
Environmental Health Criteria 227
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization or the World Health Organization.
First draft prepared by Dr R. Liteplo and Ms R. Gomes, Health Canada, Ottawa, Canada and Mr P. Howe and Mr H. Malcolm, Centre for Ecology and Hydrology, Cambridgeshire, United Kingdom
Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization Geneva, 2002
The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO) and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals.
The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment.
WHO Library Cataloguing-in-Publication Data
Fluorides.
(Environmental health criteria ; 227)
1.Fluorides - adverse effects 2.Environmental exposure 3.Occupational exposure
4.Risk assessment I.International Programme for Chemical Safety II.Series
ISBN 92 4 157227 2 (NLM classification: QV 282)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available.
©World Health Organization 2002
Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters.
The Federal Ministry for the Environment, Nature Conservation and Nuclear Safety, Germany, provided financial support for, and undertook the printing of this publication.
ENVIRONMENTAL HEALTH CRITERIA FOR FLUORIDES
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria monographs as accurately as possible without unduly delaying their publication. In the interest of all users of the Environmental Health Criteria monographs, readers are requested to communicate any errors that may have occurred to the Director of the International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland, in order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the International Register of Potentially Toxic Chemicals, Case postale 356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41 22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number 5 U01 ES02617-15 from the National Institute of Environmental Health Sciences, National Institutes of Health, USA, and by financial support from the Federal Ministry for the Environment, Nature conservation and Nuclear Safety, Germany.
Environmental Health Criteria
Objectives
In 1973, the WHO Environmental Health Criteria Programme was initiated with the following objectives:
The first Environmental Health Criteria (EHC) monograph, on mercury, was published in 1976, and since that time an ever-increasing number of assessments of chemicals and of physical effects have been produced. In addition, many EHC monographs have been devoted to evaluating toxicological methodology, e.g., for genetic, neurotoxic, teratogenic and nephrotoxic effects. Other publications have been concerned with epidemiological guidelines, evaluation of short-term tests for carcinogens, biomarkers, effects on the elderly and so forth.
Since its inauguration, the EHC Programme has widened its scope, and the importance of environmental effects, in addition to health effects, has been increasingly emphasized in the total evaluation of chemicals.
The original impetus for the Programme came from World Health Assembly resolutions and the recommendations of the 1972 UN Conference on the Human Environment. Subsequently, the work became an integral part of the International Programme on Chemical Safety (IPCS), a cooperative programme of UNEP, ILO and WHO. In this manner, with the strong support of the new partners, the importance of occupational health and environmental effects was fully recognized. The EHC monographs have become widely established, used and recognized throughout the world.
The recommendations of the 1992 UN Conference on Environment and Development and the subsequent establishment of the Intergovernmental Forum on Chemical Safety with the priorities for action in the six programme areas of Chapter 19, Agenda 21, all lend further weight to the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews on the effects on human health and the environment of chemicals and of combinations of chemicals and physical and biological agents. As such, they include and review studies that are of direct relevance for the evaluation. However, they do not describe every study carried out. Worldwide data are used and are quoted from original studies, not from abstracts or reviews. Both published and unpublished reports are considered, and it is incumbent on the authors to assess all the articles cited in the references. Preference is always given to published data. Unpublished data are used only when relevant published data are absent or when they are pivotal to the risk assessment. A detailed policy statement is available that describes the procedures used for unpublished proprietary data so that this information can be used in the evaluation without compromising its confidential nature (WHO (1999) Revised Guidelines for the Preparation of Environmental Health Criteria Monographs. PCS/99.9, Geneva, World Health Organization).
In the evaluation of human health risks, sound human data, whenever available, are preferred to animal data. Animal and in vitro studies provide support and are used mainly to supply evidence missing from human studies. It is mandatory that research on human subjects is conducted in full accord with ethical principles, including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and international authorities in making risk assessments and subsequent risk management decisions. They represent a thorough evaluation of risks and are not, in any sense, recommendations for regulation or standard setting. These latter are the exclusive purview of national and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized meetings of scientists to establish lists of priority chemicals for subsequent evaluation. Such meetings have been held in: Ispra, Italy, 1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North Carolina, USA, 1995. The selection of chemicals has been based on the following criteria: the existence of scientific evidence that the substance presents a hazard to human health and/or the environment; the possible use, persistence, accumulation or degradation of the substance shows that there may be significant human or environmental exposure; the size and nature of populations at risk (both human and other species) and risks for the environment; international concern, i.e., the substance is of major interest to several countries; adequate data on the hazards are available.
If an EHC monograph is proposed for a chemical not on the priority list, the IPCS Secretariat consults with the cooperating organizations and all the Participating Institutions before embarking on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC monograph is shown in the flow chart on the next page. A designated staff member of IPCS, responsible for the scientific quality of the document, serves as Responsible Officer (RO). The IPCS Editor is responsible for layout and language. The first draft, prepared by consultants or, more usually, staff from an IPCS Participating Institution, is based initially on data provided from the International Register of Potentially Toxic Chemicals and from reference databases such as Medline and Toxline.
The draft document, when received by the RO, may require an initial review by a small panel of experts to determine its scientific quality and objectivity. Once the RO finds the document acceptable as a first draft, it is distributed, in its unedited form, to well over 150 EHC contact points throughout the world who are asked to comment on its completeness and accuracy and, where necessary, provide additional material. The contact points, usually designated by governments, may be Participating Institutions, IPCS Focal Points or individual scientists known for their particular expertise. Generally, some four months are allowed before the comments are considered by the RO and author(s). A second draft incorporating comments received and approved by the Director, IPCS, is then distributed to Task Group members, who carry out the peer review, at least six weeks before their meeting.
The Task Group members serve as individual scientists, not as representatives of any organization, government or industry. Their function is to evaluate the accuracy, significance and relevance of the information in the document and to assess the health and environmental risks from exposure to the chemical. A summary and recommendations for further research and improved safety aspects are also required. The composition of the Task Group is dictated by the range of expertise required for the subject of the meeting and by the need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the important role played by nongovernmental organizations. Representatives from relevant national and international associations may be invited to join the Task Group as observers. While observers may provide a valuable contribution to the process, they can speak only at the invitation of the Chairperson. Observers do not participate in the final evaluation of the chemical; this is the sole responsibility of the Task Group members. When the Task Group considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers participate in the preparation of the EHC monograph must, in addition to serving in their personal capacity as scientists, inform the RO if at any time a conflict of interest, whether actual or potential, could be perceived in their work. They are required to sign a conflict of interest statement. Such a procedure ensures the transparency and probity of the process.
When the Task Group has completed its review and the RO is satisfied as to the scientific correctness and completeness of the document, the document then goes for language editing, reference checking and preparation of camera-ready copy. After approval by the Director, IPCS, the monograph is submitted to the WHO Office of Publications for printing. At this time, a copy of the final draft is sent to the Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the updating of an EHC monograph: new data are available that would substantially change the evaluation; there is public concern for health or environmental effects of the agent because of greater exposure; an appreciable time period has elapsed since the last evaluation.
All Participating Institutions are informed, through the EHC progress report, of the authors and institutions proposed for the drafting of the documents. A comprehensive file of all comments received on drafts of each EHC monograph is maintained and is available on request. The Chairpersons of Task Groups are briefed before each meeting on their role and responsibility in ensuring that these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR FLUORIDES
Members
Professor Peter Aggett, Lancashire Postgraduate School of Medicine and Health, University of Central Lancashire, Preston, Lancashire, United Kingdom
Dr Roberto Belmar, Environmental Health Division, Ministry of Health, Santiago, Chile (Chairman)
Dr John Bucher, Environmental Toxicology Program, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, USA
Dr Julio Camargo, Ecology Department, Faculty of Science, University of Alcalá, Madrid, Spain
Dr Jane Cauley, Department of Epidemiology, University of Pittsburgh, Pittsburgh, PA, USA
Professor Jan Ekstrand, Department of Basic Oral Sciences, Karolinska Institute, Stockholm, Sweden
Mr Paul Howe, Centre for Ecology and Hydrology, Monks Wood, Abbots Ripton, Huntingdon, Cambridgeshire, United Kingdom (Co-Rapporteur)
Dr Gopalakrishnan Karthikeyan, Department of Chemistry, Gandhigram Rural Institute, Gandhigram, Tamil Nadu, India
Dr Uwe Kierdorf, Institute of General and Systematic Zoology, Justus-Liebig-University of Giessen, Giessen, Germany
Dr Päivi Kurttio, Radiation and Nuclear Safety Authority, Helsinki, Finland
Dr Robert Liteplo, Existing Substances Division, Bureau of Environmental Contaminants, Health Canada, Ottawa, Ontario, Canada (Co-Rapporteur)
Dr Akiyoshi Nishikawa, National Institute of Health Sciences, Tokyo, Japan
Mr Daryl Stevens, Land and Water, Commonwealth Scientific and Industrial Research Organisation, Adelaide, Australia
Professor Paolo Vineis, Department of Biomedical Science and Human Oncology, University of Torino, Torino, Italy
Dr Jin Yinlong, Institute of Environmental Health and Engineering, Chinese Academy of Preventive Medicine, Beijing, People’s Republic of China
Secretariat
Dr Antero Aitio, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland (Secretary)
Dr Bing Heng Chen, Department of Environmental Health, School of Public Health, Shanghai Medical University, Shanghai, People’s Republic of China
Mr John Fawell, Director, Environmental Division, Warren Associates, Devizes, Wiltshire, United Kingdom
Ms Rose Gomes, Existing Substances Division, Bureau of Environmental Contaminants, Health Canada, Ottawa, Ontario, Canada
Dr Philip Jenkins, International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland
ENVIRONMENTAL HEALTH CRITERIA FOR FLUORIDES
A WHO Task Group on Environmental Health Criteria for Fluorides met at the Institute of Environmental Health and Engineering of the Chinese Academy of Preventive Medicine in Beijing, People’s Republic of China, on 28 May – 1 June 2001. The group reviewed the draft document and the peer review comments and revised and further updated the draft, including the evaluation of the risks for human health and the environment from exposure to fluorides.
The first and second drafts of this monograph were prepared by Dr R. Liteplo, Health Canada, Canada, and Mr P. Howe, Centre for Ecology and Hydrology, United Kingdom. The document was sent for peer review to the IPCS contact points and additional experts on fluoride. The authors, in collaboration with the IPCS Secretariat, revised the document based on the comments received. Following an updating at the end of 2000, the document was sent for review to the Task Group members and further revised based on these comments.
Peer review comments were received from the following:
Dr J. Ahlers, Umwelt Bundes Amt, Germany
Dr R. Benson, Region VIII, US Environmental Protection Agency, USA
Professor G.B. Bliss, N.N. Petrov’s Research Institute of Oncology, Russian Federation
Dr M. Bolger, US Food and Drug Administration, USA
Dr J. Bucher, National Institute of Environmental Health Sciences, USA
Dr J. Camargo, University of Alcalá, Spain
Dr S. Cao, Chinese Academy of Preventive Medicine, People’s Republic of China
Dr F.M. Carpanini, European Centre for Ecotoxicology and Toxicology of Chemicals, Belgium
Dr J. Cauley, University of Pittsburgh, USA
Dr L.K. Cohen, National Institute of Dental Research, USA
Dr A. Conacher, Health Canada, Canada
Dr P. Dargan, National Poison Information Service, United Kingdom
Professor I. Dési, Albert Szent-Györgyi University, Hungary
Dr J. Donohue, US Environmental Protection Agency, USA
Dr J. Ekstrand, Karolinska Institute, Sweden
Dr L. Friberg, Karolinska Institute, Sweden
Dr R. Hertel, Federal Institute for Health Protection of Consumers and Veterinary Medicine, Germany
Dr C. Hiremath, National Center for Environmental Assessment, US Environmental Protection Agency, USA
Dr B.L. Johnson, Agency for Toxic Substances and Disease Registry, USA
Dr G. Karthikeyan, Gandhigram Rural Institute, India
Dr U. Kierdorf, Justus-Liebig-University of Giessen, Germany
Dr J. Kriz, National Institute of Public Health, Czech Republic
Dr P. Kurttio, Radiation and Nuclear Safety Authority, Finland
Dr P. Lundberg, National Institute for Working Life, Sweden
Dr E. Ohanian, Office of Water, US Environmental Protection Agency, USA
Dr Y.A. Rakhmanine, Sysin Research Institute of Human Ecology and Environmental Health, Russian Federation
Dr D. Renshaw, Department of Health, United Kingdom
Dr J.M. Rice, International Agency for Research on Cancer, France
Dr T.G. Rossman, New York University School of Medicine, USA
Dr U. Schlottman, Federal Ministry for the Environment, Nature Conservation and Nuclear Safety, Germany
Dr P.A. Schulte, National Institute for Occupational Safety and Health, USA
Dr D. Stevens, Commonwealth Scientific and Industrial Research Organisation, Australia
Dr G. Ungváry, National Institute of Occupational Health, Hungary
Dr P. Vineis, University of Torino, Italy
Ms J. Walter, Swedish Poisons Information Centre, Sweden
Dr G. Whitford, Medical College of Georgia, USA
Dr A. Aitio of the IPCS central unit was responsible for the scientific aspects of the monograph, and Ms M. Sheffer, Ottawa, Canada, for the technical editing.
The efforts of all who helped in the preparation and finalization of the monograph are gratefully acknowledged.
|
ATP |
adenosine triphosphate |
|
ATPase |
adenosine triphosphatase |
|
CAS |
Chemical Abstracts Service |
|
CI |
confidence interval |
|
DNA |
deoxyribonucleic acid |
|
EC50 |
median effective concentration |
|
EHC |
Environmental Health Criteria monograph |
|
FAO |
Food and Agriculture Organization of the United Nations |
|
HMDS |
hexamethyldisiloxane |
|
IARC |
International Agency for Research on Cancer |
|
ILO |
International Labour Organization |
|
IPCS |
International Programme on Chemical Safety |
|
IQ |
intelligence quotient |
|
JECFA |
Joint FAO/WHO Expert Meeting on Food Additives |
|
JMPR |
Joint FAO/WHO Meeting on Pesticide Residues |
|
LC50 |
median lethal concentration |
|
LD50 |
median lethal dose |
|
LOEC |
lowest-observed-effect concentration |
|
LOEL |
lowest-observed-effect level |
|
LT50 |
median lethal time |
|
MATC |
maximum acceptable toxicant concentration |
|
NOEC |
no-observed-effect concentration |
|
NTP |
National Toxicology Program (USA) |
|
OR |
odds ratio |
|
RO |
Responsible Officer |
|
RR |
relative risk |
|
SD |
standard deviation |
|
UN |
United Nations |
|
UNEP |
United Nations Environment Programme |
|
WHO |
World Health Organization |
This document focuses on environmental exposure to fluoride derived mostly from inorganic sources and its effects on humans, animals and other biota. Data on hydrogen fluoride, calcium fluoride, sodium fluoride, sulfur hexafluoride and silicofluorides are covered, as these compounds are considered to be the most relevant of the inorganic fluorides on the basis of quantities released to the environment, environmental concentrations and toxicological effects on living organisms.
Hydrogen fluoride (HF) is a colourless, pungent liquid or gas that is highly soluble in organic solvents and in water, in which it forms hydrofluoric acid. Calcium fluoride (CaF2) is a colourless solid that is relatively insoluble in water and dilute acids and bases. Sodium fluoride (NaF) is a colourless to white solid that is moderately soluble in water. Sulfur hexafluoride (SF6) is a colourless, odourless, inert gas that is slightly soluble in water and readily soluble in ethanol and bases.
The most common procedure used to quantify free fluoride anion is the fluoride ion-selective electrode. Microdiffusion techniques are considered to be the most accurate methods of sample preparation (i.e., liberation of free ionic fluoride from organic and inorganic complexes).
Fluorides are released into the environment naturally through the weathering and dissolution of minerals, in emissions from volcanoes and in marine aerosols. Fluorides are also released into the environment via coal combustion and process waters and waste from various industrial processes, including steel manufacture, primary aluminium, copper and nickel production, phosphate ore processing, phosphate fertilizer production and use, glass, brick and ceramic manufacturing, and glue and adhesive production. The use of fluoride-containing pesticides as well as the controlled fluoridation of drinking-water supplies also contribute to the release of fluoride from anthropogenic sources. Based on available data, phosphate ore production and use as well as aluminium manufacture are the major industrial sources of fluoride release into the environment.
Hydrogen fluoride is an important industrial compound that is used mainly in the production of synthetic cryolite (Na3AlF6), aluminium fluoride (AlF3), motor gasoline alkylates and chlorofluorocarbons, with an annual world consumption in excess of 1 million tonnes. It is also used in etching semiconductor devices, cleaning and etching glass, cleaning brick and aluminium and tanning leather, as well as in commercial rust removers. Calcium fluoride is used as a flux in steel, glass and enamel production, as the raw material for the production of hydrofluoric acid and anhydrous hydrogen fluoride, and as an electrolyte in aluminium production. Sodium fluoride is used in the controlled fluoridation of drinking-water, as a preservative in glues, in glass and enamel production, as a flux in steel and aluminium production, as an insecticide and as a wood preservative. Sulfur hexafluoride is used extensively in various electronic components and in the production of magnesium and aluminium. Fluorosilicic acid (H2SiF6) and sodium hexafluorosilicate (Na2SiF6) are used for the fluoridation of drinking-water supplies.
Fluorides in the atmosphere may be in gaseous or particulate form. Atmospheric fluorides can be transported over large distances as a result of wind or atmospheric turbulence or can be removed from the atmosphere via wet and dry deposition or hydrolysis. Fluoride compounds, with the exception of sulfur hexafluoride, are not expected to remain in the troposphere for long periods or to migrate to the stratosphere. Sulfur hexafluoride has an atmospheric residence time ranging from 500 to several thousand years.
The transport and transformation of fluoride in water are influenced by pH, water hardness and the presence of ion-exchange materials such as clays. Fluoride is usually transported through the water cycle complexed with aluminium.
The transport and transformation of fluoride in soil are influenced by pH and the formation of predominantly aluminium and calcium complexes. Adsorption to the soil solid phase is stronger at slightly acidic pH values (5.5–6.5). Fluoride is not readily leached from soils.
The uptake of fluoride by biota is determined by the route of exposure, the bioavailability of the fluoride and the uptake/excretion kinetics in the organism. Soluble fluorides are bioaccumulated by some aquatic and terrestrial biota. However, no information was identified concerning the biomagnification of fluoride in aquatic or terrestrial food-chains.
Terrestrial plants may accumulate fluorides following airborne deposition and uptake from soil.
Fluoride levels in surface waters vary according to location and proximity to emission sources. Surface water concentrations generally range from 0.01 to 0.3 mg/litre. Seawater contains more fluoride than fresh water, with concentrations ranging from 1.2 to 1.5 mg/litre. Higher levels of fluoride have been measured in areas where the natural rock is rich in fluoride, and elevated inorganic fluoride levels are often seen in regions where there is geothermal or volcanic activity (e.g., 25–50 mg fluoride/litre in hot springs and geysers and as much as 2800 mg/litre in certain East African Rift Valley lakes). Anthropogenic discharges can also lead to increased levels of fluoride in the environment.
Airborne fluoride exists in gaseous and particulate forms, which are emitted from both natural and anthropogenic sources. Fluoride released as gaseous and particulate matter is deposited in the general vicinity of an emission source, although some particulates may react with other atmospheric constituents. The distribution and deposition of airborne fluoride are dependent upon emission strength, meteorological conditions, particulate size and chemical reactivity. In areas not in the direct vicinity of emission sources, the mean concentrations of fluoride in ambient air are generally less than 0.1 µg/m3. Levels may be slightly higher in urban than in rural locations; however, even in the vicinity of emission sources, the levels of airborne fluoride usually do not exceed 2–3 µg/m3. In areas of China where fluoride-rich coal is used as a source of fuel, reported concentrations of fluoride in ambient air have reached 6 µg/m3.
Fluoride is a component of most types of soil, with total fluoride concentrations ranging from 20 to 1000 µg/g in areas without natural phosphate or fluoride deposits and up to several thousand micrograms per gram in mineral soils with deposits of fluoride. Airborne gaseous and particulate fluorides tend to accumulate within the surface layer of soils but may be displaced throughout the root zone, even in calcareous soils. The clay and organic carbon content as well as the pH of soil are primarily responsible for the retention of fluoride in soils. Fluoride in soil is primarily associated with the soil colloid or clay fraction. For all soils, it is the soluble fluoride content that is biologically important to plants and animals.
Fluorides can be taken up by aquatic organisms directly from the water or to a lesser extent via food. Fluorides tend to accumulate in the exoskeleton or bone tissue of aquatic animals. Mean fluoride concentrations of >2000 mg/kg have been measured in the exoskeleton of krill; mean bone fluoride concentrations in aquatic mammals, such as seals and whales, ranged from 135 to 18 600 mg/kg dry weight.
Fluoride levels in terrestrial biota are higher in areas with high fluoride levels from natural and anthropogenic sources. Lichens have been used extensively as biomonitors for fluorides. Mean fluoride concentrations of 150–250 mg/kg were measured in lichens growing within 2–3 km of fluoride emission sources, compared with a background level of <1 mg fluoride/kg.
Most of the fluoride in the soil is insoluble and, therefore, less available to plants. However, high soil fluoride concentrations or low pH, clay and/or organic matter can increase fluoride levels in soil solution, increasing uptake via the plant root. If fluoride is taken up through the root, its concentrations are often higher in the root than in the shoot, due to the low mobility of fluoride in the plant. Most fluorides enter plant tissues as gases through the stomata and accumulate in leaves. Small amounts of airborne particulate fluoride can enter the plant through the epidermis and cuticle. Vegetation has been widely monitored in the vicinity of anthropogenic fluoride emission sources. Correlations between fluoride concentrations in vegetation and annual growth increments, wind pattern, distance from fluoride source and hydrogen fluoride concentrations in aerial emissions have been observed.
Fluoride accumulates in the bone tissue of terrestrial vertebrates, depending on factors such as diet and the proximity of fluoride emission sources. For example, mean fluoride concentrations of 7000–8000 mg/kg have been measured in the bones of small mammals in the vicinity of an aluminium smelter.
Fluoride is ubiquitous in the environment; therefore, sources of drinking-water are likely to contain at least some small amount of fluoride. The amount of fluoride present naturally in non-fluoridated drinking-water (i.e., drinking-water to which fluoride has not been intentionally added for the prevention of dental caries) is highly variable, being dependent upon the individual geological environment from which the water is obtained. Levels may range up to approximately 2.0 mg/litre; however, in areas of the world in which endemic fluorosis of the skeleton and/or teeth has been well documented, levels of fluoride in drinking-water supplies range from 3 to more than 20 mg/litre. In areas in which drinking-water is fluoridated (i.e., fluoride is intentionally added for the prevention of dental caries), the concentration of fluoride in drinking-water generally ranges from 0.7 to 1.2 mg/litre.
Virtually all foodstuffs contain at least trace amounts of fluoride. Elevated levels are present in fish. Tea leaves are particularly rich in fluoride; the amount of fluoride in brewed tea is dependent upon the concentration of soluble fluoride in the tea leaves, the level of fluoride in the water used in its preparation and the length of the brewing period. The concentration of fluoride in food products is not significantly increased by the addition of superphosphate fertilizers, which contain significant concentrations of fluoride (1–3%) as impurities, to agricultural soil, due to the generally low transfer coefficient from soil to plant material. However, a recent study suggests that, given the right soil conditions and application of sufficient fluoride as an impurity in phosphate fertilizers to soils, plant uptake of fluoride can be increased. The use of water containing relatively low (<3.1 mg/litre) levels of fluoride for crop irrigation generally does not increase fluoride concentrations in foodstuffs. However, this is dependent on plant species and fluoride concentrations in soil and water. The level of fluoride in foods is significantly affected by the fluoride content of the water used in preparation or processing, most notably in beverages and dry foodstuffs — for example, powdered baby formula — to which water is added prior to consumption. The concentrations of fluoride in unwashed or unprocessed foods grown in the vicinity of industrial sources (emissions) of fluoride may be greater than the levels in the same foods grown in other non-industrially exposed areas. In commercially available infant formulas sold in the USA, soy-based ready-to-use and liquid concentrate formulas contained higher levels of fluoride than the equivalent milk-based products; however, no significant difference was observed between soy- and milk-based powdered infant formulas. Fluoride has been detected in breast milk; reported levels range from <2 to about 100 µg/litre, with most values being between 5 and 10 µg/litre.
Available data on the concentrations of fluoride in indoor air are limited. In the Netherlands, concentrations of gaseous fluoride ranged from <2 to 49 µg/m3 in the indoor air of five homes constructed with wood treated with a preservative containing 56% fluoride. In China, concentrations as high as 155 µg/m3 have been reported for samples of indoor air collected from homes where coal containing high amounts of fluoride was burned indoors.
Dentifrice products for adults that are commercially available in many countries generally contain fluoride at concentrations ranging from 1000 to 1500 µg/g; some products designed for use by children contain lower levels, ranging from 250 to 500 µg/g. Dental products such as toothpaste, mouthwash and fluoride supplements have been identified as significant sources of fluoride. Mouth rinses marketed for daily home use usually contain between 230 and 500 mg fluoride/litre, whereas mouthwash products intended for weekly or biweekly use may contain 900–1000 mg fluoride/litre.
Although individual exposure to fluoride is likely to be highly variable, the inhalation of airborne fluoride generally makes a minor contribution to the total intake of this substance. For adults, the consumption of foodstuffs and drinking-water is the principal route for the intake of fluoride. In areas of the world in which coal rich in fluoride is used for heating and food preparation, the inhalation of indoor air and consumption of foodstuffs containing increased levels of fluoride also contribute to elevated intakes. Infants fed formula receive 50–100 times more fluoride than exclusively breast-fed infants. The ingestion of dentifrice by young children makes a significant contribution to their total intake of fluoride. In general, estimated intakes of fluoride in children and adolescents do not exceed approximately 2 mg/day. Although adults may have a higher absolute daily intake of fluoride in milligrams, the daily intake of fluoride by children, expressed on a milligram per kilogram body weight basis, may exceed that of adults. In certain areas worldwide in which the concentration of fluoride in the surrounding environment may be exceedingly high and/or where diets are composed of foodstuffs rich in fluoride, estimated intakes of fluoride in adults as high as 27 mg/day have been reported, the principal source being drinking-water obtained from groundwater sources located in geological areas rich in fluoride.
Occupational exposure to fluoride via inhalation or dermal contact likely occurs in individuals involved in the operation of welding equipment or in the processing of aluminium, iron ore or phosphate ore. In relatively recent studies, reported concentrations of airborne fluoride in the potrooms of aluminium smelters have been in the order of 1 mg/m3.
In humans and laboratory animals, the absorption of ingested fluoride into the general circulation occurs primarily in the stomach and intestine and is dependent upon the relative aqueous solubility of the form consumed. Soluble fluorides are almost completely absorbed from the gastrointestinal tract; however, the extent of absorption may be reduced by complex formation with aluminium, phosphorus, magnesium or calcium. There is partial to complete absorption of gaseous and particulate fluorides from the respiratory tract, with the extent of absorption dependent upon solubility and particle size.
Fluoride is rapidly distributed by the systemic circulation to the intracellular and extracellular water of tissues; however, in humans and laboratory animals, approximately 99% of the total body burden of fluoride is retained in bones and teeth. In teeth and skeletal tissue, fluoride becomes incorporated into the crystal lattice.
Fluoride crosses the placenta and is transferred from mother to fetus. Fluoride is eliminated from the body primarily in the urine. In infants, about 80–90% of a fluoride dose is retained; in adults, the corresponding figure is approximately 60%. These values can be altered by alterations in urinary flow and urinary pH.
Fluoride is present in body organs, tissues and fluids. Concentrations of fluoride in whole blood of individuals residing in a community in the USA receiving fluoridated drinking-water ranged from 20 to 60 µg/litre. The mean plasma level in 127 subjects with 5.03 mg fluoride/litre in their drinking-water was 106 ± 76 (SD) µg/litre. Serum and plasma contain virtually the same amount of fluoride. Levels of fluoride in calcified tissues are generally highest in bone, dentine and enamel. The concentration of fluoride in bone varies with age, sex and the type and specific part of bone and is believed to reflect an individual’s long-term exposure to fluoride. The concentration of fluoride in dental enamel decreases exponentially with the distance from the surface and varies with site, surface attrition, systemic exposure and exposure to topically applied fluoride. The concentration of fluoride in soft tissues is reflected by that in blood. Levels of fluoride in the urine of healthy individuals are related to the intake of fluoride. Increased levels of urinary fluoride have been measured in individuals following occupational exposure to airborne fluoride and among those residing in areas associated with endemic fluorosis.
Effects on the skeleton, such as inhibition of bone mineralization and formation, delayed fracture healing and reductions in bone volume and collagen synthesis, have been observed in a variety of studies in which rats received fluoride orally for periods of 3–5 weeks. In medium-term exposure studies, altered bone remodelling, hepatic megalocytosis, nephrosis, mineralization of the myocardium, necrosis and/or degeneration of the seminiferous tubules in the testis were observed in mice administered fluoride in drinking-water (>4.5 mg/kg body weight per day) over a period of 6 months.
In a comprehensive carcinogenicity bioassay in which groups of male and female F344/N rats and B6C3F1 mice were administered drinking-water containing up to 79 mg fluoride/litre as sodium fluoride for a period of 2 years, there was no statistically significant increase in the incidence of any tumour in any single exposed group. There was a statistically significant trend of an increased incidence of osteosarcomas in male rats with increasing exposure to fluoride. However, the incidence was within the range of historical controls.
Another 2-year carcinogenicity bioassay involving Sprague-Dawley rats exposed to up to 11.3 mg/kg body weight per day in the diet also found no statistically significant increase in the incidence of osteosarcoma or other tumours. Another study, which reported an increased incidence of osteomas in mice receiving up to 11.3 mg/kg body weight per day, is difficult to interpret, because the animals were infected with Type C retrovirus.
In general, fluoride is not mutagenic in prokaryotic cells. Although fluoride has been shown to increase the frequency of mutations at specific loci in cultured mouse lymphoma and human lymphoblastoid cells, these mutations are likely due to chromosomal damage rather than point mutations. Fluoride has been shown to be clastogenic in a variety of cell types. The mechanism of clastogenicity has been attributed to the effect of fluoride upon the synthesis of proteins involved in DNA synthesis and/or repair, rather than direct interaction between fluoride and DNA. In most studies in which fluoride was administered orally to rodents, there was no effect upon sperm morphology or the frequency of chromosomal aberrations, micronuclei, sister chromatid exchange or DNA strand breaks. However, cytogenetic damage in bone marrow or alterations in sperm cell morphology were reported when the substance was administered to rodents by intraperitoneal injection.
Reproductive or developmental effects were not observed in recent studies in which laboratory animals were administered fluoride in drinking-water. However, histopathological changes in reproductive organs have been reported in male rabbits administered (orally) 4.5 mg fluoride/kg body weight per day for 18–29 months, in male mice administered (orally) >4.5 mg fluoride/kg body weight per day for 30 days and in female rabbits injected subcutaneously with >10 mg fluoride/kg body weight per day for 100 days. Adverse effects on reproductive function have been reported in female mice administered (orally) >5.2 mg fluoride/kg body weight per day on days 6–15 after mating and in male rabbits administered (orally) >9.1 mg fluoride/kg body weight per day for 30 days.
Epidemiological investigations on the effects of fluoride on human health have examined occupationally exposed workers employed primarily in the aluminium smelting industry and populations consuming fluoridated drinking-water. In a number of analytical epidemiological studies of workers occupationally exposed to fluoride, an increased incidence of lung and bladder cancer and increased mortality due to cancer of these and other sites have been observed. In general, however, there has been no consistent pattern; in some of these epidemiological studies, the increased morbidity or mortality due to cancer can be attributed to the workers’ exposure to substances other than fluoride.
The relationship between the consumption of fluoridated drinking-water and morbidity or mortality due to cancer has been examined in a large number of epidemiological studies, performed in many countries. There is no consistent evidence of an association between the consumption of controlled fluoridated drinking-water and increased morbidity or mortality due to cancer.
Fluoride has both beneficial and detrimental effects on tooth enamel. The prevalence of dental caries is inversely related to the concentration of fluoride in drinking-water. The prevalence of dental fluorosis is highly associated with the concentration of fluoride, with a positive dose–response relationship.
Cases of skeletal fluorosis associated with the consumption of drinking-water containing elevated levels of fluoride continue to be reported. A number of factors, such as nutritional status and diet, climate (related to fluid intake), concomitant exposure to other substances and the intake of fluoride from sources other than drinking-water, are believed to play a significant role in the development of this disease. Skeletal fluorosis may develop in workers occupationally exposed to elevated levels of airborne fluoride; however, only limited new information was identified.
Evidence from several ecological studies has suggested that there may be an association between the consumption of fluoridated water and hip fractures. Other studies, however, including analytical epidemiological investigations, have not supported this finding. In some cases, a protective effect of fluoride on fracture has been reported.
Two studies permit an evaluation of fracture risk across a range of fluoride intakes. In one study, the relative risks of all fractures and of hip fracture were elevated in groups drinking water with >1.45 mg fluoride/litre (total intake >6.5 mg/day); this difference reached statistical significance for the group drinking water containing >4.32 mg fluoride/litre (total intake 14 mg/day). In the other study, an increased incidence of fractures was observed in one age group of women exposed to fluoride in drinking-water in a non-dose-dependent manner.
Epidemiological studies show no evidence of an association between the consumption of fluoridated drinking-water by mothers and increased risk of spontaneous abortion or congenital malformation. Other epidemiological investigations of occupationally exposed workers have provided no reasonable evidence of genotoxic effects or systemic effects upon the respiratory, haematopoietic, hepatic or renal systems that may be directly attributable to fluoride exposure per se.
Fluoride did not affect growth or chemical oxygen demand degrading capacity of activated sludge at concentrations of 100 mg/litre. The EC50 for inhibition of bacterial nitrification was 1218 mg fluoride/litre. Ninety-six-hour EC50s, based on growth, for freshwater and marine algae were 123 and 81 mg fluoride/litre, respectively.
Forty-eight-hour LC50s for aquatic invertebrates range from 53 to 304 mg/litre. The most sensitive freshwater invertebrates were the fingernail clam (Musculium transversum), with statistically significant mortality (50%) observed at a concentration of 2.8 mg fluoride/litre in an 8-week flow-through experiment, and several net-spinning caddisfly species (freshwater; family: Hydropsychidae), with "safe concentrations" (8760-h EC0.01s) ranging from 0.2 to 1.2 mg fluoride/litre. The brine shrimp (Artemia salina) was the most sensitive marine species tested. In a 12-day static renewal test, statistically significant growth impairment occurred at 5.0 mg fluoride/litre.
Ninety-six-hour LC50s for freshwater fish range from 51 mg/litre (rainbow trout, Oncorhynchus mykiss) to 460 mg/litre (threespine stickleback, Gasterosteus aculeatus). All of the acute toxicity tests (96 h) on marine fish gave results greater than 100 mg/litre. Inorganic fluoride toxicity to freshwater fish appears to be negatively correlated with water hardness (calcium carbonate) and positively correlated with temperature. The symptoms of acute fluoride intoxication include lethargy, violent and erratic movement and death. Twenty-day LC50s for rainbow trout ranged from 2.7 to 4.7 mg fluoride/litre in static renewal tests. "Safe concentrations" (infinite hours LC0.01s) have been estimated for rainbow trout and brown trout (Salmo trutta) at 5.1 and 7.5 mg fluoride/litre, respectively. At concentrations of >3.2 (effluent) or >3.6 (sodium fluoride) mg fluoride/litre, the hatching of catla (Catla catla) fish eggs was delayed by 1–2 h.
Behavioural experiments on adult Pacific salmon (Oncorhynchus sp.) in soft-water rivers indicate that changes in water chemistry resulting from an increase in the fluoride concentration to 0.5 mg/litre can adversely affect migration; migrating salmon are extremely sensitive to changes in the water chemistry of their river of origin. In laboratory studies, fluoride seems to be toxic for microbial processes at concentrations found in moderately fluoride polluted soils; similarly, in the field, accumulation of organic matter in the vicinity of smelters has been attributed to severe inhibition of microbial activity by fluoride.
Signs of inorganic fluoride phytotoxicity (fluorosis), such as chlorosis, necrosis and decreased growth rates, are most likely to occur in the young, expanding tissues of broadleaf plants and elongating needles of conifers. The induction of fluorosis has been clearly demonstrated in laboratory, greenhouse and controlled field plot experiments. A large number of the papers published on fluoride toxicity to plants concern glasshouse fumigation with hydrogen fluoride. Foliar necrosis was first observed on grapevines (Vitis vinifera) exposed to 0.17 and 0.27 µg/m3 after 99 and 83 days, respectively. The lowest-observed-effect level for leaf necrosis (65% of leaves) in the snow princess gladiolus (Gladiolus grandiflorus) was 0.35 µg fluoride/m3. Airborne fluoride can also affect plant disease development, although the type and magnitude of the effects are dependent on the specific plant–pathogen combination.
Several short-term solution culture studies have identified a toxic threshold for fluoride ion activity ranging from approximately 50 to 2000 µmol fluoride/litre. Toxicity is specific not only to plant species, but also to ionic species of fluoride; some aluminium fluoride complexes present in solution culture may be toxic at activities of 22–357 µmol fluoride/litre, whereas hydrogen fluoride is toxic at activities of 71–137 µmol fluoride/litre. A few studies have been carried out in which the fluoride exposures have been via the soil. The type of soil can greatly affect the uptake and potential toxicity of fluorides.
In birds, the 24-h LD50 was 50 mg/kg body weight for 1-day-old European starling (Sturnus vulgaris) chicks and 17 mg/kg body weight for 16-day-old nestlings. Growth rates were significantly reduced at 13 and 17 mg fluoride/kg body weight (the highest doses at which growth was monitored). Most of the early work on mammals was carried out on domesticated ungulates. Fluorosis has been observed in cattle and sheep. The lowest dietary level observed to cause an effect on wild ungulates was in a controlled captive study with white-tailed deer (Odocoileus virginianus) in which a general mottling of the incisors characteristic of dental fluorosis was noted in the animals at the 35 mg/kg diet dose.
Aluminium smelters, brickworks, phosphorus plants and fertilizer and fibreglass plants have all been shown to be sources of fluoride that are correlated with damage to local plant communities. Vegetation in the vicinity of a phosphorus plant revealed that the degree of damage and fluoride levels in soil humus were inversely related to the distance from the plant. Average levels of fluoride in vegetation ranged from 281 mg/kg in severely damaged areas to 44 mg/kg in lightly damaged areas; at a control site, the fluoride concentration was 7 mg/kg. Plant communities near an aluminium smelter showed differences in community composition and structure due partly to variations in fluoride tolerance. However, it must be noted that, in the field, one of the main problems with the identification of fluoride effects is the presence of confounding variables such as other atmospheric pollutants. Therefore, care must be taken when interpreting the many field studies on fluoride pollution.
The original findings of fluoride effects on mammals were from studies in the field on domestic animals such as sheep and cattle. Fluoride can be taken up from vegetation, soil and drinking-water. Tolerance levels have been identified for domesticated animals, with the lowest values for dairy cattle at 30 mg/kg feed or 2.5 mg/litre drinking-water. Incidents involving domesticated animals have originated both from natural fluoride sources, such as volcanic eruptions and the underlying geology, and from anthropogenic sources, such as mineral supplements, fluoride-emitting industries and power stations. Symptoms of fluoride toxicity include emaciation, stiffness of joints and abnormal teeth and bones. Other effects include lowered milk production and detrimental effects on the reproductive capacity of animals. The lowest dietary concentration of fluoride to cause fluorosis in wild deer was 35 mg/kg. Investigations of the effects of fluoride on wildlife have focused on impacts on the structural integrity of teeth and bone. In the vicinity of smelters, fluoride-induced effects, such as lameness, dental disfigurement and tooth damage, have been found.
Fluoride has both positive and negative effects on human health, but there is a narrow range between intakes that are associated with these effects. Exposure to all sources of fluoride, including drinking-water and foodstuffs, is important.
There is little information to characterize the dose–response relationships for the different adverse effects. In particular, there are few data on total exposure, particularly with respect to intake and fluoride absorption.
The most serious effect is the skeletal accumulation of fluoride from long-term excessive exposure to fluoride and its effect on non-neoplastic bone disease — specifically, skeletal fluorosis and bone fractures. There is clear evidence from India and China that skeletal fluorosis and an increased risk of bone fractures occur at total intakes of 14 mg fluoride/day and evidence suggestive of an increased risk of bone effects at total intakes above about 6 mg fluoride/day.
In the freshwater environment, natural fluoride concentrations are usually lower than those expected to cause toxicity in aquatic organisms. However, aquatic organisms might be adversely affected in the vicinity of anthropogenic discharges. Fluoride toxicity is dependent on water hardness.
Sensitive plant species growing near anthropogenic sources of fluoride are at risk. The release of fluoride from anthropogenic sources is associated with damage to local terrestrial plant communities, but it is often difficult to attribute these effects to fluoride alone, due to the presence of other atmospheric pollutants. Fluoride is generally strongly adsorbed by soils. Consequently, plant uptake via this pathway is relatively low, and leaching of fluoride through soil is minimal.
Concentrations of fluoride in vegetation in the vicinity of fluoride emission sources, such as aluminium smelters, can be higher than the lowest dietary effect concentration reported for mammals in laboratory experiments. Fluorosis in domesticated animals has been reported. There are still some areas reporting fluorosis incidents in livestock due to uptake of fluoride-rich mineral supplements and drinking-water. Furthermore, there is a potential risk from fluoride-contaminated pasture and soil ingestion due to the long-term use of phosphate fertilizers containing fluoride as an impurity. Fluoride-induced effects, such as lameness and tooth damage, have also been reported in wild mammals close to anthropogenic sources.
All organisms are exposed to fluoride from natural and/or anthropogenic sources. Very high intakes have been observed in areas worldwide in which the environment is rich in fluoride and where groundwater high in fluoride is consumed by humans. Increased exposure might occur in the vicinity of point sources. Fluoride in dental products is an additional source for many people.
Fluoride has both beneficial and detrimental effects on human health, with a narrow range between the intakes at which these occur.
Effects on the teeth and skeleton may be observed at exposures below those associated with the development of other organ- or tissue-specific adverse health effects.
Effects on the bone (e.g., skeletal fluorosis and fracture) are considered the most relevant outcomes in assessing the adverse effects of long-term exposure of humans to fluoride.
Skeletal fluorosis is a crippling disability that has a major public health and socioeconomic impact, affecting millions of people in various regions of Africa, China and India.
Intake of fluoride in water and foodstuffs is the primary causative factor for endemic skeletal fluorosis. In some regions, the indoor burning of fluoride-rich coal also serves as an important source of fluoride.
There are few data from which to estimate total exposure to and the bioavailability of fluoride, and there are inconsistencies in reports on the characterization of its adverse effects.
There is clear evidence from India and China that skeletal fluorosis and an increased risk of bone fractures occur at a total intake of 14 mg fluoride/day and evidence suggestive of an increased risk of bone effects at total intakes above about 6 mg fluoride/day.
Excess exposure to bioavailable fluoride constitutes a risk to aquatic and terrestrial biota.
Fluoride-sensitive species can be used as sentinels for the identification of fluoride hazards to the environment.
There is a need to improve knowledge on the accumulation of fluoride in organisms and on how to monitor and control this.
The biological effects associated with fluoride exposure should be better characterized.
This document focuses on environmental exposure to fluoride derived mostly from inorganic sources and its effects on humans, animals and other biota. Data on hydrogen fluoride, calcium fluoride, sodium fluoride, sulfur hexafluoride and silicofluorides are emphasized, as these compounds are considered the most relevant of the inorganic fluorides on the basis of quantities released to the environment, environmental concentrations and toxicological effects on living organisms.
There is one stable isotope of fluorine (F), with an atomic mass of 18.9984. There are also several radioactive isotopes (17F, 18F, 20F, 21F and 22F), with 18F having the longest half-life (109.7 min) (Weast, 1986).
At room temperature, hydrogen fluoride (HF) (relative molecular mass 20.01; density 0.991 g/litre; CAS No.
Calcium fluoride (CaF2) (relative molecular mass 78.08; CAS No.
Sodium fluoride (NaF) (relative molecular mass 41.99; CAS No.
Fluorosilicic acid (H2SiF6) (relative molecular mass 144.08; CAS No.
Sodium hexafluorosilicate (Na2SiF6) (relative molecular mass 188.05; CAS No.
Sulfur hexafluoride (SF6) (relative molecular mass 146.05; density 6.16 g/litre; CAS No.
Methods employed for the quantification of fluoride in biological samples and environmental media generally rely on the detection of fluoride ion (F–). Perhaps the most widely used method of fluoride quantification has involved potentiometry employing the fluoride ion-selective electrode (Neumüller, 1981; Harzdorf et al., 1986; ATSDR, 1993). This method has been used for the quantification of fluoride in biological tissues and fluids (e.g., urine, serum and plasma, organs, bone, teeth), foodstuffs and environmental media (e.g., air, water, soil). Owing to variations in the efficacy of sample preparation procedures, detection limits using the fluoride ion-selective electrode may range from 0.1 to 300 ng/m3 in air, from 1 to 1000 µg/litre in water and from 0.05 to 20 mg/kg in tissues (Harzdorf et al., 1986; ATSDR, 1993). Other approaches for the quantification of fluoride have included spectrophotometry, gas chromatography, ion chromatography, capillary electrophoresis, atomic absorption and photon activation (Neumüller, 1981; ATSDR, 1993; Wen et al., 1996).
Appropriate sample preparation is a critical step in the accurate quantification of fluoride, especially where only the free fluoride ion is measured. For analyses involving biological materials, the most accurate method is the microdiffusion technique, such as the acid-hexamethyldisiloxane (HMDS) diffusion method by Taves (1968), since methods involving acid or alkali digestion may not convert all complex inorganic and organic fluorides into an ionic form that can be conveniently measured (Venkateswarlu, 1983). Open ashing methods may result in the loss of volatile fluoride compounds or of fluoride itself at temperatures in excess of 550 °C, or they may result in contamination with extraneous fluoride (Venkateswarlu, 1975; Campbell, 1987).
Fluorides are released into the environment naturally through the weathering of minerals, in emissions from volcanoes and in marine aerosols (Symonds et al., 1988; ATSDR, 1993). Estimates of the annual global release of hydrogen fluoride from volcanic sources through passive degassing and eruptions range from 60 to 6000 kilotonnes, of which approximately 10% may be introduced directly into the stratosphere (Symonds et al., 1988). Annually, approximately 20 kilotonnes of fluoride may be released in marine aerosols (Symonds et al., 1988).
The main natural source of inorganic fluorides in soil is the parent rock (WHO, 1984). During weathering, some fluoride minerals (e.g., cryolite, or Na3AlF6) are rapidly broken down, especially under acidic conditions (Fuge & Andrews, 1988). Other minerals, such as fluorapatite (Ca5(PO4)3F) and calcium fluoride, are dissolved more slowly (Kabata-Pendias & Pendias, 1984). The mineral fluorophlogopite (mica; KMg3(AlSi3O10)F2) is stable in alkaline and calcareous soils (Elrashidi & Lindsay, 1986). However, its solubility is affected by pH and the activities of silicic acid (H4SiO4) and aluminium (Al3+), potassium (K+) and magnesium (Mg2+) ions.
Hydrogen fluoride (hydrofluoric acid) is an important industrial compound, with an estimated annual world consumption in excess of 1 million tonnes (Greenwood & Earnshaw, 1984). Hydrogen fluoride is manufactured from calcium fluoride and is used mainly in the production of synthetic cryolite, aluminium fluoride (AlF3), motor gasoline alkylates and chlorofluorocarbons; however, the demand for chlorofluorocarbons is decreasing as a result of efforts to restrict their use. Hydrogen fluoride is also used in the synthesis of uranium tetrafluoride (UF4) and uranium hexafluoride (UF6), both of which are used in the nuclear industry (Neumüller, 1981). It is also used in etching semiconductor devices, cleaning and etching glass, cleaning brick and aluminium and tanning leather, as well as in petrochemical manufacturing processes. Hydrogen fluoride may also be found in commercial rust removers (Upfal & Doyle, 1990).
Industrially, calcium fluoride is the principal fluoride-containing mineral used (WHO, 1984). Identified production data were confined to the USA, where the average annual production of calcium fluoride was estimated to range from 118 000 to 225 000 tonnes during 1972–1978 (ATSDR, 1993). The consumption of calcium fluoride (as fluorspar) in Canada in 1989 was estimated at 180 000 tonnes (Government of Canada, 1993); in 1977, the estimated consumption of calcium fluoride in the USA was 1 063 000 tonnes (ATSDR, 1993). Calcium fluoride is used as a flux in steel, glass and enamel production and as the raw material for the production of hydrofluoric acid and anhydrous hydrogen fluoride (Neumüller, 1981). Calcium fluoride is also used as a molten electrolyte for the separation of oxygen and alumina in aluminium production.
Data concerning the total annual consumption or production of sodium fluoride worldwide were not identified. Sodium fluoride is usually prepared from hydrofluoric acid and sodium carbonate or sodium hydroxide (Neumüller, 1981); it is used in the controlled fluoridation of drinking-water, as a preservative in certain glues, in glass and enamel production, as a flux in steel and aluminium production, as an insecticide and as a wood preservative (Neumüller, 1981).
Fluorosilicic acid is an aqueous solution that is most commonly manufactured as a co-product from the manufacture of phosphate fertilizers. It is used widely for the fluoridation of drinking-water, in which it hydrolyses to release fluoride ions. When used for the fluoridation of drinking-water, fluorosilicic acid should meet appropriate standards, such as those published by the American Water Works Association and the European Committee for Standardization or other approved schemes for drinking-water chemicals.
Sodium hexafluorosilicate, like fluorosilicic acid, is used in the fluoridation of drinking-water. It is normally completely dissolved in water prior to dosing, when it hydrolyses to give fluoride ions. When used for drinking-water fluoridation, it too should meet appropriate standards of purity for drinking-water chemicals.
More than 110 tonnes of sulfur hexafluoride are imported into Canada annually (Government of Canada, 1993). This substance is used extensively as an insulation and current interruption medium in electrical switchgear, such as power circuit breakers, in various components in electrical substations (Government of Canada, 1993) and as a protective inert gas over molten metals, such as magnesium and aluminium (Neumüller, 1987). Over 90% of the total amount of sulfur hexafluoride imported into Canada is used in the production of magnesium; the remainder is used in electrical switchgear (Government of Canada, 1993).
Fluorapatite, an important calcium- and fluoride-containing mineral, is used as a source of phosphates in the fertilizer industry (Neumüller, 1981).
Phosphate fertilizers are the major source of fluoride contamination of agricultural soils. They are manufactured from rock phosphates, which generally contain around 3.5% fluorine (Hart et al., 1934). However, during the manufacture of phosphate fertilizers, part of the fluoride is lost into the atmosphere during the acidulation process, and the concentration of fluoride in the final fertilizer is lowered further through dilution with sulfur (superphosphates) or ammonium ion (ammoniated phosphates); the final product commonly contains between 1.3 and 3.0% fluorine (McLaughlin et al., 1996). In Australia, an average annual addition of fluoride to soil through fertilization has been estimated to be 1.1 kg/ha.
Available quantitative information concerning the release of fluoride into the environment (air, water and soil) from industrial sources is limited. Fluoride is released into the environment via exhaust fumes, process waters and waste from various industrial processes, including steel manufacture, primary aluminium, copper and nickel production, phosphate fertilizer production and use, glass, brick and ceramic manufacturing, and glue and adhesive production. The use of fluoride-containing pesticides as well as the fluoridation of drinking-water supplies also contribute to the release of fluoride from anthropogenic sources.
The total annual amount of fluoride released to the environment from industrial sources was estimated to be in excess of 23 500 tonnes in Canada and 46 600 tonnes in the Netherlands (Sloof et al., 1989; Government of Canada, 1993). The relative contribution of various anthropogenic sources to total emissions of fluoride to air, water and soil in Canada are estimated at 48% for phosphate fertilizer production, 20% for chemical production, 19% for aluminium production, 8% for steel and oil production and 5% for coal burning (Government of Canada, 1993). In the Netherlands, 93% of total fluoride emissions to air, water and soil are derived from phosphate ore production and use, with smaller amounts emitted via mineral processing (2%), the metal industry (4%) and "other industry" (1%) (Sloof et al., 1989). The total amounts of hydrogen fluoride released to air, surface water, underground injection and land in the USA during 1999 were 33 000, 7.7, 1800 and 64 tonnes, respectively. Total amounts of fluorine released to air, surface water and land were 39, 24 and 500 tonnes, respectively (US EPA, 1999).
The cycling of fluoride through the biogeosphere is summarized in Figure 1.
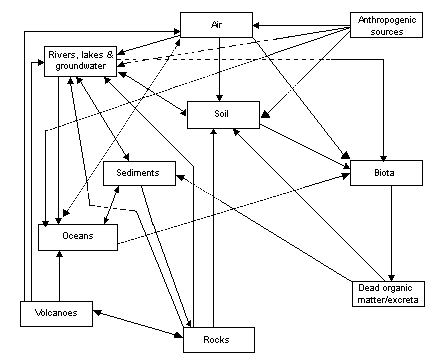
Fig. 1. Cycling of fluoride through the biogeosphere
The fate of inorganic fluorides in the atmosphere is primarily influenced by vaporization, aerosol formation, wet and dry deposition and hydrolysis (Environment Canada, 1994). Non-volatile inorganic fluoride particulates are removed from the atmosphere via condensation or nucleation processes.
Atmospheric fluorides emitted from both natural and anthropogenic sources may be in gaseous or particulate form (Kirk & Lester, 1986). Gaseous forms include hydrogen fluoride, silicon tetrafluoride (SiF4), fluorosilicic acid and sulfur hexafluoride. Particulate forms include sodium aluminium fluoride (cryolite), aluminium fluoride, calcium fluoride, sodium hexafluorosilicate, lead fluoride (PbF2) and calcium phosphate fluoride (fluorapatite). Globally, hydrogen fluoride and inorganic fluoride particulates (sodium and calcium fluoride) account for approximately 75% and 25%, respectively, of inorganic fluorides present in the atmosphere (Health Council of the Netherlands, 1990). Fluorine and the silicon fluorides are hydrolysed in the atmosphere to form hydrogen fluoride. Hydrogen fluoride may combine with water vapour to produce an aerosol or fog of aqueous hydrofluoric acid.
Fluorides adsorbed on particulate matter in the atmosphere are generally stable and are not readily hydrolysed, although they may be degraded by radiation if they persist in the atmosphere (US NAS, 1971).
Hydrofluoric acid is approximately 5 orders of magnitude less soluble than hydrochloric acid and will therefore be degassed from marine aerosols more readily than hydrochloric acid. Hydrofluoric acid is expected to be depleted in aged marine aerosols, and this may be a significant source of hydrogen fluoride in the troposphere (Brimblecombe & Clegg, 1988).
Schotte (1987) used a dispersion model to predict the formation and behaviour of the fog formed from the release of hydrogen fluoride to the atmosphere. Initially, the hydrogen fluoride will cool significantly due to depolymerization. The fog will therefore stay near ground level, since it is more dense than ambient air. As the fog mixes with more air, it will begin to warm up and it may rise, depending on the ambient air temperature and the relative humidity.
Davison et al. (1973) reported that between 60 and 74% of atmospheric fluoride in urban coal-burning areas in the United Kingdom was in gaseous form. Similarly, approximately 60% of the fluorides in the atmosphere in the Netherlands are in the gaseous state (Sloof et al., 1989).
Based upon available data, inorganic fluoride compounds, with the exception of sulfur hexafluoride, are not expected to remain in the troposphere for long periods or to migrate to the stratosphere. Estimates of the residence time of sulfur hexafluoride in the atmosphere range from 500 to several thousand years (Ramanathan et al., 1985; Chu, 1991).
Fluoride in aerosols can be transported over large distances by wind or as a result of atmospheric turbulence. The distance travelled is determined by the deposition velocity of both the gaseous hydrogen fluoride and the fluorides in particulate form. The transportation of particles with a diameter greater than 10 µm is determined by the particle falling speed, and the dispersion of such particles is generally limited to the immediate vicinity of the source. Smaller particles are less restricted by the falling speed and can be transported over larger distances (Sloof et al., 1989).
Atmospheric fluorides may be transported to soils and surface waters through both wet and dry deposition processes (US NAS, 1971). Seasonal climatic conditions are expected to influence the rate at which and mode by which atmospheric fluorides are deposited; for example, in the Tamar Valley, Tasmania, wet deposition dominates during winter (high precipitation; June to August), and dry deposition dominates during summer (low precipitation; December to February) (Low & Bloom, 1988).
Wet deposition of fluoride may occur as washout from plumes below cloud or rainout of particulates taken up by clouds. The washout process is of particular importance for the removal of soluble fractions such as hydrogen fluoride aerosols at short distances from the source. It is assumed that all irreversibly soluble gases such as hydrogen fluoride are washed out during showers. The rainout process is more important for the removal of fluorides distant from the source when the plume is situated at least partially in the clouds. The scavenging ratio, the ratio between measured concentrations in rainwater and the atmosphere, was calculated to be 0.15 × 106 (Sloof et al., 1989). For large-scale dispersion of fluorides, the annual average wet deposition rate was 1.4% per hour for fluoride aerosol and 5.9% per hour for gaseous fluorides. These values give an atmospheric residence time of 12 h for gaseous fluoride and 50 h for particulates.
The dry deposition rate for fluoride in the Agra region of India was highest between December and June, when atmospheric fluoride concentrations were highest (Saxena et al., 1994). Seasonally averaged dry deposition rates at the four sites ranged from 0.14 to 0.15 mg/m2 per day for summer (March to June), from 0.08 to 0.21 mg/m2 per day for winter (October to February) and from 0.008 to 0.03 mg/m2 per day for the monsoon season (July to September). Similar patterns of dry deposition were recorded by Chandrawanshi & Patel (1999) for central India during 1995; however, higher deposition rates were reported, with values of up to 1.1 mg/m2 per day being recorded during the winter months. The overall mean fluoride flux deposited with dust and rainwater during 1995 in central India was 474 kg/km2.
Several studies have been conducted to determine whether fluoride in rainwater was derived from anthropogenic emissions or natural sources such as sea salt cycling. Barnard & Nordstrom (1982) stated that fluoride should not be regarded as a cyclical sea salt, because the fluoride concentrations in rain from areas with no local anthropogenic emissions were not correlated with sea salt availability (as determined by the sodium concentration). Mass balance considerations suggested that the majority of fluoride samples in the rainwater were of anthropogenic origin. Similarly, Saether et al. (1995) calculated that more than 90% of fluoride in precipitation samples collected in southern Italy were of non-marine origin.
The ratio between total fluorine and chloride in rainwater from Wales was greater than the ratio in seawater (Neal et al., 1990). This implied enrichment of total fluorine relative to chloride, reflecting complex fractionation processes in the transport of fluorine from the sea to the atmosphere and back to land as precipitation. The total fluorine/chloride ratio in streamwater was higher than it was in rainwater, suggesting a net release of total fluorine from the catchment to the stream. The source of the release was uncertain, since the total fluorine concentration in baseflow waters was not significantly higher than stormflow values.
Mahadevan et al. (1986) reported a strong correlation between fluoride and sodium concentrations in precipitation samples collected from marine, coastal and inland sites in India. The authors suggested that fluoride in precipitation was derived from the cycling of sea salt. The correlation was not as strong in samples from urban areas, where the majority of fluoride was derived from anthropogenic sources.
The deposition of fluoride emitted from a phosphorus plant was reported to decrease with increasing distance from the source (Sidhu, 1982). The rate of deposition at 1.4 km from the source was calculated to be 2.61 g fluoride/ha per millimetre of rain and 3.10 g fluoride/ha per millimetre of snow water. These data corresponded to an annual deposition rate of 3.43 kg/ha. Fluoride deposition on soil from leaf litter also decreased with increasing distance from the source. Fluoride input to soil ranged from 10 to 720 g/ha per year. Input from precipitation was 5–10 times greater than it was for leaf litter.
Davison & Blakemore (1980) determined the deposition of fluoride at field sites near areas of industrial and urban sources of fluorides. The mean total fluoride deposited from wet and dry deposition and sedimentation was 38.0 µg/dm2 per week. Deposition of gaseous fluoride was 23.4 µg/dm2 per week.
The average large-scale deposition velocity for total soluble fluoride in the Netherlands was calculated to be 1.4 cm/s (Sloof et al., 1989). This figure was based upon 70% of the soluble fluoride being in a gaseous state and an atmospheric residence time of 14 h for gaseous fluorides and 12 days for aerosol fluorides. The average deposition velocity calculated does not apply to the area surrounding a point source. Under stable atmospheric conditions, a low deposition velocity will be accompanied by high atmospheric concentrations. The deposition velocity of fluoride depends heavily on atmospheric conditions. The deposition velocity for hydrogen fluoride can vary by more than 7 orders of magnitude; for particulate fluoride, it varies by less than 10%. The annual average effective deposition velocity varies with height of the emission source and was calculated to be 1.2 and 2.5 cm/s for low and high source heights, respectively, in the Netherlands.
In water, the transport and transformation of inorganic fluorides are influenced by pH, water hardness and the presence of ion-exchange materials such as clays (Environment Canada, 1994). Fluoride is usually transported through the water cycle complexed with aluminium (Ares, 1990).
In areas of extreme acidity and alkalinity, inorganic fluorides may leach from fluoride-containing minerals into surface water or groundwater (Cuker & Shilts, 1979). Solubilization of inorganic fluorides from minerals may also be enhanced by the presence of ion-exchange materials (e.g., bentonite clays and humic acid) (Pickering et al., 1988). Once dissolved, inorganic fluorides remain in solution under conditions of low pH and hardness and in the presence of ion-exchange material (Cuker & Shilts, 1979; Sahu & Karim, 1989). Soluble inorganic fluorides may also form aerosols at the air–water interface or vaporize into the atmosphere (Brimblecombe & Clegg, 1988), whereas undissolved species generally undergo sedimentation (Drury et al., 1980).
Kudo et al. (1987) calculated the fluoride mass balance for the once severely polluted Maurienne Valley in the French Alps. Fluoride emission into the valley from aluminium production plants was 500 tonnes per year in 1980. Fluoride output by the river was calculated to be 680 tonnes per year, of which 665 tonnes were due to water flow and 15 tonnes to sediment movements.
Chamblee et al. (1984) analysed approximately 100 estuarine water samples and reported that 10.5% of fluoride originated from fluoride complexes with trivalent cations such as Fe3+ and Al3+. The proportion of fluoride in the form of magnesium fluoride (MgF2) ranged from 0.4 to 33.7%. Fluoride concentrations were reported to increase with salinity (within a salinity range of 0.1–16%).
Fluoride in seawater is divided between the following fractions (Stumm & Morgan, 1981):
|
Fraction |
Proportion (%) |
Concentration (mol/litre) |
|
F– |
51 |
4.1 × 10–5 |
|
MgF+ |
47 |
3.7 × 10–5 |
|
CaF+ |
2 |
1.6 × 10–6 |
In seawater, fluorides are removed by the formation of complexes with calcium compounds, principally carbonate and phosphate (Carpenter, 1969). Undissolved fluoride is generally removed from the aquatic phase by sedimentation (US EPA, 1980). Carpenter (1969) calculated a residence time for fluoride in ocean sediment to be 2–3 million years.
Fluorosilicic acid and hydrofluoric acid in high aquatic concentrations such as may be found in industrial waste ponds may volatilize, releasing silicon tetrafluoride and hydrogen fluoride into the atmosphere (US NAS, 1971).
Factors that influence the mobility of inorganic fluorides in soil are pH and the formation of aluminium and calcium complexes (Pickering, 1985; Environment Canada, 1994).
In more acidic soils, concentrations of inorganic fluoride were considerably higher in the deeper horizons. The low affinity of fluorides for organic material results in leaching from the more acidic surface horizon and increased retention by clay minerals and silts in the more alkaline, deeper horizons (Davison, 1983; Kabata-Pendias & Pendias, 1984). This distribution profile is not observed in either alkaline or saline soils (Gilpin & Johnson, 1980; Davison, 1983). The fate of inorganic fluorides released to soil also depends on the chemical form, rate of deposition, soil chemistry and climate (Davison, 1983).
Fluoride in soil is mainly bound in complexes. The maximum adsorption of fluoride to soil was reported to occur at pH 5.5 (Barrow & Ellis, 1986). In acidic soils with pH below 6, most of the fluoride is in complexes with either aluminium or iron (e.g., AlF2+, AlF2+, AlF30, AlF4–, FeF2+, FeF2+, FeF30) (Perrott et al., 1976; Murray, 1984b; Elrashidi & Lindsay, 1986). Fluoride in alkaline soils at pH 6.5 and above is almost completely fixed in soils as calcium fluoride, if sufficient calcium carbonate is available (Brewer, 1966).
Fluoride binds to clay by displacing hydroxide from the surface of the clay (Huang & Jackson, 1965; Bower & Hatcher, 1967; Meeussen et al., 1996). The adsorption follows Langmuir adsorption equations and is strongly dependent upon pH and fluoride concentration. It is most significant at pH 3–4, and it decreases above pH 6.5.
Pickering et al. (1988) determined changes in free fluoride ions and total fluoride levels following equilibration of either poorly soluble fluoride species, such as calcium fluoride and aluminium fluoride, or wastes from aluminium smelters. The experiments were carried out on materials that had different cation-exchange capacities, such as synthetic resins, clay minerals, manganese oxide and a humic acid. Increased amounts of fluoride were released from fluoride salts and fluoride-rich wastes when solids capable of exchanging cations were present. The effect was greatest when there were more exchange sites available and when the fluoride compound cation had greater affinity for the exchange material. In a few cases, soluble complex ions were formed when the released fluoride attacked the substrate, such as illite or alumina wastes.
Fluoride is extremely immobile in soil, as determined by lysimeter experiments. MacIntire et al. (1955) reported that 75.8–99.6% of added fluoride was retained by loam soil for 4 years. Fluoride retention was correlated with the soil aluminium content. The leaching of fluoride occurred simultaneously with the leaching of aluminium, iron and organic material from soil (Polomski et al., 1982). Soil phosphate may contribute to the mobility of inorganic fluoride (Kabata-Pendias & Pendias, 1984). Oelschläger (1971) reported that approximately 0.5–6.0% of the annual addition of fluoride (atmospheric pollution and artificial fertilizers) to a forest and agricultural areas was leached from the surface to lower soil horizons. Arnesen & Krogstad (1998) found that fluoride (added as sodium fluoride) accumulation was high in the upper 0–10 cm of soil columns, where 50–90% of the accumulated fluoride was found. The B-horizons sorbed considerably more fluoride than the Ah-horizons, due to higher content of aluminium oxides/hydroxides. A study by McLaughlin et al. (2001) involving long-term application of phosphate fertilizers has shown a large portion of fluoride applied as impurities in the fertilizer to remain in the 0- to 10-cm depth of the soil profile.
In sandy acidic soils, fluoride tends to be present in water-soluble forms (Shacklette et al., 1974). Street & Elwali (1983) determined the activity of the fluoride ion in acid sandy soils that had been limed. Fluorite was shown to be the solid phase controlling fluoride ion activity in soils between pH 5.5 and 7.0. At pH values below 5.0, the fluoride ion activity indicated supersaturation with respect to fluorite. These data indicate that liming of acid soils may precipitate fluorite, with a subsequent reduction in the concentration of fluoride ion in solution.
Murray (1984b) reported that low amounts of fluoride were leached from a highly disturbed sandy podzol soil of no distinct structure. Even at high fluoride application rates (3.2–80 g per soil column of diameter 0.1 m with a depth of 2 m), only 2.6–4.6% of the fluoride applied was leached in the water-soluble form. The pH of the eluate increased with increasing fluoride application, and this was probably due to adsorption of fluoride, releasing hydroxide ions from the soil metal hydroxides. Over time, the concentration of water-soluble fluoride decreased due to increased adsorption on soil particles.
Mean soil concentrations in Pennsylvania, USA, were 377, 0.38 and 21.7 mg/kg for total fluoride, water-soluble fluoride and resin-exchangeable fluoride, respectively. The authors suggested that fluoride is relatively immobile in soil, since most of the fluoride was not readily soluble or exchangeable (Gilpin & Johnson, 1980).
The water-soluble fluoride in sodic surface soil treated with gypsum increased with increasing exchangeable sodium per cent (Chhabra et al., 1979). The increase in exchangeable sodium per cent also caused an increase in soil pH, which in turn caused an increase in water-soluble fluoride. Incubation studies revealed that a major portion of the added fluoride was adsorbed to soil within the first 8 days. Adsorption to soil followed Langmuir isotherms up to an equilibrium soluble fluoride concentration (11.4 mg/litre), with precipitation at higher concentrations.
Calcium fluoride was formed in soils irrigated with fluoride solutions. Calcium fluoride is formed when the fluoride adsorption capacity is exceeded and the fluoride and calcium ion activities exceed the ion activity product of calcium fluoride (Tracy et al., 1984). Less than 2% of applied fluoride was measured in the leachate, and between 15 and 20% of added fluoride was precipitated as calcium fluoride. Fluoride was precipitated in the upper profile, although the authors expected that once the adsorption mechanisms were exceeded, soluble fluoride would leach deeper into the soil with continued irrigation.
A large fraction of the fluoride in topsoil sampled at a distance of 0.5–1.0 km from an aluminium smelter was reported to be in water-soluble form (Polomski et al., 1982). The authors concluded that the fluoride was present as calcium fluoride.
Breimer et al. (1989) determined the vertical distribution of fluoride in the soil profiles sampled near an industrial region. In calcareous soils, fluoride (as extractable with hydrochloric acid) was restricted to the top 40–50 cm, probably due to the precipitation of calcium fluoride in the presence of lime. A slight leaching of fluoride into the Bt and C horizons was reported in non-calcareous soils. Water-extractable fluoride showed an increase with depth in the A horizons and subsequently decreased to base levels in the lower subsoil.
The adsorption of fluoride from the water phase may be an important transport characteristic in calcareous soils at low flow rates, but this exchange may be rate-limited at high flow rates (Flühler et al., 1982). Dissolved fluoride concentrations may be high around the root zone in soils with a high fluoride input such as from atmospheric deposition. The high concentrations exist only for a limited time until the fluoride is withdrawn from the solution. The adsorption isotherm was reported to be non-linear between initial concentrations of 10 and 50 mg fluoride/litre. Retention of fluoride in uncontaminated calcareous soil was higher than retention in calcareous soil from areas with fluoride contamination. The adsorption and desorption of fluoride in acidic soil were not related to previous fluoride contamination.
Fluoride-containing solutions increased the mobilization and leaching of aluminium from soils. Leaching of aluminium was reported to be greater from soil contaminated from an aluminium smelter than from uncontaminated soil (Haidouti, 1995). In the uncontaminated soil, losses of aluminium from the acid soil were higher than those from the calcareous soil. Arnesen (1998) also found that fluoride can solubilize aluminium, iron and organic material and can increase soil pH through exchange with hydroxide ions.
Unlike other soluble salts, fluoride was not leached from naturally salinized salt-affected soil. It was redistributed within the soil profile (Lavado et al., 1983). The adsorption of fluoride to soils increased with decreasing pH within the pH range 8.5–6. Retention of fluoride in the soil was positively correlated with ammonium acetate extractable iron.
Molecular fluorine hydrolyses to form hydrogen fluoride, the most stable form of fluorine in the atmosphere (ATSDR, 1993). Silicon tetrafluoride reacts with moisture in the atmosphere, forming hydrated silica and fluorosilicic acid. Uranium hexafluoride, which is used in nuclear power applications, is hydrolysed to hydrogen fluoride and uranyl fluoride (UO2F2), which are removed from the atmosphere by condensation or nucleation processes.
In dilute solutions at neutral pH, dissolved fluorides are predominantly present as the fluoride ion (Bell et al., 1970). As the pH decreases below pH 5.5, the proportion of fluoride ions decreases, and the proportion of non-dissociated hydrogen fluoride increases. However, if sufficient aluminium is present in solution, aluminium–fluoride complexes (AlF2+, AlF2+ and AlF3) generally dominate below pH 5.5 until pH 1, where hydrogen fluoride begins to dominate as pH decreases further (Parker et al., 1995). Aluminium solubility is low between pH 5.2 and pH 8.8, whereas at higher pH values, aluminium solubilizes as the aluminate ion (Al(OH)4–).
Soluble fluorides are bioaccumulated by some aquatic and terrestrial biota. However, no information was identified concerning the biomagnification of fluoride in aquatic or terrestrial food-chains (Hemens & Warwick, 1972; Barbaro et al., 1981; ATSDR, 1993). Inorganic fluorides tend to accumulate preferentially in the skeletal and dental hard tissues of vertebrates, exoskeletons of invertebrates and cell walls of plants (Hemens & Warwick, 1972; Davison, 1983; Michel et al., 1984; Kierdorf et al., 1997; Sands et al., 1998).
Bioconcentration factors greater than 10 (expressed on a wet weight basis) were reported in both aquatic plants and animals following exposure to solutions of up to 50 mg fluoride/litre (Sloof et al., 1989). Twenty-four hours after sodium fluoride (22 mg fluoride/litre) was released into an experimental pond, the concentration of fluoride in aquatic vascular plants was increased 35-fold, and uptake was also increased in algae (14-fold), molluscs (12-fold) and fish (7-fold) (Kudo & Garrec, 1983).
The water hyacinth (Eichhornia crissipes) has been reported to take up fluoride from water. Plants were exposed to fluoride solutions of 6–26 mg/litre for 4 weeks, and fluoride uptake was related to exposure, ranging from 0.8 to 5 mg/kg (Rao et al., 1973).
The fluoride concentration in Sydney rock oyster (Saccostrea commercialis) sprat increased with increasing fluoride exposure. The sprat were exposed to 0.7–30.7 mg fluoride/litre for 3 weeks and had whole-body concentrations ranging from 45 to 204 mg/kg dry weight (Nell & Livanos, 1988).
Following exposure to inorganic fluoride levels of 52 mg/litre for 72 days, prawn (Penaeus indicus), crab (Tylodiplax blephariskios), shrimp (Palaemon pacificus) and mullet (Mugil cephalus) had whole-animal (ash) concentrations of 3248, 1414, 3116 and 7743 mg fluoride/kg, respectively (Hemens & Warwick, 1972). The fluoride was accumulated from the water and not via ingested plant food. No differences in total-body fluoride levels were reported in the same species when exposed to 5.7 or 5.9 mg/litre for either 68 or 113 days (Hemens et al., 1975). Fluoride was accumulated mainly in the calcified skeletal structures of fish and crustaceans, with higher accumulation rates reported during the early stages of growth in fish and during periods of deposition of new skeletal material (ecdysis) in crustaceans.
Wright (1977) exposed brown trout (Salmo trutta) fry to inorganic fluoride (5, 10 or 20 mg/litre) for 200 h (12 °C; pH 6.8; hardness 73 mg/litre). Fry accumulated 10, 18 and 30 mg fluoride/kg at the three fluoride concentrations, respectively.
Terrestrial plants may accumulate inorganic fluorides following airborne deposition and uptake from the soil (Davison, 1983). Sloof et al. (1989) reported that the main route of uptake of fluoride by plants is from aerial deposition on the plant surface. Plant uptake from soil is generally low (except for accumulators) unless the fluoride has been added suddenly, such as following amendment with sludge or phosphate fertilizer. The availability to plants tends to decrease with time following application of fluoride. The degree of accumulation depends on several factors, including soil type and, most prominently, pH. At acidic pH (below pH 5.5), fluoride becomes more phytoavailable through complexation with soluble aluminium fluoride species, which are themselves taken up by plants or increase the potential for the fluoride ion to be taken up by the plant (Stevens et al., 1997). Plant uptake of fluoride from solution culture is dependent on plant species and positively related to the ionic strength of the growth solution. Once a threshold fluoride ion activity in nutrient solutions is reached, fluoride concentrations in plants increase rapidly (Stevens et al., 1998a).
The uptake of fluoride by ryegrass (Lolium multiflorum) grown in soil amended with a fluoride-rich sludge increased with increasing fluoride application (84–672 kg/ha) (Davis, 1980). Fluoride concentrations in the plant tissue exceeded the 30 mg/kg dry matter threshold of toxicity to cattle in the first cuts of grass exposed to 300 kg sludge/ha or greater. Concentrations in the second cut of grass exceeded the threshold value at 672 kg sludge/ha, and the third cut of grass was below the threshold at all concentrations tested.
The amount of fluoride taken up from soil by sunflower seedlings (Helianthus annuus) was proportional to the fluoride concentration of the solution added to the soil (10–100 µg/ml). Highest concentrations were found in the roots, with reduced concentrations in successive leaf ranks (Cooke et al., 1978). Similarly, concentrations of fluoride were high in roots and low in shoots in tomatoes (Lycopersicon esculentum) and oats (Avena sativa) grown on fluoride-enriched media (Stevens et al., 2000).
Reynolds & Laurence (1990) found that exposure of kidney bean (Phaseolus vulgaris) plants to intermittent high levels of atmospheric hydrogen fluoride (3 µg fluoride/m3 for 5 consecutive days or 5 µg fluoride/m3 for 3 consecutive days) caused higher accumulation in leaves than did exposure to a continuous dose at a lower level (1 µg fluoride/m3 for 15 consecutive days), even though the plants received the same total dose. They also noted that the rate of uptake from the air over a 15-day period, particularly for the highest dose (5 µg fluoride/m3), was most significant at first exposure and declined during subsequent exposures.
Walton (1987a) demonstrated that untreated earthworms (Lumbricus terrestris) from which gut soil had been removed (depurated) contained only 6% of the amount of fluoride reported in whole worms. Worms kept in soil were exposed to a sodium fluoride concentration of 1000 mg/litre for 30 days. The fluoride content of the worms was established either at the end of exposure or following an 8-day starvation period. The fluoride concentration in treated worms with their gut contents washed out was more than 6 times greater than the fluoride concentration of untreated worms. Even after an 8-day starvation period, the concentration in exposed worms was 4 times greater than the concentration in the unexposed worms. The fluoride concentration in the worms was always lower than the concentration in the soil.
The deposition of fluoride in the bones of white leghorn (Gallus gallus) chicks exposed to 252 mg fluoride/kg in the diet from hatching for 3–10 weeks was 3–7 times greater than for birds fed a basal diet. Fluoride was deposited in the bones of exposed females following sexual maturity and was shown to be associated with physiological factors associated with egg production, since birds with restricted ovulation had fluoride levels similar to those of males (Michel et al., 1984).
The bioavailability of fluoride to sheep (Ovis aries) from plants such as sea aster (Aster tripolium), sand couch (Elymus pycnanthus), fescue (Festuca rubra) and common salt-marsh grass (Puccinellia maritima) grown in an industrial area was calculated to range from 65 to 90% (Baars et al., 1987).
Dairy calves (Bos sp.) were fed on continuous (1.5 mg/kg body weight), periodic (1.5 mg/kg body weight for 6 months; control diet for 6 months) and alternate (3 mg/kg body weight for 4 months; 0.75 mg/kg body weight for 8 months) diets for 6 years. Vertebral fluoride levels were 550 mg/kg ash weight for controls and 15 000, 7700 and 14 000 mg/kg for the three treatment groups, respectively (Suttie, 1983).
Suttie et al. (1985) maintained white-tailed deer (Odocoileus virginianus) on diets containing fluoride at 25 and 50 mg/kg for 1 year. Fluoride levels in antlers ranged from 3000 to 5800 mg/kg ash weight and in vertebrae from 4200 to 7400 mg/kg.
Fluoride levels in surface waters vary according to geographical location and proximity to emission sources. Surface water concentrations generally range from 0.01 to 0.3 mg/litre (ATSDR, 1993). Ambient surface water fluoride levels are summarized in Table 1. Seawater contains more fluoride than fresh water, with concentrations ranging from 1.2 to 1.5 mg/litre.
Table 1. Concentrations of fluoride in unpolluted water
|
Location |
Fluoride concentration |
Reference |
|
Areas with low natural fluoride |
||
|
Canada |
0.05 |
Environment Canada (1994) |
|
USA |
0.035–0.052b |
Overton et al. (1986) |
|
USA (Cache la Poudre River) |
(0.3–0.5) |
Camargo et al. (1992) |
|
Belgium (River Meuse) |
0.13–0.2 |
Van Craenenbroeck & Marivoet (1987) |
|
France (rivers) |
0.08–0.25 |
Martin & Salvadori (1983) |
|
Norway (lakes) |
0.037 (<0.005–0.56) |
Skjelkvåle (1994a) |
|
Spain (Duratón River) |
0.1 |
Camargo (1996a) |
|
United Kingdom (rivers) |
<0.05–0.4 |
Fuge & Andrews (1988) |
|
Wales, United Kingdom (streams) |
0.02–0.22 |
Neal (1989) |
|
Nigeria (River Niger) |
0.1–0.12 |
Nriagu (1986) |
|
India (rivers) |
0.2–0.25 |
Zingde & Mandalia (1988) |
|
India (rivers) |
0.038–0.21 |
Datta et al. (2000) |
|
Tibet, China (rivers) |
0.04 |
Cao et al. (2000) |
|
Areas with high natural fluoride |
||
|
Hot springs and geysers, Yellowstone National Park, USA |
(25–50) |
Neuhold & Sigler (1960) |
|
Firehole and Madison rivers, USA |
(1–14) |
Neuhold & Sigler (1960) |
|
Walker and Pyramid lakes, Nevada, USA |
(up to 13) |
Sigler & Neuhold (1972) |
|
Kenya (lakes) |
(up to 2800) |
Nair et al. (1984) |
a
Mean and range of fluoride concentrations, unless otherwise stated.b
Range of means from four different regions of the eastern USA.Higher levels of fluoride have been measured in areas where the natural rock is rich in fluoride and near industrial outfalls. Skjelkvåle (1994a) measured fluoride in Norwegian lakes and found concentrations ranging from <0.005 to 0.56 mg/litre, with a mean value of 0.037 mg/litre. Fluoride content in the bedrock was the most important factor determining fluoride levels in lake waters. Elevated fluoride concentrations were found in acidified areas compared with other regions with similar geology (Skjelkvåle, 1994a). Similarly, Hirayama et al. (1996) reported increased levels of fluoride (up to 1 mg/litre) in some rivers flowing into Lake Biwa, Japan, because of the local geology.
Elevated inorganic fluoride levels in surface water are often seen in regions where there is geothermal or volcanic activity, at the foot of high mountains and in areas with geological deposits of marine origin. Thus, hot springs and geysers in Yellowstone National Park, USA, contain 25–50 mg fluoride/litre (Neuhold & Sigler, 1960), whereas the Firehole and Madison rivers, also in Yellowstone National Park, contain 1–14 mg fluoride/litre. Walker and Pyramid lakes, in Nevada, USA, contain levels as high as 13 mg fluoride/litre (Sigler & Neuhold, 1972). The most well documented area of high-fluoride surface water associated with volcanic activity follows the East African Rift Valley system from the Jordan Valley down through Sudan, Ethiopia, Uganda, Kenya and Tanzania. Many of the lakes of the Rift Valley system have extremely high fluoride concentrations — for example, 1640 mg/litre and 2800 mg/litre, respectively, in the Kenyan lakes Elmentaita and Nakura (Nair et al., 1984).
Anthropogenic sources can also lead to increased local levels of fluoride. Harbo et al. (1974) found increased concentrations of fluoride in the surface waters of Kitimat Harbour, Canada, in the vicinity of a large smelter. Fluoride concentrations in the River Meuse, Belgium, increased from a maximum of 0.2 mg/litre upriver to a mean value of 0.65 mg/litre near the outfall of a phosphate fertilizer plant (Van Craenenbroeck & Marivoet, 1987). Zingde & Mandalia (1988) found that fluoride levels were below 0.3 mg/litre in unpolluted surface waters; however, levels were higher close to a plant manufacturing fluoride chemicals in India. Surface water concentrations ranged from 0.8 to 2.8 mg fluoride/litre, with a mean value of 1.6 mg fluoride/litre. Similarly, Somashekar & Ramaswamy (1983) reported a mean level of 2.97 mg fluoride/litre (range 0.34–5.12 mg/litre) in the River Cauvery, India, near discharges from both fertilizer and paper factories. Mean fluoride levels had declined to 0.64 mg/litre 8 km downstream of the factory discharges. Fluoride concentrations in the Uppanar Estuary, South India, ranged from 0.2 to 23.2 mg/litre in the vicinity of a factory producing fluoride compounds (Karunagaran & Subramanian, 1992). Camargo (1996a) monitored the Duratón River, Spain, downstream from an effluent discharge point. The mean fluoride concentration in the effluent was 25.3 mg/litre; fluoride concentrations at 0.1, 2.2 and 7.3 km downstream were 6.8, 2.7 and 1.3 mg/litre, respectively.
Skjelkvåle (1994b) found that streams close to Norwegian aluminium smelters contained 10 times more fluoride than natural background levels due to 40 years of fluoride emissions. The levels had decreased to background within 5 km. The author pointed out that despite the higher levels near the smelters, the fluoride concentrations are still within the general range of concentrations for Norwegian surface waters. Similar results were reported by Roy et al. (2000) for an aluminium smelter on the Saguenay River, Quebec, Canada. Mean fluoride concentrations 300 m downstream of the smelter outfalls ranged from 0.2 to 0.28 mg/litre. Camargo et al. (1992) analysed the fluoride concentration in the Cache la Poudre River, USA, downstream from a wastewater treatment plant. The mean fluoride concentration downstream was 1.17 mg/litre, about 4 times the mean fluoride concentration upstream.
Airborne fluoride exists in gaseous and particulate forms emitted from both natural and anthropogenic sources. The gaseous fluorides include hydrogen fluoride, carbon tetrafluoride (CF4), hexafluoroethane (C2F6) and silicon tetrafluoride. Particulate fluorides include cryolite, chiolite (Na5Al3F14), calcium fluoride, aluminium fluoride and sodium fluoride. Fluoride released as gaseous and particulate matter is deposited in the general vicinity of an emission source (Sidhu, 1979; Low & Bloom, 1988), although some particulates may react with other atmospheric constituents. The distribution and deposition of airborne fluoride are dependent upon emission strength, meteorological conditions, particulate size and chemical reactivity (Low & Bloom, 1988).
Levels of gaseous and particulate fluoride in ambient air and in air near industrial sources are summarized in Tables 2 and 3, respectively. In areas not in the direct vicinity of emission sources, the mean concentrations of fluoride in ambient air are generally less than 0.1 µg/m3. Levels may be slightly higher in urban than in rural locations; however, even in the vicinity of emission sources, the levels of airborne fluoride usually do not exceed 2–3 µg/m3. In Canada, mean concentrations of fluoride in samples of outdoor air collected between 1980 and 1991 in the vicinity of industrial emission sources ranged up to 0.85 µg/m3 (Health Canada, 1993). In the Netherlands (Groningen, Sloe and Sas van Gent), mean concentrations of fluoride in samples of outdoor air collected during 1985 and 1986 in the vicinity of various industrial emission sources (brickworks, aluminium plant and a glass fibre factory) ranged from 0.25 to 0.4 µg/m3 (Sloof et al., 1989). Median concentrations of fluoride in the air surrounding a Norwegian aluminium smelter in the spring and summer of 1994 ranged from 1.3 to 3.8 µg/m3 (Sr yseth et al., 1996). In areas of China where fluoride-rich coal is used as a source of fuel, concentrations of fluoride in ambient air have reached 6 µg/m3 (Liu, 1995).
Table 2. Concentration of fluoride in ambient air
|
Location |
Detection limit (µg/m3) |
Number of samples |
Concentrationa (µg/m3) |
Reference |
|
Arctic, Canada |
0.001–0.004 |
nab |
0.002–0.007c |
Barrie & Hoff (1985) |
|
|
0.001–0.004 |
na |
0.006–0.008c |
Barrie & Hoff (1985) |
|
Toronto, Ontario, Canada |
na |
na |
0.03 |
Health Canada (1993) |
|
147 non-industrial urban locations, USA |
0.05 |
3687 |
<0.05 |
Thompson et al. (1971) |
|
29 rural locations, USA |
0.05 |
724 |
<0.05 |
Thompson et al. (1971) |
|
1 non-industrial urban location, United Kingdom |
0.1 |
na |
<0.1 |
Bennett & Barratt (1980) |
|
4 non-industrial urban locations, The Netherlands |
na |
na |
0.07 |
Sloof et al. (1989) |
|
a |
Mean and range of concentrations of total (gaseous and particulate) fluoride in ambient air, unless otherwise stated. |
|
b |
na = not available. |
|
c |
Range of means. |
|
d |
nd = not detectable. |
Table 3. Concentration of fluoride in outdoor air near industrial sources
|
Location |
Distance from source (km) |
Concentrationa (µg/m3) |
Reference |
|
Cornwall Island, Ontario, Canada |
1.65 |
0.79–0.85 |
Environment Canada (1989) |
|
|
4.0 |
0.43 |
|
|
Brampton, Ontario, Canada |
<1.0 |
0.73 |
Health Canada (1993) |
|
Trail, British Columbia, Canada |
<5.0 |
0.59 |
Environment Canada (1994) |
|
Various locations, Quebec, Canada |
0.5–2.0 |
0.1–0.67 |
Environment Canada (1994) |
|
South African Highveld |
Various |
0.08–0.14c |
Scheifinger & Held (1997) |
|
Norway |
|
1.3–3.8 |
Sr yseth et al. (1996) |
|
Groningen, The Netherlands |
1.3 |
0.4 |
Sloof et al. (1989) |
|
Zeeland (Sloe area), The Netherlands |
6.0 |
0.25 |
Sloof et al. (1989) |
|
China |
|
2–6 |
Liu (1995) |
|
a |
Mean and range of fluoride concentrations, unless otherwise stated. |
|
b |
nd = not detectable. |
|
c |
Range of means. |
Data on the levels of total and water-soluble fluoride in soil are presented in Table 4. Fluoride is a component of most types of soil, with total concentrations ranging from 20 to 1000 µg/g in areas without natural phosphate or fluoride deposits and up to several thousand micrograms per gram in mineral soils with deposits of fluoride (Davison, 1983). Airborne gaseous and particulate fluorides tend to accumulate within the surface layer of soils but may be displaced throughout the root zone, even in calcareous soils (Polomski et al., 1982). The clay and organic carbon content as well as the pH of soil are primarily responsible for the origin and/or retention of fluoride in soils. Fluoride in soil is primarily associated with the soil colloid or clay (Omueti & Jones, 1977). For all soils, it is the soluble fluoride content that is biologically important to plants and animals. It has also been reported that in saline soils, the bioavailability of fluoride to plants is related to the water-soluble component of the fluoride present (Davison, 1983).
Table 4. Concentration of fluoride in soil
|
Location |
Soil description |
Soil depth (cm) |
Total fluoride (mg/kg)a |
Water-soluble fluoride (mg/kg)a |
Reference |
|
Canada |
3 reference soil samples from the Canadian Soil Survey Committee |
0–15 |
160 |
nab |
Schuppli (1985) |
|
Canada |
23 reference soil samples from the Canadian Soil Survey Committee |
0–130 |
309 |
na |
Schuppli (1985) |
|
Newfoundland, Canada |
forest soil |
na |
6 |
2 (na) |
Sidhu (1982) |
|
|
soil humus from various forests |
0–3 |
(6–11) |
na (0.95–2.72) |
Sidhu (1979) |
|
|
0.7 km north-east of an elemental phosphorus plant |
0–3 |
1138–1915 |
9.7–60.2 |
Sidhu (1979) |
|
|
18.7 km north-east of an elemental phosphorus plant |
0–3 |
18.7–26.1 |
0.93–1.9 |
Sidhu (1979) |
|
Pennsylvania, USA |
55 samples of silt and clay |
0–10 |
377 |
0.38 (<0.05–1.5) |
Gilpin & Johnson (1980) |
|
Illinois, USA |
201 surface soil samples from various locations |
na |
271 |
na |
Omueti & Jones (1977) |
|
Montana, USA |
forest soil samples from an area exposed to atmospheric fluoride emissions from an aluminium smelter |
na |
330–1747 |
15.6–704 |
Polomski et al. (1982) |
|
Ohio, USA |
silt loam (0–5 cm) collected from 7 sites located in the vicinity of an aluminium plant |
0.5–4 |
353–371 |
na |
McClenahen (1976) |
|
Oklahoma, USA |
reference site |
0–2 |
121 |
0.2 |
Schroder et al. (1999) |
|
|
petrochemical waste site |
0–2 |
1954 |
4.8 (1.2–12.6) |
|
|
Greece |
0–4 km from an aluminium plant |
0–5 |
823 |
na |
Tsiros et al. (1998) |
|
|
5–15 km from an aluminium plant |
0–5 |
570 |
na |
|
|
|
8–15 km from an aluminium plant |
0–5 |
339 |
na |
|
|
Austria |
0.5 km from an aluminium smelter |
0–10 |
– |
84–124c |
Tscherko & Kandeler (1997) |
|
|
1 km from an aluminium smelter |
0–10 |
– |
48–54c |
|
|
|
4 km from an aluminium smelter |
0–10 |
– |
21–33c |
|
|
|
15 km from an aluminium smelter |
0–10 |
– |
9.8–10c |
|
|
The Netherlands |
72 samples of agricultural soil |
na |
na (39–679) |
na (0.5–13) |
Sloof et al. (1989) |
|
Guangdong Province, China |
19 samples of cultivated, acidic soil (pH 3.8–4.5) |
0–20 |
186–387c |
0.76–2.71c |
Fung et al. (1999) |
|
Tibet |
40 samples of agricultural, forest and urban soils |
na |
– |
0.45 |
Cao et al. (2000) |
a
Mean and range of fluoride concentrations, unless otherwise stated.b
na = not available.c
Range of means.Inorganic fluorides have been measured in a wide variety of organisms; therefore, the data summarized here will be only a representative sample of the available literature.
Fluorides can be taken up by aquatic organisms directly from the water or, to a lesser extent, via food. Uptake of fluorides by biota will very much depend on the proximity of anthropogenic sources, the local geology and the physicochemical conditions, which will determine the bioavailability of fluorides to any given organism.
Hocking et al. (1980) indicated that although there was some accumulation of inorganic fluoride in marine vegetation, the rate of accumulation was far lower than that of airborne inorganic fluoride by terrestrial plants. In the vicinity of an aluminium smelter, samples of seaweed were analysed for their inorganic fluoride concentrations. For the seaweeds Fucus distichus and Ectocarpus sp., inorganic fluoride concentrations were determined for both low- and mid-tide sites at 150 and 500 m from the outfall. At the low-tide sites, F. distichus had values of 62 and 48 mg fluoride/kg at 150 and 500 m, respectively. Ectocarpus sp. had a level of 143 mg fluoride/kg at 500 m from the outfall. At the mid-tide sites at 150 and 500 m, Ectocarpus sp. had levels of 317 and 140 mg fluoride/kg, respectively, while F. distichus had a level of 18 mg fluoride/kg at 500 m from the outfall.
Four species of sea grass, a marine higher plant, were sampled in the Antikyra Gulf, Greece, near an aluminium factory. Mean fluoride concentrations ranged from 23.4 to 34.4 mg/kg dry weight (Malea, 1995).
Aquatic invertebrates and fish tend to accumulate fluoride in the exoskeleton and in bone, respectively. Adelung et al. (1987) sampled two krill species, Euphausia superba and Meganyctiphanes norvegica, and found mean whole-body fluoride concentrations of 1058 and 2153 mg/kg dry weight, respectively. Most of the fluoride was concentrated in the exoskeleton, with levels of <6 mg/kg being found in soft tissues. Similar whole-body and exoskeleton values were found by Boone & Manthey (1983) and Christians & Leinemann (1980); however, they found higher levels in soft tissues, which Adelung et al. (1987) suggested was due to contamination from the exoskeleton during analysis. Sands et al. (1998) monitored a range of Antarctic crustaceans and found that euphausiids accumulated the highest fluoride concentrations, with levels up to 5477 mg/kg in the exoskeleton of Euphausia crystallorophias. The mouthparts of E. superba contained almost 13 000 mg fluoride/kg dry weight. Copepods contained the lowest fluoride levels (0.87 mg fluoride/kg whole body).
Wright & Davison (1975) studied the accumulation of fluoride in a number of marine and intertidal animals found close to an aluminium smelter at Lynemouth, United Kingdom. The seawater was found to contain approximately 3.4 mg fluoride/litre (1.9 mg fluoride/litre above background levels). The skeletal tissue of both vertebrates and invertebrates was found to contain elevated fluoride levels. The mean exoskeleton fluoride concentrations for the swimming crab Portunus depurator, the shrimp Crangon vulgaris and the prawn Leander serratus were 11.6, 11.5 and 11.3 mg/kg, respectively. Tissue accumulation of fluoride in the Atlantic cod (Gadus morhua) was highest in the skin (mean 29.6 mg fluoride/kg); in haddock (G. aeglefinus) and dabs (Pleuronectes limanda), the mean tissue values were highest in the axial skeleton, 49.3 and 99.7 mg fluoride/kg, respectively.
Fish, predominantly mullet (Mugil auratus, Mugil cephalus and Mugil labrosus), caught in Gabes Bay, South Tunisia, where fluoride-rich effluents were discharged (inorganic fluoride levels in water were about 2–3 mg/litre), were found to have tissue fluoride levels 4–5 times higher than fish obtained from the Tunis Bay, which is remote from point sources (fluoride levels in water were about 1.4 mg/litre) (Milhaud et al., 1981). The mullet caught in Gabes Bay had mean inorganic fluoride values of 320, 9.6 and 14.6 mg/kg wet weight for bone, muscle and muscle/skin, respectively. Comparatively, the mean levels of fluoride in control fish were 73, 1.8 and 3.8 mg/kg wet weight, respectively. Similar values to those found in polluted environments were reported by Gikunju (1992) for fish inhabiting Lake Naivasha, Kenya (water fluoride concentrations ranged from 2.4 to 2.6 mg/litre). Tilapia (Oreochromis leucostictus) contained fluoride concentrations of 1.97, 4.96, 143.1 and 210.6 mg/kg wet weight for muscle, skin, gills and bone, respectively.
Adelung et al. (1985) analysed bone and soft tissue from Antarctic seals (Weddel seal, Leptochynotus weddelli, and crabeater seal, Lobodon carcinophagus). Mean bone fluoride concentrations ranged from 135 to 6380 mg/kg dry weight; soft tissues contained mean concentrations ranging from 2.3 (kidney) to 9.1 (brain) mg fluoride/kg. Bone samples taken from fin whales (Balaenoptera physalus) in the North Atlantic contained fluoride concentrations ranging from 4300 to 18 600 mg/kg (Landy et al., 1991); fluoride concentrations were positively correlated with age. Mikaelian et al. (1999) found mean bone fluoride levels of 10 365 mg/kg in beluga whales (Delphinapterus leucas) from Hudson Bay, Canada, compared with mean levels of 4539 mg fluoride/kg in whales from the St. Lawrence estuary, Canada; fluoride levels were positively correlated with age only in the latter population.
Fluoride levels in terrestrial biota tend to be increased in areas with high fluoride levels due to both natural and anthropogenic sources.
Lichens have been used extensively as biomonitors for fluorides. Davies & Notcutt (1988) sampled lichens from the slopes of the Mount Etna volcano in 1985 and 1987 and found fluoride levels ranging from 2 to 141 mg/kg (lichen from control sites contained <2 mg fluoride/kg). Similarly, Davies & Notcutt (1989) found that lichens growing in the Canary Islands accumulated fluorides from minor volcanic eruptions. Fluoride levels of up to 23 mg/kg were measured, compared with a background level of <1 mg/kg. Lichens have also been used to monitor anthropogenic outputs of fluorides from both brickfields (Davies, 1982, 1986) and an aluminium smelter (Perkins et al., 1980; Perkins & Millar, 1987). Levels of fluoride were found to be related to site emissions, prevailing winds and distance from source. Mean fluoride concentrations of 150–250 mg/kg were measured in lichens growing within 2–3 km of the pollution source.
Most of the inorganic fluoride in the soil is insoluble and therefore less available to plants. The capacity for a plant to absorb inorganic fluoride from the soil will also depend on the species of plant and, to some extent, the ionic species of fluoride present in solution (Stevens et al., 1997, 1998a, 1998b) (see section 4.2). For example, some genera (e.g., Dichapetalum, Thea, Camellia, Oxylobium, Acacia and Palicoure) accumulate fluoride and show none of the symptoms of fluoride toxicity, with fluoride concentrations up to 4000 µg/g dry weight (Jacobson et al., 1966; Weinstein & Alscher-Herman, 1982). In samples obtained near a reclaimed fluorspar mining site, the levels of inorganic fluorides in plants were higher than those in samples obtained from a control site (Andrews et al., 1982). High total soil fluoride (10 000 mg/kg) has been measured in the metalliferous fluorspar tailings and is reflected by elevated concentrations in vegetation (300–1000 mg/kg) (Andrews et al., 1989). Other genera are non-accumulators of fluoride and have shown signs of fluoride toxicity at much lower fluoride concentrations in their tissues. For example, gladiolus (Gladiolus sp.) plants may become necrotic with 20 µg fluoride/g dry weight (Jacobson et al., 1966). Normal concentrations of fluoride in plant leaves usually range from 0.1 to 15 µg/g (Cholak, 1959; Cooke et al., 1976; Hara et al., 1977).
Several plant species have the ability to convert soluble fluorides taken up from soil into carbon–fluoride compounds, such as monofluoroacetic acid, monofluoro-oleic acid, monofluoropalmitic acid and monofluoromyristic acid (Marais, 1944; Ward et al., 1964). Two well known Australian species that synthesize monofluoroacetates are Gastrolobium spp. (Aplin, 1968) and Georgina gidgee (Acacia georginae F.M. Bailey) (Oelrichs & McEwan, 1961); other species include Oxylobium spp. (Australia), Dichapetalum spp. (Africa) and Palicourea marcgravii (reviewed by Weinstein, 1977). The function of monofluoroacetate in the plant is unknown. The assumption that its synthesis by the plant is a response to high concentrations of available inorganic fluoride in soil or water (Preuss et al., 1970) is not supported by the finding that some plants containing monofluoroacetate grow in soils with low fluoride concentrations (Hall, 1972).
Most fluorides enter plant tissues as gases through the stomata and accumulate in leaves. Small amounts of airborne particulate fluoride can enter the plant through the epidermis and cuticle (Weinstein, 1977). Vegetation has been widely monitored in the vicinity of anthropogenic emission sources of fluoride. Rice et al. (1984) obtained background fluoride concentrations for 17 plant species indigenous to the Northern Great Plains in the USA. Mean fluoride concentrations ranged from 1.5 to 3.3 mg/kg. Nine of these species were also monitored in polluted areas, and mean fluoride concentrations ranged from 2.8 to 11.3 mg/kg, with Rocky Mountain juniper (Juniperus scopulorum) accumulating the highest amounts of fluoride.
Leece et al. (1986) found increased levels of fluoride in basal leaves of grapevines (Vitus vinifera), 9–25 km downwind of an aluminium smelter in New South Wales, Australia. Inorganic fluoride concentrations of 4–42 mg/kg were recorded, with the highest levels 9–11 km from the smelter. Bowen (1988) monitored vegetation around two aluminium smelters in Hunter Valley, Australia. Foliar concentrations were greater than background levels at distances of up to 3 km from one smelter and up to 1 km from the other. Close to the smelters, tree species accumulated more foliar fluoride than shrub species, which in turn accumulated more foliar fluoride than herb species.
Fluoride levels in trees near an aluminium smelter in Kitimat, British Columbia, Canada, have been measured since 1954. From 1954 to 1973, western hemlock (Tsuga heterophylla) tree foliage from the vicinity of the smelter contained a mean fluoride concentration of 271 mg/kg; the surrounding area had mean levels of 104 mg/kg. For the 5-year period 1974–1979, during which time there was a 64% decline in smelter emissions, foliage from the inner, outer and surrounding zones contained mean inorganic fluoride levels of 87, 29 and 22 mg/kg, respectively (Bunce, 1985). Similarly, Amundson et al. (1990) found significantly higher levels of foliar fluoride in pine species (Pinus sp.) growing within 0.8 km of a smelter than in those growing at a distance of 1.8 km. Mean fluoride levels in vegetation in the vicinity of a phosphorus plant ranged from 44 mg/kg in lightly damaged areas to 281 mg/kg in severely damaged areas (Thompson et al., 1979). Horntvedt (1995) monitored pine and spruce (Picea sp.) needles for 15–25 years on permanent plots around eight aluminium smelters and found a high correlation between needle fluoride levels and smelter emissions.
Taylor & Basabe (1984) established correlations between fluoride concentrations in needles of Douglas-fir (Pseudotsuga menziesii) and annual growth increments, wind pattern, distance from fluoride source (aluminium smelter) and hydrogen fluoride concentrations in aerial effluents. Mean fluoride concentrations in needles ranged from 27.6 to 60.9 mg/kg dry weight for the period 1972–1975, with an overall range of 2–590 mg/kg. Needles collected from a control site contained mean fluoride concentrations ranging from 3.2 to 5.8 mg/kg.
Several invertebrate species have been sampled in the vicinity of fluoride sources such as aluminium smelters. Walton (1986) analysed earthworms collected at different distances from an aluminium smelter. The mean whole-body fluoride concentration was 112 mg/kg within 1 km of the plant and ranged from 19 to 48 mg/kg at a distance of 3–15 km. Breimer et al. (1989) found a highly significant correlation between total fluoride in earthworms (Lumbricus sp.) and hydrochloric acid-extractable fluoride in topsoil. Worm tissue (without gut) contained 6–14 mg/kg at control sites and up to 135 mg/kg at contaminated sites (topsoil contained >200 mg fluoride/kg). Vogel & Ottow (1991) sampled a variety of earthworm species in the vicinity of a chemical factory. They found a significant difference in fluoride accumulation between species. Lumbricus terrestris showed the lowest accumulation; the authors pointed out that this species tends to migrate to deeper, less contaminated mineral horizons. Those species that remain in the upper soil horizons tended to accumulate larger amounts of fluoride.
Buse (1986) monitored a range of invertebrates near an aluminium smelter and found that scavengers, such as millipedes and woodlice, accumulated the highest concentrations (mean 1100 mg fluoride/kg). Predatory spiders (Arachnida: Araneae), harvestmen (Arachnida: Opiliones), slugs and snails (Gastropoda: Pulmonata), earthworms (Oligochaeta), beetles (Insecta: Coleoptera) and centipedes (Chilopoda) contained mean fluoride concentrations of 393, 258, 190, 148, 50 and 48 mg/kg, respectively. The lowest mean fluoride value was found in grasshoppers, at 20 mg/kg.
Fluoride has been measured in the femurs of 12 species of predatory birds in the British Isles. Mean fluoride concentrations ranged from 112 mg/kg (common barn-owl, Tyto alba) to 2690 mg/kg (merlin, Falco columbarius). Raptors that feed predominantly on small birds accumulated the highest fluoride concentrations (Seel & Thomson, 1984). Henny & Burke (1990) measured fluoride in femurs of black-crowned night-herons (Nycticorax nycticorax) near a phosphate-processing complex. Fluoride concentrations ranged from 540 to 11 000 mg/kg and increased with age. Vikr ren & Stuve (1996a) found that the mean eggshell fluoride concentration was significantly increased in mew gull (Larus canus) and herring gull (Larus argentatus) eggs collected near aluminium smelters (120–212 mg fluoride/kg ash weight) compared with a control site (83–143 mg fluoride/kg).
Several authors have monitored bone fluoride concentrations in small mammals (voles, mice and shrews) in the vicinity of anthropogenic sources of fluoride (Walton, 1988). At a heavily polluted site near an aluminium smelter in the United Kingdom, field voles (Microtus agrestis) contained bone fluoride concentrations ranging from 910 to 11 000 mg/kg (mean 7148 mg/kg), and wood mice (Apodemus sylvaticus) contained bone fluoride concentrations ranging from 1800 to 17 200 mg/kg (mean 8430 mg/kg) (Walton, 1987b). Small mammals (field vole, M. agrestis, and common shrew, Sorex araneus) sampled from metalliferous fluorspar tailings contained mean fluoride concentrations ranging from 120 to 360 mg/kg. Individuals collected from a control site contained mean fluoride concentrations ranging from 11 to 85 mg/kg (Andrews et al., 1989).
Thomson (1987) studied the bone fluoride in the prey of common barn-owls (T. alba), field voles (M. agrestis) and common shrews (S. araneus). Specimens were collected from owl pellets 0.9 km from an aluminium smelter and another site 22 km from the plant. Voles were also trapped at two locations, and shrews were trapped at six sites. A negative correlation was found between distance from the smelter and inorganic fluoride levels in the vole, and inorganic fluoride levels did not widely differ between trapped specimens and owl pellet specimens. Fluoride concentrations in bones of the trapped voles ranged from 1581 to 2318 mg/kg at the plant site and from 111 to 257 mg/kg at the control site. Walton (1985) found a downward trend in bone fluoride concentrations in mice and voles at increasing distances from the smelter. Walton (1986) analysed bones from common shrews (S. araneus) and moles (Talpa europaea) collected at different distances from the emission source. Fluoride concentrations in shrews decreased with distance from the smelter. Mean fluoride concentrations in shrews were 2093 mg/kg close to the plant and ranged from 770 to 1705 mg/kg at a distance of 4–15 km. Moles from within 1 km of the plant had a mean bone fluoride content of 7740 mg/kg; however, moles collected at distances from 3 to 15 km showed no significant relationship with distance, with mean concentrations ranging from 935 to 1853 mg fluoride/kg. Shore (1995) established that it was possible to predict the bone fluoride content of field voles (M. agrestis), common shrews (S. araneus) or wood mice (A. sylvaticus) at a particular site based on data from any one of the other species.
Walton (1984) monitored red foxes (Vulpes vulpes) in the United Kingdom and found mean bone fluoride concentrations ranging from 283 mg/kg at an unpolluted site to 1650 mg/kg near a smelter. There was a positive correlation between bone fluoride content and age.
Larger mammals have also been extensively monitored for bone fluoride accumulation. Kierdorf et al. (1989) found mandibular fluoride in roe deer (Capreolus capreolus) to be positively correlated with age, the increase in concentration being higher in younger animals and declining in older deer. Similarly, positive correlations between age and bone fluoride content in the same species (Walton & Ackroyd, 1988), in red deer (Cervus elaphus) (Dabkowska & Machoy, 1989) and in European elk (Alces alces) (Machoy et al., 1995) have been found. Several studies have found that bone fluoride is increased in larger mammals in the vicinity of fluoride emission sources. Newman & Yu (1976) found that the bones of deer from an area near an aluminium plant in northwest Washington, USA, contained 10–35 times more fluoride than bones of control deer. The ribs of control deer contained 157 and 465 mg fluoride/kg, whereas the ribs of exposed deer contained 2820 and 6809 mg fluoride/kg. H. Kierdorf & Kierdorf (1999) found a significant reduction in the bone fluoride content of roe deer (C. capreolus) near a coal-fired power plant (Germany) when comparing deer monitored between 1973 and 1986 (mean 1529 mg fluoride/kg dry weight) with animals monitored between 1987 and 1998 (mean 452 mg fluoride/kg). Kierdorf & Kierdorf (2000) carried out a historical biomonitoring study on red deer (C. elaphus) antlers. Antlers collected prior to 1860 contained fluoride levels ranging from 28 to 79 mg/kg ash weight. The highest fluoride concentrations (up to 1392 mg/kg) were found during the 1970s, with a pronounced decline during the late 1980s and 1990s to fluoride concentrations ranging from 159 to 367 mg/kg.
Fluoride is ubiquitous in the environment, and, therefore, sources of drinking-water are likely to contain at least some small amount of fluoride. The amount of fluoride present naturally in non-controlled fluoridated drinking-water (i.e., drinking-water to which fluoride has not been intentionally added for the prevention of dental caries) is highly variable, being dependent upon the individual geological environment from which the water is obtained. Available data from national surveys conducted in a number of countries are summarized in Table 5.
Table 5. National surveys of fluoride in drinking-water
|
Location |
Fluoride concentration (mg/litre) |
Comment |
References |
|
Canada |
0.05–0.21 |
Range of mean concentrations in non-fluoridateda samples collected between 1984 and 1989 from 67 communities in 5 provinces |
Health Canada (1993) |
|
|
0.73–1.25 |
Range of mean concentrations in fluoridatedb samples collected between 1986 and 1989 from 320 communities in 8 provinces |
Health Canada (1993) |
|
Czech Republic |
0.05–3.0 |
Range of concentrations in more than 4000 samples of drinking-water collected between 1994 and 1996 from 36 districts within the Czech Republic |
NIPH (1996) |
|
Finland |
<0.1–3.0 |
Range of concentrations in 5900 groundwater samples |
Lahermo et al. (1990) |
|
|
<0.1–5.8 |
Range of concentrations in 1421 well-water samples |
Korkka-Niemi et al. (1993) |
|
|
<0.1–6.0 |
Range of concentrations in groundwater database of Geological Survey of Finland, 7229 shallow wells |
Lahermo & Backman (2000) |
|
|
<0.1–9.3 |
Range of concentrations in groundwater database of Geological Survey of Finland, 571 drilled wells |
Lahermo & Backman (2000) |
|
Germany |
0.02–0.17 |
Range of concentrations of drinking-water collected from various facilities in Germany between 1975 and 1986 |
Bergmann (1995) |
|
The Netherlands |
0.04–0.23 |
Public drinking-water samples collected in 1985 from water treatment plants in 12 provinces |
Sloof et al. (1989) |
|
Poland |
0.02–3.0 |
Range of mean concentrations in samples of drinking-water collected from 94 localities in central and northern Poland between 1993 and 1995 |
Czarnowski et al. (1996) |
|
Location |
Fluoride concentration (mg/litre) |
Comment |
References |
|
USA |
<0.1–1.0 |
Fluoride levels in drinking-water of approximately 62% of the US population served by public supplies range from <0.1 to 1.2 mg/litre; levels of fluoride in drinking-water of approximately 14% of the US population served by public supplies range from 1 to 2 mg/litre |
US EPA (1985); US DHHS (1991) |
|
a |
Drinking-water in which inorganic fluoride was not intentionally added for the prevention of dental caries. |
|
b |
Drinking-water in which inorganic fluoride was intentionally added for the prevention of dental caries. |
In 1986, approximately 40% of the population of Canada was served with controlled fluoridated drinking-water (Droste, 1987). Throughout Canada, there are a number of communities where sources of drinking-water contain elevated levels of fluoride (as high as 4.3 mg/litre) from natural sources; however, those identified communities represent only a very small proportion (0.8%) of the total population (Droste, 1987). Approximately 62% of the US population that is served by public water systems is supplied with drinking-water containing fluoride at concentrations at or below 1.2 mg/litre (US EPA, 1985; US DHHS, 1991). Approximately 0.4% of the population in the USA is supplied with drinking-water (from groundwater sources) containing natural fluoride that exceeds a concentration of 2.0 mg/litre (US EPA, 1985; US DHHS, 1991). In other countries, such as Poland, Finland and the Czech Republic, levels of fluoride in drinking-water as high as 3 mg/litre have also been reported (Lahermo et al., 1990; Bergmann, 1995; Czarnowski et al., 1996; NIPH, 1996).
In areas of the world in which endemic fluorosis of the skeleton and/or teeth has been well documented, levels of fluoride in groundwater supplies have been reported to range from 3 to more than 20 mg/litre (WHO, 1984; Krishnamachari, 1987; Kaminsky et al., 1990; US DHHS, 1991). In Tanzania, 30% of waters used for drinking-water contained fluoride at concentrations above 1.5 mg/litre, levels in the Rift Valley being up to 45 mg/litre (Latham & Gretch, 1967).
Mean concentrations of fluoride ranged from 0.19 to 0.33 mg/litre in more than 24 varieties of domestic and imported bottled waters purchased in the USA in 1989 (Nowak & Nowak, 1989; Stannard et al., 1990). In a study of 52 varieties of bottled water purchased in the vicinity of Houston, Texas, USA, between 1989 and 1991, the levels of fluoride ranged from <0.1 to 0.72 mg/litre (Tate & Chan, 1994). Similar findings were reported in a study of bottled water sold in Cleveland, Ohio, USA (Lalumandier & Ayers, 2000). The concentration of fluoride in 24 samples of mineral water collected in Germany between 1984 and 1989 ranged from 0.017 to 2.70 mg/litre (Bergmann, 1995).
Since publication of the first EHC document on fluoride (WHO, 1984), additional information on the levels of fluoride in foodstuffs has become available. Virtually all foodstuffs contain at least trace amounts of fluoride. A summary of various studies in which the levels of fluoride in foods have been assessed is presented in Table 6.
Table 6. Levels of fluoride in foodstuffs
|
Food |
Fluoride concentration (mg/kg)a |
Comment |
References |
|
Milk and milk products |
0.01–0.8 |
Range of concentrations in 12 varieties of dairy products in Canada |
Dabeka & McKenzie (1995) |
|
0.045–0.51 |
Range of mean concentrations in 13 varieties of dairy products in Hungary |
Schamschula et al. (1988a) |
|
|
0.019–0.16 |
Range of concentrations in milk and milk products sampled between 1981 and 1989 in Germany |
Bergmann (1995) |
|
|
Meat and poultry |
0.04–1.2 |
Range of concentrations in 17 varieties of (cooked and raw) meat and poultry in Canada |
Dabeka & McKenzie (1995) |
|
0.01–1.7 |
Range of mean concentrations in 7 varieties of meat and poultry in Hungary |
Schamschula et al. (1988a) |
|
|
0.29 |
Mean concentration in canned meat and sausage sampled between 1981 and 1989 in Germany |
Bergmann (1995) |
|
|
Fish |
0.21–4.57 |
Range of concentrations in 4 varieties of fish available in Canada |
Dabeka & McKenzie (1995) |
|
|
0.06–1.7 |
Range of concentrations in 6 varieties of fish available in the USA |
Whitford (1996) |
|
Soups |
0.41–0.84 |
Range of concentrations in 4 varieties of soup available in Canada |
Dabeka & McKenzie (1995) |
|
|
0.42–0.94 |
Range of mean concentrations in 7 varieties of soup available in Hungary |
Schamschula et al. (1988a) |
|
Baked goods and cereals |
0.04–1.02 |
Range of concentrations in 24 varieties of baked goods and cereals available in Canada |
Dabeka & McKenzie (1995) |
|
1.27–1.85 |
Range of mean concentrations in rice consumed in three villages in China |
Chen et al. (1996) |
|
|
0.06–0.49 |
Range of mean concentrations in 13 varieties of baked goods and cereals available in Hungary |
Schamschula et al. (1988a) |
|
|
|
0.05–0.39 |
Range of concentrations in bread and grains sampled between 1981 and 1989 in Germany |
Bergmann (1995) |
|
Vegetables |
0.01– 0.68 |
Range of concentrations in 38 varieties of raw, cooked and canned vegetables in Canada |
Dabeka & McKenzie (1995) |
|
|
0.28–1.34 |
Range of mean concentrations in three staple vegetables consumed in three villages in China |
Chen et al. (1996) |
|
|
0.01–0.86 |
Range of mean concentrations in 24 varieties of vegetables available in Hungary |
Schamschula et al. (1988a) |
|
|
0.023 |
Mean concentration in some vegetables sampled between 1981 and 1989 in Germany |
Bergmann (1995) |
|
Fruits and fruit juices |
0.01–0.58 |
Range of concentrations in 25 varieties of fruit and fruit juices available in Canada |
Dabeka & McKenzie (1995) |
|
0.03–0.19 |
Range of mean concentrations in 16 varieties of fruits and fruit juices available in Hungary |
Schamschula et al. (1988a) |
|
|
0.02–2.8 |
Range of concentrations in 532 varieties of fruit juice and juice-flavoured beverages in the USA |
Kiritsy et al. (1996) |
|
|
0.027 |
Mean concentration in some fruits sampled between 1981 and 1989 in Germany |
Bergmann (1995) |
|
|
0.014–0.35 |
Range of concentrations in some fruit juices sampled between 1984 and 1989 in Germany |
Bergmann (1995) |
|
|
Fats and oils |
0.05–0.13 |
Range of concentrations in 3 varieties of fats and oils available in Canada |
Dabeka & McKenzie (1995) |
|
Sugars and candies |
0.01–0.28 |
Range of concentrations in 7 varieties of sugar-containing products available in Canada |
Dabeka & McKenzie (1995) |
|
|
0.01–0.31 |
Range of mean concentrations in 12 varieties of sugar-containing foods available in Hungary |
Schamschula et al. (1988a) |
|
Beverages |
0.21–0.96 |
Range of concentrations in 6 varieties of beer, wines, coffee and soft drinks available in Canada |
Dabeka & McKenzie (1995) |
|
|
0.19–0.78 |
Range of mean concentrations in 3 varieties of coffee and soft drinks available in Hungary |
Schamschula et al. (1988a) |
|
|
0.003–0.39 |
Range of concentrations in some soft drinks sampled between 1984 and 1989 in Germany |
Bergmann (1995) |
|
|
0.02–1.28 |
Range of concentrations in 332 samples of soft drinks sold in Iowa, USA, between 1995 and 1997 |
Heilman et al. (1999) |
|
Tea |
4.97 |
Concentration in tea available in Canada |
Dabeka & McKenzie (1995) |
|
|
90.94–287.9 |
Range of mean concentrations of tea consumed in 3 villages in China |
Chen et al. (1996) |
|
|
243.7 |
Mean concentration in 4 samples of tea leaves used in Hungary |
Schamschula et al. (1988a) |
|
|
82–371 |
Range of concentrations in samples of 32 tea leaves purchased in Hong Kong |
Wei et al. (1989) |
|
|
0.005–0.174 |
Range of concentrations in herbal and children’s teas sampled between 1984 and 1989 in Germany |
Bergmann (1995) |
|
0.37–2.07 |
Range of concentrations in black tea sampled between 1984 and 1989 in Germany |
Bergmann (1995) |
a
For liquid items, concentrations are in mg/litre.Tea leaves are particularly rich in fluoride (see Table 6). The concentration of fluoride in brewed tea is dependent upon the concentration of soluble fluoride in the tea leaves, the level of fluoride in the water used in its preparation and the length of the brewing period (Smid & Kruger, 1985). Chan & Koh (1996) reported that the mean concentration of fluoride in 21 brands of caffeinated tea, 11 brands of decaffeinated tea and 12 brands of herbal tea purchased in the USA and brewed (with water containing <0.02 mg fluoride/litre) for 5–120 min was approximately 1.5–2.4, 3.2–4.2 and 0.05–0.1 mg/litre, respectively. The levels of fluoride in 40 brands of brewed and instant coffee purchased in the USA ranged from 0.1 to 0.58 mg/litre, with no significant difference between caffeinated and decaffeinated coffees (Warren et al., 1996).
The concentration of fluoride in food products is usually not significantly increased by the addition of superphosphate fertilizers to agricultural soil (Oelschläger, 1971), due to the generally low transfer coefficient from soil to plant material. However, a recent study by McLaughlin et al. (2001) found significant increases in fluoride concentrations in herbage harvested from plots fertilized with phosphate fertilizers (537 kg total phosphorus added over 59 years) over a long period (22 mg fluoride/kg) compared with concentrations in herbage harvested from unfertilized plots (11 mg fluoride/kg). These data suggest that, given the right soil conditions and application of sufficient fluoride as an impurity in phosphate fertilizers to soils, plant uptake of fluoride can be increased. The use of water containing relatively low (<3.1 mg/litre) levels of fluoride for crop irrigation generally does not increase foodstuff fluoride concentrations (Schamschula et al., 1988a). However, this is dependent on plant species and fluoride concentrations in soil and water. For example, Kabasakalis & Tsolaki (1994) showed that fluoride concentrations in vegetables irrigated with water containing 10 mg fluoride/litre were increased compared with fluoride concentrations in vegetables grown with irrigation water containing low fluoride concentrations (0.15 mg/litre). They also commented that fluoride has a tendency to accumulate in the vegetable leaves rather than in the fruits.
Levels of fluoride in foods are significantly affected by the fluoride content of the water used in preparation or processing, most notably in beverages and dry foodstuffs to which water is added prior to consumption (Kumpulainen & Koivistoinen, 1977; Schamschula et al., 1988a). In a study conducted in a rural area of China, immersion of vegetables in hot spring water containing elevated levels of fluoride (approximately 20.3 mg/litre) reportedly led to a significant increase in the fluoride content of the food product (Xu et al., 1995). The concentration of fluoride in water used to parboil rice influenced the level in the final product (Anasuya & Paranjape, 1996). The concentrations of fluoride in unwashed or unprocessed foods grown in the vicinity of industrial sources (emissions) of fluoride in Japan (Sakurai et al., 1983; Tsunoda & Tsunoda, 1986; Muramoto et al., 1991) and the United Kingdom (Jones et al., 1971) have been up to 100-fold greater than the levels in the same foods grown in other non-industrially exposed areas.
In commercially available infant formulas sold in the USA, soy-based ready-to-use and liquid concentrate formulas contained higher levels of fluoride than the equivalent milk-based products; however, no significant difference was observed between soy- and milk-based powdered infant formulas (McKnight-Hanes et al., 1988) (Table 7). The mean concentrations of fluoride in liquid milk- and soy-based concentrated formulas diluted with water containing 1 mg fluoride/litre were 0.62 and 0.74 mg/litre, respectively. The mean concentrations in powdered milk- and soy-based formulas diluted with water containing 1 mg fluoride/litre were 0.83 and 0.85 mg/litre, respectively (McKnight-Hanes et al., 1988). For infant formulas available in Australia, the average concentrations of fluoride in powdered milk- and soy-based formulas diluted with water containing 1 mg fluoride/litre were 1.2 and 1.34 mg/litre, respectively (Silva & Reynolds, 1996). Studies of fluoride levels in infant foods available in Germany revealed mean concentrations generally of 0.02–0.3 mg/litre (Bergmann, 1995). In other studies (see Table 7), levels of fluoride in infant formulas have ranged from 100 to 1600 µg/litre (Fomon & Ekstrand, 1993). A study of a variety of infant food products available in the USA indicated concentrations of fluoride ranging from 0.01 to 8.38 µg/g; the highest concentrations were in products containing chicken (Heilman et al., 1997). In other infant food and beverage products (see Table 7), fluoride levels ranged from 10 to 6000 µg/litre (Fomon & Ekstrand, 1993). Reported levels of fluoride in breast milk have ranged from <2 to 100 µg/litre, with most values being between 5 and 10 µg/litre (Ekstrand, 1981; Esala et al., 1982; Dabeka et al., 1986; Bergmann, 1995).
Table 7. Concentrations of fluoride in infant foodsa
|
Food item |
Fluoride concentration (µg/litre)b |
Reference |
|
Human milk |
5–10 |
Esala et al. (1982); Spak et al. (1983); Ekstrand et al. (1984) |
|
Cow’s milk |
30–60 |
J. Ekstrand (unpublished data) |
|
Formula |
|
Johnson & Bawden (1987); McKnight-Hanes et al. (1988) |
|
Ready to feed |
100–300 |
|
|
Concentrated liquid |
|
|
|
Milk-based |
100–300 |
|
|
Isolated soybean-based |
100–400 |
|
|
Powdered |
|
|
|
Milk-based |
400–1000 |
|
|
Isolated soybean-based |
1000–1600 |
|
|
Most products other than dry cereals |
100–300 |
Singer & Ophaug (1979); Dabeka et al. (1982) |
|
Fruit juices |
|
Singer & Ophaug (1979); Dabeka et al. (1982) |
|
Produced with non-fluoridated water |
10–200 |
|
|
Produced with fluoridated water |
100–1700 |
|
|
Dry cereals |
|
Singer & Ophaug (1979); Dabeka et al. (1982) |
|
Produced with non-fluoridated water |
90–200 |
|
|
Produced with fluoridated water |
4000–6000 |
|
|
Wet-pack cereal fruit products |
2000–3000 |
Singer & Ophaug (1979); Dabeka et al. (1982) |
|
Poultry-containing products |
100–5000 |
Singer & Ophaug (1979); Dabeka et al. (1982) |
|
a |
From Fomon & Ekstrand (1993); Fomon et al. (2000). |
|
b |
Concentration ranges have been rounded off. Most reported values fall within the ranges listed in the table. |
Available data on the concentrations of fluoride in indoor air are limited. In the Netherlands, concentrations of gaseous fluoride ranged from <2 to 49 µg/m3 in the indoor air of five homes constructed with wood treated with a preservative containing 56% fluoride (Sloof et al., 1989). Gu et al. (1990) reported a mean concentration of fluoride of approximately 60 µg/m3 for samples of indoor air collected from homes in China where coal containing high amounts of fluoride was burned indoors. In other studies conducted in China, the levels of fluoride in the indoor air of homes where fluoride-rich coal was burned have ranged from 2.1 to 46.1 µg/m3 (Liu, 1995) and from 15 to 155 µg/m3 (Zhang & Cao, 1996). (See section 5.1.2 for concentrations of fluoride in outdoor air.)
Dentifrice products for adults that are commercially available in many countries generally contain fluoride at concentrations ranging from 1000 to 1500 µg/g (Whitford, 1987; Sloof et al., 1989; Newbrun, 1992). Some dentifrices designed for use by children contain lower levels of fluoride, ranging from 250 to 500 µg/g (Newbrun, 1992). Dental products such as toothpaste, mouthwash and fluoride supplements have been identified as significant sources of fluoride (Ekstrand, 1987; Drummond et al., 1990). Topical mouth rinses marketed for daily home use may contain between 230 and 500 mg fluoride/litre, while mouthwash products intended for weekly or biweekly use may contain 900–1000 mg fluoride/litre (Sloof et al., 1989; Grad, 1990; Whitford, 1996).
Published estimates for the intake of fluoride have been summarized in Table 8. Although individual exposure to fluoride is likely to be highly variable, the inhalation of airborne fluoride in most cases makes a minor contribution to the total intake of this element. Infants may receive small quantities of fluoride from breast milk. For adults, the consumption of foodstuffs and drinking-water is the principal route for the intake of fluoride. The ingestion of dentifrice by young children makes a significant contribution to their total intake of fluoride. Studies have indicated that children between 2 and 5 years of age may ingest between 0.11 and 0.39 g of dentifrice per brushing (Bruun & Thylstrup, 1988; Simard et al., 1989; Naccache et al., 1990, 1992). In general, estimated intakes of fluoride in children and adolescents do not exceed approximately 2 mg/day.
Table 8. Estimated intakes of fluoride
|
Sources of fluoride exposure |
Age group |
Estimated fluoride intake, mg/day (mg/kg body weight per day)a |
Comment |
References |
|
Foodstuffs |
Adults |
0.6 |
Intakes based upon levels of fluoride and consumption of major foodstuffs |
Varo & Koivistoinen (1980) |
|
Foodstuffs and drinking-water in four regions of the USA |
infants (6 months): |
|
Intakes based on fluoride levels in market basket survey of foods and drinking-water and estimated consumption |
Ophaug et al. (1985) |
|
Foodstuffs (including infant formulas) and beverages, fluoridated or non-fluoridated drinking-water in North America |
children (up to 6 years of age) |
0.05–1.23 (0.01–0.16) |
Summary of 8 studies published between 1943 and 1988 on the estimated intakes of fluoride from food and beverages by North American children |
Levy (1994) |
|
Ambient air, foodstuffs (including infant formula or breast-fed), fluoridated or non-fluoridated drinking-water, soil, dentifrice in Canada |
infants (up to 6 months of age) |
<0.01–0.65 (<0.001–0.09) |
Estimated total intakes by multimedia exposure analysis based upon ranges of mean concentrations of fluoride in ambient air, fluoridated or non-fluoridated drinking-water and soil; levels of fluoride in survey of 109 foodstuffs in Canada, breast milk, infant formula and average level of fluoride in dentifrice available in Canada, as well as assigned reference values for body weight, inhalation of air, and consumption of water, soil and foodstuffs, by various age groups of the population of Canada |
Government of Canada (1993) |
|
(Infant formula or breast-fed), cereal, juices, fluoridated or non-fluoridated drinking-water, dentifrice, fluoride supplements in the USA |
infants (6 months of age) |
0.4–1.4 (0.05–0.19) |
Estimated intakes based upon concentrations of fluoride in breast milk or various infant formulas reconstituted with fluoridated or non-fluoridated drinking-water, levels in juices and cereals, as well as estimated intakes from dentifrice and fluoride supplements by children in the USA |
Levy et al. (1995) |
|
Various infant formulas reconstituted with fluoridated or non-fluoridated drinking-water in Australia |
infants (6 months of age) |
0.13–1.35 (0.02–0.17) |
Estimated intakes based upon levels of fluoride in various infant formulas available in Australia reconstituted with either fluoridated or non-fluoridated drinking-water |
Silva & Reynolds (1996) |
|
Ambient air, drinking-water and limited variety of foodstuffs in China |
adolescents (7–15 years) |
1.16–4.57 |
Estimated intakes based upon levels of fluoride in ambient air, local supplies of drinking-water and levels in a limited variety of locally grown foodstuffs in an area of China with known elevated levels of fluoride in local water supplies |
Liu (1995) |
|
Ambient air, drinking-water and limited variety of foodstuffs in China |
adolescents (8–15 years) |
1.51–10.6 |
Estimated intakes based upon levels of fluoride in ambient air, indoor air, local supplies of drinking-water and levels of fluoride in a limited variety of locally grown foodstuffs in four areas of China where fluoride-containing coal is burned for heating and cooking |
Liu (1995) |
|
Principal foods consumed by Tibetan and Han peoples residing in Sichuan province in China (levels of fluoride in drinking-water were low [0.1 mg/litre]) |
Tibetan |
|
Increased intake of fluoride by Tibetans based upon their consumption of a local type of prepared barley and brick tea; foodstuffs not consumed by Han residing in this area; prevalence of dental and skeletal fluorosis greater among Tibetans than among Han |
Cao et al. (1996) |
|
Ambient air, beverages, food and drinking-water in Hungary |
children with a mean age of 3.9 years |
0.22–1.11 |
Estimated intakes based upon levels in available foodstuffs, beverages, air and drinking-water containing levels of fluoride ranging from 0.06 to 3.1 mg/litre |
Schamschula et al. (1988b) |
|
Drinking-water and food in India |
children (age not specified) |
1.5–20 |
Estimated range of mean intakes based upon levels in foodstuffs and local supplies of drinking-water that ranged in concentration from 0.32 to 9.6 mg/litre |
Karthikeyan et al. (1996) |
|
Drinking-water and food in normal or fluorotic villages in India |
adults (age not specified) |
0.84–4.69 (normal) |
Range of intakes based upon consumed foodstuffs and local supplies of drinking-water from rural areas in India considered either normal or fluorotic, based upon the absence or occurrence of endemic skeletal fluorosis in these areas, respectively |
Anasuya et al. (1996) |
|
Diet, beverages and toothpaste in New Zealand |
children (3–4 years of age) |
0.17–1.31 (0.01–0.07) |
Range of intakes based on duplicate-diet survey of foodstuffs and beverages (non-fluoridated or fluoridated) consumed as well as calculated intake from toothpaste, in a study of 66 children |
Guha-Chowdhury et al. (1996) |
|
Commercially available foods and drinking-water in Germany |
infants 1–12 months of age |
0.099–0.205 |
Estimated intake based upon consumption of commercially available food and drinking-water containing 0.13 mg fluoride/litre |
Bergmann (1995); Bergmann & Bergmann (1995) |
|
Breast milk and homemade food in Germany |
infants 1–12 months of age |
0.002–0.075 (0.0005–0.007) |
Estimated intakes by infants receiving breast milk as well as homemade foods |
|
|
Food, beverages and drinking-water in Germany |
children 1–15 years of age |
0.112–0.264 |
Estimated intake based upon consumed foodstuffs, beverages and drinking-water |
Bergmann (1995); Bergmann & Bergmann (1995) |
|
Foods, beverages and drinking-water in Germany |
adolescents (15–18 years of age) |
0.523 (males) (0.008) |
Estimated intake based upon consumed foodstuffs, beverages and drinking-water |
|
|
Foods, beverages and drinking-water in Germany |
Adults |
0.560 (males) (0.007) |
Estimated intake based upon consumed foodstuffs, beverages and drinking-water |
|
|
16- to 40-month-old children consuming drinking-water containing 0.3 mg fluoride/litre |
0.965 (0.073) |
Estimated intake based upon consumed foodstuffs, beverages and drinking-water |
|
|
|
Foods, beverages and dentifrice in the USA |
16- to 40-month-old children consuming drinking-water containing 0.8–1.2 mg fluoride/litre |
0.965 (0.07) |
Estimated intake based upon consumed foods, beverages and dentifrice |
Rojas-Sanchez et al. (1999) |
|
a |
Data in parentheses are the estimated intakes of fluoride, expressed as mg/kg body weight per day, when presented in the reference cited. |
Although adults may have a higher absolute daily intake of fluoride in milligrams, the daily intake of fluoride by children on a milligram per kilogram body weight basis may exceed that of adults. In certain areas of the world in which the concentration of fluoride in the surrounding environment (mainly groundwater) may be exceedingly high and/or diets are composed of foods rich in fluoride, estimated intakes of fluoride in adults as high as 27 mg/day have been reported (Liu, 1995; Anasuya et al., 1996; Cao et al., 1996; Karthikeyan et al., 1996). In certain areas of China in which coal rich in fluoride is used for heating and food preparation (e.g., cooking, food drying), the inhalation of indoor air and consumption of foodstuffs containing increased levels of fluoride also contribute to elevated intakes (Zhang & Cao, 1996; Liang et al., 1997).
The information presented in this section focuses on data that have become available since the last EHC document on fluoride was published (WHO, 1984). Occupational exposure to fluoride via inhalation or dermal contact likely occurs for individuals involved in the operation of welding equipment or in the processing of aluminium, iron ore or phosphate ore (Sloof et al., 1989). In the Netherlands, concentrations of fluoride in the workroom air of seven machine shops and shipyards where welding operations were conducted ranged from 30 to 16 500 µg/m3; levels were highest in areas where welding activities were conducted in small enclosed spaces (Sloof et al., 1989). The mean concentration of fluoride in the workroom air of an aluminium plant in Sweden was 900 µg/m3, with 34% of the fluoride present in the gaseous form (Sloof et al., 1989). The concentration of total airborne fluoride in the potroom of an aluminium smelter in British Columbia, Canada, was approximately 0.48 mg/m3 (Chan-Yeung et al., 1983a). Sr yseth et al. (1995) reported that between 1986 and 1988, the average concentration of total fluoride in the potroom of an aluminium smelter in western Norway was approximately 0.5 mg/m3. Rr nneberg (1995) indicated that, based upon monitoring studies conducted at a Norwegian aluminium smelter in 1962, time-weighted-average concentrations of airborne fluoride ranged from 1.5 to 2.7 mg/m3. The time-weighted-average concentrations of either dust-borne fluoride or hydrogen fluoride at two aluminium production facilities in the Netherlands operating since the mid-1960s were reported to be approximately 0.5 mg/m3 (Sorgdrager et al., 1995). The mean level of total airborne fluoride in the potroom of an Iranian aluminium smelter was reportedly 0.93 mg/m3 (Akbar-Khanzadeh, 1995). During 1987–1988 and 1990–1994, the mean levels of gaseous fluoride in the air of a hydrofluoric acid production facility located in Mexico were reportedly 1.78 and 0.21 mg/m3, respectively (Calderon et al., 1995). Mean levels of hydrogen fluoride up to 3 mg/m3 were reported for some areas of a phosphate fertilizer production facility in Poland (Czarnowski & Krechniak, 1990).
In humans, the dominating route of fluoride absorption is via the gastrointestinal tract. Airborne fluoride may also be inhaled.
Fluoride ions are released from readily soluble fluoride compounds, such as sodium fluoride, hydrogen fluoride, fluorosilicic acid and sodium monofluorophosphate (Na2PO3F), and almost completely absorbed (Ekstrand et al., 1978; Spak et al., 1982). Fluoride compounds with low solubility, on the other hand, including calcium fluoride, magnesium fluoride and aluminium fluoride, are poorly absorbed.
After intake of sodium fluoride as tablets or a solution, fluoride is rapidly absorbed. Only a few minutes after intake, there is a rise in the plasma fluoride concentration, and the plasma peak usually occurs within 30 min. The height of the plasma peak is proportional to the fluoride dose ingested (Ekstrand et al., 1977b).
The absorptive process occurs by passive diffusion, and fluoride is absorbed principally from both the stomach and the intestine. There is no convincing evidence that active transport processes are involved. The mechanism and the rate of gastric absorption of fluoride are related to gastric acidity.
Fluoride is mainly absorbed in the form of hydrogen fluoride, which has a pKa of 3.45. That is, when ionic fluoride enters the acidic environment of the stomach lumen, it is largely converted into hydrogen fluoride (Whitford & Pashley, 1984). Most of the fluoride that is not absorbed from the stomach will be rapidly absorbed from the small intestine.
In a bioavailability study in which young volunteers were given 4 mg fluoride as calcium fluoride (a poorly soluble fluoride compound) and plasma fluoride was followed for 6 h, no increase in the plasma fluoride concentration was detected following intake, indicating no fluoride absorption (Afseth et al., 1987).
Fluoride compounds that occur naturally or are added to drinking-water yield fluoride ions, which are almost completely absorbed from the gastrointestinal tract. Thus, fluoride in drinking-water is generally bioavailable.
There is only limited information on the bioavailability of fluoride from fluoride-containing diets. In a balance study in infants, it was found that the bioavailability of the fluoride in the infants’ diet was about 90% (Ekstrand et al., 1994).
The ingestion of fluoride with food retards its absorption and reduces its bioavailability. When fluoride was ingested as sodium fluoride tablets on a fasting stomach, the bioavailability of fluoride was almost 100%. When the same dose was taken together with a glass of milk, the bioavailability decreased to 70%; when it was taken together with a calcium-rich breakfast, the bioavailability was further reduced to 60% (Ekstrand & Ehrnebo, 1979; Trautner & Einwag, 1989; Shulman & Vallejo, 1990). The decrease in absorption associated with the ingestion of milk or food is probably due to binding of fluoride with certain food constituents, including calcium and other divalent and trivalent cations. When this occurs, the faecal excretion of fluoride will increase. The timing of fluoride ingestion in relation to a meal is critical with respect to fluoride bioavailability. When a few grams of a fluoride dentifrice are swallowed on a fasting stomach, the plasma peak is recorded within 30 min; however, when the dentifrice is swallowed 15 min after a meal, the peak does not occur until after 1 h. The fluoride from most dental products intended for oral application is almost completely absorbed when swallowed (Ekstrand, 1987).
There is partial to complete absorption of gaseous and particulate fluorides from the respiratory tract (McIvor, 1990), with the extent of absorption dependent upon solubility and particle size. Particulate fluorides deposited in the bronchioles and nasopharynx may be swallowed (via ciliary clearance and/or coughing) and reach the gastrointestinal tract, where they are absorbed; relatively insoluble particulate fluorides deposited deep within the lungs may be absorbed gradually over time.
Available information on the absorption of fluoride through the skin is limited to cases of acute dermal exposure to hydrofluoric acid. Although hydrofluoric acid appears to be rapidly absorbed following dermal exposure, in view of the extremely corrosive nature of this compound, absorption into the general circulation could also be a consequence of damage to the vascular system.
Experimental studies performed in vivo with rats (Sato et al., 1986) and in vitro with isolated segments of dog jejunum (Messer et al., 1989) have indicated that fluoride (as sodium fluoride) is rapidly absorbed from the stomach and intestinal tract in animals. The rate at which fluoride is absorbed from the stomach is inversely related to the pH of the stomach contents (Whitford, 1990; Messer & Ophaug, 1993).
Although most of the fluoride ingested by laboratory animals is absorbed through the gastrointestinal tract, small amounts may also be absorbed from the oral cavity. In female Fischer F-344 rats intubated endotracheally (with oesophageal ligation) with 200 µl of a solution of sodium fluoride (Na18F), approximately 7% of the administered material was absorbed from the oral cavity within 2.5 h (Patten et al., 1978). The absorption of fluoride (as sodium fluoride in solution) from the oral cavity of Syrian hamsters increased with decreasing pH of the solution (Whitford et al., 1982).
In laboratory animals, the presence of food (Rao, 1984) and fluoride-binding ions (i.e., aluminium, calcium, magnesium) in the gastrointestinal tract (Spencer et al., 1981) significantly reduces the amount of fluoride absorbed into the general circulation. The absorption of ingested fluoride by female Wistar rats was reduced from 76 to 47% when the level of calcium in their diet was increased from 0.5 to 2% (Harrison et al., 1984). In male albino rabbits administered drinking-water containing 25 or 250 mg fluoride/litre (as sodium fluoride) for 4 weeks, the levels of fluoride in the serum (and femoral bone) were approximately 2-fold higher in animals administered a diet low in calcium (0.4%) than in controls administered a diet containing 1.6% calcium (Tsuchida et al., 1992).
Other than the observation by Morris & Smith (1982) that gaseous hydrogen fluoride was almost completely absorbed from the upper respiratory tract following the exposure of rats to concentrations ranging from 30 to 176 mg fluoride/m3, no additional quantitative data on the absorption of fluorides following inhalation exposure were identified.
Quantitative information on the dermal absorption of fluorides in animals was not identified.
Fluoride is rapidly distributed by the systemic circulation to the intracellular and extracellular water of tissues; however, the ion normally accumulates only in calcified tissues, such as bone and teeth. Fluoride is not bound to any plasma proteins (Taves, 1968; Ekstrand et al., 1977a). In the blood, the ion is asymmetrically distributed between plasma and the blood cells, so that the plasma concentration is approximately twice as high as that associated with the cells (Whitford, 1996).
There is no homeostatic regulation of the fluoride concentration in the blood. In a short-term human pharmacokinetic study, fluoride doses ranging from 3 to 10 mg were given orally in the form of tablets. The data were interpreted using an open three-compartment model, and the plasma half-life was determined from the final terminal phase, ranging from 3 to 10 h. The plasma level rose and fell during each dosage interval. That is, the fluctuation of the plasma fluoride concentration depended on the fluoride dose ingested, dose frequency and the plasma half-life of fluoride (Ekstrand et al., 1977b).
The literature contains a wide range (0.4–2.4 µmol/litre) for "normal" plasma fluoride concentrations. Some of the results may have been due to the use of fasting individuals in some studies and non-fasting in others. It is virtually certain also that problems with the analysis of fluoride have been contributory. This is especially the case when only the fluoride electrode is used for the analysis of samples with low concentrations. The use of preparative methods that concentrate the fluoride in small volumes, such as the acid-HMDS microdiffusion method, is necessary when such low levels of fluoride are to be measured.
Under steady conditions of exposure via drinking-water, the concentration of fluoride in plasma collected from fasting subjects is directly related to the concentration in the drinking-water consumed (Guy et al., 1976; Ekstrand, 1978). The mean concentration of fluoride in the blood plasma of 30 residents of communities in the USA served by drinking-water containing low concentrations of fluoride (i.e., <0.1 mg/litre) was 0.4 µmol/litre, while the mean concentration in plasma from individuals consuming drinking-water containing higher amounts of fluoride (i.e., 0.9–1.0 mg/litre) was reportedly 1 µmol/litre (Guy et al., 1976). Serum and plasma contain virtually the same amount of fluoride (Guy et al., 1976; Kono et al., 1986). There is a considerable variation during the day in plasma fluoride levels in subjects living in an area with a high fluoride concentration in the drinking-water, particularly in adults (Ekstrand, 1978).
In addition to recent fluoride intake and the level of chronic fluoride intake, plasma fluoride levels are influenced by the relative rates of bone accretion and dissolution and by the renal clearance rate of fluoride (Waterhouse et al., 1980). In the long term, there is a positive relationship between the concentration of fluoride in plasma and bone. Also, a positive relationship between plasma fluoride and age has been reported (Parkins et al., 1974).
The levels of fluoride in plasma, serum and urine have been considered useful biomarkers for fluoride exposure (Ekstrand & Ehrnebo, 1983; Ekstrand et al., 1983; Ehrnebo & Ekstrand, 1986; Whitford, 1996; WHO, 1996a; Lund et al., 1997). The concentrations of fluoride in parotid saliva have also been used to assess plasma levels of fluoride (Ekstrand, 1977; Whitford et al., 1999a). There are, however, ethical and technical limitations of the use of these fluids for large-scale monitoring of the body burden of fluoride in humans. It has been suggested that the fluoride concentration in nails and hair can be used as a marker of fluoride exposure (Czarnowski & Krechniak, 1990; Kono et al., 1990; Whitford et al., 1999b). However, there is only limited information on the reliability of these methods and the risks of contamination during exposure in field conditions. Information on appropriate preparation methods as well as analytical techniques needs to be further evaluated before these methods can be used on a large-scale basis.
Fluoride is distributed from the plasma to all tissues and organs. The rates of delivery are generally determined by the blood flow to the tissues in question. Consequently, steady-state fluoride concentrations are achieved more rapidly between plasma and well perfused tissues, such as the heart, lungs and liver, than between plasma and less well perfused tissues, such as resting skeletal muscle, skin and adipose tissue. In general, steady-state tissue-to-plasma fluoride concentration ratios fall between 0.4 and 0.9, regardless of the rates at which the steady-state levels are achieved (Whitford et al., 1979). Exceptions to this range include the kidney, pineal gland, brain and adipose tissue. Fluoride is concentrated to high levels within the kidney tubules, so this organ has a higher concentration than plasma.
Most information on the levels of fluoride in soft tissues of humans is derived from a report published by Taves et al. (1983), in which the amounts in a number of tissues (from autopsies of 13 cases of sudden death in the USA) were quantified (see Table 9 in section 6.2.5 below).
The rate of clearance of fluoride from plasma by bone is higher than that of calcium. In humans and laboratory animals, approximately 99% of the total body burden of fluoride is retained in bones and teeth (Kaminsky et al., 1990; Hamilton, 1992), with the remainder distributed in highly vascularized soft tissues and the blood (McIvor, 1990). The degree to which fluoride is stored in the skeletal tissue is related to the turnover rate of skeletal components and the level of previous exposure (Caraccio et al., 1983). Levels of fluoride in calcified tissues are generally highest in bone, dentine and enamel. The concentration of fluoride in bone varies with age, sex and the type and specific part of bone and is believed to reflect an individual’s long-term exposure to fluoride.
During the growth phase of the skeleton, a relatively high portion of an ingested fluoride dose will be deposited in the skeleton. In infants and children with skeletal growth or individuals not consuming fluoridated drinking-water, up to 75% of the daily amount of fluoride that is absorbed may be incorporated into skeletal tissue (US DHHS, 1991). When a fluoride dose (e.g., a fluoride tablet or an infant formula diluted with fluoridated drinking-water) is given to infants, the retention will be strongly correlated with the absorbed fluoride dose per kilogram body weight: the higher the fluoride dose, the higher the fluoride retention. Retention of fluoride following intake of a fluoride supplement of 0.25 mg given to infants was shown to be as high as 80–90%. In a study with adults (aged 23–27 years) in which fluoride was given as a single intravenous injection, about 60% of the injected dose (3 mg fluoride as sodium fluoride) was retained (Ekstrand et al., 1978).
The selective affinity of fluoride for mineralized tissues is, in the short term, due to uptake on the surface of bone crystallites by the processes of isoionic and heteroionic exchange. In the long run, fluoride is incorporated into the crystal lattice structure of teeth and skeletal tissue by replacing some hydroxyl ions within the unit cells of hydroxyapatite, producing partially fluoridated hydroxyapatite (i.e., fluorapatite or fluorhydroxyapatite) (WHO, 1994).
Fluoride is not irreversibly bound to bone. This has been demonstrated in persons who had lived in an area with a high fluoride concentration in the drinking-water and then moved to an area with a low water fluoride level. The urinary fluoride concentration in these individuals fell slowly for long periods, indicating that fluoride was being mobilized continuously from the skeleton and subsequently excreted (Hodge et al., 1970). Similar findings have been noted among workers with chronic occupational exposure to fluoride who were subsequently employed elsewhere (Boillat et al., 1979). Their plasma fluoride levels reflected their bone fluoride concentrations (based upon iliac crest biopsies) for long periods (2–3 years) after the industrial fluoride exposure had stopped.
The "balance" of fluoride in the body (i.e., the difference between the amount of fluoride ingested and the amount of fluoride excreted in the urine and the faeces) can be positive or negative. When the fluoride is derived from human milk or cow’s milk, biological fluids with a low fluoride content, urinary excretion generally exceeds intake (i.e., there is a negative fluoride balance). In infants, when fluoride intakes are low, sufficient fluoride is released from bone to extracellular fluid to result in urinary excretion being higher than intake (Ekstrand et al., 1994).
Hence, the plasma concentrations and the urinary excretions mirror a physiological balance that is determined by earlier fluoride exposure, the degree of accumulation of the ion in bone, the mobilization rate from bone and the efficiency of the kidneys in excreting fluoride.
Human studies have shown that the placenta is not in any sense a barrier to the passage of fluoride to the fetus. There is a direct relationship between the serum fluoride concentration of the mother and that of the fetus; the cord serum concentration is 75% that of the maternal fluoride concentration. From the fetal blood, fluoride is readily taken up by the calcifying fetal bones and teeth (Shen & Taves, 1974).
Information on the levels of fluoride in the tissues and organs of individuals is summarized in Table 9. The concentration of fluoride in the plasma of healthy individuals may range from 7.6 to 28.5 µg/litre (Guy, 1979). The mean plasma level in 127 subjects with 5.03 mg fluoride/litre in drinking-water was 106 ± 76 (SD) µg/litre (Li et al., 1995). A study of 250 healthy individuals residing in Spain revealed levels of fluoride in serum ranging from 1 to 47 µg/litre (Torra et al., 1998).
Table 9. Concentration of fluoride in human tissues and organs
|
Tissue/organ |
Number of samplesa |
Concentrationb (mean [range]) |
Reference |
|
Whole blood |
nac |
na (20–60) µg/litre |
Kissa (1987) |
|
Serum |
1088 |
14.4 (na) µg/litre |
Kono et al. (1986) |
|
Serum |
250 |
17.4 (1–47) µg/litre |
Torra et al. (1998) |
|
Plasma |
72 |
14.3 (2.7–79.8) µg/litre |
Guy et al. (1976) |
|
Urine |
na |
780 (na) µg/litre |
Kono et al. (1986) |
|
Saliva |
98 |
8.74 (na) ng/g |
Schamschula et al. (1985) |
|
Tooth enameld |
na |
na (740 000–2 100 000) ng/g |
Berndt & Stearns (1979) |
|
Iliac creste |
78 |
720 500 (106 000–2 360 000) ng/g |
Alhava et al. (1980) |
|
Iliac crestf |
10 |
1 500 000 (na) ng/g |
Zipkin et al. (1960) |
|
Ribf |
9 |
1 600 000 (na) ng/g |
Zipkin et al. (1960) |
|
Vertebraef |
10 |
2 200 000 (na) ng/g |
Zipkin et al. (1960) |
|
Hair |
53 |
2505 (na) ng/g |
Tagaki et al. (1986) |
|
Braing |
7 |
31.2 (na) ng/g |
Taves et al. (1983) |
|
Body fatg |
5 |
28.5 (na) ng/g |
Taves et al. (1983) |
|
Liverg |
5 |
19.6 (na) ng/g |
Taves et al. (1983) |
|
Muscleg |
9 |
40.3 (na) ng/g |
Taves et al. (1983) |
|
Lungg |
9 |
71.4 (na) ng/g |
Taves et al. (1983) |
|
Thymusg |
4 |
9005.2 (na) ng/g |
Taves et al. (1983) |
|
Aortag |
8 |
4331.6 (na) ng/g |
Taves et al. (1983) |
|
Kidneyg |
9 |
159.2 (na) ng/g |
Taves et al. (1983) |
|
Fingernails |
22 |
8800 (na) ng/g |
Czarnowski & Krechniak (1990) |
|
Fingernails |
6–9 |
na (1590–7570) ng/g |
Whitford et al. (1999b) |
|
a |
Total number of tissue samples analysed. |
|
b |
Mean and range of concentrations of fluoride in the tissues and organs of individuals. |
|
c |
na = not available. |
|
d |
Samples of surface tooth enamel from adults 20 years of age. Samples were taken from individuals consuming drinking-water containing up to 1 mg fluoride/litre. |
|
e |
Samples of iliac crest bone from adults (mean age 60 years). Samples were taken from individuals consuming drinking-water containing up to 0.97 mg fluoride/litre over a period of 14–18 years. |
|
f |
Samples of bone from adults 26–90 years of age. Samples were taken from individuals consuming drinking-water containing up to 1 mg fluoride/litre over a period of 10–87 years. |
|
g |
Tissues (analysed on a wet weight basis) were obtained from autopsies of cases of sudden death in the USA (mean age 29 years). Some tissue samples may have been prepared with water containing 0.9–1 mg fluoride/litre. |
Levels of fluoride in calcified tissues are generally highest in bone, dentine and enamel, although they are highly variable, owing, at least in part, to the methods of analysis used (Weidmann & Weatherell, 1970).
The concentration of fluoride in dental enamel decreases exponentially with the distance from the surface and varies with site, age, surface attrition, systemic exposure and exposure to topically applied fluoride (Weatherell et al., 1977; Schamschula et al., 1982). The average concentration of fluoride in dentine is approximately 2- to 3-fold higher than that in enamel and, like enamel, is higher in surface regions, increases with age and is related to the levels of fluoride in the drinking-water consumed (Weidmann & Weatherell, 1970; US NAS, 1971).
Whitford et al. (1991) examined a number of pharmacokinetic parameters (i.e., plasma, renal and extrarenal clearances) in mongrel dogs, Sprague-Dawley rats, cats, rabbits and hamsters intravenously administered a single dose (0.5 mg fluoride/kg body weight) of sodium fluoride and concluded that dogs most resembled humans with respect to their elimination of fluoride; plasma clearance rates were approximately 2-fold higher in rats than in dogs.
The major route for the removal of fluoride from the body is by the kidneys.
The renal clearance of fluoride in the adult typically ranges from 30 to 50 ml/min, whereas clearance rates of the other halogens (chloride, iodide and bromide) are usually less than 1.0 ml/min. The percentage of the filtered fluoride reabsorbed from the renal tubules can range from about 10 to 90%. The degree of reabsorption depends largely on the pH of the tubular fluid, urinary flow and renal function (Ekstrand et al., 1980, 1982; Whitford, 1996). The excretion of fluoride in urine is reduced in individuals with impaired renal function (Schiffl & Binswanger, 1980; Spak et al., 1985; Kono et al., 1986).
The literature contains a wide range (0.1–5 µmol/litre) for fluoride levels in breast milk. It is probable that problems with the analysis of fluoride have been contributory. The concentration of fluoride in colostrum and mature breast milk is reported to be the same — about 0.4 µmol/litre (Spak et al., 1983). The same investigators found no significant difference in fluoride concentrations of milk from mothers living in areas with fluoride concentrations in drinking-water of 1 or 0.2 mg/litre, even though their plasma concentrations reflected this difference. Further, they found no diurnal variation in the fluoride concentration (Spak et al., 1983). Studies of lactating mothers have shown that there is a limited transfer of fluoride from plasma to breast milk (Ekstrand et al., 1981). Dabeka et al. (1986), on the other hand, reported that the concentration of fluoride in breast milk was related to the fluoride content of the drinking-water consumed by the women; the mean concentration of fluoride in breast milk obtained from 32 women consuming drinking-water containing <0.16 mg fluoride/litre was 0.23 µmol/litre, whereas breast milk obtained from 112 women consuming drinking-water containing 1 mg fluoride/litre reportedly contained 0.48 µmol fluoride/litre. Concentrations of fluoride in samples of breast milk collected in Finland (Esala et al., 1982) ranged from 0.1 to 2.7 µmol/litre. In the Finnish study, the fluoride in the milk (both ionic and total fluoride) was measured after ashing the samples. Taves (1983) showed no difference between inorganic (microdiffusion) and total (ashing) fluoride in cow’s milk. Obviously, there is some uncer-tainty as to whether there really is a non-ionic fraction of fluoride in milk or whether the discrepancy is due to an analytical overestimation.
The fluoride content of human breast milk represents the natural daily fluoride intake during the first 6 months of life. This is especially important when comparing the daily fluoride intake by formula-fed and breast-fed infants.
It is generally accepted that most of the fluoride in the faeces is not absorbed. Faecal fluoride usually accounts for less than 10% of the amount ingested each day (Ekstrand et al., 1984).
The quantitative role of sweating in fluoride balance studies has not been explored using modern analytical methods in controlled studies. Based on some preliminary data from Henschler et al. (1975) on predicted losses in workers whose urinary fluoride levels are about 5 mg/litre and who lose 6 litres of sweat containing 0.2–0.3 mg fluoride during a shift, the sweat could account for approximately 5% of the fluoride excreted. In tropical climates, during periods of prolonged and heavy exercise or in occupational exposure to elevated temperatures, the fluid loss from the body via sweat may approach as much as 4–6 litres a day.
The concentration of fluoride in saliva is about two-thirds of the plasma fluoride concentration and seems to be independent of flow rate, in contrast to the situation for most electrolytes (Ekstrand, 1977; Oliveby et al., 1989; Whitford et al., 1999a). A diurnal fluctuation in the saliva fluoride concentration is also seen in an area with fluoridated drinking-water containing 1 mg fluoride/litre (Oliveby et al., 1990). The levels of fluoride in saliva are important, because this potentially provides further topical exposure of the teeth to fluoride.
LD50s for the oral administration of sodium fluoride, sodium monofluorophosphate and stannous fluoride (SnF2) to rats include values of 31–101 mg fluoride/kg body weight, 75–102 mg fluoride/kg body weight and 45.7 mg fluoride/kg body weight, respectively (IARC, 1982; Whitford, 1987, 1990; ATSDR, 1993). LD50s for the oral administration of sodium fluoride, sodium monofluorophosphate and stannous fluoride to mice include values of 44.3 and 58 mg fluoride/kg body weight, 54 and 94 mg fluoride/kg body weight and 25.5 and 31.2 mg fluoride/kg body weight, respectively (IARC, 1982; Whitford, 1990).
The acute oral exposure of laboratory animals to fluoride produces salivation, lacrimation, vomiting and diarrhoea, as well as respiratory arrest and cardiac depression (WHO, 1984).
Nephrotoxic effects produced in laboratory animals following acute oral exposure to fluoride have been reviewed (WHO, 1984). Subsequent studies have indicated that the severity of the acute renal toxicity of sodium fluoride in rats appears to be related to the age of the animal. In a study in which single doses of sodium fluoride (13.6 or 21.8 mg fluoride/kg body weight) were injected intraperitoneally into 1-, 8-, 15- or 29-day-old male and female Sprague-Dawley rats, proximal tubular necrosis and marked changes in kidney weight, urine osmolarity and pH, and chloride excretion were observed in the 29-day-old animals; effects in the younger animals were considered mild and less severe (Daston et al., 1985).
In laboratory animals, adverse effects within the gastrointestinal tract have been reported following the ingestion of acutely toxic doses of fluoride. In a study in which solutions of 1, 10 and 50 mmol fluoride/litre (as sodium fluoride dissolved in 0.1 N HCl) were introduced into the stomachs of anaesthetized female Wistar rats, histopathological effects on the gastric mucosa (i.e., desquamation of surface epithelial cells, cell loss and damage) were observed in animals administered 10 and 50 mmol/litre within 30 min of exposure (Easmann et al., 1984). Subsequently, Easmann et al. (1985) reported that following intubation of Holtzman rats with 1.5 ml of 100 mmol sodium fluoride/litre (approximately 17.8 mg fluoride/kg body weight), histopathological effects (i.e., haemorrhage, disruption of epithelial integrity and glandular structure, lysis and loss of epithelial cells) within the gastric mucosa were observed. After 48 h, there was recovery of gastric mucosal integrity.
Hydrogen fluoride is acutely toxic when administered via inhalation. LC50s ranging from 4065 to 14 400 mg fluoride/m3 have been reported for the 5-min exposure of rats to hydrogen fluoride (WHO, 1984; ATSDR, 1993). An LC50 of approximately 1084 mg fluoride/m3 has been reported for the 60-min exposure of rats to hydrogen fluoride (Rosenholtz et al., 1963). LC50s of approximately 5000 and 280 mg fluoride/m3 have been reported for the 5- or 60-min exposure of mice to hydrogen fluoride, respectively (WHO, 1984; ATSDR, 1993).
The inhalation exposure of laboratory animals to levels of hydrogen fluoride near the LC50 produces severe ocular and nasal irritation, pulmonary congestion, oedema, respiratory distress and erythema of the exposed skin (ATSDR, 1993).
Direct exposure of the skin to hydrofluoric acid produced severe burns and tissue damage, with the severity of these effects dependent upon the length of the exposure and concentration (Derelanko et al., 1985). The direct application of sodium fluoride (0.5 and 1.0% in distilled water) to the abraded skin of rats produced effects ranging from superficial necrosis to oedema and inflammation (Essman et al., 1981).
Application of an aqueous solution of 2% sodium fluoride to the eyes of rabbits caused corneal epithelial defects and necrosis in the conjunctiva (Grant & Schuman, 1993).
In short-term studies, survival was reduced in male and female F344/N rats and in male, but not female, B6C3F1 mice administered drinking-water containing 363.2 mg fluoride/litre for a period of 14 days (NTP, 1990). In other investigations, fluoride-induced effects on the skeleton included inhibition of trabecular bone mineralization in female Wistar rats administered drinking-water containing 113.5 or 136.2 mg fluoride/litre for a period of 5 weeks (Harrison et al., 1984); inhibition of endosteal bone formation and a reduction in cancellous bone volume in male Holtzman rats administered drinking-water containing 85.5 mg fluoride/litre for a period of 21 days (Turner et al., 1989); delayed fracture healing and a reduction in collagen synthesis in male albino rats receiving 14 mg fluoride/kg body weight per day for 30 days (Uslu, 1983); an increase in dermatan sulfate and chondroitin-6-sulfate in the tibia of male Sprague-Dawley rats receiving 17.5 mg fluoride/kg body weight per day for a period of 1–2 months (Prince & Navia, 1983); and a 20% increase in bone matrix formation in male C57BL/6 mice receiving 0.8 mg fluoride/kg body weight per day for 4 weeks (Marie & Mott, 1986). Ultrastructural changes were observed in skeletal muscle of male Wistar rats administered drinking-water containing 100 mg fluoride/litre (as sodium fluoride) for 8 weeks (Pang et al., 1996).
Although male Swiss mice administered (orally) 5.2 mg fluoride/kg body weight per day over a period of 35 days reportedly had reduced red blood cell counts and numbers of lymphocytes (39%) and increased numbers of monocytes, eosinophils and basophils compared with controls (Pillai et al., 1988), the levels of red blood cells, lymphocytes, neutrophils, monocytes and eosinophils in male B6C3F1 mice receiving 8.1 mg fluoride/kg body weight per day (from drinking-water for 24 weeks) were similar to those in controls receiving 0.6 mg fluoride/kg body weight per day (NTP, 1990).
In medium-term exposure studies, femoral bone bending strength was increased (approximately 38%) or decreased (approximately 20%) in adult rats administered drinking-water containing 16 mg fluoride/litre or 64–128 mg fluoride/litre, respectively, over a period of 16 weeks (Turner et al., 1992). Vertebral bone quality (compression resistance normalized for ash content) was reportedly reduced in female Wistar rats administered drinking-water containing 100 or 150 mg fluoride/litre for a period of 90 days (Sr gaard et al., 1995). Altered bone remodelling, based on thickening of the osteoid seams in the tibia and femur, was observed in male and female B6C3F1 mice administered drinking-water containing >22.7 and 45.4 mg fluoride/litre, respectively, over a period of 6 months (NTP, 1990).
Compared with unexposed controls, other effects observed in animals administered drinking-water containing added fluoride for 6 months included hyperplasia of the stomach and pathological effects (i.e., lymphocytic infiltration, hyperplasia, necrosis) in the glandular stomach of F344/N rats administered drinking-water containing 45.4 and 136 mg fluoride/litre, respectively (NTP, 1990); and reduced survival (82% and 44% in female and male B6C3F1 mice, respectively), hepatic megalocytosis, renal nephrosis, mineralization of the myocardium, necrosis and/or degeneration of the seminiferous tubules in the testis of B6C3F1 mice receiving drinking-water containing 272.4 mg fluoride/litre (NTP, 1990).
The daily intragastric administration of 22.7 mg fluoride/kg body weight per day (as sodium fluoride) to rabbits for 136 days interfered with the maturation and metabolism of collagen (Sharma, 1982a).
In early carcinogenicity bioassays conducted by Tannenbaum & Silverstone (1949), Taylor (1954) and Kanisawa & Schroeder (1969), the incidence of tumours in mice administered sodium fluoride (in either their diet or drinking-water) was, in general, not markedly greater than that observed in the unexposed controls. However, the documentation and protocols of these studies were inadequate. Deficiencies in design included small groups of animals of single sexes or various ages exposed to single dose levels for short periods of time with inadequate examination of possible target tissues.
In a comprehensive study of the carcinogenicity of sodium fluoride in laboratory animals (NTP, 1990), groups of male and female F344/N rats were administered drinking-water containing 0, 25, 100 or 175 mg sodium fluoride/litre (0, 11, 45 and 79 mg fluoride/litre) for a period of 2 years. There were 100 rats in each of the control and 175 mg/litre groups, while 70 rats per group were administered drinking-water containing 25 or 100 mg sodium fluoride/litre. Groups of 10 rats of each sex at each level of exposure were sacrificed after 27 and 66 weeks. In the high-dose group, 42 males and 54 females survived until terminal sacrifice. The estimated total intakes of fluoride from food and drinking-water by the male and female F344/N rats administered drinking-water containing 0 (control), 25, 100 or 175 mg sodium fluoride/litre were approximately 0.2, 0.8, 2.5 and 4.1 mg/kg body weight per day and 0.2, 0.8, 2.7 and 4.5 mg/kg body weight per day, respectively. At the end of the 2-year study, the levels of fluoride in the humeral bone of the control, low-, mid- and high-dose male and female rats were approximately 0.44, 0.98, 3.65 and 5.26 µg/mg bone ash and 0.55, 1.34, 3.72 and 5.55 µg/mg bone ash, respectively (NTP, 1990).
In male F344/N rats receiving 0.2, 0.8, 2.5 or 4.1 mg fluoride/kg body weight per day, the incidence of osteosarcomas (three tumours in the vertebra and one in the humerus) was 0/80, 0/51, 1/50 and 3/80, respectively (NTP, 1990). A pairwise comparison of the incidence in the high-dose group versus controls was not statistically significant (P = 0.099); if an extraskeletal osteosarcoma, located in the subcutis of the flank of one high-dose male rat, was included in the total tumour incidence in this group of animals, the pairwise comparison with the control group remained statistically insignificant (P = 0.057). However, the osteosarcomas occurred with a statistically significant (P = 0.027, by logistic regression) dose–response trend (NTP, 1990). The incidence of osteosarcoma at any site was within the range of historical controls; however, the amount of fluoride in the diets in previous National Toxicology Program (NTP) studies of other chemicals was uncontrolled and was estimated to be approximately 3.5- to 5.9-fold higher than in the NTP sodium fluoride study (NTP, 1990). In male F344/N rats receiving 0.2, 0.8, 2.5 or 4.1 mg fluoride/kg body weight per day, the incidence of oral cavity lesions (squamous papillomas or squamous cell carcinomas) was 0/80, 1/51, 2/50 and 3/80, respectively; the incidence in the fluoride-exposed groups was not significantly different from the controls and was not considered to be compound-related. The incidence of (thyroid gland) follicular cell tumours (adenomas and carcinomas) was 1/80, 1/51, 1/50 and 4/80 in the control, low-, medium- and high-dose males, respectively; the incidence in the high-dose group was not significantly different from controls and was not considered to be compound-related. There was no increase in the incidence of osteosarcomas in female F344/N rats receiving fluoride; the incidence of oral cavity neoplasms (squamous papillomas or squamous cell carcinomas) was 1/80, 1/50, 1/50 and 3/81 in female F344/N rats receiving 0.2, 0.8, 2.7 and 4.5 mg/kg body weight per day, respectively; the incidence in the high-dose group was not significantly different from the controls (NTP, 1990).
In the NTP carcinogenicity bioassay with male and female F344/N rats, no significant compound-related effects upon survival, body weight or weights of major internal organs were observed, compared with controls (NTP, 1990); however, the incidence of osteosclerosis in female rats administered drinking-water containing 175 mg sodium fluoride/litre (18/81) was significantly (P = 0.04) increased, compared with controls (6/80) (NTP, 1990). It was concluded (NTP, 1990) that the osteosarcoma finding was "equivocal evidence of carcinogenic activity" in male rats, and that there was "no evidence of carcinogenic activity" in the female rats exposed to fluoride under these conditions.
In a carcinogenicity bioassay conducted with Sprague-Dawley rats, groups of 70 animals of each sex were administered diets supplemented with various amounts of sodium fluoride for a period of 95–99 weeks (Maurer et al., 1990). The estimated total intake of fluoride by these groups of animals was 0.1, 1.8, 4.5 or 11.3 mg/kg body weight per day, respectively. After 26 weeks on study, as many as 10 animals of each sex per group were sacrificed, and after 53 weeks, 10 animals of each sex per group were sacrificed (Maurer et al., 1990). At terminal sacrifice (i.e., after 95 and 99 weeks on study for males and females, respectively), there were 26 males and 12 females remaining in the high-dose groups. At study termination, the concentration of fluoride in the bone (radius and ulna) of the males and females receiving 0.1, 1.8, 4.5 or 11.3 mg fluoride/kg body weight per day was 0.5, 5.0, 8.8 and 16.7 µg/mg bone ash and 0.5, 4.5, 8.3 and 14.4 µg/mg bone ash, respectively.
In this study, the incidence of bone tumours was 0/70, 0/58, 2/70 (one chordoma and one chondroma) and 1/70 (fibroblastic sarcoma with areas of osteoid formation) in male rats and 0/70, 2/52 (one osteosarcoma and one chondroma), 0/70 and 0/70 in female rats receiving 0.1, 1.8, 4.5 and 11.3 mg fluoride/kg body weight per day, respectively (Maurer et al., 1990). However, detailed information on the incidence of tumours in tissues or organs other than the bone and stomach were not presented, and histological examination of bone from both mid-dose groups was limited. The cranium, femur, premaxilla, maxilla, mandible, cervical vertebra, stomach, liver, kidney, incisors, adrenals, brain, heart, lungs, ovaries, uterus, pancreas, pituitary, prostate, seminal vesicles, spleen, bladder, testes, epididymides, thyroids and parathyroids obtained at the interim and terminal sacrifices from all animals receiving 0.1 or 11.3 mg fluoride/kg body weight per day were examined microscopically. The stomach, bones and teeth from animals receiving 4.5 mg fluoride/kg body weight per day and sacrificed after 26 weeks and from animals receiving 1.8 or 4.5 mg fluoride/kg body weight per day and sacrificed after 53 weeks were also examined microscopically. Not all of the bones (i.e., cranium, femur, premaxilla, maxilla, mandible, cervical vertebra) from each of the remaining animals (i.e., those alive after the interim sacrifices) receiving 1.8 or 4.5 mg fluoride/kg body weight per day were examined microscopically; however, tissues with gross lesions obtained from dead and moribund animals were evaluated.
Sprague-Dawley rats receiving 11.3 mg fluoride/kg body weight per day (administered in the diet) over a period of 95–99 weeks had reduced weight gain; animals receiving 4.5 or 11.3 mg fluoride/kg body weight per day had an increased incidence of subperiosteal hyperostosis in the skull and hyperkeratosis and acanthosis in the stomach, compared with controls receiving 0.1 mg/kg body weight per day (Maurer et al., 1990).
In the NTP (1990) carcinogenicity bioassay, male and female B6C3F1 mice were also administered drinking-water containing 0, 25, 100 or 175 mg sodium fluoride/litre (0, 11, 45 and 79 mg fluoride/litre) for 2 years. There were 100 animals in each of the control and 175 mg/litre groups, and 70 mice per group were administered drinking-water containing 25 or 100 mg sodium fluoride/litre; the total intake of fluoride from water and the diet for the males and females in these groups was estimated to be approximately 0.6, 1.7, 4.9 and 8.1 mg/kg body weight per day and 0.6, 1.9, 5.7 and 9.1 mg/kg body weight per day, respectively. At the end of the 2-year study (groups of 10 animals of each sex at each level of exposure were sacrificed after 24 and 66 weeks), the levels of fluoride in the humeral bone of the control, low-, mid- and high-dose male and female mice were approximately 0.72, 1.61, 3.58 and 5.69 µg/mg bone ash and 0.92, 1.52, 4.37 and 6.24 µg/mg bone ash, respectively (NTP, 1990).
In male B6C3F1 mice receiving 0.6, 1.7, 4.9 or 8.1 mg fluoride/kg body weight per day, the incidence of hepatoblastoma was 0/79, 1/50, 1/51 and 3/80, respectively (NTP, 1990). The overall incidence of hepatic neoplasms (adenoma, carcinoma, hepatoblastoma) was similar among all groups, and the incidence of liver tumours in all groups (control and exposed) of male mice (73–78%) was higher than observed (16–58%) in previous NTP carcinogenicity bioassays (NTP, 1990). In female B6C3F1 mice receiving 0.6, 1.9, 5.7 or 9.1 mg fluoride/kg body weight per day, the incidence of hepatoblastoma was 0/80, 1/52, 0/50 and 2/80, respectively. The overall incidence of hepatic neoplasms (adenoma, carcinoma, hepatoblastoma) was similar among all groups, and the incidence of liver tumours in all groups (control and exposed) of female mice (52–69%) was higher than observed (3–20%) in previous NTP carcinogenicity bioassays (NTP, 1990). The incidence of malignant lymphoma was 11/80, 5/52, 11/50 and 19/80 (P = 0.051), respectively. The incidence of malignant lymphoma in the control and low-dose groups was less than the lowest incidence observed in nine other investigations conducted at the study laboratory. In addition, the incidence in the high-dose group was less than the average incidence observed in historical controls (35%) and within the range of incidence observed in historical controls (10–74%) (NTP, 1990).
The administration of drinking-water containing 25, 100 or 175 mg sodium fluoride/litre to male or female B6C3F1 mice over a period of 2 years had no significant compound-related adverse effects upon survival, body weight or weights of major internal organs, compared with controls (NTP, 1990). It was concluded (NTP, 1990) that there was "no evidence of carcinogenic activity" in male and female mice exposed to fluoride under these conditions.
In a carcinogenicity bioassay in which sodium fluoride was administered in the diet to groups of 60 male and 60 female CD-1 mice over a period of 95 and 97 weeks, respectively (10 mice of each sex per group were sacrificed after 40 weeks), the incidence of osteomas in male and female controls and mice receiving 1.8, 4.5 or 11.3 mg fluoride/kg body weight per day was 1/50, 0/42, 2/44 and 13/50 and 2/50, 4/42, 2/44 and 13/50, respectively (no statistical analysis provided) (Maurer et al., 1993). The incidence of this type of tumour was increased in the high-dose groups compared with the controls. These animals were infected with a Type C retrovirus, and the role of fluoride in the etiology of these tumours is not clear (Maurer et al., 1993). Moreover, there is some controversy concerning whether they should be classified as neoplasms (US NRC, 1993). The concentration of fluoride in the bone (radius or ulna) of male controls and mice receiving 1.8, 4.5 or 11.3 mg fluoride/kg body weight per day was approximately 1.5, 4.4, 7.2 and 13.2 µg/mg bone ash, respectively; the concentration of fluoride in the (radius or ulna) bone of female controls and mice receiving 1.8, 4.5 or 11.3 mg fluoride/kg body weight per day was approximately 1.0, 3.4, 6.2 and 10.6 µg/mg bone ash, respectively.
In studies on non-neoplastic effects associated with the chronic toxicity of fluoride, bone mineralization (based on microscopic analysis) was inhibited in male and female rats administered drinking-water containing 22.7 and 36.3 mg fluoride/litre (as sodium fluoride) for a period of 250 days (Qiu et al., 1987). Femoral bone strength was reduced in male Sprague-Dawley rats administered drinking-water containing 50 mg fluoride/litre for 18 months (Turner et al., 1995). Compared with controls, the oral administration of a single dose of 4.5 mg fluoride/kg body weight per day (as sodium fluoride) to rabbits for periods ranging from 6 to 24 months produced slight alterations in ATPase (sodium and potassium) activity within erythrocytes, the activity of serum acid and alkaline phosphatase (Jain & Susheela, 1987a), the levels of dermatan sulfate, chondroitin-4-sulfate and chondroitin-6-sulfate in cancellous bone (Sharma & Susheela, 1988a), the number of haematopoietic cells in the blood (Susheela & Jain, 1983) and the disaccharide content of glycosaminoglycans isolated from cancellous bone (Sharma & Susheela, 1988b).
Evidence of mineralization of the aorta (Susheela & Kharb, 1990), morphological changes within the duodenum (Susheela & Das, 1988) and renal glomerulus (Bhatnagar & Susheela, 1998), and alterations in skin collagen metabolism (Sharma, 1982b) have also been observed in rabbits administered 4.5 mg fluoride/kg body weight per day for periods ranging from 6 to 24 months. Other effects produced following the long-term exposure of rabbits to this dose of fluoride included alterations in the proportion of erythrocytes with abnormal morphology (Susheela & Jain, 1986), the level of cortisol and corticosterone in the plasma (Das & Susheela, 1991), the level of sialic acid and glycosaminoglycans in the serum (Jha et al., 1982) and the amount of hydroxyproline present in collagen derived from tendons and cortical bone (Susheela & Sharma, 1982).
Alterations in bone remodelling (based on histomorphometric analysis) have been observed in pigs (Mosekilde et al., 1987; Kragstrup et al., 1989) administered (orally) 2 mg fluoride/kg body weight per day (as sodium fluoride) and dogs (Snow & Anderson, 1986) receiving 0.32 mg fluoride/kg body weight per day (from drinking-water containing sodium fluoride) over a period of 6 months.
The genotoxicity of fluoride has been examined in a large number of in vitro and in vivo assays, in which a wide range of end-points has been assessed. Since publication of the first EHC document on fluoride (WHO, 1984), the results from a number of studies have confirmed that, in general, fluoride is not mutagenic in prokaryotic cells (Li et al., 1987a; Tong et al., 1988; NTP, 1990; Zeiger et al., 1993); however, Ahn & Jeffery (1994) reported no significant uptake of fluoride into S. typhimurium TA98 cells.
Fluoride has been shown to increase the frequency of mutations at the thymidine kinase locus in cultured mouse lymphoma and human lymphoblastoid cells (Cole et al., 1986; Caspary et al., 1987, 1988; Crespi et al., 1990). In the studies by Cole et al. (1986) and Caspary et al. (1987), exposure to sodium fluoride produced a preferential increase in the frequency of "small mutant colonies," a phenomenon generally associated with chromosomal damage rather than point mutations (Moore et al., 1985). Indeed, the failure of sodium fluoride to increase the frequency of cells resistant to ouabain (which is produced only by a specific point mutation within the Na+, K+-ATPase gene locus) (Cole et al., 1986) is consistent with this result. In human lymphoblastoid cells, the mutagenic response at the thymidine kinase locus following a 20-day exposure to sodium fluoride was non-linear and lower than predicted from extrapolation of the 28-h exposure data, a result suggested to be indicative of a threshold for the mutagenicity of (sodium) fluoride at concentrations above 50 mg/litre (Crespi et al., 1990). Sodium fluoride did not increase the frequency of mutations at the hypoxanthine–guanine phosphoribosyl transferase locus in rat liver epithelial cells (Tong et al., 1988), Chinese hamster ovary cells (Oberly et al., 1990) or Chinese hamster lung cells exposed under neutral (Slamenova et al., 1992) or acidic (Slamenova et al., 1996) conditions.
Sodium fluoride was clastogenic in many, but not all, in vitro cytogenetic assays. The frequency of chromosomal aberrations was increased (compared with unexposed controls) following exposure of Chinese hamster lung (Don) cells (Bale & Mathew, 1987), Chinese hamster ovary cells (Aardema et al., 1989a; NTP, 1990), Syrian hamster embryo cells (Tsutsui et al., 1984a), rat vertebral body-derived cells (Mihashi & Tsutsui, 1996), rat bone marrow cells (Khalil, 1995), human leukocytes (Jachimczak & Skotarczak, 1978), human peripheral blood lymphocytes (Kishi & Tonomura, 1984; Albanese, 1987), human fibroblasts (Tsutsui et al., 1984b; Scott, 1986; Scott & Roberts, 1987; Suzuki & Tsutsui, 1989), human amnion (F1) cells (Aliev et al., 1988), human and chimpanzee lymphoid cells (Kishi & Ishida, 1993) and human oral keratinocytes (Tsutsui et al., 1991) to sodium fluoride. The chromosomal aberrations induced by sodium fluoride consisted primarily of breaks/deletions and gaps, with very few exchanges. No significant increase in chromosomal aberrations was observed in human fibroblasts, Chinese hamster ovary cells or human diploid lung cells exposed to sodium fluoride at concentrations at or below 10 mg/litre (Scott, 1986; Scott & Roberts, 1987; Aardema et al., 1989a; Oguro et al., 1995; Tsutsui et al., 1995) or in Chinese hamster lung cells at concentrations at or below 500 mg/litre (Ishidate, 1987).
Although no increase in the frequency of chromosomal aberrations was observed in human lymphocytes exposed to fluoride (Gebhart et al., 1984; Thomson et al., 1985; Gadhia & Joseph, 1997), these results are likely attributable to a number of variables related to the methodology used to assess clastogenic activity (i.e., the method of classifying chromosomal aberrations, phase of the cell cycle during which the cells were exposed and the concentration of fluoride). The pattern of induced chromosomal aberrations, the increased formation of endoreduplicated cells (Aardema et al., 1989a, 1989b), the increased delay in the cell cycle and the increased sensitivity of cells in the G2 phase to sodium fluoride are all consistent with a mechanism of clastogenicity involving an inhibition of DNA synthesis and/or repair, and it has been suggested that the effect of fluoride is upon the synthesis of proteins involved in DNA synthesis and/or repair rather than involving direct interaction between fluoride and DNA.
Although no increase in sister chromatid exchange was reported following the exposure of human peripheral blood lymphocytes (Kishi & Tonomura, 1984; Thomson et al., 1985; Tong et al., 1988; Gadhia & Joseph, 1997) and Chinese hamster ovary cells (Li et al., 1987b; Tong et al., 1988) or rat bone marrow cells (Khalil and Da’dara, 1994) to sodium fluoride, increased sister chromatid exchange was observed in Syrian hamster embryo cells (Tsutsui et al., 1984a) and Chinese hamster ovary cells (NTP, 1990) exposed to sodium fluoride. Differences in harvest times used to accommodate cell cycle delay may be, at least in part, responsible for the inconsistency in the results observed for the induction of sister chromatid exchange by fluoride.
Although the results of some studies have indicated that (sodium) fluoride increases unscheduled DNA synthesis in Syrian hamster embryo cells, human foreskin fibroblasts and human keratinocytes (Tsutsui et al., 1984a, 1984b, 1984c), Skare et al. (1986a) suggested that these results might be an artefact, possibly due to the formation of precipitable complexes of magnesium, fluoride and [3H]thymidine triphosphate.
Sodium fluoride has been reported to induce the morphological transformation of Syrian hamster embryo cells (Tsutsui et al., 1984a; Jones et al., 1988a, 1988b; Lasne et al., 1988), but not murine BALB/3T3 embryo cells (Lasne et al., 1988).
With sodium monofluorophosphate, both negative and positive results on cytogenetic change (mostly chromosomal aberrations) in human lymphocytes and leukocytes have been reported, without a clear explanation for why some studies were negative and others positive (Zeiger et al., 1993).
Fluoride induces chromosomal aberrations in several plant species, including onions (Allium spp.), broadbean (Vicia faba) and barley (Hordeum vulgare) (Zeiger et al., 1993).
Sodium fluoride was reported to induce recessive lethal mutations in the germ cells of male fruitfly (Drosophila melanogaster) (Dominok & Miller, 1990). The results of studies on the genotoxic potential of fluoride following its in vivo administration to laboratory animals have varied, principally depending upon the route of administration.
Sodium fluoride induced cytogenetic damage in bone marrow or sperm cells (i.e., chromosomal aberrations, micronuclei, alterations in sperm morphology) when administered to rodents by intraperitoneal injection (Ma et al., 1986; Pati & Bhunya, 1987); negative results have, however, also been reported (Hayashi et al., 1988). While some positive results have been reported (Akhundow et al., 1981; Aliev & Babaev, 1981; Mohammed & Chandler, 1982; Ma et al., 1986; Pati & Bhunya, 1987), in most studies in which (sodium) fluoride was administered orally (either acutely or chronically) to rodents, there was no effect upon sperm morphology or the frequency of chromosomal aberrations, micronuclei, sister chromatid exchange or DNA strand breaks (Martin et al., 1979; Skare et al., 1986b; Albanese, 1987; Li et al., 1987b, 1987c, 1987d, 1989; Pillai et al., 1988; Dunipace et al., 1989, 1998; Lu et al., 1989; Zeiger et al., 1994).
After administration of sodium monofluorophosphate, cytogenetic studies have also mostly given negative results (Voroshilin et al., 1973; Gocke et al., 1981; Albanese, 1987; Li et al., 1987b, 1989; Hayashi et al., 1988).
No evidence of pregnancy or implantation of embryos within the uterus was observed in female mice administered (orally) >5.2 mg fluoride/kg body weight per day on days 6–15 after mating (Pillai et al., 1989), although few experimental details were provided. Reductions in fertility have been observed in male rabbits administered (orally) >9.1 mg fluoride/kg body weight per day and in male mice given 4.5 mg fluoride/kg body weight per day for 30 days (Chinoy et al., 1991; Chinoy & Sharma, 1998). Histopathological changes within the organs of the reproductive system have been observed in the testes of male rabbits administered (orally) 4.5 mg fluoride/kg body weight per day for 18–29 months (Susheela & Kumar, 1991), in the ovaries of female rabbits injected subcutaneously with >10 mg fluoride/kg body weight per day for 100 days (Shashi, 1990) and in the testes of male mice administered (orally) >4.5 mg fluoride/kg body weight per day for 30 days (Chinoy & Sequeira, 1989a, 1989b). Sperm motility and viability were reduced in rats and mice administered 4.5 mg fluoride/kg body weight per day (as sodium fluoride) orally for 30 days (Chinoy et al., 1995; Chinoy & Sharma, 1998). A single injection of sodium fluoride (0.05 ml of a solution containing 250 mg sodium fluoride/litre; equivalent to an injection of approximately 5.6 µg fluoride) directly into the testes of male Sprague-Dawley rats reportedly had no significant effect upon the morphology of this organ (Sprando et al., 1996). Sprando et al. (1997) also reported that spermatogenic effects (e.g., sperm count, testes weight, histopathology) and endocrine effects (serum testosterone, luteinizing hormone and follicle stimulating hormone) were not observed in rats administered drinking-water containing 0, 25, 100, 175 or 200 mg sodium fluoride/litre for 14 weeks or in their F1 offspring exposed in utero and after parturition to fluoride. In a follow-up study, morphometric analysis revealed no changes in the testes of F1 rats exposed in utero and after birth to similar levels of fluoride in drinking-water (Sprando et al., 1998).
In an older multigeneration study conducted with female Swiss-Webster mice administered a low-fluoride diet and drinking-water containing 0, 50, 100 or 200 mg fluoride/litre, reproductive function appeared normal in the group receiving drinking-water supplemented with 50 mg fluoride/litre, based on litter production, infertility proportions, age at delivery of first litter, time interval between litters and frequency of postpartum conception; maternal toxicity and impaired reproduction were observed at higher doses (Messer et al., 1973). Tao & Suttie (1976) reported that various parameters of reproductive function (i.e., reproduction rate, litter size and weight) were not significantly different in female Webster mice administered diets containing either <0.5 or approximately 2 or 100 mg fluoride/kg for up to three generations. In an investigation in which rats were fed dog food containing high fluoride levels (56 or 460 mg fluoride/kg), no adverse reproductive effects were observed; however, there was uncertainty about the precise dose of fluoride due to a lack of data on the intake and bioavailability of fluoride in the diet (Marks et al., 1984). Although significant bone resorption was observed in breeding female rats exposed to drinking-water containing 150 mg fluoride/litre (as sodium fluoride) prior to breeding, during pregnancy and during lactation, no effects were observed in the bones of weanling pups from these females (Ream et al., 1983a, 1983b).
Adverse effects on fetal development were not observed in a study conducted with Charles River rats, in which dams received up to approximately 25 mg fluoride/kg body weight per day from drinking-water on days 0–20 of gestation (Collins et al., 1995). Fetal development was similar in a study in which pregnant CD rats and New Zealand White rabbits were administered drinking-water containing sodium fluoride on days 6 through 15 and 6 through 19 of gestation, respectively, at concentrations up to 300 and 400 mg/litre, respectively (Heindel et al., 1996). In this study, the total intake of fluoride from feed and drinking-water (and intake of fluoride from feed alone) for the rats and rabbits in the highest exposure groups was approximately 13.2 (1.0) and 13.7 (0.7) mg/kg body weight per day, respectively.
Compared with controls, T-cell mitogenesis was significantly increased (84%) while B-cell activity (antibody production) was significantly reduced (10%) in female C57BL/6N mice administered (intragastrically) 13.6 mg fluoride/kg body weight per day (as sodium fluoride dissolved in distilled water) for 10 weeks (Sein, 1988). Antibody production was also inhibited in female rabbits administered (orally) 4.5 mg fluoride/kg body weight per day over a 6- to 9-month period (Jain & Susheela, 1987b). Evidence of fluoride’s activity as a gut and systemic adjuvant was presented by Butler et al. (1990), based upon the results of a study in which rats were administered (intragastrically) 5 ml of a 100 mmol/litre solution of sodium fluoride twice a week for 2–3 weeks and given an antigen (ovalbumin) in their drinking-water.
Tissue- or organ-specific effects linked with repeated exposure to fluorides may involve metabolic pathways associated with lipid, carbohydrate, bone and energy metabolism, as well as signal transduction pathways (Kaminsky et al., 1990). Fluoride influences a number of enzymatic activities. The results of studies carried out in vitro have indicated that fluoride inhibits the synthesis of DNA and protein, inhibits cell proliferation and is cytotoxic in sufficiently high doses (Gilman, 1987; Elsair & Khelfat, 1988; Godfrey & Watson, 1988; Kaminsky et al., 1990). Effects produced by the exposure of laboratory animals and humans to fluoride may be attributable to one or more of these metabolic and biochemical effects. The stimulatory effect of fluoride on osteoblast proliferation may proceed, at least in part, through the inhibition of phosphotyrosyl protein phosphatases (Thomas et al., 1996; Lau & Baylink, 1998).
In a study designed to assess the effects of fluoride on aluminium distribution and toxicity, Stevens et al. (1987) reported that while the concurrent administration of 1 mg fluoride/day (as sodium fluoride) to male Sprague-Dawley rats receiving 0.5 mg aluminium/day (as aluminium chloride) (both compounds administered by subcutaneous injection) over a period of 30 days increased the amount of aluminium deposited in the liver (3.7-fold, P < 0.005), spleen (2-fold, P < 0.005) and adrenal glands (1.6-fold, P < 0.05) compared with animals receiving aluminium alone, the co-administration of fluoride had no effect upon the amount of aluminium deposited in the brain. Deficits in some tests of open field behaviour as well as reductions in body weight gain were greater in animals co-administered fluoride and aluminium than in animals administered either alone.
In a study conducted by Ahn et al. (1995) in which groups of male New Zealand rabbits were administered drinking-water containing 0–50 mg fluoride/litre (as sodium fluoride), 0–500 mg aluminium/litre (as aluminium chloride) or both combined for a period of 10 weeks, the accumulation of aluminium in the liver was not significantly affected by the concurrent consumption of fluoride. However, the accumulation of aluminium in the tibia (which was not influenced by the addition of aluminium to the drinking-water) was increased when the drinking-water contained 50 mg fluoride/litre. The accumulation of fluoride in the teeth and bone (tibia) was reduced with the concurrent consumption of aluminium. The aluminium content of the sterna of high-dose and control rats from the NTP (1990) sodium fluoride study described above (see sections 7.2 and 7.3) was also measured and found to be approximately 8-fold higher in rats receiving 79 mg fluoride/litre in drinking-water than in controls receiving deionized water (Ahn et al., 1995).
The level of aluminium in the brains of male Long-Evans rats given laboratory chow and distilled water or distilled water containing 2.1 mg sodium fluoride/litre or 0.5 mg aluminium fluoride/litre for 52 weeks was reported to be approximately 2-fold higher in the sodium fluoride group and about 2.5 higher in the aluminium fluoride group than in animals given the same feed and distilled water for drinking. There was significant mortality in the aluminium fluoride group. In the kidney, there was no increased aluminium in the sodium fluoride group but about twice as much in the aluminium fluoride group. Levels in the liver were not different between the three groups. Pathology studies of the brain revealed increased neuronal abnormalities in both fluoride-exposed groups in parts of the right and left hemispheres, alterations in the neuronal density in the left hemisphere and alterations of beta-amyloid, amyloid A and IgM in various parts of the brain (Varner et al., 1998). In an earlier study, the authors reported greater mortality and effects at the dose of aluminium fluoride used in this study than at 10- and 100-fold greater levels of aluminum fluoride. No dose–response in changes in the brain was apparent (Varner et al., 1993).
In a study on the neurotoxicity of sodium fluoride in rats (Mullenix et al., 1995), changes in behaviour were monitored in groups of Sprague-Dawley rats after exposure to fluoride at three different experimental stages. A number of behavioural patterns were examined, and pairs of control and experimental animals were observed at the same time. Some of the statistical methods were not fully described. In the first group, dams were administered subcutaneous injections of 0.13 mg sodium fluoride/kg on gestation days 14–18 or 17–19, 2–3 times daily. Nine weeks after delivery, they were examined, and measures of time–structure changes were depressed in exposed animals compared with controls. The second group was exposed to 0, 75, 100, 125 or 175 mg fluoride/litre in their drinking-water for 6 or 20 weeks immediately after weaning. The top group was discontinued because of mortality and dehydration, and the second top group showed significant deficits in body weight. Plasma fluoride was elevated over controls in all groups, and low-level behavioural effects from noise were correlated with plasma fluoride levels but not dose. Some specific behavioural changes compared with controls were reported for 100 and 125 mg/litre females and 125 mg/litre males, but there was considerable variation in the controls, and data for some dose groups were not reported. Plasma fluoride did not show a dose–response trend. The third group was given 0 or 100 mg fluoride/litre in drinking-water for 5–6 weeks. Testing after 10 weeks was reported to show a depression in low-level behavioural effects in females but not males. It is difficult to interpret the results of this study without more detailed analysis of the data, some of which are not presented; consequently, the significance of these data remains uncertain.
Acute oral exposure to fluoride may produce effects including nausea, vomiting, abdominal pain, diarrhoea, fatigue, drowsiness, coma, convulsions, cardiac arrest and death (Kaminsky et al., 1990; Whitford, 1990; Augenstein et al., 1991; ATSDR, 1993). Severe tissue damage, respiratory effects, cardiac arrest and deaths have been noted in case reports of individuals exposed accidentally to hydrofluoric acid through dermal contact (Buckingham, 1988; Upfal & Doyle, 1990; Bordelon et al., 1993).
The lethal dose of sodium fluoride to the average adult has been estimated to be between 5 and 10 g (32–64 mg fluoride/kg body weight); an acute dose of 5 mg fluoride/kg body weight has been considered to be the minimum that might lead to adverse health effects (Whitford, 1996). Gessner et al. (1994) reported the case of a death due to acute fluoride poisoning resulting from improperly fluoridated drinking-water; the individual was estimated to have ingested approximately 17.9 mg fluoride/kg body weight prior to death.
The toxicity of fluoride is dependent upon the type or species of the compound ingested. Generally, the more soluble salts of inorganic fluorides (e.g., sodium fluoride) are more toxic that those that are either weakly soluble or insoluble (e.g., calcium fluoride) (WHO, 1984).
Gastrointestinal effects produced following the acute ingestion of toxic amounts of fluoride likely arise from the corrosive action of hydrofluoric acid, which is produced within the acidic environment of the stomach (Spak et al., 1990; Whitford, 1990; Augenstein et al., 1991).1 Damage to the gastric mucosa (e.g., haemorrhage, loss of epithelium) has also been observed in human volunteers administered acidulated phosphate fluoride gels (Spak et al., 1990) or sodium fluoride solutions (Spak et al., 1989). Some individuals may be unusually hypersensitive to stannous fluoride, as manifested by ulcerations in the oral cavity after topical treatment (Razak & Latifah, 1988).
Cardiac arrest following accidental exposure to high levels of fluoride has been attributed to the development of hypocalcaemia and/or hyperkalaemia (Cummings & McIvor, 1988; Augenstein et al., 1991; ATSDR, 1993). The acute effects of fluoride upon the central nervous system may be due to fluoride-induced hypocalcaemia and the inhibition of cellular enzymes (Augenstein et al., 1991).
Respiratory effects (e.g., haemorrhage, pulmonary oedema, tracheobronchitis, shortness of breath) have been observed in individuals following inhalation of hydrogen fluoride (Dayal et al., 1992; ATSDR, 1993).
Since the 1960s, fluoride (usually as sodium fluoride or sodium monofluorophosphate, in combination with added calcium and, sometimes, vitamin D) has been used for the treatment of age-dependent osteoporosis at high dose levels (approximately 20–30 mg fluoride/day). While it seems apparent that such treatment increases the trabecular bone density, it apparently has no similar beneficial effect on cortical bone. Although such treatment may be protective against vertebral fractures, data on other fractures, including femoral bone, are very controversial. The assessment of the efficacy of fluoride therapies for osteoporosis is outside the scope of this document. The most often encountered side-effects of high-dose fluoride therapy include gastrointestinal pains and painful stress microfractures of feet (Bayley et al., 1990; Delmas et al., 1990; Gutteridge et al., 1990; Murray et al., 1990; Orcel et al., 1990; Riggs et al., 1990; Schnitzler et al., 1990; Riggs, 1991; Pak et al., 1994; Rizzoli et al., 1995; Meunier et al., 1998; Reginster et al., 1998; Alexandersen et al., 1999). Exposure to fluoride during treatment for osteoporosis may lead to calcium deficiency, owing to the stimulation of bone growth, even in cases where patients are given supplemental calcium as part of the therapeutic protocol (Dure-Smith et al., 1996).
No evidence of significantly adverse haematological, hepatic or renal effects was reported in a study of osteoporotic patients administered approximately 60 mg sodium fluoride/day (equivalent to a dose of 389 µg fluoride/kg body weight per day in an adult weighing 70 kg) over a period of 5 years (Hasling et al., 1987). In a study with elderly postmenopausal osteoporotic females receiving 23 mg elemental fluoride/day for a mean period of 4.2 years (ranging from 1.4 to 12.6 years) (equivalent to an intake of 470 µg/kg body weight per day in an individual weighing 58 kg), no clinically adverse alterations in blood and urine chemistries or in the frequency of sister chromatid exchange in peripheral blood lymphocytes were observed in the fluoride-exposed patients (n = 25) compared with controls not administered fluoride (Jackson et al., 1994).
The relationship between the consumption of fluoridated drinking-water and morbidity or mortality due to cancer has been examined in a large number of epidemiological studies, performed in many countries. These studies were largely prompted by a report by Yiamouyiannis & Burk (1977) that found an increase in overall cancer mortality in two of several broad age groups in 10 US cities following the implementation of drinking-water fluoridation. Although dismissed based on a variety of methodological flaws (see Doll & Kinlen, 1977; Smith, 1980; Kinlen & Doll, 1981; Chilvers, 1982; IARC, 1982, 1987; Knox, 1985), the Yiamouyiannis & Burk (1977) study stimulated a large number of other ecological studies, performed in Australia, Canada, China and the Province of Taiwan, England and Wales, New Zealand, Norway and the USA (Hoover et al., 1976; IARC, 1982, 1987; Knox, 1985; Hrudey et al., 1990; Mahoney et al., 1991; Cohn, 1992; Freni & Gaylor, 1992; Yang et al., 2000), that found no consistent relationship between deaths due to any type of cancer and the consumption of fluoride-containing (fluoridated or with naturally high fluoride content) drinking-water. Although some age- and sex-related increases in tumour incidence over time for cancer of the bones and joints and osteosarcomas were observed in one study (US DHHS, 1991), these increases were not related to the length of time that the water had been "fluoridated." Another report of an age- and sex-related increase in the rate of osteosarcoma in areas of New Jersey, USA, served with fluoridated drinking-water was based on only 10 and 7 cases in the fluoridated and non-fluoridated areas, respectively (Cohn, 1992).
Although there has been no consistent evidence of an association between the consumption of fluoridated drinking-water and increased morbidity or mortality due to cancer, most of these epidemiological studies were primarily geographic or ecological correlational investigations. Such studies are characterized by the use of aggregate units of observation, such as counties, states or provinces, rather than individuals, and usually involve the comparison of mortality or morbidity rates between exposed and unexposed areas or among areas with varying degrees of exposure. Generally, studies of this nature are limited, since the movement of individuals in and out of the exposed and non-exposed groups and other variable factors (e.g., industrialization, personal habits, etc.) that have an influence on the development of adverse health effects are not taken into account, and the statistical power may be insufficient to reveal small differences in health-related effects (e.g., increases in the incidence of rare tumours). Moreover, in such ecological studies, the intake of fluoride from other sources such as food and dental care products is not taken into account. Notably, the total intake of fluoride by individuals consuming fluoridated or non-fluoridated drinking-water may not be markedly different, due to the intake of significant amounts of fluoride from food and dental care products (US DHHS, 1991; Burt, 1992).
However, in a case–control study conducted in New York State (but excluding New York City), USA, information on the intake of fluoride from drinking-water and dental care products (e.g., fluoride tablets, mouth rinses, toothpaste and dental treatments), but not foodstuffs, was obtained for males and females <24 years of age having been diagnosed with osteosarcoma between 1978 and 1988 (Gelberg et al., 1995). The analyses were based on 130 cases and as many controls (matched by years of birth and gender). Interviews on exposure patterns were made with the subjects themselves and/or with their parents. No significant risks were observed for any individual fluoride exposure group, using either the parents’ or the children’s exposure assessments. Nor was any significant trend observed when using the parents’ exposure assessments. However, using the subjects’ exposure estimates, there was a monotonous increase in the odds ratios (ORs) for osteosarcoma with increasing exposure estimates: 1, 1.16 (95% confidence interval [CI] = 0.44–3.04), 1.72 (95% CI = 0.55–5.39) and 1.88 (95% CI = 0.64–5.55) for the exposure categories of 0–1250, 1251–2338, 2339–3987 and 3988–9291 mg total fluoride.
In a case–control study, Moss et al. (1995) found no evidence of a significantly increased risk of osteosarcoma among males or females (or both genders combined) residing in Wisconsin, USA, associated with the consumption of fluoridated drinking-water. Cases of osteosarcoma (and brain and digestive system cancers) and their controls were studied in relation to their consumption of fluoridated drinking-water (i.e., containing >0.7 mg fluoride/litre) between 1979 and 1989. Controls (approximately four per case) were matched to cases based upon age, sex and race. Odds ratios (also adjusted by conditional logistic regression for likely natural exposure to waterborne radiation and population size) for risk of osteosarcoma with consumption of fluoridated drinking-water were 1.0 (95% CI = 0.6–1.5; 110 exposed cases), 0.9 (95% CI = 0.5–1.6; 56 exposed cases) and 1.1 (95% CI = 0.6–1.9; 54 exposed cases) for both genders combined, females and males, respectively.
In a further hospital-based case–control study on osteosarcoma (22 cases and 22 matched controls), the odds ratio for "high" (>0.7 mg/litre) average lifetime or childhood drinking-water fluoride concentration was 0.33 (McGuire et al., 1991).
Skeletal fluorosis is a pathological condition that may arise following long-term exposure (either by inhalation or by ingestion) to elevated levels of fluoride. Although the incorporation of fluoride into bone may increase the stability of the crystal lattice and render the bone less soluble, bone mineralization is delayed or inhibited (Grynpas, 1990), and consequently the bones may become brittle and their tensile strength may be reduced. The severity of the effects associated with skeletal fluorosis is related to the amount of fluoride incorporated into bone. In a preclinical phase, the fluorotic patient may be relatively asymptomatic, with only a slight increase in bone mass, detected radiographically. Sporadic pain and stiffness of the joints, chronic joint pain, osteosclerosis of cancellous bone and calcification of ligaments are associated with the first and second clinical stages of skeletal fluorosis. Crippling skeletal fluorosis (clinical phase III) may be associated with limited movement of the joints, skeletal deformities, intense calcification of ligaments, muscle wasting and neurological deficits (Krishnamachari, 1987; Kaminsky et al., 1990; US DHHS, 1991). A consistent finding in cases of chronically elevated fluoride uptake is an increase in mineralization lag time of bone, which can be demonstrated by dynamic histomorphometry (Boivin et al., 1989). Osteomalacia may be observed in fluorotic individuals with a reduced or suboptimal intake of calcium; secondary hyperparathyroidism may also be observed in a subset of patients (Krishnamachari, 1987; US DHHS, 1991). Apparently in combination with nutritional deficiencies, high intakes of fluoride and the subsequent osteomalacia may also lead in children to bone deformities such as genu valgum, originally described as Kenhardt bone disease (Jackson, 1962; Krishnamachari & Krishnaswamy, 1973; Krishnamachari, 1976; Chakma et al., 2000). In osteoporotic patients, fluoride can stimulate bone formation to such an extent that, despite calcium supplementation, calcium deficiency, secondary hyperparathyroidism and osteomalacia occur (Dure-Smith et al., 1996).
The concentration of fluoride in the bone of individuals in the preclinical or crippling stages of skeletal fluorosis may be between 3500 and 5500 mg/kg bone or greater than 8400 mg/kg bone, respectively, compared with the reference values of 500–1000 mg/kg bone ash weight (US DHHS, 1991).
A number of factors, such as age, nutritional status, renal function and calcium intake, in addition to the extent and duration of exposure, can influence the amount of fluoride deposited in bone and, consequently, the development of skeletal fluorosis (US DHHS, 1991). Individuals with impaired renal function, such as those with diabetes, may be more prone to developing fluoride-related toxicological effects (i.e., fluorosis) due to their diminished excretion of fluoride (Kaminsky et al., 1990; US DHHS, 1991). Skeletal fluorosis may be reversible to some degree in a manner that is dependent upon the extent of bone remodelling (Grandjean & Thomsen, 1983).
Felsenfeld & Roberts (1991) reported the case of a 54-year-old woman who, after having consumed drinking-water containing approximately 8 mg fluoride/litre over a period of 7 years, had osteosclerosis and stiffness in her knees and hips. Five cases of crippling skeletal fluorosis in the USA have been reported over the past 40 years; the total intake of fluoride by some of these individuals over a 20-year period was estimated to be approximately 15–20 mg/day (US DHHS, 1991) (equivalent to a daily intake of 230–310 µg fluoride/kg body weight per day in an adult weighing 64 kg). There have been no systematic studies of the prevalence of this disease in the USA.
The occurrence of endemic skeletal fluorosis has been well documented in case reports and surveys of individuals residing in certain areas of the world (e.g., India, China, northern, eastern, central and southern Africa), where the intake of fluoride may be inordinately high as a result of the often significant consumption of drinking-water containing substantial amounts of naturally occurring fluoride, the indoor burning of fluoride-rich coal for heating and cooking, the preparation of foodstuffs in water containing increased fluoride and/or the consumption of specific foodstuffs naturally rich in fluoride (Haimanot et al., 1987; Krishnamachari, 1987; Pettifor et al., 1989; Kaminsky et al., 1990; Tobayiwa et al., 1991; Mithal et al., 1993; Wang et al., 1994; Abdennebi et al., 1995; Liu, 1995; Michael et al., 1996; Zhao et al., 1996; Teotia et al., 1998). Large numbers of individuals residing in India and China are afflicted with skeletal fluorosis, which in some cases may be severely crippling. In addition to an increased intake of fluoride from foodstuffs and drinking-water with high levels of fluoride, other factors, such as nutritional status as well as climate and other factors influencing fluid intake, may possibly play a significant role in the development of endemic skeletal fluorosis (Krishnamachari, 1987; Haimanot, 1990; Zang et al., 1996). These issues make it difficult to characterize the exposure–response relationship in studies of skeletal fluorosis, such as those outlined below.
In China, endemic fluorosis associated with coal burning has been identified in epidemiological investigations. Local residents absorb high doses of fluoride through inhalation and/or ingestion, as a consequence of the indoor use of high-fluoride coal in cooking, heating and drying of food (the average concentrations of fluoride in coal were 200–1500 mg/kg, with the highest up to 3000 mg/kg). In those areas, the intakes of fluoride via drinking-water were relatively low (range from 0.1 to 0.52 mg/day per person) (Table 10). The number of cases of coal-burning-type skeletal fluorosis has been estimated to be 1.5 million (Hou, 1997; Liang et al., 1997).
Table 10. Daily fluoride intake in different endemic areas in China using high-fluoride coal for cooking and drying foodstuffs indoors
|
Endemic area |
Coal type |
Daily intake (mg/person) |
|||
|
Food |
Drinking-water |
Air |
Total |
||
|
Sichuan |
Soft coal |
8.86 |
0.1 |
0.67 |
9.63 |
|
Hubei |
Anthracite |
4.12 |
0.45 |
0.55 |
6.12 |
|
Jiangxi |
Anthracite |
2.54 |
0.5 |
0.24 |
3.28 |
|
Hunan |
Anthracite |
1.81 |
0.52 |
0.31 |
2.64 |
|
Hubei |
Anthracite |
1.86 |
0.42 |
0.15 |
2.43 |
|
Jiangxi |
Firewood |
1.14 |
0.24 |
0.11 |
1.49 |
In a short communication with few details, the relationship between air, well-water and dietary fluoride and skeletal fluorosis (no information on diagnostic criteria provided) was studied among 6792 individuals in Inner Mongolia (Xu et al., 1997) (Table 11). The concentrations of fluoride in air and diet were low, and that in drinking-water showed a correlation of 0.87 with the prevalence of fluorosis. The skeletal fluorosis prevalence was <0.21% for fluoride concentrations of 0.65 mg/litre or lower and reached 19.9% for fluoride concentrations of 6.9 mg/litre.
Table 11. Prevalence of skeletal fluorosis in villages in Inner Mongolia, Chinaa
|
Fluoride content of drinking-water |
Skeletal fluorosis |
|
|
Cases |
% |
|
|
0.4 |
0 |
0 |
|
0.65 |
2 |
0.21 |
|
1.4 |
93 |
7.72 |
|
1.6 |
109 |
12.3 |
|
3.2 |
101 |
12.7 |
|
3.4 |
132 |
15.2 |
|
4.7 |
42 |
19.6 |
|
6.9 |
166 |
19.9 |
a From Xu et al. (1997).
The relationship between fluoride concentrations in drinking-water and the prevalence of skeletal fluorosis was also reported in another Chinese study (Liang et al., 1997) (Table 12).
Table 12. Prevalence of skeletal fluorosis in China as a function of drinking-water fluoride concentrationa
|
Fluoride concentration in drinking-water (mg/litre) |
Prevalence of skeletal fluorosis (%) among individuals with normal nutritionb |
Prevalence of skeletal fluorosis (%) among individuals with deficient nutritionc |
|
<0.3 |
0 |
0 |
|
0.6–1.0 |
0 |
0 |
|
>4.0 |
43.8 |
69.2 |
|
a |
From Liang et al. (1997). |
|
b |
Normal nutrition = protein >75 g/day, high-quality protein >20% of total protein, calcium >600 mg/day. |
|
c |
Deficient nutrition = protein <20 g/day, high-quality protein <10% of total protein, calcium <400 mg/day. |
According to an early estimate, the number of persons at risk of developing skeletal fluorosis was 5 million in Punjab and more in Andhra Pradesh and Madras, India (Siddiqui, 1970).
The frequency of skeletal fluorosis (as identified by the clinical picture) among children 3–10 years of age was 39% (18/46) in a village in India, where the fluoride concentrations in the three wells were 0.6, 4.0 and 1.34 mg/litre (Shivashankara et al., 2000). It was not possible to discern, from the information available, the contribution of each well to the drinking-water of the residents.
In a clinical survey for fluorosis in a random sample of residents in five areas in Tamil Nadu, South India, the drinking-water fluoride concentration was directly related to the prevalence of dental fluorosis in children (8–15 years of age) and adults. Among children, no skeletal fluorosis (no information on diagnostic criteria provided) was observed; among adults, the prevalence of fluorosis was 34% (157 individuals surveyed) in the area with the highest drinking-water fluoride concentration (summer month average 6.8 mg/litre, non-summer month average 5.6 mg/litre) (estimated total daily fluoride intake 20 mg), while no skeletal fluorosis was observed in the other areas, where the mean fluoride concentrations were 2.2 (summer months) and 1.8 (non-summer months) mg/litre or lower, with estimated total daily fluoride intakes less than 10 mg (Karthikeyan et al., 1996).
A correlation between average water fluoride concentration and prevalence of skeletal fluorosis (assessed by X-ray) was found among adults in 15 villages in Dungapur district in Rajasthan, India (Choubisa et al., 1997) (Table 13). The prevalence ranged from 4.4% at a water fluoride level of 1.4 mg/litre to 63.0% at the level of 6.0 mg/litre. Crippling fluorosis was consistently observed in villages with fluoride concentrations of >3 mg/litre.
Table 13. Relationship between drinking-water fluoride concentration and skeletal fluorosis in Rajasthan, Indiaa
|
Village |
Fluoride concentration (mg/litre) |
Prevalence of fluorosis |
% |
Crippling fluorosis |
|
|
Mean |
Range |
||||
|
Amaliya Fala |
1.4 |
0.5–1.8 |
4/92 |
4.3 |
No |
|
Doja |
2 |
1.2–2.0 |
10/102 |
9.8 |
No |
|
Selaj |
2 |
1.5–2.5 |
4/85 |
4.7 |
No |
|
Anturi |
2.3 |
0.3–3.5 |
16/180 |
8.9 |
No |
|
Batikada |
2.4 |
1.3–3.1 |
16/120 |
13.3 |
No |
|
Devsomnath |
2.5 |
0.0–3.1 |
14/188 |
7.4 |
No |
|
Masania |
2.6 |
1.0–2.6 |
28/176 |
15.9 |
No |
|
Dora |
3 |
1.2–3.9 |
22/96 |
22.9 |
Yes |
|
Dolwaniya Ka Oda |
3 |
2.5–3.5 |
14/78 |
17.9 |
Yes |
|
Palvasi |
3.4 |
2.3–4.2 |
38/114 |
33.3 |
Yes |
|
Jogiwara |
3.4 |
1.0–4.3 |
27/106 |
25.5 |
Yes |
|
Kolkhanda |
4.5 |
4.2–4.7 |
52/106 |
49.1 |
Yes |
|
Banda Ghanti |
5.7 |
3.8–6.7 |
61/115 |
53.0 |
Yes |
|
Hadmatiya |
6 |
3.9–8.3 |
118/205 |
57.6 |
Yes |
|
Pantali |
6 |
2.4–10.8 |
121/192 |
63.0 |
Yes |
a
From Choubisa et al. (1997).In four villages in the Faridabad district, North India, the percentage of people with skeletal fluorosis (assessed by the clinical appearance of impaired mobility) was 57, 43, 18 and 17, while the respective means (ranges) of the water fluoride contents were 3.2 (0.25–8.0), 3.7 (0.3–7.0), 2.5 (0.3–5.4) and 1.0 (0.7–1.6) mg/litre (Susheela et al., 1993).
A cross-sectional study in Punjab also reported the relationship between water fluoride and the prevalence of skeletal fluorosis (Jolly et al., 1968). Skeletal fluorosis was rare (assessed by X-ray) (2.4%) in a village where the average well-water fluoride concentration was 1.4 mg/litre, but its incidence was 71% in the village with the highest water fluoride concentration, 9.7 mg/litre. In five villages where the water fluoride concentration was 3–4 mg/litre, the prevalence of skeletal fluorosis was 10–42%. It was suggested that lower calcium concentrations in some water sources were associated with higher skeletal fluorosis prevalence.
The dependence of skeletal fluorosis on duration of exposure and age was studied in a village in Andhra Pradesh, India, where the drinking-water was derived from five wells, in which the fluoride concentrations were between 7.2 and 10.7 mg/litre (average 9.0 mg/litre); it was estimated that the daily water consumption was 4.6 litres, which would lead to a daily dose of 36–54 mg fluoride (Saralakumari & Ramakrishna Rao, 1993). Skeletal fluorosis started to appear after 10 years of residence in the village and reached 100% after 20 years.
In a cross-sectional study of 50 randomly selected adults who had been residents of one of two villages for all their lives in Senegal, kyphosis was used as an indicator of skeletal fluorosis (and was radiologically confirmed to be fluorosis in three cases) (Brouwer et al., 1988). The prevalence of severe kyphosis was 4 out of 55 (7%) in Guinguineo, where the well-water fluoride concentration was 3.9 mg/litre, and 11 out of 42 (26%) in Darou Rahmarie Fall, where the fluoride concentration was 7.4 mg/litre.
Several ecological and cross-sectional studies have been performed to investigate the relationship between fluoride in drinking-water (from natural sources or from fluoridation) and bone fractures or bone density (McClure, 1944; Goggin et al., 1965; Bernstein et al., 1966; Korns, 1969; Madans et al., 1983; Simonen & Laitinen, 1985; Arnala et al., 1986; Avorn & Nielssen, 1986; Sowers et al., 1986; Danielson et al., 1992; Jacobsen et al., 1992, 1993; Suarez-Almazor et al., 1993; Kröger et al., 1994; Jacqmin-Gadda et al., 1995; Lan et al., 1995; Czarnowski et al., 1999; Fabiani et al., 1999). Limitations in exposure assessment and study design preclude firm conclusions from being drawn from these studies.
Based on an ecological study of the rates of skeletal fracture among men and women between 65 and 90 years of age in the USA, the rates of hip fracture were not significantly increased among those residing in fluoridated areas (i.e., >0.7 mg fluoride/litre) compared with rates in those residing in non-fluoridated areas (i.e., <0.3 mg fluoride/litre); however, among males, the relative rates of fracture of the proximal humerus and distal forearm were significantly increased by approximately 23% and 16%, respectively (Karagas et al., 1996).
Other studies have reported either no increase or a reduced risk of hip (or skeletal) fracture associated with the consumption of fluoridated drinking-water (Madans et al., 1983; Simonen & Laitinen, 1985; Arnala et al., 1986; Jacobsen et al., 1993; Suarez-Almazor et al., 1993; Lehmann et al., 1998; Fabiani et al., 1999).
The relative risk of hip, wrist or spinal fracture was 2.2 (95% CI = 1.07–4.69) in women 55–80 years of age residing in an "elevated fluoride community" (with drinking-water containing 4 mg/litre) compared with those in a control community, with drinking-water containing 1 mg fluoride/litre (Sowers et al., 1986, 1991). The estimated mean intake of fluoride (from water-based beverages only) by women in the "elevated-fluoride community" was approximately 72 µg/kg body weight per day.
In a retrospective cohort study of 144 627 persons who had lived at least during 1967–1980 in a Finnish village outside the municipal water system, the relationship between hip fracture in 1981–1994 (from hospital discharge records) and drinking-water fluoride concentration, as estimated from a nationwide groundwater survey of 8927 wells, was studied (Kurttio et al., 1999). The fluoride levels in the wells were estimated from a national database and were approximately 1.4 times higher than the actually measured fluoride levels. The median fluoride concentration in the well-water was 0.1 mg/litre; the maximum value was 2.4 mg/litre. No association was observed between hip fractures in either men or women and the well-water fluoride concentration, when all age groups were considered. However, in women 50–65 years old at the start of follow-up, age- and area-adjusted relative risks (RR) for hip fracture increased with increasing well-water concentration in the categories 0.11–0.3, 0.3–0.5, 0.5–1.0, 1.1–1.5 and >1.5 mg/litre (RR = 1.16, 95% CI = 0.93–1.43; RR = 1.31, 95% CI = 0.86–1.99; RR = 1.53 [P < 0.05], 95% CI = 1.08–2.16; RR = 1.24, 95% CI = 0.77–2.01; RR = 2.09 [P < 0.05], 95% CI = 1.16–3.76), in comparison with a fluoride concentration of <0.1 mg/litre.
In a case–control study, Feskanich et al. (1998) assessed the risk of forearm and hip fracture among a group of female US nurses in relation to their long-term exposure to fluoride, assessed on the basis of the levels of fluoride in toenails. Women in the three highest quartiles of fluoride exposure (i.e., levels of fluoride in toenails of 2.00–3.35, 3.36–5.50 and >5.50 mg/kg) had increased and reduced risks (not statistically significant) of forearm and hip fracture, respectively, compared with women in the lowest quartile of fluoride exposure (i.e., <2.00 mg/kg).
Bone mineral density was investigated in a random stratified sample of 3222 perimenopausal women in eastern Finland (Kröger et al., 1994). Of these women, 969 had used artificially fluoridated drinking-water (1.0–1.2 mg/litre) for more than 10 years, 2024 had never used it (drinking-water fluoride concentration <0.3 mg/litre) and 229 had used fluoridated water for less than 10 years. There was no significant difference in the 10-year incidence of self-reported bone fractures between the first group and the second and third groups combined. Bone mineral densities of the spine and femoral neck, measured by dual X-ray densitometry and adjusted for age, weight, menopausal status, calcium intake, physical activity class, deliveries, alcohol consumption and oestrogen use, were statistically significantly (approximately 1%) higher among women who had used fluoridated drinking-water longer than 10 years than among the rest.
In a 5-year follow-up study (Jacqmin-Gadda et al., 1998) of 3216 men and women aged 65 years or more, risk factors of fractures of the hip or elsewhere were studied. The fluoride exposure was estimated from analyses of fluoride in water in 78 areas and by weighting the concentrations by duration of water supply use during the last 10 years and the relative contributions of different water sources in the drinking-water. Fracture rates were assessed from questionnaires and verified by the doctor of each patient reporting a fracture, when the site or multiplicity of the fracture was questionable. Logistic regression analysis was used to assess the influence of different variables on the fracture risk; the analysis considered age, sex, body mass index, smoking status, alcohol consumption, use of psychotropic drugs and fluoride exposure. No relationship was observed between fluoride exposure and fractures other than at the hip. For hip fractures, participants in the two highest drinking-water fluoride quartiles (0.11–0.25 mg/litre and >0.25 mg/litre) were at a higher risk (OR = 3.25, 95% CI = 1.66–6.38; and OR = 2.43, 95% CI = 1.11–5.33, respectively) than those below these levels. However, participants at drinking-water fluoride levels >0.7 mg/litre were not at a higher risk than those below this level (OR = 0.77, 95% CI = 0.37–1.62). When divided at the 1 mg/litre level, no increased risk appeared either.
In a multicentre prospective study on the risk factors of osteoporosis in postmenopausal women (n = 7129) (Phipps et al., 2000) (in which the earlier study, Cauley et al., 1995, is subsumed), the relationship between drinking-water fluoride exposure during the years 1950–1994 and density of lumbar spine, proximal femur, radius and calcaneus, plus incident fractures of vertebrae, hip, wrist and humerus in 1971–1990, was investigated. In a multivariate analysis, the results were adjusted for age, weight, education, muscle strength, surgical menopause, calcium intake, drinks per week, current oestrogen use, current thiazide use, non-insulin-dependent diabetes, current thyroid hormone use, walking for exercise and smoking status. The bone density was 2% higher among women continuously exposed to fluoride in drinking-water for 20 years in lumbar spine and proximal femur but lower in distal and proximal radius; for calcaneus, the difference was not statistically significant. Risk of incident fracture of the hip (RR = 0.69, 95% CI = 0.50–0.99) and spine (RR = 0.73, 95% CI = 0.55–0.97) was significantly lower among those continuously exposed to fluoride than among the non-exposed; for fractures of the humerus, the difference was not significant (RR = 0.85, 95% CI = 0.58–1.23), and for fractures of the wrist, the risk among those exposed to fluoride was elevated (RR = 1.32, 95% CI = 1.00–1.71).
In a population-based case–control study on the relationship between naturally occurring fluoride in drinking-water and fractures of the hip, lifetime drinking-water fluoride concentration was determined from residential history and data from water suppliers on 514 cases and 527 sex- and age-matched controls in the United Kingdom (Hillier et al., 2000). After adjustment for sex and age, body mass index, physical activity, age at menopause, current alcohol consumption, smoking, current treatment with oral corticosteroids and dietary calcium, the odds ratio for lifetime drinking-water containing >0.9 mg fluoride/litre was 1.0 (95% CI = 0.7–1.5) in comparison with lifetime drinking-water containing lower fluoride concentrations. No effect of fluoride concentration early or late in life on hip fracture risk was observed either. However, based on estimated total fluoride intakes in the United Kingdom, the contribution of fluoride in drinking-water was rather small (probably less than one-third).
The relationship between drinking-water fluoride concentration and hip fracture, as well as all bone fractures since the age of 20 years, was studied among 8266 Chinese men and women, residents of six villages with different drinking-water fluoride concentrations (Li et al., 2001). The studied persons had lived in the same village for no less than 25 years, most for their whole lives, and were no less than 50 years of age at the time of the study. Altogether, 531 subjects reported one or more fractures, and for 526 of them the fracture was confirmed by X-ray; the number of hip fractures was 56. In a multiple logistic regression analysis, adjusted for age and gender, the odds ratio for any fracture was 1.50, 1.25, 1.00, 1.17, 1.18 and 1.47 for drinking-water fluoride concentrations of 0.25–0.34, 0.58–0.73, 1.00–1.06, 1.45–2.19, 2.62–3.56 and 4.32–7.97 mg/litre, respectively (Table 14). From analysis of the fluoride concentrations in tea and air and anamnestic information on the use of fluoride-containing toothpastes or mouth rinses, it was concluded that drinking-water was the only significant source of fluoride exposure, and the estimated daily fluoride intake from drinking-water was estimated to be 0.73, 1.62, 3.37, 6.54, 7.85 and 14.13 mg in the six exposure categories. The difference between the group with the lowest odds ratio was significant (P < 0.01) for the lowest and highest exposure group. The observation remained qualitatively similar when only fractures after the age of 50 years were considered, but reached statistical significance only for the high exposure group. For hip fractures, the prevalence was similar for the three lowest exposure groups, but elevated for the three high exposure groups (OR = 2.13, 1.73 and 3.26); the difference was significant for the high exposure group.
Table 14. Water fluoride concentration and risk of skeletal fracturea
|
Water fluoride concentration (mg/litre) |
Total fluoride intake |
Odds ratio for all fractures |
Odds ratio for hip fracture |
|
0.25–0.34 |
0.73 |
1.50 (0.01) |
0.99 (0.99) |
|
0.58–0.73 |
1.62 |
1.25 (0.17) |
1.12 (0.85) |
|
1.00–1.06 |
3.37 |
1.00 |
1.00 |
|
1.45–2.19 |
6.54 |
1.17 (0.33) |
2.13 (0.15) |
|
2.62–3.56 |
7.85 |
1.18 (0.35) |
1.73 (0.34) |
|
4.32–7.97 |
14.13 |
1.47 (0.01) |
3.26 (0.02) |
a
From Li et al. (2001).In a meta-analysis (Jones et al., 1999) of ecological, cross-sectional and cohort studies on water fluoridation and bone fractures published in 1966–1997, water fluoridation was not observed to have an effect on fracture incidence (RR = 1.02, 95% CI = 0.96–1.09). Out of 26 studies (published in English), 18 contained information that could be used in the meta-analysis and were included. The meta-analysis correctly assigned a qualitative score to the different studies (by which the studies were weighted) and considered sources of bias such as publication bias. Although overall no relationship between fluoridation and fracture risk was observed, there was considerable heterogeneity between the studies. Four of the studies also reported on the relationship between water fluoridation and bone mass. A significant increase in lumbar spine bone mass was observed; for femoral neck, the change was positive, but statistically not significant, and for distal radius, it was negative and statistically not significant.
In case–control studies, no evidence of an association between the consumption of fluoridated drinking-water by mothers and increased risk of spontaneous abortion (Aschengrau et al., 1989), late adverse pregnancy outcome (Aschengrau et al., 1993) or congenital cardiac disease in children (Zierler et al., 1988) was identified. These findings confirm previous observations of no apparent relationship between the rates of Down syndrome or congenital malformation and the consumption of fluoridated drinking-water (see Berry, 1962; Needleman et al., 1974; Erickson et al., 1976; Berglund et al., 1980; Erickson, 1980; all cited previously in WHO, 1984).
Freni (1994) reported evidence suggesting that exposure to fluoride in drinking-water may be associated with reduced fertility rates, based upon the results of an ecological study that evaluated the "total fertility rate" (i.e., total age-specific births per 1000 women) between 1970 and 1988 for white women, 10–49 years of age, residing in 30 regions in nine US states. However, this study was based upon population means, rather than individual data.
Ernst et al. (1986) examined respiratory function in a group of male and female adolescents with "high" or "low" exposure to the emissions from an aluminium plant located near Cornwall, Ontario, Canada, and reported evidence of respiratory abnormalities (i.e., a 49% increase [P = 0.05] in the ratio of the closing volume/vital capacity) in males, but not females, in the "high" exposure group, compared with those with "low" exposure. Individuals were considered to have "high" and "low" exposure if they resided more than 60% of their lives within approximately 4 and 17 km of the plant, respectively.
Lung X-ray findings were reported in 45 cases with skeletal fluorosis in an area of China contaminated by coal combustion. Chronic bronchitis with diffuse interstitial fibrosis and pulmonary emphysema were reported to occur (Liu, 1996).
Since the residents in both studies were likely exposed to other airborne contaminants and particulates, it is difficult to attribute the effects on respiratory function solely to fluorides per se.
Two studies in two different regions of China have considered the potential effects of fluoride from drinking-water on children’s intelligence.
The first study (Li et al., 1995) was conducted on 907 children (ages 8–13) of the Guizhou province. Intelligence was measured by the China Rui Wen’s Scaler for Rural Areas. Children lived in four areas with different degrees of fluorosis prevalence (urinary fluoride ranging from 1.02 to 2.69 mg/litre). The average IQ was 89.9 in the non-fluorosis area and 80.3 in the high prevalence area. The trend among the four areas was statistically significant (P < 0.01).
The second study was carried out in Shanxi (Zhao et al., 1996) and involved 160 children (ages 7–14) randomly selected from each of two villages, one with a high level of fluoride contamination of the water (4.12 mg/litre, with 86% of the population having signs of dental fluorosis), and one with low contamination (0.91 mg/litre and 14% dental fluorosis). The "official intelligence quotient (IQ)" was measured. The average IQ in the first village was 97.69, and in the second it was 105.21 (P < 0.01). (The much higher IQ in comparison with the Li et al. (1995) study is noteworthy, but it is probably justified by the use of a different scale [Zhao et al., 1996].)
These two studies deserve similar comments:
The significance of these studies remains uncertain, but in a study of 197 children between the ages of 7 and 11 years in which childhood behavioural problems were assessed in relation to different levels of dental fluorosis, there was no association between behavioural problems and dental fluorosis (Morgan et al., 1998).
Based on an analysis of the frequency of sister chromatid exchanges in peripheral blood lymphocytes obtained from groups of approximately 100 male and female Chinese adults consuming drinking-water containing from 0.11 to 5.03 mg fluoride/litre, Li et al. (1995) concluded that fluoride was not genotoxic at these levels of exposure. The average estimated daily intake of fluoride, based upon levels in food, water and average body weight, ranged from about 20 to 280 µg/kg body weight per day. The plasma fluoride level in the 5.03 mg/litre area was 5.56 µmol/litre.
Although Jackson et al. (1997) observed a slight (7–15%) although statistically significant (P < 0.001) increase in the frequency of sister chromatid exchange in peripheral blood lymphocytes collected from individuals in the USA (n = 68, males and females) residing in an area served with drinking-water containing 4.0 mg fluoride/litre (compared with that from individuals in other areas served with water containing 0.2 and 1.0 mg fluoride/litre [n = 64 and 59, respectively]), a subsequent comparison among individuals residing in the high-fluoride area having consumed either city water (4 mg/litre; n = 30) or well-water (<0.3 mg/litre; n = 28) revealed no significant difference in the frequency of sister chromatid exchange in blood lymphocytes (Jackson et al., 1997).
In a report with scanty details on the selection of persons to be studied, an increased peripheral lymphocyte micronucleus and sister chromatid exchange frequency was observed among fluorosis patients (n = 53), compared with healthy referents (n = 20) from a fluorosis-endemic area and non-fluoride exposed referents (n = 30) in Inner Mongolia, China (Wu & Wu, 1995). In another study with limited reporting, an increased sister chromatid exchange frequency was reported among residents of a fluorosis-endemic area in North Gujarat, India (n = 100), in comparison with referents from a non-endemic area in Ahmedabad (n = 20) (Sheth et al., 1994). In a further similar study, sister chromatid exchange frequencies were elevated among residents of one out of three fluorosis-endemic villages, and chromosomal aberrations were elevated in all three villages, compared with referents from a nearby township in south Gujarat, India (Joseph & Gadhia, 2000). In all these studies, the interpretation is complicated by the limited details provided on subject selection and by possible confounding variables.
However, no effect on chromosomal aberrations or micronuclei in lymphocytes was observed in a randomized double-blind study on seven non-smoking female osteoporosis patients treated with sodium fluoride or sodium monofluorophosphate (average dose 29 mg fluoride/day) for an average of 29 months, in comparison with seven matched non-treated controls (van Asten et al., 1998).
It has been recognized for over five decades that fluoride may have both beneficial and potentially harmful effects on dental health. While the prevalence of dental caries is inversely related to a range of concentrations of fluoride in drinking-water consumed, the prevalence of dental fluorosis has been shown to be positively related to fluoride intake from many sources (Fejerskov et al., 1988, 1996). Public health programmes seeking to maximize the beneficial effects of fluoride on dental health through the introduction of fluoridated drinking-water have, at the same time, strived to minimize its adverse fluorotic effects on teeth. Based upon the studies conducted by Dean and colleagues five decades ago, the "optimum" level of fluoride in drinking-water, associated with the maximum level of dental caries protection and minimum level of dental fluorosis, was considered to be approximately 1 mg/litre. The effects of fluoride on dental health were examined by a WHO Expert Committee (WHO, 1994).
1) Dental caries
Since the first reports by Dean and colleagues published in the 1930s, oral fluoride is still considered an effective means of reducing dental caries. Historically, populations consuming fluoridated drinking-water had a much lower prevalence of dental caries than did those consuming non-fluoridated drinking-water. Over time, the difference in caries prevalence among those consuming fluoridated and non-fluoridated drinking-water has narrowed significantly. This apparent diminution in the cariostatic effectiveness of fluoridated drinking-water is likely attributable to a "diffusion" in which individuals consuming non-fluoridated drinking-water may consume significant amounts of beverages prepared in other locales with fluoridated drinking-water, as well as exposure to fluoride through the use of dental care products — mainly fluoridated toothpaste. It has been estimated that whereas approximately 210 million individuals throughout the world consume drinking-water containing levels of fluoride considered adequate for the prevention of dental caries, approximately 500 million people use fluoridated toothpastes (WHO, 1994).
Historically, it was believed that fluoride needed to become incorporated into the crystal lattice of enamel in order to effectively prevent the development of dental caries. Fluoride was considered to improve lattice stability and render the enamel less soluble to acid demineralization. Since the incorporation of fluoride into enamel, as partially fluoridated hydroxyapatite, was believed to be essential for its action, fluoride was thought best ingested. There is now, however, an increasing body of evidence to suggest that a substantial part of the cariostatic activity of fluoride is due to its effects on erupted teeth, and that the continual presence of fluoride in the saliva and in the fluid phase of dental plaque is critical to its mechanism of action. There is a growing consensus that through its interaction with the surface of enamel, fluoride in saliva and dental plaque inhibits the demineralization and promotes the remineralization taking place at the surface of the tooth.
Since the introduction of controlled fluoridated drinking-water, efforts to reduce dental caries have been extended to include the use of fluoridated toothpaste, mouth rinses and topically applied dental treatments (e.g., gels, varnishes, solutions), as well as through the use of fluoride supplements, fluoridated milk and fluoridated salt. The effectiveness and factors affecting the implementation of these various exposure regimens have been reviewed in detail by WHO (1994), and therefore only a brief summary is presented here.
The controlled fluoridation of community drinking-water to an optimum level is one of the most cost-effective means of delivering fluoride to large numbers of individuals. This method of fluoride delivery requires a suitable community-wide drinking-water delivery system along with a reasonable level of technological development (e.g., infrastructure, equipment and appropriately trained support personnel). Depending upon the annual average maximum daily air temperature, recommended levels of fluoride in drinking-water considered useful for the prevention of dental caries have ranged from 0.5 to 1.2 mg/litre.
Some countries have introduced controlled fluoridated salt as a means of reducing the prevalence of dental caries among their respective populations. Unlike controlled fluoridated drinking-water and toothpastes, there is little quantitative information on the cariostatic action of fluoridated salt, although it is considered to act in a manner like that of fluoridated drinking-water. The optimum concentration of fluoride in salt needed to reduce the incidence of dental caries must take into account the level of salt intake and the concentration of fluoride in drinking-water in individual geographical areas; however, 200 mg fluoride/kg salt has been suggested to be a minimum value (WHO, 1994).
Formerly, the administration of fluoridated milk to children was considered to be a suitable means of increasing their intake of fluoride; however, little quantitative information is available on the efficacy of this delivery system in the prevention of dental caries. To be effective as a means of delivering fluoride to children, implementation of a fluoridated milk programme requires close cooperation with the dairy industry as well as a widespread system of distribution.
It has been estimated that, worldwide, almost twice as many people are exposed to fluoride for the prevention of dental caries through the use of toothpastes as from the consumption of controlled fluoridated drinking-water. In many countries, fluoridated toothpastes, which usually contain approximately 1000 mg fluoride/kg, represent more than 95% of total dentifrice sold. The use of these products is considered to be one of the major factors responsible for the gradual decline in the prevalence of dental caries in most industrialized countries. In areas where the prevention of dental caries through the widespread use of fluoridated drinking-water, salt or milk may not be feasible, the use of fluoridated toothpastes remains an effective means of improving dental health.
Fluoride supplements, in the form of tablets, liquid drops or lozenges, are intended to provide a systemic source of fluoride when fluoridated drinking-water is not available. Problems associated with the widespread use of such supplements include poor compliance among socially and economically disadvantaged groups and the potentially inappropriate use of these products by those already consuming drinking-water containing optimal amounts of fluoride. Moreover, the results of studies suggest that fluoride is most effective when continually present at low levels in saliva and plaque fluid. In a number of jurisdictions, the recommended daily dosage of fluoride supplements is linked to the level of fluoride in the drinking-water as well as the age of the child. Although fluoride supplements may be appropriate for use in certain areas where the prevalence of dental caries is high, available data on the efficacy of this fluoride delivery system in preventing dental caries are equivocal, and there appears to be a growing consensus that fluoride supplements have a limited public health role in improving dental health.
The use of fluoridated mouth rinses has achieved a significant level of popularity among publicly based health care programmes, particularly those involving school-aged children. The levels of fluoride in these mouth rinses (0.05 or 0.2%) are related to whether they are recommended for daily or weekly use. The efficacy of fluoridated mouth rinses in the prevention of dental caries is related to the frequency of use, the level of compliance and exposure to other sources of fluoride, most notably in drinking-water. The use of fluoridated mouth rinses may be recommended for individuals with an elevated risk of dental caries, although the mouth rinses may not be appropriate for use by children younger than 6 years of age, due to their propensity to swallow significant amounts of such material, thereby increasing their risk of developing dental fluorosis.
Solutions, gels or varnishes usually applied infrequently over the course of a year by dental care professionals may be most efficacious in individuals with an elevated risk of dental caries. Owing to the level of fluoride in these materials (e.g., up to 22 300 mg/kg), health care professionals must follow protocols that reduce inadvertent ingestion of significant amounts of these products by younger children, which could cause acute toxic effects.
2) Dental fluorosis
Since the publication of the WHO (1994) assessment on the quantitative relationship between dental fluorosis and fluoride intake, a large number of further studies have been published on the matter. A recent meta-analysis (McDonagh et al., 2000) of such studies is presented in Figures 2 and 3.
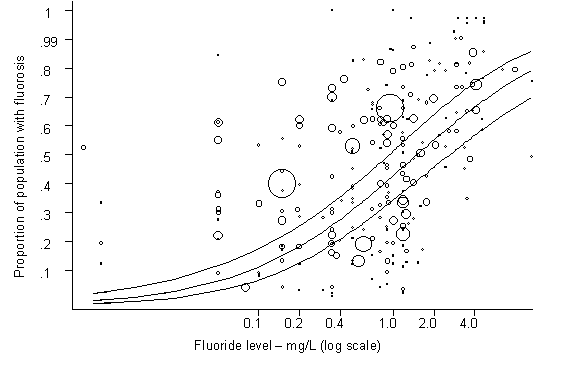
|
Note: Fluoride level plotted on the log scale because there was an approximately linear association between this and the log (odds) of fluorosis. |
|
Fig. 2. Proportion of the population with dental fluorosis by water fluoride level together with the 95% upper and lower confidence limits for the proportion |
|
[Reproduced with permission from Br Med J (2000) 321: 855–859] |
Dental fluorosis is a condition that results from the intake of excess levels of fluoride during the period of tooth development, usually from birth to approximately 6–8 years of age. It has been termed a hypoplasia or hypomineralization of dental enamel and dentine and is associated with the excessive incorporation of fluoride into these structures. The severity of this condition, generally characterized as ranging from very mild to severe, is related to the extent of fluoride exposure during the period of tooth development. Mild dental fluorosis is usually typified by the appearance of small white areas in the enamel; individuals with severe dental fluorosis have teeth that are stained and pitted ("mottled") in appearance. In human fluorotic teeth, the most prominent feature is a hypomineralization of the enamel. In contrast to many animal species, fluoride-induced enamel hypoplasia (indicating severe fluoride disturbance of enamel matrix production) seems to be rare in fluorosed human enamel. The staining and pitting of fluorosed dental enamel are both posteruptive phenomena (i.e., acquired after tooth eruption and occur as a consequence of the enamel hypomineralization). The incorporation of excessive amounts of fluoride into enamel is believed to interfere with its normal maturation, as a result of alterations in the rheologic structure of the enamel matrix and/or effects on cellular metabolic processes associated with normal enamel development (WHO, 1994; Aoba, 1997; Whitford, 1997). Experimental animal studies suggest that this hypomineralization results from fluoride disturbance of the process of enamel maturation (Richards et al., 1986).
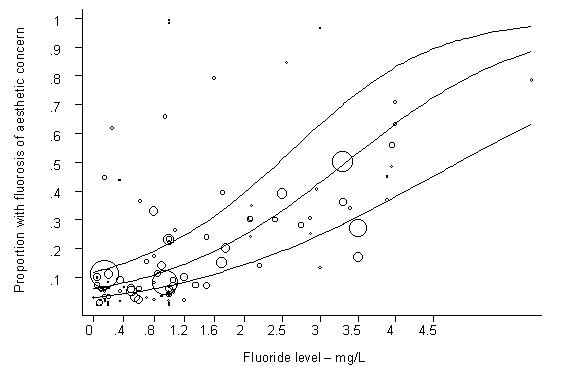
|
Note: Fluoride level plotted on the untransformed scale because there was an approximately linear association between this and the log (odds) of "aesthetic fluorosis." |
|
Fig. 3. Proportion of the population with fluorosis of aesthetic concern by water fluoride level [Reproduced with permission from Br Med J (2000) 321: 855–859] |
Unlike skeletal fluorosis, which is considered to be a marker of long-term exposure to fluoride (due to the ongoing process of bone remodelling), dental fluorosis is considered to be indicative of the level of exposure to fluoride only during the period of enamel formation. Exposure to excessive levels of fluoride after tooth development appears to have little influence on the extent of fluorosis. Re-evaluation of classical fluorosis data (Dean et al., 1941, 1942; Richards et al., 1967; Butler et al., 1985) has shown that even at low fluoride intake from water, a certain level of dental fluorosis will be found (Fejerskov et al., 1996). A dose–response relationship was also demonstrated. The data demonstrated an increase of the fluorosis community index by 0.2 for every dose increase of 0.01 mg fluoride/kg body weight.
Over the past 30–40 years, there has been an increase in the prevalence of dental fluorosis among populations consuming either fluoridated or non-fluoridated drinking-water. Although greater numbers of individuals are now being served by fluoridated drinking-water, for the most part this increased prevalence in dental fluorosis has been attributed to the widespread intake of fluoride from sources other than drinking-water, especially in areas served by non-fluoridated drinking-water. Unlike the situation in the 1930s, when the primary sources of exposure to fluoride were limited to drinking-water and foodstuffs, now there is potential exposure to fluoride from a variety of additional sources, such as toothpastes, mouth rinses, fluoride supplements and topically applied dental gels, solutions and varnishes. Exposure to fluoride may also result from the ingestion of fluoridated salt or fluoridated milk.
The prevalence of dental fluorosis is also elevated in certain areas of the world where the intake of fluoride may be inordinately high, due in large part to the elevated fluoride content of the surrounding geological environment. In China, large numbers of people exhibit dental fluorosis (Liu, 1995). In addition to the actual consumption of often large amounts of drinking-water containing naturally occurring elevated levels of fluoride, the indoor burning of coal rich in fluoride, the preparation of foodstuffs in water containing increased fluoride levels and the consumption of specific foodstuffs naturally rich in fluoride, such as tea, are believed to contribute to the elevated intake of fluoride, with the resultant development of dental fluorosis (Chen et al., 1993, 1996; Grimaldo et al., 1995; Han et al., 1995; Liu, 1995; Xu et al., 1995).
In a cross-over study, six volunteers were exposed to drinking-water with high fluoride (2.3 mg/litre), high arsenic (0.47 mg/litre) or high fluoride and high arsenic (4.05 mg fluoride/litre and 0.58 mg arsenic/litre). All volunteers followed one of these three regimes for 5 days, drank distilled water for 3 days and then were exposed to another regime. Exposure to this level of arsenic did not influence the rate of urinary excretion of fluoride of the volunteers (Wu et al., 1998).
In the area of Kuitun, Xinjiang, in China, it was found that among 619 wells investigated, 102 wells contained high levels of arsenic and fluoride. It was found that arsenic dermatosis manifested when the water arsenic level was at or above 0.12 mg/litre, with no association with the fluoride level in the water; dental fluorosis developed when the water fluoride level was at or above 3 mg/litre, regardless of the water arsenic level (Wang et al., 1997).
The health of workers occupationally exposed to fluorides, particularly those employed in the aluminium smelting industry, has been examined in a variety of epidemiological studies. Workers in this industrial setting are also exposed to a number of other substances, such as ammonia, carbon monoxide, sulfur dioxide, distillation products of tar, pitch and coal, aluminium oxide (alumina), cyanides, dust and metals such as nickel, chromium and vanadium (IARC, 1984; Sr yseth & Kongerud, 1992).
Severe tissue damage, respiratory effects, cardiac arrest and deaths have often been noted in case reports of workers exposed accidentally to hydrofluoric acid through dermal contact (Mayer & Gross, 1985; Chan et al., 1987; Mullet et al., 1987; Greco et al., 1988; Upfal & Doyle, 1990; Gubbay & Fitzpatrick, 1997). Following the exposure of two male workers to breakdown products (sulfur tetrafluoride, thionyl fluoride, hydrogen fluoride) of sulfur hexafluoride, the men became semicomatose and developed cyanosis and pulmonary oedema (Pilling & Jones, 1988). Eye and throat irritation, chest tightness, nausea, vomiting, headache and fatigue have also been observed in electrical workers exposed to the breakdown products of sulfur hexafluoride (Kraut & Lilis, 1990).
In a number of analytical epidemiological studies, an increased incidence of lung and bladder cancer and increased mortality due to cancer of the lung, liver, bladder, stomach, oesophagus, pancreas, lymphatic–haematopoietic system, prostate or brain (central nervous system) have been observed in fluoride-exposed workers employed in the aluminium smelting industry (Giovanazzi & D’Andrea, 1981; Andersen et al., 1982; Rockette & Arena, 1983; Theriault et al., 1984; Gibbs, 1985; Armstrong et al., 1986, 1994; Mur et al., 1987; Siemiatycki et al., 1991; Spinelli et al., 1991; Rr nneberg & Andersen, 1995; Romundstad et al., 2000). However, in general, there has been no consistent pattern, and bone cancer was not usually assessed. Although increases in lung cancer were observed in several studies, it is not possible to attribute these increases to fluoride exposure per se due to concomitant exposure to other substances. Indeed, in some of these epidemiological studies, the increased morbidity or mortality due to cancer was attributed to the workers’ exposure to aromatic hydrocarbons.
In a cohort study of cryolite mill workers in Denmark, Grandjean et al. (1992) indicated that a portion of the increase in the incidence of bladder cancer (17 observed versus 9.2 expected cases) among the workers may have been attributable to occupational exposure to fluoride; however, the workers were also exposed to other substances, such as quartz, siderite and small amounts of metal sulfides (Grandjean et al., 1985).
Generally, most data on the occurrence of skeletal fluorosis in occupationally exposed workers have come from older studies. Based upon a review of older studies involving aluminium smelter workers in which the number of workers examined was usually small and quantitative data on exposure to airborne fluoride were not always provided, Hodge & Smith (1977) concluded that the "incidence of detectable osteosclerosis was often high" when the levels of fluoride in the air exceeded 2.5 mg/m3 and/or levels of fluoride in the urine of these workers were greater than 9 mg/litre; at airborne concentrations below 2.5 mg/m3 (and levels in the urine of below 5 mg/litre), " years of exposure in potrooms did not produce osteosclerosis." The development of skeletal fluorosis in cryolite workers in Copenhagen was attributed to the intake (from occupational exposure) of between 20 and 80 mg fluoride/day (Grandjean, 1982). Chan-Yeung et al. (1983a) reported finding no definitive signs of skeletal fluorosis in potroom workers employed in an aluminium smelter and exposed to 0.48 mg fluoride/m3.
In a more recent study in which skeletal changes in 2258 workers employed at an aluminium plant in Poland were assessed (clinically and radiologically), the occurrence of fluorosis (multiple joint pain, initial ossification, osteosclerosis) was reported to increase with increasing duration of employment (Czerwinski et al., 1988). The occurrence of these skeletal changes was related not to quantitative data on the concentration of airborne fluoride per se, but to a qualitative "exposure index," calculated on the basis of the years of employment and the extent to which the concentration of fluoride in the air in different areas of the plant exceeded the highest permitted level of 0.5 mg hydrogen fluoride/m3. The prevalence of skeletal fluorosis reportedly increased with this "index of exposure-years," the more severe effects being observed in older workers.
Since publication of the first EHC on fluorides (WHO, 1984), additional studies have reported adverse effects on the respiratory system (e.g., reduced lung capacity, irritation of the respiratory tract, asthma, cough, bronchitis, shortness of breath and/or emphysema) in workers, predominantly those employed in aluminium smelters, occupationally exposed to airborne fluorides (Chan-Yeung et al., 1983b; Larsson et al., 1989; Sr yseth & Kongerud, 1992; Rr nneberg, 1995; Sr yseth et al., 1995; Radon et al., 1999; Romundstad et al., 2000). Owing to the exposure of these workers to other airborne contaminants and particulates, it may not be possible to attribute the effects on respiratory function solely to fluoride per se.
No evidence of haematopoietic, hepatic or renal dysfunction was observed in potroom workers employed at an aluminium smelter and exposed to 0.48 mg fluoride/m3 (Chan-Yeung et al., 1983a).
The mean frequency of sister chromatid exchange in peripheral blood lymphocytes obtained from 40 Chinese fertilizer production workers was significantly (P < 0.01) increased by approximately 50%, compared with that in a similarly sized group of controls matched for age, sex and smoking habits (Meng et al., 1995). During the period of analysis, workers were exposed to levels of fluoride (mostly hydrofluoric acid and silicon tetrafluoride) ranging from 0.5 to 0.8 mg/m3, as well as to phosphate fog, ammonia and sulfur dioxide. Among the workers, the average frequency of sister chromatid exchange was approximately 27% higher (P < 0.01) in smokers than in non-smokers. Information on the total intake of fluoride was not presented.
In a further study, an increased frequency of both chromosomal aberrations and micronuclei in circulating blood lymphocytes was also observed among the fertilizer plant workers (n = 40), in comparison with 40 controls working and studying in Shanxi University, situated in the same city as the factory, matched for sex, age and smoking habits (Meng & Zhang, 1997); the microscopic analysis was performed on coded slides.
Fluoride (100 mg/litre) did not affect growth or the chemical oxygen demand degrading capacity of activated sludge when added either as a pulse or continuously. Sludge settleability did not change following a pulse addition of fluoride; however, continuous addition of fluoride resulted in poor settling, indicated by 100–200% increases in settled sludge volume. The authors concluded that the poor settling was probably due to enhanced growth of filamentous organisms (Singh & Kar, 1989). Beg (1982) found the EC50 for inhibition of bacterial nitrification to be 1218 mg fluoride/litre.
McNulty & Lords (1960) exposed the green alga Chlorella pyrenoidosa to hydrogen fluoride in 110-min tests. Significant increases in oxygen consumption and total phosphorylated nucleotides were observed at fluoride concentrations of 2, 20 and 200 mg/litre (0.105, 1.05 and 10.5 mmol/litre). The authors concluded that, based on measurements of gas exchange at several pH values, the stimulation was probably due to the undissociated hydrogen fluoride in the medium. Smith & Woodson (1965) found that fluoride concentrations greater than 19 mg/litre (1 mmol/litre) caused growth inhibition in the same species. However, Nichol et al. (1987) found no effect of fluoride at concentrations ranging from 5 to 150 mg/litre at a variety of pH levels (5.9–8.0).
LeBlanc (1984) calculated 96-h EC50s, based on growth, to be 123 and 81 mg fluoride/litre for the freshwater green alga Selenastrum capricornutum and the marine alga Skeletonema costatum, respectively.
Rai et al. (1998) calculated 15-day LC50s for the alga Chlorella vulgaris to be 380 and 266 mg fluoride/litre (14 and 20 mmol/litre) at pH 6.8 and pH 6.0, respectively; at pH 4.5, the 3-day LC50 was 133 mg fluoride/litre (7 mmol/litre). Non-inhibitory concentrations were 66.5, 28.5 and 9.5 mg fluoride/litre (3.5, 1.5 and 0.5 mmol/litre) at pH 6.8, 6.0 and 4.5, respectively.
Antia & Klut (1981) exposed five euryhaline phytoplankton species to fluoride concentrations ranging from 50 to 200 mg/litre (14–15% salinity). The growth rate and maximum growth density of the chlorophyte Duniella tertiolecta and the diatom Thalassiosira weissflogii were unaffected at all exposure concentrations. The growth of the diatom Chaetoceros gracilis appeared to be stimulated by the presence of fluoride. The haptophyte Pavlova lutheri was 35–50% inhibited at fluoride concentrations of >150 mg/litre. The dinoflagellate Amphidinium carteri was 20–25% inhibited at 150 mg/litre and more than 90% inhibited at 200 mg/litre. Hekman et al. (1984) studied the effect of dissolved fluoride concentrations of up to 150 mg/litre on six phytoplankton species. Growth and photosynthetic oxygen evolution were unaffected at fluoride concentrations up to 50 mg/litre in all algae except Synechococcus leopoliensis. S. leopoliensis growth ceased for a period followed by growth at a reduced rate at 50 mg fluoride/litre; the threshold for growth effects and inhibition of photosynthesis in this species was 25 mg/litre. Nichol et al. (1987) found that fluoride concentrations of >100 mg/litre (5.2 mmol/litre) caused a growth lag in S. leopoliensis at neutral pH. The effect of fluoride on the growth lag was pH-dependent, with the growth lag increasing with decreasing pH. At pH 5.9, there was a measurable growth lag at 5 mg fluoride/litre. Fluoride resistance was induced by prior growth in the medium at non-inhibitory levels of sodium fluoride. It was suggested that fluoride-resistant cells retain less fluoride (taken up as undissociated hydrogen fluoride) by developing increased permeability to the fluoride anion. No effect on growth of 12 species of marine phytoplankton was observed at fluoride concentrations ranging from 10 to 50 mg/litre. At the highest concentration (100 mg/litre), no effect on growth was observed in 9 of the 12 species tested; however, 25–30% inhibition of growth was found in a diatom (Nitzschia angularis affinis), a dinoflagellate (A. carteri) and a haptophyte (P. lutheri) (Oliveira et al., 1978).
Joy & Balakrishnan (1990) studied the effect of fluoride on the diatoms Nitzschia palea (freshwater) and Amphora coffeaeformis (brackish water) during 96-h exposures. Significant enhancement of growth, compared with controls (no added fluoride), was observed at fluoride concentrations ranging from 30 to 110 mg/litre with N. palea and at a concentration of 70 mg/litre in A. coffeaeformis. A. coffeaeformis did not show significant growth differences from controls at fluoride concentrations of 90 and 110 mg/litre.
The marine dinoflagellate Amphidinium carteri was unable to grow in nutrient-enriched seawater containing 200 mg fluoride/litre. However, the organism could be adapted to grow under these conditions after repeatedly being cultured with stepwise increases in subinhibitory fluoride concentrations. Electron microscopic investigation of the adapted dinoflagellate cells revealed abnormal ultrastructural features in the chloroplast, mitochondria and nucleus. Fluoride adaptation seemed to confer a prolamellar-like configuration on the thylakoids (extensive membrane system containing the components of photosynthesis) in the centre of the pyrenoid (protein structure associated with formation and storage of starch); in organisms that had not been adapted, fluoride caused extreme disorganization of thylakoid formation (Klut et al., 1981).
Van Wensem & Adema (1991) studied the effect of potassium fluoride (32.3–3230 mg fluoride/kg [1.7–170 µmol fluoride/g]) on Carolina poplar (Populus canadensis) litter. After 4 weeks, total respiration was increased and the phosphate concentration was decreased at the highest fluoride concentration. Ammonium, nitrate and phosphate concentrations were decreased by fluoride after 9 weeks. No-observed-effect concentrations (NOECs) for the effect of fluoride on net mineralization of ammonium, nitrate and phosphate were 323, 100.7 and 1007 mg/kg dry weight (17, 5.3 and 53 µmol/g), respectively. The authors concluded that fluoride seems to be toxic for microbial processes at concentrations found in moderately fluoride polluted areas.
Wang (1986) exposed the common duckweed (Lemna minor) to fluoride and calculated an EC50, based on a reduction in frond growth, to be >60 mg fluoride/litre.
The acute toxicity of fluoride to aquatic invertebrates is summarized in Table 15. Forty-eight-hour LC50s/EC50s range from 53 to 304 mg/litre.
Table 15. Acute toxicity of sodium fluoride to aquatic invertebratesa
|
Organism |
Size/age |
Temperature |
Hardness (mg/litre)b |
pH |
Salinity |
Parameterc |
Concentration |
Reference |
|
Water flea (Daphnia magna) |
Neonates |
20 |
|
|
|
24-h EC50 |
205 n |
Dave (1984) |
|
<24 h |
20 |
|
8 |
|
24-h EC50 |
352 n |
Kühn et al. (1989) |
|
|
<24 h |
|
173 |
8 |
|
24-h LC50 |
308 n |
LeBlanc (1980) |
|
|
<24 h |
|
173 |
8 |
|
48-h LC50 |
154 n |
LeBlanc (1980) |
|
|
Neonates |
20 |
|
|
|
48-h EC50 |
98 n |
Dave (1984) |
|
|
<24 h |
15 |
169.3 |
8.14 |
|
48-h LC50 |
304 m |
Fieser et al. (1986) |
|
|
<24 h |
20 |
169.3 |
8.14 |
|
48-h LC50 |
251 m |
Fieser et al. (1986) |
|
|
<24 h |
25 |
169.3 |
8.14 |
|
48-h LC50 |
200 m |
Fieser et al. (1986) |
|
|
Caddisfly (Chimarra marginata) |
Last instar |
15.2 |
15.6 |
7.5 |
|
48-h LC50 |
120 m |
Camargo (1991) |
|
Last instar |
15.2 |
15.6 |
7.5 |
|
72-h LC50 |
79.7 m |
Camargo (1991) |
|
|
Last instar |
15.2 |
15.6 |
7.5 |
|
96-h LC50 |
44.9 m |
Camargo & Tarazona (1990) |
|
|
Caddisfly (Hydropsyche bulbifera) |
Last instar |
15.2 |
15.6 |
7.5 |
|
48-h LC50 |
79.2 m |
Camargo (1991) |
|
Last instar |
15.2 |
15.6 |
7.5 |
|
72-h LC50 |
44.9 m |
Camargo (1991) |
|
|
Last instar |
15.2 |
15.6 |
7.5 |
|
96-h LC50 |
26.3 m |
Camargo & Tarazona (1990) |
|
|
Caddisfly |
Last instar |
15.2 |
15.6 |
7.5 |
|
48-h LC50 |
86.6 m |
Camargo (1991) |
|
Last instar |
15.2 |
15.6 |
7.5 |
|
72-h LC50 |
43.7 m |
Camargo (1991) |
|
|
Last instar |
15.2 |
15.6 |
7.5 |
|
96-h LC50 |
26.5 m |
Camargo & Tarazona (1990) |
|
|
Caddisfly (Hydropsyche lobata) |
Last instar |
15.2 |
15.6 |
7.5 |
|
72-h LC50 |
78.2 m |
Camargo (1991) |
|
Last instar |
15.2 |
15.6 |
7.5 |
|
96-h LC50 |
48.2 m |
Camargo & Tarazona (1990) |
|
|
Caddisfly (Hydropsyche pellucidula) |
Last instar |
15.2 |
15.6 |
7.5 |
|
48-h LC50 |
112 m |
Camargo (1991) |
|
Last instar |
15.2 |
15.6 |
7.5 |
|
72-h LC50 |
59.1 m |
Camargo (1991) |
|
|
Last instar |
15.2 |
15.6 |
7.5 |
|
96-h LC50 |
38.5 m |
Camargo & Tarazona (1990) |
|
|
Caddisfly (Hydropsyche bronta) |
Last instar |
18 |
40.2 |
7.8 |
|
48-h LC50 |
52.6 m |
Camargo et al. (1992) |
|
Last instar |
18 |
40.2 |
7.8 |
|
72-h LC50 |
25.8 m |
Camargo et al. (1992) |
|
|
Last instar |
18 |
40.2 |
7.8 |
|
96-h LC50 |
17 m |
Camargo et al. (1992) |
|
|
Caddisfly (Hydropsyche occidentalis) |
Last instar |
18 |
40.2 |
7.8 |
|
48-h LC50 |
102 m |
Camargo et al. (1992) |
|
Last instar |
18 |
40.2 |
7.8 |
|
72-h LC50 |
53.5 m |
Camargo et al. (1992) |
|
|
Last instar |
18 |
40.2 |
7.8 |
|
96-h LC50 |
34.7 m |
Camargo et al. (1992) |
|
|
Caddisfly (Cheumatopsyche pettiti) |
Last instar |
18 |
40.2 |
7.8 |
|
48-h LC50 |
128 m |
Camargo et al. (1992) |
|
Last instar |
18 |
40.2 |
7.8 |
|
72-h LC50 |
73.2 m |
||
|
Last instar |
18 |
40.2 |
7.8 |
|
96-h LC50 |
42.5 m |
||
|
Prawn (Penaeus indicus) |
|
|
|
35 |
96-h LC50 |
1118 ne |
McClurg (1984) |
|
|
Mysid shrimp (Mysidopsis bahia) |
|
|
|
|
|
96-h LC50 |
10.5 n |
LeBlanc (1984) |
|
a |
All tests were conducted under static conditions (water unchanged for duration of test). |
|
b |
Hardness expressed as mg calcium carbonate/litre. |
|
c |
EC50s based on immobilization. |
|
d |
n = based on nominal concentrations; m = based on measured concentrations. |
|
e |
Since seawater (35%) was used in this test, it must be assumed that only approximately 100 mg fluoride/litre was, in fact, dissolved. |
Hemens & Warwick (1972) found that fluoride concentrations of up to 100 mg/litre caused no mortality in prawns exposed for 96 h. However, the brown mussel (Perna perna) showed up to 30% mortality at an initial fluoride concentration of 7.2 mg/litre during a 5-day test (the background fluoride concentration was 1 mg/litre).
LeBlanc (1980) found a 48-h NOEC for Daphnia magna of 50 mg fluoride/litre, whereas Kühn et al. (1989) reported a no-effect concentration of 231 mg fluoride/litre (24 h). Camargo & La Point (1995) calculated "safe concentrations" (8760-h EC0.01s) for the last-instar larvae of several net-spinning caddisfly species. "Safe concentrations" ranged from 0.39 mg fluoride/litre (Hydropsyche pellucidula) to 1.18 (Hydropsyche lobata) and 1.79 mg fluoride/litre (Chimarra marginata). Further "safe concentrations" were calculated for the caddisfly species Hydropsyche bronta, at 0.2 mg fluoride/litre, and H. occidentalis and Cheumatopsyche pettiti, at 0.7 mg fluoride/litre (Camargo, 1996b).
Pankhurst et al. (1980) performed toxicity tests on the blue mussel (Mytilus edulis) (160 h), the anemone Anthopleura aureoradiata (144 h) and the red krill (Munida gregaria) (259 h) using seawater dilutions of both fluoride-loaded effluent and sodium fluoride. Effluent solutions of 50 and 100 mg fluoride/litre produced high mortality, whereas sodium fluoride at concentrations up to 100 mg fluoride/litre caused negligible mortality.
Nell & Livanos (1988) found that weight gains in Sydney rock oysters (Saccostrea commercialis) decreased linearly (P < 0.01) with increasing fluoride additions from 0 to 30 mg/litre, and there was a 20% growth depression at the highest fluoride concentration. The background level of fluoride in this study was 0.7 mg/litre. Fluoride concentrations of up to 11 mg/litre had no significant effect on growth of prawns (Penaeus indicus) over a 20-day exposure period (McClurg, 1984). Fluoride (5.88 mg/litre) had no effect on the survival of mud crabs (Tylodiplax blephariskios) or the survival and growth of penaeid shrimps (Penaeus indicus) during 68-day exposures when compared with controls (0.89 mg fluoride/litre). In a 113-day test, shrimps gained significantly more weight during exposure to fluoride (5.5 mg/litre) than did controls. The authors attributed this to variations in the food source within the tanks (Hemens et al., 1975).
The fingernail clam (Musculium transversum) appears to be one of the most sensitive freshwater species tested. Sparks et al. (1983) conducted an 8-week flow-through experiment in which statistically significant mortality (50%) was observed at a concentration of 2.8 mg fluoride/litre. The brine shrimp (Artemia salina) was the most sensitive marine species tested. In a 12-day static renewal test using brine shrimp larvae, statistically significant growth impairment (measured as body length increase relative to controls) occurred at 5.0 mg fluoride/litre (Pankhurst et al., 1980). In a flow-through 90-day life cycle test, the amphipods Grandidierella lutosa and G. lignorum showed maximum population increase at a mean fluoride concentration of 2.6 mg/litre (background seawater levels ranged from 1.3 to 1.7 mg fluoride/litre). Population performance was comparable to controls at 5 mg fluoride/litre. A maximum acceptable toxicant concentration (MATC) was set at between 5.0 and 6.2 mg fluoride/litre. However, female fecundity appeared to be the most sensitive parameter, and a mean MATC based on this parameter was 4.2 mg fluoride/litre (Connell & Airey, 1982).
In a 3-week exposure test, survival and reproduction of Daphnia magna were studied (Fieser et al., 1986). Impairment of reproduction was observed at fluoride concentrations greater than 26 mg/litre. A concentration of 35 mg fluoride/litre reduced the neonate production to 44% of controls. The average number of live young dropped by more than 98% at 49 mg fluoride/litre. However, no statistics were performed in the study. Dave (1984) calculated that the NOEC for growth in Daphnia magna after 7 and 21 days was between 3.7 and 7.4 mg fluoride/litre. Similarly, for parthenogenic reproduction, the 21-day NOEC was between 3.7 and 7.4 mg fluoride/litre. The "safe" concentration, equivalent to the geometric mean of the NOEC or MATC in hard water, was 4.4 mg fluoride/litre. Kühn et al. (1989) reported a 21-day NOEC of 14 mg fluoride/litre for Daphnia magna; the most sensitive parameter was reproduction rate.
The acute toxicity of fluoride to fish is summarized in Table 16. Ninety-six-hour LC50s for freshwater fish range from 51 mg fluoride/litre (rainbow trout, Oncorhynchus mykiss) to 460 mg fluoride/litre (threespine stickleback, Gasterosteus aculeatus). All of the acute toxicity tests (96 h) on marine fish gave results greater than 100 mg fluoride/litre.
Table 16. Acute toxicity of fluoride to fisha
|
Organism |
Size/age |
Temperature |
Hardness (mg/litre)b |
pH |
Salinity |
Parameter |
Concentration |
Reference |
|
Rainbow trout (Oncorhynchus mykiss) |
<3 g |
15 |
23–62 |
7.4–8.0 |
|
96-h LC50 |
200 n |
Smith et al. (1985) |
|
1.8 g |
12 |
17 |
7.2 |
|
96-h LC50 |
51 m |
Pimentel & Bulkley (1983) |
|
|
1.8 g |
12 |
49 |
8.3 |
|
96-h LC50 |
128 m |
Pimentel & Bulkley (1983) |
|
|
1.8 g |
12 |
182 |
8.3 |
|
96-h LC50 |
140 m |
Pimentel & Bulkley (1983) |
|
|
|
1.8 g |
12 |
385 |
8.7 |
|
96-h LC50 |
193 m |
Pimentel & Bulkley (1983) |
|
|
fry |
15.1–15.5 |
20.6–24.2 |
7.5–7.8 |
|
96-h LC50 |
107.5 m |
Camargo & Tarazona (1991) |
|
Brown trout (Salmo trutta) |
fry |
15.1–15.5 |
20.6–24.2 |
7.5–7.8 |
|
96-h LC50 |
164.5 m |
Camargo & Tarazona (1991) |
|
Fathead minnow (Pimephales promelas) |
<1 g |
15–19 |
10–44 |
7.5–8.0 |
|
96-h LC50 |
315 n |
Smith et al. (1985) |
|
Threespine stickleback (Gasterosteus aculeatus) |
<1 g |
20 |
78 |
7.4–7.9 |
|
96-h LC50 |
340 n |
Smith et al. (1985) |
|
<1 g |
20 |
146 |
7.4–7.9 |
|
96-h LC50 |
380 n |
Smith et al. (1985) |
|
|
<1 g |
20 |
300 |
7.4–7.9 |
|
96-h LC50 |
460 n |
Smith et al. (1985) |
|
|
Bluegill (Lepomis macrochirus) |
|
|
|
|
|
96-h LC50 |
>240 n |
LeBlanc (1984) |
|
Sheepshead minnow (Cyprinodon variegatus) |
8–15 mm |
25–31 |
|
|
10–31 |
96-h LC50 |
>225 n |
Heitmuller et al. (1981) |
|
Ambassid (Ambassis safgha) |
adult |
20–21 |
|
|
10–28 |
96-h LC50 |
>100 n |
Hemens & Warwick (1972) |
|
Crescent perch (Therapon |
adult |
20–21 |
10–28 |
96-h LC50 |
>100 n |
Hemens & Warwick (1972) |
||
|
Mullet (Mugil cephalus) |
juvenile |
20–21 |
|
|
10–28 |
96-h LC50 |
>100 n |
Hemens & Warwick (1972) |
|
a |
All tests were conducted under static conditions (water unchanged for duration of test). |
|
b |
Hardness expressed as mg calcium carbonate/litre. |
|
c |
n = based on nominal concentrations; m = based on measured concentrations. |
Inorganic fluoride toxicity to freshwater fish appears to be negatively correlated with water hardness (due to the complexation of fluoride ions with calcium) and positively correlated with temperature (Angelovic et al., 1961; Pimentel & Bulkley, 1983; Smith et al., 1985). The 96-h LC50 for rainbow trout exposed to sodium fluoride in soft water (17 mg calcium carbonate/litre) was 51 mg fluoride/litre. Increasing the water hardness to 49 mg calcium carbonate/litre doubled the LC50 to 128 mg fluoride/litre; a further increase in water hardness to 182 mg calcium carbonate/litre led to an LC50 of 140 mg fluoride/litre (Pimentel & Bulkley, 1983). Angelovic et al. (1961) found that increasing temperature significantly increased the toxicity of fluoride to rainbow trout at temperatures ranging from 7.2 to 23.9 °C.
Camargo & Tarazona (1991) exposed rainbow trout (O. mykiss) and brown trout (Salmo trutta) to fluoride in soft water (22 mg calcium carbonate/litre) during short-term toxicity tests. For rainbow trout, 120-, 144-, 168- and 192-h LC50s were 92.4, 85.1, 73.4 and 64.1 mg fluoride/litre, respectively; for brown trout, they were 135.6, 118.5, 105.1 and 97.5 mg fluoride/litre, respectively. The addition of chloride ions (sodium chloride) decreases the toxicity of fluoride. LC50s (120 h) for rainbow trout were 6 mg fluoride/litre in the absence of chloride ions and 22 mg fluoride/litre at a chloride ion concentration of 34 mg/litre (Neuhold & Sigler, 1962). LT50 values for brown trout exposed to fluoride at a water hardness of 73 mg calcium carbonate/litre have been calculated. LT50 values ranged from ~12 h at 60 mg fluoride/litre to ~80 h at 20 mg/litre; at concentrations of <15 mg/litre, LT50 values were >240 h (Wright, 1977).
Neuhold & Sigler (1960) reported that 20-day LC50s for rainbow trout (O. mykiss) (10–20 cm) ranged from 2.7 to 4.7 mg fluoride/litre in static renewal tests at 13 °C under soft-water conditions (<3 mg calcium carbonate/litre). The authors noted that there was no further mortality after 10 days. The symptoms of acute fluoride intoxication included lethargy, violent and erratic movement and death. The authors postulated that the variation in the response of fish to fluoride could be due to a chloride–fluoride excretion mechanism over the epithelial tissues (Sigler & Neuhold, 1972). Camargo (1996a) calculated "safe concentrations" (infinite hours LC0.01s) for rainbow trout and brown trout at fluoride concentrations of 5.1 and 7.5 mg fluoride/litre, respectively.
Hemens et al. (1975) found that fluoride (5.9 or 5.5 mg/litre) had no effect on survival of mullet (Mugil cephalus) during 68- or 113-day exposures when compared with controls (0.9 mg fluoride/litre). Fluoride had no significant effect on the growth of larger mullet (50–55 mm); however, smaller mullet (16–18 mm) showed a significant decrease in growth during the 68-day test.
The eggs of the freshwater catla (Catla catla) were exposed to fluoride concentrations ranging from 1.9 to >16 mg/litre from both effluent and sodium fluoride dilutions. Hatching occurred after 6 h in both controls and those exposed to 1.9 mg fluoride/litre. At concentrations of >3.2 mg fluoride/litre for effluent dilutions and >3.6 mg fluoride/litre for sodium fluoride dilutions, hatching was delayed by 1–2 h. The authors stated that the low pH (4.1) of the effluent may have contributed to its toxicity. The weight of eggs exposed to fluoride decreased with increasing fluoride concentration and exposure time. Significant decreases in egg protein were observed at fluoride concentrations causing delayed hatching. The toxicity of fluoride to eggs was more related to the availability of fluoride ions than to total fluoride in the media (Pillai & Mane, 1984).
Kaplan et al. (1964) found little external evidence of toxicity in adult leopard frogs (Rana pipiens) exposed to fluoride concentrations ranging from 5 to 50 mg/litre for 30 days. Total red and white cell counts were reduced at all fluoride exposure concentrations; however, no statistical analysis was carried out. At fluoride concentrations ranging from 50 to 300 mg/litre, the survival time decreased with increasing exposure concentration. All frogs died within 30 days at both 250 and 300 mg fluoride/litre. Kuusisto & Telkkä (1961) exposed frog (Rana temporaria) larvae to sodium fluoride concentrations of 1, 2 and 10 mg/litre. All fluoride concentrations caused a delay in metamorphosis and reduced the activity of the thyroids revealed by histological examination. No statistical analysis of the results was carried out.
Fluoride toxicity to terrestrial plants has been studied extensively (Weinstein, 1977). Signs of inorganic fluoride phytotoxicity (fluorosis), such as chlorosis, necrosis and decreased growth rates, are most likely to occur in the young, expanding tissues of broadleaf plants and elongating needles of conifers (Pushnik & Miller, 1990). The induction of fluorosis has been clearly demonstrated in laboratory, greenhouse and controlled field plot experiments (Weinstein, 1977; Hill & Pack, 1983; Staniforth & Sidhu, 1984; Doley, 1986, 1989; McCune et al., 1991). Weinstein & Alscher-Herman (1982) reviewed the physiological responses of plants to fluoride. They concluded that calcium and magnesium play a central role in the response of plants to fluoride. A detoxification mechanism appears to consist of the sequestration and insolubilization of fluoride by reaction with calcium. The effects of atmospheric fluoride on a wide variety of terrestrial plant species have been extensively reviewed (VDI, 1989); the studies that follow have been chosen to reflect the lowest adverse effect levels cited in the literature.
A large number of the papers published on fluoride toxicity to plants concern greenhouse fumigation with hydrogen fluoride. Wolting (1975) observed leaf necrosis on freesia (Freesia sp.) cultivars during continuous fumigation at 0.5 µg fluoride/m3 for 5 months and during intermittent fumigation with 0.3 µg fluoride/m3 (6 h/day, 3–4 times/week) for 18 weeks. Leaf tip necrosis was identified in a variety of tulip (Tulipa sp.) cultivars exposed to fluoride for 6 h/day for 3 days at concentrations ranging from 0.15 to 0.67 µg fluoride/m3 (Wolting, 1978). Hitchcock et al. (1962) reported necrotic leaf tips in gladiolus (Gladiolus sp.) cultivars exposed to 0.17 µg fluoride/m3 for 9 days. In 40-day exposures of gladiolus cultivars to 0.76 µg/m3, Hill et al. (1959) found 46% necrosis and a significant increase in respiration (39%). Long-term greenhouse experiments (2–10 growing seasons) were conducted to determine the effects (necrosis, photosynthesis and growth) of hydrogen fluoride on 16 varieties of flower, fruit, vegetable and forage crops (Hill & Pack, 1983). Three identical greenhouses were used to represent a control (filtered ambient air; mean hydrogen fluoride level was 0.03 µg fluoride/m3), a hydrogen fluoride treatment (filtered ambient air; mean hydrogen fluoride levels ranged from 0.3 to 1.9 µg fluoride/m3, depending on the exposure duration for gladioli) and an industrial treatment (unfiltered ambient air; located 1.6 km downwind of a steel plant; mean hydrogen fluoride levels ranged from 0.2 to 1.9 µg fluoride/m3). The most sensitive species tested (based on measurement of necrosis over two growing seasons, following 117 days of hydrogen fluoride exposure) was the snow princess gladiolus (Gladiolus grandiflorus). The lowest-observed-effect level (LOEL) for leaf necrosis (65% of leaves) in this plant was 0.35 µg fluoride/m3. The extent of necrosis was positively correlated with increased hydrogen fluoride concentration and increased exposure duration. Less severe necrosis (2%) occurred in the industrial greenhouse chamber. The results from other test species included reduced growth or necrosis at hydrogen fluoride exposure concentrations of 0.44, 0.54 and 21.3 µg fluoride/m3 for apples (Malis domestica borkh), pole beans (Phaseolus vulgaris) and forage (red clover, Trifolium pratense, and alfalfa, Medicago sativa), respectively.
Murray (1984a) exposed grapevines (Vitis vinifera) to hydrogen fluoride concentrations of 0.07 (control), 0.17 and 0.27 µg fluoride/m3 for 189 days. Foliar necrosis was first observed on vines exposed to 0.17 and 0.27 µg fluoride/m3 after 99 and 83 days, respectively. Fluoride had no significant effect on bunch weight, number of bunches, grape yield, grape water or potential alcohol content, leaf chlorophyll b or leaf protein concentration. Both chlorophyll a and total chlorophyll were significantly reduced by fluoride exposure. Doley (1986) fumigated three varieties of grapevine with hydrogen fluoride for four successive growing seasons (the duration of exposure varied from 54 to 159 days for each season). Leaf size during the first growth of the season was not affected by fluoride concentrations of up to 1.5 µg/m3. However, leaf size was significantly reduced during the middle and latter portion of the fourth season of fumigation in two of the varieties at 0.64 µg fluoride/m3.
Suture red spot is a serious physiological disorder of the peach (Prunus persica) fruit. In open-top field chambers, hydrogen fluoride levels causing an induction of this disorder were 0.3 µg fluoride/m3 in continuous exposures of 80 days and 1.0 µg fluoride/m3 in intermittent exposures for three 6-h periods each week for 9 weeks (MacLean et al., 1984b).
Wheat (Triticum aestivum) and barley (Hordeum vulgare) exposed to hydrogen fluoride (0.38 µg/m3) for 90 days showed no effect of treatment on yield. There was, however, a significant increase in the grain protein concentration of fluoride-exposed barley plants (Murray & Wilson, 1988c). MacLean & Schneider (1981) found a significant reduction (20%) in the mean dry mass of wheat plants (T. aestivum) exposed to 0.9 µg fluoride/m3 for 4 days. MacLean et al. (1984a) exposed wheat (T. aestivum) and two sorghum (Sorghum sp.) hybrids to hydrogen fluoride at concentrations ranging from 1.6 to 3.3 µg/m3 for three successive 3-day periods. Anthesis (the maturing of the stamens) was the most sensitive stage, and this occurred during the first exposure period in wheat and the third exposure in sorghum. Hydrogen fluoride-induced foliar damage did not occur in wheat; however, both sorghum hybrids developed foliar damage in proportion to the exposure concentration. Pack (1971) found no effect of hydrogen fluoride on the progeny of bean plants (no species stated) grown at 0.58 µg fluoride/m3. However, the primary leaves of some F1 progeny of plants grown at >2.1 µg fluoride/m3 were severely stunted and distorted. MacLean et al. (1977) observed a significant reduction (25%) in the fresh mass of pods from bean plants (Phaseolus vulgaris) exposed to 0.6 µg fluoride/m3 for 43 days (emergence to harvest) in open-top field chambers. There was no effect of hydrogen fluoride exposure (0.6 µg fluoride/m3) on growth or fruiting in tomato (Lycopersicon esculentum) plants.
Madkour & Weinstein (1988) found that hydrogen fluoride at nominal concentrations of 1, 3 and 5 µg fluoride/m3 inhibited the active loading of [14C]sucrose into the minor veins of soybean (Glycine max) leaf discs during 8- to 11-day exposures. Similar results were obtained for both controlled-environment and field conditions.
Several species of eucalypt (Eucalyptus spp.) have shown sensitivity to fluoride. Murray & Wilson (1988b) exposed E. tereticornis to hydrogen fluoride (0.38 µg fluoride/m3) for 90 days in open-top chambers. Fluoride significantly reduced leaf surface area and weight in mature and immature leaves. The same significant effects on immature leaves were noted for marri (E. calophylla), tuart (E. gomphocephala) and jarrah (E. marginata) at 0.39 µg fluoride/m3 for 120 days. However, in mature leaves, leaf surface area and weight were reduced in tuart, and surface area was reduced in marri. In jarrah, these two end-points were unaffected (Murray & Wilson, 1988a).
Coniferous trees have also been identified as sensitive plant species. In field exposure chambers, significant dose–response relationships were observed between hydrogen fluoride exposure and development of needle necrosis in 2-year-old black spruce (Picea mariana) and 3-year-old white spruce (P. glauca) (McCune et al., 1991). Four test chambers, one of which served as a control, were used for different hydrogen fluoride exposure regimens for each coniferous species. Measurements of tissue necrosis were recorded 10 days following hydrogen fluoride exposure for 78 h with black spruce and 20 days following hydrogen fluoride exposure for 50 h with white spruce. Lowest-observed-effect concentrations (LOECs) for necrosis were 4.4 and 13.2 µg fluoride/m3 for black spruce and white spruce, respectively.
Airborne fluoride can also affect plant disease development, although the type and magnitude of the effects are dependent on the specific plant–pathogen combination (Laurence, 1983). Van Bruggen & Reynolds (1988) found that exposure of soybean (Glycine max) plants to hydrogen fluoride (2 µg fluoride/m3) in a controlled chamber led to significantly larger hypocotyl lesions after inoculation with either Rhizoctonia solani or Phytophthora megasperma. Plants were exposed to hydrogen fluoride for 1 week before and after the infestation. However, Laurence & Reynolds (1984) found no significant effect of hydrogen fluoride (1 or 3 µg fluoride/m3 for 5 days) on lesion characteristics of the bacterium Xanthomonas campestris on the leaves of red kidney bean (Phaseolus vulgaris) plants. Similar findings were reported by Reynolds & Laurence (1990) during both continuous (1, 3 or 5 µg fluoride/m3 for 15, 5 or 3 days, respectively) and intermittent (3 or 5 µg fluoride/m3 for 15 days) exposures.
Several studies on fluoride have been carried out in culture media. Belandria et al. (1989) studied the effect of sodium fluoride on the germination of lichen ascospores. They found that 20% of the Xanthoria parietina spores were able to germinate in the presence of 19 mg fluoride/litre (1000 µmol/litre), and similar results were obtained for Physconia distorta and Peltigera canina. P. distorta was found to be 37% inhibited at a concentration of 0.95 mg fluoride/litre (50 µmol/litre). The most resistant species was Lecanora conizaeoides, which was only weakly inhibited (44% inhibition) at 38 mg fluoride/litre (2000 µmol/litre). Ballantyne (1984) found that exposure of pea (Pisum sativum) shoots to solutions containing 19 mg fluoride/litre (1 mmol/litre) for up to 3 days caused significant increases in ATP levels in the youngest, fully expanded leaves and in entire shoots. These increases occurred before significant decreases in fresh weight, water content or water uptake and stem elongation. Neither in vitro exposure of spinach (Spinacia oleracea) petioles to 38 mg fluoride/litre (2 mmol/litre) for 3 h nor in vivo exposure of plants to gaseous hydrogen fluoride at 5 µg fluoride/m3 for 6 days resulted in visible injury. However, there was evidence from experiments on chlorophyll a fluorescence of a reduced ability to develop or maintain a trans-thylakoid proton gradient in chloroplasts containing elevated levels of fluoride (Boese et al., 1995).
Fluoride at concentrations of 190 mg/litre (10 mmol/litre) significantly reduced the in vitro photosynthetic capacity of azalea (Rhododendron sp.) cultivars (Ballantyne, 1991).
Stevens et al. (1998a) grew tomato (Lycopersicon esculentum) and oat (Avena sativa) plants in nutrient solutions (12–13 days) at nominal sodium fluoride concentrations ranging from 1 to 128 mg fluoride/litre. Fluoride ion concentrations greater than 28 mg/litre (1473 µmol/litre) caused significant decreases in the dry weights of tomato shoots and roots; oat plants were unaffected at all fluoride exposure concentrations. Stevens et al. (1998b) found that dry weights of tomato and oat shoots and roots were significantly decreased as the pH of the solution culture decreased below 4.3 at hydrogen fluoride concentrations of 1.4 mg fluoride/litre (71 µmol/litre) and 2.6 mg fluoride/litre (137 µmol/litre) for tomatos and oats, respectively. Fluoride concentrations in tomato and oat shoots that corresponded with significant reductions in plant dry weights were 228 and 125 mg/kg, respectively. Aluminium–fluoride complexation increased fluoride uptake (see chapter 4) and toxicity in oats relative to the free fluoride ion; shoot and root dry weights were significantly limited at AlF2+ concentrations of 11–22 µmol/litre and AlF2+ concentrations of 126–357 µmol/litre (Stevens et al., 1997).
Ratsch & Johndro (1986) calculated EC50s, based on inhibition of root growth, for lettuce (Lactuca sativa) plants exposed to fluoride. For the 115-h paper substrate method, an EC50 value of 450 mg fluoride/litre was found, and for the 5-day solution method, the EC50 was 660 mg fluoride/litre.
Cooke (1976) studied the effect of fluoride (200 mg/litre) on common sunflower (Helianthus annus) seeds grown in sand culture. No effect on total dry weight was observed; however, there were significant reductions in leaf growth. Keller (1980) grew Norway spruce (Picea abies) cuttings in sand and watered with 100 mg fluoride/litre during winter until bud break. Watering with sodium fluoride significantly depressed the carbon dioxide uptake of shoots. Although the previous year’s needles did not show signs of injury, most of the new needles were killed immediately after flushing with fluoride. Exposure to fluoride significantly increased the susceptibility of plants to sulfur dioxide in subsequent fumigation experiments. Zwiazek & Shay (1987) grew jack pine (Pinus banksiana) seedlings in sand culture at 3 or 15 mg fluoride/kg dry weight. Wilting was the first sign of fluoride injury and occurred in approximately 50% of plants after 25–26 h at 15 mg fluoride/kg and 2–6 h later in only 7% of plants exposed to 3 mg/kg. Fluoride-induced injuries to mesophyll and guard cells were similar to those caused by drought and included the appearance of lipid material in the cytoplasm during early stages of injury, suggesting cell membrane damage. Plants exposed to 3 mg/kg for up to 168 h showed significant reductions in water content. Respiration was significantly reduced after 24 h, but not after longer exposure times, while photosynthetic oxygen release was significantly reduced at 48 and 91 h but had recovered after 168 h (Zwiazek & Shay, 1988a). Zwiazek & Shay (1988b) reported that 3 mg fluoride/kg significantly reduced growth (as measured by fresh weight) and acid phosphatase activity and increased total organic acid content of jack pine (P. banksiana) seedlings.
A few studies have been carried out in which the fluoride exposures have been via the soil. However, the type of soil can greatly affect the uptake and subsequent toxicity of fluorides. For example, Conover & Poole (1971) found that calcined clay prevented the uptake of water-soluble fluoride, whereas sphagnum peat did not have the same capability.
In pot experiments, Singh et al. (1979) grew rice (Oryza sativa) plants in soil (pH >9.4) amended with 25–200 mg fluoride (added as sodium fluoride); the rice plants were harvested, and the soil was subsequently sown with wheat (Triticum aestivum). The authors found that the critical water-extractable fluoride concentration in soil for grain yield in wheat was 22 mg fluoride/kg, which related to a fluoride content of 35 mg/kg in mature wheat straw.
There are limited data available to determine the critical value for fluoride in soil with respect to plant toxicity. The data are also complicated by plant species ranges of sensitivities, heterogeneity of soils, soil chemical parameters that can influence fluoride availability and toxicity, and the relative toxicity of ionic species of fluoride found in the soil solution. However, more controlled solution culture studies have attempted to define toxic concentrations of fluoride exposed to the plant roots and are perhaps the best first approximation of toxic concentrations of fluoride in soil solution.
Elrashidi et al. (1998) found that an application of 100 mg fluoride/kg significantly reduced dry matter yield of barley (Hordeum vulgare) grown on both acid (pH 4.75) and neutral (pH 6.6) soils for 40 days; however, plants growing on alkaline soil (pH 7.5) were unaffected at 1000 mg/kg. The authors found no clear influence of phosphate (50–550 mg/kg soil) on the adverse effect of fluoride on dry matter yield.
Davis & Barnes (1973) found that an added soil fluoride concentration of 380 mg/kg (20 mmol/litre) significantly reduced the growth of loblolly pine (Pinus taeda) and red maple (Acer rubrum) seedlings.
Johansson & Johansson (1972) found that egg production and survival were adversely affected in flour beetles (Tribolium confusum) exposed to flour containing 4524 mg fluoride/kg (0.1% sodium fluoride) for up to 27 days. However, short-term (1–7 days) exposure to fluoride concentrations of 452.4 mg/kg (0.01% sodium fluoride) produced significant stimulation of egg production.
Hughes et al. (1985) fed cabbage looper (Trichoplusia ni) larvae on a wheat germ diet dosed with sodium fluoride (50 and 187 mg fluoride/kg dry weight) or potassium fluoride (48 and 235 mg fluoride/kg dry weight) or a diet of hydrogen fluoride-fumigated leaves (40–187 mg fluoride/kg dry weight). Larval feeding, growth and rate of development were generally reduced on diets containing sodium fluoride and potassium fluoride. The same parameters were generally greater with or unaffected by diets containing fumigated leaves.
Wang & Bian (1988) exposed silkworms (Bombyx mori) to mulberry (Morus alba) leaves containing fluoride concentrations ranging from 10 to 200 mg/kg. The threshold concentration for mortality was 30 mg/kg; 30% mortality was observed at concentrations of 30–50 mg fluoride/kg, 70% at 50–120 mg fluoride/kg and 95% at 120–200 mg fluoride/kg. There was a close correlation between cocoon development and the fluoride content of leaves, with >80 mg fluoride/kg severely inhibiting cocoon production.
Survival of isopods (woodlouse, Porcellio scaber) was not affected after 4 weeks of exposure to fluoride levels in litter up to 3230 mg/kg (170 µmol/g) (Van Wensem & Adema, 1991).
Davies et al. (1998) found no effect on the reproduction of aphids (Aphis fabae) feeding on bean (Vicia faba) plants (12 days) that had been previously exposed to either sodium fluoride in nutrient solution (15 µg fluoride/cm3) or hydrogen fluoride via fumigation (6.5 µg fluoride/m3). Plants accumulated more fluoride in the shoots during fumigation and at levels of up to 200 mg fluoride/kg; aphids accumulated up to 300 mg fluoride/kg from these plants. Similarly, Port et al. (1998) found no effect of hydrogen fluoride on the growth and survival of cabbage white caterpillars (Pieris brassicae) at a dietary concentration of 178.3 mg fluoride/kg.
Guenter & Hahn (1986) fed white leghorn hens on a diet containing up to 1300 mg fluoride/kg for 252 days. Concentrations of >1000 mg/kg resulted in significant depression of feed intake, body weight gain, feed efficiency and egg quality. In a second experiment, pullets were fed 1300 mg/kg for 49 days; similar results were obtained, but the addition of aluminium (1040 mg/kg) to the diet reduced the effects. Chan et al. (1973) found no effect on growth in Japanese quail (Coturnix coturnix japonica) fed diets resulting in average tibial fluoride concentrations of 13–2223 mg/kg ash weight.
Fleming et al. (1987) dosed nestling European starlings (Sturnus vulgaris) with daily oral doses of 6–160 mg fluoride/kg body weight. Dosing began 24–48 h after hatching and continued for 16 days. The 24-h LD50 was 50 mg/kg body weight for 1-day-old chicks and 17 mg/kg body weight for 16-day-old nestlings. Growth rates were significantly reduced at 13 and 17 mg fluoride/kg body weight (the highest doses at which growth was monitored). No histological damage due to fluoride treatments was found in liver, spleen or kidney.
Carrière et al. (1987) fed nesting American kestrels (Falco sparverius) on a diet containing fluoride levels of 62.4 (background), 4512 or 7690 mg/kg for 10 days. The diet consisted of cockerels (Gallus gallus) that had been exposed to fluoride. Fluoride levels in femurs and eggshells of treated kestrels were significantly higher than those in controls. There were no significant effects of fluoride on per cent fertility or hatchability of eggs. Clutch sizes tended to be smaller with increasing fluoride in the diet; however, the difference was not statistically significant. Seven-day-old kestrel chicks were fed on a cockerel mash diet containing 0, 1120 or 2240 mg/kg for 27 days. Growth of chicks and weights of internal organs were not significantly affected by fluoride in the diet. Per cent bone ash was significantly increased, while bone-breaking strength was significantly decreased by fluoride (Bird et al., 1992). Bird & Massari (1983) fed kestrels on a diet to which flour contaminated with fluoride (10, 50 or 500 mg/kg) had been applied. The birds receiving the highest fluoride dose died within 6 days. Fluoride at the lower doses had no effect on clutch size, hatchability or fledging success but was associated with a higher fertility. Eggs laid by kestrels at 50 mg/kg had significantly thicker shells. Lower reproductive success of eastern screech-owls (Otus asio) was noted when birds were fed 90 mg fluoride/kg diet wet weight (as sodium fluoride), but not when fed 18 mg fluoride/kg diet (Hoffman et al., 1985; Pattee et al., 1988).
Most of the early work on mammals was carried out on domesticated ungulates (Suttie, 1983). Fluorosis has been observed in experimental studies on cattle and sheep exposed to fluoride. Cattle are less tolerant of fluoride toxicity than are other livestock (Phillips & Suttie, 1960). In long-term experiments with beef cattle, 30 mg/kg of dietary fluoride caused excessive wear and staining of teeth (Hobbs & Merriman, 1959). In a further study, beginning with young calves and lasting 7 years, the tolerance for soluble fluoride was also 30 mg/kg dry diet (Shupe et al., 1963). Suttie et al. (1957) found that lactating cows could tolerate 30 mg/kg, with a rate of 50 mg/kg causing fluorosis within 3–5 years.
Tolerance levels have been identified for domesticated animals based on clinical signs and lesions. Tolerance levels in feed range from 30–40 mg/kg dry weight in dairy cattle to 150 mg/kg in lambs; in water, tolerance levels range from 2.5–4.0 mg/litre in dairy cattle to 12–15 mg/litre in lambs (Shupe & Olson, 1983; Cronin et al., 2000). The lowest dietary level observed to cause an effect on wild ungulates was in a controlled captive study, where white-tailed deer (Odocoileus virginianus) were exposed to 10 (control feed), 35 or 60 mg fluoride/kg wet weight (as sodium fluoride) in their diet for 2 years (Suttie et al., 1985). A general mottling of the incisors characteristic of dental fluorosis was noted in the animals at the 35 mg/kg diet dose; those on the higher dose also experienced minor increased wear of the molars, as well as mild hyperostoses of the long bones of the leg. No gross abnormalities of the mandible were observed. The mean fluoride content of the mandibles was approximately 1700 mg/kg ash weight for the control, 4550 mg/kg for the low-dose group and 6600 mg/kg for the high-dose group, the latter two levels being similar to those observed in affected wild deer near sources of industrial pollution (Karstad, 1967; Kay et al., 1975; Newman & Yu, 1976).
Deer mice (Peromyscus maniculatus) fed diets of 38 (control), 1065, 1355 or 1936 mg fluoride/kg diet dry weight (as sodium fluoride) for 8 weeks exhibited, at all concentrations above the control, marked weight loss, mortality, changes in femur size and dental disfigurement (Newman & Markey, 1976). Bank voles (Clethrionomys glareolus) showed a reduction in the number of litters per female, an increase in the number of days from mating to producing the first litter, increased mortality of offspring and a changed sex ratio (greater number of males) in offspring of animals fed 97 mg/kg diet (wet or dry basis not specified). Animals fed 47 mg/kg diet also showed these effects, but the differences were not significant from the control (Krasowska, 1989).
Boulton et al. (1995) exposed short-tailed field voles (Microtus agrestis), bank voles (Clethrionomys glareolus) and wood mice (Apodemus sylvaticus) to drinking-water containing 0, 40 or 80 mg fluoride/litre for up to 84 days. Both fluoride exposures induced premature mortalities in both vole species. Severe dental lesions were observed in animals surviving the highest dose. Voles were also fed on a diet containing 100 mg fluoride/kg (vegetation contaminated by atmospheric fluorides). Offspring of the voles fed the contaminated diet had significantly lower growth rates and body weights during stages of infancy and early adulthood. The incisors of newly born young were not significantly different between the contaminated and control diets; however, incisors of offspring weaned on to the contaminated diet showed marked morphological changes and severe dental lesions (Boulton et al., 1994a).
Fluoride was suggested as the cause of reduced milk production with subsequent mortality of kits in farm-raised red foxes (Vulpes vulpes) fed a diet containing 98–137 mg fluoride/kg dry weight (Eckerlin et al., 1986).
Aulerich et al. (1987) fed mink (Mustela vison) diets containing 35–385 mg fluoride/kg for 382 days. No significant differences were observed in body weight gains or fur quality between dosed and control mink. No adverse effects of fluoride on breeding, gestation, whelping or lactation were found. Survival was adversely affected at 385 mg fluoride/kg; some of the surviving mink at this concentration had weakened frontal, parietal and femoral bones. Fluoride did not affect haematological parameters or serum calcium concentrations, but serum alkaline phosphatase activity was significantly increased at fluoride concentrations of >229 mg/kg.
Mink (Mustela vison) kits and adult male mink were fed diets containing fluoride (as sodium fluoride) at concentrations ranging from 26 to 287 mg fluoride/kg wet weight for up to 8 months. Gross, radiographic and microradiographic changes were seen in bones from animals ingesting the higher levels of fluoride. Chemical analyses for fluoride generally reflected the levels ingested. After data were evaluated, a tolerance level in the feed of 50 mg fluoride/kg for breeding stock was recommended (Shupe et al., 1987).
Rao & Pal (1978) found a positive correlation between concentrations of fluoride in soil and soil organic matter content at eight sites near an aluminium factory in India and inferred that fluoride decreased the activity of soil microorganisms responsible for litter decomposition. Significant increases in the organic matter of soils were not found until total soil fluoride concentrations were greater than approximately 1000 mg/kg.
Tscherko & Kandeler (1997) studied the influence of atmospheric fluoride deposits on soil microbial biomass and its enzyme activities near an aluminium smelter at Ranshofen, Upper Austria. Mean water-extractable fluoride concentrations ranged from 10 to 124 mg/kg dry soil and were inversely correlated with microbial activity. In the most contaminated soil (up to 189 mg/kg), the microbial activities were only 5–20% of those in unpolluted soil. At a fluoride concentration above 100 mg/kg, microbial biomass and dehydrogenase activity decreased substantially, whereas arylsulfatase activity was inhibited at only 20 mg/kg. The accumulation of organic matter near the smelter (124 mg fluoride/kg) also indicated severe inhibition of microbial activity by fluoride.
Kudo & Garrec (1983) simulated the accidental release of ammonium fluoride into a pond. Mean fluoride concentrations increased from 0.2 to 7.3 mg/litre following the release. Fluoride levels returned to background concentrations within 30 days. No visible toxic effects on plants, algae, molluscs or fish were observed during the 30-day period. No details regarding the chemical characteristics of the pond water were given.
Ares et al. (1983) monitored fluoride concentrations and diatom populations in seawater near an aluminium smelter in southern Argentina. Fluoride concentrations primarily ranged from 1.1 to 1.3 mg/litre. Anthropogenic emissions accounted for 6–8% of the variance of fluoride levels in the waters. The correlations observed between fluoride concentrations and some structural characteristics of the diatom community did not show significant effects of the discharged fluoride.
Fluoride-loaded effluent adversely affected the species richness of a marine encrusting community for up to 400 m from the point of discharge. Fluoride concentrations greater than 50 mg/litre (the concentration of fluoride in effluent at which mortality was observed in the laboratory) seldom extended for more than 20 m from the outfall. The authors concluded that a sublethal effect of fluoride and/or some other effluent component appeared to be producing the observed distribution (Pankhurst et al., 1980).
High mortality and delayed migration were observed in Pacific salmon (Oncorhynchus sp.) migrating upstream near an aluminium plant on the Columbia River, USA (Washington/Oregon border). Fluoride levels in the water ranged between 0.3 and 0.5 mg/litre during 1982. Bioassay experiments on adult salmon suggested that fluoride concentrations of 0.5 mg/litre would adversely affect migration. Between 1983 and 1986, discharges from the aluminium plant were reduced, there was a corresponding drop in fluoride concentrations, and fish mortality and migration delays were decreased (Damkaer & Dey, 1989).
Mishra & Mohapatra (1998) monitored toads (Bufo melanosticus) at a fluoride-contaminated site at Hirakud, India. Mean haemoglobin content, total red blood cell count and haematocrit in blood samples were found to be significantly reduced, whereas mean corpuscular concentration and volume were significantly elevated when compared with toads at an uncontaminated site. Mean bone fluoride concentrations were 2736 mg/kg at the contaminated site and 241 mg/kg at the control site.
Aluminium smelters, brickworks, phosphorus plants and fertilizer and fibreglass plants have all been shown to be sources of fluoride that are correlated with damage to local plant communities. In 1970, vegetation in the vicinity of a phosphorus plant in Newfoundland, Canada, began to show symptoms of damage (tip burn and margin burn). Samples collected during 1973–1975 revealed that the degree of damage and fluoride levels in soil humus were inversely related to the distance from the plant. Average levels of fluoride in vegetation ranged from 281 mg/kg in severely damaged areas to 44 mg/kg in lightly damaged areas; at a control site, the fluoride concentration was 7 mg/kg (Thompson et al., 1979). Klumpp et al. (1994, 1996a) reported that plants downwind of fertilizer industries in Brazil showed damage (necrotic bands or tip burn). Bioindicator plants have revealed that severe damage corresponded to high leaf fluoride concentrations (no statistical analysis performed). Klumpp et al. (1996b) found a highly significant linear regression between leaf damage and fluoride accumulation in Gladiolus plants; the plants developed typical fluoride-induced leaf lesions. Murray (1981) found that plant communities near an aluminium smelter showed differences in community composition and structure due partly to variations in fluoride tolerance.
However, it must be noted that in the field, one of the main problems with the identification of fluoride effects is the presence of confounding variables, such as other atmospheric pollutants. McClenahen (1978) examined stands of vegetation along pollution gradients recording species richness, evenness of stand, diversity index and concentrations of three pollutants. Most vegetation characteristics were altered in areas of high pollution. Statistical analysis showed a significant positive correlation between fluorides and shrub density; however, chloride appeared to have the greatest impact on the same parameter. Therefore, care must be taken when interpreting the many field studies on fluoride pollution.
Lichens have been used widely as biomonitors of fluoride pollution. LeBlanc et al. (1971) transplanted epiphytic lichens from an unpolluted area to various distances from an aluminium factory. Lichens accumulated 600–900 mg fluoride/kg during periods of 4 or 12 months within 8 km of the factory compared with 70 mg/kg at a control site. Lichen thalli exposed for 4 months at 1 km from the factory contained some extractable chlorophyll; however, those exposed for 12 months did not. Soredia (viable structures with a potential to develop into parent lichen thalli under favourable ecological conditions) were absent in lichens growing within 4 km of the plant. Davies (1982) found that visible damage to lichen (Xanthoria parietina) thalli (growing within a 30-km radius of the Bedfordshire brickfields, United Kingdom) was observed when internal fluoride concentrations exceeded 68 mg/kg, and internal damage occurred at >90 mg fluoride/kg.
Gilbert (1985) monitored lichens near an aluminium smelter during its 11-year operational life. Epiphytic lichens were severely damaged (>50% injury) over a 25-km2 area close to the smelter. Fluoride concentrations in Ramalina farinacea near the smelter increased from 9–11 mg/kg prior to the start of the smelting operation to 120–156 mg/kg within 4 months and had reached these higher levels up to 3 km from the smelter within 3 years.
Perkins & Millar (1987) studied the effect of airborne fluoride emitted from an aluminium works on previously unpolluted assemblages of saxicolous lichens. Prior to emissions, lichens contained a mean concentration of 16 mg fluoride/kg. Fruticose (shrubby) lichens, such as Ramalina species, were the most sensitive to fluoride emissions, with lichens containing >100 mg fluoride/kg showing severe damage. Some foliose (leaf-like) lichens tolerated >200 mg fluoride/kg, and crustose (crust-like) lichens were the most tolerant group, with overall survival at 32% and 70% for the two groups, respectively, within 1 km of the works. Damage to lichens was greatest less than 2 km from the works, where 41% of Ramalina thalli showed >50% chlorosis or necrosis. Loss of lichens decreased with increasing distance from the works. The authors reported that lichens containing 300, 100 and 50 mg fluoride/kg dry weight lost 46, 15 and 10% of cover per year, respectively (Perkins, 1992).
Several studies have been carried out on the effects of fluorides on a variety of tree species and the subsequent damage to forests. Infrared aerial photographs of forest near an aluminium plant in northwestern Montana, USA, revealed widespread damage to nearly 75 000 ha of pine (Pinus sp.) trees. The authors identified several factors that suggest fluoride as the cause, including proximity to the plant, high fluoride concentrations in conifer leaves and the presence of typical fluoride-induced leaf lesions (Carlson, 1978). Bunce (1984) reported that the growth reduction of forest (2800 m3/year) estimated for the years from the establishment of an aluminium smelter (Kitimat, British Columbia, Canada) to 1974 declined by 78% for the period 1974–1979. The improvement in growth corresponded to a 79% decline in fluoride content of foliage and a 64% decline in fluoride emissions.
The effects of fluoride emissions from a phosphorus plant (the plant closed in 1989) in Long Harbour, Newfoundland, Canada, on the conifers balsam fir (Abies balsamea), black spruce (Picea mariana) and larch (Larix laricina) were monitored at sites downwind from the plant during the summer of 1982 (Sidhu & Staniforth, 1986). Mean fluoride concentrations ranged between 11.4 µg/m3 at a distance of 1.4 km from the source and 0.08 µg/m3 18.7 km from the source. At the closest site, seed production in balsam fir, black spruce and larch was impaired by 76.4%, 87.4% and 100%, respectively, compared with controls. At 10.3 km from the source (mean ambient air concentration 0.9 µg fluoride/m3), the effects included reduction of 3–10% in seed size and 23–30% in cone size and a decrease of 17–72% (varied with species) in the number of cones per tree. A significant negative correlation was observed between atmospheric fluoride levels and seed production for all three species. A significant negative correlation was also observed between foliar injury in the three species (chlorosis and necrosis) and mean atmospheric fluoride levels. Toxic effects of fluoride on these conifer species have resulted in their replacement by more tolerant hardwood species, such as birch (Betula sp.) and alder (Alnus sp.). Reproductive and vegetative characteristics of raspberry (Rubus sp.) and blueberry (Vaccinium sp.) were monitored at six sites downwind from the plant. Within 1.4 km of the plant, flower mortality was 89% for blueberry and 78% for raspberry, compared with 27% and 26%, respectively, for a control site; there were also significant decreases in size, number and dry weight of fruit. Fluoride concentrations in foliage were 403 mg/kg for raspberry and 216 mg/kg for blueberry, compared with 8 and 9 mg/kg, respectively, for the control site (Staniforth & Sidhu, 1984).
Taylor & Basabe (1984) established correlations between fluoride concentrations in pine needles (Douglas-fir, Pseudotsuga menziesii) and annual growth increments, wind pattern, distance from fluoride source (aluminium smelter) and hydrogen fluoride concentrations in emissions. The authors noted that visible fluoride symptoms and over 40% growth reduction occurred in trees that were accumulating fluorides below established "injury threshold levels." They suggested that synergism between hydrogen fluoride and sulfur dioxide may have given rise to these reduced threshold levels.
Vike & Håbjr rg (1995) recorded fluoride content and leaf injury in a variety of plant species growing in the vicinity of aluminium smelters in Norway. Scots pine (Pinus sylvestris) was the most sensitive species, showing leaf injury symptoms at a leaf fluoride content of less than 50 mg/kg at the northern-most locality and <100 mg/kg under southern maritime conditions. Broad-leaved species such as downy birch (Betula pubescens), goat willow (Salix caprea) and European mountain-ash (Sorbus aucuparia) showed similar symptoms at concentrations of 100 and 170 mg/kg for the two sites, respectively. In a later study, Vike (1999) reported that leaf injury appeared at leaf fluoride content levels as low as 30 mg/kg, and damage was restricted to within 2 km of the emission sources. Regression analysis showed a positive correlation between leaf injury and fluoride content of leaves within a locality, but great variation between localities. The author concluded that the establishment of tolerable emission levels must take into account local dispersal patterns and climatic conditions.
Ivinskis & Murray (1984) found that reductions in photosynthetic capacity, chlorophyll a and b and leaf area of grey gum (Eucalyptus punctata) were all significantly correlated with leaf fluoride content, fluoride in air and distance from an aluminium smelter. The authors also found that dusky-leaved ironbark (Eucalyptus fibrosa), a species believed to be tolerant of fluoride, showed no significant differences between sites for any of the variables except leaf area.
Mayer et al. (1988) conducted a 3-year study on honeybees (Apis mellifera) in Puyallup Valley, Washington, USA, near an aluminium plant. The mean fluoride content of bees from a site 0.8 km downwind of the plant ranged from 82 to 261 mg/kg dry weight. No significant effect of fluoride on brood survival, brood development or honey production was found.
Van Toledo (1978) found that the number of avian species near fluoride-emitting aluminium factories in Europe was depressed. They speculated that the reductions were due to fluoride emissions. Newman (1977) believed that the nesting density of house martins (Delichon urbica) was reduced near an aluminium plant with high fluoride emissions. However, there were many other air pollutants present, and so it is difficult to establish a causal relationship with fluoride in particular. Henny & Burke (1990) monitored black-crowned night herons (Nycticorax nycticorax) living near a phosphate-processing complex in Idaho, USA. Fluoride in femurs ranged from 540 to 11 000 mg/kg ash weight and increased with age. The authors studied bone strength, but, because of the strong relationship between age and fluoride concentrations, it proved difficult to separate the effects of fluoride from those of age.
The original findings of fluoride effects on mammals were from studies in the field on domestic animals such as sheep (Moule, 1944; Harvey, 1952; Peirce, 1952) and cattle (Rand & Schmidt, 1952; Neeley & Harbaugh, 1954; Burns & Allcroft, 1964; Allcroft & Burns, 1968; Krook & Maylin, 1979), and more recent studies have shown this to be an ongoing problem in some areas (Jubb et al., 1993; Fidanci et al., 1998; Swarup et al., 1998; Choubisa, 1999; Patra et al., 2000). Fluoride can be taken up from vegetation, soil and drinking-water. Incidents involving domesticated animals have originated both from natural fluoride sources, such as volcanic eruptions and the underlying geology, and from anthropogenic sources, such as mineral supplements, fluoride-emitting industries and power stations. Symptoms of chronic fluoride toxicity include emaciation, stiffness of joints and dental and skeletal fluorosis. Other effects include lowered milk production and detrimental effects on the reproductive capacity of animals. Guidance is available on fluoride tolerance levels in feed and water for domesticated animals based on clinical signs and lesions (Shupe & Olson, 1983; Cronin et al., 2000) (see section 9.1.3.3).
Investigations of the effects of fluoride on wildlife have focused on impacts on the structural integrity of teeth and bone. Most observations have involved large herbivores. For example, dental and skeletal lesions were found in mule deer (Odocoileus hemionus), elk (Cervus canadensis) and American bison (Bison bison) exposed to elevated levels (no specific anthropogenic sources identified) of fluoride in Utah, Idaho, Montana and Wyoming, USA (Shupe et al., 1984). Black-tailed deer (Odocoileus hemionus columbianus) near an aluminium smelter in Washington, USA, were found to have dental disfigurement, with the premolars of one individual deer being worn down to the gumline. Fluoride levels in ribs ranged from 2800 to 6800 mg/kg on a fat-free basis, compared with control deer, which had levels ranging from 160 to 460 mg/kg (Newman & Yu, 1976). Lameness of deer adjacent to an aluminium smelter in Montana, USA, was noted by Kay et al. (1975). They speculated that fluorosis was the cause of a change in age structure of the mule deer population. Levels in mandibles of the mule deer and white-tailed deer (Odocoileus virginianus) ranged from 1100 to 7000 and from 3400 to 9800 mg/kg, respectively, on a fat-free basis, whereas those of control deer were less than 200 mg/kg. White-tailed deer inhabiting an area adjacent to an aluminium smelter in South Carolina, USA, exhibited dental fluorosis but no osteofluorosis (Suttie et al., 1987). Mean levels in mandibles of deer older than 2.5 years were 286 mg/kg ash weight before opening of the smelter and 1275 mg/kg 3 years after start-up.
Karstad (1967) found that mandibular bone fluoride contents of 4300–7125 mg/kg in mule deer (Odocoileus hemionus) near an industrial complex were associated with pitting and black discoloration of teeth, abnormal tooth wear and fractures of teeth and jaw bones. All red deer (Cervus elaphus) yearlings, sampled near Norwegian aluminium smelters, with mandibular fluoride levels exceeding 2000 mg/kg were found to have dental fluorosis. Gross osteofluorosis was observed in deer with mandibular fluoride residues of greater than 8000 mg/kg (Vikr ren & Stuve, 1996b). Pathological alterations in the teeth of roe deer (Capreolus capreolus) in Germany were diagnosed as dental fluorosis. The diagnosis was confirmed by analysis of mandibles, which contained significant levels of fluoride (Kierdorf, 1988). The dental fluorosis was found to occur mainly in the heavily industrialized Ruhr area of Germany. H. Kierdorf et al. (1996) and U. Kierdorf et al. (1996) monitored mandibular bone fluoride concentrations and frequency and intensity of fluoride-induced dental lesions in red deer in the Czech Republic and Germany (Figure 4). The frequency and severity of dental fluorosis were positively correlated with bone fluoride levels. Detailed macroscopic, microradiographic and scanning electron microscopic methods have revealed structural changes in fluorosed dental enamel of both roe and red deer (U. Kierdorf et al., 1993, 1996). Corresponding changes have also been described in fluorosed enamel of wild boars (Sus scrofa) from fluoride-polluted areas (H. Kierdorf et al., 2000). Kierdorf & Kierdorf (1997) concluded that periods of especially elevated plasma fluoride levels in chronically fluoride-stressed deer can cause a disruption in the function of secretory ameloblasts similar to that following acute fluoride dosing in laboratory rodents. Morphological imaging has revealed a decreased width and an increased porosity of the antler cortex in deer from highly fluoride polluted areas (U. Kierdorf et al., 2000). Kierdorf et al. (1997) concluded that increased fluoride exposure of deer leads to reduced mineral content and mineral density of antler bone and that it is the rapidity of their growth and mineralization that makes antlers especially susceptible to fluoride action. The occurrence of osteofluorotic alterations has also been diagnosed in red deer (C. elaphus) with bone fluoride concentrations greater than 4000 mg/kg dry weight (Schultz et al., 1998). The authors concluded that in addition to fluoride-induced dental lesions, the occurrence of marked periodontal disease and tooth loss is an important factor responsible for a reduction of life expectancy in severely fluorotic wild red deer. It has been demonstrated that in the case of regional, long-term fluoride pollution, dental fluorosis can be used as a sensitive biomarker of fluoride exposure in deer and thus as an indicator of the level of environmental contamination by fluorides (U. Kierdorf & Kierdorf, 1999).
|
Fig. 4. Row of permanent mandibular cheek teeth (from right to left, 2nd, 3rd, 4th premolar, 1st, 2nd, 3rd molar) of a red deer (Cervus elaphus) exhibiting pronounced dental fluorosis. The premolars and the M3 exhibit enamel discoloration and increased wear. Note destruction of the tooth crown in the P3 (asterisk) and dysfunctional crown shape of the M3 (arrow). Due to the increased wear, the alveolar process has regressed in the region of P3 and P4 (arrowheads). |
Exposure of predatory wildlife is often minimal, as fluoride is largely unavailable to those mammalian and avian predators that do not digest bone. For example, barn-owls regurgitate pellets that contain virtually intact skeletons of their prey (Thomson, 1987).
Walton (1987c) found that severe dental damage in field voles (Microtus agrestis) and wood mice (Apodemus sylvaticus) was evident only within 200–300 m (downwind) of an aluminium smelter. A significant increase in tooth wear was noted in moles (Talpa europaea) at between 4 and 15 km from the smelter. Radiographs of the skeletons of voles and mice with bone fluoride levels of up to 15 000 mg/kg dry weight showed no changes when compared with rodents from control sites. Paranjpe et al. (1994) identified fluorosis in cotton rats (Sigmodon hispidus) at a petrochemical waste site. Approximately 80% of the cotton rats collected by Schroder et al. (1999) at the same site were found to have dental fluorosis. Mean bone fluoride and mean soil total fluoride levels were 1515 and 1954 mg/kg, respectively. Bone fluoride was found to be an accurate predictor of dental fluorosis when it was low (<1000 mg fluoride/kg; no fluorosis) or high (>3000 mg fluoride/kg; fluorosis), but not when it was intermediate (Schroder et al., 2000). Field voles (M. agrestis) and bank voles (Clethrionomys glareolus) showed severe dental lesions on incisor and molar teeth near a chemical works (549 mg total fluoride/kg dry weight of vegetation) and an aluminium smelter (187 mg fluoride/kg vegetation), with less marked damage at a mine tailings dam (80 mg fluoride/kg vegetation) (Boulton et al., 1994b). Boulton et al. (1999) reported that the severity of lesions in both incisor and molar teeth of field voles (M. agrestis) was positively correlated with the respective tissue fluoride concentrations.
Individuals are exposed to some level of fluoride from natural and/or anthropogenic sources. The consumption of foodstuffs, drinking-water and other beverages is generally the principal route of exposure. Significant intake may also occur in younger children from the ingestion of fluoridated dentifrice and supplements. Infants can receive small amounts of fluoride from breast milk. The inhalation of fluorides in air usually makes only a minor contribution to the overall total intake. High intakes of fluoride have been noted in geographical areas worldwide where the surrounding environment is rich in natural fluoride. In these areas, drinking-water, foodstuffs and the inhalation of indoor air where coal rich in fluoride is used for heating and cooking all contribute to the overall intake. Exposures might be increased somewhat in the vicinity of certain anthropogenic sources of fluoride. In certain occupational environments, the inhalation of workroom air containing fluoride would contribute to higher than typical ambient exposures.
Estimates of daily fluoride intake by children in Canada range from <0.01 mg (<1 µg/kg body weight for infants) to 2.1 mg (160 µg/kg body weight for children up to 4 years of age and 80 µg/kg body weight for adolescents aged 5–11 years). For adults, estimates of daily fluoride intake range from 2.2 to 4.1 mg (approximately 30 to 60 µg/kg body weight, assuming an average body weight of 64 kg). For individuals residing in certain geographical areas rich in natural fluoride, daily intakes as high as 27 mg (420 µg/kg body weight, assuming an average body weight of 64 kg) have been reported. The estimates of total fluoride intake usually do not take into account its bioavailability (e.g., the degree of fluoride absorption from fluoride-containing food items). Such information is needed in order to derive accurate estimates of daily total fluoride intake in individuals living in fluoridated as well as non-fluoridated areas and in areas of endemic fluorosis.
Fluoride has both positive and negative effects on human health, similar to essential elements. Compared with many other chemicals, there is a relatively narrow range between intakes associated with beneficial effects and exposures causing adverse effects. The exposure to low levels of fluoride in drinking-water (i.e., approximately 1 mg/litre) has long been known to have a beneficial effect on the reduction of dental caries. Clinical use of fluoride in the treatment of osteoporosis (at doses generally greater than 25 mg/day) has been investigated in a number of studies.
Based on the available data, it is evident that skeletal and dental effects related to fluoride exposure in humans occur at doses or intakes lower than those associated with other potential adverse tissue- or organ-specific effects. This is likely a consequence of the accumulation of fluoride almost exclusively in bones and teeth. In children, intakes of fluoride associated with beneficial effects on dentition overlap with those that lead to an increased prevalence of dental fluorosis. Consequently, public health programmes have sought to maximize the beneficial effects of fluoride on dental health, while at the same time minimizing its adverse fluorotic effects on the teeth.
Effects on the skeleton are the most consistent and best characterized of the toxic responses to fluoride and are considered to have direct public health relevance. The skeletal changes that have been observed in a number of animal species (i.e., cattle, sheep, poultry, rodents) exposed to fluoride are not inconsistent with effects observed in humans, although data from such studies have not been utilized in the assessment of human exposure–response.
Case reports or descriptive ecological human studies of skeletal fluorosis or osteosclerosis or studies on the occurrence of endemic (crippling) skeletal fluorosis in which the levels of fluoride are naturally very high provide little quantitative information useful in establishing an adverse effect level associated with total fluoride intake. In such studies, nutrition, the intake of fluoride (and additional minerals) from other sources and potentially confounding concomitant exposures, all of which have been suggested to play a role in the etiology of this disease (for a review, see WHO, 1984; US EPA, 1985), were not adequately documented. There is also limited quantitative information on exposure to fluoride and the development of skeletal effects (osteosclerosis or fluorosis) in occupationally exposed workers (see Hodge & Smith, 1977; Grandjean, 1982; Chan-Yeung et al., 1983a; Czerwinski et al., 1988). Inconsistent results of inherently limited cross-sectional studies of small populations exposed to often unspecified concentrations of fluoride in the vicinity of industrial sources (cited in Hodge & Smith, 1977) contribute little to an assessment of exposure–response for skeletal effects associated with exposure to fluoride. Information obtained from clinical studies, in which sodium fluoride was administered to patients for the treatment of osteoporosis, is inadequate, owing to the nature of the protocols, characteristics of the patients in these studies and inconsistencies in the results. Moreover, these clinical studies were undertaken to assess the considered beneficial effects of fluoride (i.e., its capacity to increase bone mass and decrease vertebral fractures), rather than its potential to produce adverse effects after long-term exposure.
Estimating a putative effect level for the development of skeletal fluorosis (or related changes) in humans exposed to fluoride is further complicated by differences in the radiological diagnosis of early-stage skeletal fluorosis among health care professionals (Chan-Yeung et al., 1983a), as well as by the multiplicity of factors that may influence the amount of fluoride deposited in the bone, and hence the severity of the disease. In case accounts, the development of crippling skeletal fluorosis was attributed to an intake of approximately 230–310 µg fluoride/kg body weight per day in adults weighing 64 kg (US DHHS, 1991). Skeletal fluorosis has been observed in cryolite workers having estimated (occupational) intakes of approximately 310–1250 µg fluoride/kg body weight per day in adults weighing 64 kg (Grandjean, 1982).
Evidence from ecological studies suggests that there may be an association between the consumption of fluoridated drinking-water and an increased incidence of hip fracture (based on hospitalization rates), particularly among the elderly. These results should be interpreted with caution, however, in view of the limitations of epidemiological investigations of this design. Moreover, owing to the lack of data on individual exposure in such studies, it is difficult to derive meaningful conclusions concerning the exposure–response relationship for possible skeletal effects associated with exposure to fluoride from these studies.
Although the relative risk of hip, wrist or spinal fracture was increased in some groups of women residing in an elevated-fluoride community (with drinking-water containing 4 mg/litre, and having an estimated [mean] intake of approximately 72 µg fluoride/kg body weight per day) compared with those in a control community (with drinking-water containing 1 mg fluoride/litre) (Sowers et al., 1986, 1991), the intake of fluoride by women in the elevated-fluoride community was likely underestimated (since it was derived solely from the amount of water-based beverages consumed), and the level of calcium in the drinking-water from the elevated-fluoride community was approximately 25% of the level in the control community.
Other studies, including analytical investigations, have not identified an increased risk of skeletal fracture associated with the consumption of drinking-water containing elevated levels of fluoride (Madans et al., 1983; Simonen & Laitinen, 1985; Arnala et al., 1986; Jacobsen et al., 1993; Suarez-Almazor et al., 1993; Kröger et al., 1994; Cauley et al., 1995; Lehmann et al., 1998; Hillier et al., 2000).
Only two analytical studies evaluated the risk of fracture across a range of fluoride exposures. Among 144 627 Finnish men and women, no association was observed between hip fractures and the well-water fluoride concentration, when all age groups were considered. However, in women 50–65 years of age at the start of follow-up, the relative risk of hip fracture increased with increasing well-water fluoride concentrations in the categories <0.1, 0.1–0.3, 0.3–0.5, 0.5–1.0, 1.1–1.5 and >1.5 mg/litre. The respective relative risk of hip fracture was 1.0, 1.16, 1.31, 1.53 (P < 0.05), 1.24 and 2.09 (P < 0.05) (Kurttio et al., 1999).
Among 8266 Chinese men and women, the risk of all fractures and hip fractures was examined across a range of fluoride exposures (see Table 14 in chapter 8) (Li et al., 2001). There was an increased risk of overall fracture and hip fracture that became statistically significant at drinking-water levels in excess of 4.32 mg fluoride/litre.
Owing to the widespread controlled addition of fluoride to public drinking-water supplies, numerous epidemiological and experimental studies have attempted to address concerns related to the potential carcinogenicity of this substance. There has been no consistent evidence of an association between the consumption of fluoridated drinking-water and the incidence of, or mortality due to, cancer in a large number of ecological studies performed in many countries. Although these results do not support the hypothesis of an association, their considerable limitations preclude firm conclusions from being drawn regarding the carcinogenicity of fluoride in humans. For example, cancer of the bone was not assessed in the majority of these human ecological studies. Case–control studies (Gelberg et al., 1995; Moss et al., 1995) have not identified a relationship between bone cancer and exposure to fluoride.
The incidence of, or death due to, cancer has also been investigated in a number of historic cohort studies of workers exposed to fluoride predominantly in the aluminium smelting industry. While excesses of cancers of different types have been reported in various studies, the only site for which there was excess risk in several investigations is lung cancer; due to confounding by concomitant exposure to known human carcinogens in analytical studies of occupationally exposed workers, the observed excesses cannot be attributed to fluoride exposure per se. Bone cancer was not assessed in the majority of these studies.
Although the weight of evidence does not support the view that fluoride causes cancer in humans, the data on bone cancer are relatively limited.
In early carcinogenicity bioassays conducted by Kanisawa & Schroeder (1969), Taylor (1954) and Tannenbaum & Silverstone (1949), the incidence of tumours in mice administered sodium fluoride (in either the diet or drinking-water) was, in general, not markedly greater than that observed in controls. Owing to inadequate documentation and to numerous methodological shortcomings, however, the results of these investigations do not contribute meaningfully to an assessment of the weight of evidence of the carcinogenicity of (sodium) fluoride.
The administration of drinking-water containing sodium fluoride (in amounts estimated to provide intakes ranging from 0.6 to 9.1 mg fluoride/kg body weight per day) to male and female B6C3F1 mice over a period of 2 years produced a marginal (statistically insignificant) increase in the incidence of hepatoblastoma, compared with the incidence in groups of controls administered drinking-water without added fluoride; however, this minor increase was not considered "biologically significant," since the overall incidence of hepatic tumours (adenoma, carcinoma, hepatoblastoma) was not increased in animals receiving sodium fluoride, and the incidence of all hepatic tumours in these groups of mice was higher than that in previous NTP carcinogenicity bioassays (NTP, 1990). The marginal increase in the incidence of malignant lymphoma in female B6C3F1 mice administered drinking-water containing sodium fluoride was considered not to be compound-related, since the incidence in the high-dose group was similar to that observed in historical controls (NTP, 1990). No other increases in tumour incidence were observed; however, failure to attain the maximum tolerated dose may have reduced somewhat the sensitivity of this study in mice. In a less extensive carcinogenicity bioassay in which the incidence of osteomas in male and female CD-1 mice receiving 11.3 mg fluoride/kg body weight per day in the diet was increased compared with controls (Maurer et al., 1993), the specific role of fluoride in the etiology of the tumours cannot be determined with certainty, owing to the infection of these animals with Type C retrovirus (US DHHS, 1991; Maurer et al., 1993; US NRC, 1993).
The administration of drinking-water containing sodium fluoride (in amounts estimated to provide intakes ranging from 0.2 to 4.5 mg fluoride/kg body weight per day) to F344/N rats over a period of 2 years produced a marginal (not statistically significant) increase in the incidence of oral cavity neoplasms (in males and females) and tumours in the thyroid gland (in males) (NTP, 1990). The squamous cell tumours of the oral cavity were not considered to be compound-related, since the incidence of tumours in the high-dose group was not significantly different from the controls, the incidence of these neoplasms was within the range observed in historical controls and there was no supporting evidence of focal hyperplasia of the oral mucosa (NTP, 1990). The marginal increase in thyroid (follicular cell) tumours was also considered not to be compound-related, since the incidence of these tumours in the high-dose group was not significantly different from the controls, the incidence of these neoplasms was within the range observed in historical controls and the incidence of follicular cell hyperplasia was not increased in fluoride-exposed animals (NTP, 1990). The incidence of osteosarcoma in rats with intakes ranging from 0.8 to 4.5 mg fluoride/kg body weight per day was not significantly increased, compared with controls receiving approximately 0.2 mg fluoride/kg body weight per day (NTP, 1990); however, for the male F344/N rats, it was reported that "the osteosarcomas occurred with a significant dose response trend (P = 0.027, by logistic regression)" (NTP, 1990). In a more limited carcinogenicity bioassay conducted by Maurer et al. (1990), the administration of diets containing sodium fluoride (in amounts estimated to provide intakes ranging from 1.8 to 11.3 mg fluoride/kg body weight per day) to male and female Sprague-Dawley rats over a period of 95–99 weeks produced no significant increase in the incidence of any types of tumours, compared with groups of controls receiving approximately 0.1 mg fluoride/kg body weight per day, although a small number of malignant tumours of the bone were observed.
In assessing the evidence for the carcinogenicity of fluoride derived from studies conducted with laboratory animals, some significance might be attributed to the observation of a dose–response trend in the occurrence of osteosarcomas in male F344/N rats administered sodium fluoride in drinking-water (NTP, 1990). Such a trend associated with the occurrence of a rare tumour in the tissue in which fluoride is known to accumulate cannot be casually dismissed. Moreover, the level of fluoride in the bones of the high-dose group of male rats in the NTP carcinogenicity bioassay, in which a non-significant increase in osteosarcomas was observed, is similar to that measured in humans with a preclinical phase of skeletal fluorosis. However, the biological significance of this dose–response trend is tempered by the lack of statistical significance of the observed excess in the high-dose males in comparison with controls, as well as by the absence of a comparable statistically significant trend in the incidence of osteosarcomas in female F344/N rats or male and female B6C3F1 mice receiving comparable amounts of inorganic fluoride (NTP, 1990). Indeed, the levels of fluoride in the bone of (male and female) F344/N rats and B6C3F1 mice administered sodium fluoride in drinking-water were similar (NTP, 1990). No dose–response trend in the incidence of osteosarcomas was observed in groups of male and female Sprague-Dawley rats administered diets containing sodium fluoride (Maurer et al., 1990), even though the levels of fluoride in the bone in the high-dose animals were greater than those in the male F344/N rats in which there was an increase in osteosarcomas in the NTP (1990) carcinogenicity bioassay; however, there may be variations in sensitivity of the two strains to the effects of fluoride. There is controversy concerning whether or not the osteomas in male and female CD-1 mice observed in the carcinogenicity bioassay conducted by Maurer et al. (1993) should be classified as neoplasms, and a retrovirus (in addition to fluoride) has been implicated in their etiology; however, the significant increases in (the highest-dose) fluoride-exposed versus control groups (in animals infected with retrovirus), in a tissue in which fluoride is known to accumulate, adds some weight, albeit weak, to the evidence of carcinogenicity. Overall, the evidence regarding the carcinogenicity of fluoride in laboratory animals is inconclusive.
There is evidence that fluoride is genotoxic based on the outcome of in vitro studies. Fluoride is not mutagenic in bacterial cells but is clastogenic in human and animal cells exposed in vitro. Sodium fluoride induced recessive lethal mutations in Drosphila melanogaster and cytogenetic damage after intraperitoneal injection in rodents. Generally, however, in studies in which fluoride was administered to laboratory animals by routes of exposure similar to those by which humans are normally exposed (i.e., orally), fluoride had no effect upon the frequency of chromosomal aberrations, micronuclei, sister chromatid exchange, DNA strand breaks or sperm morphology. The mechanism by which fluoride could induce genetic alterations is not known; however, it is not likely due to an interaction between the fluoride ion and DNA. Rather, it may be a secondary effect of the actions of fluoride that result from its inhibition of enzymes involved in DNA synthesis and/or repair.
A few investigations on skeletal fluorosis or the risk of fractures include quantitative estimates of the dose–response relationship. Studies in China and India report an increased prevalence of skeletal fluorosis above the level of 1.4 mg fluoride/litre in drinking-water (Jolly et al., 1968; Choubisa et al., 1997; Xu et al., 1997). However, such studies suffer from limitations: the diagnostic criteria are not always specified or consist of self-reported symptoms, and only drinking-water is considered as a source of exposure. The latter problem is likely to be important, since other studies (Liang et al., 1997; Ando et al., 1998) estimate that, at least in some regions of China and India, the contribution from food can greatly exceed that from water. Therefore, one cannot rule out that high rates of skeletal fluorosis associated with a level greater than 1.4 mg/litre in drinking-water are due to other exposures. While there is a clear excess of skeletal fluorosis in these studies for a total intake of 14 mg/day, the quantitative relationship between total intake of fluoride from different sources and the risk of skeletal fluorosis cannot be estimated because of substantial uncertainties in the prevalence of effects in the range of intakes between 3 and 14 mg/day.
The studies on fractures are also difficult to interpret, but for different reasons:
One exception is represented by a study in China (Li et al., 2001) in which different sources of exposure have been considered and an estimate of total intake is presented. In this study, there is an upward trend for the risk of total fractures above an exposure of 1.45 mg fluoride/litre in drinking-water, but only for the highest level of exposure (i.e., >4.32 mg fluoride/litre in drinking-water) was the relative risk statistically significant (RR = 1.47; P = 0.01). In the concentration range of 1.45–2.19 mg fluoride/litre in drinking-water, corresponding to a total intake of 6.54 mg/day, there was a relative risk for all fractures of 1.17 and for hip fractures of 2.13 (both not statistically significant).
In summary, estimates based on studies from China and India indicate that:
Fluorides are released into the environment naturally through the weathering and dissolution of minerals, in emissions from volcanoes and in marine aerosols. Fluorides are also released into the environment via coal combustion and process waters and waste from various industrial processes, including steel manufacture, primary aluminium, copper and nickel production, phosphate ore processing, fertilizer production and use, glass, brick and ceramic manufacturing, and glue and adhesive production.
Atmospheric fluorides can be transported over large distances as a result of wind or atmospheric turbulence or removed from the atmosphere via wet or dry deposition or hydrolysis. Fluoride concentrations in groundwater are primarily due to dissolution from local geological sources. The transport of fluorides in water is influenced by pH and water hardness and the presence of ion-exchange materials such as clays, and fluoride is usually complexed with aluminium and calcium. Fluoride is not readily leached from soils, with adsorption being strongest at pH 5.5–6.5. Soluble fluorides are bioaccumulated by some aquatic and terrestrial biota; however, no information was identified concerning the biomagnification of fluoride. Terrestrial plants may accumulate fluorides from airborne deposition and, to a lesser extent, via uptake from soil, as fluoride adsorbs strongly to soil. Fluoride accumulates in the exoskeleton of invertebrates and the bone and dental hard tissues of vertebrates.
Surface freshwater fluoride concentrations tend to range from 0.01 to 0.3 mg/litre, whereas typical marine concentrations range from 1.2 to 1.5 mg/litre (Figure 5). Higher concentrations are found in areas where there is geothermal or volcanic activity. Anthropogenic discharges lead to increased levels of fluoride in the environment. Airborne fluoride exists in gaseous and particulate forms, which are emitted from both natural and anthropogenic sources. Mean fluoride concentrations in ambient air are generally less than 0.1 µg/m3. Levels may be slightly higher in urban than in rural locations; however, even in the vicinity of emission sources, the levels of airborne fluoride usually do not exceed 2–3 µg/m3 (Figure 6). Fluoride is a component of most types of soil, with concentrations ranging from 20 to 1000 mg total fluoride/kg in areas without natural phosphate or fluoride deposits and up to several thousand milligrams per kilogram in mineral soils with deposits of fluoride (Figure 7). Reported concentrations of fluoride in vegetation are plotted in Figure 8.
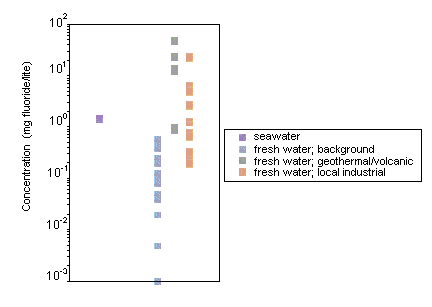
Fig. 5. Reported concentrations of fluoride in surface waters
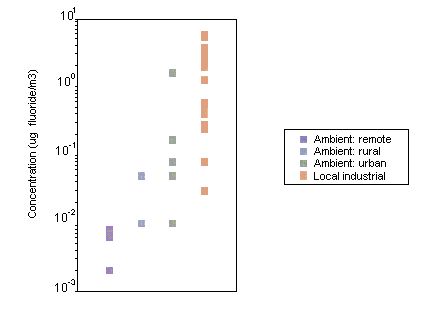
Fig. 6. Reported concentrations of fluoride in air
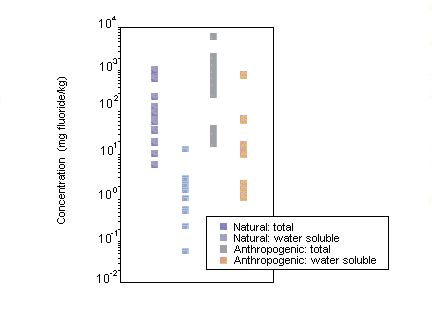
Fig. 7. Reported concentrations of fluoride in soil
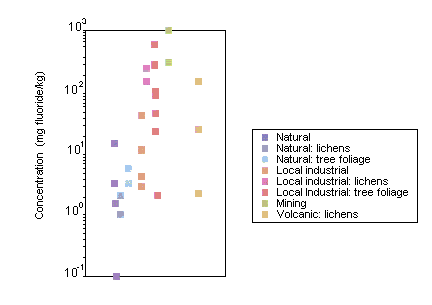
Fig. 8. Reported concentrations of fluoride in vegetation
The most sensitive aquatic invertebrate species tested were the freshwater fingernail clam (Musculium transversum), with an 8-week LC50 of 2.8 mg fluoride/litre, freshwater net-spinning caddisflies (Hydropyschidae), with estimated "safe concentrations" ranging from 0.2 to 1.2 mg fluoride/litre, and the marine brine shrimp (Artemia salina), with significant impairment of growth being observed in a 12-day static test at 5 mg/litre. For fish, 20-day LC50s for rainbow trout (Oncorhynchus mykiss) ranged from 2.7 to 4.7 mg fluoride/litre; in another study under different conditions (such as higher water hardness), a "safe concentration" of 5.1 mg/litre was estimated. The hatching of fish eggs (Catla catla) was delayed at fluoride concentrations greater than or equal to 3.6 mg fluoride/litre. Behavioural experiments on adult Pacific salmon in soft-water rivers indicate that changes in water chemistry, due to an increase in the fluoride concentration to 0.5 mg/litre, can adversely affect migration; migrating salmon are extremely sensitive to changes in the water chemistry of their river of origin. (Reported toxicity test results for a range of freshwater organisms are plotted in Figure 9.)
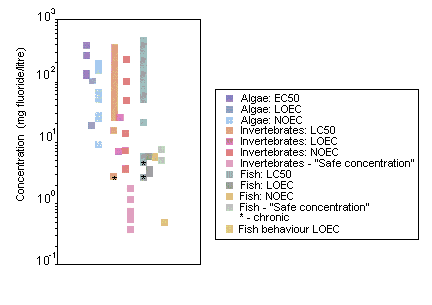
Fig. 9. Reported toxicity of fluoride to freshwater organisms
In laboratory studies, fluoride seems to be toxic for microbial processes at concentrations found in moderately fluoride-polluted soils; similarly, in the field, accumulation of organic matter in the vicinity of smelters has been attributed to severe inhibition of microbial activity by fluoride.
The LOELs for leaf necrosis in terrestrial plants were at fluoride concentrations of 0.2–0.4 µg/m3. On the basis of effect concentrations and no-effect concentrations for fumigation experiments, a relationship was derived between the NOEC and the exposure period. If this relationship is extrapolated to two growing seasons, the derived NOEC is approximately 0.2 µg fluoride/m3. Several short-term solution culture studies have identified a toxic threshold for fluoride ion activity ranging from approximately 50 to 2000 µmol fluoride/litre. Toxicity is specific not only to plant species, but also to ionic species of fluoride, where some aluminium–fluoride complexes present in solution culture may be toxic at activities of 22–357 µmol fluoride/litre, whereas hydrogen fluoride is toxic at activities of 71–137 µmol/litre. Due to the complexity of fluoride availability in soil and the high concentrations generally required for plant toxicity, no soil effect range was defined.
The growth rate of young starlings was significantly reduced at a fluoride concentration of 13 mg/kg body weight. Fluoride has been shown to cause adverse effects on domesticated animals in both the laboratory and the field. Recent studies have shown this to be an ongoing problem. Tolerance levels have been identified for domesticated animals, with the lowest values for dairy cattle at 30 mg/kg feed or 2.5 mg/litre drinking-water. The lowest dietary concentration of fluoride to cause fluorosis in white-tailed deer (Odocoileus virginianus) was 35 mg/kg. Field surveys have found that bone fluoride concentrations of >2000 mg/kg in young deer and >3000 mg/kg in cotton rats (Sigmodon hispidus) are associated with fluorosis in all individuals. Osteofluorotic alterations have been diagnosed in deer with bone fluoride concentrations of >4000 mg/kg.
In the freshwater environment, natural fluoride concentrations are usually lower than those expected to cause toxicity in aquatic organisms. However, aquatic organisms might be adversely affected in the vicinity of anthropogenic discharges. Fluoride toxicity is dependent on water hardness, with organisms living in soft-water environments at greater risk than those in hard-water areas. Because of the particular sensitivity of some freshwater invertebrates and Pacific salmon, fluoride concentrations in soft-water ecosystems (low in calcium) should not be increased by anthropogenic inputs to values exceeding 0.5 mg fluoride/litre. Aquatic organisms that inhabit areas with high natural fluoride levels from geothermal or volcanic activity would be expected to be adapted to such conditions.
Comparing the long-term NOEC for plants with air concentrations reveals that sensitive species growing near anthropogenic sources of fluoride are at risk. In the field, anthropogenic sources of fluoride have been shown to be correlated with damage to local terrestrial plant communities. However, these adverse effects are often difficult to attribute to fluoride alone, due to the presence of other atmospheric pollutants. Fluoride is generally strongly adsorbed by soils. Consequently, plant uptake via this pathway is relatively low, and leaching of fluoride through soil is minimal. However, high fluoride concentrations in soil from natural and anthropogenic sources, combined with soil properties that minimize fluoride adsorption (below pH 5.5, low carbon and clay content), can lead to increased plant availability of fluoride. Plant availability is also dependent on the ionic species of fluoride present in the soil solution. Above soil pH 5.5, fluoride in solution is predominantly the free fluoride ion, CaF+ or MgF+, which is relatively less available to uptake via the plant root. Below soil pH 5.5, aluminium–fluoride complexes can become dominant in soil solution, potentially increasing plant availability of fluoride. However, plant toxicity from uptake of fluoride from soil is rare.
Concentrations of fluoride in vegetation in the vicinity of fluoride emission sources, such as aluminium smelters, can be higher than the lowest dietary effect concentration reported for mammals in laboratory experiments. Fluorosis in domesticated animals has been reported. Incidents have originated both from natural fluoride sources, such as volcanic eruptions and the underlying geology, and from anthropogenic sources, such as fluoride-emitting industries and power stations. Guidance is available on fluoride tolerance levels in feed and water; however, there are still some areas reporting fluorosis incidents in livestock due to uptake of fluoride-rich mineral supplements and drinking-water. Furthermore, there is a potential risk from fluoride-contaminated pasture and soil ingestion due to the long-term use of phosphate fertilizers containing fluoride as an impurity. Fluoride-induced effects, such as lameness and tooth damage, have also been reported in wild ungulates, such as deer, and in small mammals close to anthropogenic sources of fluoride. Several studies have demonstrated that wild deer and rodents are sensitive bioindicators of environmental pollution by fluorides and can, therefore, be used as sentinels for the identification of fluoride hazards to the environment. Within some areas of Europe, where effective emission control measures have been introduced following clean air legislation, the bone fluoride levels in deer have fallen by around 70% in the last 15–20 years. Based on the correlation between bone fluoride levels and fluorosis, it can be assumed that the prevalence and severity of fluoride-induced effects in the wild deer population in these areas will probably have fallen at a similar rate. However, it should be noted that in other regions of Europe, fluoride discharges to the environment have not shown the same downward trend during this period, and this is reflected in the bone fluoride levels of mammals from these areas.
All organisms are exposed to various levels of fluoride from natural and/or anthropogenic sources. Very high intakes have been observed in areas worldwide in which the environment is rich in natural fluoride and where groundwater high in fluoride is consumed.
Fluoride in dental products may form a source of exposure for many individuals. Increased exposures might be experienced in the vicinity of anthropogenic point sources.
Fluoride has both beneficial and detrimental effects on human health, with a narrow range between the intakes associated with its beneficial and adverse health effects.
Effects on the teeth and skeleton are observed at exposures below those associated with the development of other organ- or tissue-specific adverse health effects. Effects on the bone are considered the most relevant to assessing the adverse effects of long-term exposure of humans to fluoride.
Skeletal fluorosis is a crippling disability affecting millions of people in various regions of Africa, India and China, which has major public health and socioeconomic impact. Intake of fluoride in the water and/or foodstuffs is the primary causative factor in the incidence of endemic skeletal fluorosis. In parts of China, a large number of people have been shown to be impacted by the indoor burning of coal with a high fluoride content. There are few data from which to estimate total exposure to and the bioavailability of fluoride, and there are inconsistencies in the characterization of its adverse effects.
There is clear evidence from India and China that skeletal fluorosis and an increased risk of bone fractures occur at a total intake of 14 mg fluoride/day, and there is evidence suggestive of an increased risk of bone effects at total intakes above about 6 mg fluoride/day.
Excess exposure to bioavailable fluoride constitute a risk to aquatic and terrestrial biota.
Fluoride-sensitive species can be used as sentinels for the identification of fluoride hazards to the environment.
Considerations should be given to the levels of fluoride and the means of application required to maximize the beneficial effects of fluoride while minimizing the potential for adverse effects on the skeleton and teeth.
It is recommended that international and national agencies identify areas in which health effects related to fluoride are found, identify the primary sources of fluoride exposure and take appropriate action(s) to reduce exposure.
It is recommended that international and national agencies support research to better characterize total fluoride exposure, exposure-health relationships and the various factors that modify and influence these.
In areas exposed to increased fluoride from anthropogenic sources, fluoride levels in the environment should be monitored for changes using appropriate bioindicators.
There is a need to improve knowledge on the accumulation of fluoride in organisms and on how to monitor and control this.
The biological effects associated with fluoride exposure should be better characterized.
There is a need:
Additional research needs include studies on the interactions of fluoride with nuclear proteins (i.e., histones) and polyamines as well as on factors influencing the membrane transport of fluoride.
There is a need:
Fluoride (used in drinking-water) is considered to be "not classifiable as to its carcinogenicity to humans" (Group 3) in the classification scheme of the International Agency for Research on Cancer (IARC, 1987).
The WHO recommended guideline value for fluoride in drinking-water is 1.5 mg/litre (WHO, 1993, 1996b). It was also noted that "in setting national standards for fluoride, it is particularly important to consider climatic conditions, water intake and intake of fluoride from other sources (e.g., from food and air). In areas with high natural fluoride levels, it is recognized that the guideline value may be difficult to achieve in some circumstances, with the treatment technology available" (WHO, 1996b).
An expert consultation of the WHO on trace elements in human nutrition and health (WHO, 1996c) categorized fluoride among "potentially toxic elements, some of which may nevertheless have some essential functions at low levels." Fluoride was regarded as "essential," since the consultation "considered resistance to dental caries to be a physiologically important function." The consultation indicated that total intakes at 1, 2 and 3 years of age "should, if possible, be limited to 0.5, 1.0 and 1.5 mg/day, respectively," with not more than 75% coming in the form of soluble fluorides from drinking-water. It was also noted that "adult intakes exceeding 5 mg of fluoride per day from all sources probably pose a significant risk of skeletal fluorosis."
Aardema M, Gibson D, & LeBoeuf R (1989a) Sodium fluoride induced chromosome aberrations in different stages of the cell cycle: a proposed mechanism. Mutat Res, 223: 191–203.
Aardema M, Gibson D, & LeBoeuf R (1989b) The kinetics of sodium fluoride-induced endoreduplication in Chinese hamster ovary cells. Environ Mol Mutagen, 14(suppl 15): 3 (abstract).
Abdennebi EH, Fandi R, & Lamnaouer D (1995) Human fluorosis in Morocco: analytical and clinical investigations. Vet Hum Toxicol, 37: 465–468.
Adelung D, Bossmann K, & Rossler D (1985) The distribution of fluoride in some Antarctic seals. Polar Biol, 5: 31–34.
Adelung D, Buchholz F, Culik B, & Keck A (1987) Fluoride in tissues of krill Euphausia superba Dana and Meganyctiphanes norvegica M. Sars in relation to the moult cycle. Polar Biol, 7(1): 43–50.
Afseth J, Ekstrand J, & Hagelid P (1987) Dissolution of calcium fluoride tablets in vitro and bioavailability in man. Scand J Dent Res, 95: 191–192.
Ahn HW & Jeffery EH (1994) Effect of aluminum on fluoride uptake by Salmonella typhimurium TA98; implications for the Ames mutagenicity assay. J Toxicol Environ Health, 41: 357–368.
Ahn H, Fulton D, Moxon D, & Jeffery E (1995) Interactive effects of fluoride and aluminum uptake and accumulation in bones of rabbits administered both agents in their drinking water. J Toxicol Environ Health, 44: 337–350.
Akbar-Khanzadeh F (1995) Exposure to particulates and fluorides and respiratory health of workers in an aluminum production potroom with limited control measures. Am Ind Hyg Assoc J, 56: 1008–1015.
Akhundow WY, Aliew AA, Kulgawin AH, & Sariva TN (1981) [Effect of complex and separate exogenous administration of vitamins on the level of chromosome aberrations in rats induced by sodium fluoride in a subacute experiment.] Izv Akad Nauk Az SSR, Ser Biol Nauk, 4: 3–5 (in Russian).
Albanese R (1987) Sodium fluoride and chromosome damage (in vitro human lymphocytes and in vivo micronucleus assays). Mutagenesis, 2: 497–499.
Alexandersen P, Riis BJ, & Christiansen C (1999) Monofluorophosphate combined with hormone replacement therapy induces a synergistic effect on bone mass by dissociating bone formation and resorption in postmenopausal women: a randomised study. J Clin Endocrinol Metab, 84: 3013–3020.
Alhava E, Olkkonen H, Kauranen P, & Kari T (1980) The effect of drinking water fluoridation on the fluoride content, strength and mineral density of human bone. Acta Orthop Scand, 51: 413–420.
Aliev AA & Babaev DA (1981) Cytogenetic activity of vitamins in bone marrow cells of rat femurs under conditions of induction of mutations by sodium fluoride. Cytol Cytogen (USSR), 15: 15–18.
Aliev A, Dzhafarova S, Medzhidov M, & Asadova A (1988) Investigation of the phenomenon and substantiation of the mechanism of antimutagenesis under conditions of the effect of sodium fluoride. Cytol Genet, 22: 24–27.
Allcroft R & Burns KN (1968) Fluorosis in cattle in England and Wales: incidence and sources. Fluoride, 1: 50–53.
Amundson RG, Belsky AJ, & Dickie RC (1990) Fluoride deposition via litterfall in a coastal-plain pine plantation in South Carolina. Water Air Soil Pollut, 50: 301–310.
Anasuya A & Paranjape PK (1996) Effect of parboiling on fluoride content of rice. Fluoride, 29: 193–201.
Anasuya A, Bapurao S, & Paranjape PK (1996) Fluoride and silicon intake in normal and endemic fluorotic areas. J Trace Elem Med Biol, 10: 149–155.
Andersen A, Dahlberg B, Magnus K, & Wannag A (1982) Risk of cancer in the Norwegian aluminum industry. Int J Cancer, 29: 295–298.
Ando M, Tadano M, Asanuma S, Tamura K, Matsushima S, Watanabe T, Kondo T, Sakurai S, Ji R, Liang C, & Cao S (1998) Health effects of indoor fluoride pollution from coal burning in China. Environ Health Perspect, 106: 239–244.
Andrews S, Cooke J, & Johnson M (1982) Fluoride in small mammals and their potential food sources in contaminated grasslands. Fluoride, 15: 56–63.
Andrews SM, Cooke JA, & Johnson MS (1989) Distribution of trace element pollutants in a contaminated ecosystem established on metalliferous fluorspar tailings. 3: Fluoride. Environ Pollut, 60: 165–179.
Angelovic JW, Sigler WF, & Neuhold JM (1961) Temperature and fluorosis in rainbow trout. J Water Pollut Control Fed, 33(4): 371–381.
Antia NJ & Klut ME (1981) Fluoride addition effects on euryhaline phytoplankter growth in nutrient-enriched seawater at an estuarine level of salinity. Bot Mar, 24: 147–152.
Aoba T (1997) The effect of fluoride on apatite structure and growth. Crit Rev Oral Biol Med, 8: 136–153.
Aplin TEH (1968) Poison plants of Western Australia. The toxic species of the genera Gastrolobium and Oxylobium heart-leaf poison (Gastrolobium bilobum), river poison (Gastrolobium forrestii), Stirling Range poison (Gastrolobium velutinumi). J Dep Agric West Aust, 9(2): 69–74.
Ares J (1990) Fluoride–aluminium water chemistry in forest ecosystems of central Europe. Chemosphere, 21(4/5): 597–612.
Ares JO, Villa A, & Gayoso AM (1983) Chemical and biological indicators of fluoride input in the marine environment near an industrial source (Argentina). Arch Environ Contam Toxicol, 12: 589–602.
Armstrong B, Tremblay C, Cyr D, & Thériault G (1986) Estimating the relationship between exposure to tar volatiles and the incidence of bladder cancer in aluminum smelter workers. Scand J Work Environ Health, 12: 486–493.
Armstrong B, Tremblay C, Baris D, & Theriault G (1994) Lung cancer mortality and polynuclear aromatic hydrocarbons: a case–cohort study of aluminum production workers in Arvida, Quebec, Canada. Am J Epidemiol, 1: 250–262.
Arnala I, Alhava E, Kivivuoir R, & Kauranen P (1986) Hip fracture not affected by fluoridation. Acta Orthop Scand, 57: 344–346.
Arnesen AKM (1998) Effect of fluoride pollution on pH and solubility of Al, Fe, Ca, Mg, K and organic matter in soil from Årdal (western Norway). Water Air Soil Pollut, 103: 375–388.
Arnesen AKM & Krogstad T (1998) Sorption and desorption of fluoride in soil polluted from the aluminium smelter at Årdal in western Norway. Water Air Soil Pollut, 103: 357–373.
Aschengrau A, Zierler S, & Cohen A (1989) Quality of community drinking water and the occurrence of spontaneous abortion. Arch Environ Health, 44: 283–290.
Aschengrau A, Zierler S, & Cohen A (1993) Quality of community drinking water and the occurrence of late adverse pregnancy outcomes. Arch Environ Health, 48: 105–113.
ATSDR (1993) Toxicological profile for fluorides, hydrogen fluoride, and fluorine. Atlanta, Georgia, US Department of Health and Human Services, Agency for Toxic Substances and Disease Registry (TP-91/17).
Augenstein W, Spoerke D, Kulig K, Hall A, Hall P, Riggs B, Saadi M, & Rumack B (1991) Fluoride ingestion in children: A review of 87 cases. Pediatrics, 88: 907–912.
Aulerich RJ, Napolitano AC, & Bursian SJ (1987) Chronic toxicity of dietary fluorine to mink. J Anim Sci, 65: 1759–1767.
Avorn J & Nielssen LC (1986) Relationship between long bone fractures and water fluoridation. Gerodontics, 2: 175–179.
Baars AJ, van Beek H, Spierenburg TJ, de Graf GJ, Beeftink WGJN, Boom J, & Pekelder JJ (1987) Fluoride pollution in a salt marsh: movement between soil, vegetation, and sheep. Bull Environ Contam Toxicol, 39: 945–952.
Bale S & Mathew M (1987) Analysis of chromosomal abnormalities at anaphase–telophase induced by sodium fluoride in vitro. Cytologia, 52: 889–893.
Ballantyne DJ (1984) ATP and growth in fluoride-treated pea shoots. Environ Exp Bot, 24(3): 277–282.
Ballantyne DJ (1991) Fluoride and photosynthetic capacity of azalea (Rhododendron) cultivars. Fluoride Q Rep, 24(1): 11–16.
Barbaro A, Francescon A, & Polo B (1981) Fluorine accumulation in aquatic organisms in the lagoon of Venice. Fluoride, 14: 102–107.
Barnard WR & Nordstrom DK (1982) Fluoride in precipitation. II. Implications for the geochemical cycling of fluoride. Atmos Environ, 16(1): 105–111.
Barrie LA & Hoff RM (1985) Five years of air chemistry observations in the Canadian arctic. Atmos Environ, 19(12): 1995–2010.
Barrow NJ & Ellis AS (1986) Testing a mechanistic model — III. The effects of pH on fluoride retention by a soil. J Soil Sci, 37: 287–293.
Bayley TA, Harrison JE, Murray TM, Josse RG, Sturtridge W, Pritzker KP, Strauss A, Vieth R, & Goodwin S (1990) Fluoride-induced fractures: relation to osteogenic effect. J Bone Miner Res, 5(suppl 1): S217–S222.
Beg SA (1982) Inhibition of nitrification by arsenic, chromium and fluoride. J Water Pollut Control Fed, 54: 482–488.
Belandria G, Asta J, & Nurit F (1989) Effects of sulphur dioxide and fluoride on ascospore germination of several lichens. Lichenologist, 21(1): 79–86.
Bell ME, Largent EJ, Ludwig TG, Muhler JC, & Stookey GK (1970) The supply of fluorine to man. In: Fluorides and human health. Geneva, World Health Organization, pp 17–74 (WHO Monograph Series No. 59).
Bennett A & Barratt R (1980) Some observations on atmospheric fluoride concentrations in Stoke-on-Trent. J R Soc Health, 100: 84–89.
Berglund LK, Iselius L, Lindsten J, Marks L, & Ryman N (1980) [Incidence of Down’s syndrome in Sweden during the years 1968–1977.] Stockholm, National Swedish Board of Health and Welfare (Report to the Reference Group for Malformations and Developmental Disorders) (in Swedish).
Bergmann KE & Bergmann RL (1995) Salt fluoridation and general health. Adv Dent Res, 9: 138–142.
Bergmann R (1995) Fluorid in der Ernährung des Menschen. Biologische Bedeutung für den wachsenden Organismus. Habilitationsschrift. Berlin, Virchow-Klinikum der Humboldt-Universität, 133 pp.
Berndt A & Stearns R (1979) Dental fluoride chemistry. Springfield, Illinois, Charles C. Thomas.
Bernstein D, Sadowskuy N, & Hegsted D (1966) Prevalence of osteoporosis in high- and low-fluoride areas of North Dakota. J Am Med Assoc, 198: 499–504.
Berry WTC (1962) Fluoridation. Med Off, 108: 204–205.
Bhatnagar M & Susheela AK (1998) Chronic fluoride toxicity: an ultrastructural study of the glomerulus of the rabbit kidney. Environ Sci, 6: 43–54.
Bird DM & Massari C (1983) Effects of dietary sodium fluoride on bone fluoride levels and reproductive performance of captive American kestrels. Environ Pollut, A31: 67–76.
Bird DM, Carrière D, & Lacombe D (1992) The effect of dietary sodium fluoride on internal organs, breast muscle, and bones in captive American kestrels (Falco sparverius). Arch Environ Contam Toxicol, 22: 242–246.
Boese SR, MacLean DC, & El-Mogazi D (1995) Effects of fluoride on chlorophyll a fluorescence in spinach. Environ Pollut, 89(2): 203–208.
Boillat MA, Baud CA, Lagier R, Garcia J, Rey P, Bang S, Boivin G, Demeurisse C, Gossi M, Tochon-Danguy HJ, Very JM, Burckhardt P, Voinier B, Donath A, & Courvoisier B (1979) [Industrial fluorosis. Multidisciplinary study on 43 workmen in the aluminium industry.] Schweiz Med Wochenschr, Suppl 8: 1–28 (in French).
Boivin G, Chavassieux P, Chapuy MC, Baud CA, & Meunier PJ (1989) Skeletal fluorosis: histomorphometric analysis of bone changes and bone fluoride content in 29 patients. Bone, 10: 89–99.
Boone RJ & Manthey M (1983) The anatomical distribution of fluoride within various body segments and organs of Antarctic krill (Euphausia superba Dana). Arch Fischereiwiss, 34(1): 81–85.
Bordelon BM, Saffle JR, & Morris SE (1993) Systemic fluoride toxicity in a child with hydrofluoric acid burns: a case report. J Trauma, 34: 437–439.
Boulton I, Cooke J, & Johnson M (1994a) Age-accumulation of fluoride in an experimental population of short-tailed field voles (Microtus agrestis L.). Sci Total Environ, 154: 29–37.
Boulton IC, Cooke JA, & Johnson MS (1994b) Fluoride accumulation and toxicity in wild small mammals. Environ Pollut, 85: 161–167.
Boulton IC, Cooke JA, & Johnson MS (1995) Fluoride accumulation and toxicity in laboratory populations of wild small mammals and white mice. J Appl Toxicol, 15(6): 423–431.
Boulton IC, Cooke JA, & Johnson MS (1999) Lesion scoring in field vole teeth: Application to the biological monitoring of environmental fluoride contamination. Environ Monit Assess, 55: 409–422.
Bowen SE (1988) Spatial and temporal patterns in the fluoride content of vegetation around two aluminium smelters in the Hunter Valley, New South Wales. Sci Total Environ, 68: 97–111.
Bower CA & Hatcher JT (1967) Adsorption of fluoride by soils and minerals. Soil Sci, 103: 151–154.
Breimer RF, Vogel J, & Ottow JCG (1989) Fluorine contamination of soils and earthworms (Lumbricus spp.) near a site of long-term industrial emission in southern Germany. Biol Fertil Soils, 7(4): 297–302.
Brewer RF (1966) Fluorine. In: Chapman HD ed. Diagnostic criteria for plants and soils. Riverside, California, University of California, Division of Agricultural Science, pp 180–195.
Brimblecombe P & Clegg SL (1988) The solubility and behaviour of acid gases in the marine aerosol. J Atmos Chem, 7: 1–18.
Brouwer ID, DeBruin A, Backer Dirks O, & Hautvast JGAJ (1988) Unsuitability of World Health Organisation guidelines for fluoride concentrations in drinking water in Senegal. Lancet, 1: 223–225.
Bruun C & Thylstrup A (1988) Dentifrice usage among Danish children. J Dent Res, 67: 1114–1117.
Buckingham FM (1988) Surgery: a radical approach to severe hydrofluoric acid burns. J Occup Med, 30: 873–874.
Bunce HWF (1984) Fluoride emissions and forest survival, growth and regeneration. Environ Pollut, A35: 169–188.
Bunce HWF (1985) Apparent stimulation of tree growth by low ambient levels of fluoride in the atmosphere. J Air Pollut Control Assoc, 35(1): 46–48.
Burns KN & Allcroft R (1964) Fluorosis in cattle. 1. Occurrence and effects in industrial areas of England and Wales, 1954–57. London, HMSO.
Burt B (1992) The changing patterns of systemic fluoride intake. J Dent Res, 71: 1228–1237.
Buse A (1986) Fluoride accumulation in invertebrates near an aluminium reduction plant in Wales. Environ Pollut, A41: 199–217.
Butler JE, Satam M, & Ekstrand J (1990) Fluoride: An adjuvant for mucosal and systemic immunity. Immunol Lett, 26: 217–220.
Butler WJ, Segreto V, & Collins E (1985) Prevalence of dental mottling in school-aged lifetime residents of 16 Texas communities. Am J Public Health, 75: 1408–1412.
Calderon J, Romieu I, Gromaldo M, Hernandez H, & Diaz-Barriga F (1995) Endemic fluorosis in San Luis Potosi, Mexico. II. Identification of risk factors associated with occupational exposure to fluoride. Fluoride, 4: 203–208.
Camargo JA (1991) Ecotoxicological study of the influence of an industrial effluent on a net-spinning caddisfly assemblage in a regulated river. Water Air Soil Pollut, 60: 263–277.
Camargo JA (1996a) Comparing levels of pollutants in regulated rivers with safe concentrations of pollutants for fishes: a case study. Chemosphere, 33(1): 81–90.
Camargo JA (1996b) Estimating safe concentrations of fluoride for three species of nearctic freshwater invertebrates: Multifactor probit analysis. Bull Environ Contam Toxicol, 56(4): 643–648.
Camargo JA & La Point TW (1995) Fluoride toxicity to aquatic life: a proposal of safe concentrations for five species of palearctic freshwater invertebrates. Arch Environ Contam Toxicol, 29: 159–163.
Camargo JA & Tarazona JV (1990) Acute toxicity to freshwater benthic macroinvertebrates of fluoride ion (F–) in soft water. Bull Environ Contam Toxicol, 45(6): 883–887.
Camargo JA & Tarazona JV (1991) Short-term toxicity of fluoride ion (F–) in soft water to rainbow trout and brown trout. Chemosphere, 22(5/6): 605–611.
Camargo JA, Ward JV, & Martin KL (1992) The relative sensitivity of competing hydropsychid species to fluoride toxicity in the Cache la Poudre River (Colorado). Arch Environ Contam Toxicol, 22: 107–113.
Campbell A (1987) Determination of fluoride in various matrices. Pure Appl Chem, 59: 695–702.
Cao J, Bai X, Zhao Y, Zhou D, Fang S, Jia M, & Wu J (1996) The relationship of fluorosis and brick tea drinking in Chinese Tibetans. Environ Health Perspect, 104: 1340–1343.
Cao J, Zhao Y, Liu J, Xirao R, & Danzeng S (2000) Environmental fluoride content in Tibet. Environ Res, 83(3): 333–337.
Caraccio T, Greensher J, & Mofenson H (1983) The toxicology of fluoride. In: Haddad L & Winchester J ed. Clinical management of poisoning and drug overdose. Philadelphia, Pennsylvania, W.B. Saunders.
Carlson CE (1978) The use of infrared aerial photography in determining fluoride damage to forest ecosystems near an aluminum plant in northwestern Montana, USA. Fluoride, 11(3): 135–141.
Carpenter R (1969) Factors controlling the marine geochemistry of fluorine. Geochim Cosmochim Acta, 33: 1153–1167.
Carrière D, Bird DM, & Stamm JW (1987) Influence of a diet of fluoride-fed cockerels on reproductive performance of captive American kestrels. Environ Pollut, 46: 151–159.
Caspary W, Myhr B, Bowers L, McGregor D, Riach C, & Brown A (1987) Mutagenic activity of fluorides in mouse lymphoma cells. Mutat Res, 187: 165–180.
Caspary W, Langenbach R, Penman B, Crespi C, Myhr B, & Mitchell A (1988) The mutagenic activity of selected compounds at the TK locus: rodent vs. human cells. Mutat Res, 196: 61–81.
Cauley JA, Murphy PA, Riley TJ, & Buhari AM (1995) Effects of fluoridated drinking water on bone mass and fractures: The study of osteoporotic fractures. J Bone Miner Res, 10: 1076–1086.
Chakma T, Vinay Rao P, Singh SB, & Tiwary RS (2000) Endemic genu valgum and other bone deformities in two villages of Mandla district in Central India. Fluoride, 33: 187–195.
Chamblee JW, Arey FK, & Heckel E (1984) Free fluoride of the Pamlico River in North Carolina. A new method to localize the discharge of polluted water into an estuary. Water Res, 18(10): 1225–1233.
Chan JT & Koh SH (1996) Fluoride content in caffeinated, decaffeinated and herbal teas. Caries Res, 30: 88–92.
Chan K-M, Svancarek WP, & Creer M (1987) Fatality due to hydrofluoric acid exposure. Clin Toxicol, 25: 333–339.
Chan MM, Rucker RB, Zeman F, & Riggins RS (1973) Effect of fluoride on bone formation and strength in Japanese quail. J Nutr, 103: 1431–1440.
Chandrawanshi CK & Patel KS (1999) Fluoride deposition in central India. Environ Monit Assess, 55: 251–265.
Chan-Yeung M, Wong R, Earnson D, Schulzer M, Subbarao K, Knickerbocker J, & Grzybowski S (1983a) Epidemiological health study of workers in an aluminum smelter in Kitimat, B.C. II. Effects on musculoskeletal and other systems. Arch Environ Health, 38: 34–40.
Chan-Yeung M, Wong R, MacLean L, Tan F, Schulzer M, Enarson D, Martin A, Dennis R, & Grzybowski S (1983b) Epidemiologic health study of workers in an aluminum smelter in British Columbia. Effects of the respiratory system. Am Rev Respir Dis, 127: 465–469.
Chen Y, Lin M, He Z, Xie X, Liu Y, Xiao Y, Zhou J, Fan Y, Xiao X, & Xu F (1993) Air pollution-type fluorosis in the region of Pingxiang, Jiangxi, People’s Republic of China. Arch Environ Health, 48: 246–249.
Chen YX, Lin MQ, He ZL, Chen C, Min D, Liu YQ, & Yu MH (1996) Relationship between total fluoride intake and dental fluorosis in areas polluted by airborne fluoride. Fluoride, 29: 7–12.
Chhabra R, Singh A, & Abrol IP (1979) Fluorine in sodic soils. Soil Sci Soc Am J, 44: 33–36.
Chilvers C (1982) Cancer mortality by site and fluoridated water supplies. J Epidemiol Community Health, 36: 237–242.
Chinoy N & Sequeira E (1989a) Effects of fluoride on the histoarchitecture of the reproductive organs of the male mouse. Reprod Toxicol, 3: 261–267.
Chinoy N & Sequeira E (1989b) Fluoride induced biochemical changes in reproductive organs of male mice. Fluoride, 22: 79–85.
Chinoy NJ & Sharma A (1998) Amelioration of fluoride toxicity by vitamins E and D in reproductive functions of male mice. Fluoride, 31: 203–216.
Chinoy N, Sequeira E, & Narayama M (1991) Effect of vitamin C and calcium on the reversibility of fluoride-induced alterations in spermatozoa of rabbits. Fluoride, 24: 29–39.
Chinoy NJ, Narayama MV, Dalal V, Rawat M, & Patel D (1995) Amelioration of fluoride toxicity in some accessory reproductive glands and spermatozoa of rat. Fluoride, 28: 75–86.
Cholak J (1959) Fluorides: A critical review. I. The occurrence of fluoride in air, food, and water. J Occup Med, 1: 501–511.
Choubisa SL (1999) Some observations on endemic fluorosis in domestic animals in southern Rajsthan (India). Vet Res Commun, 23: 457–465.
Choubisa SL, Choubisa DK, Joshi SC, & Choubisa L (1997) Fluorosis in some tribal villages of Dungapur district of Rajasthan, India. Fluoride, 30: 223–228.
Christians O & Leinemann M (1980) [Untersuchungen über Fluor im Krill (E. superba).] Inf Fischwirtsch Ausl, 6: 254–260.
Chu F (1991) SF6 and the atmosphere. Toronto, Ontario, Ontario Hydro Research Division, Electrical Research Department (91-58-K).
Cohn P (1992) An epidemiologic report on drinking water and fluoridation. Environmental Health Service, New Jersey Department of Health, November.
Cole J, Muriel W, & Bridges B (1986) The mutagenicity of sodium fluoride to L5178Y [wild-type and TK+/- (3.7.2C)] mouse lymphoma cells. Mutagenesis, 1: 157–167.
Collins TFX, Sprando RL, Shackelford ME, Black TN, Ames MJ, Welsh JJ, Balmer MF, Olejnik N, & Ruggles DI (1995) Developmental toxicity of sodium fluoride in rats. Food Chem Toxicol, 33: 951–960.
Connell AD & Airey DD (1982) The chronic effects of fluoride on the estuarine amphipods Grandidierella lutosa and G. lignorum. Water Res, 16: 1313–1317.
Conover CA & Poole RT (1971) Influence of fluoride on foliar necrosis of Cordyline terminalis Cv Baby Doll during propagation. Proc Fla State Hortic Soc, 84: 380–383.
Cooke JA (1976) The uptake of sodium monofluoroacetate by plants and its physiological effects. Fluoride, 9(4): 204–212.
Cooke JA, Johnson MS, Davison AW, & Bradshaw AD (1976) Fluoride in plant colonizing fluorspar mine waste in the Peak District and Weardale. Environ Pollut, 11: 10–23.
Cooke JA, Johnson MS, & Davison AV (1978) Uptake and translocation of fluoride in Helianthus annus L. grown in sand culture. Fluoride, 11: 76–88.
Crespi C, Seixas G, Turner T, & Penman B (1990) Sodium fluoride is a less efficient human cell mutagen at low concentrations. Environ Mol Mutagen, 15: 71–77.
Cronin SJ, Manoharan V, Hedley MJ, & Loganathan P (2000) Fluoride — a review of its fate and hazard potential in grazed pasture systems. N Z J Agric Res, 43: 295–321.
Cuker W & Shilts W (1979) Lacustrine geochemistry around the north shore of Lake Superior: Implications for evaluation of the effects of acid precipitation. Current Research Part C, Geological Survey of Canada (79-1C).
Cummings C & McIvor M (1988) Fluoride-induced hyperkalemia: the role of Ca2+-dependent K+ channels. Am J Emerg Med, 6: 1–3.
Czarnowski W & Krechniak J (1990) Fluoride in urine, hair and nails of phosphate fertilizer workers. Br J Ind Med, 47: 349–351.
Czarnowski W, Wrzenioska K, & Krechniak J (1996) Fluoride in drinking water and human urine in northern and central Poland. Sci Total Environ, 191: 177–184.
Czarnowski W, Krechniak J, Urbanska B, Stolarska K, Taraszewska-Czarnowska M, & Muraszko-Klaudel A (1999) The impact of water-borne fluoride on bone density. Fluoride, 32: 91–95.
Czerwinski E, Nowack J, Dabrowska D, Skolarczyk A, Kita B, & Ksiezyk M (1988) Bone and joint pathology in fluoride-exposed workers. Arch Environ Health, 43: 340–343.
Dabeka RW & McKenzie AD (1995) Survey of lead, cadmium, fluoride, nickel, and cobalt in food composites and estimation of dietary intakes of these elements by Canadians in 1986–1988. J Assoc Off Anal Chem Int, 78: 897–909.
Dabeka RW, McKenzie AD, Conacher HBS, & Kirkpatrick BS (1982) Determination of fluoride in Canadian infant foods and calculation of fluoride intake by infants. Can J Public Health, 73: 188–191.
Dabeka R, Karpinski K, McKenzie A, & Bajdik C (1986) Survey of lead, cadmium and fluoride in human milk and correlation of levels with environmental and food factors. Food Chem Toxicol, 24: 913–921.
Dabkowska E & Machoy Z (1989) Effects of fluoride pollution on calcium and magnesium content of mandibles (lower jaws) of wild game. Fluoride Q Rep, 22(1): 29–32.
Damkaer DM & Dey DB (1989) Evidence for fluoride effects on salmon passage at John Day Dam, Columbia River, 1982–1986. North Am J Fish Manage, 9: 154–162.
Danielson C, Lyon J, Egger M, & Goodenough G (1992) Hip fracture and fluoridation in Utah’s elderly population. J Am Med Assoc, 268: 746–748.
Das T & Susheela A (1991) Effect of chronic fluoride toxicity on glucocorticoid levels in plasma and urine. Fluoride, 24: 23–28.
Das TK, Susheela AK, Gupta IP, Dasarathy S, & Tandon RK (1994) Toxic effects of chronic fluoride ingestion on the upper gastrointestinal tract. J Clin Gastroenterol, 18: 194–199.
Dasarathy S, Das TK, Gupta IP, Susheela AK, & Tandon RK (1996) Gastroduodenal manifestations in patients with skeletal fluorosis. J Gastroenterol, 31: 333–337.
Daston G, Rehnberg B, Carver B, & Kavelock B (1985) Toxicity of sodium fluoride to the postnatally developing rat kidney. Environ Res, 37: 461–474.
Datta DK, Gupta LP, & Subramanian V (2000) Dissolved fluoride in the Lower Ganges–Brahmaputra–Meghna river system in the Bengal Basin, Bangladesh. Environ Geol, 39(10): 1163–1168.
Dave G (1984) Effects of fluoride on growth, reproduction and survival in Daphnia magna. Comp Biochem Physiol, 78C(2): 425–431.
Davies FBM (1982) Accumulation of fluoride by Xanthoria parietina growing in the vicinity of the Bedfordshire brickfields. Environ Pollut, 29: 189–196.
Davies FBM (1986) The long-term changes in fluoride content of Xanthoria parietina growing in the vicinity of the Bedfordshire brickfields. Environ Pollut, A42: 201–207.
Davies FBM & Notcutt G (1988) Accumulation of fluoride by lichens in the vicinity of Etna volcano. Water Air Soil Pollut, 42: 365–371.
Davies FBM & Notcutt G (1989) Accumulation of volcanogenic fluorides by lichens. Fluoride Q Rep, 22(2): 59–65.
Davies MT, Port GR, & Davison AW (1998) Effects of dietary and gaseous fluoride on the aphid Aphis fabae. Environ Pollut, 99: 405–409.
Davis JB & Barnes RL (1973) Effects of soil-applied fluoride and lead on growth of loblolly pine and red maple. Environ Pollut, 5: 35–44.
Davis RD (1980) Uptake of fluoride by ryegrass grown in soil treated with sewage sludge. Environ Pollut, B1: 277–284.
Davison A (1983) Uptake, transport and accumulation of soil and airborne fluorides by vegetation. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 61–82.
Davison AW & Blakemore J (1980) Rate of deposition, and resistance to deposition, of fluorides on alkali-impregnated papers. Environ Pollut, B1: 305–319.
Davison AW, Rand AW, & Betts WE (1973) Measurement of atmospheric fluoride concentrations in urban areas. Environ Pollut, 5: 23–33.
Dayal HH, Brodwick M, Morris R, Baranowski T, Trieff N, Harrison JA, Lisse JR, & Ansari GAS (1992) A community-based epidemiological study of health sequela of exposure to hydrofluoric acid. Ann Epidemiol, 2: 213–220.
Dean HT, Jay P, Arnold FA, & Elvove E (1941) Domestic waters and dental caries. II. A study of 2832 white children ages 12–14 years of eight suburban Chicago communities, including Lactobacillus acidophilus studies of 1761 children. Public Health Rep, 56: 761–792.
Dean HT, Arnold FA, & Elvove E (1942) Domestic waters and dental caries. V. Additional studies of the relation of fluoride domestic waters to dental caries in 4425 white children, age 12–14 years, of 13 cities in 4 states. Public Health Rep, 57: 1155–1179.
Delmas PD, Dupuis J, Duboeuf F, Chapuy MC, & Meunier PJ (1990) Treatment of vertebral osteoporosis with disodium monofluorophosphate: comparison with sodium fluoride. J Bone Miner Res, 5(suppl 1): S143–S147.
Derelanko M, Gad S, Gavigan F, & Dunn B (1985) Acute dermal toxicity of dilute hydrofluoric acid. J Toxicol Cutan Ocular Toxicol, 4: 73–85.
Doley D (1986) Experimental analysis of fluoride susceptibility of grapevine (Vitis vinifera L.): leaf development during four successive seasons of fumigation. New Phytol, 103: 325–340.
Doley D (1989) Fluoride-induced enhancement and inhibition of shoot growth in four taxa of Pinus. New Phytol, 112: 543–552.
Doll R & Kinlen L (1977) Fluoridation of water and cancer mortality in the U.S.A. Lancet, 1: 1300–1302.
Dominok B & Miller G (1990) Effects of fluoride on Drosophila melanogaster in relation to survival and mutagenicity. Fluoride, 23: 83–91.
Droste RL (1987) Fluoridation in Canada as of December 31, 1986. Ottawa, Ontario, Health and Welfare Canada (IWD-AR WQB-89-154).
Drummond B, Curzon M, & Strong M (1990) Estimation of fluoride absorption from swallowed fluoride toothpastes. Caries Res, 24: 211–215.
Drury JS, Ensminger JT, Hammons AS, Holleman JW, Lewis EB, Preston EL, Shriner CR, & Towill LE (1980) Reviews of the environmental effects of pollutants. IX. Fluoride. Oak Ridge, Tennessee, Oak Ridge National Laboratory (EPA 600/1-78/050).
Dunipace A, Zhang W, Noblitt T, Li Y, & Stookey G (1989) Genotoxic evaluation of chronic fluoride exposure: micronucleus and sperm morphology studies. J Dent Res, 68: 1525–1528.
Dunipace A, Brizendine E, Wilson M, Zhang W, Wilson C, Katz B, & Kafrawy A (1998) Effect of chronic fluoride exposure in uremic rats. Nephron, 78: 96–103.
Dure-Smith BA, Farley SM, Linkhart SG, Farley JR, & Baylink DJ (1996) Calcium deficiency in fluoride-treated osteoporotic patients despite calcium supplementation. J Clin Endocrinol Metab, 81: 269–275.
Easmann R, Steflik D, Pashley D, McKinney R, & Whitford G (1984) Surface changes in rat gastric mucosa induced by sodium fluoride: a scanning electron microscopy study. J Oral Pathol, 13: 255–264.
Easmann R, Pashley D, Birdsong N, McKinney R, & Whitford G (1985) Recovery of rat gastric mucosa following single fluoride dosing. J Oral Pathol, 14: 779–792.
Eckerlin RH, Krook L, Maylin GA, & Carmichael D (1986) Toxic effects of food-borne fluoride in silver foxes. Cornell Vet, 76: 395–402.
Ehrnebo M & Ekstrand J (1986) Occupational fluoride exposure and plasma fluoride levels in man. Int Arch Environ Health, 58: 179–190.
Ekstrand J (1977) Fluoride concentrations in saliva after single oral doses and their relation to plasma fluoride. Scand J Dent Res, 85: 16–17.
Ekstrand J (1978) Relationship between fluoride in the drinking water and the plasma fluoride concentration in man. Caries Res, 12: 123–127.
Ekstrand J (1981) No evidence of transfer of fluoride from plasma to breast milk. Br Med J, 283: 761–762.
Ekstrand J (1987) Pharmacokinetic aspects of topical fluorides. J Dent Res, 66: 1061–1065.
Ekstrand J & Ehrnebo M (1979) Influence of milk products on fluoride bioavailability in man. Eur J Clin Pharmacol, 16: 211–215.
Ekstrand J & Ehrnebo M (1983) The relationship between plasma fluoride, urinary excretion rate and urine fluoride concentrations in man. J Occup Med, 25: 745–748.
Ekstrand J, Ericsson Y, & Rosell S (1977a) Absence of protein-bound fluoride from human blood. Arch Oral Biol, 22: 229–232.
Ekstrand J, Alván G, Boréus L, & Norlin A (1977b) Pharmacokinetics of fluoride in man after single and multiple oral doses. Eur J Clin Pharmacol, 12: 311–317.
Ekstrand J, Ehrnebo M, & Boreus L (1978) Fluoride bioavailability after intravenous and oral administration: importance of renal clearance and urine flow. Clin Pharmacol Ther, 23: 329–371.
Ekstrand J, Ehrnebo M, Whitford GM, & Järnberg PO (1980) Fluoride pharmacokinetics during acid–base changes in man. Eur J Clin Pharmacol, 18: 189–194.
Ekstrand J, Boréus LO, & de Chateau P (1981) No evidence of transfer of fluoride from plasma to breast milk. Br Med J, 283: 761–762.
Ekstrand J, Spak C, & Ehrnebo M (1982) Renal clearance of fluoride in a steady state condition in man: influence of urinary flow and pH changes by diet. Acta Pharmacol Toxicol, 50: 321–325.
Ekstrand J, Odont LDS, & Ehrnebo M (1983) The relationship between plasma fluoride, urinary excretion rate and urine fluoride concentration in man. J Occup Med, 25: 745–748.
Ekstrand J, Hardell LI, & Spak CJ (1984) Fluoride balance studies on infants in a 1-ppm water fluoride area. Caries Res, 18: 87–92.
Ekstrand J, Ziegler EE, Nelson SE, & Fomon SJ (1994) Absorption and retention of dietary and supplemental fluoride by infants. Adv Dent Res, 8 :175–180.
Elrashidi MA & Lindsay WL (1986) Chemical equilibria of fluorine in soils. A theoretical development. Soil Sci, 141: 274–280.
Elrashidi MA, Persaud N, & Baliger VC (1998) Effect of fluoride and phosphate on yield and mineral composition of barley grown on three soils. Commun Soil Sci Plant Anal, 29(3&4): 269–283.
Elsair J & Khelfat K (1988) Subcellular effects of fluoride. Fluoride, 21: 93–99.
Environment Canada (1989) Ambient air levels of fluoride at Cornwall Island, Ontario, April 14–October 12, 1988. Ottawa, Ontario, Environment Canada, Conservation and Protection Service, Environmental Protection.
Environment Canada (1994) Inorganic fluorides. Ottawa, Ontario, Environment Canada, Ecosystem Science and Evaluation Directorate, Eco-Health Branch.
Erickson JD (1980) Down’s syndrome, water fluoridation and maternal age. Teratology, 21: 177.
Erickson JD, Oakley GP, Flynt JW, & Hay S (1976) Water fluoridation and congenital malformations: No association. J Am Dent Assoc, 93: 981–984.
Ernst P, Thomas D, & Becklake M (1986) Respiratory survey of North American Indian children living in proximity to an aluminum smelter. Am Rev Respir Dis, 133: 307–312.
Esala S, Vuori E, & Helle A (1982) Effect of maternal fluorine intake on breast milk fluorine content. Br J Nutr, 48: 201–204.
Essman E, Essman W, & Valderrama E (1981) Histaminergic mediation of the response of rat skin to topical fluorides. Arch Dermatol Res, 271: 325–340.
Fabiani L, Leoni V, & Vitali M (1999) Bone-fracture incidence rate in two Italian regions with different fluoride concentration levels in drinking water. J Trace Elem Med Biol, 13: 232–237.
Fejerskov O, Manji F, Baelum V, & Mr ller IJ (1988) Dental fluorosis — a handbook for health workers. Copenhagen, Munksgaard, 123 pp.
Fejerskov O, Baelum V, & Richards A (1996) Dose–response and dental fluorosis. In: Fejerskov O, Ekstrand J, & Burt BA ed. Fluoride in dentistry, 2nd ed. Copenhagen, Munksgaard, pp 153–166.
Felsenfeld A & Roberts M (1991) A report of fluorosis in the United States secondary to drinking well water. J Am Med Assoc, 265: 486–488.
Feskanich D, Owusu W, Hunter DJ, Willett W, Ascherio A, Spiegelman D, Morris S, Spate VL, & Colditz G (1998) Use of toenail fluoride levels as an indicator for the risk of hip and forearm fractures in women. Epidemiology, 9: 412–416.
Fidanci UR, Salmanoglu B, Marasli S, & Marasli N (1998) [The natural and industrial fluorosis in the Middle Anatolia and its effects on animal health.] Tr J Vet Anim Sci, 22: 537–544 (in Turkish).
Fieser AH, Sykora JL, Kostalos MS, Wu YC, & Weyel DW (1986) Effect of fluorides on survival and reproduction of Daphnia magna. J Water Pollut Control Fed, 58(1): 82–86.
Fleming WJ, Grue CE, Schuler CA, & Bunck CM (1987) Effects of oral doses of fluoride on nestling European starlings. Arch Environ Contam Toxicol, 16: 483–489.
Flühler H, Polomski J, & Blaser P (1982) Retention and movement of fluoride in soils. J Environ Qual, 11: 461–468.
Fomon SJ & Ekstrand J (1993) Fluoride. In: Fomon SJ ed. Nutrition of normal infants. St Louis, Missouri, Mosby, pp 299–310.
Fomon SJ, Ekstrand J, & Ziegler EE (2000) Fluoride intake and prevalence of dental fluorosis: Trends in fluoride intake with special attention to infants. J Public Health Dent, 60: 131–139.
Freni S (1994) Exposure to high fluoride concentrations in drinking water is associated with decreased birth rates. J Toxicol Environ Health, 42: 109–121.
Freni S & Gaylor D (1992) International trends in the incidence of bone cancer are not related to drinking water fluoridation. Cancer, 70: 611–618.
Fuge R & Andrews MJ (1988) Fluorine in the UK environment. Environ Geochem Health, 10(3/4): 96–104.
Fung KF, Zhang ZQ, Wong JWC, & Wong MH (1999) Fluoride contents in tea and soil from tea plantations and the release of fluoride into tea liquor during infusion. Environ Pollut, 104: 197–205.
Gadhia PK & Joseph S (1997) Sodium fluoride induced chromosome aberrations and sister chromatid exchange in cultured human lymphocytes. Fluoride, 30: 153–156.
Gebhart E, Wagner H, & Behnsen H (1984) The action of anticlastogens in human lymphocyte cultures and its modification by rat liver S9 mix. Studies with AET and sodium fluoride. Mutat Res, 129: 195–206.
Gelberg KH, Fitzgerald EF, Hwang S, & Dubrow R (1995) Fluoride exposure and childhood osteosarcoma: A case–control study. Am J Public Health, 85: 1678–1683.
Gessner B, Beller M, Middaugh J, & Whitford G (1994) Acute fluoride poisoning from a public water system. N Engl J Med, 330: 95–99.
Gibbs G (1985) Mortality of aluminum reduction plant workers, 1950 through 1977. J Occup Med, 27: 761–770.
Gikunju JK (1992) Fluoride concentration in tilapia fish (Oreochromis leucostictus) from Lake Naivasha, Kenya. Fluoride Q Rep, 25(1): 37–43.
Gilbert OL (1985) Environmental effects of airborne fluorides from aluminium smelting at Invergordon, Scotland, 1971–1983. Environ Pollut, 39: 293–302.
Gilman A (1987) G proteins: transducers of receptor-generated signals. Annu Rev Biochem, 56: 615–649.
Gilpin L & Johnson A (1980) Fluorine in agricultural soils of southeastern Pennsylvania. Soil Sci Soc Am J, 44: 255–258.
Giovanazzi A & D’Andrea F (1981) [Mortality of workers in a primary aluminum plant.] Med Lav, 4: 277–282 (in Italian).
Gocke E, King M-T, Eckhardt K, & Wild D (1981) Mutagenicity of cosmetics ingredients licensed by the European Communities. Mutat Res, 90: 91–109.
Godfrey P & Watson S (1988) Fluoride inhibits agonist-induced formation of inositol phosphates in rat cortex. Biochem Biophys Res Commun, 155: 664–669.
Goggin JE, Haddon W, Hambly GS, & Hoveland JR (1965) Incidence of femoral fractures in postmenopausal women. Public Health Rep, 80: 1005–1012.
Government of Canada (1993) Canadian Environmental Protection Act. Priority Substances List assessment report for inorganic fluorides. Prepared by Health Canada and Environment Canada. Ottawa, Ontario, Canada Communication Group (ISBN 0-662-21070-9).
Grad H (1990) Fluorides: old and new perspectives. Univ Toronto Dent J, 3: 27–31.
Grandjean P (1982) Occupational fluorosis through 50 years; clinical and epidemiological experiences. Am J Occup Med, 3: 227–236.
Grandjean P & Thomsen G (1983) Reversibility of skeletal fluorosis. Br J Ind Med, 40: 456–461.
Grandjean P, Juel K, & Moller-Jensen O (1985) Mortality and morbidity after occupational fluoride exposure. Am J Epidemiol, 121: 57–64.
Grandjean P, Olsen J, Moller-Jensen O, & Juel K (1992) Cancer incidence and mortality in workers exposed to fluoride. J Natl Cancer Inst, 84: 1903–1909.
Grant MW & Schuman JS (1993) Toxicology of the eye: effects on the eyes and visual system from chemicals, drugs, metals and minerals, plants, toxins and venoms: also, systemic side effects from eye medications, 4th ed. Springfield, Illinois, Charles C. Thomas.
Greco RJ, Hartford CE, Haith LR, & Patton ML (1988) Hydrofluoric acid-induced hypocalcemia. J Trauma, 28: 1593–1596.
Greenwood N & Earnshaw A (1984) Chemistry of the elements. Toronto, Ontario, Pergamon Press.
Grimaldo M, Borja-Aburto VH, Ramirez AL, Ponce M, Rosas M, & Diaz-Barriga F (1995) Endemic fluorosis in San Luis Potosi. I. Identification of risk factors associated with human exposure to fluoride. Environ Res, 68: 25–30.
Grynpas M (1990) Fluoride effects on bone crystals. J Bone Miner Res, 5: S169–S175.
Gu S, Rongli J, & Shouren C (1990) The physical and chemical characteristics of particles in indoor air where high fluoride coal burning takes place. Biomed Environ Sci, 3: 384–390.
Gubbay AD & Fitzpatrick RI (1997) Dermal hydrofluoric acid burns resulting in death. Aust N Z J Surg, 67: 304–306.
Guenter W & Hahn PHB (1986) Fluorine toxicity and laying hen performance. Poult Sci, 65: 769–778.
Guha-Chowdhury N, Drummond BK, & Smillie AC (1996) Total fluoride intake in children aged 3 to 4 years — a longitudinal study. J Dent Res, 75: 1451–1457.
Gupta IP, Das TK, Susheela AK, Dasarathy S, & Tandon RK (1992) Fluoride as a possible aetiological factor in non-ulcer dyspepsia. J Gastroenterol Hepatol, 7: 355–359.
Gutteridge DH, Price RI, Kent GN, Prince RL, & Michell PA (1990) Spontaneous hip fractures in fluoride-treated patients: potential causative factors. J Bone Miner Res, 5(suppl 1): S205–S215.
Guy W (1979) Inorganic and organic fluorine in human blood. In: Johansen E, Taves D, & Olsen T ed. Continuing evaluation of the use of fluorides. Boulder, Colorado, Westview Press for the American Association for the Advancement of Science, pp 125–147 (AAAS Selected Symposium 11).
Guy W, Taves D, & Brey W (1976) Organic fluorocompounds in human plasma: prevalence and characterization. Am Chem Soc Symp Ser, 28: 117–134.
Haidouti C (1995) Effects of fluoride pollution on the mobilization and leaching of aluminum in soil. Sci Total Environ, 166: 157–160.
Haimanot R (1990) Neurological complications of endemic skeletal fluorosis, with special emphasis on radiculo-myelopathy. Paraplegia, 28: 244–251.
Haimanot RT, Fekadu A, & Bushra B (1987) Endemic fluorosis in the Ethiopian Rift Valley. Trop Geogr Med, 39: 209–217.
Hall RJ (1972) The distribution of organic fluorine in some toxic tropical plants. New Phytol, 71: 855–871.
Hamilton M (1992) Water fluoridation: a risk assessment perspective. J Environ Health, 54: 27–32.
Han YZ, Zhang JQ, Liu XY, Zhang XH, Yu XH, & Dai JA (1995) High fluoride content and endemic fluorosis. Fluoride, 28: 201–202.
Hara T, Sonoda Y, & Iwai I (1977) Growth response of cabbage plant to sodium halides under water culture conditions. Soil Sci Plant Nutr, 23: 77–84.
Harbo RM, Comas FT, & Thompson JAJ (1974) Fluoride concentration in two Pacific coast inlets — an indication of industrial contamination. J Fish Res Board Can, 31: 1151–1154.
Harrison J, Hitchman A, Hassany S, Hitchman A, & Tam C (1984) The effect of diet on fluoride toxicity in growing rats. Can J Physiol Pharmacol, 62: 259–265.
Hart EB, Phillips PH, & Bohstedt G (1934) Relationship of soil fertilisation with superphosphates and rock phosphate to the fluorine content of plants and drainage waters. Am J Public Health, 24: 936–940.
Harvey JM (1952) Chronic endemic fluorosis of merino sheep in Queensland. Queensland J Agric Sci, 9: 47–141.
Harzdorf C, Kinwel N, Marangoni L, Ogleby J, Ravier J, & Roost F (1986) The determination of fluoride in environmentally relevant matrices. Anal Chim Acta, 182: 1–16.
Hasling C, Nielsen H, Melsen F, & Mosekilde L (1987) Safety of osteoporosis treatment with sodium fluoride, calcium phosphate and vitamin D. Miner Electrol Metab, 13: 96–103.
Hayashi M, Kishi M, Sofuni T, & Ishidate MJ (1988) Micronucleus test in mice on 39 food additives and eight miscellaneous chemicals. Food Chem Toxicol, 26: 487–500.
Health Canada (1993) Canadian Environmental Protection Act. Priority Substances List supporting documentation. Health-related sections for inorganic fluorides. Prepared by Priority Substances Section, Environmental Health Directorate, Health Canada, Ottawa, Ontario (unpublished).
Health Council of the Netherlands (1990) Fluorides. Assessment of integrated criteria document. Advisory report submitted to Minister and State Secretary of Health, Welfare and Cultural Affairs and the Minister of Housing, Physical Planning and Environment. The Hague, Health Council of the Netherlands.
Heilman JR, Kiritsy MC, Levy SM, & Wefel JS (1997) Fluoride concentrations of infant foods. J Am Dent Assoc, 128: 857–863.
Heilman JR, Kiritsy MC, Levy SM, & Wefel JS (1999) Assessing fluoride levels of carbonated soft drinks. J Am Dent Assoc, 130: 1593–1599.
Heindel JJ, Bates HK, Price KJ, Marr MC, Myers CB, & Schwetz BA (1996) Developmental toxicity evaluation of sodium fluoride administered to rats and rabbits in drinking water. Fundam Appl Toxicol, 30: 162–177.
Heitmuller PT, Hollister TA, & Parrish PR (1981) Acute toxicity of 54 industrial chemicals to sheepshead minnows (Cyprinodon variegatus). Bull Environ Contam Toxicol, 27: 596–604.
Hekman WE, Budd K, Palmer GR, & MacArthur JD (1984) Responses of certain freshwater planktonic algae to fluoride. J Phycol, 20: 243–249.
Hemens J & Warwick RJ (1972) The effects of fluoride on estuarine organisms. Water Res, 6: 1301–1308.
Hemens J, Warwick RJ, & Oliff WD (1975) Effect of extended exposure to low fluoride concentrations on estuarine fish and crustacea. Prog Water Technol, 7(3/4): 579–585.
Henny CJ & Burke PM (1990) Fluoride accumulation and bone strength in wild black-crowned night-herons. Arch Environ Contam Toxicol, 19: 132–137.
Henschler D, Büttner W, & Patz J (1975) Absorption, distribution in body fluid and bioavailability of fluoride. In: Kuhlencordt F & Kruse HP ed. Calcium metabolism, bone and metabolic diseases. Berlin, Springer Verlag, pp 111–121.
Hill A & Pack M (1983) Effects of atmospheric fluoride on plant growth. In: Shupe J, Peterson H, & Leone N ed. Fluoride effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 105–113.
Hill AC, Pack MR, Transtrum LG, & Winters WS (1959) Effects of atmospheric fluorides and various types of injury on the respiration of leaf tissue. Plant Physiol, 34(1): 11–16.
Hillier S, Cooper C, Kellingray S, Russell G, Hughes H, & Coggon D (2000) Fluoride in drinking water and risk of hip fracture in the UK: a case–control study. Lancet, 355(9200): 265–269.
Hirayama N, Maruo M, Obata H, Shiota A, Enya T, & Kuwamoto T (1996) Measurement of fluoride ion in the river-water flowing into Lake Biwa. Water Res, 30(4): 865–868.
Hitchcock AE, Zimmerman PW, & Coe RR (1962) Results of ten years’ work (1951–1960) on the effects of fluorides on gladiolus. Contrib Boyce Thompson Inst, 21: 303–344.
Hobbs CS & Merriman GM (1959) The effect of eight years continuous feeding of different levels of fluorine and alleviators on feed consumption, teeth, bones and production of cows. J Anim Sci, 18: 1526–1529.
Hocking MB, Hocking D, & Smyth TA (1980) Fluoride distribution and dispersion processes about an industrial point source in a forested coastal zone. Water Air Soil Pollut, 14: 133–157.
Hodge H & Smith F (1977) Occupational fluoride exposure. J Occup Med, 19: 12–39.
Hodge HC, Smith FA, & Gedalia I (1970) Excretion of fluorides. In: Fluorides and human health. Geneva, World Health Organization, pp 158–159 (WHO Monograph Series No. 59).
Hoffman DJ, Pattee OH, & Wiemeyer SN (1985) Effects of fluoride on screech owl reproduction: teratological evaluation, growth, and blood chemistry in hatchlings. Toxicol Lett, 26: 19–24.
Hoover RN, McKay FW, & Fraumeni FJF (1976) Fluoridated drinking water and the occurrence of cancer. J Natl Cancer Inst, 57: 757–768 [cited in US DHHS, 1991].
Horntvedt R (1995) Fluoride uptake in conifers related to emissions from aluminium smelters in Norway. Sci Total Environ, 163: 35–37.
Hou P (1997) The control of coal-burning fluorosis in China. Fluoride, 30: 229–232.
Hrudey SE, Soskolne CL, Berkel J, & Fincham S (1990) Drinking water fluoridation and osteosarcoma. Can J Public Health, 81: 415–416.
Huang PM & Jackson ML (1965) Mechanism of reaction of neutral fluoride solution with layer silicates and oxides of soils. Proc Soil Sci Soc Am, 29: 661–665.
Hughes PR, Weinstein LH, Johnson LM, & Braun AR (1985) Fluoride transfer in the environment: accumulation and effects on cabbage looper Trichoplusia ni of fluoride from water soluble salts and HF-fumigated leaves. Environ Pollut, A37: 175–192.
IARC (1982) Some aromatic amines, anthroquinones and nitroso compounds, and inorganic fluorides used in drinking-water and dental preparations. Lyon, International Agency for Research on Cancer, pp 237–303 (IARC Monographs on the Evaluation of Carcinogenic Risks of Chemicals to Humans, Volume 27).
IARC (1984) Polynuclear aromatic compounds. Part 3. Industrial exposures in aluminum production, coal gassification, coke production and iron and steel making. Lyon, International Agency for Research on Cancer, pp 37–64 (IARC Monographs on the Evaluation of Carcinogenic Risks of Chemicals to Humans, Volume 34).
IARC (1987) Overall evaluations of carcinogenicity: An updating of IARC monographs, volumes 1–42. Lyon, International Agency for Research on Cancer, pp 208–210 (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Supplement 7).
Ishidate MJ (1987) Data book on chromosomal aberration test in vitro, rev. ed. Tokyo, L.I.C. Inc.
Ivinskis M & Murray F (1984) Associations between metabolic injury and fluoride susceptibility in two species of Eucalyptus. Environ Pollut, A34: 207–223.
Jachimczak D & Skotarczak B (1978) The effect of fluorine and lead ions on the chromosomes of human leucocytes in vitro. Genet Pol, 19: 353–358.
Jackson R, Kelly S, Noblitt T, Zhang W, & Dunipace A (1994) The effect of fluoride therapy on blood chemistry parameters in osteoporotic females. Bone Miner, 27: 13–23.
Jackson RD, Kelly SA, Noblitt TW, Zhang W, Wilson ME, Dunipace AJ, Li Y, Katz BP, Brizendine EJ, & Stookey GK (1997) Lack of effect of long-term fluoride ingestion on blood chemistry and frequency of sister chromatid exchange in human lymphocytes. Environ Mol Mutagen, 29: 265–271.
Jackson WPU (1962) Further observations on the Kenhardt bone disease and its relation to fluorosis. S Afr Med J, 36: 932–936.
Jacobsen S, Goldberg J, Cooper C, & Lockwood S (1992) The association between water fluoridation and hip fracture among white women and men aged 65 years and older. A national ecologic study. Ann Epidemiol, 2: 617–626.
Jacobsen S, O’Fallon M, & Melton L (1993) Hip fracture incidence before and after fluoridation of the public water supply, Rochester, Minnesota. Am J Public Health, 83: 743–745.
Jacobson JS, Weinstein LH, McCune DC, & Hitchcock AE (1966) The accumulation of fluorine by plants. J Air Pollut Control Assoc, 16: 412–417.
Jacqmin-Gadda H, Commenges D, & Dartigues JF (1995) Fluorine concentration in drinking water and fractures in the elderly. J Am Med Assoc, 273: 775–776.
Jacqmin-Gadda H, Fourrier A, Commenges D, & Dartigues JF (1998) Risk factors for fractures in the elderly. Epidemiology, 9: 417–423.
Jain S & Susheela S (1987a) Effect of sodium fluoride on erythrocyte membrane function — with reference to metal ion transport in rabbits. Chemosphere, 16: 1087–1094.
Jain S & Susheela A (1987b) Effect of sodium fluoride on antibody formation in rabbits. Environ Res, 44: 117–125.
Jha M, Susheela A, Krishna N, Rajyalakshmi K, & Venkiah K (1982) Excessive ingestion of fluoride and the significance of sialic acid: Glycosaminoglycans in the serum of rabbit and human subjects. J Toxicol Clin Toxicol, 19: 1023–1030.
Johansson TSK & Johansson MP (1972) Sublethal doses of sodium fluoride affecting fecundity of confused flour beetles. J Econ Entomol, 65(2): 356–357.
Johnson J Jr & Bawden JW (1987) The fluoride content of infant formulas available in 1985. Pediatr Dent, 9: 33–37.
Jolly SS, Singh BM, Mathur OC, & Malhotra KC (1968) Epidemiological, clinical and biochemical study of endemic dental and skeletal fluorosis in Punjab. Br Med J, 4: 427–429.
Jones C, Harries J, & Martin A (1971) Fluorine in leafy vegetables. J Sci Food Agric, 22: 602–605.
Jones C, Callahan M, & Huberman E (1988a) Sodium fluoride promotes morphological transformation of Syrian hamster embryo cells. Carcinogenesis, 9: 2279–2284.
Jones C, Huberman E, Callaham M, Tu A, Halloween W, Pallota S, Sivak A, Lubet R, Avery M, Kouri R, Spalding J, & Tennant R (1988b) An inter-laboratory evaluation of the Syrian hamster embryo cell transformation assay using fourteen coded chemicals. Toxicol In Vitro, 2: 103–116.
Jones G, Riley M, Couper D, & Dwyer T (1999) Water fluoridation, bone mass and fracture: a quantitative overview of the literature. Aust N Z J Public Health, 23: 34–40.
Joseph S & Gadhia PK (2000) Sister chromatid exchange frequency and chromosome aberrations in residents of fluoride endemic regions of south Gujarat. Fluoride, 33: 154–158.
Joy CM & Balakrishnan KP (1990) Effect of fluoride on axenic cultures of diatoms. Water Air Soil Pollut, 49: 241–249.
Jubb TF, Annand TE, Main DC, & Murphy GM (1993) Phosphorus supplements and fluorosis in cattle — a northern Australian experience. Aust Vet J, 70: 379–383.
Kabasakalis V & Tsolaki A (1994) Fluoride content of vegetables irrigated with waters of high fluoride levels. Fresenius Environ Bull, 3(6): 377–380.
Kabata-Pendias A & Pendias H (1984) Fluorine. In: Trace elements in soil and plants. Boca Raton, Florida, CRC Press, pp 209–215.
Kaminsky L, Mahoney M, Leach J, Melius J, & Miller M (1990) Fluoride: Benefits and risks of exposure. Crit Rev Oral Biol Med, 1: 261–281.
Kanisawa M & Schroeder H (1969) Life term studies on the effect of trace elements on spontaneous tumours in mice and rats. Cancer Res, 29: 892–895.
Kaplan HM, Yee N, & Glaczenski SS (1964) Toxicity of fluoride for frogs. Lab Anim Care, 14(3): 185–188.
Karagas MR, Baron JA, Barrett JA, & Jacobsen SJ (1996) Patterns of fracture among United States elderly: geographic and fluoride effects. Ann Epidemiol, 6: 209–216.
Karstad L (1967) Fluorosis in deer (Odoceileus virginianus). Bull Wildl Dis Assoc, 3: 42–46.
Karthikeyan G, Pius S, & Apparao BV (1996) Contribution of fluoride in water and food to the prevalence of fluorosis in areas of Tamil Nadu in south India. Fluoride, 29: 151–155.
Karunagaran VM & Subramanian A (1992) Fluoride pollution in the Uppanar Estuary, Cuddalore, South India. Mar Pollut Bull, 24(10): 515–517.
Kay CE, Tourangeau PC, & Gordon CC (1975) Industrial fluorosis in wild mule and whitetail deer from western Montana. Fluoride, 8(4): 182–191.
Keller T (1980) The simultaneous effect of soil-borne NaF and air pollutant SO2 on CO2-uptake and pollutant accumulation. Oecologia, 44: 283–285.
Khalil A (1995) Chromosome aberrations in cultured rat bone marrow cells treated with inorganic fluorides. Mutat Res, 343: 67–74.
Khalil AM & Da’dara AA (1994) The genotoxic and cytotoxic activities of inorganic fluoride in cultured rat bone marrow cells. Arch Environ Contam Toxicol, 26: 60–63.
Kierdorf H & Kierdorf U (1997) Disturbances of the secretory stage of amelogenesis in fluorosed deer teeth: a scanning electron-microscopic study. Cell Tissue Res, 289: 125–135.
Kierdorf H & Kierdorf U (1999) Reduction of fluoride deposition in the vicinity of a brown coal-fired power plant as indicated by bone fluoride concentrations of roe deer (Capreolus capreolus). Bull Environ Contam Toxicol, 63: 473–477.
Kierdorf H, Kierdorf U, Sedlacek F, & Erdelen M (1996) Mandibular bone fluoride levels and occurrence of fluoride induced dental lesions in populations of wild deer (Cervus elaphus) from central Europe. Environ Pollut, 93(1): 75–81.
Kierdorf H, Kierdorf U, Richards A, & Sedlacek F (2000) Disturbed enamel formation in wild boars (Sus scrofa) from fluoride polluted areas in Central Europe. Anat Rec, 259: 12–24.
Kierdorf U (1988) [A study on chronic fluoride intoxication in roe deer (Capreolus capreolus L.) caused by emissions.] Z Jagdwiss, 34: 192–204 (in German).
Kierdorf U & Kierdorf H (1999) Dental fluorosis in wild deer: its use as a biomarker of increased fluoride exposure. Environ Monit Assess, 57: 265–275.
Kierdorf U & Kierdorf H (2000) The fluoride content of antlers as an indicator of fluoride exposure in red deer (Cervus elaphus): A historical biomonitoring study. Arch Environ Contam Toxicol, 38: 121–127.
Kierdorf U, Kierdorf H, Erdelen M, & Korsch JP (1989) Mandibular fluoride concentration and its relation to age in roe deer (Capreolus capreolus L.). Comp Biochem Physiol, 94A(4): 783–785.
Kierdorf U, Kierdorf H, & Fejerskov O (1993) Fluoride-induced developmental changes in enamel and dentine of European roe deer (Capreolus capreolus L.) as a result of environmental pollution. Arch Oral Biol, 38(12): 1071–1081.
Kierdorf U, Kierdorf H, Sedlacek F, & Fejerskov O (1996) Structural changes in fluorosed dental enamel of red deer (Cervus elaphus L.) from a region with severe environmental pollution by fluorides. J Anat, 188: 183–195.
Kierdorf U, Richards A, Sedlacek F, & Kierdorf H (1997) Fluoride content and mineralization of red deer (Cervus elaphus ) antlers and pedicles from fluoride polluted and uncontaminated regions. Arch Environ Contam Toxicol, 32: 222–227.
Kierdorf U, Kierdorf H, & Boyde A (2000) Structure and mineralisation density of antler and pedicle bone in red deer (Cervus elaphus L.) exposed to different levels of environmental fluoride: a quantitative backscattered electron imaging study. J Anat, 196: 71–83.
Kinlen L & Doll R (1981) Fluoridation of water supplies and cancer mortality. III. A re-examination of mortality in cities in the USA. J Epidemiol Community Health, 35: 239–244.
Kiritsy MC, Levy SM, Warren JJ, Guha-chowdhury M, Heilman JR, & Marshall T (1996) Assessing fluoride concentrations of juices and juice-flavoured drinks. J Am Dental Assoc, 127: 895–902.
Kirk PWW & Lester JN (1986) Halogen compounds. In: Harrison RM & Perry R ed. Handbook of air pollution analysis, 2nd ed. London, Chapman & Hall, pp 425–462.
Kishi K & Ishida T (1993) Clastogenic activity of sodium fluoride in great ape cells. Mutat Res, 301: 183–188.
Kishi K & Tonomura A (1984) Cytogenetic effects of sodium fluoride. Mutat Res, 130: 367 (abstract).
Kissa E (1987) Determination of inorganic fluoride in blood with a fluoride ion-selective electrode. Clin Chem, 33: 253–255.
Klumpp A, Klumpp G, & Domingos M (1994) Plants as bioindicators of air pollution at the Serra do Mar near the industrial complex of Cubatao, Brazil. Environ Pollut, 85: 109–116.
Klumpp A, Klumpp G, Domingos M, & Dias Da Silva M (1996a) Fluoride impact on native tree species of the Atlantic forest near Cubatao, Brazil. Water Air Soil Pollut, 87(1–4): 57–71.
Klumpp A, Dominos M, & Klumpp G (1996b) Assessment of the vegetation risk by fluoride emissions from fertiliser industries at Cubatao, Brazil. Sci Total Environ, 192: 219–228.
Klut ME, Bisalputra T, & Antia NJ (1981) Abnormal ultrastructural features of a marine dinoflagellate adapted to grow successfully in the presence of inhibitory fluoride concentration. J Protozool, 28(4): 406–414.
Knox E (1985) Fluoridation of water and cancer: A review of the epidemiological evidence. Report of the British Working Party. London, HMSO.
Kono K, Yoshida Y, Yamagata H, Tanimura Y, Takeda Y, Harada A, & Doi K (1986) Fluoride clearance in the aging kidney. Fluoride Research 1985. Stud Environ Sci, 27: 407–414.
Kono K, Yoshida Y, Watanabe M, Watanabe H, Inoue S, Murao M, & Doi K (1990) Elemental analysis of hair among hydrofluoric acid exposed workers. Int Arch Occup Environ Health, 62: 85–88.
Korkka-Niemi K, Sipilä A, Hatva T, Hiisvirta L, Lahti K, & Alfthan G (1993) Nation-wide rural well water survey. The quality of household water and factors influencing it. Helsinki, National Board of Waters and the Environment, 225 pp.
Korns R (1969) Relationship of water fluoridation to bone density in two NY towns. Public Health Rep, 84: 815–825.
Kragstrup J, Richards A, & Fejerskov O (1989) Effects of fluoride on cortical bone remodelling in the growing domestic pig. Bone, 10: 421–424.
Krasowska A (1989) Influence of low-chitin krill meal on reproduction of Clethrionomys glareolus (Schreber, 1780). Comp Biochem Physiol, 94C(1): 313–320.
Kraut A & Lilis R (1990) Pulmonary effects of acute exposure to degradation products of sulphur hexafluoride during electrical cable work. Br J Ind Med, 47: 828–832.
Krishnamachari KAVR (1976) Further observations on the syndrome of genu valgum of South India. Ind J Med Res, 64: 284–291.
Krishnamachari K (1987) Fluorine. Trace Elem Hum Anim Nutr, 1: 365–415.
Krishnamachari KAVR & Krishnaswamy K (1973) Genu valgum and osteoporosis in an area of endemic fluorosis. Lancet, 2: 887–889.
Kröger H, Alhava E, Honkanen R, Tupperainen M, & Saarikoski S (1994) The effect of fluoridated drinking water on axial bone mineral density — a population-based study. Bone Miner, 27: 33–41.
Krook L & Maylin GA (1979) Industrial fluoride pollution. Chronic fluoride poisoning in Cornwall Island cattle. Cornell Vet, 69(suppl 8): 4–70.
Kudo A & Garrec J-P (1983) Accidental release of fluoride into experimental pond and accumulation in sediments, plants, algae, molluscs and fish. Regul Toxicol Pharmacol, 3: 189–198.
Kudo A, Garrec JP, & Plebin R (1987) Fluoride distribution and transport along rivers in the French Alps. Ecotoxicol Environ Saf, 13(3): 263–273.
Kühn R, Pattard M, Pernak K-D, & Winter A (1989) Results of the harmful effects of water pollutants to Daphnia magna in the 21 day reproduction test. Water Res, 23(4): 501–510.
Kumpulainen J & Koivistoinen P (1977) Fluorine in foods. Residue Rev, 68: 37–57.
Kurttio P, Gustavsson N, Vartiainen T, & Pekkanen J (1999) Exposure to natural fluoride in well water and hip fracture: a cohort analysis in Finland. Am J Epidemiol, 150: 817–824.
Kuusisto AN & Telkkä A (1961) The effect of sodium fluoride on the metamorphosis of tadpoles. Acta Odontol Scand, 19: 121–127.
Lahermo P & Backman B (2000) The occurrence and geochemistry of fluorides with special reference to natural waters in Finland. Espoo, Geological Survey of Finland, 40 pp. (Research Report 149).
Lahermo P, Ilmasti M, Juntunen R, & Taka M (1990) The geochemical atlas of Finland. Part 1: The hydrogeochemical mapping of Finnish groundwater. Espoo, Geological Survey of Finland.
Lalumandier JA & Ayers LW (2000) Fluoride and bacterial content of bottled water vs tap water. Arch Fam Med, 9: 246–250.
Lan CF, Lin IF, & Wang SJ (1995) Fluoride in drinking water and the bone mineral density of women in Taiwan. Int J Epidemiol, 24: 1182–1187.
Landy RB, Lambertsen RH, Palsson PA, Krook L, Nevius A, & Eckerlin R (1991) Fluoride in the bone and diet of fin whales, Balaenoptera physalus. Mar Environ Res, 31: 241–247.
Laroche M & Mazières B (1991) Side-effects of fluoride therapy. Baillière’s Clin Rheumatol, 5: 61–76.
Larsson K, Eklund A, Arns R, Lowgren H, Nystrom J, Sundstrom G, & Tornling H (1989) Lung function and bronchial reactivity in aluminum potroom workers. Scand J Work Environ Health, 15: 296–301.
Lasne C, Lu Y, & Chouroulinkov I (1988) Transforming activities of sodium fluoride in cultured Syrian hamster embryo and BALB/3T3 cells. Cell Biol Toxicol, 4: 311–324.
Latham MC & Gretch P (1967) The effects of excessive fluoride intake. Am J Public Health, 57: 651–660.
Lau KH & Baylink DJ (1998) Molecular mechanism of action of fluoride on bone cells. J Bone Miner Res, 13: 1660–1667.
Laurence JA (1983) Effects of fluoride on plant–pathogen interactions. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 145–149.
Laurence JA & Reynolds KL (1984) Growth and lesion development of Xanthomonas campestris pv. phaseoli on leaves of red kidney bean plants exposed to hydrogen fluoride. Phytopathology, 74: 578–580.
Lavado RS, Reinaudi NB, & Parodi AA (1983) Fluoride retention and leach possibilities in Argentine salt-affected soils. Fluoride Q Rep, 16(4): 247–251.
LeBlanc GA (1980) Acute toxicity of priority pollutants to water flea (Daphnia magna). Bull Environ Contam Toxicol, 24: 684–691.
LeBlanc GA (1984) Interspecies relationships in acute toxicity of chemicals to aquatic organisms. Environ Toxicol Chem, 3: 47–60.
LeBlanc F, Comeau G, & Rao DN (1971) Fluoride injury symptoms in epiphytic lichens and mosses. Can J Bot, 49: 1691–1698.
Leece DR, Scheltema JH, Anttonen T, & Weir RG (1986) Fluoride accumulation and toxicity in grapevines Vitis vinifera L. in New South Wales. Environ Pollut, A40: 145–172.
Lehmann R, Wapniarz M, Hofmann B, Pieper B, Haubitz I, & Allolio B (1998) Drinking water fluoridation: bone mineral density and hip fracture incidence. Bone, 22: 273–278.
Levy SM (1994) Review of fluoride exposures and ingestion. Community Dent Oral Epidemiol, 22: 173–180.
Levy SM, Kiritsy MC, & Warren JJ (1995) Sources of fluoride intake in children. J Public Health Dent, 55: 39–52.
Li Y, Dunipace A, & Stookey G (1987a) Absence of mutagenic or antimutagenic activities of fluoride in Ames Salmonella assays. Mutat Res, 190: 229–236.
Li Y, Heerema N, Dunipace A, & Stokey G (1987b) Genotoxic effects of fluoride evaluated by sister-chromatid exchange. Mutat Res, 192: 191–201.
Li Y, Dunipace A, & Stookley G (1987c) Lack of genotoxic effects of fluoride in the mouse bone-marrow micronucleus test. J Dent Res, 66: 1687–1690.
Li Y, Dunipace A, & Stookey G (1987d) Effects of fluoride on the mouse sperm morphology test. J Dent Res, 66: 1509–1511.
Li Y, Zhang W, Noblitt T, Dunipace A, & Stokey G (1989) Genotoxic evaluation of chronic fluoride exposure: sister-chromatid exchange study. Mutat Res, 227: 159–165.
Li Y, Liang CK, Katz BP, Brizendine EJ, & Stookey GK (1995) Long-term exposure to fluoride in drinking water and sister chromatid exchange frequency in human blood lymphocytes. J Dent Res, 74: 1468–1474.
Li Y, Liang C, Slemenda CW, Ji R, Sun S, Cao J, Emsley C, Ma F, Wu Y, Ying P, Zhang Y, Gao S, Zhang W, Katz B, Niu S, Cao S, & Johnston C (2001) Effect of long-term exposure to fluoride in drinking water on risks of bone fractures. J Bone Miner Res, 16(5): 932–939.
Liang CK, Ji R, & Cao SR (1997) Epidemiological analysis of endemic fluorosis in China. Environ Carcinogen Ecotoxicol Rev, C15(2): 123–138.
Liu BK (1996) Lung X-ray changes in skeletal fluorosis caused by coal combustion. Fluoride, 29: 33–35.
Liu Y ed. (1995) Human exposure assessment of fluoride. An international study within the WHO/UNEP Human Exposure Assessment Location (HEAL) Programme. Beijing, Chinese Academy of Preventive Medicine, Institute of Environmental Health Monitoring, Technical Cooperation Centre of Fluoride/HEAL Programme, 64 pp.
Low PS & Bloom H (1988) Atmospheric deposition of fluoride in the lower Tamar Valley, Tasmania. Atmos Environ, 22(9): 2049–2056.
Lu D, Zheng Z, Wang J, & Chen K (1989) [Study of the mutagenic effect of sodium fluoride.] Yichuan [Hereditas (Beijing)], 11: 23 (in Chinese).
Lund K, Ekstrand J, Boe J, Sostrand P, & Kongerud J (1997) Exposure to hydrogen fluoride: an experimental study in humans of concentrations of fluoride in plasma, symptoms, and lung function. Occup Environ Med, 54: 32–37.
Ma J, Cheng L, Bai W, & Wu H (1986) [The effects of sodium fluoride on SCEs of the mice and on the micronucleus of the bone marrow of pregnant mice and its fetus liver.] Yichuan [Hereditas (Beijing)], 8: 39–41 (in Chinese).
Machoy Z, Dabkowska E, Samujlo D, Ogonski T, Raczynski J, & Gebczynska Z (1995) Relationship between fluoride content in bones and the age in European elk (Alces alces L.). Comp Biochem Physiol, 111C(1): 117–120.
MacIntire WH, Sterges AJ, & Shaw WM (1955) Fate and effects of hydrofluoric acid added to four Tennessee soils in a four year lysimeter study. J Agric Food Chem, 3(9): 777–782.
MacLean DC & Schneider RE (1981) Effects of gaseous hydrogen fluoride on the yield of field-grown wheat. Environ Pollut, 24: 39–44.
MacLean DC, Schneider RE, & McCune DC (1977) Effects of chronic exposure to gaseous fluoride on yield of field-grown bean and tomato plants. J Am Soc Hortic Sci, 102(3): 297–299.
MacLean DC, McCune DC, & Schneider RE (1984a) Growth and yield of wheat and sorghum after sequential exposures of hydrogen fluoride. Environ Pollut, A36: 351–365.
MacLean DC, Weinstein LH, McCune DC, & Schneider RE (1984b) Fluoride-induced suture red spot in "Elberta" peach. Environ Exp Bot, 24(4): 353–367.
Madans J, Kleinman J, & Cornoni-Huntley J (1983) The relationship between hip fracture and water fluoridation: an analysis of national data. Am J Public Health, 73: 296–298.
Madkour S & Weinstein LH (1988) Effects of fumigation with hydrogen fluoride on the loading of [14C]sucrose into the phloem of soybean leaves. Environ Toxicol Chem, 7: 317–320.
Mahadevan TN, Meenakshy V, & Mishra UC (1986) Fluoride cycling in nature through precipitation. Atmos Environ, 20: 1745–1749.
Mahoney MC, Nasca PC, Burnett WS, & Melius JM (1991) Bone cancer incidence rates in New York State: Time trends and fluoridated drinking water. Am J Public Health, 81: 475–479.
Malea P (1995) Fluoride content in three seagrasses of the Antikyra Gulf (Greece) in the vicinity of an aluminum factory. Sci Total Environ, 166: 11–17.
Marais JSC (1944) Monofluoracetic acid, the toxic principle of the "Grifblaar" Dichapetabum cymosum (Hook) Engl Onderstepoort. J Vet Sci Anim Ind, 20(1): 67–73.
Marie P & Mott M (1986) Short-term effects of fluoride and strontium on bone formation and resorption in the mouse. Metabolism, 35: 547–561.
Marks TA, Schellenberg D, Metzler CM, Oostveen J, & Morey MJ (1984) Effect of dog food containing 460 ppm fluoride on rat reproduction. J Toxicol Environ Health, 14: 707–714.
Martin GR, Brown KS, Matheson DW, Lebowitz H, Singer L, & Ophaug R (1979) Lack of cytogenetic effects in mice or mutations in Salmonella receiving sodium fluoride. Mutat Res, 66: 159–167.
Martin J-M & Salvadori F (1983) Fluoride pollution in French rivers and estuaries. Estuarine Coastal Shelf Sci, 17: 231–242.
Maurer J, Cheng M, Boysen B, & Anderson R (1990) Two-year carcinogenicity study of sodium fluoride in rats. J Natl Cancer Inst, 82: 1118–1126.
Maurer J, Cheng M, Boysen B, Squire R, Strandberg J, Weisbrode J, & Anderson R (1993) Confounded carcinogenicity study of sodium fluoride in CD-1 mice. Regul Toxicol Pharmacol, 18: 154–168.
Mayer DF, Lunden JD, & Weinstein LH (1988) Evaluation of fluoride levels and effects on honey bees (Apis mellifera L.) (Hymenoptera: Apidae). Fluoride Q Rep, 21(3): 113–120.
Mayer TG & Gross PL (1985) Fatal systemic fluorosis due to hydrofluoric acid burns. Ann Emerg Med, 14: 149–153.
McClenahen J (1976) Distribution of soil fluorides near an airborne fluoride source. J Environ Qual, 5: 472–475.
McClenahen JR (1978) Community changes in a deciduous forest exposed to air pollution. Can J For Res, 8: 432–438.
McClure FJ (1944) Fluoride domestic water and systemic effects. I. Relation to bone fracture experience, height and weight of high school boys and young selectees of the armed forces of the United States. Public Health Rep, 59: 1543–1558.
McClurg TP (1984) Effects of fluoride, cadmium and mercury on the estuarine prawn Penaeus indicus. Water SA, 10(1): 40–45.
McCune DC, Lauver TL, & Hansen KS (1991) Relationship of concentration of gaseous hydrogen fluoride to incidence and severity of foliar lesions in black spruce and white spruce. Can J For Res, 21: 756–761.
McDonagh MS, Whiting PF, Wilson PM, Sutton AJ, Chestnutt I, Cooper J, Misso K, Bradley M, Treasure E, & Kleijnen J (2000) Systematic review of water fluoridation. Br Med J, 321: 855–859.
McGuire S, Vanable E, McGuire M, Buckwalter JA, & Douglass CW (1991) Is there a link between fluoridated water and osteosarcoma? J Am Dent Assoc, 122: 38–45.
McIvor M (1990) Acute fluoride toxicity: pathophysiology and management. Drug Saf, 5: 79–85.
McKnight-Hanes M, Leverett D, Adair S, & Shields C (1988) Fluoride content of infant formulas: soy-based formulas as a potential factor in dental fluorosis. Pediatr Dent, 10: 189–194.
McLaughlin MJ, Tiller KG, Naidu R, & Stevens DP (1996) Review: the behaviour and environmental impact of contaminants in fertilizers. Aust J Soil Res, 34: 1–54.
McLaughlin MJ, Stevens DP, Keerthisinghe G, Cayley JWD, & Ridley AM (2001) Contamination of soil with fluoride by long-term application of superphosphates to pastures and risks to grazing animals. Aust J Soil Res, 39(3): 627–640.
McNulty IB & Lords JL (1960) Possible explanation of fluoride-induced respiration in Chlorella pyrenoidosa. Science, 132: 1553–1554.
Meeussen JCL, Scheidegger A, Hiemstra T, Van Riemsdijk WH, & Borkovec M (1996) Predicting multicomponent adsorption and transport of fluoride at variable pH in a goethite–silica sand system. Environ Sci Technol, 30(2): 481–488.
Meng Z & Zhang B (1997) Chromosomal aberrations and micronuclei in lymphocytes of workers at a phosphate fertilizer factory. Mutat Res, 393: 283–288.
Meng Z, Meng H, & Cao X (1995) Sister-chromatid exchange in lymphocytes of workers at a phosphate fertilizer factory. Mutat Res, 334: 243–246.
Messer H & Ophaug R (1993) Influence of gastric acidity on fluoride absorption in rats. J Dent Res, 72: 619–622.
Messer HH, Armstrong WD, & Singer L (1973) Influence of fluoride intake on reproduction in mice. J Nutr, 103: 1319–1326.
Messer H, Nopakum J, & Rudney J (1989) Influence of pH on intestinal fluoride transport in vitro. J Dent Assoc Thailand, 39: 226–232.
Meunier PJ, Severt JL, Reginster JY, Briancon D, Apelboom T, Netter P, Loeb G, Rouillon A, Barry S, Evreus JC, Avouac B, & Marchandise X (1998) Fluoride salts are no better at preventing new vertebral fractures than calcium–vitamin D in postmenopausal osteoporosis: the FAVO Study. Osteoporosis Int, 8: 4–12.
Michael M, Barot VV, & Chinoy NJ (1996) Investigations of soft tissue functions in fluorotic individuals of North Gujarat. Fluoride, 29: 63–71.
Michel J, Suttie J, & Sunde M (1984) Fluorine deposition in bone as related to physiological state. Poult Sci, 63: 1407–1411.
Mihashi M & Tsutsui T (1996) Clastogenic activity of sodium fluoride to rat vertebral body-derived cells in culture. Mutat Res, 368: 7–13.
Mikaelian I, Qualls CW, De Guise S, Whaley MW, & Martineau D (1999) Bone fluoride concentrations in beluga whales from Canada. J Wildl Dis, 35(2): 356–360.
Milhaud G, El Bahri L, & Dridi A (1981) The effects of fluoride on fish in Gabes Gulf. Fluoride, 14(4): 161–168.
Mishra PC & Mohapatra AK (1998) Haematological characteristics and bone fluoride content in Bufo melanostictus from an aluminium industrial site. Environ Pollut, 99: 421–423.
Mithal A, Trivedi N, Gupta SK, Kumar S, & Gupta RK (1993) Radiological spectrum of endemic fluorosis: relationship with calcium intake. Skeletal Radiol, 22: 257–261.
Mohammed A & Chandler M (1982) Cytological effects of sodium fluoride on mice. Fluoride, 15: 110–118.
Moore M, Clive D, Hozier J, Howard B, Batsun A, Turner N, & Sawyer J (1985) Analysis of trifluorothymidine resistant (TFTr) mutants of L5178Y/TK± mouse lymphoma cells. Mutat Res, 151: 161–174.
Morgan L, Allred E, Tavares M, Bellinger D, & Needleman H (1998) Investigation of the possible associations between fluorosis, fluoride exposure, and childhood behaviour problems. Am Acad Pediatr Dent, 20(4): 244–252.
Morris JB & Smith FA (1982) Regional deposition and absorption of inhaled hydrogen fluoride in the rat. Toxicol Appl Pharmacol, 62: 81–89.
Mosekilde I, Kragstrup J, & Richards A (1987) Compression strength, ash weight and volume of vertebral trabecular bone in experimental fluorosis in pigs. Calcif Tissue Int, 40: 318–322.
Moss ME, Kanarek MS, Anderson HA, Hanrahan LP, & Remington PL (1995) Osteosarcoma, seasonality, and environmental factors in Wisconsin, 1979–1989. Arch Environ Health, 50: 235–241.
Moule GR (1944) Fluoride poisoning of livestock. Queensl Agric J, 58: 54–55.
Mullenix PJ, Denbesten K, Schunior A, & Keernan WJ (1995) Neurotoxicity of sodium fluoride in rats. Neurotoxicol Teratol, 17(2): 169–177.
Mullet T, Zoeller T, Bingham H, Pepine CJ, Prida XE, Castenholz R, & Kirby R (1987) Fatal hydrofluoric acid cutaneous exposure with refractory ventricular fibrillation. J Burn Care Rehab, 8: 216–219.
Mur J, Moulin J, Meyer-Bisch C, Massin N, Coulon J, & Loulergue J (1987) Mortality of aluminum reduction plant workers in France. Int J Epidemiol, 16: 257–264.
Muramoto S, Nishizaki H, & Aoyama I (1991) Effects of fluorine emissions on agricultural products surrounding an aluminum factory. J Environ Sci Health, B26: 351–356.
Murray F (1981) Effects of fluorides on plant communities around an aluminium smelter. Environ Pollut, A24: 45–56.
Murray F (1984a) Effect of long term exposure to hydrogen fluoride on grapevines. Environ Pollut, A36: 337–349.
Murray F (1984b) Fluoride retention in highly leached disturbed soils. Environ Pollut, B7: 83–95.
Murray F & Wilson S (1988a) Effects of sulphur dioxide, hydrogen fluoride and their combination on three Eucalyptus species. Environ Pollut, 52: 265–279.
Murray F & Wilson S (1988b) Joint action of sulfur dioxide and hydrogen fluoride on growth of Eucalyptus tereticornis. Environ Exp Bot, 28(4): 343–349.
Murray F & Wilson S (1988c) The joint action of sulphur dioxide and hydrogen fluoride on the yield and quality of wheat and barley. Environ Pollut, 55: 239–249.
Murray TM, Harrison JE, Bayley TA, Josse RG, Sturtridge WC, Chow R, Budden F, Laurier L, Pritzker KP, Kandel R, Vieth R, Strauss A, & Goodwin S (1990) Fluoride treatment of postmenopausal osteoporosis: age, renal function, and other clinical factors in the osteogenic response. J Bone Miner Res, 5(suppl 1): S27–S35.
Naccache H, Simard P, Trahan L, Demers M, Lapointe C, & Brodeur J (1990) Variability in the ingestion of toothpaste by preschool children. Caries Res, 24: 359–363.
Naccache H, Simard P, Trahan L, Brodeur JM, Demers M, Lachapelle D, & Bernard PM (1992) Factors affecting the ingestion of fluoride dentifrice by children. J Public Health Dent, 52: 222–226.
Nair KR, Manji F, & Gitonga JN (1984) The occurrence and distribution of fluoride in groundwaters in Kenya. East Afr J Med, 61: 503–512.
Neal C (1989) Fluorine variations in Welsh streams and soil waters. Sci Total Environ, 80: 213–223.
Neal C, Smith CJ, Walls J, Billingham P, Hill S, & Neal M (1990) Comments on the hydrological regulation of the halogen elements in rainfall, stemflow, throughfall and stream waters at an acidic forested area in mid-Wales. Sci Total Environ, 91: 1–11.
Needleman HL, Fueschel SM, & Rothman KJ (1974) Fluoridation and the occurrence of Down’s syndrome. N Engl J Med, 291: 821–823.
Neeley KL & Harbaugh FG (1954) Effects of fluoride ingestion on a herd of dairy cattle in the Lubbock, Texas, area. J Am Vet Med Assoc, 124: 344–350.
Nell JA & Livanos G (1988) Effects of fluoride concentration in seawater on growth and fluoride accumulation by Sydney rock oyster (Saccostrea commercialis) and flat oyster (Ostrea angasi). Spat Water Res, 22(6): 749–753.
Neuhold JM & Sigler WF (1960) Effects of sodium fluoride on carp and rainbow trout. Trans Am Fish Soc, 89: 358–370.
Neuhold JM & Sigler WF (1962) Chlorides affect the toxicity of fluorides to rainbow trout. Science, 135: 732–733.
Neumüller O (1981) Römpps Chemie Lexikon, 8th ed. Vol. 2. Stuttgart, Franck’sch Verlagshandlung.
Neumüller O (1987) Römpps Chemie Lexikon, 8th ed. Vol. 5. Stuttgart, Franck’sch Verlagshandlung.
Newbrun E (1992) Current regulations and recommendations concerning water fluoridation, fluoride supplements, and topical fluoride agents. J Dent Res, 71: 1255–1265.
Newman JR (1977) Sensitivity of the house martin, Delichon urbica, to fluoride emissions. Fluoride, 10: 73–76.
Newman JR & Markey D (1976) Effects of elevated levels of fluoride on deer mice (Peromyscus maniculatus). Fluoride, 9(1): 47–52.
Newman JR & Yu M-H (1976) Fluorosis in black-tailed deer. J Wildl Dis, 12: 39–41.
Nichol BE, Budd K, Palmer GR, & MacArthur JD (1987) The mechanisms of fluoride toxicity and fluoride resistance in Synechococcus leopoliensis (Cyanophyceae). J Phycol, 23(4): 535–541.
NIPH (1996) System of monitoring the environmental impact on population health of the Czech Republic. Summary report — 1995. Prague, National Institute of Public Health.
Nowak A & Nowak M (1989) Fluoride concentration of bottled and processed waters. Iowa Dent J, 75: 28.
Nriagu JO (1986) Chemistry of the River Niger. 1. Major ions. Sci Total Environ, 58: 81–88.
NTP (1990) Toxicology and carcinogenesis studies of sodium fluoride (CAS No.
Oberly T, Rexroat M, Bewsey B, Richardson K, & Michalis K (1990) An evaluation of the CHO/HGPRT mutation assay involving suspension cultures and soft agar cloning: Results for 33 chemicals. Environ Mol Mutagen, 16: 260–271.
Oelrichs RB & McEwan T (1961) Isolation of the toxic principle in Acacia georginae. Nature, 190: 808–809.
Oelschläger W (1971) Fluoride uptake in soil and its depletion. Fluoride, 4: 80–84.
Oguro A, Cervenka J, & Horii K-I (1995) Effect of sodium fluoride on chromosomal ploidy and breakage in cultured human diploid cells (IMR-90): an evaluation of continuous and short-time treatment. Pharmacol Toxicol, 76: 292–296.
Oliveby A, Lagerlöf F, Ekstrand J, & Dawes C (1989) Studies on fluoride excretion in human whole saliva and its relation to flow rate and plasma fluoride levels. Caries Res, 23: 243–246.
Oliveby A, Twetman S, & Ekstrand J (1990) Diurnal fluoride concentration in whole saliva in children living in a high- and a low-fluoride area. Caries Res, 24: 44–47.
Oliveira L, Antia NJ, & Bisalputra T (1978) Culture studies on the effects from fluoride pollution on the growth of marine phytoplankters. J Fish Res Board Can, 35: 1500–1504.
Omueti J & Jones R (1977) Regional distribution of fluorine in Illinois soils. Soil Sci Soc Am J, 41: 771–777.
Ophaug RH, Singer L, & Harland BF (1985) Dietary fluoride intake of 6-month and 2-year-old children in four dietary regions of the United States. Am J Clin Nutr, 42: 701–707.
Orcel P, de Vernejoul MC, Prier A, Miravet L, Kuntz D, & Kaplan G (1990) Stress fractures of the lower limbs in osteoporotic patients treated with fluoride. J Bone Miner Res, 5(suppl 1): S191–S194.
Overton WS, Kanciruk P, Hook LA, Eilers JM, Landers DH, Brakke DF, Blick DJ, Linthurst RA, De Haan MD, & Omernik JM (1986) Characteristics of lakes in eastern United States. Vol. II. Washington, DC, US Environmental Protection Agency (EPA/600/4-86/007c).
Pack MR (1971) Effects of hydrogen fluoride on bean reproduction. J Air Pollut Control Assoc, 21: 133–137.
Pak CYC, Sakhaee K, Piziak V, Peterson RD, Breslau NA, Boyd P, Poindexter JR, Herzog J, Heard-Sakhaee A, Haynes S, Adams-Huet B, & Reisch JS (1994) Slow-release sodium fluoride in the management of postmenopausal osteoporosis. A randomized controlled trial. Ann Intern Med, 120: 625–632.
Pang YX, Guo YQ, Fu KW, Sun YF, & Tang RQ (1996) The effects of fluoride, alone and in combination with selenium, on the morphology and histochemistry of skeletal muscle. Fluoride, 29: 59–62.
Pankhurst NW, Boyden CR, & Wilson JB (1980) The effect of a fluoride effluent on marine organisms. Environ Pollut, A23: 299–312.
Paranjpe MG, Chandra AMS, Qualls CW, McMurry ST, Rohrer MD, Whaley MM, Lochmiller RL, & McBee K (1994) Fluorosis in a wild cotton rat (Sigmodon hispidus) population inhabiting a petrochemical waste site. Toxicol Pathol, 22(6): 569–578.
Parker DR, Norvell WA, & Chaney RL (1995) GEOCHEM-PC: a chemical speciation program for IBM and compatible personal computers. Madison, Wisconsin, Soil Science Society of America (Special Publication 42).
Parkins FM, Tinaoff N, Moutinho M, Anstey MB, & Waziri MH (1974) Relationship of human plasma fluoride and bone fluoride to age. Calcif Tissue Res, 16: 335–338.
Pati P & Bhunya S (1987) Genotoxic effect of an environmental pollutant, sodium fluoride, in mammalian in vivo test system. Caryologia, 40: 79–87.
Patra RC, Dwivedi SK, Bhardwaj B, & Swarup D (2000) Industrial fluorosis in cattle and buffalo around Udaipur, India. Sci Total Environ, 253: 145–150.
Pattee OH, Wiemeyer SN, & Swineford DM (1988) Effects of dietary fluoride on reproduction in eastern screech-owls. Arch Environ Contam Toxicol, 17: 213–218.
Patten J, Whitford G, Stringer G, & Pashley D (1978) Oral absorption of radioactive fluoride and iodide in rats. Arch Oral Biol, 23: 215–217.
Peirce AW (1952) Studies on fluorosis of sheep. I. The toxicity of water-borne fluoride for sheep maintained in pens. Aust J Agric Res, 3(3): 326–340.
Perkins DF (1992) Relationship between fluoride contents and loss of lichens near an aluminium works. Water Air Soil Pollut, 64: 503–510.
Perkins DF & Millar RO (1987) Effects of airborne fluoride emissions near an aluminium works in Wales: Part 2 — Saxicolous lichens growing on rocks and walls. Environ Pollut, 48: 185–196.
Perkins DF, Millar RO, & Neep PE (1980) Accumulation of airborne fluoride by lichens in the vicinity of an aluminium reduction plant. Environ Pollut, 21: 155–168.
Perrott KW, Smith BFL, & Inkson RHE (1976) The reaction of fluoride with soils and soil minerals. J Soil Sci, 27: 58–67.
Pettifor JM, Schnitzler CM, Ross FP, & Moodley GP (1989) Endemic skeletal fluorosis in children: hypocalcemia and the presence of renal resistance to parathyroid hormone. Bone Miner, 7: 275–288.
Phillips PH & Suttie JW (1960) The significance of time in intoxication of domestic animals by fluoride. Arch Ind Health, 21: 343–345.
Phipps K, Orwoll ES, Mason JD, & Cauley JA (2000) Community water fluoridation, bone mineral density, and fractures: prospective study of effects in older women. Br Med J, 321: 860–864.
Pickering WF (1985) The mobility of soluble fluoride in soils. Environ Pollut, B9: 281–308.
Pickering WF, Slavek J, & Waller P (1988) The effect of ion exchange on the solubility of fluoride compounds. Water Air Soil Pollut, 39: 323–336.
Pillai KS & Mane UH (1984) The effect of fluoride on fertilized eggs of a freshwater fish, Catla catla (Hamilton). Toxicol Lett, 22: 139–144.
Pillai K, Mathai A, & Deshmuhk P (1988) Effect of subacute dosage of fluoride on male mice. Toxicol Lett, 44: 21–29.
Pillai K, Mathai A, & Deshmuhk P (1989) Effect of fluoride on reproduction in mice. Fluoride, 22: 165–168.
Pilling K & Jones H (1988) Inhalation of degraded sulphur hexafluoride resulting in pulmonary oedema. J Soc Occup Med, 38: 82–84.
Pimentel R & Bulkley RV (1983) Influence of water hardness on fluoride toxicity to rainbow trout. Environ Toxicol Chem, 2: 381–386.
Polomski J, Fluhler H, & Blaser P (1982) Accumulation of airborne fluoride in soils. J Environ Qual, 11: 457–461.
Port GR, Davies MT, & Davison, AW (1998) Fluoride loading of a lepidopteran larva (Pieris brassicae) fed on treated diets. Environ Pollut, 99: 233–239.
Preuss PW, Colavito L, & Weinstein LH (1970) The synthesis of monofluoracetic acid by a tissue culture of Acacia georginae. Experientia, 26: 1059–1060.
Prince C & Navia J (1983) Glycosaminoglycan alterations in rat bone due to growth and fluorosis. J Nutr, 113: 1576–1582.
Pushnik JC & Miller GW (1990) The influences of elevated environmental fluoride on the physiology and metabolism of higher plants. Fluoride Q Rep, 23(1): 5–19.
Qiu M, Zhu X, Li S, Sun G, Ni A, Song W-Z, & Zhang J (1987) Bone dynamic changes in experimental fluorosis of rats. Chinese Med J, 100: 879–885.
Radon K, Nowak D, Heinrich-Ramm R, & Szadkowski D (1999) Respiratory health and fluoride exposure in different parts of the modern primary aluminum industry. Int Arch Occup Environ Health, 72: 297–303.
Rai LC, Husaini Y, & Mallick N (1998) pH-altered interaction of aluminium and fluoride on nutrient uptake, photosynthesis and other variables of Chlorella vulgaris. Aquat Toxicol, 42: 67–84.
Ramanathan V, Cicerone RJ, Singh HB, & Kiehl JT (1985) Trace gas trends and their potential role in climate change. J Geophys Res, 90(D3): 5547–5566.
Rand WE & Schmidt HJ (1952) The effect upon cattle of Arizona waters of high fluoride content. Am J Vet Res, 13: 50–61.
Rao DN & Pal D (1978) Effect of fluoride pollution on the organic matter content of soil. Plant Soil, 49: 653–656.
Rao G (1984) Dietary intake and bioavailability of fluoride. Ann Rev Nutr, 4: 115–136.
Rao KV, Khandekar AK, & Vaidyanadham D (1973) Uptake of fluoride by water hyacinth, Eichhornia crassipes. Ind J Exp Biol, 11: 68–69.
Ratsch HC & Johndro D (1986) Comparative toxicity of six test chemicals to lettuce using two root elongation test methods. Environ Monit Assess, 6: 267–276.
Razak I & Latifah R (1988) Unusual hypersensitivity to stannous fluoride. Ann Dent, 47: 37–39.
Ream L, Hull D, Scott J, & Pendergrass P (1983a) Fluoride ingestion during multiple pregnancies and lactations: microscopic observations on bone of the rat. Virchows Arch [Cell Pathol], 44: 35–44.
Ream L, Scott J, & Pendergrass P (1983b) Bone morphology of weanling rats from dams exposed to fluoride. Cell Tissue Res, 233: 689–691.
Reginster JY, Meurmans L, Zegels B, Rovati LC, Minne HW, Giacovelli G, Taquet AN, Setnikar I, Collette J, & Gosset C (1998) The effect of sodium monofluorophosphate plus calcium on vertebral fracture rate in postmenopausal women with moderate osteoporosis. A randomized, controlled trial. Ann Intern Med, 129: 1–8.
Reynolds KL & Laurence JA (1990) Development of common blight and accumulation of fluoride in red kidney bean plants exposed continuously or intermittently to hydrogen fluoride. Phytopathology, 80(2): 211–216.
Rice PM, Tourangeau PC, Johns C, & Gordon CC (1984) Baseline sulphur and fluoride concentrations in indigenous plants common in the Northern Great Plains. Environ Pollut, B7: 233–246.
Richards A, Kragstrup J, Josephsen K, & Fejerskov O (1986) Dental fluorosis developed in post-secretory enamel. J Dent Res, 65: 1406–1409.
Richards LF, Westmoreland WW, Tashiro M, McKay CM, & Morrison TJ (1967) Determination of optimum fluoride levels for community water supplies in relation to temperature. J Am Dent Assoc, 74: 389–397.
Riggs BL (1991) Treatment of osteoporosis with sodium fluoride or parathyroid hormone. Am J Med, 91: 37S–41S.
Riggs B, Hodgson S, O’Fallon W, Chao E, Wahner H, Muhs J, Cedel S, & Melton L (1990) Effect of fluoride treatment on the fracture rate in postmenopausal women with osteoporosis. N Engl J Med, 322: 802–809.
Rizzoli R, Chevalley T, Slosman DO, & Bonjour JP (1995) Sodium monofluorophosphate increases vertebral bone mineral density in patients with corticosteroid-induced osteoporosis. Osteoporosis Int, 5: 39–46.
Rockette H & Arena V (1983) Mortality studies of aluminum reduction plant workers: Potroom and carbon plant. J Occup Med, 25: 549–557.
Rojas-Sanchez F, Kelly SA, Drake KM, Eckert GJ, Stookey GK, & Dunipace AJ (1999) Fluoride intake from foods, beverages and dentifrice by young children in communities with negligibly and optimally fluoridated water: a pilot study. Community Dent Oral Epidemiol, 27: 288–297.
Romundstad P, Haldorsen T, & Andersen A (2000) Cancer incidence and cause specific mortality among workers in two Norwegian aluminum reduction plants. Am J Ind Med, 37: 175–183.
Rr nneberg A (1995) Mortality and cancer morbidity in workers from an aluminum smelter with pre-baked carbon anodes. 1: Exposure assessment. Occup Environ Med, 52: 242–249.
Rr nneberg A & Andersen A (1995) Mortality and cancer morbidity in workers from an aluminum smelter with pre-baked carbon anodes. 2: Cancer morbidity. Occup Environ Med, 52: 250–254.
Rosenholtz M, Carson T, Weeks M, Wilinski F, Ford D, & Oberst F (1963) A toxicopathologic study in animals after brief single exposures to hydrogen fluoride. Am Ind Hyg Assoc J, 24: 253–261.
Roy RL, Campbell PGC, Prémont S, & Labrie J (2000) Geochemistry and toxicity of aluminium in the Saguenay River, Québec, Canada, in relation to discharges from an aluminium smelter. Environ Toxicol Chem, 19(10): 2457–2466.
Saether OM, Andreassen BT, & Semb A (1995) Amounts and sources of fluoride in precipitation over southern Norway. Atmos Environ, 29(15): 1785–1793.
Sahu NK & Karim MA (1989) Fluoride incidence in natural waters in Amreli district, Gujarat. J Geol Soc Ind, 33: 450–456.
Sakurai S, Itai K, & Tsunoda H (1983) Effects of airborne fluoride on the fluoride content of rice and vegetables. Fluoride, 16: 175–180.
Sands M, Nicol S, & McMinn A (1998) Fluoride in Antarctic marine crustaceans. Mar Biol, 132: 591–598.
Saralakumari D & Ramakrishna Rao P (1993) Endemic fluorosis in the village Ralla Ananthapuram in Andhra Pradesh, an epidemiological study. Fluoride, 26: 177–180.
Sato T, Yoshitake K, & Hitomi G (1986) Mechanisms of fluoride absorption from the gastrointestinal tract in rats. Fluoride Research 1985. Stud Environ Sci, 27: 325–332.
Saxena A, Kulshrestha UC, Kumar N, Kumari KM, & Srivastava SS (1994) Distribution of air-borne fluoride: vapour phase, particulate, precipitation and dry deposition. Environ Technol, 15: 51–59.
Schamschula R, Sugar E, Agus H, Un P, & Toth K (1982) The fluoride content of human enamel in relation to environmental exposure to fluoride. Aust Dent J, 27: 243–247.
Schamschula R, Sugar E, Un P, Toth K, Barmes D, & Adkins B (1985) Physiological indicators of fluoride exposure and utilization: an epidemiological study. Community Dent Oral Epidemiol, 13: 104–107.
Schamschula R, Duppenthaler J, Sugar E, Toth K, & Barmes D (1988a) Fluoride intake and utilization by Hungarian children: associations and interrelationships. Acta Physiol Hung, 72: 253–261.
Schamschula R, Un P, Sugar E, & Duppenthaler J (1988b) The fluoride content of selected foods in relation to the fluoride concentration of water. Acta Physiol Hung, 72: 217–227.
Scheifinger H & Held G (1997) Aerosol behaviour on the South African Highveld. Atmos Environ, 31(12): 3497–3509.
Schiffl H & Binswanger U (1980) Human urinary fluoride excretion as influenced by renal functional impairment. Nephron, 26: 69–72.
Schnitzler CM, Wing JR, Mesquita JM, Gear AK, Robson HJ, & Smyth AE (1990) Risk factors for the development of stress fractures during fluoride therapy for osteoporosis. J Bone Miner Res, 5(suppl 1): S195–S200.
Schotte W (1987) Fog formation of hydrogen fluoride in air. Ind Eng Chem Res, 26(2): 300–306.
Schroder JL, Basta NT, Rafferty DP, Lochmiller RL, Kim S, Qualls W, & McBee K (1999) Soil and vegetation fluoride exposure pathways to cotton rats on a petrochemical-contaminated landfarm. Environ Toxicol Chem, 18(9): 2028–2033.
Schroder JL, Basta NT, Lochmiller RL, Rafferty DP, Payton M, Kim S, & Qualls CW (2000) Soil contamination and bioaccumulation of inorganics on petrochemical sites. Environ Toxicol Chem, 19(8): 2066–2072.
Schultz M, Kierdorf U, Sedlacek F, & Kierdorf H (1998) Pathological bone changes in the mandibles of wild red deer (Cervus elaphus L.) exposed to high environmental levels of fluoride. J Anat, 193: 431–442.
Schuppli P (1985) Total fluorine in CSSC reference soil samples. Can J Soil Sci, 65: 605–607.
Scott D (1986) Cytogenetic effects of sodium fluoride in cultured human fibroblasts. Mutagenesis, 1: 69 (abstract).
Scott D & Roberts S (1987) Extrapolation from in vitro tests to human risk: experience with sodium fluoride clastogenicity. Mutat Res, 189: 47–58.
Seel DC & Thomson AG (1984) Bone fluoride in predatory birds in the British Isles. Environ Pollut, A36: 367–374.
Sein G (1988) The effects of sodium fluoride on the immunological responses in mice. Med Sci Res, 16: 39.
Shacklette HT, Boerngen JG, & Keith JRX (1974) Selenium, fluorine and arsenic in superficial materials of the conterminous United States. Washington, DC, US Department of the Interior (Geological Survey Circular 692).
Sharma Y (1982a) Effect of sodium fluoride on collagen cross-link precursors. Toxicol Lett, 10: 97–100.
Sharma Y (1982b) Variations in the metabolism and maturation of collagen after fluoride ingestion. Biochim Biophys Acta, 715: 137–141.
Sharma K & Susheela A (1988a) Effect of fluoride on molecular weight, charge density and age related changes in the sulphated isomers of glycosaminoglycans of the rabbit cancellous bone. Int J Tissue React, 10: 327–334.
Sharma K & Susheela A (1988b) Fluoride ingestion in excess and its effect on the disaccharide profile of glycosaminoglycans of cancellous bone of the rabbit. Med Sci Res, 16: 349–350.
Shashi A (1990) Histopathological changes in rabbit ovary during experimental fluorosis. Indian J Pathol Microbiol, 33: 113–117.
Shen YW & Taves DR (1974) Fluoride concentrations in the human placenta and maternal and cord blood. Am J Obstet Gynecol, 119: 205–207.
Sheth FJ, Multani AS, & Chinoy NJ (1994) Sister chromatid exchanges: A study in fluorotic individuals in North Gujarat. Fluoride, 27: 215–219.
Shivashankara AR, Shivajara Shankara YM, Hanumanth Rao S, & Gopalakrishna Bhat P (2000) A clinical and biochemical study of chronic fluoride toxicity in children of Kheru Thanda of Gulbara district, Karnataka, India. Fluoride, 33: 66–73.
Shore RF (1995) Predicting cadmium, lead and fluoride levels in small mammals from soil residues and by species–species extrapolation. Environ Pollut, 88: 333–340.
Shulman E & Vallejo M (1990) Effects of gastric contents on the bioavailability of fluoride in humans. Pediatr Dent, 12: 237–240.
Shupe JL & Olson AE (1983) Clinical and pathological aspects of fluoride toxicosis in animals. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 319–338.
Shupe JL, Miner ML, Greenwood DA, Harris LE, & Stoddard GE (1963) The effect of fluorine on dairy cattle. 2. Clinical and pathologic effects. Am J Vet Res, 24: 964–979.
Shupe JL, Olson AE, Peterson HB, & Low JB (1984) Fluoride toxicosis in wild ungulates. J Am Vet Med Assoc, 185(11): 1295–1300.
Shupe JL, Larsen AE, & Olsen AE (1987) Effects of diets containing sodium fluoride on mink. J Wildl Dis, 23: 606–613.
Siddiqui AH (1970) Fluorosis in areas of India with a high natural content of water fluoride. In: Fluorides and human health. Geneva, World Health Organization, pp 284–294 (WHO Monograph Series No. 59).
Sidhu S (1979) Fluoride levels in air, vegetation and soil in the vicinity of a phosphorous plant. J Air Pollut Control Assoc, 29: 1069–1072.
Sidhu S (1982) Fluoride deposition through precipitation and leaf litter in a boreal forest in the vicinity of a phosphorous plant. Sci Total Environ, 23: 205–214.
Sidhu SS & Staniforth RJ (1986) Effects of atmospheric fluorides on foliage, and cone and seed production in balsam fir, larch spruce, and larch. Can J Bot, 64: 923–931.
Siemiatycki J, Gerin M, Dewar R, Nadon L, Lakhani R, Begin D, & Richardson L (1991) Associations between occupational circumstances and cancer. In: Siemiatycki J ed. Risk factors for cancer in the workplace. Boca Raton, Florida, CRC Press, pp 141–295.
Sigler WF & Neuhold JM (1972) Fluoride intoxication in fish: a review. J Wildl Dis, 8: 252–254.
Silva M & Reynolds EC (1996) Fluoride content of infant formulae in Australia. Aust Dent J, 41: 37–42.
Simard P, Lachapelle D, Trahan L, Naccache H, Demers M, & Brodeur J (1989) The ingestion of fluoride dentifrice by young children. J Dent Child, 56: 177–181.
Simonen O & Laitinen O (1985) Does fluoridation of drinking-water prevent bone fragility and prevent osteoporosis. Lancet, 2: 432–434.
Singer L & Ophaug R (1979) Total fluoride intake of infants. Pediatrics, 63: 460–466.
Singh M & Kar D (1989) Effect of fluoride on the settleability of biomass from an activated sludge plant treating petrochemical effluent. J Ferment Bioeng, 67(5): 366–368.
Singh A, Chhabra R, & Abrol IP (1979) Effect of fluorine and phosphorus applied to a sodic soil on their availability and on yield and chemical composition of wheat. Soil Sci, 128(2): 90–97.
Skare J, Wong T, Evans L, & Cody D (1986a) DNA repair studies with sodium fluoride: comparative evaluation using density gradient ultracentrifugation and autoradiography. Mutat Res, 172: 77–87.
Skare J, Schrotel K, & Nixon G (1986b) Lack of DNA strand breaks in rat testicular cells after in vivo treatment with sodium fluoride. Mutat Res, 170: 85–92.
Skjelkvåle BL (1994a) Factors influencing fluoride concentrations in Norwegian lakes. Water Air Soil Pollut, 77: 151–167.
Skjelkvåle BL (1994b) Water chemistry in areas with high deposition of fluoride. Sci Total Environ, 152: 105–112.
Slamenova D, Gabelova A, & Ruppova K (1992) Cytotoxic and genotoxic testing of sodium fluoride on Chinese hamster V79 and human EUE cells. Mutat Res, 279: 109–115.
Slamenova D, Ruppova K, Gabelova A, & Wsolova L (1996) Evaluation of mutagenic and cytotoxic effects of sodium fluoride on mammalian cells influenced by an acidic environment. Cell Biol Toxicol, 12: 11–17.
Sloof W, Eerens H, Janus J, & Ros J (1989) Integrated criteria document: Fluorides. Bilthoven, National Institute of Public Health and Environmental Protection (Report No. 758474010).
Smid J & Kruger B (1985) The fluoride content of some teas available in Australia. Aust Dent J, 30: 25–28.
Smith A (1980) An examination of the relationship between fluoridation of water and cancer mortality in 20 large US cities. N Z Med J, 91: 413–416.
Smith AO & Woodson BR (1965) The effects of fluoride on the growth of Chlorella pyrenoidosa. Va J Sci, 16(1): 1–8.
Smith LR, Holsen TM, Ibay NC, Block RM, & De Leon AB (1985) Studies on the acute toxicity of fluoride ion to stickleback, fathead minnow, and rainbow trout. Chemosphere, 14(9): 1383–1389.
Snow G & Anderson C (1986) Short-term chronic fluoride administration and trabecular bone remodelling in beagles: A pilot study. Calcif Tissue Int, 38: 217–221.
Sr gaard CH, Mosekilde L, Schwartz W, Leidig G, Minne HW, & Ziegler R (1995) Effects of fluoride on rat vertebral body biomechanical competence and bone mass. Bone, 16: 163–169.
Somashekar RK & Ramaswamy SN (1983) Fluoride concentration in the water of the river Cauvery, Karnataka, India. Int J Environ Stud, 21: 325–327.
Sorgdrager B, Pal TM, de Loof AJA, Dubois AEJ, & de Monchy JGR (1995) Occupational asthma in aluminum potroom workers related to pre-employment eosinophil count. Eur Respir J, 8: 1520–1524.
Sowers M, Wallace R, & Lemke J (1986) The relationship of bone mass and fracture history to fluoride and calcium intake: a study of three communities. Am J Clin Nutr, 44: 889–898.
Sowers M, Clark M, Jannausch M, & Wallace R (1991) A prospective study of bone mineral content and fracture in communities with differential fluoride content. Am J Epidemiol, 133: 649–660.
Sr yseth V & Kongerud J (1992) Prevalence of respiratory disorders among aluminum potroom workers in relation to exposure to fluoride. Br J Ind Med, 49: 125–130.
Sr yseth V, Kongerud J, Aalen O, Botten G, & Boe J (1995) Bronchial responsiveness decreases in relocated aluminum potroom workers compared with workers who continue their potroom exposure. Int Arch Occup Environ Health, 67: 53–57.
Sr yseth V, Kongerud J, & Boe J (1996) Allergen sensitization and exposure to irritants in infancy. Allergy, 51: 719–723.
Spak CJ, Ekstrand J, & Zylberstein D (1982) Bioavailability of fluoride added to baby formula and milk. Caries Res, 16: 249–256.
Spak CJ, Hardell LI, & de Chateau P (1983) Fluoride in human milk. Acta Paediatr Scand, 72: 699–701.
Spak C, Berg U, & Ekstrand J (1985) Renal clearance of fluoride in children and adolescents. Pediatrics, 75: 575–579.
Spak C, Sjostedt S, Eleborg L, Veress B, Perbeck L, & Ekstrand J (1989) Tissue response of gastric mucosa after ingestion of fluoride. Br Med J, 298: 1688–1689.
Spak C, Sjostedt S, Eleborg L, Veress B, Perbeck L, & Ekstrand J (1990) Studies of human gastric mucosa after application of 0.042% fluoride gel. J Dent Res, 69: 426–429.
Sparks R, Sandusky M, & Paparo A (1983) Identification of the water quality factors which prevent fingernail clams from recolonizing the Illinois River Phase III. Urbana, Illinois, University of Illinois, Water Resources Center.
Spencer H, Osis D, & Lender M (1981) Studies of fluoride metabolism in man: a review and report of original data. Sci Total Environ, 17: 1–12.
Spinelli J, Band P, Svirchev L, & Gallagher R (1991) Mortality and cancer incidence in aluminum reduction plant workers. J Occup Med, 33: 1150–1155.
Sprando RL, Black TN, Ames MJ, Rorie JI, & Collins TFX (1996) Effect of intratesticular injection of sodium fluoride on spermatogenesis. Food Chem Toxicol, 43: 377–384.
Sprando RL, Collins FX, Black TN, Rorie J, Ames MJ, & O’Donnell M (1997) Testing the potential of sodium fluoride to affect spermatogenesis in the rat. Food Chem Toxicol, 35: 881–890.
Sprando RL, Collins TF, Black T, Olejnik N, & Rorie J (1998) Testing the potential of sodium fluoride to affect spermatogenesis: a morphometric study. Food Chem Toxicol, 36: 1117–1124.
Staniforth RJ & Sidhu SS (1984) Effects of atmospheric fluorides on foliage, flower, fruit, and seed production in wild raspberry and blueberry. Can J Bot, 62: 2827–2834.
Stannard J, Rovero J, Tsamtsouris A, & Gavris V (1990) Fluoride content of some bottled waters and recommendations for fluoride supplementation. J Pedodontics, 14: 103–107.
Stevens B, Willis G, Humphrey T, & Atkins R (1987) Combined toxicity of aluminum and fluoride in the rat: possible implications in hemodialysis. Trace Elem Med, 4: 61–66.
Stevens DP, McLaughlin MJ, & Alston AM (1997) Phytotoxicity of the aluminium–fluoride complexes and their uptake from solution culture by Avena sativa and Lycopersicon esculentum. Plant Soil, 192: 81–93.
Stevens DP, McLaughlin MJ, & Alston AM (1998a) Phytotoxicity of the fluoride ion and its uptake from solution culture by Avena sativa and Lycopersicon esculentum. Plant Soil, 200: 119–129.
Stevens DP, McLaughlin MJ, & Alston AM (1998b) Phytotoxicity of hydrogen fluoride and fluoroborate and their uptake from solution culture by Avena sativa and Lycopersicon esculentum. Plant Soil, 200: 175–184.
Stevens DP, McLaughlin MJ, Randall PJ, & Keerthisinghe G (2000) Effect of fluoride supply on fluoride concentrations in five pasture species: Levels required to reach phytotoxic or potentially zootoxic concentrations in plant tissue. Plant Soil, 227: 223–233.
Street JJ & Elwali AMO (1983) Fluorite solubility in limed acid sandy soils. Soil Sci Soc Am J, 47(3): 483–485.
Stumm W & Morgan JJ (1981) Aquatic chemistry, 2nd ed. New York, John Wiley & Sons.
Suarez-Almazor M, Flowerdew G, Saunders D, Soskolne C, & Russel A (1993) The fluoridation of drinking water and hip fracture hospitalization rates in two Canadian communities. Am J Public Health, 83: 689–693.
Susheela A & Das T (1988) Chronic fluoride toxicity: A scanning electron microscopic study of duodenal mucosa. Clin Toxicol, 26: 467–476.
Susheela A & Jain S (1983) Fluoride-induced haematological changes in rabbits. Bull Environ Contam Toxicol, 30: 388–393.
Susheela A & Jain S (1986) Fluoride toxicity: Erythrocyte membrane abnormality and "echinocyte" formation. Fluoride Research 1985. Stud Environ Sci, 27: 231–239.
Susheela A & Kharb P (1990) Aortic calcification in chronic fluoride poisoning: Biochemical and electron-microscopic evidence. Exp Mol Pathol, 53: 72–80.
Susheela S & Kumar A (1991) A study of the effect of high concentrations of fluoride on the reproductive organs of male rabbits, using light and scanning electron microscopy. J Reprod Fertil, 92: 353–360.
Susheela A & Sharma Y (1982) Certain facets of F– action on collagen protein in osseous and non-osseous tissues. Fluoride, 15: 177–190.
Susheela AK, Kumar A, Bhatnakar M, & Bahadur R (1993) Prevalence of endemic fluorosis with gastrointestinal manifestations in people living in some North-Indian villages. Fluoride, 26: 97–104.
Suttie JS, Dickie R, Clay AB, Nielsen P, Mahan WE, Baumann DP, & Hamilton RJ (1987) Effects of fluoride emissions from a modern primary aluminum smelter on a local population of white-tailed deer (Odocoileus virginianus). J Wildl Dis, 23(1): 135–143.
Suttie JW (1983) The influence of nutrition and other factors on fluoride tolerance. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 291–303.
Suttie JW, Miller RF, & Phillips PH (1957) Studies of the dietary NaF on dairy cows. I. The physiological effects and the development of fluorosis. J Nutr, 63: 211–224.
Suttie JW, Hamilton RJ, Clay AC, Tobin ML, & Moore WG (1985) Effects of fluoride ingestion on white-tailed deer (Odocoileus virginianus). J Wildl Dis, 21(3): 283–288.
Suzuki N & Tsutsui T (1989) [Dependence of lethality of chromosome aberrations induced by treatment of synchronized human diploid fibroblasts with sodium fluoride on different periods of cell cycle.] Shigaku, 77: 436–447 (in Japanese).
Swarup D, Dwivedi SK, Dey S, & Ray SK (1998) Fluoride intoxication in bovines due to industrial pollution. Ind J Anim Sci, 68(7): 605–608.
Symonds R, Rose W, & Reed M (1988) Contribution of Cl- and F-bearing gases to the atmosphere by volcanos. Nature, 334: 415–418.
Tagaki Y, Ohkubo Y, Asada T, & Matsuda S (1986) An international comparison of trace amounts of fluoride contained in human hair. Fluoride Research 1985. Stud Environ Sci, 27: 423–429.
Tannenbaum A & Silverstone H (1949) Effect of low environmental temperature, dinitrophenol, or sodium fluoride on the formation of tumours in mice. Cancer Res, 9: 403–410.
Tao S & Suttie JW (1976) Evidence for lack of an effect of dietary fluoride level on reproduction in mice. J Nutr, 106: 1115–1122.
Tate WH & Chan JT (1994) Fluoride concentrations in bottled and filtered waters. Gen Dent, 42: 362–366.
Taves DR (1968) Electrophoretic mobility of serum fluoride. Nature, 220: 582–583.
Taves DR (1983) Dietary intake of fluoride ashed (total fluoride) v. unashed (inorganic fluoride) analysis of individual foods. Br J Nutr, 49: 295–301.
Taves D, Forbes N, Silverman D, & Hicks D (1983) Inorganic fluoride concentrations in human and animal tissues. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 189–193.
Taylor A (1954) Sodium fluoride in the drinking water of mice. Dent Dig, 60: 170–172.
Taylor RJ & Basabe FA (1984) Patterns of fluoride accumulation and growth reduction exhibited by Douglas fir in the vicinity of an aluminum reduction plant. Environ Pollut, A33: 221–235.
Teotia M, Teotia SP, & Singh KP (1998) Endemic chronic fluoride toxicity and dietary calcium deficiency interaction syndromes of metabolic bone disease and deformities in India: year 2000. Indian J Pediatr, 65: 371–381.
Theriault G, Gingras S, & Provencher S (1984) Telangiectasia in aluminum workers: a follow up. Br J Med, 41: 367–372.
Thomas AB, Hashomoto H, Baylink D, & Lau K-H (1996) Fluoride at mitogenic concentrations increases the steady state phosphotyrosyl phosphorylation level of cellular proteins in human bone cells. J Clin Endocrinol Metab, 81: 2570–2578.
Thompson LK, Sidhu SS, & Roberts BA (1979) Fluoride accumulations in soil and vegetation in the vicinity of a phosphorus plant. Environ Pollut, 18: 221–234.
Thompson R, McMullen T, & Morgan G (1971) Fluoride concentrations in the ambient air. J Air Pollut Control Assoc, 21: 484–487.
Thomson AG (1987) Fluoride in the prey of barn owls (Tyto alba). Environ Pollut, 44: 177–192.
Thomson E, Kilanowski F, & Perry P (1985) The effect of fluoride on chromosome aberration and sister chromatid exchange frequencies in cultured human lymphocytes. Mutat Res, 144: 89–92.
Tobayiwa C, Musiyambiri M, Chironga L, Mazorodze O, & Sapahla S (1991) Fluoride levels and dental fluorosis in two districts in Zimbabwe. Cent Afr J Med, 37: 353–361.
Tong C, McQueen C, Ved Brat S, & Williams G (1988) The lack of genotoxicity of sodium fluoride in a battery of cellular tests. Cell Biol Toxicol, 4: 173–186.
Torra M, Rodamilans M, & Corbella J (1998) Serum and urine ionic fluoride: normal range in a nonexposed population. Biol Trace Elem Res, 63: 67–71.
Tracy PW, Robbins CW, & Lewis GC (1984) Fluorite precipitation in a calcareous soil irrigated with high fluoride water. Soil Sci Soc Am J, 48: 1013–1016.
Trautner K & Einwag J (1989) Influence of milk and food on fluoride bioavailability from NaF and Na2FPO3 in man. J Dent Res, 68: 72–77.
Tscherko D & Kandeler E (1997) Ecotoxicological effects of fluorine deposits on microbial biomass and enzyme activity in grassland. Eur J Soil Sci, 48: 329–335.
Tsiros JX, Haidouti C, & Chronopoulou A (1998) Airborne fluoride contamination of soils and olive trees near an aluminium plant. Measurements and simulations. J Environ Sci Health, A33(7): 1309–1324.
Tsuchida M, Horiuchi T, & Mineshita S (1992) Metabolic effects of fluoride with and without calcium deprivation. Trace Elem Med, 9: 176–182.
Tsunoda H & Tsunoda N (1986) Fluoride absorption and excretion in human subjects following ingestion of F-contaminated agricultural products. Fluoride Research 1985. Stud Environ Sci, 27: 107–112.
Tsutsui T, Suzuki N, & Ohmori M (1984a) Sodium fluoride-induced morphological and neoplastic transformation, chromosome aberrations, sister chromatid exchanges and unscheduled DNA synthesis in cultured Syrian hamster embryo cells. Cancer Res, 44: 938–941.
Tsutsui T, Suzuki N, Ohmori M, & Maizumi H (1984b) Cytotoxicity, chromosome aberrations and unscheduled DNA synthesis in cultured human diploid fibroblasts induced by sodium fluoride. Mutat Res, 139: 193–198.
Tsutsui T, Koichi I, & Maizumi H (1984c) Induction of unscheduled DNA synthesis in cultured human oral keratinocytes by sodium fluoride. Mutat Res, 140: 43–48.
Tsutsui T, Kawamoto Y, Suzuki N, Gladen B, & Barrett J (1991) Cytotoxicity and chromosome aberrations in normal human oral keratinocytes induced by chemical carcinogens: comparison of inter-individual variations. Toxicol In Vitro, 5: 353–361.
Tsutsui T, Tanaka Y, Matsudo Y, Uehama A, Someya T, Hamaguchi F, Yamamoto H, & Takahashi M (1995) No increases in chromosome aberrations in human diploid fibroblasts following exposure to low concentrations of sodium fluoride for long times. Mutat Res, 335: 15–20.
Turner C, Akhter M, & Heaney R (1992) The effects of fluoridated water on bone strength. J Orthop Res, 10: 581–587.
Turner CH, Hasegawa K, Zhang W, Wilson M, Li Y, & Dunipace AJ (1995) Fluoride reduces bone strength in older rats. J Dent Res, 74: 1475–1481.
Turner R, Francis R, Brown D, Garand J, Hannon K, & Bell N (1989) The effects of fluoride on bone and implant histomorphometry in growing rats. J Bone Miner Res, 4: 477–484.
Upfal M & Doyle C (1990) Medical management of hydrofluoric acid exposure. J Occup Med, 32: 726–731.
US DHHS (1991) Review of fluoride. Benefits and risks. Report of the Ad Hoc Subcommittee on Fluoride of the Committee to Coordinate Environmental Health and Related Programs. Washington, DC, US Department of Health and Human Services, Public Health Service.
US EPA (1980) Reviews of the environmental effects of pollutants: IX. Fluoride. Cincinnati, Ohio, US Environmental Protection Agency, Office of Research and Development, Health Effects Research Laboratory (EPA-600/1-78-050).
US EPA (1985) Drinking water criteria document on fluoride. Cincinnati, Ohio, US Environmental Protection Agency, Office of Drinking Water (Contract 68-03-3279).
US EPA (1999) Chemical-specific TRI (Toxic Release Inventory) release and waste management data 1999. Washington, DC, US Environmental Protection Agency, Office of Environmental Information (2810).
Uslu B (1983) Effect of fluoride on collagen synthesis in the rat. Res Exp Med, 182: 7–12.
US NAS (1971) Fluorides, biological effects of atmospheric pollutants. Washington, DC, US National Academy of Sciences.
US NRC (1993) Health effects of ingested fluoride. Subcommittee on Health Effects of Ingested Fluoride, US National Research Council. Washington, DC, National Academy Press, 181 pp.
van Asten P, Darroudi F, Natarajan AT, Terpstra IJ, & Duursma SA (1998) Cytogenetic effects on lymphocytes in osteoporotic patients on long-term fluoride therapy. Pharm World Sci, 20: 214–218.
Van Bruggen AHC & Reynolds KL (1988) Effects of hydrogen fluoride on susceptibility of soybeans to Rhizoctonia solani and Phytophthora megasperma f. sp. glycinea. Agric Ecosyst Environ, 24: 443–452.
Van Craenenbroeck W & Marivoet J (1987) A comparison of simple methods for estimating the mass flow of fluoride discharged into rivers. Water Sci Technol, 19: 729–740.
Van Toledo B (1978) Fluoride content in eggs of wild birds (Parus major L. and Strix alueo L.) and the common house hen (Gallus domesticus). Fluoride, 11: 198–207.
Van Wensem J & Adema T (1991) Effects of fluoride on soil fauna mediated litter decomposition. Environ Pollut, 72: 239–251.
Varner JA, Huie CW, Horvath W, Jensen KF, & Isaacson RL (1993) Chronic AlF3 administration: II. Selected histological observations. Neurosci Res Commun, 13: 99–104.
Varner JA, Jensen KF, Horvath W, & Isaacson RL (1998) Chronic administration of aluminum-fluoride or sodium-fluoride to rats in drinking water: Alterations in neuronal and cerebrovascular integrity. Brain Res, 784: 284–298.
Varo P & Koivistoinen P (1980) Mineral composition of Finnish foods. XII. General discussion and nutritional evaluation. Acta Agric Scand, 22(suppl): 165–171.
VDI (1989) Maximum emission values to protect vegetation: maximum emission values for hydrogen fluoride. Düsseldorf, Verein Duetscher Ingenieure, 48 pp (in German with English translation).
Venkateswarlu P (1975) Determination of total fluorine in serum and other biological material by oxygen bomb and reverse extraction techniques. Anal Biochem, 68: 512–521.
Venkateswarlu P (1983) Overview of analytical methods for fluorine in air, water, soil vegetation, body fluids and tissues. In: Shupe J, Peterson H, & Leone N ed. Fluorides: Effects on vegetation, animals and humans. Salt Lake City, Utah, Paragon Press, pp 19–52.
Vike E (1999) Air-pollutant dispersal patterns and vegetation damage in the vicinity of three aluminium smelters in Norway. Sci Total Environ, 236: 75–90.
Vike E & Håbjr rg A (1995) Variation in fluoride content and leaf injury on plants associated with three aluminium smelters in Norway. Sci Total Environ, 163: 25–34.
Vikr ren T & Stuve G (1996a) Fluoride exposure and selected characteristics of eggs and bones of the herring gull (Larus argentatus) and the common gull (Larus canus). J Wildl Dis, 32(2): 190–198.
Vikr ren T & Stuve G (1996b) Fluoride exposure in cervids inhabiting areas adjacent to aluminium smelters in Norway. II. Fluorosis. J Wildl Dis, 32(2): 181–189.
Vogel J & Ottow JCG (1991) Fluoride accumulation in different earthworm species near an industrial emission source in southern Germany. Bull Environ Contam Toxicol, 47: 515–520.
Voroshilin SI, Plotko EG, Gatiyatullina EZ, & Gileva EA (1973) Cytogenetic effect of inorganic fluorine compounds on human and animal cells in vivo and in vitro. Sov Genet, 9: 492–496.
Walton KC (1984) Fluoride in fox bone near an aluminium reduction plant in Anglesey, Wales, and elsewhere in the United Kingdom. Environ Pollut, B7: 273–280.
Walton KC (1985) Fluoride in bones of small rodents near an aluminium reduction plant. Water Air Soil Pollut, 26(1): 65–70.
Walton KC (1986) Fluoride in moles, shrews and earthworms near an aluminium reduction plant. Environ Pollut, A42: 361–371.
Walton KC (1987a) Effects of treatment with sodium fluoride and subsequent starvation on fluoride content of earthworms. Bull Environ Contam Toxicol, 38: 163–170.
Walton KC (1987b) Fluoride in bones of small rodents living in areas with different pollution levels. Water Air Soil Pollut, 32: 113–122.
Walton KC (1987c) Tooth damage in field voles, wood mice and moles in areas polluted by fluoride from an aluminium reduction plant. Sci Total Environ, 65: 257–260.
Walton KC (1988) Environmental fluoride and fluorosis in mammals. Mammal Rev, 18(2): 77–90.
Walton KC & Ackroyd S (1988) Fluoride in mandibles and antlers of roe and red deer from different areas of England and Scotland. Environ Pollut, 54: 17–27.
Wang GQ, Huang YZ, Xiao BY, Qian XC, Yao H, Hu Y, Gu YL, Zhang C, & Liu KT (1997) Toxicity from water containing arsenic and fluoride in Xinjiang. Fluoride, 30(2): 81–84.
Wang J-X & Bian Y-M (1988) Fluoride effects on the mulberry–silkworm system. Environ Pollut, 52: 11–18.
Wang W (1986) Toxicity tests of aquatic pollutants by using common duckweed. Environ Pollut, B11: 1–14.
Wang Y, Yin Y, Gilula LA, & Wilson AJ (1994) Endemic fluorosis of the skeleton: radiographic features in 127 patients. Am J Roentgenol, 162: 93–98.
Ward PFB, Hall RJ, & Peters RA (1964) Fluoro-fatty acids in the seeds of Dichapetabum toxicarium. Nature, 201: 611–612.
Warren DP, Henson HA, & Chan JT (1996) Comparison of fluoride content in caffeinated, decaffeinated and instant coffee. Fluoride, 29: 147–150.
Waterhouse C, Taves D, & Munzer A (1980) Serum inorganic fluoride: changes related to previous fluoride intake, renal function and bone resorption. Clin Sci (Lond), 58(2): 145–152.
Weast R (1986) CRC handbook of chemistry and physics, 1985–1986. Boca Raton, Florida, CRC Press.
Weatherell J, Deutsch D, Robinson C, & Hallsworth A (1977) Assimilation of fluoride by enamel throughout the life of the tooth. Caries Res, 11: 85–115.
Wei SHY, Hattab FN, & Mellberg JR (1989) Concentration of fluoride and other selected elements in teas. Nutrition, 5: 237–240.
Weidmann S & Weatherell J (1970) Distribution in hard tissues. In: Fluorides and human health. Geneva, World Health Organization, pp 104–128 (WHO Monograph Series No. 59).
Weinstein L (1977) Fluoride and plant life. J Occup Med, 19: 49–78.
Weinstein LH & Alscher-Herman R (1982) Physiological responses of plants to fluorine. In: Unsworth M & Ormrod DP ed. Effects of gaseous air pollution in agriculture and horticulture. London, Butterworth Scientific, pp 139–167.
Wen MK, Li QC, & Wang CY (1996) Developments in the analysis of fluoride 1993–1995. Fluoride, 29: 82–88.
Whitford G (1987) Fluoride in dental products: safety considerations. J Dent Res, 66: 1056–1060.
Whitford G (1990) The physiological and toxicological characteristics of fluoride. J Dent Res, 69: 539–549.
Whitford G (1996) The metabolism and toxicity of fluoride, 2nd rev. ed. Basel, Karger, 156 pp (Monographs in Oral Science, Volume 16).
Whitford GM (1997) Determinants and mechanisms of enamel fluorosis. Ciba Found Symp, 205: 226–245.
Whitford GM & Pashley DH (1984) Fluoride absorption: the influence of gastric acidity. Calcif Tissue Int, 36: 302–307.
Whitford G, Pashley D, & Reynolds K (1979) Fluoride tissue distribution: short-term kinetics. Am J Physiol, 236: F141–F148.
Whitford G, Callan R, & Wang H (1982) Fluoride absorption through the hamster cheek pouch: a pH-dependent event. J Appl Toxicol, 2: 303–306.
Whitford G, Biles E, & Birdsong-Whitford N (1991) A comparative study of fluoride pharmacokinetics in five species. J Dent Res, 70: 948–951.
Whitford GM, Thomas JE, & Adair SM (1999a) Fluoride in whole saliva, parotid ductal saliva and plasma in children. Arch Oral Biol, 44: 785–788.
Whitford GM, Sampaio FC, Arneberg P, & von der Fehr FR (1999b) Fingernail fluoride: a method for monitoring fluoride exposure. Caries Res, 33: 462–467.
WHO (1984) Fluorine and fluorides. Geneva, World Health Organization (Environmental Health Criteria 36).
WHO (1993) Guidelines for drinking-water quality, 2nd ed. Vol. 1. Recommendations. Geneva, World Health Organization.
WHO (1994) Fluorides and oral health. Report of a WHO Expert Committee on Oral Health Status and Fluoride Use. Geneva, World Health Organization (WHO Technical Report Series 846).
WHO (1996a) Biological monitoring of chemical exposure in the workplace. Vol. 1. Geneva, World Health Organization.
WHO (1996b) Guidelines for drinking-water quality, 2nd ed. Vol. 2. Health criteria and other supporting information. Geneva, World Health Organization.
WHO (1996c) Trace elements in human nutrition and health. Geneva, World Health Organization.
Wolting HG (1975) Synergism of hydrogen fluoride and leaf necrosis on freesias. Neth J Plant Pathol, 81: 71–77.
Wolting HG (1978) Gevoeligheid van kastulphen voor chronische inwerking van fluorhoudende luchtverontreiniging. Bloembollencultur, 31: 728–730.
Wright DA (1977) Toxicity of fluoride to brown trout fry (Salmo trutta). Environ Pollut, 12: 57–62.
Wright DA & Davison AW (1975) The accumulation of fluoride by marine and intertidal animals. Environ Pollut, 8: 1–13.
Wu DQ & Wu Y (1995) Micronucleus and sister chromatid exchange frequency in endemic fluorosis. Fluoride, 28: 125–127.
Wu JY, Wu L, Liu KT, & Jiang P (1998) Study on absorption and excretion of joint exposure to fluoride and arsenic. Endemic Dis Bull, 13(2): 5–9.
Xu RH, Yuan HH, & Fan A (1995) Endemic fluorosis in China from ingestion of food immersed in hot spring water. Bull Environ Contam Toxicol, 54: 337–341.
Xu RQ, Wu DQ, & Xu RY (1997) Relations between environment and endemic fluorosis in Hohot region, Inner Mongolia. Fluoride, 30: 26–28.
Yang CY, Cheng MF, Tsai SS, & Hung CF (2000) Fluoride in drinking water and cancer mortality in Taiwan. Environ Res, 82: 189–193.
Yiamouyiannis J & Burk D (1977) Fluoridation and cancer: Age-dependence of cancer mortality related to artificial fluoridation. Fluoride, 10: 102–125.
Zang ZY, Fan JY, Yen W, Tian JY, Wang JG, Li XX, & Wang EL (1996) The effect of nutrition on the development of endemic osteomalacia in patients with skeletal fluorosis. Fluoride, 29: 20–24.
Zeiger E, Shelby MD, & Witt KL (1993) Genetic toxicity of fluoride. Environ Mol Mutagen, 21: 309–318.
Zeiger E, Gulati D, Kuar P, Mohamed H, Revazova J, & Deaton T (1994) Cytogenic studies of sodium fluoride in mice. Mutagenesis, 9: 467–471.
Zhang Y & Cao SR (1996) Coal burning induced endemic fluorosis in China. Fluoride, 29: 207–211.
Zhao ZP, Yaun MB, & Liu GF (1996) X-ray analysis of 80 patients with severe endemic fluorosis caused by coal burning. Fluoride, 29: 79–81.
Zierler S, Theodore M, Cohen A, & Rothman K (1988) Chemical quality of maternal drinking water and congenital heart disease. Int J Epidemiol, 17: 589–594.
Zingde MD & Mandalia AV (1988) Study of fluoride in polluted and unpolluted estuarine environments. Estuarine Coastal Shelf Sci, 27: 707–712.
Zipkin I, McClure F, & Lee W (1960) Relation of the fluoride content of human bone to its chemical composition. Arch Oral Biol, 2: 190–195.
Zwiazek JJ & Shay JM (1987) Fluoride- and drought-induced structural alterations of mesophyll and guard cells in cotyledons of jack pine (Pinus banksiana). Can J Bot, 65(11): 2310–2317.
Zwiazek JJ & Shay JM (1988a) Sodium fluoride induced metabolic changes in jack pine seedlings. I. Effect on gas exchange, water content, and carbohydrates. Can J For Res, 18(10): 1305–1310.
Zwiazek JJ & Shay JM (1988b) Sodium fluoride induced metabolic changes in jack pine seedlings. II. Effect on growth, acid phosphatase, cytokinins, and pools of soluble proteins, amino acids, and organic acids. Can J For Res, 18(10): 1311–1317.
Ce document est consacré à l’exposition environnementale aux fluorures, minéraux principalement, et à ses effets sur l’Homme, les animaux et les autres êtres vivants. Les données prises en compte concernent le fluorure d’hydrogène, le fluorure de calcium, le fluorure de sodium, l’hexafluorure de soufre et les silicofluorures étant donné que ce sont les fluorures minéraux les plus importants du point de vue des quantités libérées et des concentrations dans l’environnement ainsi que des effets toxicologiques exercés sur les organismes vivants.
Le fluorure d’hydrogène (HF) se présente sous la forme d’un liquide ou d’un gaz incolore, à l’odeur âcre, qui est très soluble dans les solvants organiques et dans l’eau avec laquelle il donne de l’acide fluorhydrique. Le fluorure de calcium (CaF2) est un solide incolore, relativement insoluble dans l’eau et les acides ou bases diluées. Le fluorure de sodium (NaF) est un solide incolore à blanc, modérément soluble dans l’eau. L’hexafluorure de soufre (SF6) est un gaz incolore et inodore, chimiquement inerte, qui est légèrement soluble dans l’eau et facilement soluble dans l’éthanol et les bases.
Le dosage des fluorures libres s’effectue le plus couramment par voie électrochimique au moyen d’une électrode sélective. On estime que les techniques de microdiffusion constituent le moyen le plus fiable pour la préparation des échantillons (c’est-à-dire par libération de d’anions fluorure libres à partir de complexes organiques ou minéraux).
Il y a libération naturelle de fluorures dans l’environnement par suite de l’érosion et de la dissolution des roches et des minéraux, à la faveur d’émissions par les volcans ou par l’intermédiaire d’aérosols marins. La combustion du charbon ainsi que les eaux usées provenant de diverses opérations industrielles, comme la fabrication de l’acier, la production d’aluminium, de cuivre et de nickel, le traitement des minerais de phosphates, la production et l’utilisation d’engrais phosphatés, la fabrication de verre, de briques et de céramiques ou encore la production de colles et d’adhésifs. L’épandage de pesticides fluorés ainsi que la fluoration de l’eau destinée à la boisson sont également des sources anthropogéniques de fluor. Les données disponibles montrent que l’extraction et l’utilisation des phosphates ainsi que la production de l’aluminium sont les sources industrielles qui rejettent le plus de fluorures dans l’environnement.
Le fluorure d’hydrogène est un produit industriel important qui est principalement utilisé pour la production de cryolithe synthétique (Na3AlF6), de fluorure d’aluminium (AlF3), des alkylates ou isoparaffines qui entrent dans la composition de l’essence automobile et de chlorofluorocarbures, avec une consommation annuelle mondiale de plus de 1 million de tonnes. On l’emploie également pour le décapage des dispositifs à semi-conducteurs, le décapage, la gravure et le dépolissage du verre, le décapage de la brique et de l’aluminium, le tannage du cuir ainsi que pour la préparation des produits antirouille vendus dans le commerce. Le fluorure de calcium est utilisé comme fondant dans la production de l’acier, du verre et de l’émail et il sert de matière première pour la préparation de l’acide fluorhydrique et du fluorure d’hydrogène anhydre ou encore d’électrolyte dans la préparation de l’aluminium. Le fluorure de sodium est utilisé pour la fluoration réglementée de l’eau de boisson, comme conservateur dans les colles, le verre et l’émail, comme fondant dans la production de l’acier et de l’aluminium et comme agent conservateur du bois. L’acide fluorosilicique (H2SiF6) et l’hexafluorosilicate de disodium (Na2SiF6) sont utilisés pour la fluoration de l’eau.
Les fluorures présents dans l’atmosphère peuvent se trouver sous forme de gaz ou de particules. Ils sont susceptibles d’être transportés sur de grandes distances par les vents ou par suite des turbulences atmosphériques ou être éliminés de l’atmosphère par dépôt à sec, par les précipitations ou encore par hydrolyse. A l’exception de l’hexafluorure de soufre, on estime que les fluorures ne restent vraisemblablement pas très longtemps dans la troposphère et qu’ils me migrent pas vers la stratosphère. La durée de séjour atmosphérique de l’hexafluorure de soufre va de 500 ans à plusieurs milliers d’années.
Le transport et la transformation des fluorures dans l’eau dépendent du pH, de la dureté de l’eau et de la présence de substances échangeuses d’ions comme les argiles. Le transport des fluorures s’effectue par le cycle de l’eau sous forme de complexes avec l’aluminium.
Dans le sol, le transport et la transformation des fluorures dépendent également du pH, mais aussi de la formation de complexes, notamment avec l’aluminium et le calcium. L’adsorption à la phase solide du sol est plus forte lorsque le pH est acide (5.5-6.5). Le lessivage des fluorures présents dans le sol ne se fait pas spontanément.
L’absorption des fluorures par les êtres vivants dépend de la voie d’exposition, de la biodisponibilité des composés et de la cinétique d’absorption et d’excrétion dans les divers organismes. Certains biotes aquatiques et terrestres accumulent les fluorures solubles. On ne possède cependant aucune information sur la bioamplification des fluorures dans les chaînes alimentaires aquatiques et terrestres.
Les plantes terrestres peuvent accumuler des fluorures aéroportés qui se sont déposés ou fixer ceux qui étaient déjà présents dans le sol.
La teneur des eaux de surface en fluorures peut varier selon le lieu et la proximité des sources d’émission. Elle va généralement de 0,01 à 0,3 mg/litre. L’eau de mer contient plus de fluorures que l’eau douce, avec une concentration comprise entre 1,2 et 1,5 mg/litre. Des concentrations plus fortes ont été relevées dans des zones où les roches sont riches en fluorures et une teneur élevée en fluorure minéral s’observe généralement dans les région où existe une activité géothermique ou volcanique (par ex. 25-50 mg/litre dans les sources chaudes et les geysers et même 2800 mg/litre dans certains lacs de la vallée du Rift, en Afrique orientale). Les décharges résultant de l’activité humaine peuvent également entraîner la présence de concentrations élevées de fluorures dans l’environnement.
Les fluorures peuvent être présents dans l’air sous forme gazeuse ou particulaire par suite d’émissions naturelles ou anthropogéniques. Les fluorures émis sous forme gazeuse ou particulaire se déposent généralement au voisinage de la source de l’émission, encore que ceux qui sont présents dans certaines particules puissent réagir sur d’autres constituants atmosphériques. La distribution et le dépôt des fluorures aéroportés dépend de l’intensité de l’émission, des conditions météorologiques, de la granulométrie des particules et de leur réactivité chimique. Dans les zones qui ne sont pas situées au voisinage immédiat des sources d’émissions, la concentration moyenne en fluorures dans l’air ambiant est généralement inférieure à 0,1 µg/m3. Elle peut être légèrement plus élevée en ville qu’à la campagne. Toutefois, même à proximité de sources d’émissions, la teneur de l’air en fluorures ne dépasse généralement pas 2 à 3 µg/m3. Dans les régions de la Chine où l’on utilise de la houille riche en fluorures comme combustible, on fait état de concentrations dans l’air ambiant pouvant atteindre 6 µg/m3.
Les fluorures sont des constituants de presque tous les types de sols, avec des teneurs totales allant de 20 à 1000 µg/g dans les régions où il n’y a pas de gisements naturels de phosphates ou de fluorures et à des concentrations de plusieurs milliers de microgrammes par gramme dans les sols où existent de tels gisements. Les fluorures présents dans l’atmosphère à l’état gazeux ou particulaire ont tendance à s’accumuler dans les couches superficielles du sol, mais peuvent migrer vers la zone radiculaire, même dans les sols calcaires. La rétention des fluorures dans le sol dépend essentiellement de la teneur en argile et en carbone organique ainsi que du pH. Les fluorures présents dans le sol y sont surtout associés à la fraction colloïdale ou argileuse. Pour tous les types de sols, c’est la teneur en fluorures solubles qui est biologiquement importante pour la faune et la flore.
Les organismes aquatiques peuvent absorber des fluorures soit directement à partir de l’eau, soit, dans une moindre mesure, à partir de leur nourriture. Les fluorures ont tendance à s’accumuler dans l’exosquelette ou dans les tissus osseux des animaux aquatiques, selon le cas. Chez le krill, par exemple, on a mesuré des concentrations moyennes de fluorures supérieures à 2000 mg/kg dans l’exosquelette. Chez les mammifères marins comme le phoque ou la baleine, la teneur moyenne des os en fluorures peut aller de 135 à 18 600 mg/kg de poids sec.
Chez les biotes terrestres, la concentration en fluorures est plus élevée dans les zones où la présence de fluorures d’origine naturelle ou anthropogénique est plus importante. On utilise beaucoup les lichens comme bioindicateurs de la présence de fluorures. Dans des lichens poussant dans un rayon de 2 à 3 km autour de sources d’émission de fluorures, on a trouvé des concentrations de l’ordre de 150-250 mg/kg, alors que la concentration de fond est de moins de 1 mg/kg.
La majorité des fluorures présents dans le sol sont insolubles et par conséquent, peu biodisponibles pour les végétaux. Toutefois, une forte teneur du sol en fluorures, un pH acide ou la présence d’argile ou de matières organiques peuvent accroître la teneur de fluorures en solution, ce qui va en favoriser leur fixation par le système radiculaire de la plante. Si des fluorures sont captés au niveau de la racine, leur concentration sera souvent plus élevée dans cette dernière que dans les pousses en raison de leur faible mobilité dans la plante. La plupart des fluorures pénètrent dans les tissus végétaux sous forme gazeuse au nivau des stomates et ils s’accumulent dans les feuilles. De petites quantités de particules fluorées peuvent également pénétrer dans la plante en traversant l’épiderme et la cuticule. Une très large surveillance s’exerce sur la végétation située aux alentours des sources d’émission anthropogéniques. On a constaté l’existence d’une corrélation entre la concentration de fluorures dans la végétation et la croissance annuelle, le régime des vents, la distance à la source d’émission de fluorures et la teneur en fluorure d’hydrogène des rejets atmosphériques.
Les fluorures s’accumulent dans les tissus osseux des vertébrés terrestres en fonction d’un certain nombre de facteurs tels que le régime alimentaire et la présence d’émissions de fluorures dans le voisinage. Par exemple, on a trouvé des une concentration moyenne de fluorures comprise entre 7000 et 8000 mg/kg dans les os de petits animaux vivant à proximité d’une usine de production d’aluminium.
Les fluorures se retrouvent dans tout l’environnement, aussi les sources d’eau potable ont-elles des chances d’en contenir au moins une petite quantité. La teneur de l’eau de boisson non fluorée en fluorures d’origine naturelle (il s’agit de l’eau à laquelle on n’a pas ajouté de flurorures pour prévenir les caries) varie dans d’importantes proportions, en fonction de l’environnement géologique de la source. La concentration peut atteindre 2,0 mg/litre; toutefois, dans les régions du monde où existe une endémie fluorotique osseuse ou dentaire parfaitement attestée, la concentration des fluorures dans l’eau de consommation va de 3 à plus de 20 mg/litre. Là où l’eau de consommation est fluorée intentionnellement pour prévenir les caries, la teneur en fluorures est de 0,7 à 1,2 mg/litre.
Presque toutes les denrées alimentaires contiennent au moins des traces de fluorures. Ils sont présents à forte concentration dans le poisson et les feuilles de thé en sont particulièrement riches. La quantité de fluorures dans le thé infusé dépend de la concentration en fluorures solubles dans les feuilles et de la durée d’infusion. L’utilisation d’engrais à base de superphosphates, qui contiennent une forte proportion de fluorures (1 à 3 %) comme impuretés, pour traiter les terres agricoles, n’accroît pas sensiblement la teneur des produits alimentaires en fluorures, car le coefficient de transfert du sol à la plante est généralement faible. Toutefois, selon une étude récente, si les conditions pédologiques s’y prêtent et que l’on traite les sols avec des phosphates suffisamment riches en impuretés flurorées, la fixation de fluorures par la plante peut augmenter. En revanche, l’irrigation des cultures avec de l’eau relativement pauvre en fluorures (moins de 3,1 mg/litre), n’entraîne généralement pas d’augmentation de la teneur des produits alimentaires en fluorures. Cela dépend cependant de l’espèce végétale en cause et de la concentration des fluorures dans le sol et l’eau. La concentration des fluorures dans les aliments est sensiblement modifiée par la teneur en fluorures de l’eau utilisée pour leur préparation ou leur fabrication, notamment dans le cas des boissons et des aliments secs - comme par exemple les aliments pour nourrissons que l’on dilue dans l’eau avant consommation. La concentration des fluorures dans les cultures vivrières situées à proximité de sources d’émission industrielles peut être plus élevée, lorsque les produits ne sont ni lavés ni transformés, que dans des cultures identiques mais situées dans des zones où il n’y a pas d’exposition d’origine industrielle. On a constaté que dans des aliments pour nourrissons vendus aux Etats-Unis et qui se présentaient sous la forme de produits à base de soja prêts à être consommés ou encore de concentrés liquides, la concentration en fluorures était plus élevée que dans les produits à base de lait; on n’a cependant constaté aucune différence entre ces derniers et les aliments pour nourrissons à base de poudre de soja. La présence de fluorures a été décelée dans le lait maternel à des concentrations comprises entre <2 à et environ 100 µg/litre, la plupart des valeurs se situant entre 5 et 10 µg/litre.
On ne dispose que de données limitées sur la teneur de l’air intérieur en fluorures. Aux Pays-Bas, on a observé une concentration en fluorures gazeux qui allait de <2 à 49 µg/m3 dans l’air de cinq habitations construites avec du bois traité par un conservateur contenant 56 % de fluorure. En Chine, on a trouvé des concentrations pouvant atteindre 155 µg/m3 dans des échantillons d’air prélevés dans des maisons où l’on brûlait du charbon à forte teneur en fluorures.
Les produits dentifrices destinés aux adultes qui sont commercialisés dans de nombreux pays contiennent généralement des fluorures à des concentrations comprises entre 1000 et 1500 µg/g; certains produits plus spécialement destinés aux enfants ont une teneur plus faible en fluorures, qui va de 250 à 500 µg/g. Les produits d’hygiène bucco-dentaire tels que pâtes dentifrices, bains de bouche et suppléments fluorés, se révèlent être une source importante de fluorures. Les bains de bouche destinés à un usage quotidien contiennent généralement de 230 à 500 mg de fluorures par litre, alors que ceux qui sont destinés à être utilisés toutes les semaines ou tous les 14 jours peuvent en contenir entre 900 et 1000 mg/litre.
L’exposition individuelle aux fluorures varie probablement beaucoup, mais l’inhalation de fluorures présents dans l’atmosphère ne contribue en général que faiblement à l’apport total de ces substances. Chez l’adulte, ce sont les produits alimentaires et l’eau de boisson qui en constituent la principale voie d’apport. Dans les régions du monde où l’on utilise du charbon riche en fluor pour se chauffer et faire la cuisine, l’air intérieur et les aliments, qui contiennent dès lors une proportion plus importante de fluorures, contribuent également pour une part importante à l’apport de ces substances. Les nourrissons alimentés à l’aide de produits formulés reçoivent 50 à 100 fois plus de fluorures que ceux qui sont nourris exclusivement au sein. L’ingestion de dentifrice par les jeunes enfants représente une fraction importante de la dose totale de fluor qu’ils absorbent. On estime en général que l’absorption de fluorures par les enfants et les adolescents ne dépasse pas environ 2 mg/jour. Bien que la dose de fluorures absorbée quotidiennement par un adulte puisse être supérieure en valeur absolue à celle qui est absorbée par un enfant, cette dernière, exprimée en mg par kg de poids corporel, peut être plus élevée que pour un adulte. Dans les régions du monde où la concentration des fluorures dans l’environnement peut être excessivement élevée et où le régime alimentaire peut comporter des produits riches en fluor, on estime qu’un adulte peut en absorber jusqu’à 27 mg par jour, la principale source étant l’eau de boisson provenant de sources souterraines captées dans des couches géologiques riches en fluor.
Les personnes qui utilisent du matériel de soudage ou qui sont employées au traitement des minerais d’aluminium, de fer ou de phosphates subissent vraisemblablement une exposition professionnelle par inhalation ou contact cutané. Selon des études relativement récentes, la concentration en fluorures dans l’air des halls d’électrolyse des usines de production d’aluminium est de l’ordre de 1 mg/m3.
Chez l’Homme comme chez les animaux de laboratoire, c’est principalement au niveau de l’estomac et de l’intestin que les fluorures ingérés passent dans la circulation générale et cette résorption dépend de la solubilité aqueuse relative de la substance en cause. Les fluorures solubles sont presque entièrement résorbés dans les voies digestives, mais le taux de résorption peut être réduit par la formation de complexes avec l’aluminium, le phosphore, le magnésium ou le calcium. Les fluorures particulaires ou gazeux qui pénètrent dans les voies respiratoires sont partiellement à totalement résorbés, le taux de résorption étant fonction de la solubilité et de la granulométrie.
Les fluorures sont rapidement amenés par le courant sanguin dans l’eau intracellulaire et extracellulaire des différents tissus; cependant chez l’Homme et les animaux de laboratoire, environ 99 % de la charge totale de l’organisme en fluorures est retenue par les os et les dents. Les fluorures sont incorporés dans le réseau cristallin des tissus dentaire et osseux.
Les fluorures traversent la barrière placentaire et il passent ainsi de la mère au foetus. Ils sont principalement éliminés par la voie urinaire. Chez le nourrisson, le fluor est retenu à environ 80-90 %; chez l’adulte, le chiffre correspondant est d’environ 60 %. Une modification du débit urinaire ou du pH peut faire varier ces valeurs.
Les fluorures sont présents dans tous les organes, tissus et liquides de l’organisme. Dans une communauté des Etats-Unis alimentée en eau fluorée, on a relevé des concentrations dans le sang total qui allaient de 20 à 60 µg/litre. Chez 127 sujets dont l’eau de boisson avait une teneur en fluorures de 5,03 mg/litre, on a trouvé un taux plasmatique moyen de 106 ± 76 (sigma) µg/litre. Le plasma et le sérum ont pratiquement la même teneur en fluorures. En ce qui concerne les tissus calcifiés, c’est généralement dans les os, la dentine et l’émail que la concentration est la plus élevée. Dans les os, cette concentration varie en fonction de l’âge et du sexe ainsi que de la nature et de la partie de l’os concernée. On estime qu’elle est le reflet de l’exposition à long terme du sujet. Dans l’émail dentaire, la concentration en fluorures diminue exponentiellement avec la distance à la surface et elle dépend de la localisation, de l’usure des faces de la dent, de l’exposition par la voie générale et des applications topiques de fluorures auxquelles il a pu être procédé. La teneur en fluorures des tissus mous correspond à la concentration sanguine. Chez un sujet en bonne santé, la concentration urinaire des fluorures est liée à l’apport de fluor. Chez des sujets exposés de par leur profession à des fluorures présents dans l’air ou résidant dans des zones où la fluorose est endémique, on constate une élévation de la teneur des urines en fluorures.
Chez des rats recevant des fluorures par voie orale pendant des périodes allant de 3 à 5 semaines, diverses études ont mis en évidence des effets au niveau du squelette consistant en une inhibition de la minéralisation et de la formation du tissus osseux, un retard dans la guérison des fractures ainsi qu’une réduction du volume osseux et de la synthèse du collagène. Des études à moyen terme au cours desquelles des souris ont reçu pendant 6 mois une eau de boisson contenant des fluorures à une concentration supérieure à 4,5 mg/kg de poids corporel, ont révélé les effets suivants : anomalies du remodelage osseux, mégalocytose hépatique, néphrose, minéralisation du myocarde, nécrose ou dégénérescence des tubes séminifères du testicule.
Une étude de cancérogénicité très complète au cours de laquelle des groupes de rats F344/N et des souris B6C3F1 des deux sexes ont reçu pendant 2 ans une eau de boisson contenant jusqu’à 79 mg/litre de fluorure de sodium, n’a révélé aucune augmentation significative, dans l’un et l’autre groupe, de l’incidence des tumeurs, quelles qu’elles soient. On a bien observé une tendance statistiquement significative à l’accroissement des ostéosarcomes chez les mâles lorsqu’on augmentait l’exposition au fluorure, mais l’incidence restait cependant dans les mêmes limites que chez les témoins historiques.
Une autre étude de cancérogénicité de 2 ans a été effectuée sur des rats Sprague-Dawley, qui ont été exposés par voie alimentaire à des doses quotidiennes de fluorures allant jusqu’à 11,3 mg/kg p.c.; aucune augmentation statistiquement significative n’a été relevée dans l’incidence des ostéosarcomes ou d’autres tumeurs. Il existe une autre étude, qui fait état d’un accroissement de l’incidence des ostéosarcomes chez des souris qui avaient reçu des doses quotidienne de fluorures allant jusqu’à 11,3 mg/kg p.c., mais les résultats sont difficiles à interpréter car les animaux étaient infectés par un rétrovirus de type C.
En règle générale, les fluorures n’ont pas d’action mutagène sur les cellules procaryotes. On a montré qu’ils augmentaient la fréquence des mutations au niveau de certains locus dans des cellules de lymphome murin et des cellules lymphoblastoïdes humaines en culture, mais ces mutations ne semblent pas être des mutations ponctuelles et sont probablement plutôt dues à des lésions chromosomiques. Les fluorures ont des effets clastogènes sur divers types de cellules. On pense que cette activité clastogène s’explique par un effet sur la synthèse ou la réparation de l’ADN, plutôt que par une interaction directe avec cette molécule. Dans la plupart des études au cours desquelles des fluorures ont été administrés par voie orale à des rongeurs, on n’a relevé aucun effet sur la morphologie des spermatozoïdes, ni sur la fréquence des aberrations chromosomiques, des micronoyaux, des échanges entre chromatides-soeurs ou des ruptures des brins de l’ADN. On a toutefois observé des lésions cytogénétiques au niveau de la moelle osseuse ainsi que des anomalies affectant la morphologie des spermatozoïdes lorsque ces substances étaient administrées par voie intrapéritonéale à des rongeurs.
Des études récentes au cours desquelles des animaux de laboratoire ont reçu des fluorures ajoutés à leur eau de boisson, n’ont pas mis en évidence d’effets sur la reproduction ou le développement. Des anomalies histopathologiques ont cependant été relevées au niveau des organes reproducteurs chez des lapins mâles qui avaient reçu par voie orale pendant 18 à 29 mois une dose quotidienne de 4,5 mg de fluorures par kg de poids corporel, chez des souris mâles soumises quotidiennement pendant 30 jours à une dose orale de fluorures supérieure ou égale à 4,5 par kg p.c. ainsi que chez des lapines qui en avaient reçu pendant 100 jours une dose quotidienne supérieure ou égale à 10 mg/kg p.c. administrée par voie sous-cutanée. Des effets génésiques indésirables ont également été observés chez des souris femelles à qui on avait administré par voie orale une dose de fluorures supérieure ou égale à 5.2 mg/kg p.c. 6 à 15 jours après l’accouplement ainsi que chez des lapins mâles qui en avaient reçu une dose quotidienne supérieure ou égale à 9.1 mg/kg p.c. pendant 30 jours.
Les études épidémiologiques consacrées aux effets des fluorures sur la santé humaine concernent l’exposition professionnelle des travailleurs, notamment ceux qui sont affectés à la production de l’aluminium, ainsi que l’exposition des populations consommant de l’eau fluorée. Un certain nombre d’études d’épidémiologie analytique portant sur des travailleurs exposés à des fluorures de par leur profession, font ressortir une augmentation de l’incidence des cancers du poumon et de la vessie ainsi qu’un accroissement de la mortalité due à ces cancers ou à des tumeurs d’autre localisation. Il n’y a toutefois généralement pas de tendance cohérente et dans certaines de ces études l’augmentation constatée de la morbidité et de la mortalité par cancer peut être attribuée à l’exposition des travailleurs à d’autres substances que les fluorures.
Un grand nombre d’études épidémiologiques ont été menée dans de nombreux pays pour étudier la relation entre la consommation d’eau fluorée et la morbidité ou la mortalité par cancer. Il n’existe aucune preuve solide d’un lien entre la consommation d’eau réglementairement fluorée une augmentation de la morbidité ou de la mortalité dues au cancer.
Les effets des fluorures sur l’émail dentaire peuvent être bénéfiques mais également nocifs. La prévalence des caries dentaires est inversement proportionnelle à la teneur de l’eau de boisson en fluorures. La prévalence de la fluorose dentaire est en rapport étroit avec cette teneur, avec une relation dose-réponse positive.
On continue à observer des cas de fluorose osseuse liés à la consommation d’eau à forte teneur en fluorures. Un certain nombre de facteurs tels que l’état nutritionnel, le régime alimentaire, le climat (qui influe sur la prise de liquides), une exposition concomitante à d’autres substances et un apport de fluorures provenant d’autres sources que l’eau de boisson jouent, semble-t-il, un rôle important dans l’apparition de cette maladie. Des cas de fluorose osseuse peuvent survenir parmi les travailleurs que leur profession expose à des concentrations élevées de fluorures aéroportés, mais on n’a guère obtenu d’informations nouvelles à ce sujet.
Les résultats d’un certain nombre d’études écologiques incitent à penser qu’il existe un lien entre la consommation d’eau fluorée et les fractures du col du fémur. Ces conclusions ne sont pas confirmées par d’autres travaux notamment par l’analyse épidémiologique du problème. Dans certains cas, on a même constaté un effet protecteur des fluorures contre ces fractures.
Deux études permettent d’évaluer le risque de fracture pour toute une gamme d’apports en fluor. La première a montré que le risque relatif de fractures de toute nature ainsi que de fracture du col du fémur était élevé dans des groupes consommant de l’eau dont la teneur en fluorures était >1,45 mg/litre (apport total >6,5 mg/jour); la différence atteignait le seuil de signification statistique pour le groupe dont l’eau de boisson avait une teneur en fluorures >4,32 mg/litre (apport total 14 mg/jour). Dans l’autre étude, on a observé un accroissement de l’incidence des fractures indépendant de la dose dans un groupe d’âge constitué de femmes exposées aux fluorures par l’eau qu’elles consommaient.
Selon les études épidémiologiques, rien n’indique que la consommation d’eau fluorée par les femmes enceintes augmente le risque d’avortement spontané ou de malformations congénitales. D’autres études épidémiologiques portant sur des travailleurs exposés de par leur profession n’ont pas décelé de véritable preuve d’effets génotoxiques ou systémiques au niveau du système respiratoire, des organes hématopoïétiques, du foie ou du rein qui puisse être directement attribué à l’exposition aux fluorures en tant que telle.
La présence de fluorures à la concentration de 100 mg/litre n’a pas affecté la croissance bactérienne ni la capacité de dégradation de boues activées mesurée par la demande chimique d’oxygène. La CE50 relative à l’inhibition de la nitrification bactérienne a été trouvée égale à 1218 mg de fluorures/litre. La CE50 à 96 h relative à la croissance des algues a été trouvée égale à 123 mg/litre pour les algues d’eau douce et à 81 mg/litre pour les algues marines.
Les valeurs de la CL50 à 48 h pour les invertébrés aquatiques vont de 53 à 304 mg/litre. Les invertébrés d’eau douce les plus sensibles sont le sphéridé Musculium transversum avec une mortalité statistiquement significative (50 %) observée à la concentration de 2,8 mg de fluorures par litre lors d’une étude de 8 semaines, ainsi que plusieurs phryganes (dulçaquicoles; famille : hydropsychidés) pour lesquelles les valeurs « sans danger » de la concentration (CE0,01 à 8760 h) vont de 0,2 à 1,2 mg/litre. L’artémia (Artemia salina) a été l’espèce marine la plus sensible observée. Lors d’un test de 12 jours avec renouvellement statique de l’eau, on a constaté une réduction statistiquement significative de la croissance à la concentration de 5,0 mg/litre.
Les valeurs de la CL50 à 96 h pour divers poissons d’eau douce vont de 51 mg/litre (truite arc-en-ciel, Oncorhynchus mykiss) à 460 mg/litre (épinoche à trois épines, Gasterosteus aculeatus). Dans tous les tests de toxicité aiguë à 96 h, on a trouvé des valeurs supérieures à 100 mg/litre pour les poissons de mer. Il existe une corrélation négative entre la toxicité des fluorures minéraux pour les poissons d’eau douce et la dureté de l’eau (carbonate de calcium) et une corrélation positive entre cette toxicité et la température. Les symptômes d’une intoxication aiguë par les fluorures consistent notamment en léthargie, mouvements violents et erratiques et mort. Lors de tests de 20 jours avec renouvellement statique de l’eau, on a trouvé une CE50 de 2,7 à 4,7 mg/litre pour la truite arc-en-ciel. On estime que la concentration « sans danger » (CL0,01 à durée infinie) est de 5,1 mg/litre pour la truite arc-en-ciel et de 7,5 mg/litre pour la truite brune (truite d’Europe, Salmo trutta). A des concentrations >3,2 (effluents) ou >3,6 (fluorure de sodium) de fluorure par litre, l’éclosion des oeufs de la carpe indienne Catla catla a été retardée de 1 à 2 h.
Les études éthologiques effectuées en rivière (eau douce) sur des saumons coho adultes (Oncorhynchus sp.) montrent qu’une modification de la composition chimique des eaux consistant à augmenter la concentration en fluorures pour la faire passer à 0,5 mg/litre peut avoir un effet négatif sur la migration; les saumons en migration sont extrêmement sensibles aux variation de composition chimique de leur rivière d’origine. Au laboratoire, les fluorures semblent avoir des effets toxiques sur les processus microbiens à des concentrations qui sont celles des sols dont la pollution fluorée est modérée; de même, l’accumulation de matières organiques dans les zones situées au voisinage d’usines de production d’aluminium est attribuée à une forte inhibition de l’activité microbienne par les fluorures.
C’est au niveau des jeunes tissus en développement des arbres feuillus et des aiguilles de conifères en phase d’allongement que l’on a le plus de chances de constater des signes de phytotoxicité fluorotique (fluorose) tels que chlorose, nécrose et réduction du rythme de croissance. Le déclenchement de la fluorose a été clairement mis en évidence en laboratoire, en serre ainsi que lors d’essais contrôlés sur des parcelles expérimentales de plein champ. Une proportion importante des articles publiés au sujet de la toxicité des fluorures pour les plantes portent sur la fumigation des serres avec du fluorure d’hydrogène. La nécrose foliaire a été observée pour la première fois sur des plants de vigne (Vitis vinifera) exposés à des concentrations de 0,17 et 0,27 µg/m3, au bout de 99 et 83 jours respectivement. La dose la plus faible produisant un effet nécrotique observable (65 % des feuilles) sur le glaïeul à grandes fleurs (Gladiolus grandiflorus) a été trouvée égale à 0,35 µg de fluorure par m3. Les fluorures aéroportés peuvent également avoir un effet sur l’apparition des maladies végétales, mais la nature et l’ampleur de cet effet dépendent du couple plante-germe pathogène en cause.
Plusieurs études de brève durée portant sur des cultures en solution nutritive ont permis de déterminer le seuil de toxicité de l’ion fluor, qui se situerait dans les limites approximatives de 50 à 2000 µmol/litre. La spécificité toxicologique observée n’est pas uniquement fonction de l’espèce végétale, elle dépend également de la nature des ions fluor en cause; ainsi, certains fluorures complexes d’aluminium présents dans des cultures en solution nutritive peuvent se révéler toxiques à des concentration de 22-357 µmol de fluorure par litre, alors que le fluorure d’hydrogène est toxique aux concentrations de 71-137 µmol/litre. Quelques études ont été consacrées à des cas d’exposition aux fluorures présents dans le sol. Elles ont permis de déterminer que la fixation et la toxicité potentielle de ces substances dépendait beaucoup de la nature du sol.
Dans le cas des oiseaux, on a obtenu une valeur de 50 mg/kg de poids corporel pour la DL50 à 24 h d’étourneaux sansonnets (Sturnus vulgaris) âgés de 1 jour et de 17 mg/kg p.c. chez des oisillons de 16 jours. Le taux de croissance a été sensiblement réduit aux doses de 13 et de 17 mg de fluorure par kg p.c. (doses les plus fortes auxquelles ont a mesuré le taux de croissance). La plupart des premiers travaux consacrés aux mammifères portent sur les ongulés domestiques. On a ainsi observé des cas de fluorose chez des bovins et des ovins. La dose alimentaire la plus faible ayant causé un effet sur des ongulés sauvages a été déterminée dans le cadre d’une étude contrôlée sur des cerfs de Virginie en captivité (Odocoileus virginianus); il s’agissait de l’apparition de tachetures sur l’ensemble des incisives des animaux à partir de 35 mg/kg de nourriture.
On a montré que les usines de production d’aluminium, de phosphates et d’engrais, les briquetteries, ainsi que les usines de production de fibres de verre constituaient toutes des sources de pollution fluorée et qu’il existait une corrélation entre leur présence et les dommages constatés sur la végétation du voisinage. L’étude de la végétation située au voisinage d’une usine de production de phosphore a montré que l’ampleur des dommages infligés aux plantes et la concentration en fluorures dans le sol étaient inversement proportionnelles à la distance de l’installation. La concentration moyenne des fluorures allait de 281 mg/kg dans les secteurs où la végétation était très endommagée, à 44 mg/kg dans les zones où les dommages étaient moindres; sur le site témoin, la concentration des fluorures était de 7 mg/kg. Dans des communautés végétales situées à proximité d’une usine de production d’aluminium, on a constaté des différences de composition et de structure dues en partie à des variations dans la tolérance au fluor. Il est toutefois à noter que, dans la nature, l’un des principaux problèmes qui se posent pour l’identification des effets de la pollution fluorée tient à la présence d’autres polluants atmosphériques qui jouent de rôle de variables de confusion. La prudence doit donc être de règle lors de l’interprétation des nombreuses études consacrées à la pollution fluorée dans l’environnement naturel.
Les premières observations d’effets de la pollution fluorée sur des mammifères ont été faites dans le cadre d’études de terrain consacrées à des animaux domestiques tels que les bovins et les ovins. Les fluorures peuvent passer chez l’animal à partir de la végétation, du sol et de l’eau qu’il boit. Les niveaux de tolérance pour les animaux domestiques se situent à 30 mg par kg de nourriture ou à 2,5 mg/litre d’eau de boisson pour les animaux laitiers. Les incidents dont ont été victimes des animaux domestiques avaient pour origine soit des sources naturelles comme les éruptions volcaniques ou la nature géologique du sol, soit des sources anthropogéniques, telles que l’administration au bétail de suppléments minéraux ou la présence d’industries responsables d’émissions de fluorures et de centrales électriques. Les symptômes constatés consistent en émaciation, raideur des articulations et anomalies des dents et du squelette. Parmi les autres effets, on peut citer une baisse de la production de lait et des effets nocifs sur la capacité de reproduction des animaux. La concentration alimentaire la plus faible ayant causé une fluorose chez des cervidés sauvages était égale à 35 mg/kg. Les études consacrées aux effets du fluor sur la faune sauvage concernent son action sur l’intégrité de la denture et du squelette. Au voisinage des usines de production d’aluminium, les effets de la pollution fluorée se traduisent par de la boiterie et une atteinte lésionnelle des dents.
Les effets des fluorures sur la santé humaine peuvent être positifs ou négatifs avec un faible écart entre les doses correspondantes. Il existe une exposition importante à l’ensemble des sources de fluor, parmi lesquelles les aliments et l’eau de consommation.
On possède peu de données qui permettraient de caractériser les relations dose-réponse concernant les divers effets indésirables. En particulier, les données relatives à l’exposition totale aux fluorures sont peu nombreuses, notamment concernant l’apport et l’absorption.
Les effets les plus graves résultent d’une accumulation de fluor dans le squelette par suite d’une exposition excessive et de longue durée et ils se traduisent par une ostéopathie non maligne, en particulier une fluorose osseuse avec tendance aux fractures. Des données en provenance de Chine et du sous-continent indien montrent à l’évidence que la fluorose osseuse et l’augmentation du risque de fractures se manifestent lorsque la dose totale de fluor atteint 14 mg/jour et on est fondé à penser que pour un apport fluoré supérieur à environ 6 mg/jour, le risque de fracture est déjà augmenté.
Dans les eaux douces, la concentration naturelle des fluorures est généralement inférieure au seuil de toxicité pour les organismes aquatiques. Ces derniers peuvent cependant souffrir de la proximité de décharges anthropogéniques. La toxicité des fluorures dépend de la dureté de l’eau.
Il existe un risque pour les espèces végétales sensibles qui poussent à proximité des sources anthropogéniques de fluorures. La libération de ces substances dans l’environnement à partir de telles sources cause des dégâts à la végétation terrestre locale, mais il est difficile de savoir quelle est la part des seuls fluorures dans ces effets, car d’autres polluants sont également présents dans l’atmosphère. Les fluorures sont habituellement fortement adsorbés aux particules du sol. Il en résulte que les plantes fixent relativement peu de fluorures par cette voie et que le lessivage est minime.
La concentration en fluorures dans la végétation au voisinage de diverses sources d’émission comme les usines de production d’aluminium, peut être supérieure au seuil de toxicité alimentaire pour les mammifères déterminé par l’expérimentation animale. On observe des cas de fluorose parmi les animaux domestiques. Il existe des régions où l’on signale encore des problèmes de fluorose dans le bétail à qui l’on donne des suppléments minéraux et une eau de boisson riche en fluor. En outre, l’utilisation prolongée d’engrais phosphatés contenant des impuretés fluorées fait courir un risque d’intoxication aux animaux qui broutent dans des pâturages contaminés ou ingèrent de la terre polluée. Des effets due à une intoxication par le fluor, tels que boiterie et lésions dentaires, s’observent également chez des mammifères sauvages vivant à proximité de sources d’émission anthropogéniques.
Tous les êtres vivants sont exposés aux fluorures provenant de sources naturelles ou anthropogéniques. L’apport en fluor est très important dans les régions du monde où l’environnement est riche en cet élément et où l’Homme consomme l’eau de sources souterraines à forte teneur en fluorures. L’exposition peut être encore plus importante à proximité de sources d’émission ponctuelles. La fluoration des produits d’hygiène dentaire constitue pour beaucoup de gens une source supplémentaire d’exposition.
Les effets des fluorures peuvent être bénéfiques ou indésirables et l’écart entre les doses correspondantes est étroit.
Des effets sur les dents et les os peuvent s’observer à des doses inférieures à celles qui produisent des effets indésirables spécifiques sur d’autres organes ou tissus.
Chez l’Homme, les effets sur les os (fluorose osseuse et tendance aux fractures) sont considérés comme les plus significatifs pour l’évaluation des effets indésirables d’une exposition prolongée aux fluorures.
La fluorose osseuse est une maladie invalidante qui touche des millions de gens dans diverses régions d’Afrique, de Chine et de l’Inde et dont les conséquences sanitaires et socio-économiques sont très importantes.
La principale cause de fluorose osseuse endémique est l’absorption de fluorures présents dans l’eau de boisson et les aliments. Dans certaines régions, l’utilisation de charbon riche en fluor comme combustible à l’intérieur des habitations est également une source importante d’exposition.
On ne possède guère de données à partir desquelles estimer l’exposition totale aux fluorures ou leur biodisponibilité et les documents relatifs à la caractérisation de leurs effets indésirables contiennent un certain nombre d’incohérences.
Il apparaît clairement, à la lumière des données en provenance de Chine et du sous-continent indien, que la fluorose osseuse et la tendance aux fractures se manifestent pour des doses totales de 14 mg de fluorure/jour et on est incité à penser qu’à partir d’environ 6 mg/jour, le risque d’atteinte osseuse est d’ores et déjà augmenté.
Une exposition excessive aux fluorures biodisponibles constitue un risque pour la faune et la flore aquatiques et terrestres.
On peut utiliser les espèces fluoro-sensibles comme sentinelles pour attirer l’attention sur les dangers dus à la présence de fluorures dans l’environnement.
Une meilleure connaissance de l’accumulation des fluorures par les êtres vivants est nécessaire et il faut également déterminer comment surveiller et réduire cette accumulation.
Il convient de mieux caractériser les effets biologiques de l’exposition aux fluorures.
Este documento se centra en la exposición en el medio ambiente a los fluoruros derivados fundamentalmente de fuentes inorgánicas y sus efectos en las personas, los animales y otra biota. Contiene datos sobre el fluoruro de hidrógeno, el fluoruro de calcio, el fluoruro de sodio, el hexafluoruro de azufre y los silicofluoruros, puesto que se considera que estos compuestos son los fluoruros inorgánicos más importantes, teniendo en cuenta el volumen de las emisiones en el medio ambiente, las concentraciones en éste y los efectos toxicológicos en los organismos vivos.
El fluoruro de hidrógeno (HF) es un líquido o gas incoloro picante muy soluble en disolventes orgánicos y en agua, con la que forma ácido fluorhídrico. El fluoruro de calcio (CaF2) es un sólido incoloro relativamente insoluble en agua y ácidos y bases diluidos. El fluoruro de sodio (NaF) es un sólido entre incoloro y blanco moderadamente soluble en agua. El hexafluoruro de azufre (SF6) es un gas inerte incoloro e inodoro ligeramente soluble en agua y fácilmente soluble en etanol y en bases.
El procedimiento más común utilizado para cuantificar la concentración de aniones fluoruro libres es el electrodo selectivo de iones fluoruro. Se considera que las técnicas de microdifusión son los métodos más exactos de preparación de muestras (es decir, la liberación de fluoruro iónico a partir de complejos orgánicos e inorgánicos).
Los fluoruros se liberan en el medio ambiente de manera natural a través de la meteorización y disolución de minerales, las emisiones de volcanes y los aerosoles marinos. También se liberan a través de la combustión del carbón y las aguas industriales y los desechos de diversos procesos industriales, en particular la fabricación de acero, la producción primaria de aluminio, de cobre y de níquel, la elaboración de minerales de fosfato, la producción y uso de fertilizantes fosfatados, la fabricación de vidrio, ladrillos y cerámica y la producción de cola y adhesivos. La utilización de plaguicidas que contienen fluoruros, así como la fluoración del abastecimiento de agua potable contribuyen a la emisión de fluoruros a partir de actividades humanas. Según los datos disponibles, la obtención y uso de minerales de fluoruro, así como la fabricación de aluminio, son las principales fuentes industriales de emisiones de fluoruros en el medio ambiente.
El fluoruro de hidrógeno es un producto industrial importante que se utiliza fundamentalmente en la producción de criolita sintética (Na3AlF6), trifluoruro de aluminio (AlF3) y los alquilatos y los clorofluorocarburos de la gasolina para motores, con un consumo anual mundial superior a un millón de toneladas. Se utiliza también en el tratamiento químico de los dispositivos semiconductores, la limpieza y el tratamiento químico del vidrio, la limpieza de los ladrillos y del aluminio y el curtido de pieles, así como en los anticorrosivos comerciales. El fluoruro de calcio se utiliza como fundente en la producción de acero, vidrio y esmalte, como materia prima para la producción de ácido fluorhídrico y fluoruro de hidrógeno anhidro y como electrolito en la producción de aluminio. El fluoruro de sodio se utiliza en la fluoración controlada del agua de bebida, como conservante en la cola, en la producción de vidrio y esmalte, como fundente en la producción de acero y aluminio, como insecticida y como conservante de la madera. El hexafluoruro de azufre se utiliza ampliamente en diversos componentes electrónicos y en la producción de magnesio y aluminio. El ácido hexafluorosilícico (H2SiF6) y el hexafluorosilicato disódico (Na2SiF6) se utilizan para la fluoración de los sistemas de abastecimiento de agua potable.
Los fluoruros se pueden encontrar en la atmósfera en forma gaseosa o particulada. Los fluoruros atmosféricos pueden recorrer largas distancias transportados por el viento o por turbulencias atmosféricas, o eliminarse mediante deposición húmeda y seca o hidrólisis. No cabe esperar de los compuestos de fluoruro, con la excepción del hexafluoruro de azufre, una permanencia prolongada en la troposfera o su desplazamiento hacia la estratosfera. El hexafluoruro de azufre tiene un tiempo de permanencia en la atmósfera que oscila entre 500 y varios miles años.
El transporte y la transformación de los fluoruros en el agua dependen del pH, la dureza del agua y la presencia de materiales intercambiadores de iones, como la arcilla. Los fluoruros se suelen transportar a través del ciclo hidrológico formando complejos con el alu minio.
El transporte y la transformación de los fluoruros en el suelo dependen del pH y de la formación de complejos, sobre todo con el aluminio y el calcio. La adsorción a la fase sólida del suelo es más fuerte con un pH ligeramente ácido (5,5-6,5). No es fácil su lixiviación del suelo.
La absorción de los fluoruros por la biota depende de la vía de exposición, de su biodisponibilidad y de la cinética de absorción/excreción del organismo. Cierta biota acuática y terrestre bioacumula fluoruros solubles. Sin embargo, no se encontró información relativa a la bioamplificación de los fluoruros en las cadenas alimentarias acuática o terrestre.
Las plantas terrestres pueden acumular fluoruros a través de la deposición de partículas suspendidas en el aire y la absorción del suelo.
Los niveles de fluoruros en las aguas superficiales varían en función del lugar y de la proximidad a fuentes de emisión. Las concentraciones en aguas superficiales generalmente oscilan entre 0,01 y 0,3 mg/l. El agua marina contiene más fluoruros que el agua dulce, con concentraciones que van de 1,2 a 1,5 mg/l. Se han registrado niveles más altos de fluoruros en zonas cuyas rocas naturales los contienen en una elevada proporción, y se observan con frecuencia niveles altos de fluoruros inorgánicos en regiones con actividad geotérmica o volcánica (p. ej., 25-50 mg/l en fuentes termales y géiseres y hasta 2800 mg/l en ciertos lagos del valle del Rift, en la región oriental de África). Las descargas de origen humano pueden provocar también un aumento de su concentración en el medio ambiente.
Los fluoruros suspendidos en el aire se encuentran en forma gaseosa y particulada, procedentes de fuentes tanto naturales como humanas. Estos fluoruros emitidos como materia gaseosa y particulada se suelen depositar en las proximidades de la fuente de emisión, aunque algunas partículas pueden reaccionar con otros componentes de la atmósfera. La distribución y deposición de los fluoruros suspendidos en el aire dependen de la intensidad de la emisión, las condiciones meteorológicas, el tamaño de las partículas y la reactividad química. En zonas no situadas en las inmediaciones de las fuentes de emisión, las concentraciones medias de fluoruros en el aire son generalmente inferiores a 0,1 µg/m3. Los niveles pueden ser ligeramente más altos en las zonas urbanas que en las rurales; sin embargo, incluso en las proximidades de las fuentes de emisión los niveles de fluoruros suspendidos en el aire no suelen ser superiores a 2-3 µg/m3. En algunas zonas de China donde se utiliza carbón con un contenido elevado de fluoruros como fuente de combustible, se han notificado concentraciones de fluoruros en el aire de hasta 6 µg/m3.
Los fluoruros forman parte de la mayoría de los tipos de suelos, con concentraciones totales de entre 20 y 1000 µg/g en zonas sin depósitos naturales de fosfatos o fluoruros y de hasta varios miles de µg por gramo en suelos minerales con depósitos de fluoruros. Los fluoruros gaseosos y particulados suspendidos en el aire tienden a acumularse en la capa superficial del suelo, pero pueden desplazarse por toda la rizosfera, incluso en suelos calcáreos. La retención de fluoruros en el suelo depende fundamentalmente del contenido de arcilla y carbono orgánico, así como del pH del suelo. Los fluoruros del suelo están asociados fundamentalmente con su fracción coloidal o arcillosa. En todos los tipos de suelos, los fluoruros biológicamente importantes para las plantas y los animales son los solubles.
Los organismos acuáticos pueden absorber fluoruros directamente del agua o en menor medida a través de los alimentos. Los fluoruros tienden a acumularse en el exoesqueleto o en el tejido óseo de los animales acuáticos. Se han registrado concentraciones medias de fluoruros > 2000 mg/kg en el exoesqueleto del krill; las concentraciones medias de fluoruros en los huesos de mamíferos acuáticos, como focas y ballenas, oscilan entre 135 y 18 600 mg/kg de peso seco.
Las concentraciones de fluoruros en la biota terrestre son superiores en las zonas con niveles de fluoruros más elevados procedentes de fuentes naturales y humanas. Con frecuencia se han utilizado líquenes como bioindicadores de fluoruros. En los líquenes que crecían en un radio de 2-3 km de las fuentes de emisión se determinaron concentraciones medias de fluoruros de 150-250 mg/kg, en comparación con un nivel de fondo < 1 mg de fluoruros/kg.
La mayor parte de los fluoruros presentes en el suelo son insolubles y, por consiguiente, están menos disponibles para las plantas. Sin embargo, factores como una concentración alta de fluoruros en el suelo o un pH bajo y la presencia de arcilla y/o materia orgánica pueden aumentar sus niveles en solución, aumentando la absorción a través de las raíces de las plantas. Si se absorben fluoruros a través de las raíces, sus concentraciones son con frecuencia más altas en éstas que en los brotes, debido a su baja movilidad en la planta. La mayor parte de los fluoruros se introducen en los tejidos de las plantas en forma de gases por los estomas y se acumulan en las hojas. Pueden entrar en la planta pequeñas cantidades de fluoruros particulados suspendidos en el aire a través de la epidermis y la cutícula. Se ha vigilado de cerca la vegetación de las proximidades de fuentes de emisión de fluoruros de origen humano. Se ha observado una correlación entre la concentración de fluoruros en la vegetación y los incrementos del crecimiento anual, las características del viento, la distancia de la fuente de fluoruros y la concentración de fluoruro de hidrógeno en las emisiones aéreas.
Los fluoruros se acumulan en el tejido óseo de los vertebrados terrestres, en función de factores como la alimentación y la proximidad de las fuentes de emisión. Por ejemplo, se han registrado concentraciones medias de fluoruros de 7000-8000 mg/kg en los huesos de pequeños mamíferos en las cercanías de una fundición de aluminio.
Los fluoruros son ubicuos en el medio ambiente; por ello, es frecuente que las fuentes de agua de bebida los contengan por lo menos en pequeñas cantidades. La cantidad de fluoruros presentes de manera natural en el agua potable no fluorada (es decir, agua de bebida a la cual no se han añadido deliberadamente fluoruros para prevenir la caries dental) es muy variable, dependiendo del entorno geológico concreto de procedencia del agua. Los niveles pueden alcanzar hasta unos 2,0 mg/l; sin embargo, en las zonas del mundo con fluorosis endémica del esqueleto y/o los dientes bien documentada, las concentraciones de fluoruros en la red de abastecimiento de agua potable van de 3 a más de 20 mg/l. En zonas con agua potable fluorada (es decir, con adición deliberada de fluoruros para la prevención de la caries dental), la concentración de fluoruros en ella generalmente oscila entre 0,7 y 1,2 mg/l.
Prácticamente todos los productos alimenticios contienen por lo menos cantidades ínfimas de fluoruros. Se observan concentraciones elevadas en los peces. Las hojas de té son particularmente ricas en ellos; la cantidad en el té en infusión depende de la concentración de fluoruros solubles en las hojas de té, del nivel de fluoruros en el agua utilizada en su preparación y del tiempo de permanencia de las hojas en el agua. La concentración de fluoruros en los productos alimenticios no aumenta de manera significativa por el uso de fertilizantes superfosfatados, que contienen concentraciones importantes de fluoruros (1%-3%) como impurezas, en las tierras agrícolas, debido a que el coeficiente de transferencia del suelo al material de las plantas es generalmente bajo. Sin embargo, un estudio reciente parece indicar que, en condiciones apropiadas del suelo y con la aplicación de suficientes fluoruros como impurezas en los fertilizantes fosfatados, puede aumentar la absorción de fluoruros por las plantas. El uso de agua de riego con niveles relativamente bajos (< 3,1 mg/l) de fluoruros en los cultivos no suele aumentar sus concentraciones en los productos alimenticios. Sin embargo, esto depende de la especie de la planta y de las concentraciones de fluoruros en el suelo y el agua. El nivel de fluoruros en los alimentos depende sobre todo del contenido de fluoruros del agua utilizada en su preparación o elaboración, sobre todo en las bebidas y los productos alimenticios secos - por ejemplo, las preparaciones en polvo para lactantes - a los cuales se añade agua antes del consumo. Las concentraciones de fluoruros en los alimentos no lavados o no elaborados en las cercanías de fuentes industriales (emisiones) de fluoruros pueden ser superiores a las de los mismos alimentos cultivados en otras zonas no industriales. En las preparaciones infantiles disponibles en el comercio en los Estados Unidos, los concentrados a base de soja listos para el consumo y líquidos contenían niveles de fluoruros superiores a los de los productos lácteos equivalentes; sin embargo, no se observó una diferencia significativa entre las preparaciones infantiles en polvo a base de soja y de leche. Se han detectado fluoruros en la leche materna, habiéndose notificado niveles comprendidos entre < 2 y aproximadamente 100 µg/l, con la mayoría de los valores situados en el intervalo de 5 a 10 µg/l.
Los datos disponibles sobre la concentración de fluoruros en el aire de espacios cerrados son limitados. En los Países Bajos, las concentraciones de fluoruros gaseosos eran de < 2 a 49 µg/m3 en el aire del interior de cinco viviendas construidas con madera tratada con un conservante que contenía un 56% de fluoruros. En China, se han notificado concentraciones de hasta 155 µg/m3 en muestras de aire recogido en el interior de viviendas donde se utilizaba como combustible carbón que contenía concentraciones elevadas de fluoruros.
Los productos dentífricos para adultos presentes en el mercado de muchos países suelen contener fluoruros en concentraciones de 1000 a 1500 µg/g; algunos productos infantiles contienen niveles más bajos, de 250 a 500 µg/g. Se han identificado productos dentales, por ejemplo pasta de dientes, colutorios y suplementos de fluoruros, como fuentes importantes de éstos. Los enjuagues bucales comercializados para uso doméstico cotidiano suelen contener entre 230 y 500 mg de fluoruros/l, mientras que los colutorios destinados a un uso semanal o quincenal suelen contener 900-1000 mg de fluoruros/l.
Aunque es probable que la exposición individual al fluoruro sea muy variable, la inhalación de fluoruros suspendidos en el aire generalmente representa una contribución de escasa importancia a la ingesta total de esta sustancia. En adultos, la vía principal de ingesta de fluoruros es el consumo de productos alimenticios y agua de bebida. En las zonas del mundo en las cuales se utiliza carbón con un alto contenido de fluoruros en la calefacción y la preparación de los alimentos, la inhalación del aire interno y el consumo de productos alimenticios con niveles altos de fluoruros también contribuyen a aumentar la ingesta. Los lactantes alimentados con preparaciones reciben 50-100 veces más fluoruros que los que se nutren exclusivamente con leche materna. La ingestión de dentífrico por los niños pequeños representa una contribución importante a su ingesta total. En general, la ingesta estimada de fluoruros en niños y adolescentes no sobrepasa los 2 mg/día. Aunque las personas adultas pueden tener una ingesta diaria absoluta más alta de fluoruros en mg, la ingesta diaria de fluoruros por los niños, expresada en mg por kg de peso corporal, puede ser superior a la de los adultos. Se ha notificado que en ciertas zonas del mundo en las cuales la concentración de fluoruros en el ambiente circundante puede ser sumamente alta y/o donde el régimen alimenticio se basa en productos que los contienen en abundancia, se estimó una ingesta de fluoruros en adultos de hasta 27 mg/día, siendo la fuente principal el agua de bebida obtenida de fuentes freáticas situadas en zonas geológicas ricas en fluoruros.
Probablemente se produce exposición ocupacional a los fluoruros por inhalación o contacto cutáneo en las personas que trabajan con equipo de soldadura o en la elaboración del aluminio, el mineral de hierro o el de fosfato. En estudios relativamente recientes se notificaron concentraciones de fluoruros suspendidos en el aire del cuarto de crisoles de las fundiciones de aluminio del orden de 1 mg/m3.
En las personas y los animales de laboratorio, el paso de los fluoruros ingeridos a la circulación general se produce fundamentalmente en el estómago y el intestino y depende de la solubilidad acuosa relativa de la forma consumida. Los fluoruros solubles se absorben casi completamente del tracto gastrointestinal; sin embargo, el grado de absorción se puede ver reducido por la formación de complejos con el aluminio, el fósforo, el magnesio o el calcio. En función de la solubilidad y del tamaño de las partículas, la absorción de los fluoruros gaseosos y particulados del tracto respiratorio puede ser parcial o completa.
Los fluoruros se distribuyen rápidamente por la circulación sistémica al agua intracelular y extracelular de los tejidos; sin embargo, en las personas y los animales de laboratorio, los huesos y los dientes retienen alrededor del 99% de la carga corporal total de fluoruros. En los dientes y el tejido esquelético, los fluoruros se incorporan al retículo cristalino.
Los fluoruros atraviesan la placenta y pasan de la madre al feto. Se eliminan del organismo fundamentalmente en la orina. En los lactantes, se retiene alrededor del 80%-90% de la dosis de fluoruros; en los adultos, la cifra correspondiente es de alrededor del 60%. Estos valores pueden verse modificados por alteraciones del flujo urinario y el pH de la orina.
Hay fluoruros presentes en los órganos, los tejidos y los fluidos del cuerpo. Las concentraciones de fluoruros en la sangre entera de personas residentes en una comunidad de los Estados Unidos que recibía agua de bebida fluorada eran de 20 a 60 µg/l. La concentración media en el plasma de 127 personas con 5,03 mg de fluoruros/l en su agua de bebida era de 106 Å 76 (SD) µg/l. El suero y el plasma contienen prácticamente la misma cantidad de fluoruros. Las concentraciones más altas de fluoruros en los tejidos calcificados se suelen dar en el hueso, la dentina y el esmalte. La concentración de fluoruros en los huesos varía con la edad, el sexo y el tipo y parte específica de hueso y se considera que corresponde a una exposición prolongada de la persona a los fluoruros. La concentración de fluoruros en el esmalte dental disminuye exponencialmente con la distancia desde la superficie y varía en función del lugar, el desgaste superficial, la exposición sistémica y la exposición a los fluoruros de aplicación externa. La concentración de fluoruros en los tejidos blandos está relacionada con la de la sangre. Los niveles de fluoruros en la orina de los individuos sanos dependen de su ingesta. Se han registrado aumentos de la concentración de fluoruros en la orina en personas tras la exposición ocupacional a fluoruros suspendidos en el aire y entre los residentes en zonas asociadas con fluorosis endémica.
En varios estudios con ratas tratadas con fluoruros por vía oral durante periodos de tres a cinco semanas se observaron en el esqueleto efectos como una inhibición de la mineralización y la formación de los huesos, retraso en la curación de las fracturas y reducción del volumen de hueso y la síntesis de colágeno. En estudios de duración media con ratones tratados con fluoruros en el agua de bebida (> 4,5 mg/kg de peso corporal al día) durante un periodo de seis meses se observó una remodelación alterada de los huesos, megalocitosis hepática, nefrosis y/o degeneración de los túbulos seminíferos de los testículos.
En una biovaloración amplia de la carcinogenicidad con grupos de ratas F344/N y ratones B6C3F1 macho y hembra a los que se administró en el agua de bebida una concentración de hasta 79 mg de fluor uros/l en forma de fluoruro de sodio durante un periodo de dos años no se observó un aumento estadísticamente significativo de la incidencia de tumores en ninguno de los grupos expuestos. Se detectó una tendencia estadísticamente significativa al aumento de la incidencia de osteosarcomas en ratas macho con la exposición creciente al fluoruro. Sin embargo, la incidencia quedaba dentro del margen de los testigos históricos.
En otra biovaloración de la carcinogenicidad de dos años con ratas Sprague-Dawley expuestas a concentraciones de hasta 11,3 mg/kg de peso corporal al día con los alimentos tampoco se observó un aumento estadísticamente significativo de los osteosarcomas u otros tumores. Otro estudio, en el que se notificó un aumento de la incidencia de osteomas en ratones que recibieron hasta 11,3 mg/kg de peso corporal al día, es difícil de interpretar, porque los animales estaban infectados por un retrovirus de tipo C.
En general, los fluoruros no son mutagénicos en células procarióticas. Aunque se ha demostrado que en cultivos de células de linfoma de ratón y linfoblastoides humanas los fluoruros aumentan la frecuencia de mutaciones en loci específicos, estas mutaciones probablemente se deben más al daño cromosómico que a mutaciones puntuales. Se ha demostrado también que los fluoruros son clastogénicos en diversos tipos de células. El mecanismo de la clastogenicidad se ha atribuido al efecto de los fluoruros en la formación de proteínas que intervienen en la síntesis y/o reparación del ADN, más que a la interacción directa entre los fluoruros y el ADN. En la mayoría de los estudios en los cuales se administraron fluoruros por vía oral a roedores, no se observaron efectos en la morfología del esperma o en la frecuencia de aberraciones cromosómicas, micronúcleos, intercambio de cromátidas hermanas o rotura de la cadena del ADN. Sin embargo, se notificaron daños citogenéticos en la médula ósea o alteraciones en la morfología de las células espermáticas cuando se administraba la sustancia a roedores por inyección intraperitoneal.
En estudios recientes con animales de laboratorio a los que se administraba fluoruro en el agua de bebida no se observaron efectos reproductivos o en el desarrollo. Sin embargo, se han notificado cambios histopatológicos en los órganos reproductivos de conejos macho tratados (por vía oral) con 4,5 mg de fluoruros/kg de peso corporal al día durante 18-29 meses, de ratones macho tratados (por vía oral) con > 4,5 mg de fluoruros/kg de peso corporal al día durante 30 días y de conejos a los que se administraron por inyección subcutánea > 10 mg de fluoruros/kg de peso corporal al día durante 100 días. Se han notificado efectos adversos en la función reproductiva de ratones hembra tratados (por vía oral) con > 5,2 mg de fluoruros/kg de peso corporal al día durante los días 6-15 después del apareamiento y de conejos macho tratados (por vía oral) con > 9,1 mg de fluoruros/kg de peso corporal al día durante 30 días.
En las investigaciones epidemiológicas sobre los efectos de los fluoruros en la salud humana se han examinado trabajadores expuestos en el lugar de trabajo empleados fundamentalmente en la industria de la fundición del aluminio y poblaciones consumidoras de agua potable fluorada. En algunos estudios epidemiológicos analíticos de trabajadores expuestos en el lugar de trabajo a fluoruros, se ha detectado una mayor incidencia de cáncer de pulmón y de vejiga y un aumento de la mortalidad debida al cáncer de estos y otros órganos. En general, sin embargo, no se ha observado una pauta uniforme; en algunos de estos estudios epidemiológicos, el aumento de la morbilidad o la mortalidad debidas al cáncer se puede atribuir a la exposición de los trabajadores a sustancias distintas de los fluoruros.
Se ha examinado en un gran número de estudios epidemiológicos realizados en numerosos países la relación entre el consumo de agua potable fluorada y la morbilidad o la mortalidad por cáncer. No hay pruebas convincentes de una asociación entre el consumo de agua potable fluorada controlada y el aumento de la morbilidad o la mortalidad por cáncer.
Los fluoruros tienen efectos tanto beneficiosos como perjudiciales en el esmalte dental. La prevalencia de la caries dental es inversamente proporcional a la concentración de fluoruros en el agua potable. La prevalencia de la fluorosis crónica está muy asociada con la concentración de fluoruros, con una relación dosis-respuesta positiva.
Se siguen notificando casos de fluorosis esquelética asociada con el consumo de agua potable que contiene niveles elevados de fluoruros. Se considera que algunos factores, como el estado nutricional y el régimen alimenticio, el clima (en relación con el consumo de líquidos), la exposición simultánea a otras sustancias y la ingesta de fluoruros de fuentes distintas del agua de bebida, desempeñan una función importante en la aparición de esta enfermedad. La fluorosis esquelética puede aparecer en trabajadores ocupacionalmente expuestos a concentraciones elevadas de fluoruros suspendidos en el aire; sin embargo, sólo se ha encontrado información nueva limitada.
Algunas pruebas obtenidas en varios estudios ecológicos parecen indicar que podría haber una asociación entre el consumo de agua fluorada y las fracturas de cadera. Sin embargo, otros estudios, incluidas algunas investigaciones epidemiológicas, no respaldan esta conclusión. En algunos casos se ha notificado un efecto protector de los fluoruros en las fracturas.
Hay dos estudios que permiten evaluar el riesgo de fractura teniendo en cuenta una serie de ingestas de fluoruros. En un estudio, los riesgos relativos de todas las fracturas y de la fractura de cadera eran elevados en los grupos que utilizaban agua de bebida con > 1,45 mg de fluoruros/l (ingesta total > 6,5 mg/día); esta diferencia alcanzó significación estadística para el grupo expuesto al agua de bebida con > 4,32 mg de fluoruros/l (ingesta total, 14 mg/día). En el otro estudio se observó un aumento no dependiente de la dosis de la incidencia de fracturas en un grupo de edad de mujeres expuestas a fluoruros en el agua de bebida.
Según los estudios epidemiológicos, no hay pruebas de una asociación entre el consumo de agua de bebida fluorada por las madres y el aumento del riesgo de aborto espontáneo o malformación congénita. Otras investigaciones epidemiológicas de trabajadores expuestos en el lugar de trabajo no han proporcionado pruebas razonables de efectos genotóxicos o efectos sistémicos en los sistemas respiratorio, hematopoyético, hepático o renal que puedan ser directamente atribuibles a la exposición en sí a los fluoruros.
El fluoruro no afectó al crecimiento o a la capacidad de reducción de la demanda química de oxígeno de los lodos activados a concentraciones de 100 mg/l. La CE50 para la inhibición de la tasa de nitrificación bacteriana fue de 1218 mg de fluoruros/l. La CE50 a las 96 horas, basada en el crecimiento, para algas de agua dulce y marinas fue de 123 y 81 mg de fluoruros/l, respectivamente.
La CL50 a las 48 horas para invertebrados acuáticos fue del orden de 53 a 304 mg/l. Los invertebrados de agua dulce más sensibles fueron Musculium transversum, habiéndose observado una mortalidad estadísticamente significativa (50%) a una concentración de 2,8 mg de fluoruros/l en un experimento de flujo continuo de ocho semanas, y varias especies de insectos de agua dulce de la familia Hydropsychidae, con «concentraciones inocuas» (CE0,01 a las 8760 horas) del orden de 0,2 a 1,2 mg de fluoruros/l. La especie marina más sensible sometida a prueba fue la artemia (Artemia salina). En una prueba estática con renovación de 12 días, se produjo un trastorno del crecimiento estadísticamente significativo con 5,0 mg de fluoruros/l.
La CL50 a las 96 horas para los peces de agua dulce osciló entre 51 mg/l (trucha arco iris, Oncorhynchus mykiss) y 460 mg/l (espinoso, Gasterosteus aculeatus). Todas las pruebas de toxicidad aguda (96 horas) en peces marinos dieron resultados superiores a 100 mg/l. La toxicidad del fluoruro inorgánico para los peces de agua dulce parece tener una correlación negativa con la dureza del agua (carbonato de calcio) y positiva con la temperatura. Entre los síntomas de intoxicación aguda por fluoruros cabe mencionar el letargo, el movimiento violento e incierto y la muerte. En pruebas estáticas con renovación la CL50 a los 20 días para la trucha arco iris osciló entre 2,7 y 4,7 mg de fluoruros/l. Se han estimado «concentraciones inocuas» (CL0,01 en un número indeterminado de horas) para la trucha arco iris y el reo (Salmo trutta) de 5,1 y 7,5 mg de fluoruros/l, respectivamente. A concentraciones > 3,2 (efluente) o > 3,6 (fluoruro de sodio) mg de fluoruros/l, la eclosión de los huevos del pez catla (Catla catla) se retrasó 1-2 horas.
Los experimentos sobre comportamiento realizados en el salmón del Pacífico adulto (Oncorhynchus sp.) en ríos de aguas blandas indican que los cambios en la química del agua, debido a un aumento de la concentración de fluoruros hasta 0,5 mg/l, puede afectar negativamente a la migración; durante la migración los salmones son enormemente sensibles a los cambios de la química del agua de sus ríos de origen. En estudios de laboratorio, los fluoruros parecen ser tóxicos para los procesos microbianos a concentraciones presentes en suelos moderadamente contaminados con fluoruros; análogamente, la acumulación de materia orgánica sobre el terreno en las cercanías de fundiciones se ha atribuido a una inhibición grave de la actividad microbiana por esta sustancia.
Los signos de fitotoxicidad de los fluoruros inorgánicos (fluorosis) como la clorosis, la necrosis y la disminución de la tasa de crecimiento es más probable que se produzcan en los tejidos jóvenes en expansión de las plantas latifolias y las agujas en fase de alargamiento de las coníferas. La inducción de fluorosis se ha demostrado claramente en experimentos realizados en laboratorios, en invernaderos y en parcelas de terreno controladas. Se han publicado numerosos estudios sobre la toxicidad de los fluoruros para las plantas en relación con la fumigación de los invernaderos con fluoruro de hidrógeno. Se observó por primera vez necrosis foliar en vides (Vitis vinifera) expuestas a 0,17 y 0,27 µg/m3 a los 99 y los 83 días, respectivamente. El nivel más bajo con efectos observados para la necrosis foliar (65% de las hojas) en el gladiolo (Gladiolus grandiflorus) fue de 0,35 µg de fluoruros/m3. Los fluoruros suspendidos en el aire pueden afectar también al desarrollo de la enfermedad de la planta, aunque el tipo y la magnitud de los efectos dependen de la combinación específica planta-patógeno.
En varios estudios breves de cultivos en solución se ha determinado un umbral tóxico para la actividad del ión fluoruro que oscila entre alrededor de 50 y 2000 µmoles de fluoruro/l. La toxicidad es específica no sólo para las especies de plantas, sino también para las especies iónicas de fluoruro; algunos complejos de fluoruro de aluminio presentes en cultivos en solución pueden ser tóxicos con actividades de 22-357 µmoles de fluoruro/l, mientras que el fluoruro de hidrógeno es tóxico con actividades de 71-137 µmoles de fluoruro/l. Se ha realizado un pequeño número de estudios sobre la exposición a los fluoruros a través del suelo. El tipo de suelo puede afectar enormemente a la absorción y la posible toxicidad de los fluoruros.
En las aves, la DL50 en 24 horas fue de 50 mg/kg de peso corporal para las crías de un día de estornino (Sturnus vulgaris) y de 17 mg/kg de peso corporal para los pajaritos de 16 días. Las tasas de crecimiento registraron una reducción significativa con 13 y 17 mg de fluoruros/kg de peso corporal (las dosis más altas a las cuales se vigiló el crecimiento). Gran parte del trabajo inicial sobre mamíferos se realizó en ungulados domesticados. Se ha observado fluorosis en el ganado bovino y ovino. La concentración más baja con los alimentos con efecto en los ungulados salvajes se obtuvo en un estudio controlado en cautividad con el venado de cola blanca (Odocoileus virginianus), en el cual se observó un moteado general de los incisivos característico de la fluorosis dental con la dosis de 35 mg/kg de alimentos.
Se ha demostrado que las fundiciones de aluminio, las fábricas de ladrillos, las fábricas de fósforo y las instalaciones de fertilizantes y de fibra de vidrio son fuentes de fluoruros que están en correlación con el daño a las comunidades de plantas locales. En la vegetación en las proximidades de una fábrica de fósforo se observó que el grado del daño y las concentraciones de fluoruros en el humus del suelo eran inversamente proporcionales a la distancia de la fábrica. El promedio de las concentraciones de fluoruros en la vegetación oscilaba entre 281 mg/kg en las zonas gravemente dañadas y 44 mg/kg en las zonas menos dañadas; en una zona testigo, la concentración de fluoruro fue de 7 mg/kg. Las comunidades de plantas en las cercanías de una fundición de aluminio mostraban diferencias en la composición y estructura de la comunidad debido en parte a variaciones en la tolerancia a los fluoruros. Sin embargo, hay que señalar que uno de los principales problemas con la identificación de los efectos de los fluoruros en la naturaleza es la presencia de variables confundidoras, como por ejemplo otros contaminantes atmosféricos. Por consiguiente, se ha de actuar con prudencia a la hora de interpretar los numerosos estudios sobre el terreno relativos a la contaminación por fluoruros.
Las conclusiones iniciales de los efectos de los fluoruros en los mamíferos procedían de estudios sobre animales domésticos, como el ganado ovino y bovino. Los fluoruros se pueden recibir de la vegetación, el suelo y el agua de bebida. Se han determinado los niveles de tolerancia para los animales domesticados, siendo los niveles más bajos para las vacas lecheras de 30 mg/kg de pienso o 2,5 mg/l de agua de bebida. Se han producido incidentes que afectaban a animales domesticados tanto a causa de fuentes naturales de fluoruros, como las erupciones volcánicas y la geología subyacente, como por fuentes de origen humano, por ejemplo los suplementos minerales, las industrias emisoras de fluoruro y las centrales eléctricas. Entre los síntomas de la toxicidad por fluoruros cabe mencionar la emaciación, la rigidez de las articulaciones y anomalías en dientes y huesos. Otros efectos son una reducción de la producción de leche y efectos negativos en la capacidad reproductiva de los animales. La concentración más baja de fluoruros en los alimentos que provocó fluorosis en el ciervo salvaje fue de 35 mg/kg. Las investigaciones acerca de los efectos de los fluoruros en la flora y fauna silvestres se han concentrado en las repercusiones sobre la integridad estructural de los dientes y los huesos. En las proximidades de las fundiciones se han observado efectos inducidos por los fluoruros, por ejemplo cojera, afeamiento dental y daños en los dientes.
Los fluoruros tienen efectos tanto positivos como negativos para la salud humana, pero el margen entre las ingestas asociadas con estos efectos es reducido. Es importante la exposición a todas las fuentes de fluoruros, en particular el agua de bebida y los productos alimenticios.
Se dispone de poca información para caracterizar la relación dosis-respuesta relativa a los distintos efectos adversos. Son escasos, sobre todo, los datos acerca de la exposición total, en particular la ingesta y la absorción de fluoruros.
El efecto más grave es la acumulación esquelética de fluoruros debida a una exposición excesiva prolongada y sus efectos en las enfermedades óseas no neoplásicas, en particular la fluorosis esquelética y las fracturas óseas. Hay pruebas claras obtenidas en la India y en China de que una ingesta total de 14 mg de fluoruros/día provoca fluorosis esquelética y un aumento del riesgo de fracturas óseas y otras pruebas hacen pensar en un mayor riesgo de efectos óseos con una ingesta total superior a unos 6 mg de fluoruros/día.
En el entorno de agua dulce, las concentraciones de fluoruros naturales suelen ser inferiores a las supuestamente tóxicas para los organismos acuáticos. Sin embargo, estos organismos podrían verse afectados negativamente en las proximidades de descargas de origen humano. La toxicidad de los fluoruros depende de la dureza del agua.
Las especies de plantas sensibles que crecen cerca de fuentes de fluoruros de origen humano están expuestas a riesgos. La emisión de fluoruro de fuentes humanas está asociada con daños para las comunidades de plantas terrestres locales, pero con frecuencia es difícil atribuir estos efectos exclusivamente a los fluoruros, debido a la presencia de otros contaminantes atmosféricos. Los suelos suelen adsorber fuertemente los fluoruros. En consecuencia, la absorción por las plantas mediante esta vía es relativamente baja y la lixiviación de los fluoruros a través del suelo mínima.
Las concentraciones de fluoruros en la vegetación cercana a fuentes de emisión, como las fundiciones de aluminio, pueden ser superiores a la concentración más baja con efecto por vía alimentaria notificada para los mamíferos en experimentos de laboratorio. Se ha notificado fluorosis en animales domesticados. Todavía hay algunas zonas que notifican incidentes de fluorosis en el ganado bovino debido al consumo de suplementos minerales y agua de bebida con un contenido elevado de fluoruros. Además, existe un posible riesgo de ingestión de pastos y suelo contaminados por fluoruros debido al uso prolongado de fertilizantes fosfatados que contienen fluoruros como impureza. Se han notificado efectos inducidos por los fluoruros, como cojera y daños en los dientes, en mamíferos salvajes de las cercanías de fuentes humanas.
Todos los organismos están expuestos a fluoruros de fuentes naturales y/o humanas. Se han observado ingestas muy elevadas en zonas de todo el mundo donde abundan los fluoruros en la naturaleza y donde las personas utilizan agua freática con un contenido elevado de fluoruros como agua de bebida. Podría haber una mayor exposición en las cercanías de fuentes puntuales. Los fluoruros de los productos dentales son una fuente adicional para muchas personas.
Los fluoruros tienen efectos tanto beneficiosos como perjudiciales en la salud humana, con un margen de ingesta reducido entre ambos.
Se pueden observar efectos en los dientes y en el esqueleto con exposiciones inferiores a las asociadas con la aparición de otros efectos adversos para la salud específicos de órganos o tejidos.
Se considera que los efectos en los huesos (por ejemplo, la fluorosis esquelética y las fracturas) son los resultados más importantes en la evaluación de los efectos adversos de la exposición prolongada de las personas a los fluoruros.
La fluorosis esquelética es una discapacidad invalidante que afecta a millones de personas en diversas regiones de África, China y la India, con repercusiones importantes para la salud pública y socioeconómicas.
La ingestión de fluoruros con el agua y los productos alimenticios es el factor causal primordial de la fluorosis esquelética endémica. En algunas regiones, la combustión en recintos cerrados de carbón con un contenido elevado de fluoruros puede contribuir asimismo como una fuente importante.
Hay pocos datos para estimar la exposición total a los fluoruros y su biodisponibilidad, y se han detectado contradicciones en los informes sobre la caracterización de sus efectos adversos.
Hay pruebas convincentes obtenidas en la India y en China de que una ingesta total de 14 mg de fluoruros/día produce fluorosis esquelética y un mayor riesgo de fracturas óseas, y pruebas que parecen indicar un mayor riesgo de efectos óseos con ingestas totales superiores a unos 6 mg de fluoruros/día.
Una exposición excesiva a fluoruros biodisponibles representa un riesgo para la biota acuática y terrestre.
Se pueden utilizar especies sensibles a los fluoruros como indicadores a fin de identificar sus peligros para el medio ambiente.
Es necesario mejorar los conocimientos sobre la acumulación de fluoruros en los organismos y sobre su vigilancia y control.
Se deben caracterizar mejor los efectos biológicos asociados con la exposición a los fluoruros.
1 Gastrointestinal effects linked to longer-term exposure to fluoride have been reported in some older studies of occupationally exposed workers (effects included gastritis, duodenal ulcers, abdominal distention, nausea) (see US DHHS, 1991; ATSDR, 1993) and observed in some individuals residing in areas of endemic skeletal fluorosis (effects included dyspepsia, abnormal surface morphology of gastrointestinal mucosa ) (Gupta et al., 1992; Dasarathy et al., 1996) or in some patients receiving fluoride for the treatment of osteoporosis (effects included pain, nausea, vomiting, diarrhoea, histological changes in gastrointestinal mucosa) (Laroche & Mazières, 1991; Das et al., 1994).
2 This section was based primarily on a review prepared by WHO (1994).
3 The effects of fluoride on dental health were reviewed in depth by WHO (1994), and therefore only a brief summary is presented here.
See Also:
Toxicological Abbreviations
Fluorides (IARC Summary & Evaluation, Supplement7, 1987)