
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 182
THALLIUM
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
First draft prepared by Professor G. Schaub, Institute of Zoology and
Parasitology, Ruhr University, Bochum, Germany
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation, and the
World Health Organization
World Health Organization
Geneva, 1996
The International Programme on Chemical Safety (IPCS) is a joint
venture of the United Nations Environment Programme, the International
Labour Organisation, and the World Health Organization. The main
objective of the IPCS is to carry out and disseminate evaluations of
the effects of chemicals on human health and the quality of the
environment. Supporting activities include the development of
epidemiological, experimental laboratory, and risk-assessment methods
that could produce internationally comparable results, and the
development of manpower in the field of toxicology. Other activities
carried out by the IPCS include the development of know-how for coping
with chemical accidents, coordination of laboratory testing and
epidemiological studies, and promotion of research on the mechanisms
of the biological action of chemicals.
WHO Library Cataloguing in Publication Data
Thallium
(Environmental health criteria ; 182)
1.Thallium - toxicity I.Series
ISBN 92 4 157182 9 (NLM Classification: QV 618)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1996
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved. The designations
employed and the presentation of the material in this publication do
not imply the expression of any opinion whatsoever on the part of the
Secretariat of the World Health Organization concerning the legal
status of any country, territory, city or area or of its authorities,
or concerning the delimitation of its frontiers or boundaries. The
mention of specific companies or of certain manufacturers' products
does not imply that they are endorsed or recommended by the World
Health Organization in preference to others of a similar nature that
are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR THALLIUM
PREAMBLE
1. SUMMARY
1.1. Identity, physical and chemical properties, and
analytical methods
1.2. Sources of human and environmental exposure
1.3. Environmental transport, distribution and transformation
1.4. Environmental levels and human exposure
1.5. Kinetics and metabolism in laboratory animals and humans
1.6. Effects on laboratory mammals and in vitro test systems
1.7. Effects on humans
1.8. Human dose-response relationship
1.9. Effects on other organisms in the laboratory and field
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL
METHODS
2.1. Identity
2.2. Physical and chemical properties
2.3. Conversion factor
2.4. Analytical methods
2.4.1. Sampling and sample preparation
2.4.2. Methods of determination
2.4.2.1 Atomic absorption spectrometry
2.4.2.2 Inductively coupled plasma - mass
spectrometry
2.4.2.3 Other methods
2.4.3. Quality control and quality assurance
2.4.4. Conclusions
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1. Natural occurrence
3.2. Anthropogenic sources
3.2.1. Production levels and processes
3.2.2. Uses
3.2.3. Emissions from industrial sources
3.2.3.1 Metal production industries
3.2.3.2 Power-generating plants
3.2.3.3 Brickworks and cement plants
3.2.3.4 Sulfuric acid plants
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1. Transport and distribution between media
4.1.1. Transport and distribution in air, water
and soil
4.1.2. Soil-vegetation transfer
4.1.2.1 Factors affecting soil-vegetation
transfer
4.1.2.2 Absorption by plants
4.1.2.3 Distribution in plants
4.2. Biotransformation
4.3. Interaction with other physical, chemical, or biological
factors
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1. Environmental levels
5.1.1. Air
5.1.2. Water
5.1.2.1 Areas not contaminated by thallium
5.1.2.2 Areas contaminated by thallium from
industrial sources
5.1.3. Rocks, soil and sediment
5.1.3.1 Areas not contaminated by thallium
5.1.3.2 Areas contaminated by thallium from
industrial sources
5.1.4. Plants and animals
5.1.4.1 Plants
5.1.4.2 Animals
5.2. General population exposure
5.3. Occupational exposure during manufacture, formulation
or use
6. KINETICS AND METABOLISM
6.1. Absorption
6.1.1. Animals
6.1.1.1 Aquatic animals
6.1.1.2 Terrestrial animals
6.1.2. Humans
6.2. Distribution
6.2.1. Animals
6.2.1.1 Distribution after administration of
a single dose
6.2.1.2 Distribution after long-term sublethal
administration
6.2.1.3 Transplacental transfer of thallium
6.2.2. Humans
6.2.2.1 Increased concentrations after lethal
poisoning
6.2.2.2 Increased concentrations after
long-term sublethal poisoning
6.2.2.3 Transplacental transfer of thallium
6.3. Metabolic transformation
6.4. Elimination and excretion
6.4.1. Animals
6.4.2. Humans
6.4.3. Methods to estimate daily intake of thallium
6.5. Retention and turnover (Biological half-life)
6.5.1. Animals
6.5.2. Humans
6.6. Kinetics at the cellular level
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
7.1. Single exposure
7.1.1. Toxicity and symptoms
7.1.2. Effects on various organs
7.2. Short-term exposure
7.2.1. Toxicity and symptoms
7.2.2. Effects on various organs
7.3. Long-term exposure: chronic toxicity
7.3.1. Toxicity and symptoms
7.3.2. Effects on various organs
7.4. Skin and eye irritation
7.4.1. Skin and hair
7.4.2. Eye
7.5. Reproductive toxicity, embryotoxicity and teratogenicity
7.5.1. Gonadotoxic effects
7.5.2. Embryotoxicity and teratogenicity
7.5.2.1 Chickens
7.5.2.2 Mammals
7.5.2.3 Delayed effects on development of
offspring
7.6. Mutagenicity and related end-points
7.7. Carcinogenicity
7.8. Neurotoxicity
7.8.1. Central nervous system
7.8.1.1 Histology and ultrastructure
7.8.1.2 Electrophysiological and biochemical
investigations
7.8.1.3 Behavioural toxicology
7.8.2. Peripheral nervous system
7.8.2.1 Histology and ultrastructure
7.8.2.2 Electrophysiological and biochemical
investigations
7.9. In vitro test systems: cell lines
7.10. Factors modifying toxicity
7.10.1. Enhancement of elimination
7.10.2. Selenium
7.11. Mechanisms of toxicity - mode of action
8. EFFECTS ON HUMANS
8.1. General population exposure
8.1.1. Acute toxicity
8.1.2. Effects of long-term exposure: chronic
toxicity
8.2. Occupational exposure
8.3. Subpopulations at special risk
8.4. Target organs in intoxicated humans: pathomorphology
and pathophysiology
8.4.1. Gastrointestinal tract and renal system
8.4.2. Cardiovascular system
8.4.3. Skin and hair
8.4.4. Nervous system
8.4.4.1 Central nervous system
8.4.4.2 Peripheral nervous system
8.4.5. Other organs
8.5. Special effects
8.5.1. Reproduction and developmental effects
8.5.2. Carcinogenicity
8.5.3. Immunotoxicological effects
8.6. Factors modifying toxicity: enhancement of
elimination
8.7. Protective measures against excessive occupational
exposure
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
9.1. Microorganisms
9.2. Aquatic organisms
9.2.1. Plants
9.2.2. Animals
9.3. Terrestrial organisms
9.3.1. Plants
9.3.1.1 Plant photosynthesis
9.3.1.2 Cytotoxic effects
9.3.1.3 Growth of plants
9.3.1.4 Different sensitivities to thallium(I)
and thallium (III)
9.3.1.5 Concentration of trace elements
9.3.1.6 Sensitivity of plants
9.3.2. Wild animals
9.3.3. Household pets and farm animals
10. EVALUATION
10.1. Evaluation of human health risks
10.1.1. Exposure levels
10.1.2. Kinetics
10.1.3. Toxic effects
10.1.4. Dose-response relationship (animals)
10.1.5. Dose-response relationship (humans)
10.2. Evaluation of the effects of thallium on the
environment
11. CONCLUSIONS AND RECOMMENDATIONS
12. FURTHER RESEARCH
REFERENCES
RESUME
RESUMEN
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (Telephone No. 9799111).
* * *
This publication was made possible by grant number 5 U01 ES02617-15
from the National Institute of Environmental Health Sciences, National
Institutes of Health, USA, and by financial support from the European
Commission.
Environmental Health Criteria
PREAMBLE
Objectives
In 1973 the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976 and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effect on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As
such, they include and review studies that are of direct relevance for
the evaluation. However, they do not describe every study carried
out. Worldwide data are used and are quoted from original studies,
not from abstracts or reviews. Both published and unpublished reports
are considered and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are only used when relevant
published data are absent or when they are pivotal to the risk
assessment. A detailed policy statement is available that describes
the procedures used for unpublished proprietary data so that this
information can be used in the evaluation without compromising its
confidential nature (WHO (1990) Revised Guidelines for the Preparation
of Environmental Health Criteria Monographs. PCS/90.69, Geneva, World
Health Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and
in vitro studies provide support and are used mainly to supply
evidence missing from human studies. It is mandatory that research on
human subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary - a review of the salient facts and the risk evaluation
of the chemical
* Identity - physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC,
JECFA, JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for environment; international concern, i.e.
the substance is of major interest to several countries; adequate data
on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the Cooperating
Organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for
layout and language. The first draft, prepared by consultants or,
more usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals, and reference data bases such as Medline
and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points, or
individual scientists known for their particular expertise. Generally
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can only
speak at the invitation of the Chairperson.
 Observers do not participate in the final evaluation of the
chemical; this is the sole responsibility of the Task Group members.
When the Task Group considers it to be appropriate, it may meet in
camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR THALLIUM
Members
Professor M. Balali-Mood, Poison Control Centre, Imam Reza Hospital,
Mashhad University of Medical Sciences, Mashhad, Islamic Republic
of Iran
Dr P. Doyle, Chemicals Evaluation Division, Environment Canada,
Ottawa, Ontario, Canada
Professor G. Kazantzis, Imperial College of Science, Technology and
Medicine, Centre for Environmental Technology, Royal School of
Mines, London, United Kingdom (Joint Rapporteur)
Dr M. Kiilunen, Department of Industrial Hygiene & Toxicology,
Institute of Occupational Health, Helsinki, Finland
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Nordberg, Department of Environmental Hygiene, Umea University,
Umea, Sweden (Chairman)
Professor G. Schaub, Department of Zoology, Institute for Zoology and
Parasitology, Ruhr University, Bochum, Germany (Joint Rapporteur)
Dr S. Velazquez, Environmental Criteria and Assessment Office, US
Environmental Protection Agency, Cincinnati, Ohio, USA
Representatives of other organizations
Dr P. Montuschi, Department of Pharmacology, Catholic University of
the Sacred Heart, Rome, Italy (representing the International Union
of Toxicology)
Observers
Dr R. Cornelis, Institute for Nuclear Sciences, State University of
Gent, Gent, Belgium
Secretariat
Dr P.G. Jenkins, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
ENVIRONMENTAL HEALTH CRITERIA FOR THALLIUM
A WHO Task Group on Environmental Health Criteria for Thallium
met in Geneva from 12 to 16 December 1994. Dr P.G. Jenkins, IPCS,
welcomed the participants on behalf of Dr M. Mercier, Director of the
IPCS, and the three IPCS cooperating organizations (UNEP/ILO/WHO).
The Group reviewed and revised the draft and made an evaluation of the
risks for human health and the environment from exposure to thallium.
The first draft was prepared by Professor G. Schaub, Institute
for Zoology and Parasitology, Ruhr University, Bochum, Germany. He
also prepared the second draft, incorporating comments received
following circulation of the first draft to the IPCS contact points
for Environmental Health Criteria monographs.
Dr P.G. Jenkins, IPCS, was responsible for both the overall
scientific content and the technical editing.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
AAS atomic absorption spectrometry
AES atomic emission spectrometry
AMP amperometric titration
CRMs certified reference materials
DPASV differential pulse anodic stripping voltametry
EDL electrode discharge lamp
EDTA ethylenediaminetetraacetic acid
GABA gamma-aminobutyric acid
GDMS glow discharge mass spectrometry
GFAAS graphite furnace atomic absorption spectrometry
GLP good laboratory practice
ICP inductively coupled plasma
IDMS isotope dilution mass spectrometry
LOEL lowest-observed-effect level
MED minimum effective dose
MIBK methyl isobutyl ketone
MS mass spectrometry
NAA neutron activation analysis
NaDDC sodium diethyldithiocarbamate
NADP nicotinamide adenine dinucleotide phosphate
NOEL no-observed-effect level
PAA photon activation analysis
TLV threshold limit value
tRNA transfer ribonucleic acid
1. SUMMARY
1.1 Identity, physical and chemical properties, and analytical
methods
Elemental thallium is a soft and malleable metal with a
bluish-white colour. When exposed to humid air or water, thallium is
oxidized rapidly on the surface or the hydroxide is formed,
respectively. Thallium has two important oxidation states,
thallium(I) and thallium(III). Monovalent (thallous) compounds behave
like alkali metals, e.g. potassium, whereas the trivalent (thallic)
compounds are less basic, resembling aluminium. In contrast to
inorganic compounds in which the thallium(I) ion is more stable in
aqueous solutions than the thallium(III) ion, the latter is more
stable in organic compounds.
The determination of thallium in environmental samples is
somewhat difficult as concentrations are in the µg/kg range or less.
Generally the limits of determination for minerals, soils and dusts
are about 20 µg/kg, for aqueous solutions about 0.1 µg/litre, and for
biological materials a few µg/kg, when no pre-concentration of
thallium is applied.
Graphite furnace atomic absorption spectrometry (GFAAS) is an
analytical method well suited for applications where high sensitivity
is required from small sample amounts with thallium present at
concentrations of a few µg/kg. Isotope dilution mass spectrometry
(IDMS) and inductively coupled plasma-mass spectrometry (ICP-MS),
possibly combined with isotope dilution, are excellent methods for
determinations offering good precision and accuracy at the µg/kg
level.
1.2 Sources of human and environmental exposure
Thallium is present in the environment as a result of natural
processes and from man-made sources. It is ubiquitous in nature and
occurs especially in sulfide ores of various heavy metals, but
normally in low concentrations. There are only a few areas with a
naturally very high thallium concentration.
Thallium is produced industrially only in small quantities (the
worldwide industrial consumption in 1991 was 10-15 tonnes/year).
Thallium and its compounds have a wide variety of industrial uses.
Its uses as a depilatory agent for humans and as a rodenticide and
insecticide are now severely restricted. The main uses are in the
electrical and electronic industries and in the production of special
glasses. Another important field of application is the use of
radioisotopes in medicine for scintigraphy and the diagnosis of
melanoma and the use of arylthallium(III) compounds in biochemistry.
Losses to the environment mainly occur from mineral smelters
(deposits of waste material and emissions into the atmosphere),
coal-burning power-generating plants, brickworks, and cement plants
(all emissions into the atmosphere). From 2000 to 5000 tonnes/year
are estimated to be mobilized world-wide by industrial processes.
Emissions of thallium from industrial processes vary widely according
to the type of industry.
Emissions from coal-fired power-generating plants can contain a
thallium concentration of 700 µg/m3 exhaust air and those from
cement plants up to 2500 µg/m3. The latter value may be reduced to
< 25 µg/m3 by using other raw materials and changing the production
process. Thallium volatilizes during the burning of coal or raw
material for cement production and recondenses on the surface of ash
particles in cooler parts of the system. These particles contain up to
50 mg thallium/kg fly-ash and are often of small size, so that only
50% of them are held back by filters in cement plants. Also, about
one third of emitted particles from power-generating plants are of the
small particle size which can be deposited in the lower respiratory
tract.
Effluent from mine tailing ponds containing up to 1620 and
36 µg/litre caused elevated levels of 88 and 1 µg/litre, respectively,
in connecting rivers. Rainwater ponds around a cement plant contained
up to 37 µg/litre. In soil maximal concentrations of 60 mg/kg have
been found near waste materials from mines; 2, 0.6 and 27 mg/kg have
been found in the vicinity of base metal smelters, brickworks and
cement plants, respectively.
In contaminated areas the majority of vegetables, fruits and meat
contain less than 1 mg thallium/kg fresh weight. Concentrations are
higher in cabbages (Brassicaceae), with up to 45 mg/kg reported in
green kale. Concentrations of thallium in the tissues of farm animals
correlate with concentrations in the fodder. In the vicinity of some
cement plants, increased concentrations in fodder (e.g., up to
1000 mg/kg in rape) and beef and rabbit meat (up to 1.5 and 5.8 mg/kg,
respectively) have been reported.
1.3 Environmental transport, distribution and transformation
Near point sources such as coal-fired power-generating stations,
some cement plants and metal smelting operations, the major source of
thallium in air is emission of fly ash. The results of one study
indicate that nearly all of the thallium in fly dust from a cement
plant was present as soluble thallium(I) chloride.
The fate of thallium added to soil (in deposited fly ash, for
example) depends largely on soil type. Retention will be greatest in
soils that contain large amounts of clay, organic matter and
iron/manganese oxides. Incorporation into stable complexes causes
enhanced thallium concentrations only in the upper levels of soils.
The uptake of thallium by vegetation increases as soil pH decreases.
In some strongly acid soils significant amounts of thallium can be
leached to local ground and surface water.
Most dissolved thallium in freshwater is expected to be in the
monovalent form. However, in strongly oxidized fresh water and most
seawater trivalent thallium may predominate. Thallium can be removed
from the water column and accumulate in sediment by various exchange,
complexation or precipitation reactions.
Although thallium can bioconcentrate, it is not likely to
biomagnify in aquatic or terrestrial food webs.
1.4 Environmental levels and human exposure
In areas not contaminated by thallium, concentrations in air are
usually < 1 ng/m3, those in water < 1 µg/litre, and those in water
sediments < 1 mg/kg. Mean concentrations in the earth's crust range
from 0.1 to 1.7 mg/kg, but very high concentrations are possible,
e.g., in coal up to 1000 mg/kg, and the rarely found minerals of
thallium consist of up to 60% of the element. Food of plant and
animal origin usually contains < 1 mg/kg dry weight and the human
average dietary intake of thallium appears to be less than 5 µg/day.
Uptake via the respiratory system is estimated to be < 0.005 µg
thallium/day.
There are only limited data about the actual thallium content of
workplace air. The most recent (1980s) concentrations of thallium
observed were < 22 µg thallium/m3 (in the production of a special
thallium alloy and in a thallium smelter). Average urinary
concentrations were determined to be in the range of 0.3-8 µg/litre
for cement workers and 0.3-10.5 µg/litre for foundry workers.
1.5 Kinetics and metabolism in laboratory animals and humans
Thallium is rapidly and well absorbed through the gastro
intestinal and respiratory tracts and is also taken up through the
skin. It is rapidly distributed to all organs and passes the placenta
(as indicated by the rapid fetal uptake) and the blood-brain barrier.
Because of its rapid accumulation in cells, concentrations of thallium
in whole blood do not reflect the levels in tissues. In acute
poisoning of experimental animals or humans, initially high
concentrations of thallium appear in the kidney, low concentrations in
fat tissue and brain, and intermediate concentrations in the other
organs; later the thallium concentration of the brain also increases.
Elimination of thallium may occur through the gastrointestinal
tract (mainly by mechanisms independent of biliary excretion), kidney,
hair, skin, sweat and breast milk. Intestinal reabsorption (mainly
from the colon) may occur with a consequent decrease in total body
clearance. In rats, the main routes of thallium elimination are
gastrointestinal (about two thirds) and renal (about one third), in
rabbits the contribution of the two routes is about equal. Thallium
is also secreted in saliva.
As with many other substances, the excretion of thallium in
humans differs from that in laboratory animals, since the rate of
excretion is generally much lower in humans (rate constant =
0.023-0.069 day-1) than in laboratory animals (average rate constant =
0.18 day-1). Another major difference between humans and animals is
the relative contribution of the different routes of excretion. In
humans, renal excretion seems to be much more important than in
animals, although its relative contribution to the total body
clearance has not been definitively established, due principally to
the lack of sufficient human data. Moreover, exposure levels,
duration of exposure, impairment of excretory organ function,
potassium intake and concomitant treatment of acute poisoning may
considerably influence the results.
In one study renal excretion of thallium was reported to be about
73%, whereas that through the gastrointestinal tract was about 3.7% of
the daily excreted amount. Excretion through hair and skin, and sweat
has been estimated to be 19.5% and 3.7%, respectively.
The biological half-life of thallium in laboratory animals
generally ranges from 3 to 8 days; in humans it is about 10 days but
values up to 30 days have been reported.
No data on the biotransformation of thallium are available.
1.6 Effects on laboratory mammals and in vitro test systems
There are no striking species-specific differences in the
toxicity of thallium(I) salts. Usually an oral intake of 20 to 60 mg
thallium/kg body weight is lethal within one week. Guinea-pigs are
slightly more sensitive than other experimental animals. The
water-insoluble thallium(III) oxide shows a somewhat lower acute
toxicity by oral or parenteral administration than thallium(I) salts.
Comparison of acute toxicity data indicates a high degree of
bioavailability from all exposure routes. Most organs are affected,
but the signs of poisoning and the sequence in which they occur reveal
some intra- and interspecies variability.
The symptoms of acute intoxication generally follow the following
sequence: firstly anorexia, vomiting and depression, later diarrhoea,
skin changes (inflammation at body orifices, skin furuncles, hair
loss), and then dyspnoea and nervous disorders. Finally, respiratory
failure leads to death.
Symptoms of chronic intoxication are similar to those of acute
intoxication. Loss of hair regularly occurs.
Histological examination reveals necrosis or other cell damage.
Necrotic changes have been observed in the kidneys, liver, intestine,
heart and the nervous system. Swelling of mitochondria and loss of
cristae, dilatations of smooth endoplasmic reticulum, increased
numbers of autophagic vacuoles and lipofuscin granules, and loss of
microvilli have been observed in many cells. The thallium-induced
alterations of functional processes may arise from physical disruption
of the membranes of subcellular organelles. In the heart,
arrhythmogenic effects are restricted to the sinus node.
Thallium intoxication causes selective impairment of certain
behavioural elements, which are correlated with biochemical effects
(which indicate cellular damage) in certain regions of the brain.
Some neurological effects seem to be caused by direct action, e.g.
ataxia and tremor by cerebellar alterations or alterations in
endocrine activity through changes in the hypothalamus. The autonomic
nervous system, mainly the adrenergic, may be activated by thallium.
In peripheral nerves, thallium seems to interfere presynaptically,
with the spontaneous release of transmitter, by antagonizing these
calcium-dependent processes.
The exact mechanism of thallium toxicity is still unknown.
Several, perhaps interconnected, mechanisms have been postulated. An
important aspect of thallium intoxication is the significant increase
in lipid peroxidation and in the activity of the lysosomal enzyme
ß-galactosidase. The resulting deficiency of glutathione leads to the
accumulation of lipid peroxides in the brain and, presumably, finally
to lipofuscin granules. The mode of action of thallium seems to be
mainly due to a disturbance of the function of the mitochondria.
Sexual activity is usually reduced in chronically poisoned
animals, and gonadotoxic effects of thallium are evident in the male
reproductive system. In the testes of rats given 10 mg thallium/litre
in the drinking-water for 16 days, the Sertoli cells were most
sensitive, and desquamation of the spermatogenic epithelium led to
immature sperm cells in the semen. This could explain the decreased
survival rate of embryos or reduced life span of offspring after
sublethal thallium-poisoning of the fathers.
Teratogenic effects, growth inhibition and disturbances in the
development of bones were found to occur in chicken embryos after
injection of thallium into the egg, but such effects in mammals, even
at maternotoxic doses, are controversial. Although transplacental
transfer has been demonstrated, many strains of mice and rats show no
or only slight teratogenic effects.
Two microbiological mutagenicity tests in Salmonella typhimurium
were negative and in vivo tests on sister chromatid exchange were
controversial. However, single studies report chromosomal aberrations
or a significant increase of single-stranded DNA breaks.
Long-term studies on the carcinogenicity of thallium are lacking.
1.7 Effects on humans
Since thallium salts are tasteless, odourless, colourless, highly
toxic, were easily obtainable in the past and still are in some
developing countries, thallium has often been used for suicide,
homicide and attempts at illegal abortion, causing acute thallium
poisoning. Indeed, thallium intoxication is considered one of the
most frequent causes, on a worldwide scale, of purposeful or
accidental human poisoning. Knowledge of chronic thallium
intoxication is limited to occupational exposure, to population groups
in contaminated areas and to cases of homicide involving multiple low
doses.
Symptoms of acute thallium toxicity depend on age, route of
administration and dose. Doses which have proved lethal vary between
6 and 40 mg/kg, being on average 10 to 15 mg/kg. Without therapy this
average dose usually results in death within 10 to 12 days, but death
occurring within 8-10 h has also been reported.
The triad of gastroenteritis, polyneuropathy and alopecia is
regarded as the classic syndrome of thallium poisoning, but in some
cases gastroenteritis and alopecia were not observed. Several other
signs and symptoms also occur, varying in order, extent and intensity.
Symptoms of thallium intoxication are often diffuse and initially
include anorexia, nausea, vomiting, metallic taste, salivation,
retrosternal and abdominal pain and occasionally gastrointestinal
haemorrhage (blood in faeces). Later, constipation is commonly seen
and may be resistant to treatment, thus interfering with antidotal
treatment.
After 2 to 5 days some of the typical thallium disorders slowly
develop, irrespective of the route of exposure. Effects on the
central and peripheral nervous system vary, but a consistent and
characteristic feature of thallium intoxication in humans is the
extreme sensitivity of the legs, followed by the "burning feet
syndrome" and paraesthesia. Involvement of the central nervous system
(CNS) is indicated by symptoms like hallucinations, lethargy,
delirium, convulsions and coma. Common circulatory symptoms are
hypertension, tachycardia and, in severe cases, cardiac failure. Loss
of head hair and sometimes body hair occurs after the second week of
poisoning; dystrophy of the nails is manifested by the appearance of
white lunular stripes (Mee's lines) 3 to 4 weeks after intoxication.
The black regions found in hair papillae are not caused by deposition
of pigments or thallium but are due to small amounts of air entering
the shaft.
In lethal cases the time until death occurs may vary from hours
to several weeks, but most commonly death occurs within 10 to 12 days.
Causes of death are mainly renal, CNS and cardiac failure.
In sublethal poisonings, recovery often requires months.
Sometimes neurological and mental disturbances as well as
electroencephalographic abnormalities and blindness can remain.
Additionally, intellectual functions seem to be adversely affected in
survivors.
In cases of chronic poisoning, symptoms are similar but in
general milder than in cases of acute intoxication. Sometimes
permanent blindness occurs. Complete recovery takes months and can be
interrupted by relapses.
In a well-investigated case of thallium emission around a cement
plant in Lengerich, Germany, thallium concentrations in the hair and
urine of exposed people did not correlate with certain features which
are known to be usually associated with chronic thallium poisoning,
but only with subjective neurological symptoms.
Postmortem examinations or biopsies following thallium poisoning
reveal damage of various organs. For example, after ingestion of
lethal doses, haemorrhages in the mucosa of the intestine, lung,
endocrine glands and heart, fatty infiltrations in liver and heart
tissue, and degenerative changes to glomeruli and renal tubules occur.
In the brain, fatty degeneration of ganglion cells, damage to axons
and disintegration of myelin sheaths can be observed.
Variations in blood pressure may be caused by direct effects of
thallium on the autonomic nervous system. Thallium intoxication
causes symmetric, mixed peripheral neuropathy. Distal nerves are
affected more than proximal nerves, and earlier but lesser degrees of
damage occur in nerves with shorter axons, e.g., cranial nerves.
Axons are swollen and contain vacuoles and distended mitochondria. In
lethal poisoning, severe damage of the vagus nerve, denervation of the
carotid sinus and lesions of the sympathetic ganglia have been
observed. In sublethal poisoning, affected nerves may undergo axonal
degeneration with no or only partial recovery within 2 years.
Retrobulbar neuritis and resulting visual disorders can develop
and persist for months after terminating treatment with thallium-
containing depilatories, and even optic atrophy may occur.
Limited data are available on the effects of thallium on human
reproduction. Menstrual cycle, libido and male potency may be
adversely affected. Effects on sperm are known to occur following
chronic intoxication. As in animal studies, transplacental transfer
occurs; this was seen following a thallium-induced abortion. However,
apart from a relatively low weight and alopecia of newborn babies,
fetal development was not affected in about 20 cases of thallium
intoxication during pregnancy.
No reports of any carcinogenic effects or data on immunological
effects of thallium are available. There is no adequate evidence of
genotoxic effects.
Therapies of thallium intoxication combine forced diuresis, use
of activated charcoal and prevention of re-absorption in the colon by
administration of Prussian blue, potassium ferric hexacyano
ferrate(II).
1.8 Human dose-response relationship
The mean urinary thallium concentration in unexposed populations
is 0.3 to 0.4 µg/litre. As thallium has a short biological half-life,
measured in days, and assuming steady-state conditions, this urinary
concentration can be taken as an indicator of total dose following
inhalation and dietary intake.
The mean urinary thallium concentration in a population sample
living near a thallium atmospheric emission source was 5.2 µg/litre.
A clear dose-response relationship was found between urinary thallium
concentration and the prevalence of tiredness, weakness, sleep
disorders, headache, nervousness, paraesthesia, and muscle and joint
pain. A similar dose-response relationship was also reported when
thallium in hair was used as an indicator of exposure.
The Task Group considered that exposures causing urinary thallium
concentrations below 5 µg/litre are unlikely to cause adverse health
effects. In the range of 5-500 µg/litre the magnitude of risk and
severity of adverse effects are uncertain, while exposure giving
values over 500 µg/litre have been associated with clinical poisoning.
1.9 Effects on other organisms in the laboratory and field
Thallium affects all organisms, but species- and also strain-
specific differences are evident. Different inorganic thallium(I) and
thallium(III) compounds and organothallium compounds can show
different toxicities.
The most important effect of thallium on microorganisms seems to
be inhibition of nitrification by soil bacteria. Results of one study
suggest that microbial community structure is disturbed at soil
concentrations in the range of 1-10 mg/kg dry weight, but the form of
thallium used in this experiment was not identified.
Thallium is taken up by all plant parts, but principally by the
roots. After uptake into the cell, it is concentrated unevenly in the
cytosol, probably bound to a peptide. Thallium concentrations found
in plants depend on soil properties (especially pH, clay and organic
matter content), as well as on the developmental stage and on the part
of the plant. Thallium accumulates in chlorophyll-containing
regions, but to a lesser degree in thallium-resistant plants. Oxygen
production is reduced by thallium, presumably by direct action on
electron transfer in photosystem II. Interference with the pigments
is indicated by the occurrence of chlorosis. In addition, impaired
uptake of trace elements seems to be involved in the mechanism of
toxicity. Growth is also affected, roots reacting more sensitively
than leaves or stems. These effects have been reported at
concentrations as low as 1 mg thallium/kg of dry plant tissue, after
exposure to monovalent forms of thallium.
Most studies of effects on aquatic organisms have used soluble
monovalent thallium compounds. The lowest thallium concentration
reported to affect aquatic species is 8 µg/litre, which caused a
reduction in growth of aquatic plants. Invertebrates are often
affected at lower concentration than fish (96-h LC50 values are
2.2 mg thallium/litre for daphnids and 120 mg/litre for a freshwater
fish). The lowest LC50 value, reported after exposure for about 40
days, was 40 µg/litre for fish.
Many cases of thallium intoxication of wildlife have been caused
by its large scale application as a rodenticide. In seed-eating
animals and predators the CNS and/or the gastrointestinal tract are
most severely affected. These effects can also be observed in farm
animals. In addition, thallium causes a loss of dorsal feathers in
ducks, salivation from the nose and mouth of cattle, and reduced
growth in broilers, laying hens, sheep and steers.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
Thallium is a soft, malleable, heavy metal with a bluish-white
colour and the chemical symbol Tl. The name thallium derives from the
beautiful green spectral line ( thallos, green shoot), which
identified the element.
An overview on the properties, synonyms and chemical formulae of
pure thallium and some of its compounds is given in Table 1.
2.1 Identity
Thallium is the fifth element in Group IIIB of the Periodic
Table. It occurs naturally as two isotopes thallium-203 and
thallium-205 with abundancies of 29.52 and 70.467%, respectively
(Aderjan et al., in press). The relative atomic mass of thallium is
204.383, the atomic number is 81, and the electron configuration is
(Xe) 4f14 5d10 6s2 6p. Due to its high specific gravity of
11.85 g/cm3, thallium belongs to the heavy metal group, which
comprises all metals with a specific gravity of over 4.5 g/cm3
(Micke et al., 1983).
2.2 Physical and chemical properties
The physical properties of elemental thallium are similar to
those of lead; it is very soft and malleable. Thallium exists in both
the monovalent (thallous) and the trivalent (thallic) form. Because
the 6s electrons possess only a low tendency to be released or bound
covalently, the thallous form is more common and stable and forms
numerous stable salts. Thallium(III) is easily reduced to thallium(I)
by reducing agents at high temperatures (Tl+/Tl3+ = +1.12v) (Micke
et al., 1983; Schoer, 1984; Stokinger, 1987).
Below 234°C the metal crystallizes in a hexagonal close-packed
form (alpha-thallium), while at 234°C it converts to the ß-form, a
cubic body-centred lattice. Thallium begins to volatilize at 174°C.
It has a melting point of 303°C, a boiling point of 1457°C and a
normal potential of Tl/Tl+ -0.335v (Micke et al., 1983). Thallium
is a very reactive metal. When exposed to air and moisture, it is
superficially oxidized, forming a coating of thallium(I) oxide
(Tl2O). At higher temperatures it reacts with a lovely green flame
to form thallium(III) oxide (Tl2O3). Thallium carbonate
(Tl2CO3) is the only heavy metal carbonate that is very soluble in
water (Micke et al., 1983; Stokinger, 1987).
Table 1. Physical and chemical properties of thallium and some selected thallium compoundsa
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium Tl 7440-28-0 204.38 11.85 303.5 1457 bluish-white, insoluble
metallic
Thallium(I) TlC2H3O2 563-68-8 263.43 3.765 131 - silky white very
acetate soluble
Thallium TlAl(SO4)2*12H2O 52238-56-9 639.66 2.306 91 - colourless 117.8
aluminium
sulfate
Thallium(I) TlBr 7789-40-0 284.29 7.557 480 815 pale yellow 0.5
bromide (17.3°C) (25°C)
Thallium(I) Tl2CO3 29809-42-5 468.78 7.110 273 - white 40.3
carbonate (15.5°C)
Thallium(I) TlCl 7791-12-0 239.84 7.004 430 720 white 2.9
chloride (30°C) (15.5°C)
Table 1 (contd).
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium(III) TlCl3 13453-32-2 310.74 - 25 decomposes colourless, very
trichloride hygroscopic soluble
TlCl3*4H2O 13453-33-3 382.80 - 37 100 (-H2O) colourless 862
Thallium TlOC2H5 20398-06-5 249.44 3.493 -3 130 colourless -
ethylate (20°C) (decomposes)
Thallium(I) TlF 7789-27-7 223.38 8.23 327 655 colourless 786
fluoride (4°C) (15°C)
Thallium(III) TlF3 7783-57-5 261.38 8.36 550 - olive decomposes
trifluoride (25°C) (decomposes) green to TlOH
Thallium TlOH 1310-83-4 221.39 - 139 - pale 259
hydroxide (decomposes) yellow
Thallium(I) TlI 7790-30-9 331.29 7.29 440 (ß) 823 (ß) yellow 0.006
iodide (alpha) (20°C)
Table 1 (contd).
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium(I) TlNO3 10102-45-1 266.39 - 206 430 white 95.5
nitrate (alpha) (20°C)
Thallium (III) Tl(NO3)3*3H2O 13453-38-8 444.44 - 105-107 decomposes colourless decomposes
nitrate
trihydrate
Thallium(I) Tl2O 1314-12-1 424.77 9.52 300 1080 (-O) black decomposes
oxide (16°C) to TlOH
Thallium(III) Tl2O3 1314-32-5 456.76 10.19 717 ± 5 875 (-O2) black insoluble
oxide (22°C)
Thallium(I) Tl2SO4 7446-18-6 504.82 6.77 632 decomposes white 48.7
sulfate (20°C)
Thallium(I) Tl2S 1314-97-2 440.85 8.46 448.5 0.2
sulfide (20°C)
a From: Stokinger (1987); Budavari (1989); Lide (1990)
In contact with water, thallium(I) hydroxide is formed from the
metal. Thallium is very soluble in HNO3 and H2SO4, but only
slow dissolution takes place in HCl, because of the low solubility of
the halides. It is insoluble in alkali bases. Thallium combines with
fluorine, chlorine and bromine at room temperature, and reacts with
iodine, sulfur, phosphorus, selenium and tellurium after heating. The
metal does not react with molecular hydrogen, nitrogen or carbon. It
forms alloys with other metals and readily amalgamates with mercury
(Micke et al., 1983).
The ionic radii and the electronegativity constant of monovalent
thallium are very similar to those of other alkali metals.
Thallium(I) hydroxide, carbonate and sulfate, like the corresponding
potassium compounds are very soluble in water. With respect to their
physical and chemical properties, e.g., poor water solubility,
thallium(I) oxide, sulfide and halides show similarities to the
corresponding compounds of silver, mercury and lead
(Trotman-Dickenson, 1973). In contrast to inorganic thallium
compounds, covalent organothallium compounds are only stable in the
trivalent form (McKillop & Taylor, 1973). Thallium(I) is not strongly
complexed by humic acids, whereas thallium(III) forms stable complexes
of the [TlX4]- or [TlX6]3- type (Schoer, 1984).
2.3 Conversion factor
1 g thallium = 0.0049 mol
1 mol thallium = 204.38 g
2.4 Analytical methods
Classical analytical methods, the introduction of new techniques
and a combination of both with enrichment or separation processes
provide suitable methods for the quantitative detection of thallium in
various media. Because thallium concen trations in environmental
samples are very low, determination directly from the sample or from
the digestion solution usually lacks sufficient accuracy. Therefore,
preconcentration procedures are necessary (Schoer, 1984; Sager & Tölg,
1984).
2.4.1 Sampling and sample preparation
Thallium losses during sampling, sample preparation and
determination are a major source of analytical error. Contamination
hazards need to be anticipated, as thallium is present in laboratory
ware and is leached out by solutions (Kosta, 1982). Glass contains
about 1-10 µg thallium/kg. Leaching of polythene containers with 6M
HCl for 1 week brought 1-10 ng thallium/cm2 into solution. In
addition, thallium(I) in 0.1M HNO3 solution adsorbs onto container
walls made of polyethene, polypropene, glassware or rubber. This
effect depends on the chemical properties of the surface of the
container walls and on the concentration of matrix ions. At a
thallium concentration of 1 mg/litre, no losses to borosilicate
surfaces at pH < 4 were reported, but extensive adsorption occurred
at pH > 10 (Sager & Tölg, 1984).
For determinations with spectrophotometric, mass spectrometric,
voltametric and other methods, digested samples are needed. With
respect to the high volatility of the metal and the low boiling points
of some of its compounds, only closed systems are recommended for the
digestion of organic matrices to prevent thallium losses. Fusion, dry
ashing and fuming with HF and H2SO4 or HClO4 may lead to severe
losses (up to 40%) of the thallium present (Matthews & Riley, 1969).
High-pressure digestion in closed quartz vessels with concentrated
acids, e.g., HNO3 or HNO3 and HF, at temperatures up to 300°C is
the most suitable procedure for nearly all matrices (Knapp, 1985). HF
interferes with analysis by GFAAS or ICP-AES and needs to be removed
by heating to dryness with H3BO3 (Han et al., 1982).
The volatility of thallium and its oxide or chloride makes it
possible to separate these with a gas stream of O2, H2 or HCl from
other elements that do not form volatile components under the same
conditions and subsequently capture them in a cool trap. This
procedure can be used as a preconcentration step when large quantities
of sample are available (Geilmann & Neeb, 1959; Han et al., 1982;
Sager, 1984).
Other methods of preconcentration are coprecipitation, anodic
electrolysis, ion exchange and liquid-liquid extraction.
Coprecipitation is not selective, but it leads to a high concentration
factor and results in a definite matrix, which might be useful in some
methods (Griepink et al., 1988). For example, coprecipitation with
Fe(OH)3 leads to separation from a salt matrix (K+, NH4+).
Electrolytic deposition or cementation with zinc powder yields an
excellent separation, although this procedure is time-consuming. Ion
exchange, which gives a specific separation in certain cases, is also
time-consuming. Liquid-liquid extraction with chelating agents is
virtually nonspecific, but it is a fast and easy method. A
disadvantage is the relatively low concentration factor (Sager & Tölg,
1984).
Isotope dilution methods have been applied to avoid ionization
matrix effects. Thallium is measured as thallium-205; the thallium-203
isotope can be used as a spike for isotope dilution (Sager, 1986).
2.4.2 Methods of determination
Thallium is almost always determined as total metal, rather than
as specific thallium compounds. Among the analytical techniques that
can be used are spectrophotometry, mass spectrophotometry (MS), atomic
absorption spectrometry (AAS), voltametry, neutron activation analysis
(NAA), X-ray fluorimetry, and inductively coupled plasma (ICP)
techniques (Sharma et al., 1986). A selection of analytical methods
is summarized in Tables 2 and 3.
2.4.2.1 Atomic absorption spectrometry
The most widely used method of thallium determination is atomic
absorption analysis, using measurement at 276.8 nm with a thallium
hollow cathode lamp. The sensitivity can be improved by the use of an
electrode discharge lamp (EDL), owing to its higher intensity.
Graphite furnace atomic absorption spectrometry (GFAAS) is a
well-established technique for the monitoring of trace elements in
nearly all kinds of matrices. The technique has sufficiently low
detection limits and is well-suited to applications where high
sensitivity is required for small sample amounts. In Table 3 some
methods for GFAAS are summarized.
The platform furnace concept in the temperature-stabilized mode,
together with Zeeman effect background correction, allows almost
interference-free determinations of many elements. Sample
pretreatment is not necessary, which greatly reduces the risk of
substance losses or contamination of the sample prior to analysis
(Minoia et al., 1990).
Matrix modifiers permit higher pyrolysis temperatures, so that
the desired element can be isolated from matrix elements and compounds
in an ideal case. Letourneau et al. (1987) found that additions of
H2SO4 as a matrix modifier were inadequate and that interferences
could not be corrected by Zeeman background compensation. Modifying
the matrix with palladium and magnesium nitrate has been suggested to
be generally applicable, but this is not as effective for thallium as
it is for other elements (Welz et al., 1988a). A combination of 6 mg
palladium with 100 mg ammonium nitrate allows the direct determination
of thallium in ten-fold diluted blood against matrix-free standards
(Yang & Smeyers-Verbeke, 1991).
Paschal & Bailey (1986) determined thallium concentrations in
urine. The samples were diluted 1:1 with a matrix modifier consisting
of magnesium nitrate, HNO3, Triton X-100 and water. The detection
limit was calculated to be 0.5 µg/litre.
Table 2. Instrumental methods for the determination of thallium
Methoda Matrix Oxidation Sample Parameters Interferences Detection Reference
state pretreatment of method limit
PAA metals - 203Tl (gamma,n) 202Tl gamma440 keV Segebade &
30 MeV bremsstrahlung Schmitt (1987)
post-irradiation
separation of Tl
from the matrix
NAA biological post- drying, 2-M 1013 n/cm2.sec 3-7 other isotopes 1 µg absolute Itawi &
material irradiation days 203Tl (n,gamma) than 204Tl Turel (1987)
extraction 204Tl 0.77 MeV ß-
measurement
AMP water Tl (I) Na2CO3, NaHCO3, -0.47 vs sat. calomel Mn(VII), Co(II), - Agrawal &
thiomalic acid electrode Sn(II), Tl(III) Khatkar (1988)
DPASV urine, - Na acetate, HClO4, -1.0 vs sat. calomel Cd, Pb 0.2 µg/litre Vandenbalck &
saliva EDTA electrode Patriarche
(1987)
ICP-MS rocks - HNO3, HF, H2O2 - polyatomic 70 ng/litre Date et al.
interferences (1988)
Table 2 (contd).
Methoda Matrix Oxidation Sample Parameters Interferences Detection Reference
state pretreatment of method limit
GDMS indium - - at pressure 3.10-4 mbar - 30 µg/kg Guidoboni &
discharge voltage 1 kV Leipziger
discharge current 3 mA (1988)
accelerating voltage
8 kV resolution 4000
ICP-AES air - HNO3/HClO4 (4:1) 190.9 nm F- 17 µg/litre NIOSH (1984)
particulates leachate
ICP-AES biological - Parr bomb F- 0.05-0.1 Que Hee &
material mg/litre Boyle (1988)
ICP-MS water - HNO3 205Tl - 0.1 µg/litre Henshaw et al.
(1989)
ICP-MS tissues - HNO3 - - 18 µg/kg Templeton
et al. (1989)
Spectrometry environmental Tl (I) dithizone, CHCl3, - Ag, Hg 1 µg/litre Sager (1986)
EDTA, citrate,
cyanide
a AMP = amperometric titration; DPASV = differential pulse anodic stripping voltametry; GDMS = glow discharge mass spectrometry;
ICP-AES =inductively coupled plasma - atomic emission spectrometry; ICP-MS = inductively coupled plasma - mass spectrometry;
NAA = neutron activation analysis; PAA = photon activation analysis; PPS = proton-induced prompt low energy photon high resolution
spectrometry
Table 3. Methods for determining thallium (Tl) with graphite furnace atomic absorption spectrometry (GFAAS)
Sample Separation Injected solution Detection limit Interferences Reference
Fly ash, soil digestion and preconcentration diluted H2SO4, HNO3 3.3 ng/litre HBr De Ruck et
by extraction of Tl(III) with including the 400 × al. (1989)
diisopropylether evaporation preconcentration step)
Urine complex with tri-n-octylamine, organic layer diluted 0.3 µg/litre max. charring Flanjak &
extraction with ethanol into 5 mg with ethanol and H2SO4 temp. 400°C Hodda (1988)
metallic n-butyl acetate gallium
Gallium - gallium 200 µg/kg - Hiltenkamp &
Jackwerth (1988)
Urine - spiked urine, diluted 2 µg/litre NaCl Berndt &
Sopczak (1987)
Urine chelation with NaDDC, extraction MIBK extract 0.05 µg/litre - Apostoli et
with MIBK al. (1988)
Mineralized - HNO3, H2SO4, ascorbic 5 µg/litre NaCl Leloux et al.
faeces and acid, Triton X-100 (1987a)
tissues
Table 3 (contd).
Sample Separation Injected solution Detection limit Interferences Reference
Blood, serum - HNO3 10 µg/litre Leloux et al.
(1987a)
Erythrocytes - HNO3 12 µg/litre Leloux et al.
(1987a)
Soil, extraction with 20 µg/kg Cu, Zn, Pb Ebarvia et
sediments tri-octyl-methylammonium, MIBK al. (1988)
Coal fly ash - HNO3 - - Bettinelli et
al. (1988)
MIBK = methyl isobutyl ketone; NaDDC = sodium diethyl dithiocarbamate
Chemical interferences due to chloride ions are important. These
interferences are caused by volatilization of thallium chloride in the
pyrolysis stage and, in part, by formation of TlCl(g) during the
atomization stage. Even matrix modification gives unsatisfactory
results. Welz et al. (1988b) showed that addition of palladium
nitrate as a modifier and application of argon with 5% H2 as a purge
gas leads to interference-free determination with, for instance, NaCl
loads of up to 100 mg. A special pre-pyrolysis step is necessary to
reduce palladium to the metal state, thus enabling adsorbed H2 to
react with the chloride compounds to form volatile HCl. Similar
results were obtained by Manning & Slavin (1988).
De Ruck et al. (1987) reported an oxidation technique for natural
waters with cerium(IV) sulfate and a subsequent preconcentration step
on an anion-exchange column. A preconcentration factor of 400 was
achieved, and the resultant detection limit was 3.3 ng/litre using
GFAAS. Flame atomic absorption is a reliable method for measurement
of thallium concentrations at the level of mg/litre or more. The
determination is easy and free from interference (Welz, 1983; Griepink
et al., 1988).
2.4.2.2 Inductively coupled plasma - mass spectrometry
ICP-MS is a promising method for concentrations in the µg/kg
range or less, and has good precision and accuracy. It is a multi-
element technique with sub-ppb detection limits for many elements.
Additional advantages of mass discrimination include its suitability
for isotope ratio analysis and stable isotope tracer analysis, and the
extended range of elements that can be studied. Some ICP-MS methods
are summarized in Table 2.
The application of ICP-MS to the analysis of thallium in
iron-rich ores was described by Date et al. (1988). No polyatomic
interferences for iron were detected in acid solutions. The addition
of 500 mg iron/litre to a solution of 1 mg thallium/litre in 1% HNO3
resulted in a 0.1% increase in the thallium peak. The detection limit
was found to be 0.07 µg/litre.
Templeton et al. (1989) examined thallium concentrations in rat
liver and blood plasma samples which were submitted to acid digestion
and reported a detection limit of 0.09 µmol/kg (18 µg/kg).
More than 250 water samples from lakes were analysed for thallium
(thallium-205) by ICP-MS after acidification with HNO3. The
detection limit was found to be 0.1 µg/litre; the recovery of spiked
analytes amounted to 112 ± 4% (Henshaw et al., 1989).
2.4.2.3 Other methods
Methods other than AAS and ICP-MS are summarized in Table 2.
Spectrophotometric determination with rhodamin B after liquid/liquid
extraction is a quick and easy method, but it is less sensitive and
has a high incidence of interference. The method is suitable for a
quick visual test, when a massive intoxication with thallium compounds
is suspected. Determinations down to 10 µg/litre are possible in
environmental matrices (Griepink et al., 1988).
Inductively coupled plasma - atomic emission spectrometry
(ICP-AES) is a rapid multi-element technique, but it does not provide
the detection limits required to measure thallium concentration in
uncontaminated samples. The NIOSH method for determining thallium in
air particulates has a detection limit of 17 µg/litre of leaching
solution (NIOSH, 1984).
Differential pulse anodic stripping voltametry (DPASV) is a
sensitive method for the quantitative determination of thallium in
water samples or urine. Voltametric methods also offer the advantage
of simultaneous determination of several metals from one solution.
The lower limit of detection for thallium(I) is 10-100 ng/litre
(Klahre et al., 1978; Vandenbalck & Patriarche, 1987; Griepink et al.,
1988).
Neutron activation analysis (NAA) is applicable for the
determination of thallium in various environmental samples, but it is
relatively slow and impractical for the routine analysis of large
numbers of samples. The detection limit is determined by the
irradiation time, neutron flux, the choice of a radiochemical
separation of the radio-isotope to remove interfering matrix
radio-isotopes and the measurement time. Levels down to the absolute
amount of ng of thallium can be determined (Schoer, 1984). This
method can therefore be used for the determination of low thallium
concentrations in biological samples. In bovine liver a detection
limit of 1.5 µg/kg was found after digestion, separation and
concentration procedures (Henke, 1991).
Thallium(I)-sensitive electrodes are not sensitive enough for
trace determinations, and high concentrations of alkali ions reduce
the selectivity. Sensitivity problems must also be considered for the
usual X-ray fluorimetry techniques. Other methods, like excitation
with charged particles and photon activation radiochemical isotope
dilution, are seldom used.
2.4.3 Quality control and quality assurance
Sample collection, analysis and data presentation should be
carried out according to a protocol which ensures adequate validation
of biological monitoring procedures (Vesterberg et al., 1993).
There is an urgent need for strict quality control and quality
assurance of the analytical data on thallium in clinical and
environmental samples. It is only when proof is given for the accuracy
of the published data that they become unequivocally useful to
establish critical concentrations and dose-response relationships in a
given population or ecosystem. General considerations of quality
control and quality assurance have been recommended by WHO (WHO, 1986;
Aitio, 1988).
To date, very few of the many studies on thallium have provided
the necessary evidence concerning the quality of the data throughout
the analytical procedure. The recognized way to control and ensure
this involves good laboratory practice (GLP), including intra- and
inter-laboratory analysis of materials with certified concentrations
of thallium. Such Certified Reference Materials (CRMs) should have
the same (or a similar) matrix as the sample to be analysed and be
certified for thallium concentrations (similar to those in the sample)
by an internationally recognized body. This implies suitable levels
for thallium in serum, whole blood, urine, faeces, animal tissues and
plants, as well as levels typical for exposed individuals, animal
studies or eco-systems (Cornelis, 1988).
Available reference materials with clinical and environmental
interest are listed in Table 4. This immediately reveals the very
poor picture for CRMs certified for thallium. Whole blood and serum
samples are totally lacking, while urine of exposed individuals is
handled by the BI CUM 2 and 3 products with assigned values for
thallium only. The BCR milk powders and the NBS liver samples carry a
reference value. Thallium has also been reported in some
environmental samples (fly ash, etc.) without being certified.
There appears to have been only one inter-laboratory survey on
thallium in two spiked urine samples (Geldmacher-von Malinckrodt et
al., 1984). The 35 participating laboratories used one of the three
routine methods, AAS, DPASV or photometry, after thallium extraction.
The samples were also analysed by IDMS (isotope dilution mass
spectrometry) and attributed reference values of 66.3 and 483 µg
thallium/litre, respectively. The evaluation of this inter-laboratory
survey revealed that about 70% of the laboratories met the goal.
2.4.4 Conclusions
There are several methods available for the determination of
thallium in biological and environmental samples. As routine methods
these are GFAAS (the most widely used), DPASV, ICP-MS and photometry.
They all require a very careful sample pretreatment and, in the case
of DPASV and photometry, perfect mineralization of the sample without
losses due to volatilization or adsorption onto the container walls.
The same remarks apply to the methods including a preconcentration
step. In the case of GFAAS and ICP-MS, direct analysis of the diluted
sample is feasible. It is strongly recommended that all analyses be
accompanied by a quality assurance programme. At present, it is
possible to determine thallium concentrations of about 0.1 µg/litre or
0.1 µg/kg.
Table 4. Reference materials for thallium determinations in biological and
environmental materialsa
Matrix Originb Code Thallium Remarks
concentration
Liver NBS SRM 1577 50 µg/kg lyophilized bovine liver
SRM 1577A 3 µg/kg lyophilized bovine liver
Milk BCR CRM 63 1.3 µg/kg natural skim milk powder
powder CRM 150 1.0 µg/kg spiked milk powder
CRM 151 0.8 µg/kg spiked milk powder
Urine BI CUM 2 93 ± 13 µg/litrec lyophilized synthetic urine
CUM 3 603 ± 78 µg/litrec lyophilized synthetic urine
City BCR BCR-CRM- 2850 µg/kg certified; error 6.7%
waste 176
Coal IRANT IRANT-ECO 14 000 µg/kg not certified
fly ash
Coal NIST NIST-SRM- 5700 µg/kg certified; error 3.5%
fly ash 1633a
Gas coal BCR BCR-CRM- 2200 µg/kg not certified
180
Steel IRANT IRANT-OK < 3000 µg/kg not certified
plant
flue dust
a According to Muramatsu & Parr (1985) and Cortes Toro et al. (1990)
b BCR: Measurement and Testing Programme, DG XII, BCR, Commission of the
European Union, Wetstraat 200, B-1049 Brussels, Belgium BI: Behring
Institute, PO box 140, D-3350 Marburg 1, Germany IRANT: Institute of
Radioecology and Applied Nuclear Techniques (CSSR) NBS (new name NIST):
Room B 311, Chemistry Building, National Institute for Standardization
and Testing, Gaithersburg, MD 20899, USA NIST: National Bureau of Standards
(USA)
c assigned values for a particular lot only
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Thallium is ubiquitous in nature, but occurs at low
concentrations (< 2 mg/kg) (section 5.1), especially in sulfide ores
of various heavy metals (zinc, copper, iron and lead) and in minerals
of potassium, caesium and rubidium (Micke et al., 1983; Kemper &
Bertram, 1984; Ohnesorge, 1985; Stokinger, 1987; Manzo & Sabbioni,
1988). Although the concentration of thallium is low, about 700 000
tonnes of thallium are contained in worldwide identified resources of
coal and 19 000 tonnes in zinc resources (US BM, 1989). There are
only a few areas with a naturally very high thallium concentration,
e.g., the Alsar in the Former Yugoslav Republic of Macedonia (Zyka,
1972). Minerals of thallium, e.g., lorandite (TlAsS2) and
crookesite ((Cu,Ag,Tl)2Se), with thallium concentrations of up to
60%, are rarely found and usually not used for production of thallium
(Micke et al., 1983; Kemper & Bertram, 1984; Briese et al., 1985;
Kazantzis, 1986).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
Since thallium is used only in small amounts by industry,
worldwide production of pure thallium is low. In 1975 about 8 tonnes
were produced in Germany and 2 to 3 tonnes in the USA (Zitko, 1975a),
while in 1987 and 1988 worldwide production was about 17 tonnes (US
BM, 1992). In 1981 the production of thallium in the USA was
discontinued. Sources for the production of thallium are zinc, lead
and sometimes copper or iron smelters and sulfuric acid plants. Flue
dust in particular is used as a thallium source (Zitko, 1975a; Smith &
Carson, 1977; Micke et al., 1983; Briese et al., 1985). Procedures
for the separation of thallium from other metals depend on the
proportions of the different minerals and, therefore, vary
considerably between the different smelters (Sanderson, 1952; Smith &
Carson, 1977; Micke et al., 1983; Kemper & Bertram, 1984; Briese et
al., 1985).
3.2.2 Uses
Thallium(I) sulfate was once used in medicine to reduce sweating
and to cure various infections, e.g., venereal diseases, ringworm of
the scalp, typhus, tuberculosis and malaria, and as a depilatory
agent, which caused many intoxications (Munch, 1934b; Smith & Carson,
1977; Emsley, 1978; Briese & Nessler, 1985a). However, therapeutic
uses of thallium have been discontinued because of its toxicity.
Since 1920, thallium(I) sulfate has been used as a rodenticide, in
Europe chiefly against rats and in the USA chiefly against ground
squirrels (Howe, 1971; Smith & Carson, 1977). Formerly it was used as
an insecticide (against ants and cockroaches). However, thallium is
no longer on sale as a rodenticide in most industrial countries
(Bruère et al., 1990), but is still used in developing countries
because of its cheapness.
Other areas in which thallium is used (Howe, 1971; Smith &
Carson, 1977; Micke et al., 1983; Briese et al., 1985; Sharma et al.,
1986; Kazantzis, 1986; Manzo & Sabbioni, 1988; ATSDR, 1992) are as
follows:
a) low temperature thermometers (down to -59°C) made from a
mixture of mercury and thallium;
b) special glasses with a high resistance and a low melting
point, containing thallium and selenium;
c) mixed crystals for infrared instruments, composed of arsenic
or thallium(I) salts and halogenides (TlI-TlBr), and
Tl3VS4, Tl3NbS4, and Tl3PSe4 for acusto-optic
and laser equipment;
d) electronic devices, e.g. thallium(I) sulfide for
semiconductors and scintillation counters;
e) mercury lamps (addition of thallium(I) halogenids increases
the yield of light and changes its spectrum);
f) alloys with lead, zinc, silver and antimony enhance
resistance to corrosion;
g) catalysing organic reactions, e.g., oxidations of
hydrocarbons and olefins (thallium compounds are being
increasingly used for organic synthesis); patents summarized
by Smith & Carson (1977);
h) radioactive isotopes, used in physics for measurement of
exact time periods (thallium-205), in industry for measuring
the thickness of material (thallium-204), and in medicine
for scintigraphy of heart, liver, thyroid and testes, and
for the diagnosis of melanoma (thallium-201) (Rao et al.,
1983; Müller-Brand et al., 1984; Urbain et al., 1986);
i) other uses, e.g., in the production of imitation jewellery,
fireworks, pigments and dyes, the impregnation of wood and
leather against bacteria and fungi, and in mineralogical
analysis;
j) minor amounts of thallium are used in biochemistry, e.g.,
arylthallium(III) compounds for modification of proteins and
tRNA (Douglas et al., 1990).
Worldwide industrial consumption in 1991 was estimated to be 10
to 15 tonnes. Between 1940 and 1980 consumption in the USA varied
considerably between 0.5 and 11 tonnes/year (Schoer, 1984), and
between 1984 and 1988 it was 1.1-1.5 tonnes/year (US BM, 1985, 1989).
In the USA it is used mainly in the electrical and electronic
industries and the 650 kg used in 1983 in the German Democratic
Republic was mainly for making special glass (Smith & Carson, 1977;
Micke et al., 1983; Briese et al., 1985; Kazantzis, 1986; Kemper &
Bertram, 1991).
3.2.3 Emissions from industrial sources
There is an enormous difference between the amount of thallium
mobilized (released into air, water or disposed of on land) and the
thallium consumption of 12 tonnes/year (section 3.2.2). Worldwide a
total of 2000-5000 tonnes of thallium is estimated to be mobilized per
year, especially through the combustion of fossil fuels, refinement of
oil fractions, the smelting of ferrous and non-ferrous ores, and also
by some other industrial processes such as cement production (Gorbauch
et al., 1984; Ewers, 1988; Nriagu & Pacyna, 1988). Smith & Carson
(1977) estimated that about 15% (240 tonnes) of total mobilized
thallium is transferred annually to the atmosphere. However, only a
small fraction is released into the atmosphere or wastewater during
production processes or from waste materials (Table 5). Summarizing
estimations for the USA by Smith & Carson (1977), Schoer (1984)
emphasized that in the USA each year nearly 1000 tonnes of thallium
are released into the environment, of which 350 tonnes are emitted in
vapours and dusts, 60 tonnes bound to non-ferrous metals, and more
than 500 tonnes contained in fluid and solid wastes.
3.2.3.1 Metal production industries
It has been estimated that worldwide over 600 tonnes of thallium
are processed per year during the smelting of lead, copper and zinc
ores (Micke et al., 1983). Thallium emissions from smelters can vary
greatly from plant to plant, depending upon the thallium content of
the raw materials and the technology used. For this reason, and
because of the lack of recent emission data, global releases can be
only roughly quantified. On the basis of the data in Table 5, a total
of about 90 tonnes of thallium may be released each year into the
atmosphere from non-ferrous metal production operations in the USA,
Canada and Germany. Dust in one zinc smelter was reported to contain
380-3700 mg thallium/kg before and 60-9700 mg/kg after starting the
production of thallium (Briese et al., 1985). Although it is not
possible to estimate the losses of thallium from mineral waste
materials, releases from these materials are generally expected to be
small.
Table 5. Estimated emissions of thallium (tonnes/year) into the environment
Emission source USA Canada Germany Europe World
Coal combustion
into air 180a 7.5b 7c 54d
140e 4f 80e 600e
6g
into soil/water 170a
into total environment 240c
Coal combustion (into air)
from electric utilities 155-620h
from industry and domestic 495-990h
Ferroalloy production
using manganese ores
into air 140a
into soil/water 220a
Raw iron production and
related coal combustion
into air 6a 35g 30d
into total environment 205a
Production of nonferrous
metals
into air 38a 44i 11g
total emission 496a
Potash-derived fertilizers
into total environment 5a
Cement plants
into air 25g 2670-5340h
Brick works 28b
Table 5. (cont'd).
Emission source USA Canada Germany Europe World
Oil fuel combustion, mining
and processing of oil shales
into soil/water 8a
total emission 8a
Waste combustion < 1g
a Smith & Carson (1977) f Brumsack et al. (1984)
b Brumsack (1977) g Davids et al. (1980)
c Sabbioni et al. (1984b) h Nriagu & Pacyna (1988)
d Bowen (1979) i Kogan (1970)
e Schoer (1984)
Data from the USA (Smith & Carson, 1977) indicate that relatively
large amounts of thallium are present in waste materials from
non-ferrous metal (mainly copper) and iron and steel production
(Table 5). Although no precise data were available on thallium levels
in waste from ferroalloy production using manganese ores, Smith &
Carson (1977) suggested that emissions from this source could be
significant. Atmospheric releases resulting from the production of
iron and steel in the USA were estimated to be relatively small (about
5 tonnes from steelmaking and 1 tonne in iron blastfurnace gases). In
the main area of iron and steel production in Germany, annual thallium
emissions into air have been estimated to be about 0.8 tonnes (Ewers,
1988).
3.2.3.2 Power-generating plants
Power-generating plants represent a major source of thallium
emissions, especially those using some brown coal or coal of the
Jurassic period. Most coals contain only about 0.5 to 3 mg/kg, mainly
incorporated in sulfide inclusions. Some of these impurities can be
removed by washing and mechanical cleaning. It has been estimated
that about half of the thallium content of coal is emitted into the
atmosphere and represents the biggest anthropogenic source (Smith &
Carson, 1977) (Table 5). In such estimations, losses from collected
fly ash are not taken into consideration, because its use may vary.
Only a minor amount is used in cement making. If it is used as a soil
stabilizer, contami nation of the environment is much higher (Smith &
Carson, 1977).
Natusch et al. (1974) found that coal-fired power-generating
plants emitted about 700 µg thallium/m3 flue gases, resulting in a
local level of air emission of about 700 ng/m3. This would result
in an estimated daily absorbed amount of 4.9 µg airborne thallium per
person (US EPA, 1980). In the European Union, coal-fired
power-generating plants were estimated to have caused a total
mobilization of 240 tonnes of thallium during 1990, about one third of
this being concentrated in the smallest particles, and atmospheric
emissions of 7 tonnes (Sabbioni et al., 1984b).
In coal burners, thallium volatilizes and recondenses onto the
surface of ash particles in cooler parts of the system. As a result,
2 to 10 times higher concentrations of thallium may occur in the
fly-ash than was present in the coal (Galba, 1982). Fly-ash thallium
content is negatively correlated with particle size (Manzo & Sabbioni,
1988). Thus, thallium and other toxic trace elements are concentrated
in the smallest particles, which pass through conventional
power-generating plant filters, remain suspended in the atmosphere for
long periods and are respirable. For instance, particles with a
diameter of 1.1-2.1 µm contain 76 mg thallium/kg fly-ash, those with a
diameter of 2.1-7.3 µm contain 62-67 mg/kg and those with a diameter
of 7.3-11.3 and > 11.3 µm contain 40 and 29 mg/kg, respectively.
Particles with a diameter of less than 74 µm contain only 7 mg
thallium/kg (Natusch et al., 1974). These particles are highly toxic,
since thallium and other heavy metals are preferentially concentrated
on the particle surfaces and therefore are relatively bioavailable
(Linton et al., 1976; Natusch, 1982).
3.2.3.3 Brickworks and cement plants
Total thallium emissions from brickworks in Germany have been
estimated to be 28 tonnes/year. This compares with emissions of
7.5 tonnes/year from the burning of coal (Brumsack, 1977).
The emission potential of cement plants was not recognized until
1979. The first effects on vegetation around a cement plant in
Lengerich, Germany were observed in 1977 (Pielow, 1979; LIS, 1980),
but only the gradual hair-loss in a rabbit led to the suspicion that
thallium was the cause of the toxic effects (LIS, 1980; Brockhaus et
al., 1981b; Dolgner et al., 1983). The source of thallium was found
to be residues of pyrite roasting added as a ferric oxide additive to
powdered limestone in order to produce special qualities of cement and
the addition of the filter fly-dust (LIS, 1980). Studies at other
plants showed much lower emission levels, so that the emission at
Lengerich was caused by the exceptional circumstances. Production
alterations in Lengerich caused a reduction in the emissions of more
than 99% (Pielow, 1979; Prinz et al., 1979; LIS, 1980).
Like power-generating plants, cement plants emit thallium mainly
bound to particles with a diameter of 0.2-0.8 µm (LIS, 1980).
Thallium concentrations in fly-dust emitted by the cement plant in
Lengerich were about 2.5 mg/m3 air, of which nearly all was
water-soluble thallium(I) chloride. Whereas the filter efficiency was
99% with respect to cement dust, it was only 50% with respect to the
thallium-containing particles. As a result, about 140 to 200 g
thallium/hour was emitted (Pielow, 1979; Prinz et al., 1979;
Weisweiler et al., 1985). Changing the production process reduced the
thallium content to less than 25 µg/m3 (< 200 mg/kg dust). In
other cement plants the concentrations in the filter dust were reduced
from 3066 mg/kg to about 100 mg/kg, and after this reduction only 13%
of the thallium was soluble in water (LIS, 1980).
3.2.3.4 Sulfuric acid plants
The sulfuric acid plant which had been the source of the roasted
pyrite used in the cement plant in Lengerich used pyrite containing
about 400 mg thallium/kg. However, in the roasted pyrite about 7% of
the thallium was water-soluble. During production of sulfuric acid, a
100-fold enrichment of thallium was found (LIS, 1980). As a
consequence, increased levels of thallium were found in Duisburg,
Germany around the sulfuric acid plant but never such high
concentrations as around the cement plant (Gubernator et al., 1979).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Transport and distribution in air, water and soil
Near point sources such as coal-fired power-generating stations,
cement plants using pyrite and some metal smelting operations, the
major source of thallium in air is emission of fly-ash (section
5.1.1). Although data on the forms of thallium in these emissions are
limited, results of one study indicate that nearly all of the thallium
in fly dust from a cement plant (in Lengerich, Germany) was present as
soluble thallium(I) chloride (LIS, 1980). No data on the amounts or
forms of thallium transported from soil into air during the dry season
were identified.
Assuming that 4 × 1012 kg of crustal rocks weather each year,
Bowen (1979) calculated that 2.4 × 106 kg thallium/year become
available to plants. However, as Smith & Carson (1977) have noted,
thallium tends to be retained during rock weathering, and
concentrations in soils tend to be somewhat enriched in soils compared
to the original bedrock.
The fate of thallium added to soil in deposited fly-ash probably
depends greatly on soil type (Crössmann, 1984). Data from Smith &
Carson (1977) suggest that retention should be greatest in soils that
contain large amounts of clay, organic matter and iron/manganese
oxides. According to McCool (1933) significant amounts of thallium
can be removed from solution in soils by ion exchange. Thallium can
also be incorporated into stable humus complexes (Crössmann, 1984),
which are resistant to rapid "wash-out" (Schoer, 1984).
Results of studies in several areas indicate that thallium
deposited from the atmosphere tends to accumulate in the surface
layers of soils (Smith & Carson, 1977; Heinrichs & Mayer, 1977; LIS,
1980). For example, after prolonged emissions from a cement plant in
Germany (LIS, 1980), thallium was found to remain in the upper levels
of soil (Schoer, 1984); material from depths 0-10, 40-50 and 60-70 cm
contained 4.2, 1.3 and 0.1 mg thallium/kg, respectively (Schoer,
1984). Retention processes will, however, be less effective in acidic
soil. For example, results of studies by Heinrichs & Mayer (1977)
indicate that about 40% of the thallium deposited from the atmosphere
onto relatively uncontaminated acidic (pH = 3.9-4.3) forest soil in
Germany was leached from the top 50 cm to lower soil horizons.
Elevated concentrations of thallium in groundwater (up to 40 µg/litre)
and in an irrigation canal (up to 96 µg/litre) in China, near a site
where waste materials from the mining of mercuric ore and coal
containing 25 to 106 mg thallium/kg were deposited (Zhou & Liu, 1985),
indicate that under some circumstances significant amounts of thallium
can move from soil into local water.
Although there is little information on the forms of thallium in
natural water, most dissolved thallium in fresh water is expected to
be present as the monovalent Tl+ ion (Smith & Carson, 1977). In
strongly oxidizing fresh water and in most seawater (Sager & Tölg,
1984), however, trivalent thallium is probably the predominant
dissolved form. Both forms of thallium can be removed from solution
by exchange and complexing reactions with suspended solid phases.
Trivalent thallium is also susceptible to reduction and precipitation
processes. According to Cotton & Wilkinson (1988), trivalent thallium
is extensively hydrolysed to form the colloidal oxide over the pH
range of natural water. Depending upon the relative kinetics of
reduction and hydrolysis, precipitation of thallium(III) hydroxide may
be an effective mechanism for removing thallium from solution. When
thallium(III) (precipitated as the oxide or hydroxide) settles into
organic-rich anaerobic sediment, it will be reduced to the monovalent
form, which can in turn be fixed in the sediment by reaction with
sulfide to form insoluble Tl2S (US EPA, 1978). Thallium is thus
relatively depleted in seawater where thallium(III) predominates and
can be enriched in sediments where organic matter accumulates under
undisturbed, anaerobic conditions (Smith & Carson, 1977).
The partitioning of thallium among the water, sediment and biotic
compartments of aquatic systems has rarely been investigated. In one
study, however, in which thallium (100 µg/litre as thallium(I)
nitrate) was added to a 7-litre glass aquarium containing washed sea
sand, goldfish and submergent aquatic angiosperms, thallium was
distributed among all of the compartments. Concentrations in water
decreased gradually, while those in the fish and vegetation increased,
throughout the 9-day duration of the experiment, indicating that
thallium was being exchanged among these media (Wallwork-Barber et
al., 1985). Concentrations in the sand increased rapidly to a
relatively low value (0.05 mg thallium/kg), and remained relatively
stable thereafter, suggesting that there was little exchange between
the sediment and the other compartments. The limited accumulation of
thallium in the sediment was attributed in part to the short duration
of the study and to the absence of organic matter and clay in the
sand.
4.1.2 Soil-vegetation transfer
4.1.2.1 Factors affecting soil-vegetation transfer
In general, the solubility of thallium compounds governs the
availability of the metal to vegetation (discussed in detail by
Cataldo & Wildung, 1978). Crössmann (1984) mentioned that so far no
method had been developed to quantify the amount of thallium in soil
that is easily available for plants. However, Schoer & Nagel (1980)
emphasized the good correlation between soil-vegetation transfer and
the concentration determined following ammonium acetate extraction
from soil. Other authors favour an EDTA/ammonium acetate extraction
(Scholl & Metzger, 1982).
Transfer is influenced by various factors, e.g., pH (section
5.1.3.2) and the type of the contaminated soil. Green rape, bush
beans and rye grass were found to take up less thallium from weakly
acidic soil (pH 6.2) than from more acidic soil (pH 5.6), and thallium
supplied by cement factory dust was more available to plants than
thallium in soil (Makridis & Amberger, 1989a). Rape plants grown on
two samples of soil from a contaminated area, one sample (A)
containing a 3-fold higher concentration of thallium than the other,
showed identical concentrations of thallium, while other vegetables
grown on sample A even showed a lower thallium content. It was
concluded, that plant availability cannot be correlated to total soil
thallium content as determined after extraction with concentrated
nitric acid (Hoffmann et al., 1982). Only 4.4% (± 2.7%) of the
thallium content of soil from a lead-zinc mining waste material area
was available to vegetation, compared to 17.5% (± 10.7%) in soil from
a cement plant area (Schoer & Nagel, 1980). In a similar study with
soil from a cement plant and with stream sediments from a mining
district (Wiesloch, Germany), rape plants took up about 20% of soil
thallium from the cement plant sample but only 1.4 to 5.1% from the
stream sediments, although the latter contained 2- to 3-fold higher
thallium concentrations; 8- to 80-fold higher concentrations of
plant-available thallium were calculated for the soil from the cement
plant (Scholl & Metzger, 1982). Comparing the uptake of thallium by
rape seedlings from soil contaminated by emissions from a cement plant
(mainly with thallium(I) chloride or iodide) with that from
uncontaminated soil (traces of thallium(I) sulfide), a 7.5-fold higher
uptake from the contaminated soil was found (Lehn & Bopp, 1987).
At lower thallium concentrations, some plant species took up a
higher percentage of the available thallium than at higher
concentrations, perhaps in part because of the stronger toxic effects
at higher concentrations. However, the total amount of thallium found
in the plants and the thallium content of the artificial soil
solutions were correlated, reaching up to 1000 mg/kg dry weight in
green kale following one week's exposure to a concentration of
10 mg/litre (Schweiger & Hoffmann, 1983).
The transfer from soil to plant also depends on a number of
factors relating to the plant, e.g., root system, kinetics of membrane
transport, metabolism of thallium (Cataldo & Wildung, 1978), so that
the total amount of thallium taken up is species-specific (section
5.1.4.2). This is shown by the bioconcentration factor (concentration
of thallium in the plant (fresh or dry weight) in relation to its
concentration in dry soil) found for different plants grown in soil
contaminated by mining waste materials or collected from sites with
naturally high concentrations (Table 6) (Schoer & Nagel, 1980; Lehn &
Bopp, 1987). Calculations based on the concentrations in plant ash
and dry soil show that the concentration factor is usually less than
20 (Smith & Carson, 1977). The concentrations of thallium in
vegetables reported by these authors are one to two orders of
magnitude higher than those found by Geilmann et al. (1960) in
vegetation grown on uncontaminated soil (Schoer & Nagel, 1980)
(sections 5.1.4.1 and 5.1.4.2). Trees can be a long-term reservoir of
thallium. As a result of emission by cement plants, the bark and
lichens of several trees contained 2-23.8 mg thallium/kg dry weight.
The use of ground-up bark from these trees for mulching can lead to
considerable uptake of thallium by other plants (Arndt et al., 1987).
4.1.2.2 Absorption by plants
Uptake of thallium(I) ions occurs via all parts of the plant,
presumably by using the uptake mechanisms for potassium. However,
uptake of fly-dust by the leaves of sunflowers is minimal (Schweiger &
Hoffmann, 1983). Although the majority of the thallium-containing
particles have a diameter less than 2 µm, they cannot be absorbed by
transpiration through the stomata (Pallaghy, 1972; LIS, 1980). In
numerous laboratory studies using nutrient solutions, a positive
correlation between plant uptake and thallium concentration in the
solution has been demonstrated (e.g., Al-Attar et al., 1988).
Comparable results have been obtained from the cultivation of mycelium
of higher fungi in thallium-enriched agar medium (Seeger & Gross,
1981).
According to Cataldo & Wildung (1978), absorption of thallium by
plants seems to be under metabolic regulation, and potassium is a
non-competitive inhibitor. Sunflowers with a deficiency of potassium
and supplied with 1 or 10 mg thallium nitrate/litre possessed a 2 to 3
times higher concentration of thallium per gram dry weight than those
supplied with potassium (Schweiger & Hoffmann, 1983). Metabolically
controlled uptake seemed to occur only with thallium(I), supplied as
the acetate, while thallium(III), supplied as the chloride, was
presumably taken up by passive processes such as cation exchange
(Logan et al., 1983, 1984). Since increasing concentrations of
potassium decrease the uptake of thallium(I), this uptake was
postulated to be mediated by the (Na+/K+) ATPase system. During a
3-h exposure to a solution concentration of 5 mg/litre, excised barley
seed roots took up about 6009 (± 185) mg thallium(I)/kg dry weight and
only 870 (± 44) mg thallium(III)/kg. Thallium(III) ions were easily
desorbed, presumably because of a large extracellular component,
whereas the thallium(I) ions were unavailable for exchange. The
different uptake mechanisms are also reflected in the sensitivity of
thallium(I), but not of thallium(III), towards temperature and
metabolic inhibitors. Using whole plants (maize), the differences in
uptake could not be confirmed, but the authors suggested that, prior
to the uptake, thallium(III) may be reduced in the soil to thallium(I)
(Logan et al., 1984).
Table 6. Bioconcentration factor for plants grown on contaminated soils
Plant Bioconcentration factora Reference
Fresh weight Dry weight
Barley (Hordeum vulgare) 0.14 Lehn & Bopp (1987)
Cabbage species:
Green kale < 0.1 Schoer & Nagel (1980)
Brussels sprouts < 0.1 Schoer & Nagel (1980)
Celeriac (Apium graveolens) < 0.1 Schoer & Nagel (1980)
Cress (Lepidium sativum) 33 Lehn & Bopp (1987)
Cress (Lepidium sativum) 0.45-0.59 Schoer & Nagel (1980)
Horse-radish (Armoracia) 0.33 Schoer & Nagel (1980)
Maize (Zea mays) 0.05 Lehn & Bopp (1987)
Mushrooms 2.9 Schoer & Nagel (1980)
Mustard (Sinapis alba) 1.07 Lehn & Bopp (1987)
Parsley (Petroselinum 0.15-0.21 Schoer & Nagel (1980)
crispum lapathifolia)
Rape (Brassica napus) 66 Lehn & Bopp (1987)
Rape (Brassica napus) 0.26-0.29 Schoer & Nagel (1980)
Spinach 594 Maier-Reiter et al.
(1987)
Wheat (Triticum aestivum) 0.05 Lehn & Bopp (1987)
a Concentration of thallium in fresh or dry weight of the plant in relation
to its concentration in dry soil
4.1.2.3 Distribution in plants
Thallium distribution at the cellular level has been investigated
with rape grown both on uncontaminated soil and on soil spiked with
non-toxic amounts of thallium (Günther & Umland, 1989). At each test
concentration about 70% of the thallium was concentrated in the
cytosol (comparable to human data in section 6.6). In the exposed
plants nearly all the thallium was in the form of free thallium(I)
ions; no thallium(III) ions or dimethylthallium compounds were
detected. However, in all the rape grown on uncontaminated soil, the
cytosolic thallium was bound, probably to a peptide. This native
thallium-complexing agent lacked sulfur-containing amino acids and
could not be induced in rape by the application of thallium (Günther &
Umland, 1989).
In addition to its varied distribution at the subcellular level,
thallium distribution in green plants depends on the developmental
stage and the part of the plant. Only in mushrooms was no specific
distribution pattern found to exist (Seeger & Gross, 1981). Rape
seedlings grown on soil contaminated by a cement plant (1 to 3 mg
thallium/kg dry soil) contained 3 to 5 times higher concentrations of
thallium than full-grown plants. The concentrations in different parts
of full-grown rape (leaf, 47 mg/kg dry weight; shoot, 5.5 mg/kg; seed,
2.1 mg/kg) (Lehn & Bopp, 1987) indicate that thallium concentrations
are higher in the chlorophyll-containing regions, a fact also known
from plants grown on uncontaminated soils (Weinig & Zink, 1967). In
rape grown on artificially contaminated soil (1 mg thallium
nitrate/plant), yellowing leaves showed higher concentrations (up to
200 mg/kg dry weight) than green leaves, while the seeds contained
only about 1 to 2% of the concentration found in the yellow leaves.
However, in rape grown in the field near a cement plant, the leaves
contained up to 85 mg thallium/kg dry weight and the seeds about
20 mg/kg (Arndt et al., 1987).
Experimentally, thallium concentrations of 0.0001 to 2.5 mg/litre
substrate increased the concentration in the shoots of the grass
Lolium perenne from < 0.075 mg/kg dry weight to 144.05 mg/kg and in
the roots from 0.42 to 576 mg/kg (Al-Attar et al., 1988).
The distribution of thallium also varies in different vegetables.
For instance, in gardens around Lengerich, leaves of kohlrabi
contained a 350-fold higher concentration than the tubes, while in
other vegetables the differences in concentrations between leaves and
other parts ranged from 3 to 45 times (see Table 13) (Hoffmann et al.,
1982). In studies with bush beans and green rape, differences in
thallium accumulation in the plants were evident (Makridis & Amberger,
1989b): after incubation in a liquid culture medium (1 mg
thallium(III) trichloride/litre) for 10 days, roots and shoots of
beans contained 742 and 62 mg/kg and those of rape 57 and 244 mg/kg,
respectively. At higher concentrations the difference between roots
and leaves disappeared in both species, the concentration in the roots
of rape increasing more strongly than in the shoots, which, in part,
was an effect of reduced growth. Kaplan et al. (1990), using
thallium(I) sulfate (0.55 and 1 mg/litre), observed at least 4-fold
higher concentrations of thallium in the roots of soya beans than in
the pods or the lower or higher leaves.
These data indicate that plants which are more resistant to
thallium do not have a reduced uptake, but a reduced transport of
thallium to the leaves (section 9.3.1.6).
4.2 Biotransformation
Laboratory experiments indicate that organothallium derivates may
originate from the biomethylation processes of anaerobic bacteria in
lake sediments (Manzo & Sabbioni, 1988). However, according to Craig
(1980), there is no firm evidence for environmental methylation. The
methylation of thallium and other heavy metals is a vitamin
B12-(cobalamin-)dependent reaction (Hill et al., 1970; Agnes et al.,
1971). Due to its reduction potential, thallium(III) is methylated by
methylcobalamin (Ridley et al., 1977). Transfer of the methyl group
to thallium(III) seems to occur by electrophilic attack of the Co-C
bond (Wood et al., 1978; Wood, 1984, 1987).
Monovalent thallium seems to be simultaneously oxidized and
methylated by specific anaerobic microorganisms to methylthallium(III)
moieties which are stabilized by complexation (Huber et al., 1978).
Oxidation of thallium(I) ions to thallium(III) oxide in yeast
mitochondria (Lindegren, 1971; Lindegren & Lindegren, 1973b) confirms
an in vivo oxidation, but specific culture conditions are necessary
to obtain this detoxification phenomenon in which thallium oxide is
deposited between cell wall and plasma membrane.
4.3 Interaction with other physical, chemical, or biological factors
In the atmosphere, chemical reactions involving thallium are not
very likely to occur (Schoer, 1984).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
Because of the limited industrial uses of thallium, emission on a
global scale resulting from the production and use of thallium
compounds is unlikely. However, thallium is present in relatively
large amounts in the raw materials used in various industrial
processes (e.g., smelting of sulfide ores, power generation using
coal, brick and cement manufacturing) (Table 5), which when released
can significantly increase environmental exposure to thallium on both
a local and regional scale.
5.1.1 Air
Bowen (1979) reported mean values of 0.06 ng particulate
thallium/m3 air for Europe and 0.22 ng/m3 for North America, and
Arnold (1986) a range of 0.1 to 30 ng/m3. The air of six large
American cities contained < 0.04 to 0.1 ng thallium/m3 (Ohnesorge,
1985). In a detailed study at Chadron, Nebraska, USA, Struempler
(1975) found yearly mean values for thallium of 0.22 ± 0.08 (range
0.07 to 0.48 ng/m3) and of 0.15 ± 0.04 ng/m3 during the summers of
1973 and 1974, respectively.
In industrial and urban areas of Genoa, Italy, the geometric mean
concentrations of thallium have been found to be 15 and 14 ng/m3
air, respectively, with maximal values of 58 ng/m3, but often values
were below 1 ng/m3 (Valerio et al., 1988, 1989). In London, levels
of 0.07 to 6 mg thallium/kg dust were measured (Bowen, 1979).
Air emission by thallium is mainly caused by mineral smelters,
power-generating plants and cement plants (ATSDR, 1992). Thallium
compounds are volatile at high temperatures and are not efficiently
retained by most emission control facilities. In consequence, large
amounts of thallium are released into the atmosphere if the raw
material (coal or ores) is not selected for a low thallium content.
In fly-ash from a power plant, only 0.08% of particles were
< 5 µm in diameter with a thallium concentration of 45 mg/kg ash
(Natusch et al., 1974).
5.1.2 Water
In the majority of reports, the authors did not specify whether
they determined dissolved and/or particulate thallium in samples of
water. This information was included if possible, according to the
methodology used in the study.
5.1.2.1 Areas not contaminated by thallium
Seawater contains < 0.01 to 0.02 µg/litre, and river water
< 0.01 to 1 µg/litre (Mason, 1966; Smith & Carson, 1977; Bowen, 1979;
Kemper & Bertram, 1984; Wachs, 1988).
In volcanic springs, low concentrations have been found
(0.25 µg/litre) (Arnold, 1986) and in three samples of hydrothermal
water it was below the detection limit of 0.6 µg/litre (Korkisch &
Steffan, 1979). Although, in these and the wastewater investigation,
dissolved and particulate thallium were not determined separately,
Henshaw et al. (1989) found concentrations of up to 0.41 µg
thallium/litre in filtered water from freshwater lakes. In three
wastewater treatment facilities in Massachusetts, USA, that had no
major industrial waste inputs, the thallium concentration in the
influent was below the detection limit of 5 µg/litre (Aulenbach et
al., 1987).
However, wastewater from oilfields (oil-well brines) in the USA
contained 12.9 to 672 µg thallium/litre, 5 out of 13 samples
containing > 400 µg thallium/litre (Korkisch & Steffan, 1979).
5.1.2.2 Areas contaminated by thallium from industrial sources
Data on thallium emission in water are available for areas with
oilfields, mineral industry and cement plants (Table 7).
Increased concentrations of thallium in well water and in water
from an irrigation canal in China resulted from old waste materials
from the mining of mercury and coal (Zhou & Liu, 1985) (section
5.1.3.2). Effluents from tailing ponds of base-metal mining
operations in New Brunswick, Canada contained 27 and 1620 µg dissolved
thallium/litre and up to 88 µg/litre was found in connecting rivers
(Zitko et al., 1975; Zitko, 1975b).
Raw wastewater from a pyrite ore mine at Lennestadt, Germany
(which was the source of the pyrite roasting residues used by the
cement plant in Lengerich, Germany) showed a thallium concentration of
160 µg/litre (LIS, 1980). Treatment in sedimentation ponds and with
lime and chlorine, reduced the concentration to 2-35 µg/litre. In a
stream, used as main drainage channel, the thallium concentration rose
from < 1 µg/litre (detection limit) to 1 µg/litre after the inlet.
This level is similar to that found in the River Rhine, Germany (0.5
and 2.5 µg/litre) (LIS, 1980).
Table 7. Concentrations of thallium in water from contaminated areasa
Locality Source Concentration of Reference
thallium (µg/litre)
Underground water, zinc smelter 13-820 BGA (1979)
Düsseldorf, Germany
Wastewater, different smeltersb < 0.1-2400 Smith & Carson
locations, USA (1977)
Tailing ponds, New mining 27; 1620 Zitko et al. (1975)
Brunswick, Canada
Rivers, different mining 21-30 US EPA (1980)
locations, USA
Rivers, New mining 1-88 Zitko et al. (1975)
Brunswick, Canada
River, mining < 1-1 LIS (1980)
Lennestadt, Germany
Wastewater, mining 2-160 LIS (1980)
Lennestadt, Germany
Well, China mining 17-40 Zhou & Liu (1985)
Irrigation canal, mining 6-96 Zhou & Liu (1985)
China
Wells, Lengerich cement plant < 1 LIS (1980)
Germany
Surface water, cement plant < 1-1 LIS (1980)
Lengerich, Germany
Water, cement plant 7.3 Mathys (1981)
Lengerich, Germany (distance 1 km)
Wastewater, cement plant < 1-37 LIS (1980)
Lengerich, Germany
Wells, Erwitte cement plant < 5-< 50 LIS (1980)
etc., Germany
Table 7 (contd).
Locality Source Concentration of Reference
thallium (µg/litre)
Surface water, cement plant < 50-130 LIS (1980)
Erwitte etc., Germany
Wastewater, cement plant 800 LIS (1980)
Erwitte etc., Germany (flue dust)
Wastewater, oil drilling 12.9-672 Korkisch & Steffan
USA (natural brines) (1979)
Wastewater, iron and steel mean = 60 MISA (1991a)
Canada plants
pulp and paper 230; MISA (1991b)
mills mean = 52
petroleum 310; MISA (1989)
refineries mean = 19
a Further literature summarized by Schoer (1984)
b Detailed data of sulfide mineral processors in Smith & Carson (1977)
Groundwater directly below a depot for pyrite roasting residues
in Duisburg, Germany contained 17 µg thallium/litre, and, at a
distance of some 100 m, up to 6 µg/litre (LIS, 1980).
In the vicinity of the cement plant in Lengerich, Germany (see
section 4.2.1.4) thallium levels were monitored in wells, rivers and
wastewater (LIS, 1980). In rivers, levels of 7.3 µg/litre 1 km from
the plant decreased to 1.8 µg/litre at a distance of 5 km and to
1.0 µg/litre at a distance of 10 km (Mathys, 1981). In all private
wells and water works, thallium concentrations were below the
detection limit of 1 µg/litre. At the purification plant of the
cement plant in Lengerich, the water from the inlet showed a
concentration of 17 µg/litre and that from the outlet of the rainwater
collection pond 37 µg/litre. A rainwater pond used for watering
cattle contained 3 µg thallium/litre.
Around other cement plants, thallium concentrations in private
wells, water works and surface water were below the detection limit
(< 5 and < 50 µg/litre). Pond water from the vicinity contained
130 µg/litre. In drip-water from the storage of flue dust, a
concentration of 800 µg/litre was determined (LIS, 1980).
The elimination of thallium from wastewater varies. Only 28% of
the thallium could be removed by conventional wastewater treatment
(liming) (Zitko et al., 1975) whereas 80 to 98% was removed in
Lennestadt (LIS, 1980). It has been suggested that Prussian Blue
could be used to eliminate thallium from wastewater (Rauws & Canton,
1976). Wool cannot be used as a filter to remove thallium from
contaminated water, since, in contrast to other metallic ions, only
minor amounts of thallium are adsorbed (Masri, 1976).
5.1.3 Rocks, soil and sediment
5.1.3.1 Areas not contaminated by thallium
Mean concentrations in the earth's crust range from 0.1 to
1.7 mg/kg. Higher values (up to 3 mg/kg) have been determined for
granite and shale; intermediate values for basalt, limestone,
sandstone and most coals, and lowest values for dunite (Table 8)
(Mason, 1966; Bowen, 1966, 1979; Brumsack, 1977; Smith & Carson, 1977;
Kemper & Bertram, 1984, 1991; Schoer, 1984; Arnold, 1986). Much
higher concentrations can occur in organic-rich shales such as the
Pierre Shale in the USA (25 mg/kg) and in coals of the Jurassic period
in Tadzhikistan (100 to 1000 mg/kg) (Smith & Carson, 1977).
Total thallium concentrations in soil typically range from 0.1 to
about 1.0 mg/kg (Geilmann et al., 1960; Bowen, 1966, 1979;
Chattopadhyay & Jervis, 1974; Brumsack, 1977; Smith & Carson, 1977;
Schoer, 1984; OMEE, 1994), but in China are around 0.011 mg/kg in
garden soil (range 0 to 0.02 mg/kg) (Zhou & Liu, 1985). Higher
concentrations (up to 5 mg total thallium/kg) have been reported,
however, in Poland (Staszyc et al., 1986), in soil on shale (Hoffmann
et al., 1982) and near some metallic ore deposits (Smith & Carson,
1977).
Marine sediments have been found to contain 0.95 mg thallium
per kg (Bowen, 1979) or, according to McLaren et al. (1987), using
isotope dilution inductively coupled plasma mass spectrometry, 0.6 to
0.7 mg/kg. Data summarized by Smith & Carson (1977) show a range of
0.14 to 1.13 mg/kg and in manganese nodules up to 614 mg/kg.
Table 8. Concentrations of thallium in uncontaminated geological samplesa
Source Concentration of Reference
thallium (mg/kg)
Mean Range
Basalt < 0.2-0.7 Smith & Carson (1977)
Basalt 0.08 Bowen (1979)
Basalt 0.02-0.06 Arnold (1986)
Clay 0.3 Smith & Carson (1977)
Clay 440-470b Smith & Carson (1977)
Clay 0.9 Bowen (1979)
Coal 0.38 < 0.2-1.4 Smith & Carson (1977)
Coal 0.2 0.01-2 Bowen (1979)
Coal 0.6 0.12-1.3 Gluskoter et al. (1977)
Brown coal (18% ash) 0.027 Brumsack et al. (1984)
Hard coal (8.7% ash) 0.51 Brumsack et al. (1984)
Hard coal (13.9% ash) 0.72 Brumsack et al. (1984)
Dunite 0.0005 Bowen (1979)
Granite 3.1 0.3-6.4 Smith & Carson (1977)
Granite 1.1 Bowen (1979)
Limestone 1.7 Smith & Carson (1977)
Limestone 0.14 Bowen (1979)
Limestone 0.1-0.9 Arnold (1986)
Sandstone 0.8 Smith & Carson (1977)
Sandstone 0.36 Bowen (1979)
Sandstone 0.05-0.4 Arnold (1986)
Shale (low in
organic carbon) 0.68 Brumsack et al. (1984)
Shale 1.2 Bowen (1979)
Shale 3.1 Smith & Carson (1977)
Black shale (rich
in organic carbon) 25 Smith & Carson (1977)
a Selected data; detailed data summarized in Smith & Carson (1977);
Bowen (1966, 1979); Schoer (1984)
b Very fine inclusions of plant matter
Uncontaminated sediments from lakes and small streams in various
parts of Canada typically contain about 0.35 mg thallium per kg (range
0.02 to 3.2 mg/kg) (G. Bonham-Carter, Geological Survey of Canada,
Applied Geochemistry Subdivision, personal communication to the IPCS),
with lowest values occurring in areas with underlying basaltic rock.
Thallium levels in the sediment of small streams in an uncontaminated
area of Münsterland, Germany contained from 0.03 to 0.1 mg/kg (LIS,
1980). In another investigation of small rivers in the same area and
in Sauerland, Germany, concentrations of 0.01 to 0.07 mg/kg dry weight
were determined (Mathys, 1981). This is also in the range of the data
summarized by Smith & Carson (1977).
5.1.3.2 Areas contaminated by thallium from industrial sources
Cases of contamination of sediment and soil by thallium are
mainly caused by mineral mining and smelters and by dust fall-out from
emissions of power-generating plants, brickworks and cement plants
(ATSDR, 1992) (Table 9).
Emissions from the cement plant in Lengerich, Germany caused a
remarkable increase in thallium concentrations in sediments of rivers
and brooks (Mathys, 1981). Sediment levels of 18 mg thallium/kg dry
weight found in a brook 1 km from the plant decreased to 8.7 mg/kg
within 4 km and then to 7.5 mg/kg in the River Glane into which the
brook flowed. Sediments of the following River Ems contained 5.0, 2.7
and 0.8 mg/kg at distances of 30, 70 and 100 km, respectively. In
comparison, river sediments from industrialized areas contained 0.05
to 1.8 mg/kg dry weight. Very high thallium levels were detected in
sediments from areas with zinc mining or iron ore industry, e.g.,
40.0 mg/kg in the River Lenne. After transport to the River Ruhr,
sediment thallium levels were 3 mg/kg dry weight (Mathys, 1981).
Large amounts of contaminated waste materials from the mining of
mercuric ore and coal containing 25 to 106 mg thallium per kg resulted
in chronic thallium poisoning in China. As a result of dispersal of
the waste materials, the garden soil of poisoned owners showed levels
from 28 to 61 mg/kg (mean: 43 mg/kg), whereas the soil levels in
gardens of unaffected families contained 6 to 11 mg/kg (mean,
8 mg/kg); this was still much higher than in other villages (mean,
0.011 mg/kg). In the affected village, the concentration of soluble
thallium salts decreased with increasing pH, with 8.0, 2.0 and
< 0.15 mg/kg at soil pH values of 1-2, 3-4 and 6-7, respectively.
The lower soil pH in the dry season (3.5-4.5 compared to pH 6-7 in the
rainy season) correlated with an increase in the number of
intoxications during these months, presumably due to an increase in
the thallium concentration in cabbage. After the experimental
addition of lime to contaminated soil, the thallium concentration in
cabbage was reduced (Zhou & Liu, 1985).
Table 9. Concentrations of thallium in soil from the vicinity of factories in Germany
Locality Source Distance Concentration of Reference
(m) thallium (mg/kg)
Göttingen lead-zinc -a 1.07 Brumsack
smelter (1977)
Duisburg copper smelter 500-1400 < 0.2-2.1 LIS (1980)
Leimen, Wiesloch mining and -a 5.5-21 Hoffmann
cement plant et al. (1982)
Lengerich cement plant 500-5000 < 0.1-6.9 LIS (1980)
Erwitte cement plant 350-1800 0.1-10.5 LIS (1980)
Schelklingenb cement plant < 3200 0.1-0.5 Arndt et
al. (1987)
Mergelstettenb cement plant < 800 0.1-0.5 Arndt et
al. (1987)
Duisburg sulfuric acid 0-1000 < 0.2-10.5 LIS (1980)
plant
Duisburg sulfuric acid 350-1100 < 0.2-2.3 LIS (1980)
plant
Göttingen brickwork -a 0.6 Brumsack
(1977)
a No data given;
b The investigation was performed 6 years after a ban on the use of iron pyrite
residues with high thallium contaminations. Owing to the method used (acid
digestion with concentrated nitric acid for 2 h at 90-95°C and addition of 10%
sulfuric acid), the concentrations measured correspond to the extractable soluble
emitted thallium and not the total thallium in the soil.
Sabbioni et al. (1984b) calculated the emissions from a
hypothetical coal-fired power-generating plant for a period of 40
years. They deduced an air-borne deposition of thallium around the
power-generating plants of 0.005 mg/kg, and the factor of increase
over the background level was estimated to be 0.001.
Around four small brickworks, samples of soil were digested by
strong acids and analysed for total concentration of thallium
(Brumsack, 1977). The proportion that was bioavailable is unknown.
Compared to a soil background level of 0.2 mg/kg, the contaminated
samples of soil showed a maximum accumulation factor of 3, while for
samples taken directly around the factory the factor was about 5.
Clear effects were found when the weather side of a hill was just
opposite the smoke stack. (Interestingly, shale from uncontaminated
areas showed a similarly high content (0.99 mg/kg)).
Thallium emission by the cement plant in Lengerich, Germany
caused an increase of thallium concentrations in the soil over an area
of 1 to 2 km radius from the plant, with a maximal level of 6.9 mg/kg
dry soil (LIS, 1980). Up to 4 mg/kg soil was determined in
agricultural soil and up to 6 mg/kg in the soil of house gardens
(Crössmann, 1984). Samples of soil taken at different depths always
showed highest thallium contaminations in the upper layers, decreasing
with increasing depth (LIS, 1980). Only small amounts of the thallium
in the upper layers were washed out (section 4.1) (Scholl & Metzger,
1982). The soil around the two plants that had produced the residues
from pyrite roasting was also highly contaminated, with maximal levels
of up to 10.5 and 2.3 mg/kg, respectively (LIS, 1980).
Soil from the vicinity of two other cement plants in Germany
contained only slightly elevated concentrations of thallium, up to
0.5 mg/kg soil, in the upper layers (see section 3.2.3.3 and 5.1.4.2)
(Table 9) (Arndt et al., 1987).
5.1.4 Plants and animals
Thallium occurs in low amounts in almost all living organisms,
including humans (Mason, 1966) (Tables 10 and 14). It seems to be a
non-essential cation in animals and plants (Yopp et al., 1974). Some
species accumulate this element.
5.1.4.1 Plants
a) Areas not contaminated by thallium
Usually thallium concentrations in plants are much less than
0.1 mg/kg dry weight (Geilmann et al., 1960) or 1 mg/kg ash (Dvornikov
et al., 1973, 1976), and levels exceeding 2 mg/kg ash are unusual
(Smith & Carson, 1977) (Table 10). However, such high thallium
concentrations have been found in plants from areas with a naturally
very high thallium concentration, e.g., the Alsar in Macedonia,
Yugoslavia (Zyka, 1972) (Table 11). Data from this area are
considered in sections 5.1.4.2 and 9.3.1.
No thallium could be detected in cabbage or grain from areas of
China not contaminated by thallium (Zhou & Liu, 1985). Plants used
for teas (e.g., Anisi, Betulae, Hibisci and Menthae) contained very
low concentrations of thallium (< 0.01 mg/kg), whereas higher
concentrations of other heavy metals and pesticides often occurred
(Ali & Blume, 1983). The majority (85%) of 421 investigated species
of wild mushrooms, which often accumulate heavy metals, contained
concentrations below the detection limit of < 0.25 mg/kg dry weight
(range < 0.25 to 5.5 mg/kg). A transfer factor of < 0.1
(concentration of thallium in the mushroom (fresh or dry weight) in
relation to its concentration in dry soil) indicates that no
accumulation took place (Seeger & Gross, 1981). However, in other
plants and using soil from a contaminated area, a much higher transfer
factor of 2.9 was determined (Schoer & Nagel, 1980). The thallophilic
Brassicaceae can contain higher amounts of thallium (1.5 mg/kg fresh
weight) than other plants, which usually contain 0.007 (detection
limit) to 0.1 mg/kg wet weight (Crössmann, 1984).
Wild plants normally contain only traces of thallium, whereas the
levels in garden plants can be increased by repeated use of sewage
sludge or potash fertilizers, which can contain 100 to 210 µg/kg
sludge or 15 to 310 µg/kg fertilizer (Geilmann et al., 1960;
Heinrichs, 1982). Also phosphate and copper fertilizers may contain up
to 400 µg thallium/kg (Boysen, 1992).
Thallium concentrations of up to 17 g/kg ash have been found in
plants from Alsar in Macedonia, Yugoslavia (Table 11), an area with
very high geogenic thallium levels (Zyka, 1972).
b) Areas contaminated by thallium from industrial sources
Soils contaminated through mineral smelters, power-generating
plants, brickworks or cement plants can greatly increase the
concentrations of thallium found in food of plant origin (Tables 12
and 13), which are the major route of entry of thallium into the food
chain. Data on bioconcentration factors are listed in Table 6. In
the contaminated area of Lengerich, Germany, consumption of home-grown
food was correlated with high levels of thallium in urine and hair,
and possibly with thallium-related health disorders among local people
(Brockhaus et al., 1981b; Dolgner et al., 1983). The importance of
these findings is underlined by the similarly elevated levels of
thallium found in the urine of family members consuming the home-grown
vegetables (Ewers, 1988).
Table 10. Concentrations of thallium in plants from uncontaminated areasa
Sourceb Concentration of thallium Reference
(µg/kg dry weight) (mg/kg ash)
Achillea millefolium 0.01-0.04 Dvornikov et al. (1973)
Achillea setacea 0.04-0.9 Dvornikov et al. (1976)
Alpine fir (L) 2-100 Shacklette et al. (1978)
Alpine fir (S) 2-70 Shacklette et al. (1978)
Anthemis tinctoria < 0.1-0.5 Dvornikov et al. (1976)
Artemisia absinthum 0.03 Dvornikov et al. (1973)
0.02-0.6 Dvornikov et al. (1976)
Artemisia campestris 0.057 Dvornikov et al. (1973)
0.04-0.8 Dvornikov et al. (1976)
Asperula humifusa 0.1-1.0 Dvornikov et al. (1976)
Clover 8-10 Geilmann et al. (1960)
Echium vulgare 0.1-0.3 Dvornikov et al. (1976)
Endive 80 Geilmann et al. (1960)
Engelmann's spruce (L) 2-10 Shacklette et al. (1978)
Engelmann's spruce (S) 15 Shacklette et al. (1978)
Euphorbia virgata 0.022-0.027 Dvornikov et al. (1973)
0.03-0.3 Dvornikov et al. (1976)
Festuca sulcata 0.2 Dvornikov et al. (1973)
0.2-0.6 Dvornikov et al. (1976)
Green cabbage 125 Geilmann et al. (1960)
Hay 20-25 Geilmann et al. (1960)
Head-lettuce 21 Geilmann et al. (1960)
Herbaceous vegetables 30-300 Bowen (1979)
Kale 150 Bowen (1979)
Leek 75 Geilmann et al. (1960)
Limber pine (L) 2-5 Shacklette et al. (1978)
Limber pine (S) 3-5 Shacklette et al. (1978)
Lodgepole pine (L) 2-5 Shacklette et al. (1978)
Lodgepole pine (S) 3-7 Shacklette et al. (1978)
Mushrooms < 0.25-5.5 Seeger & Gross (1981)
Myrtle blueberry (L, S) 2-7 Shacklette et al. (1978)
Ponderosa pine (S) 15 Shacklette et al. (1978)
Potato (L, S) 25-30 Geilmann et al. (1960)
(T) 5 Geilmann et al. (1960)
Rape (L) 25-30 Geilmann et al. (1960)
Red cabbage 40 Geilmann et al. (1960)
Table 10 (contd).
Sourceb Concentration of thallium Reference
(µg/kg dry weight) (mg/kg ash)
Salvia nemorosa 0.04 Dvornikov et al. (1973)
0.04-0.8 Dvornikov et al. (1976)
Stinging nettle (L) 28.8 Weinig & Zink (1967)
Tanacetum vulgare 0.06-0.2 Dvornikov et al. (1976)
Tobacco (L) 24-100 Geilmann et al. (1960)
Verbascum ovalifolium 0.01-0.7 Dvornikov et al. (1976)
Woody gymnosperms 50 Bowen (1979)
a Further data summarized by Dvornikov et al. (1973, 1976), Gough et al. (1979) and
Smith & Carson (1977)
b L = leaves, needles; S = stems; T = tubers
Table 11. Concentrations of thallium in plants from the Alsar region in Yugoslavia
possessing a high natural concentration of thallium in the soil
Source Concentration of thallium
(mg/kg ash weight)
Campanula sp. (L, S, F) 5990
Centaurea sp. (P) 75
Centaurea sp. (L, S) 105
Dianthus sp. (F) 5200
Echinops sp. (L) 15
Eryngium sp. (L) 3
Eryngium sp. (F) 10
Galium sp. (F) 17 000
Lavatera sp. (L, S) 125
Lavatera sp. (F) 45
Linaria triphylla 3000
Linaria triphylla 3800
According to Zyka (1972)
F = flowers; L = leaves, needles; P = pods and seeds; S = stems
Waste materials from the mining of mercuric ore and coal in China
(section 5.1.3.2) increased the concentration of thallium in cabbage
and grain. Cabbage from gardens of affected families contained
42 mg/kg fresh weight (range 39 to 49), whereas cabbage eaten by
healthy families contained 5.6 mg/kg (range 3 to 11). Other vegetables
from the gardens of affected families usually contained less than
10 mg/kg (Zhou & Liu, 1985).
An accumulation in vegetables of the genus Brassica was also
observed in Lengerich. In the area with the highest contamination, the
majority of plants and fruits contained < 0.1 to 0.4 mg/kg fresh
weight. Higher thallium levels were sometimes found in strawberries,
potatoes, beans, tomatoes, carrots and leeks, while in parsley,
celery, red currants, and all Brassicaceae high levels were usual
(LIS, 1980). Within this genus uptake of thallium varied: the
bioconcentration factor of white and red cabbage was relatively low;
it was 5- to 10-fold higher in the stems of kohlrabi. Savoy cabbage
and green kale were found to contain the highest thallium
concentrations, exceeding those of the soil (Crössmann, 1984). The
maximal value of 45.2 mg/kg fresh weight was found in green kale (LIS,
1980). Most forage plants, e.g., turnips, hay, grass and fodder corn,
contained < 5 mg/kg dry weight, but 46% of rape plants contained >
100 mg/kg (up to 1095 mg/kg dry weight) and 22% of the maize 10 to
50 mg/kg (LIS, 1980). Vegetables from Lengerich, grown in soil with
4.5 mg thallium/kg dry weight, could be classified according to their
mean thallium concentration (mg/kg fresh weight) into five groups.
These were I: green cabbage (22.6 mg/kg) and savoy cabbage
(8.5 mg/kg); II: turnip, broccoli, kohlrabi and white cabbage
(3.1 mg/kg); III: stock beet and other vegetables (1.4 mg/kg); IV: red
beet, rhubarb and spinach (0.7 mg/kg); V: the majority of fruits and
vegetables (0.5 mg/kg), e.g., red cabbage, Brussels sprouts, onion,
salad, carrot, bean, tomato, cucumber and potato (Scholl & Metzger,
1982).
The accumulating capacity of rape also became evident in an
investigation at cement factories in Schelklingen and Mergelstetten,
Germany, 6 years after the use of the same iron pyrite residues that
had been used in Lengerich was banned. The soil contained only
slightly elevated concentrations of thallium (section 5.1.3.2), and in
the majority of the plants, four of them Brassicaceae, no thallium was
detectable (Arndt et al., 1987). However, rape contained increased
levels of 2.4 to 679.6 mg thallium/kg dry weight at Mergelstetten and
1.8 to 19.1 mg/kg dry weight at Schelklingen (Table 12). The highest
levels detected in single rape plants were found within an area
extending 150-400 m downwind from the cement plant. The majority of
the rape grown in that area contained more than 5 mg thallium/kg dry
weight and could not be used as animal feed.
Table 12. Concentrations in plants from thallium-contaminated areas
Organism Concentration of thallium (mg/kg) Source of Localitya Reference
emission
Dry weight Fresh weight
Algae 9.5-43.4 mining New Brunswick Zitko et al. (1975)
Algae 0.665 cement plant Lengerich LIS (1980)
Berula 100.3 cement plant Lengerich Mathys (1981)
Berula 0.585; 0.654 cement plant Lengerich LIS (1980)
Caltha 187.3 cement plant Lengerich Mathys (1981)
Elodea 87.4 cement plant Lengerich Mathys (1981)
Elodea 0.29; 6.5 cement plant Lengerich LIS (1980)
Grass 52.0 sulfuric acid plant Duisburg LIS (1980)
Moss 125; 162 mining New Brunswick Zitko et al. (1975)
Rape 29.2 cement plant Lengerich LIS (1980)
Rape 23.7 cement plant Lengerich Kemper & Bertram (1984)
Rape 1095 cement plant Lengerich LIS (1980)
Rape 679.6 cement plant Mergelstetten Arndt et al. (1987)
Rape 19.1 cement plant Schelklingen Arndt et al. (1987)
Sparganium 0.265 cement plant Lengerich LIS (1980)
a All the localities are in Germany, except for New Brunswick (Canada)
Table 13. Concentrations in vegetables and fruits from thallium-contaminated areas
Plant Part Concentration of thallium (mg/kg)a Source of Reference
emission
Dry weight Fresh weight
Apple fruit 0.2 cement plant LIS (1980)
Bean fruit 0.7 cement plant LIS (1980)
Blackberry fruit 0.5 cement plant LIS (1980)
Black-currant fruit 0.527 cement plant Kemper & Bertram (1984)
Brussels sprout leaf 0.5 cement plant LIS (1980)
Carrot root 1.0 cement plant LIS (1980)
Carrot leaf 0.30 mining and Hoffmann et al. (1982)
root 0.10 cement plant
Celeriac stem 0.8 cement plant LIS (1980)
Cucumber leaf 0.70 mining and Hoffmann et al. (1982)
fruit 0.10 cement plant
Green cabbage leaf 14.9 cement plant Kemper & Bertram (1984)
Green cabbage leaf 45.2 cement plant LIS (1980)
Green cabbage leaf 22.6b cement plant Scholl & Metzger (1982)
Kohlrabi leaf 35.00 mining and Hoffmann et al. (1982)
stem 0.10 cement plant
Kohlrabi stem 3.1b cement plant Scholl & Metzger (1982)
Kohlrabi stem 4.9 cement plant LIS (1980)
Onion stalk 0.10 mining and Hoffmann et al. (1982)
bulb 0.01 cement plant
Onion stalk 0.4 cement plant LIS (1980)
Parsley leaf 1.2 cement plant LIS (1980)
Pear fruit 0.5 cement plant LIS (1980)
Table 13 (contd).
Plant Part Concentration of thallium (mg/kg)a Source of Reference
emission
Dry weight Fresh weight
Potato tuber 0.8 cement plant LIS (1980)
Radish leaf 5.90 mining and Hoffmann et al. (1982)
root 0.40 cement plant
Red beet leaf 2.40 mining and Hoffmann et al. (1982)
root 0.60 cement plant
Red beet root 0.7 cement plant LIS (1980)
Red-currant fruit 1.1 cement plant LIS (1980)
Savoy cabbage leaf 8.5b cement plant Scholl & Metzger (1982)
Strawberry fruit 0.9 cement plant LIS (1980)
Tomato fruit 0.6 cement plant LIS (1980)
White cabbage leaf 3.1b cement plant Scholl & Metzger (1982)
Zucchini leaf 0.90 mining and Hoffmann et al. (1982)
stem 0.02 cement plant
a Individual value, unless otherwise stated
b Mean value
5.1.4.2 Animals
a) Areas not contaminated by thallium
Investigations of three species of freshwater fish, the
omnivorous white sucker (Catostomus commersoni) and the more
carnivorous yellow perch (Perca flavescens) and brook trout
(Salvelinus fontinalis), show them to have similar average
concentrations of thallium in their axial muscle (< 0.07 to 3.0 mg/kg
dry weight), which were independent of water pH (Heit, 1985)
(Table 14). Extensive studies of different marine shellfish and fish
revealed average concentrations of 0.14 mg/kg; only in three species
(occasionally Clupanodon punctatus and Trachurus japonicus and
often Penaeus japonicus) were concentrations above 1 mg/kg found
(Hamaguchi, 1960).
In marine invertebrates concentrations were even lower, but,
owing to the low concentration in the seawater, the concentration
factor (concentration in the organism divided by the concentration in
the seawater) calculated by Smith & Carson (1977) was > 700.
Thallium concentrations in marine mammals have rarely been
investigated. In the blubber, liver, kidney, spleen and muscle of
bowhead whales, concentrations are nearly always below 0.01 mg/kg
fresh weight (Byrne et al., 1985).
Meat from farm animals contains very low levels of thallium
(Table 14).
b) Areas contaminated by thallium from industrial sources
Different animals as well as different organs vary with respect
to their accumulation capacity for thallium (Table 15).
In a case of fish poisoning, three species were found to contain
77 to 96 mg/kg muscle (Palermo et al., 1983). The liver and kidneys
of fish from a pond contaminated by a cement plant contained 1.6 and
1.3 mg/kg fresh weight, respectively (LIS, 1980). In the same area,
fish from other ponds and waters usually contained < 0.1 mg/kg
muscle.
Table 14. Concentrations of thallium in animals from uncontaminated areas
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Invertebrates
Colorado beetle whole 1 18 Geilmann et al. (1960)
Different marine invertebrates 0.001-0.03 Noddack & Noddack (1939)
Echinoderms (hard parts) 110 Bowen (1979)
Molluscs (soft parts) 340 Bowen (1979)
Fish
Various fish species 80 Bowen (1979)
Various marine 139 < 2930 Hamaguchi (1960)
shellfish and fish
Brook trout muscle 5 < 3.0 Heit (1985)
(Salvelinus fontinalis)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
White sucker muscle 28 < 2.0 Heit (1985)
(Catostomus commersoni)
Yellow perch muscle 27 < 3.0 Heit (1985)
(Perca flavescens)
Birds
Ducka kidney 15 0.03 Holm et al. (1987)
Duckb kidney 10 0.129 Holm et al. (1987)
Ducka liver 15 0.022 Holm et al. (1987)
Duckb liver 10 0.207 Holm et al. (1987)
Hen liver 2 < 50 LIS (1980)
Hen muscle 2 < 50 LIS (1980)
Mammals
Cattle hair 1 20 Geilmann et al. (1960)
Cattle hoof 1 16 Geilmann et al. (1960)
Cattle horn 1 10 Geilmann et al. (1960)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Fox intestine 25 < 2.7 Munch et al. (1974)
Fox kidney 27 0.01-1.5 Munch et al. (1974)
Fox liver 27 0.01-1.6 Munch et al. (1974)
Goat hair 1 7 Geilmann et al. (1960)
Goat hoof 1 9 Geilmann et al. (1960)
Hare hair 1 17 Geilmann et al. (1960)
Horse hair 1 7 Geilmann et al. (1960)
Horse hoof 1 4 Geilmann et al. (1960)
Marten brain 7 < 0.1-0.7 Clausen & Karlog (1974)
Marten intestine 13 < 0.01-0.57 Clausen & Karlog (1974)
Marten kidney 17 < 0.01-3.5 Clausen & Karlog (1974)
Marten liver 36 < 0.01-1.4 Clausen & Karlog (1974)
Pig hair 1 9 Geilmann et al. (1960)
Pig hoof 1 11 Geilmann et al. (1960)
Pig muscle 1 0.028 Kemper & Bertram (1984)
Pig muscle 43 < 70 Konermann et al. (1982)
Pig kidney 43 < 70 Konermann et al. (1982)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Pig kidney 6 < 50 LIS (1980)
Pig liver 43 < 70 Konermann et al. (1982)
Pig liver 6 < 50 LIS (1980)
Rabbit hair 1 60 Geilmann et al. (1960)
Rabbit hair 1 < 1.5 LIS (1980)
Roe deer liver 19 approx. 30 Holm et al. (1987)
Roe deer kidney 19 approx. 30 Holm et al. (1987)
Sheep hair 1 9 Geilmann et al. (1960)
Sheep hoof 1 12 Geilmann et al. (1960)
Sheep kidney 3 50-60 Hapke et al. (1980)
Sheep liver 3 < 50 Hapke et al. (1980)
Sheep muscle 3 50-60 Hapke et al. (1980)
a Cuxhaven, Germany (coast)
b Cuxhaven, Germany (inland)
In farm animals intake of thallium mainly occurs through
contaminated feed. In broilers and laying hens, tissue thallium
concentrations were linearly correlated with feed levels for
concentrations between 2 to 40 mg/kg fresh weight of feed (Ueberschär
et al., 1986). The accumulation factor (concentration of thallium in
the tissue in relation to its concentration in the feed) was 2 to 3
times higher for the tissues of broilers than for those of hens (Table
16). In contrast to the situation in sheep, cattle and pigs, thallium
accumulates in hens to a greater degree in the muscle than in the
liver. The concentration in the kidneys is about 90% lower than in
the egg shell. Thallium half-life is about 2 to 4 days for the
various hen tissues (Ueberschär et al., 1986).
Whereas maximal permitted concentrations of lead, mercury,
arsenic and fluoride in animal fodder have been established in
Germany, this has not been done for thallium (Crössmann, 1985).
Thallium poisoning in cattle has been caused by silage (41 mg
thallium/kg fresh weight) bought from a farm in a contaminated area
(Frerking et al., 1990). Thallium mainly accumulates in the kidneys,
liver and bones (section 6.2). Steers fed for at least 6 months with
fodder originating from the thallium-contaminated area around
Lengerich, Germany, containing about 1.25 mg/kg dry weight (daily
uptake: 0.025 mg/kg body weight), contained 0.10 ± 0.02, 1.66 ± 0.55,
0.52 ± 0.28 and 0.40 ± 0.15 mg thallium/kg fresh weight, respectively,
in muscles, kidneys, livers and testes (Hapke et al., 1980). In pigs
fed with 1.45 mg thallium/kg food (dry weight) for 5 months, muscle,
kidneys and liver contained 0.18 ± 0.04, 0.44 ± 0.06 and 0.31 ±
0.09 mg thallium/kg fresh weight, respectively. Feeding with 2.71 mg
thallium/kg dry weight resulted in 0.39 ± 0.07 (muscle), 0.7 ± 0.2
(kidney) and 0.53 ± 0.1 (liver) mg thallium/kg fresh weight. Since
0.5 mg/kg fresh weight is the limit set by the federal state of North
Rhine-Westphalia for thallium concentrations in human food, a critical
level for pigs seems to be a daily intake corresponding to 1.9 mg
thallium/kg dry matter of food (Konermann et al., 1982).
Exposure of farm animals to thallium in the vicinity of the
cement plant in Lengerich, Germany, resulted in increased thallium
levels in the liver and kidneys of various animals (LIS, 1980): 0.8%
of the samples of internal organs contained > 10 mg/kg fresh weight,
1.3% contained 5 to 10 mg/kg, 12.6% contained 1 to 5 mg/kg and 14.5%
contained 0.5 to 1 mg/kg. In 0.2%, 3% and 4.4% of meat from various
farm animals, 5-10, 1-5 and 0.5-1 mg/kg were found, respectively.
Concentrations above 0.5 mg/kg fresh weight were sometimes also found
in eggs and chicken meat (up to 0.8 mg/kg), rabbit meat (up to
5.8 mg/kg) and roe deer (1.6 mg/kg) (Table 15) (LIS, 1980). In whole
eggs with a concentration of 1.26 mg/kg fresh weight, the
concentration in albumin and yolk was 0.394 mg/kg, while in the shell
it was 4.94 mg/kg (Kemper & Bertram, 1984).
Table 15. Concentrations in animals from thallium-contaminated areas
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Fish
Morone muscle -d Taranto, Italy - 77 Palermo et al.
labraxc (1983)
Eelc muscle - Taranto, Italy - 96 Palermo et al.
(1983)
Salmon muscle mining New Brunswick, 3-4 5.1e; 14.6f Zitko et al.
liver Canada 3-4 6.8; 23.5 (1975)
gill 3-4 1.2; 30.0
Salmon muscle mining New Brunswick, 3-4 3.6-34g Zitko et al.
liver Canada 3-4 5.7-46 (1975)
gill 3-4 7-89
Silver-scaled liver cement plant Lengerich, Germany 1 1.6 LIS (1980)
fish kidney 1 1.3
brain 1 0.46
Mugil muscle - Taranto, Italy - 84 Palermo et al.
cephalusc (1983)
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Trout liver cement plant Lengerich, Germany 4 0.13 LIS (1980)
kidney 4 0.885
muscle 3 0.09
Birds
Duck liver industry Harburg, Germany 30 0.191 < 0.075-0.86 Holm et al.
Duck kidney industry Harburg, Germany 30 0.076 < 0.075-0.43 (1987)
Duck liver industry Stade, Germany 24 0.072 Holm
Duck liver industry Stade, Germany 10 0.186 et al.
Duck kidney industry Stade, Germany 10 0.042 (1987)
Duck liver cement plant Lengerich, Germany 4 0.4 LIS (1980)
muscle 4 0.4
Geesec muscle poison USA 17 29 4-57 Shaw (1932)
Hen egg cement plant Lengerich, Germany 24 1.26 Kemper &
Bertram (1984)
Hen egg cement plant Lengerich, Germany 26 1.6 LIS (1980)
liver 17 0.8
muscle 26 0.8
heart 5 0.7
stomach 9 0.9
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Pigeon liver cement plant Lengerich, Germany 1 0.6 LIS (1980)
kidney 3 0.6
muscle 5 0.4
heart 1 0.3
Mammals
Cattle kidney cement plant Lengerich, Germany 58 2.2 LIS (1980)
muscle 61 1.5
Cattle kidney not specified Germany 2 24.0 Frerking et al.
liver (presumably 2 2.3 (1990)
muscle cement plant) 1 0.4
urine 3 1.35h
Foxc liver poison Denmark 27 64.0 Munch et al.
kidney 16 34 (1974)
intestine 7 55
Martenc liver poison Denmark 15 57 Clausen &
kidney 16 92 Karlog (1974)
intestine 9 42
brain 5 8.2
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Pig liver cement plant Lengerich, Germany 3 1.2 LIS (1980)
kidney 296 1.3
muscle 300 0.6
Rabbit liver cement plant Lengerich, Germany 49 5.8 LIS (1980)
kidney 44 29.0
Roe deer liver cement plant Lengerich, Germany 1 2.6 LIS (1980)
kidney 3 14.0
muscle 2 1.6
heart 3 2.9
Sheep liver cement plant Lengerich, Germany 4 0.6 LIS (1980)
kidney 10 1.1
muscle 14 1.1
a Number of measurements (animals) e,f The two thallium concentrations of 45e and 100f µg/litre water are in the range
b mg/kg fresh weight unless otherwise stated of the natural concentrations at that locality; thallium concentration in the
c Fatal poisoning gill was higher (25.6 mg/kg fresh weight) at a lower concentration in water
d No data given (17.9 µg/litre)
g Lethal dose of 100 to 10 000 µg/litre
h mg/litre
5.2 General population exposure
The US Environmental Protection Agency calculated the typical
value for the exposure of the general population to be 0.48 ng
thallium/m3 air (US EPA, 1980). US EPA (1980) calculated an absorbed
amount of 3.4 ng/day assuming an inspired volume of 20 m3/day and
35% deposition in the lungs. BGA (1979) calculated the daily uptake
via the respiratory system to be < 5 ng thallium per day.
Table 16. Bioconcentration factora of thallium in broilers and
laying hensb and half-life in the tissues of laying hens
Bioconcentration factor
Organ Broiler Laying hen Half-life
(days)
Bone 0.54 0.26 2.0
Egg yolk - 0.26 4.1
Egg albumen - 0.14 1.6
Egg shell - 3.72 2.5
Fat 0.006 0.001 -
Feather 0.074 0.006 -
Kidney 0.77 0.38 3.6
Liver 0.19c 0.1 4.0
0.11d
Muscle 0.46 0.18 3.8
Skin 0.2 0.08 -
a Concentration of thallium in the respective tissue divided by
the concentration of thallium in the food
b From: Ueberschär et al. (1986)
c After 3 weeks
d After 6 weeks
More than 99% of samples of drinking-water in the USA contained
no thallium (detection limit, 0.3 µg/litre), and the positive samples
contained about 0.89 µg/litre. With a water consumption of
2 litre/day, this would result in an intake of < 1 µg thallium/day
for most adults (US EPA, 1980). The thallium concentration in 17
bottled mineral waters ranged between < 0.6 and 3.5 µg/litre, but
only 4 contained > 2 µg/litre (Korkisch & Steffan, 1979).
In the United Kingdom, Sherlock & Smart (1986) reported the total
dietary intake of thallium, based on the analysis of 13 diets. Four
(meat, fish, fats and green vegetables) out of nine food groups
contained samples with concentrations of thallium above the limit of
determination, ranging from 10 to 50 µg/kg fresh weight depending on
the food commodity. The average dietary intake of thallium for adults
was estimated to be 0.005 mg/day with a range of 0-0.01 mg/day,
assuming that concentrations less than the limit of determination were
equal to zero. The daily intake of thallium from vegetables alone is
estimated to be about 3.8 µg for an average adult in the USA (US EPA,
1980). Food of plant origin often contains more thallium than food of
animal origin, a 4-fold higher concentration of thallium being
eliminated in the urine of vegetarians than in that of humans eating
food of varying origin (Ohnesorge, 1985).
A minor route of thallium uptake can be sodium-free dietetic salt
(KCl), which contains up to 420 µg thallium/kg salt (Toots & Parker,
1977), but not sodium chloride, which only contains 0.08 µg/kg
(Geilmann et al., 1960). Wine has also been found to contain small
amounts of thallium (0.056 to 0.684 µg/litre) (Geilmann et al., 1960).
5.3 Occupational exposure during manufacture, formulation or use
Few data on occupational exposure to thallium are available. An
industrial plant in the USA used concentrated thallium salt solutions
for separations by centrifugation. Considerable variations in the
thallium content of the air occurred during the day, depending on the
emission potential of the different steps of the procedure (Hill &
Murphy, 1959), although no exact data were reported. In a plant in
the United Kingdom manufacturing special alloy anodes for use in
magnesium seawater batteries, air samples from two working areas
contained a maximum level of 0.022 mg thallium/m3 and 0.014 mg/m3
(Marcus, 1985). Detailed data are available from a Russian plant
producing thallium before and after changes in processes. The air
concentration varied from 0.12 to 0.18 mg thallium/m3, but peak
concentrations of 13.5 to 17.4 mg/m3 during the smelting process
were observed. During dissolving and packing of thallium salts, the
air thallium concentrations were 0.117 and 0.274 mg/m3, respectively.
After changes in the smelting process were instituted, the air
thallium content decreased to 0.0036-0.0072 mg/m3 (Tikhova, 1964).
Floating dust in a German thallium smelter contained 60 to 9700 mg
thallium/kg dust; the air-suspended dust concentration was 6-50 µg/m3
(Briese et al., 1985).
Working with thallium causes dust contamination of the hands,
this increasing with the duration of work (Tikhova, 1964). The dust
concentration on the hands has been reported to be in the range of
0.04 to 10.6 mg/m2 (Shabalina & Spiridonova, 1979).
6. KINETICS AND METABOLISM
Investigations into the kinetics and metabolism of thallium in
aquatic and terrestrial animals have mainly made use of radioactive
compounds, especially thallium-201. The investigations cited in this
chapter have been performed with various thallium salts, but to
facilitate comparison concentrations have been generally expressed as
µg (or mg) thallium/litre.
6.1 Absorption
6.1.1 Animals
6.1.1.1 Aquatic animals
Clams and mussels reached equilibrium within 12 to 19 days when
exposed to 50 or 100 µg thallium/litre. Depending on the exposure
level, the clams contained 5 or 9 mg/kg dry weight and the mussels 3
or 5 mg/kg (Zitko & Carson, 1975).
Very poor absorption of thallium salts from water containing
0.1 mg thallium/litre was found in isolated gill preparations of the
mussel Mytilus galloprovincialis (Nolan et al., 1984). In the first
10 min about 10% of the dose was absorbed.
Accumulation of thallium in juvenile salmon exposed for 300 h to
different concentrations of thallium (17.9 to 200 µg thallium/litre)
varied in different organs (Table 15). In muscle, thallium levels
increased almost linearly with the water thallium concentration from
2.3 to 27.0 mg/kg tissue (wet weight). Data on the liver show no
consistent trend, but in gills an obvious maximal accumulation
capacity of about 30 mg/kg was reached even at the lowest
concentration of 17.9 µg/litre water (Zitko et al., 1975). The mean
accumulation factor (mg thallium per kg tissue wet weight divided by
mg thallium per litre water) of the gills (up to 1430) is about three
to ten times higher than that of muscle or liver.
6.1.1.2 Terrestrial animals
Using different routes of administration of thallium(I) nitrate
solution (oral, intratracheal, subcutaneous, intraperitoneal,
intramuscular and intravenous), thallium was rapidly and almost
completely absorbed in rats (Lie et al., 1960). High concentrations
of thallium were detected in the blood within just 1-2 h after oral
administration of thallium(I) malonate and thallium(I) sulfate
(Aoyama, 1989), and as little as 1 h after oral or parenteral
administration of thallium(I) sulfate it was found in the urine and
faeces of rats (Lund, 1956a).
Only a few experimental studies on intestinal absorption are
available. In sheep and cows about 2% of the thallium ingested with
contaminated food was retained, while about 98% was eliminated
(Crössmann, 1984). Sabbioni et al. (1980b) found no obvious
differences using various doses of thallium(I) or thallium(III)
sulfate or bromide, but, after oral uptake of dimethyl thallium(III)
bromide in rats, the organs contained only 1 to 10% of the
concentrations found after the uptake of inorganic thallium,
indicating reduced absorption. In rats the absorptive capacity of
different ligated regions of the intestinal tract varies strongly:
201thallium (sulfate) was rapidly absorbed from the colon, but more
slowly from the ileum and jejunum and slowest from the stomach
(Sabbioni et al., 1984a). Within 60 min the ligated colon absorbed
about 75%. Lower values were obtained for the other regions. Voltage
clamp experiments on the mucosa of rat descending colon showed
exclusive transport by diffusion to the serosal side (Schäfer et al.,
1981).
Absorption of thallium through the skin of rats is indicated by
the determination of a cutaneous LD50 of 117 mg/kg for thallium(I)
carbonate (Shabalina et al., 1980).
6.1.2 Humans
Increased levels of thallium have been observed in the lungs of
coal miners, but no data are available concerning the absorption of
thallium salts after inhalation exposure (section 6.2.2.2) (Weinig &
Zink, 1967). Generally it is assumed that about 35% of respirable
dust is deposited in the lung (Ohnesorge, 1985; IPCS, 1992) and that
up to 100% of the deposited thallium is absorbed (Gubernator et al.,
1979). The rate of deposition and absorption is high because thallium
concentrations increase markedly with decreasing particle size
(sections 3.2.3.2 and 5.1.1.2), and small particles become deposited
in the lung whereas larger particles are deposited in the upper
respiratory system (Natusch & Wallace, 1974). In addition, nearly all
the thallium chloride in the dust emitted from the cement plant in
Lengerich was water-soluble (LIS, 1980). In human broncho-alveolar
lavage fluids, 0.258 ng thallium per 1000 cells was found in a
silicosis patient, but only 0.009-0.05 ng in patients suffering from
other lung diseases (Maier et al., 1986).
From several intoxication cases, e.g., after oral and topical
application of thallium(I) sulfate during depilatory treatment
(Barckow & Jenss, 1976; Schmidbauer & Klingler, 1979), it can be
assumed that both percutaneous and gastrointestinal absorption occur,
but no data on absorption are available. High blood thallium
concentrations have been reported following human poisoning (see
Table 20).
6.2 Distribution
6.2.1 Animals
No effect of the route of administration (oral, intratracheal,
subcutaneous, intraperitoneal, intramuscular and intravenous) on
distribution was observed by Lie et al. (1960). After intravenous
injection, an initial increase in the thallium concentration of the
blood is followed by a steep decrease within 5 to 15 min (Gehring &
Hammond, 1967; Lameijer & van Zwieten, 1977a,b). A similar trend is
observed when concentrations are compared 1.5 and 24 h after oral
administration of 10 mg thallium/kg body weight to rats or intravenous
injection into rabbits (Careaga-Olivares & Morales-Aguilera, 1993).
Thallium is distributed by the blood stream to all organs.
Data on the distribution of thallium in the main blood
compartments, e.g., serum and erythrocytes, have been reported. In
vitro measurements by Lund (1956a) and Witschi (1965) indicate that
thallium is distributed equally in blood plasma and red cells,
presumably without any direct binding, while Gregus & Klaassen (1986)
found that 2 h after intravenous injection of 1 to 30 mg/kg more than
80% of blood thallium was in the plasma. According to Ducket et al.
(1983), 24 h after intraperitoneal injection of a very low dose of
thallium, nearly 90% was located in the red blood cells and only about
10% in the plasma. An intermediate result was obtained by Ulrich &
Long (1955): after intraperitoneal injection of about 20 µg
radioactive thallium, about one third of the thallium concentration in
the whole blood was located in the plasma. This ratio did not change
over the period 0.5 h to 96 h after injection, although the
concentration in the blood decreased with time. Similar results were
obtained in human whole blood (in vitro) and in rabbit whole blood
( in vitro and in vivo) (Careaga-Olivares & Morales-Aguilera,
1990). A slightly higher concentration in the erythrocytes was also
found by Leloux et al. (1987), but during redistribution phases the
concentrations in erythrocytes were increased, suggesting that
erythrocytes may be involved in thallium uptake.
6.2.1.1 Distribution after administration of a single dose
The distribution of thallium after administration of single doses
of thallium compounds has been investigated in a number of studies,
using either subtoxic doses (0.2 µg/kg to 8 mg thallium/kg body
weight) (e.g., Ulrich & Long, 1955; Lie et al., 1960; Bradley-Moore et
al., 1975; Edel Rade et al., 1982; Ziskoven et al., 1983; Ducket et
al., 1983) or toxic doses (e.g., Lund, 1956a; Fitzek & Henning, 1976;
Achenbach et al., 1980; Leloux et al., 1987b; Aoyama, 1989).
Comparisons of dose-dependent distribution (Emara & Soliman, 1950;
Gehring & Hammond, 1967; Sabbioni et al., 1980a,b, 1982; Talas &
Wellhöner, 1983; Gregus & Klaassen, 1986; Ríos et al., 1989) revealed
only slight differences in the distribution pattern, even at
intraperitoneally administered concentrations as different as 0.00004,
2, 20 and 2000 µg thallium/rat (Sabbioni et al., 1980a).
The distribution of thallium in the organs is a time-dependent
process (Tables 17 and 18). Summarizing the investigations on rats,
rabbits, dogs and goats, in an initial phase after a single dose of an
inorganic thallium compound, e.g., thallium sulfate or thallium
chloride, maximal concentrations occurred in the kidneys and nearly
equal levels were found in the testes, myocardium, salivary glands,
muscle, liver, intestines, adrenals and thyroid; fat and brain
contained very low levels of thallium (Lund, 1956a; Gehring & Hammond,
1967; Bradley-Moore et al., 1975; Sabbioni et al., 1982; Talas &
Wellhöner, 1983). About 24 h after administration, the relative
thallium content of all organs, with the exception of the kidneys,
decreased and that of brain, muscle and testes increased. In
guinea-pigs administered lethal doses of thallium, kidney and liver
finally contained about equal levels, presumably due to kidney damage
(Weinig & Walz, 1971). With respect to the affinity and
redistribution of thallium in rat organs, Leloux et al. (1987b)
distinguished three compartments according to affinity and
redistribution which are not completely in agreement with the
experimental data shown in Table 17.
Intraperitoneal application of 16, 32 and 48 mg thallium
sulfate/kg body weight to male rats resulted in peak levels occurring
in various regions of the brain 24 h after injection, except in the
hypothalamus where the peak was reached earlier. After 24 h, the
regional concentrations decreased in the following order:
hypothalamus, midbrain, hippocampus, thalamus, pons, cerebellum,
corpus striatum and cerebral cortex. In the hypothalamus, a region
with low blood-brain barrier protective mechanisms, thallium
concentration was significantly higher than in the corpus striatum,
whereas in the cerebral cortex it was significantly lower than in all
other regions (Ríos et al., 1989). Whereas weanling rats showed a
region-dependant distribution of thallium after a single sublethal
intraperitoneal injection (16 mg thallium/kg body weight), thallium
concentrations were similar in different brain regions of newborn rats
(Galvan-Arzate & Ríos, 1994).
Table 17. Alterations in the distribution of thallium in different organs of experimental animals at different times after
administration (mean ± standard deviation)a
Rats (4; intraperitoneal)b Syrian hamsters (5; oral)c
Experiment No.: 1 2 3 4
2 h 40 h 4 h 24 h 1 h 24 h 1 h 24 h
(% of dose) (% of dose of thallium/g wet weight) (mg thallium/kg wet weight)
Blood 0.05 ± 0.01d 0.08 ± 0.05d 0.023 ± 0.001 0.027 ± 0.017 1.5 ± 0.6 1.0 ± 0.1 1.7 ± 0.6 0.8 ± 0.1
Bone - - 0.239 ± 0.036 0.216 ± 0.064 - - - -
Brain 0.027 ± 0.003 0.28 ± 0.05 0.047 ± 0.009 0.090 ± 0.023 < 0.1 3.7 ± 0.3 0.6 ± 0.1 3.0 ± 0.3
Heart 0.57 ± 0.07 0.33 ± 0.03 0.546 ± 0.064 0.337 ± 0.006 11.4 ± 1.7 7.2 ± 0.7 21.0 ± 3.4 6.6 ± 0.7
Intestine 1.1 ± 0.26 - - - - - - -
Kidney 5.65 ± 0.35 9.75 ± 0.97 3.354 ± 0.535 1.899 ± 0.253 58.5 ± 8.4 51.3 ± 14.0 88.4 ± 21.8 41.5 ± 0.9
Liver 4.44 ± 0.55 0.95 ± 0.19 0.373 ± 0.040 0.228 ± 0.028 14.3 ± 7.6 6.6 ± 2.1 39.7 ± 6.2 5.8 ± 0.4
Lung 0.55 ± 0.35 0.45 ± 0.04 0.289 ± 0.076 0.230 ± 0.081 - - - -
Muscle 0.6 ± 0.02e 0.54 ± 0.2e 0.168 ± 0.031 0.234 ± 0.091 < 0.1 9.1 ± 2.4 1.2 ± 0.5 9.1 ± 1.2
Table 17 (contd).
Rats (4; intraperitoneal)b Syrian hamsters (5; oral)c
Experiment No.: 1 2 3 4
2 h 40 h 4 h 24 h 1 h 24 h 1 h 24 h
(% of dose) (% of dose of thallium/g wet weight) (mg thallium/kg wet weight)
Pancreas 0.86 ± 0.34 0.57 ± 0.13 0.505 ± 0.163 0.310 ± 0.206 - - - -
Salivary 0.76 ± 0.06 0.7 ± 0.32 0.595 ± 0.078 0.370 ± 0.090 - - - -
gland
Spleen 0.32 ± 0.03 0.18 ± 0.02 0.338 ± 0.039 0.249 ± 0.054 - - - -
Testes/ 0.56 ± 0.04 0.93 ± 0.34 0.388 ± 0.077 0.200 ± 0.010 < 0.1 14.7 ± 0.7 1.0 ± 0.1 14.0 ± 1.0
Ovary
a - = no data given
b No. 1: Sabbioni et al. (1980b) and No. 2: Edel Rade et al. (1982): injection of thallium(I) sulfate (2 µg thallium/rat)
c Nos. 3 and 4: Aoyama (1989): administration of 12.5 mg thallium(I) sulfate/kg (No. 3) or 12.35 mg thallium(I) malonate/kg (No. 4)
d % dose/ml
e % dose/g wet weight
Table 18. Distribution of thallium in different organs of experimental animals after different
periods of exposurea
Experiment no:b 1a 1b 2 3 4a 4b 5
72 h 72 h 4 h 24 h 48 h 48 h 48 h
Blood 0.007 0.001 0.02 0.02 0.02 0.02 -
Bone 0.016 0.028 0.37 0.42 0.23c 0.23c 0.67
Brain 0.006 0.008 0.05 0.13 0.17 0.14 0.32
Fat 0.0008 0.001 - 0 0.01 0.02 -
Heart 0.027 0.030 0.55 0.20 0.34 0.25 0.63
Intestine 0.012d 0.017d - 0.51 0.49e 0.76e 0.50
Kidney 0.205 0.210 3.35 2.15 2.58 0.53 5.36
Liver 0.019 0.026 0.37 0.33 0.19 0.16 0.53
Lung 0.016 0.018 0.29 0.55 0.25 0.24 0.51
Muscle 0.014 0.025 0.17 0.49 0.25f 0.27f 0.76
Table 18 (contd).
Experiment no:b 1a 1b 2 3 4a 4b 5
72 h 72 h 4 h 24 h 48 h 48 h 48 h
Pancreas 0.010 0.014 0.51 - 0.44 0.33 -
Salivary gland - - 0.59 - 0.42 0.44 1.06
Skin - - 0.20 - - 0.32
Spleen 0.016 0.021 0.34 0.20 0.24 0.21 0.51
Testes/ 0.023 0.024 0.39 0.39 0.53 0.53 0.81
Ovary
a Thallium content in % of dose per kg wet weight; - = no data given
b No. 1: Talas & Wellhöner (1983): mean of 2 rabbits after intravenous injection of < 2 µg
(No. 1a) or 1.1 mg thallium/kg (No. 1b); No. 2: Sabbioni et al. (1982): mean of 4 rats
after intraperitoneal injection of 2 µg/rat; No. 3: Lund (1956a): data of 1 rat after
intraperitoneal injection of 10 mg/kg; No. 4: Barclay et al. (1953): mean of 4 rats after
intravenous injection of 23 µg thallium nitrate (No. 4a) or mean of 3 rats after
intravenous injection of 10.023 mg thallium sulfate (No. 4b); No. 5: Lie et al. (1960):
mean of 18 rats after injection of thallium by various routes
c Femur
d Mean of values of small and large intestine
e Lower bowel
f Abdominal muscle
Although the endocrine organs were thought to be involved in the
mechanism of toxicity, no accumulation was observed in
autoradiographic studies of low-dosed adult mice and rats (Barclay et
al., 1953; André et al., 1960). Leloux et al. (1987b) found no
obvious deviation from the levels found in other organs 5 days after
subacute or acute intoxication with 4 or 20 mg thallium nitrate/kg,
respectively. Thyresson (1951), however, reported a high
concentration of thallium (96.9 mg/kg wet weight) in the thyroid gland
of rats 24 h after administration of 40 mg thallium nitrate/kg body
weight. According to Ulrich & Long (1955), treatment of rats with
thyrotropic hormone subsequent to the administration of thallium did
not affect the uptake of thallium by the thyroid, whereas pretreatment
(2 days prior to administration) significantly increased the initial
uptake. In addition, the authors reported that adrenals, thyroid and
pituitary contained similar concentrations of thallium.
Different modes of application and different thallium compounds
hardly affect the distribution pattern, as shown by Sabbioni et al.
(1980b) with intravenous or oral administration of thallium(I),
thallium(III) and dimethyl thallium(III) in rats. Another
investigation showed that after administration of thallium(I) malonate
and thallium(I) sulfate the distribution pattern between different
organs varied only initially. Later, both patterns were similar
(Aoyama, 1989).
6.2.1.2 Distribution after long-term sublethal administration
The distribution pattern of thallium in chronically poisoned rats
shows a strong similarity to the final pattern after a single dose,
there being wide distribution throughout the body. Lameijer & van
Zwieten (1977a,b) determined thallium concentrations in urine, blood
and 19 different tissues of rats exposed to thallium (10 mg/litre) in
drinking-water for 9 or 24 weeks. No statistically significant
differences in the concentration of thallium were observed for any
tissues except kidney. The thallium concentration in the renal medulla
was about 7 times higher than in the heart, liver, muscle, brain or
skin. If rats received 30 mg/litre in drinking-water, they died
within 9 to 11 days, but after 7 days this administration had not
affected the rapid decline of the thallium blood level following
administration of an additional intravenous dose of 1 mg/kg (Lameijer
& van Zwieten, 1977a).
6.2.1.3 Transplacental transfer of thallium
Mice were gavaged with thallium(I) sulfate (8 mg thallium/kg body
weight) on gestation day 9 and tissue concentrations were determined
0.5 to 24 h later. Thallium levels in fetuses and maternal kidneys
rose during the first hour, then levelled off to a plateau which did
not change during the following 22 h. No indication of a specific
placental barrier was detected (Ziskoven et al., 1980). Ziskoven et
al. (1983) repeated the study, additionally including rats. After 10,
20 and 30 min, increasing thallium concentrations were found in the
maternal kidneys, reaching a plateau after 30 to 60 min, which was
stable for at least 50 h (last measurement). A similar time course of
thallium uptake was observed in the fetal tissues: the initial uptake
was comparable to that of the maternal kidney, but the resultant
concentrations were 10-fold lower. There were no specific differences
between mice and rats.
A slightly delayed transfer to fetuses was observed in an
autoradiographic study with mice (gestation day 15), dosed with an
intraperitoneal injection of thallium sulfate. After just 15 min,
thallium was observed in the fetuses, but the maximum level was
reached within 2 to 4 h, when some thallium elimination took effect.
During the whole observation period, fetal thallium levels were lower
than those of the placenta (Olsen & Jonsen, 1982). The authors also
reported on the influence of the stage of gestation. Thallium crossed
the placenta throughout gestation; during the early stages it was
concentrated in the visceral yolk sac placenta and during late
gestation additionally in the chorioallantoic placenta and amnion.
When near-term mice and rats (gestation day 17-18) were given
subcutaneous injections of 204thallium sulfate, thallium
concentrations in the mouse fetuses rose during the following 8 h and
in the rat fetuses during the following 16 h. Thereafter, the
fetal/maternal ratios in tissues remained constant at 0.84 in rats and
considerably lower (0.46) in mice (Gibson et al., 1967).
Intra-arterial infusion of 0.2 to 6.4 mg 204thallium
sulfate/min per kg body weight on day 20 of pregnancy in rats resulted
in an initially restricted transplacental transfer. In the lowest and
highest dosage groups, 32 min after administration, thallium levels in
fetal tissues corresponded to only about 7% of those in the maternal
plasma, perhaps because two-thirds of the whole blood thallium was
located in the erythrocytes and did not pass the placental barrier
(Gibson & Becker, 1970).
Sabbioni et al. (1982) compared placental transfer in rats after
intraperitoneal injection of a low dose (2 µg thallium-201/rat;
gestation day 13) with that after administration of a toxic dose by
gavage (10 mg thallium/kg body weight; gestation day 16). Thallium
concentrations in maternal and fetal brain were similar 4 h after
injection of the low dose. In fetal liver they were 80% lower than in
the maternal liver, and in the placenta and fetal organs they were
higher than in the blood of the dams and fetuses. Very low
concentrations were found in the amniotic fluid. After 8 days,
concentrations in most of the maternal organs were about 10 times
lower than the initial levels, while in muscle, cerebellum and brain
of the dams, reductions of only 60, 40 and 5%, respectively, were
found during the same period. In contrast to the variable decline in
maternal organs, a 10-fold decline was observed in the whole fetus and
its liver, brain and blood. Preliminary results of the authors
indicated that this was due to a much stronger thallium accumulation
in the mitochondria of the adult rat brain than in those of the fetal
brain. Furthermore, identical low-dose experiments (2 µg/rat) also
showed a faster decline of thallium concentrations in fetal than in
maternal brain, after an initially more rapid entry into the fetal
brain, whereas declines in the liver levels were nearly identical
(Edel Rade et al., 1982). Administering a toxic dose of 10 mg/kg to
the dams resulted in 60% lethality within the following 3 days. After
3 days, concentrations in the liver and brain of the surviving dams
were similar and about 2-fold higher than in the corresponding fetal
organs. Therapeutic oral dosing with Prussian Blue (100 mg/kg body
weight, twice daily) starting 8 h after administration of thallium,
significantly reduced the thallium concentrations in maternal and
fetal tissues and only 1 of 12 adult rats died (Sabbioni et al.,
1982).
Transplacental transfer of thallium has also been observed in a
cat (Fitzek & Henning, 1976). The cat showed signs of a strong
thallium intoxication and was killed after abortion of approximately
5-week-old fetuses. Thallium levels in maternal blood and fetal
tissues were similar, but the concentrations in fetal heart and lungs
were two to three times higher than in the corresponding maternal
organs.
6.2.2 Humans
Background thallium concentrations found in human body fluids and
tissues are given in Table 19. After poisoning, thallium
concentrations ranging up to nearly 36 mg/litre in blood, 25 mg/litre
in urine and 8 mg/kg in hair have been found (Table 20).
6.2.2.1 Increased concentrations after lethal poisoning
In reports of postmortem examinations after suicide or homicide,
data on the distribution of thallium in different organs are rarely
included with data on dose and application routes (Table 20). The
distribution pattern shows no consistent trend. In a single
individual, concentrations in bones, fat and muscles from different
parts of the body may vary, e.g., in vertebrae (12.7 mg/kg), sternum
(7.0 mg/kg), femur (16.4 mg/kg) and tibia (9.0 mg/kg) (case 7 in
Table 21) (Arnold, 1986). The distribution of thallium differs
considerably from that reported for potassium in humans (Davis et al.,
1981). Endocrine glands, kidneys, liver and intestine (without
content) showed the highest concentrations (Table 21).
With respect to the total amount per organ, liver or lung were
found to contain 2 to 6 times and the brain about 1.5 to 2 times more
thallium than the kidneys (Curry et al., 1969; Arnold, 1986).
A comparison between the white and grey matter of the brain
revealed that in the latter the concentration was three times higher
(Cavanagh et al., 1974). Detailed data of thallium concentrations in
different regions of the nervous system were given by Davis et al.
(1981). The authors showed that areas of the brain rich in neurons
tend to accumulate twice as much thallium as areas devoid of neurons,
and that the grey matter contains some of the highest thallium levels
of any body organ.
6.2.2.2 Increased concentrations after long-term sublethal poisoning
Thallium levels in urine (Table 22), blood or saliva of
chronically exposed people offer better indications of the actual
burden than those derived from hair samples, since elevated levels in
hair can be caused by exogenous dust (Bertram et al., 1985).
People consuming food grown in private gardens and living at a
distance of more than 3 km from the cement plant at Lengerich, Germany
showed significantly higher concentrations of thallium in their urine,
decreasing with increasing distance from the plant, than people who
did not consume food from their gardens. Thallium concentrations in
the urine of people living near the plant (< 1 km) and consuming food
grown in private gardens were about five times higher (3.95 µg/litre)
(Brockhaus et al., 1980). Peak values were 76.5 µg/litre in urine and
565 µg/kg in hair (Ewers & Brockhaus, 1982). In this area a medical
survey was carried out immediately after the occurrence of thallium
emissions had been recognized; urine thallium levels in about 80% of
the population were found to exceed the upper normal limit of
1 µg/litre (Brockhaus et al., 1980; Dolgner et al., 1983). The
recommendation to avoid home-grown vegetables was followed by many
people and resulted in a significant decrease in urine thallium
levels. However, in some residents, even 8 years later, increased
levels of > 20 µg/litre urine could be found. Probably there was
still a significant contamination of soil and thus of home-grown
vegetables (Ewers, 1988).
Subsequent studies were carried out at other cement factories.
About 70% of employees at two cement plants in Middle and Lower
Franconia, Germany were found to have normal thallium concentrations
in their urine. However, at a third factory in the same area only 30%
of employees showed normal thallium urine levels, presumably because
of the higher thallium content in the raw material used (Schaller et
al., 1980). The population around the three cement plants showed
normal urine thallium levels: of 238 people tested, 194 had thallium
concentrations below 2 µg/litre, 36 were in the range of 2 to
5 µg/litre, and 5 were between 5 and 10 µg/litre. Higher
concentrations were found in the urine of three people (11.5, 14.5 and
19.5 µg/litre) (Steuer, 1980).
Table 19. Background concentrations of thallium in humans
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Blood, 2 0.33-0.59 µg/litre Weinig & Zink (1967)
whole < 20 Bowen (1966)
13 0.47-9 Iyengar et al. (1978)
320 < 5-80 Singh et al. (1975)
0.05 Kemper (1979)
418 0.39 ± 0.05 0.1-1.1 Minoia et al. (1990)
0.5-2 Kemper & Bertram (1991)
plasma < 2.5 Bowen (1966)
1 < 2.5 Iyengar et al. (1978)
Bone 2 0.84-2.51 µg/kg fresh weight Weinig & Zink (1967)
1 2 Iyengar et al. (1978)
5 < 0.1-0.1 Goenechea & Sellier (1967)
Bonea 1 0.7; 0.9 Goenechea & Sellier (1967)
Brain < 0.5 mg/kg dry weight Bowen (1966)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Bronchoalveolar 1b 0.258 ng/1000 cells Maier et al. (1986)
lavage fluids 1c 0.009
1c 0.011
1d 0.016
1d 0.050
Faeces 5 < 0.02-3.0 µg/kg fresh weight Goenechea & Sellier (1967)
Hair 7 18.6 ± 14.9 7-51 µg/kg fresh weight Geilmann et al. (1960)
6 10.4 ± 4.3 4.8-15.8 Weinig & Zink (1967)
1 < 20 Ziegler & Ziegler (1984)
Heart < 0.4 mg/kg dry weight Bowen (1966)
Kidney < 0.4 mg/kg dry weight Bowen (1966)
Kidney 6 2.7 ± 1.1 1.44-4.1 µg/kg fresh weight Weinig & Zink (1967)
8 < 3 Iyengar et al. (1978)
259 0.03-8.6 Bösche & Magureanu (1983)
Liver 0.4 mg/kg dry weight Bowen (1966)
11 0.47 ± 0.13 < 0.4-0.9 Johnson (1976)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Liver 6 1.1 ± 0.9 0.55-2.85 µg/kg fresh weight Weinig & Zink (1967)
1 0.4 Goenechea & Sellier (1967)
6 1-9 Iyengar et al. (1978)
0.5-3 Kemper & Bertram (1991)
Lung < 0.3 mg/kg dry weight Bowen (1966)
Lung 4 1.1 ± 0.7 0.36-1.8 µg/kg fresh weight Weinig & Zink (1967)
Muscle < 0.4 mg/kg dry weight Bowen (1966)
Muscle 6 2.1 ± 2.1 0.52-7.05 µg/kg fresh weight Weinig & Zink (1967)
3 15-100 Iyengar et al. (1978)
Musclee 1 0.4 µg/kg fresh weight Goenechea & Sellier (1967)
Nail 6 51.2 ± 12.1 40-74 µg/kg fresh weight Geilmann et al. (1960)
6 2.6 ± 1.4 0.72-4.93 Weinig & Zink (1967)
Skin < 0.2 mg/kg dry weight Bowen (1966)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Urine 10 < 0.02-1.0 µg/kg fresh weight Goenechea & Sellier (1967)
Urine 0.05-0.1 µg/litre Geilmann et al. (1960)
14 0.7 ± 0.5 0.07-1.69 Weinig & Zink (1967)
0.05-20 Kemper (1979)
31 0.4 ± 0.2 < 0.1-1.2 Brockhaus et al. (1981b)
10 0.3 ± 0.2 < 0.1-0.9 Brockhaus et al. (1981b)
149 0.3 ± 0.14 0.02-0.7 Dolgner et al. (1983)
72 0.22 ± 0.14 0.06-0.61 Apostoli et al. (1988)
496 0.42 ± 0.09 0.06-0.82 Minoia et al. (1990)
0.05-1.5 Kemper & Bertram (1991)
Urine 20 < 0.3-1.1 mg/kg creatinine Schaller et al. (1980)
10 2.2 ± 1.6 Briese et al. (1985)
a 1.5 months after death
b silicosis patient
c saw setters suffering pneumoconiosis
d welders suffering emphysema
e 6 months after death
f Additional values for other tissues have been compilated by Iyengar et al. (1978)
Table 20. Concentrations of thallium in cases of poisoning
Material Number of Range of thallium Concentration unit Reference
cases concentrations
Blood, whole 50-6000a µg/litre Kemper (1979)
Blood 2 29; 7700 Alarcón-Segovia et al. (1989)
Blood, whole 3 350-36 000 Heath et al. (1983)
Blood, plasma 2 300; 1500 Heath et al. (1983)
Blood, erythrocyte 2 400; 2300 Heath et al. (1983)
Bone 1b 0.9-2.1 µg/kg fresh weight Goenechea & Sellier (1967)
Faeces 1 6500-38 400 Paulson et al. (1972)
Hair 1 650 Geilmann et al. (1960)
Hair 1b 6.8 Goenechea & Sellier (1967)
Hair 1 420-1800 Hagedorn-Götz & Stoeppler (1975)
Hair 250-8000a Kemper (1979); Kemper & Bertram (1984)
Heart 1 3600 Munch et al. (1933)
Intestine 1 3600 Munch et al. (1933)
Intestine 1 0.8; 4.0 Goenechea & Sellier (1967)
Table 20 (contd).
Material Number of Range of thallium Concentration unit Reference
cases concentrations
Kidney 5 2700-11 600 Munch et al. (1933)
Kidney 1 106 000 Heath et al. (1983)
Liver 1 75 000 Heath et al. (1983)
Liver 2 3700; 5500 Munch et al. (1933)
Lung 2 3300; 7700 Munch et al. (1933)
Muscle 1b < 0.02; 1.3 Goenechea & Sellier (1967)
Nails 1 2400 Geilmann et al. (1960)
Spleen 3 2900-6600 Munch et al. (1933)
Urine 50-25 000a µg/litre Kemper (1979)
Urine 15 500-20 400 Klöppel & Weiler (1978)
Urine 1 3100 Gastel (1978)
Urine 3 10-13 800 Alarcón-Segovia et al. (1989)
Urine 2 2700-30 000 µg/litre fresh weight Heath et al. (1983)
Urine 1 0.65 mg/kg creatinine Hagedorn-Götz & Stoeppler (1975)
a Concentrations indicative for poisoning
b 3 years after death
Table 21. Concentrations of thallium in individual cases of human poisoning
Thallium concentration (mg/kg wet weight or mg/litre)
Case no:a 1 2 3 4 5 6 7
Durationb: > 14 days > 21 days 9 days 8 days 11 days 12 days 13 days
Dose: -c - 5-10 g 0.75 gd - - -
Adrenal - - 83.6 - - - -
Blood 5.1 3.4 5.1 - - 3.0 -
Bone - 5.0 - 0.92 1.9 8.0 7.0-16.4
Brain 8.5 - 62-140 0.15 - - -
grey - 10.0 - - - - -
white - 3.0 66.1 - - - -
cerebellum - - 103.3e - 1.5 5.0 -
cerebrum - - 102.0e - 1.0 - -
Fat - - < 1.0 - 0.4-1.2 - -
Heart - 13.3 - 0.19 1.5 13.0 6.2
left - - 26.8 - - - -
right - - 131.6 - - - -
Intestine - - - 0.20 - - -
small 4.4f - - - 0.5-0.9 8.0f 6.4
colon 71.0f 120.0f 126.0 - 2.0 500.0f 8.5
Kidney 26.7 20.0 74.1 0.26 3.0 28.0 12.5
Liver 8.6 5.0 77.3 0.82 1.8 15.0 14.7
Table 21 (contd).
Thallium concentration (mg/kg wet weight or mg/litre)
Case no:a 1 2 3 4 5 6 7
Durationb: > 14 days > 21 days 9 days 8 days 11 days 12 days 13 days
Dose: -c - 5-10 g 0.75 gd - - -
Lung - 1.8 - 0.15 0.9 4.0 -
Muscle 10.1 5.0 26.8 0.21 0.4-2.0 - 3.6
Pancreas - - 71.7 - - - -
Parathyroid - - 38.1 - - - -
Pituitary - - 114.5 - - - -
Salivary gland - - 32.1 - - - -
Skin - 6.0 32.1 - 0.3 - -
Spleen - - - 0.35 1.9 - -
Testes/Ovaries - - 152.0 - - - -
Thyroid - - 33.5 - 4.6 - -
Urine 15.6 5.9 3.3 - - 3.0 -
a Case no. 1: Curry et al. (1969); no. 2: Cavanagh et al. (1974); no. 3: Davis et al. (1981); no. 4: Graben et al.
(1980); nos. 5-7: Arnold (1986); nos. 3-7: poisoning by one uptake of thallium
b Period of time from uptake of thallium to death or determination of concentrations
c - = no data given
d 15-week-old embryo; dose of mother
e Cortex
f Content
Table 22. Concentrations of thallium following environmental or occupational exposure
Material Number of Concentration of thallium Concentration unit Source of Reference
people thallium
Mean ± SD Range
(Median)
Hair 1163 20.3 ± 42.7 0.6-565 µg/kg fresh weight cement plant Brockhaus et al. (1981b)
Urine 50 (0.6) < 0.3-4.9 mg/kg creatinine cement plant Schaller et al. (1980)
47 (1.65) 0.4-6.3 mg/kg creatinine cement plant Schaller et al. (1980)
21 (0.34) < 0.3-2.9 mg/kg creatinine cement plant Schaller et al. (1980)
10 7.1 ± 6.0 mg/kg creatinine zinc smelter Briese et al. (1985)
1265 5.2 ± 8.3 < 0.1-76.5 µg/litre cement plant Brockhaus et al. (1981b)
82 2.4 ± 4.3 < 0.1-35.8 µg/litre cement plant Dolgner et al. (1983)
117 3.0 ± 5.6 0.2-37.7 µg/litre cement plant Dolgner et al. (1983)
34a 3.4 ± 3.5 0.4-14.8 µg/litre cement plant Dolgner et al. (1983)
30 0.38 ± 0.30 0.08-1.22 µg/litre cement plant Apostoli et al. (1988)
20 0.40 ± 0.34 0.08-1.22 µg/litre cement plant Apostoli et al. (1988)
10 0.33 ± 0.16 0.09-0.60 µg/litre cement plant Apostoli et al. (1988)
Table 22 (contd).
Material Number of Concentration of thallium Concentration unit Source of Reference
people thallium
Mean ± SD Range
(Median)
Urine
(contd) 9 0.38 ± 0.29 0.10-1.04 µg/litre iron smelter Apostoli et al. (1988)
74b 16.0 ± 16.9 0.2-76.5 µg/litre cement plant Brockhaus et al. (1981a)
74 7.9 ± 8.8 0.2-42.6 µg/litre cement plant Dolgner et al. (1983)
21 0.33 ± 0.27 0.06-1.04 µg/litre iron smelter Apostoli et al. (1988)
12 0.29 ± 0.21 0.06-0.70 µg/litre iron smelter Apostoli et al. (1988)
a children
b Data of people with high concentrations of thallium or possibly thallium-related disorders determined in the first survey and about
1 year later in the following line
Only a few cases resulting from industrial exposure have been
reported and seem to be mainly a result of skin contact or inhalation
(Kazantzis, 1986; Ewers, 1988).
At a zinc smelter in eastern Germany, increased thallium levels
were not only found in the urine of men working in the production
process, but also in men working in the administration. During the
production of thallium in this plant, the levels were further
increased (maximal value: 28.6 µg/litre) (Briese et al., 1985).
High concentrations of thallium were found in lung tissue from
two coal miners (20.2 and 29.5 µg/kg wet weight). Concentrations in
most other tissues were normal (Weinig & Zink, 1967).
In Italy, slight but significant increases in thallium levels
were found in the urine of cement workers (0.4 µg/litre) and cast iron
workers (0.3 µg/litre), compared with a non-exposed group
(0.2 µg/litre). There was no correlation with age or the duration of
exposure (Apostoli et al., 1988).
Weinig & Zink (1967) reported a slight elevation of urine
thallium levels of vegetarians (and smokers) compared to controls.
However, it should be noted that each group comprised only three
people and the levels were far below those of thallium-affected
people. Geilmann et al. (1960) estimated that more than 60% of the
thallium content of a cigarette (62 µg/kg) is inhaled, but no data are
available on the amount absorbed. Assuming an absorption of 50% and a
consumption of 20 cigarettes/day, 375 ng/day would be absorbed (BGA,
1979). Based on data of the thallium concentration in urine of about
120 people, non-exposed individuals and workers with suspected
industrial exposure to thallium, Apostoli et al. (1988) found no
evidence of a difference between smokers and non-smokers, all about 40
years old.
Comparing a total of 128 men, no correlation was found between
duration of employment at a cement plant (1 to 42 years) or age (16 to
62 years) and thallium concentrations in the urine (< 0.3 to 6.3 µg
thallium/g creatinine) (Schaller et al., 1980). Therefore, it can be
concluded that the uptake of low amounts of thallium does not cause
accumulation in the body.
6.2.2.3 Transplacental transfer of thallium
Abortion was produced in the fourth month of pregnancy 8 days
after ingestion of approximately 750 mg thallium sulfate. Starting 2
days after the ingestion, the mother was treated (haemodialysis,
forced diuresis, Prussian Blue) for 92 h and survived. Before the
start of the therapy, the blood of the mother and of the fetus
contained 0.07 and 0.01 mg/litre and the urine 0.4 and 0.1 mg/litre,
respectively. One day after the end of the treatment, the bones,
liver and kidney of the fetus contained 0.9, 0.8 and 0.3 mg
thallium/litre, respectively, and the blood of the mother
0.08 mg/litre (Graben et al., 1980). Additional evidence for the
transplacental transfer of thallium is provided by studies
demonstrating effects in infants exposed in utero (section 8.5.1).
6.3 Metabolic transformation
Data on the transformation and the equilibrium between the two
oxidation states of thallium ions(I and III) in body fluids and
tissues of mammals are not available. The two ions show a similar
intracellular distribution (Sabbioni et al., 1980b).
6.4 Elimination and excretion
6.4.1 Animals
In a study on the accumulation and excretion of thallium in
mussels and clams (section 6.1.1.1), the bivalves needed 7 and 30
days, respectively, to excrete all absorbed thallium. This is rapid
in comparison to other heavy metals, such as cadmium, copper, lead and
mercury, so that no significant amounts of thallium should enter the
food web in this way (Zitko & Carson, 1975).
Within 25 days after parenteral administration of 10 mg thallium
sulfate/kg body weight, rats eliminated 26% in the urine and 51% in
the faeces (Lund, 1956a). Elimination via urine started within hours
after oral application and persisted for up to 3 months. Faeces were
not found to contain thallium until the fourth day, and thallium was
still present after 1 month (Oehme, 1978). After injections of low
doses of 204thallium nitrate by different routes, the ratio of
faecal to urinary elimination of rats increased with time from 2 to 5
(Lie et al., 1960). Gregus & Klaassen (1986) reported that faecal
elimination is always greater than renal elimination. Biliary
elimination is of minor importance (Schäfer & Forth, 1980; Gregus &
Klaassen, 1986). Within 4 h after an intravenous injection, less than
0.3% of the injected thallium was eliminated in the bile of rats, but
up to 8% into the gut (Sabbioni et al., 1984a). An even lesser degree
of elimination occurred in tears, sweat, and milk (Oehme, 1978).
In contrast to absorption, secretion of thallium into the gut of
rats (given as 201thallium sulfate intravenously or directly into
the individual ligated gastrointestinal segments) is highest in
segments of the jejunum, followed by the ileum, colon and stomach
(Sabbioni et al., 1984a). Similar results were obtained after
intravenous administration to rats: in situ, the jejunum showed the
highest excretory activity, followed by the ascending colon (Henning &
Forth, 1982). The ileum and descending colon each excreted about half
of the amount of the jejunum; excretion into the stomach was
negligible. An increased dose (4 to 400 µg of thallium(I) sulfate/kg
body weight) caused increased excretion into the jejunum and
descending colon (Henning & Forth, 1982). Since thallium is also
absorbed in the colon (section 6.1), only a proportion of the secreted
thallium appears finally in the faeces.
Thallium ions are secreted against an electrochemical or
concentration gradient by an active transport mechanism, as shown in
experiments on the isolated mucosa of the descending rat colon
(Schäfer et al., 1981; Schäfer & Forth, 1987). Thallium(I) ions use,
at least in part, the same transport systems as potassium (Henning &
Forth, 1977; Schäfer & Forth, 1987), and thallium secretion is reduced
when the concentration of potassium is increased on the serosal side
(Henning et al., 1982).
6.4.2 Humans
The normal daily total elimination in humans is estimated to be
in the range of 1.64 µg thallium (urine: 1.2 µg; hair: 0.32 µg;
faeces: 0.06 µg; skin and sweat: 0.06 µg) (US EPA, 1980). About 50%
of total urinary elimination occurs within 9 to 11 days (Weinig &
Schmidt, 1966).
In lethal cases of human poisoning, postmortem examination has
always demonstrated high concentrations of thallium, especially in the
contents of the colon (Table 21). Thallium levels in human saliva are
up to 15 times higher than in the urine during the initial 2 weeks
(Richelmi et al., 1980). Minor amounts are eliminated via hair and
nails, both of which show the highest thallium concentrations of any
tissue among human populations in uncontaminated areas (Table 19).
Usually mother's milk is not an important route of elimination for
heavy metals (Hapke, 1988). However, 2 weeks after a suicide attempt
using thallium sulfate following the birth of her child (about 500 mg
thallium), the milk of a mother contained 0.25 mg thallium/litre,
while her blood only contained 0.07 mg/litre (Graben et al., 1980).
6.4.3 Methods to estimate daily intake of thallium
There are two ways to estimate daily intake of thallium, one
based on total daily excretion and the other on the total amount of
thallium in the body. In the former case, the total amount of
thallium excreted daily under steady-state conditions (a model which
may be reasonably applied to long-term exposure to low doses of
thallium) should reflect accurately the daily intake of thallium.
Using a mean urinary concentration of 0.4 µg/litre (which has been
frequently reported in unexposed populations), a daily urinary
excretion of 0.6 µg may be calculated assuming a urinary volume of
1.5 litre per day. Since we have assumed that renal excretion may
account for about 70% of the total daily excretion of thallium,
another 0.3 µg/day would be excreted by other routes, giving a value
for total thallium daily intake of about 0.9 µg. A similar procedure
leads to an estimated thallium daily intake of about 11 µg in
chronically exposed populations (using a mean urinary concentration of
5 µg/litre).
The other method for estimating daily intake assumes that the
following relationship exists between the total amount of thallium in
the body (Ab), the daily intake of thallium (Ad) and the
elimination rate constant (K):
Ad = KAb
Since the total amount of thallium in the body has been estimated
to be 100 µg per 75 kg body weight in an unexposed population (Weinig
& Zink, 1967), a daily intake of 2.3 µg may be calculated, assuming an
elimination rate constant of 0.023 day-1.
6.5 Retention and turnover (biological half-life)
6.5.1 Animals
The biological half-life of thallium in experimental animals is 3
to 8 days. Accordingly, the elimination of 70 to 90% of the
administered dose takes about 4 weeks (Oehme, 1978).
Using different routes of administration of thallium-204 in rats,
Lie et al. (1960) found a biological half-life of 3.3 days during the
first 21 days or until about 1% of the administered dose remained in
the body. This body clearance was not affected by the route of
administration and did not differ between various organs, except for
the hair, which contained up to 60% of the body burden after 21 days.
A biological half-life of 24 h was determined in pregnant mice and
rats (Gibson et al., 1967).
Durbin et al. (1957) determined a biological half-life in rats of
5.2 days and calculated half-lives of 7 and 6 days for removal from
the kidney and muscle, respectively. The half-lives in various organs
(brain, spinal cord, sciatic nerve, kidney, liver and spleen) were
lower in young rats than in adult rats and varied in different organs
of young and adult rats. Ducket et al. (1983) found that half-life
values in young rats ranged from 1.2 days for the sciatic nerve to 5.1
days for the liver (average for all tissues: 2.6 days) and in adult
rats from 2.7 days for the brain to 6.0 days for the spleen (average:
3.8 days).
6.5.2 Humans
Several investigators have reported on the half-life of thallium
in plasma and whole blood of humans acutely poisoned. Hologgitas et
al. (1980) reported the half-life in the blood of one patient to be
1.9 days. Heath et al. (1983) reported a half-life in the blood of
one patient of 1.9 days and a range of 21-24 h for the half-life in
blood for three patients. Treatment for thallium toxicosis has been
found to decrease the half-life of thallium in blood. In a review by
de Groot & van Heijst (1988), the half-life in blood decreased as
follows:
Treatment Thallium half-life in blood
No treatment (n=2) 9.5; 15 days
Prussian Blue (PB) (n=5) 3.0 ± 0.7 days
PB + forced dialysis (FD) (n=7) 2.0 ± 0.3 days
PB + FD + haemoperfusion (n=3) 1.4 ± 0.3 days
In cases of poisoning, the half-life of thallium in blood is
found to increase somewhat with time. Starting measurements at 42
days after toxic ingestion of thallium, Chandler et al. (1990)
reported a blood half-life of 3.7 days in a patient treated with PB
and intravenous potassium. The Gauss-Newton optimization model was
used in this calculation. Wainwright et al. (1988) and Schwartz et
al. (1988) presented data showing similar half-lives for thallium in
urine and in serum, but no quantitative analysis was performed.
There has only been one study of the whole-body half-life of
thallium in normal (i.e. unpoisoned) humans. In an investigation into
the use of radiolabelled thallium for medical imaging, Atkins et al.
(1977) administered thallium-200 to three volunteers. Using a
whole-body counter, the biological half-life for thallium was found to
be 9.8 days (range = 7.4-12.4 days). This determination is of much
greater value than the determinations of plasma or whole blood
half-lives for evaluating total excretion of thallium from the body.
6.6 Kinetics at the cellular level
The cellular uptake of thallium has been investigated in various
systems. Due to the similarity in ionic radius of thallium(I) and
potassium, thallium can substitute for potassium in a variety of
potassium-dependent transport processes, as indicated by studies with
microorganisms and frog skin (Norris et al., 1976; Zeiske & van
Driessche, 1986). In rats and dogs, data indicate that "the mechanism
involved in the active transport of potassium cannot differentiate
between potassium and thallium" (Gehring & Hammond, 1967)
(section 7.11).
The cytosol contains most of the intracellular thallium. In
rats, autoradiography revealed the presence of thallium in the
cytoplasm of nervous tissues during the first few days after injection
(Ducket et al., 1983), a phenomenon also evident in kidney, liver and
testis homogenates of rats treated with oral or intraperitoneal doses
of 0.00004, 2, 20, 2000 or 3150 µg thallium(I) per rat (Sabbioni et
al., 1980a,b) and in mussels (Nolan et al., 1984).
In a postmortem examination of a fatal case of thallium
poisoning, 87% of the thallium was present in the cytosol (Davis et
al., 1981) (for data on plants see section 4.1.2.3).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
The reported toxic effects of thallium are not always comparable,
since no standardized procedure is used by the different authors and
the duration of the experiments is not always stated. However, the
effective dose (ED) of thallium at which minimal adverse effects
(LDmin) occur, or at which 50% or all organisms are killed (LD50 and
LD100, respectively), are clearly correlated with the duration of
the experiment. This correlation also exists for the period of time
at which, for example, 50% of the organisms are killed.
The sections on single exposure (section 7.1.2), short-term
exposure (section 7.2.2) and long-term exposure (section 7.3.2) cover
the effects on various organs, except those on skin and eye (section
7.4) and the nervous system (section 7.8).
7.1 Single exposure
7.1.1 Toxicity and symptoms
The acute toxicity data for thallium compounds are listed in
Table 23. They vary considerably with observation time, e.g., a
7-fold higher LD50 value (2000 mg/kg) for mice is obtained if the
period of observation is reduced from 24 h to 1 h (Achenbach et al.,
1980).
There exist only insignificant differences in the toxicity of
various water-soluble thallium(I) salts to mice, rats, rabbits and
dogs. In general, for most laboratory species and an observation
period of 1 week, the LD50 or minimum effective dose (MED) values
range between 20 and 60 mg/kg body weight for thallium(I) salts,
independent of the application route, with the exception of
guinea-pigs (5 to 15 mg/kg). The toxicity of water-soluble thallium
compounds is similar for oral and parenteral routes of administration,
indicating a high degree of gastrointestinal absorption (Table 23).
The toxicity of water-insoluble thallium(III) oxide in rats and
rabbits is 2 to 4.5 times higher following oral administration than
following parenteral administration (Downs et al., 1960).
Acute toxicity is characterized by severe symptoms and/or death,
which may be caused by single exposure or by multiple lower doses
administered within 24 h. These symptoms are associated with
disorders of the digestive (vomiting, diarrhoea) and nervous system,
inflammation at body orifices, skin furuncles, tremor, loss of hair, a
necrotizing renal papillitis, and death by respiratory failure (BGA,
1979; Hapke, 1984; Kazantzis, 1986; Bruère et al., 1990).
Table 23. Toxicity of different thallium compounds in experimental animals after single exposure
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Mouse (I) carbonate subcutaneous LDmin 14 day 18 15.7 Sanotskii (1961)
Mouse (I) chloride intraperitoneal LD50 -c 24 20.5 Luckey & Venugopal (1977)
Mouse (I) sulfate oral LD50 1 h 2470 2000 Achenbach et al. (1980)
Mouse (I) sulfate oral LD50 2 h 1358 1100
Mouse (I) sulfate oral LD50 4 h 988 800
Mouse (I) sulfate oral LD50 12 h 432 350
Mouse (I) sulfate oral LD50 24 h 370 300
Mouse (I) sulfate oral LD50 36 h 235 190
Mouse (I) sulfate oral LD50 - 30 24.3 IPS (1982)
Mouse (I) sulfate intraperitoneal LD50 10 days 47.5 38.5d Stavinoha et al. (1959)
Mouse (I) sulfate intraperitoneal LD10 10 days 37 30
Rat (I) acetate oral LDmin 14 days 37.4 29 Downs et al. (1960)
Rat (I) acetate oral LD50 7 days 41.2 32
Rat (I) acetate intraperitoneal LDmin 14 days 25.8 20
Rat (I) acetate intraperitoneal LD50 7 days 29.6 23
Rat (I) nitrate intravenous LD50 - 16.3 12.5e Gehring & Hammond (1967)
Rat (I) nitrate intravenous LD50 - 18.9 14.5e Gehring & Hammond (1967)
Table 23 (contd).
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Rat (III) oxide oral LDmin 14 days 22.3 20 Downs et al. (1960)
Rat (III) oxide oral LD50 7 days 43.6 39
Rat (III) oxide intraperitoneal LDmin 14 days 103 92
Rat (III) oxide intraperitoneal LD50 7 days 80.5 72
Rat (I) sulfate oral LD50 - 10-25 8.1-20.2 IPS (1982)
Rat (I) sulfate dermal LD50 7 days 500 405 IPS (1982)
Rat (I) sulfate intraperitoneal LD100 2-6 days 33.3 27 Nachman & Hartley (1975)
Rat (R. (I) sulfate oral LD50 - 15 12.1 Wegler (1970)
norvegicus)
Rat (R. rattus) (I) sulfate oral LD50 - 76 61.5 Wegler (1970)
Hamster (I) malonate oral LD50 > 7 days 50 40 Aoyama et al. (1988)
Hamster (I) malonate oral LD100 5-7 days 62.5 50
Rabbit (I) acetate oral LDmin 14 days 24.5 19 Downs et al. (1960)
Rabbit (I) acetate intravenous LDmin 14 days 25.8 20
Rabbit (I) acetate intraperitoneal LDmin 14 days 16.8 13
Rabbit (III) oxide oral LDmin 14 days 33.5 30 Downs et al. (1960)
Rabbit (III) oxide intravenous LDmin 14 days 43.6 39
Rabbit (III) oxide intraperitoneal LDmin 14 days 67 60
Table 23 (contd).
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Guinea-pig (I) acetate subcutaneous LD100 5-9 days 19.3 15 Kaeser & Lambert (1962)
Guinea-pig (I) acetate oral LDmin 14 days 15.5 12 Downs et al. (1960)
Guinea-pig (I) acetate intraperitoneal LDmin 14 days 9.0 7
Guinea-pig (III) oxide oral LDmin 14 days 5.6 5 Downs et al. (1960)
Guinea-pig (III) oxide intraperitoneal LDmin 14 days 33.5 30
Dog (I) acetate oral LDmin 14 days 25.8 20 Downs et al. (1960)
Dog (III) oxide oral LDmin 14 days 33.5 30 Downs et al. (1960)
a LDmin = minimal dose at which lethality was observed; LD10 = dose at which 10% of the animals were killed; LD50 = dose at which 50%
of the animals were killed; LD100 = dose at which all animals were killed
b Selected data; detailed data summarized in Negherbon (1959), Christensen et al., (1973), Zitko (1975a, b),Smith & Carson (1977),
Venugopal & Luckey (1978), Schoer (1984), Nessler (1985a), Manzo & Sabbioni (1988), ATSDR (1992)
c No data given
d Graphical determination based on data of authors
e Rats fed a low or high potassium diet. An increase in dietary potassium had a small protective effect.
In rats oral administration of a toxic dose of thallium sulfate
(10 mg thallium/kg) caused reduced food intake, diarrhoea, lethargy
and ocular haemorrhage regardless of whether or not the animal
survived the first 72 h (Sabbioni et al., 1982).
Summarizing 34 cases of canine thallotoxicosis, Zook & Gilmore
(1967) emphasized the variability of the sequence and the severity of
symptoms, partly due to the intoxication stage. Frequent symptoms
were vomiting (82%), cutaneous alterations (71%), depression (62%),
anorexia (53%), nervous disorders (47%), diarrhoea (44%), respiratory
difficulty (44%), conjunctivitis (41%), dehydration (24%) and
oesophageal paralysis (6%). The sequence in which the symptoms of
intoxication occurred was generally as follows: first anorexia,
vomiting and depression, then skin changes, dyspnoea and nervous
disorders. Usually, rectal temperature was not elevated but was later
often subnormal. After 3 to 7 days of illness, erythematous lesions
occurred, which were most severe near mucocutaneous junctions and on
the foot pads. The haematological findings were leukocytosis
(neutrophilia, lymphopenia, eosinopenia) and haemoconcentration.
Proteinuria and bilirubinuria were commonly observed. Autopsy and
histo pathological examination of 15 and 12 dogs, respectively,
revealed increased heart weight (presumably caused by systemic
hypertension), myocardial necrosis, congestion of the kidney with
tubular nephrosis, pulmonary oedema, enlarged spleen, enlarged or
oedematous lymph nodes, dilatation and areas of erosion of the
oesophagus and necrosis of skeletal muscles. Some myelinated nerves
showed focal distensions of their myelin sheaths, and lesions of the
cerebrum and cerebellum were evident in dogs with neurological
disorders (Zook & Gilmore, 1967). In addition, alopecia, anorexia,
emesis and tenesmus (Coyle, 1980) and haematemesis (Waters et al.,
1992) have been reported in cases of acute poisonings in dogs.
In cats similar signs of thallium poisoning have been found,
e.g., skin alterations, apathy, lack of appetite, vomiting and signs
of peripheral and central neuropathy (Zook et al., 1968; Fitzek &
Henning, 1976). Haematological and histopathological findings were
similar to those obtained with dogs. In older cats that died early,
haemorrhagic gastroenteritis and hepatic or renal damage were evident
(Zook et al., 1968).
Implantation of a pellet of pure thallium (3 to 5 mm diameter)
into the motor cortex of a monkey, Macaca mulatta, resulted in death
within 6 days (Chusid & Kopeloff, 1962).
7.1.2 Effects on various organs
Effects on the various organs have been summarized by Sabbioni &
Manzo (1980) and ATSDR (1992). In nearly all affected organs direct
cytotoxic effects as well as indirect effects due to damage of the
nervous system have been found.
In acutely poisoned rats (single subcutaneous injection of 20 to
50 mg thallium acetate/kg), there was mild to moderate enteritis,
including oedema of the submucosa and muscularis layers, and moderate
to severe colitis (Herman & Bensch, 1967). Ultra structural
degenerative changes in the liver were frequently present, especially
in the mitochondria. These were also indicated by increased numbers
of autophagic lysosomes and lipid droplets (Herman & Bensch, 1967) and
were evident 16 h after intraperitoneal injection of 50, 100 or 200 mg
thallium(III) chloride/kg into rats (Woods & Fowler, 1986). It was
concluded that "thallium-induced alteration of hepatic functional
processes may arise from physical disruption of the membranal
integrity of subcellular organelles with which those processes are
functionally associated" (Woods & Fowler, 1986). In an extreme case
of a poisoned dog, all the parenchymal hepatocytes were necrotic and
the adrenals, thyroid, pituitary and pancreas had degenerated (Larson
et al., 1939).
The kidney is considerably affected in poisoned animals (section
9.3.3). Since the concentration of thallium in the kidney is higher
than in other organs, it would seem to be a specific target organ.
Light microscopy of rat and mouse kidney tissue up to 48 h after
administration of lethal doses of thallium (30 to 40 mg thallium
sulfate/kg body weight) showed stromal oedema, necrosis of the loops
of Henle and hydropic degeneration, as well as swelling and focal
necrosis of the epithelium of proximal convoluted tubules (Danilewicz
et al., 1979, 1980). Electron microscopy revealed degenerative
changes in the epithelial cells of the glomeruli and tubules. The
same changes in the kidneys of acutely poisoned rats have been
described by Herman & Bensch (1967) (section 7.1.2).
Oral and intravenous administration of thallium sulfate (20 to
40 mg/kg) to the dog, cat, rabbit, goat and pigeon also caused direct
effects on the respiratory apparatus, in addition to decreasing
vasomotor reactivity (Rossi et al., 1981).
In in vitro studies, addition of 30 to 50 µg thallium sulfate
reduced the beat frequency in isolated frog hearts (Buschke &
Jacobsohn, 1922). Over a range of thallium concentrations, from those
encountered after uptake from a contaminated environment (2 µg/litre)
to those seen after suicide or homicide (and also higher levels of up
to 204 mg/litre), the contractility of sheep interventricular cordis
muscles exhibited three types of response, but they were not
correlated to the thallium concentrations or period of incubation
(Ziskoven et al., 1982). Using guinea-pig papillary muscles and low
concentrations (0.02 to 2 mg thallium/litre), positive inotropic
transients were followed by an inotropic decay (Ziskoven et al.,
1982). In contrast, thallium produced concentration- and
time-dependent positive inotropic effects in guinea-pig atrial
preparations, but also inhibition of the sodium pump in ventricular
slices (Ku et al., 1978). However, there is no discrepancy between
these two effects, assuming that thallium inactivates the already
fully activated pump and stimulates the inactivated pump (Ziskoven et
al., 1982). The authors suggested that at low concentrations the
effects of thallium are not associated with changes of membrane
activity but with energy supply. Parameters of the slow inward
current at the membrane level were not specifically altered by
thallium (Wiemer et al., 1982). Another investigation involving
guinea-pig papillary muscles and sheep Purkinje fibres indicated that
the arrhythmogenic effects of thallium are restricted to the sinus
node (Achenbach et al., 1982). High concentrations (200 mg/litre)
depolarize the muscle fibre membrane and lead to irreversible damage
(Mullins & Moore, 1960). Within the muscle, thallium seems to compete
for the adsorption sites normally occupied by potassium, being
adsorbed on to myosin and thus being localized primarily in the A band
(Ling, 1977).
7.2 Short-term exposure
7.2.1 Toxicity and symptoms
In mice the daily supplementation of food with 400 µg thallium
acetate induced alopecia after about 14 days, followed by increasing
apathy and death within 16 to 18 days after the beginning of the
treatment (Buschke, 1900).
Daily intraperitoneal injection of thallium(I) acetate (5 mg/kg)
for 7 days in rats caused anorexia, reduced growth, irritability and
tenderness during handling, lethargy, diarrhoea, dragging of the hind
limbs, fits of abnormal rotation of head and neck and curving of the
body. About 15% of the rats died (Hasan et al., 1977c).
7.2.2 Effects on various organs
In subacutely poisoned rats (2 to 3 injections of 10 to 15 mg
thallium acetate/kg at intervals of 1 week), only slight colitis and
enlargements of mitochondrial granules in the liver occurred (Herman &
Bensch, 1967). Electron microscopy of the kidney revealed similar
changes to those observed in acutely poisoned animals (section 7.1).
7.3 Long-term exposure: chronic toxicity
7.3.1 Toxicity and symptoms
The chronic toxicity data on thallium compounds are listed in
Table 24.
Table 24. Toxicity of different thallium compounds in experimental animals after several administrations
Species Thallium compound Route of Toxicitya Period of Dose Reference
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Mouse (I) chloride intraperitoneal LD50 30 days 1.2 0.1 Bienvenu et al. (1963)b
Mouse (III) chloride intraperitoneal LD10 30 days 6.0 4.0 Hart & Adamson (1971)b;
LD50 30 days 6.9 4.5 Adamson et al. (1975)
Mouse (I) nitrate intraperitoneal LD50 14 days 37.5 28.6 Williams et al. (1982)b
Rat (III) chloride intraperitoneal LD10 30 days 4.85 3.2 Hart & Adamson (1971)b;
LD50 30 days 5.66 3.7 Adamson et al. (1975)
Rat (I) sulfate oral LD20 15 days 1.25 1.0 Tikhova (1964)
LDmin 15 days 0.6
Rabbit (I) carbonate oral LDmin 180 days 0.25 0.2 Tikhova (1967)c
Rabbit (I) carbonate subcutaneous LDmin 180 days 0.25 0.2
Rabbit (I) sulfate oral LDmin 180 days 0.25 0.2 Tikhova (1967)c
Rabbit (I) sulfate subcutaneous LDmin 180 days 0.25 0.2
a LDmin = minimal dose at which lethality was observed; LD10 = dose at which 10% of the animals were killed; LD50 = dose at which 50%
of the animals were killed; LD100 = dose at which all animals were killed
b Daily injection for 10 consecutive days
c Value in winter was lower than in summer
In rabbits daily administration of thallium(I) sulfate or
carbonate (0.25 mg/kg) for 6 months caused disturbed behaviour,
aggressiveness, diarrhoea, and loss of hair (Tikhova, 1967). In rats
daily ingestion of 7.5 mg thallium was lethal (Bowen, 1966).
In a study of weanling albino rats (50-80 g) fed ad libitum for
1 month on a diet containing 2, 10, 50, 100, 500 or 5000 mg
thallium(I) acetate per kg diet (5 rats/group), dietary levels of 2
and 10 mg/kg diet caused no effects on growth and survival within the
feeding period, whereas the other concentrations resulted in
mortalities of 60 to 100% within 10 days. When rats were fed for 15
weeks with 5, 15, 30 or 50 mg thallium acetate/kg diet (5 males and 5
females/group), the two lowest doses did not affect growth of males or
females. The 15 and 30 mg/kg doses caused hair loss starting after 2
weeks, and after 15 weeks the rats were almost free of hair. Within
the fourth and eighth week and after intakes of 30 mg/kg diet, 80% of
males and 60% of females died. At 50 mg/kg all males died within 2
weeks and all females within 8 weeks. No specific pathological
alterations were found in any organ. A dose of 30 mg/kg diet resulted
in moderate growth depression in males but not in females, while in
both sexes increased mortality was observed. For thallium(III) oxide
the effective concentrations were similar, and males also reacted more
sensitively than females. No specific pathological alterations were
found in any organ except the skin, where atrophy of hair follicles
and sebaceous glands were seen at both higher dose levels. The exact
concentration of thallium ingested by the rats could not be determined
but was estimated to be in the range of 1 to 3 mg thallium acetate/kg
body weight per day for the diet containing 15 mg thallium acetate/kg
food (Downs et al., 1960).
US EPA (1986) conducted a 90-day study in which male and female
Sprague-Dawley rats (20 of each sex per group) were administered
aqueous thallium sulfate by gavage at doses of 0.01, 0.05 or
0.25 mg/kg body weight per day. Both untreated controls and vehicle
(water) treated controls were included. Clinical observations were
recorded daily and neurotoxicological examinations were performed 3
times per week on selected animals. Haematological and clinical
chemistry parameters were measured on days 0, 30 and 90, and
ophthalmological examinations were performed on days 0 and 90. Upon
necropsy, selected organs were weighed. No significant differences
were seen in any group for body or organ weights. Several changes
were reported for blood chemistry parameters. Statistics were
reported for male and female rats at 30 and 90 days for each dose
group compared with both untreated controls and vehicle-treated
controls. In both males and females, small increases were seen for
serum glutamic-oxaloacetic transaminase, lactic acid dehydrogenase and
sodium levels, with statistical significance at many points. At the
lowest dose, statistically significant changes were seen in male rats
for all three of these parameters, but only when compared with
untreated controls. Higher doses resulted in statistical significance
when compared with vehicle controls. Similar patterns were seen in
female rats.
In addition, there was a dose-related increase in the incidence
of alopecia, lacrimation and exophthalmos. No treatment-related
changes were seen in the eye. Gross necropsy revealed only alopecia;
this occurred in a dose-related manner for females and was apparent at
the lowest dose level. In males, alopecia was also apparent in all
dose groups but was not strictly dose-related.
In a study with female Sprague-Dawley rats (180-200 g), thallium
sulfate was given via the drinking-water (10 mg thallium/litre) over a
period of 40 weeks and with a total intake of about 80 mg thallium/rat
(Manzo et al., 1983). First symptoms, i.e. poor hair lustre,
periorbital redness and irritability, were observed on days 20 to 25.
Hair loss first appeared after 32 days in some rats, and in several
rats was almost complete by the end of the 36-week period of
treatment. However, in other rats no hair loss occurred. Starting
from day 40 the number of rats showing mild or severe cutaneous
disorders increased strongly. After 40 days and a total ingestion of
about 10 mg thallium/rat, lethality amounted to 15%, and the surviving
rats showed no electrophysio logical abnormalities. After 240 to 280
days and the ingestion of about 70 to 80 mg/rat only an additional 6%
of the rats died; about two-thirds of the rats showed
electrophysiological effects and reduced motor and sensory action
potentials.
7.3.2 Effects on various organs
In chronically poisoned rats (initial injection of 10 to
20 mg/kg, followed by weekly subcutaneous injections of 5 mg/kg for 4
to 26 weeks), only slight colitis sometimes occurred (Herman & Bensch,
1967), but degeneration in the liver was similar to the severe effects
in acutely poisoned rats (section 7.1.2) and liver enzymes were also
affected (Bulavintseva & Bulavintsev, 1982). In the stomach of rats
the production of hydrochloric acid was reduced (Buschke, 1929).
Kidney weight was increased (Downs et al., 1960) and ultrastructure
affected in the same way as in acutely poisoned animals (Herman &
Bensch, 1967) (section 7.1.1). The greater accumulation of thallium
in the renal medulla than in the renal cortex of chronically poisoned
rats indicates a firmer binding of thallium, which might impede
thallium elimination (André et al., 1960; Lameijer & van Zwieten,
1977a).
Intratracheal administration of 0.5 or 5 mg thallium(I) salts
(iodide, bromide and chloride or mixtures of them) caused dose- and
time-dependent changes in the lungs of rats, iodide being the most
toxic salt (Spiridonova et al., 1978).
In chronically poisoned guinea-pigs, the adrenaline and lipoid
contents of the adrenal glands were considerably, sometimes totally,
reduced. Examination of chronically poisoned rats revealed a reduction
in the size of the thyroid gland, follicular atrophy and some pycnotic
nuclei (Buschke & Peiser, 1922a,b; Buschke, 1929).
7.4 Skin and eye irritation
7.4.1 Skin and hair
Thallium intoxications in dogs caused striking effects in all
layers of the skin. The dermal changes were characterized by oedema
and disruption of collagen bundles. In erythematous patches massive
parakeratosis (much more pronounced than in other dermatoses) and
occasionally a granular layer were found. In the follicles, of which
only 60% contained hairs, the external root sheath was hyperplastic,
showing an excess of parakeratotic horny material. Follicular
plugging was prominent (Schwartzman & Kirschbaum, 1962). The progress
of the erythematous lesions towards scaling and crusting included
varying degrees of necrolysis and, characteristically for thallium
intoxications, spongiform abscesses, the latter occurring also in the
hair follicles (Schwartzman & Kirschbaum, 1962) where thallium binds
strongly to melanin (Tjälve et al., 1982).
The mechanism of depilation is unclear. According to Truhaut
(1960), this major sign of thallium intoxication, alopecia, is caused
by its antimitotic activity, since hair follicles (and testes) are
normally characterized by marked mitotic activity. This adverse
effect is prevented by glutathione and cysteine. Counting the mitotic
rate per hair follicle after a subcutaneous injection of 30 mg
thallium(I) sulfate into young rats at 4 and 7 days of age, Cavanagh &
Gregson (1978) observed an initial decline in mitotic rate followed by
cell deaths in the matrix zone.
On the basis of results from early experiments on mice, rats,
guinea-pigs, cats and rabbits performed by Buschke (1900, 1903, 1911),
Buschke & Peiser (1922b,c, 1926) and Buschke et al. (1928), the
authors put forward the hypothesis that the depilatory effect is
mediated by the activation of sympathetic nerves, since light
microscopy never revealed a direct effect on the hair follicles and
since sensory hairs, innervated by cerebrospinal nerves, were never
affected, in contrast to the smooth muscles of other hairs which are
innervated by the sympathetic system.
In chronically poisoned rats (initial injection of 10 to 20 mg
thallium acetate/kg followed by weekly subcutaneous injections of
5 mg/kg for 4 to 26 weeks) (Herman & Bensch, 1967), black speckling of
periorbital hairs occurred occasionally. In other chronically
poisoned rats (section 7.3) showing complete depilation, light
microscopy revealed effects similar to those after acute intoxication,
i.e. a keratinized epidermis, decrease in the size of sebaceous glands
and reduction of the number of hair shafts and follicles, the latter
becoming atrophic or replaced by scars, collagen or fat (Downs et al.,
1960).
7.4.2 Eye
Autoradiography of adult mice demonstrated a relative
accumulation of thallium in the lens of the eye (André et al., 1960).
A biochemical investigation showed that in addition to binding to
melanin in the hair follicles, thallium also binds strongly to melanin
in the mouse eye. This might result in iritis and retrobulbar
neuritis, which are regularly observed symptoms of thallium
intoxication (Tjälve et al., 1982).
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
Thallium affects reproduction in various ways. Buschke & Peiser
(1922a) found a total reduction of sexual activity in intoxicated rats
and mice, which did not develop after administration to young animals
(Buschke & Peiser, 1922b,c) (section 7.3). This effect could be
antagonized by the administration of an ovarial hormone and
hypophyseal tissue (Buschke, 1929). Smith & Carson (1977) also
emphasized that sexual activity is usually lessened in chronically
poisoned animals.
7.5.1 Gonadotoxic effects
Several early, contradictory reports dealt with the question of
whether or not thallium affects ovarian function or the estrous cycle
(Smith & Carson, 1977). Buschke et al. (1927a) and Buschke & Berman
(1927) described strong inhibition of the estrous cycle in mice.
More attention has been directed to the effects on the male
reproductive system. Cytotoxic and perhaps mutagenic effects (section
7.6) can affect the offspring.
In several laboratory species, acute or repeated treatment with
thallium(I) salts resulted in similar or even higher concentrations of
thallium being found in the testes compared to other organs (section
6.2), pointing perhaps to a special susceptibility (Gehring & Hammond,
1967; Krassowski et al., 1977, 1980; Sabbioni et al., 1980a,b; Talas &
Wellhöner, 1983; Aoyama, 1989). However, an effect on offspring has
not been investigated in detail. Landauer (1930) demonstrated effects
on chickens, but only two cocks were included. A second
investigation, a dominant lethal test using rats, is discussed in
section 7.6.
Several authors have reported histological findings. In acutely
poisoned rats the epithelial cells of the seminal vesicle contained
numerous autophagic vacuoles (Herman & Bensch, 1967), and in acutely
intoxicated rabbits and dogs spermatogenesis was inhibited (Larson et
al., 1939; Truhaut, 1960; Zook & Gilmore, 1967). Chronic poisoning
caused total atrophy of testes in some rats (Buschke & Peiser,
1922a,b,c) or considerably disturbed spermatogenesis, which, however,
returned to normal after termination of thallium administration
(Buschke, 1929). A 6-month administration of thallium carbonate in
the drinking-water of mice (0.001 mg/litre or 0.01 mg/litre) caused a
decrease in sperm fertility at the high dose level and reduced sperm
motility at the lower dose level (Wei, 1987). Increased desquamation
of spermatogenic epithelium in rats was observed after oral
administration of 0.001 mg thallium carbonate/kg (Shabalina et al.,
1980). In rats treated with 10 mg thallium/litre in their
drinking-water for 2 months, but not in those treated for only for 1
month, there was rearrangement of the germinative epithelium,
premature release of germinal cells into the seminiferous tubules, low
epididymal sperm motility, the appearance of immature elements in the
semen and a high susceptibility in the Sertoli cells (Formigli et al.,
1985, 1986; Grégotti et al., 1985), the latter also reacting very
sensitively in in vitro studies (Grégotti et al., 1993). However,
thallium treatment did not affect the absolute or relative weight of
the testes or the plasma levels of testosterone. The mechanisms
underlying the findings for thallium may involve its antimitotic
effect or its effect on energy metabolism (Formigli et al., 1985,
1986). The use of thallium-201 (1-10 µCi), which is also utilized for
diagnosis in medicine, for testicular imaging in mice caused loss of
testicular weight and reduction in the number of sperm heads. These
effects were less severe when thallium-204 (1-10 µCi) was used. This
must have been due to the different radioisotopes (probably the
low-energy Auger electrons of thallium-201), rather than to the
physicochemical properties of thallium that determine uptake (Rao et
al., 1983).
7.5.2 Embryotoxicity and teratogenicity
7.5.2.1 Chickens
Injections of thallium (1 or 2 mg per egg) into the yolk sac of
4-day-old chick embryos strongly affected growth and survival.
Injections of thallium sulfate (0.7 mg/egg) into the chorioallantoic
membrane caused similar effects (Karnofsky et al., 1950; Ridgway &
Karnofsky, 1952).
More striking were the effects on the development of bones of the
embryonic chick: achondroplasia, leg-bone curvature, parrot beak
deformity and microcephaly (Karnofsky et al., 1950; Karnofsky & Lacon,
1964). The achondroplasia did not occur after injections of 0.2 mg
thallium sulfate/egg into the yolk sac, but with the larger doses
increasing percentages of the embryos showed shorter bones than
normal, and the tibia and femora were strikingly curved. Thallium
sulfate and nitrate showed a similar ability to produce achondroplasia
(Karnofsky et al., 1950).
Achondroplasia was only produced if thallium treatment was
performed during a critical, sensitive period (Hall, 1977, 1985).
This period began on day 5 of incubation and ended after 205 to 207 h
of incubation, coinciding with a 66% decrease in growth rate of the
embryos. During this period thallium bound rapidly to skeletal
tissues. Light and electron microscopy showed the primary site of
action to be the cartilage, maturing hypertrophic chondrocytes being
most affected (Ford et al., 1968; Hall, 1972; Skrovina et al., 1973).
An increase of glucosamine and a decrease of the mucopolysaccharides
without a simultaneous loss of collagen indicated an interference with
cartilage metabolism (Ford et al., 1968). According to Hall (1972),
thallium treatment caused an "abnormal distribution of the acid
mucopolysaccharides in otherwise normal cartilage matrix and the
formation of necrotic areas within the maturing hypertrophic
chondrocytes". The reduced secretion of the acid mucopolysaccharide
into the intercellular matrix did not affect ossification.
7.5.2.2 Mammals
In mammals, the results are inconsistent and there seems to be
great variation between species and strains (Claussen et al., 1981).
Intraperitoneal injection in rats on days 8 to 10 (2.5 mg thallium
sulfate/kg) or on days 12 to 14 (2.5 or 10.0 mg/kg) of gestation had
no effect on resorption rates but significantly reduced the body
weight of fetuses examined on day 21 of gestation. Hydronephrosis and
non-ossification of vertebral bodies were observed. The effects on
fetal body weight and vertebrae were also induced by a low potassium
diet and were not increased by an additional dose of thallium (Gibson
& Becker, 1970). According to Barlow & Sullivan (1982), the increase
in hydronephrosis may not be related to thallium. Adverse effects on
the cartilage of long bones in 6- and 9-day-old rats treated
intraperitoneally with thallium sulfate were described by Nogami &
Terashima (1973). These results might explain the reduced growth of
the suckling rats of thallium-intoxicated dams (Ehrhardt, 1927).
Ossification of rat skulls was also reduced (Buschke et al., 1927b).
In a preliminary study on mice, oral administration of thallium
sulfate (0.3, 1 and 8 mg thallium/kg body weight) to 3-6 pregnant SWS
mice (gestation day 9) caused achondroplasia in 12.5, 14 and 50% of
the offspring, respectively (Achenbach et al., 1979a,b). Earlier
administration of thallium induced miscarriage, while older fetuses
were only slightly affected. In other substrains of the SWS mice,
administration of 10 to 20 mg/kg induced no teratological effects
(Claussen et al., 1981). Under similar conditions, but using 29 NMRI
mice and oral doses of 0.8 and 8 mg thallium/kg body weight, the
weights of fetuses and placentas were not affected by the treatment
with thallium sulfate, but a slight, statistically non-significant
increase in the intra-uterine mortality rate was found in embryos
examined 18 days after fertilization. A significantly increased rate
of double placentas and of effusions of blood in the thigh of the
fetuses was observed at 8 mg/kg (Török & Schmahl, 1982). Oral
administration of 3 or 6 mg thallium chloride or acetate per kg per
day to NMRI mice (about 30 per group; day 6 to 15 of gestation) had no
observable effects on skeleton or organs at day 18 of gestation
(Claussen et al., 1981). However, post-implantation losses were
increased after administration of 6 mg thallium chloride/kg, but only
young embryonic stages were affected. Administration of 6 mg thallium
acetate/kg caused a reduction in the weight of embryos. In a parallel
study with rats, 6 or 4.5 mg thallium chloride or acetate/kg was
lethal to the dams (gestation day 6 to 15), while 3 mg/kg induced no
embryotoxic effects but increased malformations of ribs and vertebrae
(Claussen et al., 1981).
Studies of mouse embryos cultured in vitro showed that low
levels of thallium (0.2 mg/litre) affected the pre-implantation stage
(summarized by Formigli et al., 1985).
In vitro cultivation of mouse limb buds (day 11 of gestation)
in medium containing 10 mg/litre for a period of 3 days, followed by
cultivation in normal medium for another 3 days, caused impaired
development of the hand skeleton (Barrach & Neubert, 1985). In a
similar study, the extent of this effect was also increased by
decreasing the potassium concentrations of the medium, and vice versa
(Neubert & Bluth, 1985). The authors suggested that an interference
of thallium with mammalian embryonic development is possible if its
concentration in embryonic tissues reaches levels above 2-3 mg/kg.
When rat embryos (10.5 days after fertilization) were cultured
for 48 h in media containing thallium sulfate (3, 10, 30 or 100 mg
thallium/litre), embryotoxic effects were evident at all concen
trations. Increasingly deleterious effects occurred on growth and on
mesoderm and entoderm at concentrations from 10 to 100 mg/litre.
Whereas macroscopically no or only minor abnormalities could be
detected, histological examination of the embryos revealed that cell
necroses in the brain developed even at the lowest concentration. At
higher concentrations these effects increased, and necrosis was
complete at 30 mg/litre. The authors doubted whether a suggested
embryotoxic concentration of 1 mg/litre can be achieved without severe
maternal poisoning (Anschütz et al., 1981).
7.5.2.3 Delayed effects on development of offspring
Many offspring of chronically poisoned rats showing total
alopecia died during the first two days after birth. In the other
pups, hair development was severely impaired (section 7.4), and these
pups died within 4 to 5 weeks after birth. In a second litter all
offspring showed alopecia, but none of them died so early (Buschke,
1911).
In studies by Claussen et al. (1981), oral administration of 6 mg
thallium chloride/kg to NMRI mice dams (day 6 to 15 of gestation)
reduced the initial increase in weight of the offspring and slightly
increased the mortality rate during the first 3 weeks. In a parallel
study with rats, oral administration of 3 mg thallium chloride or
acetate per kg caused similar effects.
In rats, exposed prenatally by treating the dams with 0.01 g
thallium sulfate/litre in their drinking-water, gestation and weight
of offspring were not affected, but hair development was retarded
during the first 60 days of life. At an age of 30 or 60 days, the
hypotensive and hypertensive cardiovascular responses to acetylcholine
or to isoprenaline and noradrenaline, respectively, were lower than
the responses of control animals (Matera et al., 1986; Rossi et al.,
1988). Comparisons with postnatally exposed pups showed that the
reactivity of the developing vascular autonomic nervous system was
also lowered (Rossi et al., 1988).
Oral doses of 0.1, 0.5 or 1.0 mg thallium/kg body weight
(thallium sulfate) given to rats on days 6, 7, 8 and 9 of gestation
impaired the learning ability of their adult offspring in operant
behaviour tests. Postnatal administration of thallium (same doses)
had no such effect (Bornhausen & Hagen, 1984).
7.6 Mutagenicity and related end-points
Only two microbiological investigations have been performed;
these indicated no mutagenic effects (Claussen et al., 1981). Both
used the Ames test. Dehnen, whose unpublished data were described by
Claussen et al. (1981), investigated the effects of thallium(I)
acetate and used 3.1 µg-29.2 mg thallium/plate and the Salmonella
typhimurium strains TA98, 1535, 1537 and 1538. Similarly, the use
of Ames tests with thallium chloride and acetate (doses not given) and
S. typhimurium strains TA98, 100, 1535, 1537 and 1538, with and
without metabolic activation, indicated no mutagenic effects (Claussen
et al., 1981). According to these authors, no sister-chromatid
exchange was found in bone marrow cells after oral administration of
thallium chloride to Chinese hamsters (5 or 10 mg thallium/kg body
weight; twice after 24 h; 8 animals per dose).
However, thallium(I) carbonate induced sister-chromatid exchange
and chromosomal aberrations in one cell line and hypoxanthine-guanine
phosphoribosyl-transferase gene mutation in another cell line (Zhang,
1988). In studies with embryonic fibroblasts of various mouse and rat
strains, the same thallium(I) compound (0.5 to 46.9 mg/litre) caused a
significant increase in the single-stranded DNA fraction after
incubation of rat fibroblasts and cells of one mouse strain (C57Bl/6),
whereas cells of the CBA mouse strain were resistant to the same
concentrations. In a test of survival and mutability of vaccinia
virus in both mouse cell lines, the CBA cells showed increased
survival of the virus, suggesting greater efficiency of the repair
systems (Zasukhina et al., 1980, 1983).
In a dominant lethal test, white rat males received daily oral
doses of 5, 50 and 500 ng thallium carbonate/kg body weight over 8
months. Thereafter they were mated with untreated females. On
gestation day 20, 18 dams were killed. At the two higher doses there
was a tendency towards an increase in embryonic mortality, whereas at
the lowest dose the number of resorptions and post-implantation deaths
were increased (Zasukhina et al., 1983). Using these data in a
T-test, the differences between the means for the total embryonic
death of treated and untreated mice were statistically significantly
different (p < 0.05).
A mutagenic effect on sperm cells of rats was reported after oral
administration of 0.0001 mg thallium carbonate/kg (Shabalina et al.,
1980). However, the report lacked detail concerning the experimental
set-up and the Task Group considered that it could not be used for
evaluation of the health effects of thallium.
7.7 Carcinogenicity
No standardized carcinogenicity studies have so far been
performed (ATSDR, 1992). Owing possibly to their cytotoxic effects,
thallium salts may have a local antineoplastic effect in mice and rats
(Hart et al., 1971; Hart & Adamson, 1971; Adamson et al., 1975).
7.8 Neurotoxicity
7.8.1 Central nervous system
7.8.1.1 Histology and ultrastructure
In the brains of subacutely poisoned rats, Herman & Bensch (1967)
(section 7.1.2) occasionally found foci of perivascular cuffing, while
the mesencephalon of two of the four rats contained an extensive
region of acute necrosis. In addition, numerous lipofuscin bodies
were sometimes present in neuron cytoplasm. This was also found to
occur following chronic poisoning; electron microscopy showed effects
on mitochondria (Herman & Bensch, 1967).
The brain of acutely poisoned guinea-pigs (subcutaneous injection
of 15 or 18 mg thallium sulfate/kg) showed slight microscopic
alterations, e.g., swelling of cells and vacuolization in the
perikaryon (Tackmann & Lehmann, 1971). In the right parietal cortex
of rats, microglia cells and alpha astrocytes were affected 24 h after
an intraperitoneal injection of 40 mg/kg (Reyners et al., 1981). At
24 h after intraperitoneal injection of 32 mg/kg into newborn rats,
the encephalon showed oedema and congestion; even after an additional
50 days, there was focal destruction of neurons and irregular fibrosis
of the capillary vessels (Barroso-Moguel et al., 1990).
Ataxia and tremors are known to be associated with cerebellar
lesions and both neurological disorders occur in cases of thallium
intoxication. Ultrastructural alterations of the cerebellum,
especially of the mitochondria, were evident in rats after poisoning
by daily intraperitoneal injections of thallium acetate (5 mg
thallium/kg for 7 days) (Hasan et al., 1978a). Effects in other brain
regions indicate that the effects of thallium on the activity of
endocrine glands may be mediated via changes in hypothalamic control
(Hasan et al., 1977b). Using identical conditions, Hasan et al.
(1977a) observed an apparent increase in the number of
oligodendrocytes and suggested a correlation with thallium-induced
neuronal chromatolysis described by other authors, since the usual
functions of oligodendrocytes are the formation of myelin and the
nutrition of neurons.
7.8.1.2 Electrophysiological and biochemical investigations
In addition to the direct effects of thallium on the cardio
vascular and respiratory apparatus of the dog, cat, rabbit, goat and
pigeon, indirect effects involving higher vasomotor and respiratory
centres (but mostly dependent on a decrease in vasomotor reactivity)
were found following acute poisoning (section 7.1.2) (Rossi et al.,
1981).
Extra- and intracellular recording of central neuronal activity
in hippocampal slice preparations from guinea-pigs and rats showed
that thallium (20 to 40 mg/litre) seems to have a predominantly
postsynaptic effect in hippocampal slice preparations, perhaps by
exerting an unspecific influence on the intracellular metabolic
mechanisms of the CA1 pyramidal cells (Lohmann et al., 1989). Daily
intraperitoneal injections of thallium(I) acetate (4 mg/kg for 7 days)
indicated that the electrophysiological parameters of noradrenergic
transmission in rat cerebellum were reduced (Marwaha et al., 1980).
The association of the corpus striatum with the pathogenesis of
the abnormal movements that have been reported after thallium
intoxication is shown by an increased firing rate of the caudate
neurons of rats, 3 to 4 h after intravenous injection of 10 mg
thallium sulfate/kg (Hasan et al., 1977c). After daily
intraperitoneal injections of 5 mg thallium(I) acetate/kg for 7 days
(section 7.8.1), the protein content of the corpus striatum was
significantly increased and the respective breakdown enzymes were
depleted. However, the latter did not occur in the cerebrum (Hasan et
al., 1977b,c).
Certain motor dysfunctions are known to be associated with a
decrease in the level of brain dopamine, an aspect supported by the
data of Hasan et al. (1978b). Convulsive disorders may also be
related to a brain deficiency of gamma-aminobutyric acid (GABA)-ergic
mechanisms. Data obtained by Hasan et al. (1977d), but not those by
Nisticò et al. (1984), support this interpretation. Neurotoxicity
could also result from changes in the concentrations of amino acids
and other neurotransmitters (Ali et al., 1990) or an acceleration of
monoamine catabolism (Osorio-Rico et al., 1994).
Further important mechanistic aspects were increases found in the
lipid peroxidation rates and in the activity of the lysosomal enzyme
ß-galactosidase, especially in the cerebellum, brainstem and cortex,
after daily intraperitoneal injection of 8 mg thallium(I) acetate/kg
for 6 days in rats (Brown et al., 1985). Also a lower dose of 4 mg/kg
selectively altered patterns of behaviour (section 7.8.1.3). An
increased deposition of lipofuscin-like pigment granules in the
cerebellar neurons and an increase in lipid peroxidation rates in the
cerebrum and brainstem of rats (which was even exceeded by that in the
cerebellum) were described in a previous study (Hasan & Ali, 1981).
This seems to be an important mechanism of toxicity (section 7.9).
7.8.1.3 Behavioural toxicology
Intraperitoneal injections of 10 or 20 mg thallium sulfate/kg
produced only a slight conditioned flavour aversion in rats, perhaps
due to the delayed onset of symptoms (Nachman & Hartley, 1975; Peele
et al., 1986). Oral administration induced a dose-dependent aversion
to saccharin at all but the lowest dose of 2.5 mg/kg (Peele at al.,
1986, 1987). This difference might be explained by irritation of the
gastrointestinal tract after oral uptake of thallium.
In a detailed investigation of the effects of daily
intraperitoneal injections of 4 or 8 mg thallium(I) acetate/kg for 6
days on several behavioural patterns of rats, changes of behaviour
were intensified with increased dose. Some of the changes correlated
with biochemical effects (section 7.8.1.2) (indicating cellular
damage) in certain regions of the brain (Brown et al., 1985).
In pest control campaigns against wild rodents, the dying animals
left their hiding places and came to the surface, presumably due to
extreme thirst and breathing disturbances (Larson et al., 1939).
7.8.2 Peripheral nervous system
In the final phase of lethal intoxication and in vitro, a
parasympathetic stimulation seems to occur. Because thallium
diminishes the effects of adrenaline on isolated hearts or intestine,
even after parasympathetic blockage, it is thought that it may destroy
adrenaline (Truhaut, 1960).
In the initial phase of intoxication, sympathetic nerves are
stimulated (Buschke & Peiser, 1922b). Investigating the effects of
0.2 to 204 mg thallium(I) and thallium(III) ions/litre on the ATPases
of the amine-storing granules from bovine adrenal medulla and splenic
nerves, a specific inhibition by thallium(III), but not by
thallium(I), was observed at concentrations which might occur in the
tissues after intoxication with thallium(I). The authors suggested
that thallium(I) is oxidized to thallium(III) in the organism.
Because the ATPase of the nerve granula which store noradrenaline was
nearly ten-fold more sensitive than that of the adrenal medulla, which
stores mainly adrenaline, this might explain the strongly increased
elimination of noradrenaline (Burger & Starke, 1969).
7.8.2.1 Histology and ultrastructure
In the peripheral and optic nerves of acutely, subacutely and
chronically poisoned rats (Herman & Bensch, 1967) (section 7.8.1), no
consistent or even slight changes were revealed by light or electron
microscopy. However, partial atrophy of the optic nerve was found by
Buschke et al. (1928). In addition, acute poisoning of a dog (Greving
& Gagel, 1928) and guinea-pigs (subcutaneous injection of 15 or 18 mg
thallium sulfate/kg) (Tackmann & Lehmann, 1971) caused alterations of
the axons and myelin sheaths which were evident under the light
microscope. In a 36-week study (Manzo et al., 1983) (section 7.3), in
which rats were given thallium sulfate in their drinking-water (10 mg
thallium/litre), about 50% of the animals developed Wallerian
degeneration (myelin debris and vacuolization); lamination of the
myelin sheath of the sciatic nerve fibres was confined to some large
and medium-sized fibres. Degenerative lesions found in the white
matter of the spinal cord of poisoned rabbits may account for the
paralysis of the hind legs (Truhaut, 1960).
In a number of in vitro studies, thallium affected nerves,
e.g., cell outgrowth was inhibited (Sharma & Obersteiner, 1981;
Windebank, 1986) or the myelin sheath disintegrated (Peterson &
Murray, 1965).
7.8.2.2 Electrophysiological and biochemical investigations
Following a subcutaneous injection of 15 mg thallium sulfate/kg
to guinea-pigs, within days the larger and faster conducting nerve
fibres degenerated before the slower fibres and became inexcitable
(Kaeser & Lambert, 1962; Tackmann & Lehmann, 1971). In a subchronic
study (section 7.3) on rats, which received thallium sulfate via their
drinking-water (10 mg thallium/litre), the motor and sensory action
potential amplitudes were unaffected after 40 days of poisoning but
decreased after 240 days (Manzo et al., 1983). Then the motor action
potential latency was increased, and no fibrillation activity was
observed in the tibialis anterior muscle.
Neuromuscular transmission in thallium-treated rats and mice has
been investigated in detail using phrenic nerve-diaphragm preparations
(Wiegand et al., 1983, 1984a, 1986). The relationship between
thallium concentration (z, in mM) and duration of paral ysis (y, in
minutes) is approximated by the equation y = 4.6 × e8.4z (Csicsaky &
Wiegand, 1981). It has been suggested that thallium interferes
presynaptically with spontaneous transmitter release by antagonizing
these calcium-dependent processes, rather than by interfering with the
presynaptic influx of calcium ions (Wiegand et al., 1983, 1984a,b;
Wiegand, 1988). Additional data indicate that thallium, like other
heavy metals (Cooper et al., 1984), irreversibly blocks phasic
transmitter release, while spontaneous transmitter release is
reversibly enhanced (Wiegand et al., 1986; Csicsaky et al., 1988).
The sequence of the toxic effects indicate that thallium needs to be
transported across the cell membrane before it can finally interfere
with the release mechanisms. This rather indirect mode of action of
thallium was also found in the recordings of presynaptic ion currents.
Perineural recording techniques and the blocking of potassium
channels excluded the possibility that presynaptic potassium or
calcium channels were influenced by thallium in acute superfusion
experiments. Thus, the mechanisms that cause enhancement of the
spontaneous release of acetylcholine and the reduction of phasic
transmitter release at the neuromuscular junction, both of which are
induced by thallium, remain unknown at present (Wiegand et al., 1990).
7.9 In vitro test systems: cell lines
Ultrastructural studies with cultured fetal mouse heart cells
showed swollen mitochondria with loss of cristae, disintegration of
the membrane system, and a protective effect of selenium (Liu, 1986)
(section 7.10.2).
A cytotoxic effect on ovary cells was observed in in vitro
experiments on Chinese hamster ovary cells. After a 16-h incubation
with thallium(I) nitrate (40 mg/litre), 50% of the cells did not form
colonies during the following 7 days (section 7.9) (Hsie et al., 1984;
Tan et al., 1984).
Using three mammalian cell lines (human diploid embryonic
fibroblasts, HeLa cells and mouse fibroblasts) to test 11 heavy
metals, thallium was found to belong to the group of metals with a
strong inhibitory (on proliferation) or lethal effect. After
treatment for 7 days, the minimal inhibitory concentrations of
thallium(I) and thallium(III) for all three cell lines were 4 mg/litre
and 2 mg/litre, respectively, while the 50% inhibitory concen
trations were 10-15 mg/litre and 5-15 mg/litre, respectively. Half of
the cells were killed by 20-40 mg/litre (Fischer, 1981).
7.10 Factors modifying toxicity
7.10.1 Enhancement of elimination
Various substances have been evaluated for their ability to
enhance faecal or renal elimination of thallium (e.g., Lund, 1956b).
In rats, potent diuretic agents such as furosemide and ethacrynic acid
increased renal elimination of thallium, but did not further increase
the elimination induced by feeding a potassium-rich diet (Lameijer &
van Zwieten, 1977a,b). A sodium-rich diet did not promote the renal
elimination of thallium (Lameijer & van Zwieten, 1979). In rats, oral
application of activated charcoal increased faecal elimination by
about 80% but did not affect urinary elimination (e.g., Lund, 1956b).
In contrast, potassium chloride and cystine only increased renal
elimination by 47% and 60%, respectively. Meyer & Tal (1957) reported
that some compounds containing sulfur and labile methyl groups seem to
reduce the toxicity of thallium in rats.
Dithizone (diphenylthiocarbazone), which forms a firm complex
with thallium, increased faecal elimination by 33% and urinary
elimination by 75% in rats (Lund, 1956b). After treatment with
diethyldithiocarbamate "dithiocarb", the resulting lipophilic
thallium(I) chelate readily passed the blood-brain barrier and was
rapidly decomposed in the brain (Kamerbeek et al., 1971a). However,
it did not protect animals from death after administration of lethal
doses of thallium (Danilewicz et al., 1980). The benefit of other
agents, such as the diuretic 2,3-dimercapto-succinic acid (Liang et
al., 1980) or other compounds with mercapto groups (Oehme, 1972), has
still to be proven in the treatment of thallotoxicosis.
Comparing different antidotal treatments in rats, Lehmann &
Favari (1985) found an increase in thallium elimination to 99% by
dithizone, 93% by activated charcoal, 64% by the diuretic agent
furosemide, 82% by Prussian Blue, and 92% by combining Prussian Blue
and furosemide, whereas the untreated controls had only eliminated 53%
of the administered dose (2 mg) within 8 days. After treatment with
Prussian Blue and D-penicillamine in combination, the dangerous
redistribution of D-penicillamine did not occur and elimination of
thallium was better than after administration of Prussian Blue alone
(Rios & Monroy-Noyola, 1992). The efficiency of Prussian Blue could
be increased by synthesizing batches with a smaller crystal size (Rios
et al., 1991; Kravzov et al., 1993). New compounds, such as rhyolith,
N-acetylcysteine and dimercaprol, were no more, or even less,
effective (Dvorák, 1973; Henderson et al., 1985).
7.10.2 Selenium
Selenium not only protected isolated fetal hearts against damage
by thallium (section 7.9), but also decreased its lethal effect in
young rats; however, thallium-induced hair-loss was not prevented
(Ewan, 1978; Ostádalová & Babicky, 1987). Thallium inhibited
pulmonary elimination of volatile selenium compounds, increased the
retention of selenium in kidneys and liver, and did not protect
against chronic selenosis (Levander & Argrett, 1969). Thallium did
not affect the distribution of selenium in the body of mice; in
in vitro systems it seemed to interact with selenite in glutathione
solution and in erythrocytes (Naganuma et al., 1983).
7.11 Mechanisms of toxicity - mode of action
Although several (perhaps interconnected) mechanisms have often
been postulated, the exact mechanism of thallium toxicity is still
unknown (Cavanagh et al., 1974; Prick, 1979; Sabbioni & Manzo, 1980;
Nessler & Briese, 1985; Chandler & Scott, 1986; Cavanagh, 1988).
Conflicting results have been obtained with respect to the
effects of thallium on sodium/potassium ATPase activity in vitro
(stimulation) (Ivashchenko & Balmukhanov, 1974) and in vivo
(inhibition) (Mourelle et al., 1988). The irreversible inhibition of
the unidirectional transport of sodium may be due to an inhibition of
transport energy (Skulskii & Lapin, 1983). The affinity of the
thallium ion for the sodium/potassium ATPase is 9-10 times greater
than that of the potassium ion (Britten & Blank, 1968; Inturrisi,
1969). Since the thallium permeability of biological membranes is
usually 10 to 100 times greater than that of potassium and since a
similar activation of membrane sodium/potassium ATPase in human
erythrocytes and rat liver cells is caused by thallium concentrations
that are ten times lower than those of potassium and higher
concentrations of ouabain are needed to inhibit thallium-activated
ATPase, a high selectivity of thallium(I) ions for potassium transport
pathways exists (Skulskii et al., 1973, 1975; Gutknecht, 1983; Favari
& Mourelle, 1985; Zeiske & van Driessche, 1986). However, in vitro
studies must take into account the fact that concentrations of only
0.01 g thallium/litre are likely to occur in cells after lethal
poisoning with 10 mg/kg body weight, assuming uniform distribution and
no elimination (Burger & Starke, 1969).
Owing to the similarity in ionic radii of potassium and thallium,
and since the affinity of the thallium ion for sodium/potassium ATPase
is greater than that of the potassium ion, thallium accumulates within
the cell at the expense of potassium. The strong interaction of
thallium with sites normally occupied by potassium may block cycles
that depend on recurrent potassium translocation (Sabbioni & Manzo,
1980).
Once inside the cell, various mechanisms are evident, e.g.,
effects on other enzymes (Sabbioni & Manzo, 1980), the inhibition of
protein synthesis (Hultin & Näslund, 1974), the antimitotic effect of
thallium compounds (section 7.9) (Sabbioni & Manzo, 1980), especially
in the testes (sections 7.5 and 8.5.1), and/or the involvement of
riboflavin vitamin B12, which is coenzyme to a number of enzymes
(Emsley, 1978; Nessler & Briese, 1985). Any effect on riboflavin or
on enzymes containing sulfhydryl groups (see below) should result in a
disturbance of pyruvate metabolism (summarized by Nessler & Briese,
1985). Experimental animals suffering from riboflavin deficiency show
symptoms similar to those of thallium intoxication (Schoental &
Cavanagh, 1977).
Another postulated mechanism considers the general capacity of
thallium to react with thiol groups, thus interfering with a variety
of processes (Zeiske & van Driessche, 1986). Although the toxic
effect of thallium is reduced by diets high in cystine, methionine and
betaine (Oehme, 1972), interference with the metabolism of
sulfur-containing amino acids does not seem to be directly involved in
toxicity (Garcia Bugarin et al., 1989) and strong reactions with
thiols were observed for thallium(III) but not for thallium(I)
compounds (Douglas et al., 1990). An additional aspect of the
reaction of thallium with sulfhydryl groups is the induction of free
radical formation (section 7.8). This is indicated by increased lipid
peroxidation rates in the brain. Processes leading to lipid
peroxidation finally damage cell membranes by subsequent reactions of
free radicals with sulfhydryl enzymes (section 7.11). Lipid
peroxidation results in a deficiency of glutathione and leads to the
accumulation of lipid peroxides in the brain, liver and kidney and,
presumably, finally to lipofuscin granules (Herman & Bensch, 1967;
Aoyama et al., 1988). The possibility that this mechanism is
responsible for the neurotoxic effects of thallium was supported by
the simultaneous administration of thallium and acetyl-homocysteine
thiolactone, which prevented reduction in the level of sulfhydryl
radicals in the cerebellum and significantly increased glutathione
levels (Hasan & Haider, 1989). This hypothesis is further supported
by the protective effect of 1) silymarin against thallium hepato
toxicity (Mourelle et al., 1988), 2) selenium, demonstrated with the
thallium-induced ultrastructural changes found in isolated fetal mouse
heart cell (sections 7.9 and 7.10.2.) (Liu, 1986), and 3) selenium and
vitamin E against membrane damage by uncontrolled lipid peroxidation
in vivo (Hasan & Ali, 1981).
The mode of action of thallium seems to be mainly based on a
disturbance in the function of the mitochondria (Barckow & Jenss,
1976), although they are affected by high concentrations of almost all
heavy metals (Byczkowski & Sorenson, 1984). The thallium(I) cation
may either enter isolated rat liver mitochondria passively, i.e. in an
energy-independent manner (Barrera & Gómez-Puyou, 1975), or penetrate
the mitochondrial membrane electrophoreti cally (Skulskii et al.,
1978). The entry of thallium into the intramitochondrial space and
the interaction of cytosolic thallium with mitochondria membranes may
explain the deleterious effects (Skulskii et al., 1984). In isolated
mitochondria, thallium(I) acetate caused an uncoupling of oxidative
phosphorylation and swelling of the isolated mitochondria (Melnick et
al., 1976). Using ascite tumour cells in vitro, Ivashchenko et al.
(1973) found a strong, thallium-induced increase in oxygen consumption
and lactic acid production, which were inhibited by ouabain and sodium
fluoride. However, whereas oxygen consumption and anaerobic
glycolysis of tissues were affected in vitro, tissues from rats with
chronic or acute (first day) poisoning did not differ from those of
controls (Thyresson, 1950).
Comparing the effects of monovalent and trivalent thallium on
isolated rat liver mitochondria, only thallium(III) nitrate uncoupled
oxidation from phosphorylation (Hollunger, 1960). This effect could
not be reversed by adding edetic acid or dimercaprol (sections 7.10.1
and 8.6).
8. EFFECTS ON HUMANS
The toxicology of thallium is summarized in Fig. 1.
8.1 General population exposure
Thallium concentration in early-morning urine samples of nine
non-exposed subjects ranged from 0.13 to 1.69 µg/litre (Weinig & Zink,
1967). Smith & Carson (1977) gave a range of 0.6 to 2.0 µg/litre with
a mean urinary thallium concentration of 1.3 µg/litre.
The initial use of thallium in medicine, mainly as a depilatory
drug, caused many cases of intoxication. The observation of side
effects led to more detailed toxicological studies after 1918, and its
medical use was abandoned after 1945 (Emsley, 1978; Briese & Nessler,
1985a). Since thallium is tasteless, odourless, without colour and
highly toxic, and used to be easily obtainable, it was often used for
suicide, homicide and attempts at illegal abortion (Kemper & Bertram,
1984; Manzo & Sabbioni, 1988). Also large numbers of accidental
intoxications have occurred, e.g., in Berlin, Germany there were 110
cases of accidental ingestion and 39 attempted suicides by children
involving corn poisoned with thallium sulfate from 1967 to 1976; three
of the children died (von Mühlendahl et al., 1978). Munch (1934b)
summarized thallium intoxications prior to 1934. He found reports on
8006 children who had been treated with thallium as a depilatory
agent. Intoxication occurred in 447 cases and 8 children died. In
connection with its use as a rodenticide or insecticide, 21 poisonings
and 5 deaths occurred.
Recently, intoxications occurred after ingestion of a Chinese
herbal medication/nutritional supplement containing 30 g thallium per
litre, but other samples contained no thallium (Schaumburg et al.,
1992).
8.1.1 Acute toxicity
Cases of acute intoxication by thallium salts in humans, which
always cause severe symptoms, have been reported for single or
multiple oral doses of the order of 100 mg or more for adults, i.e.
1.5 to 2 mg/kg body weight (Table 25).
Symptoms of acute thallium toxicity vary with age, dose and route
of administration (Venugopal & Luckey, 1978). According to Sharma et
al. (1986), one-tenth (2-10 mg/person) of the lethal thallium dose for
adults causes death in children. However, reports of the therapeutic
use of thallium in which children tolerated larger doses than adults
indicate the contrary (Ormerod, 1928; Sessions & Goren, 1947; Prick,
1979). Of 75 children who had accidentally ingested thallium sulfate,
Observers do not participate in the final evaluation of the
chemical; this is the sole responsibility of the Task Group members.
When the Task Group considers it to be appropriate, it may meet in
camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, it then goes for language editing, reference checking, and
preparation of camera-ready copy. After approval by the Director,
IPCS, the monograph is submitted to the WHO Office of Publications for
printing. At this time a copy of the final draft is sent to the
Chairperson and Rapporteur of the Task Group to check for any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR THALLIUM
Members
Professor M. Balali-Mood, Poison Control Centre, Imam Reza Hospital,
Mashhad University of Medical Sciences, Mashhad, Islamic Republic
of Iran
Dr P. Doyle, Chemicals Evaluation Division, Environment Canada,
Ottawa, Ontario, Canada
Professor G. Kazantzis, Imperial College of Science, Technology and
Medicine, Centre for Environmental Technology, Royal School of
Mines, London, United Kingdom (Joint Rapporteur)
Dr M. Kiilunen, Department of Industrial Hygiene & Toxicology,
Institute of Occupational Health, Helsinki, Finland
Mr H. Malcolm, Institute of Terrestrial Ecology, Monks Wood
Experimental Station, Huntingdon, Cambridgeshire, United Kingdom
Dr G. Nordberg, Department of Environmental Hygiene, Umea University,
Umea, Sweden (Chairman)
Professor G. Schaub, Department of Zoology, Institute for Zoology and
Parasitology, Ruhr University, Bochum, Germany (Joint Rapporteur)
Dr S. Velazquez, Environmental Criteria and Assessment Office, US
Environmental Protection Agency, Cincinnati, Ohio, USA
Representatives of other organizations
Dr P. Montuschi, Department of Pharmacology, Catholic University of
the Sacred Heart, Rome, Italy (representing the International Union
of Toxicology)
Observers
Dr R. Cornelis, Institute for Nuclear Sciences, State University of
Gent, Gent, Belgium
Secretariat
Dr P.G. Jenkins, International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland (Secretary)
ENVIRONMENTAL HEALTH CRITERIA FOR THALLIUM
A WHO Task Group on Environmental Health Criteria for Thallium
met in Geneva from 12 to 16 December 1994. Dr P.G. Jenkins, IPCS,
welcomed the participants on behalf of Dr M. Mercier, Director of the
IPCS, and the three IPCS cooperating organizations (UNEP/ILO/WHO).
The Group reviewed and revised the draft and made an evaluation of the
risks for human health and the environment from exposure to thallium.
The first draft was prepared by Professor G. Schaub, Institute
for Zoology and Parasitology, Ruhr University, Bochum, Germany. He
also prepared the second draft, incorporating comments received
following circulation of the first draft to the IPCS contact points
for Environmental Health Criteria monographs.
Dr P.G. Jenkins, IPCS, was responsible for both the overall
scientific content and the technical editing.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
ABBREVIATIONS
AAS atomic absorption spectrometry
AES atomic emission spectrometry
AMP amperometric titration
CRMs certified reference materials
DPASV differential pulse anodic stripping voltametry
EDL electrode discharge lamp
EDTA ethylenediaminetetraacetic acid
GABA gamma-aminobutyric acid
GDMS glow discharge mass spectrometry
GFAAS graphite furnace atomic absorption spectrometry
GLP good laboratory practice
ICP inductively coupled plasma
IDMS isotope dilution mass spectrometry
LOEL lowest-observed-effect level
MED minimum effective dose
MIBK methyl isobutyl ketone
MS mass spectrometry
NAA neutron activation analysis
NaDDC sodium diethyldithiocarbamate
NADP nicotinamide adenine dinucleotide phosphate
NOEL no-observed-effect level
PAA photon activation analysis
TLV threshold limit value
tRNA transfer ribonucleic acid
1. SUMMARY
1.1 Identity, physical and chemical properties, and analytical
methods
Elemental thallium is a soft and malleable metal with a
bluish-white colour. When exposed to humid air or water, thallium is
oxidized rapidly on the surface or the hydroxide is formed,
respectively. Thallium has two important oxidation states,
thallium(I) and thallium(III). Monovalent (thallous) compounds behave
like alkali metals, e.g. potassium, whereas the trivalent (thallic)
compounds are less basic, resembling aluminium. In contrast to
inorganic compounds in which the thallium(I) ion is more stable in
aqueous solutions than the thallium(III) ion, the latter is more
stable in organic compounds.
The determination of thallium in environmental samples is
somewhat difficult as concentrations are in the µg/kg range or less.
Generally the limits of determination for minerals, soils and dusts
are about 20 µg/kg, for aqueous solutions about 0.1 µg/litre, and for
biological materials a few µg/kg, when no pre-concentration of
thallium is applied.
Graphite furnace atomic absorption spectrometry (GFAAS) is an
analytical method well suited for applications where high sensitivity
is required from small sample amounts with thallium present at
concentrations of a few µg/kg. Isotope dilution mass spectrometry
(IDMS) and inductively coupled plasma-mass spectrometry (ICP-MS),
possibly combined with isotope dilution, are excellent methods for
determinations offering good precision and accuracy at the µg/kg
level.
1.2 Sources of human and environmental exposure
Thallium is present in the environment as a result of natural
processes and from man-made sources. It is ubiquitous in nature and
occurs especially in sulfide ores of various heavy metals, but
normally in low concentrations. There are only a few areas with a
naturally very high thallium concentration.
Thallium is produced industrially only in small quantities (the
worldwide industrial consumption in 1991 was 10-15 tonnes/year).
Thallium and its compounds have a wide variety of industrial uses.
Its uses as a depilatory agent for humans and as a rodenticide and
insecticide are now severely restricted. The main uses are in the
electrical and electronic industries and in the production of special
glasses. Another important field of application is the use of
radioisotopes in medicine for scintigraphy and the diagnosis of
melanoma and the use of arylthallium(III) compounds in biochemistry.
Losses to the environment mainly occur from mineral smelters
(deposits of waste material and emissions into the atmosphere),
coal-burning power-generating plants, brickworks, and cement plants
(all emissions into the atmosphere). From 2000 to 5000 tonnes/year
are estimated to be mobilized world-wide by industrial processes.
Emissions of thallium from industrial processes vary widely according
to the type of industry.
Emissions from coal-fired power-generating plants can contain a
thallium concentration of 700 µg/m3 exhaust air and those from
cement plants up to 2500 µg/m3. The latter value may be reduced to
< 25 µg/m3 by using other raw materials and changing the production
process. Thallium volatilizes during the burning of coal or raw
material for cement production and recondenses on the surface of ash
particles in cooler parts of the system. These particles contain up to
50 mg thallium/kg fly-ash and are often of small size, so that only
50% of them are held back by filters in cement plants. Also, about
one third of emitted particles from power-generating plants are of the
small particle size which can be deposited in the lower respiratory
tract.
Effluent from mine tailing ponds containing up to 1620 and
36 µg/litre caused elevated levels of 88 and 1 µg/litre, respectively,
in connecting rivers. Rainwater ponds around a cement plant contained
up to 37 µg/litre. In soil maximal concentrations of 60 mg/kg have
been found near waste materials from mines; 2, 0.6 and 27 mg/kg have
been found in the vicinity of base metal smelters, brickworks and
cement plants, respectively.
In contaminated areas the majority of vegetables, fruits and meat
contain less than 1 mg thallium/kg fresh weight. Concentrations are
higher in cabbages (Brassicaceae), with up to 45 mg/kg reported in
green kale. Concentrations of thallium in the tissues of farm animals
correlate with concentrations in the fodder. In the vicinity of some
cement plants, increased concentrations in fodder (e.g., up to
1000 mg/kg in rape) and beef and rabbit meat (up to 1.5 and 5.8 mg/kg,
respectively) have been reported.
1.3 Environmental transport, distribution and transformation
Near point sources such as coal-fired power-generating stations,
some cement plants and metal smelting operations, the major source of
thallium in air is emission of fly ash. The results of one study
indicate that nearly all of the thallium in fly dust from a cement
plant was present as soluble thallium(I) chloride.
The fate of thallium added to soil (in deposited fly ash, for
example) depends largely on soil type. Retention will be greatest in
soils that contain large amounts of clay, organic matter and
iron/manganese oxides. Incorporation into stable complexes causes
enhanced thallium concentrations only in the upper levels of soils.
The uptake of thallium by vegetation increases as soil pH decreases.
In some strongly acid soils significant amounts of thallium can be
leached to local ground and surface water.
Most dissolved thallium in freshwater is expected to be in the
monovalent form. However, in strongly oxidized fresh water and most
seawater trivalent thallium may predominate. Thallium can be removed
from the water column and accumulate in sediment by various exchange,
complexation or precipitation reactions.
Although thallium can bioconcentrate, it is not likely to
biomagnify in aquatic or terrestrial food webs.
1.4 Environmental levels and human exposure
In areas not contaminated by thallium, concentrations in air are
usually < 1 ng/m3, those in water < 1 µg/litre, and those in water
sediments < 1 mg/kg. Mean concentrations in the earth's crust range
from 0.1 to 1.7 mg/kg, but very high concentrations are possible,
e.g., in coal up to 1000 mg/kg, and the rarely found minerals of
thallium consist of up to 60% of the element. Food of plant and
animal origin usually contains < 1 mg/kg dry weight and the human
average dietary intake of thallium appears to be less than 5 µg/day.
Uptake via the respiratory system is estimated to be < 0.005 µg
thallium/day.
There are only limited data about the actual thallium content of
workplace air. The most recent (1980s) concentrations of thallium
observed were < 22 µg thallium/m3 (in the production of a special
thallium alloy and in a thallium smelter). Average urinary
concentrations were determined to be in the range of 0.3-8 µg/litre
for cement workers and 0.3-10.5 µg/litre for foundry workers.
1.5 Kinetics and metabolism in laboratory animals and humans
Thallium is rapidly and well absorbed through the gastro
intestinal and respiratory tracts and is also taken up through the
skin. It is rapidly distributed to all organs and passes the placenta
(as indicated by the rapid fetal uptake) and the blood-brain barrier.
Because of its rapid accumulation in cells, concentrations of thallium
in whole blood do not reflect the levels in tissues. In acute
poisoning of experimental animals or humans, initially high
concentrations of thallium appear in the kidney, low concentrations in
fat tissue and brain, and intermediate concentrations in the other
organs; later the thallium concentration of the brain also increases.
Elimination of thallium may occur through the gastrointestinal
tract (mainly by mechanisms independent of biliary excretion), kidney,
hair, skin, sweat and breast milk. Intestinal reabsorption (mainly
from the colon) may occur with a consequent decrease in total body
clearance. In rats, the main routes of thallium elimination are
gastrointestinal (about two thirds) and renal (about one third), in
rabbits the contribution of the two routes is about equal. Thallium
is also secreted in saliva.
As with many other substances, the excretion of thallium in
humans differs from that in laboratory animals, since the rate of
excretion is generally much lower in humans (rate constant =
0.023-0.069 day-1) than in laboratory animals (average rate constant =
0.18 day-1). Another major difference between humans and animals is
the relative contribution of the different routes of excretion. In
humans, renal excretion seems to be much more important than in
animals, although its relative contribution to the total body
clearance has not been definitively established, due principally to
the lack of sufficient human data. Moreover, exposure levels,
duration of exposure, impairment of excretory organ function,
potassium intake and concomitant treatment of acute poisoning may
considerably influence the results.
In one study renal excretion of thallium was reported to be about
73%, whereas that through the gastrointestinal tract was about 3.7% of
the daily excreted amount. Excretion through hair and skin, and sweat
has been estimated to be 19.5% and 3.7%, respectively.
The biological half-life of thallium in laboratory animals
generally ranges from 3 to 8 days; in humans it is about 10 days but
values up to 30 days have been reported.
No data on the biotransformation of thallium are available.
1.6 Effects on laboratory mammals and in vitro test systems
There are no striking species-specific differences in the
toxicity of thallium(I) salts. Usually an oral intake of 20 to 60 mg
thallium/kg body weight is lethal within one week. Guinea-pigs are
slightly more sensitive than other experimental animals. The
water-insoluble thallium(III) oxide shows a somewhat lower acute
toxicity by oral or parenteral administration than thallium(I) salts.
Comparison of acute toxicity data indicates a high degree of
bioavailability from all exposure routes. Most organs are affected,
but the signs of poisoning and the sequence in which they occur reveal
some intra- and interspecies variability.
The symptoms of acute intoxication generally follow the following
sequence: firstly anorexia, vomiting and depression, later diarrhoea,
skin changes (inflammation at body orifices, skin furuncles, hair
loss), and then dyspnoea and nervous disorders. Finally, respiratory
failure leads to death.
Symptoms of chronic intoxication are similar to those of acute
intoxication. Loss of hair regularly occurs.
Histological examination reveals necrosis or other cell damage.
Necrotic changes have been observed in the kidneys, liver, intestine,
heart and the nervous system. Swelling of mitochondria and loss of
cristae, dilatations of smooth endoplasmic reticulum, increased
numbers of autophagic vacuoles and lipofuscin granules, and loss of
microvilli have been observed in many cells. The thallium-induced
alterations of functional processes may arise from physical disruption
of the membranes of subcellular organelles. In the heart,
arrhythmogenic effects are restricted to the sinus node.
Thallium intoxication causes selective impairment of certain
behavioural elements, which are correlated with biochemical effects
(which indicate cellular damage) in certain regions of the brain.
Some neurological effects seem to be caused by direct action, e.g.
ataxia and tremor by cerebellar alterations or alterations in
endocrine activity through changes in the hypothalamus. The autonomic
nervous system, mainly the adrenergic, may be activated by thallium.
In peripheral nerves, thallium seems to interfere presynaptically,
with the spontaneous release of transmitter, by antagonizing these
calcium-dependent processes.
The exact mechanism of thallium toxicity is still unknown.
Several, perhaps interconnected, mechanisms have been postulated. An
important aspect of thallium intoxication is the significant increase
in lipid peroxidation and in the activity of the lysosomal enzyme
ß-galactosidase. The resulting deficiency of glutathione leads to the
accumulation of lipid peroxides in the brain and, presumably, finally
to lipofuscin granules. The mode of action of thallium seems to be
mainly due to a disturbance of the function of the mitochondria.
Sexual activity is usually reduced in chronically poisoned
animals, and gonadotoxic effects of thallium are evident in the male
reproductive system. In the testes of rats given 10 mg thallium/litre
in the drinking-water for 16 days, the Sertoli cells were most
sensitive, and desquamation of the spermatogenic epithelium led to
immature sperm cells in the semen. This could explain the decreased
survival rate of embryos or reduced life span of offspring after
sublethal thallium-poisoning of the fathers.
Teratogenic effects, growth inhibition and disturbances in the
development of bones were found to occur in chicken embryos after
injection of thallium into the egg, but such effects in mammals, even
at maternotoxic doses, are controversial. Although transplacental
transfer has been demonstrated, many strains of mice and rats show no
or only slight teratogenic effects.
Two microbiological mutagenicity tests in Salmonella typhimurium
were negative and in vivo tests on sister chromatid exchange were
controversial. However, single studies report chromosomal aberrations
or a significant increase of single-stranded DNA breaks.
Long-term studies on the carcinogenicity of thallium are lacking.
1.7 Effects on humans
Since thallium salts are tasteless, odourless, colourless, highly
toxic, were easily obtainable in the past and still are in some
developing countries, thallium has often been used for suicide,
homicide and attempts at illegal abortion, causing acute thallium
poisoning. Indeed, thallium intoxication is considered one of the
most frequent causes, on a worldwide scale, of purposeful or
accidental human poisoning. Knowledge of chronic thallium
intoxication is limited to occupational exposure, to population groups
in contaminated areas and to cases of homicide involving multiple low
doses.
Symptoms of acute thallium toxicity depend on age, route of
administration and dose. Doses which have proved lethal vary between
6 and 40 mg/kg, being on average 10 to 15 mg/kg. Without therapy this
average dose usually results in death within 10 to 12 days, but death
occurring within 8-10 h has also been reported.
The triad of gastroenteritis, polyneuropathy and alopecia is
regarded as the classic syndrome of thallium poisoning, but in some
cases gastroenteritis and alopecia were not observed. Several other
signs and symptoms also occur, varying in order, extent and intensity.
Symptoms of thallium intoxication are often diffuse and initially
include anorexia, nausea, vomiting, metallic taste, salivation,
retrosternal and abdominal pain and occasionally gastrointestinal
haemorrhage (blood in faeces). Later, constipation is commonly seen
and may be resistant to treatment, thus interfering with antidotal
treatment.
After 2 to 5 days some of the typical thallium disorders slowly
develop, irrespective of the route of exposure. Effects on the
central and peripheral nervous system vary, but a consistent and
characteristic feature of thallium intoxication in humans is the
extreme sensitivity of the legs, followed by the "burning feet
syndrome" and paraesthesia. Involvement of the central nervous system
(CNS) is indicated by symptoms like hallucinations, lethargy,
delirium, convulsions and coma. Common circulatory symptoms are
hypertension, tachycardia and, in severe cases, cardiac failure. Loss
of head hair and sometimes body hair occurs after the second week of
poisoning; dystrophy of the nails is manifested by the appearance of
white lunular stripes (Mee's lines) 3 to 4 weeks after intoxication.
The black regions found in hair papillae are not caused by deposition
of pigments or thallium but are due to small amounts of air entering
the shaft.
In lethal cases the time until death occurs may vary from hours
to several weeks, but most commonly death occurs within 10 to 12 days.
Causes of death are mainly renal, CNS and cardiac failure.
In sublethal poisonings, recovery often requires months.
Sometimes neurological and mental disturbances as well as
electroencephalographic abnormalities and blindness can remain.
Additionally, intellectual functions seem to be adversely affected in
survivors.
In cases of chronic poisoning, symptoms are similar but in
general milder than in cases of acute intoxication. Sometimes
permanent blindness occurs. Complete recovery takes months and can be
interrupted by relapses.
In a well-investigated case of thallium emission around a cement
plant in Lengerich, Germany, thallium concentrations in the hair and
urine of exposed people did not correlate with certain features which
are known to be usually associated with chronic thallium poisoning,
but only with subjective neurological symptoms.
Postmortem examinations or biopsies following thallium poisoning
reveal damage of various organs. For example, after ingestion of
lethal doses, haemorrhages in the mucosa of the intestine, lung,
endocrine glands and heart, fatty infiltrations in liver and heart
tissue, and degenerative changes to glomeruli and renal tubules occur.
In the brain, fatty degeneration of ganglion cells, damage to axons
and disintegration of myelin sheaths can be observed.
Variations in blood pressure may be caused by direct effects of
thallium on the autonomic nervous system. Thallium intoxication
causes symmetric, mixed peripheral neuropathy. Distal nerves are
affected more than proximal nerves, and earlier but lesser degrees of
damage occur in nerves with shorter axons, e.g., cranial nerves.
Axons are swollen and contain vacuoles and distended mitochondria. In
lethal poisoning, severe damage of the vagus nerve, denervation of the
carotid sinus and lesions of the sympathetic ganglia have been
observed. In sublethal poisoning, affected nerves may undergo axonal
degeneration with no or only partial recovery within 2 years.
Retrobulbar neuritis and resulting visual disorders can develop
and persist for months after terminating treatment with thallium-
containing depilatories, and even optic atrophy may occur.
Limited data are available on the effects of thallium on human
reproduction. Menstrual cycle, libido and male potency may be
adversely affected. Effects on sperm are known to occur following
chronic intoxication. As in animal studies, transplacental transfer
occurs; this was seen following a thallium-induced abortion. However,
apart from a relatively low weight and alopecia of newborn babies,
fetal development was not affected in about 20 cases of thallium
intoxication during pregnancy.
No reports of any carcinogenic effects or data on immunological
effects of thallium are available. There is no adequate evidence of
genotoxic effects.
Therapies of thallium intoxication combine forced diuresis, use
of activated charcoal and prevention of re-absorption in the colon by
administration of Prussian blue, potassium ferric hexacyano
ferrate(II).
1.8 Human dose-response relationship
The mean urinary thallium concentration in unexposed populations
is 0.3 to 0.4 µg/litre. As thallium has a short biological half-life,
measured in days, and assuming steady-state conditions, this urinary
concentration can be taken as an indicator of total dose following
inhalation and dietary intake.
The mean urinary thallium concentration in a population sample
living near a thallium atmospheric emission source was 5.2 µg/litre.
A clear dose-response relationship was found between urinary thallium
concentration and the prevalence of tiredness, weakness, sleep
disorders, headache, nervousness, paraesthesia, and muscle and joint
pain. A similar dose-response relationship was also reported when
thallium in hair was used as an indicator of exposure.
The Task Group considered that exposures causing urinary thallium
concentrations below 5 µg/litre are unlikely to cause adverse health
effects. In the range of 5-500 µg/litre the magnitude of risk and
severity of adverse effects are uncertain, while exposure giving
values over 500 µg/litre have been associated with clinical poisoning.
1.9 Effects on other organisms in the laboratory and field
Thallium affects all organisms, but species- and also strain-
specific differences are evident. Different inorganic thallium(I) and
thallium(III) compounds and organothallium compounds can show
different toxicities.
The most important effect of thallium on microorganisms seems to
be inhibition of nitrification by soil bacteria. Results of one study
suggest that microbial community structure is disturbed at soil
concentrations in the range of 1-10 mg/kg dry weight, but the form of
thallium used in this experiment was not identified.
Thallium is taken up by all plant parts, but principally by the
roots. After uptake into the cell, it is concentrated unevenly in the
cytosol, probably bound to a peptide. Thallium concentrations found
in plants depend on soil properties (especially pH, clay and organic
matter content), as well as on the developmental stage and on the part
of the plant. Thallium accumulates in chlorophyll-containing
regions, but to a lesser degree in thallium-resistant plants. Oxygen
production is reduced by thallium, presumably by direct action on
electron transfer in photosystem II. Interference with the pigments
is indicated by the occurrence of chlorosis. In addition, impaired
uptake of trace elements seems to be involved in the mechanism of
toxicity. Growth is also affected, roots reacting more sensitively
than leaves or stems. These effects have been reported at
concentrations as low as 1 mg thallium/kg of dry plant tissue, after
exposure to monovalent forms of thallium.
Most studies of effects on aquatic organisms have used soluble
monovalent thallium compounds. The lowest thallium concentration
reported to affect aquatic species is 8 µg/litre, which caused a
reduction in growth of aquatic plants. Invertebrates are often
affected at lower concentration than fish (96-h LC50 values are
2.2 mg thallium/litre for daphnids and 120 mg/litre for a freshwater
fish). The lowest LC50 value, reported after exposure for about 40
days, was 40 µg/litre for fish.
Many cases of thallium intoxication of wildlife have been caused
by its large scale application as a rodenticide. In seed-eating
animals and predators the CNS and/or the gastrointestinal tract are
most severely affected. These effects can also be observed in farm
animals. In addition, thallium causes a loss of dorsal feathers in
ducks, salivation from the nose and mouth of cattle, and reduced
growth in broilers, laying hens, sheep and steers.
2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, AND ANALYTICAL METHODS
Thallium is a soft, malleable, heavy metal with a bluish-white
colour and the chemical symbol Tl. The name thallium derives from the
beautiful green spectral line ( thallos, green shoot), which
identified the element.
An overview on the properties, synonyms and chemical formulae of
pure thallium and some of its compounds is given in Table 1.
2.1 Identity
Thallium is the fifth element in Group IIIB of the Periodic
Table. It occurs naturally as two isotopes thallium-203 and
thallium-205 with abundancies of 29.52 and 70.467%, respectively
(Aderjan et al., in press). The relative atomic mass of thallium is
204.383, the atomic number is 81, and the electron configuration is
(Xe) 4f14 5d10 6s2 6p. Due to its high specific gravity of
11.85 g/cm3, thallium belongs to the heavy metal group, which
comprises all metals with a specific gravity of over 4.5 g/cm3
(Micke et al., 1983).
2.2 Physical and chemical properties
The physical properties of elemental thallium are similar to
those of lead; it is very soft and malleable. Thallium exists in both
the monovalent (thallous) and the trivalent (thallic) form. Because
the 6s electrons possess only a low tendency to be released or bound
covalently, the thallous form is more common and stable and forms
numerous stable salts. Thallium(III) is easily reduced to thallium(I)
by reducing agents at high temperatures (Tl+/Tl3+ = +1.12v) (Micke
et al., 1983; Schoer, 1984; Stokinger, 1987).
Below 234°C the metal crystallizes in a hexagonal close-packed
form (alpha-thallium), while at 234°C it converts to the ß-form, a
cubic body-centred lattice. Thallium begins to volatilize at 174°C.
It has a melting point of 303°C, a boiling point of 1457°C and a
normal potential of Tl/Tl+ -0.335v (Micke et al., 1983). Thallium
is a very reactive metal. When exposed to air and moisture, it is
superficially oxidized, forming a coating of thallium(I) oxide
(Tl2O). At higher temperatures it reacts with a lovely green flame
to form thallium(III) oxide (Tl2O3). Thallium carbonate
(Tl2CO3) is the only heavy metal carbonate that is very soluble in
water (Micke et al., 1983; Stokinger, 1987).
Table 1. Physical and chemical properties of thallium and some selected thallium compoundsa
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium Tl 7440-28-0 204.38 11.85 303.5 1457 bluish-white, insoluble
metallic
Thallium(I) TlC2H3O2 563-68-8 263.43 3.765 131 - silky white very
acetate soluble
Thallium TlAl(SO4)2*12H2O 52238-56-9 639.66 2.306 91 - colourless 117.8
aluminium
sulfate
Thallium(I) TlBr 7789-40-0 284.29 7.557 480 815 pale yellow 0.5
bromide (17.3°C) (25°C)
Thallium(I) Tl2CO3 29809-42-5 468.78 7.110 273 - white 40.3
carbonate (15.5°C)
Thallium(I) TlCl 7791-12-0 239.84 7.004 430 720 white 2.9
chloride (30°C) (15.5°C)
Table 1 (contd).
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium(III) TlCl3 13453-32-2 310.74 - 25 decomposes colourless, very
trichloride hygroscopic soluble
TlCl3*4H2O 13453-33-3 382.80 - 37 100 (-H2O) colourless 862
Thallium TlOC2H5 20398-06-5 249.44 3.493 -3 130 colourless -
ethylate (20°C) (decomposes)
Thallium(I) TlF 7789-27-7 223.38 8.23 327 655 colourless 786
fluoride (4°C) (15°C)
Thallium(III) TlF3 7783-57-5 261.38 8.36 550 - olive decomposes
trifluoride (25°C) (decomposes) green to TlOH
Thallium TlOH 1310-83-4 221.39 - 139 - pale 259
hydroxide (decomposes) yellow
Thallium(I) TlI 7790-30-9 331.29 7.29 440 (ß) 823 (ß) yellow 0.006
iodide (alpha) (20°C)
Table 1 (contd).
Name Chemical CAS registry Relative Specific Melting Boiling Colour Solubility
formula number atomic/ gravity point point in water
molecular mass (g/cm3) (°C) (°C) (g/litre)
Thallium(I) TlNO3 10102-45-1 266.39 - 206 430 white 95.5
nitrate (alpha) (20°C)
Thallium (III) Tl(NO3)3*3H2O 13453-38-8 444.44 - 105-107 decomposes colourless decomposes
nitrate
trihydrate
Thallium(I) Tl2O 1314-12-1 424.77 9.52 300 1080 (-O) black decomposes
oxide (16°C) to TlOH
Thallium(III) Tl2O3 1314-32-5 456.76 10.19 717 ± 5 875 (-O2) black insoluble
oxide (22°C)
Thallium(I) Tl2SO4 7446-18-6 504.82 6.77 632 decomposes white 48.7
sulfate (20°C)
Thallium(I) Tl2S 1314-97-2 440.85 8.46 448.5 0.2
sulfide (20°C)
a From: Stokinger (1987); Budavari (1989); Lide (1990)
In contact with water, thallium(I) hydroxide is formed from the
metal. Thallium is very soluble in HNO3 and H2SO4, but only
slow dissolution takes place in HCl, because of the low solubility of
the halides. It is insoluble in alkali bases. Thallium combines with
fluorine, chlorine and bromine at room temperature, and reacts with
iodine, sulfur, phosphorus, selenium and tellurium after heating. The
metal does not react with molecular hydrogen, nitrogen or carbon. It
forms alloys with other metals and readily amalgamates with mercury
(Micke et al., 1983).
The ionic radii and the electronegativity constant of monovalent
thallium are very similar to those of other alkali metals.
Thallium(I) hydroxide, carbonate and sulfate, like the corresponding
potassium compounds are very soluble in water. With respect to their
physical and chemical properties, e.g., poor water solubility,
thallium(I) oxide, sulfide and halides show similarities to the
corresponding compounds of silver, mercury and lead
(Trotman-Dickenson, 1973). In contrast to inorganic thallium
compounds, covalent organothallium compounds are only stable in the
trivalent form (McKillop & Taylor, 1973). Thallium(I) is not strongly
complexed by humic acids, whereas thallium(III) forms stable complexes
of the [TlX4]- or [TlX6]3- type (Schoer, 1984).
2.3 Conversion factor
1 g thallium = 0.0049 mol
1 mol thallium = 204.38 g
2.4 Analytical methods
Classical analytical methods, the introduction of new techniques
and a combination of both with enrichment or separation processes
provide suitable methods for the quantitative detection of thallium in
various media. Because thallium concen trations in environmental
samples are very low, determination directly from the sample or from
the digestion solution usually lacks sufficient accuracy. Therefore,
preconcentration procedures are necessary (Schoer, 1984; Sager & Tölg,
1984).
2.4.1 Sampling and sample preparation
Thallium losses during sampling, sample preparation and
determination are a major source of analytical error. Contamination
hazards need to be anticipated, as thallium is present in laboratory
ware and is leached out by solutions (Kosta, 1982). Glass contains
about 1-10 µg thallium/kg. Leaching of polythene containers with 6M
HCl for 1 week brought 1-10 ng thallium/cm2 into solution. In
addition, thallium(I) in 0.1M HNO3 solution adsorbs onto container
walls made of polyethene, polypropene, glassware or rubber. This
effect depends on the chemical properties of the surface of the
container walls and on the concentration of matrix ions. At a
thallium concentration of 1 mg/litre, no losses to borosilicate
surfaces at pH < 4 were reported, but extensive adsorption occurred
at pH > 10 (Sager & Tölg, 1984).
For determinations with spectrophotometric, mass spectrometric,
voltametric and other methods, digested samples are needed. With
respect to the high volatility of the metal and the low boiling points
of some of its compounds, only closed systems are recommended for the
digestion of organic matrices to prevent thallium losses. Fusion, dry
ashing and fuming with HF and H2SO4 or HClO4 may lead to severe
losses (up to 40%) of the thallium present (Matthews & Riley, 1969).
High-pressure digestion in closed quartz vessels with concentrated
acids, e.g., HNO3 or HNO3 and HF, at temperatures up to 300°C is
the most suitable procedure for nearly all matrices (Knapp, 1985). HF
interferes with analysis by GFAAS or ICP-AES and needs to be removed
by heating to dryness with H3BO3 (Han et al., 1982).
The volatility of thallium and its oxide or chloride makes it
possible to separate these with a gas stream of O2, H2 or HCl from
other elements that do not form volatile components under the same
conditions and subsequently capture them in a cool trap. This
procedure can be used as a preconcentration step when large quantities
of sample are available (Geilmann & Neeb, 1959; Han et al., 1982;
Sager, 1984).
Other methods of preconcentration are coprecipitation, anodic
electrolysis, ion exchange and liquid-liquid extraction.
Coprecipitation is not selective, but it leads to a high concentration
factor and results in a definite matrix, which might be useful in some
methods (Griepink et al., 1988). For example, coprecipitation with
Fe(OH)3 leads to separation from a salt matrix (K+, NH4+).
Electrolytic deposition or cementation with zinc powder yields an
excellent separation, although this procedure is time-consuming. Ion
exchange, which gives a specific separation in certain cases, is also
time-consuming. Liquid-liquid extraction with chelating agents is
virtually nonspecific, but it is a fast and easy method. A
disadvantage is the relatively low concentration factor (Sager & Tölg,
1984).
Isotope dilution methods have been applied to avoid ionization
matrix effects. Thallium is measured as thallium-205; the thallium-203
isotope can be used as a spike for isotope dilution (Sager, 1986).
2.4.2 Methods of determination
Thallium is almost always determined as total metal, rather than
as specific thallium compounds. Among the analytical techniques that
can be used are spectrophotometry, mass spectrophotometry (MS), atomic
absorption spectrometry (AAS), voltametry, neutron activation analysis
(NAA), X-ray fluorimetry, and inductively coupled plasma (ICP)
techniques (Sharma et al., 1986). A selection of analytical methods
is summarized in Tables 2 and 3.
2.4.2.1 Atomic absorption spectrometry
The most widely used method of thallium determination is atomic
absorption analysis, using measurement at 276.8 nm with a thallium
hollow cathode lamp. The sensitivity can be improved by the use of an
electrode discharge lamp (EDL), owing to its higher intensity.
Graphite furnace atomic absorption spectrometry (GFAAS) is a
well-established technique for the monitoring of trace elements in
nearly all kinds of matrices. The technique has sufficiently low
detection limits and is well-suited to applications where high
sensitivity is required for small sample amounts. In Table 3 some
methods for GFAAS are summarized.
The platform furnace concept in the temperature-stabilized mode,
together with Zeeman effect background correction, allows almost
interference-free determinations of many elements. Sample
pretreatment is not necessary, which greatly reduces the risk of
substance losses or contamination of the sample prior to analysis
(Minoia et al., 1990).
Matrix modifiers permit higher pyrolysis temperatures, so that
the desired element can be isolated from matrix elements and compounds
in an ideal case. Letourneau et al. (1987) found that additions of
H2SO4 as a matrix modifier were inadequate and that interferences
could not be corrected by Zeeman background compensation. Modifying
the matrix with palladium and magnesium nitrate has been suggested to
be generally applicable, but this is not as effective for thallium as
it is for other elements (Welz et al., 1988a). A combination of 6 mg
palladium with 100 mg ammonium nitrate allows the direct determination
of thallium in ten-fold diluted blood against matrix-free standards
(Yang & Smeyers-Verbeke, 1991).
Paschal & Bailey (1986) determined thallium concentrations in
urine. The samples were diluted 1:1 with a matrix modifier consisting
of magnesium nitrate, HNO3, Triton X-100 and water. The detection
limit was calculated to be 0.5 µg/litre.
Table 2. Instrumental methods for the determination of thallium
Methoda Matrix Oxidation Sample Parameters Interferences Detection Reference
state pretreatment of method limit
PAA metals - 203Tl (gamma,n) 202Tl gamma440 keV Segebade &
30 MeV bremsstrahlung Schmitt (1987)
post-irradiation
separation of Tl
from the matrix
NAA biological post- drying, 2-M 1013 n/cm2.sec 3-7 other isotopes 1 µg absolute Itawi &
material irradiation days 203Tl (n,gamma) than 204Tl Turel (1987)
extraction 204Tl 0.77 MeV ß-
measurement
AMP water Tl (I) Na2CO3, NaHCO3, -0.47 vs sat. calomel Mn(VII), Co(II), - Agrawal &
thiomalic acid electrode Sn(II), Tl(III) Khatkar (1988)
DPASV urine, - Na acetate, HClO4, -1.0 vs sat. calomel Cd, Pb 0.2 µg/litre Vandenbalck &
saliva EDTA electrode Patriarche
(1987)
ICP-MS rocks - HNO3, HF, H2O2 - polyatomic 70 ng/litre Date et al.
interferences (1988)
Table 2 (contd).
Methoda Matrix Oxidation Sample Parameters Interferences Detection Reference
state pretreatment of method limit
GDMS indium - - at pressure 3.10-4 mbar - 30 µg/kg Guidoboni &
discharge voltage 1 kV Leipziger
discharge current 3 mA (1988)
accelerating voltage
8 kV resolution 4000
ICP-AES air - HNO3/HClO4 (4:1) 190.9 nm F- 17 µg/litre NIOSH (1984)
particulates leachate
ICP-AES biological - Parr bomb F- 0.05-0.1 Que Hee &
material mg/litre Boyle (1988)
ICP-MS water - HNO3 205Tl - 0.1 µg/litre Henshaw et al.
(1989)
ICP-MS tissues - HNO3 - - 18 µg/kg Templeton
et al. (1989)
Spectrometry environmental Tl (I) dithizone, CHCl3, - Ag, Hg 1 µg/litre Sager (1986)
EDTA, citrate,
cyanide
a AMP = amperometric titration; DPASV = differential pulse anodic stripping voltametry; GDMS = glow discharge mass spectrometry;
ICP-AES =inductively coupled plasma - atomic emission spectrometry; ICP-MS = inductively coupled plasma - mass spectrometry;
NAA = neutron activation analysis; PAA = photon activation analysis; PPS = proton-induced prompt low energy photon high resolution
spectrometry
Table 3. Methods for determining thallium (Tl) with graphite furnace atomic absorption spectrometry (GFAAS)
Sample Separation Injected solution Detection limit Interferences Reference
Fly ash, soil digestion and preconcentration diluted H2SO4, HNO3 3.3 ng/litre HBr De Ruck et
by extraction of Tl(III) with including the 400 × al. (1989)
diisopropylether evaporation preconcentration step)
Urine complex with tri-n-octylamine, organic layer diluted 0.3 µg/litre max. charring Flanjak &
extraction with ethanol into 5 mg with ethanol and H2SO4 temp. 400°C Hodda (1988)
metallic n-butyl acetate gallium
Gallium - gallium 200 µg/kg - Hiltenkamp &
Jackwerth (1988)
Urine - spiked urine, diluted 2 µg/litre NaCl Berndt &
Sopczak (1987)
Urine chelation with NaDDC, extraction MIBK extract 0.05 µg/litre - Apostoli et
with MIBK al. (1988)
Mineralized - HNO3, H2SO4, ascorbic 5 µg/litre NaCl Leloux et al.
faeces and acid, Triton X-100 (1987a)
tissues
Table 3 (contd).
Sample Separation Injected solution Detection limit Interferences Reference
Blood, serum - HNO3 10 µg/litre Leloux et al.
(1987a)
Erythrocytes - HNO3 12 µg/litre Leloux et al.
(1987a)
Soil, extraction with 20 µg/kg Cu, Zn, Pb Ebarvia et
sediments tri-octyl-methylammonium, MIBK al. (1988)
Coal fly ash - HNO3 - - Bettinelli et
al. (1988)
MIBK = methyl isobutyl ketone; NaDDC = sodium diethyl dithiocarbamate
Chemical interferences due to chloride ions are important. These
interferences are caused by volatilization of thallium chloride in the
pyrolysis stage and, in part, by formation of TlCl(g) during the
atomization stage. Even matrix modification gives unsatisfactory
results. Welz et al. (1988b) showed that addition of palladium
nitrate as a modifier and application of argon with 5% H2 as a purge
gas leads to interference-free determination with, for instance, NaCl
loads of up to 100 mg. A special pre-pyrolysis step is necessary to
reduce palladium to the metal state, thus enabling adsorbed H2 to
react with the chloride compounds to form volatile HCl. Similar
results were obtained by Manning & Slavin (1988).
De Ruck et al. (1987) reported an oxidation technique for natural
waters with cerium(IV) sulfate and a subsequent preconcentration step
on an anion-exchange column. A preconcentration factor of 400 was
achieved, and the resultant detection limit was 3.3 ng/litre using
GFAAS. Flame atomic absorption is a reliable method for measurement
of thallium concentrations at the level of mg/litre or more. The
determination is easy and free from interference (Welz, 1983; Griepink
et al., 1988).
2.4.2.2 Inductively coupled plasma - mass spectrometry
ICP-MS is a promising method for concentrations in the µg/kg
range or less, and has good precision and accuracy. It is a multi-
element technique with sub-ppb detection limits for many elements.
Additional advantages of mass discrimination include its suitability
for isotope ratio analysis and stable isotope tracer analysis, and the
extended range of elements that can be studied. Some ICP-MS methods
are summarized in Table 2.
The application of ICP-MS to the analysis of thallium in
iron-rich ores was described by Date et al. (1988). No polyatomic
interferences for iron were detected in acid solutions. The addition
of 500 mg iron/litre to a solution of 1 mg thallium/litre in 1% HNO3
resulted in a 0.1% increase in the thallium peak. The detection limit
was found to be 0.07 µg/litre.
Templeton et al. (1989) examined thallium concentrations in rat
liver and blood plasma samples which were submitted to acid digestion
and reported a detection limit of 0.09 µmol/kg (18 µg/kg).
More than 250 water samples from lakes were analysed for thallium
(thallium-205) by ICP-MS after acidification with HNO3. The
detection limit was found to be 0.1 µg/litre; the recovery of spiked
analytes amounted to 112 ± 4% (Henshaw et al., 1989).
2.4.2.3 Other methods
Methods other than AAS and ICP-MS are summarized in Table 2.
Spectrophotometric determination with rhodamin B after liquid/liquid
extraction is a quick and easy method, but it is less sensitive and
has a high incidence of interference. The method is suitable for a
quick visual test, when a massive intoxication with thallium compounds
is suspected. Determinations down to 10 µg/litre are possible in
environmental matrices (Griepink et al., 1988).
Inductively coupled plasma - atomic emission spectrometry
(ICP-AES) is a rapid multi-element technique, but it does not provide
the detection limits required to measure thallium concentration in
uncontaminated samples. The NIOSH method for determining thallium in
air particulates has a detection limit of 17 µg/litre of leaching
solution (NIOSH, 1984).
Differential pulse anodic stripping voltametry (DPASV) is a
sensitive method for the quantitative determination of thallium in
water samples or urine. Voltametric methods also offer the advantage
of simultaneous determination of several metals from one solution.
The lower limit of detection for thallium(I) is 10-100 ng/litre
(Klahre et al., 1978; Vandenbalck & Patriarche, 1987; Griepink et al.,
1988).
Neutron activation analysis (NAA) is applicable for the
determination of thallium in various environmental samples, but it is
relatively slow and impractical for the routine analysis of large
numbers of samples. The detection limit is determined by the
irradiation time, neutron flux, the choice of a radiochemical
separation of the radio-isotope to remove interfering matrix
radio-isotopes and the measurement time. Levels down to the absolute
amount of ng of thallium can be determined (Schoer, 1984). This
method can therefore be used for the determination of low thallium
concentrations in biological samples. In bovine liver a detection
limit of 1.5 µg/kg was found after digestion, separation and
concentration procedures (Henke, 1991).
Thallium(I)-sensitive electrodes are not sensitive enough for
trace determinations, and high concentrations of alkali ions reduce
the selectivity. Sensitivity problems must also be considered for the
usual X-ray fluorimetry techniques. Other methods, like excitation
with charged particles and photon activation radiochemical isotope
dilution, are seldom used.
2.4.3 Quality control and quality assurance
Sample collection, analysis and data presentation should be
carried out according to a protocol which ensures adequate validation
of biological monitoring procedures (Vesterberg et al., 1993).
There is an urgent need for strict quality control and quality
assurance of the analytical data on thallium in clinical and
environmental samples. It is only when proof is given for the accuracy
of the published data that they become unequivocally useful to
establish critical concentrations and dose-response relationships in a
given population or ecosystem. General considerations of quality
control and quality assurance have been recommended by WHO (WHO, 1986;
Aitio, 1988).
To date, very few of the many studies on thallium have provided
the necessary evidence concerning the quality of the data throughout
the analytical procedure. The recognized way to control and ensure
this involves good laboratory practice (GLP), including intra- and
inter-laboratory analysis of materials with certified concentrations
of thallium. Such Certified Reference Materials (CRMs) should have
the same (or a similar) matrix as the sample to be analysed and be
certified for thallium concentrations (similar to those in the sample)
by an internationally recognized body. This implies suitable levels
for thallium in serum, whole blood, urine, faeces, animal tissues and
plants, as well as levels typical for exposed individuals, animal
studies or eco-systems (Cornelis, 1988).
Available reference materials with clinical and environmental
interest are listed in Table 4. This immediately reveals the very
poor picture for CRMs certified for thallium. Whole blood and serum
samples are totally lacking, while urine of exposed individuals is
handled by the BI CUM 2 and 3 products with assigned values for
thallium only. The BCR milk powders and the NBS liver samples carry a
reference value. Thallium has also been reported in some
environmental samples (fly ash, etc.) without being certified.
There appears to have been only one inter-laboratory survey on
thallium in two spiked urine samples (Geldmacher-von Malinckrodt et
al., 1984). The 35 participating laboratories used one of the three
routine methods, AAS, DPASV or photometry, after thallium extraction.
The samples were also analysed by IDMS (isotope dilution mass
spectrometry) and attributed reference values of 66.3 and 483 µg
thallium/litre, respectively. The evaluation of this inter-laboratory
survey revealed that about 70% of the laboratories met the goal.
2.4.4 Conclusions
There are several methods available for the determination of
thallium in biological and environmental samples. As routine methods
these are GFAAS (the most widely used), DPASV, ICP-MS and photometry.
They all require a very careful sample pretreatment and, in the case
of DPASV and photometry, perfect mineralization of the sample without
losses due to volatilization or adsorption onto the container walls.
The same remarks apply to the methods including a preconcentration
step. In the case of GFAAS and ICP-MS, direct analysis of the diluted
sample is feasible. It is strongly recommended that all analyses be
accompanied by a quality assurance programme. At present, it is
possible to determine thallium concentrations of about 0.1 µg/litre or
0.1 µg/kg.
Table 4. Reference materials for thallium determinations in biological and
environmental materialsa
Matrix Originb Code Thallium Remarks
concentration
Liver NBS SRM 1577 50 µg/kg lyophilized bovine liver
SRM 1577A 3 µg/kg lyophilized bovine liver
Milk BCR CRM 63 1.3 µg/kg natural skim milk powder
powder CRM 150 1.0 µg/kg spiked milk powder
CRM 151 0.8 µg/kg spiked milk powder
Urine BI CUM 2 93 ± 13 µg/litrec lyophilized synthetic urine
CUM 3 603 ± 78 µg/litrec lyophilized synthetic urine
City BCR BCR-CRM- 2850 µg/kg certified; error 6.7%
waste 176
Coal IRANT IRANT-ECO 14 000 µg/kg not certified
fly ash
Coal NIST NIST-SRM- 5700 µg/kg certified; error 3.5%
fly ash 1633a
Gas coal BCR BCR-CRM- 2200 µg/kg not certified
180
Steel IRANT IRANT-OK < 3000 µg/kg not certified
plant
flue dust
a According to Muramatsu & Parr (1985) and Cortes Toro et al. (1990)
b BCR: Measurement and Testing Programme, DG XII, BCR, Commission of the
European Union, Wetstraat 200, B-1049 Brussels, Belgium BI: Behring
Institute, PO box 140, D-3350 Marburg 1, Germany IRANT: Institute of
Radioecology and Applied Nuclear Techniques (CSSR) NBS (new name NIST):
Room B 311, Chemistry Building, National Institute for Standardization
and Testing, Gaithersburg, MD 20899, USA NIST: National Bureau of Standards
(USA)
c assigned values for a particular lot only
3. SOURCES OF HUMAN AND ENVIRONMENTAL EXPOSURE
3.1 Natural occurrence
Thallium is ubiquitous in nature, but occurs at low
concentrations (< 2 mg/kg) (section 5.1), especially in sulfide ores
of various heavy metals (zinc, copper, iron and lead) and in minerals
of potassium, caesium and rubidium (Micke et al., 1983; Kemper &
Bertram, 1984; Ohnesorge, 1985; Stokinger, 1987; Manzo & Sabbioni,
1988). Although the concentration of thallium is low, about 700 000
tonnes of thallium are contained in worldwide identified resources of
coal and 19 000 tonnes in zinc resources (US BM, 1989). There are
only a few areas with a naturally very high thallium concentration,
e.g., the Alsar in the Former Yugoslav Republic of Macedonia (Zyka,
1972). Minerals of thallium, e.g., lorandite (TlAsS2) and
crookesite ((Cu,Ag,Tl)2Se), with thallium concentrations of up to
60%, are rarely found and usually not used for production of thallium
(Micke et al., 1983; Kemper & Bertram, 1984; Briese et al., 1985;
Kazantzis, 1986).
3.2 Anthropogenic sources
3.2.1 Production levels and processes
Since thallium is used only in small amounts by industry,
worldwide production of pure thallium is low. In 1975 about 8 tonnes
were produced in Germany and 2 to 3 tonnes in the USA (Zitko, 1975a),
while in 1987 and 1988 worldwide production was about 17 tonnes (US
BM, 1992). In 1981 the production of thallium in the USA was
discontinued. Sources for the production of thallium are zinc, lead
and sometimes copper or iron smelters and sulfuric acid plants. Flue
dust in particular is used as a thallium source (Zitko, 1975a; Smith &
Carson, 1977; Micke et al., 1983; Briese et al., 1985). Procedures
for the separation of thallium from other metals depend on the
proportions of the different minerals and, therefore, vary
considerably between the different smelters (Sanderson, 1952; Smith &
Carson, 1977; Micke et al., 1983; Kemper & Bertram, 1984; Briese et
al., 1985).
3.2.2 Uses
Thallium(I) sulfate was once used in medicine to reduce sweating
and to cure various infections, e.g., venereal diseases, ringworm of
the scalp, typhus, tuberculosis and malaria, and as a depilatory
agent, which caused many intoxications (Munch, 1934b; Smith & Carson,
1977; Emsley, 1978; Briese & Nessler, 1985a). However, therapeutic
uses of thallium have been discontinued because of its toxicity.
Since 1920, thallium(I) sulfate has been used as a rodenticide, in
Europe chiefly against rats and in the USA chiefly against ground
squirrels (Howe, 1971; Smith & Carson, 1977). Formerly it was used as
an insecticide (against ants and cockroaches). However, thallium is
no longer on sale as a rodenticide in most industrial countries
(Bruère et al., 1990), but is still used in developing countries
because of its cheapness.
Other areas in which thallium is used (Howe, 1971; Smith &
Carson, 1977; Micke et al., 1983; Briese et al., 1985; Sharma et al.,
1986; Kazantzis, 1986; Manzo & Sabbioni, 1988; ATSDR, 1992) are as
follows:
a) low temperature thermometers (down to -59°C) made from a
mixture of mercury and thallium;
b) special glasses with a high resistance and a low melting
point, containing thallium and selenium;
c) mixed crystals for infrared instruments, composed of arsenic
or thallium(I) salts and halogenides (TlI-TlBr), and
Tl3VS4, Tl3NbS4, and Tl3PSe4 for acusto-optic
and laser equipment;
d) electronic devices, e.g. thallium(I) sulfide for
semiconductors and scintillation counters;
e) mercury lamps (addition of thallium(I) halogenids increases
the yield of light and changes its spectrum);
f) alloys with lead, zinc, silver and antimony enhance
resistance to corrosion;
g) catalysing organic reactions, e.g., oxidations of
hydrocarbons and olefins (thallium compounds are being
increasingly used for organic synthesis); patents summarized
by Smith & Carson (1977);
h) radioactive isotopes, used in physics for measurement of
exact time periods (thallium-205), in industry for measuring
the thickness of material (thallium-204), and in medicine
for scintigraphy of heart, liver, thyroid and testes, and
for the diagnosis of melanoma (thallium-201) (Rao et al.,
1983; Müller-Brand et al., 1984; Urbain et al., 1986);
i) other uses, e.g., in the production of imitation jewellery,
fireworks, pigments and dyes, the impregnation of wood and
leather against bacteria and fungi, and in mineralogical
analysis;
j) minor amounts of thallium are used in biochemistry, e.g.,
arylthallium(III) compounds for modification of proteins and
tRNA (Douglas et al., 1990).
Worldwide industrial consumption in 1991 was estimated to be 10
to 15 tonnes. Between 1940 and 1980 consumption in the USA varied
considerably between 0.5 and 11 tonnes/year (Schoer, 1984), and
between 1984 and 1988 it was 1.1-1.5 tonnes/year (US BM, 1985, 1989).
In the USA it is used mainly in the electrical and electronic
industries and the 650 kg used in 1983 in the German Democratic
Republic was mainly for making special glass (Smith & Carson, 1977;
Micke et al., 1983; Briese et al., 1985; Kazantzis, 1986; Kemper &
Bertram, 1991).
3.2.3 Emissions from industrial sources
There is an enormous difference between the amount of thallium
mobilized (released into air, water or disposed of on land) and the
thallium consumption of 12 tonnes/year (section 3.2.2). Worldwide a
total of 2000-5000 tonnes of thallium is estimated to be mobilized per
year, especially through the combustion of fossil fuels, refinement of
oil fractions, the smelting of ferrous and non-ferrous ores, and also
by some other industrial processes such as cement production (Gorbauch
et al., 1984; Ewers, 1988; Nriagu & Pacyna, 1988). Smith & Carson
(1977) estimated that about 15% (240 tonnes) of total mobilized
thallium is transferred annually to the atmosphere. However, only a
small fraction is released into the atmosphere or wastewater during
production processes or from waste materials (Table 5). Summarizing
estimations for the USA by Smith & Carson (1977), Schoer (1984)
emphasized that in the USA each year nearly 1000 tonnes of thallium
are released into the environment, of which 350 tonnes are emitted in
vapours and dusts, 60 tonnes bound to non-ferrous metals, and more
than 500 tonnes contained in fluid and solid wastes.
3.2.3.1 Metal production industries
It has been estimated that worldwide over 600 tonnes of thallium
are processed per year during the smelting of lead, copper and zinc
ores (Micke et al., 1983). Thallium emissions from smelters can vary
greatly from plant to plant, depending upon the thallium content of
the raw materials and the technology used. For this reason, and
because of the lack of recent emission data, global releases can be
only roughly quantified. On the basis of the data in Table 5, a total
of about 90 tonnes of thallium may be released each year into the
atmosphere from non-ferrous metal production operations in the USA,
Canada and Germany. Dust in one zinc smelter was reported to contain
380-3700 mg thallium/kg before and 60-9700 mg/kg after starting the
production of thallium (Briese et al., 1985). Although it is not
possible to estimate the losses of thallium from mineral waste
materials, releases from these materials are generally expected to be
small.
Table 5. Estimated emissions of thallium (tonnes/year) into the environment
Emission source USA Canada Germany Europe World
Coal combustion
into air 180a 7.5b 7c 54d
140e 4f 80e 600e
6g
into soil/water 170a
into total environment 240c
Coal combustion (into air)
from electric utilities 155-620h
from industry and domestic 495-990h
Ferroalloy production
using manganese ores
into air 140a
into soil/water 220a
Raw iron production and
related coal combustion
into air 6a 35g 30d
into total environment 205a
Production of nonferrous
metals
into air 38a 44i 11g
total emission 496a
Potash-derived fertilizers
into total environment 5a
Cement plants
into air 25g 2670-5340h
Brick works 28b
Table 5. (cont'd).
Emission source USA Canada Germany Europe World
Oil fuel combustion, mining
and processing of oil shales
into soil/water 8a
total emission 8a
Waste combustion < 1g
a Smith & Carson (1977) f Brumsack et al. (1984)
b Brumsack (1977) g Davids et al. (1980)
c Sabbioni et al. (1984b) h Nriagu & Pacyna (1988)
d Bowen (1979) i Kogan (1970)
e Schoer (1984)
Data from the USA (Smith & Carson, 1977) indicate that relatively
large amounts of thallium are present in waste materials from
non-ferrous metal (mainly copper) and iron and steel production
(Table 5). Although no precise data were available on thallium levels
in waste from ferroalloy production using manganese ores, Smith &
Carson (1977) suggested that emissions from this source could be
significant. Atmospheric releases resulting from the production of
iron and steel in the USA were estimated to be relatively small (about
5 tonnes from steelmaking and 1 tonne in iron blastfurnace gases). In
the main area of iron and steel production in Germany, annual thallium
emissions into air have been estimated to be about 0.8 tonnes (Ewers,
1988).
3.2.3.2 Power-generating plants
Power-generating plants represent a major source of thallium
emissions, especially those using some brown coal or coal of the
Jurassic period. Most coals contain only about 0.5 to 3 mg/kg, mainly
incorporated in sulfide inclusions. Some of these impurities can be
removed by washing and mechanical cleaning. It has been estimated
that about half of the thallium content of coal is emitted into the
atmosphere and represents the biggest anthropogenic source (Smith &
Carson, 1977) (Table 5). In such estimations, losses from collected
fly ash are not taken into consideration, because its use may vary.
Only a minor amount is used in cement making. If it is used as a soil
stabilizer, contami nation of the environment is much higher (Smith &
Carson, 1977).
Natusch et al. (1974) found that coal-fired power-generating
plants emitted about 700 µg thallium/m3 flue gases, resulting in a
local level of air emission of about 700 ng/m3. This would result
in an estimated daily absorbed amount of 4.9 µg airborne thallium per
person (US EPA, 1980). In the European Union, coal-fired
power-generating plants were estimated to have caused a total
mobilization of 240 tonnes of thallium during 1990, about one third of
this being concentrated in the smallest particles, and atmospheric
emissions of 7 tonnes (Sabbioni et al., 1984b).
In coal burners, thallium volatilizes and recondenses onto the
surface of ash particles in cooler parts of the system. As a result,
2 to 10 times higher concentrations of thallium may occur in the
fly-ash than was present in the coal (Galba, 1982). Fly-ash thallium
content is negatively correlated with particle size (Manzo & Sabbioni,
1988). Thus, thallium and other toxic trace elements are concentrated
in the smallest particles, which pass through conventional
power-generating plant filters, remain suspended in the atmosphere for
long periods and are respirable. For instance, particles with a
diameter of 1.1-2.1 µm contain 76 mg thallium/kg fly-ash, those with a
diameter of 2.1-7.3 µm contain 62-67 mg/kg and those with a diameter
of 7.3-11.3 and > 11.3 µm contain 40 and 29 mg/kg, respectively.
Particles with a diameter of less than 74 µm contain only 7 mg
thallium/kg (Natusch et al., 1974). These particles are highly toxic,
since thallium and other heavy metals are preferentially concentrated
on the particle surfaces and therefore are relatively bioavailable
(Linton et al., 1976; Natusch, 1982).
3.2.3.3 Brickworks and cement plants
Total thallium emissions from brickworks in Germany have been
estimated to be 28 tonnes/year. This compares with emissions of
7.5 tonnes/year from the burning of coal (Brumsack, 1977).
The emission potential of cement plants was not recognized until
1979. The first effects on vegetation around a cement plant in
Lengerich, Germany were observed in 1977 (Pielow, 1979; LIS, 1980),
but only the gradual hair-loss in a rabbit led to the suspicion that
thallium was the cause of the toxic effects (LIS, 1980; Brockhaus et
al., 1981b; Dolgner et al., 1983). The source of thallium was found
to be residues of pyrite roasting added as a ferric oxide additive to
powdered limestone in order to produce special qualities of cement and
the addition of the filter fly-dust (LIS, 1980). Studies at other
plants showed much lower emission levels, so that the emission at
Lengerich was caused by the exceptional circumstances. Production
alterations in Lengerich caused a reduction in the emissions of more
than 99% (Pielow, 1979; Prinz et al., 1979; LIS, 1980).
Like power-generating plants, cement plants emit thallium mainly
bound to particles with a diameter of 0.2-0.8 µm (LIS, 1980).
Thallium concentrations in fly-dust emitted by the cement plant in
Lengerich were about 2.5 mg/m3 air, of which nearly all was
water-soluble thallium(I) chloride. Whereas the filter efficiency was
99% with respect to cement dust, it was only 50% with respect to the
thallium-containing particles. As a result, about 140 to 200 g
thallium/hour was emitted (Pielow, 1979; Prinz et al., 1979;
Weisweiler et al., 1985). Changing the production process reduced the
thallium content to less than 25 µg/m3 (< 200 mg/kg dust). In
other cement plants the concentrations in the filter dust were reduced
from 3066 mg/kg to about 100 mg/kg, and after this reduction only 13%
of the thallium was soluble in water (LIS, 1980).
3.2.3.4 Sulfuric acid plants
The sulfuric acid plant which had been the source of the roasted
pyrite used in the cement plant in Lengerich used pyrite containing
about 400 mg thallium/kg. However, in the roasted pyrite about 7% of
the thallium was water-soluble. During production of sulfuric acid, a
100-fold enrichment of thallium was found (LIS, 1980). As a
consequence, increased levels of thallium were found in Duisburg,
Germany around the sulfuric acid plant but never such high
concentrations as around the cement plant (Gubernator et al., 1979).
4. ENVIRONMENTAL TRANSPORT, DISTRIBUTION AND TRANSFORMATION
4.1 Transport and distribution between media
4.1.1 Transport and distribution in air, water and soil
Near point sources such as coal-fired power-generating stations,
cement plants using pyrite and some metal smelting operations, the
major source of thallium in air is emission of fly-ash (section
5.1.1). Although data on the forms of thallium in these emissions are
limited, results of one study indicate that nearly all of the thallium
in fly dust from a cement plant (in Lengerich, Germany) was present as
soluble thallium(I) chloride (LIS, 1980). No data on the amounts or
forms of thallium transported from soil into air during the dry season
were identified.
Assuming that 4 × 1012 kg of crustal rocks weather each year,
Bowen (1979) calculated that 2.4 × 106 kg thallium/year become
available to plants. However, as Smith & Carson (1977) have noted,
thallium tends to be retained during rock weathering, and
concentrations in soils tend to be somewhat enriched in soils compared
to the original bedrock.
The fate of thallium added to soil in deposited fly-ash probably
depends greatly on soil type (Crössmann, 1984). Data from Smith &
Carson (1977) suggest that retention should be greatest in soils that
contain large amounts of clay, organic matter and iron/manganese
oxides. According to McCool (1933) significant amounts of thallium
can be removed from solution in soils by ion exchange. Thallium can
also be incorporated into stable humus complexes (Crössmann, 1984),
which are resistant to rapid "wash-out" (Schoer, 1984).
Results of studies in several areas indicate that thallium
deposited from the atmosphere tends to accumulate in the surface
layers of soils (Smith & Carson, 1977; Heinrichs & Mayer, 1977; LIS,
1980). For example, after prolonged emissions from a cement plant in
Germany (LIS, 1980), thallium was found to remain in the upper levels
of soil (Schoer, 1984); material from depths 0-10, 40-50 and 60-70 cm
contained 4.2, 1.3 and 0.1 mg thallium/kg, respectively (Schoer,
1984). Retention processes will, however, be less effective in acidic
soil. For example, results of studies by Heinrichs & Mayer (1977)
indicate that about 40% of the thallium deposited from the atmosphere
onto relatively uncontaminated acidic (pH = 3.9-4.3) forest soil in
Germany was leached from the top 50 cm to lower soil horizons.
Elevated concentrations of thallium in groundwater (up to 40 µg/litre)
and in an irrigation canal (up to 96 µg/litre) in China, near a site
where waste materials from the mining of mercuric ore and coal
containing 25 to 106 mg thallium/kg were deposited (Zhou & Liu, 1985),
indicate that under some circumstances significant amounts of thallium
can move from soil into local water.
Although there is little information on the forms of thallium in
natural water, most dissolved thallium in fresh water is expected to
be present as the monovalent Tl+ ion (Smith & Carson, 1977). In
strongly oxidizing fresh water and in most seawater (Sager & Tölg,
1984), however, trivalent thallium is probably the predominant
dissolved form. Both forms of thallium can be removed from solution
by exchange and complexing reactions with suspended solid phases.
Trivalent thallium is also susceptible to reduction and precipitation
processes. According to Cotton & Wilkinson (1988), trivalent thallium
is extensively hydrolysed to form the colloidal oxide over the pH
range of natural water. Depending upon the relative kinetics of
reduction and hydrolysis, precipitation of thallium(III) hydroxide may
be an effective mechanism for removing thallium from solution. When
thallium(III) (precipitated as the oxide or hydroxide) settles into
organic-rich anaerobic sediment, it will be reduced to the monovalent
form, which can in turn be fixed in the sediment by reaction with
sulfide to form insoluble Tl2S (US EPA, 1978). Thallium is thus
relatively depleted in seawater where thallium(III) predominates and
can be enriched in sediments where organic matter accumulates under
undisturbed, anaerobic conditions (Smith & Carson, 1977).
The partitioning of thallium among the water, sediment and biotic
compartments of aquatic systems has rarely been investigated. In one
study, however, in which thallium (100 µg/litre as thallium(I)
nitrate) was added to a 7-litre glass aquarium containing washed sea
sand, goldfish and submergent aquatic angiosperms, thallium was
distributed among all of the compartments. Concentrations in water
decreased gradually, while those in the fish and vegetation increased,
throughout the 9-day duration of the experiment, indicating that
thallium was being exchanged among these media (Wallwork-Barber et
al., 1985). Concentrations in the sand increased rapidly to a
relatively low value (0.05 mg thallium/kg), and remained relatively
stable thereafter, suggesting that there was little exchange between
the sediment and the other compartments. The limited accumulation of
thallium in the sediment was attributed in part to the short duration
of the study and to the absence of organic matter and clay in the
sand.
4.1.2 Soil-vegetation transfer
4.1.2.1 Factors affecting soil-vegetation transfer
In general, the solubility of thallium compounds governs the
availability of the metal to vegetation (discussed in detail by
Cataldo & Wildung, 1978). Crössmann (1984) mentioned that so far no
method had been developed to quantify the amount of thallium in soil
that is easily available for plants. However, Schoer & Nagel (1980)
emphasized the good correlation between soil-vegetation transfer and
the concentration determined following ammonium acetate extraction
from soil. Other authors favour an EDTA/ammonium acetate extraction
(Scholl & Metzger, 1982).
Transfer is influenced by various factors, e.g., pH (section
5.1.3.2) and the type of the contaminated soil. Green rape, bush
beans and rye grass were found to take up less thallium from weakly
acidic soil (pH 6.2) than from more acidic soil (pH 5.6), and thallium
supplied by cement factory dust was more available to plants than
thallium in soil (Makridis & Amberger, 1989a). Rape plants grown on
two samples of soil from a contaminated area, one sample (A)
containing a 3-fold higher concentration of thallium than the other,
showed identical concentrations of thallium, while other vegetables
grown on sample A even showed a lower thallium content. It was
concluded, that plant availability cannot be correlated to total soil
thallium content as determined after extraction with concentrated
nitric acid (Hoffmann et al., 1982). Only 4.4% (± 2.7%) of the
thallium content of soil from a lead-zinc mining waste material area
was available to vegetation, compared to 17.5% (± 10.7%) in soil from
a cement plant area (Schoer & Nagel, 1980). In a similar study with
soil from a cement plant and with stream sediments from a mining
district (Wiesloch, Germany), rape plants took up about 20% of soil
thallium from the cement plant sample but only 1.4 to 5.1% from the
stream sediments, although the latter contained 2- to 3-fold higher
thallium concentrations; 8- to 80-fold higher concentrations of
plant-available thallium were calculated for the soil from the cement
plant (Scholl & Metzger, 1982). Comparing the uptake of thallium by
rape seedlings from soil contaminated by emissions from a cement plant
(mainly with thallium(I) chloride or iodide) with that from
uncontaminated soil (traces of thallium(I) sulfide), a 7.5-fold higher
uptake from the contaminated soil was found (Lehn & Bopp, 1987).
At lower thallium concentrations, some plant species took up a
higher percentage of the available thallium than at higher
concentrations, perhaps in part because of the stronger toxic effects
at higher concentrations. However, the total amount of thallium found
in the plants and the thallium content of the artificial soil
solutions were correlated, reaching up to 1000 mg/kg dry weight in
green kale following one week's exposure to a concentration of
10 mg/litre (Schweiger & Hoffmann, 1983).
The transfer from soil to plant also depends on a number of
factors relating to the plant, e.g., root system, kinetics of membrane
transport, metabolism of thallium (Cataldo & Wildung, 1978), so that
the total amount of thallium taken up is species-specific (section
5.1.4.2). This is shown by the bioconcentration factor (concentration
of thallium in the plant (fresh or dry weight) in relation to its
concentration in dry soil) found for different plants grown in soil
contaminated by mining waste materials or collected from sites with
naturally high concentrations (Table 6) (Schoer & Nagel, 1980; Lehn &
Bopp, 1987). Calculations based on the concentrations in plant ash
and dry soil show that the concentration factor is usually less than
20 (Smith & Carson, 1977). The concentrations of thallium in
vegetables reported by these authors are one to two orders of
magnitude higher than those found by Geilmann et al. (1960) in
vegetation grown on uncontaminated soil (Schoer & Nagel, 1980)
(sections 5.1.4.1 and 5.1.4.2). Trees can be a long-term reservoir of
thallium. As a result of emission by cement plants, the bark and
lichens of several trees contained 2-23.8 mg thallium/kg dry weight.
The use of ground-up bark from these trees for mulching can lead to
considerable uptake of thallium by other plants (Arndt et al., 1987).
4.1.2.2 Absorption by plants
Uptake of thallium(I) ions occurs via all parts of the plant,
presumably by using the uptake mechanisms for potassium. However,
uptake of fly-dust by the leaves of sunflowers is minimal (Schweiger &
Hoffmann, 1983). Although the majority of the thallium-containing
particles have a diameter less than 2 µm, they cannot be absorbed by
transpiration through the stomata (Pallaghy, 1972; LIS, 1980). In
numerous laboratory studies using nutrient solutions, a positive
correlation between plant uptake and thallium concentration in the
solution has been demonstrated (e.g., Al-Attar et al., 1988).
Comparable results have been obtained from the cultivation of mycelium
of higher fungi in thallium-enriched agar medium (Seeger & Gross,
1981).
According to Cataldo & Wildung (1978), absorption of thallium by
plants seems to be under metabolic regulation, and potassium is a
non-competitive inhibitor. Sunflowers with a deficiency of potassium
and supplied with 1 or 10 mg thallium nitrate/litre possessed a 2 to 3
times higher concentration of thallium per gram dry weight than those
supplied with potassium (Schweiger & Hoffmann, 1983). Metabolically
controlled uptake seemed to occur only with thallium(I), supplied as
the acetate, while thallium(III), supplied as the chloride, was
presumably taken up by passive processes such as cation exchange
(Logan et al., 1983, 1984). Since increasing concentrations of
potassium decrease the uptake of thallium(I), this uptake was
postulated to be mediated by the (Na+/K+) ATPase system. During a
3-h exposure to a solution concentration of 5 mg/litre, excised barley
seed roots took up about 6009 (± 185) mg thallium(I)/kg dry weight and
only 870 (± 44) mg thallium(III)/kg. Thallium(III) ions were easily
desorbed, presumably because of a large extracellular component,
whereas the thallium(I) ions were unavailable for exchange. The
different uptake mechanisms are also reflected in the sensitivity of
thallium(I), but not of thallium(III), towards temperature and
metabolic inhibitors. Using whole plants (maize), the differences in
uptake could not be confirmed, but the authors suggested that, prior
to the uptake, thallium(III) may be reduced in the soil to thallium(I)
(Logan et al., 1984).
Table 6. Bioconcentration factor for plants grown on contaminated soils
Plant Bioconcentration factora Reference
Fresh weight Dry weight
Barley (Hordeum vulgare) 0.14 Lehn & Bopp (1987)
Cabbage species:
Green kale < 0.1 Schoer & Nagel (1980)
Brussels sprouts < 0.1 Schoer & Nagel (1980)
Celeriac (Apium graveolens) < 0.1 Schoer & Nagel (1980)
Cress (Lepidium sativum) 33 Lehn & Bopp (1987)
Cress (Lepidium sativum) 0.45-0.59 Schoer & Nagel (1980)
Horse-radish (Armoracia) 0.33 Schoer & Nagel (1980)
Maize (Zea mays) 0.05 Lehn & Bopp (1987)
Mushrooms 2.9 Schoer & Nagel (1980)
Mustard (Sinapis alba) 1.07 Lehn & Bopp (1987)
Parsley (Petroselinum 0.15-0.21 Schoer & Nagel (1980)
crispum lapathifolia)
Rape (Brassica napus) 66 Lehn & Bopp (1987)
Rape (Brassica napus) 0.26-0.29 Schoer & Nagel (1980)
Spinach 594 Maier-Reiter et al.
(1987)
Wheat (Triticum aestivum) 0.05 Lehn & Bopp (1987)
a Concentration of thallium in fresh or dry weight of the plant in relation
to its concentration in dry soil
4.1.2.3 Distribution in plants
Thallium distribution at the cellular level has been investigated
with rape grown both on uncontaminated soil and on soil spiked with
non-toxic amounts of thallium (Günther & Umland, 1989). At each test
concentration about 70% of the thallium was concentrated in the
cytosol (comparable to human data in section 6.6). In the exposed
plants nearly all the thallium was in the form of free thallium(I)
ions; no thallium(III) ions or dimethylthallium compounds were
detected. However, in all the rape grown on uncontaminated soil, the
cytosolic thallium was bound, probably to a peptide. This native
thallium-complexing agent lacked sulfur-containing amino acids and
could not be induced in rape by the application of thallium (Günther &
Umland, 1989).
In addition to its varied distribution at the subcellular level,
thallium distribution in green plants depends on the developmental
stage and the part of the plant. Only in mushrooms was no specific
distribution pattern found to exist (Seeger & Gross, 1981). Rape
seedlings grown on soil contaminated by a cement plant (1 to 3 mg
thallium/kg dry soil) contained 3 to 5 times higher concentrations of
thallium than full-grown plants. The concentrations in different parts
of full-grown rape (leaf, 47 mg/kg dry weight; shoot, 5.5 mg/kg; seed,
2.1 mg/kg) (Lehn & Bopp, 1987) indicate that thallium concentrations
are higher in the chlorophyll-containing regions, a fact also known
from plants grown on uncontaminated soils (Weinig & Zink, 1967). In
rape grown on artificially contaminated soil (1 mg thallium
nitrate/plant), yellowing leaves showed higher concentrations (up to
200 mg/kg dry weight) than green leaves, while the seeds contained
only about 1 to 2% of the concentration found in the yellow leaves.
However, in rape grown in the field near a cement plant, the leaves
contained up to 85 mg thallium/kg dry weight and the seeds about
20 mg/kg (Arndt et al., 1987).
Experimentally, thallium concentrations of 0.0001 to 2.5 mg/litre
substrate increased the concentration in the shoots of the grass
Lolium perenne from < 0.075 mg/kg dry weight to 144.05 mg/kg and in
the roots from 0.42 to 576 mg/kg (Al-Attar et al., 1988).
The distribution of thallium also varies in different vegetables.
For instance, in gardens around Lengerich, leaves of kohlrabi
contained a 350-fold higher concentration than the tubes, while in
other vegetables the differences in concentrations between leaves and
other parts ranged from 3 to 45 times (see Table 13) (Hoffmann et al.,
1982). In studies with bush beans and green rape, differences in
thallium accumulation in the plants were evident (Makridis & Amberger,
1989b): after incubation in a liquid culture medium (1 mg
thallium(III) trichloride/litre) for 10 days, roots and shoots of
beans contained 742 and 62 mg/kg and those of rape 57 and 244 mg/kg,
respectively. At higher concentrations the difference between roots
and leaves disappeared in both species, the concentration in the roots
of rape increasing more strongly than in the shoots, which, in part,
was an effect of reduced growth. Kaplan et al. (1990), using
thallium(I) sulfate (0.55 and 1 mg/litre), observed at least 4-fold
higher concentrations of thallium in the roots of soya beans than in
the pods or the lower or higher leaves.
These data indicate that plants which are more resistant to
thallium do not have a reduced uptake, but a reduced transport of
thallium to the leaves (section 9.3.1.6).
4.2 Biotransformation
Laboratory experiments indicate that organothallium derivates may
originate from the biomethylation processes of anaerobic bacteria in
lake sediments (Manzo & Sabbioni, 1988). However, according to Craig
(1980), there is no firm evidence for environmental methylation. The
methylation of thallium and other heavy metals is a vitamin
B12-(cobalamin-)dependent reaction (Hill et al., 1970; Agnes et al.,
1971). Due to its reduction potential, thallium(III) is methylated by
methylcobalamin (Ridley et al., 1977). Transfer of the methyl group
to thallium(III) seems to occur by electrophilic attack of the Co-C
bond (Wood et al., 1978; Wood, 1984, 1987).
Monovalent thallium seems to be simultaneously oxidized and
methylated by specific anaerobic microorganisms to methylthallium(III)
moieties which are stabilized by complexation (Huber et al., 1978).
Oxidation of thallium(I) ions to thallium(III) oxide in yeast
mitochondria (Lindegren, 1971; Lindegren & Lindegren, 1973b) confirms
an in vivo oxidation, but specific culture conditions are necessary
to obtain this detoxification phenomenon in which thallium oxide is
deposited between cell wall and plasma membrane.
4.3 Interaction with other physical, chemical, or biological factors
In the atmosphere, chemical reactions involving thallium are not
very likely to occur (Schoer, 1984).
5. ENVIRONMENTAL LEVELS AND HUMAN EXPOSURE
5.1 Environmental levels
Because of the limited industrial uses of thallium, emission on a
global scale resulting from the production and use of thallium
compounds is unlikely. However, thallium is present in relatively
large amounts in the raw materials used in various industrial
processes (e.g., smelting of sulfide ores, power generation using
coal, brick and cement manufacturing) (Table 5), which when released
can significantly increase environmental exposure to thallium on both
a local and regional scale.
5.1.1 Air
Bowen (1979) reported mean values of 0.06 ng particulate
thallium/m3 air for Europe and 0.22 ng/m3 for North America, and
Arnold (1986) a range of 0.1 to 30 ng/m3. The air of six large
American cities contained < 0.04 to 0.1 ng thallium/m3 (Ohnesorge,
1985). In a detailed study at Chadron, Nebraska, USA, Struempler
(1975) found yearly mean values for thallium of 0.22 ± 0.08 (range
0.07 to 0.48 ng/m3) and of 0.15 ± 0.04 ng/m3 during the summers of
1973 and 1974, respectively.
In industrial and urban areas of Genoa, Italy, the geometric mean
concentrations of thallium have been found to be 15 and 14 ng/m3
air, respectively, with maximal values of 58 ng/m3, but often values
were below 1 ng/m3 (Valerio et al., 1988, 1989). In London, levels
of 0.07 to 6 mg thallium/kg dust were measured (Bowen, 1979).
Air emission by thallium is mainly caused by mineral smelters,
power-generating plants and cement plants (ATSDR, 1992). Thallium
compounds are volatile at high temperatures and are not efficiently
retained by most emission control facilities. In consequence, large
amounts of thallium are released into the atmosphere if the raw
material (coal or ores) is not selected for a low thallium content.
In fly-ash from a power plant, only 0.08% of particles were
< 5 µm in diameter with a thallium concentration of 45 mg/kg ash
(Natusch et al., 1974).
5.1.2 Water
In the majority of reports, the authors did not specify whether
they determined dissolved and/or particulate thallium in samples of
water. This information was included if possible, according to the
methodology used in the study.
5.1.2.1 Areas not contaminated by thallium
Seawater contains < 0.01 to 0.02 µg/litre, and river water
< 0.01 to 1 µg/litre (Mason, 1966; Smith & Carson, 1977; Bowen, 1979;
Kemper & Bertram, 1984; Wachs, 1988).
In volcanic springs, low concentrations have been found
(0.25 µg/litre) (Arnold, 1986) and in three samples of hydrothermal
water it was below the detection limit of 0.6 µg/litre (Korkisch &
Steffan, 1979). Although, in these and the wastewater investigation,
dissolved and particulate thallium were not determined separately,
Henshaw et al. (1989) found concentrations of up to 0.41 µg
thallium/litre in filtered water from freshwater lakes. In three
wastewater treatment facilities in Massachusetts, USA, that had no
major industrial waste inputs, the thallium concentration in the
influent was below the detection limit of 5 µg/litre (Aulenbach et
al., 1987).
However, wastewater from oilfields (oil-well brines) in the USA
contained 12.9 to 672 µg thallium/litre, 5 out of 13 samples
containing > 400 µg thallium/litre (Korkisch & Steffan, 1979).
5.1.2.2 Areas contaminated by thallium from industrial sources
Data on thallium emission in water are available for areas with
oilfields, mineral industry and cement plants (Table 7).
Increased concentrations of thallium in well water and in water
from an irrigation canal in China resulted from old waste materials
from the mining of mercury and coal (Zhou & Liu, 1985) (section
5.1.3.2). Effluents from tailing ponds of base-metal mining
operations in New Brunswick, Canada contained 27 and 1620 µg dissolved
thallium/litre and up to 88 µg/litre was found in connecting rivers
(Zitko et al., 1975; Zitko, 1975b).
Raw wastewater from a pyrite ore mine at Lennestadt, Germany
(which was the source of the pyrite roasting residues used by the
cement plant in Lengerich, Germany) showed a thallium concentration of
160 µg/litre (LIS, 1980). Treatment in sedimentation ponds and with
lime and chlorine, reduced the concentration to 2-35 µg/litre. In a
stream, used as main drainage channel, the thallium concentration rose
from < 1 µg/litre (detection limit) to 1 µg/litre after the inlet.
This level is similar to that found in the River Rhine, Germany (0.5
and 2.5 µg/litre) (LIS, 1980).
Table 7. Concentrations of thallium in water from contaminated areasa
Locality Source Concentration of Reference
thallium (µg/litre)
Underground water, zinc smelter 13-820 BGA (1979)
Düsseldorf, Germany
Wastewater, different smeltersb < 0.1-2400 Smith & Carson
locations, USA (1977)
Tailing ponds, New mining 27; 1620 Zitko et al. (1975)
Brunswick, Canada
Rivers, different mining 21-30 US EPA (1980)
locations, USA
Rivers, New mining 1-88 Zitko et al. (1975)
Brunswick, Canada
River, mining < 1-1 LIS (1980)
Lennestadt, Germany
Wastewater, mining 2-160 LIS (1980)
Lennestadt, Germany
Well, China mining 17-40 Zhou & Liu (1985)
Irrigation canal, mining 6-96 Zhou & Liu (1985)
China
Wells, Lengerich cement plant < 1 LIS (1980)
Germany
Surface water, cement plant < 1-1 LIS (1980)
Lengerich, Germany
Water, cement plant 7.3 Mathys (1981)
Lengerich, Germany (distance 1 km)
Wastewater, cement plant < 1-37 LIS (1980)
Lengerich, Germany
Wells, Erwitte cement plant < 5-< 50 LIS (1980)
etc., Germany
Table 7 (contd).
Locality Source Concentration of Reference
thallium (µg/litre)
Surface water, cement plant < 50-130 LIS (1980)
Erwitte etc., Germany
Wastewater, cement plant 800 LIS (1980)
Erwitte etc., Germany (flue dust)
Wastewater, oil drilling 12.9-672 Korkisch & Steffan
USA (natural brines) (1979)
Wastewater, iron and steel mean = 60 MISA (1991a)
Canada plants
pulp and paper 230; MISA (1991b)
mills mean = 52
petroleum 310; MISA (1989)
refineries mean = 19
a Further literature summarized by Schoer (1984)
b Detailed data of sulfide mineral processors in Smith & Carson (1977)
Groundwater directly below a depot for pyrite roasting residues
in Duisburg, Germany contained 17 µg thallium/litre, and, at a
distance of some 100 m, up to 6 µg/litre (LIS, 1980).
In the vicinity of the cement plant in Lengerich, Germany (see
section 4.2.1.4) thallium levels were monitored in wells, rivers and
wastewater (LIS, 1980). In rivers, levels of 7.3 µg/litre 1 km from
the plant decreased to 1.8 µg/litre at a distance of 5 km and to
1.0 µg/litre at a distance of 10 km (Mathys, 1981). In all private
wells and water works, thallium concentrations were below the
detection limit of 1 µg/litre. At the purification plant of the
cement plant in Lengerich, the water from the inlet showed a
concentration of 17 µg/litre and that from the outlet of the rainwater
collection pond 37 µg/litre. A rainwater pond used for watering
cattle contained 3 µg thallium/litre.
Around other cement plants, thallium concentrations in private
wells, water works and surface water were below the detection limit
(< 5 and < 50 µg/litre). Pond water from the vicinity contained
130 µg/litre. In drip-water from the storage of flue dust, a
concentration of 800 µg/litre was determined (LIS, 1980).
The elimination of thallium from wastewater varies. Only 28% of
the thallium could be removed by conventional wastewater treatment
(liming) (Zitko et al., 1975) whereas 80 to 98% was removed in
Lennestadt (LIS, 1980). It has been suggested that Prussian Blue
could be used to eliminate thallium from wastewater (Rauws & Canton,
1976). Wool cannot be used as a filter to remove thallium from
contaminated water, since, in contrast to other metallic ions, only
minor amounts of thallium are adsorbed (Masri, 1976).
5.1.3 Rocks, soil and sediment
5.1.3.1 Areas not contaminated by thallium
Mean concentrations in the earth's crust range from 0.1 to
1.7 mg/kg. Higher values (up to 3 mg/kg) have been determined for
granite and shale; intermediate values for basalt, limestone,
sandstone and most coals, and lowest values for dunite (Table 8)
(Mason, 1966; Bowen, 1966, 1979; Brumsack, 1977; Smith & Carson, 1977;
Kemper & Bertram, 1984, 1991; Schoer, 1984; Arnold, 1986). Much
higher concentrations can occur in organic-rich shales such as the
Pierre Shale in the USA (25 mg/kg) and in coals of the Jurassic period
in Tadzhikistan (100 to 1000 mg/kg) (Smith & Carson, 1977).
Total thallium concentrations in soil typically range from 0.1 to
about 1.0 mg/kg (Geilmann et al., 1960; Bowen, 1966, 1979;
Chattopadhyay & Jervis, 1974; Brumsack, 1977; Smith & Carson, 1977;
Schoer, 1984; OMEE, 1994), but in China are around 0.011 mg/kg in
garden soil (range 0 to 0.02 mg/kg) (Zhou & Liu, 1985). Higher
concentrations (up to 5 mg total thallium/kg) have been reported,
however, in Poland (Staszyc et al., 1986), in soil on shale (Hoffmann
et al., 1982) and near some metallic ore deposits (Smith & Carson,
1977).
Marine sediments have been found to contain 0.95 mg thallium
per kg (Bowen, 1979) or, according to McLaren et al. (1987), using
isotope dilution inductively coupled plasma mass spectrometry, 0.6 to
0.7 mg/kg. Data summarized by Smith & Carson (1977) show a range of
0.14 to 1.13 mg/kg and in manganese nodules up to 614 mg/kg.
Table 8. Concentrations of thallium in uncontaminated geological samplesa
Source Concentration of Reference
thallium (mg/kg)
Mean Range
Basalt < 0.2-0.7 Smith & Carson (1977)
Basalt 0.08 Bowen (1979)
Basalt 0.02-0.06 Arnold (1986)
Clay 0.3 Smith & Carson (1977)
Clay 440-470b Smith & Carson (1977)
Clay 0.9 Bowen (1979)
Coal 0.38 < 0.2-1.4 Smith & Carson (1977)
Coal 0.2 0.01-2 Bowen (1979)
Coal 0.6 0.12-1.3 Gluskoter et al. (1977)
Brown coal (18% ash) 0.027 Brumsack et al. (1984)
Hard coal (8.7% ash) 0.51 Brumsack et al. (1984)
Hard coal (13.9% ash) 0.72 Brumsack et al. (1984)
Dunite 0.0005 Bowen (1979)
Granite 3.1 0.3-6.4 Smith & Carson (1977)
Granite 1.1 Bowen (1979)
Limestone 1.7 Smith & Carson (1977)
Limestone 0.14 Bowen (1979)
Limestone 0.1-0.9 Arnold (1986)
Sandstone 0.8 Smith & Carson (1977)
Sandstone 0.36 Bowen (1979)
Sandstone 0.05-0.4 Arnold (1986)
Shale (low in
organic carbon) 0.68 Brumsack et al. (1984)
Shale 1.2 Bowen (1979)
Shale 3.1 Smith & Carson (1977)
Black shale (rich
in organic carbon) 25 Smith & Carson (1977)
a Selected data; detailed data summarized in Smith & Carson (1977);
Bowen (1966, 1979); Schoer (1984)
b Very fine inclusions of plant matter
Uncontaminated sediments from lakes and small streams in various
parts of Canada typically contain about 0.35 mg thallium per kg (range
0.02 to 3.2 mg/kg) (G. Bonham-Carter, Geological Survey of Canada,
Applied Geochemistry Subdivision, personal communication to the IPCS),
with lowest values occurring in areas with underlying basaltic rock.
Thallium levels in the sediment of small streams in an uncontaminated
area of Münsterland, Germany contained from 0.03 to 0.1 mg/kg (LIS,
1980). In another investigation of small rivers in the same area and
in Sauerland, Germany, concentrations of 0.01 to 0.07 mg/kg dry weight
were determined (Mathys, 1981). This is also in the range of the data
summarized by Smith & Carson (1977).
5.1.3.2 Areas contaminated by thallium from industrial sources
Cases of contamination of sediment and soil by thallium are
mainly caused by mineral mining and smelters and by dust fall-out from
emissions of power-generating plants, brickworks and cement plants
(ATSDR, 1992) (Table 9).
Emissions from the cement plant in Lengerich, Germany caused a
remarkable increase in thallium concentrations in sediments of rivers
and brooks (Mathys, 1981). Sediment levels of 18 mg thallium/kg dry
weight found in a brook 1 km from the plant decreased to 8.7 mg/kg
within 4 km and then to 7.5 mg/kg in the River Glane into which the
brook flowed. Sediments of the following River Ems contained 5.0, 2.7
and 0.8 mg/kg at distances of 30, 70 and 100 km, respectively. In
comparison, river sediments from industrialized areas contained 0.05
to 1.8 mg/kg dry weight. Very high thallium levels were detected in
sediments from areas with zinc mining or iron ore industry, e.g.,
40.0 mg/kg in the River Lenne. After transport to the River Ruhr,
sediment thallium levels were 3 mg/kg dry weight (Mathys, 1981).
Large amounts of contaminated waste materials from the mining of
mercuric ore and coal containing 25 to 106 mg thallium per kg resulted
in chronic thallium poisoning in China. As a result of dispersal of
the waste materials, the garden soil of poisoned owners showed levels
from 28 to 61 mg/kg (mean: 43 mg/kg), whereas the soil levels in
gardens of unaffected families contained 6 to 11 mg/kg (mean,
8 mg/kg); this was still much higher than in other villages (mean,
0.011 mg/kg). In the affected village, the concentration of soluble
thallium salts decreased with increasing pH, with 8.0, 2.0 and
< 0.15 mg/kg at soil pH values of 1-2, 3-4 and 6-7, respectively.
The lower soil pH in the dry season (3.5-4.5 compared to pH 6-7 in the
rainy season) correlated with an increase in the number of
intoxications during these months, presumably due to an increase in
the thallium concentration in cabbage. After the experimental
addition of lime to contaminated soil, the thallium concentration in
cabbage was reduced (Zhou & Liu, 1985).
Table 9. Concentrations of thallium in soil from the vicinity of factories in Germany
Locality Source Distance Concentration of Reference
(m) thallium (mg/kg)
Göttingen lead-zinc -a 1.07 Brumsack
smelter (1977)
Duisburg copper smelter 500-1400 < 0.2-2.1 LIS (1980)
Leimen, Wiesloch mining and -a 5.5-21 Hoffmann
cement plant et al. (1982)
Lengerich cement plant 500-5000 < 0.1-6.9 LIS (1980)
Erwitte cement plant 350-1800 0.1-10.5 LIS (1980)
Schelklingenb cement plant < 3200 0.1-0.5 Arndt et
al. (1987)
Mergelstettenb cement plant < 800 0.1-0.5 Arndt et
al. (1987)
Duisburg sulfuric acid 0-1000 < 0.2-10.5 LIS (1980)
plant
Duisburg sulfuric acid 350-1100 < 0.2-2.3 LIS (1980)
plant
Göttingen brickwork -a 0.6 Brumsack
(1977)
a No data given;
b The investigation was performed 6 years after a ban on the use of iron pyrite
residues with high thallium contaminations. Owing to the method used (acid
digestion with concentrated nitric acid for 2 h at 90-95°C and addition of 10%
sulfuric acid), the concentrations measured correspond to the extractable soluble
emitted thallium and not the total thallium in the soil.
Sabbioni et al. (1984b) calculated the emissions from a
hypothetical coal-fired power-generating plant for a period of 40
years. They deduced an air-borne deposition of thallium around the
power-generating plants of 0.005 mg/kg, and the factor of increase
over the background level was estimated to be 0.001.
Around four small brickworks, samples of soil were digested by
strong acids and analysed for total concentration of thallium
(Brumsack, 1977). The proportion that was bioavailable is unknown.
Compared to a soil background level of 0.2 mg/kg, the contaminated
samples of soil showed a maximum accumulation factor of 3, while for
samples taken directly around the factory the factor was about 5.
Clear effects were found when the weather side of a hill was just
opposite the smoke stack. (Interestingly, shale from uncontaminated
areas showed a similarly high content (0.99 mg/kg)).
Thallium emission by the cement plant in Lengerich, Germany
caused an increase of thallium concentrations in the soil over an area
of 1 to 2 km radius from the plant, with a maximal level of 6.9 mg/kg
dry soil (LIS, 1980). Up to 4 mg/kg soil was determined in
agricultural soil and up to 6 mg/kg in the soil of house gardens
(Crössmann, 1984). Samples of soil taken at different depths always
showed highest thallium contaminations in the upper layers, decreasing
with increasing depth (LIS, 1980). Only small amounts of the thallium
in the upper layers were washed out (section 4.1) (Scholl & Metzger,
1982). The soil around the two plants that had produced the residues
from pyrite roasting was also highly contaminated, with maximal levels
of up to 10.5 and 2.3 mg/kg, respectively (LIS, 1980).
Soil from the vicinity of two other cement plants in Germany
contained only slightly elevated concentrations of thallium, up to
0.5 mg/kg soil, in the upper layers (see section 3.2.3.3 and 5.1.4.2)
(Table 9) (Arndt et al., 1987).
5.1.4 Plants and animals
Thallium occurs in low amounts in almost all living organisms,
including humans (Mason, 1966) (Tables 10 and 14). It seems to be a
non-essential cation in animals and plants (Yopp et al., 1974). Some
species accumulate this element.
5.1.4.1 Plants
a) Areas not contaminated by thallium
Usually thallium concentrations in plants are much less than
0.1 mg/kg dry weight (Geilmann et al., 1960) or 1 mg/kg ash (Dvornikov
et al., 1973, 1976), and levels exceeding 2 mg/kg ash are unusual
(Smith & Carson, 1977) (Table 10). However, such high thallium
concentrations have been found in plants from areas with a naturally
very high thallium concentration, e.g., the Alsar in Macedonia,
Yugoslavia (Zyka, 1972) (Table 11). Data from this area are
considered in sections 5.1.4.2 and 9.3.1.
No thallium could be detected in cabbage or grain from areas of
China not contaminated by thallium (Zhou & Liu, 1985). Plants used
for teas (e.g., Anisi, Betulae, Hibisci and Menthae) contained very
low concentrations of thallium (< 0.01 mg/kg), whereas higher
concentrations of other heavy metals and pesticides often occurred
(Ali & Blume, 1983). The majority (85%) of 421 investigated species
of wild mushrooms, which often accumulate heavy metals, contained
concentrations below the detection limit of < 0.25 mg/kg dry weight
(range < 0.25 to 5.5 mg/kg). A transfer factor of < 0.1
(concentration of thallium in the mushroom (fresh or dry weight) in
relation to its concentration in dry soil) indicates that no
accumulation took place (Seeger & Gross, 1981). However, in other
plants and using soil from a contaminated area, a much higher transfer
factor of 2.9 was determined (Schoer & Nagel, 1980). The thallophilic
Brassicaceae can contain higher amounts of thallium (1.5 mg/kg fresh
weight) than other plants, which usually contain 0.007 (detection
limit) to 0.1 mg/kg wet weight (Crössmann, 1984).
Wild plants normally contain only traces of thallium, whereas the
levels in garden plants can be increased by repeated use of sewage
sludge or potash fertilizers, which can contain 100 to 210 µg/kg
sludge or 15 to 310 µg/kg fertilizer (Geilmann et al., 1960;
Heinrichs, 1982). Also phosphate and copper fertilizers may contain up
to 400 µg thallium/kg (Boysen, 1992).
Thallium concentrations of up to 17 g/kg ash have been found in
plants from Alsar in Macedonia, Yugoslavia (Table 11), an area with
very high geogenic thallium levels (Zyka, 1972).
b) Areas contaminated by thallium from industrial sources
Soils contaminated through mineral smelters, power-generating
plants, brickworks or cement plants can greatly increase the
concentrations of thallium found in food of plant origin (Tables 12
and 13), which are the major route of entry of thallium into the food
chain. Data on bioconcentration factors are listed in Table 6. In
the contaminated area of Lengerich, Germany, consumption of home-grown
food was correlated with high levels of thallium in urine and hair,
and possibly with thallium-related health disorders among local people
(Brockhaus et al., 1981b; Dolgner et al., 1983). The importance of
these findings is underlined by the similarly elevated levels of
thallium found in the urine of family members consuming the home-grown
vegetables (Ewers, 1988).
Table 10. Concentrations of thallium in plants from uncontaminated areasa
Sourceb Concentration of thallium Reference
(µg/kg dry weight) (mg/kg ash)
Achillea millefolium 0.01-0.04 Dvornikov et al. (1973)
Achillea setacea 0.04-0.9 Dvornikov et al. (1976)
Alpine fir (L) 2-100 Shacklette et al. (1978)
Alpine fir (S) 2-70 Shacklette et al. (1978)
Anthemis tinctoria < 0.1-0.5 Dvornikov et al. (1976)
Artemisia absinthum 0.03 Dvornikov et al. (1973)
0.02-0.6 Dvornikov et al. (1976)
Artemisia campestris 0.057 Dvornikov et al. (1973)
0.04-0.8 Dvornikov et al. (1976)
Asperula humifusa 0.1-1.0 Dvornikov et al. (1976)
Clover 8-10 Geilmann et al. (1960)
Echium vulgare 0.1-0.3 Dvornikov et al. (1976)
Endive 80 Geilmann et al. (1960)
Engelmann's spruce (L) 2-10 Shacklette et al. (1978)
Engelmann's spruce (S) 15 Shacklette et al. (1978)
Euphorbia virgata 0.022-0.027 Dvornikov et al. (1973)
0.03-0.3 Dvornikov et al. (1976)
Festuca sulcata 0.2 Dvornikov et al. (1973)
0.2-0.6 Dvornikov et al. (1976)
Green cabbage 125 Geilmann et al. (1960)
Hay 20-25 Geilmann et al. (1960)
Head-lettuce 21 Geilmann et al. (1960)
Herbaceous vegetables 30-300 Bowen (1979)
Kale 150 Bowen (1979)
Leek 75 Geilmann et al. (1960)
Limber pine (L) 2-5 Shacklette et al. (1978)
Limber pine (S) 3-5 Shacklette et al. (1978)
Lodgepole pine (L) 2-5 Shacklette et al. (1978)
Lodgepole pine (S) 3-7 Shacklette et al. (1978)
Mushrooms < 0.25-5.5 Seeger & Gross (1981)
Myrtle blueberry (L, S) 2-7 Shacklette et al. (1978)
Ponderosa pine (S) 15 Shacklette et al. (1978)
Potato (L, S) 25-30 Geilmann et al. (1960)
(T) 5 Geilmann et al. (1960)
Rape (L) 25-30 Geilmann et al. (1960)
Red cabbage 40 Geilmann et al. (1960)
Table 10 (contd).
Sourceb Concentration of thallium Reference
(µg/kg dry weight) (mg/kg ash)
Salvia nemorosa 0.04 Dvornikov et al. (1973)
0.04-0.8 Dvornikov et al. (1976)
Stinging nettle (L) 28.8 Weinig & Zink (1967)
Tanacetum vulgare 0.06-0.2 Dvornikov et al. (1976)
Tobacco (L) 24-100 Geilmann et al. (1960)
Verbascum ovalifolium 0.01-0.7 Dvornikov et al. (1976)
Woody gymnosperms 50 Bowen (1979)
a Further data summarized by Dvornikov et al. (1973, 1976), Gough et al. (1979) and
Smith & Carson (1977)
b L = leaves, needles; S = stems; T = tubers
Table 11. Concentrations of thallium in plants from the Alsar region in Yugoslavia
possessing a high natural concentration of thallium in the soil
Source Concentration of thallium
(mg/kg ash weight)
Campanula sp. (L, S, F) 5990
Centaurea sp. (P) 75
Centaurea sp. (L, S) 105
Dianthus sp. (F) 5200
Echinops sp. (L) 15
Eryngium sp. (L) 3
Eryngium sp. (F) 10
Galium sp. (F) 17 000
Lavatera sp. (L, S) 125
Lavatera sp. (F) 45
Linaria triphylla 3000
Linaria triphylla 3800
According to Zyka (1972)
F = flowers; L = leaves, needles; P = pods and seeds; S = stems
Waste materials from the mining of mercuric ore and coal in China
(section 5.1.3.2) increased the concentration of thallium in cabbage
and grain. Cabbage from gardens of affected families contained
42 mg/kg fresh weight (range 39 to 49), whereas cabbage eaten by
healthy families contained 5.6 mg/kg (range 3 to 11). Other vegetables
from the gardens of affected families usually contained less than
10 mg/kg (Zhou & Liu, 1985).
An accumulation in vegetables of the genus Brassica was also
observed in Lengerich. In the area with the highest contamination, the
majority of plants and fruits contained < 0.1 to 0.4 mg/kg fresh
weight. Higher thallium levels were sometimes found in strawberries,
potatoes, beans, tomatoes, carrots and leeks, while in parsley,
celery, red currants, and all Brassicaceae high levels were usual
(LIS, 1980). Within this genus uptake of thallium varied: the
bioconcentration factor of white and red cabbage was relatively low;
it was 5- to 10-fold higher in the stems of kohlrabi. Savoy cabbage
and green kale were found to contain the highest thallium
concentrations, exceeding those of the soil (Crössmann, 1984). The
maximal value of 45.2 mg/kg fresh weight was found in green kale (LIS,
1980). Most forage plants, e.g., turnips, hay, grass and fodder corn,
contained < 5 mg/kg dry weight, but 46% of rape plants contained >
100 mg/kg (up to 1095 mg/kg dry weight) and 22% of the maize 10 to
50 mg/kg (LIS, 1980). Vegetables from Lengerich, grown in soil with
4.5 mg thallium/kg dry weight, could be classified according to their
mean thallium concentration (mg/kg fresh weight) into five groups.
These were I: green cabbage (22.6 mg/kg) and savoy cabbage
(8.5 mg/kg); II: turnip, broccoli, kohlrabi and white cabbage
(3.1 mg/kg); III: stock beet and other vegetables (1.4 mg/kg); IV: red
beet, rhubarb and spinach (0.7 mg/kg); V: the majority of fruits and
vegetables (0.5 mg/kg), e.g., red cabbage, Brussels sprouts, onion,
salad, carrot, bean, tomato, cucumber and potato (Scholl & Metzger,
1982).
The accumulating capacity of rape also became evident in an
investigation at cement factories in Schelklingen and Mergelstetten,
Germany, 6 years after the use of the same iron pyrite residues that
had been used in Lengerich was banned. The soil contained only
slightly elevated concentrations of thallium (section 5.1.3.2), and in
the majority of the plants, four of them Brassicaceae, no thallium was
detectable (Arndt et al., 1987). However, rape contained increased
levels of 2.4 to 679.6 mg thallium/kg dry weight at Mergelstetten and
1.8 to 19.1 mg/kg dry weight at Schelklingen (Table 12). The highest
levels detected in single rape plants were found within an area
extending 150-400 m downwind from the cement plant. The majority of
the rape grown in that area contained more than 5 mg thallium/kg dry
weight and could not be used as animal feed.
Table 12. Concentrations in plants from thallium-contaminated areas
Organism Concentration of thallium (mg/kg) Source of Localitya Reference
emission
Dry weight Fresh weight
Algae 9.5-43.4 mining New Brunswick Zitko et al. (1975)
Algae 0.665 cement plant Lengerich LIS (1980)
Berula 100.3 cement plant Lengerich Mathys (1981)
Berula 0.585; 0.654 cement plant Lengerich LIS (1980)
Caltha 187.3 cement plant Lengerich Mathys (1981)
Elodea 87.4 cement plant Lengerich Mathys (1981)
Elodea 0.29; 6.5 cement plant Lengerich LIS (1980)
Grass 52.0 sulfuric acid plant Duisburg LIS (1980)
Moss 125; 162 mining New Brunswick Zitko et al. (1975)
Rape 29.2 cement plant Lengerich LIS (1980)
Rape 23.7 cement plant Lengerich Kemper & Bertram (1984)
Rape 1095 cement plant Lengerich LIS (1980)
Rape 679.6 cement plant Mergelstetten Arndt et al. (1987)
Rape 19.1 cement plant Schelklingen Arndt et al. (1987)
Sparganium 0.265 cement plant Lengerich LIS (1980)
a All the localities are in Germany, except for New Brunswick (Canada)
Table 13. Concentrations in vegetables and fruits from thallium-contaminated areas
Plant Part Concentration of thallium (mg/kg)a Source of Reference
emission
Dry weight Fresh weight
Apple fruit 0.2 cement plant LIS (1980)
Bean fruit 0.7 cement plant LIS (1980)
Blackberry fruit 0.5 cement plant LIS (1980)
Black-currant fruit 0.527 cement plant Kemper & Bertram (1984)
Brussels sprout leaf 0.5 cement plant LIS (1980)
Carrot root 1.0 cement plant LIS (1980)
Carrot leaf 0.30 mining and Hoffmann et al. (1982)
root 0.10 cement plant
Celeriac stem 0.8 cement plant LIS (1980)
Cucumber leaf 0.70 mining and Hoffmann et al. (1982)
fruit 0.10 cement plant
Green cabbage leaf 14.9 cement plant Kemper & Bertram (1984)
Green cabbage leaf 45.2 cement plant LIS (1980)
Green cabbage leaf 22.6b cement plant Scholl & Metzger (1982)
Kohlrabi leaf 35.00 mining and Hoffmann et al. (1982)
stem 0.10 cement plant
Kohlrabi stem 3.1b cement plant Scholl & Metzger (1982)
Kohlrabi stem 4.9 cement plant LIS (1980)
Onion stalk 0.10 mining and Hoffmann et al. (1982)
bulb 0.01 cement plant
Onion stalk 0.4 cement plant LIS (1980)
Parsley leaf 1.2 cement plant LIS (1980)
Pear fruit 0.5 cement plant LIS (1980)
Table 13 (contd).
Plant Part Concentration of thallium (mg/kg)a Source of Reference
emission
Dry weight Fresh weight
Potato tuber 0.8 cement plant LIS (1980)
Radish leaf 5.90 mining and Hoffmann et al. (1982)
root 0.40 cement plant
Red beet leaf 2.40 mining and Hoffmann et al. (1982)
root 0.60 cement plant
Red beet root 0.7 cement plant LIS (1980)
Red-currant fruit 1.1 cement plant LIS (1980)
Savoy cabbage leaf 8.5b cement plant Scholl & Metzger (1982)
Strawberry fruit 0.9 cement plant LIS (1980)
Tomato fruit 0.6 cement plant LIS (1980)
White cabbage leaf 3.1b cement plant Scholl & Metzger (1982)
Zucchini leaf 0.90 mining and Hoffmann et al. (1982)
stem 0.02 cement plant
a Individual value, unless otherwise stated
b Mean value
5.1.4.2 Animals
a) Areas not contaminated by thallium
Investigations of three species of freshwater fish, the
omnivorous white sucker (Catostomus commersoni) and the more
carnivorous yellow perch (Perca flavescens) and brook trout
(Salvelinus fontinalis), show them to have similar average
concentrations of thallium in their axial muscle (< 0.07 to 3.0 mg/kg
dry weight), which were independent of water pH (Heit, 1985)
(Table 14). Extensive studies of different marine shellfish and fish
revealed average concentrations of 0.14 mg/kg; only in three species
(occasionally Clupanodon punctatus and Trachurus japonicus and
often Penaeus japonicus) were concentrations above 1 mg/kg found
(Hamaguchi, 1960).
In marine invertebrates concentrations were even lower, but,
owing to the low concentration in the seawater, the concentration
factor (concentration in the organism divided by the concentration in
the seawater) calculated by Smith & Carson (1977) was > 700.
Thallium concentrations in marine mammals have rarely been
investigated. In the blubber, liver, kidney, spleen and muscle of
bowhead whales, concentrations are nearly always below 0.01 mg/kg
fresh weight (Byrne et al., 1985).
Meat from farm animals contains very low levels of thallium
(Table 14).
b) Areas contaminated by thallium from industrial sources
Different animals as well as different organs vary with respect
to their accumulation capacity for thallium (Table 15).
In a case of fish poisoning, three species were found to contain
77 to 96 mg/kg muscle (Palermo et al., 1983). The liver and kidneys
of fish from a pond contaminated by a cement plant contained 1.6 and
1.3 mg/kg fresh weight, respectively (LIS, 1980). In the same area,
fish from other ponds and waters usually contained < 0.1 mg/kg
muscle.
Table 14. Concentrations of thallium in animals from uncontaminated areas
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Invertebrates
Colorado beetle whole 1 18 Geilmann et al. (1960)
Different marine invertebrates 0.001-0.03 Noddack & Noddack (1939)
Echinoderms (hard parts) 110 Bowen (1979)
Molluscs (soft parts) 340 Bowen (1979)
Fish
Various fish species 80 Bowen (1979)
Various marine 139 < 2930 Hamaguchi (1960)
shellfish and fish
Brook trout muscle 5 < 3.0 Heit (1985)
(Salvelinus fontinalis)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
White sucker muscle 28 < 2.0 Heit (1985)
(Catostomus commersoni)
Yellow perch muscle 27 < 3.0 Heit (1985)
(Perca flavescens)
Birds
Ducka kidney 15 0.03 Holm et al. (1987)
Duckb kidney 10 0.129 Holm et al. (1987)
Ducka liver 15 0.022 Holm et al. (1987)
Duckb liver 10 0.207 Holm et al. (1987)
Hen liver 2 < 50 LIS (1980)
Hen muscle 2 < 50 LIS (1980)
Mammals
Cattle hair 1 20 Geilmann et al. (1960)
Cattle hoof 1 16 Geilmann et al. (1960)
Cattle horn 1 10 Geilmann et al. (1960)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Fox intestine 25 < 2.7 Munch et al. (1974)
Fox kidney 27 0.01-1.5 Munch et al. (1974)
Fox liver 27 0.01-1.6 Munch et al. (1974)
Goat hair 1 7 Geilmann et al. (1960)
Goat hoof 1 9 Geilmann et al. (1960)
Hare hair 1 17 Geilmann et al. (1960)
Horse hair 1 7 Geilmann et al. (1960)
Horse hoof 1 4 Geilmann et al. (1960)
Marten brain 7 < 0.1-0.7 Clausen & Karlog (1974)
Marten intestine 13 < 0.01-0.57 Clausen & Karlog (1974)
Marten kidney 17 < 0.01-3.5 Clausen & Karlog (1974)
Marten liver 36 < 0.01-1.4 Clausen & Karlog (1974)
Pig hair 1 9 Geilmann et al. (1960)
Pig hoof 1 11 Geilmann et al. (1960)
Pig muscle 1 0.028 Kemper & Bertram (1984)
Pig muscle 43 < 70 Konermann et al. (1982)
Pig kidney 43 < 70 Konermann et al. (1982)
Table 14 (contd).
Source Part Number of Concentration of thallium Reference
measurements
µg/kg wet mg/kg dry
weight weight
Pig kidney 6 < 50 LIS (1980)
Pig liver 43 < 70 Konermann et al. (1982)
Pig liver 6 < 50 LIS (1980)
Rabbit hair 1 60 Geilmann et al. (1960)
Rabbit hair 1 < 1.5 LIS (1980)
Roe deer liver 19 approx. 30 Holm et al. (1987)
Roe deer kidney 19 approx. 30 Holm et al. (1987)
Sheep hair 1 9 Geilmann et al. (1960)
Sheep hoof 1 12 Geilmann et al. (1960)
Sheep kidney 3 50-60 Hapke et al. (1980)
Sheep liver 3 < 50 Hapke et al. (1980)
Sheep muscle 3 50-60 Hapke et al. (1980)
a Cuxhaven, Germany (coast)
b Cuxhaven, Germany (inland)
In farm animals intake of thallium mainly occurs through
contaminated feed. In broilers and laying hens, tissue thallium
concentrations were linearly correlated with feed levels for
concentrations between 2 to 40 mg/kg fresh weight of feed (Ueberschär
et al., 1986). The accumulation factor (concentration of thallium in
the tissue in relation to its concentration in the feed) was 2 to 3
times higher for the tissues of broilers than for those of hens (Table
16). In contrast to the situation in sheep, cattle and pigs, thallium
accumulates in hens to a greater degree in the muscle than in the
liver. The concentration in the kidneys is about 90% lower than in
the egg shell. Thallium half-life is about 2 to 4 days for the
various hen tissues (Ueberschär et al., 1986).
Whereas maximal permitted concentrations of lead, mercury,
arsenic and fluoride in animal fodder have been established in
Germany, this has not been done for thallium (Crössmann, 1985).
Thallium poisoning in cattle has been caused by silage (41 mg
thallium/kg fresh weight) bought from a farm in a contaminated area
(Frerking et al., 1990). Thallium mainly accumulates in the kidneys,
liver and bones (section 6.2). Steers fed for at least 6 months with
fodder originating from the thallium-contaminated area around
Lengerich, Germany, containing about 1.25 mg/kg dry weight (daily
uptake: 0.025 mg/kg body weight), contained 0.10 ± 0.02, 1.66 ± 0.55,
0.52 ± 0.28 and 0.40 ± 0.15 mg thallium/kg fresh weight, respectively,
in muscles, kidneys, livers and testes (Hapke et al., 1980). In pigs
fed with 1.45 mg thallium/kg food (dry weight) for 5 months, muscle,
kidneys and liver contained 0.18 ± 0.04, 0.44 ± 0.06 and 0.31 ±
0.09 mg thallium/kg fresh weight, respectively. Feeding with 2.71 mg
thallium/kg dry weight resulted in 0.39 ± 0.07 (muscle), 0.7 ± 0.2
(kidney) and 0.53 ± 0.1 (liver) mg thallium/kg fresh weight. Since
0.5 mg/kg fresh weight is the limit set by the federal state of North
Rhine-Westphalia for thallium concentrations in human food, a critical
level for pigs seems to be a daily intake corresponding to 1.9 mg
thallium/kg dry matter of food (Konermann et al., 1982).
Exposure of farm animals to thallium in the vicinity of the
cement plant in Lengerich, Germany, resulted in increased thallium
levels in the liver and kidneys of various animals (LIS, 1980): 0.8%
of the samples of internal organs contained > 10 mg/kg fresh weight,
1.3% contained 5 to 10 mg/kg, 12.6% contained 1 to 5 mg/kg and 14.5%
contained 0.5 to 1 mg/kg. In 0.2%, 3% and 4.4% of meat from various
farm animals, 5-10, 1-5 and 0.5-1 mg/kg were found, respectively.
Concentrations above 0.5 mg/kg fresh weight were sometimes also found
in eggs and chicken meat (up to 0.8 mg/kg), rabbit meat (up to
5.8 mg/kg) and roe deer (1.6 mg/kg) (Table 15) (LIS, 1980). In whole
eggs with a concentration of 1.26 mg/kg fresh weight, the
concentration in albumin and yolk was 0.394 mg/kg, while in the shell
it was 4.94 mg/kg (Kemper & Bertram, 1984).
Table 15. Concentrations in animals from thallium-contaminated areas
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Fish
Morone muscle -d Taranto, Italy - 77 Palermo et al.
labraxc (1983)
Eelc muscle - Taranto, Italy - 96 Palermo et al.
(1983)
Salmon muscle mining New Brunswick, 3-4 5.1e; 14.6f Zitko et al.
liver Canada 3-4 6.8; 23.5 (1975)
gill 3-4 1.2; 30.0
Salmon muscle mining New Brunswick, 3-4 3.6-34g Zitko et al.
liver Canada 3-4 5.7-46 (1975)
gill 3-4 7-89
Silver-scaled liver cement plant Lengerich, Germany 1 1.6 LIS (1980)
fish kidney 1 1.3
brain 1 0.46
Mugil muscle - Taranto, Italy - 84 Palermo et al.
cephalusc (1983)
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Trout liver cement plant Lengerich, Germany 4 0.13 LIS (1980)
kidney 4 0.885
muscle 3 0.09
Birds
Duck liver industry Harburg, Germany 30 0.191 < 0.075-0.86 Holm et al.
Duck kidney industry Harburg, Germany 30 0.076 < 0.075-0.43 (1987)
Duck liver industry Stade, Germany 24 0.072 Holm
Duck liver industry Stade, Germany 10 0.186 et al.
Duck kidney industry Stade, Germany 10 0.042 (1987)
Duck liver cement plant Lengerich, Germany 4 0.4 LIS (1980)
muscle 4 0.4
Geesec muscle poison USA 17 29 4-57 Shaw (1932)
Hen egg cement plant Lengerich, Germany 24 1.26 Kemper &
Bertram (1984)
Hen egg cement plant Lengerich, Germany 26 1.6 LIS (1980)
liver 17 0.8
muscle 26 0.8
heart 5 0.7
stomach 9 0.9
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Pigeon liver cement plant Lengerich, Germany 1 0.6 LIS (1980)
kidney 3 0.6
muscle 5 0.4
heart 1 0.3
Mammals
Cattle kidney cement plant Lengerich, Germany 58 2.2 LIS (1980)
muscle 61 1.5
Cattle kidney not specified Germany 2 24.0 Frerking et al.
liver (presumably 2 2.3 (1990)
muscle cement plant) 1 0.4
urine 3 1.35h
Foxc liver poison Denmark 27 64.0 Munch et al.
kidney 16 34 (1974)
intestine 7 55
Martenc liver poison Denmark 15 57 Clausen &
kidney 16 92 Karlog (1974)
intestine 9 42
brain 5 8.2
Table 15 (contd).
Animal Organ Source Locality na Concentration of thallium Reference
(mg/kg fresh weight)b
Mean Range Highest value
Pig liver cement plant Lengerich, Germany 3 1.2 LIS (1980)
kidney 296 1.3
muscle 300 0.6
Rabbit liver cement plant Lengerich, Germany 49 5.8 LIS (1980)
kidney 44 29.0
Roe deer liver cement plant Lengerich, Germany 1 2.6 LIS (1980)
kidney 3 14.0
muscle 2 1.6
heart 3 2.9
Sheep liver cement plant Lengerich, Germany 4 0.6 LIS (1980)
kidney 10 1.1
muscle 14 1.1
a Number of measurements (animals) e,f The two thallium concentrations of 45e and 100f µg/litre water are in the range
b mg/kg fresh weight unless otherwise stated of the natural concentrations at that locality; thallium concentration in the
c Fatal poisoning gill was higher (25.6 mg/kg fresh weight) at a lower concentration in water
d No data given (17.9 µg/litre)
g Lethal dose of 100 to 10 000 µg/litre
h mg/litre
5.2 General population exposure
The US Environmental Protection Agency calculated the typical
value for the exposure of the general population to be 0.48 ng
thallium/m3 air (US EPA, 1980). US EPA (1980) calculated an absorbed
amount of 3.4 ng/day assuming an inspired volume of 20 m3/day and
35% deposition in the lungs. BGA (1979) calculated the daily uptake
via the respiratory system to be < 5 ng thallium per day.
Table 16. Bioconcentration factora of thallium in broilers and
laying hensb and half-life in the tissues of laying hens
Bioconcentration factor
Organ Broiler Laying hen Half-life
(days)
Bone 0.54 0.26 2.0
Egg yolk - 0.26 4.1
Egg albumen - 0.14 1.6
Egg shell - 3.72 2.5
Fat 0.006 0.001 -
Feather 0.074 0.006 -
Kidney 0.77 0.38 3.6
Liver 0.19c 0.1 4.0
0.11d
Muscle 0.46 0.18 3.8
Skin 0.2 0.08 -
a Concentration of thallium in the respective tissue divided by
the concentration of thallium in the food
b From: Ueberschär et al. (1986)
c After 3 weeks
d After 6 weeks
More than 99% of samples of drinking-water in the USA contained
no thallium (detection limit, 0.3 µg/litre), and the positive samples
contained about 0.89 µg/litre. With a water consumption of
2 litre/day, this would result in an intake of < 1 µg thallium/day
for most adults (US EPA, 1980). The thallium concentration in 17
bottled mineral waters ranged between < 0.6 and 3.5 µg/litre, but
only 4 contained > 2 µg/litre (Korkisch & Steffan, 1979).
In the United Kingdom, Sherlock & Smart (1986) reported the total
dietary intake of thallium, based on the analysis of 13 diets. Four
(meat, fish, fats and green vegetables) out of nine food groups
contained samples with concentrations of thallium above the limit of
determination, ranging from 10 to 50 µg/kg fresh weight depending on
the food commodity. The average dietary intake of thallium for adults
was estimated to be 0.005 mg/day with a range of 0-0.01 mg/day,
assuming that concentrations less than the limit of determination were
equal to zero. The daily intake of thallium from vegetables alone is
estimated to be about 3.8 µg for an average adult in the USA (US EPA,
1980). Food of plant origin often contains more thallium than food of
animal origin, a 4-fold higher concentration of thallium being
eliminated in the urine of vegetarians than in that of humans eating
food of varying origin (Ohnesorge, 1985).
A minor route of thallium uptake can be sodium-free dietetic salt
(KCl), which contains up to 420 µg thallium/kg salt (Toots & Parker,
1977), but not sodium chloride, which only contains 0.08 µg/kg
(Geilmann et al., 1960). Wine has also been found to contain small
amounts of thallium (0.056 to 0.684 µg/litre) (Geilmann et al., 1960).
5.3 Occupational exposure during manufacture, formulation or use
Few data on occupational exposure to thallium are available. An
industrial plant in the USA used concentrated thallium salt solutions
for separations by centrifugation. Considerable variations in the
thallium content of the air occurred during the day, depending on the
emission potential of the different steps of the procedure (Hill &
Murphy, 1959), although no exact data were reported. In a plant in
the United Kingdom manufacturing special alloy anodes for use in
magnesium seawater batteries, air samples from two working areas
contained a maximum level of 0.022 mg thallium/m3 and 0.014 mg/m3
(Marcus, 1985). Detailed data are available from a Russian plant
producing thallium before and after changes in processes. The air
concentration varied from 0.12 to 0.18 mg thallium/m3, but peak
concentrations of 13.5 to 17.4 mg/m3 during the smelting process
were observed. During dissolving and packing of thallium salts, the
air thallium concentrations were 0.117 and 0.274 mg/m3, respectively.
After changes in the smelting process were instituted, the air
thallium content decreased to 0.0036-0.0072 mg/m3 (Tikhova, 1964).
Floating dust in a German thallium smelter contained 60 to 9700 mg
thallium/kg dust; the air-suspended dust concentration was 6-50 µg/m3
(Briese et al., 1985).
Working with thallium causes dust contamination of the hands,
this increasing with the duration of work (Tikhova, 1964). The dust
concentration on the hands has been reported to be in the range of
0.04 to 10.6 mg/m2 (Shabalina & Spiridonova, 1979).
6. KINETICS AND METABOLISM
Investigations into the kinetics and metabolism of thallium in
aquatic and terrestrial animals have mainly made use of radioactive
compounds, especially thallium-201. The investigations cited in this
chapter have been performed with various thallium salts, but to
facilitate comparison concentrations have been generally expressed as
µg (or mg) thallium/litre.
6.1 Absorption
6.1.1 Animals
6.1.1.1 Aquatic animals
Clams and mussels reached equilibrium within 12 to 19 days when
exposed to 50 or 100 µg thallium/litre. Depending on the exposure
level, the clams contained 5 or 9 mg/kg dry weight and the mussels 3
or 5 mg/kg (Zitko & Carson, 1975).
Very poor absorption of thallium salts from water containing
0.1 mg thallium/litre was found in isolated gill preparations of the
mussel Mytilus galloprovincialis (Nolan et al., 1984). In the first
10 min about 10% of the dose was absorbed.
Accumulation of thallium in juvenile salmon exposed for 300 h to
different concentrations of thallium (17.9 to 200 µg thallium/litre)
varied in different organs (Table 15). In muscle, thallium levels
increased almost linearly with the water thallium concentration from
2.3 to 27.0 mg/kg tissue (wet weight). Data on the liver show no
consistent trend, but in gills an obvious maximal accumulation
capacity of about 30 mg/kg was reached even at the lowest
concentration of 17.9 µg/litre water (Zitko et al., 1975). The mean
accumulation factor (mg thallium per kg tissue wet weight divided by
mg thallium per litre water) of the gills (up to 1430) is about three
to ten times higher than that of muscle or liver.
6.1.1.2 Terrestrial animals
Using different routes of administration of thallium(I) nitrate
solution (oral, intratracheal, subcutaneous, intraperitoneal,
intramuscular and intravenous), thallium was rapidly and almost
completely absorbed in rats (Lie et al., 1960). High concentrations
of thallium were detected in the blood within just 1-2 h after oral
administration of thallium(I) malonate and thallium(I) sulfate
(Aoyama, 1989), and as little as 1 h after oral or parenteral
administration of thallium(I) sulfate it was found in the urine and
faeces of rats (Lund, 1956a).
Only a few experimental studies on intestinal absorption are
available. In sheep and cows about 2% of the thallium ingested with
contaminated food was retained, while about 98% was eliminated
(Crössmann, 1984). Sabbioni et al. (1980b) found no obvious
differences using various doses of thallium(I) or thallium(III)
sulfate or bromide, but, after oral uptake of dimethyl thallium(III)
bromide in rats, the organs contained only 1 to 10% of the
concentrations found after the uptake of inorganic thallium,
indicating reduced absorption. In rats the absorptive capacity of
different ligated regions of the intestinal tract varies strongly:
201thallium (sulfate) was rapidly absorbed from the colon, but more
slowly from the ileum and jejunum and slowest from the stomach
(Sabbioni et al., 1984a). Within 60 min the ligated colon absorbed
about 75%. Lower values were obtained for the other regions. Voltage
clamp experiments on the mucosa of rat descending colon showed
exclusive transport by diffusion to the serosal side (Schäfer et al.,
1981).
Absorption of thallium through the skin of rats is indicated by
the determination of a cutaneous LD50 of 117 mg/kg for thallium(I)
carbonate (Shabalina et al., 1980).
6.1.2 Humans
Increased levels of thallium have been observed in the lungs of
coal miners, but no data are available concerning the absorption of
thallium salts after inhalation exposure (section 6.2.2.2) (Weinig &
Zink, 1967). Generally it is assumed that about 35% of respirable
dust is deposited in the lung (Ohnesorge, 1985; IPCS, 1992) and that
up to 100% of the deposited thallium is absorbed (Gubernator et al.,
1979). The rate of deposition and absorption is high because thallium
concentrations increase markedly with decreasing particle size
(sections 3.2.3.2 and 5.1.1.2), and small particles become deposited
in the lung whereas larger particles are deposited in the upper
respiratory system (Natusch & Wallace, 1974). In addition, nearly all
the thallium chloride in the dust emitted from the cement plant in
Lengerich was water-soluble (LIS, 1980). In human broncho-alveolar
lavage fluids, 0.258 ng thallium per 1000 cells was found in a
silicosis patient, but only 0.009-0.05 ng in patients suffering from
other lung diseases (Maier et al., 1986).
From several intoxication cases, e.g., after oral and topical
application of thallium(I) sulfate during depilatory treatment
(Barckow & Jenss, 1976; Schmidbauer & Klingler, 1979), it can be
assumed that both percutaneous and gastrointestinal absorption occur,
but no data on absorption are available. High blood thallium
concentrations have been reported following human poisoning (see
Table 20).
6.2 Distribution
6.2.1 Animals
No effect of the route of administration (oral, intratracheal,
subcutaneous, intraperitoneal, intramuscular and intravenous) on
distribution was observed by Lie et al. (1960). After intravenous
injection, an initial increase in the thallium concentration of the
blood is followed by a steep decrease within 5 to 15 min (Gehring &
Hammond, 1967; Lameijer & van Zwieten, 1977a,b). A similar trend is
observed when concentrations are compared 1.5 and 24 h after oral
administration of 10 mg thallium/kg body weight to rats or intravenous
injection into rabbits (Careaga-Olivares & Morales-Aguilera, 1993).
Thallium is distributed by the blood stream to all organs.
Data on the distribution of thallium in the main blood
compartments, e.g., serum and erythrocytes, have been reported. In
vitro measurements by Lund (1956a) and Witschi (1965) indicate that
thallium is distributed equally in blood plasma and red cells,
presumably without any direct binding, while Gregus & Klaassen (1986)
found that 2 h after intravenous injection of 1 to 30 mg/kg more than
80% of blood thallium was in the plasma. According to Ducket et al.
(1983), 24 h after intraperitoneal injection of a very low dose of
thallium, nearly 90% was located in the red blood cells and only about
10% in the plasma. An intermediate result was obtained by Ulrich &
Long (1955): after intraperitoneal injection of about 20 µg
radioactive thallium, about one third of the thallium concentration in
the whole blood was located in the plasma. This ratio did not change
over the period 0.5 h to 96 h after injection, although the
concentration in the blood decreased with time. Similar results were
obtained in human whole blood (in vitro) and in rabbit whole blood
( in vitro and in vivo) (Careaga-Olivares & Morales-Aguilera,
1990). A slightly higher concentration in the erythrocytes was also
found by Leloux et al. (1987), but during redistribution phases the
concentrations in erythrocytes were increased, suggesting that
erythrocytes may be involved in thallium uptake.
6.2.1.1 Distribution after administration of a single dose
The distribution of thallium after administration of single doses
of thallium compounds has been investigated in a number of studies,
using either subtoxic doses (0.2 µg/kg to 8 mg thallium/kg body
weight) (e.g., Ulrich & Long, 1955; Lie et al., 1960; Bradley-Moore et
al., 1975; Edel Rade et al., 1982; Ziskoven et al., 1983; Ducket et
al., 1983) or toxic doses (e.g., Lund, 1956a; Fitzek & Henning, 1976;
Achenbach et al., 1980; Leloux et al., 1987b; Aoyama, 1989).
Comparisons of dose-dependent distribution (Emara & Soliman, 1950;
Gehring & Hammond, 1967; Sabbioni et al., 1980a,b, 1982; Talas &
Wellhöner, 1983; Gregus & Klaassen, 1986; Ríos et al., 1989) revealed
only slight differences in the distribution pattern, even at
intraperitoneally administered concentrations as different as 0.00004,
2, 20 and 2000 µg thallium/rat (Sabbioni et al., 1980a).
The distribution of thallium in the organs is a time-dependent
process (Tables 17 and 18). Summarizing the investigations on rats,
rabbits, dogs and goats, in an initial phase after a single dose of an
inorganic thallium compound, e.g., thallium sulfate or thallium
chloride, maximal concentrations occurred in the kidneys and nearly
equal levels were found in the testes, myocardium, salivary glands,
muscle, liver, intestines, adrenals and thyroid; fat and brain
contained very low levels of thallium (Lund, 1956a; Gehring & Hammond,
1967; Bradley-Moore et al., 1975; Sabbioni et al., 1982; Talas &
Wellhöner, 1983). About 24 h after administration, the relative
thallium content of all organs, with the exception of the kidneys,
decreased and that of brain, muscle and testes increased. In
guinea-pigs administered lethal doses of thallium, kidney and liver
finally contained about equal levels, presumably due to kidney damage
(Weinig & Walz, 1971). With respect to the affinity and
redistribution of thallium in rat organs, Leloux et al. (1987b)
distinguished three compartments according to affinity and
redistribution which are not completely in agreement with the
experimental data shown in Table 17.
Intraperitoneal application of 16, 32 and 48 mg thallium
sulfate/kg body weight to male rats resulted in peak levels occurring
in various regions of the brain 24 h after injection, except in the
hypothalamus where the peak was reached earlier. After 24 h, the
regional concentrations decreased in the following order:
hypothalamus, midbrain, hippocampus, thalamus, pons, cerebellum,
corpus striatum and cerebral cortex. In the hypothalamus, a region
with low blood-brain barrier protective mechanisms, thallium
concentration was significantly higher than in the corpus striatum,
whereas in the cerebral cortex it was significantly lower than in all
other regions (Ríos et al., 1989). Whereas weanling rats showed a
region-dependant distribution of thallium after a single sublethal
intraperitoneal injection (16 mg thallium/kg body weight), thallium
concentrations were similar in different brain regions of newborn rats
(Galvan-Arzate & Ríos, 1994).
Table 17. Alterations in the distribution of thallium in different organs of experimental animals at different times after
administration (mean ± standard deviation)a
Rats (4; intraperitoneal)b Syrian hamsters (5; oral)c
Experiment No.: 1 2 3 4
2 h 40 h 4 h 24 h 1 h 24 h 1 h 24 h
(% of dose) (% of dose of thallium/g wet weight) (mg thallium/kg wet weight)
Blood 0.05 ± 0.01d 0.08 ± 0.05d 0.023 ± 0.001 0.027 ± 0.017 1.5 ± 0.6 1.0 ± 0.1 1.7 ± 0.6 0.8 ± 0.1
Bone - - 0.239 ± 0.036 0.216 ± 0.064 - - - -
Brain 0.027 ± 0.003 0.28 ± 0.05 0.047 ± 0.009 0.090 ± 0.023 < 0.1 3.7 ± 0.3 0.6 ± 0.1 3.0 ± 0.3
Heart 0.57 ± 0.07 0.33 ± 0.03 0.546 ± 0.064 0.337 ± 0.006 11.4 ± 1.7 7.2 ± 0.7 21.0 ± 3.4 6.6 ± 0.7
Intestine 1.1 ± 0.26 - - - - - - -
Kidney 5.65 ± 0.35 9.75 ± 0.97 3.354 ± 0.535 1.899 ± 0.253 58.5 ± 8.4 51.3 ± 14.0 88.4 ± 21.8 41.5 ± 0.9
Liver 4.44 ± 0.55 0.95 ± 0.19 0.373 ± 0.040 0.228 ± 0.028 14.3 ± 7.6 6.6 ± 2.1 39.7 ± 6.2 5.8 ± 0.4
Lung 0.55 ± 0.35 0.45 ± 0.04 0.289 ± 0.076 0.230 ± 0.081 - - - -
Muscle 0.6 ± 0.02e 0.54 ± 0.2e 0.168 ± 0.031 0.234 ± 0.091 < 0.1 9.1 ± 2.4 1.2 ± 0.5 9.1 ± 1.2
Table 17 (contd).
Rats (4; intraperitoneal)b Syrian hamsters (5; oral)c
Experiment No.: 1 2 3 4
2 h 40 h 4 h 24 h 1 h 24 h 1 h 24 h
(% of dose) (% of dose of thallium/g wet weight) (mg thallium/kg wet weight)
Pancreas 0.86 ± 0.34 0.57 ± 0.13 0.505 ± 0.163 0.310 ± 0.206 - - - -
Salivary 0.76 ± 0.06 0.7 ± 0.32 0.595 ± 0.078 0.370 ± 0.090 - - - -
gland
Spleen 0.32 ± 0.03 0.18 ± 0.02 0.338 ± 0.039 0.249 ± 0.054 - - - -
Testes/ 0.56 ± 0.04 0.93 ± 0.34 0.388 ± 0.077 0.200 ± 0.010 < 0.1 14.7 ± 0.7 1.0 ± 0.1 14.0 ± 1.0
Ovary
a - = no data given
b No. 1: Sabbioni et al. (1980b) and No. 2: Edel Rade et al. (1982): injection of thallium(I) sulfate (2 µg thallium/rat)
c Nos. 3 and 4: Aoyama (1989): administration of 12.5 mg thallium(I) sulfate/kg (No. 3) or 12.35 mg thallium(I) malonate/kg (No. 4)
d % dose/ml
e % dose/g wet weight
Table 18. Distribution of thallium in different organs of experimental animals after different
periods of exposurea
Experiment no:b 1a 1b 2 3 4a 4b 5
72 h 72 h 4 h 24 h 48 h 48 h 48 h
Blood 0.007 0.001 0.02 0.02 0.02 0.02 -
Bone 0.016 0.028 0.37 0.42 0.23c 0.23c 0.67
Brain 0.006 0.008 0.05 0.13 0.17 0.14 0.32
Fat 0.0008 0.001 - 0 0.01 0.02 -
Heart 0.027 0.030 0.55 0.20 0.34 0.25 0.63
Intestine 0.012d 0.017d - 0.51 0.49e 0.76e 0.50
Kidney 0.205 0.210 3.35 2.15 2.58 0.53 5.36
Liver 0.019 0.026 0.37 0.33 0.19 0.16 0.53
Lung 0.016 0.018 0.29 0.55 0.25 0.24 0.51
Muscle 0.014 0.025 0.17 0.49 0.25f 0.27f 0.76
Table 18 (contd).
Experiment no:b 1a 1b 2 3 4a 4b 5
72 h 72 h 4 h 24 h 48 h 48 h 48 h
Pancreas 0.010 0.014 0.51 - 0.44 0.33 -
Salivary gland - - 0.59 - 0.42 0.44 1.06
Skin - - 0.20 - - 0.32
Spleen 0.016 0.021 0.34 0.20 0.24 0.21 0.51
Testes/ 0.023 0.024 0.39 0.39 0.53 0.53 0.81
Ovary
a Thallium content in % of dose per kg wet weight; - = no data given
b No. 1: Talas & Wellhöner (1983): mean of 2 rabbits after intravenous injection of < 2 µg
(No. 1a) or 1.1 mg thallium/kg (No. 1b); No. 2: Sabbioni et al. (1982): mean of 4 rats
after intraperitoneal injection of 2 µg/rat; No. 3: Lund (1956a): data of 1 rat after
intraperitoneal injection of 10 mg/kg; No. 4: Barclay et al. (1953): mean of 4 rats after
intravenous injection of 23 µg thallium nitrate (No. 4a) or mean of 3 rats after
intravenous injection of 10.023 mg thallium sulfate (No. 4b); No. 5: Lie et al. (1960):
mean of 18 rats after injection of thallium by various routes
c Femur
d Mean of values of small and large intestine
e Lower bowel
f Abdominal muscle
Although the endocrine organs were thought to be involved in the
mechanism of toxicity, no accumulation was observed in
autoradiographic studies of low-dosed adult mice and rats (Barclay et
al., 1953; André et al., 1960). Leloux et al. (1987b) found no
obvious deviation from the levels found in other organs 5 days after
subacute or acute intoxication with 4 or 20 mg thallium nitrate/kg,
respectively. Thyresson (1951), however, reported a high
concentration of thallium (96.9 mg/kg wet weight) in the thyroid gland
of rats 24 h after administration of 40 mg thallium nitrate/kg body
weight. According to Ulrich & Long (1955), treatment of rats with
thyrotropic hormone subsequent to the administration of thallium did
not affect the uptake of thallium by the thyroid, whereas pretreatment
(2 days prior to administration) significantly increased the initial
uptake. In addition, the authors reported that adrenals, thyroid and
pituitary contained similar concentrations of thallium.
Different modes of application and different thallium compounds
hardly affect the distribution pattern, as shown by Sabbioni et al.
(1980b) with intravenous or oral administration of thallium(I),
thallium(III) and dimethyl thallium(III) in rats. Another
investigation showed that after administration of thallium(I) malonate
and thallium(I) sulfate the distribution pattern between different
organs varied only initially. Later, both patterns were similar
(Aoyama, 1989).
6.2.1.2 Distribution after long-term sublethal administration
The distribution pattern of thallium in chronically poisoned rats
shows a strong similarity to the final pattern after a single dose,
there being wide distribution throughout the body. Lameijer & van
Zwieten (1977a,b) determined thallium concentrations in urine, blood
and 19 different tissues of rats exposed to thallium (10 mg/litre) in
drinking-water for 9 or 24 weeks. No statistically significant
differences in the concentration of thallium were observed for any
tissues except kidney. The thallium concentration in the renal medulla
was about 7 times higher than in the heart, liver, muscle, brain or
skin. If rats received 30 mg/litre in drinking-water, they died
within 9 to 11 days, but after 7 days this administration had not
affected the rapid decline of the thallium blood level following
administration of an additional intravenous dose of 1 mg/kg (Lameijer
& van Zwieten, 1977a).
6.2.1.3 Transplacental transfer of thallium
Mice were gavaged with thallium(I) sulfate (8 mg thallium/kg body
weight) on gestation day 9 and tissue concentrations were determined
0.5 to 24 h later. Thallium levels in fetuses and maternal kidneys
rose during the first hour, then levelled off to a plateau which did
not change during the following 22 h. No indication of a specific
placental barrier was detected (Ziskoven et al., 1980). Ziskoven et
al. (1983) repeated the study, additionally including rats. After 10,
20 and 30 min, increasing thallium concentrations were found in the
maternal kidneys, reaching a plateau after 30 to 60 min, which was
stable for at least 50 h (last measurement). A similar time course of
thallium uptake was observed in the fetal tissues: the initial uptake
was comparable to that of the maternal kidney, but the resultant
concentrations were 10-fold lower. There were no specific differences
between mice and rats.
A slightly delayed transfer to fetuses was observed in an
autoradiographic study with mice (gestation day 15), dosed with an
intraperitoneal injection of thallium sulfate. After just 15 min,
thallium was observed in the fetuses, but the maximum level was
reached within 2 to 4 h, when some thallium elimination took effect.
During the whole observation period, fetal thallium levels were lower
than those of the placenta (Olsen & Jonsen, 1982). The authors also
reported on the influence of the stage of gestation. Thallium crossed
the placenta throughout gestation; during the early stages it was
concentrated in the visceral yolk sac placenta and during late
gestation additionally in the chorioallantoic placenta and amnion.
When near-term mice and rats (gestation day 17-18) were given
subcutaneous injections of 204thallium sulfate, thallium
concentrations in the mouse fetuses rose during the following 8 h and
in the rat fetuses during the following 16 h. Thereafter, the
fetal/maternal ratios in tissues remained constant at 0.84 in rats and
considerably lower (0.46) in mice (Gibson et al., 1967).
Intra-arterial infusion of 0.2 to 6.4 mg 204thallium
sulfate/min per kg body weight on day 20 of pregnancy in rats resulted
in an initially restricted transplacental transfer. In the lowest and
highest dosage groups, 32 min after administration, thallium levels in
fetal tissues corresponded to only about 7% of those in the maternal
plasma, perhaps because two-thirds of the whole blood thallium was
located in the erythrocytes and did not pass the placental barrier
(Gibson & Becker, 1970).
Sabbioni et al. (1982) compared placental transfer in rats after
intraperitoneal injection of a low dose (2 µg thallium-201/rat;
gestation day 13) with that after administration of a toxic dose by
gavage (10 mg thallium/kg body weight; gestation day 16). Thallium
concentrations in maternal and fetal brain were similar 4 h after
injection of the low dose. In fetal liver they were 80% lower than in
the maternal liver, and in the placenta and fetal organs they were
higher than in the blood of the dams and fetuses. Very low
concentrations were found in the amniotic fluid. After 8 days,
concentrations in most of the maternal organs were about 10 times
lower than the initial levels, while in muscle, cerebellum and brain
of the dams, reductions of only 60, 40 and 5%, respectively, were
found during the same period. In contrast to the variable decline in
maternal organs, a 10-fold decline was observed in the whole fetus and
its liver, brain and blood. Preliminary results of the authors
indicated that this was due to a much stronger thallium accumulation
in the mitochondria of the adult rat brain than in those of the fetal
brain. Furthermore, identical low-dose experiments (2 µg/rat) also
showed a faster decline of thallium concentrations in fetal than in
maternal brain, after an initially more rapid entry into the fetal
brain, whereas declines in the liver levels were nearly identical
(Edel Rade et al., 1982). Administering a toxic dose of 10 mg/kg to
the dams resulted in 60% lethality within the following 3 days. After
3 days, concentrations in the liver and brain of the surviving dams
were similar and about 2-fold higher than in the corresponding fetal
organs. Therapeutic oral dosing with Prussian Blue (100 mg/kg body
weight, twice daily) starting 8 h after administration of thallium,
significantly reduced the thallium concentrations in maternal and
fetal tissues and only 1 of 12 adult rats died (Sabbioni et al.,
1982).
Transplacental transfer of thallium has also been observed in a
cat (Fitzek & Henning, 1976). The cat showed signs of a strong
thallium intoxication and was killed after abortion of approximately
5-week-old fetuses. Thallium levels in maternal blood and fetal
tissues were similar, but the concentrations in fetal heart and lungs
were two to three times higher than in the corresponding maternal
organs.
6.2.2 Humans
Background thallium concentrations found in human body fluids and
tissues are given in Table 19. After poisoning, thallium
concentrations ranging up to nearly 36 mg/litre in blood, 25 mg/litre
in urine and 8 mg/kg in hair have been found (Table 20).
6.2.2.1 Increased concentrations after lethal poisoning
In reports of postmortem examinations after suicide or homicide,
data on the distribution of thallium in different organs are rarely
included with data on dose and application routes (Table 20). The
distribution pattern shows no consistent trend. In a single
individual, concentrations in bones, fat and muscles from different
parts of the body may vary, e.g., in vertebrae (12.7 mg/kg), sternum
(7.0 mg/kg), femur (16.4 mg/kg) and tibia (9.0 mg/kg) (case 7 in
Table 21) (Arnold, 1986). The distribution of thallium differs
considerably from that reported for potassium in humans (Davis et al.,
1981). Endocrine glands, kidneys, liver and intestine (without
content) showed the highest concentrations (Table 21).
With respect to the total amount per organ, liver or lung were
found to contain 2 to 6 times and the brain about 1.5 to 2 times more
thallium than the kidneys (Curry et al., 1969; Arnold, 1986).
A comparison between the white and grey matter of the brain
revealed that in the latter the concentration was three times higher
(Cavanagh et al., 1974). Detailed data of thallium concentrations in
different regions of the nervous system were given by Davis et al.
(1981). The authors showed that areas of the brain rich in neurons
tend to accumulate twice as much thallium as areas devoid of neurons,
and that the grey matter contains some of the highest thallium levels
of any body organ.
6.2.2.2 Increased concentrations after long-term sublethal poisoning
Thallium levels in urine (Table 22), blood or saliva of
chronically exposed people offer better indications of the actual
burden than those derived from hair samples, since elevated levels in
hair can be caused by exogenous dust (Bertram et al., 1985).
People consuming food grown in private gardens and living at a
distance of more than 3 km from the cement plant at Lengerich, Germany
showed significantly higher concentrations of thallium in their urine,
decreasing with increasing distance from the plant, than people who
did not consume food from their gardens. Thallium concentrations in
the urine of people living near the plant (< 1 km) and consuming food
grown in private gardens were about five times higher (3.95 µg/litre)
(Brockhaus et al., 1980). Peak values were 76.5 µg/litre in urine and
565 µg/kg in hair (Ewers & Brockhaus, 1982). In this area a medical
survey was carried out immediately after the occurrence of thallium
emissions had been recognized; urine thallium levels in about 80% of
the population were found to exceed the upper normal limit of
1 µg/litre (Brockhaus et al., 1980; Dolgner et al., 1983). The
recommendation to avoid home-grown vegetables was followed by many
people and resulted in a significant decrease in urine thallium
levels. However, in some residents, even 8 years later, increased
levels of > 20 µg/litre urine could be found. Probably there was
still a significant contamination of soil and thus of home-grown
vegetables (Ewers, 1988).
Subsequent studies were carried out at other cement factories.
About 70% of employees at two cement plants in Middle and Lower
Franconia, Germany were found to have normal thallium concentrations
in their urine. However, at a third factory in the same area only 30%
of employees showed normal thallium urine levels, presumably because
of the higher thallium content in the raw material used (Schaller et
al., 1980). The population around the three cement plants showed
normal urine thallium levels: of 238 people tested, 194 had thallium
concentrations below 2 µg/litre, 36 were in the range of 2 to
5 µg/litre, and 5 were between 5 and 10 µg/litre. Higher
concentrations were found in the urine of three people (11.5, 14.5 and
19.5 µg/litre) (Steuer, 1980).
Table 19. Background concentrations of thallium in humans
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Blood, 2 0.33-0.59 µg/litre Weinig & Zink (1967)
whole < 20 Bowen (1966)
13 0.47-9 Iyengar et al. (1978)
320 < 5-80 Singh et al. (1975)
0.05 Kemper (1979)
418 0.39 ± 0.05 0.1-1.1 Minoia et al. (1990)
0.5-2 Kemper & Bertram (1991)
plasma < 2.5 Bowen (1966)
1 < 2.5 Iyengar et al. (1978)
Bone 2 0.84-2.51 µg/kg fresh weight Weinig & Zink (1967)
1 2 Iyengar et al. (1978)
5 < 0.1-0.1 Goenechea & Sellier (1967)
Bonea 1 0.7; 0.9 Goenechea & Sellier (1967)
Brain < 0.5 mg/kg dry weight Bowen (1966)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Bronchoalveolar 1b 0.258 ng/1000 cells Maier et al. (1986)
lavage fluids 1c 0.009
1c 0.011
1d 0.016
1d 0.050
Faeces 5 < 0.02-3.0 µg/kg fresh weight Goenechea & Sellier (1967)
Hair 7 18.6 ± 14.9 7-51 µg/kg fresh weight Geilmann et al. (1960)
6 10.4 ± 4.3 4.8-15.8 Weinig & Zink (1967)
1 < 20 Ziegler & Ziegler (1984)
Heart < 0.4 mg/kg dry weight Bowen (1966)
Kidney < 0.4 mg/kg dry weight Bowen (1966)
Kidney 6 2.7 ± 1.1 1.44-4.1 µg/kg fresh weight Weinig & Zink (1967)
8 < 3 Iyengar et al. (1978)
259 0.03-8.6 Bösche & Magureanu (1983)
Liver 0.4 mg/kg dry weight Bowen (1966)
11 0.47 ± 0.13 < 0.4-0.9 Johnson (1976)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Liver 6 1.1 ± 0.9 0.55-2.85 µg/kg fresh weight Weinig & Zink (1967)
1 0.4 Goenechea & Sellier (1967)
6 1-9 Iyengar et al. (1978)
0.5-3 Kemper & Bertram (1991)
Lung < 0.3 mg/kg dry weight Bowen (1966)
Lung 4 1.1 ± 0.7 0.36-1.8 µg/kg fresh weight Weinig & Zink (1967)
Muscle < 0.4 mg/kg dry weight Bowen (1966)
Muscle 6 2.1 ± 2.1 0.52-7.05 µg/kg fresh weight Weinig & Zink (1967)
3 15-100 Iyengar et al. (1978)
Musclee 1 0.4 µg/kg fresh weight Goenechea & Sellier (1967)
Nail 6 51.2 ± 12.1 40-74 µg/kg fresh weight Geilmann et al. (1960)
6 2.6 ± 1.4 0.72-4.93 Weinig & Zink (1967)
Skin < 0.2 mg/kg dry weight Bowen (1966)
Table 19 (contd).
Material Number of Concentration of thallium Concentration unit Referencef
measurements
Mean ± SD Range
Urine 10 < 0.02-1.0 µg/kg fresh weight Goenechea & Sellier (1967)
Urine 0.05-0.1 µg/litre Geilmann et al. (1960)
14 0.7 ± 0.5 0.07-1.69 Weinig & Zink (1967)
0.05-20 Kemper (1979)
31 0.4 ± 0.2 < 0.1-1.2 Brockhaus et al. (1981b)
10 0.3 ± 0.2 < 0.1-0.9 Brockhaus et al. (1981b)
149 0.3 ± 0.14 0.02-0.7 Dolgner et al. (1983)
72 0.22 ± 0.14 0.06-0.61 Apostoli et al. (1988)
496 0.42 ± 0.09 0.06-0.82 Minoia et al. (1990)
0.05-1.5 Kemper & Bertram (1991)
Urine 20 < 0.3-1.1 mg/kg creatinine Schaller et al. (1980)
10 2.2 ± 1.6 Briese et al. (1985)
a 1.5 months after death
b silicosis patient
c saw setters suffering pneumoconiosis
d welders suffering emphysema
e 6 months after death
f Additional values for other tissues have been compilated by Iyengar et al. (1978)
Table 20. Concentrations of thallium in cases of poisoning
Material Number of Range of thallium Concentration unit Reference
cases concentrations
Blood, whole 50-6000a µg/litre Kemper (1979)
Blood 2 29; 7700 Alarcón-Segovia et al. (1989)
Blood, whole 3 350-36 000 Heath et al. (1983)
Blood, plasma 2 300; 1500 Heath et al. (1983)
Blood, erythrocyte 2 400; 2300 Heath et al. (1983)
Bone 1b 0.9-2.1 µg/kg fresh weight Goenechea & Sellier (1967)
Faeces 1 6500-38 400 Paulson et al. (1972)
Hair 1 650 Geilmann et al. (1960)
Hair 1b 6.8 Goenechea & Sellier (1967)
Hair 1 420-1800 Hagedorn-Götz & Stoeppler (1975)
Hair 250-8000a Kemper (1979); Kemper & Bertram (1984)
Heart 1 3600 Munch et al. (1933)
Intestine 1 3600 Munch et al. (1933)
Intestine 1 0.8; 4.0 Goenechea & Sellier (1967)
Table 20 (contd).
Material Number of Range of thallium Concentration unit Reference
cases concentrations
Kidney 5 2700-11 600 Munch et al. (1933)
Kidney 1 106 000 Heath et al. (1983)
Liver 1 75 000 Heath et al. (1983)
Liver 2 3700; 5500 Munch et al. (1933)
Lung 2 3300; 7700 Munch et al. (1933)
Muscle 1b < 0.02; 1.3 Goenechea & Sellier (1967)
Nails 1 2400 Geilmann et al. (1960)
Spleen 3 2900-6600 Munch et al. (1933)
Urine 50-25 000a µg/litre Kemper (1979)
Urine 15 500-20 400 Klöppel & Weiler (1978)
Urine 1 3100 Gastel (1978)
Urine 3 10-13 800 Alarcón-Segovia et al. (1989)
Urine 2 2700-30 000 µg/litre fresh weight Heath et al. (1983)
Urine 1 0.65 mg/kg creatinine Hagedorn-Götz & Stoeppler (1975)
a Concentrations indicative for poisoning
b 3 years after death
Table 21. Concentrations of thallium in individual cases of human poisoning
Thallium concentration (mg/kg wet weight or mg/litre)
Case no:a 1 2 3 4 5 6 7
Durationb: > 14 days > 21 days 9 days 8 days 11 days 12 days 13 days
Dose: -c - 5-10 g 0.75 gd - - -
Adrenal - - 83.6 - - - -
Blood 5.1 3.4 5.1 - - 3.0 -
Bone - 5.0 - 0.92 1.9 8.0 7.0-16.4
Brain 8.5 - 62-140 0.15 - - -
grey - 10.0 - - - - -
white - 3.0 66.1 - - - -
cerebellum - - 103.3e - 1.5 5.0 -
cerebrum - - 102.0e - 1.0 - -
Fat - - < 1.0 - 0.4-1.2 - -
Heart - 13.3 - 0.19 1.5 13.0 6.2
left - - 26.8 - - - -
right - - 131.6 - - - -
Intestine - - - 0.20 - - -
small 4.4f - - - 0.5-0.9 8.0f 6.4
colon 71.0f 120.0f 126.0 - 2.0 500.0f 8.5
Kidney 26.7 20.0 74.1 0.26 3.0 28.0 12.5
Liver 8.6 5.0 77.3 0.82 1.8 15.0 14.7
Table 21 (contd).
Thallium concentration (mg/kg wet weight or mg/litre)
Case no:a 1 2 3 4 5 6 7
Durationb: > 14 days > 21 days 9 days 8 days 11 days 12 days 13 days
Dose: -c - 5-10 g 0.75 gd - - -
Lung - 1.8 - 0.15 0.9 4.0 -
Muscle 10.1 5.0 26.8 0.21 0.4-2.0 - 3.6
Pancreas - - 71.7 - - - -
Parathyroid - - 38.1 - - - -
Pituitary - - 114.5 - - - -
Salivary gland - - 32.1 - - - -
Skin - 6.0 32.1 - 0.3 - -
Spleen - - - 0.35 1.9 - -
Testes/Ovaries - - 152.0 - - - -
Thyroid - - 33.5 - 4.6 - -
Urine 15.6 5.9 3.3 - - 3.0 -
a Case no. 1: Curry et al. (1969); no. 2: Cavanagh et al. (1974); no. 3: Davis et al. (1981); no. 4: Graben et al.
(1980); nos. 5-7: Arnold (1986); nos. 3-7: poisoning by one uptake of thallium
b Period of time from uptake of thallium to death or determination of concentrations
c - = no data given
d 15-week-old embryo; dose of mother
e Cortex
f Content
Table 22. Concentrations of thallium following environmental or occupational exposure
Material Number of Concentration of thallium Concentration unit Source of Reference
people thallium
Mean ± SD Range
(Median)
Hair 1163 20.3 ± 42.7 0.6-565 µg/kg fresh weight cement plant Brockhaus et al. (1981b)
Urine 50 (0.6) < 0.3-4.9 mg/kg creatinine cement plant Schaller et al. (1980)
47 (1.65) 0.4-6.3 mg/kg creatinine cement plant Schaller et al. (1980)
21 (0.34) < 0.3-2.9 mg/kg creatinine cement plant Schaller et al. (1980)
10 7.1 ± 6.0 mg/kg creatinine zinc smelter Briese et al. (1985)
1265 5.2 ± 8.3 < 0.1-76.5 µg/litre cement plant Brockhaus et al. (1981b)
82 2.4 ± 4.3 < 0.1-35.8 µg/litre cement plant Dolgner et al. (1983)
117 3.0 ± 5.6 0.2-37.7 µg/litre cement plant Dolgner et al. (1983)
34a 3.4 ± 3.5 0.4-14.8 µg/litre cement plant Dolgner et al. (1983)
30 0.38 ± 0.30 0.08-1.22 µg/litre cement plant Apostoli et al. (1988)
20 0.40 ± 0.34 0.08-1.22 µg/litre cement plant Apostoli et al. (1988)
10 0.33 ± 0.16 0.09-0.60 µg/litre cement plant Apostoli et al. (1988)
Table 22 (contd).
Material Number of Concentration of thallium Concentration unit Source of Reference
people thallium
Mean ± SD Range
(Median)
Urine
(contd) 9 0.38 ± 0.29 0.10-1.04 µg/litre iron smelter Apostoli et al. (1988)
74b 16.0 ± 16.9 0.2-76.5 µg/litre cement plant Brockhaus et al. (1981a)
74 7.9 ± 8.8 0.2-42.6 µg/litre cement plant Dolgner et al. (1983)
21 0.33 ± 0.27 0.06-1.04 µg/litre iron smelter Apostoli et al. (1988)
12 0.29 ± 0.21 0.06-0.70 µg/litre iron smelter Apostoli et al. (1988)
a children
b Data of people with high concentrations of thallium or possibly thallium-related disorders determined in the first survey and about
1 year later in the following line
Only a few cases resulting from industrial exposure have been
reported and seem to be mainly a result of skin contact or inhalation
(Kazantzis, 1986; Ewers, 1988).
At a zinc smelter in eastern Germany, increased thallium levels
were not only found in the urine of men working in the production
process, but also in men working in the administration. During the
production of thallium in this plant, the levels were further
increased (maximal value: 28.6 µg/litre) (Briese et al., 1985).
High concentrations of thallium were found in lung tissue from
two coal miners (20.2 and 29.5 µg/kg wet weight). Concentrations in
most other tissues were normal (Weinig & Zink, 1967).
In Italy, slight but significant increases in thallium levels
were found in the urine of cement workers (0.4 µg/litre) and cast iron
workers (0.3 µg/litre), compared with a non-exposed group
(0.2 µg/litre). There was no correlation with age or the duration of
exposure (Apostoli et al., 1988).
Weinig & Zink (1967) reported a slight elevation of urine
thallium levels of vegetarians (and smokers) compared to controls.
However, it should be noted that each group comprised only three
people and the levels were far below those of thallium-affected
people. Geilmann et al. (1960) estimated that more than 60% of the
thallium content of a cigarette (62 µg/kg) is inhaled, but no data are
available on the amount absorbed. Assuming an absorption of 50% and a
consumption of 20 cigarettes/day, 375 ng/day would be absorbed (BGA,
1979). Based on data of the thallium concentration in urine of about
120 people, non-exposed individuals and workers with suspected
industrial exposure to thallium, Apostoli et al. (1988) found no
evidence of a difference between smokers and non-smokers, all about 40
years old.
Comparing a total of 128 men, no correlation was found between
duration of employment at a cement plant (1 to 42 years) or age (16 to
62 years) and thallium concentrations in the urine (< 0.3 to 6.3 µg
thallium/g creatinine) (Schaller et al., 1980). Therefore, it can be
concluded that the uptake of low amounts of thallium does not cause
accumulation in the body.
6.2.2.3 Transplacental transfer of thallium
Abortion was produced in the fourth month of pregnancy 8 days
after ingestion of approximately 750 mg thallium sulfate. Starting 2
days after the ingestion, the mother was treated (haemodialysis,
forced diuresis, Prussian Blue) for 92 h and survived. Before the
start of the therapy, the blood of the mother and of the fetus
contained 0.07 and 0.01 mg/litre and the urine 0.4 and 0.1 mg/litre,
respectively. One day after the end of the treatment, the bones,
liver and kidney of the fetus contained 0.9, 0.8 and 0.3 mg
thallium/litre, respectively, and the blood of the mother
0.08 mg/litre (Graben et al., 1980). Additional evidence for the
transplacental transfer of thallium is provided by studies
demonstrating effects in infants exposed in utero (section 8.5.1).
6.3 Metabolic transformation
Data on the transformation and the equilibrium between the two
oxidation states of thallium ions(I and III) in body fluids and
tissues of mammals are not available. The two ions show a similar
intracellular distribution (Sabbioni et al., 1980b).
6.4 Elimination and excretion
6.4.1 Animals
In a study on the accumulation and excretion of thallium in
mussels and clams (section 6.1.1.1), the bivalves needed 7 and 30
days, respectively, to excrete all absorbed thallium. This is rapid
in comparison to other heavy metals, such as cadmium, copper, lead and
mercury, so that no significant amounts of thallium should enter the
food web in this way (Zitko & Carson, 1975).
Within 25 days after parenteral administration of 10 mg thallium
sulfate/kg body weight, rats eliminated 26% in the urine and 51% in
the faeces (Lund, 1956a). Elimination via urine started within hours
after oral application and persisted for up to 3 months. Faeces were
not found to contain thallium until the fourth day, and thallium was
still present after 1 month (Oehme, 1978). After injections of low
doses of 204thallium nitrate by different routes, the ratio of
faecal to urinary elimination of rats increased with time from 2 to 5
(Lie et al., 1960). Gregus & Klaassen (1986) reported that faecal
elimination is always greater than renal elimination. Biliary
elimination is of minor importance (Schäfer & Forth, 1980; Gregus &
Klaassen, 1986). Within 4 h after an intravenous injection, less than
0.3% of the injected thallium was eliminated in the bile of rats, but
up to 8% into the gut (Sabbioni et al., 1984a). An even lesser degree
of elimination occurred in tears, sweat, and milk (Oehme, 1978).
In contrast to absorption, secretion of thallium into the gut of
rats (given as 201thallium sulfate intravenously or directly into
the individual ligated gastrointestinal segments) is highest in
segments of the jejunum, followed by the ileum, colon and stomach
(Sabbioni et al., 1984a). Similar results were obtained after
intravenous administration to rats: in situ, the jejunum showed the
highest excretory activity, followed by the ascending colon (Henning &
Forth, 1982). The ileum and descending colon each excreted about half
of the amount of the jejunum; excretion into the stomach was
negligible. An increased dose (4 to 400 µg of thallium(I) sulfate/kg
body weight) caused increased excretion into the jejunum and
descending colon (Henning & Forth, 1982). Since thallium is also
absorbed in the colon (section 6.1), only a proportion of the secreted
thallium appears finally in the faeces.
Thallium ions are secreted against an electrochemical or
concentration gradient by an active transport mechanism, as shown in
experiments on the isolated mucosa of the descending rat colon
(Schäfer et al., 1981; Schäfer & Forth, 1987). Thallium(I) ions use,
at least in part, the same transport systems as potassium (Henning &
Forth, 1977; Schäfer & Forth, 1987), and thallium secretion is reduced
when the concentration of potassium is increased on the serosal side
(Henning et al., 1982).
6.4.2 Humans
The normal daily total elimination in humans is estimated to be
in the range of 1.64 µg thallium (urine: 1.2 µg; hair: 0.32 µg;
faeces: 0.06 µg; skin and sweat: 0.06 µg) (US EPA, 1980). About 50%
of total urinary elimination occurs within 9 to 11 days (Weinig &
Schmidt, 1966).
In lethal cases of human poisoning, postmortem examination has
always demonstrated high concentrations of thallium, especially in the
contents of the colon (Table 21). Thallium levels in human saliva are
up to 15 times higher than in the urine during the initial 2 weeks
(Richelmi et al., 1980). Minor amounts are eliminated via hair and
nails, both of which show the highest thallium concentrations of any
tissue among human populations in uncontaminated areas (Table 19).
Usually mother's milk is not an important route of elimination for
heavy metals (Hapke, 1988). However, 2 weeks after a suicide attempt
using thallium sulfate following the birth of her child (about 500 mg
thallium), the milk of a mother contained 0.25 mg thallium/litre,
while her blood only contained 0.07 mg/litre (Graben et al., 1980).
6.4.3 Methods to estimate daily intake of thallium
There are two ways to estimate daily intake of thallium, one
based on total daily excretion and the other on the total amount of
thallium in the body. In the former case, the total amount of
thallium excreted daily under steady-state conditions (a model which
may be reasonably applied to long-term exposure to low doses of
thallium) should reflect accurately the daily intake of thallium.
Using a mean urinary concentration of 0.4 µg/litre (which has been
frequently reported in unexposed populations), a daily urinary
excretion of 0.6 µg may be calculated assuming a urinary volume of
1.5 litre per day. Since we have assumed that renal excretion may
account for about 70% of the total daily excretion of thallium,
another 0.3 µg/day would be excreted by other routes, giving a value
for total thallium daily intake of about 0.9 µg. A similar procedure
leads to an estimated thallium daily intake of about 11 µg in
chronically exposed populations (using a mean urinary concentration of
5 µg/litre).
The other method for estimating daily intake assumes that the
following relationship exists between the total amount of thallium in
the body (Ab), the daily intake of thallium (Ad) and the
elimination rate constant (K):
Ad = KAb
Since the total amount of thallium in the body has been estimated
to be 100 µg per 75 kg body weight in an unexposed population (Weinig
& Zink, 1967), a daily intake of 2.3 µg may be calculated, assuming an
elimination rate constant of 0.023 day-1.
6.5 Retention and turnover (biological half-life)
6.5.1 Animals
The biological half-life of thallium in experimental animals is 3
to 8 days. Accordingly, the elimination of 70 to 90% of the
administered dose takes about 4 weeks (Oehme, 1978).
Using different routes of administration of thallium-204 in rats,
Lie et al. (1960) found a biological half-life of 3.3 days during the
first 21 days or until about 1% of the administered dose remained in
the body. This body clearance was not affected by the route of
administration and did not differ between various organs, except for
the hair, which contained up to 60% of the body burden after 21 days.
A biological half-life of 24 h was determined in pregnant mice and
rats (Gibson et al., 1967).
Durbin et al. (1957) determined a biological half-life in rats of
5.2 days and calculated half-lives of 7 and 6 days for removal from
the kidney and muscle, respectively. The half-lives in various organs
(brain, spinal cord, sciatic nerve, kidney, liver and spleen) were
lower in young rats than in adult rats and varied in different organs
of young and adult rats. Ducket et al. (1983) found that half-life
values in young rats ranged from 1.2 days for the sciatic nerve to 5.1
days for the liver (average for all tissues: 2.6 days) and in adult
rats from 2.7 days for the brain to 6.0 days for the spleen (average:
3.8 days).
6.5.2 Humans
Several investigators have reported on the half-life of thallium
in plasma and whole blood of humans acutely poisoned. Hologgitas et
al. (1980) reported the half-life in the blood of one patient to be
1.9 days. Heath et al. (1983) reported a half-life in the blood of
one patient of 1.9 days and a range of 21-24 h for the half-life in
blood for three patients. Treatment for thallium toxicosis has been
found to decrease the half-life of thallium in blood. In a review by
de Groot & van Heijst (1988), the half-life in blood decreased as
follows:
Treatment Thallium half-life in blood
No treatment (n=2) 9.5; 15 days
Prussian Blue (PB) (n=5) 3.0 ± 0.7 days
PB + forced dialysis (FD) (n=7) 2.0 ± 0.3 days
PB + FD + haemoperfusion (n=3) 1.4 ± 0.3 days
In cases of poisoning, the half-life of thallium in blood is
found to increase somewhat with time. Starting measurements at 42
days after toxic ingestion of thallium, Chandler et al. (1990)
reported a blood half-life of 3.7 days in a patient treated with PB
and intravenous potassium. The Gauss-Newton optimization model was
used in this calculation. Wainwright et al. (1988) and Schwartz et
al. (1988) presented data showing similar half-lives for thallium in
urine and in serum, but no quantitative analysis was performed.
There has only been one study of the whole-body half-life of
thallium in normal (i.e. unpoisoned) humans. In an investigation into
the use of radiolabelled thallium for medical imaging, Atkins et al.
(1977) administered thallium-200 to three volunteers. Using a
whole-body counter, the biological half-life for thallium was found to
be 9.8 days (range = 7.4-12.4 days). This determination is of much
greater value than the determinations of plasma or whole blood
half-lives for evaluating total excretion of thallium from the body.
6.6 Kinetics at the cellular level
The cellular uptake of thallium has been investigated in various
systems. Due to the similarity in ionic radius of thallium(I) and
potassium, thallium can substitute for potassium in a variety of
potassium-dependent transport processes, as indicated by studies with
microorganisms and frog skin (Norris et al., 1976; Zeiske & van
Driessche, 1986). In rats and dogs, data indicate that "the mechanism
involved in the active transport of potassium cannot differentiate
between potassium and thallium" (Gehring & Hammond, 1967)
(section 7.11).
The cytosol contains most of the intracellular thallium. In
rats, autoradiography revealed the presence of thallium in the
cytoplasm of nervous tissues during the first few days after injection
(Ducket et al., 1983), a phenomenon also evident in kidney, liver and
testis homogenates of rats treated with oral or intraperitoneal doses
of 0.00004, 2, 20, 2000 or 3150 µg thallium(I) per rat (Sabbioni et
al., 1980a,b) and in mussels (Nolan et al., 1984).
In a postmortem examination of a fatal case of thallium
poisoning, 87% of the thallium was present in the cytosol (Davis et
al., 1981) (for data on plants see section 4.1.2.3).
7. EFFECTS ON LABORATORY MAMMALS AND IN VITRO TEST SYSTEMS
The reported toxic effects of thallium are not always comparable,
since no standardized procedure is used by the different authors and
the duration of the experiments is not always stated. However, the
effective dose (ED) of thallium at which minimal adverse effects
(LDmin) occur, or at which 50% or all organisms are killed (LD50 and
LD100, respectively), are clearly correlated with the duration of
the experiment. This correlation also exists for the period of time
at which, for example, 50% of the organisms are killed.
The sections on single exposure (section 7.1.2), short-term
exposure (section 7.2.2) and long-term exposure (section 7.3.2) cover
the effects on various organs, except those on skin and eye (section
7.4) and the nervous system (section 7.8).
7.1 Single exposure
7.1.1 Toxicity and symptoms
The acute toxicity data for thallium compounds are listed in
Table 23. They vary considerably with observation time, e.g., a
7-fold higher LD50 value (2000 mg/kg) for mice is obtained if the
period of observation is reduced from 24 h to 1 h (Achenbach et al.,
1980).
There exist only insignificant differences in the toxicity of
various water-soluble thallium(I) salts to mice, rats, rabbits and
dogs. In general, for most laboratory species and an observation
period of 1 week, the LD50 or minimum effective dose (MED) values
range between 20 and 60 mg/kg body weight for thallium(I) salts,
independent of the application route, with the exception of
guinea-pigs (5 to 15 mg/kg). The toxicity of water-soluble thallium
compounds is similar for oral and parenteral routes of administration,
indicating a high degree of gastrointestinal absorption (Table 23).
The toxicity of water-insoluble thallium(III) oxide in rats and
rabbits is 2 to 4.5 times higher following oral administration than
following parenteral administration (Downs et al., 1960).
Acute toxicity is characterized by severe symptoms and/or death,
which may be caused by single exposure or by multiple lower doses
administered within 24 h. These symptoms are associated with
disorders of the digestive (vomiting, diarrhoea) and nervous system,
inflammation at body orifices, skin furuncles, tremor, loss of hair, a
necrotizing renal papillitis, and death by respiratory failure (BGA,
1979; Hapke, 1984; Kazantzis, 1986; Bruère et al., 1990).
Table 23. Toxicity of different thallium compounds in experimental animals after single exposure
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Mouse (I) carbonate subcutaneous LDmin 14 day 18 15.7 Sanotskii (1961)
Mouse (I) chloride intraperitoneal LD50 -c 24 20.5 Luckey & Venugopal (1977)
Mouse (I) sulfate oral LD50 1 h 2470 2000 Achenbach et al. (1980)
Mouse (I) sulfate oral LD50 2 h 1358 1100
Mouse (I) sulfate oral LD50 4 h 988 800
Mouse (I) sulfate oral LD50 12 h 432 350
Mouse (I) sulfate oral LD50 24 h 370 300
Mouse (I) sulfate oral LD50 36 h 235 190
Mouse (I) sulfate oral LD50 - 30 24.3 IPS (1982)
Mouse (I) sulfate intraperitoneal LD50 10 days 47.5 38.5d Stavinoha et al. (1959)
Mouse (I) sulfate intraperitoneal LD10 10 days 37 30
Rat (I) acetate oral LDmin 14 days 37.4 29 Downs et al. (1960)
Rat (I) acetate oral LD50 7 days 41.2 32
Rat (I) acetate intraperitoneal LDmin 14 days 25.8 20
Rat (I) acetate intraperitoneal LD50 7 days 29.6 23
Rat (I) nitrate intravenous LD50 - 16.3 12.5e Gehring & Hammond (1967)
Rat (I) nitrate intravenous LD50 - 18.9 14.5e Gehring & Hammond (1967)
Table 23 (contd).
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Rat (III) oxide oral LDmin 14 days 22.3 20 Downs et al. (1960)
Rat (III) oxide oral LD50 7 days 43.6 39
Rat (III) oxide intraperitoneal LDmin 14 days 103 92
Rat (III) oxide intraperitoneal LD50 7 days 80.5 72
Rat (I) sulfate oral LD50 - 10-25 8.1-20.2 IPS (1982)
Rat (I) sulfate dermal LD50 7 days 500 405 IPS (1982)
Rat (I) sulfate intraperitoneal LD100 2-6 days 33.3 27 Nachman & Hartley (1975)
Rat (R. (I) sulfate oral LD50 - 15 12.1 Wegler (1970)
norvegicus)
Rat (R. rattus) (I) sulfate oral LD50 - 76 61.5 Wegler (1970)
Hamster (I) malonate oral LD50 > 7 days 50 40 Aoyama et al. (1988)
Hamster (I) malonate oral LD100 5-7 days 62.5 50
Rabbit (I) acetate oral LDmin 14 days 24.5 19 Downs et al. (1960)
Rabbit (I) acetate intravenous LDmin 14 days 25.8 20
Rabbit (I) acetate intraperitoneal LDmin 14 days 16.8 13
Rabbit (III) oxide oral LDmin 14 days 33.5 30 Downs et al. (1960)
Rabbit (III) oxide intravenous LDmin 14 days 43.6 39
Rabbit (III) oxide intraperitoneal LDmin 14 days 67 60
Table 23 (contd).
Species Thallium compound Route of Toxicitya Period of Dose Referenceb
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Guinea-pig (I) acetate subcutaneous LD100 5-9 days 19.3 15 Kaeser & Lambert (1962)
Guinea-pig (I) acetate oral LDmin 14 days 15.5 12 Downs et al. (1960)
Guinea-pig (I) acetate intraperitoneal LDmin 14 days 9.0 7
Guinea-pig (III) oxide oral LDmin 14 days 5.6 5 Downs et al. (1960)
Guinea-pig (III) oxide intraperitoneal LDmin 14 days 33.5 30
Dog (I) acetate oral LDmin 14 days 25.8 20 Downs et al. (1960)
Dog (III) oxide oral LDmin 14 days 33.5 30 Downs et al. (1960)
a LDmin = minimal dose at which lethality was observed; LD10 = dose at which 10% of the animals were killed; LD50 = dose at which 50%
of the animals were killed; LD100 = dose at which all animals were killed
b Selected data; detailed data summarized in Negherbon (1959), Christensen et al., (1973), Zitko (1975a, b),Smith & Carson (1977),
Venugopal & Luckey (1978), Schoer (1984), Nessler (1985a), Manzo & Sabbioni (1988), ATSDR (1992)
c No data given
d Graphical determination based on data of authors
e Rats fed a low or high potassium diet. An increase in dietary potassium had a small protective effect.
In rats oral administration of a toxic dose of thallium sulfate
(10 mg thallium/kg) caused reduced food intake, diarrhoea, lethargy
and ocular haemorrhage regardless of whether or not the animal
survived the first 72 h (Sabbioni et al., 1982).
Summarizing 34 cases of canine thallotoxicosis, Zook & Gilmore
(1967) emphasized the variability of the sequence and the severity of
symptoms, partly due to the intoxication stage. Frequent symptoms
were vomiting (82%), cutaneous alterations (71%), depression (62%),
anorexia (53%), nervous disorders (47%), diarrhoea (44%), respiratory
difficulty (44%), conjunctivitis (41%), dehydration (24%) and
oesophageal paralysis (6%). The sequence in which the symptoms of
intoxication occurred was generally as follows: first anorexia,
vomiting and depression, then skin changes, dyspnoea and nervous
disorders. Usually, rectal temperature was not elevated but was later
often subnormal. After 3 to 7 days of illness, erythematous lesions
occurred, which were most severe near mucocutaneous junctions and on
the foot pads. The haematological findings were leukocytosis
(neutrophilia, lymphopenia, eosinopenia) and haemoconcentration.
Proteinuria and bilirubinuria were commonly observed. Autopsy and
histo pathological examination of 15 and 12 dogs, respectively,
revealed increased heart weight (presumably caused by systemic
hypertension), myocardial necrosis, congestion of the kidney with
tubular nephrosis, pulmonary oedema, enlarged spleen, enlarged or
oedematous lymph nodes, dilatation and areas of erosion of the
oesophagus and necrosis of skeletal muscles. Some myelinated nerves
showed focal distensions of their myelin sheaths, and lesions of the
cerebrum and cerebellum were evident in dogs with neurological
disorders (Zook & Gilmore, 1967). In addition, alopecia, anorexia,
emesis and tenesmus (Coyle, 1980) and haematemesis (Waters et al.,
1992) have been reported in cases of acute poisonings in dogs.
In cats similar signs of thallium poisoning have been found,
e.g., skin alterations, apathy, lack of appetite, vomiting and signs
of peripheral and central neuropathy (Zook et al., 1968; Fitzek &
Henning, 1976). Haematological and histopathological findings were
similar to those obtained with dogs. In older cats that died early,
haemorrhagic gastroenteritis and hepatic or renal damage were evident
(Zook et al., 1968).
Implantation of a pellet of pure thallium (3 to 5 mm diameter)
into the motor cortex of a monkey, Macaca mulatta, resulted in death
within 6 days (Chusid & Kopeloff, 1962).
7.1.2 Effects on various organs
Effects on the various organs have been summarized by Sabbioni &
Manzo (1980) and ATSDR (1992). In nearly all affected organs direct
cytotoxic effects as well as indirect effects due to damage of the
nervous system have been found.
In acutely poisoned rats (single subcutaneous injection of 20 to
50 mg thallium acetate/kg), there was mild to moderate enteritis,
including oedema of the submucosa and muscularis layers, and moderate
to severe colitis (Herman & Bensch, 1967). Ultra structural
degenerative changes in the liver were frequently present, especially
in the mitochondria. These were also indicated by increased numbers
of autophagic lysosomes and lipid droplets (Herman & Bensch, 1967) and
were evident 16 h after intraperitoneal injection of 50, 100 or 200 mg
thallium(III) chloride/kg into rats (Woods & Fowler, 1986). It was
concluded that "thallium-induced alteration of hepatic functional
processes may arise from physical disruption of the membranal
integrity of subcellular organelles with which those processes are
functionally associated" (Woods & Fowler, 1986). In an extreme case
of a poisoned dog, all the parenchymal hepatocytes were necrotic and
the adrenals, thyroid, pituitary and pancreas had degenerated (Larson
et al., 1939).
The kidney is considerably affected in poisoned animals (section
9.3.3). Since the concentration of thallium in the kidney is higher
than in other organs, it would seem to be a specific target organ.
Light microscopy of rat and mouse kidney tissue up to 48 h after
administration of lethal doses of thallium (30 to 40 mg thallium
sulfate/kg body weight) showed stromal oedema, necrosis of the loops
of Henle and hydropic degeneration, as well as swelling and focal
necrosis of the epithelium of proximal convoluted tubules (Danilewicz
et al., 1979, 1980). Electron microscopy revealed degenerative
changes in the epithelial cells of the glomeruli and tubules. The
same changes in the kidneys of acutely poisoned rats have been
described by Herman & Bensch (1967) (section 7.1.2).
Oral and intravenous administration of thallium sulfate (20 to
40 mg/kg) to the dog, cat, rabbit, goat and pigeon also caused direct
effects on the respiratory apparatus, in addition to decreasing
vasomotor reactivity (Rossi et al., 1981).
In in vitro studies, addition of 30 to 50 µg thallium sulfate
reduced the beat frequency in isolated frog hearts (Buschke &
Jacobsohn, 1922). Over a range of thallium concentrations, from those
encountered after uptake from a contaminated environment (2 µg/litre)
to those seen after suicide or homicide (and also higher levels of up
to 204 mg/litre), the contractility of sheep interventricular cordis
muscles exhibited three types of response, but they were not
correlated to the thallium concentrations or period of incubation
(Ziskoven et al., 1982). Using guinea-pig papillary muscles and low
concentrations (0.02 to 2 mg thallium/litre), positive inotropic
transients were followed by an inotropic decay (Ziskoven et al.,
1982). In contrast, thallium produced concentration- and
time-dependent positive inotropic effects in guinea-pig atrial
preparations, but also inhibition of the sodium pump in ventricular
slices (Ku et al., 1978). However, there is no discrepancy between
these two effects, assuming that thallium inactivates the already
fully activated pump and stimulates the inactivated pump (Ziskoven et
al., 1982). The authors suggested that at low concentrations the
effects of thallium are not associated with changes of membrane
activity but with energy supply. Parameters of the slow inward
current at the membrane level were not specifically altered by
thallium (Wiemer et al., 1982). Another investigation involving
guinea-pig papillary muscles and sheep Purkinje fibres indicated that
the arrhythmogenic effects of thallium are restricted to the sinus
node (Achenbach et al., 1982). High concentrations (200 mg/litre)
depolarize the muscle fibre membrane and lead to irreversible damage
(Mullins & Moore, 1960). Within the muscle, thallium seems to compete
for the adsorption sites normally occupied by potassium, being
adsorbed on to myosin and thus being localized primarily in the A band
(Ling, 1977).
7.2 Short-term exposure
7.2.1 Toxicity and symptoms
In mice the daily supplementation of food with 400 µg thallium
acetate induced alopecia after about 14 days, followed by increasing
apathy and death within 16 to 18 days after the beginning of the
treatment (Buschke, 1900).
Daily intraperitoneal injection of thallium(I) acetate (5 mg/kg)
for 7 days in rats caused anorexia, reduced growth, irritability and
tenderness during handling, lethargy, diarrhoea, dragging of the hind
limbs, fits of abnormal rotation of head and neck and curving of the
body. About 15% of the rats died (Hasan et al., 1977c).
7.2.2 Effects on various organs
In subacutely poisoned rats (2 to 3 injections of 10 to 15 mg
thallium acetate/kg at intervals of 1 week), only slight colitis and
enlargements of mitochondrial granules in the liver occurred (Herman &
Bensch, 1967). Electron microscopy of the kidney revealed similar
changes to those observed in acutely poisoned animals (section 7.1).
7.3 Long-term exposure: chronic toxicity
7.3.1 Toxicity and symptoms
The chronic toxicity data on thallium compounds are listed in
Table 24.
Table 24. Toxicity of different thallium compounds in experimental animals after several administrations
Species Thallium compound Route of Toxicitya Period of Dose Reference
administration observation (mg/kg body weight)
Thallium Thallium
compound ion
Mouse (I) chloride intraperitoneal LD50 30 days 1.2 0.1 Bienvenu et al. (1963)b
Mouse (III) chloride intraperitoneal LD10 30 days 6.0 4.0 Hart & Adamson (1971)b;
LD50 30 days 6.9 4.5 Adamson et al. (1975)
Mouse (I) nitrate intraperitoneal LD50 14 days 37.5 28.6 Williams et al. (1982)b
Rat (III) chloride intraperitoneal LD10 30 days 4.85 3.2 Hart & Adamson (1971)b;
LD50 30 days 5.66 3.7 Adamson et al. (1975)
Rat (I) sulfate oral LD20 15 days 1.25 1.0 Tikhova (1964)
LDmin 15 days 0.6
Rabbit (I) carbonate oral LDmin 180 days 0.25 0.2 Tikhova (1967)c
Rabbit (I) carbonate subcutaneous LDmin 180 days 0.25 0.2
Rabbit (I) sulfate oral LDmin 180 days 0.25 0.2 Tikhova (1967)c
Rabbit (I) sulfate subcutaneous LDmin 180 days 0.25 0.2
a LDmin = minimal dose at which lethality was observed; LD10 = dose at which 10% of the animals were killed; LD50 = dose at which 50%
of the animals were killed; LD100 = dose at which all animals were killed
b Daily injection for 10 consecutive days
c Value in winter was lower than in summer
In rabbits daily administration of thallium(I) sulfate or
carbonate (0.25 mg/kg) for 6 months caused disturbed behaviour,
aggressiveness, diarrhoea, and loss of hair (Tikhova, 1967). In rats
daily ingestion of 7.5 mg thallium was lethal (Bowen, 1966).
In a study of weanling albino rats (50-80 g) fed ad libitum for
1 month on a diet containing 2, 10, 50, 100, 500 or 5000 mg
thallium(I) acetate per kg diet (5 rats/group), dietary levels of 2
and 10 mg/kg diet caused no effects on growth and survival within the
feeding period, whereas the other concentrations resulted in
mortalities of 60 to 100% within 10 days. When rats were fed for 15
weeks with 5, 15, 30 or 50 mg thallium acetate/kg diet (5 males and 5
females/group), the two lowest doses did not affect growth of males or
females. The 15 and 30 mg/kg doses caused hair loss starting after 2
weeks, and after 15 weeks the rats were almost free of hair. Within
the fourth and eighth week and after intakes of 30 mg/kg diet, 80% of
males and 60% of females died. At 50 mg/kg all males died within 2
weeks and all females within 8 weeks. No specific pathological
alterations were found in any organ. A dose of 30 mg/kg diet resulted
in moderate growth depression in males but not in females, while in
both sexes increased mortality was observed. For thallium(III) oxide
the effective concentrations were similar, and males also reacted more
sensitively than females. No specific pathological alterations were
found in any organ except the skin, where atrophy of hair follicles
and sebaceous glands were seen at both higher dose levels. The exact
concentration of thallium ingested by the rats could not be determined
but was estimated to be in the range of 1 to 3 mg thallium acetate/kg
body weight per day for the diet containing 15 mg thallium acetate/kg
food (Downs et al., 1960).
US EPA (1986) conducted a 90-day study in which male and female
Sprague-Dawley rats (20 of each sex per group) were administered
aqueous thallium sulfate by gavage at doses of 0.01, 0.05 or
0.25 mg/kg body weight per day. Both untreated controls and vehicle
(water) treated controls were included. Clinical observations were
recorded daily and neurotoxicological examinations were performed 3
times per week on selected animals. Haematological and clinical
chemistry parameters were measured on days 0, 30 and 90, and
ophthalmological examinations were performed on days 0 and 90. Upon
necropsy, selected organs were weighed. No significant differences
were seen in any group for body or organ weights. Several changes
were reported for blood chemistry parameters. Statistics were
reported for male and female rats at 30 and 90 days for each dose
group compared with both untreated controls and vehicle-treated
controls. In both males and females, small increases were seen for
serum glutamic-oxaloacetic transaminase, lactic acid dehydrogenase and
sodium levels, with statistical significance at many points. At the
lowest dose, statistically significant changes were seen in male rats
for all three of these parameters, but only when compared with
untreated controls. Higher doses resulted in statistical significance
when compared with vehicle controls. Similar patterns were seen in
female rats.
In addition, there was a dose-related increase in the incidence
of alopecia, lacrimation and exophthalmos. No treatment-related
changes were seen in the eye. Gross necropsy revealed only alopecia;
this occurred in a dose-related manner for females and was apparent at
the lowest dose level. In males, alopecia was also apparent in all
dose groups but was not strictly dose-related.
In a study with female Sprague-Dawley rats (180-200 g), thallium
sulfate was given via the drinking-water (10 mg thallium/litre) over a
period of 40 weeks and with a total intake of about 80 mg thallium/rat
(Manzo et al., 1983). First symptoms, i.e. poor hair lustre,
periorbital redness and irritability, were observed on days 20 to 25.
Hair loss first appeared after 32 days in some rats, and in several
rats was almost complete by the end of the 36-week period of
treatment. However, in other rats no hair loss occurred. Starting
from day 40 the number of rats showing mild or severe cutaneous
disorders increased strongly. After 40 days and a total ingestion of
about 10 mg thallium/rat, lethality amounted to 15%, and the surviving
rats showed no electrophysio logical abnormalities. After 240 to 280
days and the ingestion of about 70 to 80 mg/rat only an additional 6%
of the rats died; about two-thirds of the rats showed
electrophysiological effects and reduced motor and sensory action
potentials.
7.3.2 Effects on various organs
In chronically poisoned rats (initial injection of 10 to
20 mg/kg, followed by weekly subcutaneous injections of 5 mg/kg for 4
to 26 weeks), only slight colitis sometimes occurred (Herman & Bensch,
1967), but degeneration in the liver was similar to the severe effects
in acutely poisoned rats (section 7.1.2) and liver enzymes were also
affected (Bulavintseva & Bulavintsev, 1982). In the stomach of rats
the production of hydrochloric acid was reduced (Buschke, 1929).
Kidney weight was increased (Downs et al., 1960) and ultrastructure
affected in the same way as in acutely poisoned animals (Herman &
Bensch, 1967) (section 7.1.1). The greater accumulation of thallium
in the renal medulla than in the renal cortex of chronically poisoned
rats indicates a firmer binding of thallium, which might impede
thallium elimination (André et al., 1960; Lameijer & van Zwieten,
1977a).
Intratracheal administration of 0.5 or 5 mg thallium(I) salts
(iodide, bromide and chloride or mixtures of them) caused dose- and
time-dependent changes in the lungs of rats, iodide being the most
toxic salt (Spiridonova et al., 1978).
In chronically poisoned guinea-pigs, the adrenaline and lipoid
contents of the adrenal glands were considerably, sometimes totally,
reduced. Examination of chronically poisoned rats revealed a reduction
in the size of the thyroid gland, follicular atrophy and some pycnotic
nuclei (Buschke & Peiser, 1922a,b; Buschke, 1929).
7.4 Skin and eye irritation
7.4.1 Skin and hair
Thallium intoxications in dogs caused striking effects in all
layers of the skin. The dermal changes were characterized by oedema
and disruption of collagen bundles. In erythematous patches massive
parakeratosis (much more pronounced than in other dermatoses) and
occasionally a granular layer were found. In the follicles, of which
only 60% contained hairs, the external root sheath was hyperplastic,
showing an excess of parakeratotic horny material. Follicular
plugging was prominent (Schwartzman & Kirschbaum, 1962). The progress
of the erythematous lesions towards scaling and crusting included
varying degrees of necrolysis and, characteristically for thallium
intoxications, spongiform abscesses, the latter occurring also in the
hair follicles (Schwartzman & Kirschbaum, 1962) where thallium binds
strongly to melanin (Tjälve et al., 1982).
The mechanism of depilation is unclear. According to Truhaut
(1960), this major sign of thallium intoxication, alopecia, is caused
by its antimitotic activity, since hair follicles (and testes) are
normally characterized by marked mitotic activity. This adverse
effect is prevented by glutathione and cysteine. Counting the mitotic
rate per hair follicle after a subcutaneous injection of 30 mg
thallium(I) sulfate into young rats at 4 and 7 days of age, Cavanagh &
Gregson (1978) observed an initial decline in mitotic rate followed by
cell deaths in the matrix zone.
On the basis of results from early experiments on mice, rats,
guinea-pigs, cats and rabbits performed by Buschke (1900, 1903, 1911),
Buschke & Peiser (1922b,c, 1926) and Buschke et al. (1928), the
authors put forward the hypothesis that the depilatory effect is
mediated by the activation of sympathetic nerves, since light
microscopy never revealed a direct effect on the hair follicles and
since sensory hairs, innervated by cerebrospinal nerves, were never
affected, in contrast to the smooth muscles of other hairs which are
innervated by the sympathetic system.
In chronically poisoned rats (initial injection of 10 to 20 mg
thallium acetate/kg followed by weekly subcutaneous injections of
5 mg/kg for 4 to 26 weeks) (Herman & Bensch, 1967), black speckling of
periorbital hairs occurred occasionally. In other chronically
poisoned rats (section 7.3) showing complete depilation, light
microscopy revealed effects similar to those after acute intoxication,
i.e. a keratinized epidermis, decrease in the size of sebaceous glands
and reduction of the number of hair shafts and follicles, the latter
becoming atrophic or replaced by scars, collagen or fat (Downs et al.,
1960).
7.4.2 Eye
Autoradiography of adult mice demonstrated a relative
accumulation of thallium in the lens of the eye (André et al., 1960).
A biochemical investigation showed that in addition to binding to
melanin in the hair follicles, thallium also binds strongly to melanin
in the mouse eye. This might result in iritis and retrobulbar
neuritis, which are regularly observed symptoms of thallium
intoxication (Tjälve et al., 1982).
7.5 Reproductive toxicity, embryotoxicity and teratogenicity
Thallium affects reproduction in various ways. Buschke & Peiser
(1922a) found a total reduction of sexual activity in intoxicated rats
and mice, which did not develop after administration to young animals
(Buschke & Peiser, 1922b,c) (section 7.3). This effect could be
antagonized by the administration of an ovarial hormone and
hypophyseal tissue (Buschke, 1929). Smith & Carson (1977) also
emphasized that sexual activity is usually lessened in chronically
poisoned animals.
7.5.1 Gonadotoxic effects
Several early, contradictory reports dealt with the question of
whether or not thallium affects ovarian function or the estrous cycle
(Smith & Carson, 1977). Buschke et al. (1927a) and Buschke & Berman
(1927) described strong inhibition of the estrous cycle in mice.
More attention has been directed to the effects on the male
reproductive system. Cytotoxic and perhaps mutagenic effects (section
7.6) can affect the offspring.
In several laboratory species, acute or repeated treatment with
thallium(I) salts resulted in similar or even higher concentrations of
thallium being found in the testes compared to other organs (section
6.2), pointing perhaps to a special susceptibility (Gehring & Hammond,
1967; Krassowski et al., 1977, 1980; Sabbioni et al., 1980a,b; Talas &
Wellhöner, 1983; Aoyama, 1989). However, an effect on offspring has
not been investigated in detail. Landauer (1930) demonstrated effects
on chickens, but only two cocks were included. A second
investigation, a dominant lethal test using rats, is discussed in
section 7.6.
Several authors have reported histological findings. In acutely
poisoned rats the epithelial cells of the seminal vesicle contained
numerous autophagic vacuoles (Herman & Bensch, 1967), and in acutely
intoxicated rabbits and dogs spermatogenesis was inhibited (Larson et
al., 1939; Truhaut, 1960; Zook & Gilmore, 1967). Chronic poisoning
caused total atrophy of testes in some rats (Buschke & Peiser,
1922a,b,c) or considerably disturbed spermatogenesis, which, however,
returned to normal after termination of thallium administration
(Buschke, 1929). A 6-month administration of thallium carbonate in
the drinking-water of mice (0.001 mg/litre or 0.01 mg/litre) caused a
decrease in sperm fertility at the high dose level and reduced sperm
motility at the lower dose level (Wei, 1987). Increased desquamation
of spermatogenic epithelium in rats was observed after oral
administration of 0.001 mg thallium carbonate/kg (Shabalina et al.,
1980). In rats treated with 10 mg thallium/litre in their
drinking-water for 2 months, but not in those treated for only for 1
month, there was rearrangement of the germinative epithelium,
premature release of germinal cells into the seminiferous tubules, low
epididymal sperm motility, the appearance of immature elements in the
semen and a high susceptibility in the Sertoli cells (Formigli et al.,
1985, 1986; Grégotti et al., 1985), the latter also reacting very
sensitively in in vitro studies (Grégotti et al., 1993). However,
thallium treatment did not affect the absolute or relative weight of
the testes or the plasma levels of testosterone. The mechanisms
underlying the findings for thallium may involve its antimitotic
effect or its effect on energy metabolism (Formigli et al., 1985,
1986). The use of thallium-201 (1-10 µCi), which is also utilized for
diagnosis in medicine, for testicular imaging in mice caused loss of
testicular weight and reduction in the number of sperm heads. These
effects were less severe when thallium-204 (1-10 µCi) was used. This
must have been due to the different radioisotopes (probably the
low-energy Auger electrons of thallium-201), rather than to the
physicochemical properties of thallium that determine uptake (Rao et
al., 1983).
7.5.2 Embryotoxicity and teratogenicity
7.5.2.1 Chickens
Injections of thallium (1 or 2 mg per egg) into the yolk sac of
4-day-old chick embryos strongly affected growth and survival.
Injections of thallium sulfate (0.7 mg/egg) into the chorioallantoic
membrane caused similar effects (Karnofsky et al., 1950; Ridgway &
Karnofsky, 1952).
More striking were the effects on the development of bones of the
embryonic chick: achondroplasia, leg-bone curvature, parrot beak
deformity and microcephaly (Karnofsky et al., 1950; Karnofsky & Lacon,
1964). The achondroplasia did not occur after injections of 0.2 mg
thallium sulfate/egg into the yolk sac, but with the larger doses
increasing percentages of the embryos showed shorter bones than
normal, and the tibia and femora were strikingly curved. Thallium
sulfate and nitrate showed a similar ability to produce achondroplasia
(Karnofsky et al., 1950).
Achondroplasia was only produced if thallium treatment was
performed during a critical, sensitive period (Hall, 1977, 1985).
This period began on day 5 of incubation and ended after 205 to 207 h
of incubation, coinciding with a 66% decrease in growth rate of the
embryos. During this period thallium bound rapidly to skeletal
tissues. Light and electron microscopy showed the primary site of
action to be the cartilage, maturing hypertrophic chondrocytes being
most affected (Ford et al., 1968; Hall, 1972; Skrovina et al., 1973).
An increase of glucosamine and a decrease of the mucopolysaccharides
without a simultaneous loss of collagen indicated an interference with
cartilage metabolism (Ford et al., 1968). According to Hall (1972),
thallium treatment caused an "abnormal distribution of the acid
mucopolysaccharides in otherwise normal cartilage matrix and the
formation of necrotic areas within the maturing hypertrophic
chondrocytes". The reduced secretion of the acid mucopolysaccharide
into the intercellular matrix did not affect ossification.
7.5.2.2 Mammals
In mammals, the results are inconsistent and there seems to be
great variation between species and strains (Claussen et al., 1981).
Intraperitoneal injection in rats on days 8 to 10 (2.5 mg thallium
sulfate/kg) or on days 12 to 14 (2.5 or 10.0 mg/kg) of gestation had
no effect on resorption rates but significantly reduced the body
weight of fetuses examined on day 21 of gestation. Hydronephrosis and
non-ossification of vertebral bodies were observed. The effects on
fetal body weight and vertebrae were also induced by a low potassium
diet and were not increased by an additional dose of thallium (Gibson
& Becker, 1970). According to Barlow & Sullivan (1982), the increase
in hydronephrosis may not be related to thallium. Adverse effects on
the cartilage of long bones in 6- and 9-day-old rats treated
intraperitoneally with thallium sulfate were described by Nogami &
Terashima (1973). These results might explain the reduced growth of
the suckling rats of thallium-intoxicated dams (Ehrhardt, 1927).
Ossification of rat skulls was also reduced (Buschke et al., 1927b).
In a preliminary study on mice, oral administration of thallium
sulfate (0.3, 1 and 8 mg thallium/kg body weight) to 3-6 pregnant SWS
mice (gestation day 9) caused achondroplasia in 12.5, 14 and 50% of
the offspring, respectively (Achenbach et al., 1979a,b). Earlier
administration of thallium induced miscarriage, while older fetuses
were only slightly affected. In other substrains of the SWS mice,
administration of 10 to 20 mg/kg induced no teratological effects
(Claussen et al., 1981). Under similar conditions, but using 29 NMRI
mice and oral doses of 0.8 and 8 mg thallium/kg body weight, the
weights of fetuses and placentas were not affected by the treatment
with thallium sulfate, but a slight, statistically non-significant
increase in the intra-uterine mortality rate was found in embryos
examined 18 days after fertilization. A significantly increased rate
of double placentas and of effusions of blood in the thigh of the
fetuses was observed at 8 mg/kg (Török & Schmahl, 1982). Oral
administration of 3 or 6 mg thallium chloride or acetate per kg per
day to NMRI mice (about 30 per group; day 6 to 15 of gestation) had no
observable effects on skeleton or organs at day 18 of gestation
(Claussen et al., 1981). However, post-implantation losses were
increased after administration of 6 mg thallium chloride/kg, but only
young embryonic stages were affected. Administration of 6 mg thallium
acetate/kg caused a reduction in the weight of embryos. In a parallel
study with rats, 6 or 4.5 mg thallium chloride or acetate/kg was
lethal to the dams (gestation day 6 to 15), while 3 mg/kg induced no
embryotoxic effects but increased malformations of ribs and vertebrae
(Claussen et al., 1981).
Studies of mouse embryos cultured in vitro showed that low
levels of thallium (0.2 mg/litre) affected the pre-implantation stage
(summarized by Formigli et al., 1985).
In vitro cultivation of mouse limb buds (day 11 of gestation)
in medium containing 10 mg/litre for a period of 3 days, followed by
cultivation in normal medium for another 3 days, caused impaired
development of the hand skeleton (Barrach & Neubert, 1985). In a
similar study, the extent of this effect was also increased by
decreasing the potassium concentrations of the medium, and vice versa
(Neubert & Bluth, 1985). The authors suggested that an interference
of thallium with mammalian embryonic development is possible if its
concentration in embryonic tissues reaches levels above 2-3 mg/kg.
When rat embryos (10.5 days after fertilization) were cultured
for 48 h in media containing thallium sulfate (3, 10, 30 or 100 mg
thallium/litre), embryotoxic effects were evident at all concen
trations. Increasingly deleterious effects occurred on growth and on
mesoderm and entoderm at concentrations from 10 to 100 mg/litre.
Whereas macroscopically no or only minor abnormalities could be
detected, histological examination of the embryos revealed that cell
necroses in the brain developed even at the lowest concentration. At
higher concentrations these effects increased, and necrosis was
complete at 30 mg/litre. The authors doubted whether a suggested
embryotoxic concentration of 1 mg/litre can be achieved without severe
maternal poisoning (Anschütz et al., 1981).
7.5.2.3 Delayed effects on development of offspring
Many offspring of chronically poisoned rats showing total
alopecia died during the first two days after birth. In the other
pups, hair development was severely impaired (section 7.4), and these
pups died within 4 to 5 weeks after birth. In a second litter all
offspring showed alopecia, but none of them died so early (Buschke,
1911).
In studies by Claussen et al. (1981), oral administration of 6 mg
thallium chloride/kg to NMRI mice dams (day 6 to 15 of gestation)
reduced the initial increase in weight of the offspring and slightly
increased the mortality rate during the first 3 weeks. In a parallel
study with rats, oral administration of 3 mg thallium chloride or
acetate per kg caused similar effects.
In rats, exposed prenatally by treating the dams with 0.01 g
thallium sulfate/litre in their drinking-water, gestation and weight
of offspring were not affected, but hair development was retarded
during the first 60 days of life. At an age of 30 or 60 days, the
hypotensive and hypertensive cardiovascular responses to acetylcholine
or to isoprenaline and noradrenaline, respectively, were lower than
the responses of control animals (Matera et al., 1986; Rossi et al.,
1988). Comparisons with postnatally exposed pups showed that the
reactivity of the developing vascular autonomic nervous system was
also lowered (Rossi et al., 1988).
Oral doses of 0.1, 0.5 or 1.0 mg thallium/kg body weight
(thallium sulfate) given to rats on days 6, 7, 8 and 9 of gestation
impaired the learning ability of their adult offspring in operant
behaviour tests. Postnatal administration of thallium (same doses)
had no such effect (Bornhausen & Hagen, 1984).
7.6 Mutagenicity and related end-points
Only two microbiological investigations have been performed;
these indicated no mutagenic effects (Claussen et al., 1981). Both
used the Ames test. Dehnen, whose unpublished data were described by
Claussen et al. (1981), investigated the effects of thallium(I)
acetate and used 3.1 µg-29.2 mg thallium/plate and the Salmonella
typhimurium strains TA98, 1535, 1537 and 1538. Similarly, the use
of Ames tests with thallium chloride and acetate (doses not given) and
S. typhimurium strains TA98, 100, 1535, 1537 and 1538, with and
without metabolic activation, indicated no mutagenic effects (Claussen
et al., 1981). According to these authors, no sister-chromatid
exchange was found in bone marrow cells after oral administration of
thallium chloride to Chinese hamsters (5 or 10 mg thallium/kg body
weight; twice after 24 h; 8 animals per dose).
However, thallium(I) carbonate induced sister-chromatid exchange
and chromosomal aberrations in one cell line and hypoxanthine-guanine
phosphoribosyl-transferase gene mutation in another cell line (Zhang,
1988). In studies with embryonic fibroblasts of various mouse and rat
strains, the same thallium(I) compound (0.5 to 46.9 mg/litre) caused a
significant increase in the single-stranded DNA fraction after
incubation of rat fibroblasts and cells of one mouse strain (C57Bl/6),
whereas cells of the CBA mouse strain were resistant to the same
concentrations. In a test of survival and mutability of vaccinia
virus in both mouse cell lines, the CBA cells showed increased
survival of the virus, suggesting greater efficiency of the repair
systems (Zasukhina et al., 1980, 1983).
In a dominant lethal test, white rat males received daily oral
doses of 5, 50 and 500 ng thallium carbonate/kg body weight over 8
months. Thereafter they were mated with untreated females. On
gestation day 20, 18 dams were killed. At the two higher doses there
was a tendency towards an increase in embryonic mortality, whereas at
the lowest dose the number of resorptions and post-implantation deaths
were increased (Zasukhina et al., 1983). Using these data in a
T-test, the differences between the means for the total embryonic
death of treated and untreated mice were statistically significantly
different (p < 0.05).
A mutagenic effect on sperm cells of rats was reported after oral
administration of 0.0001 mg thallium carbonate/kg (Shabalina et al.,
1980). However, the report lacked detail concerning the experimental
set-up and the Task Group considered that it could not be used for
evaluation of the health effects of thallium.
7.7 Carcinogenicity
No standardized carcinogenicity studies have so far been
performed (ATSDR, 1992). Owing possibly to their cytotoxic effects,
thallium salts may have a local antineoplastic effect in mice and rats
(Hart et al., 1971; Hart & Adamson, 1971; Adamson et al., 1975).
7.8 Neurotoxicity
7.8.1 Central nervous system
7.8.1.1 Histology and ultrastructure
In the brains of subacutely poisoned rats, Herman & Bensch (1967)
(section 7.1.2) occasionally found foci of perivascular cuffing, while
the mesencephalon of two of the four rats contained an extensive
region of acute necrosis. In addition, numerous lipofuscin bodies
were sometimes present in neuron cytoplasm. This was also found to
occur following chronic poisoning; electron microscopy showed effects
on mitochondria (Herman & Bensch, 1967).
The brain of acutely poisoned guinea-pigs (subcutaneous injection
of 15 or 18 mg thallium sulfate/kg) showed slight microscopic
alterations, e.g., swelling of cells and vacuolization in the
perikaryon (Tackmann & Lehmann, 1971). In the right parietal cortex
of rats, microglia cells and alpha astrocytes were affected 24 h after
an intraperitoneal injection of 40 mg/kg (Reyners et al., 1981). At
24 h after intraperitoneal injection of 32 mg/kg into newborn rats,
the encephalon showed oedema and congestion; even after an additional
50 days, there was focal destruction of neurons and irregular fibrosis
of the capillary vessels (Barroso-Moguel et al., 1990).
Ataxia and tremors are known to be associated with cerebellar
lesions and both neurological disorders occur in cases of thallium
intoxication. Ultrastructural alterations of the cerebellum,
especially of the mitochondria, were evident in rats after poisoning
by daily intraperitoneal injections of thallium acetate (5 mg
thallium/kg for 7 days) (Hasan et al., 1978a). Effects in other brain
regions indicate that the effects of thallium on the activity of
endocrine glands may be mediated via changes in hypothalamic control
(Hasan et al., 1977b). Using identical conditions, Hasan et al.
(1977a) observed an apparent increase in the number of
oligodendrocytes and suggested a correlation with thallium-induced
neuronal chromatolysis described by other authors, since the usual
functions of oligodendrocytes are the formation of myelin and the
nutrition of neurons.
7.8.1.2 Electrophysiological and biochemical investigations
In addition to the direct effects of thallium on the cardio
vascular and respiratory apparatus of the dog, cat, rabbit, goat and
pigeon, indirect effects involving higher vasomotor and respiratory
centres (but mostly dependent on a decrease in vasomotor reactivity)
were found following acute poisoning (section 7.1.2) (Rossi et al.,
1981).
Extra- and intracellular recording of central neuronal activity
in hippocampal slice preparations from guinea-pigs and rats showed
that thallium (20 to 40 mg/litre) seems to have a predominantly
postsynaptic effect in hippocampal slice preparations, perhaps by
exerting an unspecific influence on the intracellular metabolic
mechanisms of the CA1 pyramidal cells (Lohmann et al., 1989). Daily
intraperitoneal injections of thallium(I) acetate (4 mg/kg for 7 days)
indicated that the electrophysiological parameters of noradrenergic
transmission in rat cerebellum were reduced (Marwaha et al., 1980).
The association of the corpus striatum with the pathogenesis of
the abnormal movements that have been reported after thallium
intoxication is shown by an increased firing rate of the caudate
neurons of rats, 3 to 4 h after intravenous injection of 10 mg
thallium sulfate/kg (Hasan et al., 1977c). After daily
intraperitoneal injections of 5 mg thallium(I) acetate/kg for 7 days
(section 7.8.1), the protein content of the corpus striatum was
significantly increased and the respective breakdown enzymes were
depleted. However, the latter did not occur in the cerebrum (Hasan et
al., 1977b,c).
Certain motor dysfunctions are known to be associated with a
decrease in the level of brain dopamine, an aspect supported by the
data of Hasan et al. (1978b). Convulsive disorders may also be
related to a brain deficiency of gamma-aminobutyric acid (GABA)-ergic
mechanisms. Data obtained by Hasan et al. (1977d), but not those by
Nisticò et al. (1984), support this interpretation. Neurotoxicity
could also result from changes in the concentrations of amino acids
and other neurotransmitters (Ali et al., 1990) or an acceleration of
monoamine catabolism (Osorio-Rico et al., 1994).
Further important mechanistic aspects were increases found in the
lipid peroxidation rates and in the activity of the lysosomal enzyme
ß-galactosidase, especially in the cerebellum, brainstem and cortex,
after daily intraperitoneal injection of 8 mg thallium(I) acetate/kg
for 6 days in rats (Brown et al., 1985). Also a lower dose of 4 mg/kg
selectively altered patterns of behaviour (section 7.8.1.3). An
increased deposition of lipofuscin-like pigment granules in the
cerebellar neurons and an increase in lipid peroxidation rates in the
cerebrum and brainstem of rats (which was even exceeded by that in the
cerebellum) were described in a previous study (Hasan & Ali, 1981).
This seems to be an important mechanism of toxicity (section 7.9).
7.8.1.3 Behavioural toxicology
Intraperitoneal injections of 10 or 20 mg thallium sulfate/kg
produced only a slight conditioned flavour aversion in rats, perhaps
due to the delayed onset of symptoms (Nachman & Hartley, 1975; Peele
et al., 1986). Oral administration induced a dose-dependent aversion
to saccharin at all but the lowest dose of 2.5 mg/kg (Peele at al.,
1986, 1987). This difference might be explained by irritation of the
gastrointestinal tract after oral uptake of thallium.
In a detailed investigation of the effects of daily
intraperitoneal injections of 4 or 8 mg thallium(I) acetate/kg for 6
days on several behavioural patterns of rats, changes of behaviour
were intensified with increased dose. Some of the changes correlated
with biochemical effects (section 7.8.1.2) (indicating cellular
damage) in certain regions of the brain (Brown et al., 1985).
In pest control campaigns against wild rodents, the dying animals
left their hiding places and came to the surface, presumably due to
extreme thirst and breathing disturbances (Larson et al., 1939).
7.8.2 Peripheral nervous system
In the final phase of lethal intoxication and in vitro, a
parasympathetic stimulation seems to occur. Because thallium
diminishes the effects of adrenaline on isolated hearts or intestine,
even after parasympathetic blockage, it is thought that it may destroy
adrenaline (Truhaut, 1960).
In the initial phase of intoxication, sympathetic nerves are
stimulated (Buschke & Peiser, 1922b). Investigating the effects of
0.2 to 204 mg thallium(I) and thallium(III) ions/litre on the ATPases
of the amine-storing granules from bovine adrenal medulla and splenic
nerves, a specific inhibition by thallium(III), but not by
thallium(I), was observed at concentrations which might occur in the
tissues after intoxication with thallium(I). The authors suggested
that thallium(I) is oxidized to thallium(III) in the organism.
Because the ATPase of the nerve granula which store noradrenaline was
nearly ten-fold more sensitive than that of the adrenal medulla, which
stores mainly adrenaline, this might explain the strongly increased
elimination of noradrenaline (Burger & Starke, 1969).
7.8.2.1 Histology and ultrastructure
In the peripheral and optic nerves of acutely, subacutely and
chronically poisoned rats (Herman & Bensch, 1967) (section 7.8.1), no
consistent or even slight changes were revealed by light or electron
microscopy. However, partial atrophy of the optic nerve was found by
Buschke et al. (1928). In addition, acute poisoning of a dog (Greving
& Gagel, 1928) and guinea-pigs (subcutaneous injection of 15 or 18 mg
thallium sulfate/kg) (Tackmann & Lehmann, 1971) caused alterations of
the axons and myelin sheaths which were evident under the light
microscope. In a 36-week study (Manzo et al., 1983) (section 7.3), in
which rats were given thallium sulfate in their drinking-water (10 mg
thallium/litre), about 50% of the animals developed Wallerian
degeneration (myelin debris and vacuolization); lamination of the
myelin sheath of the sciatic nerve fibres was confined to some large
and medium-sized fibres. Degenerative lesions found in the white
matter of the spinal cord of poisoned rabbits may account for the
paralysis of the hind legs (Truhaut, 1960).
In a number of in vitro studies, thallium affected nerves,
e.g., cell outgrowth was inhibited (Sharma & Obersteiner, 1981;
Windebank, 1986) or the myelin sheath disintegrated (Peterson &
Murray, 1965).
7.8.2.2 Electrophysiological and biochemical investigations
Following a subcutaneous injection of 15 mg thallium sulfate/kg
to guinea-pigs, within days the larger and faster conducting nerve
fibres degenerated before the slower fibres and became inexcitable
(Kaeser & Lambert, 1962; Tackmann & Lehmann, 1971). In a subchronic
study (section 7.3) on rats, which received thallium sulfate via their
drinking-water (10 mg thallium/litre), the motor and sensory action
potential amplitudes were unaffected after 40 days of poisoning but
decreased after 240 days (Manzo et al., 1983). Then the motor action
potential latency was increased, and no fibrillation activity was
observed in the tibialis anterior muscle.
Neuromuscular transmission in thallium-treated rats and mice has
been investigated in detail using phrenic nerve-diaphragm preparations
(Wiegand et al., 1983, 1984a, 1986). The relationship between
thallium concentration (z, in mM) and duration of paral ysis (y, in
minutes) is approximated by the equation y = 4.6 × e8.4z (Csicsaky &
Wiegand, 1981). It has been suggested that thallium interferes
presynaptically with spontaneous transmitter release by antagonizing
these calcium-dependent processes, rather than by interfering with the
presynaptic influx of calcium ions (Wiegand et al., 1983, 1984a,b;
Wiegand, 1988). Additional data indicate that thallium, like other
heavy metals (Cooper et al., 1984), irreversibly blocks phasic
transmitter release, while spontaneous transmitter release is
reversibly enhanced (Wiegand et al., 1986; Csicsaky et al., 1988).
The sequence of the toxic effects indicate that thallium needs to be
transported across the cell membrane before it can finally interfere
with the release mechanisms. This rather indirect mode of action of
thallium was also found in the recordings of presynaptic ion currents.
Perineural recording techniques and the blocking of potassium
channels excluded the possibility that presynaptic potassium or
calcium channels were influenced by thallium in acute superfusion
experiments. Thus, the mechanisms that cause enhancement of the
spontaneous release of acetylcholine and the reduction of phasic
transmitter release at the neuromuscular junction, both of which are
induced by thallium, remain unknown at present (Wiegand et al., 1990).
7.9 In vitro test systems: cell lines
Ultrastructural studies with cultured fetal mouse heart cells
showed swollen mitochondria with loss of cristae, disintegration of
the membrane system, and a protective effect of selenium (Liu, 1986)
(section 7.10.2).
A cytotoxic effect on ovary cells was observed in in vitro
experiments on Chinese hamster ovary cells. After a 16-h incubation
with thallium(I) nitrate (40 mg/litre), 50% of the cells did not form
colonies during the following 7 days (section 7.9) (Hsie et al., 1984;
Tan et al., 1984).
Using three mammalian cell lines (human diploid embryonic
fibroblasts, HeLa cells and mouse fibroblasts) to test 11 heavy
metals, thallium was found to belong to the group of metals with a
strong inhibitory (on proliferation) or lethal effect. After
treatment for 7 days, the minimal inhibitory concentrations of
thallium(I) and thallium(III) for all three cell lines were 4 mg/litre
and 2 mg/litre, respectively, while the 50% inhibitory concen
trations were 10-15 mg/litre and 5-15 mg/litre, respectively. Half of
the cells were killed by 20-40 mg/litre (Fischer, 1981).
7.10 Factors modifying toxicity
7.10.1 Enhancement of elimination
Various substances have been evaluated for their ability to
enhance faecal or renal elimination of thallium (e.g., Lund, 1956b).
In rats, potent diuretic agents such as furosemide and ethacrynic acid
increased renal elimination of thallium, but did not further increase
the elimination induced by feeding a potassium-rich diet (Lameijer &
van Zwieten, 1977a,b). A sodium-rich diet did not promote the renal
elimination of thallium (Lameijer & van Zwieten, 1979). In rats, oral
application of activated charcoal increased faecal elimination by
about 80% but did not affect urinary elimination (e.g., Lund, 1956b).
In contrast, potassium chloride and cystine only increased renal
elimination by 47% and 60%, respectively. Meyer & Tal (1957) reported
that some compounds containing sulfur and labile methyl groups seem to
reduce the toxicity of thallium in rats.
Dithizone (diphenylthiocarbazone), which forms a firm complex
with thallium, increased faecal elimination by 33% and urinary
elimination by 75% in rats (Lund, 1956b). After treatment with
diethyldithiocarbamate "dithiocarb", the resulting lipophilic
thallium(I) chelate readily passed the blood-brain barrier and was
rapidly decomposed in the brain (Kamerbeek et al., 1971a). However,
it did not protect animals from death after administration of lethal
doses of thallium (Danilewicz et al., 1980). The benefit of other
agents, such as the diuretic 2,3-dimercapto-succinic acid (Liang et
al., 1980) or other compounds with mercapto groups (Oehme, 1972), has
still to be proven in the treatment of thallotoxicosis.
Comparing different antidotal treatments in rats, Lehmann &
Favari (1985) found an increase in thallium elimination to 99% by
dithizone, 93% by activated charcoal, 64% by the diuretic agent
furosemide, 82% by Prussian Blue, and 92% by combining Prussian Blue
and furosemide, whereas the untreated controls had only eliminated 53%
of the administered dose (2 mg) within 8 days. After treatment with
Prussian Blue and D-penicillamine in combination, the dangerous
redistribution of D-penicillamine did not occur and elimination of
thallium was better than after administration of Prussian Blue alone
(Rios & Monroy-Noyola, 1992). The efficiency of Prussian Blue could
be increased by synthesizing batches with a smaller crystal size (Rios
et al., 1991; Kravzov et al., 1993). New compounds, such as rhyolith,
N-acetylcysteine and dimercaprol, were no more, or even less,
effective (Dvorák, 1973; Henderson et al., 1985).
7.10.2 Selenium
Selenium not only protected isolated fetal hearts against damage
by thallium (section 7.9), but also decreased its lethal effect in
young rats; however, thallium-induced hair-loss was not prevented
(Ewan, 1978; Ostádalová & Babicky, 1987). Thallium inhibited
pulmonary elimination of volatile selenium compounds, increased the
retention of selenium in kidneys and liver, and did not protect
against chronic selenosis (Levander & Argrett, 1969). Thallium did
not affect the distribution of selenium in the body of mice; in
in vitro systems it seemed to interact with selenite in glutathione
solution and in erythrocytes (Naganuma et al., 1983).
7.11 Mechanisms of toxicity - mode of action
Although several (perhaps interconnected) mechanisms have often
been postulated, the exact mechanism of thallium toxicity is still
unknown (Cavanagh et al., 1974; Prick, 1979; Sabbioni & Manzo, 1980;
Nessler & Briese, 1985; Chandler & Scott, 1986; Cavanagh, 1988).
Conflicting results have been obtained with respect to the
effects of thallium on sodium/potassium ATPase activity in vitro
(stimulation) (Ivashchenko & Balmukhanov, 1974) and in vivo
(inhibition) (Mourelle et al., 1988). The irreversible inhibition of
the unidirectional transport of sodium may be due to an inhibition of
transport energy (Skulskii & Lapin, 1983). The affinity of the
thallium ion for the sodium/potassium ATPase is 9-10 times greater
than that of the potassium ion (Britten & Blank, 1968; Inturrisi,
1969). Since the thallium permeability of biological membranes is
usually 10 to 100 times greater than that of potassium and since a
similar activation of membrane sodium/potassium ATPase in human
erythrocytes and rat liver cells is caused by thallium concentrations
that are ten times lower than those of potassium and higher
concentrations of ouabain are needed to inhibit thallium-activated
ATPase, a high selectivity of thallium(I) ions for potassium transport
pathways exists (Skulskii et al., 1973, 1975; Gutknecht, 1983; Favari
& Mourelle, 1985; Zeiske & van Driessche, 1986). However, in vitro
studies must take into account the fact that concentrations of only
0.01 g thallium/litre are likely to occur in cells after lethal
poisoning with 10 mg/kg body weight, assuming uniform distribution and
no elimination (Burger & Starke, 1969).
Owing to the similarity in ionic radii of potassium and thallium,
and since the affinity of the thallium ion for sodium/potassium ATPase
is greater than that of the potassium ion, thallium accumulates within
the cell at the expense of potassium. The strong interaction of
thallium with sites normally occupied by potassium may block cycles
that depend on recurrent potassium translocation (Sabbioni & Manzo,
1980).
Once inside the cell, various mechanisms are evident, e.g.,
effects on other enzymes (Sabbioni & Manzo, 1980), the inhibition of
protein synthesis (Hultin & Näslund, 1974), the antimitotic effect of
thallium compounds (section 7.9) (Sabbioni & Manzo, 1980), especially
in the testes (sections 7.5 and 8.5.1), and/or the involvement of
riboflavin vitamin B12, which is coenzyme to a number of enzymes
(Emsley, 1978; Nessler & Briese, 1985). Any effect on riboflavin or
on enzymes containing sulfhydryl groups (see below) should result in a
disturbance of pyruvate metabolism (summarized by Nessler & Briese,
1985). Experimental animals suffering from riboflavin deficiency show
symptoms similar to those of thallium intoxication (Schoental &
Cavanagh, 1977).
Another postulated mechanism considers the general capacity of
thallium to react with thiol groups, thus interfering with a variety
of processes (Zeiske & van Driessche, 1986). Although the toxic
effect of thallium is reduced by diets high in cystine, methionine and
betaine (Oehme, 1972), interference with the metabolism of
sulfur-containing amino acids does not seem to be directly involved in
toxicity (Garcia Bugarin et al., 1989) and strong reactions with
thiols were observed for thallium(III) but not for thallium(I)
compounds (Douglas et al., 1990). An additional aspect of the
reaction of thallium with sulfhydryl groups is the induction of free
radical formation (section 7.8). This is indicated by increased lipid
peroxidation rates in the brain. Processes leading to lipid
peroxidation finally damage cell membranes by subsequent reactions of
free radicals with sulfhydryl enzymes (section 7.11). Lipid
peroxidation results in a deficiency of glutathione and leads to the
accumulation of lipid peroxides in the brain, liver and kidney and,
presumably, finally to lipofuscin granules (Herman & Bensch, 1967;
Aoyama et al., 1988). The possibility that this mechanism is
responsible for the neurotoxic effects of thallium was supported by
the simultaneous administration of thallium and acetyl-homocysteine
thiolactone, which prevented reduction in the level of sulfhydryl
radicals in the cerebellum and significantly increased glutathione
levels (Hasan & Haider, 1989). This hypothesis is further supported
by the protective effect of 1) silymarin against thallium hepato
toxicity (Mourelle et al., 1988), 2) selenium, demonstrated with the
thallium-induced ultrastructural changes found in isolated fetal mouse
heart cell (sections 7.9 and 7.10.2.) (Liu, 1986), and 3) selenium and
vitamin E against membrane damage by uncontrolled lipid peroxidation
in vivo (Hasan & Ali, 1981).
The mode of action of thallium seems to be mainly based on a
disturbance in the function of the mitochondria (Barckow & Jenss,
1976), although they are affected by high concentrations of almost all
heavy metals (Byczkowski & Sorenson, 1984). The thallium(I) cation
may either enter isolated rat liver mitochondria passively, i.e. in an
energy-independent manner (Barrera & Gómez-Puyou, 1975), or penetrate
the mitochondrial membrane electrophoreti cally (Skulskii et al.,
1978). The entry of thallium into the intramitochondrial space and
the interaction of cytosolic thallium with mitochondria membranes may
explain the deleterious effects (Skulskii et al., 1984). In isolated
mitochondria, thallium(I) acetate caused an uncoupling of oxidative
phosphorylation and swelling of the isolated mitochondria (Melnick et
al., 1976). Using ascite tumour cells in vitro, Ivashchenko et al.
(1973) found a strong, thallium-induced increase in oxygen consumption
and lactic acid production, which were inhibited by ouabain and sodium
fluoride. However, whereas oxygen consumption and anaerobic
glycolysis of tissues were affected in vitro, tissues from rats with
chronic or acute (first day) poisoning did not differ from those of
controls (Thyresson, 1950).
Comparing the effects of monovalent and trivalent thallium on
isolated rat liver mitochondria, only thallium(III) nitrate uncoupled
oxidation from phosphorylation (Hollunger, 1960). This effect could
not be reversed by adding edetic acid or dimercaprol (sections 7.10.1
and 8.6).
8. EFFECTS ON HUMANS
The toxicology of thallium is summarized in Fig. 1.
8.1 General population exposure
Thallium concentration in early-morning urine samples of nine
non-exposed subjects ranged from 0.13 to 1.69 µg/litre (Weinig & Zink,
1967). Smith & Carson (1977) gave a range of 0.6 to 2.0 µg/litre with
a mean urinary thallium concentration of 1.3 µg/litre.
The initial use of thallium in medicine, mainly as a depilatory
drug, caused many cases of intoxication. The observation of side
effects led to more detailed toxicological studies after 1918, and its
medical use was abandoned after 1945 (Emsley, 1978; Briese & Nessler,
1985a). Since thallium is tasteless, odourless, without colour and
highly toxic, and used to be easily obtainable, it was often used for
suicide, homicide and attempts at illegal abortion (Kemper & Bertram,
1984; Manzo & Sabbioni, 1988). Also large numbers of accidental
intoxications have occurred, e.g., in Berlin, Germany there were 110
cases of accidental ingestion and 39 attempted suicides by children
involving corn poisoned with thallium sulfate from 1967 to 1976; three
of the children died (von Mühlendahl et al., 1978). Munch (1934b)
summarized thallium intoxications prior to 1934. He found reports on
8006 children who had been treated with thallium as a depilatory
agent. Intoxication occurred in 447 cases and 8 children died. In
connection with its use as a rodenticide or insecticide, 21 poisonings
and 5 deaths occurred.
Recently, intoxications occurred after ingestion of a Chinese
herbal medication/nutritional supplement containing 30 g thallium per
litre, but other samples contained no thallium (Schaumburg et al.,
1992).
8.1.1 Acute toxicity
Cases of acute intoxication by thallium salts in humans, which
always cause severe symptoms, have been reported for single or
multiple oral doses of the order of 100 mg or more for adults, i.e.
1.5 to 2 mg/kg body weight (Table 25).
Symptoms of acute thallium toxicity vary with age, dose and route
of administration (Venugopal & Luckey, 1978). According to Sharma et
al. (1986), one-tenth (2-10 mg/person) of the lethal thallium dose for
adults causes death in children. However, reports of the therapeutic
use of thallium in which children tolerated larger doses than adults
indicate the contrary (Ormerod, 1928; Sessions & Goren, 1947; Prick,
1979). Of 75 children who had accidentally ingested thallium sulfate,
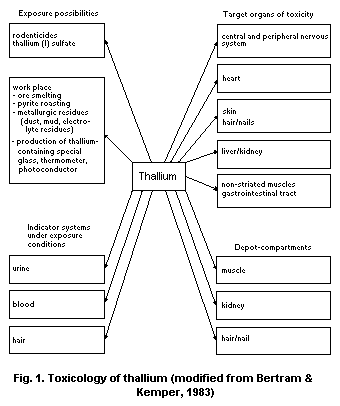 only two showed symptoms of thallium intoxication: a 2-year-old child
who had ingested 3.5 mg thallium/kg and eliminated 130 µg
thallium/litre in the urine showed no reflex movements of the legs in
the second week, while another child with 3.9 mg thallium/litre in its
urine and 40 µg/litre in its blood showed no neurological symptoms
apart from a massive alopecia in the second week. In one of two
lethal cases, the urine contained 10.3 mg/litre, and the death of both
children occurred within 2 days (von Mühlendahl et al., 1978). The
influence of age is unclear, but this may be due to the reporting of
doses either as total dose or as mg/kg body weight. Therapeutic uses
of thallium in the 1930s (8 mg/kg) resulted in peripheral neuritis in
about 10% of the patients (Cavanagh, 1979).
Table 25. Acute toxicity of thallium
Toxicitya Dose Adult/children Reference
TT > 1.5 mg/kg not specified Schoer (1984)
MLD 10-12 mg/kg adult Kazantzis (1994)
2-10 mg/kg children
MLD 14-18 mg/kg not specified Sessions & Goren (1947)
Lethality 20-100 mg per person adult Sharma et al. (1986)
2-10 mg per person children
a MLD = minimal lethal dose; TT = toxicity threshold
In adults lethal doses vary between 6 and 40 mg/kg (500 to
3000 mg/person), with an average dose of 10-15 mg/kg body weight
(Schoer, 1984). Without therapy the average dose usually results in
death within 10-12 days (Kemper, 1979), but, summarizing 150 mostly
suicidal cases of thallium intoxication, most of the patients who had
ingested about 600 mg and had no gastric lavage within the initial 2 h
died within 8-10 h (Potes-Gutierrez & Del Real, 1966). After
accidental ingestion of 10-fold overdoses of thallium acetate
(80 mg/kg), given as a depilatory in ringworm disease (Tinea), four
children died after 1-2 days (Lynch et al., 1930). Sessions & Goren
(1947) had previously suggested 14 to 18 mg thallium/kg body weight to
be the minimal fatal dose.
The triad of gastroenteritis, polyneuropathy and alopecia is
regarded as the classic syndrome of thallium poisoning (Gastel, 1978),
but in some cases gastroenteritis and alopecia are not observed.
Other symptoms also develop in varying sequence. Both lethal and
sublethal doses give rise to most of the symptoms, but these same
symptoms vary in intensity and time, probably in a dose-dependent way.
The following general trends have been summarized from numerous
reviews and case reports, e.g., Muller (1961), Moeschlin (1965, 1986),
Potes-Gutierrez & Del Real (1966), Venugopal & Luckey (1978), Gastel
(1978), Möllhoff et al. (1979), Sabbioni & Manzo (1980), Davis et al.
(1981), Saddique & Peterson (1983), Kemper & Bertram (1984), Le Quesne
(1984), Schoer (1984), Ohnesorge (1985), Nessler & Briese (1985),
Chandler & Scott (1986), Arnold (1986), Hayes & Laws (1991) and ATSDR
(1992).
People who died within 8-10 h showed increasing tachycardia,
progressive hypotension, early hyporeflexia and peripheral cyanosis.
The ingestion of lower lethal doses causes gastrointestinal
haemorrhages (blood in faeces), gastroenteritis, metallic taste,
salivation, nausea and vomiting. Neurological disorders become
apparent within 2-5 days irrespective of the route of administration.
Within 5-7 days hallucination, lethargy, delirium, convulsions, a
tingling pain in the extremities and muscular weakness are followed by
coma. The cause of death is respiratory failure or cardiac arrest.
The sequence of symptoms in less severe intoxications is outlined
in Fig. 2. Similar clinical symptoms develop after ingestion of
lethal doses, which are promptly treated by enhancing elimination
(Graben et al., 1978). Within hours after ingestion, thallium often
induces nausea or vomiting, which may also appear in the next 2 days.
Other initial symptoms, e.g., diarrhoea, abdominal pain and a dull
feeling in body extremities, occasionally occur. Constipation is
common and may be difficult to treat, thus interfering with antidotal
treatment with Prussian Blue (section 8.1). Starting at day 4, a dark
region, resembling melanin pigment, may appear in the hair roots (this
could be of diagnostic value).
Following this latent period, in the early phase lasting about 1
week, some of the typical thallium disorders slowly develop
(culminating in the third or fourth week). Firstly, retrosternal and
abdominal colic-like pains, as well as pain and tenderness in the
legs, often become prominent. Excessive thirst, sleeplessness,
restlessness, hysteriform behaviour and electro-encephalographic
abnormalities indicate involvement of the central nervous system. A
characteristic symptom of sensory neuropathy is the extreme
sensitivity of the lower extremities. The neurological syndrome can
also include optic neuritis, numbness of fingers and toes with loss of
sensation to pin-prick and touch, and the "burning feet syndrome". As
an additional sign of a mixed sensory-motor neuropathy, ankle reflexes
are lost early, while other reflexes may be maintained for a time or
even increased. During this phase, thallium intoxication can mimic a
systemic lupus erythematosus or a pseudobulbar paralysis
(Guillain-Barré syndrome or Landry's ascending paralysis) (Gastel,
1978; Alarcón-Segovia et al., 1989; Cavanagh, 1991). The renal
function is generally not affected in the early course of poisoning;
only a slight albuminuria with formed elements in the urine may be
found. Urinary elimination of porphyrins and porphyrin precursors may
be greatly increased during this early phase (Merguet et al., 1969;
Paulson et al., 1972; Bank et al., 1972; Graben et al., 1978).
During the second week hypertension and tachycardia are
frequently observed symptoms (Romero Romero et al., 1989). Sometimes
peroneal paralysis and atrophy of other muscles may develop. After a
short phase of perspiration, the skin becomes dry and scaly (probably
due to an effect on the sweat and sebaceous glands) and sometimes
necrotic. Damage to hair papillae seems to be responsible for loss of
head hair. This frequently begins during the second week. Complete
depilation occurs within about one month and regrowth begins some time
later, often without any pigment.
About 3 to 4 weeks after poisoning, dystrophy of the nails is
shown by the appearance of white lunular stripes (Mees's stripes),
which are also observed in cases of arsenic poisoning (Buschke &
Langer, 1927; Greving & Gagel, 1928).
After 4 to 5 weeks, survival of the patient is likely, but
recovery requires months. Sometimes neurological and mental
disturbances, as well as electro-encephalographic abnormalities and,
rarely, forms of paranoia, persist. Occasionally, cataract (opacity
of the eye lens) has been described. In children a high percentage of
the neurological disorders were still present after 4 years. Double
optic atrophy in one patient after 3 months was reported (Munch,
1934).
8.1.2 Effects of long-term exposure: chronic toxicity
Studies of long-term exposure to thallium resulting in chronic
poisoning have been summarized by Buschke & Langer (1927), Moeschlin
(1965), Gefel et al. (1970), Schoer (1984) and Goldblatt (1989)
without any information about doses. The symptoms show strong
variation and are in general milder than in cases of acute
intoxication. Depending on the level of exposure, a relatively long
latent period (several weeks) may be followed by just a few symptoms.
Peripheral sensorial disturbances, mental aberrations, loss of weight
and sleeplessness seem to be the most common (Valentin et al., 1971;
Sabbioni & Manzo, 1980; Nessler, 1985b). In more severe cases,
disturbances of vision, pain without marked polyneuritis, and loss of
only two showed symptoms of thallium intoxication: a 2-year-old child
who had ingested 3.5 mg thallium/kg and eliminated 130 µg
thallium/litre in the urine showed no reflex movements of the legs in
the second week, while another child with 3.9 mg thallium/litre in its
urine and 40 µg/litre in its blood showed no neurological symptoms
apart from a massive alopecia in the second week. In one of two
lethal cases, the urine contained 10.3 mg/litre, and the death of both
children occurred within 2 days (von Mühlendahl et al., 1978). The
influence of age is unclear, but this may be due to the reporting of
doses either as total dose or as mg/kg body weight. Therapeutic uses
of thallium in the 1930s (8 mg/kg) resulted in peripheral neuritis in
about 10% of the patients (Cavanagh, 1979).
Table 25. Acute toxicity of thallium
Toxicitya Dose Adult/children Reference
TT > 1.5 mg/kg not specified Schoer (1984)
MLD 10-12 mg/kg adult Kazantzis (1994)
2-10 mg/kg children
MLD 14-18 mg/kg not specified Sessions & Goren (1947)
Lethality 20-100 mg per person adult Sharma et al. (1986)
2-10 mg per person children
a MLD = minimal lethal dose; TT = toxicity threshold
In adults lethal doses vary between 6 and 40 mg/kg (500 to
3000 mg/person), with an average dose of 10-15 mg/kg body weight
(Schoer, 1984). Without therapy the average dose usually results in
death within 10-12 days (Kemper, 1979), but, summarizing 150 mostly
suicidal cases of thallium intoxication, most of the patients who had
ingested about 600 mg and had no gastric lavage within the initial 2 h
died within 8-10 h (Potes-Gutierrez & Del Real, 1966). After
accidental ingestion of 10-fold overdoses of thallium acetate
(80 mg/kg), given as a depilatory in ringworm disease (Tinea), four
children died after 1-2 days (Lynch et al., 1930). Sessions & Goren
(1947) had previously suggested 14 to 18 mg thallium/kg body weight to
be the minimal fatal dose.
The triad of gastroenteritis, polyneuropathy and alopecia is
regarded as the classic syndrome of thallium poisoning (Gastel, 1978),
but in some cases gastroenteritis and alopecia are not observed.
Other symptoms also develop in varying sequence. Both lethal and
sublethal doses give rise to most of the symptoms, but these same
symptoms vary in intensity and time, probably in a dose-dependent way.
The following general trends have been summarized from numerous
reviews and case reports, e.g., Muller (1961), Moeschlin (1965, 1986),
Potes-Gutierrez & Del Real (1966), Venugopal & Luckey (1978), Gastel
(1978), Möllhoff et al. (1979), Sabbioni & Manzo (1980), Davis et al.
(1981), Saddique & Peterson (1983), Kemper & Bertram (1984), Le Quesne
(1984), Schoer (1984), Ohnesorge (1985), Nessler & Briese (1985),
Chandler & Scott (1986), Arnold (1986), Hayes & Laws (1991) and ATSDR
(1992).
People who died within 8-10 h showed increasing tachycardia,
progressive hypotension, early hyporeflexia and peripheral cyanosis.
The ingestion of lower lethal doses causes gastrointestinal
haemorrhages (blood in faeces), gastroenteritis, metallic taste,
salivation, nausea and vomiting. Neurological disorders become
apparent within 2-5 days irrespective of the route of administration.
Within 5-7 days hallucination, lethargy, delirium, convulsions, a
tingling pain in the extremities and muscular weakness are followed by
coma. The cause of death is respiratory failure or cardiac arrest.
The sequence of symptoms in less severe intoxications is outlined
in Fig. 2. Similar clinical symptoms develop after ingestion of
lethal doses, which are promptly treated by enhancing elimination
(Graben et al., 1978). Within hours after ingestion, thallium often
induces nausea or vomiting, which may also appear in the next 2 days.
Other initial symptoms, e.g., diarrhoea, abdominal pain and a dull
feeling in body extremities, occasionally occur. Constipation is
common and may be difficult to treat, thus interfering with antidotal
treatment with Prussian Blue (section 8.1). Starting at day 4, a dark
region, resembling melanin pigment, may appear in the hair roots (this
could be of diagnostic value).
Following this latent period, in the early phase lasting about 1
week, some of the typical thallium disorders slowly develop
(culminating in the third or fourth week). Firstly, retrosternal and
abdominal colic-like pains, as well as pain and tenderness in the
legs, often become prominent. Excessive thirst, sleeplessness,
restlessness, hysteriform behaviour and electro-encephalographic
abnormalities indicate involvement of the central nervous system. A
characteristic symptom of sensory neuropathy is the extreme
sensitivity of the lower extremities. The neurological syndrome can
also include optic neuritis, numbness of fingers and toes with loss of
sensation to pin-prick and touch, and the "burning feet syndrome". As
an additional sign of a mixed sensory-motor neuropathy, ankle reflexes
are lost early, while other reflexes may be maintained for a time or
even increased. During this phase, thallium intoxication can mimic a
systemic lupus erythematosus or a pseudobulbar paralysis
(Guillain-Barré syndrome or Landry's ascending paralysis) (Gastel,
1978; Alarcón-Segovia et al., 1989; Cavanagh, 1991). The renal
function is generally not affected in the early course of poisoning;
only a slight albuminuria with formed elements in the urine may be
found. Urinary elimination of porphyrins and porphyrin precursors may
be greatly increased during this early phase (Merguet et al., 1969;
Paulson et al., 1972; Bank et al., 1972; Graben et al., 1978).
During the second week hypertension and tachycardia are
frequently observed symptoms (Romero Romero et al., 1989). Sometimes
peroneal paralysis and atrophy of other muscles may develop. After a
short phase of perspiration, the skin becomes dry and scaly (probably
due to an effect on the sweat and sebaceous glands) and sometimes
necrotic. Damage to hair papillae seems to be responsible for loss of
head hair. This frequently begins during the second week. Complete
depilation occurs within about one month and regrowth begins some time
later, often without any pigment.
About 3 to 4 weeks after poisoning, dystrophy of the nails is
shown by the appearance of white lunular stripes (Mees's stripes),
which are also observed in cases of arsenic poisoning (Buschke &
Langer, 1927; Greving & Gagel, 1928).
After 4 to 5 weeks, survival of the patient is likely, but
recovery requires months. Sometimes neurological and mental
disturbances, as well as electro-encephalographic abnormalities and,
rarely, forms of paranoia, persist. Occasionally, cataract (opacity
of the eye lens) has been described. In children a high percentage of
the neurological disorders were still present after 4 years. Double
optic atrophy in one patient after 3 months was reported (Munch,
1934).
8.1.2 Effects of long-term exposure: chronic toxicity
Studies of long-term exposure to thallium resulting in chronic
poisoning have been summarized by Buschke & Langer (1927), Moeschlin
(1965), Gefel et al. (1970), Schoer (1984) and Goldblatt (1989)
without any information about doses. The symptoms show strong
variation and are in general milder than in cases of acute
intoxication. Depending on the level of exposure, a relatively long
latent period (several weeks) may be followed by just a few symptoms.
Peripheral sensorial disturbances, mental aberrations, loss of weight
and sleeplessness seem to be the most common (Valentin et al., 1971;
Sabbioni & Manzo, 1980; Nessler, 1985b). In more severe cases,
disturbances of vision, pain without marked polyneuritis, and loss of
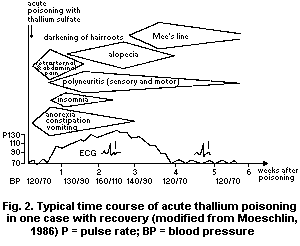 hair were reported. Later, severe polyneuritis may develop, with an
inability to walk, amaurosis (blindness) and pronounced cachexia.
Cardiac disorders include hypertension, irregular pulse and
angina-like pain. Renal dysfunction is indicated by albuminuria and
haematuria. Other symptoms are gastric anacidity, lack of appetite,
loss of weight, endocrine disorders, psychoses and encephalitis.
Complete rehabilitation takes months and can be interrupted by
relapses, probably caused by remobilization of thallium from tissue
depots.
Epidemiological studies carried out in the contaminated area of
Lengerich, Germany, comprising about 1200 people, revealed positive
correlations between the concentration of thallium found in urine or
hair samples and polyneuritic symptoms such as paraesthesias and pain
in muscles and joints, as well as psychasthenic symptoms such as
headache, sleep disorders and fatigue. No correlation was found with
respect to gastrointestinal troubles or skin disorders. Surprisingly,
a negative correlation with hair loss was found. Only one of 51
people with > 20 µg thallium/litre urine showed lunular stripes in
the nails (LIS, 1980; Brockhaus et al., 1980, 1981b; Dolgner &
Wiegand, 1982; Schoer, 1984). Strong individual variation in
sensitivity prevents an estimation of the thallium concentration in
the urine at which no effects occur (Dolgner & Wiegand, 1982).
8.2 Occupational exposure
There have been numerous reports of factory workers with thallium
poisoning, but no fatal cases have occurred. Peripheral sensorial
disturbances, mental changes, loss of weight, and sleeplessness are
the symptoms which seem to prevail (Munch, 1934b; Muller, 1961;
Malcolm, 1979; Saddique & Peterson, 1983; Triebig & Büttner, 1983;
Schoer, 1984; Nessler, 1985b; Junghans & Nessler, 1985; Ohnesorge,
1985; Kazantzis, 1986). In Germany, the United Kingdom and some other
countries, thallium poisoning represents an occupational disease
entitling the victim to compensation. In Germany, such compensation
was granted in three cases between 1970 and 1985 (Ewers, 1988).
Increased thallium concentrations in the urine of workers have
often been found. For example, the urine of workers in a company
producing alloy anode plates for use in magnesium sea water batteries
contained up to 236 µg thallium/litre, but no differences in medical
records of exposed and unexposed workers could be demonstrated
(Marcus, 1985). In his review of thallium, Ohnesorge (1985)
summarized several reports of industrial poisoning. Exposure over
several months or years resulted in typical thallium symptoms, e.g.,
leg pains, tiredness, alopecia and psychological disorders, but also
(in one case) blindness. Permanent blindness was also reported in
another review by McDonald (1941). Exposure to more than 0.01 mg
thallium/m3 for 16 to 17 years caused disorders of the vascular
system, as well as neurological symptoms (Ohnesorge, 1985). From the
triad of gastroenteritis, polyneuropathy and alopecia, only disorders
of the gastrointestinal tract were not reported.
Glomme (1983) emphasized that objective symptoms of polyneuritis
may not be demonstrable for some time. In addition to the changes in
the superficially provoked tendon reflexes, a pronounced weakness and
a fall-off in the speed of pupillary reflexes can occur.
In a further study on cement plant workers, 36, selected at
random, were subjected to thallium analyses of blood, urine and hair,
together with a neurological examination and electrophysiological
investigation including sensory and motor nerve conductive velocities,
evoked potentials and electro-encephalography (Ludolph et al., 1986).
One half of the workers examined suffered from concurrent disorders,
including diabetes mellitus. Although multiple symptoms and signs of
neurological disorders were detected, no correlation was found between
the electrophysiological findings and thallium levels in blood, urine
and hair. Urinary thallium levels were above 5 µg/litre in five of
the examined workers. Blood thallium levels above 2 µg/litre were
found in 16 workers and hair thallium levels above 20 µg/kg in four
workers. The investigators concluded that more thorough
epidemiological techniques would be required to reveal a possible
causal relationship between chronic low-dose thallium exposure and
neurological deficits.
8.3 Subpopulations at special risk
There are no subpopulations at special risk of thallium
intoxication except workers in the respective industries and
populations living in thallium-contaminated areas. There are no good
data to suggest that infants or pregnant women are more sensitive to
the effects of thallium than the general population. The available
data, however, are inadequate to fully assess these subpopulations.
Because thallium is eliminated in both urine and faeces, any
subpopulations with diminished excretory capabilities (e.g., renal
insufficiency) may be at increased risk of thallium poisoning. It has
been recommended that workers be excluded from working with thallium
if they suffer from renal or hepatic disease, anaemia, blood
dyscrasias, hypertension, alcoholism, chronic infections or endocrine
gland dysfunction. It has also been recommended that workers
potentially exposed to thallium should be encouraged to eat
potassium-rich food, as thallium and potassium ions can mimic each
other in vivo. Accordingly, potassium-deficient individuals may
also be at increased risk from thallium toxicity.
8.4 Target organs in intoxicated humans: pathomorphology and
pathophysiology
Effects on the different organs have been summarized by Prick
(1979), Sabbioni & Manzo (1980) and ATSDR (1992). In nearly all
affected organs direct cytotoxic effects, as well as indirect effects,
caused by damage to the nervous system, have been found (Prick, 1979).
8.4.1 Gastrointestinal tract and renal system
In a fatal case of thallium poisoning, in which the woman died
after at least 14 days, there was gross dilatation of the stomach and
a thin "blue line" was evident at the margin of the gingiva of the
lower incisors, but no alterations of the intestinal wall were
apparent (Curry et al., 1969). Other patients who died 1 to 16 days
after oral poisoning showed hyperaemia, congestion of the gut,
punctate haemorrhages in the mucosa of the stomach and upper
intestinal tract, and swelling of the mucosal cells (Lynch et al.,
1930; Munch et al., 1933; Heath et al., 1983). As a result of
depilatory treatment in children, gastric hypoacidity was reported
(Buschke, 1929), an effect also observed after a suicide attempt
(Greving & Gagel, 1928).
In several cases of oral poisoning, usually fatal, the liver was
usually found to be congested, greyish yellow or yellow in colour, had
microscopic fatty infiltrations of the hepatocytes and a tendency to
central necrosis (Lynch et al., 1930; Munch et al., 1933; Curry et
al., 1969).
At least 6 weeks after intoxication, renal biopsy of a patient
with 13.8 mg thallium/litre in his urine showed diffuse proliferative
glomerulonephritis with granular immunofluorescence for IgG, IgM and
C3 (Alarcón-Segovia et al., 1989). In the postmortem examinations by
Lynch et al. (1930), Munch et al. (1933) and Curry et al. (1969),
sections of kidney were dull red or congested and showed marked
hyperaemia, cloudy swelling of tubules and degenerative changes of
glomeruli. Weinig & Schmidt (1966) also reported kidney damage (but
perhaps from a previous attempt at poisoning) in a woman and her son
who died after taking thallium. This kidney damage may have been
responsible for the relatively low thallium concentrations in the
son's kidney in comparison to concentrations in other tissues (section
6.2.2).
8.4.2 Cardiovascular system
Accidental poisoning of three children, who died within two days,
caused fatty degenerations in the victims' hearts. These were more
marked and more dispersed in the youngest child, who survived longest
(Lynch et al., 1930). In some cases of postmortem-diagnosed thallium
poisoning, resulting in death within 4-14 days, fresh haemorrhagic
myocardial lesions were found (Heath et al., 1983; Andersen, 1984),
while in another case only a few focal haemorrhages were present
(Curry et al., 1969). Haematological changes, e.g., anaemia,
leucocytosis, eosinophilia, thrombocytopenia (at least partly
resulting from a toxic effect on bone marrow) and lymphopenia, have
been summarized by Saddique & Peterson (1983) and Luckit et al.
(1990).
In five patients suffering from severe and protracted thallium
poisoning, cardiovascular changes were recorded (Machtey & Bandmann,
1961; Franke et al., 1979). The patients' blood pressure showed marked
fluctuations, even in the course of one day, but systolic and
diastolic hypertension occurred only on a temporary basis. The
authors believed that these changes and also the observed tachycardia
and electro-cardiographic changes were caused by direct effects of
thallium on the autonomic nervous system. Tachycardia can appear
about a week after intoxication and last for 5 weeks (Franke et al.,
1979). Involvement of the autonomic nervous system is also indicated
by changes in renal function and by the urinary concentrations of
various metabolites (brenzcatecholamines, vanillin-mandelic acid,
ß-aminolaevulinic acid, porphobilinogen, coproporphyrin and total
porphyrins) during hypertension and tachycardia resulting from
thallium poisoning (Bock et al., 1968). Concentrations of
brenzcatechinamines and porphobilinogen were greatly increased, and
hypertension and tachycardia could be influenced by administration of
alpha- and ß-receptor blockers. In addition, increased elimination of
brenzcatecholamines, which presumably originate not only from the
adrenal medulla but also from the sympathetic nervous system,
indicates a strong stimulation of the adrenergic system (Bock et al.,
1968).
8.4.3 Skin and hair
Five young men suffering from thallium poisoning showed
follicular plugging of the skin on the nose and cheeks and in the
nasolabial folds by keratinous material, crusted eczematous lesions
and acneiform eruptions on the face, dry scaling on palms and soles,
and alopecia, not only of the scalp but sometimes also of the
eyelashes, lateral eyebrows, arms and legs. Histological examination
of skin biopsies from both scalp and cheek showed disintegrating
hairshafts, gross follicular plugging and eosinophilic keratohyaline
granules in the adjacent granular layer of the epidermis. Sebaceous
glands were sometimes necrotic. Biopsies of the pustular lesions on
the face showed folliculitis and necrosis of the follicles, while in
those from the feet marked hyperkeratosis and hypergranulosis were
evident (Heyl & Barlow, 1989). Effects on the follicles are also
reported by Hausman & Wilson (1964) and Bonnet & Pedace (1979), but in
a woman who died at least 14 days after intoxication no hyperkeratosis
in any part of the skin was found (Curry et al., 1969).
As is the case in experimental animals (section 7.4.1), the
reason for the different sensitivities of different types of hair
(lanugo, pubic and axillary hair is much less or is later affected
than hair of the head) in humans is unclear (Buschke & Peiser, 1926;
Buschke, 1929; Cavanagh, 1988). Cavanagh et al. (1974) emphasized a
direct effect on the keratinocytes, and Cavanagh (1988) finally
suggested that the difference is due to the fact that hair follicle
cells are only affected when they are mitotically active.
The depilatory effect generally does not result in permanent hair
loss. Since the new hairs which grow following thallium-induced
alopecia are stronger than those lost and also develop in regions
which had been hairless prior to the thallium poisoning, Buschke
successfully used thallium in therapy of alopecia induced by hair
disease (Buschke & Curth, 1928).
Soon after poisoning, hair papilla are seen to contain black
regions and the growing end is tapered (e.g., Hausman & Wilson, 1964;
Curry et al., 1969; Saddique & Peterson, 1983). In some reviews this
phenomenon is interpreted as black pigmentation. However, Ludwig
(1961) had already shown that these regions do not contain deposits of
pigments or thallium but small amounts of air which had entered the
shaft. Later investigations demonstrated that the gaseous inclusions
result from a trophic disorder in keratin formation (Kijewski, 1984;
Metter & Vock, 1984). In both investigations, scanning electron
microscopy demonstrated a loosening of the elements of the fibre layer
of the hairs.
8.4.4 Nervous system
Neurological disorders showing strong variability are one of the
three major symptoms of thallium poisoning (Möllhoff et al., 1979;
Prick, 1979; Sabbioni & Manzo, 1980; Le Quesne, 1984; Manzo &
Sabbioni, 1988). In contrast to the other disorders, neurological
deficits usually persist. Ataxia, mild spastic paraparesis and
impairment of intellectual powers developed after treatment of scalp
ringworm with thallium and persisted for 36 years, and it is possible
that increasing problems with mobility after 33 years were also due to
the treatment (Barnes et al., 1984).
8.4.4.1 Central nervous system
Like some other heavy metal intoxications, those caused by
thallium are usually associated with subacute and chronic (but rarely
with acute) encephalopathy (Rosenstock & Cullen, 1986). In one patient
who died 9 days after ingesting 5-10 g thallium nitrate, no
abnormalities were evident in histological or ultrastructural
examinations of the central nervous system (Davis et al., 1981). The
brain of another patient, who died within 4 days of intoxication and
had an extreme postmortem concentration of 36 mg/litre in his blood,
was moderately swollen (Andersen, 1984).
Seven people who died 11 to 16 days after accidental ingestion of
thallium had localized oedema and various grades of chromatolysis in
their neurons, especially those of the pyramidal tract, the third
nucleus, the substantia nigra and the pyramidal cells of the globus
pallidus. Blood vessels were distended with blood (Munch et al.,
1933).
In a fatal case of thallium poisoning, the brain of the dead
person was slightly swollen and oedematous about 4 weeks after the
ingestion of around 33 mg thallium sulfate/kg body weight. Petechial
haemorrhages were found in the white matter, particularly in the
parietal regions and subthalamic areas. The brain stem and cerebellum
showed a normal appearance. Axonal swelling and fragmentation in the
cortico-spinal tracts could be traced through the mid-brain, pons and
medulla into the spinal cords. Chromatolysis of brain stem nuclei was
only marked in facial and hypoglossal nuclei and nerve fibre
degeneration only in the spinal tract of the 5th nerve (Kennedy &
Cavanagh, 1976).
In a suicidal case, general degeneration of ganglion cells,
damage to axons and disintegration of myelin sheaths were observed in
the brain of the person, who died 21 days after intoxication. Fatty
degeneration of ganglion cells, acute swelling of oligodendroglia, a
spongy appearance of the basal ganglia and a particular concentration
of lesions in the calcarine cortex were prominent (Karkos, 1971). In
another fatal case, autopsy showed degeneration of ganglion cells in
the brain and spinal cord (Gefel et al., 1970).
Detrimental effects on intellectual functions were assumed not
only in a patient suffering from ringworm treatment (section 8.4.4)
(Barnes et al., 1984), but also in a student of chemistry who
eliminated 60 mg thallium/litre in his urine after poisoning in the
laboratory (Thompson et al., 1988). The data obtained from
intelligence tests on the student, performed 7 and 13 months after the
near fatal intoxication, were compared with those of his non-identical
twin brother who was of a similar educational background. Although
the brothers are not totally comparable, the tests indicated severe
deterioration particularly in memory and performance abilities and, 13
months later, there was only little general improvement.
8.4.4.2 Peripheral nervous system
Histological and ultrastructural examination of postmortem
samples can produce inconsistent results, presumably because of the
different periods of time between intoxication and sampling and
because of differences in dose size. In general, clinical symptoms
and signs can be correlated to neuropathological findings (Cavanagh,
1979).
Damage to the autonomic nervous system accounts for many of the
effects on various organs, e.g., fever, tachycardia, labile blood
pressure, orthostatic hypotension, urinary retention, constipation and
cardiac arrhythmias (Gastel, 1978; Prick, 1979). Thallium
intoxication causes symmetric, mixed peripheral neuropathy (Rosenstock
& Cullen, 1986). Distal nerves are more strongly affected than more
proximal nerves, and earlier but lesser degrees of change occur in
nerves with shorter axons, e.g., the cranial nerves (Cavanagh et al.,
1974; Cavanagh, 1979, 1988).
a) Histology and ultrastructure
Neuropathological findings vary. Little evidence of neuronal
degeneration in the sciatic nerve or spinal cord were found in a
person who died about 14 days after intoxication (Cavanagh et al.,
1974). In a fatal case of poisoning, in which the patient died just 9
days after intoxication (section 8.4.4.1), a sural nerve biopsy was
obtained 2 days before death. In addition, postmortem samples of
peripheral and cranial nerves and sections from various parts of the
central nervous system were taken. Ultrastructural examination of the
sural nerve showed that the myelin sheaths had often disintegrated
into a series of ovoids along the course of the axon (Davis et al.,
1981). Similar findings have also been reported from other sural
nerve biopsies, taken, for example, 3 days in one case and at least 5
to 6 weeks after thallium poisoning, from patients who survived (Bank
et al., 1972; Paulson et al., 1972; Alarcón-Segovia et al., 1989;
Dumitru & Kalantri, 1990). Degenerated myelin sheaths contained
myelin figures and electron-dense granules, whereas axons usually had
a normal appearance and rarely contained densely packed neurofilaments
(Bank et al., 1972). Munch et al. (1933) and Davis et al. (1981)
found axon degeneration in peripheral nerves, even in axons with
ultrastructurally normal myelin sheaths; axons were swollen and
contained vacuoles and distended mitochondria. Non-myelinated axons
on the other hand showed only slight or no abnormalities. Beadings of
axons were not only present in distal portions of peripheral nerves,
but also in some cranial nerves, whereas the other cranial nerves and
the proximal portions of peripheral nerves were histologically normal.
In another postmortem examination of a patient who died about 4
weeks after intoxication (section 8.4.4.1) (Kennedy & Cavanagh, 1976),
the nerve fibres of several peripheral nerves were severely reduced,
long fibres in particular being more severely affected. Changes in
neurons of the spinal cord were evident in all regions but most
strikingly in the lumbosacral region, where many neurons clearly
showed the classical chromatolytic changes which indicate attempted
regeneration. Dorsal column changes in the spinal tracts could
clearly be correlated in time with the peripheral nerve symptoms, and
the slight changes in the lateral cortico-spinal tracts could be
traced to the recent necrotic lesions in the diencephalon (Kennedy &
Cavanagh, 1976). Demyelination of the dorsal columns in sections of
cervical spinal cord was also observed during the postmortem
examination of a women who died at least 14 days after intoxication
(Curry et al., 1969).
The severe damage to the vagus, denervation of the carotid sinus,
and lesions of the sympathetic ganglia found in postmortem
examinations indicate the involvement of the autonomic nervous system
(Gastel, 1978).
b) Electrophysiological investigations
In a case of thallium poisoning in which the patient survived, a
sural nerve biopsy was obtained and nerve conduction and serial
electromyographic studies were carried out, beginning 10 days after
onset of the symptoms and ending 24 months later (Dumitru & Kalantri,
1990). Initially, the plantar nerves of the foot showed profound
axonal loss, from which there was no recovery, as shown by conduction
studies over the next 2 years. During the initial 4 months, sural and
peroneal nerves also underwent axonal loss but recovered within 2
years. In other cases of thallium intoxication, nerve conduction
studies gave normal results or revealed retarded latencies of nerves
of the upper (more than the lower) extremities, as well as temporal
dispersion indicating demyelination (Alarcón-Segovia et al., 1989).
Sensory fibres of the nervus medianus were examined in a patient
with acute thallium poisoning in order to assess the effects on the
conduction velocities of faster and slower nerve fibres. Two months
after the onset of symptoms the patient showed evidence of distal
sensorimotor neuropathy, but only the conduction velocities of faster
fibres were below the normal lower limit. Nine months later, symptoms
had almost disappeared and conduction velocities of both slower and
faster fibres were within the normal range (Yokoyama et al., 1990).
Only a slight electrophysiological correlation with the symptoms of a
persistent polyneuropathy were reported from an examination carried
out 3 years after a case of intoxication (Feudell, 1982).
c) Visual disorders
Retrobulbar neuritis and resulting visual impairment can develop
or persist months after termination of treatment with
thallium-containing depilatories, and even optic atrophy may occur
(e.g., Buschke & Langer, 1927; Lillie & Parker, 1932; Mahoney, 1933;
Bank et al., 1972; Bahiga et al., 1978; Tabandeh et al., 1978;
Schmidbauer & Klingler, 1979). An ascendent (retinal) atrophy of the
optic nerve may result from the toxic effects of thallium on the
retina (Hennekes, 1983). Nerve fibres in oculomotor muscles can also
show degenerative changes (Cavanagh et al., 1974). In patients with
optic neuritis some reduction in visual acuity always persists
(Goldblatt, 1989). About 10 months after thallium intoxication, a
keratoconjunctivitis sicca was found to have developed
(Alarcón-Segovia et al., 1989).
8.4.5 Other organs
Effects on the lung and endocrine glands were found in six
postmortem examinations (death occurred 11 or 15/16 days after
ingestion of thallium). Light microscopy showed the alveoli distended
with serum, marked hyperaemia and a few areas with bronchopneumonia
(Munch et al., 1933). In another fatal case the pleurae were free
from haemorrhages and adhesions (Curry et al., 1969). Of the
endocrine glands only the adrenals were affected. They showed marked
hyperaemia, small haemorrhages in the medulla, areas of necrosis and
nuclear disintegration (Munch et al., 1933). In other lethal cases
the adrenals were enlarged but without haemorrhages (Curry et al.,
1969), or were haemorrhagic (Gefel et al., 1970), or the concentration
of lipoids was reduced (Buschke, 1929).
A biopsy, taken 50 days after intoxication, showed marked areas
of atrophy of muscle tissue (Franke et al., 1979). Muscle fibrosis
was reported by Gefel et al. (1970) in a fatal case of thallium
poisoning.
8.5 Special effects
8.5.1 Reproduction and developmental effects
Few data are available with respect to the effects of thallium on
human reproduction (Schardein & Keller, 1989). Female cycles are
arrested, and libido and potency of males decrease (Buschke & Langer,
1927; Greving & Gagel, 1928). Effects on sperm are known to occur in
cases of chronic intoxication (Cottier, 1980). It should be noted that
minor amounts of thallium accumulate in the testis after diagnostic
scintigraphy, but possible effects have not been investigated.
There are no reports of any teratogenic effects in humans and an
extrapolation of animal data to humans is somewhat problematical (Kolb
Meyers, 1983; Mottet, 1985).
Reviews of more than 20 cases of thallium intoxication during
pregnancy by Petersohn (1960), Moeschlin (1965), Stevens & Barbier
(1976), Graben et al. (1980) and Barlow & Sullivan (1982) can be
summarized as follows: all attempts at illegal abortion were in vain;
the prolonged use of a depilatory cream seems to have been the cause
of 1 neonatal death. Two attempts at illegal abortion with thallium
in the first trimester of pregnancy did not affect the development of
the fetuses, although rather low birth weights were recorded. In four
additional cases of intoxication during this period of pregnancy, the
outcome was not reported.
Intoxication occurring after the first trimester can induce in
the newborn baby some symptoms of acute intoxication seen in adults,
e.g., rash and alopecia. Two babies born after the intoxication of
their mothers in the 5th and 6th months of pregnancy showed reduced
weight or no effects, respectively. Also no effects were found in a
case of intoxication (0.35 g thallium) in the 7th month of pregnancy
or in an additional six cases. However, in two cases during this
period (0.15 g thallium in one case), premature births occurred,
showing alopecia areata and low weight of one baby. Alopecia areata
and lunular stripes in the nails were observed in two newborn babies.
Low birth weight was common.
Petersohn (1960) reported an attempt at illegal abortion by
ingesting 0.5 g thallium 8 weeks before term. However, the fetus
developed normally. The child had well-developed hair and apart from
being relatively underweight showed no signs of thallium poisoning,
whereas the mother developed alopecia and polyneuritis (Erbslöh,
1960). A suicide attempt with about 1.2 g thallium 2 days before
birth caused the death of a newborn girl after 5 days; fresh blood in
the faeces was observed from the 3rd day onwards.
In the population living around the cement plant in Lengerich,
Germany, 300 women gave birth in the years 1978 and 1979. Eleven
children exhibited congenital malformations or abnormalities, five
showing major malformations (e.g., cleft lip and palate, clubfoot, hip
dislocation and ventricular septum defect). The rate of malformation
was higher than expected, but the authors suggested that the real
frequency of malformation in unaffected populations is underestimated.
It was difficult to correlate the effects with the intensity of
exposure, since the degree of exposure to which the mothers were
subjected during pregnancy could not be ascertained (Dolgner et al.,
1983). It should be noted that the fathers were not included in the
investigations. Embryotoxic effects were not considered in the
investigation at Lengerich (Claussen et al., 1981).
8.5.2 Carcinogenicity
The carcinogenicity of thallium has not been adequately evaluated
in humans. A study by Marcus (1985) on occupationally exposed workers
showed that the incidence of benign neoplasms (not further
characterized) was not significantly increased in the workers.
However, only 86 thallium-exposed and 79 controls were included in
this study and the length of observation time was not stated. The
study was also limited by the availability of medical records. Other
reports involving human exposure to thallium did not include an
investigation of carcinogenicity.
8.5.3 Immunotoxicological effects
Reduced resistance against secondary infections has been reported
only by Moeschlin (1965), but actual data on the possible
immunological effects of thallium are not available.
8.6 Factors modifying toxicity: enhancement of elimination
In studies on laboratory mammals (section 7.10.1) and in tests
with patients, enhancement of elimination was attempted (Stevens et
al., 1974). This might be achieved, provided that the thallium is not
fixed intracellularly (Barckow & Jenss, 1976). Sodium salts were
previously used as an antidote for human thallotoxicosis (Munch,
1934a), but intravenous injection of sodium thiosulfate (Sessions &
Goren, 1947) often increased the severity of symptoms (Munch, 1934a).
Although increased urinary elimination of thallium theoretically
should reduce its fatal effects, treatment with potassium salts caused
a worsening of the symptoms of thallotoxicosis in humans (Papp et al.,
1969). This was presumably due to a mobilization of intracellular
thallium, an increase in plasma levels, and redistribution (Bank et
al., 1972; Gastel, 1978).
Dithizone has also been used to treat cases of human poisoning
(summarized by Bendl, 1969 and Papp et al., 1969), in spite of its
goitrogenic and perhaps diabetogenic effects in experimental animals.
Clinical therapy with dithizone is often more effective than treatment
with potassium chloride and charcoal (Paulson et al., 1972).
Respiratory distress, confusion and diplopia have been cited as
examples of negative side effects by Barckow & Jenss (1976) and were
also reported by Saddique & Peterson (1983), but they were not
attributed to the dithizone treatment by Paulson et al. (1972).
Dithizone presumably mobilizes thallium from the compartments with
maximal concentrations, thus increasing the toxic load of the nervous
system (Cavanagh et al., 1974; Ghezzi & Bozza Marrubini, 1979).
Other agents, D-penicillamine and the chelating
diethyldithio-carbamate ("dithiocarb"), have also been used as
antidotes (Sunderman, 1967; Montoya Cabrera et al., 1979). Dithiocarb
caused a three-fold increase in urinary elimination during therapy of
a woman (Sunderman, 1967). D-penicillamine was used to treat a
patient who initially had 1200 µg thallium/litre in her urine, as well
as two other people with thallium poisoning. The authors emphasized
that no adverse effects occurred (Alarcón-Segovia et al., 1989). In a
detailed comparative survey by Cavanagh et al. (1974), it was stated
that for neither of these two antidotes (nor for several other
antidotes) was formal proof of benefit available. Negative effects of
dithiocarb therapy, such as deterioration of cerebral function, have
been observed in patients (Kamerbeek et al., 1971a).
Haemoperfusion does not affect the course of thallium
intoxication, according to Heath et al. (1983). Successful treatment
by haemodialysis was reported by Barckow & Jenss (1976) and Piazolo et
al. (1971). Elimination of thallium by haemoperfusion or
haemofiltration should be restricted to intoxications with high doses
of thallium during the previous 24 h (Briese & Nessler, 1985b).
A very effective oral antidote in experimental animals and humans
is Prussian Blue, potassium ferric hexacyanoferrate(II), an inorganic
pigment which is not absorbed by the gut (Heydlauf, 1969; Dvorák,
1970; Kamerbeek et al., 1971b; Günther, 1971; Barbier, 1974; Ghezzi &
Bozza Marrubini, 1979; Lehmann & Favari, 1984, 1985). Potassium ions
in the molecule are exchanged for thallium ions. Thus, absorption in
the intestine is prevented and the thallium-loaded molecule is
eliminated with the faeces (Forth & Henning, 1979). This therapy
results in faecal elimination greatly exceeding urinary elimination
(Stevens et al., 1974). Prussian Blue is now the main therapeutic
agent (Forth & Henning, 1979; Lehmann & Favari, 1984; Kazantzis, 1986;
Pai, 1987; Chandler et al., 1990), the colloidally soluble form being
preferable (de Groot & van Heijst, 1988).
Prussian Blue therapy and forced diuresis with furosemide and
mannitol (10 g of the soluble form dissolved in 100 ml 1.5% mannitol
as a laxative, twice daily orally or intraduodenally, until urinary
thallium elimination is < 0.6 mg/24 h), perhaps supplemented by
haemodialysis, is currently considered the optimal therapy for
thallium intoxication (Barckow & Jenss, 1976; Forth & Henning, 1979;
Briese & Nessler, 1985b; Chandler & Scott, 1986; Wainwright et al.,
1988; IPCS, 1992; Aderjan et al., 1994). If Prussian Blue is not
available, activated charcoal can be used (IPCS, 1992). The effects
on target organs, for instance neurotoxic effects, must be treated
symptomatically (Forth & Henning, 1979; Kemper, 1979; Briese &
Nessler, 1985b). In laboratory experiments on rats the hepatotoxicity
of thallium was prevented by treatment with silymarin, which has been
shown to have a hepatoprotective effect in man against several toxic
substances (Mourelle et al., 1988).
8.7 Protective measures against excessive occupational exposure
The high toxic potency of thallium has been considered in its TLV
or MAK value (threshold limit value or maximum concentration at the
workplace) of 0.1 mg/m3 (Schaller et al., 1980; MT, 1983; Marcus,
1985; DFG, 1990). This value is the limit for a 40-h working week in
the USA, France, Germany, United Kingdom and other western countries,
and has been reduced in the former-USSR to 0.01 mg/m3 air (Sabbioni
& Manzo, 1980; Nessler, 1985b). The MAK value is the mean value
during the normal 8-h working day, and, during this period, only once
may a ten-fold higher concentration occur for a period of 30 min (DFG,
1990). According to the West German General Administration Regulation
on Air Pollution Control, thallium concentration in dust fall-out
should not exceed 0.01 mg/m2 per day (Ohnesorge, 1985; Ewers, 1988).
On the basis of several reports of recommendations for the
protection of employees in industrial plants using thallium, e.g., by
Hill & Murphy (1959), Malcolm (1979), Glomme (1983), Nessler (1985b)
and a very detailed one by Sessions & Goren (1947), the following
protective measures are advisable.
a) General recommendations
i) Access to rooms in which thallium is used should be restricted
to a limited number of employees.
ii) Employees should repeatedly be informed about risk and
industrial hygiene, in a similar way to employees working with
radioisotopes. They should be instructed to report any unusual
health symptoms.
iii) Employees should be encouraged to eat potassium-rich food.
b) Medical control
i) By means of a preplacement examination, people suffering from
renal, hepatic or neurological diseases, anaemia, blood
dyscrasias, hypertension, alcoholism, chronic infections of
endocrine gland dysfunction should be excluded from working with
thallium.
ii) Urinary thallium should be periodically determined as a means of
showing the effects of education programmes and improving
industrial hygiene. The intervals will depend on the degree of
exposure.
iii) Periodic examinations should pay particular attention to the
early toxic effects of thallium, e.g., renal function,
gastrointestinal disturbances, the presence of paraesthesia and
alopecia.
c) Engineering control
i) Dust scattering should be avoided and handling of thallium
should be conducted under exhaust ventilation.
ii) Floors and tables should be wet-mopped.
iii) Dust samplers should be installed for environmental monitoring
to permit the evaluation of possible sources of contamination.
d) Personal protective equipment and hygienic measures
i) Employees should be required to use protective work clothes
including gloves.
ii) When indicated, personal exposure monitoring should be
performed.
iii) Complete sets of personal work clothes should be kept in
accommodation separate from that employed for street clothes.
Before changing clothes, gloves should be thoroughly washed and
then hands, using separate towels.
iv) Depending on the level of exposure, work clothing should be
washed periodically.
v) Clothes should be changed before eating, drinking and smoking,
all of which should be prohibited at the workplace.
vi) Washing and shower facilities should be provided and their use
enforced.
vii) Individual respirators should be worn in all operations
producing dust or fumes.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Since the majority of results have been obtained in laboratory
experiments, field observations on plants (mainly near the cement
plant in Lengerich) have not been considered under a special
subheading but have been included in sections 4.1.2.2, 9.3.1.2 and
9.3.1.6, and those on vertebrates have been included in sections 9.3.2
and 9.3.3.
The toxicity of thallium has been considered to be comparable
with that of mercury and lead, which are its neighbours in the
Periodic Table (Emsley, 1978).
9.1 Microorganisms
Buschke & Jacobsohn (1922) observed a sterilizing effect of
metallic thallium on various bacteria placed on agar plates. The
antibiotic effect of thallium was also recognized during postmortem
examination of thallium-poisoned humans, which were less decomposed
than other corpses (Muller, 1961).
The most important effect of thallium on microorganisms seems to
be the complete or partial inhibition of nitrate formation by
Nitrobacter agilis, observed at concentrations of 0.8 to 16 mg
thallium/litre (Tandon & Mishra, 1969). However, nitrification in
soils was only reduced at high thallium concentrations, which also
affected plants considerably (McCool, 1933). Direct effects on soil
microflora were demonstrated by Drucker et al. (1979). The numbers of
total aerobic bacteria and the distribution of other microorganisms
were affected at concentrations as low as 1 and 10 mg/kg soil, whereas
soil respiration was only reduced at 100 mg/kg soil. Thallium was
less toxic than silver, mercury, chromium, cadmium, copper, nickel and
zinc (in descending order of toxicity) but more toxic than other
elements, e.g., arsenic and lead (Drucker et al., 1979). However, the
properties of the soil and the form of thallium used in this study
were not reported.
The only known positive effect of thallium on organisms has been
described by Richards (1932), who obtained a higher yield of yeast in
the presence of 0.1, 1 or 10 mg thallium/litre culture medium. Higher
concentrations inhibited the growth of Saccharomyces cerevisiae.
This positive effect of thallium needs to be verified, since it could
have been caused by contamination of the thallium, which would not
have been detected by the analytical methods available at that time.
Using the same microorganism, Norris et al. (1976) found a growth
inhibition of 50% after the addition of 153 mg/litre to liquid culture
(which contained 31.3 g potassium/litre) or 10.2 mg/litre to agar
medium. The same inhibitory effect with Escherichia coli was obtained
after the addition of 184.0 mg/litre liquid culture or 10.2 mg/litre
agar and with Bacillus megaterium after adding 3.1 or 5.1 mg/litre,
respectively. The concentrations of thallium (added to the agar
medium), which prevented colony formation, i.e. 15.3, 20.4 and
40.8 mg/litre for B. megaterium, S. cerevisiae and E. coli,
respectively, also show interspecies differences. Constant thallium
concentrations of about 30% of those concentrations which inhibited
growth rates by 50%, but with lower concentrations of potassium in the
liquid medium, caused a decrease in the growth rates of B. megaterium
and S. cerevisiae (Norris et al., 1976).
Potassium/thallium antagonism has been observed using
Thiobacillus ferrooxidans. Iron oxidation by growing cultures in a
potassium-free medium, but with 0.204 to 204.39 mg thallium (as
sulfate) per litre, was normal at 20.4 mg thallium/litre but inhibited
by 204.39 mg/litre. However, this inhibitory concentration did not
affect iron oxidation in the normal medium containing about 180 mg
potassium/litre (Tuovinen & Kelly, 1974).
In inhibitory tests with Azotobacter chroococcum and
A. vinelandii on agar plates, 20 mg thallium sulfate produced very
strong inhibition, comparable to that of gold or chromate, and
stronger than that of zinc or copper (den Dooren de Jong & Roman,
1971). The two latter heavy metals were found to be also less
inhibitory in tests using the bacterium Klebsiella pneumoniae
(Sikka et al., 1987). However, in a previous study this bacterium was
much more sensitive to sulfates of zinc, copper and cadmium than to
that of thallium (Wilson & Dean, 1977). The different toxicity
mechanisms were also shown by strains resistant to these heavy metals.
Thallium-resistant strains showed the opposite reaction and were more
sensitive to gentamycin and chloramphenicol than the wild strain. In
liquid medium, up to 0.2 mg thallium/litre reduced the doubling time
(correlated to the concentration) but induced no lag, whereas higher
concentrations also induced a lag of initial development. On agar
plates, concentrations of up to 0.3 g/litre were not toxic, but with
0.4 g/litre bacterial colonies developed only rarely. A reduction in
the potassium concentrations increased the toxicity of thallium in
both media. A synergistic effect, and not a purely additive effect,
with zinc and cadmium occurred only at concentrations above
0.1 g/litre (Wilson & Dean, 1977).
The enhanced isolation of mycoplasms in the presence of
considerable numbers of other bacteria from the human urogenital tract
(Tully et al., 1983) might be caused by a species-specific differences
in the sensitivity to thallium, which had been added to the culture
medium in addition to penicillin. Such a selective cultivation of
bacteria through the addition of different concentrations of thallium
to the agar can be used as a taxonomic criterion and should be
considered when using clinical material (Kunze 1972a,b).
Different toxicities of thallium(I), thallium(III) and
organothallium compounds were shown in a study on bacteria and fungi
by Srivastava et al. (1973). At low concentrations (1 and
5 mg/litre), the growth of Colletotrichum falcatum, the causative
fungus of a sugarcane disease, was affected more by thallium(III)
chloride than by thallium(I) chloride. However, at higher
concentrations the effect of the thallium(III) compound did not
increase, and the fungus was affected more by the monovalent thallium.
With both compounds the inhibition did not exceed 20% compared to
controls, even after the addition of 80 mg/kg. The organothallium
chlorides (diphenyl derivatives) were considerably more toxic than the
thallium chlorides, causing a reduction of mycelial growth of 50%,
even at concentrations of 13 or 16 mg/kg. This inhibition was
obtained with < 1 mg/kg for most ditolyl derivatives (Srivastava et
al., 1973).
Five fungal species of the genus Aspergillus were similarly
sensitive to thallium nitrate. Their sensitivity to cadmium and
mercury nitrate was similar, but they were less sensitive to the
nitrates of nine other metals (Filimonova et al., 1973).
Anaerobic bacteria are more sensitive to thallium(I) than to
organothallium compounds (Huber et al., 1978). In several cases the
inhibition of bacterial growth was greater at lower concentrations of
the organothallium compounds.
9.2 Aquatic organisms
Little is known about the toxicology of thallium in aquatic
systems. Wachs (1988) classified thallium, together with lead and
zinc, into the toxic class II. Because of a lack of complex
formation, its toxicity is not affected by water hardness, copper,
etc. (Zitko et al., 1975). Acute and chronic toxicities to freshwater
aquatic life are reported to occur at 1400 and 40 µg/litre,
respectively. One species of fish is even affected at 20 µg/litre
after 2600 h of exposure. Marine aquatic life seems to be affected at
2130 µg/litre, and as in the case of freshwater organisms, it is clear
that more sensitive species than those tested might exist (US EPA,
1980).
9.2.1 Plants
Results from studies on algae and higher aquatic plants are given
in Table 26. Photosynthesis is affected, as shown by the reduced
oxygen evolution of the algal endosymbiont of the ciliate Paramecium
bursaria (Di Gaudio & Hirshfield, 1976). In comparison to other
heavy metals, higher concentrations of thallium were needed to reduce
the light-induced oxygen evolution of the freshwater alga
Chlamydomonas reinhardii (Overnell, 1975); about 2 mg thallium/litre
buffer caused a reduction of about 50%. Thallium strongly affects
NADP reduction or the dark stage reactions of photosynthesis.
However, it seems to inhibit photosystem I only slightly, and this
could not be confirmed in a very detailed later investigation with the
green alga Chlorella saccharophila, which also showed a reduced
oxygen evolution (Wystrcil et al., 1987). Measurements of the
variable fluorescence indicated a primary action of thallium on
electron transfer in photosystem II. At low concentrations of
thallium (2 mg/litre), disturbances of the thylakoid membranes could
explain the altered variable fluorescence. A change in the colour of
the algal suspension to a greyish green and the alterations of the
fluorescence intensity indicated effects on the pigments (Wystrcil et
al., 1987). This has also been found for the alga Chlamydomonas
reinhardtii. ß-Carotin showed the greatest sensitivity, followed by
chlorophyll a and then chlorophyll b (Maier-Reiter et al., 1987). In
addition to an increase in the degradation rate of the pigments, the
biosynthesis rate was reduced (section 9.3.1.2).
In a comparison of the toxicity of seven heavy metals for the
marine diatom Ditylum brightwellii, thallium (thallium(I) chloride)
possessed the lowest toxicity, which was not affected by the chelating
agent EDTA (Canterford & Canterford, 1980). Within 5 days a 50%
growth reduction was obtained with 330 to 350 µg total thallium/litre,
corresponding to 0.15-0.17 mg free thallium per litre. To obtain
total inhibition, concentrations of about 2.2 times higher were needed
(Table 26), but the cells showed an abnormal appearance, i.e.
pseudo-resting spore-like cells and shrinkage of protoplast and
concentration of chromophores, within about 3 days (Canterford &
Canterford, 1980; Canterford, 1980).
Species-specific sensitivities, but also different effects of the
monovalent and the trivalent forms of thallium, were evident in two
marine algae, Phaeodactylum tricornutum and Dunaliella tertiolecta
(Table 26) (Puddu et al., 1985, 1988). Complex formation with EDTA
reduced the toxicity of the thallium(III) compound. Using different
strains of three acidophilic algae, the type and level of inhibition
of growth after culture in a medium containing thallium sulfate was
very similar in the strains of Cyanidium caldarium and Cyanidioschyzon
merolae, whereas strains of Galdieria sulphuraria showed different
effects (Albertano & Pinto, 1986).
Generally, freshwater algae are affected at concentrations as low
as 100 µg/litre (US EPA, 1980). In the submerged macrophyte Elodea
canadensis, 1.4 and 2.8 mg thallium/litre water (as thallium(I)
sulfate) reduced the photosynthetic oxygen evolution during 24 h by
50% and 90%, respectively (Brown & Rattigan, 1979). In parallel
experiments, the free-floating Lemna minor was much more sensitive
(Table 26). The uptake and accumulation of thallium (thallium(I)
acetate) and its effects on growth parameters of L. minor (frond area,
Table 26. Toxicity of thalliuma to aquatic plants
Species Parameter Exposure time Concentration of thalliumb Reference
LOELc EC50d TECe
Algae
Chlamydomonas oxygen 0.25 h 2 Overnell (1975)
reinhardii evolution
C. reinhardii concentration 22 h > 0.2 20 Maier-Reiter et al. (1987)
of pigments
Ditylum brightwellii growth 5 days 0.34 0.73 Canterford & Canterford (1980)
Dunaliella tertiolecta growth - 0.08 Puddu et al. (1988)
0.18g
D. tertiolecta growth - 0.18g Puddu et al. (1985)
Elodea canadensis damage 28 days 2.0 Brown & Rattigan (1979)
E. canadensis oxygen evolution 24 h 1.4 Brown & Rattigan (1979)
Phaeodactylum growth - 0.14 Puddu et al. (1988)
tricornutum 0.22g
P. tricornutum growth - 0.24g Puddu et al. (1985)
Table 26 (contd).
Species Parameter Exposure time Concentration of thalliumb Reference
LOELc EC50d TECe
Selenastrum concentration 96 h 0.11 US EPA (1978)
capricornutum of pigments
S. capricornutum growth 96 h 0.10 US EPA (1978)
Higher Plants
Lemna minor growth (weight) 10 days 0.014 0.04 Kwan & Smith (1988)
281.5h 510.0h
L. minor growth 10 days 0.016 0.047 Kwan & Smith (1988)
(frond number) 293.8h 550.1h
L. minor growth 10 days 0.008 0.033 Kwan & Smith (1988)
(frond area) 195.8h 443.7h
L. minor damage 28 days 0.008 Brown & Rattigan (1979)
a Thallium(I), unless otherwise stated
b mg thallium/litre nutrition solution, unless otherwise stated
c LOEL = low-observed-effect level
d EC50 = concentration at which the life parameters were reduced by 50%
e TEC = concentration at which life parameters were totally inhibited
f - = no data given
g Thallium(III)
h mg thallium/kg dry weight of plant
fresh weight, frond number) were determined by Kwan & Smith (1988).
High concentrations of 1 and 2 mg/litre water induced chlorosis by the
eighth day, and 2 days later the fronds were completely devoid of any
colour. Comparing the number of fronds at different concentrations of
thallium during a period of 10 days, a stimulatory effect was evident
after exposure to 2 µg/litre and 10 µg/litre at the end of the period.
However, smaller fronds were produced and, therefore, the surface area
covered by Lemna also decreased at the lowest thallium concentrations.
At 20 µg/litre or more, all growth parameters were reduced.
Subsequent culture in thallium-free water resulted in good recovery
from the previous thallium exposure, provided that the concentration
had not exceeded 20 µg/litre. The bioconcentration factor (based on
plant weight) was 88 000 at the lowest exposure (2 µg/litre) and 6000
at an exposure of 153 µg/litre.
Using 40 µg/litre growth medium, the thallium concentrations in
L. minor did not further increase after 140 h, and over 80% of the
thallium remained in the plant (Smith & Kwan, 1989). In plant
homogenates, thallium showed little association with proteins, and the
reactions of the thallium in the soluble fraction were comparable to
those of the free metal ion. Like potassium, thallium accumulates in
the cell vacuole. When various concentrations of thallium were added
to the medium (0.02 to 0.2 mg/litre), about 0.04 mg/litre caused a 50%
reduction of growth after a period of 10 days (Smith & Kwan, 1989).
Since concentrations of 0.05 to 0.09 mg/litre were found in rivers
contaminated by mining (Zitko et al., 1975), significant effects on
macrophytes in such rivers would be expected, given the long exposure
period.
9.2.2 Animals
Toxicity data on aquatic animals are summarized in Table 27. In
an immobilization test on Daphnia magna, it was shown that the
toxicity of thallium(I) nitrate was higher than that of nickel or
cadmium but lower than that of copper, silver or mercury (Bringmann &
Kühn, 1982). In general, arthropods were affected at lower
concentrations than fish. The 96-h LC50 values were much lower for
the daphnids (water fleas) Daphnia magna and Mysidopsis bahia
(2.2 mg/litre) than for the freshwater fish Lepomis macrochirus
(bluegill) (120 mg/litre), although 50% of fathead minnows (Pimephales
promelas) were killed by 0.86 mg/litre (LeBlanc, 1984).
Comparing vertebrate with invertebrate marine species, about 10%
of the concentration needed to kill 50% of sheepshead minnows or
tidewater silversides was sufficient to kill 50% of marine shrimps (US
EPA, 1980). Acute values were 3- to 32-fold higher than chronic
values (US EPA, 1980).
Considerable differences between species were also evident in a
study by Nehring (1962/63). This study also showed that in perch
(Perca fluviatilis) lethal thallium effects depend on the length of
exposure. The fish were killed by 200 or 500 mg/litre within 10 h,
while concentrations of 100, 90-50, 40 and 20 mg/litre caused death
within 2, 2.5-3.5, 7.5 and 14 days, respectively. At 15 mg/litre, the
perch survived at least 17 days. Trout (Salmo gairdneri) and roach
(Rutilus rutilus) were more sensitive and died within 8 and 14 days,
respectively, at 4 mg/litre. In these species no effects were
observed within 17 days at concentrations of 2 mg/litre.
Juvenile Atlantic salmon (Salmo salar) are exceptionally
sensitive to thallium contamination (Zitko et al., 1975). The 18-day
LC50 is 0.1 mg/litre. The mortality of fish exposed to 0.03 mg/litre
was reported to be equal to that of the controls. The authors
suggested that 0.02 mg/litre be regarded as a no-observed-effect level
(NOEL), and that 0.04 mg/litre be regarded as a low-observed-effect
level (LOEL). Mixtures with copper or zinc did not alter thallium
toxicity.
Behavioural alterations in perch were reported by Nehring
(1962/63), even at low concentrations. Initially, following exposure
to thallium, food consumption increased, but after one or two days
neuronal damage occurred. This was indicated by uncoordinated
movement, paralysis of gills and disturbance of balance. Similar
effects on movement and respiration in fish had already been observed
by Swain & Bateman (1909/10).
Larvae of fathead minnows (Pimephales promelas) were much more
sensitive than the embryos to thallium sulfate (LeBlanc & Dean, 1984).
No effect on the rate of hatching was observed at concentrations up to
200 µg/litre, but at 720 µg/litre no hatching occurred. At
350 µg/litre no larvae survived. Growth was affected at 120 µg/litre.
In a study by Birge (1978), rainbow trout (Salmo gairdneri) eggs
were exposed to thallium from fertilization to 4 days after hatching
(total of 28 days). The exposure water was renewed every 12 h. An
LC50 of 0.17 mg/litre and an LC1 of 0.0084 mg/litre were reported.
Goldfish (Carassius auratus) eggs were also exposed to thallium from
fertilization to 4 days after hatching (total of 7 days). The
exposure water was renewed every 12 h. An LC50 of 7.00 and an LC1
of 0.0525 mg/litre were reported (Birge, 1978).
Table 27. Toxicity of thalliuma to aquatic animals
Species Stage Parameter Exposure time Concentration of thallium Reference
LOELb EC50c TECd
Invertebrates
Daphnids -e survival 48 h 2.2 LeBlanc (1984)
Daphnia magna juvenile mobility 24 h > 0.003 0.11 0.95 Bringmann & Kühn (1982)
Daphnia sp. adult survival 72 h 2-4 Nehring (1962/63)
Gammarus sp. adult survival 72 h 4 Nehring (1962/63)
Mysid shrimp survival 96 h 2.1 US EPA (1978)
(Mysidopsis bahia)
Vertebrates
Atlantic salmon juvenile survival 47 h 10 Zitko et al. (1975)
(Salmo salar) 112 h 1
435 h 0.1
2600 h 0.03
Sheepshead minnow adult survival 96 h 20.9 US EPA (1978)
(Cyprinodon variegatus)
Bluegill adult survival 96 h 132 US EPA (1980)
(Lepomis macrochirus)
adult survival 96 h 120 LeBlanc (1984)
Table 27 (contd).
Species Stage Parameter Exposure time Concentration of thallium Reference
LOELb EC50c TECd
Perch adult survival 72 h 60 Nehring (1962/63)
(Perca fluviatilis)
Fathead minnow embryo hatch > 0.2 < 0.72 LeBlanc & Dean (1984)
(Pimephales promelas) larva survival 30 day < 0.04 < 0.35
larva growth 30 day > 0.04 > 0.2
adult survival 96 h 0.86 LeBlanc (1984)
adult survival 96 h 0.08 US EPA (1980)
adult survival 96 h 1.8 US EPA (1980)
Roach adult survival 72 h 40-60 Nehring (1962/63)
(Rutilus rutilus)
Rainbow trout adult survival 72 h 10-15 Nehring (1962/63)
(Salmo gairdneri)
Tidewater silverside survival 96 h 24 Dawson et al. (1975/77)
(Mendia berrylina)
Toad adult survival - 16.7g Swain & Bateman (1909/10)
a mg thallium/litre water
b LOEL = low-observed-effect level
c EC50 = concentration at which the life parameters were reduced by 50%
d TEC = concentration at which life parameters were totally inhibited
e - = data not given
f 12 days in experiments with other species
g mg/kg injected into a lymph sinus
Amphibia are also affected by thallium. Development of frog
spawn was unaffected by concentrations of 40.8 and 200 mg/litre, but a
concentration of 0.4 mg/litre killed all tadpoles on hatching (Dilling
& Healey, 1926). This indicates that the absorption of thallium by
the eggs was minimal. Injections of lethal concentrations of thallium
acetate (> 0.005 g) into the lymph sinus of adult toads (Table 27)
caused loss of control of the hindlimbs and death by asphyxia (Swain &
Bateman, 1909/10). In a study on the narrow-mouth toad (Gastrophryne
carolinensis), eggs were exposed to thallium from fertilization to 4
days post-hatch (total 7 days). The exposure water was changed every
12 h. An LC50 of 0.11 and an LC1 of 0.0024 mg/litre were reported
(Birge, 1978).
9.3 Terrestrial organisms
9.3.1 Plants
Early investigations into the toxicity of thallium to plants were
summarized by Scharrer (1955). The most obvious effects are decreased
productivity, inhibition of photosynthesis and direct cytotoxicity.
Toxicity data are listed in Table 28.
9.3.1.1 Plant photosynthesis
In a study by Bazzaz et al. (1974), the net photosynthesis of
excised tops of sunflowers (Helianthus annuus) decreased both with
time and with the concentration of thallium in the nutrient solution
(2 to 200 mg/litre). At the highest concentration, photosynthesis was
inhibited by about 70% after 1 day, and the plants began to wilt. At
the lower concentrations, these visible symptoms appeared 4 days
later. There was a strong log-linear relationship between
photosynthesis and the thallium content of the plants. Stomatal
opening was reduced by 30 and 90%, respectively, at concentrations of
0.2 and 2 mg thallium/litre, but increased concentrations only caused
a slight additional effect (Bazzaz et al., 1974). In a direct
comparison Carlson et al. (1975) observed that the effects of thallium
sulfate on photosynthesis and transpiration in sunflowers were similar
to those in maize (Zea mays) at low thallium concentrations (up to
2 mg thallium/litre solution). Higher concentrations, up to
10 mg/litre, induced further inhibition in maize but not in
sunflowers. Using the data of Carlson et al. (1975), there was a good
linear correlation for maize between the occurrence of stomatal
opening (y) and thallium content of the solution (x):
y = 66.4 - 0.4 x; regression coefficient = 0.93
This was greater than the correlation calculated using the data for
sunflowers:
y = 45.1 - 0.4 x; regression coefficient = 0.66.
Table 28. Toxicity of thalliuma to terrestrial plantsb
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Brassica napus shoot growth 10 days > 20h,n Makridis & Amberger (1989b)
B. napus shoot growth 18 days 800i,n Makridis & Amberger (1989b)
< 2h,n
B. napus shoot growth 28 days > 2669 Allus et al. (1987)
> 10.0h
B. napus root growth 28 days > 760 Allus et al. (1987)
> 10.0h
Cucumis sativus growth 10 days < 10h Puerner & Siegel (1972)
Garden lettuce growth 7 days > 100 Schweiger & Hoffmann (1983)
10h
Garden lettuce growth summer 30 Hoffmann et al. (1982)
< 10k > 500k
Green kale growth 7 days > 500 Schweiger & Hoffmann (1983)
10h
Helianthus annuusg photosynthesis 4-5 days 63 Bazzaz et al. (1974)
(sunflower)
H. annuus photosynthesis 4-9 days 82 Carlson et al. (1975)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
H. annuus growth 7 days > 100 Schweiger & Hoffmann (1983)
10h
H. annuusg stomata opening 8 h < 0.2h approx. Bazzaz et al. (1974)
0.8h
Hordeum vulgare shoot growth 20 Davis et al. (1978)
0.5h
H. vulgare shoot growth 28 days > 21 Allus et al. (1987)
< 0.2h
H. vulgare root growth 28 days < 86 Allus et al. (1987)
< 0.2h
Kohlrabi growth summer > 600 Hoffmann et al. (1982)
(young) 500k
Lolium perenne shoot growth 21 days 0.71 251.2 Al-Attar et al. (1988)
L. perenne root growth 21 days 2.1 1990 Al-Attar et al. (1988)
Nicotiana tabacump survival 24 h 0.02h Siegel (1977)
N. tabacum germination 24 h 0.02h Siegel (1977)
Phaseolus vulgaris growth 33 days < 0.55h Kaplan et al. (1990)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
P. vulgaris shoot growth 10 days 130i,n Makridis & Amberger (1989b)
< 1h,n
Pisum sativum stem growth 28 days 5-10 Pötsch & Austenfeld (1985)
210-360h
5-10n Pieper & Austenfeld (1985)
115-123h,n
Pisum sativum leaf growth 28 days 1-5 Pötsch & Austenfeld (1985)
30-75h
> 10n Pieper & Austenfeld (1985)
30-43h,n
P. sativum root growth 28 days > 10 Pötsch & Austenfeld (1985)
> 180h
> 10n Pieper & Austenfeld (1985)
> 80h,n
Radishl growth summer 35 Hoffmann et al. (1982)
< 500k
Radishm growth summer < 300
> 500k
Rape growth summer > 500 Hoffmann et al. (1982)
200k 500k
Spinach concentration 14 days < 150 Maier-Reiter et al. (1987)
of pigments > 0.2h
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Spinach growth 9 days 280i Schweiger & Hoffmann (1983)
2h,i
Vicia faba stem growth 28 days > 10 Pötsch & Austenfeld (1985)
> 222h
5-10n Pieper & Austenfeld (1985)
36-76h,n
V. faba leaf growth 28 days > 10 Pötsch & Austenfeld (1985)
> 8h
5-10n Pieper & Austenfeld (1985)
5-7h,n
V. faba root growth 28 days > 10 Pötsch & Austenfeld (1985)
> 1320h
> 10n Pieper & Austenfeld (1985)
> 575h,n
Zea mays growth 7 days > 100 Schweiger & Hoffmann (1983)
(corn) 10h
Z. mays root growth 28 days 1h Logan et al. (1984)
Z. mays shoot growth 28 days 1h Logan et al. (1984)
Z. mays root growth 28 days 1h,n Logan et al. (1984)
Z. mays shoot growth 28 days 1h,n Logan et al. (1984)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Z. mays photosynthesis 4-9 days 82 Carlson et al. (1975)
Z. mayso stomata opening 8 h 2h Carlson et al. (1975)
a Thallium(I), unless otherwise stated
b Whole plants, unless otherwise stated
c mg thallium/kg dry weight of plant tissue, unless otherwise stated
d LOEL = low-observed-effect level
e EC50 = concentration at which the life parameters were reduced by 50%
f TEC = concentration at which life parameters were totally inhibited
g Tops
h mg thallium/litre nutrition solution
i Concentration at which the life parameters were reduced by 10%
k mg thallium/kg soil
l Root
m Leaf
n Thallium(III)
o Epiderm
p Protoplast
q Seed
The effects of thallium on the photosynthesis of spinach
chloroplasts have been investigated by Wystrcil et al. (1987). Some
alterations of variable fluorescence indicated a primary action of
thallium on electron transfer in photosystem II, which was also
evident in green algae (section 9.2.1). In photosystem I, superoxide
dismutase might also be affected by thallium (Wystrcil et al., 1987).
9.3.1.2 Cytotoxic effects
Chlorosis, followed by marginal necrosis of the leaves, is the
most prominent sign of thallium toxicity in plants. Different courses
of thallium poisoning in various plant species were reported by McCool
(1933) and later by Spencer (1937) in tobacco, by Carlson et al.
(1975) in corn and sunflowers, by Davis et al. (1978) in barley, by
Makridis & Amberger (1989b) in bushbeans and rape, and by Kaplan et
al. (1990) in beans. Similar observations on the leaves of trees in
the vicinity of the cement plant in Lengerich, Germany were reported
by LIS (1980).
The course and location of chlorosis seemingly depend on thallium
concentrations in the substrate and presumably correspond to the
distribution of thallium in the plant (Schweiger & Hoffmann, 1983).
In isolated protoplasts of Nicotiana tabacum, a cytotoxic
effect was also observed; 10% and 50% had lysed after a 24-h
incubation in 4 (± 0.4) and 20 (± 2) µg thallium/litre, respectively,
irrespective of the age of the protoplasts (Siegel, 1977). These
values are nearly identical to the percentages of seed in which
germination was inhibited (Siegel, 1977).
Chlorosis indicates a reduced concentration of pigments (section
9.2.1). In spinach, it is firstly the concentration of ß-carotene
which is reduced, then that of chlorophyll a and finally that of
chlorophyll b. The concentrations of ß-carotene and chlorophyll a
were about half the normal value after 2 weeks of incubation in a
hydroculture medium containing thallium nitrate (0.2 mg
thallium/litre) (Maier-Reiter et al., 1987).
9.3.1.3 Growth of plants
Adverse effects of thallium on the growth of plants have been
reported for various test systems (Table 28). In a comparison of the
effects of three heavy metals on the growth of cabbage seedlings,
cadmium and thallium were found to be less toxic than silver (Allus et
al., 1988).
Initial mycelial growth of three fungal species was inhibited on
agar plates containing 0.25 or 0.50 mg thallium/litre (Seeger & Gross,
1981).
In tobacco plants, concentrations as low as 5 mg thallium(I) per
litre inhibited terminal growth and caused a temporary outgrowth of
axillary buds all resembling natural frenching, i.e. a reticulate
interveinal chlorosis. In hydrocultures the root system was strongly
affected after 12 days at 0.067 mg thallium/litre. Thallium(I)
nitrate and sulfate were similarly toxic. The toxic effects of lower
concentrations were reduced by the addition of aluminium sulfate,
nitrogen and potassium iodide. In other species of Nicotiana, only
terminal growth, chlorophyll formation or roots were affected, and the
level of sensitivity to thallium corresponded to the level of
susceptibility to frenching in the field (Spencer, 1937).
After being watered for 15 days with 20 or 200 mg thallium per
litre, the growth of cucumber seedlings was unaffected, but growth was
reduced by 2000 mg thallium/litre. Toxicity was increased by limiting
the uptake of potassium. The higher sensitivity of the epicotyls
compared to the hypocotyls indicated that cell multiplication
processes are more sensitive than those entailing cell enlargement and
differentiation, a phenomenon known from many other stress factors and
toxic substances (Siegel & Siegel, 1976). In corn, production of top
and root biomass was severely reduced to between 50 and 60% of the
controls (Carlson et al., 1975).
From the differential reduction in weight in parts of the bean
plant, it has been shown to be possible to rank them according to
their increasing sensitivity to thallium(I): roots >> upper leaves
> lower leaves = upper stems > lower stems. Results from hydroponic
cultures were similar to those from field studies (Kaplan et al.,
1990). In another variety of bean, the weights of leaves and stems,
but not those of roots, were affected by exposure to thallium(III) (up
to 2 mg/kg). However, thallium(I) had no effect (Pötsch & Austenfeld,
1985; Pieper & Austenfeld, 1985).
Garden lettuce and radish growing in soils treated with TlNO3
exhibited considerably reduced growth at concentrations in dry plant
tissue of 30-35 mg thallium/kg (Hoffmann et al., 1982). Growth of
perennial rye grass (Lolium perenne) was adversely affected when
concentrations of thallium exceeded about 0.7 mg/kg dry weight in
shoots and 2.0 mg/kg dry weight in roots (Al-Attar et al., 1988).
9.3.1.4 Different sensitivities to thallium(I) and thallium(III)
Only small differences were observed between the toxic effects of
thallium(I) and thallium(III) on the dry weight of roots and shoots of
maize. Growth was slightly more reduced after application of
thallium(I) (Logan et al., 1984). Similarly, in two detailed studies
of the effects of thallium(I) and thallium(III) nitrate (0.2, 1, 2 mg
thallium/litre nutrient solution) on the dry weight of pea plants,
growth was found to be affected more after exposure to thallium(I).
However, completely opposite results were obtained for field beans
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985). Following
complexation of thallium(III) and thallium(I) nitrate with EDTA,
plants reacted differently to the two compounds. In bean stems and
roots, but not in leaves, concentrations of thallium were increased by
thallium(III) EDTA compared to those resulting from thallium(III) on
its own, while thallium(I) EDTA resulted in similar or lower thallium
concentrations than thallium(I) on its own in all three plant organs.
In the stems and leaves of peas, thallium(III) EDTA resulted in lower
thallium concentrations than thallium(III) on its own, while in the
roots thallium levels were the same for both salts. Consistently
higher thallium concentrations were found in leaves, stems and roots
of peas after exposure to thallium(I) EDTA, compared to thallium(I) on
its own (Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985).
9.3.1.5 Concentration of trace elements
The effects of thallium on plants could be caused mainly by an
imbalance of essential cellular monovalent cations or by a disturbed
uptake of trace elements (Yopp et al., 1974; Schweiger & Hoffmann,
1983). As can be concluded from the data summarized in Table 29,
thallium seems to have no uniform effect on the trace element content;
the differences between the two investigations using beans are
striking. In most studies the concentration of magnesium was found to
be reduced.
Exposure to thallium(III) chloride reduced the concentrations of
potassium and trace elements such as copper, zinc and iron in bush
beans by up to 20%; calcium, magnesium and manganese were only
slightly affected (Makridis & Amberger, 1989b). Rape was less
sensitive; uptake was decreased for potassium and copper, but
increased for zinc (and for calcium, magnesium, manganese and iron by
the reduced growth).
Complex effects on trace elements were found in studies with
Pisum sativum and Vicia faba in which thallium(I) and thallium(III)
nitrate and their respective EDTA complexes were used (Table 29)
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985).
9.3.1.6 Sensitivity of plants
Differing sensitivities among plant species, strains and
individuals have been reported for a number of air-borne contaminants
(Guderian, 1977). Species differences were also evident in many
investigations of thallium, including the detailed studies carried out
at Lengerich, Germany (LIS, 1980). There the residues of pyrite
roasting had been used for six years before effects were obvious
Table 29. Thallium-induced changes in uptakea or concentrations of trace elements in plants
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Thallium(I)
Compound TlNO3 shoot - - d - - i - d Schweiger & Hoffmann (1983)
Concentrationb 10
Exposure time 7 days
Species sunflower
Compound Tl2SO4 root u u i u d i u i Kaplan et al. (1990)
Concentration 1 leaf ic d u u d u ic ic
Exposure time 33 days
Species bean
Compound TlNO3 root - - u u - d - d Pötsch & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - u
Exposure time 28 days leaf - - u u - u - u
Compound +EDTA root - - u u - d - u
Species bean stem - - u u - u - u
leaf - - u u - u - u
Compound TlNO3 root - - d u - d - d Pötsch & Austenfeld (1985)
Concentration 0.2-2 stem - - u i - d - u
Exposure time 28 days leaf - - u u - u - u
Table 29 (cont'd).
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Compound +EDTA root - - u u - d - u
Species pea stem - - u i - i - u
leaf - - i i - u - i
Thallium(III)
Compound Tl(NO3)3 root - - u u - d - d Pieper & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - i
Exposure time 28 days leaf - - u u - d - u
Compound +EDTA root - - u u - d - u
Species pea stem - - u i - i - u
leaf - - i i - i - i
Compound Tl(NO3)3 root - - u u - d - d Pieper & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - u
Exposure time 28 days leaf - - u u - d - d
Compound +EDTA root - - u u - d - u
Species bean stem - - u u - u - u
leaf - - u u - u - u
Compound TlCl3 - i d d u i - d Makridis & Amberger (1989b)
Concentration 10-20
Exposure time 10 days
Species bean
Table 29 (cont'd).
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Compound TlCl3 - id d id id id - i Makridis & Amberger (1989b)
Concentration 10-20
Exposure time 10 days
Species rape
a d = decrease; u = unchanged; i = increase; - = not determined
b mg/litre solution
c Only upper not lower leaves
d Increase by reduced growth
(Gubernator et al., 1979). Coniferous trees were not affected, oaks
only slightly, and summer lime trees far more than winter ones. Sweet
cherry trees were more sensitive to thallium than sour cherry trees.
The leaves of pear trees still appeared healthy when apple and plum
trees had already lost theirs. Very sensitive vegetables included
beans, cucumber and potatoes. The sensitivity of fodder plants varied
too; the yield of maize was strongly reduced, but not that of rape or
turnip. There seems to be a tendency for plants with a "hard" leaf
surface to be less affected than those with a soft, hairy surface
(LIS, 1980).
Mechanisms of resistance to thallium seem to vary. Comparing the
growth data of beans (section 9.3.1.3) and peas, the higher tolerance
of beans to thallium(I) and thallium(III) (section 9.3.1.4)
corresponds to a higher concentration of thallium in the roots than in
the stems, which in turn contained more thallium than the leaves
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985). The more
sensitive peas possessed high thallium concentrations in the stems,
followed by those in the roots and leaves. Just the opposite
distribution is evident when barley and rape are compared: a higher
concentration in roots than shoots in the susceptible barley and the
opposite in the tolerant rape (Allus et al., 1987).
Green rape possesses a much higher resistance than beans after
application with thallium(III) chloride. Roots were more sensitive
than shoots and became brown. Using a 2 µg/litre nutrition solution,
the growth of bush beans was increasingly reduced after 3 days. The
first signs of reduced growth of rape were observed after 8 days
incubation in a solution of 5 mg/litre; other symptoms could only be
recognized after the thallium concentration was increased to
10 mg/litre. Then growth of the roots decreased and they became brown
(Makridis & Amberger 1989b). Thus, two mechanisms seem to enable
survival at high thallium concentrations: beans (and perhaps barley,
but not peas) reduce the amount of thallium which is transported to
the leaves, whereas the leaves of rape are only affected at very high
concentrations of thallium. Tolerance to relatively high
concentrations of thallium may be a result of complexation in plant
tissue, a phenomenon observed with other heavy metals (Cataldo &
Wildung, 1978).
In addition, selective pressure can increase tolerance. The
morphology of plants in the Alsar region of Yugoslavia, with a
naturally high soil thallium concentration, is not affected (Zyka,
1972), whereas such concentrations cause severe damage at other
locations (Schoer, 1984). In the Alsar region, zonation of plant
species with respect to soil thallium concentration can be observed
(Zyka, 1972), indicating the levels which are toxic to the various
species.
9.3.2 Wild animals
The effects of thallium on invertebrates have rarely been
investigated. After ant workers ingested about 0.2 mg thallium
chloride or thallium acetate per insect over a period of 2 months, all
survived (Jeantet et al., 1977). Field baits containing thallium
sulfate or acetate have been used against the fire ant and Pharaoh's
ant and destroyed about 90% of the colonies. These studies and the
toxic effects on crickets have been summarized by Negherbon (1959).
Corn poisoned with thallium sulfate has been used on a large
scale to control rodents. Such pest control carried out in the field
can affect various seed-eating animals and their predators (Munch et
al., 1974). An oral LC50 of 35 mg/kg fresh weight in starlings
(Sturnus vulgaris) was reported by Schafer (1972). This LC50 was
calculated following a single dose of thallium sulfate administered
via gavage, with a 7-day observation period. Thallium-poisoned baits
have also been used to control predatory birds. It was presumed that
9 out of 37 bald and golden eagles, which were collected sick or dead
in the USA, died from thallium poisoning in 1971-1972. Their kidneys
contained high concentrations of thallium (14 to 63 mg/kg) (Cromartie
et al., 1975). In experimentally poisoned eagles, which died from a
single oral dose of 120 mg thallium/kg body weight, the kidneys
contained 39 and 104 mg/kg (Bean & Hudson, 1976). Linsdale (1931)
reported the toxic effects of excessive use of thallium in California
for "ground squirrel control" on 58 species of game birds, song birds
and other wild animals. In 1972 all use of thallium in pesticides was
banned in the USA (Smith & Carson, 1977); in a study on birds carried
out between 1977 and 1981 no elevated thallium levels could be
detected (Wiemeyer et al., 1986).
In Denmark, partridges, pheasants, red foxes, badgers and martens
were found to be killed by direct ingestion of thallium-containing
rodenticides or poisoned prey (summarized by Munch et al., 1974;
Clausen & Karlog, 1974). Patho-anatomical findings from 1963 to 1971
indicated that 55 out of 299 red foxes and 5 out of 17 badgers
examined had suffered from poisoning. Determinations of thallium
concentrations in the kidney, liver and intestines demonstrated that
27 foxes and 1 badger had presumably been killed by thallium (Table
15). In most of the foxes not suspected of thallium poisoning,
thallium concentrations were < 0.1 mg/kg. Several of the poisoned
foxes showed abnormal behaviour, but only one fox showed clear
hair-loss. A typical sign in poisoned foxes was an empty stomach
(Munch et al., 1974). This was not observed in poisoned martens and
badgers. In addition, there were no skin lesions. Before death, many
of the martens showed uncoordinated movements and loss of balance.
The thallium concentrations in the inner organs of martens and badgers
ranged up to 92 mg/kg wet weight (Table 15) (Clausen & Karlog, 1974).
9.3.3 Household pets and farm animals
Accidental poisoning of pet animals (dogs and cats), ducks and
pigeons has been reported repeatedly (Zook & Gilmore, 1967). The
first detailed investigations considering toxic effects in dogs and
cats were performed to evaluate the risk of rodent poisoning campaigns
with thallium sulfate. Such cases used to be numerous, but in recent
years only occasional poisonings have occurred, due to the reduced use
of thallium as a rodenticide. For example, in Baden-Württemberg,
Germany, only 6 dogs, 4 cats and some ducks and pigeons were found to
have been poisoned by thallium from 1977 to 1989, and then no case
occurred up to 1992 (F. Baum, 1993, Institute of Animal Hygiene,
Freiburg, Germany; personal communication to the IPCS).
Symptoms of chronic intoxications in pets and farm animals are
similar to those of acute intoxications and can usually best be
observed in dogs. In ruminants uncharacteristic symptoms develop
(Hapke, 1984).
In some areas with naturally very high thallium levels, e.g., in
Yugoslavia and Israel, natural poisoning of farm animals after
consumption of vegetation has occurred (summarized by Gough et al.,
1979).
Table 30 summarizes early investigations by Ward (1931) and Shaw
(1932) on the toxicity of thallium sulfate to farm animals (quails,
geese, ducks and cattle), carried out in order to assess the risks of
its use as a rodenticide. In ducks an intraperitoneal injection of up
to 10 mg thallium/kg did not affect the birds, 15-25 mg/kg caused loss
of feathers on the back and 35-100 mg/kg was lethal within 24-63 h
(Ward, 1931). After feeding barley contaminated with 35, 50, or 75 mg
thallium/kg, ducks survived, or died in 12 days, or overnight,
respectively. Those ducks which died showed a mucous clogging of the
nasal passages (resulting in a marked gasping for breath), profuse and
green-coloured diarrhoea, loss of accommodation, wobbly gait and
extreme exhaustion. Death was due to respiratory failure and occurred
within 2 h after the beginning of intermittent asphyxial spasms.
Dissection of the dead ducks demonstrated that the intestinal tract
was plugged with a thick yellowish mucous. In addition, irritations
and ulcerations were present in the small intestine, and the livers
were enlarged and degenerated (Ward, 1931).
The same author investigated the effect on cattle (Ward, 1936).
Using thallium(I) sulfate, one cow received 50 mg thallium/kg body
weight and 3 heifers 35, 25 and 15 mg/kg, the latter two additional
doses of 20 and 35 mg/kg at 69 and 31 days after the first
administration. Two of the animals defaecated small quantities of
bloody faeces, all showed muscular twitching of flank and drooling of
a stringy mucous from nose and mouth. The cow died 5 days after
Table 30. Acute toxicity of thallium(I) sulfate for farm animals
Species Route of Period of Toxicitya Dose (mg thallium/kg Reference
administration observation body weight)
Quail oral 7 days LC100 approx. 12 Shaw (1932)
Goose oral 14 days LC100 approx. 15 Shaw (1932)
oral 2-3 days LC100 approx. 30-45 Shaw (1932)
Duck oral 14-21 days LC100 approx. 30 Shaw (1932)
oral > 15 days LOEL approx. 50 Ward (1931)
intraperitoneal > 15 days LOEL approx. 25 Ward (1931)
Cow oral 14 days LOEL approx. 25 Ward (1936)
a LC100 = concentration at which all animals were killed; LOEL = low-observed-effect level
administration, and the two heifers with the highest doses lived 11
and 14 days. The last animal was killed 3 days after the last
administration. Pathological changes were evident in the lymphatic
vessels (congested and oedematous), the liver (pale or congested) and
kidney (congested) and the walls of the digestive tract (haemorrhages,
ulcerations). Other organs appeared normal, and no significant
depilatory effect occurred. Recently Frerking et al. (1990) reported
thallium poisoning in cattle caused by the use of contaminated silage.
Symptoms were muscular twitching, colic, nervous behaviour, extreme
thirst, drooling from nose and mouth, loss of hair at the tail and,
later, erosion of nasal epithelium. The authors estimated that over a
period of 6 weeks the cows had ingested 0.75 mg thallium/kg body
weight daily (a total of 17 g thallium).
Anthropogenic contamination, especially from cement plants in
Germany, led to detailed studies of the effect of
thallium-contaminated fodder on the development of farm animals.
Continuous supplementation of maize-soybean fodder with 2, 4, 15 or
40 mg thallium(I) nitrate/kg in a 42-day broiler test and a
280(322)-day laying hen test caused obvious effects only at the
highest concentration (Ueberschär et al., 1986). In comparison to
control animals provided with uncontaminated feed, the body weight of
the treated broilers and hens was reduced by about 14 and 32%,
respectively; in the latter, laying rate (18%), feed efficiency (10%)
and eggshell thickness (2%) were also reduced. In broilers fed with
the two higher concentrations of thallium, gizzard erosion occurred.
In a detailed feeding study with fattening pigs with respect to
performance, health and meat residues, low concentrations of thallium
(daily intake of 0.05 and 0.1 mg thallium/kg body weight) were without
any effects on weight gain, carcass quality, health, or haematological
and biochemical parameters (Konermann et al., 1982). Daily
administration of 0.3 or 1.0 mg/kg body weight in drinking-water was
toxic to sheep, and the animals had to be killed after 4 and 6 weeks,
respectively (Hapke et al., 1980). Administration of daily doses of
0.03 and 0.1 mg thallium/kg body weight to sheep (for 11 weeks in
drinking-water) and 0.025 mg/kg body weight to steers (for 6 months in
fodder) caused no deaths (Hapke et al., 1980). However, in both
species daily uptake of 0.1 mg/kg body weight affected the animals
after several weeks or months; fodder should therefore contain less
than 0.5 mg/kg dry weight. Protein-rich food reduced the toxic action
of thallium (Hapke, 1984).
10. EVALUATION
10.1 Evaluation of human health risks
10.1.1 Exposure levels
Since thallium is a naturally occurring element, humans are
exposed to low levels in drinking-water, food and ambient air.
Drinking-water concentrations are often below the level of detection
(0.3 µg/litre) and rarely contribute more than 1 µg/litre. The total
intake of thallium from drinking-water has been estimated to be
< 1 µg/day for the vast majority of humans. In uncontaminated areas,
the dietary contribution of thallium has been estimated to be less
than 5 µg/day, with most of this coming from vegetables. Increased
dietary intakes have been reported for individuals living in areas
with thallium-contaminated soils; vegetables in these areas have been
found to contain thallium concentrations 1-2 orders of magnitude
higher than those grown in uncontaminated areas. However, the actual
dietary intakes for individuals consuming contaminated vegetables have
not been determined. In areas where there are no point sources of
thallium, ambient air concentrations are very low (< 1 ng/m3),
typically contributing less than 0.005 µg/day to the total intake.
Concentrations of thallium in workplace air can be several orders of
magnitude higher than those in ambient air, resulting in a
significantly increased total thallium intake. At the level of the
threshold limit values (TLVs) in some countries (0.1 mg/m3), the
thallium intake from inhalation alone would be of the order of
1000 µg/day (assuming inhalation of 10 m3 during a workshift). This
intake from inhalation alone (which may be even higher in some
workplaces) is about 500-fold higher than the total intake from
non-occupationally exposed humans living in non-contaminated areas.
There are only limited data about the actual thallium content of
workplace air. The most recent (1980s) concentrations of thallium
observed were < 22 µg thallium/m3 (in the production of a special
thallium alloy and in a thallium smelter). Average urinary
concentrations were determined to be in the range of 0.3-8 µg/litre
for cement workers and 0.3-10.5 µg/litre for foundry workers.
10.1.2 Kinetics
Thallium is rapidly and well absorbed through the
gastro-intestinal and respiratory tracts and is also taken up through
the skin. It is rapidly distributed to all organs and passes the
placenta, as indicated by the rapid fetal uptake, and the blood-brain
barrier. Because of its rapid accumulation in cells, concentrations
of thallium in whole blood do not reflect the levels in tissues. In
acute poisoning of experimental animals or humans, initially high
concentrations of thallium appear in the kidney, low concentrations in
fat tissue and brain, and intermediate concentrations in the other
organs; later the thallium concentration of the brain also increases.
Elimination of thallium may occur through the gastrointestinal
tract (mainly by mechanisms independent of biliary excretion), kidney,
hair, skin, sweat and breast milk. Intestinal reabsorption (mainly
from the colon) may occur with a consequent decrease in total body
clearance. In rats, the main routes of thallium elimination are
gastrointestinal (about 2/3) and renal (about 1/3), while in rabbits
the contribution of the two routes is about equal. Thallium is also
secreted in saliva.
As with many other substances, the excretion of thallium in
humans differs from that in laboratory animals since the rate of
excretion is generally much lower in humans (rate constant =
0.023-0.069 day-1) than in animals (average rate constant = 0.18
day-1). Another major difference between humans and animals is the
relative contribution of the different routes of excretion. In
humans, renal excretion seems to be much more important than in
animals, although its relative contribution to the total body
clearance has not been definitively established, due principally to
the lack of sufficient human data. Moreover, exposure levels,
duration of exposure, impairment of excretory organ function,
potassium intake and concomitant treatment of acute poisoning may
widely influence the results.
In a case report in which radioactive thallium (2.3 mg) was
therapeutically administered, urinary excretion of thallium within
72 h after dosing was 11% of the administered concentration, whereas
the gastrointestinal elimination was 0.5% during the same time period.
According to this study, renal excretion of thallium is about 73%,
whereas that through the gastrointestinal tract is about 3.7% of the
daily excreted amount. Elimination through hair has been estimated to
be 19.5% and that through skin and sweat 3.7%.
On the basis of the total daily excretion value, the daily intake
of thallium has been estimated to be about 11 µg and 0.9 µg in
chronically exposed and unexposed population, respectively; based on
the total amount of thallium in the body, a daily intake of about
2.3 µg may be calculated in unexposed populations.
The biological half-life of thallium in laboratory animals
generally ranges from 3 to 8 days. In humans it is about 10 days but
values of up to 30 days have been reported.
No data on the biotransformation of thallium are available.
10.1.3 Toxic effects
Thallium salts are mainly tasteless, odourless, colourless and
highly toxic. They were easily obtainable as rodenticides in the past
and are still available in some developing countries. Acute thallium
poisoning has resulted from accidental ingestion of thallium sulfate
and its use for suicide, homicide and attempts at illegal abortion.
Cases of homicide involving multiple low doses can induce chronic
intoxication. Chronic thallium intoxication has been observed in
occupationally exposed workers, and symptoms suggestive of thallium
poisoning have been seen in population groups in contaminated areas.
Clinical manifestations of acute thallium poisoning may occur
within hours or several days after exposure. Symptoms are often
diffuse and initially may include anorexia, metallic taste, nausea,
vomiting, retrosternal and abdominal pain, pain in the limbs, and
paraesthesia. Gastrointestinal haemorrhage occasionally occurs; later
on constipation is a common symptom. After the second day of thallium
poisoning, effects on the central and peripheral nervous systems,
skin, kidneys, eyes, cardiovascular and respiratory systems
progressively develop. Extreme sensitivity and pain in the legs,
later followed by the "burning feet" syndrome and paraesthesia, are
common manifestations. Insomnia, depression, hallucination, lethargy,
delirium, convulsions and coma may be followed by death, usually
between 10 and 12 days. Where survival extends beyond a week or so,
both motor and sensory neuropathy with cranial nerve involvement and
retrobulbar neuritis may develop. Common circulatory disorders, such
as hypertension, tachycardia and ischaemic cardiac changes, may also
occur. Frequently loss of head hair and sometimes also body hair
occurs after the second week of thallium poisoning. Dystrophy of the
nails is manifested by the occurrence of lunular stripes (Mee's lines)
3 to 4 weeks after intoxication. Recovery requires months and
occasionally some of the neurological and mental disturbances are
permanent. Permanent blindness may follow retrobulbar neuritis and
optic nerve atrophy.
Clinical features are generally milder in cases of chronic
poisoning than in acute thallium intoxication. Occurrence of chronic
thallium poisoning usually begins with neurological symptoms such as
tiredness, fatigue, headache and insomnia. In some cases the first
clinical findings include alopecia and constipation. The triad of
gastroenteritis, polyneuropathy and alopecia is regarded as the
classical syndrome of thallium poisoning, but in some cases
gastroenteritis and alopecia have not been reported.
Postmortem examinations following thallium poisoning reveal
damage in various organs. Haemorrhage in the mucosa of the intestine,
lungs and heart, kidney damage, fatty infiltration of the liver and
heart, and degeneration of neurons, including ganglion cells and
axons, with disintegration of myelin sheaths have all been observed.
Limited data are available on the effects of thallium on human
reproduction. Libido and male potency have been found to be adversely
affected in poisoning cases. There is no adequate evidence for a
genotoxic effect of thallium, and there have been no reports of any
carcinogenic or immunological effects.
Following low-level environmental exposure to thallium, a
dose-response relationship has been shown between thallium excretion
in urine and the prevalence of tiredness, weakness, sleep disorders,
nervousness, headache, muscle and joint pain and paraesthesia. Based
on replies to a questionnaire, a similar dose-response relationship
was seen when thallium in hair was taken as an indicator of exposure
and uptake.
10.1.4 Dose-response relationship (animals)
No lifetime studies of thallium administration have been
conducted on laboratory animals. In addition, no studies by the route
of inhalation are available. Three studies of intermediate duration
by the oral route are described in this report. A no-observed-effect
level could not be determined from any study. The lowest doses were
used in a 90-day gavage study (0, 0.01, 0.05 or 0.25 mg/kg body weight
per day). Small but statistically significant changes in some
clinical chemistry parameters were seen at the lowest dose level, as
was alopecia. From animal studies, it therefore appears that an
intake of 0.01 mg/kg per day may be associated with adverse effects.
No doses lower than this have been tested.
On the basis of LC50 values in animals and known lethal doses
in humans, it appears that humans may be more sensitive than
laboratory rodents to the toxic effects of thallium. Because of the
availability of human data and the apparently greater sensitivity of
humans, a quantitative evaluation of animal data for use in a risk
assessment has not been conducted here.
10.1.5 Dose-response relationship (humans)
Cases of acute thallium poisoning (with symptoms and signs listed
in the section 10.1.3) have occurred as a result of ingestion of doses
of thallium (in the form of soluble salts) as low as 1.5 mg/kg body
weight. Higher doses give rise to more severe symptoms. Doses that
have given rise to lethal poisoning are in the order of 10 mg/kg.
Concerning risks related to long-term exposure to lower doses of
thallium, the Task Group considered that an evaluation, although
uncertain, could best be performed on the basis of observed
relationships between urinary excretion of thallium and the occurrence
of symptoms. The urinary excretion value can be taken as an indicator
of the daily total absorbed dose from inhalation and dietary intake.
A population-based study on unexposed healthy subjects living in
northern Italy was performed with the aim of determining trace element
concentrations, including thallium, in blood, serum or plasma, and
urine, in which the collection, handling and analysis of the samples
was carried out under rigorous standardized protocols. The 496
subjects in this study, drawn from both urban and rural areas were
screened for normality by means of a questionnaire and clinical and
biochemical examination (with the exclusion of those with a history of
occupational exposure, heavy smokers, and those in a diseased state).
The mean urinary thallium concentration was 0.42 ± 0.09 µg/litre
(range 0.07-0.7 µg/litre). Other carefully controlled studies in
population samples showed similar urinary concentrations, e.g., 0.4 ±
0.2 µg/litre and 0.3 ± 0.2 µg/litre in rural and urban population
samples, respectively, and 0.3 ± 0.14 µg/litre in a sample of 149
subjects. This gives credence to a mean value of 0.3-0.4 µg/litre for
urinary thallium concentration in an unexposed population. In all
three studies, involving a total of 686 subjects, the range of urinary
thallium concentrations was 0.06-1.2 µg/litre. As thallium has a
short biological half-life, measured in days, and if a steady state
can be assumed to exist in such population-based samples, the above
urinary excretion value can be taken as an indicator of total dose in
terms of absorption following inhalation and total daily dietary
intake.
By contrast, in a population sample living in the vicinity
of thallium emission into the atmosphere, the mean urinary
thallium concentration was 5.2 µg/litre ± 8.3 µg/litre (range
0.1-76.5 µg/litre). Although a questionnaire on health effects was
compiled on each subject, no objective tests were performed. From the
replies to the questionnaire a clear dose-response relationship was
found between thallium concentration in urine and the prevalence of
tiredness, weakness, sleep disorder, headache, nervousness,
paraesthesia, muscle and joint pain. A similar dose-response
relationship was found when thallium in hair was taken as an indicator
of exposure. In a limited study on cement plant workers with thallium
exposure, where five workers showed urinary thallium levels above
5 µg/litre, but where the time interval between cessation of exposure
and urine collection was not stated, paraesthesia was reported in five
workers and distal muscle weakness in three. However, these symptoms
could not be related to thallium exposure.
From the above limited studies it is suggested that an
approximately 15-fold increase in urinary excretion of thallium above
the mean non-exposed level of 0.3 to 0.4 µg/litre may be related to
subjective symptoms which could possibly be considered as early
adverse health effects.
It is known from clinical practice that there is an increased
urinary concentration of thallium in acute poisoning cases. In 14
cases of thallium poisoning with recovery after therapy, the urinary
thallium concentrations ranged from 500 to 20 400 µg/litre. In seven
of these cases concentrations were below 2700 µg/litre. It should be
recognized that these values are not entirely comparable to those in
long-term exposure since they do not represent steady-state
conditions.
In summary, the Task Group considered that exposures causing
urinary thallium concentrations below 5 µg/litre are unlikely to cause
adverse health effects. In the range of 5-500 µg/litre the magnitude
of risk and severity of adverse effects are uncertain, while exposures
giving values over 500 µg/litre have been associated with clinical
poisoning.
10.2 Evaluation of the effects of thallium on the environment
Thallium is an element which occurs naturally in the earth's
crust, primarily in the monovalent form. In marine water and some
localized strongly oxidizing freshwater and soil, thallium may be
present primarily in the oxidized trivalent form. The major
anthropogenic sources of thallium released to the environment are
smelting of metallic ores, mining, special cement production, and the
combustion of fossil fuels, principally coal. Relatively little
thallium is released into the environment because of the production
and use of thallium compounds. Thallium levels reported in air are
generally < 1 ng/m3 although mean values up 15 ng/m3 have been
reported in industrial and urban air. Thallium may be released
directly to the environment following its use as a rodenticide,
although such use has been restricted or banned in many countries.
Thallium tends to persist in soil, although it may be leached to
water under acidic conditions. Monovalent thallium is relatively
stable in solution whereas trivalent thallium may be removed from the
water column by precipitation as the oxide or hydroxide. Although
thallium can bioconcentrate, it is unlikely to biomagnify in aquatic
or terrestrial food webs. Thallium concentrations in water tend to be
low, a maximum concentration of 2.4 mg/litre having been reported for
industrial waste water. Thallium concentrations in surface water from
industrial regions have been reported to range from 1 to 100 µg/litre,
while surface water in uncontaminated areas normally contains lower
levels. Concentrations of thallium in seawater range from < 0.01 to
0.02 µg/litre.
Most studies of effects on aquatic organisms have used soluble
monovalent thallium compounds. Acute toxic effects have been reported
in freshwater algae exposed to thallium at 100 µg/litre. Reduced
growth of aquatic macrophytes was reported following a 28-day
exposure to 8 µg/litre. The 48-h LC50 reported for Daphnia was
2200 µg/litre, while a 24-h LC50 of 110 µg/litre has also been
reported. The 96-h LC50 values for freshwater fish range from 860
to 132 000 µg/litre. An LC50 of 40 µg/litre has been reported for
freshwater fish exposed to thallium for approximately 40 days. The
96-h LC50 values for marine species are 2100 µg/litre for
invertebrates and 20.9-24 mg/litre for fish. The available aquatic
toxicity data suggest that thallium can harm aquatic organisms.
However toxic effects are likely to be limited to sites adjacent to
point sources such as some metal mining and smelting operations and
cement plants.
Thallium concentrations in uncontaminated soil typically range
from about 0.1 to 1.0 mg/kg dry weight, although higher levels can
occur near natural sources such as thallium-enriched shales and some
metallic ore deposits. Levels are generally somewhat elevated near
anthropogenic sources such as cement plants using thallium-containing
pyrite (up to 21 mg/kg dry weight) and base metal smelters (up to
2.1 mg/kg dry weight) that release large quantities of thallium to the
atmosphere.
Very few data have been identified concerning the effects on
terrestrial organisms of thallium in soil. The results of one study
suggest that microbial community structure is disturbed at
concentrations in the range of 1 to 10 mg/kg dry weight. However, the
properties of the soil and the form of thallium used in this study
were not identified.
Plants growing in uncontaminated soil normally contain 0.01 to
0.3 mg thallium/kg dry weight, while those growing near cement plants
using thallium-enriched pyrite have been reported to contain much
larger amounts (100 to 1000 mg/kg dry weight). Reduced growth has
been reported in sensitive plant species at concentrations of about
1 mg thallium/kg of dry plant tissue following exposure to monovalent
thallium. Toxic effects on terrestrial plants are therefore possible
near some cement plants using thallium-enriched pyrite.
The use of thallium as a rodenticide has resulted in poisoning of
non-target organisms including foxes, badgers, martens, partridges,
pheasants and eagles. Poisoning of domestic animals, such as dogs,
cats, ducks and pigeons, has also been widely reported. The number of
wildlife poisoning incidents has declined as a result of the reduced
use of thallium as a rodenticide. In countries with naturally high
thallium levels, such as former-Yugoslavia and Israel, some farm
animals have been poisoned following ingestion of vegetation with a
high thallium content. Symptoms of thallium poisoning have been
reported in cows that were calculated to have consumed 0.75 mg
thallium/kg body weight per day for a 6-week period.
11. CONCLUSIONS AND RECOMMENDATIONS
The currently limited industrial uses of thallium are unlikely to
pose a threat to the general environment. At industrial facilities
such as metal mining and smelting operations and cement plants using
pyrite, which can release significant amounts of thallium, the
concentration of thallium in industrial raw materials as well as stack
gases and waste water should be monitored and, if necessary,
controlled. Waste materials containing water-soluble thallium
compounds should be sealed and marked to avoid leaching and pollution
by dust.
In the general population, environmental exposure to thallium
does not pose a health threat. The total intake has been estimated to
be less than 5 µg/day, with the vast majority coming from foodstuffs;
drinking-water and air generally contribute very small amounts of
thallium.
Due to its toxicity to both humans and non-target environmental
species, the use of thallium as a rodenticide has been prohibited in
many countries. Where thallium is still available for such use,
however, the potential for accidental poisoning or for its use in
homicide or suicide remains a significant concern. It is recommended
that the use of thallium as a rodenticide be prohibited worldwide,
particularly as less hazardous methods of rodent control are
available.
Atmospheric emissions from industrial sources (e.g., cement
plants using thallium-containing pyrite) have resulted in increased
concentrations of thallium in biological samples (e.g., urine and
hair) from the population living in the vicinity. A relationship was
found between thallium concentrations in urine and hair and the
prevalence of symptoms possibly indicating early health effects of
thallium. The limited available data are not sufficient for
determining an acceptable limit for emissions. Steps should be taken,
however, to limit emissions to the greatest extent possible. Where
thallium may be released into the environment, monitoring of both
atmospheric emissions and resulting dust deposition rates should be
performed. Where environmental monitoring reveals thallium levels
significantly above background, it is recommended that biomonitoring
of the population living in the vicinity of the point source be
carried out. If biomonitoring reveals excessive exposure to thallium,
emissions from the point source should be re-evaluated and an effort
made to reduce them.
Since current occupational exposures to thallium may be of
concern to health, it is recommended that measures be taken to reduce
occupational exposure (as described in section 8.7 of this monograph).
Furthermore, revision of threshold limit values for thallium warrants
consideration.
All thallium analyses should be accompanied by a quality
assurance programme. This requires certified reference materials of
one matrix and of a similar concentration range to the sample to be
analysed and participation in an inter-laboratory comparison
programme. There is a need to make such reference materials
available.
Since thallium is rapidly and well absorbed and its excretion is
mainly renal, concentrations of thallium in urine may be considered a
relatively reliable indicator of exposure. Exposure to thallium
causing urinary concentrations below 5 µg/litre is unlikely to cause
adverse human health effects. For thallium exposure giving rise to
urinary concentrations in the range 5-500 µg/litre, the magnitude of
risk and the severity of adverse effects on human health are
uncertain, while exposure giving rise to 500 µg/litre or more has been
associated with clinical poisoning. The estimated daily oral intake
corresponding to a urinary thallium concentration of 5 µg/litre is
approximately 10 µg thallium in the form of a soluble compound.
In view of the considerable uncertainties in the evaluation, the
Task Group concluded that it was not possible to recommend a
health-based exposure limit. Until better information on the
dose-response relationship becomes available, it seems prudent to keep
exposures at levels that lead to urinary concentrations of less than
5 µg/litre.
12. FURTHER RESEARCH
a) Follow-up epidemiological studies of populations exposed
chronically to high levels of thallium (e.g., in the vicinity of
cement plants and natural sources of high concentrations of
thallium) should be performed to determine whether there is an
increased risk of pathological effects, e.g., cancer, effects on
reproduction (especially sperm cells) and congenital
malformations. Such follow-up studies are also required in order
to assess objectively the largely subjective symptom complex
experienced by certain populations in thallium-contaminated
areas.
b) Toxicokinetics of thallium, with particular respect to
distribution and excretion, should be studied in people with
low-level environmental exposure (in both uncontaminated and
contaminated areas) and in those exposed occupationally.
Adequate information from acutely poisoned patients should also
be compiled to determine toxicokinetics and the relationship with
the clinical findings. In addition, an attempt should be made to
correlate clinical findings with thallium concentrations in
biological samples. Patients should be followed for several
years to ascertain the long-term consequences of acute
poisonings.
c) Early indicators of an effect of thallium on glomerular or
tubular kidney function, on liver function, on haem biosynthesis
and on nerve conduction velocity should be sought in asymptomatic
population samples and in occupationally exposed workers where
there is evidence of excessive thallium exposure.
d) Early indicators of an effect of thallium absorption should be
sought. The effects of thallium on the haem biosynthetic pathway
suggest that estimation of porphyrins in blood and/or urine may
be a useful approach to detecting specific early effect of
thallium cellular toxicity. Similarly observations on the
endoplasmic reticulum structure with respect to protein synthesis
and polysome function suggest a need for further studies on liver
function.
e) Chronic bioassays in laboratory animals are needed to assess the
carcinogenic activity of thallium at low doses.
f) Lifetime studies in laboratory animals should be conducted using
several low doses by the routes of both inhalation and ingestion
to help determine a dose-response relationship and a
no-observed-effect level.
g) Multi-generation studies of thallium in laboratory animals are
needed to ascertain the reproductive and teratogenic effects of
thallium.
h) Sample collection, analysis and data presentation should be
carried out according to a protocol which ensures adequate
validation of biological monitoring procedures.
i) The concentrations of thallium in environmental compartments
adjacent to point sources should be determined using current
analytical techniques. The bioavailability of thallium from
various matrices should also be determined.
j) Total dietary studies should be conducted in areas with high
thallium concentrations (either natural or from industrial
sources) to determine the oral intake in populations living in
these regions.
k) Thallium concentrations in soil and vegetables from regions with
extensive acid rain should be determined.
REFERENCES
ACGHI (1980) Documentation of the threshold limit values, 4th ed.
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists.
Achenbach C, Ziskoven R, Koehler F, Bahr U, & Schulten H-R (1979a)
[Quantitative trace analysis of thallium in biological material.]
Angew Chem, 91: 944-945 (in German).
Achenbach C, Hauswirth O, Heindrichs C, Ziskoven R, Köhler F, Smend J,
& Kowalewski S (1979b) [Toxicity and teratogenicity of thallium.]
Dtsch Ärztebl, 76: 3189-3192 (in German).
Achenbach C, Hauswirth O, Heindrichs C, Ziskoven R, Köhler F, Bahr U,
Heindrichs A, & Schulten H-R (1980) Quantitative measurement of
time-dependent thallium distribution in organs of mice by field
desorption mass spectrometry. J Toxicol Environ Health, 6: 519-528.
Achenbach C, Wiemer J, Ziskoven R, & Winter U (1982) Tl+-Ions:
effects on the automaticity of sinoatrial tissue and on dV/dtmax and
iK2 of cardiac Purkinje fibres. Z Naturforsch, 37c: 1006-1014.
Adamson RH, Canellos GP, & Sieber SM (1975) Studies on the antitumor
activity of gallium nitrate (NSC-15200) and other group IIIa metal
salts. Cancer Chemother Rep, 59: 599-610.
Aderjan R, von Clarmann M, Daldrup T, Geldmacher-von Mallinckrodt M,
van Heijst ANP, Machbert G, Schütz H, & Stöppler M (in press)
[Thallium - clinical toxicological analysis after acute intoxications.
Communications of the Senate Committee on clinical and toxicological
analysis.] German Research Council (in German).
Agnes G, Hill HAO, Pratt JM, Ridsdale SC, Kennedy FS, & Williams RJP
(1971) Methyl transfer from methyl vitamin B12. Biochim Biophys
Acta, 252: 207-211.
Agrawal OP & Khatkar VB (1988) Amperometric determination of Ga(III),
In(III), Tl(I) and Pb(II) with thiomalic acid. J Electrochem Soc
India, 37: 289-292.
Aitio A (1988) Biological monitoring. In: Clarkson T, Friberg L,
Nordberg G, & Sager P ed. Biological monitoring of toxic metals. New
York, London, Plenum Press, pp 75-83.
Alarcón-Segovia D, Amigo MC, & Reyes PA (1989) Connective tissue
disease features after thallium poisoning. J Rheumatol, 16: 171-174.
Al-Attar AF, Martin MH, & Nickless G (1988) Uptake and toxicity of
cadmium, mercury and thallium to Lolium perenne seedlings.
Chemosphere, 17: 1219-1225.
Albertano P & Pinto G (1986) The action of heavy metals on the growth
of three acidophilic algae. Boll Soc Nat Napoli, 95: 319-328.
Ali SL & Blume H (1983) [Determination of the pesticide residues and
toxic heavy metal traces in crude drugs.] Pharm Ztg, 128: 2248-2252
(in German).
Ali SF, Jairaj K, Newport GD, Lipe GW, & Slikker W (1990) Thallium
intoxication produces neurochemical alterations in rat brain.
Neurotoxicology, 11: 381-390.
Allus MA, Martin MH, & Nickless G (1987) Comparative toxicity of
thallium to two plant species. Chemosphere, 16: 929-932.
Allus MA, Brereton RG, & Nickless G (1988) Chemometric studies of the
effect of toxic metals on plants: the use of response surface
methodology to investigate the influence of Tl, Cd and Ag on the
growth of cabbage seedlings. Environ Pollut, 52: 169-181.
Andersen O (1984) Clinical evidence and therapeutic indications in
neurotoxicology, exemplified by thallotoxicosis. Acta Neurol Scand,
70(suppl 100): 185-192.
André T, Ullberg S, & Winqvist G (1960) The accumulation and retention
of thallium in tissues of the mouse. Acta Pharmacol Toxicol,
16: 229-234.
Anschütz M, Herken R, & Neubert D (1981) Studies on embryotoxic
effects of thallium using the whole-embryo culture technique. In:
Neubert D & Merker H-J ed. Culture techniques: applicability for
studies on prenatal differentiation and toxicity. Proceedings of the
5th Symposium on Prenatal Development. Berlin, De Gruyter, pp 57-66.
Aoyama H (1989) Distribution and excretion of thallium after oral and
intraperitoneal administration of thallous malonate and thallous
sulfate in hamsters. Bull Environ Contam Toxicol, 42: 456-463.
Aoyama H, Yoshida M, & Yamamura Y (1988) Induction of lipid
peroxidation in tissues of thallous malonate-treated hamster.
Toxicology, 53: 11-18.
Apostoli P, Maranelli G, Minoia C, Massola A, Baldi C, & Marchiori L
(1988) Urinary thallium: critical problems, reference values and
preliminary results of an investigation in workers with suspected
industrial exposure. Sci Total Environ, 71: 513-518.
Arndt U, Finkbeiner M, Maier-Reiter W, & Wystrcil H-G (1987)
[Ecotoxicology of thallium. Behaviour and location in phytocenosis.]
Agrar-Umweltforsch Baden-Württemberg, 16: 5-21 (in German).
Arnold W (1986) [Excretion and distribution of thallium in human
poisoning.] In: Anke M, Baumann W, Bräunlich H, Brückner C, & Groppel
B ed. [5th Symposium of Trace Elements, Jena, 14-17 July 1986.] Jena,
University of Jena, pp 1241-1253 (in German).
Atkins HL, Budinger TF, Lebowitz E, Ansari AN, Greene MW, Fairchild
RG, & Ellis KJ (1977) Thallium-201 for medical use. Part 3: Human
distribution and physical imaging properties. J Nucl Med, 18: 133-140.
ATSDR (1992) Toxicological profile for thallium. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry,
pp 1-90 (ATSDR/TP-91/26).
Aulenbach DB, Meyer MA, Beckwith E, Joshi S, Vasudevan C, & Clesceri
NL (1987) Removal of heavy metals in POTW. Environ Prog, 6: 91-98.
Auler H (1926) [Therapy of cancer.] Z Krebsforsch, 23: 153-156
(in German).
Bahiga LM, Kotb NA, & El-Dessoukey EA (1978) Neurological syndromes
produced by some toxic metals encountered industrially or
environmentally. Z Ernähr.wiss, 17: 84-88.
Bank WJ, Pleasure DE, Suzuki K, Nigro M, & Katz R (1972) Thallium
poisoning. Arch Neurol, 26: 456-464.
Barbier F (1974) Treatment of thallium poisoning. Lancet,
2(7886): 965.
Barckow J & Jenss H (1976) [Thallium intoxication. Combined treatment
with haemodialysis, forced diuresis and antidote.] Med Klin,
71: 1377-1382 (in German).
Barclay RK, Peacock WC, & Karnofsky DA (1953) Distribution and
excretion of radioactive thallium in the chick embryo, rat, and man.
J Pharm Exp Ther, 107: 178-187.
Barlow SM & Sullivan FM (1982) Thallium and its compounds. In :
Reproductive hazards of industrial chemicals: An evaluation of animal
and human data. New York, Academic Press, chapter 41, pp 530-537.
Barnes MP, Murray K, & Tilley PJ (1984) Neurological deficit more than
thirty years after chronic thallium intoxication (letter). Lancet,
1: 562.
Barrach H-J & Neubert D (1985) The moderating influence of potassium
on the thallium effect in tissue differentiation in vitro.
Teratology, 31: 42A.
Barrera H & Gómez-Puyou A (1975) Characteristics of the movement of K+
across the mitochondrial membrane and the inhibitory action of Tl+.
J Biol Chem, 250: 5370-5374.
Barroso-Moguel R, Ríos-Castañeda C, Villeda-Hernández J,
Méndez-Armenta M, & Galván-Arzate S (1990) Neurotoxicity of thallium:
biochemical and morphological study of organic lesions. Arch Invest
Med, 21: 115-122.
Bazzaz FA, Carlson RW, & Rolfe GL (1974) The effect of heavy metals on
plants: Part I. Inhibition of gas exchange in sunflower by Pb, Cd, Ni
and Tl. Environ Pollut, 7: 241-246.
Bean JR & Hudson RH (1976) Acute oral toxicity and tissue residues of
thallium sulfate in golden eagles, Aquila chrysaetos. Bull Environ
Contam Toxicol, 15: 118-121.
Bell W, Hendry RA, & Annett HE (1925) The specific action of lead on
the chorion epithelium of the rabbit, contrasted with the action of
copper, thallium and thorium. J Obstet Gynaecol Br Emp, 32: 1-16.
Bendl BJ (1969) Thallium poisoning. Report of a case successfully
treated with dithizone. Arch Dermatol, 100: 443-446.
Berndt H & Sopczak D (1987) [Error recognition and programme
optimization in ET-AAS by time-resolved signals (direct determination
of element traces in urine: Cd,Co, Cr, Ni, Tl, Pb).] Fresenius Z Anal
Chem, 329: 18-26 (in German).
Bertram HP, Kemper FH, Müller C, & Cuellar JA (1985) Trace metal
patterns of human blood, saliva and hair: Environmental specimen
banking. Proceedings of the 5th International Conference on Heavy
Metals in the Environment, Toronto, vol 2, pp 102-104.
Bettinelli M, Baroni U, & Pastorelli N (1988) Determination of
arsenic, cadmium, lead, antimony, selenium and thallium in coal fly
ash using the stabilised temperature platform furnace and
Zeeman-effect background correction. J Anal At Spectrom, 3: 1005-1011.
BGA (1979) [Thallium.] Berlin, The Federal Environment Agency (Report
No. 63/28/7180) (in German).
Bienvenu P, Nofre C, & Cier A (1963) Toxicité générale comparée des
ions métalliques. Relation avec la classification périodique. C R Acad
Sci (Paris), 256: 1043-1044.
Birge WJ (1978) Aquatic toxicology of trace elements of coal and fly
ash. In: Proceedings of the Symposium on Energy and Environmental
Stress in Aquatic Systems, Augusta, Georgia, 2-4 November 1977,
pp 219-240.
Bock KD, Merguet P, Schley G, Schümann HJ, Rausch-Stroomann J-G,
Hocevar V, Schröder E, & Murata T (1968) [Pathogenesis of hypertension
and tachycardia in the acute thallium poisoning and acute intermittent
porphyria.] Dtsch Med Wochenschr, 93: 2119-2124 (in German).
Bonnet EFP & Pedace EA (1979) [Forensic anatomical histopathology of
thallium poisoning.] Prensa Med Argent, 66: 29-35 (in Spanish).
Bornhausen M & Hagen U (1984) Operant behavior performance changes in
rats after prenatal and postnatal exposure to heavy metals. Int Res
Commun Syst Med Sci, 12: 805-806.
Bösche J & Magureanu I (1983) [Heavy metal burden in Nordbaden by
thallium and cadmium - Results of investigations of kidneys.] In: Barz
J, Bösche J, Frohberg H, Joachim H, Käppner R, & Mattern R ed.
[Advances in forensic medicine. Festschrift for Georg Schmidt.]
Berlin, Springer Verlag, pp 270-275 (in German).
Bowen HJM (1966) Trace elements in biochemistry. New York, London,
Academic Press.
Bowen HJM (1979) Environmental chemistry of the elements. New York,
London, Academic Press.
Boysen P (1992) [Heavy metals and other substances in fertilizers -
Environmental research plan of the Federal Minister for Environment,
Nature Conservation and Reactor Safety, research report 107 01 016/1.]
Berlin, Erich Schmidt Verlag (Report No. UBA-FB 92-104) (in German).
Bradley-Moore PR, Lebowitz E, Greene MW, Atkins HL, & Ansari AN (1975)
Thallium-201 for medical use. II: biologic behavior. J Nucl Med,
16: 156-160.
Briese C & Nessler F (1985a) [Historical data on the discovery of
thallium and its first applications.] Wiss Z Humboldt-Univ (Berlin)
Math-Naturwiss Reihe, 34: 750-751 (in German).
Briese C & Nessler F (1985b) [Therapy of thallium poisoning.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 786-788 (in German).
Briese C, Nessler F, & Liebscher R (1985) [Production of thallium and
its significance in the different political economies.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 756-760 (in German).
Bringmann G & Kühn R (1982) [Results of toxic action of water
pollutants on Daphnia magna tested by an improved standardized
procedure.] Z Wasser Abwasser Forsch, 15: 1-6 (in German).
Britten JS & Blank M (1968) Thallium activation of the
(Na+-K+)-activated ATPase of rabbit kidney. Biochim Biophys Acta,
159: 160-166.
Brockhaus A, Dolgner R, Ewers U, Wiegand H, Freier I, Jerman E, &
Krämer U (1980) Excessive thallium absorption among a population
living near a thallium emitting cement plant. In: Holmstedt B,
Lauwerys R, Mercier M, & Robberfroid M ed. Mechanisms of toxicity and
hazard evaluation. Proceedings of the 2nd International Congress on
Toxicology, Brussels, 1980. Dev Toxicol Environ Sci, 8: 565-568.
Brockhaus A, Dolgner R, Ewers U, Freier I, Jermann E, & Wiegand H
(1981a) Repeated surveillance of exposure to thallium in a population
living around a cement plant emitting thallium containing dust. In:
Proceedings of the International Conference on Heavy Metals in the
Environment, Amsterdam, September 1981. Luxembourg, Commission of the
European Communities, pp 482-485.
Brockhaus A, Dolgner R, Ewers U, Krämer U, Soddemann H, & Wiegand H
(1981b) Intake and health effects of thallium among a population
living in the vicinity of a cement plant emitting thallium containing
dust. Int Arch Occup Environ Health, 48: 375-389.
Brodersen HP, Theilmeier A, Korsten S, Arendt U, Larbig D, & Reis HE
(1992) Acute poisoning by a potentially lethal dose of thallium:
quantitative comparison of different methods of elimination. Intensiv
Notfallbehandl, 17: 204-207.
Brown BE (1982) The form and function of metal-containing "granules"
in invertebrate tissues. Biol Rev, 57: 621-667.
Brown BT & Rattigan BM (1979) Toxicity of soluble copper and other
metal ions to Elodea canadensis. Environ Pollut, 20: 303-314.
Brown DR, Callahan BG, Cleaves MA, & Schatz RA (1985) Thallium induced
changes in behavioral patterns: correlation with altered lipid
peroxidation and lysosomal enzyme activity in brain regions of male
rats. Toxicol Ind Health, 1: 81-98.
Brumsack H-J (1977) Potential metal pollution in grass and soil
samples around brickworks. Environ Geol, 2: 33-41.
Brumsack H, Heinrichs H, & Lange H (1984) West German coal power
plants as sources of potentially toxic emissions. Environ Technol
Lett, 5: 7-22.
Budavari S ed. (1989) The Merck index: An encyclopedia of chemicals,
drugs, and biologicals, 11th ed. Rahway, New Jersey, Merck & Co.
Bulavintseva OA & Bulavintsev AG (1982) [Histoenzymic characteristics
in rats chronically poisoned with thallium sulfate.] In: Sedov KR ed.
[Geochemical environment: The problem of the health of the population
of the BAM railway zone], pp 68-69 (in Russian).
Burger A & Starke K (1969) [Influence of thallium on the ATPases of
the amine storing granules from the adrenal medulla and splenic
nerves.] Experientia (Basel), 25: 578-579 (in German).
Buschke A (1900) [Experiments on alopecia.] Berl Klin Wochenschr,
37: 1235-1236 (in German).
Buschke A (1903) [Further experiments on alopecia and the localization
of dermatoses.] Berl Klin Wochenschr, 40: 888-890 (in German).
Buschke A (1911) [Clinical and experimental observations of alopecia
(hypotrichosis) congenita.] Arch Dermatol Syph, 108: 27-40 (+ plates
IV and V) (in German). Buschke A (1929) [Thallium.] Dtsch Med
Wochenschr, 55: 1546-1547 (in German).
Buschke A & Berman L (1927) [The chemical and biological relation of
thallium and lead.] Klin Wochenschr, 6: 2428-2429 (in German).
Buschke A & Curth W (1928) [Therapeutical experiments with thallium,
apart from its use against fungal diseases of the hair of the head.]
Derm Z, 53: 113-117 (in German).
Buschke A & Jacobsohn F (1922) [Experiments on the action of
thallium.] Dtsch Med Wochenschr, 48: 859-860 (in German).
Buschke A & Langer E (1927) [The forensic and the industrial-hygienic
importance of thallium.] Münch Med Wochenschr, 74: 1494-1497
(in German).
Buschke A & Peiser B (1922a) [The effect of thallium on the endocrine
system.] Klin Wochenschr, 1: 995 (in German).
Buschke A & Peiser B (1922b) [Experimental observations of the
influence of thallium on the endocrine system.] Med Klin, 18: 731-732
(in German).
Buschke A & Peiser B (1922c) [The endocrine effect and the practical
importance of thallium.] Dermatol Wochenschr, 74: 443-445 (in German).
Buschke A & Peiser B (1926) [Thallium alopecia and sensory hairs.]
Klin Wochenschr, 5: 977-979 (in German).
Buschke A, Zondek B, & Berman L (1927a) [The inhibitory effect of
thallium on the estrous cycle of mice.] Klin Wochenschr, 6: 683-685
(in German).
Buschke A, Christeller E, & Loewenstein L (1927b) [Alterations of the
skull by experimental chronic thallium poisoning.] Klin Wochenschr,
6: 1088-1089 (in German).
Buschke A, Löwenstein L, & Joel W (1928) [Further histological results
of experimental chronic thallium poisoning.] Klin Wochenschr,
7: 1515 (in German).
Byczkowski JZ & Sorenson JRJ (1984) Effects of metal compounds on
mitochondrial function: a review. Sci Total Environ, 37: 133-162.
Byrne C, Balasubramanian R, Overton EB, & Albert TF (1985)
Concentrations of trace metals in the bowhead whale. Mar Pollut Bull,
16: 497-498.
Canterford GS (1980) Formation and regeneration of abnormal cells of
the marine diatom Ditylum brightwelli (West) Grunow. J Mar Biol Assoc
(UK), 60: 243-254.
Canterford GS & Canterford DR (1980) Toxicity of heavy metals to the
marine diatom Ditylum brightwelli (West) Grunow: correlation between
toxicity and metal speciation. J Mar Biol Assoc (UK), 60: 227-242.
Careaga-Olivares J & Morales-Aguilera A (1990) The distribution of
thallous ion between blood cells and plasma at equilibrium. Arch
Invest Med (Mex), 1990: 21-23.
Careaga-Olivares J & Morales-Aguilera A (1993) Differential
accumulation of thallous ion by diverse rabbit and rat muscles. Bull
Environ Contam Toxicol, 51: 764-771.
Carlson RW, Bazzaz FA, & Rolfe GL (1975) The effect of heavy metals on
plants. II. Net photosynthesis and transpiration of whole corn and
sunflower plants treated with Pb, Cd, Ni, and Tl. Environ Res,
10: 113-120.
Cataldo DA & Wildung RE (1978) Soil and plant factors influencing the
accumulation of heavy metals by plants. Environ Health Perspect,
27: 149-159.
Cavanagh JB (1979) Metallic toxicity and the nervous system. In: Smith
WT & Cavanagh JB ed. Recent advances in neuropathology. Edinburgh,
Churchill Livingstone, pp 247-275.
Cavanagh JB (1988) Neurotoxic effects of metals and their
interactions. Recent Adv Nerv Syst Toxicol, 100: 177-202.
Cavanagh JB (1991) Annotation. What have we learnt from Graham
Frederick Young? Reflections on the mechanism of thallium
neurotoxicity. Neuropathol Appl Neurobiol, 17: 3-9.
Cavanagh JB & Gregson M (1978) Some effects of thallium salts on the
proliferation of hair follicle cells. J Pathol, 125: 179-191.
Cavanagh JB, Fuller NH, Johnson HRM, & Rudge P (1974) The effects of
thallium salts, with particular reference to the nervous system
changes. Q J Med New Ser, 43: 293-319.
Chandler HA & Scott M (1986) A review of thallium toxicology. J R Navy
Med Serv, 72: 75-79.
Chandler HA, Archbold GPR, Gibeon JM, O'Callaghan P, Marks JN, &
Petghybridge RJ (1990) Excretion of a toxic dose of thallium. Clin
Chem, 36: 1506-1509.
Chattopadhyay A & Jervis RE (1974) Multielement determination in
market-garden soils by instrumental photon activation analysis. Anal
Chem, 46: 1630-1639.
Christensen HE, Luginbyhl TT, Hill RH Jr, McGrath DJ, Georgevich M,
Mitchell FL, & May JR ed. (1973) The toxic substances list, 1973
edition. Washington, DC, US Department of Health, Education, and
Welfare, Public Health Service, US Government Printing Office.
Chusid JG & Kopeloff LM (1962) Epileptogenic effects of pure metals
implanted in motor cortex of monkeys. J Appl Physiol, 17: 697-700.
Clausen B & Karlog O (1974) Loading with thallium among wild animals
of the marten genus and badgers in Denmark. Nord Vet.med, 26: 339-350.
Claussen U, Roll R, Dolger R, Matthiaschk G, Majewski F, Stoll B, &
Röhrborn G (1981) [Mutagenicity and teratogenicity of thallium - with
special consideration of the situation in Lengerich.] Rhein Ärztebl,
16: 469-475 (in German).
Cooper GP, Suszkiw JB, & Manalis RS (1984) Presynaptic effects of
heavy metals. In: Narahashi T ed. Cellular and molecular
neurotoxicology. New York, Raven Press, pp 1-21.
Cornelis R (1988) Analytical procedures and clinical reference
materials in monitoring human exposures to trace metals with special
reference to Cr, Pb and Tl. Sci Total Environ, 71: 269-283.
Cortes Toro E, Parr RM, & Clements SA (1990) Biological and
environmental reference materials for trace elements, nuclides and
organic microcontaminants. Vienna, International Atomic Energy Agency
(IAEA/RL/128 Rev.1).
Cottier H (1980) [Pathogenesis.] Berlin, Springer Verlag (in German).
Cotton FA & Wilkinson G (1988) Advanced inorganic chemistry, 5th ed.
New York, Chichester, Brisbane, Toronto, John Wiley and Sons.
Coyle V (1980) Diagnosis and treatment of thallium toxicosis in a dog.
J Small Anim Pract, 21: 391-397.
Craig PJ (1980) Metal cycles and biological methylation. In: Hutzinger
O ed. The handbook of environmental chemistry. Volume 1: The natural
environment and the biogeochemical cycles - Part A. Berlin, Springer
Verlag, pp 169-227.
Cromartie E, Reichel WL, Locke LN, Belisle AA, Kaiser TE, Lamont TG,
Mulhern BM, Prouty RM, & Swineford DM (1975) Residues of
organochlorine pesticides and polychlorinated biphenyls and autopsy
data for bald eagles, 1971-72. Pestic Monit J, 9: 11-14.
Crössmann G (1984) [Thallium - a new environmental contamination?]
Angew Bot, 58: 3-10 (in German).
Crössmann G (1985) [Toxic elements in feed.] Dtsch Tierärztl
Wochenschr, 92: 232-233 (in German).
Csicsaky M & Wiegand H (1981) [Neurotoxical effects of thallium on the
peripheral and central nervous systems of the rat.] Umwelthygiene,
13: 180 (in German).
Csicsaky M, Wiegand H, Uhlig S, Lohmann H, & Papadopoulos R (1988)
Miniature endplate potentials as a tool in neurotoxicology.
Toxicology, 49: 121-129.
Curry AS, Grech JL, Spiteri L, & Vassallo L (1969) Death from thallium
poisoning: A case report. Eur J Toxicol, 2: 260-269.
Danilewicz M, Danilewicz M, & Kurnatowski A (1979) [Dynamics of
histopathological changes in the kidney of experimental animals after
lethal doses of thallium.] Med Pracy, 30: 257-265 (in Polish).
Danilewicz M, Kurnatowski A, & Wagrowska-Danilewicz M (1980) [Dynamics
of pathomorphological changes in some internal organs of rats poisoned
with lethal doses of thallous sulfate and treated with dithiocarb,
with special attention to renal changes.] Patol Pol, 31: 557-565
(in Polish).
Date AR, Cheung YY, & Stuart ME (1988) Application of inductively
coupled plasma mass spectrometry to the analysis of iron ores. J Anal
At Spectrom, 3: 653-658.
Davids P, Güthner G, Lange M, Lohrer W, & Vahrenholt F (1980)
[Situation and perspectives of the techniques for purification of
air.] Staub-Reinhalt Luft, 40: 392-402 (in German).
Davis RD, Beckett PHT, & Wollan E (1978) Critical levels of twenty
potentially toxic elements in young spring barley. Plant Soil,
49: 395-408.
Davis LE, Standefer JC, Kornfeld M, Abercrombie DM, & Butler C (1981)
Acute thallium poisoning: toxicological and morphological studies of
the nervous system. Ann Neurol, 10: 38-44.
Dawson GW, Jennings AL, Drozdowski D, & Rider E (1975/77) The acute
toxicity of 47 industrial chemicals to fresh and saltwater fishes.
J Hazard Mater, 1: 303-318.
De Groot G & van Heijst ANP (1988) Toxicokinetic aspects of thallium
poisoning. Methods of treatment by toxin elimination. Sci Total
Environ, 71: 411-418.
Den Dooren de Jong LE & Roman WB (1971) Tolerance of Azotobacter for
metallic and non-metallic ions. Antonie van Leeuwenhoek J Microbiol
Serol, 37: 119-124.
De Ruck A, Vandecasteele C, & Dams R (1987) Determination of thallium
in natural waters by electrothermal atomic absorption spectrometry.
Mikrochim Acta, II: 187-193.
De Ruck A, Vandecasteele C, & Dams R (1989) Determination of thallium
in solid environmental samples by electrothermal atomic absorption
spectrometry. Anal Lett, 22: 469-479.
DFG (German Research Council) (1990) [Maximal concentrations at the
place of work and biological tolerance values of material for work
1990.] Weinheim, Verlag Chemie (in German).
Di Gaudio MR & Hirshfield HI (1976) Respiratory toxicity of thallium
in Paramecium bursaria. J Cell Biol, 70: 60A.
Dilling WJ & Healey CW (1926) Influence of lead and the metallic ions
of copper, zinc, thorium, beryllium and thallium on the germination of
frogs' spawn and on the growth of tadpoles. Ann Appl Biol,
13: 177-188.
Dolgner R & Wiegand H (1982) [Biological monitoring of thallium -
medical aspects.] Staub-Reinhalt Luft, 42: 151-152 (in German).
Dolgner R, Brockhaus A, Ewers U, Wiegand H, Majewski F, & Soddemann H
(1983) Repeated surveillance of exposure to thallium in a population
living in the vicinity of a cement plant emitting dust containing
thallium. Int Arch Occup Environ Health, 52: 79-94.
Douglas KT, Bunni MA, & Baindur SR (1990) Thallium in biochemistry.
Int J Biochem, 22: 429-438.
Downs WL, Scott JK, Steadman LT, & Maynard EA (1960) Acute and
sub-acute toxicity studies of thallium compounds. Am Ind Hyg Assoc J,
21: 399-406.
Drucker H, Garland TR, & Wildung RE (1979) Metabolic response of
microbiota to chromium and other metals. In: Kharasch N ed. Trace
metals in health and disease. New roles of metals in biochemistry, the
environment, and clinical/nutritional studies. New York, Raven Press,
pp 1-25.
Ducket S, Hiller D, & Ballas SK (1983) Quantitation and localization
of 204thallium in the central and peripheral nervous system of adult
and young rats. Neurotoxicology, 4: 227-234.
Dumitru D & Kalantri A (1990) Electrophysiologic investigation of
thallium poisoning. Muscle Nerve, 13: 433-437.
Durbin PW, Scott KG, & Hamilton JG (1957) The distribution of
radioisotopes of some heavy metals in the rat. Berkeley, University of
California Press, pp 3: 1-34 (University of California Publications in
Pharmacology, Volume 3).
Dvorák P (1970) [Hexacyanoferrates (II) as thallium antidotes.
Preparation and properties.] Arzneimittelforschung, 20: 1886-1888
(in German).
Dvorák P (1973) [Removal by rhyolitic pumice of internally deposited
thallium from the rat.] Res Exp Med, 162: 63-66 (in German).
Dvornikov AG, Ovsyannikov LB, & Sidenko OG (1973) [Biogeochemical
dispersion haloes of chalcophyllite elements in gold ore
manifestations of the Nagolnyi Ridge (The Donbas).] Dopov Akad Nauk
Ukr RSR, B35: 490-494 (in Russian).
Dvornikov AG, Ovsyannikova LB, & Sidenko OG (1976) [Some
characteristics of the coefficients of biological absorption and the
biogeochemical coefficients at hydrothermal deposits of the Donets
Basin in relation to predicting hidden ore mineralization.]
Geokhimiya, 4: 626-633 (in Russian).
Ebarvia B, Macalalad E, & Rogue N (1988) Determination of silver,
cadmium, selenium, tellurium and thallium in geochemical exploration
samples by atomic absorption spectrometry. J Anal At Spectrom,
3: 199-203.
Edel Rade J, Marafante E, Sabbioni E, Gregotti C, Di Nucci A, & Manzo
L (1982) Placental transfer and retention of 201Tl-thallium in the
rat. Toxicol Lett, 11: 275-280.
EC (1987) [Legal regulations on hazardous substances.] Luxembourg,
Office for Official Publications of the European Community, vol 2
(in German).
Ehrhardt K (1927) [The effect of maternal intoxication with thallium
on the progeny.] Klin Wochenschr, 6: 1374-1375 (in German).
Emara M & Soliman MA (1950) The distribution of ingested thallium in
the tissues of animals. J Egypt Med Assoc, 33: 1-15.
Emsley J (1978) The trouble with thallium. New Sci, 79: 392-394.
Erbslöh J (1960) [Thallium poisoning in the second half of pregnancy.]
Arch Toxicol, 18: 156-159 (in German).
Ewan RC (1978) Toxicology and adverse effects of mineral imbalance
with emphasis on selenium and other minerals. In: Oehme FW ed.
Toxicity of heavy metals in the environment. New York, Marcel Dekker
Inc., vol 1, pp 445-489.
Ewers U (1988) Environmental exposure to thallium. Sci Total Environ,
71: 285-292.
Ewers U & Brockhaus A (1982) [Biological monitoring of thallium -
analysis and results.] Staub-Reinhalt Luft, 42: 149-151 (in German).
Favari L & Mourelle M (1985) Thallium replaces potassium in activation
of the (Na+ K+)-ATPase of rat liver plasma membranes.
J Appl Toxicol, 5: 32-34.
Feudell P (1982) [Thallium polyneuropathology.] Psychiatr Neurol Med
Psychol, 34: 23-25 (in German).
Fischer AB (1981) Comparative studies of heavy metal toxicity at the
cellular level. In: Proceedings of the 3rd International Conference on
Heavy Metals in the Environment, Toronto, pp 486-490.
Fitzek A & Henning A (1976) [Distribution of thallium in the body of a
pregnant cat.] Dtsch Tierärztl Wochenschr, 83: 66-68 (in German).
Flanjak J & Hodda AE (1988) Simultaneous determination of thallium,
silver and gold in urine by flame and graphite-furnace atomic
absorption spectrometry after extraction of the iodide complexes as
ion-pairs with tri- n-octylamine. Anal Chim Acta, 207: 283-289.
Ford JK, Eyring EJ, & Anderson CE (1968) Thallium chondrodystrophy in
chick embryos. An histological and biochemical investigation. J Bone
Jt Surg, 50A: 687-700.
Formigli L, Grégotti C, Di Nucci A, Edel J, Sabbioni E, & Manzo L
(1985) Reproductive toxicity of thallium. Experimental and clinical
data. Proceedings of the 5th International Conference on Heavy Metals
in the Environment, Toronto, vol 2, pp 15-18.
Formigli L, Scelsi R, Poggi P, Grégotti C, Di Nucci A, Sabbioni E,
Gottardi L, & Manzo L (1986) Thallium-induced testicular toxicity in
the rat. Environ Res, 40: 531-539.
Forth W & Henning CH (1979) [Thallium poisoning and the therapy.]
Dtsch Ärztebl, 76: 2803-2807 (in German).
Fowler BA (1988) Mechanism of indium, thallium, and arsine gas
toxicity. In: Clarkson TW, Friberg L, Nordberg GF, & Sager PR ed.
Biological monitoring of toxic metals. New York, London, Plenum Press,
pp 469-478.
Frerking H, Osterkamp A, & Lindfeld KA (1990) [Adverse effects in
cattle due to vaccination against foot-and mouth disease or poisoning
by thallium.] Dtsch Tierärztl Wochenschr, 97: 206-207 (in German).
Galba J (1982) [Analysis of trace concentrations of phytotoxic
elements in solid emissions from the thermal power plant ENO in soil
and plants.] Pol'nohospodárstvo, 28: 293-306 (in Czechoslovakian).
Galvan-Arzate S & Rios C (1994) Thallium distribution in organs and
brain regions of developing rats. Toxicology, 90: 63-69.
Garcia Bugarin M, Casas JB, Sordo J, & Filella M (1989) Thallium(I)
interactions in biological fluids: a potentiometric investigation of
thallium(I) complex equilibria with some sulfur-containing amino
acids. J Inorg Biochem, 35: 95-105.
Gastel B (1978) Clinical conferences at the Johns Hopkins hospital:
thallium poisoning. Johns Hopkins Med J, 142: 27-31.
Gefel A, Liron M, & Hirsch W (1970) Chronic thallium poisoning.
Isr J Med Sci, 6: 380-382.
Gehring PJ & Hammond PB (1967) The interrelation between thallium and
potassium in animals. J Pharmacol Exp Ther, 155: 187-201.
Geilmann W & Neeb KH (1959) [Application of the evaporation analysis
for minor amounts of material. II. The proof and determination of
minor contents of thallium.] Fresenius Z Anal Chem, 165: 251-268
(in German).
Geilmann W, Beyermann K, Neeb K-H, & Neeb R (1960) [Thallium, a
regularly occurring trace element in animals and plants.] Biochem Z,
333: 62-70 (in German).
Geldmacher-von Malinckrodt M, Stamm D, & Enders P (1984)
Interlaboratory survey on thallium in urine. Z Rechtsmed, 93: 219-225.
Ghezzi R & Bozza Marrubini M (1979) Prussian blue in the treatment of
thallium intoxication. Vet Hum Toxicol, 21: 64-66.
Gibson JE & Becker BA (1970) Placental transfer, embryotoxicity, and
teratogenicity of thallium sulfate in normal and potassium-deficient
rats. Toxicol Appl Pharmacol, 16: 120-132.
Gibson JE, Sigdestad CP, & Becker BA (1967) Placental transport and
distribution of thallium-204 sulfate in newborn rats and mice. Toxicol
Appl Pharmacol, 10: 378-411.
Glomme J (1983) Thallium and compounds. In: Encyclopaedia of
occupational health and safety, 3rd ed. Geneva, International Labour
Office, vol 2, pp 2170-2171.
Gluskoter HJ, Ruch RR, Miller WG, Cahill RA, Dreher GB, & Kuhn JK
(1977) Trace elements in coal: occurrence and distribution - Final
report. Urbana, Illinois, Illinois State Geological Survey
(EPA 600/7-77-064).
Goenechea S & Sellier K (1967) [The natural concentration of thallium
in humans.] Dtsch Z Gesamte Gerichtl Med, 60: 135-141 (in German).
Goldblatt D (1989) Pollutants and industrial hazards. In: Rowland LP
ed. Merritt's textbook of neurology, 8th ed. Philadelphia,
Pennsylvania, Lea and Febinger, pp 919-928.
Gorbauch H, Rump HH, Alter G, & Schmitt-Henco CH (1984) [Determination
of thallium in raw materials and environmental samples.] Fresenius Z
Anal Chem, 317: 236-240 (in German).
Gough LP, Shacklette HT, & Case AA (1979) Element concentrations toxic
to plants, animals, and man. US Geol Surv Bull, 1466: 1-80.
Graben N, Doss M, & Klöppel HA (1978) [Disturbances of porphyrin
metabolism by thallium poisoning.] Med Klin (Munich), 73: 1114-1116
(in German).
Graben N, Klöppel H-A, Heidemann H, & Weiler G (1980) [Therapy of
thallium poisoning with special consideration of pregnancy and
lactation.] Med Welt, 31: 1391-1394 (in German).
Grégotti C, di Nucci A, Formigli L, Sabbioni E, & Manzo L (1985)
Altered testicular enzyme patterns in rats after long-term exposure to
thallium sulfate. J Toxicol Clin Exp, 5: 265-271.
Grégotti C, Ponce R, & Faustman EM (1993) Mechanism of toxicity of Tl,
B and Pb on testicular cells in vitro. Abstracts of the 32nd Annual
Meeting of the Society of Toxicology. Toxicologist, 13: 298.
Gregus Z & Klaassen CD (1986) Disposition of metals in rats: a
comparative study of fecal, urinary, and biliary excretion and tissue
distribution of eighteen metals. Toxicol Appl Pharmacol, 85: 24-38.
Greving R & Gagel O (1928) [Polyneuritis after acute thallium
poisoning.] Klin Wochenschr, 7: 1323-1325 (in German).
Griepink B, Sager M, & Tölg G (1988) Determination of traces of
thallium in various matrices. Pure Appl Chem, 60: 1425-1436.
Gubernator K, Moroni W, Munder S, Franke B, Ruske B, & Teufel D (1979)
[The ambiguity of thallium contaminations in the neighbourhood of
cement plants - Heidelberg Seminar on Environmental Protection,
Freiburg, Germany, October 1979.] Heidelberg, Heidelberg Institute for
Research on Energy and Environment, Institute for Applied Ecology
(in German).
Guderian R (1977) Air pollution. Berlin, Springer Verlag.
Guidoboni RJ & Leipziger FD (1988) Glow discharge mass spectrometry -
the newest tool for high purity materials analysis. J Cryst Growth,
89: 16-20.
Günther M (1971) [Distribution and toxicity of thallium (I) in the rat
as influenced by colloidal ferrihexacyanoferrate (II).] Arch Toxicol,
28: 39-45 (in German).
Günther K & Umland F (1989) Bonding states of thallium and cadmium in
thallium-treated and native rape. J Inorg Biochem, 36: 63-74.
Gutknecht J (1983) Cadmium and thallous ion permeabilities through
lipid bilayer membranes. Biochim Biophys Acta, 735: 185-188.
Hagedorn-Götz H & Stoeppler M (1975) [The forensic proof of thallium
in human hairs by flameless atomic absorption spectrometry.] Arch
Toxicol, 34: 17-26 (in German).
Hajdu S & Nessler F (1985) [Chemistry, physics and occurrence of
thallium and its compounds.] Wiss Z Humboldt-Univ (Berlin)
Math-Naturwiss Reihe, 34: 752-755 (in German).
Hall BK (1972) Thallium-induced achondroplasia in the embryonic chick.
Dev Biol, 28: 47-60.
Hall BK (1977) Thallium-induced achondroplasia in chicken embryos and
the concept of critical periods during development. Teratology,
15: 1-15.
Hall BK (1985) Critical periods during development as assessed by
thallium-induced inhibition of growth of embryonic chick tibiae
in vitro. Teratology, 31: 353-361.
Hamaguchi H (1960) [Inorganic constituents in biological materials.
XIV. Thallium, selenium, and arsenic contents in Minamata districts.]
Nippon Kagaku Zasshi, 81: 920-927 (in Japanese).
Han HB, Kaiser G, & Tölg G (1982) Decomposition of biological
materials, rocks sand soils with simultaneous volatilization of trace
elements in pure oxygen under dynamic conditions. Anal Chim Acta,
134: 3-11.
Hapke H-J (1984) [The effects of metals on domestic animals.] In:
Merian E ed. [Metals in the environment.] Weinheim, Verlag Chemie,
pp 181-194 (in German).
Hapke H-J, Barke E, & Spikermann A (1980) [Accumulation of thallium in
the edible tissues of sheep and cattle according to the amount of
thallium in the feed.] Dtsch Tierärztl Wochenschr, 87: 376-378
(in German).
Hart MM & Adamson RH (1971) Antitumor activity and toxicity of salts
of inorganic group IIIa metals: aluminium, gallium, indium, and
thallium. Proc Natl Acad Sci (USA), 68: 1623-1626.
Hart MM, Smith CF, Yancey ST, & Adamson RH (1971) Toxicity and
antitumor activity of gallium nitrate and periodically related metal
salts. J Natl Cancer Inst, 47: 1121-1127.
Hasan M & Ali F (1981) Effects of thallium, nickel and cobalt
administration on the lipid peroxidation in different regions of the
rat brain. Toxicol Appl Pharmacol, 57: 8-13.
Hasan M & Haider SS (1989) Acetyl-homocystein thiolactone protects
against some neurotoxic effects of thallium. Neurotoxicology,
10: 257-261.
Hasan M, Bajpai VK, & Shipstone AC (1977a) Electron microscopic study
of thallium-induced alterations in the oligodendrocytes of the rat
area postrema. Exp Pathol, 13: 338-345.
Hasan M, Chandra SV, Bajpai VK, & Ali SF (1977b) Electron microscopic
effects of thallium poisoning on the rat hypothalamus and hippocampus:
biochemical changes in the cerebrum. Brain Res Bull, 2: 255-262.
Hasan M, Chandra SV, Dua PR, Raghubir R, & Ali SF (1977c) Biochemical
and electrophysiologic effects of thallium poisoning on the rat corpus
striatum. Toxicol Appl Pharmacol, 41: 353-359.
Hasan M, Ashraf I, & Ali SF (1977d) Effect of thallium intoxication on
the amino acid concentration of different regions of the rat brain.
Curr Sci, 46: 554-556.
Hasan M, Ashraf I, & Bajpai VK (1978a) Electron microscopic study of
the effects of thallium poisoning on the rat cerebellum. Forensic Sci,
11: 139-146.
Hasan M, Ali SF, & Tariq M (1978b) Levels of dopamine, norepinephrine
and 5-hydroxytryptamine in different regions of the rat brain in
thallium toxicosis. Acta Pharmacol Toxicol, 43: 169-173.
Hausman R & Wilson WJ jr (1964) Thallotoxicosis: a social menace.
J Forensic Sci, 9: 72-88.
Hayes WJ & Laws ER (1991) Handbook of pesticide toxicology - Volume 2:
Classes of pesticides. New York, London, Academic Press.
Heath A, Ahlmén J, Branegård B, Lindstedt S, Wickström I, & Andersen O
(1983) Thallium poisoning - Toxin elimination and therapy in three
cases. J Toxicol Clin Toxicol, 20: 451-463.
Heinrichs H (1982) [Metals in sewage sludge from municipal water
purification plants.] Naturwissenschaften, 69: 88-89 (in German).
Heinrichs H & Mayer R (1977) Distribution and cycling of major and
trace elements in two central European forest ecosystems. J Environ
Qual, 6(4): 402-407.
Heit M (1985) Concentrations of potentially toxic trace elements in
the muscle tissue of fish from acidic and circumneutral Adirondack
Lakes. Proceedings of the 5th International Conference on Heavy Metals
in the Environment, Toronto, vol 1, pp 655-657.
Henderson P, Hale TW, Shum S, & Habersang RW (1985) N-acetylcysteine
therapy of acute heavy metal poisoning in mice. Vet Hum Toxicol,
27: 522-525.
Henke G (1991) Thallium determination in biological materials by
radiochemical neuron activation analysis. Fresenius J Anal Chem,
339: 245-248.
Hennekes R (1983) [Impairment of retinal function in acute thallium
poisoning.] Klin Mon.bl Augenheilkd, 182: 334-336 (in German).
Henning CH & Forth W (1977) Absorption and secretion of thallous ions
on isolated intestinal segments in vitro of rats.
Naunyn-Schmiedeberg's Arch Pharmacol, 297(suppl II): R2.
Henning CH & Forth W (1982) The excretion of thallium(I) ions into the
gastrointestinal tract in situ of rats. Arch Toxicol, 49: 149-158.
Henning CH, Becker G, & Forth W (1982) Movement of thallium(I) ions
across the epithelium of intestinal segments in vitro of rats.
Naunyn-Schmiedeberg's Arch Pharmacol, 321: 157-163.
Henshaw JM, Heithmar EM, & Hinners TA (1989) Inductively coupled
plasma mass spectrometric determination of trace elements in surface
waters subject to acidic deposition. Anal Chem, 61: 335-342.
Herman MM & Bensch KG (1967) Light and electron microscopic studies of
acute and chronic thallium intoxication in rats. Toxicol Appl
Pharmacol, 10: 199-222.
Heydlauf H (1969) Ferric-cyanoferrate(II): an effective antidote in
thallium poisoning. Eur J Pharmacol, 6: 340-344.
Heyl T & Barlow RJ (1989) Thallium poisoning: a dermatological
perspective. Br J Dermatol, 121: 787-791.
Hill WH & Murphy MA (1959) An appraisal of an industrial air
contaminant by electrochromatography. Am Ind Hyg Assoc J, 20: 387-391.
Hill HAO, Pratt JM, Ridsdale S, Williams FR, & Williams RJP (1970)
Kinetics of substitution of co-ordinated carbanions in cobalt(III)
corrinoids. Chem Commun, 124: 341.
Hiltenkamp E & Jackwerth E (1988) [Investigations on the determination
of Bi, Cd, Hg, Pb and Tl in high-purity gallium by graphite furnace
AAS with atomization of metallic samples.] Fresenius Z Anal Chem,
332: 134-139 (in German).
Hoffmann G, Schweiger P, & School W (1982) [Uptake of thallium by
agriculturally and horticulturally useful plants.] Landwirtsch Forsch,
35: 45-54 (in German).
Hollunger G (1960) The effect of trivalent thallium on oxidative
phosphorylation. Acta Pharmacol Toxicol, 16: 347-356.
Holm J, Brehmer R-D, Müller S, & Wester D (1987) [Bioindication of
harmful substances taking roe deer and mallard as examples.]
Fleischwirtschaft, 67: 1145-1149 (in German).
Hologgitas J, Ullucci P, Driscoll J, Grauerholz J, & Martin H (1980)
Thallium elimination kinetics in acute thallotoxicosis. J Anal
Toxicol, 4: 68-73.
Howe HE (1971) Chapter 27. Thallium. In: Hampel CA ed. Rare metals
handbook. Huntinston, New York, Huntinston Publishing Company,
pp 529-535.
Hsie AW, Schenley RL, Tan E-L, Perdue SW, Williams MW, Hayden TL, &
Turner JE (1984) The toxicity of sixteen metallic compounds in Chinese
hamster ovary cells: a comparison with mice and Drosophila. Altern
Methods Toxicol, 2: 115-125.
Huber F, Schmidt U, & Kirchmann H (1978) Aqueous chemistry of
organolead and organothallium compounds in the presence of
microorganisms. In: Organometals and organometalloids: occurrence and
fate in the environment). Washington, DC, American Chemical Society,
pp 65-81 (ACS Symposium Series No. 82).
Hultin T & Näslund PH (1974) Effects of thallium(I) on the structure
and functions of mammalian ribosomes. Chem-Biol Interact, 8: 315-328.
Inturrisi CE (1969) Thallium activation of K+-activated phosphatases
from beef brain. Biochim Biophys Acta, 173: 567-569.
IPCS (International Programme on Chemical Safety) (1992) Poisons
information monograph: Thallium. Geneva, World Health Organization
(IPCS/INTOX/PIM.525).
IPS (Industrial Association for the Protection of Plants) ed. (1982)
[Active compounds in agents for plant protection and pest control.]
Offenbach, Bintz Verlag (in German).
Itawi RK & Turel ZR (1987) Determination of thallium in biological
samples by thermal neutron activation analysis and low-level
beta-counting. J Radianal Nucl Chem, 115: 141-147.
Ivashchenko AT & Balmukhanov BS (1974) [Effect of monovalent ions on
the adenosine triphosphatase activity of ascites tumor cells.]
Izv Akad Nauk Kaz SSR Ser Biol, 12: 73-78 (in Russian).
Ivashchenko AT, Balmukhanov BS, & Toktamysova ZS (1973) [Effects of
monovalent ions on the glycolysis and respiration of ascite tumor
cells.] Tsitologiya, 15: 1487-1491 (in Russian).
Iyengar GV, Kollmer WE, & Bowen HJM (1978) The elemental composition
of human tissues and body fluids. Weinheim, Verlag Chemie.
Jeantet AY, Ballan-Dufrançais C, & Martoja R (1977) Insect resistance
to mineral pollution. Importance of spherocrystal in ionic regulation.
Rev Ecol Biol Sol, 14: 563-582.
Johnson CA (1976) The determination of some toxic metals in human
liver as a guide to normal levels in New Zealand. Part I.
Determination of Bi, Cd, Cr, Co, Cu, Pb, Mn, Ni, Ag, Tl and Zn. Anal
Chim Acta, 81: 69-74.
Junghans V & Nessler F (1985) [The clinical signs of chronic thallium
intoxication shown in occupational thallium poisonings.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 778-780 (in German).
Kaeser HE & Lambert EH (1962) Nerve function in experimental
polyneuritis. Electroencephalogr Clin Neurophysiol Prog Electromyogr,
22(suppl): 29-35.
Kamerbeek HH, Rauws AG, ten Ham M, & van Heijst ANP (1971a) Dangerous
redistribution of thallium by treatment with sodium
diethyldithiocarbamate. Acta Med Scand, 189: 149-154.
Kamerbeek HH, Rauws AG, Ten Ham M, & van Heijst ANP (1971b) Prussian
blue in therapy of thallotoxicosis: An experimental and clinical
investigation. Acta Med Scand, 189: 321-324.
Kaplan DI, Adriano DC, & Sajwan KS (1990) Thallium toxicity in bean.
J Environ Qual, 19: 359-365.
Karkos J (1971) [Neuropathological findings in thallium
encephalopathy.] Neurol Neurochir Pol, 21: 911-915 (in Polish).
Karnofsky DA & Lacon CR (1964) Effects of drugs on the skeletal
development of the chick embryo. Clin Orthop Relat Res, 33: 59-70.
Karnofsky DA, Ridgway LP, & Patterson PA (1950) Production of
achondroplasia in the chick embryo with thallium. Proc Soc Exp Biol
Med, 73: 255-259.
Kazantzis G (1986) Thallium. In: Friberg L, Nordberg GF, & Vouk V ed.
Handbook on the toxicology of metals, 2nd ed. Amsterdam, Oxford, New
York, Elsevier Science Publishers, pp 549-567.
Kazantzis G (1994) Thallium and tin. In: Alessio L, Berlin A, Roi R, &
van der Venne, M-Th ed. Biological indicators for the assessment of
human exposure to industrial chemicals. Luxembourg, European
Commission, Health and Safety Directorate, pp 41-92 (Report
EUR 14815/EN).
Kemper FH (1979) [Thallium poisonings.] Münch Med Wochenschr,
121: 1357-1358 (in German).
Kemper FH & Bertram HP (1984) [Thallium.] In: Merian E ed. [Metals in
the environment.] Weinheim, Verlag Chemie, pp 571-583.
Kemper FH & Bertram HP (1991) Thallium. In: Merian E ed. Metals and
their compounds in the environment. Weinheim, Verlag Chemie,
pp 1227-1241.
Kennedy P & Cavanagh JB (1976) Spinal changes in the neuropathy of
thallium poisoning. J Neurol Sci, 29: 295-301.
Kijewski H (1984) Nature of the black zones in hair after ingestion of
thallium. Arch Kriminol, 173: 36-44.
Klahre P, Valenta P, & Nürnberg HW (1978) [A standardized pulse
inverse voltametric analytical method for testing drinking water for
toxic metals. Part 1: Simultaneous determination of copper, cadmium,
lead and zinc, and lead and thallium.] Vom Wasser, 51: 199-219
(in German)
Klöppel A & Weiler G (1978) [Contribution to heavy metal poisonings,
especially by thallium.] Dtsch Med Wochenschr, 103: 75-76 (in German).
Knapp G (1985) Sample preparation techniques - an important part in
trace element analysis for environmental research and monitoring. Int
J Environ Anal Chem, 22: 71-83.
Kogan BI (1970) [Thallium.] Redk Elem, 5: 27-48 (in Russian).
Kolb Meyers V (1983) Chemicals which cause birth defects - teratogens:
a special concern of research chemists. Sci Total Environ, 32: 1-12.
Konermann H, Crössann G, & Hoppenbrock KH (1982) [Investigations of
the influence of thallium-contaminated food rations on performance,
health and meat residues in fattening pigs.] Tierärztl Umsch,
37: 8-21 (in German).
Korkisch J & Steffan J (1979) Determination of thallium in natural
waters. Int J Environ Anal Chem, 6: 111-118.
Kosta L (1982) Contamination as a limiting parameter in trace
analysis. Talanta, 29: 985-992.
Krassowsky GN, Yurasova OI, Charyev OG, Skrypnikov AI, & Varshavskaya
SP (1977) [Prognosis of the gonadotoxic action of heavy metals judging
by the primary effect of material accumulation.] Gig i Sanit, 7: 11-16
(in Russian).
Krassowsky GN, Wasjukowitsch LJ, Tscharyjew OG, & Kenessarijew UI
(1980) [Experimental studies on making quick prognoses of a
gonadotoxic effect of inorganic compounds.] Z Gesamte Hyg Grenzgeb,
26: 336-338 (in German).
Kravzov J, Rios C, Altagracia M, Monroy-Noyola A, & López F (1993)
Relationship between physicochemical properties of Prussian blue and
its efficacy as antidote against thallium poisoning. J Appl Toxicol,
31: 213-216.
Ku DD, Akera T, Oldaard MK, & Brody TM (1978) Effects of lithium and
thallous ions on sodium. Activity in the guinea pig heart and their
relationship to the positive inotropic action. Naunyn-Schmiedeberg's
Arch Pharmacol, 304: 167-173.
Kwan KHM & Smith S (1988) The effect of thallium on the growth of
Lemna minor and plant tissue concentrations in relation to both
exposure and toxicity. Environ Pollut, 52: 203-220.
Lameijer W & van Zwieten PA (1977a) Kinetic behaviour of thallium in
the rat. Accelerated elimination of thallium owing to treatment with
potent diuretic agents. Arch Toxicol, 37: 265-273.
Lameijer W & van Zwieten PA (1977b) Kinetics of thallium elimination
in the rat after acute and subacute intoxication; influence of
diuretic treatment. Proc Eur Soc Toxicol, 18: 163-164.
Lameijer W & van Zwieten PA (1979) The efficacy of a potassium-rich
diet compared to diuretic treatment on the renal elimination of
thallium in the rat. Arch Toxicol, 42: 33-41.
Landauer W (1930) [Influence of thallium poisoning of the father on
his descendants. Experiments on roosters.] Arch Gewerbepathol
Gewerbehyg, 1: 791-792 (in German).
Larson CP, Keller WN, & Manges JD (1939) Accidental canine
thallotoxicosis and dangers of thallium used as a rodenticidal agent.
J Am Vet Med Assoc, 95: 486-489.
LeBlanc GA (1984) Interspecies relationships in acute toxicity of
chemicals to aquatic organisms. Environ Toxicol Chem, 3: 47-60.
LeBlanc GA & Dean JW (1984) Antimony and thallium toxicity to embryos
and larvae of fathead minnows (Pimephales promelas). Bull Environ
Contam Toxicol, 32: 565-569.
Lehmann FPA & Favari L (1984) Parameters for the adsorption of
thallium ions by activated charcoal and Prussian blue. J Toxicol Clin
Toxicol, 22: 331-339.
Lehmann FPA & Favari L (1985) Acute thallium intoxication: kinetic
study of the relative efficacy of several antidotal treatments in
rats. Arch Toxicol, 57: 56-60.
Lehn H & Bopp M (1987) [Heavy metals in soil and the determination of
the availability for plants.] Angew Bot, 61: 467-481 (in German).
Leloux MS, Lich NP, & Claude JR (1987a) Flame and graphite furnace
atomic absorption spectroscopy methods for thallium - a review. At
Spectrom, 8: 71-77.
Leloux MS, Lich NP, & Claude JR (1987b) Experimental studies on
thallium toxicity in rats. I. Localization and elimination of thallium
after oral acute and sub-acute intoxication. J Toxicol Clin Exp,
7: 247-257.
Le Quesne PM (1984) Toxic neuropathies. In: Asbury AK & Gilliatt RW
ed. Butterworths international medical reviews: Neurology. Volume 4:
Peripheral nerve disorders - a practical approach. London,
Butterworths, pp 184-204.
Letourneau VA, Joshi BM, & Butler LC (1987) Comparison between Zeeman
and continuum background correction for graphite furnace AAS on
environmental samples. At Spectrom, 8: 145-149.
Levander OA & Argrett LC (1969) Effects of arsenic, mercury, thallium,
and lead on selenium metabolism in rats. Toxicol Appl Pharmacol,
14: 308-314.
Liang Y, Shi J, Chen L, & Ding G (1980) [Dimercaptosuccinic acid per
os promoted the excretions of Pb, Cu, Sb, Sr, Tl and Pm.] Acta Pharm
Sinica, 15: 335-340 (in Chinese).
Lide DR (1990) CRC handbook of chemistry and physics: A ready
reference book of chemical and physical data, 71st ed. Boca Raton,
Florida, CRC Press.
Lie R, Thomas RG, & Scott JK (1960) The distribution and excretion of
thallium-204 in the rat, with suggested MPC's and a bio-assay
procedure. Health Phys, 2: 334-340.
Lillie WI & Parker HL (1932) Retrobulbar neuritis due to thallium
poisoning. J Am Med Assoc, 98: 1347-1349.
Lindegren CC (1971) Mitochondria in intoxication and detoxication.
Physiol Chem Phys, 3: 499-500.
Lindegren CC & Lindegren G (1973a) Mitochondrial modification and
respiratory deficiency in the yeast cell caused by cadmium poisoning.
Mutat Res, 21: 315-322.
Lindegren CC & Lindegren G (1973b) Oxidative detoxication of thallium
in the yeast mitochondria. Antonie van Leeuwenhoek J Microbiol Serol,
39: 351-353.
Ling GN (1977) Thallium and cesium in muscle cells compete for the
adsorption sites normally occupied by K+. Physiol Chem Phys,
9: 217-225.
Linsdale JM (1931) Facts concerning the use of thallium in California
to poison rodents - Its destructiveness to game birds, song birds and
other valuable wild life. Condor, 33: 92-106.
Linton RW, Loh A, Natusch DFS, Evans CA Jr, & Williams P (1976)
Surface predominance of trace elements in airborne particles. Science,
191: 852-854.
LIS (Land Institute for Protection against Emissions) (1980)
[Environmental burden by thallium. Investigations in the neighbourhood
of the Dyckerhoff cement plant in Lengerich and other
thallium-emitting plants in the country NW.] Bonn, Bonn University
Press (in German).
Liu G (1986) [The protective effect of selenium on myocardial cells.]
Chung-hua Ping Li Hsueh Tsa Chih, 15: 252-254 (in Chinese).
Lockwood TH (1974) The analysis of asbestos for trace metals. Am Ind
Hyg Assoc J, 35: 245-251.
Logan PG, Lepp NW, & Phipps DA (1983) Thallium uptake by higher
plants. In: Proceedings of the 4th International Conference on Heavy
Metals in the Environment, Toronto, vol 1, pp 642-645.
Logan PG, Lepp NW, & Phipps DA (1984) Some aspects of thallium uptake
by higher plants. In: Proceedings of the 18th Trace Substances in
Environmental Health Conference, Columbia, 4-7 June 1984, pp 570-575.
Lohmann H, Csissaky M, & Wiegand H (1989) The action of thallium on
the excitability of CA1 pyramidal cells in hippocampal slices.
Neurotoxicol Teratol, 11: 545-549.
Luckey TD & Venugopal B (1977) Metal toxicity in mammals. Volume 1:
Physiologic and chemical basis for metal toxicity. New York, Plenum
Press.
Luckit J, Mir N, Hargreaves M, Costello C, & Gazzard B (1990)
Thrombocytopenia associated with thallium poisoning. Hum Exp Toxicol,
9: 47-48.
Ludolph A, Elger CE, Sennhenn R, & Bertram HP (1986) Chronic thallium
exposure in cement plant workers: clinical and electrophysiological
data. Trace Elem Med, 3: 121-125.
Ludwig E (1961) [Pathognomonic hair findings in thallium poisoning and
their significance.] Hautarzt, 12: 456-459 (in German).
Lund A (1956a) Distribution of thallium in the organism and its
elimination. Acta Pharmacol Toxicol, 12: 251-259.
Lund A (1956b) The effect of various substances on the excretion and
the toxicity of thallium in the rat. Acta Pharmacol Toxicol,
12: 260-268.
Lynch GR, Lond MB, & Scovell JMS (1930) The toxicology of thallium.
Lancet, 219(5599): 1340-1344.
Machtey I & Bandmann M (1961) The cardiovascular system in thallium
poisoning. Proc Staff Meet Beilinson Hosp, 10(suppl): 140-142.
McCool MM (1933) Effect of thallium sulphate on the growth of several
plants and on nitrification in soils. Contrib Boyce Thompson Inst,
5: 289-296.
McDonald JM (1941) Metallic poisons. Ind Med, 10: 447-452.
McKillop A & Taylor EC (1973) Thallium in organic synthesis. Chem Br,
9: 4-11.
McLaren JW, Beauchemin D, & Berman SS (1987) Application of isotope
dilution inductively coupled plasma mass spectrometry to the analysis
of marine sediments. Anal Chem, 59: 610-613.
Mahoney W (1933) Further notes on retrobulbar neuritis due to thallium
poisoning. Yale J Biol Med, 6: 583-597.
Maier EA, Dietemann-Molard A, Rastegar F, Heimburger R, Ruch C, Maier
A, Roegel E, & Leroy MJF (1986) Simultaneous determination of trace
elements in lavage fluids from human bronchial alveoli by
energy-dispersive x-ray fluorescence. 2. Determination of abnormal
lavage contents and verification of the results. Clin Chem,
32: 664-668.
Maier-Reiter W, Blind G, Klump A, Wystrcil HG, & Arndt U (1987)
[Ecotoxicology of thallium. Effect on the pigment system of plants.]
Agrar-Umweltforsch Baden-Württemberg, 16: 22-36 (in German).
Makridis H & Amberger A (1989a) [Uptake of thallium from cement
factory dust in pot trials with green rape, bush beans and rye grass.]
Landwirtsch Forsch, 42: 324-332 (in German).
Makridis H & Amberger A (1989b) [Effect of increasing levels of
thallium on yield and uptake of thallium and further mineral nutrients
by bush beans and green rape grown in nutrient solution]. Landwirtsch
Forsch, 42: 333-343 (in German).
Malcolm D (1979) Toxicology of metals other than lead. In: Gardner AW
ed. Current approaches to occupational medicine. Bristol, John Wright
and Sons Ltd., pp 18-43.
Manning DC & Slavin W (1988) The determination of thallium with the
stabilized temperature platform furnace and Zeeman background
correction. Spectrochim Acta, 43B: 1157-1165.
Manzo L & Sabbioni E (1988) Thallium toxicity and the nervous system.
In: Bondy SC & Prasad KN ed. Metal neurotoxicity. Boca Raton, Florida,
CRC Press, pp 35-54.
Manzo L, Scelsi R, Moglia A, Poggi P, Alfonsi E, Pietra R, Mousty F, &
Sabbioni E (1983) Long-term toxicity of thallium in the rat. In: Brown
SS & Savory J ed. Chemical toxicology and clinical chemistry of
metals. Proceedings of 2nd International Conference, Montreal, Canada.
New York, London, Academic Press, pp 401-405.
Marcus RL (1985) Investigation of a working population exposed to
thallium. J Soc Occup Med, 35: 4-9.
Marwaha J, Freedman R, & Hoffer B (1980) Electrophysiological changes
at a central noradrenergic synapse during thallium toxicosis. Toxicol
Appl Pharmacol, 56: 345-352.
Mason B (1966) Principles of geochemistry. New York, Chichester,
Brisbane, Toronto, John Wiley and Sons.
Masri MS (1976) Binding of metallic ions to keratins. Proceedings of
the 5th International Wool Textile Research Conference,
vol 3, pp 1-12.
Matera MG, Marrazzo R, Berrino L, Fillippelli W, Russo S, Rossi F,
Cilento M, & Marmo E (1986) [Vascular effects of perinatal exposure of
rats to thallium.] Rend Atti Accad Sci Med Chir, 140: 177-202
(in Italian).
Mathis BJ & Kevern NR (1973) Distribution of mercury, cadmium, lead
and thallium in a eutrophic lake. Washington, DC, US Department of
Commerce, National Technical Information Service (NTIS PB-221993).
Mathis BJ & Kevern NR (1975) Distribution of mercury, cadmium, lead
and thallium in a eutrophic lake. Hydrobiologia, 46: 207-222.
Mathys W (1981) Thallium in river sediments of North-West Germany: its
sources of input and its relation to other heavy metals. In:
Proceedings of the International Conference on Heavy Metals in the
Environment, Amsterdam, September 1981. Luxembourg, Commission of the
European communities WHO, pp 347-350.
Matthews AD & Riley JP (1969) The determination of thallium in
silicate rocks, marine sediments and sea water. Anal Chim Acta,
48: 25-34
Melnick RL, Monti LG, & Motzkin M (1976) Uncoupling of mitochondrial
oxidative phosphorylation by thallium. Biochem Biophys Res Commun,
69: 68-73.
Merguet P, Schümann HJ, Murata T, Rausch-Stroomann JG, Schröder E,
Parr D, & Bock KD (1969) [Investigations of the pathogenesis of
hypertension and sinus tachycardia in human thallium poisonings.]
Dtsch Arch Klin Med, 216: 1-20 (in German).
Metter D & Vock R (1984) [Investigations of the structure of hairs in
thallium poisoning.] Z Rechtsmed, 91: 201-214 (in German).
Meyer H & Tal M (1957) Studies on thallium poisoning. I. The
detoxifying action of some compounds containing sulphur and labile
methyl groups. J Sci Food Agric, 8: 516-519.
Micke H, Bertram HP, & Kemper FH (1983) [Thallium.] In: [Ullmann's
encyclopedia of technical chemistry], 4th ed. Weinheim, Verlag Chemie,
vol 23, pp 103-114 (in German).
Minoia C, Sabbioni E, Appostoli P, Pietra R, Pozzoli L, Gallorini M,
Nicolaou G, Alessio L, & Capodaglio E (1990) Trace element reference
values in tissues from inhabitants of the European Community I. A
study of 46 elements in urine, blood and serum of Italian subjects.
Sci Total Environ, 95: 89-105.
MISA (Municipal/Industrial Strategy for Abatement) (1988/89)
Preliminary report for the first six months of monitoring in the
petroleum refining sector (December 1, 1988 to May 31, 1989). Ontario,
Canada, Ministry of the Environment.
MISA (Municipal/Industrial Strategy for Abatement) (1991a) Status
report on the effluent monitoring data for the iron and steel sector
for the period from November 1, 1989 to October 31, 1990. Ontario,
Canada, Ministry of the Environment.
MISA (Municipal/Industrial Strategy for Abatement) (1991b) Preliminary
report on the first six months of process effluent monitoring in the
MISA pulp and paper sector (January 1, 1990 to June 30, 1990).
Ontario, Canada, Ministry of the Environment.
Moeschlin S (1965) [Poisoning: diagnosis and treatment.] Stuttgart,
Georg Thieme (in German).
Moeschlin S (1986) [Poisoning: diagnosis and treatment.] Stuttgart,
Georg Thieme (in German).
Möllhoff G, Schmidt G, & Bösche J (1979) [Thallium poisoning.
Neuromedical aspects.] Arch Kriminol, 163: 1-13 (in German).
Montoya Cabrera MA, Pérez Lucio C, Badillo Torre FF, Barquet Barquet
RM, & López Bárcenas F (1979) [Thallium intoxication: treatment with
D-penicillamine.] Rev. Med Inst Mex Seguro Soc, 18: 211-214
(in Spanish).
Mottet NK (1985) Morphogenic injury by chemical agents. In: Mottet NK
ed. Environmental pathology. New York, Oxford University Press,
pp 82-125.
Mourelle M, Favari L, & Amezcua JL (1988) Protection against thallium
hepatotoxicity by silymarin. J Appl Toxicol, 8: 351-354.
Muller L (1961) [Thallium poisoning.] In: Baader EW ed. [Handbook of
total occupational medicine.] Berlin, Urban & Schwarzenberg, vol II,
part 1, pp 244-250.
Müller-Brand J, Staub JJ, & Friedrich R (1984) [Thallium scintigraphy
of the thyroid as an alternative procedure for the demonstration of
functionally silent thyroid tissue.] Schweiz Med Wochenschr,
114: 1686-1689 (in German).
Mullins LJ & Moore RD (1960) The movement of thallium ions in muscle.
J Gen Physiol, 43: 759-773.
Munch JC (1934a) Antidotes. III. Thallium. J Am Pharm Assoc,
23: 91-94.
Munch JC (1934b) Human thallotoxicosis. J Am Med Assoc,
102: 1929-1934.
Munch JC, Ginsburg HM, & Nixon CE (1933) The 1932 thallotoxicosis
outbreak in California. J Am Med Assoc, 100: 1315-1319.
Munch B, Clausen B, & Karlog O (1974) Thallium poisoning in red foxes
(Vulpes vulpes) and badgers (Meles meles) in Denmark. Nord
Veterinärmed, 26: 323-338.
Muramatsu Y & Parr RM (1985) Survey of currently available reference
materials for use in connection with the determination of trace
elements in biological and environmental materials. Vienna,
International Atomic Energy Agency (IAEA/RL/128).
Nachman M & Hartley PL (1975) Role of illness in producing learned
taste aversions in rats. A comparison of several rodenticides. J Comp
Physiol Psychol, 89: 1010-1018.
Naganuma A, Tanaka T, Maeda K, Matsuda R, Tabata-Hanyu J, & Imura N
(1983) The interaction of selenium with various metals in vitro and
in vivo. Toxicology, 29: 77-86.
Natusch DFS (1982) Size distributions and concentrations of trace
elements in particulate emissions from industrial sources. VDI-Ber,
429: 253-260.
Natusch DFS & Wallace JR (1974) Urban aerosol toxicity: the influence
of particle size. Science, 186: 695-699.
Natusch DFS, Wallace JR, & Evans CA Jr (1974) Toxic trace elements:
preferential concentration in respirable particles. Science,
183: 202-204.
Negherbon WO (1959) Handbook of toxicology. Volume 3: Insecticides.
Philadelphia, Pennsylvania, W.B. Saunders Company, pp 750-751.
Nehring D (1962/63) [Investigations of the toxicological effects of
thallium ions on fish and animal food of fish.] Z Fischerei,
11: 557-561 (in German).
Nessler F (1985a) [Experimental studies made on animals to establish
the toxicity of thallium and its compounds.] Wiss Z Humboldt-Univ
(Berlin) Math-Naturwiss Reihe, 34: 761-766 (in German).
Nessler F (1985b) [Importance and history of occupational thallium
intoxications as a type of chronic thallium poisoning.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 773-777 (in German).
Nessler F & Briese C (1985) [Acute and subacute thallium intoxications
of humans.] Wiss Z Humboldt-Univ (Berlin) Math-Naturwiss Reihe,
34: 767-772 (in German).
Neubert D & Bluth U (1985) Effect of thallium on limb development in
organ culture. Teratology, 32: 29A.
NIOSH (1984) NIOSH manual of analytical methods: Method 7300.1-7300.5.
Cincinnati, Ohio, National Institute for Occupational Safety and
Health.
NIOSH (1985/86) RTECS - Registry of toxic effects of chemical
substances. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, p 4637.
NIOSH (1992) NIOSH recommendations for occupational safety and health:
Compendium of policy documents and statements. Cincinnati, Ohio,
National Institute for Occupational Safety and Health (Publication
No. 92-100).
Nisticò G, Faraone V, Naccari F, Ammendola D, Germanò D, & Marmo E
(1984) Lack of effects of thallium on GABAergic mechanisms in discrete
areas of the rat brain. Res Commun Chem Pathol Pharmacol, 46: 35-42.
Noddack I & Noddack W (1939) [The frequency of heavy metals in sea
organisms.] Ark Zool, A32(4): 1-35 (in German).
Nogami H & Terashima Y (1973) Thallium-induced achondroplasia in the
rat. Teratology, 8: 101-102.
Nolan C, Duke E, Lorenzon G, Sabbioni E, & Marafante E (1984) Heavy
metal uptake and intracellular binding in isolated gill preparations
of Mytilus galloprovincialis L. Sci Total Environ, 40: 83-92.
Norris P, Man WK, & Hughes MN (1976) Toxicity and accumulation of
thallium in bacteria and yeast. Arch Microbiol, 110: 279-286.
Nriagu JO & Pacyna JM (1988) Quantitative assessment of worldwide
contamination of air, water and soils by trace metals. Nature (Lond),
333: 134-139.
Oehme FW (1972) Mechanisms of heavy metal toxicities. Clin Toxicol,
5: 151-167.
Oehme FW (1978) Mechanisms of heavy metal inorganic toxicities. In:
Oehme FW ed. Toxicity of heavy metals in the environment. New York,
Marcel Dekker Inc., vol 1, pp 69-85.
Ohnesorge FK (1985) [Toxicological rating of arsenic, lead, cadmium,
nickel, thallium and zinc.] Düsseldorf, VDI-Association of German
Engineers Press (Progress Reports Series No. 15, Environmental
Technology No. 38) (in German).
Olsen I & Jonsen J (1982) Whole-body autoradiography of 204Tl in
embryos, fetuses and placentas of mice. Toxicology, 23: 353-358.
Ormerod MJ (1928) Pharmacological and toxicological aspects of
thallium. Can Med Assoc J, 19: 663-665.
Osorio-Rico L, Galván-Arzate S, & Ríos C (1994) Brain regional
monoaminergic activity after acute thallium administration. Abstracts
of the 33rd Annual Meeting of the Society of Toxicology. Toxicologist,
14: 267.
Ostádalová I & Babicky A (1987) Protective effect of thallium against
the formation of selenium cataract in rats. Biol Trace Elem Res,
14: 101-103.
Overnell J (1975) The effect of some heavy metal ions on
photosynthesis in a freshwater alga. Pestic Biochem Physiol, 5: 19-26.
Pai V (1987) Acute thallium poisoning. Prussian blue therapy in 9
cases. West Indian Med J, 36: 256-258.
Palermo D, Chiaravalle AE, D'Errico M, Milillo MA, & De Natale G
(1983) [An occurrence of thallium poisoning in fish.] Clin Vet,
106: 129-131 (in Italian).
Pallaghy CK (1972) Localization of thallium in stomata is independent
of transpiration. Aust J Biol Sci, 25: 415-417.
Papp JP, Gay PC, Dodson VN, & Pollard HM (1969) Potassium chloride
treatment in thallotoxicosis. Ann Intern Med, 71: 119-123.
Paschal DC & Bailey GG (1986) Determination of thallium in urine with
Zeeman effect graphite furnace atomic absorption. J Anal Toxicol,
10: 252-254.
Paschal DC, DiPietro ES, Phillips DL, & Gunter EW (1989) Age
dependence of metals in hair in a selected U.S. population. Environ
Res, 48: 17-28.
Paulson G, Vergara G, Young J, & Bird M (1972) Thallium intoxication
treated with dithizone and hemodialysis. Arch Intern Med,
129: 100-103.
Peele DB, MacPhail RC, & Farmer JD (1986) Flavor aversions induced by
thallium sulfate: importance of route of administration. Neurobehav
Toxicol Teratol, 8: 273-277.
Peele DB, Farmer JD, & MacPhail RC (1987) Conditioned flavor
aversions: applications in assessing the efficacy of chelators in the
treatment of heavy-metal intoxication. Toxicol Appl Pharmacol,
88: 397-410.
Peisach M, Lackay LG, & Gihwala D (1989) Measurement of proton-induced
prompt low energy photons by high resolution spectrometry. Analyst,
114: 279-286.
Petersohn KL (1960) [Thallium poisonings in pregnancy.] Arch Toxikol,
18: 160-164 (in German).
Peterson ER & Murray MR (1965) Patterns of peripheral demyelination
in vitro. Ann NY Acad Sci, 122: 39-50.
Piazolo P, Franz HE, Brech W, Walb D, & Wilk G (1971) [Treatment of
thallium poisoning by hemodialysis.] Dtsch Med Wochenschr,
96: 1217-1222 (in German).
Pielow E (1979) [Pollution by thallium in the environment of a cement
plant.] Umwelt, 5: 394-396 (in German).
Pieper B & Austenfeld F-A (1985) [Phytotoxicity of thallium (Tl) in
culture solution. Part 2: Effects of Tl(III) on the growth and heavy
metal contents of pea and field bean plants.] Z Pflanzenernähr
Bodenkd, 148: 83-91 (in German).
Potes-Gutierrez J & Del Real E (1966) Acute thallium intoxication. Ind
Med Surg, 35: 618-619.
Pötsch K & Austenfeld F-A (1985) [Phytotoxicity of thallium (Tl) in
culture solution. Part 1: Effects of Tl(I) on the growth and heavy
metal contents of pea and field bean plants.] Z Pflanzenernähr
Bodenkd, 148: 73-82 (in German).
Prick JJG (1979) Thallium poisoning. In: Vinken PJ & Bruyn GW ed.
Handbook of clinical neurology: Intoxications of the nervous system.
Amsterdam, North-Holland Publishers, vol 36, part 1, pp 239-278.
Prinz B, Krause GHM, & Stratmann H (1979) [Damages by thallium in the
neighbourhood of the Dyckerhoff cement plant in Lengerich, Westfalen.]
Staub-Reinhalt Luft, 39: 457-462 (in German).
Puddu A, Pettine M, Bacciu F, La Noce T, & Pagnotta R (1985) Thallium
and chromium toxicity in phytoplankton cultures. Proceedings of the
5th International Conference on Heavy Metals in the Environment,
Toronto. vol 2, pp 304-306.
Puddu A, Pettine M, La Noce T, Pagnotta R, & Bacciu F (1988) Factors
affecting thallium and chromium toxicity to marine algae. Sci Total
Environ, 71: 572.
Puerner NJ & Siegel SM (1972) The effects of mercury compounds on the
growth and orientation of cucumber seedlings. Physiol Plant,
26: 310-312.
Que Hee SS & Boyle JR (1988) Simultaneous multielemental analysis of
some environmental and biological samples by inductively coupled
plasma atomic emission spectrometry. Anal Chem, 60: 1033-1042.
Rao DV, Govelitz GF, & Sastry KSR (1983) Radiotoxicity of thallium-201
in mouse testes: inadequacy of conventional dosimetry. J Nucl Med,
24: 145-153.
Rauws AG & Canton JH (1976) Adsorption of thallium ions by Prussian
Blue. Bull Environ Contam Toxicol, 15: 335.
Reyners H, Gianfelici de Reyners E, Maisin JR, Winneke G, & Csicsaky M
(1981) Effects of different heavy metals (Cd, Tl, Zn and Pb) in the
central nervous system: a morphological assay. In: Proceedings of the
International Conference on Heavy Metals in the Environment,
Amsterdam, September 1981. Luxembourg, Commission of the European
communities WHO, pp 495-497.
Richards OW (1932) The stimulation of yeast growth by thallium, a
"bios" impurity of asparagine. J Biol Chem, 96: 405-418.
Richelmi P, Bono F, Guardia L, Ferrini B, & Manzo L (1980) Salivary
levels of thallium in acute human poisoning. Arch Toxicol,
43: 321-325.
Ridgway LP & Karnofsky DA (1952) The effects of metals on the chick
embryo: toxicity and production of abnormalities in development. Ann
NY Acad Sci, 55: 203-215.
Ridley WP, Dizikes LJ, & Wood JM (1977) Biomethylation of toxic
elements in the environment. Science, 197: 329-332.
Ríos C & Monroy-Noyola A (1992) D-penicillamine and Prussian blue as
antidotes against thallium intoxication in rats. Toxicology,
74: 69-76.
Ríos C, Galván-Arzate S, & Tapia R (1989) Brain regional thallium
distribution in rats acutely intoxicated with Tl2SO4. Arch
Toxicol, 63: 34-37.
Ríos C, Kravsov J, Altagracia M, Lopez-Naranjo F, & Monroy A (1991)
Efficacy of Prussian blue against thallium poisoning: effect of
particle size. Proc West Pharmacol, 34: 61-63.
Romero Romero E, Cherem JH, & Lifshitz A (1989) [Arterial hypertension
in acute thallitoxicosis.] Rev Med Inst Mex Seguro Soc, 27: 43-47
(in Spanish).
Rosenstock L & Cullen MR (1986) 9. Neurologic disease. In: Rosenstock
L & Cullen MR ed. Clinical occupational medicine. Philadelphia,
Pennsylvania, W.B. Saunders Company, pp 118-134.
Rossi F, Lampa E, Grella A, Vacca C, Giordano L, Perna D, Rosatti F,
Spaziante G, Giasi M, Imperatore A, Giusti I, & Marmo E (1981)
[Experimental study of the cardiovascular and respiratory effects of
thallium in various animal species.] Rend Atti Accad Sci Med Chir,
134: 49-84 (in Italian).
Rossi F, Marrazzo R, Berrino L, De Santis D, Lisa M, Susanna V,
Montanaro C, Fici F, & Marmo E (1988) Prenatal and postnatal thallium
exposure in rats: effect on development of vasomotor reactivity in
pups. Teratog Carcinog Mutagen, 8: 13-23.
Sabbioni E & Manzo L (1980) Metabolism and toxicity of thallium. In:
Proceedings of the International Congress on Neurotoxicology,
pp 249-270 (Advances in Neurotoxicology).
Sabbioni E, Marafante E, Rade J, Di Nucci A, Gregotti C, & Manzo L
(1980a) Metabolic patterns of low and toxic doses of thallium in the
rat. In: Holmstedt B, Lauwerys R, Mercier M, & Robberfroid M ed.
Mechanisms of toxicity and hazard evaluation. Proceedings of the
Second International Congress on Toxicoloy, Brussels, 1980. Amsterdam,
Oxford, New York, Elsevier/North Holland, pp 559-564.
Sabbioni E, Goetz L, Marafante E, Gregotti C, & Manzo L (1980b)
Metabolic fate of different inorganic and organic species of thallium
in the rat. Sci Total Environ, 15: 123-135.
Sabbioni E, Gregotti C, Edel J, Marafante E, Di Nucci A, & Manzo L
(1982) Organ/tissue disposition of thallium in pregnant rats. Arch
Toxicol, 5(suppl): 225-230.
Sabbioni E, Di Nucci A, Edel J, Gregotti C, Marafante E, & Manzo L
(1984a) Intestinal absorption and excretion of thallium (201 Tl) in
the rat. Arch Toxicol, 7(suppl): 446-450.
Sabbioni E, Goetz L, & Bignoli G (1984b) Health and environmental
implication of trace metals released from coal fired power plants: an
assessment study of the situation in the European Community. Sci Total
Environ, 40: 141-154.
Saddique A & Peterson CD (1983) Thallium poisoning: a review. Vet Hum
Toxicol, 25: 16-22.
Sager M (1984) Separation and enrichment of traces of thallium.
Mikrochim Acta, 1984: 461-466.
Sager M (1986) [Trace element analysis of thallium.] In: Hulpke H,
Hartkamp H, & Tölg G ed. [Analytical chemistry for the practise.]
Stuttgart, Georg Thieme, pp 1-103 (in German).
Sager M & Tölg G (1984) [Trace element analysis of thallium.] In:
Fresenius W, Günzler H, Huber W, Lüderwald I, & Tölg G ed. [Analyst
pocket book 4.] Berlin, Springer Verlag, pp 443-466 (in German).
Sanderson L (1952) Thallium. Can Min J, 73: 72-73.
Sanotskii IV (1961) [Toxicity of thallium compounds (carbonate,
iodide, and bromide)]. Toksikol Novykh Prom Khim Veshchestv, 2: 94-104
(in Russian).
Schafer EW (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol Appl
Pharmacol, 21: 315-330.
Schäfer SG & Forth W (1980) Excretion of metals into the intestine; a
comparative study on rats. In: Proceedings of the Second International
Congress on Toxicoloy, Brussels, 6-11 July 1980. Toxicol Lett,
95(spec issue).
Schäfer SG & Forth W (1987) Thallium(I) secretion across the isolated
mucosa of rat descending colon. Arch Toxicol, 52: 413-420.
Schäfer SG, Nell G, & Henning CH (1981) Movement of thallium(I) ions
in vitro. Arch Toxicol, 48: 271-279.
Schaller K-H, Manke G, Raithel HJ, Bühlmeyer G, Schmidt M, & Valentin
H (1980) Investigations of thallium-exposed workers in cement
factories. Int Arch Occup Environ Health, 47: 223-231.
Schardein JL & Keller KA (1989) Potential human developmental
toxicants and the role of animal testing in their identification and
characterization. Crit Rev Toxicol, 19: 251-339.
Scharrer K (1955) [Biochemistry of trace elements.] Berlin, Parey
(in German).
Schaub GA & Schnitker A (1988) Influence of Blastocrithidia triatomae
(Trypanosomatidae) on the reduviid bug Triatoma infestans: alterations
in the Malpighian tubules. Parasitol Res, 75: 88-97.
Schaumburg HH & Berger A (1992) Alopecia and sensory polyneuropathy
from thallium in a chinese herbal medication. J Am Med Assoc,
268: 3430-3431.
Schmidbauer H & Klingler D (1979) [Chronical thallium poisoning.] Wien
Med Wochenschr, 129: 334-336 (in German).
Schoental R & Cavanagh JB (1977) Mechanisms involved in the
`dying-back' process - an hypothesis implicating coenzymes.
Neuropathol Appl Neurobiol, 3: 145-157.
Schoer J (1984) Thallium. In: Hutzinger O ed. The handbook of
environmental chemistry. Volume 3: Anthropogenic compounds, Part C.
Berlin, Springer Verlag, pp 143-214.
Schoer J & Nagel U (1980) [Thallium in plants and soils.
Investigations in the southern Rhein-Neckar-Kreis.]
Naturwissenschaften, 67: 261-262 (in German).
Scholl W & Metzger F (1982) [Determinations of the thallium burden of
useful plants on contaminated soils in the region of Lengerich.]
Landwirtsch Forsch, 35: 216-223 (in German).
Schwartz JG, Stuckey JH, Kunkel SP, Dowd DC, & Kagan-Mallet KS (1988)
Poisoning from thallium. Tex Med, 84: 46-48.
Schwartzman RM & Kirschbaum JO (1962) The cutaneous histopathology of
thallium poisoning. J Invest Dermatol, 39: 169-173.
Schweiger P & Hoffmann G (1983) [Thallium uptake of different plants
in culture solution.] Kali-Briefe, 16: 391-398 (in German).
Seeger R & Gross M (1981) [Thallium in higher fungi.] Z Lebensm.Unters
Forsch, 173: 9-15 (in German).
Segebade C & Schmitt BF (1987) Analysis of high-purity material - a
comparison of photon activation analysis with other instrumental
methods. J Radioanal Nucl Chem, 113: 61-76.
Sessions HK & Goren S (1947) Report of investigation of health hazards
in connection with the industrial handling of thallium. US Navy Med
Bull, 47: 545-550.
Shabalina LP & Spiridonova VS (1979) [Thallium as an industrial poison
(Review of literature).] J Hyg Epidemiol Microbiol Immunol,
23: 247-255 (in Russian).
Shabalina LP, Spiridonova VS, Pavlenko GI, & Fomenko VN (1980) [Skin
resorptive, local, and mutagenic effects of thallium carbonate.] Gig
Tr Prof Zabol, 4: 54-55 (in Russian).
Shacklette HT, Erdmann JA, Harms TF, & Papp CSE (1978) Trace elements
in plant foodstuffs. In: Oehme FW ed. Toxicity of heavy metals in the
environment. New York, Marcel Dekker Inc., vol 1, pp 25-68.
Sharma RP & Obersteiner EJ (1981) Metals and neurotoxic effects:
cytotoxicity of selected metallic compounds on chick ganglia cultures.
J Comp Pathol, 91: 235-244.
Sharma J, Sharma RL, Singh HB, & Satake M (1986) Hazards and analysis
of thallium. A review. Toxicol Environ Chem, 11: 93-116.
Shaw PA (1932) Studies on thallium poisoning in game birds. Calif Fish
Game, 18: 29-34.
Sherlock JC & Smart GA (1986) Thallium in foods and the diet. Food
Addit Contam, 3: 363-370. Siegel SM (1977) The cytotoxic response of
Nicotiana' protoplasts to metal ions: a survey of the chemical
elements. Water Air Soil Pollut, 8: 293-304.
Siegel BZ & Siegel SM (1976) Effect of potassium on thallium toxicity
in cucumber seedlings: further evidence for potassium-thallium ion
antagonism. Bioinorg Chem, 6: 341-345.
Sikka R, Sabherwal U, & Arora DR (1987) Susceptibility of Klebsiella
pneumoniae to heavy metal ions in vitro. Indian J Med Res,
86: 437-440.
Singh NP, Bodgen JD, & Joselow MM (1975) Distribution of thallium and
lead in children's blood. Arch Environ Health, 30: 557-558.
Skrovina B, Micek J, Hronská J, & Spissák L (1973) Experimental and
human achondroplasia. Teratology, 8: 237.
Skulskii IA & Lapin AV (1983) Effect of thallium ions on sodium and
potassium transport in frog skin. Tsitologiya, 25: 1284-1288.
Skulskii IA, Manninen V, & Järnefelt J (1973) Interaction of thallous
ions with the cation transport mechanism in erythrocytes. Biochim
Biophys Acta, 298: 702-709.
Skulskii IA, Manninen V, & Järnefelt J (1975) Thallium inhibition of
ouabain-sensitive sodium transport and of the (Na++K+)-ATPase in
human erythrocytes. Biochim Biophys Acta, 394: 569-576.
Skulskii IA, Savina MV, Glasunov VV, & Saris NEL (1978)
Electrophoretic transport of Tl+ in mitochondria. J Membr Biol,
44: 187-194.
Skulskii IA, Ivanova TI, & Savina MV (1984) The effect of monovalent
thallium salts on liver mitochondria of rats. Zh Evol Biokhim Fiziol,
20: 353-355 (in Russian).
Smith IC & Carson BL (1977) Trace metals in the environment: Volume 1
- Thallium. Ann Arbor, Michigan, Ann Arbor Science Publishers, Inc.
Smith S & Kwan MKH (1989) Use of aquatic macrophytes as a bioassay
method to assess relative toxicity, uptake kinetics and accumulated
forms of trace metals. Hydrobiologia, 188/189: 345-351.
Sommer B (1987) [Evaluation of the effects of cadmium, lead, thallium
and fluoride in plants for food and fodder - Environmental research
plan of the Federal Minister for Environment, Nature Conservation and
Reactor Safety, research report 106 07 049/02.] Berlin, Erich Schmidt
Verlag (Report No. UBA-FB 87-026/8/87) (in German).
Spencer EL (1937) Frenching of tobacco and thallium toxicity. Am J
Bot, 24: 16-24.
Spiridonova VS, Shabalina LP, & Kremenetskaya LE (1978) [Fibrogenic
activity of thallium halides.] Gig Tr Prof Zabol, 11: 54 (in Russian).
Srivastava TN, Bajpai KK, & Singh K (1973) Anti-microbial activities
of diaryl gallium, indium and thallium compounds. Indian J Agric Sci,
43: 88-93.
Staszyc J, Królikowska-Prasal I, Matysiak W, Korobowicz E, Chróst L, &
Kusmierczyk S (1986) Morphological evaluation of the lungs of rats
affected by soil dust. Med Pr, 37: 65-72 (in Polish). Stavinoha WB,
Emerson GA, & Nash JB (1959) The effects of some sulfur compounds on
thallotoxicosis in mice. Toxicol Appl Pharmacol, 1: 638-646.
Steuer W (1980) [Results of thallium examinations of the human
population in Baden-Württemberg.] Öff Gesundh-Wesen, 42: 477-479
(in German).
Stevens W, van Peteghem C, Heyndrickx A, & Barbier F (1974) Eleven
cases of thallium intoxication treated with Prussian blue. Int J Clin
Pharmacol, 10: 1-22.
Stokinger HE (1987) Thallium. In: Clayton GD & Clayton FE ed. Patty's
industrial hygiene and toxicology: Volume 2A: Toxicology, 3rd ed. New
York, Chichester, Brisbane, Toronto, John Wiley and Sons,
pp 1914-1931.
Struempler AW (1975) Trace element composition in atmospheric
particulates during 1973 and the summer of 1974 at Chadron, Neb.
Environ Sci Technol, 9: 1164-1168.
Sunderman FW (1967) Diethyldithiocarbamate therapy of thallotoxicosis.
Am J Med Sci, 253: 209-220.
Swain RE & Bateman WG (1909/10) The toxicity of thallium salts. J Biol
Chem, 7: 137-151.
Tackmann W & Lehmann HJ (1971) [Refractory period and impulse sequence
recorded from the tibial nerves of guinea pigs with acute thallium
polyneuropathy.] Z Neurol, 199: 105-115 (in German).
Talas A & Wellhöner HH (1983) Dose-dependency of Tl+ kinetics as
studied in rabbits. Arch Toxicol, 53: 9-16.
Tan E-L, Williams MW, Schenley RL, Perdue SW, Hayden TL, Turner JE, &
Hsie AW (1984) The toxicity of sixteen metallic compounds in Chinese
hamster ovary cells. Toxicol Appl Pharmacol, 74: 330-336.
Tandon SP & Mishra MM (1969) Effect of some rare-elements on
nitrification by Nitrobacter agilis (in liquid culture medium). Proc
Natl Acad Sci India Sect A, 39: 209-216.
Templeton DM, Paudyn A, & Baines AD (1989) Multielement analysis of
biological samples by inductively coupled plasma-mass spectrometry.
Biol Trace Elem Res, 22: 17-33.
Terytze K (1987) [Entry and spread of chlorinated hydrocarbons and
toxic heavy metals with reference to their quantification in the
limnetic ecosystem.] Wiss Z Humboldt-Univ (Berlin) Math-Naturwiss
Reihe, 36: 860-865 (in German).
Thompson C, Dent J, & Saxby P (1988) Effects of thallium poisoning on
intellectual function. Br J Psychiatry, 153: 396-399.
Thyresson N (1950) Experimental investigation on thallium poisoning.
Influence of thallium on tissue metabolism. Acta Dermato-Venereol,
30: 417-441.
Thyresson N (1951) Experimental investigation on thallium poisoning.
Acta Dermato-Venereol, 31: 3-27.
Tikhova TS (1964) [Industrial hygiene problems in the production
metallic thallium and its salts.] Gig i Sanit, 29: 23-28 (in Russian).
Tikhova TS (1967) [Thallium and its compounds.] In: Izrael'son ZI ed.
[New data on the toxicology of rare metals and their compounds.]
oedinenii. Moscow, Meditsina Publishing House, pp 24-34 (in Russian).
Tjälve H, Nilsson M, & Larsson B (1982) Thallium-201: autoradiography
in pigmented mice and melanin-binding in vitro. Acta Pharmacol
Toxicol, 51: 147-153.
Toots H & Parker RB (1977) Thallium in salt substitutes: a possible
health hazard. Env Res, 14: 327-328.
Török P & Schmahl W (1982) [The environmental toxic substance thallium
sulfate.] Dtsch Ärztebl, 79(39): 31-32 (in German).
Triebig G & Büttner J (1983) [Occupational relevant neurotoxins: I.
Metals and their compounds - A literature review of the years 1970 to
1982.] Zbl Bakt Hyg I. Abt Orig B, 177: 11-36 (in German).
Trotman-Dickenson AF ed. (1973) Comprehensive inorganic chemistry:
Thallium. Oxford, New York, Pergamon Press, pp 1119-1172.
Truhaut R (1960) The toxicology of thallium. A summary prepared by W.
B. Deichmann of Truhaut's monograph "Recherches sur la Toxicologie du
Thallium." Institut National de Sécurité pour la Prévention des
Accidents du Travail et des Maladies Professionelles, Paris, 1959.
J Occup Med, 2: 334-336.
Tully JG, Taylor-Robinson D, Rose DL, Furr PM, & Hawkins DA (1983)
Evaluation of culture media for the recovery of Mycoplasma hominis
from the human urogenital tract. Sex Transm Dis, 10(suppl): 256-260.
Tuovinen OH & Kelly DP (1974) Studies on the growth of Thiobacillus
ferrooxidans. IV. Influence of monovalent metal cations on ferrous
iron oxidation and uranium toxicity in growing cultures. Arch
Microbiol, 98: 167-174.
UB (Federal Office for the Environment) (1992) [Pollution of
allotments and kitchen gardens in former Federal territory.] In: [Data
on the environment 1992/93.] Berlin, Erich Schmidt Verlag (in German).
Ueberschär K-H, Matthes S, & Vogt H (1986) [Influence of addition of
thallium to food of broiler and laying hens.] In: Anke M, Baumann W,
Bräunlich H, Brückner C, & Groppel B ed. [Proceedings of the 5th
Symposium of Trace Elements, Jena, 14-17 July 1986.] Jena, University
of Jena, pp 1233-1240 (in German).
Ulrich F & Long CNH (1955) The effects of propylthiouracil and
thyrotropic hormone on the uptake of radioactive thallium by the rat
thyroid. Yale J Biol Med, 27: 371-378.
Urbain D, Reding P, Georges B, Thys O, & Ham HR (1986) The clinical
value of 201Tl per rectum scintigraphy in the work-up of patients with
alcoholic liver disease. Eur J Nucl Med, 12: 267-270.
US BM (US Bureau of Mines) (1985) Minerals yearbook 1985. Volume 1:
Metals and minerals. Washington, DC, Government Printing Office. US BM
(US Bureau of Mines) (1989) Mineral commodity summaries 1989.
Washington, DC, US Bureau of Mines/US Geological Survey.
US EPA (1978) Ambient water quality criteria: thallium. Washington,
DC, US Environmental Protection Agency (PB-292444).
US EPA (1980) Ambient water quality criteria for thallium. Washington,
DC, US Environmental Protection Agency (EPA-440/5-80-074).
US EPA (1986) Subchronic (90-day) toxicity of thallium (I) sulfate
(Cas No. 7446-18-6) in Sprague-Dawley rats. Interim Report.
Washington, DC, US Environmental Protection Agency, Office of Solid
Wastes.
Valerio F, Brescianini C, Mazzucotelli A, & Frache R (1988) Seasonal
variation of thallium, lead, and chromium concentrations in airborne
particulate matter collected in an urban area. Sci Total Environ,
71: 501-509.
Valerio F, Brescianini C, & Lastraioli S (1989) Airborne metals in
urban areas. Int J Environ Anal Chem, 35: 101-110.
Vandenbalck JL & Patriarche GJ (1987) Electrochemical
micro-determinations of thallium(I) and chromium(VI) ions using DPASV
and DP polargraphy. Sci Total Environ, 60: 97-104.
Venugopal B & Luckey TD (1978) Metal toxicity in mammals. Volume 2:
Chemical toxicity of metals and metalloids. New York, Plenum Press.
Vesterberg O, Alessio L, Brune D, Gerhardsson L, Herber R, Kazantzis
G, Nordberg G, & Sabbioni E (1993) International project for producing
reference values for concentrations of trace elements in human blood
and urine (TRACY). Scand J Work Environ Health, 19(suppl 1): 19-26.
Von Mühlendahl KE, Etzold R, & Krienke EG (1978) [Thallium poisonings
of children.] Dtsch Med Wochenschr, 103: 1116-1117 (in German).
Wachs B (1988) [Relevance for bodies of water of the most toxic heavy
metals.] In: Bavarian Land Institute for Water Research ed. [Hazardous
substances in waste water and surface water.] Munich, Oldenbourg
Verlag, pp 176-243 (Report No. 42) (in German).
Wainwright AP, Kox WJ, House IM, Henry JA, Heaton R, & Seed WA (1988)
Clinical features and therapy of acute thallium poisoning.
Q J Med, 258: 939-944.
Wallwork-Barber MK, Lyall K, & Ferenbaugh RW (1985) Thallium movement
in a simple aquatic ecosystem. J Environ Sci Health, 20: 689-700.
Ward JC (1930) Thallium poisoning in sheep. J Am Pharm Assoc,
19: 556-559.
Ward JC (1931) Thallium poisoning in migratory birds. J Am Pharm
Assoc, 20: 1272-1276.
Ward JC (1936) Thallium. XIII. Symptoms and systemic action on cattle.
J Am Pharm Assoc, 25: 687-690.
Waters CB, Hawkins EC, & Knapp DW (1992) Acute thallium toxicosis in a
dog. J Am Vet Med Assoc, 201(6): 883-885.
Wei Q (1987) [Studies on spermotoxicity of thallium carbonate in
drinking water and its effect on reproductive function of mice.]
Chung-hua Yu Fang Hsueh Tsa Chih, 21: 141-143 (in Chinese).
Weinig E & Schmidt G (1966) [Distribution of thallium in the organism
after lethal thallium poisonings.] Arch Toxicol, 21: 199-215
(in German).
Weinig E & Walz W (1971) [Distribution of thallium in kidney and liver
after lethal thallium poisoning.] Arch Toxicol, 27: 217-225
(in German).
Weinig E & Zink P (1967) [Quantitative mass-spectrometrical
determination of the normal thallium content in the human organism.]
Arch Toxicol, 22: 255-274 (in German).
Weisweiler W, Blome K, & Kaeding L (1985) [Quantitative determination.
Thallium and lead cycles in rotary kilns for cement production.]
Staub-Reinhalt Luft, 45: 461-466 (in German).
Welz B (1983) [Atomic absorption spectrometry.] Weinheim, Verlag
Chemie (in German).
Welz B, Schlemmer G, & Mudakavi JK (1988a) Investigation and
elimination of chloride interference on thallium in graphite furnace
atomic absorption spectrometry. Anal Chem, 60: 2567-2572.
Welz B, Schlemmer G, & Mudakavi JK (1988b) Palladium nitrate -
magnesium nitrate modifier for graphite furnace atomic absorption
spectrometry; part 1. Determination of arsenic, antimony, selenium and
thallium in airborne particulate matter. J Anal At Spectrom, 3: 93-97.
WHO (1986) UNEP/WHO Global Environmental Monitoring System/HEAL
Project-Guidelines for integrated air, water, food and biological
exposure monitoring. Geneva, World Health Organization
(Document PEP/86.6).
Wiegand H (1988) Neurotoxicology of heavy metals: synaptic
transmission as influenced by mono- and divalent metal cations. In:
Proceedings of the 1st European Meeting of Environmental Hygiene,
pp 98-102.
Wiegand H, Csicsaky M, Krämer U, Papadopoulos R, & Willeke E (1983)
Neuromuscular transmission in the rat phrenic nerve hemidiaphragm
preparation influenced by thallium acetate. Proceedings of the 4th
International Conference on Heavy Metals in the Environment.
vol 2, pp 896-899.
Wiegand H, Csicsaky M, & Krämer U (1984a) The action of thallium
acetate on neuromuscular transmission in the rat phrenic
nerve-diaphragm preparation. Arch Toxicol, 55: 55-58.
Wiegand H, Papadopoulos R, Csicsaky M, & Krämer U (1984b) The action
of thallium acetate on spontaneous transmitter release in the rat
neuromuscular junction. Arch Toxicol, 55: 253-257.
Wiegand H, Lohmann H, Chandra SV, & Gotzsch U (1986) The action of
thallium acetate on phasic transmitter release in the mouse
neuromuscular junction. Arch Toxicol, 58: 265-270.
Wiegand H, Uhlig S, Gotzsch U, & Lohmann H (1990) The action of
cobalt, cadmium and thallium on presynaptic currents in mouse motor
nerve endings. Neurotoxicol Teratol, 12: 313-318.
Wiemer J, Ziskoven R, & Achenbach C (1982) Tl+-Ions: comparison of
the effects on the slow inward current and contractility of
ventricular tissues. Z Naturforsch, 37c: 1015-1022.
Wiemeyer SN, Jurek RM, & Moore JF (1986) Environmental contaminants in
surrogates, foods, and feathers of California condors (Gymnogyps
californianus). Environ Monit Assess, 6: 91-111.
Williams MW, Hoeschele JD, Turner JE, Jacobson KB, Christie NT, Paton
CL, Smith LH, Witschi HR, & Lee EH (1982) Chemical softness and acute
metal toxicity in mice and Drosophila. Toxicol Appl Pharmacol,
63: 461-469.
Wilson IC & Dean ACR (1977) Thallium sensitivity and tolerance in
Klebsiella pneumoniae. Microbios Lett, 5: 85-91.
Wimmer M (1985) [Mutagenic, cancerogenic, and teratogenic effects of
chemical substances on man, with special reference to thallium.] Wiss
Z Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 789-791
(in German).
Windebank AJ (1986) Specific inhibition of myelination by lead
in vitro; comparison with arsenic, thallium, and mercury. Exp Neurol,
94: 203-212.
Witschi HP (1965) Desorption of some toxic heavy metals from human
erythrocytes in vitro. Acta Haematol, 34: 101-115.
Wood JM (1984) Alkylations of metals and the activity of metal-alkyls.
Toxicol Environ Chem, 7: 229-240.
Wood JM (1987) Biological processes involved in the cycling of
elements between soil or sediments and the aqueous environment.
Hydrobiologia, 149: 31-42.
Wood JM, Cheh A, Dizikes LJ, Ridley WP, Rakow S, & Lakowicz JR (1978)
Mechanisms for the biomethylation of metals and metalloids. Fed Proc,
37: 16-21.
Woods JS & Fowler BA (1986) Alteration of hepatocellular structure and
function by thallium chloride: ultrastructural, morphometric, and
biochemical studies. Toxicol Appl Pharmacol, 83: 218-229.
Wystrcil H-G, Martini M, Maier-Reiter W, & Arndt U (1987)
[Ecotoxicology of thallium. Effect on photosynthesis.]
Agrar-Umweltforsch Baden-Württemberg, 16: 37-66 (in German).
Yang Q & Smeyers-Verbeke J (1991) Effectiveness of palladium matrix
modification for the determination of thallium by graphite furnace
atomic absorption spectrometry. Clin Chim Acta, 204: 23-26.
Yokoyama K, Araki S, & Abe H (1990) Distribution of nerve conduction
velocities in acute thallium poisoning. Muscle Nerve, 13: 117-120.
Yopp JH, Schmid WE, & Holst RW (1974) Determination of maximum
permissible levels of selected chemicals that exert toxic effects on
plants of economic importance in Illinois. Carbondale, Illinois,
Southern Illinois University (Report No. 74-33) (NTIS PB-237 654).
Zasukhina GD, Vasilyeva IM, & Sdirkova NI (1980) [Approach to the
determination of a mutagenic potential of environmental pollutants as
illustrated by the detection of the mutagenic effect of thallium
carbonate.] Dokl Acad Nauk SSSR, 250: 766-768 (in Russian).
Zasukhina GD, Vasilyeva IM, Sdirkova NI, Krasovsky GN, Vasyukovich LY,
Kenesariev UI, & Butenko PG (1983) Mutagenic effect of thallium and
mercury salts on rodent cells with different repair activities.
Mutat Res, 124: 163-173.
Zeiske W & van Driessche W (1986) Impairment of Na+ transport across
frog skin by Tl+: effects on turnover, area density and saturation
kinetics of apical Na+ channels. Pflügers Arch, 407: 145-152.
Zhang D (1988) [Bioassay of mutagenicity of thallium carbonate.]
Huanjing Kexue, 9: 29-32 (in Chinese).
Zhou D-X & Liu D-N (1985) Chronic thallium poisoning in a rural area
of Guizhou province, China. J Environ Health, 48: 14-18.
Ziegler E & Ziegler B (1984) [Preliminary results on the heavy metal
content in hair and serum.] Fortschr Atomspektrom Spurenanal,
1: 337-346 (in German).
Ziskoven R, Achenbach C, Bahr U, & Schulten H-R (1980) Quantitative
investigations on the diaplacental transfer of thallium by field
desorption mass spectrometry. Z Naturforsch, 35c: 902-906.
Ziskoven R, Achenbach C, & Wiemer J (1982) Tl+-Ions: influence on
cardic contractility. Z Naturforsch, 37c: 995-1005.
Ziskoven R, Achenbach C, Schulten H-R, & Roll R (1983) Thallium
determinations in fetal tissues and maternal brain and kidney. Toxicol
Lett, 19: 225-231.
Zitko V (1975a) Chemistry, applications, toxicity, and pollution
potential of thallium. Department of the Environment, Fisheries and
Marine Service, Research and Development Directorate (Technical Report
No. 518).
Zitko V (1975b) Toxicity and pollution potential of thallium. Sci
Total Environ, 4: 185-192.
Zitko V & Carson WV (1975) Accumulation of thallium in clams and
mussels. Bull Environ Contam Toxicol, 14: 530-533.
Zitko V, Carson WV, & Carson WG (1975) Thallium: occurrence in the
environment and toxicity to fish. Bull Environ Contam Toxicol,
13: 23-30.
Zook BC & Gilmore CE (1967) Thallium poisoning in dogs. J Am Vet Med
Assoc, 151: 206-217.
Zook BC, Holzworth J, & Thornton GW (1968) Thallium poisoning in cats.
J Am Vet Med Assoc, 153: 285-299.
Zyka V (1972) Thallium in plants from Alsar. Sb Geol Ved Technol
Geochim, 10: 91-96.
RESUME
1. Identité, propriétés physiques et chimiques, et méthodes d'analyse
Le thallium élémentaire est un métal mou et malléable de couleur
blanc bleuâtre. Exposé à l'air humide ou à l'eau, le thallium est,
selon le cas, rapidement oxydé en surface ou transformé en hydroxyde.
Le thallium possède deux importants degrés d'oxydation, le thallium(I)
et le thallium(III). Les composés du thallium monovalent (thalleux)
se comportent comme des dérivés de métaux alcalins, par exemple du
potassium, alors que les composés du thallium trivalent (dérivés
thalliques) sont moins basiques et rappellent les sels d'aluminium.
Contrairement aux dérivés minéraux dans lesquels l'ion thallium(I) est
plus stable en solution aqueuse que l'ion thallium(III), ce dernier
est plus stable dans les dérivés organiques.
Le dosage du thallium dans des échantillons provenant de
l'environnement est assez difficile étant donné que la concentration
est de l'ordre du µg/kg tout au plus. En général, la limite de dosage
dans les minéraux, les sols et les poussières est d'environ 20 µg/kg -
approximativement 0,1 µg/litre pour les solutions aqueuses - et dans
les produits biologiques, de quelques µg/kg, lorsqu'on ne procède pas
à une concentration préalable du thallium.
La spectrométrie d'absorption atomique avec four à tube de
graphite (GF-AAS) est une méthode d'analyse qui convient bien aux
applications exigeant une sensibilité élevée avec des prises d'essai
réduites où la concentration en thallium est de l'ordre de quelques
µg/kg. Pour obtenir une bonne précision et une bonne exactitude à des
teneurs de l'ordre du µg/kg, on peut utiliser la spectrométrie de
masse avec dilution isotopique (IDMS) et la spectrométrie de masse en
plasma à couplage inductif (ICP-MS), avec également possibilité de
dilution isotopique.
2. Sources d'exposition humaine et environnementale
La présence de thallium dans l'environnement résulte à la fois de
processus naturels et de l'activité humaine. Ce métal est omniprésent
dans la nature et on le rencontre en particulier dans les minerais
sulfurés de divers métaux lourds, quoiqu'en principe à faibles
concentrations. Il n'y a guère d'endroits où la concentration en
thallium d'origine naturelle soit très élevée. Le thallium n'est
produit industriellement qu'en petites quantités (la consommation
industrielle mondiale a été de 10 à 15 tonnes en 1991). Le thallium
et ses dérivés ont des applications industrielles très diverses. Ils
entrent dans la composition de crèmes épilatoires, de rodenticides et
d'insecticides, mais ces utilisations sont désormais strictement
limitées. On utilise principalement le thallium et ses dérivés dans
les industries électriques et électroniques ainsi que pour la
fabrication de verres spéciaux. La scintigraphie en général et le
diagnostic du mélanome en particulier constituent des applications
importantes du radio-thallium en médecine et l'on a également recours
aux dérivés d'arylthallium(III) en biochimie.
Du thallium peut être libéré dans l'environnement par des
fonderies (dépôts de déchets ou émissions dans l'atmosphère), des
centrales thermiques à charbon, des briqueteries et des cimenteries
(dans ce cas, exclusivement des émissions dans l'atmosphère). On
estime que l'industrie mobilise ainsi chaque année dans le monde de
2000 à 5000 tonnes de thallium. Les émissions de thallium dues aux
différentes opérations industrielles varient largement d'un type
d'industrie à l'autre.
Les émissions des centrales thermiques à charbon peuvent avoir
une teneur en thallium de 700 µg/m3 d'air et celles des cimenteries
des teneurs allant jusqu'à 2500 µg/m3. Cette dernière valeur peut
être ramenée à < 25 µg/m3 en ayant recours à d'autres matières
premières et en changeant de procédé. Le thallium se volatilise lors
de la combustion du charbon et des matières premières utilisées pour
la production de ciment puis il se recondense à la surface des
particules de cendre dans les parties de l'installation où la
température est plus basse. Ces particules peuvent contenir jusqu'à
50 mg de thallium/kg de cendres volantes et sont souvent de petite
taille, de sorte que seulement 50% d'entre elles sont retenues par les
filtres. De même, environ un tiers des particules émises par les
centrales thermiques sont de faible granulométrie et peuvent par
conséquent se déposer dans les voies respiratoires inférieures.
Les effluents provenant de bassins d'évacuation de terrils et
contenant dans un cas jusqu'à 1620 et dans un autre jusqu'à
36 µg/litre de thallium ont provoqué, dans les cours d'eau adjacents,
l'apparition de concentrations de thallium respectivement égales à 88
et 1 µg/litre. Dans les collections d'eau de pluie situées aux
alentours d'une cimenterie, on a constaté la présence de
concentrations de thallium allant jusqu'à 37 µg/litre. On a trouvé
dans le sol aux alentours de terrils de mines, des concentrations
atteignant 60 mg/kg. Au voisinage de la base de fours à métaux, et
près de briqueteries et de cimenteries, on a trouvé des concentrations
respectivement égales à 2, 0,6 et 27 mg/kg de terre.
Dans les zones contaminées, la concentration de thallium dans la
plupart des légumes, des fruits et des viandes est inférieure à
1 mg/kg de poids frais. Les teneurs sont plus élevées dans les choux
(crucifères) avec des concentrations allant jusqu'à 45 mg/kg dans les
choux frisés. Il y a corrélation entre la concentration du thallium
dans les tissus des animaux d'élevage et la concentration de ce métal
dans le fourrage. Au voisinage de certaines cimenteries, on a signalé
des teneurs plus élevées en thallium dans le fourrage (par exemple
jusqu'à 1000 mg/kg dans le colza) ainsi que dans la viande de boeuf et
de lapin (jusqu'à 1,5 et 5,8 mg/kg, respectivement).
3. Transport, distribution et transformation dans l'environnement
A proximité de sources ponctuelles telles que les centrales
thermiques à charbon, certaines cimenteries et fonderies de métaux, la
principale source de thallium atmosphérique est constituée par les
cendres volantes. Une étude a montré que près de la totalité du
thallium présent dans les poussières en suspension d'une cimenterie
s'y trouvait sous la forme de chlorure de thallium(I) soluble.
La destinée du thallium qui parvient jusqu'au sol (par exemple
sous forme de dépôt de cendres volantes) dépend en grande partie de la
nature de ce sol. C'est en particulier les sols à forte teneur en
argile, en matières organiques et en oxydes de fer et manganèse qui
retiennent le plus de thallium. L'accroissement de la teneur en
thallium par incorporation dans des complexes stables ne se produit
que dans les couches supérieures. La fixation du thallium par les
végétaux est d'autant plus importante que le pH est plus bas. Dans
certains sols fortement acides, d'importantes quantités de thallium
peuvent parvenir par lessivage jusqu'aux eaux de surface et aux eaux
souterraines.
On peut s'attendre à ce que la majeure partie du thallium présent
à l'état dissous dans les eaux douces se trouve sous sa forme
monovalente. Toutefois, dans les eaux où la teneur en oxygène est
forte et dans la plupart des eaux de mer, il peut y avoir prédominance
du thallium(III). Le thallium peut disparaître de l'eau par diverses
réactions d'échange, de complexation ou de précipitation.
Le thallium peut également subir une bioconcentration, mais il
est peu probable que sa concentration s'accroisse le long de la chaîne
alimentaire aquatique ou terrestre.
4. Concentrations dans l'environnement et exposition humaine
Dans les secteurs qui ne sont pas contaminés, les concentrations
de thallium dans l'air sont généralement inférieures à 1 ng/m3; dans
l'eau elles sont inférieures à 1 µg/litre et dans les sédiments
aquatiques, inférieures à 1 mg/kg. Dans l'écorce terrestre, la
concentration moyenne varie de 0,1 à 1,7 mg/kg, mais on peut trouver
des concentrations beaucoup plus élevées, par exemple jusqu'à
1000 mg/kg dans le charbon et les rares minerais de thallium que l'on
rencontre contiennent jusqu'à 60% de cet élément. Les produits
alimentaires d'origine végétale ou animale en contiennent généralement
moins de 1 mg/kg de poids sec et l'apport d'origine alimentaire moyen
chez l'homme se révèle inférieur à 5 µg/jour. On estime que l'apport
de thallium par la voie respiratoire est inférieur à 0,005 µg de
thallium par jour.
On ne possède que des données limitées sur la concentration
effective de thallium dans l'air des lieux de travail. Les valeurs
les plus récemment observées (dans les années 80) étaient inférieures
à 22 µg de thallium par m3 (il s'agissait d'un four à thallium
installé dans un atelier produisant un alliage spécial à base de
thallium). Chez des ouvriers d'une cimenterie, on a trouvé des
concentrations urinaires moyennes allant de 0,3 à 8 µg/litre et chez
des ouvriers d'une fonderie, des valeurs de 0,3 à 10,5 µg/litre.
5. Cinétique et métabolisme chez les animaux de laboratoire
et l'homme
La thallium est rapidement et bien absorbé au niveau des voies
digestives et respiratoires et il pénètre également à travers la peau.
Il se répartit rapidement dans l'ensemble des organes et traverse la
barrière placentaire (comme le montre sa fixation rapide dans les
tissus foetaux) ainsi que la barrière hématoencéphalique. Comme il
s'accumule rapidement dans les cellules, la concentration du thallium
dans le sang total ne reflète pas sa concentration tissulaire. Dans
les cas d'intoxication aiguë chez l'homme et l'animal d'expérience on
constate, au début, de fortes concentrations de thallium au niveau du
rein et de faibles concentrations dans les tissus adipeux et le
cerveau, les concentrations dans les autres organes se situant entre
les deux; ultérieurement la concentration de thallium dans le cerveau
augmente également.
L'élimination du thallium peut s'effectuer par la voie digestive
(essentiellement par des mécanismes indépendants de l'excrétion
biliaire), par les reins, les cheveux, la peau, la sueur et le lait
maternel. Il peut y avoir réabsorption intestinale, principalement au
niveau du côlon avec pour conséquences une diminution de la clairance
totale. Chez le rat les principales voies d'élimination sont la voie
digestive (environ les deux tiers) et la voie rénale (environ un
tiers); chez le lapin, ces deux voies sont d'une importance
sensiblement égale. Le thallium peut également être excrété par la
salive.
Comme dans le cas de nombreuses autres substances, l'excrétion du
thallium n'est pas identique chez l'homme et chez l'animal de
laboratoire, la vitesse d'excrétion étant généralement beaucoup plus
faible chez l'homme (constante de vitesse = 0,023-0,069 jour-1) que
chez l'animal d'expérience (constante de vitesse moyenne = 0,18
jour-1). Une autre différence importante que l'on observe entre
l'homme et l'animal concerne l'importance relative des diverses voies
d'excrétion. Chez l'homme, il semble que l'excrétion rénale soit
beaucoup plus importante que chez l'animal, encore que sa contribution
à la clairance totale n'ait pas encore été définitivement établie,
principalement du fait de l'insuffisance des données. En outre, le
niveau d'exposition, sa durée, les insuffisances au niveau des organes
excréteurs, ainsi que le traitement anti-poison qui est administré,
sont autant de facteurs qui peuvent influer considérablement sur les
résultats.
Une étude a montré que la quantité totale de thallium excrétée
quotidiennement était de 73% environ par la voie rénale et seulement
de 3,7% environ par la voie digestive. On a estimé à 19,5% la
proportion excrétée dans le système pileux et par la voie percutanée
et à 3,7% la proportion excrétée dans la sueur.
Chez l'animal de laboratoire, la demi-vie biologique du thallium
oscille généralement entre trois et huit jours. Chez l'homme elle est
d'environ 10 jours, mais on a fait état de valeurs allant jusqu'à 30
jours.
On ne dispose d'aucune donnée sur la biotransformation du
thallium.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
La toxicité des sels de thallium(I) ne varie pas de manière
spectaculaire d'une espèce à l'autre. Généralement, l'ingestion d'une
dose de 20 à 60 mg de thallium par kg de poids corporel est mortelle
en l'espace d'une semaine. Les cobayes sont légèrement plus sensibles
que les autres animaux de laboratoire. L'oxyde de thallium(III),
insoluble dans l'eau, présente une toxicité aiguë par voie orale ou
parentérale un peu plus faible que les sels de thallium(I). la
comparaison des données de toxicité aiguë montre que le thallium
présente une biodisponibilité élevée par toutes les voies
d'exposition. La plupart des organes sont affectés mais on constate
quelques variations intra- et interspécifiques pour ce qui concerne
les signes d'intoxication et l'ordre dans lequel ils se manifestent.
Les symptômes d'une intoxication aiguë par le thallium se
manifestent en général dans l'ordre suivant: on constate tout d'abord
une anorexie, des vomissements et une dépression, puis apparaissent
une diarrhée et des manifestations cutanées (inflammation au niveau
des divers orifices, furoncles, chute de cheveux), après quoi
apparaissent une dyspnée et des troubles nerveux. Enfin la mort
survient par insuffisance respiratoire.
les symptômes d'une intoxication chronique sont analogues. La
chute des cheveux est de règle.
L'examen histologique révèle des lésions cellulaires et en
particulier une nécrose. Cette nécrose a été observée au niveau des
reins, du foie, de l'intestin, du myocarde et du système nerveux. On
a observé dans nombreuses cellules, un gonflement des mitochondries
avec disparition des crêtes, une dilatation du réticulum endoplasmique
agranulaire, une augmentation du nombre de vacuoles autophagiques et
de granules de lipofuscine, enfin, une disparition des
microvillosités. Les altérations fonctionnelles qu'entraîne le
thallium peuvent s'expliquer par la destruction physique de la
membrane des organites subcellulaires. Au niveau du coeur, les effets
arythmogènes se limitent au noeud sinusal.
L'intoxication par le thallium provoque des anomalies sélectives
au niveau de certains comportements, qui sont corrélées aux effets
biochimiques (ce qui indique des lésions cellulaires) observés dans
certaines régions de l'encéphale. Un certain nombre d'effets
neurologiques semblent être dus à l'action directe du thallium, par
exemple l'ataxie et les tremblements qui seraient dus à des lésions
cérébelleuses ou à des modifications de l'activité endocrine provoquée
par des lésions au niveau de l'hypothalamus. Le système nerveux
autonome, et principalement les fibres adrénergiques, peuvent être
activés par le thallium. Dans les nerfs périphériques, il semble que
l'action du thallium s'exerce au niveau présynaptique, avec libération
spontanée du neurotransmetteur, par antagonisme avec ce processus qui
dépend du calcium.
On ne sait toujours pas exactement par quel mécanisme s'exerce la
toxicité du thallium. On pense qu'il en existe plusieurs, liés les
uns aux autres. Un aspect important de l'intoxication par le thallium
consiste dans une augmentation sensible de la peroxydation des lipides
et de l'activité d'un enzyme lysosomique, la ß-galactosidase. Il en
résulte un déficit en glutathion qui entraîne l'accumulation de
lipides peroxydés dans l'encéphale et vraisemblablement la formation
de granules de lipofuscine. Il semble que le mode d'action du
thallium consiste principalement dans une perturbation de la fonction
des mitochondries.
Une intoxication chronique par le thallium entraîne chez l'animal
une réduction de l'activité sexuelle et les effets gonadotoxiques du
thallium sont manifestes chez le mâle au niveau des organes
reproducteurs. Chez des rats qui avaient reçu pendant 16 jours 10 mg
de thallium par litre d'eau de boisson, on a constaté qu'au niveau des
testicules, c'étaient les cellules de Sertoli qui étaient les plus
sensibles et la desquamation de l'épithélium spermatogène a entraîné
la présence de spermatozoïdes immatures dans le sperme. Ce phénomène
pourrait expliquer le moindre taux de survie des embryons et la durée
de vie réduite de la progéniture après intoxication à doses sublétales
du père.
Après injection de thallium dans des oeufs de poule on a constaté
chez les embryons, la présence d'effets tératogènes, une inhibition de
la croissance et des anomalies dans le développement osseux; toutefois
ces effets sont controversés chez les mammifères, même à des doses
toxiques pour la mère. On a mis en évidence le passage
transplacentaire du thallium mais les nombreuses souches de souris et
de rats n'ont pour ainsi dire pas présenté d'effets tératogènes.
Deux épreuves de mutagénicité microbiologique effectuées sur
Salmonella typhimurium ont donné des résultats négatifs et les
résultats de la recherche in vivo d'échanges entre chromatides
soeurs demeurent controversés. Toutefois, selon certaines études, on
aurait observé des aberrations chromosomiques et une augmentation
sensible des brèches dans l'ADN monocaténaire.
On manque d'études à long terme sur la cancérogénicité du
thallium.
7. Effets sur l'homme
Etant donné que les sels de thallium sont inodores, incolores et
sans saveur, leur forte toxicité et la facilité avec laquelle on
pouvait s'en procurer naguère - et encore maintenant dans certains
pays en développement - ont fait qu'on les a souvent utilisés à des
fins de suicide, d'homicide, de tentatives d'avortement illicites avec
pour résultat, dans ce cas particulier, des intoxications aiguës.
D'ailleurs on considère que les intoxications par la thallium sont une
des causes les plus fréquentes, à l'échelle mondiale, des
empoisonnements volontaires ou accidentels chez l'homme. Ce que l'on
sait des intoxications chroniques par le thallium se limite aux
intoxications professionnelles, aux groupes de population vivant dans
des zones contaminées et aux cas d'homicide par absorption de
nombreuses petites doses.
Les symptômes de l'intoxication aiguë par le thallium dépendent
de l'âge, de la voie d'administration et de la dose. Les doses qui se
sont révélées mortelles varient entre 6 et 40 mg/kg, les valeurs
moyennes se situant entre 10 et 15 mg/kg. Si un traitement n'est pas
institué, cette dose moyenne entraîne généralement la mort en l'espace
de 10 à 12 jours, mais on a également connaissance de cas où la mort
est survenue en l'espace de 8 à 10 heures.
La triade gastro-entérite, polynévrite et alopécie est considérée
comme le symptôme classique de l'intoxication par le thallium, mais il
est arrivé qu'on n'observe ni gastroentérite ni alopécie. Il y a
également d'autres symptômes qui varient dans leur séquence, leur
ampleur et leur intensité.
Les symptômes de l'intoxication par le thallium sont souvent
diffus et commencent par de l'anorexie, des nausées, des vomissements,
une saveur métallique, de la salivation, des douleurs rétro-sternales
et abdominales et quelquefois des hémorragies gastro-intestinales
(selles sanglantes). On observe ensuite fréquemment une constipation
qui peut être rebelle au traitement et gêner par conséquent l'action
de l'antidote. Au bout de 2 à 5 jours, les troubles caractéristiques
de l'intoxication par le thallium apparaissent peu à peu, quelle soit
la voie d'exposition. Les effets sur le système nerveux central et
périphérique sont variables mais ce que l'on constate de manière
caractéristique et systématique chez les intoxiqués par le thallium,
c'est une hypersensibilité des jambes, à laquelle font suite le
syndrome des "pieds brûlants" et une paresthésie. L'atteinte du
système nerveux central (SNC) se traduit par des symptômes tels
qu'hallucinations, léthargie, délire, convulsions et coma. Les
symptômes circulatoires couramment rencontrés sont une hypertension,
une tachycardie et dans les cas graves, une défaillance cardiaque. Au
bout de la deuxième semaine, les cheveux commencent à tomber et
quelquefois même les poils du corps. La dystrophie unguéale se
manifeste par l'apparition de lunules blanches (lignes de Mee) 3 à 4
semaines après l'intoxication. Les zones noires que l'on trouve dans
la papille pileuse ne sont pas dues à des dépôts de pigment ou de
thallium mais à la présence de petites quantités d'air qui pénètrent
dans la tige du poil.
Dans les cas mortels, la mort peut survenir quelques heures à
plusieurs semaines après l'intoxication, mais la plupart du temps le
décès intervient dans les 10 à 12 jours. La cause du décès est
principalement due à une insuffisance rénale, neurologique et
cardiaque.
En cas d'intoxication sublétale, la guérison peut souvent prendre
des mois. Des séquelles peuvent subsister: problèmes neurologiques et
troubles mentaux, anomalies électroencéphalographiques et cécité. En
outre, il semble que les fonctions intellectuelles des survivants
soient affectées.
En cas d'intoxication chronique, les symptômes sont analogues
mais généralement moins prononcés qu'en cas d'intoxication aiguë. La
cécité peut quelquefois être permanente. La guérison complète prend
des mois et il peut y avoir des rechutes.
A l'occasion d'un incident dans une cimenterie de Lengerich en
Allemagne où il y avait eu émission de thallium et que l'on a bien
étudiée, on a constaté que la concentration du thallium dans le
système pileux et les urines des personnes exposées n'était pas
corrélée avec certaines caractéristiques généralement associées à
l'intoxication chronique, mais seulement avec des symptômes
neurologiques subjectifs.
L'examen de biopsies et de pièces d'autopsie après intoxication
par le thallium révèle des lésions au niveau de divers organes. Par
exemple, après l'ingestion de doses mortelles, on constate des
hémorragies de la muqueuse intestinale, des poumons, des glandes
endocrines et du coeur, des infiltrations graisseuses au niveau du
foie et du myocarde, ainsi que des altérations dégénératives des
glomérules et des tubules rénaux. Dans l'encéphale, on peut observer
une dégénérescence graisseuse des cellules glanglionnaires, des
lésions au niveau des axones et la désagrégation des gaines de
myéline.
L'action directe du thallium sur le système nerveux autonome peut
produire des variations dans la tension artérielle. L'intoxication
par le thallium provoque une névrite symétrique périphérique. Les
nerfs distaux sont davantage touchés que les nerfs proximaux et les
lésions, bien que plus précoces, sont moins prononcées dans le cas des
nerfs dont l'axone est court, par exemple les nerfs crâniens. Les
axones sont enflés et ils présentent des vacuoles et des mitochondries
distendues. Dans les cas d'intoxication mortelle, on a observé de
graves lésions du nerf vague, l'énervation du sinus carotidien et des
lésions au niveau des ganglions sympathiques. Dans les cas
d'intoxication sublétale, les nerfs atteints peuvent subir une
dégénérescence de l'axone qui peut être définitive ou s'améliorer
partiellement dans les 2 ans.
Une névrite optique rétrobulbaire entraînant des troubles visuels
peut s'installer et persister pendant des mois après un traitement par
une crème dépilatoire à base de thallium; il peut même y avoir une
atrophie du nerf optique.
On n'a guère de données concernant les effets du thallium sur la
reproduction humaine. Il peut y avoir des effets indésirables sur le
cycle menstruel, la libido et la puissance sexuelle masculine. On
sait en outre qu'une intoxication chronique peut produire des effets
sur les spermatozoïdes. Comme chez l'animal, il y a passage
transplacentaire; on l'a observé dans un cas d'avortement provoqué par
le thallium. Toutefois, on connaît environ 20 cas d'intoxication par
le thallium en cours de grossesse où, à part le fait que les
nouveaux-nés avaient un poids de naissance relativement faible et
présentaient une alopécie, leur développement foetal ne paraissait pas
avoir souffert.
On ne sait rien des effets cancérogènes et on ne possède aucune
donnée sur les effets immunologiques du thallium. On ne possède pas
non plus d'éléments suffisants en faveur d'effets génotoxiques
éventuels.
Le traitement des intoxications par le thallium associe la
diurèse provoquée, l'administration de charbon actif et la prévention
de la réabsorption par le côlon au moyen d'administration de bleu de
Prusse (hexacyanoferrate(II) de potassium et de fer(II)).
8. Relation dose-réponse chez l'homme
Dans les populations non exposées, la concentration urinaire
moyenne du thallium est de 0,3 à 0,4 µg/litre. Etant donné que le
thallium a une courte demi-vie biologique, exprimée en jours, si l'on
suppose réunies les conditions d'un état stationnaire, la
concentration urinaire peut être considérée comme un indicateur de la
dose totale après absorption de thallium par inhalation ou ingestion.
On a constaté, dans un échantillon d'une population vivant à
proximité d'une source d'émission de thallium dans l'atmosphère, une
concentration urinaire moyenne de 5,2 µg/litre. L'existence d'une
relation dose-réponse nette a été observée entre la concentration
urinaire de thallium et la prévalence des effets suivants: fatigue,
faiblesse, troubles du sommeil, migraines, nervosité, paresthésie,
douleurs musculaires et articulaires. On a également fait état d'une
relation dose-réponse analogue en utilisant la teneur des cheveux en
thallium comme indicateur de l'exposition.
Le Groupe de travail a estimé qu'une exposition entraînant une
concentration urinaire de thallium inférieure à 5 µg/litre n'est
probablement pas susceptible de causer des effets nocifs. Dans
l'intervalle 5-500 µg/litre, l'ampleur du risque et la gravité des
effets nocifs restent incertaines, mais une exposition entraînant une
concentration urinaire supérieure à 500 µg/litre correspond à une
intoxication avec des manifestations cliniques.
9. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Le thallium est nocif pour tous les organismes mais il existe des
différences évidentes entre les espèces, voire entre les souches d'une
même espèce. La toxicité peut être différente selon qu'il s'agit de
dérivés minéraux du thallium(I) ou du thallium(III) ou encore de
composés organothalliens.
L'effet le plus important du thallium sur les microorganismes
semble consister dans l'inhibition de la nitrification par les
bactéries terricoles. Les résultats d'une étude consacrée à ce
phénomène tendent à indiquer que la structure de la communauté
biologique est perturbée lorsque la concentration de thallium dans le
sol se situe dans les limites de 1 à 10 mg/kg de poids sec; toutefois
on n'a pas pu déterminer sous quelle forme le thallium avait été
utilisé dans cette expérience.
Le thallium est fixé par toutes les parties de la plante, mais
principalement par les racines. Une fois qu'il a pénétré dans la
cellule, il se concentre de manière inégale dans le cytosol,
probablement en étant lié à un peptide. La concentration de thallium
que l'on rencontre dans les végétaux dépend des propriétés du sol
(plus spécialement du pH et de la teneur en argile et en matières
organiques), ainsi que du stade de développement et de la partie du
végétal. Il s'accumule dans les zones contenant de la chlorophylle
mais il existe des végétaux résistants au thallium où cette
accumulation est moindre. La présence de thallium réduit la
production d'oxygène, probablement par action directe sur les
transferts d'électrons au niveau du photosystème II. La présence de
chlorose traduit son effet nocif sur les pigments. En outre, il
semble que la toxicité du thallium soit associée à une moindre
fixation des oligo-éléments. Il y a également une action nocive sur
la croissance, les racines étant plus sensibles que les feuilles ou
les tiges. On a observé ces effets à des concentrations ne dépassant
pas un 1 mg de thallium par kg de tissu végétal sec, après exposition
à des dérivés du thallium(I).
Dans la plupart des études consacrées aux effets du thallium sur
les organismes aquatiques, on a utilisé des dérivés solubles du
thallium(I). La concentration de thallium la plus faible pour
laquelle on a observé des effets sur les espèces aquatiques est de
8 µg/litre, concentration qui a provoqué une réduction de la
croissance de certains végétaux aquatiques. En ce qui concerne les
invertébrés, les effets nocifs se font souvent sentir à des
concentrations plus faibles que chez les poissons (les valeurs de la
CL50 à 96 heures sont de 2,2 mg de thallium par litre pour les
daphnies et de 120 mg/litre pour une espèce de poisson d'eau douce).
La valeur la plus faible de la CL50 a été observée après une
exposition d'environ 40 jours; elle était dans le cas de poissons, de
40 µg/litre.
Les nombreux cas d'intoxication par le thallium observés dans la
faune sauvage sont dus à son utilisation à grande échelle comme
rodenticide. Chez les animaux granivores et chez les prédateurs,
c'est au niveau du système nerveux central ou des voies digestives que
se produisent les effets les plus graves. On a également observés de
tels effets chez des animaux de ferme. En outre, le thallium provoque
la chute des plumes dorsales chez les canards, une salivation au
niveau du nez et de la bouche chez les bovins, et une réduction de la
croissance chez les poulets, les poules pondeuses, les moutons et les
taureaux.
RESUMEN
1. Identidad, propiedades físicas y químicas, y métodos analíticos
El talio elemental es un metal blando y maleable de color blanco
azulado. Cuando se expone al aire húmedo o al agua, se produce
respectivamente una oxidación rápida de su superficie o la formación
del hidróxido correspondiente. Tiene dos estados de oxidación
importantes: talio(I) y talio(III). Los componentes monovalentes
(talosos) se comportan de manera análoga a los metales alcalinos, como
por ejemplo el potasio, mientras que los compuestos trivalentes
(tálicos) son menos básicos, parecidos al aluminio. A diferencia de
los compuestos inorgánicos en los que el ion talio(I) es más estable
en soluciones acuosas que el ion talio(III), este último es más
estable en compuestos orgánicos.
La determinación del talio en muestras del medio ambiente es algo
difícil, porque sus concentraciones son del orden de µg/kg o
inferiores. En general, cuando no se aplica una concentración
previamente establecida de talio, los límites de la determinación en
minerales, suelos y polvo son de unos 20 µg/kg, en soluciones acuosas
de 0,1 µg/litro y en materiales biológicos de unos pocos µg/kg.
La espectrometría de absorción atómica en horno de grafito es un
método analítico idóneo para aplicaciones en las que se necesita una
alta sensibilidad debido a las pequeñas cantidades de muestra con un
contenido de talio de unos pocos µg/kg. La espectrometría de masas
con dilución isotópica y la espectrometría de plasmamasa con
acoplamiento inductivo, posiblemente combinada con la dilución
isotópica, son métodos excelentes de determinación, con buena
precisión y exactitud, del orden de µg/kg.
2. Fuentes de exposición humana y ambiental
El talio está presente en el medio ambiente como consecuencia de
procesos naturales y procedente de fuentes debidas a actividades
humanas. Está muy extendido en la naturaleza y se encuentra sobre
todo en las menas de sulfuro de diversos metales pesados, aunque suele
estar en concentraciones bajas. Sólo hay unas pocas zonas con
concentraciones naturales de talio muy elevadas.
La producción industrial es muy pequeña (el consumo industrial en
todo el mundo en 1991 fue de 10-15 toneladas/año). El talio y sus
compuestos tienen una amplia variedad de aplicaciones industriales.
Ahora se ha limitado rigurosamente su uso como depilatorio humano y
como rodenticida e insecticida. Sus principales aplicaciones están en
las industrias eléctrica y electrónica y en la producción de vidrios
especiales. Otro campo importante de aplicación es el uso de
radioisótopos en medicina para la escintigrafía, así como el
diagnóstico de melanomas y el uso de compuestos de ariltalio(III) en
bioquímica.
Las pérdidas en el medio ambiente proceden sobre todo de la
fundición de minerales (depósitos de materiales de desecho y emisiones
a la atmósfera), las centrales eléctricas alimentadas por carbón, las
fábricas de ladrillos y de cemento (todas ellas con emisiones a la
atmósfera). Se calcula que los procesos industriales movilizan en
todo el mundo de 2000 a 5000 toneladas/año. Las emisiones de talio
debidas a procesos industriales varían mucho en función del tipo de
industria.
Las emisiones de las centrales eléctricas alimentadas por carbón
pueden contener una concentración de talio de 700 µg/m3 de aire de
salida y las de las fábricas de cemento hasta 2500 µg/m3. Esta
última cifra se puede reducir hasta < 25 µg/m3 mediante el uso de
otras materias primas y cambiando el proceso de producción. El talio
se volatiliza durante la combustión del carbón o la materia prima
utilizada en la fabricación de cemento y se vuelve a condensar sobre
la superficie de las partículas de ceniza en las partes más frías del
sistema. Estas partículas contienen hasta 50 mg de talio/kg de
polvillo de ceniza y son con frecuencia de pequeño tamaño, de manera
que los filtros de las fábricas de cemento retienen sólo un 50%.
Alrededor de un tercio de las partículas que emiten las centrales
eléctricas son también de un tamaño tan pequeño que se pueden
depositar en las vías respiratorias inferiores.
Los efluentes procedentes de los depósitos de decantación de
residuos mineros, con un contenido de hasta 1620 y 36 µg/litro,
produjeron en los ríos de vertido niveles elevados de 88 y 1 µg/litro,
respectivamente. En los estanques de agua de lluvia cercanos a una
fábrica de cemento se encontraron hasta 37 µg/litro. En el suelo se
han detectado concentraciones máximas de 60 mg/kg en zonas próximas a
materiales de desecho de minas; en las cercanías de fundiciones de
metales no preciosos y de fábricas de ladrillos y de cemento se
detectaron concentraciones de 2, 0,6 y 27 mg/kg, respectivamente. En
las zonas contaminadas, la mayoría de las hortalizas, frutas y carne
contienen menos de 1 mg de talio/kg de peso fresco. Las
concentraciones son superiores en las coles (Brassicaceae), habiéndose
notificado niveles de hasta 45 mg/kg en la col rizada verde. Las
concentraciones de talio en los tejidos de los animales de granja se
corresponden con las concentraciones en el forraje. En las cercanías
de algunas fábricas de cemento, se han descrito niveles superiores en
el forraje (por ejemplo, hasta 1000 mg/kg en la colza) y en la carne
de vacuno y de conejo (hasta 1,5 y 5,8 mg/kg, respectivamente).
3. Transporte, distribución y transformación en el medio ambiente
Cerca de fuentes localizadas, como centrales eléctricas de
carbón, algunas fábricas de cemento y operaciones de fundición de
metales, la fuente principal de talio en el aire es la emisión de
polvillo de ceniza. Los resultados de un estudio indican que casi
todo el talio del polvo flotante procedente de una fábrica de cemento
estaba presente como cloruro de talio(I) soluble.
El destino final del talio que se incorpora al suelo (debido, por
ejemplo, al depósito del polvillo de ceniza) depende fundamentalmente
del tipo de suelo. La retención es máxima en suelos que contienen
grandes cantidades de arcilla, materia orgánica y óxidos de
hierro/manganeso. La incorporación de talio a complejos estables sólo
produce concentraciones más elevadas en las capas superiores del
suelo. La absorción del talio por la vegetación va aumentando a
medida que el pH del suelo disminuye. En algunos suelos fuertemente
ácidos se puede producir lixiviación de cantidades importantes de
talio al terreno y las aguas superficiales próximos.
La mayor parte del talio disuelto en agua dulce suele ser
monovalente. Sin embargo, en agua dulce muy oxidada y en la mayor
parte del agua marina puede predominar la forma trivalente. El talio
se puede eliminar de la columna de agua y acumularse en el sedimento
mediante diversas reacciones de intercambio, formación de complejos o
precipitación.
Aunque puede darse una bioconcentración del talio, la
bioamplificación del elemento en las redes alimentarias acuática o
terrestre es improbable.
4. Niveles medioambientales y exposición humana
En zonas no contaminadas por talio, las concentraciones en el
aire suelen ser < 1 ng/m3, en el agua < 1 µg/litro y en los
sedimentos del agua < 1 mg/kg. Las concentraciones medias en la
corteza terrestre oscilan entre 0,1 y 1,7 mg/kg, pero es posible
encontrar niveles muy elevados, por ejemplo hasta de 1000 mg/kg en el
carbón, y los minerales de talio que raramente se encuentran contienen
hasta un 60% del elemento. Los alimentos de origen vegetal y animal
suelen contener < 1 mg/kg de peso seco y la ingestión media humana de
talio con los alimentos parece ser inferior a 5 µg/día. Se estima que
la absorción a través del sistema respiratorio es < 0,005 µg de
talio/día.
Se dispone sólo de datos limitados sobre el contenido real de
talio en el aire de los lugares de trabajo. Las concentraciones
observadas más recientemente (decenio de 1980) fueron < 22 µg de
talio/m3 (en la producción de una aleación especial de talio y en
una fundición de talio). El promedio de la concentración determinada
en orina fue del orden de 0,3-8 µg/litro en los trabajadores del
cemento y de 0,3-10,5 µg/litro en los de funderías.
5. Cinética y metabolismo en animales de laboratorio y en el ser
humano
El talio se absorbe con rapidez y facilidad a través de los
tractos gastrointestinal y respiratorio, así como por vía cutánea. Se
distribuye en poco tiempo por todos los órganos y atraviesa la
placenta (como se demuestra por la rápida absorción fetal) y la
barrera hematoencefálica. Debido a su acumulación rápida en las
células, las concentraciones de talio en la sangre no se corresponden
con su nivel en los tejidos. En casos de intoxicación aguda de
animales experimentales o de personas, se producen al principio
concentraciones de talio elevadas en el riñón, bajas en el tejido
adiposo y en el cerebro e intermedias en los demás órganos; luego
aumentan también sus niveles en el cerebro.
La eliminación del talio se puede producir a través del tracto
gastrointestinal (básicamente mediante mecanismos independientes de la
excreción biliar), el riñón, el pelo, la piel, el sudor y la leche
materna. Se puede producir una reabsorción intestinal (sobre todo
desde el colon), con la consiguiente disminución en la eliminación
total del organismo. En la rata, las principales vías de eliminación
del talio son la gastrointestinal (unos dos tercios) y la renal
(alrededor de un tercio), siendo semejante la contribución de ambas
vías en el caso de los conejos. El talio se elimina también por la
saliva.
Al igual que con otras muchas sustancias, la excreción de talio
en el ser humano difiere de la observada en los animales de
laboratorio; en aquél la velocidad de excreción es mucho más baja
(constante de velocidad = 0,023-0,069 día-1) que en éstos (la
constante de velocidad media = 0,18 día-1). Otra diferencia
importante entre el hombre y los animales es la contribución relativa
de las distintas vías de excreción. La excreción renal parece ser
mucho más importante en el ser humano que en los animales, aunque no
se ha determinado completamente su contribución relativa a la
eliminación total del organismo, debido fundamentalmente a la falta de
suficientes datos respecto al hombre. Además, los niveles de
exposición, su duración, la alteración de la función de los órganos de
excreción, la absorción de potasio y el tratamiento correspondiente de
la intoxicación aguda pueden influir considerablemente en los
resultados.
En un estudio sobre la excreción renal de talio se notificó un
resultado de alrededor del 73%, mientras que a través del tracto
gastrointestinal fue de sólo el 3,7% de la cantidad diaria excretada.
La excreción estimada a través del pelo y la piel y del sudor fue del
19,5% y el 3,7%, respectivamente.
La semivida biológica del talio en animales de laboratorio oscila
generalmente entre 3 y 8 días; en el ser humano es de unos 10 días,
aunque se ha informado de valores superiores a los 30 días.
No se dispone de datos sobre su biotransformación.
6. Efectos en mamíferos de laboratorio y en sistemas de ensayo
in vitro
No hay diferencias específicas sorprendentes por especies en
cuanto a la toxicidad de las sales de talio(I). Normalmente, una
ingestión oral de 20 a 60 mg de talio/kg de peso corporal es letal en
un plazo de una semana. Los cobayos son ligeramente más sensibles que
otros animales de experimentación. El óxido tálico(III) insoluble en
agua muestra una toxicidad aguda algo más baja por vía oral o
parenteral que las sales de talio(I). Al comparar los datos de
toxicidad aguda se aprecia una elevada biodisponibilidad a partir de
todas las vías de exposición. Afecta a la mayor parte de los órganos,
pero los signos de intoxicación y la sucesión de los mismos indican
una cierta variabilidad intraespecífica e interespecífica.
Los síntomas de la intoxicación aguda se suceden en general de la
manera siguiente: en primer lugar anorexia, vómitos y depresión, más
tarde diarrea, cambios cutáneos (inflamación en los orificios
corporales, furúnculos cutáneos, pérdida de pelo) y luego disnea y
trastornos nerviosos. Por último, la insuficiencia respiratoria que
provoca la muerte.
Los síntomas de la intoxicación crónica son semejantes a los de
la intoxicación aguda. Se produce regularmente pérdida de pelo.
En el examen histológico se puede observar necrosis u otros daños
celulares. Se han detectado cambios necróticos en los riñones, el
hígado, el intestino, el corazón y el sistema nervioso. En numerosas
células se ha observado hinchazón de las mitocondrias y pérdida de
crestas, dilataciones del retículo endoplasmático liso, aumento del
número de vacuolas autofágicas y de gránulos de lipofucsina y pérdida
de microvellosidades. Las alteraciones de procesos funcionales
debidas al talio pueden estar provocadas por la rotura física de las
membranas de los orgánulos subcelulares. En el corazón, los efectos
arritmogénicos se limitan al nódulo sinoatrial.
La intoxicación por talio provoca la alteración selectiva de
determinados elementos de la conducta relacionados con efectos
bioquímicos (lo que indica un daño celular) en ciertas regiones
cerebrales. Algunos efectos neurológicos parecen deberse a la acción
directa, por ejemplo la ataxia y el temblor a causa de trastornos del
cerebelo o alteraciones de la actividad endocrina debidos a cambios en
el hipotálamo. El talio puede activar el sistema nervioso autónomo,
fundamentalmente el adrenérgico. En los nervios periféricos parece
interferir a nivel presináptico en la liberación espontánea del
transmisor, ejerciendo un efecto antagónico en estos procesos
dependientes del calcio.
No se conoce todavía el mecanismo exacto de la toxicidad del
talio. Se han propuesto varios mecanismos, que tal vez están
relacionados entre sí. Un aspecto importante de la intoxicación por
talio es el aumento significativo de la peroxidación de lípidos y de
la actividad de una enzima lisosómica, la ß-galactosidasa. El
resultado es una deficiencia de glutatión que provoca la acumulación
de peróxidos de lípidos en el cerebro y, al parecer, por último, la
formación de gránulos de lipofucsina. Parece que el mecanismo de
acción del talio radica fundamentalmente en una alteración de la
función mitocondrial.
Los animales con intoxicación crónica suelen presentar una
actividad sexual reducida, y en el sistema reproductor del macho son
evidentes los efectos gonadotóxicos del talio. En los testículos de
ratas que recibieron 10 mg de talio/litro de agua de beber durante 16
días, las células de Sertoli fueron las más sensibles y la descamación
del epitelio espermatogénico provocó la aparición de espermatozoides
inmaduros en el semen. Esto podría explicar el menor índice de
supervivencia de los embriones o la reducción del periodo de vida de
la descendencia tras una intoxicación subletal por talio de los
padres.
Tras la inyección de talio en huevos, se observaron en los
embriones de pollo efectos teratogénicos, inhibición del crecimiento y
trastornos del desarrollo óseo, pero en los mamíferos estos efectos
son discutibles, incluso a dosis tóxicas para la madre. Aunque se ha
demostrado que atraviesa la placenta, muchas estirpes de ratones y
ratas no muestran efectos teratogénicos en absoluto, o sólo
ligeramente.
Dos pruebas de mutagenicidad microbiológica en Salmonella
typhimurium dieron resultados negativos, y las pruebas in vivo sobre
intercambio de cromátides hermanas fueron controvertidas. Sin
embargo, en estudios aislados se han observado aberraciones
cromosómicas o un aumento significativo de la fragmentación del ADN de
cadena sencilla.
No se dispone de estudios de larga duración sobre la
carcinogenicidad del talio.
7. Efectos en el ser humano
Debido a que las sales de talio son insípidas, inodoras,
incoloras, muy tóxicas, fáciles de obtener en el pasado e incluso
ahora en algunos países en desarrollo, este elemento se ha utilizado a
menudo con fines suicidas, homicidas y de aborto ilegal, provocando
intoxicación aguda. Es más, se considera que la intoxicación por
talio es una de las causas más frecuentes, a escala mundial, de
intoxicación humana voluntaria o accidental. Los conocimientos sobre
la intoxicación crónica se limitan a la exposición profesional, a
grupos de población que viven en zonas contaminadas y a casos de
homicidio con dosis bajas múltiples.
Los síntomas de toxicidad aguda del talio dependen de la edad, la
vía de administración y la dosis. Las dosis que han resultado letales
varían entre 6 y 40 mg/kg, con un promedio de 10 a 15 mg/kg. Sin
tratamiento, esta dosis media suele producir la muerte en un plazo de
10 a 12 días, pero también se han descrito casos de defunción en 8-10
horas.
Se considera que la gastroenteritis, la polineuropatía y la
alopecia son los tres síntomas clásicos de la intoxicación por talio,
pero en algunos casos no se observó gastroenteritis ni alopecia.
También se producen otros signos y síntomas, con un orden de
aparición, amplitud e intensidad variables.
Los síntomas de la intoxicación son a menudo imprecisos y
consisten al principio en anorexia, náuseas, vómitos, sabor metálico,
salivación, dolor retrosternal y abdominal y a veces hemorragia
gastrointestinal (sangre en heces). Luego se suele observar
estreñimiento, que puede ser resistente al tratamiento, interfiriendo
así con el antídoto administrado.
Después de un periodo de dos a cinco días aparecen lentamente
algunos de los trastornos asociados normalmente al talio, con
independencia de la vía de exposición. Aunque los efectos en el
sistema nervioso central y periférico varían, un rasgo constante y
característico de la intoxicación por talio en el hombre es la
sensibilidad extrema de las piernas, a la que sigue el «síndrome de
los pies urentes» y la parestesia. Su acción sobre el sistema
nervioso central (SNC) se refleja en síntomas tales como
alucinaciones, letargia, delirio, convulsiones y coma. Los síntomas
circulatorios normales son hipertensión, taquicardia y, en los casos
graves, insuficiencia cardiaca. Después de la segunda semana de la
intoxicación se suele producir pérdida del pelo y, a veces, del vello;
la distrofia de las uñas se detecta por la aparición de rayas
semicirculares blancas (líneas de Mee) tres o cuatro semanas después
de la intoxicación. Las regiones negras que se observan en las
papilas pilosas no se producen por la deposición de pigmentos o de
talio, sino que se deben a pequeñas cantidades de aire que entran en
el tallo piloso.
En los casos letales, la muerte sobreviene en un plazo que oscila
entre unas horas y varias semanas, pero normalmente se produce a los
10 ó 12 días. Las causas del fallecimiento son generalmente
insuficiencia renal, del SNC y cardiaca.
En intoxicaciones subletales, la recuperación requiere con
frecuencia meses. A veces persisten los trastornos neurológicos y
mentales, así como las anomalías electroencefalográficas y la ceguera.
Por otra parte, parece ser que los supervivientes sufren un deterioro
de las funciones intelectuales.
En casos de intoxicación crónica los síntomas son semejantes,
pero en general más leves que en la intoxicación aguda. A veces se
produce ceguera permanente. La recuperación completa requiere meses y
se puede interrumpir por recaídas.
En un caso bien investigado de emisión de talio alrededor de una
fábrica de cemento de Lengerich, Alemania, las concentraciones de
talio en el pelo y la orina de las personas expuestas no se
correspondían con algunas características típicas que suelen estar
relacionadas con la intoxicación crónica por talio, sino sólo con
síntomas neurológicos subjetivos.
La autopsias y biopsias realizadas tras las intoxicaciones por
talio ponen de manifiesto daños en diversos órganos. Por ejemplo,
tras la ingestión de dosis letales se producen hemorragias en la
mucosa intestinal, los pulmones, las glándulas endocrinas y el
corazón, infiltraciones grasas en el hígado y el tejido cardiaco, así
como cambios degenerativos en los glomérulos y los túbulos renales.
En el cerebro se puede observar degeneración grasa de las células
ganglionares, lesiones axonales y desintegración de las vainas de
mielina.
Las variaciones de la presión sanguínea pueden deberse a los
efectos directos del talio en el sistema nervioso autónomo. La
intoxicación por talio produce neuropatía periférica mixta simétrica.
Los nervios distales sufren más daños que los proximales, y los
nervios de axón corto, por ejemplo los craneales, se ven afectados
antes, aunque en menor grado. Los axones se hinchan y contienen
vacuolas y mitocondrias dilatadas. En los casos de intoxicación
letal, se han observado daños graves del nervio vago, desnervación del
seno carotídeo y lesiones de los ganglios simpáticos. En la
intoxicación subletal, los nervios afectados pueden sufrir
degeneración axonal, con recuperación nula o sólo parcial en un plazo
de dos años.
A veces se produce una neuritis retrobulbar con los consiguientes
trastornos visuales, que puede persistir durante meses, después de un
tratamiento con productos depilatorios con talio; este trastorno puede
desembocar incluso en la atrofia óptica. Los datos sobre los efectos
del talio en la reproducción humana son limitados. Puede afectar
negativamente al ciclo menstrual, la libido y la potencia masculina.
Se sabe que la intoxicación crónica tiene efectos sobre el esperma.
Al igual que en los estudios con animales, se ha observado que
atraviesa la placenta; esto se ha puesto de manifiesto tras un aborto
inducido por el talio. Sin embargo, en unos 20 casos de intoxicación
por talio durante el embarazo no se vio afectado el desarrollo fetal,
salvo el peso relativamente bajo y la alopecia de los recién nacidos.
No se dispone de informes sobre efectos carcinógenos o datos
sobre efectos inmunológicos debidos al talio. No hay pruebas
suficientes de efectos genotóxicos.
El tratamiento de la intoxicación por talio combina la diuresis
forzada, el uso de carbón vegetal activado y la prevención de la
reabsorción en el colon mediante la administración de azul de Prusia,
hexacianoferrato(II) férrico potásico.
8. Relación dosis-respuesta en el ser humano
La concentración media de talio en orina en la población no
expuesta es de 0,3 a 0,4 µg/litro. Habida cuenta de que el talio
tiene una semivida biológica breve, establecida en días, y suponiendo
unas condiciones estables, se puede tomar esta concentración urinaria
como indicador de la dosis total tras la inhalación y la ingestión con
los alimentos.
La concentración media en la orina en una muestra de población
que vive cerca de una fuente de emisión de talio fue de 5,2 µg/litro.
Se encontró una relación dosis-respuesta clara entre las
concentraciones en la orina de talio y el predominio de cansancio,
debilidad, trastornos del sueño, dolor de cabeza, nerviosismo,
parestesia y dolor muscular y de las articulaciones. Se informó
asimismo de una relación dosis-respuesta semejante cuando se utilizó
el talio en el pelo como indicador de la exposición.
El Grupo Especial de Trabajo consideró que las exposiciones que
producen concentraciones de talio en la orina inferiores a 5 µg/litro
probablemente no son perjudiciales para la salud. En el margen de 5 a
500 µg/litro, la magnitud del riesgo y la gravedad de los efectos
adversos son inciertas, mientras que la exposición que da lugar a más
de 500 µg/litro está asociada a una intoxicación clínica.
9. Efectos en otros organismos en el laboratorio y en el medio
ambiente
El talio afecta a todos los organismos, pero hay diferencias
específicas de especies e incluso de variedades. Los diferentes
compuestos inorgánicos de talio(I) y talio(III), así como sus
compuestos orgánicos, pueden tener distinta toxicidad.
El efecto más importante del talio en los microorganismos parece
ser la inhibición de la nitrificación por las bacterias del suelo.
Los resultados de un estudio parecen indicar que la estructura de la
flora microbiana se altera a concentraciones en el suelo comprendidas
entre 1 y 10 mg/kg de peso seco, pero no se precisó la forma de talio
utilizada en este experimento.
Absorben talio todas las partes de las plantas, pero sobre todo
las raíces. Una vez que ha penetrado en las células, se concentra de
forma desigual en el citosol, probablemente unido a un péptido. Las
concentraciones de talio que se observan en las plantas dependen de
las propiedades del suelo (en particular el pH y el contenido de
arcilla y materia orgánica), así como de la fase de desarrollo y de la
parte de la planta. Se acumula en las zonas que contienen clorofila,
pero lo hace en menor grado en las plantas resistentes al talio.
Reduce la producción de oxígeno, posiblemente por acción directa sobre
la transferencia de electrones en el fotosistema II. Su interferencia
con los pigmentos se pone de manifiesto por la aparición de clorosis.
Por otra parte, en el mecanismo de la toxicidad parece intervenir una
alteración de la absorción de oligoelementos. Afecta también al
crecimiento, siendo más sensibles las raíces que las hojas o los
tallos. Estos efectos se han descrito tras la exposición a formas
monovalentes de talio con niveles de sólo 1 mg/kg de tejido vegetal
seco.
En la mayoría de los estudios de los efectos en los organismos
acuáticos se han utilizado compuestos solubles de talio monovalente.
La concentración más baja notificada capaz de afectar a las especies
acuáticas es de 8 µg/litro, con una reducción del crecimiento de las
plantas. Los invertebrados se suelen ver afectados a concentraciones
más bajas que los peces (los valores de la CL50 en 96 horas son de
2,2 mg de talio/litro para los dáfnidos y de 120 mg/litro para un pez
de agua dulce). El valor más bajo de la CL50, notificado tras la
exposición durante unos 40 días, fue de 40 µg/litro para los peces.
Muchos casos de intoxicación por talio de la flora y fauna
silvestres se han debido a su aplicación en gran escala como
rodenticida. En animales que se alimentan de semillas y en
depredadores afecta gravemente sobre todo al SNC y al aparato
gastrointestinal. Estos mismos efectos se pueden observar en los
animales de granja. A esto hay que añadir que el talio provoca una
pérdida de plumas dorsales en los patos, salivación de la nariz y la
boca del ganado vacuno y reducción del crecimiento de los pollos de
asar, las gallinas ponedoras, las ovejas y los novillos.
hair were reported. Later, severe polyneuritis may develop, with an
inability to walk, amaurosis (blindness) and pronounced cachexia.
Cardiac disorders include hypertension, irregular pulse and
angina-like pain. Renal dysfunction is indicated by albuminuria and
haematuria. Other symptoms are gastric anacidity, lack of appetite,
loss of weight, endocrine disorders, psychoses and encephalitis.
Complete rehabilitation takes months and can be interrupted by
relapses, probably caused by remobilization of thallium from tissue
depots.
Epidemiological studies carried out in the contaminated area of
Lengerich, Germany, comprising about 1200 people, revealed positive
correlations between the concentration of thallium found in urine or
hair samples and polyneuritic symptoms such as paraesthesias and pain
in muscles and joints, as well as psychasthenic symptoms such as
headache, sleep disorders and fatigue. No correlation was found with
respect to gastrointestinal troubles or skin disorders. Surprisingly,
a negative correlation with hair loss was found. Only one of 51
people with > 20 µg thallium/litre urine showed lunular stripes in
the nails (LIS, 1980; Brockhaus et al., 1980, 1981b; Dolgner &
Wiegand, 1982; Schoer, 1984). Strong individual variation in
sensitivity prevents an estimation of the thallium concentration in
the urine at which no effects occur (Dolgner & Wiegand, 1982).
8.2 Occupational exposure
There have been numerous reports of factory workers with thallium
poisoning, but no fatal cases have occurred. Peripheral sensorial
disturbances, mental changes, loss of weight, and sleeplessness are
the symptoms which seem to prevail (Munch, 1934b; Muller, 1961;
Malcolm, 1979; Saddique & Peterson, 1983; Triebig & Büttner, 1983;
Schoer, 1984; Nessler, 1985b; Junghans & Nessler, 1985; Ohnesorge,
1985; Kazantzis, 1986). In Germany, the United Kingdom and some other
countries, thallium poisoning represents an occupational disease
entitling the victim to compensation. In Germany, such compensation
was granted in three cases between 1970 and 1985 (Ewers, 1988).
Increased thallium concentrations in the urine of workers have
often been found. For example, the urine of workers in a company
producing alloy anode plates for use in magnesium sea water batteries
contained up to 236 µg thallium/litre, but no differences in medical
records of exposed and unexposed workers could be demonstrated
(Marcus, 1985). In his review of thallium, Ohnesorge (1985)
summarized several reports of industrial poisoning. Exposure over
several months or years resulted in typical thallium symptoms, e.g.,
leg pains, tiredness, alopecia and psychological disorders, but also
(in one case) blindness. Permanent blindness was also reported in
another review by McDonald (1941). Exposure to more than 0.01 mg
thallium/m3 for 16 to 17 years caused disorders of the vascular
system, as well as neurological symptoms (Ohnesorge, 1985). From the
triad of gastroenteritis, polyneuropathy and alopecia, only disorders
of the gastrointestinal tract were not reported.
Glomme (1983) emphasized that objective symptoms of polyneuritis
may not be demonstrable for some time. In addition to the changes in
the superficially provoked tendon reflexes, a pronounced weakness and
a fall-off in the speed of pupillary reflexes can occur.
In a further study on cement plant workers, 36, selected at
random, were subjected to thallium analyses of blood, urine and hair,
together with a neurological examination and electrophysiological
investigation including sensory and motor nerve conductive velocities,
evoked potentials and electro-encephalography (Ludolph et al., 1986).
One half of the workers examined suffered from concurrent disorders,
including diabetes mellitus. Although multiple symptoms and signs of
neurological disorders were detected, no correlation was found between
the electrophysiological findings and thallium levels in blood, urine
and hair. Urinary thallium levels were above 5 µg/litre in five of
the examined workers. Blood thallium levels above 2 µg/litre were
found in 16 workers and hair thallium levels above 20 µg/kg in four
workers. The investigators concluded that more thorough
epidemiological techniques would be required to reveal a possible
causal relationship between chronic low-dose thallium exposure and
neurological deficits.
8.3 Subpopulations at special risk
There are no subpopulations at special risk of thallium
intoxication except workers in the respective industries and
populations living in thallium-contaminated areas. There are no good
data to suggest that infants or pregnant women are more sensitive to
the effects of thallium than the general population. The available
data, however, are inadequate to fully assess these subpopulations.
Because thallium is eliminated in both urine and faeces, any
subpopulations with diminished excretory capabilities (e.g., renal
insufficiency) may be at increased risk of thallium poisoning. It has
been recommended that workers be excluded from working with thallium
if they suffer from renal or hepatic disease, anaemia, blood
dyscrasias, hypertension, alcoholism, chronic infections or endocrine
gland dysfunction. It has also been recommended that workers
potentially exposed to thallium should be encouraged to eat
potassium-rich food, as thallium and potassium ions can mimic each
other in vivo. Accordingly, potassium-deficient individuals may
also be at increased risk from thallium toxicity.
8.4 Target organs in intoxicated humans: pathomorphology and
pathophysiology
Effects on the different organs have been summarized by Prick
(1979), Sabbioni & Manzo (1980) and ATSDR (1992). In nearly all
affected organs direct cytotoxic effects, as well as indirect effects,
caused by damage to the nervous system, have been found (Prick, 1979).
8.4.1 Gastrointestinal tract and renal system
In a fatal case of thallium poisoning, in which the woman died
after at least 14 days, there was gross dilatation of the stomach and
a thin "blue line" was evident at the margin of the gingiva of the
lower incisors, but no alterations of the intestinal wall were
apparent (Curry et al., 1969). Other patients who died 1 to 16 days
after oral poisoning showed hyperaemia, congestion of the gut,
punctate haemorrhages in the mucosa of the stomach and upper
intestinal tract, and swelling of the mucosal cells (Lynch et al.,
1930; Munch et al., 1933; Heath et al., 1983). As a result of
depilatory treatment in children, gastric hypoacidity was reported
(Buschke, 1929), an effect also observed after a suicide attempt
(Greving & Gagel, 1928).
In several cases of oral poisoning, usually fatal, the liver was
usually found to be congested, greyish yellow or yellow in colour, had
microscopic fatty infiltrations of the hepatocytes and a tendency to
central necrosis (Lynch et al., 1930; Munch et al., 1933; Curry et
al., 1969).
At least 6 weeks after intoxication, renal biopsy of a patient
with 13.8 mg thallium/litre in his urine showed diffuse proliferative
glomerulonephritis with granular immunofluorescence for IgG, IgM and
C3 (Alarcón-Segovia et al., 1989). In the postmortem examinations by
Lynch et al. (1930), Munch et al. (1933) and Curry et al. (1969),
sections of kidney were dull red or congested and showed marked
hyperaemia, cloudy swelling of tubules and degenerative changes of
glomeruli. Weinig & Schmidt (1966) also reported kidney damage (but
perhaps from a previous attempt at poisoning) in a woman and her son
who died after taking thallium. This kidney damage may have been
responsible for the relatively low thallium concentrations in the
son's kidney in comparison to concentrations in other tissues (section
6.2.2).
8.4.2 Cardiovascular system
Accidental poisoning of three children, who died within two days,
caused fatty degenerations in the victims' hearts. These were more
marked and more dispersed in the youngest child, who survived longest
(Lynch et al., 1930). In some cases of postmortem-diagnosed thallium
poisoning, resulting in death within 4-14 days, fresh haemorrhagic
myocardial lesions were found (Heath et al., 1983; Andersen, 1984),
while in another case only a few focal haemorrhages were present
(Curry et al., 1969). Haematological changes, e.g., anaemia,
leucocytosis, eosinophilia, thrombocytopenia (at least partly
resulting from a toxic effect on bone marrow) and lymphopenia, have
been summarized by Saddique & Peterson (1983) and Luckit et al.
(1990).
In five patients suffering from severe and protracted thallium
poisoning, cardiovascular changes were recorded (Machtey & Bandmann,
1961; Franke et al., 1979). The patients' blood pressure showed marked
fluctuations, even in the course of one day, but systolic and
diastolic hypertension occurred only on a temporary basis. The
authors believed that these changes and also the observed tachycardia
and electro-cardiographic changes were caused by direct effects of
thallium on the autonomic nervous system. Tachycardia can appear
about a week after intoxication and last for 5 weeks (Franke et al.,
1979). Involvement of the autonomic nervous system is also indicated
by changes in renal function and by the urinary concentrations of
various metabolites (brenzcatecholamines, vanillin-mandelic acid,
ß-aminolaevulinic acid, porphobilinogen, coproporphyrin and total
porphyrins) during hypertension and tachycardia resulting from
thallium poisoning (Bock et al., 1968). Concentrations of
brenzcatechinamines and porphobilinogen were greatly increased, and
hypertension and tachycardia could be influenced by administration of
alpha- and ß-receptor blockers. In addition, increased elimination of
brenzcatecholamines, which presumably originate not only from the
adrenal medulla but also from the sympathetic nervous system,
indicates a strong stimulation of the adrenergic system (Bock et al.,
1968).
8.4.3 Skin and hair
Five young men suffering from thallium poisoning showed
follicular plugging of the skin on the nose and cheeks and in the
nasolabial folds by keratinous material, crusted eczematous lesions
and acneiform eruptions on the face, dry scaling on palms and soles,
and alopecia, not only of the scalp but sometimes also of the
eyelashes, lateral eyebrows, arms and legs. Histological examination
of skin biopsies from both scalp and cheek showed disintegrating
hairshafts, gross follicular plugging and eosinophilic keratohyaline
granules in the adjacent granular layer of the epidermis. Sebaceous
glands were sometimes necrotic. Biopsies of the pustular lesions on
the face showed folliculitis and necrosis of the follicles, while in
those from the feet marked hyperkeratosis and hypergranulosis were
evident (Heyl & Barlow, 1989). Effects on the follicles are also
reported by Hausman & Wilson (1964) and Bonnet & Pedace (1979), but in
a woman who died at least 14 days after intoxication no hyperkeratosis
in any part of the skin was found (Curry et al., 1969).
As is the case in experimental animals (section 7.4.1), the
reason for the different sensitivities of different types of hair
(lanugo, pubic and axillary hair is much less or is later affected
than hair of the head) in humans is unclear (Buschke & Peiser, 1926;
Buschke, 1929; Cavanagh, 1988). Cavanagh et al. (1974) emphasized a
direct effect on the keratinocytes, and Cavanagh (1988) finally
suggested that the difference is due to the fact that hair follicle
cells are only affected when they are mitotically active.
The depilatory effect generally does not result in permanent hair
loss. Since the new hairs which grow following thallium-induced
alopecia are stronger than those lost and also develop in regions
which had been hairless prior to the thallium poisoning, Buschke
successfully used thallium in therapy of alopecia induced by hair
disease (Buschke & Curth, 1928).
Soon after poisoning, hair papilla are seen to contain black
regions and the growing end is tapered (e.g., Hausman & Wilson, 1964;
Curry et al., 1969; Saddique & Peterson, 1983). In some reviews this
phenomenon is interpreted as black pigmentation. However, Ludwig
(1961) had already shown that these regions do not contain deposits of
pigments or thallium but small amounts of air which had entered the
shaft. Later investigations demonstrated that the gaseous inclusions
result from a trophic disorder in keratin formation (Kijewski, 1984;
Metter & Vock, 1984). In both investigations, scanning electron
microscopy demonstrated a loosening of the elements of the fibre layer
of the hairs.
8.4.4 Nervous system
Neurological disorders showing strong variability are one of the
three major symptoms of thallium poisoning (Möllhoff et al., 1979;
Prick, 1979; Sabbioni & Manzo, 1980; Le Quesne, 1984; Manzo &
Sabbioni, 1988). In contrast to the other disorders, neurological
deficits usually persist. Ataxia, mild spastic paraparesis and
impairment of intellectual powers developed after treatment of scalp
ringworm with thallium and persisted for 36 years, and it is possible
that increasing problems with mobility after 33 years were also due to
the treatment (Barnes et al., 1984).
8.4.4.1 Central nervous system
Like some other heavy metal intoxications, those caused by
thallium are usually associated with subacute and chronic (but rarely
with acute) encephalopathy (Rosenstock & Cullen, 1986). In one patient
who died 9 days after ingesting 5-10 g thallium nitrate, no
abnormalities were evident in histological or ultrastructural
examinations of the central nervous system (Davis et al., 1981). The
brain of another patient, who died within 4 days of intoxication and
had an extreme postmortem concentration of 36 mg/litre in his blood,
was moderately swollen (Andersen, 1984).
Seven people who died 11 to 16 days after accidental ingestion of
thallium had localized oedema and various grades of chromatolysis in
their neurons, especially those of the pyramidal tract, the third
nucleus, the substantia nigra and the pyramidal cells of the globus
pallidus. Blood vessels were distended with blood (Munch et al.,
1933).
In a fatal case of thallium poisoning, the brain of the dead
person was slightly swollen and oedematous about 4 weeks after the
ingestion of around 33 mg thallium sulfate/kg body weight. Petechial
haemorrhages were found in the white matter, particularly in the
parietal regions and subthalamic areas. The brain stem and cerebellum
showed a normal appearance. Axonal swelling and fragmentation in the
cortico-spinal tracts could be traced through the mid-brain, pons and
medulla into the spinal cords. Chromatolysis of brain stem nuclei was
only marked in facial and hypoglossal nuclei and nerve fibre
degeneration only in the spinal tract of the 5th nerve (Kennedy &
Cavanagh, 1976).
In a suicidal case, general degeneration of ganglion cells,
damage to axons and disintegration of myelin sheaths were observed in
the brain of the person, who died 21 days after intoxication. Fatty
degeneration of ganglion cells, acute swelling of oligodendroglia, a
spongy appearance of the basal ganglia and a particular concentration
of lesions in the calcarine cortex were prominent (Karkos, 1971). In
another fatal case, autopsy showed degeneration of ganglion cells in
the brain and spinal cord (Gefel et al., 1970).
Detrimental effects on intellectual functions were assumed not
only in a patient suffering from ringworm treatment (section 8.4.4)
(Barnes et al., 1984), but also in a student of chemistry who
eliminated 60 mg thallium/litre in his urine after poisoning in the
laboratory (Thompson et al., 1988). The data obtained from
intelligence tests on the student, performed 7 and 13 months after the
near fatal intoxication, were compared with those of his non-identical
twin brother who was of a similar educational background. Although
the brothers are not totally comparable, the tests indicated severe
deterioration particularly in memory and performance abilities and, 13
months later, there was only little general improvement.
8.4.4.2 Peripheral nervous system
Histological and ultrastructural examination of postmortem
samples can produce inconsistent results, presumably because of the
different periods of time between intoxication and sampling and
because of differences in dose size. In general, clinical symptoms
and signs can be correlated to neuropathological findings (Cavanagh,
1979).
Damage to the autonomic nervous system accounts for many of the
effects on various organs, e.g., fever, tachycardia, labile blood
pressure, orthostatic hypotension, urinary retention, constipation and
cardiac arrhythmias (Gastel, 1978; Prick, 1979). Thallium
intoxication causes symmetric, mixed peripheral neuropathy (Rosenstock
& Cullen, 1986). Distal nerves are more strongly affected than more
proximal nerves, and earlier but lesser degrees of change occur in
nerves with shorter axons, e.g., the cranial nerves (Cavanagh et al.,
1974; Cavanagh, 1979, 1988).
a) Histology and ultrastructure
Neuropathological findings vary. Little evidence of neuronal
degeneration in the sciatic nerve or spinal cord were found in a
person who died about 14 days after intoxication (Cavanagh et al.,
1974). In a fatal case of poisoning, in which the patient died just 9
days after intoxication (section 8.4.4.1), a sural nerve biopsy was
obtained 2 days before death. In addition, postmortem samples of
peripheral and cranial nerves and sections from various parts of the
central nervous system were taken. Ultrastructural examination of the
sural nerve showed that the myelin sheaths had often disintegrated
into a series of ovoids along the course of the axon (Davis et al.,
1981). Similar findings have also been reported from other sural
nerve biopsies, taken, for example, 3 days in one case and at least 5
to 6 weeks after thallium poisoning, from patients who survived (Bank
et al., 1972; Paulson et al., 1972; Alarcón-Segovia et al., 1989;
Dumitru & Kalantri, 1990). Degenerated myelin sheaths contained
myelin figures and electron-dense granules, whereas axons usually had
a normal appearance and rarely contained densely packed neurofilaments
(Bank et al., 1972). Munch et al. (1933) and Davis et al. (1981)
found axon degeneration in peripheral nerves, even in axons with
ultrastructurally normal myelin sheaths; axons were swollen and
contained vacuoles and distended mitochondria. Non-myelinated axons
on the other hand showed only slight or no abnormalities. Beadings of
axons were not only present in distal portions of peripheral nerves,
but also in some cranial nerves, whereas the other cranial nerves and
the proximal portions of peripheral nerves were histologically normal.
In another postmortem examination of a patient who died about 4
weeks after intoxication (section 8.4.4.1) (Kennedy & Cavanagh, 1976),
the nerve fibres of several peripheral nerves were severely reduced,
long fibres in particular being more severely affected. Changes in
neurons of the spinal cord were evident in all regions but most
strikingly in the lumbosacral region, where many neurons clearly
showed the classical chromatolytic changes which indicate attempted
regeneration. Dorsal column changes in the spinal tracts could
clearly be correlated in time with the peripheral nerve symptoms, and
the slight changes in the lateral cortico-spinal tracts could be
traced to the recent necrotic lesions in the diencephalon (Kennedy &
Cavanagh, 1976). Demyelination of the dorsal columns in sections of
cervical spinal cord was also observed during the postmortem
examination of a women who died at least 14 days after intoxication
(Curry et al., 1969).
The severe damage to the vagus, denervation of the carotid sinus,
and lesions of the sympathetic ganglia found in postmortem
examinations indicate the involvement of the autonomic nervous system
(Gastel, 1978).
b) Electrophysiological investigations
In a case of thallium poisoning in which the patient survived, a
sural nerve biopsy was obtained and nerve conduction and serial
electromyographic studies were carried out, beginning 10 days after
onset of the symptoms and ending 24 months later (Dumitru & Kalantri,
1990). Initially, the plantar nerves of the foot showed profound
axonal loss, from which there was no recovery, as shown by conduction
studies over the next 2 years. During the initial 4 months, sural and
peroneal nerves also underwent axonal loss but recovered within 2
years. In other cases of thallium intoxication, nerve conduction
studies gave normal results or revealed retarded latencies of nerves
of the upper (more than the lower) extremities, as well as temporal
dispersion indicating demyelination (Alarcón-Segovia et al., 1989).
Sensory fibres of the nervus medianus were examined in a patient
with acute thallium poisoning in order to assess the effects on the
conduction velocities of faster and slower nerve fibres. Two months
after the onset of symptoms the patient showed evidence of distal
sensorimotor neuropathy, but only the conduction velocities of faster
fibres were below the normal lower limit. Nine months later, symptoms
had almost disappeared and conduction velocities of both slower and
faster fibres were within the normal range (Yokoyama et al., 1990).
Only a slight electrophysiological correlation with the symptoms of a
persistent polyneuropathy were reported from an examination carried
out 3 years after a case of intoxication (Feudell, 1982).
c) Visual disorders
Retrobulbar neuritis and resulting visual impairment can develop
or persist months after termination of treatment with
thallium-containing depilatories, and even optic atrophy may occur
(e.g., Buschke & Langer, 1927; Lillie & Parker, 1932; Mahoney, 1933;
Bank et al., 1972; Bahiga et al., 1978; Tabandeh et al., 1978;
Schmidbauer & Klingler, 1979). An ascendent (retinal) atrophy of the
optic nerve may result from the toxic effects of thallium on the
retina (Hennekes, 1983). Nerve fibres in oculomotor muscles can also
show degenerative changes (Cavanagh et al., 1974). In patients with
optic neuritis some reduction in visual acuity always persists
(Goldblatt, 1989). About 10 months after thallium intoxication, a
keratoconjunctivitis sicca was found to have developed
(Alarcón-Segovia et al., 1989).
8.4.5 Other organs
Effects on the lung and endocrine glands were found in six
postmortem examinations (death occurred 11 or 15/16 days after
ingestion of thallium). Light microscopy showed the alveoli distended
with serum, marked hyperaemia and a few areas with bronchopneumonia
(Munch et al., 1933). In another fatal case the pleurae were free
from haemorrhages and adhesions (Curry et al., 1969). Of the
endocrine glands only the adrenals were affected. They showed marked
hyperaemia, small haemorrhages in the medulla, areas of necrosis and
nuclear disintegration (Munch et al., 1933). In other lethal cases
the adrenals were enlarged but without haemorrhages (Curry et al.,
1969), or were haemorrhagic (Gefel et al., 1970), or the concentration
of lipoids was reduced (Buschke, 1929).
A biopsy, taken 50 days after intoxication, showed marked areas
of atrophy of muscle tissue (Franke et al., 1979). Muscle fibrosis
was reported by Gefel et al. (1970) in a fatal case of thallium
poisoning.
8.5 Special effects
8.5.1 Reproduction and developmental effects
Few data are available with respect to the effects of thallium on
human reproduction (Schardein & Keller, 1989). Female cycles are
arrested, and libido and potency of males decrease (Buschke & Langer,
1927; Greving & Gagel, 1928). Effects on sperm are known to occur in
cases of chronic intoxication (Cottier, 1980). It should be noted that
minor amounts of thallium accumulate in the testis after diagnostic
scintigraphy, but possible effects have not been investigated.
There are no reports of any teratogenic effects in humans and an
extrapolation of animal data to humans is somewhat problematical (Kolb
Meyers, 1983; Mottet, 1985).
Reviews of more than 20 cases of thallium intoxication during
pregnancy by Petersohn (1960), Moeschlin (1965), Stevens & Barbier
(1976), Graben et al. (1980) and Barlow & Sullivan (1982) can be
summarized as follows: all attempts at illegal abortion were in vain;
the prolonged use of a depilatory cream seems to have been the cause
of 1 neonatal death. Two attempts at illegal abortion with thallium
in the first trimester of pregnancy did not affect the development of
the fetuses, although rather low birth weights were recorded. In four
additional cases of intoxication during this period of pregnancy, the
outcome was not reported.
Intoxication occurring after the first trimester can induce in
the newborn baby some symptoms of acute intoxication seen in adults,
e.g., rash and alopecia. Two babies born after the intoxication of
their mothers in the 5th and 6th months of pregnancy showed reduced
weight or no effects, respectively. Also no effects were found in a
case of intoxication (0.35 g thallium) in the 7th month of pregnancy
or in an additional six cases. However, in two cases during this
period (0.15 g thallium in one case), premature births occurred,
showing alopecia areata and low weight of one baby. Alopecia areata
and lunular stripes in the nails were observed in two newborn babies.
Low birth weight was common.
Petersohn (1960) reported an attempt at illegal abortion by
ingesting 0.5 g thallium 8 weeks before term. However, the fetus
developed normally. The child had well-developed hair and apart from
being relatively underweight showed no signs of thallium poisoning,
whereas the mother developed alopecia and polyneuritis (Erbslöh,
1960). A suicide attempt with about 1.2 g thallium 2 days before
birth caused the death of a newborn girl after 5 days; fresh blood in
the faeces was observed from the 3rd day onwards.
In the population living around the cement plant in Lengerich,
Germany, 300 women gave birth in the years 1978 and 1979. Eleven
children exhibited congenital malformations or abnormalities, five
showing major malformations (e.g., cleft lip and palate, clubfoot, hip
dislocation and ventricular septum defect). The rate of malformation
was higher than expected, but the authors suggested that the real
frequency of malformation in unaffected populations is underestimated.
It was difficult to correlate the effects with the intensity of
exposure, since the degree of exposure to which the mothers were
subjected during pregnancy could not be ascertained (Dolgner et al.,
1983). It should be noted that the fathers were not included in the
investigations. Embryotoxic effects were not considered in the
investigation at Lengerich (Claussen et al., 1981).
8.5.2 Carcinogenicity
The carcinogenicity of thallium has not been adequately evaluated
in humans. A study by Marcus (1985) on occupationally exposed workers
showed that the incidence of benign neoplasms (not further
characterized) was not significantly increased in the workers.
However, only 86 thallium-exposed and 79 controls were included in
this study and the length of observation time was not stated. The
study was also limited by the availability of medical records. Other
reports involving human exposure to thallium did not include an
investigation of carcinogenicity.
8.5.3 Immunotoxicological effects
Reduced resistance against secondary infections has been reported
only by Moeschlin (1965), but actual data on the possible
immunological effects of thallium are not available.
8.6 Factors modifying toxicity: enhancement of elimination
In studies on laboratory mammals (section 7.10.1) and in tests
with patients, enhancement of elimination was attempted (Stevens et
al., 1974). This might be achieved, provided that the thallium is not
fixed intracellularly (Barckow & Jenss, 1976). Sodium salts were
previously used as an antidote for human thallotoxicosis (Munch,
1934a), but intravenous injection of sodium thiosulfate (Sessions &
Goren, 1947) often increased the severity of symptoms (Munch, 1934a).
Although increased urinary elimination of thallium theoretically
should reduce its fatal effects, treatment with potassium salts caused
a worsening of the symptoms of thallotoxicosis in humans (Papp et al.,
1969). This was presumably due to a mobilization of intracellular
thallium, an increase in plasma levels, and redistribution (Bank et
al., 1972; Gastel, 1978).
Dithizone has also been used to treat cases of human poisoning
(summarized by Bendl, 1969 and Papp et al., 1969), in spite of its
goitrogenic and perhaps diabetogenic effects in experimental animals.
Clinical therapy with dithizone is often more effective than treatment
with potassium chloride and charcoal (Paulson et al., 1972).
Respiratory distress, confusion and diplopia have been cited as
examples of negative side effects by Barckow & Jenss (1976) and were
also reported by Saddique & Peterson (1983), but they were not
attributed to the dithizone treatment by Paulson et al. (1972).
Dithizone presumably mobilizes thallium from the compartments with
maximal concentrations, thus increasing the toxic load of the nervous
system (Cavanagh et al., 1974; Ghezzi & Bozza Marrubini, 1979).
Other agents, D-penicillamine and the chelating
diethyldithio-carbamate ("dithiocarb"), have also been used as
antidotes (Sunderman, 1967; Montoya Cabrera et al., 1979). Dithiocarb
caused a three-fold increase in urinary elimination during therapy of
a woman (Sunderman, 1967). D-penicillamine was used to treat a
patient who initially had 1200 µg thallium/litre in her urine, as well
as two other people with thallium poisoning. The authors emphasized
that no adverse effects occurred (Alarcón-Segovia et al., 1989). In a
detailed comparative survey by Cavanagh et al. (1974), it was stated
that for neither of these two antidotes (nor for several other
antidotes) was formal proof of benefit available. Negative effects of
dithiocarb therapy, such as deterioration of cerebral function, have
been observed in patients (Kamerbeek et al., 1971a).
Haemoperfusion does not affect the course of thallium
intoxication, according to Heath et al. (1983). Successful treatment
by haemodialysis was reported by Barckow & Jenss (1976) and Piazolo et
al. (1971). Elimination of thallium by haemoperfusion or
haemofiltration should be restricted to intoxications with high doses
of thallium during the previous 24 h (Briese & Nessler, 1985b).
A very effective oral antidote in experimental animals and humans
is Prussian Blue, potassium ferric hexacyanoferrate(II), an inorganic
pigment which is not absorbed by the gut (Heydlauf, 1969; Dvorák,
1970; Kamerbeek et al., 1971b; Günther, 1971; Barbier, 1974; Ghezzi &
Bozza Marrubini, 1979; Lehmann & Favari, 1984, 1985). Potassium ions
in the molecule are exchanged for thallium ions. Thus, absorption in
the intestine is prevented and the thallium-loaded molecule is
eliminated with the faeces (Forth & Henning, 1979). This therapy
results in faecal elimination greatly exceeding urinary elimination
(Stevens et al., 1974). Prussian Blue is now the main therapeutic
agent (Forth & Henning, 1979; Lehmann & Favari, 1984; Kazantzis, 1986;
Pai, 1987; Chandler et al., 1990), the colloidally soluble form being
preferable (de Groot & van Heijst, 1988).
Prussian Blue therapy and forced diuresis with furosemide and
mannitol (10 g of the soluble form dissolved in 100 ml 1.5% mannitol
as a laxative, twice daily orally or intraduodenally, until urinary
thallium elimination is < 0.6 mg/24 h), perhaps supplemented by
haemodialysis, is currently considered the optimal therapy for
thallium intoxication (Barckow & Jenss, 1976; Forth & Henning, 1979;
Briese & Nessler, 1985b; Chandler & Scott, 1986; Wainwright et al.,
1988; IPCS, 1992; Aderjan et al., 1994). If Prussian Blue is not
available, activated charcoal can be used (IPCS, 1992). The effects
on target organs, for instance neurotoxic effects, must be treated
symptomatically (Forth & Henning, 1979; Kemper, 1979; Briese &
Nessler, 1985b). In laboratory experiments on rats the hepatotoxicity
of thallium was prevented by treatment with silymarin, which has been
shown to have a hepatoprotective effect in man against several toxic
substances (Mourelle et al., 1988).
8.7 Protective measures against excessive occupational exposure
The high toxic potency of thallium has been considered in its TLV
or MAK value (threshold limit value or maximum concentration at the
workplace) of 0.1 mg/m3 (Schaller et al., 1980; MT, 1983; Marcus,
1985; DFG, 1990). This value is the limit for a 40-h working week in
the USA, France, Germany, United Kingdom and other western countries,
and has been reduced in the former-USSR to 0.01 mg/m3 air (Sabbioni
& Manzo, 1980; Nessler, 1985b). The MAK value is the mean value
during the normal 8-h working day, and, during this period, only once
may a ten-fold higher concentration occur for a period of 30 min (DFG,
1990). According to the West German General Administration Regulation
on Air Pollution Control, thallium concentration in dust fall-out
should not exceed 0.01 mg/m2 per day (Ohnesorge, 1985; Ewers, 1988).
On the basis of several reports of recommendations for the
protection of employees in industrial plants using thallium, e.g., by
Hill & Murphy (1959), Malcolm (1979), Glomme (1983), Nessler (1985b)
and a very detailed one by Sessions & Goren (1947), the following
protective measures are advisable.
a) General recommendations
i) Access to rooms in which thallium is used should be restricted
to a limited number of employees.
ii) Employees should repeatedly be informed about risk and
industrial hygiene, in a similar way to employees working with
radioisotopes. They should be instructed to report any unusual
health symptoms.
iii) Employees should be encouraged to eat potassium-rich food.
b) Medical control
i) By means of a preplacement examination, people suffering from
renal, hepatic or neurological diseases, anaemia, blood
dyscrasias, hypertension, alcoholism, chronic infections of
endocrine gland dysfunction should be excluded from working with
thallium.
ii) Urinary thallium should be periodically determined as a means of
showing the effects of education programmes and improving
industrial hygiene. The intervals will depend on the degree of
exposure.
iii) Periodic examinations should pay particular attention to the
early toxic effects of thallium, e.g., renal function,
gastrointestinal disturbances, the presence of paraesthesia and
alopecia.
c) Engineering control
i) Dust scattering should be avoided and handling of thallium
should be conducted under exhaust ventilation.
ii) Floors and tables should be wet-mopped.
iii) Dust samplers should be installed for environmental monitoring
to permit the evaluation of possible sources of contamination.
d) Personal protective equipment and hygienic measures
i) Employees should be required to use protective work clothes
including gloves.
ii) When indicated, personal exposure monitoring should be
performed.
iii) Complete sets of personal work clothes should be kept in
accommodation separate from that employed for street clothes.
Before changing clothes, gloves should be thoroughly washed and
then hands, using separate towels.
iv) Depending on the level of exposure, work clothing should be
washed periodically.
v) Clothes should be changed before eating, drinking and smoking,
all of which should be prohibited at the workplace.
vi) Washing and shower facilities should be provided and their use
enforced.
vii) Individual respirators should be worn in all operations
producing dust or fumes.
9. EFFECTS ON OTHER ORGANISMS IN THE LABORATORY AND FIELD
Since the majority of results have been obtained in laboratory
experiments, field observations on plants (mainly near the cement
plant in Lengerich) have not been considered under a special
subheading but have been included in sections 4.1.2.2, 9.3.1.2 and
9.3.1.6, and those on vertebrates have been included in sections 9.3.2
and 9.3.3.
The toxicity of thallium has been considered to be comparable
with that of mercury and lead, which are its neighbours in the
Periodic Table (Emsley, 1978).
9.1 Microorganisms
Buschke & Jacobsohn (1922) observed a sterilizing effect of
metallic thallium on various bacteria placed on agar plates. The
antibiotic effect of thallium was also recognized during postmortem
examination of thallium-poisoned humans, which were less decomposed
than other corpses (Muller, 1961).
The most important effect of thallium on microorganisms seems to
be the complete or partial inhibition of nitrate formation by
Nitrobacter agilis, observed at concentrations of 0.8 to 16 mg
thallium/litre (Tandon & Mishra, 1969). However, nitrification in
soils was only reduced at high thallium concentrations, which also
affected plants considerably (McCool, 1933). Direct effects on soil
microflora were demonstrated by Drucker et al. (1979). The numbers of
total aerobic bacteria and the distribution of other microorganisms
were affected at concentrations as low as 1 and 10 mg/kg soil, whereas
soil respiration was only reduced at 100 mg/kg soil. Thallium was
less toxic than silver, mercury, chromium, cadmium, copper, nickel and
zinc (in descending order of toxicity) but more toxic than other
elements, e.g., arsenic and lead (Drucker et al., 1979). However, the
properties of the soil and the form of thallium used in this study
were not reported.
The only known positive effect of thallium on organisms has been
described by Richards (1932), who obtained a higher yield of yeast in
the presence of 0.1, 1 or 10 mg thallium/litre culture medium. Higher
concentrations inhibited the growth of Saccharomyces cerevisiae.
This positive effect of thallium needs to be verified, since it could
have been caused by contamination of the thallium, which would not
have been detected by the analytical methods available at that time.
Using the same microorganism, Norris et al. (1976) found a growth
inhibition of 50% after the addition of 153 mg/litre to liquid culture
(which contained 31.3 g potassium/litre) or 10.2 mg/litre to agar
medium. The same inhibitory effect with Escherichia coli was obtained
after the addition of 184.0 mg/litre liquid culture or 10.2 mg/litre
agar and with Bacillus megaterium after adding 3.1 or 5.1 mg/litre,
respectively. The concentrations of thallium (added to the agar
medium), which prevented colony formation, i.e. 15.3, 20.4 and
40.8 mg/litre for B. megaterium, S. cerevisiae and E. coli,
respectively, also show interspecies differences. Constant thallium
concentrations of about 30% of those concentrations which inhibited
growth rates by 50%, but with lower concentrations of potassium in the
liquid medium, caused a decrease in the growth rates of B. megaterium
and S. cerevisiae (Norris et al., 1976).
Potassium/thallium antagonism has been observed using
Thiobacillus ferrooxidans. Iron oxidation by growing cultures in a
potassium-free medium, but with 0.204 to 204.39 mg thallium (as
sulfate) per litre, was normal at 20.4 mg thallium/litre but inhibited
by 204.39 mg/litre. However, this inhibitory concentration did not
affect iron oxidation in the normal medium containing about 180 mg
potassium/litre (Tuovinen & Kelly, 1974).
In inhibitory tests with Azotobacter chroococcum and
A. vinelandii on agar plates, 20 mg thallium sulfate produced very
strong inhibition, comparable to that of gold or chromate, and
stronger than that of zinc or copper (den Dooren de Jong & Roman,
1971). The two latter heavy metals were found to be also less
inhibitory in tests using the bacterium Klebsiella pneumoniae
(Sikka et al., 1987). However, in a previous study this bacterium was
much more sensitive to sulfates of zinc, copper and cadmium than to
that of thallium (Wilson & Dean, 1977). The different toxicity
mechanisms were also shown by strains resistant to these heavy metals.
Thallium-resistant strains showed the opposite reaction and were more
sensitive to gentamycin and chloramphenicol than the wild strain. In
liquid medium, up to 0.2 mg thallium/litre reduced the doubling time
(correlated to the concentration) but induced no lag, whereas higher
concentrations also induced a lag of initial development. On agar
plates, concentrations of up to 0.3 g/litre were not toxic, but with
0.4 g/litre bacterial colonies developed only rarely. A reduction in
the potassium concentrations increased the toxicity of thallium in
both media. A synergistic effect, and not a purely additive effect,
with zinc and cadmium occurred only at concentrations above
0.1 g/litre (Wilson & Dean, 1977).
The enhanced isolation of mycoplasms in the presence of
considerable numbers of other bacteria from the human urogenital tract
(Tully et al., 1983) might be caused by a species-specific differences
in the sensitivity to thallium, which had been added to the culture
medium in addition to penicillin. Such a selective cultivation of
bacteria through the addition of different concentrations of thallium
to the agar can be used as a taxonomic criterion and should be
considered when using clinical material (Kunze 1972a,b).
Different toxicities of thallium(I), thallium(III) and
organothallium compounds were shown in a study on bacteria and fungi
by Srivastava et al. (1973). At low concentrations (1 and
5 mg/litre), the growth of Colletotrichum falcatum, the causative
fungus of a sugarcane disease, was affected more by thallium(III)
chloride than by thallium(I) chloride. However, at higher
concentrations the effect of the thallium(III) compound did not
increase, and the fungus was affected more by the monovalent thallium.
With both compounds the inhibition did not exceed 20% compared to
controls, even after the addition of 80 mg/kg. The organothallium
chlorides (diphenyl derivatives) were considerably more toxic than the
thallium chlorides, causing a reduction of mycelial growth of 50%,
even at concentrations of 13 or 16 mg/kg. This inhibition was
obtained with < 1 mg/kg for most ditolyl derivatives (Srivastava et
al., 1973).
Five fungal species of the genus Aspergillus were similarly
sensitive to thallium nitrate. Their sensitivity to cadmium and
mercury nitrate was similar, but they were less sensitive to the
nitrates of nine other metals (Filimonova et al., 1973).
Anaerobic bacteria are more sensitive to thallium(I) than to
organothallium compounds (Huber et al., 1978). In several cases the
inhibition of bacterial growth was greater at lower concentrations of
the organothallium compounds.
9.2 Aquatic organisms
Little is known about the toxicology of thallium in aquatic
systems. Wachs (1988) classified thallium, together with lead and
zinc, into the toxic class II. Because of a lack of complex
formation, its toxicity is not affected by water hardness, copper,
etc. (Zitko et al., 1975). Acute and chronic toxicities to freshwater
aquatic life are reported to occur at 1400 and 40 µg/litre,
respectively. One species of fish is even affected at 20 µg/litre
after 2600 h of exposure. Marine aquatic life seems to be affected at
2130 µg/litre, and as in the case of freshwater organisms, it is clear
that more sensitive species than those tested might exist (US EPA,
1980).
9.2.1 Plants
Results from studies on algae and higher aquatic plants are given
in Table 26. Photosynthesis is affected, as shown by the reduced
oxygen evolution of the algal endosymbiont of the ciliate Paramecium
bursaria (Di Gaudio & Hirshfield, 1976). In comparison to other
heavy metals, higher concentrations of thallium were needed to reduce
the light-induced oxygen evolution of the freshwater alga
Chlamydomonas reinhardii (Overnell, 1975); about 2 mg thallium/litre
buffer caused a reduction of about 50%. Thallium strongly affects
NADP reduction or the dark stage reactions of photosynthesis.
However, it seems to inhibit photosystem I only slightly, and this
could not be confirmed in a very detailed later investigation with the
green alga Chlorella saccharophila, which also showed a reduced
oxygen evolution (Wystrcil et al., 1987). Measurements of the
variable fluorescence indicated a primary action of thallium on
electron transfer in photosystem II. At low concentrations of
thallium (2 mg/litre), disturbances of the thylakoid membranes could
explain the altered variable fluorescence. A change in the colour of
the algal suspension to a greyish green and the alterations of the
fluorescence intensity indicated effects on the pigments (Wystrcil et
al., 1987). This has also been found for the alga Chlamydomonas
reinhardtii. ß-Carotin showed the greatest sensitivity, followed by
chlorophyll a and then chlorophyll b (Maier-Reiter et al., 1987). In
addition to an increase in the degradation rate of the pigments, the
biosynthesis rate was reduced (section 9.3.1.2).
In a comparison of the toxicity of seven heavy metals for the
marine diatom Ditylum brightwellii, thallium (thallium(I) chloride)
possessed the lowest toxicity, which was not affected by the chelating
agent EDTA (Canterford & Canterford, 1980). Within 5 days a 50%
growth reduction was obtained with 330 to 350 µg total thallium/litre,
corresponding to 0.15-0.17 mg free thallium per litre. To obtain
total inhibition, concentrations of about 2.2 times higher were needed
(Table 26), but the cells showed an abnormal appearance, i.e.
pseudo-resting spore-like cells and shrinkage of protoplast and
concentration of chromophores, within about 3 days (Canterford &
Canterford, 1980; Canterford, 1980).
Species-specific sensitivities, but also different effects of the
monovalent and the trivalent forms of thallium, were evident in two
marine algae, Phaeodactylum tricornutum and Dunaliella tertiolecta
(Table 26) (Puddu et al., 1985, 1988). Complex formation with EDTA
reduced the toxicity of the thallium(III) compound. Using different
strains of three acidophilic algae, the type and level of inhibition
of growth after culture in a medium containing thallium sulfate was
very similar in the strains of Cyanidium caldarium and Cyanidioschyzon
merolae, whereas strains of Galdieria sulphuraria showed different
effects (Albertano & Pinto, 1986).
Generally, freshwater algae are affected at concentrations as low
as 100 µg/litre (US EPA, 1980). In the submerged macrophyte Elodea
canadensis, 1.4 and 2.8 mg thallium/litre water (as thallium(I)
sulfate) reduced the photosynthetic oxygen evolution during 24 h by
50% and 90%, respectively (Brown & Rattigan, 1979). In parallel
experiments, the free-floating Lemna minor was much more sensitive
(Table 26). The uptake and accumulation of thallium (thallium(I)
acetate) and its effects on growth parameters of L. minor (frond area,
Table 26. Toxicity of thalliuma to aquatic plants
Species Parameter Exposure time Concentration of thalliumb Reference
LOELc EC50d TECe
Algae
Chlamydomonas oxygen 0.25 h 2 Overnell (1975)
reinhardii evolution
C. reinhardii concentration 22 h > 0.2 20 Maier-Reiter et al. (1987)
of pigments
Ditylum brightwellii growth 5 days 0.34 0.73 Canterford & Canterford (1980)
Dunaliella tertiolecta growth - 0.08 Puddu et al. (1988)
0.18g
D. tertiolecta growth - 0.18g Puddu et al. (1985)
Elodea canadensis damage 28 days 2.0 Brown & Rattigan (1979)
E. canadensis oxygen evolution 24 h 1.4 Brown & Rattigan (1979)
Phaeodactylum growth - 0.14 Puddu et al. (1988)
tricornutum 0.22g
P. tricornutum growth - 0.24g Puddu et al. (1985)
Table 26 (contd).
Species Parameter Exposure time Concentration of thalliumb Reference
LOELc EC50d TECe
Selenastrum concentration 96 h 0.11 US EPA (1978)
capricornutum of pigments
S. capricornutum growth 96 h 0.10 US EPA (1978)
Higher Plants
Lemna minor growth (weight) 10 days 0.014 0.04 Kwan & Smith (1988)
281.5h 510.0h
L. minor growth 10 days 0.016 0.047 Kwan & Smith (1988)
(frond number) 293.8h 550.1h
L. minor growth 10 days 0.008 0.033 Kwan & Smith (1988)
(frond area) 195.8h 443.7h
L. minor damage 28 days 0.008 Brown & Rattigan (1979)
a Thallium(I), unless otherwise stated
b mg thallium/litre nutrition solution, unless otherwise stated
c LOEL = low-observed-effect level
d EC50 = concentration at which the life parameters were reduced by 50%
e TEC = concentration at which life parameters were totally inhibited
f - = no data given
g Thallium(III)
h mg thallium/kg dry weight of plant
fresh weight, frond number) were determined by Kwan & Smith (1988).
High concentrations of 1 and 2 mg/litre water induced chlorosis by the
eighth day, and 2 days later the fronds were completely devoid of any
colour. Comparing the number of fronds at different concentrations of
thallium during a period of 10 days, a stimulatory effect was evident
after exposure to 2 µg/litre and 10 µg/litre at the end of the period.
However, smaller fronds were produced and, therefore, the surface area
covered by Lemna also decreased at the lowest thallium concentrations.
At 20 µg/litre or more, all growth parameters were reduced.
Subsequent culture in thallium-free water resulted in good recovery
from the previous thallium exposure, provided that the concentration
had not exceeded 20 µg/litre. The bioconcentration factor (based on
plant weight) was 88 000 at the lowest exposure (2 µg/litre) and 6000
at an exposure of 153 µg/litre.
Using 40 µg/litre growth medium, the thallium concentrations in
L. minor did not further increase after 140 h, and over 80% of the
thallium remained in the plant (Smith & Kwan, 1989). In plant
homogenates, thallium showed little association with proteins, and the
reactions of the thallium in the soluble fraction were comparable to
those of the free metal ion. Like potassium, thallium accumulates in
the cell vacuole. When various concentrations of thallium were added
to the medium (0.02 to 0.2 mg/litre), about 0.04 mg/litre caused a 50%
reduction of growth after a period of 10 days (Smith & Kwan, 1989).
Since concentrations of 0.05 to 0.09 mg/litre were found in rivers
contaminated by mining (Zitko et al., 1975), significant effects on
macrophytes in such rivers would be expected, given the long exposure
period.
9.2.2 Animals
Toxicity data on aquatic animals are summarized in Table 27. In
an immobilization test on Daphnia magna, it was shown that the
toxicity of thallium(I) nitrate was higher than that of nickel or
cadmium but lower than that of copper, silver or mercury (Bringmann &
Kühn, 1982). In general, arthropods were affected at lower
concentrations than fish. The 96-h LC50 values were much lower for
the daphnids (water fleas) Daphnia magna and Mysidopsis bahia
(2.2 mg/litre) than for the freshwater fish Lepomis macrochirus
(bluegill) (120 mg/litre), although 50% of fathead minnows (Pimephales
promelas) were killed by 0.86 mg/litre (LeBlanc, 1984).
Comparing vertebrate with invertebrate marine species, about 10%
of the concentration needed to kill 50% of sheepshead minnows or
tidewater silversides was sufficient to kill 50% of marine shrimps (US
EPA, 1980). Acute values were 3- to 32-fold higher than chronic
values (US EPA, 1980).
Considerable differences between species were also evident in a
study by Nehring (1962/63). This study also showed that in perch
(Perca fluviatilis) lethal thallium effects depend on the length of
exposure. The fish were killed by 200 or 500 mg/litre within 10 h,
while concentrations of 100, 90-50, 40 and 20 mg/litre caused death
within 2, 2.5-3.5, 7.5 and 14 days, respectively. At 15 mg/litre, the
perch survived at least 17 days. Trout (Salmo gairdneri) and roach
(Rutilus rutilus) were more sensitive and died within 8 and 14 days,
respectively, at 4 mg/litre. In these species no effects were
observed within 17 days at concentrations of 2 mg/litre.
Juvenile Atlantic salmon (Salmo salar) are exceptionally
sensitive to thallium contamination (Zitko et al., 1975). The 18-day
LC50 is 0.1 mg/litre. The mortality of fish exposed to 0.03 mg/litre
was reported to be equal to that of the controls. The authors
suggested that 0.02 mg/litre be regarded as a no-observed-effect level
(NOEL), and that 0.04 mg/litre be regarded as a low-observed-effect
level (LOEL). Mixtures with copper or zinc did not alter thallium
toxicity.
Behavioural alterations in perch were reported by Nehring
(1962/63), even at low concentrations. Initially, following exposure
to thallium, food consumption increased, but after one or two days
neuronal damage occurred. This was indicated by uncoordinated
movement, paralysis of gills and disturbance of balance. Similar
effects on movement and respiration in fish had already been observed
by Swain & Bateman (1909/10).
Larvae of fathead minnows (Pimephales promelas) were much more
sensitive than the embryos to thallium sulfate (LeBlanc & Dean, 1984).
No effect on the rate of hatching was observed at concentrations up to
200 µg/litre, but at 720 µg/litre no hatching occurred. At
350 µg/litre no larvae survived. Growth was affected at 120 µg/litre.
In a study by Birge (1978), rainbow trout (Salmo gairdneri) eggs
were exposed to thallium from fertilization to 4 days after hatching
(total of 28 days). The exposure water was renewed every 12 h. An
LC50 of 0.17 mg/litre and an LC1 of 0.0084 mg/litre were reported.
Goldfish (Carassius auratus) eggs were also exposed to thallium from
fertilization to 4 days after hatching (total of 7 days). The
exposure water was renewed every 12 h. An LC50 of 7.00 and an LC1
of 0.0525 mg/litre were reported (Birge, 1978).
Table 27. Toxicity of thalliuma to aquatic animals
Species Stage Parameter Exposure time Concentration of thallium Reference
LOELb EC50c TECd
Invertebrates
Daphnids -e survival 48 h 2.2 LeBlanc (1984)
Daphnia magna juvenile mobility 24 h > 0.003 0.11 0.95 Bringmann & Kühn (1982)
Daphnia sp. adult survival 72 h 2-4 Nehring (1962/63)
Gammarus sp. adult survival 72 h 4 Nehring (1962/63)
Mysid shrimp survival 96 h 2.1 US EPA (1978)
(Mysidopsis bahia)
Vertebrates
Atlantic salmon juvenile survival 47 h 10 Zitko et al. (1975)
(Salmo salar) 112 h 1
435 h 0.1
2600 h 0.03
Sheepshead minnow adult survival 96 h 20.9 US EPA (1978)
(Cyprinodon variegatus)
Bluegill adult survival 96 h 132 US EPA (1980)
(Lepomis macrochirus)
adult survival 96 h 120 LeBlanc (1984)
Table 27 (contd).
Species Stage Parameter Exposure time Concentration of thallium Reference
LOELb EC50c TECd
Perch adult survival 72 h 60 Nehring (1962/63)
(Perca fluviatilis)
Fathead minnow embryo hatch > 0.2 < 0.72 LeBlanc & Dean (1984)
(Pimephales promelas) larva survival 30 day < 0.04 < 0.35
larva growth 30 day > 0.04 > 0.2
adult survival 96 h 0.86 LeBlanc (1984)
adult survival 96 h 0.08 US EPA (1980)
adult survival 96 h 1.8 US EPA (1980)
Roach adult survival 72 h 40-60 Nehring (1962/63)
(Rutilus rutilus)
Rainbow trout adult survival 72 h 10-15 Nehring (1962/63)
(Salmo gairdneri)
Tidewater silverside survival 96 h 24 Dawson et al. (1975/77)
(Mendia berrylina)
Toad adult survival - 16.7g Swain & Bateman (1909/10)
a mg thallium/litre water
b LOEL = low-observed-effect level
c EC50 = concentration at which the life parameters were reduced by 50%
d TEC = concentration at which life parameters were totally inhibited
e - = data not given
f 12 days in experiments with other species
g mg/kg injected into a lymph sinus
Amphibia are also affected by thallium. Development of frog
spawn was unaffected by concentrations of 40.8 and 200 mg/litre, but a
concentration of 0.4 mg/litre killed all tadpoles on hatching (Dilling
& Healey, 1926). This indicates that the absorption of thallium by
the eggs was minimal. Injections of lethal concentrations of thallium
acetate (> 0.005 g) into the lymph sinus of adult toads (Table 27)
caused loss of control of the hindlimbs and death by asphyxia (Swain &
Bateman, 1909/10). In a study on the narrow-mouth toad (Gastrophryne
carolinensis), eggs were exposed to thallium from fertilization to 4
days post-hatch (total 7 days). The exposure water was changed every
12 h. An LC50 of 0.11 and an LC1 of 0.0024 mg/litre were reported
(Birge, 1978).
9.3 Terrestrial organisms
9.3.1 Plants
Early investigations into the toxicity of thallium to plants were
summarized by Scharrer (1955). The most obvious effects are decreased
productivity, inhibition of photosynthesis and direct cytotoxicity.
Toxicity data are listed in Table 28.
9.3.1.1 Plant photosynthesis
In a study by Bazzaz et al. (1974), the net photosynthesis of
excised tops of sunflowers (Helianthus annuus) decreased both with
time and with the concentration of thallium in the nutrient solution
(2 to 200 mg/litre). At the highest concentration, photosynthesis was
inhibited by about 70% after 1 day, and the plants began to wilt. At
the lower concentrations, these visible symptoms appeared 4 days
later. There was a strong log-linear relationship between
photosynthesis and the thallium content of the plants. Stomatal
opening was reduced by 30 and 90%, respectively, at concentrations of
0.2 and 2 mg thallium/litre, but increased concentrations only caused
a slight additional effect (Bazzaz et al., 1974). In a direct
comparison Carlson et al. (1975) observed that the effects of thallium
sulfate on photosynthesis and transpiration in sunflowers were similar
to those in maize (Zea mays) at low thallium concentrations (up to
2 mg thallium/litre solution). Higher concentrations, up to
10 mg/litre, induced further inhibition in maize but not in
sunflowers. Using the data of Carlson et al. (1975), there was a good
linear correlation for maize between the occurrence of stomatal
opening (y) and thallium content of the solution (x):
y = 66.4 - 0.4 x; regression coefficient = 0.93
This was greater than the correlation calculated using the data for
sunflowers:
y = 45.1 - 0.4 x; regression coefficient = 0.66.
Table 28. Toxicity of thalliuma to terrestrial plantsb
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Brassica napus shoot growth 10 days > 20h,n Makridis & Amberger (1989b)
B. napus shoot growth 18 days 800i,n Makridis & Amberger (1989b)
< 2h,n
B. napus shoot growth 28 days > 2669 Allus et al. (1987)
> 10.0h
B. napus root growth 28 days > 760 Allus et al. (1987)
> 10.0h
Cucumis sativus growth 10 days < 10h Puerner & Siegel (1972)
Garden lettuce growth 7 days > 100 Schweiger & Hoffmann (1983)
10h
Garden lettuce growth summer 30 Hoffmann et al. (1982)
< 10k > 500k
Green kale growth 7 days > 500 Schweiger & Hoffmann (1983)
10h
Helianthus annuusg photosynthesis 4-5 days 63 Bazzaz et al. (1974)
(sunflower)
H. annuus photosynthesis 4-9 days 82 Carlson et al. (1975)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
H. annuus growth 7 days > 100 Schweiger & Hoffmann (1983)
10h
H. annuusg stomata opening 8 h < 0.2h approx. Bazzaz et al. (1974)
0.8h
Hordeum vulgare shoot growth 20 Davis et al. (1978)
0.5h
H. vulgare shoot growth 28 days > 21 Allus et al. (1987)
< 0.2h
H. vulgare root growth 28 days < 86 Allus et al. (1987)
< 0.2h
Kohlrabi growth summer > 600 Hoffmann et al. (1982)
(young) 500k
Lolium perenne shoot growth 21 days 0.71 251.2 Al-Attar et al. (1988)
L. perenne root growth 21 days 2.1 1990 Al-Attar et al. (1988)
Nicotiana tabacump survival 24 h 0.02h Siegel (1977)
N. tabacum germination 24 h 0.02h Siegel (1977)
Phaseolus vulgaris growth 33 days < 0.55h Kaplan et al. (1990)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
P. vulgaris shoot growth 10 days 130i,n Makridis & Amberger (1989b)
< 1h,n
Pisum sativum stem growth 28 days 5-10 Pötsch & Austenfeld (1985)
210-360h
5-10n Pieper & Austenfeld (1985)
115-123h,n
Pisum sativum leaf growth 28 days 1-5 Pötsch & Austenfeld (1985)
30-75h
> 10n Pieper & Austenfeld (1985)
30-43h,n
P. sativum root growth 28 days > 10 Pötsch & Austenfeld (1985)
> 180h
> 10n Pieper & Austenfeld (1985)
> 80h,n
Radishl growth summer 35 Hoffmann et al. (1982)
< 500k
Radishm growth summer < 300
> 500k
Rape growth summer > 500 Hoffmann et al. (1982)
200k 500k
Spinach concentration 14 days < 150 Maier-Reiter et al. (1987)
of pigments > 0.2h
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Spinach growth 9 days 280i Schweiger & Hoffmann (1983)
2h,i
Vicia faba stem growth 28 days > 10 Pötsch & Austenfeld (1985)
> 222h
5-10n Pieper & Austenfeld (1985)
36-76h,n
V. faba leaf growth 28 days > 10 Pötsch & Austenfeld (1985)
> 8h
5-10n Pieper & Austenfeld (1985)
5-7h,n
V. faba root growth 28 days > 10 Pötsch & Austenfeld (1985)
> 1320h
> 10n Pieper & Austenfeld (1985)
> 575h,n
Zea mays growth 7 days > 100 Schweiger & Hoffmann (1983)
(corn) 10h
Z. mays root growth 28 days 1h Logan et al. (1984)
Z. mays shoot growth 28 days 1h Logan et al. (1984)
Z. mays root growth 28 days 1h,n Logan et al. (1984)
Z. mays shoot growth 28 days 1h,n Logan et al. (1984)
Table 28 (contd).
Species Parameter Exposure time Concentration of thalliumc Reference
LOELd EC50e TECf
Z. mays photosynthesis 4-9 days 82 Carlson et al. (1975)
Z. mayso stomata opening 8 h 2h Carlson et al. (1975)
a Thallium(I), unless otherwise stated
b Whole plants, unless otherwise stated
c mg thallium/kg dry weight of plant tissue, unless otherwise stated
d LOEL = low-observed-effect level
e EC50 = concentration at which the life parameters were reduced by 50%
f TEC = concentration at which life parameters were totally inhibited
g Tops
h mg thallium/litre nutrition solution
i Concentration at which the life parameters were reduced by 10%
k mg thallium/kg soil
l Root
m Leaf
n Thallium(III)
o Epiderm
p Protoplast
q Seed
The effects of thallium on the photosynthesis of spinach
chloroplasts have been investigated by Wystrcil et al. (1987). Some
alterations of variable fluorescence indicated a primary action of
thallium on electron transfer in photosystem II, which was also
evident in green algae (section 9.2.1). In photosystem I, superoxide
dismutase might also be affected by thallium (Wystrcil et al., 1987).
9.3.1.2 Cytotoxic effects
Chlorosis, followed by marginal necrosis of the leaves, is the
most prominent sign of thallium toxicity in plants. Different courses
of thallium poisoning in various plant species were reported by McCool
(1933) and later by Spencer (1937) in tobacco, by Carlson et al.
(1975) in corn and sunflowers, by Davis et al. (1978) in barley, by
Makridis & Amberger (1989b) in bushbeans and rape, and by Kaplan et
al. (1990) in beans. Similar observations on the leaves of trees in
the vicinity of the cement plant in Lengerich, Germany were reported
by LIS (1980).
The course and location of chlorosis seemingly depend on thallium
concentrations in the substrate and presumably correspond to the
distribution of thallium in the plant (Schweiger & Hoffmann, 1983).
In isolated protoplasts of Nicotiana tabacum, a cytotoxic
effect was also observed; 10% and 50% had lysed after a 24-h
incubation in 4 (± 0.4) and 20 (± 2) µg thallium/litre, respectively,
irrespective of the age of the protoplasts (Siegel, 1977). These
values are nearly identical to the percentages of seed in which
germination was inhibited (Siegel, 1977).
Chlorosis indicates a reduced concentration of pigments (section
9.2.1). In spinach, it is firstly the concentration of ß-carotene
which is reduced, then that of chlorophyll a and finally that of
chlorophyll b. The concentrations of ß-carotene and chlorophyll a
were about half the normal value after 2 weeks of incubation in a
hydroculture medium containing thallium nitrate (0.2 mg
thallium/litre) (Maier-Reiter et al., 1987).
9.3.1.3 Growth of plants
Adverse effects of thallium on the growth of plants have been
reported for various test systems (Table 28). In a comparison of the
effects of three heavy metals on the growth of cabbage seedlings,
cadmium and thallium were found to be less toxic than silver (Allus et
al., 1988).
Initial mycelial growth of three fungal species was inhibited on
agar plates containing 0.25 or 0.50 mg thallium/litre (Seeger & Gross,
1981).
In tobacco plants, concentrations as low as 5 mg thallium(I) per
litre inhibited terminal growth and caused a temporary outgrowth of
axillary buds all resembling natural frenching, i.e. a reticulate
interveinal chlorosis. In hydrocultures the root system was strongly
affected after 12 days at 0.067 mg thallium/litre. Thallium(I)
nitrate and sulfate were similarly toxic. The toxic effects of lower
concentrations were reduced by the addition of aluminium sulfate,
nitrogen and potassium iodide. In other species of Nicotiana, only
terminal growth, chlorophyll formation or roots were affected, and the
level of sensitivity to thallium corresponded to the level of
susceptibility to frenching in the field (Spencer, 1937).
After being watered for 15 days with 20 or 200 mg thallium per
litre, the growth of cucumber seedlings was unaffected, but growth was
reduced by 2000 mg thallium/litre. Toxicity was increased by limiting
the uptake of potassium. The higher sensitivity of the epicotyls
compared to the hypocotyls indicated that cell multiplication
processes are more sensitive than those entailing cell enlargement and
differentiation, a phenomenon known from many other stress factors and
toxic substances (Siegel & Siegel, 1976). In corn, production of top
and root biomass was severely reduced to between 50 and 60% of the
controls (Carlson et al., 1975).
From the differential reduction in weight in parts of the bean
plant, it has been shown to be possible to rank them according to
their increasing sensitivity to thallium(I): roots >> upper leaves
> lower leaves = upper stems > lower stems. Results from hydroponic
cultures were similar to those from field studies (Kaplan et al.,
1990). In another variety of bean, the weights of leaves and stems,
but not those of roots, were affected by exposure to thallium(III) (up
to 2 mg/kg). However, thallium(I) had no effect (Pötsch & Austenfeld,
1985; Pieper & Austenfeld, 1985).
Garden lettuce and radish growing in soils treated with TlNO3
exhibited considerably reduced growth at concentrations in dry plant
tissue of 30-35 mg thallium/kg (Hoffmann et al., 1982). Growth of
perennial rye grass (Lolium perenne) was adversely affected when
concentrations of thallium exceeded about 0.7 mg/kg dry weight in
shoots and 2.0 mg/kg dry weight in roots (Al-Attar et al., 1988).
9.3.1.4 Different sensitivities to thallium(I) and thallium(III)
Only small differences were observed between the toxic effects of
thallium(I) and thallium(III) on the dry weight of roots and shoots of
maize. Growth was slightly more reduced after application of
thallium(I) (Logan et al., 1984). Similarly, in two detailed studies
of the effects of thallium(I) and thallium(III) nitrate (0.2, 1, 2 mg
thallium/litre nutrient solution) on the dry weight of pea plants,
growth was found to be affected more after exposure to thallium(I).
However, completely opposite results were obtained for field beans
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985). Following
complexation of thallium(III) and thallium(I) nitrate with EDTA,
plants reacted differently to the two compounds. In bean stems and
roots, but not in leaves, concentrations of thallium were increased by
thallium(III) EDTA compared to those resulting from thallium(III) on
its own, while thallium(I) EDTA resulted in similar or lower thallium
concentrations than thallium(I) on its own in all three plant organs.
In the stems and leaves of peas, thallium(III) EDTA resulted in lower
thallium concentrations than thallium(III) on its own, while in the
roots thallium levels were the same for both salts. Consistently
higher thallium concentrations were found in leaves, stems and roots
of peas after exposure to thallium(I) EDTA, compared to thallium(I) on
its own (Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985).
9.3.1.5 Concentration of trace elements
The effects of thallium on plants could be caused mainly by an
imbalance of essential cellular monovalent cations or by a disturbed
uptake of trace elements (Yopp et al., 1974; Schweiger & Hoffmann,
1983). As can be concluded from the data summarized in Table 29,
thallium seems to have no uniform effect on the trace element content;
the differences between the two investigations using beans are
striking. In most studies the concentration of magnesium was found to
be reduced.
Exposure to thallium(III) chloride reduced the concentrations of
potassium and trace elements such as copper, zinc and iron in bush
beans by up to 20%; calcium, magnesium and manganese were only
slightly affected (Makridis & Amberger, 1989b). Rape was less
sensitive; uptake was decreased for potassium and copper, but
increased for zinc (and for calcium, magnesium, manganese and iron by
the reduced growth).
Complex effects on trace elements were found in studies with
Pisum sativum and Vicia faba in which thallium(I) and thallium(III)
nitrate and their respective EDTA complexes were used (Table 29)
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985).
9.3.1.6 Sensitivity of plants
Differing sensitivities among plant species, strains and
individuals have been reported for a number of air-borne contaminants
(Guderian, 1977). Species differences were also evident in many
investigations of thallium, including the detailed studies carried out
at Lengerich, Germany (LIS, 1980). There the residues of pyrite
roasting had been used for six years before effects were obvious
Table 29. Thallium-induced changes in uptakea or concentrations of trace elements in plants
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Thallium(I)
Compound TlNO3 shoot - - d - - i - d Schweiger & Hoffmann (1983)
Concentrationb 10
Exposure time 7 days
Species sunflower
Compound Tl2SO4 root u u i u d i u i Kaplan et al. (1990)
Concentration 1 leaf ic d u u d u ic ic
Exposure time 33 days
Species bean
Compound TlNO3 root - - u u - d - d Pötsch & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - u
Exposure time 28 days leaf - - u u - u - u
Compound +EDTA root - - u u - d - u
Species bean stem - - u u - u - u
leaf - - u u - u - u
Compound TlNO3 root - - d u - d - d Pötsch & Austenfeld (1985)
Concentration 0.2-2 stem - - u i - d - u
Exposure time 28 days leaf - - u u - u - u
Table 29 (cont'd).
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Compound +EDTA root - - u u - d - u
Species pea stem - - u i - i - u
leaf - - i i - u - i
Thallium(III)
Compound Tl(NO3)3 root - - u u - d - d Pieper & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - i
Exposure time 28 days leaf - - u u - d - u
Compound +EDTA root - - u u - d - u
Species pea stem - - u i - i - u
leaf - - i i - i - i
Compound Tl(NO3)3 root - - u u - d - d Pieper & Austenfeld (1985)
Concentration 0.2-2 stem - - u u - d - u
Exposure time 28 days leaf - - u u - d - d
Compound +EDTA root - - u u - d - u
Species bean stem - - u u - u - u
leaf - - u u - u - u
Compound TlCl3 - i d d u i - d Makridis & Amberger (1989b)
Concentration 10-20
Exposure time 10 days
Species bean
Table 29 (cont'd).
Part of Trace elements Reference
plant
B Ca Cu Fe Mg Mn Mo Zn
Compound TlCl3 - id d id id id - i Makridis & Amberger (1989b)
Concentration 10-20
Exposure time 10 days
Species rape
a d = decrease; u = unchanged; i = increase; - = not determined
b mg/litre solution
c Only upper not lower leaves
d Increase by reduced growth
(Gubernator et al., 1979). Coniferous trees were not affected, oaks
only slightly, and summer lime trees far more than winter ones. Sweet
cherry trees were more sensitive to thallium than sour cherry trees.
The leaves of pear trees still appeared healthy when apple and plum
trees had already lost theirs. Very sensitive vegetables included
beans, cucumber and potatoes. The sensitivity of fodder plants varied
too; the yield of maize was strongly reduced, but not that of rape or
turnip. There seems to be a tendency for plants with a "hard" leaf
surface to be less affected than those with a soft, hairy surface
(LIS, 1980).
Mechanisms of resistance to thallium seem to vary. Comparing the
growth data of beans (section 9.3.1.3) and peas, the higher tolerance
of beans to thallium(I) and thallium(III) (section 9.3.1.4)
corresponds to a higher concentration of thallium in the roots than in
the stems, which in turn contained more thallium than the leaves
(Pötsch & Austenfeld, 1985; Pieper & Austenfeld, 1985). The more
sensitive peas possessed high thallium concentrations in the stems,
followed by those in the roots and leaves. Just the opposite
distribution is evident when barley and rape are compared: a higher
concentration in roots than shoots in the susceptible barley and the
opposite in the tolerant rape (Allus et al., 1987).
Green rape possesses a much higher resistance than beans after
application with thallium(III) chloride. Roots were more sensitive
than shoots and became brown. Using a 2 µg/litre nutrition solution,
the growth of bush beans was increasingly reduced after 3 days. The
first signs of reduced growth of rape were observed after 8 days
incubation in a solution of 5 mg/litre; other symptoms could only be
recognized after the thallium concentration was increased to
10 mg/litre. Then growth of the roots decreased and they became brown
(Makridis & Amberger 1989b). Thus, two mechanisms seem to enable
survival at high thallium concentrations: beans (and perhaps barley,
but not peas) reduce the amount of thallium which is transported to
the leaves, whereas the leaves of rape are only affected at very high
concentrations of thallium. Tolerance to relatively high
concentrations of thallium may be a result of complexation in plant
tissue, a phenomenon observed with other heavy metals (Cataldo &
Wildung, 1978).
In addition, selective pressure can increase tolerance. The
morphology of plants in the Alsar region of Yugoslavia, with a
naturally high soil thallium concentration, is not affected (Zyka,
1972), whereas such concentrations cause severe damage at other
locations (Schoer, 1984). In the Alsar region, zonation of plant
species with respect to soil thallium concentration can be observed
(Zyka, 1972), indicating the levels which are toxic to the various
species.
9.3.2 Wild animals
The effects of thallium on invertebrates have rarely been
investigated. After ant workers ingested about 0.2 mg thallium
chloride or thallium acetate per insect over a period of 2 months, all
survived (Jeantet et al., 1977). Field baits containing thallium
sulfate or acetate have been used against the fire ant and Pharaoh's
ant and destroyed about 90% of the colonies. These studies and the
toxic effects on crickets have been summarized by Negherbon (1959).
Corn poisoned with thallium sulfate has been used on a large
scale to control rodents. Such pest control carried out in the field
can affect various seed-eating animals and their predators (Munch et
al., 1974). An oral LC50 of 35 mg/kg fresh weight in starlings
(Sturnus vulgaris) was reported by Schafer (1972). This LC50 was
calculated following a single dose of thallium sulfate administered
via gavage, with a 7-day observation period. Thallium-poisoned baits
have also been used to control predatory birds. It was presumed that
9 out of 37 bald and golden eagles, which were collected sick or dead
in the USA, died from thallium poisoning in 1971-1972. Their kidneys
contained high concentrations of thallium (14 to 63 mg/kg) (Cromartie
et al., 1975). In experimentally poisoned eagles, which died from a
single oral dose of 120 mg thallium/kg body weight, the kidneys
contained 39 and 104 mg/kg (Bean & Hudson, 1976). Linsdale (1931)
reported the toxic effects of excessive use of thallium in California
for "ground squirrel control" on 58 species of game birds, song birds
and other wild animals. In 1972 all use of thallium in pesticides was
banned in the USA (Smith & Carson, 1977); in a study on birds carried
out between 1977 and 1981 no elevated thallium levels could be
detected (Wiemeyer et al., 1986).
In Denmark, partridges, pheasants, red foxes, badgers and martens
were found to be killed by direct ingestion of thallium-containing
rodenticides or poisoned prey (summarized by Munch et al., 1974;
Clausen & Karlog, 1974). Patho-anatomical findings from 1963 to 1971
indicated that 55 out of 299 red foxes and 5 out of 17 badgers
examined had suffered from poisoning. Determinations of thallium
concentrations in the kidney, liver and intestines demonstrated that
27 foxes and 1 badger had presumably been killed by thallium (Table
15). In most of the foxes not suspected of thallium poisoning,
thallium concentrations were < 0.1 mg/kg. Several of the poisoned
foxes showed abnormal behaviour, but only one fox showed clear
hair-loss. A typical sign in poisoned foxes was an empty stomach
(Munch et al., 1974). This was not observed in poisoned martens and
badgers. In addition, there were no skin lesions. Before death, many
of the martens showed uncoordinated movements and loss of balance.
The thallium concentrations in the inner organs of martens and badgers
ranged up to 92 mg/kg wet weight (Table 15) (Clausen & Karlog, 1974).
9.3.3 Household pets and farm animals
Accidental poisoning of pet animals (dogs and cats), ducks and
pigeons has been reported repeatedly (Zook & Gilmore, 1967). The
first detailed investigations considering toxic effects in dogs and
cats were performed to evaluate the risk of rodent poisoning campaigns
with thallium sulfate. Such cases used to be numerous, but in recent
years only occasional poisonings have occurred, due to the reduced use
of thallium as a rodenticide. For example, in Baden-Württemberg,
Germany, only 6 dogs, 4 cats and some ducks and pigeons were found to
have been poisoned by thallium from 1977 to 1989, and then no case
occurred up to 1992 (F. Baum, 1993, Institute of Animal Hygiene,
Freiburg, Germany; personal communication to the IPCS).
Symptoms of chronic intoxications in pets and farm animals are
similar to those of acute intoxications and can usually best be
observed in dogs. In ruminants uncharacteristic symptoms develop
(Hapke, 1984).
In some areas with naturally very high thallium levels, e.g., in
Yugoslavia and Israel, natural poisoning of farm animals after
consumption of vegetation has occurred (summarized by Gough et al.,
1979).
Table 30 summarizes early investigations by Ward (1931) and Shaw
(1932) on the toxicity of thallium sulfate to farm animals (quails,
geese, ducks and cattle), carried out in order to assess the risks of
its use as a rodenticide. In ducks an intraperitoneal injection of up
to 10 mg thallium/kg did not affect the birds, 15-25 mg/kg caused loss
of feathers on the back and 35-100 mg/kg was lethal within 24-63 h
(Ward, 1931). After feeding barley contaminated with 35, 50, or 75 mg
thallium/kg, ducks survived, or died in 12 days, or overnight,
respectively. Those ducks which died showed a mucous clogging of the
nasal passages (resulting in a marked gasping for breath), profuse and
green-coloured diarrhoea, loss of accommodation, wobbly gait and
extreme exhaustion. Death was due to respiratory failure and occurred
within 2 h after the beginning of intermittent asphyxial spasms.
Dissection of the dead ducks demonstrated that the intestinal tract
was plugged with a thick yellowish mucous. In addition, irritations
and ulcerations were present in the small intestine, and the livers
were enlarged and degenerated (Ward, 1931).
The same author investigated the effect on cattle (Ward, 1936).
Using thallium(I) sulfate, one cow received 50 mg thallium/kg body
weight and 3 heifers 35, 25 and 15 mg/kg, the latter two additional
doses of 20 and 35 mg/kg at 69 and 31 days after the first
administration. Two of the animals defaecated small quantities of
bloody faeces, all showed muscular twitching of flank and drooling of
a stringy mucous from nose and mouth. The cow died 5 days after
Table 30. Acute toxicity of thallium(I) sulfate for farm animals
Species Route of Period of Toxicitya Dose (mg thallium/kg Reference
administration observation body weight)
Quail oral 7 days LC100 approx. 12 Shaw (1932)
Goose oral 14 days LC100 approx. 15 Shaw (1932)
oral 2-3 days LC100 approx. 30-45 Shaw (1932)
Duck oral 14-21 days LC100 approx. 30 Shaw (1932)
oral > 15 days LOEL approx. 50 Ward (1931)
intraperitoneal > 15 days LOEL approx. 25 Ward (1931)
Cow oral 14 days LOEL approx. 25 Ward (1936)
a LC100 = concentration at which all animals were killed; LOEL = low-observed-effect level
administration, and the two heifers with the highest doses lived 11
and 14 days. The last animal was killed 3 days after the last
administration. Pathological changes were evident in the lymphatic
vessels (congested and oedematous), the liver (pale or congested) and
kidney (congested) and the walls of the digestive tract (haemorrhages,
ulcerations). Other organs appeared normal, and no significant
depilatory effect occurred. Recently Frerking et al. (1990) reported
thallium poisoning in cattle caused by the use of contaminated silage.
Symptoms were muscular twitching, colic, nervous behaviour, extreme
thirst, drooling from nose and mouth, loss of hair at the tail and,
later, erosion of nasal epithelium. The authors estimated that over a
period of 6 weeks the cows had ingested 0.75 mg thallium/kg body
weight daily (a total of 17 g thallium).
Anthropogenic contamination, especially from cement plants in
Germany, led to detailed studies of the effect of
thallium-contaminated fodder on the development of farm animals.
Continuous supplementation of maize-soybean fodder with 2, 4, 15 or
40 mg thallium(I) nitrate/kg in a 42-day broiler test and a
280(322)-day laying hen test caused obvious effects only at the
highest concentration (Ueberschär et al., 1986). In comparison to
control animals provided with uncontaminated feed, the body weight of
the treated broilers and hens was reduced by about 14 and 32%,
respectively; in the latter, laying rate (18%), feed efficiency (10%)
and eggshell thickness (2%) were also reduced. In broilers fed with
the two higher concentrations of thallium, gizzard erosion occurred.
In a detailed feeding study with fattening pigs with respect to
performance, health and meat residues, low concentrations of thallium
(daily intake of 0.05 and 0.1 mg thallium/kg body weight) were without
any effects on weight gain, carcass quality, health, or haematological
and biochemical parameters (Konermann et al., 1982). Daily
administration of 0.3 or 1.0 mg/kg body weight in drinking-water was
toxic to sheep, and the animals had to be killed after 4 and 6 weeks,
respectively (Hapke et al., 1980). Administration of daily doses of
0.03 and 0.1 mg thallium/kg body weight to sheep (for 11 weeks in
drinking-water) and 0.025 mg/kg body weight to steers (for 6 months in
fodder) caused no deaths (Hapke et al., 1980). However, in both
species daily uptake of 0.1 mg/kg body weight affected the animals
after several weeks or months; fodder should therefore contain less
than 0.5 mg/kg dry weight. Protein-rich food reduced the toxic action
of thallium (Hapke, 1984).
10. EVALUATION
10.1 Evaluation of human health risks
10.1.1 Exposure levels
Since thallium is a naturally occurring element, humans are
exposed to low levels in drinking-water, food and ambient air.
Drinking-water concentrations are often below the level of detection
(0.3 µg/litre) and rarely contribute more than 1 µg/litre. The total
intake of thallium from drinking-water has been estimated to be
< 1 µg/day for the vast majority of humans. In uncontaminated areas,
the dietary contribution of thallium has been estimated to be less
than 5 µg/day, with most of this coming from vegetables. Increased
dietary intakes have been reported for individuals living in areas
with thallium-contaminated soils; vegetables in these areas have been
found to contain thallium concentrations 1-2 orders of magnitude
higher than those grown in uncontaminated areas. However, the actual
dietary intakes for individuals consuming contaminated vegetables have
not been determined. In areas where there are no point sources of
thallium, ambient air concentrations are very low (< 1 ng/m3),
typically contributing less than 0.005 µg/day to the total intake.
Concentrations of thallium in workplace air can be several orders of
magnitude higher than those in ambient air, resulting in a
significantly increased total thallium intake. At the level of the
threshold limit values (TLVs) in some countries (0.1 mg/m3), the
thallium intake from inhalation alone would be of the order of
1000 µg/day (assuming inhalation of 10 m3 during a workshift). This
intake from inhalation alone (which may be even higher in some
workplaces) is about 500-fold higher than the total intake from
non-occupationally exposed humans living in non-contaminated areas.
There are only limited data about the actual thallium content of
workplace air. The most recent (1980s) concentrations of thallium
observed were < 22 µg thallium/m3 (in the production of a special
thallium alloy and in a thallium smelter). Average urinary
concentrations were determined to be in the range of 0.3-8 µg/litre
for cement workers and 0.3-10.5 µg/litre for foundry workers.
10.1.2 Kinetics
Thallium is rapidly and well absorbed through the
gastro-intestinal and respiratory tracts and is also taken up through
the skin. It is rapidly distributed to all organs and passes the
placenta, as indicated by the rapid fetal uptake, and the blood-brain
barrier. Because of its rapid accumulation in cells, concentrations
of thallium in whole blood do not reflect the levels in tissues. In
acute poisoning of experimental animals or humans, initially high
concentrations of thallium appear in the kidney, low concentrations in
fat tissue and brain, and intermediate concentrations in the other
organs; later the thallium concentration of the brain also increases.
Elimination of thallium may occur through the gastrointestinal
tract (mainly by mechanisms independent of biliary excretion), kidney,
hair, skin, sweat and breast milk. Intestinal reabsorption (mainly
from the colon) may occur with a consequent decrease in total body
clearance. In rats, the main routes of thallium elimination are
gastrointestinal (about 2/3) and renal (about 1/3), while in rabbits
the contribution of the two routes is about equal. Thallium is also
secreted in saliva.
As with many other substances, the excretion of thallium in
humans differs from that in laboratory animals since the rate of
excretion is generally much lower in humans (rate constant =
0.023-0.069 day-1) than in animals (average rate constant = 0.18
day-1). Another major difference between humans and animals is the
relative contribution of the different routes of excretion. In
humans, renal excretion seems to be much more important than in
animals, although its relative contribution to the total body
clearance has not been definitively established, due principally to
the lack of sufficient human data. Moreover, exposure levels,
duration of exposure, impairment of excretory organ function,
potassium intake and concomitant treatment of acute poisoning may
widely influence the results.
In a case report in which radioactive thallium (2.3 mg) was
therapeutically administered, urinary excretion of thallium within
72 h after dosing was 11% of the administered concentration, whereas
the gastrointestinal elimination was 0.5% during the same time period.
According to this study, renal excretion of thallium is about 73%,
whereas that through the gastrointestinal tract is about 3.7% of the
daily excreted amount. Elimination through hair has been estimated to
be 19.5% and that through skin and sweat 3.7%.
On the basis of the total daily excretion value, the daily intake
of thallium has been estimated to be about 11 µg and 0.9 µg in
chronically exposed and unexposed population, respectively; based on
the total amount of thallium in the body, a daily intake of about
2.3 µg may be calculated in unexposed populations.
The biological half-life of thallium in laboratory animals
generally ranges from 3 to 8 days. In humans it is about 10 days but
values of up to 30 days have been reported.
No data on the biotransformation of thallium are available.
10.1.3 Toxic effects
Thallium salts are mainly tasteless, odourless, colourless and
highly toxic. They were easily obtainable as rodenticides in the past
and are still available in some developing countries. Acute thallium
poisoning has resulted from accidental ingestion of thallium sulfate
and its use for suicide, homicide and attempts at illegal abortion.
Cases of homicide involving multiple low doses can induce chronic
intoxication. Chronic thallium intoxication has been observed in
occupationally exposed workers, and symptoms suggestive of thallium
poisoning have been seen in population groups in contaminated areas.
Clinical manifestations of acute thallium poisoning may occur
within hours or several days after exposure. Symptoms are often
diffuse and initially may include anorexia, metallic taste, nausea,
vomiting, retrosternal and abdominal pain, pain in the limbs, and
paraesthesia. Gastrointestinal haemorrhage occasionally occurs; later
on constipation is a common symptom. After the second day of thallium
poisoning, effects on the central and peripheral nervous systems,
skin, kidneys, eyes, cardiovascular and respiratory systems
progressively develop. Extreme sensitivity and pain in the legs,
later followed by the "burning feet" syndrome and paraesthesia, are
common manifestations. Insomnia, depression, hallucination, lethargy,
delirium, convulsions and coma may be followed by death, usually
between 10 and 12 days. Where survival extends beyond a week or so,
both motor and sensory neuropathy with cranial nerve involvement and
retrobulbar neuritis may develop. Common circulatory disorders, such
as hypertension, tachycardia and ischaemic cardiac changes, may also
occur. Frequently loss of head hair and sometimes also body hair
occurs after the second week of thallium poisoning. Dystrophy of the
nails is manifested by the occurrence of lunular stripes (Mee's lines)
3 to 4 weeks after intoxication. Recovery requires months and
occasionally some of the neurological and mental disturbances are
permanent. Permanent blindness may follow retrobulbar neuritis and
optic nerve atrophy.
Clinical features are generally milder in cases of chronic
poisoning than in acute thallium intoxication. Occurrence of chronic
thallium poisoning usually begins with neurological symptoms such as
tiredness, fatigue, headache and insomnia. In some cases the first
clinical findings include alopecia and constipation. The triad of
gastroenteritis, polyneuropathy and alopecia is regarded as the
classical syndrome of thallium poisoning, but in some cases
gastroenteritis and alopecia have not been reported.
Postmortem examinations following thallium poisoning reveal
damage in various organs. Haemorrhage in the mucosa of the intestine,
lungs and heart, kidney damage, fatty infiltration of the liver and
heart, and degeneration of neurons, including ganglion cells and
axons, with disintegration of myelin sheaths have all been observed.
Limited data are available on the effects of thallium on human
reproduction. Libido and male potency have been found to be adversely
affected in poisoning cases. There is no adequate evidence for a
genotoxic effect of thallium, and there have been no reports of any
carcinogenic or immunological effects.
Following low-level environmental exposure to thallium, a
dose-response relationship has been shown between thallium excretion
in urine and the prevalence of tiredness, weakness, sleep disorders,
nervousness, headache, muscle and joint pain and paraesthesia. Based
on replies to a questionnaire, a similar dose-response relationship
was seen when thallium in hair was taken as an indicator of exposure
and uptake.
10.1.4 Dose-response relationship (animals)
No lifetime studies of thallium administration have been
conducted on laboratory animals. In addition, no studies by the route
of inhalation are available. Three studies of intermediate duration
by the oral route are described in this report. A no-observed-effect
level could not be determined from any study. The lowest doses were
used in a 90-day gavage study (0, 0.01, 0.05 or 0.25 mg/kg body weight
per day). Small but statistically significant changes in some
clinical chemistry parameters were seen at the lowest dose level, as
was alopecia. From animal studies, it therefore appears that an
intake of 0.01 mg/kg per day may be associated with adverse effects.
No doses lower than this have been tested.
On the basis of LC50 values in animals and known lethal doses
in humans, it appears that humans may be more sensitive than
laboratory rodents to the toxic effects of thallium. Because of the
availability of human data and the apparently greater sensitivity of
humans, a quantitative evaluation of animal data for use in a risk
assessment has not been conducted here.
10.1.5 Dose-response relationship (humans)
Cases of acute thallium poisoning (with symptoms and signs listed
in the section 10.1.3) have occurred as a result of ingestion of doses
of thallium (in the form of soluble salts) as low as 1.5 mg/kg body
weight. Higher doses give rise to more severe symptoms. Doses that
have given rise to lethal poisoning are in the order of 10 mg/kg.
Concerning risks related to long-term exposure to lower doses of
thallium, the Task Group considered that an evaluation, although
uncertain, could best be performed on the basis of observed
relationships between urinary excretion of thallium and the occurrence
of symptoms. The urinary excretion value can be taken as an indicator
of the daily total absorbed dose from inhalation and dietary intake.
A population-based study on unexposed healthy subjects living in
northern Italy was performed with the aim of determining trace element
concentrations, including thallium, in blood, serum or plasma, and
urine, in which the collection, handling and analysis of the samples
was carried out under rigorous standardized protocols. The 496
subjects in this study, drawn from both urban and rural areas were
screened for normality by means of a questionnaire and clinical and
biochemical examination (with the exclusion of those with a history of
occupational exposure, heavy smokers, and those in a diseased state).
The mean urinary thallium concentration was 0.42 ± 0.09 µg/litre
(range 0.07-0.7 µg/litre). Other carefully controlled studies in
population samples showed similar urinary concentrations, e.g., 0.4 ±
0.2 µg/litre and 0.3 ± 0.2 µg/litre in rural and urban population
samples, respectively, and 0.3 ± 0.14 µg/litre in a sample of 149
subjects. This gives credence to a mean value of 0.3-0.4 µg/litre for
urinary thallium concentration in an unexposed population. In all
three studies, involving a total of 686 subjects, the range of urinary
thallium concentrations was 0.06-1.2 µg/litre. As thallium has a
short biological half-life, measured in days, and if a steady state
can be assumed to exist in such population-based samples, the above
urinary excretion value can be taken as an indicator of total dose in
terms of absorption following inhalation and total daily dietary
intake.
By contrast, in a population sample living in the vicinity
of thallium emission into the atmosphere, the mean urinary
thallium concentration was 5.2 µg/litre ± 8.3 µg/litre (range
0.1-76.5 µg/litre). Although a questionnaire on health effects was
compiled on each subject, no objective tests were performed. From the
replies to the questionnaire a clear dose-response relationship was
found between thallium concentration in urine and the prevalence of
tiredness, weakness, sleep disorder, headache, nervousness,
paraesthesia, muscle and joint pain. A similar dose-response
relationship was found when thallium in hair was taken as an indicator
of exposure. In a limited study on cement plant workers with thallium
exposure, where five workers showed urinary thallium levels above
5 µg/litre, but where the time interval between cessation of exposure
and urine collection was not stated, paraesthesia was reported in five
workers and distal muscle weakness in three. However, these symptoms
could not be related to thallium exposure.
From the above limited studies it is suggested that an
approximately 15-fold increase in urinary excretion of thallium above
the mean non-exposed level of 0.3 to 0.4 µg/litre may be related to
subjective symptoms which could possibly be considered as early
adverse health effects.
It is known from clinical practice that there is an increased
urinary concentration of thallium in acute poisoning cases. In 14
cases of thallium poisoning with recovery after therapy, the urinary
thallium concentrations ranged from 500 to 20 400 µg/litre. In seven
of these cases concentrations were below 2700 µg/litre. It should be
recognized that these values are not entirely comparable to those in
long-term exposure since they do not represent steady-state
conditions.
In summary, the Task Group considered that exposures causing
urinary thallium concentrations below 5 µg/litre are unlikely to cause
adverse health effects. In the range of 5-500 µg/litre the magnitude
of risk and severity of adverse effects are uncertain, while exposures
giving values over 500 µg/litre have been associated with clinical
poisoning.
10.2 Evaluation of the effects of thallium on the environment
Thallium is an element which occurs naturally in the earth's
crust, primarily in the monovalent form. In marine water and some
localized strongly oxidizing freshwater and soil, thallium may be
present primarily in the oxidized trivalent form. The major
anthropogenic sources of thallium released to the environment are
smelting of metallic ores, mining, special cement production, and the
combustion of fossil fuels, principally coal. Relatively little
thallium is released into the environment because of the production
and use of thallium compounds. Thallium levels reported in air are
generally < 1 ng/m3 although mean values up 15 ng/m3 have been
reported in industrial and urban air. Thallium may be released
directly to the environment following its use as a rodenticide,
although such use has been restricted or banned in many countries.
Thallium tends to persist in soil, although it may be leached to
water under acidic conditions. Monovalent thallium is relatively
stable in solution whereas trivalent thallium may be removed from the
water column by precipitation as the oxide or hydroxide. Although
thallium can bioconcentrate, it is unlikely to biomagnify in aquatic
or terrestrial food webs. Thallium concentrations in water tend to be
low, a maximum concentration of 2.4 mg/litre having been reported for
industrial waste water. Thallium concentrations in surface water from
industrial regions have been reported to range from 1 to 100 µg/litre,
while surface water in uncontaminated areas normally contains lower
levels. Concentrations of thallium in seawater range from < 0.01 to
0.02 µg/litre.
Most studies of effects on aquatic organisms have used soluble
monovalent thallium compounds. Acute toxic effects have been reported
in freshwater algae exposed to thallium at 100 µg/litre. Reduced
growth of aquatic macrophytes was reported following a 28-day
exposure to 8 µg/litre. The 48-h LC50 reported for Daphnia was
2200 µg/litre, while a 24-h LC50 of 110 µg/litre has also been
reported. The 96-h LC50 values for freshwater fish range from 860
to 132 000 µg/litre. An LC50 of 40 µg/litre has been reported for
freshwater fish exposed to thallium for approximately 40 days. The
96-h LC50 values for marine species are 2100 µg/litre for
invertebrates and 20.9-24 mg/litre for fish. The available aquatic
toxicity data suggest that thallium can harm aquatic organisms.
However toxic effects are likely to be limited to sites adjacent to
point sources such as some metal mining and smelting operations and
cement plants.
Thallium concentrations in uncontaminated soil typically range
from about 0.1 to 1.0 mg/kg dry weight, although higher levels can
occur near natural sources such as thallium-enriched shales and some
metallic ore deposits. Levels are generally somewhat elevated near
anthropogenic sources such as cement plants using thallium-containing
pyrite (up to 21 mg/kg dry weight) and base metal smelters (up to
2.1 mg/kg dry weight) that release large quantities of thallium to the
atmosphere.
Very few data have been identified concerning the effects on
terrestrial organisms of thallium in soil. The results of one study
suggest that microbial community structure is disturbed at
concentrations in the range of 1 to 10 mg/kg dry weight. However, the
properties of the soil and the form of thallium used in this study
were not identified.
Plants growing in uncontaminated soil normally contain 0.01 to
0.3 mg thallium/kg dry weight, while those growing near cement plants
using thallium-enriched pyrite have been reported to contain much
larger amounts (100 to 1000 mg/kg dry weight). Reduced growth has
been reported in sensitive plant species at concentrations of about
1 mg thallium/kg of dry plant tissue following exposure to monovalent
thallium. Toxic effects on terrestrial plants are therefore possible
near some cement plants using thallium-enriched pyrite.
The use of thallium as a rodenticide has resulted in poisoning of
non-target organisms including foxes, badgers, martens, partridges,
pheasants and eagles. Poisoning of domestic animals, such as dogs,
cats, ducks and pigeons, has also been widely reported. The number of
wildlife poisoning incidents has declined as a result of the reduced
use of thallium as a rodenticide. In countries with naturally high
thallium levels, such as former-Yugoslavia and Israel, some farm
animals have been poisoned following ingestion of vegetation with a
high thallium content. Symptoms of thallium poisoning have been
reported in cows that were calculated to have consumed 0.75 mg
thallium/kg body weight per day for a 6-week period.
11. CONCLUSIONS AND RECOMMENDATIONS
The currently limited industrial uses of thallium are unlikely to
pose a threat to the general environment. At industrial facilities
such as metal mining and smelting operations and cement plants using
pyrite, which can release significant amounts of thallium, the
concentration of thallium in industrial raw materials as well as stack
gases and waste water should be monitored and, if necessary,
controlled. Waste materials containing water-soluble thallium
compounds should be sealed and marked to avoid leaching and pollution
by dust.
In the general population, environmental exposure to thallium
does not pose a health threat. The total intake has been estimated to
be less than 5 µg/day, with the vast majority coming from foodstuffs;
drinking-water and air generally contribute very small amounts of
thallium.
Due to its toxicity to both humans and non-target environmental
species, the use of thallium as a rodenticide has been prohibited in
many countries. Where thallium is still available for such use,
however, the potential for accidental poisoning or for its use in
homicide or suicide remains a significant concern. It is recommended
that the use of thallium as a rodenticide be prohibited worldwide,
particularly as less hazardous methods of rodent control are
available.
Atmospheric emissions from industrial sources (e.g., cement
plants using thallium-containing pyrite) have resulted in increased
concentrations of thallium in biological samples (e.g., urine and
hair) from the population living in the vicinity. A relationship was
found between thallium concentrations in urine and hair and the
prevalence of symptoms possibly indicating early health effects of
thallium. The limited available data are not sufficient for
determining an acceptable limit for emissions. Steps should be taken,
however, to limit emissions to the greatest extent possible. Where
thallium may be released into the environment, monitoring of both
atmospheric emissions and resulting dust deposition rates should be
performed. Where environmental monitoring reveals thallium levels
significantly above background, it is recommended that biomonitoring
of the population living in the vicinity of the point source be
carried out. If biomonitoring reveals excessive exposure to thallium,
emissions from the point source should be re-evaluated and an effort
made to reduce them.
Since current occupational exposures to thallium may be of
concern to health, it is recommended that measures be taken to reduce
occupational exposure (as described in section 8.7 of this monograph).
Furthermore, revision of threshold limit values for thallium warrants
consideration.
All thallium analyses should be accompanied by a quality
assurance programme. This requires certified reference materials of
one matrix and of a similar concentration range to the sample to be
analysed and participation in an inter-laboratory comparison
programme. There is a need to make such reference materials
available.
Since thallium is rapidly and well absorbed and its excretion is
mainly renal, concentrations of thallium in urine may be considered a
relatively reliable indicator of exposure. Exposure to thallium
causing urinary concentrations below 5 µg/litre is unlikely to cause
adverse human health effects. For thallium exposure giving rise to
urinary concentrations in the range 5-500 µg/litre, the magnitude of
risk and the severity of adverse effects on human health are
uncertain, while exposure giving rise to 500 µg/litre or more has been
associated with clinical poisoning. The estimated daily oral intake
corresponding to a urinary thallium concentration of 5 µg/litre is
approximately 10 µg thallium in the form of a soluble compound.
In view of the considerable uncertainties in the evaluation, the
Task Group concluded that it was not possible to recommend a
health-based exposure limit. Until better information on the
dose-response relationship becomes available, it seems prudent to keep
exposures at levels that lead to urinary concentrations of less than
5 µg/litre.
12. FURTHER RESEARCH
a) Follow-up epidemiological studies of populations exposed
chronically to high levels of thallium (e.g., in the vicinity of
cement plants and natural sources of high concentrations of
thallium) should be performed to determine whether there is an
increased risk of pathological effects, e.g., cancer, effects on
reproduction (especially sperm cells) and congenital
malformations. Such follow-up studies are also required in order
to assess objectively the largely subjective symptom complex
experienced by certain populations in thallium-contaminated
areas.
b) Toxicokinetics of thallium, with particular respect to
distribution and excretion, should be studied in people with
low-level environmental exposure (in both uncontaminated and
contaminated areas) and in those exposed occupationally.
Adequate information from acutely poisoned patients should also
be compiled to determine toxicokinetics and the relationship with
the clinical findings. In addition, an attempt should be made to
correlate clinical findings with thallium concentrations in
biological samples. Patients should be followed for several
years to ascertain the long-term consequences of acute
poisonings.
c) Early indicators of an effect of thallium on glomerular or
tubular kidney function, on liver function, on haem biosynthesis
and on nerve conduction velocity should be sought in asymptomatic
population samples and in occupationally exposed workers where
there is evidence of excessive thallium exposure.
d) Early indicators of an effect of thallium absorption should be
sought. The effects of thallium on the haem biosynthetic pathway
suggest that estimation of porphyrins in blood and/or urine may
be a useful approach to detecting specific early effect of
thallium cellular toxicity. Similarly observations on the
endoplasmic reticulum structure with respect to protein synthesis
and polysome function suggest a need for further studies on liver
function.
e) Chronic bioassays in laboratory animals are needed to assess the
carcinogenic activity of thallium at low doses.
f) Lifetime studies in laboratory animals should be conducted using
several low doses by the routes of both inhalation and ingestion
to help determine a dose-response relationship and a
no-observed-effect level.
g) Multi-generation studies of thallium in laboratory animals are
needed to ascertain the reproductive and teratogenic effects of
thallium.
h) Sample collection, analysis and data presentation should be
carried out according to a protocol which ensures adequate
validation of biological monitoring procedures.
i) The concentrations of thallium in environmental compartments
adjacent to point sources should be determined using current
analytical techniques. The bioavailability of thallium from
various matrices should also be determined.
j) Total dietary studies should be conducted in areas with high
thallium concentrations (either natural or from industrial
sources) to determine the oral intake in populations living in
these regions.
k) Thallium concentrations in soil and vegetables from regions with
extensive acid rain should be determined.
REFERENCES
ACGHI (1980) Documentation of the threshold limit values, 4th ed.
Cincinnati, Ohio, American Conference of Governmental Industrial
Hygienists.
Achenbach C, Ziskoven R, Koehler F, Bahr U, & Schulten H-R (1979a)
[Quantitative trace analysis of thallium in biological material.]
Angew Chem, 91: 944-945 (in German).
Achenbach C, Hauswirth O, Heindrichs C, Ziskoven R, Köhler F, Smend J,
& Kowalewski S (1979b) [Toxicity and teratogenicity of thallium.]
Dtsch Ärztebl, 76: 3189-3192 (in German).
Achenbach C, Hauswirth O, Heindrichs C, Ziskoven R, Köhler F, Bahr U,
Heindrichs A, & Schulten H-R (1980) Quantitative measurement of
time-dependent thallium distribution in organs of mice by field
desorption mass spectrometry. J Toxicol Environ Health, 6: 519-528.
Achenbach C, Wiemer J, Ziskoven R, & Winter U (1982) Tl+-Ions:
effects on the automaticity of sinoatrial tissue and on dV/dtmax and
iK2 of cardiac Purkinje fibres. Z Naturforsch, 37c: 1006-1014.
Adamson RH, Canellos GP, & Sieber SM (1975) Studies on the antitumor
activity of gallium nitrate (NSC-15200) and other group IIIa metal
salts. Cancer Chemother Rep, 59: 599-610.
Aderjan R, von Clarmann M, Daldrup T, Geldmacher-von Mallinckrodt M,
van Heijst ANP, Machbert G, Schütz H, & Stöppler M (in press)
[Thallium - clinical toxicological analysis after acute intoxications.
Communications of the Senate Committee on clinical and toxicological
analysis.] German Research Council (in German).
Agnes G, Hill HAO, Pratt JM, Ridsdale SC, Kennedy FS, & Williams RJP
(1971) Methyl transfer from methyl vitamin B12. Biochim Biophys
Acta, 252: 207-211.
Agrawal OP & Khatkar VB (1988) Amperometric determination of Ga(III),
In(III), Tl(I) and Pb(II) with thiomalic acid. J Electrochem Soc
India, 37: 289-292.
Aitio A (1988) Biological monitoring. In: Clarkson T, Friberg L,
Nordberg G, & Sager P ed. Biological monitoring of toxic metals. New
York, London, Plenum Press, pp 75-83.
Alarcón-Segovia D, Amigo MC, & Reyes PA (1989) Connective tissue
disease features after thallium poisoning. J Rheumatol, 16: 171-174.
Al-Attar AF, Martin MH, & Nickless G (1988) Uptake and toxicity of
cadmium, mercury and thallium to Lolium perenne seedlings.
Chemosphere, 17: 1219-1225.
Albertano P & Pinto G (1986) The action of heavy metals on the growth
of three acidophilic algae. Boll Soc Nat Napoli, 95: 319-328.
Ali SL & Blume H (1983) [Determination of the pesticide residues and
toxic heavy metal traces in crude drugs.] Pharm Ztg, 128: 2248-2252
(in German).
Ali SF, Jairaj K, Newport GD, Lipe GW, & Slikker W (1990) Thallium
intoxication produces neurochemical alterations in rat brain.
Neurotoxicology, 11: 381-390.
Allus MA, Martin MH, & Nickless G (1987) Comparative toxicity of
thallium to two plant species. Chemosphere, 16: 929-932.
Allus MA, Brereton RG, & Nickless G (1988) Chemometric studies of the
effect of toxic metals on plants: the use of response surface
methodology to investigate the influence of Tl, Cd and Ag on the
growth of cabbage seedlings. Environ Pollut, 52: 169-181.
Andersen O (1984) Clinical evidence and therapeutic indications in
neurotoxicology, exemplified by thallotoxicosis. Acta Neurol Scand,
70(suppl 100): 185-192.
André T, Ullberg S, & Winqvist G (1960) The accumulation and retention
of thallium in tissues of the mouse. Acta Pharmacol Toxicol,
16: 229-234.
Anschütz M, Herken R, & Neubert D (1981) Studies on embryotoxic
effects of thallium using the whole-embryo culture technique. In:
Neubert D & Merker H-J ed. Culture techniques: applicability for
studies on prenatal differentiation and toxicity. Proceedings of the
5th Symposium on Prenatal Development. Berlin, De Gruyter, pp 57-66.
Aoyama H (1989) Distribution and excretion of thallium after oral and
intraperitoneal administration of thallous malonate and thallous
sulfate in hamsters. Bull Environ Contam Toxicol, 42: 456-463.
Aoyama H, Yoshida M, & Yamamura Y (1988) Induction of lipid
peroxidation in tissues of thallous malonate-treated hamster.
Toxicology, 53: 11-18.
Apostoli P, Maranelli G, Minoia C, Massola A, Baldi C, & Marchiori L
(1988) Urinary thallium: critical problems, reference values and
preliminary results of an investigation in workers with suspected
industrial exposure. Sci Total Environ, 71: 513-518.
Arndt U, Finkbeiner M, Maier-Reiter W, & Wystrcil H-G (1987)
[Ecotoxicology of thallium. Behaviour and location in phytocenosis.]
Agrar-Umweltforsch Baden-Württemberg, 16: 5-21 (in German).
Arnold W (1986) [Excretion and distribution of thallium in human
poisoning.] In: Anke M, Baumann W, Bräunlich H, Brückner C, & Groppel
B ed. [5th Symposium of Trace Elements, Jena, 14-17 July 1986.] Jena,
University of Jena, pp 1241-1253 (in German).
Atkins HL, Budinger TF, Lebowitz E, Ansari AN, Greene MW, Fairchild
RG, & Ellis KJ (1977) Thallium-201 for medical use. Part 3: Human
distribution and physical imaging properties. J Nucl Med, 18: 133-140.
ATSDR (1992) Toxicological profile for thallium. Atlanta, Georgia,
Agency for Toxic Substances and Disease Registry,
pp 1-90 (ATSDR/TP-91/26).
Aulenbach DB, Meyer MA, Beckwith E, Joshi S, Vasudevan C, & Clesceri
NL (1987) Removal of heavy metals in POTW. Environ Prog, 6: 91-98.
Auler H (1926) [Therapy of cancer.] Z Krebsforsch, 23: 153-156
(in German).
Bahiga LM, Kotb NA, & El-Dessoukey EA (1978) Neurological syndromes
produced by some toxic metals encountered industrially or
environmentally. Z Ernähr.wiss, 17: 84-88.
Bank WJ, Pleasure DE, Suzuki K, Nigro M, & Katz R (1972) Thallium
poisoning. Arch Neurol, 26: 456-464.
Barbier F (1974) Treatment of thallium poisoning. Lancet,
2(7886): 965.
Barckow J & Jenss H (1976) [Thallium intoxication. Combined treatment
with haemodialysis, forced diuresis and antidote.] Med Klin,
71: 1377-1382 (in German).
Barclay RK, Peacock WC, & Karnofsky DA (1953) Distribution and
excretion of radioactive thallium in the chick embryo, rat, and man.
J Pharm Exp Ther, 107: 178-187.
Barlow SM & Sullivan FM (1982) Thallium and its compounds. In :
Reproductive hazards of industrial chemicals: An evaluation of animal
and human data. New York, Academic Press, chapter 41, pp 530-537.
Barnes MP, Murray K, & Tilley PJ (1984) Neurological deficit more than
thirty years after chronic thallium intoxication (letter). Lancet,
1: 562.
Barrach H-J & Neubert D (1985) The moderating influence of potassium
on the thallium effect in tissue differentiation in vitro.
Teratology, 31: 42A.
Barrera H & Gómez-Puyou A (1975) Characteristics of the movement of K+
across the mitochondrial membrane and the inhibitory action of Tl+.
J Biol Chem, 250: 5370-5374.
Barroso-Moguel R, Ríos-Castañeda C, Villeda-Hernández J,
Méndez-Armenta M, & Galván-Arzate S (1990) Neurotoxicity of thallium:
biochemical and morphological study of organic lesions. Arch Invest
Med, 21: 115-122.
Bazzaz FA, Carlson RW, & Rolfe GL (1974) The effect of heavy metals on
plants: Part I. Inhibition of gas exchange in sunflower by Pb, Cd, Ni
and Tl. Environ Pollut, 7: 241-246.
Bean JR & Hudson RH (1976) Acute oral toxicity and tissue residues of
thallium sulfate in golden eagles, Aquila chrysaetos. Bull Environ
Contam Toxicol, 15: 118-121.
Bell W, Hendry RA, & Annett HE (1925) The specific action of lead on
the chorion epithelium of the rabbit, contrasted with the action of
copper, thallium and thorium. J Obstet Gynaecol Br Emp, 32: 1-16.
Bendl BJ (1969) Thallium poisoning. Report of a case successfully
treated with dithizone. Arch Dermatol, 100: 443-446.
Berndt H & Sopczak D (1987) [Error recognition and programme
optimization in ET-AAS by time-resolved signals (direct determination
of element traces in urine: Cd,Co, Cr, Ni, Tl, Pb).] Fresenius Z Anal
Chem, 329: 18-26 (in German).
Bertram HP, Kemper FH, Müller C, & Cuellar JA (1985) Trace metal
patterns of human blood, saliva and hair: Environmental specimen
banking. Proceedings of the 5th International Conference on Heavy
Metals in the Environment, Toronto, vol 2, pp 102-104.
Bettinelli M, Baroni U, & Pastorelli N (1988) Determination of
arsenic, cadmium, lead, antimony, selenium and thallium in coal fly
ash using the stabilised temperature platform furnace and
Zeeman-effect background correction. J Anal At Spectrom, 3: 1005-1011.
BGA (1979) [Thallium.] Berlin, The Federal Environment Agency (Report
No. 63/28/7180) (in German).
Bienvenu P, Nofre C, & Cier A (1963) Toxicité générale comparée des
ions métalliques. Relation avec la classification périodique. C R Acad
Sci (Paris), 256: 1043-1044.
Birge WJ (1978) Aquatic toxicology of trace elements of coal and fly
ash. In: Proceedings of the Symposium on Energy and Environmental
Stress in Aquatic Systems, Augusta, Georgia, 2-4 November 1977,
pp 219-240.
Bock KD, Merguet P, Schley G, Schümann HJ, Rausch-Stroomann J-G,
Hocevar V, Schröder E, & Murata T (1968) [Pathogenesis of hypertension
and tachycardia in the acute thallium poisoning and acute intermittent
porphyria.] Dtsch Med Wochenschr, 93: 2119-2124 (in German).
Bonnet EFP & Pedace EA (1979) [Forensic anatomical histopathology of
thallium poisoning.] Prensa Med Argent, 66: 29-35 (in Spanish).
Bornhausen M & Hagen U (1984) Operant behavior performance changes in
rats after prenatal and postnatal exposure to heavy metals. Int Res
Commun Syst Med Sci, 12: 805-806.
Bösche J & Magureanu I (1983) [Heavy metal burden in Nordbaden by
thallium and cadmium - Results of investigations of kidneys.] In: Barz
J, Bösche J, Frohberg H, Joachim H, Käppner R, & Mattern R ed.
[Advances in forensic medicine. Festschrift for Georg Schmidt.]
Berlin, Springer Verlag, pp 270-275 (in German).
Bowen HJM (1966) Trace elements in biochemistry. New York, London,
Academic Press.
Bowen HJM (1979) Environmental chemistry of the elements. New York,
London, Academic Press.
Boysen P (1992) [Heavy metals and other substances in fertilizers -
Environmental research plan of the Federal Minister for Environment,
Nature Conservation and Reactor Safety, research report 107 01 016/1.]
Berlin, Erich Schmidt Verlag (Report No. UBA-FB 92-104) (in German).
Bradley-Moore PR, Lebowitz E, Greene MW, Atkins HL, & Ansari AN (1975)
Thallium-201 for medical use. II: biologic behavior. J Nucl Med,
16: 156-160.
Briese C & Nessler F (1985a) [Historical data on the discovery of
thallium and its first applications.] Wiss Z Humboldt-Univ (Berlin)
Math-Naturwiss Reihe, 34: 750-751 (in German).
Briese C & Nessler F (1985b) [Therapy of thallium poisoning.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 786-788 (in German).
Briese C, Nessler F, & Liebscher R (1985) [Production of thallium and
its significance in the different political economies.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 756-760 (in German).
Bringmann G & Kühn R (1982) [Results of toxic action of water
pollutants on Daphnia magna tested by an improved standardized
procedure.] Z Wasser Abwasser Forsch, 15: 1-6 (in German).
Britten JS & Blank M (1968) Thallium activation of the
(Na+-K+)-activated ATPase of rabbit kidney. Biochim Biophys Acta,
159: 160-166.
Brockhaus A, Dolgner R, Ewers U, Wiegand H, Freier I, Jerman E, &
Krämer U (1980) Excessive thallium absorption among a population
living near a thallium emitting cement plant. In: Holmstedt B,
Lauwerys R, Mercier M, & Robberfroid M ed. Mechanisms of toxicity and
hazard evaluation. Proceedings of the 2nd International Congress on
Toxicology, Brussels, 1980. Dev Toxicol Environ Sci, 8: 565-568.
Brockhaus A, Dolgner R, Ewers U, Freier I, Jermann E, & Wiegand H
(1981a) Repeated surveillance of exposure to thallium in a population
living around a cement plant emitting thallium containing dust. In:
Proceedings of the International Conference on Heavy Metals in the
Environment, Amsterdam, September 1981. Luxembourg, Commission of the
European Communities, pp 482-485.
Brockhaus A, Dolgner R, Ewers U, Krämer U, Soddemann H, & Wiegand H
(1981b) Intake and health effects of thallium among a population
living in the vicinity of a cement plant emitting thallium containing
dust. Int Arch Occup Environ Health, 48: 375-389.
Brodersen HP, Theilmeier A, Korsten S, Arendt U, Larbig D, & Reis HE
(1992) Acute poisoning by a potentially lethal dose of thallium:
quantitative comparison of different methods of elimination. Intensiv
Notfallbehandl, 17: 204-207.
Brown BE (1982) The form and function of metal-containing "granules"
in invertebrate tissues. Biol Rev, 57: 621-667.
Brown BT & Rattigan BM (1979) Toxicity of soluble copper and other
metal ions to Elodea canadensis. Environ Pollut, 20: 303-314.
Brown DR, Callahan BG, Cleaves MA, & Schatz RA (1985) Thallium induced
changes in behavioral patterns: correlation with altered lipid
peroxidation and lysosomal enzyme activity in brain regions of male
rats. Toxicol Ind Health, 1: 81-98.
Brumsack H-J (1977) Potential metal pollution in grass and soil
samples around brickworks. Environ Geol, 2: 33-41.
Brumsack H, Heinrichs H, & Lange H (1984) West German coal power
plants as sources of potentially toxic emissions. Environ Technol
Lett, 5: 7-22.
Budavari S ed. (1989) The Merck index: An encyclopedia of chemicals,
drugs, and biologicals, 11th ed. Rahway, New Jersey, Merck & Co.
Bulavintseva OA & Bulavintsev AG (1982) [Histoenzymic characteristics
in rats chronically poisoned with thallium sulfate.] In: Sedov KR ed.
[Geochemical environment: The problem of the health of the population
of the BAM railway zone], pp 68-69 (in Russian).
Burger A & Starke K (1969) [Influence of thallium on the ATPases of
the amine storing granules from the adrenal medulla and splenic
nerves.] Experientia (Basel), 25: 578-579 (in German).
Buschke A (1900) [Experiments on alopecia.] Berl Klin Wochenschr,
37: 1235-1236 (in German).
Buschke A (1903) [Further experiments on alopecia and the localization
of dermatoses.] Berl Klin Wochenschr, 40: 888-890 (in German).
Buschke A (1911) [Clinical and experimental observations of alopecia
(hypotrichosis) congenita.] Arch Dermatol Syph, 108: 27-40 (+ plates
IV and V) (in German). Buschke A (1929) [Thallium.] Dtsch Med
Wochenschr, 55: 1546-1547 (in German).
Buschke A & Berman L (1927) [The chemical and biological relation of
thallium and lead.] Klin Wochenschr, 6: 2428-2429 (in German).
Buschke A & Curth W (1928) [Therapeutical experiments with thallium,
apart from its use against fungal diseases of the hair of the head.]
Derm Z, 53: 113-117 (in German).
Buschke A & Jacobsohn F (1922) [Experiments on the action of
thallium.] Dtsch Med Wochenschr, 48: 859-860 (in German).
Buschke A & Langer E (1927) [The forensic and the industrial-hygienic
importance of thallium.] Münch Med Wochenschr, 74: 1494-1497
(in German).
Buschke A & Peiser B (1922a) [The effect of thallium on the endocrine
system.] Klin Wochenschr, 1: 995 (in German).
Buschke A & Peiser B (1922b) [Experimental observations of the
influence of thallium on the endocrine system.] Med Klin, 18: 731-732
(in German).
Buschke A & Peiser B (1922c) [The endocrine effect and the practical
importance of thallium.] Dermatol Wochenschr, 74: 443-445 (in German).
Buschke A & Peiser B (1926) [Thallium alopecia and sensory hairs.]
Klin Wochenschr, 5: 977-979 (in German).
Buschke A, Zondek B, & Berman L (1927a) [The inhibitory effect of
thallium on the estrous cycle of mice.] Klin Wochenschr, 6: 683-685
(in German).
Buschke A, Christeller E, & Loewenstein L (1927b) [Alterations of the
skull by experimental chronic thallium poisoning.] Klin Wochenschr,
6: 1088-1089 (in German).
Buschke A, Löwenstein L, & Joel W (1928) [Further histological results
of experimental chronic thallium poisoning.] Klin Wochenschr,
7: 1515 (in German).
Byczkowski JZ & Sorenson JRJ (1984) Effects of metal compounds on
mitochondrial function: a review. Sci Total Environ, 37: 133-162.
Byrne C, Balasubramanian R, Overton EB, & Albert TF (1985)
Concentrations of trace metals in the bowhead whale. Mar Pollut Bull,
16: 497-498.
Canterford GS (1980) Formation and regeneration of abnormal cells of
the marine diatom Ditylum brightwelli (West) Grunow. J Mar Biol Assoc
(UK), 60: 243-254.
Canterford GS & Canterford DR (1980) Toxicity of heavy metals to the
marine diatom Ditylum brightwelli (West) Grunow: correlation between
toxicity and metal speciation. J Mar Biol Assoc (UK), 60: 227-242.
Careaga-Olivares J & Morales-Aguilera A (1990) The distribution of
thallous ion between blood cells and plasma at equilibrium. Arch
Invest Med (Mex), 1990: 21-23.
Careaga-Olivares J & Morales-Aguilera A (1993) Differential
accumulation of thallous ion by diverse rabbit and rat muscles. Bull
Environ Contam Toxicol, 51: 764-771.
Carlson RW, Bazzaz FA, & Rolfe GL (1975) The effect of heavy metals on
plants. II. Net photosynthesis and transpiration of whole corn and
sunflower plants treated with Pb, Cd, Ni, and Tl. Environ Res,
10: 113-120.
Cataldo DA & Wildung RE (1978) Soil and plant factors influencing the
accumulation of heavy metals by plants. Environ Health Perspect,
27: 149-159.
Cavanagh JB (1979) Metallic toxicity and the nervous system. In: Smith
WT & Cavanagh JB ed. Recent advances in neuropathology. Edinburgh,
Churchill Livingstone, pp 247-275.
Cavanagh JB (1988) Neurotoxic effects of metals and their
interactions. Recent Adv Nerv Syst Toxicol, 100: 177-202.
Cavanagh JB (1991) Annotation. What have we learnt from Graham
Frederick Young? Reflections on the mechanism of thallium
neurotoxicity. Neuropathol Appl Neurobiol, 17: 3-9.
Cavanagh JB & Gregson M (1978) Some effects of thallium salts on the
proliferation of hair follicle cells. J Pathol, 125: 179-191.
Cavanagh JB, Fuller NH, Johnson HRM, & Rudge P (1974) The effects of
thallium salts, with particular reference to the nervous system
changes. Q J Med New Ser, 43: 293-319.
Chandler HA & Scott M (1986) A review of thallium toxicology. J R Navy
Med Serv, 72: 75-79.
Chandler HA, Archbold GPR, Gibeon JM, O'Callaghan P, Marks JN, &
Petghybridge RJ (1990) Excretion of a toxic dose of thallium. Clin
Chem, 36: 1506-1509.
Chattopadhyay A & Jervis RE (1974) Multielement determination in
market-garden soils by instrumental photon activation analysis. Anal
Chem, 46: 1630-1639.
Christensen HE, Luginbyhl TT, Hill RH Jr, McGrath DJ, Georgevich M,
Mitchell FL, & May JR ed. (1973) The toxic substances list, 1973
edition. Washington, DC, US Department of Health, Education, and
Welfare, Public Health Service, US Government Printing Office.
Chusid JG & Kopeloff LM (1962) Epileptogenic effects of pure metals
implanted in motor cortex of monkeys. J Appl Physiol, 17: 697-700.
Clausen B & Karlog O (1974) Loading with thallium among wild animals
of the marten genus and badgers in Denmark. Nord Vet.med, 26: 339-350.
Claussen U, Roll R, Dolger R, Matthiaschk G, Majewski F, Stoll B, &
Röhrborn G (1981) [Mutagenicity and teratogenicity of thallium - with
special consideration of the situation in Lengerich.] Rhein Ärztebl,
16: 469-475 (in German).
Cooper GP, Suszkiw JB, & Manalis RS (1984) Presynaptic effects of
heavy metals. In: Narahashi T ed. Cellular and molecular
neurotoxicology. New York, Raven Press, pp 1-21.
Cornelis R (1988) Analytical procedures and clinical reference
materials in monitoring human exposures to trace metals with special
reference to Cr, Pb and Tl. Sci Total Environ, 71: 269-283.
Cortes Toro E, Parr RM, & Clements SA (1990) Biological and
environmental reference materials for trace elements, nuclides and
organic microcontaminants. Vienna, International Atomic Energy Agency
(IAEA/RL/128 Rev.1).
Cottier H (1980) [Pathogenesis.] Berlin, Springer Verlag (in German).
Cotton FA & Wilkinson G (1988) Advanced inorganic chemistry, 5th ed.
New York, Chichester, Brisbane, Toronto, John Wiley and Sons.
Coyle V (1980) Diagnosis and treatment of thallium toxicosis in a dog.
J Small Anim Pract, 21: 391-397.
Craig PJ (1980) Metal cycles and biological methylation. In: Hutzinger
O ed. The handbook of environmental chemistry. Volume 1: The natural
environment and the biogeochemical cycles - Part A. Berlin, Springer
Verlag, pp 169-227.
Cromartie E, Reichel WL, Locke LN, Belisle AA, Kaiser TE, Lamont TG,
Mulhern BM, Prouty RM, & Swineford DM (1975) Residues of
organochlorine pesticides and polychlorinated biphenyls and autopsy
data for bald eagles, 1971-72. Pestic Monit J, 9: 11-14.
Crössmann G (1984) [Thallium - a new environmental contamination?]
Angew Bot, 58: 3-10 (in German).
Crössmann G (1985) [Toxic elements in feed.] Dtsch Tierärztl
Wochenschr, 92: 232-233 (in German).
Csicsaky M & Wiegand H (1981) [Neurotoxical effects of thallium on the
peripheral and central nervous systems of the rat.] Umwelthygiene,
13: 180 (in German).
Csicsaky M, Wiegand H, Uhlig S, Lohmann H, & Papadopoulos R (1988)
Miniature endplate potentials as a tool in neurotoxicology.
Toxicology, 49: 121-129.
Curry AS, Grech JL, Spiteri L, & Vassallo L (1969) Death from thallium
poisoning: A case report. Eur J Toxicol, 2: 260-269.
Danilewicz M, Danilewicz M, & Kurnatowski A (1979) [Dynamics of
histopathological changes in the kidney of experimental animals after
lethal doses of thallium.] Med Pracy, 30: 257-265 (in Polish).
Danilewicz M, Kurnatowski A, & Wagrowska-Danilewicz M (1980) [Dynamics
of pathomorphological changes in some internal organs of rats poisoned
with lethal doses of thallous sulfate and treated with dithiocarb,
with special attention to renal changes.] Patol Pol, 31: 557-565
(in Polish).
Date AR, Cheung YY, & Stuart ME (1988) Application of inductively
coupled plasma mass spectrometry to the analysis of iron ores. J Anal
At Spectrom, 3: 653-658.
Davids P, Güthner G, Lange M, Lohrer W, & Vahrenholt F (1980)
[Situation and perspectives of the techniques for purification of
air.] Staub-Reinhalt Luft, 40: 392-402 (in German).
Davis RD, Beckett PHT, & Wollan E (1978) Critical levels of twenty
potentially toxic elements in young spring barley. Plant Soil,
49: 395-408.
Davis LE, Standefer JC, Kornfeld M, Abercrombie DM, & Butler C (1981)
Acute thallium poisoning: toxicological and morphological studies of
the nervous system. Ann Neurol, 10: 38-44.
Dawson GW, Jennings AL, Drozdowski D, & Rider E (1975/77) The acute
toxicity of 47 industrial chemicals to fresh and saltwater fishes.
J Hazard Mater, 1: 303-318.
De Groot G & van Heijst ANP (1988) Toxicokinetic aspects of thallium
poisoning. Methods of treatment by toxin elimination. Sci Total
Environ, 71: 411-418.
Den Dooren de Jong LE & Roman WB (1971) Tolerance of Azotobacter for
metallic and non-metallic ions. Antonie van Leeuwenhoek J Microbiol
Serol, 37: 119-124.
De Ruck A, Vandecasteele C, & Dams R (1987) Determination of thallium
in natural waters by electrothermal atomic absorption spectrometry.
Mikrochim Acta, II: 187-193.
De Ruck A, Vandecasteele C, & Dams R (1989) Determination of thallium
in solid environmental samples by electrothermal atomic absorption
spectrometry. Anal Lett, 22: 469-479.
DFG (German Research Council) (1990) [Maximal concentrations at the
place of work and biological tolerance values of material for work
1990.] Weinheim, Verlag Chemie (in German).
Di Gaudio MR & Hirshfield HI (1976) Respiratory toxicity of thallium
in Paramecium bursaria. J Cell Biol, 70: 60A.
Dilling WJ & Healey CW (1926) Influence of lead and the metallic ions
of copper, zinc, thorium, beryllium and thallium on the germination of
frogs' spawn and on the growth of tadpoles. Ann Appl Biol,
13: 177-188.
Dolgner R & Wiegand H (1982) [Biological monitoring of thallium -
medical aspects.] Staub-Reinhalt Luft, 42: 151-152 (in German).
Dolgner R, Brockhaus A, Ewers U, Wiegand H, Majewski F, & Soddemann H
(1983) Repeated surveillance of exposure to thallium in a population
living in the vicinity of a cement plant emitting dust containing
thallium. Int Arch Occup Environ Health, 52: 79-94.
Douglas KT, Bunni MA, & Baindur SR (1990) Thallium in biochemistry.
Int J Biochem, 22: 429-438.
Downs WL, Scott JK, Steadman LT, & Maynard EA (1960) Acute and
sub-acute toxicity studies of thallium compounds. Am Ind Hyg Assoc J,
21: 399-406.
Drucker H, Garland TR, & Wildung RE (1979) Metabolic response of
microbiota to chromium and other metals. In: Kharasch N ed. Trace
metals in health and disease. New roles of metals in biochemistry, the
environment, and clinical/nutritional studies. New York, Raven Press,
pp 1-25.
Ducket S, Hiller D, & Ballas SK (1983) Quantitation and localization
of 204thallium in the central and peripheral nervous system of adult
and young rats. Neurotoxicology, 4: 227-234.
Dumitru D & Kalantri A (1990) Electrophysiologic investigation of
thallium poisoning. Muscle Nerve, 13: 433-437.
Durbin PW, Scott KG, & Hamilton JG (1957) The distribution of
radioisotopes of some heavy metals in the rat. Berkeley, University of
California Press, pp 3: 1-34 (University of California Publications in
Pharmacology, Volume 3).
Dvorák P (1970) [Hexacyanoferrates (II) as thallium antidotes.
Preparation and properties.] Arzneimittelforschung, 20: 1886-1888
(in German).
Dvorák P (1973) [Removal by rhyolitic pumice of internally deposited
thallium from the rat.] Res Exp Med, 162: 63-66 (in German).
Dvornikov AG, Ovsyannikov LB, & Sidenko OG (1973) [Biogeochemical
dispersion haloes of chalcophyllite elements in gold ore
manifestations of the Nagolnyi Ridge (The Donbas).] Dopov Akad Nauk
Ukr RSR, B35: 490-494 (in Russian).
Dvornikov AG, Ovsyannikova LB, & Sidenko OG (1976) [Some
characteristics of the coefficients of biological absorption and the
biogeochemical coefficients at hydrothermal deposits of the Donets
Basin in relation to predicting hidden ore mineralization.]
Geokhimiya, 4: 626-633 (in Russian).
Ebarvia B, Macalalad E, & Rogue N (1988) Determination of silver,
cadmium, selenium, tellurium and thallium in geochemical exploration
samples by atomic absorption spectrometry. J Anal At Spectrom,
3: 199-203.
Edel Rade J, Marafante E, Sabbioni E, Gregotti C, Di Nucci A, & Manzo
L (1982) Placental transfer and retention of 201Tl-thallium in the
rat. Toxicol Lett, 11: 275-280.
EC (1987) [Legal regulations on hazardous substances.] Luxembourg,
Office for Official Publications of the European Community, vol 2
(in German).
Ehrhardt K (1927) [The effect of maternal intoxication with thallium
on the progeny.] Klin Wochenschr, 6: 1374-1375 (in German).
Emara M & Soliman MA (1950) The distribution of ingested thallium in
the tissues of animals. J Egypt Med Assoc, 33: 1-15.
Emsley J (1978) The trouble with thallium. New Sci, 79: 392-394.
Erbslöh J (1960) [Thallium poisoning in the second half of pregnancy.]
Arch Toxicol, 18: 156-159 (in German).
Ewan RC (1978) Toxicology and adverse effects of mineral imbalance
with emphasis on selenium and other minerals. In: Oehme FW ed.
Toxicity of heavy metals in the environment. New York, Marcel Dekker
Inc., vol 1, pp 445-489.
Ewers U (1988) Environmental exposure to thallium. Sci Total Environ,
71: 285-292.
Ewers U & Brockhaus A (1982) [Biological monitoring of thallium -
analysis and results.] Staub-Reinhalt Luft, 42: 149-151 (in German).
Favari L & Mourelle M (1985) Thallium replaces potassium in activation
of the (Na+ K+)-ATPase of rat liver plasma membranes.
J Appl Toxicol, 5: 32-34.
Feudell P (1982) [Thallium polyneuropathology.] Psychiatr Neurol Med
Psychol, 34: 23-25 (in German).
Fischer AB (1981) Comparative studies of heavy metal toxicity at the
cellular level. In: Proceedings of the 3rd International Conference on
Heavy Metals in the Environment, Toronto, pp 486-490.
Fitzek A & Henning A (1976) [Distribution of thallium in the body of a
pregnant cat.] Dtsch Tierärztl Wochenschr, 83: 66-68 (in German).
Flanjak J & Hodda AE (1988) Simultaneous determination of thallium,
silver and gold in urine by flame and graphite-furnace atomic
absorption spectrometry after extraction of the iodide complexes as
ion-pairs with tri- n-octylamine. Anal Chim Acta, 207: 283-289.
Ford JK, Eyring EJ, & Anderson CE (1968) Thallium chondrodystrophy in
chick embryos. An histological and biochemical investigation. J Bone
Jt Surg, 50A: 687-700.
Formigli L, Grégotti C, Di Nucci A, Edel J, Sabbioni E, & Manzo L
(1985) Reproductive toxicity of thallium. Experimental and clinical
data. Proceedings of the 5th International Conference on Heavy Metals
in the Environment, Toronto, vol 2, pp 15-18.
Formigli L, Scelsi R, Poggi P, Grégotti C, Di Nucci A, Sabbioni E,
Gottardi L, & Manzo L (1986) Thallium-induced testicular toxicity in
the rat. Environ Res, 40: 531-539.
Forth W & Henning CH (1979) [Thallium poisoning and the therapy.]
Dtsch Ärztebl, 76: 2803-2807 (in German).
Fowler BA (1988) Mechanism of indium, thallium, and arsine gas
toxicity. In: Clarkson TW, Friberg L, Nordberg GF, & Sager PR ed.
Biological monitoring of toxic metals. New York, London, Plenum Press,
pp 469-478.
Frerking H, Osterkamp A, & Lindfeld KA (1990) [Adverse effects in
cattle due to vaccination against foot-and mouth disease or poisoning
by thallium.] Dtsch Tierärztl Wochenschr, 97: 206-207 (in German).
Galba J (1982) [Analysis of trace concentrations of phytotoxic
elements in solid emissions from the thermal power plant ENO in soil
and plants.] Pol'nohospodárstvo, 28: 293-306 (in Czechoslovakian).
Galvan-Arzate S & Rios C (1994) Thallium distribution in organs and
brain regions of developing rats. Toxicology, 90: 63-69.
Garcia Bugarin M, Casas JB, Sordo J, & Filella M (1989) Thallium(I)
interactions in biological fluids: a potentiometric investigation of
thallium(I) complex equilibria with some sulfur-containing amino
acids. J Inorg Biochem, 35: 95-105.
Gastel B (1978) Clinical conferences at the Johns Hopkins hospital:
thallium poisoning. Johns Hopkins Med J, 142: 27-31.
Gefel A, Liron M, & Hirsch W (1970) Chronic thallium poisoning.
Isr J Med Sci, 6: 380-382.
Gehring PJ & Hammond PB (1967) The interrelation between thallium and
potassium in animals. J Pharmacol Exp Ther, 155: 187-201.
Geilmann W & Neeb KH (1959) [Application of the evaporation analysis
for minor amounts of material. II. The proof and determination of
minor contents of thallium.] Fresenius Z Anal Chem, 165: 251-268
(in German).
Geilmann W, Beyermann K, Neeb K-H, & Neeb R (1960) [Thallium, a
regularly occurring trace element in animals and plants.] Biochem Z,
333: 62-70 (in German).
Geldmacher-von Malinckrodt M, Stamm D, & Enders P (1984)
Interlaboratory survey on thallium in urine. Z Rechtsmed, 93: 219-225.
Ghezzi R & Bozza Marrubini M (1979) Prussian blue in the treatment of
thallium intoxication. Vet Hum Toxicol, 21: 64-66.
Gibson JE & Becker BA (1970) Placental transfer, embryotoxicity, and
teratogenicity of thallium sulfate in normal and potassium-deficient
rats. Toxicol Appl Pharmacol, 16: 120-132.
Gibson JE, Sigdestad CP, & Becker BA (1967) Placental transport and
distribution of thallium-204 sulfate in newborn rats and mice. Toxicol
Appl Pharmacol, 10: 378-411.
Glomme J (1983) Thallium and compounds. In: Encyclopaedia of
occupational health and safety, 3rd ed. Geneva, International Labour
Office, vol 2, pp 2170-2171.
Gluskoter HJ, Ruch RR, Miller WG, Cahill RA, Dreher GB, & Kuhn JK
(1977) Trace elements in coal: occurrence and distribution - Final
report. Urbana, Illinois, Illinois State Geological Survey
(EPA 600/7-77-064).
Goenechea S & Sellier K (1967) [The natural concentration of thallium
in humans.] Dtsch Z Gesamte Gerichtl Med, 60: 135-141 (in German).
Goldblatt D (1989) Pollutants and industrial hazards. In: Rowland LP
ed. Merritt's textbook of neurology, 8th ed. Philadelphia,
Pennsylvania, Lea and Febinger, pp 919-928.
Gorbauch H, Rump HH, Alter G, & Schmitt-Henco CH (1984) [Determination
of thallium in raw materials and environmental samples.] Fresenius Z
Anal Chem, 317: 236-240 (in German).
Gough LP, Shacklette HT, & Case AA (1979) Element concentrations toxic
to plants, animals, and man. US Geol Surv Bull, 1466: 1-80.
Graben N, Doss M, & Klöppel HA (1978) [Disturbances of porphyrin
metabolism by thallium poisoning.] Med Klin (Munich), 73: 1114-1116
(in German).
Graben N, Klöppel H-A, Heidemann H, & Weiler G (1980) [Therapy of
thallium poisoning with special consideration of pregnancy and
lactation.] Med Welt, 31: 1391-1394 (in German).
Grégotti C, di Nucci A, Formigli L, Sabbioni E, & Manzo L (1985)
Altered testicular enzyme patterns in rats after long-term exposure to
thallium sulfate. J Toxicol Clin Exp, 5: 265-271.
Grégotti C, Ponce R, & Faustman EM (1993) Mechanism of toxicity of Tl,
B and Pb on testicular cells in vitro. Abstracts of the 32nd Annual
Meeting of the Society of Toxicology. Toxicologist, 13: 298.
Gregus Z & Klaassen CD (1986) Disposition of metals in rats: a
comparative study of fecal, urinary, and biliary excretion and tissue
distribution of eighteen metals. Toxicol Appl Pharmacol, 85: 24-38.
Greving R & Gagel O (1928) [Polyneuritis after acute thallium
poisoning.] Klin Wochenschr, 7: 1323-1325 (in German).
Griepink B, Sager M, & Tölg G (1988) Determination of traces of
thallium in various matrices. Pure Appl Chem, 60: 1425-1436.
Gubernator K, Moroni W, Munder S, Franke B, Ruske B, & Teufel D (1979)
[The ambiguity of thallium contaminations in the neighbourhood of
cement plants - Heidelberg Seminar on Environmental Protection,
Freiburg, Germany, October 1979.] Heidelberg, Heidelberg Institute for
Research on Energy and Environment, Institute for Applied Ecology
(in German).
Guderian R (1977) Air pollution. Berlin, Springer Verlag.
Guidoboni RJ & Leipziger FD (1988) Glow discharge mass spectrometry -
the newest tool for high purity materials analysis. J Cryst Growth,
89: 16-20.
Günther M (1971) [Distribution and toxicity of thallium (I) in the rat
as influenced by colloidal ferrihexacyanoferrate (II).] Arch Toxicol,
28: 39-45 (in German).
Günther K & Umland F (1989) Bonding states of thallium and cadmium in
thallium-treated and native rape. J Inorg Biochem, 36: 63-74.
Gutknecht J (1983) Cadmium and thallous ion permeabilities through
lipid bilayer membranes. Biochim Biophys Acta, 735: 185-188.
Hagedorn-Götz H & Stoeppler M (1975) [The forensic proof of thallium
in human hairs by flameless atomic absorption spectrometry.] Arch
Toxicol, 34: 17-26 (in German).
Hajdu S & Nessler F (1985) [Chemistry, physics and occurrence of
thallium and its compounds.] Wiss Z Humboldt-Univ (Berlin)
Math-Naturwiss Reihe, 34: 752-755 (in German).
Hall BK (1972) Thallium-induced achondroplasia in the embryonic chick.
Dev Biol, 28: 47-60.
Hall BK (1977) Thallium-induced achondroplasia in chicken embryos and
the concept of critical periods during development. Teratology,
15: 1-15.
Hall BK (1985) Critical periods during development as assessed by
thallium-induced inhibition of growth of embryonic chick tibiae
in vitro. Teratology, 31: 353-361.
Hamaguchi H (1960) [Inorganic constituents in biological materials.
XIV. Thallium, selenium, and arsenic contents in Minamata districts.]
Nippon Kagaku Zasshi, 81: 920-927 (in Japanese).
Han HB, Kaiser G, & Tölg G (1982) Decomposition of biological
materials, rocks sand soils with simultaneous volatilization of trace
elements in pure oxygen under dynamic conditions. Anal Chim Acta,
134: 3-11.
Hapke H-J (1984) [The effects of metals on domestic animals.] In:
Merian E ed. [Metals in the environment.] Weinheim, Verlag Chemie,
pp 181-194 (in German).
Hapke H-J, Barke E, & Spikermann A (1980) [Accumulation of thallium in
the edible tissues of sheep and cattle according to the amount of
thallium in the feed.] Dtsch Tierärztl Wochenschr, 87: 376-378
(in German).
Hart MM & Adamson RH (1971) Antitumor activity and toxicity of salts
of inorganic group IIIa metals: aluminium, gallium, indium, and
thallium. Proc Natl Acad Sci (USA), 68: 1623-1626.
Hart MM, Smith CF, Yancey ST, & Adamson RH (1971) Toxicity and
antitumor activity of gallium nitrate and periodically related metal
salts. J Natl Cancer Inst, 47: 1121-1127.
Hasan M & Ali F (1981) Effects of thallium, nickel and cobalt
administration on the lipid peroxidation in different regions of the
rat brain. Toxicol Appl Pharmacol, 57: 8-13.
Hasan M & Haider SS (1989) Acetyl-homocystein thiolactone protects
against some neurotoxic effects of thallium. Neurotoxicology,
10: 257-261.
Hasan M, Bajpai VK, & Shipstone AC (1977a) Electron microscopic study
of thallium-induced alterations in the oligodendrocytes of the rat
area postrema. Exp Pathol, 13: 338-345.
Hasan M, Chandra SV, Bajpai VK, & Ali SF (1977b) Electron microscopic
effects of thallium poisoning on the rat hypothalamus and hippocampus:
biochemical changes in the cerebrum. Brain Res Bull, 2: 255-262.
Hasan M, Chandra SV, Dua PR, Raghubir R, & Ali SF (1977c) Biochemical
and electrophysiologic effects of thallium poisoning on the rat corpus
striatum. Toxicol Appl Pharmacol, 41: 353-359.
Hasan M, Ashraf I, & Ali SF (1977d) Effect of thallium intoxication on
the amino acid concentration of different regions of the rat brain.
Curr Sci, 46: 554-556.
Hasan M, Ashraf I, & Bajpai VK (1978a) Electron microscopic study of
the effects of thallium poisoning on the rat cerebellum. Forensic Sci,
11: 139-146.
Hasan M, Ali SF, & Tariq M (1978b) Levels of dopamine, norepinephrine
and 5-hydroxytryptamine in different regions of the rat brain in
thallium toxicosis. Acta Pharmacol Toxicol, 43: 169-173.
Hausman R & Wilson WJ jr (1964) Thallotoxicosis: a social menace.
J Forensic Sci, 9: 72-88.
Hayes WJ & Laws ER (1991) Handbook of pesticide toxicology - Volume 2:
Classes of pesticides. New York, London, Academic Press.
Heath A, Ahlmén J, Branegård B, Lindstedt S, Wickström I, & Andersen O
(1983) Thallium poisoning - Toxin elimination and therapy in three
cases. J Toxicol Clin Toxicol, 20: 451-463.
Heinrichs H (1982) [Metals in sewage sludge from municipal water
purification plants.] Naturwissenschaften, 69: 88-89 (in German).
Heinrichs H & Mayer R (1977) Distribution and cycling of major and
trace elements in two central European forest ecosystems. J Environ
Qual, 6(4): 402-407.
Heit M (1985) Concentrations of potentially toxic trace elements in
the muscle tissue of fish from acidic and circumneutral Adirondack
Lakes. Proceedings of the 5th International Conference on Heavy Metals
in the Environment, Toronto, vol 1, pp 655-657.
Henderson P, Hale TW, Shum S, & Habersang RW (1985) N-acetylcysteine
therapy of acute heavy metal poisoning in mice. Vet Hum Toxicol,
27: 522-525.
Henke G (1991) Thallium determination in biological materials by
radiochemical neuron activation analysis. Fresenius J Anal Chem,
339: 245-248.
Hennekes R (1983) [Impairment of retinal function in acute thallium
poisoning.] Klin Mon.bl Augenheilkd, 182: 334-336 (in German).
Henning CH & Forth W (1977) Absorption and secretion of thallous ions
on isolated intestinal segments in vitro of rats.
Naunyn-Schmiedeberg's Arch Pharmacol, 297(suppl II): R2.
Henning CH & Forth W (1982) The excretion of thallium(I) ions into the
gastrointestinal tract in situ of rats. Arch Toxicol, 49: 149-158.
Henning CH, Becker G, & Forth W (1982) Movement of thallium(I) ions
across the epithelium of intestinal segments in vitro of rats.
Naunyn-Schmiedeberg's Arch Pharmacol, 321: 157-163.
Henshaw JM, Heithmar EM, & Hinners TA (1989) Inductively coupled
plasma mass spectrometric determination of trace elements in surface
waters subject to acidic deposition. Anal Chem, 61: 335-342.
Herman MM & Bensch KG (1967) Light and electron microscopic studies of
acute and chronic thallium intoxication in rats. Toxicol Appl
Pharmacol, 10: 199-222.
Heydlauf H (1969) Ferric-cyanoferrate(II): an effective antidote in
thallium poisoning. Eur J Pharmacol, 6: 340-344.
Heyl T & Barlow RJ (1989) Thallium poisoning: a dermatological
perspective. Br J Dermatol, 121: 787-791.
Hill WH & Murphy MA (1959) An appraisal of an industrial air
contaminant by electrochromatography. Am Ind Hyg Assoc J, 20: 387-391.
Hill HAO, Pratt JM, Ridsdale S, Williams FR, & Williams RJP (1970)
Kinetics of substitution of co-ordinated carbanions in cobalt(III)
corrinoids. Chem Commun, 124: 341.
Hiltenkamp E & Jackwerth E (1988) [Investigations on the determination
of Bi, Cd, Hg, Pb and Tl in high-purity gallium by graphite furnace
AAS with atomization of metallic samples.] Fresenius Z Anal Chem,
332: 134-139 (in German).
Hoffmann G, Schweiger P, & School W (1982) [Uptake of thallium by
agriculturally and horticulturally useful plants.] Landwirtsch Forsch,
35: 45-54 (in German).
Hollunger G (1960) The effect of trivalent thallium on oxidative
phosphorylation. Acta Pharmacol Toxicol, 16: 347-356.
Holm J, Brehmer R-D, Müller S, & Wester D (1987) [Bioindication of
harmful substances taking roe deer and mallard as examples.]
Fleischwirtschaft, 67: 1145-1149 (in German).
Hologgitas J, Ullucci P, Driscoll J, Grauerholz J, & Martin H (1980)
Thallium elimination kinetics in acute thallotoxicosis. J Anal
Toxicol, 4: 68-73.
Howe HE (1971) Chapter 27. Thallium. In: Hampel CA ed. Rare metals
handbook. Huntinston, New York, Huntinston Publishing Company,
pp 529-535.
Hsie AW, Schenley RL, Tan E-L, Perdue SW, Williams MW, Hayden TL, &
Turner JE (1984) The toxicity of sixteen metallic compounds in Chinese
hamster ovary cells: a comparison with mice and Drosophila. Altern
Methods Toxicol, 2: 115-125.
Huber F, Schmidt U, & Kirchmann H (1978) Aqueous chemistry of
organolead and organothallium compounds in the presence of
microorganisms. In: Organometals and organometalloids: occurrence and
fate in the environment). Washington, DC, American Chemical Society,
pp 65-81 (ACS Symposium Series No. 82).
Hultin T & Näslund PH (1974) Effects of thallium(I) on the structure
and functions of mammalian ribosomes. Chem-Biol Interact, 8: 315-328.
Inturrisi CE (1969) Thallium activation of K+-activated phosphatases
from beef brain. Biochim Biophys Acta, 173: 567-569.
IPCS (International Programme on Chemical Safety) (1992) Poisons
information monograph: Thallium. Geneva, World Health Organization
(IPCS/INTOX/PIM.525).
IPS (Industrial Association for the Protection of Plants) ed. (1982)
[Active compounds in agents for plant protection and pest control.]
Offenbach, Bintz Verlag (in German).
Itawi RK & Turel ZR (1987) Determination of thallium in biological
samples by thermal neutron activation analysis and low-level
beta-counting. J Radianal Nucl Chem, 115: 141-147.
Ivashchenko AT & Balmukhanov BS (1974) [Effect of monovalent ions on
the adenosine triphosphatase activity of ascites tumor cells.]
Izv Akad Nauk Kaz SSR Ser Biol, 12: 73-78 (in Russian).
Ivashchenko AT, Balmukhanov BS, & Toktamysova ZS (1973) [Effects of
monovalent ions on the glycolysis and respiration of ascite tumor
cells.] Tsitologiya, 15: 1487-1491 (in Russian).
Iyengar GV, Kollmer WE, & Bowen HJM (1978) The elemental composition
of human tissues and body fluids. Weinheim, Verlag Chemie.
Jeantet AY, Ballan-Dufrançais C, & Martoja R (1977) Insect resistance
to mineral pollution. Importance of spherocrystal in ionic regulation.
Rev Ecol Biol Sol, 14: 563-582.
Johnson CA (1976) The determination of some toxic metals in human
liver as a guide to normal levels in New Zealand. Part I.
Determination of Bi, Cd, Cr, Co, Cu, Pb, Mn, Ni, Ag, Tl and Zn. Anal
Chim Acta, 81: 69-74.
Junghans V & Nessler F (1985) [The clinical signs of chronic thallium
intoxication shown in occupational thallium poisonings.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 778-780 (in German).
Kaeser HE & Lambert EH (1962) Nerve function in experimental
polyneuritis. Electroencephalogr Clin Neurophysiol Prog Electromyogr,
22(suppl): 29-35.
Kamerbeek HH, Rauws AG, ten Ham M, & van Heijst ANP (1971a) Dangerous
redistribution of thallium by treatment with sodium
diethyldithiocarbamate. Acta Med Scand, 189: 149-154.
Kamerbeek HH, Rauws AG, Ten Ham M, & van Heijst ANP (1971b) Prussian
blue in therapy of thallotoxicosis: An experimental and clinical
investigation. Acta Med Scand, 189: 321-324.
Kaplan DI, Adriano DC, & Sajwan KS (1990) Thallium toxicity in bean.
J Environ Qual, 19: 359-365.
Karkos J (1971) [Neuropathological findings in thallium
encephalopathy.] Neurol Neurochir Pol, 21: 911-915 (in Polish).
Karnofsky DA & Lacon CR (1964) Effects of drugs on the skeletal
development of the chick embryo. Clin Orthop Relat Res, 33: 59-70.
Karnofsky DA, Ridgway LP, & Patterson PA (1950) Production of
achondroplasia in the chick embryo with thallium. Proc Soc Exp Biol
Med, 73: 255-259.
Kazantzis G (1986) Thallium. In: Friberg L, Nordberg GF, & Vouk V ed.
Handbook on the toxicology of metals, 2nd ed. Amsterdam, Oxford, New
York, Elsevier Science Publishers, pp 549-567.
Kazantzis G (1994) Thallium and tin. In: Alessio L, Berlin A, Roi R, &
van der Venne, M-Th ed. Biological indicators for the assessment of
human exposure to industrial chemicals. Luxembourg, European
Commission, Health and Safety Directorate, pp 41-92 (Report
EUR 14815/EN).
Kemper FH (1979) [Thallium poisonings.] Münch Med Wochenschr,
121: 1357-1358 (in German).
Kemper FH & Bertram HP (1984) [Thallium.] In: Merian E ed. [Metals in
the environment.] Weinheim, Verlag Chemie, pp 571-583.
Kemper FH & Bertram HP (1991) Thallium. In: Merian E ed. Metals and
their compounds in the environment. Weinheim, Verlag Chemie,
pp 1227-1241.
Kennedy P & Cavanagh JB (1976) Spinal changes in the neuropathy of
thallium poisoning. J Neurol Sci, 29: 295-301.
Kijewski H (1984) Nature of the black zones in hair after ingestion of
thallium. Arch Kriminol, 173: 36-44.
Klahre P, Valenta P, & Nürnberg HW (1978) [A standardized pulse
inverse voltametric analytical method for testing drinking water for
toxic metals. Part 1: Simultaneous determination of copper, cadmium,
lead and zinc, and lead and thallium.] Vom Wasser, 51: 199-219
(in German)
Klöppel A & Weiler G (1978) [Contribution to heavy metal poisonings,
especially by thallium.] Dtsch Med Wochenschr, 103: 75-76 (in German).
Knapp G (1985) Sample preparation techniques - an important part in
trace element analysis for environmental research and monitoring. Int
J Environ Anal Chem, 22: 71-83.
Kogan BI (1970) [Thallium.] Redk Elem, 5: 27-48 (in Russian).
Kolb Meyers V (1983) Chemicals which cause birth defects - teratogens:
a special concern of research chemists. Sci Total Environ, 32: 1-12.
Konermann H, Crössann G, & Hoppenbrock KH (1982) [Investigations of
the influence of thallium-contaminated food rations on performance,
health and meat residues in fattening pigs.] Tierärztl Umsch,
37: 8-21 (in German).
Korkisch J & Steffan J (1979) Determination of thallium in natural
waters. Int J Environ Anal Chem, 6: 111-118.
Kosta L (1982) Contamination as a limiting parameter in trace
analysis. Talanta, 29: 985-992.
Krassowsky GN, Yurasova OI, Charyev OG, Skrypnikov AI, & Varshavskaya
SP (1977) [Prognosis of the gonadotoxic action of heavy metals judging
by the primary effect of material accumulation.] Gig i Sanit, 7: 11-16
(in Russian).
Krassowsky GN, Wasjukowitsch LJ, Tscharyjew OG, & Kenessarijew UI
(1980) [Experimental studies on making quick prognoses of a
gonadotoxic effect of inorganic compounds.] Z Gesamte Hyg Grenzgeb,
26: 336-338 (in German).
Kravzov J, Rios C, Altagracia M, Monroy-Noyola A, & López F (1993)
Relationship between physicochemical properties of Prussian blue and
its efficacy as antidote against thallium poisoning. J Appl Toxicol,
31: 213-216.
Ku DD, Akera T, Oldaard MK, & Brody TM (1978) Effects of lithium and
thallous ions on sodium. Activity in the guinea pig heart and their
relationship to the positive inotropic action. Naunyn-Schmiedeberg's
Arch Pharmacol, 304: 167-173.
Kwan KHM & Smith S (1988) The effect of thallium on the growth of
Lemna minor and plant tissue concentrations in relation to both
exposure and toxicity. Environ Pollut, 52: 203-220.
Lameijer W & van Zwieten PA (1977a) Kinetic behaviour of thallium in
the rat. Accelerated elimination of thallium owing to treatment with
potent diuretic agents. Arch Toxicol, 37: 265-273.
Lameijer W & van Zwieten PA (1977b) Kinetics of thallium elimination
in the rat after acute and subacute intoxication; influence of
diuretic treatment. Proc Eur Soc Toxicol, 18: 163-164.
Lameijer W & van Zwieten PA (1979) The efficacy of a potassium-rich
diet compared to diuretic treatment on the renal elimination of
thallium in the rat. Arch Toxicol, 42: 33-41.
Landauer W (1930) [Influence of thallium poisoning of the father on
his descendants. Experiments on roosters.] Arch Gewerbepathol
Gewerbehyg, 1: 791-792 (in German).
Larson CP, Keller WN, & Manges JD (1939) Accidental canine
thallotoxicosis and dangers of thallium used as a rodenticidal agent.
J Am Vet Med Assoc, 95: 486-489.
LeBlanc GA (1984) Interspecies relationships in acute toxicity of
chemicals to aquatic organisms. Environ Toxicol Chem, 3: 47-60.
LeBlanc GA & Dean JW (1984) Antimony and thallium toxicity to embryos
and larvae of fathead minnows (Pimephales promelas). Bull Environ
Contam Toxicol, 32: 565-569.
Lehmann FPA & Favari L (1984) Parameters for the adsorption of
thallium ions by activated charcoal and Prussian blue. J Toxicol Clin
Toxicol, 22: 331-339.
Lehmann FPA & Favari L (1985) Acute thallium intoxication: kinetic
study of the relative efficacy of several antidotal treatments in
rats. Arch Toxicol, 57: 56-60.
Lehn H & Bopp M (1987) [Heavy metals in soil and the determination of
the availability for plants.] Angew Bot, 61: 467-481 (in German).
Leloux MS, Lich NP, & Claude JR (1987a) Flame and graphite furnace
atomic absorption spectroscopy methods for thallium - a review. At
Spectrom, 8: 71-77.
Leloux MS, Lich NP, & Claude JR (1987b) Experimental studies on
thallium toxicity in rats. I. Localization and elimination of thallium
after oral acute and sub-acute intoxication. J Toxicol Clin Exp,
7: 247-257.
Le Quesne PM (1984) Toxic neuropathies. In: Asbury AK & Gilliatt RW
ed. Butterworths international medical reviews: Neurology. Volume 4:
Peripheral nerve disorders - a practical approach. London,
Butterworths, pp 184-204.
Letourneau VA, Joshi BM, & Butler LC (1987) Comparison between Zeeman
and continuum background correction for graphite furnace AAS on
environmental samples. At Spectrom, 8: 145-149.
Levander OA & Argrett LC (1969) Effects of arsenic, mercury, thallium,
and lead on selenium metabolism in rats. Toxicol Appl Pharmacol,
14: 308-314.
Liang Y, Shi J, Chen L, & Ding G (1980) [Dimercaptosuccinic acid per
os promoted the excretions of Pb, Cu, Sb, Sr, Tl and Pm.] Acta Pharm
Sinica, 15: 335-340 (in Chinese).
Lide DR (1990) CRC handbook of chemistry and physics: A ready
reference book of chemical and physical data, 71st ed. Boca Raton,
Florida, CRC Press.
Lie R, Thomas RG, & Scott JK (1960) The distribution and excretion of
thallium-204 in the rat, with suggested MPC's and a bio-assay
procedure. Health Phys, 2: 334-340.
Lillie WI & Parker HL (1932) Retrobulbar neuritis due to thallium
poisoning. J Am Med Assoc, 98: 1347-1349.
Lindegren CC (1971) Mitochondria in intoxication and detoxication.
Physiol Chem Phys, 3: 499-500.
Lindegren CC & Lindegren G (1973a) Mitochondrial modification and
respiratory deficiency in the yeast cell caused by cadmium poisoning.
Mutat Res, 21: 315-322.
Lindegren CC & Lindegren G (1973b) Oxidative detoxication of thallium
in the yeast mitochondria. Antonie van Leeuwenhoek J Microbiol Serol,
39: 351-353.
Ling GN (1977) Thallium and cesium in muscle cells compete for the
adsorption sites normally occupied by K+. Physiol Chem Phys,
9: 217-225.
Linsdale JM (1931) Facts concerning the use of thallium in California
to poison rodents - Its destructiveness to game birds, song birds and
other valuable wild life. Condor, 33: 92-106.
Linton RW, Loh A, Natusch DFS, Evans CA Jr, & Williams P (1976)
Surface predominance of trace elements in airborne particles. Science,
191: 852-854.
LIS (Land Institute for Protection against Emissions) (1980)
[Environmental burden by thallium. Investigations in the neighbourhood
of the Dyckerhoff cement plant in Lengerich and other
thallium-emitting plants in the country NW.] Bonn, Bonn University
Press (in German).
Liu G (1986) [The protective effect of selenium on myocardial cells.]
Chung-hua Ping Li Hsueh Tsa Chih, 15: 252-254 (in Chinese).
Lockwood TH (1974) The analysis of asbestos for trace metals. Am Ind
Hyg Assoc J, 35: 245-251.
Logan PG, Lepp NW, & Phipps DA (1983) Thallium uptake by higher
plants. In: Proceedings of the 4th International Conference on Heavy
Metals in the Environment, Toronto, vol 1, pp 642-645.
Logan PG, Lepp NW, & Phipps DA (1984) Some aspects of thallium uptake
by higher plants. In: Proceedings of the 18th Trace Substances in
Environmental Health Conference, Columbia, 4-7 June 1984, pp 570-575.
Lohmann H, Csissaky M, & Wiegand H (1989) The action of thallium on
the excitability of CA1 pyramidal cells in hippocampal slices.
Neurotoxicol Teratol, 11: 545-549.
Luckey TD & Venugopal B (1977) Metal toxicity in mammals. Volume 1:
Physiologic and chemical basis for metal toxicity. New York, Plenum
Press.
Luckit J, Mir N, Hargreaves M, Costello C, & Gazzard B (1990)
Thrombocytopenia associated with thallium poisoning. Hum Exp Toxicol,
9: 47-48.
Ludolph A, Elger CE, Sennhenn R, & Bertram HP (1986) Chronic thallium
exposure in cement plant workers: clinical and electrophysiological
data. Trace Elem Med, 3: 121-125.
Ludwig E (1961) [Pathognomonic hair findings in thallium poisoning and
their significance.] Hautarzt, 12: 456-459 (in German).
Lund A (1956a) Distribution of thallium in the organism and its
elimination. Acta Pharmacol Toxicol, 12: 251-259.
Lund A (1956b) The effect of various substances on the excretion and
the toxicity of thallium in the rat. Acta Pharmacol Toxicol,
12: 260-268.
Lynch GR, Lond MB, & Scovell JMS (1930) The toxicology of thallium.
Lancet, 219(5599): 1340-1344.
Machtey I & Bandmann M (1961) The cardiovascular system in thallium
poisoning. Proc Staff Meet Beilinson Hosp, 10(suppl): 140-142.
McCool MM (1933) Effect of thallium sulphate on the growth of several
plants and on nitrification in soils. Contrib Boyce Thompson Inst,
5: 289-296.
McDonald JM (1941) Metallic poisons. Ind Med, 10: 447-452.
McKillop A & Taylor EC (1973) Thallium in organic synthesis. Chem Br,
9: 4-11.
McLaren JW, Beauchemin D, & Berman SS (1987) Application of isotope
dilution inductively coupled plasma mass spectrometry to the analysis
of marine sediments. Anal Chem, 59: 610-613.
Mahoney W (1933) Further notes on retrobulbar neuritis due to thallium
poisoning. Yale J Biol Med, 6: 583-597.
Maier EA, Dietemann-Molard A, Rastegar F, Heimburger R, Ruch C, Maier
A, Roegel E, & Leroy MJF (1986) Simultaneous determination of trace
elements in lavage fluids from human bronchial alveoli by
energy-dispersive x-ray fluorescence. 2. Determination of abnormal
lavage contents and verification of the results. Clin Chem,
32: 664-668.
Maier-Reiter W, Blind G, Klump A, Wystrcil HG, & Arndt U (1987)
[Ecotoxicology of thallium. Effect on the pigment system of plants.]
Agrar-Umweltforsch Baden-Württemberg, 16: 22-36 (in German).
Makridis H & Amberger A (1989a) [Uptake of thallium from cement
factory dust in pot trials with green rape, bush beans and rye grass.]
Landwirtsch Forsch, 42: 324-332 (in German).
Makridis H & Amberger A (1989b) [Effect of increasing levels of
thallium on yield and uptake of thallium and further mineral nutrients
by bush beans and green rape grown in nutrient solution]. Landwirtsch
Forsch, 42: 333-343 (in German).
Malcolm D (1979) Toxicology of metals other than lead. In: Gardner AW
ed. Current approaches to occupational medicine. Bristol, John Wright
and Sons Ltd., pp 18-43.
Manning DC & Slavin W (1988) The determination of thallium with the
stabilized temperature platform furnace and Zeeman background
correction. Spectrochim Acta, 43B: 1157-1165.
Manzo L & Sabbioni E (1988) Thallium toxicity and the nervous system.
In: Bondy SC & Prasad KN ed. Metal neurotoxicity. Boca Raton, Florida,
CRC Press, pp 35-54.
Manzo L, Scelsi R, Moglia A, Poggi P, Alfonsi E, Pietra R, Mousty F, &
Sabbioni E (1983) Long-term toxicity of thallium in the rat. In: Brown
SS & Savory J ed. Chemical toxicology and clinical chemistry of
metals. Proceedings of 2nd International Conference, Montreal, Canada.
New York, London, Academic Press, pp 401-405.
Marcus RL (1985) Investigation of a working population exposed to
thallium. J Soc Occup Med, 35: 4-9.
Marwaha J, Freedman R, & Hoffer B (1980) Electrophysiological changes
at a central noradrenergic synapse during thallium toxicosis. Toxicol
Appl Pharmacol, 56: 345-352.
Mason B (1966) Principles of geochemistry. New York, Chichester,
Brisbane, Toronto, John Wiley and Sons.
Masri MS (1976) Binding of metallic ions to keratins. Proceedings of
the 5th International Wool Textile Research Conference,
vol 3, pp 1-12.
Matera MG, Marrazzo R, Berrino L, Fillippelli W, Russo S, Rossi F,
Cilento M, & Marmo E (1986) [Vascular effects of perinatal exposure of
rats to thallium.] Rend Atti Accad Sci Med Chir, 140: 177-202
(in Italian).
Mathis BJ & Kevern NR (1973) Distribution of mercury, cadmium, lead
and thallium in a eutrophic lake. Washington, DC, US Department of
Commerce, National Technical Information Service (NTIS PB-221993).
Mathis BJ & Kevern NR (1975) Distribution of mercury, cadmium, lead
and thallium in a eutrophic lake. Hydrobiologia, 46: 207-222.
Mathys W (1981) Thallium in river sediments of North-West Germany: its
sources of input and its relation to other heavy metals. In:
Proceedings of the International Conference on Heavy Metals in the
Environment, Amsterdam, September 1981. Luxembourg, Commission of the
European communities WHO, pp 347-350.
Matthews AD & Riley JP (1969) The determination of thallium in
silicate rocks, marine sediments and sea water. Anal Chim Acta,
48: 25-34
Melnick RL, Monti LG, & Motzkin M (1976) Uncoupling of mitochondrial
oxidative phosphorylation by thallium. Biochem Biophys Res Commun,
69: 68-73.
Merguet P, Schümann HJ, Murata T, Rausch-Stroomann JG, Schröder E,
Parr D, & Bock KD (1969) [Investigations of the pathogenesis of
hypertension and sinus tachycardia in human thallium poisonings.]
Dtsch Arch Klin Med, 216: 1-20 (in German).
Metter D & Vock R (1984) [Investigations of the structure of hairs in
thallium poisoning.] Z Rechtsmed, 91: 201-214 (in German).
Meyer H & Tal M (1957) Studies on thallium poisoning. I. The
detoxifying action of some compounds containing sulphur and labile
methyl groups. J Sci Food Agric, 8: 516-519.
Micke H, Bertram HP, & Kemper FH (1983) [Thallium.] In: [Ullmann's
encyclopedia of technical chemistry], 4th ed. Weinheim, Verlag Chemie,
vol 23, pp 103-114 (in German).
Minoia C, Sabbioni E, Appostoli P, Pietra R, Pozzoli L, Gallorini M,
Nicolaou G, Alessio L, & Capodaglio E (1990) Trace element reference
values in tissues from inhabitants of the European Community I. A
study of 46 elements in urine, blood and serum of Italian subjects.
Sci Total Environ, 95: 89-105.
MISA (Municipal/Industrial Strategy for Abatement) (1988/89)
Preliminary report for the first six months of monitoring in the
petroleum refining sector (December 1, 1988 to May 31, 1989). Ontario,
Canada, Ministry of the Environment.
MISA (Municipal/Industrial Strategy for Abatement) (1991a) Status
report on the effluent monitoring data for the iron and steel sector
for the period from November 1, 1989 to October 31, 1990. Ontario,
Canada, Ministry of the Environment.
MISA (Municipal/Industrial Strategy for Abatement) (1991b) Preliminary
report on the first six months of process effluent monitoring in the
MISA pulp and paper sector (January 1, 1990 to June 30, 1990).
Ontario, Canada, Ministry of the Environment.
Moeschlin S (1965) [Poisoning: diagnosis and treatment.] Stuttgart,
Georg Thieme (in German).
Moeschlin S (1986) [Poisoning: diagnosis and treatment.] Stuttgart,
Georg Thieme (in German).
Möllhoff G, Schmidt G, & Bösche J (1979) [Thallium poisoning.
Neuromedical aspects.] Arch Kriminol, 163: 1-13 (in German).
Montoya Cabrera MA, Pérez Lucio C, Badillo Torre FF, Barquet Barquet
RM, & López Bárcenas F (1979) [Thallium intoxication: treatment with
D-penicillamine.] Rev. Med Inst Mex Seguro Soc, 18: 211-214
(in Spanish).
Mottet NK (1985) Morphogenic injury by chemical agents. In: Mottet NK
ed. Environmental pathology. New York, Oxford University Press,
pp 82-125.
Mourelle M, Favari L, & Amezcua JL (1988) Protection against thallium
hepatotoxicity by silymarin. J Appl Toxicol, 8: 351-354.
Muller L (1961) [Thallium poisoning.] In: Baader EW ed. [Handbook of
total occupational medicine.] Berlin, Urban & Schwarzenberg, vol II,
part 1, pp 244-250.
Müller-Brand J, Staub JJ, & Friedrich R (1984) [Thallium scintigraphy
of the thyroid as an alternative procedure for the demonstration of
functionally silent thyroid tissue.] Schweiz Med Wochenschr,
114: 1686-1689 (in German).
Mullins LJ & Moore RD (1960) The movement of thallium ions in muscle.
J Gen Physiol, 43: 759-773.
Munch JC (1934a) Antidotes. III. Thallium. J Am Pharm Assoc,
23: 91-94.
Munch JC (1934b) Human thallotoxicosis. J Am Med Assoc,
102: 1929-1934.
Munch JC, Ginsburg HM, & Nixon CE (1933) The 1932 thallotoxicosis
outbreak in California. J Am Med Assoc, 100: 1315-1319.
Munch B, Clausen B, & Karlog O (1974) Thallium poisoning in red foxes
(Vulpes vulpes) and badgers (Meles meles) in Denmark. Nord
Veterinärmed, 26: 323-338.
Muramatsu Y & Parr RM (1985) Survey of currently available reference
materials for use in connection with the determination of trace
elements in biological and environmental materials. Vienna,
International Atomic Energy Agency (IAEA/RL/128).
Nachman M & Hartley PL (1975) Role of illness in producing learned
taste aversions in rats. A comparison of several rodenticides. J Comp
Physiol Psychol, 89: 1010-1018.
Naganuma A, Tanaka T, Maeda K, Matsuda R, Tabata-Hanyu J, & Imura N
(1983) The interaction of selenium with various metals in vitro and
in vivo. Toxicology, 29: 77-86.
Natusch DFS (1982) Size distributions and concentrations of trace
elements in particulate emissions from industrial sources. VDI-Ber,
429: 253-260.
Natusch DFS & Wallace JR (1974) Urban aerosol toxicity: the influence
of particle size. Science, 186: 695-699.
Natusch DFS, Wallace JR, & Evans CA Jr (1974) Toxic trace elements:
preferential concentration in respirable particles. Science,
183: 202-204.
Negherbon WO (1959) Handbook of toxicology. Volume 3: Insecticides.
Philadelphia, Pennsylvania, W.B. Saunders Company, pp 750-751.
Nehring D (1962/63) [Investigations of the toxicological effects of
thallium ions on fish and animal food of fish.] Z Fischerei,
11: 557-561 (in German).
Nessler F (1985a) [Experimental studies made on animals to establish
the toxicity of thallium and its compounds.] Wiss Z Humboldt-Univ
(Berlin) Math-Naturwiss Reihe, 34: 761-766 (in German).
Nessler F (1985b) [Importance and history of occupational thallium
intoxications as a type of chronic thallium poisoning.] Wiss Z
Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 773-777 (in German).
Nessler F & Briese C (1985) [Acute and subacute thallium intoxications
of humans.] Wiss Z Humboldt-Univ (Berlin) Math-Naturwiss Reihe,
34: 767-772 (in German).
Neubert D & Bluth U (1985) Effect of thallium on limb development in
organ culture. Teratology, 32: 29A.
NIOSH (1984) NIOSH manual of analytical methods: Method 7300.1-7300.5.
Cincinnati, Ohio, National Institute for Occupational Safety and
Health.
NIOSH (1985/86) RTECS - Registry of toxic effects of chemical
substances. Cincinnati, Ohio, National Institute for Occupational
Safety and Health, p 4637.
NIOSH (1992) NIOSH recommendations for occupational safety and health:
Compendium of policy documents and statements. Cincinnati, Ohio,
National Institute for Occupational Safety and Health (Publication
No. 92-100).
Nisticò G, Faraone V, Naccari F, Ammendola D, Germanò D, & Marmo E
(1984) Lack of effects of thallium on GABAergic mechanisms in discrete
areas of the rat brain. Res Commun Chem Pathol Pharmacol, 46: 35-42.
Noddack I & Noddack W (1939) [The frequency of heavy metals in sea
organisms.] Ark Zool, A32(4): 1-35 (in German).
Nogami H & Terashima Y (1973) Thallium-induced achondroplasia in the
rat. Teratology, 8: 101-102.
Nolan C, Duke E, Lorenzon G, Sabbioni E, & Marafante E (1984) Heavy
metal uptake and intracellular binding in isolated gill preparations
of Mytilus galloprovincialis L. Sci Total Environ, 40: 83-92.
Norris P, Man WK, & Hughes MN (1976) Toxicity and accumulation of
thallium in bacteria and yeast. Arch Microbiol, 110: 279-286.
Nriagu JO & Pacyna JM (1988) Quantitative assessment of worldwide
contamination of air, water and soils by trace metals. Nature (Lond),
333: 134-139.
Oehme FW (1972) Mechanisms of heavy metal toxicities. Clin Toxicol,
5: 151-167.
Oehme FW (1978) Mechanisms of heavy metal inorganic toxicities. In:
Oehme FW ed. Toxicity of heavy metals in the environment. New York,
Marcel Dekker Inc., vol 1, pp 69-85.
Ohnesorge FK (1985) [Toxicological rating of arsenic, lead, cadmium,
nickel, thallium and zinc.] Düsseldorf, VDI-Association of German
Engineers Press (Progress Reports Series No. 15, Environmental
Technology No. 38) (in German).
Olsen I & Jonsen J (1982) Whole-body autoradiography of 204Tl in
embryos, fetuses and placentas of mice. Toxicology, 23: 353-358.
Ormerod MJ (1928) Pharmacological and toxicological aspects of
thallium. Can Med Assoc J, 19: 663-665.
Osorio-Rico L, Galván-Arzate S, & Ríos C (1994) Brain regional
monoaminergic activity after acute thallium administration. Abstracts
of the 33rd Annual Meeting of the Society of Toxicology. Toxicologist,
14: 267.
Ostádalová I & Babicky A (1987) Protective effect of thallium against
the formation of selenium cataract in rats. Biol Trace Elem Res,
14: 101-103.
Overnell J (1975) The effect of some heavy metal ions on
photosynthesis in a freshwater alga. Pestic Biochem Physiol, 5: 19-26.
Pai V (1987) Acute thallium poisoning. Prussian blue therapy in 9
cases. West Indian Med J, 36: 256-258.
Palermo D, Chiaravalle AE, D'Errico M, Milillo MA, & De Natale G
(1983) [An occurrence of thallium poisoning in fish.] Clin Vet,
106: 129-131 (in Italian).
Pallaghy CK (1972) Localization of thallium in stomata is independent
of transpiration. Aust J Biol Sci, 25: 415-417.
Papp JP, Gay PC, Dodson VN, & Pollard HM (1969) Potassium chloride
treatment in thallotoxicosis. Ann Intern Med, 71: 119-123.
Paschal DC & Bailey GG (1986) Determination of thallium in urine with
Zeeman effect graphite furnace atomic absorption. J Anal Toxicol,
10: 252-254.
Paschal DC, DiPietro ES, Phillips DL, & Gunter EW (1989) Age
dependence of metals in hair in a selected U.S. population. Environ
Res, 48: 17-28.
Paulson G, Vergara G, Young J, & Bird M (1972) Thallium intoxication
treated with dithizone and hemodialysis. Arch Intern Med,
129: 100-103.
Peele DB, MacPhail RC, & Farmer JD (1986) Flavor aversions induced by
thallium sulfate: importance of route of administration. Neurobehav
Toxicol Teratol, 8: 273-277.
Peele DB, Farmer JD, & MacPhail RC (1987) Conditioned flavor
aversions: applications in assessing the efficacy of chelators in the
treatment of heavy-metal intoxication. Toxicol Appl Pharmacol,
88: 397-410.
Peisach M, Lackay LG, & Gihwala D (1989) Measurement of proton-induced
prompt low energy photons by high resolution spectrometry. Analyst,
114: 279-286.
Petersohn KL (1960) [Thallium poisonings in pregnancy.] Arch Toxikol,
18: 160-164 (in German).
Peterson ER & Murray MR (1965) Patterns of peripheral demyelination
in vitro. Ann NY Acad Sci, 122: 39-50.
Piazolo P, Franz HE, Brech W, Walb D, & Wilk G (1971) [Treatment of
thallium poisoning by hemodialysis.] Dtsch Med Wochenschr,
96: 1217-1222 (in German).
Pielow E (1979) [Pollution by thallium in the environment of a cement
plant.] Umwelt, 5: 394-396 (in German).
Pieper B & Austenfeld F-A (1985) [Phytotoxicity of thallium (Tl) in
culture solution. Part 2: Effects of Tl(III) on the growth and heavy
metal contents of pea and field bean plants.] Z Pflanzenernähr
Bodenkd, 148: 83-91 (in German).
Potes-Gutierrez J & Del Real E (1966) Acute thallium intoxication. Ind
Med Surg, 35: 618-619.
Pötsch K & Austenfeld F-A (1985) [Phytotoxicity of thallium (Tl) in
culture solution. Part 1: Effects of Tl(I) on the growth and heavy
metal contents of pea and field bean plants.] Z Pflanzenernähr
Bodenkd, 148: 73-82 (in German).
Prick JJG (1979) Thallium poisoning. In: Vinken PJ & Bruyn GW ed.
Handbook of clinical neurology: Intoxications of the nervous system.
Amsterdam, North-Holland Publishers, vol 36, part 1, pp 239-278.
Prinz B, Krause GHM, & Stratmann H (1979) [Damages by thallium in the
neighbourhood of the Dyckerhoff cement plant in Lengerich, Westfalen.]
Staub-Reinhalt Luft, 39: 457-462 (in German).
Puddu A, Pettine M, Bacciu F, La Noce T, & Pagnotta R (1985) Thallium
and chromium toxicity in phytoplankton cultures. Proceedings of the
5th International Conference on Heavy Metals in the Environment,
Toronto. vol 2, pp 304-306.
Puddu A, Pettine M, La Noce T, Pagnotta R, & Bacciu F (1988) Factors
affecting thallium and chromium toxicity to marine algae. Sci Total
Environ, 71: 572.
Puerner NJ & Siegel SM (1972) The effects of mercury compounds on the
growth and orientation of cucumber seedlings. Physiol Plant,
26: 310-312.
Que Hee SS & Boyle JR (1988) Simultaneous multielemental analysis of
some environmental and biological samples by inductively coupled
plasma atomic emission spectrometry. Anal Chem, 60: 1033-1042.
Rao DV, Govelitz GF, & Sastry KSR (1983) Radiotoxicity of thallium-201
in mouse testes: inadequacy of conventional dosimetry. J Nucl Med,
24: 145-153.
Rauws AG & Canton JH (1976) Adsorption of thallium ions by Prussian
Blue. Bull Environ Contam Toxicol, 15: 335.
Reyners H, Gianfelici de Reyners E, Maisin JR, Winneke G, & Csicsaky M
(1981) Effects of different heavy metals (Cd, Tl, Zn and Pb) in the
central nervous system: a morphological assay. In: Proceedings of the
International Conference on Heavy Metals in the Environment,
Amsterdam, September 1981. Luxembourg, Commission of the European
communities WHO, pp 495-497.
Richards OW (1932) The stimulation of yeast growth by thallium, a
"bios" impurity of asparagine. J Biol Chem, 96: 405-418.
Richelmi P, Bono F, Guardia L, Ferrini B, & Manzo L (1980) Salivary
levels of thallium in acute human poisoning. Arch Toxicol,
43: 321-325.
Ridgway LP & Karnofsky DA (1952) The effects of metals on the chick
embryo: toxicity and production of abnormalities in development. Ann
NY Acad Sci, 55: 203-215.
Ridley WP, Dizikes LJ, & Wood JM (1977) Biomethylation of toxic
elements in the environment. Science, 197: 329-332.
Ríos C & Monroy-Noyola A (1992) D-penicillamine and Prussian blue as
antidotes against thallium intoxication in rats. Toxicology,
74: 69-76.
Ríos C, Galván-Arzate S, & Tapia R (1989) Brain regional thallium
distribution in rats acutely intoxicated with Tl2SO4. Arch
Toxicol, 63: 34-37.
Ríos C, Kravsov J, Altagracia M, Lopez-Naranjo F, & Monroy A (1991)
Efficacy of Prussian blue against thallium poisoning: effect of
particle size. Proc West Pharmacol, 34: 61-63.
Romero Romero E, Cherem JH, & Lifshitz A (1989) [Arterial hypertension
in acute thallitoxicosis.] Rev Med Inst Mex Seguro Soc, 27: 43-47
(in Spanish).
Rosenstock L & Cullen MR (1986) 9. Neurologic disease. In: Rosenstock
L & Cullen MR ed. Clinical occupational medicine. Philadelphia,
Pennsylvania, W.B. Saunders Company, pp 118-134.
Rossi F, Lampa E, Grella A, Vacca C, Giordano L, Perna D, Rosatti F,
Spaziante G, Giasi M, Imperatore A, Giusti I, & Marmo E (1981)
[Experimental study of the cardiovascular and respiratory effects of
thallium in various animal species.] Rend Atti Accad Sci Med Chir,
134: 49-84 (in Italian).
Rossi F, Marrazzo R, Berrino L, De Santis D, Lisa M, Susanna V,
Montanaro C, Fici F, & Marmo E (1988) Prenatal and postnatal thallium
exposure in rats: effect on development of vasomotor reactivity in
pups. Teratog Carcinog Mutagen, 8: 13-23.
Sabbioni E & Manzo L (1980) Metabolism and toxicity of thallium. In:
Proceedings of the International Congress on Neurotoxicology,
pp 249-270 (Advances in Neurotoxicology).
Sabbioni E, Marafante E, Rade J, Di Nucci A, Gregotti C, & Manzo L
(1980a) Metabolic patterns of low and toxic doses of thallium in the
rat. In: Holmstedt B, Lauwerys R, Mercier M, & Robberfroid M ed.
Mechanisms of toxicity and hazard evaluation. Proceedings of the
Second International Congress on Toxicoloy, Brussels, 1980. Amsterdam,
Oxford, New York, Elsevier/North Holland, pp 559-564.
Sabbioni E, Goetz L, Marafante E, Gregotti C, & Manzo L (1980b)
Metabolic fate of different inorganic and organic species of thallium
in the rat. Sci Total Environ, 15: 123-135.
Sabbioni E, Gregotti C, Edel J, Marafante E, Di Nucci A, & Manzo L
(1982) Organ/tissue disposition of thallium in pregnant rats. Arch
Toxicol, 5(suppl): 225-230.
Sabbioni E, Di Nucci A, Edel J, Gregotti C, Marafante E, & Manzo L
(1984a) Intestinal absorption and excretion of thallium (201 Tl) in
the rat. Arch Toxicol, 7(suppl): 446-450.
Sabbioni E, Goetz L, & Bignoli G (1984b) Health and environmental
implication of trace metals released from coal fired power plants: an
assessment study of the situation in the European Community. Sci Total
Environ, 40: 141-154.
Saddique A & Peterson CD (1983) Thallium poisoning: a review. Vet Hum
Toxicol, 25: 16-22.
Sager M (1984) Separation and enrichment of traces of thallium.
Mikrochim Acta, 1984: 461-466.
Sager M (1986) [Trace element analysis of thallium.] In: Hulpke H,
Hartkamp H, & Tölg G ed. [Analytical chemistry for the practise.]
Stuttgart, Georg Thieme, pp 1-103 (in German).
Sager M & Tölg G (1984) [Trace element analysis of thallium.] In:
Fresenius W, Günzler H, Huber W, Lüderwald I, & Tölg G ed. [Analyst
pocket book 4.] Berlin, Springer Verlag, pp 443-466 (in German).
Sanderson L (1952) Thallium. Can Min J, 73: 72-73.
Sanotskii IV (1961) [Toxicity of thallium compounds (carbonate,
iodide, and bromide)]. Toksikol Novykh Prom Khim Veshchestv, 2: 94-104
(in Russian).
Schafer EW (1972) The acute oral toxicity of 369 pesticidal,
pharmaceutical and other chemicals to wild birds. Toxicol Appl
Pharmacol, 21: 315-330.
Schäfer SG & Forth W (1980) Excretion of metals into the intestine; a
comparative study on rats. In: Proceedings of the Second International
Congress on Toxicoloy, Brussels, 6-11 July 1980. Toxicol Lett,
95(spec issue).
Schäfer SG & Forth W (1987) Thallium(I) secretion across the isolated
mucosa of rat descending colon. Arch Toxicol, 52: 413-420.
Schäfer SG, Nell G, & Henning CH (1981) Movement of thallium(I) ions
in vitro. Arch Toxicol, 48: 271-279.
Schaller K-H, Manke G, Raithel HJ, Bühlmeyer G, Schmidt M, & Valentin
H (1980) Investigations of thallium-exposed workers in cement
factories. Int Arch Occup Environ Health, 47: 223-231.
Schardein JL & Keller KA (1989) Potential human developmental
toxicants and the role of animal testing in their identification and
characterization. Crit Rev Toxicol, 19: 251-339.
Scharrer K (1955) [Biochemistry of trace elements.] Berlin, Parey
(in German).
Schaub GA & Schnitker A (1988) Influence of Blastocrithidia triatomae
(Trypanosomatidae) on the reduviid bug Triatoma infestans: alterations
in the Malpighian tubules. Parasitol Res, 75: 88-97.
Schaumburg HH & Berger A (1992) Alopecia and sensory polyneuropathy
from thallium in a chinese herbal medication. J Am Med Assoc,
268: 3430-3431.
Schmidbauer H & Klingler D (1979) [Chronical thallium poisoning.] Wien
Med Wochenschr, 129: 334-336 (in German).
Schoental R & Cavanagh JB (1977) Mechanisms involved in the
`dying-back' process - an hypothesis implicating coenzymes.
Neuropathol Appl Neurobiol, 3: 145-157.
Schoer J (1984) Thallium. In: Hutzinger O ed. The handbook of
environmental chemistry. Volume 3: Anthropogenic compounds, Part C.
Berlin, Springer Verlag, pp 143-214.
Schoer J & Nagel U (1980) [Thallium in plants and soils.
Investigations in the southern Rhein-Neckar-Kreis.]
Naturwissenschaften, 67: 261-262 (in German).
Scholl W & Metzger F (1982) [Determinations of the thallium burden of
useful plants on contaminated soils in the region of Lengerich.]
Landwirtsch Forsch, 35: 216-223 (in German).
Schwartz JG, Stuckey JH, Kunkel SP, Dowd DC, & Kagan-Mallet KS (1988)
Poisoning from thallium. Tex Med, 84: 46-48.
Schwartzman RM & Kirschbaum JO (1962) The cutaneous histopathology of
thallium poisoning. J Invest Dermatol, 39: 169-173.
Schweiger P & Hoffmann G (1983) [Thallium uptake of different plants
in culture solution.] Kali-Briefe, 16: 391-398 (in German).
Seeger R & Gross M (1981) [Thallium in higher fungi.] Z Lebensm.Unters
Forsch, 173: 9-15 (in German).
Segebade C & Schmitt BF (1987) Analysis of high-purity material - a
comparison of photon activation analysis with other instrumental
methods. J Radioanal Nucl Chem, 113: 61-76.
Sessions HK & Goren S (1947) Report of investigation of health hazards
in connection with the industrial handling of thallium. US Navy Med
Bull, 47: 545-550.
Shabalina LP & Spiridonova VS (1979) [Thallium as an industrial poison
(Review of literature).] J Hyg Epidemiol Microbiol Immunol,
23: 247-255 (in Russian).
Shabalina LP, Spiridonova VS, Pavlenko GI, & Fomenko VN (1980) [Skin
resorptive, local, and mutagenic effects of thallium carbonate.] Gig
Tr Prof Zabol, 4: 54-55 (in Russian).
Shacklette HT, Erdmann JA, Harms TF, & Papp CSE (1978) Trace elements
in plant foodstuffs. In: Oehme FW ed. Toxicity of heavy metals in the
environment. New York, Marcel Dekker Inc., vol 1, pp 25-68.
Sharma RP & Obersteiner EJ (1981) Metals and neurotoxic effects:
cytotoxicity of selected metallic compounds on chick ganglia cultures.
J Comp Pathol, 91: 235-244.
Sharma J, Sharma RL, Singh HB, & Satake M (1986) Hazards and analysis
of thallium. A review. Toxicol Environ Chem, 11: 93-116.
Shaw PA (1932) Studies on thallium poisoning in game birds. Calif Fish
Game, 18: 29-34.
Sherlock JC & Smart GA (1986) Thallium in foods and the diet. Food
Addit Contam, 3: 363-370. Siegel SM (1977) The cytotoxic response of
Nicotiana' protoplasts to metal ions: a survey of the chemical
elements. Water Air Soil Pollut, 8: 293-304.
Siegel BZ & Siegel SM (1976) Effect of potassium on thallium toxicity
in cucumber seedlings: further evidence for potassium-thallium ion
antagonism. Bioinorg Chem, 6: 341-345.
Sikka R, Sabherwal U, & Arora DR (1987) Susceptibility of Klebsiella
pneumoniae to heavy metal ions in vitro. Indian J Med Res,
86: 437-440.
Singh NP, Bodgen JD, & Joselow MM (1975) Distribution of thallium and
lead in children's blood. Arch Environ Health, 30: 557-558.
Skrovina B, Micek J, Hronská J, & Spissák L (1973) Experimental and
human achondroplasia. Teratology, 8: 237.
Skulskii IA & Lapin AV (1983) Effect of thallium ions on sodium and
potassium transport in frog skin. Tsitologiya, 25: 1284-1288.
Skulskii IA, Manninen V, & Järnefelt J (1973) Interaction of thallous
ions with the cation transport mechanism in erythrocytes. Biochim
Biophys Acta, 298: 702-709.
Skulskii IA, Manninen V, & Järnefelt J (1975) Thallium inhibition of
ouabain-sensitive sodium transport and of the (Na++K+)-ATPase in
human erythrocytes. Biochim Biophys Acta, 394: 569-576.
Skulskii IA, Savina MV, Glasunov VV, & Saris NEL (1978)
Electrophoretic transport of Tl+ in mitochondria. J Membr Biol,
44: 187-194.
Skulskii IA, Ivanova TI, & Savina MV (1984) The effect of monovalent
thallium salts on liver mitochondria of rats. Zh Evol Biokhim Fiziol,
20: 353-355 (in Russian).
Smith IC & Carson BL (1977) Trace metals in the environment: Volume 1
- Thallium. Ann Arbor, Michigan, Ann Arbor Science Publishers, Inc.
Smith S & Kwan MKH (1989) Use of aquatic macrophytes as a bioassay
method to assess relative toxicity, uptake kinetics and accumulated
forms of trace metals. Hydrobiologia, 188/189: 345-351.
Sommer B (1987) [Evaluation of the effects of cadmium, lead, thallium
and fluoride in plants for food and fodder - Environmental research
plan of the Federal Minister for Environment, Nature Conservation and
Reactor Safety, research report 106 07 049/02.] Berlin, Erich Schmidt
Verlag (Report No. UBA-FB 87-026/8/87) (in German).
Spencer EL (1937) Frenching of tobacco and thallium toxicity. Am J
Bot, 24: 16-24.
Spiridonova VS, Shabalina LP, & Kremenetskaya LE (1978) [Fibrogenic
activity of thallium halides.] Gig Tr Prof Zabol, 11: 54 (in Russian).
Srivastava TN, Bajpai KK, & Singh K (1973) Anti-microbial activities
of diaryl gallium, indium and thallium compounds. Indian J Agric Sci,
43: 88-93.
Staszyc J, Królikowska-Prasal I, Matysiak W, Korobowicz E, Chróst L, &
Kusmierczyk S (1986) Morphological evaluation of the lungs of rats
affected by soil dust. Med Pr, 37: 65-72 (in Polish). Stavinoha WB,
Emerson GA, & Nash JB (1959) The effects of some sulfur compounds on
thallotoxicosis in mice. Toxicol Appl Pharmacol, 1: 638-646.
Steuer W (1980) [Results of thallium examinations of the human
population in Baden-Württemberg.] Öff Gesundh-Wesen, 42: 477-479
(in German).
Stevens W, van Peteghem C, Heyndrickx A, & Barbier F (1974) Eleven
cases of thallium intoxication treated with Prussian blue. Int J Clin
Pharmacol, 10: 1-22.
Stokinger HE (1987) Thallium. In: Clayton GD & Clayton FE ed. Patty's
industrial hygiene and toxicology: Volume 2A: Toxicology, 3rd ed. New
York, Chichester, Brisbane, Toronto, John Wiley and Sons,
pp 1914-1931.
Struempler AW (1975) Trace element composition in atmospheric
particulates during 1973 and the summer of 1974 at Chadron, Neb.
Environ Sci Technol, 9: 1164-1168.
Sunderman FW (1967) Diethyldithiocarbamate therapy of thallotoxicosis.
Am J Med Sci, 253: 209-220.
Swain RE & Bateman WG (1909/10) The toxicity of thallium salts. J Biol
Chem, 7: 137-151.
Tackmann W & Lehmann HJ (1971) [Refractory period and impulse sequence
recorded from the tibial nerves of guinea pigs with acute thallium
polyneuropathy.] Z Neurol, 199: 105-115 (in German).
Talas A & Wellhöner HH (1983) Dose-dependency of Tl+ kinetics as
studied in rabbits. Arch Toxicol, 53: 9-16.
Tan E-L, Williams MW, Schenley RL, Perdue SW, Hayden TL, Turner JE, &
Hsie AW (1984) The toxicity of sixteen metallic compounds in Chinese
hamster ovary cells. Toxicol Appl Pharmacol, 74: 330-336.
Tandon SP & Mishra MM (1969) Effect of some rare-elements on
nitrification by Nitrobacter agilis (in liquid culture medium). Proc
Natl Acad Sci India Sect A, 39: 209-216.
Templeton DM, Paudyn A, & Baines AD (1989) Multielement analysis of
biological samples by inductively coupled plasma-mass spectrometry.
Biol Trace Elem Res, 22: 17-33.
Terytze K (1987) [Entry and spread of chlorinated hydrocarbons and
toxic heavy metals with reference to their quantification in the
limnetic ecosystem.] Wiss Z Humboldt-Univ (Berlin) Math-Naturwiss
Reihe, 36: 860-865 (in German).
Thompson C, Dent J, & Saxby P (1988) Effects of thallium poisoning on
intellectual function. Br J Psychiatry, 153: 396-399.
Thyresson N (1950) Experimental investigation on thallium poisoning.
Influence of thallium on tissue metabolism. Acta Dermato-Venereol,
30: 417-441.
Thyresson N (1951) Experimental investigation on thallium poisoning.
Acta Dermato-Venereol, 31: 3-27.
Tikhova TS (1964) [Industrial hygiene problems in the production
metallic thallium and its salts.] Gig i Sanit, 29: 23-28 (in Russian).
Tikhova TS (1967) [Thallium and its compounds.] In: Izrael'son ZI ed.
[New data on the toxicology of rare metals and their compounds.]
oedinenii. Moscow, Meditsina Publishing House, pp 24-34 (in Russian).
Tjälve H, Nilsson M, & Larsson B (1982) Thallium-201: autoradiography
in pigmented mice and melanin-binding in vitro. Acta Pharmacol
Toxicol, 51: 147-153.
Toots H & Parker RB (1977) Thallium in salt substitutes: a possible
health hazard. Env Res, 14: 327-328.
Török P & Schmahl W (1982) [The environmental toxic substance thallium
sulfate.] Dtsch Ärztebl, 79(39): 31-32 (in German).
Triebig G & Büttner J (1983) [Occupational relevant neurotoxins: I.
Metals and their compounds - A literature review of the years 1970 to
1982.] Zbl Bakt Hyg I. Abt Orig B, 177: 11-36 (in German).
Trotman-Dickenson AF ed. (1973) Comprehensive inorganic chemistry:
Thallium. Oxford, New York, Pergamon Press, pp 1119-1172.
Truhaut R (1960) The toxicology of thallium. A summary prepared by W.
B. Deichmann of Truhaut's monograph "Recherches sur la Toxicologie du
Thallium." Institut National de Sécurité pour la Prévention des
Accidents du Travail et des Maladies Professionelles, Paris, 1959.
J Occup Med, 2: 334-336.
Tully JG, Taylor-Robinson D, Rose DL, Furr PM, & Hawkins DA (1983)
Evaluation of culture media for the recovery of Mycoplasma hominis
from the human urogenital tract. Sex Transm Dis, 10(suppl): 256-260.
Tuovinen OH & Kelly DP (1974) Studies on the growth of Thiobacillus
ferrooxidans. IV. Influence of monovalent metal cations on ferrous
iron oxidation and uranium toxicity in growing cultures. Arch
Microbiol, 98: 167-174.
UB (Federal Office for the Environment) (1992) [Pollution of
allotments and kitchen gardens in former Federal territory.] In: [Data
on the environment 1992/93.] Berlin, Erich Schmidt Verlag (in German).
Ueberschär K-H, Matthes S, & Vogt H (1986) [Influence of addition of
thallium to food of broiler and laying hens.] In: Anke M, Baumann W,
Bräunlich H, Brückner C, & Groppel B ed. [Proceedings of the 5th
Symposium of Trace Elements, Jena, 14-17 July 1986.] Jena, University
of Jena, pp 1233-1240 (in German).
Ulrich F & Long CNH (1955) The effects of propylthiouracil and
thyrotropic hormone on the uptake of radioactive thallium by the rat
thyroid. Yale J Biol Med, 27: 371-378.
Urbain D, Reding P, Georges B, Thys O, & Ham HR (1986) The clinical
value of 201Tl per rectum scintigraphy in the work-up of patients with
alcoholic liver disease. Eur J Nucl Med, 12: 267-270.
US BM (US Bureau of Mines) (1985) Minerals yearbook 1985. Volume 1:
Metals and minerals. Washington, DC, Government Printing Office. US BM
(US Bureau of Mines) (1989) Mineral commodity summaries 1989.
Washington, DC, US Bureau of Mines/US Geological Survey.
US EPA (1978) Ambient water quality criteria: thallium. Washington,
DC, US Environmental Protection Agency (PB-292444).
US EPA (1980) Ambient water quality criteria for thallium. Washington,
DC, US Environmental Protection Agency (EPA-440/5-80-074).
US EPA (1986) Subchronic (90-day) toxicity of thallium (I) sulfate
(Cas No. 7446-18-6) in Sprague-Dawley rats. Interim Report.
Washington, DC, US Environmental Protection Agency, Office of Solid
Wastes.
Valerio F, Brescianini C, Mazzucotelli A, & Frache R (1988) Seasonal
variation of thallium, lead, and chromium concentrations in airborne
particulate matter collected in an urban area. Sci Total Environ,
71: 501-509.
Valerio F, Brescianini C, & Lastraioli S (1989) Airborne metals in
urban areas. Int J Environ Anal Chem, 35: 101-110.
Vandenbalck JL & Patriarche GJ (1987) Electrochemical
micro-determinations of thallium(I) and chromium(VI) ions using DPASV
and DP polargraphy. Sci Total Environ, 60: 97-104.
Venugopal B & Luckey TD (1978) Metal toxicity in mammals. Volume 2:
Chemical toxicity of metals and metalloids. New York, Plenum Press.
Vesterberg O, Alessio L, Brune D, Gerhardsson L, Herber R, Kazantzis
G, Nordberg G, & Sabbioni E (1993) International project for producing
reference values for concentrations of trace elements in human blood
and urine (TRACY). Scand J Work Environ Health, 19(suppl 1): 19-26.
Von Mühlendahl KE, Etzold R, & Krienke EG (1978) [Thallium poisonings
of children.] Dtsch Med Wochenschr, 103: 1116-1117 (in German).
Wachs B (1988) [Relevance for bodies of water of the most toxic heavy
metals.] In: Bavarian Land Institute for Water Research ed. [Hazardous
substances in waste water and surface water.] Munich, Oldenbourg
Verlag, pp 176-243 (Report No. 42) (in German).
Wainwright AP, Kox WJ, House IM, Henry JA, Heaton R, & Seed WA (1988)
Clinical features and therapy of acute thallium poisoning.
Q J Med, 258: 939-944.
Wallwork-Barber MK, Lyall K, & Ferenbaugh RW (1985) Thallium movement
in a simple aquatic ecosystem. J Environ Sci Health, 20: 689-700.
Ward JC (1930) Thallium poisoning in sheep. J Am Pharm Assoc,
19: 556-559.
Ward JC (1931) Thallium poisoning in migratory birds. J Am Pharm
Assoc, 20: 1272-1276.
Ward JC (1936) Thallium. XIII. Symptoms and systemic action on cattle.
J Am Pharm Assoc, 25: 687-690.
Waters CB, Hawkins EC, & Knapp DW (1992) Acute thallium toxicosis in a
dog. J Am Vet Med Assoc, 201(6): 883-885.
Wei Q (1987) [Studies on spermotoxicity of thallium carbonate in
drinking water and its effect on reproductive function of mice.]
Chung-hua Yu Fang Hsueh Tsa Chih, 21: 141-143 (in Chinese).
Weinig E & Schmidt G (1966) [Distribution of thallium in the organism
after lethal thallium poisonings.] Arch Toxicol, 21: 199-215
(in German).
Weinig E & Walz W (1971) [Distribution of thallium in kidney and liver
after lethal thallium poisoning.] Arch Toxicol, 27: 217-225
(in German).
Weinig E & Zink P (1967) [Quantitative mass-spectrometrical
determination of the normal thallium content in the human organism.]
Arch Toxicol, 22: 255-274 (in German).
Weisweiler W, Blome K, & Kaeding L (1985) [Quantitative determination.
Thallium and lead cycles in rotary kilns for cement production.]
Staub-Reinhalt Luft, 45: 461-466 (in German).
Welz B (1983) [Atomic absorption spectrometry.] Weinheim, Verlag
Chemie (in German).
Welz B, Schlemmer G, & Mudakavi JK (1988a) Investigation and
elimination of chloride interference on thallium in graphite furnace
atomic absorption spectrometry. Anal Chem, 60: 2567-2572.
Welz B, Schlemmer G, & Mudakavi JK (1988b) Palladium nitrate -
magnesium nitrate modifier for graphite furnace atomic absorption
spectrometry; part 1. Determination of arsenic, antimony, selenium and
thallium in airborne particulate matter. J Anal At Spectrom, 3: 93-97.
WHO (1986) UNEP/WHO Global Environmental Monitoring System/HEAL
Project-Guidelines for integrated air, water, food and biological
exposure monitoring. Geneva, World Health Organization
(Document PEP/86.6).
Wiegand H (1988) Neurotoxicology of heavy metals: synaptic
transmission as influenced by mono- and divalent metal cations. In:
Proceedings of the 1st European Meeting of Environmental Hygiene,
pp 98-102.
Wiegand H, Csicsaky M, Krämer U, Papadopoulos R, & Willeke E (1983)
Neuromuscular transmission in the rat phrenic nerve hemidiaphragm
preparation influenced by thallium acetate. Proceedings of the 4th
International Conference on Heavy Metals in the Environment.
vol 2, pp 896-899.
Wiegand H, Csicsaky M, & Krämer U (1984a) The action of thallium
acetate on neuromuscular transmission in the rat phrenic
nerve-diaphragm preparation. Arch Toxicol, 55: 55-58.
Wiegand H, Papadopoulos R, Csicsaky M, & Krämer U (1984b) The action
of thallium acetate on spontaneous transmitter release in the rat
neuromuscular junction. Arch Toxicol, 55: 253-257.
Wiegand H, Lohmann H, Chandra SV, & Gotzsch U (1986) The action of
thallium acetate on phasic transmitter release in the mouse
neuromuscular junction. Arch Toxicol, 58: 265-270.
Wiegand H, Uhlig S, Gotzsch U, & Lohmann H (1990) The action of
cobalt, cadmium and thallium on presynaptic currents in mouse motor
nerve endings. Neurotoxicol Teratol, 12: 313-318.
Wiemer J, Ziskoven R, & Achenbach C (1982) Tl+-Ions: comparison of
the effects on the slow inward current and contractility of
ventricular tissues. Z Naturforsch, 37c: 1015-1022.
Wiemeyer SN, Jurek RM, & Moore JF (1986) Environmental contaminants in
surrogates, foods, and feathers of California condors (Gymnogyps
californianus). Environ Monit Assess, 6: 91-111.
Williams MW, Hoeschele JD, Turner JE, Jacobson KB, Christie NT, Paton
CL, Smith LH, Witschi HR, & Lee EH (1982) Chemical softness and acute
metal toxicity in mice and Drosophila. Toxicol Appl Pharmacol,
63: 461-469.
Wilson IC & Dean ACR (1977) Thallium sensitivity and tolerance in
Klebsiella pneumoniae. Microbios Lett, 5: 85-91.
Wimmer M (1985) [Mutagenic, cancerogenic, and teratogenic effects of
chemical substances on man, with special reference to thallium.] Wiss
Z Humboldt-Univ (Berlin) Math-Naturwiss Reihe, 34: 789-791
(in German).
Windebank AJ (1986) Specific inhibition of myelination by lead
in vitro; comparison with arsenic, thallium, and mercury. Exp Neurol,
94: 203-212.
Witschi HP (1965) Desorption of some toxic heavy metals from human
erythrocytes in vitro. Acta Haematol, 34: 101-115.
Wood JM (1984) Alkylations of metals and the activity of metal-alkyls.
Toxicol Environ Chem, 7: 229-240.
Wood JM (1987) Biological processes involved in the cycling of
elements between soil or sediments and the aqueous environment.
Hydrobiologia, 149: 31-42.
Wood JM, Cheh A, Dizikes LJ, Ridley WP, Rakow S, & Lakowicz JR (1978)
Mechanisms for the biomethylation of metals and metalloids. Fed Proc,
37: 16-21.
Woods JS & Fowler BA (1986) Alteration of hepatocellular structure and
function by thallium chloride: ultrastructural, morphometric, and
biochemical studies. Toxicol Appl Pharmacol, 83: 218-229.
Wystrcil H-G, Martini M, Maier-Reiter W, & Arndt U (1987)
[Ecotoxicology of thallium. Effect on photosynthesis.]
Agrar-Umweltforsch Baden-Württemberg, 16: 37-66 (in German).
Yang Q & Smeyers-Verbeke J (1991) Effectiveness of palladium matrix
modification for the determination of thallium by graphite furnace
atomic absorption spectrometry. Clin Chim Acta, 204: 23-26.
Yokoyama K, Araki S, & Abe H (1990) Distribution of nerve conduction
velocities in acute thallium poisoning. Muscle Nerve, 13: 117-120.
Yopp JH, Schmid WE, & Holst RW (1974) Determination of maximum
permissible levels of selected chemicals that exert toxic effects on
plants of economic importance in Illinois. Carbondale, Illinois,
Southern Illinois University (Report No. 74-33) (NTIS PB-237 654).
Zasukhina GD, Vasilyeva IM, & Sdirkova NI (1980) [Approach to the
determination of a mutagenic potential of environmental pollutants as
illustrated by the detection of the mutagenic effect of thallium
carbonate.] Dokl Acad Nauk SSSR, 250: 766-768 (in Russian).
Zasukhina GD, Vasilyeva IM, Sdirkova NI, Krasovsky GN, Vasyukovich LY,
Kenesariev UI, & Butenko PG (1983) Mutagenic effect of thallium and
mercury salts on rodent cells with different repair activities.
Mutat Res, 124: 163-173.
Zeiske W & van Driessche W (1986) Impairment of Na+ transport across
frog skin by Tl+: effects on turnover, area density and saturation
kinetics of apical Na+ channels. Pflügers Arch, 407: 145-152.
Zhang D (1988) [Bioassay of mutagenicity of thallium carbonate.]
Huanjing Kexue, 9: 29-32 (in Chinese).
Zhou D-X & Liu D-N (1985) Chronic thallium poisoning in a rural area
of Guizhou province, China. J Environ Health, 48: 14-18.
Ziegler E & Ziegler B (1984) [Preliminary results on the heavy metal
content in hair and serum.] Fortschr Atomspektrom Spurenanal,
1: 337-346 (in German).
Ziskoven R, Achenbach C, Bahr U, & Schulten H-R (1980) Quantitative
investigations on the diaplacental transfer of thallium by field
desorption mass spectrometry. Z Naturforsch, 35c: 902-906.
Ziskoven R, Achenbach C, & Wiemer J (1982) Tl+-Ions: influence on
cardic contractility. Z Naturforsch, 37c: 995-1005.
Ziskoven R, Achenbach C, Schulten H-R, & Roll R (1983) Thallium
determinations in fetal tissues and maternal brain and kidney. Toxicol
Lett, 19: 225-231.
Zitko V (1975a) Chemistry, applications, toxicity, and pollution
potential of thallium. Department of the Environment, Fisheries and
Marine Service, Research and Development Directorate (Technical Report
No. 518).
Zitko V (1975b) Toxicity and pollution potential of thallium. Sci
Total Environ, 4: 185-192.
Zitko V & Carson WV (1975) Accumulation of thallium in clams and
mussels. Bull Environ Contam Toxicol, 14: 530-533.
Zitko V, Carson WV, & Carson WG (1975) Thallium: occurrence in the
environment and toxicity to fish. Bull Environ Contam Toxicol,
13: 23-30.
Zook BC & Gilmore CE (1967) Thallium poisoning in dogs. J Am Vet Med
Assoc, 151: 206-217.
Zook BC, Holzworth J, & Thornton GW (1968) Thallium poisoning in cats.
J Am Vet Med Assoc, 153: 285-299.
Zyka V (1972) Thallium in plants from Alsar. Sb Geol Ved Technol
Geochim, 10: 91-96.
RESUME
1. Identité, propriétés physiques et chimiques, et méthodes d'analyse
Le thallium élémentaire est un métal mou et malléable de couleur
blanc bleuâtre. Exposé à l'air humide ou à l'eau, le thallium est,
selon le cas, rapidement oxydé en surface ou transformé en hydroxyde.
Le thallium possède deux importants degrés d'oxydation, le thallium(I)
et le thallium(III). Les composés du thallium monovalent (thalleux)
se comportent comme des dérivés de métaux alcalins, par exemple du
potassium, alors que les composés du thallium trivalent (dérivés
thalliques) sont moins basiques et rappellent les sels d'aluminium.
Contrairement aux dérivés minéraux dans lesquels l'ion thallium(I) est
plus stable en solution aqueuse que l'ion thallium(III), ce dernier
est plus stable dans les dérivés organiques.
Le dosage du thallium dans des échantillons provenant de
l'environnement est assez difficile étant donné que la concentration
est de l'ordre du µg/kg tout au plus. En général, la limite de dosage
dans les minéraux, les sols et les poussières est d'environ 20 µg/kg -
approximativement 0,1 µg/litre pour les solutions aqueuses - et dans
les produits biologiques, de quelques µg/kg, lorsqu'on ne procède pas
à une concentration préalable du thallium.
La spectrométrie d'absorption atomique avec four à tube de
graphite (GF-AAS) est une méthode d'analyse qui convient bien aux
applications exigeant une sensibilité élevée avec des prises d'essai
réduites où la concentration en thallium est de l'ordre de quelques
µg/kg. Pour obtenir une bonne précision et une bonne exactitude à des
teneurs de l'ordre du µg/kg, on peut utiliser la spectrométrie de
masse avec dilution isotopique (IDMS) et la spectrométrie de masse en
plasma à couplage inductif (ICP-MS), avec également possibilité de
dilution isotopique.
2. Sources d'exposition humaine et environnementale
La présence de thallium dans l'environnement résulte à la fois de
processus naturels et de l'activité humaine. Ce métal est omniprésent
dans la nature et on le rencontre en particulier dans les minerais
sulfurés de divers métaux lourds, quoiqu'en principe à faibles
concentrations. Il n'y a guère d'endroits où la concentration en
thallium d'origine naturelle soit très élevée. Le thallium n'est
produit industriellement qu'en petites quantités (la consommation
industrielle mondiale a été de 10 à 15 tonnes en 1991). Le thallium
et ses dérivés ont des applications industrielles très diverses. Ils
entrent dans la composition de crèmes épilatoires, de rodenticides et
d'insecticides, mais ces utilisations sont désormais strictement
limitées. On utilise principalement le thallium et ses dérivés dans
les industries électriques et électroniques ainsi que pour la
fabrication de verres spéciaux. La scintigraphie en général et le
diagnostic du mélanome en particulier constituent des applications
importantes du radio-thallium en médecine et l'on a également recours
aux dérivés d'arylthallium(III) en biochimie.
Du thallium peut être libéré dans l'environnement par des
fonderies (dépôts de déchets ou émissions dans l'atmosphère), des
centrales thermiques à charbon, des briqueteries et des cimenteries
(dans ce cas, exclusivement des émissions dans l'atmosphère). On
estime que l'industrie mobilise ainsi chaque année dans le monde de
2000 à 5000 tonnes de thallium. Les émissions de thallium dues aux
différentes opérations industrielles varient largement d'un type
d'industrie à l'autre.
Les émissions des centrales thermiques à charbon peuvent avoir
une teneur en thallium de 700 µg/m3 d'air et celles des cimenteries
des teneurs allant jusqu'à 2500 µg/m3. Cette dernière valeur peut
être ramenée à < 25 µg/m3 en ayant recours à d'autres matières
premières et en changeant de procédé. Le thallium se volatilise lors
de la combustion du charbon et des matières premières utilisées pour
la production de ciment puis il se recondense à la surface des
particules de cendre dans les parties de l'installation où la
température est plus basse. Ces particules peuvent contenir jusqu'à
50 mg de thallium/kg de cendres volantes et sont souvent de petite
taille, de sorte que seulement 50% d'entre elles sont retenues par les
filtres. De même, environ un tiers des particules émises par les
centrales thermiques sont de faible granulométrie et peuvent par
conséquent se déposer dans les voies respiratoires inférieures.
Les effluents provenant de bassins d'évacuation de terrils et
contenant dans un cas jusqu'à 1620 et dans un autre jusqu'à
36 µg/litre de thallium ont provoqué, dans les cours d'eau adjacents,
l'apparition de concentrations de thallium respectivement égales à 88
et 1 µg/litre. Dans les collections d'eau de pluie situées aux
alentours d'une cimenterie, on a constaté la présence de
concentrations de thallium allant jusqu'à 37 µg/litre. On a trouvé
dans le sol aux alentours de terrils de mines, des concentrations
atteignant 60 mg/kg. Au voisinage de la base de fours à métaux, et
près de briqueteries et de cimenteries, on a trouvé des concentrations
respectivement égales à 2, 0,6 et 27 mg/kg de terre.
Dans les zones contaminées, la concentration de thallium dans la
plupart des légumes, des fruits et des viandes est inférieure à
1 mg/kg de poids frais. Les teneurs sont plus élevées dans les choux
(crucifères) avec des concentrations allant jusqu'à 45 mg/kg dans les
choux frisés. Il y a corrélation entre la concentration du thallium
dans les tissus des animaux d'élevage et la concentration de ce métal
dans le fourrage. Au voisinage de certaines cimenteries, on a signalé
des teneurs plus élevées en thallium dans le fourrage (par exemple
jusqu'à 1000 mg/kg dans le colza) ainsi que dans la viande de boeuf et
de lapin (jusqu'à 1,5 et 5,8 mg/kg, respectivement).
3. Transport, distribution et transformation dans l'environnement
A proximité de sources ponctuelles telles que les centrales
thermiques à charbon, certaines cimenteries et fonderies de métaux, la
principale source de thallium atmosphérique est constituée par les
cendres volantes. Une étude a montré que près de la totalité du
thallium présent dans les poussières en suspension d'une cimenterie
s'y trouvait sous la forme de chlorure de thallium(I) soluble.
La destinée du thallium qui parvient jusqu'au sol (par exemple
sous forme de dépôt de cendres volantes) dépend en grande partie de la
nature de ce sol. C'est en particulier les sols à forte teneur en
argile, en matières organiques et en oxydes de fer et manganèse qui
retiennent le plus de thallium. L'accroissement de la teneur en
thallium par incorporation dans des complexes stables ne se produit
que dans les couches supérieures. La fixation du thallium par les
végétaux est d'autant plus importante que le pH est plus bas. Dans
certains sols fortement acides, d'importantes quantités de thallium
peuvent parvenir par lessivage jusqu'aux eaux de surface et aux eaux
souterraines.
On peut s'attendre à ce que la majeure partie du thallium présent
à l'état dissous dans les eaux douces se trouve sous sa forme
monovalente. Toutefois, dans les eaux où la teneur en oxygène est
forte et dans la plupart des eaux de mer, il peut y avoir prédominance
du thallium(III). Le thallium peut disparaître de l'eau par diverses
réactions d'échange, de complexation ou de précipitation.
Le thallium peut également subir une bioconcentration, mais il
est peu probable que sa concentration s'accroisse le long de la chaîne
alimentaire aquatique ou terrestre.
4. Concentrations dans l'environnement et exposition humaine
Dans les secteurs qui ne sont pas contaminés, les concentrations
de thallium dans l'air sont généralement inférieures à 1 ng/m3; dans
l'eau elles sont inférieures à 1 µg/litre et dans les sédiments
aquatiques, inférieures à 1 mg/kg. Dans l'écorce terrestre, la
concentration moyenne varie de 0,1 à 1,7 mg/kg, mais on peut trouver
des concentrations beaucoup plus élevées, par exemple jusqu'à
1000 mg/kg dans le charbon et les rares minerais de thallium que l'on
rencontre contiennent jusqu'à 60% de cet élément. Les produits
alimentaires d'origine végétale ou animale en contiennent généralement
moins de 1 mg/kg de poids sec et l'apport d'origine alimentaire moyen
chez l'homme se révèle inférieur à 5 µg/jour. On estime que l'apport
de thallium par la voie respiratoire est inférieur à 0,005 µg de
thallium par jour.
On ne possède que des données limitées sur la concentration
effective de thallium dans l'air des lieux de travail. Les valeurs
les plus récemment observées (dans les années 80) étaient inférieures
à 22 µg de thallium par m3 (il s'agissait d'un four à thallium
installé dans un atelier produisant un alliage spécial à base de
thallium). Chez des ouvriers d'une cimenterie, on a trouvé des
concentrations urinaires moyennes allant de 0,3 à 8 µg/litre et chez
des ouvriers d'une fonderie, des valeurs de 0,3 à 10,5 µg/litre.
5. Cinétique et métabolisme chez les animaux de laboratoire
et l'homme
La thallium est rapidement et bien absorbé au niveau des voies
digestives et respiratoires et il pénètre également à travers la peau.
Il se répartit rapidement dans l'ensemble des organes et traverse la
barrière placentaire (comme le montre sa fixation rapide dans les
tissus foetaux) ainsi que la barrière hématoencéphalique. Comme il
s'accumule rapidement dans les cellules, la concentration du thallium
dans le sang total ne reflète pas sa concentration tissulaire. Dans
les cas d'intoxication aiguë chez l'homme et l'animal d'expérience on
constate, au début, de fortes concentrations de thallium au niveau du
rein et de faibles concentrations dans les tissus adipeux et le
cerveau, les concentrations dans les autres organes se situant entre
les deux; ultérieurement la concentration de thallium dans le cerveau
augmente également.
L'élimination du thallium peut s'effectuer par la voie digestive
(essentiellement par des mécanismes indépendants de l'excrétion
biliaire), par les reins, les cheveux, la peau, la sueur et le lait
maternel. Il peut y avoir réabsorption intestinale, principalement au
niveau du côlon avec pour conséquences une diminution de la clairance
totale. Chez le rat les principales voies d'élimination sont la voie
digestive (environ les deux tiers) et la voie rénale (environ un
tiers); chez le lapin, ces deux voies sont d'une importance
sensiblement égale. Le thallium peut également être excrété par la
salive.
Comme dans le cas de nombreuses autres substances, l'excrétion du
thallium n'est pas identique chez l'homme et chez l'animal de
laboratoire, la vitesse d'excrétion étant généralement beaucoup plus
faible chez l'homme (constante de vitesse = 0,023-0,069 jour-1) que
chez l'animal d'expérience (constante de vitesse moyenne = 0,18
jour-1). Une autre différence importante que l'on observe entre
l'homme et l'animal concerne l'importance relative des diverses voies
d'excrétion. Chez l'homme, il semble que l'excrétion rénale soit
beaucoup plus importante que chez l'animal, encore que sa contribution
à la clairance totale n'ait pas encore été définitivement établie,
principalement du fait de l'insuffisance des données. En outre, le
niveau d'exposition, sa durée, les insuffisances au niveau des organes
excréteurs, ainsi que le traitement anti-poison qui est administré,
sont autant de facteurs qui peuvent influer considérablement sur les
résultats.
Une étude a montré que la quantité totale de thallium excrétée
quotidiennement était de 73% environ par la voie rénale et seulement
de 3,7% environ par la voie digestive. On a estimé à 19,5% la
proportion excrétée dans le système pileux et par la voie percutanée
et à 3,7% la proportion excrétée dans la sueur.
Chez l'animal de laboratoire, la demi-vie biologique du thallium
oscille généralement entre trois et huit jours. Chez l'homme elle est
d'environ 10 jours, mais on a fait état de valeurs allant jusqu'à 30
jours.
On ne dispose d'aucune donnée sur la biotransformation du
thallium.
6. Effets sur les mammifères de laboratoire et les systèmes d'épreuve
in vitro
La toxicité des sels de thallium(I) ne varie pas de manière
spectaculaire d'une espèce à l'autre. Généralement, l'ingestion d'une
dose de 20 à 60 mg de thallium par kg de poids corporel est mortelle
en l'espace d'une semaine. Les cobayes sont légèrement plus sensibles
que les autres animaux de laboratoire. L'oxyde de thallium(III),
insoluble dans l'eau, présente une toxicité aiguë par voie orale ou
parentérale un peu plus faible que les sels de thallium(I). la
comparaison des données de toxicité aiguë montre que le thallium
présente une biodisponibilité élevée par toutes les voies
d'exposition. La plupart des organes sont affectés mais on constate
quelques variations intra- et interspécifiques pour ce qui concerne
les signes d'intoxication et l'ordre dans lequel ils se manifestent.
Les symptômes d'une intoxication aiguë par le thallium se
manifestent en général dans l'ordre suivant: on constate tout d'abord
une anorexie, des vomissements et une dépression, puis apparaissent
une diarrhée et des manifestations cutanées (inflammation au niveau
des divers orifices, furoncles, chute de cheveux), après quoi
apparaissent une dyspnée et des troubles nerveux. Enfin la mort
survient par insuffisance respiratoire.
les symptômes d'une intoxication chronique sont analogues. La
chute des cheveux est de règle.
L'examen histologique révèle des lésions cellulaires et en
particulier une nécrose. Cette nécrose a été observée au niveau des
reins, du foie, de l'intestin, du myocarde et du système nerveux. On
a observé dans nombreuses cellules, un gonflement des mitochondries
avec disparition des crêtes, une dilatation du réticulum endoplasmique
agranulaire, une augmentation du nombre de vacuoles autophagiques et
de granules de lipofuscine, enfin, une disparition des
microvillosités. Les altérations fonctionnelles qu'entraîne le
thallium peuvent s'expliquer par la destruction physique de la
membrane des organites subcellulaires. Au niveau du coeur, les effets
arythmogènes se limitent au noeud sinusal.
L'intoxication par le thallium provoque des anomalies sélectives
au niveau de certains comportements, qui sont corrélées aux effets
biochimiques (ce qui indique des lésions cellulaires) observés dans
certaines régions de l'encéphale. Un certain nombre d'effets
neurologiques semblent être dus à l'action directe du thallium, par
exemple l'ataxie et les tremblements qui seraient dus à des lésions
cérébelleuses ou à des modifications de l'activité endocrine provoquée
par des lésions au niveau de l'hypothalamus. Le système nerveux
autonome, et principalement les fibres adrénergiques, peuvent être
activés par le thallium. Dans les nerfs périphériques, il semble que
l'action du thallium s'exerce au niveau présynaptique, avec libération
spontanée du neurotransmetteur, par antagonisme avec ce processus qui
dépend du calcium.
On ne sait toujours pas exactement par quel mécanisme s'exerce la
toxicité du thallium. On pense qu'il en existe plusieurs, liés les
uns aux autres. Un aspect important de l'intoxication par le thallium
consiste dans une augmentation sensible de la peroxydation des lipides
et de l'activité d'un enzyme lysosomique, la ß-galactosidase. Il en
résulte un déficit en glutathion qui entraîne l'accumulation de
lipides peroxydés dans l'encéphale et vraisemblablement la formation
de granules de lipofuscine. Il semble que le mode d'action du
thallium consiste principalement dans une perturbation de la fonction
des mitochondries.
Une intoxication chronique par le thallium entraîne chez l'animal
une réduction de l'activité sexuelle et les effets gonadotoxiques du
thallium sont manifestes chez le mâle au niveau des organes
reproducteurs. Chez des rats qui avaient reçu pendant 16 jours 10 mg
de thallium par litre d'eau de boisson, on a constaté qu'au niveau des
testicules, c'étaient les cellules de Sertoli qui étaient les plus
sensibles et la desquamation de l'épithélium spermatogène a entraîné
la présence de spermatozoïdes immatures dans le sperme. Ce phénomène
pourrait expliquer le moindre taux de survie des embryons et la durée
de vie réduite de la progéniture après intoxication à doses sublétales
du père.
Après injection de thallium dans des oeufs de poule on a constaté
chez les embryons, la présence d'effets tératogènes, une inhibition de
la croissance et des anomalies dans le développement osseux; toutefois
ces effets sont controversés chez les mammifères, même à des doses
toxiques pour la mère. On a mis en évidence le passage
transplacentaire du thallium mais les nombreuses souches de souris et
de rats n'ont pour ainsi dire pas présenté d'effets tératogènes.
Deux épreuves de mutagénicité microbiologique effectuées sur
Salmonella typhimurium ont donné des résultats négatifs et les
résultats de la recherche in vivo d'échanges entre chromatides
soeurs demeurent controversés. Toutefois, selon certaines études, on
aurait observé des aberrations chromosomiques et une augmentation
sensible des brèches dans l'ADN monocaténaire.
On manque d'études à long terme sur la cancérogénicité du
thallium.
7. Effets sur l'homme
Etant donné que les sels de thallium sont inodores, incolores et
sans saveur, leur forte toxicité et la facilité avec laquelle on
pouvait s'en procurer naguère - et encore maintenant dans certains
pays en développement - ont fait qu'on les a souvent utilisés à des
fins de suicide, d'homicide, de tentatives d'avortement illicites avec
pour résultat, dans ce cas particulier, des intoxications aiguës.
D'ailleurs on considère que les intoxications par la thallium sont une
des causes les plus fréquentes, à l'échelle mondiale, des
empoisonnements volontaires ou accidentels chez l'homme. Ce que l'on
sait des intoxications chroniques par le thallium se limite aux
intoxications professionnelles, aux groupes de population vivant dans
des zones contaminées et aux cas d'homicide par absorption de
nombreuses petites doses.
Les symptômes de l'intoxication aiguë par le thallium dépendent
de l'âge, de la voie d'administration et de la dose. Les doses qui se
sont révélées mortelles varient entre 6 et 40 mg/kg, les valeurs
moyennes se situant entre 10 et 15 mg/kg. Si un traitement n'est pas
institué, cette dose moyenne entraîne généralement la mort en l'espace
de 10 à 12 jours, mais on a également connaissance de cas où la mort
est survenue en l'espace de 8 à 10 heures.
La triade gastro-entérite, polynévrite et alopécie est considérée
comme le symptôme classique de l'intoxication par le thallium, mais il
est arrivé qu'on n'observe ni gastroentérite ni alopécie. Il y a
également d'autres symptômes qui varient dans leur séquence, leur
ampleur et leur intensité.
Les symptômes de l'intoxication par le thallium sont souvent
diffus et commencent par de l'anorexie, des nausées, des vomissements,
une saveur métallique, de la salivation, des douleurs rétro-sternales
et abdominales et quelquefois des hémorragies gastro-intestinales
(selles sanglantes). On observe ensuite fréquemment une constipation
qui peut être rebelle au traitement et gêner par conséquent l'action
de l'antidote. Au bout de 2 à 5 jours, les troubles caractéristiques
de l'intoxication par le thallium apparaissent peu à peu, quelle soit
la voie d'exposition. Les effets sur le système nerveux central et
périphérique sont variables mais ce que l'on constate de manière
caractéristique et systématique chez les intoxiqués par le thallium,
c'est une hypersensibilité des jambes, à laquelle font suite le
syndrome des "pieds brûlants" et une paresthésie. L'atteinte du
système nerveux central (SNC) se traduit par des symptômes tels
qu'hallucinations, léthargie, délire, convulsions et coma. Les
symptômes circulatoires couramment rencontrés sont une hypertension,
une tachycardie et dans les cas graves, une défaillance cardiaque. Au
bout de la deuxième semaine, les cheveux commencent à tomber et
quelquefois même les poils du corps. La dystrophie unguéale se
manifeste par l'apparition de lunules blanches (lignes de Mee) 3 à 4
semaines après l'intoxication. Les zones noires que l'on trouve dans
la papille pileuse ne sont pas dues à des dépôts de pigment ou de
thallium mais à la présence de petites quantités d'air qui pénètrent
dans la tige du poil.
Dans les cas mortels, la mort peut survenir quelques heures à
plusieurs semaines après l'intoxication, mais la plupart du temps le
décès intervient dans les 10 à 12 jours. La cause du décès est
principalement due à une insuffisance rénale, neurologique et
cardiaque.
En cas d'intoxication sublétale, la guérison peut souvent prendre
des mois. Des séquelles peuvent subsister: problèmes neurologiques et
troubles mentaux, anomalies électroencéphalographiques et cécité. En
outre, il semble que les fonctions intellectuelles des survivants
soient affectées.
En cas d'intoxication chronique, les symptômes sont analogues
mais généralement moins prononcés qu'en cas d'intoxication aiguë. La
cécité peut quelquefois être permanente. La guérison complète prend
des mois et il peut y avoir des rechutes.
A l'occasion d'un incident dans une cimenterie de Lengerich en
Allemagne où il y avait eu émission de thallium et que l'on a bien
étudiée, on a constaté que la concentration du thallium dans le
système pileux et les urines des personnes exposées n'était pas
corrélée avec certaines caractéristiques généralement associées à
l'intoxication chronique, mais seulement avec des symptômes
neurologiques subjectifs.
L'examen de biopsies et de pièces d'autopsie après intoxication
par le thallium révèle des lésions au niveau de divers organes. Par
exemple, après l'ingestion de doses mortelles, on constate des
hémorragies de la muqueuse intestinale, des poumons, des glandes
endocrines et du coeur, des infiltrations graisseuses au niveau du
foie et du myocarde, ainsi que des altérations dégénératives des
glomérules et des tubules rénaux. Dans l'encéphale, on peut observer
une dégénérescence graisseuse des cellules glanglionnaires, des
lésions au niveau des axones et la désagrégation des gaines de
myéline.
L'action directe du thallium sur le système nerveux autonome peut
produire des variations dans la tension artérielle. L'intoxication
par le thallium provoque une névrite symétrique périphérique. Les
nerfs distaux sont davantage touchés que les nerfs proximaux et les
lésions, bien que plus précoces, sont moins prononcées dans le cas des
nerfs dont l'axone est court, par exemple les nerfs crâniens. Les
axones sont enflés et ils présentent des vacuoles et des mitochondries
distendues. Dans les cas d'intoxication mortelle, on a observé de
graves lésions du nerf vague, l'énervation du sinus carotidien et des
lésions au niveau des ganglions sympathiques. Dans les cas
d'intoxication sublétale, les nerfs atteints peuvent subir une
dégénérescence de l'axone qui peut être définitive ou s'améliorer
partiellement dans les 2 ans.
Une névrite optique rétrobulbaire entraînant des troubles visuels
peut s'installer et persister pendant des mois après un traitement par
une crème dépilatoire à base de thallium; il peut même y avoir une
atrophie du nerf optique.
On n'a guère de données concernant les effets du thallium sur la
reproduction humaine. Il peut y avoir des effets indésirables sur le
cycle menstruel, la libido et la puissance sexuelle masculine. On
sait en outre qu'une intoxication chronique peut produire des effets
sur les spermatozoïdes. Comme chez l'animal, il y a passage
transplacentaire; on l'a observé dans un cas d'avortement provoqué par
le thallium. Toutefois, on connaît environ 20 cas d'intoxication par
le thallium en cours de grossesse où, à part le fait que les
nouveaux-nés avaient un poids de naissance relativement faible et
présentaient une alopécie, leur développement foetal ne paraissait pas
avoir souffert.
On ne sait rien des effets cancérogènes et on ne possède aucune
donnée sur les effets immunologiques du thallium. On ne possède pas
non plus d'éléments suffisants en faveur d'effets génotoxiques
éventuels.
Le traitement des intoxications par le thallium associe la
diurèse provoquée, l'administration de charbon actif et la prévention
de la réabsorption par le côlon au moyen d'administration de bleu de
Prusse (hexacyanoferrate(II) de potassium et de fer(II)).
8. Relation dose-réponse chez l'homme
Dans les populations non exposées, la concentration urinaire
moyenne du thallium est de 0,3 à 0,4 µg/litre. Etant donné que le
thallium a une courte demi-vie biologique, exprimée en jours, si l'on
suppose réunies les conditions d'un état stationnaire, la
concentration urinaire peut être considérée comme un indicateur de la
dose totale après absorption de thallium par inhalation ou ingestion.
On a constaté, dans un échantillon d'une population vivant à
proximité d'une source d'émission de thallium dans l'atmosphère, une
concentration urinaire moyenne de 5,2 µg/litre. L'existence d'une
relation dose-réponse nette a été observée entre la concentration
urinaire de thallium et la prévalence des effets suivants: fatigue,
faiblesse, troubles du sommeil, migraines, nervosité, paresthésie,
douleurs musculaires et articulaires. On a également fait état d'une
relation dose-réponse analogue en utilisant la teneur des cheveux en
thallium comme indicateur de l'exposition.
Le Groupe de travail a estimé qu'une exposition entraînant une
concentration urinaire de thallium inférieure à 5 µg/litre n'est
probablement pas susceptible de causer des effets nocifs. Dans
l'intervalle 5-500 µg/litre, l'ampleur du risque et la gravité des
effets nocifs restent incertaines, mais une exposition entraînant une
concentration urinaire supérieure à 500 µg/litre correspond à une
intoxication avec des manifestations cliniques.
9. Effets sur les autres êtres vivants au laboratoire et dans leur
milieu naturel
Le thallium est nocif pour tous les organismes mais il existe des
différences évidentes entre les espèces, voire entre les souches d'une
même espèce. La toxicité peut être différente selon qu'il s'agit de
dérivés minéraux du thallium(I) ou du thallium(III) ou encore de
composés organothalliens.
L'effet le plus important du thallium sur les microorganismes
semble consister dans l'inhibition de la nitrification par les
bactéries terricoles. Les résultats d'une étude consacrée à ce
phénomène tendent à indiquer que la structure de la communauté
biologique est perturbée lorsque la concentration de thallium dans le
sol se situe dans les limites de 1 à 10 mg/kg de poids sec; toutefois
on n'a pas pu déterminer sous quelle forme le thallium avait été
utilisé dans cette expérience.
Le thallium est fixé par toutes les parties de la plante, mais
principalement par les racines. Une fois qu'il a pénétré dans la
cellule, il se concentre de manière inégale dans le cytosol,
probablement en étant lié à un peptide. La concentration de thallium
que l'on rencontre dans les végétaux dépend des propriétés du sol
(plus spécialement du pH et de la teneur en argile et en matières
organiques), ainsi que du stade de développement et de la partie du
végétal. Il s'accumule dans les zones contenant de la chlorophylle
mais il existe des végétaux résistants au thallium où cette
accumulation est moindre. La présence de thallium réduit la
production d'oxygène, probablement par action directe sur les
transferts d'électrons au niveau du photosystème II. La présence de
chlorose traduit son effet nocif sur les pigments. En outre, il
semble que la toxicité du thallium soit associée à une moindre
fixation des oligo-éléments. Il y a également une action nocive sur
la croissance, les racines étant plus sensibles que les feuilles ou
les tiges. On a observé ces effets à des concentrations ne dépassant
pas un 1 mg de thallium par kg de tissu végétal sec, après exposition
à des dérivés du thallium(I).
Dans la plupart des études consacrées aux effets du thallium sur
les organismes aquatiques, on a utilisé des dérivés solubles du
thallium(I). La concentration de thallium la plus faible pour
laquelle on a observé des effets sur les espèces aquatiques est de
8 µg/litre, concentration qui a provoqué une réduction de la
croissance de certains végétaux aquatiques. En ce qui concerne les
invertébrés, les effets nocifs se font souvent sentir à des
concentrations plus faibles que chez les poissons (les valeurs de la
CL50 à 96 heures sont de 2,2 mg de thallium par litre pour les
daphnies et de 120 mg/litre pour une espèce de poisson d'eau douce).
La valeur la plus faible de la CL50 a été observée après une
exposition d'environ 40 jours; elle était dans le cas de poissons, de
40 µg/litre.
Les nombreux cas d'intoxication par le thallium observés dans la
faune sauvage sont dus à son utilisation à grande échelle comme
rodenticide. Chez les animaux granivores et chez les prédateurs,
c'est au niveau du système nerveux central ou des voies digestives que
se produisent les effets les plus graves. On a également observés de
tels effets chez des animaux de ferme. En outre, le thallium provoque
la chute des plumes dorsales chez les canards, une salivation au
niveau du nez et de la bouche chez les bovins, et une réduction de la
croissance chez les poulets, les poules pondeuses, les moutons et les
taureaux.
RESUMEN
1. Identidad, propiedades físicas y químicas, y métodos analíticos
El talio elemental es un metal blando y maleable de color blanco
azulado. Cuando se expone al aire húmedo o al agua, se produce
respectivamente una oxidación rápida de su superficie o la formación
del hidróxido correspondiente. Tiene dos estados de oxidación
importantes: talio(I) y talio(III). Los componentes monovalentes
(talosos) se comportan de manera análoga a los metales alcalinos, como
por ejemplo el potasio, mientras que los compuestos trivalentes
(tálicos) son menos básicos, parecidos al aluminio. A diferencia de
los compuestos inorgánicos en los que el ion talio(I) es más estable
en soluciones acuosas que el ion talio(III), este último es más
estable en compuestos orgánicos.
La determinación del talio en muestras del medio ambiente es algo
difícil, porque sus concentraciones son del orden de µg/kg o
inferiores. En general, cuando no se aplica una concentración
previamente establecida de talio, los límites de la determinación en
minerales, suelos y polvo son de unos 20 µg/kg, en soluciones acuosas
de 0,1 µg/litro y en materiales biológicos de unos pocos µg/kg.
La espectrometría de absorción atómica en horno de grafito es un
método analítico idóneo para aplicaciones en las que se necesita una
alta sensibilidad debido a las pequeñas cantidades de muestra con un
contenido de talio de unos pocos µg/kg. La espectrometría de masas
con dilución isotópica y la espectrometría de plasmamasa con
acoplamiento inductivo, posiblemente combinada con la dilución
isotópica, son métodos excelentes de determinación, con buena
precisión y exactitud, del orden de µg/kg.
2. Fuentes de exposición humana y ambiental
El talio está presente en el medio ambiente como consecuencia de
procesos naturales y procedente de fuentes debidas a actividades
humanas. Está muy extendido en la naturaleza y se encuentra sobre
todo en las menas de sulfuro de diversos metales pesados, aunque suele
estar en concentraciones bajas. Sólo hay unas pocas zonas con
concentraciones naturales de talio muy elevadas.
La producción industrial es muy pequeña (el consumo industrial en
todo el mundo en 1991 fue de 10-15 toneladas/año). El talio y sus
compuestos tienen una amplia variedad de aplicaciones industriales.
Ahora se ha limitado rigurosamente su uso como depilatorio humano y
como rodenticida e insecticida. Sus principales aplicaciones están en
las industrias eléctrica y electrónica y en la producción de vidrios
especiales. Otro campo importante de aplicación es el uso de
radioisótopos en medicina para la escintigrafía, así como el
diagnóstico de melanomas y el uso de compuestos de ariltalio(III) en
bioquímica.
Las pérdidas en el medio ambiente proceden sobre todo de la
fundición de minerales (depósitos de materiales de desecho y emisiones
a la atmósfera), las centrales eléctricas alimentadas por carbón, las
fábricas de ladrillos y de cemento (todas ellas con emisiones a la
atmósfera). Se calcula que los procesos industriales movilizan en
todo el mundo de 2000 a 5000 toneladas/año. Las emisiones de talio
debidas a procesos industriales varían mucho en función del tipo de
industria.
Las emisiones de las centrales eléctricas alimentadas por carbón
pueden contener una concentración de talio de 700 µg/m3 de aire de
salida y las de las fábricas de cemento hasta 2500 µg/m3. Esta
última cifra se puede reducir hasta < 25 µg/m3 mediante el uso de
otras materias primas y cambiando el proceso de producción. El talio
se volatiliza durante la combustión del carbón o la materia prima
utilizada en la fabricación de cemento y se vuelve a condensar sobre
la superficie de las partículas de ceniza en las partes más frías del
sistema. Estas partículas contienen hasta 50 mg de talio/kg de
polvillo de ceniza y son con frecuencia de pequeño tamaño, de manera
que los filtros de las fábricas de cemento retienen sólo un 50%.
Alrededor de un tercio de las partículas que emiten las centrales
eléctricas son también de un tamaño tan pequeño que se pueden
depositar en las vías respiratorias inferiores.
Los efluentes procedentes de los depósitos de decantación de
residuos mineros, con un contenido de hasta 1620 y 36 µg/litro,
produjeron en los ríos de vertido niveles elevados de 88 y 1 µg/litro,
respectivamente. En los estanques de agua de lluvia cercanos a una
fábrica de cemento se encontraron hasta 37 µg/litro. En el suelo se
han detectado concentraciones máximas de 60 mg/kg en zonas próximas a
materiales de desecho de minas; en las cercanías de fundiciones de
metales no preciosos y de fábricas de ladrillos y de cemento se
detectaron concentraciones de 2, 0,6 y 27 mg/kg, respectivamente. En
las zonas contaminadas, la mayoría de las hortalizas, frutas y carne
contienen menos de 1 mg de talio/kg de peso fresco. Las
concentraciones son superiores en las coles (Brassicaceae), habiéndose
notificado niveles de hasta 45 mg/kg en la col rizada verde. Las
concentraciones de talio en los tejidos de los animales de granja se
corresponden con las concentraciones en el forraje. En las cercanías
de algunas fábricas de cemento, se han descrito niveles superiores en
el forraje (por ejemplo, hasta 1000 mg/kg en la colza) y en la carne
de vacuno y de conejo (hasta 1,5 y 5,8 mg/kg, respectivamente).
3. Transporte, distribución y transformación en el medio ambiente
Cerca de fuentes localizadas, como centrales eléctricas de
carbón, algunas fábricas de cemento y operaciones de fundición de
metales, la fuente principal de talio en el aire es la emisión de
polvillo de ceniza. Los resultados de un estudio indican que casi
todo el talio del polvo flotante procedente de una fábrica de cemento
estaba presente como cloruro de talio(I) soluble.
El destino final del talio que se incorpora al suelo (debido, por
ejemplo, al depósito del polvillo de ceniza) depende fundamentalmente
del tipo de suelo. La retención es máxima en suelos que contienen
grandes cantidades de arcilla, materia orgánica y óxidos de
hierro/manganeso. La incorporación de talio a complejos estables sólo
produce concentraciones más elevadas en las capas superiores del
suelo. La absorción del talio por la vegetación va aumentando a
medida que el pH del suelo disminuye. En algunos suelos fuertemente
ácidos se puede producir lixiviación de cantidades importantes de
talio al terreno y las aguas superficiales próximos.
La mayor parte del talio disuelto en agua dulce suele ser
monovalente. Sin embargo, en agua dulce muy oxidada y en la mayor
parte del agua marina puede predominar la forma trivalente. El talio
se puede eliminar de la columna de agua y acumularse en el sedimento
mediante diversas reacciones de intercambio, formación de complejos o
precipitación.
Aunque puede darse una bioconcentración del talio, la
bioamplificación del elemento en las redes alimentarias acuática o
terrestre es improbable.
4. Niveles medioambientales y exposición humana
En zonas no contaminadas por talio, las concentraciones en el
aire suelen ser < 1 ng/m3, en el agua < 1 µg/litro y en los
sedimentos del agua < 1 mg/kg. Las concentraciones medias en la
corteza terrestre oscilan entre 0,1 y 1,7 mg/kg, pero es posible
encontrar niveles muy elevados, por ejemplo hasta de 1000 mg/kg en el
carbón, y los minerales de talio que raramente se encuentran contienen
hasta un 60% del elemento. Los alimentos de origen vegetal y animal
suelen contener < 1 mg/kg de peso seco y la ingestión media humana de
talio con los alimentos parece ser inferior a 5 µg/día. Se estima que
la absorción a través del sistema respiratorio es < 0,005 µg de
talio/día.
Se dispone sólo de datos limitados sobre el contenido real de
talio en el aire de los lugares de trabajo. Las concentraciones
observadas más recientemente (decenio de 1980) fueron < 22 µg de
talio/m3 (en la producción de una aleación especial de talio y en
una fundición de talio). El promedio de la concentración determinada
en orina fue del orden de 0,3-8 µg/litro en los trabajadores del
cemento y de 0,3-10,5 µg/litro en los de funderías.
5. Cinética y metabolismo en animales de laboratorio y en el ser
humano
El talio se absorbe con rapidez y facilidad a través de los
tractos gastrointestinal y respiratorio, así como por vía cutánea. Se
distribuye en poco tiempo por todos los órganos y atraviesa la
placenta (como se demuestra por la rápida absorción fetal) y la
barrera hematoencefálica. Debido a su acumulación rápida en las
células, las concentraciones de talio en la sangre no se corresponden
con su nivel en los tejidos. En casos de intoxicación aguda de
animales experimentales o de personas, se producen al principio
concentraciones de talio elevadas en el riñón, bajas en el tejido
adiposo y en el cerebro e intermedias en los demás órganos; luego
aumentan también sus niveles en el cerebro.
La eliminación del talio se puede producir a través del tracto
gastrointestinal (básicamente mediante mecanismos independientes de la
excreción biliar), el riñón, el pelo, la piel, el sudor y la leche
materna. Se puede producir una reabsorción intestinal (sobre todo
desde el colon), con la consiguiente disminución en la eliminación
total del organismo. En la rata, las principales vías de eliminación
del talio son la gastrointestinal (unos dos tercios) y la renal
(alrededor de un tercio), siendo semejante la contribución de ambas
vías en el caso de los conejos. El talio se elimina también por la
saliva.
Al igual que con otras muchas sustancias, la excreción de talio
en el ser humano difiere de la observada en los animales de
laboratorio; en aquél la velocidad de excreción es mucho más baja
(constante de velocidad = 0,023-0,069 día-1) que en éstos (la
constante de velocidad media = 0,18 día-1). Otra diferencia
importante entre el hombre y los animales es la contribución relativa
de las distintas vías de excreción. La excreción renal parece ser
mucho más importante en el ser humano que en los animales, aunque no
se ha determinado completamente su contribución relativa a la
eliminación total del organismo, debido fundamentalmente a la falta de
suficientes datos respecto al hombre. Además, los niveles de
exposición, su duración, la alteración de la función de los órganos de
excreción, la absorción de potasio y el tratamiento correspondiente de
la intoxicación aguda pueden influir considerablemente en los
resultados.
En un estudio sobre la excreción renal de talio se notificó un
resultado de alrededor del 73%, mientras que a través del tracto
gastrointestinal fue de sólo el 3,7% de la cantidad diaria excretada.
La excreción estimada a través del pelo y la piel y del sudor fue del
19,5% y el 3,7%, respectivamente.
La semivida biológica del talio en animales de laboratorio oscila
generalmente entre 3 y 8 días; en el ser humano es de unos 10 días,
aunque se ha informado de valores superiores a los 30 días.
No se dispone de datos sobre su biotransformación.
6. Efectos en mamíferos de laboratorio y en sistemas de ensayo
in vitro
No hay diferencias específicas sorprendentes por especies en
cuanto a la toxicidad de las sales de talio(I). Normalmente, una
ingestión oral de 20 a 60 mg de talio/kg de peso corporal es letal en
un plazo de una semana. Los cobayos son ligeramente más sensibles que
otros animales de experimentación. El óxido tálico(III) insoluble en
agua muestra una toxicidad aguda algo más baja por vía oral o
parenteral que las sales de talio(I). Al comparar los datos de
toxicidad aguda se aprecia una elevada biodisponibilidad a partir de
todas las vías de exposición. Afecta a la mayor parte de los órganos,
pero los signos de intoxicación y la sucesión de los mismos indican
una cierta variabilidad intraespecífica e interespecífica.
Los síntomas de la intoxicación aguda se suceden en general de la
manera siguiente: en primer lugar anorexia, vómitos y depresión, más
tarde diarrea, cambios cutáneos (inflamación en los orificios
corporales, furúnculos cutáneos, pérdida de pelo) y luego disnea y
trastornos nerviosos. Por último, la insuficiencia respiratoria que
provoca la muerte.
Los síntomas de la intoxicación crónica son semejantes a los de
la intoxicación aguda. Se produce regularmente pérdida de pelo.
En el examen histológico se puede observar necrosis u otros daños
celulares. Se han detectado cambios necróticos en los riñones, el
hígado, el intestino, el corazón y el sistema nervioso. En numerosas
células se ha observado hinchazón de las mitocondrias y pérdida de
crestas, dilataciones del retículo endoplasmático liso, aumento del
número de vacuolas autofágicas y de gránulos de lipofucsina y pérdida
de microvellosidades. Las alteraciones de procesos funcionales
debidas al talio pueden estar provocadas por la rotura física de las
membranas de los orgánulos subcelulares. En el corazón, los efectos
arritmogénicos se limitan al nódulo sinoatrial.
La intoxicación por talio provoca la alteración selectiva de
determinados elementos de la conducta relacionados con efectos
bioquímicos (lo que indica un daño celular) en ciertas regiones
cerebrales. Algunos efectos neurológicos parecen deberse a la acción
directa, por ejemplo la ataxia y el temblor a causa de trastornos del
cerebelo o alteraciones de la actividad endocrina debidos a cambios en
el hipotálamo. El talio puede activar el sistema nervioso autónomo,
fundamentalmente el adrenérgico. En los nervios periféricos parece
interferir a nivel presináptico en la liberación espontánea del
transmisor, ejerciendo un efecto antagónico en estos procesos
dependientes del calcio.
No se conoce todavía el mecanismo exacto de la toxicidad del
talio. Se han propuesto varios mecanismos, que tal vez están
relacionados entre sí. Un aspecto importante de la intoxicación por
talio es el aumento significativo de la peroxidación de lípidos y de
la actividad de una enzima lisosómica, la ß-galactosidasa. El
resultado es una deficiencia de glutatión que provoca la acumulación
de peróxidos de lípidos en el cerebro y, al parecer, por último, la
formación de gránulos de lipofucsina. Parece que el mecanismo de
acción del talio radica fundamentalmente en una alteración de la
función mitocondrial.
Los animales con intoxicación crónica suelen presentar una
actividad sexual reducida, y en el sistema reproductor del macho son
evidentes los efectos gonadotóxicos del talio. En los testículos de
ratas que recibieron 10 mg de talio/litro de agua de beber durante 16
días, las células de Sertoli fueron las más sensibles y la descamación
del epitelio espermatogénico provocó la aparición de espermatozoides
inmaduros en el semen. Esto podría explicar el menor índice de
supervivencia de los embriones o la reducción del periodo de vida de
la descendencia tras una intoxicación subletal por talio de los
padres.
Tras la inyección de talio en huevos, se observaron en los
embriones de pollo efectos teratogénicos, inhibición del crecimiento y
trastornos del desarrollo óseo, pero en los mamíferos estos efectos
son discutibles, incluso a dosis tóxicas para la madre. Aunque se ha
demostrado que atraviesa la placenta, muchas estirpes de ratones y
ratas no muestran efectos teratogénicos en absoluto, o sólo
ligeramente.
Dos pruebas de mutagenicidad microbiológica en Salmonella
typhimurium dieron resultados negativos, y las pruebas in vivo sobre
intercambio de cromátides hermanas fueron controvertidas. Sin
embargo, en estudios aislados se han observado aberraciones
cromosómicas o un aumento significativo de la fragmentación del ADN de
cadena sencilla.
No se dispone de estudios de larga duración sobre la
carcinogenicidad del talio.
7. Efectos en el ser humano
Debido a que las sales de talio son insípidas, inodoras,
incoloras, muy tóxicas, fáciles de obtener en el pasado e incluso
ahora en algunos países en desarrollo, este elemento se ha utilizado a
menudo con fines suicidas, homicidas y de aborto ilegal, provocando
intoxicación aguda. Es más, se considera que la intoxicación por
talio es una de las causas más frecuentes, a escala mundial, de
intoxicación humana voluntaria o accidental. Los conocimientos sobre
la intoxicación crónica se limitan a la exposición profesional, a
grupos de población que viven en zonas contaminadas y a casos de
homicidio con dosis bajas múltiples.
Los síntomas de toxicidad aguda del talio dependen de la edad, la
vía de administración y la dosis. Las dosis que han resultado letales
varían entre 6 y 40 mg/kg, con un promedio de 10 a 15 mg/kg. Sin
tratamiento, esta dosis media suele producir la muerte en un plazo de
10 a 12 días, pero también se han descrito casos de defunción en 8-10
horas.
Se considera que la gastroenteritis, la polineuropatía y la
alopecia son los tres síntomas clásicos de la intoxicación por talio,
pero en algunos casos no se observó gastroenteritis ni alopecia.
También se producen otros signos y síntomas, con un orden de
aparición, amplitud e intensidad variables.
Los síntomas de la intoxicación son a menudo imprecisos y
consisten al principio en anorexia, náuseas, vómitos, sabor metálico,
salivación, dolor retrosternal y abdominal y a veces hemorragia
gastrointestinal (sangre en heces). Luego se suele observar
estreñimiento, que puede ser resistente al tratamiento, interfiriendo
así con el antídoto administrado.
Después de un periodo de dos a cinco días aparecen lentamente
algunos de los trastornos asociados normalmente al talio, con
independencia de la vía de exposición. Aunque los efectos en el
sistema nervioso central y periférico varían, un rasgo constante y
característico de la intoxicación por talio en el hombre es la
sensibilidad extrema de las piernas, a la que sigue el «síndrome de
los pies urentes» y la parestesia. Su acción sobre el sistema
nervioso central (SNC) se refleja en síntomas tales como
alucinaciones, letargia, delirio, convulsiones y coma. Los síntomas
circulatorios normales son hipertensión, taquicardia y, en los casos
graves, insuficiencia cardiaca. Después de la segunda semana de la
intoxicación se suele producir pérdida del pelo y, a veces, del vello;
la distrofia de las uñas se detecta por la aparición de rayas
semicirculares blancas (líneas de Mee) tres o cuatro semanas después
de la intoxicación. Las regiones negras que se observan en las
papilas pilosas no se producen por la deposición de pigmentos o de
talio, sino que se deben a pequeñas cantidades de aire que entran en
el tallo piloso.
En los casos letales, la muerte sobreviene en un plazo que oscila
entre unas horas y varias semanas, pero normalmente se produce a los
10 ó 12 días. Las causas del fallecimiento son generalmente
insuficiencia renal, del SNC y cardiaca.
En intoxicaciones subletales, la recuperación requiere con
frecuencia meses. A veces persisten los trastornos neurológicos y
mentales, así como las anomalías electroencefalográficas y la ceguera.
Por otra parte, parece ser que los supervivientes sufren un deterioro
de las funciones intelectuales.
En casos de intoxicación crónica los síntomas son semejantes,
pero en general más leves que en la intoxicación aguda. A veces se
produce ceguera permanente. La recuperación completa requiere meses y
se puede interrumpir por recaídas.
En un caso bien investigado de emisión de talio alrededor de una
fábrica de cemento de Lengerich, Alemania, las concentraciones de
talio en el pelo y la orina de las personas expuestas no se
correspondían con algunas características típicas que suelen estar
relacionadas con la intoxicación crónica por talio, sino sólo con
síntomas neurológicos subjetivos.
La autopsias y biopsias realizadas tras las intoxicaciones por
talio ponen de manifiesto daños en diversos órganos. Por ejemplo,
tras la ingestión de dosis letales se producen hemorragias en la
mucosa intestinal, los pulmones, las glándulas endocrinas y el
corazón, infiltraciones grasas en el hígado y el tejido cardiaco, así
como cambios degenerativos en los glomérulos y los túbulos renales.
En el cerebro se puede observar degeneración grasa de las células
ganglionares, lesiones axonales y desintegración de las vainas de
mielina.
Las variaciones de la presión sanguínea pueden deberse a los
efectos directos del talio en el sistema nervioso autónomo. La
intoxicación por talio produce neuropatía periférica mixta simétrica.
Los nervios distales sufren más daños que los proximales, y los
nervios de axón corto, por ejemplo los craneales, se ven afectados
antes, aunque en menor grado. Los axones se hinchan y contienen
vacuolas y mitocondrias dilatadas. En los casos de intoxicación
letal, se han observado daños graves del nervio vago, desnervación del
seno carotídeo y lesiones de los ganglios simpáticos. En la
intoxicación subletal, los nervios afectados pueden sufrir
degeneración axonal, con recuperación nula o sólo parcial en un plazo
de dos años.
A veces se produce una neuritis retrobulbar con los consiguientes
trastornos visuales, que puede persistir durante meses, después de un
tratamiento con productos depilatorios con talio; este trastorno puede
desembocar incluso en la atrofia óptica. Los datos sobre los efectos
del talio en la reproducción humana son limitados. Puede afectar
negativamente al ciclo menstrual, la libido y la potencia masculina.
Se sabe que la intoxicación crónica tiene efectos sobre el esperma.
Al igual que en los estudios con animales, se ha observado que
atraviesa la placenta; esto se ha puesto de manifiesto tras un aborto
inducido por el talio. Sin embargo, en unos 20 casos de intoxicación
por talio durante el embarazo no se vio afectado el desarrollo fetal,
salvo el peso relativamente bajo y la alopecia de los recién nacidos.
No se dispone de informes sobre efectos carcinógenos o datos
sobre efectos inmunológicos debidos al talio. No hay pruebas
suficientes de efectos genotóxicos.
El tratamiento de la intoxicación por talio combina la diuresis
forzada, el uso de carbón vegetal activado y la prevención de la
reabsorción en el colon mediante la administración de azul de Prusia,
hexacianoferrato(II) férrico potásico.
8. Relación dosis-respuesta en el ser humano
La concentración media de talio en orina en la población no
expuesta es de 0,3 a 0,4 µg/litro. Habida cuenta de que el talio
tiene una semivida biológica breve, establecida en días, y suponiendo
unas condiciones estables, se puede tomar esta concentración urinaria
como indicador de la dosis total tras la inhalación y la ingestión con
los alimentos.
La concentración media en la orina en una muestra de población
que vive cerca de una fuente de emisión de talio fue de 5,2 µg/litro.
Se encontró una relación dosis-respuesta clara entre las
concentraciones en la orina de talio y el predominio de cansancio,
debilidad, trastornos del sueño, dolor de cabeza, nerviosismo,
parestesia y dolor muscular y de las articulaciones. Se informó
asimismo de una relación dosis-respuesta semejante cuando se utilizó
el talio en el pelo como indicador de la exposición.
El Grupo Especial de Trabajo consideró que las exposiciones que
producen concentraciones de talio en la orina inferiores a 5 µg/litro
probablemente no son perjudiciales para la salud. En el margen de 5 a
500 µg/litro, la magnitud del riesgo y la gravedad de los efectos
adversos son inciertas, mientras que la exposición que da lugar a más
de 500 µg/litro está asociada a una intoxicación clínica.
9. Efectos en otros organismos en el laboratorio y en el medio
ambiente
El talio afecta a todos los organismos, pero hay diferencias
específicas de especies e incluso de variedades. Los diferentes
compuestos inorgánicos de talio(I) y talio(III), así como sus
compuestos orgánicos, pueden tener distinta toxicidad.
El efecto más importante del talio en los microorganismos parece
ser la inhibición de la nitrificación por las bacterias del suelo.
Los resultados de un estudio parecen indicar que la estructura de la
flora microbiana se altera a concentraciones en el suelo comprendidas
entre 1 y 10 mg/kg de peso seco, pero no se precisó la forma de talio
utilizada en este experimento.
Absorben talio todas las partes de las plantas, pero sobre todo
las raíces. Una vez que ha penetrado en las células, se concentra de
forma desigual en el citosol, probablemente unido a un péptido. Las
concentraciones de talio que se observan en las plantas dependen de
las propiedades del suelo (en particular el pH y el contenido de
arcilla y materia orgánica), así como de la fase de desarrollo y de la
parte de la planta. Se acumula en las zonas que contienen clorofila,
pero lo hace en menor grado en las plantas resistentes al talio.
Reduce la producción de oxígeno, posiblemente por acción directa sobre
la transferencia de electrones en el fotosistema II. Su interferencia
con los pigmentos se pone de manifiesto por la aparición de clorosis.
Por otra parte, en el mecanismo de la toxicidad parece intervenir una
alteración de la absorción de oligoelementos. Afecta también al
crecimiento, siendo más sensibles las raíces que las hojas o los
tallos. Estos efectos se han descrito tras la exposición a formas
monovalentes de talio con niveles de sólo 1 mg/kg de tejido vegetal
seco.
En la mayoría de los estudios de los efectos en los organismos
acuáticos se han utilizado compuestos solubles de talio monovalente.
La concentración más baja notificada capaz de afectar a las especies
acuáticas es de 8 µg/litro, con una reducción del crecimiento de las
plantas. Los invertebrados se suelen ver afectados a concentraciones
más bajas que los peces (los valores de la CL50 en 96 horas son de
2,2 mg de talio/litro para los dáfnidos y de 120 mg/litro para un pez
de agua dulce). El valor más bajo de la CL50, notificado tras la
exposición durante unos 40 días, fue de 40 µg/litro para los peces.
Muchos casos de intoxicación por talio de la flora y fauna
silvestres se han debido a su aplicación en gran escala como
rodenticida. En animales que se alimentan de semillas y en
depredadores afecta gravemente sobre todo al SNC y al aparato
gastrointestinal. Estos mismos efectos se pueden observar en los
animales de granja. A esto hay que añadir que el talio provoca una
pérdida de plumas dorsales en los patos, salivación de la nariz y la
boca del ganado vacuno y reducción del crecimiento de los pollos de
asar, las gallinas ponedoras, las ovejas y los novillos.
See Also:
Toxicological Abbreviations
Thallium (ICSC)
Thallium (PIM 525)