2 min) appears to be representative of
haemoglobin clearance. The next slower component (19.8 ± 6.6 min)
is most likely related to clearance of krypton from muscle. The
two components with the slowest clearance rates can be related to
body fat compartments. A half-time of about 2.4 h is attributed to
a fat compartment not located in adipose tissue. The retention
half-time of krypton in adipose tissue is the slowest component and
is correlated significantly with the total body fat content. The
relationship is T´(h) = 0.17 (percentage fat) + 0.75 [C6].
E. DOSIMETRY
142. Krypton-85 released to the environment causes a radiation
dose to man through external irradiation from amounts in air and
through internal irradiation from amounts within the body. Tissues
are irradiated both from the activity in the organ itself and from
the activity present in the surrounding organs.
1. Dose per unit exposure
143. The equilibrium absorbed dose rates to body organs per unit
concentration of krypton-85 in air are summarized in Table IV.4
[N1]. For comparison, the recently published values of the ICRP
are also listed [I3]. The ICRP values represent minor adjustments,
except for the lungs, for which the beta dose due to 85Kr in the
airways of the lungs has been disregarded.
144. The dose equivalent rates in various organs are listed in
Table IV.5. These are the ICRP values [I3]. The quality factor
for 85Kr radiation is one. Therefore the dose equivalent rates are
numerically equal to the absorbed dose values. When combined with
the tissue weighting factors suggested by the ICRP to account for
varying incidence of health effects, the effective dose equivalent
rate is obtained, which for 85Kr is 8 10-9 Sv/a per Bq/m3.
Table IV.4 Equilibrium absorbed dose rate to body organs per unit air concentration from
immersion in a semi-infinite cloud of krypton-85 (10-9 Gy/a per Bq/m3) [N1]
------------------------------------------------------------------------------------------
Source Organ
Skin Adipose Lungs Red bone Skeleton Ovaries Testes
tissue marrow
------------------------------------------------------------------------------------------
Krypton-85 in air
Photons in air 4.1 3.2 3.0 3.8 4.1 1.3 3.5
Betas in air 490.0 - - - - - -
Bremsstrahlung in air 0.6 0.57 0.51 0.97 1.1 0.30 0.68
Bremsstrahlung in skin 0.015 0.0018 0.0011 0.0010 0.0030 0.0006 0.0027
Krypton-85 in the body
Photons in the body 0.0006 0.0006 0.0006 0.0007 0.0006 0.0008 0.0008
Betas in the body 0.10 0.30 0.10 0.21 0.10 0.10 0.10
Bremsstrahlung in the body 0.0001 0.0002 0.0002 0.0003 0.0003 0.0002 0.0003
Betas in airways of lung - - 4.9 - - - -
------------------------------------------------------------------------------------------
Total 490 4.1 8.5 5.0 5.3 1.7 4.3
------------------------------------------------------------------------------------------
Total [I3] 410 3.8 5.0 5.4 4.6
------------------------------------------------------------------------------------------
Table IV.5 Dose equivalent rates from submersion in
semi-infinite cloud of 85Kr (10-9 Sv/a per Bq/m3) [I3]
------------------------------------------------------------
Dose Weighting Effective
equivalent factor dose
rate equivalent
rate
------------------------------------------------------------
Gonads 4.6 0.25 1.1
Breast 3.9 0.15 0.6
Red bone marrow 5.0 0.12 0.6
Lungs 3.8 0.12 0.46
Bone surface 5.4 0.03 0.16
Spleen 4.0 0.06 0.24
Small intestinal wall 3.8 0.06 0.23
Kidneys 3.5 0.06 0.21
Adrenal glands 3.5 0.06 0.21
Liver 3.3 0.06 0.20
Skin 410.0 0.01 4.1
------------------------------------------------------------
Total 8.1
------------------------------------------------------------
2. Dose per unit release
145. The collective effective dose equivalent commitment per unit
release of 85Kr into the atmosphere, Sc1, can be determined, based
on the procedures previously used by UNSCEAR [U7]. In the local
region of the release, the following formula can be applied
c x 100 km r -1.5
S1 = (Q)1 km deltaN phi / (1 km) 2 pi r dr
1
A dispersion factor, x/Q, of 5 10-7 s/m3 is assumed, which agrees
with the data of Table IV.2. The population density, deltaN is
assumed to be 100 man/km2 in the local region extending to 100 km
distance. The dose factor, phi is 8 10-9 Sv/a per Bq/m3 as given
above. The concentration of 85Kr in air is assumed to decrease as
a function of the distance, r, from the release point. The
integral of the distance dependence over the local region times the
dispersion factor, the population density, and the dose factor
gives the collective effective dose equivalent commitment per unit
85Kr activity released. The result is
c
S1 (local) = 1 10-18 man Sv/Bq
146. The collective dose equivalent commitment per unit release of
85Kr to the global population can be determined from the following
formula [U7]
c infinite .---
S1 = / D(t) N(t)dt
0
.---
where D(t) is the per caput dose rate per unit activity released
and N(t) is the population size.
147. After uniform mixing in the global atmosphere, the per caput
dose rate from 85Kr is equal to the product of the concentration in
air, Co, which decreases due to radioactive decay of 85Kr, and the
dose factor, phi. The population is assumed to increase at a rate,
nu, of 2% per year. The collective effective dose equivalent
commitment is, thus,
c infinite -lambda t nu t C0 phi N0
S1 = / C0e phi N0e dt = ---------
0 lambda-nu
148. The population-weighted surface air concentrations of Table
IV.3 can be used to estimate the global collective dose. An
initial world population of 4 109 is assumed. The following
contributions are obtained in the first two years following the
release of 85Kr to the atmosphere:
c Bq a/m3 Sv/a
S1 (1st year) = 4 109 man 19.5 10-20 -------- 8 10-9 ------
Bq Bq/m3
= 6 10-18 man Sv/Bq
c Bq a/m3 Sv/a
S1 (2nd year) = 4.08 109 man 27.6 10-20 -------- 8 10-9 ------
Bq Bq/m3
= 9 10-18 man Sv/Bq
Thereafter, the formula in the preceding paragraph can be applied.
Bq a/m3 Sv/a
4.16 109 man 22 10-20 -------- 8 10-9 -----
c Bq Bq/m3
S1 (> 2 years) = -----------------------------------------------
1n 2
------- - 0.02 1/a
10.73 a
= 1.6 10-16 man Sv/Bq
The total global collective dose equivalent commitment per unit
85Kr activity released is
c
S1 (global) = 1.8 10-16 man Sv/Bq
149. A more approximate estimation, namely assuming instant mixing
of the 85Kr in the global atmosphere of 4 1018 m3, gives the same
result. The initial concentration is then 25 10-20 Bq/m3. Using
the formula in paragraph 147, with an initial population of 4 109,
gives the value of 1.8 10-16 man Sv/Bq for the global collective
dose equivalent commitment per unit 85Kr activity released.
150. It is seen by comparison that the local contribution to the
collective dose equivalent commitment per unit release of krypton-
85 is negligible. Therefore, the total dose estimate is
independent of the location of the release.
E. REFERENCES
A1 Apsimon, H.M. and A.J.H. Goddard. Modelling the atmospheric
dispersal of radioactive pollutants beyond the first few hours
of travel. p. 124-135 in The seventh International Technical
Meeting on Air pollution Modelling and its Application.
Proceedings of a symposium. Airlie, Va., U.S.A., 1976.
B1 Bernhardt, D.C., A.A. Moghissi and J.W. Cochran. Atmospheric
concentrations of fission product noble gases. p. 97-118 in
The Noble Gases (A.A. Moghissi and J.W. Cochran, eds.). U.S.
Government Printing Office, Washington, 1975.
B3 Bonka, H., R. Schulten, K. Brüssermann et al. Zukünftige
radioaktive Umweltbelastung in der BRD durch radionuklide aus
kerntechnischen Anlagen im Normalbetrieb. Berichte der
Kernforschungsanlage Jülich, Jül-1220 (1975).
B4 Bieri, R.H., M. Koide and E.D. Goldberg. The noble gas
contents of Pacific seawaters. J. Geophys. Res. 71: 5243-5247
(1966).
C2 Crouch, E.A.C. Fission product yields from neutron induced
fission, atomic data and nuclear data tables, Vol. 19, No. 5.
Academic Press Inc., New York, 1977.
C5 Commission of the European Communities. Methodology for
evaluation the radiological consequences of radioactive
effluents released in normal operations. V/3865/79 - EN, FR
Directorate of Health Protection (1979).
C6 Cohn, S.H., K.J. Ellis and H. Susskind. Evaluation of the
health hazard from inhaled krypton-85. International Symposium
on biological implications of radionuclides released from
nuclear industries. IAEA-SM-237/46 (1979).
D1 Diethorn, W.S. and W.L. Stockho. The dose to man from
atmospheric krypton-85. Health Phys. 23: 653-662 (1972).
D3 Depres, A. and J. LeGrand. Une méthode d'évaluation des
transferts atmosphériques à longue distance. p. 237-249 in La
dispersion en milieu physique naturel. Proceedings of a
seminar. Cadarache, France, 1978.
E1 Erdman, C.A. and A.B. Reynolds. Radionuclide behaviour during
normal operations of liquid metal cooled fast breeder reactors,
part 1: Production. Nucl. Saf. 16: 43-52 (1975).
H2 Heller, D., W. Roedel, K.O. Münnich et al. Decreasing release
of krypton-85 into the atmosphere. Naturwissenschaften 64(7):
383 (1977).
I1 International Atomic Energy Agency. Power reactors in member
states IAEA, Vienna, 1980.
I2 Islitzer, N.F. and D.H. Slade. Diffusion and transport
experiments in meteorology and atomic energy. U.S. Atomic
Energy Commission report USAEC-TID-24190 (1968).
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. International Commission
on Radiological Protection report 30, supplement to part I.
Pergamon Press, Oxford, 1979.
K1 Kirk, W.P. Krypton-85: A review of the literature and an
analysis of radiation hazards, USEPA report EPA-NP-19251
(1972).
K3 Kelly, G.N., J.A. Jones, P.M. Bryant et al. The predicted
radiation exposure of the population of the European community
resulting from discharge of krypton-85, tritium, carbon-14 and
iodine-129 from the nuclear power industry to the year 2000.
Commission of the European Communities, Directorate of Health
Protection. V/2676/75 (1975).
M1 Martin, A. and M. Apsimon. The forecasting of radioactive
wastes arising from nuclear fuel reprocessing. p. 55-60 in
Management of Radioactive Wastes from Fuel Reprocessing.
Proceedings of a symposium jointly organized by OECD/NEA and
International Atomic Energy Agency. Paris, 1972.
M2 Machta, L., G.J. Ferber and J.L. Hefter. Regional and global
scale dispersion of krypton-85 for population dose
calculations. p. 411-424 in Physical Behaviour of Radioactive
Contaminants in the Atmosphere. International Atomic Energy
Agency publication STI/PUB/355. Vienna, 1974.
N1 National Council on Radiation Protection and Measurements.
Krypton-85 in the atmosphere - accumulation, biological
significance and control technology. NCRP No. 44 (1975).
R1 Rozanski, K. Krypton-85 in the atmosphere 1950-1977: A data
review. Envir. International 2: 139-143 (1979).
S1 Sutton, O.G. The theory of Eddy diffusion in the atmosphere.
Proc. R. Soc. (London), Ser. A 135: 143-152 (1932).
S2 Snyder, W.S., L.T. Dillman, M.R. Ford et al. Dosimetry for a
man immersed in an infinite cloud of 85-K5, p. 119-122 in The
Noble Gases (A.A. Moghissi and R.E. Stanley eds.). U.S.
Government Printing Office, Washington, 1975.
U1 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Thirteenth Session, Supplement No. 17
(A/3838). New York, 1958.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No. 16
(A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No. 14
(A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14 (A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13 (A/7613). New York, 1969.
U6 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication No. E.72.IX.17 and
18. New York, 1972.
U7 United Nations. Sources and Effects of Ionizing Radiation.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation 1977 report to the General
Assembly, with annexes. United Nations sales publication No.
E.77.IX.1. New York, 1977.
V. STRONTIUM-90
A. INTRODUCTION
151. Strontium is element number 38 in the periodic table. It is
an alkaline earth element and is therefore similar to calcium,
barium and radium. It follows calcium through the food chains from
environment to man, but some degree of discrimination exists
against strontium. Both strontium and calcium are retained in the
body largely in bone.
152. Since the early days of atmospheric nuclear testing the
importance of 90Sr as a contributor to the radiation exposure of
man has been recognized. Strontium-90 is a radionuclide formed in
the process of nuclear fission. It has a radioactive half-life of
29.1 years and decays by beta emission. Its daughter, 90Y, is also
radioactive with a half-life of 64.0 hours, and decays by beta
emission to the stable isotope 90Zr. The decay scheme for 90Sr and
90Y is given in Figure V.I. A summary of fission yields for 90Sr
is given in Table V.1.
B. SOURCES
118. Krypton-85 is produced by cosmic ray interactions in the
atmosphere, in nuclear power reactors, and nuclear explosions. The
main release source is the dissolution step in the reprocessing of
nuclear fuel.
119. Concentrations of 85Kr in the atmosphere increased sharply
after 1955 due to the production and testing of nuclear weapons and
the development of the nuclear power industry. More recently the
input rates of 85Kr into air have decreased [H2]. There have been
reductions in plutonium production for military purposes and in
nuclear fuel reprocessing.
120. A review of 85Kr measurement data for 1950-77 has been
prepared by Rozanski [R1]. The most recent data indicate that
concentrations in air have stablized at about 0.6 Bq/m3 in the
northern hemisphere and 0.4 Bq/m3 in the southern hemisphere [R1].
The major sources are in the northern hemisphere, accounting for
the higher levels in that hemisphere.
1. Natural krypton-85
121. Krypton-85 is present in small amounts in the environment as
a result of spontaneous fission of natural uranium and interactions
of cosmic ray neutrons with atmospheric 84Kr. The steady state
environmental inventories of 85Kr from these sources have been
calculated: 7.4 1010 Bq in the upper 3 m of the total land and
water surface due to spontaneous fission of natural uranium, 3.7
1011 Bq in the atmosphere from cosmic ray production and 3.7 105 Bq
in the oceans from the atmospheric source [D1]. These estimates,
in comparison with the estimates of man-made sources of 85Kr to
follow, are negligible in contributing to the world's total 85Kr
inventory.
2. Nuclear explosions
122. Since 85Kr is produced during fission, it has been generated
by nuclear weapon tests. The total amount of 85Kr produced in
nuclear testing can be calculated from the ratio of 85Kr/90Sr
fission yield of 0.08, giving an activity ratio of 0.22 [C2].
Measurements of 90Sr activity have been reported and discussed in
the reports of UNSCEAR [U1-U7]. There have been 6 1017 Bq of 90Sr
produced in weapon testing through 1976 [U7], corresponding to
about 1.3 1017 Bq of 85Kr.
123. Another source of 85Kr associated with nuclear weapons is in
the production of plutonium in military reactors. The amount of
85Kr released from this source is estimated to be two times higher
than that from the weapon tests [D1]. Naval propulsion reactors
also contribute to the 85Kr inventory with an annual production in
the region of 1.1 to 1.9 1016 Bq [B1]. Including all sources, the
total amount of 85Kr produced in operations for military purposes
is still rather small in comparison to the prospective generation
of 85Kr by the nuclear power industry.
3. Nuclear fuel cycle
124. Krypton-85 is produced by fission in the fuel of nuclear
reactors and in very low trace amounts in the moderator or coolant,
due to contamination with fissile material. The rates of 85Kr
production are related to the type of fuel and degree of burn-up.
Production and emission rates may be conveniently normalized to
unit electrical energy generated (for power reactors) or to the
electrical energy generated by the reactors serviced (for fuel
reprocessing plants).
125. The amounts of 85Kr produced vary according to reactor type.
For thermal reactors, the range of estimated production is about
1.1 to 1.5 1013 Bq/MW(e)a. For FBRs the values are about 25%
smaller [E1, M1], for HTGRs 50% higher [B3]. A production rate of
1.4 1013 Bq/MW(e)a has been correlated with some measurements from
reprocessing plants [U7] and this value can be taken for general
evaluations.
126. An estimate of 85Kr annual generation from reactor operation
can be obtained from the installed capacity of nuclear reactors of
1.25 105 MW(e) worldwide in 1980 [I1], with the assumptions of 60%
utilization and average 85Kr generation rate of 1.4 1013 Bq/MW(e)a:
1.25 105 MW(e)a x 0.6 x 1.4 1013 Bq/MW(e)a = 1 1018 Bq/a
The actual release rate is less, since delays occur before
reprocessing and not all fuel is reprocessed.
127. Reported releases of 85Kr and other fission noble gases were
listed in the 1977 report of UNSCEAR [U7]. There are large
differences in the release values of the various plants. Although
the relevant data are not very extensive, there are indications of
improved retention of 85Kr at reactors in recent years due to the
installation of additional hold-up tanks or adsorption columns.
128. In the reprocessing plant the spent fuel elements are
dismantled and the nuclear material dissolved. Procedures to
separate 85Kr from gaseous effluents and to provide long-term
retention are under study, but current practice is to allow
controlled release to the atmosphere.
C. BEHAVIOUR IN THE ENVIRONMENT
129. Krypton-85 discharged to the environment disperses in the
atmosphere and largely remains there until decay. It can become
washed out by rain and diffuse into surface layers of soil and
oceans, but these processes account for very little transfer of
85Kr from the atmosphere.
1. Dispersion in the atmosphere
130. Materials released to the atmosphere are transported downwind
and dispersed according to atmospheric mixing processes. The
estimation of this dispersion is commonly approached by using a
diffusion-transport equation. Several models have been developed
for this purpose, using a variety of boundary conditions and
simplifying assumptions. Most of them are based on the Gaussian
plume diffusion model [S1, I2], which has been shown to be adequate
in many practical situations. The krypton concentrations in air at
various distances for a release from a 30 m high stack are shown in
Table IV.2 [C5].
Table IV.2 Krypton-85
concentration in air for a
release of 1 Bq/s (stack height
30 m, Pasquill category D)
[C5]
-------------------------------
Distance Concentration
(km) (Bq/m3)
-------------------------------
1 4.8 10-7
10 1.3 10-8
100 4.4 10-10
1000 3.2 10-11
-------------------------------
131. For estimation of dispersion at greater distances, some
shortcomings in the Gaussian model are evident in the assumptions
that the meteorologic conditions and the direction of the wind
remain constant throughout the transit of the plume. To overcome
these difficulties, long-range models have been developed [A1, D3,
M2], which follow the trajectories of masses of air passing over
the release point and take into account the changing meteorologic
conditions with time. A survey of several diffusion models and of
their applications is given in [C5].
132. The global circulation of 85Kr can be approximated by a
simple compartment model, consisting of single compartments
representing the atmosphere in the northern and in the southern
hemispheres. Following a single release, equilibrium
concentrations in the atmosphere are achieved after about two
years. Further decrease in concentrations is due to radioactive
decay. In applying this model, Kelly et al. [K3] determined that
the integral concentration in air would be 5.3 10-18 Bq a m-3 per
Bq released. The atmospheric mass was assumed to be 3.8 1021 g,
equivalent to 3.1 1018 m3 at STP.
133. The dispersion calculations of Machta et al. [M2] are based
on detailed meteorological considerations and allow population-
weighted exposures to be determined. Table IV.3 lists the average
surface air concentrations of 85Kr in latitude bands following
release of 1 Bq in the 30-50° N latitude band. Uniform
concentrations are achieved after two years, after which the
integral concentration until complete decay is
10.73 a
22 10-20 Bq/m3 ------- = 3.4 10-18 Bq a/m3
1n 2
Adding the contributions from the first two years gives
3.9 10-18 Bq a/m3 for the population weighted integral
concentration of 85Kr in air from a release of 1 Bq.
2. Removal from the atmosphere
134. There is very little removal of 85Kr from the atmosphere,
except by radio-active decay. The low solubility of krypton in
water limits the accumulation of 85Kr in rainwater. Adsorption of
85Kr on particulate matter in air and subsequent deposition of the
particles provides a removal means of very low efficiency [N1].
135. The transfer of 85Kr to soil can occur by diffusion
processes; however, estimates of this transfer can account for
only about 0.05% of the total krypton in the atmosphere [N1].
Therefore, soil in general is not an important removal sink for
85Kr.
136. The efficiency of the oceans as a sink for 85Kr can be
determined from the natural krypton content of the atmosphere and
of the mixed layer of the ocean. From estimates of the krypton
concentration in air, the atmospheric volume and the density
krypton (STP), a total mass of about 1.64 1016 g of krypton in the
atmosphere is calculated [N1]. Assuming that the mixed layer of
the ocean extends to 100 m depth and an area of 3.6 1018 cm2, and
using the measured average krypton concentration in this layer of
seawater of 5 10-8 by volume [B4], a total mass of 6.7 1012 g of
krypton in the mixed layer of the ocean is obtained. This
corresponds approximately to 0.04% of the atmospheric mass of
krypton.
Table IV.3 Average surface air concentration of krypton-85
(1 Bq emitted uniformly over one year in 30-50° N latitude band)
[M2]
---------------------------------------------------------------
Krypton-85 concentration Population
Latitude band (10-20 Bq/m3) distribution %
Year 1 Year 2 Year 3
---------------------------------------------------------------
70 - 90° N 23 32 22 -
50 - 70° N 25 31 22 12.6
30 - 50° N 23 30 22 32.0
10 - 30° N 19 27 22 39.0
10° N - 10° S 11 22 22 11.5
10 - 30° S 6.3 22 22 3.4
30 - 50° S 5.1 20 22 1.5
50 - 70° S 4.3 19 22 0.05
70 - 90° S 3.8 19 22 -
Population weighted
integral
concentration
(10-20Bq a/m3) 19.5 27.6 22.0
---------------------------------------------------------------
137. An estimate of the total mass of krypton in the oceans as a
whole is obtained using an average concentration by volume of
krypton in the oceans of 9 10-8 [B4], a total ocean volume of 1.4
1024 cm3, and a krypton density of 3.73 10-3 g/cm3 at STP. This
calculation results in a total ocean inventory of about 4.7 1014 g
of krypton, or approximately 3% of the total atmospheric krypton
[N1]. These figures clearly indicate that the oceans can serve
only as a minor sink for 85Kr discharged into the atmosphere.
D. TRANSFER TO MAN
138. Following release to the atmosphere 85Kr becomes widely
dispersed. Exposure of man occurs by external irradiation from the
passing cloud or the dispersed gas and by internal irradiation
following inhalation of 85Kr and absorption in tissues.
139. After intake, 85Kr is distributed in the body by blood and
lymph fluids and is absorbed in the various tissues. A person
immersed in an atmosphere of 85Kr at low concentration would rather
quickly come into equilibrium with it. The concentrations in body
tissues are determined by multiplying the concentration in air by a
partitioning factor, called the Ostwald's coefficient. The
relevant values reflect the rate at which tissues are perfused with
blood, the solubility of the gas in the several tissues and the
velocity of diffusion of krypton across anatomical boundaries. The
concentration of 85Kr in the body is not uniform, the concentration
in the adipose tissue being nearly 50 times higher than that in
other parts of the body.
140. As a first approximation, one may only account for a
difference in the absorption behaviour of krypton in fat and non-
fat tissues, with values of the Ostwald coefficient of 0.45 for fat
and 0.07 for non-fat tissue [N1]. Other more elaborate models use
weight-related coefficients, where the density of the absorbing
tissue is taken into account [S2].
141. The total body retention of 85Kr has been subjected to
exponential analysis. Several clearance rates have been
recognized. Recent work has suggested a model for krypton in the
body consisting of five compartments [C6]. The fastest component
probably represents the clearance from circulating blood,
particularly blood plasma (T´ = 21.5 ± 5.7 s). The second
component (4.74 ± 2 min) appears to be representative of
haemoglobin clearance. The next slower component (19.8 ± 6.6 min)
is most likely related to clearance of krypton from muscle. The
two components with the slowest clearance rates can be related to
body fat compartments. A half-time of about 2.4 h is attributed to
a fat compartment not located in adipose tissue. The retention
half-time of krypton in adipose tissue is the slowest component and
is correlated significantly with the total body fat content. The
relationship is T´(h) = 0.17 (percentage fat) + 0.75 [C6].
E. DOSIMETRY
142. Krypton-85 released to the environment causes a radiation
dose to man through external irradiation from amounts in air and
through internal irradiation from amounts within the body. Tissues
are irradiated both from the activity in the organ itself and from
the activity present in the surrounding organs.
1. Dose per unit exposure
143. The equilibrium absorbed dose rates to body organs per unit
concentration of krypton-85 in air are summarized in Table IV.4
[N1]. For comparison, the recently published values of the ICRP
are also listed [I3]. The ICRP values represent minor adjustments,
except for the lungs, for which the beta dose due to 85Kr in the
airways of the lungs has been disregarded.
144. The dose equivalent rates in various organs are listed in
Table IV.5. These are the ICRP values [I3]. The quality factor
for 85Kr radiation is one. Therefore the dose equivalent rates are
numerically equal to the absorbed dose values. When combined with
the tissue weighting factors suggested by the ICRP to account for
varying incidence of health effects, the effective dose equivalent
rate is obtained, which for 85Kr is 8 10-9 Sv/a per Bq/m3.
Table IV.4 Equilibrium absorbed dose rate to body organs per unit air concentration from
immersion in a semi-infinite cloud of krypton-85 (10-9 Gy/a per Bq/m3) [N1]
------------------------------------------------------------------------------------------
Source Organ
Skin Adipose Lungs Red bone Skeleton Ovaries Testes
tissue marrow
------------------------------------------------------------------------------------------
Krypton-85 in air
Photons in air 4.1 3.2 3.0 3.8 4.1 1.3 3.5
Betas in air 490.0 - - - - - -
Bremsstrahlung in air 0.6 0.57 0.51 0.97 1.1 0.30 0.68
Bremsstrahlung in skin 0.015 0.0018 0.0011 0.0010 0.0030 0.0006 0.0027
Krypton-85 in the body
Photons in the body 0.0006 0.0006 0.0006 0.0007 0.0006 0.0008 0.0008
Betas in the body 0.10 0.30 0.10 0.21 0.10 0.10 0.10
Bremsstrahlung in the body 0.0001 0.0002 0.0002 0.0003 0.0003 0.0002 0.0003
Betas in airways of lung - - 4.9 - - - -
------------------------------------------------------------------------------------------
Total 490 4.1 8.5 5.0 5.3 1.7 4.3
------------------------------------------------------------------------------------------
Total [I3] 410 3.8 5.0 5.4 4.6
------------------------------------------------------------------------------------------
Table IV.5 Dose equivalent rates from submersion in
semi-infinite cloud of 85Kr (10-9 Sv/a per Bq/m3) [I3]
------------------------------------------------------------
Dose Weighting Effective
equivalent factor dose
rate equivalent
rate
------------------------------------------------------------
Gonads 4.6 0.25 1.1
Breast 3.9 0.15 0.6
Red bone marrow 5.0 0.12 0.6
Lungs 3.8 0.12 0.46
Bone surface 5.4 0.03 0.16
Spleen 4.0 0.06 0.24
Small intestinal wall 3.8 0.06 0.23
Kidneys 3.5 0.06 0.21
Adrenal glands 3.5 0.06 0.21
Liver 3.3 0.06 0.20
Skin 410.0 0.01 4.1
------------------------------------------------------------
Total 8.1
------------------------------------------------------------
2. Dose per unit release
145. The collective effective dose equivalent commitment per unit
release of 85Kr into the atmosphere, Sc1, can be determined, based
on the procedures previously used by UNSCEAR [U7]. In the local
region of the release, the following formula can be applied
c x 100 km r -1.5
S1 = (Q)1 km deltaN phi / (1 km) 2 pi r dr
1
A dispersion factor, x/Q, of 5 10-7 s/m3 is assumed, which agrees
with the data of Table IV.2. The population density, deltaN is
assumed to be 100 man/km2 in the local region extending to 100 km
distance. The dose factor, phi is 8 10-9 Sv/a per Bq/m3 as given
above. The concentration of 85Kr in air is assumed to decrease as
a function of the distance, r, from the release point. The
integral of the distance dependence over the local region times the
dispersion factor, the population density, and the dose factor
gives the collective effective dose equivalent commitment per unit
85Kr activity released. The result is
c
S1 (local) = 1 10-18 man Sv/Bq
146. The collective dose equivalent commitment per unit release of
85Kr to the global population can be determined from the following
formula [U7]
c infinite .---
S1 = / D(t) N(t)dt
0
.---
where D(t) is the per caput dose rate per unit activity released
and N(t) is the population size.
147. After uniform mixing in the global atmosphere, the per caput
dose rate from 85Kr is equal to the product of the concentration in
air, Co, which decreases due to radioactive decay of 85Kr, and the
dose factor, phi. The population is assumed to increase at a rate,
nu, of 2% per year. The collective effective dose equivalent
commitment is, thus,
c infinite -lambda t nu t C0 phi N0
S1 = / C0e phi N0e dt = ---------
0 lambda-nu
148. The population-weighted surface air concentrations of Table
IV.3 can be used to estimate the global collective dose. An
initial world population of 4 109 is assumed. The following
contributions are obtained in the first two years following the
release of 85Kr to the atmosphere:
c Bq a/m3 Sv/a
S1 (1st year) = 4 109 man 19.5 10-20 -------- 8 10-9 ------
Bq Bq/m3
= 6 10-18 man Sv/Bq
c Bq a/m3 Sv/a
S1 (2nd year) = 4.08 109 man 27.6 10-20 -------- 8 10-9 ------
Bq Bq/m3
= 9 10-18 man Sv/Bq
Thereafter, the formula in the preceding paragraph can be applied.
Bq a/m3 Sv/a
4.16 109 man 22 10-20 -------- 8 10-9 -----
c Bq Bq/m3
S1 (> 2 years) = -----------------------------------------------
1n 2
------- - 0.02 1/a
10.73 a
= 1.6 10-16 man Sv/Bq
The total global collective dose equivalent commitment per unit
85Kr activity released is
c
S1 (global) = 1.8 10-16 man Sv/Bq
149. A more approximate estimation, namely assuming instant mixing
of the 85Kr in the global atmosphere of 4 1018 m3, gives the same
result. The initial concentration is then 25 10-20 Bq/m3. Using
the formula in paragraph 147, with an initial population of 4 109,
gives the value of 1.8 10-16 man Sv/Bq for the global collective
dose equivalent commitment per unit 85Kr activity released.
150. It is seen by comparison that the local contribution to the
collective dose equivalent commitment per unit release of krypton-
85 is negligible. Therefore, the total dose estimate is
independent of the location of the release.
E. REFERENCES
A1 Apsimon, H.M. and A.J.H. Goddard. Modelling the atmospheric
dispersal of radioactive pollutants beyond the first few hours
of travel. p. 124-135 in The seventh International Technical
Meeting on Air pollution Modelling and its Application.
Proceedings of a symposium. Airlie, Va., U.S.A., 1976.
B1 Bernhardt, D.C., A.A. Moghissi and J.W. Cochran. Atmospheric
concentrations of fission product noble gases. p. 97-118 in
The Noble Gases (A.A. Moghissi and J.W. Cochran, eds.). U.S.
Government Printing Office, Washington, 1975.
B3 Bonka, H., R. Schulten, K. Brüssermann et al. Zukünftige
radioaktive Umweltbelastung in der BRD durch radionuklide aus
kerntechnischen Anlagen im Normalbetrieb. Berichte der
Kernforschungsanlage Jülich, Jül-1220 (1975).
B4 Bieri, R.H., M. Koide and E.D. Goldberg. The noble gas
contents of Pacific seawaters. J. Geophys. Res. 71: 5243-5247
(1966).
C2 Crouch, E.A.C. Fission product yields from neutron induced
fission, atomic data and nuclear data tables, Vol. 19, No. 5.
Academic Press Inc., New York, 1977.
C5 Commission of the European Communities. Methodology for
evaluation the radiological consequences of radioactive
effluents released in normal operations. V/3865/79 - EN, FR
Directorate of Health Protection (1979).
C6 Cohn, S.H., K.J. Ellis and H. Susskind. Evaluation of the
health hazard from inhaled krypton-85. International Symposium
on biological implications of radionuclides released from
nuclear industries. IAEA-SM-237/46 (1979).
D1 Diethorn, W.S. and W.L. Stockho. The dose to man from
atmospheric krypton-85. Health Phys. 23: 653-662 (1972).
D3 Depres, A. and J. LeGrand. Une méthode d'évaluation des
transferts atmosphériques à longue distance. p. 237-249 in La
dispersion en milieu physique naturel. Proceedings of a
seminar. Cadarache, France, 1978.
E1 Erdman, C.A. and A.B. Reynolds. Radionuclide behaviour during
normal operations of liquid metal cooled fast breeder reactors,
part 1: Production. Nucl. Saf. 16: 43-52 (1975).
H2 Heller, D., W. Roedel, K.O. Münnich et al. Decreasing release
of krypton-85 into the atmosphere. Naturwissenschaften 64(7):
383 (1977).
I1 International Atomic Energy Agency. Power reactors in member
states IAEA, Vienna, 1980.
I2 Islitzer, N.F. and D.H. Slade. Diffusion and transport
experiments in meteorology and atomic energy. U.S. Atomic
Energy Commission report USAEC-TID-24190 (1968).
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. International Commission
on Radiological Protection report 30, supplement to part I.
Pergamon Press, Oxford, 1979.
K1 Kirk, W.P. Krypton-85: A review of the literature and an
analysis of radiation hazards, USEPA report EPA-NP-19251
(1972).
K3 Kelly, G.N., J.A. Jones, P.M. Bryant et al. The predicted
radiation exposure of the population of the European community
resulting from discharge of krypton-85, tritium, carbon-14 and
iodine-129 from the nuclear power industry to the year 2000.
Commission of the European Communities, Directorate of Health
Protection. V/2676/75 (1975).
M1 Martin, A. and M. Apsimon. The forecasting of radioactive
wastes arising from nuclear fuel reprocessing. p. 55-60 in
Management of Radioactive Wastes from Fuel Reprocessing.
Proceedings of a symposium jointly organized by OECD/NEA and
International Atomic Energy Agency. Paris, 1972.
M2 Machta, L., G.J. Ferber and J.L. Hefter. Regional and global
scale dispersion of krypton-85 for population dose
calculations. p. 411-424 in Physical Behaviour of Radioactive
Contaminants in the Atmosphere. International Atomic Energy
Agency publication STI/PUB/355. Vienna, 1974.
N1 National Council on Radiation Protection and Measurements.
Krypton-85 in the atmosphere - accumulation, biological
significance and control technology. NCRP No. 44 (1975).
R1 Rozanski, K. Krypton-85 in the atmosphere 1950-1977: A data
review. Envir. International 2: 139-143 (1979).
S1 Sutton, O.G. The theory of Eddy diffusion in the atmosphere.
Proc. R. Soc. (London), Ser. A 135: 143-152 (1932).
S2 Snyder, W.S., L.T. Dillman, M.R. Ford et al. Dosimetry for a
man immersed in an infinite cloud of 85-K5, p. 119-122 in The
Noble Gases (A.A. Moghissi and R.E. Stanley eds.). U.S.
Government Printing Office, Washington, 1975.
U1 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Thirteenth Session, Supplement No. 17
(A/3838). New York, 1958.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No. 16
(A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No. 14
(A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14 (A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13 (A/7613). New York, 1969.
U6 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication No. E.72.IX.17 and
18. New York, 1972.
U7 United Nations. Sources and Effects of Ionizing Radiation.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation 1977 report to the General
Assembly, with annexes. United Nations sales publication No.
E.77.IX.1. New York, 1977.
V. STRONTIUM-90
A. INTRODUCTION
151. Strontium is element number 38 in the periodic table. It is
an alkaline earth element and is therefore similar to calcium,
barium and radium. It follows calcium through the food chains from
environment to man, but some degree of discrimination exists
against strontium. Both strontium and calcium are retained in the
body largely in bone.
152. Since the early days of atmospheric nuclear testing the
importance of 90Sr as a contributor to the radiation exposure of
man has been recognized. Strontium-90 is a radionuclide formed in
the process of nuclear fission. It has a radioactive half-life of
29.1 years and decays by beta emission. Its daughter, 90Y, is also
radioactive with a half-life of 64.0 hours, and decays by beta
emission to the stable isotope 90Zr. The decay scheme for 90Sr and
90Y is given in Figure V.I. A summary of fission yields for 90Sr
is given in Table V.1.
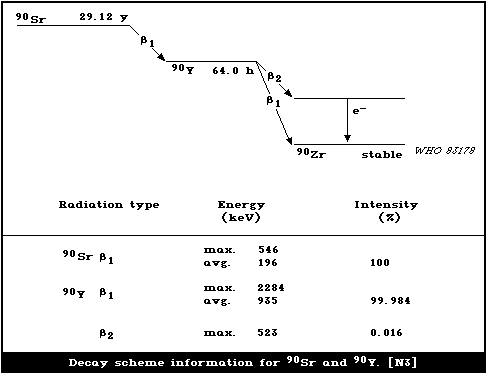 Table V.1 Fission yields of strontium-90 [C3]
-------------------------------------
Fission yield (%)
Nuclide Thermal Fast
-------------------------------------
235U 5.84 5.21
239Pu 2.12 2.05
238U 3.20
232Th 7.66
Average for
nuclear tests a/ 3.50
-------------------------------------
a/ From reference [H1].
153. Large amounts of 90Sr were released in nuclear tests and
dispersed throughout the world. Strontium-90 is also produced in
the nuclear fuel cycle, but only small amounts are released to the
environment. Strontium-90 in the environment is efficiently
transferred to human diet. The absorption of 90Sr by the body is
relatively high and it has a long biological retention time.
154. Because of the correspondence in behaviour of strontium and
calcium in the environment and in man, it has been the practice to
express measurement results in diet and bone as quotients of 90Sr
to Ca concentrations. Discrimination is reflected as ratios of
strontium to calcium quotients in samples to those in precursor
samples in the transfer chain. Expressing results in terms of the
strontium to calcium quotients has the practical advantage that for
many environmental transfer processes, such as absorption into the
body, secretion into milk and deposition in bone, the ratios remain
relatively constant and predictable. However, since the average
levels of calcium in diet and man are nearly constant, assessments
of 90Sr can also be made on the basis of amounts per unit mass or
volume of material.
B. SOURCES
1. Nuclear explosions
155. Strontium-90 is produced in nuclear explosions in the amount
of approximately 3.7 1015 Bq per Mt of fission energy. Measurements
of the fission debris from large nuclear tests gave a 90Sr fission
yield estimate of 3.5% [H1]. Assuming 1 kt of fission energy
corresponds to 1.45 1023 fissions [H1] and using the current best
estimate of the 90Sr half-life (29.12 ± 0.24 a) [N3], the 90Sr
production yield is estimated to be 3.8 ± 0.1 1015 Bq per Mt
fission energy. Large deviations are possible for individual
tests.
156. Approximate fission energy yields of nuclear weapons tests
conducted in the atmosphere have been published. The total for
tests through 1962 was 194 Mt of fission energy [F1, U3]. This
would correspond to a production of about 74 1016 Bq of 90Sr. An
additional 9 1016 Bq have been produced in atmospheric tests to the
end of 1980. A portion of the total amount of 90Sr was local
fallout, deposited in the immediate vicinity of testing regions.
Local fallout is important especially for lower yield tests
detonated on the land or water surface. The best estimates of the
activity of globally distributed 90Sr come from measurements of
90Sr deposition. This amounts to 6 1017 Bq for all tests
conducted through 1980.
2. Nuclear fuel cycle
(a) Nuclear reactors
157. Strontium-90 is produced by fission in the fuel of nuclear
reactors. The amounts produced vary depending on the fuel
composition, reactor type, and degree of fuel burn-up achieved.
The yields for various fission processes were given in Table V.1.
In fairly high burn-up fuel (33000 MW(t)d t-1) of a PWR, the 90Sr
production is estimated to be 2.83 1015 Bq per tonne of fuel,
corresponding to 9.5 1013 Bq per MW(e)a of electricity
generated [O1].
158. Small amounts of 90Sr produced in the fuel in nuclear
reactors may reach the coolant through defects in the fuel
cladding. In coolant purification or following coolant leakage,
90Sr may reach the gaseous and liquid effluent streams. In
controlled amounts, some of the effluents are released to the
environment.
159. The activity releases of 90Sr were listed in the 1977 report
of UNSCEAR [U6]. Average discharges were of the order of 0.01 to 4
106 Bq per MW(e)a in PWRs and BWRs, respectively, with most of the
release in liquid effluents. Somewhat larger releases of 90Sr in
liquid effluents from GCRs arise primarily from spent fuel storage
pools. There are considerable variations in the release amounts
from individual reactors per unit electricity generated.
160. Assuming for each reactor type that the limited data of the
90Sr activity released per unit of electrical energy generated are
representative, it is possible to obtain a very rough estimate of
the total amount of 90Sr released from reactors worldwide. Using
the installed capacities of the various reactor types in 1980 [I1]
and assuming a reactor utilization of 60%, the estimated annual
release from all reactors is about 2 1012 Bq (Table V.2). In this
calculation, it is assumed for the reactor types for which no data
are available, that the releases are similar to those from BWRs.
Table V.2 Estimated global discharges of strontium-90
from nuclear power stations in 1980
------------------------------------------------------------
Reactor Reactor Capacity Production Estimated
type number [MW(e)a] rate [Bq strontium-90
per MW(e)a] discharge (Bq)
------------------------------------------------------------
PWR 96 64239 9 104 0.3 1010
BWR 62 35170 4 106 8 1010
GCR 36 7086 4 108 170 1010
Other 47 18490 4 106 4 1010
------------------------------------------------------------
Total 241 124985 2 1012
------------------------------------------------------------
161. There have not been many reports on 90Sr measurements in the
environment surrounding nuclear reactors that can be attributed to
reactor operation. In fact, 90Sr would not be a likely
radionuclide to be investigated, and furthermore results of
measurements could hardly be made independent of fallout 90Sr. The
much more readily detectable 137Cs, for example, is released in
activity amounts up to several hundred times that of 90Sr [U6].
Caesium-134 is released from reactors in amounts about 60% less
than 137Cs and, in addition, it would not be confused with weapons
fallout in the environment. For these reasons, estimates of 90Sr
levels in the environment from reactor release must generally be
based on measured releases and environmental dispersion
calculations.
(b) Fuel reprocessing plants
162. In fuel reprocessing plants the fuel is dissolved to recover
uranium and plutonium for reuse. All the 90Sr and other fission
products as well go to waste streams. The radionuclide activities
in airborne and liquid effluents from fuel reprocessing plants have
been recorded by UNSCEAR [U6]. The data for 90Sr are summarized in
Table V.3.
Table V.3 Average normalized discharges of strontium-80
to the environment from fuel reprocessing plants
(Bq per MW(e)a) [U6]
--------------------------------------------------------------
Plant Airborne Liquid Total
effluents effluents
--------------------------------------------------------------
Windscale (United Kingdom) 5 106 2 1011 2 1011
Nuclear Fuel Services (U.S.A.) 2 106 5 108 5 108
--------------------------------------------------------------
163. Strontium-90 is released from fuel reprocessing plants
primarily in liquid effluents. Relative to the total amounts of
90Sr in spent fuel, the release amounts are not large. The
fractional releases in liquid effluents are approximately 2 10-3
from the Windscale plant in the United Kingdom and were about
5 10-6 from the small Nuclear Fuel Services plant in the U.S.A.,
which is no longer in operation.
164. The fractional release of 90Sr in airborne effluents is about
3 10-8, compared to the amounts present in spent fuel. The
experiences at Windscale and at Nuclear Fuel Services are similar
in this regard.
165. The releases of radioactivity into the Irish Sea from the
Windscale plant and the corresponding levels in the environment
have been closely studied [N1]. Estimates of inventories of
activity in the sea and sediments and of movements of the activity
in its passage into the North Sea are being made [M1]. Some
results of environmental surveys around the reprocessing plant at
La Hague have also been published [S1]. Most of the attention,
however, is focused on the greater quantities of other fission
products released.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Movement in soil
166. The downward penentration of 90Sr in soil is slow, although
it is more rapid than for 137Cs or 249Pu. Even after several years
90Sr remains in the upper few centimetres in undisturbed soil. The
rate of movement varies with soil type; a low content of clay and
humus, a high content of electrolytes and a rapid movement of water
increase penetration [U2]. The mechanism of movement is thought to
involve both leaching and diffusion.
2. Transfer to plants
167. Plants acquire 90Sr by direct deposition onto foliage and by
root uptake of 90Sr in the soil. Absorption into the leaves is
relatively slow and superficial material is readily lost by
weathering. The translocation of 90Sr from plant leaf or grain
surfaces to other parts of the plant is small.
168. Capture of 90Sr on inflorescences of plants is of importance
for entry to grain. Concentrations in husked grain will be higher
than in the milled product during periods of deposition.
169. Uptake from soil is normally the primary mode of 90Sr entry
into plants. The quantity of absorbable calcium in soil is an
important factor in determining the extent of 90Sr absorption by
plants. Uptake is greatest from soils of low calcium content.
Uptake is thus reduced by the addition of lime, but usually not by
a factor exceeding 3 [U2]. When soils contain adequate calcium for
growth and the exchange capacity is largely saturated with calcium,
the addition of lime has little or no effect.
170. Other factors which affect root uptake of 90Sr include the
clay and humus content of the soil, pH, the concentration of
electrolytes other than calcium and the moisture content. The
addition of organic matter and fertilizers to soil may have varying
effects on plant uptake, which are, however, usually not large when
these materials are applied at normal agricultural levels.
171. Plant-base absorption of 90Sr has been noted to be quite
efficient. The 90Sr trapped in the surface root mat is relatively
undiluted with the calcium in the soil and is in a particularly
favourable position for absorption. This would help to explain the
higher concentrations of 90Sr in grain in periods shortly after
deposition and would be an important process in permanent pastures.
3. Transfer to milk
172. The total quantity of ingested 90Sr secreted into the milk of
cows is variable, depending on the milk yield. Values range from
0.5 to 2% of a single oral administration [U2]. With continuous
ingestion under normal conditions of feeding, several
investigations have shown that about 0.08% of the amount given
daily is secreted per litre of milk. The transfer to goats milk
may be more than ten times greater, corresponding to a higher
proportion of dietary calcium secreted into the milk as well.
4. Transfer to diet
173. The transfer of fallout 90Sr to diet has been extensively
studied. Assessments by UNSCEAR have been based on application of
generalized transfer models [U6]. The transfer from deposition to
diet is quantitatively described by means of the transfer factor
P23 defined as the time-integrated 90Sr/Ca quotient in the diet
divided by the 90Sr integrated deposition density. The integrals
may be replaced by summations if the relevant quantities are
assessed over discrete intervals of time. In fact, annual values
are the most generally available information. The transfer factor
P23 is usually expressed in mBq a/gCa per Bq m-2.
174. The transfer to diet from a specific deposition occurs over
an extended period as long as 90Sr remains in soil available for
root uptake. The model used by UNSCEAR to describe the transfer of
90Sr from deposition to diet is
infinite -µm
C(n) = b1 f(n) + b2 f(n-1) + b3 sigma f(n-m)e
m=1
where C(n) is the 90Sr/Ca quotient in total diet, in a food group,
or in an individual food item, in the year n; f(n) is the annual
deposition density in the year n, and b1, b2, b3, and µ are factors
which can be derived from reported data by regression analysis [U5,
U6]. The first term in the equation represents the contribution to
dietary 90Sr per unit deposition density in the current year, while
the second term expresses separately the contribution from
deposition in the previous year, reflecting also the use in the
current year of stored food produced in the previous year. The
third term expresses the contribution to dietary 90Sr from the
deposition density in all previous years, resulting from root
uptake and taking into account decay and loss of availability due
to downward movement in soil or to other physical or chemical
changes which may occur. The inverse of µ is the mean life of
available 90Sr in soil, which varies for individual foods and soil
conditions.
175. The transfer factor P23 describes the cumulative transfer of
90Sr to diet per unit deposition density. Using the model
described above, the expression of the transfer factor is
infinite -µm e-µ
P23 = b1 + b2 + b3 sigma e = b1 + b2 + b3 -----
m=1 1-e-µ
The transfer factor P23 can therefore be estimated from the
parameters b1, b2, b3 and µ obtained by regression analysis from
reported data.
176. The values of the parameters obtained from the regression
fits to 90Sr deposition density and diet data from the fallout
measurement programmes in New York and Denmark for dietary
components and for the total diet have been reported by UNSCEAR
[U6]. For total diet, the values of the parameters (b1 ~ 1.0,
b2 ~ 0.9, b3 ~ 0.3, µ ~ 0.1) give estimates of the initial
transfer (b1 + b2) of 1.9 mBq a (gCa)-1 per Bq m-2 and of the
long-term transfer (last term of equation above) of 2.9 mBq a
(gCa)-1 per Bq m-2. The total transfer, the value of P23, is
4.8 mBq a (gCa)-1 per Bq m-2.
177. Similar data from Argentina have also been evaluated, with
reasonable agreement found for all three areas for individual foods
and for total diet. Some differences are noted which are due to
the different definition of the food groups or to different soil
conditions and agricultural practices in the three countries.
178. The long-term transfer of 90Sr to diet from a single input to
the environment can be illustrated from the values of the transfer
parameters obtained from component groups of diet. For example, it
is determined that 90% of the cumulative transfer from a single
release of 90Sr is completed within 9 years for meat, fish and
eggs, 12 years for grain products, 14 years for milk, 32 years for
vegetables and 77 years for fruit. More rapid transfer indicates
that direct deposition processes are more important. Slow transfer
represents primarily uptake from the slowly decaying deposit of
90Sr in soil. Since the exponential decreases of the transfer for
the various groups differ, a direct fit to total diet data with a
single exponential transfer function is expected to be less
accurate than the summation of fits for the individual components.
179. The transfer of 90Sr from deposition density to some
individual foods, particularly to milk, has been studied for a
number of different areas of the world. These results are listed
in Table V.4. The transfer factors range from 2.1 to 7.6 mBq a/gCa
per Bq m-2.
Table V.4 Parameters of the transfer function between deposition density and 90 Sr/Ca in milk
-------------------------------------------------------------------------------------------------
Parameter Northern San New United Denmark Argentina Norway Australia Faroe
a/ hemisphere Francisco York Kingdom Islands
-------------------------------------------------------------------------------------------------
b1 0.84 0.61 0.69 0.89 0.99 1.39 0.70 2.07 2.70
b2 0.54 0.61 0.23 0.47 0.46 1.24 0.44 1.27 1.38
b3 0.22 0.19 0.19 0.15 0.23 0.12 1.02 0.30 0.81
µ 0.12 0.19 0.13 0.13 0.13 0.12 0.33 0.08 0.21
P23 3.1 2.1 2.3 2.4 3.1 3.5 3.7 7.2 7.6
-------------------------------------------------------------------------------------------------
a/ The unit for parameters b1, b2, b3 is mBq a(gCa)-1 per Bq m-2
The unit for parameter µ is a-1
The unit for the transfer factor P23 is mBq a(gCa)-1 per Bq m-2.
180. Milk has been used as an indicator of the levels of 90Sr in
total diet in areas where foods other than milk were not analysed.
This must be done, however, with some caution. It is usually the
case that following a period of 90Sr deposition, the 90Sr/Ca
quotient in milk declines somewhat more rapidly than the 90Sr/Ca
quotient in total diet. Where data are available, it is seen that
the exponential factor is greater in milk than in total diet. This
means that the diet-milk ratio of 90Sr/Ca values changes with time
during and after the period in which the 90Sr deposition occurs.
181. The diet-milk ratios at particular times also show
considerable variation from one country to another [U6]. In most
countries where milk is an important component of the diet, the
diet-milk ratio has averaged about 1.4 for most of the fallout
years. A trend to increasing ratios in the future is expected if
the deposition density rate remains at a very low level. In
countries where milk is not an important component of diet, the
diet-milk ratio will have a much higher value.
5. Aquatic behaviour
182. Strontium, like calcium, appears mainly in ionic form in
water and is not strongly sorbed by suspended particulate
materials. The fraction of strontium found in the particulate
phase in several freshwater systems ranged from 1 to 10% [V1].
183. It has been of interest to determine the behaviour of 90Sr in
aquatic environments in the vicinity of nuclear installations. A
number of determinations of the concentration factors (ratios of
integrated concentrations or of equilibrium concentrations in
organisms and in the water) have been performed in recent years for
various marine biota by measurements of stable and radioactive
strontium under laboratory and field conditions [C1, F2, N2, U4,
U1]. Typical values are 100 for algae, 2 to 10 for crabs and
lobster, about 1 for the flesh of ocean fish and 5 for fresh water
fish.
184. The primary uptake of strontium and also calcium by fish
occurs directly from the water. Therefore, accumulations in
organisms are little dependent on trophic level [V1]. The
concentration factor for fish depends inversely on the
concentration in the water. Vanderploeg et al. [V1] have suggested
a quantitative relationship. The concentration factors for fish
bone are about two orders of magnitude greater than for flesh.
185. The UNSCEAR aquatic model can be used to estimate the
transfer of 90Sr from the aquatic environment to diet for
generalized discharge situations [U6]. Further discussion of this
is presented in the section on dosimetry.
D. TRANSFER TO MAN
186. Strontium-90 is acquired by man primarily through ingestion
of 90Sr contaminated food. Terrestrial pathways are generally more
important than aquatic pathways in transferring 90Sr to man. From
fallout experience it is noted that 90Sr in drinking water always
contributes less than 5% of the total ingestion intake and 90Sr in
fish is a minor contributor even in countries where consumption of
fish is high. For example, it is estimated that only about 3% of
the fallout 90Sr intake by man in Japan between 1966 and 197l came
from fish [U1].
187. Correlations of fallout 90Sr in diet with measurements of
90Sr in bone have provided a relationship which is used to evaluate
the transfer to man. The transfer function used by UNSCEAR [U5,
U6] is:
infinite -µm
Cb(n) = c Cd(n) + g sigma Cd(n-m)e
m=o
where Cb(n) is the 90Sr/Ca quotient in bone in the year n, Cd(n) is
the dietary 90Sr/Ca quotient in the year n, and c, g, and µ are
constants determined from regression analysis of the bone and diet
data. The parameter can be associated with that portion of 90Sr
intake which is retained for a short term on bone surfaces and is
readily exchanged with plasma, and the parameter g with that
portion more tightly retained in bone. The exponential term
describes the effective removal rate of 90Sr from bone due to
radioactive decay and bone remodelling.
188. The transfer factor P34 linking diet and bone, is defined as
the ratio of the time integrated 90Sr/Ca quotient in bone to that
in diet. It may also be thought of as the cumulative transfer and
retention 90Sr in bone per unit intake in diet. The expression for
the transfer factor using the above function is
infinite -µm g
P34 = c + g sigma e = c + ------
m=o 1-e-µ
Data are available for regression analysis applying this model only
for 90Sr in vertebrae. It is recognized that there is greater
initial retention in cancellous bone such as vertebrae than in
compact bone, but there is also more rapid turnover. Therefore, the
time integrated results are expected to be representative of the
skeleton as a whole.
189. The results of regression fits of fallout 90Sr diet and adult
vertebrae data for various localities have been given in UNSCEAR
reports [U5, U6]. Representative values of the parameters are 0.02
Bq a (gCa)-1 in bone per Bq a (gCa)-1 in diet for both c and g and
about 0.2 a-1 for µ. The values determined for P34 range from 0.12
to 0.16 Bq a (gCa)-1 in bone per Bq a (gCa)-1 in diet.
190. From the values of the exponential parameter µ it is possible
to infer the mean residence time of 90Sr in bone. This ranges from
3.4 to 6.7 years, corresponding to bone turnover rates of 12 to 23%
per year. There is no reason to expect that the metabolic
behaviour should differ in the various areas of the world, and as
far as is known, dietary composition of usual foods does not affect
90Sr availability. Therefore, the differences in the estimate are
attributed to variations in the survey measurements.
191. To account for the bone 90Sr/Ca quotients in children,
Bennett [B2] has used age-dependent parameters in the above
transfer function. For children under 9 years the parameter c was
found to be zero, consistent with a single exponential transfer
model as originally proposed by Rivera [R1]. The best fit was
obtained with a turnover rate of 90Sr varying from about 100% per
year down to about 40% per year in the pre-teenage years and then
falling with age to about 20% per year for adults. Beninson [B1]
had reported a similar variation in turnover rate with age in
Argentina. The fractional retention of strontium, the fraction of
dietary intake incorporated into the skeleton, was also found by
Bennett [B2] to vary with age being five to seven times higher for
infants than for adults. Additional considerations of 90Sr
metabolism as a function of age have been presented by Papworth and
Vennart [P1].
192. The initial 90Sr concentration in the newborn must be
determined from an empirical relationship with the mother's diet.
The 90Sr/Ca quotient in bone of newborn varies from 0.1 to 0.2
times the 90Sr/Ca quotient in diet of the mother during the year
prior to the birth. An average of about 0.15 is obtained from the
survey data [B1, B2].
E. DOSIMETRY
1. Dose per unit intake
193. It is useful for radiological assessments to have expressions
for the dose per unit ingestion intake and per unit inhaled amount.
The absorbed doses in bone marrow and in bone-lining cells per unit
integrated activity of 90Sr in bone have been evaluated based on
the work of Spiers [S2]. These are the transfer factors P45
relating activity in bone to the doses [U5, U6]. The values are
P45 (bone marrow) = 0.38 mGy per Bq a (gCa)-1; P45 (bone-lining
cells) = 0.53 mGy per Bq a (gCa)-1. If it may be assumed that the
dietary calcium intake rate is 1 g daily (365 g/a), the transfer
factor to bone per unit intake, P34, is 0.14/365 Bq a (gCa)-1 per
Bq. This multiplied by the factor P45 gives the absorbed doses in
bone marrow and bone-lining cells. The results are given in Table V.5.
Table V.5 Absorbed dose per unit intake of
strontium-90 (Gy/Bq)
--------------------------------------------------
Lung Bone marrow Bone-lining
cells
--------------------------------------------------
Ingestion - 1.5 10-7 2.0 10-7
Inhalation 5.8 10-9 4.9 10-7 6.9 10-7
--------------------------------------------------
194. The ICRP Task Group lung model gives guidance regarding the
disposition of inhaled radioactivity [I3, I4] in the respiratory
tract. The respiratory system is divided into the nasopharyngeal
region (NP), the tracheobronchial region (TB), and the pulmonary
region (P). For the present dosimetric assessments it will be
assumed that the 90Sr is associated with typical ambient aerosols
of average diameter 0.5 µm. The 90Sr compounds are grouped in
Class D, retention in the lung being in the order of days,
specifically 0.5 d for the portion deposited in the pulmonary
region.
195. Fractional deposition of 0.5 µm particles in the NP, TB, and
P regions are 0.14, 0.08, and 0.30 and subsequent fractional
transfers to blood for the Class D compounds are 0.5, 0.95, and
1.0, respectively. Total transfer to blood is thus 0.446 of the
inhaled amount. Fractional transfer from blood to bone is 0.3
[I2].
196. The lung dose as a function of the inhaled activity can be
calculated from the expression
_
k A E f 1.44 TB
Dlung = ----------------
M
_
where k is a dosimetric constant, A is the activity inhaled, E is
the average energy per disintegration, f is the fraction of the
activity retained in the lung, TB is the retention half-time in the
lung, and M is the mass of the lungs. The lung dose per unit
activity of 90Sr inhaled is therefore:
Gy/d MeV 1.44 0.5d
Dlung = 13.8 10-6 ------ 1.13 --- 0.52 ---------
Bq MeV dis 1000 g
g dis
= 5.8 10-9 Gy/Bq
197. Assuming that the mean residence time of 90Sr in bone is 10
years, applicable to the skeleton as a whole, the integrated
concentration of 90Sr in bone per unit inhaled amount is 0.446 x
0.3 x 10 years 100 gCa = 1.3 10-3 Bq a/gCa per Bq inhaled. The
absorbed doses in bone marrow and bone lining cells are obtained by
multiplying the values of the transfer factor P45 given above. The
results are listed in Table V.5.
2. Dose per unit release
(a) Nuclear explosions
198. The dose commitment from 90Sr released by nuclear explosions
can be assessed using the environmental compartment model outlined
in the introduction. The transfer factor of a sequence of steps in
series is the product of the transfer factors of each step. The
dose commitment, Dc, is related to the integrated deposition
density of 90Sr, F, by the following expression:
Dc = P23 P34 P45F
where P23, P34 and P45 are the transfer factors discussed
previously. Average values for these transfer factors, as assessed
in the 1977 UNSCEAR report [U6], are P23 = 5 mBq a/gCa per Bq m-2,
P34 = 0.14, and P45 as given previously.
199. The dose commitment from 90Sr ingestion per unit of
widespread deposition density of 90Sr such as from nuclear
explosions is thus 0.3 µGy per Bq m-2 for bone marrow and 0.4 µGy
per Bq m-2 for bone lining cells.
200. The dose commitment from 90Sr via the inhalation pathway can
be estimated from the time integrated concentration of 90Sr in air.
Multiplying the dose commitment per unit inhalation intake (Table
V.5) by the inhalation intake rate of 22 m3 d-1 gives the value of
1.3 10-7 Gy per Bq d m-3 for the dose commitment to lungs per unit
integrated concentration of 90Sr in air.
201. The dose commitment to lungs can also be referred to measured
values of the integrated deposition density. Dividing the
integrated deposition density (Bq m-2 by the average deposition
velocity (m s-1) gives the time integrated concentration in air.
From long-term measurements of fallout 90Sr, the average deposition
velocity is 2.2 cm/s in New York, 2.0 cm/s in Argentina, and 1.5
cm/s in Denmark [A1, C2, E1]. The values determined from annual
measurements at all three sites range from 1.2 to 2.9 cm/s. The
value of 2 cm s-1 can be taken as representative. One Bq m-2
integrated deposition density thus corresponds to 5.8 10-4 Bq d
m-3) in air. The dose commitment to lung per unit integrated
deposition density of 90Sr is thus 7.5 10-11 Gy per Bq m-2.
202. The total amount of 90Sr released to the environment by
nuclear tests, 6 1017 Bq, has given a population-weighted
integrated deposition density of 1940 Bq m-2 in the world as a
whole [U6]. The world population is 4 109. With these values the
collective dose commitments per unit activity of 90Sr released may
be estimated. The results, which are summarized later, apply to
the geographic pattern of past nuclear tests.
(b) Nuclear installations
203. The activity of 90Sr in airborne effluents is dispersed by
turbulent air movement and is eventually deposited on the ground.
It then enters the ingestion pathway. The average situation over
several years of routine discharges will result in a nearly
completed deposition within a region with a radius R. The average
integrated deposition density, for a discharged activity A is F =
A/pi R2. The number of individuals exposed to that mean integrated
deposition density, on the other hand, is N = deltaN pi R2,
provided the population density deltaN can be assumed to be
constant over the relevant area.
204. It follows that the collective dose commitment per unit
activity released, Sc1, can be assessed by the expression
Sc = P23 P34 P45 deltaN
1
Using the values for the transfer factors given previously, and
assuming a population density of 25 man km-2, the collective dose
commitments per unit activity released are estimated to be about 7
10-12 man Gy per Bq for bone marrow and about 9 10-12 man Gy per Bq
for bone-lining cells. These values apply provided that food is
locally produced and that the production suffices for the
population density under consideration. The estimates would
probably be lower in actual circumstances.
205. The contribution of inhalation to the collective dose
commitment, for effluent releases over many years, can be estimated
by integration of the functions describing the atmospheric
dispersion, assuming complete depletion by deposition within 100
km. The collective dose commitment contribution per unit activity
released would be given by the expression
c X 100 km r -1.5
S1 = (Q)1 km I deltan phi / (1 km) 2 pi r dr
1 km
X
where (Q)1 km is the dispersion factor at 1 km from the release
point, I is the individual intake rate of air, deltaN is the
population density, phi is the dose per unit activity inhaled of
90Sr and r is the distance from the release point [U6].
206. The collective dose commitment may also be estimated using
the deposition velocity, thus avoiding the need to specify the
deposition area. That is, the integrated deposition density from a
discharged activity A is A/pi R2. Dividing by the deposition
velocity gives the integrated concentration of 90Sr in air.
Multiplying by the population, deltaN pi R2, removes the areal
dependence. Using a population density of 25 man km-2, an air
intake rate of 22 m3 d-1, a deposition velocity of 0.5 cm s-1
appropriate for particulates from near surface releases, and the
dosimetric factors given in Table V.5, estimates of the collective
r dose commitments per unit release are obtained. The results are
listed in the summary Table V.6.
207. The collective dose commitment from the input of 90Sr in
water bodies, normalized per unit activity released, can be
estimated [U5] using the expression
c sigmak Nk Ik fk phi
S1 = ------------------------
V(lambda + 1/tau)
Here V is the volume of receiving waters, tau is the turnover time
of receiving waters, lambda is the decay constant of 90Sr, Nk is
the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
208. The quantity V(lambda + 1/tau) is the infinite-time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite-time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentration in water and in fish will
be high, but the population which can be served with drinking water
or by fish consumption will be limited. For inputs into larger
volume of water the concentrations will be smaller, but the
population involved will be correspondingly larger. It seems
reasonable to assume, as a first approximation, that the quantities
Nk/V are relatively constant and independent of V.
209. A summary of the values used in the assessments presented by
UNSCEAR [U6] is given in the following listing:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 0.78 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.5
fish 5 1
shellfish 1
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
Using these values and the dosimetric factors given in Table V.5,
the collective dose commitments per unit activity released in
liquid effluents are estimated to be about 9 10-12 man Gy/Bq for
bone marrow and 1 10-11 man Gy/Bq for bone lining cells for
discharges into fresh waters, and a rounded value of 4 10-16 man
Gy/Bq for both tissues for discharges into the sea. The values for
sea discharges underestimate the collective dose commitment because
they neglect the contribution from large-scale mixing with a longer
residence time. However, due to the relatively short life of 90Sr,
this contribution can only have a small effect on the estimates.
3. Summary
210. Table V.6 summarizes the values obtained above for the
collective dose commitments, normalized per unit activity released,
for releases by atmospheric nuclear explosions and in effluents of
the nuclear power industry. In both cases they are the result of
generalized assessments and substantial variations should be
expected in site-specific cases.
211. For 90Sr and 90Y radiations, the quality factor is one, and
the weighting factors are 0.12 for lung and for bone marrow and
0.03 for bone lining cells. The effective dose equivalent
commitment per unit intake of 90Sr is thus 2.4 10-8 Sv/Bq
(ingestion) and 8.0 10-8 Sv/Bq (inhalation). The collective
effective dose equivalent commitments per unit release from nuclear
installations and from nuclear tests are included in Table V.6.
Table V.6 Summary of collective dose commitments per unit
strontium-90 activity released (10-14 man Gy per Bq)
----------------------------------------------------------------
Lung Bone Bone lining Effective
marrow cells a/
----------------------------------------------------------------
Nuclear explosions
Ingestion 400 500 60
Inhalation 0.1 8 10 1
Nuclear installations
Release to air b/
Ingestion 700 900 100
Inhalation 0.7 60 90 10
Release to fresh water
Drinking water 900 1000 100
Fish 20 30 3
Release to salt water
Fish 0.03 0.04 0.005
Shellfish 0.005 0.007 0.0008
----------------------------------------------------------------
a/ Collective effective dose equivalent commitment
(10-14 man Sv/Bq).
b/ Assumes population density of 25 man/km2 which is fully
sustained by local food production.
F. REFERENCES
A. Aarkrog, A. and J. Lippert. Environmental radioactivity in
Denmark in 1971, 1972, 1973, 1974 and 1975. Danish Atomic
Energy Commission. Riso reports 265 (1972), 291 (1973), 305
(1974), 323 (1975) and 363 (1976).
B1 Beninson, D., A. Migliori de Beninson, C. Menossi et al.
Radioestroncio en el hombre en funcion de la edad. Trabajo
presentado en el Quinto Congreso International de la "Société
francaise de radioprotection". Grenoble, 1971.
B2 Bennett, B.G. Strontium-90 in human bone - 1976 results for New
York and San Francisco. p. I-69-I-84 in Health and Safety
Laboratory environmental quarterly report HASL-328. New
York,1977
C1 Cancio, D., J.A. Llauró, N.R. Ciallella et al. Incorporación de
radioescio por organismos marinos. p. 347-356 in Radioactive
Contamination of the Marine Environment. Proceedings of a
symposium, Seattle, 1972. IAEA publication STI/PUB/313.
Vienna, 1973.
C2 Comisión Nacional de Energía Atomica. Argentina. Information
submitted 1977.
C3 Crouch, E.A.C. Fission product yields from neutron induced
fission. Atomic Data and Nuclear Data Tables, Vol. 19, No. 5.
Academic Press, New York, 1977.
E1 Environmental Measurements Laboratory, U.S. Department of
Energy. Environmental quarterly and appendix. Environmental
Measurements Laboratory report EML-334. New York, 1978.
F1 Federal Radiation Council. Estimates and evaluation of fallout
in the United States from nuclear weapons testing conducted
through 1962 - Report No. 4 (1963).
F2 Foyn, L. Some marine radioecological problems at the nuclear
power station establishment at Oslofjorden. Fisheries
Directorate Sea Research Institute report series B, No. 10.
Bergen, Norway, 1973.
H1 Hallden, N.A., I.M. Fisenne, D.Y. Ong et al. Radioactive decay
of weapons debris p. 194-199 in Health and Safety Laboratory
fallout program quarterly summary report HASL-117. New York,
1961.
I1 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.12
I2 International Radiological Protection. Report of Committee II
on Permissible Dose for Internal Radiation. ICRP publication 2,
Pergamon Press, 1959.
I3 International Commission on Radiological Protection. Task
group on lung dynamics. Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.
12: 173-226 (1966).
I4 International Commission on Radiological Protection. Task
group of Committee 2. The metabolism of compounds of plutonium
and other actinides. ICRP publication 19, Pergamon Press,
1972.
M1 Mitchell, N.T. Radioactivity in surface and coastal waters of
the British Isles, 1972-1973. U.K. Ministry of Agriculture
Fisheries and Food. Fisheries Radiobiological Laboratory
report FRL-7, FRL-8, FRL-9, FRL-10 (1971, 1973, 1975).
N1 National Radiological Protection Board. The data submitted by
the United Kingdom to the United Nations Scientific Committee
on the Effects of Atomic Radiation for the 1977 Report to the
General Assembly. NRPB report R47, Harwell (1976).
N2 Norwegian Institute for Water Research. Release of radioactive
materials from nuclear power stations. Report No. 2.
Dispersal mechanisms, pathways and concentration factors for
radionuclides in the cooling waters. Report 0-177/70 (1974).
N3 National Council on Radiation Protection and Measurements. A
handbook of radioactivity measurements procedures. NCRP report
No. 58, Washington D.C. (1978).
O1 Oak Ridge National Laboratory. Siting of fuel reprocessing
plants and waste management facilities. Oak Ridge National
Laboratory report ORNL-4451 (1970).
P1 Papworth, D.G. and J. Vennart. Retention of 90-Sr in human
bone at different ages and the resulting radiation doses.
Phys. Med. Biol. 18: 169-186 (1973).
R1 Rivera, J. and J.H. Harley. The HASL bone program 1961-1964.
U.S. Atomic Energy Commission report HASL-163. New York, 1965.
S1 Scheidhauer, J., R. Ausset, J. Planet et al. Programme de
surveillance de l'environment marin du centre de La Hague.
p. 347-365 in Population Dose Evaluation and Standards for
Man and His Environment. IAEA publication STI/PUB/375. Vienna,
1974.
S2 Spiers, F.W., G.D. Zanelli, P.J. Darley et al. Beta-particle
dose rates in human and animal bone. p. 130-148 in Biomedical
Implications of Radiostrontium Exposure. U.S. Atomic Energy
Commission Symposium Series 25 (1972).
U1 Ueda, T., Y. Suzuki and R. Nakamuru. Transfer of caesium-137
and strontium-90 from the environment to the Japanese
population via the marine environment in Population Dose
Evaluation and Standards for Man and His Environment. IAEA
publication STI/PUB/375. Vienna, 1974.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No. 16
(A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Nineteenth Session, Supplement
No. 14 (A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-First Session, Supplement No.
14 (A/6314). New York. 1966.
U5 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication, No. E.72.IX.17 and
18. New York, 1977.
U6 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
V1 Vanderploeg, H.W., D.C. Porzyck, W.H. Wilcox et al.
Bioaccummulation factors for radionuclides in freshwater biota.
Oak Ridge National Laboratory report ORNL-5002 (1975).
VI. IODINE
A. INTRODUCTION
212. Iodine is a volatile element which is very mobile in the
environment. It is non-uniformly distributed in nature, its
abundance in the lithosphere being about 5 times higher than in the
ocean waters. The iodine in the sea apparently originates from
erosion of the land masses. Recycling to the terrestrial biosphere
occurs through evaporation of sea water and decomposition of
substances of marine origin.
213. There are at least 25 iodine isotopes with mass numbers
ranging from 117 to 141. All except 127I are radioactive. Omitting
the very short-lived 140I and 141I, thirteen isotopes are produced
by fission:
127I (stable), 128I (25 min), 129I (1.57 107a), 130I (12.4 h),
131I (8.06 d), 132I (2.3 h), 133I (21 h), 134I (52.8 min),
135I (6.7 h), 136I (83 s), 137I (23 s), 138I (5.9 s) and 139I
(2 s). From the point of view of environmental contamination and
resulting doses to man, the most important isotopes of iodine are
131I and 129I. They are the only radioactive isotopes of iodine
produced by fission with half-lives longer than one day. Iodine-
131 is a beta-emitter with a half-life of 8.06 days and a maximum
beta energy of 0.81 MeV emitting also gamma rays of 0.36 and 0.64
MeV and other energies. Iodine-129 has a very long half-life (1.57
107 a); it is a beta-emitter (maximum energy: 0.15 MeV) with an
accompanying gamma ray of 0.09 MeV in 8% of the disintegrations
[D1]. The two isotopes are mainly found in the environment as a
result of nuclear explosions and releases from nuclear reactors and
fuel reprocessing plants. Only these two isotopes of iodine are
considered in this report.
214. Iodine enters the metabolism of living organisms and is
selectively taken up and concentrated in the thryoid gland; it
plays a major role in the synthesis of the thyroid hormone and is
secreted in milk. Owing to its decay properties, 131I has been
extensively used in the medical field for diagnosis and treatment
of thyroid abnormalities.
B. SOURCES
1. Natural production
215. Like any other fission product, 129I and 131I are present in
the environment as a result of spontaneous fission of natural
uranium. In view of its very long half-life, 129I has accumulated
in the earth's crust and also in the ocean waters from where it is
available to disperse in the whole biosphere.
216. In 1962, Edwards [E1] predicted the natural 129I/127I atom
ratio in sea water to be about 3 10-14 from spontaneous fission of
uranium-238 and estimated that contributions of the same order of
magnitude would arise from spontaneous fission of 235U and from
production in the upper atmosphere by interaction of energetic
protons, neutrons and photons on isotopes of xenon. Experiments
later confirmed the validity of Edwards' estimate, the 129I/127I
atom ratios derived from measurements in iodine-rich minerals
ranging from 2 10-15 to 10-13 [M1, S1]. Taking the concentrations
of 127I in sea water to be 0.064 µg g-1 and the global volume of
sea water to be 6 1017 m3, a natural activity of 129I of 7 109 Bq
in the oceans is obtained from an 129I/127I atom ratio of 3 10-14.
2. Nuclear explosions
217. The activity of 131I (or of 129I) generated in nuclear
explosions can be derived from the measurements of 90Sr deposition
[U1] and from the ratios of the fission yields of 131I (or 129I)
and of 90Sr for nuclear weapons tests [H2].
218. The total activity of 90Sr produced in nuclear explosions
through 1976, which has been globally distributed, is estimated to
be 6 1017 Bq [U1]. This does not include the local fallout, which
is deposited in the vicinity of the test area and which can be
important for the lower yield tests detonated near or on the land
surface.
219. On the basis of measured fission product yields of individual
nuclides obtained by analysis of debris from megaton weapons, the
yields for 90Sr, 129I and 131I are estimated to be 0.035, 0.0126
and 0.029, respectively [H2].
220. The activities of 129I and 131I produced in nuclear tests
through 1976, which gave rise to globally distributed fallout, are
thus found to be 4 1011 Bq and 6 1020 Bq. Owing to the half-lives
of the two nuclides, practically all of the activity of 129I is
still present in the environment where it will remain for millions
of years whereas practically all of the activity of 131I has
decayed.
3. Nuclear fuel cycle
(a) Nuclear power plants
221. Iodine-131 and iodine-129 are produced by fission in the fuel
of nuclear reactors. The equilibrium activity of 131I per unit of
electrical power is established after a few weeks of irradiation in
uranium fuel at about 3 1015 Bq per MW(e) [U1] and increases
slightly from the beginning of the fuel irradiation to the end, as
the result of the larger fission yield of plutonium which
contributes increasingly to power production as the burn-up
proceeds. The corresponding activity of 131I produced per unit
energy generated is about 9 1016 Bq per MW(e)a.
222. Small amounts of 131I produced in the fuel may reach the
coolant of the nuclear reactor through defects in the fuel
cladding. In coolant purification or following coolant leakage,
131I may reach the gaseous and liquid effluent streams. The
reported 131I releases in airborne effluents, as summarized by
UNSCEAR [U1], show an extremely wide variability due mainly to
different waste treatment systems. The overall activity discharged
per unit energy generated is of the order of 107 Bq per MW(e)a from
PWRs and 108 Bq per MW(e)a from BWRs [I1, U1] while the limited
data from GCRs and HWRs indicate that the average airborne releases
are comparable to those from PWRs [U1].
223. Airborne iodine can occur in various chemical forms in the
airborne effluents. Elemental iodine (I and I2), organic iodine
with methyl iodide (CH3I) as the simplest organic compound and
hypoiodous acid (HOI) may be present in significant proportions,
iodine is also to some extent bound to particulates.
224. Few data are available on the proportions of organic and
inorganic forms of the iodine released to the atmosphere. Analyses
at power stations in the Federal Republic of Germany show that only
a very small fraction (usually less than 1%) of the iodine released
in airborne effluents is in particulate form [W1]. Recent
measurements in the U.S.A. indicate that, in airborne effluents
from PWRs, on the average 31% of 131I is organic, 40% HOI, 27%
elemental and 2% in particulate form [P1], whereas in BWRs, the
proportions were found to be somewhat different, namely 40%
organic, 20% HOI, 28% elemental and 12% particulate [P2]. These
proportions, however, are expected to vary significantly according
to the type of waste treatment in use.
225. In liquid effluents from LWRs, 131I is found in amounts
comparable to the airborne releases from BWRs (108 Bq per MW(e)a)
[U1]. It is not reported in the liquid effluents from other types
of reactors.
226. In 1980, the installed nuclear capacity was 1.25 105 MW(e) on
a worldwide scale [I2]. Assuming an average load factor of 0.6,
the energy produced was 7.5 104 MW(e)a. The global production and
release of 131I at the reactor sites in 1980 are estimated to be
about 7 1021 Bq and 2 1013 Bq, respectively, using the figures
given previously for production and release, and assuming that the
releases are similar to those from BWRs for the reactor types for
which no data are available. The estimate of the global release is
very crude, but nevertheless shows that only a tiny fraction of the
order of 10-9 of the 131I produced in the reactors is discharged
into the environment. Table VI.1 provides a break-down of the
releases from reactors according to reactor type.
227. Being a volatile element, iodine is readily released to the
atmosphere in the case of a reactor accident. The two reported
reactor accidents which have caused measurable irradiation of the
public occurred at Windscale (U.K.) in October 1957 and at Three
Mile Island in March 1979. The release of 131I to the atmosphere
was about 7 1014 Bq in the Windscale accident [L1] and 6 1011 Bq in
the Three Mile Island accident [H3].
228. The activity of 129I produced in a nuclear reactor is much
lower than that of 131I. The production of 129I per unit energy
generated is approximately 5 107 Bq per MW(e)a [U1] corresponding
to an inventory of 1.5 108 Bq per MW(e) after three years of fuel
irradiation. Iodine-129 has not been identified in routine
discharges of nuclear reactors. A rough estimate of the activity
of 129I discharged per unit energy generated can be calculated
assuming that the ratio of the release rate to the inventory in
reactor fuel is the same for 129I and 131I. As the activity of
129I in fuel is at least 2 107 times lower than that of 131I, the
release rate of 129I per unit energy generated is at most 0.1 Bq
per MW(e)a; activity of 129I discharged from nuclear reactors in
1980 would thus be of the order of 104 Bq.
(b) Fuel reprocessing plants
229. At the fuel reprocessing stage of the nuclear fuel cycle (if
it is undertaken), the elements uranium and plutonium in the
irradiated nuclear fuel are recovered for reuse in fission
reactors. Before reprocessing, the spent fuel elements are stored
under water until 131I has decayed to insignificant amounts.
Storage times of six months and one year result in the reduction of
the activities of 131I originally present in the fuel by factors of
6.5 106 and 4.3 1013, respectively.
230. In 1980, the only reprocessing plants operating commercially
were at Windscale (U.K.) and La Hague and Marcoule (France): in
addition there were several small experimental reprocessing
facilities, such as the one at Karlsruhe (Federal Republic of
Germany). The combined capacity of the reprocessing plants was
much lower than the amount of fuel discharged from reactors
worldwide.
Table VI.1 Estimated global discharges of 131I from nuclear power
stations in 1980
-------------------------------------------------------------------------
Estimated release Estimated discharges
rates in 1980
Reactor Number Capacity (Bq per MW(e)a) (Bq)
type (MW(e)a) ----------------- -------------------------
Airborne Liquid Airborne Liquid Total
-------------------------------------------------------------------------
PWR 96 64239 107 108 6 1011 6 1012 7 1012
BWR 62 35170 108 108 4 1012 4 1012 8 1012
HWR 14 5963 107 108 6 1010 6 1011 7 1011
GCR 36 7086 107 108 7 1010 7 1011 8 1011
Other 33 12527 108 108 1 1012 1 1012 2 1012
-------------------------------------------------------------------------
Total 241 124985 - - 6 1012 1 1013 2 1013
-------------------------------------------------------------------------
231. The activity of 131I released into the environment from
reprocessing plants depends critically on the storage time: in
practice with the growing backlog of fuel for reprocessing a
storage time of one year is common but even a relatively small
quantity of fuel with a short storage time will dominate the total
131I releases. Luykx and Fraser [L2] have expressed the reported
releases of 131I from Windscale, La Hague, Marcoule and Karlsruhe
during 1974-1978 in terms of activity discharged per unit of
electricity generated. The results range from less than 1.5 105 to
7 107 Bq per MW(e)a and are presented in Table VI.2. The
activities of 131I discharged in liquid effluents have not been
reported.
232. The discharges of 129I depend upon the specific waste
treatment at the reprocessing plant. With regard to airborne
effluents, the reported activites released per unit of electricity
generated were, on average during the 1975-1978 time period, 2.7
106 Bq per MW(e)a at Windscale and 4.8 105 Bq per MW(e)a at
Karlsruhe, representing about 4% and 1%, respectively, of the fuel
content [L2]. In a series of measurements from November 1975 to
August 1977 the average values for the components of 129I
discharges from Karlsruhe were reported at 74% inorganic, 23%
organic and 2% aerosol [B1].
Table VI.2 Average normalized activities of 129I and 131I
discharged into the environment by fuel reprocessing plants
(Bq per MW(e)a)
---------------------------------------------------------------------------
Iodine-129 Iodine-131
Plant ---------------------------- ------------------------------
Location Airborne Liquid Total Airborne Liquid Total
effluents effluents effluents effluents
---------------------------------------------------------------------------
Windscale 2.7 106 5.6 107 5.9 107 2.6 106 N.A. a/ >2.6 106
La Hague N.A. N.A. N.A. 1.1 107 N.A. >1.1 107
Marcoule N.A. N.A. N.A. 7.4 107 N.A. >7.4 107
Karlsruhe 4.8 105 N.A. N.A. >1.5 105 N.A. -
---------------------------------------------------------------------------
a/ N.A. = Data not available.
233. In recent years, the 129I released in liquid effluents was
only measured at Windscale. They average at 5.6 107 Bq per MW(e)a
which corresponds fairly well with the theoretical fuel content
[L2].
234. The information on 131I and 129I activities discharged per
unit electricity generated is summarized in Table VI.2. If it is
assumed for the four reprocessing plants, that all the 129I
contained in the fuel is discharged into the environment, the total
129I released in 1978 was about 3 1011 Bq, which is 7 orders of
magnitude higher than the total activity estimated to be released
from reactors. With regard to 131I, it is much more difficult to
assess the total activity released from fuel reprocessing plants,
as the activities discharged into liquid effluents have not been
reported. However, using the pessimistic assumptions that the
activity contained in the airborne effluents represents 1% of the
activity present in the fuel at the time of reprocessing and that
the rest of the activity is discharged into liquid effluents, it is
found that the total annual 131I discharges from fuel reprocessing
plants are about 5 1011 Bq, which is much less than the global
discharges from reactors.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Nuclear explosions
235. The behaviour in the environment of 131I produced in the
nuclear explosions has been extensively studied, especially the
air-vegetation-milk pathway which is generally the most significant
route by which humans are exposed. Much of the literature has been
referenced in UNSCEAR reports [U1-U5]. Environmental
concentrations of 131I following large nuclear explosions are
significant; they are easily measured and this allows the transfer
factors to be derived from observations. In contrast, the
environmental concentrations (and the resulting dose rates) of 129I
are extremely low and have only been measured in a few studies.
Although some of the aspects of environmental behaviour of 131I
following nuclear explosions apply also to 129I, the discussion in
this section will be limited to 131I.
236. Radioactive fallout is observed to circle the earth in 20-30
days on average [R1] which is approximately the mean residence time
of an aerosol in the troposphere and is longer than the mean life
of 131I. It is thus during its first pass around the earth that a
given atom of 131I formed in a nuclear explosion will either decay
in the atmosphere or deposit on the earth's surface. It is
unlikely that during such a short period the clouds of debris
become well mixed. The ground-level air concentrations of 131I at
a particular station fluctuate according to meteorological
conditions and are not necessarily representative of a larger
region nor of a latitude band [P3].
237. Information on the physical and chemical nature of fallout
131I is very limited. In the U.K., late 1961, an average 75% of
the activity was in particulate form, the rest being in the gaseous
state [E2] but in the U.S.A. in 1962 the particulate fraction was
found to vary from 10 to 90% [P4]. These large variations may be
partly explained by the physical and chemical transformations
undergone by 131I following its formation: Voillequé [V1] observed
that the fraction of the total airborne 131I associated with
particulates is about 0.5 to 0.7 in the first few days following a
nuclear explosion but that it later decreases to be approximately
0.3 after two months. In the gaseous fraction, the proportion of
organic compounds was found to increase in the same two months time
period from one fourth to about three fourths of the gaseous iodine
[V1].
238. Iodine-131 deposition on the ground and on vegetation occurs
by dry and wet deposition. The rate of dry deposition can be
characterized by the deposition velocity on the vegetation which
was derived to be about 2 10-2 m s-1 [C2, H4] from measurements
performed during the series of tests of late 1961. When
precipitation occurs, fallout 131I is deposited at a much faster
rate than in dry weather, essentially by rain-out, i.e., in-cloud
mechanisms, rather than by wash-out, i.e., below the cloud
processes [U5]. On the other hand, rain washes the surface of the
leaves and thus removes some of the radio-iodine. Chamberlain and
Chadwick in the U.K. [C2] and Hull in the U.S.A. [H4] calculated on
the basis of their measurements that in late 1961 about 50% of the
131I falling out in rain was retained on herbage.
239. Even though the observed ground-level air activity
concentrations and deposited activities of 131I vary widely from
one area to another according to meterological conditions, it is
possible to obtain a rough estimate of the total activity density
deposited, weighted over the world's population, from the average
ratio of the 131I/140Ba deposited activity densities. Data from
Argentina covering the years 1966 to 1973 [B2, B3, C3] reveal that
the 131I/140Ba ratio of the annual deposited activity densities
varied from 0.4 to 1.3 with a median value of 0.6. Relevant
information is also provided by the air concentrations of 131I and
140Ba which are measured in the stations of the global network of
the U.K. Atomic Energy Agency [C4]. The annual average of the
integrated air activity concentration 131I/140Ba ratios of nine
stations scattered over the world ranged from 0.19 to 3.1 with a
median value of 0.46. However, the corresponding ratios of the
deposited densities were higher as only the particulate fraction of
131I was measured in the air. Assuming that the particulate
fraction of 131I represents half of the total activity of that
nuclide in the air, the median value would be approximately 0.9,
which is comparable to the Argentinian value of 0.6. An
intermediate value of 0.7 will be adopted in this document.
240. As the average ratio of the 140Ba/95Zr deposited activity
density is estimated to be 0.62 [U2] and the population-weighted
global average deposition density of 95Zr from all tests is
approximately 2.4 104 Bq m-2 [U2], the population-weighted global
average deposition density of 131I is thus found to be on the order
of 104 Bq m-2.
241. Fresh milk is usually the main source of 131I in food because
of the concentration achieved by the grazing animal and the short
storage period of milk. Besides, milk plays an important worldwide
role in the diet of infants. The relationship between the
integrated cow's milk concentration and the deposition density has
been derived from measurements in Argentina [B2] to be 6.3 10-4 Bq
a 1-1 per Bq m-2 and to show little variation from year to year.
The integrated milk concentrations observed throughout the world by
the fallout network stations have been reported by UNSCEAR [U1-U5].
2. Industrial releases
242. The environmental behaviour of radio-iodines released from
nuclear facilities differs in some aspects from that of fallout as
the chemical forms are not in the same proportion and as the
releases occur at discrete points on the surface of the earth both
in the atmosphere and in the aquatic environment. As the
authorities are concerned with the total impact resulting from the
releases of radionuclides, all the important pathways leading to
man have been investigated, mainly through laboratory and field
experiments, and occasionally following unplanned releases. In
comparison to 131I and to fallout, much more 129I is released from
industrial operations and it will be considered in this section,
together with 131I in the discussion of the local and regional
aspects, and on its own in the discussion of the global aspects.
(a) Local and regional aspects
(i) Atmospheric releases
243. The behaviour in the atmosphere of the radio-iodines released
from nuclear facilities is complicated by the various forms that
iodine may take (particulate, elemental, organic, or as hypoiodous
acid). Elemental iodine readily deposits on forage and enters the
cow-milk-man pathway. Organic iodine is retained much less
efficiently by vegetation and its deposition velocity is 200 to
1000 times smaller than that of the elemental form [A1, H5].
Particulate associated iodine and hypoiodous acid will be deposited
at rates intermediate between those for the elemental and organic
forms [V2]. Physico-chemical transformations occurring during
atmospheric transport may also affect the distribution of the
various forms of iodine, since some of them are not stable in
sunlight. On the basis of photochemical considerations, the
atmospheric residence times for I2 are estimated to be less than a
minute during the day, much shorter than the residence times for
CH3I and other organic iodides in plant effluents (about 60 hours
in sunlight) [V1]. The elemental form would be expected to become
rapidly associated with airborne aerosols, so that deposition at
distances beyond the immediate vicinity of the release would be
largely governed by the particulate behaviour. It must be pointed
out, however, that the atmospheric residence times of CH3I derived
from environmental measurements is much longer than that obtained
from photochemical considerations (about 100 days to be compared
with 60 hours in sunlight) [V1].
244. Numerous field and laboratory experiments have been conducted
to determine the deposition velocity on vegetation of various forms
of iodine [B4, H6, V2]. Most of the experiments dealt with
elemental iodine, for which the deposition velocity was found to
vary with the temperature, the relative humidity of the air, the
wind speed and the vegetation density. The best fit of the
experimental data is obtained assuming that the deposition velocity
on vegetation of elemental iodine is proportional to the wind speed
and to the vegetation density and is an exponential function of the
temperature and of the relative humidity of the air [A2]. Typical
values of the deposition velocity on vegetation are 2 10-2 m s-1
for elemental iodine and 5 10-5 m s-1 for organic iodide. In the
case of particulates, a value of 10-3 m s-1 was found representative
for grass, about 2 10-3 m s-1 for clover and about 3 10-4 m s-1 for
vegetation-free soil [H7]. Since the deposition velocity varies
with the vegetation density, less variability is encountered by
normalizing the values by the mass of dry vegetation per unit area.
UNSCEAR [U1] adopted a normalized value of 5 10-3 m3 kg-1 s-1 for
the deposition velocity on grass of radio-iodine in effluents from
nuclear installations.
245. Regarding atmospheric releases of radio-iodine, the main
pathways to man are inhalation and consumption of fresh milk and
fresh leafy vegetables; consumption of beef is also taken into
consideration in the case of 129I.
246. The assessment of the transfer of iodine to milk requires the
knowledge of the value of the following parameters in addition to
the deposition velocity; the residence half-time of iodine on
vegetation, the average mass of grass consumed per cow and per day
under average agricultural conditions during the grazing season,
the fractional pasture grazing time and the fractional transfer of
the daily ingested activity by the cow per unit volume of produced
milk.
247. The residence half-time of 131I on grass is 3-6 days, most
estimates lying around 5 days [B4, B5]. This figure seems to be
valid irrespective of the iodine production source (fallout,
nuclear plants, experiments) and of the climatic characteristics of
the region [B4]. The corresponding residence half-time of stable
iodine (or of 129I) on grass is about l4 days. The depletion
mechanisms involved are: transfer to the roots; volatilization;
leaching by atmospheric precipitation; mechanical removal by wind,
rain or other agents; death or decomposition of the leaves or of
their surface layer [B4, C2]. The opinions do not agree on the
relative importance of those mechanisms [B4, C2].
248. The daily grass requirement of a lactating cow is estimated
at 10 kg dry matter [B4, H6]. The pasture grazing time varies
according to the climatic conditions and to the cattle management
practices. An average grazing time of six months per year was
assumed by UNSCEAR [U1]. During the winter months, the cows are
held in the stable and consume dry fodder in which the activity
concentration of 131I will have decayed to insignificant levels and
that of 129I can be assumed to be the same as that in herbage.
249. The transfer of iodine is usually expressed as the fraction
present in milk of the ingested activity under equilibrium
conditions. This quotient was found to be around 5 10-3 d 1-1 [B6,
U1]. The value adopted by UNSCEAR in its 1977 report is an upper
estimate of 10-2 d 1-1 [U1]. In the case of a simple
administration of 131I to the cow, Lengemann and Comar [L3]
observed that the maximum concentration is reached within one day
and that it is followed by a rapid decrease (half-time of about 1.5
days) in the first 3-4 days and a slower decrease (half-time of
about 3 days) afterwards. Among the different factors which can
have an influence on the grass to milk transfer of iodine, the two
most important may be the milk productivity by animals and the
season, a higher iodine secretion in milk occurring with a
productivity increase and in the warm season [B4, G1].
250. Taking account of all the parameters given above, an
integrated air concentration of 1 Bq a m-3 of 131I or 129I would
result in an integrated milk concentration for 131I of 160 Bq a 1-1
and for 129I of 870 Bq a 1-1. Transfer via wet deposition is
usually insignificant over the course of a year [U1].
251. The transfer of iodine from air to fresh leafy vegetables has
been assessed by UNSCEAR [U1] on the basis of the values given
above for the deposition velocity and the residence time on the
vegetation, and of a dry-to-wet vegetation weight ratio of 0.5. In
addition, a fractional removal by washing of 0.4 [U6] and an
average 7 day marketing delay (resulting in a decay factor of 0.55
for 131I and 1.0 for 129I) were taken into account. Time
integrated conditions of 340 Bq a kg-1 (fresh weight) for 131I and
1740 Bq a kg-1 (fresh weight) for 129I are calculated for the case
of a time-integrated air concentration of 1 Bq a m-3.
252. An estimate of the transfer of 129I has also been carried out
by UNSCEAR [U1]. Using the values given above for the deposition
velocity on grass, residence half-time on grass and grass
consumption rate, the resulting transfer factor is 260 Bq a kg-1
per Bq a m-3 of 129I in air if the fractional transfer of daily
ingested activity per unit mass of meat is taken to be 3 10-3
d kg-1 [P5].
253. In the assessment of the 129I concentration in grass
following deposition of that nuclide, the root uptake from soil has
not been taken into account. It has been estimated [B7] that this
pathway could contribute at equilibrium only about 20% of the
concentration in grass arising from direct deposition.
(ii) Aquatic releases
254. Information on the behaviour of iodine in the aquatic
environment is rather limited. The UNSCEAR aquatic model [U1] can
be used to estimate the transfer of 131I and 129I from the aquatic
environment to diet for generalized discharge situations. In that
model, it is assumed that iodine is not removed to sediments but
that during treatment of drinking water, 20% of the activity
contained in raw water is removed. For the two isotopes of iodine
considered, the concentration factors are taken to be 15 1 kg-1 for
fresh water fish, 20 1 kg-1 for marine fish and 100 1 kg-1 for
shellfish. Further discussion is presented in the section on
dosimetry.
(b) Global aspect
255. Because of its very long half-life (1.57 107 a) and of the
mobility of iodine in the environment, 129I may become widely
distributed on the global scale. Whether released into the
atmosphere or into the aquatic environment, 129I will eventually
reach the oceans in a time period very short in comparison with its
half-life. Iodine-129 will then be recycled to the atmosphere and
the terrestrial biosphere, mainly by evaporation of seawater.
Atmospheric water is exchanged within about 10 days and it follows
therefore that there is a rapid exchange of iodine. The specific
activity approach has been the usual method to assess the dose
commitments and the long-term environmental concentrations from
129I discharges. According to that approach, the specific activity
of 129I per unit mass of stable iodine of any environmental
material of the terrestrial biosphere (including air and food-stuffs)
will be in the long term equal to that of seawater. Assuming 6
1023 g to be the mass of ocean waters with an iodine concentration
of 0.064 µg per gram of water [T1], the specific activity of 129I
per unit mass of stable iodine obtained in sea water after a
release of 1 Bq of that radionuclide into the environment is found
to be 2.6 10-17 Bq g-1. If it is assumed that there is no
environmental sink for iodine, 129I will be recycled throughout its
mean life of 2.3 107a.
D. TRANSFER TO MAN
256. Iodine is an element of fundamental importance for the human
organism since it is an essential component of the thyroid hormone,
which is necessary for the growth and metabolism of the body. The
metbolic cycle of iodine in man, especially in the adult, is
sufficiently well known in its fundamental behaviour, as a
consequence of the large number of clinical studies carried out in
the last years with radioactive isotopes of iodine [B4].
257. The absorption by the blood from the gastro-intestinal tract
is complete and very rapid. It is absorbed at the rate of about 5%
per minute and it can be considered to be complete after two hours
[B4]. When inhaled in the form of inorganic iodide or as
methyliodide, a fraction of about 70% is absorbed [M2] whereas more
than 90% of it is absorbed when inhaled in the form of elemental
vapour [M3].
258. The uptake by the thyroid of the iodine contained in blood as
well as the size of the thyroid gland are both very dependent upon
the daily intake of stable iodine [D2]. The model adopted by ICRP
[I3] for the metabolism of iodine applicable to adults is based on
the three-compartment model of Riggs [R2]. ICRP [I3] assumes that
30% of the iodine entering the blood is translocated to the thyroid
while the remainder goes directly to excretion. Iodine in the
thyroid is assumed to be retained with a biological half-life of
120 days and to be lost from the gland in the form of organic
iodine. Organic iodine is assumed to be uniformly distributed
among all organs and tissues of the body other than the thyroid and
to be retained there with a biological half-life of 12 days. One-
tenth of this organic iodine is assumed to go directly to faecal
excretion and the rest is assumed to be returned to blood as
inorganic iodine.
259. A quantitative assessment of the transfer of iodine to man
must take into consideration the variation with age of the various
parameters involved, which are essentially the mass of the thyroid
gland, the fractional uptake by the thyroid, the effective
residence half-time in the thyroid, the breathing rate and the
consumption rate of foodstuffs. The values adopted for 129I and
131I by UNSCEAR in its 1977 report [U1] are presented in Table
VI.3. It is to be noted that the metabolic parameters for adults
are not in complete agreement with those adopted by ICRP [I3]; the
resulting differences in the thyroid absorbed doses vary according
to the pathway and the isotope considered but they are in all cases
less than 50%.
Table VI.3 Variation with age of the parameters used in the
assessment of the transfer to man of 129I and 131I [U1]
-----------------------------------------------------------------
Age
6 4 14 Adult
months years years
-----------------------------------------------------------------
Mass of the thyroid (g) 2 4 14 20
Effective half-time
in the thyroid (d) 131I 6.0 6.3 6.9 7.6
129I 23 28 48 136
Inhalation pathway
Fractional uptake by the thyroid 0.30 0.26 0.26 0.26
Breathing rate (m3 a-1) 1150 3530 6440 8030
Ingestion pathway
Fractional uptake by the thyroid 0.40 0.35 0.35 0.35
Consumption rate:
Milk (1 a-1) 330 180 150 90
Leafy vegetables (kg a-1) 0 13 20 30
Beef (kg a-1) 0 8 15 27
Drinking water (1 a-1) 438 438 438 438
River fish (kg a-1) 1 1 1 1
Ocean fish (kg a-1) 6 6 6 6
Shellfish (kg a-1) 1 1 1 1
-----------------------------------------------------------------
E. DOSIMETRY
1. Dose per unit intake
260. As iodine is selectively taken up in the thyroid gland, its
concentration in that organ is considerably higher than in the
other organs and tissues in the body. The stable iodine
concentration in the thyroid tissue of an adult is of the order of
500 µg g-1 while in the rest of the body it is much less than 1 µg
g-1 [S2]. Since the major contribution of the absorbed doses from
129I and 131I is due to the emission of the beta-particles, which
have a short range in the human tissues, the absorbed doses in the
thyroid are about 1000 times higher than those in the other organs
and tissues, which will not be considered in this document.
261. The significant variation with age of the metabolic
parameters is reflected in the thyroid absorbed doses per unit
intake. Table VI.4 presents the values of the age-dependent
thyroid absorbed doses per unit intake adopted by UNSCEAR in its
1977 report [U1]. These values are derived from the metabolic
parameters presented in Table VI.3 and from the following figures
for the energy absorbed in the thyroid (MeV) for disintegration:
0.18, 0.18, 0.19 and 0.19 MeV for 131I and 0.060, 0.061, 0.063 and
0.064 MeV for 129I for the ages of 0.5, 4, 14 years and adult,
respectively.
Table VI.4 Age-dependent absorbed doses in the thyroid gland
per unit intake of 131I and 129I (Gy Bq-1)
------------------------------------------------------------
Thyroid aborbed dose Age
per unit intake 6 months 4 years 14 years Adult
------------------------------------------------------------
Inhalation: 131I 3.2 10-6 1.5 10-6 4.9 10-7 3.8 10-7
129I 4.1 10-6 2.2 10-6 1.1 10-6 2.3 10-6
Ingestion: 131I 4.3 10-6 2.0 10-6 6.5 10-7 5.1 10-7
129I 5.4 10-6 3.0 10-6 1.5 10-6 3.0 10-6
------------------------------------------------------------
2. Dose per unit release
(a) Nuclear explosions
262. The thyroid dose commitment via the ingestion (milk) pathway
from 131I released by nuclear explosions can be assessed from the
sequential product of transfer factors
Dc = P23 P35 F
where F is the integrated deposition density, P23 is the deposition
to milk transfer factor and P35 is the milk to thyroid dose
transfer factor. The value of F weighted for the world's
population and that of P23 for fallout deposition were given above
as 104 Bq m-2 and 6.3 10-4 Bq a 1-1 per Bq m-2, respectively. The
value of P35 can be derived from the age-dependent consumption
rates of milk given in Table VI.3, the age-dependent thyroid doses
per unit ingested activity presented in Table IV.4, and from the
assumption that the three groups of children are representative of
the age groups 0-1, 1-9 and 10-19 years, respectively, and that
these groups contain respectively 2, 16, and 20% of the population
[U1]. The value of P35 (milk) is thus found to be 1.3 10-4 Gy per
Bq a 1-1. The thyroid dose commitment for the world's population
arising from 131I from global fallout of past nuclear explosions is
therefore estimated to be about 8 10-4 Gy. Most of the dose
commitment was in fact delivered in the early 1960s. Taking the
world's population at that time to be 3 109 persons, the collective
dose commitment would be about 2 106 man Gy. Since 6 1020 Bq of
131I were estimated to give rise to global fallout, the individual
and collective thyroid dose commitments per unit activity released
are found to be 1.3 10-24 Gy Bq-1 and 3 10-15 man Gy Bq-1,
respectively.
(b) Nuclear industry
(i) Local and regional contribution
263. Atmospheric releases. The contribution of the inhalation
pathway to the collective dose commitments from effluent releases
can be estimated from the integrated concentrations of 131I and
129I in ground-level air. Assuming that all the activity released
in the atmosphere will eventually deposit on the ground, the
integrated concentration in ground-level air is the total amount of
131I or 129I released per unit area of the deposition region
divided by the deposition velocity, vd. The population affected is
the population density deltaN, times the area of the deposition
region. The collective dose commitment per unit activity released
is given by the expression Sc = deltaN phi/vd where phi is the age-
weighted thyroid dose per unit integrated air concentration.
264. On the basis of a figure of 5 10-3 m3 kg-1 s-1 for the
deposition velocity on grass per unit area density of vegetation
(see paragraph 244), using a value of 0.1 kg (dry) m-2 for the mass
of grass per unit area, and assuming that the activity deposited on
grass represents one fourth of the total activity deposited on
ground, vd would be 2 10-3 m s-1 or 6 104 m a-1. From the data
contained in Tables VI.3 and VI.4 and the age distribution given
above, the age-weighted thyroid dose per unit integrated air
concentration would be 3.4 10-3 and 1.4 10-2 Gy per Bq a m-3 for
131I and 129I respectively. As the deposition area is expected to
be very large, the population density is assumed to be about 25
persons km-2, that is 2.5 10-5 persons m-2. The collective thyroid
dose commitments are thus estimated to be 1.4 10-12 and 5.8 10-12
man Gy Bq-1 for 131I and 129I, respectively.
265. The contribution of the ingestion pathway (consumption of
milk, leafy vegetables and meat) to the collective dose commitments
per unit activity released can be assessed by the expression Sc =
P13 P35 deltaN where P13 is the air to dietary product to age-
weighted thyroid dose transfer factor, and deltaN the population
density. Using the values and assumptions given previously, the
collective thyroid dose commitments are estimated to be, for 131I,
8.7 10-12 and 2.3 10-12 man Gy Bq-1 for consumption of milk and
fresh leafy vegetables, respectively, while the values for 129I
would be 1.2 10-10, 5.0 10-11 and 6.4 10-12 man Gy Bq-1 for
consumption of milk, fresh leafy vegetables and beef, respectively.
266. Aquatic releases. The collective thyroid dose commitment per
unit activity of 131I and 129I discharged into the aquatic
environment can be estimated, as in the 1977 UNSCEAR report [U1],
using the expression
Sc = N I f phi
V(lambda + 1/tau)
where V is the volume of the receiving waters, tau the turn-over
time of receiving waters, lambda the decay constant of the
radionuclide considered, N the number of individuals exposed, I the
individual consumption rate of the foodstuff considered, f the
concentration factor of the radionuclide in that foodstuff, and phi
the thyroid dose per unit activity ingested.
1
267. The quantity V(lambda + 1/tau) is the infinite time integral
of the water concentration per unit activity released, while that
quantity multiplied by f is the infinite time integral of the
concentration in the consumed item (fish, for example). For inputs
into small volumes of water, the concentrations in water and in
fish will be high, but the populations which can be served with
drinking water or by fish consumption will be limited. For inputs
into large volumes of water, the concentrations will be smaller,
but the populations involved will be larger. It is reasonable,
therefore, to assume as a first approximation that the quantities
V/N are relatively constant; they are taken to be 3 107 and 3 109
litre per man, for fresh water and sea water, respectively. Using
the values given previously in the text and in the tables, the
collective thyroid dose commitments per unit activity of 131I and
129I can be estimated. The results are presented in Table VI.5.
Table VI.5 Collective thyroid dose commitments
per unit activity of 131I and 129I released
in the aquatic environment (man Gy Bq-1)
------------------------------------------------
Type of release 131I 129I
and pathway
------------------------------------------------
Release to fresh water
- Drinking water 3.2 10-13 3.2 10-10
- Fish 1.4 10-14 1.4 10-11
Release to sea water
- Fish 1.1 10-15 1.1 10-13
- Shellfish 8.8 10-16 1.1 10-14
------------------------------------------------
(ii) Global contribution
268. Using the specific activity approach described previously,
the activity concentration of 129I per unit mass of 127I is the
same in the sea water and in the human thyroid. Assuming that the
concentration of stable iodine per unit mass of thyroid is 80, 180,
300 and 600 µg g-1 at ages 6 months, 4 years and 14 years and for
adults, respectively and using the age distribution given
previously, a specific activity of 1 Bq per gram of stable iodine
in the thyroid would lead to an age-weighted annual thyroid dose of
1.5 10-7 Gy. Since a release of 1 Bq 129I results in a long-term
concentration of 2.6 10-17 Bq g-1 stable iodine per Bq (see
paragraph 255), the collective thyroid dose commitment arising from
discharges of 129I would be about 9 10-7 man Gy Bq-1, assuming a
world population of 1010 and no sink for iodine in the environment.
269. Most of the collective dose commitment to the thyroid is
delivered in the far future, as the mean life of 129I is 2.3 107 a.
The average dose rate per unit activity released would be extremely
low (4 10-24 Gy a-1 per Bq released). The estimate of the
collective thyroid dose commitment could be significantly in error
if there exists a mechanism that efficiently removed iodine from
the biosphere at a rate significantly greater than the radioactive
decay constant of 129I of 4 10-8 a-1. Such a mechanism could be
the retention by ocean sediments.
(c) Summary
270. Table VI.6 summarizes the results given above for the thyroid
collective dose commitments per unit activity released, and
provides also the collective effective dose equivalent commitments
per unit activity released. The latter quantities are obtained by
multiplying the former by 0.03.
Table VI.6 Summary of collective thyroid dose commitments
per unit activity released and of collective effective dose
equivalent commitments per unit activity released
---------------------------------------------------------------
131I 129Ia 131I 129Ia
------------------ ------------------
(man Gy Bq-1) (man Sv Bq-1)
---------------------------------------------------------------
Weapon tests 3 10-15b 9 10-17b
Industrial releases
a) Atmosphere
Inhalation 10-12 6 10-12 3 10-14 2 10-13
Ingestion
- Milk 1 10-11 1 10-10 3 10-13 3 10-12
- Leafy vegetables 2 10-11 5 10-11 6 10-14 2 10-12
- Beef - 6 10-12 - 2 10-13
b) Rivers
Ingestion
- Water 3 10-13 3 10-10 9 10-15 9 10-12
- Fish 1 10-14 1 10-11 3 10-16 3 10-13
c) Oceans
Ingestion
- Fish 1 10-15 1 10-13 3 10-17 3 10-15
- Shellfish 9 10-16 9 10-14 3 10-17 3 10-15
---------------------------------------------------------------
a First passage estimates. The long-term, global estimates
for 129I from all sources and by all pathways are 9 10-7 man
Gy Bq-1 and 3 10-8 man Sv Bq-1.
b Milk consumption.
F. REFERENCES
A1 Atkins, D.H.F., R.C. Chadwick and A.C. Chamberlain. Deposition
of radioactive methyl iodide vegetation. Health Phys. 13: 91
(1967).
A2 Angeletti, A. and A. Sauve. Estimation de la vitesse de dépôt
de l'iode vapeur sur les végétaux en fonction des eléments du
climat. Congrés commun de radioprotection. SFRP-FS. Lausanne,
1981.
B1 Berg, R. and H. Schuettelkopf. Die Messung der Verteilung in
und der Abgabe von I-129 aus der Wiederaufarbeit-ungsanlage
Karlsruhe. p.81 in Radioactive Effluents from Nuclear Fuel
Reprocessing Plants. CEC document V/2266/78 (1978).
B2 Beninson, D., A.M. Migliori de Beninson, and C. Menossi.
Fallout radioactivo debido a las explosiones en el Pacífico sur
en el período 1966-1970. Comisíon Nacional de Energía Atómica.
Informe RS 28/49 (1971).
B3 Beninson, D., A.M. Migliori de Beninson and C. Menossi. Fallout
radioactivo debido a las explosiones en el Pacífico sur en el
período 1971-1972. Comisíon Nacional de Energia Atómica.
Informe RS 43/102 (1973).
B4 Breuer, F. and M. de Bortoli. Behaviour of radioiodine in the
environment and in man. Comitato Nazionale Energia Nucleare
report RT/PROT(73)13 (1973).
B5 Bergström, S.O.W. Transport of fallout 131-I into milk. p.
159-174 in Radiological Concentration Processes (Aberg, B.
and F.P. Hungate, eds.). Pergamon Press, 1967.
B6 Bustad, L.F., D.H. Wood, E.E. Elefson et al. 131-I in milk and
thyroid of dairy cattle following a single contamination event
and prolonged daily administration. Health Phys. 9: 1231-1234
(1963).
B7 Bouville, A. Estimation des doses dues aux rejets d'iode 129
par les installations nucléaires. Echelles locale et
régionale. p. 53-68 in I-129. Proceedings of an NEA
Specialist Meeting. OECD, Paris, 1977.
C2 Chamberlain, A.C. and R.C. Chadwick. Transport of iodine from
atmosphere to ground. United Kingdom Atomic Energy Authority
Research Group report AERE-R-4870. Harwell, Berkshire, 1965.
C3 Comisíon Nacional de Energía Atómica, Argentina. Fallout
Radioactivo debido a las explosiones en el Pacífico sur en el
período Enero-Octubre de 1973. Informe RS-47/118 (1973).
C4 Cambray, R.S. et al. Radioactive fallout in air and rain.
Reports covering the years 1955-1979. AERE-R-4094 (1962);
AERE-R-4392 (1963); AERE-R-4687 (1964); AERE-R-4997 (1965);
AERE-R-6556 (1970); AERE-R-7245 (1972); AERE-R-7832 (1974);
AERE-R-8267 (1975); AERE-R-8671 (1976); AERE-R-9018 (1978);
AERE-R-9441 (1979); AERE-R-9672 (1980).
D1 Dillman, L.T. and F.C. Von der Lage. Radionuclide decay
schemes and nuclear parameters for use in radiation-dose
estimation. NM/MIRD Pamphlet No.10. Society of Nuclear
Medicine, September 1975.
D2 Dolphin, G.W. Dietary intakes of iodine and thyroid dosimetry.
Health Phys. 21: 711-712 (1971).
E1 Edwards, R.R. Iodine-129: Its occurrence in nature and its
utility as a tracer. Science 137: 851-853 (1962).
E2 Eggleton, A.E.J., D.H. Atkins and L.B. Cousins. Chemical and
physical nature of fallout 131-I and carrier-free 131-I
released in air. An abstract. Health Phys. 9: 1111 (1963).
G1 Garner, R.J. and R. Scott Russell. Isotopes of iodine. Chapter
14 in Radioactivity and human diet (R. Scott Russell ed.).
Pergmon Press, 1966.
H2 Harley, N., I. Fisenne, L.D.Y.Ong et al. Fission yield product
decay. p. 251-260 in Health and Safety Laboratory Fallout
Program quarterly summary report HASL-164. New York, 1965.
H3 Hull, A.P. Critical evaluation of radiological measurements
and of the need for evacuation of the nearby public during the
Three Mile Island incident. Paper presented at the IAEA
International Conference on current nuclear power plant safety
issues. Stockholm, October 1980.
H4 Hull, A.P. Vegetation retention and vegetation-milk ratios of
fallout 131-I. Health Phys. 9: 1173-1177 (1963).
H5 Heinemann, K., M. Stoeppler, K.J. Vogt et al. Untersuchungen
zur Ablagerung und Desorption von Jod auf Vegetation.
Kernforschungsanlage Jülich report Jül-1287 (1976).
H6 Hoffman, F.O. A reassessment of the deposition velocity in the
prediction of the environment transport of radioiodine from air
to milk. Health Phys. 32: 437-441 (1977).
H7 Horbert, J., K.J. Vogt and L. Angeletti. Untersuchung zur
Ablagerung von Aerosolen auf Vegetation and anderen
Grenzflächen. Kernforschungsanlage Jülich report Jül-1288
(1976).
I1 International Atomic Energy Agency. Radioiodine removal in
nuclear facilities. Methods and techniques for normal and
emergency situations. Technical Reports Series No. 201.
Vienna, 1980.
I2 International Atomic Energy Agency. Power reactors in member
states. 1980 edition. Vienna, 1980.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP Publication 30, Part
1. Annals of the ICRP, Vol. 2, No. 3/4 (1979).
L1 Loutit, J.F., W.G. Marley and R.S. Russell. The nuclear
reactor accident at Windscale, October 1957. Environmental
aspects. Appendix H in The Hazards to Man of Nuclear and
Allied Radiations. A second report to the Medical Research
Council, London, 1960.
L2 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European Community. Discharge data 1974-1978. Radiological
aspects. CEC document V/4116/80 (1980).
L3 Lengemann, F.W.. Radioiodine in the milk of cows and goats
after oral administration of radioiodate and radioiodide.
Health Phys. 17: 565-569 (1969).
M1 Manuel, O.K. Iodine-129. A study of its transport in the
environment and distribution in biological systems. Report C00-
2450-3 (1976).
M2 Morgan, A., D.G. Morgan and G.M. Arkell. A study of retention
and subsequent metabolism of inhaled methyl iodide in Inhaled
Particles and Vapours II (C.N. Davies, ed.). Pergamon Press,
1967.
M3 Morgan, A., D.J. Morgan and A. Black. A study of the
deposition, translocation and excretion of radioiodine inhaled
as iodine vapour. Health Phys. 15: 313-322 (1968).
P1 Pelletier, C.A., J.E. Cline. E.D. Barefoot et al. Sources of
radioiodine at pressurized water reactors. EPRI report NP-939
(1978).
P2 Pelletier, C.A., J.E. Cline, E.D. Barefoot et al. Sources of
radioiodine at boiling water reactors. EPRI report NP-495
(1978).
P3 Peirson, D.H. and J.R. Keane. The characteristics of early
fallout from the Russian nuclear explosions of 1961. Nature
196: 801-807 (1962).
P4 Perkins, R.W. Physical and chemical form of 131-I in fallout.
Health Phys: 9: 1113-1122 (1963).
P5 Palms, J.M., V.R. Veluri and F.W. Boone. The environmental
impact of iodine-129 released by a nuclear fuel reprocessing
plant. Nucl. Saf. 16: 593-602 (1975).
R1 République française. Retombées radioactives à la suite des
tirs nucléaires en Polynésie. Mai-décembre 1970.
R2 Riggs, D.S. Quantitative aspects of iodine metabolism in man.
Pharmacol. Rev. 4: 284-370 (1952).
S1 Srinivasan, B., E.C. Alexander Jr. and O.K. Manuel. Iodine-129
in terrestrial ores. Science 173: 327-328 (1971).
S2 Spiers, F.W. Radioisotopes in the human body: physical and
biological aspects. Academic Press, 1968.
T1 Turekian, K.K. The oceans, streams and atmosphere in Handbook
of Geochemistry (K.H. Wedepohl, ed.). Springer, Berlin,
Heidelberg, New York, 1969.
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York, 1977.
U2 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication No. E.72.IX.17 and
18. New York, 1972.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13(A:7613). New York, 1969.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14(A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No.
14(A/5814). New York, 1964.
U6 United States Environmental Protection Agency. Environmental
analysis of the uranium fuel cycle. Part II. Nuclear power
reactors. U.S. Environmental Protection Agency report EPA-
520/9-73-003-C (1973).
V1 Voillequé, P.G. Iodine species in reactor effluents and in the
environment. EPRI report NP-1269 (1979).
V2 Voillequé, P.G. and J.H. Keller. Air-to-vegetation transport
of 131-I as hypoiodous acid (HOI). Health Phys. 40: 91-94
(1981).
W1 Winkleman, I. Bericht über die im Filterproben aus der
Abluftüberwashungsanlage von Kernkraftwerken in der
Bundesrepublik Deutschland in Jahre 1976 nachgewiesenen
Einzelnuklide. STH-4/77 (1977).
VII. CAESIUM-137
A. INTRODUCTION
271. Caesium is element number 55 in the periodic table. It is an
alkali metal like potassium, and it resembles potassium
metabolically. Whereas potassium is an essential element for man,
there is no evidence that caesium is also an essential trace
element. In fact, stable caesium, 133Cs, is fairly rare in the
biosphere and in geological occurrence. Average occurrence in the
earth's crust is 3 µg g-1. In specific rock types the estimated
average concentration is 1 µg g-1 in basalts and 5 µg g-1 in
granite. The K/Cs ratio in basalts is 7500 [T2]. Stable caesium
occurrence in fresh water, lakes and rivers ranges between 0.01 and
1.2 ng g-1 and is 0.5 ng g-1 in the ocean [K2]. Stable potassium
is more abundant, with usual concentrations of 0.2 to 10 µg g-1 in
fresh waters and 380 µg g-1 in the ocean [V1].
272. The radioactive isotope 137Cs is produced in nuclear fission
and is one of the more significant fission products. The fission
yield is relatively high, about 6 atoms per 100 fissions,
independent of the type of fission in uranium or plutonium (Table
VII.1). It has a radioactive half-life of 30.17 a and its beta
decay is accompanied by a gamma ray of moderate energy. Figure
VII.I shows the decay scheme and lists the primary transition
energies.
Table VII.1 Fission yields of
caesium-137 [C3]
-----------------------------
Fission yield (%)
Nuclide -----------------
Thermal Fast
-----------------------------
235U 6.21 6.12
239Pu 6.64 6.50
238U 5.93
232Th 6.73
-----------------------------
Table V.1 Fission yields of strontium-90 [C3]
-------------------------------------
Fission yield (%)
Nuclide Thermal Fast
-------------------------------------
235U 5.84 5.21
239Pu 2.12 2.05
238U 3.20
232Th 7.66
Average for
nuclear tests a/ 3.50
-------------------------------------
a/ From reference [H1].
153. Large amounts of 90Sr were released in nuclear tests and
dispersed throughout the world. Strontium-90 is also produced in
the nuclear fuel cycle, but only small amounts are released to the
environment. Strontium-90 in the environment is efficiently
transferred to human diet. The absorption of 90Sr by the body is
relatively high and it has a long biological retention time.
154. Because of the correspondence in behaviour of strontium and
calcium in the environment and in man, it has been the practice to
express measurement results in diet and bone as quotients of 90Sr
to Ca concentrations. Discrimination is reflected as ratios of
strontium to calcium quotients in samples to those in precursor
samples in the transfer chain. Expressing results in terms of the
strontium to calcium quotients has the practical advantage that for
many environmental transfer processes, such as absorption into the
body, secretion into milk and deposition in bone, the ratios remain
relatively constant and predictable. However, since the average
levels of calcium in diet and man are nearly constant, assessments
of 90Sr can also be made on the basis of amounts per unit mass or
volume of material.
B. SOURCES
1. Nuclear explosions
155. Strontium-90 is produced in nuclear explosions in the amount
of approximately 3.7 1015 Bq per Mt of fission energy. Measurements
of the fission debris from large nuclear tests gave a 90Sr fission
yield estimate of 3.5% [H1]. Assuming 1 kt of fission energy
corresponds to 1.45 1023 fissions [H1] and using the current best
estimate of the 90Sr half-life (29.12 ± 0.24 a) [N3], the 90Sr
production yield is estimated to be 3.8 ± 0.1 1015 Bq per Mt
fission energy. Large deviations are possible for individual
tests.
156. Approximate fission energy yields of nuclear weapons tests
conducted in the atmosphere have been published. The total for
tests through 1962 was 194 Mt of fission energy [F1, U3]. This
would correspond to a production of about 74 1016 Bq of 90Sr. An
additional 9 1016 Bq have been produced in atmospheric tests to the
end of 1980. A portion of the total amount of 90Sr was local
fallout, deposited in the immediate vicinity of testing regions.
Local fallout is important especially for lower yield tests
detonated on the land or water surface. The best estimates of the
activity of globally distributed 90Sr come from measurements of
90Sr deposition. This amounts to 6 1017 Bq for all tests
conducted through 1980.
2. Nuclear fuel cycle
(a) Nuclear reactors
157. Strontium-90 is produced by fission in the fuel of nuclear
reactors. The amounts produced vary depending on the fuel
composition, reactor type, and degree of fuel burn-up achieved.
The yields for various fission processes were given in Table V.1.
In fairly high burn-up fuel (33000 MW(t)d t-1) of a PWR, the 90Sr
production is estimated to be 2.83 1015 Bq per tonne of fuel,
corresponding to 9.5 1013 Bq per MW(e)a of electricity
generated [O1].
158. Small amounts of 90Sr produced in the fuel in nuclear
reactors may reach the coolant through defects in the fuel
cladding. In coolant purification or following coolant leakage,
90Sr may reach the gaseous and liquid effluent streams. In
controlled amounts, some of the effluents are released to the
environment.
159. The activity releases of 90Sr were listed in the 1977 report
of UNSCEAR [U6]. Average discharges were of the order of 0.01 to 4
106 Bq per MW(e)a in PWRs and BWRs, respectively, with most of the
release in liquid effluents. Somewhat larger releases of 90Sr in
liquid effluents from GCRs arise primarily from spent fuel storage
pools. There are considerable variations in the release amounts
from individual reactors per unit electricity generated.
160. Assuming for each reactor type that the limited data of the
90Sr activity released per unit of electrical energy generated are
representative, it is possible to obtain a very rough estimate of
the total amount of 90Sr released from reactors worldwide. Using
the installed capacities of the various reactor types in 1980 [I1]
and assuming a reactor utilization of 60%, the estimated annual
release from all reactors is about 2 1012 Bq (Table V.2). In this
calculation, it is assumed for the reactor types for which no data
are available, that the releases are similar to those from BWRs.
Table V.2 Estimated global discharges of strontium-90
from nuclear power stations in 1980
------------------------------------------------------------
Reactor Reactor Capacity Production Estimated
type number [MW(e)a] rate [Bq strontium-90
per MW(e)a] discharge (Bq)
------------------------------------------------------------
PWR 96 64239 9 104 0.3 1010
BWR 62 35170 4 106 8 1010
GCR 36 7086 4 108 170 1010
Other 47 18490 4 106 4 1010
------------------------------------------------------------
Total 241 124985 2 1012
------------------------------------------------------------
161. There have not been many reports on 90Sr measurements in the
environment surrounding nuclear reactors that can be attributed to
reactor operation. In fact, 90Sr would not be a likely
radionuclide to be investigated, and furthermore results of
measurements could hardly be made independent of fallout 90Sr. The
much more readily detectable 137Cs, for example, is released in
activity amounts up to several hundred times that of 90Sr [U6].
Caesium-134 is released from reactors in amounts about 60% less
than 137Cs and, in addition, it would not be confused with weapons
fallout in the environment. For these reasons, estimates of 90Sr
levels in the environment from reactor release must generally be
based on measured releases and environmental dispersion
calculations.
(b) Fuel reprocessing plants
162. In fuel reprocessing plants the fuel is dissolved to recover
uranium and plutonium for reuse. All the 90Sr and other fission
products as well go to waste streams. The radionuclide activities
in airborne and liquid effluents from fuel reprocessing plants have
been recorded by UNSCEAR [U6]. The data for 90Sr are summarized in
Table V.3.
Table V.3 Average normalized discharges of strontium-80
to the environment from fuel reprocessing plants
(Bq per MW(e)a) [U6]
--------------------------------------------------------------
Plant Airborne Liquid Total
effluents effluents
--------------------------------------------------------------
Windscale (United Kingdom) 5 106 2 1011 2 1011
Nuclear Fuel Services (U.S.A.) 2 106 5 108 5 108
--------------------------------------------------------------
163. Strontium-90 is released from fuel reprocessing plants
primarily in liquid effluents. Relative to the total amounts of
90Sr in spent fuel, the release amounts are not large. The
fractional releases in liquid effluents are approximately 2 10-3
from the Windscale plant in the United Kingdom and were about
5 10-6 from the small Nuclear Fuel Services plant in the U.S.A.,
which is no longer in operation.
164. The fractional release of 90Sr in airborne effluents is about
3 10-8, compared to the amounts present in spent fuel. The
experiences at Windscale and at Nuclear Fuel Services are similar
in this regard.
165. The releases of radioactivity into the Irish Sea from the
Windscale plant and the corresponding levels in the environment
have been closely studied [N1]. Estimates of inventories of
activity in the sea and sediments and of movements of the activity
in its passage into the North Sea are being made [M1]. Some
results of environmental surveys around the reprocessing plant at
La Hague have also been published [S1]. Most of the attention,
however, is focused on the greater quantities of other fission
products released.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Movement in soil
166. The downward penentration of 90Sr in soil is slow, although
it is more rapid than for 137Cs or 249Pu. Even after several years
90Sr remains in the upper few centimetres in undisturbed soil. The
rate of movement varies with soil type; a low content of clay and
humus, a high content of electrolytes and a rapid movement of water
increase penetration [U2]. The mechanism of movement is thought to
involve both leaching and diffusion.
2. Transfer to plants
167. Plants acquire 90Sr by direct deposition onto foliage and by
root uptake of 90Sr in the soil. Absorption into the leaves is
relatively slow and superficial material is readily lost by
weathering. The translocation of 90Sr from plant leaf or grain
surfaces to other parts of the plant is small.
168. Capture of 90Sr on inflorescences of plants is of importance
for entry to grain. Concentrations in husked grain will be higher
than in the milled product during periods of deposition.
169. Uptake from soil is normally the primary mode of 90Sr entry
into plants. The quantity of absorbable calcium in soil is an
important factor in determining the extent of 90Sr absorption by
plants. Uptake is greatest from soils of low calcium content.
Uptake is thus reduced by the addition of lime, but usually not by
a factor exceeding 3 [U2]. When soils contain adequate calcium for
growth and the exchange capacity is largely saturated with calcium,
the addition of lime has little or no effect.
170. Other factors which affect root uptake of 90Sr include the
clay and humus content of the soil, pH, the concentration of
electrolytes other than calcium and the moisture content. The
addition of organic matter and fertilizers to soil may have varying
effects on plant uptake, which are, however, usually not large when
these materials are applied at normal agricultural levels.
171. Plant-base absorption of 90Sr has been noted to be quite
efficient. The 90Sr trapped in the surface root mat is relatively
undiluted with the calcium in the soil and is in a particularly
favourable position for absorption. This would help to explain the
higher concentrations of 90Sr in grain in periods shortly after
deposition and would be an important process in permanent pastures.
3. Transfer to milk
172. The total quantity of ingested 90Sr secreted into the milk of
cows is variable, depending on the milk yield. Values range from
0.5 to 2% of a single oral administration [U2]. With continuous
ingestion under normal conditions of feeding, several
investigations have shown that about 0.08% of the amount given
daily is secreted per litre of milk. The transfer to goats milk
may be more than ten times greater, corresponding to a higher
proportion of dietary calcium secreted into the milk as well.
4. Transfer to diet
173. The transfer of fallout 90Sr to diet has been extensively
studied. Assessments by UNSCEAR have been based on application of
generalized transfer models [U6]. The transfer from deposition to
diet is quantitatively described by means of the transfer factor
P23 defined as the time-integrated 90Sr/Ca quotient in the diet
divided by the 90Sr integrated deposition density. The integrals
may be replaced by summations if the relevant quantities are
assessed over discrete intervals of time. In fact, annual values
are the most generally available information. The transfer factor
P23 is usually expressed in mBq a/gCa per Bq m-2.
174. The transfer to diet from a specific deposition occurs over
an extended period as long as 90Sr remains in soil available for
root uptake. The model used by UNSCEAR to describe the transfer of
90Sr from deposition to diet is
infinite -µm
C(n) = b1 f(n) + b2 f(n-1) + b3 sigma f(n-m)e
m=1
where C(n) is the 90Sr/Ca quotient in total diet, in a food group,
or in an individual food item, in the year n; f(n) is the annual
deposition density in the year n, and b1, b2, b3, and µ are factors
which can be derived from reported data by regression analysis [U5,
U6]. The first term in the equation represents the contribution to
dietary 90Sr per unit deposition density in the current year, while
the second term expresses separately the contribution from
deposition in the previous year, reflecting also the use in the
current year of stored food produced in the previous year. The
third term expresses the contribution to dietary 90Sr from the
deposition density in all previous years, resulting from root
uptake and taking into account decay and loss of availability due
to downward movement in soil or to other physical or chemical
changes which may occur. The inverse of µ is the mean life of
available 90Sr in soil, which varies for individual foods and soil
conditions.
175. The transfer factor P23 describes the cumulative transfer of
90Sr to diet per unit deposition density. Using the model
described above, the expression of the transfer factor is
infinite -µm e-µ
P23 = b1 + b2 + b3 sigma e = b1 + b2 + b3 -----
m=1 1-e-µ
The transfer factor P23 can therefore be estimated from the
parameters b1, b2, b3 and µ obtained by regression analysis from
reported data.
176. The values of the parameters obtained from the regression
fits to 90Sr deposition density and diet data from the fallout
measurement programmes in New York and Denmark for dietary
components and for the total diet have been reported by UNSCEAR
[U6]. For total diet, the values of the parameters (b1 ~ 1.0,
b2 ~ 0.9, b3 ~ 0.3, µ ~ 0.1) give estimates of the initial
transfer (b1 + b2) of 1.9 mBq a (gCa)-1 per Bq m-2 and of the
long-term transfer (last term of equation above) of 2.9 mBq a
(gCa)-1 per Bq m-2. The total transfer, the value of P23, is
4.8 mBq a (gCa)-1 per Bq m-2.
177. Similar data from Argentina have also been evaluated, with
reasonable agreement found for all three areas for individual foods
and for total diet. Some differences are noted which are due to
the different definition of the food groups or to different soil
conditions and agricultural practices in the three countries.
178. The long-term transfer of 90Sr to diet from a single input to
the environment can be illustrated from the values of the transfer
parameters obtained from component groups of diet. For example, it
is determined that 90% of the cumulative transfer from a single
release of 90Sr is completed within 9 years for meat, fish and
eggs, 12 years for grain products, 14 years for milk, 32 years for
vegetables and 77 years for fruit. More rapid transfer indicates
that direct deposition processes are more important. Slow transfer
represents primarily uptake from the slowly decaying deposit of
90Sr in soil. Since the exponential decreases of the transfer for
the various groups differ, a direct fit to total diet data with a
single exponential transfer function is expected to be less
accurate than the summation of fits for the individual components.
179. The transfer of 90Sr from deposition density to some
individual foods, particularly to milk, has been studied for a
number of different areas of the world. These results are listed
in Table V.4. The transfer factors range from 2.1 to 7.6 mBq a/gCa
per Bq m-2.
Table V.4 Parameters of the transfer function between deposition density and 90 Sr/Ca in milk
-------------------------------------------------------------------------------------------------
Parameter Northern San New United Denmark Argentina Norway Australia Faroe
a/ hemisphere Francisco York Kingdom Islands
-------------------------------------------------------------------------------------------------
b1 0.84 0.61 0.69 0.89 0.99 1.39 0.70 2.07 2.70
b2 0.54 0.61 0.23 0.47 0.46 1.24 0.44 1.27 1.38
b3 0.22 0.19 0.19 0.15 0.23 0.12 1.02 0.30 0.81
µ 0.12 0.19 0.13 0.13 0.13 0.12 0.33 0.08 0.21
P23 3.1 2.1 2.3 2.4 3.1 3.5 3.7 7.2 7.6
-------------------------------------------------------------------------------------------------
a/ The unit for parameters b1, b2, b3 is mBq a(gCa)-1 per Bq m-2
The unit for parameter µ is a-1
The unit for the transfer factor P23 is mBq a(gCa)-1 per Bq m-2.
180. Milk has been used as an indicator of the levels of 90Sr in
total diet in areas where foods other than milk were not analysed.
This must be done, however, with some caution. It is usually the
case that following a period of 90Sr deposition, the 90Sr/Ca
quotient in milk declines somewhat more rapidly than the 90Sr/Ca
quotient in total diet. Where data are available, it is seen that
the exponential factor is greater in milk than in total diet. This
means that the diet-milk ratio of 90Sr/Ca values changes with time
during and after the period in which the 90Sr deposition occurs.
181. The diet-milk ratios at particular times also show
considerable variation from one country to another [U6]. In most
countries where milk is an important component of the diet, the
diet-milk ratio has averaged about 1.4 for most of the fallout
years. A trend to increasing ratios in the future is expected if
the deposition density rate remains at a very low level. In
countries where milk is not an important component of diet, the
diet-milk ratio will have a much higher value.
5. Aquatic behaviour
182. Strontium, like calcium, appears mainly in ionic form in
water and is not strongly sorbed by suspended particulate
materials. The fraction of strontium found in the particulate
phase in several freshwater systems ranged from 1 to 10% [V1].
183. It has been of interest to determine the behaviour of 90Sr in
aquatic environments in the vicinity of nuclear installations. A
number of determinations of the concentration factors (ratios of
integrated concentrations or of equilibrium concentrations in
organisms and in the water) have been performed in recent years for
various marine biota by measurements of stable and radioactive
strontium under laboratory and field conditions [C1, F2, N2, U4,
U1]. Typical values are 100 for algae, 2 to 10 for crabs and
lobster, about 1 for the flesh of ocean fish and 5 for fresh water
fish.
184. The primary uptake of strontium and also calcium by fish
occurs directly from the water. Therefore, accumulations in
organisms are little dependent on trophic level [V1]. The
concentration factor for fish depends inversely on the
concentration in the water. Vanderploeg et al. [V1] have suggested
a quantitative relationship. The concentration factors for fish
bone are about two orders of magnitude greater than for flesh.
185. The UNSCEAR aquatic model can be used to estimate the
transfer of 90Sr from the aquatic environment to diet for
generalized discharge situations [U6]. Further discussion of this
is presented in the section on dosimetry.
D. TRANSFER TO MAN
186. Strontium-90 is acquired by man primarily through ingestion
of 90Sr contaminated food. Terrestrial pathways are generally more
important than aquatic pathways in transferring 90Sr to man. From
fallout experience it is noted that 90Sr in drinking water always
contributes less than 5% of the total ingestion intake and 90Sr in
fish is a minor contributor even in countries where consumption of
fish is high. For example, it is estimated that only about 3% of
the fallout 90Sr intake by man in Japan between 1966 and 197l came
from fish [U1].
187. Correlations of fallout 90Sr in diet with measurements of
90Sr in bone have provided a relationship which is used to evaluate
the transfer to man. The transfer function used by UNSCEAR [U5,
U6] is:
infinite -µm
Cb(n) = c Cd(n) + g sigma Cd(n-m)e
m=o
where Cb(n) is the 90Sr/Ca quotient in bone in the year n, Cd(n) is
the dietary 90Sr/Ca quotient in the year n, and c, g, and µ are
constants determined from regression analysis of the bone and diet
data. The parameter can be associated with that portion of 90Sr
intake which is retained for a short term on bone surfaces and is
readily exchanged with plasma, and the parameter g with that
portion more tightly retained in bone. The exponential term
describes the effective removal rate of 90Sr from bone due to
radioactive decay and bone remodelling.
188. The transfer factor P34 linking diet and bone, is defined as
the ratio of the time integrated 90Sr/Ca quotient in bone to that
in diet. It may also be thought of as the cumulative transfer and
retention 90Sr in bone per unit intake in diet. The expression for
the transfer factor using the above function is
infinite -µm g
P34 = c + g sigma e = c + ------
m=o 1-e-µ
Data are available for regression analysis applying this model only
for 90Sr in vertebrae. It is recognized that there is greater
initial retention in cancellous bone such as vertebrae than in
compact bone, but there is also more rapid turnover. Therefore, the
time integrated results are expected to be representative of the
skeleton as a whole.
189. The results of regression fits of fallout 90Sr diet and adult
vertebrae data for various localities have been given in UNSCEAR
reports [U5, U6]. Representative values of the parameters are 0.02
Bq a (gCa)-1 in bone per Bq a (gCa)-1 in diet for both c and g and
about 0.2 a-1 for µ. The values determined for P34 range from 0.12
to 0.16 Bq a (gCa)-1 in bone per Bq a (gCa)-1 in diet.
190. From the values of the exponential parameter µ it is possible
to infer the mean residence time of 90Sr in bone. This ranges from
3.4 to 6.7 years, corresponding to bone turnover rates of 12 to 23%
per year. There is no reason to expect that the metabolic
behaviour should differ in the various areas of the world, and as
far as is known, dietary composition of usual foods does not affect
90Sr availability. Therefore, the differences in the estimate are
attributed to variations in the survey measurements.
191. To account for the bone 90Sr/Ca quotients in children,
Bennett [B2] has used age-dependent parameters in the above
transfer function. For children under 9 years the parameter c was
found to be zero, consistent with a single exponential transfer
model as originally proposed by Rivera [R1]. The best fit was
obtained with a turnover rate of 90Sr varying from about 100% per
year down to about 40% per year in the pre-teenage years and then
falling with age to about 20% per year for adults. Beninson [B1]
had reported a similar variation in turnover rate with age in
Argentina. The fractional retention of strontium, the fraction of
dietary intake incorporated into the skeleton, was also found by
Bennett [B2] to vary with age being five to seven times higher for
infants than for adults. Additional considerations of 90Sr
metabolism as a function of age have been presented by Papworth and
Vennart [P1].
192. The initial 90Sr concentration in the newborn must be
determined from an empirical relationship with the mother's diet.
The 90Sr/Ca quotient in bone of newborn varies from 0.1 to 0.2
times the 90Sr/Ca quotient in diet of the mother during the year
prior to the birth. An average of about 0.15 is obtained from the
survey data [B1, B2].
E. DOSIMETRY
1. Dose per unit intake
193. It is useful for radiological assessments to have expressions
for the dose per unit ingestion intake and per unit inhaled amount.
The absorbed doses in bone marrow and in bone-lining cells per unit
integrated activity of 90Sr in bone have been evaluated based on
the work of Spiers [S2]. These are the transfer factors P45
relating activity in bone to the doses [U5, U6]. The values are
P45 (bone marrow) = 0.38 mGy per Bq a (gCa)-1; P45 (bone-lining
cells) = 0.53 mGy per Bq a (gCa)-1. If it may be assumed that the
dietary calcium intake rate is 1 g daily (365 g/a), the transfer
factor to bone per unit intake, P34, is 0.14/365 Bq a (gCa)-1 per
Bq. This multiplied by the factor P45 gives the absorbed doses in
bone marrow and bone-lining cells. The results are given in Table V.5.
Table V.5 Absorbed dose per unit intake of
strontium-90 (Gy/Bq)
--------------------------------------------------
Lung Bone marrow Bone-lining
cells
--------------------------------------------------
Ingestion - 1.5 10-7 2.0 10-7
Inhalation 5.8 10-9 4.9 10-7 6.9 10-7
--------------------------------------------------
194. The ICRP Task Group lung model gives guidance regarding the
disposition of inhaled radioactivity [I3, I4] in the respiratory
tract. The respiratory system is divided into the nasopharyngeal
region (NP), the tracheobronchial region (TB), and the pulmonary
region (P). For the present dosimetric assessments it will be
assumed that the 90Sr is associated with typical ambient aerosols
of average diameter 0.5 µm. The 90Sr compounds are grouped in
Class D, retention in the lung being in the order of days,
specifically 0.5 d for the portion deposited in the pulmonary
region.
195. Fractional deposition of 0.5 µm particles in the NP, TB, and
P regions are 0.14, 0.08, and 0.30 and subsequent fractional
transfers to blood for the Class D compounds are 0.5, 0.95, and
1.0, respectively. Total transfer to blood is thus 0.446 of the
inhaled amount. Fractional transfer from blood to bone is 0.3
[I2].
196. The lung dose as a function of the inhaled activity can be
calculated from the expression
_
k A E f 1.44 TB
Dlung = ----------------
M
_
where k is a dosimetric constant, A is the activity inhaled, E is
the average energy per disintegration, f is the fraction of the
activity retained in the lung, TB is the retention half-time in the
lung, and M is the mass of the lungs. The lung dose per unit
activity of 90Sr inhaled is therefore:
Gy/d MeV 1.44 0.5d
Dlung = 13.8 10-6 ------ 1.13 --- 0.52 ---------
Bq MeV dis 1000 g
g dis
= 5.8 10-9 Gy/Bq
197. Assuming that the mean residence time of 90Sr in bone is 10
years, applicable to the skeleton as a whole, the integrated
concentration of 90Sr in bone per unit inhaled amount is 0.446 x
0.3 x 10 years 100 gCa = 1.3 10-3 Bq a/gCa per Bq inhaled. The
absorbed doses in bone marrow and bone lining cells are obtained by
multiplying the values of the transfer factor P45 given above. The
results are listed in Table V.5.
2. Dose per unit release
(a) Nuclear explosions
198. The dose commitment from 90Sr released by nuclear explosions
can be assessed using the environmental compartment model outlined
in the introduction. The transfer factor of a sequence of steps in
series is the product of the transfer factors of each step. The
dose commitment, Dc, is related to the integrated deposition
density of 90Sr, F, by the following expression:
Dc = P23 P34 P45F
where P23, P34 and P45 are the transfer factors discussed
previously. Average values for these transfer factors, as assessed
in the 1977 UNSCEAR report [U6], are P23 = 5 mBq a/gCa per Bq m-2,
P34 = 0.14, and P45 as given previously.
199. The dose commitment from 90Sr ingestion per unit of
widespread deposition density of 90Sr such as from nuclear
explosions is thus 0.3 µGy per Bq m-2 for bone marrow and 0.4 µGy
per Bq m-2 for bone lining cells.
200. The dose commitment from 90Sr via the inhalation pathway can
be estimated from the time integrated concentration of 90Sr in air.
Multiplying the dose commitment per unit inhalation intake (Table
V.5) by the inhalation intake rate of 22 m3 d-1 gives the value of
1.3 10-7 Gy per Bq d m-3 for the dose commitment to lungs per unit
integrated concentration of 90Sr in air.
201. The dose commitment to lungs can also be referred to measured
values of the integrated deposition density. Dividing the
integrated deposition density (Bq m-2 by the average deposition
velocity (m s-1) gives the time integrated concentration in air.
From long-term measurements of fallout 90Sr, the average deposition
velocity is 2.2 cm/s in New York, 2.0 cm/s in Argentina, and 1.5
cm/s in Denmark [A1, C2, E1]. The values determined from annual
measurements at all three sites range from 1.2 to 2.9 cm/s. The
value of 2 cm s-1 can be taken as representative. One Bq m-2
integrated deposition density thus corresponds to 5.8 10-4 Bq d
m-3) in air. The dose commitment to lung per unit integrated
deposition density of 90Sr is thus 7.5 10-11 Gy per Bq m-2.
202. The total amount of 90Sr released to the environment by
nuclear tests, 6 1017 Bq, has given a population-weighted
integrated deposition density of 1940 Bq m-2 in the world as a
whole [U6]. The world population is 4 109. With these values the
collective dose commitments per unit activity of 90Sr released may
be estimated. The results, which are summarized later, apply to
the geographic pattern of past nuclear tests.
(b) Nuclear installations
203. The activity of 90Sr in airborne effluents is dispersed by
turbulent air movement and is eventually deposited on the ground.
It then enters the ingestion pathway. The average situation over
several years of routine discharges will result in a nearly
completed deposition within a region with a radius R. The average
integrated deposition density, for a discharged activity A is F =
A/pi R2. The number of individuals exposed to that mean integrated
deposition density, on the other hand, is N = deltaN pi R2,
provided the population density deltaN can be assumed to be
constant over the relevant area.
204. It follows that the collective dose commitment per unit
activity released, Sc1, can be assessed by the expression
Sc = P23 P34 P45 deltaN
1
Using the values for the transfer factors given previously, and
assuming a population density of 25 man km-2, the collective dose
commitments per unit activity released are estimated to be about 7
10-12 man Gy per Bq for bone marrow and about 9 10-12 man Gy per Bq
for bone-lining cells. These values apply provided that food is
locally produced and that the production suffices for the
population density under consideration. The estimates would
probably be lower in actual circumstances.
205. The contribution of inhalation to the collective dose
commitment, for effluent releases over many years, can be estimated
by integration of the functions describing the atmospheric
dispersion, assuming complete depletion by deposition within 100
km. The collective dose commitment contribution per unit activity
released would be given by the expression
c X 100 km r -1.5
S1 = (Q)1 km I deltan phi / (1 km) 2 pi r dr
1 km
X
where (Q)1 km is the dispersion factor at 1 km from the release
point, I is the individual intake rate of air, deltaN is the
population density, phi is the dose per unit activity inhaled of
90Sr and r is the distance from the release point [U6].
206. The collective dose commitment may also be estimated using
the deposition velocity, thus avoiding the need to specify the
deposition area. That is, the integrated deposition density from a
discharged activity A is A/pi R2. Dividing by the deposition
velocity gives the integrated concentration of 90Sr in air.
Multiplying by the population, deltaN pi R2, removes the areal
dependence. Using a population density of 25 man km-2, an air
intake rate of 22 m3 d-1, a deposition velocity of 0.5 cm s-1
appropriate for particulates from near surface releases, and the
dosimetric factors given in Table V.5, estimates of the collective
r dose commitments per unit release are obtained. The results are
listed in the summary Table V.6.
207. The collective dose commitment from the input of 90Sr in
water bodies, normalized per unit activity released, can be
estimated [U5] using the expression
c sigmak Nk Ik fk phi
S1 = ------------------------
V(lambda + 1/tau)
Here V is the volume of receiving waters, tau is the turnover time
of receiving waters, lambda is the decay constant of 90Sr, Nk is
the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
208. The quantity V(lambda + 1/tau) is the infinite-time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite-time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentration in water and in fish will
be high, but the population which can be served with drinking water
or by fish consumption will be limited. For inputs into larger
volume of water the concentrations will be smaller, but the
population involved will be correspondingly larger. It seems
reasonable to assume, as a first approximation, that the quantities
Nk/V are relatively constant and independent of V.
209. A summary of the values used in the assessments presented by
UNSCEAR [U6] is given in the following listing:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 0.78 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.5
fish 5 1
shellfish 1
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
Using these values and the dosimetric factors given in Table V.5,
the collective dose commitments per unit activity released in
liquid effluents are estimated to be about 9 10-12 man Gy/Bq for
bone marrow and 1 10-11 man Gy/Bq for bone lining cells for
discharges into fresh waters, and a rounded value of 4 10-16 man
Gy/Bq for both tissues for discharges into the sea. The values for
sea discharges underestimate the collective dose commitment because
they neglect the contribution from large-scale mixing with a longer
residence time. However, due to the relatively short life of 90Sr,
this contribution can only have a small effect on the estimates.
3. Summary
210. Table V.6 summarizes the values obtained above for the
collective dose commitments, normalized per unit activity released,
for releases by atmospheric nuclear explosions and in effluents of
the nuclear power industry. In both cases they are the result of
generalized assessments and substantial variations should be
expected in site-specific cases.
211. For 90Sr and 90Y radiations, the quality factor is one, and
the weighting factors are 0.12 for lung and for bone marrow and
0.03 for bone lining cells. The effective dose equivalent
commitment per unit intake of 90Sr is thus 2.4 10-8 Sv/Bq
(ingestion) and 8.0 10-8 Sv/Bq (inhalation). The collective
effective dose equivalent commitments per unit release from nuclear
installations and from nuclear tests are included in Table V.6.
Table V.6 Summary of collective dose commitments per unit
strontium-90 activity released (10-14 man Gy per Bq)
----------------------------------------------------------------
Lung Bone Bone lining Effective
marrow cells a/
----------------------------------------------------------------
Nuclear explosions
Ingestion 400 500 60
Inhalation 0.1 8 10 1
Nuclear installations
Release to air b/
Ingestion 700 900 100
Inhalation 0.7 60 90 10
Release to fresh water
Drinking water 900 1000 100
Fish 20 30 3
Release to salt water
Fish 0.03 0.04 0.005
Shellfish 0.005 0.007 0.0008
----------------------------------------------------------------
a/ Collective effective dose equivalent commitment
(10-14 man Sv/Bq).
b/ Assumes population density of 25 man/km2 which is fully
sustained by local food production.
F. REFERENCES
A. Aarkrog, A. and J. Lippert. Environmental radioactivity in
Denmark in 1971, 1972, 1973, 1974 and 1975. Danish Atomic
Energy Commission. Riso reports 265 (1972), 291 (1973), 305
(1974), 323 (1975) and 363 (1976).
B1 Beninson, D., A. Migliori de Beninson, C. Menossi et al.
Radioestroncio en el hombre en funcion de la edad. Trabajo
presentado en el Quinto Congreso International de la "Société
francaise de radioprotection". Grenoble, 1971.
B2 Bennett, B.G. Strontium-90 in human bone - 1976 results for New
York and San Francisco. p. I-69-I-84 in Health and Safety
Laboratory environmental quarterly report HASL-328. New
York,1977
C1 Cancio, D., J.A. Llauró, N.R. Ciallella et al. Incorporación de
radioescio por organismos marinos. p. 347-356 in Radioactive
Contamination of the Marine Environment. Proceedings of a
symposium, Seattle, 1972. IAEA publication STI/PUB/313.
Vienna, 1973.
C2 Comisión Nacional de Energía Atomica. Argentina. Information
submitted 1977.
C3 Crouch, E.A.C. Fission product yields from neutron induced
fission. Atomic Data and Nuclear Data Tables, Vol. 19, No. 5.
Academic Press, New York, 1977.
E1 Environmental Measurements Laboratory, U.S. Department of
Energy. Environmental quarterly and appendix. Environmental
Measurements Laboratory report EML-334. New York, 1978.
F1 Federal Radiation Council. Estimates and evaluation of fallout
in the United States from nuclear weapons testing conducted
through 1962 - Report No. 4 (1963).
F2 Foyn, L. Some marine radioecological problems at the nuclear
power station establishment at Oslofjorden. Fisheries
Directorate Sea Research Institute report series B, No. 10.
Bergen, Norway, 1973.
H1 Hallden, N.A., I.M. Fisenne, D.Y. Ong et al. Radioactive decay
of weapons debris p. 194-199 in Health and Safety Laboratory
fallout program quarterly summary report HASL-117. New York,
1961.
I1 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.12
I2 International Radiological Protection. Report of Committee II
on Permissible Dose for Internal Radiation. ICRP publication 2,
Pergamon Press, 1959.
I3 International Commission on Radiological Protection. Task
group on lung dynamics. Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.
12: 173-226 (1966).
I4 International Commission on Radiological Protection. Task
group of Committee 2. The metabolism of compounds of plutonium
and other actinides. ICRP publication 19, Pergamon Press,
1972.
M1 Mitchell, N.T. Radioactivity in surface and coastal waters of
the British Isles, 1972-1973. U.K. Ministry of Agriculture
Fisheries and Food. Fisheries Radiobiological Laboratory
report FRL-7, FRL-8, FRL-9, FRL-10 (1971, 1973, 1975).
N1 National Radiological Protection Board. The data submitted by
the United Kingdom to the United Nations Scientific Committee
on the Effects of Atomic Radiation for the 1977 Report to the
General Assembly. NRPB report R47, Harwell (1976).
N2 Norwegian Institute for Water Research. Release of radioactive
materials from nuclear power stations. Report No. 2.
Dispersal mechanisms, pathways and concentration factors for
radionuclides in the cooling waters. Report 0-177/70 (1974).
N3 National Council on Radiation Protection and Measurements. A
handbook of radioactivity measurements procedures. NCRP report
No. 58, Washington D.C. (1978).
O1 Oak Ridge National Laboratory. Siting of fuel reprocessing
plants and waste management facilities. Oak Ridge National
Laboratory report ORNL-4451 (1970).
P1 Papworth, D.G. and J. Vennart. Retention of 90-Sr in human
bone at different ages and the resulting radiation doses.
Phys. Med. Biol. 18: 169-186 (1973).
R1 Rivera, J. and J.H. Harley. The HASL bone program 1961-1964.
U.S. Atomic Energy Commission report HASL-163. New York, 1965.
S1 Scheidhauer, J., R. Ausset, J. Planet et al. Programme de
surveillance de l'environment marin du centre de La Hague.
p. 347-365 in Population Dose Evaluation and Standards for
Man and His Environment. IAEA publication STI/PUB/375. Vienna,
1974.
S2 Spiers, F.W., G.D. Zanelli, P.J. Darley et al. Beta-particle
dose rates in human and animal bone. p. 130-148 in Biomedical
Implications of Radiostrontium Exposure. U.S. Atomic Energy
Commission Symposium Series 25 (1972).
U1 Ueda, T., Y. Suzuki and R. Nakamuru. Transfer of caesium-137
and strontium-90 from the environment to the Japanese
population via the marine environment in Population Dose
Evaluation and Standards for Man and His Environment. IAEA
publication STI/PUB/375. Vienna, 1974.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No. 16
(A/5216). New York, 1962.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official
Records of the General Assembly, Nineteenth Session, Supplement
No. 14 (A/5814). New York, 1964.
U4 United Nations. Report of the United Nations Scientific
Committee on the effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-First Session, Supplement No.
14 (A/6314). New York. 1966.
U5 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication, No. E.72.IX.17 and
18. New York, 1977.
U6 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
V1 Vanderploeg, H.W., D.C. Porzyck, W.H. Wilcox et al.
Bioaccummulation factors for radionuclides in freshwater biota.
Oak Ridge National Laboratory report ORNL-5002 (1975).
VI. IODINE
A. INTRODUCTION
212. Iodine is a volatile element which is very mobile in the
environment. It is non-uniformly distributed in nature, its
abundance in the lithosphere being about 5 times higher than in the
ocean waters. The iodine in the sea apparently originates from
erosion of the land masses. Recycling to the terrestrial biosphere
occurs through evaporation of sea water and decomposition of
substances of marine origin.
213. There are at least 25 iodine isotopes with mass numbers
ranging from 117 to 141. All except 127I are radioactive. Omitting
the very short-lived 140I and 141I, thirteen isotopes are produced
by fission:
127I (stable), 128I (25 min), 129I (1.57 107a), 130I (12.4 h),
131I (8.06 d), 132I (2.3 h), 133I (21 h), 134I (52.8 min),
135I (6.7 h), 136I (83 s), 137I (23 s), 138I (5.9 s) and 139I
(2 s). From the point of view of environmental contamination and
resulting doses to man, the most important isotopes of iodine are
131I and 129I. They are the only radioactive isotopes of iodine
produced by fission with half-lives longer than one day. Iodine-
131 is a beta-emitter with a half-life of 8.06 days and a maximum
beta energy of 0.81 MeV emitting also gamma rays of 0.36 and 0.64
MeV and other energies. Iodine-129 has a very long half-life (1.57
107 a); it is a beta-emitter (maximum energy: 0.15 MeV) with an
accompanying gamma ray of 0.09 MeV in 8% of the disintegrations
[D1]. The two isotopes are mainly found in the environment as a
result of nuclear explosions and releases from nuclear reactors and
fuel reprocessing plants. Only these two isotopes of iodine are
considered in this report.
214. Iodine enters the metabolism of living organisms and is
selectively taken up and concentrated in the thryoid gland; it
plays a major role in the synthesis of the thyroid hormone and is
secreted in milk. Owing to its decay properties, 131I has been
extensively used in the medical field for diagnosis and treatment
of thyroid abnormalities.
B. SOURCES
1. Natural production
215. Like any other fission product, 129I and 131I are present in
the environment as a result of spontaneous fission of natural
uranium. In view of its very long half-life, 129I has accumulated
in the earth's crust and also in the ocean waters from where it is
available to disperse in the whole biosphere.
216. In 1962, Edwards [E1] predicted the natural 129I/127I atom
ratio in sea water to be about 3 10-14 from spontaneous fission of
uranium-238 and estimated that contributions of the same order of
magnitude would arise from spontaneous fission of 235U and from
production in the upper atmosphere by interaction of energetic
protons, neutrons and photons on isotopes of xenon. Experiments
later confirmed the validity of Edwards' estimate, the 129I/127I
atom ratios derived from measurements in iodine-rich minerals
ranging from 2 10-15 to 10-13 [M1, S1]. Taking the concentrations
of 127I in sea water to be 0.064 µg g-1 and the global volume of
sea water to be 6 1017 m3, a natural activity of 129I of 7 109 Bq
in the oceans is obtained from an 129I/127I atom ratio of 3 10-14.
2. Nuclear explosions
217. The activity of 131I (or of 129I) generated in nuclear
explosions can be derived from the measurements of 90Sr deposition
[U1] and from the ratios of the fission yields of 131I (or 129I)
and of 90Sr for nuclear weapons tests [H2].
218. The total activity of 90Sr produced in nuclear explosions
through 1976, which has been globally distributed, is estimated to
be 6 1017 Bq [U1]. This does not include the local fallout, which
is deposited in the vicinity of the test area and which can be
important for the lower yield tests detonated near or on the land
surface.
219. On the basis of measured fission product yields of individual
nuclides obtained by analysis of debris from megaton weapons, the
yields for 90Sr, 129I and 131I are estimated to be 0.035, 0.0126
and 0.029, respectively [H2].
220. The activities of 129I and 131I produced in nuclear tests
through 1976, which gave rise to globally distributed fallout, are
thus found to be 4 1011 Bq and 6 1020 Bq. Owing to the half-lives
of the two nuclides, practically all of the activity of 129I is
still present in the environment where it will remain for millions
of years whereas practically all of the activity of 131I has
decayed.
3. Nuclear fuel cycle
(a) Nuclear power plants
221. Iodine-131 and iodine-129 are produced by fission in the fuel
of nuclear reactors. The equilibrium activity of 131I per unit of
electrical power is established after a few weeks of irradiation in
uranium fuel at about 3 1015 Bq per MW(e) [U1] and increases
slightly from the beginning of the fuel irradiation to the end, as
the result of the larger fission yield of plutonium which
contributes increasingly to power production as the burn-up
proceeds. The corresponding activity of 131I produced per unit
energy generated is about 9 1016 Bq per MW(e)a.
222. Small amounts of 131I produced in the fuel may reach the
coolant of the nuclear reactor through defects in the fuel
cladding. In coolant purification or following coolant leakage,
131I may reach the gaseous and liquid effluent streams. The
reported 131I releases in airborne effluents, as summarized by
UNSCEAR [U1], show an extremely wide variability due mainly to
different waste treatment systems. The overall activity discharged
per unit energy generated is of the order of 107 Bq per MW(e)a from
PWRs and 108 Bq per MW(e)a from BWRs [I1, U1] while the limited
data from GCRs and HWRs indicate that the average airborne releases
are comparable to those from PWRs [U1].
223. Airborne iodine can occur in various chemical forms in the
airborne effluents. Elemental iodine (I and I2), organic iodine
with methyl iodide (CH3I) as the simplest organic compound and
hypoiodous acid (HOI) may be present in significant proportions,
iodine is also to some extent bound to particulates.
224. Few data are available on the proportions of organic and
inorganic forms of the iodine released to the atmosphere. Analyses
at power stations in the Federal Republic of Germany show that only
a very small fraction (usually less than 1%) of the iodine released
in airborne effluents is in particulate form [W1]. Recent
measurements in the U.S.A. indicate that, in airborne effluents
from PWRs, on the average 31% of 131I is organic, 40% HOI, 27%
elemental and 2% in particulate form [P1], whereas in BWRs, the
proportions were found to be somewhat different, namely 40%
organic, 20% HOI, 28% elemental and 12% particulate [P2]. These
proportions, however, are expected to vary significantly according
to the type of waste treatment in use.
225. In liquid effluents from LWRs, 131I is found in amounts
comparable to the airborne releases from BWRs (108 Bq per MW(e)a)
[U1]. It is not reported in the liquid effluents from other types
of reactors.
226. In 1980, the installed nuclear capacity was 1.25 105 MW(e) on
a worldwide scale [I2]. Assuming an average load factor of 0.6,
the energy produced was 7.5 104 MW(e)a. The global production and
release of 131I at the reactor sites in 1980 are estimated to be
about 7 1021 Bq and 2 1013 Bq, respectively, using the figures
given previously for production and release, and assuming that the
releases are similar to those from BWRs for the reactor types for
which no data are available. The estimate of the global release is
very crude, but nevertheless shows that only a tiny fraction of the
order of 10-9 of the 131I produced in the reactors is discharged
into the environment. Table VI.1 provides a break-down of the
releases from reactors according to reactor type.
227. Being a volatile element, iodine is readily released to the
atmosphere in the case of a reactor accident. The two reported
reactor accidents which have caused measurable irradiation of the
public occurred at Windscale (U.K.) in October 1957 and at Three
Mile Island in March 1979. The release of 131I to the atmosphere
was about 7 1014 Bq in the Windscale accident [L1] and 6 1011 Bq in
the Three Mile Island accident [H3].
228. The activity of 129I produced in a nuclear reactor is much
lower than that of 131I. The production of 129I per unit energy
generated is approximately 5 107 Bq per MW(e)a [U1] corresponding
to an inventory of 1.5 108 Bq per MW(e) after three years of fuel
irradiation. Iodine-129 has not been identified in routine
discharges of nuclear reactors. A rough estimate of the activity
of 129I discharged per unit energy generated can be calculated
assuming that the ratio of the release rate to the inventory in
reactor fuel is the same for 129I and 131I. As the activity of
129I in fuel is at least 2 107 times lower than that of 131I, the
release rate of 129I per unit energy generated is at most 0.1 Bq
per MW(e)a; activity of 129I discharged from nuclear reactors in
1980 would thus be of the order of 104 Bq.
(b) Fuel reprocessing plants
229. At the fuel reprocessing stage of the nuclear fuel cycle (if
it is undertaken), the elements uranium and plutonium in the
irradiated nuclear fuel are recovered for reuse in fission
reactors. Before reprocessing, the spent fuel elements are stored
under water until 131I has decayed to insignificant amounts.
Storage times of six months and one year result in the reduction of
the activities of 131I originally present in the fuel by factors of
6.5 106 and 4.3 1013, respectively.
230. In 1980, the only reprocessing plants operating commercially
were at Windscale (U.K.) and La Hague and Marcoule (France): in
addition there were several small experimental reprocessing
facilities, such as the one at Karlsruhe (Federal Republic of
Germany). The combined capacity of the reprocessing plants was
much lower than the amount of fuel discharged from reactors
worldwide.
Table VI.1 Estimated global discharges of 131I from nuclear power
stations in 1980
-------------------------------------------------------------------------
Estimated release Estimated discharges
rates in 1980
Reactor Number Capacity (Bq per MW(e)a) (Bq)
type (MW(e)a) ----------------- -------------------------
Airborne Liquid Airborne Liquid Total
-------------------------------------------------------------------------
PWR 96 64239 107 108 6 1011 6 1012 7 1012
BWR 62 35170 108 108 4 1012 4 1012 8 1012
HWR 14 5963 107 108 6 1010 6 1011 7 1011
GCR 36 7086 107 108 7 1010 7 1011 8 1011
Other 33 12527 108 108 1 1012 1 1012 2 1012
-------------------------------------------------------------------------
Total 241 124985 - - 6 1012 1 1013 2 1013
-------------------------------------------------------------------------
231. The activity of 131I released into the environment from
reprocessing plants depends critically on the storage time: in
practice with the growing backlog of fuel for reprocessing a
storage time of one year is common but even a relatively small
quantity of fuel with a short storage time will dominate the total
131I releases. Luykx and Fraser [L2] have expressed the reported
releases of 131I from Windscale, La Hague, Marcoule and Karlsruhe
during 1974-1978 in terms of activity discharged per unit of
electricity generated. The results range from less than 1.5 105 to
7 107 Bq per MW(e)a and are presented in Table VI.2. The
activities of 131I discharged in liquid effluents have not been
reported.
232. The discharges of 129I depend upon the specific waste
treatment at the reprocessing plant. With regard to airborne
effluents, the reported activites released per unit of electricity
generated were, on average during the 1975-1978 time period, 2.7
106 Bq per MW(e)a at Windscale and 4.8 105 Bq per MW(e)a at
Karlsruhe, representing about 4% and 1%, respectively, of the fuel
content [L2]. In a series of measurements from November 1975 to
August 1977 the average values for the components of 129I
discharges from Karlsruhe were reported at 74% inorganic, 23%
organic and 2% aerosol [B1].
Table VI.2 Average normalized activities of 129I and 131I
discharged into the environment by fuel reprocessing plants
(Bq per MW(e)a)
---------------------------------------------------------------------------
Iodine-129 Iodine-131
Plant ---------------------------- ------------------------------
Location Airborne Liquid Total Airborne Liquid Total
effluents effluents effluents effluents
---------------------------------------------------------------------------
Windscale 2.7 106 5.6 107 5.9 107 2.6 106 N.A. a/ >2.6 106
La Hague N.A. N.A. N.A. 1.1 107 N.A. >1.1 107
Marcoule N.A. N.A. N.A. 7.4 107 N.A. >7.4 107
Karlsruhe 4.8 105 N.A. N.A. >1.5 105 N.A. -
---------------------------------------------------------------------------
a/ N.A. = Data not available.
233. In recent years, the 129I released in liquid effluents was
only measured at Windscale. They average at 5.6 107 Bq per MW(e)a
which corresponds fairly well with the theoretical fuel content
[L2].
234. The information on 131I and 129I activities discharged per
unit electricity generated is summarized in Table VI.2. If it is
assumed for the four reprocessing plants, that all the 129I
contained in the fuel is discharged into the environment, the total
129I released in 1978 was about 3 1011 Bq, which is 7 orders of
magnitude higher than the total activity estimated to be released
from reactors. With regard to 131I, it is much more difficult to
assess the total activity released from fuel reprocessing plants,
as the activities discharged into liquid effluents have not been
reported. However, using the pessimistic assumptions that the
activity contained in the airborne effluents represents 1% of the
activity present in the fuel at the time of reprocessing and that
the rest of the activity is discharged into liquid effluents, it is
found that the total annual 131I discharges from fuel reprocessing
plants are about 5 1011 Bq, which is much less than the global
discharges from reactors.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Nuclear explosions
235. The behaviour in the environment of 131I produced in the
nuclear explosions has been extensively studied, especially the
air-vegetation-milk pathway which is generally the most significant
route by which humans are exposed. Much of the literature has been
referenced in UNSCEAR reports [U1-U5]. Environmental
concentrations of 131I following large nuclear explosions are
significant; they are easily measured and this allows the transfer
factors to be derived from observations. In contrast, the
environmental concentrations (and the resulting dose rates) of 129I
are extremely low and have only been measured in a few studies.
Although some of the aspects of environmental behaviour of 131I
following nuclear explosions apply also to 129I, the discussion in
this section will be limited to 131I.
236. Radioactive fallout is observed to circle the earth in 20-30
days on average [R1] which is approximately the mean residence time
of an aerosol in the troposphere and is longer than the mean life
of 131I. It is thus during its first pass around the earth that a
given atom of 131I formed in a nuclear explosion will either decay
in the atmosphere or deposit on the earth's surface. It is
unlikely that during such a short period the clouds of debris
become well mixed. The ground-level air concentrations of 131I at
a particular station fluctuate according to meteorological
conditions and are not necessarily representative of a larger
region nor of a latitude band [P3].
237. Information on the physical and chemical nature of fallout
131I is very limited. In the U.K., late 1961, an average 75% of
the activity was in particulate form, the rest being in the gaseous
state [E2] but in the U.S.A. in 1962 the particulate fraction was
found to vary from 10 to 90% [P4]. These large variations may be
partly explained by the physical and chemical transformations
undergone by 131I following its formation: Voillequé [V1] observed
that the fraction of the total airborne 131I associated with
particulates is about 0.5 to 0.7 in the first few days following a
nuclear explosion but that it later decreases to be approximately
0.3 after two months. In the gaseous fraction, the proportion of
organic compounds was found to increase in the same two months time
period from one fourth to about three fourths of the gaseous iodine
[V1].
238. Iodine-131 deposition on the ground and on vegetation occurs
by dry and wet deposition. The rate of dry deposition can be
characterized by the deposition velocity on the vegetation which
was derived to be about 2 10-2 m s-1 [C2, H4] from measurements
performed during the series of tests of late 1961. When
precipitation occurs, fallout 131I is deposited at a much faster
rate than in dry weather, essentially by rain-out, i.e., in-cloud
mechanisms, rather than by wash-out, i.e., below the cloud
processes [U5]. On the other hand, rain washes the surface of the
leaves and thus removes some of the radio-iodine. Chamberlain and
Chadwick in the U.K. [C2] and Hull in the U.S.A. [H4] calculated on
the basis of their measurements that in late 1961 about 50% of the
131I falling out in rain was retained on herbage.
239. Even though the observed ground-level air activity
concentrations and deposited activities of 131I vary widely from
one area to another according to meterological conditions, it is
possible to obtain a rough estimate of the total activity density
deposited, weighted over the world's population, from the average
ratio of the 131I/140Ba deposited activity densities. Data from
Argentina covering the years 1966 to 1973 [B2, B3, C3] reveal that
the 131I/140Ba ratio of the annual deposited activity densities
varied from 0.4 to 1.3 with a median value of 0.6. Relevant
information is also provided by the air concentrations of 131I and
140Ba which are measured in the stations of the global network of
the U.K. Atomic Energy Agency [C4]. The annual average of the
integrated air activity concentration 131I/140Ba ratios of nine
stations scattered over the world ranged from 0.19 to 3.1 with a
median value of 0.46. However, the corresponding ratios of the
deposited densities were higher as only the particulate fraction of
131I was measured in the air. Assuming that the particulate
fraction of 131I represents half of the total activity of that
nuclide in the air, the median value would be approximately 0.9,
which is comparable to the Argentinian value of 0.6. An
intermediate value of 0.7 will be adopted in this document.
240. As the average ratio of the 140Ba/95Zr deposited activity
density is estimated to be 0.62 [U2] and the population-weighted
global average deposition density of 95Zr from all tests is
approximately 2.4 104 Bq m-2 [U2], the population-weighted global
average deposition density of 131I is thus found to be on the order
of 104 Bq m-2.
241. Fresh milk is usually the main source of 131I in food because
of the concentration achieved by the grazing animal and the short
storage period of milk. Besides, milk plays an important worldwide
role in the diet of infants. The relationship between the
integrated cow's milk concentration and the deposition density has
been derived from measurements in Argentina [B2] to be 6.3 10-4 Bq
a 1-1 per Bq m-2 and to show little variation from year to year.
The integrated milk concentrations observed throughout the world by
the fallout network stations have been reported by UNSCEAR [U1-U5].
2. Industrial releases
242. The environmental behaviour of radio-iodines released from
nuclear facilities differs in some aspects from that of fallout as
the chemical forms are not in the same proportion and as the
releases occur at discrete points on the surface of the earth both
in the atmosphere and in the aquatic environment. As the
authorities are concerned with the total impact resulting from the
releases of radionuclides, all the important pathways leading to
man have been investigated, mainly through laboratory and field
experiments, and occasionally following unplanned releases. In
comparison to 131I and to fallout, much more 129I is released from
industrial operations and it will be considered in this section,
together with 131I in the discussion of the local and regional
aspects, and on its own in the discussion of the global aspects.
(a) Local and regional aspects
(i) Atmospheric releases
243. The behaviour in the atmosphere of the radio-iodines released
from nuclear facilities is complicated by the various forms that
iodine may take (particulate, elemental, organic, or as hypoiodous
acid). Elemental iodine readily deposits on forage and enters the
cow-milk-man pathway. Organic iodine is retained much less
efficiently by vegetation and its deposition velocity is 200 to
1000 times smaller than that of the elemental form [A1, H5].
Particulate associated iodine and hypoiodous acid will be deposited
at rates intermediate between those for the elemental and organic
forms [V2]. Physico-chemical transformations occurring during
atmospheric transport may also affect the distribution of the
various forms of iodine, since some of them are not stable in
sunlight. On the basis of photochemical considerations, the
atmospheric residence times for I2 are estimated to be less than a
minute during the day, much shorter than the residence times for
CH3I and other organic iodides in plant effluents (about 60 hours
in sunlight) [V1]. The elemental form would be expected to become
rapidly associated with airborne aerosols, so that deposition at
distances beyond the immediate vicinity of the release would be
largely governed by the particulate behaviour. It must be pointed
out, however, that the atmospheric residence times of CH3I derived
from environmental measurements is much longer than that obtained
from photochemical considerations (about 100 days to be compared
with 60 hours in sunlight) [V1].
244. Numerous field and laboratory experiments have been conducted
to determine the deposition velocity on vegetation of various forms
of iodine [B4, H6, V2]. Most of the experiments dealt with
elemental iodine, for which the deposition velocity was found to
vary with the temperature, the relative humidity of the air, the
wind speed and the vegetation density. The best fit of the
experimental data is obtained assuming that the deposition velocity
on vegetation of elemental iodine is proportional to the wind speed
and to the vegetation density and is an exponential function of the
temperature and of the relative humidity of the air [A2]. Typical
values of the deposition velocity on vegetation are 2 10-2 m s-1
for elemental iodine and 5 10-5 m s-1 for organic iodide. In the
case of particulates, a value of 10-3 m s-1 was found representative
for grass, about 2 10-3 m s-1 for clover and about 3 10-4 m s-1 for
vegetation-free soil [H7]. Since the deposition velocity varies
with the vegetation density, less variability is encountered by
normalizing the values by the mass of dry vegetation per unit area.
UNSCEAR [U1] adopted a normalized value of 5 10-3 m3 kg-1 s-1 for
the deposition velocity on grass of radio-iodine in effluents from
nuclear installations.
245. Regarding atmospheric releases of radio-iodine, the main
pathways to man are inhalation and consumption of fresh milk and
fresh leafy vegetables; consumption of beef is also taken into
consideration in the case of 129I.
246. The assessment of the transfer of iodine to milk requires the
knowledge of the value of the following parameters in addition to
the deposition velocity; the residence half-time of iodine on
vegetation, the average mass of grass consumed per cow and per day
under average agricultural conditions during the grazing season,
the fractional pasture grazing time and the fractional transfer of
the daily ingested activity by the cow per unit volume of produced
milk.
247. The residence half-time of 131I on grass is 3-6 days, most
estimates lying around 5 days [B4, B5]. This figure seems to be
valid irrespective of the iodine production source (fallout,
nuclear plants, experiments) and of the climatic characteristics of
the region [B4]. The corresponding residence half-time of stable
iodine (or of 129I) on grass is about l4 days. The depletion
mechanisms involved are: transfer to the roots; volatilization;
leaching by atmospheric precipitation; mechanical removal by wind,
rain or other agents; death or decomposition of the leaves or of
their surface layer [B4, C2]. The opinions do not agree on the
relative importance of those mechanisms [B4, C2].
248. The daily grass requirement of a lactating cow is estimated
at 10 kg dry matter [B4, H6]. The pasture grazing time varies
according to the climatic conditions and to the cattle management
practices. An average grazing time of six months per year was
assumed by UNSCEAR [U1]. During the winter months, the cows are
held in the stable and consume dry fodder in which the activity
concentration of 131I will have decayed to insignificant levels and
that of 129I can be assumed to be the same as that in herbage.
249. The transfer of iodine is usually expressed as the fraction
present in milk of the ingested activity under equilibrium
conditions. This quotient was found to be around 5 10-3 d 1-1 [B6,
U1]. The value adopted by UNSCEAR in its 1977 report is an upper
estimate of 10-2 d 1-1 [U1]. In the case of a simple
administration of 131I to the cow, Lengemann and Comar [L3]
observed that the maximum concentration is reached within one day
and that it is followed by a rapid decrease (half-time of about 1.5
days) in the first 3-4 days and a slower decrease (half-time of
about 3 days) afterwards. Among the different factors which can
have an influence on the grass to milk transfer of iodine, the two
most important may be the milk productivity by animals and the
season, a higher iodine secretion in milk occurring with a
productivity increase and in the warm season [B4, G1].
250. Taking account of all the parameters given above, an
integrated air concentration of 1 Bq a m-3 of 131I or 129I would
result in an integrated milk concentration for 131I of 160 Bq a 1-1
and for 129I of 870 Bq a 1-1. Transfer via wet deposition is
usually insignificant over the course of a year [U1].
251. The transfer of iodine from air to fresh leafy vegetables has
been assessed by UNSCEAR [U1] on the basis of the values given
above for the deposition velocity and the residence time on the
vegetation, and of a dry-to-wet vegetation weight ratio of 0.5. In
addition, a fractional removal by washing of 0.4 [U6] and an
average 7 day marketing delay (resulting in a decay factor of 0.55
for 131I and 1.0 for 129I) were taken into account. Time
integrated conditions of 340 Bq a kg-1 (fresh weight) for 131I and
1740 Bq a kg-1 (fresh weight) for 129I are calculated for the case
of a time-integrated air concentration of 1 Bq a m-3.
252. An estimate of the transfer of 129I has also been carried out
by UNSCEAR [U1]. Using the values given above for the deposition
velocity on grass, residence half-time on grass and grass
consumption rate, the resulting transfer factor is 260 Bq a kg-1
per Bq a m-3 of 129I in air if the fractional transfer of daily
ingested activity per unit mass of meat is taken to be 3 10-3
d kg-1 [P5].
253. In the assessment of the 129I concentration in grass
following deposition of that nuclide, the root uptake from soil has
not been taken into account. It has been estimated [B7] that this
pathway could contribute at equilibrium only about 20% of the
concentration in grass arising from direct deposition.
(ii) Aquatic releases
254. Information on the behaviour of iodine in the aquatic
environment is rather limited. The UNSCEAR aquatic model [U1] can
be used to estimate the transfer of 131I and 129I from the aquatic
environment to diet for generalized discharge situations. In that
model, it is assumed that iodine is not removed to sediments but
that during treatment of drinking water, 20% of the activity
contained in raw water is removed. For the two isotopes of iodine
considered, the concentration factors are taken to be 15 1 kg-1 for
fresh water fish, 20 1 kg-1 for marine fish and 100 1 kg-1 for
shellfish. Further discussion is presented in the section on
dosimetry.
(b) Global aspect
255. Because of its very long half-life (1.57 107 a) and of the
mobility of iodine in the environment, 129I may become widely
distributed on the global scale. Whether released into the
atmosphere or into the aquatic environment, 129I will eventually
reach the oceans in a time period very short in comparison with its
half-life. Iodine-129 will then be recycled to the atmosphere and
the terrestrial biosphere, mainly by evaporation of seawater.
Atmospheric water is exchanged within about 10 days and it follows
therefore that there is a rapid exchange of iodine. The specific
activity approach has been the usual method to assess the dose
commitments and the long-term environmental concentrations from
129I discharges. According to that approach, the specific activity
of 129I per unit mass of stable iodine of any environmental
material of the terrestrial biosphere (including air and food-stuffs)
will be in the long term equal to that of seawater. Assuming 6
1023 g to be the mass of ocean waters with an iodine concentration
of 0.064 µg per gram of water [T1], the specific activity of 129I
per unit mass of stable iodine obtained in sea water after a
release of 1 Bq of that radionuclide into the environment is found
to be 2.6 10-17 Bq g-1. If it is assumed that there is no
environmental sink for iodine, 129I will be recycled throughout its
mean life of 2.3 107a.
D. TRANSFER TO MAN
256. Iodine is an element of fundamental importance for the human
organism since it is an essential component of the thyroid hormone,
which is necessary for the growth and metabolism of the body. The
metbolic cycle of iodine in man, especially in the adult, is
sufficiently well known in its fundamental behaviour, as a
consequence of the large number of clinical studies carried out in
the last years with radioactive isotopes of iodine [B4].
257. The absorption by the blood from the gastro-intestinal tract
is complete and very rapid. It is absorbed at the rate of about 5%
per minute and it can be considered to be complete after two hours
[B4]. When inhaled in the form of inorganic iodide or as
methyliodide, a fraction of about 70% is absorbed [M2] whereas more
than 90% of it is absorbed when inhaled in the form of elemental
vapour [M3].
258. The uptake by the thyroid of the iodine contained in blood as
well as the size of the thyroid gland are both very dependent upon
the daily intake of stable iodine [D2]. The model adopted by ICRP
[I3] for the metabolism of iodine applicable to adults is based on
the three-compartment model of Riggs [R2]. ICRP [I3] assumes that
30% of the iodine entering the blood is translocated to the thyroid
while the remainder goes directly to excretion. Iodine in the
thyroid is assumed to be retained with a biological half-life of
120 days and to be lost from the gland in the form of organic
iodine. Organic iodine is assumed to be uniformly distributed
among all organs and tissues of the body other than the thyroid and
to be retained there with a biological half-life of 12 days. One-
tenth of this organic iodine is assumed to go directly to faecal
excretion and the rest is assumed to be returned to blood as
inorganic iodine.
259. A quantitative assessment of the transfer of iodine to man
must take into consideration the variation with age of the various
parameters involved, which are essentially the mass of the thyroid
gland, the fractional uptake by the thyroid, the effective
residence half-time in the thyroid, the breathing rate and the
consumption rate of foodstuffs. The values adopted for 129I and
131I by UNSCEAR in its 1977 report [U1] are presented in Table
VI.3. It is to be noted that the metabolic parameters for adults
are not in complete agreement with those adopted by ICRP [I3]; the
resulting differences in the thyroid absorbed doses vary according
to the pathway and the isotope considered but they are in all cases
less than 50%.
Table VI.3 Variation with age of the parameters used in the
assessment of the transfer to man of 129I and 131I [U1]
-----------------------------------------------------------------
Age
6 4 14 Adult
months years years
-----------------------------------------------------------------
Mass of the thyroid (g) 2 4 14 20
Effective half-time
in the thyroid (d) 131I 6.0 6.3 6.9 7.6
129I 23 28 48 136
Inhalation pathway
Fractional uptake by the thyroid 0.30 0.26 0.26 0.26
Breathing rate (m3 a-1) 1150 3530 6440 8030
Ingestion pathway
Fractional uptake by the thyroid 0.40 0.35 0.35 0.35
Consumption rate:
Milk (1 a-1) 330 180 150 90
Leafy vegetables (kg a-1) 0 13 20 30
Beef (kg a-1) 0 8 15 27
Drinking water (1 a-1) 438 438 438 438
River fish (kg a-1) 1 1 1 1
Ocean fish (kg a-1) 6 6 6 6
Shellfish (kg a-1) 1 1 1 1
-----------------------------------------------------------------
E. DOSIMETRY
1. Dose per unit intake
260. As iodine is selectively taken up in the thyroid gland, its
concentration in that organ is considerably higher than in the
other organs and tissues in the body. The stable iodine
concentration in the thyroid tissue of an adult is of the order of
500 µg g-1 while in the rest of the body it is much less than 1 µg
g-1 [S2]. Since the major contribution of the absorbed doses from
129I and 131I is due to the emission of the beta-particles, which
have a short range in the human tissues, the absorbed doses in the
thyroid are about 1000 times higher than those in the other organs
and tissues, which will not be considered in this document.
261. The significant variation with age of the metabolic
parameters is reflected in the thyroid absorbed doses per unit
intake. Table VI.4 presents the values of the age-dependent
thyroid absorbed doses per unit intake adopted by UNSCEAR in its
1977 report [U1]. These values are derived from the metabolic
parameters presented in Table VI.3 and from the following figures
for the energy absorbed in the thyroid (MeV) for disintegration:
0.18, 0.18, 0.19 and 0.19 MeV for 131I and 0.060, 0.061, 0.063 and
0.064 MeV for 129I for the ages of 0.5, 4, 14 years and adult,
respectively.
Table VI.4 Age-dependent absorbed doses in the thyroid gland
per unit intake of 131I and 129I (Gy Bq-1)
------------------------------------------------------------
Thyroid aborbed dose Age
per unit intake 6 months 4 years 14 years Adult
------------------------------------------------------------
Inhalation: 131I 3.2 10-6 1.5 10-6 4.9 10-7 3.8 10-7
129I 4.1 10-6 2.2 10-6 1.1 10-6 2.3 10-6
Ingestion: 131I 4.3 10-6 2.0 10-6 6.5 10-7 5.1 10-7
129I 5.4 10-6 3.0 10-6 1.5 10-6 3.0 10-6
------------------------------------------------------------
2. Dose per unit release
(a) Nuclear explosions
262. The thyroid dose commitment via the ingestion (milk) pathway
from 131I released by nuclear explosions can be assessed from the
sequential product of transfer factors
Dc = P23 P35 F
where F is the integrated deposition density, P23 is the deposition
to milk transfer factor and P35 is the milk to thyroid dose
transfer factor. The value of F weighted for the world's
population and that of P23 for fallout deposition were given above
as 104 Bq m-2 and 6.3 10-4 Bq a 1-1 per Bq m-2, respectively. The
value of P35 can be derived from the age-dependent consumption
rates of milk given in Table VI.3, the age-dependent thyroid doses
per unit ingested activity presented in Table IV.4, and from the
assumption that the three groups of children are representative of
the age groups 0-1, 1-9 and 10-19 years, respectively, and that
these groups contain respectively 2, 16, and 20% of the population
[U1]. The value of P35 (milk) is thus found to be 1.3 10-4 Gy per
Bq a 1-1. The thyroid dose commitment for the world's population
arising from 131I from global fallout of past nuclear explosions is
therefore estimated to be about 8 10-4 Gy. Most of the dose
commitment was in fact delivered in the early 1960s. Taking the
world's population at that time to be 3 109 persons, the collective
dose commitment would be about 2 106 man Gy. Since 6 1020 Bq of
131I were estimated to give rise to global fallout, the individual
and collective thyroid dose commitments per unit activity released
are found to be 1.3 10-24 Gy Bq-1 and 3 10-15 man Gy Bq-1,
respectively.
(b) Nuclear industry
(i) Local and regional contribution
263. Atmospheric releases. The contribution of the inhalation
pathway to the collective dose commitments from effluent releases
can be estimated from the integrated concentrations of 131I and
129I in ground-level air. Assuming that all the activity released
in the atmosphere will eventually deposit on the ground, the
integrated concentration in ground-level air is the total amount of
131I or 129I released per unit area of the deposition region
divided by the deposition velocity, vd. The population affected is
the population density deltaN, times the area of the deposition
region. The collective dose commitment per unit activity released
is given by the expression Sc = deltaN phi/vd where phi is the age-
weighted thyroid dose per unit integrated air concentration.
264. On the basis of a figure of 5 10-3 m3 kg-1 s-1 for the
deposition velocity on grass per unit area density of vegetation
(see paragraph 244), using a value of 0.1 kg (dry) m-2 for the mass
of grass per unit area, and assuming that the activity deposited on
grass represents one fourth of the total activity deposited on
ground, vd would be 2 10-3 m s-1 or 6 104 m a-1. From the data
contained in Tables VI.3 and VI.4 and the age distribution given
above, the age-weighted thyroid dose per unit integrated air
concentration would be 3.4 10-3 and 1.4 10-2 Gy per Bq a m-3 for
131I and 129I respectively. As the deposition area is expected to
be very large, the population density is assumed to be about 25
persons km-2, that is 2.5 10-5 persons m-2. The collective thyroid
dose commitments are thus estimated to be 1.4 10-12 and 5.8 10-12
man Gy Bq-1 for 131I and 129I, respectively.
265. The contribution of the ingestion pathway (consumption of
milk, leafy vegetables and meat) to the collective dose commitments
per unit activity released can be assessed by the expression Sc =
P13 P35 deltaN where P13 is the air to dietary product to age-
weighted thyroid dose transfer factor, and deltaN the population
density. Using the values and assumptions given previously, the
collective thyroid dose commitments are estimated to be, for 131I,
8.7 10-12 and 2.3 10-12 man Gy Bq-1 for consumption of milk and
fresh leafy vegetables, respectively, while the values for 129I
would be 1.2 10-10, 5.0 10-11 and 6.4 10-12 man Gy Bq-1 for
consumption of milk, fresh leafy vegetables and beef, respectively.
266. Aquatic releases. The collective thyroid dose commitment per
unit activity of 131I and 129I discharged into the aquatic
environment can be estimated, as in the 1977 UNSCEAR report [U1],
using the expression
Sc = N I f phi
V(lambda + 1/tau)
where V is the volume of the receiving waters, tau the turn-over
time of receiving waters, lambda the decay constant of the
radionuclide considered, N the number of individuals exposed, I the
individual consumption rate of the foodstuff considered, f the
concentration factor of the radionuclide in that foodstuff, and phi
the thyroid dose per unit activity ingested.
1
267. The quantity V(lambda + 1/tau) is the infinite time integral
of the water concentration per unit activity released, while that
quantity multiplied by f is the infinite time integral of the
concentration in the consumed item (fish, for example). For inputs
into small volumes of water, the concentrations in water and in
fish will be high, but the populations which can be served with
drinking water or by fish consumption will be limited. For inputs
into large volumes of water, the concentrations will be smaller,
but the populations involved will be larger. It is reasonable,
therefore, to assume as a first approximation that the quantities
V/N are relatively constant; they are taken to be 3 107 and 3 109
litre per man, for fresh water and sea water, respectively. Using
the values given previously in the text and in the tables, the
collective thyroid dose commitments per unit activity of 131I and
129I can be estimated. The results are presented in Table VI.5.
Table VI.5 Collective thyroid dose commitments
per unit activity of 131I and 129I released
in the aquatic environment (man Gy Bq-1)
------------------------------------------------
Type of release 131I 129I
and pathway
------------------------------------------------
Release to fresh water
- Drinking water 3.2 10-13 3.2 10-10
- Fish 1.4 10-14 1.4 10-11
Release to sea water
- Fish 1.1 10-15 1.1 10-13
- Shellfish 8.8 10-16 1.1 10-14
------------------------------------------------
(ii) Global contribution
268. Using the specific activity approach described previously,
the activity concentration of 129I per unit mass of 127I is the
same in the sea water and in the human thyroid. Assuming that the
concentration of stable iodine per unit mass of thyroid is 80, 180,
300 and 600 µg g-1 at ages 6 months, 4 years and 14 years and for
adults, respectively and using the age distribution given
previously, a specific activity of 1 Bq per gram of stable iodine
in the thyroid would lead to an age-weighted annual thyroid dose of
1.5 10-7 Gy. Since a release of 1 Bq 129I results in a long-term
concentration of 2.6 10-17 Bq g-1 stable iodine per Bq (see
paragraph 255), the collective thyroid dose commitment arising from
discharges of 129I would be about 9 10-7 man Gy Bq-1, assuming a
world population of 1010 and no sink for iodine in the environment.
269. Most of the collective dose commitment to the thyroid is
delivered in the far future, as the mean life of 129I is 2.3 107 a.
The average dose rate per unit activity released would be extremely
low (4 10-24 Gy a-1 per Bq released). The estimate of the
collective thyroid dose commitment could be significantly in error
if there exists a mechanism that efficiently removed iodine from
the biosphere at a rate significantly greater than the radioactive
decay constant of 129I of 4 10-8 a-1. Such a mechanism could be
the retention by ocean sediments.
(c) Summary
270. Table VI.6 summarizes the results given above for the thyroid
collective dose commitments per unit activity released, and
provides also the collective effective dose equivalent commitments
per unit activity released. The latter quantities are obtained by
multiplying the former by 0.03.
Table VI.6 Summary of collective thyroid dose commitments
per unit activity released and of collective effective dose
equivalent commitments per unit activity released
---------------------------------------------------------------
131I 129Ia 131I 129Ia
------------------ ------------------
(man Gy Bq-1) (man Sv Bq-1)
---------------------------------------------------------------
Weapon tests 3 10-15b 9 10-17b
Industrial releases
a) Atmosphere
Inhalation 10-12 6 10-12 3 10-14 2 10-13
Ingestion
- Milk 1 10-11 1 10-10 3 10-13 3 10-12
- Leafy vegetables 2 10-11 5 10-11 6 10-14 2 10-12
- Beef - 6 10-12 - 2 10-13
b) Rivers
Ingestion
- Water 3 10-13 3 10-10 9 10-15 9 10-12
- Fish 1 10-14 1 10-11 3 10-16 3 10-13
c) Oceans
Ingestion
- Fish 1 10-15 1 10-13 3 10-17 3 10-15
- Shellfish 9 10-16 9 10-14 3 10-17 3 10-15
---------------------------------------------------------------
a First passage estimates. The long-term, global estimates
for 129I from all sources and by all pathways are 9 10-7 man
Gy Bq-1 and 3 10-8 man Sv Bq-1.
b Milk consumption.
F. REFERENCES
A1 Atkins, D.H.F., R.C. Chadwick and A.C. Chamberlain. Deposition
of radioactive methyl iodide vegetation. Health Phys. 13: 91
(1967).
A2 Angeletti, A. and A. Sauve. Estimation de la vitesse de dépôt
de l'iode vapeur sur les végétaux en fonction des eléments du
climat. Congrés commun de radioprotection. SFRP-FS. Lausanne,
1981.
B1 Berg, R. and H. Schuettelkopf. Die Messung der Verteilung in
und der Abgabe von I-129 aus der Wiederaufarbeit-ungsanlage
Karlsruhe. p.81 in Radioactive Effluents from Nuclear Fuel
Reprocessing Plants. CEC document V/2266/78 (1978).
B2 Beninson, D., A.M. Migliori de Beninson, and C. Menossi.
Fallout radioactivo debido a las explosiones en el Pacífico sur
en el período 1966-1970. Comisíon Nacional de Energía Atómica.
Informe RS 28/49 (1971).
B3 Beninson, D., A.M. Migliori de Beninson and C. Menossi. Fallout
radioactivo debido a las explosiones en el Pacífico sur en el
período 1971-1972. Comisíon Nacional de Energia Atómica.
Informe RS 43/102 (1973).
B4 Breuer, F. and M. de Bortoli. Behaviour of radioiodine in the
environment and in man. Comitato Nazionale Energia Nucleare
report RT/PROT(73)13 (1973).
B5 Bergström, S.O.W. Transport of fallout 131-I into milk. p.
159-174 in Radiological Concentration Processes (Aberg, B.
and F.P. Hungate, eds.). Pergamon Press, 1967.
B6 Bustad, L.F., D.H. Wood, E.E. Elefson et al. 131-I in milk and
thyroid of dairy cattle following a single contamination event
and prolonged daily administration. Health Phys. 9: 1231-1234
(1963).
B7 Bouville, A. Estimation des doses dues aux rejets d'iode 129
par les installations nucléaires. Echelles locale et
régionale. p. 53-68 in I-129. Proceedings of an NEA
Specialist Meeting. OECD, Paris, 1977.
C2 Chamberlain, A.C. and R.C. Chadwick. Transport of iodine from
atmosphere to ground. United Kingdom Atomic Energy Authority
Research Group report AERE-R-4870. Harwell, Berkshire, 1965.
C3 Comisíon Nacional de Energía Atómica, Argentina. Fallout
Radioactivo debido a las explosiones en el Pacífico sur en el
período Enero-Octubre de 1973. Informe RS-47/118 (1973).
C4 Cambray, R.S. et al. Radioactive fallout in air and rain.
Reports covering the years 1955-1979. AERE-R-4094 (1962);
AERE-R-4392 (1963); AERE-R-4687 (1964); AERE-R-4997 (1965);
AERE-R-6556 (1970); AERE-R-7245 (1972); AERE-R-7832 (1974);
AERE-R-8267 (1975); AERE-R-8671 (1976); AERE-R-9018 (1978);
AERE-R-9441 (1979); AERE-R-9672 (1980).
D1 Dillman, L.T. and F.C. Von der Lage. Radionuclide decay
schemes and nuclear parameters for use in radiation-dose
estimation. NM/MIRD Pamphlet No.10. Society of Nuclear
Medicine, September 1975.
D2 Dolphin, G.W. Dietary intakes of iodine and thyroid dosimetry.
Health Phys. 21: 711-712 (1971).
E1 Edwards, R.R. Iodine-129: Its occurrence in nature and its
utility as a tracer. Science 137: 851-853 (1962).
E2 Eggleton, A.E.J., D.H. Atkins and L.B. Cousins. Chemical and
physical nature of fallout 131-I and carrier-free 131-I
released in air. An abstract. Health Phys. 9: 1111 (1963).
G1 Garner, R.J. and R. Scott Russell. Isotopes of iodine. Chapter
14 in Radioactivity and human diet (R. Scott Russell ed.).
Pergmon Press, 1966.
H2 Harley, N., I. Fisenne, L.D.Y.Ong et al. Fission yield product
decay. p. 251-260 in Health and Safety Laboratory Fallout
Program quarterly summary report HASL-164. New York, 1965.
H3 Hull, A.P. Critical evaluation of radiological measurements
and of the need for evacuation of the nearby public during the
Three Mile Island incident. Paper presented at the IAEA
International Conference on current nuclear power plant safety
issues. Stockholm, October 1980.
H4 Hull, A.P. Vegetation retention and vegetation-milk ratios of
fallout 131-I. Health Phys. 9: 1173-1177 (1963).
H5 Heinemann, K., M. Stoeppler, K.J. Vogt et al. Untersuchungen
zur Ablagerung und Desorption von Jod auf Vegetation.
Kernforschungsanlage Jülich report Jül-1287 (1976).
H6 Hoffman, F.O. A reassessment of the deposition velocity in the
prediction of the environment transport of radioiodine from air
to milk. Health Phys. 32: 437-441 (1977).
H7 Horbert, J., K.J. Vogt and L. Angeletti. Untersuchung zur
Ablagerung von Aerosolen auf Vegetation and anderen
Grenzflächen. Kernforschungsanlage Jülich report Jül-1288
(1976).
I1 International Atomic Energy Agency. Radioiodine removal in
nuclear facilities. Methods and techniques for normal and
emergency situations. Technical Reports Series No. 201.
Vienna, 1980.
I2 International Atomic Energy Agency. Power reactors in member
states. 1980 edition. Vienna, 1980.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP Publication 30, Part
1. Annals of the ICRP, Vol. 2, No. 3/4 (1979).
L1 Loutit, J.F., W.G. Marley and R.S. Russell. The nuclear
reactor accident at Windscale, October 1957. Environmental
aspects. Appendix H in The Hazards to Man of Nuclear and
Allied Radiations. A second report to the Medical Research
Council, London, 1960.
L2 Luykx, F. and G. Fraser. Radioactive effluents from nuclear
power stations and nuclear fuel reprocessing plants in the
European Community. Discharge data 1974-1978. Radiological
aspects. CEC document V/4116/80 (1980).
L3 Lengemann, F.W.. Radioiodine in the milk of cows and goats
after oral administration of radioiodate and radioiodide.
Health Phys. 17: 565-569 (1969).
M1 Manuel, O.K. Iodine-129. A study of its transport in the
environment and distribution in biological systems. Report C00-
2450-3 (1976).
M2 Morgan, A., D.G. Morgan and G.M. Arkell. A study of retention
and subsequent metabolism of inhaled methyl iodide in Inhaled
Particles and Vapours II (C.N. Davies, ed.). Pergamon Press,
1967.
M3 Morgan, A., D.J. Morgan and A. Black. A study of the
deposition, translocation and excretion of radioiodine inhaled
as iodine vapour. Health Phys. 15: 313-322 (1968).
P1 Pelletier, C.A., J.E. Cline. E.D. Barefoot et al. Sources of
radioiodine at pressurized water reactors. EPRI report NP-939
(1978).
P2 Pelletier, C.A., J.E. Cline, E.D. Barefoot et al. Sources of
radioiodine at boiling water reactors. EPRI report NP-495
(1978).
P3 Peirson, D.H. and J.R. Keane. The characteristics of early
fallout from the Russian nuclear explosions of 1961. Nature
196: 801-807 (1962).
P4 Perkins, R.W. Physical and chemical form of 131-I in fallout.
Health Phys: 9: 1113-1122 (1963).
P5 Palms, J.M., V.R. Veluri and F.W. Boone. The environmental
impact of iodine-129 released by a nuclear fuel reprocessing
plant. Nucl. Saf. 16: 593-602 (1975).
R1 République française. Retombées radioactives à la suite des
tirs nucléaires en Polynésie. Mai-décembre 1970.
R2 Riggs, D.S. Quantitative aspects of iodine metabolism in man.
Pharmacol. Rev. 4: 284-370 (1952).
S1 Srinivasan, B., E.C. Alexander Jr. and O.K. Manuel. Iodine-129
in terrestrial ores. Science 173: 327-328 (1971).
S2 Spiers, F.W. Radioisotopes in the human body: physical and
biological aspects. Academic Press, 1968.
T1 Turekian, K.K. The oceans, streams and atmosphere in Handbook
of Geochemistry (K.H. Wedepohl, ed.). Springer, Berlin,
Heidelberg, New York, 1969.
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York, 1977.
U2 United Nations. Ionizing Radiation: Levels and Effects.
Report of the United Nations Scientific Committee on the
Effects of Atomic Radiation to the General Assembly, with
annexes. United Nations sales publication No. E.72.IX.17 and
18. New York, 1972.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13(A:7613). New York, 1969.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14(A/6314). New York, 1966.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No.
14(A/5814). New York, 1964.
U6 United States Environmental Protection Agency. Environmental
analysis of the uranium fuel cycle. Part II. Nuclear power
reactors. U.S. Environmental Protection Agency report EPA-
520/9-73-003-C (1973).
V1 Voillequé, P.G. Iodine species in reactor effluents and in the
environment. EPRI report NP-1269 (1979).
V2 Voillequé, P.G. and J.H. Keller. Air-to-vegetation transport
of 131-I as hypoiodous acid (HOI). Health Phys. 40: 91-94
(1981).
W1 Winkleman, I. Bericht über die im Filterproben aus der
Abluftüberwashungsanlage von Kernkraftwerken in der
Bundesrepublik Deutschland in Jahre 1976 nachgewiesenen
Einzelnuklide. STH-4/77 (1977).
VII. CAESIUM-137
A. INTRODUCTION
271. Caesium is element number 55 in the periodic table. It is an
alkali metal like potassium, and it resembles potassium
metabolically. Whereas potassium is an essential element for man,
there is no evidence that caesium is also an essential trace
element. In fact, stable caesium, 133Cs, is fairly rare in the
biosphere and in geological occurrence. Average occurrence in the
earth's crust is 3 µg g-1. In specific rock types the estimated
average concentration is 1 µg g-1 in basalts and 5 µg g-1 in
granite. The K/Cs ratio in basalts is 7500 [T2]. Stable caesium
occurrence in fresh water, lakes and rivers ranges between 0.01 and
1.2 ng g-1 and is 0.5 ng g-1 in the ocean [K2]. Stable potassium
is more abundant, with usual concentrations of 0.2 to 10 µg g-1 in
fresh waters and 380 µg g-1 in the ocean [V1].
272. The radioactive isotope 137Cs is produced in nuclear fission
and is one of the more significant fission products. The fission
yield is relatively high, about 6 atoms per 100 fissions,
independent of the type of fission in uranium or plutonium (Table
VII.1). It has a radioactive half-life of 30.17 a and its beta
decay is accompanied by a gamma ray of moderate energy. Figure
VII.I shows the decay scheme and lists the primary transition
energies.
Table VII.1 Fission yields of
caesium-137 [C3]
-----------------------------
Fission yield (%)
Nuclide -----------------
Thermal Fast
-----------------------------
235U 6.21 6.12
239Pu 6.64 6.50
238U 5.93
232Th 6.73
-----------------------------
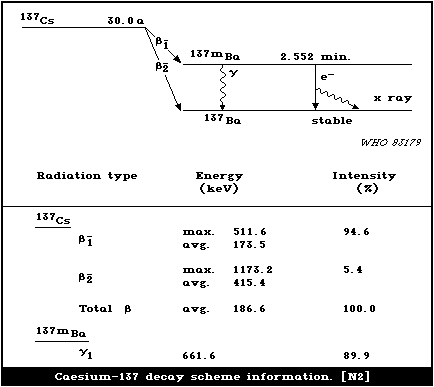 273. The chemical similarity of caesium and potassium and the
opportunity to make simultaneous measurements by gamma spectrometry
of 137Cs and naturally-occurring 40K has encouraged the expression
of 137Cs concentrations relative to the potassium concentration in
a manner analogous to that used for strontium and calcium.
However, caesium and potassium are not interdependent and do not
behave in such a regular manner in biological systems as do
strontium and calcium. As the levels of potassium in diet and man
remain roughly constant (1.4 g per litre of milk and 2 g per
kilogram of body weight), the 137Cs/K quotients can be converted
easily to 137Cs concentrations. Dietary intake of 137Cs increases
in proportion to the amount of food consumed, however the 137Cs/K
quotient in diet is relatively constant for adult and children
diets for widespread contamination situations [G4]. An additional
advantage of expressing 137Cs levels in the body in terms of the
137Cs/K quotient is that age and sex differences are minimized and
the values correlate more closely to 137Cs concentrations per unit
of lean body mass, which seems to be a more important parameter for
dosimetric purposes than the whole body mass. It is expected,
however, that assessments in the future will be presented
independently for 137Cs, without so much reliance on the stable
congener element.
274. A great deal of information has accumulated on 137Cs in the
environment, particularly the measurements of fallout 137Cs in air,
deposition, diet and man. Much of the literature has been
referenced by UNSCEAR over the years. Recent reviews of 137Cs data
have been published by Moiseev and Ramzaev [M5] and the United
States National Council on Radiation Protection and Measurements
[N1]. This document is not extensive in terms of references cited.
The representative references for most statements can be taken as
the starting points for the more extensive literature available.
B. SOURCES
1. Nuclear explosions
275. Atmospheric testing of nuclear weapons has resulted in
widespread distribution in the environment of radioactive fission
and activation products. Extensive measurements of fallout
radioactivity have been conducted. The data have been reported and
discussed in each report of UNSCEAR [U2, U3, U4, U5, U6, U7, U8].
The stratospheric inventory of 90Sr has been measured in a long-
term programme [L1]. Global networks to measure fallout deposition
have reported results for 90Sr [F1] and 137Cs [C1]. The activity
ratio of 137Cs to 90Sr in long-term deposition has been found to be
relatively constant at about 1.6 [U7], although variations for
individual samplings are encountered [S1].
276. The total amount of 90Sr produced in weapons testing through
1980, which has been globally dispersed, is estimated to be 6.0
1017 Bq [F1]. Less than 1% of this amount remained in the
stratosphere [L1]. The remainder has been deposited on the earth's
surface. This corresponds to 9.6 1017 Bq of 137Cs produced in
nuclear testing. Radioactive decay has reduced the cumulative
deposit of 137Cs to 6.9 1017 Bq [C1], 76% of which is in the
northern hemisphere and 24% in the southern hemisphere.
2. Nuclear fuel cycle
(a) Nuclear reactors
277. Caesium-137 is produced by fission in the fuel of nuclear
reactors. The amounts produced depend on the degree of fuel burn-
up achieved and to some extent on the type of fuel and the neutron
spectrum in the reactor. In fairly high burn-up fuel (33000 MW[t]d
t-1) of a pressurized water reactor, the 137Cs production is
estimated to be 3.9 1015 Bq per tonne of fuel, corresponding to 1.3
1014 Bq per MW(e)a of electricity generated [O1].
278. Small amounts of fission products produced in the fuel in
nuclear reactors may reach the coolant through defects in the fuel
cladding. In coolant purification or following coolant leakage,
these fission products may reach gaseous and liquid effluent
streams. In controlled amounts, some of the effluents are released
to the environment.
279. Reported amounts of 137Cs released to the environment from
reactors have been summarized by UNSCEAR [U8]. The averaged
release rates for some reactor types are included in Table VII.2.
Table VII.2 Estimated global discharges of 137Cs
from nuclear power stations in 1980
--------------------------------------------------------------
Reactor Reactor Capacity Release rate Estimated
type number [MW(e)a] [Bq per MW(e)a] discharge (Bq)
--------------------------------------------------------------
PWR 96 64239 6 107 2 1012
BWR 62 35170 9 108 19 1012
GCR 36 7086 2 109 9 1012
--------------------------------------------------------------
Other 47 18490 9 108 10 1012
--------------------------------------------------------------
Total 241 124985 4 1013
--------------------------------------------------------------
These are not necessarily the typical situation for a particular
reactor. During a specific year it is normally the case that a
somewhat larger release occurs from a single reactor with
insignificant releases reported from all other sites. This means
that there is a large range in release distribution, covering three
or four orders of magnitude. For all reactor types the release of
137Cs is primarily in liquid effluents; negligible amounts, in
comparison, occur in airborne effluents.
280. Assuming the averaged normalized release rates to be
representative for the reactor groups, it is possible to obtain a
very rough estimate of the total amount of 137Cs released from
reactors worldwide. Using the installed capacities of the various
reactor types as of 1980 [I4] and assuming a reactor utilization of
60%, the estimated annual release from all reactors is about 4 1013
Bq. In this calculation, it is assumed for the reactor types for
which no data are available, that the releases are similar to those
from BWRs.
(b) Fuel reprocessing plants
281. In fuel reprocessing plants the fuel is dissolved to recover
uranium and plutonium for re-use. All of the 137Cs and other
fission products as well go to the waste streams. The radionuclide
activities in airborne and liquid effluents from fueld reprocessing
plants have been recorded by UNSCEAR [U8].
282. Caesium-137 is released from fuel reprocessing plants
primarily in liquid effluents. Averaged release rates during the
years 1971-1972 were 0.6, 90 and 520 109 Bq/MW(e)a from the Nuclear
Fuel Services plant (U.S.A.) (no longer in operation), La Hague
(France) and Windscale (U.K.), respectively [U8]. The release of
caesium-137 in liquid effluents is small relative to the amount in
spent fuel. The fractional liquid releases from the Windscale
plant were approximately 4 10-3. The only data for 137Cs in
airborne effluents is for the Nuclear Fuel Services plant, which
corresponded to 4 104 Bq/MW(e)a [U8], several orders of magnitude
less than in liquid effluents.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Fixation in soil
283. Caesium is generally rather strongly fixed in soil. Downward
migration and availability to plants is thereby reduced. In
mineral soils the movement of 137Cs is appreciably less than that
of 90Sr. Three to four years after deposition on the soil surface,
the median depth to which it has penetrated is usually less than 2
cm [F5]. Its mobility may be somewhat greater in organic soils.
Much smaller amounts of 137Cs than of 90Sr are leached out of the
soil to enter rivers and lakes.
284. There are exceptional areas, however, where caesium fixation
in soil is much less, allowing enhanced transfer of caesium to
plants. Marei et al. [M2] identified regions in the USSR where the
soil is wet, peaty and podzolic, from which transfer of 137Cs into
the food chain is 10 times higher than for other areas. Other
regions of the world where the soils give rise to high 137Cs
transfer into diet have also been identified, for example, in the
Faroe Islands, New Zealand and Sweden [U8].
285. It has been reported that in clay minerals the important
factor in fixation of caesium is the ability of certain layered
silicates such as micas, vermiculites and illites to adsorb or fix
trace quantities of caesium [T1]. Caesium ions are trapped in the
interlayer regions of vermiculite or at the frayed edges of illites
and micas. Caesium is thus more strongly retained in soils
containing predominantly micaceous minerals. Soils which do not
contain large quantities of micaceous minerals, such as tropical
soils, peat soils, and podzolic soils, exhibit less retention and
allow greater uptake of caesium by plants.
286. Fixation of caesium by sediments in aquatic environments
occurs in a similar fashion to fixation in soil. The preferential
adsorption of 137Cs to the micaceous component of sediments has
been demonstrated under environmental conditions.
2. Transfer to plants
287. Caesium-137 may be transferred to plants by direct deposition
onto plant surfaces or by root uptake from accumulated deposits in
soil. In general, direct foliar absorption is the predominant mode
of plant contamination when the deposition rate is relatively high.
Root uptake is low except in those cases mentioned above, when soil
conditions allow low fixation of caesium.
288. Caesium depositing on plant surfaces is retained to the same
extent as other particulate debris. A removal half-time of l4 days
due to weathering is generally assumed. Once absorbed by the
plant, caesium is readily redistributed through the plant.
Relationships between air concentrations and subsequent
concentrations of 137Cs in pasture plants have been discussed by
Hawthorne et al. [H4] and Pelletier and Voilleque [P2].
289. Caesium may enter plants by plant base absorption before
becoming fixed in soil. Thus, retention of 137Cs in the root mat of
pastures may allow 137Cs to be relatively more available to plants
for a period of a year or more [U3]. A high level of organic
matter in soil can enhance the absorption of caesium by plants
[B1]. Sorption of organic molecules on clay surfaces prevents the
retention of caesium and also of potassium by these minerals.
Thus, for permanent pastures in temperate regions, the frequent
high organic matter content of the upper soil layer allows shallow
rooted grass to absorb 137Cs relatively more freely for a somewhat
more extended period following deposition. Mushrooms have been
shown to concentrate very effectively 137Cs from soil [G2, M1],
which may be associated with the highly organic areas of growing.
290. Uptake to plants of 137Cs from soil low in available
potassium may be somewhat increased. The addition of potassium may
decrease absorption of 137Cs in this case; however, this has no
effect when the available potassium is high [N5, F4]. Root uptake
of caesium is in general included in the range 0.01 to 1, which is
the ratio of caesium concentrations in the dry plant material to
that in dry soil [M3].
3. Transfer to milk
291. The fractional amount of 137Cs transferred into milk is
slightly greater than that of potassium. It has been shown that
some 10% of orally ingested 137Cs is secreted into the milk of
dairy cows, corresponding to 1.3 to 1.5% of the amount ingested per
litre of milk [G1, I1, L2]. The transfer of fallout 137Cs in field
conditions has been found to be somewhat less, ranging from 0.25 to
0.86% of intake per litre of milk [P2, M1, S4].
4. Transfer to meat
292. A correlation between 137Cs concentration in beef and in milk
has been noted, the ratio of concentrations in meat (Bq/kg) and in
milk (Bq/1) averaging about 4 for production in the same locality
[L5, E1, J2]. This would correspond to a transfer of about 4% of
the daily intake per kilogram of meat [F5]. Equilibrium conditions
are reached in about 30 days in the cow [F5]. It cannot generally
be expected that there will be a useful relationship between 137Cs
in meat and milk, since animals produced for the two purposes are
frequently provided with different diets and are reared in
different areas.
5. Transfer to diet
293. For general contamination situations, as for global fallout
radioactivity, the main contributions to dietary intake of 137Cs
are generally from grain products, meat and milk. Fruit and
vegetables contribute much smaller amounts of 137Cs. This has been
the pattern for western diets, such as Denmark [A1] and the United
States [G4]. In Japan the main contribution has been from cereals
[U1]. For all foods the transfer from a specific deposited amount
seems to be rapid, being essentially completed within the first two
years after deposition.
294. Marine food chains are of secondary importance in
contributing to dietary intake of fallout 137Cs, even in countries
where fish is widely consumed. In Japan between 1966 and 1971,
only about 8% of the 137Cs in diet came from the consumption of
fish products [U1]. As the contributions from other foods
decrease, however, the relative contribution from fish can
increase. During 1976 in Denmark, 15% of the 137Cs intake was
attributed to fish [A2]. It is possible that individuals consuming
large amounts of freshwater fish may acquire 137Cs burdens several
times greater than individuals eating more diversified diets [G3].
295. The transfer of 137Cs from deposition to diet has been
studied quantitatively using the following transfer function
between deposition and diet components in regression analyses of
reported data [U8]:
j j j infinite
Cj(n) = b1 f(n) + b2 f(n-1) + b3 sigma f(n-m) e-µm
m=1
where Cj(n) is the concentration of 137Cs in the diet component j
during the year (n) in mBq/gK; f(n) is the deposition density of
137Cs in the current year (n) and in previous years (n-1) in Bq/m2;
bjn are the proportionality factors, and µ is the decay constant
accounting for radioactive decay and reduced availability of
deposition in all previous years. The first term of the equations
is the rate term giving the contribution to dietary level from the
current year's deposition amount. The second term is the long term
contribution expressing separately the contribution from the
previous year's deposition including storage of foods by market
practices. The third term gives the contribution from the
cumulative deposit of 137Cs in soil.
296. The quotient of the time-integrated concentration of 137Cs in
diet and the integrated deposition density defines the transfer
factor P23. The integrals are replaced by summations if, as is
usually the case, the relevant quantities are assessed over
discrete intervals of time, such as annual averages
infinite
sigma C(n)
n=1
P23 = -----------
infinite
sigma f(n)
n=1
The transfer factor is usually expressed in mBq a/gK per Bq/m2. It
may be evaluated for total diet or for dietary components.
297. Using the model described above, the evaluation of the
transfer factor reduces to
infinite -µm e-µ
P23 = b1 + b2 + b3 sigma e = b1 + b2 + b3 -----
m=1 1-e-µ
The parameters of the transfer function, obtained by regression
fits of the 137Cs/K quotients in milk, dietary components and total
diet from several countries have been reported by UNSCEAR [U8].
Some of these results are given in Table VII.3.
298. The lowest values for the transfer factor for milk are
obtained for the U.S.A., Denmark and the U.K. Most of the transfer
of 137Cs deposition to milk is from direct deposition. Less than
15% of the transfer is from uptake of 137Cs from soil.
Intermediate values of P23milk are obtained from the USSR and
Argentina. Transfer from direct deposition is increased and uptake
from soil is more significant. The highest values of P23milk are
obtained for New Zealand, Australia, Norway and the Faroe Islands.
A high value has also been reported for Finland, which is 15.8 mBq
a (gK)-1 per Bq m-2 [C2]. These results can be explained by
efficient transfer of direct deposition and soil conditions which
allow only low fixation of 137Cs.
299. The range of values of transfer factors for milk, 3.4 to 27.5
mBq a (gK)-1 per Bq m-2, indicates that it is not easy to specify a
typical transfer situation. To estimate the 137Cs transfer to milk
in a particular region requires some indication of soil and pasture
conditions.
300. Using the comprehensive data from Denmark of 137Cs in diet
[A2], values of the parameters of the transfer function of various
components of diet and for the total diet have been determined.
For most foods, the major contribution to the value of the transfer
factor comes from direct deposition. Only a small transfer is
attributed to uptake from soil. The values of the parameters for
the fit to total diet data are: b1 = 1.6, b2 = 2.2, b3 = 0.04 mBq
a(gK)-1 per Bq m-2 µ = 0.11 a-1 and P23 = 4.1 mBq a(gK)-1 per
Bq m-2. Short-term transfer to diet is thus estimated to comprise
93% of the total transfer, (b1 + b2)/P23.
301. The general pattern of 137Cs transfer to diet can be expected
to be similar to that for Denmark, although increased transfer can
occur in specific areas, depending on soil conditions or particular
consumption habits. UNSCEAR has used a rounded value of P23 of 4
mBq a(gK)-1 per Bq m-2 to assess the dose commitments from 137Cs
for the world population [U8].
Table VII.3 Parameters of the transfer functions between deposition density and
137Cs/K in milk
-----------------------------------------------------------------------------------------
Parameter U.S.A. Denmark United USSR Argentina New Australia Norway Faroe
a/ Kingdom Zealand Islands
1973 1976 1977 1972 1976 1977 1973 1976 1976
-----------------------------------------------------------------------------------------
b1 2.3 2.2 1.7 4.4 3.2 6.8 7.3 3.5 6.8
b2 1.3 1.2 1.6 0.2 0.0 0.9 4.8 2.4 5.5
b3 0.0 0.01 0.04 0.3 1.7 3.1 0.3 2.5 2.8
µ 0.05 0.03 0.07 0.2 0.3 0.5 0.2 0.2 0.2
P23 3.4 3.7 3.9 6.2 7.5 13.0 13.4 15.6 27.5
-----------------------------------------------------------------------------------------
a/ The unit for parameters b1, b2, b3 is mBq a(gK)-1 per Bq m-2.
The unit for parameter µ is a-1.
The unit for the transfer factor P23 is mBq a(gK)-1 per Bq m-2.
6. The lichen-caribou-man foodchain
302. A terrestrial situation which allows much greater than usual
transfer of caesium to man is the lichen-caribou-man foodchain.
Caesium depositing on lichens is retained quite effectively. There
is a very slow decrease in activity with time, approximately 5 to
10% of the 137Cs being eliminated annually [U5]. Lichens provide
the food base for grazing caribou and reindeer during the winter.
A proportionality factor of 137Cs in lichen to that in reindeer
meat in northern Sweden during the winter averaged 4.9 ± 0.4 [L4].
In summer also grass and herbaceous plants are consumed by the
animals, and therefore the 137Cs levels in meat show marked
seasonal variation.
303. High concentrations of 137Cs arise in Lapp and Eskimo
populations who eat the caribou and reindeer meat. Levels of 740-
1300 Bq per kg of body weight were observed in individuals during
1964 from fallout [H1, M4], about a factor of 100 greater than
burdens of individuals from temperate northern hemisphere regions.
Other fallout radionuclides, such as 54Mn and 55Fe, and the natural
isotopes 210Pb-210Po are also concentrated along the lichen-
caribou-man foodchain.
7. Aquatic behaviour
304. Caesium in the aquatic environment is strongly adsorbed by
suspended particulate materials, especially clays. Therefore, the
amount of caesium in the soluble phase decreases with increasing
suspended solid concentrations. Potassium is also sorbed, but to a
much less degree.
305. Caesium levels in fish are inversely related to the potassium
content of the water [K1]. Because of the high concentration of
potassium in the ocean, the transfer of 137Cs to fish is of primary
concern in the freshwater environment. The activity of fresh water
fish may be 100 times that of ocean fish, given the same caesium
concentrations in water.
306. The low mineral content of fresh water also enhances the
absorption of 137Cs by aquatic plants. Aquatic plants from fresh
water areas, which are sometimes important in cattle feed, may have
increased levels of 137Cs compared to 137Cs which may have
deposited on nearby pasture ground.
307. Caesium in aquatic animals is accumulated primarily from the
food chain. Absorption efficiency of potassium and caesium from
food is high. In animals the excretion rate of potassium is about
3 times larger than that of caesium. As a result, the caesium
concentration per unit amount of potassium in tissues increases by
a factor of about 3 with each trophic level [P1].
308. The food web also accumulates caesium from suspended and
bottom sediments. Filter feeders may accumulate caesium adsorbed
to particulate matter. Benthic invertebrates obtain caesium
absorbed to ingested bottom sediments. Fish ingest those
invertebrates and also some sediment particles along with the prey.
Absorption efficiency in the fish depends on the caesium fixation
ability of the sediment minerals.
309. The concentration factors (ratio of concentration in organism
to that in the water) for caesium must be related to the potassium
concentration in the water and to the turbidity. From a literature
review of values for fresh water systems [V1], suggested values of
the concentration factors are 1000 for algae and plants, molluscs
and invertebrates in all waters and 5000/Kw and 1500/Kw for non-
piscivorous and piscivorous fish, respectively, in clear waters,
where Kw is the stable potassium concentration of water in µg/g.
The factors for fish are a factor of 5 less in turbid waters (> 50
µg/g suspended solids). The concentration factors for caesium in
the ocean are 10 for algae and molluscs, 30 for fish and 50 for
molluscs [F6].
D. TRANSFER TO MAN
1. Absorption and distribution in tissues
310. As a general rule, caesium compounds are soluble in body
fluids. Intestinal absorption is complete (100%) under
experimental conditions [R2, S3], but from normal diets is probably
less efficient, ranging from 50 to 80% [F5]. In man caesium is
secreted into the gastrointestinal tract, between the stomach and
small intestine, and is readily reabsorbed [I2]. One basis for
therapeutic treatment in internal contamination cases is to
administer solutions of Prussian blue, which binds with caesium in
the gastrointestinal tract preventing reabsorption [I2, D1].
311. Caesium migrates rapidly into cells of the body following
intake and becomes relatively uniformly distributed in soft tissues
[R2, L3, R4]. The metabolism in mothers and infants has also been
studied. There seems to be no placental discrimination, as the
newborn has 137Cs concentrations about equal to that of the mother
[B3].
312. Concentrations of caesium and potassium are low in fat
tissues. Therefore, for equal 137Cs concentrations in intake, the
concentrations of 137Cs (Bq per kg body weight) in males are higher
than in females, due to the higher average proportion of fat tissue
in the female body. However, a difference is also expected due to
longer retention time of caesium in males. Expressed in Bq 137Cs
per gK, the difference between males and females is somewhat
reduced.
313. It has been inconclusive for some time whether 137Cs
concentrates in bone. One study reported that the concentration of
137Cs in rib bones, which were free of muscle but not of marrow,
was comparable to the concentration in soft tissues [Y2]. The
results were variable, and subsequent studies both did and did not
confirm these results [A3, H3, N4]. From the results of a recent
study it appears that caesium associated with bone is present in
the marrow portion with only slight uptake by the hard tissue [H2].
314. A slower turnover of caesium in bone could allow
concentrations in bone to lag behind those in tissue, causing
higher relative concentrations in bone during periods of decreasing
intake. A longer retention half-time would eventually be noted in
whole-body measurements. However, such a component has not been
identified in over 1000 days of measurement following an acute
intake case [R4].
2. Retention half-time
315. A great many investigations of the biological half-time in
man have been conducted. Many of the references are collected in
the discussion by Lloyd [L6]. The half-time in man varies
considerably, depending on age and other factors. The half-time is
less in women than in men, and the half-time in children and
infants is less than in adults. Pregnant women have shorter
caesium half-times than in their non-pregnant conditions. Table
VII.4 shows the summary of 137Cs retention half-time reported
recently by the NCRP [N1], using the data of Lloyd et al. [L7] and
Zundel et al. [Zl].
Table VII.4 Retention half-time of 137Cs in the
human body [N1]
--------------------------------------------------
Subjects Number Age Half-time (d)
--------------------------------------------------
Men 26 23-55 a 105 ± 25
Women 15 20-51 a 84 ± 20
Pregnant women 24 16-39 a 49 ± 16
Children 7 5-17 a 57 ± 20
Infants 5 17-143 d 19 ± 8
--------------------------------------------------
316. The biological half-time for caesium in man can be considered
a function of age for juveniles and of sex for adults, but it is
not determined by body mass [L6]. Half-time and body mass may,
however, be dependent on some other common factors. The rate of
caesium turnover may be under hormonal influence or control or may
reflect the general metabolic rate [L6].
317. Shorter half-time components of 137Cs in man have been
reported, including one of only 2 to 3 hours [N3]. In general, two
components of the 137Cs half-time in man have been established: a
small fraction (10 to 15%) excreted with a short half-time (1 to
1.5 days) and the remainder excreted more slowly (50 to 150 days)
[R1, R3]. The ICRP suggests representative values of retention of
10% with a half-time of 2 days and 90% with a half-time of 110
days. Integral retention is 143 Bq d per Bq intake, contributed
almost entirely by the long-term component.
3. Transfer factor
318. The value of the transfer factor P34 relating concentrations
of 137Cs in diet and man can be derived from the integral retention
by dividing by the potassium content of the body (140 g) and
multiplying by the daily potassium intake (3.3 g d-1) [I5]. The
result is 3.4 Bq a (gK)-1 in man per Bq a (gK)-1 in diet.
319. The relatively short biological half-time of caesium in the
body makes it possible to assess the transfer factor P34 from the
measured 137Cs/K quotients in diet and man integrated over a few
years. Using this procedure, an average value of 3 Bq a (gK)-1 per
Bq a (gK)-1 diet is derived [U6, U7, U8].
E. DOSIMETRY
1. Dose per unit intake
320. The dose from 137Cs in tissue is due to the beta particles
from 137Cs decay and to the photon, x-rays, and conversion and
Auger electrons from decay of the daughter, 137mBa. A portion of
the photon energy will escape from the body, depending on the body
size. Calculations have been performed for uniform distributions
of 137Cs in the body for a range of proportions and masses
corresponding to infants, children and adults [N1, F1].
321. The average dose rate within the body for a uniform 137Cs
concentration of one Bq per kg body weight is about 3.5 nGy/d from
beta particles in both adults and infants plus 3.2 nGy/d from
photons in the adult and about 1.6 nGy/d in infants. The totals
are 6.7 nGy/d per Bq/kg in the adult and 5.1 nGy/d per Bq/kg in the
infant.
322. For 140 gK in the 70 kg adult body, the dose rate
corresponding to 1 Bq 137Cs per gK would be
µGy d-1 140 gK 365 d µGy/a
0.0067 -------- ------ ----- = 4.9 -------
Bq(kg)-1 70 kg a Bq/gK
Spiers [S2] also obtains this result and estimates the
corresponding dose rate in a child weighing 8 kg of
µGy/a
4.1 -------
Bq/gK
323. UNSCEAR [U6, U7, U8] assessed the transfer factor between
tissue and dose, P45, as 4.9 µGy per Bq a (gK)-1. The value is
nearly independent of age, being only slightly less for the child.
For a single uptake of 137Cs, the integral retention per unit
intake is 143 Bq d per Bq intake (paragraph 317). The average
absorbed dose in the body per unit intake is thus
µGy/d Bq d 1
0.0067 ----- 143 ----- ----- = 1.4 10-8 Gy per Bq intake.
Bq/kg Bq 70 kg
324. This result applies to intake by ingestion. For inhalation
the value is less by a factor of 0.63 (for particles of 1 µm size)
due to fractional deposition in the lungs of inhaled amounts.
Following inhalation there is only a short retention in the lung
(half-time = 0.5 d) for the soluble caesium compounds and a small
dose primarily from the beta particles before the 137Cs becomes
distributed throughout the body.
2. Dose per unit release
(a) Nuclear explosions
325. The dose commitment via the ingestion pathway from 137Cs
released by nuclear explosions can be assessed from the sequential
product of transfer factors
Dc = P23 P34 P45 F
where F is the integrated deposition density. The values of the
transfer factors as derived above are: P23 = 4 10-3 Bq a (gK)-1
per Bq m-2, P34 = 3 Bq a (gK)-1 per Bq a (gK)-1, and P45 = 4.9 10-6
Gy per Bq a (gK)-1. The dose commitment from 137Cs ingestion per
unit widespread deposition density, such as from nuclear
explosions, is thus 6 10-8 Gy per Bq m-2.
326. The total amount of 137Cs released to the environment by
nuclear tests, 9.6 1017 Bq, has given a population-weighted
integrated deposition density of 3100 Bq m-2 in the world as a
whole [U8]. The world population is 4 109. With these values, the
collective dose commitment per unit activity of 137Cs released is
estimated to be 8 10-13 man Gy per Bq (ingestion).
327. The dose commitment via the inhalation pathway is determined
from the integrated concentration of 137Cs in air, which is
estimated from the integrated deposition density (Bq m-2) divided
by the deposition velocity (m s-1). As for 90Sr, the average
deposition velocity can be taken to be 2 cm s-1. Thus, 1 Bq m-2
integrated deposition density corresponds to 5.8 10-4 Bq d m-3 in
air. With the above estimates of integrated deposition density,
total amount of 137Cs released by nuclear tests, world population,
breathing rate (22 m3 d-1) and dose per unit intake (8.8 10-9 Gy
Bq-1), the collective dose commitment per unit activity of 137Cs
released is estimated to be 1 10-15 man Gy per Bq (inhalation).
328. For the external exposure pathway it is assumed that the
deposited 137Cs becomes exponentially distributed in soil with a
mean depth of 3 cm. This gives a dose rate in air of 8.9 109 Gy
per Bq m-2 [B2]. The mean life of 137Cs in soil is 43.5 years,
determined by its radioactive decay. The dose to air from the
integrated deposition density is, thus, 3.9 10-7 Gy per Bq m-2.
The dose to tissue is determined by a factor of 0.8 to account for
the change of material (air to tissue) and back-scatter and
shielding afforded by other tissues of the body and a factor of 0.4
to account for building shielding and time spent indoors [U8]. The
combined factor is 0.8 x 0.4 = 0.32. The transfer factor P25
relating integrated deposition of 137Cs to tissue dose is thus 3.9
10-7 x 0.32 = 1.2 10-7 Gy per Bq m-2. Using the above values of
integrated deposition density, total amount of 137Cs released, and
world population, the collective dose commitment per unit activity
of 137Cs released is estimated to be 1.6 10-12 Gy per Bq (external
exposure).
(b) Nuclear installations
329. The contribution of the inhalation pathway to the collective
dose commitment for effluent releases can be estimated from the
integrated concentration of 137Cs in air. This can be derived
from a dispersion formula or from an estimate of the deposition
velocity. In the latter case, the integrated concentration in air
is the total amount of 137Cs released per unit area of the
deposition region divided by the deposition velocity, vd. The
population affected is the population density deltaN times the area
of the deposition region. The areal dependence is removed by the
product of these quantities. The collective dose commitment per
unit activity released is given by the expression
c
S1 = I deltaN phi/vd
where I is the individual intake rate of air and phi is the dose
per unit activity of 137Cs inhaled.
330. Using a deposition velocity of 0.5 cm s-1 for near surface
releases, a population density of 25 man km-2, an air intake rate
of 22 m3/d and the dosimetric factor of 8.8 10-9 Gy per Bq intake,
the collective dose commitment for the inhalation pathway per unit
activity released is estimated to be 1 10-14 man Gy/Bq.
331. The contribution of the ingestion pathway from airborne
effluents to the collective dose commitment per unit activity
released, Sc1, can be assessed by the expression
c
S1 = P23 P34 P45 deltaN
Using the values for the transfer factors given previously, and
assuming a constant population density of 25 man km-2 in the region
of deposition, the collective dose commitment for the ingestion
pathway per unit activity released is estimated to be 2 10-12 man
Gy/Bq. This value assumes that food is locally produced and that
the production suffices for the population density under
consideration. It also applies for the case of 137Cs becoming
relatively rapidly fixed in soil (P23 = 4 mBq a [gK]-1 per Bq m-2).
For other types of soil conditions or special consumption patterns
and also for other population densities, the estimate should be
adjusted accordingly.
332. For the external exposure pathway the transfer factor P25
relating integrated deposition of 137Cs in soil to the tissue dose
has been assessed above with regard to nuclear explosions to be 1.2
10-7 Gy per Bq m-2. This value is of general applicability.
Similarly to the ingestion pathway, the collective dose commitment
per unit activity released, Sc1, can be assessed by the expression
c
S1 = P25 deltaN
Assuming a population density of 25 man km-2, the collective dose
commitment from external exposure per unit activity released is
estimated to be 3 10-12 man Gy/Bq.
333. For aquatic ingestion pathways from the input of 137Cs to
water bodies the collective dose commitment, normalized per unit
activity released, can be estimated [U8], using the expression
c ksigma Nk Ik fk phi
S1 = -----------------------
V(lambda + 1/tau)
where V is the volume of the receiving waters, tau is the turnover
time of receiving waters, lambda is the decay constant of 137Cs, Nk
is the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
334. The quantity V(lambda+1/tau) is the infinite time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentrations in water and in fish
will be high, but the population which can be served with drinking
water or by fish consumption will be limited. For inputs into
larger volumes of water, the concentrations will be smaller, but
the populations involved will be correspondingly larger. It is
reasonable, therefore, to assume as a first approximation that the
quantities Nk/V are relatively constant, independent of V. The
values for these quantities as well as values for the other
parameters of the above expression have been extensively discussed
by UNSCEAR [U8].
335. A listing of the values used in the assessments presented by
UNSCEAR is given below [U8]:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 0.3 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.2
fish 400 30
shellfish 30
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
(c) Summary
336. Table VII.5 summarizes the values obtained above for the
collective dose commitments per unit of 137Cs activity released in
airborne and liquid effluents. These are also the values of the
collective effective dose equivalent commitments with Sv replacing
Gy, since the quality factor is one and the dose is assumed to be
uniform in all tissues. The largest collective dose commitments
result from airborne discharges due to the external exposure and
ingestion pathways. These estimates are for a generalized release
situation, and substantial variations could be expected in site-
specific cases.
Table VII.5 Summary of collective dose
commitments per unit 137Cs activity
released [10-14 man Gy per Bq]
------------------------------------------
All tissues
------------------------------------------
Nuclear explosions
External exposure 160
Ingestion 80
Inhalation 0.1
Nuclear installations
Release to air a/
External exposure 300
Ingestion 200
Inhalation 1
Release to fresh water
Fish 50
Drinking water 10
Release to salt water
Fish 0.08
Shellfish 0.02
------------------------------------------
a/ Assumes population density of 25
man km-2.
F. REFERENCES
A1 Aarkrog, A. Prediction models for strontium-90 and caesium-137
levels in the human food chain. Health Phys. 20: 297 (1971).
A2 Aarkrog, A. and J. Lippert. Environmental radioactivity in
Denmark in 1976. Riso National Laboratory report No. 361
(1977).
A3 Anderson, R.W. and P.F. Gustafson. Concentration of caesium-
137 in human rib bone. Science 137: 668 (1962).
B1 Barber, D.A. Influence of soil organic matter on the entry of
caesium-137 into plants. Nature 204: 1326-1327 (1964).
B2 Beck, H.L. and G. de Planque. The radiation field in air due
to distributed gamma-ray sources in the ground. U.S. Atomic
Energy Commission report HASL-195. New York, 1968.
B3 Bengtsson, L.G., Y. Naversten and K.G. Svensson. Maternal and
infantile metabolism of caesium. p. 21-32 in Assessment of
Radioactivity in Man. Proceedings of a symposium, Heidelberg,
May 1964. International Atomic Energy Agency publication
STI/PUB/Vol. 2. Vienna, 1964.
C1 Cambray, R.S., E.M.R. Fisher, K. Playford et al. Radioactive
fallout in air and rain: results to the end of 1977. United
Kingdom Atomic Energy Authority report AERE-R9016 (1978).
C2 Castrén, O. UNSCEAR transfer coefficients P-23 (milk) and P-
234 for 90-Sr and 137-Cs in Finland. p. 73-76 in Studies on
Environmental Radioactivity in Finland 1971-1975. Institute of
Radiation Protection report STL-A21. Helsinki, 1977.
C3 Crouch, E.A.C. Fission product yields from neutron induced
fission. Atomic Data and Nuclear Data Tables, Vol. 19, No. 5
Academic Press, New York, 1977.
D1 Ducousso, R., A. Causse and C. Pasquier. Comparative effects
of acetazalamide and Prussian blue on 137-Cs retention in the
rat. Health Phys. 28: 75-78 (1975).
E1 Ellis, F.B. and B.T. Barnes. Relationship between the
concentration of caesium-137 in beef and milk. p. 77-78 in
Agricultural Research Council Radiobiological Laboratory annual
report ARCRL-14 (1965).
F1 Feely, H.W. World-wide deposition of strontium-90 through
1977. p. 1.19-1.41 in Environmental Measurements Laboratory
environmental quarterly report EML-344. New York, 1978.
F4 Fredriksson, L., B. Eriksson and B. Rasmunson. Plant uptake of
90-Sr and 137-Cs from soils, Vol. 18. p. 449-470 in
Proceedings of the Second International Conference on Peaceful
Uses of Atomic Energy, Geneva, 1958.
F5 Fredriksson, L., R.J. Garner and R.S. Russell. Caesium-137. p.
317-352 in Radioactivity and Human Diet (R.S. Russell, ed.).
Pergamon Press, 1966.
F6 Freke, A.M. A model for the approximate calculation of safe
rates of discharge of radioactive wastes into marine
environments. Health Phys. 13: 743-758 (1967).
G1 Gragle, R.G. Uptake and excretion of caesium-134 and
potassium-42 in lactating dairy cows. J. Dairy Sci. 44: 352-
357 (1961).
G2 Grueter, H. Radioactive fission product 137-Cs in mushrooms in
West Germany during 1963-1970. Health Phys. 20: 655-656
(1971).
G3 Gustafson, P.F. Comments on radionuclides in aquatic
ecosystems. p. 853-858 in Radioecological Concentration
Processes (B. Aberg and F. Hungate, eds.). Pergamon Press,
1967.
G4 Gustafson, P.F. and J.E. Miller. The significance of 137-Cs in
man and his diet. Health Phys. 16: 167-183 (1969).
H1 Hanson, W.C. Radioecological concentration processes
characterizing arctic ecosystem. p. 183-191 in Radioecological
Concentration Processes (B. Aberg and F. Hungate, eds.).
Pergamon Press, 1967.
H2 Hardy, E.P. On 137-Cs and 90-Sr in bone. p. I.64-I.69 in
Health and Safety Laboratory Fallout Program Quarterly Summary
report HASL-278. New York, 1974.
H3 Harrison, G.E., A. Sutton, K.H. Edwards et al. Concentrations
of radioactive and stable caesium in bone and soft tissues.
Br. J. Radiol. 36: 745-748 (1963).
H4 Hawthorne, H.A., S.D. Zellner, L.L. Eberhardt et al. 137-
Caesium cycling in a Utah dairy farm. Health Phys. 30: 447-464
(1976).
I1 Ilin, D.I. and Y.I. Koskalev. On the metabolism of caesium,
strontium and a mixture of beta-emitters in cows. J. Nucl.
Energ. 5: 413-420 (1957).
I2 International Atomic Energy Agency. Manual on early medical
treatment of possible radiation injury. International Atomic
Energy Agency publication Safety Series No. 47 and
IAEA/STI/PUB/506. Vienna, 1978.
I4 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
I5 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP publication 23.
Pergamon Press, 1975.
J2 Johnson, J., G.M. Ward and A.H. Dal. Comparisons of 137-Cs
levels in feed and meat of cattle fed on pasture and in the dry
lot. Health Phys. 10: 612 (1964) (Abstract).
K1 Kohehmainen, S., E. Häsänen and J.K. Miettinen. 137-Cs in
fish, plankton and plants in Finnish lakes during 1964-1965. p.
913-919 in Radioecological Concentration Processes (B. Aberg
and F.P. Hungate, eds.). Pergamon Press, 1967.
K2 Krumholz, L., E. Goldberg and H. Boroughs. Ecological factors
involved in the uptake, accumulation and loss of radionuclides
by aquatic organisms in The Effects of Atomic Radiation on
Oceanography and Fisheries. National Academy of Sciences,
National Research Council Publication 551. Washington, 1957.
L1 Leifer, R. and L. Toonkel. Updating stratospheric inventories
to April 1977. p. I.3-I.14 in Environmental Measurements
Laboratory environmental quarterly report EML-334. New York,
1978.
L2 Lengemann, F.W. and R.A. Wentworth. The transfer coefficient
of 137-Cs into cows milk as related to the level of milk
production. Health Phys. 34: 720-722 (1978).
L3 Lidén, K. The metabolism of caesium. p. 33 in Assessment of
Radioactivity in Man. Proceedings of a symposium, Heidelberg,
May 1964. International Atomic Energy Agency publication
STI/PUB/84/Vol. 2. Vienna, 1964.
L4 Lidén, K. and M. Gustafsson. Relationship and seasonal
variation of 137-Cs in lichen, reindeer and man in northern
Sweden, 1961 to 1965. University of Helsinki, Department of
Radiochemistry annual report 1965. Helsinki, 1966.
L5 Lindell, B. and A. Magi. The occurrence of 137-Cs in Swedish
food, especially dairy milk and in the human body after the
nuclear test explosions in 1961 and 1962. Ark. Fys. 29: 69-96
(1965).
L6 Lloyd, R.D. Caesium-137 half-times in humans. Health Phys.
25: 605 (1973).
L7 Lloyd, R.D., W.S. Zundel, C.W. Mays et al. Short caesium
half-times in patients with muscular dystrophy. Nature 220:
1029-1031 (1968).
M1 Marei, A.N., R.M. Barkhudarov, N.J. Novikova et al. Effect of
natural factors on caesium-137 accumulation in the bodies of
residents in some geographical regions. Health Phys. 22: 9-15
(1972).
M2 Marei, A.N., R.M. Barkhudarov and N.J. Novikova. Radiation
safety aspects of the geochemical areas with increased transfer
of caesium-137. Manuscript No. 0020-R-4.
M3 Menzel, R.G. Soil-plant relationships of radioactive elements.
Health Phys. 11: 1325-1332 (1965).
M4 Miettinen, J.K. and E. Hasanen. 137-Cs in Finnish Lapps and
other Finns in 1962-1966. p. 221-231 in Radioecological
Concentration Processes (B. Aberga and F. Hungate, eds.).
Pergamon Press, 1967.
M5 Moiseev, A.A. and P.U. Ramzaev. Caesium-137 in the biosphere.
Atomizdat, Moscow, 1975 (in Russian).
N1 National Council on Radiation Protection and Measurements.
Caesium-137 from the environment to man: metabolism and dose.
National Council on Radiation Protection and Measurements
report No. 52. Washington D.C., 1977.
N2 National Council on Radiation Protection and Measuremmnts. A
handbook on Radioactivity measurements procedures. National
Council on Radiation Protection report No. 58. Washington,
D.C., 1978.
N3 Naversten, Y. and K. Lidén. Half-life studies of radiocaesium
in humans. p. 79-87 in Assessment of the Radioactive Body
Burdens in Man. Proceedings of a symposium, Heidelberg, May
1964. International Atomic Energy Agency publication
STI/PUB/84/Vol. 2. Vienna, 1964.
N4 Nay, U., W. Stahlhofen and A. Kaul. Distribution of caesium-
137 in samples consisting of soft tissues, bone and bone
marrow. p. 49-60 in Assessment of Radioactivity in Man.
Proceedings of a symposium, Heidelberg, May 1964.
International Atomic Energy Agency publication STI/PUB/84/Vol.2
Vienna, 1964.
N5 Nishita, H., E.M. Romney and K.H. Larson. Uptake of
radioactive fission products by crop plants. J. Agric. Food
Chem. 9: 101-106 (1961).
O1 Oak Ridge National Laboratory. Siting of fuel reprocessing
plants and waste management facilities. Oak Ridge National
Laboratory report ORNL-4451 (1970).
P1 Pendleton, R.C., C.W. Mays, R.D. Lloyd et al. A trophic level
effect on 137-Cs concentration. Health Phys. 11: 1503 (1965).
P2 Pelletier, C.A. and P.G. Voilleque. The behaviour of 137-
caesium and other fallout radionuclides on a Michigan
dairy farm. Health Phys. 21: 777-792 (1971).
R1 Richmond, C.R., J.E. Furchner and W.H. Langham. Long-term
retention of radio-caesium by man. Health Phys. 8: 201-205
(1962).
R2 Rosoff, B., S.H. Cohn and H. Spencer. Caesium-137 metabolism
in man. Radiat. Res. 19: 643-654 (1963).
R3 Rundo, J., J.I. Mason, D. Newton et al. Biological half-life
of caesium in man in acute and chronic exposure. Nature 200:
188-189 (1963).
R4 Rundo, J. The metabolism of biologically important
radionuclides. VI. A survey of the metabolism of caesium in
man. Br. J. Radiol. 37: 108 (1964).
S1 Sherill, R.D., N.G. Sumerlin, J.N. Beck et al. Variation of
the ratio of caesium-137 to strontium-90 in the atmosphere.
Health Phys. 28: 335-340 (1975).
S2 Spiers, F.W. Radioisotopes in the human body: physical and
biological aspects. Academic Press, New York, 1968.
S3 Stara, J.F. Tissue distribution and excretion of caesium-137
in the guinea pig after administration by three different
routes. Health Phys. 11: 1195-1202 (1965).
S4 Stewart, H.F., G.W. Ward and J.E. Johnson. Availability of
fallout 137-Cs to dairy cattle from different types of feed.
J. Dairy Sci. 48: 709 (1965).
T1 Tamura, T. and D.G. Jacobs. Structural implicatioms in caesium
sorption. Health Phys. 2: 391-398 (1960).
T2 Taylor, S.R. Abundance of chemical elements in the continental
crust: a new table. Geochim. Cosmochim. Acta 28: 1273-1285
(1964).
U1 Ueda, T., Y. Suzuki and R. Nakamura. Transfer of 137-Cs and
90-Sr from the environment to the Japanese population via the
marine environment. p. 501-511 in Dose Evaluation and
Standards for Man and His Environment. International Atomic
Energy Agency publication STI/PUB/375. Vienna, 1974.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Thirteenth Session, Supplement No.
17(A/3838). New York, 1958.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No.
16(A/5216). New York, 1962.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No.
14(A/5814). New York, 1964.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14(A/6314). New York, 1966.
U6 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13(A/7613). New York, 1969.
U7 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation to the General
Assembly, with annexes Volume I: Levels, Volume II: Effects.
United Nations sales publication No.E.72.IX.17 and 18. New
York, 1972.
U8 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No.E.77.IX.I. New York 1977.
V1 Vanderploeg, H.A., D.C. Parzyck, W.H. Wilcox et al.
Bioaccumulation factors for radionuclides in freshwater biota.
Oak Ridge National Laboratory report 0RNL-5002 (1975).
Y2 Yamagata, N. and T. Yamagata. The concentration of 137-Cs in
human tissues and organs. Bull. Inst. Publ. Health 9: 72-78
(1960).
Z1 Zundel, W.S., F.H. Tyler, C.W. Mays et al. Short half-times of
caesium-137 in pregnant women. Nature 221:89 (1969).
VIII. RADON
A. INTRODUCTION
337. Radon is element number 86, but the term generally refers to
the isotope 222Rn, the decay product of 226Ra. Other isotopes of
radon, 220Rn and 219Rn, are generally referred to by their
historical names, thoron and actinon, respectively. Radon has a
half-life of 3.82 days and decays by alpha-particle emission to a
polonium isotope (218Po), which by further decay through isotopes
of lead, bismuth, polonium and thallium ends the uranium (238U)
decay chain with stable lead (206Pb).
338. Radon is a chemically inert gas. It arises from decay of
radium which occurs in soil and other common materials. Part of the
radon produced diffuses into the surrounding environment.
339. The short-lived decay products of radon, called radon
daughters, are 218Po (3.05 min), 214Pb (26.8 min), 214Bi (19.7 min)
and 214Po (1.6 10-4s). They become largely attached to aerosols in
air and if inhaled they are partly deposited in the human
respiratory tract. The radiation doses caused by inhalation of
radon daughters in air constitute the main part of the natural
radiation dose to man. The radiation dose caused by radon itself
is minor by comparison with that of radon daughters.
340. The levels of radon and radon daughters in air depend on the
source and on the dilution in the air. The levels are normally
higher indoors than outdoors. Reduced ventilation may cause radon
released from building materials to build up in enclosed spaces.
The radon levels may be very high in underground mines,
particularly in uranium mines.
B. SOURCES
1. Outdoors
(a) Natural radon
341. The main source of radon outdoors is radium in the earth's
crust. The concentration of uranium and radium in the ground
varies with the types of rocks and minerals. The concentration of
radium in rocks and soil is often (but not always) the same as that
of uranium. Fractionated dissolution and transport of uranium-234
and/or radium can cause breaks in the uranium chain [S2].
342. The total amount of radium in the outer 10 km of the earth's
crust is of the order of 1024 Bq. Most of the radon produced by
decay of radium is physically attached to the radium-bearing
material and only a small part diffuses out into the air. Other
relatively less important sources of radon in air outdoors are
plants, ground water, oceans, etc. The sources contributing to the
total amount of radon are given in Table VIII.1. It is assumed
that the radon exhalation rate from the land (soil) is 0.02 Bq m-2
s-1 and from the oceans is 70 µBq m-2 s-1. The total production
rate of radon is of the order of 1020 Bq a-1; the equilibrium
inventory in the atmosphere, determined from the total production
rate divided by the decay constant (66.2 a-1) is estimated to be
1.5 1018 Bq.
Table VIII.1 Sources of radon in the global
atmosphere [H5]
---------------------------------------------
Source Radon production
per year (Bq)
---------------------------------------------
Soil 9 1019
Plants and ground water < 2 1019
Oceans 9 1017
Houses a/ 3 1016
Natural gas 3 1014
Coal 2 1013
---------------------------------------------
a/ The value for houses is estimated in this
document assuming 109 reference houses
(see Table VIII.3). The true value may be
between 5 1015-1017 Bq a-1.
(b) Mines and mine tailings
343. Sources of radon of local interest include the tailings from
uranium and phosphate mining and milling and from geothermal power
stations. The radon exhalation rate from tailings depends on the
radium content of the tailings, on the emanation factor (fractional
release of radon) and on the land reclamation (overburden). The
radon exhalation rate from uncovered uranium tailings varies from
0.5 Bq m-2 s-1 or less to 10 Bq m-2 s-1 or more [S9, U4]. The
thickness and area of the tailings per unit mass of uranium in the
ore can also vary depending on the tailings engineering and
therefore the radon exhalation rate varies in the range of about
10-100 Bq s-1 per MW(e)a [S9, U4]. By covering the tailings with a
few meters of soil the radon exhalation rate is reduced by one to
several orders of magnitude. The radon exhalation from tailings of
phosphate mining and milling also depends on land reclamation and
the type of ore. A range of exhalation rates of about 0.01-1 Bq
m-2 s-1 has been reported [R1]. Geothermal power stations may cause
radon releases into the air by releases of radon from the water
which is depressurized at the surface. A radon release of the
order of 1011 Bq per MW(e)a has been reported [M3]. The radon
release from coal-fired plants is 3 to 4 orders of magnitude less.
344. The sources of radon in underground spaces like mines are
radium in the rock and minerals of the mine and radon in water.
The total release of radon into the mine depends on many factors:
the uranium-radium concentration of the ore; the number and size of
cracks in the ore; the isolation of abandoned spaces; the random
concentration and amount of water; the isolation of water; and the
ventilation system principles. By using a normal range of total
ventilation rates (10-1000 m3 s-1) and a normal radon concentration
(0.1-10 kBq m-3) the range of radon release into the mine may be
estimated at 1-104 kBq s-1. Since there is normally an inverse
proportionality between radon concentration and ventilation rate,
lower ventilation rates are more often related to higher radon
concentrations and vice versa. There is also a radon release from
the mine to outside air and it can be compared with the radon
release from uncovered tailings. Mining of 100 tonnes uranium per
year results in 5000-15000 m2 of tailings per year depending on the
percentage of uranium in the ore and the tailings engineering. The
radon release will be in the range of 1-10 kBq s-1. In non-uranium
mines radon-rich water is often a significant source of radon to
the mine [S5]. The radon concentration of the water may be in the
range of 100-1000 kBq m-3.
2. Indoors
(a) Building materials
345. Radon in houses comes from building materials, the soil under
the house, the water and the domestic gas. Radium concentrations
in building materials have been investigated. The data indicate
that some materials such as aerated concrete with alum shale and
phospho-gypsum from sedimentary ores have significantly higher
radium concentrations than others and cause enhanced radon
concentrations indoors. If these materials are excluded, the
average concentrations of radium in building material is about 100
Bq kg-1. Materials with low activity are wood, natural gypsum,
sand and gravel. The radon exhalation rate from walls, floors and
ceilings is dependent on the radium concentration, the emanation
power, the diffusion coefficient in the material and the qualities
and thickness of any applied sealant on the surfaces.
346. The radon exhalation rate from uncovered building materials
varies by several orders of magnitude from about 10-6 Bq m-2 s-1
(gypsumboard, fiberboard, chipboard, bricks) to (0.1-10) 10-3 Bq
m-2 s-1 for concrete of different origins and qualities [J5, P3,
S17, M8, W3]. The radon exhalation rate per Bq Ra/kg varies less,
e.g., (1.6 ± 0.88) 10-5 Bq m-2 s-1 per Bq Ra/kg for some building
materials in the Federal Republic of Germany [W3]. If the same
values are normalized to an emanating power of 1%, the radon
exhalation rate is (4.4 ± 1.9) 10-6 Bq m-2 s-1 per Bq Ra/kg per
percentage emanating power. In the first case the standard
deviation is 54% and in the last case it is 43%. By painting,
plastering or application of wall-paper on the wall the radon
exhalation may be reduced by less than a factor of 5 [W3, M8].
(b) Soil
347. The contribution of radon from the soil into a building
depends on the thickness and tightness of the base structure. The
exhalation from the soil is of the order of 10-2 Bq m-2 s-1 and a
concrete floor in cellars should normally reduce the radon
exhalation from soil into the building by a factor of 10 or more.
Even so, radon from soil may contribute significantly to the radon
concentrations in a house, particularly in the cellar and in wooden
houses. In some areas houses are built on natural uranium deposits
(Canada) [L2, K2], on phosphate-related land (Florida, U.S.A.) [U3,
U13], on waste products from uranium industry (Colorado, U.S.A.)
[C6], and on waste products from alum production from radium-rich
alum shales (Sweden) [S22]. In these cases the radon exhalation
rate may be several orders of magnitude higher than from normal
soil.
(c) Water
348. Another source of radon in a house may be radon-rich water.
The relative radon release depends on the use of water. Boiling
and splashing of the water increase releases and consequently the
highest radon releases occur in washrooms, at shower-baths and in
the kitchen during cooking. The resultant radon concentration in a
house depends on the amount of water used, the volume of the house
and the ventilation. Several studies have been made to estimate
the relative significance of radon from water [P1, C2, N1], and a
typical value of the air-to-water radon concentration quotient is
about 10-4. Measurements of radon in water are most often made in
areas with suspected high concentrations because of uranium
deposits and estimates of the weighted average radon concentration
in water for a country are rare.
349. Population-weighted average concentrations of radon in
drinking water have been estimated for a few countries and are
found to be 40 kBq m-3 in Finland [A5, C3], 7 kBq m-3 in Sweden
[S8] and 0.4-4 kBq m-3 in the Federal Republic of Germany [M9].
The corresponding radon release into a house can be estimated by
assuming a daily use of 500 1 per person and 10-100% relative
release from the water.
(d) Natural gas
350. Natural gas containing radon may also be a source of radon in
houses. Gas is transported as purified gas in long transmission
lines and distributed to the homes or bottled under pressure as
propane for sale as liquified petroleum gas (LPG). The radon
concentrations in natural gas at the production wells are found to
vary from undetectable values up to about 40 kBq m-3 [U12, H6].
During supply, transit, storage and delivery the radon
concentration decreases to an approximate average of the order of 1
kBq m-3 for both natural gas and LPG (in U.S.A.) [U12, B1, G1].
(e) Summary
351. The relative contribution of different radon sources to the
total radon input in a house is estimated in Table VIII.2 with some
typical values of radon concentration and releases. The radon in
outside air is brought into the house by ventilation. The volume
of the house is assumed to be 200 m3 and the inner surface area 350
m2.
Table VIII.2 The relative significance of different radon sources
in a reference house
----------------------------------------------------------------------
Source Radon flux Comments
(Bq d-1)
----------------------------------------------------------------------
Building material 70 103 Emanation rate 2 10-3 Bq m-2 s-1
Water 4 103 1000 1 d-1 and 4 kBq m-3, 100% release
Outside air 9 103 Radon concentration outdoors 0.004 kBq
m-3; ventilation rate 0.5 per hour
Natural gas 3 103
Liquified 0.2 103
petroleum gas
----------------------------------------------------------------------
C. BEHAVIOUR IN THE ENVIRONMENT
1. Release from soil
(a) Emanation
352. The mechanism of radon release from rock, soil and other
materials is not very well understood and is probably not always
the same. The main physical phenomena are recoil and diffusion of
the radon atom through imperfections of the crystalline structures
of the radium-bearing particle followed by a secondary diffusion,
which depends on the porosity of the material [A1]. High porosity
increases the diffusion rate. The release rate from a material
depends also on its moisture content: if the moisture content is
very low the radon release is decreased by the effect of
re-adsorption of radon atoms on surfaces in the pores. If the
moisture content increases slightly, the radon release increases up
to a certain moisture content, above which the release of radon
decreases again owing to a decreasing diffusion rate in water-
filled pores [M7].
(b) Diffusion
353. Once radon has entered the air or water surrounding the
emanating radium-bearing particle, it is transported by diffusion,
earth-mechanical and convective flow, percolation of rain water and
flow of ground water. The diffusion mechanism can be expressed by
the equation
Cx = Co exp [-x/ ´(D/lambda)]
where Cx is the radon concentration at distance x in air or water
from the emanating surface; Co is the radon concentration at the
surface; D is the diffusion coefficient (gas kinetic) and lambda is
the decay constant [G2]. The diffusion constant D is about 10-2
cm2 s-1 in air and 10-5 cm2 s-1 in water. This means that it takes
on the average about 13 days for a radon atom to diffuse 5 m in air
or 5 cm in water. In that time the radon would decay by almost a
factor of 10. Accordingly, long distance transport of radon in air
and water mainly depends on the other mechanisms mentioned above,
which are the transport of air and water itself.
(c) Exhalation
354. The radon concentration Cz in soil air at depth z below the
surface depends on the diffusion coefficient D, the emanating
factor a (0 < a < 1), the fractional pore space of the soil f, the
radium activity concentration Cr (per unit volume of soil) and the
decay constant of radon g according to the equation [J1]
a x Cr
Cz = ------ [1 - exp (- ´(lambda/D) z]
f
The exhalation rate is expressed by the equation [J1]
d(Cz)
R = D [-----]
dz z=o
The combination of the above two equations gives
lambda a Cr
R = ----------- ´(D/lambda)
f
If a = 0.1; Cr = 0.07 Bq cm-3; f = 0.3 and D = 0.01 cm2 s-1;
lambda = 2 10-6 s-1, the exhalation rate R is 3 10-2 Bq m-2 s-1.
355. The diffusion rate and thereby the exhalation rate is
influenced by meteorological factors such as rainfall, snowfall,
freezing and variations in atmospheric pressure. An increase in
these parameters will decrease the exhalation rate. Measured
values of radon exhalation rate from soil vary between about 0.0002
and 0.07 Bq m-2 s-1 [G2, W1]. The radon exhalation from sea water
per unit area and time is about two orders of magnitude less.
2. Dispersion in air
356. The dispersion of radon in air is influenced by the vertical
temperature gradient, the direction and strength of the wind and
the air turbulance. The dispersion of radon daughters is also
influenced by precipitation. The vertical distribution of radon
and its daughters in air can be calculated from the following
system of differential equations [J2]:
d C1
--- (K --) - lambda1 C1 = 0
dz dz
d C1
--- (K --) + lambdai-1 Ci-1 - (lambdai + LAMBDA) Ci = 0
dz dz
where C1 is the concentration of radon atoms in air at the height
z; ni is the concentration of radon daughter i in air at the
height z; lambda1 is the decay constant of radon; lambdai is the
decay constant of radon daughter i; LAMBDA is the removal rate of
radon daughters caused by washout and rainout. Boundary conditions
to the above equations are
Ci (z=0) = 0 for i > 1 and Ci(z->infinite) = 0 for i = 1,2,3, ...
By assuming a constant radon exhalation from an infinite plane
(ground surface) which equals the radioactive decay of the total
radon content in the atmosphere it is possible to solve the first
two equations in this paragraph, which in combination with
different values of the turbulent diffusion coefficient K [J1, J2]
give the vertical distribution of radon and radon daughters for
different atmospheric stabilities.
357. Measured values of the relative distribution of radon in air
are shown in Figure VIII.I. Although measured values in this case
are found to follow the predicted vertical distribution fairly
well, the models described above should only be taken to serve as
rough guidance for the prediction of radon daughter levels. The
varying radon exhalation rate on land and on sea and varying
meteorological conditions may cause distribution patterns different
from those predicted by the model.
273. The chemical similarity of caesium and potassium and the
opportunity to make simultaneous measurements by gamma spectrometry
of 137Cs and naturally-occurring 40K has encouraged the expression
of 137Cs concentrations relative to the potassium concentration in
a manner analogous to that used for strontium and calcium.
However, caesium and potassium are not interdependent and do not
behave in such a regular manner in biological systems as do
strontium and calcium. As the levels of potassium in diet and man
remain roughly constant (1.4 g per litre of milk and 2 g per
kilogram of body weight), the 137Cs/K quotients can be converted
easily to 137Cs concentrations. Dietary intake of 137Cs increases
in proportion to the amount of food consumed, however the 137Cs/K
quotient in diet is relatively constant for adult and children
diets for widespread contamination situations [G4]. An additional
advantage of expressing 137Cs levels in the body in terms of the
137Cs/K quotient is that age and sex differences are minimized and
the values correlate more closely to 137Cs concentrations per unit
of lean body mass, which seems to be a more important parameter for
dosimetric purposes than the whole body mass. It is expected,
however, that assessments in the future will be presented
independently for 137Cs, without so much reliance on the stable
congener element.
274. A great deal of information has accumulated on 137Cs in the
environment, particularly the measurements of fallout 137Cs in air,
deposition, diet and man. Much of the literature has been
referenced by UNSCEAR over the years. Recent reviews of 137Cs data
have been published by Moiseev and Ramzaev [M5] and the United
States National Council on Radiation Protection and Measurements
[N1]. This document is not extensive in terms of references cited.
The representative references for most statements can be taken as
the starting points for the more extensive literature available.
B. SOURCES
1. Nuclear explosions
275. Atmospheric testing of nuclear weapons has resulted in
widespread distribution in the environment of radioactive fission
and activation products. Extensive measurements of fallout
radioactivity have been conducted. The data have been reported and
discussed in each report of UNSCEAR [U2, U3, U4, U5, U6, U7, U8].
The stratospheric inventory of 90Sr has been measured in a long-
term programme [L1]. Global networks to measure fallout deposition
have reported results for 90Sr [F1] and 137Cs [C1]. The activity
ratio of 137Cs to 90Sr in long-term deposition has been found to be
relatively constant at about 1.6 [U7], although variations for
individual samplings are encountered [S1].
276. The total amount of 90Sr produced in weapons testing through
1980, which has been globally dispersed, is estimated to be 6.0
1017 Bq [F1]. Less than 1% of this amount remained in the
stratosphere [L1]. The remainder has been deposited on the earth's
surface. This corresponds to 9.6 1017 Bq of 137Cs produced in
nuclear testing. Radioactive decay has reduced the cumulative
deposit of 137Cs to 6.9 1017 Bq [C1], 76% of which is in the
northern hemisphere and 24% in the southern hemisphere.
2. Nuclear fuel cycle
(a) Nuclear reactors
277. Caesium-137 is produced by fission in the fuel of nuclear
reactors. The amounts produced depend on the degree of fuel burn-
up achieved and to some extent on the type of fuel and the neutron
spectrum in the reactor. In fairly high burn-up fuel (33000 MW[t]d
t-1) of a pressurized water reactor, the 137Cs production is
estimated to be 3.9 1015 Bq per tonne of fuel, corresponding to 1.3
1014 Bq per MW(e)a of electricity generated [O1].
278. Small amounts of fission products produced in the fuel in
nuclear reactors may reach the coolant through defects in the fuel
cladding. In coolant purification or following coolant leakage,
these fission products may reach gaseous and liquid effluent
streams. In controlled amounts, some of the effluents are released
to the environment.
279. Reported amounts of 137Cs released to the environment from
reactors have been summarized by UNSCEAR [U8]. The averaged
release rates for some reactor types are included in Table VII.2.
Table VII.2 Estimated global discharges of 137Cs
from nuclear power stations in 1980
--------------------------------------------------------------
Reactor Reactor Capacity Release rate Estimated
type number [MW(e)a] [Bq per MW(e)a] discharge (Bq)
--------------------------------------------------------------
PWR 96 64239 6 107 2 1012
BWR 62 35170 9 108 19 1012
GCR 36 7086 2 109 9 1012
--------------------------------------------------------------
Other 47 18490 9 108 10 1012
--------------------------------------------------------------
Total 241 124985 4 1013
--------------------------------------------------------------
These are not necessarily the typical situation for a particular
reactor. During a specific year it is normally the case that a
somewhat larger release occurs from a single reactor with
insignificant releases reported from all other sites. This means
that there is a large range in release distribution, covering three
or four orders of magnitude. For all reactor types the release of
137Cs is primarily in liquid effluents; negligible amounts, in
comparison, occur in airborne effluents.
280. Assuming the averaged normalized release rates to be
representative for the reactor groups, it is possible to obtain a
very rough estimate of the total amount of 137Cs released from
reactors worldwide. Using the installed capacities of the various
reactor types as of 1980 [I4] and assuming a reactor utilization of
60%, the estimated annual release from all reactors is about 4 1013
Bq. In this calculation, it is assumed for the reactor types for
which no data are available, that the releases are similar to those
from BWRs.
(b) Fuel reprocessing plants
281. In fuel reprocessing plants the fuel is dissolved to recover
uranium and plutonium for re-use. All of the 137Cs and other
fission products as well go to the waste streams. The radionuclide
activities in airborne and liquid effluents from fueld reprocessing
plants have been recorded by UNSCEAR [U8].
282. Caesium-137 is released from fuel reprocessing plants
primarily in liquid effluents. Averaged release rates during the
years 1971-1972 were 0.6, 90 and 520 109 Bq/MW(e)a from the Nuclear
Fuel Services plant (U.S.A.) (no longer in operation), La Hague
(France) and Windscale (U.K.), respectively [U8]. The release of
caesium-137 in liquid effluents is small relative to the amount in
spent fuel. The fractional liquid releases from the Windscale
plant were approximately 4 10-3. The only data for 137Cs in
airborne effluents is for the Nuclear Fuel Services plant, which
corresponded to 4 104 Bq/MW(e)a [U8], several orders of magnitude
less than in liquid effluents.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Fixation in soil
283. Caesium is generally rather strongly fixed in soil. Downward
migration and availability to plants is thereby reduced. In
mineral soils the movement of 137Cs is appreciably less than that
of 90Sr. Three to four years after deposition on the soil surface,
the median depth to which it has penetrated is usually less than 2
cm [F5]. Its mobility may be somewhat greater in organic soils.
Much smaller amounts of 137Cs than of 90Sr are leached out of the
soil to enter rivers and lakes.
284. There are exceptional areas, however, where caesium fixation
in soil is much less, allowing enhanced transfer of caesium to
plants. Marei et al. [M2] identified regions in the USSR where the
soil is wet, peaty and podzolic, from which transfer of 137Cs into
the food chain is 10 times higher than for other areas. Other
regions of the world where the soils give rise to high 137Cs
transfer into diet have also been identified, for example, in the
Faroe Islands, New Zealand and Sweden [U8].
285. It has been reported that in clay minerals the important
factor in fixation of caesium is the ability of certain layered
silicates such as micas, vermiculites and illites to adsorb or fix
trace quantities of caesium [T1]. Caesium ions are trapped in the
interlayer regions of vermiculite or at the frayed edges of illites
and micas. Caesium is thus more strongly retained in soils
containing predominantly micaceous minerals. Soils which do not
contain large quantities of micaceous minerals, such as tropical
soils, peat soils, and podzolic soils, exhibit less retention and
allow greater uptake of caesium by plants.
286. Fixation of caesium by sediments in aquatic environments
occurs in a similar fashion to fixation in soil. The preferential
adsorption of 137Cs to the micaceous component of sediments has
been demonstrated under environmental conditions.
2. Transfer to plants
287. Caesium-137 may be transferred to plants by direct deposition
onto plant surfaces or by root uptake from accumulated deposits in
soil. In general, direct foliar absorption is the predominant mode
of plant contamination when the deposition rate is relatively high.
Root uptake is low except in those cases mentioned above, when soil
conditions allow low fixation of caesium.
288. Caesium depositing on plant surfaces is retained to the same
extent as other particulate debris. A removal half-time of l4 days
due to weathering is generally assumed. Once absorbed by the
plant, caesium is readily redistributed through the plant.
Relationships between air concentrations and subsequent
concentrations of 137Cs in pasture plants have been discussed by
Hawthorne et al. [H4] and Pelletier and Voilleque [P2].
289. Caesium may enter plants by plant base absorption before
becoming fixed in soil. Thus, retention of 137Cs in the root mat of
pastures may allow 137Cs to be relatively more available to plants
for a period of a year or more [U3]. A high level of organic
matter in soil can enhance the absorption of caesium by plants
[B1]. Sorption of organic molecules on clay surfaces prevents the
retention of caesium and also of potassium by these minerals.
Thus, for permanent pastures in temperate regions, the frequent
high organic matter content of the upper soil layer allows shallow
rooted grass to absorb 137Cs relatively more freely for a somewhat
more extended period following deposition. Mushrooms have been
shown to concentrate very effectively 137Cs from soil [G2, M1],
which may be associated with the highly organic areas of growing.
290. Uptake to plants of 137Cs from soil low in available
potassium may be somewhat increased. The addition of potassium may
decrease absorption of 137Cs in this case; however, this has no
effect when the available potassium is high [N5, F4]. Root uptake
of caesium is in general included in the range 0.01 to 1, which is
the ratio of caesium concentrations in the dry plant material to
that in dry soil [M3].
3. Transfer to milk
291. The fractional amount of 137Cs transferred into milk is
slightly greater than that of potassium. It has been shown that
some 10% of orally ingested 137Cs is secreted into the milk of
dairy cows, corresponding to 1.3 to 1.5% of the amount ingested per
litre of milk [G1, I1, L2]. The transfer of fallout 137Cs in field
conditions has been found to be somewhat less, ranging from 0.25 to
0.86% of intake per litre of milk [P2, M1, S4].
4. Transfer to meat
292. A correlation between 137Cs concentration in beef and in milk
has been noted, the ratio of concentrations in meat (Bq/kg) and in
milk (Bq/1) averaging about 4 for production in the same locality
[L5, E1, J2]. This would correspond to a transfer of about 4% of
the daily intake per kilogram of meat [F5]. Equilibrium conditions
are reached in about 30 days in the cow [F5]. It cannot generally
be expected that there will be a useful relationship between 137Cs
in meat and milk, since animals produced for the two purposes are
frequently provided with different diets and are reared in
different areas.
5. Transfer to diet
293. For general contamination situations, as for global fallout
radioactivity, the main contributions to dietary intake of 137Cs
are generally from grain products, meat and milk. Fruit and
vegetables contribute much smaller amounts of 137Cs. This has been
the pattern for western diets, such as Denmark [A1] and the United
States [G4]. In Japan the main contribution has been from cereals
[U1]. For all foods the transfer from a specific deposited amount
seems to be rapid, being essentially completed within the first two
years after deposition.
294. Marine food chains are of secondary importance in
contributing to dietary intake of fallout 137Cs, even in countries
where fish is widely consumed. In Japan between 1966 and 1971,
only about 8% of the 137Cs in diet came from the consumption of
fish products [U1]. As the contributions from other foods
decrease, however, the relative contribution from fish can
increase. During 1976 in Denmark, 15% of the 137Cs intake was
attributed to fish [A2]. It is possible that individuals consuming
large amounts of freshwater fish may acquire 137Cs burdens several
times greater than individuals eating more diversified diets [G3].
295. The transfer of 137Cs from deposition to diet has been
studied quantitatively using the following transfer function
between deposition and diet components in regression analyses of
reported data [U8]:
j j j infinite
Cj(n) = b1 f(n) + b2 f(n-1) + b3 sigma f(n-m) e-µm
m=1
where Cj(n) is the concentration of 137Cs in the diet component j
during the year (n) in mBq/gK; f(n) is the deposition density of
137Cs in the current year (n) and in previous years (n-1) in Bq/m2;
bjn are the proportionality factors, and µ is the decay constant
accounting for radioactive decay and reduced availability of
deposition in all previous years. The first term of the equations
is the rate term giving the contribution to dietary level from the
current year's deposition amount. The second term is the long term
contribution expressing separately the contribution from the
previous year's deposition including storage of foods by market
practices. The third term gives the contribution from the
cumulative deposit of 137Cs in soil.
296. The quotient of the time-integrated concentration of 137Cs in
diet and the integrated deposition density defines the transfer
factor P23. The integrals are replaced by summations if, as is
usually the case, the relevant quantities are assessed over
discrete intervals of time, such as annual averages
infinite
sigma C(n)
n=1
P23 = -----------
infinite
sigma f(n)
n=1
The transfer factor is usually expressed in mBq a/gK per Bq/m2. It
may be evaluated for total diet or for dietary components.
297. Using the model described above, the evaluation of the
transfer factor reduces to
infinite -µm e-µ
P23 = b1 + b2 + b3 sigma e = b1 + b2 + b3 -----
m=1 1-e-µ
The parameters of the transfer function, obtained by regression
fits of the 137Cs/K quotients in milk, dietary components and total
diet from several countries have been reported by UNSCEAR [U8].
Some of these results are given in Table VII.3.
298. The lowest values for the transfer factor for milk are
obtained for the U.S.A., Denmark and the U.K. Most of the transfer
of 137Cs deposition to milk is from direct deposition. Less than
15% of the transfer is from uptake of 137Cs from soil.
Intermediate values of P23milk are obtained from the USSR and
Argentina. Transfer from direct deposition is increased and uptake
from soil is more significant. The highest values of P23milk are
obtained for New Zealand, Australia, Norway and the Faroe Islands.
A high value has also been reported for Finland, which is 15.8 mBq
a (gK)-1 per Bq m-2 [C2]. These results can be explained by
efficient transfer of direct deposition and soil conditions which
allow only low fixation of 137Cs.
299. The range of values of transfer factors for milk, 3.4 to 27.5
mBq a (gK)-1 per Bq m-2, indicates that it is not easy to specify a
typical transfer situation. To estimate the 137Cs transfer to milk
in a particular region requires some indication of soil and pasture
conditions.
300. Using the comprehensive data from Denmark of 137Cs in diet
[A2], values of the parameters of the transfer function of various
components of diet and for the total diet have been determined.
For most foods, the major contribution to the value of the transfer
factor comes from direct deposition. Only a small transfer is
attributed to uptake from soil. The values of the parameters for
the fit to total diet data are: b1 = 1.6, b2 = 2.2, b3 = 0.04 mBq
a(gK)-1 per Bq m-2 µ = 0.11 a-1 and P23 = 4.1 mBq a(gK)-1 per
Bq m-2. Short-term transfer to diet is thus estimated to comprise
93% of the total transfer, (b1 + b2)/P23.
301. The general pattern of 137Cs transfer to diet can be expected
to be similar to that for Denmark, although increased transfer can
occur in specific areas, depending on soil conditions or particular
consumption habits. UNSCEAR has used a rounded value of P23 of 4
mBq a(gK)-1 per Bq m-2 to assess the dose commitments from 137Cs
for the world population [U8].
Table VII.3 Parameters of the transfer functions between deposition density and
137Cs/K in milk
-----------------------------------------------------------------------------------------
Parameter U.S.A. Denmark United USSR Argentina New Australia Norway Faroe
a/ Kingdom Zealand Islands
1973 1976 1977 1972 1976 1977 1973 1976 1976
-----------------------------------------------------------------------------------------
b1 2.3 2.2 1.7 4.4 3.2 6.8 7.3 3.5 6.8
b2 1.3 1.2 1.6 0.2 0.0 0.9 4.8 2.4 5.5
b3 0.0 0.01 0.04 0.3 1.7 3.1 0.3 2.5 2.8
µ 0.05 0.03 0.07 0.2 0.3 0.5 0.2 0.2 0.2
P23 3.4 3.7 3.9 6.2 7.5 13.0 13.4 15.6 27.5
-----------------------------------------------------------------------------------------
a/ The unit for parameters b1, b2, b3 is mBq a(gK)-1 per Bq m-2.
The unit for parameter µ is a-1.
The unit for the transfer factor P23 is mBq a(gK)-1 per Bq m-2.
6. The lichen-caribou-man foodchain
302. A terrestrial situation which allows much greater than usual
transfer of caesium to man is the lichen-caribou-man foodchain.
Caesium depositing on lichens is retained quite effectively. There
is a very slow decrease in activity with time, approximately 5 to
10% of the 137Cs being eliminated annually [U5]. Lichens provide
the food base for grazing caribou and reindeer during the winter.
A proportionality factor of 137Cs in lichen to that in reindeer
meat in northern Sweden during the winter averaged 4.9 ± 0.4 [L4].
In summer also grass and herbaceous plants are consumed by the
animals, and therefore the 137Cs levels in meat show marked
seasonal variation.
303. High concentrations of 137Cs arise in Lapp and Eskimo
populations who eat the caribou and reindeer meat. Levels of 740-
1300 Bq per kg of body weight were observed in individuals during
1964 from fallout [H1, M4], about a factor of 100 greater than
burdens of individuals from temperate northern hemisphere regions.
Other fallout radionuclides, such as 54Mn and 55Fe, and the natural
isotopes 210Pb-210Po are also concentrated along the lichen-
caribou-man foodchain.
7. Aquatic behaviour
304. Caesium in the aquatic environment is strongly adsorbed by
suspended particulate materials, especially clays. Therefore, the
amount of caesium in the soluble phase decreases with increasing
suspended solid concentrations. Potassium is also sorbed, but to a
much less degree.
305. Caesium levels in fish are inversely related to the potassium
content of the water [K1]. Because of the high concentration of
potassium in the ocean, the transfer of 137Cs to fish is of primary
concern in the freshwater environment. The activity of fresh water
fish may be 100 times that of ocean fish, given the same caesium
concentrations in water.
306. The low mineral content of fresh water also enhances the
absorption of 137Cs by aquatic plants. Aquatic plants from fresh
water areas, which are sometimes important in cattle feed, may have
increased levels of 137Cs compared to 137Cs which may have
deposited on nearby pasture ground.
307. Caesium in aquatic animals is accumulated primarily from the
food chain. Absorption efficiency of potassium and caesium from
food is high. In animals the excretion rate of potassium is about
3 times larger than that of caesium. As a result, the caesium
concentration per unit amount of potassium in tissues increases by
a factor of about 3 with each trophic level [P1].
308. The food web also accumulates caesium from suspended and
bottom sediments. Filter feeders may accumulate caesium adsorbed
to particulate matter. Benthic invertebrates obtain caesium
absorbed to ingested bottom sediments. Fish ingest those
invertebrates and also some sediment particles along with the prey.
Absorption efficiency in the fish depends on the caesium fixation
ability of the sediment minerals.
309. The concentration factors (ratio of concentration in organism
to that in the water) for caesium must be related to the potassium
concentration in the water and to the turbidity. From a literature
review of values for fresh water systems [V1], suggested values of
the concentration factors are 1000 for algae and plants, molluscs
and invertebrates in all waters and 5000/Kw and 1500/Kw for non-
piscivorous and piscivorous fish, respectively, in clear waters,
where Kw is the stable potassium concentration of water in µg/g.
The factors for fish are a factor of 5 less in turbid waters (> 50
µg/g suspended solids). The concentration factors for caesium in
the ocean are 10 for algae and molluscs, 30 for fish and 50 for
molluscs [F6].
D. TRANSFER TO MAN
1. Absorption and distribution in tissues
310. As a general rule, caesium compounds are soluble in body
fluids. Intestinal absorption is complete (100%) under
experimental conditions [R2, S3], but from normal diets is probably
less efficient, ranging from 50 to 80% [F5]. In man caesium is
secreted into the gastrointestinal tract, between the stomach and
small intestine, and is readily reabsorbed [I2]. One basis for
therapeutic treatment in internal contamination cases is to
administer solutions of Prussian blue, which binds with caesium in
the gastrointestinal tract preventing reabsorption [I2, D1].
311. Caesium migrates rapidly into cells of the body following
intake and becomes relatively uniformly distributed in soft tissues
[R2, L3, R4]. The metabolism in mothers and infants has also been
studied. There seems to be no placental discrimination, as the
newborn has 137Cs concentrations about equal to that of the mother
[B3].
312. Concentrations of caesium and potassium are low in fat
tissues. Therefore, for equal 137Cs concentrations in intake, the
concentrations of 137Cs (Bq per kg body weight) in males are higher
than in females, due to the higher average proportion of fat tissue
in the female body. However, a difference is also expected due to
longer retention time of caesium in males. Expressed in Bq 137Cs
per gK, the difference between males and females is somewhat
reduced.
313. It has been inconclusive for some time whether 137Cs
concentrates in bone. One study reported that the concentration of
137Cs in rib bones, which were free of muscle but not of marrow,
was comparable to the concentration in soft tissues [Y2]. The
results were variable, and subsequent studies both did and did not
confirm these results [A3, H3, N4]. From the results of a recent
study it appears that caesium associated with bone is present in
the marrow portion with only slight uptake by the hard tissue [H2].
314. A slower turnover of caesium in bone could allow
concentrations in bone to lag behind those in tissue, causing
higher relative concentrations in bone during periods of decreasing
intake. A longer retention half-time would eventually be noted in
whole-body measurements. However, such a component has not been
identified in over 1000 days of measurement following an acute
intake case [R4].
2. Retention half-time
315. A great many investigations of the biological half-time in
man have been conducted. Many of the references are collected in
the discussion by Lloyd [L6]. The half-time in man varies
considerably, depending on age and other factors. The half-time is
less in women than in men, and the half-time in children and
infants is less than in adults. Pregnant women have shorter
caesium half-times than in their non-pregnant conditions. Table
VII.4 shows the summary of 137Cs retention half-time reported
recently by the NCRP [N1], using the data of Lloyd et al. [L7] and
Zundel et al. [Zl].
Table VII.4 Retention half-time of 137Cs in the
human body [N1]
--------------------------------------------------
Subjects Number Age Half-time (d)
--------------------------------------------------
Men 26 23-55 a 105 ± 25
Women 15 20-51 a 84 ± 20
Pregnant women 24 16-39 a 49 ± 16
Children 7 5-17 a 57 ± 20
Infants 5 17-143 d 19 ± 8
--------------------------------------------------
316. The biological half-time for caesium in man can be considered
a function of age for juveniles and of sex for adults, but it is
not determined by body mass [L6]. Half-time and body mass may,
however, be dependent on some other common factors. The rate of
caesium turnover may be under hormonal influence or control or may
reflect the general metabolic rate [L6].
317. Shorter half-time components of 137Cs in man have been
reported, including one of only 2 to 3 hours [N3]. In general, two
components of the 137Cs half-time in man have been established: a
small fraction (10 to 15%) excreted with a short half-time (1 to
1.5 days) and the remainder excreted more slowly (50 to 150 days)
[R1, R3]. The ICRP suggests representative values of retention of
10% with a half-time of 2 days and 90% with a half-time of 110
days. Integral retention is 143 Bq d per Bq intake, contributed
almost entirely by the long-term component.
3. Transfer factor
318. The value of the transfer factor P34 relating concentrations
of 137Cs in diet and man can be derived from the integral retention
by dividing by the potassium content of the body (140 g) and
multiplying by the daily potassium intake (3.3 g d-1) [I5]. The
result is 3.4 Bq a (gK)-1 in man per Bq a (gK)-1 in diet.
319. The relatively short biological half-time of caesium in the
body makes it possible to assess the transfer factor P34 from the
measured 137Cs/K quotients in diet and man integrated over a few
years. Using this procedure, an average value of 3 Bq a (gK)-1 per
Bq a (gK)-1 diet is derived [U6, U7, U8].
E. DOSIMETRY
1. Dose per unit intake
320. The dose from 137Cs in tissue is due to the beta particles
from 137Cs decay and to the photon, x-rays, and conversion and
Auger electrons from decay of the daughter, 137mBa. A portion of
the photon energy will escape from the body, depending on the body
size. Calculations have been performed for uniform distributions
of 137Cs in the body for a range of proportions and masses
corresponding to infants, children and adults [N1, F1].
321. The average dose rate within the body for a uniform 137Cs
concentration of one Bq per kg body weight is about 3.5 nGy/d from
beta particles in both adults and infants plus 3.2 nGy/d from
photons in the adult and about 1.6 nGy/d in infants. The totals
are 6.7 nGy/d per Bq/kg in the adult and 5.1 nGy/d per Bq/kg in the
infant.
322. For 140 gK in the 70 kg adult body, the dose rate
corresponding to 1 Bq 137Cs per gK would be
µGy d-1 140 gK 365 d µGy/a
0.0067 -------- ------ ----- = 4.9 -------
Bq(kg)-1 70 kg a Bq/gK
Spiers [S2] also obtains this result and estimates the
corresponding dose rate in a child weighing 8 kg of
µGy/a
4.1 -------
Bq/gK
323. UNSCEAR [U6, U7, U8] assessed the transfer factor between
tissue and dose, P45, as 4.9 µGy per Bq a (gK)-1. The value is
nearly independent of age, being only slightly less for the child.
For a single uptake of 137Cs, the integral retention per unit
intake is 143 Bq d per Bq intake (paragraph 317). The average
absorbed dose in the body per unit intake is thus
µGy/d Bq d 1
0.0067 ----- 143 ----- ----- = 1.4 10-8 Gy per Bq intake.
Bq/kg Bq 70 kg
324. This result applies to intake by ingestion. For inhalation
the value is less by a factor of 0.63 (for particles of 1 µm size)
due to fractional deposition in the lungs of inhaled amounts.
Following inhalation there is only a short retention in the lung
(half-time = 0.5 d) for the soluble caesium compounds and a small
dose primarily from the beta particles before the 137Cs becomes
distributed throughout the body.
2. Dose per unit release
(a) Nuclear explosions
325. The dose commitment via the ingestion pathway from 137Cs
released by nuclear explosions can be assessed from the sequential
product of transfer factors
Dc = P23 P34 P45 F
where F is the integrated deposition density. The values of the
transfer factors as derived above are: P23 = 4 10-3 Bq a (gK)-1
per Bq m-2, P34 = 3 Bq a (gK)-1 per Bq a (gK)-1, and P45 = 4.9 10-6
Gy per Bq a (gK)-1. The dose commitment from 137Cs ingestion per
unit widespread deposition density, such as from nuclear
explosions, is thus 6 10-8 Gy per Bq m-2.
326. The total amount of 137Cs released to the environment by
nuclear tests, 9.6 1017 Bq, has given a population-weighted
integrated deposition density of 3100 Bq m-2 in the world as a
whole [U8]. The world population is 4 109. With these values, the
collective dose commitment per unit activity of 137Cs released is
estimated to be 8 10-13 man Gy per Bq (ingestion).
327. The dose commitment via the inhalation pathway is determined
from the integrated concentration of 137Cs in air, which is
estimated from the integrated deposition density (Bq m-2) divided
by the deposition velocity (m s-1). As for 90Sr, the average
deposition velocity can be taken to be 2 cm s-1. Thus, 1 Bq m-2
integrated deposition density corresponds to 5.8 10-4 Bq d m-3 in
air. With the above estimates of integrated deposition density,
total amount of 137Cs released by nuclear tests, world population,
breathing rate (22 m3 d-1) and dose per unit intake (8.8 10-9 Gy
Bq-1), the collective dose commitment per unit activity of 137Cs
released is estimated to be 1 10-15 man Gy per Bq (inhalation).
328. For the external exposure pathway it is assumed that the
deposited 137Cs becomes exponentially distributed in soil with a
mean depth of 3 cm. This gives a dose rate in air of 8.9 109 Gy
per Bq m-2 [B2]. The mean life of 137Cs in soil is 43.5 years,
determined by its radioactive decay. The dose to air from the
integrated deposition density is, thus, 3.9 10-7 Gy per Bq m-2.
The dose to tissue is determined by a factor of 0.8 to account for
the change of material (air to tissue) and back-scatter and
shielding afforded by other tissues of the body and a factor of 0.4
to account for building shielding and time spent indoors [U8]. The
combined factor is 0.8 x 0.4 = 0.32. The transfer factor P25
relating integrated deposition of 137Cs to tissue dose is thus 3.9
10-7 x 0.32 = 1.2 10-7 Gy per Bq m-2. Using the above values of
integrated deposition density, total amount of 137Cs released, and
world population, the collective dose commitment per unit activity
of 137Cs released is estimated to be 1.6 10-12 Gy per Bq (external
exposure).
(b) Nuclear installations
329. The contribution of the inhalation pathway to the collective
dose commitment for effluent releases can be estimated from the
integrated concentration of 137Cs in air. This can be derived
from a dispersion formula or from an estimate of the deposition
velocity. In the latter case, the integrated concentration in air
is the total amount of 137Cs released per unit area of the
deposition region divided by the deposition velocity, vd. The
population affected is the population density deltaN times the area
of the deposition region. The areal dependence is removed by the
product of these quantities. The collective dose commitment per
unit activity released is given by the expression
c
S1 = I deltaN phi/vd
where I is the individual intake rate of air and phi is the dose
per unit activity of 137Cs inhaled.
330. Using a deposition velocity of 0.5 cm s-1 for near surface
releases, a population density of 25 man km-2, an air intake rate
of 22 m3/d and the dosimetric factor of 8.8 10-9 Gy per Bq intake,
the collective dose commitment for the inhalation pathway per unit
activity released is estimated to be 1 10-14 man Gy/Bq.
331. The contribution of the ingestion pathway from airborne
effluents to the collective dose commitment per unit activity
released, Sc1, can be assessed by the expression
c
S1 = P23 P34 P45 deltaN
Using the values for the transfer factors given previously, and
assuming a constant population density of 25 man km-2 in the region
of deposition, the collective dose commitment for the ingestion
pathway per unit activity released is estimated to be 2 10-12 man
Gy/Bq. This value assumes that food is locally produced and that
the production suffices for the population density under
consideration. It also applies for the case of 137Cs becoming
relatively rapidly fixed in soil (P23 = 4 mBq a [gK]-1 per Bq m-2).
For other types of soil conditions or special consumption patterns
and also for other population densities, the estimate should be
adjusted accordingly.
332. For the external exposure pathway the transfer factor P25
relating integrated deposition of 137Cs in soil to the tissue dose
has been assessed above with regard to nuclear explosions to be 1.2
10-7 Gy per Bq m-2. This value is of general applicability.
Similarly to the ingestion pathway, the collective dose commitment
per unit activity released, Sc1, can be assessed by the expression
c
S1 = P25 deltaN
Assuming a population density of 25 man km-2, the collective dose
commitment from external exposure per unit activity released is
estimated to be 3 10-12 man Gy/Bq.
333. For aquatic ingestion pathways from the input of 137Cs to
water bodies the collective dose commitment, normalized per unit
activity released, can be estimated [U8], using the expression
c ksigma Nk Ik fk phi
S1 = -----------------------
V(lambda + 1/tau)
where V is the volume of the receiving waters, tau is the turnover
time of receiving waters, lambda is the decay constant of 137Cs, Nk
is the number of individuals exposed by pathway k, Ik is the
individual consumption rate of pathway item k, fk is the
concentration factor for the consumed item in pathway k, and phi is
the collective dose per unit activity ingested collectively by the
exposed group.
1
334. The quantity V(lambda+1/tau) is the infinite time integral
of the water concentration per unit of activity released, while the
quantity multiplied by fk is the infinite time integral of the
concentration in the consumed item k. For radionuclide inputs into
small volumes of water, the concentrations in water and in fish
will be high, but the population which can be served with drinking
water or by fish consumption will be limited. For inputs into
larger volumes of water, the concentrations will be smaller, but
the populations involved will be correspondingly larger. It is
reasonable, therefore, to assume as a first approximation that the
quantities Nk/V are relatively constant, independent of V. The
values for these quantities as well as values for the other
parameters of the above expression have been extensively discussed
by UNSCEAR [U8].
335. A listing of the values used in the assessments presented by
UNSCEAR is given below [U8]:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 0.3 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.2
fish 400 30
shellfish 30
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
(c) Summary
336. Table VII.5 summarizes the values obtained above for the
collective dose commitments per unit of 137Cs activity released in
airborne and liquid effluents. These are also the values of the
collective effective dose equivalent commitments with Sv replacing
Gy, since the quality factor is one and the dose is assumed to be
uniform in all tissues. The largest collective dose commitments
result from airborne discharges due to the external exposure and
ingestion pathways. These estimates are for a generalized release
situation, and substantial variations could be expected in site-
specific cases.
Table VII.5 Summary of collective dose
commitments per unit 137Cs activity
released [10-14 man Gy per Bq]
------------------------------------------
All tissues
------------------------------------------
Nuclear explosions
External exposure 160
Ingestion 80
Inhalation 0.1
Nuclear installations
Release to air a/
External exposure 300
Ingestion 200
Inhalation 1
Release to fresh water
Fish 50
Drinking water 10
Release to salt water
Fish 0.08
Shellfish 0.02
------------------------------------------
a/ Assumes population density of 25
man km-2.
F. REFERENCES
A1 Aarkrog, A. Prediction models for strontium-90 and caesium-137
levels in the human food chain. Health Phys. 20: 297 (1971).
A2 Aarkrog, A. and J. Lippert. Environmental radioactivity in
Denmark in 1976. Riso National Laboratory report No. 361
(1977).
A3 Anderson, R.W. and P.F. Gustafson. Concentration of caesium-
137 in human rib bone. Science 137: 668 (1962).
B1 Barber, D.A. Influence of soil organic matter on the entry of
caesium-137 into plants. Nature 204: 1326-1327 (1964).
B2 Beck, H.L. and G. de Planque. The radiation field in air due
to distributed gamma-ray sources in the ground. U.S. Atomic
Energy Commission report HASL-195. New York, 1968.
B3 Bengtsson, L.G., Y. Naversten and K.G. Svensson. Maternal and
infantile metabolism of caesium. p. 21-32 in Assessment of
Radioactivity in Man. Proceedings of a symposium, Heidelberg,
May 1964. International Atomic Energy Agency publication
STI/PUB/Vol. 2. Vienna, 1964.
C1 Cambray, R.S., E.M.R. Fisher, K. Playford et al. Radioactive
fallout in air and rain: results to the end of 1977. United
Kingdom Atomic Energy Authority report AERE-R9016 (1978).
C2 Castrén, O. UNSCEAR transfer coefficients P-23 (milk) and P-
234 for 90-Sr and 137-Cs in Finland. p. 73-76 in Studies on
Environmental Radioactivity in Finland 1971-1975. Institute of
Radiation Protection report STL-A21. Helsinki, 1977.
C3 Crouch, E.A.C. Fission product yields from neutron induced
fission. Atomic Data and Nuclear Data Tables, Vol. 19, No. 5
Academic Press, New York, 1977.
D1 Ducousso, R., A. Causse and C. Pasquier. Comparative effects
of acetazalamide and Prussian blue on 137-Cs retention in the
rat. Health Phys. 28: 75-78 (1975).
E1 Ellis, F.B. and B.T. Barnes. Relationship between the
concentration of caesium-137 in beef and milk. p. 77-78 in
Agricultural Research Council Radiobiological Laboratory annual
report ARCRL-14 (1965).
F1 Feely, H.W. World-wide deposition of strontium-90 through
1977. p. 1.19-1.41 in Environmental Measurements Laboratory
environmental quarterly report EML-344. New York, 1978.
F4 Fredriksson, L., B. Eriksson and B. Rasmunson. Plant uptake of
90-Sr and 137-Cs from soils, Vol. 18. p. 449-470 in
Proceedings of the Second International Conference on Peaceful
Uses of Atomic Energy, Geneva, 1958.
F5 Fredriksson, L., R.J. Garner and R.S. Russell. Caesium-137. p.
317-352 in Radioactivity and Human Diet (R.S. Russell, ed.).
Pergamon Press, 1966.
F6 Freke, A.M. A model for the approximate calculation of safe
rates of discharge of radioactive wastes into marine
environments. Health Phys. 13: 743-758 (1967).
G1 Gragle, R.G. Uptake and excretion of caesium-134 and
potassium-42 in lactating dairy cows. J. Dairy Sci. 44: 352-
357 (1961).
G2 Grueter, H. Radioactive fission product 137-Cs in mushrooms in
West Germany during 1963-1970. Health Phys. 20: 655-656
(1971).
G3 Gustafson, P.F. Comments on radionuclides in aquatic
ecosystems. p. 853-858 in Radioecological Concentration
Processes (B. Aberg and F. Hungate, eds.). Pergamon Press,
1967.
G4 Gustafson, P.F. and J.E. Miller. The significance of 137-Cs in
man and his diet. Health Phys. 16: 167-183 (1969).
H1 Hanson, W.C. Radioecological concentration processes
characterizing arctic ecosystem. p. 183-191 in Radioecological
Concentration Processes (B. Aberg and F. Hungate, eds.).
Pergamon Press, 1967.
H2 Hardy, E.P. On 137-Cs and 90-Sr in bone. p. I.64-I.69 in
Health and Safety Laboratory Fallout Program Quarterly Summary
report HASL-278. New York, 1974.
H3 Harrison, G.E., A. Sutton, K.H. Edwards et al. Concentrations
of radioactive and stable caesium in bone and soft tissues.
Br. J. Radiol. 36: 745-748 (1963).
H4 Hawthorne, H.A., S.D. Zellner, L.L. Eberhardt et al. 137-
Caesium cycling in a Utah dairy farm. Health Phys. 30: 447-464
(1976).
I1 Ilin, D.I. and Y.I. Koskalev. On the metabolism of caesium,
strontium and a mixture of beta-emitters in cows. J. Nucl.
Energ. 5: 413-420 (1957).
I2 International Atomic Energy Agency. Manual on early medical
treatment of possible radiation injury. International Atomic
Energy Agency publication Safety Series No. 47 and
IAEA/STI/PUB/506. Vienna, 1978.
I4 International Atomic Energy Agency. Power reactors in member
states. IAEA, Vienna, 1980.
I5 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP publication 23.
Pergamon Press, 1975.
J2 Johnson, J., G.M. Ward and A.H. Dal. Comparisons of 137-Cs
levels in feed and meat of cattle fed on pasture and in the dry
lot. Health Phys. 10: 612 (1964) (Abstract).
K1 Kohehmainen, S., E. Häsänen and J.K. Miettinen. 137-Cs in
fish, plankton and plants in Finnish lakes during 1964-1965. p.
913-919 in Radioecological Concentration Processes (B. Aberg
and F.P. Hungate, eds.). Pergamon Press, 1967.
K2 Krumholz, L., E. Goldberg and H. Boroughs. Ecological factors
involved in the uptake, accumulation and loss of radionuclides
by aquatic organisms in The Effects of Atomic Radiation on
Oceanography and Fisheries. National Academy of Sciences,
National Research Council Publication 551. Washington, 1957.
L1 Leifer, R. and L. Toonkel. Updating stratospheric inventories
to April 1977. p. I.3-I.14 in Environmental Measurements
Laboratory environmental quarterly report EML-334. New York,
1978.
L2 Lengemann, F.W. and R.A. Wentworth. The transfer coefficient
of 137-Cs into cows milk as related to the level of milk
production. Health Phys. 34: 720-722 (1978).
L3 Lidén, K. The metabolism of caesium. p. 33 in Assessment of
Radioactivity in Man. Proceedings of a symposium, Heidelberg,
May 1964. International Atomic Energy Agency publication
STI/PUB/84/Vol. 2. Vienna, 1964.
L4 Lidén, K. and M. Gustafsson. Relationship and seasonal
variation of 137-Cs in lichen, reindeer and man in northern
Sweden, 1961 to 1965. University of Helsinki, Department of
Radiochemistry annual report 1965. Helsinki, 1966.
L5 Lindell, B. and A. Magi. The occurrence of 137-Cs in Swedish
food, especially dairy milk and in the human body after the
nuclear test explosions in 1961 and 1962. Ark. Fys. 29: 69-96
(1965).
L6 Lloyd, R.D. Caesium-137 half-times in humans. Health Phys.
25: 605 (1973).
L7 Lloyd, R.D., W.S. Zundel, C.W. Mays et al. Short caesium
half-times in patients with muscular dystrophy. Nature 220:
1029-1031 (1968).
M1 Marei, A.N., R.M. Barkhudarov, N.J. Novikova et al. Effect of
natural factors on caesium-137 accumulation in the bodies of
residents in some geographical regions. Health Phys. 22: 9-15
(1972).
M2 Marei, A.N., R.M. Barkhudarov and N.J. Novikova. Radiation
safety aspects of the geochemical areas with increased transfer
of caesium-137. Manuscript No. 0020-R-4.
M3 Menzel, R.G. Soil-plant relationships of radioactive elements.
Health Phys. 11: 1325-1332 (1965).
M4 Miettinen, J.K. and E. Hasanen. 137-Cs in Finnish Lapps and
other Finns in 1962-1966. p. 221-231 in Radioecological
Concentration Processes (B. Aberga and F. Hungate, eds.).
Pergamon Press, 1967.
M5 Moiseev, A.A. and P.U. Ramzaev. Caesium-137 in the biosphere.
Atomizdat, Moscow, 1975 (in Russian).
N1 National Council on Radiation Protection and Measurements.
Caesium-137 from the environment to man: metabolism and dose.
National Council on Radiation Protection and Measurements
report No. 52. Washington D.C., 1977.
N2 National Council on Radiation Protection and Measuremmnts. A
handbook on Radioactivity measurements procedures. National
Council on Radiation Protection report No. 58. Washington,
D.C., 1978.
N3 Naversten, Y. and K. Lidén. Half-life studies of radiocaesium
in humans. p. 79-87 in Assessment of the Radioactive Body
Burdens in Man. Proceedings of a symposium, Heidelberg, May
1964. International Atomic Energy Agency publication
STI/PUB/84/Vol. 2. Vienna, 1964.
N4 Nay, U., W. Stahlhofen and A. Kaul. Distribution of caesium-
137 in samples consisting of soft tissues, bone and bone
marrow. p. 49-60 in Assessment of Radioactivity in Man.
Proceedings of a symposium, Heidelberg, May 1964.
International Atomic Energy Agency publication STI/PUB/84/Vol.2
Vienna, 1964.
N5 Nishita, H., E.M. Romney and K.H. Larson. Uptake of
radioactive fission products by crop plants. J. Agric. Food
Chem. 9: 101-106 (1961).
O1 Oak Ridge National Laboratory. Siting of fuel reprocessing
plants and waste management facilities. Oak Ridge National
Laboratory report ORNL-4451 (1970).
P1 Pendleton, R.C., C.W. Mays, R.D. Lloyd et al. A trophic level
effect on 137-Cs concentration. Health Phys. 11: 1503 (1965).
P2 Pelletier, C.A. and P.G. Voilleque. The behaviour of 137-
caesium and other fallout radionuclides on a Michigan
dairy farm. Health Phys. 21: 777-792 (1971).
R1 Richmond, C.R., J.E. Furchner and W.H. Langham. Long-term
retention of radio-caesium by man. Health Phys. 8: 201-205
(1962).
R2 Rosoff, B., S.H. Cohn and H. Spencer. Caesium-137 metabolism
in man. Radiat. Res. 19: 643-654 (1963).
R3 Rundo, J., J.I. Mason, D. Newton et al. Biological half-life
of caesium in man in acute and chronic exposure. Nature 200:
188-189 (1963).
R4 Rundo, J. The metabolism of biologically important
radionuclides. VI. A survey of the metabolism of caesium in
man. Br. J. Radiol. 37: 108 (1964).
S1 Sherill, R.D., N.G. Sumerlin, J.N. Beck et al. Variation of
the ratio of caesium-137 to strontium-90 in the atmosphere.
Health Phys. 28: 335-340 (1975).
S2 Spiers, F.W. Radioisotopes in the human body: physical and
biological aspects. Academic Press, New York, 1968.
S3 Stara, J.F. Tissue distribution and excretion of caesium-137
in the guinea pig after administration by three different
routes. Health Phys. 11: 1195-1202 (1965).
S4 Stewart, H.F., G.W. Ward and J.E. Johnson. Availability of
fallout 137-Cs to dairy cattle from different types of feed.
J. Dairy Sci. 48: 709 (1965).
T1 Tamura, T. and D.G. Jacobs. Structural implicatioms in caesium
sorption. Health Phys. 2: 391-398 (1960).
T2 Taylor, S.R. Abundance of chemical elements in the continental
crust: a new table. Geochim. Cosmochim. Acta 28: 1273-1285
(1964).
U1 Ueda, T., Y. Suzuki and R. Nakamura. Transfer of 137-Cs and
90-Sr from the environment to the Japanese population via the
marine environment. p. 501-511 in Dose Evaluation and
Standards for Man and His Environment. International Atomic
Energy Agency publication STI/PUB/375. Vienna, 1974.
U2 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Thirteenth Session, Supplement No.
17(A/3838). New York, 1958.
U3 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Seventeenth Session, Supplement No.
16(A/5216). New York, 1962.
U4 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Nineteenth Session, Supplement No.
14(A/5814). New York, 1964.
U5 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-first Session, Supplement No.
14(A/6314). New York, 1966.
U6 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation. Official Records
of the General Assembly, Twenty-fourth Session, Supplement No.
13(A/7613). New York, 1969.
U7 United Nations. Report of the United Nations Scientific
Committee on the Effects of Atomic Radiation to the General
Assembly, with annexes Volume I: Levels, Volume II: Effects.
United Nations sales publication No.E.72.IX.17 and 18. New
York, 1972.
U8 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No.E.77.IX.I. New York 1977.
V1 Vanderploeg, H.A., D.C. Parzyck, W.H. Wilcox et al.
Bioaccumulation factors for radionuclides in freshwater biota.
Oak Ridge National Laboratory report 0RNL-5002 (1975).
Y2 Yamagata, N. and T. Yamagata. The concentration of 137-Cs in
human tissues and organs. Bull. Inst. Publ. Health 9: 72-78
(1960).
Z1 Zundel, W.S., F.H. Tyler, C.W. Mays et al. Short half-times of
caesium-137 in pregnant women. Nature 221:89 (1969).
VIII. RADON
A. INTRODUCTION
337. Radon is element number 86, but the term generally refers to
the isotope 222Rn, the decay product of 226Ra. Other isotopes of
radon, 220Rn and 219Rn, are generally referred to by their
historical names, thoron and actinon, respectively. Radon has a
half-life of 3.82 days and decays by alpha-particle emission to a
polonium isotope (218Po), which by further decay through isotopes
of lead, bismuth, polonium and thallium ends the uranium (238U)
decay chain with stable lead (206Pb).
338. Radon is a chemically inert gas. It arises from decay of
radium which occurs in soil and other common materials. Part of the
radon produced diffuses into the surrounding environment.
339. The short-lived decay products of radon, called radon
daughters, are 218Po (3.05 min), 214Pb (26.8 min), 214Bi (19.7 min)
and 214Po (1.6 10-4s). They become largely attached to aerosols in
air and if inhaled they are partly deposited in the human
respiratory tract. The radiation doses caused by inhalation of
radon daughters in air constitute the main part of the natural
radiation dose to man. The radiation dose caused by radon itself
is minor by comparison with that of radon daughters.
340. The levels of radon and radon daughters in air depend on the
source and on the dilution in the air. The levels are normally
higher indoors than outdoors. Reduced ventilation may cause radon
released from building materials to build up in enclosed spaces.
The radon levels may be very high in underground mines,
particularly in uranium mines.
B. SOURCES
1. Outdoors
(a) Natural radon
341. The main source of radon outdoors is radium in the earth's
crust. The concentration of uranium and radium in the ground
varies with the types of rocks and minerals. The concentration of
radium in rocks and soil is often (but not always) the same as that
of uranium. Fractionated dissolution and transport of uranium-234
and/or radium can cause breaks in the uranium chain [S2].
342. The total amount of radium in the outer 10 km of the earth's
crust is of the order of 1024 Bq. Most of the radon produced by
decay of radium is physically attached to the radium-bearing
material and only a small part diffuses out into the air. Other
relatively less important sources of radon in air outdoors are
plants, ground water, oceans, etc. The sources contributing to the
total amount of radon are given in Table VIII.1. It is assumed
that the radon exhalation rate from the land (soil) is 0.02 Bq m-2
s-1 and from the oceans is 70 µBq m-2 s-1. The total production
rate of radon is of the order of 1020 Bq a-1; the equilibrium
inventory in the atmosphere, determined from the total production
rate divided by the decay constant (66.2 a-1) is estimated to be
1.5 1018 Bq.
Table VIII.1 Sources of radon in the global
atmosphere [H5]
---------------------------------------------
Source Radon production
per year (Bq)
---------------------------------------------
Soil 9 1019
Plants and ground water < 2 1019
Oceans 9 1017
Houses a/ 3 1016
Natural gas 3 1014
Coal 2 1013
---------------------------------------------
a/ The value for houses is estimated in this
document assuming 109 reference houses
(see Table VIII.3). The true value may be
between 5 1015-1017 Bq a-1.
(b) Mines and mine tailings
343. Sources of radon of local interest include the tailings from
uranium and phosphate mining and milling and from geothermal power
stations. The radon exhalation rate from tailings depends on the
radium content of the tailings, on the emanation factor (fractional
release of radon) and on the land reclamation (overburden). The
radon exhalation rate from uncovered uranium tailings varies from
0.5 Bq m-2 s-1 or less to 10 Bq m-2 s-1 or more [S9, U4]. The
thickness and area of the tailings per unit mass of uranium in the
ore can also vary depending on the tailings engineering and
therefore the radon exhalation rate varies in the range of about
10-100 Bq s-1 per MW(e)a [S9, U4]. By covering the tailings with a
few meters of soil the radon exhalation rate is reduced by one to
several orders of magnitude. The radon exhalation from tailings of
phosphate mining and milling also depends on land reclamation and
the type of ore. A range of exhalation rates of about 0.01-1 Bq
m-2 s-1 has been reported [R1]. Geothermal power stations may cause
radon releases into the air by releases of radon from the water
which is depressurized at the surface. A radon release of the
order of 1011 Bq per MW(e)a has been reported [M3]. The radon
release from coal-fired plants is 3 to 4 orders of magnitude less.
344. The sources of radon in underground spaces like mines are
radium in the rock and minerals of the mine and radon in water.
The total release of radon into the mine depends on many factors:
the uranium-radium concentration of the ore; the number and size of
cracks in the ore; the isolation of abandoned spaces; the random
concentration and amount of water; the isolation of water; and the
ventilation system principles. By using a normal range of total
ventilation rates (10-1000 m3 s-1) and a normal radon concentration
(0.1-10 kBq m-3) the range of radon release into the mine may be
estimated at 1-104 kBq s-1. Since there is normally an inverse
proportionality between radon concentration and ventilation rate,
lower ventilation rates are more often related to higher radon
concentrations and vice versa. There is also a radon release from
the mine to outside air and it can be compared with the radon
release from uncovered tailings. Mining of 100 tonnes uranium per
year results in 5000-15000 m2 of tailings per year depending on the
percentage of uranium in the ore and the tailings engineering. The
radon release will be in the range of 1-10 kBq s-1. In non-uranium
mines radon-rich water is often a significant source of radon to
the mine [S5]. The radon concentration of the water may be in the
range of 100-1000 kBq m-3.
2. Indoors
(a) Building materials
345. Radon in houses comes from building materials, the soil under
the house, the water and the domestic gas. Radium concentrations
in building materials have been investigated. The data indicate
that some materials such as aerated concrete with alum shale and
phospho-gypsum from sedimentary ores have significantly higher
radium concentrations than others and cause enhanced radon
concentrations indoors. If these materials are excluded, the
average concentrations of radium in building material is about 100
Bq kg-1. Materials with low activity are wood, natural gypsum,
sand and gravel. The radon exhalation rate from walls, floors and
ceilings is dependent on the radium concentration, the emanation
power, the diffusion coefficient in the material and the qualities
and thickness of any applied sealant on the surfaces.
346. The radon exhalation rate from uncovered building materials
varies by several orders of magnitude from about 10-6 Bq m-2 s-1
(gypsumboard, fiberboard, chipboard, bricks) to (0.1-10) 10-3 Bq
m-2 s-1 for concrete of different origins and qualities [J5, P3,
S17, M8, W3]. The radon exhalation rate per Bq Ra/kg varies less,
e.g., (1.6 ± 0.88) 10-5 Bq m-2 s-1 per Bq Ra/kg for some building
materials in the Federal Republic of Germany [W3]. If the same
values are normalized to an emanating power of 1%, the radon
exhalation rate is (4.4 ± 1.9) 10-6 Bq m-2 s-1 per Bq Ra/kg per
percentage emanating power. In the first case the standard
deviation is 54% and in the last case it is 43%. By painting,
plastering or application of wall-paper on the wall the radon
exhalation may be reduced by less than a factor of 5 [W3, M8].
(b) Soil
347. The contribution of radon from the soil into a building
depends on the thickness and tightness of the base structure. The
exhalation from the soil is of the order of 10-2 Bq m-2 s-1 and a
concrete floor in cellars should normally reduce the radon
exhalation from soil into the building by a factor of 10 or more.
Even so, radon from soil may contribute significantly to the radon
concentrations in a house, particularly in the cellar and in wooden
houses. In some areas houses are built on natural uranium deposits
(Canada) [L2, K2], on phosphate-related land (Florida, U.S.A.) [U3,
U13], on waste products from uranium industry (Colorado, U.S.A.)
[C6], and on waste products from alum production from radium-rich
alum shales (Sweden) [S22]. In these cases the radon exhalation
rate may be several orders of magnitude higher than from normal
soil.
(c) Water
348. Another source of radon in a house may be radon-rich water.
The relative radon release depends on the use of water. Boiling
and splashing of the water increase releases and consequently the
highest radon releases occur in washrooms, at shower-baths and in
the kitchen during cooking. The resultant radon concentration in a
house depends on the amount of water used, the volume of the house
and the ventilation. Several studies have been made to estimate
the relative significance of radon from water [P1, C2, N1], and a
typical value of the air-to-water radon concentration quotient is
about 10-4. Measurements of radon in water are most often made in
areas with suspected high concentrations because of uranium
deposits and estimates of the weighted average radon concentration
in water for a country are rare.
349. Population-weighted average concentrations of radon in
drinking water have been estimated for a few countries and are
found to be 40 kBq m-3 in Finland [A5, C3], 7 kBq m-3 in Sweden
[S8] and 0.4-4 kBq m-3 in the Federal Republic of Germany [M9].
The corresponding radon release into a house can be estimated by
assuming a daily use of 500 1 per person and 10-100% relative
release from the water.
(d) Natural gas
350. Natural gas containing radon may also be a source of radon in
houses. Gas is transported as purified gas in long transmission
lines and distributed to the homes or bottled under pressure as
propane for sale as liquified petroleum gas (LPG). The radon
concentrations in natural gas at the production wells are found to
vary from undetectable values up to about 40 kBq m-3 [U12, H6].
During supply, transit, storage and delivery the radon
concentration decreases to an approximate average of the order of 1
kBq m-3 for both natural gas and LPG (in U.S.A.) [U12, B1, G1].
(e) Summary
351. The relative contribution of different radon sources to the
total radon input in a house is estimated in Table VIII.2 with some
typical values of radon concentration and releases. The radon in
outside air is brought into the house by ventilation. The volume
of the house is assumed to be 200 m3 and the inner surface area 350
m2.
Table VIII.2 The relative significance of different radon sources
in a reference house
----------------------------------------------------------------------
Source Radon flux Comments
(Bq d-1)
----------------------------------------------------------------------
Building material 70 103 Emanation rate 2 10-3 Bq m-2 s-1
Water 4 103 1000 1 d-1 and 4 kBq m-3, 100% release
Outside air 9 103 Radon concentration outdoors 0.004 kBq
m-3; ventilation rate 0.5 per hour
Natural gas 3 103
Liquified 0.2 103
petroleum gas
----------------------------------------------------------------------
C. BEHAVIOUR IN THE ENVIRONMENT
1. Release from soil
(a) Emanation
352. The mechanism of radon release from rock, soil and other
materials is not very well understood and is probably not always
the same. The main physical phenomena are recoil and diffusion of
the radon atom through imperfections of the crystalline structures
of the radium-bearing particle followed by a secondary diffusion,
which depends on the porosity of the material [A1]. High porosity
increases the diffusion rate. The release rate from a material
depends also on its moisture content: if the moisture content is
very low the radon release is decreased by the effect of
re-adsorption of radon atoms on surfaces in the pores. If the
moisture content increases slightly, the radon release increases up
to a certain moisture content, above which the release of radon
decreases again owing to a decreasing diffusion rate in water-
filled pores [M7].
(b) Diffusion
353. Once radon has entered the air or water surrounding the
emanating radium-bearing particle, it is transported by diffusion,
earth-mechanical and convective flow, percolation of rain water and
flow of ground water. The diffusion mechanism can be expressed by
the equation
Cx = Co exp [-x/ ´(D/lambda)]
where Cx is the radon concentration at distance x in air or water
from the emanating surface; Co is the radon concentration at the
surface; D is the diffusion coefficient (gas kinetic) and lambda is
the decay constant [G2]. The diffusion constant D is about 10-2
cm2 s-1 in air and 10-5 cm2 s-1 in water. This means that it takes
on the average about 13 days for a radon atom to diffuse 5 m in air
or 5 cm in water. In that time the radon would decay by almost a
factor of 10. Accordingly, long distance transport of radon in air
and water mainly depends on the other mechanisms mentioned above,
which are the transport of air and water itself.
(c) Exhalation
354. The radon concentration Cz in soil air at depth z below the
surface depends on the diffusion coefficient D, the emanating
factor a (0 < a < 1), the fractional pore space of the soil f, the
radium activity concentration Cr (per unit volume of soil) and the
decay constant of radon g according to the equation [J1]
a x Cr
Cz = ------ [1 - exp (- ´(lambda/D) z]
f
The exhalation rate is expressed by the equation [J1]
d(Cz)
R = D [-----]
dz z=o
The combination of the above two equations gives
lambda a Cr
R = ----------- ´(D/lambda)
f
If a = 0.1; Cr = 0.07 Bq cm-3; f = 0.3 and D = 0.01 cm2 s-1;
lambda = 2 10-6 s-1, the exhalation rate R is 3 10-2 Bq m-2 s-1.
355. The diffusion rate and thereby the exhalation rate is
influenced by meteorological factors such as rainfall, snowfall,
freezing and variations in atmospheric pressure. An increase in
these parameters will decrease the exhalation rate. Measured
values of radon exhalation rate from soil vary between about 0.0002
and 0.07 Bq m-2 s-1 [G2, W1]. The radon exhalation from sea water
per unit area and time is about two orders of magnitude less.
2. Dispersion in air
356. The dispersion of radon in air is influenced by the vertical
temperature gradient, the direction and strength of the wind and
the air turbulance. The dispersion of radon daughters is also
influenced by precipitation. The vertical distribution of radon
and its daughters in air can be calculated from the following
system of differential equations [J2]:
d C1
--- (K --) - lambda1 C1 = 0
dz dz
d C1
--- (K --) + lambdai-1 Ci-1 - (lambdai + LAMBDA) Ci = 0
dz dz
where C1 is the concentration of radon atoms in air at the height
z; ni is the concentration of radon daughter i in air at the
height z; lambda1 is the decay constant of radon; lambdai is the
decay constant of radon daughter i; LAMBDA is the removal rate of
radon daughters caused by washout and rainout. Boundary conditions
to the above equations are
Ci (z=0) = 0 for i > 1 and Ci(z->infinite) = 0 for i = 1,2,3, ...
By assuming a constant radon exhalation from an infinite plane
(ground surface) which equals the radioactive decay of the total
radon content in the atmosphere it is possible to solve the first
two equations in this paragraph, which in combination with
different values of the turbulent diffusion coefficient K [J1, J2]
give the vertical distribution of radon and radon daughters for
different atmospheric stabilities.
357. Measured values of the relative distribution of radon in air
are shown in Figure VIII.I. Although measured values in this case
are found to follow the predicted vertical distribution fairly
well, the models described above should only be taken to serve as
rough guidance for the prediction of radon daughter levels. The
varying radon exhalation rate on land and on sea and varying
meteorological conditions may cause distribution patterns different
from those predicted by the model.
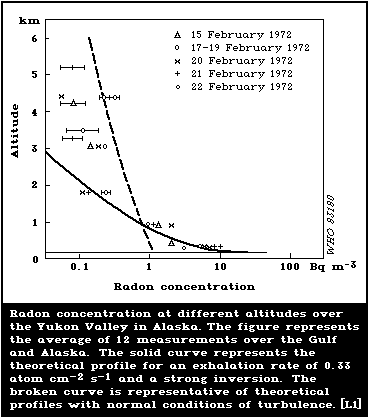 358. At ground level the time-variation of radon concentrations
depends on the variation of the radon exhalation rate and of the
vertical dispersion of radon. The effect of increased vertical
dispersion of radon by turbulence during spring, as compared with
autumn, outweighs the greater exhalation rate of radon during late
spring and summer, as compared with autumm and winter. The overall
effect is a seasonal variation of the radon concentration at ground
level with a minimum in the spring and summer and a maximum in the
autumn and winter observed in several measurements [M1, B2, R2,
Me]. Diurnal variations of the radon concentration in air
at ground level also occur because of different varying turbulent
mixing: the concentrations are maximum in the early morning and
minimum in the afternoon. The variations are generally less than
one order of magnitude [R2, J3].
359. For estimation of the dispersion of radon released from a
point (for instance, a geothermal plant or a mine ventilation
outlet) the most commonly used statistical model is the Gaussian
plume equation [S18, P2, G4]. The estimated concentrations at
different distances are therefore dependent on local meteorological
conditions, terrain roughness, etc. However, in the case of a
continuous release, the daily variations are smoothed out and an
annual average is obtained, which differs from place to place only
according to persistent and substantial local differences.
360. The dispersion of radon released from extended sources like
mill tailings can be estimated from dispersion formulas, assuming
the extended source to consist of a number of small point sources.
The relative concentration of radon released from a point source is
approximately inversely proportional to the p-power of the distance
d from the source. If the concentration Cd at distance d is
expressed relative to the concentration C1 at the reference
distance d1 the expression is
d -p
Cd = (--) C1
d1
The formula approximately gives the relative concentration at
distances more than 1 km if the reference distance d1 = 1km and
p = 1.2 - 1.5.
361. The dispersion and relative vertical distribution of the
radon daughters in air mainly follow the behaviour of radon. Owing
to deviating atmospheric parameters for radon daughters as compared
with radon (e.g., precipitation by rainout and washout) there is
seldom equilibrium between radon daughters and radon and between
the different radon daughters. The long-lived decay products of
radon (210Pb, 210Bi and 210Po) behave in the troposphere as
aerosols with residence times of the order of ten days and more.
Because of their long physical half-life there is no simple
correlation between these nuclides and radon.
3. Indoor behaviour
362. For closed spaces (e.g., a mine or a house) a theoretical
correlation may be established between radon concentration in air
and radon input (exhalation and transport by inlet air) and
ventilation rate. The change of the radon concentration in the
enclosure is given by the following equation
d C(t) = R (S) + Ak + Colambdanu - C(t) (lambda + lambdanu)
dt V V
where C(t) is the radon concentration in the air at the time t; R
is the radon exhalation rate from unit surface in the room; S is
the emanating surface area; V is the volume of the space, Ak is
the radon release from the source (water, gas); Co is the radon
concentration in the inlet air; lambdanu is the ventilation rate
(h-1); and lambda is the decay constant of radon.
363. At equilibrium the radon concentration in the enclosure is
R(S) + Ak + Co lambdanu
C = V V
lambda + lambdanu
In homes 0.1 < lambdanu < 3 h-1, and since lambda = 7.6 10-3
h-1 and lambdanu >> lambda, the above equation takes the form
R(S) + Ak
C = V V + Co
lambdanu
As long as lambdanu >> lambda and Co is negligible, the radon
concentration indoors increases in direct proportion to the
decrease in ventilation rate. As the ventilation rate increases
from 0 to 0.1 and to 1 h-1, the radon concentration decreases by
factors of 13 and 10, respectively.
364. In view of the strong influence of the ventilation rate,
there are great variations of the radon levels as the effective
ventilation of a room is changed. This is caused by meteorological
conditions (wind, pressure, temperature) and by human activities
like opening doors and windows. There may be variations of the
radon concentration in air caused by changes of the radon
exhalation rate from surfaces, which in turn can be caused by
changes of atmospheric pressure [J6]. Diurnal variation in houses
have been studied in several long-term measurements [S11, D1, S21,
H4, J7, S12, M6] and variations of the order of ten and more may
occur. Maxima during the night and early morning and minima at
noon are usually found, but for several reasons that is not always
the case. Only a few studies have been reported on the seasonal
variations of radon concentration indoors. The variations of the
monthly averages are found to be less than a factor 3 [F1, S13].
Examples of measured variations of radon concentration in houses
are shown in Figure VIII.II [M10].
358. At ground level the time-variation of radon concentrations
depends on the variation of the radon exhalation rate and of the
vertical dispersion of radon. The effect of increased vertical
dispersion of radon by turbulence during spring, as compared with
autumn, outweighs the greater exhalation rate of radon during late
spring and summer, as compared with autumm and winter. The overall
effect is a seasonal variation of the radon concentration at ground
level with a minimum in the spring and summer and a maximum in the
autumn and winter observed in several measurements [M1, B2, R2,
Me]. Diurnal variations of the radon concentration in air
at ground level also occur because of different varying turbulent
mixing: the concentrations are maximum in the early morning and
minimum in the afternoon. The variations are generally less than
one order of magnitude [R2, J3].
359. For estimation of the dispersion of radon released from a
point (for instance, a geothermal plant or a mine ventilation
outlet) the most commonly used statistical model is the Gaussian
plume equation [S18, P2, G4]. The estimated concentrations at
different distances are therefore dependent on local meteorological
conditions, terrain roughness, etc. However, in the case of a
continuous release, the daily variations are smoothed out and an
annual average is obtained, which differs from place to place only
according to persistent and substantial local differences.
360. The dispersion of radon released from extended sources like
mill tailings can be estimated from dispersion formulas, assuming
the extended source to consist of a number of small point sources.
The relative concentration of radon released from a point source is
approximately inversely proportional to the p-power of the distance
d from the source. If the concentration Cd at distance d is
expressed relative to the concentration C1 at the reference
distance d1 the expression is
d -p
Cd = (--) C1
d1
The formula approximately gives the relative concentration at
distances more than 1 km if the reference distance d1 = 1km and
p = 1.2 - 1.5.
361. The dispersion and relative vertical distribution of the
radon daughters in air mainly follow the behaviour of radon. Owing
to deviating atmospheric parameters for radon daughters as compared
with radon (e.g., precipitation by rainout and washout) there is
seldom equilibrium between radon daughters and radon and between
the different radon daughters. The long-lived decay products of
radon (210Pb, 210Bi and 210Po) behave in the troposphere as
aerosols with residence times of the order of ten days and more.
Because of their long physical half-life there is no simple
correlation between these nuclides and radon.
3. Indoor behaviour
362. For closed spaces (e.g., a mine or a house) a theoretical
correlation may be established between radon concentration in air
and radon input (exhalation and transport by inlet air) and
ventilation rate. The change of the radon concentration in the
enclosure is given by the following equation
d C(t) = R (S) + Ak + Colambdanu - C(t) (lambda + lambdanu)
dt V V
where C(t) is the radon concentration in the air at the time t; R
is the radon exhalation rate from unit surface in the room; S is
the emanating surface area; V is the volume of the space, Ak is
the radon release from the source (water, gas); Co is the radon
concentration in the inlet air; lambdanu is the ventilation rate
(h-1); and lambda is the decay constant of radon.
363. At equilibrium the radon concentration in the enclosure is
R(S) + Ak + Co lambdanu
C = V V
lambda + lambdanu
In homes 0.1 < lambdanu < 3 h-1, and since lambda = 7.6 10-3
h-1 and lambdanu >> lambda, the above equation takes the form
R(S) + Ak
C = V V + Co
lambdanu
As long as lambdanu >> lambda and Co is negligible, the radon
concentration indoors increases in direct proportion to the
decrease in ventilation rate. As the ventilation rate increases
from 0 to 0.1 and to 1 h-1, the radon concentration decreases by
factors of 13 and 10, respectively.
364. In view of the strong influence of the ventilation rate,
there are great variations of the radon levels as the effective
ventilation of a room is changed. This is caused by meteorological
conditions (wind, pressure, temperature) and by human activities
like opening doors and windows. There may be variations of the
radon concentration in air caused by changes of the radon
exhalation rate from surfaces, which in turn can be caused by
changes of atmospheric pressure [J6]. Diurnal variation in houses
have been studied in several long-term measurements [S11, D1, S21,
H4, J7, S12, M6] and variations of the order of ten and more may
occur. Maxima during the night and early morning and minima at
noon are usually found, but for several reasons that is not always
the case. Only a few studies have been reported on the seasonal
variations of radon concentration indoors. The variations of the
monthly averages are found to be less than a factor 3 [F1, S13].
Examples of measured variations of radon concentration in houses
are shown in Figure VIII.II [M10].
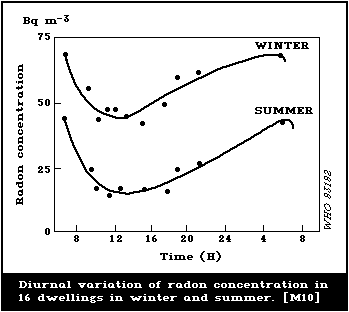 365. In mines and other underground spaces there are also diurnal
and seasonal variations of the radon concentration. The diurnal
variations are most often minor if the ventilation is unchanged by
the seasonal variations may be large with maxima during the summer
and minima during the winter. This is caused by the change from
winter to summer in the temperature gradient from outside to inside
the mine (Figure VIII.III).
365. In mines and other underground spaces there are also diurnal
and seasonal variations of the radon concentration. The diurnal
variations are most often minor if the ventilation is unchanged by
the seasonal variations may be large with maxima during the summer
and minima during the winter. This is caused by the change from
winter to summer in the temperature gradient from outside to inside
the mine (Figure VIII.III).
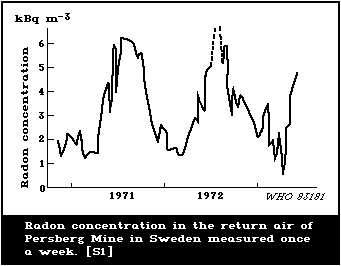 4. Radon daughter concentrations
(a) Concentration expression
366. The concentration of radon daughters can be expressed in
terms of their activity or of their potential alpha energy, the
latter being the total alpha energy emitted during the decay of the
atoms present down to 210Pb. For any mixture of radon daughters in
air the potential alpha energy is the sum of the potential alpha
energy of all daughter atoms in the air. A unit of exposure which
is used in mines is the working level (WL). It is defined as any
combination of short-lived radon daughters per litre of air that
will result in the emission of 1.3 105 MeV of alpha energy in their
decay to 210Pb.
367. Another quantity of interest in connection with radon
daughters is the equilibrium factor F defined as the ratio of the
total potential alpha energy for the given daughter concentration
to the total potential alpha energy of the daughters if they are in
equilibrium with radon. If the unit WL is used, the equilibrium
factor F can be calculated as
a Calpha
F = ---------
C
where Calpha is the potential alpha-energy concentration in WL of
radon daughters; C is the radon activity concentration in Bq 1-1;
and a is a constant (a = 3.7 Bq 1-1/WL).
368. For a room having a known ventilation rate lambdanu (air
changes per hour) it is possible to calculate the equilibrium
factor F. The relationship between F and lambdanu is shown in
Figure VIII.IV.
4. Radon daughter concentrations
(a) Concentration expression
366. The concentration of radon daughters can be expressed in
terms of their activity or of their potential alpha energy, the
latter being the total alpha energy emitted during the decay of the
atoms present down to 210Pb. For any mixture of radon daughters in
air the potential alpha energy is the sum of the potential alpha
energy of all daughter atoms in the air. A unit of exposure which
is used in mines is the working level (WL). It is defined as any
combination of short-lived radon daughters per litre of air that
will result in the emission of 1.3 105 MeV of alpha energy in their
decay to 210Pb.
367. Another quantity of interest in connection with radon
daughters is the equilibrium factor F defined as the ratio of the
total potential alpha energy for the given daughter concentration
to the total potential alpha energy of the daughters if they are in
equilibrium with radon. If the unit WL is used, the equilibrium
factor F can be calculated as
a Calpha
F = ---------
C
where Calpha is the potential alpha-energy concentration in WL of
radon daughters; C is the radon activity concentration in Bq 1-1;
and a is a constant (a = 3.7 Bq 1-1/WL).
368. For a room having a known ventilation rate lambdanu (air
changes per hour) it is possible to calculate the equilibrium
factor F. The relationship between F and lambdanu is shown in
Figure VIII.IV.
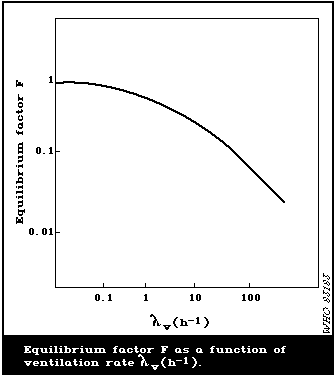 369. The product C x F, where C is the radon concentration and F
is the equilibrium factor is called the equilibrium equivalent
concentration of radon (EEC); it corresponds to a concentration of
radon for which the radon daughters in equilibrium with radon have
the same potential alpha energy as the actual daughter
concentration of interest.
(b) Attachment
370. The radon daughters in air may be unattached (free atoms or
ions) or attached to aerosols. The first daughter, 218Po, is at
the time of formation an unattached ion or neutral atom. But
within a few seconds most of the 218Po becomes attached to an
aerosol and the subsequent decay products 214Po and 214Bi are
therefore to a great extent attached to aerosols at their
formation.
371. The attachment rate of a free radon daughter depends
therefore on the number and size distribution of the aerosols in
the air. These parameters vary in different rooms and will thus
affect the attachment rate. In a house with normal aerosol
concentration (ca. 104 cm-3) and size distribution, the attachment
rate will be about 10-2 s-1, i.e., the mean life of the free radon
daughter will be about 100 s. In a mine with higher aerosol
concentration the corresponding values may be about 0.3 s-1 and
4 s, respectively.
372. Radon daughters in room air will also attach to the surfaces
in the room. The deposition rate for radon daughters attached to
aerosols is dependent on the diffusion rate of the aerosols and the
proportion between the surface area and the volume of the room. If
that proportion is 2 m-1, the mean life of the attached radon
daughters (as far as deposition is concerned) is of the order of
one hour. Unattached radon daughters have much higher diffusion
rate than aerosols and therefore the deposition rate is also
higher. The corresponding mean life is of the order of one minute.
373. The fraction of unattached radon daughters in room air is
also dependent on radioactive decay and ventilation rate. With
given values of the attachment rate of free atoms and deposition
rate of unattached atoms, the fraction of unattached daughters in
air increases with increasing decay constant and ventilation rate.
This means that the fraction of unattached 218Po atoms (lambda =
13.6 h-1) is normally higher than, e.g., that of 214Pb (lambda =
1.6 h-1). Measured values are in the range of 1-30%.
(c) Equilibrium variations
374. Ventilation and deposition to surfaces both prevent the radon
daughters reaching equilibrium with radon in air. Only the
dependence of the equilibrium factor on the ventilation rate was
considered in Figure VIII.IV. However, because of deposition,
measured values of the factor F are often lower than the predicted
value from Figure VIII.IV. The deviation is larger in air with low
aerosol concentrations. An approximate expected value of F is
obtained by multiplying the value in Figure VIII.IV by 0.5.
375. Measured values of F in houses show great variation mainly
due to differing ventilation conditions. In the UNSCEAR 1977
report [U12] an average value of F for houses of 0.5 was adopted.
In outdoor air the equilibrium factor is also dependent on
meteorological factors. Measured values indicate an average value
of 0.6, which was used by UNSCEAR in its 1977 report. For uranium
mines with good ventilation a factor of 0.3 may be appropriate.
D. TRANSFER TO MAN
376. The transfer to man of radon and radon daughters occurs from
the inhalation of air. A negligible amount arises from decay of
radium in ingested food and water. From a dose standpoint, it is
most important to know the intake amount of radon daughters in air.
377. The amount of radon daughters inhaled depends upon the
concentration in air and on the breathing rate. The breathing rate
varies with different levels of physical activity and age. For the
adult, the average breathing rates are 20 1 min-1 during light
activity, 7.5 1 min-1 resting and 12.5 1 min-1 for intermediate
activity [I1].
378. To compute the average air intake rate, it will be assumed
that the time spent indoors per day (19 h) consists of 5.5 h light
activity, 8 h resting and 5.5 h intermediate activity. The time
spent outdoors (5 h) is assumed to consist of 2 h light activity
and 3 h intermediate activity. This gives estimated intake rates
of approximately 15 m3 d-1 indoors and 5 m3 d-1 outdoors.
379. The deposition of radon daughters in the respiratory system
depends on the size distribution of the aerosols to which they are
attached and on the fraction of unattached radon daughters. The
deposition is also influenced by the manner of breathing. The
attached radon daughters are deposited in the pulmonary region.
However, the deposition is not 100 per cent. Some is exhaled and
some is transported by mucus before decay. An approximate value of
the fractional retention may be about 50% [U12] but great
variations have been reported.
380. The unattached radon daughters are mainly deposited in the
upper respiratory tract. The efficient deposition of unattached
daughters has been experimentally verified in a model lung by
Chamberlain and Dyson [C4]. However, a major part of the
unattached daughters is removed by nasal deposition [U12].
E. DOSIMETRY
1. Dose per unit exposure
381. The doses of radon gas in air are negligible in comparison
with those of the daughters. On rare occasions, when there is a
great disequilibrium between radon and radon daughters, the
relative contribution from radon to effective dose equivalent could
be significant.
382. Inhalation of radon daughters leads to inhomogeneous
irradiation of the respiratory tract. The maximum dose is received
by the basal cells in the epithelium of the upper bronchi in the
tracheobronchial region of the lungs due to deposition of
unattached radon daughters. Normally, the fraction of unattached
radon daughters in air is small (a few per cent). Attached radon
daughters are mostly deposited in the pulmonary region.
383. Jacobi [J3] has calculated the doses from radon daughters to
the tracheobronchial region of the lung (assumed mass 45 g) and to
the pulmonary region (955 g) as a function of the unattached
fraction, f, of the potential alpha energy. The absorbed energy
fractions are 0.03 (1 + 6f) joule in the tracheobronchial region
per joule inhaled and 0.38 (1 - f) joule in the pulmonary region
per joule inhaled. Total energy absorbed in the lung, reflecting
deposition and clearance for an average aerosol size and breathing
rate is of the order of 40%.
384. The value of the unattached fraction, f, of radon daughters
in air is usually in the range 0.02 to 0.1 [U12]. For a mean value
of 0.06, the absorbed doses in the lungs are 0.9 Gy per joule
inhaled in the tracheobronchial region and 0.4 Gy per joule inhaled
in the pulmonary region.
385. The dose equivalent is obtained by multiplying by a quality
factor, which for alpha radiation is 20. The effective dose
equivalent is obtained by multiplying by the tissue weighting
factor, which for the lungs is 0.12. It might be appropriate to
apply a weighting factor of 0.06 to the dose equivalent to the
basal cell layer of the tracheobronchial region and 0.06 for the
pulmonary region. The contributions to the effective dose
equivalent to the lungs per unit energy inhaled are thus 1.1 Sv J-1
(tracheobronchial region) and 0.5 Sv J-1 (pulmonary region).
386. The dosimetry of radon daughters in the lungs is, at the
moment, under review. The comparisons of various dosimetric models
may lead to recommendations for adjustments of some of these
results. For this report a value of 1.6 Sv per joule inhaled will
be used for the total effective dose equivalent to the lungs,
applicable to exposures indoors and outdoors and also to
occupational exposures in mines.
387. The potential alpha energy of an atom in the decay chain of
radon is the total alpha energy emitted during decay of the atom up
to 210Pb. Dividing by the decay constant of the radionuclide gives
the potential alpha energy per unit of activity. The values for
radon and its short-lived daughters are given in Table VIII.3.
388. The measured concentration of radon in air must be multiplied
by the equilibrium concentration (EEC). For an EEC of 1 Bq m-3 and
a breathing rate of 20 m3 d-1 (7300 m3 a-1), the effective dose
equivalent rate is
Bq m3 J Sv
1 -- 7300 34500 MeV 1.6 0-13 --- 1.6 -- = 6 10-5 Sv a-1
m3 a MeV J
The effective dose equivalent per unit integrated concentration of
radon in air is 6 10-5 Sv per Bq a m-3.
Table VIII.3 Potential alpha energy of radon and
short-lived decay products
-----------------------------------------------------
Radionuclide Energy per atom Energy per unit
(MeV) activity (MeV Bq-1)
-----------------------------------------------------
222Rn 19.2 9.15 106
218Po 13.7 3620
214Pb 7.69 17800
214Bi 7.69 13100
214Po 7.69 0.002
-----------------------------------------------------
Total a/ 34500
-----------------------------------------------------
a/ The total is the sum of the potential energies
of the daughters only.
389. In terms of WLM for occupational exposures, the potential
alpha energy inhaled is calculated as follows:
1.3 105 MeV 1-1 1200 1 170 h 1.6 10-13 d
WL h M Mev
= 4.2 10-3 J (WLM)-1
where the breathing rate is 1200 1 h-1 (20 1 min-1) during an 8 h
working day. The effective dose equivalent per unit exposure is
4.2 10-3 d 1.6 Sv = 7 10-3 Sv (WLM)-1
WLM J
The results are summarized in Table VIII.4.
Table VIII.4 Effective dose equivalent per unit exposure from
short-lived radon decay products
----------------------------------------------------------------
Public (indoors and outdoors) 5.10-5 Sv (Bq a m-3)-1
Workers 7 10-3 Sv (WLM)-1
----------------------------------------------------------------
390. The effective dose equivalent caused by inhalation of radon
without daughters at a concentration of 1 Bq m-3 is about 7 10-7 Sv
a-1 [H8]. This is only about one per cent of the effective dose
equivalent caused by inhalation of radon daughters in equilibrium
with radon.
391. Radon in water may cause a radiation dose to man by ingestion
of water and by inhalation of the radon daughters produced by decay
of the radon released to air. Consumption of 0.5 1/day of radon-
rich water with a radon concentration of 1 kBq 1-1 will lead to an
effective dose equivalent of 0.5 mSv a-1 (by ingestion) [S10].
392. The levels of radon have been measured repeatedly in
different places of the world. The levels found in outdoor air
vary between about 0.1 to 10 Bq m-3, lower values having been found
over oceans and islands, higher values over continents. For the
estimation of an average dose equivalent, a radon concentration in
air of 3.7 Bq m-3 might be used, corresponding to an equilibrium
equivalent concentration of 2.2 Bq m-3 (equilibrium factor = 0.6).
If it is assummed that people are outdoors 20% of the time as an
annual average (i.e., the occupancy factor is 0.2) the
corresponding average effective dose equivalent would be 1.5 10-2
mSv per year from radon daughters.
393. In houses the radon levels may be high because of radium-rich
building materials and/or poor ventilation. However, normal values
are of the order of 10 Bq m-3. An inhabitant-weighted average value
for countries in which comprehensive measurements have been carried
out is an equilibrium equivalent concentration of radon of 14 Bq-3.
Using the effective dose equivalent per unit exposure given in
Table VIII.4 for the public, 0.06 mSv (Bq a m-3)-1, and an
occupancy factor of 0.8, an equilibrium equivalent concentration
of 14 Bq m-3 will correspond to an effective dose equivalent of 0.7
mSv per year. A summary of the dose estimates for indoor and
outdoor exposures is given in Table VIII.5.
2. Dose per unit release
394. Estimates of the dose per unit release of radon are quite
variable, depending on the location of the release, the population
density, and the conditions of the dispersion. The most
generalized estimates of the collective effective dose equivalents
to the world population (4 109 person) may be made by combining
dose values of Table VIII.5 with the total amounts of radon
released of Table VIII.1. For a release of radon outdoors (1020 Bq
a-1 world-wide) the estimate is 1 10-15 man Sv per Bq, and for a
release indoors (3 1016 Bq a-1 world-wide) the result is 9 10-11
man Sv per Bq.
Table VIII.5 Average effective dose equivalent from inhalation
of radon daughters in air
----------------------------------------------------------------
Equilibrium Occupancy Dose factor Effective
equivalent factor (mSv per Bq dose
concentration a m-3) equivalent
(Bq m-3) (mSv a-1)
----------------------------------------------------------------
Outdoors 2.2 0.2 0.06 0.03
Indoors 14.0 0.8 0.06 0.7
----------------------------------------------------------------
395. Releases of radon from nuclear installations result from
mining and milling operations and from the residual tailings. These
generally occur in areas of low population density - from 3 man km-2
in mining areas to about 25 man km-2 around the mills [U12]. The
local collective dose per unit release can be estimated by an
integration over the distance from 1 to 100 km, assuming an
atmospheric dispersion factor of 5 10-7 s m-3 at 1 km from the
release and a reduction in concentration inversely proportional to
the 1.5 power of the distance expressed in kilometres [U12]. Using
the dose factor of 0.06 mSu per Bq a m-3 and population densities
of 3 and 25 man km-2, the range of estimates of the collective
effective dose equivalent commitment is 3 10-16 to 3 10-15 man Sv
per Bq. This does not yet account for limited outdoor occupancy.
Thus, an average estimate of the collective dose per unit release
is roughly the same as the generalized estimate obtained above for
an outdoor release, namely 1 10-15 man Sv per Bq.
F. REFERENCES
A1 Andrews, J.N. and D.F. Wood. Mechanism of radon release in
rock matrics and entry into groundwaters. Trans. Inst. Min.
Metall, (IMM) Sect. B: Applied Earth Science 81: 198-209
(1972).
A5 Asikainen, M. and H. Kahlos. Natural radioactivity of ground-
and surface-water in Finland. Institute of Radiation
Protection report STL-A24. Helsinki, 1977 (in Finnish).
B1 Barton, C.J., R.E. Moore and P.S. Rohwer. Contribution of
radon in natural gas to the natural radioactivity dose in
homes. Oak Ridge National Laboratory report 0RNL-TM-4154
(1973).
B2 Blifford, J.H. Jr., H. Friedman, L.B. Lockhart, Jr. et al.
Geographical and time distribution of radioactivity in the air.
J. Atmos. Terr. Phys. 9: 1-17 (1956).
C2 Castrén, O., M. Asikainen, M. Annanmäki et al. High natural
radioactivity of bored wells as a radiation hygienic problem in
Finland, in Proceedings of the Fourth International Congress
of the International Radiological Protection Association
(IRPA). Paris, April 1977.
C3 Castrén, P. The contribution of bored wells respiratory radon
daughter exposure in Finland. p. 1364-1370 in Natural
Radiation Environment III. CONF-780422 (1980).
C4 Chamberlain, A.C. and E.D. Dyson. The dose to the trachea and
bronchi from the decay products of radon and thoron. Br. J.
Radiol. 29: 317-329 (1956).
C6 Culot, M.V., H.G. Olson and K.J. Schiager. Radon progeny
control in buildings. Final report of the U.S. Atomic Energy
Commission C00-22731. Fort Collins, Colorado State University,
1973.
D1 Davies, B.L. and J. Forward: Measurement of atmospheric radon
in and out of doors. Health Phys. 19: 136 (1970).
F1 Fisenne, I.M. and N.H. Harley. Lung dose estimates from
natural radioactivity measured in urban air (1973).
G1 Gesell, T.F., R.H. Johnson, Jr. and D.E. Bernhardt. Assessment
of potential radiological population health effects from radon
in liquified petroleum gas. U.S. Environmental Protection
Agency report EPA-520/1-75-002 (1977).
G2 George, A.C. Environmental radon and radon daughters, U.S.
Department of Energy Environmental Measurements Laboratory
Report EML-383 New York (1980).
G4 Gyllander, C. and U. Widemo. Concentration statistics based on
experimental data of atmospheric diffusion. AB Atomenergi
Studsvik report S-447. Sweden, 1972.
H5 Harley, J.H. Environmental radon. p. 109-114 in Noble Gases
(R.E. Stanley and A.A. Moghissi, eds.). U.S. Energy Research
and Development Administration report CONF-730915 (1973).
H6 Heijde, H.B. van der, H. Beens and A.R. de Monchy. The
occurrence of radioactive elements in natural gas.
Ecotoxicology and Environmental Safety 1: 49-87 (1977).
H8 Hofmann, W., F. Steinhäusler and E. Pohl. Age-, sex- and
weight-dependent dose patterns due to inhaled natural
radionuclides, p. 1116-1144 in Natural Radiation Environment
III. CONF-780422 (1980).
H9 Holleman, D., D. Martz and K. Schiager, Total respiratory
deposition of radon daughters from inhalation of uranium mine
atmospheres. Health Phys. 17: 187-192 (1969).
I1 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP Publication 23.
Pergamon Press, Oxford, 1974.
J1 Jacobi, W. Die natürliche Radioaktivität der Atmosphäre und
ihre Bedeutung für die Strahlenbelastung des Menschen. Hahn-
Meier-Institut für Kernforschung Berlin report HMJ-B 21 (1962).
J2 Jacobi, W. and K. André. The vertical distribution of radon-
222, radon-220 and their decay products in the atmosphere. J.
Geophys. Res. 68: 3799-3814 (1963).
J3 Jacobi, W. Relations between the inhaled potential alpha-
energy of 222-Rn and 222-Rn and 220-Rn-daughter and the
absorbed alpha-energy in the bronchial and pulmonary region.
Health Phys. 23: 3-11 (1972).
J5 Jonassen, N. and J.P. McLaughlin. Exhalation of radon-222 from
building materials and walls, p. 1211-1236 in Natural
Radiation Environment III. CONF-780422 (1980).
J6 Jonassen, N. On the effect of atmospheric pressure variations
on the radon-222 concentration in unventilated rooms. Health
Phys. 29: 216-220 (1975).
K2 Knight, G.B. and C.E. Makepeace. Modification of the natural
radionuclide distribution by some human activities in Canada.
p. 1494-1560 in Natural Radiation Environment III. CONF-
780422 (1980).
L1 Larson, R.E. Radon profiles over Kilauea, the Hawaiian Islands
and Yukon snow cover. Pageoph 112: 203-208 (1974).
L2 Letourneau, E.G., R.G. McGregor and H. Taniquchi. Background
levels of radon and radon-daughters in Canadian homes. Paper
presented at the Nuclear Energy Agency (NEA) Meeting on
Personal Dosimetry and Area Monitoring Suitable for Radon and
Daughter Products. Paris, November 1978.
M1 Malakhov, S.G. and P.G. Chernyscheva. On the seasonal
variations in the concentration of radon and thoron in the
surface layer of the atmosphere. p. 60-68 in Radioactive
Isotopes in the Atmosphere and Their Use in Meteorology (J.L.
Karol et al., eds.). U.S. Atomic Energy Commission report AEC-
tr-6711 (1964).
M3 Mastinu, G.G. The radiological impact of geothermal energy.
Paper presented at the EEC/SCPRI seminar on the radiological
burden of man from natural radioactivity in the European
community. Le Vesinet, France, December 1979.
M4 Mattsson, R. Seasonal variation of short-lived radon progeny,
210-Pb and 210-Po, in ground level air in Finland. J. Geophys.
Res. 75: 1741-1744 (1970).
M7 Megromi, K. and R. Mamuro. Emanation and exhalation of radon
and thoron gases from soil particles. J. Geophys. Res. 79:
3357-3360 (1974).
M8 Mustonan, R. Measurements of the radon inhalation rates from
building materials. Presented at the Nordic Society for
Radiation Protection meeting "Radiation in our Environment".
Geilo, Norway, January 1980.
M9 Muth, H. Natürliche Radioaktivität in Trinkwasser,
Nahrungsmitteln und in Menschen in Deutschland. Paper
presented at EEC/SCPRI seminar on the radiological burden of
man from natural radioactivity in the European community. Le
Vesinet, Paris, December 1979.
M10 Mäkeläinen, I. Preliminary survey on radon in Finnish
dwellings. Presented at the Nordic Society for Radiation
Protection meeting "Radiation in our Environment". Geilo,
Norway, January 1980.
N1 Nuclear Energy Agency (OECD). Radiological implication of the
natural radioactivity in building materials - physical aspects.
Report NEA (78) 12. Paris, 1978.
P1 Partridge, J.E., T.R. Horton and E.L. Sensintaffar. A study of
radon-222 released from water during typical household
activities. U.S. Environmental Protection Agency. ORP/EERF-
79-1 (1979).
P2 Pasquill, E. The estimation of the dispersion of windborne
material. Meterorol. Magazine 90: 33 (1961).
P3 Porstendörfer, J.A., A. Schraub and A. Wiche. Bestimmung der
Radon-Exhalation aus Baumaterialien. Paper presented at the
Annual Meeting of the Deutsche Gesellschaft für Biophysick e.V.
Freiburg. Federal Republic of Germany, 1974.
R1 Raghavayya, M. and J.H. Jones. A wire screen-filter paper
combination for the measurement of fractions of unattached
radon daughters in uranium mines. Health Phys. 36: 417-429
(1974).
R2 Rangarajan, C., S.S. Gopalakrithnan and C.D. Eapen. The
diurnal and seasonal changes in short-lived radon-thoron
daughters' concentrations in the coastal and inland regions of
India and their possible relation to regional climatology.
Pageoph 112: 941-953 (1974).
S2 Sandström, O. Study on radioactive minerals occurring in
Swedish mines. Gruvforskningen Serie B, Nr. 242 (1978) (in
Swedish).
S5 Snihs, J.O. The significance of radon and its progeny as
natural radiation sources in Sweden. p. 115-130 in Nobel Gases
(R.E. Stanley and A.A. Moghissi, eds.). U.S. Energy Research
and Development Administration report CONF-730915 (1973).
S7 Snihs, J.O. The approach to radon problems in non-uranium
mines in Sweden. p. 900-912 in Proceedings of the Third
International Congress of the International Radiological
Protection Association (IRPA). Washington, 1973.
S8 Snihs, J.O. Personal communication (1979).
S9 Snihs, J.O. and P.O. Agnedal. The radiological impacts of
uranium mill tailings - a review with special emphasis on the
tailing at Ranstad in Sweden. p. 105-113 in Proceedings of the
NEA Seminar on Management, Stabilization and Environmental
Impact of Uranium Tailings. Albuquerque, 1978.
S10 Soumela, M. and H. Kahlos. Studies on the elimination rate and
the radiation exposure following ingestion of 222-Rn rich
water. Health Phys. 23: 641-652 (1972).
S11 Spitz, H.B. and M.E. Wrenn. The diurnal variation of the
radon-222 concentration in residential structures in Grand
Junction, Colorado. Report prepared for the Workshop on
Environmental Radiation, February 1974. U.S. Atomic Energy
Commission report HASL-287. New York, 1974.
S13 Steinhäusler, F. Long-term measurements of 222-Rn, 220-Rn,
214-Pb and 212-Pb concentrations in the air of private and
public buildings and their dependence on meteorological
parameters. Health Phys. 29: 705-713 (1975).
S17 Stranden, E. and L. Berteig. Radon in dwellings and
influencing factors. Health Phys. 39: 275-284 (1980).
S18 Sutton, O.G. Micrometeorology. McGraw Hill, London, 1953.
S21 Swedjemark, G. Radon in dwellings. Some preliminary results
of long-term measurements. National Institute of Radiation
Protection report SSI-1974-020. Stockholm, 1974 (in Swedish).
S22 Swedjemark, G.A. and B. Hakansson. Radon concentration and
gamma-radiation in one-family houses built with unusually large
fraction of aerated concrete based on alum shale. National
Institute of Radiation Protection report SSI: 1978-022.
Stockholm, 1978.
U3 United States Environmental Protection Agency. Preliminary
findings of radon-daughter levels in structures constructed on
reclaimed Florida phosphate land. U.S. Environmental
Protection Agency Technical note ORP/CSD-75-4. Washington
D.C., 1975.
U4 United States Environmental Protection Agency. Environmental
analysis of the uranium fuel cycle. Part I. Fuel Supply. U.S.
Environmental Protection Agency report EPA-520/9-73-003-B
(1973).
U12 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No.E.77.IX.I. New York, 1977.
U13 University of Florida. Radioactivity of lands and associated
structures. Fourth Semi-annual Technical Report submitted to
Florida Phosphate Council. Cumulative Summary report March 1976
to February 1978.
W1 Wilkening, M.H., W.E. Clemento and D. Stanley. Radon-222 flux
measurements in widely separated regions, p. 717-730 in
Natural Radiation Environment II (J.A.S. Adams and W.L. Lowden,
eds.). The University of Chicago Press, Chicago (1975).
W3 Wicke, A., J. Postendörfler and A. Schraub. Der Einfluss
unterschiedlicher Baumaterialien auf die Radon-konzentration on
Wohnräumen. Tagungsbericht Radioaktivität und Umwelt. 12.
Jahrestagung. Norderney, Oct. 1978.
IX. PLUTONIUM
A. INTRODUCTION
396. Plutonium, the element of atomic number 94, is a member of
the actinide series of elements, those of atomic number 89
(actinium) through 103. The actinide elements have similar
chemical properties and are also similar to the lanthanide or rare
earth elements of atomic number 57 (lanthanum) through 71. The
actinides are considered a second rare earth series. Elements
beyond uranium (atomic number 92) in the periodic table are called
transuranium elements. Some environmental information on another
transuranium element, americium (atomic number 95), is also
available and will be considered here.
397. Plutonium occurs naturally in very small quantities. It is
formed continuously in uranium ores by neutron capture, the
neutrons being produced by spontaneous fissioning of uranium. The
uppermost layers of the earth's crust contain a few kilograms of
239Pu and about the same amount of primordial 244Pu. Plutonium
naturally occurring can only be detected in the richest uranium
ore.
398. The most important of the 15 plutonium isotopes, 239Pu, has a
half-life of 24065 years and is produced from uranium in nuclear
reactors:
238U (n,gamma) 239U beta 239Np beta 239Pu
-> ->
One of the plutonium isotopes, 241Pu, decays by beta-particle
emission with a 14.4 year half-life to americium-241 which has a
half-life of 432.2 years. The decay properties of 238Pu, 239Pu,
240Pu, 241Pu, and 241Am are listed in Table IX.1. Americium is
produced by successive neutron capture reactions by plutonium
isotopes in a reactor:
239Pu (n,gamma) 240Pu (n,gamma) 241Pu beta 241Am
->
399. Plutonium can exist in four valence states in aqueous
solutions: III,IV,V and VI. The IV state is the most common under
physiological conditions where it will exist in solution only as a
strongly complexed ion. Weak complexes of Pu(IV) in neutral
solutions will form polymeric hydroxides. Plutonium oxidizes
rapidly and, thus, the very insoluble PuO2 is the most common form
in the environment, although Pu(VI) has been reported in oceans
[N1] and drinking water [L1]. Some plutonium will be complexed
with biological ligands and incorporated in micro-organisms or in
plant or animal tissues.
400. Americium can exist in three valence states in aqueous
solutions: III, IV and VI. The trivalent state is the stable form
under physiological conditions. The most common oxide is AmO2,
which is more soluble than PuO2. Americium appears to be more
readily incorporated into biological materials when dispersed to
the environment than is plutonium.
401. Plutonium isotopes, 238Pu, 239Pu and 240Pu and the americium
isotope 241Am, emit alpha radiation. Since the x rays accompanying
the alpha emissions are low energy, concentrations of these
isotopes that might occur in the environment would not cause
biological effects unless they are incorporated in biological
material. Deposition in the lungs and absorption from the gastro-
intestinal tract following ingestion are the most important routes
of entry into the bodies of animals and human beings.
Table IX.1 Decay information for plutonium and
americium isotopes
---------------------------------------------------
Isotope Half-life Decay Energy a/ Intensity
(years) mode (MeV) (%)
---------------------------------------------------
238Pu 87.74 alpha 5.59 0.716
5.55 0.283
5.45 0.001
239Pu 24065 alpha 5.24 0.739
5.23 0.152
5.19 0.107
240Pu 6537 alpha 5.26 0.734
5.21 0.265
5.11 0.0009
241Pu 14.4 beta 5.24 10-3 1.0
(average)
241Am 432.2 alpha 5.58 0.852
5.54 0.128
5.48 0.014
5.64 0.003
5.61 0.002
---------------------------------------------------
a/ Principal transitions.
B. SOURCES
1. Nuclear explosions
402. The major source which has introduced plutonium into the
environment has been atmospheric nuclear testing. Of nearly 1.5
1016 Bq of 239, 240Pu activity released, about 1.2 1016 Bq has been
dispersed and deposited world-wide [H2]. The remainder has been
deposited locally at the sites of the tests. The amount deposited
in the northern hemisphere, where most of the tests were conducted,
was three times that deposited in the southern hemisphere.
403. The amounts of the globally dispersed plutonium isotopes
produced in all nuclear tests are listed in Table IX.2. Of the
total mass of plutonium released world-wide (4 106 g), 96% is
comprised of 239Pu and 240Pu. These two isotopes are not
separately distinguished in alpha spectrometry and the combined
amounts are usually reported. The production data and also
analysis of environmental samples indicate that of 239, 240Pu total
amounts, 85% by mass or 60% by activity is due to 239Pu.
Table IX.2 Production of globally dispersed plutonium
isotopes in atmospheric nuclear testing
---------------------------------------------------------
Specific Amount produced
Isotope Half-life activity -------------------------
(a) (Bq/g) Activity (Bq) Mass (t)
---------------------------------------------------------
238Pu 87.74 6.3 1011 3.3 1014 0.00051
239Pu 24065 2.3 109 7.4 1015 3.26
240Pu 6537 8.4 109 5.2 1015 0.59
241Pu 14.4 3.8 1012 1.7 1017 0.041
242Pu 376000 1.5 108 1.6 1013 0.11
---------------------------------------------------------
404. Most of the alpha activity of plutonium produced in nuclear
explosions is due to 239, 240Pu. 241Pu is a beta-emitter which
decays to 241Am. Little 241Am was produced directly in the tests,
but the activity amounts are accumulating in the environment as
241Pu decays. The activity of fallout 241Am in soil is currently
about 25% of that of 239, 240Pu. Decay of 241Pu this far and
subsequent decay will result in a total production of 241Am from
nuclear tests of 5.5 1015 Bq [B4].
2. Nuclear fuel cycle
405. There are about 240 nuclear reactors used to generate
electric power throughout the world. Upon fuel discharge, for each
MW(e)a of electricity produced, it can be calculated that 1.2 1012
Bq of 239, 240Pu are produced, 4.0 1012 Bq of 238Pu, 2.1 1014 Bq of
241Pu and 2.8 1011 Bq of 241Am [H4, K1]. Generally more than half
of the 239Pu produced during reactor operation undergoes fission,
thus contributing to the energy produced by the reactor. Routine
operation of the reactors probably has not resulted in the release
to the environment, world-wide, of more than trace amounts of
plutonium and americium.
406. Discharge of plutonium to rivers and oceans from fuel
reprocessing plants can be much more significant. It has been
estimated that 0.1-1% of the plutonium throughput is released in
liquid effluent from the Windscale plant in the U.K. [M1]. Little
plutonium is released to the air from reprocessing plants. For
example, it has been estimated that future reprocessing activities
may result in the airborne release of about 40 to 4000 Bq of
plutonium and 4 to 40 Bq of americium per MW(e)a [K1].
3. Other sources
407. Processes involved in the production of nuclear weapons have
resulted in the release of plutonium to the surrounding
environment. Releases occur from fabrication and particularly from
reprocessing plants. Leakage from an oil storage area at Rocky
Flats plant in Colorado resulted in release of about 2 1011 Bq of
239Pu, half of it offsite but in the near vicinity of the plant.
At Mound Laboratory in Miamisburg, Ohio, 4 1011 Bq of 238Pu were
washed into an abandoned canal and another 2 1010 Bq was estimated
to have been released to the air. Plutonium has been released to
onsite disposal areas at several laboratories.
408. A few accidents involving nuclear weapons have been reported
that introduced plutonium into the environment. The crash of an
airplane at Thule, Greenland resulted in about 9 1011 Bq of
plutonium being deposited on the shore and in the bottom sediments.
A much smaller quantity is believed to have been carried by winds
away from the accident site [F1]. The collision of two military
planes resulted in plutonium from two weapons being dispersed at
Palomares, Spain. Much soil was removed in an attempt to clean up
the plutonium. Three aberrant missiles were deliberately destroyed
in flight and another burned on the launch pad at Johnson Island in
the Pacific [F1]. Although the launch pad was decontaminated,
undoubtedly several kilograms of plutonium were dispersed to the
ocean from the three accidents. It is estimated that from 4 1011
to 40 1011 Bq of plutonium remain available to be incorporated into
biological systems from these accidents [F1].
409. The use of plutonium in thermoelectric generation systems of
spacecraft has resulted in a relatively small amount of 238Pu and
239Pu being introduced into the environment. Of nearly 350 1014 Bq
of 238Pu with an accompanying 260 1011 Bq of 239Pu carried into
space by 19 U.S. spacecraft, 6.3 1014 Bq of 238Pu and 4.8 1011 Bq of
239Pu were dispersed into the environment when one spacecraft re-
entered the atmosphere and burned over the Indian Ocean. Nearly 80%
of this was dispersed in the stratosphere of the southern
hemisphere and about 20% in the stratosphere of the northern
hemisphere [H1, P5]. Another 16.5 1014 Bq of 238Pu and 12 1011 Bq
of 239Pu entered, with containers intact into the Pacific Ocean as
a result of an aborted flight. In another aborted flight, the
plutonium source was recovered intact from the ocean floor [D4].
410. Large usage is now being made of smoke detectors containing
241Am as an ionization source, with an average of 1 105 Bq of 241Am
in each detector. About 28 1011 Bq of 241Am has been distributed
throughout the U.S.A. in this form [U2]. Since the useful life of
these detectors is estimated to be 10 years, it can be assumed that
some of these detectors have already been disposed of in sanitary
landfills, by incineration, and by other means.
411. Other uses of transuranium elements are made in consumer or
medical devices, notably 238Pu in heart pacemakers. These are
sealed sources, which when handled properly in normal
circumstances, should not allow the contents to be released to the
environment.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Movement in soil
412. When plutonium enters soil as fallout, or is added in
solutions containing hydrolyzable Pu(IV), it is usually highly
insoluble, regardless of soil type [W4]. Diffusion coefficients for
surface soils [G1] are universally low (approximately 10-7 cm2
s-1). Therefore, the major inventory normally remains in the top cm
of undisturbed soils, even with considerable water percolation
[E6]. The relative immobility of plutonium in soils under these
circumstances may be attributed to the initial low solubility of
fallout particles and interaction of Pu(IV) hydrolysis products
with soil, mineral and organic surfaces [W4].
413. A small fraction (< 0.1%) of plutonium in soils is soluble,
accounting for limited plant uptake from soil and chemical mobility
under certain conditions in subsoils. This may be due to the
presence of complexing agents or valence states less subject than
Pu(IV) to hydrolysis and insolubilization. When Pu(IV) is added to
soil as a synthetic or natural organic complex, solubility is
initially greatly increased (several orders of magnitude) because
of reduced hydrolysis; subsequent mobility is a function of
complex stability, competition with other ligands, and resistance
of the ligand to chemical and microbial degradation [W6]. Empirical
evidence suggests the presence of Pu(III) under reduced conditions
in ground waters, and Pu(V)-Pu(VI) have been identified in certain
natural waters [W4, B6].
414. Physical processes also account for some vertical movement in
soil. Cultivation results in redistribution within the plow layer
(to 30 cm) and longterm field studies have traced plutonium
migration to 30 cm in undisturbed arid soil [N4]. In the latter
case, the increased mobility over that predicted by diffusion alone
has been attributed to biological transport and particle movement.
415. Since most plutonium is strongly absorbed on surface soils,
wind and water erosion become primary environmental transport
mechanisms [W4]. Transport distance will generally be a function
of the size of the particle with which plutonium is associated.
Particles in the fine silt-clay size range are the most likely to
contain the highest concentrations of plutonium, to be transported
the greatest distance by wind and water, and to remain attached to
biological surfaces.
416. Detailed investigations of the behaviour of americium in
soils are lacking. In contrast to plutonium, disproportionation
does not occur readily, and Am(III) would be the expected stable
species [W4]. Hydrolysis reactions also influence the behaviour of
Am(III) in soil, but the products of Am(III) hydrolysis are more
soluble than those of Pu(IV) [R3].
2. Transfer to plants
417. Principal mechanisms of plutonium and americium transport to
vegetation are foliar interception and root uptake. Foliar uptake
is dependent upon chemical form and size of the particle
intercepted, residence time and weathering reactions of the leaf.
Translocation to the seeds and roots after deposition on the leaf
of soyabeans is approximately 10-5 of intercepted amounts [W4].
The primary mode of entry into plants is root uptake, with reported
soil-to-plant concentration ratios for plutonium ranging from 10-3
to 10-8 [E5]. Increasing evidence suggests that the solubility in
soil, rather than discrimination at the plant root level, is the
limiting factor in plutonium uptake by plants [W6].
418. Evidence suggests that plutonium is transported across the
root as Pu(IV) [W6, D3]. Complexed Pu(IV) is probably the major
translocated species in plants, and several anionic and cationic
complexes of Pu(IV) have been determined in the xylem of plants
supplied with Pu(IV) [B3]. Plutonium is not uniformly distributed
in the plant. The plutonium concentration decreases up the stem of
soyabeans, and lowest plutonium concentrations occur in the seeds
of barley and soyabeans [W6]. Systematic investigations of
americium translocation and deposition in the plant after root
uptake have not been conducted.
3. Transfer to animals
419. The primary sources of plutonium and americium to domestic
animals are inhalation and consumption of plant tissues containing
plutonium in surface-absorbed particles or in tissues. In grazing
herbivores, plutonium is primarily associated with the gastro-
intestinal tract and pelt, and to a lesser degree, with the lungs
[W4]. Gastro-intestinal absorption requires the presence of
soluble plutonium and hydrolysis/complexation reactions are likely
to govern solubility. The reducing potential of the gut appears
sufficient to maintain principally the Pu(IV) state, which is
subject to insolubilization by hydrolysis. The effect is more
pronounced in the presence of additional reducing substances, such
as food residue [S8]. The fraction of ingested amount absorbed and
deposited in the bone and liver is approximately 10-4 [W6].
However, administration of Pu(VI) in solution to starved animals,
or Pu(IV) in complexed forms (synthetic chelates or plant tissues)
may increase gut absorption [W6, W5, B2].
420. The gastro-intestinal absorption of americium from gavaged
solutions is slightly greater than that of plutonium perhaps
reflecting the reduced tendency of americium for hydrolysis.
Information on absorption of americium incorporated in plant
tissues is not yet available.
4. Transfer to diet
421. The transfer of plutonium and americium to diet from fallout
has not been as extensively studied as the transfer of 90Sr and
137Cs. A complete diet sampling, conducted annually in New York,
was analysed for 239, 240Pu in 1972 [B5] and 1974 [B4]. The 1974
samples were also analysed for 241Am. A few samples of selected
foods from 1963 and 1964 were also analysed for 239, 240Pu.
422. The highest concentrations of 239, 240Pu and 241Am were found
in shellfish, followed by grain products and fresh fruits and
vegetables. The lowest concentrations were in meat, milk, eggs,
fresh fish and in processed foods. The values indicate that
external contamination is a factor in the occurrence of plutonium
in foods. For the shellfish sample, comprising clams and shrimp,
most of the plutonium and americium were found in the clams. The
muscle in the fresh fish sample, comprising halibut, snapper and
flounder, had a 239, 240Pu concentration 10 times less and a 241Am
concentration 50 times less than the shellfish sample.
423. Based on the New York sampling, the intake by ingestion
during 1974 was estimated to be 60 mBq a-1 for 239, 240Pu and 16
mBq a-1 for 241Am. The ratio of americium to plutonium was 0.27 in
the total diet indicating little increase of americium relative to
plutonium compared to the americium and plutonium in the soil.
424. A 239, 240Pu dietary intake record has been calculated based
upon the annual fallout deposition rate and the cumulative deposit
in soil and compared with the New York food sample surveys
conducted in 1963, 1964, 1972 and 1974 [B4]. Assuming no further
atmospheric injections, the 239, 240Pu dietary intake will remain
relatively constant at 0.03 Bq a-1 owing to uptake from the 81 Bq
m-2 cumulative deposit in soil. For 241Am the calculation
indicates that the uptake of 241Am from the cumulative deposit in
soil is a factor of two greater than plutonium. The estimated
241Am dietary intake continues to increase as the cumulative
deposit in soil increases owing to ingrowth from 241Pu decay. When
the cumulative deposit reaches its projected maximum of 29 Bq m-2,
the dietary intake will also be at a maximum of 0.03 Bq a-1.
425. The cumulative transfer of plutonium and americium to diet
depends very much on the assumed residence times in soil. These
times are no doubt shorter than the radioactive mean lives due to
leaching and fixation in soil. Extremes of transfer estimates are
obtained by taking the mean residence times to be 50 years in one
case and the radioactive mean lives in the other case. The
geometric mean of these extremes then gives a tentative estimate of
the transfer factor P23 from deposition density to diet. The
results are 0.6, 0.3 and 0.2 Bq per Bq m-2 for 239Pu, 240Pu and
241Am, respectively. For 238Pu and 241Pu the estimates of P23 are
based on the radioactive mean lives. Most of the transfer is
attributed to direct deposition [B4]. The values are 0.08 and 0.04
Bq per Bq m-2 for 238Pu and 241Pu, respectively.
5. Aquatic behaviour
426. Plutonium is mobilized off watersheds to rivers and coastal
waters [H1, M5, H3, S3, S2]. Estimates available for plutonium
indicate input ranges from 0.05% per year for heavily cultivated
watersheds [M5, S2] to 0.005% per year for a heavily forested
watershed, indicating a residence time of 103 to 2 104 years [W4].
427. Environmental studies have shown that in a variety of
comparatively shallow bodies of water, both fresh water and marine,
more than 96% of the total plutonium released to or deposited on
these environments is rapidly transferred to sediment [E1, L2, H5,
H6, P6, S1, N2, H3, E4, P1, W3, S2]. However, in the deep oceans
there is only slow transfer of the total plutonium in the ocean
water column to deposited sediments. It is estimated that this may
represent about 30% by 1980 of deposited fallout plutonium [B8].
(a) Freshwater systems
428. The behaviour of plutonium and americium has been studied in
a wide range of fresh water systems [S2, E3, D1, R1, B3, W3, B7].
The concentrations of plutonium in the water of these systems
varied by more than four orders of magnitude [W4]. Higher
concentrations of plutonium have been observed in lakes with low
pH, lakes with high sulfate concentrations and other acidic lakes
[W1]. Chemical analyses indicate that while the plutonium in Lake
Michigan in the U.S.A. was predominantly in the Pu(V) and (VI)
states, in all other lakes studied Pu(III) and (IV) predominated
[N1, W2]. The results strongly suggest that the solubility of
plutonium is governed by different complexing agents. In waters of
high pH, the concentration of CO3-2 and HCO3- is relatively high,
and carbonate complexes can form. In waters of low pH, such
complexes cannot exist, and the solubility must be due to
complexing with other ligands, such as natural organic compounds.
429. A relationship has been shown between the concentration of
plutonium in water and the concentration in sediments or
particulate matter [N1]. Values for the distribution constant, KD,
vary between 104 and 5 105, with most values not varying more than
fivefold. Considering the wide variety of the systems, including
sediment types, size, source terms, etc., this small range in
values suggests a commonality in the behaviour of plutonium in
these systems. There is some evidence that the plutonium absorbed
by sediment particles is predominantly in the (III) and (IV)
states, yet on re-equilibration of sediment with water, it has been
shown that there is a conversion of Pu(III) and Pu(IV) back to
Pu(V) or (VI). This strongly supports the hypothesis that the
concentration of plutonium in many fresh water lakes and rivers is
controlled by an equilibrium between water and sediment [B7].
(b) Marine systems
430. The behaviour and fate of transuranic elements in the marine
environment were given very little attention before the mid-1960s.
By far the greatest effort for the next decade was applied to
determine the residence time of plutonium in the oceans [B8, M4].
Comparison with 90Sr and 137Cs indicates that the residence time of
plutonium in the water column is less than that of both 90Sr and
137Cs. The observed distribution has been explained in terms of a
distribution of particles settling at various velocities [N2].
431. In the Irish Sea, the concentration of dissolved 239, 240Pu
is only 6% as large as that of 137Cs, normalized to a unit
discharge rate; the value for 241Am is even lower at 3%. These
values suggest that plutonium and americium leave the water phase
very rapidly. Measurements in the Irish Sea indicate that
plutonium is in solution predominantly as Pu(VI) and on particles
as Pu(III) and Pu(IV) [Nl], a situation similar to that which
exists in the Great Lakes in the U.S.A.
432. Distribution coefficients, KD, between water and suspended
sediments for plutonium in the oceans are similar to those in the
Great Lakes and do not appear to be source related. Similar KD
values (104 to 105) for sediments from the Irish Sea and Enewetak
Lagoon suggest that similar chemical reactions are occurring. In
the Irish Sea, the overall order of KD values for transuranic
nuclides is 241Am > 242Cm and 244Cm > 239, 240Pu [P2].
(c) Bioaccumulation
433. Trophic-level studies in freshwater and marine environments
indicate that plutonium concentration factors for organisms
relative to water generally decrease at higher trophic levels [H5,
H6, B7, D2, E2, P4]. Typical values for plutonium in the edible
portions used for assessment purposes are 10 for fish, 100 for
crustacea and 1000 for molluscs and algae [I1, N3]. Whole
organisms values may be 10 to 50 times higher depending upon the
degree of contamination by sediment. Limited field data indicate
increased concentration factors for 241Am over plutonium in lower
trophic levels and in fish [P3, W1, P4].
434. Laboratory studies indicate that marine teleost fish can
absorb Pu(VI) by direct uptake from sea-water with limited
absorption across the gut from labelled food or sediment. Marine
elasmobranchs on the other hand appear to absorb plutonium across
the gut relatively easily [P3]. Crustacea, such as crabs, have
high assimilation efficiency for plutonium when fed labelled food
[F3, G2], and some biomagnification in a simple laboratory
invertebrate food chain has been observed [F2].
D. TRANSFER TO MAN
435. Information on the transfer of plutonium to man is available
from autopsy measurements on persons exposed to plutonium from
weapons test fallout, from occupational sources, and from
intentionally administered plutonium in terminally ill patients.
The data from fallout plutonium are most pertinent to general
environmental considerations. In addition, the results from many
animal studies provide supporting data on the probable magnitude of
the biokinetic parameters that determine this transfer.
436. Extensive data on the fallout plutonium content of persons in
the general population have been published, including data from
several areas in the U.S.A. [M2, W7], from Finland [M3] and Japan
[O1]. Although great variability was noted from sample to sample,
particularly where only small quantities of tissue were available,
the average results agreed reasonably well with computed tissue
plutonium burdens [B5], based on estimated plutonium intake by
inhalation (ingestion was shown to be insignificant relative to
inhalation), assuming metabolic parameters as employed by ICRP.
437. Considering all of the human data now available, there would
seem to be no reason to alter the ICRP assumption of an equal
distribution of systemic plutonium between skeleton and liver (45%
in each) [I4]. The deposition and retention of fallout plutonium
in the lung seems also to be well described by the ICRP lung model,
assuming behaviour as a Class Y compound [I5]. The ICRP lung model
appears to overestimate substantially the transfer of fallout
plutonium to tracheobronchial lymph nodes. From extensive studies
in experimental animals it is known that the extent of
translocation from lung to lymph nodes varies widely with the
chemical and physical form of the particles inhaled [B1]. The
human data also suggest that gonadal deposition may be slightly
higher than the fraction of 10-5 g-1 assumed by ICRP [I2]. More
precise analytical data are required, however, to support such a
conclusion. Studies of plutonium deposition in the gonads of
several species of experimental animals support the ICRP assumption
[R2].
438. Based on comparative studies in experimental animals, ICRP
has assumed that inhaled americium will behave in humans in a
manner identical with plutonium [I4], except that all americium
compounds are assumed to follow Class W lung model kinetics [I2];
i.e., americium is more rapidly cleared from the lung and more
efficiently translocated to bone and liver than is plutonium oxide.
Applying these assumptions to the estimated intake by inhalation of
fallout americium and 241Pu (which will decay to 241Am), estimates
have been made of human americium burdens [B4]. These indicate a
241Am/239, 240Pu ratio of 0.24 in l978, which will increase to 0.38
by the year 2000 because of further decay of deposited 241Pu.
Pooled samples from 18 autopsies done from 1970 to 1974 showed a
measured 241Am/239, 240Pu ratio, in vertebrae, of 0.22 [B4]. The
close agreement between measured and calculated ratios lends
support to the ICRP assumptions.
439. Data on the gastro-intestinal absorption of plutonium and
americium are available only from studies in experimental animals.
Such studies were summarized by an ICRP task group in 1972, which
led to assumed values for the fraction absorbed of 10-6 for
plutonium oxide and 3 10-5 for other commonly occurring compounds
of plutonium [I4]. It was recognized that a much higher absorption
might be expected for complexed forms of plutonium. More recent
data obtained in a variety of animal species [S4, S6] resulted in a
modification of ICRP estimates to 10-5 for oxides and hydroxides of
plutonium, 10-4 for other commonly occurring plutonium compounds,
and 5 10-4 for all compounds of americium [I2]. Qualitative
support for the plutonium numbers is provided by autopsy data from
a group of reindeer-herding northern Finns who ingest large
quantities of plutonium-rich reindeer liver [M3]. Their plutonium
burdens seem to be no higher than those of southern Finns, who do
not ingest these relatively large quantities of plutonium. A
difference should have been apparent if the fraction absorbed from
the gastro-intestinal tract had been much greater than 10-4.
440. There is evidence from animal studies that plutonium
incorporated into alfalfa [S7] or liver [S4] may be absorbed to a
greater extent than inorganic plutonium; the effect is not large,
however, and is reversed in the case of americium [S4]. Concern has
been expressed that the gastro-intestinal absorption of plutonium
in the hexavalent state, such as may be produced by chlorination of
water supplies, may be markedly increased as compared to
tetravalent plutonium [L1]. It has since been shown, however, that
under normal conditions in the gastro-intestinal tract of
experimental animals, no such increase of gastro-intestinal
absorption is observed [S8].
441. A marked increase in the gastro-intestinal absorption of
plutonium and other actinides has been reported in neonatal animals
of several species [I4, S5]. This increase may be as much as
several hundredfold in the case of rats and several thousandfold in
the case of miniature swine. In addition to increased absorption,
there is a prolonged retention of the actinide within the mucosa of
a small intestine [S5]. It must be assumed that the human infant
will also show an increased absorption, although the magnitude of
this increase and its duration is unknown.
442. In addition to the inhalation and ingestion routes, actinides
may under unusual circumstances of occupational exposure, enter the
body by absorption through the intact or punctured skin [I4, B1].
Normally, however, intact skin is an effective barrier to plutonium
entry, and this route of entry should not be of concern for general
environmental exposure.
443. Once deposited systemically, plutonium is tenaciously
retained. This fact is qualitatively evident from extensive data
on the excretion of plutonium by occupationally exposed humans
[V1]. It has also been quantitatively evaluated in a few human
cases and in a variety of experimental animals [I4, D5]. Based on
these data, ICRP has employed a biological half-time of 100 years
for plutonium in the skeleton, and a half-time of 40 years for
plutonium in liver; plutonium in gonads is assumed to be retained
without loss [I4, I2]. The same parameters are assumed to apply in
the case of americium.
E. DOSIMETRY
1. Dose per unit intake
444. The doses to the various tissues following inhalation or
ingestion or plutonium and americium are determined using the
parameters and models suggested by ICRP. A variety of estimates
are possible, depending on particle size of inhaled particles and
the solubility class of both inhaled and ingested material. The
values given below are for the representative 1 µm aerosol size and
for the insoluble oxide or hydroxide forms of plutonium.
445. The ICRP lung model divides the respiratory system into three
compartments. Deposition fractions in each region for the 1 µm
particle size are 29% in the nasopharyngeal region, 8% in the
tracheo-bronchial region and 23% in the pulmonary region. Smaller
particles have a progressively greater deposition fraction in the
pulmonary region and less retention in the nasopharyngeal region.
446. Inhaled, insoluble plutonium particles are assigned Class Y
parameters, retention in the lungs of the order of years (500 d
half-time for 60% of the pulmonary deposition). Because of greater
mobility of americium, all of its compounds are assigned Class W
parameters, with retention in the lungs of the order of weeks (50 d
half-time for 60% of the pulmonary deposition). In both cases, 40%
of the pulmonary deposition is cleared with a half-time of 1 d by
mucocilliary action through the tracheo-bronchial region.
Translocation from the naso-pharyngeal and tracheo-bronchial region
is rapid, within one day, with most of the material swallowed and
small amounts absorbed to blood.
447. For both inhaled and ingested material reaching blood,
fractional transfer to blood and liver is assumed to be 0.45 each
and 3.5 10-4 to gonads (testes) for both plutonium and americium.
Uptake to blood following ingestion of insoluble plutonium is 10-5
and 5 10-4 for all compounds of americium.
Table IX.3 Absorbed dose equivalent commitments per unit
intake of plutonium and americium (Sv per Bq)
---------------------------------------------------------------------------
Isotope Lung Liver Gonads Red bone Bone Effective
marrow lining a/
cells
---------------------------------------------------------------------------
Inhalation
238Pu 3.2 10-4 1.8 10-4 b/ 6.6 10-5 8.3 10-4 8.2 10-5
239Pu 3.2 10-4 2.1 10-4 b/ 7.6 10-5 9.5 10-4 8.9 10-5
240Pu 3.2 10-4 2.1 10-4 b/ 7.6 10-5 9.5 10-4 8.9 10-5
241Pu 3.2 10-6 4.4 10-6 2.8 10-7 1.7 10-6 2.1 10-5 1.6 10-6
242Pu b/ 5.5 10-4 3.2 10-5 2.0 10-4 2.5 10-3 1.4 10-4
Ingestion
238Pu b/ 4.0 10-8 2.3 10-9 1.5 10-8 1.8 10-7 1.5 10-8
239Pu b/ 4.4 10-8 2.6 10-9 1.6 10-8 2.1 10-7 1.6 10-8
240Pu b/ 4.4 10-8 2.6 10-9 1.6 10-8 2.1 10-7 1.6 10-8
241Pu b/ 8.6 10-10 5.7 10-11 3.4 10-10 4.2 10-9 2.5 10-10
242Pu b/ 2.3 10-6 1.4 10-7 8.4 10-7 1.1 10-5 5.9 10-7
---------------------------------------------------------------------------
a/ Effective dose equivalent commitment per unit intake (Sv per Bq).
b/ Doses which contribute < 10% to the effective dose equivalent commitment.
Note: for plutonium - Class Y and f1 = 10-5
for americium - Class W and f1 = 3 10-4.
Reference [I3].
448. The estimates of the absorbed dose equivalents per unit
intake are given in Table IX.3. These are computed over a 50 year
period following intake. They correspond to the transfer factors
P25 and P35 relating intake in air and diet, respectively, to
tissue dose. For these dose equivalent values, the quality factor
of 20 has been used for the alpha-emitters and 1 for the beta-
emitters (241Pu).
2. Dose per unit release
(a) Nuclear explosions
449. The dose equivalent commitments for plutonium and
americium released in nuclear explosions can be assessed using
the expressions
Dc = P25 Ia (inhalation)
Dc = P23 P35 F (ingestion)
where Ia is the cumulative intake from air (the integrated
concentration in air (Bq a m-3) times the breathing rate (22 m3
d-1) and F is the integrated deposition density. The values of
the transfer factors were discussed above.
450. For the past pattern of nuclear tests the population-weighted
global integrated concentration in air and deposition density can
be determined from comparisons with 90Sr [U1], from Pu-90Sr
production ratios, and from 241Pu decay considerations [B4]. The
results are for air: 1.5, 37, 25, 830 and 1.7 µBq a m-3 and for
deposition density: 0.85, 21, 14, 470 and 16 Bq m-2 for 238Pu,
239Pu, 240Pu, 241Pu and 241Am, respectively. With this
information, the dose equivalent commitments can be determined by
using the above expressions, and the values per unit activity
released can be obtained by dividing the results by the estimated
input amounts from nuclear testing (Table IX.2).
451. Finally, the collective dose equivalent commitments are
determined by multiplying by the appropriate global population.
For inhalation, the activity in air has by now been nearly
depleted, so the present global population applies (4 109 persons).
For ingestion of the short half-life 241Pu, the present population
applies and for the other isotopes, the long-term transfer can be
assumed to apply to the projected equilibrium value of 1010
persons. The results for the collective effective dose equivalent
commitment per unit release are included in the summary Table IX.4.
(b) Nuclear installation
452. The contribution of the inhalation pathway to the collective
dose commitments for plutonium and americium in airborne effluents
from nuclear installations can be estimated from the integrated
concentrations in air in the dispersion region. It has previously
been shown that the appropriate formula is
c I deltaN P25
S1 = ------------
vd
where I is the air breathing rate (22 m3 d-1), deltaN is the
population density in the region (25 man km-2), P25 is the dose per
unit intake factors (Table IX.3) and vd is the deposition velocity
(0.5 cm s-1). The integrated concentration in air is determined by
the amount released (unit activity) per unit area of the deposition
region divided by the deposition velocity. The areal dependence is
removed by multiplying by the population of the region (times the
area). The results from evaluating this expression are summarized
in Table IX.4.
Table IX.4 Summary of collective effective dose equivalent
commitments per unit activity released of plutonium and
americium (10-14 man Sv per Bq)
---------------------------------------------------------------
238Pu 239Pu 240Pu 241Pu 241Am
---------------------------------------------------------------
Nuclear explosions
Inhalation 1000 1000 1000 30 5 a/
Ingestion 3 30 10 0.01 300
Nuclear installations
Release to air b/
Inhalation 10000 10000 10000 200 20000
Ingestion 3 20 10 0.03 300
Release to fresh water
Drinking water 20 20 20 0.2 900
Fish 5 5 5 0.06 200
Release to salt water
Fish 0.01 0.01 0.01 0.0001 0.4
Shellfish 0.2 0.2 0.2 0.002 6
---------------------------------------------------------------
a/ Per Bq of 241Pu released.
b/ Assumes population density of 25 man km-2.
453. The contribution of the ingestion pathway from airborne
effluents to the collective dose commitment per unit activity
released, Sc1, can be determined by using the expression
c
S1 = P23 P35 SN
Using the values of the transfer factors given previously and a
population density, deltaN, of 25 man km-2 in the region of
deposition, the values summarized in Table IX.4 are obtained.
454. For the aquatic ingestion pathways the generalized UNSCEAR
model is utilized [U1]
c Nk Ik fk P35
S1 = -----------------
V(lambda + 1/tau)
to be evaluated for each pathway k. The quotient of water
receiving volume, V, and the population involved, Nk, is the water
utilization factor [U1], assumed to be a constant for each pathway.
A summary of the values used in the assessments by UNSCEAR is given
in the following listing:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 1.0 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.1
fish 10 3
shellfish 300
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
455. Due to insufficient data, the values of the above listing
will be assumed to apply to all isotopes of plutonium and
americium. The values of the dose factor, P35, are given in Table
IX.3. A summary of the evaluated results is given in Table IX.4.
456. Assessments using the above model only account for that
portion of the dose given during the mean residence time of the
water in the receiving area. These are essentially complete
collective dose commitments for the release to fresh water and for
the shorter-lived isotopes (241Pu). The removal of the longer-
lived isotopes to the sediments of the deep ocean will largely
preclude any further contributions to the dose estimates.
F. REFERENCES
B1 Bair, W.J. Recent animal studies on the deposition, retention
and translocation of plutonium and other transuranic compounds.
p. 51-83 in Diagnosis and Treatment of Incorporated
Radionuclides. Proceedings of a seminar. Vienna, 1976.
B2 Ballou, J.E., K.R. Price, R.A. Gies et al. The influence of
DTPA on the biological availability of transuranics. Health
Phys. 34: 445 (1978).
B3 Bartelt, G.E., C.W. Wayman, S.E. Groves et al. 238-Pu and
239,240-Pu distribution in fish and invertebrates from the
Great Miami River, Ohio. p. 517-530 in Transuranics in Natural
Environments (M.G. White and P.B. Dunaway, eds.). Proceedings
of a symposium, Gatlinburg, Tennessee, 1976. ERDA report NV0-
178, Nevada Operations Office, NTIS, 1977.
B4 Bennett, B.G. Environmental aspects of americium.
Environmental Measurements Laboratory report EML-348, New York,
1978.
B5 Bennett, B.G. Transuranic element pathways to man. p. 367-383
in Transuranium Nuclides in the Environment. IAEA publication
STI/PUB/410, Vienna, 1976.
B6 Bondietti, E. and F.H. Sweeton. Transuranic speciation in the
environment. p. 449-476 in Transuranics in Natural
Environments (M.G. White and P.B. Dunaway, eds.). Proceedings
of a symposium, Gatlinburg, Tennessee, 1976. ERDA report NV0-
178, Nevada Operations Office, NTIS, 1977.
B7 Bowen, V.T. Plutonium and americium concentration along fresh
water chains of the Great Lakes, U.S.A. USAEC report C00-3568-
13, Woods Hole Oceanographic Institution, NTIS, 1976.
B8 Bowen, V.T., K.M. Wong and V.E. Noshkin. 239-Pu in and over
the Atlantic Ocean. J. Mar. Res. 29: 1 (1971).
D1 Dahlman, R.C. Transuranium elements in aquatic and terrestrial
environments. p. 131-147 in Environmental Sciences Division
annual progress report. USAEC report ORNL-5257, Oak Ridge
National Laboratory, NTIS, 1976.
D2 Dahlman, R.C., E.A. Bondietti and L.D. Eyman. Biological
pathways and chemical behaviour of plutonium and other
actinides in the environment. p. 47-80 in Actinides in the
Environment. ACS symposium series 35, American Chemical
Society, 1976.
D3 Delaney, M.S. and C.W. Francis. The relative uptake of 237-Pu
(IV) and Pu (VI) oxidation states from water by bushbeans.
Health Phys. 34: 492 (1978).
D4 Dobry, Jr., T.J. Transuranic elements in space nuclear power
systems. p. 83-85 in Transuranic Elements in the Environment
( W.C. Hanson, ed.). DOE publication series TID-22800,
Washington D.C., 1980.
D5 Durbin, P.W. Plutonium in man: a new look at the old data. p.
469-537 in Radiobiology of Plutonium (B.J. Stover and W.S.S.
Jee, eds.). J.W. Press, Salt Lake City, 1972
E1 Edgington, D.N. and J.A. Robbins. The behaviour of plutonium
and other long-lived radionuclides in Lake Michigan. II.
Patterns of deposition in the sediments. p. 245-260 in Impacts
of Nuclear Releases into the Aquatic Environment. Proceedings
of a symposium. Otaniemi, Finland 1975. IAEA publication
STI/PUB/406, Vienna, 1975.
E2 Edgington, D.N., M.A. Wahlgren and J.S. Marshall. The
behaviour of plutonium in aquatic ecosystems: a summary of
studies on the Great Lakes. p.45-80 in Environmental Toxicity
of Aquatic Radionuclides: Model and Mechanisms (M.W. Miller
and J.N. Stannard, eds.). Ann Arbor Science Publishers, Ann
Arbor, Michigan, 1976.
E3 Emery, R.M. and D.C. Kopfer. The distribution of transuranic
elements in a freshwater pond ecosystem. p. 269-286 in
Environmental Toxicity of Aquatic Radionuclides: Models and
Mechanisms (M.W. Miller and J.N. Stannard, eds.). Ann Arbor
Science Publishers, Ann Arbor, Michigan, 1976.
E4 Emergy, R.M., D.C. Kopfer and M.C. McShane. The migration of
plutonium from a freshwater ecosystem at Hanford in Transuranic
Elements in the Environment (W.C. Hanson, ed.). DOE
publication series, TID-22800, Washington, D.C. 1980.
E5 Energy Research and Development Administration. Environmental
research for transuranic elements in Workshop on Environmental
Research for Transuranic Elements. Proceedings of the
workshop, Battelle Seattle Research Centre, Seattle,
Washington, November 1975. ERDA report ERDA-76/134, NTIS
(1976).
E6 Essington, E.H., E.B. Fowler, R.O. Gilbert et al. Plutonium,
americium and uranium concentration in Nevada Test Site soil
profiles. p. 157-173 in Transuranium Nuclides in the
Environment. Proceedings of a symposium, San Fransisco, 1975.
IAEA publication STI/PUB/410, Vienna, 1976.
F1 Facer, G. Quantities of transuranic elements in the
environment from operations relating to nuclear weapons. p. 86-
91 in Transuranic Elements in the Environment (W.C. Hanson,
ed.). DOE publication series, TID-22800, Washington, D.C.,
1980.
F2 Fowler, S.W. and T.M. Beasley. Plutonium and americium in
fish. Nature 265: 384 (1977).
F3 Fowler, S.W. and J.C. Guary. High Absorption efficiency for
ingested plutonium in crabs. Nature 266: 827-828 (1977).
G1 Garland, T.R. and R.E. Wildung. Physiochemical
characterization of mobile plutonium species in soils, p. 254-
263 in Biological Implications of Metals in the Environment
(H. Drucker and R.E. Wildung, eds.). DOE symposium series,
Richland, Washington, 1975. CONF-750929 NTIS (1977).
G2 Guary, J.C. and A. Fraizier. Influence of trophic level and
calcification on the uptake of plutonium observed, in situ, in
marine organisms. Health Phys. 32: 21-28 (1977).
H1 Harley, J.H. Plutonium in the environment - a review. Prepared
for the 1979 annual meeting of the Radiation Research Society
of Japan, Osaka City, Japan, 1979.
H2 Hardy, E.P. World-wide distribution of plutonium. p. 115-128
in Plutonium and Other Transuranium Elements: Sources,
Environmental Distribution and Biomedical Effects (B.W.
Wachholz, ed.). Presented at the Environmental Protection
Agency Plutonium Standards Hearings, Washington, D.C., 1974.
U.S. Atomic Energy Commission, Springfield, Virginia, 1974.
H3 Hayes, D.W. and H.J. Horton. Plutonium and americium behaviour
in the Savannah River marine environment in Transuranic
Elements in the Environment (W.C. Hanson, ed.). DOE
publication series, TID-22800, Washington, D.C., 1980.
H4 Heeb, C.M. and E.T. Merrill. Significant actinide activities
in the LWR and LMFBR nuclear fuel cycle. EPA-520/2-75-006
(1974).
H5 Hetherington, J.A., D.F. Jefferies and M.B. Lovett. Some
investigations into the behaviour of plutonium in the marine
environment. p. 193-212 in Impacts of Nuclear Releases into
the Aquatic Environment. Proceedings of a symposium,
Ontaniemi, Finland, 1975. IAEA publication STI/PUB/406,
Vienna, 1975.
H6 Hetherington, J.A., D.F. Jefferies, N.T. Michell et al.
Environmental and public health consequences of the controlled
disposal of transuranic elements to the marine environment. p.
139-156 in Transuranium Nuclides in the Environment.
Proceedings of a symposium, San Fransisco, 1975. IAEA
publication STI/PUB/410, Vienna, 1976.
I1 International Atomic Energy Agency. The radiological basis of
the IAEA revised definition and recommendations concerning high
level radioactive waste unsuitable for dumping at sea. IAEA-
211 (1978).
I2 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP publication 30,
Pergamon Press, 1979.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers, supplement to Part I.
ICRP publication 30, Pergamon Press, 1979.
I4 International Commission on Radiological Protection. The
metabolism of compounds of plutonium and other actinides. ICRP
publication 19, Pergamon Press, 1972.
I5 International Commission on Radiological Protection. Task
group on lung dynamics. Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.
12: 173-226 (1966).
K1 Kreiter, M.R., J.E. Mendel and R.W. McKee. Transuranic wastes
from the commercial light-water-reactor cycle. p. 92-106 in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
L1 Larsen, R.P. and R.D. Oldham. Plutonium in drinking water:
effects of chlorination on its maximum permissible
concentration. Science 201: 1008-1009 (1978).
L2 Livingstone, H.L. and V.T. Bowen. Americium in the marine
environment - relationships to plutonium. p. 107-130 in
Environmental Toxicity of Aquatic Radionuclides: Models and
Mechanisms (M.W. Miller and J.N. Stannard, eds.). Ann Arbor
Science Publishers, Ann Arbor, Michigan, 1976.
M1 Morley, F. and G.N. Kelly. Radiological protection and
transuranic wastes from the nuclear fuel cycle. Proceedings of
a NEA/CEA Seminar on the management of plutonium contaminated
solid wastes. Marcoule, October, 1974.
M2 McInroy, J.F., E.E. Campbell, W.D. Moss et al. Plutonium in
autopsy tissue: a revision and updating of data reported in
LA-4875. Health Phys. 37: 1-136 (1979).
M3 Miettinen, J.K., H. Mussalo, M. Hakanen et al. Distribution of
plutonium and americium in human and animal tissues after
chronic exposures. p. 265-268 (Vol. III) in Book of Papers.
Fifth International Congress of the International Radiation
Protection Association, Jerusalem, 1980.
M4 Miyake, Y. and Y. Sugimura. The plutonium content of Pacific
Ocean waters. p. 91-104 in Transuranium Nuclides in the
Environment. Proceedings of a symposium, San Francisco, 1975.
IAEA publication STI/PUB/410, Vienna, 1976.
M5 Muller, R.N., D.G. Sprugel and B. Kohn. Erosional transport
and deposition of plutonium and caesium in two small midwestern
watersheds. J. Environ. Qual. 7(2): 171-174 (1978).
N1 Nelson, D.M. and M.B. Lovett. The oxidation state of plutonium
in the Irish Sea. Nature 276: 599-601 (1978).
N2 Noshkin, V.E. and V.T. Bowen. Concentrations and distributions
of long-lived fallout radionuclides in open ocean sediments. p.
671-684 in Radioactive Contamination of the Marine
Enviornment. IAEA, Vienna, 1973.
N3 National Radiological Protection Board/CEA. Methodology for
evaluating the radiological consequences of radioactive
effluents released in normal operations. Joint report of
NRPB/CEA, CEC (1979).
N4 Nyhans, J.W., F.R. Miera, Jr. and R.E. Neher. Distribution of
plutonium in trinity soils after 28 years. J. Environ. Qual.
5(4): 431-437 (1976).
O1 Okabayashi, H., H. Watanabe and Y. Takizawa. Measurement of
plutonium in Japanese human organs. J. Radiat. Res. 19: 62-69
(1978).
P1 Paine, D. Plutonium in Rocky Flats freshwater systems in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
P2 Pentreath, R.J., D.F. Jefferies, M.B. Lovett et al. The
behaviour of transuranium and other long-lived radionuclides in
the Irish Sea and its relevance to the deep sea disposal of
radioactive wastes. Proceedings of the Third NEA Seminar on
Marine Radioecology, OECD, Paris, 1980.
P3 Pentreath, R.J. and M.B. Lovett. Transuranic nuclides in
plaice, pleuronectes platessa from the northeastern Irish Sea.
Marine Biol. 48: 19-26 (1978).
P4 Pentreath, R.J., D.S. Woodhead, B.R. Harvey et al. A
preliminary assessment of some naturally-occurring
radionuclides in marine organisms (including deep sea fish) and
the absorbed dose resulting from them. Proceedings of the Third
NEA Seminar on Marine Radioecology, OECD, Paris, 1980.
P5 Perkins, R.W. and C.W. Thomas. World-wide fallout. p. 53-82 in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
P6 Pillai, K.C. and E. Mathew. Plutonium in the aquatic
environment, its behaviour, distribution and significance. p.
25-45 in Transuranium Nuclides in the Environment. Proceedings
of a symposium, San Francisco, 1975. IAEA publication
STI/PUB/410, Vienna, 1976.
R1 Rees, T.F., J.M. Cleveland and W.C. Gottschall. Dispersion of
plutonium from contaminated pond sediments. Environ. Sci.
Technol. 12(9): 1085-1087 (1978).
R2 Richmond, C.R. and R.L. Thomas. Plutonium and other actinide
elements in gonadal tissue of man and animals. Health Phys. 29:
241-250 (1975).
R3 Routson, R.C., G. Jansen and A.V. Robinson. 99-Tc, 237-Np and
241-Am sorption on two United States subsoils from differing
weathering intensity areas. Health Phys. 33: 311-317 (1977).
S1 Schell, W.R., F.G. Lowman and R.P. Marshall. Geochemistry of
transuranic elements at Bikini Atoll in Transuranic Elements
in the Environments (W.C. Hanson, ed.). DOE publication
series, TID-22800, Washington D.C., 1980.
S2 Simpson, H.J., R.M. Trier and C.R. Olsen. Transport of
plutonium by rivers in Transuranic Elements in the Environment
(W.C. Hanson, ed.). DOE publication series, TID-22800,
Washington, D.C., 1980.
S3 Sprugel, D.H. and G.E. Bartelt. Erosional removal of fallout
plutonium from a large midwestern watershed. J. Environ. Qual.
7(2): 175-177 (1978).
S4 Stather, J.W., J.D. Harrison, P. Rodwell et al. The
gastrointestinal absorption of plutonium and americium in the
hamster. Phys. Med. Biol. 24: 396-407 (1979).
S5 Sullivan, M.F. Absorption of actinide elements from the
gastrointestinal tract of neonatal animals. Health Phys. 38:
173-185 (1980).
S6 Sullivan, M.F. Absorption of actinide elements from the
gastrointestinal tract of rats, guinea pigs and dogs. Health
Phys. 38: 159-171 (1980).
S7 Sullivan, M.F., T.R. Garland, D.A. Cataldo et al. Absorption of
plutonium from the gastrointestinal tracts of rats and guinea
pigs after ingestion of alfalfa containing 238-Pu. Health
Phys. 38: 215-221 (1980).
S8 Sullivan, M.F., J.L. Ryan, L.S. Gorham et al. The influence of
oxidation state on the absorption of plutonium from the
gastrointestinal tract. Radiat. Res. 80: 116-121 (1979).
T1 Till, J.E., S.V. Kaye and J.R. Trabalka. Toxicity of uranium
and plutonium to the developing embryos of fish. ERDA report
ORNL-5160, Oak Ridge National Laboratory, NTIS, 1976.
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation, 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
U2 U.S. Nuclear Regulatory Commission. Environmental Assessment
of Ionization Chamber Smoke Detectors Containing Am-24l (R.
Belanger, D.W. Buckley and J.B. Swenson, eds.). Prepared for
the U.S. Nuclear Regulatory Commission by Science Applications,
Inc., La Jolla, California, 1979.
V1 Voelz, G.L., L.H. Hempleman, J.N.P. Lawrence et al. A 32-year
medical follow-up of Manhattan project plutonium workers.
Health Phys. 37: 445-485 (1979).
W1 Wahlgren, M.A., J.J. Alberts, K.A. Orlandini et al. A
comparison of the concentrations of fallout-derived plutonium
in a series of freshwater lakes. p. 92-94 in Radiological and
Environmental Research Division annual report, (1977). ERDA
report ANL-77-65 (Part 3), Argonne National Laboratory, NTIS
(1977).
W2 Wahlgren, M.A., J.J. Alberts, D.M. Nelson et al. Study of the
occurrence of multiple oxidation states of plutonium in natural
water systems. p. 95-98 in Radiological and Environmental
Research Division annual report (1977). ERDA report ANL-77-65
(Part 3), Argonne National Laboratory, NTIS (1977).
W3 Wahlgren, M.A., J.A. Robbins and D.N. Edgington. Plutonium in
the Great Lakes in Transuranic Elements in the Environment
(W.C. Hanson, ed.). DOE publication series, TID-22800,
Washington, D.C., 1980.
W4 Watters, R.L., D.N. Edgington, T.E. Hakonson et al. Synthesis
of the research literature in Transuranic Elements in the
Environment (W.C. Hanson, ed.). DOE publication series, TID-
22800, Washington, D.C., 1980.
W5 Weeks, M.H., J. Katz, W.O. Oakley et al. Further studies on
the gastrointestinal absorption of plutonium. Radiat. Res. 4:
339 (1956).
W6 Wildung, R.E., T.R. Garland and D.A. Cataldo. Environmental
processes leading to the presence of organically-bound
plutonium in plant tissues consumed by animals in Biological
Implications of Radionuclides Released from Nuclear Industries.
Proceedings of a symposium, Vienna, 1978. IAEA, Vienna, 1979.
W7 Wrenn, M.E. and N. Cohen. Determination of Pu-239/240 tissue
concentrations in non-occupationally exposed residents of New
York City. Annual report to the Department of Energy. New
York University Medical Centre report C00-2968-2. New
York,1978.
X. RADIATION EFFECTS
457. The interaction of radiation with matter results in the
liberation of energy carried by the alpha-particle, beta-particle,
gamma or x-rays and the ionization or excitation of the irradiated
material. In biological material, damage may be caused directly to
cell components by the radiation interactions or indirectly by the
actions of free radicals, the charged fragments of ionization
events. The damage may result in cell death or in cell
transformations, which at some later time may cause harmful effects
in the irradiated individual or in his offspring. The amount of
damage depends on the amount of radiation, which may be from
external irradiation or from radionuclides within the body, on the
type of radiation and on the sensitivity of the tissue.
458. Radiation effects in man are usually classified as somatic
and genetic or hereditary, according to whether they affect somatic
or germinal cells. Somatic damage is expressed therefore by
definition within the lifetime of the irradiated individual, while
genetic damage is expressed at some stage in his progeny. Somatic
effects are - somewhat loosely - further distinguished as immediate
or late depending on the time of their appearance.
A. SOMATIC EFFECTS
1. Early somatic effects
459. The immediate or early somatic effects are expressed in man
within a few days or a few weeks after exposure as a result of
damage to one or more of the self-renewing tissues. These effects
are also called functional because they are due to the inactivation
of a great number of functional cells of a given differentiative
line. Selective irradiation of a given tissue (as in the case of
exposure to some internal emitters) usually leads to effects
localized in that tissue; whole-body irradiation above a given
dose results, on the contrary, in the appearance of generalized
effects, usually under the form of a specific syndrome which
depends on dose. The clinical severity of the immediate effects
changes considerably with the dose, dose rate, type and energy of
the absorbed radiation and part of the body irradiated.
460. The nature of immediate effects is non-stochastic or
deterministic, in the sense that these effects are expected to
occur in an exposed individual absorbing doses in excess of a
minimum amount of radiation referred to as the threshold dose. The
threshold dose is extremely variable depending on the effect and
the tissue considered. It is of the order of a few tenths of a Gy
for most functional effects of importance for radiation protection.
The existence of a threshold of dose is an important characteristic
of these effects; it makes it virtually impossible for them to
appear for doses below the threshold, thus allowing their complete
avoidance.
461. Another important aspect of early effects to be emphasized in
relation to irradiation from internal emitters is the variability
of the dose threshold as a function of dose rate. Low dose rate
irradiation is normally a condition leading to an increase of the
threshold. Since irradiation from radionuclides (particularly at
the levels usually present in the environment) takes place at very
low doses and dose rates it is virtually impossible, except under
exceptional conditions of emergency, that immediate effects might
be observed. They do not deserve therefore any more extended
treatment.
2. Late somatic effects
462. Late somatic effects are those appearing in irradiated
individuals after a latency period and are expressed mainly in the
form of leukaemias or solid tumours. These effects are stochastic
or statistical in nature, in so far as it is impossible to identify
a causal relationship for them in any given case. The correlation
between radiation dose and induction of these conditions may only
be shown on large populations of irradiated individuals as an
increase of the above diseases over their apparently spontaneous
background incidence.
463. Since it is impossible to establish with any certainty the
shape of the dose-induction relationship for late tumorous effects,
particularly at low doses and dose rates, it is usually assumed
that the frequency of their occurrence is linear with dose and
without threshold. The clinical severity of these conditions is
variable, but it is commonly assumed for purposes of radiation
protection that they might be of a uniform and maximum severity,
namely the death of an individual.
B. GENETIC EFFECTS
464. Radiation-induced hereditary effects may appear in the
progeny of irradiated individuals within the first generation
following irradiation - in which case the damage is called dominant
- or within subsequent generations when the genes that carry the
same mutation in the male and in the female genetic complement
happen to match in the genome of the zygote. In this latter case
the genetic damage is called recessive. Clinically, radiation-
induced hereditary conditions have a large spectrum of severity
from the relatively trivial to the very harmful.
465. As in the case of the somatic late effects, radiation
protection usually makes reference to the most severe hereditary
diseases which are either incompatible with life or very disabling
for the individual. Radiation protection also assumes that for
this class of effects a linear non-threshold induction relationship
with dose may apply.
C. DOSE-RESPONSE RELATIONSHIPS
466. In experimental animals and in man late somatic and
hereditary effects may exhibit different shapes of the dose-effect
relationships, according to a large number of physical and
biological variables operating in each particular system. Linear,
linear-quadratic, quadratic or complex relationships have been
described in various circumstances. No generalization may be gained
by the consideration of all existing experience, except perhaps
that each specific system responds according to different kinetics
of action and that biologically complex effects usually correspond
to more complex types of relationships. It would be impossible to
set up a rationale for a system of radiation protection by
considering each case separately. To overcome this difficulty the
assumption is made that late somatic and hereditary effects of
irradiation follow a non-threshold linear function of dose. This
assumption is simple and there is evidence that it is also a
conservative assumption in most cases.
467. It is important to stress the meaning of the assumption of
non-threshold linearity. It postulates, on the one hand, that
there is no dose, however small, that may in principle be
considered safe and no dose increment, however small, which could
not produce a corresponding increase of effect and therefore of
risk. The summation of doses taken as a measure of total risk and
calculations of collective doses as expression of the total
detriment in the exposed population have little meaning outside the
assumption of non-threshold linearity.
468. On the other hand, adoption of the assumption of linearity
involves implicitly the adoption of other important principles.
Actually, if there is a linear relationship between the dose and
the induction of stochastic effects, it becomes possible to use the
average dose received by a given organ or tissue as a significant
reference quantity. Under these conditions it becomes unnecessary
to consider the dose variability within the given organ or tissue
because the response of the component cells (taken to be of uniform
sensitivity) will in any case be linear with the absorbed dose.
The reactions of the component cells will in any case sum-up to
produce an overall effect corresponding to that expected from the
mean dose in that organ or tissue.
469. In case of internal irradiation there may be problems related
to the presence of point sources. The following considerations
apply in these cases. Firstly, with regard to non-stochastic
effects, cell death resulting from high doses within microscopic
volumes of the tissue are expected to produce less harm than for
the same dose uniformly distributed within large volumes: this is
because killing of transformed cells by high doses would be
expected to lead to inactivation of potentially transformed cells.
Moreover, loss of cells around the zones of highest dose absorption
would not be expected to result in significant decrease in tissue
or organ function, unless the functional reserve of the organ or
tissue is impaired for other reasons.
D. RISK ESTIMATES
470. UNSCEAR has extensively reviewed in its 1977 report most
information on the subject of tumour induction in man by ionizing
radiation. The Committee concluded that the risk for all fatal
malignancies after whole-body irradiation at low doses and dose
rates of low-LET radiation - as an average of both sexes and all
ages - is in the region of 10-2 Sv-1. The risk of inducing non-
fatal malignancies under the same conditions would probably fall in
the same range.
471. It should be emphasized that for doses of the order of those
received annually from natural sources no direct information is
available. The above estimate is derived predominantly from
observations conducted on people exposed to absorbed doses of over
1 Gy. While the rate per unit dose or doses in the region of the
natural background would unlikely be higher, it could, however, be
substantially lower. There is no evidence that irradiation from
internal sources would produce rates of tumour induction differing
from those from external irradiation, if account is taken of the
mean absorbed doses in tissues.
472. Concerning hereditary effects, the Committee estimated that
when a population is continuously exposed to low doses of low-LET
radiation at rates of the order of 10-2 Gy per generation (a
generation corresponds to about 30 years) about 50 genetic diseases
might be expected to occur per one million first generation
progeny. At equilibrium, the total genetic damage expressed over
all generations (or the value in each generation reached after
prolonged continuous exposure) would be of the order of 150 cases
per million progeny.
473. It is often convenient to be able to estimate the total
detriment to the individuals from irradiation of specific organs or
tissues, taking into account the various types of effects from
various irradiations. This could be a difficult procedure,
however, in cases when different irradiation modalities lead to
different effects. Such a situation applies for irradiation by
internally deposited radionuclides, when the nuclides produce
different doses in tissues of varying sensitivity and the effects
must be added to the effects of whole-body irradiation.
474. In order to overcome this difficulty, ICRP, in its
publication 26, has designed a system allowing combined estimates
of risk in various organs and tissues, based on their
susceptibility to various effects. These risks of effects of
irradiations of specific tissues are weighted relative to the total
effect from whole-body irradiation. The risks apply to one
individual or the whole exposed population, making use of the
hypothesis of linearity. It is realized, of course, that the
applicability of risk estimates may vary according to the
characteristics of the individual (genetic make-up, sex, age, etc.)
or to the structure of the exposed population. But it is also held
that an acceptable level of precision may be reached by assuming an
average risk value to be applied to all members of the population
irrespective of the above mentioned variability.
475. The mortality risks and weighting factors recommended by ICRP
are shown in Table X.1. These factors have been derived for the
protection of workers, but may also be applied to large population
groups, provided allowance is made for the hereditary effects which
would be expected to appear in all generations subsequent to the
second.
476. It must be emphasized that the risk estimates for induction
of somatic and genetic effects should be regarded as the best
possible numerical conclusions to be drawn from a very
heterogeneous data base affected by various types of dosimetric and
epidemiological uncertainties. Although it is felt that such
estimates are reasonably precise for the purpose of radiation
protection they are to be interpreted and used in a statistical
sense simply as illustrations of the order of magnitude of
potential risks. The actual validity of these estimates could not
possibly be tested empirically under normal circumstances owing to
the small levels of the risks compared with the far higher
"spontaneous" background of similar conditions in the general
population.
Table X.1 Mortality risk and weighting factors for
different organs (from ICRP publication 26)
------------------------------------------------------------
Tissue Mortality risk Weighting
(Sv-1) factor
------------------------------------------------------------
Breast 0.25 10-2 0.15
Red bone marrow 0.2 10-2 0.12
Lungs 0.2 10-2 0.12
Thyroid 0.05 10-2 0.03
Bone surface 0.05 10-2 0.03
Remainder 0.5 10-2 0.30
Gonads (hereditary effects in
the first two generations) 0.4 10-2 0.25
------------------------------------------------------------
Total 1.62 10-2 1.00
------------------------------------------------------------
XI. CONCLUSIONS
A. RADIONUCLIDES AND THE ENVIRONMENT
477. Radiation is a natural feature of man's environment - from the
high energy charged particles which make up cosmic radiation to the
radioactive decay of radionuclides in the earth's crust and in the
biosphere. Several of the radionuclides considered in this
document have important natural sources, including tritium, carbon-
14, krypton-85 and radon. The first three are produced mainly by
cosmic ray interactions in the atmosphere. Radon arises from the
decay of radium present in the earth's crust. All of these
radionuclides are widely dispersed in air. Tritium and 14C enter
more general bio-geochemical cycles, the hydrological cycle and the
carbon cycle, respectively.
478. Several activities of man result in the production of
radionuclides and contribute to the radiation background. An
important source has been the testing of nuclear weapons in the
atmosphere. Large scale atmospheric testing was completed prior to
the Test Ban Treaty of 1963, but additional tests by some countries
have continued. The radionuclides produced in atmospheric nuclear
explosions become widely dispersed in the atmosphere, primarily of
the hemisphere in which the test was conducted, but with some
interhemispheric mixing contribute to the global exposures to
fallout radioactivity. Important fission radionuclides in terms of
the doses delivered include 131I, 90Sr and 137Cs. In the long-
term, the radionuclides 14C and isotopes of plutonium become
important contributors to the radiation dose.
479. The generation of electricity using nuclear power reactors
also results in the production of radionuclides and in some
radiation exposure of man. The radioactive materials are largely
contained within the fuel elements in the reactor or in waste
treatment systems at the fuel reprocessing plant. Releases of
controlled amounts occur in liquid and airborne effluents from the
nuclear installations. Accidents could result in potentially
greater releases of radioactive materials.
480. The natural and man-made sources of radionuclides in the
environment have been discussed for the various radionuclides
considered in this document. The amounts of radioactive materials
released from the various sources have been reviewed and the
relevant values have been shown to depend very much on specific
past practices. Even the normalized release amounts, for example
the amounts per Mt in nuclear explosions or per MW(e)a of
electricity generated, have limited validity. The amounts released
in atmospheric nuclear testing depend on the types of devices
tested and on the geographic pattern of past testing. Releases
from nuclear power installations depend on the efficiency and
integrity of present designs and on the specific waste management
practices currently utilized.
481. The behaviour of radionuclides in the environment has been
studied rather extensively, so that by now, the dispersion of these
pollutants in the environment is fairly well understood. Values
have been derived for the various transfer factors which describe
the transport of radionuclides in the environment and their
transfer to man. For the purpose of dose assessment it is
necessary to consider only the more specific aspects of particular
release situations and to adjust the more generally valid
parameters to the local conditions.
B. DOSE ASSESSMENTS
482. Exposure of man to radionuclides in the environment occurs by
inhalation of amounts in air, ingestion of amounts incorporated
into diet, or from external exposure to radionuclides in air or
deposited on the ground. The dose assessments for the
radionuclides considered in this document have been directed toward
obtaining estimates of the collective effective dose equivalent
commitments per unit amount of activity of the radionuclide
released. This expression of dose gives the absorbed dose,
weighted for radiation type and sensitivity of irradiated tissues
to the entire population and for as long as the exposures from a
specific release continue. This quantity is expected to be most
directly related to the total health detriment which may result.
The results of the dose assessments are summarized in Table XI.1.
483. The degree of transfer of the radionuclides to man and thus
the dose estimates vary depending on the source of release of the
radionuclides. Natural production of tritium, 14C and 85Kr occurs
primarily in the upper atmosphere, following which there is
widespread dispersion, and for 3H and 14C generalized cycling
throughout the environment. Nuclear explosions conducted in the
atmosphere, particularly large scale tests, result in injection of
debris into the stratosphere, from where the radionuclides are
globally dispersed. Releases of radionuclides from nuclear
installations are near surface emissions in airborne effluents or
in liquid effluents to rivers and lakes or to the marine
environment. Exposures are primarily to the local and regional
populations, although the longer-lived radionuclides may also
become more widely dispersed.
484. Release of unit activity of a radionuclide to the environment
generally results in the lowest collective effective dose
equivalent commitment when the release is to the marine
environment. In this case, for the generalized situation
considered here, transfer of radionuclides to man occurs only
through ingestion of fish and shellfish. There may, however, be
important specialized pathways of transfer in more specific release
circumstances.
485. Somewhat greater collective doses result from releases to
freshwater systems, particularly if the water is subsequently used
for drinking. Transfer of radionuclides to fish may also be
somewhat more in fresh water than in the ocean.
Table XI.1 Collective effective dose equivalent commitments
per unit activity released (10-12 man Sv per Bq)
-------------------------------------------------------------
Source
Nuclear installations
Radio- Nuclear Airborne Release to Marine
nuclide Natural explosions release fresh water release
-------------------------------------------------------------
3H 0.0005 0.0008 0.0009 0.003 0.0008
14C 120 120 120 120 120
85Kr 0.0002 0.0002 0.0002 0.0002 0.0002
90Sr - 0.6 1 1 0.00006
129I - 30000 30000 30000 30000
131I - 0.00009 0.4 0.009 0.00006
137Cs - 2 5 0.6 0.001
222Rn 0.001 - 0.001 - -
238Pu - 10 100 0.3 0.002
239Pu - 10 100 0.3 0.002
240Pu - 10 100 0.3 0.002
241Pu - 0.3 2 0.pp3 0.00002
241Am - 3 200 10 0.06
-------------------------------------------------------------
These values are only approximations of a very generalized nature.
The text contains a full discussion of limitations and of the
specific assumptions utilized.
486. The largest doses generally result from airborne releases.
The radionuclides in air may be inhaled, although the contributions
to the dose from this pathway are usually small. More importantly,
the deposited radionuclides are available for incorporation into
the general diet. Long-term transfer of the radionuclides to man
may then occur.
487. The greatest contributors to the collective dose commitments
per unit activity released are the longest-lived radionuclides.
Specifically, the very low dose rates from 129I result in
substantial collective doses over the millions of years mean
radioactive lifetime of this radionuclide. The same is true of 14C
and some isotopes of plutonium, for which the mean radioactive
lifetimes are of the order of thousands of years. Of course, the
validity of assessments which involve such long-term projections
must be questioned. These results can not be related to
equilibrium dose rates to the world's population. For this
purpose, limiting the integration periods to the duration of the
practices, (for example a few hundred years at the most for nuclear
power production) obtaining the incomplete collective dose
commitments, has some merit.
C. EFFECTS EVALUATION
488. The final step in evaluating the consequences of releasing
radionuclides into the environment, once exposures have been
determined, is to estimate the health effects. A great deal of
study has been made of radiation exposure-response relationships
and risk estimates have been formulated for the various effects.
There is uncertainty, however, in knowing whether the risk
estimates, which are generally only obtainable at higher doses and
dose rates, will be valid for the low level chronic exposures of
environmental situations.
489. It is not the intention of this document to provide detailed
guidance regarding effects evaluations. This requires more
detailed specifications of the radionuclides present in the sources
and the amounts released and closer consideration of the
environmental conditions and the consequent exposures to the
population involved. Only a brief summary has been presented
(Chapter X) of the main aspects of radiation effects and of the
considerations involved in assigning risk estimates.
490. In the generally accepted philosophy of radiation protection,
all exposures are considered to increase the risk of harmful
effects. Increased risks are only justified when balanced by net
positive benefits from the radiation operations. The conceptual
basis for measuring benefits and accounting for acceptability of
risks are topics under review by national and international bodies.
491. The first part of the procedure of assessing the consequences
of releasing radionuclides into the environment - the specification
of sources, of environmental behaviour of radionuclides and of the
consequent exposures - appears, in general, to be well founded, as
shown by the reviews of radionuclide behaviour and dosimetry of
this document. This contributes in a general sense to the
establishment of health criteria for these radionuclides. The
usual precautions must be expressed in applying general criteria to
specific situations. Considerable judgement is always required to
make meaningful evaluations to serve as proper guides for future
actions. It should also be appreciated that the results of
radiological assessments are but one aspect of the considerations
on which decisions must be made. The better knowledge that has
developed on these aspects should not unduly condition other
factors, such as socio-economic considerations, which are also of
importance in the rational selection of various possible options.
ANNEX I
Excerpts from "Basic Safety Standards for Radiation Protection 1982
Edition", (Safety Series No. 9) IAEA
369. The product C x F, where C is the radon concentration and F
is the equilibrium factor is called the equilibrium equivalent
concentration of radon (EEC); it corresponds to a concentration of
radon for which the radon daughters in equilibrium with radon have
the same potential alpha energy as the actual daughter
concentration of interest.
(b) Attachment
370. The radon daughters in air may be unattached (free atoms or
ions) or attached to aerosols. The first daughter, 218Po, is at
the time of formation an unattached ion or neutral atom. But
within a few seconds most of the 218Po becomes attached to an
aerosol and the subsequent decay products 214Po and 214Bi are
therefore to a great extent attached to aerosols at their
formation.
371. The attachment rate of a free radon daughter depends
therefore on the number and size distribution of the aerosols in
the air. These parameters vary in different rooms and will thus
affect the attachment rate. In a house with normal aerosol
concentration (ca. 104 cm-3) and size distribution, the attachment
rate will be about 10-2 s-1, i.e., the mean life of the free radon
daughter will be about 100 s. In a mine with higher aerosol
concentration the corresponding values may be about 0.3 s-1 and
4 s, respectively.
372. Radon daughters in room air will also attach to the surfaces
in the room. The deposition rate for radon daughters attached to
aerosols is dependent on the diffusion rate of the aerosols and the
proportion between the surface area and the volume of the room. If
that proportion is 2 m-1, the mean life of the attached radon
daughters (as far as deposition is concerned) is of the order of
one hour. Unattached radon daughters have much higher diffusion
rate than aerosols and therefore the deposition rate is also
higher. The corresponding mean life is of the order of one minute.
373. The fraction of unattached radon daughters in room air is
also dependent on radioactive decay and ventilation rate. With
given values of the attachment rate of free atoms and deposition
rate of unattached atoms, the fraction of unattached daughters in
air increases with increasing decay constant and ventilation rate.
This means that the fraction of unattached 218Po atoms (lambda =
13.6 h-1) is normally higher than, e.g., that of 214Pb (lambda =
1.6 h-1). Measured values are in the range of 1-30%.
(c) Equilibrium variations
374. Ventilation and deposition to surfaces both prevent the radon
daughters reaching equilibrium with radon in air. Only the
dependence of the equilibrium factor on the ventilation rate was
considered in Figure VIII.IV. However, because of deposition,
measured values of the factor F are often lower than the predicted
value from Figure VIII.IV. The deviation is larger in air with low
aerosol concentrations. An approximate expected value of F is
obtained by multiplying the value in Figure VIII.IV by 0.5.
375. Measured values of F in houses show great variation mainly
due to differing ventilation conditions. In the UNSCEAR 1977
report [U12] an average value of F for houses of 0.5 was adopted.
In outdoor air the equilibrium factor is also dependent on
meteorological factors. Measured values indicate an average value
of 0.6, which was used by UNSCEAR in its 1977 report. For uranium
mines with good ventilation a factor of 0.3 may be appropriate.
D. TRANSFER TO MAN
376. The transfer to man of radon and radon daughters occurs from
the inhalation of air. A negligible amount arises from decay of
radium in ingested food and water. From a dose standpoint, it is
most important to know the intake amount of radon daughters in air.
377. The amount of radon daughters inhaled depends upon the
concentration in air and on the breathing rate. The breathing rate
varies with different levels of physical activity and age. For the
adult, the average breathing rates are 20 1 min-1 during light
activity, 7.5 1 min-1 resting and 12.5 1 min-1 for intermediate
activity [I1].
378. To compute the average air intake rate, it will be assumed
that the time spent indoors per day (19 h) consists of 5.5 h light
activity, 8 h resting and 5.5 h intermediate activity. The time
spent outdoors (5 h) is assumed to consist of 2 h light activity
and 3 h intermediate activity. This gives estimated intake rates
of approximately 15 m3 d-1 indoors and 5 m3 d-1 outdoors.
379. The deposition of radon daughters in the respiratory system
depends on the size distribution of the aerosols to which they are
attached and on the fraction of unattached radon daughters. The
deposition is also influenced by the manner of breathing. The
attached radon daughters are deposited in the pulmonary region.
However, the deposition is not 100 per cent. Some is exhaled and
some is transported by mucus before decay. An approximate value of
the fractional retention may be about 50% [U12] but great
variations have been reported.
380. The unattached radon daughters are mainly deposited in the
upper respiratory tract. The efficient deposition of unattached
daughters has been experimentally verified in a model lung by
Chamberlain and Dyson [C4]. However, a major part of the
unattached daughters is removed by nasal deposition [U12].
E. DOSIMETRY
1. Dose per unit exposure
381. The doses of radon gas in air are negligible in comparison
with those of the daughters. On rare occasions, when there is a
great disequilibrium between radon and radon daughters, the
relative contribution from radon to effective dose equivalent could
be significant.
382. Inhalation of radon daughters leads to inhomogeneous
irradiation of the respiratory tract. The maximum dose is received
by the basal cells in the epithelium of the upper bronchi in the
tracheobronchial region of the lungs due to deposition of
unattached radon daughters. Normally, the fraction of unattached
radon daughters in air is small (a few per cent). Attached radon
daughters are mostly deposited in the pulmonary region.
383. Jacobi [J3] has calculated the doses from radon daughters to
the tracheobronchial region of the lung (assumed mass 45 g) and to
the pulmonary region (955 g) as a function of the unattached
fraction, f, of the potential alpha energy. The absorbed energy
fractions are 0.03 (1 + 6f) joule in the tracheobronchial region
per joule inhaled and 0.38 (1 - f) joule in the pulmonary region
per joule inhaled. Total energy absorbed in the lung, reflecting
deposition and clearance for an average aerosol size and breathing
rate is of the order of 40%.
384. The value of the unattached fraction, f, of radon daughters
in air is usually in the range 0.02 to 0.1 [U12]. For a mean value
of 0.06, the absorbed doses in the lungs are 0.9 Gy per joule
inhaled in the tracheobronchial region and 0.4 Gy per joule inhaled
in the pulmonary region.
385. The dose equivalent is obtained by multiplying by a quality
factor, which for alpha radiation is 20. The effective dose
equivalent is obtained by multiplying by the tissue weighting
factor, which for the lungs is 0.12. It might be appropriate to
apply a weighting factor of 0.06 to the dose equivalent to the
basal cell layer of the tracheobronchial region and 0.06 for the
pulmonary region. The contributions to the effective dose
equivalent to the lungs per unit energy inhaled are thus 1.1 Sv J-1
(tracheobronchial region) and 0.5 Sv J-1 (pulmonary region).
386. The dosimetry of radon daughters in the lungs is, at the
moment, under review. The comparisons of various dosimetric models
may lead to recommendations for adjustments of some of these
results. For this report a value of 1.6 Sv per joule inhaled will
be used for the total effective dose equivalent to the lungs,
applicable to exposures indoors and outdoors and also to
occupational exposures in mines.
387. The potential alpha energy of an atom in the decay chain of
radon is the total alpha energy emitted during decay of the atom up
to 210Pb. Dividing by the decay constant of the radionuclide gives
the potential alpha energy per unit of activity. The values for
radon and its short-lived daughters are given in Table VIII.3.
388. The measured concentration of radon in air must be multiplied
by the equilibrium concentration (EEC). For an EEC of 1 Bq m-3 and
a breathing rate of 20 m3 d-1 (7300 m3 a-1), the effective dose
equivalent rate is
Bq m3 J Sv
1 -- 7300 34500 MeV 1.6 0-13 --- 1.6 -- = 6 10-5 Sv a-1
m3 a MeV J
The effective dose equivalent per unit integrated concentration of
radon in air is 6 10-5 Sv per Bq a m-3.
Table VIII.3 Potential alpha energy of radon and
short-lived decay products
-----------------------------------------------------
Radionuclide Energy per atom Energy per unit
(MeV) activity (MeV Bq-1)
-----------------------------------------------------
222Rn 19.2 9.15 106
218Po 13.7 3620
214Pb 7.69 17800
214Bi 7.69 13100
214Po 7.69 0.002
-----------------------------------------------------
Total a/ 34500
-----------------------------------------------------
a/ The total is the sum of the potential energies
of the daughters only.
389. In terms of WLM for occupational exposures, the potential
alpha energy inhaled is calculated as follows:
1.3 105 MeV 1-1 1200 1 170 h 1.6 10-13 d
WL h M Mev
= 4.2 10-3 J (WLM)-1
where the breathing rate is 1200 1 h-1 (20 1 min-1) during an 8 h
working day. The effective dose equivalent per unit exposure is
4.2 10-3 d 1.6 Sv = 7 10-3 Sv (WLM)-1
WLM J
The results are summarized in Table VIII.4.
Table VIII.4 Effective dose equivalent per unit exposure from
short-lived radon decay products
----------------------------------------------------------------
Public (indoors and outdoors) 5.10-5 Sv (Bq a m-3)-1
Workers 7 10-3 Sv (WLM)-1
----------------------------------------------------------------
390. The effective dose equivalent caused by inhalation of radon
without daughters at a concentration of 1 Bq m-3 is about 7 10-7 Sv
a-1 [H8]. This is only about one per cent of the effective dose
equivalent caused by inhalation of radon daughters in equilibrium
with radon.
391. Radon in water may cause a radiation dose to man by ingestion
of water and by inhalation of the radon daughters produced by decay
of the radon released to air. Consumption of 0.5 1/day of radon-
rich water with a radon concentration of 1 kBq 1-1 will lead to an
effective dose equivalent of 0.5 mSv a-1 (by ingestion) [S10].
392. The levels of radon have been measured repeatedly in
different places of the world. The levels found in outdoor air
vary between about 0.1 to 10 Bq m-3, lower values having been found
over oceans and islands, higher values over continents. For the
estimation of an average dose equivalent, a radon concentration in
air of 3.7 Bq m-3 might be used, corresponding to an equilibrium
equivalent concentration of 2.2 Bq m-3 (equilibrium factor = 0.6).
If it is assummed that people are outdoors 20% of the time as an
annual average (i.e., the occupancy factor is 0.2) the
corresponding average effective dose equivalent would be 1.5 10-2
mSv per year from radon daughters.
393. In houses the radon levels may be high because of radium-rich
building materials and/or poor ventilation. However, normal values
are of the order of 10 Bq m-3. An inhabitant-weighted average value
for countries in which comprehensive measurements have been carried
out is an equilibrium equivalent concentration of radon of 14 Bq-3.
Using the effective dose equivalent per unit exposure given in
Table VIII.4 for the public, 0.06 mSv (Bq a m-3)-1, and an
occupancy factor of 0.8, an equilibrium equivalent concentration
of 14 Bq m-3 will correspond to an effective dose equivalent of 0.7
mSv per year. A summary of the dose estimates for indoor and
outdoor exposures is given in Table VIII.5.
2. Dose per unit release
394. Estimates of the dose per unit release of radon are quite
variable, depending on the location of the release, the population
density, and the conditions of the dispersion. The most
generalized estimates of the collective effective dose equivalents
to the world population (4 109 person) may be made by combining
dose values of Table VIII.5 with the total amounts of radon
released of Table VIII.1. For a release of radon outdoors (1020 Bq
a-1 world-wide) the estimate is 1 10-15 man Sv per Bq, and for a
release indoors (3 1016 Bq a-1 world-wide) the result is 9 10-11
man Sv per Bq.
Table VIII.5 Average effective dose equivalent from inhalation
of radon daughters in air
----------------------------------------------------------------
Equilibrium Occupancy Dose factor Effective
equivalent factor (mSv per Bq dose
concentration a m-3) equivalent
(Bq m-3) (mSv a-1)
----------------------------------------------------------------
Outdoors 2.2 0.2 0.06 0.03
Indoors 14.0 0.8 0.06 0.7
----------------------------------------------------------------
395. Releases of radon from nuclear installations result from
mining and milling operations and from the residual tailings. These
generally occur in areas of low population density - from 3 man km-2
in mining areas to about 25 man km-2 around the mills [U12]. The
local collective dose per unit release can be estimated by an
integration over the distance from 1 to 100 km, assuming an
atmospheric dispersion factor of 5 10-7 s m-3 at 1 km from the
release and a reduction in concentration inversely proportional to
the 1.5 power of the distance expressed in kilometres [U12]. Using
the dose factor of 0.06 mSu per Bq a m-3 and population densities
of 3 and 25 man km-2, the range of estimates of the collective
effective dose equivalent commitment is 3 10-16 to 3 10-15 man Sv
per Bq. This does not yet account for limited outdoor occupancy.
Thus, an average estimate of the collective dose per unit release
is roughly the same as the generalized estimate obtained above for
an outdoor release, namely 1 10-15 man Sv per Bq.
F. REFERENCES
A1 Andrews, J.N. and D.F. Wood. Mechanism of radon release in
rock matrics and entry into groundwaters. Trans. Inst. Min.
Metall, (IMM) Sect. B: Applied Earth Science 81: 198-209
(1972).
A5 Asikainen, M. and H. Kahlos. Natural radioactivity of ground-
and surface-water in Finland. Institute of Radiation
Protection report STL-A24. Helsinki, 1977 (in Finnish).
B1 Barton, C.J., R.E. Moore and P.S. Rohwer. Contribution of
radon in natural gas to the natural radioactivity dose in
homes. Oak Ridge National Laboratory report 0RNL-TM-4154
(1973).
B2 Blifford, J.H. Jr., H. Friedman, L.B. Lockhart, Jr. et al.
Geographical and time distribution of radioactivity in the air.
J. Atmos. Terr. Phys. 9: 1-17 (1956).
C2 Castrén, O., M. Asikainen, M. Annanmäki et al. High natural
radioactivity of bored wells as a radiation hygienic problem in
Finland, in Proceedings of the Fourth International Congress
of the International Radiological Protection Association
(IRPA). Paris, April 1977.
C3 Castrén, P. The contribution of bored wells respiratory radon
daughter exposure in Finland. p. 1364-1370 in Natural
Radiation Environment III. CONF-780422 (1980).
C4 Chamberlain, A.C. and E.D. Dyson. The dose to the trachea and
bronchi from the decay products of radon and thoron. Br. J.
Radiol. 29: 317-329 (1956).
C6 Culot, M.V., H.G. Olson and K.J. Schiager. Radon progeny
control in buildings. Final report of the U.S. Atomic Energy
Commission C00-22731. Fort Collins, Colorado State University,
1973.
D1 Davies, B.L. and J. Forward: Measurement of atmospheric radon
in and out of doors. Health Phys. 19: 136 (1970).
F1 Fisenne, I.M. and N.H. Harley. Lung dose estimates from
natural radioactivity measured in urban air (1973).
G1 Gesell, T.F., R.H. Johnson, Jr. and D.E. Bernhardt. Assessment
of potential radiological population health effects from radon
in liquified petroleum gas. U.S. Environmental Protection
Agency report EPA-520/1-75-002 (1977).
G2 George, A.C. Environmental radon and radon daughters, U.S.
Department of Energy Environmental Measurements Laboratory
Report EML-383 New York (1980).
G4 Gyllander, C. and U. Widemo. Concentration statistics based on
experimental data of atmospheric diffusion. AB Atomenergi
Studsvik report S-447. Sweden, 1972.
H5 Harley, J.H. Environmental radon. p. 109-114 in Noble Gases
(R.E. Stanley and A.A. Moghissi, eds.). U.S. Energy Research
and Development Administration report CONF-730915 (1973).
H6 Heijde, H.B. van der, H. Beens and A.R. de Monchy. The
occurrence of radioactive elements in natural gas.
Ecotoxicology and Environmental Safety 1: 49-87 (1977).
H8 Hofmann, W., F. Steinhäusler and E. Pohl. Age-, sex- and
weight-dependent dose patterns due to inhaled natural
radionuclides, p. 1116-1144 in Natural Radiation Environment
III. CONF-780422 (1980).
H9 Holleman, D., D. Martz and K. Schiager, Total respiratory
deposition of radon daughters from inhalation of uranium mine
atmospheres. Health Phys. 17: 187-192 (1969).
I1 International Commission on Radiological Protection. Report of
the task group on reference man. ICRP Publication 23.
Pergamon Press, Oxford, 1974.
J1 Jacobi, W. Die natürliche Radioaktivität der Atmosphäre und
ihre Bedeutung für die Strahlenbelastung des Menschen. Hahn-
Meier-Institut für Kernforschung Berlin report HMJ-B 21 (1962).
J2 Jacobi, W. and K. André. The vertical distribution of radon-
222, radon-220 and their decay products in the atmosphere. J.
Geophys. Res. 68: 3799-3814 (1963).
J3 Jacobi, W. Relations between the inhaled potential alpha-
energy of 222-Rn and 222-Rn and 220-Rn-daughter and the
absorbed alpha-energy in the bronchial and pulmonary region.
Health Phys. 23: 3-11 (1972).
J5 Jonassen, N. and J.P. McLaughlin. Exhalation of radon-222 from
building materials and walls, p. 1211-1236 in Natural
Radiation Environment III. CONF-780422 (1980).
J6 Jonassen, N. On the effect of atmospheric pressure variations
on the radon-222 concentration in unventilated rooms. Health
Phys. 29: 216-220 (1975).
K2 Knight, G.B. and C.E. Makepeace. Modification of the natural
radionuclide distribution by some human activities in Canada.
p. 1494-1560 in Natural Radiation Environment III. CONF-
780422 (1980).
L1 Larson, R.E. Radon profiles over Kilauea, the Hawaiian Islands
and Yukon snow cover. Pageoph 112: 203-208 (1974).
L2 Letourneau, E.G., R.G. McGregor and H. Taniquchi. Background
levels of radon and radon-daughters in Canadian homes. Paper
presented at the Nuclear Energy Agency (NEA) Meeting on
Personal Dosimetry and Area Monitoring Suitable for Radon and
Daughter Products. Paris, November 1978.
M1 Malakhov, S.G. and P.G. Chernyscheva. On the seasonal
variations in the concentration of radon and thoron in the
surface layer of the atmosphere. p. 60-68 in Radioactive
Isotopes in the Atmosphere and Their Use in Meteorology (J.L.
Karol et al., eds.). U.S. Atomic Energy Commission report AEC-
tr-6711 (1964).
M3 Mastinu, G.G. The radiological impact of geothermal energy.
Paper presented at the EEC/SCPRI seminar on the radiological
burden of man from natural radioactivity in the European
community. Le Vesinet, France, December 1979.
M4 Mattsson, R. Seasonal variation of short-lived radon progeny,
210-Pb and 210-Po, in ground level air in Finland. J. Geophys.
Res. 75: 1741-1744 (1970).
M7 Megromi, K. and R. Mamuro. Emanation and exhalation of radon
and thoron gases from soil particles. J. Geophys. Res. 79:
3357-3360 (1974).
M8 Mustonan, R. Measurements of the radon inhalation rates from
building materials. Presented at the Nordic Society for
Radiation Protection meeting "Radiation in our Environment".
Geilo, Norway, January 1980.
M9 Muth, H. Natürliche Radioaktivität in Trinkwasser,
Nahrungsmitteln und in Menschen in Deutschland. Paper
presented at EEC/SCPRI seminar on the radiological burden of
man from natural radioactivity in the European community. Le
Vesinet, Paris, December 1979.
M10 Mäkeläinen, I. Preliminary survey on radon in Finnish
dwellings. Presented at the Nordic Society for Radiation
Protection meeting "Radiation in our Environment". Geilo,
Norway, January 1980.
N1 Nuclear Energy Agency (OECD). Radiological implication of the
natural radioactivity in building materials - physical aspects.
Report NEA (78) 12. Paris, 1978.
P1 Partridge, J.E., T.R. Horton and E.L. Sensintaffar. A study of
radon-222 released from water during typical household
activities. U.S. Environmental Protection Agency. ORP/EERF-
79-1 (1979).
P2 Pasquill, E. The estimation of the dispersion of windborne
material. Meterorol. Magazine 90: 33 (1961).
P3 Porstendörfer, J.A., A. Schraub and A. Wiche. Bestimmung der
Radon-Exhalation aus Baumaterialien. Paper presented at the
Annual Meeting of the Deutsche Gesellschaft für Biophysick e.V.
Freiburg. Federal Republic of Germany, 1974.
R1 Raghavayya, M. and J.H. Jones. A wire screen-filter paper
combination for the measurement of fractions of unattached
radon daughters in uranium mines. Health Phys. 36: 417-429
(1974).
R2 Rangarajan, C., S.S. Gopalakrithnan and C.D. Eapen. The
diurnal and seasonal changes in short-lived radon-thoron
daughters' concentrations in the coastal and inland regions of
India and their possible relation to regional climatology.
Pageoph 112: 941-953 (1974).
S2 Sandström, O. Study on radioactive minerals occurring in
Swedish mines. Gruvforskningen Serie B, Nr. 242 (1978) (in
Swedish).
S5 Snihs, J.O. The significance of radon and its progeny as
natural radiation sources in Sweden. p. 115-130 in Nobel Gases
(R.E. Stanley and A.A. Moghissi, eds.). U.S. Energy Research
and Development Administration report CONF-730915 (1973).
S7 Snihs, J.O. The approach to radon problems in non-uranium
mines in Sweden. p. 900-912 in Proceedings of the Third
International Congress of the International Radiological
Protection Association (IRPA). Washington, 1973.
S8 Snihs, J.O. Personal communication (1979).
S9 Snihs, J.O. and P.O. Agnedal. The radiological impacts of
uranium mill tailings - a review with special emphasis on the
tailing at Ranstad in Sweden. p. 105-113 in Proceedings of the
NEA Seminar on Management, Stabilization and Environmental
Impact of Uranium Tailings. Albuquerque, 1978.
S10 Soumela, M. and H. Kahlos. Studies on the elimination rate and
the radiation exposure following ingestion of 222-Rn rich
water. Health Phys. 23: 641-652 (1972).
S11 Spitz, H.B. and M.E. Wrenn. The diurnal variation of the
radon-222 concentration in residential structures in Grand
Junction, Colorado. Report prepared for the Workshop on
Environmental Radiation, February 1974. U.S. Atomic Energy
Commission report HASL-287. New York, 1974.
S13 Steinhäusler, F. Long-term measurements of 222-Rn, 220-Rn,
214-Pb and 212-Pb concentrations in the air of private and
public buildings and their dependence on meteorological
parameters. Health Phys. 29: 705-713 (1975).
S17 Stranden, E. and L. Berteig. Radon in dwellings and
influencing factors. Health Phys. 39: 275-284 (1980).
S18 Sutton, O.G. Micrometeorology. McGraw Hill, London, 1953.
S21 Swedjemark, G. Radon in dwellings. Some preliminary results
of long-term measurements. National Institute of Radiation
Protection report SSI-1974-020. Stockholm, 1974 (in Swedish).
S22 Swedjemark, G.A. and B. Hakansson. Radon concentration and
gamma-radiation in one-family houses built with unusually large
fraction of aerated concrete based on alum shale. National
Institute of Radiation Protection report SSI: 1978-022.
Stockholm, 1978.
U3 United States Environmental Protection Agency. Preliminary
findings of radon-daughter levels in structures constructed on
reclaimed Florida phosphate land. U.S. Environmental
Protection Agency Technical note ORP/CSD-75-4. Washington
D.C., 1975.
U4 United States Environmental Protection Agency. Environmental
analysis of the uranium fuel cycle. Part I. Fuel Supply. U.S.
Environmental Protection Agency report EPA-520/9-73-003-B
(1973).
U12 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation 1977 report to the General Assembly, with annexes.
United Nations sales publication No.E.77.IX.I. New York, 1977.
U13 University of Florida. Radioactivity of lands and associated
structures. Fourth Semi-annual Technical Report submitted to
Florida Phosphate Council. Cumulative Summary report March 1976
to February 1978.
W1 Wilkening, M.H., W.E. Clemento and D. Stanley. Radon-222 flux
measurements in widely separated regions, p. 717-730 in
Natural Radiation Environment II (J.A.S. Adams and W.L. Lowden,
eds.). The University of Chicago Press, Chicago (1975).
W3 Wicke, A., J. Postendörfler and A. Schraub. Der Einfluss
unterschiedlicher Baumaterialien auf die Radon-konzentration on
Wohnräumen. Tagungsbericht Radioaktivität und Umwelt. 12.
Jahrestagung. Norderney, Oct. 1978.
IX. PLUTONIUM
A. INTRODUCTION
396. Plutonium, the element of atomic number 94, is a member of
the actinide series of elements, those of atomic number 89
(actinium) through 103. The actinide elements have similar
chemical properties and are also similar to the lanthanide or rare
earth elements of atomic number 57 (lanthanum) through 71. The
actinides are considered a second rare earth series. Elements
beyond uranium (atomic number 92) in the periodic table are called
transuranium elements. Some environmental information on another
transuranium element, americium (atomic number 95), is also
available and will be considered here.
397. Plutonium occurs naturally in very small quantities. It is
formed continuously in uranium ores by neutron capture, the
neutrons being produced by spontaneous fissioning of uranium. The
uppermost layers of the earth's crust contain a few kilograms of
239Pu and about the same amount of primordial 244Pu. Plutonium
naturally occurring can only be detected in the richest uranium
ore.
398. The most important of the 15 plutonium isotopes, 239Pu, has a
half-life of 24065 years and is produced from uranium in nuclear
reactors:
238U (n,gamma) 239U beta 239Np beta 239Pu
-> ->
One of the plutonium isotopes, 241Pu, decays by beta-particle
emission with a 14.4 year half-life to americium-241 which has a
half-life of 432.2 years. The decay properties of 238Pu, 239Pu,
240Pu, 241Pu, and 241Am are listed in Table IX.1. Americium is
produced by successive neutron capture reactions by plutonium
isotopes in a reactor:
239Pu (n,gamma) 240Pu (n,gamma) 241Pu beta 241Am
->
399. Plutonium can exist in four valence states in aqueous
solutions: III,IV,V and VI. The IV state is the most common under
physiological conditions where it will exist in solution only as a
strongly complexed ion. Weak complexes of Pu(IV) in neutral
solutions will form polymeric hydroxides. Plutonium oxidizes
rapidly and, thus, the very insoluble PuO2 is the most common form
in the environment, although Pu(VI) has been reported in oceans
[N1] and drinking water [L1]. Some plutonium will be complexed
with biological ligands and incorporated in micro-organisms or in
plant or animal tissues.
400. Americium can exist in three valence states in aqueous
solutions: III, IV and VI. The trivalent state is the stable form
under physiological conditions. The most common oxide is AmO2,
which is more soluble than PuO2. Americium appears to be more
readily incorporated into biological materials when dispersed to
the environment than is plutonium.
401. Plutonium isotopes, 238Pu, 239Pu and 240Pu and the americium
isotope 241Am, emit alpha radiation. Since the x rays accompanying
the alpha emissions are low energy, concentrations of these
isotopes that might occur in the environment would not cause
biological effects unless they are incorporated in biological
material. Deposition in the lungs and absorption from the gastro-
intestinal tract following ingestion are the most important routes
of entry into the bodies of animals and human beings.
Table IX.1 Decay information for plutonium and
americium isotopes
---------------------------------------------------
Isotope Half-life Decay Energy a/ Intensity
(years) mode (MeV) (%)
---------------------------------------------------
238Pu 87.74 alpha 5.59 0.716
5.55 0.283
5.45 0.001
239Pu 24065 alpha 5.24 0.739
5.23 0.152
5.19 0.107
240Pu 6537 alpha 5.26 0.734
5.21 0.265
5.11 0.0009
241Pu 14.4 beta 5.24 10-3 1.0
(average)
241Am 432.2 alpha 5.58 0.852
5.54 0.128
5.48 0.014
5.64 0.003
5.61 0.002
---------------------------------------------------
a/ Principal transitions.
B. SOURCES
1. Nuclear explosions
402. The major source which has introduced plutonium into the
environment has been atmospheric nuclear testing. Of nearly 1.5
1016 Bq of 239, 240Pu activity released, about 1.2 1016 Bq has been
dispersed and deposited world-wide [H2]. The remainder has been
deposited locally at the sites of the tests. The amount deposited
in the northern hemisphere, where most of the tests were conducted,
was three times that deposited in the southern hemisphere.
403. The amounts of the globally dispersed plutonium isotopes
produced in all nuclear tests are listed in Table IX.2. Of the
total mass of plutonium released world-wide (4 106 g), 96% is
comprised of 239Pu and 240Pu. These two isotopes are not
separately distinguished in alpha spectrometry and the combined
amounts are usually reported. The production data and also
analysis of environmental samples indicate that of 239, 240Pu total
amounts, 85% by mass or 60% by activity is due to 239Pu.
Table IX.2 Production of globally dispersed plutonium
isotopes in atmospheric nuclear testing
---------------------------------------------------------
Specific Amount produced
Isotope Half-life activity -------------------------
(a) (Bq/g) Activity (Bq) Mass (t)
---------------------------------------------------------
238Pu 87.74 6.3 1011 3.3 1014 0.00051
239Pu 24065 2.3 109 7.4 1015 3.26
240Pu 6537 8.4 109 5.2 1015 0.59
241Pu 14.4 3.8 1012 1.7 1017 0.041
242Pu 376000 1.5 108 1.6 1013 0.11
---------------------------------------------------------
404. Most of the alpha activity of plutonium produced in nuclear
explosions is due to 239, 240Pu. 241Pu is a beta-emitter which
decays to 241Am. Little 241Am was produced directly in the tests,
but the activity amounts are accumulating in the environment as
241Pu decays. The activity of fallout 241Am in soil is currently
about 25% of that of 239, 240Pu. Decay of 241Pu this far and
subsequent decay will result in a total production of 241Am from
nuclear tests of 5.5 1015 Bq [B4].
2. Nuclear fuel cycle
405. There are about 240 nuclear reactors used to generate
electric power throughout the world. Upon fuel discharge, for each
MW(e)a of electricity produced, it can be calculated that 1.2 1012
Bq of 239, 240Pu are produced, 4.0 1012 Bq of 238Pu, 2.1 1014 Bq of
241Pu and 2.8 1011 Bq of 241Am [H4, K1]. Generally more than half
of the 239Pu produced during reactor operation undergoes fission,
thus contributing to the energy produced by the reactor. Routine
operation of the reactors probably has not resulted in the release
to the environment, world-wide, of more than trace amounts of
plutonium and americium.
406. Discharge of plutonium to rivers and oceans from fuel
reprocessing plants can be much more significant. It has been
estimated that 0.1-1% of the plutonium throughput is released in
liquid effluent from the Windscale plant in the U.K. [M1]. Little
plutonium is released to the air from reprocessing plants. For
example, it has been estimated that future reprocessing activities
may result in the airborne release of about 40 to 4000 Bq of
plutonium and 4 to 40 Bq of americium per MW(e)a [K1].
3. Other sources
407. Processes involved in the production of nuclear weapons have
resulted in the release of plutonium to the surrounding
environment. Releases occur from fabrication and particularly from
reprocessing plants. Leakage from an oil storage area at Rocky
Flats plant in Colorado resulted in release of about 2 1011 Bq of
239Pu, half of it offsite but in the near vicinity of the plant.
At Mound Laboratory in Miamisburg, Ohio, 4 1011 Bq of 238Pu were
washed into an abandoned canal and another 2 1010 Bq was estimated
to have been released to the air. Plutonium has been released to
onsite disposal areas at several laboratories.
408. A few accidents involving nuclear weapons have been reported
that introduced plutonium into the environment. The crash of an
airplane at Thule, Greenland resulted in about 9 1011 Bq of
plutonium being deposited on the shore and in the bottom sediments.
A much smaller quantity is believed to have been carried by winds
away from the accident site [F1]. The collision of two military
planes resulted in plutonium from two weapons being dispersed at
Palomares, Spain. Much soil was removed in an attempt to clean up
the plutonium. Three aberrant missiles were deliberately destroyed
in flight and another burned on the launch pad at Johnson Island in
the Pacific [F1]. Although the launch pad was decontaminated,
undoubtedly several kilograms of plutonium were dispersed to the
ocean from the three accidents. It is estimated that from 4 1011
to 40 1011 Bq of plutonium remain available to be incorporated into
biological systems from these accidents [F1].
409. The use of plutonium in thermoelectric generation systems of
spacecraft has resulted in a relatively small amount of 238Pu and
239Pu being introduced into the environment. Of nearly 350 1014 Bq
of 238Pu with an accompanying 260 1011 Bq of 239Pu carried into
space by 19 U.S. spacecraft, 6.3 1014 Bq of 238Pu and 4.8 1011 Bq of
239Pu were dispersed into the environment when one spacecraft re-
entered the atmosphere and burned over the Indian Ocean. Nearly 80%
of this was dispersed in the stratosphere of the southern
hemisphere and about 20% in the stratosphere of the northern
hemisphere [H1, P5]. Another 16.5 1014 Bq of 238Pu and 12 1011 Bq
of 239Pu entered, with containers intact into the Pacific Ocean as
a result of an aborted flight. In another aborted flight, the
plutonium source was recovered intact from the ocean floor [D4].
410. Large usage is now being made of smoke detectors containing
241Am as an ionization source, with an average of 1 105 Bq of 241Am
in each detector. About 28 1011 Bq of 241Am has been distributed
throughout the U.S.A. in this form [U2]. Since the useful life of
these detectors is estimated to be 10 years, it can be assumed that
some of these detectors have already been disposed of in sanitary
landfills, by incineration, and by other means.
411. Other uses of transuranium elements are made in consumer or
medical devices, notably 238Pu in heart pacemakers. These are
sealed sources, which when handled properly in normal
circumstances, should not allow the contents to be released to the
environment.
C. BEHAVIOUR IN THE ENVIRONMENT
1. Movement in soil
412. When plutonium enters soil as fallout, or is added in
solutions containing hydrolyzable Pu(IV), it is usually highly
insoluble, regardless of soil type [W4]. Diffusion coefficients for
surface soils [G1] are universally low (approximately 10-7 cm2
s-1). Therefore, the major inventory normally remains in the top cm
of undisturbed soils, even with considerable water percolation
[E6]. The relative immobility of plutonium in soils under these
circumstances may be attributed to the initial low solubility of
fallout particles and interaction of Pu(IV) hydrolysis products
with soil, mineral and organic surfaces [W4].
413. A small fraction (< 0.1%) of plutonium in soils is soluble,
accounting for limited plant uptake from soil and chemical mobility
under certain conditions in subsoils. This may be due to the
presence of complexing agents or valence states less subject than
Pu(IV) to hydrolysis and insolubilization. When Pu(IV) is added to
soil as a synthetic or natural organic complex, solubility is
initially greatly increased (several orders of magnitude) because
of reduced hydrolysis; subsequent mobility is a function of
complex stability, competition with other ligands, and resistance
of the ligand to chemical and microbial degradation [W6]. Empirical
evidence suggests the presence of Pu(III) under reduced conditions
in ground waters, and Pu(V)-Pu(VI) have been identified in certain
natural waters [W4, B6].
414. Physical processes also account for some vertical movement in
soil. Cultivation results in redistribution within the plow layer
(to 30 cm) and longterm field studies have traced plutonium
migration to 30 cm in undisturbed arid soil [N4]. In the latter
case, the increased mobility over that predicted by diffusion alone
has been attributed to biological transport and particle movement.
415. Since most plutonium is strongly absorbed on surface soils,
wind and water erosion become primary environmental transport
mechanisms [W4]. Transport distance will generally be a function
of the size of the particle with which plutonium is associated.
Particles in the fine silt-clay size range are the most likely to
contain the highest concentrations of plutonium, to be transported
the greatest distance by wind and water, and to remain attached to
biological surfaces.
416. Detailed investigations of the behaviour of americium in
soils are lacking. In contrast to plutonium, disproportionation
does not occur readily, and Am(III) would be the expected stable
species [W4]. Hydrolysis reactions also influence the behaviour of
Am(III) in soil, but the products of Am(III) hydrolysis are more
soluble than those of Pu(IV) [R3].
2. Transfer to plants
417. Principal mechanisms of plutonium and americium transport to
vegetation are foliar interception and root uptake. Foliar uptake
is dependent upon chemical form and size of the particle
intercepted, residence time and weathering reactions of the leaf.
Translocation to the seeds and roots after deposition on the leaf
of soyabeans is approximately 10-5 of intercepted amounts [W4].
The primary mode of entry into plants is root uptake, with reported
soil-to-plant concentration ratios for plutonium ranging from 10-3
to 10-8 [E5]. Increasing evidence suggests that the solubility in
soil, rather than discrimination at the plant root level, is the
limiting factor in plutonium uptake by plants [W6].
418. Evidence suggests that plutonium is transported across the
root as Pu(IV) [W6, D3]. Complexed Pu(IV) is probably the major
translocated species in plants, and several anionic and cationic
complexes of Pu(IV) have been determined in the xylem of plants
supplied with Pu(IV) [B3]. Plutonium is not uniformly distributed
in the plant. The plutonium concentration decreases up the stem of
soyabeans, and lowest plutonium concentrations occur in the seeds
of barley and soyabeans [W6]. Systematic investigations of
americium translocation and deposition in the plant after root
uptake have not been conducted.
3. Transfer to animals
419. The primary sources of plutonium and americium to domestic
animals are inhalation and consumption of plant tissues containing
plutonium in surface-absorbed particles or in tissues. In grazing
herbivores, plutonium is primarily associated with the gastro-
intestinal tract and pelt, and to a lesser degree, with the lungs
[W4]. Gastro-intestinal absorption requires the presence of
soluble plutonium and hydrolysis/complexation reactions are likely
to govern solubility. The reducing potential of the gut appears
sufficient to maintain principally the Pu(IV) state, which is
subject to insolubilization by hydrolysis. The effect is more
pronounced in the presence of additional reducing substances, such
as food residue [S8]. The fraction of ingested amount absorbed and
deposited in the bone and liver is approximately 10-4 [W6].
However, administration of Pu(VI) in solution to starved animals,
or Pu(IV) in complexed forms (synthetic chelates or plant tissues)
may increase gut absorption [W6, W5, B2].
420. The gastro-intestinal absorption of americium from gavaged
solutions is slightly greater than that of plutonium perhaps
reflecting the reduced tendency of americium for hydrolysis.
Information on absorption of americium incorporated in plant
tissues is not yet available.
4. Transfer to diet
421. The transfer of plutonium and americium to diet from fallout
has not been as extensively studied as the transfer of 90Sr and
137Cs. A complete diet sampling, conducted annually in New York,
was analysed for 239, 240Pu in 1972 [B5] and 1974 [B4]. The 1974
samples were also analysed for 241Am. A few samples of selected
foods from 1963 and 1964 were also analysed for 239, 240Pu.
422. The highest concentrations of 239, 240Pu and 241Am were found
in shellfish, followed by grain products and fresh fruits and
vegetables. The lowest concentrations were in meat, milk, eggs,
fresh fish and in processed foods. The values indicate that
external contamination is a factor in the occurrence of plutonium
in foods. For the shellfish sample, comprising clams and shrimp,
most of the plutonium and americium were found in the clams. The
muscle in the fresh fish sample, comprising halibut, snapper and
flounder, had a 239, 240Pu concentration 10 times less and a 241Am
concentration 50 times less than the shellfish sample.
423. Based on the New York sampling, the intake by ingestion
during 1974 was estimated to be 60 mBq a-1 for 239, 240Pu and 16
mBq a-1 for 241Am. The ratio of americium to plutonium was 0.27 in
the total diet indicating little increase of americium relative to
plutonium compared to the americium and plutonium in the soil.
424. A 239, 240Pu dietary intake record has been calculated based
upon the annual fallout deposition rate and the cumulative deposit
in soil and compared with the New York food sample surveys
conducted in 1963, 1964, 1972 and 1974 [B4]. Assuming no further
atmospheric injections, the 239, 240Pu dietary intake will remain
relatively constant at 0.03 Bq a-1 owing to uptake from the 81 Bq
m-2 cumulative deposit in soil. For 241Am the calculation
indicates that the uptake of 241Am from the cumulative deposit in
soil is a factor of two greater than plutonium. The estimated
241Am dietary intake continues to increase as the cumulative
deposit in soil increases owing to ingrowth from 241Pu decay. When
the cumulative deposit reaches its projected maximum of 29 Bq m-2,
the dietary intake will also be at a maximum of 0.03 Bq a-1.
425. The cumulative transfer of plutonium and americium to diet
depends very much on the assumed residence times in soil. These
times are no doubt shorter than the radioactive mean lives due to
leaching and fixation in soil. Extremes of transfer estimates are
obtained by taking the mean residence times to be 50 years in one
case and the radioactive mean lives in the other case. The
geometric mean of these extremes then gives a tentative estimate of
the transfer factor P23 from deposition density to diet. The
results are 0.6, 0.3 and 0.2 Bq per Bq m-2 for 239Pu, 240Pu and
241Am, respectively. For 238Pu and 241Pu the estimates of P23 are
based on the radioactive mean lives. Most of the transfer is
attributed to direct deposition [B4]. The values are 0.08 and 0.04
Bq per Bq m-2 for 238Pu and 241Pu, respectively.
5. Aquatic behaviour
426. Plutonium is mobilized off watersheds to rivers and coastal
waters [H1, M5, H3, S3, S2]. Estimates available for plutonium
indicate input ranges from 0.05% per year for heavily cultivated
watersheds [M5, S2] to 0.005% per year for a heavily forested
watershed, indicating a residence time of 103 to 2 104 years [W4].
427. Environmental studies have shown that in a variety of
comparatively shallow bodies of water, both fresh water and marine,
more than 96% of the total plutonium released to or deposited on
these environments is rapidly transferred to sediment [E1, L2, H5,
H6, P6, S1, N2, H3, E4, P1, W3, S2]. However, in the deep oceans
there is only slow transfer of the total plutonium in the ocean
water column to deposited sediments. It is estimated that this may
represent about 30% by 1980 of deposited fallout plutonium [B8].
(a) Freshwater systems
428. The behaviour of plutonium and americium has been studied in
a wide range of fresh water systems [S2, E3, D1, R1, B3, W3, B7].
The concentrations of plutonium in the water of these systems
varied by more than four orders of magnitude [W4]. Higher
concentrations of plutonium have been observed in lakes with low
pH, lakes with high sulfate concentrations and other acidic lakes
[W1]. Chemical analyses indicate that while the plutonium in Lake
Michigan in the U.S.A. was predominantly in the Pu(V) and (VI)
states, in all other lakes studied Pu(III) and (IV) predominated
[N1, W2]. The results strongly suggest that the solubility of
plutonium is governed by different complexing agents. In waters of
high pH, the concentration of CO3-2 and HCO3- is relatively high,
and carbonate complexes can form. In waters of low pH, such
complexes cannot exist, and the solubility must be due to
complexing with other ligands, such as natural organic compounds.
429. A relationship has been shown between the concentration of
plutonium in water and the concentration in sediments or
particulate matter [N1]. Values for the distribution constant, KD,
vary between 104 and 5 105, with most values not varying more than
fivefold. Considering the wide variety of the systems, including
sediment types, size, source terms, etc., this small range in
values suggests a commonality in the behaviour of plutonium in
these systems. There is some evidence that the plutonium absorbed
by sediment particles is predominantly in the (III) and (IV)
states, yet on re-equilibration of sediment with water, it has been
shown that there is a conversion of Pu(III) and Pu(IV) back to
Pu(V) or (VI). This strongly supports the hypothesis that the
concentration of plutonium in many fresh water lakes and rivers is
controlled by an equilibrium between water and sediment [B7].
(b) Marine systems
430. The behaviour and fate of transuranic elements in the marine
environment were given very little attention before the mid-1960s.
By far the greatest effort for the next decade was applied to
determine the residence time of plutonium in the oceans [B8, M4].
Comparison with 90Sr and 137Cs indicates that the residence time of
plutonium in the water column is less than that of both 90Sr and
137Cs. The observed distribution has been explained in terms of a
distribution of particles settling at various velocities [N2].
431. In the Irish Sea, the concentration of dissolved 239, 240Pu
is only 6% as large as that of 137Cs, normalized to a unit
discharge rate; the value for 241Am is even lower at 3%. These
values suggest that plutonium and americium leave the water phase
very rapidly. Measurements in the Irish Sea indicate that
plutonium is in solution predominantly as Pu(VI) and on particles
as Pu(III) and Pu(IV) [Nl], a situation similar to that which
exists in the Great Lakes in the U.S.A.
432. Distribution coefficients, KD, between water and suspended
sediments for plutonium in the oceans are similar to those in the
Great Lakes and do not appear to be source related. Similar KD
values (104 to 105) for sediments from the Irish Sea and Enewetak
Lagoon suggest that similar chemical reactions are occurring. In
the Irish Sea, the overall order of KD values for transuranic
nuclides is 241Am > 242Cm and 244Cm > 239, 240Pu [P2].
(c) Bioaccumulation
433. Trophic-level studies in freshwater and marine environments
indicate that plutonium concentration factors for organisms
relative to water generally decrease at higher trophic levels [H5,
H6, B7, D2, E2, P4]. Typical values for plutonium in the edible
portions used for assessment purposes are 10 for fish, 100 for
crustacea and 1000 for molluscs and algae [I1, N3]. Whole
organisms values may be 10 to 50 times higher depending upon the
degree of contamination by sediment. Limited field data indicate
increased concentration factors for 241Am over plutonium in lower
trophic levels and in fish [P3, W1, P4].
434. Laboratory studies indicate that marine teleost fish can
absorb Pu(VI) by direct uptake from sea-water with limited
absorption across the gut from labelled food or sediment. Marine
elasmobranchs on the other hand appear to absorb plutonium across
the gut relatively easily [P3]. Crustacea, such as crabs, have
high assimilation efficiency for plutonium when fed labelled food
[F3, G2], and some biomagnification in a simple laboratory
invertebrate food chain has been observed [F2].
D. TRANSFER TO MAN
435. Information on the transfer of plutonium to man is available
from autopsy measurements on persons exposed to plutonium from
weapons test fallout, from occupational sources, and from
intentionally administered plutonium in terminally ill patients.
The data from fallout plutonium are most pertinent to general
environmental considerations. In addition, the results from many
animal studies provide supporting data on the probable magnitude of
the biokinetic parameters that determine this transfer.
436. Extensive data on the fallout plutonium content of persons in
the general population have been published, including data from
several areas in the U.S.A. [M2, W7], from Finland [M3] and Japan
[O1]. Although great variability was noted from sample to sample,
particularly where only small quantities of tissue were available,
the average results agreed reasonably well with computed tissue
plutonium burdens [B5], based on estimated plutonium intake by
inhalation (ingestion was shown to be insignificant relative to
inhalation), assuming metabolic parameters as employed by ICRP.
437. Considering all of the human data now available, there would
seem to be no reason to alter the ICRP assumption of an equal
distribution of systemic plutonium between skeleton and liver (45%
in each) [I4]. The deposition and retention of fallout plutonium
in the lung seems also to be well described by the ICRP lung model,
assuming behaviour as a Class Y compound [I5]. The ICRP lung model
appears to overestimate substantially the transfer of fallout
plutonium to tracheobronchial lymph nodes. From extensive studies
in experimental animals it is known that the extent of
translocation from lung to lymph nodes varies widely with the
chemical and physical form of the particles inhaled [B1]. The
human data also suggest that gonadal deposition may be slightly
higher than the fraction of 10-5 g-1 assumed by ICRP [I2]. More
precise analytical data are required, however, to support such a
conclusion. Studies of plutonium deposition in the gonads of
several species of experimental animals support the ICRP assumption
[R2].
438. Based on comparative studies in experimental animals, ICRP
has assumed that inhaled americium will behave in humans in a
manner identical with plutonium [I4], except that all americium
compounds are assumed to follow Class W lung model kinetics [I2];
i.e., americium is more rapidly cleared from the lung and more
efficiently translocated to bone and liver than is plutonium oxide.
Applying these assumptions to the estimated intake by inhalation of
fallout americium and 241Pu (which will decay to 241Am), estimates
have been made of human americium burdens [B4]. These indicate a
241Am/239, 240Pu ratio of 0.24 in l978, which will increase to 0.38
by the year 2000 because of further decay of deposited 241Pu.
Pooled samples from 18 autopsies done from 1970 to 1974 showed a
measured 241Am/239, 240Pu ratio, in vertebrae, of 0.22 [B4]. The
close agreement between measured and calculated ratios lends
support to the ICRP assumptions.
439. Data on the gastro-intestinal absorption of plutonium and
americium are available only from studies in experimental animals.
Such studies were summarized by an ICRP task group in 1972, which
led to assumed values for the fraction absorbed of 10-6 for
plutonium oxide and 3 10-5 for other commonly occurring compounds
of plutonium [I4]. It was recognized that a much higher absorption
might be expected for complexed forms of plutonium. More recent
data obtained in a variety of animal species [S4, S6] resulted in a
modification of ICRP estimates to 10-5 for oxides and hydroxides of
plutonium, 10-4 for other commonly occurring plutonium compounds,
and 5 10-4 for all compounds of americium [I2]. Qualitative
support for the plutonium numbers is provided by autopsy data from
a group of reindeer-herding northern Finns who ingest large
quantities of plutonium-rich reindeer liver [M3]. Their plutonium
burdens seem to be no higher than those of southern Finns, who do
not ingest these relatively large quantities of plutonium. A
difference should have been apparent if the fraction absorbed from
the gastro-intestinal tract had been much greater than 10-4.
440. There is evidence from animal studies that plutonium
incorporated into alfalfa [S7] or liver [S4] may be absorbed to a
greater extent than inorganic plutonium; the effect is not large,
however, and is reversed in the case of americium [S4]. Concern has
been expressed that the gastro-intestinal absorption of plutonium
in the hexavalent state, such as may be produced by chlorination of
water supplies, may be markedly increased as compared to
tetravalent plutonium [L1]. It has since been shown, however, that
under normal conditions in the gastro-intestinal tract of
experimental animals, no such increase of gastro-intestinal
absorption is observed [S8].
441. A marked increase in the gastro-intestinal absorption of
plutonium and other actinides has been reported in neonatal animals
of several species [I4, S5]. This increase may be as much as
several hundredfold in the case of rats and several thousandfold in
the case of miniature swine. In addition to increased absorption,
there is a prolonged retention of the actinide within the mucosa of
a small intestine [S5]. It must be assumed that the human infant
will also show an increased absorption, although the magnitude of
this increase and its duration is unknown.
442. In addition to the inhalation and ingestion routes, actinides
may under unusual circumstances of occupational exposure, enter the
body by absorption through the intact or punctured skin [I4, B1].
Normally, however, intact skin is an effective barrier to plutonium
entry, and this route of entry should not be of concern for general
environmental exposure.
443. Once deposited systemically, plutonium is tenaciously
retained. This fact is qualitatively evident from extensive data
on the excretion of plutonium by occupationally exposed humans
[V1]. It has also been quantitatively evaluated in a few human
cases and in a variety of experimental animals [I4, D5]. Based on
these data, ICRP has employed a biological half-time of 100 years
for plutonium in the skeleton, and a half-time of 40 years for
plutonium in liver; plutonium in gonads is assumed to be retained
without loss [I4, I2]. The same parameters are assumed to apply in
the case of americium.
E. DOSIMETRY
1. Dose per unit intake
444. The doses to the various tissues following inhalation or
ingestion or plutonium and americium are determined using the
parameters and models suggested by ICRP. A variety of estimates
are possible, depending on particle size of inhaled particles and
the solubility class of both inhaled and ingested material. The
values given below are for the representative 1 µm aerosol size and
for the insoluble oxide or hydroxide forms of plutonium.
445. The ICRP lung model divides the respiratory system into three
compartments. Deposition fractions in each region for the 1 µm
particle size are 29% in the nasopharyngeal region, 8% in the
tracheo-bronchial region and 23% in the pulmonary region. Smaller
particles have a progressively greater deposition fraction in the
pulmonary region and less retention in the nasopharyngeal region.
446. Inhaled, insoluble plutonium particles are assigned Class Y
parameters, retention in the lungs of the order of years (500 d
half-time for 60% of the pulmonary deposition). Because of greater
mobility of americium, all of its compounds are assigned Class W
parameters, with retention in the lungs of the order of weeks (50 d
half-time for 60% of the pulmonary deposition). In both cases, 40%
of the pulmonary deposition is cleared with a half-time of 1 d by
mucocilliary action through the tracheo-bronchial region.
Translocation from the naso-pharyngeal and tracheo-bronchial region
is rapid, within one day, with most of the material swallowed and
small amounts absorbed to blood.
447. For both inhaled and ingested material reaching blood,
fractional transfer to blood and liver is assumed to be 0.45 each
and 3.5 10-4 to gonads (testes) for both plutonium and americium.
Uptake to blood following ingestion of insoluble plutonium is 10-5
and 5 10-4 for all compounds of americium.
Table IX.3 Absorbed dose equivalent commitments per unit
intake of plutonium and americium (Sv per Bq)
---------------------------------------------------------------------------
Isotope Lung Liver Gonads Red bone Bone Effective
marrow lining a/
cells
---------------------------------------------------------------------------
Inhalation
238Pu 3.2 10-4 1.8 10-4 b/ 6.6 10-5 8.3 10-4 8.2 10-5
239Pu 3.2 10-4 2.1 10-4 b/ 7.6 10-5 9.5 10-4 8.9 10-5
240Pu 3.2 10-4 2.1 10-4 b/ 7.6 10-5 9.5 10-4 8.9 10-5
241Pu 3.2 10-6 4.4 10-6 2.8 10-7 1.7 10-6 2.1 10-5 1.6 10-6
242Pu b/ 5.5 10-4 3.2 10-5 2.0 10-4 2.5 10-3 1.4 10-4
Ingestion
238Pu b/ 4.0 10-8 2.3 10-9 1.5 10-8 1.8 10-7 1.5 10-8
239Pu b/ 4.4 10-8 2.6 10-9 1.6 10-8 2.1 10-7 1.6 10-8
240Pu b/ 4.4 10-8 2.6 10-9 1.6 10-8 2.1 10-7 1.6 10-8
241Pu b/ 8.6 10-10 5.7 10-11 3.4 10-10 4.2 10-9 2.5 10-10
242Pu b/ 2.3 10-6 1.4 10-7 8.4 10-7 1.1 10-5 5.9 10-7
---------------------------------------------------------------------------
a/ Effective dose equivalent commitment per unit intake (Sv per Bq).
b/ Doses which contribute < 10% to the effective dose equivalent commitment.
Note: for plutonium - Class Y and f1 = 10-5
for americium - Class W and f1 = 3 10-4.
Reference [I3].
448. The estimates of the absorbed dose equivalents per unit
intake are given in Table IX.3. These are computed over a 50 year
period following intake. They correspond to the transfer factors
P25 and P35 relating intake in air and diet, respectively, to
tissue dose. For these dose equivalent values, the quality factor
of 20 has been used for the alpha-emitters and 1 for the beta-
emitters (241Pu).
2. Dose per unit release
(a) Nuclear explosions
449. The dose equivalent commitments for plutonium and
americium released in nuclear explosions can be assessed using
the expressions
Dc = P25 Ia (inhalation)
Dc = P23 P35 F (ingestion)
where Ia is the cumulative intake from air (the integrated
concentration in air (Bq a m-3) times the breathing rate (22 m3
d-1) and F is the integrated deposition density. The values of
the transfer factors were discussed above.
450. For the past pattern of nuclear tests the population-weighted
global integrated concentration in air and deposition density can
be determined from comparisons with 90Sr [U1], from Pu-90Sr
production ratios, and from 241Pu decay considerations [B4]. The
results are for air: 1.5, 37, 25, 830 and 1.7 µBq a m-3 and for
deposition density: 0.85, 21, 14, 470 and 16 Bq m-2 for 238Pu,
239Pu, 240Pu, 241Pu and 241Am, respectively. With this
information, the dose equivalent commitments can be determined by
using the above expressions, and the values per unit activity
released can be obtained by dividing the results by the estimated
input amounts from nuclear testing (Table IX.2).
451. Finally, the collective dose equivalent commitments are
determined by multiplying by the appropriate global population.
For inhalation, the activity in air has by now been nearly
depleted, so the present global population applies (4 109 persons).
For ingestion of the short half-life 241Pu, the present population
applies and for the other isotopes, the long-term transfer can be
assumed to apply to the projected equilibrium value of 1010
persons. The results for the collective effective dose equivalent
commitment per unit release are included in the summary Table IX.4.
(b) Nuclear installation
452. The contribution of the inhalation pathway to the collective
dose commitments for plutonium and americium in airborne effluents
from nuclear installations can be estimated from the integrated
concentrations in air in the dispersion region. It has previously
been shown that the appropriate formula is
c I deltaN P25
S1 = ------------
vd
where I is the air breathing rate (22 m3 d-1), deltaN is the
population density in the region (25 man km-2), P25 is the dose per
unit intake factors (Table IX.3) and vd is the deposition velocity
(0.5 cm s-1). The integrated concentration in air is determined by
the amount released (unit activity) per unit area of the deposition
region divided by the deposition velocity. The areal dependence is
removed by multiplying by the population of the region (times the
area). The results from evaluating this expression are summarized
in Table IX.4.
Table IX.4 Summary of collective effective dose equivalent
commitments per unit activity released of plutonium and
americium (10-14 man Sv per Bq)
---------------------------------------------------------------
238Pu 239Pu 240Pu 241Pu 241Am
---------------------------------------------------------------
Nuclear explosions
Inhalation 1000 1000 1000 30 5 a/
Ingestion 3 30 10 0.01 300
Nuclear installations
Release to air b/
Inhalation 10000 10000 10000 200 20000
Ingestion 3 20 10 0.03 300
Release to fresh water
Drinking water 20 20 20 0.2 900
Fish 5 5 5 0.06 200
Release to salt water
Fish 0.01 0.01 0.01 0.0001 0.4
Shellfish 0.2 0.2 0.2 0.002 6
---------------------------------------------------------------
a/ Per Bq of 241Pu released.
b/ Assumes population density of 25 man km-2.
453. The contribution of the ingestion pathway from airborne
effluents to the collective dose commitment per unit activity
released, Sc1, can be determined by using the expression
c
S1 = P23 P35 SN
Using the values of the transfer factors given previously and a
population density, deltaN, of 25 man km-2 in the region of
deposition, the values summarized in Table IX.4 are obtained.
454. For the aquatic ingestion pathways the generalized UNSCEAR
model is utilized [U1]
c Nk Ik fk P35
S1 = -----------------
V(lambda + 1/tau)
to be evaluated for each pathway k. The quotient of water
receiving volume, V, and the population involved, Nk, is the water
utilization factor [U1], assumed to be a constant for each pathway.
A summary of the values used in the assessments by UNSCEAR is given
in the following listing:
Parameter fresh water sea water
1. tau, turnover time of 10 a 1 a
receiving water
2. Correction factor for 1.0 1.0
sediment removal
3. V, water utilization factor 3 107 1/man 3 109 1/man
N
4. fk, concentration factor
for item k
drinking water 0.1
fish 10 3
shellfish 300
5. Ik, consumption rate for
item k
drinking water 440 1/a
fish 1 kg/a 6 kg/a
shellfish 1 kg/a
455. Due to insufficient data, the values of the above listing
will be assumed to apply to all isotopes of plutonium and
americium. The values of the dose factor, P35, are given in Table
IX.3. A summary of the evaluated results is given in Table IX.4.
456. Assessments using the above model only account for that
portion of the dose given during the mean residence time of the
water in the receiving area. These are essentially complete
collective dose commitments for the release to fresh water and for
the shorter-lived isotopes (241Pu). The removal of the longer-
lived isotopes to the sediments of the deep ocean will largely
preclude any further contributions to the dose estimates.
F. REFERENCES
B1 Bair, W.J. Recent animal studies on the deposition, retention
and translocation of plutonium and other transuranic compounds.
p. 51-83 in Diagnosis and Treatment of Incorporated
Radionuclides. Proceedings of a seminar. Vienna, 1976.
B2 Ballou, J.E., K.R. Price, R.A. Gies et al. The influence of
DTPA on the biological availability of transuranics. Health
Phys. 34: 445 (1978).
B3 Bartelt, G.E., C.W. Wayman, S.E. Groves et al. 238-Pu and
239,240-Pu distribution in fish and invertebrates from the
Great Miami River, Ohio. p. 517-530 in Transuranics in Natural
Environments (M.G. White and P.B. Dunaway, eds.). Proceedings
of a symposium, Gatlinburg, Tennessee, 1976. ERDA report NV0-
178, Nevada Operations Office, NTIS, 1977.
B4 Bennett, B.G. Environmental aspects of americium.
Environmental Measurements Laboratory report EML-348, New York,
1978.
B5 Bennett, B.G. Transuranic element pathways to man. p. 367-383
in Transuranium Nuclides in the Environment. IAEA publication
STI/PUB/410, Vienna, 1976.
B6 Bondietti, E. and F.H. Sweeton. Transuranic speciation in the
environment. p. 449-476 in Transuranics in Natural
Environments (M.G. White and P.B. Dunaway, eds.). Proceedings
of a symposium, Gatlinburg, Tennessee, 1976. ERDA report NV0-
178, Nevada Operations Office, NTIS, 1977.
B7 Bowen, V.T. Plutonium and americium concentration along fresh
water chains of the Great Lakes, U.S.A. USAEC report C00-3568-
13, Woods Hole Oceanographic Institution, NTIS, 1976.
B8 Bowen, V.T., K.M. Wong and V.E. Noshkin. 239-Pu in and over
the Atlantic Ocean. J. Mar. Res. 29: 1 (1971).
D1 Dahlman, R.C. Transuranium elements in aquatic and terrestrial
environments. p. 131-147 in Environmental Sciences Division
annual progress report. USAEC report ORNL-5257, Oak Ridge
National Laboratory, NTIS, 1976.
D2 Dahlman, R.C., E.A. Bondietti and L.D. Eyman. Biological
pathways and chemical behaviour of plutonium and other
actinides in the environment. p. 47-80 in Actinides in the
Environment. ACS symposium series 35, American Chemical
Society, 1976.
D3 Delaney, M.S. and C.W. Francis. The relative uptake of 237-Pu
(IV) and Pu (VI) oxidation states from water by bushbeans.
Health Phys. 34: 492 (1978).
D4 Dobry, Jr., T.J. Transuranic elements in space nuclear power
systems. p. 83-85 in Transuranic Elements in the Environment
( W.C. Hanson, ed.). DOE publication series TID-22800,
Washington D.C., 1980.
D5 Durbin, P.W. Plutonium in man: a new look at the old data. p.
469-537 in Radiobiology of Plutonium (B.J. Stover and W.S.S.
Jee, eds.). J.W. Press, Salt Lake City, 1972
E1 Edgington, D.N. and J.A. Robbins. The behaviour of plutonium
and other long-lived radionuclides in Lake Michigan. II.
Patterns of deposition in the sediments. p. 245-260 in Impacts
of Nuclear Releases into the Aquatic Environment. Proceedings
of a symposium. Otaniemi, Finland 1975. IAEA publication
STI/PUB/406, Vienna, 1975.
E2 Edgington, D.N., M.A. Wahlgren and J.S. Marshall. The
behaviour of plutonium in aquatic ecosystems: a summary of
studies on the Great Lakes. p.45-80 in Environmental Toxicity
of Aquatic Radionuclides: Model and Mechanisms (M.W. Miller
and J.N. Stannard, eds.). Ann Arbor Science Publishers, Ann
Arbor, Michigan, 1976.
E3 Emery, R.M. and D.C. Kopfer. The distribution of transuranic
elements in a freshwater pond ecosystem. p. 269-286 in
Environmental Toxicity of Aquatic Radionuclides: Models and
Mechanisms (M.W. Miller and J.N. Stannard, eds.). Ann Arbor
Science Publishers, Ann Arbor, Michigan, 1976.
E4 Emergy, R.M., D.C. Kopfer and M.C. McShane. The migration of
plutonium from a freshwater ecosystem at Hanford in Transuranic
Elements in the Environment (W.C. Hanson, ed.). DOE
publication series, TID-22800, Washington, D.C. 1980.
E5 Energy Research and Development Administration. Environmental
research for transuranic elements in Workshop on Environmental
Research for Transuranic Elements. Proceedings of the
workshop, Battelle Seattle Research Centre, Seattle,
Washington, November 1975. ERDA report ERDA-76/134, NTIS
(1976).
E6 Essington, E.H., E.B. Fowler, R.O. Gilbert et al. Plutonium,
americium and uranium concentration in Nevada Test Site soil
profiles. p. 157-173 in Transuranium Nuclides in the
Environment. Proceedings of a symposium, San Fransisco, 1975.
IAEA publication STI/PUB/410, Vienna, 1976.
F1 Facer, G. Quantities of transuranic elements in the
environment from operations relating to nuclear weapons. p. 86-
91 in Transuranic Elements in the Environment (W.C. Hanson,
ed.). DOE publication series, TID-22800, Washington, D.C.,
1980.
F2 Fowler, S.W. and T.M. Beasley. Plutonium and americium in
fish. Nature 265: 384 (1977).
F3 Fowler, S.W. and J.C. Guary. High Absorption efficiency for
ingested plutonium in crabs. Nature 266: 827-828 (1977).
G1 Garland, T.R. and R.E. Wildung. Physiochemical
characterization of mobile plutonium species in soils, p. 254-
263 in Biological Implications of Metals in the Environment
(H. Drucker and R.E. Wildung, eds.). DOE symposium series,
Richland, Washington, 1975. CONF-750929 NTIS (1977).
G2 Guary, J.C. and A. Fraizier. Influence of trophic level and
calcification on the uptake of plutonium observed, in situ, in
marine organisms. Health Phys. 32: 21-28 (1977).
H1 Harley, J.H. Plutonium in the environment - a review. Prepared
for the 1979 annual meeting of the Radiation Research Society
of Japan, Osaka City, Japan, 1979.
H2 Hardy, E.P. World-wide distribution of plutonium. p. 115-128
in Plutonium and Other Transuranium Elements: Sources,
Environmental Distribution and Biomedical Effects (B.W.
Wachholz, ed.). Presented at the Environmental Protection
Agency Plutonium Standards Hearings, Washington, D.C., 1974.
U.S. Atomic Energy Commission, Springfield, Virginia, 1974.
H3 Hayes, D.W. and H.J. Horton. Plutonium and americium behaviour
in the Savannah River marine environment in Transuranic
Elements in the Environment (W.C. Hanson, ed.). DOE
publication series, TID-22800, Washington, D.C., 1980.
H4 Heeb, C.M. and E.T. Merrill. Significant actinide activities
in the LWR and LMFBR nuclear fuel cycle. EPA-520/2-75-006
(1974).
H5 Hetherington, J.A., D.F. Jefferies and M.B. Lovett. Some
investigations into the behaviour of plutonium in the marine
environment. p. 193-212 in Impacts of Nuclear Releases into
the Aquatic Environment. Proceedings of a symposium,
Ontaniemi, Finland, 1975. IAEA publication STI/PUB/406,
Vienna, 1975.
H6 Hetherington, J.A., D.F. Jefferies, N.T. Michell et al.
Environmental and public health consequences of the controlled
disposal of transuranic elements to the marine environment. p.
139-156 in Transuranium Nuclides in the Environment.
Proceedings of a symposium, San Fransisco, 1975. IAEA
publication STI/PUB/410, Vienna, 1976.
I1 International Atomic Energy Agency. The radiological basis of
the IAEA revised definition and recommendations concerning high
level radioactive waste unsuitable for dumping at sea. IAEA-
211 (1978).
I2 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers. ICRP publication 30,
Pergamon Press, 1979.
I3 International Commission on Radiological Protection. Limits for
intakes of radionuclides by workers, supplement to Part I.
ICRP publication 30, Pergamon Press, 1979.
I4 International Commission on Radiological Protection. The
metabolism of compounds of plutonium and other actinides. ICRP
publication 19, Pergamon Press, 1972.
I5 International Commission on Radiological Protection. Task
group on lung dynamics. Deposition and retention models for
internal dosimetry of the human respiratory tract. Health Phys.
12: 173-226 (1966).
K1 Kreiter, M.R., J.E. Mendel and R.W. McKee. Transuranic wastes
from the commercial light-water-reactor cycle. p. 92-106 in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
L1 Larsen, R.P. and R.D. Oldham. Plutonium in drinking water:
effects of chlorination on its maximum permissible
concentration. Science 201: 1008-1009 (1978).
L2 Livingstone, H.L. and V.T. Bowen. Americium in the marine
environment - relationships to plutonium. p. 107-130 in
Environmental Toxicity of Aquatic Radionuclides: Models and
Mechanisms (M.W. Miller and J.N. Stannard, eds.). Ann Arbor
Science Publishers, Ann Arbor, Michigan, 1976.
M1 Morley, F. and G.N. Kelly. Radiological protection and
transuranic wastes from the nuclear fuel cycle. Proceedings of
a NEA/CEA Seminar on the management of plutonium contaminated
solid wastes. Marcoule, October, 1974.
M2 McInroy, J.F., E.E. Campbell, W.D. Moss et al. Plutonium in
autopsy tissue: a revision and updating of data reported in
LA-4875. Health Phys. 37: 1-136 (1979).
M3 Miettinen, J.K., H. Mussalo, M. Hakanen et al. Distribution of
plutonium and americium in human and animal tissues after
chronic exposures. p. 265-268 (Vol. III) in Book of Papers.
Fifth International Congress of the International Radiation
Protection Association, Jerusalem, 1980.
M4 Miyake, Y. and Y. Sugimura. The plutonium content of Pacific
Ocean waters. p. 91-104 in Transuranium Nuclides in the
Environment. Proceedings of a symposium, San Francisco, 1975.
IAEA publication STI/PUB/410, Vienna, 1976.
M5 Muller, R.N., D.G. Sprugel and B. Kohn. Erosional transport
and deposition of plutonium and caesium in two small midwestern
watersheds. J. Environ. Qual. 7(2): 171-174 (1978).
N1 Nelson, D.M. and M.B. Lovett. The oxidation state of plutonium
in the Irish Sea. Nature 276: 599-601 (1978).
N2 Noshkin, V.E. and V.T. Bowen. Concentrations and distributions
of long-lived fallout radionuclides in open ocean sediments. p.
671-684 in Radioactive Contamination of the Marine
Enviornment. IAEA, Vienna, 1973.
N3 National Radiological Protection Board/CEA. Methodology for
evaluating the radiological consequences of radioactive
effluents released in normal operations. Joint report of
NRPB/CEA, CEC (1979).
N4 Nyhans, J.W., F.R. Miera, Jr. and R.E. Neher. Distribution of
plutonium in trinity soils after 28 years. J. Environ. Qual.
5(4): 431-437 (1976).
O1 Okabayashi, H., H. Watanabe and Y. Takizawa. Measurement of
plutonium in Japanese human organs. J. Radiat. Res. 19: 62-69
(1978).
P1 Paine, D. Plutonium in Rocky Flats freshwater systems in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
P2 Pentreath, R.J., D.F. Jefferies, M.B. Lovett et al. The
behaviour of transuranium and other long-lived radionuclides in
the Irish Sea and its relevance to the deep sea disposal of
radioactive wastes. Proceedings of the Third NEA Seminar on
Marine Radioecology, OECD, Paris, 1980.
P3 Pentreath, R.J. and M.B. Lovett. Transuranic nuclides in
plaice, pleuronectes platessa from the northeastern Irish Sea.
Marine Biol. 48: 19-26 (1978).
P4 Pentreath, R.J., D.S. Woodhead, B.R. Harvey et al. A
preliminary assessment of some naturally-occurring
radionuclides in marine organisms (including deep sea fish) and
the absorbed dose resulting from them. Proceedings of the Third
NEA Seminar on Marine Radioecology, OECD, Paris, 1980.
P5 Perkins, R.W. and C.W. Thomas. World-wide fallout. p. 53-82 in
Transuranic Elements in the Environment (W.C. Hanson, ed.).
DOE publication series, TID-22800, Washington, D.C., 1980.
P6 Pillai, K.C. and E. Mathew. Plutonium in the aquatic
environment, its behaviour, distribution and significance. p.
25-45 in Transuranium Nuclides in the Environment. Proceedings
of a symposium, San Francisco, 1975. IAEA publication
STI/PUB/410, Vienna, 1976.
R1 Rees, T.F., J.M. Cleveland and W.C. Gottschall. Dispersion of
plutonium from contaminated pond sediments. Environ. Sci.
Technol. 12(9): 1085-1087 (1978).
R2 Richmond, C.R. and R.L. Thomas. Plutonium and other actinide
elements in gonadal tissue of man and animals. Health Phys. 29:
241-250 (1975).
R3 Routson, R.C., G. Jansen and A.V. Robinson. 99-Tc, 237-Np and
241-Am sorption on two United States subsoils from differing
weathering intensity areas. Health Phys. 33: 311-317 (1977).
S1 Schell, W.R., F.G. Lowman and R.P. Marshall. Geochemistry of
transuranic elements at Bikini Atoll in Transuranic Elements
in the Environments (W.C. Hanson, ed.). DOE publication
series, TID-22800, Washington D.C., 1980.
S2 Simpson, H.J., R.M. Trier and C.R. Olsen. Transport of
plutonium by rivers in Transuranic Elements in the Environment
(W.C. Hanson, ed.). DOE publication series, TID-22800,
Washington, D.C., 1980.
S3 Sprugel, D.H. and G.E. Bartelt. Erosional removal of fallout
plutonium from a large midwestern watershed. J. Environ. Qual.
7(2): 175-177 (1978).
S4 Stather, J.W., J.D. Harrison, P. Rodwell et al. The
gastrointestinal absorption of plutonium and americium in the
hamster. Phys. Med. Biol. 24: 396-407 (1979).
S5 Sullivan, M.F. Absorption of actinide elements from the
gastrointestinal tract of neonatal animals. Health Phys. 38:
173-185 (1980).
S6 Sullivan, M.F. Absorption of actinide elements from the
gastrointestinal tract of rats, guinea pigs and dogs. Health
Phys. 38: 159-171 (1980).
S7 Sullivan, M.F., T.R. Garland, D.A. Cataldo et al. Absorption of
plutonium from the gastrointestinal tracts of rats and guinea
pigs after ingestion of alfalfa containing 238-Pu. Health
Phys. 38: 215-221 (1980).
S8 Sullivan, M.F., J.L. Ryan, L.S. Gorham et al. The influence of
oxidation state on the absorption of plutonium from the
gastrointestinal tract. Radiat. Res. 80: 116-121 (1979).
T1 Till, J.E., S.V. Kaye and J.R. Trabalka. Toxicity of uranium
and plutonium to the developing embryos of fish. ERDA report
ORNL-5160, Oak Ridge National Laboratory, NTIS, 1976.
U1 United Nations. Sources and Effects of Ionizing Radiation.
United Nations Scientific Committee on the Effects of Atomic
Radiation, 1977 report to the General Assembly, with annexes.
United Nations sales publication No. E.77.IX.I. New York,
1977.
U2 U.S. Nuclear Regulatory Commission. Environmental Assessment
of Ionization Chamber Smoke Detectors Containing Am-24l (R.
Belanger, D.W. Buckley and J.B. Swenson, eds.). Prepared for
the U.S. Nuclear Regulatory Commission by Science Applications,
Inc., La Jolla, California, 1979.
V1 Voelz, G.L., L.H. Hempleman, J.N.P. Lawrence et al. A 32-year
medical follow-up of Manhattan project plutonium workers.
Health Phys. 37: 445-485 (1979).
W1 Wahlgren, M.A., J.J. Alberts, K.A. Orlandini et al. A
comparison of the concentrations of fallout-derived plutonium
in a series of freshwater lakes. p. 92-94 in Radiological and
Environmental Research Division annual report, (1977). ERDA
report ANL-77-65 (Part 3), Argonne National Laboratory, NTIS
(1977).
W2 Wahlgren, M.A., J.J. Alberts, D.M. Nelson et al. Study of the
occurrence of multiple oxidation states of plutonium in natural
water systems. p. 95-98 in Radiological and Environmental
Research Division annual report (1977). ERDA report ANL-77-65
(Part 3), Argonne National Laboratory, NTIS (1977).
W3 Wahlgren, M.A., J.A. Robbins and D.N. Edgington. Plutonium in
the Great Lakes in Transuranic Elements in the Environment
(W.C. Hanson, ed.). DOE publication series, TID-22800,
Washington, D.C., 1980.
W4 Watters, R.L., D.N. Edgington, T.E. Hakonson et al. Synthesis
of the research literature in Transuranic Elements in the
Environment (W.C. Hanson, ed.). DOE publication series, TID-
22800, Washington, D.C., 1980.
W5 Weeks, M.H., J. Katz, W.O. Oakley et al. Further studies on
the gastrointestinal absorption of plutonium. Radiat. Res. 4:
339 (1956).
W6 Wildung, R.E., T.R. Garland and D.A. Cataldo. Environmental
processes leading to the presence of organically-bound
plutonium in plant tissues consumed by animals in Biological
Implications of Radionuclides Released from Nuclear Industries.
Proceedings of a symposium, Vienna, 1978. IAEA, Vienna, 1979.
W7 Wrenn, M.E. and N. Cohen. Determination of Pu-239/240 tissue
concentrations in non-occupationally exposed residents of New
York City. Annual report to the Department of Energy. New
York University Medical Centre report C00-2968-2. New
York,1978.
X. RADIATION EFFECTS
457. The interaction of radiation with matter results in the
liberation of energy carried by the alpha-particle, beta-particle,
gamma or x-rays and the ionization or excitation of the irradiated
material. In biological material, damage may be caused directly to
cell components by the radiation interactions or indirectly by the
actions of free radicals, the charged fragments of ionization
events. The damage may result in cell death or in cell
transformations, which at some later time may cause harmful effects
in the irradiated individual or in his offspring. The amount of
damage depends on the amount of radiation, which may be from
external irradiation or from radionuclides within the body, on the
type of radiation and on the sensitivity of the tissue.
458. Radiation effects in man are usually classified as somatic
and genetic or hereditary, according to whether they affect somatic
or germinal cells. Somatic damage is expressed therefore by
definition within the lifetime of the irradiated individual, while
genetic damage is expressed at some stage in his progeny. Somatic
effects are - somewhat loosely - further distinguished as immediate
or late depending on the time of their appearance.
A. SOMATIC EFFECTS
1. Early somatic effects
459. The immediate or early somatic effects are expressed in man
within a few days or a few weeks after exposure as a result of
damage to one or more of the self-renewing tissues. These effects
are also called functional because they are due to the inactivation
of a great number of functional cells of a given differentiative
line. Selective irradiation of a given tissue (as in the case of
exposure to some internal emitters) usually leads to effects
localized in that tissue; whole-body irradiation above a given
dose results, on the contrary, in the appearance of generalized
effects, usually under the form of a specific syndrome which
depends on dose. The clinical severity of the immediate effects
changes considerably with the dose, dose rate, type and energy of
the absorbed radiation and part of the body irradiated.
460. The nature of immediate effects is non-stochastic or
deterministic, in the sense that these effects are expected to
occur in an exposed individual absorbing doses in excess of a
minimum amount of radiation referred to as the threshold dose. The
threshold dose is extremely variable depending on the effect and
the tissue considered. It is of the order of a few tenths of a Gy
for most functional effects of importance for radiation protection.
The existence of a threshold of dose is an important characteristic
of these effects; it makes it virtually impossible for them to
appear for doses below the threshold, thus allowing their complete
avoidance.
461. Another important aspect of early effects to be emphasized in
relation to irradiation from internal emitters is the variability
of the dose threshold as a function of dose rate. Low dose rate
irradiation is normally a condition leading to an increase of the
threshold. Since irradiation from radionuclides (particularly at
the levels usually present in the environment) takes place at very
low doses and dose rates it is virtually impossible, except under
exceptional conditions of emergency, that immediate effects might
be observed. They do not deserve therefore any more extended
treatment.
2. Late somatic effects
462. Late somatic effects are those appearing in irradiated
individuals after a latency period and are expressed mainly in the
form of leukaemias or solid tumours. These effects are stochastic
or statistical in nature, in so far as it is impossible to identify
a causal relationship for them in any given case. The correlation
between radiation dose and induction of these conditions may only
be shown on large populations of irradiated individuals as an
increase of the above diseases over their apparently spontaneous
background incidence.
463. Since it is impossible to establish with any certainty the
shape of the dose-induction relationship for late tumorous effects,
particularly at low doses and dose rates, it is usually assumed
that the frequency of their occurrence is linear with dose and
without threshold. The clinical severity of these conditions is
variable, but it is commonly assumed for purposes of radiation
protection that they might be of a uniform and maximum severity,
namely the death of an individual.
B. GENETIC EFFECTS
464. Radiation-induced hereditary effects may appear in the
progeny of irradiated individuals within the first generation
following irradiation - in which case the damage is called dominant
- or within subsequent generations when the genes that carry the
same mutation in the male and in the female genetic complement
happen to match in the genome of the zygote. In this latter case
the genetic damage is called recessive. Clinically, radiation-
induced hereditary conditions have a large spectrum of severity
from the relatively trivial to the very harmful.
465. As in the case of the somatic late effects, radiation
protection usually makes reference to the most severe hereditary
diseases which are either incompatible with life or very disabling
for the individual. Radiation protection also assumes that for
this class of effects a linear non-threshold induction relationship
with dose may apply.
C. DOSE-RESPONSE RELATIONSHIPS
466. In experimental animals and in man late somatic and
hereditary effects may exhibit different shapes of the dose-effect
relationships, according to a large number of physical and
biological variables operating in each particular system. Linear,
linear-quadratic, quadratic or complex relationships have been
described in various circumstances. No generalization may be gained
by the consideration of all existing experience, except perhaps
that each specific system responds according to different kinetics
of action and that biologically complex effects usually correspond
to more complex types of relationships. It would be impossible to
set up a rationale for a system of radiation protection by
considering each case separately. To overcome this difficulty the
assumption is made that late somatic and hereditary effects of
irradiation follow a non-threshold linear function of dose. This
assumption is simple and there is evidence that it is also a
conservative assumption in most cases.
467. It is important to stress the meaning of the assumption of
non-threshold linearity. It postulates, on the one hand, that
there is no dose, however small, that may in principle be
considered safe and no dose increment, however small, which could
not produce a corresponding increase of effect and therefore of
risk. The summation of doses taken as a measure of total risk and
calculations of collective doses as expression of the total
detriment in the exposed population have little meaning outside the
assumption of non-threshold linearity.
468. On the other hand, adoption of the assumption of linearity
involves implicitly the adoption of other important principles.
Actually, if there is a linear relationship between the dose and
the induction of stochastic effects, it becomes possible to use the
average dose received by a given organ or tissue as a significant
reference quantity. Under these conditions it becomes unnecessary
to consider the dose variability within the given organ or tissue
because the response of the component cells (taken to be of uniform
sensitivity) will in any case be linear with the absorbed dose.
The reactions of the component cells will in any case sum-up to
produce an overall effect corresponding to that expected from the
mean dose in that organ or tissue.
469. In case of internal irradiation there may be problems related
to the presence of point sources. The following considerations
apply in these cases. Firstly, with regard to non-stochastic
effects, cell death resulting from high doses within microscopic
volumes of the tissue are expected to produce less harm than for
the same dose uniformly distributed within large volumes: this is
because killing of transformed cells by high doses would be
expected to lead to inactivation of potentially transformed cells.
Moreover, loss of cells around the zones of highest dose absorption
would not be expected to result in significant decrease in tissue
or organ function, unless the functional reserve of the organ or
tissue is impaired for other reasons.
D. RISK ESTIMATES
470. UNSCEAR has extensively reviewed in its 1977 report most
information on the subject of tumour induction in man by ionizing
radiation. The Committee concluded that the risk for all fatal
malignancies after whole-body irradiation at low doses and dose
rates of low-LET radiation - as an average of both sexes and all
ages - is in the region of 10-2 Sv-1. The risk of inducing non-
fatal malignancies under the same conditions would probably fall in
the same range.
471. It should be emphasized that for doses of the order of those
received annually from natural sources no direct information is
available. The above estimate is derived predominantly from
observations conducted on people exposed to absorbed doses of over
1 Gy. While the rate per unit dose or doses in the region of the
natural background would unlikely be higher, it could, however, be
substantially lower. There is no evidence that irradiation from
internal sources would produce rates of tumour induction differing
from those from external irradiation, if account is taken of the
mean absorbed doses in tissues.
472. Concerning hereditary effects, the Committee estimated that
when a population is continuously exposed to low doses of low-LET
radiation at rates of the order of 10-2 Gy per generation (a
generation corresponds to about 30 years) about 50 genetic diseases
might be expected to occur per one million first generation
progeny. At equilibrium, the total genetic damage expressed over
all generations (or the value in each generation reached after
prolonged continuous exposure) would be of the order of 150 cases
per million progeny.
473. It is often convenient to be able to estimate the total
detriment to the individuals from irradiation of specific organs or
tissues, taking into account the various types of effects from
various irradiations. This could be a difficult procedure,
however, in cases when different irradiation modalities lead to
different effects. Such a situation applies for irradiation by
internally deposited radionuclides, when the nuclides produce
different doses in tissues of varying sensitivity and the effects
must be added to the effects of whole-body irradiation.
474. In order to overcome this difficulty, ICRP, in its
publication 26, has designed a system allowing combined estimates
of risk in various organs and tissues, based on their
susceptibility to various effects. These risks of effects of
irradiations of specific tissues are weighted relative to the total
effect from whole-body irradiation. The risks apply to one
individual or the whole exposed population, making use of the
hypothesis of linearity. It is realized, of course, that the
applicability of risk estimates may vary according to the
characteristics of the individual (genetic make-up, sex, age, etc.)
or to the structure of the exposed population. But it is also held
that an acceptable level of precision may be reached by assuming an
average risk value to be applied to all members of the population
irrespective of the above mentioned variability.
475. The mortality risks and weighting factors recommended by ICRP
are shown in Table X.1. These factors have been derived for the
protection of workers, but may also be applied to large population
groups, provided allowance is made for the hereditary effects which
would be expected to appear in all generations subsequent to the
second.
476. It must be emphasized that the risk estimates for induction
of somatic and genetic effects should be regarded as the best
possible numerical conclusions to be drawn from a very
heterogeneous data base affected by various types of dosimetric and
epidemiological uncertainties. Although it is felt that such
estimates are reasonably precise for the purpose of radiation
protection they are to be interpreted and used in a statistical
sense simply as illustrations of the order of magnitude of
potential risks. The actual validity of these estimates could not
possibly be tested empirically under normal circumstances owing to
the small levels of the risks compared with the far higher
"spontaneous" background of similar conditions in the general
population.
Table X.1 Mortality risk and weighting factors for
different organs (from ICRP publication 26)
------------------------------------------------------------
Tissue Mortality risk Weighting
(Sv-1) factor
------------------------------------------------------------
Breast 0.25 10-2 0.15
Red bone marrow 0.2 10-2 0.12
Lungs 0.2 10-2 0.12
Thyroid 0.05 10-2 0.03
Bone surface 0.05 10-2 0.03
Remainder 0.5 10-2 0.30
Gonads (hereditary effects in
the first two generations) 0.4 10-2 0.25
------------------------------------------------------------
Total 1.62 10-2 1.00
------------------------------------------------------------
XI. CONCLUSIONS
A. RADIONUCLIDES AND THE ENVIRONMENT
477. Radiation is a natural feature of man's environment - from the
high energy charged particles which make up cosmic radiation to the
radioactive decay of radionuclides in the earth's crust and in the
biosphere. Several of the radionuclides considered in this
document have important natural sources, including tritium, carbon-
14, krypton-85 and radon. The first three are produced mainly by
cosmic ray interactions in the atmosphere. Radon arises from the
decay of radium present in the earth's crust. All of these
radionuclides are widely dispersed in air. Tritium and 14C enter
more general bio-geochemical cycles, the hydrological cycle and the
carbon cycle, respectively.
478. Several activities of man result in the production of
radionuclides and contribute to the radiation background. An
important source has been the testing of nuclear weapons in the
atmosphere. Large scale atmospheric testing was completed prior to
the Test Ban Treaty of 1963, but additional tests by some countries
have continued. The radionuclides produced in atmospheric nuclear
explosions become widely dispersed in the atmosphere, primarily of
the hemisphere in which the test was conducted, but with some
interhemispheric mixing contribute to the global exposures to
fallout radioactivity. Important fission radionuclides in terms of
the doses delivered include 131I, 90Sr and 137Cs. In the long-
term, the radionuclides 14C and isotopes of plutonium become
important contributors to the radiation dose.
479. The generation of electricity using nuclear power reactors
also results in the production of radionuclides and in some
radiation exposure of man. The radioactive materials are largely
contained within the fuel elements in the reactor or in waste
treatment systems at the fuel reprocessing plant. Releases of
controlled amounts occur in liquid and airborne effluents from the
nuclear installations. Accidents could result in potentially
greater releases of radioactive materials.
480. The natural and man-made sources of radionuclides in the
environment have been discussed for the various radionuclides
considered in this document. The amounts of radioactive materials
released from the various sources have been reviewed and the
relevant values have been shown to depend very much on specific
past practices. Even the normalized release amounts, for example
the amounts per Mt in nuclear explosions or per MW(e)a of
electricity generated, have limited validity. The amounts released
in atmospheric nuclear testing depend on the types of devices
tested and on the geographic pattern of past testing. Releases
from nuclear power installations depend on the efficiency and
integrity of present designs and on the specific waste management
practices currently utilized.
481. The behaviour of radionuclides in the environment has been
studied rather extensively, so that by now, the dispersion of these
pollutants in the environment is fairly well understood. Values
have been derived for the various transfer factors which describe
the transport of radionuclides in the environment and their
transfer to man. For the purpose of dose assessment it is
necessary to consider only the more specific aspects of particular
release situations and to adjust the more generally valid
parameters to the local conditions.
B. DOSE ASSESSMENTS
482. Exposure of man to radionuclides in the environment occurs by
inhalation of amounts in air, ingestion of amounts incorporated
into diet, or from external exposure to radionuclides in air or
deposited on the ground. The dose assessments for the
radionuclides considered in this document have been directed toward
obtaining estimates of the collective effective dose equivalent
commitments per unit amount of activity of the radionuclide
released. This expression of dose gives the absorbed dose,
weighted for radiation type and sensitivity of irradiated tissues
to the entire population and for as long as the exposures from a
specific release continue. This quantity is expected to be most
directly related to the total health detriment which may result.
The results of the dose assessments are summarized in Table XI.1.
483. The degree of transfer of the radionuclides to man and thus
the dose estimates vary depending on the source of release of the
radionuclides. Natural production of tritium, 14C and 85Kr occurs
primarily in the upper atmosphere, following which there is
widespread dispersion, and for 3H and 14C generalized cycling
throughout the environment. Nuclear explosions conducted in the
atmosphere, particularly large scale tests, result in injection of
debris into the stratosphere, from where the radionuclides are
globally dispersed. Releases of radionuclides from nuclear
installations are near surface emissions in airborne effluents or
in liquid effluents to rivers and lakes or to the marine
environment. Exposures are primarily to the local and regional
populations, although the longer-lived radionuclides may also
become more widely dispersed.
484. Release of unit activity of a radionuclide to the environment
generally results in the lowest collective effective dose
equivalent commitment when the release is to the marine
environment. In this case, for the generalized situation
considered here, transfer of radionuclides to man occurs only
through ingestion of fish and shellfish. There may, however, be
important specialized pathways of transfer in more specific release
circumstances.
485. Somewhat greater collective doses result from releases to
freshwater systems, particularly if the water is subsequently used
for drinking. Transfer of radionuclides to fish may also be
somewhat more in fresh water than in the ocean.
Table XI.1 Collective effective dose equivalent commitments
per unit activity released (10-12 man Sv per Bq)
-------------------------------------------------------------
Source
Nuclear installations
Radio- Nuclear Airborne Release to Marine
nuclide Natural explosions release fresh water release
-------------------------------------------------------------
3H 0.0005 0.0008 0.0009 0.003 0.0008
14C 120 120 120 120 120
85Kr 0.0002 0.0002 0.0002 0.0002 0.0002
90Sr - 0.6 1 1 0.00006
129I - 30000 30000 30000 30000
131I - 0.00009 0.4 0.009 0.00006
137Cs - 2 5 0.6 0.001
222Rn 0.001 - 0.001 - -
238Pu - 10 100 0.3 0.002
239Pu - 10 100 0.3 0.002
240Pu - 10 100 0.3 0.002
241Pu - 0.3 2 0.pp3 0.00002
241Am - 3 200 10 0.06
-------------------------------------------------------------
These values are only approximations of a very generalized nature.
The text contains a full discussion of limitations and of the
specific assumptions utilized.
486. The largest doses generally result from airborne releases.
The radionuclides in air may be inhaled, although the contributions
to the dose from this pathway are usually small. More importantly,
the deposited radionuclides are available for incorporation into
the general diet. Long-term transfer of the radionuclides to man
may then occur.
487. The greatest contributors to the collective dose commitments
per unit activity released are the longest-lived radionuclides.
Specifically, the very low dose rates from 129I result in
substantial collective doses over the millions of years mean
radioactive lifetime of this radionuclide. The same is true of 14C
and some isotopes of plutonium, for which the mean radioactive
lifetimes are of the order of thousands of years. Of course, the
validity of assessments which involve such long-term projections
must be questioned. These results can not be related to
equilibrium dose rates to the world's population. For this
purpose, limiting the integration periods to the duration of the
practices, (for example a few hundred years at the most for nuclear
power production) obtaining the incomplete collective dose
commitments, has some merit.
C. EFFECTS EVALUATION
488. The final step in evaluating the consequences of releasing
radionuclides into the environment, once exposures have been
determined, is to estimate the health effects. A great deal of
study has been made of radiation exposure-response relationships
and risk estimates have been formulated for the various effects.
There is uncertainty, however, in knowing whether the risk
estimates, which are generally only obtainable at higher doses and
dose rates, will be valid for the low level chronic exposures of
environmental situations.
489. It is not the intention of this document to provide detailed
guidance regarding effects evaluations. This requires more
detailed specifications of the radionuclides present in the sources
and the amounts released and closer consideration of the
environmental conditions and the consequent exposures to the
population involved. Only a brief summary has been presented
(Chapter X) of the main aspects of radiation effects and of the
considerations involved in assigning risk estimates.
490. In the generally accepted philosophy of radiation protection,
all exposures are considered to increase the risk of harmful
effects. Increased risks are only justified when balanced by net
positive benefits from the radiation operations. The conceptual
basis for measuring benefits and accounting for acceptability of
risks are topics under review by national and international bodies.
491. The first part of the procedure of assessing the consequences
of releasing radionuclides into the environment - the specification
of sources, of environmental behaviour of radionuclides and of the
consequent exposures - appears, in general, to be well founded, as
shown by the reviews of radionuclide behaviour and dosimetry of
this document. This contributes in a general sense to the
establishment of health criteria for these radionuclides. The
usual precautions must be expressed in applying general criteria to
specific situations. Considerable judgement is always required to
make meaningful evaluations to serve as proper guides for future
actions. It should also be appreciated that the results of
radiological assessments are but one aspect of the considerations
on which decisions must be made. The better knowledge that has
developed on these aspects should not unduly condition other
factors, such as socio-economic considerations, which are also of
importance in the rational selection of various possible options.
ANNEX I
Excerpts from "Basic Safety Standards for Radiation Protection 1982
Edition", (Safety Series No. 9) IAEA