
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 205
POLYBROMINATED DIBENZO-p-DIOXINS AND DIBENZOFURANS
This report contains the collective views of an international group
of experts and does not necessarily represent the decisions or the
stated policy of the United Nations Environment Programme, the
International Labour Organisation, or the World Health
Organization.
First draft prepared by Dr C. Melber and Dr J. Kielhorn, Fraunhofer
Institute of Toxicology and Aerosol Research, Hanover, Germany
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 1998
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO), and the World Health Organization (WHO). The overall objectives
of the IPCS are to establish the scientific basis for assessment of
the risk to human health and the environment from exposure to
chemicals, through international peer review processes, as a
prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization, the United Nations
Institute for Training and Research, and the Organisation for Economic
Co-operation and Development (Participating Organizations), following
recommendations made by the 1992 UN Conference on Environment and
Development to strengthen cooperation and increase coordination in the
field of chemical safety. The purpose of the IOMC is to promote
coordination of the policies and activities pursued by the
Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing in Publication Data
Polybrominated dibenzo- p-dioxins and dibenzofurans.
(Environmental health criteria ; 205)
1.Dioxins 2.Benzofurans
3.Environmental exposure 4.Occupational exposure
I.International Programme on Chemical Safety II.Series
ISBN 92 4 157205 1 (NLM Classification: QD 405)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1998
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city, or area
or of its authorities, or concerning the delimitation of its frontiers
or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR POLYBROMINATED DIBENZO- p-DIOXINS
AND DIBENZOFURANS
PREAMBLE
ABBREVIATIONS
1. SUMMARY
1.1. Identity, physical and chemical properties,
and analytical methods
1.2. Formation and sources of human and
environmental exposure
1.3. Environmental transport, distribution,
and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism
1.6. Effects on laboratory mammals and in vitro
test systems
1.7. Effects on humans
1.8. Effects on other organisms in the laboratory
and field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.2.1. Appearance, melting and boiling
points, water solubility, vapour pressure,
octanol/water partition coefficient,
and sorption coefficient
2.2.2. Stability of PBDDs/PBDFs
2.2.2.1 Photolysis
2.2.2.2 Thermolytic degradation
of PBDDs/PBDFs
2.2.3. Chemical reactions
2.3. Conversion factors
2.4. Analytical methods
2.4.1. General aspects
2.4.2. Sampling and extraction
2.4.2.1 Ambient air, airborne dust,
automobile exhaust, flue gas,
and products of thermolysis
2.4.2.2 Water and aqueous samples
2.4.2.3 Environmental samples: soil,
sediment, and sewage sludge
2.4.2.4 Flame retardants, polymers,
fly ash samples, dust, soot,
and fire residues
2.4.2.5 Biological matrices: human
milk, blood/plasma, tissues,
and fish samples
2.4.3. Sample clean-up
2.4.4. Separation
2.4.5. Detection, quantification, and confirmation
of PBDDs/PBDFs by MS techniques
2.4.6. The need for analysis of
2,3,7,8-substituted congeners
2.4.7. Interfering substances
2.4.8. Standards
3. FORMATION AND SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Synthesis and use
3.2. By-products of brominated organic chemicals
(including flame retardants)
3.3. Formation from the photochemical degradation
of brominated organic chemicals
3.4. Formation from the laboratory thermolysis of
bromine-containing flame retardants
3.5. Formation during production of plastic materials
and presence in consumer products containing
flame retardants
3.5.1. Formation during production processes
3.5.2. Presence in resins and polymer products
3.6. Emissions from flame-retarded consumer products
3.7. Presence in fire residues, smoke condensates,
and gases after fires
3.7.1. Experimental fires
3.7.2. Accidental fires
3.8. Formation from incineration of fuels
3.9. Formation during waste disposal and treatment
3.9.1. Incineration
3.9.2. Disposal
3.9.3. Recycling
3.9.3.1 Plastics
3.9.3.2 Metals
3.10. Presence in automotive exhaust
3.11. Formation during textile processing
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Air
4.1.2. Water and sediments
4.1.3. Soil
4.1.4. Biota
4.2. Environmental transformation
4.2.1. Photochemical degradation
4.2.2. Microbial degradation
4.3. Bioaccumulation and biomagnification
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.1.1 Ambient air
5.1.1.2 Indoor air
5.1.2. Water and sediment
5.1.3. Soil, sewage sludge, and biocompost
5.1.4. Food and feed
5.1.5. Other products
5.1.6. Terrestrial and aquatic organisms
5.1.6.1 Plants
5.1.6.2 Animals
5.2. General population exposure
5.2.1. Exposure data
5.2.2. Monitoring of human tissues and fluids
5.3. Occupational exposure
5.3.1. Workplace monitoring data
5.3.1.1 Flame retardant/polymer industry
5.3.1.2 Offices/studios
5.3.1.3 Recycling plants
5.3.1.4 Other workplaces
5.3.2. Monitoring of human tissues and fluids
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Dibenzo- p-dioxins
6.1.2. Dibenzofurans
6.2. Distribution
6.2.1. Levels in organs and blood
6.2.1.1 Dibenzo- p-dioxins
6.2.1.2 Dibenzofurans
6.2.2. Transfer to offspring
6.3. Metabolic transformation
6.3.1. Dibenzo- p-dioxins
6.3.2. Dibenzofurans
6.4. Elimination and excretion
6.4.1. Dibenzo- p-dioxins
6.4.2. Dibenzofurans
6.5. Retention and turnover
6.5.1. Animal studies
6.5.2. Human studies
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Dibenzo- p-dioxins
7.1.2. Dibenzofurans
7.1.3. Remarks on the lethality of PBDDs/PBDFs
7.2. Short-term exposure
7.2.1. Dibenzo- p-dioxins
7.2.2. Dibenzofurans
7.3. Long-term exposure
7.4. Skin and eye irritation, sensitization, dermal
lesions, and acne
7.5. Reproductive and developmental toxicity
7.5.1. Reproductive toxicity
7.5.2. Developmental toxicity
7.6. Mutagenicity and related end-points
7.7. Carcinogenicity
7.7.1. Short-term studies
7.7.2. Long-term studies
7.8. Other special studies
7.8.1. Immunotoxicity
7.8.1.1 Dibenzo- p-dioxins
7.8.1.2 Dibenzofurans
7.8.2. Effects on intermediary metabolism:
Porphyrin effects
7.8.3. Effects on vitamin A storage
7.8.4. Endocrine interactions
7.8.5. Interaction with drugs and toxicants
7.8.6. Induction of microsomal enzymes
7.8.6.1 Dibenzo- p-dioxins
7.8.6.2 Dibenzofurans
7.8.6.3 Combustion products
7.9. Mechanisms of toxicity -- mode of action
7.10. Experimental data on selected PBDDs/PBDFs
and their relevance to the toxicity equivalency
factor (TEF) concept
8. EFFECTS ON HUMANS
8.1. General population exposure
8.2. Occupational/accidental exposure
8.3. Subpopulations at special risk
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE
ENVIRONMENT
10.1. Hazard evaluation
10.2. Exposure evaluation
10.3. Risk evaluation
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
AND THE ENVIRONMENT
11.1. Conclusions
11.2. Recommendations
12. FURTHER RESEARCH
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
APPENDICES
RÉSUMÉ
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41
22 - 9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
* * *
This publication was made possible by grant number 5 U01
ES02617-15 from the National Institute of Environmental Health
Sciences, National Institutes of Health, USA, and by financial support
from the European Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure
to environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects
of pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As such,
they include and review studies that are of direct relevance for the
evaluation. However, they do not describe every study carried out.
Worldwide data are used and are quoted from original studies, not from
abstracts or reviews. Both published and unpublished reports are
considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant published
data are absent or when they are pivotal to the risk assessment. A
detailed policy statement is available that describes the procedures
used for unpublished proprietary data so that this information can be
used in the evaluation without compromising its confidential nature
(WHO (1990) Revised Guidelines for the Preparation of Environmental
Health Criteria Monographs. PCS/90.69, Geneva, World Health
Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and in vitro
studies provide support and are used mainly to supply evidence
missing from human studies. It is mandatory that research on human
subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
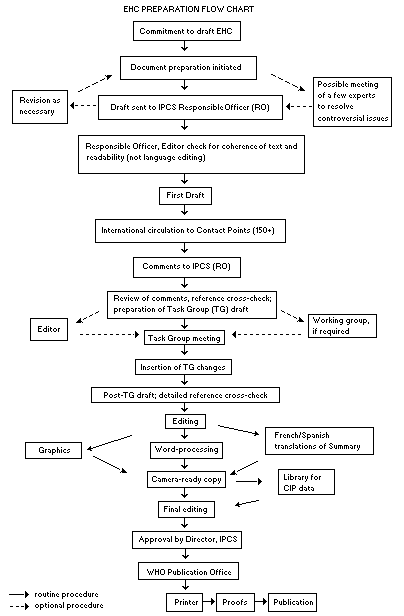
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for layout
and language. The first draft, prepared by consultants or, more
usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR POLYBROMINATED
DIBENZO- p- DIOXINS AND DIBENZOFURANS
Members
Dr A.P.J.M. de Jong, Laboratory of Organic Analytical Chemistry,
National Institute of Public Health and Environment, Bilthoven, The
Netherlands
Ms J. Diliberto, National Health and Environmental Effects
Research Laboratory, Experimental Toxicology Division, US
Environmental Protection Agency, Research Triangle Park, North
Carolina, USA
Dr M. Feeley, Toxicology Evaluation Section, Bureau of Chemical
Safety, Health Canada, Tunney's Pasture, Ottawa, Ontario, Canada
(Rapporteur)
Dr H. Fiedler, Bayerisches Institut für Abfallforschung BIFA
GmbH, Augsburg, Germany
Professor B. Jansson, Institute of Applied Environmental Research,
Stockholm University, Stockholm, Sweden (Chairman)
Dr Y. Kurokawa, Biological Safety Research Center, National
Institute of Health Sciences, Tokyo, Japan
Dr C. Melber, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Professor D. Neubert, Institute for Clinical Pharmacology and
Toxicology, Berlin, Germany
Professor C. Rappe, Institute of Environmental Chemistry,
University of Umea, Umea, Sweden
Observer
Dr B. Schatowitz, Environmental and Trace Analysis Consumer
Care Division, Ciba-Geigy AG, Basel, Switzerland (Representing the
European Centre for Ecotoxicology and Toxicology of Chemicals)
Secretariat
Dr H. Galal-Gorchev, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
Dr J. Kielhorn, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr I. Mangelsdorf, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr F.X.R. van Leeuwen, Chemical Safety, European Centre for
Environment and Health, Bilthoven Division, De Bilt, The Netherlands
ENVIRONMENTAL HEALTH CRITERIA FOR POLYBROMINATED DIBENZO- p-DIOXINS
AND DIBENZOFURANS
A WHO Task Group on Environmental Health Criteria for
Polybrominated Dibenzo- p-dioxins and Dibenzofurans met at the
Fraunhofer Institute for Toxicology and Aerosol Research, Hanover,
Germany from 11 to 15 November 1996. Professor U. Heinrich opened the
meeting and welcomed the participants on behalf of the host institute.
Dr H. Galal-Gorchev, IPCS, welcomed the participants on behalf of the
Director, IPCS, and the three IPCS cooperating organizations
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft criteria
monograph and made an evaluation of the risks for human health and the
environment from exposure to polybrominated dibenzo- p-dioxins and
dibenzofurans.
Dr J. Kielhorn and Dr C. Melber, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany, prepared the first
draft of this monograph. They also prepared the second draft,
incorporating comments received following the circulation of the first
draft to the IPCS Contact Points for Environmental Health Criteria
monographs.
Dr H. Galal-Gorchev, IPCS Central Unit, was responsible for the
overall scientific content and Ms M. Sheffer, Scientific Editor,
Ottawa, Canada, for the linguistic editing.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
ABS acrylonitrile-butadiene-styrene
Ah aromatic hydrocarbon
AHH arylhydrocarbon hydroxylase
BB bromobiphenyl
BDE bromodiphenyl ether
CAS Chemical Abstracts Service
CYP cytochrome P-450
DBB/decaBB decabromobiphenyl
DBDE/decaBDE decabromodiphenyl ether
DD/DF dibenzo- p-dioxin/dibenzofuran
DiBDD dibromodibenzo- p-dioxin
DiBDF dibromodibenzofuran
DiXDF mixed dihalogenated dibenzofuran
EC50 median effective concentration
ED50 median effective dose
EI-SIM-MS electron impact-selective ion monitoring-mass
spectrometry
EPA Environmental Protection Agency (USA)
EROD ethoxyresorufin- O-deethylase
GC gas chromatography
HexaBB hexabromobiphenyl
HIPS high-impact polystyrene
HpBDD/heptaBDD heptabromodibenzo- p-dioxin
HpBDF/heptaBDF heptabromodibenzofuran
HPLC high-performance liquid chromatography
HRGC high-resolution gas chromatography
HRMS high-resolution mass spectrometry
HxBDD/hexaBDD hexabromodibenzo- p-dioxin
HxBDF/hexaBDF hexabromodibenzofuran
HxCDD/hexaCDD hexachlorodibenzo- p-dioxin
I-TEF international toxicity equivalency factor
I-TEQ international toxic equivalent
LD50 median lethal dose
LOAEL lowest-observed-adverse-effect level
LOEL lowest-observed-effect level
MI-IR matrix isolation infrared spectrometry
MoBDD/monoBDD monobromodibenzo- p-dioxin
MoBDF/monoBDF monobromodibenzofuran
MS mass spectrometry
n sample size
NCI negative ion chemical ionization
n.d. not detected
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
n.sp. not specified
OBDE/octaBDE octabromodiphenyl ether
OcBDD/octaBDD octabromodibenzo- p-dioxin
OcBDF/octaBDF octabromodibenzofuran
OCDD/OcCDD/octaCDD octachlorodibenzo- p-dioxin
PAH polycyclic aromatic hydrocarbon
PBB polybrominated biphenyl
PBDD polybrominated dibenzo- p-dioxin
PBDE polybrominated diphenyl ether
PBDF polybrominated dibenzofuran
PBT polybutylene terephthalate
PCB polychlorinated biphenyl
PCDD polychlorinated dibenzo- p-dioxin
PCDE polychlorinated diphenyl ether
PCDF polychlorinated dibenzofuran
PeBDD/pentaBDD pentabromodibenzo- p-dioxin
PeBDE/pentaBDE pentabromodiphenyl ether
PeBDF/pentaBDF pentabromodibenzofuran
PeCDF/pentaCDF pentachlorodibenzofuran
PeHDD pentahalogenated dibenzo- p-dioxin
PeHDF pentahalogenated dibenzofuran
PHDD polyhalogenated dibenzo- p-dioxin (used as
collective term including PCDD, PBDD, PXDD)
PHDF polyhalogenated dibenzofuran (used as collective
term including PCDF, PBDF, PXDF)
PVC polyvinyl chloride
PXDD mixed (brominated/chlorinated) halogenated
dibenzo- p-dioxin
PXDF mixed (brominated/chlorinated) halogenated
dibenzofuran
RI retention index
RIA radioimmunoassay
RMM relative molecular mass
SD standard deviation
T3 triiodothyronine
T4 thyroxin
TBBPA tetrabromobisphenol A
TBCDD 2,3-dibromo-7,8-dichlorodibenzo- p- dioxin
TBDD/2,3,7,8-TeBDD 2,3,7,8-tetrabromodibenzo- p-dioxin
TBDF/2,3,7,8-TeBDF 2,3,7,8-tetrabromodibenzofuran
TBPI bis-tetrabromo-phthalimide ethylene
TCDD/2,3,7,8-TeCDD 2,3,7,8-tetrachlorodibenzo- p-dioxin
TCDF/2,3,7,8-TeCDF 2,3,7,8-tetrachlorodibenzofuran
TeBDD/tetraBDD tetrabromodibenzo- p-dioxin
TeBDF/tetraBDF tetrabromodibenzofuran
TEF toxicity equivalency factor
TeHDD tetrahalogenated dibenzo- p-dioxin
TEQ toxic equivalent
TeXDD/tetraXDD mixed tetrahalogenated dibenzo- p- dioxin
THDF 2,3,7,8-tetrahalogenated dibenzofuran
TrBDD/triBDD tribromodibenzo- p-dioxin
TrBDF/triBDF tribromodibenzofuran
TrHDD/triHDD trihalogenated dibenzo- p-dioxin
TV television
TxDD 2,3,7,8-substituted mixed tetrahalogenated
dibenzo- p-dioxin
UV ultraviolet
WHO World Health Organization
1. SUMMARY
1.1 Identity, physical and chemical properties, and analytical
methods
Polybrominated dibenzo- p-dioxins (PBDDs) and polybrominated
dibenzofurans (PBDFs) are almost planar tricyclic aromatic compounds.
Theoretically, 75 PBDDs and 135 PBDFs are possible. In addition, a
large number of mixed halogenated congeners -- 1550
brominated/chlorinated dibenzo- p-dioxins (PXDDs) and 3050
brominated/chlorinated dibenzofurans (PXDFs) -- are theoretically
possible. Because of the complexity of the analytical procedures and
paucity of analytical reference standards, it has been possible to
characterize and determine only a small number of these compounds. The
most toxic congeners are those substituted at positions 2, 3, 7, and
8. There are 7 2,3,7,8-substituted PBDDs and 10 2,3,7,8-substituted
PBDFs, as well as 337 possible 2,3,7,8-substituted PXDDs and 647
possible 2,3,7,8-substituted PXDFs.
PBDDs/PBDFs have higher molecular weights than their chlorinated
analogues, high melting points, low vapour pressures, and low water
solubilities. They are generally soluble in fats, oils, and organic
solvents. There are very few experimental data on the physical and
chemical properties of PBDDs/PBDFs.
Photolysis occurs at a more rapid rate for PBDDs/PBDFs than for
polychlorinated dibenzo- p-dioxins (PCDDs) and polychlorinated
dibenzofurans (PCDFs). PBDDs/PBDFs are thermostable. The temperatures
of formation and destruction of PBDDs/PBDFs depend on several
conditions, including the presence or absence of oxygen, polymers, and
flame retardant additives, such as antimony trioxide (Sb2O3).
In the presence of excess chlorine, bromine is substituted by
chlorine to give PXDDs/PXDFs.
Because of the toxic nature of these compounds and their
photolytic properties, care must be taken during sampling and
analysis. Highly sensitive, selective, and specific analytical methods
(gas chromatography/mass spectrometry, or GC/MS) are required because
of the large number of PBDD/PBDF congeners. Sampling procedures are
identical for all polyhalogenated dibenzo- p-dioxins (PHDDs) and
polyhalogenated dibenzofurans (PHDFs), but separation and
determination of PBDDs/PBDFs (and PXDDs/PXDFs) differ slightly from
those of their chlorinated analogues. PBDDs/PBDFs have higher
molecular weights and longer GC retention times than the chlorinated
analogues, as well as different MS isotopic cluster patterns and
interference compounds. Exact identification of specific brominated
congeners is very limited owing to the small number of reference
standards currently available. For the same reason, determination of
mixed halogenated congeners is almost impossible.
1.2 Formation and sources of human and environmental exposure
PBDDs/PBDFs are not known to occur naturally. They are not
intentionally produced (except for scientific purposes) but are
generated as undesired by-products in various processes. They can be
formed by chemical, photochemical, or thermal reactions from
precursors and by so-called de novo synthesis.
PBDDs/PBDFs have been found as contaminants in brominated organic
chemicals (e.g. bromophenols) and, in particular, in flame retardants,
such as polybrominated diphenyl ethers (PBDEs), decabromobiphenyl
(decaBB or DBB), 1,2-bis(tribromophenoxy)ethane, tetrabromobisphenol A
(TBBPA), and others. They have been detected in distillation residues
of some bromophenols and bromoanilines and in wastes from chemical
laboratories.
PBDFs and, to a lesser extent, PBDDs have been detected as
photochemical degradation products of brominated organic chemicals,
such as PBDEs and bromophenols.
Laboratory thermolysis experiments showed the formation of
PBDDs/PBDFs from bromophenols, PBDEs, polybrominated biphenyls (PBBs),
and other brominated flame retardants (pure or in a polymer matrix).
There was a broad range of yields, from zero to maximum values
(reached from PBDEs) in the g/kg range. Generally, PBDFs were much
more abundant than PBDDs. The optimum PBDF formation temperature of a
series of pure flame retardants was in the range of 600-900°C. The
presence of polymers or synergists (e.g. Sb2O3) resulted in a
decrease in the optimum formation temperature (down to 400°C). In
addition to temperature and the presence of polymer matrix or
synergists, several other factors, such as metals, metal oxides,
water, oxygen, and the type of combustion apparatus used, influenced
the yield and pattern of PBDDs/PBDFs. In ternary mixtures of PBDE,
polymer matrix, and Sb2O3, tetrabromodibenzo furans (tetraBDFs or
TeBDFs) were frequently the most abundant homologue group.
2,3,7,8-Substituted PBDDs/PBDFs (tetra to hepta) were found at varying
concentrations; for example, 2,3,7,8-TeBDF was found at up to 2000
mg/kg in pyrolysates of polymers containing octabromodiphenyl ether
(octaBDE or OBDE).
In the manufacture of plastics, elevated temperatures (150-300°C)
occur during several processes. Studies of the exhaust streams from
machines processing polymers -- such as
acrylonitrile-butadiene-styrene (ABS) and polybutylene terephthalate
(PBT) -- containing different types of brominated flame retardants
showed that PBDDs/PBDFs (di to octa) can be formed at these
temperatures. OBDE and decabromodiphenyl ether (decaBDE or DBDE)
produced the highest amounts of PBDDs/PBDFs, the major portion
consisting of PBDFs. Levels observed with TBBPA or
bis-tetrabromo-phthalimide ethylene (TBPI) were several orders of
magnitude lower. No PBDDs/PBDFs were detected during processing of ABS
flame-retarded by brominated styrene or
1,2-bis(tribromophenoxy)ethane. 2,3,7,8-Substituted congeners were not
determined (processing of DBDE), were detected at trace levels
(processing of OBDE), or were not detected (processing of TBBPA and
TBPI).
Various plastic materials at several processing stages were
analysed for PBDDs/PBDFs. These included (granulated) resins and
moulded parts whose flame retardant additives were known as well as
samples from commercial electrical appliances (television sets,
printers, computers) whose flame retardant additives were unknown. The
highest levels of PBDDs/PBDFs were found in materials flame-retarded
with PBDEs and were in the range of several thousand µg/kg, thus
exceeding the levels of other flame retardant/polymer systems by
orders of magnitude. Factors influencing the extent of formation are
temperature and the duration of such processes as blending, extrusion,
and moulding. Again PBDFs dominated, with some exceptions, over PBDDs,
with the highly brominated (>tetra) derivatives being prevalent. Peak
concentrations were seen with pentabromodibenzofurans (pentaBDFs or
PeBDFs) and hexabromodibenzofurans (hexaBDFs or HxBDFs). The latter
reached levels as high as 3000 µg/kg in casing parts. Printed circuit
boards contained tetra- and pentaBDFs at maximum concentrations of
1300 and 1400 µg/kg, respectively. Total PBDF (mono to hexa)
concentrations were in the range of 3.6-3430 µg/kg.
2,3,7,8-Substituted PBDDs/PBDFs were not determined, were not
detectable, or were present at relatively low concentrations. Maximum
concentrations of 2,3,7,8-substituted PBDFs (tetra to hexa) in casings
or printed circuit boards ranged from 11 µg/kg (tetra) to 203 µg/kg
(hexa).
Experiments to determine whether PBDFs were released from
television sets or similar appliances during use showed air levels
ranging from not detected to 1800 pg total PBDFs (tetra to hexa) per
appliance.
Burning of products containing brominated compounds caused
emission of PBDDs/PBDFs. In experimental fire tests simulating real
fire conditions with electrical appliances such as television sets,
printers, computer terminals, and their casings, high PBDF (mono to
hexa) concentrations were detected in the combustion residues
(thousands of mg/kg), in smoke condensate (hundreds of µg/m2), and in
smoke (up to 1700 µg/m3). PBDD concentrations amounted to about 3% of
the detected levels of PBDDs/PBDFs. The 2,3,7,8-substituted isomer was
mostly below 3% of the total tetraBDFs. 2,3,7,8-Substituted penta- and
hexaBDFs yielded between 1 and 16% of the corresponding totals.
Burning of test vehicles resulted in PBDF (mono to octa)
concentrations of up to 4.3 µg/kg in the fire residues.
During real fire accidents in private residences (television sets
involved), offices (computers involved), and other buildings,
concentrations measured were in most cases below the values found in
the model experiments described above, but the qualitative composition
of the samples was similar. PBDFs were found in almost all samples,
but PBDDs were not always detected; if present, their concentrations
were low. The PBDF concentrations in combustion residues were mainly
in the µg/kg range (low to high), but single maximum values (sum of
mono to hexa) of up to 107 mg/kg were also observed. The PBDF (mono to
hexa) area contaminant concentrations in close vicinity to the fire
site ranged between 0.1 and 13 µg/m2 in most cases. Additionally,
relevant concentrations of PXDDs/PXDFs could be detected. The
proportion of 2,3,7,8-substituted PBDDs/PBDFs was relatively low in
most of the samples examined. For example, maximum proportions of 3,
10, or 18% of the corresponding totals of tetra-, penta-, or hexaBDFs,
respectively, were reported from fire accidents with television sets.
Soot samples collected after a fire in a computer room contained
2,3,7,8-substituted tetra- and pentabromodibenzo- p-dioxins
(tetra/pentaBDDs or TeBDD/PeBDD) and tetra- and pentaBDFs, with a
maximum concentration of 48 µg/kg for 2,3,7,8-TeBDF (TBDF).
PXDDs were detected in ash from a wood-fired boiler. However, the
sort of wood (treated or untreated) was not specified. No data were
available on the incineration of other fuels, such as coal, peat, or
fuel oil.
The presence of PBDDs/PBDFs and/or PXDDs/PXDFs has been reported
in fly ash and/or flue gas of municipal, hospital, or hazardous waste
incinerators. The majority of these compounds are probably produced in
the incinerator itself, by formation from precursors at high
temperatures in the flame or by de novo synthesis at low
temperatures in the post-combustion zone of the incinerator. The
formation of PXDDs/PXDFs is explained by the extensive
bromine-chlorine exchange reactions (with chlorine donors in waste)
observed under several test conditions. The quantities of PBDDs/PBDFs
and PXDDs/ PXDFs measured in fly ash of incinerators were in the range
of ng/kg to µg/kg. In most cases, the concentrations of
dibenzo- p-dioxins exceeded those of dibenzofurans, and PXDDs/PXDFs
were more abundant than PBDDs/PBDFs. Of 2,3,7,8-substituted congeners,
a mixed tetrahalogenated dibenzo- p-dioxin (tetraXDD or TeXDD)
(Br2Cl2DD) was found.
Analyses of waste samples from some disposal sites showed the
presence of PBDDs/PBDFs and PXDDs/PXDFs at concentrations of several
hundred to several thousand ng/kg dry weight. The concentration of
dibenzo- p-dioxins (up to 580 ng/kg) was below that of dibenzofurans
(up to 4230 ng/kg). Generally, the homologue profile was dominated by
the lower halogenated (mono to tetra) derivatives. Chemical laboratory
waste contained PBDDs/PBDFs, with a peak concentration of 15 500 ng/kg
for hexaBDFs.
PBDDs/PBDFs were present in plastic materials (with or without
metals) of several recycling stages. The samples originated mainly
from office machines, printed circuit boards, and other electronic
scrap. In some cases, the sum concentration of eight selected PBDD/
PBDF congeners having the 2,3,7,8-substitution was as high as 65
µg/kg. Metal reclamation was also found to be a source of PBDDs and/or
PXDDs/PXDFs. PBDDs/PBDFs have also been detected in textile industries
where brominated flame retardants have been used. PBDFs were found in
the exhaust air, in the textiles before and after processing, and in
the chimney depositions.
PBDDs/PBDFs and PXDDs/PXDFs (along with PCDDs/PCDFs) have been
detected in emissions of motors using leaded petrol, in emissions of
motors using unleaded petrol with and without catalytic converters,
and in emissions of diesel engines. Because of the brominated and
chlorinated scavengers (dibromo- and dichloroethane) used in leaded
petrol, the highest levels of PHDDs/PHDFs (several thousand ng/m3)
were found with this type of petrol. Unleaded petrol produced much
lower emissions of PHDDs/PHDFs (approximately two orders of magnitude
lower). A further reduction was seen after catalytic gas cleaning. The
values for diesel engines were somewhat higher than those found with
the Otto motors (spark ignition engines) run on unleaded petrol. In
exhaust gases from combustion of leaded petrol, PBDDs/PBDFs were more
abundant than PXDDs/PXDFs and PCDDs/PCDFs. Generally, the
concentrations of dibenzofurans exceeded those of dibenzo- p-dioxins,
and there was a dominance of lower substituted homologues (mono to
tri). Similar patterns were seen in residues adhering to mufflers.
1.3 Environmental transport, distribution, and transformation
There are very few data available on the environmental transport
and distribution of PBDDs/PBDFs. Generally, their physicochemical
properties suggest similarities to PCDDs/PCDFs. Therefore, if released
to the environment, they may be preferably distributed into carbon- or
fat-rich compartments, as with PCDDs/PCDFs.
Airborne PBDDs/PBDFs were found to be transported in both the
particulate and vapour phase, the partitioning ratio depending on the
degree of bromination.
No experimental data are available on the movement of PBDDs/
PBDFs in water or soil. For PBDFs (tri to penta), adsorption to
sediment was reported. Owing to the low water solubility of PBDDs/
PBDFs, leaching through the soil may be limited but may be increased
in the presence of organic solvents or humic acids.
There are no experimental data on processes for the transport and
distribution of PBDDs/PBDFs between environmental media and biota or
within biota. Based on the similar high octanol/water partition
coefficients calculated for selected PCDDs/PCDFs, PBDDs/PBDFs, and
PXDDs/PXDFs, a bioavailability comparable to that of PCDDs/ PCDFs is
expected.
Photolysis of PBDDs/PBDFs and PXDDs/PXDFs was studied in organic
solvents and on quartz surfaces in the laboratory, as well as in soil
and on soot (and dust) particles under outdoor conditions. The slowest
photolytic reactions were observed under the latter, more
environmentally relevant, conditions. Reductive debromination was
found to be a major photochemical pathway. The rate of decomposition
of different congeners depended on their bromine substitution pattern.
Generally, higher brominated congeners and those with lateral bromines
had shorter half-lives. Calculated half-lives were in the order of
minutes (use of direct sunlight or ultraviolet [UV] light and quartz
vials), hours (use of solid films or soot or dust particles and
sunlight), or hundreds to thousands of hours (use of soil and
sunlight). For example, the estimated sunlight-induced half-lives for
2,3,7,8-TeBDD (TBDD) were 0.8 min (in organic solution) or 32 h
(dispersed as solid films). A half-life of 3-6 months was estimated
for tetraBDD isomers in surface soil. Compared with PCDDs/PCDFs, the
brominated counterparts were photochemically less stable. PXDDs/PXDFs
preferentially lost their bromine atoms during photolysis and
therefore were transformed into PCDDs/PCDFs, which had longer
photolytic half-lives. Such a transformation of PXDDs/PXDFs to
PCDDs/PCDFs also occurs during incineration processes.
PBDDs/PBDFs seem to be poorly degradable by microorganisms.
The presence of PBDDs/PBDFs in animals and in humans, as seen in
a few studies, is indicative of their accumulation potential.
2,3,7,8-TeBDD accumulated in rats during subchronic administration.
Bioaccumulation, bioconcentration, or biomagnification factors for
PBDDs/PBDFs or PXDDs/PXDFs are not available.
1.4 Environmental levels and human exposure
To date, in contrast to PCDDs/PCDFs, PBDDs/PBDFs have not been
frequently included in monitoring programmes. The few studies
performed indicate a limited occurrence.
In ambient air, PBDFs were found more frequently than PBDDs. Only
lower brominated PBDDs (mono to tetra) were detected at concentrations
ranging from not detected (n.d.) to about 0.85 pg/m3 for
monobromodibenzo- p-dioxins (monoBDDs or MoBDDs) in a motor way
tunnel and an underground garage. Of PBDFs, mono- to hexabrominated
homologues were found, their concentrations ranging from n.d. to 74
pg/m3. The concentrations (mean values) of total PBDDs/ PBDFs (tri to
hexa) measured, for example, in Germany in a motorway tunnel, in a
city, and in a suburban area amounted to 23 pg/m3, 2 pg/m3, and 0.59
pg/m3, respectively; 2,3,7,8-TeBDD was not detected, and the maximum
concentrations of 2,3,7,8-TeBDF and 1,2,3,7,8-PeBDF were 0.28 pg/m3
and 0.08 pg/m3, respectively. PXDFs were identified in
traffic-related air samples at concentrations up to 41 pg/m3
(Cl1Br1DFs). Outdoor dust samples (mainly from motorways) also
showed a predominance of PBDFs/PXDFs (maxima of several thousand
ng/kg) over PBDDs/PXDDs (maxima of up to some hundred ng/kg).
Indoor air samples taken from rooms equipped with a number of
operating electronic appliances (television and/or computer monitors)
showed the presence of PBDFs (tetra to hepta) at total concentrations
ranging from 0.23 to 1.27 pg/m3. PBDDs were not detected. Dust
samples collected in computer rooms yielded total PBDF levels of
2.4-5.5 µg/kg dust. In contrast to air, the homologue pattern in dust
was dominated by hexaBDFs and heptabromodibenzofurans (heptaBDFs or
HpBDFs). Only in dust samples were low concentrations of tetraBDDs (up
to 1 µg/kg) and of 2,3,7,8-substituted tetra- and pentaBDFs (up to
0.07 µg/kg) detectable. PBDF concentrations in the one sample of house
dust were lower by a factor of 10. The sum concentration of
PBDDs/PBDFs equalled that of PCDDs/PCDFs in dust from computer rooms
but was lower than that of PCDDs/PCDFs in house dust. Dust from an
underground garage contained lower halogenated PBDFs (mono and di) and
PXDFs (di to tetra), with a maximum concentration of 4.3 µg/kg for
mixed dihalogenated dibenzofurans (DiXDFs).
No data are available on levels of PBDDs/PBDFs in water samples.
In river and marine sediment samples from an industrialized zone,
tetraBDDs (up to 0.006 µg/kg dry weight) and tetra- to hexaBDFs (sum
up to 0.37 µg/kg dry weight) were detected. Sediment from road
drainage contained PBDFs (sum of mono to tri: 2.5 µg/kg; sum of tetra
to hepta: 0.3 µg/kg) and PXDFs (sum of di and tri: 1.85 µg/kg), but no
PBDDs.
Similarly, soil samples taken near a motorway contained
monobromodibenzofurans (monoBDFs or MoBDFs) and dibromodibenzofurans
(DiBDFs) (sum: 1.3 µg/kg), tetra- and pentaBDFs (sum: 0.02 µg/kg), and
PXDFs (sum: 1 µg/kg), but no PBDDs. Soil samples taken from an
incineration field and near a metal reclamation factory gave total
PBDF concentrations of up to 100 µg/kg, but no PBDDs were detected. In
a series of sewage sludge samples from municipal wastewater treatment
plants, total PBDF concentrations ranged from n.d. to 3 µg/kg. In one
case, traces of tetraBDDs and 2,3,7,8-TeBDF were detected. A
biocompost sample was nearly free of PBDDs/ PBDFs (tetraBDFs: <0.003
µg/kg).
There are no quantitative data on levels of PBDDs/PBDFs in food.
In grass and pine needle samples collected near motorways, lower
halogenated PBDFs/PXDFs (mono to tetra) and traces of PBDDs/ PXDDs
(mono to tri) were detectable.
No PBDDs/PBDFs were found in the few wildlife samples tested.
In cow's milk collected at dairy farms in the vicinity of a
municipal waste incinerator, tribromodibenzofurans (triBDFs or
TrBDFs), a tetraBDF, and a pentaBDF (not having the
2,3,7,8-substitution pattern) were tentatively identified.
PBDDs/PBDFs have not been detected in the few tested samples of
human adipose tissues or milk samples from the general public.
Contamination by PBDDs/PBDFs is possible at a variety of
workplaces involved in the production, processing, use, or disposal of
certain flame retardants or their products, especially where processes
involve elevated temperatures. The magnitude of worker exposure
depends not only on the compounds involved but also on the quality of
the air and ventilation conditions. There are only limited workplace
monitoring data from plastic producing or processing facilities, from
offices/studios with large numbers of electrical appliances
continuously in use, and from recycling workplaces (including
secondary copper plants). Generally, PBDFs were more abundant than
PBDDs, and PBDF air concentrations were highest at workplaces where
DBDE-containing polymers were produced. In many samples,
2,3,7,8-substituted PBDFs/PBDDs were detectable. PBDD/PBDF
contamination was also found at the work area under the fume hood of a
chemical laboratory. Monitoring data at waste incineration facilities
are lacking.
1.5 Kinetics and metabolism
Most of the studies refer to 2,3,7,8-TeBDD and, to a lesser
extent, 1,2,7,8-TeBDF. Half-life calculations have included some
additional congeners.
2,3,7,8-TeBDD was absorbed in rats after oral, intratracheal, and
dermal administration, the percent absorption varying with route and
dose. Single doses of 1 nmol 2,3,7,8-TeBDD/kg body weight led to an
absorption of 80% (oral and intratracheal routes) or 12% (dermal
route) of the administered dose. The dermal absorption of 1 nmol
1,2,7,8-TeBDF/kg body weight was about 29%. Oral absorption of
2,3,7,8-TeBDD appeared to be comparable to that of
2,3,7,8-tetrachlorodibenzo- p-dioxin (2,3,7,8-TeCDD or TCDD).
However, dermal absorption of 2,3,7,8-TeBDD was about one-third that
of an equimolar dose of 2,3,7,8-TeCDD.
2,3,7,8-TeBDD or 1,2,7,8-TeBDF administered to rats, by any
route, was distributed throughout the entire body, with major deposits
found in liver and adipose tissue, followed by skin and muscle. For
example, 3 days after single oral doses of 2,3,7,8-TeBDD (1 nmol/kg
body weight), the portions in these tissues amounted to 20%, 20%, 11%,
and 4%, respectively, whereas thymus and adrenals contained 0.03% and
0.4%, respectively, of the administered dose. The partitioning of
2,3,7,8-TeBDD between liver and adipose tissue of rats was found to be
influenced by dose, route of exposure, and time post-dosing. The
ratios of liver : fat concentrations measured under different
conditions ranged from 0.2 to 6.5 (range for single doses of
2,3,7,8-TeBDD in rats). No experimental data were available on the
transfer of PBDDs/PBDFs to offspring.
TetraBDD/BDF metabolites were detected in bile and faeces from
rats. They were mainly formed by aromatic hydroxylation and hydrolytic
debromination. The rate of metabolism (indirectly determined as the
rate of biliary excretion) differed between 2,3,7,8-TeBDD (about 7%)
and 1,2,7,8-TeBDF (about 50%). Three days after an intravenous dose of
2,3,7,8-TeBDD (1 nmol/kg body weight), 14% of the administered dose
was found as metabolites in the faeces of rats.
Elimination and excretion of 2,3,7,8-TeBDD were studied in rats
using oral, intravenous, intratracheal, and dermal routes of
administration. In all studies, the major route of elimination was
through the faeces, the eliminated radioactivity ranging from 2%
(dermal route) to 42% (oral route) of the administered dose (1 nmol
[3H]2,3,7,8- TeBDD/kg body weight) in faeces samples, and from 0.2 to
1% in urine samples. Similarly, in studies with 1,2,7,8-TeBDF in rats,
excretion was mainly through the faeces, only 2-3% of the intravenous,
oral, or dermal doses being excreted in urine. During the first days
following oral doses, unabsorbed material and biliary excretion
appeared to be the major sources of eliminated compound in faeces. The
portions of parent 2,3,7,8-TeBDD found in faeces of rats after
administration of 1 nmol 2,3,7,8-TeBDD/kg body weight were 53% (oral
route), 43% (intratracheal route), and 10-20% (intravenous route). A
few days after oral application of 2,3,7,8-TeBDD (1 nmol/kg body
weight), about 20% of the dose administered was eliminated as parent
compound.
Data on retention and turnover are available for some PBDDs/
PBDFs. The relative body burden of 2,3,7,8-TeBDD (and other congeners)
in rats depends on the route of exposure and on the dose administered,
reflecting differences in absorption. Half-lives were calculated for
several PBDDs/PXDDs and PBDFs in various tissues and faeces of rats.
They ranged between 1 day (1,2,7,8-TeBDF from body) and 99 days
(2,3,4,7,8-PeBDF from liver). The estimated half-lives of 17, 18, and
58 days for 2,3,7,8-TeBDD in liver, faeces, and adipose tissue,
respectively, were similar to those reported for 2,3,7,8-TeCDD in
liver and faeces, but higher (by a factor of >2) than those reported
for 2,3,7,8-TeCDD in adipose tissue. Despite differences in early
retention, half-lives of 2,3,7,8-TeBDF and
2,3,7,8-tetrachlorodibenzofuran (2,3,7,8-TeCDF or TCDF) in liver were
comparable.
As with PCDDs/PCDFs, half-lives calculated for humans are much
longer than those for rats. There are estimations of 3-11 years (mean:
5.9 years) for 2,3,7,8-TeBDD and of 1-2 years (mean: 1.5 years) for
2,3,7,8-TeBDF. The persistence of these compounds in humans was also
seen in the case of a chemist who had synthesized 2,3,7,8-TeBDD and
2,3,7,8-TeCDD in 1956. Thirty-five years after exposure, markedly
elevated levels of 2,3,7,8-TeBDD were found in his blood.
1.6 Effects on laboratory mammals and in vitro test systems
Most studies were concerned with the toxicity of 2,3,7,8-TeBDD,
but some information was also available on other PBDDs/PBDFs and
PXDDs/PXDFs.
2,3,7,8-TeBDD caused typical 2,3,7,8-TeCDD-like effects,
including wasting syndrome, thymus atrophy, and liver toxicity.
Additionally, liver damage described as peliosis hepatis, which has
not been reported after exposure of rats to 2,3,7,8-TeCDD, was
observed. The pattern of lesions (lethality, histopathology, liver and
thymus weights) found in guinea-pigs after a single exposure and in
rats after short-term exposure to 2,3,7,8-TeBDF was similar to that of
2,3,7,8-TeCDF.
2,3,7,8-TeBDD interacts with the endocrine system. In rats,
dose-related changes in circulating thyroid hormones and impairment of
spermatogenic activity have been observed.
The oral LD50 (28-day observation period) of 2,3,7,8-TeBDD in
Wistar rats was about 100 µg/kg body weight for females and about 300
µg/kg body weight for males. Oral LD50 values for 2,3,7,8- TeCDD
obtained from other studies ranged between 22 and >3000 µg/kg body
weight. Equimolar doses of 2,3,7,8-TeBDF and 2,3,7,8-TeCDF resulted in
comparable mortality rates in guinea-pigs. For example, 100% mortality
was seen after treatment with both 2,3,7,8-TeBDF (0.03 µmol/kg body
weight, 15.8 µg/kg body weight) and 2,3,7,8-TeCDF (0.03 µmol/kg body
weight, 10 µg/kg body weight). Pre-peliotic lesions and changes in
thyroid hormones were seen in rats after a single dose of 100 µg
2,3,7,8-TeBDD/kg body weight.
In Wistar rats administered 2,3,7,8-TeBDD orally for 13 weeks,
evidence for decreased spermatogenic activity, defective and necrotic
spermatocytes, signs of severe peliosis hepatis, and changes in
circulating thyroid hormones and organ weights were observed. The
no-observed-adverse-effect level (NOAEL) was 0.01 µg/kg body weight
per day.
2,3,7,8-TeBDF administered orally to Sprague-Dawley rats for 4
weeks caused dose-dependent growth retardation and histopathological
changes in liver and thymus. The NOAEL was 1 µg/kg body weight per
day.
Developmental toxicity of some 2,3,7,8-substituted PBDDs/ PBDFs
occurred in mice at subcutaneous and oral doses that produced no
maternal toxicity and no fetal mortality. The lowest-observed-effect
levels (LOELs) (in µg/kg body weight) for hydronephrosis and cleft
palate after a single oral exposure of pregnant mice were,
respectively, as follows: 3 and 48 for 2,3,7,8-TeBDD, 25 and 200 for
2,3,7,8-TeBDF, 400 and 2400 for 2,3,4,7,8-PeBDF, and 500 and 3000-4000
for 1,2,3,7,8-PeBDF. Compared on a molar basis, 2,3,7,8-TeBDD and
2,3,7,8-TeCDD were almost equipotent in induction of hydronephrosis.
Compared on a weight basis, generally the brominated isomers were
slightly less potent than the chlorinated ones in induction of
hydronephrosis and cleft palate. However, 2,3,7,8-TeBDF was more
active than 2,3,7,8-TeCDF.
No information was found on the mutagenicity of PBDDs/PBDFs or
related end-points.
No long-term toxicity and carcinogenicity studies with PBDDs/
PBDFs were available. 2,3,7,8-TeBDD tested positive in a cell
transformation assay using murine peritoneal macrophages. However, the
transforming potency of 2,3,7,8-TeBDD was seven times less than that
of 2,3,7,8-TeCDD. Later, tumours developed in nude mice after
subcutaneous injection of the resulting established cell lines.
A series of several PBDDs and PXDDs (tetra and penta) given
intraperitoneally to immature male Wistar rats caused body weight
losses 14 days after injection. On the basis of molar ED50 values,
the most toxic compounds tested were 2,3,7,8-TeBDD,
2-Br1-3,7,8-Cl3- DD, and 2,3-Br2-7,8-Cl2-DD (TBCDD), which are
substituted only in the four lateral positions. The relative potencies
of the other PBDDs examined followed the order 2,3,7,8- >
1,2,3,7,8- > 1,2,4,7,8- > 1,3,7,8-DD. In other experiments, there
were only slight differences in the ED50 values (on a molar basis)
for body weight loss, thymic atrophy, and hepatic enzyme induction
between 2,3,7,8-TeCDD and 2,3,7,8-TeBDD.
Thymic atrophy and other signs of immunotoxicity (e.g.
haematological parameters, alterations of certain lymphocyte
subpopulations) were seen with several PBDDs/PXDDs and 2,3,7,8-TeBDF
in the rat and with 2,3,7,8-TeBDD and TBCDD in the marmoset monkey
(Callithrix jacchus). It was concluded that, on a molar basis, the
potency of 2,3,7,8-TeBDD was comparable to that of 2,3,7,8-TeCDD in
rats and monkeys. For example, a significant effect on certain
lymphocyte subpopulations in monkeys was found after a single
subcutaneous dose of 30 ng 2,3,7,8-TeBDD/kg body weight versus 10 ng
2,3,7,8-TeCDD/kg body weight. Effects on immunotoxicity after
perinatal exposure to PBDDs/PBDFs have not been investigated.
After subchronic dosing of either 2,3,7,8-TeBDD or 2,3,7,8-TeCDD
by oral gavage in mice, there was a dose-dependent increase in total
hepatic porphyrins.
After single oral doses of 2,3,7,8-TeBDD and 2,3,7,8-TeCDD,
reductions in concentration and total amount of vitamin A were
observed in the liver of rats, with 2,3,7,8-TeBDD being slightly less
potent than 2,3,7,8-TeCDD (on a molar basis).
2,3,7,8-TeBDD and 2,3,7,8-TeBDF produced hyperkeratosis in the
rabbit ear assay at a dose of 100 µg/rabbit, but not at 10 µg/rabbit.
A no-observed-effect level (NOEL) for 2,3,7,8-TeCDD was 0.01
µg/rabbit.
Several tetra- (Br1Cl3DDs, Br2Cl2DDs) and penta- (Br1Cl4DD)
halogenated congeners with 2,3,7,8-substitution were found to have an
antiestrogenic potency similar to that of 2,3,7,8-TeCDD, as examined
in cultures of human breast cancer cells.
In rats, 2,3,7-tribromodibenzo- p-dioxin (2,3,7-triBDD/TrBDD)
depressed the disappearance of ouabain from plasma, its excretion into
bile, and bile flow to a slightly lesser extent than 2,3,7,8-TeCDD.
PBDDs/PBDFs and PXDDs/PXDFs are potent inducers of certain
cytochrome P-450 (CYP)-dependent microsomal enzymes. ED50 values of
0.8-1 nmol/kg body weight for CYP1A1 induction and about 0.2 nmol/kg
body weight for CYP1A2 induction in rat liver were estimated after
single oral doses of 2,3,7,8-TeBDD. CYP1A1 induction (arylhydrocarbon
hydroxylase [AHH] and/or ethoxyresorufin- O-deethylase [EROD]
induction) was observed in a variety of species and tissues
in vivo and in rat cell cultures in vitro. A lot of different
congeners were found to be active, as well as pyrolysates from certain
flame retardants. Generally, enzyme induction proceeded
dose-dependently at non-toxic concentrations, started soon after
exposure, and was long-lasting. It was measurable at exposures as low
as the pmol range. The induction potency varied over several orders of
magnitude for different congeners, depending on their chemical
structure. The most potent inducers were TCDD, TBDD, and TBCDD.
Compared (on a molar basis) with their chlorinated analogues, the
PBDDs and PXDDs had more or less similar potency. In contrast to TCDD,
whose relative induction potency was independent of the tissue
examined, TBDD was five times more potent at inducing EROD activity in
the liver than in skin and lung following subchronic exposure of mice.
The ranking order for induction of EROD activity in marmoset monkeys
was TCDD > 2,3,4,7,8-pentachlorodibenzofuran
(2,3,4,7,8-pentaCDF/PeCDF) > 2,3,4,7,8-PeBDF when enzyme activities
were compared with the hepatic concentrations. In vitro tests with
rat cell cultures resulted in similar molar EC50 values of AHH and
EROD induction potencies between corresponding PXDFs and PCDFs.
PBDDs/PBDFs are believed to share a common mechanism of action
with PCDDs/PCDFs and other related halogenated aromatic hydrocarbons
(Ah). Binding to the cytosolic Ah receptor, which plays a central role
in mediating 2,3,7,8-TeCDD-like toxicity, was confirmed for several
PBDDs and PXDDs/PXDFs. Their receptor-binding affinities varied by
several orders of magnitude but were comparable to those of their
chlorinated analogues.
1.7 Effects on humans
There are no data on the exposure of humans to PBDDs/PBDFs or on
their effects on the health of the general population.
Two cases of acute health problems due to 2,3,7,8-TeBDD/ TeCDD
exposure have been reported, with symptoms including chloracne.
In another study, male personnel of a chemical plant with
documented exposure to PBDDs/PBDFs originating from the use of
brominated flame retardants (OBDE and DBDE) were subjected to
immunological and additional clinical laboratory tests. Although there
were indications of minor changes in immunological parameters, the
overall evaluation of their health status did not reveal an impact of
2,3,7,8-TeBDD/TeBDF body burden on the immune system.
There are no reports on cancer mortality caused by PBDDs/ PBDFs.
1.8 Effects on other organisms in the laboratory and field
There is only limited information on the effects of PBDDs/PBDFs
on microorganisms, plants, or invertebrate or vertebrate wildlife
species.
Using the rainbow trout (Oncorhynchus mykiss) sac fry early
life stage mortality bioassay, a series of PBDD/PBDF congeners were
tested and found to be active. This bioassay also demonstrated that
for both PBDDs and PBDFs, there was a decreased potency with increased
bromine substitution. Both 2,3,7,8-TeBDD and 2,3,7,8-TeBDF were more
potent than their chlorinated analogues.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
PHDDs/PHDFs are almost planar tricyclic aromatic compounds. There
are eight positions on both the dibenzo- p-dioxin and the
dibenzofuran molecules where halogen substitution can occur. The
positions are numbered as shown in Fig. 1 for PBDDs and PBDFs.
Each series consists of eight homologous groups (groups of
congeners having the same number of bromine atoms), and in each group
there are different numbers of isomers (see Table 1). Theoretically,
75 PBDDs and 135 PBDFs are possible, as well as a large number of
mixed halogenated congeners -- 1550 PXDDs and 3050 PXDFs (Buser,
1987a). There are 7 2,3,7,8-substituted PBDDs and 10
2,3,7,8-substituted PBDFs (see Table 2), as well as 337 possible
2,3,7,8-substituted PXDDs and 647 possible 2,3,7,8-substituted PXDFs
(Ballschmiter & Bacher, 1996). PCDDs/PCDFs are discussed in a separate
Environmental Health Criteria monograph (WHO, 1989).
Because of the complexity of the analytical procedures (see
section 2.4), it has been possible to characterize only a small number
of PBDDs/PBDFs and PXDDs/PXDFs. Tables 3 and 4 show the Chemical
Abstracts Service (CAS) numbers that have been allocated to some of
these compounds.
2.2 Physical and chemical properties
2.2.1 Appearance, melting and boiling points, water solubility, vapour
pressure, octanol/water partition coefficient, and sorption coefficient
Experimental data on the physical and chemical properties of
PBDDs/PBDFs are scarce (see Table 5). In many cases, only predicted
values are available. It should be noted that for PCDDs, experimental
data are often lower than the calculated values (Shiu et al., 1988;
Fiedler & Schramm, 1990). This is also to be expected for the
brominated and for the mixed halogenated compounds (Fiedler & Schramm,
1990). Measured values for the aqueous solubility of PCDDs decrease
dramatically with increase in chlorine substitution and temperature
(Shiu et al., 1988).
PBDDs/PBDFs have higher molecular weights than their chlorinated
analogues, high melting points, and low water solubilities, but they
are generally soluble in fats, oils, and organic solvents (see Table
5). PBDDs/PBDFs have, like their chlorinated analogues, very low
vapour pressures, and at ambient temperatures they are mostly found
bound to particles. For the lower substituted compounds, PBDDs/PBDFs
have higher calculated p Kow values than the chlorinated congeners
(Fiedler & Schramm, 1990) and are therefore more lipophilic.
Table 1. Number of isomers, elemental composition, and molecular weight for PBDDs/PBDFs
Compound Number of isomers Elemental Molecular
Total 2,3,7,8-Substituted composition weight
MoBDD 2 - C12H7O2Br 263.1
DiBDD 10 - C12H6O2Br2 342.0
TrBDD 14 - C12H5O2Br3 420.9
TeBDD 22 1 C12H4O2Br4 499.8
PeBDD 14 1 C12H3O2Br5 578.7
HxBDD 10 3 C12H2O2Br6 657.6
HpBDD 2 1 C12HO2Br7 736.5
OcBDD 1 1 C12O2Br8 815.4
MoBDF 4 - C12H7OBr 247.1
DiBDF 16 - C12H6OBr2 326.0
TrBDF 28 - C12H5OBr3 404.9
TeBDF 38 1 C12H4OBr4 483.8
PeBDF 28 2 C12H3OBr5 562.7
HxBDF 16 4 C12H2OBr6 641.6
HpBDF 4 2 C12HOBr7 720.5
OcBDF 1 1 C12OBr8 799.4
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR POLYBROMINATED
DIBENZO- p- DIOXINS AND DIBENZOFURANS
Members
Dr A.P.J.M. de Jong, Laboratory of Organic Analytical Chemistry,
National Institute of Public Health and Environment, Bilthoven, The
Netherlands
Ms J. Diliberto, National Health and Environmental Effects
Research Laboratory, Experimental Toxicology Division, US
Environmental Protection Agency, Research Triangle Park, North
Carolina, USA
Dr M. Feeley, Toxicology Evaluation Section, Bureau of Chemical
Safety, Health Canada, Tunney's Pasture, Ottawa, Ontario, Canada
(Rapporteur)
Dr H. Fiedler, Bayerisches Institut für Abfallforschung BIFA
GmbH, Augsburg, Germany
Professor B. Jansson, Institute of Applied Environmental Research,
Stockholm University, Stockholm, Sweden (Chairman)
Dr Y. Kurokawa, Biological Safety Research Center, National
Institute of Health Sciences, Tokyo, Japan
Dr C. Melber, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Professor D. Neubert, Institute for Clinical Pharmacology and
Toxicology, Berlin, Germany
Professor C. Rappe, Institute of Environmental Chemistry,
University of Umea, Umea, Sweden
Observer
Dr B. Schatowitz, Environmental and Trace Analysis Consumer
Care Division, Ciba-Geigy AG, Basel, Switzerland (Representing the
European Centre for Ecotoxicology and Toxicology of Chemicals)
Secretariat
Dr H. Galal-Gorchev, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
Dr J. Kielhorn, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr I. Mangelsdorf, Fraunhofer Institute for Toxicology and Aerosol
Research, Hanover, Germany
Dr F.X.R. van Leeuwen, Chemical Safety, European Centre for
Environment and Health, Bilthoven Division, De Bilt, The Netherlands
ENVIRONMENTAL HEALTH CRITERIA FOR POLYBROMINATED DIBENZO- p-DIOXINS
AND DIBENZOFURANS
A WHO Task Group on Environmental Health Criteria for
Polybrominated Dibenzo- p-dioxins and Dibenzofurans met at the
Fraunhofer Institute for Toxicology and Aerosol Research, Hanover,
Germany from 11 to 15 November 1996. Professor U. Heinrich opened the
meeting and welcomed the participants on behalf of the host institute.
Dr H. Galal-Gorchev, IPCS, welcomed the participants on behalf of the
Director, IPCS, and the three IPCS cooperating organizations
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft criteria
monograph and made an evaluation of the risks for human health and the
environment from exposure to polybrominated dibenzo- p-dioxins and
dibenzofurans.
Dr J. Kielhorn and Dr C. Melber, Fraunhofer Institute for
Toxicology and Aerosol Research, Hanover, Germany, prepared the first
draft of this monograph. They also prepared the second draft,
incorporating comments received following the circulation of the first
draft to the IPCS Contact Points for Environmental Health Criteria
monographs.
Dr H. Galal-Gorchev, IPCS Central Unit, was responsible for the
overall scientific content and Ms M. Sheffer, Scientific Editor,
Ottawa, Canada, for the linguistic editing.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
ABS acrylonitrile-butadiene-styrene
Ah aromatic hydrocarbon
AHH arylhydrocarbon hydroxylase
BB bromobiphenyl
BDE bromodiphenyl ether
CAS Chemical Abstracts Service
CYP cytochrome P-450
DBB/decaBB decabromobiphenyl
DBDE/decaBDE decabromodiphenyl ether
DD/DF dibenzo- p-dioxin/dibenzofuran
DiBDD dibromodibenzo- p-dioxin
DiBDF dibromodibenzofuran
DiXDF mixed dihalogenated dibenzofuran
EC50 median effective concentration
ED50 median effective dose
EI-SIM-MS electron impact-selective ion monitoring-mass
spectrometry
EPA Environmental Protection Agency (USA)
EROD ethoxyresorufin- O-deethylase
GC gas chromatography
HexaBB hexabromobiphenyl
HIPS high-impact polystyrene
HpBDD/heptaBDD heptabromodibenzo- p-dioxin
HpBDF/heptaBDF heptabromodibenzofuran
HPLC high-performance liquid chromatography
HRGC high-resolution gas chromatography
HRMS high-resolution mass spectrometry
HxBDD/hexaBDD hexabromodibenzo- p-dioxin
HxBDF/hexaBDF hexabromodibenzofuran
HxCDD/hexaCDD hexachlorodibenzo- p-dioxin
I-TEF international toxicity equivalency factor
I-TEQ international toxic equivalent
LD50 median lethal dose
LOAEL lowest-observed-adverse-effect level
LOEL lowest-observed-effect level
MI-IR matrix isolation infrared spectrometry
MoBDD/monoBDD monobromodibenzo- p-dioxin
MoBDF/monoBDF monobromodibenzofuran
MS mass spectrometry
n sample size
NCI negative ion chemical ionization
n.d. not detected
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
n.sp. not specified
OBDE/octaBDE octabromodiphenyl ether
OcBDD/octaBDD octabromodibenzo- p-dioxin
OcBDF/octaBDF octabromodibenzofuran
OCDD/OcCDD/octaCDD octachlorodibenzo- p-dioxin
PAH polycyclic aromatic hydrocarbon
PBB polybrominated biphenyl
PBDD polybrominated dibenzo- p-dioxin
PBDE polybrominated diphenyl ether
PBDF polybrominated dibenzofuran
PBT polybutylene terephthalate
PCB polychlorinated biphenyl
PCDD polychlorinated dibenzo- p-dioxin
PCDE polychlorinated diphenyl ether
PCDF polychlorinated dibenzofuran
PeBDD/pentaBDD pentabromodibenzo- p-dioxin
PeBDE/pentaBDE pentabromodiphenyl ether
PeBDF/pentaBDF pentabromodibenzofuran
PeCDF/pentaCDF pentachlorodibenzofuran
PeHDD pentahalogenated dibenzo- p-dioxin
PeHDF pentahalogenated dibenzofuran
PHDD polyhalogenated dibenzo- p-dioxin (used as
collective term including PCDD, PBDD, PXDD)
PHDF polyhalogenated dibenzofuran (used as collective
term including PCDF, PBDF, PXDF)
PVC polyvinyl chloride
PXDD mixed (brominated/chlorinated) halogenated
dibenzo- p-dioxin
PXDF mixed (brominated/chlorinated) halogenated
dibenzofuran
RI retention index
RIA radioimmunoassay
RMM relative molecular mass
SD standard deviation
T3 triiodothyronine
T4 thyroxin
TBBPA tetrabromobisphenol A
TBCDD 2,3-dibromo-7,8-dichlorodibenzo- p- dioxin
TBDD/2,3,7,8-TeBDD 2,3,7,8-tetrabromodibenzo- p-dioxin
TBDF/2,3,7,8-TeBDF 2,3,7,8-tetrabromodibenzofuran
TBPI bis-tetrabromo-phthalimide ethylene
TCDD/2,3,7,8-TeCDD 2,3,7,8-tetrachlorodibenzo- p-dioxin
TCDF/2,3,7,8-TeCDF 2,3,7,8-tetrachlorodibenzofuran
TeBDD/tetraBDD tetrabromodibenzo- p-dioxin
TeBDF/tetraBDF tetrabromodibenzofuran
TEF toxicity equivalency factor
TeHDD tetrahalogenated dibenzo- p-dioxin
TEQ toxic equivalent
TeXDD/tetraXDD mixed tetrahalogenated dibenzo- p- dioxin
THDF 2,3,7,8-tetrahalogenated dibenzofuran
TrBDD/triBDD tribromodibenzo- p-dioxin
TrBDF/triBDF tribromodibenzofuran
TrHDD/triHDD trihalogenated dibenzo- p-dioxin
TV television
TxDD 2,3,7,8-substituted mixed tetrahalogenated
dibenzo- p-dioxin
UV ultraviolet
WHO World Health Organization
1. SUMMARY
1.1 Identity, physical and chemical properties, and analytical
methods
Polybrominated dibenzo- p-dioxins (PBDDs) and polybrominated
dibenzofurans (PBDFs) are almost planar tricyclic aromatic compounds.
Theoretically, 75 PBDDs and 135 PBDFs are possible. In addition, a
large number of mixed halogenated congeners -- 1550
brominated/chlorinated dibenzo- p-dioxins (PXDDs) and 3050
brominated/chlorinated dibenzofurans (PXDFs) -- are theoretically
possible. Because of the complexity of the analytical procedures and
paucity of analytical reference standards, it has been possible to
characterize and determine only a small number of these compounds. The
most toxic congeners are those substituted at positions 2, 3, 7, and
8. There are 7 2,3,7,8-substituted PBDDs and 10 2,3,7,8-substituted
PBDFs, as well as 337 possible 2,3,7,8-substituted PXDDs and 647
possible 2,3,7,8-substituted PXDFs.
PBDDs/PBDFs have higher molecular weights than their chlorinated
analogues, high melting points, low vapour pressures, and low water
solubilities. They are generally soluble in fats, oils, and organic
solvents. There are very few experimental data on the physical and
chemical properties of PBDDs/PBDFs.
Photolysis occurs at a more rapid rate for PBDDs/PBDFs than for
polychlorinated dibenzo- p-dioxins (PCDDs) and polychlorinated
dibenzofurans (PCDFs). PBDDs/PBDFs are thermostable. The temperatures
of formation and destruction of PBDDs/PBDFs depend on several
conditions, including the presence or absence of oxygen, polymers, and
flame retardant additives, such as antimony trioxide (Sb2O3).
In the presence of excess chlorine, bromine is substituted by
chlorine to give PXDDs/PXDFs.
Because of the toxic nature of these compounds and their
photolytic properties, care must be taken during sampling and
analysis. Highly sensitive, selective, and specific analytical methods
(gas chromatography/mass spectrometry, or GC/MS) are required because
of the large number of PBDD/PBDF congeners. Sampling procedures are
identical for all polyhalogenated dibenzo- p-dioxins (PHDDs) and
polyhalogenated dibenzofurans (PHDFs), but separation and
determination of PBDDs/PBDFs (and PXDDs/PXDFs) differ slightly from
those of their chlorinated analogues. PBDDs/PBDFs have higher
molecular weights and longer GC retention times than the chlorinated
analogues, as well as different MS isotopic cluster patterns and
interference compounds. Exact identification of specific brominated
congeners is very limited owing to the small number of reference
standards currently available. For the same reason, determination of
mixed halogenated congeners is almost impossible.
1.2 Formation and sources of human and environmental exposure
PBDDs/PBDFs are not known to occur naturally. They are not
intentionally produced (except for scientific purposes) but are
generated as undesired by-products in various processes. They can be
formed by chemical, photochemical, or thermal reactions from
precursors and by so-called de novo synthesis.
PBDDs/PBDFs have been found as contaminants in brominated organic
chemicals (e.g. bromophenols) and, in particular, in flame retardants,
such as polybrominated diphenyl ethers (PBDEs), decabromobiphenyl
(decaBB or DBB), 1,2-bis(tribromophenoxy)ethane, tetrabromobisphenol A
(TBBPA), and others. They have been detected in distillation residues
of some bromophenols and bromoanilines and in wastes from chemical
laboratories.
PBDFs and, to a lesser extent, PBDDs have been detected as
photochemical degradation products of brominated organic chemicals,
such as PBDEs and bromophenols.
Laboratory thermolysis experiments showed the formation of
PBDDs/PBDFs from bromophenols, PBDEs, polybrominated biphenyls (PBBs),
and other brominated flame retardants (pure or in a polymer matrix).
There was a broad range of yields, from zero to maximum values
(reached from PBDEs) in the g/kg range. Generally, PBDFs were much
more abundant than PBDDs. The optimum PBDF formation temperature of a
series of pure flame retardants was in the range of 600-900°C. The
presence of polymers or synergists (e.g. Sb2O3) resulted in a
decrease in the optimum formation temperature (down to 400°C). In
addition to temperature and the presence of polymer matrix or
synergists, several other factors, such as metals, metal oxides,
water, oxygen, and the type of combustion apparatus used, influenced
the yield and pattern of PBDDs/PBDFs. In ternary mixtures of PBDE,
polymer matrix, and Sb2O3, tetrabromodibenzo furans (tetraBDFs or
TeBDFs) were frequently the most abundant homologue group.
2,3,7,8-Substituted PBDDs/PBDFs (tetra to hepta) were found at varying
concentrations; for example, 2,3,7,8-TeBDF was found at up to 2000
mg/kg in pyrolysates of polymers containing octabromodiphenyl ether
(octaBDE or OBDE).
In the manufacture of plastics, elevated temperatures (150-300°C)
occur during several processes. Studies of the exhaust streams from
machines processing polymers -- such as
acrylonitrile-butadiene-styrene (ABS) and polybutylene terephthalate
(PBT) -- containing different types of brominated flame retardants
showed that PBDDs/PBDFs (di to octa) can be formed at these
temperatures. OBDE and decabromodiphenyl ether (decaBDE or DBDE)
produced the highest amounts of PBDDs/PBDFs, the major portion
consisting of PBDFs. Levels observed with TBBPA or
bis-tetrabromo-phthalimide ethylene (TBPI) were several orders of
magnitude lower. No PBDDs/PBDFs were detected during processing of ABS
flame-retarded by brominated styrene or
1,2-bis(tribromophenoxy)ethane. 2,3,7,8-Substituted congeners were not
determined (processing of DBDE), were detected at trace levels
(processing of OBDE), or were not detected (processing of TBBPA and
TBPI).
Various plastic materials at several processing stages were
analysed for PBDDs/PBDFs. These included (granulated) resins and
moulded parts whose flame retardant additives were known as well as
samples from commercial electrical appliances (television sets,
printers, computers) whose flame retardant additives were unknown. The
highest levels of PBDDs/PBDFs were found in materials flame-retarded
with PBDEs and were in the range of several thousand µg/kg, thus
exceeding the levels of other flame retardant/polymer systems by
orders of magnitude. Factors influencing the extent of formation are
temperature and the duration of such processes as blending, extrusion,
and moulding. Again PBDFs dominated, with some exceptions, over PBDDs,
with the highly brominated (>tetra) derivatives being prevalent. Peak
concentrations were seen with pentabromodibenzofurans (pentaBDFs or
PeBDFs) and hexabromodibenzofurans (hexaBDFs or HxBDFs). The latter
reached levels as high as 3000 µg/kg in casing parts. Printed circuit
boards contained tetra- and pentaBDFs at maximum concentrations of
1300 and 1400 µg/kg, respectively. Total PBDF (mono to hexa)
concentrations were in the range of 3.6-3430 µg/kg.
2,3,7,8-Substituted PBDDs/PBDFs were not determined, were not
detectable, or were present at relatively low concentrations. Maximum
concentrations of 2,3,7,8-substituted PBDFs (tetra to hexa) in casings
or printed circuit boards ranged from 11 µg/kg (tetra) to 203 µg/kg
(hexa).
Experiments to determine whether PBDFs were released from
television sets or similar appliances during use showed air levels
ranging from not detected to 1800 pg total PBDFs (tetra to hexa) per
appliance.
Burning of products containing brominated compounds caused
emission of PBDDs/PBDFs. In experimental fire tests simulating real
fire conditions with electrical appliances such as television sets,
printers, computer terminals, and their casings, high PBDF (mono to
hexa) concentrations were detected in the combustion residues
(thousands of mg/kg), in smoke condensate (hundreds of µg/m2), and in
smoke (up to 1700 µg/m3). PBDD concentrations amounted to about 3% of
the detected levels of PBDDs/PBDFs. The 2,3,7,8-substituted isomer was
mostly below 3% of the total tetraBDFs. 2,3,7,8-Substituted penta- and
hexaBDFs yielded between 1 and 16% of the corresponding totals.
Burning of test vehicles resulted in PBDF (mono to octa)
concentrations of up to 4.3 µg/kg in the fire residues.
During real fire accidents in private residences (television sets
involved), offices (computers involved), and other buildings,
concentrations measured were in most cases below the values found in
the model experiments described above, but the qualitative composition
of the samples was similar. PBDFs were found in almost all samples,
but PBDDs were not always detected; if present, their concentrations
were low. The PBDF concentrations in combustion residues were mainly
in the µg/kg range (low to high), but single maximum values (sum of
mono to hexa) of up to 107 mg/kg were also observed. The PBDF (mono to
hexa) area contaminant concentrations in close vicinity to the fire
site ranged between 0.1 and 13 µg/m2 in most cases. Additionally,
relevant concentrations of PXDDs/PXDFs could be detected. The
proportion of 2,3,7,8-substituted PBDDs/PBDFs was relatively low in
most of the samples examined. For example, maximum proportions of 3,
10, or 18% of the corresponding totals of tetra-, penta-, or hexaBDFs,
respectively, were reported from fire accidents with television sets.
Soot samples collected after a fire in a computer room contained
2,3,7,8-substituted tetra- and pentabromodibenzo- p-dioxins
(tetra/pentaBDDs or TeBDD/PeBDD) and tetra- and pentaBDFs, with a
maximum concentration of 48 µg/kg for 2,3,7,8-TeBDF (TBDF).
PXDDs were detected in ash from a wood-fired boiler. However, the
sort of wood (treated or untreated) was not specified. No data were
available on the incineration of other fuels, such as coal, peat, or
fuel oil.
The presence of PBDDs/PBDFs and/or PXDDs/PXDFs has been reported
in fly ash and/or flue gas of municipal, hospital, or hazardous waste
incinerators. The majority of these compounds are probably produced in
the incinerator itself, by formation from precursors at high
temperatures in the flame or by de novo synthesis at low
temperatures in the post-combustion zone of the incinerator. The
formation of PXDDs/PXDFs is explained by the extensive
bromine-chlorine exchange reactions (with chlorine donors in waste)
observed under several test conditions. The quantities of PBDDs/PBDFs
and PXDDs/ PXDFs measured in fly ash of incinerators were in the range
of ng/kg to µg/kg. In most cases, the concentrations of
dibenzo- p-dioxins exceeded those of dibenzofurans, and PXDDs/PXDFs
were more abundant than PBDDs/PBDFs. Of 2,3,7,8-substituted congeners,
a mixed tetrahalogenated dibenzo- p-dioxin (tetraXDD or TeXDD)
(Br2Cl2DD) was found.
Analyses of waste samples from some disposal sites showed the
presence of PBDDs/PBDFs and PXDDs/PXDFs at concentrations of several
hundred to several thousand ng/kg dry weight. The concentration of
dibenzo- p-dioxins (up to 580 ng/kg) was below that of dibenzofurans
(up to 4230 ng/kg). Generally, the homologue profile was dominated by
the lower halogenated (mono to tetra) derivatives. Chemical laboratory
waste contained PBDDs/PBDFs, with a peak concentration of 15 500 ng/kg
for hexaBDFs.
PBDDs/PBDFs were present in plastic materials (with or without
metals) of several recycling stages. The samples originated mainly
from office machines, printed circuit boards, and other electronic
scrap. In some cases, the sum concentration of eight selected PBDD/
PBDF congeners having the 2,3,7,8-substitution was as high as 65
µg/kg. Metal reclamation was also found to be a source of PBDDs and/or
PXDDs/PXDFs. PBDDs/PBDFs have also been detected in textile industries
where brominated flame retardants have been used. PBDFs were found in
the exhaust air, in the textiles before and after processing, and in
the chimney depositions.
PBDDs/PBDFs and PXDDs/PXDFs (along with PCDDs/PCDFs) have been
detected in emissions of motors using leaded petrol, in emissions of
motors using unleaded petrol with and without catalytic converters,
and in emissions of diesel engines. Because of the brominated and
chlorinated scavengers (dibromo- and dichloroethane) used in leaded
petrol, the highest levels of PHDDs/PHDFs (several thousand ng/m3)
were found with this type of petrol. Unleaded petrol produced much
lower emissions of PHDDs/PHDFs (approximately two orders of magnitude
lower). A further reduction was seen after catalytic gas cleaning. The
values for diesel engines were somewhat higher than those found with
the Otto motors (spark ignition engines) run on unleaded petrol. In
exhaust gases from combustion of leaded petrol, PBDDs/PBDFs were more
abundant than PXDDs/PXDFs and PCDDs/PCDFs. Generally, the
concentrations of dibenzofurans exceeded those of dibenzo- p-dioxins,
and there was a dominance of lower substituted homologues (mono to
tri). Similar patterns were seen in residues adhering to mufflers.
1.3 Environmental transport, distribution, and transformation
There are very few data available on the environmental transport
and distribution of PBDDs/PBDFs. Generally, their physicochemical
properties suggest similarities to PCDDs/PCDFs. Therefore, if released
to the environment, they may be preferably distributed into carbon- or
fat-rich compartments, as with PCDDs/PCDFs.
Airborne PBDDs/PBDFs were found to be transported in both the
particulate and vapour phase, the partitioning ratio depending on the
degree of bromination.
No experimental data are available on the movement of PBDDs/
PBDFs in water or soil. For PBDFs (tri to penta), adsorption to
sediment was reported. Owing to the low water solubility of PBDDs/
PBDFs, leaching through the soil may be limited but may be increased
in the presence of organic solvents or humic acids.
There are no experimental data on processes for the transport and
distribution of PBDDs/PBDFs between environmental media and biota or
within biota. Based on the similar high octanol/water partition
coefficients calculated for selected PCDDs/PCDFs, PBDDs/PBDFs, and
PXDDs/PXDFs, a bioavailability comparable to that of PCDDs/ PCDFs is
expected.
Photolysis of PBDDs/PBDFs and PXDDs/PXDFs was studied in organic
solvents and on quartz surfaces in the laboratory, as well as in soil
and on soot (and dust) particles under outdoor conditions. The slowest
photolytic reactions were observed under the latter, more
environmentally relevant, conditions. Reductive debromination was
found to be a major photochemical pathway. The rate of decomposition
of different congeners depended on their bromine substitution pattern.
Generally, higher brominated congeners and those with lateral bromines
had shorter half-lives. Calculated half-lives were in the order of
minutes (use of direct sunlight or ultraviolet [UV] light and quartz
vials), hours (use of solid films or soot or dust particles and
sunlight), or hundreds to thousands of hours (use of soil and
sunlight). For example, the estimated sunlight-induced half-lives for
2,3,7,8-TeBDD (TBDD) were 0.8 min (in organic solution) or 32 h
(dispersed as solid films). A half-life of 3-6 months was estimated
for tetraBDD isomers in surface soil. Compared with PCDDs/PCDFs, the
brominated counterparts were photochemically less stable. PXDDs/PXDFs
preferentially lost their bromine atoms during photolysis and
therefore were transformed into PCDDs/PCDFs, which had longer
photolytic half-lives. Such a transformation of PXDDs/PXDFs to
PCDDs/PCDFs also occurs during incineration processes.
PBDDs/PBDFs seem to be poorly degradable by microorganisms.
The presence of PBDDs/PBDFs in animals and in humans, as seen in
a few studies, is indicative of their accumulation potential.
2,3,7,8-TeBDD accumulated in rats during subchronic administration.
Bioaccumulation, bioconcentration, or biomagnification factors for
PBDDs/PBDFs or PXDDs/PXDFs are not available.
1.4 Environmental levels and human exposure
To date, in contrast to PCDDs/PCDFs, PBDDs/PBDFs have not been
frequently included in monitoring programmes. The few studies
performed indicate a limited occurrence.
In ambient air, PBDFs were found more frequently than PBDDs. Only
lower brominated PBDDs (mono to tetra) were detected at concentrations
ranging from not detected (n.d.) to about 0.85 pg/m3 for
monobromodibenzo- p-dioxins (monoBDDs or MoBDDs) in a motor way
tunnel and an underground garage. Of PBDFs, mono- to hexabrominated
homologues were found, their concentrations ranging from n.d. to 74
pg/m3. The concentrations (mean values) of total PBDDs/ PBDFs (tri to
hexa) measured, for example, in Germany in a motorway tunnel, in a
city, and in a suburban area amounted to 23 pg/m3, 2 pg/m3, and 0.59
pg/m3, respectively; 2,3,7,8-TeBDD was not detected, and the maximum
concentrations of 2,3,7,8-TeBDF and 1,2,3,7,8-PeBDF were 0.28 pg/m3
and 0.08 pg/m3, respectively. PXDFs were identified in
traffic-related air samples at concentrations up to 41 pg/m3
(Cl1Br1DFs). Outdoor dust samples (mainly from motorways) also
showed a predominance of PBDFs/PXDFs (maxima of several thousand
ng/kg) over PBDDs/PXDDs (maxima of up to some hundred ng/kg).
Indoor air samples taken from rooms equipped with a number of
operating electronic appliances (television and/or computer monitors)
showed the presence of PBDFs (tetra to hepta) at total concentrations
ranging from 0.23 to 1.27 pg/m3. PBDDs were not detected. Dust
samples collected in computer rooms yielded total PBDF levels of
2.4-5.5 µg/kg dust. In contrast to air, the homologue pattern in dust
was dominated by hexaBDFs and heptabromodibenzofurans (heptaBDFs or
HpBDFs). Only in dust samples were low concentrations of tetraBDDs (up
to 1 µg/kg) and of 2,3,7,8-substituted tetra- and pentaBDFs (up to
0.07 µg/kg) detectable. PBDF concentrations in the one sample of house
dust were lower by a factor of 10. The sum concentration of
PBDDs/PBDFs equalled that of PCDDs/PCDFs in dust from computer rooms
but was lower than that of PCDDs/PCDFs in house dust. Dust from an
underground garage contained lower halogenated PBDFs (mono and di) and
PXDFs (di to tetra), with a maximum concentration of 4.3 µg/kg for
mixed dihalogenated dibenzofurans (DiXDFs).
No data are available on levels of PBDDs/PBDFs in water samples.
In river and marine sediment samples from an industrialized zone,
tetraBDDs (up to 0.006 µg/kg dry weight) and tetra- to hexaBDFs (sum
up to 0.37 µg/kg dry weight) were detected. Sediment from road
drainage contained PBDFs (sum of mono to tri: 2.5 µg/kg; sum of tetra
to hepta: 0.3 µg/kg) and PXDFs (sum of di and tri: 1.85 µg/kg), but no
PBDDs.
Similarly, soil samples taken near a motorway contained
monobromodibenzofurans (monoBDFs or MoBDFs) and dibromodibenzofurans
(DiBDFs) (sum: 1.3 µg/kg), tetra- and pentaBDFs (sum: 0.02 µg/kg), and
PXDFs (sum: 1 µg/kg), but no PBDDs. Soil samples taken from an
incineration field and near a metal reclamation factory gave total
PBDF concentrations of up to 100 µg/kg, but no PBDDs were detected. In
a series of sewage sludge samples from municipal wastewater treatment
plants, total PBDF concentrations ranged from n.d. to 3 µg/kg. In one
case, traces of tetraBDDs and 2,3,7,8-TeBDF were detected. A
biocompost sample was nearly free of PBDDs/ PBDFs (tetraBDFs: <0.003
µg/kg).
There are no quantitative data on levels of PBDDs/PBDFs in food.
In grass and pine needle samples collected near motorways, lower
halogenated PBDFs/PXDFs (mono to tetra) and traces of PBDDs/ PXDDs
(mono to tri) were detectable.
No PBDDs/PBDFs were found in the few wildlife samples tested.
In cow's milk collected at dairy farms in the vicinity of a
municipal waste incinerator, tribromodibenzofurans (triBDFs or
TrBDFs), a tetraBDF, and a pentaBDF (not having the
2,3,7,8-substitution pattern) were tentatively identified.
PBDDs/PBDFs have not been detected in the few tested samples of
human adipose tissues or milk samples from the general public.
Contamination by PBDDs/PBDFs is possible at a variety of
workplaces involved in the production, processing, use, or disposal of
certain flame retardants or their products, especially where processes
involve elevated temperatures. The magnitude of worker exposure
depends not only on the compounds involved but also on the quality of
the air and ventilation conditions. There are only limited workplace
monitoring data from plastic producing or processing facilities, from
offices/studios with large numbers of electrical appliances
continuously in use, and from recycling workplaces (including
secondary copper plants). Generally, PBDFs were more abundant than
PBDDs, and PBDF air concentrations were highest at workplaces where
DBDE-containing polymers were produced. In many samples,
2,3,7,8-substituted PBDFs/PBDDs were detectable. PBDD/PBDF
contamination was also found at the work area under the fume hood of a
chemical laboratory. Monitoring data at waste incineration facilities
are lacking.
1.5 Kinetics and metabolism
Most of the studies refer to 2,3,7,8-TeBDD and, to a lesser
extent, 1,2,7,8-TeBDF. Half-life calculations have included some
additional congeners.
2,3,7,8-TeBDD was absorbed in rats after oral, intratracheal, and
dermal administration, the percent absorption varying with route and
dose. Single doses of 1 nmol 2,3,7,8-TeBDD/kg body weight led to an
absorption of 80% (oral and intratracheal routes) or 12% (dermal
route) of the administered dose. The dermal absorption of 1 nmol
1,2,7,8-TeBDF/kg body weight was about 29%. Oral absorption of
2,3,7,8-TeBDD appeared to be comparable to that of
2,3,7,8-tetrachlorodibenzo- p-dioxin (2,3,7,8-TeCDD or TCDD).
However, dermal absorption of 2,3,7,8-TeBDD was about one-third that
of an equimolar dose of 2,3,7,8-TeCDD.
2,3,7,8-TeBDD or 1,2,7,8-TeBDF administered to rats, by any
route, was distributed throughout the entire body, with major deposits
found in liver and adipose tissue, followed by skin and muscle. For
example, 3 days after single oral doses of 2,3,7,8-TeBDD (1 nmol/kg
body weight), the portions in these tissues amounted to 20%, 20%, 11%,
and 4%, respectively, whereas thymus and adrenals contained 0.03% and
0.4%, respectively, of the administered dose. The partitioning of
2,3,7,8-TeBDD between liver and adipose tissue of rats was found to be
influenced by dose, route of exposure, and time post-dosing. The
ratios of liver : fat concentrations measured under different
conditions ranged from 0.2 to 6.5 (range for single doses of
2,3,7,8-TeBDD in rats). No experimental data were available on the
transfer of PBDDs/PBDFs to offspring.
TetraBDD/BDF metabolites were detected in bile and faeces from
rats. They were mainly formed by aromatic hydroxylation and hydrolytic
debromination. The rate of metabolism (indirectly determined as the
rate of biliary excretion) differed between 2,3,7,8-TeBDD (about 7%)
and 1,2,7,8-TeBDF (about 50%). Three days after an intravenous dose of
2,3,7,8-TeBDD (1 nmol/kg body weight), 14% of the administered dose
was found as metabolites in the faeces of rats.
Elimination and excretion of 2,3,7,8-TeBDD were studied in rats
using oral, intravenous, intratracheal, and dermal routes of
administration. In all studies, the major route of elimination was
through the faeces, the eliminated radioactivity ranging from 2%
(dermal route) to 42% (oral route) of the administered dose (1 nmol
[3H]2,3,7,8- TeBDD/kg body weight) in faeces samples, and from 0.2 to
1% in urine samples. Similarly, in studies with 1,2,7,8-TeBDF in rats,
excretion was mainly through the faeces, only 2-3% of the intravenous,
oral, or dermal doses being excreted in urine. During the first days
following oral doses, unabsorbed material and biliary excretion
appeared to be the major sources of eliminated compound in faeces. The
portions of parent 2,3,7,8-TeBDD found in faeces of rats after
administration of 1 nmol 2,3,7,8-TeBDD/kg body weight were 53% (oral
route), 43% (intratracheal route), and 10-20% (intravenous route). A
few days after oral application of 2,3,7,8-TeBDD (1 nmol/kg body
weight), about 20% of the dose administered was eliminated as parent
compound.
Data on retention and turnover are available for some PBDDs/
PBDFs. The relative body burden of 2,3,7,8-TeBDD (and other congeners)
in rats depends on the route of exposure and on the dose administered,
reflecting differences in absorption. Half-lives were calculated for
several PBDDs/PXDDs and PBDFs in various tissues and faeces of rats.
They ranged between 1 day (1,2,7,8-TeBDF from body) and 99 days
(2,3,4,7,8-PeBDF from liver). The estimated half-lives of 17, 18, and
58 days for 2,3,7,8-TeBDD in liver, faeces, and adipose tissue,
respectively, were similar to those reported for 2,3,7,8-TeCDD in
liver and faeces, but higher (by a factor of >2) than those reported
for 2,3,7,8-TeCDD in adipose tissue. Despite differences in early
retention, half-lives of 2,3,7,8-TeBDF and
2,3,7,8-tetrachlorodibenzofuran (2,3,7,8-TeCDF or TCDF) in liver were
comparable.
As with PCDDs/PCDFs, half-lives calculated for humans are much
longer than those for rats. There are estimations of 3-11 years (mean:
5.9 years) for 2,3,7,8-TeBDD and of 1-2 years (mean: 1.5 years) for
2,3,7,8-TeBDF. The persistence of these compounds in humans was also
seen in the case of a chemist who had synthesized 2,3,7,8-TeBDD and
2,3,7,8-TeCDD in 1956. Thirty-five years after exposure, markedly
elevated levels of 2,3,7,8-TeBDD were found in his blood.
1.6 Effects on laboratory mammals and in vitro test systems
Most studies were concerned with the toxicity of 2,3,7,8-TeBDD,
but some information was also available on other PBDDs/PBDFs and
PXDDs/PXDFs.
2,3,7,8-TeBDD caused typical 2,3,7,8-TeCDD-like effects,
including wasting syndrome, thymus atrophy, and liver toxicity.
Additionally, liver damage described as peliosis hepatis, which has
not been reported after exposure of rats to 2,3,7,8-TeCDD, was
observed. The pattern of lesions (lethality, histopathology, liver and
thymus weights) found in guinea-pigs after a single exposure and in
rats after short-term exposure to 2,3,7,8-TeBDF was similar to that of
2,3,7,8-TeCDF.
2,3,7,8-TeBDD interacts with the endocrine system. In rats,
dose-related changes in circulating thyroid hormones and impairment of
spermatogenic activity have been observed.
The oral LD50 (28-day observation period) of 2,3,7,8-TeBDD in
Wistar rats was about 100 µg/kg body weight for females and about 300
µg/kg body weight for males. Oral LD50 values for 2,3,7,8- TeCDD
obtained from other studies ranged between 22 and >3000 µg/kg body
weight. Equimolar doses of 2,3,7,8-TeBDF and 2,3,7,8-TeCDF resulted in
comparable mortality rates in guinea-pigs. For example, 100% mortality
was seen after treatment with both 2,3,7,8-TeBDF (0.03 µmol/kg body
weight, 15.8 µg/kg body weight) and 2,3,7,8-TeCDF (0.03 µmol/kg body
weight, 10 µg/kg body weight). Pre-peliotic lesions and changes in
thyroid hormones were seen in rats after a single dose of 100 µg
2,3,7,8-TeBDD/kg body weight.
In Wistar rats administered 2,3,7,8-TeBDD orally for 13 weeks,
evidence for decreased spermatogenic activity, defective and necrotic
spermatocytes, signs of severe peliosis hepatis, and changes in
circulating thyroid hormones and organ weights were observed. The
no-observed-adverse-effect level (NOAEL) was 0.01 µg/kg body weight
per day.
2,3,7,8-TeBDF administered orally to Sprague-Dawley rats for 4
weeks caused dose-dependent growth retardation and histopathological
changes in liver and thymus. The NOAEL was 1 µg/kg body weight per
day.
Developmental toxicity of some 2,3,7,8-substituted PBDDs/ PBDFs
occurred in mice at subcutaneous and oral doses that produced no
maternal toxicity and no fetal mortality. The lowest-observed-effect
levels (LOELs) (in µg/kg body weight) for hydronephrosis and cleft
palate after a single oral exposure of pregnant mice were,
respectively, as follows: 3 and 48 for 2,3,7,8-TeBDD, 25 and 200 for
2,3,7,8-TeBDF, 400 and 2400 for 2,3,4,7,8-PeBDF, and 500 and 3000-4000
for 1,2,3,7,8-PeBDF. Compared on a molar basis, 2,3,7,8-TeBDD and
2,3,7,8-TeCDD were almost equipotent in induction of hydronephrosis.
Compared on a weight basis, generally the brominated isomers were
slightly less potent than the chlorinated ones in induction of
hydronephrosis and cleft palate. However, 2,3,7,8-TeBDF was more
active than 2,3,7,8-TeCDF.
No information was found on the mutagenicity of PBDDs/PBDFs or
related end-points.
No long-term toxicity and carcinogenicity studies with PBDDs/
PBDFs were available. 2,3,7,8-TeBDD tested positive in a cell
transformation assay using murine peritoneal macrophages. However, the
transforming potency of 2,3,7,8-TeBDD was seven times less than that
of 2,3,7,8-TeCDD. Later, tumours developed in nude mice after
subcutaneous injection of the resulting established cell lines.
A series of several PBDDs and PXDDs (tetra and penta) given
intraperitoneally to immature male Wistar rats caused body weight
losses 14 days after injection. On the basis of molar ED50 values,
the most toxic compounds tested were 2,3,7,8-TeBDD,
2-Br1-3,7,8-Cl3- DD, and 2,3-Br2-7,8-Cl2-DD (TBCDD), which are
substituted only in the four lateral positions. The relative potencies
of the other PBDDs examined followed the order 2,3,7,8- >
1,2,3,7,8- > 1,2,4,7,8- > 1,3,7,8-DD. In other experiments, there
were only slight differences in the ED50 values (on a molar basis)
for body weight loss, thymic atrophy, and hepatic enzyme induction
between 2,3,7,8-TeCDD and 2,3,7,8-TeBDD.
Thymic atrophy and other signs of immunotoxicity (e.g.
haematological parameters, alterations of certain lymphocyte
subpopulations) were seen with several PBDDs/PXDDs and 2,3,7,8-TeBDF
in the rat and with 2,3,7,8-TeBDD and TBCDD in the marmoset monkey
(Callithrix jacchus). It was concluded that, on a molar basis, the
potency of 2,3,7,8-TeBDD was comparable to that of 2,3,7,8-TeCDD in
rats and monkeys. For example, a significant effect on certain
lymphocyte subpopulations in monkeys was found after a single
subcutaneous dose of 30 ng 2,3,7,8-TeBDD/kg body weight versus 10 ng
2,3,7,8-TeCDD/kg body weight. Effects on immunotoxicity after
perinatal exposure to PBDDs/PBDFs have not been investigated.
After subchronic dosing of either 2,3,7,8-TeBDD or 2,3,7,8-TeCDD
by oral gavage in mice, there was a dose-dependent increase in total
hepatic porphyrins.
After single oral doses of 2,3,7,8-TeBDD and 2,3,7,8-TeCDD,
reductions in concentration and total amount of vitamin A were
observed in the liver of rats, with 2,3,7,8-TeBDD being slightly less
potent than 2,3,7,8-TeCDD (on a molar basis).
2,3,7,8-TeBDD and 2,3,7,8-TeBDF produced hyperkeratosis in the
rabbit ear assay at a dose of 100 µg/rabbit, but not at 10 µg/rabbit.
A no-observed-effect level (NOEL) for 2,3,7,8-TeCDD was 0.01
µg/rabbit.
Several tetra- (Br1Cl3DDs, Br2Cl2DDs) and penta- (Br1Cl4DD)
halogenated congeners with 2,3,7,8-substitution were found to have an
antiestrogenic potency similar to that of 2,3,7,8-TeCDD, as examined
in cultures of human breast cancer cells.
In rats, 2,3,7-tribromodibenzo- p-dioxin (2,3,7-triBDD/TrBDD)
depressed the disappearance of ouabain from plasma, its excretion into
bile, and bile flow to a slightly lesser extent than 2,3,7,8-TeCDD.
PBDDs/PBDFs and PXDDs/PXDFs are potent inducers of certain
cytochrome P-450 (CYP)-dependent microsomal enzymes. ED50 values of
0.8-1 nmol/kg body weight for CYP1A1 induction and about 0.2 nmol/kg
body weight for CYP1A2 induction in rat liver were estimated after
single oral doses of 2,3,7,8-TeBDD. CYP1A1 induction (arylhydrocarbon
hydroxylase [AHH] and/or ethoxyresorufin- O-deethylase [EROD]
induction) was observed in a variety of species and tissues
in vivo and in rat cell cultures in vitro. A lot of different
congeners were found to be active, as well as pyrolysates from certain
flame retardants. Generally, enzyme induction proceeded
dose-dependently at non-toxic concentrations, started soon after
exposure, and was long-lasting. It was measurable at exposures as low
as the pmol range. The induction potency varied over several orders of
magnitude for different congeners, depending on their chemical
structure. The most potent inducers were TCDD, TBDD, and TBCDD.
Compared (on a molar basis) with their chlorinated analogues, the
PBDDs and PXDDs had more or less similar potency. In contrast to TCDD,
whose relative induction potency was independent of the tissue
examined, TBDD was five times more potent at inducing EROD activity in
the liver than in skin and lung following subchronic exposure of mice.
The ranking order for induction of EROD activity in marmoset monkeys
was TCDD > 2,3,4,7,8-pentachlorodibenzofuran
(2,3,4,7,8-pentaCDF/PeCDF) > 2,3,4,7,8-PeBDF when enzyme activities
were compared with the hepatic concentrations. In vitro tests with
rat cell cultures resulted in similar molar EC50 values of AHH and
EROD induction potencies between corresponding PXDFs and PCDFs.
PBDDs/PBDFs are believed to share a common mechanism of action
with PCDDs/PCDFs and other related halogenated aromatic hydrocarbons
(Ah). Binding to the cytosolic Ah receptor, which plays a central role
in mediating 2,3,7,8-TeCDD-like toxicity, was confirmed for several
PBDDs and PXDDs/PXDFs. Their receptor-binding affinities varied by
several orders of magnitude but were comparable to those of their
chlorinated analogues.
1.7 Effects on humans
There are no data on the exposure of humans to PBDDs/PBDFs or on
their effects on the health of the general population.
Two cases of acute health problems due to 2,3,7,8-TeBDD/ TeCDD
exposure have been reported, with symptoms including chloracne.
In another study, male personnel of a chemical plant with
documented exposure to PBDDs/PBDFs originating from the use of
brominated flame retardants (OBDE and DBDE) were subjected to
immunological and additional clinical laboratory tests. Although there
were indications of minor changes in immunological parameters, the
overall evaluation of their health status did not reveal an impact of
2,3,7,8-TeBDD/TeBDF body burden on the immune system.
There are no reports on cancer mortality caused by PBDDs/ PBDFs.
1.8 Effects on other organisms in the laboratory and field
There is only limited information on the effects of PBDDs/PBDFs
on microorganisms, plants, or invertebrate or vertebrate wildlife
species.
Using the rainbow trout (Oncorhynchus mykiss) sac fry early
life stage mortality bioassay, a series of PBDD/PBDF congeners were
tested and found to be active. This bioassay also demonstrated that
for both PBDDs and PBDFs, there was a decreased potency with increased
bromine substitution. Both 2,3,7,8-TeBDD and 2,3,7,8-TeBDF were more
potent than their chlorinated analogues.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
2.1 Identity
PHDDs/PHDFs are almost planar tricyclic aromatic compounds. There
are eight positions on both the dibenzo- p-dioxin and the
dibenzofuran molecules where halogen substitution can occur. The
positions are numbered as shown in Fig. 1 for PBDDs and PBDFs.
Each series consists of eight homologous groups (groups of
congeners having the same number of bromine atoms), and in each group
there are different numbers of isomers (see Table 1). Theoretically,
75 PBDDs and 135 PBDFs are possible, as well as a large number of
mixed halogenated congeners -- 1550 PXDDs and 3050 PXDFs (Buser,
1987a). There are 7 2,3,7,8-substituted PBDDs and 10
2,3,7,8-substituted PBDFs (see Table 2), as well as 337 possible
2,3,7,8-substituted PXDDs and 647 possible 2,3,7,8-substituted PXDFs
(Ballschmiter & Bacher, 1996). PCDDs/PCDFs are discussed in a separate
Environmental Health Criteria monograph (WHO, 1989).
Because of the complexity of the analytical procedures (see
section 2.4), it has been possible to characterize only a small number
of PBDDs/PBDFs and PXDDs/PXDFs. Tables 3 and 4 show the Chemical
Abstracts Service (CAS) numbers that have been allocated to some of
these compounds.
2.2 Physical and chemical properties
2.2.1 Appearance, melting and boiling points, water solubility, vapour
pressure, octanol/water partition coefficient, and sorption coefficient
Experimental data on the physical and chemical properties of
PBDDs/PBDFs are scarce (see Table 5). In many cases, only predicted
values are available. It should be noted that for PCDDs, experimental
data are often lower than the calculated values (Shiu et al., 1988;
Fiedler & Schramm, 1990). This is also to be expected for the
brominated and for the mixed halogenated compounds (Fiedler & Schramm,
1990). Measured values for the aqueous solubility of PCDDs decrease
dramatically with increase in chlorine substitution and temperature
(Shiu et al., 1988).
PBDDs/PBDFs have higher molecular weights than their chlorinated
analogues, high melting points, and low water solubilities, but they
are generally soluble in fats, oils, and organic solvents (see Table
5). PBDDs/PBDFs have, like their chlorinated analogues, very low
vapour pressures, and at ambient temperatures they are mostly found
bound to particles. For the lower substituted compounds, PBDDs/PBDFs
have higher calculated p Kow values than the chlorinated congeners
(Fiedler & Schramm, 1990) and are therefore more lipophilic.
Table 1. Number of isomers, elemental composition, and molecular weight for PBDDs/PBDFs
Compound Number of isomers Elemental Molecular
Total 2,3,7,8-Substituted composition weight
MoBDD 2 - C12H7O2Br 263.1
DiBDD 10 - C12H6O2Br2 342.0
TrBDD 14 - C12H5O2Br3 420.9
TeBDD 22 1 C12H4O2Br4 499.8
PeBDD 14 1 C12H3O2Br5 578.7
HxBDD 10 3 C12H2O2Br6 657.6
HpBDD 2 1 C12HO2Br7 736.5
OcBDD 1 1 C12O2Br8 815.4
MoBDF 4 - C12H7OBr 247.1
DiBDF 16 - C12H6OBr2 326.0
TrBDF 28 - C12H5OBr3 404.9
TeBDF 38 1 C12H4OBr4 483.8
PeBDF 28 2 C12H3OBr5 562.7
HxBDF 16 4 C12H2OBr6 641.6
HpBDF 4 2 C12HOBr7 720.5
OcBDF 1 1 C12OBr8 799.4
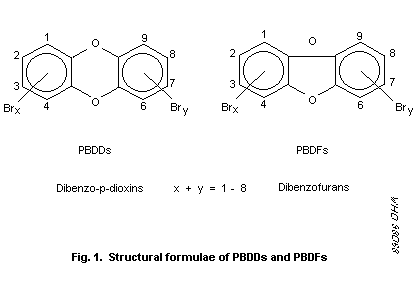 Table 2. PBDDs/PBDFs brominated at the
2,3,7,8-positions
PBDD congenera PBDF congenera
2,3,7,8-TeBDD* 2,3,7,8-TeBDF*
1,2,3,7,8-PeBDD* 1,2,3,7,8-PeBDF*
2,3,4,7,8-PeBDF*
1,2,3,4,7,8-HxBDD* 1,2,3,4,7,8-HxBDF
1,2,3,6,7,8-HxBDD* 1,2,3,6,7,8-HxBDF
1,2,3,7,8,9-HxBDD* 1,2,3,7,8,9-HxBDF
2,3,4,6,7,8-HxBDF
1,2,3,4,6,7,8-HpBDD 1,2,3,4,6,7,8-HpBDF
1,2,3,4,7,8,9-HpBDF
OcBDD OcBDF
a The congeners marked with an asterisk (*)
are cited in the German Dioxin Directive (1994)
(see Appendix I).
2.2.2 Stability of PBDDs/PBDFs
2.2.2.1 Photolysis
In the presence of laboratory light or sunlight, photolysis
occurs at a more rapid rate for PBDDs/PBDFs than for PCDDs/PCDFs
(Buser, 1988; Chatkittikunwong & Creaser, 1994a; for details, see
section 4.2.1). This should be taken into consideration when analyses
of these compounds are carried out (see sections 2.4.1 and 4.2.1).
Photolysis on quartz surfaces under sunlight is a much slower
process than photolysis in organic solvents (Buser, 1988). PBDDs/
PBDFs adsorbed on incinerator soot particles remained relatively
stable and degraded only slowly during a 6-h period (Lutes et al.,
1990, 1992a,b). Studies of PBDDs in soil showed that for the same
congeners, the half-lives in this matrix are four times longer than in
solution (Chatkittikunwong & Creaser, 1994a).
Under conditions of ambient temperature and protection from
light, there is no appreciable (>1%) degradation of crystalline
PBDDs/ PBDFs and no significant change (0.6%, with the exception of
octaBDD [9.7%]) in standard solution (solvent: n-nonane)
concentrations over a period of 3 years (Re et al., 1995).
Table 3. CAS numbers for some PBDDs/PBDFs
PBDD congenera CAS number PBDF congenera CAS number
Br1DD 103456-34-4 Br1DF 103456-35-5
1-Br1DD 105908-71-2 2-Br1DF 86-76-0
2-Br1DD 105906-36-3
Br2DD 103456-37-7 Br2DF 103456-40-2
1,6-Br2DD 91371-14-1 2,7-Br2DF 65489-80-7
2,7-Br2DD 39073-07-9 2,8-Br2DF 10016-52-1
2,8-Br2DD 105836-96-2
Br3DD 103456-38-8 Br3DF 103456-41-3
1,2,8-Br3DF 84761-81-9
2,3,8-Br3DF 84761-82-0
Br4DD 103456-39-9 Br4DF 106340-44-7
1,2,3,4-Br4DD 104549-41-9 1,2,7,8-Br4DF 84761-80-8
2,3,7,8-Br4DD 50585-41-6 2,3,7,8-Br4DF 67733-57-7
Br5DD 103456-36-6 Br5DF 68795-14-2
1,2,3,7,8-Br5DD 109333-34-8 1,2,3,7,8-Br5DF 107555-93-1
2,3,4,6,7-Br5DF 124388-77-8
2,3,4,7,8-Br5DF 131166-92-2
Br6DD 103456-42-4 Br6DF 103456-33-3
1,2,3,4,7,8-Br6DD 110999-44-5 1,2,3,4,6,7-Br6DF 124388-78-9
1,2,3,6,7,8-Br6DD 110999-45-6 1,2,3,6,7,8-Br6DF 107555-94-2
1,2,3,7,8,9-Br6DD 110999-46-7
Br7DD 103456-43-5 Br7DF 62994-32-5
1,2,3,4,6,7,8-Br7DF 107555-95-3
Br8DD 2170-45-8 Br8DF 103582-29-2
a The homologue groups are underlined.
2.2.2.2 Thermolytic degradation of PBDDs/PBDFs
As discussed in chapter 3, the temperature of formation and
destruction of PBDDs/PBDFs depends on several conditions, such as
residence time, the presence/absence of oxygen, polymers, and
additives such as Sb2O3, as well as the efficiency of the apparatus
used for the thermal degradation. In laboratory experiments on the
thermolysis of polybrominated flame retardants (see section 3.4; Table
11), the PBDDs/PBDFs formed were destroyed at 800°C in an air
atmosphere after a 2.0-second residence time (Striebich et al., 1991).
PBDDs/PBDFs formed at 600°C from the thermolysis of plastics
containing DBDE or PBDE were no longer detectable at 800°C (Lahaniatis
et al., 1991). However, Thoma et al. (1987b) found that PBDDs/PBDFs
are still formed at 900°C. There is thus no definitive information on
the temperature needed to destroy PBDDs/PBDFs.
Table 4. CAS numbers for some PXDDs/PXDFs
PXDD congenera CAS number PXDF congenera CAS number
Br1Cl1DD 109007-09-02 Br1Cl1DF 109264-70-2
Br1Cl2DD 107227-59-8 Br1Cl2DF 107227-60-1
Br1Cl3DD 107227-75-8 Br1Cl3DF 107227-56-5
8-Br1-2,3,4-Cl3DF n.g.b
Br1Cl4DD 109264-61-1 Br1Cl4DF 109302-36-5
1-Br1-2,3,7,8-Cl4DF 104549-43-1
4-Br1-2,3,7,8-Cl4DF 115656-08-1
Br1Cl5DD 109264-65-5 Br1Cl5DF 107103-81-1
Br1Cl6DD 109264-67-7 Br1Cl6DF 107207-47-6
Br1Cl7DD 109264-69-9 Br1Cl7DF 109302-40-1
Br2Cl1DD 107227-58-7 Br2Cl1DF 107227-57-6
Br2Cl2DD 107227-74-7 Br2Cl2DF 107227-55-4
Br2Cl3DD 109031-99-4 Br2Cl3DF 107227-53-2
Br2Cl4DD 109264-62-2 Br2Cl4DF 107207-48-7
Br2Cl5DD 109264-66-6 Br2Cl5DF 107207-45-4
Br2Cl6DD 109264-68-8 Br2Cl6DF 109302-39-8
Br3Cl1DD n.g.b Br3Cl1DF 107227-54-3
Br3Cl2DD n.g.b Br3Cl2DF 107227-52-1
Br3Cl3DD n.g.b Br3Cl3DF 107207-46-5
Br3Cl4DD n.g.b Br3Cl4DF 107207-42-1
Br3Cl5DD n.g.b Br3Cl5DF n.g.b
Br4Cl1DD n.g.b Br4Cl1DF 107227-51-0
Br4Cl2DD n.g.b Br4Cl2DF 107207-44-3
1,2,3,4-Br4-7,8-Cl2DD 134974-39-3
Br4Cl3DD n.g.b Br4Cl3DF 107207-41-0
Br4Cl4DD n.g.b Br4Cl4DF n.g.b
1,2,3,4-Br4-6,7,8,9-Cl4DD 124728-12-7
Br5ClxDD n.g.b Br5Cl1DF 107207-49-8
other Br5ClxDF n.g.b
Br6Cl1DD 107207-38-8 Br6Cl1DF n.g.b
Br6Cl2DD n.g.b Br6Cl2DF 107207-36-3
1,2,4,6,7,9-Br6-3,8-Cl2DD 2170-44-7
Br7Cl1DD n.g.b Br7Cl1DF 107207-37-4
a The homologue groups are underlined.
b n.g. = CAS numbers not found (probably not yet allocated).
Table 5. Physical and chemical properties of some PBDDs/PBDFs
Compound Appearance Melting point Boiling point Water Vapour Octanol/water Sorption
(°C) (observed) (°C) solubility pressure partition coefficient
(predicted) [log S] [log P] coefficient [log Koc]
(mol/litre) (Pa at 25°C) [log Kow] (mol/litre)
(predicted) (predicted) (predicted) (predicted)
PBDDs
1-MoBDD white needles 104-106a 338.2b 3.5 × 10-3b
2-MoBDD n.g.c 93-94.5a 338.2b -6.12d 4.0 × 10-3b 5.62d 4.39d
(90-92)a
1,6-DiBDD 207e 375b 1.5 × 10-4b
2,3-DiBDD n.g.c 157.2-158f 375b -6.90d 1.6 × 10-4b 6.25d 4.74d
2,7-DiBDD 174-176a 375b 1.5 × 10-4b
193-194e
2,8-DiBDD 149.5-151a 375b 1.7 × 10-4b
(145-150)a
3,7-DiBDD -7.24d 6.53d 4.89d
-7.99d 7.14d 5.22d
1,2,3,4-TeBDD 6 × 10-7g
2,3,7,8-TeBDD white granules 334-336a,f 438.3b -8.72d 6.4 × 10-7b 7.74d 5.54d
6.50h
7.73i
1,2,3,7,8-PeBDD -9.45d 8.32d 5.87d
1,2,3,4,6,7, -10.89d 9.50d 6.50d
8-HpBDD
OcBDD 376j 523.2b -11.69d 4.1 × 10-11b 10.08d 6.82d
9.3 × 10-16g
PBDFs
monoBDF 2.89-3.26k
2-MoBDF -5.42d 5.05d 4.08d
diBDF 4.35-4.46k 5.58-6.09k
2,7-DiBDF -6.25d 5.95d 4.47d
triBDF 5.36-5.47k 6.49-6.79k
Table 5. (Continued)
Compound Appearance Melting point Boiling point Water Vapour Octanol/water Sorption
(°C) (observed) (°C) solubility pressure partition coefficient
(predicted) [log S] [log P] coefficient [log Koc]
(mol/litre) (Pa at 25°C) [log Kow] (mol/litre)
(predicted) (predicted) (predicted) (predicted)
1,2,8-TrBDF + colourless 144-148l
2,3,8-TrBDF prismsl
2,3,7-TrBDF -7.26d 6.55d 4.90d
tetraBDF 6.35-6.41k 7.72-8.72k
1,2,7,8-TeBDF colourless needlesl 240.5-242l 6.20h
2,3,7,8-TeBDF colourless needlesl 301-302l -7.99d 7.14d 5.22d
5.98h
2,3,4,6-TeBDF -7.99d 7.14d 5.22d
pentaBDF 7.25-7.45k
1,2,3,7,8-PeBDF 7.04h
7.56i
2,3,4,7,8-PeBDF -8.71d 7.73d 5.54d
hexaBDF 8.34k
2,3,4,6,7, -9.43d 8.31d 5.86d
8-HxBDF
1,2,3,4,6,7, 9 × 10-11g
8-HpBDF
a From Gilman & Dietrich (1957). Melting points in parentheses are values from other sources
reported by these authors.
b From Rordorf (1987).
c n.g. = not given.
d Predicted; from Fiedler & Schramm (1990). Sorption coefficient [log Koc] = distribution
coefficient between compound adsorbed to soil organic carbon and the compound in solution.
e From Tomita et al. (1959)
f From Kende & Wade (1973).
g From Rordorf et al. (1990).
h From Jackson et al. (1993), estimated from measured reverse-phase high-performance liquid
chromatography (HPLC) retention times.
Table 5. (Continued)
i From Jackson et al. (1993), calculated.
j From Denivelle et al. (1960).
k From Watanabe & Tatsukawa (1990).
l From Tashiro & Yoshiya (1982).
2.2.3 Chemical reactions
Aromatic carbon-bromine bonds are generally weaker than similar
carbon-chlorine bonds, and, consequently, bromine can be substituted
more easily. In general, the reductive substitution of halogens in
aromatic structures becomes easier as the halogen atoms' size
increases (Wania & Lenoir, 1990).
In the presence of excess chlorine, bromine can be substituted by
chlorine to give PXDDs/PXDFs -- for example, under conditions such as
those present in municipal incinerators (Wilken et al., 1990; Luijk et
al., 1992a).
Wania & Lenoir (1990) investigated the effect of heating
1,2,3,4-TeBDD (20 µg) in the presence of copper (1 g) at 100, 120,
150, or 210°C for a duration of 30 seconds to 1 h. With increasing
heating time, the spectrum of PBDD shifted from tetraBDD to lower
brominated congeners, and the sum of the quantities decreased. The
reaction rate increased with increasing temperature. At 210°C for 30
min, all PBDDs had disappeared, but the dibenzo- p-dioxin ring
structure remained intact.
In a further experiment, it was shown that the presence of water
(10 or 100 µg/litre) considerably increased the yield of debrominated
products.
On heating monoBDD and 1 g copper to 150°C, the debrominated
product dibenzo- p-dioxin and dimers of this compound were
identified. On heating hexaBDD and octaBDD to 150°C in the presence of
copper, it was found that appreciable quantities of the original
compounds could still be detected after 1 h. The reaction was
considerably slower than in the comparable experiment with tetraBDD.
The debromination reactions proceed faster than the respective
dechlorination reactions with PCDDs (Hagenmaier et al., 1987).
2.3 Conversion factors
At 25°C and 101.3 kPa, conversion factors for converting airborne
concentrations from ppm to mg/m3 for a particular PBDD/ PBDF congener
can be calculated from the relative molecular mass (RMM):
1 ppm = RMM/24.45 mg/m3
1 mg/m3 = 24.45/RMM ppm
For example, for monoBDF, 1 ppm = 247.1/24.45 = 10.1 mg/m3.
Similarly, 1 mg/m3 = 0.099 ppm.
2.4 Analytical methods
2.4.1 General aspects
Some PBDD/PBDF congeners are highly toxic (see chapter 7). Using
the principles of Good Laboratory Practice, great precautions should
be taken in handling the samples. Additionally, precautions must be
taken owing to the photochemical instability of the brominated and
mixed brominated/chlorinated congeners (see also sections 2.2.2.1 and
4.2.1). The use of amber-coloured glassware and filters on lamps and
windows is mandatory.
Sampling, sample treatment (extraction and clean-up), and
analysis for PBDDs/PBDFs and PXDDs/PXDFs follow largely the methods
and techniques currently used for PCDDs/PCDFs (Donnelly et al.,
1989a,b, 1990; US EPA, 1990, 1992 [Methods 1613 and 8290]; Maier et
al., 1994; Ballschmiter & Bacher, 1996). The large number of isomers
in some homologous groups (see Table 1) makes the separation and
quantification of individual congeners difficult. Using highly
selective, specific, and sensitive analytical methods,
2,3,7,8-substituted PBDDs/PBDFs can be detected, although co-elution
with other isomers cannot be excluded.
Accurate identification of specific congeners is limited by the
small number of reference standards available. The large number of
PXDDs/PXDFs (see section 2.1) makes it impossible to identify and
quantify individual congeners. Homologue groups, however, can be
analysed semi-quantitatively. Major steps in the analytical procedures
are as follows:
- spiking of the homologous sample with labelled standards
- use of matrix-specific extraction procedures (pretreatment of the
sample before extraction where necessary)
- clean-up by column chromatography, liquid-liquid extraction, HPLC
- concentration of the eluate (addition of a high-boiling solvent
as a keeper where necessary); addition of a recovery standard
- analysis by GC/MS.
Many of the analytical methods for PCDDs/PCDFs have been
validated in the past decade in interlaboratory studies organized,
among others, by: the World Health Organization (WHO) for biological
samples (WHO/EURO, 1989, 1991; Stephens et al., 1992; WHO/ECEH, 1996);
the European Community Bureau of Reference for environmental samples,
including fly ash (Maier et al., 1994), and for milk powder (Schimmel
et al., 1994; Tuinstra et al., 1996); and the European Committee for
Standardization for emissions by stationary sources (Bröker, 1996). To
avoid systematic errors in the individual steps from sampling to
analysis, recoveries must be controlled by the addition of appropriate
stable isotope labelled standards before extraction of the sample,
clean-up, and final quantification.
2.4.2 Sampling and extraction
The sampling procedures recommended for PCDDs/PCDFs (WHO, 1989
and citations in second paragraph of section 2.4.1) also apply for the
brominated congeners.
2.4.2.1 Ambient air, airborne dust, automobile exhaust, flue gas, and
products of thermolysis
Experience from PCDD/PCDF analysis has shown critical or weak
points in current gas sampling (ambient air, indoor air, exhaust gas)
techniques. Requirements are as follows:
- representativeness of samples; special attention must be given to
isokinetic sampling of particles in emission samples
- stability of the sample on the sampling medium during the
sampling period; the filter should be kept below 120°C and
protected from light
- recovery of the analytes from the sampling train (as checked by
appropriate spiked standards)
- use of clean equipment to avoid contamination of the sample (as
checked by appropriate blanks)
- complete trapping of gas and particle phases (aerosols) to avoid
sample losses.
Quartz fibre filters with polyurethane foam plugs have been used
to sample ambient air up to 1000 m3 (Wagel et al., 1989; Päpke et
al., 1990; Harless et al., 1992; Watanabe et al., 1992). (Note: There
may be interferences from brominated organic aromatic flame retardants
in polyurethane foam.)
Haglund et al. (1988) described a method to collect both the
particulate phase (using a Teflon-coated filter) and the gas phase
(cryotechnique) in vehicle exhaust.
Hutzinger et al. (1990) used the so-called Grimmer apparatus to
sample automobile exhaust: the sampling train consists of a large
glass condenser and a non-impregnated fibreglass filter. Typically, 50
m3 of automobile exhaust were taken per sample; the temperature at
the muffler outlet was kept below 50°C. The experiments were carried
out as stationary motor tests. The total emissions of the motor were
sucked through the sampling train by a pressure-controlled blower.
Emissions from a laboratory furnace experiment were collected by
a sampling train, including a high-efficiency quartz fibre filter (to
collect organic-laden particulate material) and an XAD-2 resin (to
adsorb semivolatile organic compounds) (Riggs et al., 1992).
Thermolytic products have been collected as condensate in a
quartz-wool-filled condenser tube (Neupert et al., 1989b).
2.4.2.2 Water and aqueous samples
Analysis of water samples should follow a different approach. If
the samples are free of particles, a normal liquid-liquid extraction
is sufficient. If, however, the samples contain particles, both the
particles and the water phase should be extracted separately -- the
solids by methods recommended for solids, the water phase as described
above.
2.4.2.3 Environmental samples: soil, sediment, and sewage sludge
For environmental samples, problems arise in obtaining a
representative sample. For soil sampling, a method was described by
Fortunati et al. (1994).
Prior to the extraction, appropriate measures should be taken to
ensure that PHDDs/PHDFs in the sample material are fully accessible to
the extraction solvent. In a number of applications, this includes a
digestion of the sample (solids) and/or the complete removal of water
(wet solid samples) prior to extraction clean-up; a chemical
destruction of non-persistent chemicals can be useful by incubating
the sample in neat sulfuric acid (H2SO4). PHDDs/PHDFs are shown to
be stable. Treatment with (strong) bases should be avoided, as
PHDDs/PHDFs may degrade.
For the study of sewage samples, Hagenmaier et al. (1992) dried,
powdered, and extracted the samples with toluene for 18 h. After
concentration, the extracts were treated with concentrated sulfuric
acid.
Proven digestion and water removal methods are treatment with
hydrochloric acid (10% HCl) and Dean Stark collector (US EPA, 1990;
Rappe et al., 1996), respectively. Sediments should be treated with
copper powder to eliminate sulfur (Kjeller et al., 1993).
2.4.2.4 Flame retardants, polymers, fly ash samples, dust, soot, and
fire residues
In general, the analysis of plastics is performed by dissolving
the polymer in a suitable solvent. Non-dissolvable plastics should be
powdered and Soxhlet-extracted.
Dibromomethane was used to dissolve samples of PBDE (Tondeur et
al., 1990). Ranken et al. (1994) noted that this solvent must first be
specially purified before use to remove the colour, which caused
quantitative interferences in the mass spectrometer. TBBPA can be
dissolved in methanol (Tondeur et al., 1990; Ranken et al., 1994).
PBT resins (extruded beads/powder) were extracted best with
1,1,1-trichloroethane/phenol followed by water partitioning of phenol;
powdered high-impact polystyrene (HIPS) samples by toluene/reflux; and
powdered ABS samples by dichloromethane (Donnelly et al., 1989a).
Kieper (1996) used toluene for Soxhlet extraction from samples of
flame-retarded polymers: DBDE (with polystyrene/polystyrenebutadiene),
1,2-bis(tribromophenoxy)ethane (with polystyrene), TBBPA-carbonate
oligomer (with PBT), dibromostyrene, and tribromostyrene (both with
polyamide 66).
Samples of burnt plastic, PBT material, ash/slag, and soil were
Soxhlet-extracted with dichloromethane (Neupert & Pump, 1992). Clausen
et al. (1987) used Soxhlet extraction with hexane. ABS was extracted
under reflux with methylene chloride (Donnelly et al., 1990).
Dry fly ash was treated with 10% HCl, dried, and neutralized.
After further drying, the sample was Soxhlet-extracted with toluene
(Hosseinpour et al., 1989). Similar procedures were used by Tong et
al. (1991) and Huang et al. (1992a,b).
PBDDs/PBDFs from dust samples and smoke condensate were
Soxhlet-extracted with toluene (UBA, 1992; Funcke et al., 1995).
Samples from fire residues were ground and extracted with toluene;
wipe samples of soot were extracted with hexane (Harms et al., 1995).
2.4.2.5 Biological matrices: human milk, blood/plasma, tissues, and
fish samples
For biological samples, most appropriate extraction methods are
those giving the highest yields (or recovery) for the lipids in the
sample (i.e. milk, blood, tissue).
Neupert et al. (1989a,b) quantified PBDDs/PBDFs in rat liver,
adipose tissues, and faeces. After homogenization with sodium sulfate,
extraction was performed on a multiple-layer column using
dichloromethane/hexane.
Fish samples were ground with sodium sulfate and homogenized.
Methanol and sodium oxalate were added to milk samples (De Jong et
al., 1992). Diethyl ether and hexane were used to extract PHDDs/ PHDFs
from the fat fraction of the milk and fish samples.
For PBDD/PBDF determination, samples of human adipose tissue were
homogenized, extracted with dichloromethane, dried with sodium
sulfate, and solvent-exchanged into hexane (Cramer et al., 1990a).
This method was also used by Zober et al. (1992).
Fat removal can be performed utilizing a semipermeable membrane
technique (Bergqvist et al., 1993), which enables larger amounts of
fat (sample size up to 200 g) to be eliminated from the sample matrix
(>95%) and improved detection limits.
Lyophilization has been used successfully in the analysis of TBDD
in biological matrices such as rat livers or marmoset monkey tissues
(Schulz-Schalge et al., 1991a,b; Schulz et al., 1993; Nagao et al.,
1995/96).
2.4.3 Sample clean-up
Sample clean-up is carried out to remove those materials that
might otherwise interfere with the analysis. A variety of liquid
chromatography separations have been used, including silica, florisil,
alumina, and various combinations of these columns. Usually an
acid/base wash followed by alumina column chromatography is used to
remove the bulk of interferences, and carbon column chromatography is
used to remove residual interferences (Donnelly et al., 1986, 1987,
1989b).
Where PBDEs are likely contaminants, a modification of separation
techniques is necessary. Alumina columns are ineffective in separating
PBDFs from PBDEs. Carbon columns were found to be more effective, but
the higher brominated PBDFs could be removed from the column only by
back-flushing with an aromatic solvent (Donnelly et al., 1987; Hileman
et al., 1989). Bonilla et al. (1990) introduced an HPLC step to the
clean-up procedure. The sample was passed through an AX21 carbon
column. The column was washed in the forward direction with
dichloromethane/cyclohexane and dichloromethane/methanol/benzene and
back-flushed with toluene. This procedure decreased the PBDE
concentration in the final sample by six orders of magnitude.
Depending on the aim of the analysis (general surveying or
specific search and quantification of 2,3,7,8-substituted congeners),
a number of 13C standards are required to be added at several stages
during sampling and analysis.
In the WHO interlaboratory calibration study for the analysis of
PCDDs/PCDFs in human blood and milk, the basic clean-up/ separation
methods used by some laboratories were activated carbon as the primary
PCDD/PCDF isolation step followed by alumina; the remaining
laboratories used other procedures, mostly H2SO4 followed by
alumina. Nearly all methods used a step involving some type of carbon
chromatography (Stephens et al., 1992).
2.4.4 Separation
GC is used for the separation of PBDDs/PBDFs. PBDDs/PBDFs have
much higher retention times and elution temperatures (30-40°C higher)
than their chlorinated analogues (Buser, 1991). The higher brominated
congeners have extremely long retention times, so non-polar (SE 54),
medium-length (up to 25-m) columns are generally used. Such a column
is suitable for separating the PBDD and PBDF homologues. Elution
temperatures on a 25-m SE 54 high-resolution gas chromatography (HRGC)
column range from 184-188°C for monoBDDs/BDFs to 260-273°C for
pentaBDDs/BDFs. Hexa-, hepta-, and octa-homologues elute during the
isothermal phase at 280°C (Buser, 1986a, 1991). Cross-linked columns
allow higher temperatures, reducing analysis times (Hutzinger et al.,
1990). Table 6 gives retention indices (RIs) of some PBDDs/PBDFs as
well as PBDEs, which are possible contaminants (Donnelly et al.,
1987). A 30-m DB-5 or DB-5MS fused capillary column has been found to
be quite useful for the GC/MS analysis of tetra- through
hepta-substituted PBDDs/PBDFs (Ranken et al., 1994).
Table 6. Retention indices (RIs) of PBDDs,
PBDFs, and PBDEsa,b
Congener RI
PBDDs
2-MoBDD 1868
2,8-DiBDD 2174
1,3,7-TrBDD 2423
2,3,7-TrBDD 2475
2,3,7,8-TeBDD 2800
1,2,7,8-TeBDD 2811
1,2,4,7,8-PeBDD 3072
1,2,3,7,8-PeBDD 3145
1,2,3,4,7,8-HxBDD 3412
1,2,3,6,7,8-HxBDD 3475
1,2,3,7,8,9-HxBDD 3798
1,2,3,4,6,7,8-HpBDD 3763
OcBDD 4219
PBDFs
2-MoBDF 1834
2,8-DiBDF 2133
1,2,8-TrBDF 2416
2,3,8-TrBDF 2433
1,2,7,8-TeBDF 2740
2,3,7,8-TeBDF 2791
1,2,3,7,8-PeBDF 3103
1,2,3,6,7,8-HxBDF 3479
1,2,3,4,6,7,8-HpBDF 3806
OcBDF 4231
PBDEs
HexaBDE 2888
HexaBDE 3004
HexaBDE 3015
HexaBDE 3030
HexaBDE 3051
HexaBDE 3095
HexaBDE 3286
HexaBDE 3314
HexaBDE 3369
HexaBDE 3411
OctaBDE 3525
OctaBDE 3577
OctaBDE 3601
OctaBDE 3627
Table 6. (Continued)
Congener RI
OctaBDE 3654
OctaBDE 3737
OctaBDE 3786
NonaBDE 3951
NonaBDE 4003
DecaBDE 4310
a From Donnelly et al. (1987).
b Chromatographic conditions:
30 m x 0.32 mm DB-5 GC column;
He carrier gas at ca. 7 psi head
pressure; temperature programmed
from 10 min at 170-320 °C at
8 °C/min.
The elution of PCDDs/PCDFs and PBDDs/PBDFs occurs in the order of
the molecular weights. PXDDs/PXDFs elute between the corresponding
chloro- and bromo-analogues (Buser, 1987a). However, mixed congeners
containing bromine elute earlier than expected on a molecular weight
basis, relative to the chloro-compounds (e.g. BrCl5 before Cl7)
(Buser, 1991). (For isomer-specific analysis, columns of different
polarity should be used.)
Owing to the lack of PBDD/PBDF (and PXDD/PXDF) standards, it has
not been possible to identify all congeners. Instead, a combination of
MS and GC RI identification has to be used for the analysis of
2,3,7,8-substituted PBDDs/PBDFs, PCDDs/PCDFs, and PXDDs/ PXDFs. An RI
model has been developed to predict the GC retention times for 1700 of
these compounds (Donnelly & Sovocool, 1991; Donnelly et al., 1991a,b).
2.4.5 Detection, quantification, and confirmation of PBDDs/PBDFs
by MS techniques
Detection, quantification, and confirmation are usually performed
by MS, as only this technique shows sufficient selectivity to
distinguish PBDDs/PBDFs from other halogenated compounds (e.g. PBDEs)
that are present in the sample. MS allows the determination of the
number and type of halogens present from characteristic isotope
distribution patterns, but it does not give any information about
which isomer is present (Buser, 1991). (Among the MS methods,
high-resolution mass spectrometry [HRMS] is preferred owing to higher
selectivity, and the tandem MS or negative ion chemical ionization
[NCI] techniques are a useful screening method because of the
diagnostic Br [79/81] fragment.)
Donnelly et al. (1987) and Sovocool et al. (1987) developed and
refined US Environmental Protection Agency (EPA) Method 8280 to
measure PBDFs/PBDDs by GC/MS. Significant features of the mass spectra
reported by these authors include the sequential losses of Br*, COBr*,
(Br*+COBr*), and (2Br*+COBr*). PBDEs may interfere with the
determination of PBDFs (and PBDDs), as the (M-2Br)+* fragment has
the same m/z composition cluster as that of a PBDF with two fewer
bromines; additional fragmentation mimics the PBDF containing two
fewer bromines (Donnelly et al., 1987). This potential for co-elution
(see Tables 6 and 7) must be considered in the evaluation of reports
of PBDF (and PBDD) formation where PBDE interference is likely.
Confirmation criteria for the detection and quantification of
PBDDs/PBDFs have been proposed (Donnelly et al., 1987):
* The retention time/RI must be correct for that analyte (standards
are needed).
* Recovery of the "surrogate" standard should be in the 40-120%
range.
* All m/z monitored for a given analyte must maximize
simultaneously ± 1 second, with a signal to noise ratio greater
than or equal to 2.5 for each. The M+* cluster is relatively
intense for all congeners. For confirmation, two additional ions
(m/z) should be monitored in electron impact-selective ion
monitoring-mass spectrometry (EI-SIM-MS).
* The ratio between the two ions of the M+* cluster must be
within 20% (relative) of the theoretical.
* When monitoring for PBDFs, the absence of PBDE should be
demonstrated (see Table 7).
The identification and determination of positional isomers are
very complex. Complementary methods based on other instrumental
techniques such as GC/matrix isolation infrared spectrometry (MI-IR)
have been developed to allow the unambiguous identification of each
individual compound at high concentrations. For 2,3,7,8-TeBDD and
2,3,7,8-TeBDF, the most intense matrix isolation infrared band is
given by frequencies 1478 and 1434, respectively (Wurrey et al., 1989;
Childers et al., 1992). Childers et al. (1992) gave additional
frequencies for some PXDDs/PXDFs.
2.4.6 The need for analysis of 2,3,7,8-substituted congeners
Table 2 (section 2.1) gives the PBDD/PBDF congeners substituted
with bromine in the 2,3,7,8-positions. As these are the most toxic
congeners, in some investigations only these congeners are determined.
Table 7. Molecular ions (M+, 79Br isotope) of PBDDs, PBDFs, and
PBDEs showing possible interference during monitoring and determinationa
Compound Brominated congeners
mono- di- tri- tetra- penta- hexa- hepta- octa- nona- deca-
PBDFs 246 324 402 480 556 636 714 792
PBDEs 248 326 404 482 560 638 716 794 872 950
PBDDs 262 340 418 496 574 652 730 808
a From Buser (1986a) and Donnelly et al. (1987).
The German Dioxin Directive (1994) established limitations on the
concentrations of certain 2,3,7,8-substituted PBDDs/PBDFs in products
to be placed on the market (see Appendix I). In 1987, the US EPA
issued a Test Rule requiring manufacturers and importers of certain
halogen-containing chemicals to analyse their products for
2,3,7,8-substituted PHDDs/PHDFs (US EPA, 1987) (see Appendix I).
Owing to these actions, activities in PBDD/PBDF analysis have
improved and 2,3,7,8-substituted standards have been synthesized; in
1995, 12 of the 17 2,3,7,8-substituted isotopically labelled and 11 of
the 17 2,3,7,8-substituted native PBDDs/PBDFs were available.
2.4.7 Interfering substances
For a general discussion on the problem of interfering
substances, see Buser (1991). Substances possibly interfering with
PBDF determinations include PBDEs, as discussed above. This is of
particular importance in the analysis of substances containing this
flame retardant, their thermolytic products, and environmental samples
where this flame retardant is implicated. PBBs can be separated from
PBDFs/PBDDs and are not a source of cross-contamination (Donnelly et
al., 1987).
2.4.8 Standards
As mentioned in section 2.4.6, a number of PBDD/PBDF standards
have been made commercially available in recent years, in particular
the 2,3,7,8-substituted congeners. The following is therefore more of
historical than of practical interest.
For use as standards, samples of mono- through octaBDDs (Munslow
et al., 1987) and mono- through octaBDFs (Sovocool et al., 1987), as
well as PXDDs/PXDFs (Donnelly et al., 1987, 1989b), can be synthesized
by the electrophilic bromination of dibenzo- p-dioxin and
dibenzofuran. Special reaction conditions optimize the selectivity and
the corresponding yield. Extended reaction times result in a higher
degree of bromination; elevated temperature and increasing amount of
iron/iron (III) chloride (FeCl3) catalyst can be used to accelerate
the reaction (Hutzinger et al., 1989).
Donnelly et al. (1991a) prepared over 100 PHDDs using
self-condensation of halogenated phenols, coupling of halogenated
catechols with halobenzenes or halonitrobenzenes, and electrophilic
halogenation.
Ramalingam et al. (1986) synthesized PBDDs from bromocatechol and
polybromonitrobenzene in the presence of anhydrous potassium carbonate
(K2CO3) in acetone. For PXDDs, bromocatechol or chlorocatechol was
refluxed with polychloronitrobenzene or polybromonitrobenzene in the
presence of anhydrous K2CO3 in acetone.
Mixed halogenated compounds can be prepared by the bromination of
PCDDs and PCDFs, by the chlorination of PBDDs and PBDFs, or by halogen
exchange (Buser, 1987a).
Chatkittikunwong & Creaser (1994b) described the synthesis of
PBDDs/PBDFs and PXDDs/PXDFs by electrophilic halogenation of
substituted precursors with iron (III) halides (in the absence of
halogen).
Jay & Stieglitz (1996) prepared PBDFs and PCDFs from reaction of
copper (II) bromide (CuBr2) or copper (II) chloride, dihydrate
(CuCl2*2H2O), respectively, with dibenzofuran. For synthesis of
PXDFs, the brominated reaction mixture was further reacted with
CuCl2*2H2O.
Nestrick et al. (1989) developed a procedure for synthesizing
13C12-labelled PBDDs/PBDFs from their chlorinated analogues.
3. FORMATION AND SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
PBDDs/PBDFs are not known to occur naturally. They are not
intentionally produced but are formed as undesired by-products in
various processes. They can be formed by chemical, photochemical, or
thermal reactions from precursors or by so-called de novo synthesis
(from organic materials with bromine). PBDDs/PBDFs have been found as
contaminants in brominated organic chemicals. Thermolysis of
brominated flame retardants, in particular PBDEs, has been implicated
as an obvious source of PBDDs/PBDFs. Heating and burning of products
containing such brominated compounds can cause emission of
PBDDs/PBDFs. PBDDs/PBDFs have also been detected in traffic-related
emissions.
The formation of PXDDs/PXDFs is possible in combustion processes
if both bromine and chlorine are present (Buser, 1987a,b), such as in
waste incineration, in particular of old computer/business machines,
and in motor combustion processes.
3.1 Synthesis and use
PBDDs/PBDFs have no commercial use and are synthesized for
research purposes only and as standards for analytical determination
(see section 2.4.8). As an exception, a patent was awarded for the use
of heptaBDF as a flame retardant (Richtzenhain & Schrage, 1977).
3.2 By-products of brominated organic chemicals (including
flame retardants)
Theoretically, some 40 brominated organic chemicals may be
contaminated with PBDDs/PBDFs. Such chemicals include flame retardants
and fire extinguishers, pesticides (e.g. bromophenols, bromophos,
bromoxynil, profonofos), solvents, and chemical intermediates or
additives (Esposito et al., 1980; Lee et al., 1986, 1987; Johnson et
al., 1989; Bretthauer et al., 1991). Possible PBDD/PBDF formation
pathways have been suggested. Chemicals considered as being important
are TBBPA and its derivatives, penta- (PeBDE), octa-, and decaBDE,
2,4,6-tribromophenol, and 1,2-bis(tribromophenoxy)ethane (Johnson et
al., 1989).
Analytical data on the occurrence of PBDDs/PBDFs in brominated
organic chemicals are scarce. Tables 8 and 9 (concentrations of PBDFs
and PBDDs, respectively) include data on PBDEs, PBBs, TBBPA,
1,2-bis(tribromophenoxy)ethane, brominated phenols, brominated
anilines, brominated styrenes, and others.
The highest concentrations of PBDFs were found in PBDEs (up to
8000 µg/kg). Maximum PBDF values measured in DBB, TBBPA, bromophenols,
and bromoanilines were approximately 115 µg/kg, 64 µg/kg, 31 µg/kg,
and 2 µg/kg, respectively. PBDF levels ranging from about 92 to 500
µg/kg were observed in distillation residues of bromophenols and
bromoanilines (Table 8). This is of importance, particularly in
synthetic and analytical laboratories and laboratory waste disposal
(Vogt et al., 1994a,b).
The highest concentration of PBDDs (approx. 8500 µg/kg) was found
in 1,2-bis(tribromophenoxy)ethane, followed by 86 µg/kg in
2,4,6-tribromophenol and 8 µg/kg in TBBPA. As seen with PBDFs, PBDDs
were strongly enriched in distillation residues of selected
bromophenols (Table 9). Additionally, Ritterbusch et al. (1994a)
reported the occurrence of PBDFs (mono to penta: 12.1 µg/kg) and PBDDs
(mono to penta: 1.1 µg/kg) in solvent wastes of chemical laboratories.
2,3,7,8-Substituted PBDDs/PBDFs were not detected in TBBPA (Thies
et al., 1990; Tondeur et al., 1990; Brenner & Knies, 1993a,b; Ranken
et al., 1994), TBPI (Brenner & Knies, 1994), or 2,4,6-tribromophenol
(Tondeur et al., 1990; Vogt et al., 1994a). A sample of commercial
decaBDE was found to contain 1,2,3,7,8-PeBDF (1.6 µg/kg) and
1,2,3,4,7,8-HxBDF (37 µg/kg). Other 2,3,7,8-substituted tetra- to
hexaBDDs/BDFs did not exceed 0.1-5.1 µg/kg. All concentrations given
were maximum values, because co-elution was possible (UBA, 1992).
2,3,7,8-Substituted PBDDs/PBDFs were not detected in multiple samples
(n = 21; three companies) of commercial decaBDE at target limits of
quantitation (according to the US EPA Test Rule; see Appendix I)
ranging from 0.1 to 1.0 µg/kg, from 0.5 to 5 µg/kg, from 2.5 to 25
µg/kg, and from 100 to 1000 µg/kg for tetra-, penta-, hexa-, and
hepta-substitution, respectively. 1,2,3,4,6,7,8-HpBDF was found in all
samples at concentrations of 56-300 µg/kg, which were well below the
target limit of quantitation of 1000 µg/kg for this congener (Ranken
et al., 1994). Another study (Kieper, 1996) analysed five different
flame retardants (DBDE, 1,2-bis(tribromophenoxy)ethane,
TBBPA-carbonate oligomer, di- and tribromostyrene) for a total of
eight 2,3,7,8-substituted PBDDs/PBDFs (tetra to hexa). Contents, if
any, were below the respective detection limits. If detection limits
were included in the evaluation (as a concentration of half the
detection limit), the sum concentrations would range from 0.2 to 18.5
µg/kg. (A problem may arise in evaluating the sum of PBDDs/PBDFs if
the analytical limit of detection is rather high in some of the
congeners. From a scientific point of view, values below the limit of
quantification should not be used. However, for administrative
purposes, sometimes a value of one-half the detection limit is assumed
and included in the sum of PBDDs/PBDFs. A considerable difference may
occur between a scientific and this "administrative" approach. It is
highly recommended that the approach for the calculation be stated if
values that have not been measured are included in the summation.)
2,3,7,8-TeBDD was absent in bromophenols, whereas 2,3,7,8-TeBDF
was identified in crude 4-bromophenol and 2,4-dibromophenol at levels
of 0.12 and 0.15 µg/kg, respectively, and in their distillation
residues at 8.3 and 9.8 µg/kg, respectively (Vogt et al., 1994a). Both
2,3,7,8-substituted TeBDF and TeBDD were present (at 40 and 10 ng/kg,
respectively) in solvent wastes of chemical laboratories (Ritterbusch
et al., 1994a).
Table 8. Concentrations of PBDFs found in brominated organic chemicals
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
PBDE (tetra- to 8000 - - - 2000 4000 2000 - - Hileman et al.
hexaBDE) (commercial) (1989)
PBDE (hexa- to >4000 - - - n.d. 2000-4000 2000-4000 presentb - Hileman et al.
nonaBDE) (commercial) (approx. (1989)
200)
DBDE (commercial) - - - - n.d. n.d. 200 presentb,c - Hileman et al.
(approx. (approx. (1989)
200) 200)
DBDE (commercial) 286.3 - - - - - 2.3 250 34 Donnelly et al.
(concentrate) 6880 - - - 23 107 3470 2700 580 (1989a)
DBDE (commercial) 2037 - 0.04 <0.9 0.15 <0.01 <0.2 1842 195 Brenner
& Knies (1990)
DBDE (commercial) 79.3 0.4 0.3 0.3 0.8 10.5 67.0 - - UBA (1992)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.08) (0.08) (0.08) (0.8) (1.5) (1.8)
DBB (commercial) 115 99.9 9.0 5.7 <1 <5 <5 <10 <10 Atochem
(1990)
TBBPA + derivatives 63.6 - n.d. n.d. n.d. 1.0 12.2 31.5 18.9 Thoma et al.
(technical grade) (1986b);
Dumler et al.
(1990c)
TBBPA + derivatives
(commercial) >3 2 1 n.d. n.d. n.d. <14 - - Thies et al.
(<0.5) (<1) (<2) (1990)
Table 8. (Continued)
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
TBBPA + derivatives n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Brenner & Knies
(BC 52) (commercial) (0.001-0.4) (1993a,b)
TBBPA-oligocarbonate 1.46 n.d. n.d. n.d. 0.07 0.33 1.06 - - Kieper (1996)
(0.01) (0.01) (0.01)
TBPI 0.21 - 0 0 0.21 0 0 0 - Brenner & Knies
(Saytex BT 93) (1994)
Hexabromocyclo 50 - <10 <10 20 30 <10 <10 - Brenner (1993)
dodecane
(technical)
4-Bromophenol Ritterbusch et
(crude) 1.56 0.08 0.72 0.54 0.22 - - - - al. (1994a);
(distilled) 1.19 0.44 0.75 n.d. n.d. - - - - Vogt et al.
(distillation 378.54 63.47 230.46 69.17 15.44 - - - - (1994a)
residue)
(commercial) 0.37 0.06 0.31 n.d. n.d. - - - -
2,4-Dibromophenol Ritterbusch et
(crude) 3.30 0.16 1.43 1.44 0.12 - - - - al. (1994a);
(distilled) 0.85 0.36 0.49 n.d. n.d. - - - - Vogt et al.
(distillation residue) 498.99 62.47 353.51 68.91 4.3 - - - - (1994a)
2,4,6-Tribromo phenol Thoma et al.
(technical grade) 31.4 - 2.2 16.2 12.0 1.0 n.d. n.d. n.d. (1986b);
(crude) 4.6 0.79 2.66 1.19 n.d. n.d. n.d. n.d. - Dumler et al.
(1990c);
Vogt et al.
(1994a)
Pentabromophenol n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
(analytical grade) (1986b);
Dumler et al.
(1990c)
Table 8. (Continued)
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
Tetrabromophthalic n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
anhydride (analytical (1986b);
grade) Dumler et al.
(1990c)
2,4,6-Tribromo Vogt et al.
aniline (crude) 1.88 0.24 0.28 n.d. 0.25 1.01 - - - (1994b)
(recrystallized) 0.47 n.d. 0.47 n.d. n.d. n.d. - - -
(distillation 92.35 n.d. 3.15 8.40 15.90 64.90 - - -
residue)
2,6-Dibromo-4-nitroaniline
(crude) 0.90 0.33 0.57 n.d. n.d. n.d. - - - Vogt et al.
(1994b)
1,2-Bis(tribromo-phenoxy)ethane n.d. n.d. n.d. n.d. n.d. n.d. n.d. - -- Kieper (1996)
(0.5) (0.5) (0.5) (0.5) (1.0) (4.0)
Polytribromostyrene 16.74 n.d. 4.03 1.2 n.d. 0.36 3.29 3.01 4.85 Kieper (1996)
(0.06) (0.06)
Polydibromostyrene 24.09 0.09 0.26 0.21 n.d. n.d. 2.33 2.93 18.27 Kieper (1996)
(0.03) (0.04)
a - = no information; n.d. = not detected (detection limits in parentheses, if specified).
b Not quantifiable because of lack of standards.
c Major component.
Table 9. Concentrations of PBDDs found in brominated organic chemicals
Chemical Concentrations of PBDDs (µg/kg)a Reference
Sum MoBDDs DiBDDs TrBDDs TeBDDs PeBDDs HxBDDs HpBDDs OcBDD
DBDE (commercial) 0.4 - - - 0.05 0.35 - - - Brenner & Knies
(1990)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - UBA (1992)
(0.1-<5.1) (<0.1) (<0.1) (<0.1) (<0.2) (<0.7) (<5.1)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.03) (0.03) (0.03) (0.03) (0.1) (0.35)
TBBPA + derivatives n.d. - - n.d. n.d. - - - - Thoma et al.
(technical grade) (1986b);
Dumler et al.
(1990c)
TBBPA + derivatives 8 n.d. n.d. n.d. 1 2 5 - - Thies et al.
(commercial) (<0.5) (<0.5) (<0.5) (1990)
TBBPA + derivatives 0.006 - n.d. n.d. 0.006 n.d. n.d. n.d. n.d. Brenner & Knies
(BC 52) (commercial) (0.001) (0.4) (1993a,b)
TBBPA-oligocarbonate n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.01) (0.01) (0.01) (0.01) (0.02) (0.06)
Hexabromocyclo - - <10 <10 <10 <10 <10 <10 - Brenner (1993)
dodecane (technical)
4-Bromophenol Ritterbusch et
(crude) 0.40 0.04 0.15 0.21 n.d. - - - - al. (1994a);
(distilled) 0.14 0.07 0.07 n.d. n.d. - - - - Vogt et al.
(distillation residue) 39.0 14.41 12.84 11.75 n.d. - - - - (1994a)
(commercial) n.d. n.d. n.d. n.d. n.d. - - - -
2,4-Dibromophenol Vogt et al.
(crude) 0.16 0.03 0.13 n.d. n.d. - - - - (1994a);
(distilled) 0.08 0.04 0.04 n.d. n.d. - - - - Ritterbusch et
(distillation residue) 18.75 1.37 3.13 10.76 3.49 - - - - al. (1994a)
Table 9. (cont'd)
Chemical Concentrations of PBDDs (µg/kg)a Reference
Sum MoBDDs DiBDDs TrBDDs TeBDDs PeBDDs HxBDDs HpBDDs OcBDD
2,4,6-Tribromo phenol Thoma et al.
(technical grade) 85.5 - - 1.5 84.0 - - - - (1986b);
(crude) n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. Dumler et al.
(1990c);
Vogt et al.
(1994a)
Pentabromophenol n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
(analytical grade) (1986b);
Dumler et al.
(1990c)
Tetrabromophthalic Thoma et al.
anhydride (analytical grade) n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. (1986b);
Dumler et al.
(1990c)
2,4,6-Tribromo aniline Vogt et al.
(crude) n.d. - - - n.d. - - - - (1994b)
(recrystallized) n.d. - - - n.d. - - - -
(distillation residue) 5.45 - - - 5.45 - - - -
2,6-Dibromo-4-nitroaniline
(crude) n.d. - - - n.d. - - - - Vogt et al.
(1994b)
1,2-Bis(tribromophenoxy)ethane 8455 n.d. n.d. 107 8348 n.d. n.d. - - Kieper (1996)
(1.0) (1.0) (2.0) (5.0)
Polytribromostyrene 5.63 1.78 3.85 n.d. n.d. n.d. n.d. n.d. <0.38 Kieper (1996)
(0.02) (0.02) (0.03) (0.11) (0.19)
Polydibromostyrene n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. <0.17 Kieper (1996)
(0.02) (0.02) (0.02) (0.02) (0.04) (0.08) (0.12)
a - = no information; n.d. = not detected (detection limits in parentheses, if specified).
3.3 Formation from the photochemical degradation of brominated organic
chemicals
The formation of PBDDs/PBDFs was observed under laboratory
conditions after irradiation of PBDEs (Watanabe & Tatsukawa, 1987) or
of bromophenols (Ritterbusch et al., 1994a) and is also believed to
occur after outdoor exposure of PBDEs (Birla & Kamens, 1994).
The major photoproducts of the flame retardant DBDE (technical
grade) irradiated in hexane solution by UV light and sunlight were
lower brominated PBDEs and mono- to hexa-substituted PBDFs. PBDDs were
not detected. Based on the kinetics of the reactions, the formation of
PBDFs occurred secondarily from debrominated PBDEs as photoproducts of
DBDE, but not directly from DBDE. UV irradiation of DBDE for 16 h gave
about a 20% yield of total PBDFs, with tetraBDFs, but not
2,3,7,8-TeBDF, being the main components (Watanabe & Tatsukawa, 1987).
The concentrations of PBDDs/PBDFs (mono to penta) in several
bromophenol samples were drastically increased, up to three orders of
magnitude, after UV irradiation for 60 min. Apparently, the rate of
photochemical PBDD/PBDF formation was greater than the rate of
degradation (see section 4.2.1). An exception was 2,3,7,8-TeBDF, which
disappeared after irradiation. Both bromophenols themselves as well as
their contaminants (PBDEs) may act as PBDD/PBDF precursors
(Ritterbusch et al., 1994a). The highest levels were found with the
mono- to tetrabrominated homologues. The total concentrations of
dibenzofurans exceeded those of dibenzo- p-dioxins: 315.7 µg/kg
versus 14.2 µg/kg after irradiation of crude 4-bromophenol, and 1750.7
µg/kg versus 301.0 µg/kg after irradiation of crude 2,4-dibromophenol
(Ritterbusch et al., 1994a).
The atmospheric stability of PBDDs/PBDFs that resulted after
combustion of polyurethane foam (containing PBDEs) at a range of
temperatures was examined under sunlight conditions. The formation of
PBDFs, primarily tetra- and pentaBDFs (isomers not examined), in the
presence of sunlight was seen with the products of low-temperature
(400-470°C) combustion. The formation of the PBDF compounds in this
case was thought to be the result of the photolysis of unburned PBDEs
(Birla & Kamens, 1994).
For photochemical transformations of higher brominated
PBDDs/PBDFs to lower brominated congeners, see section 4.2.1.
3.4 Formation from the laboratory thermolysis of bromine-containing
flame retardants
The potential of typical brominated flame retardants to form
PBDDs/PBDFs was examined under various conditions in a series of
laboratory thermolysisa experiments (see also Tables 10-12). The
flame retardants were thermally treated either alone (Buser et al.,
a For definitions of terms referring to thermal treatment, see
Appendix II.
1978; Thoma & Hutzinger, 1987a,b; Dumler et al., 1989a, 1990b;
Zacharewski et al., 1989) or blended with polymer matrices (Dumler et
al., 1990b,c; Riggs et al., 1990; Lahaniatis et al., 1991; Lorenz &
Bahadir, 1993). PBDFs were found in most of the samples, but both the
concentration and the degree of bromination varied greatly. PBDDs were
detected to a lesser extent. Owing to the different conditions used,
it is difficult to compare these studies quantitatively -- except for
results from the same experimental series.
The largest yields of PBDDs/PBDFs were obtained from PBDEs
(especially in combination with polymers) and from bromophenols, both
reaching values in the g/kg range. In contrast to PBDEs, the
bromophenols that are flame retardants of only limited use (BMU, 1989)
were not tested in polymer matrices. About an order of magnitude lower
yields of PBDDs/PBDFs were observed upon thermolysis of certain PBBs.
Again, lower but significant amounts of PBDDs/ PBDFs in the mg/kg
range were generated by TBBPA and 1,2-bis-(tribromophenoxy)ethane.
Formation of PBDDs/PBDFs from all other flame retardants tested was
very low or undetectable (Tables 10 and 11). Among the PBDEs, the
yield of PBDDs/PBDFs in thermolytic residues decreased from pentaBDE
to octaBDE to decaBDE (e.g. Buser, 1986a; Thoma et al., 1987a; Luijk
et al., 1991).
The optimum PBDF formation temperatures of flame retardants
thermally treated alone were found to be in the range of 600-900°C.
For example, bromophenols and TBBPA showed PBDF formation maxima at
800°C, pentaBDE at 700-800°C, and decaBDE at 800-900°C, whereas PBBs
(hexabromobiphenyl, or hexaBB) had no clear peak concentration between
700°C and 900°C on pyrolysis in quartz tubes (Thoma et al., 1986a,
1987a). Under other experimental conditions, decaBDE (alone) produced
maxima at 600°C (Dumler et al., 1990a) or 700°C (Dumler et al.,
1989a,c, 1990b,c). When decaBDE was burned in a polymer matrix, the
PBDF formation maximum was shifted to lower temperatures (Thoma et
al., 1987a; Dumler et al., 1990b). This effect was not observed with
pentaBDE (Thoma et al., 1987a; Dumler, 1989; Hutzinger, 1990).
Plastic/flame retardant mixtures showed maxima of PBDF formation at
600°C (decaBDE/PBT: Dumler et al., 1990b; decaBB/PBT: Luijk & Govers,
1992), 700°C (pentaBDE/polyurethane: Dumler, 1989; Hutzinger, 1990;
TBBPA/ABS: Luijk & Govers, 1992), or 800°C (TBBPA/ epoxide laminate:
Dumler, 1989; Hutzinger, 1990). The presence of Sb2O3 in the polymer
matrices resulted in a further decrease of the optimum formation
temperature (down to 400°C) of PBDFs from octa- or decaBDE (Clausen et
al., 1987; Bieniek et al., 1989; Dumler, 1989; Dumler et al., 1989a,c,
1990a,b; Hutzinger, 1990; Zier et al., 1990; Luijk et al., 1991). An
example referring to decaBDE is given in Table 12.
The polymer matrix and the synergistic action of Sb2O3
influenced both the optimum temperature range and the yield of PBDFs
(e.g. with hexaBB and decaBDE). Additionally, thermolysis in a polymer
matrix changed the ratio of PBDF congeners to lower brominated
compounds (e.g. Thoma et al., 1987a; Dumler et al., 1990b; see also
Table 12). Frequently, tetraBDFs were the most abundant homologue
Table 10. Survey on the generation of PBDFs and PBDDs during thermolysis of bromoorganic flame retardants
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
PBDEs
Technical PBDE quartz minivials Buser (1986a)
in air 510 °C mono to penta mono to tetra Sigma PBDDs/PBDFs:
5000-10 000
in air 630 °C mono to hexa mono to penta Sigma PBDDs/PBDFs:
100 000
Technical PBDE quartz tubes
(Bromkal 70-5 DE, 700-900 °C mono to penta mono to tetra TeBDFs: up to 330 400 (700 °C) Thoma et al.
70 DE, and G1) TeBDDs: up to 15 400 (700 °C) (1987a)
800 °C mono to penta mono to tetra Sigma PBDFs/PBDDs: Zacharewski et
up to 610 393 al. (1988)
Technical PeBDE
(Bromkal 70-5 DE) various types of Dumler et al.
ovens (DIN, BIS, (1987);
VCI) 600 °C di to penta di to tetra TeBDFs: up to 87 827 Hutzinger et
TeBDDs: up to 12 374 al. (1989)
Technical PeBDE quartz minivials
500 °C tetra, penta tetra, penta Sigma PBDFs/PBDDs: 12 000 Luijk et al.
600 °C tetra to hexa tetra to hexa Sigma PBDFs/PBDDs: 270 000 (1990, 1991)
Technical PeBDE pyrojector, Thoma & Hutzinger
(Bromkal 70-5 DE) absence of di to tetra none small amounts (1987b, 1989)
oxygen (helium)
700-900 °C
Technical OBDE quartz minivials Buser (1986a)
630 °C tetra to hepta tri to hepta Sigma PBDDs/PBDFs: 50 000
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical OBDE quartz minivials Luijk et al.
600 °C tetra to hexa tetra to hexa Sigma PBDDs/PBDFs: 56 000 (1990, 1991)
Technical DBDE quartz minivials Buser (1986a)
630 °C tetra to hepta tetra to octa Sigma PBDDs/PBDFs:
10 000-20 000
Technical DBDE quartz tubes
(FR 300 BA) 700 °C tetra to octa hepta, octa OcBDD/BDF: 2690 Thoma et al.
800 °C tetra, hexa hepta, octa OcBDD/BDF: 9230 (1987a)
to octa
900 °C penta to octa hepta, octa OcBDD/BDF:13 413
800 °C tetra, hexa hepta, octa Sigma PBDDs/BDFs: Zacharewski et
to octa 10 935 al. (1988)
Technical DBDE VCI oven
400-1000 °C hepta, octa hepta, octa n.sp. Klusmeier et
al. (1988)
Technical DBDE VCI oven
300-800 °C mono to hepta tetra, hexa, Sigma PBDFs: up to Dumler et al.
hepta 7222 (700 °C) (1989c);
Sigma PBDDs: up to Hutzinger
588 (800 °C) (1990)
Technical DBDE DIN oven
400 °C hexa to octa hexa to octa Sigma PBDDs/PBDFs: Dumler (1989);
470/364
600 °C hexa to octa hexa to octa Sigma PBDDs/PBDFs: Dumler et al.
2756/447 (1990a)
800 °C tri to hepta tri to octa Sigma PBDDs/PBDFs:
1114/690
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical DBDE quartz minivials
600 °C tetra to hexa tetra to hexa Sigma PBDDs/PBDFs: 1700 Luijk et al.
(1990, 1991)
Technical DBDE pyrojector, Thoma &
absence of Hutzinger
oxygen
(Fr 300 BA) 700 °C hepta, octa none n.sp. (1987b, 1989)
Two technical PBDE high-temperature Striebich et al.
mixtures (Br3-Br10) flow reactor (1990, 1991)
in nitrogen
650 °C di to tetra none Sigma PBDFs: 900
in air 625 °C tri, tetra di to tetra Sigma PBDFs: 600
Sigma PBDDs: 900
PBBs
Technical hexaBB glass tubes O'Keefe
(FireMaster(R) FF-1) (open) in air
380-400 °C tetra, penta n.a. TeBDFs/PeBDFs: 40/4 (1978)
(sealed) in
nitrogen
380-400 °C traces (tetra) n.a.
Technical hexaBB quartz tubes
(FireMaster(R) BP-6) 700, 800,
900 °C di to hepta none TeBDFs: up to 1523 Thoma et al.
(1987a)
800 °C tri to hepta none Sigma PBDFs: 2070 Zacharewski
et al. (1988)
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical hexaBB pyrojector,
(FireMaster(R) BP-6) absence of
oxygen
600-900 °C none none Thoma & Hutzinger
(1987b, 1989)
Technical decaBB glass tubes Atochem
(Adine 0102) (loosely plugged) (1987)
800 °C none none -
Bromophenols
2-Bromophenol 3 different types
of ovens Dumler et al.
600 °C mono to tri mono, di Sigma PBDFs: up to 215 425 (1987);
Sigma PBDDs: up to 60 634 Hutzinger et
al. (1989)
2,4,6-Tribromophenol quartz tubes Thoma et al.
700, 800, 900 °C di to penta di to hexa TeBDDs: up to 896 000 (1986a)
TeBDFs: up to 8950
2,4,6-Tribromophenol 3 different types Dumler et al.
of ovens (1987);
600 °C tri to penta di to penta Sigma PBDFs: up to 8820
Sigma PBDDs: up to 880 503 Hutzinger et
al. (1989)
2,4,6-Tribromophenol pyrojector, Thoma & Hutzinger
absence of oxygen (1987b, 1989)
600-900 °C none di to penta n.sp.
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
2,4,6-Tribromophenol high-temperature Striebich et al.
flow reactor (1990, 1991);
in nitrogen 625 °C n. a. none n.sp. Dellinger et
in air 500 °C none tetra al. (1993)
2,4,6-Tribromophenol high-temperature
flow reactor
300-800 °C n.sp. tri, tetra 1,3,6,8- and 1,3,7,9-TeBDD:
310 000 and 250 000 (500 °C) Sidhu et al. (1995)
Pentabromophenol quartz tubes
700, 800, 900 °C penta to hepta penta to octa Sigma PBDFs: up to 7042
Sigma PBDDs: up to 7508 Thoma et al. (1986a)
Pentabromophenol 3 different types
of ovens
600 °C tri, tetra tetra, hepta TeBDFs: up to 3307
TeBDDs: up to 3567 Dumler et al. (1987);
Hutzinger et al. (1989)
Pentabromophenol pyrojector, absence
of oxygen 700 °C none hepta, octa small amounts Thoma & Hutzinger
(1987b, 1989)
Others
TBBPA quartz tubes
700, 800, 900 °C mono to tetra mono to tetra Sigma PBDDs/PBDFs:
up to 1150/498 Thoma et al. (1986a)
TBBPA BIS oven
240 °C di di low levels Thies et al. (1990)
Hexabromocyclododecane quartz tubes
700 °C tri to hexa tri, tetra Sigma PBDFs: 0.25
Sigma PBDDs: 0.05 Brenner (1993)
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
1,2-Bis(tribromophenoxy)ethane high-temperature
flow reactor
in nitrogen 475 °C none none
in air 450 °C none tetra n.sp. Striebich et al.
(1990, 1991)
Tetrabromophthalic
anhydride quartz tubes
700, 800, 900 °C none none Thoma et al. (1986a)
2,4,6-Tribromoaniline sealed tubes
640 °C tetra tetra n.sp. Alsabbagh et al. (1992)
N-(tribromophenyl)-maleimide sealed tubes
630 °C tetra tetra n.sp. Alsabbagh et al. (1992)
a For definitions and descriptions of apparatuses used for thermolysis experiments, see Merz et al. (1986) or Appendix II.
b n.a. = not analysed; n.sp. = not specified.
c Owing to the different conditions used, different studies should not be compared quantitatively.
Table 11. Survey on the generation of PBDFs and PBDDs during thermolysis of bromoorganic flame retardants in polymer matrices
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBDEs
PentaBDE Thoma et al.
(Bromkal 70-5 DE) polystyrene quartz tube (1987a)
700-900 °C 420 000 (Br1-Br5) - -
polyethylene quartz tube
700-900 °C 200 000 (Br1-Br5) - -
PentaBDE polyurethane 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C approx. 50 000c,d x
PentaBDE polyurethane DIN oven Hutzinger
300-800 °C 42 000 (Br1-Br6)c x (1990)
PentaBDE polyurethane foam ignition vessel
670-780 °C n.sp. x Birla & Kamens
(1994)
PentaBDE laminate (SiO2) BIS oven Lenoir et al.
600 °C 2000 (Br1-Br5) x (1994)
laminate (TiO2) BIS oven
600 °C 2.6 (Br1-Br3) x
OctaBDE ABS (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
OctaBDE ABS (Sb2O3) DIN oven
300-800 °C 280 000 (Br1-Br7)c x Hutzinger (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
OctaBDE ABS (Sb2O3) DIN oven Neupert et al.
600 °C 9000 (Br3-Br6) x (1989b)
DecaBDE Thoma et al.
(FR 300 BA) polystyrene quartz tube 7000 (Br1-Br7) - - (1987a)
700-900 °C
polyethylene quartz tube 170 000 (Br1-Br8) - -
700-900 °C
DecaBDE polystyrene (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
DecaBDE polystyrene (Sb2O3) DIN oven 228 000 Hutzinger
(Br1-Br8)c (1990)
300-800 °C x
DecaBDE polypropylene DIN oven Dumler
400-800 °C 255 000 (Br1-Br7)c x (1989)
DecaBDE polypropylene closed glass 393 000 (Br1-Br5)c x Dumler
vials 600 °C (1989)
DecaBDE polypropylene 3 different ovens Dumler et al.
(Sb2O3) (VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
DecaBDE polypropylene DIN oven Hutzinger
(Sb2O3) 300-800 °C 290 000 (Br1-Br8)c x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
DecaBDE HIPS (Sb2O3) mass burning rate Pinkerton et
apparatus al. (1989)
(21% O2)
500-800 °C 1600 (Br1-Br8) x
DecaBDE HIPS (Sb2O3) quartz tube Luijk et al.
reactor (1990, 1991)
in nitrogen
275-825 °C 4300 (Br2-Br8)e x
in air
500-700 °C 710 (Br2-Br6)e x
DecaBDE PBT quartz tube Lahaniatis
apparatus et al. (1989)
400-800 °C none x
DecaBDE PBT VCI oven Sovocool et
400-600 °C n.sp. (presence al. (1990)
of Br1-Br6) x
DecaBDE PBT (Sb2O3) quartz tube Clausen et
apparatus al. (1989)
400-800 °C 13 100 (Br1-Br6) - -
DecaBDE PBT (Sb2O3) VCI oven Dumler et
300-800 °C 160 000 (Br1-Br7)c - - al. (1989c)
PBT (Sb2O3) DIN oven Hutzinger
300-800 °C 228 000 (Br1-Br8)c x (1990)
PBT (Sb2O3) BIS oven Zier et al.
400-1000 °C 13 800 (Br1-Br5)e x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBT (Sb2O3) BIS oven Lenoir et al.
in nitrogen + H2O 29 500 (Br1-Br7) (1994)
600 °C
epoxide resin quartz tube Clausen et
(Sb2O3) apparatus al. (1987)
400-800 °C none x
plastic sheets quartz tube n.sp. Lahaniatis
apparatus (presence of et al. (1989)
600 °C various PBDFs) x
PBBs
HexaBB
(FM BP-6) polystyrene quartz tube 8900 (Br1-Br4) - - Thoma et al.
700-900 °C (1987a)
polyethylene quartz tube 43 000 (Br1-Br4) - -
700-900 °C
DecaBB PBT quartz tube Luijk &
reactor in Govers (1992)
nitrogen
+ 10% O2
400-700 °C 100 (Br3-Br8)e x
Others
TBBPA PBT 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBT (Sb2O3) VCI oven Hutzinger
800 °C 41 (Br1, Br2)e x (1990)
PBT (Sb2O3) BIS oven Thies et al.
600 °C 0.11 (Br1-Br4)e x (1990)
epoxide laminate 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <50d,e x
epoxide laminate DIN oven Dumler (1989);
600-800 °C 10 (Br1-Br3)e x Hutzinger (1990)
epoxide laminate VCI oven
800 °C 23 (Br1-Br3)e x Hutzinger (1990)
epoxide laminate 3 different ovens Dumler et al.
(Cu) (VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x
epoxide laminate VCI oven Hutzinger
800 °C 40 (Br1-Br2)e x (1990)
polycarbonate 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <10d,e x
polycarbonate DIN oven Hutzinger
600 °C 8.9 (Br1-Br3)e x (1990)
TBBPA polycarbonate BIS oven Thies et al.
600 °C 5.5 (Br1-Br6)e x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
ABS BIS oven Thies et al.
600 °C 0.4 (Br2-Br4)e x (1990)
ABS quartz tube Luijk & Govers
reactor in (1992)
nitrogen + 10% O2
400-700 °C 3.1 (Br1-Br5)e x
ABS (Sb2O3) VCI oven Hutzinger
800 °C 5.4 (Br1, Br2)e x (1990)
BIS and DIN ovens Thies et al.
600 °C 2.1 (Br1-Br4)e x (1990)
1,2-Bis(tribromophenoxy)ethane ABS (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <1000c,d x
ABS (Sb2O3) DIN oven Hutzinger
600 °C 500 (Br1-Br4)c x (1990)
Tetrabromophthalic polyurethane 3 different ovens Dumler et al.
anhydride (VCI, BIS,DIN) (1989b)
600-800 °C <100c,d x
polyurethane VCI oven Hutzinger
800 °C 44 (Br1)c x (1990)
DIN oven
800 °C 0.4 (Br4)c x
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
Hexabromocyclo polystyrene 3 different ovens Dumler et al.
dodecane (VCI, BIS, DIN) (1989b)
800 °C 10c,d x Hutzinger
DIN oven (1990)
800 °C 5.5 (Br2-Br4)c x
Hexabromocyclo polystyrene quartz tube Brenner (1993)
dodecane apparatus
700 °C 0.11 (Br4-Br6) x
polystyrene quartz tube Brenner (1993)
insulation foam apparatus
700 °C 0.38 (Br2-Br8) x
Polybrominated polyester 3 different ovens Dumler et al.
polystyrene (VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x Hutzinger
DIN oven (1990)
600 °C 36 (Br1-Br4)e x
Polytribromostyrene PBT (Sb2O3) quartz tube Clausen et al.
400-800 °C none x (1987)
Dibromopropyldian polypropylene 3 different ovens Dumler et al.
(Sb2O3) (VCI, BIS, DIN) (1989b)
600-800 °C <100c,d x
polypropylene DIN oven Hutzinger
(Sb2O3) 600 °C 28 (Br2, Br3)c x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
1,2-Bis(tetrabromo ABS (Sb2O3) 3 different ovens Dumler et al.
phthalimido)ethane (VCI, BIS, DIN) (1989b)
600-800 °C <500c,d x
ABS (Sb2O3) DIN oven Hutzinger
800 °C 118 (Br1,Br2)c x (1990)
PBT (Sb2O3) quartz tube Clausen et al.
400-800 °C none x (1987)
Tetrabromobenz PBT quartz tube Clausen et al.
imidazolone 400-800 °C none x (1987)
Polypentabromo PBT quartz tube Clausen et al.
benzylacrylate 400-800 °C none x (1987)
a For definitions and descriptions of apparatuses used for thermolysis experiments, see Merz et al. (1986) or Appendix II.
b - = no information; n.sp. = not specified.
c Related to flame retardant.
d Figures only.
e Related to blend.
group in these ternary mixtures (e.g. Clausen et al., 1987; Bieniek et
al., 1989; Dumler, 1989; Hutzinger, 1990; Luijk et al., 1991).
Formation of PBDDs from decaBDE, octaBDE, decaBB, and TBBPA
showed a similar dependence on temperature and/or matrix as seen with
PBDFs, but the yields were much lower (Dumler et al., 1990b;
Hutzinger, 1990; Luijk & Govers, 1992). Thermolysis of pentaBDEs,
bromophenols, 1,2-bis(tribromophenoxy)ethane, and some samples of
TBBPA resulted in an increase in the relative proportion of PBDDs
(e.g. Dumler, 1989; Hutzinger, 1990; see also Table 10). Some smaller
differences in the temperature profiles between PBDDs and PBDFs can
also occur (e.g. thermolysis of TBBPA/laminate: formation maximum of
PBDFs at 800°C, of PBDDs at 400°C and 700°C; Dumler, 1989).
The influence of various metals (tin, iron, zinc, copper) and
metal oxides (oxides of zinc, copper, iron, and antimony; silica,
SiO2, and titanium dioxide, TiO2) on yield and pattern of
PBDFs/PBDDs was studied in thermolysis of decaBDE or pentaBDE in
polymer matrices (Lenoir et al., 1994). During thermolysis of decaBDE
in PBT (500°C, BIS oven), all metals and the oxides of copper (I),
iron (III), and antimony (III) caused an increase in PBDD
concentrations. PBDF concentrations were enhanced by oxides of iron
and antimony. Oxides of zinc and copper (II) strongly reduced yields
of PBDDs/PBDFs. During thermolysis of pentaBDE in laminate (600°C, BIS
oven), SiO2 was found to be nearly inert, whereas addition of TiO2
(3%) led to a significant reduction in PBDD/PBDF levels.
Water also plays an important role, as seen in thermolysis
experiments with decaBDE in PBT/Sb2O3 performed in a nitrogen
atmosphere with and without water at 600°C (Zier et al., 1990; Lenoir
et al., 1994). Water increased the PBDF, PBDD, and 2,3,7,8-TeBDF
concentrations by a factor of 7.5, 2.8, and 10, respectively.
Other factors influencing yields and pattern of PBDDs/PBDFs
formed in thermolysis of flame retardants are oxygen (O'Keefe, 1978;
Thoma & Hutzinger, 1987a,b; Bieniek et al., 1989; Luijk & Govers,
1992), air flow rates (Klusmeier et al., 1988), types of combustion
apparatuses (types frequently used are described in Appendix II), and
residence time. Thermolysis of a flame-retarded resin (glass fibre
reinforced PBT/decaBDE/Sb2O3) under simulated municipal waste
incineration conditions confirmed the formation of PBDDs/PBDFs (Riggs
et al., 1990).
2,3,7,8-TeBDF was identified after pyrolysis of a technical-grade
flame retardant (alone) consisting primarily of 2,2',4,4',5,5'-hexaBB
(Buser et al., 1978; O'Keefe, 1978). Thies et al. (1990) found
2,3,7,8- substituted congeners from experiments where TBBPA (alone)
was pyrolysed. 2,3,7,8-Substituted congeners were also found after
pyrolysis of plastics containing decaBB (Luijk & Govers, 1992), PBDEs
(Dumler, 1989; Lahaniatis et al., 1989, 1991; Dumler et al., 1990c;
Hutzinger, 1990; Zier et al., 1990), and TBBPA (Thies et al., 1990;
Lorenz & Bahadir, 1993). Maximum concentrations of 2,3,7,8-TeBDF of up
to 2000 mg/kg of flame retardant were found for pyrolysed polymers
Table 12. Yields of PBDFs from combustion of DBDE, alone and in a polypropylene matrixa,b
PBDF yield (mg/kg) from combustion PBDF yield (mg/kg) from combustion of DBDE in a
of DBDE alone polypropylene matrixc
PBDFs 400 °C 600 °C 800 °C 400 °C 600 °C 800 °C
MonoBDFs - - - 14 432 10 676 4192
DiBDFs - - - 26 462 14 845 4850
TriBDFs - - 11 39 997 24 036 8354
TetraBDFs - - 28 107 517 49 677 29 147
PentaBDFs - - 35 37 419 18 458 6867
HexaBDFs 96 447 81 24 432 5465 948
HeptaBDFs 204 1449 959 4762 1033 353
OctaBDF 107 860 - - - -
Total 407 2756 1114 255 021 124 190 54 711
a Adapted from Dumler et al. (1990a,b).
b Combustion of commercial DBDE samples (pure or mixture of polypropylene/12.5%
DBDE/7.5% Sb2O3) in DIN oven.
c Yield related to DBDE.
containing octaBDE (Dumler, 1989). The highest values of
1,2,3,7,8-PeBDF measured following thermolysis of plastics containing
decaBDE, decaBB, or TBBPA were 63 mg/kg of plastic blend (Zier et al.,
1990), 1.2 mg/kg (Luijk & Govers, 1992), and 50 µg/kg (Thies et al.,
1990), respectively. Maximum levels of 2,3,7,8-substituted tetra- and
pentaBDDs were below 1 mg/kg of blend. For example, 2,3,7,8-TeBDD was
formed during thermolysis of a decaBDE/epoxide resin at 0.8 mg/kg
(Lahaniatis et al., 1991). Because of a lack of reference substances,
the higher brominated PBDDs/ PBDFs with the 2,3,7,8-substitution
pattern were not quantified. (However, a tentative value of 21 µg/kg
for 1,2,3,4,6,7,8-HpBDF was found by Brenner [1993] after thermolysis
of an insulating board consisting of polystyrene foam blended with
hexabromocyclododecane.)
Pyrolysis of PBBs (hexaBB) and PBDEs (PeBDE) at 800°C in the
presence of polyvinyl chloride (PVC) did not result in the formation
of PBDFs/PBDDs but led to mixed brominated/chlorinated biphenyls or
diphenyl ethers and to fully chlorinated compounds. Apparently, the
substitution reactions were favoured over ring-closing reactions
(Thoma et al., 1987b). Moreover, such bromine-chlorine exchange
reactions were also observed during pyrolysis (at 800°C and 900°C) of
PBDDs/PBDFs, leading to PXDDs/PXDFs and PCDDs/ PCDFs. The chlorine
source could be either organic or inorganic (Thoma et al., 1987b,c,d,
1989) (see also section 3.9.1).
Possible pathways for the thermolytic formation of PBDDs/ PBDFs
from flame retardants have been discussed by several authors (Buser,
1986a; Bieniek et al., 1989; Dumler, 1989; Hutzinger, 1990; Lahaniatis
et al., 1991; Luijk et al., 1991; Luijk & Govers, 1992).
Other related polycyclic compounds identified after thermolysis
of brominated flame retardants included bromomethyldibenzofurans
(Sovocool et al., 1990; Lenoir et al., 1994), brominated
benzo[b]naphtho[2,3-d]furans (Lenoir, 1994), brominated phenazines
(Alsabbagh et al., 1992), and hexabromonaphthalene (O'Keefe, 1978).
Detailed descriptions of thermolysis experiments involving PBDEs
and TBBPA are given in the Environmental Health Criteria monographs
for these compounds (WHO, 1994b, 1995).
3.5 Formation during production of plastic materials and presence in
consumer products containing flame retardants
Brominated flame retardants are or were routinely added, at
levels up to 20%, to a number of commercial products, such as
plastics, textiles, carpets, and other materials (Buser, 1986a; WHO,
1994a,b, 1995). They also have applications in a variety of
industries, such as the electronic, electrical engineering, building,
and transport industries (BMU, 1989; Troitzsch, 1990).
PBDDs/PBDFs, as contaminants of certain flame retardants (section
3.2), would be transferred to the flame-retarded products and, under
thermolytic stress (section 3.4), could additionally be formed from
these chemicals during manufacturing processes.
3.5.1 Formation during production processes
PBDD/PBDF levels were monitored during typical processes
(extrusion, injection moulding, etc.) used in the production and
processing of flame-retarded polymers (Donnelly et al., 1989a; Bonilla
et al., 1990; Brenner & Knies, 1990, 1992, 1993a,b, 1994; Thies et
al., 1990). Extruder temperatures of 150-300°C were noted (Donnelly et
al., 1989a; Brenner & Knies, 1990). The polymers used were ABS and
PBT, and the flame retardants examined included OBDE, DBDE,
TBBPA-carbonate oligomer (BC 52), TBPI, brominated styrene, and
1,2-bis(tribromophenoxy)ethane. Results from different studies
(monitoring exhaust streams) are compiled in Table 13. It can be seen
that OBDE and DBDE produced the highest amounts of PBDDs/ PBDFs, the
major portion consisting of PBDFs. PBDF concentrations of about 73 000
ng/m3 air or 7.7 ng/g extruded resin were measured during extrusion
of resins containing DBDE, and 1850 ng/g extruded resin in the case of
OBDE. The levels observed with TBBPA and TBPI were lower by several
orders of magnitude. No PBDDs/PBDFs were formed from brominated
styrene or brominated phenoxyethane. Homologue groups present included
monoBDFs through octaBDF (Table 13). 2,3,7,8-Substituted congeners
were not determined with DBDE (Brenner & Knies, 1990) and were not
detected with TBBPA-carbonate oligomer and TBPI, at detection limits
ranging from 1 to 58 pg/m3 (Brenner & Knies, 1992, 1993a,b, 1994).
However, PBDFs with 2,3,7,8-substitution (0.012 ng/g extruded resin;
but co-elution was possible) were found for OBDE just above the
detection limit (Bonilla et al., 1990).
The actual worker exposure to PBDDs/PBDFs depends on the
ventilation and exhaust conditions around the machines. In some
studies, parallel measurements of the workplace atmosphere were
undertaken, results of which are discussed in section 5.3 (Brenner &
Knies, 1990, 1993a,b; Thies et al., 1990).
3.5.2 Presence in resins and polymer products
PBDDs/PBDFs were determined in various plastic materials at
several processing stages (BMU, 1989; Donnelly et al., 1989a; Bonilla
et al., 1990; Brenner & Knies, 1990, 1992, 1993a,b, 1994; Hutzinger,
1990; McAllister et al., 1990; Thies et al., 1990; Luijk et al.,
1992c; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et al., 1993; Kieper,
1996). These examinations included granulated resins, moulded parts
whose flame retardant additives were known, and parts of commercial
electrical appliances (television sets, printers, computers) whose
flame retardants were unknown (see also Tables 14 and 15). Polymers
used were ABS, HIPS, polystyrene, polyamide, PBT, polypropylene, or
polyurethane in combination with about 5-20% PBDEs, TBBPA,
bromopolystyrenes, TBPI, or other compounds as flame retardants (see
Table 14). The highest levels of PBDDs/PBDFs were found in materials
Table 13. Formation of PBDDs/PBDFs during manufacturing processes
(production of flame-retarded polymers in the chemical industry)
Concentrations (ng/m3)a
Process Sample PBDDs PBDFs PBDF homologue groups Reference
Manufacture of PBT/ Exhaust stream 1 72 904 DiBDFs: 322; TrBDFs: 705; Brenner & Knies
glass fibre resin from extruder head TeBDFs: 980; PeBDFs: 3910; (1990)
blended with HxBDFs: 22 162; HpBDFs: 39 550;
DBDE/Sb2O3 OcBDF: 5275
Manufacture of PBT/glass Exhaust stream 1.1 1.3 DiBDFs: 0.42 (0.16); TrBDFs: Brenner & Knies
fibre resin blended with from extruder (0.87)b 0.48 (0.31);TeBDFs: 0.24 (0.06); (1993a,b)
TBBPA (BC 52)/Sb2O3 head PeBDFs: 0.04 (0.33);
HxBDFs: 0.15 (0.01)b
Exhaust stream 0.2 0.7 DiBDFs: 0.23 (0.2); TrBDFs:
from granulator (1.01)b 0.29 (0.49); TeBDFs:
0.17 (0.08); PeBDFs: 0.02
(0.07); HxBDFs: 0 (0.24)b
Exhaust stream n.d. 0.08 DiBDFs: 0.004; TrBDFs:
from injection (0.001-0.2) 0.012; TeBDFs: 0.014;
moulding machine PeBDFs: 0.013; HxBDFs:
0.039
Exhaust stream n.d. 0.006 DiBDFs: 0.004; TeBDFs: 0.002
from storage hood (0.001-0.2)
Manufacture of PBT/glass Exhaust stream 0.05 0.78 DiBDFs: 0.18; TrBDFs: 0.34; Brenner & Knies
fibre resin blended with from extruder TeBDFs: 0.18; PeBDFs: 0.03; (1994)
TBPI head HxBDFs: 0.06
Processing of ABS/TBBPA Off-gas near a 6 213 MoBDFs: 13; DiBDFs: 200 Thies et al.
material compounding (1990)
machine
Table 13 (Continued)
Concentrations (ng/g of extruded resin)
Process Sample PBDDs PBDFs PBDF homologue groups Reference
Processing of PBT/DBDE Fumes generated n.a. 7.69 TeBDFs: 1.35; PeBDFs: 2.47; Donnelly et al.
+ Sb2O3 resin during extrusion HxBDFs: 1.67; HpBDFs: 2.20 (1989a)
Processing of Fumes generated Bonilla et al.
* ABS/brominated styrene during extrusion n.d. n.d. - (1990)
terpolymer resin
* ABS/1,2-bis(tribromophenoxy)ethane n.d. n.d. -
resin
* ABS/TBBPA resin 0.006 0.020 n.sp.
* ABS/OBDE resin 0.54 1850 n.sp.
a n.a. = not analysed; n.d. = not detected (detection limits in parentheses, if specified); n.sp. = not specified.
b Values in parentheses were obtained when PBT was blended with BC 52 (TBBPA)-PBT-batch (approx. 50% BC 52) instead of BC 52 powder.
flame-retarded with PBDEs and were in the range of several thousand
µg/kg, thus exceeding the levels in any other systems by orders of
magnitude. The range of PBDF concentrations found is given in Table
14. PBDFs were, with few exceptions, the predominant components. The
contamination of electrical appliances having an unknown flame
retardant equipment is shown in Table 15.
Generally, the concentrations of PBDDs/PBDFs were higher in the
typical resin/PBDE products than in the flame retardants alone (see
also section 3.2). For comparisons, it should be noted that the
concentrations shown in Table 14 were calculated on the basis of the
weight of the resins. They would be higher if calculated on the basis
of the weight of flame retardant in the resins. Values obtained using
the latter calculation were presented by Hutzinger (1990), who found
PBDF concentrations ranging from 112 to 1888 mg/kg in polymers
(polypropylene, PBT, ABS, or polyurethane) containing PBDEs as flame
retardants.
Frequently, additional processing resulted in a further increase
in total PBDFs (see Table 14). For example, the sum of mono- to
hexaBDF levels measured in casing parts was higher by a factor of
about 5 compared with that found in the corresponding polymer-flame
retardant blend (polystyrene/DBDE) actually used in manufacturing
(Table 14; UBA, 1992). Factors influencing the extent of formation of
PBDFs are temperature and the duration of such processes as blending,
extrusion, and moulding (Table 14; Donnelly et al., 1989a; McAllister
et al., 1990).
Within the PBDF homologue groups, the highly brominated (>tetra)
derivatives were generally prevalent. Frequently, peak concentrations
were seen with penta- and hexaBDFs (see Tables 14 and 15). In casing
parts, hexaBDFs reached levels as high as 2950 µg/kg; in printed
circuit boards, maximum concentrations (>1000 µg/kg) were seen with
tetra- and pentaBDFs (Table 15; UBA, 1992). Concentrations of mono-,
di-, and triBDFs were in the low µg/kg range, <30 µg/kg for polymers
and casings (BMU, 1989; Brenner & Knies, 1990; Thies et al., 1990;
UBA, 1992) and <450 µg/kg for printed circuit boards (UBA, 1992).
2,3,7,8-Substituted PBDFs were not analysed (Brenner & Knies,
1990), were not detectable (Thies et al., 1990; Brenner & Knies,
1993a,b, 1994; Kieper, 1996), or were detected in some formulations at
low concentrations (Donnelly et al., 1989a; Bonilla et al., 1990;
McAllister et al., 1990; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et
al., 1993; Kieper, 1996). A moulded part consisting of ABS/PBDE
contained 2,3,7,8-TeBDF at 2 µg/kg (Meyer et al., 1993). Plastics for
casings and printed circuit boards of electrical appliances (n = 14)
were found to be contaminated with tetra-, penta-, and hexaBDFs having
the 2,3,7,8-substitution pattern (Table 15; UBA, 1992). The maximum
concentrations of 2,3,7,8-TeBDF were <4% of the total tetraBDFs in
the case of casings and <17% in the case of printed circuit boards.
Higher percentages were found for the two pentaBDFs and for
1,2,3,4,7,8-HxBDF, usually <7-22%, but exceptionally reaching as high
as 75% of the respective homologue groups (UBA, 1992). As co-elution
Table 14. Concentrations of PBDFs in several flame-retarded plastic materials
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
ABS/OBDE
* Granulate - 2100 24 000 50 000 3900 1700 BMU (1989)
* Normal extrusionb
(n = 17-22) - 2.8-3.6 870-1800 2100-2380 500-780 26-64 Donnelly et al.
(1989a)
* Abusive extrusionb - 150-170 29 000-34 000 8200-10 000 500-920 19 Donnelly et al.
(n = 1-2) (1989a)
* Pre-extrusion resin 38 300 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 84 500 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Normal mouldingc - 3 1100 <135 000 - - McAllister et al.
(1990)
* Abusive mouldingc - 170 <14 000 <118 000 - - McAllister et al.
(1990)
ABS/TBBPA
* Pre-extrusion resin 1090 - - - - - Bonilla et al.
(Br4-Br7)
(1990)
* Post-extrusion resin n.d. - - - - - Bonilla et al.
(1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Commercial polymer - n.d. n.d. n.d. Thies et al.
(<2) (<3) (<20) - - (1990)
ABS/brominated
styrene terpolymer
* Pre-extrusion resin 37.5 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 84.0 - - - - - Bonilla et al.
(Br4-Br7) (1990)
ABS/1,2-bis(tribromo-phenoxy)ethane
* Pre-extrusion resin 44.5 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 16.2 - - - - - Bonilla et al.
(Br4-Br7) (1990)
HIPS/DBDE
* Normal extrusiond - - 4.5 950 720 150 Donnelly et al.
(1989a)
* Abusive extrusiond - 2.3 22.6 107 78 0.5 Donnelly et al.
(1989a)
* Extreme extrusiond - 0.01 8.6 200 2100 3200 Donnelly et al.
(1989a)
* Base resin - 10 40 <5300 - - McAllister et al.
(1990)
* Normal mouldinge - 10 50 <14 300 - - McAllister et al.
(1990)
* Abusive mouldinge - 10 60 <5500 - - McAllister et al.
(1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Extreme mouldinge - 20 200 <34 100 - - McAllister et al.
(1990)
* Pre-extrusion resin - - - - approx. approx. Luijk et al.
1500 4500 (1992c)
* Post-extrusion resin - - - - approx. approx. Luijk et al.
(4 cycles at 275 °C) 9000 45 000 (1992c)
Polystyrene/DBDE
* Compound 194 2.7 14.6 174 - - UBA (1992)
* 2 casing parts manufactured from
the above compound 640; 1313 54; 39 147; 106 1092; 409 - - UBA (1992)
(Br1-Br6)
Polystyrenebutadiene/DBDE
* Compound n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.2) (1.0) (50)
Polystyrene
1,2-bis-(tribromophenoxy)ethane
* Compound n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.7) (2.5) (9.7)
Polyamide/polytribromostyrene
* Compound 15.3 n.d. 0.28 1.81 3.43 6.21 Kieper (1996)
(Br1-Br8) (0.2)
Polyamide/polydibromostyrene
* Compound 4.18 0.64 0.38 0.37 0.46 2.15 Kieper (1996)
(Br1-Br8)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
PBT/DBDE
* Granulate (n = 7) - 6-501 20-920 59-65 000 66-136 000 n.d.-2600 BMU (1989)
* Normal extrusionf - 1-26 18-130 71-1600 180-3800 410-4100 Donnelly et al.
(n = 17-22) (1989a)
* Abusive extrusionf - 76-240 13 000-43 000 69 000-180 000 48 000-94 000 1200-11 000 Donnelly et al.
(n = 5) (1989a)
* Extreme extrusionf
(n = 3) - 1020-2590 68 200-82 800 272 000- 72 500- - Donnelly et al.
708 000 108 000 (1989a)
* Blend - 6.2 27 151 approx. 560 approx. 280 Brenner & Knies
(1990)
* Base resin - 3 20 110 - - McAllister et al.
(1990)
* Normal mouldingg - 3 2 13 - - McAllister et al.
(1990)
* Abusive mouldingg - 30 >7800 >16 100 - - McAllister et al.
(1990)
* Extreme mouldingg - 1000 >54 000 >7000 - - McAllister et al.
(1990)
PBT/TBBPA
* Commercial polymer - n.d. n.d. n.d. - - Thies et al.
(<0.2) (<0.1) (<1) (1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Extruder granulate Brenner & Knies
(n = 3) - n.d. n.d. 0.4-0.8 0.6-3.5 - (1990)
* Moulded test articles Brenner & Knies
(n = 2) - 0.17-0.2 n.d.-0.06 1.5-2.2 1.9-3.8 - (1993a,b)
* Compound 8.41 0.14 2.13 6.14 - - Kieper (1996)
(Br1-Br6)
PBT/bromopolystyrene
* Granulate (n = 2) - n.d.-5 2-10 34-130 11-460 - BMU (1989)
PBT/bis-tetrabromophthalimide
* Granulate - - 5 35 31 - BMU (1989)
PBT/TBPI
* Polymer - 0.57 0.07 0.02 3.4 - Brenner & Knies
(1994)
* Granulate (n = 2) - up to 0.8 0 0 0 - Brenner & Knies
(1994)
* Moulded test article - 0 0 0 0 - Brenner & Knies
(1994)
Polypropylene/DBDE
* Granulate - 53 191 10 000 1370 2600 BMU (1989)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
Polyurethane/PeBDE
* Granulate - 18 000 57 000 44 000 - - BMU (1989)
a - = not mentioned; n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified.
b Normal/abusive extrusion conditions: 227 °C/246 °C; 1 min/10 min cycle.
c Normal/abusive moulding conditions: 225 °C/245 °C; 1 min/10 min cycle.
d Normal/abusive/extreme extrusion conditions: 216-218 °C/238-243 °C/266-271 °C; 30 second/5 min/7 min cycle.
e Normal/abusive/extreme moulding conditions: 215-220 °C/235-245 °C/265-270 °C; 30 second/5 min/7 min cycle.
f Normal/abusive/extreme extrusion conditions: 250-254 °C/254 °C/254 °C; 23 second/5 min/10 min cycle.
g Normal/abusive/extreme moulding conditions: 255 °C/255 °C/255 °C; 23 second/5 min/10 min cycle.
cannot be excluded, all concentrations of 2,3,7,8-substituted
congeners may be overestimated (UBA, 1992; Meyer et al., 1993; Kieper,
1996).
Table 15. PBDF/PBDD concentrations found in plastics from commercial
electrical appliances with unknown polymer/flame retardant systema
PBDFs/PBDDs Concentrationsb (µg/kg) in plastics for
casings printed circuit boards
(n = 8) (n = 6)
MonoBDFs n.d.-0.5 n.d.-19.8
DiBDFs n.d.-3.1 n.d.-149
TriBDFs n.d.-13.1 0.2-441
TetraBDFs n.d.-48.9 0.6-1264
2,3,7,8-TeBDFc <0.1-1.2 <0.1-11.1
PentaBDFs n.d.-1126 n.d.-1372
1,2,3,7,8-PeBDFc <0.1-16.4 <0.1-24
2,3,4,7,8-PeBDFc <9.1-31.5 <0.1-6.5
HexaBDFs n.d.-2952 n.d.-185
1,2,3,4,7,8-HxBDFc <0.7-203 <1.5-9.9
Total PBDFs n.d.-4125 3.6-3430
Total PBDDs n.d.-113.6d 1.9-1974e
a Adapted from UBA (1992).
b n.d. = not detected. Detection limits: <0.1 µg/kg for
mono- to triBDFs/BDDs; <0.1-<0.3 µg/kg for tetra- and
pentaBDFs/BDDs; <0.7-<2.1 µg/kg for hexaBDFs; and <1.0-<15.5
µg/kg for hexaBDDs.
c Maximum values, because co-elution could not be excluded.
d Seven of eight samples = n.d.
e Five of six samples = 1.9-13.9 µg/kg.
PBDDs were not routinely detected in the samples examined
(Donnelly et al., 1989a; Brenner & Knies, 1990, 1992, 1993a,b;
Hutzinger, 1990; Thies et al., 1990; Kieper, 1996). If present, their
maximum concentrations in several thermoplastic resins ranged from
0.006 to 4500 µg/kg (Bonilla et al., 1990; McAllister et al., 1990;
Lorenz & Bahadir, 1993; Meyer et al., 1993; Kieper, 1996) and from 1.9
to 1974 µg/kg in plastics taken from electrical appliances (UBA,
1992). Whereas some samples contained PBDDs, mainly tetraBDDs, only
trace amounts of PBDFs were present in these samples (Lorenz &
Bahadir, 1993; Kieper, 1996). With plastics from electrical
appliances, one out of eight casing parts and all six printed circuit
boards examined gave positive results for PBDDs (UBA, 1992). The PBDD
concentration measured in the one positive casing part was 114 µg/kg
(consisting only of hexaBDDs). In five out of seven positive samples,
the percentage of PBDDs was low, amounting to a maximum of 2.7% of the
total PBDD/PBDF content. In the remaining two samples (showing
concentrations of about 3 and 2000 µg/kg, the latter being mainly
tetraBDDs), the percentages of PBDDs were 46.3 and 99.7% of the total
PBDD/PBDF levels (UBA, 1992; see also Table 15). Altogether, the PBDD
homologue distribution pattern was somewhat irregular and included
mono- to hexaBDDs (UBA, 1992).
Although 2,3,7,8-substituted penta- and hexaBDDs were present (up
to 25 µg/kg) in a few samples (McAllister et al., 1990; UBA, 1992;
Meyer et al., 1993), 2,3,7,8-TeBDD could not be detected at detection
limits mostly below 0.3 µg/kg (Bonilla et al., 1990; McAllister et
al., 1990; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et al., 1993;
Kieper, 1996).
3.6 Emissions from flame-retarded consumer products
Some experiments were performed to clarify whether or not PBDFs
were released from television sets, computers, or similar appliances
(Bruckmann et al., 1990; Ranken et al., 1990; Thies et al., 1990; UBA,
1992). Positive (Bruckmann et al., 1990; Thies et al., 1990; UBA,
1992) and negative (Ranken et al., 1990; UBA, 1992) results were
obtained.
None of three (Ranken et al., 1990) and three of four (UBA, 1992)
units (television sets, computer monitors) tested in experimental
chambers were found to release PBDFs during 3 × 8 h of operation
(Ranken et al., 1990) or continuously for 72 h (UBA, 1992).
In the experiments conducted by UBA (1992), maximum total
emissions of PBDFs (tetra through hexa) were 1800 pg/unit tested, with
tetraBDFs being most prevalent. 2,3,7,8-TeBDF was detected in one
appliance (see Table 16). Concentrations of PBDEs concomitantly
measured ranged between <1.4 and 890 ng/unit. No PBDFs (detection
limit: 3-10 pg/printer) and only low levels of PBDEs were observed
with the three printers tested.
In another study (Bruckmann et al., 1990), air samples were
collected in a closed room (27 m3) at different distances from a new
television set operating for 3 days for approximately 17 h/day. Total
concentrations of PBDFs (tri through penta) were found to be 155
pg/m3 air at a distance of 0.15 m above the television set and 28
pg/m3 air in the centre of the room (2.2 m distant from the
television set). According to a calculation of UBA (1992), the
concentrations of tetraBDFs (11 pg/m3), pentaBDFs (0.5 pg/m3), and
hexaBDFs (<0.1 pg/m3) measured above the television set correspond
to 732, 33, and <7 pg emitted per television set, respectively. For
comparison, ambient air concentrations were 0.16 pg/m3 (tetraBDFs)
and <0.05 pg/m3 (penta- plus hexaBDFs). Bruckmann et al. (1990) did
not analyse for any 2,3,7,8-substituted congeners.
Table 16. Emissions of PBDFs from television sets and computer monitors
as measured in a test chambera,b
PBDFs Emissions (pg/appliance)c
Television sets (n = 2) Computer monitors (n = 2)
Colour Monochrome
TetraBDFsd 320 n.d. 1045 605
2,3,7,8-TeBDFe n.d. (3) n.d. (5) 15 n.d. (3)
PentaBDFs n.d. n.d. 330 165
1,2,3,7,8-PeBDFe n.d. (7) n.d. (8) <5 n.d. (5)
2,3,4,7,8-PeBDFe n.d. (7) n.d. (8) <5 n.d. (5)
HexaBDFs n.d. (10) n.d. (10) 424 80
HeptaBDFs n.d. (10) n.d. (10) n.d. (15) n.d. (10)
OctaBDF n.a. n.a. n.a. n.a.
Sum tetra- to hexaBDFsf 320 n.d. 1799 850
a Adapted from UBA (1992).
b Volume of test chamber: 1.17 m3; sampling time: continuously, 72 h; sucking
speed: 1.5 m3/h.
c n.d. = not detected (detection limits in parentheses, where specified);
n.a. = not analysed.
d Emissions correspond to about 3, 10, and 6 pg/m3, respectively.
e Maximum value, as co-elution with internal standard cannot be excluded.
f Emissions less than the detection/quantification limit are set as 50% of the limit
when calculating the sum of homologues.
Thies et al. (1990) detected tetraBDFs and pentaBDFs at
concentrations of 3 and 8 pg/m3, respectively, in an air sample
(sampling volume about 500 m3) taken 15 cm above a television cabinet
being installed and kept at 46°C in an office room (45 m3). No PBDFs
could be detected in an air sample collected at a distance of 2.2 m
from the television set.
The presence of PBDFs in "non-experimental" rooms equipped with
monitors and other appliances is discussed in sections 5.1.1 and
5.3.1.
3.7 Presence in fire residues, smoke condensates, and gases after
fires
3.7.1 Experimental fires
Experimental fire tests simulating real fire conditions were
performed with electrical appliances such as television sets,
printers, computer terminals, and their casings (Fabarius et al.,
1990; Hamm & Theisen, 1992; UBA, 1992). High PBDF concentrations can
be produced under these conditions (see Table 17). The total PBDD/
PBDF concentrations in combustion residues reached values of between 1
and 1930 mg/kg (up to 0.2%) for electrical appliances and between 8000
and 9000 mg/kg (almost 1%) for the casing parts (see Table 17).
PBDD/PBDF levels determined in some appliances before burning were in
the range n.d.-4.2 mg/kg (Hamm & Theisen, 1992; UBA, 1992). Analysis
of smoke condensate from the fire test room gave area contaminant
concentrations ranging from 6 to 1610 µg/m2 (Table 17), most values
being in the range of 100-400 µg/m2 (Hamm & Theisen, 1992; UBA,
1992). The smoke samples collected during the fire tests contained
0.8-1700 µg PBDDs/PBDFs/m3 (Table 17).
With some exceptions -- where penta- or decaBDE was specified --
the flame retardants being included in the test materials were unknown
(Fabarius et al., 1990; Hamm & Theisen, 1992; UBA, 1992).
As seen in Table 17, the total PBDD concentrations were low in
combustion residues (n.d.-2.0 mg/kg), smoke condensate (n.d.-1.2
µg/m2), and smoke (n.d.-3.9 µg/m3), with a maximum of 3% of the
corresponding total PBDD/PBDF concentrations (Fabarius et al., 1990;
Hamm & Theisen, 1992; UBA, 1992).
In one study (Fabarius et al., 1990), the most abundant homologue
groups were the tri- and tetraBDDs/BDFs. Other studies (Hamm &
Theisen, 1992; UBA, 1992) showed a homologue distribution pattern
dominated by tetra- through hexaBDDs/BDFs.
The proportion of 2,3,7,8-TeBDF was mostly under 3% of the total
of tetraBDFs. 2,3,7,8-Substituted congeners of penta- and hexaBDFs
yielded between 1% and a maximum of 16% of the corresponding totals.
For example, concentrations measured in fire residues (n = 8) ranged
from 0.005 to 18 mg/kg for 2,3,7,8-TeBDF, from 0.005 to 116 mg/kg for
the two 2,3,7,8-substituted PeBDFs, and from <0.014 to 567 mg/kg for
1,2,3,4,7,8-HxBDF (UBA, 1992). PBDDs with 2,3,7,8-substitution were
not detected; however, their detection limits were relatively high
(0.001-1.9 mg/kg in combustion residues) (UBA, 1992).
During preliminary experiments intended to improve sampling
techniques, test vehicles (one car, one subway wagon) were burnt in a
traffic tunnel (Wichmann et al., 1993). Noticeable amounts of PBDFs
(Br1-Br6) and PCDDs (Cl4-Cl8) were released at both fires. The
measurements performed gave values in the high ng/m3 range (graphics
given only). Fire residue samples from different materials (e.g.
paint, floor coverings, cables, etc.) taken from the burnt-out
vehicles (one car: n = 4, one subway wagon: n = 5) showed
concentrations of up to 4000 ng/kg for mono- to triBDFs and up to 250
ng/kg for tetra- to octaBDFs. PBDD concentrations were lower, having
peak concentrations of 120 ng/kg for mono- to triBDDs and of <22
ng/kg for tetra- to octaBDDs (Zelinski et al., 1994).
Table 17. PBDF/PBDD concentrations in samples from fire tests with electrical appliances
Fire object (n) Fire site Homologue groups Total PBDF/PBDD concentrationsa,b
analysed/abundant Combustion residue Smoke condensate Smoke/gas Reference
(mg/kg) (µg/m2) (µg/m3)
Television barrel Br3-Br8/Br3, Br4 79.27 n.a. 0.825 Fabarius et al.
set (2) (2.0) (0.02) (1990)
Casing parts of room Br1-Br6/Br4-Br6 7750-8700 177-1610 n.a. Hamm & Theisen
electrical (n.d.) (n.d.) (1992);
appliances (2) UBA (1992)
Electrical room Br1-Br6/Br4-Br6 1-1930 6-323 11-1700 Hamm & Theisen
appliances (6) (n.d. -0.7) (n.d.-1.2) (n.d.-3.9) (1992);
UBA (1992)
a PBDD concentration in parentheses.
b n.a. = not analysed, n.d. = not detected.
(It is very difficult to extrapolate values from gases, smoke, or
flue gases [ng/m3] to areal contamination [ng/m2]. The indoor and
outdoor dispersion vary greatly and are quite difficult to predict.
Furthermore, human exposure and resulting body burdens cannot be
predicted.)
3.7.2 Accidental fires
Analyses of fire residues, smoke condensates, gases, and
firemen's trousers confirmed the expected release of PBDDs/PBDFs
during real fire accidents (Buser, 1986b; Bruckmann et al., 1990;
Fabarius et al., 1990; Hamm & Theisen, 1992; Neupert & Pump, 1992;
UBA, 1992; Zelinski et al., 1993, 1994; Schacht et al., 1995; see also
Table 18). With some exceptions (bowling hall, stockhouse, computer
room), all fire cases examined occurred in private residences with
television sets being involved. Concentrations measured were mostly
below the values found in the model experiments described above, but
the qualitative compositions of the samples were similar (see section
3.7.1).
PBDFs dominated clearly over PBDDs. In contrast to PBDFs, which
were found in almost all samples, PBDDs were not regularly detected;
if present, their concentrations were low. For example, PBDDs could be
identified in three of nine television fire incidents. In these three
incidents, only four of nine samples contained PBDDs, the maximum
concentrations being less than 1.5% of the sum of PBDF/ PBDD (mono
through hexa) levels (UBA, 1992). Likewise, the sum of PBDD
concentrations in two soot samples collected after a fire in a
computer room were in the range of 0.1-30 µg/kg (Schacht et al.,
1995).
As seen in Table 18, PBDFs covered a wide range of
concentrations. The PBDF levels of combustion residues were mainly in
the µg/kg range, but two maximum values of 107 mg/kg (Br1-Br6; UBA,
1992) and 17 mg/kg (Br1-Br8; Zelinski et al., 1993) were also
observed (see Table 18). The PBDF (Br1-Br6) area contaminant
concentrations (caused by smoke condensates) in close vicinity to the
fire site ranged between 0.1 and 13.1 µg/m2 in most cases (UBA,
1992). In the adjoining areas, the levels measured were usually lower
by a factor of 2-34. Interestingly, there were no systematic
correlations between PBDF concentrations found in combustion residues
(fire site) and those found in wipe samples (area contamination). It
is thus difficult to predict the area contamination from residue
analysis (Hamm & Theisen, 1992; UBA, 1992). Zelinski et al. (1993)
found that not only the distance from the source but also the surface
characteristics of the objects influence the PBDF content of the
samples. The maximum gaseous emissions of PBDFs (Br2-Br8) measured
in other cases amounted to 3.5 ng/m3 (Fabarius et al., 1990). One
study provided tentative information on contamination of a fireman's
trousers after fighting a fire. PBDFs (Br2-Br8) were found at a
concentration of 2.01 µg/kg (Fabarius et al., 1990).
Table 18. Release of PBDFs during accidental fires in private residences or other buildings
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
MonoBDFs house (3) TV set (1) n.d.-0.1 8.1-16.9 n.d.-2.2 n.a. UBA (1992)
house (1) TV set (1) 17 n.d. n.d. n.a. UBA (1992)
flat (5) TV set (1) n.d. n.d.-9.3 n.d.-14.7 n.a. UBA (1992)
DiBDFs residence (4-5) n.sp. 0.1-2.3 n.a. n.a. <0.1 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-2.7 43.7-146 n.d.-36 n.a. UBA (1992)
house (1) TV set (1) 595 2 15 n.a. UBA (1992)
flat (5) TV set (1) n.d.-0.7 n.d.-75.8 n.d.-94 n.a. UBA (1992)
TriBDFs residence (4-5) n.sp. 0.06-7.31 n.a. n.a. <0.1-2.0 Fabarius et al.
(1990)
house (3) TV set (1) 0.2-17.9 122-1131 24-110 n.a. UBA (1992)
house (1) TV set (1) 3491 272 85 n.a. UBA (1992)
flat (5) TV set (1) n.d.-3.4 n.d.-756 n.d.-587 n.a. UBA (1992)
TetraBDFs stock house (1) n.sp. n.sp. 4.0; 32 8.9; 123 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. 0.12-8.6 n.a. n.a. <0.1-1.5 Fabarius et al.
(1990)
house (3) TV set (1) 0.4-93.2 532-4396 64-216 n.a. UBA (1992)
house (1) TV set (1) 16 063 2392 225 n.a. UBA (1992)
flat (5) TV set (1) n.d.-6.6 n.d.-2432 n.d.-1505 n.a. UBA (1992)
computer room (1) equipment 13.6-2700 n.a. n.a. n.a. Schacht et al.
(2 soot samples) (1995)
Table 18. (Continued)
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
PentaBDFs stock house (1) n.sp. n.sp. 0.8; 4.4 9.2; 78 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-5.7 n.a. n.a. <0.01 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-64 1597-3919 142-232 n.a. UBA (1992)
house (1) TV set (1) 64 932 4263 401 n.a. UBA (1992)
flat (5) TV set (1) 0.1-6.5 n.d.-3671 n.d.-1670 n.a. UBA (1992)
computer room (1) equipment
(2 soot samples) 15-2100 n.a. n.a. n.a. Schacht et al.
(1995)
HexaBDFs stock house (1) n.sp. n.sp. 4.5; 27 15; 52 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-2.34 n.a. n.a. 0.1 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-102 2210-4840 69-981 n.a. UBA (1992)
house (1) TV set (1) 21 740 544 121 n.a. UBA (1992)
flat (5) TV set (1) n.d.-7.7 n.d.-3574 n.d.-1504 n.a. UBA (1992)
computer room (1) equipment 0.8-711 n.a. n.a. n.a. Schacht et al.
(2 soot (1995)
samples)
HeptaBDFs stock house (1) n.sp. n.sp. 0.25; 1.2 7; 13 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-0.87 n.a. n.a. <0.1 Fabarius et al.
(1990)
computer room (1) equipment n.d.-6.2 n.a. n.a. n.a. Schacht et al.
(2 soot (1995)
samples)
OctaBDF stock house (1) n.sp. n.sp. 1.1; 5.6 0.5; 8.5 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05 n.a. n.a. <0.1 Fabarius et al.
(1990)
Table 18. (Continued)
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
Sum MoBDFs-TrBDFs flat (1) TV set (1) 1894 n.a. n.a. n.a. Zelinski et al.
(1993)
other things 1.2-203 n.a. n.a. n.a. Zelinski et al.
(11) (1993)
flat (1) window frame, 2.3-67.8 n.a. n.a. n.a.
etc. (4)
flat (1) wallpaper n.d.-20.7 n.a. n.a. n.a. Zelinski et al.
above TV, (1993)
etc. (5)
Sum MoBDFs- HxBDFs house/flat (8) TV set (1) 0.5-235 n.d.-13 054 n.d.-5374 n.a. UBA (1992)
house (1) TV set (1) 106 838 7473 847 n.a. UBA (1992)
Sum DiBDFs- OcBDF residence (4-5) n.sp. 0.3-27.3 n.a. n.a. <0.1-3.5 Fabarius et al.
(1990)
Sum TeBDFs- HpBDFs computer room (1) equipment 29-5600 n.a. n.a. n.a. Schacht et al.
(2 soot samples) (1995)
Sum TeBDFs- OcBDF stock house (1) n.sp. (6) n.d.-1.4 n.sp. n.sp. n.a. Bruckmann et al.
flat (1) TV set (1) 14 910 n.a. n.a. n.a. (1990)
other things
(11) 0.4-63.6 n.a. n.a. n.a. Zelinski et al.
(1993)
flat (1) window frame, 1.0-14.4 n.a. n.a. n.a. Zelinski et al.
etc. (4) (1993)
flat (1) wallpaper above 2.7-169 n.a. n.a. n.a. Zelinski et al.
TV, etc. (5) (1993)
a Sampling time after fire: 0-7 days (if specified).
b n.a. = not analysed; n.d. = not detectable; n.sp. = not specified.
c Samples taken close to fire site (A) or distant from fire site (B).
The distribution pattern of the PBDF homologue groups was, in
most cases, dominated by tetra- through hexabrominated homologues (see
Table 18). On the other hand, peak concentrations were found for the
di- through tetraBDFs (Zelinski et al., 1993). The following order
resulted from ranking the dibenzofuran concentrations detected in fire
residues from a computer room: TeBDFs > PeBDFs > HxBDFs > HpBDFs
(Schacht et al., 1995).
The proportion of 2,3,7,8-substituted isomers was low in the
samples examined (Fabarius et al., 1990; UBA, 1992; Zelinski et al.,
1993). Single maximum proportions of 3, 10, or 18% of the
corresponding totals of tetra-, penta-, or hexaBDFs, respectively,
were reported from fire accidents with television sets (UBA, 1992; see
also Table 19). Neupert & Pump (1992) reported on the occurrence of
2,3,7,8-substituted tetra- and pentaBDFs in residue samples collected
after a fire in the store of a plastics production plant. Maximum
concentrations found were 0.4, 1, and 2 µg/kg for 2,3,7,8-TeBDF,
1,2,3,7,8-PeBDF, and 2,3,4,7,8-PeBDF, respectively. 2,3,7,8-TeBDD,
which usually has detection limits of 0.1 µg/kg (residue samples) or
0.8 ng/m2 (wipe samples), was identified at 0.3 µg/kg in one residue
sample of a television set (UBA, 1992). In four out of five samples
from the warehouse fire, 2,3,7,8-TeBDD was detected at concentrations
of 0.3-0.5 µg/kg (Neupert & Pump, 1992). Soot samples (n = 2)
collected after a fire in a computer room (Schacht et al., 1995)
contained 2,3,7,8-TeBDD (n.d./0.6 µg/kg), 1,2,3,7,8-PeBDD (n.d./
<0.04 µg/kg), 2,3,7,8-TeBDF (0.03/48.3 µg/kg), 1,2,3,7,8-PeBDF
(0.2/15 µg/kg), and 2,3,4,7,8-PeBDF (0.4/14 µg/kg).
Analyses of a wipe sample close to the fire site (television)
showed the presence of PXDFs (tri through octa; up to three bromines
per molecule). The total concentration was 3240 ng/m2, or about half
the amount found for PBDFs in this sample (UBA, 1992). Small
quantities of PXDFs were also detected in a soot sample from a fire of
a bowling hall in which plastic, wood, and other materials burnt
(Buser, 1986b). Significant concentrations of PXDFs (about 30-200
µg/kg) occurred in fire residues from a department store (Wilken &
Schanne, 1994).
PCDDs/PCDFs were concomitantly present in many samples, but
mostly at lower total concentrations than PBDDs/PBDFs (Bruckmann et
al., 1990; Fabarius et al., 1990; UBA, 1992; Wichmann et al., 1992a,b;
Wilken & Schanne, 1994; Schacht et al., 1995). Exceptions were gas
samples and firemen's trousers containing a higher proportion of
PCDDs/PCDFs (Fabarius et al., 1990). The estimated PBDD/PBDF
concentrations in the soot sample from a fire in a bowling hall were
also lower than those of PCDDs/PCDFs (Buser, 1986b). Other
contaminants additionally identified in fire residues were polycyclic
aromatic hydrocarbons (PAHs) (Hamm & Theisen, 1992), chlorinated and
brominated benzenes, bromophenols, polychlorinated biphenyls (PCBs),
PBBs, PBDEs, and TBBPA (Buser, 1986b; Fabarius et al., 1990; Zelinski
et al., 1993).
Table 19. 2,3,7,8-Substituted PBDFs found in residue and wipe samples after accidental
fires of television sets in private residencesa
Congeners Residenceb PBDF concentrationsc
Fire residues Smoke condensated,e
(µg/kg) (ng/m2)
A B
2,3,7,8-TeBDF house (3) <0.1-1.0 17-40.9 1.8-6.8
house (1) 264 n.r. 1.8
flat (5) <0.1-0.1 <0.2-22 <0.1-12.2
1,2,3,7,8-PeBDF house (3) <0.1-3.1 22.5-143 <2.5-6.1
house (1) 1863 n.r. <1.3
flat (5) <0.1-0.1 <0.2-80.7 <0.3-38.2
2,3,4,7,8-PeBDF house (3) <0.1-1.8 17.7-87.7 <0.4-<2.9
house (1) 848 n.r. <1.3
flat (5) <0.1-<0.3 <0.4-31.5 <0.3-15.9
1,2,3,4,7,8-HxBDF house (3) <0.3-17.7 107-851 <6-<17.5
house (1) 1932 n.r. <8.5
flat (5) <0.5-<8.0 <8.5-173 <4.5-69.5
a Adapted from UBA (1992).
b Sampling time after fire: 0.5-4 days.
c Maximum values, as co-elution cannot be excluded.
d Samples taken close to fire site (A) or distant from fire site (B).
e n.r. = not recorded.
3.8 Formation from incineration of fuels
PXDDs (Br1Cl5DDs) and hexachlorodibenzo- p-dioxins (hexa CDDs)
were detected in ash from a wood-fired boiler (nature of "wood" not
specified) at concentrations of 55 µg/kg and 418 µg/kg, respectively
(Harless et al., 1989).
No data were available on incineration of coal, peat, or fuel oil
in power plants.
3.9 Formation during waste disposal and treatment
3.9.1 Incineration
The formation of PCDDs/PCDFs in fly ash of waste incinerators was
recognized in the 1970s (Olie et al., 1977); the additional presence
of PXDDs/PXDFs was first reported in 1986 (Schäfer & Ballschmiter,
1986). Since then, a number of studies (Nakano et al., 1987; Öberg et
al., 1987; Schwind et al., 1988, 1989; Sovocool et al., 1988, 1989;
Harless et al., 1989; Hosseinpour et al., 1989; Huang et al., 1991,
1992a,b; Tong et al., 1991; Funcke & Hemminghaus, 1993; Hartenstein,
1993; Chatkittikunwong & Creaser, 1994c; Takasuga et al., 1994) have
documented the presence of PBDDs/PBDFs and/or PXDDs/PXDFs in fly ash
and/or flue gas of municipal, clinical, or hazardous waste
incinerators (see also Table 20).
The amount of PHDDs/PHDFs formed critically depends on the
combustion conditions and on the extent to which the combustion can be
controlled. In the past, it was customary in many industrial countries
-- and may still be usual in many developing countries -- to burn the
waste in landfills by open fires, with incomplete combustion forming
toxic by-products. Such conditions may be especially favourable for
the formation of PHDDs/PHDFs. When the problem was recognized, the
technology of waste incineration was greatly improved, and it has at
present reached a high degree of sophistication. An important
reduction can be achieved by optimizing the burning conditions, by
increasing the temperature, residence time, and turbulence. Energy can
be recovered by boilers and can be converted to heat or electricity. A
quick quench to temperatures below 250°C has been found to minimize
the formation of PCDDs/PCDFs.
The flue gas emissions can be controlled by scrubbing the gases
with dry, semi-dry, or wet technologies. Addition of lime and charcoal
before filtering in a baghouse has been successful. Further reduction
can be achieved by the use of catalysts, which destroy the remaining
PCDDs/PCDFs. This indicates that control of environmental hazards may
be achieved by appropriate but, of course, more expensive measures.
The improved technology should not exclude initiatives for waste
minimization and the development of new technologies for recycling of
plastics and wastes.
Several possibilities for the origin of PHDDs/PHDFs exist. Some
PHDDs/PHDFs may be introduced in trace amounts by the feedstock and
may resist combustion. Far larger amounts can be produced in the
incinerator itself, by formation from precursors at high temperatures
in the flame (see section 3.4 and, for example, Sidhu et al., 1995) or
by de novo synthesis at low temperatures in the post-combustion zone
of the incinerator through gas-solid interactions on fly ash. The
latter hypothesis has been proved by several studies (Stieglitz et
al., 1989; Stieglitz & Vogg, 1990; Heinbuch & Stieglitz, 1992, 1993;
Luijk et al., 1992b, 1994). The formation of PXDDs/PXDFs is explained
by the extensive bromine-chlorine exchange reactions observed under
several test conditions (Thoma et al., 1987b,c, 1989; Zier et al.,
1991; Luijk et al., 1992a, 1994). It is assumed that because of the
large quantities of chlorine donors in waste, these reactions
ultimately result in the formation of completely chlorinated
compounds. On the other hand, irreversible bromination of PCDDs/PCDFs
may occur as the fly ash moves from hotter to cooler regions of the
incinerator (Sovocool et al., 1989; Huang et al., 1992b).
Table 20. Detection of PBDDs/PBDFs and PXDDs/PXDFs in fly ash or
flue gas from waste incinerators
Waste Sample (n) Homologue groups detecteda Concentrations Reference
(country)a PBDDs PBDFs PXDDs PXDFs
Municipal fly ash (1) n.a. TeBDFs Br1Cl3-7DDs Br1Cl3-7DFs TeBDFs: 16 ng/kg Schwind et al.
(Germany) Br2Cl2-6DDs Br2Cl2-3DFs Sigma PXDDs: 5535 ng/kg (1988, 1989);
Br3Cl1DFs Sigma PXDFs: 3157 ng/kg Hosseinpour et al.
(1989)
Municipal fly ash (1) n.a. n.a. Br1Cl3-7DDs Br1Cl3-7DFs Sigma PXDDs: 108 µg/kgb Sovocool et al.
(USA) Sigma PXDFs: 9.8 µg/kgb (1988, 1989)
Municipal fly ash (1) n.sp. n.sp. Br1Cl5DDs n.sp. Br1Cl5DDs: 31 µg/kg Harless et al.
(USA) (1989)
Municipal fly ash (1) n.a. n.a. Br1Cl3-7DDs Br1Cl3-7DFs Sigma PXDDs: 56 µg/kg Tong et al.
(USA) Sigma PXDFs: 47 µg/kg (1991)
Municipal fly ash (3) n.a. n.a. Br1Cl3-7DDs n.a. Sigma PXDDs: 0.5-163 Huang et al.
(n.sp.) µg/kg (1992a)
Municipal fly ash (3) n.a. n.a. Br2Cl2-6DDs Br2Cl2-6DFs Sigma PXDDs: 772-2602
(USA) ng/kg
Sigma PXDFs: 334-1513 Huang et al.
ng/kg (1992b)
Municipal fly ash (1) n.a. n.a. Br2Cl2DDs n.d. Br2Cl2DDs: 0.4 ng/kg Huang et al.
(Japan) (1992b)
Municipal fly ash (1) n.a. n.a. Br2Cl2DDs Br2Cl2-6DFs Sigma PXDDs: 1704 ng/kg Huang et al.
(Canada) Sigma PXDFs: 1335 ng/kg (1992b)
Table 20. (Continued)
Waste Sample (n) Homologue groups detecteda Concentrations Reference
(country)a PBDDs PBDFs PXDDs PXDFs
Municipal fly ash (3) Di-, TrBDDs Mo-, DiBDFs Br1Cl1,4,5DDs Br1Cl1,3,4DFs Sigma PBDDs: 145-436 Chatkittikunwong
(United ng/kg & Creaser (1994c)
Kingdom) Br2Cl1-2DDs Br2Cl1-3DFs Sigma PBDFs: 12-325
ng/kg
Sigma PXDDs: 406-1005
ng/kg
Sigma PXDFs: 1347-2922
ng/kg
Clinical fly ash (1) n.d. MoBDFs Br1Cl1,4,5DDs Br1Cl1,3DFs MoBDFs: 77 ng/kg Chatkittikunwong
(United (Di, Br2Cl1-2DDs Br2Cl1DFs Sigma PXDDs: 705 ng/kg & Creaser (1994c)
Kingdom) TrBDDs) Sigma PXDFs: 427 ng/kg
Hazardous flue gas (6) n.d. n.a. Br1Cl3DDs Br1Cl3DFs Br1Cl3DDs: Öberg et al.
(Sweden) (TeBDDs) n.d.-1.3 ng/m3 (1987)
Br1Cl3DFs:
n.d.-4.5 ng/m3
Hazardous flue gas (2) n.sp. n.sp. n.a. n.a. Sigma PBDDs: Hartenstein
(Germany) 0.76-0.82 ng/m3 (1993)
Sigma PBDFs:
0.76-0.82 ng/m3
a n.a. = not analysed; n.d. = not detected; n.sp. = not specified.
b Estimated concentration.
There are some reports on the consequences of an increase in
bromine input during test operations in incinerators. In a large-scale
experiment at the municipal waste incinerator at Bielefeld-Herford
(Germany), material containing 4.8% pentaBDE was added to the normal
fuel (Lahl et al., 1991). The fly ash from the electrostatic
precipitator was analysed for PCDDs/PCDFs and PXDDs/PXDFs as well as
for inorganic bromine. The bromine content in the samples ranged from
0.37 to 0.59 mg Br-/kg. Of the mixed PXDD/PXDF congeners, only
monobromopolychlorinated PXDDs/PXDFs (Br1ClxDDs/ Br1ClxDFs) could
be detected in the five fly ash samples. The concentrations of
PXDDs/PXDFs (Br1Cl2 to Br1Cl7) ranged from 1.547 to 10.163 µg/kg.
Interestingly, the concentrations of the purely chlorinated compounds
(Cl4-Cl8DDs/Cl4-Cl8DFs) were much higher (up to 406.17 µg/kg) than
normally detected (50-150 µg/kg). Although the highest concentrations
of all three parameters analysed (Br-, PXDDs/PXDFs, and PCDDs/PCDFs)
were found in the same sample, a quantitative relationship could not
be established. Whereas concentrations of PCDDs were higher than
concentrations of PCDFs in all samples, concentrations of PXDFs were
higher than concentrations of PXDDs (Lahl et al., 1991). An increase
in concentrations of monobromopolychlorinated PXDDs/PXDFs in the crude
gas was also found after addition of tetrabromomethane to the furnace
of a municipal waste incinerator (Wilken et al., 1990). Wanke et al.
(1996) studied the influence of additional bromine input (up to
sixfold) into municipal solid waste incinerators at a pilot plant
(nominal throughput: 200 kg/h) at combustion temperatures of 850 or
950°C. Extruded polystyrene foams and rigid polyurethane foams (2-4%
Br by weight) were introduced as bromine source. In the raw gases of
the polyurethane foam combustion, no increase in PCDDs/PCDFs was
detected when compared with "normal" fuel. The concentrations of
PBDDs/PBDFs were very low in all experiments. Both test series, the
experiments with extruded polystyrene and rigid polyurethane foams,
showed elevated levels of PXDDs/PXDFs. As was reported from the
Bielefeld-Herford incinerator, of the PXDDs/PXDFs, those congeners
containing only one bromine were the most abundant; dibrominated
species could hardly be detected. More than two bromine atoms could
not be identified in any sample. Similarly, the PXDDs/PXDFs
contributed only 20-30% of the total sum of all halogenated (PCDDs/
PCDFs + PXDDs/PXDFs) dibenzo- p-dioxins and dibenzofurans. This
finding was not found earlier by Hartenstein (1993), who reported much
higher concentrations of PBDDs/PBDFs than of PCDDs/PCDFs in flue gas
samples of a hazardous waste incinerator. The results of Wanke et al.
(1996) showed that, at least for the PXDFs, there is a correlation
between the content of bromide in the fly ash (up to 5% by weight) and
the concentrations of PXDFs. Above 5% Br-, no further increase in
concentrations of PXDFs could be determined; in other words,
saturation was obtained. For the PBDDs, such a correlation could not
be established, as the concentrations of the PBDDs were too low and
the standard deviation too large.
Once the PHDDs/PHDFs have been formed, they partition between
stack gas (gas phase) and fly ash (solid phase) (Schramm et al.,
1990).
The quantities of PBDDs/PBDFs and PXDDs/PXDFs measured in fly ash
of incinerators were in the range of ng/kg to µg/kg (see Table 20).
The few flue gas measurements gave values in the low ng/m3 range (see
Table 20).
In most cases, the concentrations of dibenzo- p-dioxins exceeded
those of dibenzofurans (see Table 20). PXDDs/PXDFs were more abundant
than PBDDs/PBDFs (Chatkittikunwong & Creaser, 1994c; see also Table
20) but were present in fly ash or chimney residues at lower levels
than their fully chlorinated counterparts, reaching 1-20% of the
levels of PCDDs/PCDFs (Schäfer & Ballschmiter, 1986; Sovocool et al.,
1988, 1989; Harless et al., 1989; Tong et al., 1991; Chatkittikunwong
& Creaser, 1994c). For example, the total PCDD/ PCDF levels ranged
from 37 to 62 µg/kg, and the total PBDD/PBDF plus PXDD/PXDF levels
amounted to 1.2-3.5 µg/kg in fly ash from municipal (n = 3) and
clinical (n = 1) incinerators (Chatkittikunwong & Creaser, 1994c).
However, flue gas of a hazardous waste incinerator contained more
PBDDs/PBDFs than PCDDs/PCDFs (1.6 versus 0.05 ng/m3) (Hartenstein,
1993). No information was provided on the ratio of chlorinated to
brominated compounds in the feedstock.
The main homologues of PXDDs/PXDFs that were detected consisted
of mono- and dibromopolychlorinated PXDDs/PXDFs (see Table 20).
Generally, the highest levels were found for Br1Cl4, Br1Cl5, or
Br1Cl6 congeners (Schwind et al., 1988, 1989; Sovocool et al., 1988,
1989; Hosseinpour et al., 1989; Tong et al., 1991; Chatkittikunwong &
Creaser, 1994c).
The isomer distribution patterns of PXDDs/PXDFs were similar to
those found for PCDDs/PCDFs and similar among different samples,
indicating common mechanisms of formation, regardless of the
incinerator conditions and nature of the feedstock (Harless et al.,
1989; Huang et al., 1992b; Chatkittikunwong & Creaser, 1994c).
Of the 2,3,7,8-substituted congeners, 2,3-Br2-7,8-Cl2DD was
found in several fly ash samples at concentrations ranging from 4 to
12 ng/kg (maximum values because the degree of co-elution was not
known) (Huang et al., 1992b). In another test series, 2,3,7,8-TeBDD
and 2,3,7,8-TeBDF could not be detected (detection limits: 16 and 8
ng/kg, respectively) (Chatkittikunwong & Creaser, 1994c).
There are several methods to minimize the emissions of
dibenzo- p-dioxins and dibenzofurans from incinerators. They are
mostly described for PCDDs/PCDFs, but they may also be valid for
PBDDs/ PBDFs and PXDDs/PXDFs (Boyd & Mortland, 1985; Hagenmaier et
al., 1987a,b; Vogg et al., 1987; Vogg, 1989; Wania & Lenoir, 1990;
Acharya et al., 1991; Spahl et al., 1993; Gullett et al., 1994;
Schreiber, 1994; van de Plassche et al., 1994; Vehlow, 1995).
Flue gas monitored after flue gas cleaning in the stack of a
Swedish municipal solid waste incinerator operating at a high
combustion efficiency did not contain certain PBDDs/PBDFs and
PXDDs/PXDFs (Öberg & Bergström, 1990). The detection limits for the
compounds examined were 0.4 ng/m3 (tetraBDFs/BDDs, pentaBDDs) and
0.03 ng/m3 (Br1Cl3DDs/DFs, Br2Cl2DDs).
3.9.2 Disposal
Disposal sites (dumps, landfills) are expected to be an important
source of brominated dibenzo- p-dioxins/dibenzofurans (Sovocool et
al., 1988; Donnelly et al., 1990; Öberg & Bergström, 1990) because
they receive plastic waste, municipal incinerator fly ash, automotive
fluff (ground-up waste residue from junked cars, which remains after
the bulk metals have been reclaimed), etc. and can also be subject to
occasional fires.
A detailed investigation of waste samples from three German
disposal sites confirmed the occurrence of PBDDs/PBDFs and PXDDs/PXDFs
along with PCDDs/PCDFs (Dawidowsky, 1993). The sum of the
concentrations of PBDDs/PBDFs and PXDDs/PXDFs was in the range of
several hundred to thousands of ng/kg dry weight (see Table 21). The
concentration of dibenzo- p-dioxins was low (PBDDs/ PXDDs: 6-580
ng/kg) in relation to the concentration of dibenzofurans (PBDFs/PXDFs:
217-4229 ng/kg). PBDFs may be prevalent over PBDDs (maximum
concentrations >3000 ng/kg versus >300 ng/kg) because they originate
from PBDEs, which were found at high concentrations in the same
samples (sum of mono- to decaBDEs: 4400-17 500 ng/kg dry weight,
n = 6).
The homologue profile was dominated by lower halogenated
derivatives (up to Br4/X4; Table 21). This pattern contrasted to
that of PCDDs/PCDFs, which had peak concentrations of higher
chlorinated homologues (Dawidowsky, 1993). These waste samples showed
high total concentrations of PCDDs/PCDFs, with PCDDs being the most
abundant components. PCDD values ranged from 3000 to 9000 ng/kg (n =
5) or to nearly 30 000 ng/kg (n = 6); PCDFs reached levels of
2000-5600 ng/kg (n = 6) (Dawidowsky, 1993).
Another study determined PBDDs/PBDFs (and PCDDs/PCDFs) in several
waste samples of an analytical "dioxin" laboratory (Ritterbusch et
al., 1994b). The sum of PBDD/PBDF concentrations in waste oil samples
(n = 3) from the GC/MS system ranged from <150 to 14 000 ng/kg (sum
of PCDDs/PCDFs: <200-13 600 ng/kg). Other laboratory waste samples
(n = 4) also contained PBDDs/PBDFs (mono to hexa), with a peak
concentration of 15 500 ng/kg for hexaBDFs.
3.9.3 Recycling
3.9.3.1 Plastics
Granulated parts of office machine casings, which were
reprocessed once or several times, were analysed for the eight
2,3,7,8- substituted PBDDs/PBDFs of the 1994 German Dioxin Directive
(Meyer et al., 1993). The polymer was ABS flame-retarded with either
PBDE or TBBPA, as well as mixed electronic waste with unknown flame
retardants. Materials flame-retarded with PBDE yielded the highest
Table 21. Occurrence of PBDDs/PBDFs and PXDDs/PXDFs in waste samples from three disposal sitesa
Compounds Concentrations (ng/kg dry weight)b
Disposal site A Disposal site B Disposal site C
Sample 1 Sample 2 Sample 3 Sample 4 Sample 5 Sample 6
Dibenzo-p-dioxins
PBDDs 9 24 155 313 2 1
(Br1, Br3) (Br1, Br3) (Br2) (Br1, Br2) (Br1, Br3) (Br3)
PXDDs 43 33 29 267 5 5
(X2, X4) (X2, X4) (X3, X4) (X3-X4) (X4) (X4)
Total 52 57 184 580 7 6
Dibenzofurans
PBDFs 1672 2675 386 3262 170 285
(Br1-Br4) (Br1-Br3) (Br1-Br4) (Br1-Br4) (Br1-Br3) (Br1-Br4)
PXDFs 1261 1554 682 539 47 9
(X2-X4) (X2-X4) (X2-X4) (X2-X4) (X2-X4) (X4)
Total 2933 4229 1068 3801 217 294
a Adapted from Dawidowsky (1993).
b Homologue groups quantified are given in parentheses.
concentrations of these eight PBDDs/PBDFs (16-65 µg/kg), depending on
processing. Mixed electronic waste was contaminated by lower
concentrations (4.9-26 µg/kg). Concentrations of the PBDDs/PBDFs in
TBBPA flame-retarded material were lower still (n.d.-2.4 µg/kg).
Recycling of printed circuits containing TBBPA flame retardant
can lead to the formation of PBDDs/PBDFs (Lorenz & Bahadir, 1993).
Whereas untreated basic recovered material contained total
concentrations of PBDDs/PBDFs (mono to octa) of 0.22 µg/kg, shredded
material was contaminated with PBDDs/PBDFs at levels ranging from 0.03
(metal fraction) to 1.13 µg/kg (mixed and plastic fraction) following
treatments with hammer mill, impact grinder, separation, and
granulation processes. The main components were monoBDFs (0.05-0.32
µg/kg), diBDFs (0.23 µg/kg), and tetraBDDs (0.03-0.73 µg/kg).
2,3,7,8-TeBDD/TeBDF were not detected (detection limits: 0.01-0.05
µg/kg). The contamination was probably due to thermal decomposition of
TBBPA.
Scrap of electronic devices (printed circuit boards with
electronic components) and other flame-retarded plastics were
subjected to various recycling activities (Dumler-Gradl et al., 1995).
After mechanical processing (hammer mill) of TBBPA-containing
plastics, no PBDDs/PBDFs could be detected. After (laboratory-scale)
pyrolysis and solvolysis procedures, especially of chopped printed
circuit boards, high amounts of PBDDs/PBDFs were found in the
condensed (a) or extracted (b) material (e.g. in (a), sums of tetra-,
penta-, hexa-, and heptaBDFs were 7035, 5470, 213, and 31 µg/g,
respectively; 2,3,7,8-TeBDF: 29 µg/kg; 2,3,4,7,8-PeBDF: 24 µg/kg; or
in (b), sums of tetra-, penta-, and hexaBDDs were 0.23, 0.20, and 1.98
µg/kg, respectively; 2,3,7,8-TeBDD: 0.06 µg/kg; sums of tetra-,
penta-, hexa-, and heptaBDFs were 230, 309, 97.5, and 0.9 µg/kg,
respectively; 2,3,7,8-TeBDF: 5 2,3,4,7,8-PeBDF: 12.6 µg/kg). Only
small amounts of mono- and diBDFs were detected at (pilot-scale)
pyrolysis of printed circuit boards and mixed flame-retarded plastics.
Plastic material recovered from cables and subsequently burned
contained PBDDs/PBDFs (mono to hexa) at a concentration of 36.5 µg/kg
(Lorenz, 1994).
3.9.3.2 Metals
The formation of PCDDs/PCDFs (WHO, 1989) and other organochlorine
compounds (Sinkkonen et al., 1994) during metal reclamation is well
known. However, the occurrence of the brominated analogues is
documented in only a few cases. PBDDs/PBDFs (tri to hexa) were
analysed in ash samples (n = 2) from a metal reclamation factory in
southern Taiwan (Watanabe et al., 1993), and PXDDs/PXDFs were
identified in ash samples (n = 2) from a secondary copper furnace in
the USA (Harless et al., 1989).
Whereas PBDDs were not detected (detection limits: <0.25-<1.0
µg/kg) in the Taiwanese study, PBDFs reached total concentrations of
15-45 µg/kg (composed of triBDFs: 5-9 µg/kg, tetraBDFs: 4-15 µg/kg,
pentaBDFs: 3-9 µg/kg, and hexaBDFs: 3-12 µg/kg). PXDDs/ PXDFs were
also observed, but not quantified, in these samples. The ratio of
PBDFs to PCDFs was about 1 : 100. PBDEs were also present at somewhat
lower levels than PCDFs (Watanabe et al., 1993).
The ash from a secondary copper furnace contained Br1Cl6DDs
(1-34 µg/kg) and Br1Cl6DFs (17 µg/kg). These concentrations were
approximately one order of magnitude lower than those of PCDDs (27-411
µg/kg) and PCDFs (89-173 µg/kg) (Harless et al., 1989).
3.10 Presence in automotive exhaust
The combustion processes occurring in motors of automobiles can
lead to the formation of PBDDs/PBDFs and PXDDs/PXDFs (Buser,
1987a,b,c). Simulation experiments using iso-octane (plus additives)
as a defined fuel (Ballschmiter et al., 1990; Bacher et al., 1991) and
single measurements using commercial petrol (Haglund et al., 1988)
gave positive results. The same was true for a joint project between
three German universities, which was initiated to provide more and
representative data on the emission of PHDDs/PHDFs from motor vehicles
under realistic conditions (Hagenmaier et al., 1990a; Hutzinger et
al., 1990; Weberruß, 1990; Schwind et al., 1991; Dawidowsky, 1993). In
this study, 46 exhaust samples were taken from Otto (spark-ignition or
Otto-cycle engine) and diesel motors running with different commercial
fuels. The experiments were carried out mostly as stationary motor
tests.
PHDDs/PHDFs were detected in emissions of motors running on
leaded petrol and on unleaded petrol with and without catalytic
converters (Haglund et al., 1988; Hagenmaier et al., 1990a; Hutzinger
et al., 1990; Dawidowsky, 1993) and in emissions of diesel engines
(Hagenmaier et al., 1990a; Hutzinger et al., 1990; Dawidowsky, 1993)
(see also Table 22). Because of the brominated and chlorinated
scavengers (dibromo- and dichloroethane) used in leaded petrol, the
highest levels of PHDDs/PHDFs were found with this type of petrol. The
emissions reached several thousand ng/m3 (e.g. >6000 ng/m3 in
exhaust air or 90 000 ng/litre fuel used). Unleaded petrol, which
contained only trace amounts of halogenated compounds, caused much
lower emissions of PHDDs/PHDFs (approximately two orders of magnitude
lower). A further reduction to below 5% of the emissions from
non-halogenated petrol was achieved in experiments using catalytic
cleaning of the exhaust. The values for diesel engines were a little
higher than those found with the Otto motors run on unleaded petrol
and equipped with a catalytic converter. In the case of diesel
combustion, the sources of the halogens needed for the PHDD/PHDF
formation could not be clearly identified (e.g. Hagenmaier et al.,
1990a). The negative results of Haglund et al. (1988) obtained for a
heavy-duty diesel truck are thought to be due to their higher
detection limits compared with the later studies (Dawidowsky, 1993)
showing positive results for both diesel cars (n = 8) and trucks
(n= 2). Differences in results involving diesel fuel combustion can
also be due in part to differences in diesel fuel compositions
(seasonal, manufacture).
A considerable portion of the PHDDs/PHDFs consisted of
PBDDs/PBDFs and PXDDs/PXDFs (Dawidowsky, 1993; see also Table 22). In
exhaust gases from combustion of leaded petrol, they were more
abundant than PCDDs/PCDFs: PBDDs/PBDFs > PXDDs/ PXDFs approx.
PCDDs/PCDFs.
Generally, in all studies performed, the concentrations of
dibenzofurans exceeded those of the dibenzo- p-dioxins (see also
Table 22). There was a dominance of lower substituted homologues (mono
to tri), as can be seen, for example, from the homologue profile of
PBDFs and PXDFs shown in Table 23.
Brominated and mixed brominated/chlorinated congeners with
2,3,7,8-substitution were not determined. However, they may be present
at trace amounts as well as their chlorinated counterparts (Marklund
et al., 1987, 1990; Bingham et al., 1989; Hagenmaier et al., 1990a;
Bacher et al., 1991).
PHDDs/PHDFs were detected not only in exhaust samples but also in
residues adhering to mufflers. An absorption in the mufflers was
observed for PBDDs/PBDFs and PXDFs (Ballschmiter et al., 1990) and for
PCDDs/PCDFs (Ballschmiter et al., 1990; Marklund et al., 1990). The
two samples tested by Ballschmiter et al. (1990) showed some
correlations to the exhaust samples (dominance of lower brominated
homologues, prevalence of PBDFs over PBDDs and of PBDFs over PCDFs).
Results from traffic-related environmental samples are discussed
in chapter 5.
The increasing use of lead-free petrol will reduce the input of
PHDDs/PHDFs into the environment from this source (cf. Hagenmeier,
1994).
3.11 Formation during textile processing
Different textile processes (resin finish on the basis of
magnesium chloride [MgCl2], flame-proof finishes on the basis of
Sb2O3/hexa bromocyclododecane, ammonium bromide [NH4Br], and PVC)
were tested for the occurrence and formation of PBDDs/PBDFs (Sedlak et
al., 1996). The eight 2,3,7,8-substituted congeners of the 1994 German
Dioxin Directive (see Appendix I) were determined in the exhaust air,
the textiles before and after processing, and the chimney depositions.
Exhaust air concentrations were between 29 and 102 pg/m3.
Concentrations in textiles before processing were 1.35-5.65 ng/kg and
after processing 1.80-41.0 ng/kg. The only significant increase was
observed for the PVC process (5.65 versus 41.0 ng/kg). Chimney
depositions showed concentrations of 92.3-6618 ng/kg. Only traces of
PXDDs/PXDFs were detected in the textiles, but chimney depositions
contained up to 17 µg/kg.
Table 22. Emissions of PHDDs/PHDFs from automobile combustion engines under
various motor/fuel conditionsa,b
PHDDs/PHDFs Mean emissions (ng/litre fuel)c
Leaded petrol Unleaded petrol Unleaded petrol with Diesel fuel
catalytic converter
(n = 4) (n = 6) (n = 3) (n = 8)
Dibenzo-p-dioxins
PBDDs 1576 (Br1-Br4) 18 (Br1-Br4) 0.8 (Br1-Br3) 1.9 (Br1-Br3)
PXDDs 742 (up to X5) 4 (up to X4) 0.6 (up to X4) 0.4 (up to X3)
PCDDs 606 (Cl1-Cl8) 42 (Cl1-Cl8) 0.9 (Cl1-Cl8) 3.4 (Cl1-Cl8)
Total PHDDs 2924 64 2.3 5.7
Dibenzofurans
PBDFs 45 428 (Br1-Br6) 364 (Br1-Br5) 22.6 (Br1-Br5) 136 (Br1-Br4)
PXDFs 22 418 (up to X5) 101 (up to X5) 6.9 (up to X5) 24 (up to X4)
PCDFs 21 698 (Cl1-Cl8) 502 (Cl1-Cl8) 7.9 (Cl1-Cl8) 167 (Cl1-Cl8)
Total PHDFs 89 544 967 37.4 327
a Adapted from Dawidowsky (1993).
b The exhaust samples were obtained from Otto motors run on leaded petrol and unleaded petrol with and
without catalytic converters and from diesel engines. Br/Cl content of fuels: leaded petrol, 76/70 mg/kg;
unleaded petrol and diesel, <1 mg/kg.
c Homologue groups present are given in parentheses.
Table 23. Homologue distribution pattern of PBDFs
and PXDFs detected in exhaust samples (n = 4) from
vehicle motors run on leaded petrola
Homologue groups Mean emissions
(ng/litre fuel)
PBDFs
MonoBDFs 27 501
DiBDFs 17 496
TriBDFs 354
TetraBDFs 70
PentaBDFs 6.6
HexaBDFs 0.2
HeptaBDFs n.d.b
PXDFs
Br1Cl1DFs 18 941
Br1Cl2DFs 377
Br2Cl1DFs 2649
Br1Cl3DFs 67
Br2Cl2DFs 174
Br3Cl1DFs 138
Br3Cl2DFs 19.5
Br4Cl1DFs 7.4
a Adapted from Dawidowsky (1993).
b n.d. = not detected.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
Because of their physicochemical properties (see chapter 2),
PBDDs/PBDFs are expected to be preferably distributed into carbon- or
fat-rich compartments.
4.1.1 Air
Airborne PBDDs/PBDFs released from various sources (see chapter
3) are transported in both the particulate and vapour phase. In
traffic-related air samples, the lower halogenated PBDDs/PBDFs (Br1,
Br2), PCDDs/PCDFs (Cl1 through Cl3), and PXDDs/PXDFs (X2) were detected
predominantly in the gaseous phase (Ballschmiter et al., 1990). In
contrast, Lutes et al. (1992a) found that tetra- and pentaBDDs/BDFs
generated by combustion of polyurethane foam containing PBDEs
partitioned primarily to the particulate phase. The ratio of
concentrations between particulate phase and particulate plus vapour
phase was 0.95-0.99. Monitoring of ambient city air (Harless et al.,
1992) for PBDDs/PBDFs (tetra through hexa), PCDDs/PCDFs (tetra through
octa), and PXDDs/PXDFs (tetra: Br1Cl3) revealed that most of the
penta- and hexa- and about 60% of the tetraBDDs/BDFs were associated
with the particulate phase. Most of the hepta- and octaCDDs/CDFs were
also particle-bound, whereas the lower chlorinated congeners,
including the mixed tetrahalogenated compounds, were distributed to
the gaseous phase.
During atmospheric transport, photochemical transformation can
occur (see section 4.2.1). There are no data on deposition of
PBDDs/PBDFs to soil, vegetation, or water.
4.1.2 Water and sediments
Few data are available on the movement of PBDDs/PBDFs through
water and associated sediment.
Watanabe (1988) determined adsorption coefficients on sediment of
several halogenated organic compounds, including PBDFs. Adsorption
coefficients (log Kd; where Kd = [µg/g sediment]/[µg/ml water]) were
4.51, 4.62, and 4.79 for triBDFs, tetraBDFs, and pentaBDFs,
respectively.
4.1.3 Soil
To date, no experimental data on the behaviour of PBDDs/PBDFs in
soil are available. Mobility of PBDDs/PBDFs in soil is assumed to be
governed by their low water solubility (see chapter 2) and their
strong adsorption to particulate matter. Their mobility depends on
soil type (organic matter content, dissolved humic acids, pH, etc.),
weather conditions, and congeners studied. In general, mobility is
expected to be rather low; however, in special cases, such as at waste
disposal sites where organic solvents are concomitantly present,
significant leaching could occur. As shown for PCDDs (Webster et al.,
1986), the presence of dissolved humic substances in water can also
enhance the solubility of such compounds.
Other transport mechanisms to be considered include transport via
dust particles or volatilization (to air and vegetation), via eroded
soil (to surface waters), and via biomass removal, as reported for
PCDDs/PCDFs (Young, 1983; WHO, 1989).
The persistence of PCDDs/PCDFs (tetra to octa) in soil
environments was found to be high (e.g. Orazio et al., 1992), and
movement of 2,3,7,8-TeCDD was primarily associated with liquid carrier
contaminants such as petroleum oil (Kapila et al., 1989).
2,3,7,8-TeCDD half-lives in soil were calculated to be as high as
10-12 years (di Domenico et al., 1980; Young, 1983).
4.1.4 Biota
Isolated reports have been published on the presence of
PBDDs/PBDFs in animals and plants (see chapter 5). However, no data
are available on the transport of PBDDs/PBDFs and their distribution
between environmental media and biota. The similar high octanol/water
partition coefficients calculated for selected PCDDs/PCDFs,
PBDDs/PBDFs, and PXDDs/PXDFs (Fiedler & Schramm, 1990; see also Table
5 in section 2.2.1) indicate that PBDDs/PBDFs may have a
bioavailability qualitatively comparable to that reported for
PCDDs/PCDFs (de Wit, 1993; Rappe, 1993). A factor influencing the
range of bioavailability both within the homologue groups and between
analogues is the molecule size.
There are no data on the transfer of PBDDs/PBDFs to plants via
deposition processes or uptake from soil or on their transfer along
the terrestrial food-chain. However, it may be expected that the
brominated congeners behave qualitatively like their chlorinated
analogues.
PCDDs/PCDFs can also enter aquatic biota, primarily via sediment
(Fairchild et al., 1992; de Wit, 1993; Fletcher & McKay, 1993; Pruell
et al., 1993). PBDDs/PBDFs are expected to have a similar potential
(see also chapter 5).
4.2 Environmental transformation
4.2.1 Photochemical degradation
Photolysis of PBDDs/PBDFs was studied in organic solvents and on
quartz surfaces in the laboratory as well as in soil and on soot
particles under outdoor conditions. The slowest photolytic reactions
were observed under the latter, more environmentally relevant,
conditions. From experiments with octaCDD, it is known that
photochemical dechlorination on soil takes place in the axial
positions, resulting in 2,3,7,8-TeCDD; in solutions, the lateral
chlorines are removed, resulting in 1,4,6,9-TeCDD (Kieatiwong et al.,
1990).
Laboratory studies showed that PBDDs/PBDFs and PXDDs/ PXDFs
degrade in organic solvents after irradiation with sunlight,
artificial light, or UV light (Buser, 1988; Neupert et al., 1988;
Lahaniatis et al., 1991; Lenoir et al., 1991; Chatkittikunwong &
Creaser, 1994a; Ritterbusch et al., 1994a; Watanabe et al., 1994). The
major photochemical pathway is a reductive debromination, resulting in
the formation of lower brominated congeners (Buser, 1988; Neupert et
al., 1988; Lenoir et al., 1991; Chatkittikunwong & Creaser, 1994a;
Ritterbusch et al., 1994a) and, finally, in the formation of
unsubstituted dibenzo- p-dioxin and dibenzofuran (Buser, 1988). Other
products also observed after photolysis were diaryl ethers, which were
generated by ring fission of mono- and diBDDs but not of higher
brominated PBDDs (Lenoir et al., 1991), and, occasionally, benzyl
derivatives. The latter were formed by reaction of photoproducts of
2,3,7,8-TeBDD with the solvent toluene (Neupert et al., 1988). Studies
of different PBDD/PBDF congeners indicated that the rate of
decomposition depends on the bromine substitution pattern. Generally,
higher brominated congeners and those with lateral bromines had
shorter half-lives (Buser, 1988; Neupert et al., 1988; Lenoir et al.,
1991); in one experiment (Lenoir et al., 1991), however, octaBDD was
more stable than a hexaBDD. Two dibenzofurans (2,3,7,8-TeBDF and
octaBDF) dissolved in toluene and irradiated by fluorescent light for
several days were found to decompose more rapidly than the
corresponding dibenzo- p-dioxins (Neupert et al., 1988). The rate of
photolysis decreased with increasing polarity of the solvent, as
tested for 2,3,7,8-TeBDD (Lahaniatis et al., 1991) and 1,2,3,4-TeBDD
(Lenoir et al., 1991).
The calculated half-lives (assuming a first-order kinetic scheme)
were in the range of minutes (use of direct sunlight or UV light and
quartz vials) or of the order of 100-1000 h (use of laboratory
daylight or artificial light and glass vials). For example, half-lives
of 0.8 and 0.7 min were estimated for TBDD and TBDF, respectively,
after 60 min of sunlight irradiation in organic solution in quartz
vials (Buser, 1988). The mean half-lives found for PBDDs and PXDDs
exposed to laboratory daylight in dodecane solution stored in glass
vials were 480 h for tetraBDDs, 150 h for pentaBDDs, and 300-995 h for
various PXDDs (tetra- to hexahalogenated congeners with various Br/Cl
combinations: Br1-3/Cl1-5) (Chatkittikunwong & Creaser, 1994a).
Compared with the chlorinated analogues, PBDDs/PBDFs had a faster
photolytic reaction in iso-octane, with half-lives of 3 min for
1,2,3,4-TeBDD and 380 min for 1,2,3,4-TeCDD (Buser, 1988; Lenoir et
al., 1991). The easier loss of bromine than of chlorine from the
parent molecule has important consequences for the PXDDs/PXDFs, in
that they undergo photolytic degradation to form PCDDs/PCDFs.
Consistently short photolytic half-lives (0.5 - 4 min) were observed
for the mixed mono- and dibromotetrachloroDDs/DFs tested, whereas the
resulting tetraCDDs/CDFs were much more stable (Buser, 1988).
The possibility of removing PBDDs/PBDFs from laboratory wastes by
UV photolysis was examined by Ritterbusch et al. (1994a,b). This
method of decontamination was successful when solutions with a low
concentration of other photochemically active species were applied. It
was unsuitable for the degradation of the PBDD/PBDF contamination of
bromophenols, because the rate of photochemical PBDD/PBDF formation
was greater than the rate of degradation. The rate of photolytic
degradation was slower in waste solutions than in PBDD/PBDF standard
solutions (Ritterbusch et al., 1994a). The degradation of PBDDs/PBDFs
occurred faster than that of PCDDs/PCDFs (Ritterbusch et al., 1994b).
Photolysis of PBDDs/PBDFs (Br4) and PXDDs/PXDFs (X5, X6: mono- and
dibromo-2,3,7,8-TeCDDs/CDFs) occurred much more slowly on quartz
surfaces, under sunlight, than in organic solvents (Buser, 1988). The
photolytic half-lives of the tetrabrominated congeners tested were in
the range of 30 h; those for the chlorinated analogues were in the
range of 65-300 h (see Table 24).
These solid-phase experiments appeared to predict the real
environmental behaviour of PBDDs/PBDFs far better than the organic
solution-phase experiments. Photodegradation studied in soil
(Chatkittikunwong & Creaser, 1994a) and on airborne particles (Lutes
et al., 1990, 1992a,b) under outdoor conditions was found to be a slow
process. Half-lives of PBDDs and PXDDs in soil samples that were
placed outdoors over a 3-month period were in the range of 600 - 4000
h for tri- to hexahalogenated congeners. These half-lives were, on
average, four times longer than those estimated for the same congeners
in solution in the laboratory. The half-life of tetraBDD isomers in
surface soil was estimated to be 3 - 6 months (Chatkittikunwong &
Creaser, 1994a). Other tests conducted in Teflon film chambers under
realistic outdoor conditions showed that PBDDs/PBDFs (tetra through
hexa) adsorbed on incinerator soot particles remained relatively
stable or degraded only slowly during 6 h. No significant decay of the
2,3,7,8-substituted congeners was found (Lutes et al., 1990, 1992a,b).
The photodegradation, if occurring, was believed to have a half-life
of at least 3 h and probably much longer (Lutes et al., 1992a,b).
Similarly, particle-bound emissions of tetraBDDs, tetraBDFs, and
pentaBDFs (isomers not determined), produced from high-temperature
(670 - 780°C) combustion of polyurethane foam containing PBDEs and
monitored in outdoor Teflon chambers in the presence of sunlight, were
stable over several hours. In contrast, a decay of tetraBDDs was
observed after low-temperature (400 - 470°C) combustion of the
polyurethane foam (Birla & Kamens, 1994). Little disappearance of
higher halogenated PXDFs was seen in preliminary tests monitoring
halogenated dibenzofurans associated with airborne dust collected on a
glass filter and exposed to sunlight (Watanabe et al., 1994).
Table 24. Sunlight-induced photolysis of tetrahalogenated
dibenzo-p-dioxins and dibenzofurans dispersed as solid filmsa
Compound Estimated half-lifeb
(h)
Dibenzo-p-dioxins
1,2,3,4-TeBDD 26
2,3,7,8-TeBDD 32
1,2,3,4-TeCDD 65
2,3,7,8-TeCDD 300
Dibenzofurans
2,3,7,8-TeBDF 35
2,3,7,8-TeCDF 120
a From Buser (1988).
b Values derived from few data points
obtained after a total exposure time of
10 and 20 h; estimated accuracy of half-lives + 50%.
4.2.2 Microbial degradation
Like other halogenated aromatics, PBDDs/PBDFs seem to be very
recalcitrant against microbial degradation. Only a monobrominated
dibenzofuran (2-bromodibenzofuran) was tested and found to be degraded
by bacteria. Bacteria of the genus Pseudomonas isolated from water of
the river Rhine or of industrial wastewater treatment plants and
cultured in the laboratory on 1,2-dichlorobenzene or 4-chlorophenol as
single-carbon source were able to oxidize 2-bromodibenzofuran
(Springer & Rast, 1988).
There are some reports on the degradation of the parent,
non-halogenated dibenzo- p-dioxin and/or dibenzofuran by microorganisms
in soil and by white rot fungi (Cerniglia et al., 1979; Hammel et al.,
1986; Bumpus, 1989; Hofmann et al., 1992). Some active bacterial
strains were found in several genera - for example, Beijerinckia
(Klecka & Gibson, 1980), Brevibacterium (Strubel et al., 1989, 1991),
Pseudomonas (Klecka & Gibson, 1979; Foght & Westlake, 1988; Springer &
Rast, 1988; Fortnagel et al., 1989a,b, 1990; Harms et al., 1990; Figge
et al., 1991), Sphingomonas (Wittich et al., 1992; Figge et al., 1993;
Happe et al., 1993), and Staphylococcus (Monna et al., 1993).
4.3 Bioaccumulation and biomagnification
At present, bioaccumulation, bioconcentration, or
biomagnification factors for specific PBDD/PBDF congeners are not
available. The presence of PBDDs/PBDFs in animals (section 5.1.6.2)
and in humans (section 5.3.2), as seen in a few isolated studies, is
indicative of their accumulation potential (see also section 6.5.1).
This is to be expected from the lipophilic properties of PBDDs/PBDFs
and the high accumulation potential of better-studied related
compounds, such as PCDDs/PCDFs (e.g. WHO, 1989; Cook et al., 1991).
The extent of accumulation and biomagnification may vary depending on
species and congeners tested, as found for PCDDs/PCDFs (e.g. de Wit,
1993; de Wit et al., 1993; Rappe, 1993; Walker & Peterson, 1994).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
Limited data are available on levels of PBDDs/PBDFs in the
environment. Most monitoring data available for PBDDs/PBDFs have been
collected near identified sources (e.g. roadways).
5.1.1 Air
5.1.1.1 Ambient air
Sources of PBDDs/PBDFs are complex and may have changed in recent
years. Therefore, temporal differences and changes must be taken into
account. In some European countries, the use of leaded petrol (and the
use of scavengers) has been largely abandoned; in the USA, the use of
leaded petrol with scavengers declined even earlier than in Europe. In
other countries, this type of petrol may still be in use. Thus, some
of the data given may not be representative of the present situation
(e.g. in motorway tunnels, urban areas, etc.). Furthermore, the type
of waste incineration has drastically changed within the last decade
in industrialized countries. These variables have to be kept in mind
when evaluating the following compilations and Table 25.
1) Air samples
PBDD/PBDF levels analysed in urban air, more or less close to
traffic, and in air collected at industrial sites are compiled in
Table 25. The sampling methods used covered particulate-associated and
vapour-phase PBDDs/PBDFs. Only low concentrations of PBDDs (mono
through tetra) were detected, with maximum concentrations of about
0.85 pg monoBDDs/m3 in a motorway tunnel and an underground garage.
Higher brominated (penta through octa) homologues were not detected or
were not analysed (Table 25). No PXDDs were found (Ballschmiter et
al., 1990).
PBDFs were found to a greater extent than PBDDs (Table 25). Of
the homologue groups, mono- to hexaBDFs could be detected; hepta- and
octaBDFs were not identified. Because of the small database, only
trends in the homologue pattern can be seen. It appears that lower
brominated homologues (mono through tetra) dominate, particularly in
samples related to traffic. The highest concentration was measured for
monoBDFs in an air sample from a motorway tunnel and amounted to 74
pg/m3 (Table 25). However, Harless et al. (1992) found pentaBDFs and
hexaBDFs (0.22 and 0.30 pg/m3, respectively) in addition to tetraBDFs
(0.19 pg/m3 air) only after long-term sampling (7 days; 2660 m3 air) of
ambient air (at Research Triangle Park, NC, USA). Eight tetraBDF, two
pentaBDF, and one hexaBDF isomers were detected. Using shorter
sampling periods (24 h), only tetraBDFs were detected, at
concentrations ranging from 0.13 to 0.20 pg/m3. The lower
concentrations of PBDFs reported from this study would probably be due
in part to the absence of scavengers in motor fuel in North America.
The highest concentrations of PBDF homologue groups were found in
motorway tunnels (Table 25). No data are available on concentrations
of lower brominated homologues (Br1 through Br3) in samples from
industrial areas. The highest concentrations of tetraBDFs were
reported in the vicinity of a resource recycling centre in Taiwan
(2.1 - 6.6 pg/m3) and in the German motorway tunnels (n.d. - 3.4 pg/m3)
(see Table 25).
The sums of total PBDDs/PBDFs (tri to hexa) in the air of a
motorway tunnel, of a city, and of a suburban area in Germany were
22.3/0.7 pg/m3 (means; n = 3), 1.97/0.08 pg/m3 (means; n = 6), and 0.59
pg/m3/n.d. (means; n = 3), respectively. The concentrations of
2,3,7,8-TeBDF ranged from n.d. to 0.28 (n = 12) and those of
1,2,3,7,8-PeBDF from n.d. to 0.08 pg/m3 (n = 12) (Päpke et al., 1990;
Hiester, 1992).
PBDFs and their possible precursors, PBDEs, were concomitantly
found in air samples from industrial areas of Taiwan and Japan. Tri-,
tetra-, penta-, and hexaBDE concentrations ranged from 6 to 34, from
10 to 55, from 5 to 34, and from 6 to 81 pg/m3, respectively (Watanabe
et al., 1992).
Of the PXDFs, dihalogenated dibenzofurans were detected in
traffic-related air samples at concentrations of up to 40.8 pg/m3
(Cl1Br1DFs), which is higher than found for the brominated congeners.
Concentrations of monoBDFs exceeded the levels of monoCDFs
(Ballschmiter et al., 1990).
2) Dust samples
Outdoor dust samples were collected in motorway tunnels in
Germany and, as a control, in the eaves of a house in a German rural
area remote from the source (traffic). These and samples obtained from
the USA (roadside dust) and from Japan (dust of a motorway tunnel)
were analysed for PCDDs/PCDFs, PBDDs/PBDFs (Br1 through Br4), and
PXDDs/PXDFs (up to X4) (Ballschmiter et al., 1990). The results are
summarized in Table 26 (PBDDs/PBDFs) and Table 27 (PXDDs/PXDFs).
Whereas the control sample (from eaves) contained no PBDDs (detection
limit: 20 ng/kg dust) and only 30 ng monoBDFs/kg dust, the samples
taken close to traffic showed a complex pattern of homologues.
Table 25. Concentrations of PBDDs/PBDFs in ambient air
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
Dibenzo-p-dioxins
MonoBDDs Germany motorway tunnel (1) n.sp. 0.85 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 0.50-0.86 Ballschmiter et al. (1990)
DiBDDs Germany motorway tunnel (1) n.sp. <0.15 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d.-<0.15 Ballschmiter et al. (1990)
TriBDDs Germany motorway tunnel in Essen (3) 1990 0.37-0.75c Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.05-0.09 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.03) Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d.-0.09 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 0.3-0.5 Watanabe et al. (1992)
TetraBDDs Germany motorway tunnel in Essen (3) 1990 n.d.-0.18 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d.-0.04 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d.-0.3 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d.-0.2 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
PentaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d. Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d. Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
HexaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. (0.1-0.2) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.1-0.4) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.2) Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d. Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d. Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
HeptaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. (0.3-0.5) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.5-0.7) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.5) Päpke et al. (1990)
Table 25. (Continued)
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
OctaBDD Germany motorway tunnel in Essen (3) 1990 n.d. (0.7-0.8) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (1) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (1) Päpke et al. (1990)
Dibenzofurans
MonoBDFs Germany motorway tunnel (1) n.sp. 73.72 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 37.26-42.12 Ballschmiter et al. (1990)
DiBDFs Germany motorway tunnel (1) n.sp. 28.50 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 2.12-6.20 Ballschmiter et al. (1990)
TriBDFs Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 17-25 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.71-2.0 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 0.40-0.82 Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.3-1.0 Watanabe et al. (1992)
TetraBDFs Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 1.50-3.35 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.15-0.53 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.2-2.3 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 2.1-6.6 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 0.13-0.20 Harless et al. (1992)
PentaBDFs Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.2-3.7 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 1.8-7.7 Watanabe et al. (1992)
Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 0.32-O.41 Päpke et al. (1990)
Germany urban area of Düsseldorf(6) 1990 0.08-0.22 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d.-0.14 Päpke et al. (1990)
USA Research Triangle Park, NC (5) 1990/91 n.d.-0.22 Harless et al. (1992)
HexaBDFs Germany motorway tunnel in Essen (3) 1990 n.d. (0.1-0.2) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.2-0.4) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.2) Päpke et al. (1990)
Table 25. (Continued)
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
HexaBDFs Japan urban area of Osaka (5) n.sp. 0.3-5.1 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 1.1-3.4 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d.-0.30 Harless et al. (1992)
HeptaBDFs Germany motorway tunnel in Essen (3) 1990 n.d. (0.4-0.5) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.3-0.7) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.5) Päpke et al. (1990)
OctaBDF Germany motorway tunnel in Essen (3) 1990 n.d. (0.7-0.8) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (1) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (1) Päpke et al. (1990)
a n.sp. = not specified.
b n.d. = not detected (detection limits in parentheses, if specified).
c Containing possibly confounding components.
Concentrations of PBDDs in the dust samples were low, ranging
from n.d. to 690 ng/kg, from n.d. to 960 ng/kg, from n.d. to 170
ng/kg, and from n.d. to 110 ng/kg for mono-, di-, tri-, and tetraBDDs,
respectively (Ballschmiter et al., 1990).
PBDFs were present at higher levels than PBDDs and reached
maximum values of 8860 ng monoBDFs/kg dust (motorway tunnel in
Germany), 22 280 ng diBDFs/kg dust (motorway tunnel in Japan), 5680 ng
triBDFs/kg dust (highway in the USA), and 650 ng tetraBDFs/kg dust
(highway in the USA). Dust samples (n = 7) taken from motorway tunnels
in Germany showed decreasing levels from mono- through tetraBDFs.
Concentrations ranged from 330 to 8860 ng/kg, from 300 to 6730 ng/kg,
and from n.d. to 920 ng/kg for mono-, di-, and triBDFs, respectively;
tetraBDFs were not detected (Table 26).
Airborne dust collected at an urban area in Osaka (Japan)
contained PBDFs (tetra to hexa) and PCDDs/PCDFs (tetra to octa) at
concentrations ranging from 4.2 to 17 pg/m3 (n = 7) and from 30 to 250
pg/m3, respectively. Monobrominated PXDD/PXDF (tetra to octa)
concentrations were roughly estimated at one-tenth to one-quarter
those of PCDDs/PCDFs in the same samples (Watanabe et al., 1995).
PXDDs (Cl1Br1DDs) were identified at a maximum concentration of
170 ng/kg in a motorway tunnel (Table 27). PXDFs were found in dust
samples from motorway tunnels, highways, and roads at concentrations
ranging from 300 to 9600 ng/kg for X2DFs, from n.d. to 7870 ng/kg for
X3DFs, and from n.d. to 830 ng/kg for X4DFs (Table 27).
There are no data available for the 2,3,7,8-substituted
PBDDs/PBDFs (Ballschmiter et al., 1990).
Generally, the dust samples from the German motorway tunnels
contained higher concentrations of PBDFs than of PCDFs, both
consisting of lower halogenated homologues (e.g. 16 510 ng mono- to
triBDFs/kg versus 5610 ng mono- to tetraCDFs/kg). The relation between
concentrations of PBDDs and PCDDs was varying. Whereas within PBDDs
only mono- and dibrominated homologues could be detected (at sum
concentrations of up to about 1200 ng/kg), there was a dominance of
hepta- and octaCDDs within PCDDs (total concentrations of up to about
1700 ng/kg). Altogether, these patterns may be reflective of
contributions from automobile exhaust. The sample from the eaves
(remote from traffic, long-term residue) showed lower concentrations
of PBDFs (30 ng monoBDFs/kg) than of PCDFs (sum of mono- to hexaCDFs:
735 ng/kg), no PBDDs, and hepta- to octaCDDs (at 180 ng/kg). The lower
proportion of brominated compounds in the latter sample may be partly
due to an easier photo-dehalogenation compared with the chlorinated
ones (Ballschmiter et al., 1990).
Table 26. Concentrations of PBDDs/PBDFs in outdoor dust samplesa
Congener Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Dibenzo-p-dioxins
MonoBDDs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 n.d.-180
Germany motorway tunnel (outside of city) (2) 1989 n.d.-690
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 <20
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 100
DiBDDs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 n.d.-120
Germany motorway tunnel (outside of city) (2) 1989 n.d.-540
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 960
TriBDDs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 170
TetraBDDs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months)(1)d
Japan motorway tunnel (1)e 1978 110
Dibenzofurans
MonoBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 1050-2570
Germany motorway tunnel (outside of city) (2) 1989 330-8860
Table 26. (Continued)
Congener Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
MonoBDFs Germany eaves (rural area) (1) n.sp. 30
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 510
USA (Washington, DC) highway (inside of city; pooled sample 1982 2100
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 1370
DiBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 1560-5030
Germany motorway tunnel (outside of city) (2) 1989 300-6730
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 690
USA (Washington, DC) highway (inside of city; pooled sample 1982 2700
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 22 280
TriBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 75-310
Germany motorway tunnel (outside of city) (2) 1989 n.d.-920
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 530
USA (Washington, DC) highway (inside of city; pooled sample 1982 5680
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 4820
TetraBDFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 650
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 310
a Adapted from Ballschmiter et al. (1990).
b n.sp. = not specified.
c n.d. = not detected (detection limit 20 ng/kg).
d Reference sample from US National Institute of Standards.
e Reference sample from Japanese National Institute for Environmental Studies.
Table 27. Concentrations of PXDDs/PXDFs in outdoor dust from various sourcesa
PXDDs/PXDFs Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Dibenzo-p-dioxins
Cl1Br1DDs Germany motorway tunnel (inside of city) (5) 1988/89 n.d.
Germany motorway tunnel (outside of city) (2) 1989 n.d.-170
Germany eaves (rural area) (1) n.sp. n.d.
USA street (2)d 1978 and 1982 n.d.
Japan motorway tunnel (1)e 1978 n.d.
Dibenzofurans
X2DFs
Cl1Br1DFs Germany motorway tunnel (inside of city) (5) 1988/89 1150-4340
Germany motorway tunnel (outside of city) (2) 1989 300-9600
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 1280
USA (Washington, DC) highway (inside of city; pooled sample 1982 4150
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 7220
X3DFs
Cl1Br2DFs Germany motorway tunnel (inside of city) (5) 1988/89 180-830
Germany motorway tunnel (outside of city) (2) 1989 n.d.-1300
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 2270
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 1130
Table 27. (Continued)
PXDDs/PXDFs Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Cl2Br1DFs Germany motorway tunnel (inside of city) (5) 1988/89 n.d.-180
Germany motorway tunnel (outside of city) (2) 1989 n.d.-970
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 1160
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 7870
X4DFs
Cl2Br2DFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
Japan motorway tunnel (1)e 1978 n.d.
Cl3Br1DFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 830
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 370
Cl3Br1DFS Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 380
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 170
a Adapted from Ballschmiter et al. (1990).
b n.sp. = not specified.
c n.d. = not detected (detection limit 20 ng/kg).
d Reference sample from US National Institute of Standards.
e Reference sample from Japanese National Institute for Environmental Studies.
Another sample of roadside dust (n = 1; collected after the ban
of halogenated scavengers in petrol) from Germany was analysed for
tetra- to heptaBDDs/BDFs and tetra- to octaCDDs (Schacht et al.,
1995). PBDDs were not detected, whereas the sum of PBDFs amounted to
52 ng/kg (no 2,3,7,8-substituted congeners). The sum concentration of
PCDDs/PCDFs was 581 ng/kg.
5.1.1.2 Indoor air
PBDDs/PBDFs were determined in rooms equipped with a number of
operating electronic appliances (computers, monitors, printers; Tables
28, 29, and 30; Schacht et al., 1995), in an underground garage (Table
31), in a private room, and in rooms after accidental fires (see
chapter 3).
PBDDs (tetra through octa) were not detected in air samples from
rooms equipped with electronic appliances (Chriske et al., 1990), but
they were present in dust samples (n = 3) from computer rooms (mainly
tetraBDDs: 0.03 - 1 µg/kg; 2,3,7,8-TeBDD: n.d. - <2 µg/kg; Schacht et
al., 1995). Dust samples from an underground garage tested for
mono- to tetraBDDs contained no or only traces of monoBDDs and no
PXDDs (Table 31) (Ballschmiter et al., 1990).
Total concentrations of PBDFs (Br4 through Br7) measured in air
from "computer"-related rooms ranged from 0.23 to 1.27 pg/m3, whereas
dust samples collected in the same rooms yielded total levels of
2.43 - 5.48 µg/kg dust (Tables 28 and 30; UBA, 1992). The homologue
pattern was also different. In contrast to air (see Table 28), the
homologue pattern in dust was dominated by hexa- and heptaBDFs (Table
30; UBA, 1992). In addition, only in dust samples were there
indications for the presence of 2,3,7,8-substituted tetra- and
pentaBDFs (Table 30; UBA, 1992). Another study (Schacht et al., 1995)
found comparable PBDF concentrations (sum PBDFs [Br4-Br8]: 3.6 - 3.8
µg/kg; 2,3,7,8-TeBDF: 0.01 - 0.07 µg/kg) and profiles in dust samples
(n = 3), which were obtained from vacuum cleaning and from the air
conditioning system of computer rooms. The PBDF concentrations in
house dust (n = 1) were lower by a factor of 10. The sum concentration
of PBDDs/PBDFs equalled that of PCDDs/PCDFs in dust from computer
rooms (3.6 - 4.8 µg/kg versus 0.5 - 4.5 µg/kg) but was lower than that
of PCDDs/PCDFs in house dust (0.3 µg/kg versus 34.2 µg/kg) (Schacht et
al., 1995). Chemicals of low volatility tend to accumulate on solid
surfaces and in house dust (Gebefügi, 1989; Gebefügi & Kreuzig, 1989).
It should be noted that concentrations of PBDEs concomitantly
measured in the air and dust samples of the offices were considerably
higher than those of PBDFs. Total PBDE concentrations (Br4 through
Br10) ranged from 97 to 969 pg/m3 air and from 507 to 2939 µg/kg dust
(UBA, 1992).
Table 28. Indoor air concentrations of PBDFs
Congener groups Type of room Number of Equipment in room Concentrationa Reference
samples (number per room) (µg/m3)
TetraBDFs office 6 display and computer monitors (2-8) 0.21-0.66 Chriske et al. (1990)
office (police central 1 monitors (approximately 50) 0.41 UBA (1992)
traffic control)
office (direction rooms 3 display and computer monitors <0.1-0.47 UBA (1992)
of a TV studio) (approximately 50)
2,3,7,8-TeBDF office 4 monitors (approximately 50) n.d. (0.03-0.08) UBA (1992)
PentaBDFs office 6 display and computer monitors (2-8) 0.03-0.61 Chriske et al. (1990)
office (police central 1 monitors (approximately 50) n.d. (n.sp.) UBA (1992)
traffic control)
office (direction rooms 3 display and computer monitors 0.1-0.5 UBA (1992)
of a TV studio) (approximately 50)
1,2,3,7,8-PeBDF office 4 monitors (approximately 50) n.d. (0.05-0.1) UBA (1992)
2,3,4,7,8-PeBDF office 4 monitors (approximately 50) n.d. (0.05-0.1) UBA (1992)
HexaBDFs office 10 monitors (2-8/approximately 50) n.d.-0.4 (0.1) Chriske et al. (1990);
UBA (1992)
HeptaBDFs office 10 monitors (2-8/approximately 50) n.d. (0.1-0.2) Chriske et al. (1990);
UBA (1992)
OctaBDF office 6 display and computer monitors (2-8) n.d. (n.sp.) Chriske et al. (1990)
Sum PBDFs office 6 display and computer monitors (2-8) 0.23-1.18 Chriske et al. (1990)
office 4 monitors (approximately 50) 0.25-1.27 UBA (1992)
a n.d. = not detected (detection limit in parentheses); n.sp. = not specified.
Table 29. Correlation between number of monitors operating
in a room and sum of concentrations of tetra- and pentaBDFsa
Number of monitors per room Sum of concentrations of
tetraBDFs and pentaBDFs
(µg/m3)b
0 (ambient air) <0.1
0 0.2-0.39
2 0.42-0.47
4 0.57-0.80
5c 0.23
8 1.18
a Adapted from Chriske et al. (1990).
b 2,3,7,8-Substituted congeners not determined:
c Equipment from one manufacturer.
Table 30. Concentrations of PBDFs in indoor dust samples collected
in rooms equipped with a number of display and/or computer monitorsa
PBDFs Concentration (µg/kg)b
Sample 1 Sample 2 Sample 3 Sample 4
(Police traffic (TV studio) (TV studio) (TV studio)
control office)
TetraBDFs 0.351 0.196 0.265 0.295
2,3,7,8-TeBDFc n.d. (0.001) n.d. (0.001) 0.002 0.005
PentaBDFs 0.159 0.331 0.691 0.456
1,2,3,7,8-PeBDFc 0.004 0.012 0.020 0.015
2,3,4,7,8-PeBDFc 0.003 0.004 0.006 0.005
HexaBDFs 1.71 1.44 1.78 0.982
HeptaBDFs 2.41 3.51 0.744 0.693
Sum PBDFs 4.63 5.477 3.48 2.426
a Adapted from UBA (1992).
b n.d. = not detected (detection limit in parentheses).
c Maximum value given; co-elution with internal standard, etc., cannot be excluded.
Table 31. Concentrations of PBDDs/PBDFs and PXDDs/PXDFs in dust samples
collected in Germany in 1988/89 from an underground garagea
Concentrationb (ng/kg)
Floor Wall Waste air motors
(n = 6) (n = 1) (n = 1)
Dibenzo-p-dioxins
MonoBDDs n.d. n.d. 40
DiBDDs-tetraBDDs n.d. n.d. n.d.
Cl1Br1DDs n.d. n.d. n.d.
Dibenzofurans
MonoBDFs 150-390 550 2800
DiBDFs 280-560 820 3500
TriBDFs n.d. n.d. 500
TetraBDFs n.d. n.d. n.d.
Cl1Br1DFsc 180-630 4300
Cl1Br2DFsc n.d. 790
Cl2Br1DFsc n.d. 520
Cl1Br3DFsc n.d. n.d.
Cl2Br2DFsc n.d. n.d.
Cl3BhDFsc n.d. n.d.
a Adapted from Ballschmiter et al. (1990).
b n.d. = not detected (detection limit 20 ng/kg).
c Concentrations for floor and wall dust samples combined.
The PBDF profile (Br1 through Br4) found in dust from an
underground garage was dominated by the lower brominated homologues
(Ballschmiter et al., 1990). Concentrations ranged from 150 to 560
ng/kg dust for mono- and diBDFs in samples from the floor, but they
increased at special sampling sites (wall, ventilation motors) to a
maximum of 3500 ng diBDFs/kg dust (Table 31).
PXDFs were analysed for and detected in dust samples from the
underground garage (Ballschmiter et al., 1990). Their concentrations
ranged from n.d. to 4300 ng/kg dust for di- through tetrahalogenated
PXDFs (Table 31).
5.1.2 Water and sediment
No information is available on contamination of water with
PBDDs/PBDFs.
River and marine sediment samples (n = 5) from Japan monitored
for tetra- to hexaBDDs/BDFs contained tetraBDDs (n.d. - 0.006 µg/kg
dry weight) and tetra- to hexaBDFs at total concentrations of
0.03 - 0.37 µg/kg dry weight. There were also data on PXDDs/PXDFs
(Watanabe et al., 1995).
Ballschmiter et al. (1990) investigated sediment (sludge)
collected from a drain that received runoff water from a German
motorway crossing. They found no PBDDs (Br1 through Br4), but PBDFs and
PXDFs were found, their concentrations ranging from 180 to 1690 ng/kg
dry sludge (di- and trihalogenated homologues; sum of PBDFs/PXDFs:
2500/1850 ng/kg). Although this was a single sample, it showed,
together with the results for PCDDs/PCDFs, a pattern recognized as
typical for automobile-derived contamination - that is, predominance
of PBDFs over PCDFs, prevalence of dibenzofurans over
dibenzo- p-dioxins, and increased concentrations of lower halogenated
dibenzo- p-dioxins and dibenzofurans (Ballschmiter et al., 1990).
Similar correlations were observed with samples of soil (section
5.1.3) and grass (section 5.1.6.1) collected near motorways
(Ballschmiter et al., 1990).
Sediments recently sampled from road sewers (n = 2) in Germany
(Schacht et al., 1995) had lower concentrations of PBDFs (sum of tetra
to hepta: up to 300 ng/kg) than of PCDFs (sum of tetra to octa: up to
1300 ng/kg).
Analyses of laminated sediment core from the Baltic Proper
revealed PBDEs in sediment layers dating from the 1950s and later and
made evident a dramatic increase, ranging from 4- to 20-fold, in PBDEs
in the 1980s. Analyses for PBDFs were not performed (Nylund et al.,
1992).
5.1.3 Soil, sewage sludge, and biocompost
One soil sample taken near a motorway (depth: 0 - 2 cm) in
Germany contained 0.74 µg monoBDFs/kg, 0.58 µg diBDFs/kg, and 1 µg
Cl1Br1DFs/kg soil. Other PBDFs or PXDFs (up to Br4/X4) were not
detected, and no PBDDs (Br1 through Br4) were found (detection limit:
0.02 µg/kg). PCDFs (Cl1 through Cl5) were present at concentrations
ranging from 0.04 to 0.38 µg/kg, and PCDDs (Cl1 through Cl8) were
detected at 0.1 µg/kg (Ballschmiter et al., 1990). Another soil sample
(n = 1; no details given) collected near a motorway in Germany also
did not contain PBDDs, and the concentrations of PBDFs were low (sum
of tetra- and pentaBDFs: 0.02 µg/kg; 2,3,7,8-TeBDF: <0.001 µg/kg).
The sum of PCDDs/PCDFs was 0.3 µg/kg (Schacht et al., 1995).
Watanabe et al. (1992) reported the presence of PBDFs (about 100
µg total PBDFs/kg), together with PBDEs and PCDDs/PCDFs, in a soil
sample (depth not given) taken from an incineration field in Taiwan
that was contaminated with large amounts of ash from plastic
materials. They also found a large number of PXDFs but no PXDDs in the
same sample.
Further samples from disposal sites are discussed in chapter 3.
Soil samples (n = 2) collected at a metal reclamation factory
area in southern Taiwan had total PBDF concentrations of 48 and 87
µg/kg. The PBDFs included triBDFs (16-30 µg/kg), tetraBDFs (16 - 34
µg/kg), pentaBDFs (11 - 18 µg/kg), and hexaBDFs (5 µg/kg). PBDDs were
not detected (detection limits ranged from <0.25 to 1 µg/kg)
(Watanabe et al., 1993).
Soil samples (n = 3; 0 - 10 cm depth, if necessary after removal
of plants and grass; at distances of >1000 - <2000 m, main wind
direction) were collected a few months after a fire in a warehouse
where bromine-containing plastic pellets were stored (Neupert & Pump,
1992). In one of three sites (samples), 2,3,7,8-substituted
PBDDs/PBDFs were found (detection limit 0.5 ng/kg) at 3.5 ng/kg (sum
of five tetra- to hexaBDDs and of three tetra- to pentaBDFs).
A series of sewage sludge samples (n = 13) from municipal
wastewater treatment plants in Germany were analysed for PBDDs/PXDDs
and PBDFs/PXDFs as well as for other polyhalogenated compounds
(Hagenmaier et al., 1992). The analytical data on these sludges, which
were destined to be discharged on fields for fertilization, are
summarized in Tables 32 and 33. PBDFs were detected in all samples,
their concentrations (sum of mono- to pentaBDFs) reaching a maximum of
about 3 µg/kg. Of the homologue groups (Table 32), the diBDFs were
predominant, with concentrations ranging from 0.27 to 1.99 µg/kg.
PBDDs, PXDDs, and PXDFs were not found in any samples. The ratio of
median concentrations for PBDFs and PCDDs/PCDFs was 1 : 9 (Table 33).
PBDEs (tri- to heptaBDEs), the possible precursors of PBDFs, were
detected in all samples at higher levels than PBDFs (Table 33).
Another study group (Ballschmiter et al., 1990) did not find any
PBDDs/PBDFs (mono through tetra) in sewage sludge samples from rural
(n = 12) and urban (n = 5) areas (detection limit not given).
Traces of tetraBDDs (0.006 µg/kg) and tetra- to heptaBDFs (sum:
0.32 µg/kg; 2,3,7,8-TeBDF: 0.003 µg/kg) together with PCDDs/PCDFs
(sum: 8.34 µg/kg) were found in a single sewage sludge sample (Schacht
et al., 1995).
Biocompost (n = 1) containing tetra- to octaCDDs/CDFs (sum: 3.2
µg/kg) showed no detectable PBDDs (detection limit not given); of
PBDFs (tetra to hepta), only tetraBDFs (<0.003 µg/kg; no
2,3,7,8-TeBDF) were present (Schacht et al., 1995).
Table 32. PBDFs detected in sewage sludge samples from municipal
wastewater treatment plants in Germanya
PBDFs Number of samples Concentrationb
(µg/kg)
MonoBDFs 9 0.05-0.67
DiBDFs 8 0.27-1.99
TriBDFs 9 0.07-0.20
TetraBDFs 9 0.03-0.23
PentaBDFs 9 n.d.-0.01
Sum of mono to pentaBDFs 0.29-3.05
a Adapted from Hagenmeier et al. (1992).
b n.d. = not detected.
Table 33. Concentrations of PBDFs and other polyhalogenated aromatic
compounds in sewage sludgea
Compounds Number of Concentration (µg/kg)
samples Range Median Mean Standard
deviation
Mono- to pentaBDFs 13 0.21-3.05 1.11 1.17 0.92
Tri- to heptaBDEs 13 0.49-17.73 8.37 8.58 5.51
PCBs 17 233-3456 674 911 767
Total PCDDs 13 3.27-27.82 9.20 10.71 6.69
Total PCDFs 13 0.18-7.09 0.53 1.07 1.83
a Adapted from Hagenmaier et al. (1992).
5.1.4 Food and feed
Little information was found in the literature. Market basket
surveys are not available.
Crops and other vegetation growing in the vicinity of potential
emitters can be contaminated, as shown by an analysis of grass
collected near a motorway. The pattern and levels of PBDDs/PBDFs and
PXDDs/PXDFs found in plants are described in section 5.1.6.1.
The occurrence of PBDDs/PBDFs in seafood and milk is discussed in
section 5.1.6.2.
5.1.5 Other products
For levels of PBDDs/PBDFs in consumer products such as electrical
appliances, and emissions from them, see chapter 3.
5.1.6 Terrestrial and aquatic organisms
5.1.6.1 Plants
Grass samples collected in the vicinity of a motorway contained
lower brominated PBDFs (mono- through triBDFs), PXDFs (up to X3), and
traces of monoBDDs (Ballschmiter et al., 1990; Table 34). Between the
homologue groups, concentrations peaked at mono- and diBDFs and
Cl1Br1DFs (>2000 ng/kg). This pattern paralleled that found in the
soil sample from the same sampling site, but the levels in the grass
sample were higher (Table 34).
PBDDs/PBDFs (mono through tetra), PXDDs/PXDFs (up to tetra), and
PCDDs/PCDFs (mono to octa) have been analysed in needles from a pine
near a highway (Schwind, 1991). Again, concentration peaks were seen
with the mono- and diBDFs (see Table 34) and with monoCDFs (4380 ng/kg
dry weight). The sum of the PHDFs (mono to octa) was about a factor of
100 higher than the sum of PHDDs (owing to the high concentration of
mono- to trihalogenated dibenzofurans [>12 500 ng/kg dry weight]).
5.1.6.2 Animals
1) Wildlife
In a randomly selected pooled sample consisting of shrimp (380 g)
and mussels (600 g) as well as fish (cod: 1500 g; plaice: 300 g), a
tetraBDD and a tetraBDF could be detected, both
non-2,3,7,8-substituted (De Jong et al., 1992). A quantitative
analysis was not performed.
Neither PBDDs/PBDFs nor PXDDs/PXDFs were found in one homogenate
of muscle from Baltic salmon (Salmo salar; Sweden) at detection limits
of 0.2 - 20 ng/kg fresh weight (Wiberg et al., 1992). PBDDs/PBDFs were
not detected (detection limits of 2, 3, and 8 ng/kg for tetra-,
penta-, and hexaBDDs/BDFs, respectively) in pooled muscle samples of
young (n = 15), middle-aged (n = 15), and old (n = 15) carp (Cyprinus
carpio) collected from the Buffalo River, NY, USA (Loganathan et al.,
1995). No PBDDs/PBDFs or PXDDs/PXDFs (tetra to hexa) were detected
(detection limit not specified) in fish (n = 4) captured in 1993 in
rivers near Osaka in Japan (Watanabe et al., 1995).
One composite sample of muscle from osprey (Pandion haliaetus;
Sweden; n = 35; cf. Jansson et al., 1993) was analysed for PBDDs/PBDFs
and for PXDDs/PXDFs. The results were negative at detection limits of
0.2 - 20 ng/kg fresh weight (Wiberg et al., 1992).
Table 34. Concentrations of PBDDs/PBDFs and PXDDs/PXDFs in
environmental samples taken near motorways in Germany
Homologue groups Concentrationa (ng/kg)
Grassb Soilb Pine needlesc
(after 1 month (depth: 0-2 cm) (dry weight)
of dryness)
Dibenzo-p-dioxins
MonoBDDs 60 n.d. 28.3
DiBDDs n.d. n.d. 25.5
TriBDDs n.d. n.d. 5.5
TetraBDDs n.d. n.d. <4
Cl1Br1DDs - - +
Cl1Br2DDs - - 5.6
Cl2Br1DDs - - 1.7
X4DDs - - <4d
Dibenzofurans
MonoBDFs 2530 740 5491
DiBDFs 2170 580 1053
TriBDFs 40 n.d. 258
TetraBDFs n.d. n.d. 53
Cl1Br1DFs 2420 1000 788
Cl1Br2DFs 240 n.d. 358
Cl2Br2DFs 190 n.d. 116
Cl2Br2DFs n.d. n.d. 58
Cl3Br1DFs n.d. n.d. 55
Cl1Br3DFs - - 74
a n.d. = not detected (detection limit 20 ng/kg); - = not analysed;
+ = detectable, but not quantifiable.
b Adapted from Ballschmiter et al. (1990).
c Adapted from Schwind (1991).
d For each of the three possible combinations (Cl2Br2, Cl3Br1, Cl1Br3).
PCDDs/PCDFs were present in all samples mentioned above. For
example, carp from the Buffalo River (Loganathan et al., 1995) were
found to contain noticeable concentrations of total PCDDs (27 - 146
ng/kg wet weight) and total PCDFs (22 - 99 ng/kg) along with total
PCBs (>2 mg/kg) and total PBDEs (13 - 23 µg/kg).
2) Farm animals
Cow's milk collected at dairy farms in the deposition area of an
"old technology" municipal waste incinerator in the Netherlands was
analysed for PBDDs/PBDFs and PXDDs/PXDFs. The pooled (n = 11) milk
sample contained compounds that were tentatively identified (but not
quantified) as two triBDFs, one tetraBDF, and one pentaBDF, all four
not having the 2,3,7,8-substitution pattern (De Jong et al., 1992).
The same sample contained high levels of PCDDs/PCDFs.
5.2 General population exposure
5.2.1 Exposure data
There is no quantitative information available on exposure of the
general population, special subpopulations, or infants to PBDDs/ PBDFs
from several sources (see chapter 3 and section 5.1).
5.2.2 Monitoring of human tissues and fluids
Few studies have monitored PBDDs/PBDFs in human tissues or milk.
On behalf of the National Human Adipose Tissue Survey, the US EPA
initiated a study in 1987 to analyse 2,3,7,8-substituted tetra-through
hexaBDDs/BDFs in adipose tissue of the general population. Eight
hundred and sixty-five individual tissue specimens were collected and
combined into 48 composite samples referring to the nine US census
divisions and three age groups. None of the six targeted PBDDs/PBDFs
was detected at average detection limits of approximately 1 ng/kg for
2,3,7,8-TeBDD/TeBDF (range: 0.4 - 8.9 ng/kg), 10 ng/kg for
1,2,3,7,8-PeBDD/PeBDF and for 1,2,3,4,7,8-HxBDD (range: 0.8 - 54
ng/kg), and 40 ng/kg for 1,2,3,4,7,8-HxBDF (range: 2.5 - 120 ng/kg) on
a lipid weight basis (Cramer et al., 1990a,b). In all samples
analysed, there were indications of the presence of PBDEs (hexa
through octa) (Cramer et al., 1990a,b).
Neither PBDDs/PBDFs nor PXDDs/PXDFs were found in two human
adipose tissue samples (male: 70 years of age; female: 60 years of
age) examined in a German study (Dawidowsky, 1993). The detection
limits for tetra-, penta-, and hexa-substituted congeners were
0.2 - 1.6 ng/kg, 0.9 - 3.5 ng/kg, and 4.6 - 14 ng/kg on a lipid weight
basis, respectively.
One composite sample of human milk (38 g) from Sweden was
examined for PBDDs/PBDFs and PXDDs/PXDFs (Wiberg et al., 1992).
Results of this study, which did not give any information on the
number of original specimens or sampling strategy, were negative. The
detection limits were reported to be in the range of 0.2 - 20 ng/kg on
a lipid weight basis. PCDDs/PCDFs were detectable in this sample.
Another investigation from Germany (Dawidowsky, 1993) led to similar
results. Although PCDDs/PCDFs were present in three human milk
samples, PBDDs/PBDFs and PXDDs/PXDFs were not detectable at detection
limits of 0.8 - 5.5 ng/kg (tetra substitution), 3.2 - 13 ng/kg (penta
substitution), and 13 - 53 ng/kg (hexa substitution) on a lipid weight
basis. At present, the presence of PBDDs/PBDFs in human milk at very
low concentrations cannot be ruled out (Somogyi & Beck, 1993).
5.3 Occupational exposure
5.3.1 Workplace monitoring data
Contamination by PBDDs/PBDFs is possible in a variety of
workplaces involved in producing, processing, using, or disposing of
certain flame retardants or products containing them (see chapter 3),
especially where the processes involve elevated temperatures. There
are only limited workplace monitoring data.
5.3.1.1 Flame retardant/polymer industry
A set of data is available on workplaces in the chemical industry
producing flame-retarded polymers (Brenner & Knies, 1990, 1992,
1993a,b, 1994; Thies et al., 1990; Brenner, 1993; Kieper, 1996). Air
samples were taken during extrusion production and moulding of PBT,
ABS, polystyrene, or polyamide resins blended with various brominated
flame retardants (see Table 35).
PBDF concentrations measured near the extruder and injection
moulding machines, in the whole building, in the storage and refilling
area, and at other sites are summarized in Table 35. Within the
homologue groups measured, a maximum concentration of about 600 ng/m3
was found for the sum of hexaBDFs. In one experimental series, PBDF
concentrations in workplace air were higher by a factor of about 1000
where DBDE was used, compared with TBBPA. The difference was explained
by the different properties of DBDE and TBBPA, as well as different
exhaust and ventilation conditions (Brenner & Knies, 1993a). In the
study involving DBDE/PBT, PBDF concentrations near the extruder
workplace and in the air of the whole room were similar, indicating
their general distribution in the building. Lower levels were found in
the storage and refilling area (Brenner & Knies, 1990).
2,3,7,8-TeBDF was not detected in a lot of samples (detection
limits 1 - 100 pg/m3 air) (Brenner & Knies, 1990, 1993a, 1994; Thies
et al., 1990; Kieper, 1996) but was detectable in some others (Kieper,
1996). Low amounts of penta- and hexaBDFs substituted in the
2,3,7,8-position (0.3 - 2.6 ng/m3 air) were tentatively identified at
DBDE/PBT workplaces (Brenner & Knies, 1990).
Concentrations of PBDDs in the DBDE/PBT study were two orders of
magnitude below those of PBDFs, with di-, tri-, tetra-, penta-, and
hexaBDD concentrations being <0.05, 0.35, 2.04, 8.37, and 17 ng/m3
air, respectively (Brenner & Knies, 1990). The tentative analysis for
2,3,7,8-substituted PBDDs showed the presence of 2,3,7,8-TeBDD (<0.5
ng/m3 air), of 1,2,3,7,8-PeBDD (1.3 ng/m3 air), and of two hexaBDDs (1
and 1.6 ng/m3) (Brenner & Knies, 1990). In contrast, no PBDDs were
found in air samples of the workplace area in a TBBPA study, although
low amounts of di- (0.94 ng/m3), tri-(0.07 ng/m3), and tetraBDDs (0.08
Table 35. Air contamination by PBDFs measured at workplaces where flame-retarded thermoplastic resins
are produced and processed
PBDFs Resin/flame retardanta Number of Air volumeb Concentrationc Reference
sampling (m3) (ng/m3)
stations
MonoBDFs PBT/TBBPA 3 13-18 n.d.-0.26 (<0.006) Kieper (1996)
PS-PS-butadiene/DBDE 5 20-30 0.01-0.16 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.012-0.017 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.017-0,049 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.026-0.129 Kieper (1996)
DiBDFs PBT-glass fibre/DBDE 3 30-150 0.2-1.3 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-0.34 (<0.004) Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.19-1.02 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.14 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (< 1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.040-0.223 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.043-0.139 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.070-0.21 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.094-0.24 Kieper (1996)
TriBDFs PBT-glass fibre/DBDE 3 30-150 1.1-13 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-0.11 (<0.012) Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.065-3.04 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.18 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.077-0.484 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.063-0.337 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.049-0.274 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.075-0.169 Kieper (1996)
TetraBDFs PBT-glass fibre/DBDE 3 30-150 5.1-34 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 0.03-0.05 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.157-6.92 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.14 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS/hexabromocyclododecane 1 n.sp. 0.02 Brenner (1993)
PS-PS-butadiene/DBDE 5 20-30 0.173-0.40 Kieper (1996)
Table 35. (Continued)
PBDFs Resin/flame retardanta Number of Air volumeb Concentrationc Reference
sampling (m3) (ng/m3)
stations
TetraBDFs PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.136-0.30 Kieper (1996)
(cont'd) PA/polytribromostyrene 5 19-29 0.017-0.43 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.051-0.15 Kieper (1996)
PentaBDFs PBT-glass fibre/DBDE 3 30-150 8.6-143 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 0.07-0.19 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.11-5.63 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0,12 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS/hexabromocyclododecane 1 n.sp. 1 Brenner (1993)
PS-PS-butadiene/DBDE 5 20-30 0.27-0.49 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.16-0.18 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.04-0.30 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.035-0.12 Kieper (1996)
HexaBDFs PBT-glass fibre/DBDE 3 30-150 13-594 Brenner & Knies (1990)
PBT-giass fibre/TBBPA 2 185-260 0.05-0.26 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.06-3.61 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.11 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.81-6.37 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.33-0.73 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.06-0.29 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.028-0.099 Kieper (1996)
HeptaBDFs PBT-glass fibre/DBDE 3 30-150 -88-260 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-<0.04 (<0.013) Brenner & Knies (1993a)
PA/polytribromostyrene 5 1 9-29 0.07-0.30 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.04-0.07 Kieper (1996)
OctaBDF PBT-glass fibre/DBDE 3 30-150 n.d. approx. -7 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-<0.08 (0.026) Brenner & Knies (1993a)
PA/polytribromostyrene 5 19-29 0.03-0.20 Kieper (1996)
PA/polydibromostyrene 5 6-12 <0.004-0.09 Kieper (1996)
a PA = polyamide 66; PS = polystyrene; TBBPA = tetrabromobisphenol A or its derivatives.
b n.sp. = not specified.
c n.d. = not detected (detection limits in parentheses, if specified).
ng/m3) were emitted by the extruder equipment. The detection limits of
di- through octaBDDs ranged from 0.001 to 0.4 ng/m3 air. No
2,3,7,8-substituted PBDDs (tetra- through hexaBDDs) were found
(detection limits ranged from 0.001 to 0.1 ng/m3 air) (Brenner &
Knies, 1993a). Preliminary tests at the workplace during injection
moulding of polystyrene blended with hexabromocyclododecane showed
tetraBDD concentrations of 2.5 ng/m3, consisting of two isomers with
no 2,3,7, 8-substitution (Brenner, 1993). During extruder experiments
with PBT/TBPI (Brenner & Knies, 1994), the sum concentration of PBDDs
was in the low pg/m3 range, and no 2,3,7,8-substituted congeners were
detected (detection limits ranged from 1 to 240 pg/m3; tetra to octa).
Air samples from selected workplaces (operated permanently or
periodically) of three plastic processing plants were monitored for
mono- to hexaBDFs/BDDs (resin/flame retardant used:
polystyrene-polystyrene-butadiene/DBDE;
polystyrene/1,2-bis(tribromophenoxy)-ethane; PBT/TBBPA-carbonate
oligomer) and for mono- to octaBDFs/BDDs (resin/flame retardant used:
polyamide 66/polytribromostyrene; polyamide 66/polydibromostyrene),
including eight or more 2,3,7,8-substituted congeners (Kieper, 1996).
Depending on workplace and flame retardant used, the sum of PBDF/PBDD
concentrations (mono to hexa) ranged from 258 to 77 414 pg/m3. The
highest sum concentrations within workplaces permanently operated
(range: 258 - 10 018 pg/m3) were found at workplaces processing
1,2-bis(tribromo-phenoxy)ethane. Although samples from the latter did
not contain any 2,3,7,8-substituted congeners, many other samples gave
positive results, with maximum sum concentrations of some thousand
pg/m3. The highest concentrations of 2,3,7,8-substituted PBDFs/PBDDs
were seen at the TBBPA-carbonate oligomer/PBT workplaces (operated
permanently and periodically). The ranges of concentrations (n = 3) in
pg/m3 were as follows:
2,3,7,8-TeBDF: <4 - <165 (detection limit
elevated
owing to interfering
components)
1,2,3,7,8-PeBDF: 2 - 100
2,3,4,7,8-PeBDF: 6 - 313
1,2,3,4,7,8-HxBDF: 7 - 445
2,3,7,8-TeBDD: <6 - <293 (detection limit
elevated
owing to interfering
components)
1,2,3,7,8-PeBDD: 23 - 1137
1,2,3,4,7,8-/1,2,3,6,7,8-HxBDD: 25 - 1161
1,2,3,7,8,9-HxBDD: 9 - 578
Results for 2,3,7,8-substituted heptaBDFs/BDDs were obtained at
workplaces processing polyamide flame-retarded by polytribromo-styrene
(n = 5) or polydibromostyrene (n = 5). The concentrations measured (in
pg/m3) ranged in the following manner (n = 10):
1,2,3,4,6,7,8-HpBDF: 26 - 280
1,2,3,4,7,8,9-HpBDF: <1 - 13
1,2,3,4,6,7,8-HpBDD: <1.4 - 11
5.3.1.2 Offices/studios
Some monitoring results are available for workplaces (offices;
television studios) equipped with a number of electrical appliances
continually in use, such as display and computer monitors (Chriske et
al., 1990; UBA, 1992; see also section 5.1.1). Maximum air
concentrations measured were 0.56 pg/m3, 0.61 pg/m3, and 0.4 pg/m3 for
tetra-, penta-, and hexaBDFs (see Table 28). Although heptaBDFs could
not be detected in air samples (Table 28), they were found in the
corresponding dust samples at concentrations ranging from 0.7 to 3.5
µg/kg (Table 30). Similarly, no detectable amounts of tetra- and
pentaBDFs substituted in the 2,3,7,8-position were found in air
samples (Table 28), but they were found in dust samples, with
concentrations ranging from n.d. to 0.005 µg/kg (2,3,7,8-TeBDF) and
from 0.003 to 0.020 µg/kg (two pentaBDFs) (Table 30). PBDDs were not
detected (Chriske et al., 1990).
5.3.1.3 Recycling plants
Recycling of plastic materials (pure or in combination with other
materials, e.g. metals, that can have catalytic effects) may be a
source of PHDDs/PHDFs, depending - inter alia - on the type of flame
retardants blended with them (see chapter 3). Workplace air was
monitored in a pilot plant recycling defective printed circuits
(Lorenz & Bahadir, 1993). These printed circuits contained copper and
TBBPA, a flame retardant with a relatively low potency for generation
of PBDDs/PBDFs. Air samples (n = 2) taken near the running shredding
systems did not contain PBDFs, although small amounts of mono- and
diBDFs were found in the shredded material (0.05 - 0.32 µg/kg).
However, owing to short sampling time and resulting low air volumes
sampled (6 - 7 m3), the detection limits were only 0.02 - 0.1 ng/m3
(mono- through pentaBDFs) and 0.2 - 0.4 ng/m3 (hexa- through
heptaBDFs). No PBDDs (mono- through octaBDDs) were found in the air
samples (detection limits 0.02 - 2 ng/m3 for mono- through heptaBDDs),
but residues of tetraBDDs (0.03 - 0.73 µg/kg) were present in samples
of the processed waste. Neither 2,3,7,8-TeBDD nor 2,3,7,8-TeBDF was
found in any of the samples tested (Lorenz & Bahadir, 1993).
In 1991, air samples (air volume: 20 - 30 m3; sampling period: 6
- 10 h) from three workplace stations in an operating secondary copper
plant and ground dust samples (pooled sample from five sites)
collected in another secondary copper plant (shut down in 1990) in
Germany were monitored for PBDDs/PBDFs (Kieper, 1996). The sum of
mono- to hexaBDF concentrations in the workplace air samples ranged
from 8 to 190 pg/m3. 2,3,7,8-TeBDF was present in one sample at 0.4
pg/m3 but was not detected in either of the other air samples
(detection limits 0.1 - 0.8 pg/m3). PBDDs (mono to hexa) were not
detectable (detection limits 0.1 - 1.4 pg/m3). The dust sample
contained 21.02 µg mono- to hexaBDFs/kg, with a maximum of 8.4 µg/kg
for tetraBDFs. Concentrations of 2,3,7,8-TeBDF, 1,2,3,7,8-PeBDF,
2,3,4,7,8-PeBDF, and 1,2,3,4,7,8-HxBDF were 0.09, 0.10, 0.12, and 0.09
µg/kg (maximum values), respectively. PBDDs (mono to hexa) were not
found (detection limits 0.01 - 0.03 µg/kg).
5.3.1.4 Other workplaces
Monitoring data from workplaces in waste incineration facilities
and disposal sites are lacking.
PBDF concentrations of up to 100 mg/kg (see section 3.7.2) were
found in combustion residues of accidental fires; therefore, firemen
and other workers coming in contact with fire fume, dust, and residues
could be exposed to these substances.
The work area under the fume hood (waste oil from air pumps) of a
laboratory was found to be contaminated with PBDDs/PBDFs and
PCDDs/PCDFs (Ritterbusch et al., 1994b). Wipe tests showed a sum
concentration of 580 ng/m2 for PBDDs/PBDFs (mono to hexa) and of 360
ng/m2 for PCDDs/PCDFs (tetra to octa).
5.3.2 Monitoring of human tissues and fluids
A chemist who suffered from acute intoxication after synthesizing
several grams of 2,3,7,8-TeBDD and 2,3,7,8-TeCDD in 1956 without using
a hood or protective clothing (Schecter & Ryan, 1990, 1991, 1992;
Schecter, 1992; see also section 8.2) was examined 35 years after
exposure. His blood contained 625 ng TBDD/kg blood lipid (concomitant
with 18 ng TCDD/kg blood lipid) (Schecter, 1992; Schecter & Ryan,
1992). For comparison, the average blood lipid concentration in the
general population of the USA was reported as 3 - 5 ng/kg for TCDD and
not detectable for TBDD (Schecter et al., 1994a). (Blood analysis of a
Japanese student who had developed chloracne [see section 8.2] 1 month
after exposure to PCDFs and PBDFs showed no detectable amounts of
PCDFs [or PCBs]. PBDFs were not included in this analysis, performed
in 1982, about half a year after exposure [Asahi & Urabe, 1987].)
Zober et al. (1992) investigated employees of a chemical plant
that had produced thermoplastic resins (PBT) blended with the flame
retardants OBDE or DBDE. The potential for exposure to PBDDs/PBDFs
including the homologue groups di- through octaBDDs/BDFs was
established by workplace air measurements, as described by Brenner &
Knies (1990) (see also section 5.3.1). However, analysis of the blood
lipids of the personnel focused mainly on 2,3,7,8-TeBDD/TeBDF.
Elevated levels of both congeners were found in the blood samples of
potentially exposed workers, although both isomers could not (TBDF) or
could hardly (TBDD) be identified in the previous workplace air
samples (see section 5.3.1). The concentrations of TBDD in venous
blood exceeded those of TBDF (ranges: n.d. - 478 ng/kg blood lipid
versus n.d. - 112 ng/kg blood lipid) among the male study group (see
Table 36). As seen in Table 36, there was a correlation between blood
levels of TBDF/TBDD measured and job type, working conditions, and
working period. Highest median (TBDF/TBDD: 18/91 ng/kg blood lipid)
and maximum (TBDF/TBDD: 112/478 ng/kg blood lipid) values were found
in the 18 extruder operators who had been first engaged before 1986.
The lower levels observed in the 11 operators employed during and
after 1986 may be due to the shorter exposure time, changes in
production process, or technical improvements, as suggested by the
authors of this study. Other long-term employees (n - 5) showed
intermediate blood levels ranging from <7 to 26 ng TBDF/kg blood
lipid and from 7 to 48 ng TBDD/kg blood lipid. The lowest values were
seen in the technical support personnel (see Table 36). Data from a
referent group were not provided, but among referents (n = 5) of a
preceding study, PBDFs/PBDDs were either not detected or marginally
present (Zober et al., 1992).
Table 36. Concentrations of 2,3,7,8-TeBDF and 2,3,7,8-TeBDD in blood of personnel from industry
using PBDEa
Job type/first year on job Number Concentration (ng/kg blood lipid)b,c
2,3,7,8-TeBDF 2,3,7,8-TeBDD
Median Minimum Maximum Median Minimum Maximum
Extruder operators 29 8 n.d. 112 40 n.d. 478
1975-1985 18 18 n.d. 112 91 16 478
1986-1988 11 4 n.d. 11 n.d. n.d. 11
Maintenance mechanics
(1975-1983) 3 16 <7 26 17 17 22
Production employees;
other areas (1976-1982) 2 7 7 7 28 7 48
Technical support personnel 8 2 n.d. 11 n.d. n.d. 5
a From Zober et al. (1992).
b Samples collected in 1990/91.
c n.d. = not detected (detection limit not specified).
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Dibenzo-p-dioxins
All studies available on absorption of PBDDs refer to absorption
of 2,3,7,8-TeBDD in rats (see Table 37). It was absorbed after oral,
dermal, and intratracheal administration, the percent absorption
varying with route and dose.
After administration of a single dose of 1 nmol/kg body weight,
absorption was about 80% by the oral and intratracheal routes, whereas
only about 12% was absorbed through the skin. Oral absorption of
single doses declined from 80% at lower doses (1 - 10 nmol/kg body
weight) to about 50% at higher doses (500 nmol/kg body weight), thus
suggesting non-linear absorption.
Oral and pulmonary absorption of an equimolar dose of
2,3,7,8-TeCDD using identical experimental conditions as with TBDD
were 88% and 95%, respectively (Diliberto et al., 1996). Dermal
absorption of TBDD was about one-third that of an equimolar dose of
TCDD (Jackson et al., 1991; Diliberto et al., 1993). Differences
between TBDD and TCDD pulmonary and dermal absorption may be explained
by the octanol/water partition coefficients and the size of the
halogen substituents.
As enteral absorption for many PHDDs/PHDFs is known to be
variable and incomplete (especially demonstrated for the higher
chlorinated congeners), subcutaneous application has been used in
several of the relevant studies. For the majority of the chlorinated
congeners, a significant degree of absorption was reported within a
few days (exception: octachlorodibenzo- p-dioxin, or OCDD). A 99%
absorption rate has been reported by this route for TBDD (600 ng/kg
body weight) as well as for TCDD (300 ng/kg body weight) in the rat
(Nagao et al., 1995/96).
6.1.2 Dibenzofurans
Dermal absorption of 1,2,7,8-TeBDF was examined in male Fischer
344 rats following a single dose of 1 nmol/kg body weight (Kedderis et
al., 1994). About 29% of the administered dose was absorbed,
quantified on the basis of the amount found in tissues (4%, excluding
the skin site) and excreted within 72 h. This dermal absorption was
intermediate, compared with that of TBDD (12%: see section 6.1.1),
TCDD (41%: Banks & Birnbaum, 1991), and TCDF (48%: Brewster et al.,
1989) after single equimolar doses.
Table 37. Absorption of 2,3,7,8-TeBDD in rats
Strain (sex) Route (vehicle) Dosing regimen Absorption,a time Method References
Wistar oral (arachis oil with single dose 80% (male), 48 h faeces Ivens et al.
(female, male) 5% toluene) 100 µg/kg body weight 83% (female), 48 h analysis (1992)
(n = 4) (= 0.2 pmol/kg body weight) [TBDD]
Fischer 344 oral (water: ethanol: single dose faeces Diliberto et al.
(male) Emulphor(R) = 3: 1: 1) and tissue (1990a,b, 1993);
(n = 3-4) analysis
0.5 µg/kg body weight 78%, 72 h [3H-TBDD]
(= 0.001 µmol/kg body weight) Kedderis et al.
(1992a)
5 µg/kg body weight 82%, 72 h
(= 0.01 µmol/kg body weight)
50 µg/kg body weight 60%, 72 h
(= 0.1 µmol/kg body weight)
250 µg/kg body weight 47%, 72 h
(= 0.5 µmol/kg body weight)
Fischer 344 intratracheal (water: single dose 80%, 72 h faeces and Diliberto et al.
(male) ethanol: Emulphor(R) 0.5 µg/kg body weight tissue (1991, 1993);
(n = 3-4) = 3: 1: 1) (= 0.001 µmol/kg body weight) analysis Kedderis et al.
[3H-TBDD] (1992a)
Fischer 344 dermal (acetone) single dose 12%, 72 h faeces and Jackson et al.
(male) 0.5 µg/kg body weight tissue (1991);
(n = 3-4) (= 0.001 pmollkg body weight) analysis Kedderis et al.
(= 0.2 nmol/1.8 cm2) [3H-TBDD] (1992a);
Diliberto et al.
(1993)
a Values based on concentration of 2,3,7,8-TeBDD [TBDD] or on 3H activity ([3H-TBDD]).
6.2 Distribution
6.2.1 Levels in organs and blood
6.2.1.1 Dibenzo-p-dioxins
Almost all studies available on PBDD distribution refer to
disposition of 2,3,7,8-TeBDD in the rat. As shown in Table 38, TBDD is
distributed throughout the whole body, with major deposits found in
liver and adipose tissue, followed by skin and muscle (Kedderis et
al., 1991a; Diliberto et al., 1993). Appreciable amounts of [3H]TBDD
were also found in adrenals and thymus (Diliberto et al., 1993). Three
days after oral exposure to [3H]TBDD, liver and adipose tissue
contained more than 65% of the body burden (Diliberto et al., 1993).
Distribution of TBDD can be described by a physiologically based
pharmacokinetic model consisting of a blood compartment and five
tissue compartments: liver, fat, skin, slowly perfused tissues, and
richly perfused tissues (Kedderis et al., 1992b, 1993; Buckley, 1995).
The partitioning of TBDD between liver and adipose tissue was
studied in Fischer 344 rats exposed to [3H]TBDD (Diliberto et al.,
1990a,b, 1991, 1993; Kedderis et al., 1990, 1991a,b, 1992a, 1993;
Jackson et al., 1991) and found to be influenced by dose, route of
exposure, and time post-dosing (see Table 39 for representative data).
Dose-dependent changes in partition ratios were seen in the
intravenous and oral studies (see Table 39). Liver concentrations of
TBDD were disproportionately increased at the higher doses compared
with the lower dose of 1 nmol/kg body weight (liver: fat concentration
ratios: 2.6 and 0.2 by the intravenous route at high and low dose,
respectively, and >5 and 2.9 by the oral route). However, whereas
liver concentrations of TBDD were disproportionately increased at 10
nmol/kg body weight compared with the 1 nmol/kg body weight oral dose,
the increase was related to dose in the 10 - 100 nmol/kg body weight
dose range. Factors influencing the dose-dependent nature of TBDD
tissue distribution are discussed by Kedderis et al. (1993).
Intravenous, oral, intratracheal, and dermal treatment with 1
nmol [3H]TBDD/kg body weight resulted 3 days later in liver: fat
concentration ratios of 3.4, 2.9, 2.0, and 1.5, respectively (see
Table 39). The lower ratio observed for the dermal exposure is
explained by differences in absorbed dose (low internal exposure; see
section 6.1) and dose-related tissue distribution.
Time-dependent changes in the distribution pattern were
demonstrated in the intravenous study. Partition ratios (liver : fat)
of 3.4 and 0.2, respectively, observed 3 and 56 days after single
intravenous exposure, indicated an increased distribution in adipose
tissue with increasing time after dosing (Table 39). In addition to
redistribution, tissue-specific elimination may also be occurring.
Table 38. Distribution of TBDD-derived radioactivity in Fischer 344 rats
3 days after oral, dermal, or intratracheal administration of 1 nmol
[3H]TBDD/kg body weighta,b
Tissue % administered dosec,d % absorbed dose/g tissued,e
Oral Dermal Intratracheal Oral Dermal Intratracheal
Liver 20.3 2.4 19.5 2.4 2.4 0.3
Adipose tissue 19.6 3.8 24.7 0.8 1.6 1.2
Skin 10.9 1.8 8.3 0.3 0.5 0.3
Muscle 3.5 0.8 3.0 0.04 0.07 0.04
Blood 0.4 0.06 0.2 0.03 0.03 0.01
Thymus 0.03f 0.03 0.08 0.2 1.1 0.4
Adrenals 0.4f 0.01 0.02 0.5 1.2 0.4
Kidneys - 0.05 0.1 - 0.2 0.1
Spleen - 0.01 0.02 - 0.2 0.06
Lungs - 0.06 0.1 - 0.5 0.2
Heart - 0,02 0.03 - 0.2 0.06
Testes - 0.02 0.05 - 0.06 0.03
Brain - 0.01 0.02 - 0.05 0.02
Stomach - 0.03 0.1 - 0.3 0.2
Table 38. (Continued)
Tissue % administered dosec,d % absorbed dose/g tissued,e
Oral Dermal Intratracheal Oral Dermal Intratracheal
Small intestines - 0.04 0.2 - 0.2 0.1
Large intestines - 0.04 0.2 - 0.3 0.1
a Adapted from Diliberto et al. (1993); oral absorption = 79%; dermal absorption
= 12%; intratracheal absorption = 78%.
b Mean values; n = 3-4; standard deviation and statistical details omitted.
c Percentage of the administered dose normalized to 100% recovery.
d - = not analysed.
e Percentage adjusted to 100% absorption.
f n = 1.
Table 39. Partition of [3H]TBDD-derived radioactivity between liver and
adipose tissue of ratsa,b
Route of Dosec Observation TBDD concentration Liver: fat
exposure (nmol/kg period (pmol/g) concentration
body weight) (days) Liver Fat ratio
Intravenous 1 3 8.1 2.4 3.4
1 56 0.2 1.1 0.2
100 56 117.2 45.3 2.6
Oral 1 3 4.9 1.7 2.9
10 3 79.9 13.6 5.9
100 3 518.3 93,4 5.6
500 3 2216.3 340.1 6.5
Intratracheal 1 3 4.1 2.1 2.0
Dermal 1 3 0.6 0.4 1.5
a Adapted from Kedderis et al. (1992a); Diliberto et al. (1993).
b Male Fischer 344 rats; n = 3-4; single doses; vehicle: ethanol: Emulphor(R):
water = 1: 1: 3 (oral, intravenous, intratracheal exposure), acetone (dermal exposure).
c 1 nmol/kg body weight corresponds to 0.5 µg/kg body weight.
Other studies using non-labelled TBDD (vehicle: arachis oil with
5% toluene) and another rat strain (Wistar) also found higher
concentrations of TBDD in the liver than in adipose tissue 2 days
after single oral doses (Neupert et al., 1989; Ivens et al., 1992) and
after daily oral exposure for 91 days (Ivens et al., 1990, 1993).
Both TCDD and TBDD appeared to be distributed in a similar
manner, and differences (e.g. after dermal exposure) can be attributed
to the higher lipophilic nature of TBDD (Diliberto et al., 1993;
Kedderis et al., 1993). Concerning distribution between adipose tissue
and blood, a 2.7-fold higher fat/blood partition coefficient was
assumed for TBDD compared with TCDD (Kedderis et al., 1993).
Tissue concentrations and concentration ratios (liver : adipose
tissue) have been compared under identical experimental conditions for
TBDD (single subcutaneous injection, Wistar rats, 600 ng/kg body
weight) and TCDD (300 ng/kg body weight) (Nagao et al., 1995/96). As
shown in Table 40, the liver : adipose tissue concentration ratio
increases with increasing doses for both congeners. In contrast, the
concentration ratios for TCDD/TBDD were rather dose-independent in
adipose tissue and also in the liver. Whereas hepatic tissue
concentrations were very similar at the doses used for TBDD and TCDD,
concentrations in adipose tissue were higher for TBDD over the entire
dose range. When increasing the dose 100-fold (from 30 to 3000 ng/kg
body weight), hepatic concentrations increased 174 times for TCDD and
256 times for TBDD. In contrast, concentrations in adipose tissue
increased only 26 times (TCDD) and 21 times (TBDD).
When the two corresponding chlorinated and brominated
1,2,3,7,8-pentahalogenated dibenzo- p-dioxins (PeHDDs) were given as a
mixture (2 nmol/kg body weight, each) subcutaneously to Wistar rats,
the same tissue distribution (liver : adipose tissue concentration
ratio of about 7) was found for both congeners at the maximal tissue
concentrations (Golor et al., 1993).
Following administration of 2,3,7-trihalogenated
dibenzo- p-dioxins (TrHDDs: Cl3DD, Br3DD, Cl2BrDD) to female Wistar rats
(single intravenous injections of mixtures containing 3, 10, or 50
µg/kg body weight for each congener), dose- and time-dependent changes
in tissue concentrations (liver, adipose tissue, thymus) were seen. It
is remarkable that concentrations of all three congeners were highest
in adipose tissue and lowest in liver (about two orders of magnitude
lower a few hours after the injection). Surprisingly high
concentrations were found in the thymus, almost an order of magnitude
higher than in liver (Golor et al., 1995).
6.2.1.2 Dibenzofurans
Disposition studies on dibenzofurans were conducted in rats using
[4,6-3H]-1,2,7,8-TeBDF (Kedderis et al., 1994). As with TBDD, the
major tissue depots included liver, adipose tissue, and skin (1 - 72 h
after single intravenous, oral, and dermal doses of 1 nmol/kg body
weight). Relatively high concentrations of 1,2,7,8-TeBDF were also
observed in the adrenal glands. Generally, concentrations in the liver
exceeded those in fat, and perirenal fat contained higher amounts than
epididymal fat. For example, 72 h following oral administration, there
was a liver : fat concentration ratio of about 2. A decline in liver
concentrations of 1,2,7,8-TeBDF was seen from 1 to 24 h after
intravenous treatment. This was related to metabolic elimination and
to a slight accumulation in adipose tissue.
Tissue levels of 1,2,7,8-TeBDF in lungs, small intestine, heart,
stomach, spleen, and thymus of Fischer 344 rats 1 h after intravenous
dosing were in the range of 0.2 - 1.3% of the dose/g of tissue. The
corresponding results for liver, kidneys, perirenal fat, adrenals, and
skin were 4.9, 0.5, 0.1, 5.1 and 0.1%, respectively. The tissue levels
of 1,2,7,8-TeBDF 72 h after oral administration were 0.11% (liver),
0.07% (perirenal fat), 0.10% (adrenals), and 0.03% (skin) of the
dose/g of tissue (Kedderis et al., 1994).
Seven days after a single subcutaneous dose of 2,3,4,7,8-PeBDF
(420 ng/kg body weight) was given to marmoset monkeys (n = 3), a
liver : fat concentration ratio of 12.2 was observed (Schulz et al.,
1993). A similar high deposition rate in liver was found with the
chlorinated analogue, whereas TCDD showed a liver : fat concentration
ratio of about 1.
6.2.2 Transfer to offspring
There are no experimental data available on transfer of
PBDDs/PBDFs to offspring.
However, transfer of various PCDDs/PCDFs via placenta and/or
through milk has been documented in rats, mice, goats, cows (WHO,
1989), marmoset monkeys (Hagenmaier et al., 1990b), and humans
(Schecter & Ryan, 1994; Schecter et al., 1994b, 1995, 1996a,b). The
bioavailability of PCDDs/PCDFs from breast milk was found to be high
(up to >95%) in human infants (Jödicke et al., 1992; McLachlan, 1993;
Pluim et al., 1993).
6.3 Metabolic transformation
6.3.1 Dibenzo-p-dioxins
Metabolism of PBDDs has been studied in rats given 2,3,7,8-TeBDD
orally or intravenously. No metabolites were found in liver (Kedderis
et al., 1991a), but metabolites were detected in bile from rats (male
Fischer 344 or female Sprague-Dawley) (Kedderis et al., 1991a; De
Jongh et al., 1992, 1993).
Three days after an intravenous dose of 1 nmol [3H]TBDD/kg body
weight, faeces of F344 rats (see also section 6.4) contained about 3%
of the administered dose as parent compound and about 14% of the dose
as metabolites (Kedderis et al., 1991a). About 80 - 90% of faecal and
biliary radioactivity excreted following intravenous dosing was
attributed to TBDD metabolites (Kedderis et al., 1991a,b).
Table 40. Comparison of tissue concentration and of liver: adipose tissue concentration ratio after a single subcutaneous
injection of 2,3,7,8-TeBDD or 2,3,7,8-TeCDD in ratsa,b
Dose Liver tissue Adipose tissue Liver: adipose tissue
(ng/kg body weight) concentration ratio
TCDD TBDD TCDD: TBDD TCDD TBDD TCDD: TBDD TCDD TBDD
(ng/g) (ng/g) concentration ratio (ng/g) (ng/g) concentration ratio
30 0.16 0.08 2.1 0.14 0.6 0.2 1.2 0.2
300 3.38 3.60 0.9 0.82 2.7 0.3 4.1 1.4
3000 27.9 20.5 1.4 3.7 12.5 0.3 7.7 1.9
Increase: 30-3000 174 x 256 x 26 x 21 x
a Adapted from Nagao et al. (1995/96).
b Female Wistar rats; n = 3 or 6; single subcutaneous doses; vehicle: toluene/DMSO (dimethyl sulfoxide); observation: day
7 after treatment.
If biliary excretion of TBDD-derived radioactivity is considered
as an indirect assessment of metabolism, TBDD is relatively slowly
metabolized. Within a 5-h period, 6.6% of a radiolabelled intravenous
dose (1 nmol [3H]TBDD/kg body weight) was excreted in bile of male
Fischer 344 rats (Kedderis et al., 1991b).
Studies with pretreated or untreated rats showed that TBDD and
TCDD did not induce their own metabolism in vivo (Kedderis et al.,
1991b, 1992a).
The main metabolites identified (three
hydroxybromodibenzo- p-dioxins and one dihydroxytetrabromoether) were
formed by aromatic hydroxylation and hydrolytic debromination and
suggest the metabolic pathway shown in Fig. 2 (De Jongh et al., 1993).
Similarities and differences in the metabolic pathways of TBDD and
TCDD are discussed by De Jongh et al. (1993).
In summary, several of the metabolites are similar.
Quantitatively, the dioxin ring-opening route seems to be favoured
somewhat more in TCDD (Poiger & Buser, 1984) than in TBDD metabolism;
qualitatively, the absence of a second methoxytribromodibenzodioxin
differs from TCDD metabolism.
6.3.2 Dibenzofurans
Information on the metabolism of [3H]1,2,7,8-TeBDF is available
from a study determining biliary elimination of
[3H]1,2,7,8-TeBDF-derived radioactivity (Kedderis et al., 1994).
Approximately 50% of the administered dose of [3H]1,2,7,8-TeBDF was
excreted in the bile of rats in 8 h. HPLC analysis confirmed the
presence of metabolites of 1,2,7,8-TeBDF in the bile (Kedderis et al.,
1994). If biliary excretion of PHDDs/PHDFs is used as an indirect
measure of metabolism, as is assumed by several authors (Kedderis et
al., 1991b; McKinley et al., 1993), this result is indicative of a
considerable metabolism of 1,2,7,8-TeBDF, in contrast to
2,3,7,8-TeBDD, which was more slowly metabolized (see above). The
differences between both congeners can be explained by their different
structures, 2,3,7,8- versus 1,2,7,8-substitution, the latter being
more susceptible to metabolism owing to the presence of two adjacent
unsubstituted carbon atoms in the 1,2-bromine ring (Kedderis et al.,
1994).
6.4 Elimination and excretion
Elimination of PHDDs/PHDFs occurs predominantly after conversion
to more polar metabolites in the liver and excretion of these
metabolites via the bile. There is apparently no or very little
excretion of unchanged congeners with the bile. However, for several
of the chlorinated congeners, secretion of the unchanged substances
into the intestinal lumen and subsequent excretion via the faeces have
been described (Abraham et al., 1989). It can be expected that the
lipophilic PBDDs/PBDFs may also be secreted into the intestinal lumen.
Table 2. PBDDs/PBDFs brominated at the
2,3,7,8-positions
PBDD congenera PBDF congenera
2,3,7,8-TeBDD* 2,3,7,8-TeBDF*
1,2,3,7,8-PeBDD* 1,2,3,7,8-PeBDF*
2,3,4,7,8-PeBDF*
1,2,3,4,7,8-HxBDD* 1,2,3,4,7,8-HxBDF
1,2,3,6,7,8-HxBDD* 1,2,3,6,7,8-HxBDF
1,2,3,7,8,9-HxBDD* 1,2,3,7,8,9-HxBDF
2,3,4,6,7,8-HxBDF
1,2,3,4,6,7,8-HpBDD 1,2,3,4,6,7,8-HpBDF
1,2,3,4,7,8,9-HpBDF
OcBDD OcBDF
a The congeners marked with an asterisk (*)
are cited in the German Dioxin Directive (1994)
(see Appendix I).
2.2.2 Stability of PBDDs/PBDFs
2.2.2.1 Photolysis
In the presence of laboratory light or sunlight, photolysis
occurs at a more rapid rate for PBDDs/PBDFs than for PCDDs/PCDFs
(Buser, 1988; Chatkittikunwong & Creaser, 1994a; for details, see
section 4.2.1). This should be taken into consideration when analyses
of these compounds are carried out (see sections 2.4.1 and 4.2.1).
Photolysis on quartz surfaces under sunlight is a much slower
process than photolysis in organic solvents (Buser, 1988). PBDDs/
PBDFs adsorbed on incinerator soot particles remained relatively
stable and degraded only slowly during a 6-h period (Lutes et al.,
1990, 1992a,b). Studies of PBDDs in soil showed that for the same
congeners, the half-lives in this matrix are four times longer than in
solution (Chatkittikunwong & Creaser, 1994a).
Under conditions of ambient temperature and protection from
light, there is no appreciable (>1%) degradation of crystalline
PBDDs/ PBDFs and no significant change (0.6%, with the exception of
octaBDD [9.7%]) in standard solution (solvent: n-nonane)
concentrations over a period of 3 years (Re et al., 1995).
Table 3. CAS numbers for some PBDDs/PBDFs
PBDD congenera CAS number PBDF congenera CAS number
Br1DD 103456-34-4 Br1DF 103456-35-5
1-Br1DD 105908-71-2 2-Br1DF 86-76-0
2-Br1DD 105906-36-3
Br2DD 103456-37-7 Br2DF 103456-40-2
1,6-Br2DD 91371-14-1 2,7-Br2DF 65489-80-7
2,7-Br2DD 39073-07-9 2,8-Br2DF 10016-52-1
2,8-Br2DD 105836-96-2
Br3DD 103456-38-8 Br3DF 103456-41-3
1,2,8-Br3DF 84761-81-9
2,3,8-Br3DF 84761-82-0
Br4DD 103456-39-9 Br4DF 106340-44-7
1,2,3,4-Br4DD 104549-41-9 1,2,7,8-Br4DF 84761-80-8
2,3,7,8-Br4DD 50585-41-6 2,3,7,8-Br4DF 67733-57-7
Br5DD 103456-36-6 Br5DF 68795-14-2
1,2,3,7,8-Br5DD 109333-34-8 1,2,3,7,8-Br5DF 107555-93-1
2,3,4,6,7-Br5DF 124388-77-8
2,3,4,7,8-Br5DF 131166-92-2
Br6DD 103456-42-4 Br6DF 103456-33-3
1,2,3,4,7,8-Br6DD 110999-44-5 1,2,3,4,6,7-Br6DF 124388-78-9
1,2,3,6,7,8-Br6DD 110999-45-6 1,2,3,6,7,8-Br6DF 107555-94-2
1,2,3,7,8,9-Br6DD 110999-46-7
Br7DD 103456-43-5 Br7DF 62994-32-5
1,2,3,4,6,7,8-Br7DF 107555-95-3
Br8DD 2170-45-8 Br8DF 103582-29-2
a The homologue groups are underlined.
2.2.2.2 Thermolytic degradation of PBDDs/PBDFs
As discussed in chapter 3, the temperature of formation and
destruction of PBDDs/PBDFs depends on several conditions, such as
residence time, the presence/absence of oxygen, polymers, and
additives such as Sb2O3, as well as the efficiency of the apparatus
used for the thermal degradation. In laboratory experiments on the
thermolysis of polybrominated flame retardants (see section 3.4; Table
11), the PBDDs/PBDFs formed were destroyed at 800°C in an air
atmosphere after a 2.0-second residence time (Striebich et al., 1991).
PBDDs/PBDFs formed at 600°C from the thermolysis of plastics
containing DBDE or PBDE were no longer detectable at 800°C (Lahaniatis
et al., 1991). However, Thoma et al. (1987b) found that PBDDs/PBDFs
are still formed at 900°C. There is thus no definitive information on
the temperature needed to destroy PBDDs/PBDFs.
Table 4. CAS numbers for some PXDDs/PXDFs
PXDD congenera CAS number PXDF congenera CAS number
Br1Cl1DD 109007-09-02 Br1Cl1DF 109264-70-2
Br1Cl2DD 107227-59-8 Br1Cl2DF 107227-60-1
Br1Cl3DD 107227-75-8 Br1Cl3DF 107227-56-5
8-Br1-2,3,4-Cl3DF n.g.b
Br1Cl4DD 109264-61-1 Br1Cl4DF 109302-36-5
1-Br1-2,3,7,8-Cl4DF 104549-43-1
4-Br1-2,3,7,8-Cl4DF 115656-08-1
Br1Cl5DD 109264-65-5 Br1Cl5DF 107103-81-1
Br1Cl6DD 109264-67-7 Br1Cl6DF 107207-47-6
Br1Cl7DD 109264-69-9 Br1Cl7DF 109302-40-1
Br2Cl1DD 107227-58-7 Br2Cl1DF 107227-57-6
Br2Cl2DD 107227-74-7 Br2Cl2DF 107227-55-4
Br2Cl3DD 109031-99-4 Br2Cl3DF 107227-53-2
Br2Cl4DD 109264-62-2 Br2Cl4DF 107207-48-7
Br2Cl5DD 109264-66-6 Br2Cl5DF 107207-45-4
Br2Cl6DD 109264-68-8 Br2Cl6DF 109302-39-8
Br3Cl1DD n.g.b Br3Cl1DF 107227-54-3
Br3Cl2DD n.g.b Br3Cl2DF 107227-52-1
Br3Cl3DD n.g.b Br3Cl3DF 107207-46-5
Br3Cl4DD n.g.b Br3Cl4DF 107207-42-1
Br3Cl5DD n.g.b Br3Cl5DF n.g.b
Br4Cl1DD n.g.b Br4Cl1DF 107227-51-0
Br4Cl2DD n.g.b Br4Cl2DF 107207-44-3
1,2,3,4-Br4-7,8-Cl2DD 134974-39-3
Br4Cl3DD n.g.b Br4Cl3DF 107207-41-0
Br4Cl4DD n.g.b Br4Cl4DF n.g.b
1,2,3,4-Br4-6,7,8,9-Cl4DD 124728-12-7
Br5ClxDD n.g.b Br5Cl1DF 107207-49-8
other Br5ClxDF n.g.b
Br6Cl1DD 107207-38-8 Br6Cl1DF n.g.b
Br6Cl2DD n.g.b Br6Cl2DF 107207-36-3
1,2,4,6,7,9-Br6-3,8-Cl2DD 2170-44-7
Br7Cl1DD n.g.b Br7Cl1DF 107207-37-4
a The homologue groups are underlined.
b n.g. = CAS numbers not found (probably not yet allocated).
Table 5. Physical and chemical properties of some PBDDs/PBDFs
Compound Appearance Melting point Boiling point Water Vapour Octanol/water Sorption
(°C) (observed) (°C) solubility pressure partition coefficient
(predicted) [log S] [log P] coefficient [log Koc]
(mol/litre) (Pa at 25°C) [log Kow] (mol/litre)
(predicted) (predicted) (predicted) (predicted)
PBDDs
1-MoBDD white needles 104-106a 338.2b 3.5 × 10-3b
2-MoBDD n.g.c 93-94.5a 338.2b -6.12d 4.0 × 10-3b 5.62d 4.39d
(90-92)a
1,6-DiBDD 207e 375b 1.5 × 10-4b
2,3-DiBDD n.g.c 157.2-158f 375b -6.90d 1.6 × 10-4b 6.25d 4.74d
2,7-DiBDD 174-176a 375b 1.5 × 10-4b
193-194e
2,8-DiBDD 149.5-151a 375b 1.7 × 10-4b
(145-150)a
3,7-DiBDD -7.24d 6.53d 4.89d
-7.99d 7.14d 5.22d
1,2,3,4-TeBDD 6 × 10-7g
2,3,7,8-TeBDD white granules 334-336a,f 438.3b -8.72d 6.4 × 10-7b 7.74d 5.54d
6.50h
7.73i
1,2,3,7,8-PeBDD -9.45d 8.32d 5.87d
1,2,3,4,6,7, -10.89d 9.50d 6.50d
8-HpBDD
OcBDD 376j 523.2b -11.69d 4.1 × 10-11b 10.08d 6.82d
9.3 × 10-16g
PBDFs
monoBDF 2.89-3.26k
2-MoBDF -5.42d 5.05d 4.08d
diBDF 4.35-4.46k 5.58-6.09k
2,7-DiBDF -6.25d 5.95d 4.47d
triBDF 5.36-5.47k 6.49-6.79k
Table 5. (Continued)
Compound Appearance Melting point Boiling point Water Vapour Octanol/water Sorption
(°C) (observed) (°C) solubility pressure partition coefficient
(predicted) [log S] [log P] coefficient [log Koc]
(mol/litre) (Pa at 25°C) [log Kow] (mol/litre)
(predicted) (predicted) (predicted) (predicted)
1,2,8-TrBDF + colourless 144-148l
2,3,8-TrBDF prismsl
2,3,7-TrBDF -7.26d 6.55d 4.90d
tetraBDF 6.35-6.41k 7.72-8.72k
1,2,7,8-TeBDF colourless needlesl 240.5-242l 6.20h
2,3,7,8-TeBDF colourless needlesl 301-302l -7.99d 7.14d 5.22d
5.98h
2,3,4,6-TeBDF -7.99d 7.14d 5.22d
pentaBDF 7.25-7.45k
1,2,3,7,8-PeBDF 7.04h
7.56i
2,3,4,7,8-PeBDF -8.71d 7.73d 5.54d
hexaBDF 8.34k
2,3,4,6,7, -9.43d 8.31d 5.86d
8-HxBDF
1,2,3,4,6,7, 9 × 10-11g
8-HpBDF
a From Gilman & Dietrich (1957). Melting points in parentheses are values from other sources
reported by these authors.
b From Rordorf (1987).
c n.g. = not given.
d Predicted; from Fiedler & Schramm (1990). Sorption coefficient [log Koc] = distribution
coefficient between compound adsorbed to soil organic carbon and the compound in solution.
e From Tomita et al. (1959)
f From Kende & Wade (1973).
g From Rordorf et al. (1990).
h From Jackson et al. (1993), estimated from measured reverse-phase high-performance liquid
chromatography (HPLC) retention times.
Table 5. (Continued)
i From Jackson et al. (1993), calculated.
j From Denivelle et al. (1960).
k From Watanabe & Tatsukawa (1990).
l From Tashiro & Yoshiya (1982).
2.2.3 Chemical reactions
Aromatic carbon-bromine bonds are generally weaker than similar
carbon-chlorine bonds, and, consequently, bromine can be substituted
more easily. In general, the reductive substitution of halogens in
aromatic structures becomes easier as the halogen atoms' size
increases (Wania & Lenoir, 1990).
In the presence of excess chlorine, bromine can be substituted by
chlorine to give PXDDs/PXDFs -- for example, under conditions such as
those present in municipal incinerators (Wilken et al., 1990; Luijk et
al., 1992a).
Wania & Lenoir (1990) investigated the effect of heating
1,2,3,4-TeBDD (20 µg) in the presence of copper (1 g) at 100, 120,
150, or 210°C for a duration of 30 seconds to 1 h. With increasing
heating time, the spectrum of PBDD shifted from tetraBDD to lower
brominated congeners, and the sum of the quantities decreased. The
reaction rate increased with increasing temperature. At 210°C for 30
min, all PBDDs had disappeared, but the dibenzo- p-dioxin ring
structure remained intact.
In a further experiment, it was shown that the presence of water
(10 or 100 µg/litre) considerably increased the yield of debrominated
products.
On heating monoBDD and 1 g copper to 150°C, the debrominated
product dibenzo- p-dioxin and dimers of this compound were
identified. On heating hexaBDD and octaBDD to 150°C in the presence of
copper, it was found that appreciable quantities of the original
compounds could still be detected after 1 h. The reaction was
considerably slower than in the comparable experiment with tetraBDD.
The debromination reactions proceed faster than the respective
dechlorination reactions with PCDDs (Hagenmaier et al., 1987).
2.3 Conversion factors
At 25°C and 101.3 kPa, conversion factors for converting airborne
concentrations from ppm to mg/m3 for a particular PBDD/ PBDF congener
can be calculated from the relative molecular mass (RMM):
1 ppm = RMM/24.45 mg/m3
1 mg/m3 = 24.45/RMM ppm
For example, for monoBDF, 1 ppm = 247.1/24.45 = 10.1 mg/m3.
Similarly, 1 mg/m3 = 0.099 ppm.
2.4 Analytical methods
2.4.1 General aspects
Some PBDD/PBDF congeners are highly toxic (see chapter 7). Using
the principles of Good Laboratory Practice, great precautions should
be taken in handling the samples. Additionally, precautions must be
taken owing to the photochemical instability of the brominated and
mixed brominated/chlorinated congeners (see also sections 2.2.2.1 and
4.2.1). The use of amber-coloured glassware and filters on lamps and
windows is mandatory.
Sampling, sample treatment (extraction and clean-up), and
analysis for PBDDs/PBDFs and PXDDs/PXDFs follow largely the methods
and techniques currently used for PCDDs/PCDFs (Donnelly et al.,
1989a,b, 1990; US EPA, 1990, 1992 [Methods 1613 and 8290]; Maier et
al., 1994; Ballschmiter & Bacher, 1996). The large number of isomers
in some homologous groups (see Table 1) makes the separation and
quantification of individual congeners difficult. Using highly
selective, specific, and sensitive analytical methods,
2,3,7,8-substituted PBDDs/PBDFs can be detected, although co-elution
with other isomers cannot be excluded.
Accurate identification of specific congeners is limited by the
small number of reference standards available. The large number of
PXDDs/PXDFs (see section 2.1) makes it impossible to identify and
quantify individual congeners. Homologue groups, however, can be
analysed semi-quantitatively. Major steps in the analytical procedures
are as follows:
- spiking of the homologous sample with labelled standards
- use of matrix-specific extraction procedures (pretreatment of the
sample before extraction where necessary)
- clean-up by column chromatography, liquid-liquid extraction, HPLC
- concentration of the eluate (addition of a high-boiling solvent
as a keeper where necessary); addition of a recovery standard
- analysis by GC/MS.
Many of the analytical methods for PCDDs/PCDFs have been
validated in the past decade in interlaboratory studies organized,
among others, by: the World Health Organization (WHO) for biological
samples (WHO/EURO, 1989, 1991; Stephens et al., 1992; WHO/ECEH, 1996);
the European Community Bureau of Reference for environmental samples,
including fly ash (Maier et al., 1994), and for milk powder (Schimmel
et al., 1994; Tuinstra et al., 1996); and the European Committee for
Standardization for emissions by stationary sources (Bröker, 1996). To
avoid systematic errors in the individual steps from sampling to
analysis, recoveries must be controlled by the addition of appropriate
stable isotope labelled standards before extraction of the sample,
clean-up, and final quantification.
2.4.2 Sampling and extraction
The sampling procedures recommended for PCDDs/PCDFs (WHO, 1989
and citations in second paragraph of section 2.4.1) also apply for the
brominated congeners.
2.4.2.1 Ambient air, airborne dust, automobile exhaust, flue gas, and
products of thermolysis
Experience from PCDD/PCDF analysis has shown critical or weak
points in current gas sampling (ambient air, indoor air, exhaust gas)
techniques. Requirements are as follows:
- representativeness of samples; special attention must be given to
isokinetic sampling of particles in emission samples
- stability of the sample on the sampling medium during the
sampling period; the filter should be kept below 120°C and
protected from light
- recovery of the analytes from the sampling train (as checked by
appropriate spiked standards)
- use of clean equipment to avoid contamination of the sample (as
checked by appropriate blanks)
- complete trapping of gas and particle phases (aerosols) to avoid
sample losses.
Quartz fibre filters with polyurethane foam plugs have been used
to sample ambient air up to 1000 m3 (Wagel et al., 1989; Päpke et
al., 1990; Harless et al., 1992; Watanabe et al., 1992). (Note: There
may be interferences from brominated organic aromatic flame retardants
in polyurethane foam.)
Haglund et al. (1988) described a method to collect both the
particulate phase (using a Teflon-coated filter) and the gas phase
(cryotechnique) in vehicle exhaust.
Hutzinger et al. (1990) used the so-called Grimmer apparatus to
sample automobile exhaust: the sampling train consists of a large
glass condenser and a non-impregnated fibreglass filter. Typically, 50
m3 of automobile exhaust were taken per sample; the temperature at
the muffler outlet was kept below 50°C. The experiments were carried
out as stationary motor tests. The total emissions of the motor were
sucked through the sampling train by a pressure-controlled blower.
Emissions from a laboratory furnace experiment were collected by
a sampling train, including a high-efficiency quartz fibre filter (to
collect organic-laden particulate material) and an XAD-2 resin (to
adsorb semivolatile organic compounds) (Riggs et al., 1992).
Thermolytic products have been collected as condensate in a
quartz-wool-filled condenser tube (Neupert et al., 1989b).
2.4.2.2 Water and aqueous samples
Analysis of water samples should follow a different approach. If
the samples are free of particles, a normal liquid-liquid extraction
is sufficient. If, however, the samples contain particles, both the
particles and the water phase should be extracted separately -- the
solids by methods recommended for solids, the water phase as described
above.
2.4.2.3 Environmental samples: soil, sediment, and sewage sludge
For environmental samples, problems arise in obtaining a
representative sample. For soil sampling, a method was described by
Fortunati et al. (1994).
Prior to the extraction, appropriate measures should be taken to
ensure that PHDDs/PHDFs in the sample material are fully accessible to
the extraction solvent. In a number of applications, this includes a
digestion of the sample (solids) and/or the complete removal of water
(wet solid samples) prior to extraction clean-up; a chemical
destruction of non-persistent chemicals can be useful by incubating
the sample in neat sulfuric acid (H2SO4). PHDDs/PHDFs are shown to
be stable. Treatment with (strong) bases should be avoided, as
PHDDs/PHDFs may degrade.
For the study of sewage samples, Hagenmaier et al. (1992) dried,
powdered, and extracted the samples with toluene for 18 h. After
concentration, the extracts were treated with concentrated sulfuric
acid.
Proven digestion and water removal methods are treatment with
hydrochloric acid (10% HCl) and Dean Stark collector (US EPA, 1990;
Rappe et al., 1996), respectively. Sediments should be treated with
copper powder to eliminate sulfur (Kjeller et al., 1993).
2.4.2.4 Flame retardants, polymers, fly ash samples, dust, soot, and
fire residues
In general, the analysis of plastics is performed by dissolving
the polymer in a suitable solvent. Non-dissolvable plastics should be
powdered and Soxhlet-extracted.
Dibromomethane was used to dissolve samples of PBDE (Tondeur et
al., 1990). Ranken et al. (1994) noted that this solvent must first be
specially purified before use to remove the colour, which caused
quantitative interferences in the mass spectrometer. TBBPA can be
dissolved in methanol (Tondeur et al., 1990; Ranken et al., 1994).
PBT resins (extruded beads/powder) were extracted best with
1,1,1-trichloroethane/phenol followed by water partitioning of phenol;
powdered high-impact polystyrene (HIPS) samples by toluene/reflux; and
powdered ABS samples by dichloromethane (Donnelly et al., 1989a).
Kieper (1996) used toluene for Soxhlet extraction from samples of
flame-retarded polymers: DBDE (with polystyrene/polystyrenebutadiene),
1,2-bis(tribromophenoxy)ethane (with polystyrene), TBBPA-carbonate
oligomer (with PBT), dibromostyrene, and tribromostyrene (both with
polyamide 66).
Samples of burnt plastic, PBT material, ash/slag, and soil were
Soxhlet-extracted with dichloromethane (Neupert & Pump, 1992). Clausen
et al. (1987) used Soxhlet extraction with hexane. ABS was extracted
under reflux with methylene chloride (Donnelly et al., 1990).
Dry fly ash was treated with 10% HCl, dried, and neutralized.
After further drying, the sample was Soxhlet-extracted with toluene
(Hosseinpour et al., 1989). Similar procedures were used by Tong et
al. (1991) and Huang et al. (1992a,b).
PBDDs/PBDFs from dust samples and smoke condensate were
Soxhlet-extracted with toluene (UBA, 1992; Funcke et al., 1995).
Samples from fire residues were ground and extracted with toluene;
wipe samples of soot were extracted with hexane (Harms et al., 1995).
2.4.2.5 Biological matrices: human milk, blood/plasma, tissues, and
fish samples
For biological samples, most appropriate extraction methods are
those giving the highest yields (or recovery) for the lipids in the
sample (i.e. milk, blood, tissue).
Neupert et al. (1989a,b) quantified PBDDs/PBDFs in rat liver,
adipose tissues, and faeces. After homogenization with sodium sulfate,
extraction was performed on a multiple-layer column using
dichloromethane/hexane.
Fish samples were ground with sodium sulfate and homogenized.
Methanol and sodium oxalate were added to milk samples (De Jong et
al., 1992). Diethyl ether and hexane were used to extract PHDDs/ PHDFs
from the fat fraction of the milk and fish samples.
For PBDD/PBDF determination, samples of human adipose tissue were
homogenized, extracted with dichloromethane, dried with sodium
sulfate, and solvent-exchanged into hexane (Cramer et al., 1990a).
This method was also used by Zober et al. (1992).
Fat removal can be performed utilizing a semipermeable membrane
technique (Bergqvist et al., 1993), which enables larger amounts of
fat (sample size up to 200 g) to be eliminated from the sample matrix
(>95%) and improved detection limits.
Lyophilization has been used successfully in the analysis of TBDD
in biological matrices such as rat livers or marmoset monkey tissues
(Schulz-Schalge et al., 1991a,b; Schulz et al., 1993; Nagao et al.,
1995/96).
2.4.3 Sample clean-up
Sample clean-up is carried out to remove those materials that
might otherwise interfere with the analysis. A variety of liquid
chromatography separations have been used, including silica, florisil,
alumina, and various combinations of these columns. Usually an
acid/base wash followed by alumina column chromatography is used to
remove the bulk of interferences, and carbon column chromatography is
used to remove residual interferences (Donnelly et al., 1986, 1987,
1989b).
Where PBDEs are likely contaminants, a modification of separation
techniques is necessary. Alumina columns are ineffective in separating
PBDFs from PBDEs. Carbon columns were found to be more effective, but
the higher brominated PBDFs could be removed from the column only by
back-flushing with an aromatic solvent (Donnelly et al., 1987; Hileman
et al., 1989). Bonilla et al. (1990) introduced an HPLC step to the
clean-up procedure. The sample was passed through an AX21 carbon
column. The column was washed in the forward direction with
dichloromethane/cyclohexane and dichloromethane/methanol/benzene and
back-flushed with toluene. This procedure decreased the PBDE
concentration in the final sample by six orders of magnitude.
Depending on the aim of the analysis (general surveying or
specific search and quantification of 2,3,7,8-substituted congeners),
a number of 13C standards are required to be added at several stages
during sampling and analysis.
In the WHO interlaboratory calibration study for the analysis of
PCDDs/PCDFs in human blood and milk, the basic clean-up/ separation
methods used by some laboratories were activated carbon as the primary
PCDD/PCDF isolation step followed by alumina; the remaining
laboratories used other procedures, mostly H2SO4 followed by
alumina. Nearly all methods used a step involving some type of carbon
chromatography (Stephens et al., 1992).
2.4.4 Separation
GC is used for the separation of PBDDs/PBDFs. PBDDs/PBDFs have
much higher retention times and elution temperatures (30-40°C higher)
than their chlorinated analogues (Buser, 1991). The higher brominated
congeners have extremely long retention times, so non-polar (SE 54),
medium-length (up to 25-m) columns are generally used. Such a column
is suitable for separating the PBDD and PBDF homologues. Elution
temperatures on a 25-m SE 54 high-resolution gas chromatography (HRGC)
column range from 184-188°C for monoBDDs/BDFs to 260-273°C for
pentaBDDs/BDFs. Hexa-, hepta-, and octa-homologues elute during the
isothermal phase at 280°C (Buser, 1986a, 1991). Cross-linked columns
allow higher temperatures, reducing analysis times (Hutzinger et al.,
1990). Table 6 gives retention indices (RIs) of some PBDDs/PBDFs as
well as PBDEs, which are possible contaminants (Donnelly et al.,
1987). A 30-m DB-5 or DB-5MS fused capillary column has been found to
be quite useful for the GC/MS analysis of tetra- through
hepta-substituted PBDDs/PBDFs (Ranken et al., 1994).
Table 6. Retention indices (RIs) of PBDDs,
PBDFs, and PBDEsa,b
Congener RI
PBDDs
2-MoBDD 1868
2,8-DiBDD 2174
1,3,7-TrBDD 2423
2,3,7-TrBDD 2475
2,3,7,8-TeBDD 2800
1,2,7,8-TeBDD 2811
1,2,4,7,8-PeBDD 3072
1,2,3,7,8-PeBDD 3145
1,2,3,4,7,8-HxBDD 3412
1,2,3,6,7,8-HxBDD 3475
1,2,3,7,8,9-HxBDD 3798
1,2,3,4,6,7,8-HpBDD 3763
OcBDD 4219
PBDFs
2-MoBDF 1834
2,8-DiBDF 2133
1,2,8-TrBDF 2416
2,3,8-TrBDF 2433
1,2,7,8-TeBDF 2740
2,3,7,8-TeBDF 2791
1,2,3,7,8-PeBDF 3103
1,2,3,6,7,8-HxBDF 3479
1,2,3,4,6,7,8-HpBDF 3806
OcBDF 4231
PBDEs
HexaBDE 2888
HexaBDE 3004
HexaBDE 3015
HexaBDE 3030
HexaBDE 3051
HexaBDE 3095
HexaBDE 3286
HexaBDE 3314
HexaBDE 3369
HexaBDE 3411
OctaBDE 3525
OctaBDE 3577
OctaBDE 3601
OctaBDE 3627
Table 6. (Continued)
Congener RI
OctaBDE 3654
OctaBDE 3737
OctaBDE 3786
NonaBDE 3951
NonaBDE 4003
DecaBDE 4310
a From Donnelly et al. (1987).
b Chromatographic conditions:
30 m x 0.32 mm DB-5 GC column;
He carrier gas at ca. 7 psi head
pressure; temperature programmed
from 10 min at 170-320 °C at
8 °C/min.
The elution of PCDDs/PCDFs and PBDDs/PBDFs occurs in the order of
the molecular weights. PXDDs/PXDFs elute between the corresponding
chloro- and bromo-analogues (Buser, 1987a). However, mixed congeners
containing bromine elute earlier than expected on a molecular weight
basis, relative to the chloro-compounds (e.g. BrCl5 before Cl7)
(Buser, 1991). (For isomer-specific analysis, columns of different
polarity should be used.)
Owing to the lack of PBDD/PBDF (and PXDD/PXDF) standards, it has
not been possible to identify all congeners. Instead, a combination of
MS and GC RI identification has to be used for the analysis of
2,3,7,8-substituted PBDDs/PBDFs, PCDDs/PCDFs, and PXDDs/ PXDFs. An RI
model has been developed to predict the GC retention times for 1700 of
these compounds (Donnelly & Sovocool, 1991; Donnelly et al., 1991a,b).
2.4.5 Detection, quantification, and confirmation of PBDDs/PBDFs
by MS techniques
Detection, quantification, and confirmation are usually performed
by MS, as only this technique shows sufficient selectivity to
distinguish PBDDs/PBDFs from other halogenated compounds (e.g. PBDEs)
that are present in the sample. MS allows the determination of the
number and type of halogens present from characteristic isotope
distribution patterns, but it does not give any information about
which isomer is present (Buser, 1991). (Among the MS methods,
high-resolution mass spectrometry [HRMS] is preferred owing to higher
selectivity, and the tandem MS or negative ion chemical ionization
[NCI] techniques are a useful screening method because of the
diagnostic Br [79/81] fragment.)
Donnelly et al. (1987) and Sovocool et al. (1987) developed and
refined US Environmental Protection Agency (EPA) Method 8280 to
measure PBDFs/PBDDs by GC/MS. Significant features of the mass spectra
reported by these authors include the sequential losses of Br*, COBr*,
(Br*+COBr*), and (2Br*+COBr*). PBDEs may interfere with the
determination of PBDFs (and PBDDs), as the (M-2Br)+* fragment has
the same m/z composition cluster as that of a PBDF with two fewer
bromines; additional fragmentation mimics the PBDF containing two
fewer bromines (Donnelly et al., 1987). This potential for co-elution
(see Tables 6 and 7) must be considered in the evaluation of reports
of PBDF (and PBDD) formation where PBDE interference is likely.
Confirmation criteria for the detection and quantification of
PBDDs/PBDFs have been proposed (Donnelly et al., 1987):
* The retention time/RI must be correct for that analyte (standards
are needed).
* Recovery of the "surrogate" standard should be in the 40-120%
range.
* All m/z monitored for a given analyte must maximize
simultaneously ± 1 second, with a signal to noise ratio greater
than or equal to 2.5 for each. The M+* cluster is relatively
intense for all congeners. For confirmation, two additional ions
(m/z) should be monitored in electron impact-selective ion
monitoring-mass spectrometry (EI-SIM-MS).
* The ratio between the two ions of the M+* cluster must be
within 20% (relative) of the theoretical.
* When monitoring for PBDFs, the absence of PBDE should be
demonstrated (see Table 7).
The identification and determination of positional isomers are
very complex. Complementary methods based on other instrumental
techniques such as GC/matrix isolation infrared spectrometry (MI-IR)
have been developed to allow the unambiguous identification of each
individual compound at high concentrations. For 2,3,7,8-TeBDD and
2,3,7,8-TeBDF, the most intense matrix isolation infrared band is
given by frequencies 1478 and 1434, respectively (Wurrey et al., 1989;
Childers et al., 1992). Childers et al. (1992) gave additional
frequencies for some PXDDs/PXDFs.
2.4.6 The need for analysis of 2,3,7,8-substituted congeners
Table 2 (section 2.1) gives the PBDD/PBDF congeners substituted
with bromine in the 2,3,7,8-positions. As these are the most toxic
congeners, in some investigations only these congeners are determined.
Table 7. Molecular ions (M+, 79Br isotope) of PBDDs, PBDFs, and
PBDEs showing possible interference during monitoring and determinationa
Compound Brominated congeners
mono- di- tri- tetra- penta- hexa- hepta- octa- nona- deca-
PBDFs 246 324 402 480 556 636 714 792
PBDEs 248 326 404 482 560 638 716 794 872 950
PBDDs 262 340 418 496 574 652 730 808
a From Buser (1986a) and Donnelly et al. (1987).
The German Dioxin Directive (1994) established limitations on the
concentrations of certain 2,3,7,8-substituted PBDDs/PBDFs in products
to be placed on the market (see Appendix I). In 1987, the US EPA
issued a Test Rule requiring manufacturers and importers of certain
halogen-containing chemicals to analyse their products for
2,3,7,8-substituted PHDDs/PHDFs (US EPA, 1987) (see Appendix I).
Owing to these actions, activities in PBDD/PBDF analysis have
improved and 2,3,7,8-substituted standards have been synthesized; in
1995, 12 of the 17 2,3,7,8-substituted isotopically labelled and 11 of
the 17 2,3,7,8-substituted native PBDDs/PBDFs were available.
2.4.7 Interfering substances
For a general discussion on the problem of interfering
substances, see Buser (1991). Substances possibly interfering with
PBDF determinations include PBDEs, as discussed above. This is of
particular importance in the analysis of substances containing this
flame retardant, their thermolytic products, and environmental samples
where this flame retardant is implicated. PBBs can be separated from
PBDFs/PBDDs and are not a source of cross-contamination (Donnelly et
al., 1987).
2.4.8 Standards
As mentioned in section 2.4.6, a number of PBDD/PBDF standards
have been made commercially available in recent years, in particular
the 2,3,7,8-substituted congeners. The following is therefore more of
historical than of practical interest.
For use as standards, samples of mono- through octaBDDs (Munslow
et al., 1987) and mono- through octaBDFs (Sovocool et al., 1987), as
well as PXDDs/PXDFs (Donnelly et al., 1987, 1989b), can be synthesized
by the electrophilic bromination of dibenzo- p-dioxin and
dibenzofuran. Special reaction conditions optimize the selectivity and
the corresponding yield. Extended reaction times result in a higher
degree of bromination; elevated temperature and increasing amount of
iron/iron (III) chloride (FeCl3) catalyst can be used to accelerate
the reaction (Hutzinger et al., 1989).
Donnelly et al. (1991a) prepared over 100 PHDDs using
self-condensation of halogenated phenols, coupling of halogenated
catechols with halobenzenes or halonitrobenzenes, and electrophilic
halogenation.
Ramalingam et al. (1986) synthesized PBDDs from bromocatechol and
polybromonitrobenzene in the presence of anhydrous potassium carbonate
(K2CO3) in acetone. For PXDDs, bromocatechol or chlorocatechol was
refluxed with polychloronitrobenzene or polybromonitrobenzene in the
presence of anhydrous K2CO3 in acetone.
Mixed halogenated compounds can be prepared by the bromination of
PCDDs and PCDFs, by the chlorination of PBDDs and PBDFs, or by halogen
exchange (Buser, 1987a).
Chatkittikunwong & Creaser (1994b) described the synthesis of
PBDDs/PBDFs and PXDDs/PXDFs by electrophilic halogenation of
substituted precursors with iron (III) halides (in the absence of
halogen).
Jay & Stieglitz (1996) prepared PBDFs and PCDFs from reaction of
copper (II) bromide (CuBr2) or copper (II) chloride, dihydrate
(CuCl2*2H2O), respectively, with dibenzofuran. For synthesis of
PXDFs, the brominated reaction mixture was further reacted with
CuCl2*2H2O.
Nestrick et al. (1989) developed a procedure for synthesizing
13C12-labelled PBDDs/PBDFs from their chlorinated analogues.
3. FORMATION AND SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
PBDDs/PBDFs are not known to occur naturally. They are not
intentionally produced but are formed as undesired by-products in
various processes. They can be formed by chemical, photochemical, or
thermal reactions from precursors or by so-called de novo synthesis
(from organic materials with bromine). PBDDs/PBDFs have been found as
contaminants in brominated organic chemicals. Thermolysis of
brominated flame retardants, in particular PBDEs, has been implicated
as an obvious source of PBDDs/PBDFs. Heating and burning of products
containing such brominated compounds can cause emission of
PBDDs/PBDFs. PBDDs/PBDFs have also been detected in traffic-related
emissions.
The formation of PXDDs/PXDFs is possible in combustion processes
if both bromine and chlorine are present (Buser, 1987a,b), such as in
waste incineration, in particular of old computer/business machines,
and in motor combustion processes.
3.1 Synthesis and use
PBDDs/PBDFs have no commercial use and are synthesized for
research purposes only and as standards for analytical determination
(see section 2.4.8). As an exception, a patent was awarded for the use
of heptaBDF as a flame retardant (Richtzenhain & Schrage, 1977).
3.2 By-products of brominated organic chemicals (including
flame retardants)
Theoretically, some 40 brominated organic chemicals may be
contaminated with PBDDs/PBDFs. Such chemicals include flame retardants
and fire extinguishers, pesticides (e.g. bromophenols, bromophos,
bromoxynil, profonofos), solvents, and chemical intermediates or
additives (Esposito et al., 1980; Lee et al., 1986, 1987; Johnson et
al., 1989; Bretthauer et al., 1991). Possible PBDD/PBDF formation
pathways have been suggested. Chemicals considered as being important
are TBBPA and its derivatives, penta- (PeBDE), octa-, and decaBDE,
2,4,6-tribromophenol, and 1,2-bis(tribromophenoxy)ethane (Johnson et
al., 1989).
Analytical data on the occurrence of PBDDs/PBDFs in brominated
organic chemicals are scarce. Tables 8 and 9 (concentrations of PBDFs
and PBDDs, respectively) include data on PBDEs, PBBs, TBBPA,
1,2-bis(tribromophenoxy)ethane, brominated phenols, brominated
anilines, brominated styrenes, and others.
The highest concentrations of PBDFs were found in PBDEs (up to
8000 µg/kg). Maximum PBDF values measured in DBB, TBBPA, bromophenols,
and bromoanilines were approximately 115 µg/kg, 64 µg/kg, 31 µg/kg,
and 2 µg/kg, respectively. PBDF levels ranging from about 92 to 500
µg/kg were observed in distillation residues of bromophenols and
bromoanilines (Table 8). This is of importance, particularly in
synthetic and analytical laboratories and laboratory waste disposal
(Vogt et al., 1994a,b).
The highest concentration of PBDDs (approx. 8500 µg/kg) was found
in 1,2-bis(tribromophenoxy)ethane, followed by 86 µg/kg in
2,4,6-tribromophenol and 8 µg/kg in TBBPA. As seen with PBDFs, PBDDs
were strongly enriched in distillation residues of selected
bromophenols (Table 9). Additionally, Ritterbusch et al. (1994a)
reported the occurrence of PBDFs (mono to penta: 12.1 µg/kg) and PBDDs
(mono to penta: 1.1 µg/kg) in solvent wastes of chemical laboratories.
2,3,7,8-Substituted PBDDs/PBDFs were not detected in TBBPA (Thies
et al., 1990; Tondeur et al., 1990; Brenner & Knies, 1993a,b; Ranken
et al., 1994), TBPI (Brenner & Knies, 1994), or 2,4,6-tribromophenol
(Tondeur et al., 1990; Vogt et al., 1994a). A sample of commercial
decaBDE was found to contain 1,2,3,7,8-PeBDF (1.6 µg/kg) and
1,2,3,4,7,8-HxBDF (37 µg/kg). Other 2,3,7,8-substituted tetra- to
hexaBDDs/BDFs did not exceed 0.1-5.1 µg/kg. All concentrations given
were maximum values, because co-elution was possible (UBA, 1992).
2,3,7,8-Substituted PBDDs/PBDFs were not detected in multiple samples
(n = 21; three companies) of commercial decaBDE at target limits of
quantitation (according to the US EPA Test Rule; see Appendix I)
ranging from 0.1 to 1.0 µg/kg, from 0.5 to 5 µg/kg, from 2.5 to 25
µg/kg, and from 100 to 1000 µg/kg for tetra-, penta-, hexa-, and
hepta-substitution, respectively. 1,2,3,4,6,7,8-HpBDF was found in all
samples at concentrations of 56-300 µg/kg, which were well below the
target limit of quantitation of 1000 µg/kg for this congener (Ranken
et al., 1994). Another study (Kieper, 1996) analysed five different
flame retardants (DBDE, 1,2-bis(tribromophenoxy)ethane,
TBBPA-carbonate oligomer, di- and tribromostyrene) for a total of
eight 2,3,7,8-substituted PBDDs/PBDFs (tetra to hexa). Contents, if
any, were below the respective detection limits. If detection limits
were included in the evaluation (as a concentration of half the
detection limit), the sum concentrations would range from 0.2 to 18.5
µg/kg. (A problem may arise in evaluating the sum of PBDDs/PBDFs if
the analytical limit of detection is rather high in some of the
congeners. From a scientific point of view, values below the limit of
quantification should not be used. However, for administrative
purposes, sometimes a value of one-half the detection limit is assumed
and included in the sum of PBDDs/PBDFs. A considerable difference may
occur between a scientific and this "administrative" approach. It is
highly recommended that the approach for the calculation be stated if
values that have not been measured are included in the summation.)
2,3,7,8-TeBDD was absent in bromophenols, whereas 2,3,7,8-TeBDF
was identified in crude 4-bromophenol and 2,4-dibromophenol at levels
of 0.12 and 0.15 µg/kg, respectively, and in their distillation
residues at 8.3 and 9.8 µg/kg, respectively (Vogt et al., 1994a). Both
2,3,7,8-substituted TeBDF and TeBDD were present (at 40 and 10 ng/kg,
respectively) in solvent wastes of chemical laboratories (Ritterbusch
et al., 1994a).
Table 8. Concentrations of PBDFs found in brominated organic chemicals
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
PBDE (tetra- to 8000 - - - 2000 4000 2000 - - Hileman et al.
hexaBDE) (commercial) (1989)
PBDE (hexa- to >4000 - - - n.d. 2000-4000 2000-4000 presentb - Hileman et al.
nonaBDE) (commercial) (approx. (1989)
200)
DBDE (commercial) - - - - n.d. n.d. 200 presentb,c - Hileman et al.
(approx. (approx. (1989)
200) 200)
DBDE (commercial) 286.3 - - - - - 2.3 250 34 Donnelly et al.
(concentrate) 6880 - - - 23 107 3470 2700 580 (1989a)
DBDE (commercial) 2037 - 0.04 <0.9 0.15 <0.01 <0.2 1842 195 Brenner
& Knies (1990)
DBDE (commercial) 79.3 0.4 0.3 0.3 0.8 10.5 67.0 - - UBA (1992)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.08) (0.08) (0.08) (0.8) (1.5) (1.8)
DBB (commercial) 115 99.9 9.0 5.7 <1 <5 <5 <10 <10 Atochem
(1990)
TBBPA + derivatives 63.6 - n.d. n.d. n.d. 1.0 12.2 31.5 18.9 Thoma et al.
(technical grade) (1986b);
Dumler et al.
(1990c)
TBBPA + derivatives
(commercial) >3 2 1 n.d. n.d. n.d. <14 - - Thies et al.
(<0.5) (<1) (<2) (1990)
Table 8. (Continued)
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
TBBPA + derivatives n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Brenner & Knies
(BC 52) (commercial) (0.001-0.4) (1993a,b)
TBBPA-oligocarbonate 1.46 n.d. n.d. n.d. 0.07 0.33 1.06 - - Kieper (1996)
(0.01) (0.01) (0.01)
TBPI 0.21 - 0 0 0.21 0 0 0 - Brenner & Knies
(Saytex BT 93) (1994)
Hexabromocyclo 50 - <10 <10 20 30 <10 <10 - Brenner (1993)
dodecane
(technical)
4-Bromophenol Ritterbusch et
(crude) 1.56 0.08 0.72 0.54 0.22 - - - - al. (1994a);
(distilled) 1.19 0.44 0.75 n.d. n.d. - - - - Vogt et al.
(distillation 378.54 63.47 230.46 69.17 15.44 - - - - (1994a)
residue)
(commercial) 0.37 0.06 0.31 n.d. n.d. - - - -
2,4-Dibromophenol Ritterbusch et
(crude) 3.30 0.16 1.43 1.44 0.12 - - - - al. (1994a);
(distilled) 0.85 0.36 0.49 n.d. n.d. - - - - Vogt et al.
(distillation residue) 498.99 62.47 353.51 68.91 4.3 - - - - (1994a)
2,4,6-Tribromo phenol Thoma et al.
(technical grade) 31.4 - 2.2 16.2 12.0 1.0 n.d. n.d. n.d. (1986b);
(crude) 4.6 0.79 2.66 1.19 n.d. n.d. n.d. n.d. - Dumler et al.
(1990c);
Vogt et al.
(1994a)
Pentabromophenol n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
(analytical grade) (1986b);
Dumler et al.
(1990c)
Table 8. (Continued)
Chemical Concentrations of PBDFs (µg/kg)a Reference
Sum MoBDFs DiBDFs TrBDFs TeBDFs PeBDFs HxBDFs HpBDFs OcBDF
Tetrabromophthalic n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
anhydride (analytical (1986b);
grade) Dumler et al.
(1990c)
2,4,6-Tribromo Vogt et al.
aniline (crude) 1.88 0.24 0.28 n.d. 0.25 1.01 - - - (1994b)
(recrystallized) 0.47 n.d. 0.47 n.d. n.d. n.d. - - -
(distillation 92.35 n.d. 3.15 8.40 15.90 64.90 - - -
residue)
2,6-Dibromo-4-nitroaniline
(crude) 0.90 0.33 0.57 n.d. n.d. n.d. - - - Vogt et al.
(1994b)
1,2-Bis(tribromo-phenoxy)ethane n.d. n.d. n.d. n.d. n.d. n.d. n.d. - -- Kieper (1996)
(0.5) (0.5) (0.5) (0.5) (1.0) (4.0)
Polytribromostyrene 16.74 n.d. 4.03 1.2 n.d. 0.36 3.29 3.01 4.85 Kieper (1996)
(0.06) (0.06)
Polydibromostyrene 24.09 0.09 0.26 0.21 n.d. n.d. 2.33 2.93 18.27 Kieper (1996)
(0.03) (0.04)
a - = no information; n.d. = not detected (detection limits in parentheses, if specified).
b Not quantifiable because of lack of standards.
c Major component.
Table 9. Concentrations of PBDDs found in brominated organic chemicals
Chemical Concentrations of PBDDs (µg/kg)a Reference
Sum MoBDDs DiBDDs TrBDDs TeBDDs PeBDDs HxBDDs HpBDDs OcBDD
DBDE (commercial) 0.4 - - - 0.05 0.35 - - - Brenner & Knies
(1990)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - UBA (1992)
(0.1-<5.1) (<0.1) (<0.1) (<0.1) (<0.2) (<0.7) (<5.1)
DBDE (commercial) n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.03) (0.03) (0.03) (0.03) (0.1) (0.35)
TBBPA + derivatives n.d. - - n.d. n.d. - - - - Thoma et al.
(technical grade) (1986b);
Dumler et al.
(1990c)
TBBPA + derivatives 8 n.d. n.d. n.d. 1 2 5 - - Thies et al.
(commercial) (<0.5) (<0.5) (<0.5) (1990)
TBBPA + derivatives 0.006 - n.d. n.d. 0.006 n.d. n.d. n.d. n.d. Brenner & Knies
(BC 52) (commercial) (0.001) (0.4) (1993a,b)
TBBPA-oligocarbonate n.d. n.d. n.d. n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.01) (0.01) (0.01) (0.01) (0.02) (0.06)
Hexabromocyclo - - <10 <10 <10 <10 <10 <10 - Brenner (1993)
dodecane (technical)
4-Bromophenol Ritterbusch et
(crude) 0.40 0.04 0.15 0.21 n.d. - - - - al. (1994a);
(distilled) 0.14 0.07 0.07 n.d. n.d. - - - - Vogt et al.
(distillation residue) 39.0 14.41 12.84 11.75 n.d. - - - - (1994a)
(commercial) n.d. n.d. n.d. n.d. n.d. - - - -
2,4-Dibromophenol Vogt et al.
(crude) 0.16 0.03 0.13 n.d. n.d. - - - - (1994a);
(distilled) 0.08 0.04 0.04 n.d. n.d. - - - - Ritterbusch et
(distillation residue) 18.75 1.37 3.13 10.76 3.49 - - - - al. (1994a)
Table 9. (cont'd)
Chemical Concentrations of PBDDs (µg/kg)a Reference
Sum MoBDDs DiBDDs TrBDDs TeBDDs PeBDDs HxBDDs HpBDDs OcBDD
2,4,6-Tribromo phenol Thoma et al.
(technical grade) 85.5 - - 1.5 84.0 - - - - (1986b);
(crude) n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. Dumler et al.
(1990c);
Vogt et al.
(1994a)
Pentabromophenol n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. Thoma et al.
(analytical grade) (1986b);
Dumler et al.
(1990c)
Tetrabromophthalic Thoma et al.
anhydride (analytical grade) n.d. - n.d. n.d. n.d. n.d. n.d. n.d. n.d. (1986b);
Dumler et al.
(1990c)
2,4,6-Tribromo aniline Vogt et al.
(crude) n.d. - - - n.d. - - - - (1994b)
(recrystallized) n.d. - - - n.d. - - - -
(distillation residue) 5.45 - - - 5.45 - - - -
2,6-Dibromo-4-nitroaniline
(crude) n.d. - - - n.d. - - - - Vogt et al.
(1994b)
1,2-Bis(tribromophenoxy)ethane 8455 n.d. n.d. 107 8348 n.d. n.d. - - Kieper (1996)
(1.0) (1.0) (2.0) (5.0)
Polytribromostyrene 5.63 1.78 3.85 n.d. n.d. n.d. n.d. n.d. <0.38 Kieper (1996)
(0.02) (0.02) (0.03) (0.11) (0.19)
Polydibromostyrene n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. <0.17 Kieper (1996)
(0.02) (0.02) (0.02) (0.02) (0.04) (0.08) (0.12)
a - = no information; n.d. = not detected (detection limits in parentheses, if specified).
3.3 Formation from the photochemical degradation of brominated organic
chemicals
The formation of PBDDs/PBDFs was observed under laboratory
conditions after irradiation of PBDEs (Watanabe & Tatsukawa, 1987) or
of bromophenols (Ritterbusch et al., 1994a) and is also believed to
occur after outdoor exposure of PBDEs (Birla & Kamens, 1994).
The major photoproducts of the flame retardant DBDE (technical
grade) irradiated in hexane solution by UV light and sunlight were
lower brominated PBDEs and mono- to hexa-substituted PBDFs. PBDDs were
not detected. Based on the kinetics of the reactions, the formation of
PBDFs occurred secondarily from debrominated PBDEs as photoproducts of
DBDE, but not directly from DBDE. UV irradiation of DBDE for 16 h gave
about a 20% yield of total PBDFs, with tetraBDFs, but not
2,3,7,8-TeBDF, being the main components (Watanabe & Tatsukawa, 1987).
The concentrations of PBDDs/PBDFs (mono to penta) in several
bromophenol samples were drastically increased, up to three orders of
magnitude, after UV irradiation for 60 min. Apparently, the rate of
photochemical PBDD/PBDF formation was greater than the rate of
degradation (see section 4.2.1). An exception was 2,3,7,8-TeBDF, which
disappeared after irradiation. Both bromophenols themselves as well as
their contaminants (PBDEs) may act as PBDD/PBDF precursors
(Ritterbusch et al., 1994a). The highest levels were found with the
mono- to tetrabrominated homologues. The total concentrations of
dibenzofurans exceeded those of dibenzo- p-dioxins: 315.7 µg/kg
versus 14.2 µg/kg after irradiation of crude 4-bromophenol, and 1750.7
µg/kg versus 301.0 µg/kg after irradiation of crude 2,4-dibromophenol
(Ritterbusch et al., 1994a).
The atmospheric stability of PBDDs/PBDFs that resulted after
combustion of polyurethane foam (containing PBDEs) at a range of
temperatures was examined under sunlight conditions. The formation of
PBDFs, primarily tetra- and pentaBDFs (isomers not examined), in the
presence of sunlight was seen with the products of low-temperature
(400-470°C) combustion. The formation of the PBDF compounds in this
case was thought to be the result of the photolysis of unburned PBDEs
(Birla & Kamens, 1994).
For photochemical transformations of higher brominated
PBDDs/PBDFs to lower brominated congeners, see section 4.2.1.
3.4 Formation from the laboratory thermolysis of bromine-containing
flame retardants
The potential of typical brominated flame retardants to form
PBDDs/PBDFs was examined under various conditions in a series of
laboratory thermolysisa experiments (see also Tables 10-12). The
flame retardants were thermally treated either alone (Buser et al.,
a For definitions of terms referring to thermal treatment, see
Appendix II.
1978; Thoma & Hutzinger, 1987a,b; Dumler et al., 1989a, 1990b;
Zacharewski et al., 1989) or blended with polymer matrices (Dumler et
al., 1990b,c; Riggs et al., 1990; Lahaniatis et al., 1991; Lorenz &
Bahadir, 1993). PBDFs were found in most of the samples, but both the
concentration and the degree of bromination varied greatly. PBDDs were
detected to a lesser extent. Owing to the different conditions used,
it is difficult to compare these studies quantitatively -- except for
results from the same experimental series.
The largest yields of PBDDs/PBDFs were obtained from PBDEs
(especially in combination with polymers) and from bromophenols, both
reaching values in the g/kg range. In contrast to PBDEs, the
bromophenols that are flame retardants of only limited use (BMU, 1989)
were not tested in polymer matrices. About an order of magnitude lower
yields of PBDDs/PBDFs were observed upon thermolysis of certain PBBs.
Again, lower but significant amounts of PBDDs/ PBDFs in the mg/kg
range were generated by TBBPA and 1,2-bis-(tribromophenoxy)ethane.
Formation of PBDDs/PBDFs from all other flame retardants tested was
very low or undetectable (Tables 10 and 11). Among the PBDEs, the
yield of PBDDs/PBDFs in thermolytic residues decreased from pentaBDE
to octaBDE to decaBDE (e.g. Buser, 1986a; Thoma et al., 1987a; Luijk
et al., 1991).
The optimum PBDF formation temperatures of flame retardants
thermally treated alone were found to be in the range of 600-900°C.
For example, bromophenols and TBBPA showed PBDF formation maxima at
800°C, pentaBDE at 700-800°C, and decaBDE at 800-900°C, whereas PBBs
(hexabromobiphenyl, or hexaBB) had no clear peak concentration between
700°C and 900°C on pyrolysis in quartz tubes (Thoma et al., 1986a,
1987a). Under other experimental conditions, decaBDE (alone) produced
maxima at 600°C (Dumler et al., 1990a) or 700°C (Dumler et al.,
1989a,c, 1990b,c). When decaBDE was burned in a polymer matrix, the
PBDF formation maximum was shifted to lower temperatures (Thoma et
al., 1987a; Dumler et al., 1990b). This effect was not observed with
pentaBDE (Thoma et al., 1987a; Dumler, 1989; Hutzinger, 1990).
Plastic/flame retardant mixtures showed maxima of PBDF formation at
600°C (decaBDE/PBT: Dumler et al., 1990b; decaBB/PBT: Luijk & Govers,
1992), 700°C (pentaBDE/polyurethane: Dumler, 1989; Hutzinger, 1990;
TBBPA/ABS: Luijk & Govers, 1992), or 800°C (TBBPA/ epoxide laminate:
Dumler, 1989; Hutzinger, 1990). The presence of Sb2O3 in the polymer
matrices resulted in a further decrease of the optimum formation
temperature (down to 400°C) of PBDFs from octa- or decaBDE (Clausen et
al., 1987; Bieniek et al., 1989; Dumler, 1989; Dumler et al., 1989a,c,
1990a,b; Hutzinger, 1990; Zier et al., 1990; Luijk et al., 1991). An
example referring to decaBDE is given in Table 12.
The polymer matrix and the synergistic action of Sb2O3
influenced both the optimum temperature range and the yield of PBDFs
(e.g. with hexaBB and decaBDE). Additionally, thermolysis in a polymer
matrix changed the ratio of PBDF congeners to lower brominated
compounds (e.g. Thoma et al., 1987a; Dumler et al., 1990b; see also
Table 12). Frequently, tetraBDFs were the most abundant homologue
Table 10. Survey on the generation of PBDFs and PBDDs during thermolysis of bromoorganic flame retardants
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
PBDEs
Technical PBDE quartz minivials Buser (1986a)
in air 510 °C mono to penta mono to tetra Sigma PBDDs/PBDFs:
5000-10 000
in air 630 °C mono to hexa mono to penta Sigma PBDDs/PBDFs:
100 000
Technical PBDE quartz tubes
(Bromkal 70-5 DE, 700-900 °C mono to penta mono to tetra TeBDFs: up to 330 400 (700 °C) Thoma et al.
70 DE, and G1) TeBDDs: up to 15 400 (700 °C) (1987a)
800 °C mono to penta mono to tetra Sigma PBDFs/PBDDs: Zacharewski et
up to 610 393 al. (1988)
Technical PeBDE
(Bromkal 70-5 DE) various types of Dumler et al.
ovens (DIN, BIS, (1987);
VCI) 600 °C di to penta di to tetra TeBDFs: up to 87 827 Hutzinger et
TeBDDs: up to 12 374 al. (1989)
Technical PeBDE quartz minivials
500 °C tetra, penta tetra, penta Sigma PBDFs/PBDDs: 12 000 Luijk et al.
600 °C tetra to hexa tetra to hexa Sigma PBDFs/PBDDs: 270 000 (1990, 1991)
Technical PeBDE pyrojector, Thoma & Hutzinger
(Bromkal 70-5 DE) absence of di to tetra none small amounts (1987b, 1989)
oxygen (helium)
700-900 °C
Technical OBDE quartz minivials Buser (1986a)
630 °C tetra to hepta tri to hepta Sigma PBDDs/PBDFs: 50 000
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical OBDE quartz minivials Luijk et al.
600 °C tetra to hexa tetra to hexa Sigma PBDDs/PBDFs: 56 000 (1990, 1991)
Technical DBDE quartz minivials Buser (1986a)
630 °C tetra to hepta tetra to octa Sigma PBDDs/PBDFs:
10 000-20 000
Technical DBDE quartz tubes
(FR 300 BA) 700 °C tetra to octa hepta, octa OcBDD/BDF: 2690 Thoma et al.
800 °C tetra, hexa hepta, octa OcBDD/BDF: 9230 (1987a)
to octa
900 °C penta to octa hepta, octa OcBDD/BDF:13 413
800 °C tetra, hexa hepta, octa Sigma PBDDs/BDFs: Zacharewski et
to octa 10 935 al. (1988)
Technical DBDE VCI oven
400-1000 °C hepta, octa hepta, octa n.sp. Klusmeier et
al. (1988)
Technical DBDE VCI oven
300-800 °C mono to hepta tetra, hexa, Sigma PBDFs: up to Dumler et al.
hepta 7222 (700 °C) (1989c);
Sigma PBDDs: up to Hutzinger
588 (800 °C) (1990)
Technical DBDE DIN oven
400 °C hexa to octa hexa to octa Sigma PBDDs/PBDFs: Dumler (1989);
470/364
600 °C hexa to octa hexa to octa Sigma PBDDs/PBDFs: Dumler et al.
2756/447 (1990a)
800 °C tri to hepta tri to octa Sigma PBDDs/PBDFs:
1114/690
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical DBDE quartz minivials
600 °C tetra to hexa tetra to hexa Sigma PBDDs/PBDFs: 1700 Luijk et al.
(1990, 1991)
Technical DBDE pyrojector, Thoma &
absence of Hutzinger
oxygen
(Fr 300 BA) 700 °C hepta, octa none n.sp. (1987b, 1989)
Two technical PBDE high-temperature Striebich et al.
mixtures (Br3-Br10) flow reactor (1990, 1991)
in nitrogen
650 °C di to tetra none Sigma PBDFs: 900
in air 625 °C tri, tetra di to tetra Sigma PBDFs: 600
Sigma PBDDs: 900
PBBs
Technical hexaBB glass tubes O'Keefe
(FireMaster(R) FF-1) (open) in air
380-400 °C tetra, penta n.a. TeBDFs/PeBDFs: 40/4 (1978)
(sealed) in
nitrogen
380-400 °C traces (tetra) n.a.
Technical hexaBB quartz tubes
(FireMaster(R) BP-6) 700, 800,
900 °C di to hepta none TeBDFs: up to 1523 Thoma et al.
(1987a)
800 °C tri to hepta none Sigma PBDFs: 2070 Zacharewski
et al. (1988)
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
Technical hexaBB pyrojector,
(FireMaster(R) BP-6) absence of
oxygen
600-900 °C none none Thoma & Hutzinger
(1987b, 1989)
Technical decaBB glass tubes Atochem
(Adine 0102) (loosely plugged) (1987)
800 °C none none -
Bromophenols
2-Bromophenol 3 different types
of ovens Dumler et al.
600 °C mono to tri mono, di Sigma PBDFs: up to 215 425 (1987);
Sigma PBDDs: up to 60 634 Hutzinger et
al. (1989)
2,4,6-Tribromophenol quartz tubes Thoma et al.
700, 800, 900 °C di to penta di to hexa TeBDDs: up to 896 000 (1986a)
TeBDFs: up to 8950
2,4,6-Tribromophenol 3 different types Dumler et al.
of ovens (1987);
600 °C tri to penta di to penta Sigma PBDFs: up to 8820
Sigma PBDDs: up to 880 503 Hutzinger et
al. (1989)
2,4,6-Tribromophenol pyrojector, Thoma & Hutzinger
absence of oxygen (1987b, 1989)
600-900 °C none di to penta n.sp.
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
2,4,6-Tribromophenol high-temperature Striebich et al.
flow reactor (1990, 1991);
in nitrogen 625 °C n. a. none n.sp. Dellinger et
in air 500 °C none tetra al. (1993)
2,4,6-Tribromophenol high-temperature
flow reactor
300-800 °C n.sp. tri, tetra 1,3,6,8- and 1,3,7,9-TeBDD:
310 000 and 250 000 (500 °C) Sidhu et al. (1995)
Pentabromophenol quartz tubes
700, 800, 900 °C penta to hepta penta to octa Sigma PBDFs: up to 7042
Sigma PBDDs: up to 7508 Thoma et al. (1986a)
Pentabromophenol 3 different types
of ovens
600 °C tri, tetra tetra, hepta TeBDFs: up to 3307
TeBDDs: up to 3567 Dumler et al. (1987);
Hutzinger et al. (1989)
Pentabromophenol pyrojector, absence
of oxygen 700 °C none hepta, octa small amounts Thoma & Hutzinger
(1987b, 1989)
Others
TBBPA quartz tubes
700, 800, 900 °C mono to tetra mono to tetra Sigma PBDDs/PBDFs:
up to 1150/498 Thoma et al. (1986a)
TBBPA BIS oven
240 °C di di low levels Thies et al. (1990)
Hexabromocyclododecane quartz tubes
700 °C tri to hexa tri, tetra Sigma PBDFs: 0.25
Sigma PBDDs: 0.05 Brenner (1993)
Table 10. (Continued)
Flame retardant Conditions of PBDFsb PBDDsb Concentrations (mg/kg)b,c Reference
thermolysisa
1,2-Bis(tribromophenoxy)ethane high-temperature
flow reactor
in nitrogen 475 °C none none
in air 450 °C none tetra n.sp. Striebich et al.
(1990, 1991)
Tetrabromophthalic
anhydride quartz tubes
700, 800, 900 °C none none Thoma et al. (1986a)
2,4,6-Tribromoaniline sealed tubes
640 °C tetra tetra n.sp. Alsabbagh et al. (1992)
N-(tribromophenyl)-maleimide sealed tubes
630 °C tetra tetra n.sp. Alsabbagh et al. (1992)
a For definitions and descriptions of apparatuses used for thermolysis experiments, see Merz et al. (1986) or Appendix II.
b n.a. = not analysed; n.sp. = not specified.
c Owing to the different conditions used, different studies should not be compared quantitatively.
Table 11. Survey on the generation of PBDFs and PBDDs during thermolysis of bromoorganic flame retardants in polymer matrices
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBDEs
PentaBDE Thoma et al.
(Bromkal 70-5 DE) polystyrene quartz tube (1987a)
700-900 °C 420 000 (Br1-Br5) - -
polyethylene quartz tube
700-900 °C 200 000 (Br1-Br5) - -
PentaBDE polyurethane 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C approx. 50 000c,d x
PentaBDE polyurethane DIN oven Hutzinger
300-800 °C 42 000 (Br1-Br6)c x (1990)
PentaBDE polyurethane foam ignition vessel
670-780 °C n.sp. x Birla & Kamens
(1994)
PentaBDE laminate (SiO2) BIS oven Lenoir et al.
600 °C 2000 (Br1-Br5) x (1994)
laminate (TiO2) BIS oven
600 °C 2.6 (Br1-Br3) x
OctaBDE ABS (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
OctaBDE ABS (Sb2O3) DIN oven
300-800 °C 280 000 (Br1-Br7)c x Hutzinger (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
OctaBDE ABS (Sb2O3) DIN oven Neupert et al.
600 °C 9000 (Br3-Br6) x (1989b)
DecaBDE Thoma et al.
(FR 300 BA) polystyrene quartz tube 7000 (Br1-Br7) - - (1987a)
700-900 °C
polyethylene quartz tube 170 000 (Br1-Br8) - -
700-900 °C
DecaBDE polystyrene (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
DecaBDE polystyrene (Sb2O3) DIN oven 228 000 Hutzinger
(Br1-Br8)c (1990)
300-800 °C x
DecaBDE polypropylene DIN oven Dumler
400-800 °C 255 000 (Br1-Br7)c x (1989)
DecaBDE polypropylene closed glass 393 000 (Br1-Br5)c x Dumler
vials 600 °C (1989)
DecaBDE polypropylene 3 different ovens Dumler et al.
(Sb2O3) (VCI, BIS, DIN) (1989b)
600-800 °C >100 000c,d x
DecaBDE polypropylene DIN oven Hutzinger
(Sb2O3) 300-800 °C 290 000 (Br1-Br8)c x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
DecaBDE HIPS (Sb2O3) mass burning rate Pinkerton et
apparatus al. (1989)
(21% O2)
500-800 °C 1600 (Br1-Br8) x
DecaBDE HIPS (Sb2O3) quartz tube Luijk et al.
reactor (1990, 1991)
in nitrogen
275-825 °C 4300 (Br2-Br8)e x
in air
500-700 °C 710 (Br2-Br6)e x
DecaBDE PBT quartz tube Lahaniatis
apparatus et al. (1989)
400-800 °C none x
DecaBDE PBT VCI oven Sovocool et
400-600 °C n.sp. (presence al. (1990)
of Br1-Br6) x
DecaBDE PBT (Sb2O3) quartz tube Clausen et
apparatus al. (1989)
400-800 °C 13 100 (Br1-Br6) - -
DecaBDE PBT (Sb2O3) VCI oven Dumler et
300-800 °C 160 000 (Br1-Br7)c - - al. (1989c)
PBT (Sb2O3) DIN oven Hutzinger
300-800 °C 228 000 (Br1-Br8)c x (1990)
PBT (Sb2O3) BIS oven Zier et al.
400-1000 °C 13 800 (Br1-Br5)e x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBT (Sb2O3) BIS oven Lenoir et al.
in nitrogen + H2O 29 500 (Br1-Br7) (1994)
600 °C
epoxide resin quartz tube Clausen et
(Sb2O3) apparatus al. (1987)
400-800 °C none x
plastic sheets quartz tube n.sp. Lahaniatis
apparatus (presence of et al. (1989)
600 °C various PBDFs) x
PBBs
HexaBB
(FM BP-6) polystyrene quartz tube 8900 (Br1-Br4) - - Thoma et al.
700-900 °C (1987a)
polyethylene quartz tube 43 000 (Br1-Br4) - -
700-900 °C
DecaBB PBT quartz tube Luijk &
reactor in Govers (1992)
nitrogen
+ 10% O2
400-700 °C 100 (Br3-Br8)e x
Others
TBBPA PBT 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
PBT (Sb2O3) VCI oven Hutzinger
800 °C 41 (Br1, Br2)e x (1990)
PBT (Sb2O3) BIS oven Thies et al.
600 °C 0.11 (Br1-Br4)e x (1990)
epoxide laminate 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <50d,e x
epoxide laminate DIN oven Dumler (1989);
600-800 °C 10 (Br1-Br3)e x Hutzinger (1990)
epoxide laminate VCI oven
800 °C 23 (Br1-Br3)e x Hutzinger (1990)
epoxide laminate 3 different ovens Dumler et al.
(Cu) (VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x
epoxide laminate VCI oven Hutzinger
800 °C 40 (Br1-Br2)e x (1990)
polycarbonate 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <10d,e x
polycarbonate DIN oven Hutzinger
600 °C 8.9 (Br1-Br3)e x (1990)
TBBPA polycarbonate BIS oven Thies et al.
600 °C 5.5 (Br1-Br6)e x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
ABS BIS oven Thies et al.
600 °C 0.4 (Br2-Br4)e x (1990)
ABS quartz tube Luijk & Govers
reactor in (1992)
nitrogen + 10% O2
400-700 °C 3.1 (Br1-Br5)e x
ABS (Sb2O3) VCI oven Hutzinger
800 °C 5.4 (Br1, Br2)e x (1990)
BIS and DIN ovens Thies et al.
600 °C 2.1 (Br1-Br4)e x (1990)
1,2-Bis(tribromophenoxy)ethane ABS (Sb2O3) 3 different ovens Dumler et al.
(VCI, BIS, DIN) (1989b)
600-800 °C <1000c,d x
ABS (Sb2O3) DIN oven Hutzinger
600 °C 500 (Br1-Br4)c x (1990)
Tetrabromophthalic polyurethane 3 different ovens Dumler et al.
anhydride (VCI, BIS,DIN) (1989b)
600-800 °C <100c,d x
polyurethane VCI oven Hutzinger
800 °C 44 (Br1)c x (1990)
DIN oven
800 °C 0.4 (Br4)c x
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
Hexabromocyclo polystyrene 3 different ovens Dumler et al.
dodecane (VCI, BIS, DIN) (1989b)
800 °C 10c,d x Hutzinger
DIN oven (1990)
800 °C 5.5 (Br2-Br4)c x
Hexabromocyclo polystyrene quartz tube Brenner (1993)
dodecane apparatus
700 °C 0.11 (Br4-Br6) x
polystyrene quartz tube Brenner (1993)
insulation foam apparatus
700 °C 0.38 (Br2-Br8) x
Polybrominated polyester 3 different ovens Dumler et al.
polystyrene (VCI, BIS, DIN) (1989b)
600-800 °C <100d,e x Hutzinger
DIN oven (1990)
600 °C 36 (Br1-Br4)e x
Polytribromostyrene PBT (Sb2O3) quartz tube Clausen et al.
400-800 °C none x (1987)
Dibromopropyldian polypropylene 3 different ovens Dumler et al.
(Sb2O3) (VCI, BIS, DIN) (1989b)
600-800 °C <100c,d x
polypropylene DIN oven Hutzinger
(Sb2O3) 600 °C 28 (Br2, Br3)c x (1990)
Table 11. (Continued)
Flame retardant Polymer (additive) Conditions of Maximum yields PBDDs Reference
thermolysisa (mg/kg) of PBDFs presentb
(sum of homologue
groups detected)b Yes No
1,2-Bis(tetrabromo ABS (Sb2O3) 3 different ovens Dumler et al.
phthalimido)ethane (VCI, BIS, DIN) (1989b)
600-800 °C <500c,d x
ABS (Sb2O3) DIN oven Hutzinger
800 °C 118 (Br1,Br2)c x (1990)
PBT (Sb2O3) quartz tube Clausen et al.
400-800 °C none x (1987)
Tetrabromobenz PBT quartz tube Clausen et al.
imidazolone 400-800 °C none x (1987)
Polypentabromo PBT quartz tube Clausen et al.
benzylacrylate 400-800 °C none x (1987)
a For definitions and descriptions of apparatuses used for thermolysis experiments, see Merz et al. (1986) or Appendix II.
b - = no information; n.sp. = not specified.
c Related to flame retardant.
d Figures only.
e Related to blend.
group in these ternary mixtures (e.g. Clausen et al., 1987; Bieniek et
al., 1989; Dumler, 1989; Hutzinger, 1990; Luijk et al., 1991).
Formation of PBDDs from decaBDE, octaBDE, decaBB, and TBBPA
showed a similar dependence on temperature and/or matrix as seen with
PBDFs, but the yields were much lower (Dumler et al., 1990b;
Hutzinger, 1990; Luijk & Govers, 1992). Thermolysis of pentaBDEs,
bromophenols, 1,2-bis(tribromophenoxy)ethane, and some samples of
TBBPA resulted in an increase in the relative proportion of PBDDs
(e.g. Dumler, 1989; Hutzinger, 1990; see also Table 10). Some smaller
differences in the temperature profiles between PBDDs and PBDFs can
also occur (e.g. thermolysis of TBBPA/laminate: formation maximum of
PBDFs at 800°C, of PBDDs at 400°C and 700°C; Dumler, 1989).
The influence of various metals (tin, iron, zinc, copper) and
metal oxides (oxides of zinc, copper, iron, and antimony; silica,
SiO2, and titanium dioxide, TiO2) on yield and pattern of
PBDFs/PBDDs was studied in thermolysis of decaBDE or pentaBDE in
polymer matrices (Lenoir et al., 1994). During thermolysis of decaBDE
in PBT (500°C, BIS oven), all metals and the oxides of copper (I),
iron (III), and antimony (III) caused an increase in PBDD
concentrations. PBDF concentrations were enhanced by oxides of iron
and antimony. Oxides of zinc and copper (II) strongly reduced yields
of PBDDs/PBDFs. During thermolysis of pentaBDE in laminate (600°C, BIS
oven), SiO2 was found to be nearly inert, whereas addition of TiO2
(3%) led to a significant reduction in PBDD/PBDF levels.
Water also plays an important role, as seen in thermolysis
experiments with decaBDE in PBT/Sb2O3 performed in a nitrogen
atmosphere with and without water at 600°C (Zier et al., 1990; Lenoir
et al., 1994). Water increased the PBDF, PBDD, and 2,3,7,8-TeBDF
concentrations by a factor of 7.5, 2.8, and 10, respectively.
Other factors influencing yields and pattern of PBDDs/PBDFs
formed in thermolysis of flame retardants are oxygen (O'Keefe, 1978;
Thoma & Hutzinger, 1987a,b; Bieniek et al., 1989; Luijk & Govers,
1992), air flow rates (Klusmeier et al., 1988), types of combustion
apparatuses (types frequently used are described in Appendix II), and
residence time. Thermolysis of a flame-retarded resin (glass fibre
reinforced PBT/decaBDE/Sb2O3) under simulated municipal waste
incineration conditions confirmed the formation of PBDDs/PBDFs (Riggs
et al., 1990).
2,3,7,8-TeBDF was identified after pyrolysis of a technical-grade
flame retardant (alone) consisting primarily of 2,2',4,4',5,5'-hexaBB
(Buser et al., 1978; O'Keefe, 1978). Thies et al. (1990) found
2,3,7,8- substituted congeners from experiments where TBBPA (alone)
was pyrolysed. 2,3,7,8-Substituted congeners were also found after
pyrolysis of plastics containing decaBB (Luijk & Govers, 1992), PBDEs
(Dumler, 1989; Lahaniatis et al., 1989, 1991; Dumler et al., 1990c;
Hutzinger, 1990; Zier et al., 1990), and TBBPA (Thies et al., 1990;
Lorenz & Bahadir, 1993). Maximum concentrations of 2,3,7,8-TeBDF of up
to 2000 mg/kg of flame retardant were found for pyrolysed polymers
Table 12. Yields of PBDFs from combustion of DBDE, alone and in a polypropylene matrixa,b
PBDF yield (mg/kg) from combustion PBDF yield (mg/kg) from combustion of DBDE in a
of DBDE alone polypropylene matrixc
PBDFs 400 °C 600 °C 800 °C 400 °C 600 °C 800 °C
MonoBDFs - - - 14 432 10 676 4192
DiBDFs - - - 26 462 14 845 4850
TriBDFs - - 11 39 997 24 036 8354
TetraBDFs - - 28 107 517 49 677 29 147
PentaBDFs - - 35 37 419 18 458 6867
HexaBDFs 96 447 81 24 432 5465 948
HeptaBDFs 204 1449 959 4762 1033 353
OctaBDF 107 860 - - - -
Total 407 2756 1114 255 021 124 190 54 711
a Adapted from Dumler et al. (1990a,b).
b Combustion of commercial DBDE samples (pure or mixture of polypropylene/12.5%
DBDE/7.5% Sb2O3) in DIN oven.
c Yield related to DBDE.
containing octaBDE (Dumler, 1989). The highest values of
1,2,3,7,8-PeBDF measured following thermolysis of plastics containing
decaBDE, decaBB, or TBBPA were 63 mg/kg of plastic blend (Zier et al.,
1990), 1.2 mg/kg (Luijk & Govers, 1992), and 50 µg/kg (Thies et al.,
1990), respectively. Maximum levels of 2,3,7,8-substituted tetra- and
pentaBDDs were below 1 mg/kg of blend. For example, 2,3,7,8-TeBDD was
formed during thermolysis of a decaBDE/epoxide resin at 0.8 mg/kg
(Lahaniatis et al., 1991). Because of a lack of reference substances,
the higher brominated PBDDs/ PBDFs with the 2,3,7,8-substitution
pattern were not quantified. (However, a tentative value of 21 µg/kg
for 1,2,3,4,6,7,8-HpBDF was found by Brenner [1993] after thermolysis
of an insulating board consisting of polystyrene foam blended with
hexabromocyclododecane.)
Pyrolysis of PBBs (hexaBB) and PBDEs (PeBDE) at 800°C in the
presence of polyvinyl chloride (PVC) did not result in the formation
of PBDFs/PBDDs but led to mixed brominated/chlorinated biphenyls or
diphenyl ethers and to fully chlorinated compounds. Apparently, the
substitution reactions were favoured over ring-closing reactions
(Thoma et al., 1987b). Moreover, such bromine-chlorine exchange
reactions were also observed during pyrolysis (at 800°C and 900°C) of
PBDDs/PBDFs, leading to PXDDs/PXDFs and PCDDs/ PCDFs. The chlorine
source could be either organic or inorganic (Thoma et al., 1987b,c,d,
1989) (see also section 3.9.1).
Possible pathways for the thermolytic formation of PBDDs/ PBDFs
from flame retardants have been discussed by several authors (Buser,
1986a; Bieniek et al., 1989; Dumler, 1989; Hutzinger, 1990; Lahaniatis
et al., 1991; Luijk et al., 1991; Luijk & Govers, 1992).
Other related polycyclic compounds identified after thermolysis
of brominated flame retardants included bromomethyldibenzofurans
(Sovocool et al., 1990; Lenoir et al., 1994), brominated
benzo[b]naphtho[2,3-d]furans (Lenoir, 1994), brominated phenazines
(Alsabbagh et al., 1992), and hexabromonaphthalene (O'Keefe, 1978).
Detailed descriptions of thermolysis experiments involving PBDEs
and TBBPA are given in the Environmental Health Criteria monographs
for these compounds (WHO, 1994b, 1995).
3.5 Formation during production of plastic materials and presence in
consumer products containing flame retardants
Brominated flame retardants are or were routinely added, at
levels up to 20%, to a number of commercial products, such as
plastics, textiles, carpets, and other materials (Buser, 1986a; WHO,
1994a,b, 1995). They also have applications in a variety of
industries, such as the electronic, electrical engineering, building,
and transport industries (BMU, 1989; Troitzsch, 1990).
PBDDs/PBDFs, as contaminants of certain flame retardants (section
3.2), would be transferred to the flame-retarded products and, under
thermolytic stress (section 3.4), could additionally be formed from
these chemicals during manufacturing processes.
3.5.1 Formation during production processes
PBDD/PBDF levels were monitored during typical processes
(extrusion, injection moulding, etc.) used in the production and
processing of flame-retarded polymers (Donnelly et al., 1989a; Bonilla
et al., 1990; Brenner & Knies, 1990, 1992, 1993a,b, 1994; Thies et
al., 1990). Extruder temperatures of 150-300°C were noted (Donnelly et
al., 1989a; Brenner & Knies, 1990). The polymers used were ABS and
PBT, and the flame retardants examined included OBDE, DBDE,
TBBPA-carbonate oligomer (BC 52), TBPI, brominated styrene, and
1,2-bis(tribromophenoxy)ethane. Results from different studies
(monitoring exhaust streams) are compiled in Table 13. It can be seen
that OBDE and DBDE produced the highest amounts of PBDDs/ PBDFs, the
major portion consisting of PBDFs. PBDF concentrations of about 73 000
ng/m3 air or 7.7 ng/g extruded resin were measured during extrusion
of resins containing DBDE, and 1850 ng/g extruded resin in the case of
OBDE. The levels observed with TBBPA and TBPI were lower by several
orders of magnitude. No PBDDs/PBDFs were formed from brominated
styrene or brominated phenoxyethane. Homologue groups present included
monoBDFs through octaBDF (Table 13). 2,3,7,8-Substituted congeners
were not determined with DBDE (Brenner & Knies, 1990) and were not
detected with TBBPA-carbonate oligomer and TBPI, at detection limits
ranging from 1 to 58 pg/m3 (Brenner & Knies, 1992, 1993a,b, 1994).
However, PBDFs with 2,3,7,8-substitution (0.012 ng/g extruded resin;
but co-elution was possible) were found for OBDE just above the
detection limit (Bonilla et al., 1990).
The actual worker exposure to PBDDs/PBDFs depends on the
ventilation and exhaust conditions around the machines. In some
studies, parallel measurements of the workplace atmosphere were
undertaken, results of which are discussed in section 5.3 (Brenner &
Knies, 1990, 1993a,b; Thies et al., 1990).
3.5.2 Presence in resins and polymer products
PBDDs/PBDFs were determined in various plastic materials at
several processing stages (BMU, 1989; Donnelly et al., 1989a; Bonilla
et al., 1990; Brenner & Knies, 1990, 1992, 1993a,b, 1994; Hutzinger,
1990; McAllister et al., 1990; Thies et al., 1990; Luijk et al.,
1992c; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et al., 1993; Kieper,
1996). These examinations included granulated resins, moulded parts
whose flame retardant additives were known, and parts of commercial
electrical appliances (television sets, printers, computers) whose
flame retardants were unknown (see also Tables 14 and 15). Polymers
used were ABS, HIPS, polystyrene, polyamide, PBT, polypropylene, or
polyurethane in combination with about 5-20% PBDEs, TBBPA,
bromopolystyrenes, TBPI, or other compounds as flame retardants (see
Table 14). The highest levels of PBDDs/PBDFs were found in materials
Table 13. Formation of PBDDs/PBDFs during manufacturing processes
(production of flame-retarded polymers in the chemical industry)
Concentrations (ng/m3)a
Process Sample PBDDs PBDFs PBDF homologue groups Reference
Manufacture of PBT/ Exhaust stream 1 72 904 DiBDFs: 322; TrBDFs: 705; Brenner & Knies
glass fibre resin from extruder head TeBDFs: 980; PeBDFs: 3910; (1990)
blended with HxBDFs: 22 162; HpBDFs: 39 550;
DBDE/Sb2O3 OcBDF: 5275
Manufacture of PBT/glass Exhaust stream 1.1 1.3 DiBDFs: 0.42 (0.16); TrBDFs: Brenner & Knies
fibre resin blended with from extruder (0.87)b 0.48 (0.31);TeBDFs: 0.24 (0.06); (1993a,b)
TBBPA (BC 52)/Sb2O3 head PeBDFs: 0.04 (0.33);
HxBDFs: 0.15 (0.01)b
Exhaust stream 0.2 0.7 DiBDFs: 0.23 (0.2); TrBDFs:
from granulator (1.01)b 0.29 (0.49); TeBDFs:
0.17 (0.08); PeBDFs: 0.02
(0.07); HxBDFs: 0 (0.24)b
Exhaust stream n.d. 0.08 DiBDFs: 0.004; TrBDFs:
from injection (0.001-0.2) 0.012; TeBDFs: 0.014;
moulding machine PeBDFs: 0.013; HxBDFs:
0.039
Exhaust stream n.d. 0.006 DiBDFs: 0.004; TeBDFs: 0.002
from storage hood (0.001-0.2)
Manufacture of PBT/glass Exhaust stream 0.05 0.78 DiBDFs: 0.18; TrBDFs: 0.34; Brenner & Knies
fibre resin blended with from extruder TeBDFs: 0.18; PeBDFs: 0.03; (1994)
TBPI head HxBDFs: 0.06
Processing of ABS/TBBPA Off-gas near a 6 213 MoBDFs: 13; DiBDFs: 200 Thies et al.
material compounding (1990)
machine
Table 13 (Continued)
Concentrations (ng/g of extruded resin)
Process Sample PBDDs PBDFs PBDF homologue groups Reference
Processing of PBT/DBDE Fumes generated n.a. 7.69 TeBDFs: 1.35; PeBDFs: 2.47; Donnelly et al.
+ Sb2O3 resin during extrusion HxBDFs: 1.67; HpBDFs: 2.20 (1989a)
Processing of Fumes generated Bonilla et al.
* ABS/brominated styrene during extrusion n.d. n.d. - (1990)
terpolymer resin
* ABS/1,2-bis(tribromophenoxy)ethane n.d. n.d. -
resin
* ABS/TBBPA resin 0.006 0.020 n.sp.
* ABS/OBDE resin 0.54 1850 n.sp.
a n.a. = not analysed; n.d. = not detected (detection limits in parentheses, if specified); n.sp. = not specified.
b Values in parentheses were obtained when PBT was blended with BC 52 (TBBPA)-PBT-batch (approx. 50% BC 52) instead of BC 52 powder.
flame-retarded with PBDEs and were in the range of several thousand
µg/kg, thus exceeding the levels in any other systems by orders of
magnitude. The range of PBDF concentrations found is given in Table
14. PBDFs were, with few exceptions, the predominant components. The
contamination of electrical appliances having an unknown flame
retardant equipment is shown in Table 15.
Generally, the concentrations of PBDDs/PBDFs were higher in the
typical resin/PBDE products than in the flame retardants alone (see
also section 3.2). For comparisons, it should be noted that the
concentrations shown in Table 14 were calculated on the basis of the
weight of the resins. They would be higher if calculated on the basis
of the weight of flame retardant in the resins. Values obtained using
the latter calculation were presented by Hutzinger (1990), who found
PBDF concentrations ranging from 112 to 1888 mg/kg in polymers
(polypropylene, PBT, ABS, or polyurethane) containing PBDEs as flame
retardants.
Frequently, additional processing resulted in a further increase
in total PBDFs (see Table 14). For example, the sum of mono- to
hexaBDF levels measured in casing parts was higher by a factor of
about 5 compared with that found in the corresponding polymer-flame
retardant blend (polystyrene/DBDE) actually used in manufacturing
(Table 14; UBA, 1992). Factors influencing the extent of formation of
PBDFs are temperature and the duration of such processes as blending,
extrusion, and moulding (Table 14; Donnelly et al., 1989a; McAllister
et al., 1990).
Within the PBDF homologue groups, the highly brominated (>tetra)
derivatives were generally prevalent. Frequently, peak concentrations
were seen with penta- and hexaBDFs (see Tables 14 and 15). In casing
parts, hexaBDFs reached levels as high as 2950 µg/kg; in printed
circuit boards, maximum concentrations (>1000 µg/kg) were seen with
tetra- and pentaBDFs (Table 15; UBA, 1992). Concentrations of mono-,
di-, and triBDFs were in the low µg/kg range, <30 µg/kg for polymers
and casings (BMU, 1989; Brenner & Knies, 1990; Thies et al., 1990;
UBA, 1992) and <450 µg/kg for printed circuit boards (UBA, 1992).
2,3,7,8-Substituted PBDFs were not analysed (Brenner & Knies,
1990), were not detectable (Thies et al., 1990; Brenner & Knies,
1993a,b, 1994; Kieper, 1996), or were detected in some formulations at
low concentrations (Donnelly et al., 1989a; Bonilla et al., 1990;
McAllister et al., 1990; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et
al., 1993; Kieper, 1996). A moulded part consisting of ABS/PBDE
contained 2,3,7,8-TeBDF at 2 µg/kg (Meyer et al., 1993). Plastics for
casings and printed circuit boards of electrical appliances (n = 14)
were found to be contaminated with tetra-, penta-, and hexaBDFs having
the 2,3,7,8-substitution pattern (Table 15; UBA, 1992). The maximum
concentrations of 2,3,7,8-TeBDF were <4% of the total tetraBDFs in
the case of casings and <17% in the case of printed circuit boards.
Higher percentages were found for the two pentaBDFs and for
1,2,3,4,7,8-HxBDF, usually <7-22%, but exceptionally reaching as high
as 75% of the respective homologue groups (UBA, 1992). As co-elution
Table 14. Concentrations of PBDFs in several flame-retarded plastic materials
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
ABS/OBDE
* Granulate - 2100 24 000 50 000 3900 1700 BMU (1989)
* Normal extrusionb
(n = 17-22) - 2.8-3.6 870-1800 2100-2380 500-780 26-64 Donnelly et al.
(1989a)
* Abusive extrusionb - 150-170 29 000-34 000 8200-10 000 500-920 19 Donnelly et al.
(n = 1-2) (1989a)
* Pre-extrusion resin 38 300 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 84 500 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Normal mouldingc - 3 1100 <135 000 - - McAllister et al.
(1990)
* Abusive mouldingc - 170 <14 000 <118 000 - - McAllister et al.
(1990)
ABS/TBBPA
* Pre-extrusion resin 1090 - - - - - Bonilla et al.
(Br4-Br7)
(1990)
* Post-extrusion resin n.d. - - - - - Bonilla et al.
(1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Commercial polymer - n.d. n.d. n.d. Thies et al.
(<2) (<3) (<20) - - (1990)
ABS/brominated
styrene terpolymer
* Pre-extrusion resin 37.5 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 84.0 - - - - - Bonilla et al.
(Br4-Br7) (1990)
ABS/1,2-bis(tribromo-phenoxy)ethane
* Pre-extrusion resin 44.5 - - - - - Bonilla et al.
(Br4-Br7) (1990)
* Post-extrusion resin 16.2 - - - - - Bonilla et al.
(Br4-Br7) (1990)
HIPS/DBDE
* Normal extrusiond - - 4.5 950 720 150 Donnelly et al.
(1989a)
* Abusive extrusiond - 2.3 22.6 107 78 0.5 Donnelly et al.
(1989a)
* Extreme extrusiond - 0.01 8.6 200 2100 3200 Donnelly et al.
(1989a)
* Base resin - 10 40 <5300 - - McAllister et al.
(1990)
* Normal mouldinge - 10 50 <14 300 - - McAllister et al.
(1990)
* Abusive mouldinge - 10 60 <5500 - - McAllister et al.
(1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Extreme mouldinge - 20 200 <34 100 - - McAllister et al.
(1990)
* Pre-extrusion resin - - - - approx. approx. Luijk et al.
1500 4500 (1992c)
* Post-extrusion resin - - - - approx. approx. Luijk et al.
(4 cycles at 275 °C) 9000 45 000 (1992c)
Polystyrene/DBDE
* Compound 194 2.7 14.6 174 - - UBA (1992)
* 2 casing parts manufactured from
the above compound 640; 1313 54; 39 147; 106 1092; 409 - - UBA (1992)
(Br1-Br6)
Polystyrenebutadiene/DBDE
* Compound n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.2) (1.0) (50)
Polystyrene
1,2-bis-(tribromophenoxy)ethane
* Compound n.d. n.d. n.d. n.d. - - Kieper (1996)
(0.7) (2.5) (9.7)
Polyamide/polytribromostyrene
* Compound 15.3 n.d. 0.28 1.81 3.43 6.21 Kieper (1996)
(Br1-Br8) (0.2)
Polyamide/polydibromostyrene
* Compound 4.18 0.64 0.38 0.37 0.46 2.15 Kieper (1996)
(Br1-Br8)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
PBT/DBDE
* Granulate (n = 7) - 6-501 20-920 59-65 000 66-136 000 n.d.-2600 BMU (1989)
* Normal extrusionf - 1-26 18-130 71-1600 180-3800 410-4100 Donnelly et al.
(n = 17-22) (1989a)
* Abusive extrusionf - 76-240 13 000-43 000 69 000-180 000 48 000-94 000 1200-11 000 Donnelly et al.
(n = 5) (1989a)
* Extreme extrusionf
(n = 3) - 1020-2590 68 200-82 800 272 000- 72 500- - Donnelly et al.
708 000 108 000 (1989a)
* Blend - 6.2 27 151 approx. 560 approx. 280 Brenner & Knies
(1990)
* Base resin - 3 20 110 - - McAllister et al.
(1990)
* Normal mouldingg - 3 2 13 - - McAllister et al.
(1990)
* Abusive mouldingg - 30 >7800 >16 100 - - McAllister et al.
(1990)
* Extreme mouldingg - 1000 >54 000 >7000 - - McAllister et al.
(1990)
PBT/TBBPA
* Commercial polymer - n.d. n.d. n.d. - - Thies et al.
(<0.2) (<0.1) (<1) (1990)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
* Extruder granulate Brenner & Knies
(n = 3) - n.d. n.d. 0.4-0.8 0.6-3.5 - (1990)
* Moulded test articles Brenner & Knies
(n = 2) - 0.17-0.2 n.d.-0.06 1.5-2.2 1.9-3.8 - (1993a,b)
* Compound 8.41 0.14 2.13 6.14 - - Kieper (1996)
(Br1-Br6)
PBT/bromopolystyrene
* Granulate (n = 2) - n.d.-5 2-10 34-130 11-460 - BMU (1989)
PBT/bis-tetrabromophthalimide
* Granulate - - 5 35 31 - BMU (1989)
PBT/TBPI
* Polymer - 0.57 0.07 0.02 3.4 - Brenner & Knies
(1994)
* Granulate (n = 2) - up to 0.8 0 0 0 - Brenner & Knies
(1994)
* Moulded test article - 0 0 0 0 - Brenner & Knies
(1994)
Polypropylene/DBDE
* Granulate - 53 191 10 000 1370 2600 BMU (1989)
Table 14. (Continued)
Resin/flame retardant Concentrations (µg/kg)a Reference
Sum
(homologue
groups) TetraBDFs PentaBDFs HexaBDFs HeptaBDFs OctaBDF
Polyurethane/PeBDE
* Granulate - 18 000 57 000 44 000 - - BMU (1989)
a - = not mentioned; n.d. = not detected (detection limit in parentheses, if specified); n.sp. = not specified.
b Normal/abusive extrusion conditions: 227 °C/246 °C; 1 min/10 min cycle.
c Normal/abusive moulding conditions: 225 °C/245 °C; 1 min/10 min cycle.
d Normal/abusive/extreme extrusion conditions: 216-218 °C/238-243 °C/266-271 °C; 30 second/5 min/7 min cycle.
e Normal/abusive/extreme moulding conditions: 215-220 °C/235-245 °C/265-270 °C; 30 second/5 min/7 min cycle.
f Normal/abusive/extreme extrusion conditions: 250-254 °C/254 °C/254 °C; 23 second/5 min/10 min cycle.
g Normal/abusive/extreme moulding conditions: 255 °C/255 °C/255 °C; 23 second/5 min/10 min cycle.
cannot be excluded, all concentrations of 2,3,7,8-substituted
congeners may be overestimated (UBA, 1992; Meyer et al., 1993; Kieper,
1996).
Table 15. PBDF/PBDD concentrations found in plastics from commercial
electrical appliances with unknown polymer/flame retardant systema
PBDFs/PBDDs Concentrationsb (µg/kg) in plastics for
casings printed circuit boards
(n = 8) (n = 6)
MonoBDFs n.d.-0.5 n.d.-19.8
DiBDFs n.d.-3.1 n.d.-149
TriBDFs n.d.-13.1 0.2-441
TetraBDFs n.d.-48.9 0.6-1264
2,3,7,8-TeBDFc <0.1-1.2 <0.1-11.1
PentaBDFs n.d.-1126 n.d.-1372
1,2,3,7,8-PeBDFc <0.1-16.4 <0.1-24
2,3,4,7,8-PeBDFc <9.1-31.5 <0.1-6.5
HexaBDFs n.d.-2952 n.d.-185
1,2,3,4,7,8-HxBDFc <0.7-203 <1.5-9.9
Total PBDFs n.d.-4125 3.6-3430
Total PBDDs n.d.-113.6d 1.9-1974e
a Adapted from UBA (1992).
b n.d. = not detected. Detection limits: <0.1 µg/kg for
mono- to triBDFs/BDDs; <0.1-<0.3 µg/kg for tetra- and
pentaBDFs/BDDs; <0.7-<2.1 µg/kg for hexaBDFs; and <1.0-<15.5
µg/kg for hexaBDDs.
c Maximum values, because co-elution could not be excluded.
d Seven of eight samples = n.d.
e Five of six samples = 1.9-13.9 µg/kg.
PBDDs were not routinely detected in the samples examined
(Donnelly et al., 1989a; Brenner & Knies, 1990, 1992, 1993a,b;
Hutzinger, 1990; Thies et al., 1990; Kieper, 1996). If present, their
maximum concentrations in several thermoplastic resins ranged from
0.006 to 4500 µg/kg (Bonilla et al., 1990; McAllister et al., 1990;
Lorenz & Bahadir, 1993; Meyer et al., 1993; Kieper, 1996) and from 1.9
to 1974 µg/kg in plastics taken from electrical appliances (UBA,
1992). Whereas some samples contained PBDDs, mainly tetraBDDs, only
trace amounts of PBDFs were present in these samples (Lorenz &
Bahadir, 1993; Kieper, 1996). With plastics from electrical
appliances, one out of eight casing parts and all six printed circuit
boards examined gave positive results for PBDDs (UBA, 1992). The PBDD
concentration measured in the one positive casing part was 114 µg/kg
(consisting only of hexaBDDs). In five out of seven positive samples,
the percentage of PBDDs was low, amounting to a maximum of 2.7% of the
total PBDD/PBDF content. In the remaining two samples (showing
concentrations of about 3 and 2000 µg/kg, the latter being mainly
tetraBDDs), the percentages of PBDDs were 46.3 and 99.7% of the total
PBDD/PBDF levels (UBA, 1992; see also Table 15). Altogether, the PBDD
homologue distribution pattern was somewhat irregular and included
mono- to hexaBDDs (UBA, 1992).
Although 2,3,7,8-substituted penta- and hexaBDDs were present (up
to 25 µg/kg) in a few samples (McAllister et al., 1990; UBA, 1992;
Meyer et al., 1993), 2,3,7,8-TeBDD could not be detected at detection
limits mostly below 0.3 µg/kg (Bonilla et al., 1990; McAllister et
al., 1990; UBA, 1992; Lorenz & Bahadir, 1993; Meyer et al., 1993;
Kieper, 1996).
3.6 Emissions from flame-retarded consumer products
Some experiments were performed to clarify whether or not PBDFs
were released from television sets, computers, or similar appliances
(Bruckmann et al., 1990; Ranken et al., 1990; Thies et al., 1990; UBA,
1992). Positive (Bruckmann et al., 1990; Thies et al., 1990; UBA,
1992) and negative (Ranken et al., 1990; UBA, 1992) results were
obtained.
None of three (Ranken et al., 1990) and three of four (UBA, 1992)
units (television sets, computer monitors) tested in experimental
chambers were found to release PBDFs during 3 × 8 h of operation
(Ranken et al., 1990) or continuously for 72 h (UBA, 1992).
In the experiments conducted by UBA (1992), maximum total
emissions of PBDFs (tetra through hexa) were 1800 pg/unit tested, with
tetraBDFs being most prevalent. 2,3,7,8-TeBDF was detected in one
appliance (see Table 16). Concentrations of PBDEs concomitantly
measured ranged between <1.4 and 890 ng/unit. No PBDFs (detection
limit: 3-10 pg/printer) and only low levels of PBDEs were observed
with the three printers tested.
In another study (Bruckmann et al., 1990), air samples were
collected in a closed room (27 m3) at different distances from a new
television set operating for 3 days for approximately 17 h/day. Total
concentrations of PBDFs (tri through penta) were found to be 155
pg/m3 air at a distance of 0.15 m above the television set and 28
pg/m3 air in the centre of the room (2.2 m distant from the
television set). According to a calculation of UBA (1992), the
concentrations of tetraBDFs (11 pg/m3), pentaBDFs (0.5 pg/m3), and
hexaBDFs (<0.1 pg/m3) measured above the television set correspond
to 732, 33, and <7 pg emitted per television set, respectively. For
comparison, ambient air concentrations were 0.16 pg/m3 (tetraBDFs)
and <0.05 pg/m3 (penta- plus hexaBDFs). Bruckmann et al. (1990) did
not analyse for any 2,3,7,8-substituted congeners.
Table 16. Emissions of PBDFs from television sets and computer monitors
as measured in a test chambera,b
PBDFs Emissions (pg/appliance)c
Television sets (n = 2) Computer monitors (n = 2)
Colour Monochrome
TetraBDFsd 320 n.d. 1045 605
2,3,7,8-TeBDFe n.d. (3) n.d. (5) 15 n.d. (3)
PentaBDFs n.d. n.d. 330 165
1,2,3,7,8-PeBDFe n.d. (7) n.d. (8) <5 n.d. (5)
2,3,4,7,8-PeBDFe n.d. (7) n.d. (8) <5 n.d. (5)
HexaBDFs n.d. (10) n.d. (10) 424 80
HeptaBDFs n.d. (10) n.d. (10) n.d. (15) n.d. (10)
OctaBDF n.a. n.a. n.a. n.a.
Sum tetra- to hexaBDFsf 320 n.d. 1799 850
a Adapted from UBA (1992).
b Volume of test chamber: 1.17 m3; sampling time: continuously, 72 h; sucking
speed: 1.5 m3/h.
c n.d. = not detected (detection limits in parentheses, where specified);
n.a. = not analysed.
d Emissions correspond to about 3, 10, and 6 pg/m3, respectively.
e Maximum value, as co-elution with internal standard cannot be excluded.
f Emissions less than the detection/quantification limit are set as 50% of the limit
when calculating the sum of homologues.
Thies et al. (1990) detected tetraBDFs and pentaBDFs at
concentrations of 3 and 8 pg/m3, respectively, in an air sample
(sampling volume about 500 m3) taken 15 cm above a television cabinet
being installed and kept at 46°C in an office room (45 m3). No PBDFs
could be detected in an air sample collected at a distance of 2.2 m
from the television set.
The presence of PBDFs in "non-experimental" rooms equipped with
monitors and other appliances is discussed in sections 5.1.1 and
5.3.1.
3.7 Presence in fire residues, smoke condensates, and gases after
fires
3.7.1 Experimental fires
Experimental fire tests simulating real fire conditions were
performed with electrical appliances such as television sets,
printers, computer terminals, and their casings (Fabarius et al.,
1990; Hamm & Theisen, 1992; UBA, 1992). High PBDF concentrations can
be produced under these conditions (see Table 17). The total PBDD/
PBDF concentrations in combustion residues reached values of between 1
and 1930 mg/kg (up to 0.2%) for electrical appliances and between 8000
and 9000 mg/kg (almost 1%) for the casing parts (see Table 17).
PBDD/PBDF levels determined in some appliances before burning were in
the range n.d.-4.2 mg/kg (Hamm & Theisen, 1992; UBA, 1992). Analysis
of smoke condensate from the fire test room gave area contaminant
concentrations ranging from 6 to 1610 µg/m2 (Table 17), most values
being in the range of 100-400 µg/m2 (Hamm & Theisen, 1992; UBA,
1992). The smoke samples collected during the fire tests contained
0.8-1700 µg PBDDs/PBDFs/m3 (Table 17).
With some exceptions -- where penta- or decaBDE was specified --
the flame retardants being included in the test materials were unknown
(Fabarius et al., 1990; Hamm & Theisen, 1992; UBA, 1992).
As seen in Table 17, the total PBDD concentrations were low in
combustion residues (n.d.-2.0 mg/kg), smoke condensate (n.d.-1.2
µg/m2), and smoke (n.d.-3.9 µg/m3), with a maximum of 3% of the
corresponding total PBDD/PBDF concentrations (Fabarius et al., 1990;
Hamm & Theisen, 1992; UBA, 1992).
In one study (Fabarius et al., 1990), the most abundant homologue
groups were the tri- and tetraBDDs/BDFs. Other studies (Hamm &
Theisen, 1992; UBA, 1992) showed a homologue distribution pattern
dominated by tetra- through hexaBDDs/BDFs.
The proportion of 2,3,7,8-TeBDF was mostly under 3% of the total
of tetraBDFs. 2,3,7,8-Substituted congeners of penta- and hexaBDFs
yielded between 1% and a maximum of 16% of the corresponding totals.
For example, concentrations measured in fire residues (n = 8) ranged
from 0.005 to 18 mg/kg for 2,3,7,8-TeBDF, from 0.005 to 116 mg/kg for
the two 2,3,7,8-substituted PeBDFs, and from <0.014 to 567 mg/kg for
1,2,3,4,7,8-HxBDF (UBA, 1992). PBDDs with 2,3,7,8-substitution were
not detected; however, their detection limits were relatively high
(0.001-1.9 mg/kg in combustion residues) (UBA, 1992).
During preliminary experiments intended to improve sampling
techniques, test vehicles (one car, one subway wagon) were burnt in a
traffic tunnel (Wichmann et al., 1993). Noticeable amounts of PBDFs
(Br1-Br6) and PCDDs (Cl4-Cl8) were released at both fires. The
measurements performed gave values in the high ng/m3 range (graphics
given only). Fire residue samples from different materials (e.g.
paint, floor coverings, cables, etc.) taken from the burnt-out
vehicles (one car: n = 4, one subway wagon: n = 5) showed
concentrations of up to 4000 ng/kg for mono- to triBDFs and up to 250
ng/kg for tetra- to octaBDFs. PBDD concentrations were lower, having
peak concentrations of 120 ng/kg for mono- to triBDDs and of <22
ng/kg for tetra- to octaBDDs (Zelinski et al., 1994).
Table 17. PBDF/PBDD concentrations in samples from fire tests with electrical appliances
Fire object (n) Fire site Homologue groups Total PBDF/PBDD concentrationsa,b
analysed/abundant Combustion residue Smoke condensate Smoke/gas Reference
(mg/kg) (µg/m2) (µg/m3)
Television barrel Br3-Br8/Br3, Br4 79.27 n.a. 0.825 Fabarius et al.
set (2) (2.0) (0.02) (1990)
Casing parts of room Br1-Br6/Br4-Br6 7750-8700 177-1610 n.a. Hamm & Theisen
electrical (n.d.) (n.d.) (1992);
appliances (2) UBA (1992)
Electrical room Br1-Br6/Br4-Br6 1-1930 6-323 11-1700 Hamm & Theisen
appliances (6) (n.d. -0.7) (n.d.-1.2) (n.d.-3.9) (1992);
UBA (1992)
a PBDD concentration in parentheses.
b n.a. = not analysed, n.d. = not detected.
(It is very difficult to extrapolate values from gases, smoke, or
flue gases [ng/m3] to areal contamination [ng/m2]. The indoor and
outdoor dispersion vary greatly and are quite difficult to predict.
Furthermore, human exposure and resulting body burdens cannot be
predicted.)
3.7.2 Accidental fires
Analyses of fire residues, smoke condensates, gases, and
firemen's trousers confirmed the expected release of PBDDs/PBDFs
during real fire accidents (Buser, 1986b; Bruckmann et al., 1990;
Fabarius et al., 1990; Hamm & Theisen, 1992; Neupert & Pump, 1992;
UBA, 1992; Zelinski et al., 1993, 1994; Schacht et al., 1995; see also
Table 18). With some exceptions (bowling hall, stockhouse, computer
room), all fire cases examined occurred in private residences with
television sets being involved. Concentrations measured were mostly
below the values found in the model experiments described above, but
the qualitative compositions of the samples were similar (see section
3.7.1).
PBDFs dominated clearly over PBDDs. In contrast to PBDFs, which
were found in almost all samples, PBDDs were not regularly detected;
if present, their concentrations were low. For example, PBDDs could be
identified in three of nine television fire incidents. In these three
incidents, only four of nine samples contained PBDDs, the maximum
concentrations being less than 1.5% of the sum of PBDF/ PBDD (mono
through hexa) levels (UBA, 1992). Likewise, the sum of PBDD
concentrations in two soot samples collected after a fire in a
computer room were in the range of 0.1-30 µg/kg (Schacht et al.,
1995).
As seen in Table 18, PBDFs covered a wide range of
concentrations. The PBDF levels of combustion residues were mainly in
the µg/kg range, but two maximum values of 107 mg/kg (Br1-Br6; UBA,
1992) and 17 mg/kg (Br1-Br8; Zelinski et al., 1993) were also
observed (see Table 18). The PBDF (Br1-Br6) area contaminant
concentrations (caused by smoke condensates) in close vicinity to the
fire site ranged between 0.1 and 13.1 µg/m2 in most cases (UBA,
1992). In the adjoining areas, the levels measured were usually lower
by a factor of 2-34. Interestingly, there were no systematic
correlations between PBDF concentrations found in combustion residues
(fire site) and those found in wipe samples (area contamination). It
is thus difficult to predict the area contamination from residue
analysis (Hamm & Theisen, 1992; UBA, 1992). Zelinski et al. (1993)
found that not only the distance from the source but also the surface
characteristics of the objects influence the PBDF content of the
samples. The maximum gaseous emissions of PBDFs (Br2-Br8) measured
in other cases amounted to 3.5 ng/m3 (Fabarius et al., 1990). One
study provided tentative information on contamination of a fireman's
trousers after fighting a fire. PBDFs (Br2-Br8) were found at a
concentration of 2.01 µg/kg (Fabarius et al., 1990).
Table 18. Release of PBDFs during accidental fires in private residences or other buildings
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
MonoBDFs house (3) TV set (1) n.d.-0.1 8.1-16.9 n.d.-2.2 n.a. UBA (1992)
house (1) TV set (1) 17 n.d. n.d. n.a. UBA (1992)
flat (5) TV set (1) n.d. n.d.-9.3 n.d.-14.7 n.a. UBA (1992)
DiBDFs residence (4-5) n.sp. 0.1-2.3 n.a. n.a. <0.1 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-2.7 43.7-146 n.d.-36 n.a. UBA (1992)
house (1) TV set (1) 595 2 15 n.a. UBA (1992)
flat (5) TV set (1) n.d.-0.7 n.d.-75.8 n.d.-94 n.a. UBA (1992)
TriBDFs residence (4-5) n.sp. 0.06-7.31 n.a. n.a. <0.1-2.0 Fabarius et al.
(1990)
house (3) TV set (1) 0.2-17.9 122-1131 24-110 n.a. UBA (1992)
house (1) TV set (1) 3491 272 85 n.a. UBA (1992)
flat (5) TV set (1) n.d.-3.4 n.d.-756 n.d.-587 n.a. UBA (1992)
TetraBDFs stock house (1) n.sp. n.sp. 4.0; 32 8.9; 123 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. 0.12-8.6 n.a. n.a. <0.1-1.5 Fabarius et al.
(1990)
house (3) TV set (1) 0.4-93.2 532-4396 64-216 n.a. UBA (1992)
house (1) TV set (1) 16 063 2392 225 n.a. UBA (1992)
flat (5) TV set (1) n.d.-6.6 n.d.-2432 n.d.-1505 n.a. UBA (1992)
computer room (1) equipment 13.6-2700 n.a. n.a. n.a. Schacht et al.
(2 soot samples) (1995)
Table 18. (Continued)
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
PentaBDFs stock house (1) n.sp. n.sp. 0.8; 4.4 9.2; 78 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-5.7 n.a. n.a. <0.01 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-64 1597-3919 142-232 n.a. UBA (1992)
house (1) TV set (1) 64 932 4263 401 n.a. UBA (1992)
flat (5) TV set (1) 0.1-6.5 n.d.-3671 n.d.-1670 n.a. UBA (1992)
computer room (1) equipment
(2 soot samples) 15-2100 n.a. n.a. n.a. Schacht et al.
(1995)
HexaBDFs stock house (1) n.sp. n.sp. 4.5; 27 15; 52 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-2.34 n.a. n.a. 0.1 Fabarius et al.
(1990)
house (3) TV set (1) n.d.-102 2210-4840 69-981 n.a. UBA (1992)
house (1) TV set (1) 21 740 544 121 n.a. UBA (1992)
flat (5) TV set (1) n.d.-7.7 n.d.-3574 n.d.-1504 n.a. UBA (1992)
computer room (1) equipment 0.8-711 n.a. n.a. n.a. Schacht et al.
(2 soot (1995)
samples)
HeptaBDFs stock house (1) n.sp. n.sp. 0.25; 1.2 7; 13 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05-0.87 n.a. n.a. <0.1 Fabarius et al.
(1990)
computer room (1) equipment n.d.-6.2 n.a. n.a. n.a. Schacht et al.
(2 soot (1995)
samples)
OctaBDF stock house (1) n.sp. n.sp. 1.1; 5.6 0.5; 8.5 n.a. Bruckmann et al.
(1990)
residence (4-5) n.sp. <0.05 n.a. n.a. <0.1 Fabarius et al.
(1990)
Table 18. (Continued)
PBDFs Buildinga Major fire PBDF concentrationsb Reference
(n) objectb (n) Fire residues Smoke condensatec Gases
(µg/kg) (ng/m2) (ng/m3)
A B
Sum MoBDFs-TrBDFs flat (1) TV set (1) 1894 n.a. n.a. n.a. Zelinski et al.
(1993)
other things 1.2-203 n.a. n.a. n.a. Zelinski et al.
(11) (1993)
flat (1) window frame, 2.3-67.8 n.a. n.a. n.a.
etc. (4)
flat (1) wallpaper n.d.-20.7 n.a. n.a. n.a. Zelinski et al.
above TV, (1993)
etc. (5)
Sum MoBDFs- HxBDFs house/flat (8) TV set (1) 0.5-235 n.d.-13 054 n.d.-5374 n.a. UBA (1992)
house (1) TV set (1) 106 838 7473 847 n.a. UBA (1992)
Sum DiBDFs- OcBDF residence (4-5) n.sp. 0.3-27.3 n.a. n.a. <0.1-3.5 Fabarius et al.
(1990)
Sum TeBDFs- HpBDFs computer room (1) equipment 29-5600 n.a. n.a. n.a. Schacht et al.
(2 soot samples) (1995)
Sum TeBDFs- OcBDF stock house (1) n.sp. (6) n.d.-1.4 n.sp. n.sp. n.a. Bruckmann et al.
flat (1) TV set (1) 14 910 n.a. n.a. n.a. (1990)
other things
(11) 0.4-63.6 n.a. n.a. n.a. Zelinski et al.
(1993)
flat (1) window frame, 1.0-14.4 n.a. n.a. n.a. Zelinski et al.
etc. (4) (1993)
flat (1) wallpaper above 2.7-169 n.a. n.a. n.a. Zelinski et al.
TV, etc. (5) (1993)
a Sampling time after fire: 0-7 days (if specified).
b n.a. = not analysed; n.d. = not detectable; n.sp. = not specified.
c Samples taken close to fire site (A) or distant from fire site (B).
The distribution pattern of the PBDF homologue groups was, in
most cases, dominated by tetra- through hexabrominated homologues (see
Table 18). On the other hand, peak concentrations were found for the
di- through tetraBDFs (Zelinski et al., 1993). The following order
resulted from ranking the dibenzofuran concentrations detected in fire
residues from a computer room: TeBDFs > PeBDFs > HxBDFs > HpBDFs
(Schacht et al., 1995).
The proportion of 2,3,7,8-substituted isomers was low in the
samples examined (Fabarius et al., 1990; UBA, 1992; Zelinski et al.,
1993). Single maximum proportions of 3, 10, or 18% of the
corresponding totals of tetra-, penta-, or hexaBDFs, respectively,
were reported from fire accidents with television sets (UBA, 1992; see
also Table 19). Neupert & Pump (1992) reported on the occurrence of
2,3,7,8-substituted tetra- and pentaBDFs in residue samples collected
after a fire in the store of a plastics production plant. Maximum
concentrations found were 0.4, 1, and 2 µg/kg for 2,3,7,8-TeBDF,
1,2,3,7,8-PeBDF, and 2,3,4,7,8-PeBDF, respectively. 2,3,7,8-TeBDD,
which usually has detection limits of 0.1 µg/kg (residue samples) or
0.8 ng/m2 (wipe samples), was identified at 0.3 µg/kg in one residue
sample of a television set (UBA, 1992). In four out of five samples
from the warehouse fire, 2,3,7,8-TeBDD was detected at concentrations
of 0.3-0.5 µg/kg (Neupert & Pump, 1992). Soot samples (n = 2)
collected after a fire in a computer room (Schacht et al., 1995)
contained 2,3,7,8-TeBDD (n.d./0.6 µg/kg), 1,2,3,7,8-PeBDD (n.d./
<0.04 µg/kg), 2,3,7,8-TeBDF (0.03/48.3 µg/kg), 1,2,3,7,8-PeBDF
(0.2/15 µg/kg), and 2,3,4,7,8-PeBDF (0.4/14 µg/kg).
Analyses of a wipe sample close to the fire site (television)
showed the presence of PXDFs (tri through octa; up to three bromines
per molecule). The total concentration was 3240 ng/m2, or about half
the amount found for PBDFs in this sample (UBA, 1992). Small
quantities of PXDFs were also detected in a soot sample from a fire of
a bowling hall in which plastic, wood, and other materials burnt
(Buser, 1986b). Significant concentrations of PXDFs (about 30-200
µg/kg) occurred in fire residues from a department store (Wilken &
Schanne, 1994).
PCDDs/PCDFs were concomitantly present in many samples, but
mostly at lower total concentrations than PBDDs/PBDFs (Bruckmann et
al., 1990; Fabarius et al., 1990; UBA, 1992; Wichmann et al., 1992a,b;
Wilken & Schanne, 1994; Schacht et al., 1995). Exceptions were gas
samples and firemen's trousers containing a higher proportion of
PCDDs/PCDFs (Fabarius et al., 1990). The estimated PBDD/PBDF
concentrations in the soot sample from a fire in a bowling hall were
also lower than those of PCDDs/PCDFs (Buser, 1986b). Other
contaminants additionally identified in fire residues were polycyclic
aromatic hydrocarbons (PAHs) (Hamm & Theisen, 1992), chlorinated and
brominated benzenes, bromophenols, polychlorinated biphenyls (PCBs),
PBBs, PBDEs, and TBBPA (Buser, 1986b; Fabarius et al., 1990; Zelinski
et al., 1993).
Table 19. 2,3,7,8-Substituted PBDFs found in residue and wipe samples after accidental
fires of television sets in private residencesa
Congeners Residenceb PBDF concentrationsc
Fire residues Smoke condensated,e
(µg/kg) (ng/m2)
A B
2,3,7,8-TeBDF house (3) <0.1-1.0 17-40.9 1.8-6.8
house (1) 264 n.r. 1.8
flat (5) <0.1-0.1 <0.2-22 <0.1-12.2
1,2,3,7,8-PeBDF house (3) <0.1-3.1 22.5-143 <2.5-6.1
house (1) 1863 n.r. <1.3
flat (5) <0.1-0.1 <0.2-80.7 <0.3-38.2
2,3,4,7,8-PeBDF house (3) <0.1-1.8 17.7-87.7 <0.4-<2.9
house (1) 848 n.r. <1.3
flat (5) <0.1-<0.3 <0.4-31.5 <0.3-15.9
1,2,3,4,7,8-HxBDF house (3) <0.3-17.7 107-851 <6-<17.5
house (1) 1932 n.r. <8.5
flat (5) <0.5-<8.0 <8.5-173 <4.5-69.5
a Adapted from UBA (1992).
b Sampling time after fire: 0.5-4 days.
c Maximum values, as co-elution cannot be excluded.
d Samples taken close to fire site (A) or distant from fire site (B).
e n.r. = not recorded.
3.8 Formation from incineration of fuels
PXDDs (Br1Cl5DDs) and hexachlorodibenzo- p-dioxins (hexa CDDs)
were detected in ash from a wood-fired boiler (nature of "wood" not
specified) at concentrations of 55 µg/kg and 418 µg/kg, respectively
(Harless et al., 1989).
No data were available on incineration of coal, peat, or fuel oil
in power plants.
3.9 Formation during waste disposal and treatment
3.9.1 Incineration
The formation of PCDDs/PCDFs in fly ash of waste incinerators was
recognized in the 1970s (Olie et al., 1977); the additional presence
of PXDDs/PXDFs was first reported in 1986 (Schäfer & Ballschmiter,
1986). Since then, a number of studies (Nakano et al., 1987; Öberg et
al., 1987; Schwind et al., 1988, 1989; Sovocool et al., 1988, 1989;
Harless et al., 1989; Hosseinpour et al., 1989; Huang et al., 1991,
1992a,b; Tong et al., 1991; Funcke & Hemminghaus, 1993; Hartenstein,
1993; Chatkittikunwong & Creaser, 1994c; Takasuga et al., 1994) have
documented the presence of PBDDs/PBDFs and/or PXDDs/PXDFs in fly ash
and/or flue gas of municipal, clinical, or hazardous waste
incinerators (see also Table 20).
The amount of PHDDs/PHDFs formed critically depends on the
combustion conditions and on the extent to which the combustion can be
controlled. In the past, it was customary in many industrial countries
-- and may still be usual in many developing countries -- to burn the
waste in landfills by open fires, with incomplete combustion forming
toxic by-products. Such conditions may be especially favourable for
the formation of PHDDs/PHDFs. When the problem was recognized, the
technology of waste incineration was greatly improved, and it has at
present reached a high degree of sophistication. An important
reduction can be achieved by optimizing the burning conditions, by
increasing the temperature, residence time, and turbulence. Energy can
be recovered by boilers and can be converted to heat or electricity. A
quick quench to temperatures below 250°C has been found to minimize
the formation of PCDDs/PCDFs.
The flue gas emissions can be controlled by scrubbing the gases
with dry, semi-dry, or wet technologies. Addition of lime and charcoal
before filtering in a baghouse has been successful. Further reduction
can be achieved by the use of catalysts, which destroy the remaining
PCDDs/PCDFs. This indicates that control of environmental hazards may
be achieved by appropriate but, of course, more expensive measures.
The improved technology should not exclude initiatives for waste
minimization and the development of new technologies for recycling of
plastics and wastes.
Several possibilities for the origin of PHDDs/PHDFs exist. Some
PHDDs/PHDFs may be introduced in trace amounts by the feedstock and
may resist combustion. Far larger amounts can be produced in the
incinerator itself, by formation from precursors at high temperatures
in the flame (see section 3.4 and, for example, Sidhu et al., 1995) or
by de novo synthesis at low temperatures in the post-combustion zone
of the incinerator through gas-solid interactions on fly ash. The
latter hypothesis has been proved by several studies (Stieglitz et
al., 1989; Stieglitz & Vogg, 1990; Heinbuch & Stieglitz, 1992, 1993;
Luijk et al., 1992b, 1994). The formation of PXDDs/PXDFs is explained
by the extensive bromine-chlorine exchange reactions observed under
several test conditions (Thoma et al., 1987b,c, 1989; Zier et al.,
1991; Luijk et al., 1992a, 1994). It is assumed that because of the
large quantities of chlorine donors in waste, these reactions
ultimately result in the formation of completely chlorinated
compounds. On the other hand, irreversible bromination of PCDDs/PCDFs
may occur as the fly ash moves from hotter to cooler regions of the
incinerator (Sovocool et al., 1989; Huang et al., 1992b).
Table 20. Detection of PBDDs/PBDFs and PXDDs/PXDFs in fly ash or
flue gas from waste incinerators
Waste Sample (n) Homologue groups detecteda Concentrations Reference
(country)a PBDDs PBDFs PXDDs PXDFs
Municipal fly ash (1) n.a. TeBDFs Br1Cl3-7DDs Br1Cl3-7DFs TeBDFs: 16 ng/kg Schwind et al.
(Germany) Br2Cl2-6DDs Br2Cl2-3DFs Sigma PXDDs: 5535 ng/kg (1988, 1989);
Br3Cl1DFs Sigma PXDFs: 3157 ng/kg Hosseinpour et al.
(1989)
Municipal fly ash (1) n.a. n.a. Br1Cl3-7DDs Br1Cl3-7DFs Sigma PXDDs: 108 µg/kgb Sovocool et al.
(USA) Sigma PXDFs: 9.8 µg/kgb (1988, 1989)
Municipal fly ash (1) n.sp. n.sp. Br1Cl5DDs n.sp. Br1Cl5DDs: 31 µg/kg Harless et al.
(USA) (1989)
Municipal fly ash (1) n.a. n.a. Br1Cl3-7DDs Br1Cl3-7DFs Sigma PXDDs: 56 µg/kg Tong et al.
(USA) Sigma PXDFs: 47 µg/kg (1991)
Municipal fly ash (3) n.a. n.a. Br1Cl3-7DDs n.a. Sigma PXDDs: 0.5-163 Huang et al.
(n.sp.) µg/kg (1992a)
Municipal fly ash (3) n.a. n.a. Br2Cl2-6DDs Br2Cl2-6DFs Sigma PXDDs: 772-2602
(USA) ng/kg
Sigma PXDFs: 334-1513 Huang et al.
ng/kg (1992b)
Municipal fly ash (1) n.a. n.a. Br2Cl2DDs n.d. Br2Cl2DDs: 0.4 ng/kg Huang et al.
(Japan) (1992b)
Municipal fly ash (1) n.a. n.a. Br2Cl2DDs Br2Cl2-6DFs Sigma PXDDs: 1704 ng/kg Huang et al.
(Canada) Sigma PXDFs: 1335 ng/kg (1992b)
Table 20. (Continued)
Waste Sample (n) Homologue groups detecteda Concentrations Reference
(country)a PBDDs PBDFs PXDDs PXDFs
Municipal fly ash (3) Di-, TrBDDs Mo-, DiBDFs Br1Cl1,4,5DDs Br1Cl1,3,4DFs Sigma PBDDs: 145-436 Chatkittikunwong
(United ng/kg & Creaser (1994c)
Kingdom) Br2Cl1-2DDs Br2Cl1-3DFs Sigma PBDFs: 12-325
ng/kg
Sigma PXDDs: 406-1005
ng/kg
Sigma PXDFs: 1347-2922
ng/kg
Clinical fly ash (1) n.d. MoBDFs Br1Cl1,4,5DDs Br1Cl1,3DFs MoBDFs: 77 ng/kg Chatkittikunwong
(United (Di, Br2Cl1-2DDs Br2Cl1DFs Sigma PXDDs: 705 ng/kg & Creaser (1994c)
Kingdom) TrBDDs) Sigma PXDFs: 427 ng/kg
Hazardous flue gas (6) n.d. n.a. Br1Cl3DDs Br1Cl3DFs Br1Cl3DDs: Öberg et al.
(Sweden) (TeBDDs) n.d.-1.3 ng/m3 (1987)
Br1Cl3DFs:
n.d.-4.5 ng/m3
Hazardous flue gas (2) n.sp. n.sp. n.a. n.a. Sigma PBDDs: Hartenstein
(Germany) 0.76-0.82 ng/m3 (1993)
Sigma PBDFs:
0.76-0.82 ng/m3
a n.a. = not analysed; n.d. = not detected; n.sp. = not specified.
b Estimated concentration.
There are some reports on the consequences of an increase in
bromine input during test operations in incinerators. In a large-scale
experiment at the municipal waste incinerator at Bielefeld-Herford
(Germany), material containing 4.8% pentaBDE was added to the normal
fuel (Lahl et al., 1991). The fly ash from the electrostatic
precipitator was analysed for PCDDs/PCDFs and PXDDs/PXDFs as well as
for inorganic bromine. The bromine content in the samples ranged from
0.37 to 0.59 mg Br-/kg. Of the mixed PXDD/PXDF congeners, only
monobromopolychlorinated PXDDs/PXDFs (Br1ClxDDs/ Br1ClxDFs) could
be detected in the five fly ash samples. The concentrations of
PXDDs/PXDFs (Br1Cl2 to Br1Cl7) ranged from 1.547 to 10.163 µg/kg.
Interestingly, the concentrations of the purely chlorinated compounds
(Cl4-Cl8DDs/Cl4-Cl8DFs) were much higher (up to 406.17 µg/kg) than
normally detected (50-150 µg/kg). Although the highest concentrations
of all three parameters analysed (Br-, PXDDs/PXDFs, and PCDDs/PCDFs)
were found in the same sample, a quantitative relationship could not
be established. Whereas concentrations of PCDDs were higher than
concentrations of PCDFs in all samples, concentrations of PXDFs were
higher than concentrations of PXDDs (Lahl et al., 1991). An increase
in concentrations of monobromopolychlorinated PXDDs/PXDFs in the crude
gas was also found after addition of tetrabromomethane to the furnace
of a municipal waste incinerator (Wilken et al., 1990). Wanke et al.
(1996) studied the influence of additional bromine input (up to
sixfold) into municipal solid waste incinerators at a pilot plant
(nominal throughput: 200 kg/h) at combustion temperatures of 850 or
950°C. Extruded polystyrene foams and rigid polyurethane foams (2-4%
Br by weight) were introduced as bromine source. In the raw gases of
the polyurethane foam combustion, no increase in PCDDs/PCDFs was
detected when compared with "normal" fuel. The concentrations of
PBDDs/PBDFs were very low in all experiments. Both test series, the
experiments with extruded polystyrene and rigid polyurethane foams,
showed elevated levels of PXDDs/PXDFs. As was reported from the
Bielefeld-Herford incinerator, of the PXDDs/PXDFs, those congeners
containing only one bromine were the most abundant; dibrominated
species could hardly be detected. More than two bromine atoms could
not be identified in any sample. Similarly, the PXDDs/PXDFs
contributed only 20-30% of the total sum of all halogenated (PCDDs/
PCDFs + PXDDs/PXDFs) dibenzo- p-dioxins and dibenzofurans. This
finding was not found earlier by Hartenstein (1993), who reported much
higher concentrations of PBDDs/PBDFs than of PCDDs/PCDFs in flue gas
samples of a hazardous waste incinerator. The results of Wanke et al.
(1996) showed that, at least for the PXDFs, there is a correlation
between the content of bromide in the fly ash (up to 5% by weight) and
the concentrations of PXDFs. Above 5% Br-, no further increase in
concentrations of PXDFs could be determined; in other words,
saturation was obtained. For the PBDDs, such a correlation could not
be established, as the concentrations of the PBDDs were too low and
the standard deviation too large.
Once the PHDDs/PHDFs have been formed, they partition between
stack gas (gas phase) and fly ash (solid phase) (Schramm et al.,
1990).
The quantities of PBDDs/PBDFs and PXDDs/PXDFs measured in fly ash
of incinerators were in the range of ng/kg to µg/kg (see Table 20).
The few flue gas measurements gave values in the low ng/m3 range (see
Table 20).
In most cases, the concentrations of dibenzo- p-dioxins exceeded
those of dibenzofurans (see Table 20). PXDDs/PXDFs were more abundant
than PBDDs/PBDFs (Chatkittikunwong & Creaser, 1994c; see also Table
20) but were present in fly ash or chimney residues at lower levels
than their fully chlorinated counterparts, reaching 1-20% of the
levels of PCDDs/PCDFs (Schäfer & Ballschmiter, 1986; Sovocool et al.,
1988, 1989; Harless et al., 1989; Tong et al., 1991; Chatkittikunwong
& Creaser, 1994c). For example, the total PCDD/ PCDF levels ranged
from 37 to 62 µg/kg, and the total PBDD/PBDF plus PXDD/PXDF levels
amounted to 1.2-3.5 µg/kg in fly ash from municipal (n = 3) and
clinical (n = 1) incinerators (Chatkittikunwong & Creaser, 1994c).
However, flue gas of a hazardous waste incinerator contained more
PBDDs/PBDFs than PCDDs/PCDFs (1.6 versus 0.05 ng/m3) (Hartenstein,
1993). No information was provided on the ratio of chlorinated to
brominated compounds in the feedstock.
The main homologues of PXDDs/PXDFs that were detected consisted
of mono- and dibromopolychlorinated PXDDs/PXDFs (see Table 20).
Generally, the highest levels were found for Br1Cl4, Br1Cl5, or
Br1Cl6 congeners (Schwind et al., 1988, 1989; Sovocool et al., 1988,
1989; Hosseinpour et al., 1989; Tong et al., 1991; Chatkittikunwong &
Creaser, 1994c).
The isomer distribution patterns of PXDDs/PXDFs were similar to
those found for PCDDs/PCDFs and similar among different samples,
indicating common mechanisms of formation, regardless of the
incinerator conditions and nature of the feedstock (Harless et al.,
1989; Huang et al., 1992b; Chatkittikunwong & Creaser, 1994c).
Of the 2,3,7,8-substituted congeners, 2,3-Br2-7,8-Cl2DD was
found in several fly ash samples at concentrations ranging from 4 to
12 ng/kg (maximum values because the degree of co-elution was not
known) (Huang et al., 1992b). In another test series, 2,3,7,8-TeBDD
and 2,3,7,8-TeBDF could not be detected (detection limits: 16 and 8
ng/kg, respectively) (Chatkittikunwong & Creaser, 1994c).
There are several methods to minimize the emissions of
dibenzo- p-dioxins and dibenzofurans from incinerators. They are
mostly described for PCDDs/PCDFs, but they may also be valid for
PBDDs/ PBDFs and PXDDs/PXDFs (Boyd & Mortland, 1985; Hagenmaier et
al., 1987a,b; Vogg et al., 1987; Vogg, 1989; Wania & Lenoir, 1990;
Acharya et al., 1991; Spahl et al., 1993; Gullett et al., 1994;
Schreiber, 1994; van de Plassche et al., 1994; Vehlow, 1995).
Flue gas monitored after flue gas cleaning in the stack of a
Swedish municipal solid waste incinerator operating at a high
combustion efficiency did not contain certain PBDDs/PBDFs and
PXDDs/PXDFs (Öberg & Bergström, 1990). The detection limits for the
compounds examined were 0.4 ng/m3 (tetraBDFs/BDDs, pentaBDDs) and
0.03 ng/m3 (Br1Cl3DDs/DFs, Br2Cl2DDs).
3.9.2 Disposal
Disposal sites (dumps, landfills) are expected to be an important
source of brominated dibenzo- p-dioxins/dibenzofurans (Sovocool et
al., 1988; Donnelly et al., 1990; Öberg & Bergström, 1990) because
they receive plastic waste, municipal incinerator fly ash, automotive
fluff (ground-up waste residue from junked cars, which remains after
the bulk metals have been reclaimed), etc. and can also be subject to
occasional fires.
A detailed investigation of waste samples from three German
disposal sites confirmed the occurrence of PBDDs/PBDFs and PXDDs/PXDFs
along with PCDDs/PCDFs (Dawidowsky, 1993). The sum of the
concentrations of PBDDs/PBDFs and PXDDs/PXDFs was in the range of
several hundred to thousands of ng/kg dry weight (see Table 21). The
concentration of dibenzo- p-dioxins was low (PBDDs/ PXDDs: 6-580
ng/kg) in relation to the concentration of dibenzofurans (PBDFs/PXDFs:
217-4229 ng/kg). PBDFs may be prevalent over PBDDs (maximum
concentrations >3000 ng/kg versus >300 ng/kg) because they originate
from PBDEs, which were found at high concentrations in the same
samples (sum of mono- to decaBDEs: 4400-17 500 ng/kg dry weight,
n = 6).
The homologue profile was dominated by lower halogenated
derivatives (up to Br4/X4; Table 21). This pattern contrasted to
that of PCDDs/PCDFs, which had peak concentrations of higher
chlorinated homologues (Dawidowsky, 1993). These waste samples showed
high total concentrations of PCDDs/PCDFs, with PCDDs being the most
abundant components. PCDD values ranged from 3000 to 9000 ng/kg (n =
5) or to nearly 30 000 ng/kg (n = 6); PCDFs reached levels of
2000-5600 ng/kg (n = 6) (Dawidowsky, 1993).
Another study determined PBDDs/PBDFs (and PCDDs/PCDFs) in several
waste samples of an analytical "dioxin" laboratory (Ritterbusch et
al., 1994b). The sum of PBDD/PBDF concentrations in waste oil samples
(n = 3) from the GC/MS system ranged from <150 to 14 000 ng/kg (sum
of PCDDs/PCDFs: <200-13 600 ng/kg). Other laboratory waste samples
(n = 4) also contained PBDDs/PBDFs (mono to hexa), with a peak
concentration of 15 500 ng/kg for hexaBDFs.
3.9.3 Recycling
3.9.3.1 Plastics
Granulated parts of office machine casings, which were
reprocessed once or several times, were analysed for the eight
2,3,7,8- substituted PBDDs/PBDFs of the 1994 German Dioxin Directive
(Meyer et al., 1993). The polymer was ABS flame-retarded with either
PBDE or TBBPA, as well as mixed electronic waste with unknown flame
retardants. Materials flame-retarded with PBDE yielded the highest
Table 21. Occurrence of PBDDs/PBDFs and PXDDs/PXDFs in waste samples from three disposal sitesa
Compounds Concentrations (ng/kg dry weight)b
Disposal site A Disposal site B Disposal site C
Sample 1 Sample 2 Sample 3 Sample 4 Sample 5 Sample 6
Dibenzo-p-dioxins
PBDDs 9 24 155 313 2 1
(Br1, Br3) (Br1, Br3) (Br2) (Br1, Br2) (Br1, Br3) (Br3)
PXDDs 43 33 29 267 5 5
(X2, X4) (X2, X4) (X3, X4) (X3-X4) (X4) (X4)
Total 52 57 184 580 7 6
Dibenzofurans
PBDFs 1672 2675 386 3262 170 285
(Br1-Br4) (Br1-Br3) (Br1-Br4) (Br1-Br4) (Br1-Br3) (Br1-Br4)
PXDFs 1261 1554 682 539 47 9
(X2-X4) (X2-X4) (X2-X4) (X2-X4) (X2-X4) (X4)
Total 2933 4229 1068 3801 217 294
a Adapted from Dawidowsky (1993).
b Homologue groups quantified are given in parentheses.
concentrations of these eight PBDDs/PBDFs (16-65 µg/kg), depending on
processing. Mixed electronic waste was contaminated by lower
concentrations (4.9-26 µg/kg). Concentrations of the PBDDs/PBDFs in
TBBPA flame-retarded material were lower still (n.d.-2.4 µg/kg).
Recycling of printed circuits containing TBBPA flame retardant
can lead to the formation of PBDDs/PBDFs (Lorenz & Bahadir, 1993).
Whereas untreated basic recovered material contained total
concentrations of PBDDs/PBDFs (mono to octa) of 0.22 µg/kg, shredded
material was contaminated with PBDDs/PBDFs at levels ranging from 0.03
(metal fraction) to 1.13 µg/kg (mixed and plastic fraction) following
treatments with hammer mill, impact grinder, separation, and
granulation processes. The main components were monoBDFs (0.05-0.32
µg/kg), diBDFs (0.23 µg/kg), and tetraBDDs (0.03-0.73 µg/kg).
2,3,7,8-TeBDD/TeBDF were not detected (detection limits: 0.01-0.05
µg/kg). The contamination was probably due to thermal decomposition of
TBBPA.
Scrap of electronic devices (printed circuit boards with
electronic components) and other flame-retarded plastics were
subjected to various recycling activities (Dumler-Gradl et al., 1995).
After mechanical processing (hammer mill) of TBBPA-containing
plastics, no PBDDs/PBDFs could be detected. After (laboratory-scale)
pyrolysis and solvolysis procedures, especially of chopped printed
circuit boards, high amounts of PBDDs/PBDFs were found in the
condensed (a) or extracted (b) material (e.g. in (a), sums of tetra-,
penta-, hexa-, and heptaBDFs were 7035, 5470, 213, and 31 µg/g,
respectively; 2,3,7,8-TeBDF: 29 µg/kg; 2,3,4,7,8-PeBDF: 24 µg/kg; or
in (b), sums of tetra-, penta-, and hexaBDDs were 0.23, 0.20, and 1.98
µg/kg, respectively; 2,3,7,8-TeBDD: 0.06 µg/kg; sums of tetra-,
penta-, hexa-, and heptaBDFs were 230, 309, 97.5, and 0.9 µg/kg,
respectively; 2,3,7,8-TeBDF: 5 2,3,4,7,8-PeBDF: 12.6 µg/kg). Only
small amounts of mono- and diBDFs were detected at (pilot-scale)
pyrolysis of printed circuit boards and mixed flame-retarded plastics.
Plastic material recovered from cables and subsequently burned
contained PBDDs/PBDFs (mono to hexa) at a concentration of 36.5 µg/kg
(Lorenz, 1994).
3.9.3.2 Metals
The formation of PCDDs/PCDFs (WHO, 1989) and other organochlorine
compounds (Sinkkonen et al., 1994) during metal reclamation is well
known. However, the occurrence of the brominated analogues is
documented in only a few cases. PBDDs/PBDFs (tri to hexa) were
analysed in ash samples (n = 2) from a metal reclamation factory in
southern Taiwan (Watanabe et al., 1993), and PXDDs/PXDFs were
identified in ash samples (n = 2) from a secondary copper furnace in
the USA (Harless et al., 1989).
Whereas PBDDs were not detected (detection limits: <0.25-<1.0
µg/kg) in the Taiwanese study, PBDFs reached total concentrations of
15-45 µg/kg (composed of triBDFs: 5-9 µg/kg, tetraBDFs: 4-15 µg/kg,
pentaBDFs: 3-9 µg/kg, and hexaBDFs: 3-12 µg/kg). PXDDs/ PXDFs were
also observed, but not quantified, in these samples. The ratio of
PBDFs to PCDFs was about 1 : 100. PBDEs were also present at somewhat
lower levels than PCDFs (Watanabe et al., 1993).
The ash from a secondary copper furnace contained Br1Cl6DDs
(1-34 µg/kg) and Br1Cl6DFs (17 µg/kg). These concentrations were
approximately one order of magnitude lower than those of PCDDs (27-411
µg/kg) and PCDFs (89-173 µg/kg) (Harless et al., 1989).
3.10 Presence in automotive exhaust
The combustion processes occurring in motors of automobiles can
lead to the formation of PBDDs/PBDFs and PXDDs/PXDFs (Buser,
1987a,b,c). Simulation experiments using iso-octane (plus additives)
as a defined fuel (Ballschmiter et al., 1990; Bacher et al., 1991) and
single measurements using commercial petrol (Haglund et al., 1988)
gave positive results. The same was true for a joint project between
three German universities, which was initiated to provide more and
representative data on the emission of PHDDs/PHDFs from motor vehicles
under realistic conditions (Hagenmaier et al., 1990a; Hutzinger et
al., 1990; Weberruß, 1990; Schwind et al., 1991; Dawidowsky, 1993). In
this study, 46 exhaust samples were taken from Otto (spark-ignition or
Otto-cycle engine) and diesel motors running with different commercial
fuels. The experiments were carried out mostly as stationary motor
tests.
PHDDs/PHDFs were detected in emissions of motors running on
leaded petrol and on unleaded petrol with and without catalytic
converters (Haglund et al., 1988; Hagenmaier et al., 1990a; Hutzinger
et al., 1990; Dawidowsky, 1993) and in emissions of diesel engines
(Hagenmaier et al., 1990a; Hutzinger et al., 1990; Dawidowsky, 1993)
(see also Table 22). Because of the brominated and chlorinated
scavengers (dibromo- and dichloroethane) used in leaded petrol, the
highest levels of PHDDs/PHDFs were found with this type of petrol. The
emissions reached several thousand ng/m3 (e.g. >6000 ng/m3 in
exhaust air or 90 000 ng/litre fuel used). Unleaded petrol, which
contained only trace amounts of halogenated compounds, caused much
lower emissions of PHDDs/PHDFs (approximately two orders of magnitude
lower). A further reduction to below 5% of the emissions from
non-halogenated petrol was achieved in experiments using catalytic
cleaning of the exhaust. The values for diesel engines were a little
higher than those found with the Otto motors run on unleaded petrol
and equipped with a catalytic converter. In the case of diesel
combustion, the sources of the halogens needed for the PHDD/PHDF
formation could not be clearly identified (e.g. Hagenmaier et al.,
1990a). The negative results of Haglund et al. (1988) obtained for a
heavy-duty diesel truck are thought to be due to their higher
detection limits compared with the later studies (Dawidowsky, 1993)
showing positive results for both diesel cars (n = 8) and trucks
(n= 2). Differences in results involving diesel fuel combustion can
also be due in part to differences in diesel fuel compositions
(seasonal, manufacture).
A considerable portion of the PHDDs/PHDFs consisted of
PBDDs/PBDFs and PXDDs/PXDFs (Dawidowsky, 1993; see also Table 22). In
exhaust gases from combustion of leaded petrol, they were more
abundant than PCDDs/PCDFs: PBDDs/PBDFs > PXDDs/ PXDFs approx.
PCDDs/PCDFs.
Generally, in all studies performed, the concentrations of
dibenzofurans exceeded those of the dibenzo- p-dioxins (see also
Table 22). There was a dominance of lower substituted homologues (mono
to tri), as can be seen, for example, from the homologue profile of
PBDFs and PXDFs shown in Table 23.
Brominated and mixed brominated/chlorinated congeners with
2,3,7,8-substitution were not determined. However, they may be present
at trace amounts as well as their chlorinated counterparts (Marklund
et al., 1987, 1990; Bingham et al., 1989; Hagenmaier et al., 1990a;
Bacher et al., 1991).
PHDDs/PHDFs were detected not only in exhaust samples but also in
residues adhering to mufflers. An absorption in the mufflers was
observed for PBDDs/PBDFs and PXDFs (Ballschmiter et al., 1990) and for
PCDDs/PCDFs (Ballschmiter et al., 1990; Marklund et al., 1990). The
two samples tested by Ballschmiter et al. (1990) showed some
correlations to the exhaust samples (dominance of lower brominated
homologues, prevalence of PBDFs over PBDDs and of PBDFs over PCDFs).
Results from traffic-related environmental samples are discussed
in chapter 5.
The increasing use of lead-free petrol will reduce the input of
PHDDs/PHDFs into the environment from this source (cf. Hagenmeier,
1994).
3.11 Formation during textile processing
Different textile processes (resin finish on the basis of
magnesium chloride [MgCl2], flame-proof finishes on the basis of
Sb2O3/hexa bromocyclododecane, ammonium bromide [NH4Br], and PVC)
were tested for the occurrence and formation of PBDDs/PBDFs (Sedlak et
al., 1996). The eight 2,3,7,8-substituted congeners of the 1994 German
Dioxin Directive (see Appendix I) were determined in the exhaust air,
the textiles before and after processing, and the chimney depositions.
Exhaust air concentrations were between 29 and 102 pg/m3.
Concentrations in textiles before processing were 1.35-5.65 ng/kg and
after processing 1.80-41.0 ng/kg. The only significant increase was
observed for the PVC process (5.65 versus 41.0 ng/kg). Chimney
depositions showed concentrations of 92.3-6618 ng/kg. Only traces of
PXDDs/PXDFs were detected in the textiles, but chimney depositions
contained up to 17 µg/kg.
Table 22. Emissions of PHDDs/PHDFs from automobile combustion engines under
various motor/fuel conditionsa,b
PHDDs/PHDFs Mean emissions (ng/litre fuel)c
Leaded petrol Unleaded petrol Unleaded petrol with Diesel fuel
catalytic converter
(n = 4) (n = 6) (n = 3) (n = 8)
Dibenzo-p-dioxins
PBDDs 1576 (Br1-Br4) 18 (Br1-Br4) 0.8 (Br1-Br3) 1.9 (Br1-Br3)
PXDDs 742 (up to X5) 4 (up to X4) 0.6 (up to X4) 0.4 (up to X3)
PCDDs 606 (Cl1-Cl8) 42 (Cl1-Cl8) 0.9 (Cl1-Cl8) 3.4 (Cl1-Cl8)
Total PHDDs 2924 64 2.3 5.7
Dibenzofurans
PBDFs 45 428 (Br1-Br6) 364 (Br1-Br5) 22.6 (Br1-Br5) 136 (Br1-Br4)
PXDFs 22 418 (up to X5) 101 (up to X5) 6.9 (up to X5) 24 (up to X4)
PCDFs 21 698 (Cl1-Cl8) 502 (Cl1-Cl8) 7.9 (Cl1-Cl8) 167 (Cl1-Cl8)
Total PHDFs 89 544 967 37.4 327
a Adapted from Dawidowsky (1993).
b The exhaust samples were obtained from Otto motors run on leaded petrol and unleaded petrol with and
without catalytic converters and from diesel engines. Br/Cl content of fuels: leaded petrol, 76/70 mg/kg;
unleaded petrol and diesel, <1 mg/kg.
c Homologue groups present are given in parentheses.
Table 23. Homologue distribution pattern of PBDFs
and PXDFs detected in exhaust samples (n = 4) from
vehicle motors run on leaded petrola
Homologue groups Mean emissions
(ng/litre fuel)
PBDFs
MonoBDFs 27 501
DiBDFs 17 496
TriBDFs 354
TetraBDFs 70
PentaBDFs 6.6
HexaBDFs 0.2
HeptaBDFs n.d.b
PXDFs
Br1Cl1DFs 18 941
Br1Cl2DFs 377
Br2Cl1DFs 2649
Br1Cl3DFs 67
Br2Cl2DFs 174
Br3Cl1DFs 138
Br3Cl2DFs 19.5
Br4Cl1DFs 7.4
a Adapted from Dawidowsky (1993).
b n.d. = not detected.
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION, AND TRANSFORMATION
4.1 Transport and distribution between media
Because of their physicochemical properties (see chapter 2),
PBDDs/PBDFs are expected to be preferably distributed into carbon- or
fat-rich compartments.
4.1.1 Air
Airborne PBDDs/PBDFs released from various sources (see chapter
3) are transported in both the particulate and vapour phase. In
traffic-related air samples, the lower halogenated PBDDs/PBDFs (Br1,
Br2), PCDDs/PCDFs (Cl1 through Cl3), and PXDDs/PXDFs (X2) were detected
predominantly in the gaseous phase (Ballschmiter et al., 1990). In
contrast, Lutes et al. (1992a) found that tetra- and pentaBDDs/BDFs
generated by combustion of polyurethane foam containing PBDEs
partitioned primarily to the particulate phase. The ratio of
concentrations between particulate phase and particulate plus vapour
phase was 0.95-0.99. Monitoring of ambient city air (Harless et al.,
1992) for PBDDs/PBDFs (tetra through hexa), PCDDs/PCDFs (tetra through
octa), and PXDDs/PXDFs (tetra: Br1Cl3) revealed that most of the
penta- and hexa- and about 60% of the tetraBDDs/BDFs were associated
with the particulate phase. Most of the hepta- and octaCDDs/CDFs were
also particle-bound, whereas the lower chlorinated congeners,
including the mixed tetrahalogenated compounds, were distributed to
the gaseous phase.
During atmospheric transport, photochemical transformation can
occur (see section 4.2.1). There are no data on deposition of
PBDDs/PBDFs to soil, vegetation, or water.
4.1.2 Water and sediments
Few data are available on the movement of PBDDs/PBDFs through
water and associated sediment.
Watanabe (1988) determined adsorption coefficients on sediment of
several halogenated organic compounds, including PBDFs. Adsorption
coefficients (log Kd; where Kd = [µg/g sediment]/[µg/ml water]) were
4.51, 4.62, and 4.79 for triBDFs, tetraBDFs, and pentaBDFs,
respectively.
4.1.3 Soil
To date, no experimental data on the behaviour of PBDDs/PBDFs in
soil are available. Mobility of PBDDs/PBDFs in soil is assumed to be
governed by their low water solubility (see chapter 2) and their
strong adsorption to particulate matter. Their mobility depends on
soil type (organic matter content, dissolved humic acids, pH, etc.),
weather conditions, and congeners studied. In general, mobility is
expected to be rather low; however, in special cases, such as at waste
disposal sites where organic solvents are concomitantly present,
significant leaching could occur. As shown for PCDDs (Webster et al.,
1986), the presence of dissolved humic substances in water can also
enhance the solubility of such compounds.
Other transport mechanisms to be considered include transport via
dust particles or volatilization (to air and vegetation), via eroded
soil (to surface waters), and via biomass removal, as reported for
PCDDs/PCDFs (Young, 1983; WHO, 1989).
The persistence of PCDDs/PCDFs (tetra to octa) in soil
environments was found to be high (e.g. Orazio et al., 1992), and
movement of 2,3,7,8-TeCDD was primarily associated with liquid carrier
contaminants such as petroleum oil (Kapila et al., 1989).
2,3,7,8-TeCDD half-lives in soil were calculated to be as high as
10-12 years (di Domenico et al., 1980; Young, 1983).
4.1.4 Biota
Isolated reports have been published on the presence of
PBDDs/PBDFs in animals and plants (see chapter 5). However, no data
are available on the transport of PBDDs/PBDFs and their distribution
between environmental media and biota. The similar high octanol/water
partition coefficients calculated for selected PCDDs/PCDFs,
PBDDs/PBDFs, and PXDDs/PXDFs (Fiedler & Schramm, 1990; see also Table
5 in section 2.2.1) indicate that PBDDs/PBDFs may have a
bioavailability qualitatively comparable to that reported for
PCDDs/PCDFs (de Wit, 1993; Rappe, 1993). A factor influencing the
range of bioavailability both within the homologue groups and between
analogues is the molecule size.
There are no data on the transfer of PBDDs/PBDFs to plants via
deposition processes or uptake from soil or on their transfer along
the terrestrial food-chain. However, it may be expected that the
brominated congeners behave qualitatively like their chlorinated
analogues.
PCDDs/PCDFs can also enter aquatic biota, primarily via sediment
(Fairchild et al., 1992; de Wit, 1993; Fletcher & McKay, 1993; Pruell
et al., 1993). PBDDs/PBDFs are expected to have a similar potential
(see also chapter 5).
4.2 Environmental transformation
4.2.1 Photochemical degradation
Photolysis of PBDDs/PBDFs was studied in organic solvents and on
quartz surfaces in the laboratory as well as in soil and on soot
particles under outdoor conditions. The slowest photolytic reactions
were observed under the latter, more environmentally relevant,
conditions. From experiments with octaCDD, it is known that
photochemical dechlorination on soil takes place in the axial
positions, resulting in 2,3,7,8-TeCDD; in solutions, the lateral
chlorines are removed, resulting in 1,4,6,9-TeCDD (Kieatiwong et al.,
1990).
Laboratory studies showed that PBDDs/PBDFs and PXDDs/ PXDFs
degrade in organic solvents after irradiation with sunlight,
artificial light, or UV light (Buser, 1988; Neupert et al., 1988;
Lahaniatis et al., 1991; Lenoir et al., 1991; Chatkittikunwong &
Creaser, 1994a; Ritterbusch et al., 1994a; Watanabe et al., 1994). The
major photochemical pathway is a reductive debromination, resulting in
the formation of lower brominated congeners (Buser, 1988; Neupert et
al., 1988; Lenoir et al., 1991; Chatkittikunwong & Creaser, 1994a;
Ritterbusch et al., 1994a) and, finally, in the formation of
unsubstituted dibenzo- p-dioxin and dibenzofuran (Buser, 1988). Other
products also observed after photolysis were diaryl ethers, which were
generated by ring fission of mono- and diBDDs but not of higher
brominated PBDDs (Lenoir et al., 1991), and, occasionally, benzyl
derivatives. The latter were formed by reaction of photoproducts of
2,3,7,8-TeBDD with the solvent toluene (Neupert et al., 1988). Studies
of different PBDD/PBDF congeners indicated that the rate of
decomposition depends on the bromine substitution pattern. Generally,
higher brominated congeners and those with lateral bromines had
shorter half-lives (Buser, 1988; Neupert et al., 1988; Lenoir et al.,
1991); in one experiment (Lenoir et al., 1991), however, octaBDD was
more stable than a hexaBDD. Two dibenzofurans (2,3,7,8-TeBDF and
octaBDF) dissolved in toluene and irradiated by fluorescent light for
several days were found to decompose more rapidly than the
corresponding dibenzo- p-dioxins (Neupert et al., 1988). The rate of
photolysis decreased with increasing polarity of the solvent, as
tested for 2,3,7,8-TeBDD (Lahaniatis et al., 1991) and 1,2,3,4-TeBDD
(Lenoir et al., 1991).
The calculated half-lives (assuming a first-order kinetic scheme)
were in the range of minutes (use of direct sunlight or UV light and
quartz vials) or of the order of 100-1000 h (use of laboratory
daylight or artificial light and glass vials). For example, half-lives
of 0.8 and 0.7 min were estimated for TBDD and TBDF, respectively,
after 60 min of sunlight irradiation in organic solution in quartz
vials (Buser, 1988). The mean half-lives found for PBDDs and PXDDs
exposed to laboratory daylight in dodecane solution stored in glass
vials were 480 h for tetraBDDs, 150 h for pentaBDDs, and 300-995 h for
various PXDDs (tetra- to hexahalogenated congeners with various Br/Cl
combinations: Br1-3/Cl1-5) (Chatkittikunwong & Creaser, 1994a).
Compared with the chlorinated analogues, PBDDs/PBDFs had a faster
photolytic reaction in iso-octane, with half-lives of 3 min for
1,2,3,4-TeBDD and 380 min for 1,2,3,4-TeCDD (Buser, 1988; Lenoir et
al., 1991). The easier loss of bromine than of chlorine from the
parent molecule has important consequences for the PXDDs/PXDFs, in
that they undergo photolytic degradation to form PCDDs/PCDFs.
Consistently short photolytic half-lives (0.5 - 4 min) were observed
for the mixed mono- and dibromotetrachloroDDs/DFs tested, whereas the
resulting tetraCDDs/CDFs were much more stable (Buser, 1988).
The possibility of removing PBDDs/PBDFs from laboratory wastes by
UV photolysis was examined by Ritterbusch et al. (1994a,b). This
method of decontamination was successful when solutions with a low
concentration of other photochemically active species were applied. It
was unsuitable for the degradation of the PBDD/PBDF contamination of
bromophenols, because the rate of photochemical PBDD/PBDF formation
was greater than the rate of degradation. The rate of photolytic
degradation was slower in waste solutions than in PBDD/PBDF standard
solutions (Ritterbusch et al., 1994a). The degradation of PBDDs/PBDFs
occurred faster than that of PCDDs/PCDFs (Ritterbusch et al., 1994b).
Photolysis of PBDDs/PBDFs (Br4) and PXDDs/PXDFs (X5, X6: mono- and
dibromo-2,3,7,8-TeCDDs/CDFs) occurred much more slowly on quartz
surfaces, under sunlight, than in organic solvents (Buser, 1988). The
photolytic half-lives of the tetrabrominated congeners tested were in
the range of 30 h; those for the chlorinated analogues were in the
range of 65-300 h (see Table 24).
These solid-phase experiments appeared to predict the real
environmental behaviour of PBDDs/PBDFs far better than the organic
solution-phase experiments. Photodegradation studied in soil
(Chatkittikunwong & Creaser, 1994a) and on airborne particles (Lutes
et al., 1990, 1992a,b) under outdoor conditions was found to be a slow
process. Half-lives of PBDDs and PXDDs in soil samples that were
placed outdoors over a 3-month period were in the range of 600 - 4000
h for tri- to hexahalogenated congeners. These half-lives were, on
average, four times longer than those estimated for the same congeners
in solution in the laboratory. The half-life of tetraBDD isomers in
surface soil was estimated to be 3 - 6 months (Chatkittikunwong &
Creaser, 1994a). Other tests conducted in Teflon film chambers under
realistic outdoor conditions showed that PBDDs/PBDFs (tetra through
hexa) adsorbed on incinerator soot particles remained relatively
stable or degraded only slowly during 6 h. No significant decay of the
2,3,7,8-substituted congeners was found (Lutes et al., 1990, 1992a,b).
The photodegradation, if occurring, was believed to have a half-life
of at least 3 h and probably much longer (Lutes et al., 1992a,b).
Similarly, particle-bound emissions of tetraBDDs, tetraBDFs, and
pentaBDFs (isomers not determined), produced from high-temperature
(670 - 780°C) combustion of polyurethane foam containing PBDEs and
monitored in outdoor Teflon chambers in the presence of sunlight, were
stable over several hours. In contrast, a decay of tetraBDDs was
observed after low-temperature (400 - 470°C) combustion of the
polyurethane foam (Birla & Kamens, 1994). Little disappearance of
higher halogenated PXDFs was seen in preliminary tests monitoring
halogenated dibenzofurans associated with airborne dust collected on a
glass filter and exposed to sunlight (Watanabe et al., 1994).
Table 24. Sunlight-induced photolysis of tetrahalogenated
dibenzo-p-dioxins and dibenzofurans dispersed as solid filmsa
Compound Estimated half-lifeb
(h)
Dibenzo-p-dioxins
1,2,3,4-TeBDD 26
2,3,7,8-TeBDD 32
1,2,3,4-TeCDD 65
2,3,7,8-TeCDD 300
Dibenzofurans
2,3,7,8-TeBDF 35
2,3,7,8-TeCDF 120
a From Buser (1988).
b Values derived from few data points
obtained after a total exposure time of
10 and 20 h; estimated accuracy of half-lives + 50%.
4.2.2 Microbial degradation
Like other halogenated aromatics, PBDDs/PBDFs seem to be very
recalcitrant against microbial degradation. Only a monobrominated
dibenzofuran (2-bromodibenzofuran) was tested and found to be degraded
by bacteria. Bacteria of the genus Pseudomonas isolated from water of
the river Rhine or of industrial wastewater treatment plants and
cultured in the laboratory on 1,2-dichlorobenzene or 4-chlorophenol as
single-carbon source were able to oxidize 2-bromodibenzofuran
(Springer & Rast, 1988).
There are some reports on the degradation of the parent,
non-halogenated dibenzo- p-dioxin and/or dibenzofuran by microorganisms
in soil and by white rot fungi (Cerniglia et al., 1979; Hammel et al.,
1986; Bumpus, 1989; Hofmann et al., 1992). Some active bacterial
strains were found in several genera - for example, Beijerinckia
(Klecka & Gibson, 1980), Brevibacterium (Strubel et al., 1989, 1991),
Pseudomonas (Klecka & Gibson, 1979; Foght & Westlake, 1988; Springer &
Rast, 1988; Fortnagel et al., 1989a,b, 1990; Harms et al., 1990; Figge
et al., 1991), Sphingomonas (Wittich et al., 1992; Figge et al., 1993;
Happe et al., 1993), and Staphylococcus (Monna et al., 1993).
4.3 Bioaccumulation and biomagnification
At present, bioaccumulation, bioconcentration, or
biomagnification factors for specific PBDD/PBDF congeners are not
available. The presence of PBDDs/PBDFs in animals (section 5.1.6.2)
and in humans (section 5.3.2), as seen in a few isolated studies, is
indicative of their accumulation potential (see also section 6.5.1).
This is to be expected from the lipophilic properties of PBDDs/PBDFs
and the high accumulation potential of better-studied related
compounds, such as PCDDs/PCDFs (e.g. WHO, 1989; Cook et al., 1991).
The extent of accumulation and biomagnification may vary depending on
species and congeners tested, as found for PCDDs/PCDFs (e.g. de Wit,
1993; de Wit et al., 1993; Rappe, 1993; Walker & Peterson, 1994).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
Limited data are available on levels of PBDDs/PBDFs in the
environment. Most monitoring data available for PBDDs/PBDFs have been
collected near identified sources (e.g. roadways).
5.1.1 Air
5.1.1.1 Ambient air
Sources of PBDDs/PBDFs are complex and may have changed in recent
years. Therefore, temporal differences and changes must be taken into
account. In some European countries, the use of leaded petrol (and the
use of scavengers) has been largely abandoned; in the USA, the use of
leaded petrol with scavengers declined even earlier than in Europe. In
other countries, this type of petrol may still be in use. Thus, some
of the data given may not be representative of the present situation
(e.g. in motorway tunnels, urban areas, etc.). Furthermore, the type
of waste incineration has drastically changed within the last decade
in industrialized countries. These variables have to be kept in mind
when evaluating the following compilations and Table 25.
1) Air samples
PBDD/PBDF levels analysed in urban air, more or less close to
traffic, and in air collected at industrial sites are compiled in
Table 25. The sampling methods used covered particulate-associated and
vapour-phase PBDDs/PBDFs. Only low concentrations of PBDDs (mono
through tetra) were detected, with maximum concentrations of about
0.85 pg monoBDDs/m3 in a motorway tunnel and an underground garage.
Higher brominated (penta through octa) homologues were not detected or
were not analysed (Table 25). No PXDDs were found (Ballschmiter et
al., 1990).
PBDFs were found to a greater extent than PBDDs (Table 25). Of
the homologue groups, mono- to hexaBDFs could be detected; hepta- and
octaBDFs were not identified. Because of the small database, only
trends in the homologue pattern can be seen. It appears that lower
brominated homologues (mono through tetra) dominate, particularly in
samples related to traffic. The highest concentration was measured for
monoBDFs in an air sample from a motorway tunnel and amounted to 74
pg/m3 (Table 25). However, Harless et al. (1992) found pentaBDFs and
hexaBDFs (0.22 and 0.30 pg/m3, respectively) in addition to tetraBDFs
(0.19 pg/m3 air) only after long-term sampling (7 days; 2660 m3 air) of
ambient air (at Research Triangle Park, NC, USA). Eight tetraBDF, two
pentaBDF, and one hexaBDF isomers were detected. Using shorter
sampling periods (24 h), only tetraBDFs were detected, at
concentrations ranging from 0.13 to 0.20 pg/m3. The lower
concentrations of PBDFs reported from this study would probably be due
in part to the absence of scavengers in motor fuel in North America.
The highest concentrations of PBDF homologue groups were found in
motorway tunnels (Table 25). No data are available on concentrations
of lower brominated homologues (Br1 through Br3) in samples from
industrial areas. The highest concentrations of tetraBDFs were
reported in the vicinity of a resource recycling centre in Taiwan
(2.1 - 6.6 pg/m3) and in the German motorway tunnels (n.d. - 3.4 pg/m3)
(see Table 25).
The sums of total PBDDs/PBDFs (tri to hexa) in the air of a
motorway tunnel, of a city, and of a suburban area in Germany were
22.3/0.7 pg/m3 (means; n = 3), 1.97/0.08 pg/m3 (means; n = 6), and 0.59
pg/m3/n.d. (means; n = 3), respectively. The concentrations of
2,3,7,8-TeBDF ranged from n.d. to 0.28 (n = 12) and those of
1,2,3,7,8-PeBDF from n.d. to 0.08 pg/m3 (n = 12) (Päpke et al., 1990;
Hiester, 1992).
PBDFs and their possible precursors, PBDEs, were concomitantly
found in air samples from industrial areas of Taiwan and Japan. Tri-,
tetra-, penta-, and hexaBDE concentrations ranged from 6 to 34, from
10 to 55, from 5 to 34, and from 6 to 81 pg/m3, respectively (Watanabe
et al., 1992).
Of the PXDFs, dihalogenated dibenzofurans were detected in
traffic-related air samples at concentrations of up to 40.8 pg/m3
(Cl1Br1DFs), which is higher than found for the brominated congeners.
Concentrations of monoBDFs exceeded the levels of monoCDFs
(Ballschmiter et al., 1990).
2) Dust samples
Outdoor dust samples were collected in motorway tunnels in
Germany and, as a control, in the eaves of a house in a German rural
area remote from the source (traffic). These and samples obtained from
the USA (roadside dust) and from Japan (dust of a motorway tunnel)
were analysed for PCDDs/PCDFs, PBDDs/PBDFs (Br1 through Br4), and
PXDDs/PXDFs (up to X4) (Ballschmiter et al., 1990). The results are
summarized in Table 26 (PBDDs/PBDFs) and Table 27 (PXDDs/PXDFs).
Whereas the control sample (from eaves) contained no PBDDs (detection
limit: 20 ng/kg dust) and only 30 ng monoBDFs/kg dust, the samples
taken close to traffic showed a complex pattern of homologues.
Table 25. Concentrations of PBDDs/PBDFs in ambient air
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
Dibenzo-p-dioxins
MonoBDDs Germany motorway tunnel (1) n.sp. 0.85 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 0.50-0.86 Ballschmiter et al. (1990)
DiBDDs Germany motorway tunnel (1) n.sp. <0.15 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d.-<0.15 Ballschmiter et al. (1990)
TriBDDs Germany motorway tunnel in Essen (3) 1990 0.37-0.75c Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.05-0.09 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.03) Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d.-0.09 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 0.3-0.5 Watanabe et al. (1992)
TetraBDDs Germany motorway tunnel in Essen (3) 1990 n.d.-0.18 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d.-0.04 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d.-0.3 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d.-0.2 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
PentaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d. Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d. Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
HexaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. (0.1-0.2) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.1-0.4) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.2) Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. n.d. Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. n.d. Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d. Harless et al. (1992)
HeptaBDDs Germany motorway tunnel in Essen (3) 1990 n.d. (0.3-0.5) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.5-0.7) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.5) Päpke et al. (1990)
Table 25. (Continued)
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
OctaBDD Germany motorway tunnel in Essen (3) 1990 n.d. (0.7-0.8) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (1) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (1) Päpke et al. (1990)
Dibenzofurans
MonoBDFs Germany motorway tunnel (1) n.sp. 73.72 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 37.26-42.12 Ballschmiter et al. (1990)
DiBDFs Germany motorway tunnel (1) n.sp. 28.50 Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. 2.12-6.20 Ballschmiter et al. (1990)
TriBDFs Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 17-25 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.71-2.0 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 0.40-0.82 Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.3-1.0 Watanabe et al. (1992)
TetraBDFs Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 1.50-3.35 Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 0.15-0.53 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.2-2.3 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 2.1-6.6 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 0.13-0.20 Harless et al. (1992)
PentaBDFs Germany suburban area of the small city Borken (3) 1990 n.d. Päpke et al. (1990)
Japan urban area of Osaka (5) n.sp. 0.2-3.7 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 1.8-7.7 Watanabe et al. (1992)
Germany motorway tunnel (1) n.sp. n.d. (0.1) Ballschmiter et al. (1990)
Germany air outlet from an underground garage (2) n.sp. n.d. (0.05-0.1) Ballschmiter et al. (1990)
Germany motorway tunnel in Essen (3) 1990 0.32-O.41 Päpke et al. (1990)
Germany urban area of Düsseldorf(6) 1990 0.08-0.22 Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d.-0.14 Päpke et al. (1990)
USA Research Triangle Park, NC (5) 1990/91 n.d.-0.22 Harless et al. (1992)
HexaBDFs Germany motorway tunnel in Essen (3) 1990 n.d. (0.1-0.2) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.2-0.4) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.2) Päpke et al. (1990)
Table 25. (Continued)
Congener Country Sampling site (n) Year of Concentrationb Reference
samplinga (µg/m3)
HexaBDFs Japan urban area of Osaka (5) n.sp. 0.3-5.1 Watanabe et al. (1992)
Taiwan recycling resource centre (3) n.sp. 1.1-3.4 Watanabe et al. (1992)
USA Research Triangle Park, NC (5) 1990/91 n.d.-0.30 Harless et al. (1992)
HeptaBDFs Germany motorway tunnel in Essen (3) 1990 n.d. (0.4-0.5) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (0.3-0.7) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (0.5) Päpke et al. (1990)
OctaBDF Germany motorway tunnel in Essen (3) 1990 n.d. (0.7-0.8) Päpke et al. (1990)
Germany urban area of Düsseldorf (6) 1990 n.d. (1) Päpke et al. (1990)
Germany suburban area of the small city Borken (3) 1990 n.d. (1) Päpke et al. (1990)
a n.sp. = not specified.
b n.d. = not detected (detection limits in parentheses, if specified).
c Containing possibly confounding components.
Concentrations of PBDDs in the dust samples were low, ranging
from n.d. to 690 ng/kg, from n.d. to 960 ng/kg, from n.d. to 170
ng/kg, and from n.d. to 110 ng/kg for mono-, di-, tri-, and tetraBDDs,
respectively (Ballschmiter et al., 1990).
PBDFs were present at higher levels than PBDDs and reached
maximum values of 8860 ng monoBDFs/kg dust (motorway tunnel in
Germany), 22 280 ng diBDFs/kg dust (motorway tunnel in Japan), 5680 ng
triBDFs/kg dust (highway in the USA), and 650 ng tetraBDFs/kg dust
(highway in the USA). Dust samples (n = 7) taken from motorway tunnels
in Germany showed decreasing levels from mono- through tetraBDFs.
Concentrations ranged from 330 to 8860 ng/kg, from 300 to 6730 ng/kg,
and from n.d. to 920 ng/kg for mono-, di-, and triBDFs, respectively;
tetraBDFs were not detected (Table 26).
Airborne dust collected at an urban area in Osaka (Japan)
contained PBDFs (tetra to hexa) and PCDDs/PCDFs (tetra to octa) at
concentrations ranging from 4.2 to 17 pg/m3 (n = 7) and from 30 to 250
pg/m3, respectively. Monobrominated PXDD/PXDF (tetra to octa)
concentrations were roughly estimated at one-tenth to one-quarter
those of PCDDs/PCDFs in the same samples (Watanabe et al., 1995).
PXDDs (Cl1Br1DDs) were identified at a maximum concentration of
170 ng/kg in a motorway tunnel (Table 27). PXDFs were found in dust
samples from motorway tunnels, highways, and roads at concentrations
ranging from 300 to 9600 ng/kg for X2DFs, from n.d. to 7870 ng/kg for
X3DFs, and from n.d. to 830 ng/kg for X4DFs (Table 27).
There are no data available for the 2,3,7,8-substituted
PBDDs/PBDFs (Ballschmiter et al., 1990).
Generally, the dust samples from the German motorway tunnels
contained higher concentrations of PBDFs than of PCDFs, both
consisting of lower halogenated homologues (e.g. 16 510 ng mono- to
triBDFs/kg versus 5610 ng mono- to tetraCDFs/kg). The relation between
concentrations of PBDDs and PCDDs was varying. Whereas within PBDDs
only mono- and dibrominated homologues could be detected (at sum
concentrations of up to about 1200 ng/kg), there was a dominance of
hepta- and octaCDDs within PCDDs (total concentrations of up to about
1700 ng/kg). Altogether, these patterns may be reflective of
contributions from automobile exhaust. The sample from the eaves
(remote from traffic, long-term residue) showed lower concentrations
of PBDFs (30 ng monoBDFs/kg) than of PCDFs (sum of mono- to hexaCDFs:
735 ng/kg), no PBDDs, and hepta- to octaCDDs (at 180 ng/kg). The lower
proportion of brominated compounds in the latter sample may be partly
due to an easier photo-dehalogenation compared with the chlorinated
ones (Ballschmiter et al., 1990).
Table 26. Concentrations of PBDDs/PBDFs in outdoor dust samplesa
Congener Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Dibenzo-p-dioxins
MonoBDDs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 n.d.-180
Germany motorway tunnel (outside of city) (2) 1989 n.d.-690
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 <20
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 100
DiBDDs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 n.d.-120
Germany motorway tunnel (outside of city) (2) 1989 n.d.-540
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 960
TriBDDs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 170
TetraBDDs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (1) n.sp. n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 n.d.
over 12 months)(1)d
Japan motorway tunnel (1)e 1978 110
Dibenzofurans
MonoBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 1050-2570
Germany motorway tunnel (outside of city) (2) 1989 330-8860
Table 26. (Continued)
Congener Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
MonoBDFs Germany eaves (rural area) (1) n.sp. 30
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 510
USA (Washington, DC) highway (inside of city; pooled sample 1982 2100
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 1370
DiBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 1560-5030
Germany motorway tunnel (outside of city) (2) 1989 300-6730
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 690
USA (Washington, DC) highway (inside of city; pooled sample 1982 2700
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 22 280
TriBDFs Germany (Ulm) motorway tunnel (inside of city) (5) 1988/89 75-310
Germany motorway tunnel (outside of city) (2) 1989 n.d.-920
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 530
USA (Washington, DC) highway (inside of city; pooled sample 1982 5680
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 4820
TetraBDFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 650
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 310
a Adapted from Ballschmiter et al. (1990).
b n.sp. = not specified.
c n.d. = not detected (detection limit 20 ng/kg).
d Reference sample from US National Institute of Standards.
e Reference sample from Japanese National Institute for Environmental Studies.
Table 27. Concentrations of PXDDs/PXDFs in outdoor dust from various sourcesa
PXDDs/PXDFs Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Dibenzo-p-dioxins
Cl1Br1DDs Germany motorway tunnel (inside of city) (5) 1988/89 n.d.
Germany motorway tunnel (outside of city) (2) 1989 n.d.-170
Germany eaves (rural area) (1) n.sp. n.d.
USA street (2)d 1978 and 1982 n.d.
Japan motorway tunnel (1)e 1978 n.d.
Dibenzofurans
X2DFs
Cl1Br1DFs Germany motorway tunnel (inside of city) (5) 1988/89 1150-4340
Germany motorway tunnel (outside of city) (2) 1989 300-9600
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 1280
USA (Washington, DC) highway (inside of city; pooled sample 1982 4150
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 7220
X3DFs
Cl1Br2DFs Germany motorway tunnel (inside of city) (5) 1988/89 180-830
Germany motorway tunnel (outside of city) (2) 1989 n.d.-1300
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 2270
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 1130
Table 27. (Continued)
PXDDs/PXDFs Country Sampling site Year of samplingb Concentrationc
(n) (ng/kg)
Cl2Br1DFs Germany motorway tunnel (inside of city) (5) 1988/89 n.d.-180
Germany motorway tunnel (outside of city) (2) 1989 n.d.-970
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 1160
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 7870
X4DFs
Cl2Br2DFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
Japan motorway tunnel (1)e 1978 n.d.
Cl3Br1DFs Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 830
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 370
Cl3Br1DFS Germany motorway tunnel (7) 1988/89 n.d.
Germany eaves (rural area) (1) n.sp. n.d.
USA (St. Louis, MO) street (outskirts of the city) (1)d 1978 n.d.
USA (Washington, DC) highway (inside of city; pooled sample 1982 380
over 12 months) (1)d
Japan motorway tunnel (1)e 1978 170
a Adapted from Ballschmiter et al. (1990).
b n.sp. = not specified.
c n.d. = not detected (detection limit 20 ng/kg).
d Reference sample from US National Institute of Standards.
e Reference sample from Japanese National Institute for Environmental Studies.
Another sample of roadside dust (n = 1; collected after the ban
of halogenated scavengers in petrol) from Germany was analysed for
tetra- to heptaBDDs/BDFs and tetra- to octaCDDs (Schacht et al.,
1995). PBDDs were not detected, whereas the sum of PBDFs amounted to
52 ng/kg (no 2,3,7,8-substituted congeners). The sum concentration of
PCDDs/PCDFs was 581 ng/kg.
5.1.1.2 Indoor air
PBDDs/PBDFs were determined in rooms equipped with a number of
operating electronic appliances (computers, monitors, printers; Tables
28, 29, and 30; Schacht et al., 1995), in an underground garage (Table
31), in a private room, and in rooms after accidental fires (see
chapter 3).
PBDDs (tetra through octa) were not detected in air samples from
rooms equipped with electronic appliances (Chriske et al., 1990), but
they were present in dust samples (n = 3) from computer rooms (mainly
tetraBDDs: 0.03 - 1 µg/kg; 2,3,7,8-TeBDD: n.d. - <2 µg/kg; Schacht et
al., 1995). Dust samples from an underground garage tested for
mono- to tetraBDDs contained no or only traces of monoBDDs and no
PXDDs (Table 31) (Ballschmiter et al., 1990).
Total concentrations of PBDFs (Br4 through Br7) measured in air
from "computer"-related rooms ranged from 0.23 to 1.27 pg/m3, whereas
dust samples collected in the same rooms yielded total levels of
2.43 - 5.48 µg/kg dust (Tables 28 and 30; UBA, 1992). The homologue
pattern was also different. In contrast to air (see Table 28), the
homologue pattern in dust was dominated by hexa- and heptaBDFs (Table
30; UBA, 1992). In addition, only in dust samples were there
indications for the presence of 2,3,7,8-substituted tetra- and
pentaBDFs (Table 30; UBA, 1992). Another study (Schacht et al., 1995)
found comparable PBDF concentrations (sum PBDFs [Br4-Br8]: 3.6 - 3.8
µg/kg; 2,3,7,8-TeBDF: 0.01 - 0.07 µg/kg) and profiles in dust samples
(n = 3), which were obtained from vacuum cleaning and from the air
conditioning system of computer rooms. The PBDF concentrations in
house dust (n = 1) were lower by a factor of 10. The sum concentration
of PBDDs/PBDFs equalled that of PCDDs/PCDFs in dust from computer
rooms (3.6 - 4.8 µg/kg versus 0.5 - 4.5 µg/kg) but was lower than that
of PCDDs/PCDFs in house dust (0.3 µg/kg versus 34.2 µg/kg) (Schacht et
al., 1995). Chemicals of low volatility tend to accumulate on solid
surfaces and in house dust (Gebefügi, 1989; Gebefügi & Kreuzig, 1989).
It should be noted that concentrations of PBDEs concomitantly
measured in the air and dust samples of the offices were considerably
higher than those of PBDFs. Total PBDE concentrations (Br4 through
Br10) ranged from 97 to 969 pg/m3 air and from 507 to 2939 µg/kg dust
(UBA, 1992).
Table 28. Indoor air concentrations of PBDFs
Congener groups Type of room Number of Equipment in room Concentrationa Reference
samples (number per room) (µg/m3)
TetraBDFs office 6 display and computer monitors (2-8) 0.21-0.66 Chriske et al. (1990)
office (police central 1 monitors (approximately 50) 0.41 UBA (1992)
traffic control)
office (direction rooms 3 display and computer monitors <0.1-0.47 UBA (1992)
of a TV studio) (approximately 50)
2,3,7,8-TeBDF office 4 monitors (approximately 50) n.d. (0.03-0.08) UBA (1992)
PentaBDFs office 6 display and computer monitors (2-8) 0.03-0.61 Chriske et al. (1990)
office (police central 1 monitors (approximately 50) n.d. (n.sp.) UBA (1992)
traffic control)
office (direction rooms 3 display and computer monitors 0.1-0.5 UBA (1992)
of a TV studio) (approximately 50)
1,2,3,7,8-PeBDF office 4 monitors (approximately 50) n.d. (0.05-0.1) UBA (1992)
2,3,4,7,8-PeBDF office 4 monitors (approximately 50) n.d. (0.05-0.1) UBA (1992)
HexaBDFs office 10 monitors (2-8/approximately 50) n.d.-0.4 (0.1) Chriske et al. (1990);
UBA (1992)
HeptaBDFs office 10 monitors (2-8/approximately 50) n.d. (0.1-0.2) Chriske et al. (1990);
UBA (1992)
OctaBDF office 6 display and computer monitors (2-8) n.d. (n.sp.) Chriske et al. (1990)
Sum PBDFs office 6 display and computer monitors (2-8) 0.23-1.18 Chriske et al. (1990)
office 4 monitors (approximately 50) 0.25-1.27 UBA (1992)
a n.d. = not detected (detection limit in parentheses); n.sp. = not specified.
Table 29. Correlation between number of monitors operating
in a room and sum of concentrations of tetra- and pentaBDFsa
Number of monitors per room Sum of concentrations of
tetraBDFs and pentaBDFs
(µg/m3)b
0 (ambient air) <0.1
0 0.2-0.39
2 0.42-0.47
4 0.57-0.80
5c 0.23
8 1.18
a Adapted from Chriske et al. (1990).
b 2,3,7,8-Substituted congeners not determined:
c Equipment from one manufacturer.
Table 30. Concentrations of PBDFs in indoor dust samples collected
in rooms equipped with a number of display and/or computer monitorsa
PBDFs Concentration (µg/kg)b
Sample 1 Sample 2 Sample 3 Sample 4
(Police traffic (TV studio) (TV studio) (TV studio)
control office)
TetraBDFs 0.351 0.196 0.265 0.295
2,3,7,8-TeBDFc n.d. (0.001) n.d. (0.001) 0.002 0.005
PentaBDFs 0.159 0.331 0.691 0.456
1,2,3,7,8-PeBDFc 0.004 0.012 0.020 0.015
2,3,4,7,8-PeBDFc 0.003 0.004 0.006 0.005
HexaBDFs 1.71 1.44 1.78 0.982
HeptaBDFs 2.41 3.51 0.744 0.693
Sum PBDFs 4.63 5.477 3.48 2.426
a Adapted from UBA (1992).
b n.d. = not detected (detection limit in parentheses).
c Maximum value given; co-elution with internal standard, etc., cannot be excluded.
Table 31. Concentrations of PBDDs/PBDFs and PXDDs/PXDFs in dust samples
collected in Germany in 1988/89 from an underground garagea
Concentrationb (ng/kg)
Floor Wall Waste air motors
(n = 6) (n = 1) (n = 1)
Dibenzo-p-dioxins
MonoBDDs n.d. n.d. 40
DiBDDs-tetraBDDs n.d. n.d. n.d.
Cl1Br1DDs n.d. n.d. n.d.
Dibenzofurans
MonoBDFs 150-390 550 2800
DiBDFs 280-560 820 3500
TriBDFs n.d. n.d. 500
TetraBDFs n.d. n.d. n.d.
Cl1Br1DFsc 180-630 4300
Cl1Br2DFsc n.d. 790
Cl2Br1DFsc n.d. 520
Cl1Br3DFsc n.d. n.d.
Cl2Br2DFsc n.d. n.d.
Cl3BhDFsc n.d. n.d.
a Adapted from Ballschmiter et al. (1990).
b n.d. = not detected (detection limit 20 ng/kg).
c Concentrations for floor and wall dust samples combined.
The PBDF profile (Br1 through Br4) found in dust from an
underground garage was dominated by the lower brominated homologues
(Ballschmiter et al., 1990). Concentrations ranged from 150 to 560
ng/kg dust for mono- and diBDFs in samples from the floor, but they
increased at special sampling sites (wall, ventilation motors) to a
maximum of 3500 ng diBDFs/kg dust (Table 31).
PXDFs were analysed for and detected in dust samples from the
underground garage (Ballschmiter et al., 1990). Their concentrations
ranged from n.d. to 4300 ng/kg dust for di- through tetrahalogenated
PXDFs (Table 31).
5.1.2 Water and sediment
No information is available on contamination of water with
PBDDs/PBDFs.
River and marine sediment samples (n = 5) from Japan monitored
for tetra- to hexaBDDs/BDFs contained tetraBDDs (n.d. - 0.006 µg/kg
dry weight) and tetra- to hexaBDFs at total concentrations of
0.03 - 0.37 µg/kg dry weight. There were also data on PXDDs/PXDFs
(Watanabe et al., 1995).
Ballschmiter et al. (1990) investigated sediment (sludge)
collected from a drain that received runoff water from a German
motorway crossing. They found no PBDDs (Br1 through Br4), but PBDFs and
PXDFs were found, their concentrations ranging from 180 to 1690 ng/kg
dry sludge (di- and trihalogenated homologues; sum of PBDFs/PXDFs:
2500/1850 ng/kg). Although this was a single sample, it showed,
together with the results for PCDDs/PCDFs, a pattern recognized as
typical for automobile-derived contamination - that is, predominance
of PBDFs over PCDFs, prevalence of dibenzofurans over
dibenzo- p-dioxins, and increased concentrations of lower halogenated
dibenzo- p-dioxins and dibenzofurans (Ballschmiter et al., 1990).
Similar correlations were observed with samples of soil (section
5.1.3) and grass (section 5.1.6.1) collected near motorways
(Ballschmiter et al., 1990).
Sediments recently sampled from road sewers (n = 2) in Germany
(Schacht et al., 1995) had lower concentrations of PBDFs (sum of tetra
to hepta: up to 300 ng/kg) than of PCDFs (sum of tetra to octa: up to
1300 ng/kg).
Analyses of laminated sediment core from the Baltic Proper
revealed PBDEs in sediment layers dating from the 1950s and later and
made evident a dramatic increase, ranging from 4- to 20-fold, in PBDEs
in the 1980s. Analyses for PBDFs were not performed (Nylund et al.,
1992).
5.1.3 Soil, sewage sludge, and biocompost
One soil sample taken near a motorway (depth: 0 - 2 cm) in
Germany contained 0.74 µg monoBDFs/kg, 0.58 µg diBDFs/kg, and 1 µg
Cl1Br1DFs/kg soil. Other PBDFs or PXDFs (up to Br4/X4) were not
detected, and no PBDDs (Br1 through Br4) were found (detection limit:
0.02 µg/kg). PCDFs (Cl1 through Cl5) were present at concentrations
ranging from 0.04 to 0.38 µg/kg, and PCDDs (Cl1 through Cl8) were
detected at 0.1 µg/kg (Ballschmiter et al., 1990). Another soil sample
(n = 1; no details given) collected near a motorway in Germany also
did not contain PBDDs, and the concentrations of PBDFs were low (sum
of tetra- and pentaBDFs: 0.02 µg/kg; 2,3,7,8-TeBDF: <0.001 µg/kg).
The sum of PCDDs/PCDFs was 0.3 µg/kg (Schacht et al., 1995).
Watanabe et al. (1992) reported the presence of PBDFs (about 100
µg total PBDFs/kg), together with PBDEs and PCDDs/PCDFs, in a soil
sample (depth not given) taken from an incineration field in Taiwan
that was contaminated with large amounts of ash from plastic
materials. They also found a large number of PXDFs but no PXDDs in the
same sample.
Further samples from disposal sites are discussed in chapter 3.
Soil samples (n = 2) collected at a metal reclamation factory
area in southern Taiwan had total PBDF concentrations of 48 and 87
µg/kg. The PBDFs included triBDFs (16-30 µg/kg), tetraBDFs (16 - 34
µg/kg), pentaBDFs (11 - 18 µg/kg), and hexaBDFs (5 µg/kg). PBDDs were
not detected (detection limits ranged from <0.25 to 1 µg/kg)
(Watanabe et al., 1993).
Soil samples (n = 3; 0 - 10 cm depth, if necessary after removal
of plants and grass; at distances of >1000 - <2000 m, main wind
direction) were collected a few months after a fire in a warehouse
where bromine-containing plastic pellets were stored (Neupert & Pump,
1992). In one of three sites (samples), 2,3,7,8-substituted
PBDDs/PBDFs were found (detection limit 0.5 ng/kg) at 3.5 ng/kg (sum
of five tetra- to hexaBDDs and of three tetra- to pentaBDFs).
A series of sewage sludge samples (n = 13) from municipal
wastewater treatment plants in Germany were analysed for PBDDs/PXDDs
and PBDFs/PXDFs as well as for other polyhalogenated compounds
(Hagenmaier et al., 1992). The analytical data on these sludges, which
were destined to be discharged on fields for fertilization, are
summarized in Tables 32 and 33. PBDFs were detected in all samples,
their concentrations (sum of mono- to pentaBDFs) reaching a maximum of
about 3 µg/kg. Of the homologue groups (Table 32), the diBDFs were
predominant, with concentrations ranging from 0.27 to 1.99 µg/kg.
PBDDs, PXDDs, and PXDFs were not found in any samples. The ratio of
median concentrations for PBDFs and PCDDs/PCDFs was 1 : 9 (Table 33).
PBDEs (tri- to heptaBDEs), the possible precursors of PBDFs, were
detected in all samples at higher levels than PBDFs (Table 33).
Another study group (Ballschmiter et al., 1990) did not find any
PBDDs/PBDFs (mono through tetra) in sewage sludge samples from rural
(n = 12) and urban (n = 5) areas (detection limit not given).
Traces of tetraBDDs (0.006 µg/kg) and tetra- to heptaBDFs (sum:
0.32 µg/kg; 2,3,7,8-TeBDF: 0.003 µg/kg) together with PCDDs/PCDFs
(sum: 8.34 µg/kg) were found in a single sewage sludge sample (Schacht
et al., 1995).
Biocompost (n = 1) containing tetra- to octaCDDs/CDFs (sum: 3.2
µg/kg) showed no detectable PBDDs (detection limit not given); of
PBDFs (tetra to hepta), only tetraBDFs (<0.003 µg/kg; no
2,3,7,8-TeBDF) were present (Schacht et al., 1995).
Table 32. PBDFs detected in sewage sludge samples from municipal
wastewater treatment plants in Germanya
PBDFs Number of samples Concentrationb
(µg/kg)
MonoBDFs 9 0.05-0.67
DiBDFs 8 0.27-1.99
TriBDFs 9 0.07-0.20
TetraBDFs 9 0.03-0.23
PentaBDFs 9 n.d.-0.01
Sum of mono to pentaBDFs 0.29-3.05
a Adapted from Hagenmeier et al. (1992).
b n.d. = not detected.
Table 33. Concentrations of PBDFs and other polyhalogenated aromatic
compounds in sewage sludgea
Compounds Number of Concentration (µg/kg)
samples Range Median Mean Standard
deviation
Mono- to pentaBDFs 13 0.21-3.05 1.11 1.17 0.92
Tri- to heptaBDEs 13 0.49-17.73 8.37 8.58 5.51
PCBs 17 233-3456 674 911 767
Total PCDDs 13 3.27-27.82 9.20 10.71 6.69
Total PCDFs 13 0.18-7.09 0.53 1.07 1.83
a Adapted from Hagenmaier et al. (1992).
5.1.4 Food and feed
Little information was found in the literature. Market basket
surveys are not available.
Crops and other vegetation growing in the vicinity of potential
emitters can be contaminated, as shown by an analysis of grass
collected near a motorway. The pattern and levels of PBDDs/PBDFs and
PXDDs/PXDFs found in plants are described in section 5.1.6.1.
The occurrence of PBDDs/PBDFs in seafood and milk is discussed in
section 5.1.6.2.
5.1.5 Other products
For levels of PBDDs/PBDFs in consumer products such as electrical
appliances, and emissions from them, see chapter 3.
5.1.6 Terrestrial and aquatic organisms
5.1.6.1 Plants
Grass samples collected in the vicinity of a motorway contained
lower brominated PBDFs (mono- through triBDFs), PXDFs (up to X3), and
traces of monoBDDs (Ballschmiter et al., 1990; Table 34). Between the
homologue groups, concentrations peaked at mono- and diBDFs and
Cl1Br1DFs (>2000 ng/kg). This pattern paralleled that found in the
soil sample from the same sampling site, but the levels in the grass
sample were higher (Table 34).
PBDDs/PBDFs (mono through tetra), PXDDs/PXDFs (up to tetra), and
PCDDs/PCDFs (mono to octa) have been analysed in needles from a pine
near a highway (Schwind, 1991). Again, concentration peaks were seen
with the mono- and diBDFs (see Table 34) and with monoCDFs (4380 ng/kg
dry weight). The sum of the PHDFs (mono to octa) was about a factor of
100 higher than the sum of PHDDs (owing to the high concentration of
mono- to trihalogenated dibenzofurans [>12 500 ng/kg dry weight]).
5.1.6.2 Animals
1) Wildlife
In a randomly selected pooled sample consisting of shrimp (380 g)
and mussels (600 g) as well as fish (cod: 1500 g; plaice: 300 g), a
tetraBDD and a tetraBDF could be detected, both
non-2,3,7,8-substituted (De Jong et al., 1992). A quantitative
analysis was not performed.
Neither PBDDs/PBDFs nor PXDDs/PXDFs were found in one homogenate
of muscle from Baltic salmon (Salmo salar; Sweden) at detection limits
of 0.2 - 20 ng/kg fresh weight (Wiberg et al., 1992). PBDDs/PBDFs were
not detected (detection limits of 2, 3, and 8 ng/kg for tetra-,
penta-, and hexaBDDs/BDFs, respectively) in pooled muscle samples of
young (n = 15), middle-aged (n = 15), and old (n = 15) carp (Cyprinus
carpio) collected from the Buffalo River, NY, USA (Loganathan et al.,
1995). No PBDDs/PBDFs or PXDDs/PXDFs (tetra to hexa) were detected
(detection limit not specified) in fish (n = 4) captured in 1993 in
rivers near Osaka in Japan (Watanabe et al., 1995).
One composite sample of muscle from osprey (Pandion haliaetus;
Sweden; n = 35; cf. Jansson et al., 1993) was analysed for PBDDs/PBDFs
and for PXDDs/PXDFs. The results were negative at detection limits of
0.2 - 20 ng/kg fresh weight (Wiberg et al., 1992).
Table 34. Concentrations of PBDDs/PBDFs and PXDDs/PXDFs in
environmental samples taken near motorways in Germany
Homologue groups Concentrationa (ng/kg)
Grassb Soilb Pine needlesc
(after 1 month (depth: 0-2 cm) (dry weight)
of dryness)
Dibenzo-p-dioxins
MonoBDDs 60 n.d. 28.3
DiBDDs n.d. n.d. 25.5
TriBDDs n.d. n.d. 5.5
TetraBDDs n.d. n.d. <4
Cl1Br1DDs - - +
Cl1Br2DDs - - 5.6
Cl2Br1DDs - - 1.7
X4DDs - - <4d
Dibenzofurans
MonoBDFs 2530 740 5491
DiBDFs 2170 580 1053
TriBDFs 40 n.d. 258
TetraBDFs n.d. n.d. 53
Cl1Br1DFs 2420 1000 788
Cl1Br2DFs 240 n.d. 358
Cl2Br2DFs 190 n.d. 116
Cl2Br2DFs n.d. n.d. 58
Cl3Br1DFs n.d. n.d. 55
Cl1Br3DFs - - 74
a n.d. = not detected (detection limit 20 ng/kg); - = not analysed;
+ = detectable, but not quantifiable.
b Adapted from Ballschmiter et al. (1990).
c Adapted from Schwind (1991).
d For each of the three possible combinations (Cl2Br2, Cl3Br1, Cl1Br3).
PCDDs/PCDFs were present in all samples mentioned above. For
example, carp from the Buffalo River (Loganathan et al., 1995) were
found to contain noticeable concentrations of total PCDDs (27 - 146
ng/kg wet weight) and total PCDFs (22 - 99 ng/kg) along with total
PCBs (>2 mg/kg) and total PBDEs (13 - 23 µg/kg).
2) Farm animals
Cow's milk collected at dairy farms in the deposition area of an
"old technology" municipal waste incinerator in the Netherlands was
analysed for PBDDs/PBDFs and PXDDs/PXDFs. The pooled (n = 11) milk
sample contained compounds that were tentatively identified (but not
quantified) as two triBDFs, one tetraBDF, and one pentaBDF, all four
not having the 2,3,7,8-substitution pattern (De Jong et al., 1992).
The same sample contained high levels of PCDDs/PCDFs.
5.2 General population exposure
5.2.1 Exposure data
There is no quantitative information available on exposure of the
general population, special subpopulations, or infants to PBDDs/ PBDFs
from several sources (see chapter 3 and section 5.1).
5.2.2 Monitoring of human tissues and fluids
Few studies have monitored PBDDs/PBDFs in human tissues or milk.
On behalf of the National Human Adipose Tissue Survey, the US EPA
initiated a study in 1987 to analyse 2,3,7,8-substituted tetra-through
hexaBDDs/BDFs in adipose tissue of the general population. Eight
hundred and sixty-five individual tissue specimens were collected and
combined into 48 composite samples referring to the nine US census
divisions and three age groups. None of the six targeted PBDDs/PBDFs
was detected at average detection limits of approximately 1 ng/kg for
2,3,7,8-TeBDD/TeBDF (range: 0.4 - 8.9 ng/kg), 10 ng/kg for
1,2,3,7,8-PeBDD/PeBDF and for 1,2,3,4,7,8-HxBDD (range: 0.8 - 54
ng/kg), and 40 ng/kg for 1,2,3,4,7,8-HxBDF (range: 2.5 - 120 ng/kg) on
a lipid weight basis (Cramer et al., 1990a,b). In all samples
analysed, there were indications of the presence of PBDEs (hexa
through octa) (Cramer et al., 1990a,b).
Neither PBDDs/PBDFs nor PXDDs/PXDFs were found in two human
adipose tissue samples (male: 70 years of age; female: 60 years of
age) examined in a German study (Dawidowsky, 1993). The detection
limits for tetra-, penta-, and hexa-substituted congeners were
0.2 - 1.6 ng/kg, 0.9 - 3.5 ng/kg, and 4.6 - 14 ng/kg on a lipid weight
basis, respectively.
One composite sample of human milk (38 g) from Sweden was
examined for PBDDs/PBDFs and PXDDs/PXDFs (Wiberg et al., 1992).
Results of this study, which did not give any information on the
number of original specimens or sampling strategy, were negative. The
detection limits were reported to be in the range of 0.2 - 20 ng/kg on
a lipid weight basis. PCDDs/PCDFs were detectable in this sample.
Another investigation from Germany (Dawidowsky, 1993) led to similar
results. Although PCDDs/PCDFs were present in three human milk
samples, PBDDs/PBDFs and PXDDs/PXDFs were not detectable at detection
limits of 0.8 - 5.5 ng/kg (tetra substitution), 3.2 - 13 ng/kg (penta
substitution), and 13 - 53 ng/kg (hexa substitution) on a lipid weight
basis. At present, the presence of PBDDs/PBDFs in human milk at very
low concentrations cannot be ruled out (Somogyi & Beck, 1993).
5.3 Occupational exposure
5.3.1 Workplace monitoring data
Contamination by PBDDs/PBDFs is possible in a variety of
workplaces involved in producing, processing, using, or disposing of
certain flame retardants or products containing them (see chapter 3),
especially where the processes involve elevated temperatures. There
are only limited workplace monitoring data.
5.3.1.1 Flame retardant/polymer industry
A set of data is available on workplaces in the chemical industry
producing flame-retarded polymers (Brenner & Knies, 1990, 1992,
1993a,b, 1994; Thies et al., 1990; Brenner, 1993; Kieper, 1996). Air
samples were taken during extrusion production and moulding of PBT,
ABS, polystyrene, or polyamide resins blended with various brominated
flame retardants (see Table 35).
PBDF concentrations measured near the extruder and injection
moulding machines, in the whole building, in the storage and refilling
area, and at other sites are summarized in Table 35. Within the
homologue groups measured, a maximum concentration of about 600 ng/m3
was found for the sum of hexaBDFs. In one experimental series, PBDF
concentrations in workplace air were higher by a factor of about 1000
where DBDE was used, compared with TBBPA. The difference was explained
by the different properties of DBDE and TBBPA, as well as different
exhaust and ventilation conditions (Brenner & Knies, 1993a). In the
study involving DBDE/PBT, PBDF concentrations near the extruder
workplace and in the air of the whole room were similar, indicating
their general distribution in the building. Lower levels were found in
the storage and refilling area (Brenner & Knies, 1990).
2,3,7,8-TeBDF was not detected in a lot of samples (detection
limits 1 - 100 pg/m3 air) (Brenner & Knies, 1990, 1993a, 1994; Thies
et al., 1990; Kieper, 1996) but was detectable in some others (Kieper,
1996). Low amounts of penta- and hexaBDFs substituted in the
2,3,7,8-position (0.3 - 2.6 ng/m3 air) were tentatively identified at
DBDE/PBT workplaces (Brenner & Knies, 1990).
Concentrations of PBDDs in the DBDE/PBT study were two orders of
magnitude below those of PBDFs, with di-, tri-, tetra-, penta-, and
hexaBDD concentrations being <0.05, 0.35, 2.04, 8.37, and 17 ng/m3
air, respectively (Brenner & Knies, 1990). The tentative analysis for
2,3,7,8-substituted PBDDs showed the presence of 2,3,7,8-TeBDD (<0.5
ng/m3 air), of 1,2,3,7,8-PeBDD (1.3 ng/m3 air), and of two hexaBDDs (1
and 1.6 ng/m3) (Brenner & Knies, 1990). In contrast, no PBDDs were
found in air samples of the workplace area in a TBBPA study, although
low amounts of di- (0.94 ng/m3), tri-(0.07 ng/m3), and tetraBDDs (0.08
Table 35. Air contamination by PBDFs measured at workplaces where flame-retarded thermoplastic resins
are produced and processed
PBDFs Resin/flame retardanta Number of Air volumeb Concentrationc Reference
sampling (m3) (ng/m3)
stations
MonoBDFs PBT/TBBPA 3 13-18 n.d.-0.26 (<0.006) Kieper (1996)
PS-PS-butadiene/DBDE 5 20-30 0.01-0.16 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.012-0.017 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.017-0,049 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.026-0.129 Kieper (1996)
DiBDFs PBT-glass fibre/DBDE 3 30-150 0.2-1.3 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-0.34 (<0.004) Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.19-1.02 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.14 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (< 1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.040-0.223 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.043-0.139 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.070-0.21 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.094-0.24 Kieper (1996)
TriBDFs PBT-glass fibre/DBDE 3 30-150 1.1-13 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-0.11 (<0.012) Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.065-3.04 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.18 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.077-0.484 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.063-0.337 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.049-0.274 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.075-0.169 Kieper (1996)
TetraBDFs PBT-glass fibre/DBDE 3 30-150 5.1-34 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 0.03-0.05 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.157-6.92 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.14 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS/hexabromocyclododecane 1 n.sp. 0.02 Brenner (1993)
PS-PS-butadiene/DBDE 5 20-30 0.173-0.40 Kieper (1996)
Table 35. (Continued)
PBDFs Resin/flame retardanta Number of Air volumeb Concentrationc Reference
sampling (m3) (ng/m3)
stations
TetraBDFs PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.136-0.30 Kieper (1996)
(cont'd) PA/polytribromostyrene 5 19-29 0.017-0.43 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.051-0.15 Kieper (1996)
PentaBDFs PBT-glass fibre/DBDE 3 30-150 8.6-143 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 0.07-0.19 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.11-5.63 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0,12 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS/hexabromocyclododecane 1 n.sp. 1 Brenner (1993)
PS-PS-butadiene/DBDE 5 20-30 0.27-0.49 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.16-0.18 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.04-0.30 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.035-0.12 Kieper (1996)
HexaBDFs PBT-glass fibre/DBDE 3 30-150 13-594 Brenner & Knies (1990)
PBT-giass fibre/TBBPA 2 185-260 0.05-0.26 Brenner & Knies (1993a)
PBT/TBBPA 3 13-18 0.06-3.61 Kieper (1996)
PBT-glass fibre/TBPI 3-4 185-260 up to 0.11 Brenner & Knies (1994)
ABS/TBBPA 1 4-5 n.d. (<1) Thies et al. (1990)
PS-PS-butadiene/DBDE 5 20-30 0.81-6.37 Kieper (1996)
PS/1,2-bis(tribromophenoxy)ethane 3 17-24 0.33-0.73 Kieper (1996)
PA/polytribromostyrene 5 19-29 0.06-0.29 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.028-0.099 Kieper (1996)
HeptaBDFs PBT-glass fibre/DBDE 3 30-150 -88-260 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-<0.04 (<0.013) Brenner & Knies (1993a)
PA/polytribromostyrene 5 1 9-29 0.07-0.30 Kieper (1996)
PA/polydibromostyrene 5 6-12 0.04-0.07 Kieper (1996)
OctaBDF PBT-glass fibre/DBDE 3 30-150 n.d. approx. -7 Brenner & Knies (1990)
PBT-glass fibre/TBBPA 2 185-260 n.d.-<0.08 (0.026) Brenner & Knies (1993a)
PA/polytribromostyrene 5 19-29 0.03-0.20 Kieper (1996)
PA/polydibromostyrene 5 6-12 <0.004-0.09 Kieper (1996)
a PA = polyamide 66; PS = polystyrene; TBBPA = tetrabromobisphenol A or its derivatives.
b n.sp. = not specified.
c n.d. = not detected (detection limits in parentheses, if specified).
ng/m3) were emitted by the extruder equipment. The detection limits of
di- through octaBDDs ranged from 0.001 to 0.4 ng/m3 air. No
2,3,7,8-substituted PBDDs (tetra- through hexaBDDs) were found
(detection limits ranged from 0.001 to 0.1 ng/m3 air) (Brenner &
Knies, 1993a). Preliminary tests at the workplace during injection
moulding of polystyrene blended with hexabromocyclododecane showed
tetraBDD concentrations of 2.5 ng/m3, consisting of two isomers with
no 2,3,7, 8-substitution (Brenner, 1993). During extruder experiments
with PBT/TBPI (Brenner & Knies, 1994), the sum concentration of PBDDs
was in the low pg/m3 range, and no 2,3,7,8-substituted congeners were
detected (detection limits ranged from 1 to 240 pg/m3; tetra to octa).
Air samples from selected workplaces (operated permanently or
periodically) of three plastic processing plants were monitored for
mono- to hexaBDFs/BDDs (resin/flame retardant used:
polystyrene-polystyrene-butadiene/DBDE;
polystyrene/1,2-bis(tribromophenoxy)-ethane; PBT/TBBPA-carbonate
oligomer) and for mono- to octaBDFs/BDDs (resin/flame retardant used:
polyamide 66/polytribromostyrene; polyamide 66/polydibromostyrene),
including eight or more 2,3,7,8-substituted congeners (Kieper, 1996).
Depending on workplace and flame retardant used, the sum of PBDF/PBDD
concentrations (mono to hexa) ranged from 258 to 77 414 pg/m3. The
highest sum concentrations within workplaces permanently operated
(range: 258 - 10 018 pg/m3) were found at workplaces processing
1,2-bis(tribromo-phenoxy)ethane. Although samples from the latter did
not contain any 2,3,7,8-substituted congeners, many other samples gave
positive results, with maximum sum concentrations of some thousand
pg/m3. The highest concentrations of 2,3,7,8-substituted PBDFs/PBDDs
were seen at the TBBPA-carbonate oligomer/PBT workplaces (operated
permanently and periodically). The ranges of concentrations (n = 3) in
pg/m3 were as follows:
2,3,7,8-TeBDF: <4 - <165 (detection limit
elevated
owing to interfering
components)
1,2,3,7,8-PeBDF: 2 - 100
2,3,4,7,8-PeBDF: 6 - 313
1,2,3,4,7,8-HxBDF: 7 - 445
2,3,7,8-TeBDD: <6 - <293 (detection limit
elevated
owing to interfering
components)
1,2,3,7,8-PeBDD: 23 - 1137
1,2,3,4,7,8-/1,2,3,6,7,8-HxBDD: 25 - 1161
1,2,3,7,8,9-HxBDD: 9 - 578
Results for 2,3,7,8-substituted heptaBDFs/BDDs were obtained at
workplaces processing polyamide flame-retarded by polytribromo-styrene
(n = 5) or polydibromostyrene (n = 5). The concentrations measured (in
pg/m3) ranged in the following manner (n = 10):
1,2,3,4,6,7,8-HpBDF: 26 - 280
1,2,3,4,7,8,9-HpBDF: <1 - 13
1,2,3,4,6,7,8-HpBDD: <1.4 - 11
5.3.1.2 Offices/studios
Some monitoring results are available for workplaces (offices;
television studios) equipped with a number of electrical appliances
continually in use, such as display and computer monitors (Chriske et
al., 1990; UBA, 1992; see also section 5.1.1). Maximum air
concentrations measured were 0.56 pg/m3, 0.61 pg/m3, and 0.4 pg/m3 for
tetra-, penta-, and hexaBDFs (see Table 28). Although heptaBDFs could
not be detected in air samples (Table 28), they were found in the
corresponding dust samples at concentrations ranging from 0.7 to 3.5
µg/kg (Table 30). Similarly, no detectable amounts of tetra- and
pentaBDFs substituted in the 2,3,7,8-position were found in air
samples (Table 28), but they were found in dust samples, with
concentrations ranging from n.d. to 0.005 µg/kg (2,3,7,8-TeBDF) and
from 0.003 to 0.020 µg/kg (two pentaBDFs) (Table 30). PBDDs were not
detected (Chriske et al., 1990).
5.3.1.3 Recycling plants
Recycling of plastic materials (pure or in combination with other
materials, e.g. metals, that can have catalytic effects) may be a
source of PHDDs/PHDFs, depending - inter alia - on the type of flame
retardants blended with them (see chapter 3). Workplace air was
monitored in a pilot plant recycling defective printed circuits
(Lorenz & Bahadir, 1993). These printed circuits contained copper and
TBBPA, a flame retardant with a relatively low potency for generation
of PBDDs/PBDFs. Air samples (n = 2) taken near the running shredding
systems did not contain PBDFs, although small amounts of mono- and
diBDFs were found in the shredded material (0.05 - 0.32 µg/kg).
However, owing to short sampling time and resulting low air volumes
sampled (6 - 7 m3), the detection limits were only 0.02 - 0.1 ng/m3
(mono- through pentaBDFs) and 0.2 - 0.4 ng/m3 (hexa- through
heptaBDFs). No PBDDs (mono- through octaBDDs) were found in the air
samples (detection limits 0.02 - 2 ng/m3 for mono- through heptaBDDs),
but residues of tetraBDDs (0.03 - 0.73 µg/kg) were present in samples
of the processed waste. Neither 2,3,7,8-TeBDD nor 2,3,7,8-TeBDF was
found in any of the samples tested (Lorenz & Bahadir, 1993).
In 1991, air samples (air volume: 20 - 30 m3; sampling period: 6
- 10 h) from three workplace stations in an operating secondary copper
plant and ground dust samples (pooled sample from five sites)
collected in another secondary copper plant (shut down in 1990) in
Germany were monitored for PBDDs/PBDFs (Kieper, 1996). The sum of
mono- to hexaBDF concentrations in the workplace air samples ranged
from 8 to 190 pg/m3. 2,3,7,8-TeBDF was present in one sample at 0.4
pg/m3 but was not detected in either of the other air samples
(detection limits 0.1 - 0.8 pg/m3). PBDDs (mono to hexa) were not
detectable (detection limits 0.1 - 1.4 pg/m3). The dust sample
contained 21.02 µg mono- to hexaBDFs/kg, with a maximum of 8.4 µg/kg
for tetraBDFs. Concentrations of 2,3,7,8-TeBDF, 1,2,3,7,8-PeBDF,
2,3,4,7,8-PeBDF, and 1,2,3,4,7,8-HxBDF were 0.09, 0.10, 0.12, and 0.09
µg/kg (maximum values), respectively. PBDDs (mono to hexa) were not
found (detection limits 0.01 - 0.03 µg/kg).
5.3.1.4 Other workplaces
Monitoring data from workplaces in waste incineration facilities
and disposal sites are lacking.
PBDF concentrations of up to 100 mg/kg (see section 3.7.2) were
found in combustion residues of accidental fires; therefore, firemen
and other workers coming in contact with fire fume, dust, and residues
could be exposed to these substances.
The work area under the fume hood (waste oil from air pumps) of a
laboratory was found to be contaminated with PBDDs/PBDFs and
PCDDs/PCDFs (Ritterbusch et al., 1994b). Wipe tests showed a sum
concentration of 580 ng/m2 for PBDDs/PBDFs (mono to hexa) and of 360
ng/m2 for PCDDs/PCDFs (tetra to octa).
5.3.2 Monitoring of human tissues and fluids
A chemist who suffered from acute intoxication after synthesizing
several grams of 2,3,7,8-TeBDD and 2,3,7,8-TeCDD in 1956 without using
a hood or protective clothing (Schecter & Ryan, 1990, 1991, 1992;
Schecter, 1992; see also section 8.2) was examined 35 years after
exposure. His blood contained 625 ng TBDD/kg blood lipid (concomitant
with 18 ng TCDD/kg blood lipid) (Schecter, 1992; Schecter & Ryan,
1992). For comparison, the average blood lipid concentration in the
general population of the USA was reported as 3 - 5 ng/kg for TCDD and
not detectable for TBDD (Schecter et al., 1994a). (Blood analysis of a
Japanese student who had developed chloracne [see section 8.2] 1 month
after exposure to PCDFs and PBDFs showed no detectable amounts of
PCDFs [or PCBs]. PBDFs were not included in this analysis, performed
in 1982, about half a year after exposure [Asahi & Urabe, 1987].)
Zober et al. (1992) investigated employees of a chemical plant
that had produced thermoplastic resins (PBT) blended with the flame
retardants OBDE or DBDE. The potential for exposure to PBDDs/PBDFs
including the homologue groups di- through octaBDDs/BDFs was
established by workplace air measurements, as described by Brenner &
Knies (1990) (see also section 5.3.1). However, analysis of the blood
lipids of the personnel focused mainly on 2,3,7,8-TeBDD/TeBDF.
Elevated levels of both congeners were found in the blood samples of
potentially exposed workers, although both isomers could not (TBDF) or
could hardly (TBDD) be identified in the previous workplace air
samples (see section 5.3.1). The concentrations of TBDD in venous
blood exceeded those of TBDF (ranges: n.d. - 478 ng/kg blood lipid
versus n.d. - 112 ng/kg blood lipid) among the male study group (see
Table 36). As seen in Table 36, there was a correlation between blood
levels of TBDF/TBDD measured and job type, working conditions, and
working period. Highest median (TBDF/TBDD: 18/91 ng/kg blood lipid)
and maximum (TBDF/TBDD: 112/478 ng/kg blood lipid) values were found
in the 18 extruder operators who had been first engaged before 1986.
The lower levels observed in the 11 operators employed during and
after 1986 may be due to the shorter exposure time, changes in
production process, or technical improvements, as suggested by the
authors of this study. Other long-term employees (n - 5) showed
intermediate blood levels ranging from <7 to 26 ng TBDF/kg blood
lipid and from 7 to 48 ng TBDD/kg blood lipid. The lowest values were
seen in the technical support personnel (see Table 36). Data from a
referent group were not provided, but among referents (n = 5) of a
preceding study, PBDFs/PBDDs were either not detected or marginally
present (Zober et al., 1992).
Table 36. Concentrations of 2,3,7,8-TeBDF and 2,3,7,8-TeBDD in blood of personnel from industry
using PBDEa
Job type/first year on job Number Concentration (ng/kg blood lipid)b,c
2,3,7,8-TeBDF 2,3,7,8-TeBDD
Median Minimum Maximum Median Minimum Maximum
Extruder operators 29 8 n.d. 112 40 n.d. 478
1975-1985 18 18 n.d. 112 91 16 478
1986-1988 11 4 n.d. 11 n.d. n.d. 11
Maintenance mechanics
(1975-1983) 3 16 <7 26 17 17 22
Production employees;
other areas (1976-1982) 2 7 7 7 28 7 48
Technical support personnel 8 2 n.d. 11 n.d. n.d. 5
a From Zober et al. (1992).
b Samples collected in 1990/91.
c n.d. = not detected (detection limit not specified).
6. KINETICS AND METABOLISM
6.1 Absorption
6.1.1 Dibenzo-p-dioxins
All studies available on absorption of PBDDs refer to absorption
of 2,3,7,8-TeBDD in rats (see Table 37). It was absorbed after oral,
dermal, and intratracheal administration, the percent absorption
varying with route and dose.
After administration of a single dose of 1 nmol/kg body weight,
absorption was about 80% by the oral and intratracheal routes, whereas
only about 12% was absorbed through the skin. Oral absorption of
single doses declined from 80% at lower doses (1 - 10 nmol/kg body
weight) to about 50% at higher doses (500 nmol/kg body weight), thus
suggesting non-linear absorption.
Oral and pulmonary absorption of an equimolar dose of
2,3,7,8-TeCDD using identical experimental conditions as with TBDD
were 88% and 95%, respectively (Diliberto et al., 1996). Dermal
absorption of TBDD was about one-third that of an equimolar dose of
TCDD (Jackson et al., 1991; Diliberto et al., 1993). Differences
between TBDD and TCDD pulmonary and dermal absorption may be explained
by the octanol/water partition coefficients and the size of the
halogen substituents.
As enteral absorption for many PHDDs/PHDFs is known to be
variable and incomplete (especially demonstrated for the higher
chlorinated congeners), subcutaneous application has been used in
several of the relevant studies. For the majority of the chlorinated
congeners, a significant degree of absorption was reported within a
few days (exception: octachlorodibenzo- p-dioxin, or OCDD). A 99%
absorption rate has been reported by this route for TBDD (600 ng/kg
body weight) as well as for TCDD (300 ng/kg body weight) in the rat
(Nagao et al., 1995/96).
6.1.2 Dibenzofurans
Dermal absorption of 1,2,7,8-TeBDF was examined in male Fischer
344 rats following a single dose of 1 nmol/kg body weight (Kedderis et
al., 1994). About 29% of the administered dose was absorbed,
quantified on the basis of the amount found in tissues (4%, excluding
the skin site) and excreted within 72 h. This dermal absorption was
intermediate, compared with that of TBDD (12%: see section 6.1.1),
TCDD (41%: Banks & Birnbaum, 1991), and TCDF (48%: Brewster et al.,
1989) after single equimolar doses.
Table 37. Absorption of 2,3,7,8-TeBDD in rats
Strain (sex) Route (vehicle) Dosing regimen Absorption,a time Method References
Wistar oral (arachis oil with single dose 80% (male), 48 h faeces Ivens et al.
(female, male) 5% toluene) 100 µg/kg body weight 83% (female), 48 h analysis (1992)
(n = 4) (= 0.2 pmol/kg body weight) [TBDD]
Fischer 344 oral (water: ethanol: single dose faeces Diliberto et al.
(male) Emulphor(R) = 3: 1: 1) and tissue (1990a,b, 1993);
(n = 3-4) analysis
0.5 µg/kg body weight 78%, 72 h [3H-TBDD]
(= 0.001 µmol/kg body weight) Kedderis et al.
(1992a)
5 µg/kg body weight 82%, 72 h
(= 0.01 µmol/kg body weight)
50 µg/kg body weight 60%, 72 h
(= 0.1 µmol/kg body weight)
250 µg/kg body weight 47%, 72 h
(= 0.5 µmol/kg body weight)
Fischer 344 intratracheal (water: single dose 80%, 72 h faeces and Diliberto et al.
(male) ethanol: Emulphor(R) 0.5 µg/kg body weight tissue (1991, 1993);
(n = 3-4) = 3: 1: 1) (= 0.001 µmol/kg body weight) analysis Kedderis et al.
[3H-TBDD] (1992a)
Fischer 344 dermal (acetone) single dose 12%, 72 h faeces and Jackson et al.
(male) 0.5 µg/kg body weight tissue (1991);
(n = 3-4) (= 0.001 pmollkg body weight) analysis Kedderis et al.
(= 0.2 nmol/1.8 cm2) [3H-TBDD] (1992a);
Diliberto et al.
(1993)
a Values based on concentration of 2,3,7,8-TeBDD [TBDD] or on 3H activity ([3H-TBDD]).
6.2 Distribution
6.2.1 Levels in organs and blood
6.2.1.1 Dibenzo-p-dioxins
Almost all studies available on PBDD distribution refer to
disposition of 2,3,7,8-TeBDD in the rat. As shown in Table 38, TBDD is
distributed throughout the whole body, with major deposits found in
liver and adipose tissue, followed by skin and muscle (Kedderis et
al., 1991a; Diliberto et al., 1993). Appreciable amounts of [3H]TBDD
were also found in adrenals and thymus (Diliberto et al., 1993). Three
days after oral exposure to [3H]TBDD, liver and adipose tissue
contained more than 65% of the body burden (Diliberto et al., 1993).
Distribution of TBDD can be described by a physiologically based
pharmacokinetic model consisting of a blood compartment and five
tissue compartments: liver, fat, skin, slowly perfused tissues, and
richly perfused tissues (Kedderis et al., 1992b, 1993; Buckley, 1995).
The partitioning of TBDD between liver and adipose tissue was
studied in Fischer 344 rats exposed to [3H]TBDD (Diliberto et al.,
1990a,b, 1991, 1993; Kedderis et al., 1990, 1991a,b, 1992a, 1993;
Jackson et al., 1991) and found to be influenced by dose, route of
exposure, and time post-dosing (see Table 39 for representative data).
Dose-dependent changes in partition ratios were seen in the
intravenous and oral studies (see Table 39). Liver concentrations of
TBDD were disproportionately increased at the higher doses compared
with the lower dose of 1 nmol/kg body weight (liver: fat concentration
ratios: 2.6 and 0.2 by the intravenous route at high and low dose,
respectively, and >5 and 2.9 by the oral route). However, whereas
liver concentrations of TBDD were disproportionately increased at 10
nmol/kg body weight compared with the 1 nmol/kg body weight oral dose,
the increase was related to dose in the 10 - 100 nmol/kg body weight
dose range. Factors influencing the dose-dependent nature of TBDD
tissue distribution are discussed by Kedderis et al. (1993).
Intravenous, oral, intratracheal, and dermal treatment with 1
nmol [3H]TBDD/kg body weight resulted 3 days later in liver: fat
concentration ratios of 3.4, 2.9, 2.0, and 1.5, respectively (see
Table 39). The lower ratio observed for the dermal exposure is
explained by differences in absorbed dose (low internal exposure; see
section 6.1) and dose-related tissue distribution.
Time-dependent changes in the distribution pattern were
demonstrated in the intravenous study. Partition ratios (liver : fat)
of 3.4 and 0.2, respectively, observed 3 and 56 days after single
intravenous exposure, indicated an increased distribution in adipose
tissue with increasing time after dosing (Table 39). In addition to
redistribution, tissue-specific elimination may also be occurring.
Table 38. Distribution of TBDD-derived radioactivity in Fischer 344 rats
3 days after oral, dermal, or intratracheal administration of 1 nmol
[3H]TBDD/kg body weighta,b
Tissue % administered dosec,d % absorbed dose/g tissued,e
Oral Dermal Intratracheal Oral Dermal Intratracheal
Liver 20.3 2.4 19.5 2.4 2.4 0.3
Adipose tissue 19.6 3.8 24.7 0.8 1.6 1.2
Skin 10.9 1.8 8.3 0.3 0.5 0.3
Muscle 3.5 0.8 3.0 0.04 0.07 0.04
Blood 0.4 0.06 0.2 0.03 0.03 0.01
Thymus 0.03f 0.03 0.08 0.2 1.1 0.4
Adrenals 0.4f 0.01 0.02 0.5 1.2 0.4
Kidneys - 0.05 0.1 - 0.2 0.1
Spleen - 0.01 0.02 - 0.2 0.06
Lungs - 0.06 0.1 - 0.5 0.2
Heart - 0,02 0.03 - 0.2 0.06
Testes - 0.02 0.05 - 0.06 0.03
Brain - 0.01 0.02 - 0.05 0.02
Stomach - 0.03 0.1 - 0.3 0.2
Table 38. (Continued)
Tissue % administered dosec,d % absorbed dose/g tissued,e
Oral Dermal Intratracheal Oral Dermal Intratracheal
Small intestines - 0.04 0.2 - 0.2 0.1
Large intestines - 0.04 0.2 - 0.3 0.1
a Adapted from Diliberto et al. (1993); oral absorption = 79%; dermal absorption
= 12%; intratracheal absorption = 78%.
b Mean values; n = 3-4; standard deviation and statistical details omitted.
c Percentage of the administered dose normalized to 100% recovery.
d - = not analysed.
e Percentage adjusted to 100% absorption.
f n = 1.
Table 39. Partition of [3H]TBDD-derived radioactivity between liver and
adipose tissue of ratsa,b
Route of Dosec Observation TBDD concentration Liver: fat
exposure (nmol/kg period (pmol/g) concentration
body weight) (days) Liver Fat ratio
Intravenous 1 3 8.1 2.4 3.4
1 56 0.2 1.1 0.2
100 56 117.2 45.3 2.6
Oral 1 3 4.9 1.7 2.9
10 3 79.9 13.6 5.9
100 3 518.3 93,4 5.6
500 3 2216.3 340.1 6.5
Intratracheal 1 3 4.1 2.1 2.0
Dermal 1 3 0.6 0.4 1.5
a Adapted from Kedderis et al. (1992a); Diliberto et al. (1993).
b Male Fischer 344 rats; n = 3-4; single doses; vehicle: ethanol: Emulphor(R):
water = 1: 1: 3 (oral, intravenous, intratracheal exposure), acetone (dermal exposure).
c 1 nmol/kg body weight corresponds to 0.5 µg/kg body weight.
Other studies using non-labelled TBDD (vehicle: arachis oil with
5% toluene) and another rat strain (Wistar) also found higher
concentrations of TBDD in the liver than in adipose tissue 2 days
after single oral doses (Neupert et al., 1989; Ivens et al., 1992) and
after daily oral exposure for 91 days (Ivens et al., 1990, 1993).
Both TCDD and TBDD appeared to be distributed in a similar
manner, and differences (e.g. after dermal exposure) can be attributed
to the higher lipophilic nature of TBDD (Diliberto et al., 1993;
Kedderis et al., 1993). Concerning distribution between adipose tissue
and blood, a 2.7-fold higher fat/blood partition coefficient was
assumed for TBDD compared with TCDD (Kedderis et al., 1993).
Tissue concentrations and concentration ratios (liver : adipose
tissue) have been compared under identical experimental conditions for
TBDD (single subcutaneous injection, Wistar rats, 600 ng/kg body
weight) and TCDD (300 ng/kg body weight) (Nagao et al., 1995/96). As
shown in Table 40, the liver : adipose tissue concentration ratio
increases with increasing doses for both congeners. In contrast, the
concentration ratios for TCDD/TBDD were rather dose-independent in
adipose tissue and also in the liver. Whereas hepatic tissue
concentrations were very similar at the doses used for TBDD and TCDD,
concentrations in adipose tissue were higher for TBDD over the entire
dose range. When increasing the dose 100-fold (from 30 to 3000 ng/kg
body weight), hepatic concentrations increased 174 times for TCDD and
256 times for TBDD. In contrast, concentrations in adipose tissue
increased only 26 times (TCDD) and 21 times (TBDD).
When the two corresponding chlorinated and brominated
1,2,3,7,8-pentahalogenated dibenzo- p-dioxins (PeHDDs) were given as a
mixture (2 nmol/kg body weight, each) subcutaneously to Wistar rats,
the same tissue distribution (liver : adipose tissue concentration
ratio of about 7) was found for both congeners at the maximal tissue
concentrations (Golor et al., 1993).
Following administration of 2,3,7-trihalogenated
dibenzo- p-dioxins (TrHDDs: Cl3DD, Br3DD, Cl2BrDD) to female Wistar rats
(single intravenous injections of mixtures containing 3, 10, or 50
µg/kg body weight for each congener), dose- and time-dependent changes
in tissue concentrations (liver, adipose tissue, thymus) were seen. It
is remarkable that concentrations of all three congeners were highest
in adipose tissue and lowest in liver (about two orders of magnitude
lower a few hours after the injection). Surprisingly high
concentrations were found in the thymus, almost an order of magnitude
higher than in liver (Golor et al., 1995).
6.2.1.2 Dibenzofurans
Disposition studies on dibenzofurans were conducted in rats using
[4,6-3H]-1,2,7,8-TeBDF (Kedderis et al., 1994). As with TBDD, the
major tissue depots included liver, adipose tissue, and skin (1 - 72 h
after single intravenous, oral, and dermal doses of 1 nmol/kg body
weight). Relatively high concentrations of 1,2,7,8-TeBDF were also
observed in the adrenal glands. Generally, concentrations in the liver
exceeded those in fat, and perirenal fat contained higher amounts than
epididymal fat. For example, 72 h following oral administration, there
was a liver : fat concentration ratio of about 2. A decline in liver
concentrations of 1,2,7,8-TeBDF was seen from 1 to 24 h after
intravenous treatment. This was related to metabolic elimination and
to a slight accumulation in adipose tissue.
Tissue levels of 1,2,7,8-TeBDF in lungs, small intestine, heart,
stomach, spleen, and thymus of Fischer 344 rats 1 h after intravenous
dosing were in the range of 0.2 - 1.3% of the dose/g of tissue. The
corresponding results for liver, kidneys, perirenal fat, adrenals, and
skin were 4.9, 0.5, 0.1, 5.1 and 0.1%, respectively. The tissue levels
of 1,2,7,8-TeBDF 72 h after oral administration were 0.11% (liver),
0.07% (perirenal fat), 0.10% (adrenals), and 0.03% (skin) of the
dose/g of tissue (Kedderis et al., 1994).
Seven days after a single subcutaneous dose of 2,3,4,7,8-PeBDF
(420 ng/kg body weight) was given to marmoset monkeys (n = 3), a
liver : fat concentration ratio of 12.2 was observed (Schulz et al.,
1993). A similar high deposition rate in liver was found with the
chlorinated analogue, whereas TCDD showed a liver : fat concentration
ratio of about 1.
6.2.2 Transfer to offspring
There are no experimental data available on transfer of
PBDDs/PBDFs to offspring.
However, transfer of various PCDDs/PCDFs via placenta and/or
through milk has been documented in rats, mice, goats, cows (WHO,
1989), marmoset monkeys (Hagenmaier et al., 1990b), and humans
(Schecter & Ryan, 1994; Schecter et al., 1994b, 1995, 1996a,b). The
bioavailability of PCDDs/PCDFs from breast milk was found to be high
(up to >95%) in human infants (Jödicke et al., 1992; McLachlan, 1993;
Pluim et al., 1993).
6.3 Metabolic transformation
6.3.1 Dibenzo-p-dioxins
Metabolism of PBDDs has been studied in rats given 2,3,7,8-TeBDD
orally or intravenously. No metabolites were found in liver (Kedderis
et al., 1991a), but metabolites were detected in bile from rats (male
Fischer 344 or female Sprague-Dawley) (Kedderis et al., 1991a; De
Jongh et al., 1992, 1993).
Three days after an intravenous dose of 1 nmol [3H]TBDD/kg body
weight, faeces of F344 rats (see also section 6.4) contained about 3%
of the administered dose as parent compound and about 14% of the dose
as metabolites (Kedderis et al., 1991a). About 80 - 90% of faecal and
biliary radioactivity excreted following intravenous dosing was
attributed to TBDD metabolites (Kedderis et al., 1991a,b).
Table 40. Comparison of tissue concentration and of liver: adipose tissue concentration ratio after a single subcutaneous
injection of 2,3,7,8-TeBDD or 2,3,7,8-TeCDD in ratsa,b
Dose Liver tissue Adipose tissue Liver: adipose tissue
(ng/kg body weight) concentration ratio
TCDD TBDD TCDD: TBDD TCDD TBDD TCDD: TBDD TCDD TBDD
(ng/g) (ng/g) concentration ratio (ng/g) (ng/g) concentration ratio
30 0.16 0.08 2.1 0.14 0.6 0.2 1.2 0.2
300 3.38 3.60 0.9 0.82 2.7 0.3 4.1 1.4
3000 27.9 20.5 1.4 3.7 12.5 0.3 7.7 1.9
Increase: 30-3000 174 x 256 x 26 x 21 x
a Adapted from Nagao et al. (1995/96).
b Female Wistar rats; n = 3 or 6; single subcutaneous doses; vehicle: toluene/DMSO (dimethyl sulfoxide); observation: day
7 after treatment.
If biliary excretion of TBDD-derived radioactivity is considered
as an indirect assessment of metabolism, TBDD is relatively slowly
metabolized. Within a 5-h period, 6.6% of a radiolabelled intravenous
dose (1 nmol [3H]TBDD/kg body weight) was excreted in bile of male
Fischer 344 rats (Kedderis et al., 1991b).
Studies with pretreated or untreated rats showed that TBDD and
TCDD did not induce their own metabolism in vivo (Kedderis et al.,
1991b, 1992a).
The main metabolites identified (three
hydroxybromodibenzo- p-dioxins and one dihydroxytetrabromoether) were
formed by aromatic hydroxylation and hydrolytic debromination and
suggest the metabolic pathway shown in Fig. 2 (De Jongh et al., 1993).
Similarities and differences in the metabolic pathways of TBDD and
TCDD are discussed by De Jongh et al. (1993).
In summary, several of the metabolites are similar.
Quantitatively, the dioxin ring-opening route seems to be favoured
somewhat more in TCDD (Poiger & Buser, 1984) than in TBDD metabolism;
qualitatively, the absence of a second methoxytribromodibenzodioxin
differs from TCDD metabolism.
6.3.2 Dibenzofurans
Information on the metabolism of [3H]1,2,7,8-TeBDF is available
from a study determining biliary elimination of
[3H]1,2,7,8-TeBDF-derived radioactivity (Kedderis et al., 1994).
Approximately 50% of the administered dose of [3H]1,2,7,8-TeBDF was
excreted in the bile of rats in 8 h. HPLC analysis confirmed the
presence of metabolites of 1,2,7,8-TeBDF in the bile (Kedderis et al.,
1994). If biliary excretion of PHDDs/PHDFs is used as an indirect
measure of metabolism, as is assumed by several authors (Kedderis et
al., 1991b; McKinley et al., 1993), this result is indicative of a
considerable metabolism of 1,2,7,8-TeBDF, in contrast to
2,3,7,8-TeBDD, which was more slowly metabolized (see above). The
differences between both congeners can be explained by their different
structures, 2,3,7,8- versus 1,2,7,8-substitution, the latter being
more susceptible to metabolism owing to the presence of two adjacent
unsubstituted carbon atoms in the 1,2-bromine ring (Kedderis et al.,
1994).
6.4 Elimination and excretion
Elimination of PHDDs/PHDFs occurs predominantly after conversion
to more polar metabolites in the liver and excretion of these
metabolites via the bile. There is apparently no or very little
excretion of unchanged congeners with the bile. However, for several
of the chlorinated congeners, secretion of the unchanged substances
into the intestinal lumen and subsequent excretion via the faeces have
been described (Abraham et al., 1989). It can be expected that the
lipophilic PBDDs/PBDFs may also be secreted into the intestinal lumen.
 6.4.1 Dibenzo-p-dioxins
Elimination was studied in rats with TBDD and with
1,2,3,7,8-PeBDD (faecal excretion was monitored in rats only with
TBDD). The animals were exposed to single doses of TBDD or
[1,6-3H]TBDD (see Table 41) by the oral (Diliberto et al., 1990a,b,
1993; Ivens et al., 1992), intravenous (Kedderis et al., 1991a),
intratracheal (Diliberto et al., 1991, 1993), or dermal (Jackson et
al., 1991; Diliberto et al., 1993) route or to subcutaneous doses of
1,2,3,7,8-PeBDD (Golor et al., 1993).
In all studies, the major route of elimination was through the
faeces (see Table 41). Eliminated radioactivity (after 2 - 3 days) in
faeces ranged from 2 to 42% of the administered dose of 1 nmol
[3H]TBDD/kg body weight and from 0.2 to 1% in urine. Unabsorbed
material and biliary excretion appeared to be the major source of
eliminated compound in faeces.
Based on the intravenous and oral studies (Table 41), the
cumulative elimination of radioactivity in faeces was dose-dependent.
Higher doses tended to result in higher elimination rates. The
dose-related differences following oral administration were a
consequence of differences in amounts eliminated on days 1 and 2 in
each group and are likely due to differences in percent absorption
(maybe due to the competing processes of uptake versus transit or to
limited aqueous solubility of TBDD at high doses). In the 56-day
intravenous study, a disproportionately greater elimination of
radioactivity at the high (100 nmol/kg body weight) versus the low (1
nmol/kg body weight) dose was observed beginning 3 weeks after
treatment (Kedderis et al., 1991a, 1992a).
Faecal elimination curves of the intravenous study were analysed
and found to be tri-exponential for the low dose (1 nmol/kg body
weight) and bi-exponential for the high dose (100 nmol/kg body
weight), with estimated initial and terminal half-lives of <1 and 18
days, respectively (see Table 45 in section 6.5.1).
From the oral study (Table 41), it was shown that 0.3% or less of
the administered dose was eliminated in the urine for all dose groups.
However, the relative amounts of urinary elimination were found to be
higher at the two low doses, consistent with enhanced absorption at
these dose levels (Diliberto et al., 1993).
Elimination of radioactivity in faeces over the first 3 days was
comparatively high after oral and intratracheal administration but was
lower after intravenous and dermal exposure to 1 nmol [3H]TBDD/kg body
weight (see Table 41). The faecal excretion of only 2% of the
administered dermal dose implies a very low elimination following
dermal exposure. However, based on the percentage of the dermally
absorbed dose, a value of 17% is obtained, which is comparable to the
faecal elimination of an equimolar intravenous dose (Kedderis et al.,
1991a; Diliberto et al., 1993).
Table 41. Elimination of 2,3,7,8-TeBDD in rats after single radiolabelled and unlabelled dosesa
Strain Route Dose Observation Test Eliminationb Reference
(sex) (vehicle) period (days) (% of administered dose)
nmol/kg µg/kg Faeces Urine
body weight body weight
Fischer 344 intravenous 1 0.5 1 R 8-10 n.sp. Kedderis et al.
(male) (water: 1 0.5 56 50 4.5 (1991a)
(n = 3-4) ethanol: 100 50 56 70 7.6
Emulphor(R)
= 3: 1: 1)
Fischer 344 oral 1 0.5 3 R 42 ± 2 0.3 Diliberto et al.
(male) (water: 10 5 3 39 ± 1 0.3 (1993)
(n = 3-4) ethanol: 100 50 3 58 ± 5 0.2
Emulphor(R) 500 250 3 72 ± 5 0.2
= 3: 1: 1)
Fischer 344 intratracheal 1 0.5 3 R 41 ± 2 1 Diliberto et al.
(male) (water: (1993)
(n = 3-4) ethanol:
Emulphor(R)
= 3 : 1 : 1)
Fischer 344 dermal 1 0.5 3 R 2 0.2 Diliberto et al.
(male) (acetone) (1993)
(n: 3-4)
Wistar ora] 200 100 2 U 20 (male) n.sp. Ivens et al.
(female, male) (arachis oil 17 (female) (1992)
(n: 5) with 5% 3-7 1 n.sp.
toluene)
a R = administration of [1,6-3H]-2,3,7,8-TeBDD (purity = >98%); elimination refers to eliminated radioactivity. U = administration of
unlabelled 2,3,7,8-TeBDD (purity = 98%); elimination refers to recovery of TBDD.
b n.sp. = not specified.
According to Kedderis et al. (1992b), the large percentage of the
dose excreted in urine and faeces after a single intravenous dose of
[3H]TBDD within the first few days could be attributed to a rapidly
excreted impurity in the radiolabelled TBDD, which is not detectable
by conventional radio-HPLC techniques. On the other hand, HPLC in
combination with preceding hexane extraction of faeces was reported to
distinguish successfully between parent TBDD and TBDD metabolites in
analysing TBDD-derived radioactivity in faeces (Kedderis et al.,
1991a; Diliberto et al., 1993). Results are compiled in Table 42.
Three days after oral, intratracheal, and intravenous administration
to rats of 1 nmol [3H]TBDD/kg body weight, approximately 22, 18, and
3%, respectively, of the administered dose could be attributed to the
parent compound extracted from faeces. In relation to the total
radioactivity measured in faeces over 3 days, the content of parent
TBDD ranged from 10 to 67% (see Table 43) after intravenous,
intratracheal, and oral exposure (Kedderis et al., 1991a; Diliberto et
al., 1993). Possible metabolites account for approximately 18 - 24%
(oral study) and 14% (intravenous study) of the administered dose
(Diliberto et al., 1993). Most of the parent TBDD was found in faeces
collected at days 1 and 2 (see Table 42 and Ivens et al., 1992 in
Table 41), which is consistent (at least for the oral and
intratracheal studies) with the assumption that a portion of the
material is not absorbed during passage through the gastrointestinal
tract. Excretion of absorbed TBDD is thought to be limited by the rate
of metabolism (Kedderis et al., 1991a).
6.4.2 Dibenzofurans
Elimination of 1,2,7,8-TeBDF in rats was primarily via biliary
excretion of metabolite(s) (see section 6.3) in the faeces (Kedderis
et al., 1994). Following intravenous administration of 1 nmol
[3H]-1,2,7,8-TeBDF/kg body weight, 39% of the dose was found in faeces
and 55% in intestinal contents after 24 h. After administration of
identical oral and dermal doses, 58 and 23%, respectively, of the
doses were excreted into the faeces within 72 h. Excretion in the
urine was only 2 - 3% of the intravenous, oral, or dermal dose
(Kedderis et al., 1994).
6.5 Retention and turnover
Data on retention and turnover are available for some PBDDs (tri
to penta), a tetraXDD, and some PBDFs (tetra, penta) in rats and for
2,3,7,8-TeBDD in humans.
6.5.1 Animal studies
Apparent elimination half-lives are complex for several
PHDDs/PHDFs, for various reasons:
* As is well known for TCDD (Abraham et al., 1988), elimination
from the main organs (liver and adipose tissue) does not proceed
in the expected semi-logarithmic form, but is at least biphasic.
Table 42. Percent administered dose of parent [3H]TBDD recovered in faeces of ratsa,b
Route Dose % administered dose excreted in faeces
(nmol/kg characterized as parent [3H]TBDDC
body Day 1 Day 2 Day 3 Cumulative
weight) after dosing after dosing after dosing (days 1-3)
Oral 1 11.7 ± 3.6 7.9 ± 2.1 2.5 ± 1.6 22.2 ± 2.1
10 6.9 ± 4.9 12.5 ± 3.8 2.0 ± 1.2 21.4 ± 1.8
100 16.1 ± 9.6 16.7 ± 9.0 2.6 ± 1.6 35.4 ± 1.8d
500 26.4 ± 11.2 18.3 ± 9.7 3.6 ± 3.4 48.3 ± 3.0d
Intratracheal 1 12.4 ± 1.7 4.6 ± 0.7 0.6 ± 0.02 17.6d
Intravenouse 1 1.6 ± 0.3 0.7 ± 0.3 0.5 ± 0.3 2.8d
a Adapted from Diliberto et al. (1993).
b Fischer 344 rats.
c Mean ± SD; n = 3 or 4; faecal extraction with hexane followed by HPLC
characterization of the extract.
d Statistically different from 1 nmol/kg oral close group (p < 0.05).
e Kedderis et al. (1991a).
Table 43. Contents of parent [3H]TBDD in faeces of ratsa
Route Dose % total radioactivity in faeces Reference
(nmol/kg characterized as parent
body [3H]TBDD (cumulative
weight) percentages days 1-3)
Oral 1 53b Diliberto et al.
10 55 (1993)
100 60
500 67
Intratracheal 1 43
Intravenous 1 and 100 10-20 Kedderis et al.
(1991a)
a Group size: n = 3-4.
b Percentage represents the amount of parent TBDD that was excreted via
faeces (days 1-3) as a result of unabsorbed TBDD and/or gastrointestinal
transluminal excretion of TBDD.
* The elimination half-lives from liver and adipose tissue (and
also from some tissues like the thymus) are often not identical.
This is due to metabolic conversion in and excretion from the
liver and a more or less rapid equilibrium in the adipose tissue
(probably dependent on lipid solubility).
* Tissue distribution is dose-dependent. The liver : adipose
tissue concentration ratio may change by orders of magnitude
when the dose is greatly increased (or decreases during
elimination).
* The role of elimination may be different in various rodent
strains, and it is dramatically different between various
species (e.g. orders of magnitude difference for TCDD between
rodents and humans). For this reason, the rate of cumulation to
a steady state (for a given dose and dose interval) differs by a
factor of about 100 for the rat and humans in the case of TCDD.
Although no comparative data (rat versus human) exist for the
elimination half-lives for PBDDs/PBDFs, similar differences must
be expected.
The relative body burden of radiolabelled TBDD in rats depends on
the route of exposure and on the dose administered (see Table 44),
reflecting differences in absorption. However, if per cent body
burdens are adjusted for the percent absorbed dose, they become more
similar after oral, intratracheal, dermal, and intravenous
administration of 1 nmol [3H]TBDD/kg body weight (see Table 44). More
than half of the body burden was found in the liver and adipose tissue
of rats (Diliberto et al., 1993; see also section 6.2).
Distribution patterns of TBDD between the major tissue depots
changed with dose and time (see also section 6.2), as seen in
single-dose studies (Kedderis et al., 1991a, 1993; Diliberto et al.
1993) and in short-term studies (Ivens et al., 1990). For example,
liver : adipose tissue TBDD concentration ratios declined from a
maximum of 30 to 0.2 during a period of 56 days after a single
intravenous administration of 1 nmol TBDD/kg body weight (Kedderis et
al., 1991). At this dose, radioactivity levels in the liver peaked by
7 h and then gradually declined, concomitantly with a slow
accumulation in adipose tissue, which reached the maximum
concentration by 14 days (Kedderis et al., 1990). Levels of
TBDD-derived radioactivity in blood declined rapidly to <2% of the
administered dose by day 1 after dosing (Kedderis et al., 1991a).
Liver concentrations of TBDD in rats, 1 - 78 days following a single
subcutaneous injection of 600 ng (1.2 nmol)/kg body weight, were
highest on day 3 after administration (Nagao et al., 1990c). In a
91-day oral study, the concentration of TBDD increased in the liver
and adipose tissue of rats during treatment. In the subsequent
recovery phase, a biphasic decline in TBDD concentrations in the liver
was observed, whereas the concentration of TBDD in adipose tissue
remained fairly constant, until a decrease began after a 30-day
recovery period (Ivens et al., 1990).
Table 44. Body burden of [3H]TBDD-derived radioactivity in ratsa 3 days after administration of a single dose
Route Dose % body burden Reference
(vehicle) (nmol/kg body weight)
Administered dose Absorbed dose
Oral 1 58 73 Diliberto et al. (1993)
(water: ethanol: 10 61 75
Emulphor(R) = 3: 1 : 1) 100 41 67
500 28 59
Intratracheal 1 59 ± 2 76 ± 2 Diliberto et al. (1993)
(water: ethanol:
Emulphor(R) = 3: 1: 1 )
Dermal 1 10 ± 1 82 ± 18 Diliberto et al. (1993)
(acetone)
Intravenous 1 82 ± 2 - Diliberto et al. (1993);
(water: ethanol:
Emulphor(R) = 3: 1: 1) Kedderis et al. (1991a)
a Fischer 344 rats, n = 3-4,
Estimated half-lives for TBDD and other PBDD/PBDF congeners are
compiled in Table 45.
Half-lives calculated for TBDD were as high as 58 days in adipose
tissue and skin. Shorter half-lives (up to 27 days) were found in
blood, muscle, liver, and whole body (Table 45). Compared with TCDD,
half-lives of TBDD were similar for liver and whole body but higher
(39 - 58 days versus 17 - 25 days) for adipose tissue (Kedderis et
al., 1991a and references therein; Nagao et al., 1995/96 and
references therein).
To compare the kinetics of three pairs of corresponding
poly-chlorinated and polybrominated PHDDs/PHDFs, a mixture of the six
substances (2,3,7,8-tetrahalogenated dibenzofuran [THDF],
2,3,4,7,8-pentahalogenated dibenzofuran [2,3,4,7,8-PeHDF], and
1,2,3,7,8-PeHDD) was given subcutaneously (single dose) to Wistar rats
(1 - 2 nmol/kg body weight each). Concentration changes in liver and
adipose tissue were monitored over a period of 95 days (Golor et al.,
1993). Kinetics of both the chlorinated and brominated 2,3,4,7,8 PeHDF
were similar in liver and also in adipose tissue, but levels were more
than 10 times higher in the liver. The rate of decline was also very
similar for the chlorinated and brominated 1,2,3,7,8-PeHDD in liver
and also in adipose tissue, but the profile of the kinetics was
different in the two tissues. Besides the level being more than one
order of magnitude higher in the liver, there was a rather steady
decline in the concentration in the hepatic tissue, while the
concentration in adipose tissue increased within the first month
(possibly because of redistribution phenomena) and then slowly
declined thereafter.
The most remarkable difference between the chlorinated and
brominated congeners was found in the case of THDF. The chlorinated
congener (TCDF) is known to be rapidly eliminated from liver as well
as adipose tissue in the rat. This was also found in these studies,
the rate of decline from the liver clearly being biphasic. In
contrast, the brominated congener (TBDF) was much more slowly
eliminated from both the liver and adipose tissue. While in the liver,
the overall elimination rate resembled that of the chlorinated
congener at the second elimination phase (about 2 weeks after
administration); the elimination rate in the adipose tissue was
comparatively slow (after an initial increase in the concentration
during the first 2 weeks after administration). Thus, the THDF
exhibited the larger kinetic difference between the chlorinated and
the brominated forms, the TBDF being much more persistent in the rat.
No comparative data are available for this pair of congeners in other
species.
6.5.2 Human studies
Some data on retention and turnover in humans are available for
2,3,7,8-TeBDD and 2,3,7,8-TeBDF.
Table 45. Biological half-lives of several PBDD/PBDF congeners in rats after single doses
Strain Congenera Route Dose Elimination from Calculated half-life Reference
(sex) (solvent) (observation period) (days) (kinetic phase)
Dibenzo-p-dioxins
Fischer 344 [3H]TBDD intravenous 1 nmol/kg whole body 0.7 (1st phase) Kedderis et al.
(female) (water: ethanol: (56 days) body weight 2.9 (2nd phase) (1991a)
(n = 3-4) Emulphor(R) 17.8 (3rd phase)
= 3: 1 : 1)
100 nmol/kg whole body 0.6 (1st phase)
body weight 17.8 (2nd phase)
1 nmol/kg liver 4.5 (1st phase)
body weight 16.5 (2nd phase)
adipose tissue 57.8
skin 2.5 (1st phase)
57.8 (2nd phase)
muscle 1.6 (1st phase)
26.7 (2nd phase)
blood 18.2
Wistar TBDD subcutaneous 60 ng/kg liver 13.3 Nagao et al.
(female) (toluene/DMSO (78 days) body weight (12.0-14.9)b (1995/96)
(n = 3-10) = 1+2; v/v) (1.2 nmol/kg adipose tissue 39.4
body weight) (26-82)b
Wistar 1,2,3,7, 8-PeBDD subcutaneous 2.2 nmol/kg liver 21 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (17-27)b (1993)
(n = n.sp. ) = 14-2; v/v) adipose tissue 55
(39-97)b
Table 45. (Continued)
Strain Congenera Route Dose Elimination from Calculated half-life Reference
(sex) (solvent) (observation period) (days) (kinetic phase)
Wistar 2,3,7-TrBDD intravenous 50 µg/kg liver 2 (3rd phase) Golor et al.
(female) (<5% toluene in 14 days body weightc (47 h) (1995)
(n = 3) peanut oil/0.9% (119 nmol/kg adipose tissue 2-3 (3rd phase)
NaCl, 1+9, v/v) body weight) (43 h)
thymus 3-4 (3rd phase)
(91 h)
2,3-Cl2,7-Br1DD intravenous 50 µg/kg liver 3-4 (3rd phase)
(<5% toluene in 14 days body weightc (72 h)
peanut oil/0.9% (151 nmol/kg adipose tissue 1.5 (3rd phase)
NaCl, 1+9, v/v) body weight) (36 h)
thymus 3-4 (3rd phase)
(92 h)
Dibenzofurans
Wistar TBDF subcutaneous 1.7 nmol/kg liver 20 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (17-25)b (1993)
(n = n.sp.) = 1+2; v/v) adipose tissue 30
(26-36)b
Wistar 2,3,4,7,8-PeBDF subcutaneous 1.1 nmol/kg liver 99 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (59-302)b (1993)
(n = n.sp.) = 1+2; v/v) adipose tissue 80
(49-220)b
Fischer 344 [3H]1,2,7,8-TeBDF intravenous 1 nmol/kg body 1 Kedderis et al.
(male) (water: ethanol: (24 h) body weight (1994)
(n = 3-4) Emulphor(R)
= 3: 1 : 1)
a n.sp. = not specified.
b 95% confidence interval in days.
c Given in a mixture together with other brominated and chlorinated PHDD/PHDF congeners.
The first report of a PBDD in human tissue is indicative of the
very long persistence of these compounds. Thirty-five years after
exposure, markedly elevated levels of TBDD (625 pg TBDD/g blood lipid)
were found in the blood of a chemist (see also section 5.3) who had
synthesized TBDD and TCDD in 1956 (Schecter & Ryan, 1990, 1991, 1992;
Schecter, 1992). It was not possible to calculate the actual half-life
because of the lack of earlier measurements.
Another study (Zober et al., 1992) provided data for estimating
the apparent half-lives of TBDD and TBDF. Employees of a chemical
plant who had PBDD/PBDF body burdens resulting from processing
brominated flame retardants (OBDE and DBDE) were monitored over a
3-year period from 1989 to 1991 (see also section 5.3). Based on data
from three subjects, the following half-lives were calculated:
2,3,7,8-TeBDD: 2.9 - 10.8 years (mean: 5.9 years)
2,3,7,8-TeBDF: 1.1 - 1.9 years (mean: 1.5 years)
These half-lives are much longer than those reported in rats
(section 6.5.1), but they are consistent with findings on the
chlorinated analogues. Estimated half-lives of TCDD in humans ranged
between 5 and 11 years (Poiger & Schlatter, 1986; Pirkle et al., 1989;
Wolfe et al., 1994).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Dibenzo-p-dioxins
The toxicity of PBDDs and PXDDs was studied in rats and mice
after single oral (Yang et al., 1983; Ivens-Kohl et al., 1990; Ivens
et al., 1992), intraperitoneal (Mason et al., 1987a,b), or
subcutaneous (Nagao et al., 1990a) doses.
Single oral doses of 10, 33, 100, or 300 µg 2,3,7,8-TeBDD/kg body
weight given to Wistar rats (5/sex per group) caused severe decreases
in body weight gain (males) or weight loss (females) and deaths (see
Table 46) at 100 and 300 µg/kg body weight during the 28 days of
observation. At 33 µg/kg body weight, rats of both sexes showed slight
decreases in body weight; at 10 µg/kg body weight, only the weight of
females was slightly reduced. Emaciation, piloerection, and poor
general health were seen in females of the 100 µg/kg body weight dose
group and in most animals of the highest dose group (Ivens et al.,
1992). Food and water intake were reduced in rats administered 100 µg
TBDD/kg body weight (Ivens-Kohl et al., 1990). Absolute and relative
thymus weights were decreased with increasing dose from 10 to 100
µg/kg body weight. At the highest dose of 300 µg/kg body weight, the
thymus was not detectable in either male or female rats. The
liver-to-body-weight ratio was increased in all dose groups (males:
10 - 25%, females: 4 - 15%). A dose-dependent increase in the
testes-to-body-weight ratio began at 33 µg/kg body weight. Nearly all
animals dying before term showed signs of haemorrhage in the
gastrointestinal tract (Ivens et al., 1992).
Histological alterations were consistently seen in the thymus of
TBDD-treated rats. Early thymic atrophy (characterized by the
phagocytosis of lymphocytes by histiocytes) was observed at doses of
10 and 33 µg TBDD/kg body weight, and severe atrophy was seen at 100
µg TBDD/kg body weight. Lymphocytic depletion was also observed in the
spleen at and above 33 µg TBDD/kg body weight and in Peyer's patches
of the ileum (folliculi lymphatici aggregati) at 300 µg/kg body
weight. Apart from lymphatic tissue, the organ most affected by TBDD
treatment was the liver. First signs of dose-dependent hepatotoxicity
such as cytoplasmic vacuolation and rarefaction and cellular
hypertrophy were seen after a single administration of 10 µg TBDD/kg
body weight. The cellular hypertrophy was accompanied by swelling of
the nuclei, accentuated nucleoli, and cytoplasmic transformations.
Pre-peliotic foci in the liver were seen at doses of 100 and 300 µg
TBDD/kg body weight (Ivens et al., 1992).
Haematological investigations (in rats administered single oral
doses of 10 - 300 µg TBDD/kg body weight) revealed a dose-dependent
but marginal decline in haemoglobin content and cell number
(thrombocytes, leukocytes, erythrocytes). Electrophoresis of serum
proteins showed small dose-dependent changes in alpha- and
Table 46. Mortality associated with oral administration of PBDDs/PBDFs
PBDD/PBDF Species Sex (number Dose Details Observation Mortality Time to deathb Reference
(carrier) (strain) per group) (µg/kg body perioda (number dead/ (days)
weight) number treated)
2,3,7,8-TeBDD rat female, male 10, 33 single dose 28 days no mortality - Ivens et al.
(arachis oil (Wistar) (5) 100 single dose 28 days female: 3/5 n.r. (1992)
with 0.5-5% 300 single dose 28 days female: 5/5 11-19
toluene) male: 3/5 16-22
2,3,7,8-TeBDD rat female, male 0.01 dally doses 90 days female: 1/10 n.r. Löser &
(arachis oil) (Wistar) (10) (for 90 days) Ivens
0.10 daily doses 90 days female: 1/10 n.r. (1989);
(for 90 days) Ivens et al.
1 daily doses 90 days female: 1/10 n.r. (1993)
(for 90 days) male: 2/10
3 daily doses 90 days female, male: n.r.
(for 90 days) 5/10
10 daily doses 90 days female, male: up to 35 days
(for 90 days) 10/10 (dead or
moribund)
2,3,7,8-TeBDF rat female, male 1, 10, 50 daily doses 4 weeks no mortality - Hardy et al.
(corn oil/ (Sprague- (5) (5 days/week (1990)
acetone) Dawley) for 4 weeks)
150 daily doses 4 weeks female:4/5 18-24
(5 days/week male: 3/5 (dead
for 4 weeks) or moribund)
500 daily doses 4 weeks female, male: 17-25
(5 days/week 5/5 (dead or
for 4 weeks) moribund)
2,3,7,8-TeBDF guinea-pig male (6) 0.47 single dose 30 days no mortality - Moore et al.
(corn oil) (Hartley) 1.58 single dose 30 days no mortality - (1979)
4.74 single dose 30 days 1/6 26
15.84 single dose 30 days 6/6 10-13
a After first dosing.
b n.r. = not recorded.
beta-globulins. Thyroid hormone concentrations were reduced
dose-dependently in serum (Ivens et al., 1992) (for details, see
section 7.8.4).
A series of several PBDDs and PXDDs (tetra through penta) given
intraperitoneally to immature male Wistar rats (n = 4) caused body
weight losses 14 days after injection (Mason et al., 1987a,b). The
most toxic compounds tested were 2,3,7,8-TeBDD, 2-Br1-3,7,8-Cl3-DD, and
TBCDD, which are substituted only in the four lateral positions. The
latter analogue exhibited the highest activity in this series (Mason
et al., 1987a). The relative potencies of PBDDs examined followed the
order 2,3,7,8- > 1,2,3,7,8- > 1,2,4,7,8- > 1,3,7,8- (Mason et al.,
1987a,b). There were slight differences in the ED50 values for body
weight loss (on a molar basis) between TCDD (Mason et al., 1986) and
TBDD (Mason et al., 1987a,b).
Further effects following a single dose of PBDD/PBDF or PXDD/PXDF
congeners are described in sections 7.8.1 (thymic atrophy) and 7.8.6
(hepatic enzyme induction).
Significant increases in relative liver weights were observed in
male Sprague-Dawley rats given single oral doses of 25 µg
2,3,7-TrBDD/kg body weight (Yang et al., 1983) and in pregnant mice
administered single subcutaneous doses of 5 - 90 µg TBDD/kg body
weight (Nagao et al., 1990a).
7.1.2 Dibenzofurans
2,3,7,8-TeBDF given to six guinea-pigs in single oral doses of
0.47, 1.58, 4.74, or 15.84 µg/kg body weight (equivalent on a molar
basis to 0.3 - 10 µg/kg 2,3,7,8-TeCDF) was lethal at 4.74 and 15.84
µg/kg body weight (see also Table 46). No deaths or body weight
effects were seen in the lower dose groups. Animals found dead showed
at necropsy a marked reduction in the size of the thymus, lack of body
fat, and reduction of muscle mass. Histopathological findings in
tissues of lethally intoxicated guinea-pigs included a loss of
lymphoid cells in the thymic cortex and hyperplasia of epithelial
cells in the renal pelvis, ureter, and urinary bladder. In addition,
hypocellularity of bone marrow and seminiferous tubules, lymphoid
elements in spleen, Peyer's patches, and adrenal haemorrhage were
seen. In contrast to other rodents and rabbits, whose primary target
organ after dibenzo- p-dioxin/dibenzofuran exposure is the liver, there
was a lack of liver damage in the guinea-pigs. Mild thymus lymphoid
hypoplasia was the only histological alteration in animals of the
highest non-lethal dose group that survived the 30-day observation
period (Moore et al., 1979). Altogether, the pattern of lesions was
similar to those described for guinea-pigs exposed to PCDDs/PCDFs
(Moore et al., 1979; Kociba & Schwetz, 1982; WHO, 1989).
7.1.3 Remarks on the lethality of PBDDs/PBDFs
It is characteristic for TCDD and related compounds to have a
significant latency period between the time of exposure and the time
of death. Further, these compounds are relatively persistent and
produce similar effects (e.g. wasting syndrome) regardless of single
or short-term exposure as a result of prolonged internal exposure
(e.g. McConnell, 1989; WHO, 1989, 1994a). For these reasons, the few
mortality data available for PBDDs/PBDFs from single and multiple
dosing studies are compiled in this section.
All information refers to 2,3,7,8-TeBDD (Loeser & Ivens, 1989;
Pinkerton et al., 1989; Ivens-Kohl et al., 1990; Ivens et al., 1992,
1993) and to 2,3,7,8-TeBDF (Moore et al., 1979; Pinkerton et al.,
1989; Hardy et al., 1990).
The oral LD50 of TBDD in Wistar rats was about 100 µg/kg body
weight for females and 300 µg/kg body weight for males (Ivens-Kohl et
al., 1990). After single oral exposure of Sprague-Dawley rats to TBDD
and TBDF, the LD50 was >500 µg/kg body weight (Brominated Flame
Retardants Information Panel, 1987, cited in Pinkerton et al., 1989
and in Hardy et al., 1990, the latter specifying >500 µg/kg for TBDD
and >5000 µg/kg for TBDF, n = n.sp.). Additional data on mortality
are summarized in Table 46.
Although differences in the dosage regimen make a direct
comparison impossible, it can be seen from Table 46 that guinea-pigs
are more sensitive than rats to the lethal action of TBDF, which is
consistent with findings observed with TCDD/TCDF (WHO, 1989).
Generally, there are large differences in sensitivity between species
and strains. For example, a more than 500-fold difference in acute LD50
values between the most TCDD-susceptible (Long-Evans) and the most
TCDD-resistant (Han/Wistar) rat strains was reported for TCDD (WHO,
1989; Pohjanvirta et al., 1993, 1994). The oral LD50 was reported to
range from 22 to >3000 µg TCDD/kg body weight in different rat
strains (WHO, 1989).
In a study in guinea-pigs (Moore et al., 1979), equimolar doses
of TCDF and TBDF resulted in comparable mortality rates (see Table
46). None (0/6) of the animals died following oral exposure to 1 µg
TCDF/kg body weight (equivalent to 1.58 µg TBDF/kg body weight),
whereas all six animals died at 10 µg TCDF/kg body weight (equivalent
to 15.84 µg TBDF/kg body weight). Similarly, the mean time to death
was about 12 days for both chemicals.
7.2 Short-term exposure
7.2.1 Dibenzo-p-dioxins
In a 3-month toxicity study (Ivens-Kohl et al., 1989; Löser &
Ivens, 1989; Ivens et al., 1993), 2,3,7,8-TeBDD was administered daily
by gavage to Wistar rats (10/sex per group) at doses of 0.01, 0.1, 1,
3, or 10 µg/kg body weight. No overt signs of toxicity were seen at
0.01 and 0.1 µg/kg body weight per day. Doses of 3 and 10 µg/kg body
weight per day caused a high mortality (see Table 46) and wasting
syndrome. Mean body weight gain, feed intake, and water intake were
reduced dose-dependently from 1 µg/kg body weight per day.
Changes in haemoglobin content, packed cell volume, and number of
thrombocytes were seen mainly in rats given the 1 and 3 µg TBDD/kg
body weight per day doses. The prothrombin time was markedly prolonged
at 3 µg/kg body weight per day. Clinical chemistry showed slight
increases in plasma alkaline phosphatase, aspartate aminotransferase,
and total blood bilirubin in males and females receiving a dose of 1
µg/kg body weight per day. These changes were significant at 3 µg/kg
body weight per day. Alanine aminotransferase was increased in females
only at 3 µg/kg body weight per day. There was also a decrease in
serum triglyceride levels, mainly at 1 and 3 µg/kg body weight per
day. Dose-dependent changes in thyroid hormone concentrations in serum
were observed in male and female rats. Triiodothyronine (T3) levels
were increased and thyroxin (T4) levels were decreased at doses of 0.1
µg/kg body weight per day and higher. The effects at 0.1 µg/kg body
weight per day, however, were considered to be marginal (see also
section 7.8.4). Activities of microsomal enzymes were dose-dependently
elevated (see also section 7.8.6). Protein excretion in urine
increased in males and females at doses higher than 3 µg/kg body
weight per day.
Changes in relative organ weights (increase in liver, lung,
kidney; reduction in thymus) were generally observed at doses of 1
µg/kg body weight per day and higher. Relative liver weights were
significantly increased at doses of 0.1 µg/kg body weight per day and
higher; relative thymus weights were significantly decreased at doses
of 0.01 µg/kg body weight per day and higher. Histopathological
examination revealed dose-dependent changes, mainly beginning at the 1
µg/kg body weight per day dose. These included severe atrophy of
lymphatic tissue in thymus and spleen and liver damage described as
peliosis hepatis parenchymatosa (irregular-shaped cavernous and
blood-filled spaces in the liver, lack of epithelial lining, blood
cysts in the sinusoidal lumen and in Disse's spaces, etc.) (Bannasch
et al., 1985). Spermatogenesis in the testes was adversely affected,
and defective or necrotic spermatocytes were found in the epididymis
(Ivens et al., 1993; see also section 7.5).
The NOAEL in this study was considered to be 0.01 µg TBDD/kg body
weight per day.
Compared with TCDD, TBDD elicits a similar spectrum of toxic
effects following subchronic exposure but appears to be less active
than TCDD. A subchronic NOAEL for TCDD (dosing for only 5 days/ week)
in rats was reported to be 0.01 µg/kg body weight per day (Kociba et
al., 1976). However, peliosis hepatis was not reported to occur after
treatment with TCDD (Ivens et al., 1993 and references therein).
7.2.2 Dibenzofurans
2,3,7,8-TeBDF was administered to Sprague-Dawley rats (5/sex per
group) at daily oral doses of 1, 10, 50, 150, or 500 µg/kg body
weight, 5 days/week for 4 weeks (Fulfs, 1989; Hardy et al., 1990).
Most animals in the 150 and 500 µg/kg body weight per day dose groups
died or were in a moribund condition between study days 17 and 24. At
150 µg/kg body weight per day, 3 of 10 rats survived through day 28
(see Table 46). Animals from the two highest dose groups had yellowish
urine beginning around day 15. Group mean body weight was depressed in
a time- and dose-dependent manner. The mean relative thymus weights of
males at 150 µg/kg body weight per day and of females at 10 and 50
µg/kg body weight per day were decreased. No significant changes in
relative mean liver, adrenal, or spleen weights were detected. In this
study, treatment-related histopathological alterations were noted in
the liver and thymus of animals in the 50 µg/kg body weight per day
dose group and, to a lesser extent, in the 10 µg/kg body weight per
day dose group. Liver changes consisted of panlobular hypertrophy of
the hepatocytes with associated hepatocyte vacuolation and focal
necrosis. Thymic atrophy consisting of overall depletion of the
lymphoid elements was present in all 50 µg/kg body weight per day rats
from which thymus was available and in most animals from the 10 µg/kg
body weight per day dose group. No treatment-related alterations were
observed in the 1 µg/kg body weight per day group (Hardy et al.,
1990), which can be considered as the NOAEL for this study.
The results do not indicate a lower potency of TBDF compared with
TBDD, as the studies differed in their experimental design (dosing for
4 weeks, 5 days/week versus 13 weeks, 7 days/week; Wistar versus
Sprague-Dawley rats; different animal numbers/dose group).
7.3 Long-term exposure
No long-term exposure studies with PBDDs/PBDFs were available.
7.4 Skin and eye irritation, sensitization, dermal lesions, and
acne
A common feature of toxicity of dioxin-like compounds such as
PCDDs/PCDFs, PCBs, and PBBs is their hyperkeratotic activity in humans
and some animal species (WHO, 1989, 1993, 1994a).
A standard test method for acnegenic activity, the rabbit ear
assay first described by Adams et al. (1941), was applied to
2,3,7,8-TeBDD and 2,3,7,8-TeBDF (Pinkerton et al., 1989). Both
congeners produced hyperkeratosis at a total dose of 100 µg/rabbit,
but not at 10 µg/rabbit (probably repeated application over a 4-week
period). The solvent used was not specified. Under the same
conditions, combustion residues (soot/char) from a HIPS/DBDE/Sb2O3
sample (for the PBDF content, see Table 11) were found to have no
acnegenic activity (Pinkerton et al., 1989).
In the case of TCDD, the minimum dose inducing hyperkeratosis
after a single administration ranged from 1 µg TCDD/ear to 160 µg
TCDD/ear, depending on the vehicle used (Poiger & Schlatter, 1980).
During a 4-week test with TCDD, no effect was observed at a total dose
of 8 ng/rabbit, and dose-dependent responses were obtained at
0.08 - 800 µg/rabbit (Schwetz et al., 1973).
7.5 Reproductive and developmental toxicity
7.5.1 Reproductive toxicity
A dose-dependent increase in testes-to-body-weight ratio was seen
28 days after oral administration of single doses of 2,3,7,8-TeBDD (0,
10, 33, 100, or 300 µg/kg body weight; solvent: arachis oil with 5%
toluene) to male Wistar rats (n = 5). This effect was seen from 33
µg/kg body weight onwards. Body weight gain was reduced
dose-dependently (marginally at 33 µg/kg body weight), but there was
no loss of body weight (Ivens et al., 1992).
Decreased spermatogenic activity in the testes and defective or
necrotic spermatocytes in the epididymis were found in Wistar rats
(n = 10) after daily oral administration of TBDD (in arachis oil) at 3
or 10 µg/kg body weight per day, 7 days/week for 13 weeks. Severe
effects were observed at 10 µg/kg body weight per day, and moderate
effects at 1 µg/kg body weight per day. The NOEL was 0.1 µg/kg body
weight per day (Ivens-Kohl et al., 1989; Ivens et al., 1993).
Adverse effects on the male reproductive system (e.g. reduction
in number, size, and organelle content of Leydig cells in adult rat
testes) were observed following single intraperitoneal injections of
12.5 - 50 µg TCDD/kg body weight (Johnson et al., 1994). However, at
perinatal exposure, a single oral dose as low as 64 ng TCDD/kg body
weight (the lowest maternal dose tested) given to mothers at day 15 of
gestation was sufficient to reduce sperm production in the male
offspring (Mably et al., 1992). Reduced sperm numbers were also
observed in offspring of pregnant rats administered 1 µg TCDD/kg body
weight on gestation day 8 or 15 and in offspring of Syrian hamsters
dosed with 2 µg/kg body weight on gestation day 11 (Gray et al.,
1995).
7.5.2 Developmental toxicity
Several PBDDs/PBDFs were found to be inducers of cleft palate and
hydronephrosis in mice. These effects occurred at doses (see Table 47)
that produce no or only marginal general maternal toxicity and no
fetal mortality (Nagao et al., 1990a,d; Birnbaum et al., 1991).
Single subcutaneous injections of TBDD or TCDD (5 - 90 µg/kg body
weight) were administered to NMRI mice on day 9 of pregnancy. Maternal
and fetal toxicity were assessed on day 18 (Nagao et al., 1990a,d).
TBDD and TCDD caused significant dose-related increases in the
incidence of cleft palate in the total number of viable fetuses and in
the number of litters with cleft palate. On a molar basis, the potency
of TBDD for cleft palate induction relative to that of TCDD was found
to be 0.6: ED50 values were 61.7 µg/kg body weight per day
(corresponding to 0.124 µmol/kg body weight per day) for TBDD and 24
µg/kg body weight per day (corresponding to 0.075 µmol/kg body weight
per day) for TCDD. Both TBDD and TCDD at the doses given increased
maternal liver-to-body-weight ratios. There were no significant
effects on maternal weight gain or the number of viable fetuses per
litter.
In another study (Birnbaum et al., 1991), the teratogenic effects
of TBDD and three PBDFs (TBDF, 1,2,3,7,8-PeBDF, and 2,3,4,7,8-PeBDF)
were examined in C57BL/6N mice. Pregnant dams were treated on
gestation day 10 with single oral doses of each congener and
sacrificed on gestation day 18. Doses ranged from 0 to 192 µg/kg body
weight for TBDD and from 0 to 4000 µg/kg body weight for the three
dibenzofurans. All compounds produced hydronephrosis at doses below
that at which cleft palate occurred (see also Table 47). The LOELs
(µg/kg body weight) for hydronephrosis and cleft palate, respectively,
were as follows: TBDD: 3 and 48; TBDF: 25 and 200; 1,2,3,7,8-PeBDF:
500 and 3000 - 4000; and 2,3,4,7,8-PeBDF: 400 and 2400. Embryo/fetal
mortality was significantly increased at 500 µg TBDF/kg body weight
and higher doses. At 3000 µg TBDF/kg body weight, the few survivors
were oedematous. After exposure to TBDD, the number of live fetuses
showed a "decreasing trend," which was not significant. Dose-related
increases in fetal weights were observed with TBDD and TBDF. However,
the increases were significant only for TBDF at doses of 500 µg/kg
body weight and higher. Maternal liver weight increased at all dose
levels examined for all four compounds. Maternal weight gain was
elevated at the highest dose of TBDF (4000 µg/kg body weight), and
this increase was due to subcutaneous oedema.
Birnbaum et al. (1991) compared the relative toxicities of the
brominated congeners with those of the chlorinated ones. Based on the
approximate ED50 values, TBDD appeared to be almost half as potent as
TCDD in the induction of hydronephrosis in offspring of treated dams
(4 µg/kg body weight versus 9 µg/kg body weight; see also Table 47).
However, compared on a molar basis, TBDD and TCDD were almost
equipotent. A survey of the relative potencies (on a weight basis) of
PBDD/PCDD and PBDF/PCDF congeners for the induction of cleft palate is
given in Table 48. It can be seen that bromination decreased the
activity of TBDD relative to TCDD but increased the potency of TBDF
relative to TCDF. The pentaBDFs tested, however, were slightly less
potent than the pentaCDFs.
7.6 Mutagenicity and related end-points
No information was found on the mutagenicity of PBDDs/PBDFs or on
related end-points.
Whereas there is limited or conflicting evidence demonstrating
the positive mutagenic potential of PCDDs (WHO, 1989), co-mutagenic or
co-recombinogenic effects of PCDDs have been demonstrated in vivo in
the mouse spot test (Fahrig, 1993).
Table 47. Development at toxicity of PBDDs/PBDFs
PBDDs/PBDFs Species Route Dose (µg/kg Effects Remarks Reference
(vehicle) (strain) (dosing body weight)
(n)a regimen)b
2,3,7,8-TeBDD mouse subcutaneous 0-90 from 5 µg/kg body weight: no effect on the Nagao et al.
(toluene/DMSO (NMRI) (single dose (5, 10, 30, F0: increase in relative number of viable (1990a,b)
= 1+2, v/v) (n.sp.) on gd 9) 50, 90) liver weight, fetuses per
F1: cleft palate, litter, fetal
ED50: 62 µg/kg body weight weight, or fetal
(0.123 µmol/kg body weight) deaths
2,3,7,8-TeBDD mouse oral 0-192 from 3 µg/kg body weight: trend for a Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (3, 6, 12, F0: increase in relative decrease in (1991)
(11-20) on gd 10) 24, 48, 96, liver weight, number of live
192) fetuses and an
F1: hydronephrosis, increase in fetal body
ED50:9 µg/kg body weight weight with increasing
(0.018 µmol/kg body weight) dose
from 48 µg/kg body weight:
F1 cleft palate,
ED50: 65 µg/kg body weight
(0.13 µmol/kg body weight)
2,3,7,8-TeBDF mouse oral 0-4000 from 25 µg/kg body weight: Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (25, 50,100, F0: increase in relative (1991)
(7-22) on gd 10) 200, 250, liver weight,
500, 1000, F1: hydronephrosis,
3000,4000) ED50: approx. 12 µg/kg body
weight (0.024 µmol/kg
body weight) from 200 µg/kg
body weight: F1: cleft palate,
ED50:154 µg/kg body weight
(0.31 µmol/kg body weight)
from 500 µg/kg body weight:
F1 increase in fetal
mortality and live fetal body
weight 4000 µg/kg body weight:
F0: subcutaneous oedema
1,2,3,7,8-PeBDF mouse oral 0-4000 from 250 µg/kg body weight: no increase in Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (250, 500, F0: increase in relative fetal mortality (1991)
(5-11) on gd 10) 1000, 2000, liver weight or fetal body weight
3000, 4000) from 500 µg/kg body weight:
F1: hydronephrosis,
ED50: approx. 340 µg/kg body
weight from 3000c µg/kg body
weight: F1: cleft palate,
ED50:4088 µg/kg body weight
2,3,4,7,8-PeBDF mouse oral 0-4000 from 25 µg/kg body weight: no effect on fetal Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (25, 50,100, F0: increase in relative liver mortality or fetal body (1991)
(9-16) on gd 10) 200, 400, weight weight
800, 1600, from 400 µg/kg body weight:
2400, 4000) F1: hydronephrosis,
ED50: approx. 437 µg/kg body
weight from 2400 µg/kg body
weight: F1: cleft palate,
ED50: 3024 µg/kg body weight
a Number of litters; n.sp. = not specified.
b gd= gestation day (first day of gestation designated day zero).
c First significant at 3000 µg/kg body weight, statistically (p < 0.01) significant at 4000 µg/kg body weight.
Table 48. Relative potencies of PBDD/PCDD and PBDF/PCDF congeners relative
to TCDD for the induction of cleft palate in micea
Congener Relative potencies for cleft palateb
H=Br H=Cl
Dibenzo-p-dioxins
2,3,7,8-TeHDD 0.24 1
Dibenzofurans
2,3,7,8.TeHDF 0.10 0.05
1,2,3,7,8-PeHDF 0.004 0.03
2,3,4,7,8-PeHDF 0.005 0.09
a Adapted from Birnbaum et al. (1991).
b Derived from ED50 values: on a weight basis.
The cell-transforming potential of TBDD has been demonstrated in
a host-mediated in vivo/in vitro assay with peritoneal murine
macro-phages (Massa et al., 1990). NMRI mice were intraperitoneally
administered 0.39 nmol TBDD (corresponding to 195 ng) or TCDD
(corresponding to 125 ng) per mouse. Isolation of resident macrophages
4 days later, cultivation in soft agar for 5 - 6 days, and evaluation
of the clones indicated that the transforming capacity of TBDD was
seven times less than that of TCDD (Massa et al., 1991).
7.7 Carcinogenicity
7.7.1 Short-term studies
A permanent cell line was established from peritoneal macrophages
of mice treated with TBDD. These cells were tested for their
tumorigenicity in athymic nude (nu/nu) mice. Animals given
subcutaneous injections of these cells (1 x 106 cells) developed
tumours at the injection site 3 weeks later (Massa et al., 1991,
1992a,b).
7.7.2 Long-term studies
PBDDs/PBDFs have not been tested for carcinogenicity in long-term
studies.
TCDD was shown to be a multisite carcinogen in both sexes of rats
and mice at doses below the maximum tolerated dose (WHO, 1989).
7.8 Other special studies
7.8.1 Immunotoxicity
Thymus atrophy and other signs of immunotoxicity were found to be
the main characteristic toxic effects (besides body weight loss or
decrease in the rate of weight gain) after exposure to TCDD and were
observed in almost all laboratory animals (Vos & Luster, 1989; WHO,
1989). The limited data available for PBDDs/PBDFs from studies with
rats, guinea-pigs, and monkeys confirmed the expected immunotoxic
potential. Parameters examined were influences on lymphoid tissues,
effects on serum protein levels or other haematological parameters,
and alterations of certain lymphocyte subpopulations in peripheral
blood. Effects on immunotoxicity after perinatal exposure to
PBDDs/PBDFs have not been investigated.
7.8.1.1 Dibenzo-p-dioxins
Dose-dependent decreases in thymus weights and atrophy of thymus
and other lymphatic tissues were observed in rats after single
exposures to 2,3,7,8-TeBDD (Mason et al., 1987a,b; Ivens et al., 1992;
see also section 7.1) and to a series of other PBDD or PXDD congeners
(Mason et al., 1987a,b) and after subchronic exposure to 2,3,7,8-TeBDD
(Ivens-Kohl et al., 1989; Löser & Ivens, 1989; Ivens et al., 1993; see
also section 7.2). Effects were found after single doses of 10 µg/kg
body weight or higher and after daily doses of 1 µg/kg body weight and
higher for 3 months (Ivens-Kohl et al., 1989; Löser & Ivens, 1989;
Ivens et al., 1992, 1993). The potency of TBDD for causing thymic
atrophy was comparable to that of TCDD (Mason et al., 1987b).
1,3,7,8-TeBDD, 1,2,3,7,8-PeBDD, and 1,2,4,7,8-PeBDD were less active
(Mason et al., 1987b). The mixed 2-Br1-3,7,8-Cl3DD was as active as
TBDD and TCDD; the mixed 2,3-Br2-7,8-Cl2DD was the most toxic analogue
in this series (Mason et al., 1987a).
Some routine haematological parameters, including serum protein
electrophoresis (reduction in number of thrombocytes, prolongation of
prothrombin time, slight anisocytosis, poikilocytosis, slight
reduction in gamma globulins, etc.), examined in the subchronic (3
months) rat study (doses applied: 0.01 - 10 µg/kg body weight) might
suggest an impact on cellular and, possibly, humoral immunity
beginning at daily doses of 1 µg TBDD/kg body weight (Ivens-Kohl et
al., 1989).
It has been shown that very low doses of PBDDs and PCDDs affect
the immune system of the marmoset monkey (Neubert R. et al., 1990,
1991, 1992, 1993; Neubert, 1991, 1993a). After dibenzo- p-dioxin
exposure, alterations were seen in lymphocytes from peripheral blood
of mature marmosets. The lymphocyte subpopulations showing the most
pronounced effects were the "helper-inducer" or "memory" T cells
(CD4+CDw29+ in the marmoset, probably corresponding to the
CD4+CD45RO+CDw29+ cells in humans) and certain B cell subsets (e.g.
CD20+) (Neubert, 1993a; Neubert et al., 1993). Several weeks after a
single subcutaneous dose of 30 ng TBDD/kg body weight (corresponding
to 60 pmol TBDD/kg body weight), there was a significant decrease in
the percentage and absolute number of the T helper-inducer
subpopulation (CD4+CDw29+) in venous blood of treated marmosets (n =
3). This effect on cell-mediated immunity is pronounced in young
animals (Vos, 1993). In addition, a significant decrease in the number
of B cells (CD20+) was observed (Neubert, 1993a). This is considered
to be less dependent on the age of animals (Vos, 1993). Injection of 3
ng TBDD/kg body weight (corresponding to 6 pmol TBDD/kg body weight)
did not induce any changes (Neubert, 1993a; Neubert et al., 1993).
With TCDD, a clear dose-response relationship was found at doses
of 10 ng/kg body weight and higher, and a questionable response
(confined to single individuals) was seen at 3 ng/kg body weight. No
effect was detectable with single doses of 1 ng TCDD/kg body weight or
lower. It was concluded that, on a molar basis, the potencies of TBDD
and TCDD may be similar in this experimental approach (Neubert, 1993a;
Neubert et al., 1993).
One mixed halogenated congener, namely TBCDD, was also
investigated. No changes in the subpopulations tested were found
following a single subcutaneous dose of 3 ng/kg body weight. Treatment
with 30 TBCDD/kg body weight decreased the percentage of CD24+CDw29+
cells and of CD20+ lymphocytes. However, no significant effect was
seen in the small number of marmosets used (n = 3) on the absolute
number of cells per µl of blood. From these data, it appeared that
this congener may be less potent than TCDD and TBDD (Neubert, 1993a;
Neubert et al., 1993).
7.8.1.2 Dibenzofurans
A marked reduction in the size of the thymus, loss of lymphoid
cells in the thymic cortex, hypocellularity of bone marrow, lymphoid
elements in spleen, and Peyer's patches were seen in guinea-pigs after
single oral doses of TBDF (4.7 - 15.8 µg/kg body weight) that also
induced mortality. At a non-lethal dose of 1.6 µg/kg body weight, only
mild evidence of thymus lymphoid hypoplasia was noted after the 30-day
observation period (Moore et al., 1979; see also section 7.1).
Short-term exposure of Sprague-Dawley rats to daily oral doses of
1-500 µg TBDF/kg body weight (5 days/week for 4 weeks) caused
dose-dependent reductions in thymus weights and thymic atrophy at the
10 µg/kg body weight per day and higher doses (Hardy et al., 1990; see
also section 7.2).
7.8.2 Effects on intermediary metabolism: Porphyrin effects
Hepatic porphyrin accumulation was studied after subchronic
dosing (5 days/week for 13 weeks) of female B6C3F1 mice by oral gavage
with individual congeners of PBDDs/PCDDs, including 2,3,7,8-TeBDD and
2,3,7,8-TeCDD (Table 49). Dose-dependent increases in total hepatic
porphyrins were found for both TBDD and TCDD. The relative
porphyrinogenic potencies were determined by the authors using TCDD as
a reference (TCDD = 1 and TBDD = 0.4).
Table 49. Total hepatic porphyrin accumulation in female B6C3F1 mice
after 13 weeks of exposure to TBDD or TODDa
TBDD TCDD
Dose Hepatic porphyrin Dose Hepatic porphyrin
(ng/kg body accumulation (ng/kg body accumulation
weight per day) (µg/g) weight per day) (µg/g)
0 0.207 0 0.196
30 0.296 0.15 0.204
90 0.346 0.45 0.212
300 0.429 1,5 0.212
900 13.4 4.5 0.222
3000 26.2 15 0.256
45 0.592
150 13.2
450 18.6
a Adapted from van Birgelen et al. (1996).
7.8.3 Effects on vitamin A storage
Various brominated or chlorinated aromatic compounds are able to
reduce the vitamin A content of the liver (WHO, 1989, 1993, 1994a). A
single oral dose of 10 µg TBDD/kg body weight decreased both the
concentration and the total amount of vitamin A (retinol) in the liver
of adult male Sprague-Dawley rats (n = 5) 4 weeks after the start of
the experiment. Reductions in concentration and total amount of
vitamin A were 45 and 51%, respectively (Thunberg et al., 1984). A
more pronounced effect was elicited by TCDD tested in the same study,
the reductions being 88 and 87%, respectively. However, on a molar
basis, TCDD was only slightly more potent than TBDD (Thunberg et al.,
1984).
7.8.4 Endocrine interactions
Thyroid hormones were affected in rats after single (Ivens et
al., 1992; see also section 7.1) and subchronic (Löser & Ivens, 1989;
Ivens et al., 1993; see also section 7.2) exposures to 2,3,7,8-TeBDD.
Four weeks after single oral doses of 10, 33, or 100 µg TBDD/kg body
weight, T3 was increased and T4 was reduced dose-dependently in the
serum of female and male Wistar rats ( n = 5) (Ivens et al., 1992).
Wistar rats (10/sex per group) treated with daily doses of 0.01 - 3.0
µg/kg body weight per day for 3 months had reduced T4 and increased T3
levels at 0.1 µg/kg body weight per day and higher (highest dose group
of 10 µg/kg body weight per day not examined) (Löser & Ivens, 1989;
Ivens et al., 1993; see also section 7.2.1).
The antiestrogenic potency of a series of PXDDs/PXDFs was
examined in cultures of MCF-7 human breast cells (Spink et al., 1994).
Two effects, stimulation of the metabolism of 17 ß-estradiol and
inhibition of the estrogen-dependent formation of multicellular foci,
were measured as indices of antiestrogenicity. Several tetra(Br1Cl3DDs,
Br2Cl2DDs) and penta- (Br1Cl4DD) halogenated congeners with
2,3,7,8-substitution stimulated estradiol metabolism with a potency
similar to that of TCDD. The EC50 values of these PXDDs were 0.6 - 0.8
nmol (TCDD: 0.8 nmol). The focus formation was inhibited by the same
congeners, with EC50 values ranging from 0.5 to 1.2 nmol (TCDD: 0.3
nmol). Dibenzo- p-dioxins and dibenzofurans with other substitution
patterns were markedly less active (EC50 values in nmol for inhibition
of focus formation: 17, 8-Br1-2,3,4-Cl3DF and 7-Br1-2,3-Cl2-DD; 1700,
2,3,7-Cl3-8-methylDD; >1700, 2,7-Br2DF and several methyl-substituted
PCDDs/PCDFs).
7.8.5 Interaction with drugs and toxicants
A single oral dose of 2,3,7-TrBDD (25 µg/kg body weight)
depressed the plasma disappearance of ouabain and its excretion in
bile of male Sprague-Dawley rats 10 days after treatment. In addition,
the bile flow was decreased by 2,3,7-TrBDD. TCDD elicited the same
effects but more markedly than 2,3,7-TrBDD (Yang et al., 1983).
7.8.6 Induction of microsomal enzymes
PBDDs/PBDFs, like TCDD and other environmental contaminants, are
potent inducers of certain CYP-dependent enzymes. Most frequently, the
activity of the cytochrome CYP1A1 was examined (direct measurements by
radioimmunoassay [RIA] or determination of marker enzymes [mostly AHH
and EROD]).
The isoenzyme CYP1A2 (determined by RIA) was also found to be
inducible. ED50 values of 0.8 - 1 nmol/kg body weight for CYP1A1
induction and 0.2 nmol/kg body weight for CYP1A2 induction in rat
liver were estimated after single oral doses of TBDD (Kedderis et al.,
1991b, 1992a, 1993). Induction of CYP1A2 enzyme activity has been
determined in mouse liver after subchronic dosing by oral gavage with
TBDD (Birnbaum & DeVito, 1995) and in liver of marmoset monkeys after
single subcutaneous doses of TBDD (Schulz et al., 1996).
Induction of the monooxygenases AHH and/or EROD was observed
following single exposure of chicken embryos (Kende & Wade, 1973;
Poland & Glover, 1973; Kende et al., 1974; Ramalingam et al., 1986),
guinea-pigs (Schmidt & Ivens-Kohl, 1990b), rats (Thunberg et al.,
1984; Magon et al., 1987a,b; Nagao et al., 1990b,e; Schmidt &
Ivens-Kohl, 1990a,b; Schulz-Schalge et al., 1990, 1991a,b; Kedderis et
al., 1991b, 1992a, 1993; Ivens et al., 1992), and marmoset monkeys
(Schulz et al., 1993, 1996) and after subchronic dosing by oral gavage
in mice (Birnbaum et al., 1993; Birnbaum & DeVito, 1995; van Birgelen
et al., 1996) and rats (Ivens et al., 1993), as well as in primary
cultures of rat hepatocytes (Blankenburg et al., 1990) and in rat
hepatoma H-4-II E cells (Bradlaw & Casterline, 1979; Bradlaw et al.,
1980; Denomme et al., 1985, 1986; Mason et al., 1987a; Zacharewski et
al., 1988, 1989; Safe et al., 1989a,b). Almost all experiments
referred to the tetra- and pentaBDDs/XDDs. Only a few studies (Poland
& Glover, 1973; Kende et al., 1974; Denomme et al., 1985) included
tri- or diBDDs/XDDs. There was one study (Schulz et al., 1993) on
dibenzofurans (pentaBDF) and another (Denomme et al., 1986) on PXDFs.
Several aspects of enzyme induction by PHDDs/PHDFs, with emphasis
on the chlorinated congeners, are addressed in Goldstein & Safe (1989)
and Neubert (1993b). The general mechanisms of induction of the CYP1A1
enzyme system were discussed by Okey (1990, 1992).
7.8.6.1 Dibenzo-p-dioxins
Enzyme induction proceeded dose-dependently at non-toxic
concentrations. It was measurable at exposures as low as the pmol
range (Schulz-Schalge et al., 1990, 1991b: <100 pmol TBDD/kg body
weight in the rat). Induction started soon after exposure and was
long-lasting, 1 - 98 days after a single subcutaneous administration
of TBDD, TCDD, and tetraXDDs (Nagao et al., 1990c; Schulz-Schalge et
al., 1991a). Maximum EROD induction was seen approximately 7 days
after a single subcutaneous exposure of rats to several
2,3,7,8-substituted dibenzo- p-dioxins (Nagao et al., 1990c;
Schulz-Schalge et al., 1991a). Oral exposure of rats to 1 µg TBDD/kg
body weight per day for 91 days also showed maximum induction after 7
days of treatment (Ivens et al., 1993). In contrast, during low-dose
oral treatment of mice (30 - 3000 ng TBDD/kg body weight per day for
13 weeks), a biphasic response was observed, not leading to maximum
enzyme induction (Birnbaum et al., 1993).
In a comparison of the decline in EROD activity after single
doses of 600 µg TBDD/kg body weight and 300 µg TCDD/kg body weight,
the curves were superimposable over a period of 90 days (Nagao et al.,
1995/96). When the hepatic EROD activity was related to the molar
concentrations of TBDD and TCDD, the same relationship was also
demonstrated. This indicates that both TBDD and TCDD exhibit identical
potencies with respect to the induction of EROD, when compared on a
molar basis. When compared on a weight basis (as is done for an
international toxic equivalent [I-TEQ] approach), TBDD is less potent
with respect to this end-point.
Although the liver was the most important organ for enzyme
induction, EROD induction by TBDD was also observed in other tissues
(lung, skin, kidney) of rats (Ivens et al., 1993) and mice (Birnbaum
et al., 1993) and in lung of marmoset monkeys (Schulz et al., 1996).
The induction potency for different PBDD congeners varied over
several orders of magnitude. For example, the activities for in vivo
hepatic AHH induction differed by four orders of magnitude between
four brominated congeners, the most active being 2,3,7,8-TeBDD and the
least 1,3,7,8-TeBDD (see Tables 50 and 51). Similar structure-activity
correlations were seen in other experimental systems ( in vitro
studies, EROD activity, toxic effects) and were discussed in several
reviews (Goldstein & Safe, 1989; Safe et al., 1989a,b; Safe, 1990). As
with PCDDs (WHO, 1989), the most potent compounds were those having
the 2,3,7,8-substitution pattern. In summary, the most effective
inducers of in vitro and/or in vivo AHH or EROD activity were TCDD,
TBDD, and TBCDD.
Compared on a molar basis to their chlorinated analogues, the
PBDDs and PXDDs have more or less similar potencies (see Tables 50 and
51; Kende & Wade, 1973; Kende et al., 1974; Bradlaw & Casterline,
1979; Thunberg et al., 1984; Denomme et al., 1985; Abraham et al.,
1988; Blankenburg et al., 1990; Nagao et al., 1990c; Schulz-Schalge et
al., 1990, 1991b). Nevertheless, some differences are apparent between
TBDD and TCDD, the congeners most intensively studied. In contrast to
TCDD, whose relative induction potency was independent of the tissue
examined, TBDD was five times more potent in inducing EROD activity in
the liver than in skin and lung following subchronic exposure of mice
(Birnbaum et al., 1993).
Only limited data are available on possible species-dependent
variations. The estimated potency for EROD reduction of TBDD relative
to TCDD ranged from 0.75 to 5.3 in rats (calculated by Birnbaum et
al., 1993 from data of Safe, 1990) and from 0.04 to 0.2 in mice
(Birnbaum et al., 1993). According to the authors, it is possible that
these differences may be indicative of species differences, despite
the different experimental designs used in the rat and mice studies.
7.8.6.2 Dibenzofurans
2,3,4,7,8-PeBDF was investigated together with its chlorinated
counterpart and TCDD in the marmoset monkey. Six days after a single
subcutaneous dose of 420 ng 2,3,4,7,8-PeBDF/kg body weight (TCDD:
50 - 500 ng/kg body weight; 2,3,4,7,8-PeCDF: 240 - 2400 ng/kg body
weight), the caffeine breath test was performed in vivo; 1 day later,
the animals were sacrificed, and EROD activity was determined in liver
microsomes. There was a good correlation between EROD activity in
hepatic microsomes and caffeine N-demethylation. All three compounds
showed inducing capacity, and the ranking order was TCDD >
2,3,4,7,8-PeCDF > 2,3,4,7,8-PeBDF when enzyme activities were
compared with the hepatic concentrations (Schulz et al., 1993).
Table 50. In vivo ED50 values for AHH induction and toxic effects
of several PHDDs in the rata,b
Congeners In vivo ED50 (µmol/kg body weight)
Inhibition of body Thymic atrophy Hepatic AHH
weight gain induction
2,3,7,8-TeBDD 0.068 0.034 0.0076
2,3,7,8-TeCDD 0.05 0.09 0.004
2,3-BR2-7,8-Cl2-DD 0.012 0.0073 0.000 49
2-BR2-3,7,8-Cl3-DD 0.12 0.035 0.0025
1,2,3,7,8-PeBDD 0.87 0.39 0.025
1,2,3,7,8-PeCDD 0.62 0.17 0.031
1,2,4,7,8-PeBDD 12.9 6.17 0.195
1,2,4,7,8-PeCDD 34.0 11.2 2.82
1,3,7,8-TeBDD 252.0 35.5 6.50
1,3,7,8-TeCDD 132.0 100.0 31.2
a Adapted from Mason et al. (1987a); Safe et al. (1989b).
b Immature male Wistar rats (n = 4), 14 days after single intraperitoneal doses.
For PXDFs, the AHH and EROD enzyme induction potencies of
8-Br1-2,3-Cl2DF and 8-Br1-2,3,4-Cl3DF were tested in vitro in rat
hepatoma H-4-II E cells (Denomme et al., 1986). The EC50 values
(mol/litre) were similar to those of the fully chlorinated analogues.
7.8.6.3 Combustion products
Pyrolysates of several brominated flame retardants, which
contained relatively high levels of PBDFs/PBDDs (see also section 3),
produced dose-dependent EROD and AHH activity in vitro (rat hepatoma
H-4-II E cells) (Zacharewski et al., 1988, 1989), as well as in liver
microsomes of rats (Zacharewski et al., 1988, 1989; Schmidt &
Ivens-Kohl, 1990b) and guinea-pigs (Schmidt & Ivens-Kohl, 1990b)
sacrificed 14 days or 4 weeks after single intraperitoneal or oral
doses.
7.9 Mechanisms of toxicity - mode of action
The mechanisms underlying the diversity of biochemical and toxic
effects of TCDD are being extensively studied, but they are complex
and not yet fully understood. Because of similarities in chemical
structure and in the pattern of responses, PBDDs/PBDFs are believed to
Table 51. Comparative survey on CYP1A1 induction (measured as EROD activity in vitro or in vivo in liver)
by 2,3,7,8-substituted TeHDDs
Species Details Parameter Resultsa Reference
(strain) TBDD TCDD TBCDD
Rat in vitro: hepatoma H-4-11 E EC50 (nmol/litre) 0.235 0.080 0.055 Mason et al. (1987a); Safe
cells (1987); Goldstein & Safe
(1989); Safe et al. (1989b)
Rat (Wistar) single intraperitoneal doses ED50 (nmol/kg body weight) 0.355 3 0.347 Mason et al. (1986,
1987a); Safe (1990)
pED50b 9.45 8.16 n.sp. Mason et al. (1987b)
Rat single oral doses ED50 (nmol/kg body weight) 0.8-1 n.a. n.a. Kedderis et al. (1992a)
(Fischer 344)
Rat (Wistar) single subcutaneous dose resorufin (pmol/mg protein Schulz-Schalge et al.
of 2 nmol/kg body weight per min) after: (1991)
- 7 days 6740 5210 4330
- 98 days 410 162 105
Mouse multiple oral doses relative potency 0.2 1 n.a. Birnbaum et al. (1993)
(B6C3F1)
Chick embryo single injections ED50 (pmol/egg) 9.4 11.1 7.35 Ramalingam et al. (1986)
a n.a. = not analysed; n.sp. = not specified.
b pED50 = -log ED50.
share a common mechanism of action with PCDDs/PCDFs and other related
halogenated aromatic hydrocarbons (Poland & Knutson, 1982; Goldstein &
Safe, 1989; Mennear & Lee, 1994). Most information is derived from
studies with TCDD, the prototypical and best examined compound of this
class of chemicals. PHDDs/PHDFs act as multisite toxicants in
laboratory animals and elicit species-, sex-, and tissue-dependent
responses (Vanden Heuvel & Lucier, 1993). In carcinogenic processes,
TCDD and related compounds function as tumour promoters (reviewed in
Lucier et al., 1993b; Huff, 1994).
On a cellular basis, TCDD and related compounds cause alterations
in cell proliferation and differentiation without a mutational effect
on DNA (Goldstein & Safe, 1989; Silbergeld & Gasiewicz, 1989; Nebert
et al., 1991; Nebert, 1994). However, there is a possibility that TCDD
may influence the DNA-damaging potential of endogenous compounds
(Lucier et al., 1993b). Examples of impairment of normal cellular
regulatory systems by PCDDs/PCDFs have been compiled (WHO, 1989).
Recently, inhibition of gap-junctional intercellular communication by
TCDD was reported (De Haan et al., 1993).
At the molecular level, there is growing evidence that most, if
not all, biochemical or toxic effects, including carcinogenicity, are
mediated through an intracytoplasmic protein, the Ah receptor (Couture
et al., 1990; Denison, 1990, 1991; Whitlock, 1990, 1993; Landers &
Bunce, 1991; Lucier, 1991; Nebert et al., 1991, 1993; Safe et al.,
1991; Hahn & Stegeman, 1992; Poellinger et al., 1992; Andersen et al.,
1993; Birnbaum, 1993, 1994; Lucier et al., 1993a,b; Poellinger, 1993;
Safe, 1993a; Vanden Heuvel & Lucier, 1993; Whitelaw et al., 1993; Okey
et al., 1994; Fernandez-Salguero et al., 1996). However, although the
central role of the Ah receptor in mediating TCDD toxicity is
generally accepted, some findings may indicate the existence of other
mechanisms operating independently of the Ah receptor (Skene et al.,
1989; WHO, 1989; Holsapple et al., 1991; Nebert et al., 1993; Okey et
al., 1994).
The potential of binding to the cytosolic Ah receptor was
confirmed for several mono- through penta-substituted PBDDs and PXDDs
(Denomme et al., 1985; Mason et al., 1987a,b; Romkes et al., 1987) and
PXDFs (Denomme et al., 1986). Their receptor-binding affinities varied
by several orders of magnitude (depending on structure) but were
comparable to those of their PCDD analogues (Goldstein & Safe, 1989).
As an example, the in vitro EC50 values for rat hepatic cytosolic
receptor-binding potencies of TCDD, TBDD, and TBCDD were found to be
1.00 x 10-9, 1.50 x 10-9, and 1.48 x 10-9 mol/litre, respectively (Mason
et al., 1987a).
Limited information is available on possible additive,
synergistic (overadditive), or antagonistic interactions between
different PBDDs/PBDFs and other xenobiotics. As seen in a rainbow
trout early life stage mortality bioassay (see chapter 9), selected
PBDD, PBDF, and PBB congeners act additively (Hornung et al., 1996a).
All kinds of interactions could be found between PCDDs, PCDFs, and
PCBs (summarized by Skene et al., 1989; Safe, 1990; Hornung et al.,
1996a).
There have been no studies identified detailing the possible
actions of non-2,3,7,8-substituted PBDD/PBDF congeners.
7.10 Experimental data on selected PBDDs/PBDFs and their relevance
to the toxicity equivalency factor (TEF) concept
The TEF method is used as a procedure to facilitate the
evaluation of complex mixtures of related chemicals. The real
concentrations of different congeners in a sample are changed to (by
multiplying by a factor called the TEF) and expressed as uniform
"toxic" concentrations, as related to a reference substance. A major
application of this method is for risk management or regulatory
purposes. Advantages and limitations in the use of TEFs were discussed
intensively with regard to PCDDs/PCDFs (e.g. Kutz et al., 1990;
Ahlborg et al., 1992; Neubert D. et al., 1992; Ahlborg, 1994; DeVito &
Birnbaum, 1994; Silbergeld & deFur, 1994). For the 2,3,7,8-substituted
derivatives within these compounds, a series of TEF schemes was
developed by several agencies in different countries. The schemes
differ in the weighting factors for certain congeners. In 1988, an
internationally elaborated TEF scheme was published (NATO-CCMS, 1988),
presenting the so-called international TEFs (I-TEFs) (see Table 52).
It was followed by a modified proposal of Safe (1990, 1993a,b).
Additionally, his concept included (besides PCBs, PBBs, and
poly-chlorinated diphenyl ethers [PCDEs]) the PBDDs/PBDFs and PXDDs/
PXDFs to which the same factors were attributed as described for the
chlorinated analogues. Currently, there are no TEFs for
2,3,7,8-substituted PBDDs/PBDFs that have international agreement.
Most of the toxicologically relevant information available refers
to the 2,3,7,8-substituted dibenzo- p-dioxin analogue pair. Data
summarized in Table 53 support the concept of using corresponding TEF
values for both analogues. Even if for some end-points slightly lower
potencies (on a molar basis, and somewhat more pronounced on a weight
basis) were found for 2,3,7,8-TeBDD than for 2,3,7,8-TeCDD, kinetic
differences (especially the longer half-life of 2,3,7,8-TeBDD in
adipose tissue) may favour the use of identical TEFs for 2,3,7,8-TeBDD
and 2,3,7,8-TeCDD (e.g. Neubert, 1993b).
Table 52. I-TEFs for PCDDs/PCDFs
Congeners I.TEFsa
2,3,7,8-TeCDD 1
2,3,7,8-TeCDF 0.1
1,2,3,7,8-PeCDD 0.5
1,2,3,7,8-PeCDF 0.05
2,3,4,7,8.PeCDF 0.5
1,2,3,4,7,8-HxCDD 0.1
1,2,3,4,7,8-HxCDF 0.1
1,2,3,6,7,8-HxCDD 0.1
1,2,3,6,7,8-HxCDF 0.1
1,2,3,7,8,9-HxCDD 0.1
1,2,3,7,8,9-HxCDF 0.1
2,3,4,6,7,8-HxCDF 0.1
1,2,3,4,6,7,8-HpCDD 0.01
1,2,3,4,6,7,8-HpCDF 0.01
1,2,3,4.7,8,9-HpCDF 0.01
1,2,3,4,6,7,8,9-OcCDD 0.001
1,2,3,4,6,7,8,9-OcCDF 0.001
a From NATO/CCMS (1988).
For another analogue pair, namely 2,3,7,8-TeBDF and
2,3,7,8-TeCDF, it was suggested that a higher TEF than that chosen for
2,3,7,8-TeCDF (0.1) be used, possibly around 0.3 on a molar basis or
0.2 on a weight basis (Neubert, 1993b). This proposal was also based
on kinetic data - that is, the much longer elimination half-life of
2,3,7,8-TeBDF than of 2,3,7,8-TeCDF observed in rat liver (Neubert,
1993b). Additionally, 2,3,7,8-TeBDF was found to be more potent than
2,3,7,8-TeCDF in producing cleft palate and hydronephrosis in mice
(Birnbaum et al., 1991 and references therein). In a fish toxicity
assay (see chapter 9), 2,3,7,8-TeBDF was ninefold (molar basis) more
potent than 2,3,7,8-TeCDF (Hornung et al., 1996b).
Considering the few data available on the higher halogenated
congeners and the wide range of potencies leading to the established
TEFs, the (preliminary) use of the same TEF values for the other
PBDD/PBDF or PXDD/PXDF congeners as described for the chlorinated
analogues appears to be justified.
Table 53. A comparative survey of several biological and toxicological parameters for 2,3,7,8-TeBDD and 2,3,7,8-TeCDD
(tested in parallel-running experiments)
Parameter Details TBDD TCDDa Reference
Kinetics
Absorption rat, oral route up to 80% _b Ivens et al. (1992);
Diliberto et al. (1993)
Metabolism rat about 7% about 10% Kedderis et al.
(1991b)
Half-life rat (faeces) 18 days _c Kedderis et al.
(1991a)
rat (adipose 56 days _d Kedderis et al.
tissue) (1991a)
human 3-11 years _d Zober et al. (1992)
Receptor binding incubation of 1.5 x 10-9 mol/litre 1.0 x 10.9 Mason et al. (1987b)
(EC50) cytosolic mol/litre
receptor protein
Microsomal enzyme
induction
Binding affinity rat liver 9.0 nmol 6.5 nmol Kedderis et al. (1993)
of CYP1A2
AHH induction rat liver 9.12 8.41 Mason et al. (1987b)
(pED50)f
EROD induction rat liver 9.45 8.16 Mason et al. (1987b)
(pED50)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
EROD induction rat liver Schulz-Schalge et al.
(molar basis) -after 7 days 6740 pmol resorufin/mg 5210 pmol resorufin/mg (1991a,b)g
protein per min protein per min
- after 98 days 410 pmol resorufin/mg 162 pmol resorufin/mg
protein per min protein per min
EROD induction rat liver identical dose-effect enzyme concentration Nagao et al.
(relative potency, and curves (1995/96)
molar basis)
EROD induction chick embryo liver 9.4 pmol/egg 11.1 pmol/egg Ramalingam et al.
(ED50) (1986)
EROD induction mouse liver 0.2 1 Birnbaum et al.
(relative potency, (subchronic exposure) (1993); Birnbaum &
molar basis) DeVito (1995)
ACOHh induction mouse liver 0.2 1 Birnbaum & DeVito
(relative potency, (subchronic exposure) (1995)
molar basis)
EROD induction mouse lung 0.1 1 Birnbaum & DeVito
(relative potency, (subchronic exposure) (1995)
molar basis)
EROD induction mouse skin 0.04 1 Birnbaum et al.
(relative potency, (subchronic exposure) (1993); Birnbaum &
molar basis) DeVito (1995)
EROD induction mouse liver 0.31 1 Van Birgelen et al.
(relative potency) (subchronic exposure) (1996)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
ACOH induction mouse liver 0.11 1 Van Birgelen et al.
(relative potency) (subchronic exposure) (1996)
Hepatic porphyrin mouse liver 0.4 1 Van Birgelen et al.
accumulation (subchronic exposure) (1996)
(relative potency)
Body weight loss rat 7.17 7.28 Mason et al. (1987b)
(pED50)
Immunotoxicity
Thymic atrophy rat 7.47 7.03 Mason et al. (1987b)
(pED50)
Changes in certain marmoset monkey 3 ng/kg body weight 1 ng/kg body weight Neubert (1993a)
lymphocyte (6 pmol/kg) (3 pmol/kg)
subpopulations
(NOEL)
Developmental toxicity
Cleft palate (ED50) mouse (NMRI) 62 ± 5 µg/kg body 24 ± 1 µg/kg body weight Nagao et al. (1990a)
weight (0.075 µmol/kg body
(0.123 pmol/kg body weight)
weight)
mouse (C57BL/6N) 65 µg/kg body weight (15 µg/kg body weight)i Birnbaum et al,
(1991)
Hydronephrosis mouse (C57BL/6N) 9 µg/kg body weight 4 µg/kg body weight Birnbaum et al.
(ED50) (estimated) (1991)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
Tumorigenicity in vivo/in vitro 0.14 1 Massa et al. (1991)
(short-term test: assay with murine
relative macrophages
cell-transforming
potential, molar
basis)
Mortality Rainbow trout 1.14-2.54 1 Hornung et al.
(LD50, relative (early life stage) (1996b)
potency, molar
basis)
a - = not performed.
b Comparable data from other TCDD studies (WHO, 1989); 88% measured by Diliberto et al. (1996).
c Comparable data from other TCDD studies (references in Kedderis et al., 1991a; Weber et al., 1993).
d Less than values from other TCDD studies (17-25 days; references in Kedderis et al., 1991a).
e Comparable data from other TCDD studies (5-11 years, references cited in chapter 8).
f pED50 = -log ED50 (molar basis).
g Data in agreement with results of Abraham et al. (1988) and Nagao et al. (1990b).
h ACOH = acetanilide-4-hydroxylase.
i Measured by the same laboratory (Birnbaum et al., 1989).
j See chapter 9 for information about the study.
8. EFFECTS ON HUMANS
There is little information available on the effects of
PBDDs/PBDFs on human health. The main human health features discussed
in connection with PCDDs/PCDFs are immunotoxicity (Lorenzen & Okey,
1991; Vos, 1993), developmental toxicity (Sweeney, 1994),
neurotoxicity (Peper et al., 1993), carcinogenicity (Kogevinas et al.,
1993; Bertazzi & di Domenico, 1994; Hardell et al., 1994), and skin,
liver, and gastrointestinal toxicity (Mennear & Lee, 1994). Recently,
a brief critical review of the short- and long-term non-cancer health
effects of PCDDs/PCDFs was given by Sweeney et al. (1993). The
relevance of biochemical effects of PCDDs/PCDFs has also been
addressed (Lucier, 1991).
8.1 General population exposure
There are no data for the general population on exposure to or
effects of PBDDs/PBDFs.
8.2 Occupational/accidental exposure
Two cases of acute health problems due to 2,3,7,8-TeBDD/TeCDD
exposure have been described (see also section 5.3.2). The first case
refers to an American chemist who developed serious illness, including
chloracne, headaches, and back and leg pain, after synthesizing
TBDD/TCDD (Schecter, 1992). The second case was that of a Japanese
student who suffered from very severe acne-like eruptions on his
cheeks and chin following synthesis of PCDFs and PBDFs (Asahi & Urabe,
1987).
In the course of a morbidity study, male personnel of a chemical
plant with documented exposure to PBDDs/PBDFs (see section 5.3)
originating from the use of brominated flame retardants (OBDE and
DBDE) were subjected to a general health examination, including
special immunological tests (Zober et al., 1992). The measurements of
the exposed (n = 21; exposed for up to 13 years) and the control (n =
42; employees of a similar resin production plant but with no use of
PBDEs within the plant) groups included cellular and humoral
parameters (distribution of lymphocyte subsets, concentrations of
immunoglobulins, immune complexes, complements C3 and C4, and
antinuclear antibodies). Functional abnormalities of the immune system
were not investigated. Only one person with the highest TBDD/TBDF
burden (478/112 pg/g blood lipid) showed some changes (high
concentrations of complement C4; low total lymphocyte, T cell, T
helper cell, and natural killer cell counts) but did not have clinical
symptoms attributable to an immunodeficiency disease. The health
problems of this 54-year-old worker (hypertension, low back pain,
hyperuricaemia, and signs of an old inactive tuberculosis) were
thought to be not dioxin-dependent. No notable differences from the
control group were found with the other participants with lower blood
lipid levels (<208 pg TBDD/g blood lipid; <58 pg TBDF/g blood
lipid). Evaluating the group results as a whole, complement C4
concentrations increased significantly with increasing concentrations
of both TBDD and TBDF. Marginal associations independent of the person
with the highest exposure were seen with C3 concentrations (increased
with TBDF concentrations). However, retesting of the one person at a
later date showed that his C4 levels had dropped. Altogether, the
effects observed were not considered to be indicative of an impact of
PBDDs/PBDFs on the immune system.
A shortcoming of this study was a lack of measurements of
PBDD/PBDF concentrations (and concentrations of related compounds,
such as PCDDs/PCDFs or PCBs) in the control group. In addition, the
internal exposure was related to only two congeners, namely TBDD and
TBDF, although a lot of other homologues were identified at the
corresponding workplace (see section 5.3.1). This was partly due to
analytical limitations (lack of reference substances) and partly due
to the high weighting factor of TBDD in toxic equivalent (TEQ)
calculations.
Additional clinical laboratory tests (liver function tests, lipid
and glucose measures, thyroid function parameters, haematological and
coagulation indicators) were performed within the same study cohort.
They did not reveal any remarkable differences between the exposed
(n = 38 - 42) and the control (n = 40 - 42) groups (Ott & Zober,
1996).
Although there are indications for excesses of cancer mortality
seen in workers exposed to TCDD (e.g. Zober et al., 1990; Fingerhut et
al., 1991; Manz et al., 1991; Becher et al., 1996), there are no
reports of cancer mortality caused by PBDDs/PBDFs.
8.3 Subpopulations at special risk
No data are available with regard to the effects of PBDDs/PBDFs
on high-risk subpopulations.
As discussed for PCDDs/PCDFs (Helge, 1993), fetuses, newborn
babies, and children are at a higher risk of exposure for several
reasons (placental and milk transfer, contact with soil and dust,
immunological and physiological immaturity, lack of fat depots, etc.),
and developing systems, based on experimental evidence, may be more
sensitive to the adverse effects of halogenated aromatic compounds
than developed systems of adults.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
There are limited reports on the effects of PBDDs/PBDFs on
microorganisms, plants or invertebrate species. Regarding vertebrates,
data from a fish early life stage mortality bioassay are available
(Hornung et al., 1996a,b).
Newly fertilized rainbow trout eggs were injected with a series
of PBDDs, PXDDs, and PBDFs (see Table 54). All congeners tested but
2,7-DiBDF caused mortality in the rainbow trout sac fry by a blue sac
syndrome. The signs of toxicity were identical to those produced by
TCDD and included yolk sac oedema, pericardial oedema, multifocal
haemorrhages, reduced growth, and craniofacial malformations.
2,3,7,8-TeBDD showed the highest potency among the brominated
congeners and was also more potent than 2,3,7,8-TeCDD. Similarly,
2,3,7,8-TeBDF was more potent than 2,3,7,8-TeCDD, whereas the other
PBDDs/PBDFs were equipotent (or less potent) than identically
substituted PCDDs/PCDFs (Hornung et al., 1996b).
The interactions between pairs of PBDD and PBDF congeners were
also studied using the rainbow trout sac fry (early life stage)
mortality bioassay (Hornung et al., 1996a). The tested pairs were as
follows: 2,3,7,8-TeBDD and 1,2,3,7,8-PeBDF; 2,3,7,8-TeBDD and
1,2,3,7,8-PeBDD; and 1,2,3,7,8-PeBDF and 1,2,3,7,8-PeBDD. As with the
individual PBDD and PBDF congeners, their mixtures also produced
TCDD-like toxicity and mortality in the rainbow trout sac fry. The
rank order for LD50 in the individual congeners tested, from lowest to
highest, was as follows: 2,3,7,8-TeBDD < 1,2,3,7,8-PeBDD
< 1,2,3,7,8-PeBDF. The interactions between each of the tested pairs
were additive in causing sac fry mortality.
Laboratory studies with the related PCDDs/PCDFs showed adverse
effects on fish and avian species, especially on the early life stages
(Cook et al., 1991; Walker & Peterson, 1991, 1994; Peterson, 1993;
Peterson et al., 1993). Correlations between environmental exposure to
polyhalogenated aromatics (PCDDs/PCDFs, PCBs) and the decline of some
populations of marine and freshwater mammals, fish, and fish-eating
birds or symptoms such as reproductive dysfunction or other lesions
were investigated in a number of field studies (Gilbertson, 1989;
Rappe, 1993).
Recent results of the research on the effects of
dibenzo- p-dioxins and dioxin-like compounds on wildlife support the
conclusion that there are real-world adverse effects on wildlife
caused by these compounds. Although PCDD/PCDF have contributed to
these effects, in most locations the major effects are due to the
dioxin-like non- and mono-ortho-substituted PCB congeners. Effects
have been reported in mammals and birds as well as in fish. While the
current effects are subtle, there is no assimilative capacity for TEQs
in the global environment. Trends in TEQs in industrialized regions
are continuing to decline. Concentrations of TEQs in remote areas,
such as the Arctic and open ocean, may not yet have reached their
maximum. The contribution of PBDDs/PBDFs to these effects is unknown
(Giesy et al., 1994; Fiedler & Van den Berg, 1996).
Table 54. Early life stage mortality in rainbow trout (Oncorhynchus
mykiss) caused by PBDDs, PXDDs, and PBDFsa
Congener Blue sac syndromeb LD50c Rainbow trout
(ng/g egg) strain
Dibenzo-p-dioxins
2,3,7-TrBDD + 18.9 Erwin
+ 15.6 McConoughy
2,3,7,8-TeBDDd + 0.222 Eagle Lake
+ 0.264 Eagle Lake
+ 0.158 Erwin
+ 0.122 Arlee
1,3,7,8-TeBDD + 29 Erwin
1,2,3,7,8-PeBDD + 4.16 Eagle Lake
+ 4.92 Erwin
1,2,3,4,7,8-HxBDD + 63.7 Arlee
2,8-Cl2-3,7-Br2-DD + 0.448 Erwin
2,3,7-Cl3-8-Br1-DD + 0.410 Erwin
Dibenzofurans
2,7-DiBDF - >597e Erwin
2,3,7,8-TeBDF + 1.5 Erwin
2,3,4,7,8-PeBDF + 6.19 Erwin
1,2,3,7,8-PeBDF + 9.56 Erwin
1,2,3,4,7,8-HxBDF + 247 Erwin
a Modified from Hornung et al. (1996b).
Table 54 (Continued)
b TCDD-like toxicity grossly identical to blue sac syndrome was
characterized by sac fry mortality that was preceded by yolk sac
oedema, pericardial oedema, multifocal haemorrhages, growth
retardation, and craniofacial malformations.
c Based on cumulative hatching and sac fry mortality (for fiducial
limits, see original); eggs (n = 30 per dose) injected with
seven graded doses of congener incorporated into phosphatidylcholine
liposomes.
d For comparison: LD50 values for 2,3,7,8-TeCDD: 0.171 ng/g egg
(Shasta strain); 0.374 ng/g egg (Adee strain) (Walker & Peterson,
1991; Zabel et al., 1995).
e No signs of toxicity occurred at the highest egg dose tested.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
There is much less information on PBDDs/PBDFs than on their
chlorinated analogues. The analytical methods for separating and
identifying the individual brominated congeners are less advanced than
those for their chlorinated analogues, and only a few standards are
available. Present analytical methods are able to quantify total
brominated homologue groups and are able to detect but not quantify
the mixed brominated/chlorinated congeners.
10.1 Hazard evaluation
Data on bioconcentration and biomagnification are lacking. The
physicochemical properties of PBDDs/PBDFs and extrapolations from
PCDDs/PCDFs suppose that PBDDs/PBDFs are enriched in carbon- and
fat-rich environmental compartments in a manner similar to
PCDDs/PCDFs. Atmospheric transport occurs in the vapour and
particulate phases, the ratio depending on molecular weight.
Photochemical degradation of PBDDs/PBDFs and PXDDs/ PXDFs
adsorbed on surfaces was much slower than that in organic solvents.
Under environmental conditions, debromination was found to be a slow
process (several months in soil).
From the available data, it is apparent that PBDDs/PBDFs have a
toxic potential similar to that of PCDDs/PCDFs.
Generally, the studies performed with 2,3,7,8-substituted
PBDDs/PBDFs showed typical TCDD-like effects in experimental animals
and a persistence in animal and possibly human tissues apparently
similar to that seen with PCDDs/PCDFs. For some end-points,
quantitative differences were found. For example, under certain
experimental conditions, 2,3,7,8-TeBDF was more potent than
2,3,7,8-TeCDF in inducing cleft palate and hydronephrosis in mice
following prenatal exposure. 2,3,7,8-TeBDD was somewhat less potent
than its chlorinated counterpart with regard to short-term toxicity,
cell-transforming capacity, antiestrogenic activity, and decreasing
vitamin A storage. Both brominated congeners had longer elimination
half-lives in rat adipose tissues than the chlorinated ones. More or
less comparable activities of these and some other PBDDs/PBDFs with
their chlorinated analogues were seen for receptor binding, enzyme
induction, and immunotoxic effects.
The few data available on trisubstituted PBDDs suggest a higher
metabolic rate and lower short-term toxicity compared with the
2,3,7,8-substituted congeners. Limited data are available for other
non-2,3,7,8-substituted PBDD/PBDF congeners. Studies with
1,2,7,8-TeBDF demonstrated higher rates of metabolic elimination in
rats compared with 2,3,7,8-TeCDF or 2,3,7,8-TeBDD.
Long-term toxicity studies as well as perinatal exposure and
multigeneration studies are also lacking for the 2,3,7,8-substituted
PBDDs/PBDFs. Nevertheless, the possibility of adverse effects - for
example, carcinogenicity, reproductive/developmental toxicity, and
neurotoxicity - cannot be excluded, based on the similarity in results
obtained in short-term studies with PBDDs/PBDFs compared with
PCDDs/PCDFs. However, in a comparison of relative potencies,
2,3,7,8-TeCDD was approximately two times more potent than
2,3,7,8-TeBDD in inducing the accumulation of total hepatic porphyrin
in mice following subchronic (13-week) exposure.
Congeners with a high degree of bromination (>penta) were
usually not included in toxicity studies (owing to a lack of pure
congeners), although they were detectable in workplace air and other
samples.
Exposure to single congeners of PBDDs/PBDFs and PXDDs/ PXDFs has
resulted in TCDD-like toxic effects and mortality in the rainbow trout
sac fry early life stage mortality bioassay. Pairs of PBDDs/PBDFs have
been shown to have additive effects on the mortality of rainbow trout
fry. In these bioassay studies, single exposures to 2,3,7,8-TeBDD and
2,3,7,8-TeBDF were more potent than single exposures to the
chlorinated analogues.
There are no pertinent data on microbial degradation.
Photochemical debromination is thought to be the major transformation
process for PBDDs/PBDFs and PXDDs/PXDFs. As was shown in laboratory
experiments, PBDDs/PBDFs and PXDDs/PXDFs were degraded in organic
solvents after irradiation with sunlight or UV light. The easier loss
of bromine than of chlorine resulted in the formation of the
PCDDs/PCDFs when PXDDs/PXDFs were exposed to light.
10.2 Exposure evaluation
As PBDDs/PBDFs are not known to occur naturally, their presence
is indicative of thermal or photolytic degradation or thermal
transformation of brominated chemicals, many of which are used as
flame retardants. The occurrence of such chemicals in consumer
products (e.g. electrical appliances, textiles, petrol) may, in some
cases, result in a risk for the population.
The database on the occurrence of PBDDs/PBDFs in ambient air and
dust is too small for detailed comparisons with PCDDs/PCDFs and
estimations of their impact on human health. Only one
2,3,7,8-substituted PBDF congener was detected in ambient air.
Automobile exhaust was found to be a diffuse source of PBDDs/PBDFs and
PXDDs/PXDFs (along with PCDDs/PCDFs), if halogenated scavengers are
used in the petrol.
Because typical municipal waste generally contains much more
chlorine than bromine, the formation of mixed and completely
chlorinated compounds from brominated precursors is possible and was
confirmed experimentally. Modern waste incinerators are capable of
realizing very low emissions of PCDDs/PCDFs; however, this modern
technology is not applied in all countries.
Emissions (and effluents) of PBDDs/PBDFs into the environment
from plants (processing organic chemicals, recycling plastics and
metals), waste incinerators, etc. are hardly documented, but may vary
depending on the industrial hygienic standards (including waste
disposal practices). A limit value of 0.1 ng TEQ/m3 for PCDDs/PCDFs in
exhaust gases of municipal waste incinerators was established by some
countries.
A critical source for release of PBDDs/PBDFs and PXDDs/ PXDFs
into the environment is accidental fires of materials containing
brominated compounds. Concentrations measured depend on the specific
fire situation and can be very high. The highest concentrations are
found in the solid residues.
PBDDs/PBDFs have been found in indoor air (maximum PBDF sum
concentration, up to hepta: 1.27 pg/m3) and dust samples of rooms
equipped with electronic appliances and in house dust. Whereas
2,3,7,8-substituted congeners were not detected in the air samples,
they were detectable in dust samples (tetra- to pentaBDFs: up to 0.07
µg/kg). Dust collected in computer rooms contained comparable amounts
of PBDDs/PBDFs and PCDDs/PCDFs (e.g. maximum sum concentrations: each
about 5 µg/kg).
Potential exposure pathways for the general population, clean-up
personnel, and fire personnel may originate from accidental fires
where bromine-containing plastics are involved. Gas, smoke, and
residue samples as well as firemen's trousers were found to contain
PBDDs/PBDFs (mainly PBDFs), including a portion of up to 20%
2,3,7,8-substituted congeners, and PXDDs/PXDFs. Generally, the
analyses of fire incidents showed a wide variation of contamination in
the µg/kg (residue), ng/m2 (smoke condensate), or ng/m3 (gas) range,
with very high peak concentrations possible. In several residue
samples, the sum concentrations of seven 2,3,7,8-substituted congeners
(2,3,7,8-TeBDF, 2,3,4,7,8-PeBDF, and five tetra- to hexaBDDs) were
greater than 5 µg/kg. In one television fire incident, the sum
concentrations of these congeners in residues amounted to 1100 µg/kg.
Additionally, this sample contained high concentrations of
1,2,3,7,8-PeBDF (1860 µg/kg) and of 1,2,3,4,7,8-HxBDF (1900 µg/kg).
PBDF concentrations measured at experimental fires generally were
higher than those measured at real fires and may be considered as
worst-case examples. The area contaminant concentrations of PBDFs
resulting from experimental or real television fires were in a similar
range as found for PCDDs/PCDFs after fires involving their precursors.
Fires involving televisions or computers may produce higher PBDD/PBDF
than PCDD/PCDF concentrations - for example, maximum sum
concentrations of 5600 µg/kg (13 µg TEQa/kg) versus 320 µg/kg (2.3 µg
I-TEQ/kg) in soot samples after a fire in a computer room.
a Calculated by Schacht et al. (1995) using PCDD/PCDF I-TEFs for
the brominated congeners.
Potential issues of concern are fires and suspected leaching
processes at waste disposal sites (additional presence of organic
solvents and other contaminants).
One study reported on the analysis of PBDDs/PBDFs and PXDDs/PXDFs
in a sample of muscle from a salmon from the Baltic Sea and a pooled
sample of human milk from Sweden. The concentrations were found to be
below the detection limit (0.3 ng 2,3,7,8-TeBDF/kg and 0.4 ng
2,3,7,8-TeBDD/kg), suggesting a very low exposure to PBDDs/PBDFs and
PXDDs/PXDFs. On the other hand, a series of 2,3,7,8-substituted
PCDDs/PCDFs were detected in these samples.
Workplaces identified as involving a risk of exposure to
PBDDs/PBDFs (and PXDDs/PXDFs) include mainly those in the plastic and
recycling industry using brominated flame retardants or products
containing them and those of firemen and clean-up personnel associated
with fires.
Recent monitoring data from three plastic plants showed
concentrations between 260 and >10 000 pg/m3 (sum of mono- to
hexaBDDs/BDFs). Concentrations of eight 2,3,7,8-substituted congeners
(three tetra- to pentaBDFs and five tetra- to hexaBDDs) ranged from
0.11 to 18 pg TEQb/m3 at permanently operated workplaces. At
periodically operated workplaces (maximal stay: 1 h/day), a maximum
concentration of the eight congeners of 954 pg TEQb/m3 has been
measured. There are no occupational threshold limits for PBDDs/PBDFs.
PBDD/PBDF concentrations currently measured in plastics, recycled
products, electronic scrap, and other waste samples were considerable.
In view of the growing worldwide production and use of brominated
flame retardants (estimated worldwide demand in 1992: 150 000 tonnes;
OECD, 1994) as additives to a series of polymers, it can be assumed
that the amount of bromine-containing waste will be increasing in the
future. In particular, electronic scrap from casings and printed
circuit boards of computers, etc., flame-retarded with brominated
compounds will reach the waste streams and then be a potentially major
source of PBDDs/PBDFs (and PXDDs/PXDFs and PCDDs/PCDFs).
A potential hazard can arise in chemical research laboratories
performing special syntheses. Distillation residues, other wastes, and
equipment were found to be contaminated by PBDDs/PBDFs and/or
PXDDs/PXDFs.
Few human monitoring data are available. One study published
blood monitoring data of personnel from an industry using PBDEs. The
analyses showed (1) uptake of PBDD/PBDF congeners, (2) the presence of
2,3,7,8-substituted congeners, which were not or hardly detectable in
the corresponding workplace air samples, and (3) estimated half-lives
typical for dioxin-like compounds.
b Calculated by Kieper (1996) using PCDD/PCDF I-TEFs for the
brominated congeners.
The main route of human exposure to PCDDs/PCDFs for the general
population is via food intake (more than 95%). Whereas the presence of
PCDDs/PCDFs was confirmed in most foodstuffs, PBDDs/PBDFs (>tetra)
could only be detected but not quantified in shellfish, fish, and
cow's milk. 2,3,7,8-Substituted congeners could not be identified.
For the general environment, it was found that the concentrations
of PBDDs/PBDFs are much lower than those of the chlorinated analogues
(tetra- through octahalogenated), the homologues with four or more
bromine atoms being hardly detected in environmental samples. At
present, there are only few data, but these data do not indicate an
accumulation of PBDDs/PBDFs or PXDDs/PXDFs along the terrestrial or
the aquatic food-chain. However, the limited data set does not allow
for environmental trend analysis.
Currently, most PBDDs/PBDFs and their precursors are bound into
products and have not yet reached waste streams and the environment.
10.3 Risk evaluation
PBDDs/PBDFs have been detected in air, dust, soil, sediment,
sewage sludge, grass, and fish, but not in the general human
population. This limited occurrence is in contrast to the ubiquitous
PCDDs/PCDFs. Generally, concentrations measured were low.
The major risk group for exposure to PBDDs/PBDFs has apparently
been workers involved in the production and application of brominated
flame retardants (e.g. extruder personnel). In these persons, clearly
increased body burdens of 2,3,7,8-TeBDD and 2,3,7,8-TeBDF have been
measured. Another possible source of exposure may be automobile
exhaust, as a result of incomplete combustion of bromine-containing
materials. No data on increased body burdens have been reported (and
apparently body burdens have not been measured). For the general
population, there seems to be a very low risk of exposure, compared
with the risk of exposure to PCDDs/PCDFs. In the few samples measured
(breast milk), no PBDDs/PBDFs were detected, whereas at least 100
times higher PCDD/PCDF levels were present.
Within the group of workers with clearly increased PBDD/PBDF body
burdens, no clinical adverse health effects were reported, and only a
few data for laboratory values of the volunteers were outside the
normal reference range. Although no exact data on elimination
half-lives have been reported for humans, the data available indicated
a considerable persistence of the 2,3,7,8-TeBDD/TeBDF congeners within
the human organism.
From all the information currently available, it can be concluded
that the potential of the PBDDs/PBDFs for biological (e.g. enzyme
induction) and toxic actions is very similar to that of the
PCDDs/PCDFs.
It is difficult to compare the potency of PBDDs/PBDFs with that
of their chlorinated analogues, as the database for the individual
PBDDs/PBDFs is very small, with respect to both data from animal
studies and observations in humans. With a few exceptions in most of
the systems tested, the few evaluated PBDD/PBDF congeners were less
potent than the corresponding chlorinated congeners. Kinetically,
however, some of the brominated substances exhibited a higher
persistence within the mammalian organism compared with the
corresponding chlorinated substances. Therefore, as judged from data
on 2,3,7,8-TeBDD, a TEF of 1.0 (equal to that of TCDD) would represent
a conservative approach. For 2,3,7,8-TeBDF, a somewhat higher TEF may
be suggested, as the substance has been shown in rodents to have a
clearly longer elimination half-life compared with 2,3,7,8-TeCDF; a
TEF of 0.2 - 0.3 may be justified.
The current limited experimental database does not allow a
complete hazard assessment and the recommendation of a safe level of
exposure to PBDDs/PBDFs for the general population.
However, if a comparison of the health impact of PBDDs/PBDFs with
their chlorinated analogues is needed, the data published on
2,3,7,8-TeBDD may be taken as an example. For 2,3,7,8-TeBDD, a NOAEL
of 10 ng/kg body weight per day may be established in a 13-week study
in rats. This value compares with the NOAEL of 10 ng/kg body weight
per day for 2,3,7,8-TeCDD, as derived from the 13-week study in rats.
The very limited data set available for concentrations of
PBDDs/PBDFs in environmental compartments makes it impossible to
conduct a proper risk evaluation for the environment. However, the few
data on levels of these substances indicate that they are much lower
than levels of their chlorinated counterparts. The assumption that
both brominated and chlorinated dibenzo- p-dioxins and dibenzofurans
act through a common mechanism and that their potency is not greatly
dependent on the nature of the halogen atom (chlorine or bromine) will
lead to the conclusion that the PBDDs/PBDFs will contribute marginally
to the total "dioxin" effect.
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
AND THE ENVIRONMENT
11.1 Conclusions
PBDDs/PBDFs are contaminants that are more or less similar to
PCDDs/PCDFs in their persistence and toxicity. Therefore, humans and
the environment should be protected from them. Exposure of the general
population to PCDDs/PCDFs appears to be greater than exposure to
PBDDs/PBDFs. Limited biomonitoring information indicates very low
residues, compared with PCDDs/PCDFs. Brominated flame retardants and
their precursors appear to be a main source of PBDDs/PBDFs.
A limited experimental database exists for PBDDs/PBDFs and would
therefore exclude an attempt at a complete hazard identification.
Current information does not allow a quantitative risk assessment,
although toxicological similarities appear to exist between certain
PBDD/PBDF congeners and their corresponding chlorinated homologues. On
an interim basis, it is suggested that current I-TEFs for the 17
2,3,7,8-substituted PCDD/PCDF congeners be applied to the comparable
brominated and mixed halogenated congeners.
11.2 Recommendations
Owing to the accumulating and toxic potential of some
PBDDs/PBDFs, every effort should be made to prevent exposure of humans
to, and pollution of the environment by, these compounds.
Brominated flame retardants should not be used where suitable
replacements are available, and future efforts should encourage the
development of further substitutes.
Appropriate precautions, including monitoring, should be taken
both to protect workers from exposure to PBDDs/PBDFs and to prevent
their release into the environment in emissions and effluents.
Disposal of industrial wastes, fire residues, and consumer
products containing brominated compounds should be controlled to
minimize environmental contamination by PBDDs/PBDFs and their
precursors. All products flame-retarded with bromine compounds should
be labelled and disposed of only in properly constituted waste
incinerators working at consistent operating conditions, to avoid the
release of PBDDs/PBDFs.
The use of leaded petrol, which necessitates the use of
halogenated scavengers, should be avoided.
Selected PBDD/PBDF congeners (2,3,7,8-TeBDD/TeBDF) should be
included in ongoing dioxin monitoring programmes to enhance the
existing database.
12. FURTHER RESEARCH
Analytical methods, including screening techniques, should be
improved. Interlaboratory comparisons should be undertaken to validate
methodologies.
As the experimental database is limited, comparative
toxicological and ecotoxicological studies with selected PBDD/PBDF
congeners should be performed with respect to both identifying
appropriate adverse- and no-adverse-effect levels and improving the
interim TEF recommendation.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
There have been no previous evaluations by international bodies.
(A toxicological evaluation of PBDDs/PBDFs was prepared by the
German Federal Health Office [Appel, 1991, 1993].)
REFERENCES
Abraham K, Krowke R, & Neubert D (1988) Pharmacokinetics and
biological activity of 2,3,7,8-tetrachlorodibenzo-para-dioxin: 1.
Dose-dependent tissue distribution and induction of hepatic
ethoxyresorufin O-deethylase in rats following a single injection.
Arch Toxicol, 62: 359-368.
Abraham K, Wiesmüller T, Brunner H, Krowke R, Hagenmaier H, & Neubert
D (1989) Elimination of various polychlorinated dibenzo- p-dioxins
and dibenzofurans (PCDDs and PCDFs) in rat faeces. Arch Toxicol, 63:
75-78.
Acharya P, DeCicco SG, & Novak RG (1991) Factors that can influence
and control the emissions of dioxins and furans from hazardous waste
incinerators. J Air Waste Manage Assoc, 41: 1605-1615.
Adams EM, Irish DD, Spencer HC, & Rove VK (1941) The response of
rabbit skin to compounds reported to have caused acneform dermatitis.
Ind Med Surg, 2: 1-4.
Ahlborg UG (1994) Toxic equivalency factors for polychlorinated
dioxins, dibenzofurans and biphenyls -- pro's and con's and
consequences. In: Fiedler H, Hutzinger O, Clement R, & Sakai S ed.
Dioxin'94: 14th International Symposium on Chlorinated Dioxins, PCB
and Related Compounds, Kyoto, Japan, 21-25 November 1994 (Short
papers). Kyoto, Kyoto University, Department of Environmental and
Sanitary Engineering, pp 11-14 (Organohalogen Compounds, Volume 21).
Ahlborg U, Brouwer A, Fingerhut M, Jacobson J, Jacobson S, Kennedy S,
Kettrup A, Koeman J, Poiger H, Rappe C, Safe S, Seegal R, Tuomisto J,
& Van den Berg M (1992) Impact of polychlorinated dibenzo- p-dioxins,
dibenzofurans, and biphenyls on human and environmental health, with
special emphasis on application of the toxic equivalency factor
concept. Eur J Pharmacol, 228: 179-199.
Alsabbagh AM, Aldous KM, Narang RS, & O'Keefe PW (1992) Formation of
brominated dioxins and brominated phenazines from pyrolyses of
2,4,6-tribromoaniline and N-(tribromophenyl) maleimide. Chemosphere,
24: 1625-1632.
Andersen ME, Mills JJ, Gargas ML, Kedderis L, Birnbaum LS, Neubert D,
& Greenlee WF (1993) Modeling receptor-mediated processes with dioxin:
implications for pharmacokinetics and risk assessment. Risk Anal, 13:
25-36.
Anon (1996) German ordinance on forbidden chemicals.
Bundesgesetzblatt, 1(39): 1151-1166.
Appel KE (1991) [Polybrominated dibenzodioxins and dibenzofurans:
Toxicological evaluation.] Bundesgesundheitsblatt, 10: 460-470 (in
German).
Appel KE (1993) Polybrominated dibenzodioxins and dibenzofurans:
Toxicological evaluation. In: The Toxicology Forum: Current views on
the impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1992. Washington DC, Toxicology Forum, Inc., pp
147-174.
Asahi M & Urabe H (1987) A case of "Yusho"-like skin eruptions due to
halogenated PCB-analogue compounds. Chemosphere, 16: 2069-2072.
ATOCHEM (1987) Decabromobiphenyl (Adine 0102): Pyrolysis testing.
Paris La Defense, France, ATOCHEM, 7 pp (Unpublished report).
ATOCHEM (1990) Decabromobiphenyl (Adine 0102). Paris La Defense,
France, ATOCHEM, 3 pp (Unpublished report).
Bacher R, Riehle U, Swerev M, & Ballschmiter K (1991) Patterns and
levels of polyhalogenated (Br-, Cl-) dibenzodioxins and dibenzofurans
in automobile traffic related samples. Chemosphere, 23: 1151-1171.
Ballschmiter KH & Bacher R (1996) [Dioxins: Chemistry, analysis,
occurrence, environmental behaviour and toxicology of halogenated
dibenzo- p-dioxins and dibenzofurans.] Weinheim, Germany, VCH
Publishers, 507 pp (in German).
Ballschmiter K, Bacher R, Riehle U, & Swerev M (1990) [Environmental
research report: Contribution of automobile exhaust to the general
environmental burden through dibenzodioxins (PHaIDD) and dibenzofurans
(PHaIDF).] Bonn, German Federal Ministry for Research and Technology,
234 pp (in German).
Banks YB & Birnbaum LS (1991) Absorption of
2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) after low dose dermal
exposure. Toxicol Appl Pharmacol, 107: 302-310.
Bannasch P, Wayss K, & Zerban H (1985) Peliosis hepatis, rodents. In:
Jones TC, Mohr U, & Hunt RD ed. Pathology of laboratory animals,
digestive system. Berlin, Springer Verlag, pp 110-115 (ILSI
Monograph).
Becher H, Flesch-Janys D, Kauppinen T, Kogevinas M, Steindorf K, Manz
A, & Wahrendorf J (1996) Cancer mortality in German male workers
exposed to phenoxy herbicides and dioxins. Cancer Causes Control, 7:
312-321.
Bergqvist PA, Strandberg B, Bergek S, & Rappe S (1993) Lipid reduction
during the analyses of PCDDs, PCDFs and PCBs in environmental samples
using semipermeable membrane technique. Organohalogen Compds, 11:
41-44.
Bertazzi PA & di Domenico A (1994) Chemical, environmental and health
aspects of the Seveso, Italy, accident. In: Schecter A ed. Dioxins and
health. New York, London, Plenum Press, pp 587-632.
Bieniek D, Bahadir M, & Korte F (1989) Formation of heterocyclic
hazardous compounds by thermal degradation of organic compounds.
Heterocycles, 28: 719-722.
Bingham AG, Edmunds CJ, Graham BWL, & Jones MT (1989) Determination of
PCDDs and PCDFs in car exhaust. Chemosphere, 19: 669-673.
Birla P & Kamens RM (1994) Effect of combustion temperature on the
atmospheric stability of polybrominated dibenzo- p-dioxins and
dibenzofurans. Environ Sci Technol, 28: 1437-1443.
Birnbaum LS (1993) EPA's reassessment of dioxin risk: Directed health
research. Chemosphere, 27: 469-475.
Birnbaum LS (1994) The mechanism of dioxin toxicity: Relationship to
risk assessment. Environ Health Perspect, 102: 157-167.
Birnbaum LS & DeVito MJ (1995) Use of toxic equivalency factors for
risk assessment for dioxins and related compounds. Toxicology, 105:
391-401.
Birnbaum LS, Harris MW, Stocking LM, Clark AM, & Morrissey RE (1989)
Retinoic acid and 2,3,7,8-tetrachlorodibenzo- p-dioxin selectively
enhance teratogenesis in C57BL/6N mice. Toxicol Appl Pharmacol, 98:
487-500.
Birnbaum LS, Morrissey RE, & Harris MW (1991) Teratogenic effects of
2,3,7,8-tetrabromodibenzo- p-dioxin and three polybrominated
dibenzofurans in C57BL/6N mice. Toxicol Appl Pharmacol, 107: 141-152.
Birnbaum LS, Ross DG, & DeVito MJ (1993) Dose response relationships
for EROD induction in liver, lung and skin for dioxin and
dibenzofurans. In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss
A, & Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 237-240 (Organohalogen
Compounds, Volume 13).
Blankenburg G, Hutzinger O, & Neubert D (1990) Preliminary assessment
of the potency of 2,3,7,8-tetra(bromo/chloro)dibenzo- p-dioxins to
induce EROD activity in primary cell cultures of rat hepatocytes. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 77-81 (Organohalogen Compounds, Volume
4).
BMU (1989) [Information from the Federal Minister for the Environment,
Nature Conservation and Nuclear Safety. Polybrominated dibenzodioxins
and dibenzofurans (PBDD/PBDF) from brominated flame retardants: Risk
assessment and suggestions for action.] Bonn, Federal Ministry for the
Environment, Nature Conservation and Nuclear Safety, 245 pp (in
German).
Bonilla JV, Munro HE, Mitchum RK, & Bauer MR (1990) Analysis of
brominated dibenzo- p-dioxins and dibenzofurans in ABS resins
containing brominated flame retardants. J Fire Sci, 8: 395-404.
Boyd SA & Mortland MM (1985) Dioxin radical formation and
polymerization on Cu(II) smectite. Nature (Lond), 316: 532-535.
Bradlaw JA & Casterline JL Jr (1979) Induction of enzyme activity in
cell culture: A rapid screen for detection of planar polychlorinated
organic compounds. J Assoc Off Anal Chem, 62: 904-916.
Bradlaw JA, Garthoff LH, Hurley NE, & Firestone D (1980) Comparative
induction of aryl hydrocarbon hydroxylase activity in vitro by
analogues of dibenzo- p-dioxin. Food Cosmet Toxicol, 18: 627-636.
Brenner KS (1993) Polystyrene/- and extruded polystyrene foam
(XPS)/-hexabromo-cyclododecane-blends under thermolytic stress; PBDD &
PBDF-determination. In: Fiedler H, Frank H, Hutzinger O, Parzefall W,
Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Environmental Agency, pp 381-386
(Organohalogen Compounds, Volume 11).
Brenner KS & Knies H (1990) Formation of polybrominated dibenzofurans
(PBDF's) and -dioxins (PBDD's) during extrusion production of a
polybutyleneterphthalate (PBTP)/glassfibre resin blended with
decabromodiphenylether (DBDPE)/Sb2O3: Product and workplace
analysis. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 319-325 (Organohalogen
Compounds, Volume 2).
Brenner KS & Knies H (1992) Workplace monitoring of PBDF's and PBDD's
during extrusion production and injection molding of a
polybutyleneterephthalate(PBTP)/glass fibre/tetrabromo bisphenol A
carbonate oligomer (BC52)/Sb2O3-resin. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 39-32 (Organohalogen Compounds, Volume 9).
Brenner KS & Knies H (1993a) Workplace monitoring of polybrominated
dibenzofurans (PBDF's) and -dioxins (PBDD's) during extrusion
production and molding of a poly (butylene-terephthalate) (PBTP)/glass
fibre resin blended with tetrabromobisphenol A carbonate oligomer (BC
52)/Sb2O3; air sampling train and product analysis. Toxicol Environ
Chem, 38: 81-94.
Brenner KS & Knies H (1993b) Workplace monitoring of PBDFs and PBDDs
during extrusion production and injection molding of a
polybutyleneterephthalate (PBTP)/glass fiber/tetrabromo-bisphenol A
carbonate oligomer (BC52*)/Sb2O3-resin: Part II. Chemosphere, 26:
1953-1963.
Brenner KS & Knies H (1994) Workplace monitoring of polybrominated
dibenzofurans (PBDF's) and -dioxins (PBDDs), Part III Extruder
production and injection molding of bis-tetrabromophthalimide ethylene
(TBPI)/Sb2O3-blended polybutylene-terephthalate (PBTB). In: Fiedler
H, Hutzinger O, Clement R, & Sakai S ed.Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 335-340
(Organohalogen Compounds, Volume 20).
Bretthauer EW, Kraus HW, & di Domenico A (1991) Dioxin perspectives: A
pilot study on international information exchange on dioxins and
related compounds, Volume 16. New York, London, Plenum Press, 772 pp.
Brewster DW, Banks YB, Clark AM, & Birnbaum LS (1989) Comparative
dermal absorption of 2,3,7,8-tetrachlorodibenzo- p-dioxin and three
polychlorinated dibenzofurans. Toxicol Appl Pharmacol, 97: 156-166.
Bröker G (1996) Comparative and validation PCDD/F-measurements with
different sampling systems. Organohalogen Compds, 27: 433-439.
Bruckmann P, Hackhe K, Ball M, Lis A, & Päpke O (1990) Degassing of
PBDD/PBDFs from a television set -- PBDD/PBDF levels after a fire in a
stock house -- two case studies. In: Freiji L ed. Proceedings of the
Workshop on Brominated Aromatic Flame Retardants, Skokloster, Sweden,
24-26 October 1989. Solna, National Chemicals Inspectorate (KEMI), pp
99-104.
Buckley LA (1995) Biologically-based models of dioxin
pharmacokinetics. Toxicology, 102: 125-131.
Bumpus JA (1989) Biodegradation of polycyclic aromatic hydrocarbons by
Phanerochaete chrysosporium. Appl Environ Microbiol, 55: 154-158.
Buser HR (1986a) Polybrominated dibenzofurans and dibenzo- p-dioxins:
Thermal reaction products of polybrominated diphenyl ether flame
retardants. Environ Sci Technol, 20: 404-408.
Buser HR (1986b) Selective detection of brominated aromatic compounds
using gas chromatography/negative chemical ionization mass
spectrometry. Anal Chem, 58: 2913-2919.
Buser HR (1987a) Brominated and brominated/chlorinated dibenzodioxins
and dibenzofurans: Potential environmental contaminants. Chemosphere,
16: 713-732.
Buser HR (1987b) [Brominated and mixed brominated/chlorinated
dibenzodioxins and dibenzofurans.] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
243-256 (VDI-Report No. 634) (in German).
Buser HR (1987c) Brominated and brominated/chlorinated dibenzodioxins
and dibenzofurans: Potential environmental contaminants. Chemosphere,
16: 1873-1876.
Buser HR (1988) Rapid photolytic decomposition of brominated and
brominated/chlorinated dibenzodioxins and dibenzofurans. Chemosphere,
17: 889-903.
Buser HR (1991) Review of methods of analysis for polychlorinated
dibenzodioxins and dibenzofurans. In: Rappe C, Buser HR, Dodet B, &
O'Neill IK ed. Environmental carcinogens: Methods of analysis and
exposure measurement. Lyon, International Agency for Research on
Cancer, pp 105-146 (IARC Scientific Publications No. 108).
Buser HR, Bosshardt HP, & Rappe C (1978) Formation of polychlorinated
dibenzofurans (PCDFs) from the pyrolysis of PCBs. Chemosphere, 1:
109-119.
Cerniglia CE, Morgan JC, & Gibson DT (1979) Bacterial and fungal
oxidation of dibenzofuran. Biochem J, 180: 175-185.
Chatkittikunwong W & Creaser CS (1994a) Stability of bromo- and
bromochloro-dibenzo- p-dioxins under laboratory and environmental
conditions. Chemosphere, 29: 547-557.
Chatkittikunwong W & Creaser CS (1994b) Microscale synthesis of
bromo- and bromochloro-dibenzo- p-dioxins and dibenzofurans.
Chemosphere, 28: 11-21.
Chatkittikunwong W & Creaser CS (1994c) Bromo-, bromochloro- and
dibenzo- p-dioxins and dibenzofurans in incinerator flyash.
Chemosphere, 29: 559-566.
Childers JW, Wilson NK, Harless RL, & Barbour RK (1992)
Characterization of brominated and bromo/chloro dibenzo- p-dioxins
and dibenzofurans by gas chromatography/matrix isolation- infrared
spectrometry. Chemosphere, 25: 1285-1290.
Chriske HW, Weilburg W, Drösemeier E, & Päpke O (1990) [Exposure to
dioxins and furans at computer workplaces?] Arbeitsmed Sozialmed
Präventivmed, 25: 302-306 (in German).
Clausen E, Lahaniatis ES, Bahadir M, & Bieniek D (1987) [Determination
of brominated dibenzofurans formed by thermolysis of polymers
containing decabromodiphenylether as flame retardant.] Fresenius Z
Anal Chem, 327: 297-300 (in German).
Cook PM, Kuehl DW, Walker MK, & Peterson RE (1991) Bioaccumulation and
toxicity of TCDD and related compounds in aquatic ecosystems. In:
Gallo MA, Scheuplein RJ, & van der Heijden CA ed. Biological basis for
risk assessment of dioxins and related compounds. Cold Spring Harbor,
New York, Cold Spring Harbor Laboratory Press, pp 143-167 (Banbury
Report 35).
Couture LA, Abbott BD, & Birnbaum LS (1990) A critical review of the
developmental toxicity and teratogenicity of
2,3,7,8-tetrachlorodibenzo- p-dioxin: Recent advances toward
understanding the mechanism. Teratology, 42: 619-627.
Cramer PH, Ayling RE, Thornburg KR, Stanley JS, Remmers JC, Breen JJ,
& Schwemberger J (1990a) Evaluation of an analytical method for the
determination of polybrominated dibenzo- p-dioxins/dibenzofurans
(PBDD/PBDF) in human adipose tissue. Chemosphere, 20: 821-827.
Cramer PH, Stanley JS, Bauer KM, Ayling RE, Thornburg KR, &
Schwemberger J (1990b) Final report on brominated dioxins and
dibenzofurans in human adipose tissue. Washington, DC, US
Environmental Protection Agency, 75 pp (EPA-560/5-90-005; PB
91-103507).
Dawidowsky N (1993) [Origin and occurrence of polyhalogenated
dibenzodioxins and dibenzofurans in the environment.] Tübingen,
Eberhard-Karls University, Faculty of Chemistry and Pharmacy, 188 pp
(Dissertation) (in German).
De Haan LHJ, Bos TA, Aarts JMMJG, & Brouwer A (1993) Inhibition of
gap-junctional intercellular communication by
tetrachlorodibenzo- p -dioxin (TCDD): Possible role of the Ah
receptor. In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, &
Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 157-161 (Organohalogen
Compounds, Volume 13).
De Jong APJM, van der Heeft E, Marsman JA, & Liem AKD (1992)
Investigation on the occurrence of polyhalogenated (Br/Cl)
dibenzodioxins and dibenzofurans in cow's milk and fish tissue.
Chemosphere, 25: 1551-1557.
De Jongh J, Buser HR, & Poiger H (1992) The metabolism of
2,3,7,8-tetrabromo-dibenzodioxin in the rat. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 33-36 (Organohalogen Compounds, Volume 10).
De Jongh J, Buser HR, & Poiger H (1993) The metabolism of
2,3,7,8-tetrabromo-dibenzodioxin in the rat. Xenobiotica, 23: 19-26.
Dellinger B, Maqsud L, & Sidhu S (1993) Kinetics of toxic combustion
by-product formation during brominated flame retardant incineration.
In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed.
Dioxin'93: 13th International Symposium on Chlorinated Dioxins and
Related Compounds, Vienna, 20-24 September 1993. Vienna, Austrian
Federal Environmental Agency, pp 261-264 (Organohalogen Compounds,
Volume 11).
Denison MS (1990) The molecular mechanism of action of
2,3,7,8-tetrachlorodibenzo- p -dioxin and related halogenated aromatic
hydrocarbons. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar:
10th International Symposium on Chlorinated Dioxins and Related
Compounds. Bayreuth, Germany, Ecoinforma Press, pp 95-98
(Organohalogen Compounds, Volume 4).
Denison MS (1991) The molecular mechanism of action of
2,3,7,8-tetrachlorodibenzo- p-dioxin and related halogenated aromatic
hydrocarbons. Chemosphere, 23: 1825-1830.
Denivelle L, Fort R, & Van Hai P (1960) Sur les dibenzo- p-dioxines
octochlorée et octobromée et sur les oxydes de phényle décachloré et
décabromé. Bull Soc Chim Fr, 11/12: 1538-1543.
Denomme MA, Homonoko K, Fujita T, Sawyer T, & Safe S (1985) Effects of
substituents on the cytosolic receptor-binding activities and aryl
hydrocarbon hydroxylase induction potencies of 7-substituted
2,3-dichlorodibenzo- p-dioxins. Mol Pharmacol, 27: 656-661.
Denomme MA, Homonoko K, Fujita T, Sawyer T, & Safe S (1986)
Substituted polychlorinated dibenzofuran receptor binding affinities
and aryl hydrocarbon hydroxylase induction potencies -- A QSAR
analysis. Chem-Biol Interact, 57: 175-187.
DeVito MJ & Birnbaum LS (1994) Toxicology of dioxins and related
chemicals. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 139-162.
De Wit C (1993) Bioaccumulation in aquatic and terrestrial organisms.
In: The Toxicology Forum: Current views on the impact of dioxins and
furans on human health and the environment, Berlin, 9-11 November
1992. Washington, DC, Toxicology Forum, Inc., pp 353-363.
De Wit CA, Järnberg UG, Asplund LT, Jansson B, Olsson M, Lindstedt LL,
Bergek S, Hjelt M, Rappe C, & Andersson Ö (1993) Biomagnification of
PCDD/PCDF, non-ortho and mono-ortho PCB in predators from aquatic and
terrestrial ecosystems. In: Fiedler H, Frank H, Hutzinger O, Parzefall
W, Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Environmental Agency, pp 325-328
(Organohalogen Compounds, Volume 12).
Di Domenico A, Silano A, Viviano V, & Zapponi G (1980) Accidental
release of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) at Seveso,
Italy. Ecotoxicol Environ Saf, 4: 339-345.
Diliberto JJ, Kedderis LB, & Birnbaum LS (1990a) Absorption of
2,3,7,8-tetrabromo-dibenzo- p-dioxin (TBDD) in male rats.
Toxicologist, 10: 54.
Diliberto JJ, Kedderis LB, & Birnbaum LS (1990b) Acute oral exposure
to 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD). In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 309-311 (Organohalogen Compounds, Volume 1).
Diliberto JJ, Jackson JA, & Birnbaum LS (1991) Acute pulmonary
absorption of 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in rats.
Toxicologist, 11: 272.
Diliberto JJ, Kedderis LB, Jackson JA, & Birnbaum LS (1993) Effects of
dose and routes of exposure on the disposition of
2,3,7,8-[3H]tetrabromodibenzo- p-dioxin (TBDD) in the rat. Toxicol
Appl Pharmacol, 120: 315-326.
Diliberto JJ, Jackson JA, & Birnbaum LS (1996) Comparison of
2,3,7,8-tetrachloro-dibenzo- p-dioxin (TCDD) disposition following
pulmonary, oral, dermal, and parenteral exposures to rats. Toxicol
Appl Pharmacol, 138: 158-168.
Donnelly JR & Sovocool GW (1991) Predictions of bromo- and
bromochloro-dioxin GC elution properties. Chemosphere, 22: 455-460.
Donnelly JR, Vonnahme TL, Hedin CM, & Niederhut WJ (1986) Evaluation
of RCRA method 8280 for analysis of dioxins and dibenzofurans. In:
Rappe C, Choudhary G, & Keith LH ed. Chlorinated dioxins and
dibenzofurans in perspective. Chelsea, Michigan, Lewis Publishers,
Inc., chapter 3, pp 399-435.
Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Hedin CM, Sovocool GW,
& Mitchum RK (1987) The chemistry and mass spectrometry of brominated
dibenzo- p-dioxins and dibenzofurans. Biomed Environ Mass Spectrom,
14: 465-472.
Donnelly JR, Grange AH, Nunn NJ, Sovocool GW, Brumley WC, & Mitchum RK
(1989a) Analysis of thermoplastic resins for brominated dibenzofurans.
Biomed Environ Mass Spectrom, 18: 884-896.
Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Sovocool GW, & Mitchum
RK (1989b) Preparation of bromochlorodibenzo- p-dioxins and
dibenzofurans and analysis by EPA RCRA method 8280. Chemosphere, 18:
209-216.
Donnelly JR, Grange AH, Nunn NJ, Sovocool GW, & Breen JJ (1990)
Bromo- and bromochloro-dibenzo- p-dioxins and dibenzofurans in the
environment. Chemosphere, 20: 1423-1430.
Donnelly JR, Munslow WD, Grange AH, Pettit TL, Simmons RD, Kumar KS, &
Sovocool GW (1991a) A gas chromatographic/mass spectrometric approach
for isomer-specific environmental monitoring of the 1700 bromo-,
chloro-, and bromochloro-dibenzo- p-dioxins. Biol Mass Spectrom, 20:
329-337.
Donnelly JR, Munslow WD, Grange AH, Pettit TL, Simmons RD, & Sovocool
GW (1991b) Correlation of the structure with linear retention index
for bromo- and bromochlorodibenzo- p-dioxins and bromodibenzofurans.
J Chromatogr, 540: 290-310.
Dumler R (1989) [Fire tests to study formation of brominated
dibenzofurans and -dioxins from fllame retarded plastics.] Bayreuth,
University of Bayreuth, Faculty of Biology, Chemistry and Geology, 132
pp (Dissertation) (in German).
Dumler R, Teufl C, Lenoir D, & Hutzinger O (1987) [Formation and
quantities of PBDF and PBDD from decomposition of flame retardants
using different pyrolysis apparata (poster).] In: [Dioxin -- A
technical, analytical, ecological and toxicological challenge:
Mannheim Colloquium, 5-7 May 1987.] Düsseldorf, Society of German
Engineers, pp 287-292 (VDI-Report No. 634) (in German).
Dumler R, Thoma H, Lenoir D, & Hutzinger O (1989a) Thermal formation
of polybrominated dibenzodioxins (PBDD) and dibenzofurans (PBDF) from
bromine containing flame retardants. Chemosphere, 19: 305-308.
Dumler R, Lenoir HTD, & Hutzinger O (1989b) PBDF and PBDD from the
combustion of bromine containing flame retarded polymers: A survey.
Chemosphere, 19: 2023-2031.
Dumler R, Lenoir D, Thoma H, & Hutzinger O (1989c) Thermal formation
of polybrominated dibenzofurans from decabromodiphenyl ether in a
polybutylene-terephthalate polymer matrix. J Anal Appl Pyrolysis, 16:
153-158.
Dumler R, Lenoir D, & Hutzinger O (1990a) Formation of brominated
dibenzofurans and -dioxins from the combustion of the flame retardant
decabromodiphenyl ether under different conditions. In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 325-328 (Organohalogen Compounds, Volume 2).
Dumler R, Lenoir D, Thoma H, & Hutzinger O (1990b) Thermal formation
of polybrominated dibenzofurans and dioxins from decabromodiphenyl
ether flame retardant: Influence of antimony(III) oxide and the
polymer matrix. Chemosphere, 20: 1867-1873.
Dumler R, Thoma H, & Hutzinger O (1990c) Content and formation of
toxic products in flame retardants. In: Freiji L ed. Proceedings of
the Workshop on Brominated Aromatic Flame Retardants, Skokloster,
Sweden, 24-26 October 1989. Solna, National Chemicals Inspectorate
(KEMI), pp 93-98.
Dumler-Gradl R, Tartler D, Thoma H, & Vierle O (1995) Detection of
polybrominated diphenylethers (PBDE), dibenzofurans (PBDF) and
dibenzodioxins (PBDD) in scrap of electronics and recycled products.
In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, & Reiner
E ed. Dioxin'95: 15th International Symposium on Chlorinated Dioxins
and Related Compounds, Edmonton, Canada, 21-25 August 1995 (Short
papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 101-104
(Organohalogen Compounds, Volume 24).
Esposito MP, Tiernan TO, & Dryden FE (1980) Dioxins (Final Report).
Cincinnati, Ohio, US Environmental Protection Agency, 371 pp
(EPA-600/2-80-197; PB 82-136847) .
Fabarius G, Wilken M, Bergas M, & Zeschmar-Lahl B (1990) Release of
organic pollutants during accidental fires. In: Hutzinger O & Fiedler
H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 373-377 (Organohalogen Compounds, Volume 3).
Fahrig R (1993) Genetic effects of dioxins in the spot test with mice.
Environ Health Perspect, 101(suppl 3): 257-261.
Fairchild WL, Muir DCG, Currie RS, & Yarechewski AL (1992) Emerging
insects as a biotic pathway for movement of
2,3,7,8-tetrachlorodibenzofuran from lake sediments. Environ Toxicol
Chem, 11: 867-872.
Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, & Gonzalez FJ
(1996) Aryl-hydrocarbon receptor-deficient mice are resistant to
2,3,7,8-tetrachlorodibenzo- p-dioxin-induced toxicity. Toxicol Appl
Pharmacol, 140: 173-179.
Fiedler H & Schramm KW (1990) QSAR generated octanol-water partition
coefficients of selected mixed halogenated dibenzodioxins and
dibenzofurans. Chemosphere, 20: 1597-1602.
Fiedler H & Van den Berg M (1996) Polychlorinated dibenzo- p-dioxins,
polychlorinated dibenzofurans, and related compounds. Update and
recent developments. ESPR-Environ Sci Pollut Res, 3: 122-128.
Figge K, Wernitz A, Fortnagel P, Wittich R-M, & Harms H (1991)
[Dibenzofuran: Bacterial mineralization -- kinetics of degradation in
heterogenous systems.] UWSF-Z Umweltchem Ökotoxikol, 3: 201-205 (in
German).
Figge K, Metzdorf U, Nevermann J, Schmiese J, Keskin M, Fortnagel P, &
Wittich RM (1993) [Bacterial mineralization of dibenzofuran,
dibenzo- p-dioxin, and 1,2,4,5-tetrachlorobenzene in soils.] UWSF-Z
Umweltchem Ökotoxikol, 5: 122-130 (in German).
Fingerhut MA, Halperin WE, Marlow BS, Piacitelli LA, Honchar PA,
Sweeny MH, Greife AL, Dill PA, Steenland K, & Surunda AJ (1991) Cancer
mortality in workers exposed to 2,3,7,8-tetrachlorodibenzo- p-dioxin.
N Engl J Med, 324: 212-218.
Fletcher CL & McKay WA (1993) Polychlorinated dibenzo- p-dioxins
(PCDDs) and dibenzofurans (PCDFs) in the aquatic environment -- A
literature review. Chemosphere, 26: 1041-1069.
Foght JM & Westlake DWS (1988) Degradation of polycyclic aromatic
hydrocarbons and aromatic heterocycles by a Pseudomonas species. Can
J Microbiol, 34: 1135-1141.
Fortnagel P, Harms H, Wittich R-M, Francke W, Krohn S, & Meyer H
(1989a) Cleavage of dibenzofuran and dibenzodioxin ring systems by a
Pseudomonas bacterium. Naturwissenschaften, 76: 222-223.
Fortnagel P, Wittich R-M, Harms H, Schmidt S, Franke S, Sinnwell V,
Wilkes H, & Francke W (1989b) New bacterial degradation of the biaryl
ether structure. Regioselective dioxygenation prompts cleavage of
ether bonds. Naturwissenschaften, 76: 523-524.
Fortnagel P, Harms H, Wittich R-M, Krohn S, Meyer H, Sinnwell V,
Wilkes H, & Francke W (1990) Metabolism of dibenzofuran by
Pseudomonas sp. strain HH69 and the mixed culture HH27. Appl Environ
Microbiol, 56: 1148-1156.
Fortunati GU, Banfi C, & Pasturenzi M (1994) Soil sampling. Fresenius
J Anal Chem, 348: 86-100.
Fulfs JC (1989) IRI Study 119.048 28, day range finding study
2,3,7,8-tetrabromo-dibenzofuran (TBDF) in the rat. Test article:
2,3,7,8-tetrabromodibenzofuran. Fort Collins, Colorado, Inhausen
Research Institute, Inc., 6 pp .
Funcke W & Hemminghaus HJ (1993) Evaluation of PCDF/D, PBDF/D and
PBCDF/D profiles in flue gas of combustion facilities using a
statistical distribution function. In: Fiedler H, Frank H, Hutzinger
O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th International
Symposium on Chlorinated Dioxins and Related Compounds, Vienna, 20-24
September 1993. Vienna, Austrian Federal Environmental Agency, pp
345-350 (Organohalogen Compounds, Volume 11).
Funcke W, Mayer J & Wilken M (1995) Analysis of polyhalogenated
dibenzofurans and dibenzo(p)dioxins (PXDF/Ds) in the scope of an
interlaboratory test. In: Bolt D, Clement R, Fiedler H, Harrison B,
Ramamoorthy S, & Reiner E ed. Dioxin'95: 15th International Symposium
on Chlorinated Dioxins and Related Compounds, Edmonton, Canada, 21-25
August 1995 (Short papers). Edmonton, Alberta, Dioxin'95 Secretariat,
pp 311-314 (Organohalogen Compounds, Volume 23).
Gebefügi I (1989) Chemical exposure in enclosed environments. Toxicol
Environ Chem, 20-21: 121-127.
Gebefügi I & Kreuzig G (1989) [Surface concentration of halogenated
compounds in indoor rooms.] In: [Halogenated organic compounds in the
environment -- Source, measurement, effect, corrective measures:
Mannheim Colloquium, 25-27 April 1989.] Düsseldorf, Society of German
Engineers, vol 1, pp 503-510 (VDI Report No. 745) (in German).
German Dioxin Directive (1994) [First directive for the changing of
the directive on forbidden chemicals.] Bundesgesetzblatt, 1: 1493-1495
(in German).
Giesy JP, Ludwig JP, & Tillitt DE (1994) Dioxins, dibenzofurans, PCBs
and colonial, fish-eating water birds. In: Schecter A ed. Dioxins and
health. New York, London, Plenum Press, pp 249-307.
Gilbertson M (1989) Effects on fish and wildlife populations. In:
Kimbrough RD & Jensen AA ed. Halogenated biphenyls, terphenyls,
naphthalenes, dibenzodioxins and related products, 2nd ed. Amsterdam,
Oxford, New York, Elsevier Science Publishers, chapter 4, pp 103-127.
Gilman H & Dietrich J (1957) Halogen derivatives of
dibenzo- p-dioxin. J Am Chem Soc, 79: 1439-1441.
Goldstein JA & Safe S (1989) Mechanism of action and
structure-activity relationship for the chlorinated
dibenzo- p-dioxins and related compounds. In: Kimbrough RD & Jensen
AA ed. Halogenated biphenyls, terphenyls, naphthalenes, dibenzodioxins
and related products, 2nd ed. Amsterdam, Oxford, New York, Elsevier
Science Publishers, chapter 9, pp 239-293.
Golor G, Yamashita K, McLachlan M, Hutzinger O, & Neubert D (1993)
Comparison of the kinetics of chlorinated and brominated dioxins and
furans in the rat. In: Fiedler H, Frank H, Hutzinger O, Parzefall W,
Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Governmental Agency, pp 203-206
(Organohalogen Compounds, Volume 12).
Golor G, Kociok O, Persaud TNV, Hinkel M, & Petrick K (1995)
Toxicokinetic properties of trihalogenated dibenzo- p-dioxins in the
rat. In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, &
Reiner E ed. Dioxin'95: 15th International Symposium on Chlorinated
Dioxins and Related Compounds, Edmonton, Canada, 21-25 August 1995
(Short papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 269-272
(Organohalogen Compounds, Volume 25).
Gray LE, Kelce WR, Monosson E, Ostby JS, & Birnbaum LS (1995) Exposure
to TCDD during development permanently alters reproductive function in
male Long Evans rats and hamsters: reduced ejaculated and epididymal
sperm numbers and sex accessory gland weights in offspring with normal
androgenic status. Toxicol Appl Pharmacol, 131: 108-118.
Gullett BK, Lemieux PM, & Dunn JE (1994) Role of combustion and
sorbent parameters in prevention of polychlorinated dibenzo- p-dioxin
and polychlorinated dibenzofuran formation during waste combustion.
Environ Sci Technol, 28: 107-118.
Hagenmeier H (1994) Contributions of diesel-powered vehicles and wood
burning to overall PCDD/PCDF compounds. In: Fiedler H, Hutzinger O,
Clement R, & Sakai S ed. Dioxin'94: 14th International Symposium on
Chlorinated Dioxins, PCB and Related Compounds, Kyoto, Japan, 21-25
November 1994 (Short papers). Kyoto, Kyoto University, Department of
Environmental and Sanitary Engineering, pp 267-270 (Organohalogen
Compounds, Volume 20).
Hagenmaier H, Brunner H, Haag R, & Kraft M (1987a) Copper-catalyzed
dechlorination-hydrogenation of polychlorinated dibenzofurans and
other chlorinated aromatic compounds. Environ Sci Technol, 21:
1085-1088.
Hagenmaier H, Brunner H, Haag R, & Kraft M (1987b) [The significance
of catalytic effects in the formation and destruction of
polychlorinated dibenzodioxins and polychlorinated dibenzofurans.]
Düsseldorf, Society of German Engineers, pp 557-584 (VDI-Report No.
634) (in German).
Hagenmaier H, Dawidowsky N, Weberruß U, Hutzinger O, Schwind KH, Thoma
H, Essers U, Bühler U, & Greiner R (1990a) Emission of polyhalogenated
dibenzodioxins and dibenzofurans from combustion-engines. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 329-334 (Organohalogen Compounds, Volume
2).
Hagenmaier H, Wiesmueller T, Golor G, Krowke R, Helge H, & Neubert D
(1990b) Transfer of various polychlorinated dibenzo- p-dioxins and
dibenzofurans (PCDDs and PCDFs) via placenta and through milk in a
marmoset monkey. Arch Toxicol, 64: 601-615.
Hagenmaier H, She J, Benz T, Dawidowsky N, Düsterhöft L, & Linding C
(1992) Analysis of sewage sludge for polyhalogenated
dibenzo- p-dioxins, dibenzofurans, and diphenylethers. Chemosphere,
25: 1457-1462.
Haglund P, Egebäck KE, & Jansson B (1988) Analysis of polybrominated
dioxins and furans in vehicle exhaust. Chemosphere, 17: 2129-2140.
Hahn ME & Stegeman JJ (1992) Phylogenetic distribution of the Ah
receptor in non-mammalian species: Implications for dioxin toxicity
and Ah receptor evolution. Chemosphere, 25: 931-937.
Hamm S & Theisen J (1992) Formation of polybrominated dibenzofurans
and polybrominated dibenzo(p)dioxins at fires of electrical
appliances. Poster presented at Dioxin'92: 12th International
Symposium on Dioxins and Related Compounds, Tampere, Finland, 24-28
August 1992, 9 pp.
Hammel KE, Kalyanaraman B, & Kirk TK (1986) Oxidation of polycyclic
aromatic hydrocarbons and dibenzo[p]-dioxins by Phanerochaete
chrysosporium ligninase. J Biol Chem, 261: 16948-16952.
Happe B, Eltis LD, Poth H, Hedderich R, & Timmis KN (1993)
Characterization of 2,2',3-trihydroxybiphenyl dioxygenase, an
extradiol dioxygenase from dibenzofuran- and
dibenzo- p-dioxin-degrading bacterium Sphingomonas sp. strain RW1.
J Bacteriol, 175: 7313-7320.
Hardell L, Eriksson M, Axelson O, & Zahm SH (1994) Cancer
epidemiology. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 525-547.
Hardy ML, Sistrunk PH, Eldan M, & McFadden DJ (1990)
2,3,7,8-tetrabromodibenzo-furan (TBDF): 4 week subchronic toxicity
study in the rat. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
313-316 (Organohalogen Compounds, Volume 1).
Harless RL, Lewis RG, McDaniel DD, & Dupuy AE Jr (1989) Identification
of bromochlorodibenzo- p-dioxins and dibenzofurans in ash samples.
Chemosphere, 18: 201-208.
Harless RL, Lewis RG, McDaniel DD, Gibson JF, & Dupuy AE (1992)
Evaluation of a sampling and analysis method for determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in ambient air.
Chemosphere, 25: 1317-1322.
Harms H, Wittich R-M, Sinnwell V, Meyer H, Fortnagel P, & Francke W
(1990) Transformation of dibenzo- p-dioxin by Pseudomonas sp.
strain HH69. Appl Environ Microbiol, 56: 1157-1159.
Harms H, Lorenz W, & Bahadir M (1995) [Rapid determination of
polyhalogenated dioxins and furans at fire residues.] GIT Fachz Lab,
39: 724-729 (in German).
Hartenstein H-U (1993) Fixed bed activated coke filters for the
control of toxic metals and organics from waste incinerators. In:
Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed.
Dioxin'93: 13th International Symposium on Chlorinated Dioxins and
Related Compounds, Vienna, 20-24 September 1993. Vienna, Austrian
Federal Environmental Agency, pp 11-16 (Organohalogen Compounds,
Volume 12).
Heinbuch D & Stieglitz L (1992) Formation of brominated compounds on
fly ash. In: Dioxin'92: 12th International Symposium on Chlorinated
Dioxins and Related Compounds, Tampere, Finland, 24-28 August 1992.
Helsinki, Finnish Institute of Occupational Health, pp 257-260
(Organohalogen Compounds, Volume 8).
Heinbuch D & Stieglitz L (1993) Formation of brominated compounds on
fly ash. Chemosphere, 27: 317-324.
Helge H (1993) A pediatrician's view of studies on dioxin-attributed
effects in children. In: The Toxicology Forum: Current views on the
impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1993. Washington, DC, Toxicology Forum, Inc., pp
272-287.
Hiester E (1992) [Measurement of polyhalogenated dioxins and furans in
air.] Essen, Germany, Institute for Air Pollution Control for the
State of Northrhine-Westfalia, pp 87-93 (LIS Report) (in German).
Hileman F, Wehler J, Wendling J, Orth R, Ritchie C, & McKenzie D
(1989) Dibenzofuran in diphenyl oxide and the relationship to
brominated dibenzofurans in brominated diphenyl oxide. Chemosphere,
18: 217-224.
Hofmann KH, Polnisch E, Kreisel H, Mach H, & Schubert M (1992)
Degradation of dibenzofuran and dibenzodioxins by fungi. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 93-96 (Organohalogen
Compounds, Volume 9).
Holsapple MP, Morris DL, Wood SC, & Snyder NK (1991)
2,3,7,8-Tetrachlorodibenzo- p-dioxin induced changes in
immunocompetence: Possible mechanisms. Annu Rev Pharmacol Toxicol, 31:
73-100.
Hornung MW, Zabel EW, & Peterson RE (1996a) Additive interactions
between pairs of polybrominated dibenzo- p-dioxin, dibenzofuran, and
biphenyl congeners in a rainbow trout early life stage mortality
bioassay. Toxicol Appl Pharmacol, 140: 345-355.
Hornung MW, Zabel EW, & Peterson RE (1996b) Toxic equivalency factors
of polybrominated dibenzo- p-dioxin, dibenzofuran, biphenyl, and
polyhalogenated diphenyl ether congeners based on rainbow trout early
life stage mortality. Toxicol Appl Pharmacol, 140: 227-234.
Hosseinpour J, Schwind KH, & Thoma H (1989) Brominated-chlorinated
dibenzo- p-dioxins and furans: Synthesis of standards and detection
in fly ash from a municipal waste incinerator. Chemosphere, 19:
109-114.
Huang LQ, Moore C, McGown S, Tomer KB, & Tong HY (1991) The
application of high-resolution MS and hybrid MS/MS to the analysis of
flyash for monobrominated polychlorinated dibenzo- p-dioxins and
dibenzofurans. Abstr Papers Am Chem Soc, 202: analytical section,
abstract 50.
Huang LQ, Paiva A, Tong H, Monson SJ, & Gross ML (1992a) Application
of gas chromatography high-resolution mass spectrometry to the
determination of trace monobromopolychlorodibenzo- p-dioxins in
environmental samples. J Am Soc Mass Spectrom, 3: 248-259.
Huang LQ, Tong H, & Donnelly JR (1992b) Characterization of
dibromopolychlorodibenzo- p-dioxins and
dibromopolychlorodibenzofurans in municipal waste incinerator fly ash
using gas chromatography/mass spectrometry. Anal Chem, 64: 1034-1040.
Huff J (1994) Dioxins and mammalian carcinogenesis. In: Schecter A ed.
Dioxins and health. New York, London, Plenum Press, pp 389-408.
Hutzinger O (1990) [Studies on the possible emission of polybrominated
dibenzodioxins and dibenzofurans during fires with flame retarded
plastics.] Bayreuth, University of Bayreuth, Department of
Environmental Chemistry and Geochemistry, 68 pp (Research Report No.
10403362 prepared for the German Federal Ministry of Environment) (in
German).
Hutzinger O, Dumler R, Lenoir D, Teufl C, & Thoma H (1989) PBDD and
PBDF from brominated flame retardants: Combustion equipment,
analytical methodology and synthesis of standards. Chemosphere, 18:
1235-1242.
Hutzinger O, Hagenmaier H, & Essers U (1990) [Studies on the emission
of halogenated dibenzodioxins and dibenzofurans from combustion motors
using commercial fuels.] Bonn, German Federal Ministry for Research
and Technology, 224 pp (in German).
Ivens I, Neupert M, Löser E, & Thies J (1990) Storage and elimination
of 2,3,7,8-tetrabromodibenzo- p-dioxin in liver and adipose tissue of
the rat. Chemosphere, 20: 1209-1214.
Ivens IA, Löser E, Rinke M, Schmidt U, & Neupert M (1992) Toxicity of
2,3,7,8-tetrabromodibenzo- p-dioxin in rats after single oral
administration. Toxicology, 73: 53-69.
Ivens IA, Löser E, Rinke M, Schmidt U, & Mohr U (1993) Subchronic
toxicity of 2,3,7,8-tetrabromodibenzo- p-dioxin in rats. Toxicology,
83: 181-201.
Ivens-Kohl I, Mohr U, & Löser E (1989)
[2,3,7,8-Tetrabromodibenzo- p-dioxin: subchronic toxicological study
on rats (oral application via stomach tube for 3 months with
subsequent observation period of 3 months).] Wuppertal, Germany, Bayer
AG, 1298 pp (Unpublished report No. 18477) (in German).
Ivens-Kohl I, Rinke M, & Löser E (1990)
[2,3,7,8-Tetrabromodibenzo- p-dioxin: Studies on acute oral toxicity
in male and female Wistar rats.] Wuppertal, Germany, Bayer AG, 205 pp
(Unpublished report No. 18668) (in German).
Jackson JA, Diliberto JJ, Kedderis LB, & Birnbaum LS (1991) Dermal
absorption and disposition of 2,3,7,8-tetrabromodibenzo- p-dioxin
(TBDD) in rats. Toxicologist, 11: 271.
Jackson JA, Diliberto JJ, & Birnbaum LS (1993) Estimation of
octanol-water partition coefficients and correlation with dermal
absorption for several polyhalogenated aromatic hydrocarbons. Fundam
Appl Toxicol, 21: 334-344.
Jansson B, Andersson R, Asplund L, Litzen K, Nylund K, Sellström U,
Uvemo U-B, Wahlberg C, Wideqvist U, Odsjö T, & Olsson M (1993)
Chlorinated and brominated persistent organic compounds in biological
samples from the environment. Environ Toxicol Chem, 12: 1163-1174.
Jay K & Stieglitz L (1996) Synthesis of mixed halogenated
dibenzofurans (X = Cl, Br). Chemosphere, 33: 1041-1045.
Jödicke B, Ende M, Helge H, & Neubert D (1992) Fecal excretion of
PCDDs/PCDFs in a 3-month-old breast-fed infant. Chemosphere, 25:
1061-1065.
Johnson J, Breen JJ, Murray TM, Glatz JA, Steele DH, & Stanley JS
(1989) Polyhalogenated dibenzo- p-dioxins/dibenzofurans testing and
reporting requirements under the Toxic Substances Control Act (TSCA).
Chemosphere, 19: 849-852.
Johnson L, Wilker CE, Safe SH, Scott B, Dean D, & White PH (1994)
2,3,7,8-Tetrachlorodibenzo- p-dioxin reduces the number, size and
organelle content of Leydig cells in adult rat testes. Toxicology, 89:
49-65.
Kapila S, Yanders AF, Orazio CE, Meadows JE, Cerlesi S, & Clevenger TE
(1989) Field and laboratory studies on the movement and fate of
tetrachlorodibenzo- p-dioxin in soil. Chemosphere, 18: 1297-1304.
Kedderis LB, Diliberto JJ, & Birnbaum LS (1990) Disposition of
intravenous 2,3,7,8-tetrabromodibenzodioxin (TBDD) in rats.
Toxicologist, 10: 310.
Kedderis LB, Diliberto JJ, & Birnbaum LS (1991a) Disposition and
excretion of intravenous 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD)
in rats. Toxicol Appl Pharmacol, 108: 397-406.
Kedderis LB, Diliberto JJ, Linko P, Goldstein JA, & Birnbaum LS
(1991b) Disposition of 2,3,7,8-tetrabromodibenzo- p-dioxin and
2,3,7,8-tetrachlorodibenzo- p-dioxin in the rat: Biliary excretion
and induction of cytochromes CYP1A1 and CYP1A2. Toxicol Appl
Pharmacol, 111: 163-172.
Kedderis LB, Diliberto JJ, Jackson JA, Linko P, Goldstein JA, &
Birnbaum LS (1992a) Effects of dose and route of exposure on dioxin
disposition. Chemosphere, 25: 7-10.
Kedderis LB, Mills JJ, Andersen ME, & Birnbaum LS (1992b) A
physiologically-based pharmacokinetic model for
2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in the rat. In: Dioxin'92:
12th International Symposium on Chlorinated Dioxins and Related
Compounds, Tampere, Finland, 24-28 August 1992. Helsinki, Finnish
Institute of Occupational Health, pp 113-116 (Organohalogen Compounds,
Volume 10).
Kedderis LB, Mills JJ, Andersen ME, & Birnbaum LS (1993) A
physiologically based pharmacokinetic model for
2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in the rat: Tissue
distribution and CYP1A induction. Toxicol Appl Pharmacol, 121: 87-98.
Kedderis LB, Jackson JA, Patterson DG, Grainger J, Diliberto JJ, &
Birnbaum LS (1994) Chemical characterization and disposition studies
with 1,2,7,8-tetrabromodibenzofuran in the rat. J Toxicol Environ
Health, 41: 53-69.
Kende AS & Wade JJ (1973) Synthesis of new steric and electronic
analogs of 2,3,7,8-tetrachlorodibenzo- p-dioxin. Environ Health
Perspect, 5: 49-57.
Kende AS, Wade JJ, Ridge D, & Poland A (1974) Synthesis and Fourier
transform carbon-13 spectroscopy of new toxic
polyhalodibenzo- p-dioxins. J Org Chem, 39: 931-937.
Kieatiwong S, Nguyen LV, Hebert VR, Hackett M, Miller G, Miille MJ, &
Mitzel R (1990) Photolysis of chlorinated dioxins in organic solvents
and on soils. Environ Sci Technol, 24: 1575-1580.
Kieper H (1996) [Exposure to polybrominated dibenzofurans and -dioxins
in copper plants and during the production of flame retarded
plastics.] Bremerhaven, Wirtschaftsverlag NW, pp 1-115 (in German).
Kjeller L-O, Kulp SE, Jonsson B, & Rappe C (1993) Methodology for the
determination of polychlorinated dibenzo- p-dioxins and dibenzofurans
in sediment samples. Toxicol Environ Chem, 39: 1-12.
Klecka GM & Gibson DT (1979) Metabolism of dibenzo- p-dioxin by a
Pseudomonas species. Biochem J, 180: 639-645.
Klecka GM & Gibson DT (1980) Metabolism of dibenzo- p-dioxin and
chlorinated dibenzo- p-dioxins by a Beijerinckia species. Appl
Environ Microbiol, 39: 288-296.
Klusmeier W, Vögler P, Ohrbach KH, Weber H, & Kettrup A (1988) Thermal
decomposition of decabromodiphenyl ether. J Anal Appl Pyrolysis, 13:
277-285.
Kociba RJ & Schwetz BA (1982) Toxicity of
2,3,7,8-tetrachloro- p-dioxin (TCDD). Drug Metab Rev, 13: 387-406.
Kociba RJ, Keeler PA, Park CN, & Gehring PJ (1976)
2,3,7,8-Tetrachlorodibenzo- p-dioxin (TCDD): Results of a 13-week
oral toxicity study in rats. Toxicol Appl Pharmacol, 35: 553-574.
Kogevinas M, Parkin DM, Cordier S, Cung TA, Hung L, Cao Dai L,
Rivera-Pomar E, Raphael M, & Stellman S (1993) Case-control studies on
soft-tissue sarcoma and non-Hodgkin lymphoma in Vietnam. In: Fiedler
H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93:
13th International Symposium on Chlorinated Dioxins and Related
Compounds, Vienna, 20-24 September 1993. Vienna, Austrian Federal
Environmental Agency, pp 375-378 (Organohalogen Compounds, Volume 13).
Kutz FW, Barnes DG, Bretthauer EW, Bottimore DP, & Greim H (1990) The
international toxicity equivalency factor (I-TEF) method for
estimating risks associated with exposures to complex mixtures of
dioxins and related compounds. Toxicol Environ Chem, 26: 99-109.
Lahaniatis ES, Bergheim W, & Rainer C (1989) Hazardous halogenated
substances formed during combustion processes. Toxicol Environ Chem,
20/21: 501-506.
Lahaniatis E, Bergheim W, & Bieniek D (1991) Formation of
2,3,7,8-tetrabromo-dibenzodioxin and -furan by thermolysis of polymers
containing brominated flame retardants. Toxicol Environ Chem, 31/32:
521-526.
Lahl U, Wilken M, & Wiebe A (1991) [Polybrominated diphenylether in
waste incineration.] Müll Abfall, 23: 83-87 (in German).
Landers JP & Bunce NJ (1991) The Ah-receptor and the mechanism of
dioxin toxicity. Biochem J, 276: 273-287.
Lee A, Campbell B, & Kelly W (1986) Dioxin and furan contamination in
the manufacture of halogenated organic chemicals (Contract No.
68-03-3274). Cincinnati, Ohio, US Environmental Protection Agency, 71
pp (EPA/600/2-86/101; PB 87-119905).
Lee A, Campbell B, & Kelly W (1987) Project summary: Dioxin and furan
contamination in the manufacture of halogenated organic chemicals
(Contract No. 68-03-3274). Cincinnati, Ohio, US Environmental
Protection Agency, 7 pp (EPA/600/S2-86/101).
Lenoir D (1994) Behaviour and fate of aromatic bromine compounds in
the environment. In: Advances in organobromine chemistry. Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp 363-386.
Lenoir D, Schramm K-W, Hutzinger O, & Schedel G (1991) Photochemical
degradation of brominated dibenzo- p-dioxins and -furans in organic
solvents. Chemosphere, 22: 821-834.
Lenoir D, Zier B, Bieniek D, & Kettrup A (1994) The influence of water
and metals on PBDD/F concentration in incineration of
decabromobiphenyl ether in polymeric matrices. Chemosphere, 28:
1921-1928.
Loganathan BG, Kannan K, Watanabe I, Kawano M, Irvine K, Kumar S, &
Sikka HC (1995) Isomer-specific determination and toxic evaluation of
polychlorinated biphenyls, polychlorinated/ brominated
dibenzo- p-dioxins and dibenzofurans, polybrominated biphenyl ethers,
and extractable organic halogen in carp from the Buffalo River, New
York. Environ Sci Technol, 29: 1832-1838.
Lorenz W (1994) [Emissions from halogen-containing plastics in certain
treatment processes.] Abfallwirtsch Lichte neuen Vorschr, 9: 143-157
(in German).
Lorenz W & Bahadir M (1993) Recycling of flame retardants containing
printed circuits: A study of the possible formation of polyhalogenated
dibenzodioxins/-furans. Chemosphere, 26: 2221-2229.
Lorenzen A & Okey AB (1991) Detection and characterization of Ah
receptor in tissue and cells from human tonsils. Toxicol Appl
Pharmacol, 107: 203-214.
Löser E & Ivens I (1989) Preliminary results of a 3 month toxicity
study on rats with 2,3,7,8-tetrabromodibenzo- p-dioxin
(2,3,7,8-TBDD). Chemosphere, 19: 759-764.
Lucier GW (1991) Humans are a sensitive species to some of the
biochemical effects of structural analogs of dioxin. Environ Toxicol
Chem, 10: 727-735.
Lucier GW, Portier CJ, & Gallo MA (1993a) Receptor mechanisms and
dose-response models for the effects of dioxins. Environ Health
Perspect, 101: 37-44.
Lucier GW, Clark G, Hiermath C, Tritscher A, Sewall C, & Huff J
(1993b) Carcinogenicity of TCDD in laboratory animals: Implications
for risk assessment. Toxicol Ind Health, 9: 631-668.
Luijk R & Govers HAJ (1992) The formation of polybrominated
dibenzo- p-dioxins (PBDDs) and dibenzofurans (PBDFs) during pyrolysis
of polymer blends containing brominated flame retardants. Chemosphere,
25: 361-374.
Luijk R, Wever H, Olie K, & Govers HAJ (1990) Formation of
polybrominated dibenzo- p-dioxins and -dibenzofurans during pyrolysis
of polybrominated diphenylethers and high impact polystyrene. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 335-338 (Organohalogen Compounds, Volume
2).
Luijk R, Wever H, Olie K, Govers HAJ, & Boon JJ (1991) The influence
of the polymer matrix on the formation of polybrominated
dibenzo- p-dioxins (PBDDs) and polybrominated dibenzofurans (PBDFs).
Chemosphere, 23: 1173-1183.
Luijk R, Jansen J, & Govers HAJ (1992a) The exchange of bromine and
chlorine in 2,3,7,8-tetrabromo-dibenzo- p-dioxin. In: Dioxin'92: 12th
International Symposium on Chlorinated Dioxins and Related Compounds,
Tampere, Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 99-101 (Organohalogen Compounds, Volume 8).
Luijk R, Dorland K, Smit P, & Govers HAJ (1992b) The halogenation of
dibenzo- p-dioxin and dibenzofuran in a model fly ash system. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 273-276 (Organohalogen
Compounds, Volume 8).
Luijk R, Govers HAJ, & Nellssen L (1992c) Formation of polybrominated
dibenzofurans during extrusion of high-impact
polystyrene/decabromodiphenyl ether/antimony(III)oxide. Environ Sci
Technol, 26: 2191-2198.
Luijk R, Dorland C, Smit P, Jansen J, & Govers HAJ (1994) The role of
bromine in the de novo synthesis in a model fly ash system.
Chemosphere, 28: 1299-1309.
Lutes CC, Charles MJ, & Kamens RM (1990) The atmospheric stability of
polybrominated dibenzo- p-dioxins and dibenzofurans. In: Jayanty RKM
& Gay BW Jr ed. Measurement of toxic and related air pollutants:
Proceedings of the 1990 EPA/A and WMA International Symposium,
Raleigh, North Carolina, 1-4 May 1990. Research Triangle Park, North
Carolina, Research Triangle Institute, pp 38-44 (EPA/600/9-90/026; PB
91-120279).
Lutes CC, Charles MJ, & Kamens RM (1992a) The atmospheric stability of
polybrominated dibenzo- p-dioxins and dibenzofurans. Chemosphere, 25:
99-102.
Lutes CC, Charles MJ, Odum JR, & Kamens RM (1992b) Chamber aging
studies on the atmospheric stability of polybrominated
dibenzo- p-dioxins and dibenzofurans. Environ Sci Technol, 26:
991-998.
Mably TA, Bjerke DL, Moore RW, Gendron-Fitzpatrick A, & Peterson RE
(1992) In utero and lactational exposure of male rats to
2,3,7,8-tetrachlorodibenzo- p-dioxin: 3. Effects of spermatogenesis
and reproductive capability. Toxicol Appl Pharmacol, 114: 118-126.
Maier EA, Griepink B, & Fortunati U (1994) Round table discussions.
Outcome and recommendations. Fresenius J Anal Chem, 348: 171-179.
Manz A, Berger J, Dwyer JH, Flesch-Janys D, Nagel S, & Waltsgott H
(1991) Cancer mortality among workers in chemical plant contaminated
with dioxin. Lancet, 338: 959-964.
Marklund S, Rappe C, Tysklind M, & Egebäck KE (1987) Identification of
polychlorinated dibenzofurans and dioxins in exhausts from cars run on
leaded gasoline. Chemosphere, 16: 29-36.
Marklund S, Andersson R, Tysklind M, Rappe C, Egebäck KE, Björkman E,
& Grigoriadis V (1990) Emissions of PCDD's and PCDF's in gasoline and
diesel fueled cars. Chemosphere, 20: 553-561.
Mason G, Farrell B, Keys B, Piskorska-Pliszczynsksa J, Safe L, & Safe
S (1986) Polychlorinated dibenzo- p-dioxins: Quantitative in vivo
and in vitro structure-activity relationships. Toxicology, 41:
21-31.
Mason G, Zacharewski T, Denomme MA, Safe L, & Safe S (1987a)
Polybrominated dibenzo- p-dioxins and related compounds:
Quantitative in vivo and in vitro structure-activity
relationships. Toxicology, 44: 245-255.
Mason G, Denomme MA, Safe L, & Safe S (1987b) Polybrominated and
chlorinated dibenzo- p-dioxins: Synthesis biologic and toxic effects
and structure-activity relationships. Chemosphere, 16: 1729-1731.
Massa T, Gerber T, Pfaffenholz V, Chandra A, Schlatterer B, & Chandra
B (1990) A host-mediated in vivo/ in vitro assay with peritoneal
murine macrophages for the detection of carcinogenic chemicals. J
Cancer Res Clin Oncol, 116: 357-364.
Massa T, Esmaeili A, Schlatterer B, Hagenmaier H, & Chandra P (1991)
Carcinogenic and co-carcinogenic potential of
2,3,7,8-tetrachlorodibenzo- p-dioxin in a host-mediated in vivo/
in vitro assay. Chemosphere, 23: 1855-1868.
Massa T, Esmaeili A, Fortmeyer H, Schlatterer B, Hagenmaier H, &
Chandra P (1992a) Carcinogenic and co-carcinogenic potential of
2,3,7,8-tetrachlorodibenzodioxin in a host-mediated in vivo/
in vitro assay. Chemosphere, 25: 1085-1090.
Massa T, Esmaeili A, Fortmeyer H, Schlatterer B, Hagenmaier H, &
Chandra P (1992b) Cell transforming and oncogenic activity of
2,3,7,8-tetrachloro- and 2,3,7,8-tetrabromodibenzo- p-dioxin.
Anticancer Res, 12: 2053-2060.
McAllister DL, Mazac CJ, Gorsich R, Freiberg M, & Tondeur Y (1990)
Analysis of polymers containing brominated diphenyl ethers as flame
retardants after molding under various conditions. Chemosphere, 20:
1537-1541.
McConnell EE (1989) Acute and chronic toxicity and carcinogenesis in
animals. In: Kimbrough RD & Jensen AA ed. Halogenated biphenyls,
terphenyls, naphthalenes, dibenzodioxins and related products, 2nd ed.
Amsterdam, Oxford, New York, Elsevier Science Publishers, chapter 6,
pp 161-193.
McKinley MK, Kedderis LB, & Birnbaum LS (1993) The effect of
pretreatment on the biliary excretion of
2,3,7,8-tetrachlorodibenzo- p-dioxin,
2,3,7,8-tetrachlorodibenzofuran, and 3,3',4,4'- tetrachlorobiphenyl in
the rat. Fundam Appl Toxicol, 21: 425-432.
McLachlan MJ (1993) Digestive tract absorption of polychlorinated
dibenzo- p-dioxins, dibenzofurans, and biphenyls in a nursing infant.
Toxicol Appl Pharmacol, 123: 68-72.
Mennear JH & Lee CC (1994) Polybrominated dibenzo- p-dioxins and
dibenzofurans: Literature review and health assessment. Environ Health
Perspect, 102(suppl 1): 265-274.
Merz W, Neu H-J, Kuck M, Winkler K, Gorbach S, & Muffler H (1986) [A
process for the generation and analytical characterization of gaseous
combustion products.] Fresenius Z Anal Chem, 325: 449-460 (in German).
Meyer H, Neupert M, Pump W, & Willenberg B (1993) [Flame retardants
determine recyclability.] Kunststoffe, 83: 253-257 (in German).
Monna L, Omori T, & Kodama T (1993) Microbial degradation of
dibenzofuran, fluorene, and dibenzo- p-dioxin by Staphylococcus
auriculans DBF63. Appl Environ Microbiol, 59: 285-289.
Moore JA, McConnell EE, Dalgard DW, & Harris MW (1979) Comparative
toxicity of three halogenated dibenzofurans in guinea pigs, mice, and
rhesus monkeys. Ann NY Acad Sci, 320: 151-163.
Munslow WD, Sovocool GW, Donnelly JR, & Mitchum RK (1987)
Electrophilic bromination of dibenzo- p-dioxin. Chemosphere, 16:
1661-1666.
Nagao T, Golor G, Krowke R, & Neubert D (1990a) Comparison of cleft
palate frequency induced by 2,3,7,8-tetrabromodibenzo- p-dioxin and
2,3,7,8-tetrachlorodibenzo- p-dioxin in mice. In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 317-319 (Organohalogen Compounds, Volume 1).
Nagao T, Neubert D, & Löser E (1990b) Comparative studies on the
induction of ethoxyresorufin O-deethylase by 2,3,7,8-TCDD and
2,3,7,8-TBrDD. Chemosphere, 20: 1189-1192.
Nagao T, Golor G, Bittmann H, & Löser E (1990c) Induction of hepatic
monooxygenases by 2,3,7,8-tetrabromodibenzo- p-dioxin and liver
tissue concentrations in rats. Naunyn- Schmiedeberg's Arch Pharmacol,
341: R19.
Nagao T, Golor G, Krowke R, & Neubert D (1990d) Comparative study on
the teratogenic potency of 2,3,7,8-TBrDD and 2,3,7,8-TCDD in mice.
Teratology, 42: 27A.
Nagao T, Yamashita K, Golor G, Bittmann H, Körner W, Hagenmeier H, &
Neubert D (1995/96) Tissue distribution after a single subcutaneous
administration of 2,3,7,8-tetrabromodibenzo- p-dioxin in comparison
with toxicokinetics of 2,3,7,8-tetrachlorodibenzo- p-dioxin in female
Wistar rats. Life Sci, 58: 325-336.
Nakano T, Tsuji M, & Okuno T (1987) Level of chlorinated organic
compounds in the atmosphere. Chemosphere, 16: 1781-1786.
NATO/CCMS (1988) Pilot study on international information exchange on
dioxins and related compounds. Scientific basis for the development of
the international toxicity equivalence factor (I-TEF) method of risk
assessment for complex mixtures of dioxins and related compounds.
Brussels, North Atlantic Treaty Organization, Committee on the
Challenges of Modern Society, 56 pp (Report No. 176).
Nebert DW (1994) Drug-metabolizing enzymes in ligand-modulated
transcription. Biochem Pharmacol, 47: 25-37.
Nebert DW, Petersen DD, & Puga A (1991) Human Ah locus polymorphism
and cancer: Inducibility of CYP1A1 and other genes by combustion
products and dioxin. Pharmacogenetics, 1: 68-78.
Nebert DW, Puga A, & Vasiliou V (1993) Role of the Ah receptor and the
dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal
transduction. Ann NY Acad Sci, 685: 624-640.
Nestrick TJ, Lamparski LL, & Peters TL (1989) Micro-scale bromination
procedure for synthesis of 2,3,7,8-substituted brominated
dibenzo- p-dioxins and dibenzofurans from [13C] labeled precursors.
Chemosphere, 18: 1761-1770.
Neubert D (1991) Peculiarities of the toxicity of polyhalogenated
dibenzo- p-dioxins and dibenzofurans in animals and man. Chemosphere,
23: 1869-1893.
Neubert D (1993a) Survey on the influence of PHDDs/PHDFs on
lymphocytes in venous blood. In: The Toxicology Forum: Current views
on the impact of dioxins and furans on human health and the
environment, Berlin, 9-11 November 1992. Washington, DC, Toxicology
Forum, Inc., pp 299-315.
Neubert D (1993b) Some data on the induction of monooxygenases by
PHDDs/PHDFs. In: The Toxicology Forum: Current views on the impact of
dioxins and furans on the human health and the environment, Berlin,
9-11 November 1992. Washington, DC, Toxicology Forum, Inc., pp 72-90.
Neubert R, Jacob-Müller U, Stahlmann R, Helge H, & Neubert D (1990)
Polyhalogenated dibenzo- p-dioxins and dibenzofurans and the immune
system: 1. Effects on peripheral lymphocytes of a non-human primate
(Callithrix jacchus) after treatment with
2,3,7,8-tetrachlorodibenzo- p-dioxin. Arch Toxicol, 64: 345-359.
Neubert R, Jacob-Müller U, & Helge H (1991) Polyhalogenated
dibenzo- p-dioxins and dibenzofurans and the immune system: 2.
In vitro effects of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) on
lymphocytes of venous blood from man and a non-human primate
(Callithrix jacchus). Arch Toxicol, 65: 213-219.
Neubert D, Golor G, & Neubert R (1992) TCDD-toxicity equivalencies for
PCDD/PCDF congeners: Prerequisites and limitations. Chemosphere, 25:
65-70.
Neubert R, Golor G, Stahlmann R, Helge H, & Neubert D (1992)
Polyhalogenated dibenzo- p- dioxins and dibenzofurans and the immune
system: 4. Effects of multiple-dose treatment with
2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) on peripheral lymphocyte
subpopulations of a non-human primate (Callithrix jacchus). Arch
Toxicol, 66: 250-259.
Neubert R, Stahlmann R, Korte M, Van Loveren H, Vos JG, Golor G, Webb
JR, Helge H, & Neubert D (1993) Effects of small doses of dioxins on
the immune system of marmosets and rats. Ann NY Acad Sci, 685:
662-686.
Neupert M & Pump W (1992) Experiences from a large scale warehouse
fire with bromine-containing polybutylene terephthalate forming
almost no polybrominated dibenzodioxins and dibenzofurans. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 163-168 (Organohalogen
Compounds, Volume 9).
Neupert M, Grupe A, & Weis H (1988) Stability of polybrominated
dibenzo- p-dioxins and dibenzofurans under laboratory handling
conditions. Chemosphere, 17: 1089-1097.
Neupert M, Weis H, Stock B, & Thies J (1989a) Analytical procedures in
connection with acute toxicity studies: I.
Tetrabromodibenzo- p-dioxin (TBDD). Chemosphere, 19: 115-120.
Neupert M, Weis H, Thies J, & Stock B (1989b) Analytical procedures in
connection with acute animal toxicity studies: II. Pyrolysis products
obtained from ABS copolymer containing octabromo-diphenylether as a
flame retardant. Chemosphere, 19: 219-224.
Nylund K, Asplung L, Jansson B, Jonsson P, Litzen K, & Sellström
(1992) Analysis of some polyhalogenated organic pollutants in sediment
and sewage sludge. Chemosphere, 24: 1721-1730.
Öberg T & Bergström J (1990) Bromine and waste incineration -- an
environmental risk? In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
339-342 (Organohalogen Compounds, Volume 2).
Öberg T, Warman K, & Bergström J (1987) Brominated aromatics from
combustion. Chemosphere, 16: 2451-2465.
OECD (1994) Risk reduction monograph No. 3: Selected brominated flame
retardants background and national experience with reducing risk.
Paris, Organisation for Economic Co-operation and Development, 152 pp
(OECD Environment Monograph Series No. 102 - OECD/GD (94)969).
O'Keefe PW (1978) Formation of brominated dibenzofurans from pyrolysis
of the polybrominated biphenyl fire retardant, Firemaster FF-1.
Environ Health Perspect, 23: 347-350.
Okey AB (1990) Enzyme induction in the cytochrome P450 system.
Pharmacol Ther, 45: 241-298.
Okey AB (1992) Enzyme induction in the cytochrome P-450 system. In:
Kalow W ed. Pharmacogenetics of drug metabolism. New York, Pergamon
Press, pp 549-608.
Okey AB, Riddick DS, & Harper PA (1994) The Ah receptor: Mediator of
the toxicity of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) and
related compounds. Toxicol Lett, 70: 1-22.
Olie K, Vermeulen PL, & Hutzinger O (1977) Chorodibenzo- p-dioxins
and chlorodibenzo-furans are trace components of fly ash and flue gas
of some municipal incinerators in the Netherlands. Chemosphere, 6:
455-459.
Orazio CE, Kapila S, Puri RK, & Yanders AF (1992) Persistence of
chlorinated dioxins and furans in the soil environment. Chemosphere,
25: 1469-1474.
Ott MG & Zober A (1996) Morbidity study of extruder personnel with
potential exposure to brominated dioxins and furans: II. Results of
clinical laboratory studies. Occup Environ Med, 53: 833-846.
Päpke O, Lis A, Priegnitz J, Helmcke K, & Ball M (1990) [Development
of methods for determination of polyhalogenated dioxins and furans in
the air -- Final report: December 1989-October 1990.] Hamburg, ERGO
Forschungsgesellschaft mbH, 24 pp + appendix (in German).
Peper M, Klett M, Frentzel-Beyme R, & Heller WD (1993)
Neuropsychological effects of chronic exposure to environmental
dioxins and furans. Environ Res, 60: 124-135.
Peterson RE (1993) Effects on aquatic and terrestrial organisms. In:
The Toxicology Forum: Current views on the impact of dioxins and
furans on human health and the environment, Berlin, 9-11 November
1992. Washington, DC, Toxicology Forum, Inc., pp 383-399.
Peterson RE, Theobald HM, & Kimmel GL (1993) Developmental and
reproductive toxicity of dioxins and related compounds: Cross-species
comparisons. Crit Rev Toxicol, 23: 283-335.
Pinkerton MN, Kociba RJ, Petrella RV, McAllister DL, Willis ML, Fulfs
JC, Thoma H, & Hutzinger O (1989) A preliminary report on the
investigation of the comparative toxicity of combustion products of
high impact polystyrene with and without
decabromo-diphenyloxide/antimony trioxide as a flame retardant using
2,3,7,8-tetrabromodibenzo- p-dioxin and 2,3,7,8-tetrabromo
dibenzofuran as positive controls. Chemosphere, 18: 1243-1249.
Pirkle JL, Wolfe WH, Patterson DG, Needham LL, Michalek JE, Miner JC,
Peterson MR, & Phillips DL (1989) Estimates of the half-life of
2,3,7,8-tetrachlorodibenzo- p-dioxin in Vietnam veterans of Operation
Ranch Hand. J Toxicol Environ Health, 27: 165-171.
Pluim HJ, Wever J, Koppe JG, Slikke JW, & Olie K (1993) Intake and
faecal excretion of chlorinated dioxins and dibenzofurans in
breast-fed infants at different ages. Chemosphere, 26: 1947-1952.
Poellinger L (1993) Molecular mechanisms of dioxins action. In: The
Toxicology Forum: Current views on the impact of dioxins and furans on
human health and the environment, Berlin, 9-11 November 1992.
Washington, DC, Toxicology Forum, Inc., pp 91-100.
Poellinger L, Göttlicher M, & Gustafsson JA (1992) The dioxin and
peroxisome proliferator-activated receptors: Nuclear receptors in
search of endogenous ligands. Trends Pharmacol Sci, 13: 241-245.
Pohjanvirta R, Unkila M, & Tuomisto J (1993) Comparative acute
lethality of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD),
1,2,3,7,8-pentachlorodibenzo- p-dioxin and
1,2,3,4,7,8-hexachlorodibenzo- p-dioxin in the most TCDD-susceptible
and the most TCDD-resistant rat strain. Pharmacol Toxicol, 73: 52-56.
Pohjanvirta R, Hirvonen MR, Unkila M, Savolainen K, & Tuomisto J
(1994) TCDD decreases brain inositol concentrations in the rat.
Toxicol Lett, 70: 363-372.
Poiger H & Buser H-R (1984) The metabolism of TCDD in the dog and rat.
In: Poland A & Kimbrough RD ed. Biological mechanisms of dioxin
action. Cold Spring Harbor, New York, Cold Spring Harbor Laboratory,
pp 39-47 (Banbury Report 18).
Poiger H & Schlatter C (1980) Influence of solvents and adsorbents on
dermal and intestinal absorption of TCDD. Food Cosmet Toxicol, 18:
477-481.
Poiger H & Schlatter C (1986) Pharmacokinetics of 2,3,7,8-TCDD in man.
Chemosphere, 15: 1489-1494.
Poland A & Glover E (1973) Chlorinated dibenzo- p-dioxins: Potent
inducers of o-aminolevulinic acid synthetase and aryl hydrocarbon hydroxylase. II.
A study of the structure-activity relationship. Mol Pharmacol, 9:
736-747.
Poland A & Knutson JC (1982) 2,3,7,8-Tetrachlorodibenzo- p-dioxin and
related halogenated aromatic hydrocarbons: Examination of the
mechanism of toxicity. Annu Rev Pharmacol Toxicol, 22: 517-554.
Pruell RJ, Taplin BK, McGovern DG, Montmarquet T, & Johnson MW (1993)
Laboratory studies on the accumulation of polychlorinated
dibenzo- p-dioxins and dibenzofurans from sediment and food by the
American lobster (Homarus americanus). In: Fiedler H, Frank H,
Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 239-242 (Organohalogen Compounds, Volume 12).
Ramalingam B, Mazer T, Wagel DJ, Malloy CM, Taylor ML, TiernanTO,
Garrett JH, & Rifkind AB (1986) Synthesis and characterization of
polybromo- and polybromochloro-dibenzo- p-dioxins and dibenzofurans.
In: Rappe C, Choudhary G, & Keith LH ed. Chlorinated dioxins and
dibenzofurans in perspective: 189th National Meeting of the American
Chemical Society, Miami Beach, Florida, USA, September 1985. Chelsea,
Michigan, Lewis Publishers, Inc., pp 485-499.
Ranken PF, Ricks GM, Lynam DR, & Ariano JM (1990) Is watching
television toxic? BFRIP-sponsored study on the emission of PBDD/PBDFS
from operating television sets. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
343-347 (Organohalogen Compounds, Volume 2).
Ranken PF, Freiberg M, Mazac CJ, Bauer MR, Varcoe FT, & Tondeur Y
(1994) Definitive study of the determination of polybrominated
dibenzo- p-dioxins and polybrominated dibenzofurans in
decabromodiphenyloxide and tetrabromobisphenol-A. Bull Soc Chim Belg,
103: 219-233.
Rappe C (1993) Environmental concentrations and ecotoxicological
effects of PCDD's, PCDF's and related compounds. In: Fiedler H, Frank
H, Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 163-170 (Organohalogen Compounds, Volume 12).
Rappe C, Andersson R, Bonner MS, Cooper KR, Fiedler H, Howell FG, &
Lau C (1996) PCDDs and PCDFs in municipal sewage sludge and effluent
from the state of Mississippi, USA. Organohalog Compds, 28: 105-110.
Re MA, Bolt DL, & Chandler RA (1995) Stability of polybrominated
dibenzo- p-dioxin and dibenzofuran standard solutions. In: Bolt D,
Clement R, Fiedler H, Harrison B, Ramamoorthy S, & Reiner E ed.
Dioxin'95: 15th International Symposium on Chlorinated Dioxins and
Related Compounds, Edmonton, Canada, 21-25 August 1995 (Short papers).
Edmonton, Alberta, Dioxin'95 Secretariat, pp 269-272 (Organohalogen
Compounds, Volume 23).
Richtzenhain H & Schrage K (1977) [Highly brominated dibenzofurans.]
Munich, German Federal Patent Office, 14 pp (Patent No. 2534381
-- Dynamit Nobel AG) (in German).
Riggs KB, Pitts GE, White JS, Mitchum RK, & Reuther JJ (1990)
Polybrominated dibenzo- p-dioxin and polybrominated dibenzofuran
emissions from incineration of flame-retarded resins in a simulated
municipal waste incinerator. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
351-356 (Organohalogen Compounds, Volume 2).
Riggs K, Reuther J, White J, & Pitts G (1992) Determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in simulated
incinerator emissions. Chemosphere, 25: 1415-1420.
Ritterbusch J, Vogt R, Lorenz W, Bahadir M, & Hopf H (1994a)
UV-photolysis of PXDD/F-contaminated bromophenols and wastes of
chemical laboratories. Chemosphere, 29: 457-464.
Ritterbusch J, Lorenz W, & Bahadir M (1994b) Determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in analytical
laboratory waste and their decomposition by UV-photolysis.
Chemosphere, 29: 1829-1838.
Romkes M, Piskorska-Pliszczynska J, Keys B, Safe S, & Fujita T (1987)
Quantitative structure-activity relationships: Analysis of
interactions of 2,3,7,8-tetrachlorodibenzo- p-dioxin and
2- substituted analogues with rat, mouse, guinea pig, and hamster
cytosolic receptor. Cancer Res, 47: 5108-5111.
Rordorf BF (1987) Prediction of vapor pressures, boiling points and
enthalpies of fusion for twenty-nine halogenated dibenzo- p-dioxins.
Thermochim Acta, 112: 117-122.
Rordorf BF, Sarna LP, Webster GRB, Safe SH, Safe LM, Lenoir D, Schwind
KH, & Hutzinger O (1990) Vapor pressure measurements on halogenated
dibenzo- p-dioxins and dibenzofurans. An extended data set for
correlation method. Chemosphere, 20: 1603-1609.
Safe S (1987) Determination of 2,3,7,8-TCDD toxic equivalent factors
(TEF's): Support for the use of the in vitro AHH induction assay.
Chemosphere, 16: 791-802.
Safe S (1990) Polychlorinated biphenyls (PCBs), dibenzo- p-dioxins
(PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental
and mechanistic considerations which support the development of toxic
equivalency factors (TEFs). Crit Rev Toxicol, 21: 51-88.
Safe S (1993a) Development of bioassays and approaches for the risk
assessment of 2,3,7,8-tetrachlorodibenzo- p-dioxin and related
compounds. Environ Health Perspect, 101(suppl 3): 317-325.
Safe S (1993b) Development, validation and limitations of toxic
equivalency factors. In: The Toxicology Forum: Current views on the
impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1992. Washington, DC, Toxicology Forum, Inc., pp
176-180.
Safe S, Davis D, Romkes M, Yao C, Keyes B, Piskorska-Pliszczynska J,
Farrell K, Mason G, Denomme MA, Safe L, Zmudzka B, & Holcomb M (1989a)
Development and validation of in vitro bioassays for 2,3,7,8-TCDD
equivalents. Chemosphere, 19: 853-860.
Safe S, Mason G, Sawyer T, Zacharewski T, Harris M, Yao C, Keys B,
Farrell K, Holcomb M, Davis D, Safe L, Piskorska-Pliszczynska J, Leece
B, Denomme MA, Hutzinger O, Thoma H, Chittim B, & Madge J (1989b)
Development and validation of in-vitro induction assays for toxic
halogenated aromatic mixtures: A review. Toxicol Ind Health, 5:
757-775.
Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, Romkes M, &
Biegel L (1991) 2,3,7,8-Tetrachlorodibenzo- p-dioxin (TCDD) and
related compounds as antiestrogens: Characterization and mechanism of
action. Pharmacol Toxicol, 69: 400-409.
Schacht U, Gras B, & Sievers S (1995) [Determination of polybrominated
and polychlorinated dibenzodioxins and -furans in various
environmentally relevant materials.] In: Dioxin-Information and
EPA-Reassessment, Bayreuth, 12-14 June 1995. Organohalog Compds, 22:
325-334 (in German).
Schäfer W & Ballschmiter K (1986) Monobromopolychloroderivatives of
benzene, biphenyl, dibenzofuran and dibenzodioxin formed in
chemical-waste burning. Chemosphere, 15: 755-763.
Schecter A (1992) Dioxins and dibenzofurans in potentially exposed
workers: Serial tissue levels in a worker exposed in a PCB transformer
fire cleanup and blood levels in three exposed chemists. Chemosphere,
25: 1117-1122.
Schecter A & Ryan JJ (1990) Chlorinated and brominated dioxin levels
in the blood of a chemist who became ill after synthesizing
2,3,7,8-TCDD and 2,3,7,8-TBDD. In: Hutzinger O & Fiedler H ed.
Dioxin'90: 10th International Symposium on Chlorinated Dioxins and
Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp 141-144
(Organohalogen Compounds, Volume 4).
Schecter A & Ryan JJ (1991) Brominated and chlorinated dioxin blood
levels in a chemist 34 years after exposure to
2,3,7,8-tetrachlorodibenzodioxin and 2,3,7,8-tetrabromo-dibenzodioxin.
Chemosphere, 23: 1921-1924.
Schecter A & Ryan JJ (1992) Persistent brominated and chlorinated
dioxin blood levels in a chemist: 35 years after dioxin exposure. J
Occup Med, 34: 702-707.
Schecter A & Ryan JJ (1994) Decrease in milk and blood dioxin levels
over time in a mother nursing twins: Estimates of decreased maternal
and increased infant dioxin body burden from nursing. In: Fiedler H,
Hutzinger O, Clement R, & Sakai S ed. Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 159-162
(Organohalogen Compounds, Volume 21).
Schecter A, Ryan JJ, Masuda Y, Brandt-Rauf P, Constable J, Dinh Cau H,
Cao Dai P, Tri Quynh H, Thi Ngoc Phuong N, & Hoang Phiet P (1994a)
Chlorinated and brominated dioxins and dibenzofurans in human tissue
following exposure. Environ Health Perspect, 102(suppl 1): 135-147.
Schecter A, Päpke O, Lis A, & Ball M (1994b) Chlorinated dioxin and
dibenzofuran levels in US human placentas and fetal tissue in
comparison with US adult population dioxin levels. In: Fiedler H,
Hutzinger O, Clement R, & Sakai S ed. Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 63-64
(Organohalogen Compounds, Volume 21).
Schecter A , Päpke O, Lis A, & Olson JR (1995) Chlorinated dioxin,
dibenzofuran and PCB levels in human fetal tissue at 8-18 weeks
gestational age, compared with placental, newborn and adult tissue
levels. In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, &
Reiner E ed. Dioxin'95: 15th International Symposium on Chlorinated
Dioxins and Related Compounds, Edmonton, Canada, 21-25 August 1995
(Short papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 167-171
(Organohalogen Compounds, Volume 25).
Schecter A, Päpke O, Lis A, Ball M, Ryan JJ, Olson JR, Li L, & Kessler
H (1996a) Decrease in milk and blood dioxin levels over two years in a
mother nursing twins: Estimates of decreased maternal and increased
infant dioxin body burden from nursing. Chemosphere, 32: 543-549.
Schecter A, Startin J, Wright C, Päpke O, Ball M, & Lis A (1996b)
Concentrations of polychlorinated dibenzo- p-dioxins and
dibenzofurans in human placental and fetal tissues from the US and in
placentas from Yu-Cheng exposed mothers. Chemosphere, 32: 551-557.
Schimmel H, Griepink B, Maier EA, Kramer GN, Roos AH, & Tuinstra LGMT
(1994) Intercomparison study on milk powder fortified with PCDD and
PCDF. Fresenius J Anal Chem, 348: 37-46.
Schmidt U & Ivens-Kohl I (1990a) [Enzyme activity in the liver after
acute application of 2,3,7,8-tetrabromodibenzodioxin on rats.]
Wuppertal-Elberfeld, Bayer AG, 26 pp (Unpublished report No. 18657)
(in German).
Schmidt U & Ivens-Kohl I (1990b) [Enzyme activity in the liver after
acute application of pyrolysis condensate Novodur L2FR on rats and
guinea pigs.] Wuppertal-Elberfeld, Bayer AG, 28 pp (Unpublished report
No. 18696) (in German).
Schramm K-W, Lenoir D, & Hutzinger O (1990) Fugacity calculations of
vapor-flyash partition of polyhalogenated dioxins and furans.
Chemosphere, 20: 563-568.
Schreiber R (1994) [Emissions reduced to a minimum: Bonn trash
incineration plant -- experiences with a plant that was built
according to the latest knowledge of modern environmental technology.
Trash utilization.] Energie, 46: 18-25 (in German).
Schulz T, Golor G, Körner W, Hagenmaier H, & Neubert D (1993)
Comparative study on enzyme induction and tissue distribution of
2,3,7,8-tetrachlorodibenzo- p-dioxin,
2,3,4,7,8-pentachloro-dibenzofuran and
2,3,4,7,8-pentabromodibenzofuran in marmoset monkeys (Callithrix
jacchus). In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A,
& Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 145-148 (Organohalogen
Compounds, Volume 13).
Schulz TG, Neubert D, Donald SD, & Edwards RJ (1996) Induction of
cytochromes P450 by dioxins in liver and lung of marmoset monkeys
(Callithrix jacchus). Adv Exp Med Biol, 387: 443-446.
Schulz-Schalge T, Koch E, Schwind K-H, Hutzinger O, & Neubert D (1990)
Comparative study on the inductive potency of TCDD and TBrDD with
three 2,3,7,8-mixed-halogenated dioxins in liver microsomes of male
rats. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 321-324 (Organohalogen
Compounds, Volume 1).
Schulz-Schalge T, Schwind K-H, & Hutzinger O (1991a) Biological
activity of TCDD, TBrDD and three 2,3,7,8-mixed halogenated
dibenzo- p-dioxins. Naunyn-Schmiedeberg's Arch Pharmacol, 343(suppl):
R21.
Schulz-Schalge T, Koch E, Schwind K-H, Hutzinger O, & Neubert D
(1991b) Inductive potency of TCDD, TBDD and three
2,3,7,8-mixed-halogenated dioxins in liver microsomes of male rats:
Enzyme kinetic considerations. Chemosphere, 23: 1925-1931.
Schwetz BA, Norris JM, Sparschu GL, Rowe VK, Gehring PJ, Emerson JL, &
Gerbig CG (1973) Toxicology of chlorinated dibenzo- p-dioxins.
Environ Health Perspect, 5: 87-99.
Schwind KH (1991) [Halogenated dibenzo- p-dioxins and dibenzofurans
from incineration processes.] Bayreuth, University of Bayreuth,
Faculty of Biology, Chemistry and Geology, 206 pp (Dissertation) (in
German).
Schwind KH, Hosseinpour J, & Thoma H (1988) Brominated/chlorinated
dibenzo- p-dioxins and dibenzofurans -- Part 1:
Brominated/chlorinated and brominated dibenzo- p-dioxins and
dibenzofurans in fly ash from a municipal waste incinerator.
Chemosphere, 17: 1875-1884.
Schwind KH, Hosseinpour J, & Thoma H (1989) [Brominated/chlorinated
dibenzo- p-dioxins and dibenzofurans from waste incineration.] UWSF-Z
Umweltchem Ökotoxikol, 1: 24 (in German).
Schwind KH, Thoma H, Hutzinger O, Dawidowsky N, Weberuß U, Hagenmaier
H, Bühler U, Greiner R, Essers U, & Bessey E (1991) [Emission of
halogenated dibenzodioxins (PXDD) und dibenzofurans (PXDF) from motors
using commercial fuels.] USWF-Z Umweltchem Ökotoxikol, 3: 291-298 (in
German).
Sedlak D, Dumler-Grad R, Thoma H, & Vierle O (1996) Formation of
polyhalogenated dibenzodioxin and dibenzofurans (PXDD/F) during
textile processings. Organohalog Compds, 27: 201-205.
Shiu WY, Doucette W, Gobas FAPC, Andren A, & Mackay D (1988)
Physical-chemical properties of chlorinated dibenzo- p-dioxins.
Environ Sci Technol, 22: 651-658.
Sidhu SS, Maqsud L, Dellinger B, & Mascolo G (1995) The homogeneous,
gas-phase formation of chlorinated and brominated dibenzo- p-dioxin
from 2,4,6-trichloro- and 2,4,6-tribromophenols. Combust Flame, 100:
11-20.
Silbergeld EK & deFur PL (1994) Risk assessment of dioxinlike
compounds. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 51-78.
Silbergeld EK & Gasiewicz TA (1989) Dioxins and the Ah receptor. Am J
Ind Med, 16: 455-474.
Sinkkonen S, Vattulainen A, Aittola J-P, Paasivirta J, Tarhanen J, &
Lahtiperä M (1994) Metal reclamation produces sulphur analogues of
toxic dioxins and furans. Chemosphere, 28: 1279-1288.
Skene SA, Dewhurst IC, & Greenberg M (1989) Polychlorinated
dibenzo- p-dioxins and polychlorinated dibenzofurans: The risks to
human health -- A review. Hum Toxicol, 8: 173-204.
Somogyi A & Beck H (1993) Nurturing and breast feeding: exposure to
chemicals in breast milk. Environ Health Perspect, 101: 45-52.
Sovocool GW, Munslow WD, Donnelly JR, & Mitchum RK (1987)
Electrophilic bromination of dibenzofuran. Chemosphere, 16: 221-224.
Sovocool GW, Mitchum RK, Tondeur Y, Munslow WD, Vonnahme TL, &
Donnelly JR (1988) Bromo- and bromochloro-polynuclear aromatic
hydrocarbons, dioxins and dibenzofurans in municipal incinerator fly
ash. Biomed Environ Mass Spectrom, 15: 669-676.
Sovocool GW, Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Tondeur Y,
& Mitchum RK (1989) Analysis of municipal incinerator fly ash for
bromo- and bromochloro-dioxins, dibenzofurans, and related compounds.
Chemosphere, 18: 193-200.
Sovocool GW, Donnelly JR, Grange AH, Simmons RD, & Munslow WD (1990)
Brominated dioxins and other bromoaromatics in plastic pyrolysates.
In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 365-368 (Organohalogen
Compounds, Volume 2).
Spahl R, Dorn IH, Horn HC, & Hess K (1993) [Catalytic decomposition of
dioxins for incineration plants.] Entsorgungspraxis, 5: 328-331 (in
German).
Spink DC, Johnson JA, Connor SP, Aldous KM, & Gierthy JF (1994)
Stimulation of 17 ß-estradiol metabolism in MCF-7 cells by
bromochloro- and chloromethyl-substituted dibenzo- p-dioxins and
dibenzofurans: Correlations with antiestrogenic activity. J Toxicol
Environ Health, 41: 451-466.
Springer W & Rast HG (1988) [Biological degradation of polyhalogenated
mono- and polycyclic aromatics.] GWF-Wasser/Abwasser, 129: 70-75 (in
German).
Stephens RD, Rappe C, Hayward DG, Nygren M, Startin J, Ersboll A,
Carlé J, & Yrjänheikki E (1992) World Health Organization
international intercalibration study on dioxins and furans in human
milk and blood. Anal Chem, 64: 3109-3137.
Stieglitz L & Vogg H (1990) The de-novo-synthesis of PCDD/PCDF and
other organohalogen compounds on fly ash. In: Hutzinger O & Fiedler H
ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, p 173 (Organohalogen Compounds, Volume 3).
Stieglitz L, Zwick G, Beck J, Bautz H, & Roth W (1989) Carbonaceous
particles in fly ash: A source for the de-novo-synthesis of
organochlorocompounds. Chemosphere, 19: 283-290.
Striebich RC, Rubey WA, Tirey DA, & Dellinger B (1990) High
temperature thermal decomposition of polybrominated flame retardant
materials. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar:
10th International Symposium on Chlorinated Dioxins and Related
Compounds. Bayreuth, Germany, Ecoinforma Press, pp 369-373
(Organohalogen Compounds, Volume 2).
Striebich RC, Rubey WA, Tirey DA, & Dellinger B (1991)
High-temperature degradation of polybrominated flame retardant
materials. Chemosphere, 23: 1197-1204.
Strubel V, Rast HG, Fietz W, Knackmuss HJ, & Engesser KH (1989)
Enrichment of dibenzofuran utilizing bacteria with high co-metabolic
potential towards dibenzodioxin and other anellated aromatics. FEMS
Microbiol Lett, 58: 233-238.
Strubel V, Engesser K-H, Fischer P, & Knackmuss H-J (1991)
3-(2-Hydroxyphenyl)catechol as substrate for proximal meta ring
cleavage in dibenzofuran degradation by Brevibacterium sp. strain
DPO 1361. J Bacteriol, 173: 1932-1937.
Sweeney A (1994) Reproductive epidemiology of dioxins. In: Schecter A
ed. Dioxins and health. New York, London, Plenum Press, pp 549-585.
Sweeney MH, Fingerhut MA, Calvert GM, Piacitelli LA, Alderfer RJ,
Davis-King K, Halperin WE, Connally LB, & Marlow DA (1993) Noncancer
health effects and exposure to 2,3,7,8-TCDD. In: Fiedler H, Frank H,
Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 369-374 (Organohalogen Compounds, Volume 13).
Takasuga T, Inoue G, Ohi E, Ireland P, Suzuki T, & Takeda N (1994)
Determination of halogenated aromatic and polycyclic aromatic
hydrocarbons formed during MSW incineration. In: Fiedler H, Hutzinger
O, Clement R, & Sakai S ed. Dioxin'94: 14th International Symposium on
Chlorinated Dioxins, PCB and Related Compounds, Kyoto, Japan, 21-25
November 1994 (Short papers). Kyoto, Kyoto University, Department of
Environmental and Sanitary Engineering, pp 41-44 (Organohalogen
Compounds, Volume 19).
Tashiro M & Yoshiya H (1982) Determination of structures of tri- and
tetrabromodibenzofurans formed in bromination of dibenzofuran.
Heterocycles, 19: 2349-2354.
Thies J, Neupert M, & Pump W (1990) Tetrabromobisphenol A (TBBA), its
derivatives and their flame retarded (FR) polymers -- Content of
polybrominated dibenzo- p-dioxins (PBDD) and dibenzofurans (PBDF)
-- PBDD/F formation under processing and smouldering (worst case)
conditions. Chemosphere, 20: 1921-1928.
Thoma H & Hutzinger O (1987a) [Pyrolysis and GC/MS-analysis of flame
retardants in on-line operation (poster).] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
293-297 (VDI-Report No. 634) (in German).
Thoma H & Hutzinger O (1987b) Pyrolysis and GC/MS-analysis of
brominated flame retardants in on-line operation. Chemosphere, 16:
1353-1360.
Thoma H & Hutzinger O (1989) Pyrolysis and GC/MS-analysis of
brominated flame retardants in on-line operation. Chemosphere, 18:
1047-1050.
Thoma H, Rist S, Hauschulz G, & Hutzinger O (1986a) Polybrominated
dibenzodioxins and -furans from the pyrolysis of some flame
retardants. Chemosphere, 15: 649-652.
Thoma H, Rist S, Hauschulz G, & Hutzinger O (1986b) Polybrominated
dibenzodioxins (PBrDD) and dibenzofurans (PBrDF) in some flame
retardant preparations. Chemosphere, 15: 2111-2113.
Thoma H, Hauschulz G, Knorr E, & Hutzinger O (1987a) Polybrominated
dibenzofurans (PBDF) and dibenzodioxins (PBDD) from the pyrolysis of
neat brominated diphenylethers, biphenyls and plastic mixtures of
these compounds. Chemosphere, 16: 277-285.
Thoma H, Hauschulz G, & Hutzinger O (1987b) PVC-induced
chlorine-bromine exchange in the pyrolysis of polybrominated diphenyl
ethers, -biphenyls, -dibenzodioxins and dibenzofurans. Chemosphere,
16: 297-307.
Thoma H, Hauschulz G, & Hutzinger O (1987c) Chlorine-bromine exchange
during pyrolysis of 1,2,3,4-tetrabromodibenzodioxin with different
chlorine donors. Chemosphere, 16: 1579-1581.
Thoma H, Hauschulz G, & Hutzinger O (1987d) [Pyrolysis of
dibenzodioxin, dibenzofuran and 1,2,3,4-T4BrDD with different chlorine
donors and catalysts (poster).] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
277-286 (VDI-Report No. 634) (in German).
Thoma H, Hauschulz G, & Hutzinger O (1989) Pyrolysis of dibenzodioxin,
dibenzofuran and 1,2,3,4-tetrabromodibenzodioxin with different
chlorine donors and catalysts. Chemosphere, 18: 1213-1217.
Thunberg T, Ahlborg UG, & Wahlstrom B (1984) Comparison between the
effects of 2,3,7,8- tetrachlorodibenzo- p-dioxin and six other
compounds on the vitamin A storage, the UDP- glucuronosyltransferase
and the aryl hydrocarbon hydroxylase activity in the rat liver. Arch
Toxicol, 55: 16-19.
Tomita M, Ueda S, & Narisada M (1959) Studies on the
dibenzo- p-dioxin (diphenylene dioxide) derivatives: XXVII. Synthesis
of polyhalodibenzo- p-dioxin. J Pharm Soc Jpn, 79: 186-192.
Tondeur Y, Gorsich R, Mazac C, Freiberg M, Hass J, & McAllister D
(1990) Analytical protocol for the analysis of polybrominated
dibenzodioxins and dibenzofurans: Data quality objectives and
single-laboratory evaluation. Chemosphere, 20: 1269-1276.
Tong HY, Monson SJ, Gross ML, & Huang LQ (1991)
Monobromopolychlorodibenzo- p-dioxins and dibenzofurans in municipal
waste incinerator flyash. Anal Chem, 23: 2697-2705.
Troitzsch JH (1990) Brominated flame retardants: Current situation in
the Federal Republic of Germany, October 1989. In: Freiji L ed.
Proceedings of the Workshop on Brominated Aromatic Flame Retardants,
Skokloster, Sweden, 24-26 October 1989. Solna, Sweden, National
Chemicals Inspectorate (KEMI), pp 117-122.
Tuinstra LGMT, Startin JR, & Maier EA (1996) Certification of the
contents of PCDDs and PCDFs in milkpowder. Organohalog Compds, 27:
459-463.
UBA (1992) [Further studies on the formation of polybrominated dioxins
and furans during thermal stress on flame retarded plastics and
textiles, Parts 1 and 2 (UBA-Texts 45/92).] Berlin, German Federal
Environmental Agency, pp 86, 151 (Research reports No. 104 03
364/01/02, UBA-FB 91-082 & 92-097) (in German).
US EPA (1987) Polyhalogenated dibenzo- p-dioxins/dibenzofurans:
Testing and reporting requirements -- Final Rule. Fed Reg, 52(108):
21412-21417.
US EPA (1990) Method 1613: Tetra- through octa-chlorinated dioxins and
furans by isotope dilution HRGC/HRMS -- Revision A. Washington, DC, US
Environmental Protection Agency, Office of Water Regulations and
Standards.
US EPA (1992) Method 8290: Polychlorinated dibenzodioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs) by high-resolution gas
chromatography/high-resolution mass spectrometry (HRMS); Washington,
DC, US Environmental Protection Agency.
Van Birgelen APJM, DeVito MJ, Akins JM, Ross DG, Diliberto JJ, &
Birnbaum LS (1996) Relative potencies of polychlorinated
dibenzo- p-dioxins, dibenzofurans, and biphenyls derived from hepatic
porphyrin accumulation in mice. Toxicol Appl Pharmacol, 138: 98-109.
Van den Heuvel JP & Lucier G (1993) Environmental toxicology of
polychlorinated dibenzo- p-dioxins and polychlorinated
dibenzofurans. Environ Health Perspect, 100: 189-200.
Van de Plassche EJ, Polder MD, & Schipper-Zablotskaja M (1994) Risk
assessment of polybrominated biphenyls and polybrominated diphenyl
ethers. Bilthoven, The Netherlands, National Institute of Public
Health and Environmental Protection (RIVM), 34 pp (Report No. 679101
016).
Vehlow J (1995) State of the art in emissions control and residue
quality: a rational point of view. In: European Conference: The future
of European thermal waste treatment, Paris, September 1995. Karlsruhe,
Research Centre, pp 1-19.
Vogg H (1989) [Topical technical questions on the law for minimization
of dioxins and furans from waste incineration processes.] Wasser Luft
Boden, 11/12: 25-27 (in German).
Vogg H, Metzger M, & Stieglitz L (1987) Recent findings on the
formation and decomposition of PCDD/PCDF in solid municipal waste
incineration. In: Emissions of trace organics from municipal solid
waste incinerators, Copenhagen, 20-22 January 1987, pp 207-216.
Vogt R, Lorenz W, Bahadir M, & Hopf H (1994a) Brominated
dibenzodioxins and -furans: Level of potential hazard during synthesis
of bromophenols carried out for educational purposes. Labor Praxis,
18: 30-36.
Vogt R, Lorenz W, Bahadir M, & Hopf H (1994b) PBDD/F formation during
bromoaniline synthesis in teaching laboratories. Fresenius Environ
Bull, 3: 746-750.
Vos (1993) TCDD effects on the immune system: an overview. In: The
Toxicology Forum: Current views on the impact of dioxins and furans on
human health and the environment, Berlin, 9-11 November 1992.
Washington DC, Toxicology Forum, Inc., pp 288-298.
Vos JG & Luster MI (1989) Immune alterations. In: Kimbrough RD &
Jensen AA ed. Halogenated biphenyls, terphenyls, naphthalenes,
dibenzodioxins and related products, 2nd ed. Amsterdam, Oxford, New
York, Elsevier Science Publishers, chapter 10, pp 295-319.
Wagel DH, Tiernan TO, Taylor ML, Garrett JH, Vanness GF, Solch JG, &
Harden LA (1989) Assessments of ambient air sampling techniques for
collecting airborne polyhalogenated dibenzo- p-dioxins (PCDD),
dibenzofurans (PCDF) and biphenyls (PCB). Chemosphere, 18: 177-184.
Walker MK & Peterson RE (1991) Potencies of polychlorinated
dibenzo- p-dioxin, dibenzofuran, and biphenyl congeners, relative to
2,3,7,8-tetrachlorodibenzo- p-dioxin for producing early life stage
mortality in rainbow trout (Oncorhynchus mykiss). Aquat Toxicol, 21:
219-238.
Walker MK & Peterson E (1994) Aquatic toxicity of dioxins and related
chemicals. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 347-387.
Wania F & Lenoir D (1990) Copper-promoted reactions of polybrominated
dibenzo-para-dioxins (PBrDD). Chemosphere, 21: 417-432.
Wanke T, Vehlow J, Mark FE, & Brenner KS (1996) The influence of flame
retarded plastic foams upon the formation of Br containing
dibenzo- p-dioxins and dibenzofurans in a MSWI. Organohalog Compds,
28: 530-535.
Watanabe I (1988) [Behavior of organobrominated compounds at the
sediment phase in the environment.] Osalka-furitsu Koshu Eisei
Kenkyusho Kenkyu Hokoku, 26: 129-133 (in Japanese with English
summary).
Watanabe I & Tatsukawa R (1987) Formation of brominated dibenzofurans
from the photolysis of flame retardant decabromobiphenyl ether in
hexane solution by UV and sun light. Bull Environ Contam Toxicol, 39:
953-959.
Watanabe I & Tatsukawa R (1990) Anthropogenic brominated aromatics in
the Japanese environment. In: Freiji L ed. Proceedings of the Workshop
on Brominated Aromatic Flame Retardants, Skokloster, Sweden, 24-26
October 1989. Solna, Sweden, National Chemicals Inspectorate (KEMI),
pp 63-71.
Watanabe I, Kawano M, Wang Y, Chen Y, & Tatsukawa R (1992)
Polybrominated dibenzo- p-dioxins (PBDDs) and dibenzofurans (PBDFs)
in atmospheric air in Taiwan and Japan. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 309-312 (Organohalogen Compounds, Volume 9).
Watanabe I, Kawano M, & Tatsukawa R (1993) Consumption trend and
environmental research on brominated flame retardants in Japan and the
formation of poly-halogenated dibenzofurans at the metal reclamation
factory. Paper presented at the OECD Flame Retardants Workshop,
Neuchâtel, February 1993, 7 pp.
Watanabe I, Kawano M, & Tatsukawa R (1994) The photolysis of
halogenated dibenzofurans in hexane solution and on airborne dust by
sunlight. In: Fiedler H, Hutzinger O, Clement R, & Sakai S ed.
Dioxin'94: 14th International Symposium on Chlorinated Dioxins, PCB
and Related Compounds, Kyoto, Japan, 21-25 November 1994 (Short
papers). Kyoto, Kyoto University, Department of Environmental and
Sanitary Engineering, pp 235-238 (Organohalogen Compounds, Volume 19).
Watanabe I, Kawano M, & Tatsukawa R (1995) Polybrominated and mixed
polybromo/chlorinated dibenzo- p-dioxins and -dibenzofurans in the
Japanese environment. In: Bolt D, Clement R, Fiedler H, Harrison B,
Ramamoorthy S, & Reiner E ed. Dioxin'95: 15th International Symposium
on Chlorinated Dioxins and Related Compounds, Edmonton, Canada, 21-25
August 1995 (Short papers). Edmonton, Alberta, Dioxin'95 Secretariat,
pp 337-340 (Organohalogen Compounds, Volume 24).
Weber LWD, Ernst SW, Stahl BU, & Rozman K (1993) Tissue distribution
and toxicokinetics of 2,3,7,8-tetra-chlorodibenzo- p-dioxin in rats
after intravenous injection. Fundam Appl Toxicol: 21: 523-534.
Weberruß U (1990) [Polyhalogenated dibenzodioxins and dibenzofurans in
the environment.] Tübingen, Eberhard-Karls University, Faculty of
Chemistry and Pharmacy, 168 pp (Dissertation) (in German).
Webster GRB, Muldrew DH, Graham JJ, Sarna LP, & Muir DCG (1986)
Dissolved organic matter mediated aquatic transport of chlorinated
dioxins. Chemosphere, 15: 1379-1386.
Whitelaw M, Pongratz I, Wilhelmsson A, Gustaffson JA, & Poellinger L
(1993) Ligand-dependent recruitment of the Arnt coregulator determines
DNA recognition by the dioxin receptor. Mol Cell Biol, 13: 2504-2514.
Whitlock JP Jr (1990) Genetic and molecular aspects of
2,3,7,8-tetrachlorodibenzo- p-dioxin. Annu Rev Pharmacol Toxicol, 30:
251-277.
Whitlock JP Jr (1993) Mechanistic aspects of dioxin action. Chem Res
Toxicol, 6: 754-763.
WHO (1989) Environmental health criteria 88: Polychlorinated
dibenzo-para-dioxins and dibenzofurans. Geneva, World Health
Organization, International Programme on Chemical Safety, 409 pp.
WHO (1993) Environmental health criteria 140: Polychlorinated
biphenyls and terphenyls, 2nd ed. Geneva, World Health Organization,
International Programme on Chemical Safety, 682 pp.
WHO (1994a) Environmental health criteria 152: Polybrominated
biphenyls. Geneva, World Health Organization, International Programme
on Chemical Safety, 577 pp.
WHO (1994b) Environmental health criteria 162: Brominated
diphenylethers. Geneva, World Health Organization, International
Programme on Chemical Safety, 347 pp.
WHO (1995) Environmental health criteria 172: Tetrabromobisphenol A
and derivatives. Geneva, World Health Organization, International
Programme on Chemical Safety.
WHO/ECEH (1996) Quality assessment of PCB, PCDD and PCDF analysis:
Third round of WHO-coordinated study. World Health Organization,
Regional Office for Europe (Environmental Health in Europe 2).
WHO/EURO (1989) Levels of PCBs, PCDDs and PCDFs in breast milk:
Results of WHO-coordinated interlaboratory quality control studies and
analytical field studies. Copenhagen, World Health Organization,
Regional Office for Europe (Environmental Health Series 34).
WHO/EURO (1991) Levels of PCBs, PCDDs and PCDFs in human milk and
blood: Second round of quality control studies. Copenhagen, World
Health Organization, Regional Office for Europe (Environment and
Health in Europe 37).
Wiberg K, Rappe C, & Haglund P (1992) Analysis of bromo-, chloro- and
mixed bromo/chloro-dibenzo- p-dioxins and dibenzofurans in salmon,
osprey and human milk. Chemosphere, 24: 1431-1439.
Wichmann H, Zelinski V, Lorenz W, & Bahadir M (1992a) [Chlorinated and
brominated pollutants in accidental fire residues from private houses
-- analytical results, waste disposal, decontamination, safety
provisions for workers.] Wiss Umwelt, 12: 75-78 (in German).
Wichmann H, Lorenz W, & Bahadir M (1992b) Chlorinated compounds in
accidental fire residues from private houses. Fresenius Environ Bull,
1: 376-381.
Wichmann H, Zelinski V, Scholz-Böttcher B, Lorenz W, & Bahadir M
(1993) Sampling strategy to investigate the distribution behaviour of
low volatile pollutants like "dioxins" during vehicle fires in traffic
tunnels. Chemosphere, 26: 1159-1166.
Wilken M & Schanne L (1994) [Brominated dioxins -- by fires: a higher
risk than PCDD and PCDF.] In: [Dioxin emissions and sources.]
Darmstadt, Eigenverlag, pp 109-127 (in German).
Wilken M, Beyer A, & Jager J (1990) Generation of brominated dioxins
and furans in a municipal waste incinerator (MWI): Results of a case
study. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 377-380 (Organohalogen
Compounds, Volume 2).
Wittich R-M, Wilkes H, Sinnwell V, Francke W, & Fortnagel P (1992)
Metabolism of dibenzo- p- dioxin by Sphingomonas sp. strain RW1.
Appl Environ Microbiol, 58: 1005-1010.
Wolfe WH, Michalek JE, Miner JC, Pirkle JL, Caudill SP, Patterson DG
Jr, & Needham LL (1994) Determinants of TCDD half-life in veterans of
Operation Ranch Hand. J Toxicol Environ Health, 41: 481-488.
Wurrey CJ, Fairless BJ, & Kimball HE (1989) Matrix isolation GC/FT-IR
spectra of octachlorinated and 2,3,7,8-tetrabrominated
dibenzo- p-dioxins and dibenzofurans. Spectroscopy, 4: 39-43.
Yang KH, Yoo BS, & Choe SY (1983) Effects of halogenated
dibenzo- p-dioxins on plasma disappearance and biliary excretion of
ouabain in rats. Toxicol Lett, 15: 259-264.
Young AL (1983) Long term studies on the persistence and movement of
TCDD in a natural ecosystem. In: Tucker RE, Young AL, & Gray AP ed.
Human and environmental risks of chlorinated dioxins and related
compounds. New York, London, Plenum Press, pp 173-190.
Zabel EW, Cook PM, & Peterson RE (1995) Toxic equivalency factors of
polychlorinated dibenzo- p-dioxin, dibenzofuran and biphenyl
congeners based on early life stage mortality in rainbow trout
(Oncorhynchus mykiss). Aquat Toxicol, 31: 315-328.
Zacharewski T, Harris M, Safe S, Thoma H, & Hutzinger O (1988)
Applications of the in vitro aryl hydrocarbon hydroxylase induction
assay for determining "2,3,7,8-tetrachloro-dibenzo- p-dioxin
equivalents": Pyrolyzed brominated flame retardants. Toxicology, 51:
177-189.
Zacharewski T, Harris M, Safe S, Thoma H, Hauschulz G, Knorr E, &
Hutzinger O (1989) Applications of the in vitro AHH induction
bioassay for determining 2,3,7,8-TCDD equivalents: Pyrolyzed flame
retardant mixtures. Chemosphere, 18: 383-387.
Zelinski V, Lorenz W, & Bahadir M (1993) Brominated flame retardants
and resulting PBDD/F in accidental fire residues from private
residences. Chemosphere, 27: 1519-1528.
Zelinski V, Wichmann H, Lorenz W, & Bahadir M (1994) Chlorinated and
brominated dioxins and furans in fire residues from private residences
and vehicles -- A comparison. Fresenius Environ Bull, 3: 449-453.
Zier B, Lahaniatis D, Bieniek D, & Kettrup A (1990) Formation of
brominated dibenzodioxins and -furans by thermolysis of
polybutylene-terephthalate containing decabromodiphenylether
-- influence of temperature, antimony trioxide and water. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 381-384 (Organohalogen Compounds, Volume
2).
Zier B, Lenoir D, Lahaniatis ES, & Kettrup A (1991) Surface catalyzed
halogenation-dehalogenation reactions of aromatic bromine compounds
adsorbed on fly ash. Chemosphere, 22: 1121-1129.
Zober A, Messerer P, & Huber P (1990) Thirty-four-year mortality
follow-up of BASF employees exposed to 2,3,7,8-TCDD after the 1953
accident. Int Arch Occup Environ Health, 62: 139-157.
Zober MA, Ott MG, Päpke O, Senft K, & Germann C (1992) Morbidity study
of extruder personnel with potential exposure to brominated dioxins
and furans: I. Results of blood monitoring and immunological tests.
Brit J Ind Med, 49: 532-544.
APPENDICES
APPENDIX I: Directives/Test Rules Concerning PBDDs/PBDFs
Table 2 (in chapter 2) gives the PBDD/PBDF congeners substituted
with bromine in the 2,3,7,8-positions. As these are the most toxic
congeners, in some investigations only these congeners are determined.
The German Dioxin Directive (1994) has established temporary and
permanent limitations on the concentrations of certain
2,3,7,8-substituted PBDDs/PBDFs in products brought to the German
marketplace (see also Anon, 1996). For the first 5 years (i.e. until
15 July 1999), the sum concentration of the four PBDD/PBDF congeners
listed in category A must be less than 10 ppb, and the sum
concentration of the eight congeners in categories A and B must be
less than 60 ppb. After this time, the limits become 1 ppb and 5 ppb,
respectively. Restrictions on PCDDs/PCDFs are already established.
Category A Category B
2,3,7,8-TeBDD 1,2,3,7,8-PeBDF
2,3,7,8-TeBDF 1,2,3,4,7,8-HxBDD
1,2,3,7,8-PeBDD 1,2,3,6,7,8-HxBDD
2,3,4,7,8-PeBDF 1,2,3,7,8,9-HxBDD
In 1987, the US EPA issued a Test Rule requiring manufacturers
and importers of certain halogen-containing chemicals to analyse their
products for 2,3,7,8-substituted PHDDs/PHDFs. Specific limits of
quantitation and analytical requirements concerning recoveries and
precision were included in the Test Rule. Among the chemicals tested
were decabromodiphenyloxide and TBBPA.
Analyte Limit of quantitation (ppb)
2,3,7,8-TeBDD 0.1
1,2,3,7,8-PeBDD 0.5
1,2,3,4,7,8-HxBDD 2.5
1,2,3,6,7,8-HxBDD 2.5
1,2,3,7,8-HxBDD 2.5
1,2,3,4,6,7,8-HpBDD 100
2,3,7,8-TeBDF 1
1,2,3,7,8-PeBDF 5
2,3,4,7,8-PeBDF 5
1,2,3,4,7,8-HxBDF 25
2,3,4,6,7,8-HxBDF 25
1,2,3,7,8,9-HxBDF 25
1,2,3,4,6,7,8-HpBDF 1000
1,2,3,4,7,8,9-HpBDF 1000
APPENDIX II: Apparatuses and Terminology Used in Thermolysis
Experiments
Apparatuses used for thermolysis experiments are given below:
DIN oven: According to German standard DIN 53436. Open horizon tal
quartz tube (1 m long, 4 cm in diameter) with a gas flow in one
direction. The sample (mg to a few g) is placed on a quartz plate
within the tube, and a ring oven moves outside along the tube (1
cm/min). The design of the DIN apparatus simulates real fire
situations ranging from smouldering to open flame.
BIS oven: Same as DIN apparatus but stationary oven.
VCI oven: Vertical combustion apparatus with two heating zones. The
oven is heated to a defined temperature, and then the sample
(approximately 50 mg) is dropped via a valve into the combustion zone.
During the experiment, an air flow is maintained. Residence times
range from a few seconds up to 10 min. Owing to the small sample size,
the limit of quantification is relatively high. The VCI apparatus
simulates an instant fire.
In general, the combustion gases are adsorbed on XAD resin, and
the solid residues can be analysed for PBDDs/PBDFs as well. Typical
temperature ranges for all experiments are from 300 to 800 _C.
Quartz tube: The sample is sealed into a quartz tube and then heated
to a defined temperature.
Terms referring to thermal treatment are used as follows:
Thermolysis: General term for treating materials, compounds, etc. at
elevated temperatures. No specification is given for oxygen content,
technology, etc.
Combustion: General term for thermal treatment, more specifically in
the presence of oxygen or air.
Incineration: Refers to large-scale plants as used, for example, in
the incineration of municipal solid waste, hazardous waste, sewage
sludge, clinical waste, etc.
Pyrolysis: Thermal treatment under exclusion of oxygen/air.
RÉSUMÉ
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Les polybromodibenzo- p-dioxines (PBDD) et les polybromodi
benzofuranes (PBDF) sont des composés aromatiques de structure
quasiment plane. Il peut y avoir théoriquement 75 PBDD et 135 PBDF. En
outre, un grand nombre de dérivés halogénés mixtes -- 1550
bromo/chloro dibenzo- p-dioxines (PXDD) et 3050 bromo/chloro
dibenzofuranes (PXDF) -- sont également envisageables sur le plan
théorique. En raison de la complexité des méthodes d'analyse et de la
rareté des substances de référence utilisables à des fins analytiques,
il n'est possible de rechercher et de doser qu'un petit nombre de ces
composés. Les dérivés les plus toxiques sont ceux qui sont substitués
en position 2, 3, 7 et 8. Il existe ainsi 7 PBDD et 10 PBDF substitués
en 2, 3, 7 et 8 et on peut également envisager 337 PXDD et 647 PXDF
substitués aux mêmes positions.
Les PBDD et les PBDF ont une masse moléculaire plus élevée que
celle de leurs homologues chlorés. Leur point de fusion est élevé et
leur tension de vapeur est faible, tout comme leur solubilité dans
l'eau. Ils sont généralement solubles dans les graisses, les huiles et
les solvants organiques. On ne possède que très peu de données
expérimentales sur les propriétés physiques et chimiques de ces
composés.
Les PBDD et les PBDF sont photolysés plus rapidement que les
dibenzo- para-dioxines polychlorées (PCDD) et les dibenzofurannes
polychlorés (PCDF). Ils présentent une bonne stabilité à la chaleur.
Les températures de formation ou de destruction des PBDD/PBDF
dépendent d'un certain nombre de facteurs, entre autres, la présence
ou l'absence d'oxygène, de polymères ou d'additifs retardateurs de
flamme comme le trioxyde d'antimoine (Sb2O3).
En présence d'un excès de chlore celui-ci se substitue au brome
pour donner des PXDD/PXDF.
En raison du caractère toxique de ces composés et de leur
sensibilité à la photolyse, des précautions sont en prendre lors de
l'échantillonnage et de l'analyse. Il faut en particulier faire appel
à des méthodes extrêmement sensibles, sélectives et spécifiques (comme
la chromatographie en phase gazeuse couplée à la spectrométrie de
masse) en raison du nombre très élevé de dérivés. Les méthodes
d'échantillonnage sont identiques pour toutes les dibenzo- p-dioxines
et tous les dibenzofuranes polyhalogénés (PHDD et PHDF), toutefois la
séparation et le dosage des PBDD et des PBDF (et des PXDD et des PXDF)
sont un peu différents de ceux de leurs homologues chlorés. Les
PBDD/PBDF ont une masse moléculaire plus élevée et un temps de
rétention chromatographique plus long que leurs homologues chlorés. En
outre, la configuration des motifs isotopiques observés en
spectrométrie de masse et les interférences sont également différents.
Le très petit nombre de substances de référence limite
considérablement les possibilités d'identification des dérivés
polybromés. Pour la même raison, la recherche et le dosage des dérivés
halogénés mixtes sont quasiment impossibles.
2. Formation et sources d'exposition humaine et environnementale
On ne connaît pas de PBDD/PBDF d'origine naturelle. On ne les
produit pas non plus délibérément (sauf à des fins scientifiques): ce
sont en général les sous-produits involontaires de divers processus.
Ils peuvent se former au cours de réactions chimiques, photochimiques
ou thermiques à partir d'un certain nombre de précurseurs ou être
synthétisés de novo.
Ils sont présents à l'état d'impuretés dans divers dérivés
organiques bromés, comme les bromophénols et en particulier, dans les
retardateurs de flamme, comme les polybromodiphényléthers (PBDE), le
décabromodiphényle (décaBB ou DBB), le 1,2-bis(tribromophénoxy)éthane,
le tétrabromobisphénol A (TBBPA) etc. On en a mis en évidence dans des
résidus de distillation de divers bromophénols et bromoanilines ainsi
que dans des déchets de laboratoires de chimie.
On a décelé la présence de PBDF et, en moins grande quantité, de
PBDD, dans les produits de photodécomposition de composés organiques
bromés tels que les PBDE et les bromophénols.
Les essais de thermolyse effectués en laboratoire ont mis en
évidence la formation de PBDD/PBDF à partir de bromophénols, de PBDE,
de polybromobiphényles (PBB) et autres dérivés bromés utilisés comme
retardateurs de flamme (à l'état pur ou dans une matrice polymère).
Les rendements obtenus variaient dans de larges proportions, allant de
zéro à des valeurs maximales de l'ordre du g/kg (dans le cas des
PBDE). En général, il y avait beaucoup plus de PBDF que de PBDD. On a
constaté que pour une série de retardateurs de flamme, la température
optimale de formation des PBDF se situait entre 600 et 900°C. La
présence de polymères ou de synergisants (par ex. Sb2O3) a eu pour
effet de diminuer la température de optimale de formation, la ramenant
aux alentours de 400°C. Outre la température et la présence d'une
matrice polymère ou de synergisants, d'autres facteurs tels que les
métaux ou oxydes métalliques, l'eau, l'oxygène et le système de
combustion utilisé ont également eu une influence sur la nature et la
proportion des PBDD ou PBDF obtenus. Dans le cas de mélanges ternaires
constitués d'un PBDE, d'une matrice polymère et de Sb2O3, ce sont
des tétrabromodibenzofuranes (tétraBDF ou TeBDF) que l'on a surtout
obtenus. On a trouvé des 2,3,7,8-PBDD/PBDF (tétra et hepta) à diverses
concentrations; par exemple du 2,3,7,8-TeBDF à des concentrations
allant jusqu'à 2000 mg/kg dans les produits de pyrolyse de polymères
contenant de l'octabromodiphényléther (octaBDE ou OBDE).
Dans l'industrie des matières plastiques, des températures
élevées (150-300°C) peuvent être atteintes au cours de divers
processus. L'étude des vapeurs qui s'échappent des presses à injecter,
extrudeuses etc. lors du thermoformage de plastiques tels que les
résines ABS (acrylonitrile-butadiène-styrène) ou PBT (téréphtalate de
polybutylène) contenant divers retardateurs de flamme bromés, montre
que des PBDD/PBDF peuvent se former à ces températures. Ce sont l'OBDE
et le décabromodiphényléther (décaBDE ou DBDE) qui donnent naissance
aux plus grandes quantités de PBDD/PBDF, les PBDF étant les plus
abondants. La quantité de TBBPA ou de TBPI
(bis-tétrabromophtalimide-éthylène) sont beaucoup plus faibles (de
plusieurs ordres de grandeur). Lors du thermoformage de résines ABS
contenant du bromostyrène ou du 1,2-bis(tribromophénoxy)éthane comme
retardateurs de flamme, on n'a pas décelé de PBDD/PBDF. En ce qui
concerne les autres homologues substitués en 2,3,7,8, soit on ne les a
pas recherchés (en présence de DBDE), soit on en a trouvé des traces
(en présence d'OBDE), soit on n'est pas parvenu à les mettre en
évidence (en présence de TBBPA et de TBPI).
On a analysé divers plastiques à différents stades de leur mise
en forme, à la recherche de PBDD/PBDF. Il s'agissait soit de poudres à
mouler sous forme de granulés soit de pièces moulées dont on
connaissait les additifs retardateurs de flamme ainsi que de divers
objets de l'électroménager gris ou brun (téléviseurs, imprimantes,
ordinateurs) dont on ignorait quels additifs ils pouvaient contenir.
C'est dans les produits contenant des PBDE comme retardateurs de
flamme que l'on a trouvé la plus grande quantité de PBDD/PBDF. Ces
teneurs étaient de l'ordre de plusieurs milliers de µg/kg, c'est à
dire qu'elles dépassaient de plusieurs ordres de grandeur la
concentration observée dans les autres systèmes polymère/retardateur
de flamme. Les quantités formées dépendaient de la température et de
la durée des divers processus: adjonction de l'additif, extrusion ou
moulage par injection. Là encore et à quelques exceptions près, ce
sont les PBDF qui prédominaient par rapport aux PBDD, les dérivés les
plus substitués (plus de quatre bromes) étant présents en abondance.
Ce sont les pentabromodibenzofuranes (pentaDBF ou PeDBF) et les
hexabromodibenzofuranes (hexaBDF ou HxBDF) dont les concentrations
étaient les plus élevées. Dans des gainages de plastique, la
concentration de ces derniers atteignait 3000 µg/kg. Les supports de
circuits imprimés contenaient des tétra- et pentaBDF aux
concentrations maximales respectives de 1300 et 1400 µg/kg. La
concentration totale en PBDF (mono à hexa) se situait dans les limites
de 3,6 à 3430 µg/kg. Les PBDF substitués en 2,3,7,8 n'ont pas été
dosés ou bien n'étaient pas décelables ou encore étaient présents à
des concentrations trop faibles. La concentration maximale des PBDF
substitués en 2,3,7,8 (tétra à hexa) dans des gainages et des supports
de circuits imprimés allait de 11 µg/kg (tétra) à 203 µg/kg (hexa).
Les mesures effectuées pour déterminer si des PBDF sont libérés
par les téléviseurs et autres appareils de ce genre pendant leur
fonctionnement ont montré que la concentration de ces composés dans
l'air allait de zéro (non décelable) à 1800 pg de PBDF totaux (tétra à
hexa) par appareil.
La combustion de produits contenant des composés bromés provoque
un dégagement de PBDD/PBDF. Lors d'essais au cours desquels on avait
reproduit les conditions d'un véritable incendie, on a constaté que
des appareils électriques tels que téléviseurs, imprimantes, terminaux
d'ordinateurs et leurs gainages ou boîtiers laissaient des résidus
contenant de fortes concentrations de PBDF (mono à hexa), atteignant
plusieurs milliers de mg/kg. Ces concentrations étaient également
élevées dans les fumées (jusqu'à 1700 µg/m3) et leurs condensats
(plusieurs centaines de µg/m2). La concentration des PBDD était égale
à 3% de celle des PBDD/PBDF. Celle de l'isomère substitués en 2,3,7,8
n'atteignait pas 3% de la concentration totale des PBDD/PBDF. Les
penta- et hexaPBDF substitués en 2,3,7,8 ont fourni entre 1 et 16% du
total correspondant. Lors d'essais d'incendie de véhicules, on a
trouvé dans les résidus, des concentrations en PBDF (mono à octa)
pouvant aller jusqu'à 4,3 µg/kg.
Au cours d'incendies réels dans des résidences privées
(téléviseur étant en cause), dans des immeubles de bureaux (ordinateur
en cause) ou d'autres bâtiments, on a trouvé des concentrations
généralement inférieures à celles que l'on avaient obtenues
expérimentalement comme indiqué ci-dessus, la composition étant
toutefois qualitativement similaire. On a mis en évidence des PBDF
dans la presque totalité des échantillons; par contre, on n'a pas
toujours trouvé des PBDD. Lorsqu'elles étaient présentes, c'était à
faible concentration. La concentration des PBDF dans les résidus de
combustion était généralement de l'ordre de plusieurs µg/kg (faible à
élevée) mais on a observé des valeurs maximales (somme des dérivés
mono à hexa) pouvant atteindre 107 mg/kg. A proximité immédiate du
lieu des incendies, on a décelé la présence d'une contamination par
des PBDF (mono à hexa) à des concentrations allant la plupart du temps
de 0,1 à 13 µg/m2. En outre, on a pu déceler la présence de PXDD/PXDF
à des concentrations significatives. La proportion des PBDD/PBDF
substitués en 2,3,7,8 était relativement faible dans la majeure partie
des échantillons étudiés. Par exemple, lors d'incendies impliquant des
téléviseurs, les proportions maximales étaient respectivement égales à
3, 10 et 18% du total des tétra-, penta- et hexaBDF. Des prélèvements
de suie effectués après l'incendie d'une salle d'ordinateurs
contenaient des tétra- et des pentabromodibenzo- p-dioxines (tétra/
pentaBDD ou TeBDD/PeBDD) ainsi que des tétra- et pentaBDF à diverses
concentrations, la plus élevée (48 µg/kg) étant celle du 2,3,7,8-TeBDF
(TBDF).
On a mis en évidence des PXDD dans les cendres d'une chaudière à
bois. On n'a toutefois pas précisé de quelle sorte de bois il
s'agissait (traité ou non traité). On ne dispose d'aucune donnée sur
l'incinération d'autres combustibles comme le charbon, la tourbe ou le
mazout.
On a signalé la présence de PBDD/PBDF ou de PXDD/PXDF dans les
cendres volantes et les gaz émis par les incinérateurs municipaux ou
hospitaliers ou encore ceux que l'on utilise pour détruire les déchets
dangereux. La plupart de ces composés prennent probablement naissance
dans l'incinérateur lui-même, soit à partir de précurseurs qui
réagissent aux températures élevées engendrées par les flammes, soit
par synthèse de novo à basse température dans la zone de post
combustion de l'appareil. On explique la formation des PXDD/PXDF par
un échange important entre atomes de brome et de chlore (échange avec
des composés chlorés présents dans les déchets), comme on peut
l'observer expérimentalement dans un certain nombre de cas. Les
concentrations de PBDD/PBDF et PXDD/PXDF mesurées dans les cendres
volantes des incinérateurs sont de l'ordre du ng/kg ou du µg/kg. Dans
la plupart des cas, on constate que la concentration des
dibenzo- p-dioxines dépasse celle des dibenzofuranes et que les
PXDD/PXDF sont plus abondants que les PBDD/PBDF. Parmi les homologues
substitués en 2,3,7,8, on a trouvé une dibenzo- p-dioxine
tétrahalogénée mixte (tétraXDD ou TeXDD) (Br2Cl2DD).
L'analyse d'échantillons de déchets provenant de décharges a
révélé la présence de PBDD/PBDF et de PXDD/PXDF à des concentrations
allant de plusieurs centaines à plusieurs milliers de ng/kg de poids à
sec. La concentration des dibenzo- p-dioxines (jusqu'à 580 ng/kg)
était inférieure à celle des dibenzofuranes (jusqu'à 4230 ng/kg). En
ce qui concerne les homologues, ce sont les dérivés les moins
halogénés (mono à tétra) qui prédominaient. Dans les déchets de
laboratoires de chimie, on a relevé la présence de PBDD/PBDF dont la
concentration maximale atteignait 15 500 ng/kg (dans le cas des
hexaBDF).
On a décelé la présence de PBDD/PBDF dans des plastiques (avec ou
sans métaux) à différents stades de leur recyclage. Les échantillons
provenaient pour la plupart de matériel de bureau, de supports de
circuits imprimés et autres types de matériel électronique au rebut.
Dans certains cas, la concentration totale des huits homologues
retenus (substitués en 2,3,7,8) atteignait 65 µg/kg. On a constaté que
la récupération des métaux pouvait aussi être une source de PBDD ou de
PXDD/PXDF. Des PBDD/PBDF ont été mis en évidence dans industries
textiles faisant usage de derivés bromés comme retardateurs de flamme.
On a également décelé des PBDF dans des gaz d'échappement, dans des
textiles avant et après traitement ainsi que dans des dépôts de
cheminée.
Des PBDD/PBDF et des PXDD/PXDF (ainsi que des PCDD/ PCDF) ont été
décelés dans les gaz d'échappement de moteurs utilisant de l'essence
au plomb, dans ceux de moteurs utilisant de l'essence sans plomb avec
ou sans catalyseur, ainsi que dans ceux de moteurs diesel. Etant donné
l'utilisation d'agents de balayage bromés ou chlorés (dibromo- et
dichloroéthane) comme additifs à l'essence au plomb, c'est dans
celle-ci que l'on trouve la plus forte concentration de PHDD/PHDF
(plusieurs milliers de ng/m3). L'essence sans plomb donne lieu à des
émissions beaucoup moins importantes de PHDD/ PHDF (inférieures
d'environ deux ordres de grandeur). L'épuration catalytique des gaz
permet de réduire encore ces valeurs. La concentration est un peu plus
élevée pour les moteurs diesel que pour les moteurs à explosion
(moteurs à allumage commandé) fonctionnant à l'essence sans plomb. On
a constaté que les PBDD/PBDF étaient plus abondants que les PXDD/PXDF
et que les PCDD/PCDF dans les gaz résultant de la combustion d'essence
au plomb. En général, la concentration des dibenzofuranes était
supérieure à celle des dibenzo- p-dioxines, avec prédominance des
homologues les moins substitués (mono à tri). On a observé une
composition analogue dans les résidus adhérant à la paroi des pots
d'échappement.
3. Transport, distribution et transformation dans l'environnement
On ne possède guère de données sur le transport et la
distribution des PBDD/PBDF dans l'environnement. En général, leurs
propriétés physicochimiques suggèrent certaines similitudes avec les
PCDD/ PCDF. On peut donc s'attendre, en cas de libération dans
l'environnement, à ce qu'ils se répartissent de préférence dans les
compartiments riches en carbone ou en corps gras, comme c'est le cas
des PCDD/PCDF.
Le transport des PBDD/PBDF aéroportés s'effectue soit sous la
forme de gouttelettes soit en phase gazeuse, le coefficient de partage
étant fonction du degré de bromation.
On ne dispose d'aucune donnée expérimentale sur la migration des
PBDD/PBDF dans l'eau ou le sol. Dans le cas des PBDF (tri à penta) on
a observé une adsorption aux sédiments. Du fait que les PBDD/PBDF sont
peu solubles dans l'eau, le lessivage à partir du sol devrait être
limité tout en étant susceptible de s'accroître en présence de
solvants organiques ou d'acides humiques.
Il n'existe pas non plus de données expérimentales sur les
processus de transport et de distribution des PBDD/PBDF entre les
divers compartiments du milieu et les biotes ou encore à l'intérieur
de ces biotes. En s'appuyant sur la valeur élevée du coefficient de
partage octanol/eau calculé pour un certain nombre de PCDD/PCDF, PBDD/
PBDF et PXDD/PXDF, on peut s'attendre à ce que la biodisponibilité des
PCDD/PCDF soit également élevée.
On a étudié la photolyse des PBDD/PBDF et celle des PXDD/ PXDF,
soit au laboratoire dans des solvants organiques ou sur des surfaces
de quartz, soit à l'extérieur sur le sol et sur des particules de suie
ou de poussière. C'est dans ces dernières conditions, plus
représentatives de la réalité environnementale, que l'on a observé les
réactions les plus lentes. La débromation réductrice s'est révélée
être l'une des principales voies de décomposition photochimique. La
vitesse de décomposition dépend du nombre et de la position des atomes
de brome. En général, les dérivés les plus substitués ou substitués
sur les chaînes latérales sont ceux dont la demi-vie est la plus
courte. Le calcul de la demi-vie donne des valeurs qui sont de l'ordre
de quelques minutes (rayonnement solaire direct ou UV et ampoules de
quartz), de quelques heures (pellicules solides, particules de suie ou
poussières et rayonnement solaire) et enfin de quelques centaines ou
milliers d'heures (échantillons de sol et rayonnement solaire). Par
exemple, la valeur calculée de la demi-vie du 2,3,7,8-TeBDD (TBDD) est
de 0,8 minutes en présence de lumière solaire (en solution dans un
solvant organique) ou de 32 h (dispersé sous la forme de pellicules
solides). On estime que la demi-vie des différents isomères du
tétraBDD est de 3 à 6 mois à la surface du sol. Si on les compare aux
PCDD/PCDF, on constate que les homologues bromés sont
photochimiquement moins stables. Dans le cas des PXDD/PXDF, ce sont
les atomes de brome qui s'éliminent préférentiellement lors de la
photolyse pour donner naissance à des PCDD/PCDF dont la demi-vie
photolytique est plus longue. Cette transformation des PXDD/ PXDF en
PCDD/PCDF se produit également pendant l'incinération.
Les PBDD/PBDF semblent être peu biodégradables.
La présence de PBDD/PBDF chez l'Homme et les animaux, comme le
révèlent un certain nombre d'études, est l'indication de leur capacité
de bioaccumulation. Lors d'études d'alimentation de type subchronique,
on a constaté que la 2,3,7,8-TeBDD s'accumulait dans l'organisme du
rat. On ne connaît pas la valeur des facteurs de bioaccumulation, de
bioconcentration ou de bioamplification des PBDD/PBDF et des
PXDD/PXDF.
4. Concentrations dans l'environnement et exposition humaine
Jusqu'ici, on ne s'est que rarement préoccupé d'inclure les
PCDD/PCDF et PBDD/PBDF dans les programmes de surveillance de
l'environnement. Les quelques études dont on dispose indiquent qu'ils
n'y sont pas très souvent présents.
Dans l'air ambiant, on trouve plus fréquemment des PBDF que des
PBDD. Seules des PBDD peu bromées (mono à tétra) ont été décelées à
des concentrations allant de non décelable à environ 0,85 pg/m3; il
s'agissait en l'occurrence de monobromodibenzo- p-dioxines (monoBDD
ou MoBDD) dans l'air d'un tunnel routier et d'un garage souterrain.
Dans le cas des PBDF, on a trouvé des homologues mono- à hexabromés à
des concentrations allant de non décelable à 74 pg/m3. Par exemple,
la concentration moyenne des PBDD/PBDF totaux (tri à hexa) mesurée en
Allemagne dans un tunnel routier, en centre ville et dans une banlieue
était respectivement égale à 23 pg/m3, 2 pg/m3 et 0,59 pg/m3; on
n'a pas constaté la présence de 2,3,7,8-TeBDD et la concentration
maximale des 2,3,7,8-TeBDF et des 1,2,3,7,8-PeBDF était respectivement
égale à 0,28 et 0,08 pg/m3. On a mis en évidence des PXDF dans des
échantillons d'air de rues à grande circulation, à des concentrations
allant jusqu'à 41 pg/m3 (Cl1Br1DF). Dans des échantillons de
poussières extérieures (provenant pour la plupart d'autoroutes), on a
également constaté la prédominance de PBDF/ PXDF (concentrations
maximales de plusieurs ng/kg) par rapport aux PBDD/PBDF
(concentrations maximales jusqu'à quelques centaines de ng/kg).
Dans des échantillons d'air prélevés dans des pièces équipées
d'un certain nombres d'appareils électroniques (téléviseurs ou
moniteurs d'ordinateurs), on a constaté la présence de PBDF (tétra à
hepta) à une concentration totale allant de 0,23 à 1,27 pg/m3. On n'a
pas décelé de PBDD. Dans des échantillons de poussière provenant de
salles d'ordinateurs, on a trouvé des PBDF à la concentration totale
de 2,4 à 5,5 µg/kg. Contrairement à ce que l'on a observé dans l'air,
il y avait prédominance des hexaBDF et des heptaBDF (HpBDF)
(heptabromodibenzofuranes). C'est seulement dans les échantillons de
poussière que l'on a relevé la présence de faibles concentrations de
tétraBDD (jusqu'à 1 µg/kg), de tétraBDF substitués en 2,3,7,8 et de
pentaBDF (jusqu'à 0,07 µg/kg). Dans un échantillon de poussière
ménagère, la concentration des PBDF était plus basse d'un facteur 10.
La concentration totale des PBDD/PBDF était égale à celle des PCDD/
PCDF dans la poussière de salles d'ordinateurs mais elle était plus
faible dans la poussière ménagère. La poussière d'un garage souterrain
contenait moins de PBDF (mono et di) et de PXDF (di à tétra)
faiblement bromés, avec une concentration maximale de 4,3 µg/kg pour
les dibenzofuranes dihalogénés mixtes (DiXDF).
On ne dispose d'aucune donnée sur la concentration des PBDD/PBDF
dans l'eau.
Dans des sédiments fluviaux et marins prélevés au voisinage d'un
site industriel, on a trouvé des tétraBDD (jusqu'à 0,006 µg/kg de
poids sec) ainsi que des PBDF (tétra à hexa) (concentration totale
allant jusqu'à 0,37 µg/kg de poids sec). Dans les sédiments provenant
d'un réseau de drainage routier, on a trouvé des PBDF (concentration
totale des mono à tri: 2,5 µg/kg; concentration totale des tétra à
hepta: 0,3 µg/kg) et des PXDF (concentration totale des di et des tri:
1,85 µg/kg), mais pas de PBDD.
De même, des échantillons de sol prélevés à proximité d'une
autoroute contenaient des monobromodibenzofuranes (monoBDF ou MoBDF)
et des dibromodibenzofuranes (DiBDF) (total: 1,3 µg/kg), des tétra- et
des pentaBDF (total: 0,02 µg/kg) et des PXDF (total: 1 µg/kg), mais
pas de PBDD. Des échantillons de sol prélevés sur un site
d'incinération à proximité d'une usine de récupération de métaux
contenaient des PBDF à une concentration totale allant jusqu'à 100
µg/kg, mais pas de PBDD non plus. Dans une série d'échantillons de
boues d'égout provenant de stations d'épuration municipales, on a
trouvé une teneur totale en PBDF allant de non décelable à 3 µg/kg.
Dans un cas, on a décelé des traces de tétraBDD et de 2,3,7,8-TeBDF.
Un échantillon de compost s'est révélé à peu près exempt de PBDD/PBDF
(tétraBDF <0,003 µg/kg).
On ne possède aucune donnée quantitative sur la teneur des
denrées alimentaires en PBDD/PBDF.
Dans de l'herbe et des aiguilles de pin prélevées à proximité
d'une autoroute, on a décelé la présence de PBDF/PXDF faiblement
halogénés (mono à tétra) et de traces de PBDD/PXDD (mono à tri).
On n'a pas trouvé de traces de PBDD/PBDF dans les rares
échantillons biologiques de faune et de flore sauvages dont on
disposait.
Dans du lait de vache provenant de fermes situées à proximité
d'une installation municipale d'incinération, on pense avoir trouvé
des dérivés halogénés qui seraient des tribromodibenzofuranes (triBDF
ou TrBDF), un tétraBDF et un pentaBDF (sans substitution en 2,3,7,8).
Dans les rares échantillons de tissus adipeux et de lait humain
provenant de la population générale qui ont été analysés à la
recherche de PBDD/PBDF, on n'a pas trouvé trace de ces produits.
Une contamination par des PBDD/PBDF est possible sur divers lieux
de travail où l'on produit, transforme, utilise ou évacue certains
retardateurs de flamme ou produits qui en contiennent, notamment quand
sont mis en oeuvre des processus nécessitant des températures élevées.
Le degré d'exposition des travailleurs dépend non seulement des
composés en cause mais encore de la qualité de l'air et de la
ventilation. On ne possède que peu de données résultant de la
surveillance de divers lieux de travail tels qu'usines de production
ou de transformation de matières plastiques, bureaux ou studios
comportant un grand nombre d'appareils électriques en fonctionnement
continu ou unités de recyclage (notamment installations de
récupération du cuivre). En général, on constate que les PBDF sont
plus abondants que les PBDD et que leur concentration est maximale
dans les ateliers où sont produits des polymères contenant des DBDE.
On a pu mettre en évidence des PBDF/PBDD substitués en 2,3,7,8 dans de
nombreux échantillons. Une contamination par ces composés a également
été constatée dans un laboratoire de chimie, dans la partie d'une
paillasse située au-dessous de la hotte. On manque de données de
contrôle relatives aux installations d'incinération des déchets.
5. Cinétique et métabolisme
La plupart des études concernent la 2,3,7,8-TeBDD et, dans une
moindre mesure, le 1,2,3,7,8-TeBDF. La demi-vie a également été
calculée pour un certain nombre d'homologues.
Après administration à des rats par voie buccale, intratrachéenne
ou par application cutanée, on a constaté que la 2,3,7,8-TeBDD était
résorbé dans une proportion qui dépendait de la voie d'administration
et de la dose. Par exemple, une dose unique de 1 nmol de 2,3,7,8-TeBDD
par kg de poids corporel a été résorbée à hauteur de 80% (voie buccale
ou intratrachéenne) ou de 12% (voie percutanée). L'absorption
percutanée de 1 nmol de 1,2,7,8-TeBDF par kg de poids corporel a été
d'environ 29%. Per os, la TeBDD est résorbée dans une proportion
comparable à celle de la 2,3,7,8-tétrachlorodibenzo- p-dioxine
(2,3,7,8- TeCDD ou TCDD). Par contre, l'absorption percutanée de ce
même composé (2,3,7,8-TeBDD) a été à peu près égale aux deux tiers de
celle d'une dose équimolaire de 2,3,7,8-TeCDD.
Lorsqu'on les administre à des rats par n'importe quelle voie, la
2,3,7,8-TeBDD ou le 1,2,7,8-TeBDF se répartissent dans tout
l'organisme, s'accumulant de préférence dans le foie et les tissus
adipeux, puis, dans l'ordre, dans la peau et les muscles. Par exemple,
3 jours après administration de doses uniques de 2,3,7,8-TeBDD (1 nmol
par kg de poids corporel) on observait, dans ces tissus, une
répartition dans les proportions respectives de 20%, 20%, 11% et 4%,
alors que le thymus et les surrénales n'en contenaient respectivement
que 0,03% et 0,4%. Chez le rat, la répartition de la 2,3,7,8-TeBDD
entre le foie et les tissus adipeux dépendait de la dose, du mode
d'exposition, et du temps écoulé depuis l'administration. Le rapport
concentration dans le foie/concentration dans les tissus adipeux
mesuré dans différentes conditions allait de 0,2 à 6,5 (doses uniques
de 2,3,7,8-TeBDD administrées à des rats). On ne possède aucune donnée
expérimentale concernant la transmission des PBDD/PBDF à la
progéniture.
On a décelé la présence de métabolites de tétraBDD/BDF dans la
bile et les matières fécales de rats. Ces métabolites se forment
principalement par hydroxylation du noyau aromatique et débromation
hydrolytique. Le taux de métabolisation (déterminé indirectement par
le taux d'excrétion biliaire) était différent selon qu'il s'agissait
de 2,3,7,8-TeBDD (environ 7%) ou de 1,2,7,8-TeBDF (environ 50%). Trois
jours après l'administration par voie intraveineuse d'une dose de
2,3,7,8-TeBDD égale à 1 nmol par kg de poids corporel, 14% de la dose
initiale étaient retrouvés sous forme de métabolites dans les matières
fécales des rats.
L'élimination et l'excrétion de la 2,3,7,8-TeBDD a été étudiée
chez le rat en utilisant différentes voies d'administration: voies
buccale, intraveineuse, intratrachéenne et percutanée. Dans toutes les
études, la principale voie d'élimination a été la voie fécale. La
radioactivité éliminée allait de 2% (voie percutanée) à 42% (voie
buccale) de la dose initiale, c'est-à-dire 1 nmol de
(3H)2,3,7,8-TeBDD par kg de poids corporel dans les échantillons de
matières fécales, et de 0,2 à 1% dans les échantillons d'urine. De
même, l'étude du 1,2,7,8-TeBDF sur des rats a également montré que
l'excrétion se fait essentiellement par la voie fécale, la dose
initiale administrée par voie intraveineuse, buccale ou percutanée
n'étant excrétée qu'à hauteur de 2 à 3% dans les urines. Au cours des
premiers jours qui ont suivi l'administration par voie buccale, les
composés ont été principalement éliminés tels quels dans les matières
fécales ainsi que dans la bile. La fraction de la dose initiale de
2,3,7,8-TeBDD retrouvée dans les matières fécales des rats après
administration de 1 nmol de ce composé par kg de poids corporel était
respectivement égale à 53% (voie buccale), 43% (voie intratrachéale)
et 10-20% (voie intraveineuse). Quelques jours après l'administration
de 2,3,7,8-TeBDD par voie buccale (1 nmol/kg de poids corporel),
environ 20% du composé initial ont été éliminés sous forme inchangée.
Pour un certain nombre de PBDD/PBDF, on possède des données sur
la rétention et la vitesse d'élimination. En particulier, on sait que
chez le rat, la charge relative de l'organisme en 2,3,7,8-TeBDD (et
homologues) dépend de la voie d'administration et de la dose
administrée, traduisant ainsi la variation du degré de résorption. On
a calculé la demi-vie d'un certain nombre de PBDD/PXDD et PBDF
présents dans divers tissus et dans les matières fécales de rats. Elle
s'échelonne entre 1 jour (élimination du 1,2,7,8-TeBDF présent d
l'organisme) et 99 jours (élimination du 2,3,4,7,8-PeBDF présent dans
le foie). Le calcul de la demi-vie de la 2,3,7,8-TeBDD présente dans
le foie, les matières fécales et les tissus adipeux donne les valeurs
respectives de 17, 18 et 58 jours, valeurs qui sont du même ordre que
celles de la 2,3,7,8-TeCDD dans le cas du foie et des matières fécales
mais plus de deux fois plus élevés que dans le cas des tissus adipeux.
Malgré des différences de rétention au cours des premiers jours, le
2,3,7,8-TeBDF et le 2,3,7,8-tétrachlorodibenzofurane (2,3,7,8-TeCDF ou
TCDF) ont une demi-vie comparable au niveau du foie.
Comme dans le cas des PCDD/PCDF, le calcul donne une demi-vie
beaucoup plus longue chez l'Homme que chez l'animal. Les estimations
sont les suivantes: 3 à 11 ans (moyenne 5,9 ans) pour la 2,3,7,8-TeBDD
et 1 à 2 ans (moyenne 1,5 ans) pour le 2,3,7,8-TeBDF. On a également
pu se rendre compte de la persistance de ces composés dans le cas d'un
chimiste qui avait préparé de la 2,3,7,8-TeBDD et de la 2,3,7,8-TeCDD
en 1956. Trente-cinq ans après, son sang contenait encore une quantité
importante de 2,3,7,8-TeBDD.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
La plupart des études ont porté sur la toxicité de la
2,3,7,8-TeBDD, mais on dispose tout de même de quelques données sur
les autres PBDD/PBDF et PXDD/PXDF.
La 2,3,7,8-TeBDD a des effets analogues à ceux de la
2,3,7,8-TeCDD, notamment un syndrome de dépérissement, une atrophie du
thymus et une action toxique sur le foie. On a observé au niveau du
foie des lésions caractéristiques d'une péliose hépatique, lésions qui
n'ont pas été observées chez le rat après exposition à la
2,3,7,8-TeCDD. De par leur nature (létalité, histopathologie, poids du
foie et du thymus), les lésions ou effets toxiques observés chez le
cobaye et le rat après une brève exposition au 2,3,7,8-TeBDF étaient
analogues à ceux observés après exposition au 2,3,7,8-TeCDF.
La 2,3,7,8-TeBDD agit sur le système endocrinien. Chez le rat, on
a observé une modification des hormones thyroïdiennes présentes dans
la circulation ainsi qu'une diminution de la spermatogénèse.
La DL50 par voie orale (période d'observation de 28 jours) de la
2,3,7,8-TeBDD pour le rat Wistar est d'environ 100 µg/kg de poids
corporel dans le cas des femelles et d'environ 300 µg/kg p.c. dans le
cas des mâles. Celle de la 2,3,7,8-TeCDD tirée d'autres études varie
de 22 à >3000 µg/kg p.c. Des doses équimolaires de 2,3,7,8-TeBDF et
de 2,3,7,8-TeCDF ont entraîné une mortalité comparable chez des
cobayes. Par exemple, on a observé une mortalité de 100% après
administration de 2,3,7,8-TeBDF (0,03 µmol/kg p.c., 15,8 µg/kg p.c.)
et de 2,3,7,8-TeCDF (0,03 µmol/kg p.c., 10 µg/kg p.c.). On a noté la
présence de lésions prépéliotiques et une modification des hormones
thyroïdiennes chez des rats qui avaient reçu une dose unique de 100 µg
de 2,3,7,8-TeBDD par kg de poids corporel.
Chez des rats Wistar ayant reçu pendant 13 semaines de la
2,3,7,8-TeBDD par voie orale, on a relevé des signes de réduction de
la spermatogénèse, la présence de spermatocytes anormaux ou nécrosés,
les signes d'une péliose hépatique grave ainsi qu'une modification des
hormones thyroïdiennes circulantes et du poids des organes. La dose
sans effet nocif observable (NOAEL) a été trouvée égale à 0,01 µg/kg
p.c. par jour.
Du 2,3,7,8-TeBDF administré par voie orale à des rats
Sprague-Dawley pendant 4 semaines a provoqué un retard de croissance
lié à la dose et des anomalies histopathologiques au niveau du foie et
du thymus. La NOAEL a été estimée 1 µg/kg p.c. par jour.
Chez des souris qui avaient reçu des 2,3,7,8-PBDD/PBDF
administrés par voie orale ou sous-cutanée on a noté, pour certains de
ces produits, des effets délétères sur le développement à des doses
non toxiques pour les mères et non létales pour les foetus. La dose la
plus faible (en µg/kg p.c.) produisant un effet observable (LOEL) - à
savoir une hydronéphrose et une fente palatine - après administration
d'une dose unique par voie orale à des souris gravides a été trouvée
respectivement égale à: 3 et 48 pour la 2,3,7,8-TeBDD, à 25 et 200
pour le 2,3,7,8-TeBDF, à 400 et 2400 pour le 2,3,4,7,8-PeBDF et à 500
et 3000-4000 pour le 1,2,3,7,8-PeBDF. On a constaté que la
2,3,7,8-TeBDD et la 2,3,7,8-TeCDD avaient pratiquement la même
aptitude à induire une hydronéphrose lorsqu'on utilisait la mole comme
unité; par contre, sur une base pondérale, les isomères bromés se
révélaient moins aptes que les isomères chlorés à produire une
hydronéphrose ou une fente palatine. Le 2,3,7,8-TeBDF, en revanche,
était plus actif que le 2,3,7,8-TeCDF.
On n'a trouvé aucune donnée sur la mutagénicité des PBDD/ PBDF ni
sur des points d'aboutissement toxicologiques en rapport avec des
propriétés mutagènes.
On ne dispose d'aucune étude sur la toxicité ou la
cancérogénicité à long terme des PBDD/PBDF. Une épreuve de
transformation cellulaire sur macrophages péritonéaux murins a donné
un résultat positif avec la 2,3,7,8-TeBDD. Cependant, l'activité
transformante de la 2,3,7,8-TeBDD était sept fois moins forte que
celle de la 2,3,7,8-TeCDD. En injectant les cellules ainsi obtenues à
des souris nude par voie sous-cutanée, on a observé l'apparition
ultérieure de tumeurs.
Après injection d'une série de PBDD et de PXDD (tétra et penta)
par voie intrapéritonéale à des rats Wistar mâles immatures, on a
constaté une perte de poids au bout de 14 jours. En se basant sur la
valeur de la DE50 (exprimée en moles), on a constaté que les composés
les plus toxiques étaient la 2,3,7,8-TeBDD, la 2-Br1-3,7,8-Cl3-DD et
la 2,3-Br2-7,8-Cl2-DD (TBCDD), qui ne sont substituées que sur les
quatre positions latérales. Pour l'activité relative des autres PBDD
étudiées, on a trouvé l'ordre suivant: 2,3,7,8- > 1,2,3,7,8- >
1,2,4,7,8- > 1,3,7,8-DD. Selon d'autres études, il n'y aurait que peu
de différence entre la 2,3,7,8-TeCDD et la 2,3,7,8-TeBDD en ce qui
concerne la valeur de la DE50 (exprimée en moles) pour la perte de
poids, l'atrophie du thymus et l'induction des enzymes hépatiques.
Une atrophie du thymus et d'autres signes d'immunotoxicité (par
ex. dans les paramètres hématologiques et aussi des modifications dans
certaines sous-populations de lymphocytes) ont été observés chez le
rat après exposition à plusieurs PBDD/PXDD et au 2,3,7,8-TeBDF ainsi
que chez le singe marmouset (Callithrix jacchus) après exposition à
la 2,3,7,8-TeBDD et à la TBCDD. On en a conclu qu'exprimée en moles,
l'activité de la 2,3,7,8-TeBDD était comparable à celle de la
2,3,7,8-TeCDD chez le rat et le singe. Par exemple, on a observé un
effet sensible sur certaines sous-populations de lymphocytes simiens
après injection sous-cutanée d'une dose unique de 30 ng de
2,3,7,8-TeBDD par kg de poids corporel, le même effet étant obtenu
avec une dose de 10 ng de 2,3,7,8-TeCDD par kg p.c. On n'a pas étudié
les effets immunotoxiques d'une exposition périnatale aux PBDD/PBDF.
Après avoir administré de manière subchronique de la
2,3,7,8-TeBDD ou de la 2,3,7,8-TeCDD par gavage à des souris, on a
constaté un accroissement des porphyrines hépatiques totales qui
dépendait de la dose.
Après administration à des rats d'une dose unique de
2,3,7,8-TeBDD et de 2,3,7,8-TeCDD, on a observé une réduction de la
concentration et de la quantité totale de vitamine A dans le foie, la
2,3,7,8-TeBDD ayant une activité (exprimée en moles) un peu inférieure
à celle de la 2,3,7,8-TeCDD.
Lors d'une épreuve sur oreille de lapin effectuée avec de la
2,3,7,8-TeBDD et du 2,3,7,8-TeBDF, on a observé une hyperkératose à la
dose de 100 µg/animal mais pas à la dose de 10 µg/animal. Dans le cas
de la 2,3,7,8-TeCDD, la dose sans effet observable (NOEL) était de
0,01 µg/animal.
On a constaté que plusieurs homologues tétra- (Br1Cl3DD,
Br2Cl2DD) et penta- (Br1Cl4DD) halogénés substitués en 2,3,7,8
avaient une activité antiestrogénique analogue à celle de la
2,3,7,8-TeCDD, comme l'a montré l'observation de cultures de cellules
humaines de cancer du sein.
Chez le rat, la 2,3,7-tribromodibenzo- p-dioxine (2,3,7-triBDD/
TrBDD) a réduit l'élimination de l'ouabaïne du plasma et son excrétion
par la voie biliaire; elle a également agi comme anticholagogue, mais
dans une moindre proportion que la 2,3,7,8-TeCDD.
Les PBDD/PBDF et les PXDD/PXDF sont de puissants inducteurs de
certaines enzymes microsomiennes dépendant du cytochrome P-450 (CYP).
On a obtenu une DE50 de 0,8-1 nmol/kg p.c. pour l'induction de la
CYP1A1 et d'environ 0,2 nmol/kg p.c. pour l'induction de la CYP1A2
dans le foie de rat après administration d'une dose unique de
2,3,7,8-TeBDD par voie orale. L'induction de la CYP1A1
(arylhydrocarbure-hydroxylase [AHH] ou de
l'ethoxyrésorufine- O-déséthylase [EROD]) a été observée chez
diverses espèces et un certain nombre de tissus in vivo ainsi que
dans des cultures de cellules de rat in vitro. Divers homologues se
sont révélés actifs à cet égard, de même que les produits de pyrolyse
de certains retardateurs de flamme. En général, l'induction des
enzymes se produisait à des concentration non toxiques, elle dépendait
de la dose, commençait peu après l'exposition et était de longue
durée. Elle était mesurable à des concentrations de l'ordre de la
picomole. L'activité inductrice différait de plusieurs ordres de
grandeur d'un homologue à l'autre, en fonction de la structure
chimique. Les inducteurs les plus actifs étaient la TCDD, la TBDD et
la TBCDD. Comparées à leurs homologues chlorés, les PBDD et les PXDD
avaient une activité (exprimée en moles) à peu près équivalente.
Contrairement à la TCDD, dont l'activité inductrice relative s'est
montrée indépendante du type de tissu examiné, la TBDD a fait preuve
d'en activité inductrice de l'EROD cinq fois plus élevée dans le foie
que dans le tissu cutané ou pulmonaire, lors d'études comportant
l'exposition de souris dans des conditions de subchronicité. Chez le
singe marmouset, l'activité inductrice de l'EROD s'établit comme suit:
TCDD > 2,3,4,7,8-pentachlorodibenzofurane (2,3,4,7,8-pentaCDF/PeCDF)
> 2,3,4,7,8- PeBDF, l'activité des enzymes étant comparée à la
concentration dans le foie. Des épreuves in vitro sur cultures de
cellules de rat ont donné des valeurs analogues pour la CE50 des PXDF
et des PCDF correspondants, l'effet examiné étant l'induction de l'AHH
et de l'EROD.
On pense que les PBDD/PBDF ont le même mode d'action que les
PCDD/PCDF et les d'autres hydrocarbures aromatiques halogénés de type
voisin. La fixation sur le récepteur cytosolique aux hydrocarbures
aromatiques, qui joue un rôle central dans la toxicité des composés du
type 2,3,7,8-TeCDD, a été confirmée pour plusieurs PBDD et PXDD/PXDF.
Leur affinité pour ce récepteur variait de plusieurs ordres de
grandeur, mais elle était comparable à celle de leurs homologues
chlorés.
7. Effets sur l'Homme
On ne dispose d'aucune donnée sur l'exposition humaine aux
PBDD/PBDF ni au sujet de leurs effets sur la santé de la population
dans son ensemble.
Deux cas d'effets aigus dus à une exposition à la 2,3,7,8-TeBDD/
TeCDD ont été rapportés, avec différents symptômes, dont une
chloracné.
Lors d'une autre étude, des employés de sexe masculin travaillant
dans une usine chimique et effectivement exposés à des PBDD/PBDF
provenant de l'utilisation de retardateurs de flamme (OBDE et DBDE),
ont été soumis à des épreuves immunologiques et autres examens de
laboratoire. Malgré la présence de légères modifications dans les
paramètres immunologiques, leur état général ne laissait en aucun cas
penser que la teneur de leur organisme en 2,3,7,8-TeBDD/TeBDF ait eu
des effets sur leur système immunitaire.
On ne connaît aucun cas de cancer mortel qui serait dû aux
PBDD/PBDF.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On ne possède que des données limitées sur les effets que les
PBDD/PBDF peuvent avoir sur les microorganismes, les plantes, les
invertébrés et les vertébrés.
On a soumis une série de PBDD/PBDF et leurs homologues chlorés à
une épreuve biologique sur alevins de truite arc-en-ciel
(Onchorhyncus mykiss) qui a mis en évidence l'activité de ces
composés. L'épreuve a montré, entre autres, que l'activité des PBDD/
PBDF diminue à mesure qu'augmente de degré de substitution par le
brome. La 2,3,7,8-TeBDD et le 2,3,7,8-TeBDF sont plus actifs que leurs
homologues chlorés respectifs.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
Las dibenzo- p-dioxinas polibromadas (PBDD) y los dibenzo
furanos polibromados (PBDF) son compuestos aromáticos tricíclicos casi
planares. En teoría existen 75 PBDD y 135 PBDF. Además es en teoría
posible la existencia de un amplio número de congéneres halogenados
mixtos: 1550 dibenzo- p-dioxinas bromadas/cloradas (PXDD) y 3050
dibenzofuranos bromados/clorados (PXDF). Dada la complejidad de los
procedimientos analíticos y la escasez de normas analíticas de
referencia, sólo se ha podido identificar y determinar un pequeño
número de estos productos. Los congéneres más tóxicos son los
sustituidos en las posiciones 2,3,7 y 8. Existen 7 PBDD sustituidos en
las posiciones 2,3,7 y 8 y 10 PBDF sustituidos en las posiciones 2,3,7
y 8, así como 337 posibles PXDD sustituidos en las posiciones 2,3,7 y
8, y 647 posibles PXDF sustituidos en las posiciones 2,3,7 y 8.
Los PBDD y PBDF tienen mayores pesos moleculares que sus análogos
clorados, altos puntos de fusión, bajas presiones de vapor y bajas
solubilidades en agua. En general son solubles en grasas, aceites y
disolventes orgánicos. Existen escasos datos experimentales sobre las
propiedades físicas y químicas de los PBDD y PBDF.
La fotolisis se produce con más rapidez en el caso de los PBDD y
PBDF que en las dibenzo- p-dioxinas policloradas (PCDD) y los
dibenzofuranos policlorados (PCDF). Los PBDD y PBDF son termoestables.
Las temperaturas de formación y destrucción de los PBDD y PBDF
dependen de varias condiciones, que incluyen la presencia o ausencia
de oxígeno, polímeros y aditivos pirorretardantes, como el trióxido de
antimonio (Sb2O3).
En presencia de cloro en exceso, el bromo es sustituido por cloro
para dar PXDD y PXDF.
Teniendo en cuenta el carácter tóxico de estos productos y sus
propiedades fotolíticas debe actuarse con cuidado en el curso del
muestreo y el análisis. Dado el alto número de congéneres de los PBDD
y PBDF se necesitan métodos de análisis muy sensibles, selectivos y
específicos (cromatografía de gases o espectrometría de masas). Los
procedimientos de muestreo son idénticos en el caso de todas las
dibenzo- p-dioxinas polihalogenadas (PHDD) y los dibenzo furanos
polihalogenados (PHDF), pero la separación y determinación de las PBDD
y los PBDF (y las PXDD y los PXDF) difiere ligeramente de las
correspondientes a sus análogos clorados. Los PBDD y PBDF tienen pesos
moleculares más altos y mayores periodos de retención en la
cromatografía de gases que los análogos clorados, así como distintos
tipos de agrupación isotópica en espectrometría de masas y de
compuestos de interferencia. La identificación exacta de determinados
congéneres bromados es muy limitada debido al escaso número de
patrones de referencia actualmente disponibles. Por el mismo motivo,
la determinación de los congéneres halogenados mixtos es casi
imposible.
2. Formación y fuentes de exposición humana y ambiental
No se conoce la presencia natural de PBDD y PBDF. No se producen
de modo intencional (excepto para fines científicos), pero aparecen
como productos indeseados en distintos procesos. Pueden formarse por
reacciones químicas, fotoquímicas o térmicas a partir de precursores y
en la llamada síntesis de novo.
Se han hallado PBDD y PBDF como contaminantes en productos
químicos orgánicos bromados (por ej., bromofenoles) y, en particular,
en pirorretardantes, como los éteres difenílicos polibromados (PBDE),
el decabromobifenilo (decaBB o DBB), el 1,2-bis(tribromofenoxi)etano,
el tetrabromobisfenol A (TBBPA) y otros. Se han hallado en residuos de
destilación de algunos bromofenoles y bromoanilinas y en desechos de
laboratorios químicos.
Los PBDF y, en menor grado, las PBDD se han hallado como
productos de degradación fotoquímica de sustancias químicas orgánicas
bromadas, como los PBDE y los bromofenoles.
En experimentos de termolisis en laboratorio se ha observado la
formación de PBDD y PBDF a partir de bromofenoles, PBDE, bifenilos
polibromados (PBB) y otros pirorretardantes bromados (puros o en
matriz de polímero). Se observó una amplia gama de rendimientos, desde
0 hasta los valores máximos (alcanzados a partir de los PBDE) en la
gama de g/kg. Por lo general, los PBDF son mucho más abundantes que
las PBDD. La temperatura óptima de formación de PBDF en una serie de
pirorretardantes puros se situó en la gama de 600-900°C. La presencia
de polímeros o productos sinérgicos (por ej., Sb2O3) produjo la
disminución de la temperatura óptima de formación (hasta 40°C). Además
de la temperatura y la presencia de productos sinérgicos o de una
matriz de polímero, varios otros factores, tales como la presencia de
metales, óxidos metálicos, agua y oxígeno, y el tipo de aparato de
combustión utilizado, influyeron en el rendimiento y el tipo de PBDD y
PBDF obtenidos. En las mezclas ternarias de PBDE, matriz de polímero y
Sb2O3, los tetrabromo dibenzofuranos (tetraBDF o TeBDF) fueron con
frecuencia el grupo homólogo más abundante. Se hallaron en
concentraciones variables PBDD y PBDF sustituidos en las posiciones
2,3,7 y 8 (tetra a hepta); por ejemplo, se halló el 2,3,7,8-TeBDF en
concentraciones de hasta 2000 mg/kg en pirolizados de polímeros que
contenían éter de octabromodifenilo (octaBDE u OBDE).
En la fabricación de plásticos se producen altas temperaturas
(150-300°C) en varios procesos. Los estudios de los vapores de escape
de máquinas de tratamiento de polímeros, como el
acrilonitrilo-butadieno-estireno (ABS) y el tereftalato de
polibutileno (PBT), que contenían distintos tipos de pirorretardantes
bromados, mostraron que pueden formarse PBDD y PBDF (di a octa) a esas
temperaturas. El OBDE y el éter de decabromodifenilo (decaBDE o DBDE)
produjeron las mayores cantidades de PBDD y PBDF, consistiendo la
porción principal en PBDF. Las concentraciones observadas en el caso
de TBBPA o de etileno de bis-tetrabromo-ftalimida (TBPI) eran
inferiores en varios órdenes de magnitud. No se hallaron PBDD ni PBDF
en el curso del tratamiento de ABS pirorretardado por medio de
estireno bromado o 1,2-bis(tribromofenoxi)etano. Los congéneres
sustituidos en las posiciones 2,3,7 y 8 no se determinaron
(fabricación de DBDE), se hallaron sólo en concentraciones
infinitesimales (fabricación de OBDE) o no se detectaron (fabricación
de TBBPA y TBPI).
Se analizó la presencia de PBDD y PBDF en varios materiales
plásticos en distintas etapas de fabricación. Comprendieron resinas
(granuladas) y partes moldeadas, cuyos aditivos pirorretardantes eran
conocidos, así como muestras de dispositivos eléctricos comerciales
(televisores, impresoras, ordenadores), cuyos aditivos
pirorretardantes eran desconocidos. Se hallaron las mayores
concentraciones de PBDD y PBDF en los materiales pirorretardados con
PBDE, en la gama de varios miles de µg/kg, excediendo así en varios
órdenes de magnitud a las concentraciones de otros
pirorretardantes/sistemas de polímero. Los factores que influyen en la
cuantía de la formación son la temperatura y la duración de procesos
tales como el mezclado, la extrusión y el moldeo. También en este caso
dominan los PBDF, con algunas excepciones, sobre las PBDD,
prevaleciendo los derivados muy bromados (>tetra). Las máximas
concentraciones se observaron en el caso de los
pentabromodibenzofuranos (pentaBDF o PeBDF) y los
hexabromodibenzofuranos (hexaBDF o HxBDF). Los últimos alcanzaron
concentraciones tan altas como 3000 µg/kg en piezas de revestimiento.
Los tableros de circuitos impresos contenían tetra- y pentaBDF en
concentraciones máximas de 1300 y 1400 µg/kg, respectivamente. Las
concentraciones totales de PBDF (mono a hexa) se hallaban en la gama
de 3,6-3430 µg/kg. Los PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 no se determinaron, eran indetectables o se hallaban en
concentraciones relativamente bajas. Las concentraciones máximas de
PBDF sustituidos en las posiciones 2,3,7 y 8 (tetra a hexa) en
revestimientos o tableros de circuitos impresos eran de 11 µg/kg
(tetra) a 203 µg/kg (hexa).
Los experimentos destinados a determinar si se liberaban PBDF de
los televisores o aparatos análogos durante el uso mostraron la
presencia de concentraciones en el aire que iban de los niveles
indetectados a 1800 pg de PBDF totales (tetra a hexa) por aparato.
La combustión de productos que contenían compuestos bromados
produjo la emisión de PBDD y PBDF. Las pruebas experimentales de
incendio que simulaban condiciones reales utilizando aparatos
eléctricos tales como televisores, impresoras, terminales de
ordenador, y sus receptáculos, permitieron hallar altas
concentraciones de PBDF (mono a hexa) en los residuos de la combustión
(miles de µg/kg), en el condensado del humo (centenares de µg/m2) y
en el humo (hasta 1700 µg/m3). Las concentraciones de PBDD fueron del
3% aproximada mente de los niveles detectados de PBDD y PBDF. El
isómero 2,3,6,8-sustituido se hallaba sobre todo por debajo del 3% del
total de tetraBDF. Los penta- y hexaBDF 2,3,7,8-sustituidos dieron del
1 al 16% de los totales correspondientes. La combustión de vehículos
de prueba produjo concentraciones de PBDF (mono a octa) de hasta 4,3
µg/kg en los residuos del incendio.
En el curso de incendios reales en residencias privadas (con
inclusión de televisores), oficinas (con inclusión de ordenadores) y
otros edificios, las concentraciones medidas se hallaban en la mayoría
de los casos por debajo de los valores observados en los modelos
experimentales antes descritos, pero la composición cualitativa de las
muestras era análoga. Se hallaron PBDF en casi todas las muestras,
pero no siempre se detectaron PBDD; si se encontraban, sus
concentraciones eran bajas. Las concentraciones de PBDF en los
residuos de la combustión se hallaban principalmente en la gama de
µg/kg (bajas a altas), pero también se observaron valores máximos
(suma de mono a hexa) de hasta 107 mg/kg. Las concentraciones
contaminantes de PBDF (mono a hexa) en las cercanías del lugar del
incendio variaban entre 0,1 y 13 µg/m2 en la mayoría de los casos.
Pudieron detectarse además concentraciones significativas de PXDD y
PXDF. La proporción de PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 era relativamente baja en la mayoría de las muestras examinadas.
Por ejemplo, se registraron proporciones máximas del 3, el 10 o el 18%
de los totales correspondientes de tetra-, penta- o hexaBDF,
respectivamente, en los incendios que comprendieron televisores. Las
muestras de cenizas recogidas después de un incendio en una sala de
ordenadores contenían tetra- y pentabromodibenzo- p-dioxinas
sustituidas en las posiciones 2,3,7 y 8 (tetra/pentaBDD o TeBDD/PeBDD)
y tetra- y pentaBDF, con una concentración máxima de 48 µg/kg en el
caso del 2,3,7,8-TeBDF (TBDF).
Se detectaron PXDD en la ceniza de una caldera de combustión de
madera. Sin embargo, no se especificó el tipo de madera (tratada o sin
tratar). No se dispuso de datos sobre la incineración de otros
combustibles, como carbón, turba o fueloil.
Se señaló la presencia de PBDD/PBDF y/o PXDD/PXDF en cenizas
volantes y en los gases de combustión de incineradores municipales, de
hospital o de desechos peligrosos. La mayor parte de esos productos
estaban formados probablemente en el propio incinerador, a partir de
precursores a altas temperaturas en la llama o por síntesis de novo
a temperaturas bajas en la zona poscombustión del incinerador. La
formación de PXDD y PXDF se explica por las amplias reacciones de
intercambio bromo-cloro (con donantes de cloro en los desechos)
observadas en varias condiciones de prueba. Las cantidades de
PBDD/PBDF y PXDD/PXDF medidas en las cenizas volantes de los
incineradores se hallaban comprendidas en la gama de ng/kg a µg/kg. En
la mayoría de los casos, las concentraciones de dibenzo- p-dioxinas
excedían a las de los dibenzofuranos, siendo los PXDD/PXDF más
abundantes que los PBDD/PBDF. Entre los congéneres 2,3,7,8-sustituidos
se halló una dibenzo- p-dioxina tetrahalogenada mixta (tetraXDD o
TeXDD) (Br2Cl2DD).
Los análisis de muestras de desechos procedentes de varios
vertederos mostraron la presencia de PBDD/PBDF y PXDD/PXDF en
concentraciones de varios centenares a varios miles de ng/kg de peso
en seco. La concentración de las dibenzo- p-dioxinas (hasta 580
ng/kg) era inferior a la de los dibenzofuranos (hasta 4230 ng/kg). Por
lo general, la gama de homólogos estaba dominada por los derivados
menos halogenados (mono a tetra). Los desechos de laboratorios
químicos contenían PBDD y PBDF con una concentración máxima de 15 500
ng/kg en el caso de los hexaBDF.
Se hallaron PBDD y PBDF en materiales plásticos (con o sin
metales) en varias etapas de reciclado. Las muestras procedían
principalmente de maquinaria de oficina, tableros de circuitos
impresos y otras chatarras electrónicas. En algunos casos, la suma de
las concentraciones de 8 congéneres PBDD/PBDF seleccionados con
sustituciones en las posiciones 2,3,7 y 8 llegaba a 65 µg/kg. También
se observó que la recuperación de metales era una fuente de PBDD y/o
PXDD/PXDF. Igualmente se detectaron PBDD y PBDF en la industria
textil, en la que se utilizaban pirorretardantes bromados. Se hallaron
PBDF en el aire de salida de industrias textiles, antes y después del
procesado, y en sedimentos de chimenea.
Se han detectado PBDD/PBDF y PXDD/PXDF (junto con PCDD/ PCDF) en
los gases de escape de motores que utilizan gasolina plomada, en los
gases de escape de motores que usan gasolina sin plomo con o sin
convertidores catalíticos y en los gases de escape de motores diesel.
Teniendo en cuenta que la gasolina plomada contiene productos de
limpieza bromados y clorados (dibromoetano y dicloroetano), las
mayores concentraciones de PHDD y PHDF (varios miles de ng/m3) se
encuentran en este tipo de gasolina. La gasolina sin plomo produce
emisiones muy inferiores de PHDD y PHDF (aproximadamente inferiores en
dos órdenes de magnitud). Tras la limpieza catalítica de los gases se
observa una nueva reducción. Los valores correspondientes a los
motores diesel eran ligeramente superiores a los hallados en los
motores Otto (motores de encendido por chispa) que funcionan con
gasolina sin plomo. En los gases de escape procedentes de la
combustión de gasolina plomada, los PBDD/PBDF eran más abundantes que
los PXDD/PXDF y PCDD/PCDF. En general, las concentraciones de los
dibenzofuranos excedían a las de las dibenzo- p- dioxinas, con un
predominio de los homólogos de baja sustitución (mono a tri). Se han
hallado distribuciones análogas en los residuos adheridos a los
silenciadores de escape.
3. Transporte, distribución y transformación en el medio ambiente
Se dispone de datos muy escasos sobre el transporte y la
distribución en el medio ambiente de los PBDD y PBDF. Por lo general,
sus propiedades fisicoquímicas permiten pensar en analogías con los
PCDD y PCDF. Por consiguiente, si pasan al medio ambiente, pueden
estar de preferencia distribuidos en compartimentos ricos en carbono y
grasas, como sucede con los PCDD y PCDF.
El transporte por el aire de PBDD y PBDF se realiza en forma de
partículas y en fase de vapor, dependiendo la relación de partición
del grado de bromación.
No se dispone de datos experimentales sobre el movimiento de los
PBDD y PBDF en el agua o el suelo. En el caso de los PBDF (tri a
penta) se ha señalado la adsorción al sedimento. Debido a la baja
hidrosolubilidad de los PBDD y PBDF, la filtración por el suelo puede
estar limitada, pero aumentar en presencia de disolventes orgánicos o
ácidos húmicos.
No se dispone de datos experimentales sobre los procesos de
transporte y distribución de los PBDD y PBDF entre el medio ambiente y
los biota o dentro del los biota. Basándose en la existencia de
análogos coeficientes elevados de partición octanol/agua, calculados
para determinados PCDD/PCDF, PBDD/PBDF y PXDD/PXDF, se supone una
biodisponibilidad comparable a la de los PCDD y PCDF.
Se estudió la fotolisis de los PBDD/PBDF y PXDD/PXDF en
disolventes orgánicos y sobre superficies de cuarzo en el laboratorio,
así como en el suelo y en partículas de hollín (y polvo) al aire
libre. Se observaron las reacciones fotolíticas más lentas en estas
últimas condiciones, más pertinentes respecto al medio ambiente. Se
observó que la desbromación reductora era la principal vía metabólica.
La tasa de descomposición de los distintos congéneres depende de su
tipo de sustitución del bromo. Por lo general, los congéneres muy
bromados y los que poseen bromo en posiciones laterales tienen
semividas más breves. Las semividas calculadas eran del orden de
minutos (empleo de luz solar directa o luz ultravioleta [UV] y de
viales de cuarzo), horas (empleo de láminas sólidas o de partículas de
hollín o polvo y luz solar) o de centenares a miles de horas (empleo
del suelo y luz solar). Por ejemplo, las semividas inducidas por la
luz solar estimadas para la 2,3,7,8-TeBDD (TBDD) eran de 0,8 min (en
solución orgánica) o de 32 horas (en dispersión como láminas sólidas).
Se calculó una semivida de 3-6 meses para los isómeros tetraBDD en el
suelo superficial. En comparación con los PCDD y PCDF, los
correspondientes compuestos bromados presentaban menos estabilidad
fotoquímica. Los PXDD y PXDF pierden de preferencia sus átomos de
bromo durante la fotolisis, siendo transformados en PCDD y PCDF, que
tienen semividas fotolíticas más largas. Esa transformación de
PXDD/PXDF en PCDD/PCDF se produce también durante los procesos de
incineración.
Los PBDD y PBDF parecen ser escasamente degradables por la acción
de los microorganismos.
Como se ha observado en algunos estudios, la presencia de PBDD y
PBDF en animales y seres humanos indica su potencial de acumulación.
La 2,3,7,8-TeBDD se acumula en ratas durante la administración
subcrónica. No se dispone de los factores de bioacumulación,
bioconcentración o bioamplificación de los PBDD/PBDF o PXDD/ PXDF.
4. Niveles ambientales y exposición humana
Hasta la fecha, en contraste con los PCDD y PCDF, los PBDD y PBDF
no se han incluido con frecuencia en programas de vigilancia. Los
pocos estudios realizados muestran una aparición limitada.
En el aire ambiental, los PBDF se encuentran con más frecuencia
que las PBDD. Sólo se han detectado PBDD bromados inferiores (mono a
tetra) en concentraciones que iban de las indetectadas a las de 0,85
pg/m3 aproximadamente para las monobromodibenzo- p-dioxinas (monoBDD
o MoBDD) en un túnel de carretera y en un garaje subterráneo. Entre
los PBDF se hallaron homólogos mono a hexabromados, en concentraciones
que iban del nivel indetectado a 74 pg/m3. Por ejemplo, en Alemania
se midieron las concentraciones (valores medios) de los PBDD y PBDF
totales (tri a hexa) en un túnel de carretera, en el centro de una
ciudad y en una zona suburbana, obteniendo valores de 23 pg/m3, 2
pg/m3 y 0,59 pg/m3, respectiva mente; no se detectó la 2,3,7,8-TeBDD
y las concentraciones máximas de 2,3,7,8-TeBDF y 1,2,3,7,8-PeBDF
fueron de 0,28 pg/m3 y 0,08 pg/m3, respectivamente. Se hallaron PXDF
en muestras de aire en zonas de tráfico en concentraciones de hasta 41
pg/m3 (Cl1Br1DF). En las muestras de polvo tomadas al aire libre
(principalmente en carreteras) se observó también un predominio de
PBDF y PXDF (valores máximos de varios miles de ng/kg) respecto a las
PBDD y PXDD (valores máximos de hasta unos centenares de ng/kg).
Las muestras de aire tomado de locales equipados con distintos
dispositivos electrónicos en funcionamiento (televisores o monitores
de ordenador) mostraron la presencia de PBDF (tetra a hepta) en
concentraciones totales que iban de 0,23 a 1,27 pg/m3. No se
detectaron PBDD. Las muestras de polvo recogidas en un local de
ordenadores dieron concentraciones totales de PBDF de 2,4-5,5 µg/kg de
polvo. En contraste con el aire, la distribución homóloga en el polvo
está dominada por los hexaBDF y los heptabromodibenzofuranos (heptaBDF
o HpBDF). Sólo en las muestras de polvo se hallaron concentraciones
bajas de tetraBDD (hasta 1 µg/kg) y de tetra y pentaBDF sustituidos en
las posiciones 2,3,7 y 8 (hasta 0,07 µg/kg) detectables. Las
concentraciones de PBDF en una muestra de polvo doméstico eran
inferiores en un factor de 10. La concentración sumada de PBDD y PBDF
fue igual a la de PCDD y PCDF en el polvo tomado de locales de
ordenadores, pero inferior a la de PCDD y PCDF en el polvo doméstico.
El polvo tomado en un garaje subterráneo contenía PBDF (mono y di) y
PXDF (di a tetra) halogenados inferiores, con una concentración máxima
de 4,3 µg/kg en el caso de los dibenzofuranos dihalogenados mixtos
(DiXDF).
No se dispone de datos sobre las concentraciones de PBDD y PBDF
en las muestras de agua.
En las muestras de sedimentos de río y mar tomados en una zona
industrializada se detectaron tetraBDD (hasta 0,006 µg/kg de peso en
seco) y tetra a hexaBDF (en conjunto hasta 0,37 µg/kg de peso en
seco). El sedimento procedente de un drenaje de carretera contenía
PBDF (suma de mono a tri: 2,5 µg/kg; suma de tetra a hepta: 0,3 µg/kg)
y PXDF (suma de di y tri: 1,85 µg/kg), pero no PBDD.
Asimismo, las muestras de suelo tomadas cerca de una carretera
contenían monobromodibenzofuranos (monoBDF o MoBDF) y
dibromodibenzofuranos (DiBDF) (suma: 1,3 µg/kg) tetra y pentaBDF
(suma: 0,02 µg/kg) y PXDF (suma: 1 µg/kg ), pero no PBDD. Las muestras
de suelo tomadas de un terreno de incineración y cerca de una fábrica
de recuperación de metales dieron concentraciones totales de PBDF de
hasta 100 µg/kg, pero sin detectar PBDD. En una serie de muestras de
fango de alcantarillado procedentes de plantas municipales de
tratamiento de aguas residuales se hallaron concentraciones totales de
PBDF comprendidas entre niveles indetectados y 3 µg/kg. En un caso se
hallaron valores infinitesimales de tetraBDD y 2,3,7,8-TeBDF. Una
muestra de abono biológico estaba casi exenta de PBDD y PBDF
(tetraBDF: <0,003 µg/kg).
No se dispone de datos cuantitativos sobre las concentraciones de
PBDD y PBDF en los alimentos.
En muestras de hierba y de agujas de pino recogidas cerca de
carreteras se encontraron PBDF y PXDF halogenados inferiores (mono a
tetra) y valores infinitesimales de PBDD y PXDD (mono a tri).
No se han encontrado PBDD ni PBDF en las escasas muestras de
animales o plantas silvestres analizados.
En la leche de vaca recogida en granjas lecheras cerca de una
instalación incineradora de desechos municipales se identificaron de
modo provisional tribromodibenzofuranos (triBDF o TrBDF), un tetraBDF
y un pentaBDF (no tenían el tipo de sustitución en las posiciones
2,3,7 y 8).
No se han detectado PBDD ni PBDF en las escasas muestras
analizadas de tejidos adiposos humanos o de muestras de leche
procedentes de la población general.
Es posible la contaminación por PBDD y PBDF en distintos lugares
de trabajo en donde se procede a producir, elaborar, utilizar o
eliminar ciertos pirorretardantes o sus productos, en particular si se
emplean altas temperaturas. La magnitud de la exposición del
trabajador depende no sólo de los productos utilizados sino también de
la calidad del aire y de las condiciones de ventilación. Se dispone de
escasos datos de vigilancia del lugar de trabajo procedentes de
instalaciones de producción o elaboración de plásticos, de oficinas o
de estudios con un alto número de dispositivos eléctricos en
funcionamiento continuo y de instalaciones de reciclado (incluidas
plantas de reciclado de cobre). Por lo general, los PBDF eran más
abundantes que las PBDD y las concentraciones en el aire de PBDF eran
superiores en los lugares de producción de polímeros que contenían
DBDE. En numerosas muestras se detectaron PBDF y PBDD con
sustituciones en las posiciones 2,3,7 y 8. También se halló
contaminación por PBDD y PBDF en la zona de trabajo comprendida debajo
de la chimenea de humos de un laboratorio químico. Se carece de datos
de vigilancia procedentes de instalaciones de incineración de
desechos.
5. Cinética y metabolismo
La mayor parte de los estudios se refieren a la 2,3,7,8-TeBDD y,
en menor cuantía, al 1,2,7,8-TeBDF. Los cálculos de la semivida han
comprendido algunos congéneres adicionales.
La 2,3,7,8-TeBDD se absorbió en ratas después de la
administración oral, intratraqueal y cutánea, variando el porcentaje
de absorción conforme a la vía y la dosis. Las dosis únicas de 1 nmol
de 2,3,7,8-TeBDD/kg de peso corporal condujeron a la absorción del 80%
(vías oral e intratraqueal) o el 12% (vía cutánea) de la dosis
administrada. La absorción cutánea de 1 nmol de 1,2,7,8-TeBDF/kg de
peso corporal fue del 29% aproximadamente. La absorción oral de
2,3,7,8-TeBDD pareció ser comparable a la de la
2,3,7,8-tetraclorodibenzo- p-dioxina (2,3,7,8-TeCDD o TCDD). Sin
embargo, la absorción cutánea de 2,3,7,8-TeBDD fue la tercera parte
aproximadamente de la dosis equimolar de 2,3,7,8-TeCDD.
La 2,3,7,8-TeBDD o el 1,2,7,8-TeBDF administrados a ratas, por
cualquier vía, se distribuyeron por todo el organismo, hallándose los
principales depósitos en los tejidos hepático y adiposo, seguidos de
la piel y el tejido muscular. Por ejemplo, 3 días después de la
administración de dosis orales únicas de 2,3,7,8-TeBDD (1 nmol/kg de
peso corporal), las porciones halladas en esos tejidos eran del 20%,
el 20%, el 11% y el 4% respectivamente, mientras que el timo y las
glándulas suprarrenales contenían el 0,03% y el 0,4%, respectivamente,
de la dosis administrada. La partición de la 2,3,7,8-TeBDD entre el
hígado y el tejido adiposo de ratas estaba influida por la dosis, la
vía de exposición y el tiempo transcurrido después de la
administración. Las relaciones entre las concentraciones del hígado y
el tejido adiposo medidas en distintas condiciones variaban entre 0,2
y 6,5 (gama para dosis únicas de 2,3,7,8-TeBDD en ratas). No se
dispuso de datos experimentales sobre la transferencia de PBDD y PBDF
a las crías.
Se hallaron metabolitos de tetraBDD/BDF en la bilis y las heces
de ratas. Se formaron principalmente por hidroxilación aromática y
debromación hidrolítica. La tasa de metabolismo (determinada
indirectamente como tasa de excreción biliar) difería entre la
2,3,7,8-TeBDD (el 7% aproximadamente) y el 1,2,7,8-TeBDF (el 50%
aproximadamente). Tres días después de la administración intravenosa
de una dosis de 2,3,7,8-TeBDD (1 nmol/kg de peso corporal), el 14% de
la dosis administrada se halló en forma de metabolitos en las heces de
ratas.
Se estudiaron en ratas la eliminación y excreción de la
2,3,7,8-TeBDD utilizando las vías de administración oral, intravenosa,
intratraqueal y cutánea. En todos los estudios, la principal vía de
eliminación fue las heces, variando la radiactividad eliminada entre
el 2% (vía cutánea) y el 42% (vía oral) de la dosis administrada (1
nmol de [3H]2,3,7,8-TeBDD/kg de peso corporal) en muestras de heces,
y entre el 0,2 y el 1% en muestras de orina. Asimismo, en estudios del
1,2,7,8-TeBDF en ratas, la excreción se produjo principalmente por las
heces y sólo se eliminó por la orina el 2-3% de las dosis intravenosa,
oral o cutánea. En los primeros días que siguieron a la administración
de las dosis orales, el material no absorbido y la excreción biliar
parecieron ser las principales fuentes de sustancia eliminada por las
heces. Las porciones de 2,3,7,8-TeBDD original hallado en heces de
ratas después de la administración de 1 nmol de 2,3,7,8-TeBDD/kg de
peso corporal fueron del 53% (vía oral), el 43% (vía intratraqueal) y
el 10-20% (vía intravenosa). Pocos días después de la administración
oral de 2,3,7,8-TeBDD (1 nmol/kg de peso corporal), el 20%
aproximadamente de la dosis administrada se eliminó como sustancia
original.
Se dispone de datos sobre la retención y el ciclo biológico en el
caso de algunos PBDD y PBDF. En las ratas, la carga corporal relativa
de 2,3,7,8-TeBDD (y otros congéneres) depende de la vía y de la dosis
administrada, mostrando diferencias en la absorción. Se calcularon las
semividas de varios PBDD/PXDD y PBDF en distintos tejidos y en heces
de ratas. Variaron entre un día (1,2,7,8-TeBDF en el organismo en
conjunto) y 99 días (2,3,4,7,8-PeBDF en el hígado). Las semividas
calculadas de 17,18 y 58 días para la 2,3,7,8-TeBDD en el hígado, las
heces y el tejido adiposo, respectivamente, fueron análogas a las
señaladas para la 2,3,7,8-TeCDD en el hígado y las heces, pero
superiores (en un factor de >2) a las registradas para la
2,3,7,8-TeCDD en el tejido adiposo. Pese a las diferencias en la
retención inicial, las semividas de la 2,3,7,8-TeBDF y el
2,3,7,8-tetraclorodibenzofurano (2,3,7,8-TeCDF o TCDF) en el hígado
fueron comparables.
En lo que respecta a los PCDD y PCDF, las semividas calculadas en
personas son mucho más largas que las correspondientes a ratas. Se
dispone de estimaciones de 3-11 años (promedio: 5,9 años) para la
2,3,7,8-TeBDD y de 1-2 años (promedio: 1,5 años) para el
2,3,7,8-TeBDF. También se observó la persistencia de esas sustancias
en el caso de un químico que sintetizó 2,3,7,8-TeBDD y 2,3,7,8-TeCDD
en 1956. A los 35 años de la exposición se hallaron en su sangre
concentraciones muy elevadas de 2,3,7,8-TeBDD.
6. Efectos en mamíferos de laboratorio y en sistemas de pruebas
in vitro
La mayor parte de los estudios se refieren a la toxicidad de la
2,3,7,8-TeBDD, pero también se dispone de alguna información sobre
otros PBDD/PBDF y PXDD/PXDF.
La 2,3,7,8-TeBDD produjo efectos típicos análogos a los de la
2,3,7,8-TeCDD, incluidos el síndrome de consunción, la atrofia tímica
y la toxicidad hepática. Se observaron además lesiones hepáticas
descritas como púrpura hepática, que no se habían registrado después
de la exposición de ratas a la 2,3,7,8-TeCDD. El tipo de lesiones
(mortalidad, histopatología, pesos del hígado y el timo) hallado en
cobayos después de una sola exposición y en ratas después de la
exposición a corto plazo al 2,3,7,8-TeBDF fue análogo al observado en
el caso del 2,3,7,8-TeCDF.
La 2,3,7,8-TeBDD mantiene una interacción con el sistema
endocrino. En ratas se han observado alteraciones relacionadas con la
dosis en las hormonas tiroideas circulantes y alteración de la
actividad espermatogénica.
La DL50 oral (periodo de observación de 28 días) de la
2,3,7,8- TeBDD en ratas Wistar fue de 100 µg/kg de peso corporal
aproximadamente en las hembras y de 300 µg/kg de peso corporal
aproximadamente en los machos. Los valores de la DL50 oral para la
2,3,7,8- TeCDD obtenidos en otros estudios variaron entre 22 y >3000
µg/kg de peso corporal. Las dosis equimolares de 2,3,7,8-TeBDF y de
2,3,7,8-TeCDF dieron tasas de mortalidad comparables en cobayos. Por
ejemplo, se observó una mortalidad del 100% después de la
administración de 2,3,7,8-TeBDF (0,03 µmol/kg de peso corporal, 15,8
µg/kg de peso corporal) y de 2,3,7,8-TeCDF (0,03 µmol/kg de peso
corporal, 10 µg/kg de peso corporal). Se observaron en ratas lesiones
prepurpúreas y modificaciones de las hormonas tiroideas después de la
administración de una sola dosis de 100 µg/kg de 2,3,7,8-TeBDD/kg de
peso corporal.
En ratas Wistar a las que se administró 2,3,7,8-TeBDD por vía
oral durante 13 semanas se observaron signos de disminución de la
espermatogénesis, presencia de espermatocitos defectuosos y
necróticos, signos de púrpura hepática grave y modificaciones de las
hormonas tiroideas circulantes y de los pesos de los órganos. El nivel
de efectos adversos no observados fue de 0,01 µg/kg de peso corporal
por día.
La administración oral de 2,3,7,8-TeBDF a ratas Sprague-Dawley
durante 4 semanas provocó retraso del crecimiento dependiente de la
dosis y lesiones histopatológicas en el hígado y el timo. El nivel de
efecto adverso no observado fue de 1 µg/kg de peso corporal por día.
En ratones se observó la aparición de toxicidad en el desarrollo
en el caso de algunos PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 administrando dosis subcutáneas y orales que no provocaron
toxicidad materna ni mortalidad fetal. Los niveles de efectos mínimos
observados (en µg/kg de peso corporal) para la hidronefrosis y el
paladar hendido, después de una sola dosis oral, en ratonas gestantes
fueron, respectivamente, los siguientes: 3 y 48 para la 2,3,7,8-TeBDD,
25 y 200 para el 2,3,7,8-TeBDF, 400 y 2400 para el 2,3,4,7,8-PeBDF, y
500 y 3000-4000 para el 1,2,3,7,8-PeBDF. En comparación con la base
molar, la 2,3,7,8-TeBDD y la 2,3,7,8-TeCDD presentaron casi la misma
actividad en la inducción de la hidronefrosis. Al efectuar la
comparación con el peso, los isómeros bromados fueron en general menos
potentes que los clorados en la inducción de la hidronefrosis y el
paladar hendido. Sin embargo, el 2,3,7,8-TeBDF fue más activo que el
2,3,7,8-TeCDF.
No se halló información sobre la mutagenicidad de los PBDD y PBDF
o puntos finales conexos.
No se dispuso de estudios sobre la toxicidad y la
carcinogenicidad a largo plazo con PBDD y PBDF. La 2,3,7,8-TeBDD
resultó positiva en una prueba de transformación celular utilizando
macrófagos peritoneales murinos. Sin embargo, la actividad
transformadora de la 2,3,7,8-TeBDD fue siete veces menor que la de la
2,3,7,8-TeCDD. Más tarde aparecieron tumores en ratones lampiños tras
la inyección subcutánea de las estirpes celulares establecidas
resultantes.
La administración intraperitoneal de una serie de varias PBDD y
PXDD (tetra y penta) a ratas Wistar inmaduras de sexo masculino
produjo pérdidas de peso 14 días después de la inyección. Basándose en
los valores de DE50 molar, las sustancias más tóxicas ensayadas
fueron las 2,3,7,8-TeBDD, 2-Br1-3,7,8-Cl3-DD y 2,3-Br2-7,8-Cl2-DD
(TBCDD), con sustituciones sólo en las cuatro posiciones laterales.
Las actividades relativas de las demás PBDD examinadas siguieron el
siguiente orden: 2,3,7,8- > 1,2,3,7,8- >1,2,4,7,8- > 1,3,7,8-DD. En
otros experimentos sólo se observaron ligeras diferencias en los
valores de la DE50 (sobre una base molar) para la pérdida de peso
total, la atrofia tímica y la inducción de las enzimas hepáticas entre
la 2,3,7,8-TeCDD y la 2,3,7,8-TeBDD.
Se observaron atrofia tímica y otros signos de immunotoxicidad
(por ej., parámetros hematológicos y alteraciones de ciertas
subpoblaciones de linfocitos) con la administración de varias
PBDD/PXDD y de 2,3,7,8-TeBDF en la rata y con las 2,3,7,8-TeBDD y
TBCDD en el mono tití (Callithrix jacchus). Se llegó a la conclusión
de que, sobre una base molar, la actividad de la 2,3,7,8-TeBDD es
comparable a la de la 2,3,7,8-TeCDD en ratas y monos. Por ejemplo, se
observó un efecto notable en cierta subpoblaciones de linfocitos en
monos después de una sola dosis subcutánea de 30 ng de
2,3,7,8-TeBDD/kg de peso corporal en relación con 10 ng de
2,3,7,8-TeCDD/kg de peso corporal. No se han investigado los efectos
sobre la immunotoxicidad después de la exposición perinatal a los PBDD
y PBDF.
Tras la administración subcrónica de 2,3,7,8-TeBDD o
2,3,7,8-TeCDD por cebado oral de ratones se produjo un aumento
dependiente de la dosis en las profirinas hepáticas totales.
Dosis orales únicas de 2,3,7,8-TeBDD y 2,3,7,8-TeCDD produjeron
reducciones en la concentración y la cantidad total de vitamina A en
el hígado de ratas, siendo la 2,3,7,8-TeBDD ligeramente menos potente
que la 2,3,7,8-TeCDD (sobre una base molar).
La 2,3,7,8-TeBDD y el 2,3,7,8-TeBDF produjeron hiperqueratosis en
la oreja del conejo en una dosis de 100 µg/conejo, pero no con 10
µg/conejo. El nivel de efecto no observado para la 2,3,7,8-TeCDD fue
de 0,01 µg/conejo.
Se observó que varios congéneres halogenados tetra (Br1Cl3DD,
Br2Cl2DD) y penta (Br1Cl4DD) con sustitución en las posiciones
2,3,7 y 8 presentaban una actividad antiestrogénica análoga a la de la
de 2,3,7,8-TeCDD, examinada en cultivos de células de cáncer mamario
humano.
En ratas, la 2,3,7-tribromodibenzo- p-dioxina (2,3,7-triBDD/
TrBDD) reducía la desaparición de la uabaína del plasma, su
eliminación por la bilis y el flujo biliar en una amplitud ligeramente
inferior a la observada con la 2,3,7,8-TeCDD.
Los PBDD/PBDF y PXDD/PXDF son potentes inductores de ciertas
enzimas microsómicas dependientes del citocromo P-450. Se calcularon
valores de DE50 de 0,8-1 nmol/kg de peso corporal para la inducción
del citocromo P-1A1 y de 0,2 nmol/kg de peso corporal aproximadamente
para la inducción del citocromo P-1A2 en el hígado de rata tras la
administración oral de dosis únicas de 2,3,7,8-TeBDD. Se observó la
inducción del citocromo P-1A1 (inducción de la hidroxilasa de
arilhidrocarbono y/o la etoxirresorrufina- O-desetilasa) en distintas
especies y tejidos in vivo y en cultivo celular de rata in vitro.
Se observó que distintos congéneres eran activos, así como los
pirolizados de ciertos pirorretardantes. Por lo general, la inducción
enzimática dependía de la dosis en concentraciones no tóxicas,
comenzaba después de la exposición y era duradera. Resultó mensurable
en exposiciones tan bajas como las situadas en la gama de pmol. La
actividad inductora varió en varios órdenes de magnitud para distintos
congéneres, en función de su estructura química. Los inductores más
potentes fueron las TCDD, TBDD y TBCDD. En comparación (sobre una base
molar) con sus análogos clorados, las PBDD y PXDD tenían más o menos
igual actividad. En contraste con la TCDD, cuya actividad inductora
relativa era independiente del tejido examinado, la TBDD era cinco
veces más activa en la inducción de la
etoxirresorrufina- O-desetilasa en el hígado que en la piel y el
pulmón después de la exposición subcrónica de ratones. La clasificación
de la inducción de la actividad de la etoxirresorrufina- O-desetilasa
en monos titís fue de TCDD > 2,3,4,7,8- pentaclorodibenzofurano >
2,3,4,7,8-pentaCDF/PeCDF > 2,3,4,7,8-PeBDF cuando se compararon las
actividades enzimáticas con las concentraciones hepáticas. En las
pruebas in vitro con cultivos de células de rata se obtuvieron
valores de la CE50 molar análogos para las actividades de inducción
de la hidroxilasa del arilhidrocarbono y de la
etoxirresorrufina- O-desetilasa entre los PXDF y PCDF correspondientes.
Se estima que los PBDD y PBDF comparten un mecanismo común de
acción con los PCDD y PCDF y otros hidrocarburos aromáticos
halogenados. Se confirmó el enlace con el receptor de hidrocarburos
aromáticos citosólico, que desempeña una función central en la
mediación de la toxicidad afin a la de la 2,3,7,8-TeCDD, en el caso de
varios PBDD y PXDD/PXDF. Sus afinidades de enlace con los receptores
variaron en varios órdenes de magnitud, pero fueron comparables a las
de sus análogos clorados.
7. Efectos en el ser humano
No se dispone de datos sobre la exposición de seres humanos a los
PBDD y PBDF o sobre sus efectos en la salud de la población general.
Se han registrado dos casos de problemas de salud agudos debidos
a la exposición a 2,3,7,8-TeBDD/TeCDD, con síntomas que comprendían el
cloroacné.
En otro estudio, el personal masculino de una fábrica de
productos químicos con exposición documentada a los PBDD y PBDF
procedentes del uso de pirorretardantes bromados (OBDE y DBDE) fue
sometido a pruebas de laboratorio inmunológicas y clínicas
adicionales. Aunque se observaron indicios de modificaciones menores
de los parámetros inmunológicos, la evaluación global de su estado de
salud no mostró un efecto de la carga corporal de 2,3,7,8-TeBDD/ TeBDF
sobre el sistema inmunitario.
No existen informes sobre la mortalidad cancerosa producida por
los PBDD y PBDF.
8. Efectos en otros organismos en el laboratorio y en el medio
ambiente
Sólo se dispone de información limitada sobre los efectos de los
PBDD y PBDF en microorganismos, plantas, invertebrados o especies
silvestres vertebradas.
En una biovaloración de la mortalidad precoz de pececillos de
trucha irisada (Oncorhynchus mykiss), se ensayó una serie de
congéneres de PBDD y PBDF, que resultaron activos. Esta biovaloración
demostró también que tanto las PBDD como los PBDF tienen menor
actividad al aumentar la sustitución por bromo. Tanto la 2,3,7,8-TeBDD
como el 2,3,7,8-TeBDF eran más activos que sus análogos clorados.
6.4.1 Dibenzo-p-dioxins
Elimination was studied in rats with TBDD and with
1,2,3,7,8-PeBDD (faecal excretion was monitored in rats only with
TBDD). The animals were exposed to single doses of TBDD or
[1,6-3H]TBDD (see Table 41) by the oral (Diliberto et al., 1990a,b,
1993; Ivens et al., 1992), intravenous (Kedderis et al., 1991a),
intratracheal (Diliberto et al., 1991, 1993), or dermal (Jackson et
al., 1991; Diliberto et al., 1993) route or to subcutaneous doses of
1,2,3,7,8-PeBDD (Golor et al., 1993).
In all studies, the major route of elimination was through the
faeces (see Table 41). Eliminated radioactivity (after 2 - 3 days) in
faeces ranged from 2 to 42% of the administered dose of 1 nmol
[3H]TBDD/kg body weight and from 0.2 to 1% in urine. Unabsorbed
material and biliary excretion appeared to be the major source of
eliminated compound in faeces.
Based on the intravenous and oral studies (Table 41), the
cumulative elimination of radioactivity in faeces was dose-dependent.
Higher doses tended to result in higher elimination rates. The
dose-related differences following oral administration were a
consequence of differences in amounts eliminated on days 1 and 2 in
each group and are likely due to differences in percent absorption
(maybe due to the competing processes of uptake versus transit or to
limited aqueous solubility of TBDD at high doses). In the 56-day
intravenous study, a disproportionately greater elimination of
radioactivity at the high (100 nmol/kg body weight) versus the low (1
nmol/kg body weight) dose was observed beginning 3 weeks after
treatment (Kedderis et al., 1991a, 1992a).
Faecal elimination curves of the intravenous study were analysed
and found to be tri-exponential for the low dose (1 nmol/kg body
weight) and bi-exponential for the high dose (100 nmol/kg body
weight), with estimated initial and terminal half-lives of <1 and 18
days, respectively (see Table 45 in section 6.5.1).
From the oral study (Table 41), it was shown that 0.3% or less of
the administered dose was eliminated in the urine for all dose groups.
However, the relative amounts of urinary elimination were found to be
higher at the two low doses, consistent with enhanced absorption at
these dose levels (Diliberto et al., 1993).
Elimination of radioactivity in faeces over the first 3 days was
comparatively high after oral and intratracheal administration but was
lower after intravenous and dermal exposure to 1 nmol [3H]TBDD/kg body
weight (see Table 41). The faecal excretion of only 2% of the
administered dermal dose implies a very low elimination following
dermal exposure. However, based on the percentage of the dermally
absorbed dose, a value of 17% is obtained, which is comparable to the
faecal elimination of an equimolar intravenous dose (Kedderis et al.,
1991a; Diliberto et al., 1993).
Table 41. Elimination of 2,3,7,8-TeBDD in rats after single radiolabelled and unlabelled dosesa
Strain Route Dose Observation Test Eliminationb Reference
(sex) (vehicle) period (days) (% of administered dose)
nmol/kg µg/kg Faeces Urine
body weight body weight
Fischer 344 intravenous 1 0.5 1 R 8-10 n.sp. Kedderis et al.
(male) (water: 1 0.5 56 50 4.5 (1991a)
(n = 3-4) ethanol: 100 50 56 70 7.6
Emulphor(R)
= 3: 1: 1)
Fischer 344 oral 1 0.5 3 R 42 ± 2 0.3 Diliberto et al.
(male) (water: 10 5 3 39 ± 1 0.3 (1993)
(n = 3-4) ethanol: 100 50 3 58 ± 5 0.2
Emulphor(R) 500 250 3 72 ± 5 0.2
= 3: 1: 1)
Fischer 344 intratracheal 1 0.5 3 R 41 ± 2 1 Diliberto et al.
(male) (water: (1993)
(n = 3-4) ethanol:
Emulphor(R)
= 3 : 1 : 1)
Fischer 344 dermal 1 0.5 3 R 2 0.2 Diliberto et al.
(male) (acetone) (1993)
(n: 3-4)
Wistar ora] 200 100 2 U 20 (male) n.sp. Ivens et al.
(female, male) (arachis oil 17 (female) (1992)
(n: 5) with 5% 3-7 1 n.sp.
toluene)
a R = administration of [1,6-3H]-2,3,7,8-TeBDD (purity = >98%); elimination refers to eliminated radioactivity. U = administration of
unlabelled 2,3,7,8-TeBDD (purity = 98%); elimination refers to recovery of TBDD.
b n.sp. = not specified.
According to Kedderis et al. (1992b), the large percentage of the
dose excreted in urine and faeces after a single intravenous dose of
[3H]TBDD within the first few days could be attributed to a rapidly
excreted impurity in the radiolabelled TBDD, which is not detectable
by conventional radio-HPLC techniques. On the other hand, HPLC in
combination with preceding hexane extraction of faeces was reported to
distinguish successfully between parent TBDD and TBDD metabolites in
analysing TBDD-derived radioactivity in faeces (Kedderis et al.,
1991a; Diliberto et al., 1993). Results are compiled in Table 42.
Three days after oral, intratracheal, and intravenous administration
to rats of 1 nmol [3H]TBDD/kg body weight, approximately 22, 18, and
3%, respectively, of the administered dose could be attributed to the
parent compound extracted from faeces. In relation to the total
radioactivity measured in faeces over 3 days, the content of parent
TBDD ranged from 10 to 67% (see Table 43) after intravenous,
intratracheal, and oral exposure (Kedderis et al., 1991a; Diliberto et
al., 1993). Possible metabolites account for approximately 18 - 24%
(oral study) and 14% (intravenous study) of the administered dose
(Diliberto et al., 1993). Most of the parent TBDD was found in faeces
collected at days 1 and 2 (see Table 42 and Ivens et al., 1992 in
Table 41), which is consistent (at least for the oral and
intratracheal studies) with the assumption that a portion of the
material is not absorbed during passage through the gastrointestinal
tract. Excretion of absorbed TBDD is thought to be limited by the rate
of metabolism (Kedderis et al., 1991a).
6.4.2 Dibenzofurans
Elimination of 1,2,7,8-TeBDF in rats was primarily via biliary
excretion of metabolite(s) (see section 6.3) in the faeces (Kedderis
et al., 1994). Following intravenous administration of 1 nmol
[3H]-1,2,7,8-TeBDF/kg body weight, 39% of the dose was found in faeces
and 55% in intestinal contents after 24 h. After administration of
identical oral and dermal doses, 58 and 23%, respectively, of the
doses were excreted into the faeces within 72 h. Excretion in the
urine was only 2 - 3% of the intravenous, oral, or dermal dose
(Kedderis et al., 1994).
6.5 Retention and turnover
Data on retention and turnover are available for some PBDDs (tri
to penta), a tetraXDD, and some PBDFs (tetra, penta) in rats and for
2,3,7,8-TeBDD in humans.
6.5.1 Animal studies
Apparent elimination half-lives are complex for several
PHDDs/PHDFs, for various reasons:
* As is well known for TCDD (Abraham et al., 1988), elimination
from the main organs (liver and adipose tissue) does not proceed
in the expected semi-logarithmic form, but is at least biphasic.
Table 42. Percent administered dose of parent [3H]TBDD recovered in faeces of ratsa,b
Route Dose % administered dose excreted in faeces
(nmol/kg characterized as parent [3H]TBDDC
body Day 1 Day 2 Day 3 Cumulative
weight) after dosing after dosing after dosing (days 1-3)
Oral 1 11.7 ± 3.6 7.9 ± 2.1 2.5 ± 1.6 22.2 ± 2.1
10 6.9 ± 4.9 12.5 ± 3.8 2.0 ± 1.2 21.4 ± 1.8
100 16.1 ± 9.6 16.7 ± 9.0 2.6 ± 1.6 35.4 ± 1.8d
500 26.4 ± 11.2 18.3 ± 9.7 3.6 ± 3.4 48.3 ± 3.0d
Intratracheal 1 12.4 ± 1.7 4.6 ± 0.7 0.6 ± 0.02 17.6d
Intravenouse 1 1.6 ± 0.3 0.7 ± 0.3 0.5 ± 0.3 2.8d
a Adapted from Diliberto et al. (1993).
b Fischer 344 rats.
c Mean ± SD; n = 3 or 4; faecal extraction with hexane followed by HPLC
characterization of the extract.
d Statistically different from 1 nmol/kg oral close group (p < 0.05).
e Kedderis et al. (1991a).
Table 43. Contents of parent [3H]TBDD in faeces of ratsa
Route Dose % total radioactivity in faeces Reference
(nmol/kg characterized as parent
body [3H]TBDD (cumulative
weight) percentages days 1-3)
Oral 1 53b Diliberto et al.
10 55 (1993)
100 60
500 67
Intratracheal 1 43
Intravenous 1 and 100 10-20 Kedderis et al.
(1991a)
a Group size: n = 3-4.
b Percentage represents the amount of parent TBDD that was excreted via
faeces (days 1-3) as a result of unabsorbed TBDD and/or gastrointestinal
transluminal excretion of TBDD.
* The elimination half-lives from liver and adipose tissue (and
also from some tissues like the thymus) are often not identical.
This is due to metabolic conversion in and excretion from the
liver and a more or less rapid equilibrium in the adipose tissue
(probably dependent on lipid solubility).
* Tissue distribution is dose-dependent. The liver : adipose
tissue concentration ratio may change by orders of magnitude
when the dose is greatly increased (or decreases during
elimination).
* The role of elimination may be different in various rodent
strains, and it is dramatically different between various
species (e.g. orders of magnitude difference for TCDD between
rodents and humans). For this reason, the rate of cumulation to
a steady state (for a given dose and dose interval) differs by a
factor of about 100 for the rat and humans in the case of TCDD.
Although no comparative data (rat versus human) exist for the
elimination half-lives for PBDDs/PBDFs, similar differences must
be expected.
The relative body burden of radiolabelled TBDD in rats depends on
the route of exposure and on the dose administered (see Table 44),
reflecting differences in absorption. However, if per cent body
burdens are adjusted for the percent absorbed dose, they become more
similar after oral, intratracheal, dermal, and intravenous
administration of 1 nmol [3H]TBDD/kg body weight (see Table 44). More
than half of the body burden was found in the liver and adipose tissue
of rats (Diliberto et al., 1993; see also section 6.2).
Distribution patterns of TBDD between the major tissue depots
changed with dose and time (see also section 6.2), as seen in
single-dose studies (Kedderis et al., 1991a, 1993; Diliberto et al.
1993) and in short-term studies (Ivens et al., 1990). For example,
liver : adipose tissue TBDD concentration ratios declined from a
maximum of 30 to 0.2 during a period of 56 days after a single
intravenous administration of 1 nmol TBDD/kg body weight (Kedderis et
al., 1991). At this dose, radioactivity levels in the liver peaked by
7 h and then gradually declined, concomitantly with a slow
accumulation in adipose tissue, which reached the maximum
concentration by 14 days (Kedderis et al., 1990). Levels of
TBDD-derived radioactivity in blood declined rapidly to <2% of the
administered dose by day 1 after dosing (Kedderis et al., 1991a).
Liver concentrations of TBDD in rats, 1 - 78 days following a single
subcutaneous injection of 600 ng (1.2 nmol)/kg body weight, were
highest on day 3 after administration (Nagao et al., 1990c). In a
91-day oral study, the concentration of TBDD increased in the liver
and adipose tissue of rats during treatment. In the subsequent
recovery phase, a biphasic decline in TBDD concentrations in the liver
was observed, whereas the concentration of TBDD in adipose tissue
remained fairly constant, until a decrease began after a 30-day
recovery period (Ivens et al., 1990).
Table 44. Body burden of [3H]TBDD-derived radioactivity in ratsa 3 days after administration of a single dose
Route Dose % body burden Reference
(vehicle) (nmol/kg body weight)
Administered dose Absorbed dose
Oral 1 58 73 Diliberto et al. (1993)
(water: ethanol: 10 61 75
Emulphor(R) = 3: 1 : 1) 100 41 67
500 28 59
Intratracheal 1 59 ± 2 76 ± 2 Diliberto et al. (1993)
(water: ethanol:
Emulphor(R) = 3: 1: 1 )
Dermal 1 10 ± 1 82 ± 18 Diliberto et al. (1993)
(acetone)
Intravenous 1 82 ± 2 - Diliberto et al. (1993);
(water: ethanol:
Emulphor(R) = 3: 1: 1) Kedderis et al. (1991a)
a Fischer 344 rats, n = 3-4,
Estimated half-lives for TBDD and other PBDD/PBDF congeners are
compiled in Table 45.
Half-lives calculated for TBDD were as high as 58 days in adipose
tissue and skin. Shorter half-lives (up to 27 days) were found in
blood, muscle, liver, and whole body (Table 45). Compared with TCDD,
half-lives of TBDD were similar for liver and whole body but higher
(39 - 58 days versus 17 - 25 days) for adipose tissue (Kedderis et
al., 1991a and references therein; Nagao et al., 1995/96 and
references therein).
To compare the kinetics of three pairs of corresponding
poly-chlorinated and polybrominated PHDDs/PHDFs, a mixture of the six
substances (2,3,7,8-tetrahalogenated dibenzofuran [THDF],
2,3,4,7,8-pentahalogenated dibenzofuran [2,3,4,7,8-PeHDF], and
1,2,3,7,8-PeHDD) was given subcutaneously (single dose) to Wistar rats
(1 - 2 nmol/kg body weight each). Concentration changes in liver and
adipose tissue were monitored over a period of 95 days (Golor et al.,
1993). Kinetics of both the chlorinated and brominated 2,3,4,7,8 PeHDF
were similar in liver and also in adipose tissue, but levels were more
than 10 times higher in the liver. The rate of decline was also very
similar for the chlorinated and brominated 1,2,3,7,8-PeHDD in liver
and also in adipose tissue, but the profile of the kinetics was
different in the two tissues. Besides the level being more than one
order of magnitude higher in the liver, there was a rather steady
decline in the concentration in the hepatic tissue, while the
concentration in adipose tissue increased within the first month
(possibly because of redistribution phenomena) and then slowly
declined thereafter.
The most remarkable difference between the chlorinated and
brominated congeners was found in the case of THDF. The chlorinated
congener (TCDF) is known to be rapidly eliminated from liver as well
as adipose tissue in the rat. This was also found in these studies,
the rate of decline from the liver clearly being biphasic. In
contrast, the brominated congener (TBDF) was much more slowly
eliminated from both the liver and adipose tissue. While in the liver,
the overall elimination rate resembled that of the chlorinated
congener at the second elimination phase (about 2 weeks after
administration); the elimination rate in the adipose tissue was
comparatively slow (after an initial increase in the concentration
during the first 2 weeks after administration). Thus, the THDF
exhibited the larger kinetic difference between the chlorinated and
the brominated forms, the TBDF being much more persistent in the rat.
No comparative data are available for this pair of congeners in other
species.
6.5.2 Human studies
Some data on retention and turnover in humans are available for
2,3,7,8-TeBDD and 2,3,7,8-TeBDF.
Table 45. Biological half-lives of several PBDD/PBDF congeners in rats after single doses
Strain Congenera Route Dose Elimination from Calculated half-life Reference
(sex) (solvent) (observation period) (days) (kinetic phase)
Dibenzo-p-dioxins
Fischer 344 [3H]TBDD intravenous 1 nmol/kg whole body 0.7 (1st phase) Kedderis et al.
(female) (water: ethanol: (56 days) body weight 2.9 (2nd phase) (1991a)
(n = 3-4) Emulphor(R) 17.8 (3rd phase)
= 3: 1 : 1)
100 nmol/kg whole body 0.6 (1st phase)
body weight 17.8 (2nd phase)
1 nmol/kg liver 4.5 (1st phase)
body weight 16.5 (2nd phase)
adipose tissue 57.8
skin 2.5 (1st phase)
57.8 (2nd phase)
muscle 1.6 (1st phase)
26.7 (2nd phase)
blood 18.2
Wistar TBDD subcutaneous 60 ng/kg liver 13.3 Nagao et al.
(female) (toluene/DMSO (78 days) body weight (12.0-14.9)b (1995/96)
(n = 3-10) = 1+2; v/v) (1.2 nmol/kg adipose tissue 39.4
body weight) (26-82)b
Wistar 1,2,3,7, 8-PeBDD subcutaneous 2.2 nmol/kg liver 21 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (17-27)b (1993)
(n = n.sp. ) = 14-2; v/v) adipose tissue 55
(39-97)b
Table 45. (Continued)
Strain Congenera Route Dose Elimination from Calculated half-life Reference
(sex) (solvent) (observation period) (days) (kinetic phase)
Wistar 2,3,7-TrBDD intravenous 50 µg/kg liver 2 (3rd phase) Golor et al.
(female) (<5% toluene in 14 days body weightc (47 h) (1995)
(n = 3) peanut oil/0.9% (119 nmol/kg adipose tissue 2-3 (3rd phase)
NaCl, 1+9, v/v) body weight) (43 h)
thymus 3-4 (3rd phase)
(91 h)
2,3-Cl2,7-Br1DD intravenous 50 µg/kg liver 3-4 (3rd phase)
(<5% toluene in 14 days body weightc (72 h)
peanut oil/0.9% (151 nmol/kg adipose tissue 1.5 (3rd phase)
NaCl, 1+9, v/v) body weight) (36 h)
thymus 3-4 (3rd phase)
(92 h)
Dibenzofurans
Wistar TBDF subcutaneous 1.7 nmol/kg liver 20 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (17-25)b (1993)
(n = n.sp.) = 1+2; v/v) adipose tissue 30
(26-36)b
Wistar 2,3,4,7,8-PeBDF subcutaneous 1.1 nmol/kg liver 99 Golor et al.
(female) (toluene/DMSO (35-95 days) body weightc (59-302)b (1993)
(n = n.sp.) = 1+2; v/v) adipose tissue 80
(49-220)b
Fischer 344 [3H]1,2,7,8-TeBDF intravenous 1 nmol/kg body 1 Kedderis et al.
(male) (water: ethanol: (24 h) body weight (1994)
(n = 3-4) Emulphor(R)
= 3: 1 : 1)
a n.sp. = not specified.
b 95% confidence interval in days.
c Given in a mixture together with other brominated and chlorinated PHDD/PHDF congeners.
The first report of a PBDD in human tissue is indicative of the
very long persistence of these compounds. Thirty-five years after
exposure, markedly elevated levels of TBDD (625 pg TBDD/g blood lipid)
were found in the blood of a chemist (see also section 5.3) who had
synthesized TBDD and TCDD in 1956 (Schecter & Ryan, 1990, 1991, 1992;
Schecter, 1992). It was not possible to calculate the actual half-life
because of the lack of earlier measurements.
Another study (Zober et al., 1992) provided data for estimating
the apparent half-lives of TBDD and TBDF. Employees of a chemical
plant who had PBDD/PBDF body burdens resulting from processing
brominated flame retardants (OBDE and DBDE) were monitored over a
3-year period from 1989 to 1991 (see also section 5.3). Based on data
from three subjects, the following half-lives were calculated:
2,3,7,8-TeBDD: 2.9 - 10.8 years (mean: 5.9 years)
2,3,7,8-TeBDF: 1.1 - 1.9 years (mean: 1.5 years)
These half-lives are much longer than those reported in rats
(section 6.5.1), but they are consistent with findings on the
chlorinated analogues. Estimated half-lives of TCDD in humans ranged
between 5 and 11 years (Poiger & Schlatter, 1986; Pirkle et al., 1989;
Wolfe et al., 1994).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1 Single exposure
7.1.1 Dibenzo-p-dioxins
The toxicity of PBDDs and PXDDs was studied in rats and mice
after single oral (Yang et al., 1983; Ivens-Kohl et al., 1990; Ivens
et al., 1992), intraperitoneal (Mason et al., 1987a,b), or
subcutaneous (Nagao et al., 1990a) doses.
Single oral doses of 10, 33, 100, or 300 µg 2,3,7,8-TeBDD/kg body
weight given to Wistar rats (5/sex per group) caused severe decreases
in body weight gain (males) or weight loss (females) and deaths (see
Table 46) at 100 and 300 µg/kg body weight during the 28 days of
observation. At 33 µg/kg body weight, rats of both sexes showed slight
decreases in body weight; at 10 µg/kg body weight, only the weight of
females was slightly reduced. Emaciation, piloerection, and poor
general health were seen in females of the 100 µg/kg body weight dose
group and in most animals of the highest dose group (Ivens et al.,
1992). Food and water intake were reduced in rats administered 100 µg
TBDD/kg body weight (Ivens-Kohl et al., 1990). Absolute and relative
thymus weights were decreased with increasing dose from 10 to 100
µg/kg body weight. At the highest dose of 300 µg/kg body weight, the
thymus was not detectable in either male or female rats. The
liver-to-body-weight ratio was increased in all dose groups (males:
10 - 25%, females: 4 - 15%). A dose-dependent increase in the
testes-to-body-weight ratio began at 33 µg/kg body weight. Nearly all
animals dying before term showed signs of haemorrhage in the
gastrointestinal tract (Ivens et al., 1992).
Histological alterations were consistently seen in the thymus of
TBDD-treated rats. Early thymic atrophy (characterized by the
phagocytosis of lymphocytes by histiocytes) was observed at doses of
10 and 33 µg TBDD/kg body weight, and severe atrophy was seen at 100
µg TBDD/kg body weight. Lymphocytic depletion was also observed in the
spleen at and above 33 µg TBDD/kg body weight and in Peyer's patches
of the ileum (folliculi lymphatici aggregati) at 300 µg/kg body
weight. Apart from lymphatic tissue, the organ most affected by TBDD
treatment was the liver. First signs of dose-dependent hepatotoxicity
such as cytoplasmic vacuolation and rarefaction and cellular
hypertrophy were seen after a single administration of 10 µg TBDD/kg
body weight. The cellular hypertrophy was accompanied by swelling of
the nuclei, accentuated nucleoli, and cytoplasmic transformations.
Pre-peliotic foci in the liver were seen at doses of 100 and 300 µg
TBDD/kg body weight (Ivens et al., 1992).
Haematological investigations (in rats administered single oral
doses of 10 - 300 µg TBDD/kg body weight) revealed a dose-dependent
but marginal decline in haemoglobin content and cell number
(thrombocytes, leukocytes, erythrocytes). Electrophoresis of serum
proteins showed small dose-dependent changes in alpha- and
Table 46. Mortality associated with oral administration of PBDDs/PBDFs
PBDD/PBDF Species Sex (number Dose Details Observation Mortality Time to deathb Reference
(carrier) (strain) per group) (µg/kg body perioda (number dead/ (days)
weight) number treated)
2,3,7,8-TeBDD rat female, male 10, 33 single dose 28 days no mortality - Ivens et al.
(arachis oil (Wistar) (5) 100 single dose 28 days female: 3/5 n.r. (1992)
with 0.5-5% 300 single dose 28 days female: 5/5 11-19
toluene) male: 3/5 16-22
2,3,7,8-TeBDD rat female, male 0.01 dally doses 90 days female: 1/10 n.r. Löser &
(arachis oil) (Wistar) (10) (for 90 days) Ivens
0.10 daily doses 90 days female: 1/10 n.r. (1989);
(for 90 days) Ivens et al.
1 daily doses 90 days female: 1/10 n.r. (1993)
(for 90 days) male: 2/10
3 daily doses 90 days female, male: n.r.
(for 90 days) 5/10
10 daily doses 90 days female, male: up to 35 days
(for 90 days) 10/10 (dead or
moribund)
2,3,7,8-TeBDF rat female, male 1, 10, 50 daily doses 4 weeks no mortality - Hardy et al.
(corn oil/ (Sprague- (5) (5 days/week (1990)
acetone) Dawley) for 4 weeks)
150 daily doses 4 weeks female:4/5 18-24
(5 days/week male: 3/5 (dead
for 4 weeks) or moribund)
500 daily doses 4 weeks female, male: 17-25
(5 days/week 5/5 (dead or
for 4 weeks) moribund)
2,3,7,8-TeBDF guinea-pig male (6) 0.47 single dose 30 days no mortality - Moore et al.
(corn oil) (Hartley) 1.58 single dose 30 days no mortality - (1979)
4.74 single dose 30 days 1/6 26
15.84 single dose 30 days 6/6 10-13
a After first dosing.
b n.r. = not recorded.
beta-globulins. Thyroid hormone concentrations were reduced
dose-dependently in serum (Ivens et al., 1992) (for details, see
section 7.8.4).
A series of several PBDDs and PXDDs (tetra through penta) given
intraperitoneally to immature male Wistar rats (n = 4) caused body
weight losses 14 days after injection (Mason et al., 1987a,b). The
most toxic compounds tested were 2,3,7,8-TeBDD, 2-Br1-3,7,8-Cl3-DD, and
TBCDD, which are substituted only in the four lateral positions. The
latter analogue exhibited the highest activity in this series (Mason
et al., 1987a). The relative potencies of PBDDs examined followed the
order 2,3,7,8- > 1,2,3,7,8- > 1,2,4,7,8- > 1,3,7,8- (Mason et al.,
1987a,b). There were slight differences in the ED50 values for body
weight loss (on a molar basis) between TCDD (Mason et al., 1986) and
TBDD (Mason et al., 1987a,b).
Further effects following a single dose of PBDD/PBDF or PXDD/PXDF
congeners are described in sections 7.8.1 (thymic atrophy) and 7.8.6
(hepatic enzyme induction).
Significant increases in relative liver weights were observed in
male Sprague-Dawley rats given single oral doses of 25 µg
2,3,7-TrBDD/kg body weight (Yang et al., 1983) and in pregnant mice
administered single subcutaneous doses of 5 - 90 µg TBDD/kg body
weight (Nagao et al., 1990a).
7.1.2 Dibenzofurans
2,3,7,8-TeBDF given to six guinea-pigs in single oral doses of
0.47, 1.58, 4.74, or 15.84 µg/kg body weight (equivalent on a molar
basis to 0.3 - 10 µg/kg 2,3,7,8-TeCDF) was lethal at 4.74 and 15.84
µg/kg body weight (see also Table 46). No deaths or body weight
effects were seen in the lower dose groups. Animals found dead showed
at necropsy a marked reduction in the size of the thymus, lack of body
fat, and reduction of muscle mass. Histopathological findings in
tissues of lethally intoxicated guinea-pigs included a loss of
lymphoid cells in the thymic cortex and hyperplasia of epithelial
cells in the renal pelvis, ureter, and urinary bladder. In addition,
hypocellularity of bone marrow and seminiferous tubules, lymphoid
elements in spleen, Peyer's patches, and adrenal haemorrhage were
seen. In contrast to other rodents and rabbits, whose primary target
organ after dibenzo- p-dioxin/dibenzofuran exposure is the liver, there
was a lack of liver damage in the guinea-pigs. Mild thymus lymphoid
hypoplasia was the only histological alteration in animals of the
highest non-lethal dose group that survived the 30-day observation
period (Moore et al., 1979). Altogether, the pattern of lesions was
similar to those described for guinea-pigs exposed to PCDDs/PCDFs
(Moore et al., 1979; Kociba & Schwetz, 1982; WHO, 1989).
7.1.3 Remarks on the lethality of PBDDs/PBDFs
It is characteristic for TCDD and related compounds to have a
significant latency period between the time of exposure and the time
of death. Further, these compounds are relatively persistent and
produce similar effects (e.g. wasting syndrome) regardless of single
or short-term exposure as a result of prolonged internal exposure
(e.g. McConnell, 1989; WHO, 1989, 1994a). For these reasons, the few
mortality data available for PBDDs/PBDFs from single and multiple
dosing studies are compiled in this section.
All information refers to 2,3,7,8-TeBDD (Loeser & Ivens, 1989;
Pinkerton et al., 1989; Ivens-Kohl et al., 1990; Ivens et al., 1992,
1993) and to 2,3,7,8-TeBDF (Moore et al., 1979; Pinkerton et al.,
1989; Hardy et al., 1990).
The oral LD50 of TBDD in Wistar rats was about 100 µg/kg body
weight for females and 300 µg/kg body weight for males (Ivens-Kohl et
al., 1990). After single oral exposure of Sprague-Dawley rats to TBDD
and TBDF, the LD50 was >500 µg/kg body weight (Brominated Flame
Retardants Information Panel, 1987, cited in Pinkerton et al., 1989
and in Hardy et al., 1990, the latter specifying >500 µg/kg for TBDD
and >5000 µg/kg for TBDF, n = n.sp.). Additional data on mortality
are summarized in Table 46.
Although differences in the dosage regimen make a direct
comparison impossible, it can be seen from Table 46 that guinea-pigs
are more sensitive than rats to the lethal action of TBDF, which is
consistent with findings observed with TCDD/TCDF (WHO, 1989).
Generally, there are large differences in sensitivity between species
and strains. For example, a more than 500-fold difference in acute LD50
values between the most TCDD-susceptible (Long-Evans) and the most
TCDD-resistant (Han/Wistar) rat strains was reported for TCDD (WHO,
1989; Pohjanvirta et al., 1993, 1994). The oral LD50 was reported to
range from 22 to >3000 µg TCDD/kg body weight in different rat
strains (WHO, 1989).
In a study in guinea-pigs (Moore et al., 1979), equimolar doses
of TCDF and TBDF resulted in comparable mortality rates (see Table
46). None (0/6) of the animals died following oral exposure to 1 µg
TCDF/kg body weight (equivalent to 1.58 µg TBDF/kg body weight),
whereas all six animals died at 10 µg TCDF/kg body weight (equivalent
to 15.84 µg TBDF/kg body weight). Similarly, the mean time to death
was about 12 days for both chemicals.
7.2 Short-term exposure
7.2.1 Dibenzo-p-dioxins
In a 3-month toxicity study (Ivens-Kohl et al., 1989; Löser &
Ivens, 1989; Ivens et al., 1993), 2,3,7,8-TeBDD was administered daily
by gavage to Wistar rats (10/sex per group) at doses of 0.01, 0.1, 1,
3, or 10 µg/kg body weight. No overt signs of toxicity were seen at
0.01 and 0.1 µg/kg body weight per day. Doses of 3 and 10 µg/kg body
weight per day caused a high mortality (see Table 46) and wasting
syndrome. Mean body weight gain, feed intake, and water intake were
reduced dose-dependently from 1 µg/kg body weight per day.
Changes in haemoglobin content, packed cell volume, and number of
thrombocytes were seen mainly in rats given the 1 and 3 µg TBDD/kg
body weight per day doses. The prothrombin time was markedly prolonged
at 3 µg/kg body weight per day. Clinical chemistry showed slight
increases in plasma alkaline phosphatase, aspartate aminotransferase,
and total blood bilirubin in males and females receiving a dose of 1
µg/kg body weight per day. These changes were significant at 3 µg/kg
body weight per day. Alanine aminotransferase was increased in females
only at 3 µg/kg body weight per day. There was also a decrease in
serum triglyceride levels, mainly at 1 and 3 µg/kg body weight per
day. Dose-dependent changes in thyroid hormone concentrations in serum
were observed in male and female rats. Triiodothyronine (T3) levels
were increased and thyroxin (T4) levels were decreased at doses of 0.1
µg/kg body weight per day and higher. The effects at 0.1 µg/kg body
weight per day, however, were considered to be marginal (see also
section 7.8.4). Activities of microsomal enzymes were dose-dependently
elevated (see also section 7.8.6). Protein excretion in urine
increased in males and females at doses higher than 3 µg/kg body
weight per day.
Changes in relative organ weights (increase in liver, lung,
kidney; reduction in thymus) were generally observed at doses of 1
µg/kg body weight per day and higher. Relative liver weights were
significantly increased at doses of 0.1 µg/kg body weight per day and
higher; relative thymus weights were significantly decreased at doses
of 0.01 µg/kg body weight per day and higher. Histopathological
examination revealed dose-dependent changes, mainly beginning at the 1
µg/kg body weight per day dose. These included severe atrophy of
lymphatic tissue in thymus and spleen and liver damage described as
peliosis hepatis parenchymatosa (irregular-shaped cavernous and
blood-filled spaces in the liver, lack of epithelial lining, blood
cysts in the sinusoidal lumen and in Disse's spaces, etc.) (Bannasch
et al., 1985). Spermatogenesis in the testes was adversely affected,
and defective or necrotic spermatocytes were found in the epididymis
(Ivens et al., 1993; see also section 7.5).
The NOAEL in this study was considered to be 0.01 µg TBDD/kg body
weight per day.
Compared with TCDD, TBDD elicits a similar spectrum of toxic
effects following subchronic exposure but appears to be less active
than TCDD. A subchronic NOAEL for TCDD (dosing for only 5 days/ week)
in rats was reported to be 0.01 µg/kg body weight per day (Kociba et
al., 1976). However, peliosis hepatis was not reported to occur after
treatment with TCDD (Ivens et al., 1993 and references therein).
7.2.2 Dibenzofurans
2,3,7,8-TeBDF was administered to Sprague-Dawley rats (5/sex per
group) at daily oral doses of 1, 10, 50, 150, or 500 µg/kg body
weight, 5 days/week for 4 weeks (Fulfs, 1989; Hardy et al., 1990).
Most animals in the 150 and 500 µg/kg body weight per day dose groups
died or were in a moribund condition between study days 17 and 24. At
150 µg/kg body weight per day, 3 of 10 rats survived through day 28
(see Table 46). Animals from the two highest dose groups had yellowish
urine beginning around day 15. Group mean body weight was depressed in
a time- and dose-dependent manner. The mean relative thymus weights of
males at 150 µg/kg body weight per day and of females at 10 and 50
µg/kg body weight per day were decreased. No significant changes in
relative mean liver, adrenal, or spleen weights were detected. In this
study, treatment-related histopathological alterations were noted in
the liver and thymus of animals in the 50 µg/kg body weight per day
dose group and, to a lesser extent, in the 10 µg/kg body weight per
day dose group. Liver changes consisted of panlobular hypertrophy of
the hepatocytes with associated hepatocyte vacuolation and focal
necrosis. Thymic atrophy consisting of overall depletion of the
lymphoid elements was present in all 50 µg/kg body weight per day rats
from which thymus was available and in most animals from the 10 µg/kg
body weight per day dose group. No treatment-related alterations were
observed in the 1 µg/kg body weight per day group (Hardy et al.,
1990), which can be considered as the NOAEL for this study.
The results do not indicate a lower potency of TBDF compared with
TBDD, as the studies differed in their experimental design (dosing for
4 weeks, 5 days/week versus 13 weeks, 7 days/week; Wistar versus
Sprague-Dawley rats; different animal numbers/dose group).
7.3 Long-term exposure
No long-term exposure studies with PBDDs/PBDFs were available.
7.4 Skin and eye irritation, sensitization, dermal lesions, and
acne
A common feature of toxicity of dioxin-like compounds such as
PCDDs/PCDFs, PCBs, and PBBs is their hyperkeratotic activity in humans
and some animal species (WHO, 1989, 1993, 1994a).
A standard test method for acnegenic activity, the rabbit ear
assay first described by Adams et al. (1941), was applied to
2,3,7,8-TeBDD and 2,3,7,8-TeBDF (Pinkerton et al., 1989). Both
congeners produced hyperkeratosis at a total dose of 100 µg/rabbit,
but not at 10 µg/rabbit (probably repeated application over a 4-week
period). The solvent used was not specified. Under the same
conditions, combustion residues (soot/char) from a HIPS/DBDE/Sb2O3
sample (for the PBDF content, see Table 11) were found to have no
acnegenic activity (Pinkerton et al., 1989).
In the case of TCDD, the minimum dose inducing hyperkeratosis
after a single administration ranged from 1 µg TCDD/ear to 160 µg
TCDD/ear, depending on the vehicle used (Poiger & Schlatter, 1980).
During a 4-week test with TCDD, no effect was observed at a total dose
of 8 ng/rabbit, and dose-dependent responses were obtained at
0.08 - 800 µg/rabbit (Schwetz et al., 1973).
7.5 Reproductive and developmental toxicity
7.5.1 Reproductive toxicity
A dose-dependent increase in testes-to-body-weight ratio was seen
28 days after oral administration of single doses of 2,3,7,8-TeBDD (0,
10, 33, 100, or 300 µg/kg body weight; solvent: arachis oil with 5%
toluene) to male Wistar rats (n = 5). This effect was seen from 33
µg/kg body weight onwards. Body weight gain was reduced
dose-dependently (marginally at 33 µg/kg body weight), but there was
no loss of body weight (Ivens et al., 1992).
Decreased spermatogenic activity in the testes and defective or
necrotic spermatocytes in the epididymis were found in Wistar rats
(n = 10) after daily oral administration of TBDD (in arachis oil) at 3
or 10 µg/kg body weight per day, 7 days/week for 13 weeks. Severe
effects were observed at 10 µg/kg body weight per day, and moderate
effects at 1 µg/kg body weight per day. The NOEL was 0.1 µg/kg body
weight per day (Ivens-Kohl et al., 1989; Ivens et al., 1993).
Adverse effects on the male reproductive system (e.g. reduction
in number, size, and organelle content of Leydig cells in adult rat
testes) were observed following single intraperitoneal injections of
12.5 - 50 µg TCDD/kg body weight (Johnson et al., 1994). However, at
perinatal exposure, a single oral dose as low as 64 ng TCDD/kg body
weight (the lowest maternal dose tested) given to mothers at day 15 of
gestation was sufficient to reduce sperm production in the male
offspring (Mably et al., 1992). Reduced sperm numbers were also
observed in offspring of pregnant rats administered 1 µg TCDD/kg body
weight on gestation day 8 or 15 and in offspring of Syrian hamsters
dosed with 2 µg/kg body weight on gestation day 11 (Gray et al.,
1995).
7.5.2 Developmental toxicity
Several PBDDs/PBDFs were found to be inducers of cleft palate and
hydronephrosis in mice. These effects occurred at doses (see Table 47)
that produce no or only marginal general maternal toxicity and no
fetal mortality (Nagao et al., 1990a,d; Birnbaum et al., 1991).
Single subcutaneous injections of TBDD or TCDD (5 - 90 µg/kg body
weight) were administered to NMRI mice on day 9 of pregnancy. Maternal
and fetal toxicity were assessed on day 18 (Nagao et al., 1990a,d).
TBDD and TCDD caused significant dose-related increases in the
incidence of cleft palate in the total number of viable fetuses and in
the number of litters with cleft palate. On a molar basis, the potency
of TBDD for cleft palate induction relative to that of TCDD was found
to be 0.6: ED50 values were 61.7 µg/kg body weight per day
(corresponding to 0.124 µmol/kg body weight per day) for TBDD and 24
µg/kg body weight per day (corresponding to 0.075 µmol/kg body weight
per day) for TCDD. Both TBDD and TCDD at the doses given increased
maternal liver-to-body-weight ratios. There were no significant
effects on maternal weight gain or the number of viable fetuses per
litter.
In another study (Birnbaum et al., 1991), the teratogenic effects
of TBDD and three PBDFs (TBDF, 1,2,3,7,8-PeBDF, and 2,3,4,7,8-PeBDF)
were examined in C57BL/6N mice. Pregnant dams were treated on
gestation day 10 with single oral doses of each congener and
sacrificed on gestation day 18. Doses ranged from 0 to 192 µg/kg body
weight for TBDD and from 0 to 4000 µg/kg body weight for the three
dibenzofurans. All compounds produced hydronephrosis at doses below
that at which cleft palate occurred (see also Table 47). The LOELs
(µg/kg body weight) for hydronephrosis and cleft palate, respectively,
were as follows: TBDD: 3 and 48; TBDF: 25 and 200; 1,2,3,7,8-PeBDF:
500 and 3000 - 4000; and 2,3,4,7,8-PeBDF: 400 and 2400. Embryo/fetal
mortality was significantly increased at 500 µg TBDF/kg body weight
and higher doses. At 3000 µg TBDF/kg body weight, the few survivors
were oedematous. After exposure to TBDD, the number of live fetuses
showed a "decreasing trend," which was not significant. Dose-related
increases in fetal weights were observed with TBDD and TBDF. However,
the increases were significant only for TBDF at doses of 500 µg/kg
body weight and higher. Maternal liver weight increased at all dose
levels examined for all four compounds. Maternal weight gain was
elevated at the highest dose of TBDF (4000 µg/kg body weight), and
this increase was due to subcutaneous oedema.
Birnbaum et al. (1991) compared the relative toxicities of the
brominated congeners with those of the chlorinated ones. Based on the
approximate ED50 values, TBDD appeared to be almost half as potent as
TCDD in the induction of hydronephrosis in offspring of treated dams
(4 µg/kg body weight versus 9 µg/kg body weight; see also Table 47).
However, compared on a molar basis, TBDD and TCDD were almost
equipotent. A survey of the relative potencies (on a weight basis) of
PBDD/PCDD and PBDF/PCDF congeners for the induction of cleft palate is
given in Table 48. It can be seen that bromination decreased the
activity of TBDD relative to TCDD but increased the potency of TBDF
relative to TCDF. The pentaBDFs tested, however, were slightly less
potent than the pentaCDFs.
7.6 Mutagenicity and related end-points
No information was found on the mutagenicity of PBDDs/PBDFs or on
related end-points.
Whereas there is limited or conflicting evidence demonstrating
the positive mutagenic potential of PCDDs (WHO, 1989), co-mutagenic or
co-recombinogenic effects of PCDDs have been demonstrated in vivo in
the mouse spot test (Fahrig, 1993).
Table 47. Development at toxicity of PBDDs/PBDFs
PBDDs/PBDFs Species Route Dose (µg/kg Effects Remarks Reference
(vehicle) (strain) (dosing body weight)
(n)a regimen)b
2,3,7,8-TeBDD mouse subcutaneous 0-90 from 5 µg/kg body weight: no effect on the Nagao et al.
(toluene/DMSO (NMRI) (single dose (5, 10, 30, F0: increase in relative number of viable (1990a,b)
= 1+2, v/v) (n.sp.) on gd 9) 50, 90) liver weight, fetuses per
F1: cleft palate, litter, fetal
ED50: 62 µg/kg body weight weight, or fetal
(0.123 µmol/kg body weight) deaths
2,3,7,8-TeBDD mouse oral 0-192 from 3 µg/kg body weight: trend for a Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (3, 6, 12, F0: increase in relative decrease in (1991)
(11-20) on gd 10) 24, 48, 96, liver weight, number of live
192) fetuses and an
F1: hydronephrosis, increase in fetal body
ED50:9 µg/kg body weight weight with increasing
(0.018 µmol/kg body weight) dose
from 48 µg/kg body weight:
F1 cleft palate,
ED50: 65 µg/kg body weight
(0.13 µmol/kg body weight)
2,3,7,8-TeBDF mouse oral 0-4000 from 25 µg/kg body weight: Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (25, 50,100, F0: increase in relative (1991)
(7-22) on gd 10) 200, 250, liver weight,
500, 1000, F1: hydronephrosis,
3000,4000) ED50: approx. 12 µg/kg body
weight (0.024 µmol/kg
body weight) from 200 µg/kg
body weight: F1: cleft palate,
ED50:154 µg/kg body weight
(0.31 µmol/kg body weight)
from 500 µg/kg body weight:
F1 increase in fetal
mortality and live fetal body
weight 4000 µg/kg body weight:
F0: subcutaneous oedema
1,2,3,7,8-PeBDF mouse oral 0-4000 from 250 µg/kg body weight: no increase in Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (250, 500, F0: increase in relative fetal mortality (1991)
(5-11) on gd 10) 1000, 2000, liver weight or fetal body weight
3000, 4000) from 500 µg/kg body weight:
F1: hydronephrosis,
ED50: approx. 340 µg/kg body
weight from 3000c µg/kg body
weight: F1: cleft palate,
ED50:4088 µg/kg body weight
2,3,4,7,8-PeBDF mouse oral 0-4000 from 25 µg/kg body weight: no effect on fetal Birnbaum et al.
(corn oil) (C57BL/6N) (single dose (25, 50,100, F0: increase in relative liver mortality or fetal body (1991)
(9-16) on gd 10) 200, 400, weight weight
800, 1600, from 400 µg/kg body weight:
2400, 4000) F1: hydronephrosis,
ED50: approx. 437 µg/kg body
weight from 2400 µg/kg body
weight: F1: cleft palate,
ED50: 3024 µg/kg body weight
a Number of litters; n.sp. = not specified.
b gd= gestation day (first day of gestation designated day zero).
c First significant at 3000 µg/kg body weight, statistically (p < 0.01) significant at 4000 µg/kg body weight.
Table 48. Relative potencies of PBDD/PCDD and PBDF/PCDF congeners relative
to TCDD for the induction of cleft palate in micea
Congener Relative potencies for cleft palateb
H=Br H=Cl
Dibenzo-p-dioxins
2,3,7,8-TeHDD 0.24 1
Dibenzofurans
2,3,7,8.TeHDF 0.10 0.05
1,2,3,7,8-PeHDF 0.004 0.03
2,3,4,7,8-PeHDF 0.005 0.09
a Adapted from Birnbaum et al. (1991).
b Derived from ED50 values: on a weight basis.
The cell-transforming potential of TBDD has been demonstrated in
a host-mediated in vivo/in vitro assay with peritoneal murine
macro-phages (Massa et al., 1990). NMRI mice were intraperitoneally
administered 0.39 nmol TBDD (corresponding to 195 ng) or TCDD
(corresponding to 125 ng) per mouse. Isolation of resident macrophages
4 days later, cultivation in soft agar for 5 - 6 days, and evaluation
of the clones indicated that the transforming capacity of TBDD was
seven times less than that of TCDD (Massa et al., 1991).
7.7 Carcinogenicity
7.7.1 Short-term studies
A permanent cell line was established from peritoneal macrophages
of mice treated with TBDD. These cells were tested for their
tumorigenicity in athymic nude (nu/nu) mice. Animals given
subcutaneous injections of these cells (1 x 106 cells) developed
tumours at the injection site 3 weeks later (Massa et al., 1991,
1992a,b).
7.7.2 Long-term studies
PBDDs/PBDFs have not been tested for carcinogenicity in long-term
studies.
TCDD was shown to be a multisite carcinogen in both sexes of rats
and mice at doses below the maximum tolerated dose (WHO, 1989).
7.8 Other special studies
7.8.1 Immunotoxicity
Thymus atrophy and other signs of immunotoxicity were found to be
the main characteristic toxic effects (besides body weight loss or
decrease in the rate of weight gain) after exposure to TCDD and were
observed in almost all laboratory animals (Vos & Luster, 1989; WHO,
1989). The limited data available for PBDDs/PBDFs from studies with
rats, guinea-pigs, and monkeys confirmed the expected immunotoxic
potential. Parameters examined were influences on lymphoid tissues,
effects on serum protein levels or other haematological parameters,
and alterations of certain lymphocyte subpopulations in peripheral
blood. Effects on immunotoxicity after perinatal exposure to
PBDDs/PBDFs have not been investigated.
7.8.1.1 Dibenzo-p-dioxins
Dose-dependent decreases in thymus weights and atrophy of thymus
and other lymphatic tissues were observed in rats after single
exposures to 2,3,7,8-TeBDD (Mason et al., 1987a,b; Ivens et al., 1992;
see also section 7.1) and to a series of other PBDD or PXDD congeners
(Mason et al., 1987a,b) and after subchronic exposure to 2,3,7,8-TeBDD
(Ivens-Kohl et al., 1989; Löser & Ivens, 1989; Ivens et al., 1993; see
also section 7.2). Effects were found after single doses of 10 µg/kg
body weight or higher and after daily doses of 1 µg/kg body weight and
higher for 3 months (Ivens-Kohl et al., 1989; Löser & Ivens, 1989;
Ivens et al., 1992, 1993). The potency of TBDD for causing thymic
atrophy was comparable to that of TCDD (Mason et al., 1987b).
1,3,7,8-TeBDD, 1,2,3,7,8-PeBDD, and 1,2,4,7,8-PeBDD were less active
(Mason et al., 1987b). The mixed 2-Br1-3,7,8-Cl3DD was as active as
TBDD and TCDD; the mixed 2,3-Br2-7,8-Cl2DD was the most toxic analogue
in this series (Mason et al., 1987a).
Some routine haematological parameters, including serum protein
electrophoresis (reduction in number of thrombocytes, prolongation of
prothrombin time, slight anisocytosis, poikilocytosis, slight
reduction in gamma globulins, etc.), examined in the subchronic (3
months) rat study (doses applied: 0.01 - 10 µg/kg body weight) might
suggest an impact on cellular and, possibly, humoral immunity
beginning at daily doses of 1 µg TBDD/kg body weight (Ivens-Kohl et
al., 1989).
It has been shown that very low doses of PBDDs and PCDDs affect
the immune system of the marmoset monkey (Neubert R. et al., 1990,
1991, 1992, 1993; Neubert, 1991, 1993a). After dibenzo- p-dioxin
exposure, alterations were seen in lymphocytes from peripheral blood
of mature marmosets. The lymphocyte subpopulations showing the most
pronounced effects were the "helper-inducer" or "memory" T cells
(CD4+CDw29+ in the marmoset, probably corresponding to the
CD4+CD45RO+CDw29+ cells in humans) and certain B cell subsets (e.g.
CD20+) (Neubert, 1993a; Neubert et al., 1993). Several weeks after a
single subcutaneous dose of 30 ng TBDD/kg body weight (corresponding
to 60 pmol TBDD/kg body weight), there was a significant decrease in
the percentage and absolute number of the T helper-inducer
subpopulation (CD4+CDw29+) in venous blood of treated marmosets (n =
3). This effect on cell-mediated immunity is pronounced in young
animals (Vos, 1993). In addition, a significant decrease in the number
of B cells (CD20+) was observed (Neubert, 1993a). This is considered
to be less dependent on the age of animals (Vos, 1993). Injection of 3
ng TBDD/kg body weight (corresponding to 6 pmol TBDD/kg body weight)
did not induce any changes (Neubert, 1993a; Neubert et al., 1993).
With TCDD, a clear dose-response relationship was found at doses
of 10 ng/kg body weight and higher, and a questionable response
(confined to single individuals) was seen at 3 ng/kg body weight. No
effect was detectable with single doses of 1 ng TCDD/kg body weight or
lower. It was concluded that, on a molar basis, the potencies of TBDD
and TCDD may be similar in this experimental approach (Neubert, 1993a;
Neubert et al., 1993).
One mixed halogenated congener, namely TBCDD, was also
investigated. No changes in the subpopulations tested were found
following a single subcutaneous dose of 3 ng/kg body weight. Treatment
with 30 TBCDD/kg body weight decreased the percentage of CD24+CDw29+
cells and of CD20+ lymphocytes. However, no significant effect was
seen in the small number of marmosets used (n = 3) on the absolute
number of cells per µl of blood. From these data, it appeared that
this congener may be less potent than TCDD and TBDD (Neubert, 1993a;
Neubert et al., 1993).
7.8.1.2 Dibenzofurans
A marked reduction in the size of the thymus, loss of lymphoid
cells in the thymic cortex, hypocellularity of bone marrow, lymphoid
elements in spleen, and Peyer's patches were seen in guinea-pigs after
single oral doses of TBDF (4.7 - 15.8 µg/kg body weight) that also
induced mortality. At a non-lethal dose of 1.6 µg/kg body weight, only
mild evidence of thymus lymphoid hypoplasia was noted after the 30-day
observation period (Moore et al., 1979; see also section 7.1).
Short-term exposure of Sprague-Dawley rats to daily oral doses of
1-500 µg TBDF/kg body weight (5 days/week for 4 weeks) caused
dose-dependent reductions in thymus weights and thymic atrophy at the
10 µg/kg body weight per day and higher doses (Hardy et al., 1990; see
also section 7.2).
7.8.2 Effects on intermediary metabolism: Porphyrin effects
Hepatic porphyrin accumulation was studied after subchronic
dosing (5 days/week for 13 weeks) of female B6C3F1 mice by oral gavage
with individual congeners of PBDDs/PCDDs, including 2,3,7,8-TeBDD and
2,3,7,8-TeCDD (Table 49). Dose-dependent increases in total hepatic
porphyrins were found for both TBDD and TCDD. The relative
porphyrinogenic potencies were determined by the authors using TCDD as
a reference (TCDD = 1 and TBDD = 0.4).
Table 49. Total hepatic porphyrin accumulation in female B6C3F1 mice
after 13 weeks of exposure to TBDD or TODDa
TBDD TCDD
Dose Hepatic porphyrin Dose Hepatic porphyrin
(ng/kg body accumulation (ng/kg body accumulation
weight per day) (µg/g) weight per day) (µg/g)
0 0.207 0 0.196
30 0.296 0.15 0.204
90 0.346 0.45 0.212
300 0.429 1,5 0.212
900 13.4 4.5 0.222
3000 26.2 15 0.256
45 0.592
150 13.2
450 18.6
a Adapted from van Birgelen et al. (1996).
7.8.3 Effects on vitamin A storage
Various brominated or chlorinated aromatic compounds are able to
reduce the vitamin A content of the liver (WHO, 1989, 1993, 1994a). A
single oral dose of 10 µg TBDD/kg body weight decreased both the
concentration and the total amount of vitamin A (retinol) in the liver
of adult male Sprague-Dawley rats (n = 5) 4 weeks after the start of
the experiment. Reductions in concentration and total amount of
vitamin A were 45 and 51%, respectively (Thunberg et al., 1984). A
more pronounced effect was elicited by TCDD tested in the same study,
the reductions being 88 and 87%, respectively. However, on a molar
basis, TCDD was only slightly more potent than TBDD (Thunberg et al.,
1984).
7.8.4 Endocrine interactions
Thyroid hormones were affected in rats after single (Ivens et
al., 1992; see also section 7.1) and subchronic (Löser & Ivens, 1989;
Ivens et al., 1993; see also section 7.2) exposures to 2,3,7,8-TeBDD.
Four weeks after single oral doses of 10, 33, or 100 µg TBDD/kg body
weight, T3 was increased and T4 was reduced dose-dependently in the
serum of female and male Wistar rats ( n = 5) (Ivens et al., 1992).
Wistar rats (10/sex per group) treated with daily doses of 0.01 - 3.0
µg/kg body weight per day for 3 months had reduced T4 and increased T3
levels at 0.1 µg/kg body weight per day and higher (highest dose group
of 10 µg/kg body weight per day not examined) (Löser & Ivens, 1989;
Ivens et al., 1993; see also section 7.2.1).
The antiestrogenic potency of a series of PXDDs/PXDFs was
examined in cultures of MCF-7 human breast cells (Spink et al., 1994).
Two effects, stimulation of the metabolism of 17 ß-estradiol and
inhibition of the estrogen-dependent formation of multicellular foci,
were measured as indices of antiestrogenicity. Several tetra(Br1Cl3DDs,
Br2Cl2DDs) and penta- (Br1Cl4DD) halogenated congeners with
2,3,7,8-substitution stimulated estradiol metabolism with a potency
similar to that of TCDD. The EC50 values of these PXDDs were 0.6 - 0.8
nmol (TCDD: 0.8 nmol). The focus formation was inhibited by the same
congeners, with EC50 values ranging from 0.5 to 1.2 nmol (TCDD: 0.3
nmol). Dibenzo- p-dioxins and dibenzofurans with other substitution
patterns were markedly less active (EC50 values in nmol for inhibition
of focus formation: 17, 8-Br1-2,3,4-Cl3DF and 7-Br1-2,3-Cl2-DD; 1700,
2,3,7-Cl3-8-methylDD; >1700, 2,7-Br2DF and several methyl-substituted
PCDDs/PCDFs).
7.8.5 Interaction with drugs and toxicants
A single oral dose of 2,3,7-TrBDD (25 µg/kg body weight)
depressed the plasma disappearance of ouabain and its excretion in
bile of male Sprague-Dawley rats 10 days after treatment. In addition,
the bile flow was decreased by 2,3,7-TrBDD. TCDD elicited the same
effects but more markedly than 2,3,7-TrBDD (Yang et al., 1983).
7.8.6 Induction of microsomal enzymes
PBDDs/PBDFs, like TCDD and other environmental contaminants, are
potent inducers of certain CYP-dependent enzymes. Most frequently, the
activity of the cytochrome CYP1A1 was examined (direct measurements by
radioimmunoassay [RIA] or determination of marker enzymes [mostly AHH
and EROD]).
The isoenzyme CYP1A2 (determined by RIA) was also found to be
inducible. ED50 values of 0.8 - 1 nmol/kg body weight for CYP1A1
induction and 0.2 nmol/kg body weight for CYP1A2 induction in rat
liver were estimated after single oral doses of TBDD (Kedderis et al.,
1991b, 1992a, 1993). Induction of CYP1A2 enzyme activity has been
determined in mouse liver after subchronic dosing by oral gavage with
TBDD (Birnbaum & DeVito, 1995) and in liver of marmoset monkeys after
single subcutaneous doses of TBDD (Schulz et al., 1996).
Induction of the monooxygenases AHH and/or EROD was observed
following single exposure of chicken embryos (Kende & Wade, 1973;
Poland & Glover, 1973; Kende et al., 1974; Ramalingam et al., 1986),
guinea-pigs (Schmidt & Ivens-Kohl, 1990b), rats (Thunberg et al.,
1984; Magon et al., 1987a,b; Nagao et al., 1990b,e; Schmidt &
Ivens-Kohl, 1990a,b; Schulz-Schalge et al., 1990, 1991a,b; Kedderis et
al., 1991b, 1992a, 1993; Ivens et al., 1992), and marmoset monkeys
(Schulz et al., 1993, 1996) and after subchronic dosing by oral gavage
in mice (Birnbaum et al., 1993; Birnbaum & DeVito, 1995; van Birgelen
et al., 1996) and rats (Ivens et al., 1993), as well as in primary
cultures of rat hepatocytes (Blankenburg et al., 1990) and in rat
hepatoma H-4-II E cells (Bradlaw & Casterline, 1979; Bradlaw et al.,
1980; Denomme et al., 1985, 1986; Mason et al., 1987a; Zacharewski et
al., 1988, 1989; Safe et al., 1989a,b). Almost all experiments
referred to the tetra- and pentaBDDs/XDDs. Only a few studies (Poland
& Glover, 1973; Kende et al., 1974; Denomme et al., 1985) included
tri- or diBDDs/XDDs. There was one study (Schulz et al., 1993) on
dibenzofurans (pentaBDF) and another (Denomme et al., 1986) on PXDFs.
Several aspects of enzyme induction by PHDDs/PHDFs, with emphasis
on the chlorinated congeners, are addressed in Goldstein & Safe (1989)
and Neubert (1993b). The general mechanisms of induction of the CYP1A1
enzyme system were discussed by Okey (1990, 1992).
7.8.6.1 Dibenzo-p-dioxins
Enzyme induction proceeded dose-dependently at non-toxic
concentrations. It was measurable at exposures as low as the pmol
range (Schulz-Schalge et al., 1990, 1991b: <100 pmol TBDD/kg body
weight in the rat). Induction started soon after exposure and was
long-lasting, 1 - 98 days after a single subcutaneous administration
of TBDD, TCDD, and tetraXDDs (Nagao et al., 1990c; Schulz-Schalge et
al., 1991a). Maximum EROD induction was seen approximately 7 days
after a single subcutaneous exposure of rats to several
2,3,7,8-substituted dibenzo- p-dioxins (Nagao et al., 1990c;
Schulz-Schalge et al., 1991a). Oral exposure of rats to 1 µg TBDD/kg
body weight per day for 91 days also showed maximum induction after 7
days of treatment (Ivens et al., 1993). In contrast, during low-dose
oral treatment of mice (30 - 3000 ng TBDD/kg body weight per day for
13 weeks), a biphasic response was observed, not leading to maximum
enzyme induction (Birnbaum et al., 1993).
In a comparison of the decline in EROD activity after single
doses of 600 µg TBDD/kg body weight and 300 µg TCDD/kg body weight,
the curves were superimposable over a period of 90 days (Nagao et al.,
1995/96). When the hepatic EROD activity was related to the molar
concentrations of TBDD and TCDD, the same relationship was also
demonstrated. This indicates that both TBDD and TCDD exhibit identical
potencies with respect to the induction of EROD, when compared on a
molar basis. When compared on a weight basis (as is done for an
international toxic equivalent [I-TEQ] approach), TBDD is less potent
with respect to this end-point.
Although the liver was the most important organ for enzyme
induction, EROD induction by TBDD was also observed in other tissues
(lung, skin, kidney) of rats (Ivens et al., 1993) and mice (Birnbaum
et al., 1993) and in lung of marmoset monkeys (Schulz et al., 1996).
The induction potency for different PBDD congeners varied over
several orders of magnitude. For example, the activities for in vivo
hepatic AHH induction differed by four orders of magnitude between
four brominated congeners, the most active being 2,3,7,8-TeBDD and the
least 1,3,7,8-TeBDD (see Tables 50 and 51). Similar structure-activity
correlations were seen in other experimental systems ( in vitro
studies, EROD activity, toxic effects) and were discussed in several
reviews (Goldstein & Safe, 1989; Safe et al., 1989a,b; Safe, 1990). As
with PCDDs (WHO, 1989), the most potent compounds were those having
the 2,3,7,8-substitution pattern. In summary, the most effective
inducers of in vitro and/or in vivo AHH or EROD activity were TCDD,
TBDD, and TBCDD.
Compared on a molar basis to their chlorinated analogues, the
PBDDs and PXDDs have more or less similar potencies (see Tables 50 and
51; Kende & Wade, 1973; Kende et al., 1974; Bradlaw & Casterline,
1979; Thunberg et al., 1984; Denomme et al., 1985; Abraham et al.,
1988; Blankenburg et al., 1990; Nagao et al., 1990c; Schulz-Schalge et
al., 1990, 1991b). Nevertheless, some differences are apparent between
TBDD and TCDD, the congeners most intensively studied. In contrast to
TCDD, whose relative induction potency was independent of the tissue
examined, TBDD was five times more potent in inducing EROD activity in
the liver than in skin and lung following subchronic exposure of mice
(Birnbaum et al., 1993).
Only limited data are available on possible species-dependent
variations. The estimated potency for EROD reduction of TBDD relative
to TCDD ranged from 0.75 to 5.3 in rats (calculated by Birnbaum et
al., 1993 from data of Safe, 1990) and from 0.04 to 0.2 in mice
(Birnbaum et al., 1993). According to the authors, it is possible that
these differences may be indicative of species differences, despite
the different experimental designs used in the rat and mice studies.
7.8.6.2 Dibenzofurans
2,3,4,7,8-PeBDF was investigated together with its chlorinated
counterpart and TCDD in the marmoset monkey. Six days after a single
subcutaneous dose of 420 ng 2,3,4,7,8-PeBDF/kg body weight (TCDD:
50 - 500 ng/kg body weight; 2,3,4,7,8-PeCDF: 240 - 2400 ng/kg body
weight), the caffeine breath test was performed in vivo; 1 day later,
the animals were sacrificed, and EROD activity was determined in liver
microsomes. There was a good correlation between EROD activity in
hepatic microsomes and caffeine N-demethylation. All three compounds
showed inducing capacity, and the ranking order was TCDD >
2,3,4,7,8-PeCDF > 2,3,4,7,8-PeBDF when enzyme activities were
compared with the hepatic concentrations (Schulz et al., 1993).
Table 50. In vivo ED50 values for AHH induction and toxic effects
of several PHDDs in the rata,b
Congeners In vivo ED50 (µmol/kg body weight)
Inhibition of body Thymic atrophy Hepatic AHH
weight gain induction
2,3,7,8-TeBDD 0.068 0.034 0.0076
2,3,7,8-TeCDD 0.05 0.09 0.004
2,3-BR2-7,8-Cl2-DD 0.012 0.0073 0.000 49
2-BR2-3,7,8-Cl3-DD 0.12 0.035 0.0025
1,2,3,7,8-PeBDD 0.87 0.39 0.025
1,2,3,7,8-PeCDD 0.62 0.17 0.031
1,2,4,7,8-PeBDD 12.9 6.17 0.195
1,2,4,7,8-PeCDD 34.0 11.2 2.82
1,3,7,8-TeBDD 252.0 35.5 6.50
1,3,7,8-TeCDD 132.0 100.0 31.2
a Adapted from Mason et al. (1987a); Safe et al. (1989b).
b Immature male Wistar rats (n = 4), 14 days after single intraperitoneal doses.
For PXDFs, the AHH and EROD enzyme induction potencies of
8-Br1-2,3-Cl2DF and 8-Br1-2,3,4-Cl3DF were tested in vitro in rat
hepatoma H-4-II E cells (Denomme et al., 1986). The EC50 values
(mol/litre) were similar to those of the fully chlorinated analogues.
7.8.6.3 Combustion products
Pyrolysates of several brominated flame retardants, which
contained relatively high levels of PBDFs/PBDDs (see also section 3),
produced dose-dependent EROD and AHH activity in vitro (rat hepatoma
H-4-II E cells) (Zacharewski et al., 1988, 1989), as well as in liver
microsomes of rats (Zacharewski et al., 1988, 1989; Schmidt &
Ivens-Kohl, 1990b) and guinea-pigs (Schmidt & Ivens-Kohl, 1990b)
sacrificed 14 days or 4 weeks after single intraperitoneal or oral
doses.
7.9 Mechanisms of toxicity - mode of action
The mechanisms underlying the diversity of biochemical and toxic
effects of TCDD are being extensively studied, but they are complex
and not yet fully understood. Because of similarities in chemical
structure and in the pattern of responses, PBDDs/PBDFs are believed to
Table 51. Comparative survey on CYP1A1 induction (measured as EROD activity in vitro or in vivo in liver)
by 2,3,7,8-substituted TeHDDs
Species Details Parameter Resultsa Reference
(strain) TBDD TCDD TBCDD
Rat in vitro: hepatoma H-4-11 E EC50 (nmol/litre) 0.235 0.080 0.055 Mason et al. (1987a); Safe
cells (1987); Goldstein & Safe
(1989); Safe et al. (1989b)
Rat (Wistar) single intraperitoneal doses ED50 (nmol/kg body weight) 0.355 3 0.347 Mason et al. (1986,
1987a); Safe (1990)
pED50b 9.45 8.16 n.sp. Mason et al. (1987b)
Rat single oral doses ED50 (nmol/kg body weight) 0.8-1 n.a. n.a. Kedderis et al. (1992a)
(Fischer 344)
Rat (Wistar) single subcutaneous dose resorufin (pmol/mg protein Schulz-Schalge et al.
of 2 nmol/kg body weight per min) after: (1991)
- 7 days 6740 5210 4330
- 98 days 410 162 105
Mouse multiple oral doses relative potency 0.2 1 n.a. Birnbaum et al. (1993)
(B6C3F1)
Chick embryo single injections ED50 (pmol/egg) 9.4 11.1 7.35 Ramalingam et al. (1986)
a n.a. = not analysed; n.sp. = not specified.
b pED50 = -log ED50.
share a common mechanism of action with PCDDs/PCDFs and other related
halogenated aromatic hydrocarbons (Poland & Knutson, 1982; Goldstein &
Safe, 1989; Mennear & Lee, 1994). Most information is derived from
studies with TCDD, the prototypical and best examined compound of this
class of chemicals. PHDDs/PHDFs act as multisite toxicants in
laboratory animals and elicit species-, sex-, and tissue-dependent
responses (Vanden Heuvel & Lucier, 1993). In carcinogenic processes,
TCDD and related compounds function as tumour promoters (reviewed in
Lucier et al., 1993b; Huff, 1994).
On a cellular basis, TCDD and related compounds cause alterations
in cell proliferation and differentiation without a mutational effect
on DNA (Goldstein & Safe, 1989; Silbergeld & Gasiewicz, 1989; Nebert
et al., 1991; Nebert, 1994). However, there is a possibility that TCDD
may influence the DNA-damaging potential of endogenous compounds
(Lucier et al., 1993b). Examples of impairment of normal cellular
regulatory systems by PCDDs/PCDFs have been compiled (WHO, 1989).
Recently, inhibition of gap-junctional intercellular communication by
TCDD was reported (De Haan et al., 1993).
At the molecular level, there is growing evidence that most, if
not all, biochemical or toxic effects, including carcinogenicity, are
mediated through an intracytoplasmic protein, the Ah receptor (Couture
et al., 1990; Denison, 1990, 1991; Whitlock, 1990, 1993; Landers &
Bunce, 1991; Lucier, 1991; Nebert et al., 1991, 1993; Safe et al.,
1991; Hahn & Stegeman, 1992; Poellinger et al., 1992; Andersen et al.,
1993; Birnbaum, 1993, 1994; Lucier et al., 1993a,b; Poellinger, 1993;
Safe, 1993a; Vanden Heuvel & Lucier, 1993; Whitelaw et al., 1993; Okey
et al., 1994; Fernandez-Salguero et al., 1996). However, although the
central role of the Ah receptor in mediating TCDD toxicity is
generally accepted, some findings may indicate the existence of other
mechanisms operating independently of the Ah receptor (Skene et al.,
1989; WHO, 1989; Holsapple et al., 1991; Nebert et al., 1993; Okey et
al., 1994).
The potential of binding to the cytosolic Ah receptor was
confirmed for several mono- through penta-substituted PBDDs and PXDDs
(Denomme et al., 1985; Mason et al., 1987a,b; Romkes et al., 1987) and
PXDFs (Denomme et al., 1986). Their receptor-binding affinities varied
by several orders of magnitude (depending on structure) but were
comparable to those of their PCDD analogues (Goldstein & Safe, 1989).
As an example, the in vitro EC50 values for rat hepatic cytosolic
receptor-binding potencies of TCDD, TBDD, and TBCDD were found to be
1.00 x 10-9, 1.50 x 10-9, and 1.48 x 10-9 mol/litre, respectively (Mason
et al., 1987a).
Limited information is available on possible additive,
synergistic (overadditive), or antagonistic interactions between
different PBDDs/PBDFs and other xenobiotics. As seen in a rainbow
trout early life stage mortality bioassay (see chapter 9), selected
PBDD, PBDF, and PBB congeners act additively (Hornung et al., 1996a).
All kinds of interactions could be found between PCDDs, PCDFs, and
PCBs (summarized by Skene et al., 1989; Safe, 1990; Hornung et al.,
1996a).
There have been no studies identified detailing the possible
actions of non-2,3,7,8-substituted PBDD/PBDF congeners.
7.10 Experimental data on selected PBDDs/PBDFs and their relevance
to the toxicity equivalency factor (TEF) concept
The TEF method is used as a procedure to facilitate the
evaluation of complex mixtures of related chemicals. The real
concentrations of different congeners in a sample are changed to (by
multiplying by a factor called the TEF) and expressed as uniform
"toxic" concentrations, as related to a reference substance. A major
application of this method is for risk management or regulatory
purposes. Advantages and limitations in the use of TEFs were discussed
intensively with regard to PCDDs/PCDFs (e.g. Kutz et al., 1990;
Ahlborg et al., 1992; Neubert D. et al., 1992; Ahlborg, 1994; DeVito &
Birnbaum, 1994; Silbergeld & deFur, 1994). For the 2,3,7,8-substituted
derivatives within these compounds, a series of TEF schemes was
developed by several agencies in different countries. The schemes
differ in the weighting factors for certain congeners. In 1988, an
internationally elaborated TEF scheme was published (NATO-CCMS, 1988),
presenting the so-called international TEFs (I-TEFs) (see Table 52).
It was followed by a modified proposal of Safe (1990, 1993a,b).
Additionally, his concept included (besides PCBs, PBBs, and
poly-chlorinated diphenyl ethers [PCDEs]) the PBDDs/PBDFs and PXDDs/
PXDFs to which the same factors were attributed as described for the
chlorinated analogues. Currently, there are no TEFs for
2,3,7,8-substituted PBDDs/PBDFs that have international agreement.
Most of the toxicologically relevant information available refers
to the 2,3,7,8-substituted dibenzo- p-dioxin analogue pair. Data
summarized in Table 53 support the concept of using corresponding TEF
values for both analogues. Even if for some end-points slightly lower
potencies (on a molar basis, and somewhat more pronounced on a weight
basis) were found for 2,3,7,8-TeBDD than for 2,3,7,8-TeCDD, kinetic
differences (especially the longer half-life of 2,3,7,8-TeBDD in
adipose tissue) may favour the use of identical TEFs for 2,3,7,8-TeBDD
and 2,3,7,8-TeCDD (e.g. Neubert, 1993b).
Table 52. I-TEFs for PCDDs/PCDFs
Congeners I.TEFsa
2,3,7,8-TeCDD 1
2,3,7,8-TeCDF 0.1
1,2,3,7,8-PeCDD 0.5
1,2,3,7,8-PeCDF 0.05
2,3,4,7,8.PeCDF 0.5
1,2,3,4,7,8-HxCDD 0.1
1,2,3,4,7,8-HxCDF 0.1
1,2,3,6,7,8-HxCDD 0.1
1,2,3,6,7,8-HxCDF 0.1
1,2,3,7,8,9-HxCDD 0.1
1,2,3,7,8,9-HxCDF 0.1
2,3,4,6,7,8-HxCDF 0.1
1,2,3,4,6,7,8-HpCDD 0.01
1,2,3,4,6,7,8-HpCDF 0.01
1,2,3,4.7,8,9-HpCDF 0.01
1,2,3,4,6,7,8,9-OcCDD 0.001
1,2,3,4,6,7,8,9-OcCDF 0.001
a From NATO/CCMS (1988).
For another analogue pair, namely 2,3,7,8-TeBDF and
2,3,7,8-TeCDF, it was suggested that a higher TEF than that chosen for
2,3,7,8-TeCDF (0.1) be used, possibly around 0.3 on a molar basis or
0.2 on a weight basis (Neubert, 1993b). This proposal was also based
on kinetic data - that is, the much longer elimination half-life of
2,3,7,8-TeBDF than of 2,3,7,8-TeCDF observed in rat liver (Neubert,
1993b). Additionally, 2,3,7,8-TeBDF was found to be more potent than
2,3,7,8-TeCDF in producing cleft palate and hydronephrosis in mice
(Birnbaum et al., 1991 and references therein). In a fish toxicity
assay (see chapter 9), 2,3,7,8-TeBDF was ninefold (molar basis) more
potent than 2,3,7,8-TeCDF (Hornung et al., 1996b).
Considering the few data available on the higher halogenated
congeners and the wide range of potencies leading to the established
TEFs, the (preliminary) use of the same TEF values for the other
PBDD/PBDF or PXDD/PXDF congeners as described for the chlorinated
analogues appears to be justified.
Table 53. A comparative survey of several biological and toxicological parameters for 2,3,7,8-TeBDD and 2,3,7,8-TeCDD
(tested in parallel-running experiments)
Parameter Details TBDD TCDDa Reference
Kinetics
Absorption rat, oral route up to 80% _b Ivens et al. (1992);
Diliberto et al. (1993)
Metabolism rat about 7% about 10% Kedderis et al.
(1991b)
Half-life rat (faeces) 18 days _c Kedderis et al.
(1991a)
rat (adipose 56 days _d Kedderis et al.
tissue) (1991a)
human 3-11 years _d Zober et al. (1992)
Receptor binding incubation of 1.5 x 10-9 mol/litre 1.0 x 10.9 Mason et al. (1987b)
(EC50) cytosolic mol/litre
receptor protein
Microsomal enzyme
induction
Binding affinity rat liver 9.0 nmol 6.5 nmol Kedderis et al. (1993)
of CYP1A2
AHH induction rat liver 9.12 8.41 Mason et al. (1987b)
(pED50)f
EROD induction rat liver 9.45 8.16 Mason et al. (1987b)
(pED50)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
EROD induction rat liver Schulz-Schalge et al.
(molar basis) -after 7 days 6740 pmol resorufin/mg 5210 pmol resorufin/mg (1991a,b)g
protein per min protein per min
- after 98 days 410 pmol resorufin/mg 162 pmol resorufin/mg
protein per min protein per min
EROD induction rat liver identical dose-effect enzyme concentration Nagao et al.
(relative potency, and curves (1995/96)
molar basis)
EROD induction chick embryo liver 9.4 pmol/egg 11.1 pmol/egg Ramalingam et al.
(ED50) (1986)
EROD induction mouse liver 0.2 1 Birnbaum et al.
(relative potency, (subchronic exposure) (1993); Birnbaum &
molar basis) DeVito (1995)
ACOHh induction mouse liver 0.2 1 Birnbaum & DeVito
(relative potency, (subchronic exposure) (1995)
molar basis)
EROD induction mouse lung 0.1 1 Birnbaum & DeVito
(relative potency, (subchronic exposure) (1995)
molar basis)
EROD induction mouse skin 0.04 1 Birnbaum et al.
(relative potency, (subchronic exposure) (1993); Birnbaum &
molar basis) DeVito (1995)
EROD induction mouse liver 0.31 1 Van Birgelen et al.
(relative potency) (subchronic exposure) (1996)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
ACOH induction mouse liver 0.11 1 Van Birgelen et al.
(relative potency) (subchronic exposure) (1996)
Hepatic porphyrin mouse liver 0.4 1 Van Birgelen et al.
accumulation (subchronic exposure) (1996)
(relative potency)
Body weight loss rat 7.17 7.28 Mason et al. (1987b)
(pED50)
Immunotoxicity
Thymic atrophy rat 7.47 7.03 Mason et al. (1987b)
(pED50)
Changes in certain marmoset monkey 3 ng/kg body weight 1 ng/kg body weight Neubert (1993a)
lymphocyte (6 pmol/kg) (3 pmol/kg)
subpopulations
(NOEL)
Developmental toxicity
Cleft palate (ED50) mouse (NMRI) 62 ± 5 µg/kg body 24 ± 1 µg/kg body weight Nagao et al. (1990a)
weight (0.075 µmol/kg body
(0.123 pmol/kg body weight)
weight)
mouse (C57BL/6N) 65 µg/kg body weight (15 µg/kg body weight)i Birnbaum et al,
(1991)
Hydronephrosis mouse (C57BL/6N) 9 µg/kg body weight 4 µg/kg body weight Birnbaum et al.
(ED50) (estimated) (1991)
Table 53. (Continued)
Parameter Details TBDD TCDDa Reference
Tumorigenicity in vivo/in vitro 0.14 1 Massa et al. (1991)
(short-term test: assay with murine
relative macrophages
cell-transforming
potential, molar
basis)
Mortality Rainbow trout 1.14-2.54 1 Hornung et al.
(LD50, relative (early life stage) (1996b)
potency, molar
basis)
a - = not performed.
b Comparable data from other TCDD studies (WHO, 1989); 88% measured by Diliberto et al. (1996).
c Comparable data from other TCDD studies (references in Kedderis et al., 1991a; Weber et al., 1993).
d Less than values from other TCDD studies (17-25 days; references in Kedderis et al., 1991a).
e Comparable data from other TCDD studies (5-11 years, references cited in chapter 8).
f pED50 = -log ED50 (molar basis).
g Data in agreement with results of Abraham et al. (1988) and Nagao et al. (1990b).
h ACOH = acetanilide-4-hydroxylase.
i Measured by the same laboratory (Birnbaum et al., 1989).
j See chapter 9 for information about the study.
8. EFFECTS ON HUMANS
There is little information available on the effects of
PBDDs/PBDFs on human health. The main human health features discussed
in connection with PCDDs/PCDFs are immunotoxicity (Lorenzen & Okey,
1991; Vos, 1993), developmental toxicity (Sweeney, 1994),
neurotoxicity (Peper et al., 1993), carcinogenicity (Kogevinas et al.,
1993; Bertazzi & di Domenico, 1994; Hardell et al., 1994), and skin,
liver, and gastrointestinal toxicity (Mennear & Lee, 1994). Recently,
a brief critical review of the short- and long-term non-cancer health
effects of PCDDs/PCDFs was given by Sweeney et al. (1993). The
relevance of biochemical effects of PCDDs/PCDFs has also been
addressed (Lucier, 1991).
8.1 General population exposure
There are no data for the general population on exposure to or
effects of PBDDs/PBDFs.
8.2 Occupational/accidental exposure
Two cases of acute health problems due to 2,3,7,8-TeBDD/TeCDD
exposure have been described (see also section 5.3.2). The first case
refers to an American chemist who developed serious illness, including
chloracne, headaches, and back and leg pain, after synthesizing
TBDD/TCDD (Schecter, 1992). The second case was that of a Japanese
student who suffered from very severe acne-like eruptions on his
cheeks and chin following synthesis of PCDFs and PBDFs (Asahi & Urabe,
1987).
In the course of a morbidity study, male personnel of a chemical
plant with documented exposure to PBDDs/PBDFs (see section 5.3)
originating from the use of brominated flame retardants (OBDE and
DBDE) were subjected to a general health examination, including
special immunological tests (Zober et al., 1992). The measurements of
the exposed (n = 21; exposed for up to 13 years) and the control (n =
42; employees of a similar resin production plant but with no use of
PBDEs within the plant) groups included cellular and humoral
parameters (distribution of lymphocyte subsets, concentrations of
immunoglobulins, immune complexes, complements C3 and C4, and
antinuclear antibodies). Functional abnormalities of the immune system
were not investigated. Only one person with the highest TBDD/TBDF
burden (478/112 pg/g blood lipid) showed some changes (high
concentrations of complement C4; low total lymphocyte, T cell, T
helper cell, and natural killer cell counts) but did not have clinical
symptoms attributable to an immunodeficiency disease. The health
problems of this 54-year-old worker (hypertension, low back pain,
hyperuricaemia, and signs of an old inactive tuberculosis) were
thought to be not dioxin-dependent. No notable differences from the
control group were found with the other participants with lower blood
lipid levels (<208 pg TBDD/g blood lipid; <58 pg TBDF/g blood
lipid). Evaluating the group results as a whole, complement C4
concentrations increased significantly with increasing concentrations
of both TBDD and TBDF. Marginal associations independent of the person
with the highest exposure were seen with C3 concentrations (increased
with TBDF concentrations). However, retesting of the one person at a
later date showed that his C4 levels had dropped. Altogether, the
effects observed were not considered to be indicative of an impact of
PBDDs/PBDFs on the immune system.
A shortcoming of this study was a lack of measurements of
PBDD/PBDF concentrations (and concentrations of related compounds,
such as PCDDs/PCDFs or PCBs) in the control group. In addition, the
internal exposure was related to only two congeners, namely TBDD and
TBDF, although a lot of other homologues were identified at the
corresponding workplace (see section 5.3.1). This was partly due to
analytical limitations (lack of reference substances) and partly due
to the high weighting factor of TBDD in toxic equivalent (TEQ)
calculations.
Additional clinical laboratory tests (liver function tests, lipid
and glucose measures, thyroid function parameters, haematological and
coagulation indicators) were performed within the same study cohort.
They did not reveal any remarkable differences between the exposed
(n = 38 - 42) and the control (n = 40 - 42) groups (Ott & Zober,
1996).
Although there are indications for excesses of cancer mortality
seen in workers exposed to TCDD (e.g. Zober et al., 1990; Fingerhut et
al., 1991; Manz et al., 1991; Becher et al., 1996), there are no
reports of cancer mortality caused by PBDDs/PBDFs.
8.3 Subpopulations at special risk
No data are available with regard to the effects of PBDDs/PBDFs
on high-risk subpopulations.
As discussed for PCDDs/PCDFs (Helge, 1993), fetuses, newborn
babies, and children are at a higher risk of exposure for several
reasons (placental and milk transfer, contact with soil and dust,
immunological and physiological immaturity, lack of fat depots, etc.),
and developing systems, based on experimental evidence, may be more
sensitive to the adverse effects of halogenated aromatic compounds
than developed systems of adults.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
There are limited reports on the effects of PBDDs/PBDFs on
microorganisms, plants or invertebrate species. Regarding vertebrates,
data from a fish early life stage mortality bioassay are available
(Hornung et al., 1996a,b).
Newly fertilized rainbow trout eggs were injected with a series
of PBDDs, PXDDs, and PBDFs (see Table 54). All congeners tested but
2,7-DiBDF caused mortality in the rainbow trout sac fry by a blue sac
syndrome. The signs of toxicity were identical to those produced by
TCDD and included yolk sac oedema, pericardial oedema, multifocal
haemorrhages, reduced growth, and craniofacial malformations.
2,3,7,8-TeBDD showed the highest potency among the brominated
congeners and was also more potent than 2,3,7,8-TeCDD. Similarly,
2,3,7,8-TeBDF was more potent than 2,3,7,8-TeCDD, whereas the other
PBDDs/PBDFs were equipotent (or less potent) than identically
substituted PCDDs/PCDFs (Hornung et al., 1996b).
The interactions between pairs of PBDD and PBDF congeners were
also studied using the rainbow trout sac fry (early life stage)
mortality bioassay (Hornung et al., 1996a). The tested pairs were as
follows: 2,3,7,8-TeBDD and 1,2,3,7,8-PeBDF; 2,3,7,8-TeBDD and
1,2,3,7,8-PeBDD; and 1,2,3,7,8-PeBDF and 1,2,3,7,8-PeBDD. As with the
individual PBDD and PBDF congeners, their mixtures also produced
TCDD-like toxicity and mortality in the rainbow trout sac fry. The
rank order for LD50 in the individual congeners tested, from lowest to
highest, was as follows: 2,3,7,8-TeBDD < 1,2,3,7,8-PeBDD
< 1,2,3,7,8-PeBDF. The interactions between each of the tested pairs
were additive in causing sac fry mortality.
Laboratory studies with the related PCDDs/PCDFs showed adverse
effects on fish and avian species, especially on the early life stages
(Cook et al., 1991; Walker & Peterson, 1991, 1994; Peterson, 1993;
Peterson et al., 1993). Correlations between environmental exposure to
polyhalogenated aromatics (PCDDs/PCDFs, PCBs) and the decline of some
populations of marine and freshwater mammals, fish, and fish-eating
birds or symptoms such as reproductive dysfunction or other lesions
were investigated in a number of field studies (Gilbertson, 1989;
Rappe, 1993).
Recent results of the research on the effects of
dibenzo- p-dioxins and dioxin-like compounds on wildlife support the
conclusion that there are real-world adverse effects on wildlife
caused by these compounds. Although PCDD/PCDF have contributed to
these effects, in most locations the major effects are due to the
dioxin-like non- and mono-ortho-substituted PCB congeners. Effects
have been reported in mammals and birds as well as in fish. While the
current effects are subtle, there is no assimilative capacity for TEQs
in the global environment. Trends in TEQs in industrialized regions
are continuing to decline. Concentrations of TEQs in remote areas,
such as the Arctic and open ocean, may not yet have reached their
maximum. The contribution of PBDDs/PBDFs to these effects is unknown
(Giesy et al., 1994; Fiedler & Van den Berg, 1996).
Table 54. Early life stage mortality in rainbow trout (Oncorhynchus
mykiss) caused by PBDDs, PXDDs, and PBDFsa
Congener Blue sac syndromeb LD50c Rainbow trout
(ng/g egg) strain
Dibenzo-p-dioxins
2,3,7-TrBDD + 18.9 Erwin
+ 15.6 McConoughy
2,3,7,8-TeBDDd + 0.222 Eagle Lake
+ 0.264 Eagle Lake
+ 0.158 Erwin
+ 0.122 Arlee
1,3,7,8-TeBDD + 29 Erwin
1,2,3,7,8-PeBDD + 4.16 Eagle Lake
+ 4.92 Erwin
1,2,3,4,7,8-HxBDD + 63.7 Arlee
2,8-Cl2-3,7-Br2-DD + 0.448 Erwin
2,3,7-Cl3-8-Br1-DD + 0.410 Erwin
Dibenzofurans
2,7-DiBDF - >597e Erwin
2,3,7,8-TeBDF + 1.5 Erwin
2,3,4,7,8-PeBDF + 6.19 Erwin
1,2,3,7,8-PeBDF + 9.56 Erwin
1,2,3,4,7,8-HxBDF + 247 Erwin
a Modified from Hornung et al. (1996b).
Table 54 (Continued)
b TCDD-like toxicity grossly identical to blue sac syndrome was
characterized by sac fry mortality that was preceded by yolk sac
oedema, pericardial oedema, multifocal haemorrhages, growth
retardation, and craniofacial malformations.
c Based on cumulative hatching and sac fry mortality (for fiducial
limits, see original); eggs (n = 30 per dose) injected with
seven graded doses of congener incorporated into phosphatidylcholine
liposomes.
d For comparison: LD50 values for 2,3,7,8-TeCDD: 0.171 ng/g egg
(Shasta strain); 0.374 ng/g egg (Adee strain) (Walker & Peterson,
1991; Zabel et al., 1995).
e No signs of toxicity occurred at the highest egg dose tested.
10. EVALUATION OF HUMAN HEALTH RISKS AND EFFECTS ON THE ENVIRONMENT
There is much less information on PBDDs/PBDFs than on their
chlorinated analogues. The analytical methods for separating and
identifying the individual brominated congeners are less advanced than
those for their chlorinated analogues, and only a few standards are
available. Present analytical methods are able to quantify total
brominated homologue groups and are able to detect but not quantify
the mixed brominated/chlorinated congeners.
10.1 Hazard evaluation
Data on bioconcentration and biomagnification are lacking. The
physicochemical properties of PBDDs/PBDFs and extrapolations from
PCDDs/PCDFs suppose that PBDDs/PBDFs are enriched in carbon- and
fat-rich environmental compartments in a manner similar to
PCDDs/PCDFs. Atmospheric transport occurs in the vapour and
particulate phases, the ratio depending on molecular weight.
Photochemical degradation of PBDDs/PBDFs and PXDDs/ PXDFs
adsorbed on surfaces was much slower than that in organic solvents.
Under environmental conditions, debromination was found to be a slow
process (several months in soil).
From the available data, it is apparent that PBDDs/PBDFs have a
toxic potential similar to that of PCDDs/PCDFs.
Generally, the studies performed with 2,3,7,8-substituted
PBDDs/PBDFs showed typical TCDD-like effects in experimental animals
and a persistence in animal and possibly human tissues apparently
similar to that seen with PCDDs/PCDFs. For some end-points,
quantitative differences were found. For example, under certain
experimental conditions, 2,3,7,8-TeBDF was more potent than
2,3,7,8-TeCDF in inducing cleft palate and hydronephrosis in mice
following prenatal exposure. 2,3,7,8-TeBDD was somewhat less potent
than its chlorinated counterpart with regard to short-term toxicity,
cell-transforming capacity, antiestrogenic activity, and decreasing
vitamin A storage. Both brominated congeners had longer elimination
half-lives in rat adipose tissues than the chlorinated ones. More or
less comparable activities of these and some other PBDDs/PBDFs with
their chlorinated analogues were seen for receptor binding, enzyme
induction, and immunotoxic effects.
The few data available on trisubstituted PBDDs suggest a higher
metabolic rate and lower short-term toxicity compared with the
2,3,7,8-substituted congeners. Limited data are available for other
non-2,3,7,8-substituted PBDD/PBDF congeners. Studies with
1,2,7,8-TeBDF demonstrated higher rates of metabolic elimination in
rats compared with 2,3,7,8-TeCDF or 2,3,7,8-TeBDD.
Long-term toxicity studies as well as perinatal exposure and
multigeneration studies are also lacking for the 2,3,7,8-substituted
PBDDs/PBDFs. Nevertheless, the possibility of adverse effects - for
example, carcinogenicity, reproductive/developmental toxicity, and
neurotoxicity - cannot be excluded, based on the similarity in results
obtained in short-term studies with PBDDs/PBDFs compared with
PCDDs/PCDFs. However, in a comparison of relative potencies,
2,3,7,8-TeCDD was approximately two times more potent than
2,3,7,8-TeBDD in inducing the accumulation of total hepatic porphyrin
in mice following subchronic (13-week) exposure.
Congeners with a high degree of bromination (>penta) were
usually not included in toxicity studies (owing to a lack of pure
congeners), although they were detectable in workplace air and other
samples.
Exposure to single congeners of PBDDs/PBDFs and PXDDs/ PXDFs has
resulted in TCDD-like toxic effects and mortality in the rainbow trout
sac fry early life stage mortality bioassay. Pairs of PBDDs/PBDFs have
been shown to have additive effects on the mortality of rainbow trout
fry. In these bioassay studies, single exposures to 2,3,7,8-TeBDD and
2,3,7,8-TeBDF were more potent than single exposures to the
chlorinated analogues.
There are no pertinent data on microbial degradation.
Photochemical debromination is thought to be the major transformation
process for PBDDs/PBDFs and PXDDs/PXDFs. As was shown in laboratory
experiments, PBDDs/PBDFs and PXDDs/PXDFs were degraded in organic
solvents after irradiation with sunlight or UV light. The easier loss
of bromine than of chlorine resulted in the formation of the
PCDDs/PCDFs when PXDDs/PXDFs were exposed to light.
10.2 Exposure evaluation
As PBDDs/PBDFs are not known to occur naturally, their presence
is indicative of thermal or photolytic degradation or thermal
transformation of brominated chemicals, many of which are used as
flame retardants. The occurrence of such chemicals in consumer
products (e.g. electrical appliances, textiles, petrol) may, in some
cases, result in a risk for the population.
The database on the occurrence of PBDDs/PBDFs in ambient air and
dust is too small for detailed comparisons with PCDDs/PCDFs and
estimations of their impact on human health. Only one
2,3,7,8-substituted PBDF congener was detected in ambient air.
Automobile exhaust was found to be a diffuse source of PBDDs/PBDFs and
PXDDs/PXDFs (along with PCDDs/PCDFs), if halogenated scavengers are
used in the petrol.
Because typical municipal waste generally contains much more
chlorine than bromine, the formation of mixed and completely
chlorinated compounds from brominated precursors is possible and was
confirmed experimentally. Modern waste incinerators are capable of
realizing very low emissions of PCDDs/PCDFs; however, this modern
technology is not applied in all countries.
Emissions (and effluents) of PBDDs/PBDFs into the environment
from plants (processing organic chemicals, recycling plastics and
metals), waste incinerators, etc. are hardly documented, but may vary
depending on the industrial hygienic standards (including waste
disposal practices). A limit value of 0.1 ng TEQ/m3 for PCDDs/PCDFs in
exhaust gases of municipal waste incinerators was established by some
countries.
A critical source for release of PBDDs/PBDFs and PXDDs/ PXDFs
into the environment is accidental fires of materials containing
brominated compounds. Concentrations measured depend on the specific
fire situation and can be very high. The highest concentrations are
found in the solid residues.
PBDDs/PBDFs have been found in indoor air (maximum PBDF sum
concentration, up to hepta: 1.27 pg/m3) and dust samples of rooms
equipped with electronic appliances and in house dust. Whereas
2,3,7,8-substituted congeners were not detected in the air samples,
they were detectable in dust samples (tetra- to pentaBDFs: up to 0.07
µg/kg). Dust collected in computer rooms contained comparable amounts
of PBDDs/PBDFs and PCDDs/PCDFs (e.g. maximum sum concentrations: each
about 5 µg/kg).
Potential exposure pathways for the general population, clean-up
personnel, and fire personnel may originate from accidental fires
where bromine-containing plastics are involved. Gas, smoke, and
residue samples as well as firemen's trousers were found to contain
PBDDs/PBDFs (mainly PBDFs), including a portion of up to 20%
2,3,7,8-substituted congeners, and PXDDs/PXDFs. Generally, the
analyses of fire incidents showed a wide variation of contamination in
the µg/kg (residue), ng/m2 (smoke condensate), or ng/m3 (gas) range,
with very high peak concentrations possible. In several residue
samples, the sum concentrations of seven 2,3,7,8-substituted congeners
(2,3,7,8-TeBDF, 2,3,4,7,8-PeBDF, and five tetra- to hexaBDDs) were
greater than 5 µg/kg. In one television fire incident, the sum
concentrations of these congeners in residues amounted to 1100 µg/kg.
Additionally, this sample contained high concentrations of
1,2,3,7,8-PeBDF (1860 µg/kg) and of 1,2,3,4,7,8-HxBDF (1900 µg/kg).
PBDF concentrations measured at experimental fires generally were
higher than those measured at real fires and may be considered as
worst-case examples. The area contaminant concentrations of PBDFs
resulting from experimental or real television fires were in a similar
range as found for PCDDs/PCDFs after fires involving their precursors.
Fires involving televisions or computers may produce higher PBDD/PBDF
than PCDD/PCDF concentrations - for example, maximum sum
concentrations of 5600 µg/kg (13 µg TEQa/kg) versus 320 µg/kg (2.3 µg
I-TEQ/kg) in soot samples after a fire in a computer room.
a Calculated by Schacht et al. (1995) using PCDD/PCDF I-TEFs for
the brominated congeners.
Potential issues of concern are fires and suspected leaching
processes at waste disposal sites (additional presence of organic
solvents and other contaminants).
One study reported on the analysis of PBDDs/PBDFs and PXDDs/PXDFs
in a sample of muscle from a salmon from the Baltic Sea and a pooled
sample of human milk from Sweden. The concentrations were found to be
below the detection limit (0.3 ng 2,3,7,8-TeBDF/kg and 0.4 ng
2,3,7,8-TeBDD/kg), suggesting a very low exposure to PBDDs/PBDFs and
PXDDs/PXDFs. On the other hand, a series of 2,3,7,8-substituted
PCDDs/PCDFs were detected in these samples.
Workplaces identified as involving a risk of exposure to
PBDDs/PBDFs (and PXDDs/PXDFs) include mainly those in the plastic and
recycling industry using brominated flame retardants or products
containing them and those of firemen and clean-up personnel associated
with fires.
Recent monitoring data from three plastic plants showed
concentrations between 260 and >10 000 pg/m3 (sum of mono- to
hexaBDDs/BDFs). Concentrations of eight 2,3,7,8-substituted congeners
(three tetra- to pentaBDFs and five tetra- to hexaBDDs) ranged from
0.11 to 18 pg TEQb/m3 at permanently operated workplaces. At
periodically operated workplaces (maximal stay: 1 h/day), a maximum
concentration of the eight congeners of 954 pg TEQb/m3 has been
measured. There are no occupational threshold limits for PBDDs/PBDFs.
PBDD/PBDF concentrations currently measured in plastics, recycled
products, electronic scrap, and other waste samples were considerable.
In view of the growing worldwide production and use of brominated
flame retardants (estimated worldwide demand in 1992: 150 000 tonnes;
OECD, 1994) as additives to a series of polymers, it can be assumed
that the amount of bromine-containing waste will be increasing in the
future. In particular, electronic scrap from casings and printed
circuit boards of computers, etc., flame-retarded with brominated
compounds will reach the waste streams and then be a potentially major
source of PBDDs/PBDFs (and PXDDs/PXDFs and PCDDs/PCDFs).
A potential hazard can arise in chemical research laboratories
performing special syntheses. Distillation residues, other wastes, and
equipment were found to be contaminated by PBDDs/PBDFs and/or
PXDDs/PXDFs.
Few human monitoring data are available. One study published
blood monitoring data of personnel from an industry using PBDEs. The
analyses showed (1) uptake of PBDD/PBDF congeners, (2) the presence of
2,3,7,8-substituted congeners, which were not or hardly detectable in
the corresponding workplace air samples, and (3) estimated half-lives
typical for dioxin-like compounds.
b Calculated by Kieper (1996) using PCDD/PCDF I-TEFs for the
brominated congeners.
The main route of human exposure to PCDDs/PCDFs for the general
population is via food intake (more than 95%). Whereas the presence of
PCDDs/PCDFs was confirmed in most foodstuffs, PBDDs/PBDFs (>tetra)
could only be detected but not quantified in shellfish, fish, and
cow's milk. 2,3,7,8-Substituted congeners could not be identified.
For the general environment, it was found that the concentrations
of PBDDs/PBDFs are much lower than those of the chlorinated analogues
(tetra- through octahalogenated), the homologues with four or more
bromine atoms being hardly detected in environmental samples. At
present, there are only few data, but these data do not indicate an
accumulation of PBDDs/PBDFs or PXDDs/PXDFs along the terrestrial or
the aquatic food-chain. However, the limited data set does not allow
for environmental trend analysis.
Currently, most PBDDs/PBDFs and their precursors are bound into
products and have not yet reached waste streams and the environment.
10.3 Risk evaluation
PBDDs/PBDFs have been detected in air, dust, soil, sediment,
sewage sludge, grass, and fish, but not in the general human
population. This limited occurrence is in contrast to the ubiquitous
PCDDs/PCDFs. Generally, concentrations measured were low.
The major risk group for exposure to PBDDs/PBDFs has apparently
been workers involved in the production and application of brominated
flame retardants (e.g. extruder personnel). In these persons, clearly
increased body burdens of 2,3,7,8-TeBDD and 2,3,7,8-TeBDF have been
measured. Another possible source of exposure may be automobile
exhaust, as a result of incomplete combustion of bromine-containing
materials. No data on increased body burdens have been reported (and
apparently body burdens have not been measured). For the general
population, there seems to be a very low risk of exposure, compared
with the risk of exposure to PCDDs/PCDFs. In the few samples measured
(breast milk), no PBDDs/PBDFs were detected, whereas at least 100
times higher PCDD/PCDF levels were present.
Within the group of workers with clearly increased PBDD/PBDF body
burdens, no clinical adverse health effects were reported, and only a
few data for laboratory values of the volunteers were outside the
normal reference range. Although no exact data on elimination
half-lives have been reported for humans, the data available indicated
a considerable persistence of the 2,3,7,8-TeBDD/TeBDF congeners within
the human organism.
From all the information currently available, it can be concluded
that the potential of the PBDDs/PBDFs for biological (e.g. enzyme
induction) and toxic actions is very similar to that of the
PCDDs/PCDFs.
It is difficult to compare the potency of PBDDs/PBDFs with that
of their chlorinated analogues, as the database for the individual
PBDDs/PBDFs is very small, with respect to both data from animal
studies and observations in humans. With a few exceptions in most of
the systems tested, the few evaluated PBDD/PBDF congeners were less
potent than the corresponding chlorinated congeners. Kinetically,
however, some of the brominated substances exhibited a higher
persistence within the mammalian organism compared with the
corresponding chlorinated substances. Therefore, as judged from data
on 2,3,7,8-TeBDD, a TEF of 1.0 (equal to that of TCDD) would represent
a conservative approach. For 2,3,7,8-TeBDF, a somewhat higher TEF may
be suggested, as the substance has been shown in rodents to have a
clearly longer elimination half-life compared with 2,3,7,8-TeCDF; a
TEF of 0.2 - 0.3 may be justified.
The current limited experimental database does not allow a
complete hazard assessment and the recommendation of a safe level of
exposure to PBDDs/PBDFs for the general population.
However, if a comparison of the health impact of PBDDs/PBDFs with
their chlorinated analogues is needed, the data published on
2,3,7,8-TeBDD may be taken as an example. For 2,3,7,8-TeBDD, a NOAEL
of 10 ng/kg body weight per day may be established in a 13-week study
in rats. This value compares with the NOAEL of 10 ng/kg body weight
per day for 2,3,7,8-TeCDD, as derived from the 13-week study in rats.
The very limited data set available for concentrations of
PBDDs/PBDFs in environmental compartments makes it impossible to
conduct a proper risk evaluation for the environment. However, the few
data on levels of these substances indicate that they are much lower
than levels of their chlorinated counterparts. The assumption that
both brominated and chlorinated dibenzo- p-dioxins and dibenzofurans
act through a common mechanism and that their potency is not greatly
dependent on the nature of the halogen atom (chlorine or bromine) will
lead to the conclusion that the PBDDs/PBDFs will contribute marginally
to the total "dioxin" effect.
11. CONCLUSIONS AND RECOMMENDATIONS FOR PROTECTION OF HUMAN HEALTH
AND THE ENVIRONMENT
11.1 Conclusions
PBDDs/PBDFs are contaminants that are more or less similar to
PCDDs/PCDFs in their persistence and toxicity. Therefore, humans and
the environment should be protected from them. Exposure of the general
population to PCDDs/PCDFs appears to be greater than exposure to
PBDDs/PBDFs. Limited biomonitoring information indicates very low
residues, compared with PCDDs/PCDFs. Brominated flame retardants and
their precursors appear to be a main source of PBDDs/PBDFs.
A limited experimental database exists for PBDDs/PBDFs and would
therefore exclude an attempt at a complete hazard identification.
Current information does not allow a quantitative risk assessment,
although toxicological similarities appear to exist between certain
PBDD/PBDF congeners and their corresponding chlorinated homologues. On
an interim basis, it is suggested that current I-TEFs for the 17
2,3,7,8-substituted PCDD/PCDF congeners be applied to the comparable
brominated and mixed halogenated congeners.
11.2 Recommendations
Owing to the accumulating and toxic potential of some
PBDDs/PBDFs, every effort should be made to prevent exposure of humans
to, and pollution of the environment by, these compounds.
Brominated flame retardants should not be used where suitable
replacements are available, and future efforts should encourage the
development of further substitutes.
Appropriate precautions, including monitoring, should be taken
both to protect workers from exposure to PBDDs/PBDFs and to prevent
their release into the environment in emissions and effluents.
Disposal of industrial wastes, fire residues, and consumer
products containing brominated compounds should be controlled to
minimize environmental contamination by PBDDs/PBDFs and their
precursors. All products flame-retarded with bromine compounds should
be labelled and disposed of only in properly constituted waste
incinerators working at consistent operating conditions, to avoid the
release of PBDDs/PBDFs.
The use of leaded petrol, which necessitates the use of
halogenated scavengers, should be avoided.
Selected PBDD/PBDF congeners (2,3,7,8-TeBDD/TeBDF) should be
included in ongoing dioxin monitoring programmes to enhance the
existing database.
12. FURTHER RESEARCH
Analytical methods, including screening techniques, should be
improved. Interlaboratory comparisons should be undertaken to validate
methodologies.
As the experimental database is limited, comparative
toxicological and ecotoxicological studies with selected PBDD/PBDF
congeners should be performed with respect to both identifying
appropriate adverse- and no-adverse-effect levels and improving the
interim TEF recommendation.
13. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
There have been no previous evaluations by international bodies.
(A toxicological evaluation of PBDDs/PBDFs was prepared by the
German Federal Health Office [Appel, 1991, 1993].)
REFERENCES
Abraham K, Krowke R, & Neubert D (1988) Pharmacokinetics and
biological activity of 2,3,7,8-tetrachlorodibenzo-para-dioxin: 1.
Dose-dependent tissue distribution and induction of hepatic
ethoxyresorufin O-deethylase in rats following a single injection.
Arch Toxicol, 62: 359-368.
Abraham K, Wiesmüller T, Brunner H, Krowke R, Hagenmaier H, & Neubert
D (1989) Elimination of various polychlorinated dibenzo- p-dioxins
and dibenzofurans (PCDDs and PCDFs) in rat faeces. Arch Toxicol, 63:
75-78.
Acharya P, DeCicco SG, & Novak RG (1991) Factors that can influence
and control the emissions of dioxins and furans from hazardous waste
incinerators. J Air Waste Manage Assoc, 41: 1605-1615.
Adams EM, Irish DD, Spencer HC, & Rove VK (1941) The response of
rabbit skin to compounds reported to have caused acneform dermatitis.
Ind Med Surg, 2: 1-4.
Ahlborg UG (1994) Toxic equivalency factors for polychlorinated
dioxins, dibenzofurans and biphenyls -- pro's and con's and
consequences. In: Fiedler H, Hutzinger O, Clement R, & Sakai S ed.
Dioxin'94: 14th International Symposium on Chlorinated Dioxins, PCB
and Related Compounds, Kyoto, Japan, 21-25 November 1994 (Short
papers). Kyoto, Kyoto University, Department of Environmental and
Sanitary Engineering, pp 11-14 (Organohalogen Compounds, Volume 21).
Ahlborg U, Brouwer A, Fingerhut M, Jacobson J, Jacobson S, Kennedy S,
Kettrup A, Koeman J, Poiger H, Rappe C, Safe S, Seegal R, Tuomisto J,
& Van den Berg M (1992) Impact of polychlorinated dibenzo- p-dioxins,
dibenzofurans, and biphenyls on human and environmental health, with
special emphasis on application of the toxic equivalency factor
concept. Eur J Pharmacol, 228: 179-199.
Alsabbagh AM, Aldous KM, Narang RS, & O'Keefe PW (1992) Formation of
brominated dioxins and brominated phenazines from pyrolyses of
2,4,6-tribromoaniline and N-(tribromophenyl) maleimide. Chemosphere,
24: 1625-1632.
Andersen ME, Mills JJ, Gargas ML, Kedderis L, Birnbaum LS, Neubert D,
& Greenlee WF (1993) Modeling receptor-mediated processes with dioxin:
implications for pharmacokinetics and risk assessment. Risk Anal, 13:
25-36.
Anon (1996) German ordinance on forbidden chemicals.
Bundesgesetzblatt, 1(39): 1151-1166.
Appel KE (1991) [Polybrominated dibenzodioxins and dibenzofurans:
Toxicological evaluation.] Bundesgesundheitsblatt, 10: 460-470 (in
German).
Appel KE (1993) Polybrominated dibenzodioxins and dibenzofurans:
Toxicological evaluation. In: The Toxicology Forum: Current views on
the impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1992. Washington DC, Toxicology Forum, Inc., pp
147-174.
Asahi M & Urabe H (1987) A case of "Yusho"-like skin eruptions due to
halogenated PCB-analogue compounds. Chemosphere, 16: 2069-2072.
ATOCHEM (1987) Decabromobiphenyl (Adine 0102): Pyrolysis testing.
Paris La Defense, France, ATOCHEM, 7 pp (Unpublished report).
ATOCHEM (1990) Decabromobiphenyl (Adine 0102). Paris La Defense,
France, ATOCHEM, 3 pp (Unpublished report).
Bacher R, Riehle U, Swerev M, & Ballschmiter K (1991) Patterns and
levels of polyhalogenated (Br-, Cl-) dibenzodioxins and dibenzofurans
in automobile traffic related samples. Chemosphere, 23: 1151-1171.
Ballschmiter KH & Bacher R (1996) [Dioxins: Chemistry, analysis,
occurrence, environmental behaviour and toxicology of halogenated
dibenzo- p-dioxins and dibenzofurans.] Weinheim, Germany, VCH
Publishers, 507 pp (in German).
Ballschmiter K, Bacher R, Riehle U, & Swerev M (1990) [Environmental
research report: Contribution of automobile exhaust to the general
environmental burden through dibenzodioxins (PHaIDD) and dibenzofurans
(PHaIDF).] Bonn, German Federal Ministry for Research and Technology,
234 pp (in German).
Banks YB & Birnbaum LS (1991) Absorption of
2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) after low dose dermal
exposure. Toxicol Appl Pharmacol, 107: 302-310.
Bannasch P, Wayss K, & Zerban H (1985) Peliosis hepatis, rodents. In:
Jones TC, Mohr U, & Hunt RD ed. Pathology of laboratory animals,
digestive system. Berlin, Springer Verlag, pp 110-115 (ILSI
Monograph).
Becher H, Flesch-Janys D, Kauppinen T, Kogevinas M, Steindorf K, Manz
A, & Wahrendorf J (1996) Cancer mortality in German male workers
exposed to phenoxy herbicides and dioxins. Cancer Causes Control, 7:
312-321.
Bergqvist PA, Strandberg B, Bergek S, & Rappe S (1993) Lipid reduction
during the analyses of PCDDs, PCDFs and PCBs in environmental samples
using semipermeable membrane technique. Organohalogen Compds, 11:
41-44.
Bertazzi PA & di Domenico A (1994) Chemical, environmental and health
aspects of the Seveso, Italy, accident. In: Schecter A ed. Dioxins and
health. New York, London, Plenum Press, pp 587-632.
Bieniek D, Bahadir M, & Korte F (1989) Formation of heterocyclic
hazardous compounds by thermal degradation of organic compounds.
Heterocycles, 28: 719-722.
Bingham AG, Edmunds CJ, Graham BWL, & Jones MT (1989) Determination of
PCDDs and PCDFs in car exhaust. Chemosphere, 19: 669-673.
Birla P & Kamens RM (1994) Effect of combustion temperature on the
atmospheric stability of polybrominated dibenzo- p-dioxins and
dibenzofurans. Environ Sci Technol, 28: 1437-1443.
Birnbaum LS (1993) EPA's reassessment of dioxin risk: Directed health
research. Chemosphere, 27: 469-475.
Birnbaum LS (1994) The mechanism of dioxin toxicity: Relationship to
risk assessment. Environ Health Perspect, 102: 157-167.
Birnbaum LS & DeVito MJ (1995) Use of toxic equivalency factors for
risk assessment for dioxins and related compounds. Toxicology, 105:
391-401.
Birnbaum LS, Harris MW, Stocking LM, Clark AM, & Morrissey RE (1989)
Retinoic acid and 2,3,7,8-tetrachlorodibenzo- p-dioxin selectively
enhance teratogenesis in C57BL/6N mice. Toxicol Appl Pharmacol, 98:
487-500.
Birnbaum LS, Morrissey RE, & Harris MW (1991) Teratogenic effects of
2,3,7,8-tetrabromodibenzo- p-dioxin and three polybrominated
dibenzofurans in C57BL/6N mice. Toxicol Appl Pharmacol, 107: 141-152.
Birnbaum LS, Ross DG, & DeVito MJ (1993) Dose response relationships
for EROD induction in liver, lung and skin for dioxin and
dibenzofurans. In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss
A, & Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 237-240 (Organohalogen
Compounds, Volume 13).
Blankenburg G, Hutzinger O, & Neubert D (1990) Preliminary assessment
of the potency of 2,3,7,8-tetra(bromo/chloro)dibenzo- p-dioxins to
induce EROD activity in primary cell cultures of rat hepatocytes. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 77-81 (Organohalogen Compounds, Volume
4).
BMU (1989) [Information from the Federal Minister for the Environment,
Nature Conservation and Nuclear Safety. Polybrominated dibenzodioxins
and dibenzofurans (PBDD/PBDF) from brominated flame retardants: Risk
assessment and suggestions for action.] Bonn, Federal Ministry for the
Environment, Nature Conservation and Nuclear Safety, 245 pp (in
German).
Bonilla JV, Munro HE, Mitchum RK, & Bauer MR (1990) Analysis of
brominated dibenzo- p-dioxins and dibenzofurans in ABS resins
containing brominated flame retardants. J Fire Sci, 8: 395-404.
Boyd SA & Mortland MM (1985) Dioxin radical formation and
polymerization on Cu(II) smectite. Nature (Lond), 316: 532-535.
Bradlaw JA & Casterline JL Jr (1979) Induction of enzyme activity in
cell culture: A rapid screen for detection of planar polychlorinated
organic compounds. J Assoc Off Anal Chem, 62: 904-916.
Bradlaw JA, Garthoff LH, Hurley NE, & Firestone D (1980) Comparative
induction of aryl hydrocarbon hydroxylase activity in vitro by
analogues of dibenzo- p-dioxin. Food Cosmet Toxicol, 18: 627-636.
Brenner KS (1993) Polystyrene/- and extruded polystyrene foam
(XPS)/-hexabromo-cyclododecane-blends under thermolytic stress; PBDD &
PBDF-determination. In: Fiedler H, Frank H, Hutzinger O, Parzefall W,
Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Environmental Agency, pp 381-386
(Organohalogen Compounds, Volume 11).
Brenner KS & Knies H (1990) Formation of polybrominated dibenzofurans
(PBDF's) and -dioxins (PBDD's) during extrusion production of a
polybutyleneterphthalate (PBTP)/glassfibre resin blended with
decabromodiphenylether (DBDPE)/Sb2O3: Product and workplace
analysis. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 319-325 (Organohalogen
Compounds, Volume 2).
Brenner KS & Knies H (1992) Workplace monitoring of PBDF's and PBDD's
during extrusion production and injection molding of a
polybutyleneterephthalate(PBTP)/glass fibre/tetrabromo bisphenol A
carbonate oligomer (BC52)/Sb2O3-resin. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 39-32 (Organohalogen Compounds, Volume 9).
Brenner KS & Knies H (1993a) Workplace monitoring of polybrominated
dibenzofurans (PBDF's) and -dioxins (PBDD's) during extrusion
production and molding of a poly (butylene-terephthalate) (PBTP)/glass
fibre resin blended with tetrabromobisphenol A carbonate oligomer (BC
52)/Sb2O3; air sampling train and product analysis. Toxicol Environ
Chem, 38: 81-94.
Brenner KS & Knies H (1993b) Workplace monitoring of PBDFs and PBDDs
during extrusion production and injection molding of a
polybutyleneterephthalate (PBTP)/glass fiber/tetrabromo-bisphenol A
carbonate oligomer (BC52*)/Sb2O3-resin: Part II. Chemosphere, 26:
1953-1963.
Brenner KS & Knies H (1994) Workplace monitoring of polybrominated
dibenzofurans (PBDF's) and -dioxins (PBDDs), Part III Extruder
production and injection molding of bis-tetrabromophthalimide ethylene
(TBPI)/Sb2O3-blended polybutylene-terephthalate (PBTB). In: Fiedler
H, Hutzinger O, Clement R, & Sakai S ed.Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 335-340
(Organohalogen Compounds, Volume 20).
Bretthauer EW, Kraus HW, & di Domenico A (1991) Dioxin perspectives: A
pilot study on international information exchange on dioxins and
related compounds, Volume 16. New York, London, Plenum Press, 772 pp.
Brewster DW, Banks YB, Clark AM, & Birnbaum LS (1989) Comparative
dermal absorption of 2,3,7,8-tetrachlorodibenzo- p-dioxin and three
polychlorinated dibenzofurans. Toxicol Appl Pharmacol, 97: 156-166.
Bröker G (1996) Comparative and validation PCDD/F-measurements with
different sampling systems. Organohalogen Compds, 27: 433-439.
Bruckmann P, Hackhe K, Ball M, Lis A, & Päpke O (1990) Degassing of
PBDD/PBDFs from a television set -- PBDD/PBDF levels after a fire in a
stock house -- two case studies. In: Freiji L ed. Proceedings of the
Workshop on Brominated Aromatic Flame Retardants, Skokloster, Sweden,
24-26 October 1989. Solna, National Chemicals Inspectorate (KEMI), pp
99-104.
Buckley LA (1995) Biologically-based models of dioxin
pharmacokinetics. Toxicology, 102: 125-131.
Bumpus JA (1989) Biodegradation of polycyclic aromatic hydrocarbons by
Phanerochaete chrysosporium. Appl Environ Microbiol, 55: 154-158.
Buser HR (1986a) Polybrominated dibenzofurans and dibenzo- p-dioxins:
Thermal reaction products of polybrominated diphenyl ether flame
retardants. Environ Sci Technol, 20: 404-408.
Buser HR (1986b) Selective detection of brominated aromatic compounds
using gas chromatography/negative chemical ionization mass
spectrometry. Anal Chem, 58: 2913-2919.
Buser HR (1987a) Brominated and brominated/chlorinated dibenzodioxins
and dibenzofurans: Potential environmental contaminants. Chemosphere,
16: 713-732.
Buser HR (1987b) [Brominated and mixed brominated/chlorinated
dibenzodioxins and dibenzofurans.] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
243-256 (VDI-Report No. 634) (in German).
Buser HR (1987c) Brominated and brominated/chlorinated dibenzodioxins
and dibenzofurans: Potential environmental contaminants. Chemosphere,
16: 1873-1876.
Buser HR (1988) Rapid photolytic decomposition of brominated and
brominated/chlorinated dibenzodioxins and dibenzofurans. Chemosphere,
17: 889-903.
Buser HR (1991) Review of methods of analysis for polychlorinated
dibenzodioxins and dibenzofurans. In: Rappe C, Buser HR, Dodet B, &
O'Neill IK ed. Environmental carcinogens: Methods of analysis and
exposure measurement. Lyon, International Agency for Research on
Cancer, pp 105-146 (IARC Scientific Publications No. 108).
Buser HR, Bosshardt HP, & Rappe C (1978) Formation of polychlorinated
dibenzofurans (PCDFs) from the pyrolysis of PCBs. Chemosphere, 1:
109-119.
Cerniglia CE, Morgan JC, & Gibson DT (1979) Bacterial and fungal
oxidation of dibenzofuran. Biochem J, 180: 175-185.
Chatkittikunwong W & Creaser CS (1994a) Stability of bromo- and
bromochloro-dibenzo- p-dioxins under laboratory and environmental
conditions. Chemosphere, 29: 547-557.
Chatkittikunwong W & Creaser CS (1994b) Microscale synthesis of
bromo- and bromochloro-dibenzo- p-dioxins and dibenzofurans.
Chemosphere, 28: 11-21.
Chatkittikunwong W & Creaser CS (1994c) Bromo-, bromochloro- and
dibenzo- p-dioxins and dibenzofurans in incinerator flyash.
Chemosphere, 29: 559-566.
Childers JW, Wilson NK, Harless RL, & Barbour RK (1992)
Characterization of brominated and bromo/chloro dibenzo- p-dioxins
and dibenzofurans by gas chromatography/matrix isolation- infrared
spectrometry. Chemosphere, 25: 1285-1290.
Chriske HW, Weilburg W, Drösemeier E, & Päpke O (1990) [Exposure to
dioxins and furans at computer workplaces?] Arbeitsmed Sozialmed
Präventivmed, 25: 302-306 (in German).
Clausen E, Lahaniatis ES, Bahadir M, & Bieniek D (1987) [Determination
of brominated dibenzofurans formed by thermolysis of polymers
containing decabromodiphenylether as flame retardant.] Fresenius Z
Anal Chem, 327: 297-300 (in German).
Cook PM, Kuehl DW, Walker MK, & Peterson RE (1991) Bioaccumulation and
toxicity of TCDD and related compounds in aquatic ecosystems. In:
Gallo MA, Scheuplein RJ, & van der Heijden CA ed. Biological basis for
risk assessment of dioxins and related compounds. Cold Spring Harbor,
New York, Cold Spring Harbor Laboratory Press, pp 143-167 (Banbury
Report 35).
Couture LA, Abbott BD, & Birnbaum LS (1990) A critical review of the
developmental toxicity and teratogenicity of
2,3,7,8-tetrachlorodibenzo- p-dioxin: Recent advances toward
understanding the mechanism. Teratology, 42: 619-627.
Cramer PH, Ayling RE, Thornburg KR, Stanley JS, Remmers JC, Breen JJ,
& Schwemberger J (1990a) Evaluation of an analytical method for the
determination of polybrominated dibenzo- p-dioxins/dibenzofurans
(PBDD/PBDF) in human adipose tissue. Chemosphere, 20: 821-827.
Cramer PH, Stanley JS, Bauer KM, Ayling RE, Thornburg KR, &
Schwemberger J (1990b) Final report on brominated dioxins and
dibenzofurans in human adipose tissue. Washington, DC, US
Environmental Protection Agency, 75 pp (EPA-560/5-90-005; PB
91-103507).
Dawidowsky N (1993) [Origin and occurrence of polyhalogenated
dibenzodioxins and dibenzofurans in the environment.] Tübingen,
Eberhard-Karls University, Faculty of Chemistry and Pharmacy, 188 pp
(Dissertation) (in German).
De Haan LHJ, Bos TA, Aarts JMMJG, & Brouwer A (1993) Inhibition of
gap-junctional intercellular communication by
tetrachlorodibenzo- p -dioxin (TCDD): Possible role of the Ah
receptor. In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, &
Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 157-161 (Organohalogen
Compounds, Volume 13).
De Jong APJM, van der Heeft E, Marsman JA, & Liem AKD (1992)
Investigation on the occurrence of polyhalogenated (Br/Cl)
dibenzodioxins and dibenzofurans in cow's milk and fish tissue.
Chemosphere, 25: 1551-1557.
De Jongh J, Buser HR, & Poiger H (1992) The metabolism of
2,3,7,8-tetrabromo-dibenzodioxin in the rat. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 33-36 (Organohalogen Compounds, Volume 10).
De Jongh J, Buser HR, & Poiger H (1993) The metabolism of
2,3,7,8-tetrabromo-dibenzodioxin in the rat. Xenobiotica, 23: 19-26.
Dellinger B, Maqsud L, & Sidhu S (1993) Kinetics of toxic combustion
by-product formation during brominated flame retardant incineration.
In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed.
Dioxin'93: 13th International Symposium on Chlorinated Dioxins and
Related Compounds, Vienna, 20-24 September 1993. Vienna, Austrian
Federal Environmental Agency, pp 261-264 (Organohalogen Compounds,
Volume 11).
Denison MS (1990) The molecular mechanism of action of
2,3,7,8-tetrachlorodibenzo- p -dioxin and related halogenated aromatic
hydrocarbons. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar:
10th International Symposium on Chlorinated Dioxins and Related
Compounds. Bayreuth, Germany, Ecoinforma Press, pp 95-98
(Organohalogen Compounds, Volume 4).
Denison MS (1991) The molecular mechanism of action of
2,3,7,8-tetrachlorodibenzo- p-dioxin and related halogenated aromatic
hydrocarbons. Chemosphere, 23: 1825-1830.
Denivelle L, Fort R, & Van Hai P (1960) Sur les dibenzo- p-dioxines
octochlorée et octobromée et sur les oxydes de phényle décachloré et
décabromé. Bull Soc Chim Fr, 11/12: 1538-1543.
Denomme MA, Homonoko K, Fujita T, Sawyer T, & Safe S (1985) Effects of
substituents on the cytosolic receptor-binding activities and aryl
hydrocarbon hydroxylase induction potencies of 7-substituted
2,3-dichlorodibenzo- p-dioxins. Mol Pharmacol, 27: 656-661.
Denomme MA, Homonoko K, Fujita T, Sawyer T, & Safe S (1986)
Substituted polychlorinated dibenzofuran receptor binding affinities
and aryl hydrocarbon hydroxylase induction potencies -- A QSAR
analysis. Chem-Biol Interact, 57: 175-187.
DeVito MJ & Birnbaum LS (1994) Toxicology of dioxins and related
chemicals. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 139-162.
De Wit C (1993) Bioaccumulation in aquatic and terrestrial organisms.
In: The Toxicology Forum: Current views on the impact of dioxins and
furans on human health and the environment, Berlin, 9-11 November
1992. Washington, DC, Toxicology Forum, Inc., pp 353-363.
De Wit CA, Järnberg UG, Asplund LT, Jansson B, Olsson M, Lindstedt LL,
Bergek S, Hjelt M, Rappe C, & Andersson Ö (1993) Biomagnification of
PCDD/PCDF, non-ortho and mono-ortho PCB in predators from aquatic and
terrestrial ecosystems. In: Fiedler H, Frank H, Hutzinger O, Parzefall
W, Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Environmental Agency, pp 325-328
(Organohalogen Compounds, Volume 12).
Di Domenico A, Silano A, Viviano V, & Zapponi G (1980) Accidental
release of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) at Seveso,
Italy. Ecotoxicol Environ Saf, 4: 339-345.
Diliberto JJ, Kedderis LB, & Birnbaum LS (1990a) Absorption of
2,3,7,8-tetrabromo-dibenzo- p-dioxin (TBDD) in male rats.
Toxicologist, 10: 54.
Diliberto JJ, Kedderis LB, & Birnbaum LS (1990b) Acute oral exposure
to 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD). In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 309-311 (Organohalogen Compounds, Volume 1).
Diliberto JJ, Jackson JA, & Birnbaum LS (1991) Acute pulmonary
absorption of 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in rats.
Toxicologist, 11: 272.
Diliberto JJ, Kedderis LB, Jackson JA, & Birnbaum LS (1993) Effects of
dose and routes of exposure on the disposition of
2,3,7,8-[3H]tetrabromodibenzo- p-dioxin (TBDD) in the rat. Toxicol
Appl Pharmacol, 120: 315-326.
Diliberto JJ, Jackson JA, & Birnbaum LS (1996) Comparison of
2,3,7,8-tetrachloro-dibenzo- p-dioxin (TCDD) disposition following
pulmonary, oral, dermal, and parenteral exposures to rats. Toxicol
Appl Pharmacol, 138: 158-168.
Donnelly JR & Sovocool GW (1991) Predictions of bromo- and
bromochloro-dioxin GC elution properties. Chemosphere, 22: 455-460.
Donnelly JR, Vonnahme TL, Hedin CM, & Niederhut WJ (1986) Evaluation
of RCRA method 8280 for analysis of dioxins and dibenzofurans. In:
Rappe C, Choudhary G, & Keith LH ed. Chlorinated dioxins and
dibenzofurans in perspective. Chelsea, Michigan, Lewis Publishers,
Inc., chapter 3, pp 399-435.
Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Hedin CM, Sovocool GW,
& Mitchum RK (1987) The chemistry and mass spectrometry of brominated
dibenzo- p-dioxins and dibenzofurans. Biomed Environ Mass Spectrom,
14: 465-472.
Donnelly JR, Grange AH, Nunn NJ, Sovocool GW, Brumley WC, & Mitchum RK
(1989a) Analysis of thermoplastic resins for brominated dibenzofurans.
Biomed Environ Mass Spectrom, 18: 884-896.
Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Sovocool GW, & Mitchum
RK (1989b) Preparation of bromochlorodibenzo- p-dioxins and
dibenzofurans and analysis by EPA RCRA method 8280. Chemosphere, 18:
209-216.
Donnelly JR, Grange AH, Nunn NJ, Sovocool GW, & Breen JJ (1990)
Bromo- and bromochloro-dibenzo- p-dioxins and dibenzofurans in the
environment. Chemosphere, 20: 1423-1430.
Donnelly JR, Munslow WD, Grange AH, Pettit TL, Simmons RD, Kumar KS, &
Sovocool GW (1991a) A gas chromatographic/mass spectrometric approach
for isomer-specific environmental monitoring of the 1700 bromo-,
chloro-, and bromochloro-dibenzo- p-dioxins. Biol Mass Spectrom, 20:
329-337.
Donnelly JR, Munslow WD, Grange AH, Pettit TL, Simmons RD, & Sovocool
GW (1991b) Correlation of the structure with linear retention index
for bromo- and bromochlorodibenzo- p-dioxins and bromodibenzofurans.
J Chromatogr, 540: 290-310.
Dumler R (1989) [Fire tests to study formation of brominated
dibenzofurans and -dioxins from fllame retarded plastics.] Bayreuth,
University of Bayreuth, Faculty of Biology, Chemistry and Geology, 132
pp (Dissertation) (in German).
Dumler R, Teufl C, Lenoir D, & Hutzinger O (1987) [Formation and
quantities of PBDF and PBDD from decomposition of flame retardants
using different pyrolysis apparata (poster).] In: [Dioxin -- A
technical, analytical, ecological and toxicological challenge:
Mannheim Colloquium, 5-7 May 1987.] Düsseldorf, Society of German
Engineers, pp 287-292 (VDI-Report No. 634) (in German).
Dumler R, Thoma H, Lenoir D, & Hutzinger O (1989a) Thermal formation
of polybrominated dibenzodioxins (PBDD) and dibenzofurans (PBDF) from
bromine containing flame retardants. Chemosphere, 19: 305-308.
Dumler R, Lenoir HTD, & Hutzinger O (1989b) PBDF and PBDD from the
combustion of bromine containing flame retarded polymers: A survey.
Chemosphere, 19: 2023-2031.
Dumler R, Lenoir D, Thoma H, & Hutzinger O (1989c) Thermal formation
of polybrominated dibenzofurans from decabromodiphenyl ether in a
polybutylene-terephthalate polymer matrix. J Anal Appl Pyrolysis, 16:
153-158.
Dumler R, Lenoir D, & Hutzinger O (1990a) Formation of brominated
dibenzofurans and -dioxins from the combustion of the flame retardant
decabromodiphenyl ether under different conditions. In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 325-328 (Organohalogen Compounds, Volume 2).
Dumler R, Lenoir D, Thoma H, & Hutzinger O (1990b) Thermal formation
of polybrominated dibenzofurans and dioxins from decabromodiphenyl
ether flame retardant: Influence of antimony(III) oxide and the
polymer matrix. Chemosphere, 20: 1867-1873.
Dumler R, Thoma H, & Hutzinger O (1990c) Content and formation of
toxic products in flame retardants. In: Freiji L ed. Proceedings of
the Workshop on Brominated Aromatic Flame Retardants, Skokloster,
Sweden, 24-26 October 1989. Solna, National Chemicals Inspectorate
(KEMI), pp 93-98.
Dumler-Gradl R, Tartler D, Thoma H, & Vierle O (1995) Detection of
polybrominated diphenylethers (PBDE), dibenzofurans (PBDF) and
dibenzodioxins (PBDD) in scrap of electronics and recycled products.
In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, & Reiner
E ed. Dioxin'95: 15th International Symposium on Chlorinated Dioxins
and Related Compounds, Edmonton, Canada, 21-25 August 1995 (Short
papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 101-104
(Organohalogen Compounds, Volume 24).
Esposito MP, Tiernan TO, & Dryden FE (1980) Dioxins (Final Report).
Cincinnati, Ohio, US Environmental Protection Agency, 371 pp
(EPA-600/2-80-197; PB 82-136847) .
Fabarius G, Wilken M, Bergas M, & Zeschmar-Lahl B (1990) Release of
organic pollutants during accidental fires. In: Hutzinger O & Fiedler
H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 373-377 (Organohalogen Compounds, Volume 3).
Fahrig R (1993) Genetic effects of dioxins in the spot test with mice.
Environ Health Perspect, 101(suppl 3): 257-261.
Fairchild WL, Muir DCG, Currie RS, & Yarechewski AL (1992) Emerging
insects as a biotic pathway for movement of
2,3,7,8-tetrachlorodibenzofuran from lake sediments. Environ Toxicol
Chem, 11: 867-872.
Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, & Gonzalez FJ
(1996) Aryl-hydrocarbon receptor-deficient mice are resistant to
2,3,7,8-tetrachlorodibenzo- p-dioxin-induced toxicity. Toxicol Appl
Pharmacol, 140: 173-179.
Fiedler H & Schramm KW (1990) QSAR generated octanol-water partition
coefficients of selected mixed halogenated dibenzodioxins and
dibenzofurans. Chemosphere, 20: 1597-1602.
Fiedler H & Van den Berg M (1996) Polychlorinated dibenzo- p-dioxins,
polychlorinated dibenzofurans, and related compounds. Update and
recent developments. ESPR-Environ Sci Pollut Res, 3: 122-128.
Figge K, Wernitz A, Fortnagel P, Wittich R-M, & Harms H (1991)
[Dibenzofuran: Bacterial mineralization -- kinetics of degradation in
heterogenous systems.] UWSF-Z Umweltchem Ökotoxikol, 3: 201-205 (in
German).
Figge K, Metzdorf U, Nevermann J, Schmiese J, Keskin M, Fortnagel P, &
Wittich RM (1993) [Bacterial mineralization of dibenzofuran,
dibenzo- p-dioxin, and 1,2,4,5-tetrachlorobenzene in soils.] UWSF-Z
Umweltchem Ökotoxikol, 5: 122-130 (in German).
Fingerhut MA, Halperin WE, Marlow BS, Piacitelli LA, Honchar PA,
Sweeny MH, Greife AL, Dill PA, Steenland K, & Surunda AJ (1991) Cancer
mortality in workers exposed to 2,3,7,8-tetrachlorodibenzo- p-dioxin.
N Engl J Med, 324: 212-218.
Fletcher CL & McKay WA (1993) Polychlorinated dibenzo- p-dioxins
(PCDDs) and dibenzofurans (PCDFs) in the aquatic environment -- A
literature review. Chemosphere, 26: 1041-1069.
Foght JM & Westlake DWS (1988) Degradation of polycyclic aromatic
hydrocarbons and aromatic heterocycles by a Pseudomonas species. Can
J Microbiol, 34: 1135-1141.
Fortnagel P, Harms H, Wittich R-M, Francke W, Krohn S, & Meyer H
(1989a) Cleavage of dibenzofuran and dibenzodioxin ring systems by a
Pseudomonas bacterium. Naturwissenschaften, 76: 222-223.
Fortnagel P, Wittich R-M, Harms H, Schmidt S, Franke S, Sinnwell V,
Wilkes H, & Francke W (1989b) New bacterial degradation of the biaryl
ether structure. Regioselective dioxygenation prompts cleavage of
ether bonds. Naturwissenschaften, 76: 523-524.
Fortnagel P, Harms H, Wittich R-M, Krohn S, Meyer H, Sinnwell V,
Wilkes H, & Francke W (1990) Metabolism of dibenzofuran by
Pseudomonas sp. strain HH69 and the mixed culture HH27. Appl Environ
Microbiol, 56: 1148-1156.
Fortunati GU, Banfi C, & Pasturenzi M (1994) Soil sampling. Fresenius
J Anal Chem, 348: 86-100.
Fulfs JC (1989) IRI Study 119.048 28, day range finding study
2,3,7,8-tetrabromo-dibenzofuran (TBDF) in the rat. Test article:
2,3,7,8-tetrabromodibenzofuran. Fort Collins, Colorado, Inhausen
Research Institute, Inc., 6 pp .
Funcke W & Hemminghaus HJ (1993) Evaluation of PCDF/D, PBDF/D and
PBCDF/D profiles in flue gas of combustion facilities using a
statistical distribution function. In: Fiedler H, Frank H, Hutzinger
O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th International
Symposium on Chlorinated Dioxins and Related Compounds, Vienna, 20-24
September 1993. Vienna, Austrian Federal Environmental Agency, pp
345-350 (Organohalogen Compounds, Volume 11).
Funcke W, Mayer J & Wilken M (1995) Analysis of polyhalogenated
dibenzofurans and dibenzo(p)dioxins (PXDF/Ds) in the scope of an
interlaboratory test. In: Bolt D, Clement R, Fiedler H, Harrison B,
Ramamoorthy S, & Reiner E ed. Dioxin'95: 15th International Symposium
on Chlorinated Dioxins and Related Compounds, Edmonton, Canada, 21-25
August 1995 (Short papers). Edmonton, Alberta, Dioxin'95 Secretariat,
pp 311-314 (Organohalogen Compounds, Volume 23).
Gebefügi I (1989) Chemical exposure in enclosed environments. Toxicol
Environ Chem, 20-21: 121-127.
Gebefügi I & Kreuzig G (1989) [Surface concentration of halogenated
compounds in indoor rooms.] In: [Halogenated organic compounds in the
environment -- Source, measurement, effect, corrective measures:
Mannheim Colloquium, 25-27 April 1989.] Düsseldorf, Society of German
Engineers, vol 1, pp 503-510 (VDI Report No. 745) (in German).
German Dioxin Directive (1994) [First directive for the changing of
the directive on forbidden chemicals.] Bundesgesetzblatt, 1: 1493-1495
(in German).
Giesy JP, Ludwig JP, & Tillitt DE (1994) Dioxins, dibenzofurans, PCBs
and colonial, fish-eating water birds. In: Schecter A ed. Dioxins and
health. New York, London, Plenum Press, pp 249-307.
Gilbertson M (1989) Effects on fish and wildlife populations. In:
Kimbrough RD & Jensen AA ed. Halogenated biphenyls, terphenyls,
naphthalenes, dibenzodioxins and related products, 2nd ed. Amsterdam,
Oxford, New York, Elsevier Science Publishers, chapter 4, pp 103-127.
Gilman H & Dietrich J (1957) Halogen derivatives of
dibenzo- p-dioxin. J Am Chem Soc, 79: 1439-1441.
Goldstein JA & Safe S (1989) Mechanism of action and
structure-activity relationship for the chlorinated
dibenzo- p-dioxins and related compounds. In: Kimbrough RD & Jensen
AA ed. Halogenated biphenyls, terphenyls, naphthalenes, dibenzodioxins
and related products, 2nd ed. Amsterdam, Oxford, New York, Elsevier
Science Publishers, chapter 9, pp 239-293.
Golor G, Yamashita K, McLachlan M, Hutzinger O, & Neubert D (1993)
Comparison of the kinetics of chlorinated and brominated dioxins and
furans in the rat. In: Fiedler H, Frank H, Hutzinger O, Parzefall W,
Riss A, & Safe S ed. Dioxin'93: 13th International Symposium on
Chlorinated Dioxins and Related Compounds, Vienna, 20-24 September
1993. Vienna, Austrian Federal Governmental Agency, pp 203-206
(Organohalogen Compounds, Volume 12).
Golor G, Kociok O, Persaud TNV, Hinkel M, & Petrick K (1995)
Toxicokinetic properties of trihalogenated dibenzo- p-dioxins in the
rat. In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, &
Reiner E ed. Dioxin'95: 15th International Symposium on Chlorinated
Dioxins and Related Compounds, Edmonton, Canada, 21-25 August 1995
(Short papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 269-272
(Organohalogen Compounds, Volume 25).
Gray LE, Kelce WR, Monosson E, Ostby JS, & Birnbaum LS (1995) Exposure
to TCDD during development permanently alters reproductive function in
male Long Evans rats and hamsters: reduced ejaculated and epididymal
sperm numbers and sex accessory gland weights in offspring with normal
androgenic status. Toxicol Appl Pharmacol, 131: 108-118.
Gullett BK, Lemieux PM, & Dunn JE (1994) Role of combustion and
sorbent parameters in prevention of polychlorinated dibenzo- p-dioxin
and polychlorinated dibenzofuran formation during waste combustion.
Environ Sci Technol, 28: 107-118.
Hagenmeier H (1994) Contributions of diesel-powered vehicles and wood
burning to overall PCDD/PCDF compounds. In: Fiedler H, Hutzinger O,
Clement R, & Sakai S ed. Dioxin'94: 14th International Symposium on
Chlorinated Dioxins, PCB and Related Compounds, Kyoto, Japan, 21-25
November 1994 (Short papers). Kyoto, Kyoto University, Department of
Environmental and Sanitary Engineering, pp 267-270 (Organohalogen
Compounds, Volume 20).
Hagenmaier H, Brunner H, Haag R, & Kraft M (1987a) Copper-catalyzed
dechlorination-hydrogenation of polychlorinated dibenzofurans and
other chlorinated aromatic compounds. Environ Sci Technol, 21:
1085-1088.
Hagenmaier H, Brunner H, Haag R, & Kraft M (1987b) [The significance
of catalytic effects in the formation and destruction of
polychlorinated dibenzodioxins and polychlorinated dibenzofurans.]
Düsseldorf, Society of German Engineers, pp 557-584 (VDI-Report No.
634) (in German).
Hagenmaier H, Dawidowsky N, Weberruß U, Hutzinger O, Schwind KH, Thoma
H, Essers U, Bühler U, & Greiner R (1990a) Emission of polyhalogenated
dibenzodioxins and dibenzofurans from combustion-engines. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 329-334 (Organohalogen Compounds, Volume
2).
Hagenmaier H, Wiesmueller T, Golor G, Krowke R, Helge H, & Neubert D
(1990b) Transfer of various polychlorinated dibenzo- p-dioxins and
dibenzofurans (PCDDs and PCDFs) via placenta and through milk in a
marmoset monkey. Arch Toxicol, 64: 601-615.
Hagenmaier H, She J, Benz T, Dawidowsky N, Düsterhöft L, & Linding C
(1992) Analysis of sewage sludge for polyhalogenated
dibenzo- p-dioxins, dibenzofurans, and diphenylethers. Chemosphere,
25: 1457-1462.
Haglund P, Egebäck KE, & Jansson B (1988) Analysis of polybrominated
dioxins and furans in vehicle exhaust. Chemosphere, 17: 2129-2140.
Hahn ME & Stegeman JJ (1992) Phylogenetic distribution of the Ah
receptor in non-mammalian species: Implications for dioxin toxicity
and Ah receptor evolution. Chemosphere, 25: 931-937.
Hamm S & Theisen J (1992) Formation of polybrominated dibenzofurans
and polybrominated dibenzo(p)dioxins at fires of electrical
appliances. Poster presented at Dioxin'92: 12th International
Symposium on Dioxins and Related Compounds, Tampere, Finland, 24-28
August 1992, 9 pp.
Hammel KE, Kalyanaraman B, & Kirk TK (1986) Oxidation of polycyclic
aromatic hydrocarbons and dibenzo[p]-dioxins by Phanerochaete
chrysosporium ligninase. J Biol Chem, 261: 16948-16952.
Happe B, Eltis LD, Poth H, Hedderich R, & Timmis KN (1993)
Characterization of 2,2',3-trihydroxybiphenyl dioxygenase, an
extradiol dioxygenase from dibenzofuran- and
dibenzo- p-dioxin-degrading bacterium Sphingomonas sp. strain RW1.
J Bacteriol, 175: 7313-7320.
Hardell L, Eriksson M, Axelson O, & Zahm SH (1994) Cancer
epidemiology. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 525-547.
Hardy ML, Sistrunk PH, Eldan M, & McFadden DJ (1990)
2,3,7,8-tetrabromodibenzo-furan (TBDF): 4 week subchronic toxicity
study in the rat. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
313-316 (Organohalogen Compounds, Volume 1).
Harless RL, Lewis RG, McDaniel DD, & Dupuy AE Jr (1989) Identification
of bromochlorodibenzo- p-dioxins and dibenzofurans in ash samples.
Chemosphere, 18: 201-208.
Harless RL, Lewis RG, McDaniel DD, Gibson JF, & Dupuy AE (1992)
Evaluation of a sampling and analysis method for determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in ambient air.
Chemosphere, 25: 1317-1322.
Harms H, Wittich R-M, Sinnwell V, Meyer H, Fortnagel P, & Francke W
(1990) Transformation of dibenzo- p-dioxin by Pseudomonas sp.
strain HH69. Appl Environ Microbiol, 56: 1157-1159.
Harms H, Lorenz W, & Bahadir M (1995) [Rapid determination of
polyhalogenated dioxins and furans at fire residues.] GIT Fachz Lab,
39: 724-729 (in German).
Hartenstein H-U (1993) Fixed bed activated coke filters for the
control of toxic metals and organics from waste incinerators. In:
Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed.
Dioxin'93: 13th International Symposium on Chlorinated Dioxins and
Related Compounds, Vienna, 20-24 September 1993. Vienna, Austrian
Federal Environmental Agency, pp 11-16 (Organohalogen Compounds,
Volume 12).
Heinbuch D & Stieglitz L (1992) Formation of brominated compounds on
fly ash. In: Dioxin'92: 12th International Symposium on Chlorinated
Dioxins and Related Compounds, Tampere, Finland, 24-28 August 1992.
Helsinki, Finnish Institute of Occupational Health, pp 257-260
(Organohalogen Compounds, Volume 8).
Heinbuch D & Stieglitz L (1993) Formation of brominated compounds on
fly ash. Chemosphere, 27: 317-324.
Helge H (1993) A pediatrician's view of studies on dioxin-attributed
effects in children. In: The Toxicology Forum: Current views on the
impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1993. Washington, DC, Toxicology Forum, Inc., pp
272-287.
Hiester E (1992) [Measurement of polyhalogenated dioxins and furans in
air.] Essen, Germany, Institute for Air Pollution Control for the
State of Northrhine-Westfalia, pp 87-93 (LIS Report) (in German).
Hileman F, Wehler J, Wendling J, Orth R, Ritchie C, & McKenzie D
(1989) Dibenzofuran in diphenyl oxide and the relationship to
brominated dibenzofurans in brominated diphenyl oxide. Chemosphere,
18: 217-224.
Hofmann KH, Polnisch E, Kreisel H, Mach H, & Schubert M (1992)
Degradation of dibenzofuran and dibenzodioxins by fungi. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 93-96 (Organohalogen
Compounds, Volume 9).
Holsapple MP, Morris DL, Wood SC, & Snyder NK (1991)
2,3,7,8-Tetrachlorodibenzo- p-dioxin induced changes in
immunocompetence: Possible mechanisms. Annu Rev Pharmacol Toxicol, 31:
73-100.
Hornung MW, Zabel EW, & Peterson RE (1996a) Additive interactions
between pairs of polybrominated dibenzo- p-dioxin, dibenzofuran, and
biphenyl congeners in a rainbow trout early life stage mortality
bioassay. Toxicol Appl Pharmacol, 140: 345-355.
Hornung MW, Zabel EW, & Peterson RE (1996b) Toxic equivalency factors
of polybrominated dibenzo- p-dioxin, dibenzofuran, biphenyl, and
polyhalogenated diphenyl ether congeners based on rainbow trout early
life stage mortality. Toxicol Appl Pharmacol, 140: 227-234.
Hosseinpour J, Schwind KH, & Thoma H (1989) Brominated-chlorinated
dibenzo- p-dioxins and furans: Synthesis of standards and detection
in fly ash from a municipal waste incinerator. Chemosphere, 19:
109-114.
Huang LQ, Moore C, McGown S, Tomer KB, & Tong HY (1991) The
application of high-resolution MS and hybrid MS/MS to the analysis of
flyash for monobrominated polychlorinated dibenzo- p-dioxins and
dibenzofurans. Abstr Papers Am Chem Soc, 202: analytical section,
abstract 50.
Huang LQ, Paiva A, Tong H, Monson SJ, & Gross ML (1992a) Application
of gas chromatography high-resolution mass spectrometry to the
determination of trace monobromopolychlorodibenzo- p-dioxins in
environmental samples. J Am Soc Mass Spectrom, 3: 248-259.
Huang LQ, Tong H, & Donnelly JR (1992b) Characterization of
dibromopolychlorodibenzo- p-dioxins and
dibromopolychlorodibenzofurans in municipal waste incinerator fly ash
using gas chromatography/mass spectrometry. Anal Chem, 64: 1034-1040.
Huff J (1994) Dioxins and mammalian carcinogenesis. In: Schecter A ed.
Dioxins and health. New York, London, Plenum Press, pp 389-408.
Hutzinger O (1990) [Studies on the possible emission of polybrominated
dibenzodioxins and dibenzofurans during fires with flame retarded
plastics.] Bayreuth, University of Bayreuth, Department of
Environmental Chemistry and Geochemistry, 68 pp (Research Report No.
10403362 prepared for the German Federal Ministry of Environment) (in
German).
Hutzinger O, Dumler R, Lenoir D, Teufl C, & Thoma H (1989) PBDD and
PBDF from brominated flame retardants: Combustion equipment,
analytical methodology and synthesis of standards. Chemosphere, 18:
1235-1242.
Hutzinger O, Hagenmaier H, & Essers U (1990) [Studies on the emission
of halogenated dibenzodioxins and dibenzofurans from combustion motors
using commercial fuels.] Bonn, German Federal Ministry for Research
and Technology, 224 pp (in German).
Ivens I, Neupert M, Löser E, & Thies J (1990) Storage and elimination
of 2,3,7,8-tetrabromodibenzo- p-dioxin in liver and adipose tissue of
the rat. Chemosphere, 20: 1209-1214.
Ivens IA, Löser E, Rinke M, Schmidt U, & Neupert M (1992) Toxicity of
2,3,7,8-tetrabromodibenzo- p-dioxin in rats after single oral
administration. Toxicology, 73: 53-69.
Ivens IA, Löser E, Rinke M, Schmidt U, & Mohr U (1993) Subchronic
toxicity of 2,3,7,8-tetrabromodibenzo- p-dioxin in rats. Toxicology,
83: 181-201.
Ivens-Kohl I, Mohr U, & Löser E (1989)
[2,3,7,8-Tetrabromodibenzo- p-dioxin: subchronic toxicological study
on rats (oral application via stomach tube for 3 months with
subsequent observation period of 3 months).] Wuppertal, Germany, Bayer
AG, 1298 pp (Unpublished report No. 18477) (in German).
Ivens-Kohl I, Rinke M, & Löser E (1990)
[2,3,7,8-Tetrabromodibenzo- p-dioxin: Studies on acute oral toxicity
in male and female Wistar rats.] Wuppertal, Germany, Bayer AG, 205 pp
(Unpublished report No. 18668) (in German).
Jackson JA, Diliberto JJ, Kedderis LB, & Birnbaum LS (1991) Dermal
absorption and disposition of 2,3,7,8-tetrabromodibenzo- p-dioxin
(TBDD) in rats. Toxicologist, 11: 271.
Jackson JA, Diliberto JJ, & Birnbaum LS (1993) Estimation of
octanol-water partition coefficients and correlation with dermal
absorption for several polyhalogenated aromatic hydrocarbons. Fundam
Appl Toxicol, 21: 334-344.
Jansson B, Andersson R, Asplund L, Litzen K, Nylund K, Sellström U,
Uvemo U-B, Wahlberg C, Wideqvist U, Odsjö T, & Olsson M (1993)
Chlorinated and brominated persistent organic compounds in biological
samples from the environment. Environ Toxicol Chem, 12: 1163-1174.
Jay K & Stieglitz L (1996) Synthesis of mixed halogenated
dibenzofurans (X = Cl, Br). Chemosphere, 33: 1041-1045.
Jödicke B, Ende M, Helge H, & Neubert D (1992) Fecal excretion of
PCDDs/PCDFs in a 3-month-old breast-fed infant. Chemosphere, 25:
1061-1065.
Johnson J, Breen JJ, Murray TM, Glatz JA, Steele DH, & Stanley JS
(1989) Polyhalogenated dibenzo- p-dioxins/dibenzofurans testing and
reporting requirements under the Toxic Substances Control Act (TSCA).
Chemosphere, 19: 849-852.
Johnson L, Wilker CE, Safe SH, Scott B, Dean D, & White PH (1994)
2,3,7,8-Tetrachlorodibenzo- p-dioxin reduces the number, size and
organelle content of Leydig cells in adult rat testes. Toxicology, 89:
49-65.
Kapila S, Yanders AF, Orazio CE, Meadows JE, Cerlesi S, & Clevenger TE
(1989) Field and laboratory studies on the movement and fate of
tetrachlorodibenzo- p-dioxin in soil. Chemosphere, 18: 1297-1304.
Kedderis LB, Diliberto JJ, & Birnbaum LS (1990) Disposition of
intravenous 2,3,7,8-tetrabromodibenzodioxin (TBDD) in rats.
Toxicologist, 10: 310.
Kedderis LB, Diliberto JJ, & Birnbaum LS (1991a) Disposition and
excretion of intravenous 2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD)
in rats. Toxicol Appl Pharmacol, 108: 397-406.
Kedderis LB, Diliberto JJ, Linko P, Goldstein JA, & Birnbaum LS
(1991b) Disposition of 2,3,7,8-tetrabromodibenzo- p-dioxin and
2,3,7,8-tetrachlorodibenzo- p-dioxin in the rat: Biliary excretion
and induction of cytochromes CYP1A1 and CYP1A2. Toxicol Appl
Pharmacol, 111: 163-172.
Kedderis LB, Diliberto JJ, Jackson JA, Linko P, Goldstein JA, &
Birnbaum LS (1992a) Effects of dose and route of exposure on dioxin
disposition. Chemosphere, 25: 7-10.
Kedderis LB, Mills JJ, Andersen ME, & Birnbaum LS (1992b) A
physiologically-based pharmacokinetic model for
2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in the rat. In: Dioxin'92:
12th International Symposium on Chlorinated Dioxins and Related
Compounds, Tampere, Finland, 24-28 August 1992. Helsinki, Finnish
Institute of Occupational Health, pp 113-116 (Organohalogen Compounds,
Volume 10).
Kedderis LB, Mills JJ, Andersen ME, & Birnbaum LS (1993) A
physiologically based pharmacokinetic model for
2,3,7,8-tetrabromodibenzo- p-dioxin (TBDD) in the rat: Tissue
distribution and CYP1A induction. Toxicol Appl Pharmacol, 121: 87-98.
Kedderis LB, Jackson JA, Patterson DG, Grainger J, Diliberto JJ, &
Birnbaum LS (1994) Chemical characterization and disposition studies
with 1,2,7,8-tetrabromodibenzofuran in the rat. J Toxicol Environ
Health, 41: 53-69.
Kende AS & Wade JJ (1973) Synthesis of new steric and electronic
analogs of 2,3,7,8-tetrachlorodibenzo- p-dioxin. Environ Health
Perspect, 5: 49-57.
Kende AS, Wade JJ, Ridge D, & Poland A (1974) Synthesis and Fourier
transform carbon-13 spectroscopy of new toxic
polyhalodibenzo- p-dioxins. J Org Chem, 39: 931-937.
Kieatiwong S, Nguyen LV, Hebert VR, Hackett M, Miller G, Miille MJ, &
Mitzel R (1990) Photolysis of chlorinated dioxins in organic solvents
and on soils. Environ Sci Technol, 24: 1575-1580.
Kieper H (1996) [Exposure to polybrominated dibenzofurans and -dioxins
in copper plants and during the production of flame retarded
plastics.] Bremerhaven, Wirtschaftsverlag NW, pp 1-115 (in German).
Kjeller L-O, Kulp SE, Jonsson B, & Rappe C (1993) Methodology for the
determination of polychlorinated dibenzo- p-dioxins and dibenzofurans
in sediment samples. Toxicol Environ Chem, 39: 1-12.
Klecka GM & Gibson DT (1979) Metabolism of dibenzo- p-dioxin by a
Pseudomonas species. Biochem J, 180: 639-645.
Klecka GM & Gibson DT (1980) Metabolism of dibenzo- p-dioxin and
chlorinated dibenzo- p-dioxins by a Beijerinckia species. Appl
Environ Microbiol, 39: 288-296.
Klusmeier W, Vögler P, Ohrbach KH, Weber H, & Kettrup A (1988) Thermal
decomposition of decabromodiphenyl ether. J Anal Appl Pyrolysis, 13:
277-285.
Kociba RJ & Schwetz BA (1982) Toxicity of
2,3,7,8-tetrachloro- p-dioxin (TCDD). Drug Metab Rev, 13: 387-406.
Kociba RJ, Keeler PA, Park CN, & Gehring PJ (1976)
2,3,7,8-Tetrachlorodibenzo- p-dioxin (TCDD): Results of a 13-week
oral toxicity study in rats. Toxicol Appl Pharmacol, 35: 553-574.
Kogevinas M, Parkin DM, Cordier S, Cung TA, Hung L, Cao Dai L,
Rivera-Pomar E, Raphael M, & Stellman S (1993) Case-control studies on
soft-tissue sarcoma and non-Hodgkin lymphoma in Vietnam. In: Fiedler
H, Frank H, Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93:
13th International Symposium on Chlorinated Dioxins and Related
Compounds, Vienna, 20-24 September 1993. Vienna, Austrian Federal
Environmental Agency, pp 375-378 (Organohalogen Compounds, Volume 13).
Kutz FW, Barnes DG, Bretthauer EW, Bottimore DP, & Greim H (1990) The
international toxicity equivalency factor (I-TEF) method for
estimating risks associated with exposures to complex mixtures of
dioxins and related compounds. Toxicol Environ Chem, 26: 99-109.
Lahaniatis ES, Bergheim W, & Rainer C (1989) Hazardous halogenated
substances formed during combustion processes. Toxicol Environ Chem,
20/21: 501-506.
Lahaniatis E, Bergheim W, & Bieniek D (1991) Formation of
2,3,7,8-tetrabromo-dibenzodioxin and -furan by thermolysis of polymers
containing brominated flame retardants. Toxicol Environ Chem, 31/32:
521-526.
Lahl U, Wilken M, & Wiebe A (1991) [Polybrominated diphenylether in
waste incineration.] Müll Abfall, 23: 83-87 (in German).
Landers JP & Bunce NJ (1991) The Ah-receptor and the mechanism of
dioxin toxicity. Biochem J, 276: 273-287.
Lee A, Campbell B, & Kelly W (1986) Dioxin and furan contamination in
the manufacture of halogenated organic chemicals (Contract No.
68-03-3274). Cincinnati, Ohio, US Environmental Protection Agency, 71
pp (EPA/600/2-86/101; PB 87-119905).
Lee A, Campbell B, & Kelly W (1987) Project summary: Dioxin and furan
contamination in the manufacture of halogenated organic chemicals
(Contract No. 68-03-3274). Cincinnati, Ohio, US Environmental
Protection Agency, 7 pp (EPA/600/S2-86/101).
Lenoir D (1994) Behaviour and fate of aromatic bromine compounds in
the environment. In: Advances in organobromine chemistry. Amsterdam,
Oxford, New York, Elsevier Science Publishers, pp 363-386.
Lenoir D, Schramm K-W, Hutzinger O, & Schedel G (1991) Photochemical
degradation of brominated dibenzo- p-dioxins and -furans in organic
solvents. Chemosphere, 22: 821-834.
Lenoir D, Zier B, Bieniek D, & Kettrup A (1994) The influence of water
and metals on PBDD/F concentration in incineration of
decabromobiphenyl ether in polymeric matrices. Chemosphere, 28:
1921-1928.
Loganathan BG, Kannan K, Watanabe I, Kawano M, Irvine K, Kumar S, &
Sikka HC (1995) Isomer-specific determination and toxic evaluation of
polychlorinated biphenyls, polychlorinated/ brominated
dibenzo- p-dioxins and dibenzofurans, polybrominated biphenyl ethers,
and extractable organic halogen in carp from the Buffalo River, New
York. Environ Sci Technol, 29: 1832-1838.
Lorenz W (1994) [Emissions from halogen-containing plastics in certain
treatment processes.] Abfallwirtsch Lichte neuen Vorschr, 9: 143-157
(in German).
Lorenz W & Bahadir M (1993) Recycling of flame retardants containing
printed circuits: A study of the possible formation of polyhalogenated
dibenzodioxins/-furans. Chemosphere, 26: 2221-2229.
Lorenzen A & Okey AB (1991) Detection and characterization of Ah
receptor in tissue and cells from human tonsils. Toxicol Appl
Pharmacol, 107: 203-214.
Löser E & Ivens I (1989) Preliminary results of a 3 month toxicity
study on rats with 2,3,7,8-tetrabromodibenzo- p-dioxin
(2,3,7,8-TBDD). Chemosphere, 19: 759-764.
Lucier GW (1991) Humans are a sensitive species to some of the
biochemical effects of structural analogs of dioxin. Environ Toxicol
Chem, 10: 727-735.
Lucier GW, Portier CJ, & Gallo MA (1993a) Receptor mechanisms and
dose-response models for the effects of dioxins. Environ Health
Perspect, 101: 37-44.
Lucier GW, Clark G, Hiermath C, Tritscher A, Sewall C, & Huff J
(1993b) Carcinogenicity of TCDD in laboratory animals: Implications
for risk assessment. Toxicol Ind Health, 9: 631-668.
Luijk R & Govers HAJ (1992) The formation of polybrominated
dibenzo- p-dioxins (PBDDs) and dibenzofurans (PBDFs) during pyrolysis
of polymer blends containing brominated flame retardants. Chemosphere,
25: 361-374.
Luijk R, Wever H, Olie K, & Govers HAJ (1990) Formation of
polybrominated dibenzo- p-dioxins and -dibenzofurans during pyrolysis
of polybrominated diphenylethers and high impact polystyrene. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 335-338 (Organohalogen Compounds, Volume
2).
Luijk R, Wever H, Olie K, Govers HAJ, & Boon JJ (1991) The influence
of the polymer matrix on the formation of polybrominated
dibenzo- p-dioxins (PBDDs) and polybrominated dibenzofurans (PBDFs).
Chemosphere, 23: 1173-1183.
Luijk R, Jansen J, & Govers HAJ (1992a) The exchange of bromine and
chlorine in 2,3,7,8-tetrabromo-dibenzo- p-dioxin. In: Dioxin'92: 12th
International Symposium on Chlorinated Dioxins and Related Compounds,
Tampere, Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 99-101 (Organohalogen Compounds, Volume 8).
Luijk R, Dorland K, Smit P, & Govers HAJ (1992b) The halogenation of
dibenzo- p-dioxin and dibenzofuran in a model fly ash system. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 273-276 (Organohalogen
Compounds, Volume 8).
Luijk R, Govers HAJ, & Nellssen L (1992c) Formation of polybrominated
dibenzofurans during extrusion of high-impact
polystyrene/decabromodiphenyl ether/antimony(III)oxide. Environ Sci
Technol, 26: 2191-2198.
Luijk R, Dorland C, Smit P, Jansen J, & Govers HAJ (1994) The role of
bromine in the de novo synthesis in a model fly ash system.
Chemosphere, 28: 1299-1309.
Lutes CC, Charles MJ, & Kamens RM (1990) The atmospheric stability of
polybrominated dibenzo- p-dioxins and dibenzofurans. In: Jayanty RKM
& Gay BW Jr ed. Measurement of toxic and related air pollutants:
Proceedings of the 1990 EPA/A and WMA International Symposium,
Raleigh, North Carolina, 1-4 May 1990. Research Triangle Park, North
Carolina, Research Triangle Institute, pp 38-44 (EPA/600/9-90/026; PB
91-120279).
Lutes CC, Charles MJ, & Kamens RM (1992a) The atmospheric stability of
polybrominated dibenzo- p-dioxins and dibenzofurans. Chemosphere, 25:
99-102.
Lutes CC, Charles MJ, Odum JR, & Kamens RM (1992b) Chamber aging
studies on the atmospheric stability of polybrominated
dibenzo- p-dioxins and dibenzofurans. Environ Sci Technol, 26:
991-998.
Mably TA, Bjerke DL, Moore RW, Gendron-Fitzpatrick A, & Peterson RE
(1992) In utero and lactational exposure of male rats to
2,3,7,8-tetrachlorodibenzo- p-dioxin: 3. Effects of spermatogenesis
and reproductive capability. Toxicol Appl Pharmacol, 114: 118-126.
Maier EA, Griepink B, & Fortunati U (1994) Round table discussions.
Outcome and recommendations. Fresenius J Anal Chem, 348: 171-179.
Manz A, Berger J, Dwyer JH, Flesch-Janys D, Nagel S, & Waltsgott H
(1991) Cancer mortality among workers in chemical plant contaminated
with dioxin. Lancet, 338: 959-964.
Marklund S, Rappe C, Tysklind M, & Egebäck KE (1987) Identification of
polychlorinated dibenzofurans and dioxins in exhausts from cars run on
leaded gasoline. Chemosphere, 16: 29-36.
Marklund S, Andersson R, Tysklind M, Rappe C, Egebäck KE, Björkman E,
& Grigoriadis V (1990) Emissions of PCDD's and PCDF's in gasoline and
diesel fueled cars. Chemosphere, 20: 553-561.
Mason G, Farrell B, Keys B, Piskorska-Pliszczynsksa J, Safe L, & Safe
S (1986) Polychlorinated dibenzo- p-dioxins: Quantitative in vivo
and in vitro structure-activity relationships. Toxicology, 41:
21-31.
Mason G, Zacharewski T, Denomme MA, Safe L, & Safe S (1987a)
Polybrominated dibenzo- p-dioxins and related compounds:
Quantitative in vivo and in vitro structure-activity
relationships. Toxicology, 44: 245-255.
Mason G, Denomme MA, Safe L, & Safe S (1987b) Polybrominated and
chlorinated dibenzo- p-dioxins: Synthesis biologic and toxic effects
and structure-activity relationships. Chemosphere, 16: 1729-1731.
Massa T, Gerber T, Pfaffenholz V, Chandra A, Schlatterer B, & Chandra
B (1990) A host-mediated in vivo/ in vitro assay with peritoneal
murine macrophages for the detection of carcinogenic chemicals. J
Cancer Res Clin Oncol, 116: 357-364.
Massa T, Esmaeili A, Schlatterer B, Hagenmaier H, & Chandra P (1991)
Carcinogenic and co-carcinogenic potential of
2,3,7,8-tetrachlorodibenzo- p-dioxin in a host-mediated in vivo/
in vitro assay. Chemosphere, 23: 1855-1868.
Massa T, Esmaeili A, Fortmeyer H, Schlatterer B, Hagenmaier H, &
Chandra P (1992a) Carcinogenic and co-carcinogenic potential of
2,3,7,8-tetrachlorodibenzodioxin in a host-mediated in vivo/
in vitro assay. Chemosphere, 25: 1085-1090.
Massa T, Esmaeili A, Fortmeyer H, Schlatterer B, Hagenmaier H, &
Chandra P (1992b) Cell transforming and oncogenic activity of
2,3,7,8-tetrachloro- and 2,3,7,8-tetrabromodibenzo- p-dioxin.
Anticancer Res, 12: 2053-2060.
McAllister DL, Mazac CJ, Gorsich R, Freiberg M, & Tondeur Y (1990)
Analysis of polymers containing brominated diphenyl ethers as flame
retardants after molding under various conditions. Chemosphere, 20:
1537-1541.
McConnell EE (1989) Acute and chronic toxicity and carcinogenesis in
animals. In: Kimbrough RD & Jensen AA ed. Halogenated biphenyls,
terphenyls, naphthalenes, dibenzodioxins and related products, 2nd ed.
Amsterdam, Oxford, New York, Elsevier Science Publishers, chapter 6,
pp 161-193.
McKinley MK, Kedderis LB, & Birnbaum LS (1993) The effect of
pretreatment on the biliary excretion of
2,3,7,8-tetrachlorodibenzo- p-dioxin,
2,3,7,8-tetrachlorodibenzofuran, and 3,3',4,4'- tetrachlorobiphenyl in
the rat. Fundam Appl Toxicol, 21: 425-432.
McLachlan MJ (1993) Digestive tract absorption of polychlorinated
dibenzo- p-dioxins, dibenzofurans, and biphenyls in a nursing infant.
Toxicol Appl Pharmacol, 123: 68-72.
Mennear JH & Lee CC (1994) Polybrominated dibenzo- p-dioxins and
dibenzofurans: Literature review and health assessment. Environ Health
Perspect, 102(suppl 1): 265-274.
Merz W, Neu H-J, Kuck M, Winkler K, Gorbach S, & Muffler H (1986) [A
process for the generation and analytical characterization of gaseous
combustion products.] Fresenius Z Anal Chem, 325: 449-460 (in German).
Meyer H, Neupert M, Pump W, & Willenberg B (1993) [Flame retardants
determine recyclability.] Kunststoffe, 83: 253-257 (in German).
Monna L, Omori T, & Kodama T (1993) Microbial degradation of
dibenzofuran, fluorene, and dibenzo- p-dioxin by Staphylococcus
auriculans DBF63. Appl Environ Microbiol, 59: 285-289.
Moore JA, McConnell EE, Dalgard DW, & Harris MW (1979) Comparative
toxicity of three halogenated dibenzofurans in guinea pigs, mice, and
rhesus monkeys. Ann NY Acad Sci, 320: 151-163.
Munslow WD, Sovocool GW, Donnelly JR, & Mitchum RK (1987)
Electrophilic bromination of dibenzo- p-dioxin. Chemosphere, 16:
1661-1666.
Nagao T, Golor G, Krowke R, & Neubert D (1990a) Comparison of cleft
palate frequency induced by 2,3,7,8-tetrabromodibenzo- p-dioxin and
2,3,7,8-tetrachlorodibenzo- p-dioxin in mice. In: Hutzinger O &
Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, pp 317-319 (Organohalogen Compounds, Volume 1).
Nagao T, Neubert D, & Löser E (1990b) Comparative studies on the
induction of ethoxyresorufin O-deethylase by 2,3,7,8-TCDD and
2,3,7,8-TBrDD. Chemosphere, 20: 1189-1192.
Nagao T, Golor G, Bittmann H, & Löser E (1990c) Induction of hepatic
monooxygenases by 2,3,7,8-tetrabromodibenzo- p-dioxin and liver
tissue concentrations in rats. Naunyn- Schmiedeberg's Arch Pharmacol,
341: R19.
Nagao T, Golor G, Krowke R, & Neubert D (1990d) Comparative study on
the teratogenic potency of 2,3,7,8-TBrDD and 2,3,7,8-TCDD in mice.
Teratology, 42: 27A.
Nagao T, Yamashita K, Golor G, Bittmann H, Körner W, Hagenmeier H, &
Neubert D (1995/96) Tissue distribution after a single subcutaneous
administration of 2,3,7,8-tetrabromodibenzo- p-dioxin in comparison
with toxicokinetics of 2,3,7,8-tetrachlorodibenzo- p-dioxin in female
Wistar rats. Life Sci, 58: 325-336.
Nakano T, Tsuji M, & Okuno T (1987) Level of chlorinated organic
compounds in the atmosphere. Chemosphere, 16: 1781-1786.
NATO/CCMS (1988) Pilot study on international information exchange on
dioxins and related compounds. Scientific basis for the development of
the international toxicity equivalence factor (I-TEF) method of risk
assessment for complex mixtures of dioxins and related compounds.
Brussels, North Atlantic Treaty Organization, Committee on the
Challenges of Modern Society, 56 pp (Report No. 176).
Nebert DW (1994) Drug-metabolizing enzymes in ligand-modulated
transcription. Biochem Pharmacol, 47: 25-37.
Nebert DW, Petersen DD, & Puga A (1991) Human Ah locus polymorphism
and cancer: Inducibility of CYP1A1 and other genes by combustion
products and dioxin. Pharmacogenetics, 1: 68-78.
Nebert DW, Puga A, & Vasiliou V (1993) Role of the Ah receptor and the
dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal
transduction. Ann NY Acad Sci, 685: 624-640.
Nestrick TJ, Lamparski LL, & Peters TL (1989) Micro-scale bromination
procedure for synthesis of 2,3,7,8-substituted brominated
dibenzo- p-dioxins and dibenzofurans from [13C] labeled precursors.
Chemosphere, 18: 1761-1770.
Neubert D (1991) Peculiarities of the toxicity of polyhalogenated
dibenzo- p-dioxins and dibenzofurans in animals and man. Chemosphere,
23: 1869-1893.
Neubert D (1993a) Survey on the influence of PHDDs/PHDFs on
lymphocytes in venous blood. In: The Toxicology Forum: Current views
on the impact of dioxins and furans on human health and the
environment, Berlin, 9-11 November 1992. Washington, DC, Toxicology
Forum, Inc., pp 299-315.
Neubert D (1993b) Some data on the induction of monooxygenases by
PHDDs/PHDFs. In: The Toxicology Forum: Current views on the impact of
dioxins and furans on the human health and the environment, Berlin,
9-11 November 1992. Washington, DC, Toxicology Forum, Inc., pp 72-90.
Neubert R, Jacob-Müller U, Stahlmann R, Helge H, & Neubert D (1990)
Polyhalogenated dibenzo- p-dioxins and dibenzofurans and the immune
system: 1. Effects on peripheral lymphocytes of a non-human primate
(Callithrix jacchus) after treatment with
2,3,7,8-tetrachlorodibenzo- p-dioxin. Arch Toxicol, 64: 345-359.
Neubert R, Jacob-Müller U, & Helge H (1991) Polyhalogenated
dibenzo- p-dioxins and dibenzofurans and the immune system: 2.
In vitro effects of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) on
lymphocytes of venous blood from man and a non-human primate
(Callithrix jacchus). Arch Toxicol, 65: 213-219.
Neubert D, Golor G, & Neubert R (1992) TCDD-toxicity equivalencies for
PCDD/PCDF congeners: Prerequisites and limitations. Chemosphere, 25:
65-70.
Neubert R, Golor G, Stahlmann R, Helge H, & Neubert D (1992)
Polyhalogenated dibenzo- p- dioxins and dibenzofurans and the immune
system: 4. Effects of multiple-dose treatment with
2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) on peripheral lymphocyte
subpopulations of a non-human primate (Callithrix jacchus). Arch
Toxicol, 66: 250-259.
Neubert R, Stahlmann R, Korte M, Van Loveren H, Vos JG, Golor G, Webb
JR, Helge H, & Neubert D (1993) Effects of small doses of dioxins on
the immune system of marmosets and rats. Ann NY Acad Sci, 685:
662-686.
Neupert M & Pump W (1992) Experiences from a large scale warehouse
fire with bromine-containing polybutylene terephthalate forming
almost no polybrominated dibenzodioxins and dibenzofurans. In:
Dioxin'92: 12th International Symposium on Chlorinated Dioxins and
Related Compounds, Tampere, Finland, 24-28 August 1992. Helsinki,
Finnish Institute of Occupational Health, pp 163-168 (Organohalogen
Compounds, Volume 9).
Neupert M, Grupe A, & Weis H (1988) Stability of polybrominated
dibenzo- p-dioxins and dibenzofurans under laboratory handling
conditions. Chemosphere, 17: 1089-1097.
Neupert M, Weis H, Stock B, & Thies J (1989a) Analytical procedures in
connection with acute toxicity studies: I.
Tetrabromodibenzo- p-dioxin (TBDD). Chemosphere, 19: 115-120.
Neupert M, Weis H, Thies J, & Stock B (1989b) Analytical procedures in
connection with acute animal toxicity studies: II. Pyrolysis products
obtained from ABS copolymer containing octabromo-diphenylether as a
flame retardant. Chemosphere, 19: 219-224.
Nylund K, Asplung L, Jansson B, Jonsson P, Litzen K, & Sellström
(1992) Analysis of some polyhalogenated organic pollutants in sediment
and sewage sludge. Chemosphere, 24: 1721-1730.
Öberg T & Bergström J (1990) Bromine and waste incineration -- an
environmental risk? In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
339-342 (Organohalogen Compounds, Volume 2).
Öberg T, Warman K, & Bergström J (1987) Brominated aromatics from
combustion. Chemosphere, 16: 2451-2465.
OECD (1994) Risk reduction monograph No. 3: Selected brominated flame
retardants background and national experience with reducing risk.
Paris, Organisation for Economic Co-operation and Development, 152 pp
(OECD Environment Monograph Series No. 102 - OECD/GD (94)969).
O'Keefe PW (1978) Formation of brominated dibenzofurans from pyrolysis
of the polybrominated biphenyl fire retardant, Firemaster FF-1.
Environ Health Perspect, 23: 347-350.
Okey AB (1990) Enzyme induction in the cytochrome P450 system.
Pharmacol Ther, 45: 241-298.
Okey AB (1992) Enzyme induction in the cytochrome P-450 system. In:
Kalow W ed. Pharmacogenetics of drug metabolism. New York, Pergamon
Press, pp 549-608.
Okey AB, Riddick DS, & Harper PA (1994) The Ah receptor: Mediator of
the toxicity of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) and
related compounds. Toxicol Lett, 70: 1-22.
Olie K, Vermeulen PL, & Hutzinger O (1977) Chorodibenzo- p-dioxins
and chlorodibenzo-furans are trace components of fly ash and flue gas
of some municipal incinerators in the Netherlands. Chemosphere, 6:
455-459.
Orazio CE, Kapila S, Puri RK, & Yanders AF (1992) Persistence of
chlorinated dioxins and furans in the soil environment. Chemosphere,
25: 1469-1474.
Ott MG & Zober A (1996) Morbidity study of extruder personnel with
potential exposure to brominated dioxins and furans: II. Results of
clinical laboratory studies. Occup Environ Med, 53: 833-846.
Päpke O, Lis A, Priegnitz J, Helmcke K, & Ball M (1990) [Development
of methods for determination of polyhalogenated dioxins and furans in
the air -- Final report: December 1989-October 1990.] Hamburg, ERGO
Forschungsgesellschaft mbH, 24 pp + appendix (in German).
Peper M, Klett M, Frentzel-Beyme R, & Heller WD (1993)
Neuropsychological effects of chronic exposure to environmental
dioxins and furans. Environ Res, 60: 124-135.
Peterson RE (1993) Effects on aquatic and terrestrial organisms. In:
The Toxicology Forum: Current views on the impact of dioxins and
furans on human health and the environment, Berlin, 9-11 November
1992. Washington, DC, Toxicology Forum, Inc., pp 383-399.
Peterson RE, Theobald HM, & Kimmel GL (1993) Developmental and
reproductive toxicity of dioxins and related compounds: Cross-species
comparisons. Crit Rev Toxicol, 23: 283-335.
Pinkerton MN, Kociba RJ, Petrella RV, McAllister DL, Willis ML, Fulfs
JC, Thoma H, & Hutzinger O (1989) A preliminary report on the
investigation of the comparative toxicity of combustion products of
high impact polystyrene with and without
decabromo-diphenyloxide/antimony trioxide as a flame retardant using
2,3,7,8-tetrabromodibenzo- p-dioxin and 2,3,7,8-tetrabromo
dibenzofuran as positive controls. Chemosphere, 18: 1243-1249.
Pirkle JL, Wolfe WH, Patterson DG, Needham LL, Michalek JE, Miner JC,
Peterson MR, & Phillips DL (1989) Estimates of the half-life of
2,3,7,8-tetrachlorodibenzo- p-dioxin in Vietnam veterans of Operation
Ranch Hand. J Toxicol Environ Health, 27: 165-171.
Pluim HJ, Wever J, Koppe JG, Slikke JW, & Olie K (1993) Intake and
faecal excretion of chlorinated dioxins and dibenzofurans in
breast-fed infants at different ages. Chemosphere, 26: 1947-1952.
Poellinger L (1993) Molecular mechanisms of dioxins action. In: The
Toxicology Forum: Current views on the impact of dioxins and furans on
human health and the environment, Berlin, 9-11 November 1992.
Washington, DC, Toxicology Forum, Inc., pp 91-100.
Poellinger L, Göttlicher M, & Gustafsson JA (1992) The dioxin and
peroxisome proliferator-activated receptors: Nuclear receptors in
search of endogenous ligands. Trends Pharmacol Sci, 13: 241-245.
Pohjanvirta R, Unkila M, & Tuomisto J (1993) Comparative acute
lethality of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD),
1,2,3,7,8-pentachlorodibenzo- p-dioxin and
1,2,3,4,7,8-hexachlorodibenzo- p-dioxin in the most TCDD-susceptible
and the most TCDD-resistant rat strain. Pharmacol Toxicol, 73: 52-56.
Pohjanvirta R, Hirvonen MR, Unkila M, Savolainen K, & Tuomisto J
(1994) TCDD decreases brain inositol concentrations in the rat.
Toxicol Lett, 70: 363-372.
Poiger H & Buser H-R (1984) The metabolism of TCDD in the dog and rat.
In: Poland A & Kimbrough RD ed. Biological mechanisms of dioxin
action. Cold Spring Harbor, New York, Cold Spring Harbor Laboratory,
pp 39-47 (Banbury Report 18).
Poiger H & Schlatter C (1980) Influence of solvents and adsorbents on
dermal and intestinal absorption of TCDD. Food Cosmet Toxicol, 18:
477-481.
Poiger H & Schlatter C (1986) Pharmacokinetics of 2,3,7,8-TCDD in man.
Chemosphere, 15: 1489-1494.
Poland A & Glover E (1973) Chlorinated dibenzo- p-dioxins: Potent
inducers of o-aminolevulinic acid synthetase and aryl hydrocarbon hydroxylase. II.
A study of the structure-activity relationship. Mol Pharmacol, 9:
736-747.
Poland A & Knutson JC (1982) 2,3,7,8-Tetrachlorodibenzo- p-dioxin and
related halogenated aromatic hydrocarbons: Examination of the
mechanism of toxicity. Annu Rev Pharmacol Toxicol, 22: 517-554.
Pruell RJ, Taplin BK, McGovern DG, Montmarquet T, & Johnson MW (1993)
Laboratory studies on the accumulation of polychlorinated
dibenzo- p-dioxins and dibenzofurans from sediment and food by the
American lobster (Homarus americanus). In: Fiedler H, Frank H,
Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 239-242 (Organohalogen Compounds, Volume 12).
Ramalingam B, Mazer T, Wagel DJ, Malloy CM, Taylor ML, TiernanTO,
Garrett JH, & Rifkind AB (1986) Synthesis and characterization of
polybromo- and polybromochloro-dibenzo- p-dioxins and dibenzofurans.
In: Rappe C, Choudhary G, & Keith LH ed. Chlorinated dioxins and
dibenzofurans in perspective: 189th National Meeting of the American
Chemical Society, Miami Beach, Florida, USA, September 1985. Chelsea,
Michigan, Lewis Publishers, Inc., pp 485-499.
Ranken PF, Ricks GM, Lynam DR, & Ariano JM (1990) Is watching
television toxic? BFRIP-sponsored study on the emission of PBDD/PBDFS
from operating television sets. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
343-347 (Organohalogen Compounds, Volume 2).
Ranken PF, Freiberg M, Mazac CJ, Bauer MR, Varcoe FT, & Tondeur Y
(1994) Definitive study of the determination of polybrominated
dibenzo- p-dioxins and polybrominated dibenzofurans in
decabromodiphenyloxide and tetrabromobisphenol-A. Bull Soc Chim Belg,
103: 219-233.
Rappe C (1993) Environmental concentrations and ecotoxicological
effects of PCDD's, PCDF's and related compounds. In: Fiedler H, Frank
H, Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 163-170 (Organohalogen Compounds, Volume 12).
Rappe C, Andersson R, Bonner MS, Cooper KR, Fiedler H, Howell FG, &
Lau C (1996) PCDDs and PCDFs in municipal sewage sludge and effluent
from the state of Mississippi, USA. Organohalog Compds, 28: 105-110.
Re MA, Bolt DL, & Chandler RA (1995) Stability of polybrominated
dibenzo- p-dioxin and dibenzofuran standard solutions. In: Bolt D,
Clement R, Fiedler H, Harrison B, Ramamoorthy S, & Reiner E ed.
Dioxin'95: 15th International Symposium on Chlorinated Dioxins and
Related Compounds, Edmonton, Canada, 21-25 August 1995 (Short papers).
Edmonton, Alberta, Dioxin'95 Secretariat, pp 269-272 (Organohalogen
Compounds, Volume 23).
Richtzenhain H & Schrage K (1977) [Highly brominated dibenzofurans.]
Munich, German Federal Patent Office, 14 pp (Patent No. 2534381
-- Dynamit Nobel AG) (in German).
Riggs KB, Pitts GE, White JS, Mitchum RK, & Reuther JJ (1990)
Polybrominated dibenzo- p-dioxin and polybrominated dibenzofuran
emissions from incineration of flame-retarded resins in a simulated
municipal waste incinerator. In: Hutzinger O & Fiedler H ed.
Dioxin'90/EPRI-Seminar: 10th International Symposium on Chlorinated
Dioxins and Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp
351-356 (Organohalogen Compounds, Volume 2).
Riggs K, Reuther J, White J, & Pitts G (1992) Determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in simulated
incinerator emissions. Chemosphere, 25: 1415-1420.
Ritterbusch J, Vogt R, Lorenz W, Bahadir M, & Hopf H (1994a)
UV-photolysis of PXDD/F-contaminated bromophenols and wastes of
chemical laboratories. Chemosphere, 29: 457-464.
Ritterbusch J, Lorenz W, & Bahadir M (1994b) Determination of
polyhalogenated dibenzo- p-dioxins and dibenzofurans in analytical
laboratory waste and their decomposition by UV-photolysis.
Chemosphere, 29: 1829-1838.
Romkes M, Piskorska-Pliszczynska J, Keys B, Safe S, & Fujita T (1987)
Quantitative structure-activity relationships: Analysis of
interactions of 2,3,7,8-tetrachlorodibenzo- p-dioxin and
2- substituted analogues with rat, mouse, guinea pig, and hamster
cytosolic receptor. Cancer Res, 47: 5108-5111.
Rordorf BF (1987) Prediction of vapor pressures, boiling points and
enthalpies of fusion for twenty-nine halogenated dibenzo- p-dioxins.
Thermochim Acta, 112: 117-122.
Rordorf BF, Sarna LP, Webster GRB, Safe SH, Safe LM, Lenoir D, Schwind
KH, & Hutzinger O (1990) Vapor pressure measurements on halogenated
dibenzo- p-dioxins and dibenzofurans. An extended data set for
correlation method. Chemosphere, 20: 1603-1609.
Safe S (1987) Determination of 2,3,7,8-TCDD toxic equivalent factors
(TEF's): Support for the use of the in vitro AHH induction assay.
Chemosphere, 16: 791-802.
Safe S (1990) Polychlorinated biphenyls (PCBs), dibenzo- p-dioxins
(PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental
and mechanistic considerations which support the development of toxic
equivalency factors (TEFs). Crit Rev Toxicol, 21: 51-88.
Safe S (1993a) Development of bioassays and approaches for the risk
assessment of 2,3,7,8-tetrachlorodibenzo- p-dioxin and related
compounds. Environ Health Perspect, 101(suppl 3): 317-325.
Safe S (1993b) Development, validation and limitations of toxic
equivalency factors. In: The Toxicology Forum: Current views on the
impact of dioxins and furans on human health and the environment,
Berlin, 9-11 November 1992. Washington, DC, Toxicology Forum, Inc., pp
176-180.
Safe S, Davis D, Romkes M, Yao C, Keyes B, Piskorska-Pliszczynska J,
Farrell K, Mason G, Denomme MA, Safe L, Zmudzka B, & Holcomb M (1989a)
Development and validation of in vitro bioassays for 2,3,7,8-TCDD
equivalents. Chemosphere, 19: 853-860.
Safe S, Mason G, Sawyer T, Zacharewski T, Harris M, Yao C, Keys B,
Farrell K, Holcomb M, Davis D, Safe L, Piskorska-Pliszczynska J, Leece
B, Denomme MA, Hutzinger O, Thoma H, Chittim B, & Madge J (1989b)
Development and validation of in-vitro induction assays for toxic
halogenated aromatic mixtures: A review. Toxicol Ind Health, 5:
757-775.
Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, Romkes M, &
Biegel L (1991) 2,3,7,8-Tetrachlorodibenzo- p-dioxin (TCDD) and
related compounds as antiestrogens: Characterization and mechanism of
action. Pharmacol Toxicol, 69: 400-409.
Schacht U, Gras B, & Sievers S (1995) [Determination of polybrominated
and polychlorinated dibenzodioxins and -furans in various
environmentally relevant materials.] In: Dioxin-Information and
EPA-Reassessment, Bayreuth, 12-14 June 1995. Organohalog Compds, 22:
325-334 (in German).
Schäfer W & Ballschmiter K (1986) Monobromopolychloroderivatives of
benzene, biphenyl, dibenzofuran and dibenzodioxin formed in
chemical-waste burning. Chemosphere, 15: 755-763.
Schecter A (1992) Dioxins and dibenzofurans in potentially exposed
workers: Serial tissue levels in a worker exposed in a PCB transformer
fire cleanup and blood levels in three exposed chemists. Chemosphere,
25: 1117-1122.
Schecter A & Ryan JJ (1990) Chlorinated and brominated dioxin levels
in the blood of a chemist who became ill after synthesizing
2,3,7,8-TCDD and 2,3,7,8-TBDD. In: Hutzinger O & Fiedler H ed.
Dioxin'90: 10th International Symposium on Chlorinated Dioxins and
Related Compounds. Bayreuth, Germany, Ecoinforma Press, pp 141-144
(Organohalogen Compounds, Volume 4).
Schecter A & Ryan JJ (1991) Brominated and chlorinated dioxin blood
levels in a chemist 34 years after exposure to
2,3,7,8-tetrachlorodibenzodioxin and 2,3,7,8-tetrabromo-dibenzodioxin.
Chemosphere, 23: 1921-1924.
Schecter A & Ryan JJ (1992) Persistent brominated and chlorinated
dioxin blood levels in a chemist: 35 years after dioxin exposure. J
Occup Med, 34: 702-707.
Schecter A & Ryan JJ (1994) Decrease in milk and blood dioxin levels
over time in a mother nursing twins: Estimates of decreased maternal
and increased infant dioxin body burden from nursing. In: Fiedler H,
Hutzinger O, Clement R, & Sakai S ed. Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 159-162
(Organohalogen Compounds, Volume 21).
Schecter A, Ryan JJ, Masuda Y, Brandt-Rauf P, Constable J, Dinh Cau H,
Cao Dai P, Tri Quynh H, Thi Ngoc Phuong N, & Hoang Phiet P (1994a)
Chlorinated and brominated dioxins and dibenzofurans in human tissue
following exposure. Environ Health Perspect, 102(suppl 1): 135-147.
Schecter A, Päpke O, Lis A, & Ball M (1994b) Chlorinated dioxin and
dibenzofuran levels in US human placentas and fetal tissue in
comparison with US adult population dioxin levels. In: Fiedler H,
Hutzinger O, Clement R, & Sakai S ed. Dioxin'94: 14th International
Symposium on Chlorinated Dioxins, PCB and Related Compounds, Kyoto,
Japan, 21-25 November 1994 (Short papers). Kyoto, Kyoto University,
Department of Environmental and Sanitary Engineering, pp 63-64
(Organohalogen Compounds, Volume 21).
Schecter A , Päpke O, Lis A, & Olson JR (1995) Chlorinated dioxin,
dibenzofuran and PCB levels in human fetal tissue at 8-18 weeks
gestational age, compared with placental, newborn and adult tissue
levels. In: Bolt D, Clement R, Fiedler H, Harrison B, Ramamoorthy S, &
Reiner E ed. Dioxin'95: 15th International Symposium on Chlorinated
Dioxins and Related Compounds, Edmonton, Canada, 21-25 August 1995
(Short papers). Edmonton, Alberta, Dioxin'95 Secretariat, pp 167-171
(Organohalogen Compounds, Volume 25).
Schecter A, Päpke O, Lis A, Ball M, Ryan JJ, Olson JR, Li L, & Kessler
H (1996a) Decrease in milk and blood dioxin levels over two years in a
mother nursing twins: Estimates of decreased maternal and increased
infant dioxin body burden from nursing. Chemosphere, 32: 543-549.
Schecter A, Startin J, Wright C, Päpke O, Ball M, & Lis A (1996b)
Concentrations of polychlorinated dibenzo- p-dioxins and
dibenzofurans in human placental and fetal tissues from the US and in
placentas from Yu-Cheng exposed mothers. Chemosphere, 32: 551-557.
Schimmel H, Griepink B, Maier EA, Kramer GN, Roos AH, & Tuinstra LGMT
(1994) Intercomparison study on milk powder fortified with PCDD and
PCDF. Fresenius J Anal Chem, 348: 37-46.
Schmidt U & Ivens-Kohl I (1990a) [Enzyme activity in the liver after
acute application of 2,3,7,8-tetrabromodibenzodioxin on rats.]
Wuppertal-Elberfeld, Bayer AG, 26 pp (Unpublished report No. 18657)
(in German).
Schmidt U & Ivens-Kohl I (1990b) [Enzyme activity in the liver after
acute application of pyrolysis condensate Novodur L2FR on rats and
guinea pigs.] Wuppertal-Elberfeld, Bayer AG, 28 pp (Unpublished report
No. 18696) (in German).
Schramm K-W, Lenoir D, & Hutzinger O (1990) Fugacity calculations of
vapor-flyash partition of polyhalogenated dioxins and furans.
Chemosphere, 20: 563-568.
Schreiber R (1994) [Emissions reduced to a minimum: Bonn trash
incineration plant -- experiences with a plant that was built
according to the latest knowledge of modern environmental technology.
Trash utilization.] Energie, 46: 18-25 (in German).
Schulz T, Golor G, Körner W, Hagenmaier H, & Neubert D (1993)
Comparative study on enzyme induction and tissue distribution of
2,3,7,8-tetrachlorodibenzo- p-dioxin,
2,3,4,7,8-pentachloro-dibenzofuran and
2,3,4,7,8-pentabromodibenzofuran in marmoset monkeys (Callithrix
jacchus). In: Fiedler H, Frank H, Hutzinger O, Parzefall W, Riss A,
& Safe S ed. Dioxin'93: 13th International Symposium on Chlorinated
Dioxins and Related Compounds, Vienna, 20-24 September 1993. Vienna,
Austrian Federal Environmental Agency, pp 145-148 (Organohalogen
Compounds, Volume 13).
Schulz TG, Neubert D, Donald SD, & Edwards RJ (1996) Induction of
cytochromes P450 by dioxins in liver and lung of marmoset monkeys
(Callithrix jacchus). Adv Exp Med Biol, 387: 443-446.
Schulz-Schalge T, Koch E, Schwind K-H, Hutzinger O, & Neubert D (1990)
Comparative study on the inductive potency of TCDD and TBrDD with
three 2,3,7,8-mixed-halogenated dioxins in liver microsomes of male
rats. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 321-324 (Organohalogen
Compounds, Volume 1).
Schulz-Schalge T, Schwind K-H, & Hutzinger O (1991a) Biological
activity of TCDD, TBrDD and three 2,3,7,8-mixed halogenated
dibenzo- p-dioxins. Naunyn-Schmiedeberg's Arch Pharmacol, 343(suppl):
R21.
Schulz-Schalge T, Koch E, Schwind K-H, Hutzinger O, & Neubert D
(1991b) Inductive potency of TCDD, TBDD and three
2,3,7,8-mixed-halogenated dioxins in liver microsomes of male rats:
Enzyme kinetic considerations. Chemosphere, 23: 1925-1931.
Schwetz BA, Norris JM, Sparschu GL, Rowe VK, Gehring PJ, Emerson JL, &
Gerbig CG (1973) Toxicology of chlorinated dibenzo- p-dioxins.
Environ Health Perspect, 5: 87-99.
Schwind KH (1991) [Halogenated dibenzo- p-dioxins and dibenzofurans
from incineration processes.] Bayreuth, University of Bayreuth,
Faculty of Biology, Chemistry and Geology, 206 pp (Dissertation) (in
German).
Schwind KH, Hosseinpour J, & Thoma H (1988) Brominated/chlorinated
dibenzo- p-dioxins and dibenzofurans -- Part 1:
Brominated/chlorinated and brominated dibenzo- p-dioxins and
dibenzofurans in fly ash from a municipal waste incinerator.
Chemosphere, 17: 1875-1884.
Schwind KH, Hosseinpour J, & Thoma H (1989) [Brominated/chlorinated
dibenzo- p-dioxins and dibenzofurans from waste incineration.] UWSF-Z
Umweltchem Ökotoxikol, 1: 24 (in German).
Schwind KH, Thoma H, Hutzinger O, Dawidowsky N, Weberuß U, Hagenmaier
H, Bühler U, Greiner R, Essers U, & Bessey E (1991) [Emission of
halogenated dibenzodioxins (PXDD) und dibenzofurans (PXDF) from motors
using commercial fuels.] USWF-Z Umweltchem Ökotoxikol, 3: 291-298 (in
German).
Sedlak D, Dumler-Grad R, Thoma H, & Vierle O (1996) Formation of
polyhalogenated dibenzodioxin and dibenzofurans (PXDD/F) during
textile processings. Organohalog Compds, 27: 201-205.
Shiu WY, Doucette W, Gobas FAPC, Andren A, & Mackay D (1988)
Physical-chemical properties of chlorinated dibenzo- p-dioxins.
Environ Sci Technol, 22: 651-658.
Sidhu SS, Maqsud L, Dellinger B, & Mascolo G (1995) The homogeneous,
gas-phase formation of chlorinated and brominated dibenzo- p-dioxin
from 2,4,6-trichloro- and 2,4,6-tribromophenols. Combust Flame, 100:
11-20.
Silbergeld EK & deFur PL (1994) Risk assessment of dioxinlike
compounds. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 51-78.
Silbergeld EK & Gasiewicz TA (1989) Dioxins and the Ah receptor. Am J
Ind Med, 16: 455-474.
Sinkkonen S, Vattulainen A, Aittola J-P, Paasivirta J, Tarhanen J, &
Lahtiperä M (1994) Metal reclamation produces sulphur analogues of
toxic dioxins and furans. Chemosphere, 28: 1279-1288.
Skene SA, Dewhurst IC, & Greenberg M (1989) Polychlorinated
dibenzo- p-dioxins and polychlorinated dibenzofurans: The risks to
human health -- A review. Hum Toxicol, 8: 173-204.
Somogyi A & Beck H (1993) Nurturing and breast feeding: exposure to
chemicals in breast milk. Environ Health Perspect, 101: 45-52.
Sovocool GW, Munslow WD, Donnelly JR, & Mitchum RK (1987)
Electrophilic bromination of dibenzofuran. Chemosphere, 16: 221-224.
Sovocool GW, Mitchum RK, Tondeur Y, Munslow WD, Vonnahme TL, &
Donnelly JR (1988) Bromo- and bromochloro-polynuclear aromatic
hydrocarbons, dioxins and dibenzofurans in municipal incinerator fly
ash. Biomed Environ Mass Spectrom, 15: 669-676.
Sovocool GW, Donnelly JR, Munslow WD, Vonnahme TL, Nunn NJ, Tondeur Y,
& Mitchum RK (1989) Analysis of municipal incinerator fly ash for
bromo- and bromochloro-dioxins, dibenzofurans, and related compounds.
Chemosphere, 18: 193-200.
Sovocool GW, Donnelly JR, Grange AH, Simmons RD, & Munslow WD (1990)
Brominated dioxins and other bromoaromatics in plastic pyrolysates.
In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 365-368 (Organohalogen
Compounds, Volume 2).
Spahl R, Dorn IH, Horn HC, & Hess K (1993) [Catalytic decomposition of
dioxins for incineration plants.] Entsorgungspraxis, 5: 328-331 (in
German).
Spink DC, Johnson JA, Connor SP, Aldous KM, & Gierthy JF (1994)
Stimulation of 17 ß-estradiol metabolism in MCF-7 cells by
bromochloro- and chloromethyl-substituted dibenzo- p-dioxins and
dibenzofurans: Correlations with antiestrogenic activity. J Toxicol
Environ Health, 41: 451-466.
Springer W & Rast HG (1988) [Biological degradation of polyhalogenated
mono- and polycyclic aromatics.] GWF-Wasser/Abwasser, 129: 70-75 (in
German).
Stephens RD, Rappe C, Hayward DG, Nygren M, Startin J, Ersboll A,
Carlé J, & Yrjänheikki E (1992) World Health Organization
international intercalibration study on dioxins and furans in human
milk and blood. Anal Chem, 64: 3109-3137.
Stieglitz L & Vogg H (1990) The de-novo-synthesis of PCDD/PCDF and
other organohalogen compounds on fly ash. In: Hutzinger O & Fiedler H
ed. Dioxin'90/EPRI-Seminar: 10th International Symposium on
Chlorinated Dioxins and Related Compounds. Bayreuth, Germany,
Ecoinforma Press, p 173 (Organohalogen Compounds, Volume 3).
Stieglitz L, Zwick G, Beck J, Bautz H, & Roth W (1989) Carbonaceous
particles in fly ash: A source for the de-novo-synthesis of
organochlorocompounds. Chemosphere, 19: 283-290.
Striebich RC, Rubey WA, Tirey DA, & Dellinger B (1990) High
temperature thermal decomposition of polybrominated flame retardant
materials. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar:
10th International Symposium on Chlorinated Dioxins and Related
Compounds. Bayreuth, Germany, Ecoinforma Press, pp 369-373
(Organohalogen Compounds, Volume 2).
Striebich RC, Rubey WA, Tirey DA, & Dellinger B (1991)
High-temperature degradation of polybrominated flame retardant
materials. Chemosphere, 23: 1197-1204.
Strubel V, Rast HG, Fietz W, Knackmuss HJ, & Engesser KH (1989)
Enrichment of dibenzofuran utilizing bacteria with high co-metabolic
potential towards dibenzodioxin and other anellated aromatics. FEMS
Microbiol Lett, 58: 233-238.
Strubel V, Engesser K-H, Fischer P, & Knackmuss H-J (1991)
3-(2-Hydroxyphenyl)catechol as substrate for proximal meta ring
cleavage in dibenzofuran degradation by Brevibacterium sp. strain
DPO 1361. J Bacteriol, 173: 1932-1937.
Sweeney A (1994) Reproductive epidemiology of dioxins. In: Schecter A
ed. Dioxins and health. New York, London, Plenum Press, pp 549-585.
Sweeney MH, Fingerhut MA, Calvert GM, Piacitelli LA, Alderfer RJ,
Davis-King K, Halperin WE, Connally LB, & Marlow DA (1993) Noncancer
health effects and exposure to 2,3,7,8-TCDD. In: Fiedler H, Frank H,
Hutzinger O, Parzefall W, Riss A, & Safe S ed. Dioxin'93: 13th
International Symposium on Chlorinated Dioxins and Related Compounds,
Vienna, 20-24 September 1993. Vienna, Austrian Federal Environmental
Agency, pp 369-374 (Organohalogen Compounds, Volume 13).
Takasuga T, Inoue G, Ohi E, Ireland P, Suzuki T, & Takeda N (1994)
Determination of halogenated aromatic and polycyclic aromatic
hydrocarbons formed during MSW incineration. In: Fiedler H, Hutzinger
O, Clement R, & Sakai S ed. Dioxin'94: 14th International Symposium on
Chlorinated Dioxins, PCB and Related Compounds, Kyoto, Japan, 21-25
November 1994 (Short papers). Kyoto, Kyoto University, Department of
Environmental and Sanitary Engineering, pp 41-44 (Organohalogen
Compounds, Volume 19).
Tashiro M & Yoshiya H (1982) Determination of structures of tri- and
tetrabromodibenzofurans formed in bromination of dibenzofuran.
Heterocycles, 19: 2349-2354.
Thies J, Neupert M, & Pump W (1990) Tetrabromobisphenol A (TBBA), its
derivatives and their flame retarded (FR) polymers -- Content of
polybrominated dibenzo- p-dioxins (PBDD) and dibenzofurans (PBDF)
-- PBDD/F formation under processing and smouldering (worst case)
conditions. Chemosphere, 20: 1921-1928.
Thoma H & Hutzinger O (1987a) [Pyrolysis and GC/MS-analysis of flame
retardants in on-line operation (poster).] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
293-297 (VDI-Report No. 634) (in German).
Thoma H & Hutzinger O (1987b) Pyrolysis and GC/MS-analysis of
brominated flame retardants in on-line operation. Chemosphere, 16:
1353-1360.
Thoma H & Hutzinger O (1989) Pyrolysis and GC/MS-analysis of
brominated flame retardants in on-line operation. Chemosphere, 18:
1047-1050.
Thoma H, Rist S, Hauschulz G, & Hutzinger O (1986a) Polybrominated
dibenzodioxins and -furans from the pyrolysis of some flame
retardants. Chemosphere, 15: 649-652.
Thoma H, Rist S, Hauschulz G, & Hutzinger O (1986b) Polybrominated
dibenzodioxins (PBrDD) and dibenzofurans (PBrDF) in some flame
retardant preparations. Chemosphere, 15: 2111-2113.
Thoma H, Hauschulz G, Knorr E, & Hutzinger O (1987a) Polybrominated
dibenzofurans (PBDF) and dibenzodioxins (PBDD) from the pyrolysis of
neat brominated diphenylethers, biphenyls and plastic mixtures of
these compounds. Chemosphere, 16: 277-285.
Thoma H, Hauschulz G, & Hutzinger O (1987b) PVC-induced
chlorine-bromine exchange in the pyrolysis of polybrominated diphenyl
ethers, -biphenyls, -dibenzodioxins and dibenzofurans. Chemosphere,
16: 297-307.
Thoma H, Hauschulz G, & Hutzinger O (1987c) Chlorine-bromine exchange
during pyrolysis of 1,2,3,4-tetrabromodibenzodioxin with different
chlorine donors. Chemosphere, 16: 1579-1581.
Thoma H, Hauschulz G, & Hutzinger O (1987d) [Pyrolysis of
dibenzodioxin, dibenzofuran and 1,2,3,4-T4BrDD with different chlorine
donors and catalysts (poster).] In: [Dioxin -- A technical,
analytical, ecological and toxicological challenge: Mannheim
Colloquium, 5-7 May 1987.] Düsseldorf, Society of German Engineers, pp
277-286 (VDI-Report No. 634) (in German).
Thoma H, Hauschulz G, & Hutzinger O (1989) Pyrolysis of dibenzodioxin,
dibenzofuran and 1,2,3,4-tetrabromodibenzodioxin with different
chlorine donors and catalysts. Chemosphere, 18: 1213-1217.
Thunberg T, Ahlborg UG, & Wahlstrom B (1984) Comparison between the
effects of 2,3,7,8- tetrachlorodibenzo- p-dioxin and six other
compounds on the vitamin A storage, the UDP- glucuronosyltransferase
and the aryl hydrocarbon hydroxylase activity in the rat liver. Arch
Toxicol, 55: 16-19.
Tomita M, Ueda S, & Narisada M (1959) Studies on the
dibenzo- p-dioxin (diphenylene dioxide) derivatives: XXVII. Synthesis
of polyhalodibenzo- p-dioxin. J Pharm Soc Jpn, 79: 186-192.
Tondeur Y, Gorsich R, Mazac C, Freiberg M, Hass J, & McAllister D
(1990) Analytical protocol for the analysis of polybrominated
dibenzodioxins and dibenzofurans: Data quality objectives and
single-laboratory evaluation. Chemosphere, 20: 1269-1276.
Tong HY, Monson SJ, Gross ML, & Huang LQ (1991)
Monobromopolychlorodibenzo- p-dioxins and dibenzofurans in municipal
waste incinerator flyash. Anal Chem, 23: 2697-2705.
Troitzsch JH (1990) Brominated flame retardants: Current situation in
the Federal Republic of Germany, October 1989. In: Freiji L ed.
Proceedings of the Workshop on Brominated Aromatic Flame Retardants,
Skokloster, Sweden, 24-26 October 1989. Solna, Sweden, National
Chemicals Inspectorate (KEMI), pp 117-122.
Tuinstra LGMT, Startin JR, & Maier EA (1996) Certification of the
contents of PCDDs and PCDFs in milkpowder. Organohalog Compds, 27:
459-463.
UBA (1992) [Further studies on the formation of polybrominated dioxins
and furans during thermal stress on flame retarded plastics and
textiles, Parts 1 and 2 (UBA-Texts 45/92).] Berlin, German Federal
Environmental Agency, pp 86, 151 (Research reports No. 104 03
364/01/02, UBA-FB 91-082 & 92-097) (in German).
US EPA (1987) Polyhalogenated dibenzo- p-dioxins/dibenzofurans:
Testing and reporting requirements -- Final Rule. Fed Reg, 52(108):
21412-21417.
US EPA (1990) Method 1613: Tetra- through octa-chlorinated dioxins and
furans by isotope dilution HRGC/HRMS -- Revision A. Washington, DC, US
Environmental Protection Agency, Office of Water Regulations and
Standards.
US EPA (1992) Method 8290: Polychlorinated dibenzodioxins (PCDDs) and
polychlorinated dibenzofurans (PCDFs) by high-resolution gas
chromatography/high-resolution mass spectrometry (HRMS); Washington,
DC, US Environmental Protection Agency.
Van Birgelen APJM, DeVito MJ, Akins JM, Ross DG, Diliberto JJ, &
Birnbaum LS (1996) Relative potencies of polychlorinated
dibenzo- p-dioxins, dibenzofurans, and biphenyls derived from hepatic
porphyrin accumulation in mice. Toxicol Appl Pharmacol, 138: 98-109.
Van den Heuvel JP & Lucier G (1993) Environmental toxicology of
polychlorinated dibenzo- p-dioxins and polychlorinated
dibenzofurans. Environ Health Perspect, 100: 189-200.
Van de Plassche EJ, Polder MD, & Schipper-Zablotskaja M (1994) Risk
assessment of polybrominated biphenyls and polybrominated diphenyl
ethers. Bilthoven, The Netherlands, National Institute of Public
Health and Environmental Protection (RIVM), 34 pp (Report No. 679101
016).
Vehlow J (1995) State of the art in emissions control and residue
quality: a rational point of view. In: European Conference: The future
of European thermal waste treatment, Paris, September 1995. Karlsruhe,
Research Centre, pp 1-19.
Vogg H (1989) [Topical technical questions on the law for minimization
of dioxins and furans from waste incineration processes.] Wasser Luft
Boden, 11/12: 25-27 (in German).
Vogg H, Metzger M, & Stieglitz L (1987) Recent findings on the
formation and decomposition of PCDD/PCDF in solid municipal waste
incineration. In: Emissions of trace organics from municipal solid
waste incinerators, Copenhagen, 20-22 January 1987, pp 207-216.
Vogt R, Lorenz W, Bahadir M, & Hopf H (1994a) Brominated
dibenzodioxins and -furans: Level of potential hazard during synthesis
of bromophenols carried out for educational purposes. Labor Praxis,
18: 30-36.
Vogt R, Lorenz W, Bahadir M, & Hopf H (1994b) PBDD/F formation during
bromoaniline synthesis in teaching laboratories. Fresenius Environ
Bull, 3: 746-750.
Vos (1993) TCDD effects on the immune system: an overview. In: The
Toxicology Forum: Current views on the impact of dioxins and furans on
human health and the environment, Berlin, 9-11 November 1992.
Washington DC, Toxicology Forum, Inc., pp 288-298.
Vos JG & Luster MI (1989) Immune alterations. In: Kimbrough RD &
Jensen AA ed. Halogenated biphenyls, terphenyls, naphthalenes,
dibenzodioxins and related products, 2nd ed. Amsterdam, Oxford, New
York, Elsevier Science Publishers, chapter 10, pp 295-319.
Wagel DH, Tiernan TO, Taylor ML, Garrett JH, Vanness GF, Solch JG, &
Harden LA (1989) Assessments of ambient air sampling techniques for
collecting airborne polyhalogenated dibenzo- p-dioxins (PCDD),
dibenzofurans (PCDF) and biphenyls (PCB). Chemosphere, 18: 177-184.
Walker MK & Peterson RE (1991) Potencies of polychlorinated
dibenzo- p-dioxin, dibenzofuran, and biphenyl congeners, relative to
2,3,7,8-tetrachlorodibenzo- p-dioxin for producing early life stage
mortality in rainbow trout (Oncorhynchus mykiss). Aquat Toxicol, 21:
219-238.
Walker MK & Peterson E (1994) Aquatic toxicity of dioxins and related
chemicals. In: Schecter A ed. Dioxins and health. New York, London,
Plenum Press, pp 347-387.
Wania F & Lenoir D (1990) Copper-promoted reactions of polybrominated
dibenzo-para-dioxins (PBrDD). Chemosphere, 21: 417-432.
Wanke T, Vehlow J, Mark FE, & Brenner KS (1996) The influence of flame
retarded plastic foams upon the formation of Br containing
dibenzo- p-dioxins and dibenzofurans in a MSWI. Organohalog Compds,
28: 530-535.
Watanabe I (1988) [Behavior of organobrominated compounds at the
sediment phase in the environment.] Osalka-furitsu Koshu Eisei
Kenkyusho Kenkyu Hokoku, 26: 129-133 (in Japanese with English
summary).
Watanabe I & Tatsukawa R (1987) Formation of brominated dibenzofurans
from the photolysis of flame retardant decabromobiphenyl ether in
hexane solution by UV and sun light. Bull Environ Contam Toxicol, 39:
953-959.
Watanabe I & Tatsukawa R (1990) Anthropogenic brominated aromatics in
the Japanese environment. In: Freiji L ed. Proceedings of the Workshop
on Brominated Aromatic Flame Retardants, Skokloster, Sweden, 24-26
October 1989. Solna, Sweden, National Chemicals Inspectorate (KEMI),
pp 63-71.
Watanabe I, Kawano M, Wang Y, Chen Y, & Tatsukawa R (1992)
Polybrominated dibenzo- p-dioxins (PBDDs) and dibenzofurans (PBDFs)
in atmospheric air in Taiwan and Japan. In: Dioxin'92: 12th
International Symposium on Dioxins and Related Compounds, Tampere,
Finland, 24-28 August 1992. Helsinki, Finnish Institute of
Occupational Health, pp 309-312 (Organohalogen Compounds, Volume 9).
Watanabe I, Kawano M, & Tatsukawa R (1993) Consumption trend and
environmental research on brominated flame retardants in Japan and the
formation of poly-halogenated dibenzofurans at the metal reclamation
factory. Paper presented at the OECD Flame Retardants Workshop,
Neuchâtel, February 1993, 7 pp.
Watanabe I, Kawano M, & Tatsukawa R (1994) The photolysis of
halogenated dibenzofurans in hexane solution and on airborne dust by
sunlight. In: Fiedler H, Hutzinger O, Clement R, & Sakai S ed.
Dioxin'94: 14th International Symposium on Chlorinated Dioxins, PCB
and Related Compounds, Kyoto, Japan, 21-25 November 1994 (Short
papers). Kyoto, Kyoto University, Department of Environmental and
Sanitary Engineering, pp 235-238 (Organohalogen Compounds, Volume 19).
Watanabe I, Kawano M, & Tatsukawa R (1995) Polybrominated and mixed
polybromo/chlorinated dibenzo- p-dioxins and -dibenzofurans in the
Japanese environment. In: Bolt D, Clement R, Fiedler H, Harrison B,
Ramamoorthy S, & Reiner E ed. Dioxin'95: 15th International Symposium
on Chlorinated Dioxins and Related Compounds, Edmonton, Canada, 21-25
August 1995 (Short papers). Edmonton, Alberta, Dioxin'95 Secretariat,
pp 337-340 (Organohalogen Compounds, Volume 24).
Weber LWD, Ernst SW, Stahl BU, & Rozman K (1993) Tissue distribution
and toxicokinetics of 2,3,7,8-tetra-chlorodibenzo- p-dioxin in rats
after intravenous injection. Fundam Appl Toxicol: 21: 523-534.
Weberruß U (1990) [Polyhalogenated dibenzodioxins and dibenzofurans in
the environment.] Tübingen, Eberhard-Karls University, Faculty of
Chemistry and Pharmacy, 168 pp (Dissertation) (in German).
Webster GRB, Muldrew DH, Graham JJ, Sarna LP, & Muir DCG (1986)
Dissolved organic matter mediated aquatic transport of chlorinated
dioxins. Chemosphere, 15: 1379-1386.
Whitelaw M, Pongratz I, Wilhelmsson A, Gustaffson JA, & Poellinger L
(1993) Ligand-dependent recruitment of the Arnt coregulator determines
DNA recognition by the dioxin receptor. Mol Cell Biol, 13: 2504-2514.
Whitlock JP Jr (1990) Genetic and molecular aspects of
2,3,7,8-tetrachlorodibenzo- p-dioxin. Annu Rev Pharmacol Toxicol, 30:
251-277.
Whitlock JP Jr (1993) Mechanistic aspects of dioxin action. Chem Res
Toxicol, 6: 754-763.
WHO (1989) Environmental health criteria 88: Polychlorinated
dibenzo-para-dioxins and dibenzofurans. Geneva, World Health
Organization, International Programme on Chemical Safety, 409 pp.
WHO (1993) Environmental health criteria 140: Polychlorinated
biphenyls and terphenyls, 2nd ed. Geneva, World Health Organization,
International Programme on Chemical Safety, 682 pp.
WHO (1994a) Environmental health criteria 152: Polybrominated
biphenyls. Geneva, World Health Organization, International Programme
on Chemical Safety, 577 pp.
WHO (1994b) Environmental health criteria 162: Brominated
diphenylethers. Geneva, World Health Organization, International
Programme on Chemical Safety, 347 pp.
WHO (1995) Environmental health criteria 172: Tetrabromobisphenol A
and derivatives. Geneva, World Health Organization, International
Programme on Chemical Safety.
WHO/ECEH (1996) Quality assessment of PCB, PCDD and PCDF analysis:
Third round of WHO-coordinated study. World Health Organization,
Regional Office for Europe (Environmental Health in Europe 2).
WHO/EURO (1989) Levels of PCBs, PCDDs and PCDFs in breast milk:
Results of WHO-coordinated interlaboratory quality control studies and
analytical field studies. Copenhagen, World Health Organization,
Regional Office for Europe (Environmental Health Series 34).
WHO/EURO (1991) Levels of PCBs, PCDDs and PCDFs in human milk and
blood: Second round of quality control studies. Copenhagen, World
Health Organization, Regional Office for Europe (Environment and
Health in Europe 37).
Wiberg K, Rappe C, & Haglund P (1992) Analysis of bromo-, chloro- and
mixed bromo/chloro-dibenzo- p-dioxins and dibenzofurans in salmon,
osprey and human milk. Chemosphere, 24: 1431-1439.
Wichmann H, Zelinski V, Lorenz W, & Bahadir M (1992a) [Chlorinated and
brominated pollutants in accidental fire residues from private houses
-- analytical results, waste disposal, decontamination, safety
provisions for workers.] Wiss Umwelt, 12: 75-78 (in German).
Wichmann H, Lorenz W, & Bahadir M (1992b) Chlorinated compounds in
accidental fire residues from private houses. Fresenius Environ Bull,
1: 376-381.
Wichmann H, Zelinski V, Scholz-Böttcher B, Lorenz W, & Bahadir M
(1993) Sampling strategy to investigate the distribution behaviour of
low volatile pollutants like "dioxins" during vehicle fires in traffic
tunnels. Chemosphere, 26: 1159-1166.
Wilken M & Schanne L (1994) [Brominated dioxins -- by fires: a higher
risk than PCDD and PCDF.] In: [Dioxin emissions and sources.]
Darmstadt, Eigenverlag, pp 109-127 (in German).
Wilken M, Beyer A, & Jager J (1990) Generation of brominated dioxins
and furans in a municipal waste incinerator (MWI): Results of a case
study. In: Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th
International Symposium on Chlorinated Dioxins and Related Compounds.
Bayreuth, Germany, Ecoinforma Press, pp 377-380 (Organohalogen
Compounds, Volume 2).
Wittich R-M, Wilkes H, Sinnwell V, Francke W, & Fortnagel P (1992)
Metabolism of dibenzo- p- dioxin by Sphingomonas sp. strain RW1.
Appl Environ Microbiol, 58: 1005-1010.
Wolfe WH, Michalek JE, Miner JC, Pirkle JL, Caudill SP, Patterson DG
Jr, & Needham LL (1994) Determinants of TCDD half-life in veterans of
Operation Ranch Hand. J Toxicol Environ Health, 41: 481-488.
Wurrey CJ, Fairless BJ, & Kimball HE (1989) Matrix isolation GC/FT-IR
spectra of octachlorinated and 2,3,7,8-tetrabrominated
dibenzo- p-dioxins and dibenzofurans. Spectroscopy, 4: 39-43.
Yang KH, Yoo BS, & Choe SY (1983) Effects of halogenated
dibenzo- p-dioxins on plasma disappearance and biliary excretion of
ouabain in rats. Toxicol Lett, 15: 259-264.
Young AL (1983) Long term studies on the persistence and movement of
TCDD in a natural ecosystem. In: Tucker RE, Young AL, & Gray AP ed.
Human and environmental risks of chlorinated dioxins and related
compounds. New York, London, Plenum Press, pp 173-190.
Zabel EW, Cook PM, & Peterson RE (1995) Toxic equivalency factors of
polychlorinated dibenzo- p-dioxin, dibenzofuran and biphenyl
congeners based on early life stage mortality in rainbow trout
(Oncorhynchus mykiss). Aquat Toxicol, 31: 315-328.
Zacharewski T, Harris M, Safe S, Thoma H, & Hutzinger O (1988)
Applications of the in vitro aryl hydrocarbon hydroxylase induction
assay for determining "2,3,7,8-tetrachloro-dibenzo- p-dioxin
equivalents": Pyrolyzed brominated flame retardants. Toxicology, 51:
177-189.
Zacharewski T, Harris M, Safe S, Thoma H, Hauschulz G, Knorr E, &
Hutzinger O (1989) Applications of the in vitro AHH induction
bioassay for determining 2,3,7,8-TCDD equivalents: Pyrolyzed flame
retardant mixtures. Chemosphere, 18: 383-387.
Zelinski V, Lorenz W, & Bahadir M (1993) Brominated flame retardants
and resulting PBDD/F in accidental fire residues from private
residences. Chemosphere, 27: 1519-1528.
Zelinski V, Wichmann H, Lorenz W, & Bahadir M (1994) Chlorinated and
brominated dioxins and furans in fire residues from private residences
and vehicles -- A comparison. Fresenius Environ Bull, 3: 449-453.
Zier B, Lahaniatis D, Bieniek D, & Kettrup A (1990) Formation of
brominated dibenzodioxins and -furans by thermolysis of
polybutylene-terephthalate containing decabromodiphenylether
-- influence of temperature, antimony trioxide and water. In:
Hutzinger O & Fiedler H ed. Dioxin'90/EPRI-Seminar: 10th International
Symposium on Chlorinated Dioxins and Related Compounds. Bayreuth,
Germany, Ecoinforma Press, pp 381-384 (Organohalogen Compounds, Volume
2).
Zier B, Lenoir D, Lahaniatis ES, & Kettrup A (1991) Surface catalyzed
halogenation-dehalogenation reactions of aromatic bromine compounds
adsorbed on fly ash. Chemosphere, 22: 1121-1129.
Zober A, Messerer P, & Huber P (1990) Thirty-four-year mortality
follow-up of BASF employees exposed to 2,3,7,8-TCDD after the 1953
accident. Int Arch Occup Environ Health, 62: 139-157.
Zober MA, Ott MG, Päpke O, Senft K, & Germann C (1992) Morbidity study
of extruder personnel with potential exposure to brominated dioxins
and furans: I. Results of blood monitoring and immunological tests.
Brit J Ind Med, 49: 532-544.
APPENDICES
APPENDIX I: Directives/Test Rules Concerning PBDDs/PBDFs
Table 2 (in chapter 2) gives the PBDD/PBDF congeners substituted
with bromine in the 2,3,7,8-positions. As these are the most toxic
congeners, in some investigations only these congeners are determined.
The German Dioxin Directive (1994) has established temporary and
permanent limitations on the concentrations of certain
2,3,7,8-substituted PBDDs/PBDFs in products brought to the German
marketplace (see also Anon, 1996). For the first 5 years (i.e. until
15 July 1999), the sum concentration of the four PBDD/PBDF congeners
listed in category A must be less than 10 ppb, and the sum
concentration of the eight congeners in categories A and B must be
less than 60 ppb. After this time, the limits become 1 ppb and 5 ppb,
respectively. Restrictions on PCDDs/PCDFs are already established.
Category A Category B
2,3,7,8-TeBDD 1,2,3,7,8-PeBDF
2,3,7,8-TeBDF 1,2,3,4,7,8-HxBDD
1,2,3,7,8-PeBDD 1,2,3,6,7,8-HxBDD
2,3,4,7,8-PeBDF 1,2,3,7,8,9-HxBDD
In 1987, the US EPA issued a Test Rule requiring manufacturers
and importers of certain halogen-containing chemicals to analyse their
products for 2,3,7,8-substituted PHDDs/PHDFs. Specific limits of
quantitation and analytical requirements concerning recoveries and
precision were included in the Test Rule. Among the chemicals tested
were decabromodiphenyloxide and TBBPA.
Analyte Limit of quantitation (ppb)
2,3,7,8-TeBDD 0.1
1,2,3,7,8-PeBDD 0.5
1,2,3,4,7,8-HxBDD 2.5
1,2,3,6,7,8-HxBDD 2.5
1,2,3,7,8-HxBDD 2.5
1,2,3,4,6,7,8-HpBDD 100
2,3,7,8-TeBDF 1
1,2,3,7,8-PeBDF 5
2,3,4,7,8-PeBDF 5
1,2,3,4,7,8-HxBDF 25
2,3,4,6,7,8-HxBDF 25
1,2,3,7,8,9-HxBDF 25
1,2,3,4,6,7,8-HpBDF 1000
1,2,3,4,7,8,9-HpBDF 1000
APPENDIX II: Apparatuses and Terminology Used in Thermolysis
Experiments
Apparatuses used for thermolysis experiments are given below:
DIN oven: According to German standard DIN 53436. Open horizon tal
quartz tube (1 m long, 4 cm in diameter) with a gas flow in one
direction. The sample (mg to a few g) is placed on a quartz plate
within the tube, and a ring oven moves outside along the tube (1
cm/min). The design of the DIN apparatus simulates real fire
situations ranging from smouldering to open flame.
BIS oven: Same as DIN apparatus but stationary oven.
VCI oven: Vertical combustion apparatus with two heating zones. The
oven is heated to a defined temperature, and then the sample
(approximately 50 mg) is dropped via a valve into the combustion zone.
During the experiment, an air flow is maintained. Residence times
range from a few seconds up to 10 min. Owing to the small sample size,
the limit of quantification is relatively high. The VCI apparatus
simulates an instant fire.
In general, the combustion gases are adsorbed on XAD resin, and
the solid residues can be analysed for PBDDs/PBDFs as well. Typical
temperature ranges for all experiments are from 300 to 800 _C.
Quartz tube: The sample is sealed into a quartz tube and then heated
to a defined temperature.
Terms referring to thermal treatment are used as follows:
Thermolysis: General term for treating materials, compounds, etc. at
elevated temperatures. No specification is given for oxygen content,
technology, etc.
Combustion: General term for thermal treatment, more specifically in
the presence of oxygen or air.
Incineration: Refers to large-scale plants as used, for example, in
the incineration of municipal solid waste, hazardous waste, sewage
sludge, clinical waste, etc.
Pyrolysis: Thermal treatment under exclusion of oxygen/air.
RÉSUMÉ
1. Identité, propriétés physiques et chimiques et méthodes
d'analyse
Les polybromodibenzo- p-dioxines (PBDD) et les polybromodi
benzofuranes (PBDF) sont des composés aromatiques de structure
quasiment plane. Il peut y avoir théoriquement 75 PBDD et 135 PBDF. En
outre, un grand nombre de dérivés halogénés mixtes -- 1550
bromo/chloro dibenzo- p-dioxines (PXDD) et 3050 bromo/chloro
dibenzofuranes (PXDF) -- sont également envisageables sur le plan
théorique. En raison de la complexité des méthodes d'analyse et de la
rareté des substances de référence utilisables à des fins analytiques,
il n'est possible de rechercher et de doser qu'un petit nombre de ces
composés. Les dérivés les plus toxiques sont ceux qui sont substitués
en position 2, 3, 7 et 8. Il existe ainsi 7 PBDD et 10 PBDF substitués
en 2, 3, 7 et 8 et on peut également envisager 337 PXDD et 647 PXDF
substitués aux mêmes positions.
Les PBDD et les PBDF ont une masse moléculaire plus élevée que
celle de leurs homologues chlorés. Leur point de fusion est élevé et
leur tension de vapeur est faible, tout comme leur solubilité dans
l'eau. Ils sont généralement solubles dans les graisses, les huiles et
les solvants organiques. On ne possède que très peu de données
expérimentales sur les propriétés physiques et chimiques de ces
composés.
Les PBDD et les PBDF sont photolysés plus rapidement que les
dibenzo- para-dioxines polychlorées (PCDD) et les dibenzofurannes
polychlorés (PCDF). Ils présentent une bonne stabilité à la chaleur.
Les températures de formation ou de destruction des PBDD/PBDF
dépendent d'un certain nombre de facteurs, entre autres, la présence
ou l'absence d'oxygène, de polymères ou d'additifs retardateurs de
flamme comme le trioxyde d'antimoine (Sb2O3).
En présence d'un excès de chlore celui-ci se substitue au brome
pour donner des PXDD/PXDF.
En raison du caractère toxique de ces composés et de leur
sensibilité à la photolyse, des précautions sont en prendre lors de
l'échantillonnage et de l'analyse. Il faut en particulier faire appel
à des méthodes extrêmement sensibles, sélectives et spécifiques (comme
la chromatographie en phase gazeuse couplée à la spectrométrie de
masse) en raison du nombre très élevé de dérivés. Les méthodes
d'échantillonnage sont identiques pour toutes les dibenzo- p-dioxines
et tous les dibenzofuranes polyhalogénés (PHDD et PHDF), toutefois la
séparation et le dosage des PBDD et des PBDF (et des PXDD et des PXDF)
sont un peu différents de ceux de leurs homologues chlorés. Les
PBDD/PBDF ont une masse moléculaire plus élevée et un temps de
rétention chromatographique plus long que leurs homologues chlorés. En
outre, la configuration des motifs isotopiques observés en
spectrométrie de masse et les interférences sont également différents.
Le très petit nombre de substances de référence limite
considérablement les possibilités d'identification des dérivés
polybromés. Pour la même raison, la recherche et le dosage des dérivés
halogénés mixtes sont quasiment impossibles.
2. Formation et sources d'exposition humaine et environnementale
On ne connaît pas de PBDD/PBDF d'origine naturelle. On ne les
produit pas non plus délibérément (sauf à des fins scientifiques): ce
sont en général les sous-produits involontaires de divers processus.
Ils peuvent se former au cours de réactions chimiques, photochimiques
ou thermiques à partir d'un certain nombre de précurseurs ou être
synthétisés de novo.
Ils sont présents à l'état d'impuretés dans divers dérivés
organiques bromés, comme les bromophénols et en particulier, dans les
retardateurs de flamme, comme les polybromodiphényléthers (PBDE), le
décabromodiphényle (décaBB ou DBB), le 1,2-bis(tribromophénoxy)éthane,
le tétrabromobisphénol A (TBBPA) etc. On en a mis en évidence dans des
résidus de distillation de divers bromophénols et bromoanilines ainsi
que dans des déchets de laboratoires de chimie.
On a décelé la présence de PBDF et, en moins grande quantité, de
PBDD, dans les produits de photodécomposition de composés organiques
bromés tels que les PBDE et les bromophénols.
Les essais de thermolyse effectués en laboratoire ont mis en
évidence la formation de PBDD/PBDF à partir de bromophénols, de PBDE,
de polybromobiphényles (PBB) et autres dérivés bromés utilisés comme
retardateurs de flamme (à l'état pur ou dans une matrice polymère).
Les rendements obtenus variaient dans de larges proportions, allant de
zéro à des valeurs maximales de l'ordre du g/kg (dans le cas des
PBDE). En général, il y avait beaucoup plus de PBDF que de PBDD. On a
constaté que pour une série de retardateurs de flamme, la température
optimale de formation des PBDF se situait entre 600 et 900°C. La
présence de polymères ou de synergisants (par ex. Sb2O3) a eu pour
effet de diminuer la température de optimale de formation, la ramenant
aux alentours de 400°C. Outre la température et la présence d'une
matrice polymère ou de synergisants, d'autres facteurs tels que les
métaux ou oxydes métalliques, l'eau, l'oxygène et le système de
combustion utilisé ont également eu une influence sur la nature et la
proportion des PBDD ou PBDF obtenus. Dans le cas de mélanges ternaires
constitués d'un PBDE, d'une matrice polymère et de Sb2O3, ce sont
des tétrabromodibenzofuranes (tétraBDF ou TeBDF) que l'on a surtout
obtenus. On a trouvé des 2,3,7,8-PBDD/PBDF (tétra et hepta) à diverses
concentrations; par exemple du 2,3,7,8-TeBDF à des concentrations
allant jusqu'à 2000 mg/kg dans les produits de pyrolyse de polymères
contenant de l'octabromodiphényléther (octaBDE ou OBDE).
Dans l'industrie des matières plastiques, des températures
élevées (150-300°C) peuvent être atteintes au cours de divers
processus. L'étude des vapeurs qui s'échappent des presses à injecter,
extrudeuses etc. lors du thermoformage de plastiques tels que les
résines ABS (acrylonitrile-butadiène-styrène) ou PBT (téréphtalate de
polybutylène) contenant divers retardateurs de flamme bromés, montre
que des PBDD/PBDF peuvent se former à ces températures. Ce sont l'OBDE
et le décabromodiphényléther (décaBDE ou DBDE) qui donnent naissance
aux plus grandes quantités de PBDD/PBDF, les PBDF étant les plus
abondants. La quantité de TBBPA ou de TBPI
(bis-tétrabromophtalimide-éthylène) sont beaucoup plus faibles (de
plusieurs ordres de grandeur). Lors du thermoformage de résines ABS
contenant du bromostyrène ou du 1,2-bis(tribromophénoxy)éthane comme
retardateurs de flamme, on n'a pas décelé de PBDD/PBDF. En ce qui
concerne les autres homologues substitués en 2,3,7,8, soit on ne les a
pas recherchés (en présence de DBDE), soit on en a trouvé des traces
(en présence d'OBDE), soit on n'est pas parvenu à les mettre en
évidence (en présence de TBBPA et de TBPI).
On a analysé divers plastiques à différents stades de leur mise
en forme, à la recherche de PBDD/PBDF. Il s'agissait soit de poudres à
mouler sous forme de granulés soit de pièces moulées dont on
connaissait les additifs retardateurs de flamme ainsi que de divers
objets de l'électroménager gris ou brun (téléviseurs, imprimantes,
ordinateurs) dont on ignorait quels additifs ils pouvaient contenir.
C'est dans les produits contenant des PBDE comme retardateurs de
flamme que l'on a trouvé la plus grande quantité de PBDD/PBDF. Ces
teneurs étaient de l'ordre de plusieurs milliers de µg/kg, c'est à
dire qu'elles dépassaient de plusieurs ordres de grandeur la
concentration observée dans les autres systèmes polymère/retardateur
de flamme. Les quantités formées dépendaient de la température et de
la durée des divers processus: adjonction de l'additif, extrusion ou
moulage par injection. Là encore et à quelques exceptions près, ce
sont les PBDF qui prédominaient par rapport aux PBDD, les dérivés les
plus substitués (plus de quatre bromes) étant présents en abondance.
Ce sont les pentabromodibenzofuranes (pentaDBF ou PeDBF) et les
hexabromodibenzofuranes (hexaBDF ou HxBDF) dont les concentrations
étaient les plus élevées. Dans des gainages de plastique, la
concentration de ces derniers atteignait 3000 µg/kg. Les supports de
circuits imprimés contenaient des tétra- et pentaBDF aux
concentrations maximales respectives de 1300 et 1400 µg/kg. La
concentration totale en PBDF (mono à hexa) se situait dans les limites
de 3,6 à 3430 µg/kg. Les PBDF substitués en 2,3,7,8 n'ont pas été
dosés ou bien n'étaient pas décelables ou encore étaient présents à
des concentrations trop faibles. La concentration maximale des PBDF
substitués en 2,3,7,8 (tétra à hexa) dans des gainages et des supports
de circuits imprimés allait de 11 µg/kg (tétra) à 203 µg/kg (hexa).
Les mesures effectuées pour déterminer si des PBDF sont libérés
par les téléviseurs et autres appareils de ce genre pendant leur
fonctionnement ont montré que la concentration de ces composés dans
l'air allait de zéro (non décelable) à 1800 pg de PBDF totaux (tétra à
hexa) par appareil.
La combustion de produits contenant des composés bromés provoque
un dégagement de PBDD/PBDF. Lors d'essais au cours desquels on avait
reproduit les conditions d'un véritable incendie, on a constaté que
des appareils électriques tels que téléviseurs, imprimantes, terminaux
d'ordinateurs et leurs gainages ou boîtiers laissaient des résidus
contenant de fortes concentrations de PBDF (mono à hexa), atteignant
plusieurs milliers de mg/kg. Ces concentrations étaient également
élevées dans les fumées (jusqu'à 1700 µg/m3) et leurs condensats
(plusieurs centaines de µg/m2). La concentration des PBDD était égale
à 3% de celle des PBDD/PBDF. Celle de l'isomère substitués en 2,3,7,8
n'atteignait pas 3% de la concentration totale des PBDD/PBDF. Les
penta- et hexaPBDF substitués en 2,3,7,8 ont fourni entre 1 et 16% du
total correspondant. Lors d'essais d'incendie de véhicules, on a
trouvé dans les résidus, des concentrations en PBDF (mono à octa)
pouvant aller jusqu'à 4,3 µg/kg.
Au cours d'incendies réels dans des résidences privées
(téléviseur étant en cause), dans des immeubles de bureaux (ordinateur
en cause) ou d'autres bâtiments, on a trouvé des concentrations
généralement inférieures à celles que l'on avaient obtenues
expérimentalement comme indiqué ci-dessus, la composition étant
toutefois qualitativement similaire. On a mis en évidence des PBDF
dans la presque totalité des échantillons; par contre, on n'a pas
toujours trouvé des PBDD. Lorsqu'elles étaient présentes, c'était à
faible concentration. La concentration des PBDF dans les résidus de
combustion était généralement de l'ordre de plusieurs µg/kg (faible à
élevée) mais on a observé des valeurs maximales (somme des dérivés
mono à hexa) pouvant atteindre 107 mg/kg. A proximité immédiate du
lieu des incendies, on a décelé la présence d'une contamination par
des PBDF (mono à hexa) à des concentrations allant la plupart du temps
de 0,1 à 13 µg/m2. En outre, on a pu déceler la présence de PXDD/PXDF
à des concentrations significatives. La proportion des PBDD/PBDF
substitués en 2,3,7,8 était relativement faible dans la majeure partie
des échantillons étudiés. Par exemple, lors d'incendies impliquant des
téléviseurs, les proportions maximales étaient respectivement égales à
3, 10 et 18% du total des tétra-, penta- et hexaBDF. Des prélèvements
de suie effectués après l'incendie d'une salle d'ordinateurs
contenaient des tétra- et des pentabromodibenzo- p-dioxines (tétra/
pentaBDD ou TeBDD/PeBDD) ainsi que des tétra- et pentaBDF à diverses
concentrations, la plus élevée (48 µg/kg) étant celle du 2,3,7,8-TeBDF
(TBDF).
On a mis en évidence des PXDD dans les cendres d'une chaudière à
bois. On n'a toutefois pas précisé de quelle sorte de bois il
s'agissait (traité ou non traité). On ne dispose d'aucune donnée sur
l'incinération d'autres combustibles comme le charbon, la tourbe ou le
mazout.
On a signalé la présence de PBDD/PBDF ou de PXDD/PXDF dans les
cendres volantes et les gaz émis par les incinérateurs municipaux ou
hospitaliers ou encore ceux que l'on utilise pour détruire les déchets
dangereux. La plupart de ces composés prennent probablement naissance
dans l'incinérateur lui-même, soit à partir de précurseurs qui
réagissent aux températures élevées engendrées par les flammes, soit
par synthèse de novo à basse température dans la zone de post
combustion de l'appareil. On explique la formation des PXDD/PXDF par
un échange important entre atomes de brome et de chlore (échange avec
des composés chlorés présents dans les déchets), comme on peut
l'observer expérimentalement dans un certain nombre de cas. Les
concentrations de PBDD/PBDF et PXDD/PXDF mesurées dans les cendres
volantes des incinérateurs sont de l'ordre du ng/kg ou du µg/kg. Dans
la plupart des cas, on constate que la concentration des
dibenzo- p-dioxines dépasse celle des dibenzofuranes et que les
PXDD/PXDF sont plus abondants que les PBDD/PBDF. Parmi les homologues
substitués en 2,3,7,8, on a trouvé une dibenzo- p-dioxine
tétrahalogénée mixte (tétraXDD ou TeXDD) (Br2Cl2DD).
L'analyse d'échantillons de déchets provenant de décharges a
révélé la présence de PBDD/PBDF et de PXDD/PXDF à des concentrations
allant de plusieurs centaines à plusieurs milliers de ng/kg de poids à
sec. La concentration des dibenzo- p-dioxines (jusqu'à 580 ng/kg)
était inférieure à celle des dibenzofuranes (jusqu'à 4230 ng/kg). En
ce qui concerne les homologues, ce sont les dérivés les moins
halogénés (mono à tétra) qui prédominaient. Dans les déchets de
laboratoires de chimie, on a relevé la présence de PBDD/PBDF dont la
concentration maximale atteignait 15 500 ng/kg (dans le cas des
hexaBDF).
On a décelé la présence de PBDD/PBDF dans des plastiques (avec ou
sans métaux) à différents stades de leur recyclage. Les échantillons
provenaient pour la plupart de matériel de bureau, de supports de
circuits imprimés et autres types de matériel électronique au rebut.
Dans certains cas, la concentration totale des huits homologues
retenus (substitués en 2,3,7,8) atteignait 65 µg/kg. On a constaté que
la récupération des métaux pouvait aussi être une source de PBDD ou de
PXDD/PXDF. Des PBDD/PBDF ont été mis en évidence dans industries
textiles faisant usage de derivés bromés comme retardateurs de flamme.
On a également décelé des PBDF dans des gaz d'échappement, dans des
textiles avant et après traitement ainsi que dans des dépôts de
cheminée.
Des PBDD/PBDF et des PXDD/PXDF (ainsi que des PCDD/ PCDF) ont été
décelés dans les gaz d'échappement de moteurs utilisant de l'essence
au plomb, dans ceux de moteurs utilisant de l'essence sans plomb avec
ou sans catalyseur, ainsi que dans ceux de moteurs diesel. Etant donné
l'utilisation d'agents de balayage bromés ou chlorés (dibromo- et
dichloroéthane) comme additifs à l'essence au plomb, c'est dans
celle-ci que l'on trouve la plus forte concentration de PHDD/PHDF
(plusieurs milliers de ng/m3). L'essence sans plomb donne lieu à des
émissions beaucoup moins importantes de PHDD/ PHDF (inférieures
d'environ deux ordres de grandeur). L'épuration catalytique des gaz
permet de réduire encore ces valeurs. La concentration est un peu plus
élevée pour les moteurs diesel que pour les moteurs à explosion
(moteurs à allumage commandé) fonctionnant à l'essence sans plomb. On
a constaté que les PBDD/PBDF étaient plus abondants que les PXDD/PXDF
et que les PCDD/PCDF dans les gaz résultant de la combustion d'essence
au plomb. En général, la concentration des dibenzofuranes était
supérieure à celle des dibenzo- p-dioxines, avec prédominance des
homologues les moins substitués (mono à tri). On a observé une
composition analogue dans les résidus adhérant à la paroi des pots
d'échappement.
3. Transport, distribution et transformation dans l'environnement
On ne possède guère de données sur le transport et la
distribution des PBDD/PBDF dans l'environnement. En général, leurs
propriétés physicochimiques suggèrent certaines similitudes avec les
PCDD/ PCDF. On peut donc s'attendre, en cas de libération dans
l'environnement, à ce qu'ils se répartissent de préférence dans les
compartiments riches en carbone ou en corps gras, comme c'est le cas
des PCDD/PCDF.
Le transport des PBDD/PBDF aéroportés s'effectue soit sous la
forme de gouttelettes soit en phase gazeuse, le coefficient de partage
étant fonction du degré de bromation.
On ne dispose d'aucune donnée expérimentale sur la migration des
PBDD/PBDF dans l'eau ou le sol. Dans le cas des PBDF (tri à penta) on
a observé une adsorption aux sédiments. Du fait que les PBDD/PBDF sont
peu solubles dans l'eau, le lessivage à partir du sol devrait être
limité tout en étant susceptible de s'accroître en présence de
solvants organiques ou d'acides humiques.
Il n'existe pas non plus de données expérimentales sur les
processus de transport et de distribution des PBDD/PBDF entre les
divers compartiments du milieu et les biotes ou encore à l'intérieur
de ces biotes. En s'appuyant sur la valeur élevée du coefficient de
partage octanol/eau calculé pour un certain nombre de PCDD/PCDF, PBDD/
PBDF et PXDD/PXDF, on peut s'attendre à ce que la biodisponibilité des
PCDD/PCDF soit également élevée.
On a étudié la photolyse des PBDD/PBDF et celle des PXDD/ PXDF,
soit au laboratoire dans des solvants organiques ou sur des surfaces
de quartz, soit à l'extérieur sur le sol et sur des particules de suie
ou de poussière. C'est dans ces dernières conditions, plus
représentatives de la réalité environnementale, que l'on a observé les
réactions les plus lentes. La débromation réductrice s'est révélée
être l'une des principales voies de décomposition photochimique. La
vitesse de décomposition dépend du nombre et de la position des atomes
de brome. En général, les dérivés les plus substitués ou substitués
sur les chaînes latérales sont ceux dont la demi-vie est la plus
courte. Le calcul de la demi-vie donne des valeurs qui sont de l'ordre
de quelques minutes (rayonnement solaire direct ou UV et ampoules de
quartz), de quelques heures (pellicules solides, particules de suie ou
poussières et rayonnement solaire) et enfin de quelques centaines ou
milliers d'heures (échantillons de sol et rayonnement solaire). Par
exemple, la valeur calculée de la demi-vie du 2,3,7,8-TeBDD (TBDD) est
de 0,8 minutes en présence de lumière solaire (en solution dans un
solvant organique) ou de 32 h (dispersé sous la forme de pellicules
solides). On estime que la demi-vie des différents isomères du
tétraBDD est de 3 à 6 mois à la surface du sol. Si on les compare aux
PCDD/PCDF, on constate que les homologues bromés sont
photochimiquement moins stables. Dans le cas des PXDD/PXDF, ce sont
les atomes de brome qui s'éliminent préférentiellement lors de la
photolyse pour donner naissance à des PCDD/PCDF dont la demi-vie
photolytique est plus longue. Cette transformation des PXDD/ PXDF en
PCDD/PCDF se produit également pendant l'incinération.
Les PBDD/PBDF semblent être peu biodégradables.
La présence de PBDD/PBDF chez l'Homme et les animaux, comme le
révèlent un certain nombre d'études, est l'indication de leur capacité
de bioaccumulation. Lors d'études d'alimentation de type subchronique,
on a constaté que la 2,3,7,8-TeBDD s'accumulait dans l'organisme du
rat. On ne connaît pas la valeur des facteurs de bioaccumulation, de
bioconcentration ou de bioamplification des PBDD/PBDF et des
PXDD/PXDF.
4. Concentrations dans l'environnement et exposition humaine
Jusqu'ici, on ne s'est que rarement préoccupé d'inclure les
PCDD/PCDF et PBDD/PBDF dans les programmes de surveillance de
l'environnement. Les quelques études dont on dispose indiquent qu'ils
n'y sont pas très souvent présents.
Dans l'air ambiant, on trouve plus fréquemment des PBDF que des
PBDD. Seules des PBDD peu bromées (mono à tétra) ont été décelées à
des concentrations allant de non décelable à environ 0,85 pg/m3; il
s'agissait en l'occurrence de monobromodibenzo- p-dioxines (monoBDD
ou MoBDD) dans l'air d'un tunnel routier et d'un garage souterrain.
Dans le cas des PBDF, on a trouvé des homologues mono- à hexabromés à
des concentrations allant de non décelable à 74 pg/m3. Par exemple,
la concentration moyenne des PBDD/PBDF totaux (tri à hexa) mesurée en
Allemagne dans un tunnel routier, en centre ville et dans une banlieue
était respectivement égale à 23 pg/m3, 2 pg/m3 et 0,59 pg/m3; on
n'a pas constaté la présence de 2,3,7,8-TeBDD et la concentration
maximale des 2,3,7,8-TeBDF et des 1,2,3,7,8-PeBDF était respectivement
égale à 0,28 et 0,08 pg/m3. On a mis en évidence des PXDF dans des
échantillons d'air de rues à grande circulation, à des concentrations
allant jusqu'à 41 pg/m3 (Cl1Br1DF). Dans des échantillons de
poussières extérieures (provenant pour la plupart d'autoroutes), on a
également constaté la prédominance de PBDF/ PXDF (concentrations
maximales de plusieurs ng/kg) par rapport aux PBDD/PBDF
(concentrations maximales jusqu'à quelques centaines de ng/kg).
Dans des échantillons d'air prélevés dans des pièces équipées
d'un certain nombres d'appareils électroniques (téléviseurs ou
moniteurs d'ordinateurs), on a constaté la présence de PBDF (tétra à
hepta) à une concentration totale allant de 0,23 à 1,27 pg/m3. On n'a
pas décelé de PBDD. Dans des échantillons de poussière provenant de
salles d'ordinateurs, on a trouvé des PBDF à la concentration totale
de 2,4 à 5,5 µg/kg. Contrairement à ce que l'on a observé dans l'air,
il y avait prédominance des hexaBDF et des heptaBDF (HpBDF)
(heptabromodibenzofuranes). C'est seulement dans les échantillons de
poussière que l'on a relevé la présence de faibles concentrations de
tétraBDD (jusqu'à 1 µg/kg), de tétraBDF substitués en 2,3,7,8 et de
pentaBDF (jusqu'à 0,07 µg/kg). Dans un échantillon de poussière
ménagère, la concentration des PBDF était plus basse d'un facteur 10.
La concentration totale des PBDD/PBDF était égale à celle des PCDD/
PCDF dans la poussière de salles d'ordinateurs mais elle était plus
faible dans la poussière ménagère. La poussière d'un garage souterrain
contenait moins de PBDF (mono et di) et de PXDF (di à tétra)
faiblement bromés, avec une concentration maximale de 4,3 µg/kg pour
les dibenzofuranes dihalogénés mixtes (DiXDF).
On ne dispose d'aucune donnée sur la concentration des PBDD/PBDF
dans l'eau.
Dans des sédiments fluviaux et marins prélevés au voisinage d'un
site industriel, on a trouvé des tétraBDD (jusqu'à 0,006 µg/kg de
poids sec) ainsi que des PBDF (tétra à hexa) (concentration totale
allant jusqu'à 0,37 µg/kg de poids sec). Dans les sédiments provenant
d'un réseau de drainage routier, on a trouvé des PBDF (concentration
totale des mono à tri: 2,5 µg/kg; concentration totale des tétra à
hepta: 0,3 µg/kg) et des PXDF (concentration totale des di et des tri:
1,85 µg/kg), mais pas de PBDD.
De même, des échantillons de sol prélevés à proximité d'une
autoroute contenaient des monobromodibenzofuranes (monoBDF ou MoBDF)
et des dibromodibenzofuranes (DiBDF) (total: 1,3 µg/kg), des tétra- et
des pentaBDF (total: 0,02 µg/kg) et des PXDF (total: 1 µg/kg), mais
pas de PBDD. Des échantillons de sol prélevés sur un site
d'incinération à proximité d'une usine de récupération de métaux
contenaient des PBDF à une concentration totale allant jusqu'à 100
µg/kg, mais pas de PBDD non plus. Dans une série d'échantillons de
boues d'égout provenant de stations d'épuration municipales, on a
trouvé une teneur totale en PBDF allant de non décelable à 3 µg/kg.
Dans un cas, on a décelé des traces de tétraBDD et de 2,3,7,8-TeBDF.
Un échantillon de compost s'est révélé à peu près exempt de PBDD/PBDF
(tétraBDF <0,003 µg/kg).
On ne possède aucune donnée quantitative sur la teneur des
denrées alimentaires en PBDD/PBDF.
Dans de l'herbe et des aiguilles de pin prélevées à proximité
d'une autoroute, on a décelé la présence de PBDF/PXDF faiblement
halogénés (mono à tétra) et de traces de PBDD/PXDD (mono à tri).
On n'a pas trouvé de traces de PBDD/PBDF dans les rares
échantillons biologiques de faune et de flore sauvages dont on
disposait.
Dans du lait de vache provenant de fermes situées à proximité
d'une installation municipale d'incinération, on pense avoir trouvé
des dérivés halogénés qui seraient des tribromodibenzofuranes (triBDF
ou TrBDF), un tétraBDF et un pentaBDF (sans substitution en 2,3,7,8).
Dans les rares échantillons de tissus adipeux et de lait humain
provenant de la population générale qui ont été analysés à la
recherche de PBDD/PBDF, on n'a pas trouvé trace de ces produits.
Une contamination par des PBDD/PBDF est possible sur divers lieux
de travail où l'on produit, transforme, utilise ou évacue certains
retardateurs de flamme ou produits qui en contiennent, notamment quand
sont mis en oeuvre des processus nécessitant des températures élevées.
Le degré d'exposition des travailleurs dépend non seulement des
composés en cause mais encore de la qualité de l'air et de la
ventilation. On ne possède que peu de données résultant de la
surveillance de divers lieux de travail tels qu'usines de production
ou de transformation de matières plastiques, bureaux ou studios
comportant un grand nombre d'appareils électriques en fonctionnement
continu ou unités de recyclage (notamment installations de
récupération du cuivre). En général, on constate que les PBDF sont
plus abondants que les PBDD et que leur concentration est maximale
dans les ateliers où sont produits des polymères contenant des DBDE.
On a pu mettre en évidence des PBDF/PBDD substitués en 2,3,7,8 dans de
nombreux échantillons. Une contamination par ces composés a également
été constatée dans un laboratoire de chimie, dans la partie d'une
paillasse située au-dessous de la hotte. On manque de données de
contrôle relatives aux installations d'incinération des déchets.
5. Cinétique et métabolisme
La plupart des études concernent la 2,3,7,8-TeBDD et, dans une
moindre mesure, le 1,2,3,7,8-TeBDF. La demi-vie a également été
calculée pour un certain nombre d'homologues.
Après administration à des rats par voie buccale, intratrachéenne
ou par application cutanée, on a constaté que la 2,3,7,8-TeBDD était
résorbé dans une proportion qui dépendait de la voie d'administration
et de la dose. Par exemple, une dose unique de 1 nmol de 2,3,7,8-TeBDD
par kg de poids corporel a été résorbée à hauteur de 80% (voie buccale
ou intratrachéenne) ou de 12% (voie percutanée). L'absorption
percutanée de 1 nmol de 1,2,7,8-TeBDF par kg de poids corporel a été
d'environ 29%. Per os, la TeBDD est résorbée dans une proportion
comparable à celle de la 2,3,7,8-tétrachlorodibenzo- p-dioxine
(2,3,7,8- TeCDD ou TCDD). Par contre, l'absorption percutanée de ce
même composé (2,3,7,8-TeBDD) a été à peu près égale aux deux tiers de
celle d'une dose équimolaire de 2,3,7,8-TeCDD.
Lorsqu'on les administre à des rats par n'importe quelle voie, la
2,3,7,8-TeBDD ou le 1,2,7,8-TeBDF se répartissent dans tout
l'organisme, s'accumulant de préférence dans le foie et les tissus
adipeux, puis, dans l'ordre, dans la peau et les muscles. Par exemple,
3 jours après administration de doses uniques de 2,3,7,8-TeBDD (1 nmol
par kg de poids corporel) on observait, dans ces tissus, une
répartition dans les proportions respectives de 20%, 20%, 11% et 4%,
alors que le thymus et les surrénales n'en contenaient respectivement
que 0,03% et 0,4%. Chez le rat, la répartition de la 2,3,7,8-TeBDD
entre le foie et les tissus adipeux dépendait de la dose, du mode
d'exposition, et du temps écoulé depuis l'administration. Le rapport
concentration dans le foie/concentration dans les tissus adipeux
mesuré dans différentes conditions allait de 0,2 à 6,5 (doses uniques
de 2,3,7,8-TeBDD administrées à des rats). On ne possède aucune donnée
expérimentale concernant la transmission des PBDD/PBDF à la
progéniture.
On a décelé la présence de métabolites de tétraBDD/BDF dans la
bile et les matières fécales de rats. Ces métabolites se forment
principalement par hydroxylation du noyau aromatique et débromation
hydrolytique. Le taux de métabolisation (déterminé indirectement par
le taux d'excrétion biliaire) était différent selon qu'il s'agissait
de 2,3,7,8-TeBDD (environ 7%) ou de 1,2,7,8-TeBDF (environ 50%). Trois
jours après l'administration par voie intraveineuse d'une dose de
2,3,7,8-TeBDD égale à 1 nmol par kg de poids corporel, 14% de la dose
initiale étaient retrouvés sous forme de métabolites dans les matières
fécales des rats.
L'élimination et l'excrétion de la 2,3,7,8-TeBDD a été étudiée
chez le rat en utilisant différentes voies d'administration: voies
buccale, intraveineuse, intratrachéenne et percutanée. Dans toutes les
études, la principale voie d'élimination a été la voie fécale. La
radioactivité éliminée allait de 2% (voie percutanée) à 42% (voie
buccale) de la dose initiale, c'est-à-dire 1 nmol de
(3H)2,3,7,8-TeBDD par kg de poids corporel dans les échantillons de
matières fécales, et de 0,2 à 1% dans les échantillons d'urine. De
même, l'étude du 1,2,7,8-TeBDF sur des rats a également montré que
l'excrétion se fait essentiellement par la voie fécale, la dose
initiale administrée par voie intraveineuse, buccale ou percutanée
n'étant excrétée qu'à hauteur de 2 à 3% dans les urines. Au cours des
premiers jours qui ont suivi l'administration par voie buccale, les
composés ont été principalement éliminés tels quels dans les matières
fécales ainsi que dans la bile. La fraction de la dose initiale de
2,3,7,8-TeBDD retrouvée dans les matières fécales des rats après
administration de 1 nmol de ce composé par kg de poids corporel était
respectivement égale à 53% (voie buccale), 43% (voie intratrachéale)
et 10-20% (voie intraveineuse). Quelques jours après l'administration
de 2,3,7,8-TeBDD par voie buccale (1 nmol/kg de poids corporel),
environ 20% du composé initial ont été éliminés sous forme inchangée.
Pour un certain nombre de PBDD/PBDF, on possède des données sur
la rétention et la vitesse d'élimination. En particulier, on sait que
chez le rat, la charge relative de l'organisme en 2,3,7,8-TeBDD (et
homologues) dépend de la voie d'administration et de la dose
administrée, traduisant ainsi la variation du degré de résorption. On
a calculé la demi-vie d'un certain nombre de PBDD/PXDD et PBDF
présents dans divers tissus et dans les matières fécales de rats. Elle
s'échelonne entre 1 jour (élimination du 1,2,7,8-TeBDF présent d
l'organisme) et 99 jours (élimination du 2,3,4,7,8-PeBDF présent dans
le foie). Le calcul de la demi-vie de la 2,3,7,8-TeBDD présente dans
le foie, les matières fécales et les tissus adipeux donne les valeurs
respectives de 17, 18 et 58 jours, valeurs qui sont du même ordre que
celles de la 2,3,7,8-TeCDD dans le cas du foie et des matières fécales
mais plus de deux fois plus élevés que dans le cas des tissus adipeux.
Malgré des différences de rétention au cours des premiers jours, le
2,3,7,8-TeBDF et le 2,3,7,8-tétrachlorodibenzofurane (2,3,7,8-TeCDF ou
TCDF) ont une demi-vie comparable au niveau du foie.
Comme dans le cas des PCDD/PCDF, le calcul donne une demi-vie
beaucoup plus longue chez l'Homme que chez l'animal. Les estimations
sont les suivantes: 3 à 11 ans (moyenne 5,9 ans) pour la 2,3,7,8-TeBDD
et 1 à 2 ans (moyenne 1,5 ans) pour le 2,3,7,8-TeBDF. On a également
pu se rendre compte de la persistance de ces composés dans le cas d'un
chimiste qui avait préparé de la 2,3,7,8-TeBDD et de la 2,3,7,8-TeCDD
en 1956. Trente-cinq ans après, son sang contenait encore une quantité
importante de 2,3,7,8-TeBDD.
6. Effets sur les mammifères de laboratoire et les systèmes
d'épreuve in vitro
La plupart des études ont porté sur la toxicité de la
2,3,7,8-TeBDD, mais on dispose tout de même de quelques données sur
les autres PBDD/PBDF et PXDD/PXDF.
La 2,3,7,8-TeBDD a des effets analogues à ceux de la
2,3,7,8-TeCDD, notamment un syndrome de dépérissement, une atrophie du
thymus et une action toxique sur le foie. On a observé au niveau du
foie des lésions caractéristiques d'une péliose hépatique, lésions qui
n'ont pas été observées chez le rat après exposition à la
2,3,7,8-TeCDD. De par leur nature (létalité, histopathologie, poids du
foie et du thymus), les lésions ou effets toxiques observés chez le
cobaye et le rat après une brève exposition au 2,3,7,8-TeBDF étaient
analogues à ceux observés après exposition au 2,3,7,8-TeCDF.
La 2,3,7,8-TeBDD agit sur le système endocrinien. Chez le rat, on
a observé une modification des hormones thyroïdiennes présentes dans
la circulation ainsi qu'une diminution de la spermatogénèse.
La DL50 par voie orale (période d'observation de 28 jours) de la
2,3,7,8-TeBDD pour le rat Wistar est d'environ 100 µg/kg de poids
corporel dans le cas des femelles et d'environ 300 µg/kg p.c. dans le
cas des mâles. Celle de la 2,3,7,8-TeCDD tirée d'autres études varie
de 22 à >3000 µg/kg p.c. Des doses équimolaires de 2,3,7,8-TeBDF et
de 2,3,7,8-TeCDF ont entraîné une mortalité comparable chez des
cobayes. Par exemple, on a observé une mortalité de 100% après
administration de 2,3,7,8-TeBDF (0,03 µmol/kg p.c., 15,8 µg/kg p.c.)
et de 2,3,7,8-TeCDF (0,03 µmol/kg p.c., 10 µg/kg p.c.). On a noté la
présence de lésions prépéliotiques et une modification des hormones
thyroïdiennes chez des rats qui avaient reçu une dose unique de 100 µg
de 2,3,7,8-TeBDD par kg de poids corporel.
Chez des rats Wistar ayant reçu pendant 13 semaines de la
2,3,7,8-TeBDD par voie orale, on a relevé des signes de réduction de
la spermatogénèse, la présence de spermatocytes anormaux ou nécrosés,
les signes d'une péliose hépatique grave ainsi qu'une modification des
hormones thyroïdiennes circulantes et du poids des organes. La dose
sans effet nocif observable (NOAEL) a été trouvée égale à 0,01 µg/kg
p.c. par jour.
Du 2,3,7,8-TeBDF administré par voie orale à des rats
Sprague-Dawley pendant 4 semaines a provoqué un retard de croissance
lié à la dose et des anomalies histopathologiques au niveau du foie et
du thymus. La NOAEL a été estimée 1 µg/kg p.c. par jour.
Chez des souris qui avaient reçu des 2,3,7,8-PBDD/PBDF
administrés par voie orale ou sous-cutanée on a noté, pour certains de
ces produits, des effets délétères sur le développement à des doses
non toxiques pour les mères et non létales pour les foetus. La dose la
plus faible (en µg/kg p.c.) produisant un effet observable (LOEL) - à
savoir une hydronéphrose et une fente palatine - après administration
d'une dose unique par voie orale à des souris gravides a été trouvée
respectivement égale à: 3 et 48 pour la 2,3,7,8-TeBDD, à 25 et 200
pour le 2,3,7,8-TeBDF, à 400 et 2400 pour le 2,3,4,7,8-PeBDF et à 500
et 3000-4000 pour le 1,2,3,7,8-PeBDF. On a constaté que la
2,3,7,8-TeBDD et la 2,3,7,8-TeCDD avaient pratiquement la même
aptitude à induire une hydronéphrose lorsqu'on utilisait la mole comme
unité; par contre, sur une base pondérale, les isomères bromés se
révélaient moins aptes que les isomères chlorés à produire une
hydronéphrose ou une fente palatine. Le 2,3,7,8-TeBDF, en revanche,
était plus actif que le 2,3,7,8-TeCDF.
On n'a trouvé aucune donnée sur la mutagénicité des PBDD/ PBDF ni
sur des points d'aboutissement toxicologiques en rapport avec des
propriétés mutagènes.
On ne dispose d'aucune étude sur la toxicité ou la
cancérogénicité à long terme des PBDD/PBDF. Une épreuve de
transformation cellulaire sur macrophages péritonéaux murins a donné
un résultat positif avec la 2,3,7,8-TeBDD. Cependant, l'activité
transformante de la 2,3,7,8-TeBDD était sept fois moins forte que
celle de la 2,3,7,8-TeCDD. En injectant les cellules ainsi obtenues à
des souris nude par voie sous-cutanée, on a observé l'apparition
ultérieure de tumeurs.
Après injection d'une série de PBDD et de PXDD (tétra et penta)
par voie intrapéritonéale à des rats Wistar mâles immatures, on a
constaté une perte de poids au bout de 14 jours. En se basant sur la
valeur de la DE50 (exprimée en moles), on a constaté que les composés
les plus toxiques étaient la 2,3,7,8-TeBDD, la 2-Br1-3,7,8-Cl3-DD et
la 2,3-Br2-7,8-Cl2-DD (TBCDD), qui ne sont substituées que sur les
quatre positions latérales. Pour l'activité relative des autres PBDD
étudiées, on a trouvé l'ordre suivant: 2,3,7,8- > 1,2,3,7,8- >
1,2,4,7,8- > 1,3,7,8-DD. Selon d'autres études, il n'y aurait que peu
de différence entre la 2,3,7,8-TeCDD et la 2,3,7,8-TeBDD en ce qui
concerne la valeur de la DE50 (exprimée en moles) pour la perte de
poids, l'atrophie du thymus et l'induction des enzymes hépatiques.
Une atrophie du thymus et d'autres signes d'immunotoxicité (par
ex. dans les paramètres hématologiques et aussi des modifications dans
certaines sous-populations de lymphocytes) ont été observés chez le
rat après exposition à plusieurs PBDD/PXDD et au 2,3,7,8-TeBDF ainsi
que chez le singe marmouset (Callithrix jacchus) après exposition à
la 2,3,7,8-TeBDD et à la TBCDD. On en a conclu qu'exprimée en moles,
l'activité de la 2,3,7,8-TeBDD était comparable à celle de la
2,3,7,8-TeCDD chez le rat et le singe. Par exemple, on a observé un
effet sensible sur certaines sous-populations de lymphocytes simiens
après injection sous-cutanée d'une dose unique de 30 ng de
2,3,7,8-TeBDD par kg de poids corporel, le même effet étant obtenu
avec une dose de 10 ng de 2,3,7,8-TeCDD par kg p.c. On n'a pas étudié
les effets immunotoxiques d'une exposition périnatale aux PBDD/PBDF.
Après avoir administré de manière subchronique de la
2,3,7,8-TeBDD ou de la 2,3,7,8-TeCDD par gavage à des souris, on a
constaté un accroissement des porphyrines hépatiques totales qui
dépendait de la dose.
Après administration à des rats d'une dose unique de
2,3,7,8-TeBDD et de 2,3,7,8-TeCDD, on a observé une réduction de la
concentration et de la quantité totale de vitamine A dans le foie, la
2,3,7,8-TeBDD ayant une activité (exprimée en moles) un peu inférieure
à celle de la 2,3,7,8-TeCDD.
Lors d'une épreuve sur oreille de lapin effectuée avec de la
2,3,7,8-TeBDD et du 2,3,7,8-TeBDF, on a observé une hyperkératose à la
dose de 100 µg/animal mais pas à la dose de 10 µg/animal. Dans le cas
de la 2,3,7,8-TeCDD, la dose sans effet observable (NOEL) était de
0,01 µg/animal.
On a constaté que plusieurs homologues tétra- (Br1Cl3DD,
Br2Cl2DD) et penta- (Br1Cl4DD) halogénés substitués en 2,3,7,8
avaient une activité antiestrogénique analogue à celle de la
2,3,7,8-TeCDD, comme l'a montré l'observation de cultures de cellules
humaines de cancer du sein.
Chez le rat, la 2,3,7-tribromodibenzo- p-dioxine (2,3,7-triBDD/
TrBDD) a réduit l'élimination de l'ouabaïne du plasma et son excrétion
par la voie biliaire; elle a également agi comme anticholagogue, mais
dans une moindre proportion que la 2,3,7,8-TeCDD.
Les PBDD/PBDF et les PXDD/PXDF sont de puissants inducteurs de
certaines enzymes microsomiennes dépendant du cytochrome P-450 (CYP).
On a obtenu une DE50 de 0,8-1 nmol/kg p.c. pour l'induction de la
CYP1A1 et d'environ 0,2 nmol/kg p.c. pour l'induction de la CYP1A2
dans le foie de rat après administration d'une dose unique de
2,3,7,8-TeBDD par voie orale. L'induction de la CYP1A1
(arylhydrocarbure-hydroxylase [AHH] ou de
l'ethoxyrésorufine- O-déséthylase [EROD]) a été observée chez
diverses espèces et un certain nombre de tissus in vivo ainsi que
dans des cultures de cellules de rat in vitro. Divers homologues se
sont révélés actifs à cet égard, de même que les produits de pyrolyse
de certains retardateurs de flamme. En général, l'induction des
enzymes se produisait à des concentration non toxiques, elle dépendait
de la dose, commençait peu après l'exposition et était de longue
durée. Elle était mesurable à des concentrations de l'ordre de la
picomole. L'activité inductrice différait de plusieurs ordres de
grandeur d'un homologue à l'autre, en fonction de la structure
chimique. Les inducteurs les plus actifs étaient la TCDD, la TBDD et
la TBCDD. Comparées à leurs homologues chlorés, les PBDD et les PXDD
avaient une activité (exprimée en moles) à peu près équivalente.
Contrairement à la TCDD, dont l'activité inductrice relative s'est
montrée indépendante du type de tissu examiné, la TBDD a fait preuve
d'en activité inductrice de l'EROD cinq fois plus élevée dans le foie
que dans le tissu cutané ou pulmonaire, lors d'études comportant
l'exposition de souris dans des conditions de subchronicité. Chez le
singe marmouset, l'activité inductrice de l'EROD s'établit comme suit:
TCDD > 2,3,4,7,8-pentachlorodibenzofurane (2,3,4,7,8-pentaCDF/PeCDF)
> 2,3,4,7,8- PeBDF, l'activité des enzymes étant comparée à la
concentration dans le foie. Des épreuves in vitro sur cultures de
cellules de rat ont donné des valeurs analogues pour la CE50 des PXDF
et des PCDF correspondants, l'effet examiné étant l'induction de l'AHH
et de l'EROD.
On pense que les PBDD/PBDF ont le même mode d'action que les
PCDD/PCDF et les d'autres hydrocarbures aromatiques halogénés de type
voisin. La fixation sur le récepteur cytosolique aux hydrocarbures
aromatiques, qui joue un rôle central dans la toxicité des composés du
type 2,3,7,8-TeCDD, a été confirmée pour plusieurs PBDD et PXDD/PXDF.
Leur affinité pour ce récepteur variait de plusieurs ordres de
grandeur, mais elle était comparable à celle de leurs homologues
chlorés.
7. Effets sur l'Homme
On ne dispose d'aucune donnée sur l'exposition humaine aux
PBDD/PBDF ni au sujet de leurs effets sur la santé de la population
dans son ensemble.
Deux cas d'effets aigus dus à une exposition à la 2,3,7,8-TeBDD/
TeCDD ont été rapportés, avec différents symptômes, dont une
chloracné.
Lors d'une autre étude, des employés de sexe masculin travaillant
dans une usine chimique et effectivement exposés à des PBDD/PBDF
provenant de l'utilisation de retardateurs de flamme (OBDE et DBDE),
ont été soumis à des épreuves immunologiques et autres examens de
laboratoire. Malgré la présence de légères modifications dans les
paramètres immunologiques, leur état général ne laissait en aucun cas
penser que la teneur de leur organisme en 2,3,7,8-TeBDD/TeBDF ait eu
des effets sur leur système immunitaire.
On ne connaît aucun cas de cancer mortel qui serait dû aux
PBDD/PBDF.
8. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
On ne possède que des données limitées sur les effets que les
PBDD/PBDF peuvent avoir sur les microorganismes, les plantes, les
invertébrés et les vertébrés.
On a soumis une série de PBDD/PBDF et leurs homologues chlorés à
une épreuve biologique sur alevins de truite arc-en-ciel
(Onchorhyncus mykiss) qui a mis en évidence l'activité de ces
composés. L'épreuve a montré, entre autres, que l'activité des PBDD/
PBDF diminue à mesure qu'augmente de degré de substitution par le
brome. La 2,3,7,8-TeBDD et le 2,3,7,8-TeBDF sont plus actifs que leurs
homologues chlorés respectifs.
RESUMEN
1. Identidad, propiedades físicas y químicas y métodos analíticos
Las dibenzo- p-dioxinas polibromadas (PBDD) y los dibenzo
furanos polibromados (PBDF) son compuestos aromáticos tricíclicos casi
planares. En teoría existen 75 PBDD y 135 PBDF. Además es en teoría
posible la existencia de un amplio número de congéneres halogenados
mixtos: 1550 dibenzo- p-dioxinas bromadas/cloradas (PXDD) y 3050
dibenzofuranos bromados/clorados (PXDF). Dada la complejidad de los
procedimientos analíticos y la escasez de normas analíticas de
referencia, sólo se ha podido identificar y determinar un pequeño
número de estos productos. Los congéneres más tóxicos son los
sustituidos en las posiciones 2,3,7 y 8. Existen 7 PBDD sustituidos en
las posiciones 2,3,7 y 8 y 10 PBDF sustituidos en las posiciones 2,3,7
y 8, así como 337 posibles PXDD sustituidos en las posiciones 2,3,7 y
8, y 647 posibles PXDF sustituidos en las posiciones 2,3,7 y 8.
Los PBDD y PBDF tienen mayores pesos moleculares que sus análogos
clorados, altos puntos de fusión, bajas presiones de vapor y bajas
solubilidades en agua. En general son solubles en grasas, aceites y
disolventes orgánicos. Existen escasos datos experimentales sobre las
propiedades físicas y químicas de los PBDD y PBDF.
La fotolisis se produce con más rapidez en el caso de los PBDD y
PBDF que en las dibenzo- p-dioxinas policloradas (PCDD) y los
dibenzofuranos policlorados (PCDF). Los PBDD y PBDF son termoestables.
Las temperaturas de formación y destrucción de los PBDD y PBDF
dependen de varias condiciones, que incluyen la presencia o ausencia
de oxígeno, polímeros y aditivos pirorretardantes, como el trióxido de
antimonio (Sb2O3).
En presencia de cloro en exceso, el bromo es sustituido por cloro
para dar PXDD y PXDF.
Teniendo en cuenta el carácter tóxico de estos productos y sus
propiedades fotolíticas debe actuarse con cuidado en el curso del
muestreo y el análisis. Dado el alto número de congéneres de los PBDD
y PBDF se necesitan métodos de análisis muy sensibles, selectivos y
específicos (cromatografía de gases o espectrometría de masas). Los
procedimientos de muestreo son idénticos en el caso de todas las
dibenzo- p-dioxinas polihalogenadas (PHDD) y los dibenzo furanos
polihalogenados (PHDF), pero la separación y determinación de las PBDD
y los PBDF (y las PXDD y los PXDF) difiere ligeramente de las
correspondientes a sus análogos clorados. Los PBDD y PBDF tienen pesos
moleculares más altos y mayores periodos de retención en la
cromatografía de gases que los análogos clorados, así como distintos
tipos de agrupación isotópica en espectrometría de masas y de
compuestos de interferencia. La identificación exacta de determinados
congéneres bromados es muy limitada debido al escaso número de
patrones de referencia actualmente disponibles. Por el mismo motivo,
la determinación de los congéneres halogenados mixtos es casi
imposible.
2. Formación y fuentes de exposición humana y ambiental
No se conoce la presencia natural de PBDD y PBDF. No se producen
de modo intencional (excepto para fines científicos), pero aparecen
como productos indeseados en distintos procesos. Pueden formarse por
reacciones químicas, fotoquímicas o térmicas a partir de precursores y
en la llamada síntesis de novo.
Se han hallado PBDD y PBDF como contaminantes en productos
químicos orgánicos bromados (por ej., bromofenoles) y, en particular,
en pirorretardantes, como los éteres difenílicos polibromados (PBDE),
el decabromobifenilo (decaBB o DBB), el 1,2-bis(tribromofenoxi)etano,
el tetrabromobisfenol A (TBBPA) y otros. Se han hallado en residuos de
destilación de algunos bromofenoles y bromoanilinas y en desechos de
laboratorios químicos.
Los PBDF y, en menor grado, las PBDD se han hallado como
productos de degradación fotoquímica de sustancias químicas orgánicas
bromadas, como los PBDE y los bromofenoles.
En experimentos de termolisis en laboratorio se ha observado la
formación de PBDD y PBDF a partir de bromofenoles, PBDE, bifenilos
polibromados (PBB) y otros pirorretardantes bromados (puros o en
matriz de polímero). Se observó una amplia gama de rendimientos, desde
0 hasta los valores máximos (alcanzados a partir de los PBDE) en la
gama de g/kg. Por lo general, los PBDF son mucho más abundantes que
las PBDD. La temperatura óptima de formación de PBDF en una serie de
pirorretardantes puros se situó en la gama de 600-900°C. La presencia
de polímeros o productos sinérgicos (por ej., Sb2O3) produjo la
disminución de la temperatura óptima de formación (hasta 40°C). Además
de la temperatura y la presencia de productos sinérgicos o de una
matriz de polímero, varios otros factores, tales como la presencia de
metales, óxidos metálicos, agua y oxígeno, y el tipo de aparato de
combustión utilizado, influyeron en el rendimiento y el tipo de PBDD y
PBDF obtenidos. En las mezclas ternarias de PBDE, matriz de polímero y
Sb2O3, los tetrabromo dibenzofuranos (tetraBDF o TeBDF) fueron con
frecuencia el grupo homólogo más abundante. Se hallaron en
concentraciones variables PBDD y PBDF sustituidos en las posiciones
2,3,7 y 8 (tetra a hepta); por ejemplo, se halló el 2,3,7,8-TeBDF en
concentraciones de hasta 2000 mg/kg en pirolizados de polímeros que
contenían éter de octabromodifenilo (octaBDE u OBDE).
En la fabricación de plásticos se producen altas temperaturas
(150-300°C) en varios procesos. Los estudios de los vapores de escape
de máquinas de tratamiento de polímeros, como el
acrilonitrilo-butadieno-estireno (ABS) y el tereftalato de
polibutileno (PBT), que contenían distintos tipos de pirorretardantes
bromados, mostraron que pueden formarse PBDD y PBDF (di a octa) a esas
temperaturas. El OBDE y el éter de decabromodifenilo (decaBDE o DBDE)
produjeron las mayores cantidades de PBDD y PBDF, consistiendo la
porción principal en PBDF. Las concentraciones observadas en el caso
de TBBPA o de etileno de bis-tetrabromo-ftalimida (TBPI) eran
inferiores en varios órdenes de magnitud. No se hallaron PBDD ni PBDF
en el curso del tratamiento de ABS pirorretardado por medio de
estireno bromado o 1,2-bis(tribromofenoxi)etano. Los congéneres
sustituidos en las posiciones 2,3,7 y 8 no se determinaron
(fabricación de DBDE), se hallaron sólo en concentraciones
infinitesimales (fabricación de OBDE) o no se detectaron (fabricación
de TBBPA y TBPI).
Se analizó la presencia de PBDD y PBDF en varios materiales
plásticos en distintas etapas de fabricación. Comprendieron resinas
(granuladas) y partes moldeadas, cuyos aditivos pirorretardantes eran
conocidos, así como muestras de dispositivos eléctricos comerciales
(televisores, impresoras, ordenadores), cuyos aditivos
pirorretardantes eran desconocidos. Se hallaron las mayores
concentraciones de PBDD y PBDF en los materiales pirorretardados con
PBDE, en la gama de varios miles de µg/kg, excediendo así en varios
órdenes de magnitud a las concentraciones de otros
pirorretardantes/sistemas de polímero. Los factores que influyen en la
cuantía de la formación son la temperatura y la duración de procesos
tales como el mezclado, la extrusión y el moldeo. También en este caso
dominan los PBDF, con algunas excepciones, sobre las PBDD,
prevaleciendo los derivados muy bromados (>tetra). Las máximas
concentraciones se observaron en el caso de los
pentabromodibenzofuranos (pentaBDF o PeBDF) y los
hexabromodibenzofuranos (hexaBDF o HxBDF). Los últimos alcanzaron
concentraciones tan altas como 3000 µg/kg en piezas de revestimiento.
Los tableros de circuitos impresos contenían tetra- y pentaBDF en
concentraciones máximas de 1300 y 1400 µg/kg, respectivamente. Las
concentraciones totales de PBDF (mono a hexa) se hallaban en la gama
de 3,6-3430 µg/kg. Los PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 no se determinaron, eran indetectables o se hallaban en
concentraciones relativamente bajas. Las concentraciones máximas de
PBDF sustituidos en las posiciones 2,3,7 y 8 (tetra a hexa) en
revestimientos o tableros de circuitos impresos eran de 11 µg/kg
(tetra) a 203 µg/kg (hexa).
Los experimentos destinados a determinar si se liberaban PBDF de
los televisores o aparatos análogos durante el uso mostraron la
presencia de concentraciones en el aire que iban de los niveles
indetectados a 1800 pg de PBDF totales (tetra a hexa) por aparato.
La combustión de productos que contenían compuestos bromados
produjo la emisión de PBDD y PBDF. Las pruebas experimentales de
incendio que simulaban condiciones reales utilizando aparatos
eléctricos tales como televisores, impresoras, terminales de
ordenador, y sus receptáculos, permitieron hallar altas
concentraciones de PBDF (mono a hexa) en los residuos de la combustión
(miles de µg/kg), en el condensado del humo (centenares de µg/m2) y
en el humo (hasta 1700 µg/m3). Las concentraciones de PBDD fueron del
3% aproximada mente de los niveles detectados de PBDD y PBDF. El
isómero 2,3,6,8-sustituido se hallaba sobre todo por debajo del 3% del
total de tetraBDF. Los penta- y hexaBDF 2,3,7,8-sustituidos dieron del
1 al 16% de los totales correspondientes. La combustión de vehículos
de prueba produjo concentraciones de PBDF (mono a octa) de hasta 4,3
µg/kg en los residuos del incendio.
En el curso de incendios reales en residencias privadas (con
inclusión de televisores), oficinas (con inclusión de ordenadores) y
otros edificios, las concentraciones medidas se hallaban en la mayoría
de los casos por debajo de los valores observados en los modelos
experimentales antes descritos, pero la composición cualitativa de las
muestras era análoga. Se hallaron PBDF en casi todas las muestras,
pero no siempre se detectaron PBDD; si se encontraban, sus
concentraciones eran bajas. Las concentraciones de PBDF en los
residuos de la combustión se hallaban principalmente en la gama de
µg/kg (bajas a altas), pero también se observaron valores máximos
(suma de mono a hexa) de hasta 107 mg/kg. Las concentraciones
contaminantes de PBDF (mono a hexa) en las cercanías del lugar del
incendio variaban entre 0,1 y 13 µg/m2 en la mayoría de los casos.
Pudieron detectarse además concentraciones significativas de PXDD y
PXDF. La proporción de PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 era relativamente baja en la mayoría de las muestras examinadas.
Por ejemplo, se registraron proporciones máximas del 3, el 10 o el 18%
de los totales correspondientes de tetra-, penta- o hexaBDF,
respectivamente, en los incendios que comprendieron televisores. Las
muestras de cenizas recogidas después de un incendio en una sala de
ordenadores contenían tetra- y pentabromodibenzo- p-dioxinas
sustituidas en las posiciones 2,3,7 y 8 (tetra/pentaBDD o TeBDD/PeBDD)
y tetra- y pentaBDF, con una concentración máxima de 48 µg/kg en el
caso del 2,3,7,8-TeBDF (TBDF).
Se detectaron PXDD en la ceniza de una caldera de combustión de
madera. Sin embargo, no se especificó el tipo de madera (tratada o sin
tratar). No se dispuso de datos sobre la incineración de otros
combustibles, como carbón, turba o fueloil.
Se señaló la presencia de PBDD/PBDF y/o PXDD/PXDF en cenizas
volantes y en los gases de combustión de incineradores municipales, de
hospital o de desechos peligrosos. La mayor parte de esos productos
estaban formados probablemente en el propio incinerador, a partir de
precursores a altas temperaturas en la llama o por síntesis de novo
a temperaturas bajas en la zona poscombustión del incinerador. La
formación de PXDD y PXDF se explica por las amplias reacciones de
intercambio bromo-cloro (con donantes de cloro en los desechos)
observadas en varias condiciones de prueba. Las cantidades de
PBDD/PBDF y PXDD/PXDF medidas en las cenizas volantes de los
incineradores se hallaban comprendidas en la gama de ng/kg a µg/kg. En
la mayoría de los casos, las concentraciones de dibenzo- p-dioxinas
excedían a las de los dibenzofuranos, siendo los PXDD/PXDF más
abundantes que los PBDD/PBDF. Entre los congéneres 2,3,7,8-sustituidos
se halló una dibenzo- p-dioxina tetrahalogenada mixta (tetraXDD o
TeXDD) (Br2Cl2DD).
Los análisis de muestras de desechos procedentes de varios
vertederos mostraron la presencia de PBDD/PBDF y PXDD/PXDF en
concentraciones de varios centenares a varios miles de ng/kg de peso
en seco. La concentración de las dibenzo- p-dioxinas (hasta 580
ng/kg) era inferior a la de los dibenzofuranos (hasta 4230 ng/kg). Por
lo general, la gama de homólogos estaba dominada por los derivados
menos halogenados (mono a tetra). Los desechos de laboratorios
químicos contenían PBDD y PBDF con una concentración máxima de 15 500
ng/kg en el caso de los hexaBDF.
Se hallaron PBDD y PBDF en materiales plásticos (con o sin
metales) en varias etapas de reciclado. Las muestras procedían
principalmente de maquinaria de oficina, tableros de circuitos
impresos y otras chatarras electrónicas. En algunos casos, la suma de
las concentraciones de 8 congéneres PBDD/PBDF seleccionados con
sustituciones en las posiciones 2,3,7 y 8 llegaba a 65 µg/kg. También
se observó que la recuperación de metales era una fuente de PBDD y/o
PXDD/PXDF. Igualmente se detectaron PBDD y PBDF en la industria
textil, en la que se utilizaban pirorretardantes bromados. Se hallaron
PBDF en el aire de salida de industrias textiles, antes y después del
procesado, y en sedimentos de chimenea.
Se han detectado PBDD/PBDF y PXDD/PXDF (junto con PCDD/ PCDF) en
los gases de escape de motores que utilizan gasolina plomada, en los
gases de escape de motores que usan gasolina sin plomo con o sin
convertidores catalíticos y en los gases de escape de motores diesel.
Teniendo en cuenta que la gasolina plomada contiene productos de
limpieza bromados y clorados (dibromoetano y dicloroetano), las
mayores concentraciones de PHDD y PHDF (varios miles de ng/m3) se
encuentran en este tipo de gasolina. La gasolina sin plomo produce
emisiones muy inferiores de PHDD y PHDF (aproximadamente inferiores en
dos órdenes de magnitud). Tras la limpieza catalítica de los gases se
observa una nueva reducción. Los valores correspondientes a los
motores diesel eran ligeramente superiores a los hallados en los
motores Otto (motores de encendido por chispa) que funcionan con
gasolina sin plomo. En los gases de escape procedentes de la
combustión de gasolina plomada, los PBDD/PBDF eran más abundantes que
los PXDD/PXDF y PCDD/PCDF. En general, las concentraciones de los
dibenzofuranos excedían a las de las dibenzo- p- dioxinas, con un
predominio de los homólogos de baja sustitución (mono a tri). Se han
hallado distribuciones análogas en los residuos adheridos a los
silenciadores de escape.
3. Transporte, distribución y transformación en el medio ambiente
Se dispone de datos muy escasos sobre el transporte y la
distribución en el medio ambiente de los PBDD y PBDF. Por lo general,
sus propiedades fisicoquímicas permiten pensar en analogías con los
PCDD y PCDF. Por consiguiente, si pasan al medio ambiente, pueden
estar de preferencia distribuidos en compartimentos ricos en carbono y
grasas, como sucede con los PCDD y PCDF.
El transporte por el aire de PBDD y PBDF se realiza en forma de
partículas y en fase de vapor, dependiendo la relación de partición
del grado de bromación.
No se dispone de datos experimentales sobre el movimiento de los
PBDD y PBDF en el agua o el suelo. En el caso de los PBDF (tri a
penta) se ha señalado la adsorción al sedimento. Debido a la baja
hidrosolubilidad de los PBDD y PBDF, la filtración por el suelo puede
estar limitada, pero aumentar en presencia de disolventes orgánicos o
ácidos húmicos.
No se dispone de datos experimentales sobre los procesos de
transporte y distribución de los PBDD y PBDF entre el medio ambiente y
los biota o dentro del los biota. Basándose en la existencia de
análogos coeficientes elevados de partición octanol/agua, calculados
para determinados PCDD/PCDF, PBDD/PBDF y PXDD/PXDF, se supone una
biodisponibilidad comparable a la de los PCDD y PCDF.
Se estudió la fotolisis de los PBDD/PBDF y PXDD/PXDF en
disolventes orgánicos y sobre superficies de cuarzo en el laboratorio,
así como en el suelo y en partículas de hollín (y polvo) al aire
libre. Se observaron las reacciones fotolíticas más lentas en estas
últimas condiciones, más pertinentes respecto al medio ambiente. Se
observó que la desbromación reductora era la principal vía metabólica.
La tasa de descomposición de los distintos congéneres depende de su
tipo de sustitución del bromo. Por lo general, los congéneres muy
bromados y los que poseen bromo en posiciones laterales tienen
semividas más breves. Las semividas calculadas eran del orden de
minutos (empleo de luz solar directa o luz ultravioleta [UV] y de
viales de cuarzo), horas (empleo de láminas sólidas o de partículas de
hollín o polvo y luz solar) o de centenares a miles de horas (empleo
del suelo y luz solar). Por ejemplo, las semividas inducidas por la
luz solar estimadas para la 2,3,7,8-TeBDD (TBDD) eran de 0,8 min (en
solución orgánica) o de 32 horas (en dispersión como láminas sólidas).
Se calculó una semivida de 3-6 meses para los isómeros tetraBDD en el
suelo superficial. En comparación con los PCDD y PCDF, los
correspondientes compuestos bromados presentaban menos estabilidad
fotoquímica. Los PXDD y PXDF pierden de preferencia sus átomos de
bromo durante la fotolisis, siendo transformados en PCDD y PCDF, que
tienen semividas fotolíticas más largas. Esa transformación de
PXDD/PXDF en PCDD/PCDF se produce también durante los procesos de
incineración.
Los PBDD y PBDF parecen ser escasamente degradables por la acción
de los microorganismos.
Como se ha observado en algunos estudios, la presencia de PBDD y
PBDF en animales y seres humanos indica su potencial de acumulación.
La 2,3,7,8-TeBDD se acumula en ratas durante la administración
subcrónica. No se dispone de los factores de bioacumulación,
bioconcentración o bioamplificación de los PBDD/PBDF o PXDD/ PXDF.
4. Niveles ambientales y exposición humana
Hasta la fecha, en contraste con los PCDD y PCDF, los PBDD y PBDF
no se han incluido con frecuencia en programas de vigilancia. Los
pocos estudios realizados muestran una aparición limitada.
En el aire ambiental, los PBDF se encuentran con más frecuencia
que las PBDD. Sólo se han detectado PBDD bromados inferiores (mono a
tetra) en concentraciones que iban de las indetectadas a las de 0,85
pg/m3 aproximadamente para las monobromodibenzo- p-dioxinas (monoBDD
o MoBDD) en un túnel de carretera y en un garaje subterráneo. Entre
los PBDF se hallaron homólogos mono a hexabromados, en concentraciones
que iban del nivel indetectado a 74 pg/m3. Por ejemplo, en Alemania
se midieron las concentraciones (valores medios) de los PBDD y PBDF
totales (tri a hexa) en un túnel de carretera, en el centro de una
ciudad y en una zona suburbana, obteniendo valores de 23 pg/m3, 2
pg/m3 y 0,59 pg/m3, respectiva mente; no se detectó la 2,3,7,8-TeBDD
y las concentraciones máximas de 2,3,7,8-TeBDF y 1,2,3,7,8-PeBDF
fueron de 0,28 pg/m3 y 0,08 pg/m3, respectivamente. Se hallaron PXDF
en muestras de aire en zonas de tráfico en concentraciones de hasta 41
pg/m3 (Cl1Br1DF). En las muestras de polvo tomadas al aire libre
(principalmente en carreteras) se observó también un predominio de
PBDF y PXDF (valores máximos de varios miles de ng/kg) respecto a las
PBDD y PXDD (valores máximos de hasta unos centenares de ng/kg).
Las muestras de aire tomado de locales equipados con distintos
dispositivos electrónicos en funcionamiento (televisores o monitores
de ordenador) mostraron la presencia de PBDF (tetra a hepta) en
concentraciones totales que iban de 0,23 a 1,27 pg/m3. No se
detectaron PBDD. Las muestras de polvo recogidas en un local de
ordenadores dieron concentraciones totales de PBDF de 2,4-5,5 µg/kg de
polvo. En contraste con el aire, la distribución homóloga en el polvo
está dominada por los hexaBDF y los heptabromodibenzofuranos (heptaBDF
o HpBDF). Sólo en las muestras de polvo se hallaron concentraciones
bajas de tetraBDD (hasta 1 µg/kg) y de tetra y pentaBDF sustituidos en
las posiciones 2,3,7 y 8 (hasta 0,07 µg/kg) detectables. Las
concentraciones de PBDF en una muestra de polvo doméstico eran
inferiores en un factor de 10. La concentración sumada de PBDD y PBDF
fue igual a la de PCDD y PCDF en el polvo tomado de locales de
ordenadores, pero inferior a la de PCDD y PCDF en el polvo doméstico.
El polvo tomado en un garaje subterráneo contenía PBDF (mono y di) y
PXDF (di a tetra) halogenados inferiores, con una concentración máxima
de 4,3 µg/kg en el caso de los dibenzofuranos dihalogenados mixtos
(DiXDF).
No se dispone de datos sobre las concentraciones de PBDD y PBDF
en las muestras de agua.
En las muestras de sedimentos de río y mar tomados en una zona
industrializada se detectaron tetraBDD (hasta 0,006 µg/kg de peso en
seco) y tetra a hexaBDF (en conjunto hasta 0,37 µg/kg de peso en
seco). El sedimento procedente de un drenaje de carretera contenía
PBDF (suma de mono a tri: 2,5 µg/kg; suma de tetra a hepta: 0,3 µg/kg)
y PXDF (suma de di y tri: 1,85 µg/kg), pero no PBDD.
Asimismo, las muestras de suelo tomadas cerca de una carretera
contenían monobromodibenzofuranos (monoBDF o MoBDF) y
dibromodibenzofuranos (DiBDF) (suma: 1,3 µg/kg) tetra y pentaBDF
(suma: 0,02 µg/kg) y PXDF (suma: 1 µg/kg ), pero no PBDD. Las muestras
de suelo tomadas de un terreno de incineración y cerca de una fábrica
de recuperación de metales dieron concentraciones totales de PBDF de
hasta 100 µg/kg, pero sin detectar PBDD. En una serie de muestras de
fango de alcantarillado procedentes de plantas municipales de
tratamiento de aguas residuales se hallaron concentraciones totales de
PBDF comprendidas entre niveles indetectados y 3 µg/kg. En un caso se
hallaron valores infinitesimales de tetraBDD y 2,3,7,8-TeBDF. Una
muestra de abono biológico estaba casi exenta de PBDD y PBDF
(tetraBDF: <0,003 µg/kg).
No se dispone de datos cuantitativos sobre las concentraciones de
PBDD y PBDF en los alimentos.
En muestras de hierba y de agujas de pino recogidas cerca de
carreteras se encontraron PBDF y PXDF halogenados inferiores (mono a
tetra) y valores infinitesimales de PBDD y PXDD (mono a tri).
No se han encontrado PBDD ni PBDF en las escasas muestras de
animales o plantas silvestres analizados.
En la leche de vaca recogida en granjas lecheras cerca de una
instalación incineradora de desechos municipales se identificaron de
modo provisional tribromodibenzofuranos (triBDF o TrBDF), un tetraBDF
y un pentaBDF (no tenían el tipo de sustitución en las posiciones
2,3,7 y 8).
No se han detectado PBDD ni PBDF en las escasas muestras
analizadas de tejidos adiposos humanos o de muestras de leche
procedentes de la población general.
Es posible la contaminación por PBDD y PBDF en distintos lugares
de trabajo en donde se procede a producir, elaborar, utilizar o
eliminar ciertos pirorretardantes o sus productos, en particular si se
emplean altas temperaturas. La magnitud de la exposición del
trabajador depende no sólo de los productos utilizados sino también de
la calidad del aire y de las condiciones de ventilación. Se dispone de
escasos datos de vigilancia del lugar de trabajo procedentes de
instalaciones de producción o elaboración de plásticos, de oficinas o
de estudios con un alto número de dispositivos eléctricos en
funcionamiento continuo y de instalaciones de reciclado (incluidas
plantas de reciclado de cobre). Por lo general, los PBDF eran más
abundantes que las PBDD y las concentraciones en el aire de PBDF eran
superiores en los lugares de producción de polímeros que contenían
DBDE. En numerosas muestras se detectaron PBDF y PBDD con
sustituciones en las posiciones 2,3,7 y 8. También se halló
contaminación por PBDD y PBDF en la zona de trabajo comprendida debajo
de la chimenea de humos de un laboratorio químico. Se carece de datos
de vigilancia procedentes de instalaciones de incineración de
desechos.
5. Cinética y metabolismo
La mayor parte de los estudios se refieren a la 2,3,7,8-TeBDD y,
en menor cuantía, al 1,2,7,8-TeBDF. Los cálculos de la semivida han
comprendido algunos congéneres adicionales.
La 2,3,7,8-TeBDD se absorbió en ratas después de la
administración oral, intratraqueal y cutánea, variando el porcentaje
de absorción conforme a la vía y la dosis. Las dosis únicas de 1 nmol
de 2,3,7,8-TeBDD/kg de peso corporal condujeron a la absorción del 80%
(vías oral e intratraqueal) o el 12% (vía cutánea) de la dosis
administrada. La absorción cutánea de 1 nmol de 1,2,7,8-TeBDF/kg de
peso corporal fue del 29% aproximadamente. La absorción oral de
2,3,7,8-TeBDD pareció ser comparable a la de la
2,3,7,8-tetraclorodibenzo- p-dioxina (2,3,7,8-TeCDD o TCDD). Sin
embargo, la absorción cutánea de 2,3,7,8-TeBDD fue la tercera parte
aproximadamente de la dosis equimolar de 2,3,7,8-TeCDD.
La 2,3,7,8-TeBDD o el 1,2,7,8-TeBDF administrados a ratas, por
cualquier vía, se distribuyeron por todo el organismo, hallándose los
principales depósitos en los tejidos hepático y adiposo, seguidos de
la piel y el tejido muscular. Por ejemplo, 3 días después de la
administración de dosis orales únicas de 2,3,7,8-TeBDD (1 nmol/kg de
peso corporal), las porciones halladas en esos tejidos eran del 20%,
el 20%, el 11% y el 4% respectivamente, mientras que el timo y las
glándulas suprarrenales contenían el 0,03% y el 0,4%, respectivamente,
de la dosis administrada. La partición de la 2,3,7,8-TeBDD entre el
hígado y el tejido adiposo de ratas estaba influida por la dosis, la
vía de exposición y el tiempo transcurrido después de la
administración. Las relaciones entre las concentraciones del hígado y
el tejido adiposo medidas en distintas condiciones variaban entre 0,2
y 6,5 (gama para dosis únicas de 2,3,7,8-TeBDD en ratas). No se
dispuso de datos experimentales sobre la transferencia de PBDD y PBDF
a las crías.
Se hallaron metabolitos de tetraBDD/BDF en la bilis y las heces
de ratas. Se formaron principalmente por hidroxilación aromática y
debromación hidrolítica. La tasa de metabolismo (determinada
indirectamente como tasa de excreción biliar) difería entre la
2,3,7,8-TeBDD (el 7% aproximadamente) y el 1,2,7,8-TeBDF (el 50%
aproximadamente). Tres días después de la administración intravenosa
de una dosis de 2,3,7,8-TeBDD (1 nmol/kg de peso corporal), el 14% de
la dosis administrada se halló en forma de metabolitos en las heces de
ratas.
Se estudiaron en ratas la eliminación y excreción de la
2,3,7,8-TeBDD utilizando las vías de administración oral, intravenosa,
intratraqueal y cutánea. En todos los estudios, la principal vía de
eliminación fue las heces, variando la radiactividad eliminada entre
el 2% (vía cutánea) y el 42% (vía oral) de la dosis administrada (1
nmol de [3H]2,3,7,8-TeBDD/kg de peso corporal) en muestras de heces,
y entre el 0,2 y el 1% en muestras de orina. Asimismo, en estudios del
1,2,7,8-TeBDF en ratas, la excreción se produjo principalmente por las
heces y sólo se eliminó por la orina el 2-3% de las dosis intravenosa,
oral o cutánea. En los primeros días que siguieron a la administración
de las dosis orales, el material no absorbido y la excreción biliar
parecieron ser las principales fuentes de sustancia eliminada por las
heces. Las porciones de 2,3,7,8-TeBDD original hallado en heces de
ratas después de la administración de 1 nmol de 2,3,7,8-TeBDD/kg de
peso corporal fueron del 53% (vía oral), el 43% (vía intratraqueal) y
el 10-20% (vía intravenosa). Pocos días después de la administración
oral de 2,3,7,8-TeBDD (1 nmol/kg de peso corporal), el 20%
aproximadamente de la dosis administrada se eliminó como sustancia
original.
Se dispone de datos sobre la retención y el ciclo biológico en el
caso de algunos PBDD y PBDF. En las ratas, la carga corporal relativa
de 2,3,7,8-TeBDD (y otros congéneres) depende de la vía y de la dosis
administrada, mostrando diferencias en la absorción. Se calcularon las
semividas de varios PBDD/PXDD y PBDF en distintos tejidos y en heces
de ratas. Variaron entre un día (1,2,7,8-TeBDF en el organismo en
conjunto) y 99 días (2,3,4,7,8-PeBDF en el hígado). Las semividas
calculadas de 17,18 y 58 días para la 2,3,7,8-TeBDD en el hígado, las
heces y el tejido adiposo, respectivamente, fueron análogas a las
señaladas para la 2,3,7,8-TeCDD en el hígado y las heces, pero
superiores (en un factor de >2) a las registradas para la
2,3,7,8-TeCDD en el tejido adiposo. Pese a las diferencias en la
retención inicial, las semividas de la 2,3,7,8-TeBDF y el
2,3,7,8-tetraclorodibenzofurano (2,3,7,8-TeCDF o TCDF) en el hígado
fueron comparables.
En lo que respecta a los PCDD y PCDF, las semividas calculadas en
personas son mucho más largas que las correspondientes a ratas. Se
dispone de estimaciones de 3-11 años (promedio: 5,9 años) para la
2,3,7,8-TeBDD y de 1-2 años (promedio: 1,5 años) para el
2,3,7,8-TeBDF. También se observó la persistencia de esas sustancias
en el caso de un químico que sintetizó 2,3,7,8-TeBDD y 2,3,7,8-TeCDD
en 1956. A los 35 años de la exposición se hallaron en su sangre
concentraciones muy elevadas de 2,3,7,8-TeBDD.
6. Efectos en mamíferos de laboratorio y en sistemas de pruebas
in vitro
La mayor parte de los estudios se refieren a la toxicidad de la
2,3,7,8-TeBDD, pero también se dispone de alguna información sobre
otros PBDD/PBDF y PXDD/PXDF.
La 2,3,7,8-TeBDD produjo efectos típicos análogos a los de la
2,3,7,8-TeCDD, incluidos el síndrome de consunción, la atrofia tímica
y la toxicidad hepática. Se observaron además lesiones hepáticas
descritas como púrpura hepática, que no se habían registrado después
de la exposición de ratas a la 2,3,7,8-TeCDD. El tipo de lesiones
(mortalidad, histopatología, pesos del hígado y el timo) hallado en
cobayos después de una sola exposición y en ratas después de la
exposición a corto plazo al 2,3,7,8-TeBDF fue análogo al observado en
el caso del 2,3,7,8-TeCDF.
La 2,3,7,8-TeBDD mantiene una interacción con el sistema
endocrino. En ratas se han observado alteraciones relacionadas con la
dosis en las hormonas tiroideas circulantes y alteración de la
actividad espermatogénica.
La DL50 oral (periodo de observación de 28 días) de la
2,3,7,8- TeBDD en ratas Wistar fue de 100 µg/kg de peso corporal
aproximadamente en las hembras y de 300 µg/kg de peso corporal
aproximadamente en los machos. Los valores de la DL50 oral para la
2,3,7,8- TeCDD obtenidos en otros estudios variaron entre 22 y >3000
µg/kg de peso corporal. Las dosis equimolares de 2,3,7,8-TeBDF y de
2,3,7,8-TeCDF dieron tasas de mortalidad comparables en cobayos. Por
ejemplo, se observó una mortalidad del 100% después de la
administración de 2,3,7,8-TeBDF (0,03 µmol/kg de peso corporal, 15,8
µg/kg de peso corporal) y de 2,3,7,8-TeCDF (0,03 µmol/kg de peso
corporal, 10 µg/kg de peso corporal). Se observaron en ratas lesiones
prepurpúreas y modificaciones de las hormonas tiroideas después de la
administración de una sola dosis de 100 µg/kg de 2,3,7,8-TeBDD/kg de
peso corporal.
En ratas Wistar a las que se administró 2,3,7,8-TeBDD por vía
oral durante 13 semanas se observaron signos de disminución de la
espermatogénesis, presencia de espermatocitos defectuosos y
necróticos, signos de púrpura hepática grave y modificaciones de las
hormonas tiroideas circulantes y de los pesos de los órganos. El nivel
de efectos adversos no observados fue de 0,01 µg/kg de peso corporal
por día.
La administración oral de 2,3,7,8-TeBDF a ratas Sprague-Dawley
durante 4 semanas provocó retraso del crecimiento dependiente de la
dosis y lesiones histopatológicas en el hígado y el timo. El nivel de
efecto adverso no observado fue de 1 µg/kg de peso corporal por día.
En ratones se observó la aparición de toxicidad en el desarrollo
en el caso de algunos PBDD y PBDF sustituidos en las posiciones 2,3,7
y 8 administrando dosis subcutáneas y orales que no provocaron
toxicidad materna ni mortalidad fetal. Los niveles de efectos mínimos
observados (en µg/kg de peso corporal) para la hidronefrosis y el
paladar hendido, después de una sola dosis oral, en ratonas gestantes
fueron, respectivamente, los siguientes: 3 y 48 para la 2,3,7,8-TeBDD,
25 y 200 para el 2,3,7,8-TeBDF, 400 y 2400 para el 2,3,4,7,8-PeBDF, y
500 y 3000-4000 para el 1,2,3,7,8-PeBDF. En comparación con la base
molar, la 2,3,7,8-TeBDD y la 2,3,7,8-TeCDD presentaron casi la misma
actividad en la inducción de la hidronefrosis. Al efectuar la
comparación con el peso, los isómeros bromados fueron en general menos
potentes que los clorados en la inducción de la hidronefrosis y el
paladar hendido. Sin embargo, el 2,3,7,8-TeBDF fue más activo que el
2,3,7,8-TeCDF.
No se halló información sobre la mutagenicidad de los PBDD y PBDF
o puntos finales conexos.
No se dispuso de estudios sobre la toxicidad y la
carcinogenicidad a largo plazo con PBDD y PBDF. La 2,3,7,8-TeBDD
resultó positiva en una prueba de transformación celular utilizando
macrófagos peritoneales murinos. Sin embargo, la actividad
transformadora de la 2,3,7,8-TeBDD fue siete veces menor que la de la
2,3,7,8-TeCDD. Más tarde aparecieron tumores en ratones lampiños tras
la inyección subcutánea de las estirpes celulares establecidas
resultantes.
La administración intraperitoneal de una serie de varias PBDD y
PXDD (tetra y penta) a ratas Wistar inmaduras de sexo masculino
produjo pérdidas de peso 14 días después de la inyección. Basándose en
los valores de DE50 molar, las sustancias más tóxicas ensayadas
fueron las 2,3,7,8-TeBDD, 2-Br1-3,7,8-Cl3-DD y 2,3-Br2-7,8-Cl2-DD
(TBCDD), con sustituciones sólo en las cuatro posiciones laterales.
Las actividades relativas de las demás PBDD examinadas siguieron el
siguiente orden: 2,3,7,8- > 1,2,3,7,8- >1,2,4,7,8- > 1,3,7,8-DD. En
otros experimentos sólo se observaron ligeras diferencias en los
valores de la DE50 (sobre una base molar) para la pérdida de peso
total, la atrofia tímica y la inducción de las enzimas hepáticas entre
la 2,3,7,8-TeCDD y la 2,3,7,8-TeBDD.
Se observaron atrofia tímica y otros signos de immunotoxicidad
(por ej., parámetros hematológicos y alteraciones de ciertas
subpoblaciones de linfocitos) con la administración de varias
PBDD/PXDD y de 2,3,7,8-TeBDF en la rata y con las 2,3,7,8-TeBDD y
TBCDD en el mono tití (Callithrix jacchus). Se llegó a la conclusión
de que, sobre una base molar, la actividad de la 2,3,7,8-TeBDD es
comparable a la de la 2,3,7,8-TeCDD en ratas y monos. Por ejemplo, se
observó un efecto notable en cierta subpoblaciones de linfocitos en
monos después de una sola dosis subcutánea de 30 ng de
2,3,7,8-TeBDD/kg de peso corporal en relación con 10 ng de
2,3,7,8-TeCDD/kg de peso corporal. No se han investigado los efectos
sobre la immunotoxicidad después de la exposición perinatal a los PBDD
y PBDF.
Tras la administración subcrónica de 2,3,7,8-TeBDD o
2,3,7,8-TeCDD por cebado oral de ratones se produjo un aumento
dependiente de la dosis en las profirinas hepáticas totales.
Dosis orales únicas de 2,3,7,8-TeBDD y 2,3,7,8-TeCDD produjeron
reducciones en la concentración y la cantidad total de vitamina A en
el hígado de ratas, siendo la 2,3,7,8-TeBDD ligeramente menos potente
que la 2,3,7,8-TeCDD (sobre una base molar).
La 2,3,7,8-TeBDD y el 2,3,7,8-TeBDF produjeron hiperqueratosis en
la oreja del conejo en una dosis de 100 µg/conejo, pero no con 10
µg/conejo. El nivel de efecto no observado para la 2,3,7,8-TeCDD fue
de 0,01 µg/conejo.
Se observó que varios congéneres halogenados tetra (Br1Cl3DD,
Br2Cl2DD) y penta (Br1Cl4DD) con sustitución en las posiciones
2,3,7 y 8 presentaban una actividad antiestrogénica análoga a la de la
de 2,3,7,8-TeCDD, examinada en cultivos de células de cáncer mamario
humano.
En ratas, la 2,3,7-tribromodibenzo- p-dioxina (2,3,7-triBDD/
TrBDD) reducía la desaparición de la uabaína del plasma, su
eliminación por la bilis y el flujo biliar en una amplitud ligeramente
inferior a la observada con la 2,3,7,8-TeCDD.
Los PBDD/PBDF y PXDD/PXDF son potentes inductores de ciertas
enzimas microsómicas dependientes del citocromo P-450. Se calcularon
valores de DE50 de 0,8-1 nmol/kg de peso corporal para la inducción
del citocromo P-1A1 y de 0,2 nmol/kg de peso corporal aproximadamente
para la inducción del citocromo P-1A2 en el hígado de rata tras la
administración oral de dosis únicas de 2,3,7,8-TeBDD. Se observó la
inducción del citocromo P-1A1 (inducción de la hidroxilasa de
arilhidrocarbono y/o la etoxirresorrufina- O-desetilasa) en distintas
especies y tejidos in vivo y en cultivo celular de rata in vitro.
Se observó que distintos congéneres eran activos, así como los
pirolizados de ciertos pirorretardantes. Por lo general, la inducción
enzimática dependía de la dosis en concentraciones no tóxicas,
comenzaba después de la exposición y era duradera. Resultó mensurable
en exposiciones tan bajas como las situadas en la gama de pmol. La
actividad inductora varió en varios órdenes de magnitud para distintos
congéneres, en función de su estructura química. Los inductores más
potentes fueron las TCDD, TBDD y TBCDD. En comparación (sobre una base
molar) con sus análogos clorados, las PBDD y PXDD tenían más o menos
igual actividad. En contraste con la TCDD, cuya actividad inductora
relativa era independiente del tejido examinado, la TBDD era cinco
veces más activa en la inducción de la
etoxirresorrufina- O-desetilasa en el hígado que en la piel y el
pulmón después de la exposición subcrónica de ratones. La clasificación
de la inducción de la actividad de la etoxirresorrufina- O-desetilasa
en monos titís fue de TCDD > 2,3,4,7,8- pentaclorodibenzofurano >
2,3,4,7,8-pentaCDF/PeCDF > 2,3,4,7,8-PeBDF cuando se compararon las
actividades enzimáticas con las concentraciones hepáticas. En las
pruebas in vitro con cultivos de células de rata se obtuvieron
valores de la CE50 molar análogos para las actividades de inducción
de la hidroxilasa del arilhidrocarbono y de la
etoxirresorrufina- O-desetilasa entre los PXDF y PCDF correspondientes.
Se estima que los PBDD y PBDF comparten un mecanismo común de
acción con los PCDD y PCDF y otros hidrocarburos aromáticos
halogenados. Se confirmó el enlace con el receptor de hidrocarburos
aromáticos citosólico, que desempeña una función central en la
mediación de la toxicidad afin a la de la 2,3,7,8-TeCDD, en el caso de
varios PBDD y PXDD/PXDF. Sus afinidades de enlace con los receptores
variaron en varios órdenes de magnitud, pero fueron comparables a las
de sus análogos clorados.
7. Efectos en el ser humano
No se dispone de datos sobre la exposición de seres humanos a los
PBDD y PBDF o sobre sus efectos en la salud de la población general.
Se han registrado dos casos de problemas de salud agudos debidos
a la exposición a 2,3,7,8-TeBDD/TeCDD, con síntomas que comprendían el
cloroacné.
En otro estudio, el personal masculino de una fábrica de
productos químicos con exposición documentada a los PBDD y PBDF
procedentes del uso de pirorretardantes bromados (OBDE y DBDE) fue
sometido a pruebas de laboratorio inmunológicas y clínicas
adicionales. Aunque se observaron indicios de modificaciones menores
de los parámetros inmunológicos, la evaluación global de su estado de
salud no mostró un efecto de la carga corporal de 2,3,7,8-TeBDD/ TeBDF
sobre el sistema inmunitario.
No existen informes sobre la mortalidad cancerosa producida por
los PBDD y PBDF.
8. Efectos en otros organismos en el laboratorio y en el medio
ambiente
Sólo se dispone de información limitada sobre los efectos de los
PBDD y PBDF en microorganismos, plantas, invertebrados o especies
silvestres vertebradas.
En una biovaloración de la mortalidad precoz de pececillos de
trucha irisada (Oncorhynchus mykiss), se ensayó una serie de
congéneres de PBDD y PBDF, que resultaron activos. Esta biovaloración
demostró también que tanto las PBDD como los PBDF tienen menor
actividad al aumentar la sustitución por bromo. Tanto la 2,3,7,8-TeBDD
como el 2,3,7,8-TeBDF eran más activos que sus análogos clorados.